6 分组分析结果
6.1 分组元数据统计 
对分组样本及其元数据进行统计。
结果文件是:group_compare.html
6.2 各分组OTU/ASVs韦恩图
OTU/ASVs比较韦恩图(样本数/分组数<=5个样本,若分组数大于5出花瓣图)
图解读:每个圈代表一个样本/分组,圈之间的重迭区域表示样本/分组间共有的 OTUs,每个区域的数字大小表示该区域对应的 OTUs 数目。
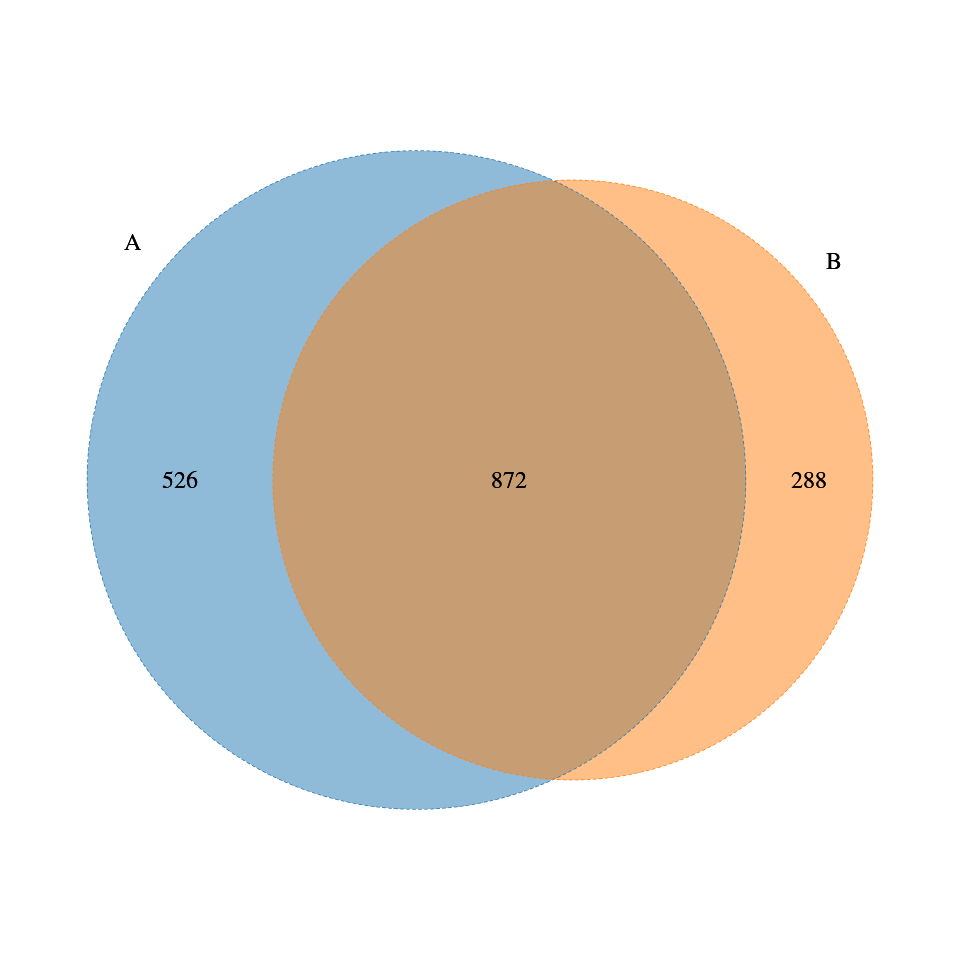 |
| Fig 6-2-1 各分组OTU/ASVs韦恩图 |
6.3 Alpha多样性结果
分组之间的alpha多样性指数使用非参数统计检验。
具体统计差异可见文件夹:/alphadiv/下
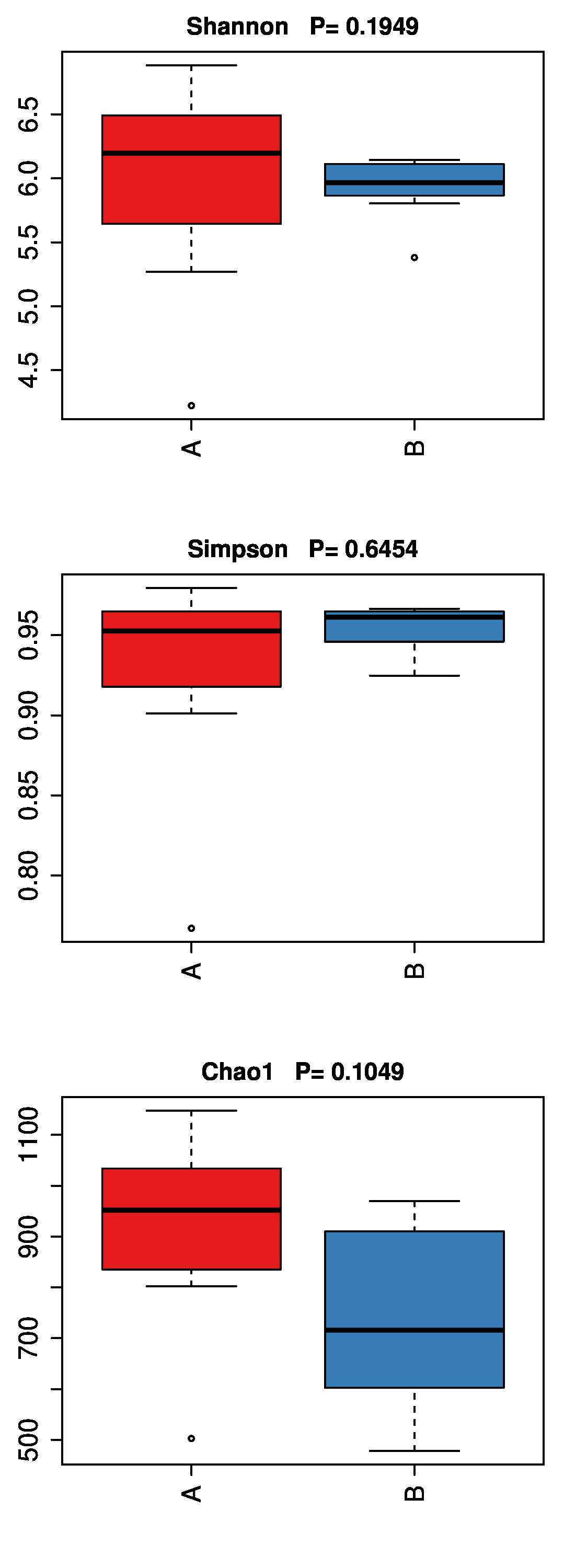 |
| Fig 6-3-1 Alpha多样性组间统计差异 |
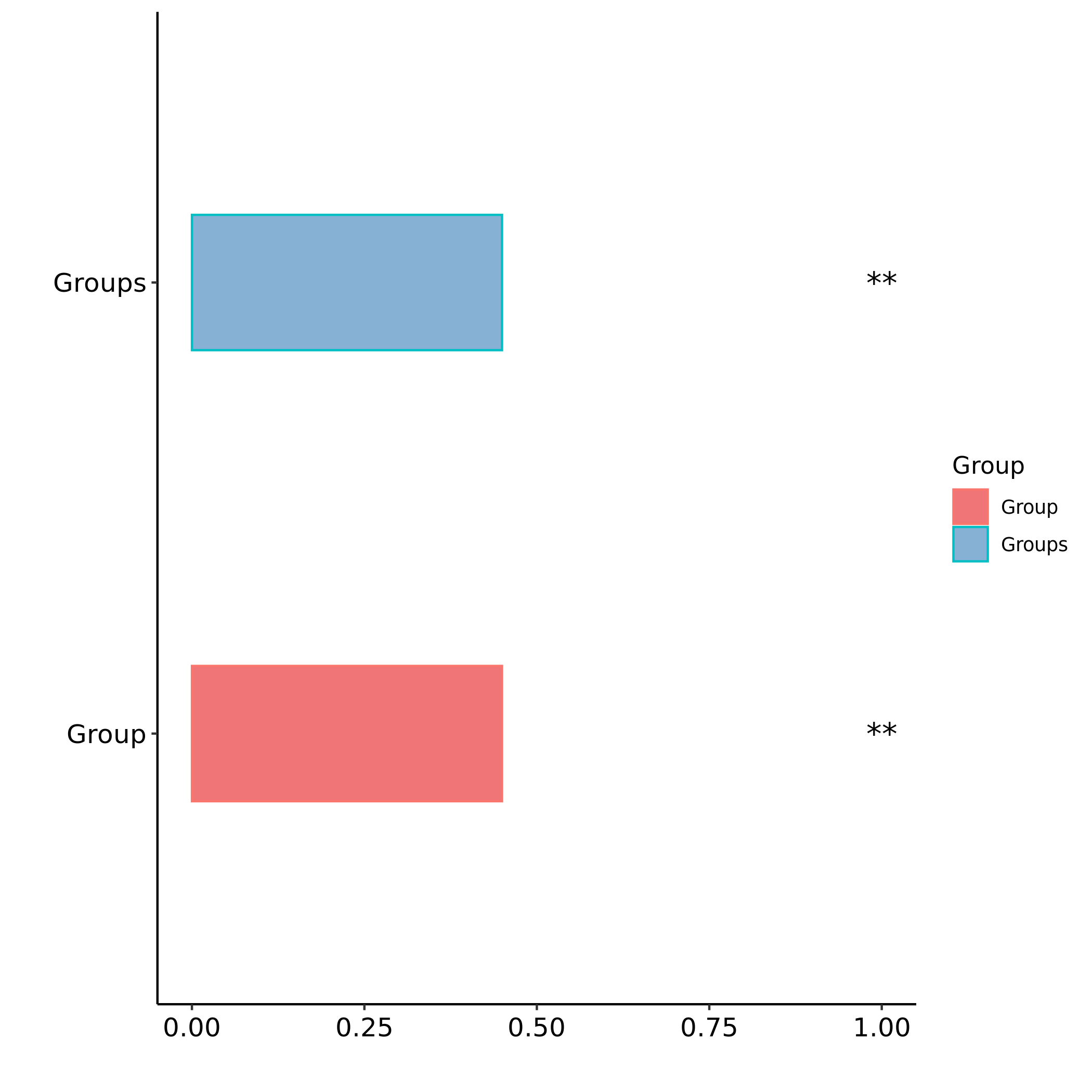
6.4 Beta多样性Anosim检验结果
Anosim分析是一种非参数检验,用来检验组间的差异是否显著大于组内差异,从而判断分组是否有意义。
图解读 :该方法主要有两个数值结果:R值,用于比较不同组间是否存在差异;P值,用于说明是否有显著差异。
R-value 介于(-1,1)之间,R-value > 0,说明组间差异大于组内差异。R-value < 0,说明组间差异小于组内差异,
R只是组间是否有差异的数值表示,并不提供显著性说明。统计分析的可信度用 P-value 表示,P< 0.05 表示统计
具有显著性。
对 Anosim 的分析结果,基于两两样本之间的距离值排序获得的秩(组间的为 between,组内的为 within),这样
任一两两组的比较可以获得三个分类的数据,并进行箱线图的展示(若两个箱的凹槽互不重迭,则表明它们的中
位数有显著差异)
做组间差异比较分析,分组内部至少要3个样本;若样本数不够或组间差异不明显则不生成该图
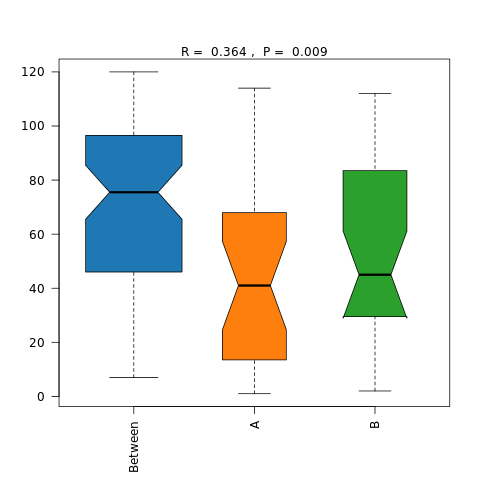 |
|
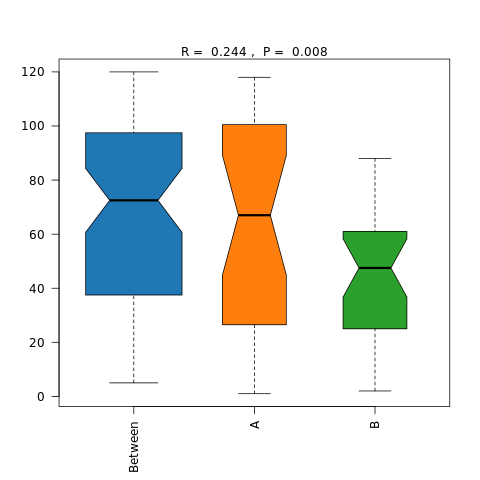 |
|
| Fig 6-4-1 非加权距离Anosim分析 |
|
Fig 6-4-2 加权距离Anosim分析 |
|
|
|
|
|
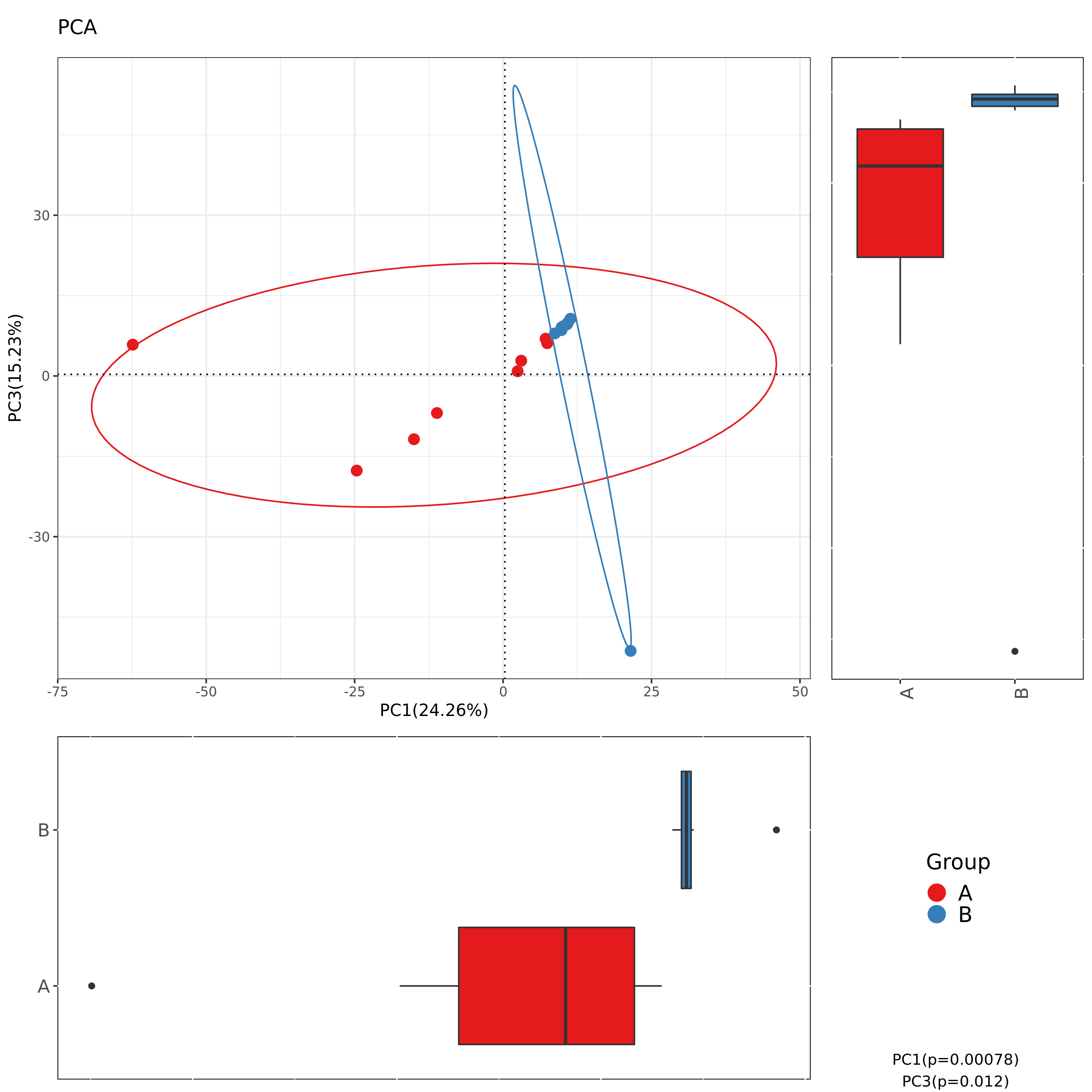
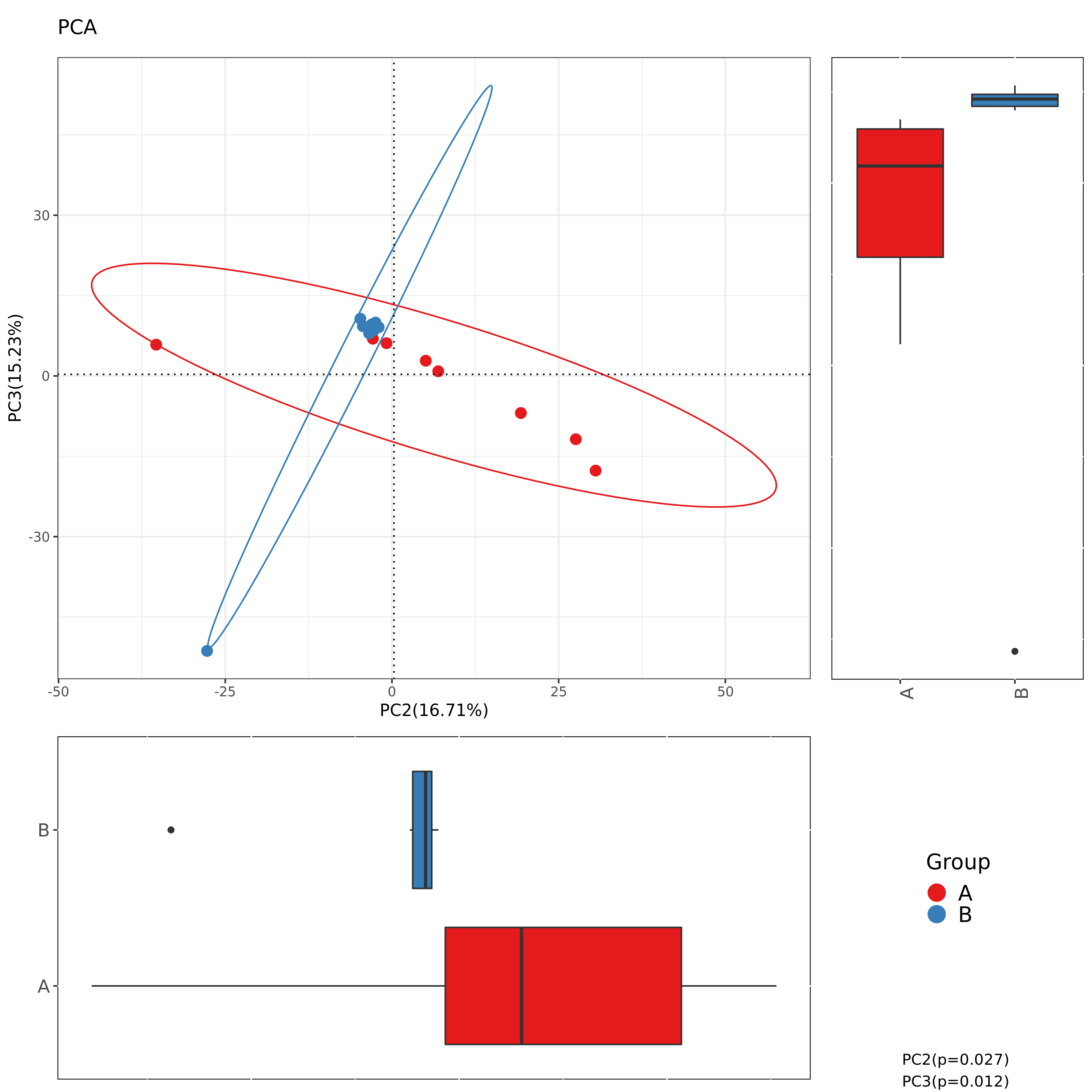
6.5 Beta多样性PCA结果
使用bray_curtis的PCA组间分布及差异
图解读:图中每一个点代表一个样本,相同颜色的点来自同一个分组。距离反应样本相似性
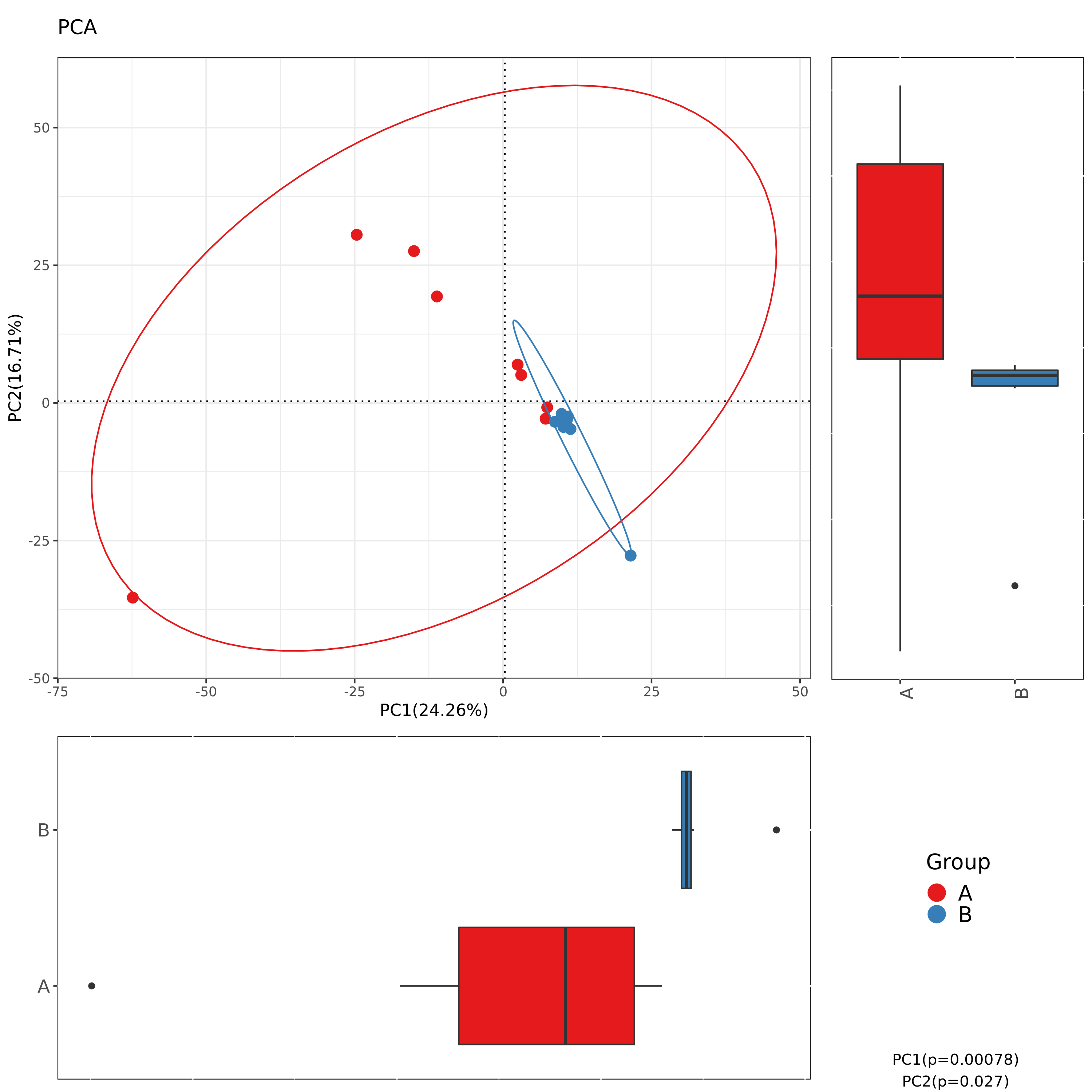 |
|
 |
|
 |
| Fig 6-5-1 bray_curtis的PCA组间分布及差异PC1-PC2 |
|
Fig 6-5-2 bray_curtis的PCA组间分布及差异PC1-PC3 |
|
Fig 6-5-3 bray_curtis的PCA组间分布及差异PC2-PC3 |
|
|
|
|
|
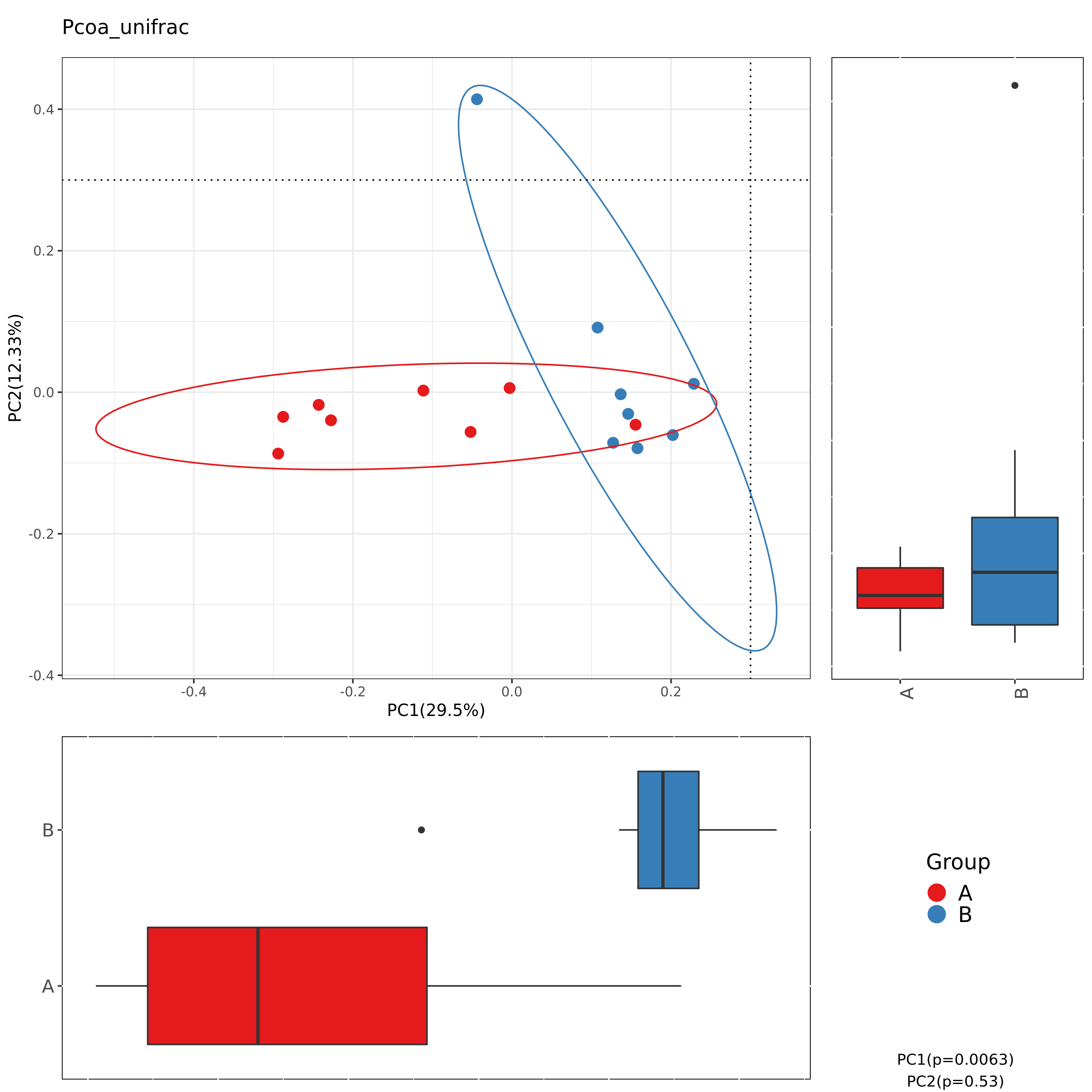
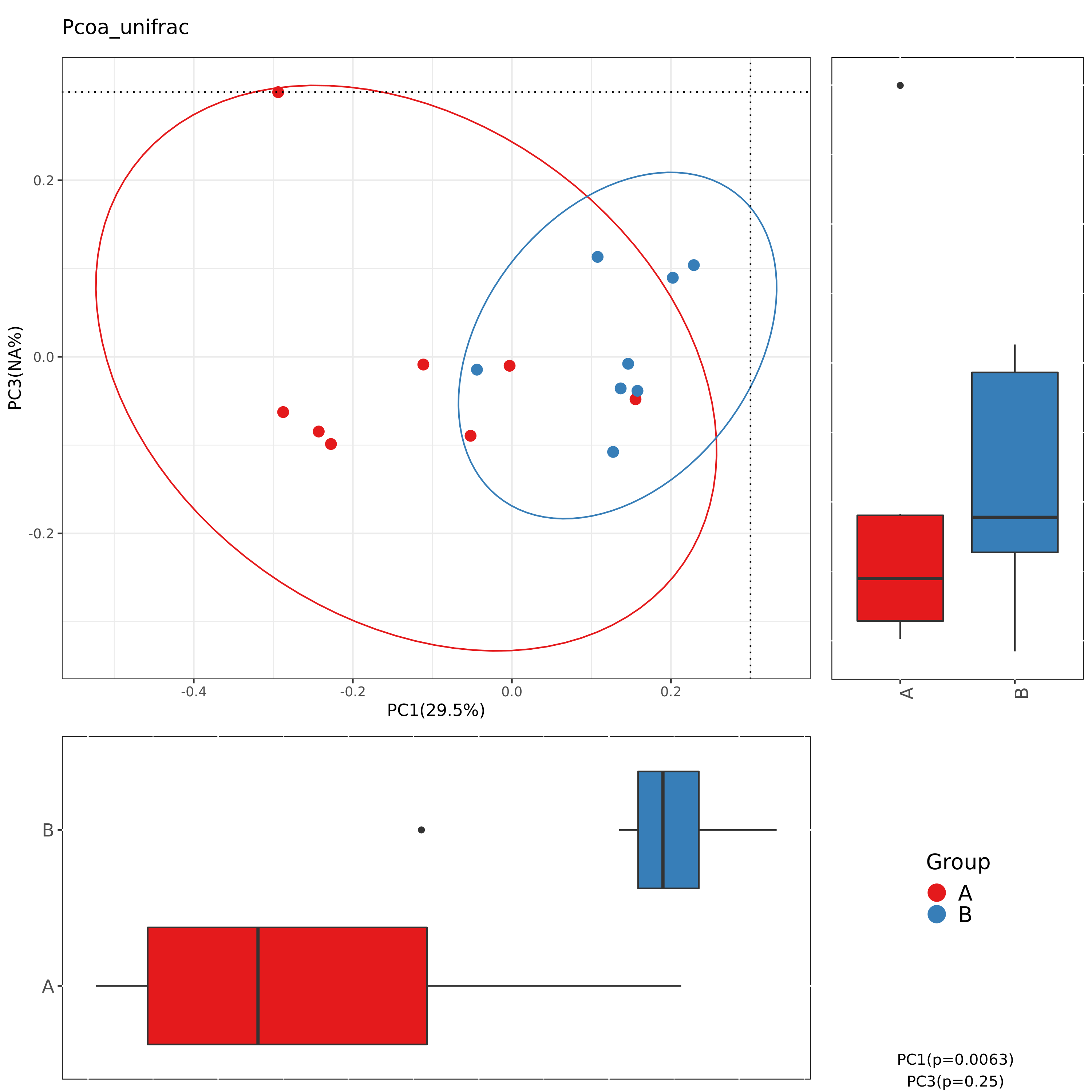
6.6 Beta多样性非加权PCoA结果
主坐标分析 PCoA (Principal component analysis)是一种研究资料相似性或差异性的可视化方法,通过一系列的特征值和特征向量排序后,通过一系列的特征值和特征向量进行排序后,选择主要排在前几位的特征值,PCoA 可以找到距离矩阵中最主要的坐标,结果是数据矩阵的一个旋转,它没有改变样品点之间的相互位置关系,只是改变了坐标系统。通过 PCoA 可以观察个体或群体间的差异。我们基于 Bray-Curtis 距离 Weighted Unifrac 距离 和Unweighted Unifrac 距离来进行 PCoA 分析。
使用unweighted_unifrac的PCoA组间分布及差异
图解读:图中每一个点代表一个样本,相同颜色的点来自同一个分组。距离反应样本相似性
 |
|
 |
|
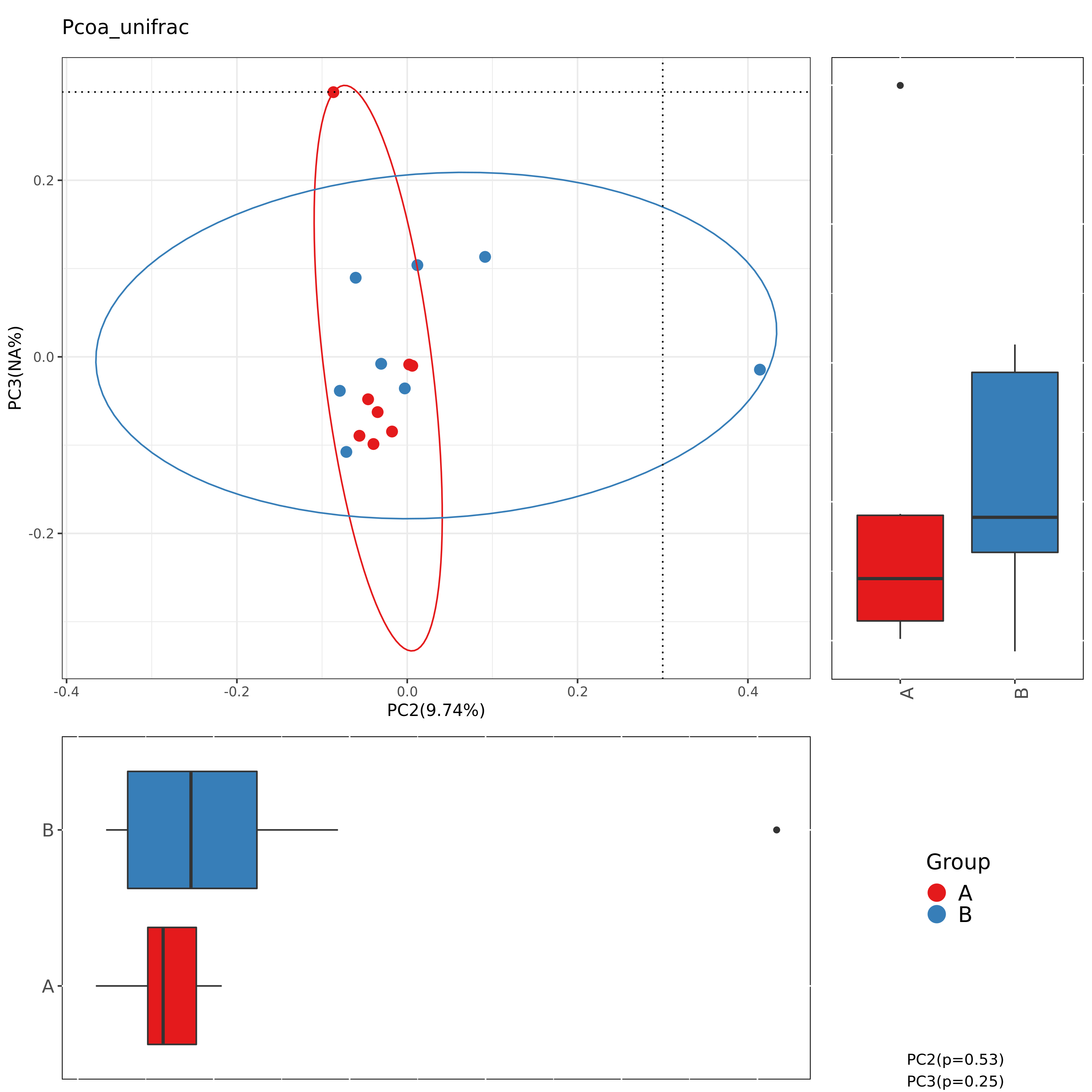 |
| Fig 6-6-1 unweighted_unifrac的PCoA组间分布及差异PC1-PC2 |
|
Fig 6-6-2 unweighted_unifrac的PCoA组间分布及差异PC1-PC3 |
|
Fig 6-6-3 unweighted_unifrac的PCoA组间分布及差异PC2-PC3 |
|
|
|
|
|
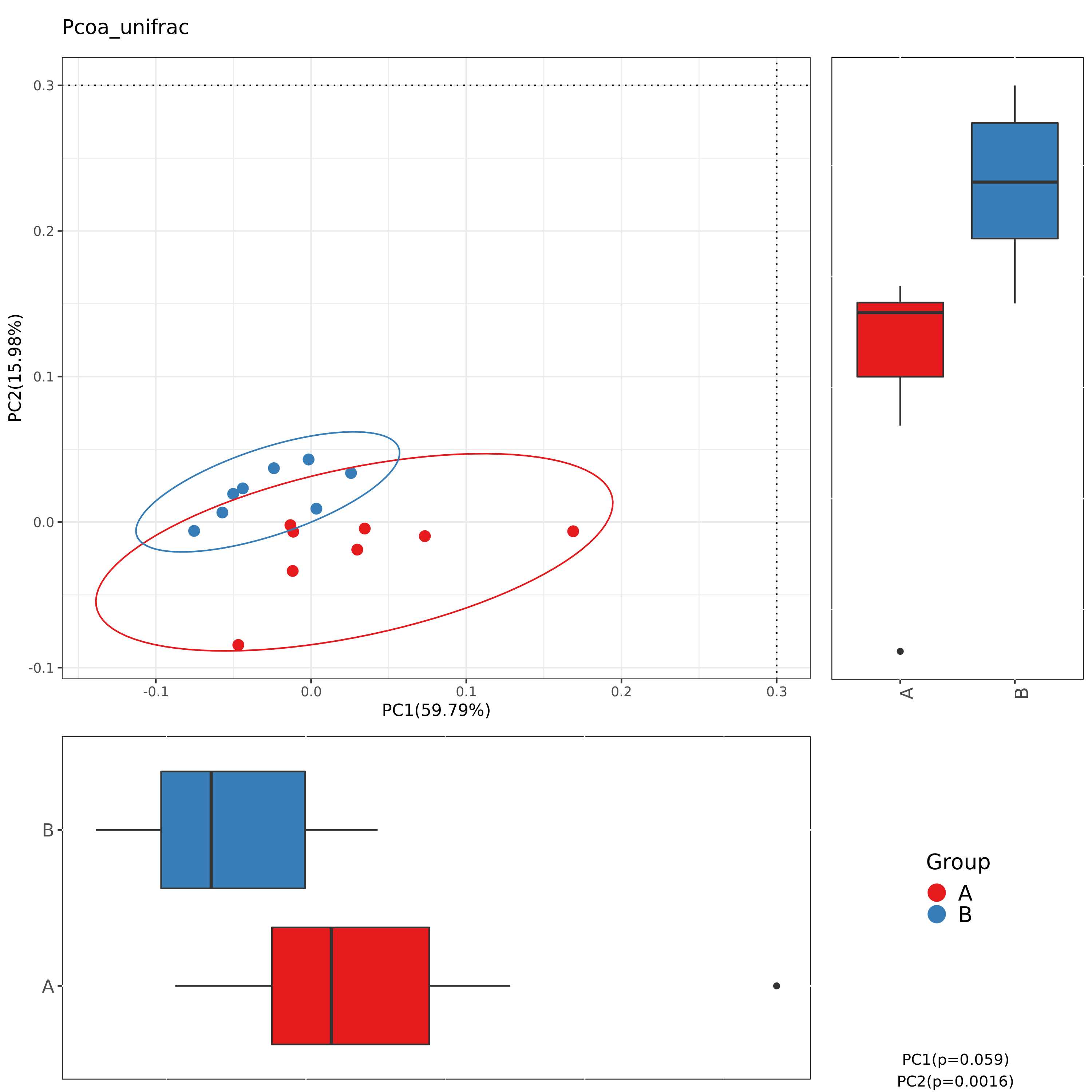
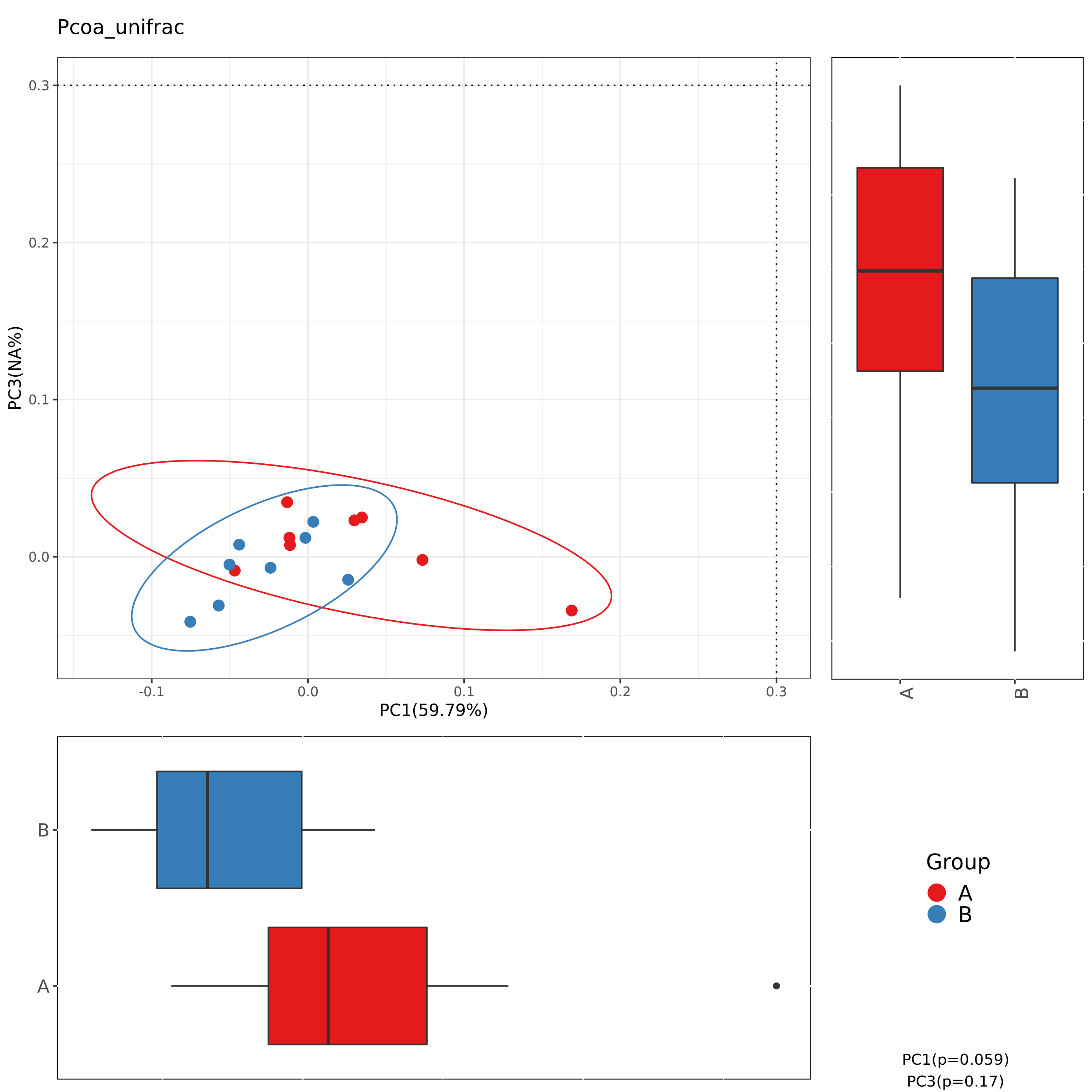
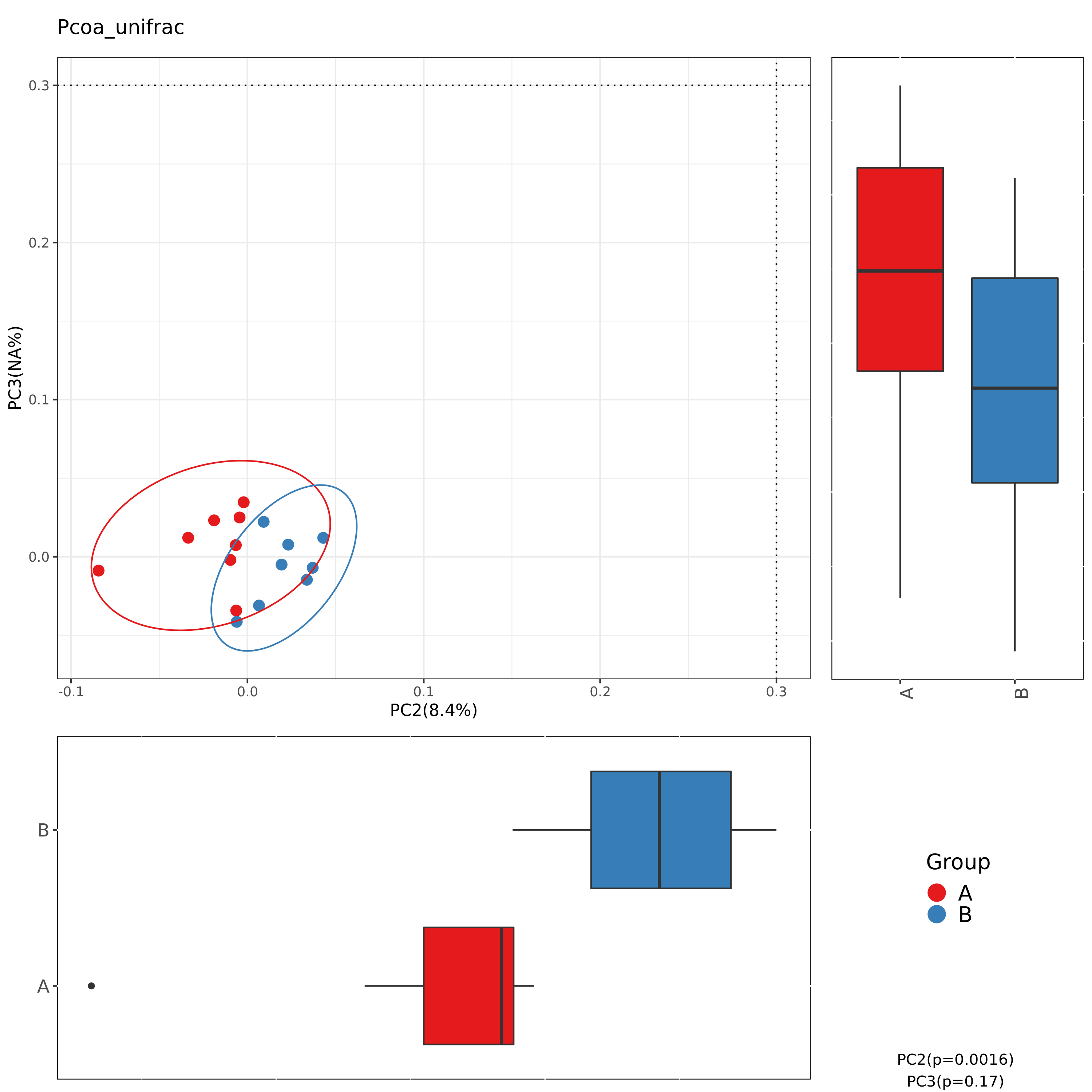
6.7 Beta多样性加权PCoA结果
使用weighted_unifrac的PCoA组间分布及差异
图解读:图中每一个点代表一个样本,相同颜色的点来自同一个分组。距离反应样本相似性
 |
|
 |
|
 |
| Fig 6-7-1 weighted_unifrac的PCoA组间分布及差异PC1-PC2 |
|
Fig 6-7-2 weighted_unifrac的PCoA组间分布及差异PC1-PC3 |
|
Fig 6-7-3 weighted_unifrac的PCoA组间分布及差异PC2-PC3 |
|
|
|
|
|
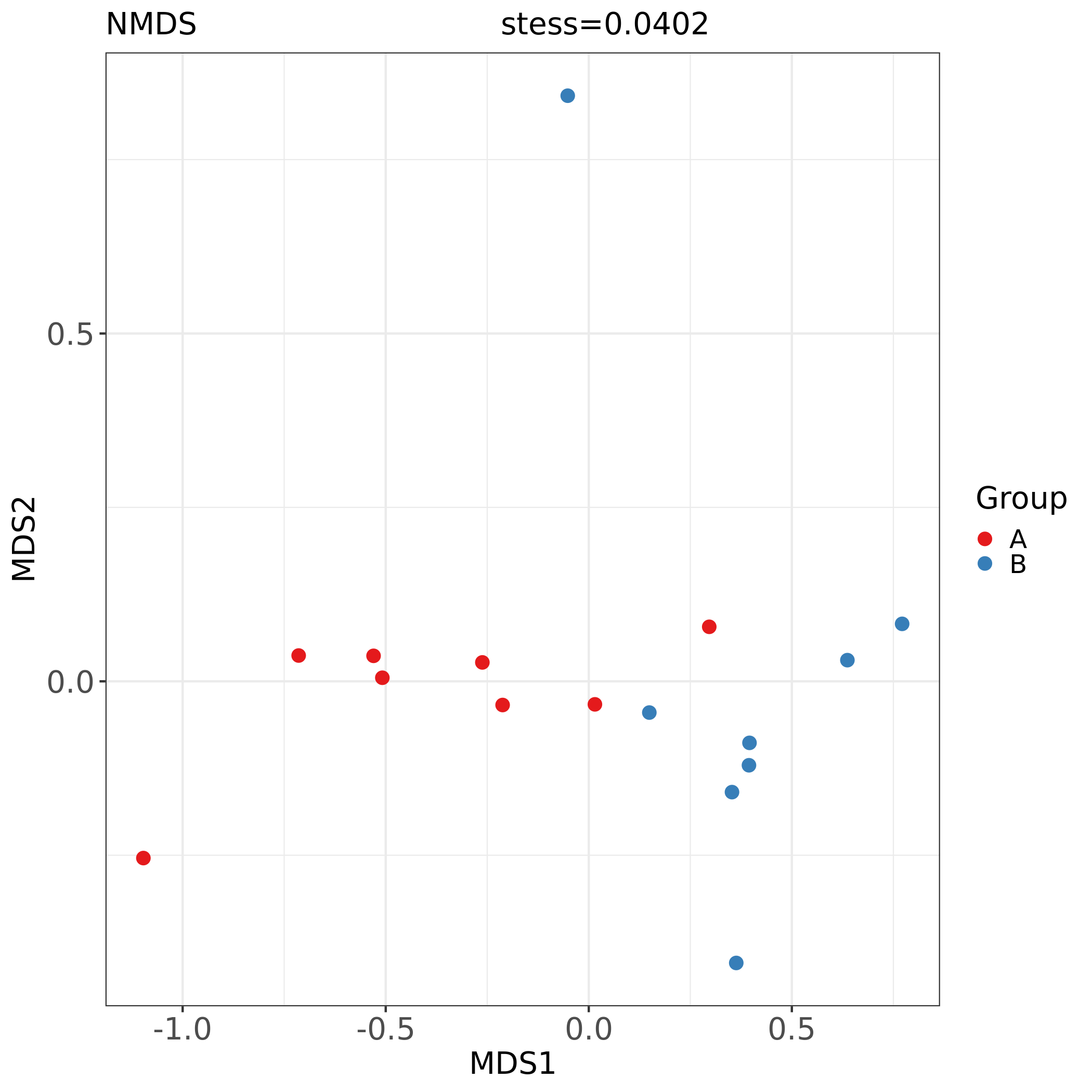
6.8 Beta多样性NMDS结果
NMDS组间分布及差异
非度量多维尺度分析 NMDS 分析(Nonmetric Multidimensional Scaling)与上述 PcoA 分析类似,也是一种基于样本距离矩阵的分析方法,通过降维处理展现样本特定的距离分布。与 PcoA 的区别是 NMDS 分析不依赖于特征根和特征向量的计算,而是通过对样本距离进行等级排序,使样本在低维空间中的排序尽可能符合彼此之间的距离远近关系(而非确切距离数值)。因此,NMDS 分析不受样本距离的数值影响,对于结构复杂的数据排序结果可能更稳定。
图解读:图中每一个点代表一个样本,相同颜色的点来自同一个分组。距离反应样本相似性。表头上的stress值是NMDS的应力函数值。在PCA、PCoA等排序分析中,对于每个排序轴,均可计算得到其对应的解释量,一般情况下我们可以根据解释量来评估排序结果是否适用;而对于NMDS分析,它的排序轴不存在解释量一说,但可以计算得到一个总的应力函数值,因此我们需要参考应力函数值来对排序结果进行评估。在PCA、PCoA等基于坐标轴解释量的评估中,解释量越高越好;而在NMDS排序分析中,尽可能选择较低的应力函数值。一般情况下,应力函数值的值不要大于0.2。
 |
|
 |
|
| Fig 6-8-1 NMDS间分布及差异 |
|
Fig 6-8-2 环境因子的Envfit统计检验 |
|
|
|
|
|
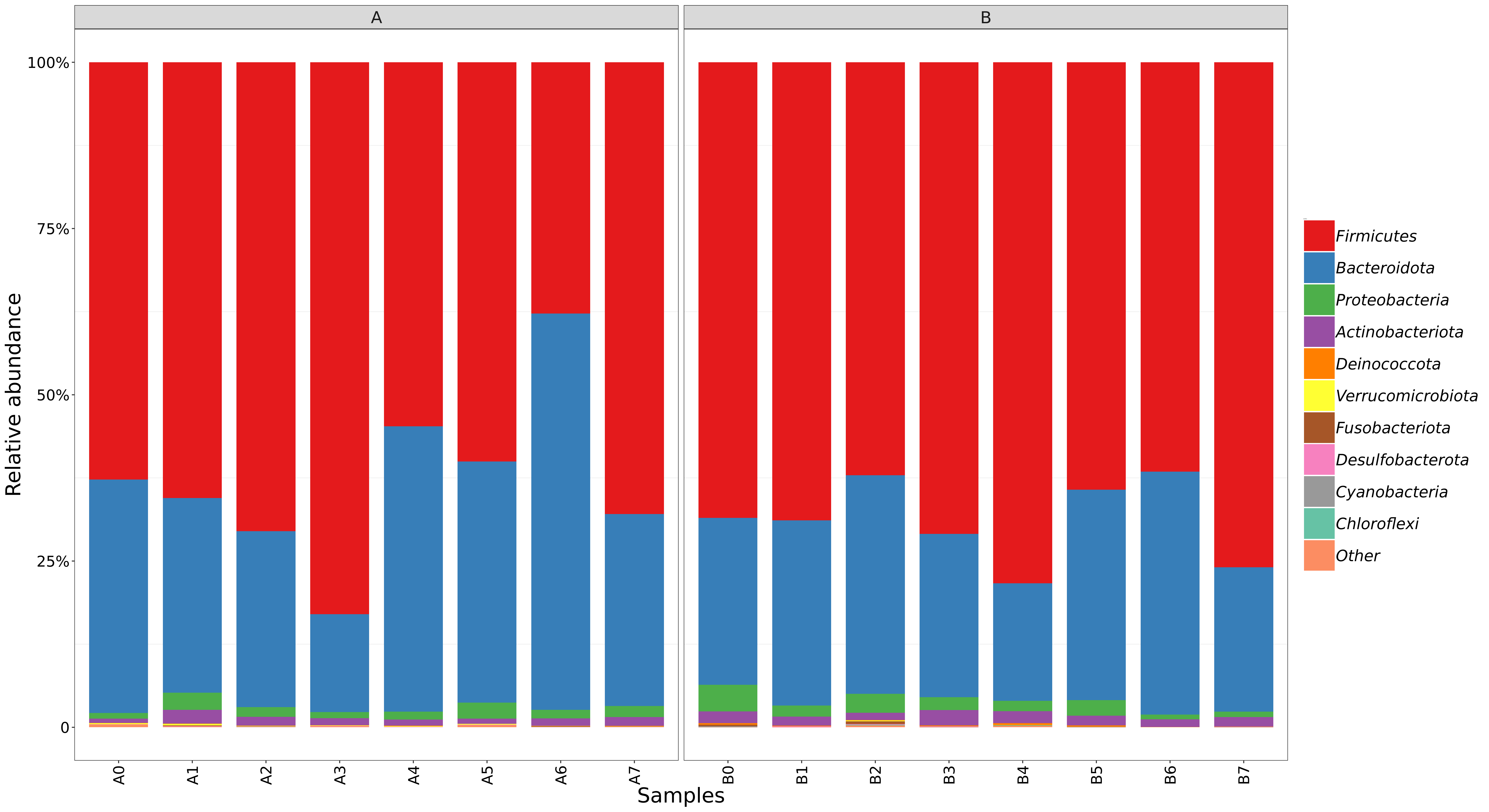
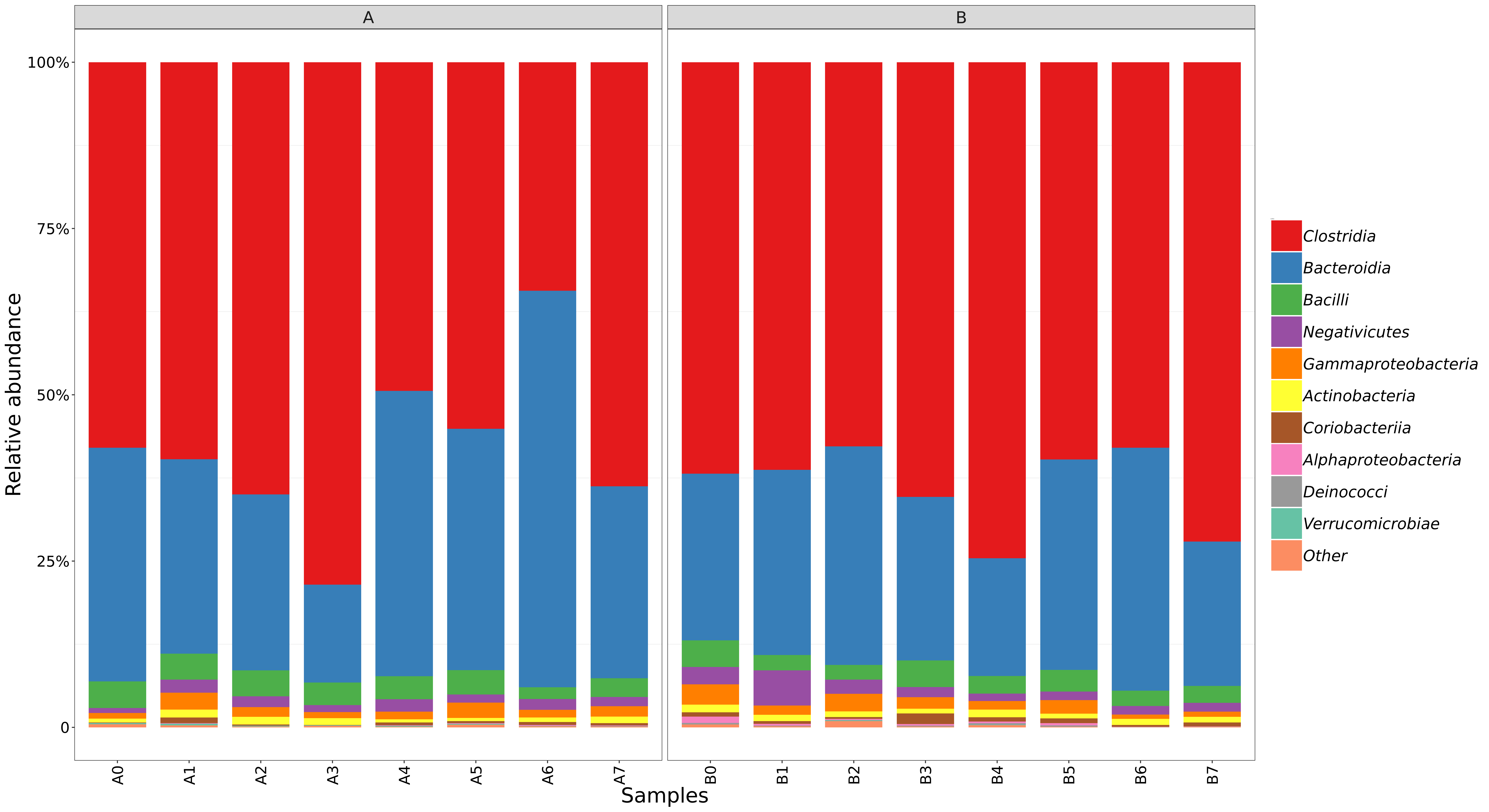
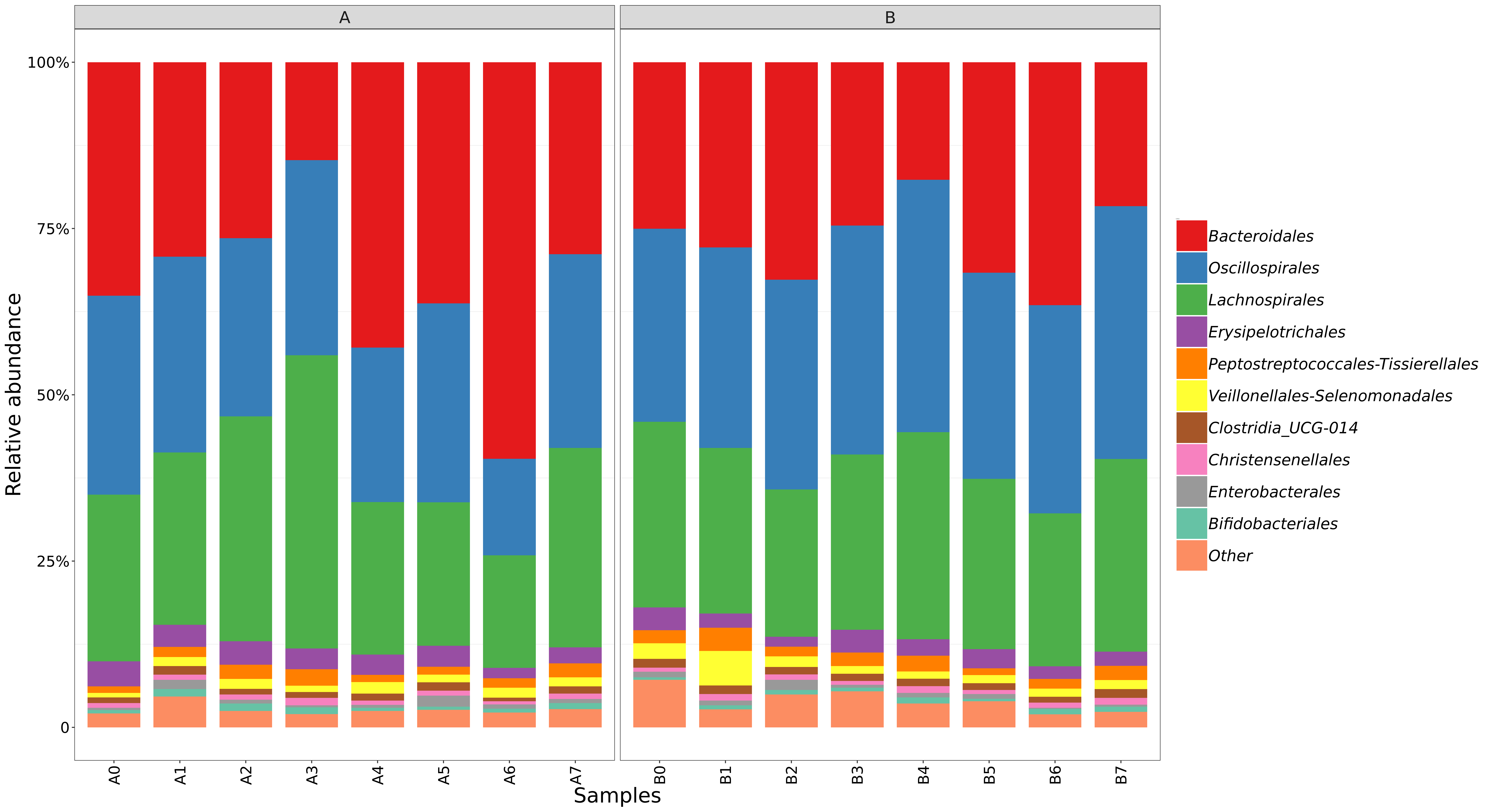
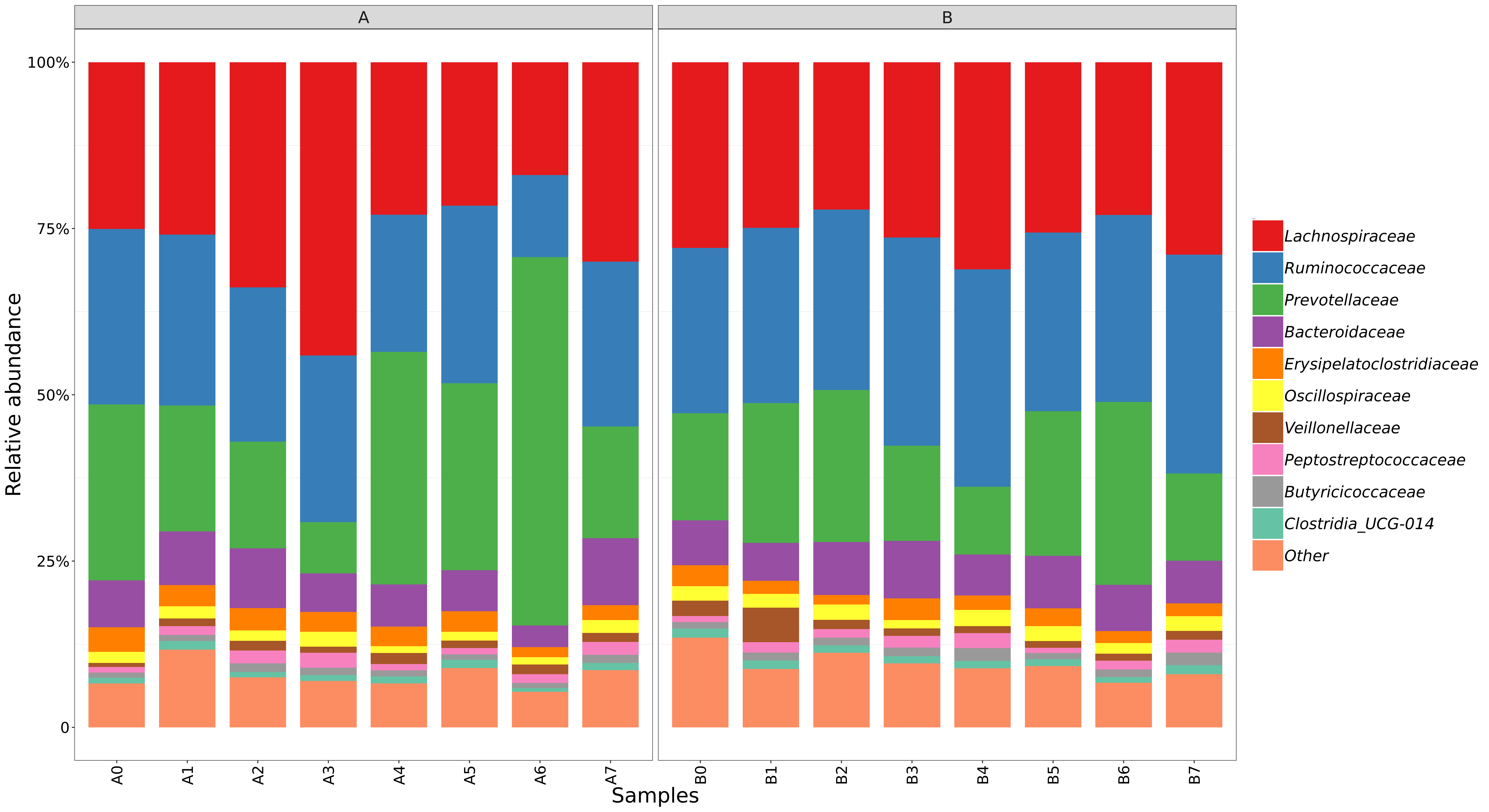
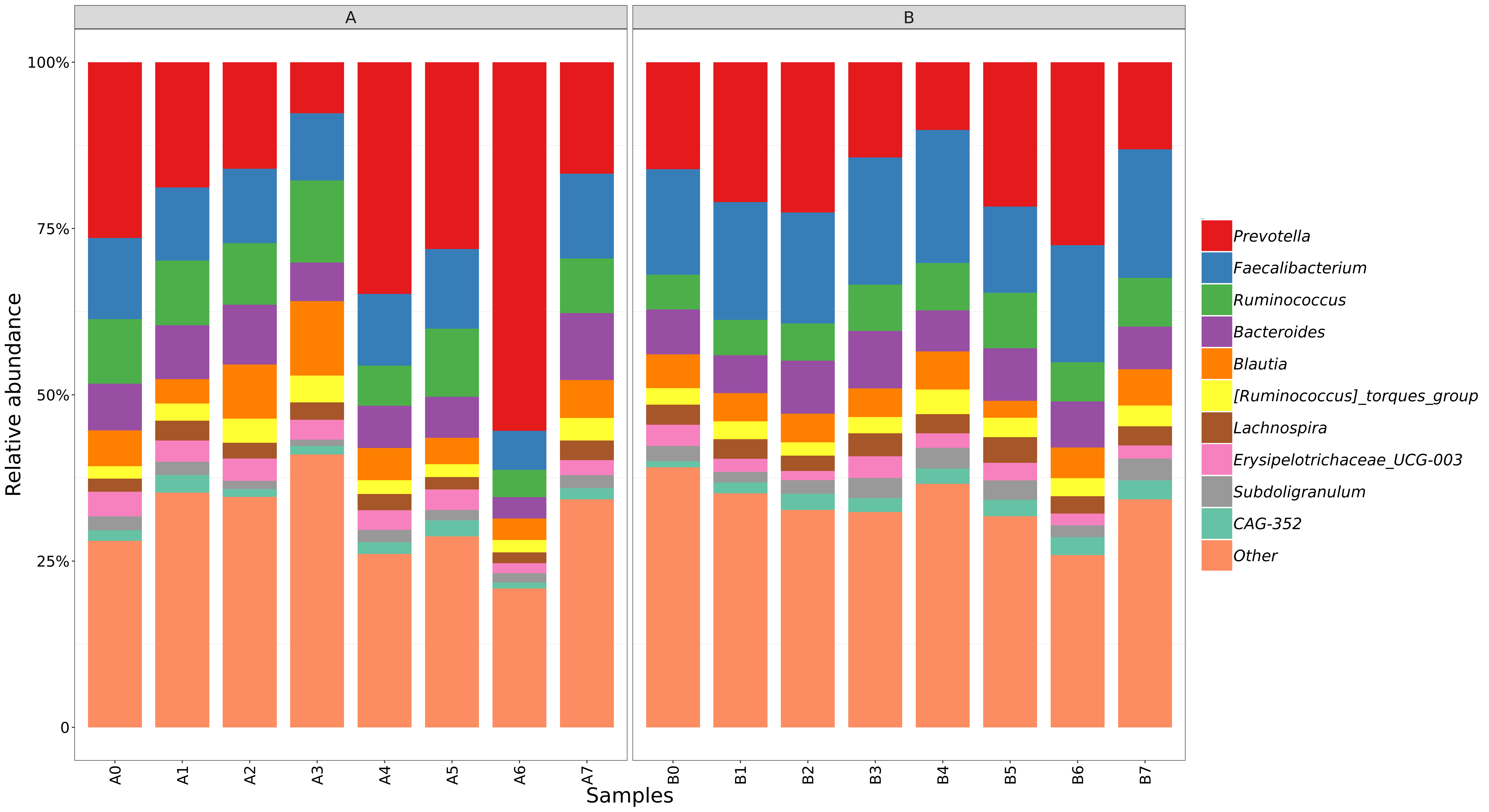
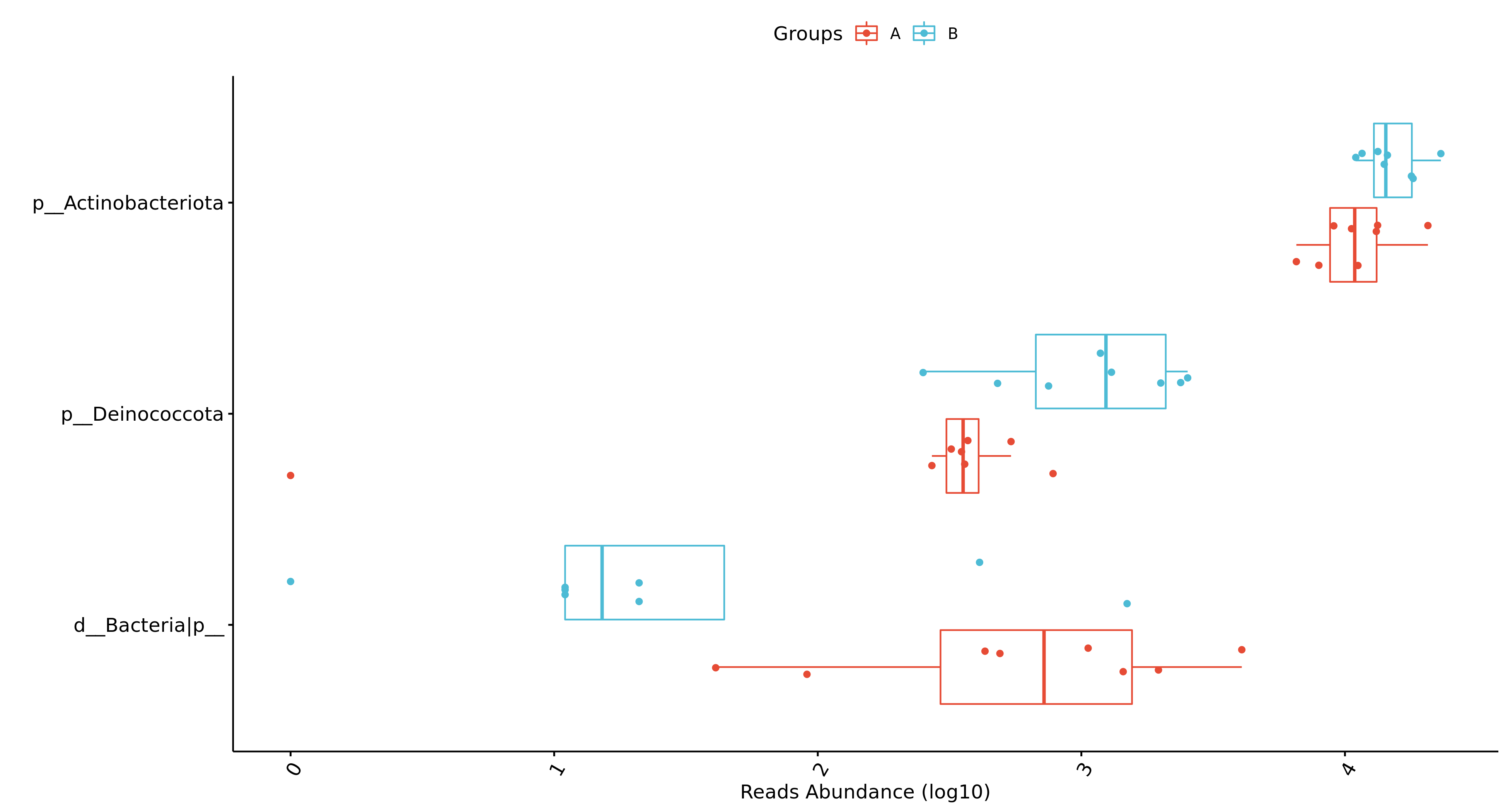
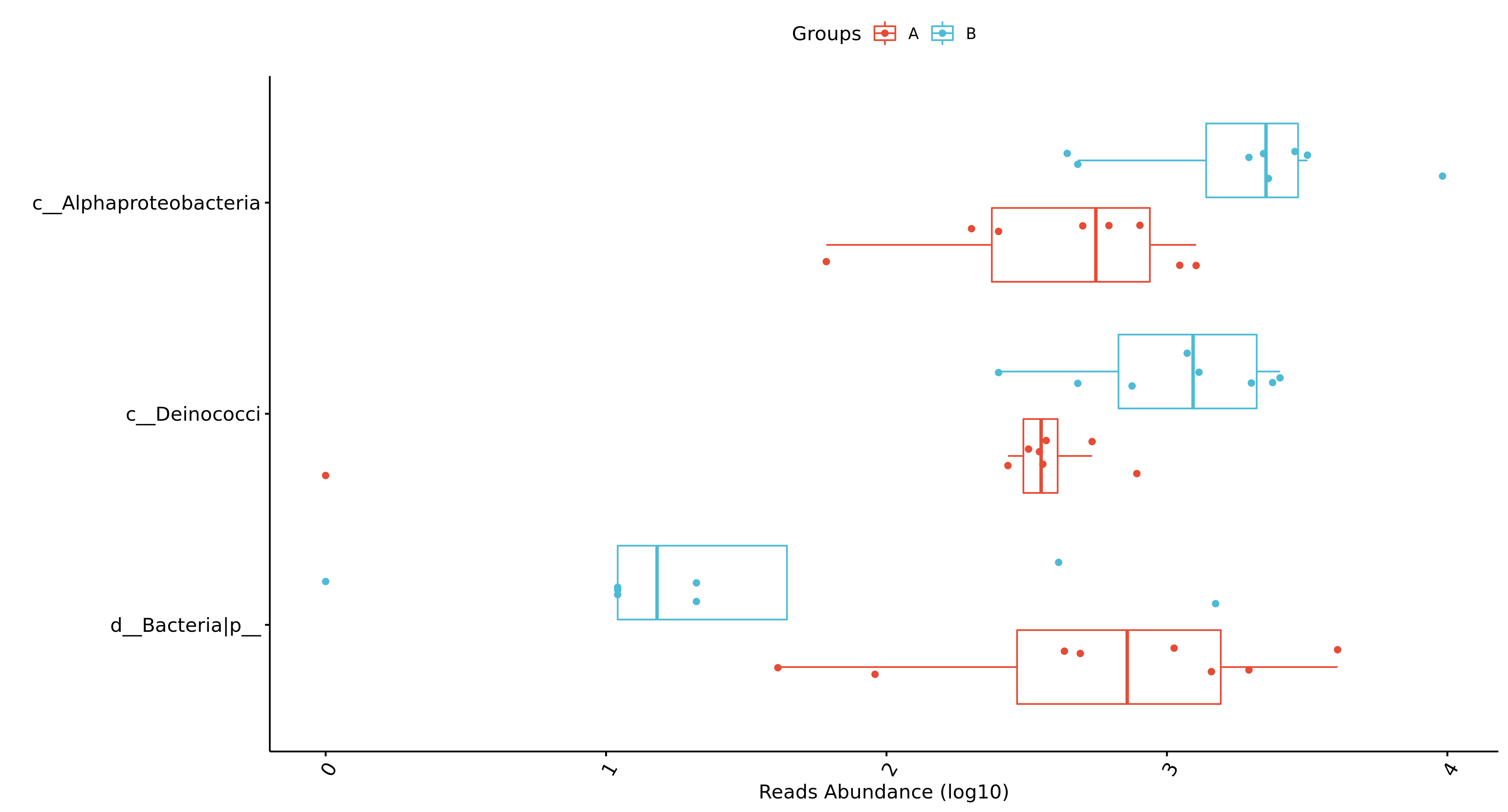
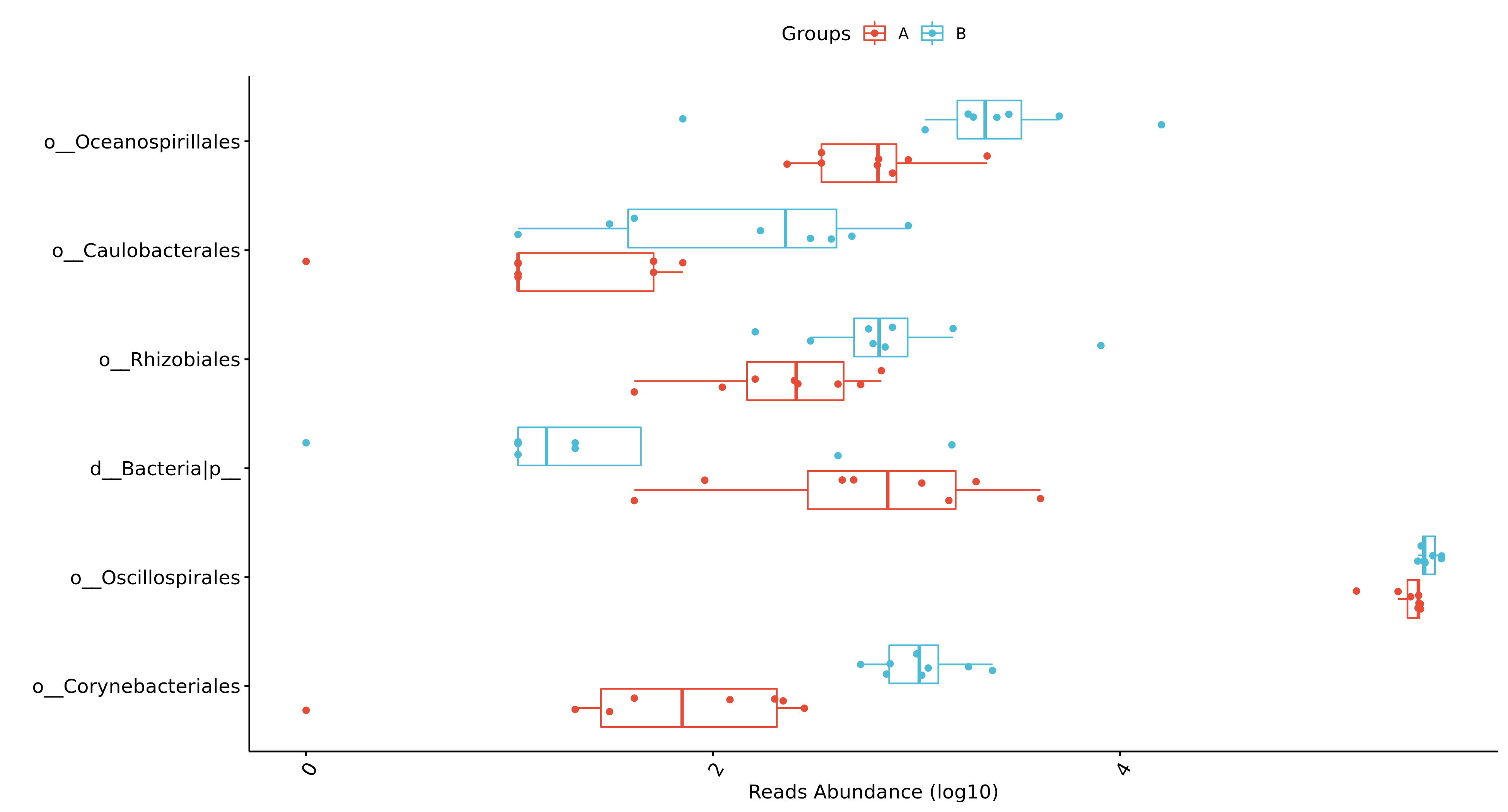
6.9 组间物种组成柱状图
基于物种丰度表,对组内样本间的物种进行组成分析,取相对丰度排名前十的物种,剩余归为other。
结果在文件夹:/taxanomyBar/taxon_hist/without_unknown/all下
- Phylum
- Class
- Order
- Family
- Genus
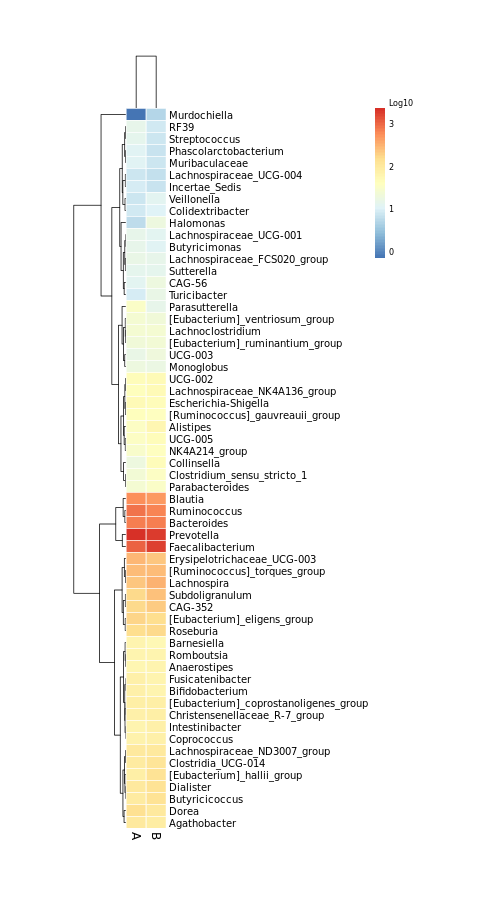
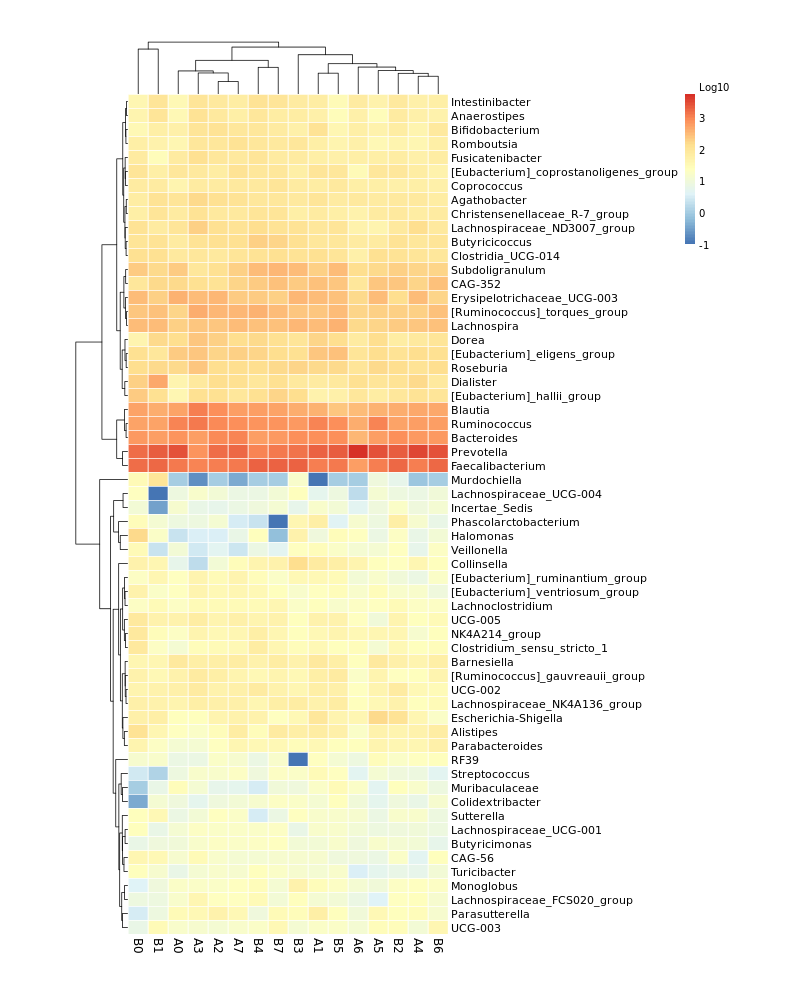
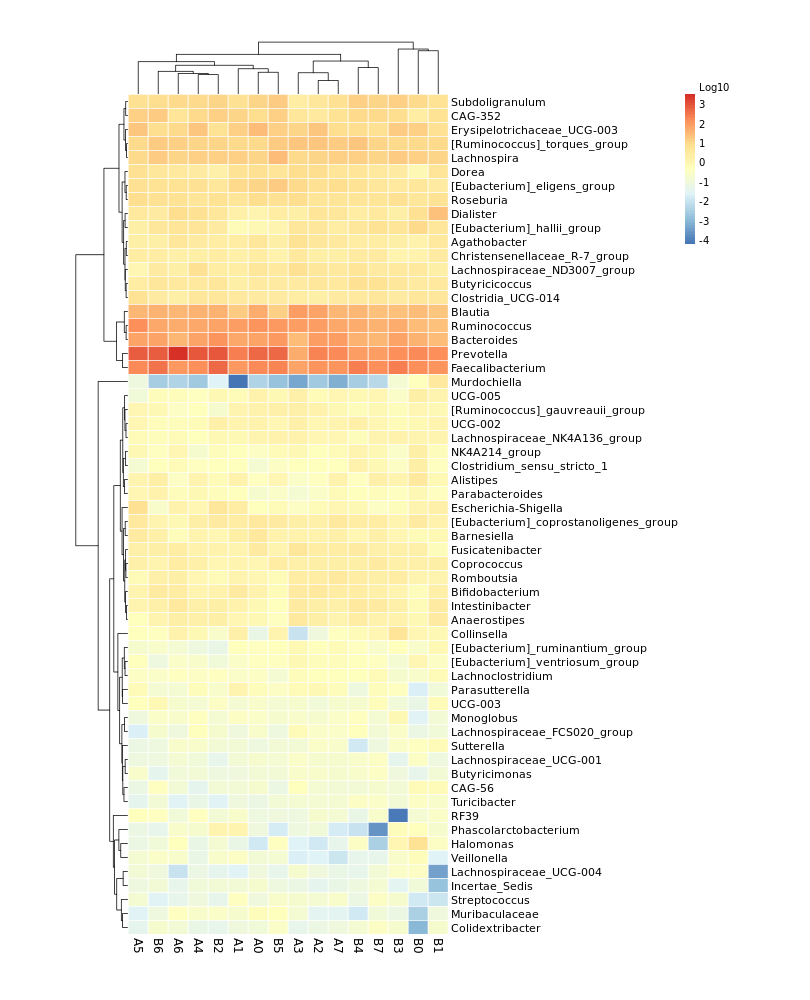
6.10 样本物种组成聚类热图
属水平物种的聚类热图,每个小方格表示每个样本对应的物种,其颜色表示该物种含量,(红色代表高丰度,蓝色代表低丰度)。每行表示每个物种在不同样本中的表达量情况,每列表示每个样品不同物种的表达量情况。上方树形图表示对不同样品的聚类分析结果,左侧树状图表示对不同物种的聚类分析结果。从图中可以了解样品之间的相似性以及属水平上的群落构成相似性。
结果在文件夹:/heatmap/下:
nogroup/ 文件夹表示:样本间聚类热图 Group/ 文件夹表示:分组间聚类热图
图片名称以 non_ 开头表示:未对数据进行标准化和中心化处理
cluster_ 表示:做聚类分析,样本按照相似度的顺序进行排列。不加cluster_ 则按照样本顺序排序
100log10 表示:属水平物种相对丰度数据 × 100 再取 log10
log10 表示:属水平物种相对丰度数据 × 10000 再取 log10
log2 表示:属水平物种相对丰度数据 × 10000 再取 log2
- 样本间聚类热图 Log10无标准化
- 样本间聚类热图 Log10标准化
Fig 6-10-1 样本属构成热图 Log10无标准化
Fig 6-10-2 样本属构成聚类热图 Log10无标准化
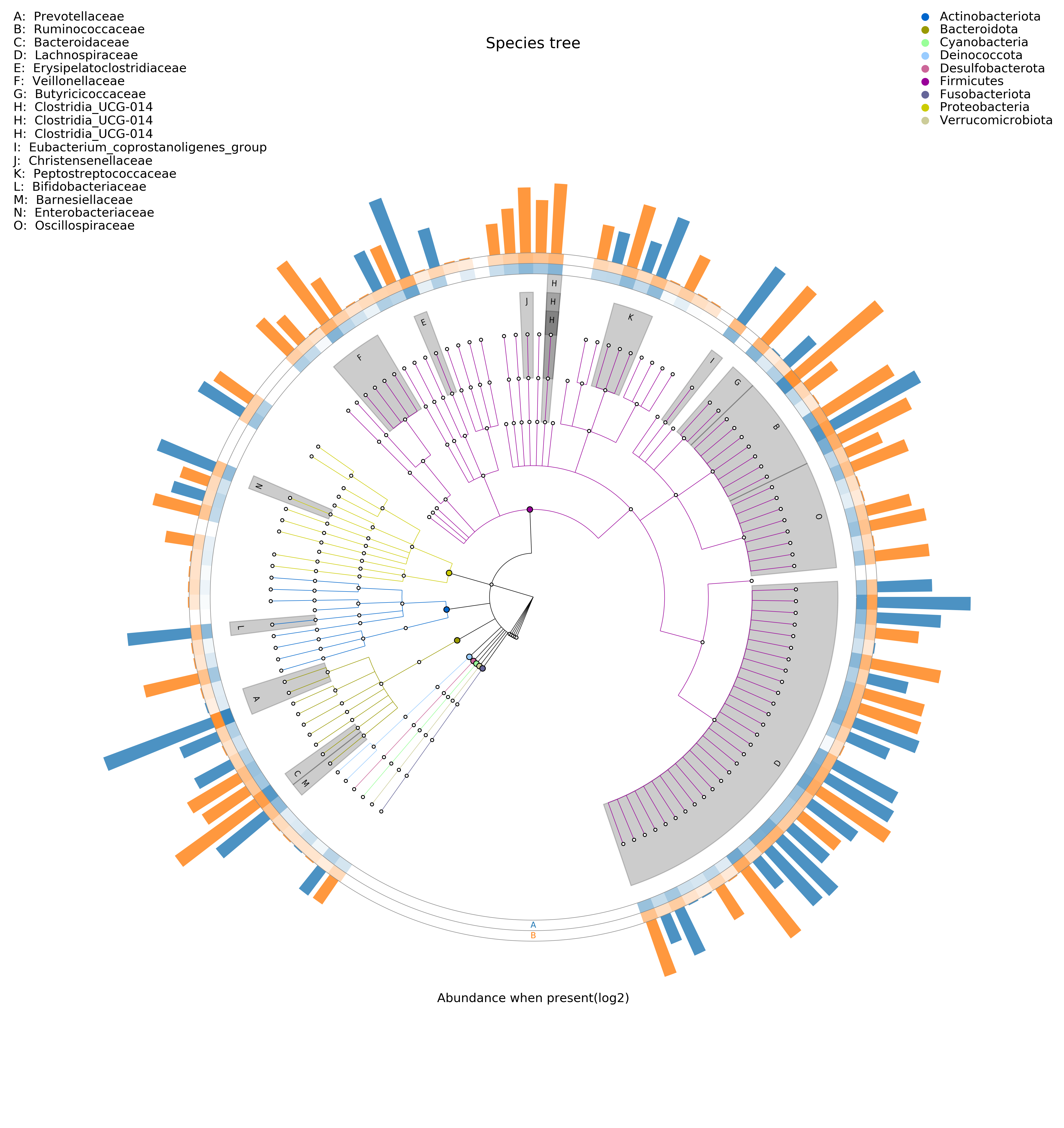
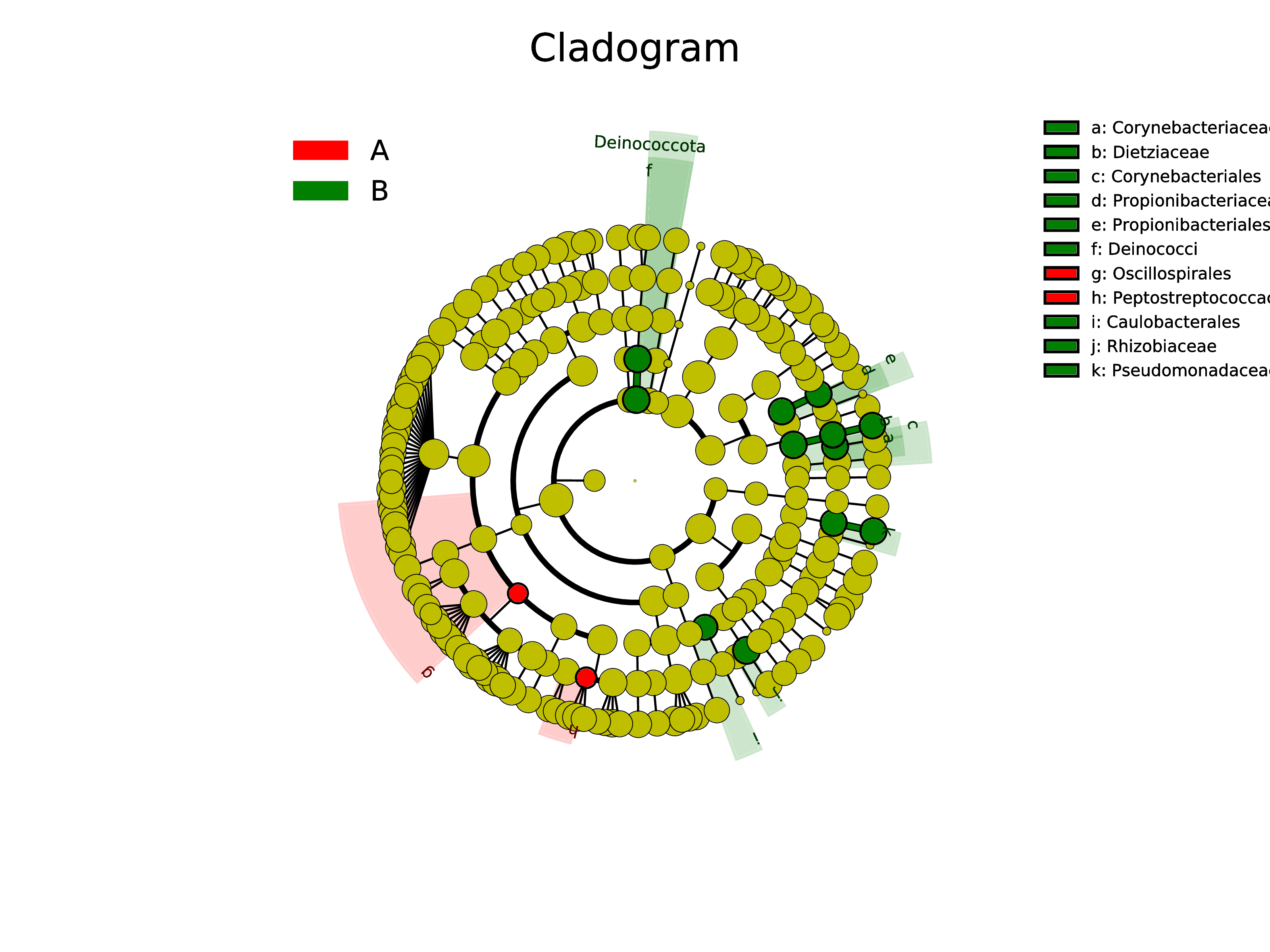
6.10-2 GraPhlan 图
物种进化树的样本群落分布图 GraPhlan 图是将不同样本的群落结构及分布以物种分类树的形式在一个环图中展示。使用 GraPhlan 结合 OTU Table对一个分组所有样本的 OTU物种注释结果进行总体展示,便于看出优势菌种。其目的是将物种之间的进化关系以及不同样本的物种分布丰度和最高分布样本的信息在一个视觉集中在换图中展示,提供的信息量较其他图更丰富。
图解读:图中中间为物种进化分类树,不同颜色的分支代表不同的纲(具体的代表颜色见右上角的图例),之后外圈的灰色标示字母的环表示的是本次研究中比例最高的 15 个科(字母代表的科参见左上角的图例)。之后的外圈提供的是热力图,如果样本数 <=10 个则绘制样本,如果样本数超过 10 个则按照分组绘制,每一环为一个样本,根据其丰度绘制的热力图。最外圈为柱状图,绘制的是该属所占比例最高的样本的丰度和样本颜色(样本颜色见环最下方的样本名字的颜色)。其中热力图和柱状图取值均为原比例值 x10000后进行 log2 转换后的值。
 |
|
| Fig 6-8-1 NMDS间分布及差异 |
|
|
|
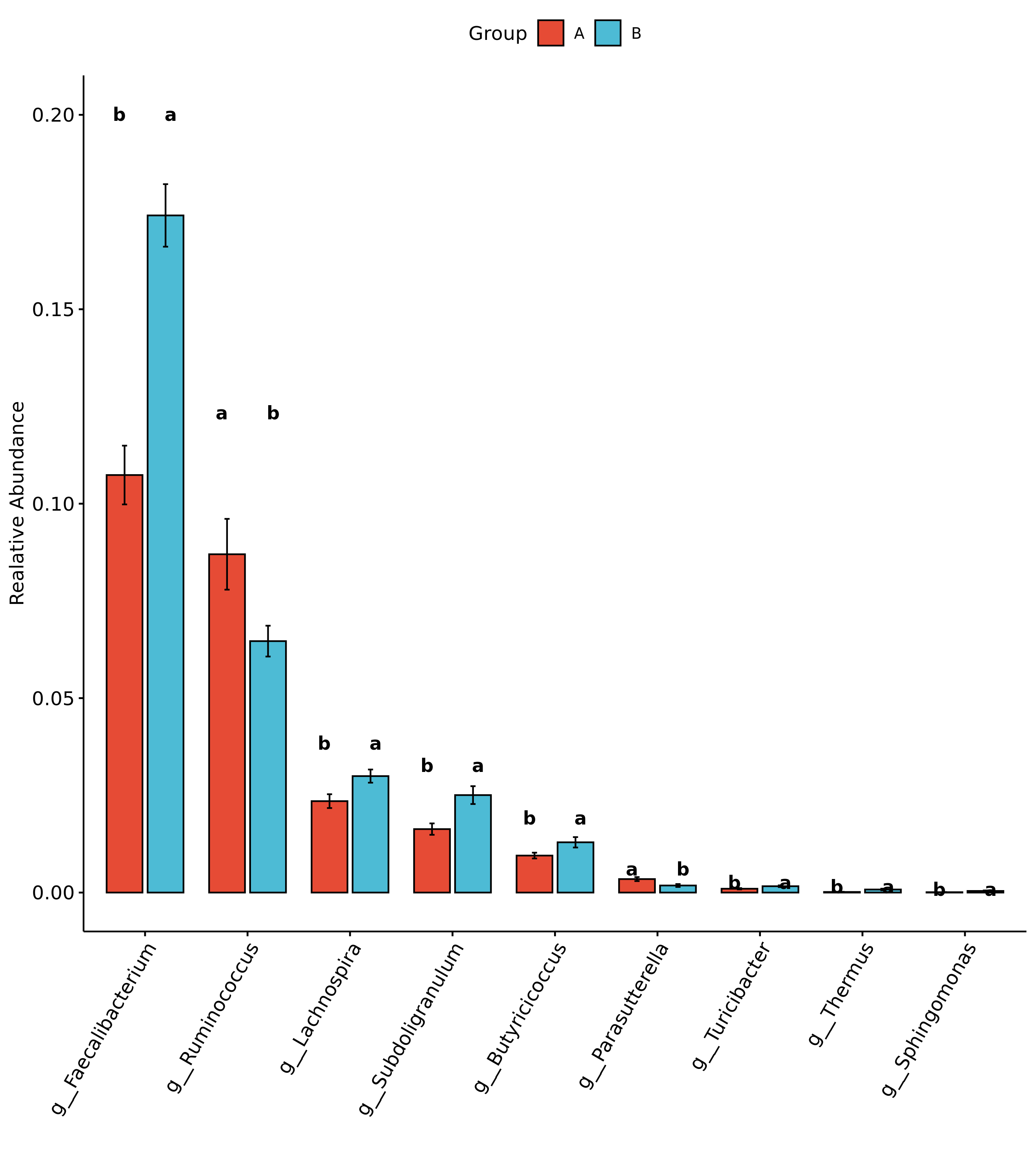
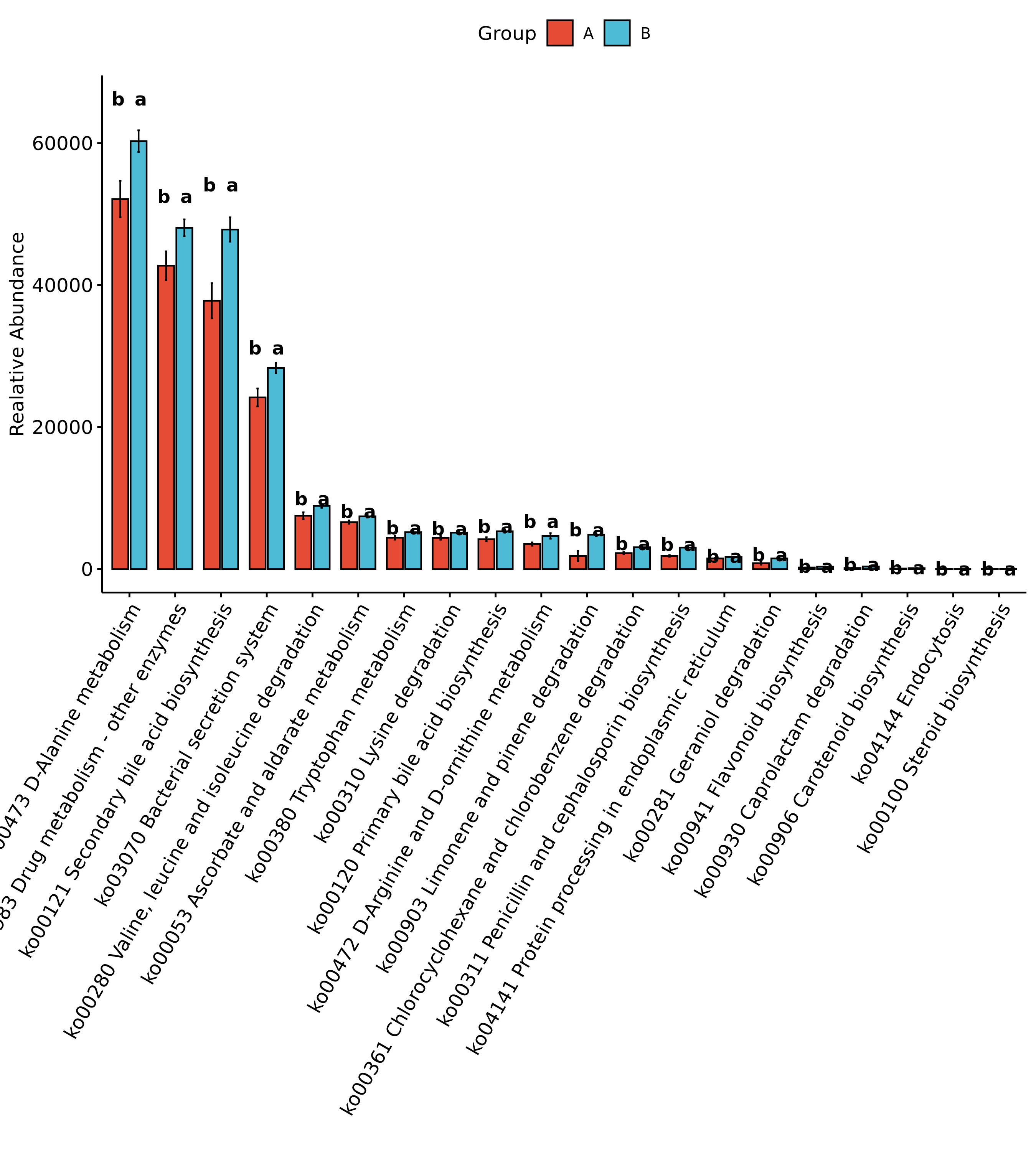
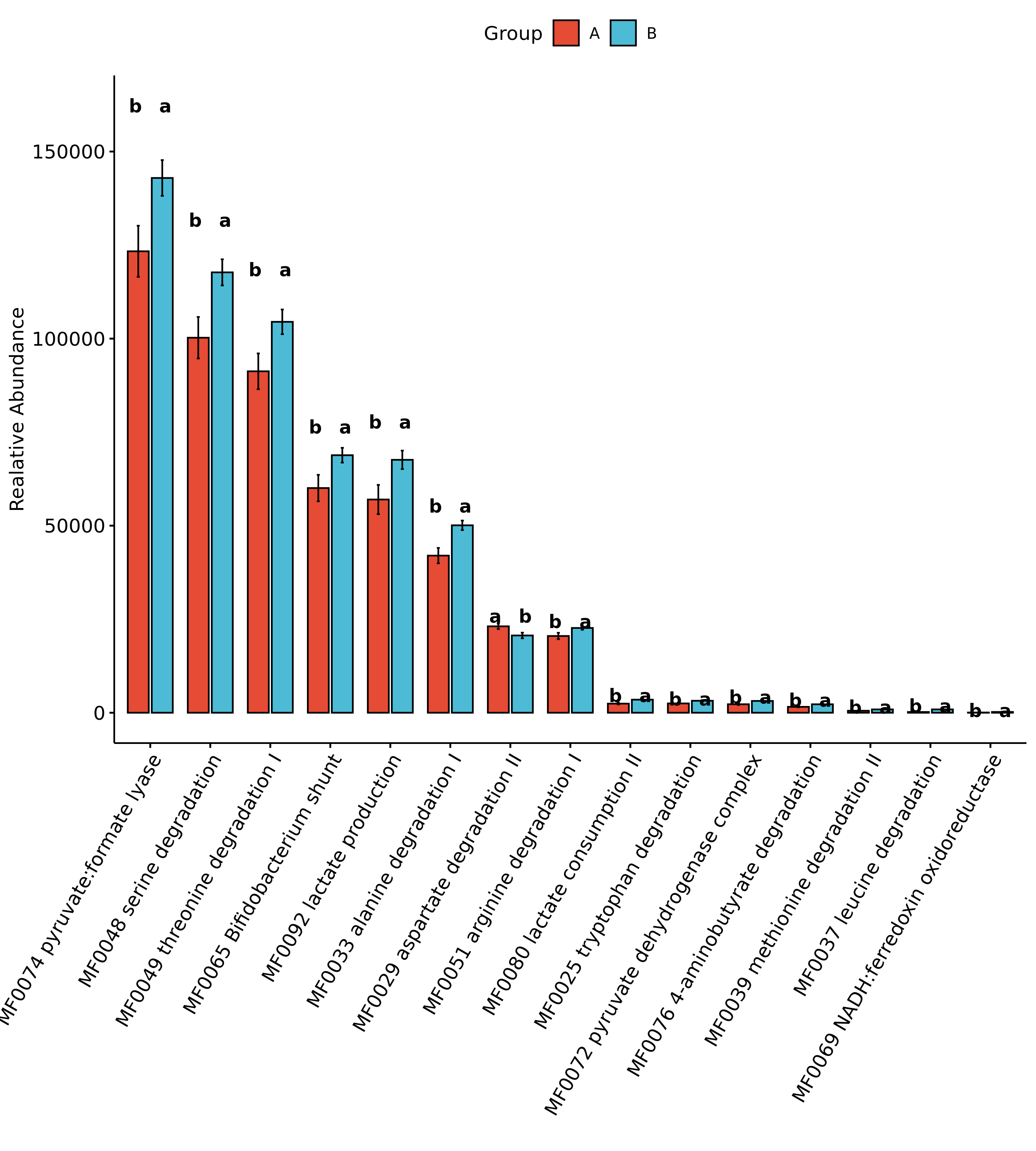
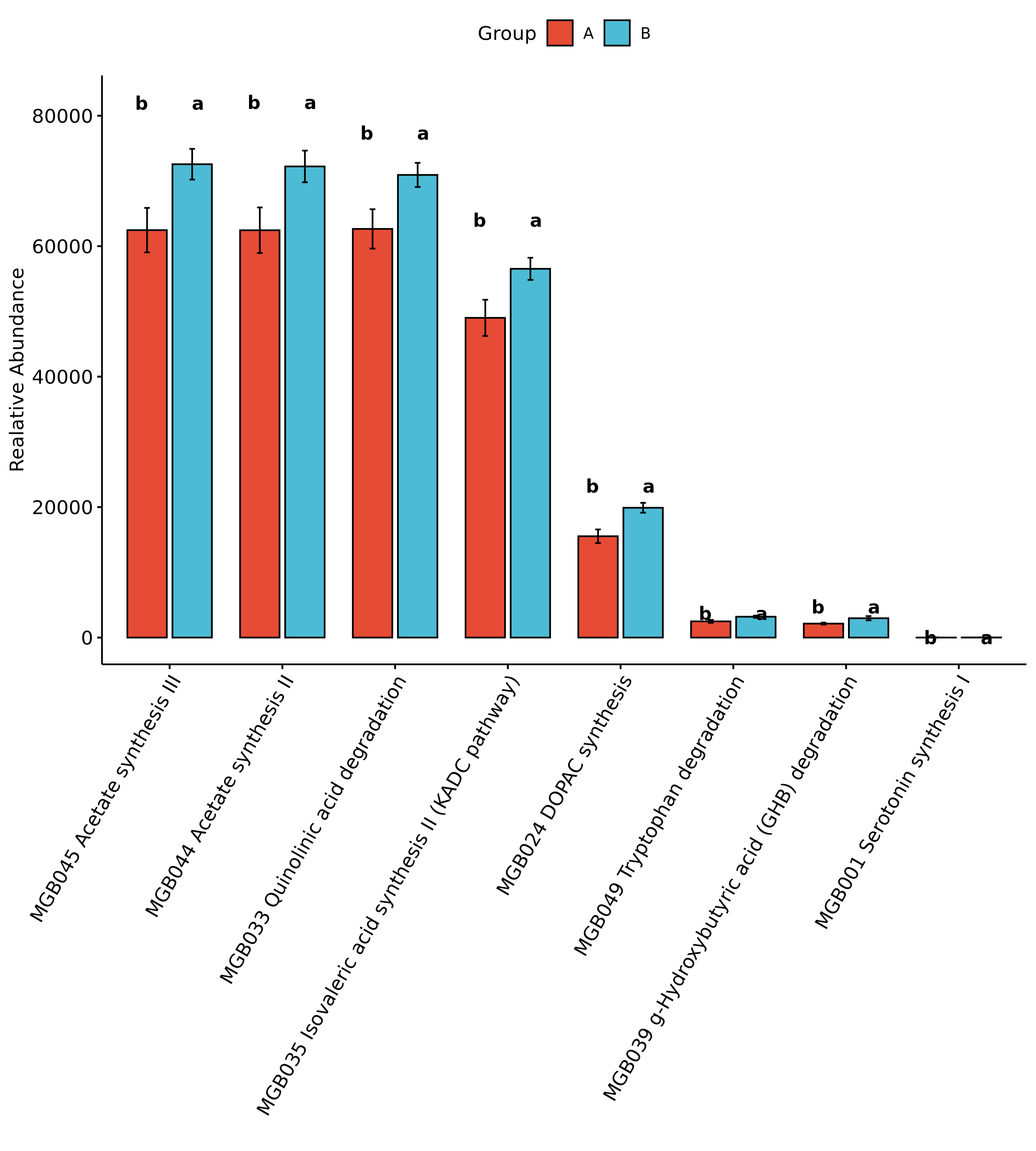
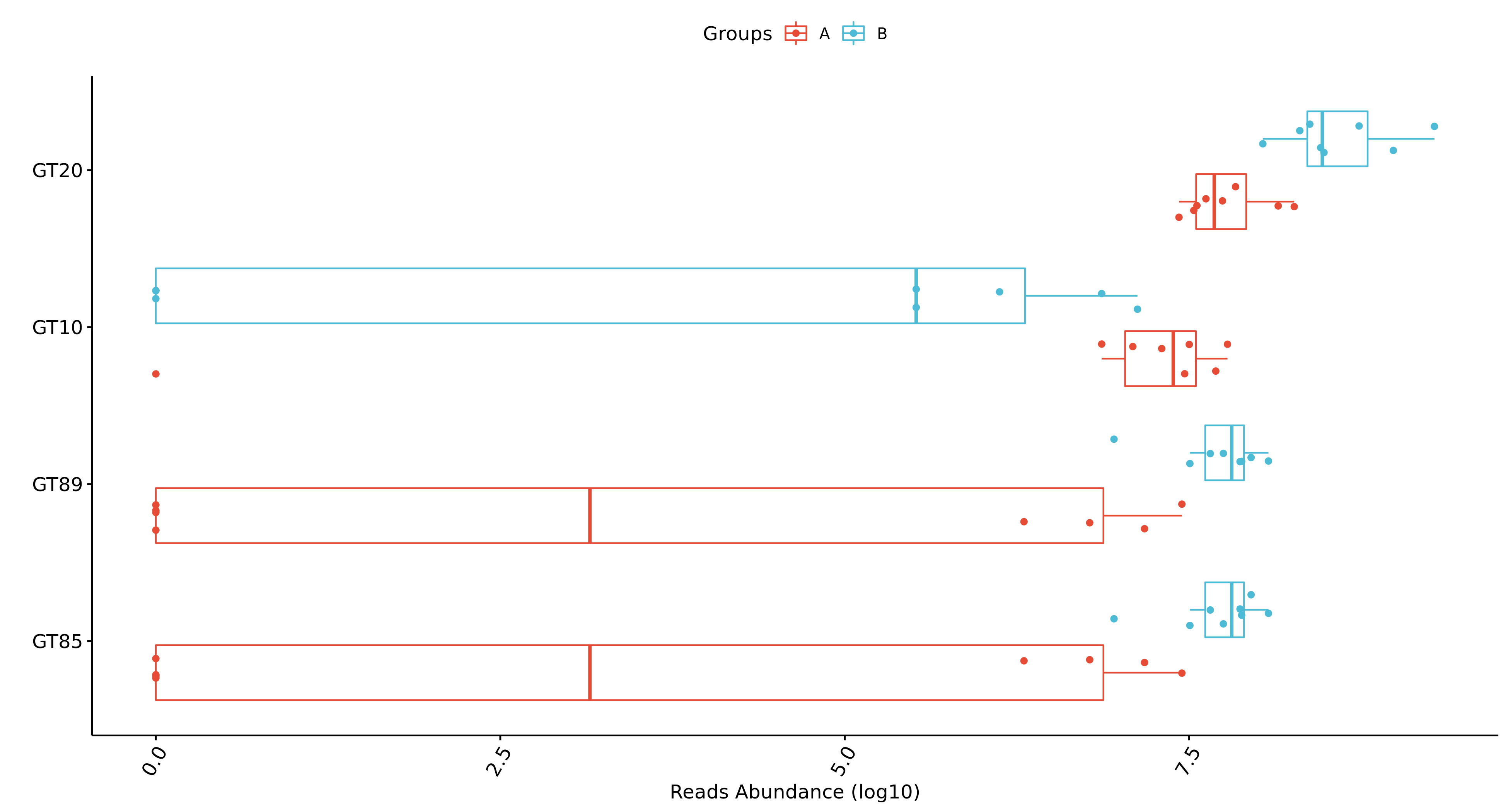
6.11 组间物种及功能差异分析 Tukey检验
该检验适合于各个分组样本数量均等的情况下。
结果在文件夹:/diff_analysis/TukeyHSD/下
每个物种分类组间差异统计结果文件在:/diff_analysis/TukeyHSD/Tukey_XXXX.result.csv
图中的字母代表显著性差异的字母表示法,只要含有相同的字母,就表明两组之间没有显著性差异。例如a和ab含有相同字母“a”,表示两组之间没有显著性差异。ab中的“b”表示这一组和其他含有字母b的组(比如bc)没有显著性差异,但是a和bc就有显著性差异了。
- Genus
- Phylum
- FAPROTAX
- METACYC
- KEGG
- GMM 代谢模块
- GBM 神经递质
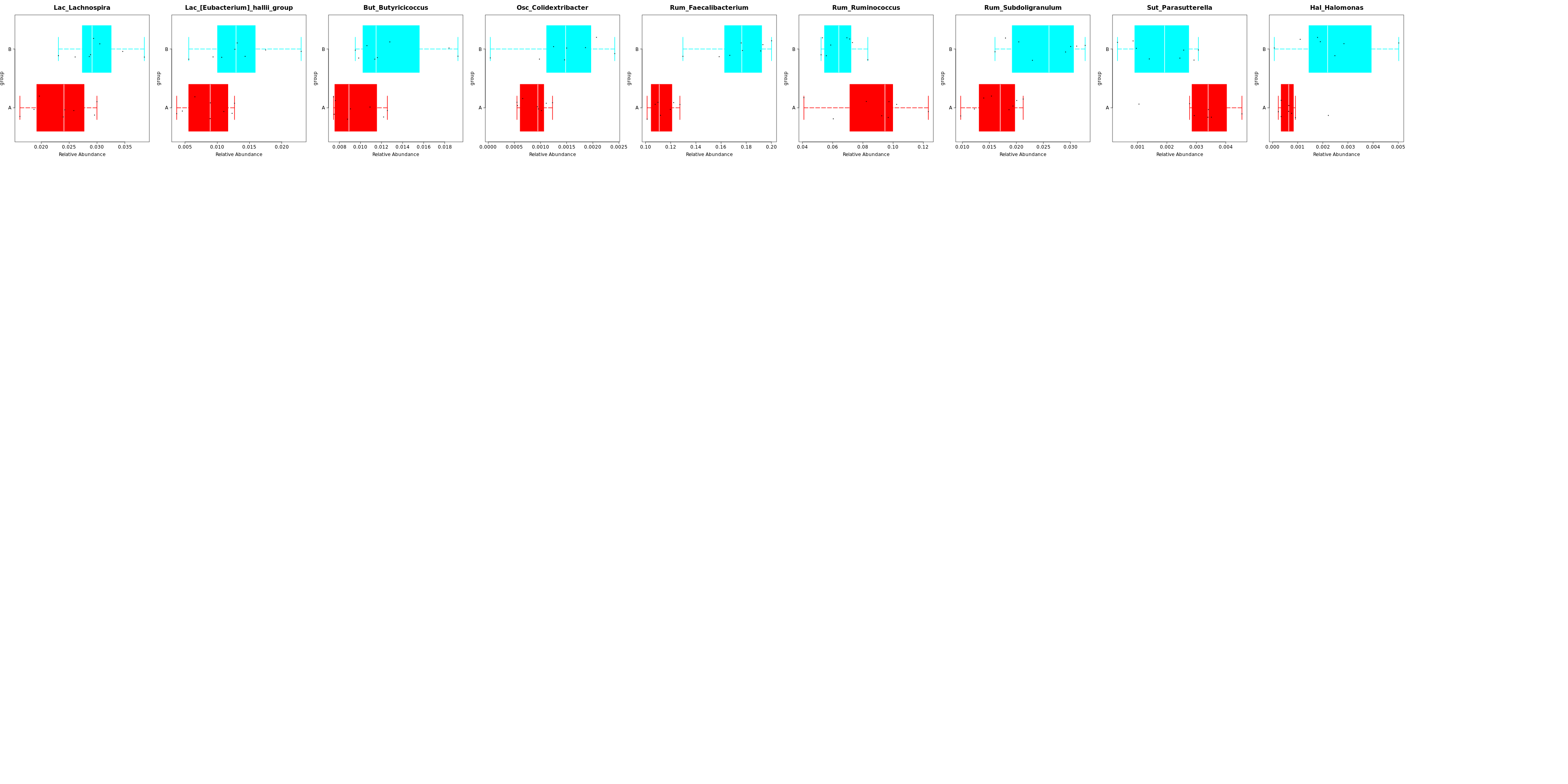
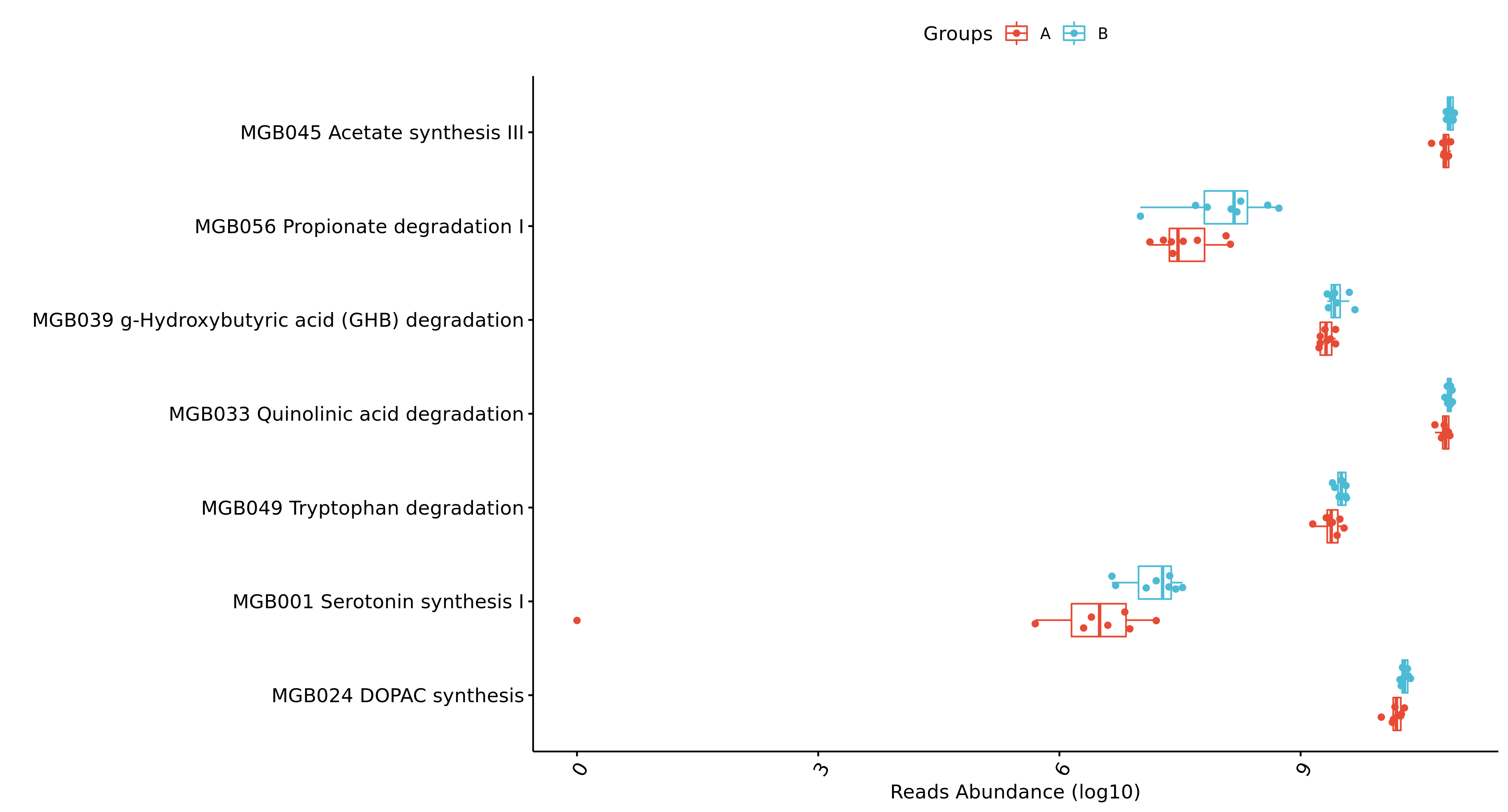
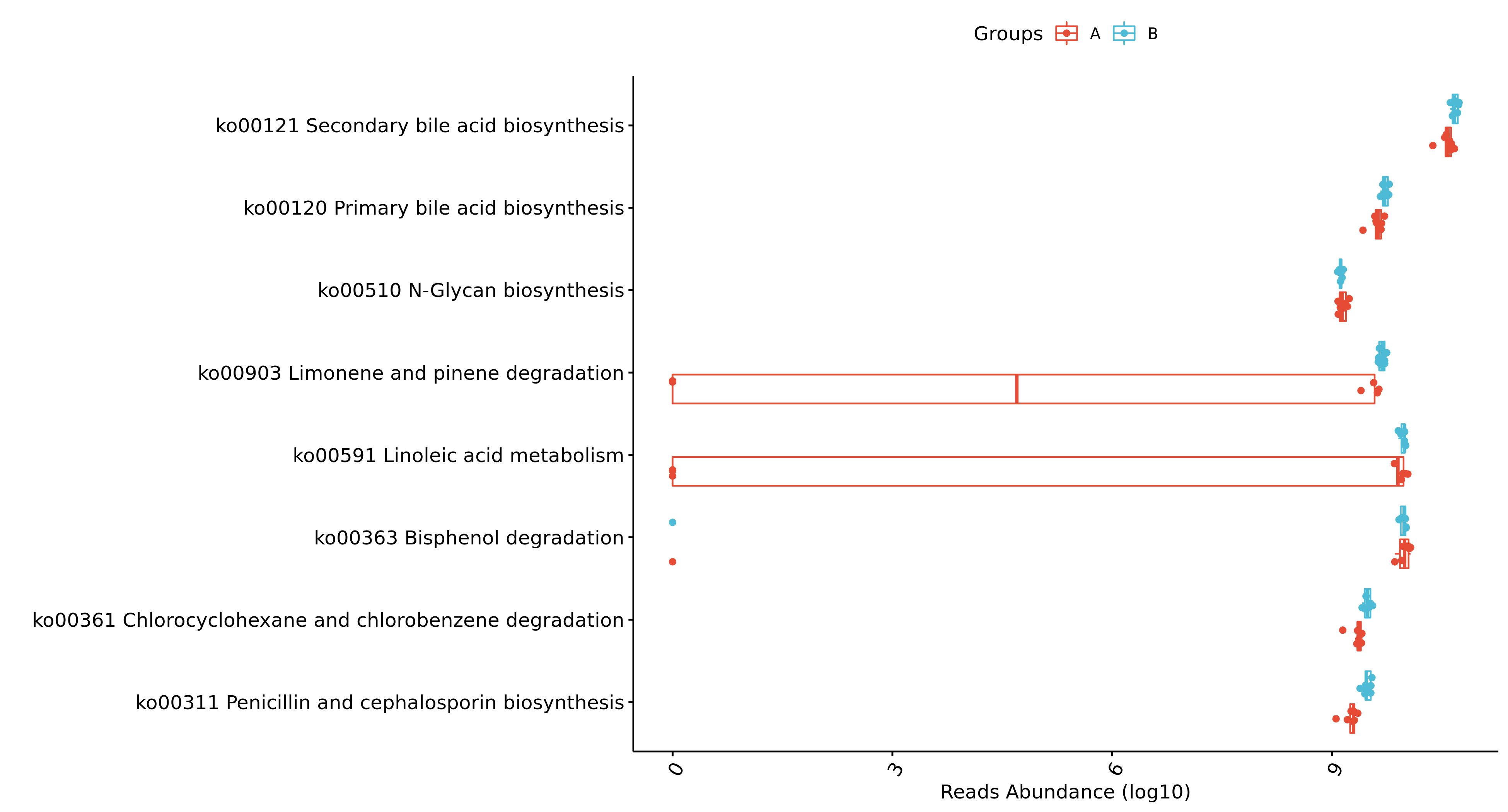
Fig 6-11-2-1 组间差异物种 (仅显示top前十的结果,top20及完整可前往目录查看)
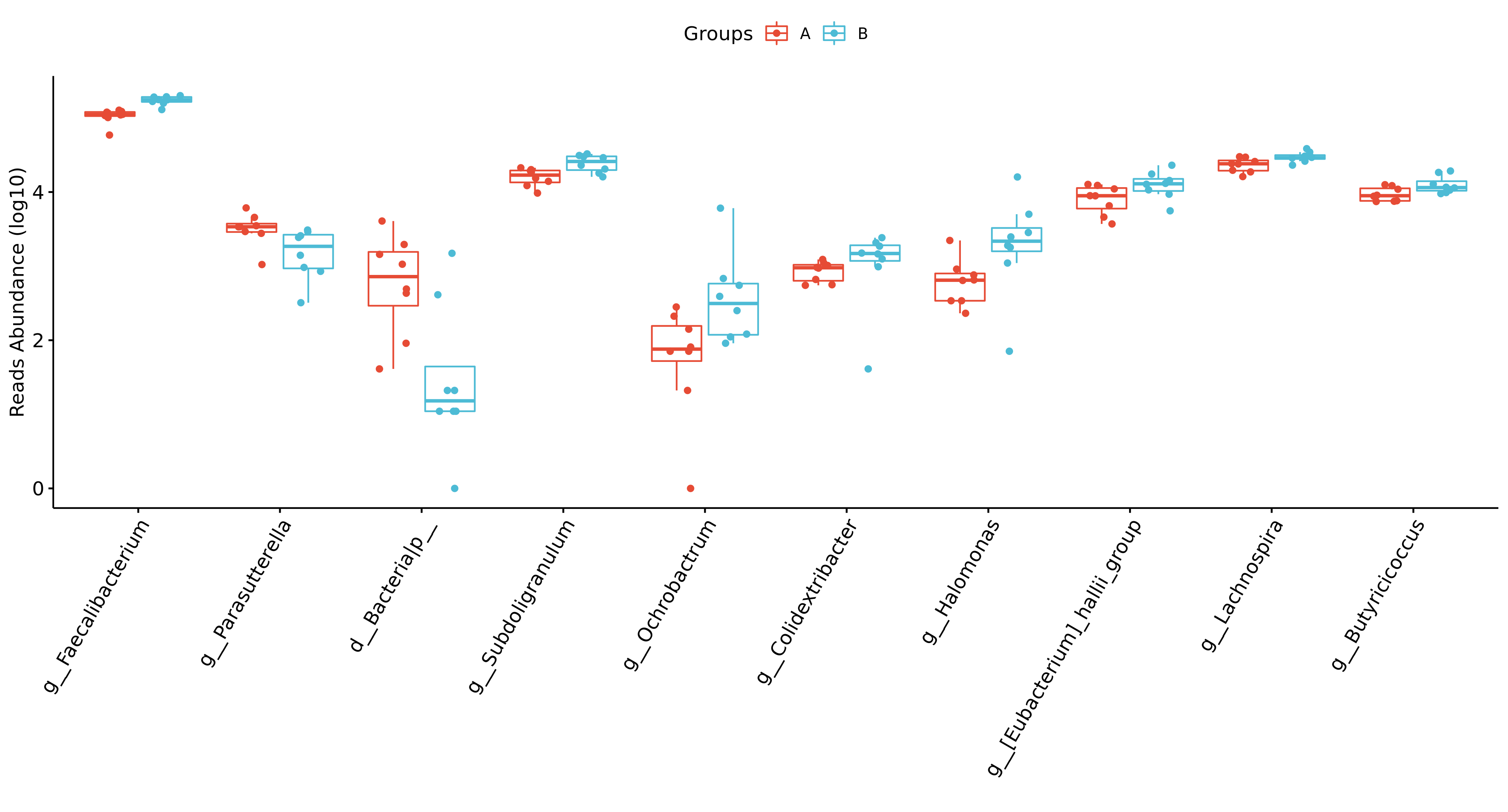
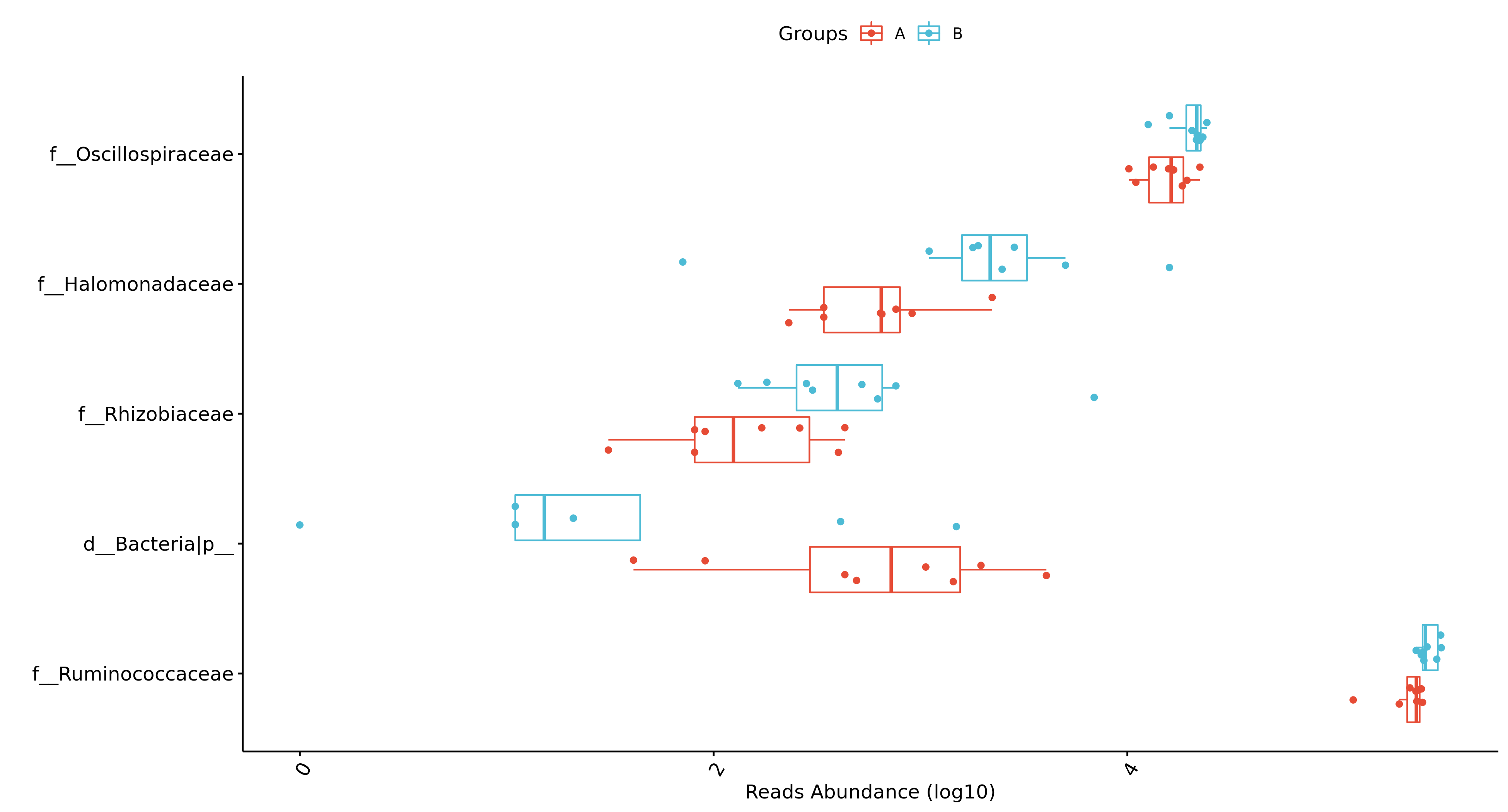
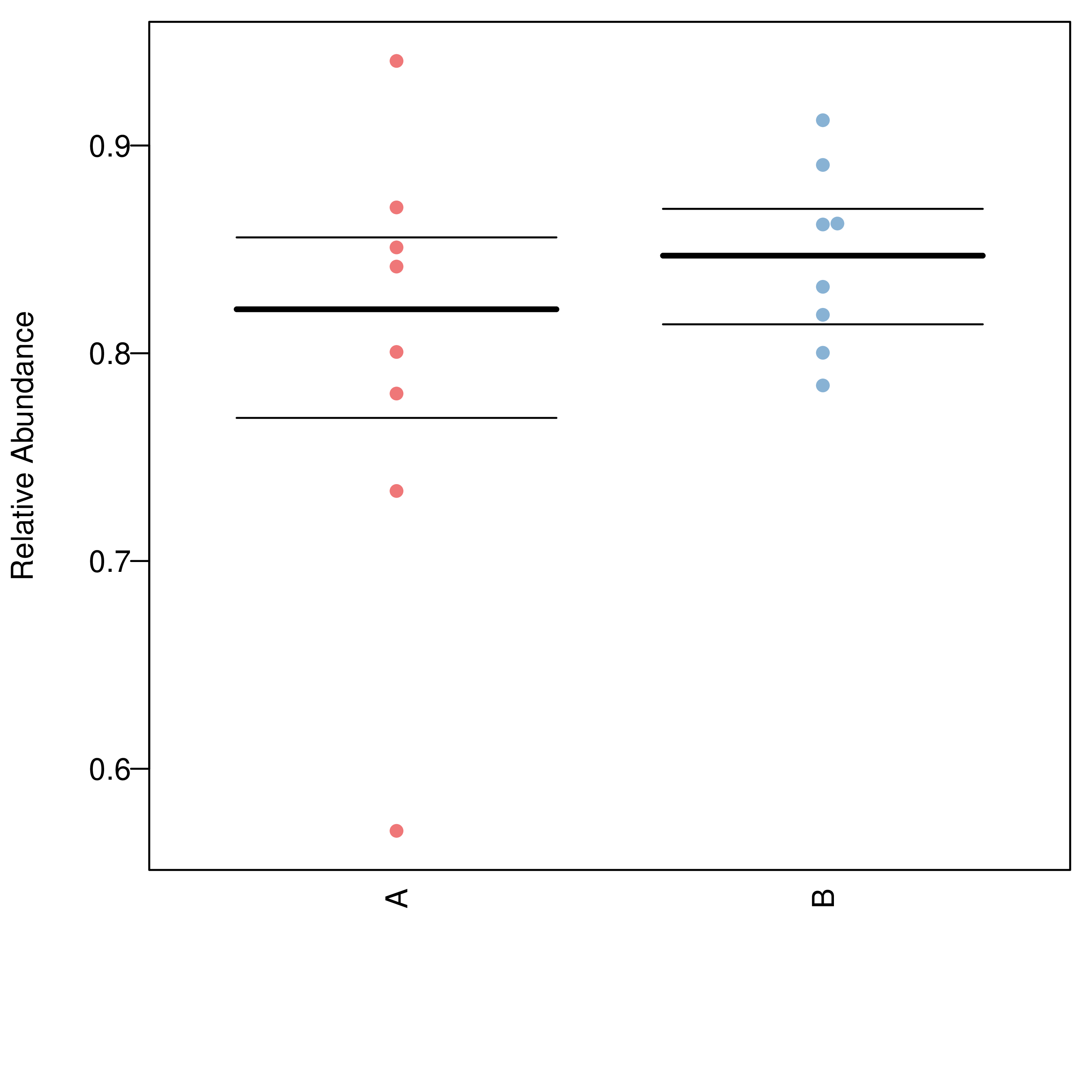
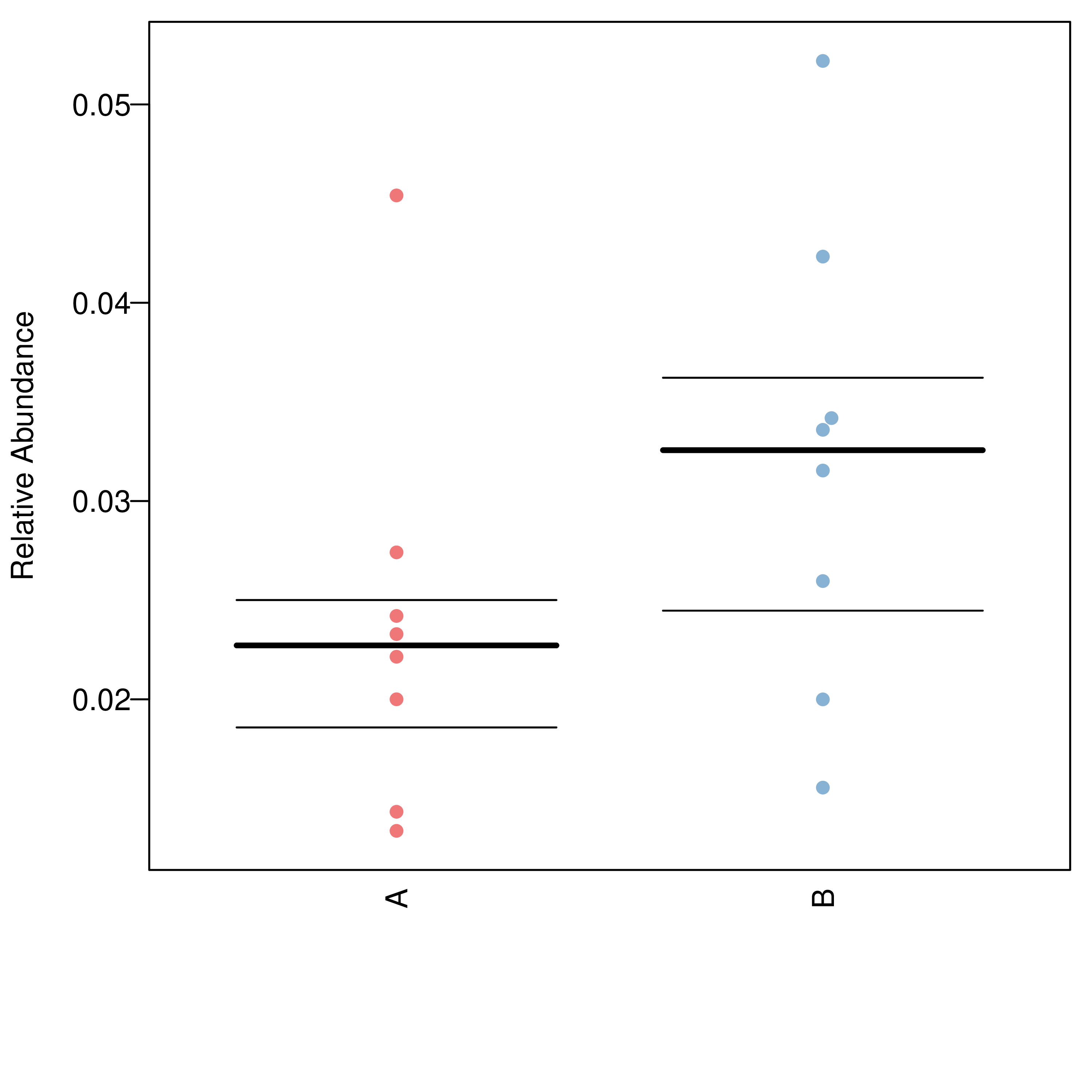
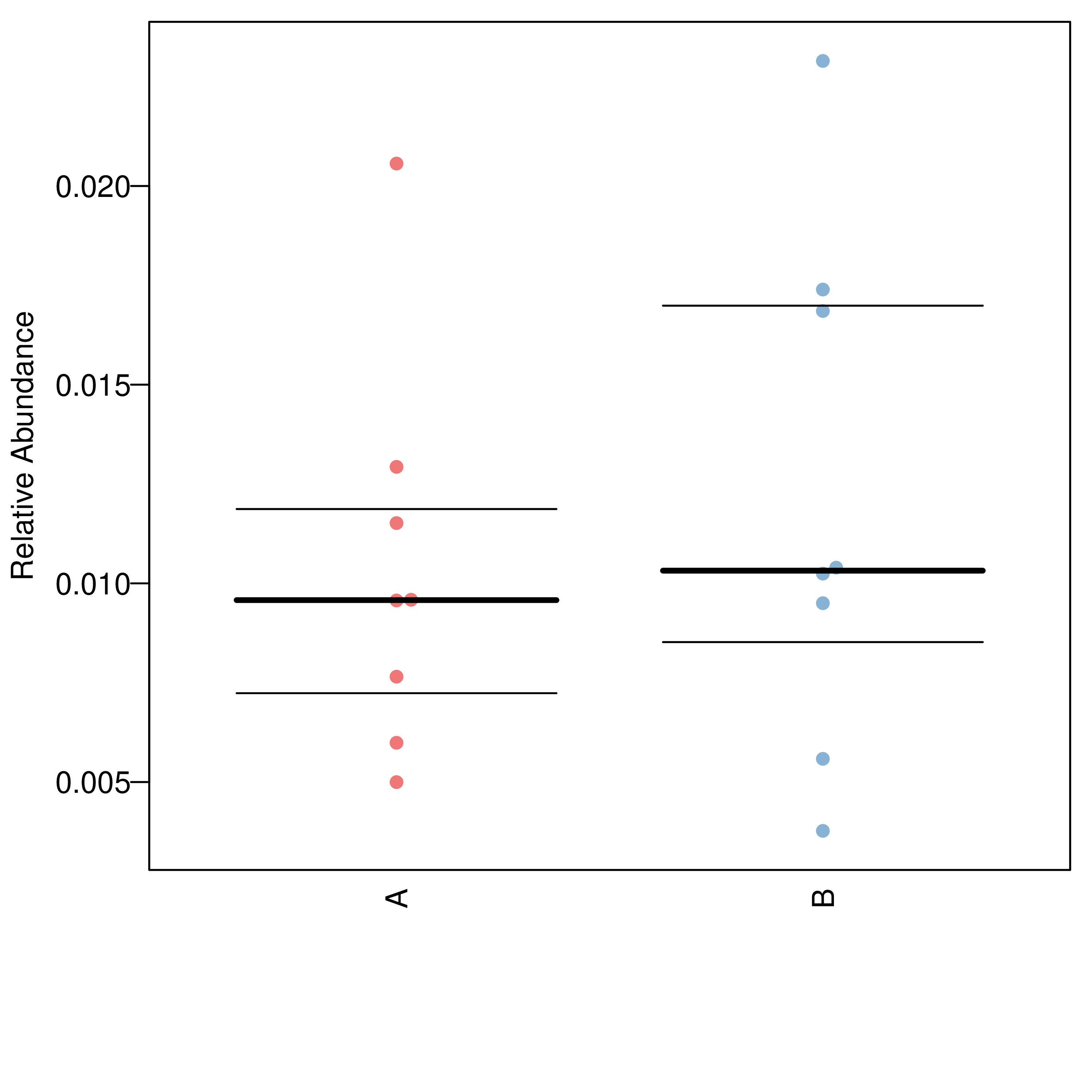
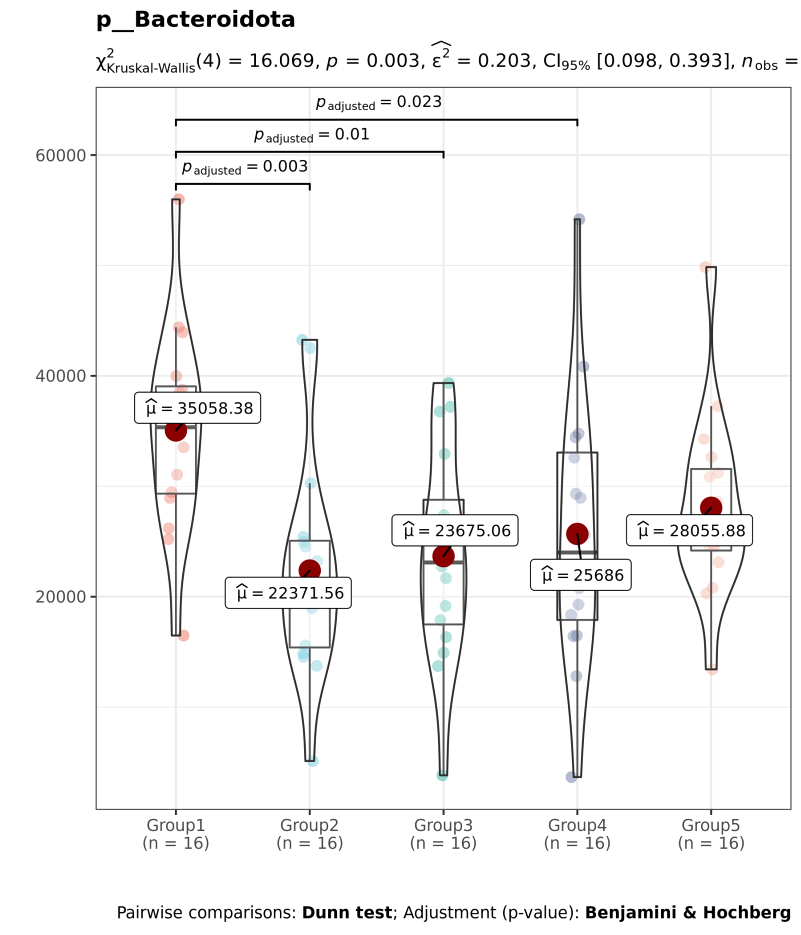
6.11-2 组间物种差异分析 非参数检验
结果在文件夹:/diff_analysis/TaxaMarkers/下
每个物种分类组间差异统计结果图在文件夹:/diff_analysis/UnivarTestXXXX/figure/single_plot下,示例如下图:
- Genus
- Phylum
- Class
- Order
- Family
- Species
Fig 6-11-2-1 组间差异物种 (仅显示top前十的结果,top20及完整可前往目录查看)
6.12 组间菌群比较选取物种标志物
注:做组间差异分析需要满足条件:每个分组的组内至少需要3个样本。若未提供分组信息或分组情况不满足要求,则不能完成第7部分 组间差异分析。
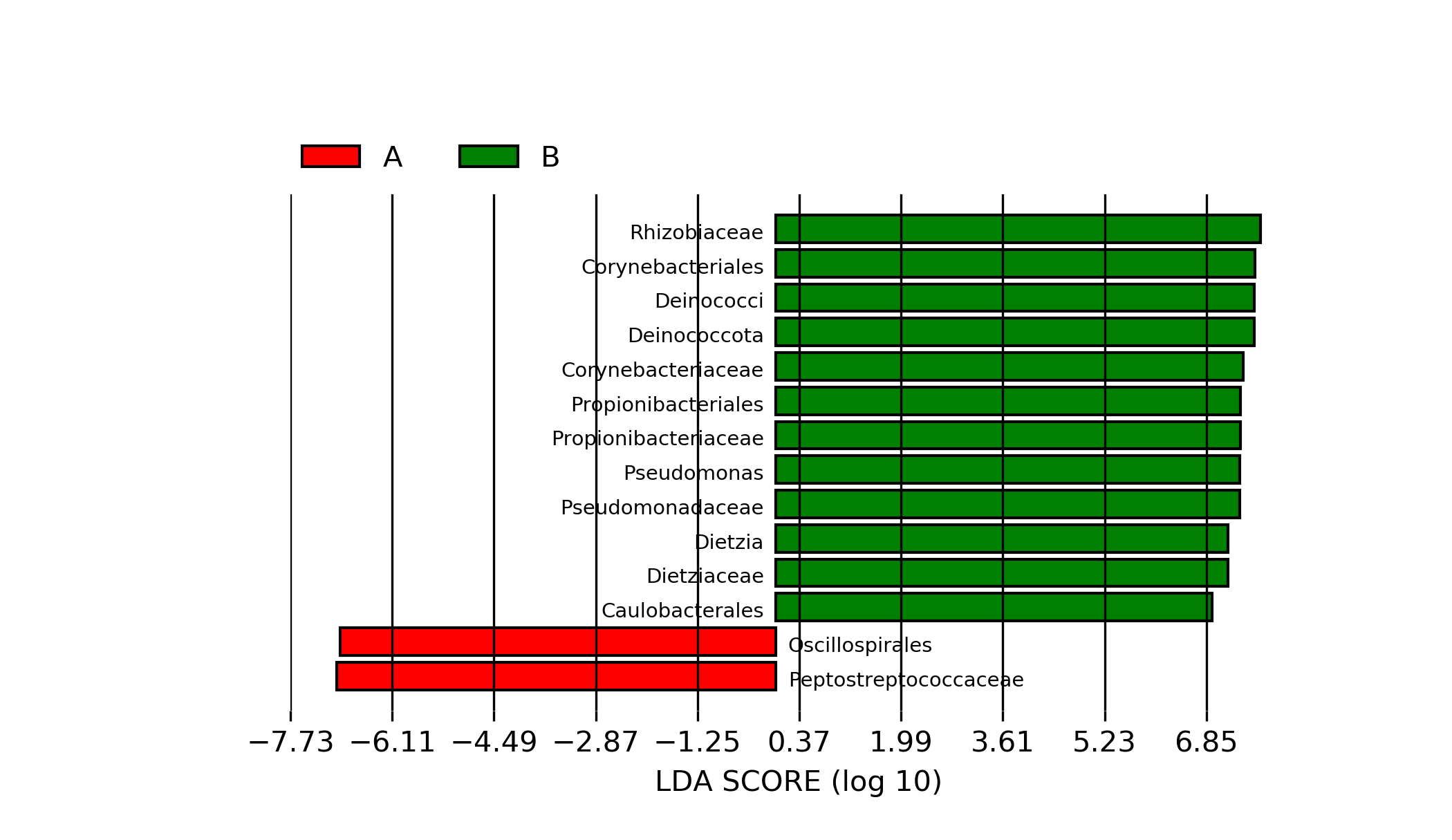
LEfSe是基于线性判别分析(Linear discriminant analysis,LDA)的分析方法,其将线性判别分析与非参数的 Kruskal-Wallis 以及 Wilcoxon 秩和检验相结合,从而筛选组与组之间生物标记物 Biomarker(组间差异显著物种)。 Lefse分析选取的组间差异标记物,是该分组相较于其他各分组具体显著差异用于区分的优势物种,所以不一定所有分组都能找出优势物种。
Lefse分析是对所有分组进行比较的,若图中只显示了部分分组的差异结果,则说明lefse分析在该分组情况下只在部分组中找到差异标记物,而其余分组没有差异标记物(即该分组没有找到相较于其他分组具有明显优势物种)。
详细标志菌结果见:Lefse_Analysis/biomarkers_raw_images目录
图解读: 图中不同颜色代表不同样本或组之间的显著差异物种。使用 LefSe 软件分析获得,其中显著差异的logarithmic LDA score设为 2。由于 LDA 会尝试寻找基于单一菌属能进行组间分类的特征菌属,同时多个分组的情况容易找不到显著的特征菌属。图中仅展示有显著差异的分组。
 |
|
 |
|
| Fig 6-12-1 LEfSe各组物种标志物进化树 |
|
Fig 6-12-2 LEfSe各组物种标志物 |
|
|
|
|
|
6.13 Bugbase菌群表型特征分析
Bugbase是对菌群表型特征预测工具,2016年发表文章公布其软件原理。该工具主要进行表型预测,其中表型类型包括革兰氏阳性、革兰氏阴性、生物膜形成、致病性、移动组件、氧需求,包括厌氧菌、好氧菌、兼性菌)及氧化胁迫耐受等7类。
参考文献:Thomas A M, Jesus E C, Lopes A, et al. Tissue-associated bacterial alterations in rectal carcinoma patients revealed by 16S rRNA community profiling[J]. Frontiers in Cellular and Infection Microbiology, 2016, 6.
各部分分布结果在文件夹:Bugbase/predicted_phenotypes下
各OTU贡献分类结果在文件夹:Bugbase/otu_contributions下
每个物种分类组间差异统计结果见:Bugbase/BugBase_pvlaue.txt

图解读:三条线自上而下分别代表上四分位、平均值及下四分位。
- Aerobic
- Anaerobic
- Contains_Mobile_Elements
- Facultatively_Anaerobic
- Forms_Biofilms
- Gram_Negative
- Gram_Positive
- Potentially_Pathogenic
- Stress_Tolerant
Fig 6-13-1 Bugbase菌群表型特征分析
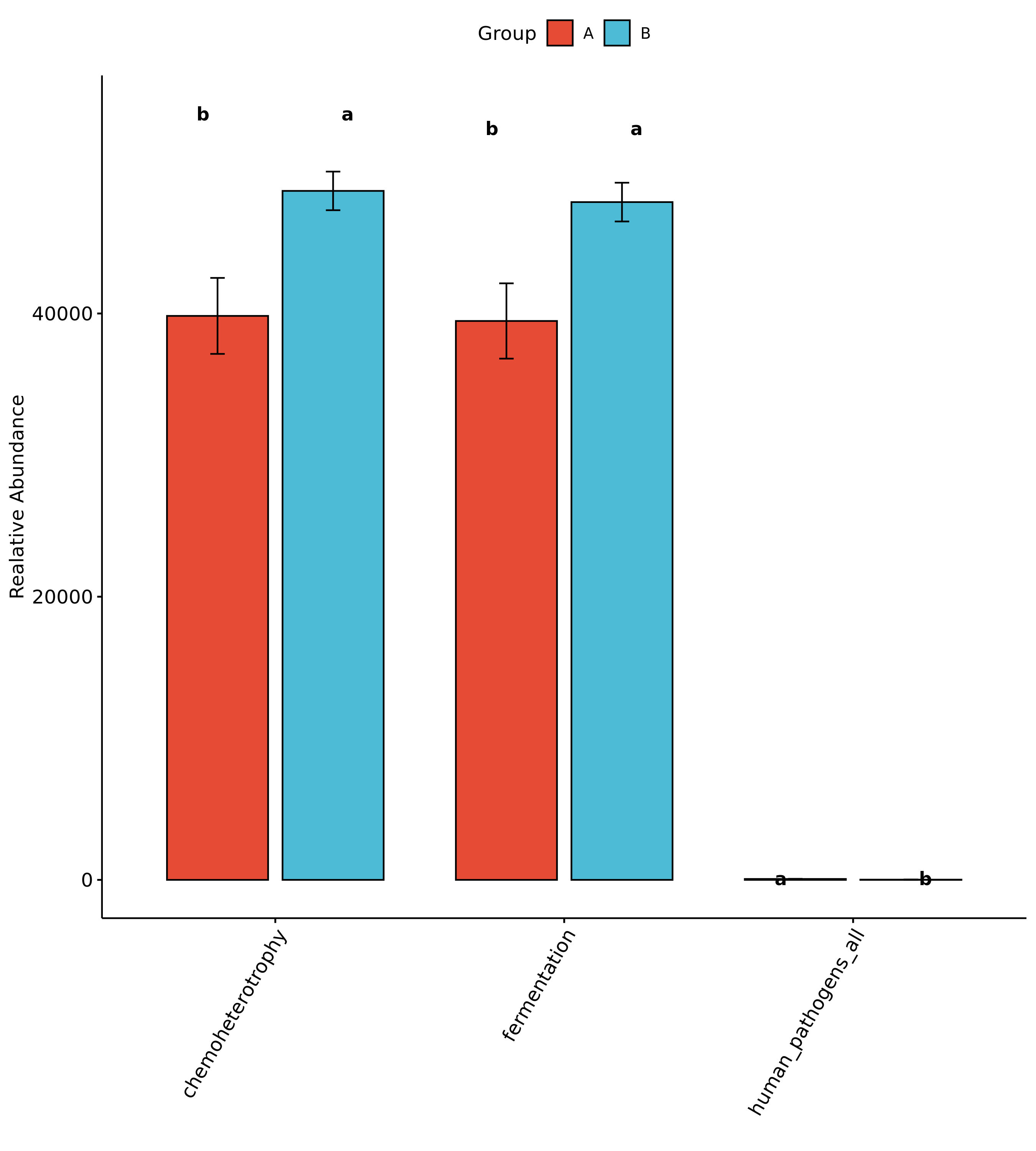
6.14 FAPROTAX生态功能预测
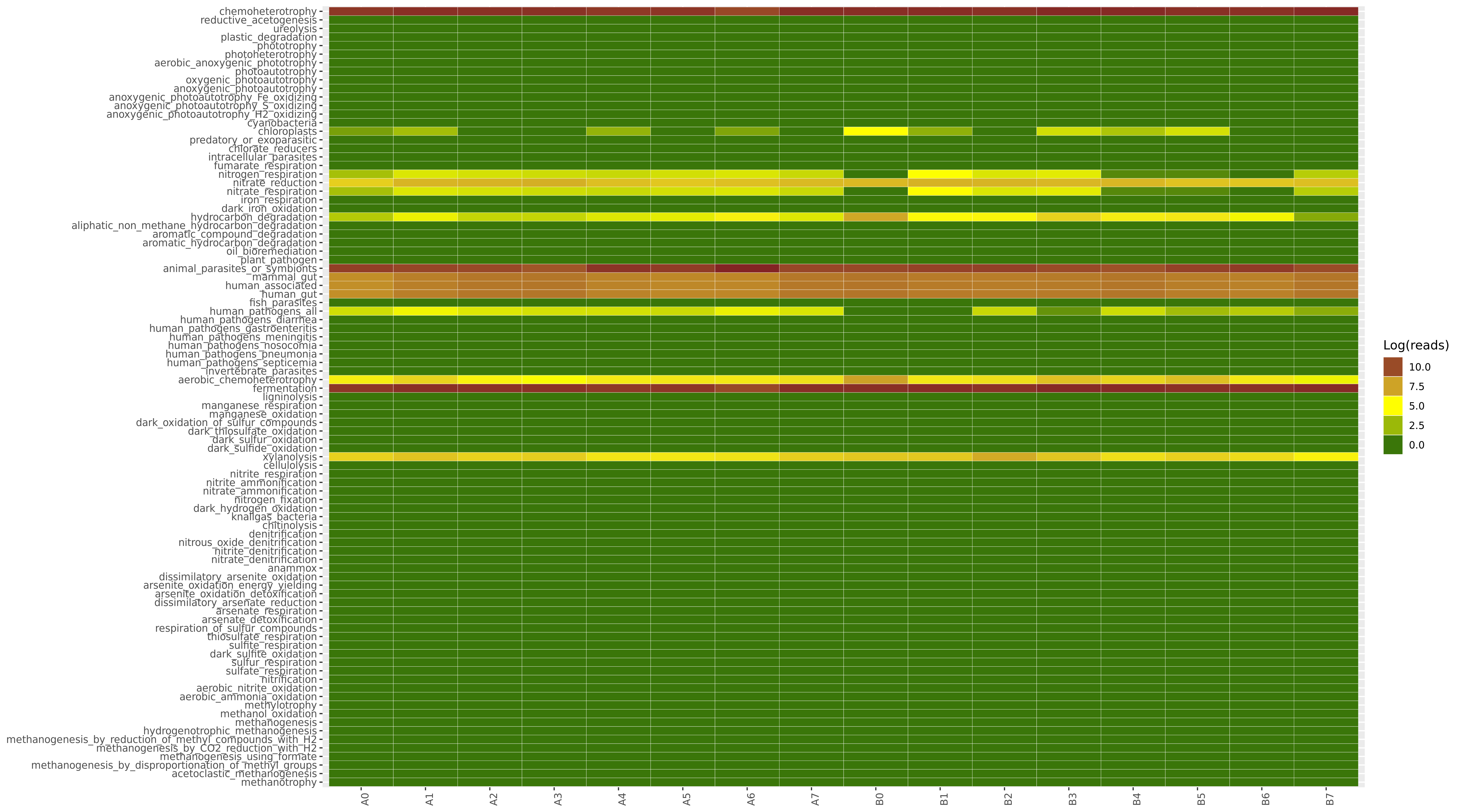
FAPROTAX是一款在2016年发表在 SCIENCE上的较新的基于16S测序的功能预测软件。它整合了多个已发表的可培养菌文章的原核功能数据库,数据库包含超过4600个物种的7600多个功能注释信息,这些信息共分为nitrate respiration, methanogenesis, fermentation 和 plant pathogenesis 等 80 多个功能分组。
FAPROTAX 是基于目前对可培养菌的文献资料手动整理的原核功能注释数据库,其包含了收集自 4600 多个原核微生物的 80 多个功能分组(如硝酸盐呼吸、产甲烷、发酵、植物病原等)的 7600 多条功能注释信息。如果PICRUSt在肠道微生物研究更为适合,那么FAPROTAX尤其适用于生态环境研究,特别是地球化学物质循环分析。FAPROTAX 适用于对环境样本(如海洋、湖泊等)的生物地球化学循环过程(特别是碳、氢、氮、磷、硫等元素循环)进行功能注释预测。因其基于已发表验证的可培养菌文献,其预测准确度可能较好,但相比于上述PICRUSt 和 Tax4Fun 来说预测的覆盖度可能会降低。参考文献: Louca, S., Parfrey, L. W. & Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277(2016).
各项分类组间差异统计结果见:/FAPROTAX/Markers/目录
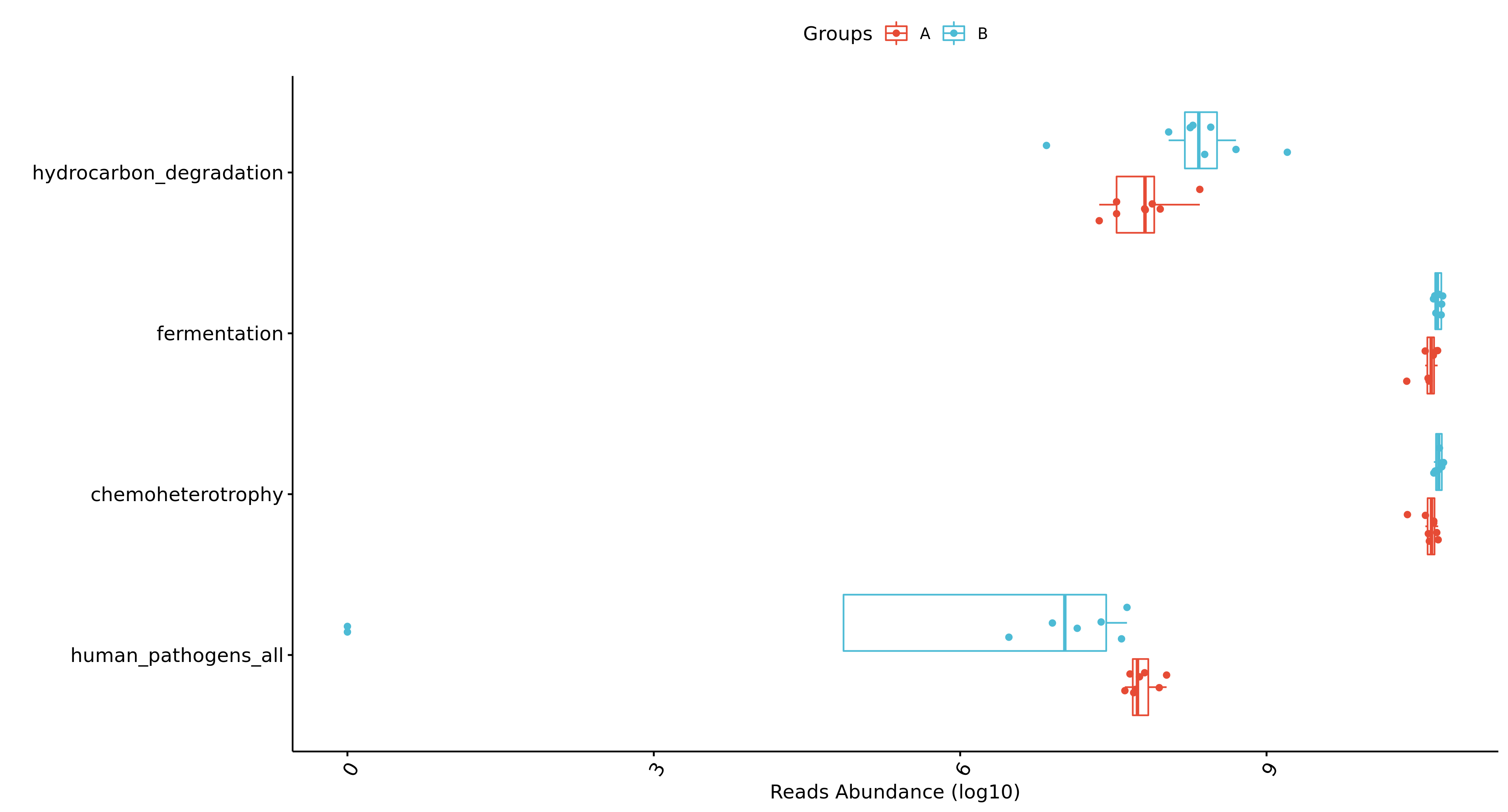
图解读:FAPROTAX 可根据 16S 序列的分类注释结果对微生物群落功能(特别是生物地化循环相关)进行注释预测。图中横坐标代表样本,纵坐标表示包括碳、氢、氮、硫等元素循环相关及其他诸多功能分组。 可快速用于评估样品来源或特征
 |
|
 |
|
| Fig 6-14-1 FAPROTAX生态功能预测热图 |
|
Fig 6-14-2 FAPROTAX生态功能非参数统计检验 |
|
|
|
|
|
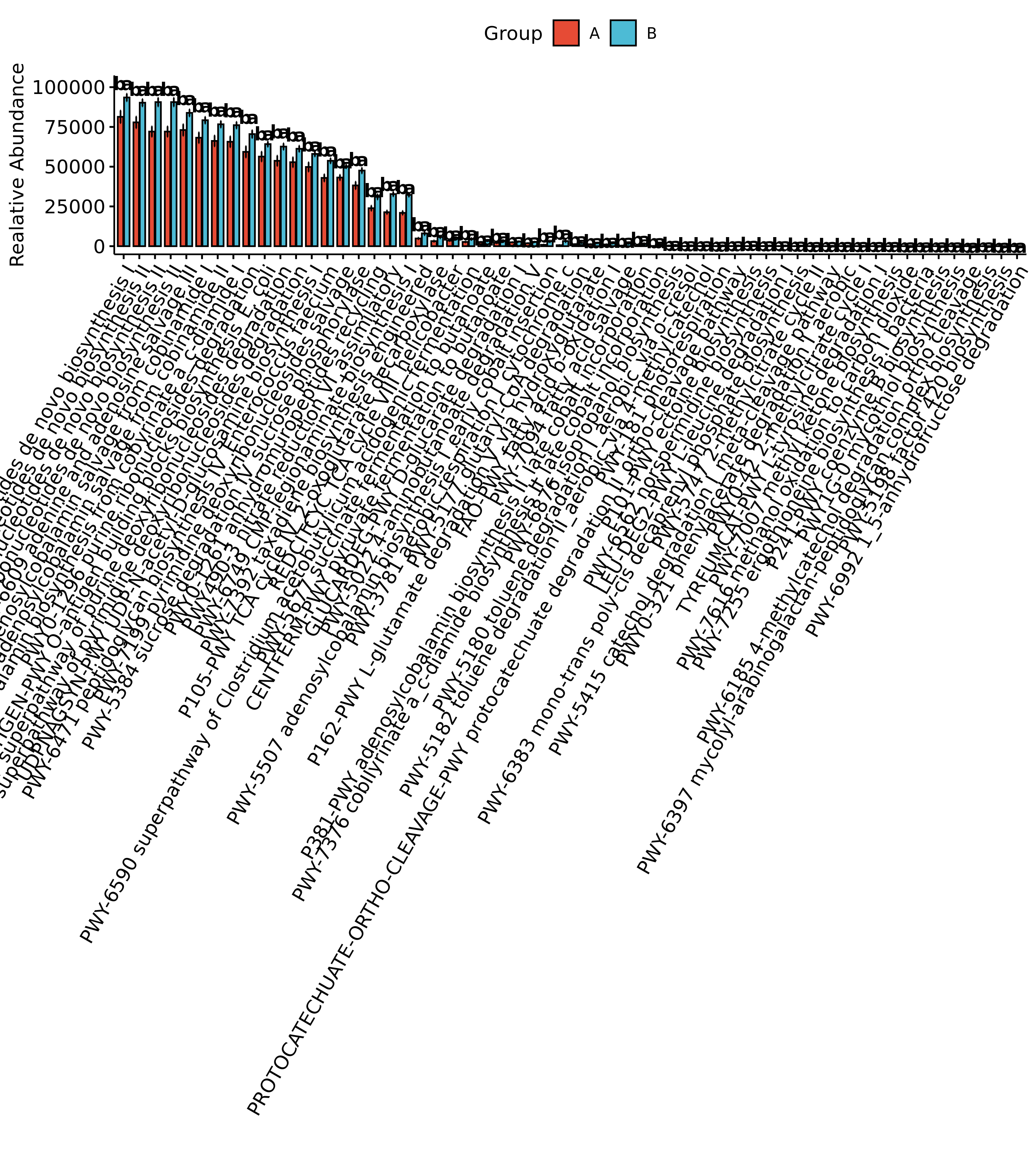
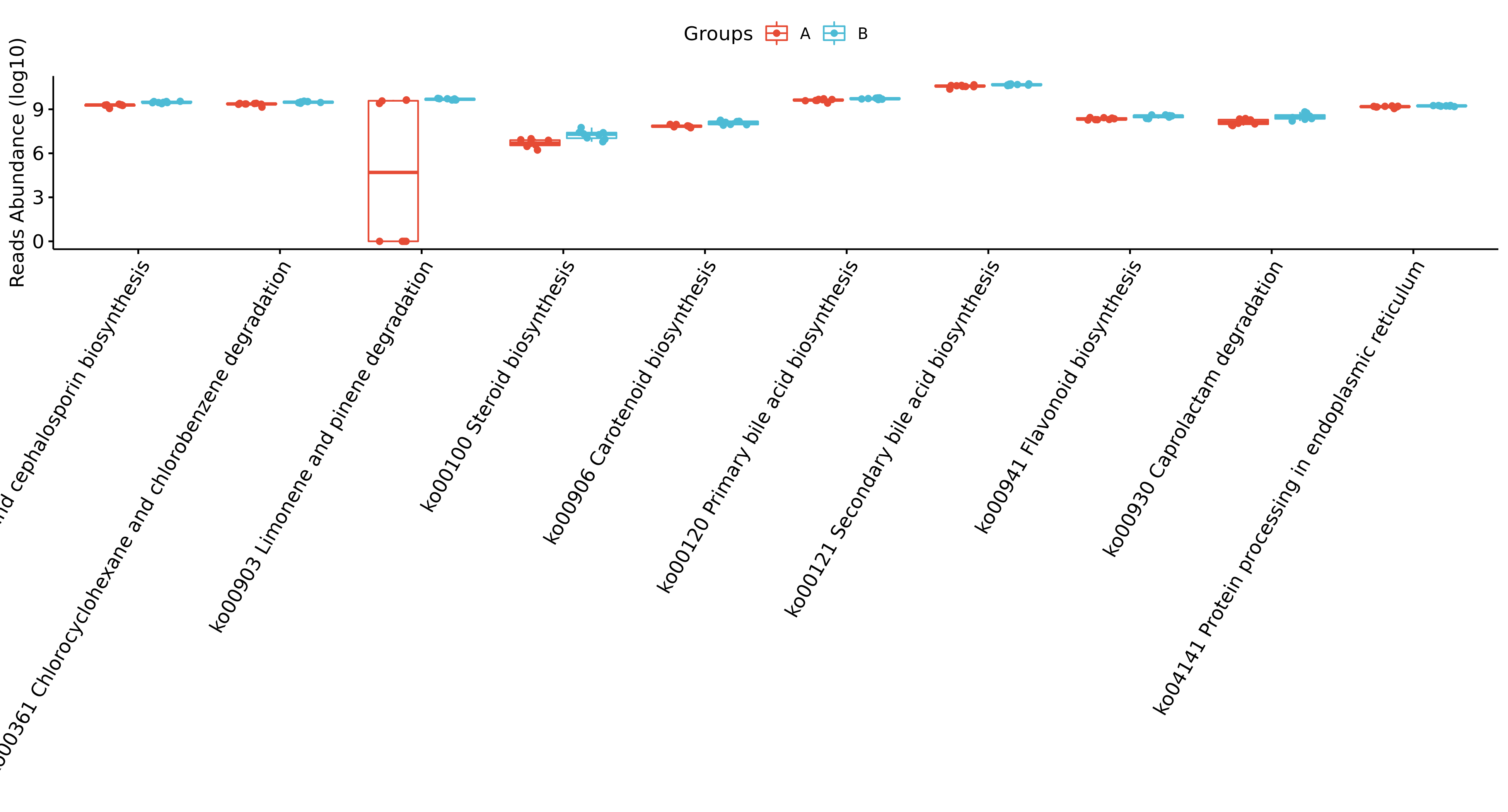
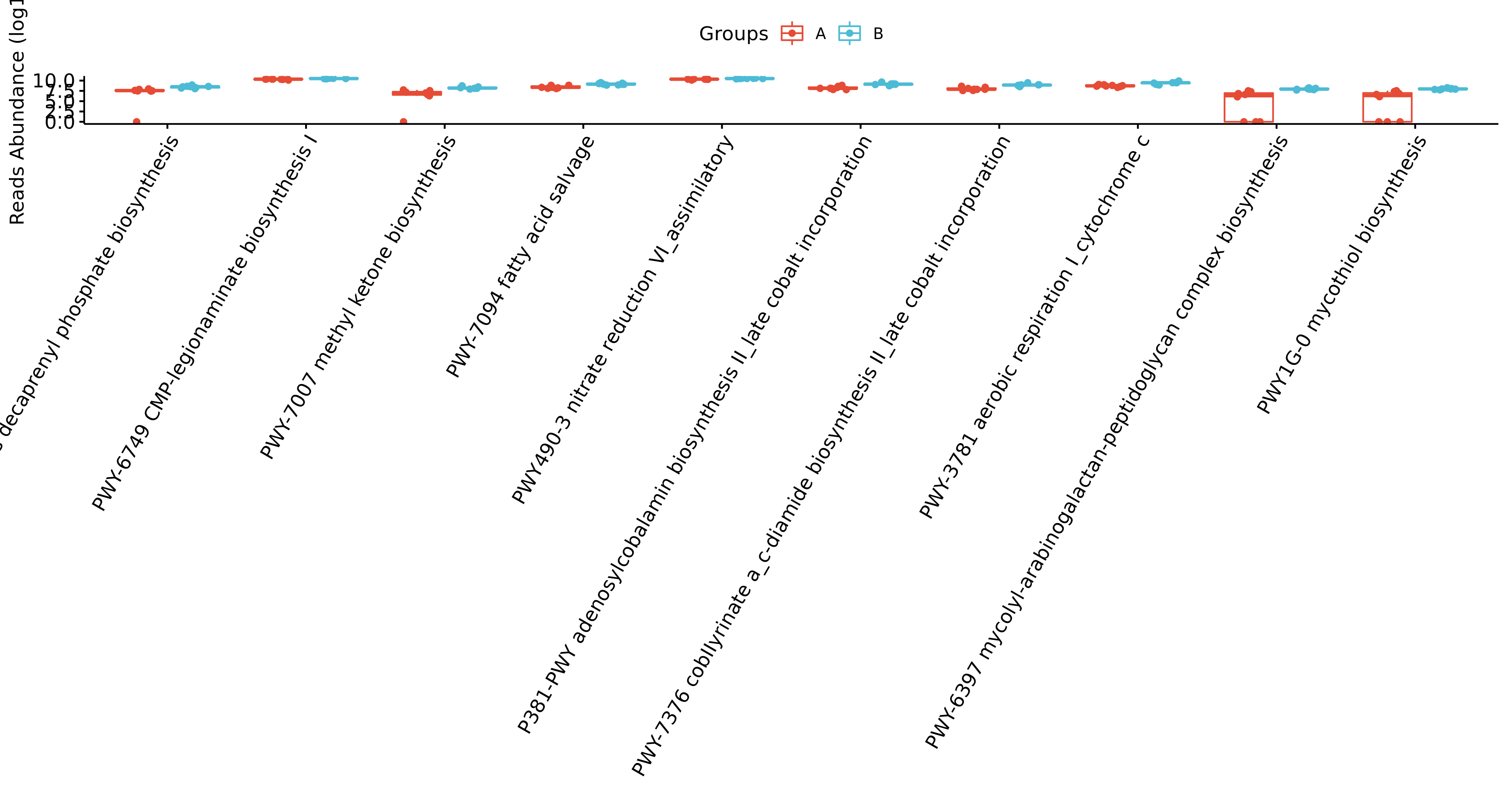
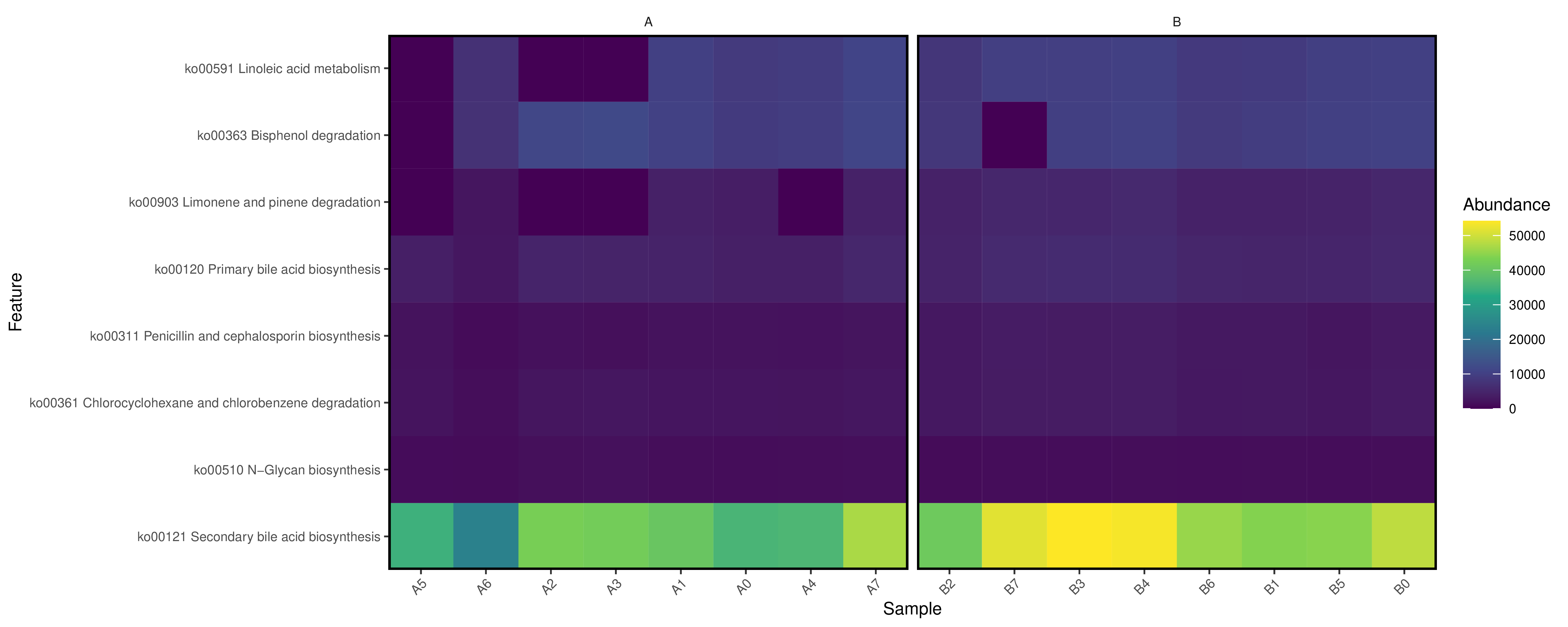
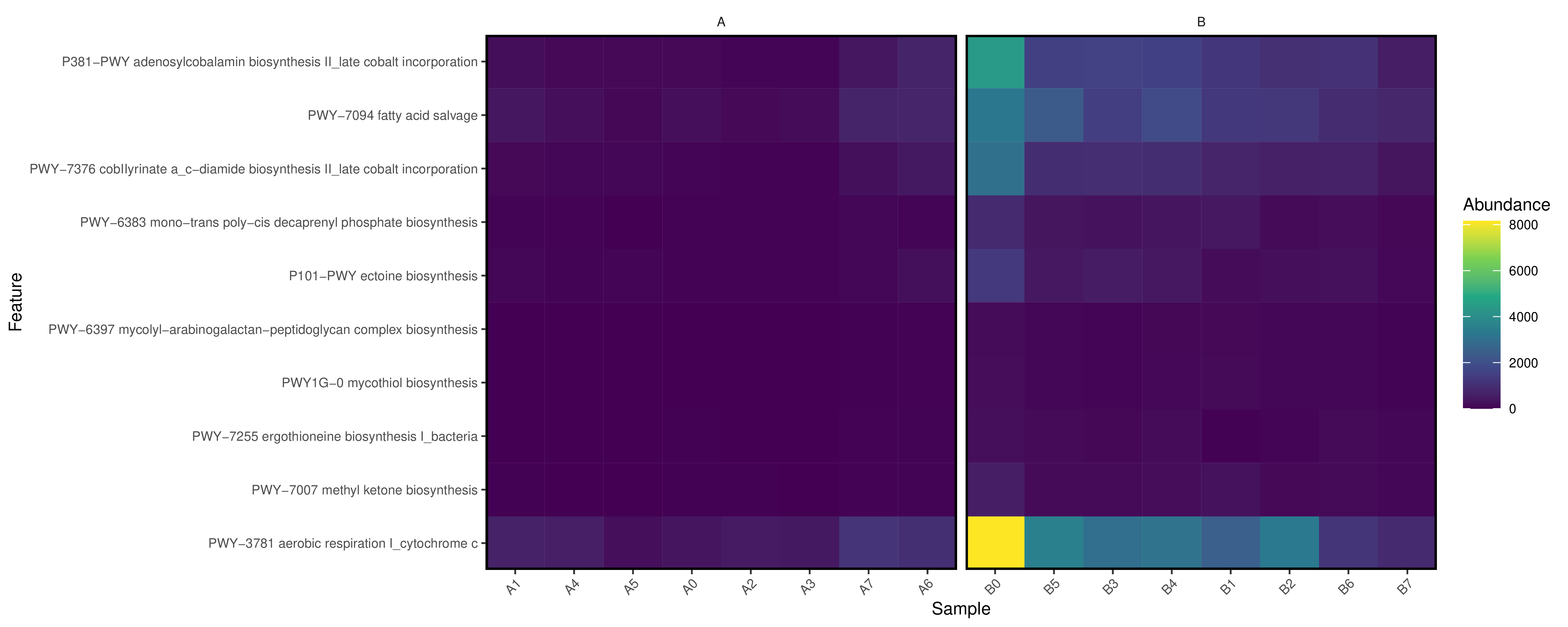
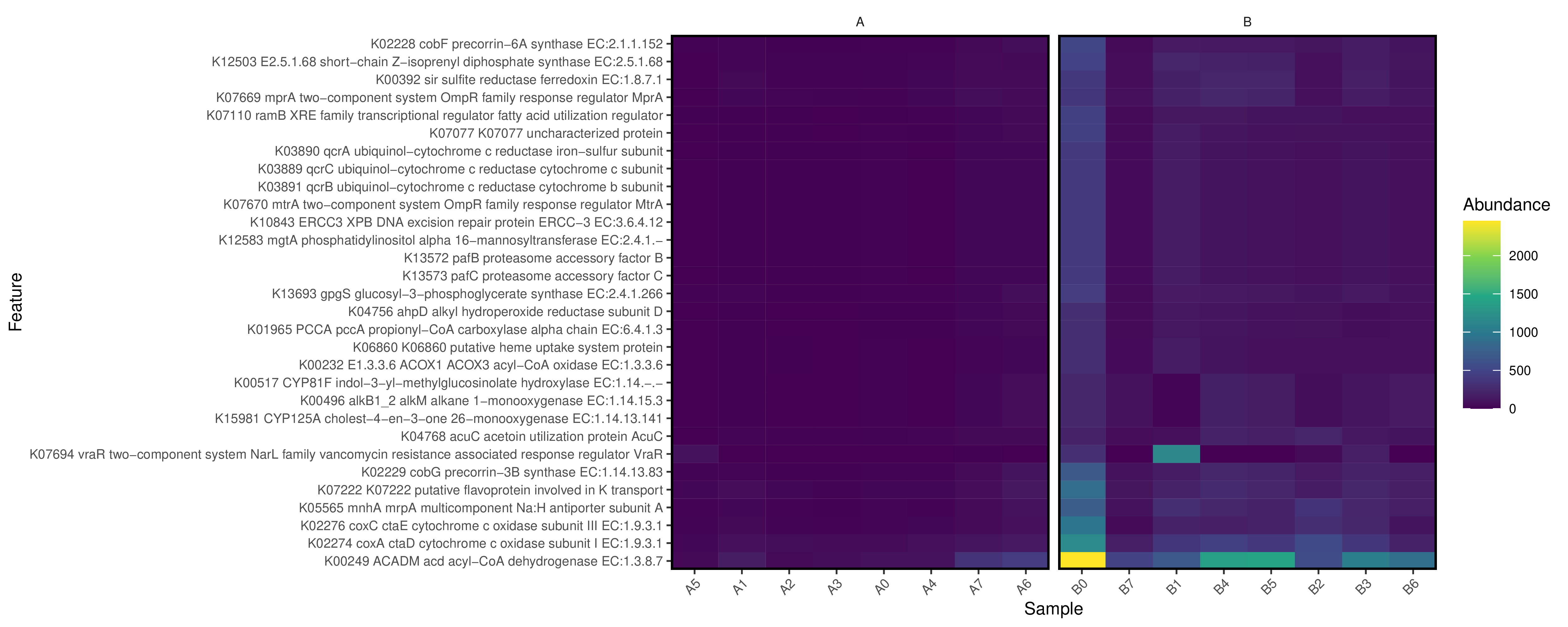
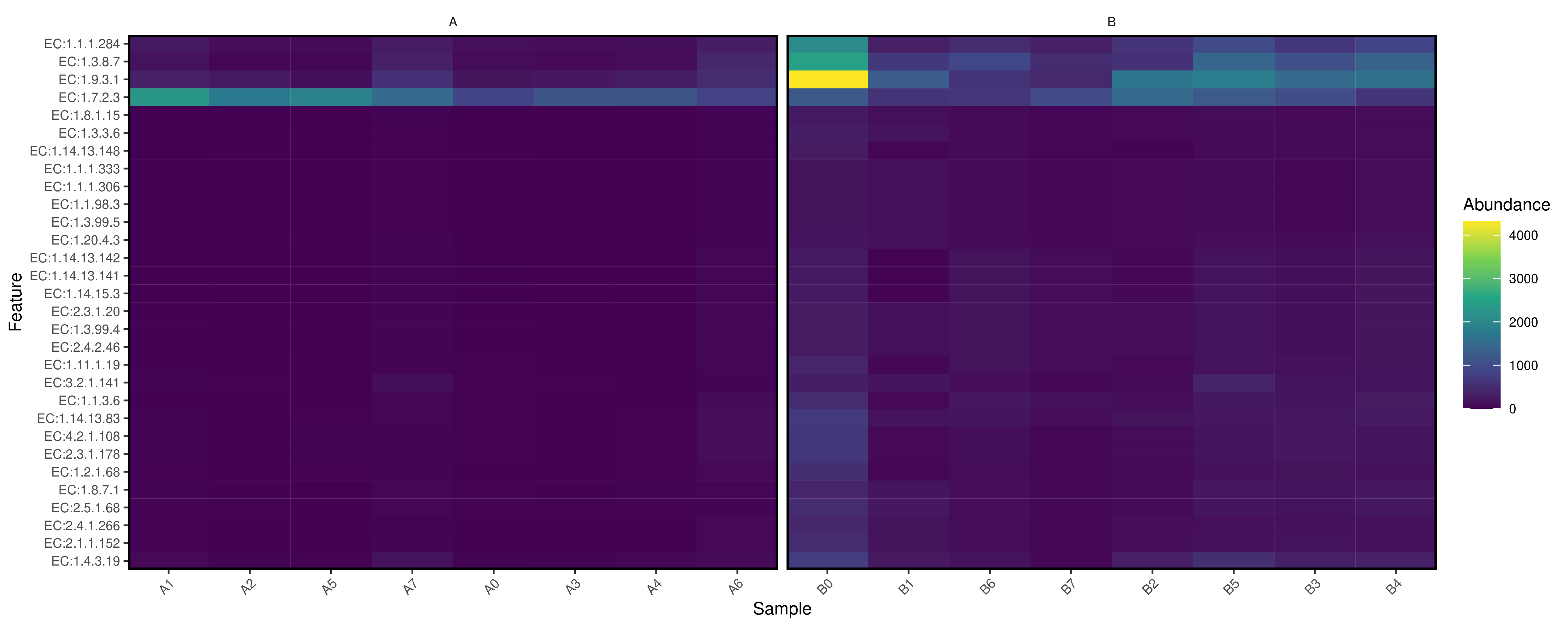
6.15 Picrust2功能预测分析
PICRUSt 是最早被开发的基于 16S rRNA 基因序列预测微生物群落功能的工具,通过对已有测序微生物基因组的基因功能的构成进行分析后,我们可以通过 16s 测序获得的物种构成推测样本中的功能基因的构成,从而分析不同样本和分组之间在功能上的差异(PICRUSt Nature Biotechnology, 1-10. 8 2013)。通过对宏基因组测序数据功能分析和对应 16s 预测功能分析结果的比较发现,此方法的准确性在 84%-95%,对肠道微生物菌群和土壤菌群的功能分析接近 95%,能非常好的反映样品中的功能基因构成。为了能够通过 16s 测序数据来准确的预测出功能构成,首先需要对原始16s测序数据的种属数量进行标准化,因为不同的种属菌包含的16s拷贝数不相同。然后将 16s 的种属构成信息通过构建好的已测序基因组的种属功能基因构成表映像获得预测的功能结果。基于 16S rDNA 序列的 PICRUSt 功能预测,可获得包括EC、以及KEGG的KO等基因注释和KEGG Pathways以及MetaCYC的代谢通路丰度富集情况。提供 COG,KO 基因预测以及 KEGG 代谢途径预测。 用户也可自行使用我们提供的文件和软件(STAMP)对不同层级以及不同分组之间进行统计分析和制图。
PICRUSt预测本报告使用PICRUSt2 v2.3.0-b版本,使用经过滤的OTU序列和默认参数进行预测。https://github.com/picrust/picrust2/wiki
此外在原有数据的基础上,我们增加了自建的CAZy数据库的注释分析。
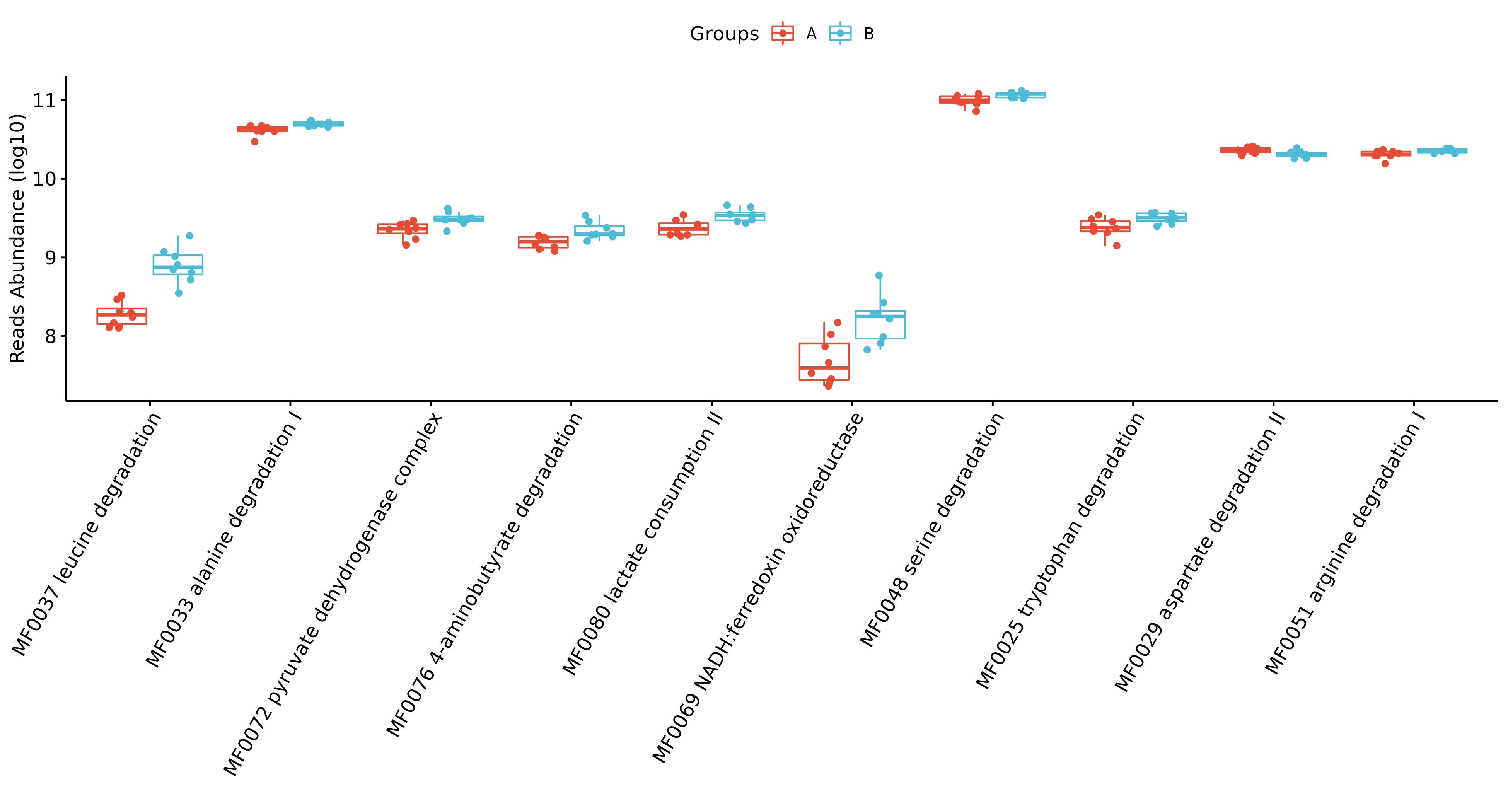
通过omixer-rpm工具,我们进一步将通路的KO基因注释转化为代谢模块GMM和神经递质模块GBM的结果,方法详见:https://github.com/raeslab/omixer-rpm,使用默认参数。
各部分预测结果在文件夹:../Picrust2下
分类组间差异统计结果见:diff_analysis/UnivarTestXXX,包括EC,KO,KEGG,METACYC以及CAZY、GMM和GBM模块。
单项结果组间差异图在各文件夹:/diff_analysis/UnivarTestXXX/figure/single_plot下
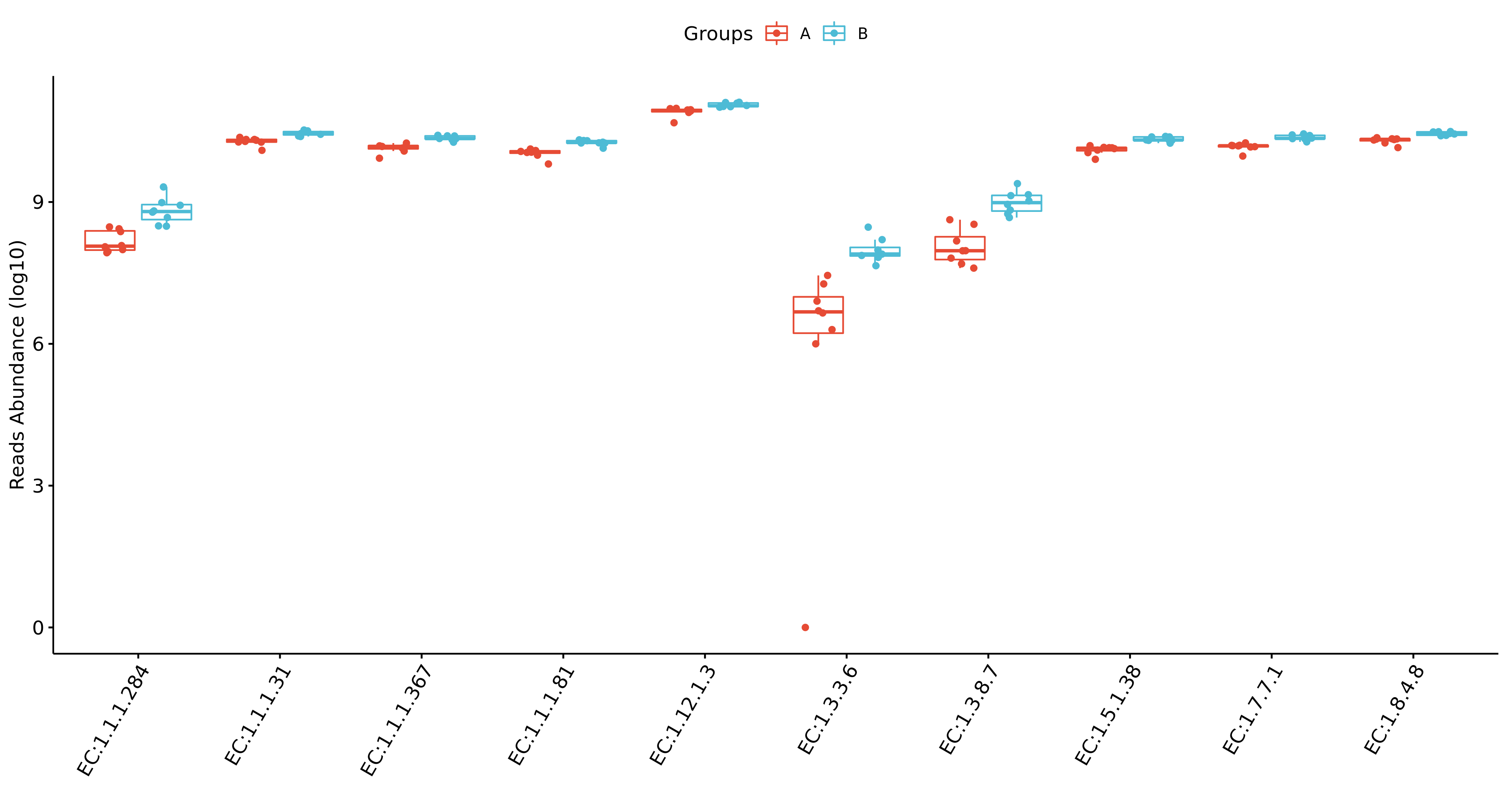
- KEGG
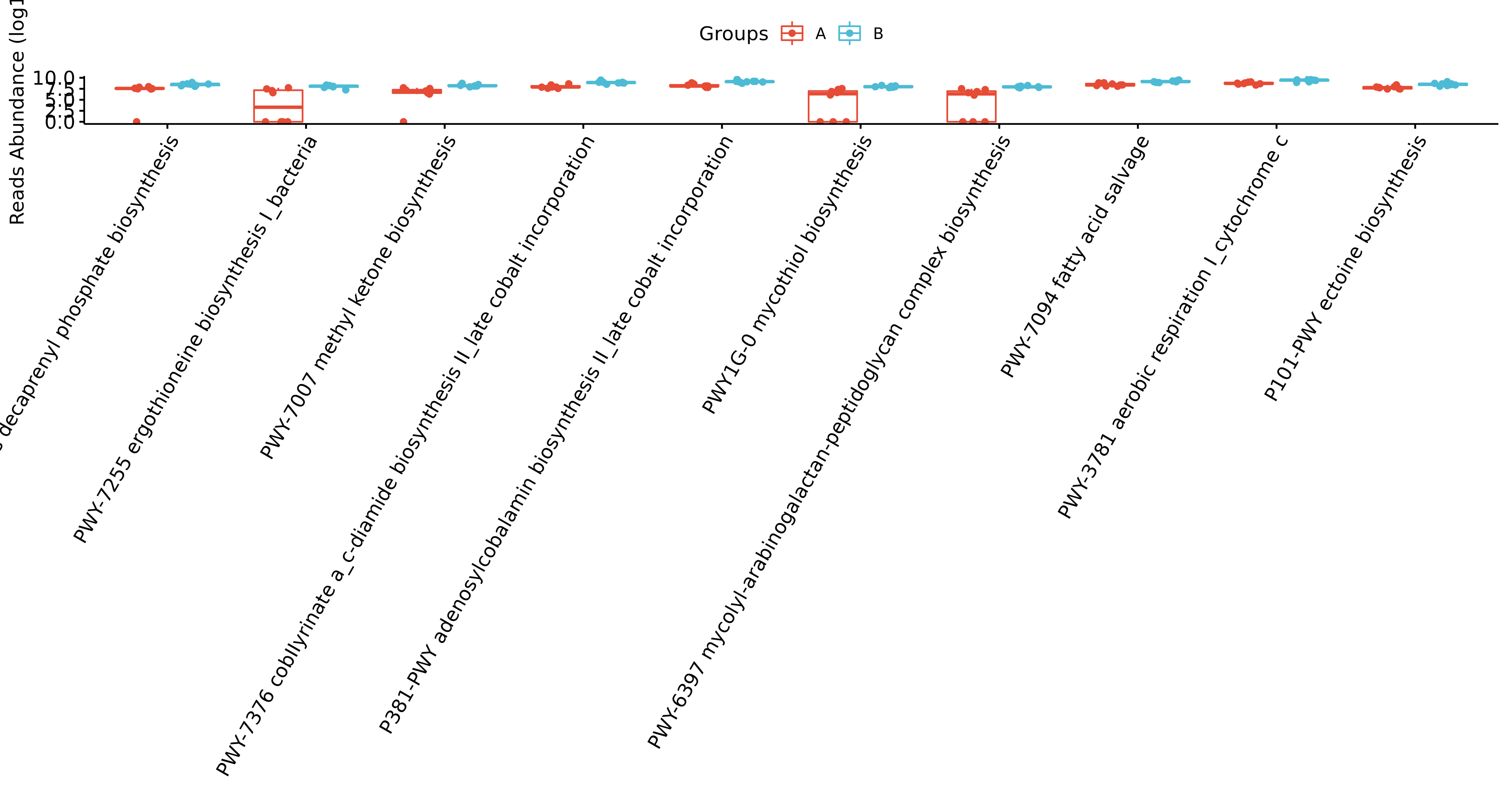
- METACYC
- CAZY
- GMM 代谢模块
- GBM 神经递质
- KO
- EC
Fig 6-15-1 功能预测组间差异分析 非参数检验 (仅显示top前十的结果,top20及完整可前往目录查看)
6.16 组间物种及功能差异分析 metagenomeSeq
metagenomeSeq是用R开发的一个包,metagenomeSeq的基本思想,用normalization实现分类注释时的biases处理,同时用零膨胀高斯分布(zero-flated Gaussian distribution)处理了测序深度所带来的影响,在此基础上,利用线性模型找到存在的差异所在。
结果在文件夹:/diff_analysis/metagenomeRXXX/下
每个物种分类组间差异统计结果图在文件夹:/diff_analysis/metagenomeRXXXX/figure/single_plot下,示例如下图:
- Genus
- Phylum
- Class
- Order
- Family
- Species
- METACYC
- KEGG
- CAZY
- GMM 代谢模块
- GBM 神经递质
- KO
- EC
Fig 6-16-1 组间差异物种 metagenomeSeq分析方法 (仅显示top前十的结果,top20及完整可前往目录查看)
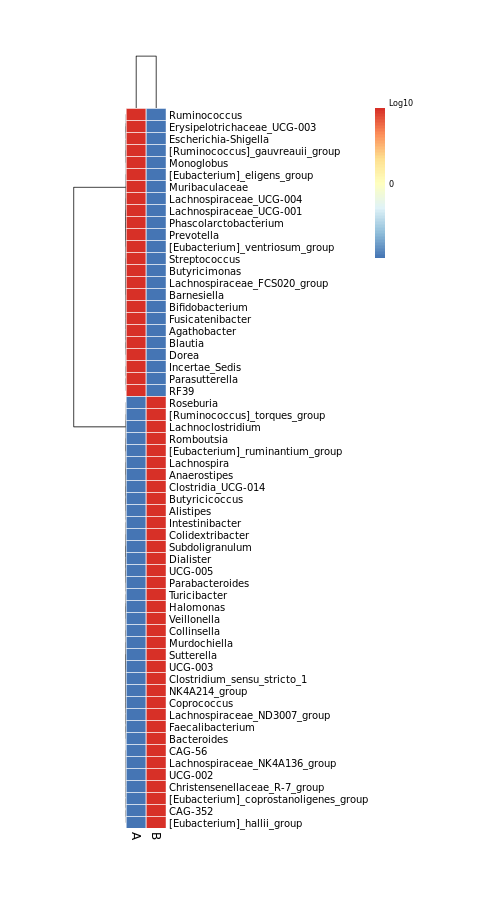
6.17 组间物种及功能差异热图 基于metagenomeSeq结果
结果在文件夹:/diff_analysis/heatmap下
- Genus
- Phylum
- Class
- Order
- Family
- Species
- Bugbase
- FAPROTAX
- METACYC
- KEGG
- CAZY
- GMM 代谢模块
- GBM 神经递质
- KO
- EC
Fig 6-17-1 组间差异物种及功能热图 metagenomeSeq分析方法
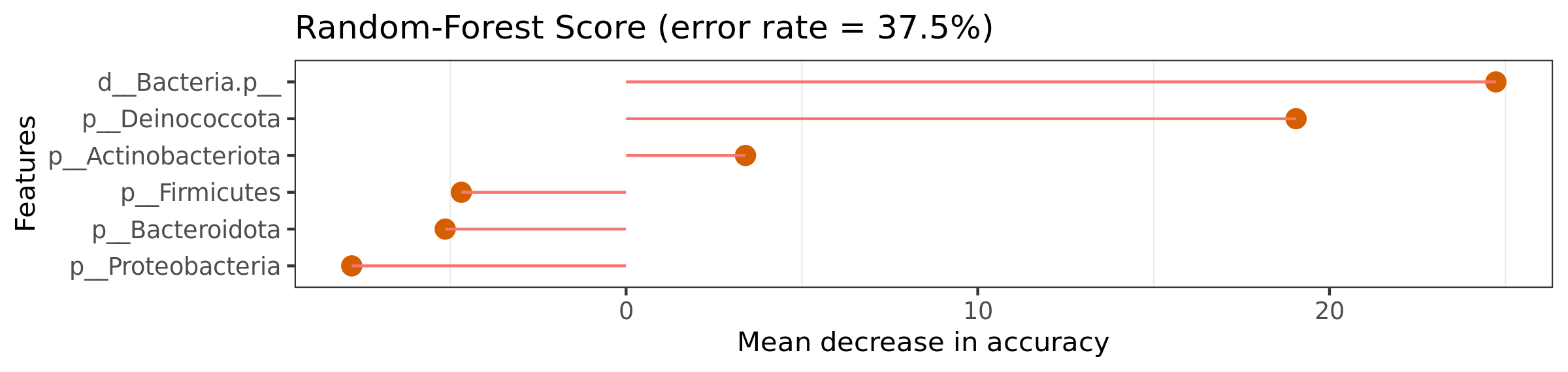
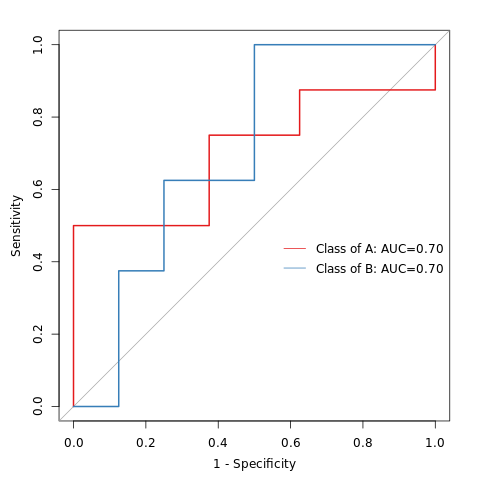
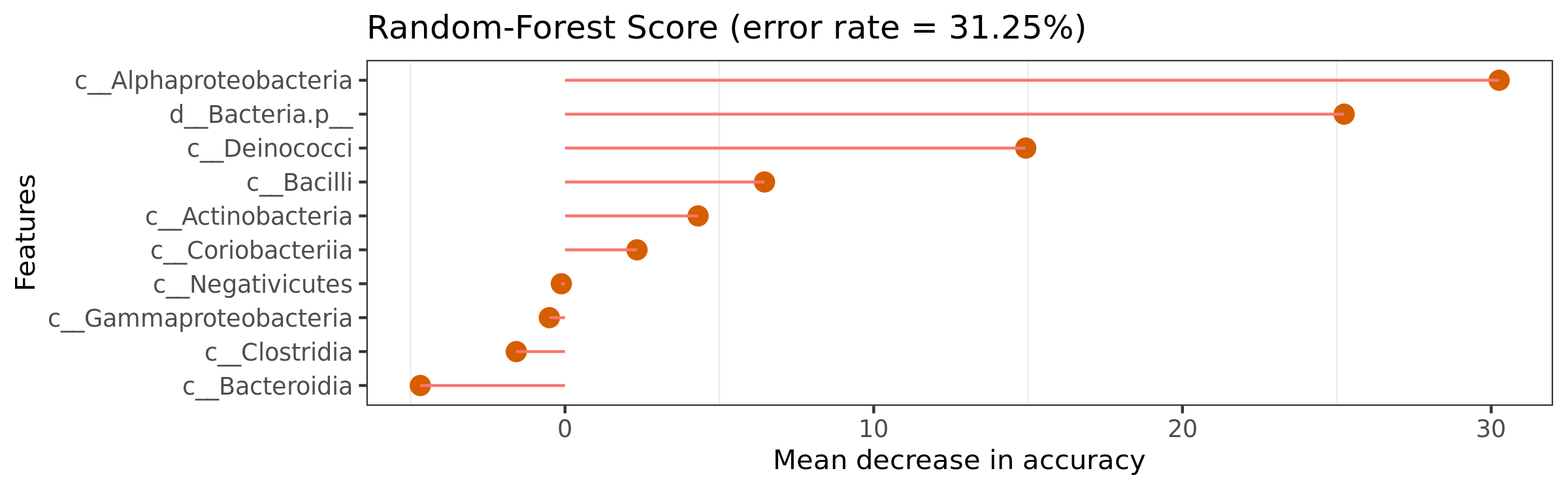
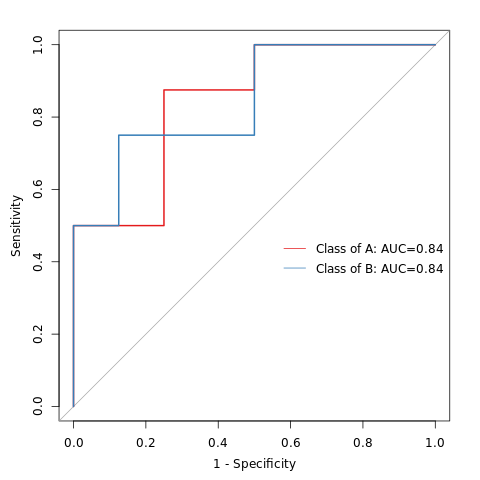
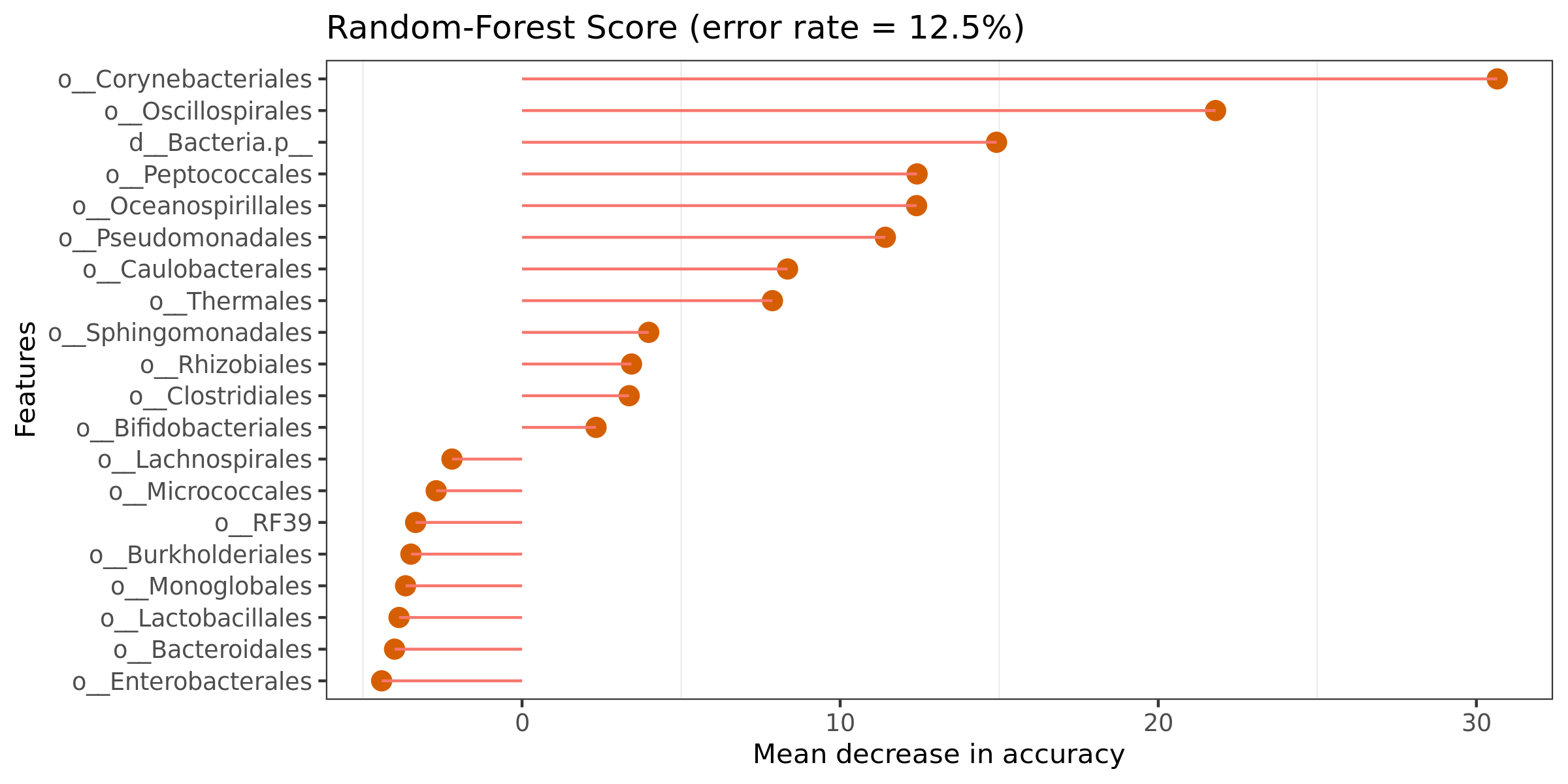
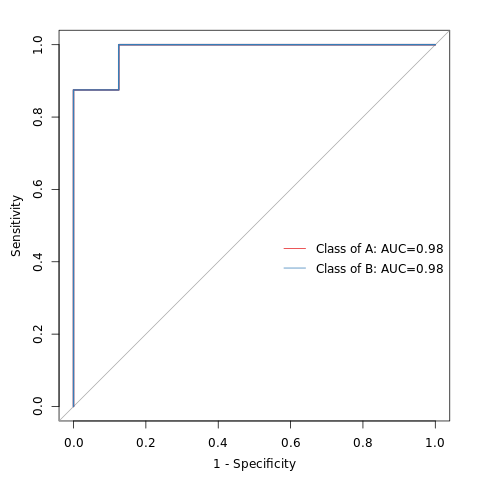
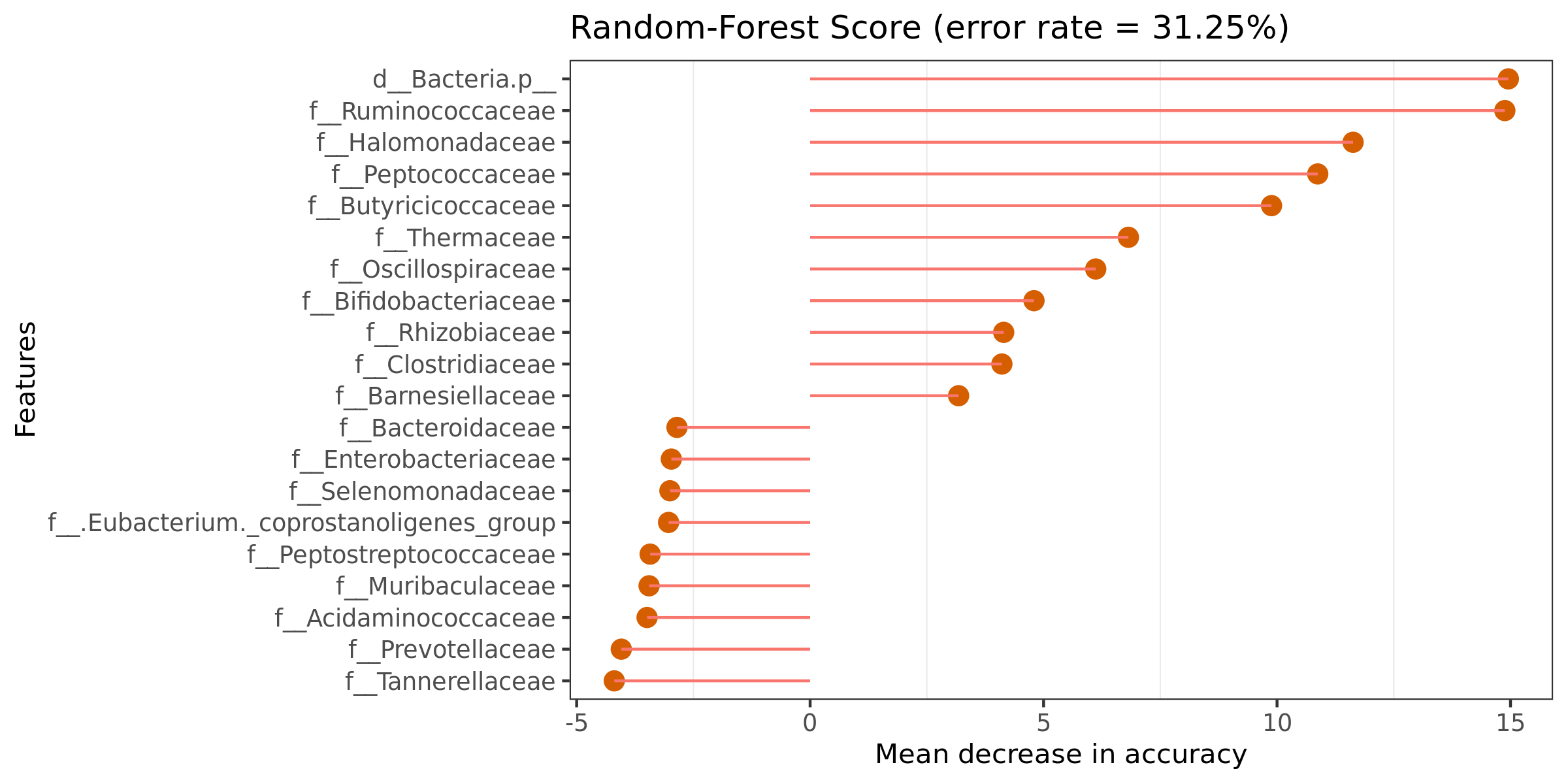
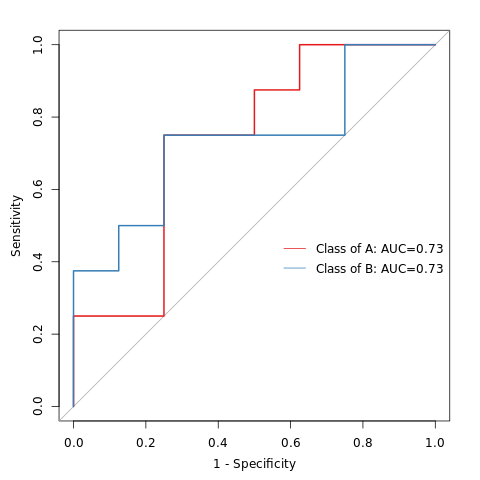
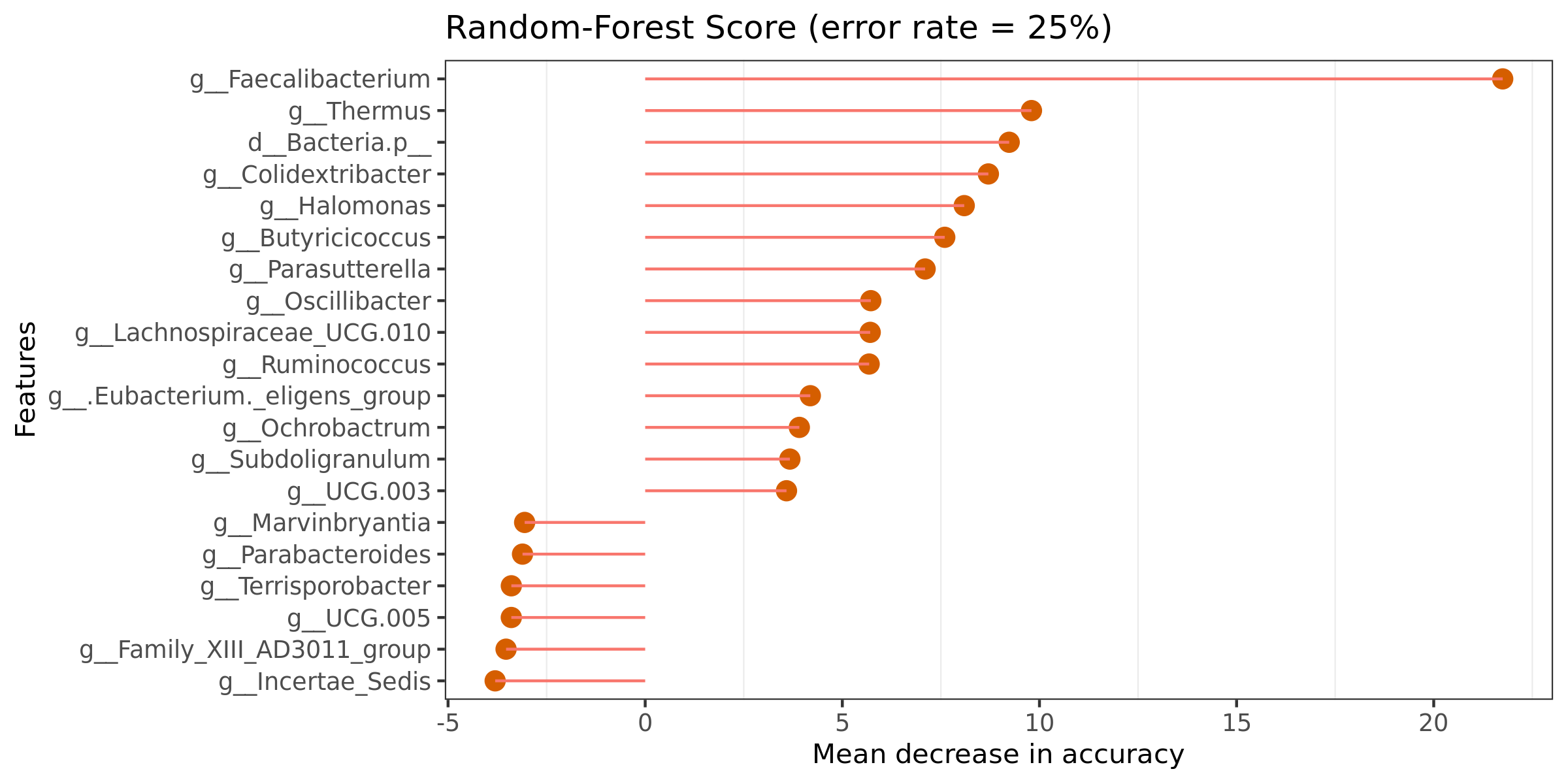
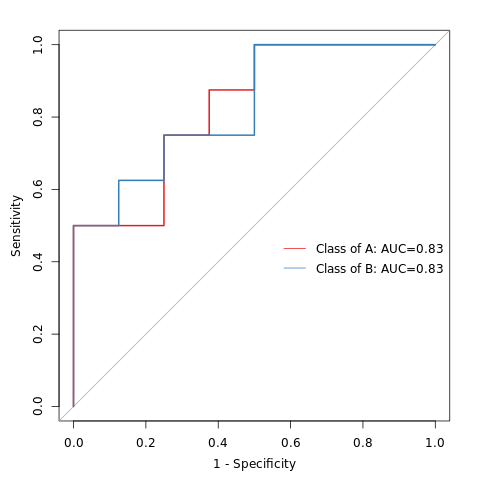
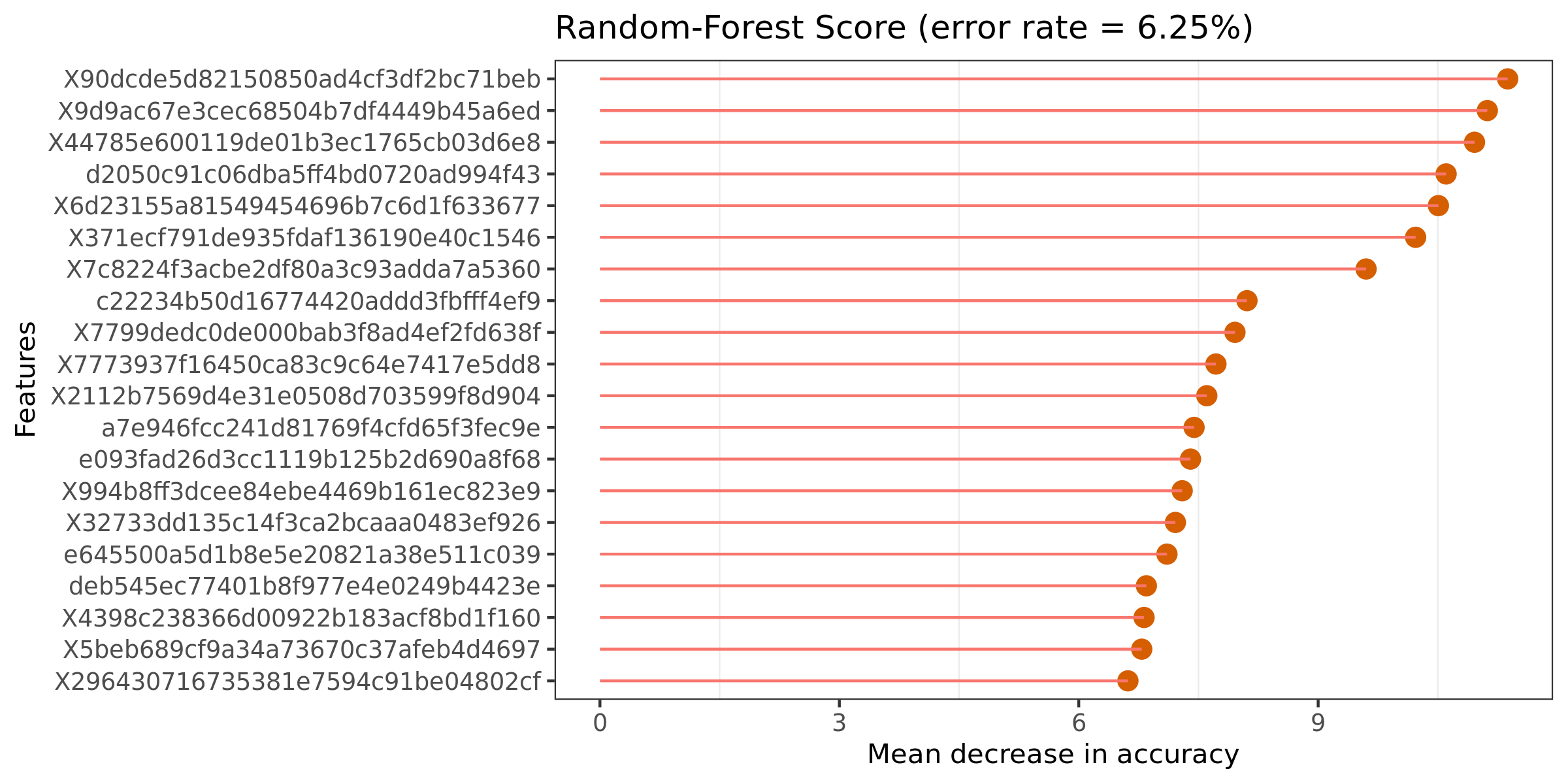
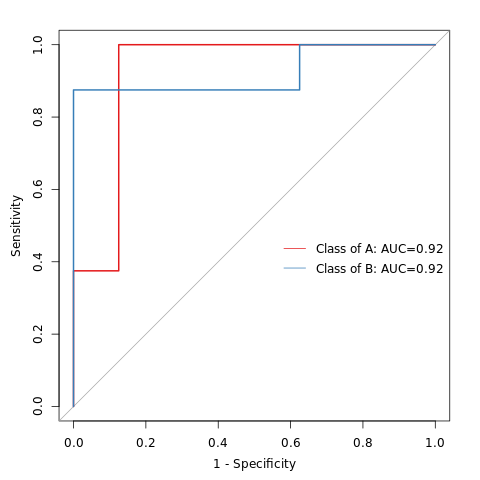
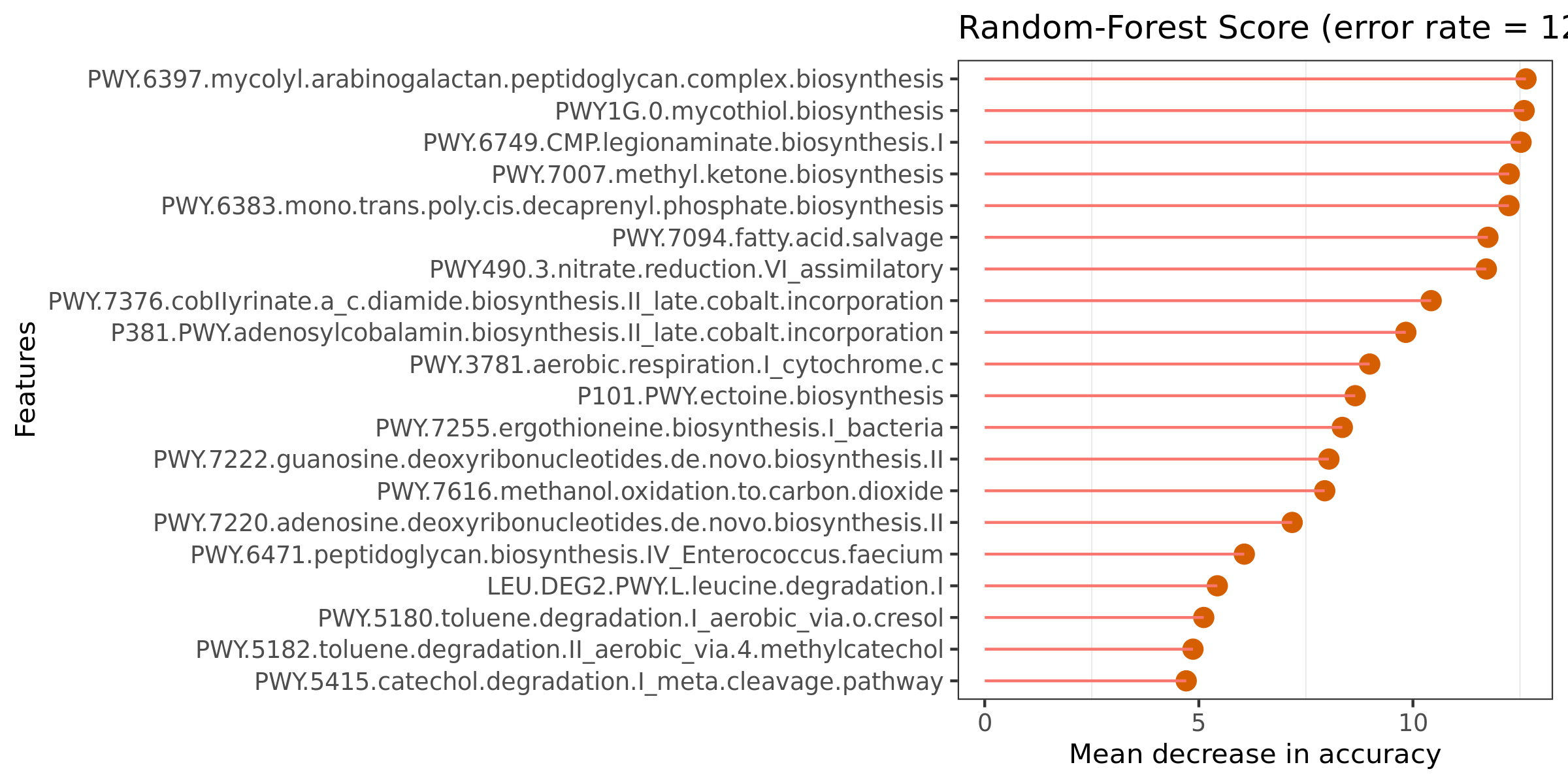
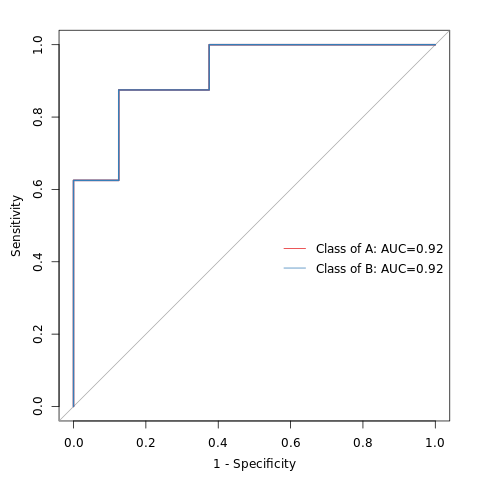
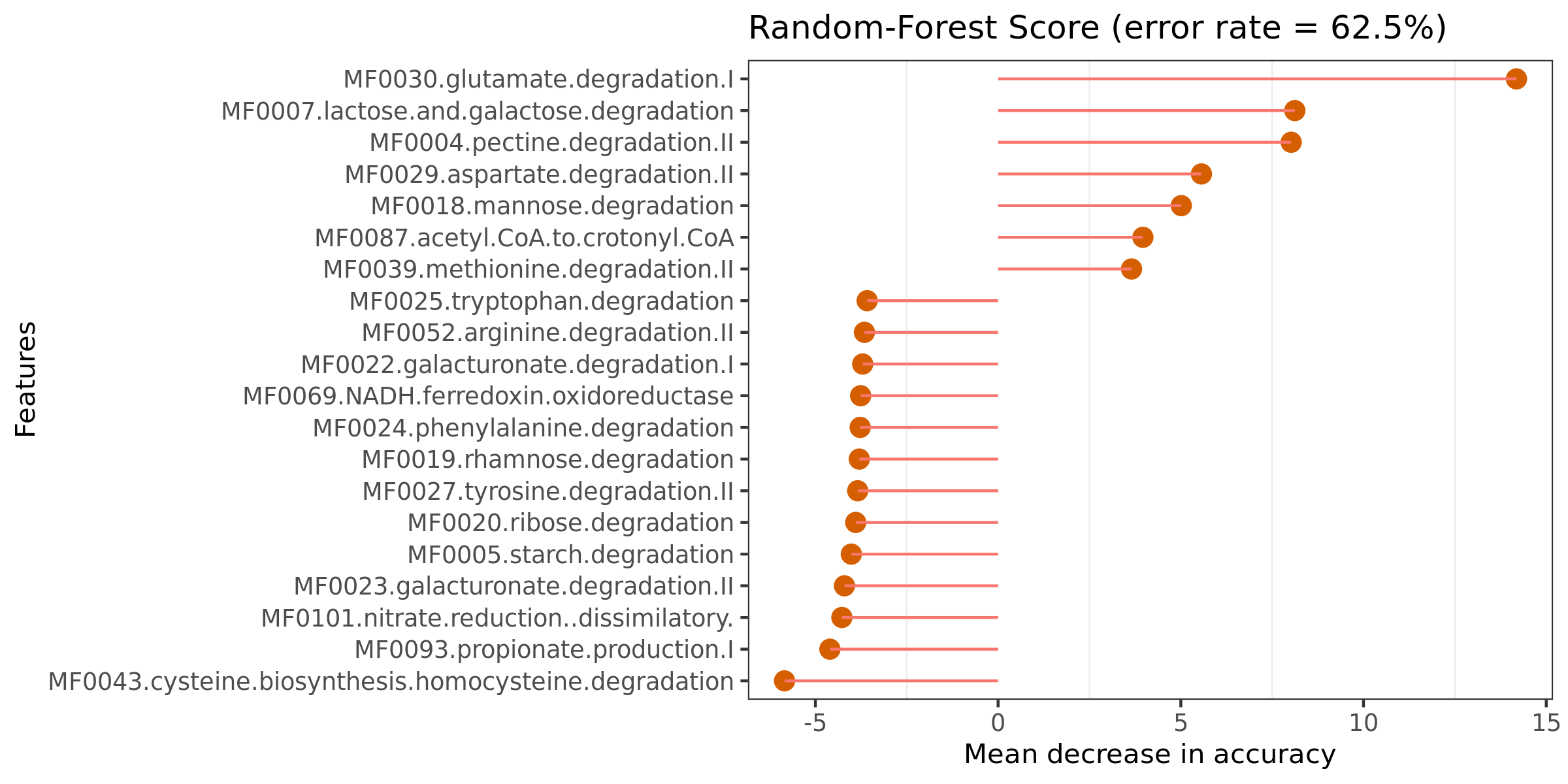
6.19 随机森林组间预测
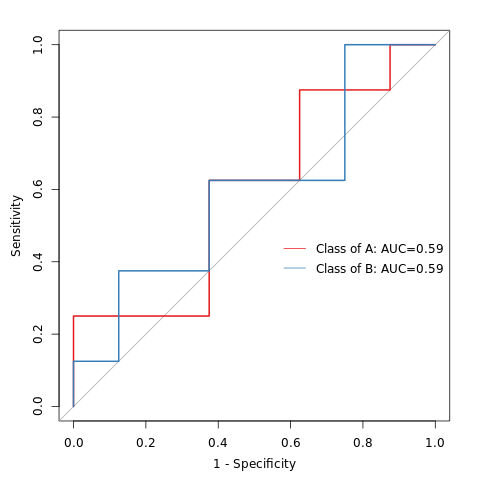
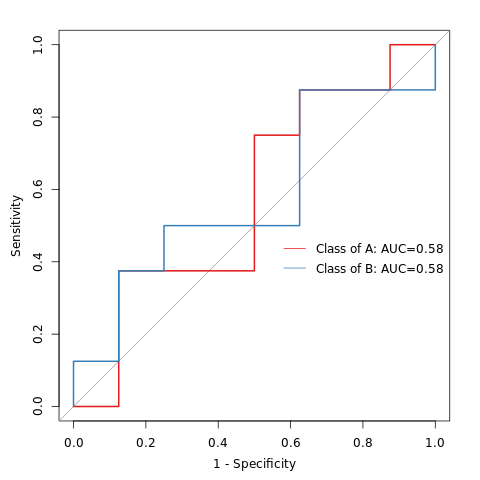
ROC曲线下的面积值在 1.0 和 0.5 之间。在 AUC>0.5 的情况下,AUC 越接近于 1,说明诊断效果越好。AUC在 0.5~0.7 时有较低准确性,AUC在 0.7~0.9 时有一定准确性,AUC 在 0.9 以上时有较高准确性。AUC=0.5 时,说明诊断方法完全不起作用,无诊断价值。AUC<0.5 不符合真实情况,在实际中极少出现。
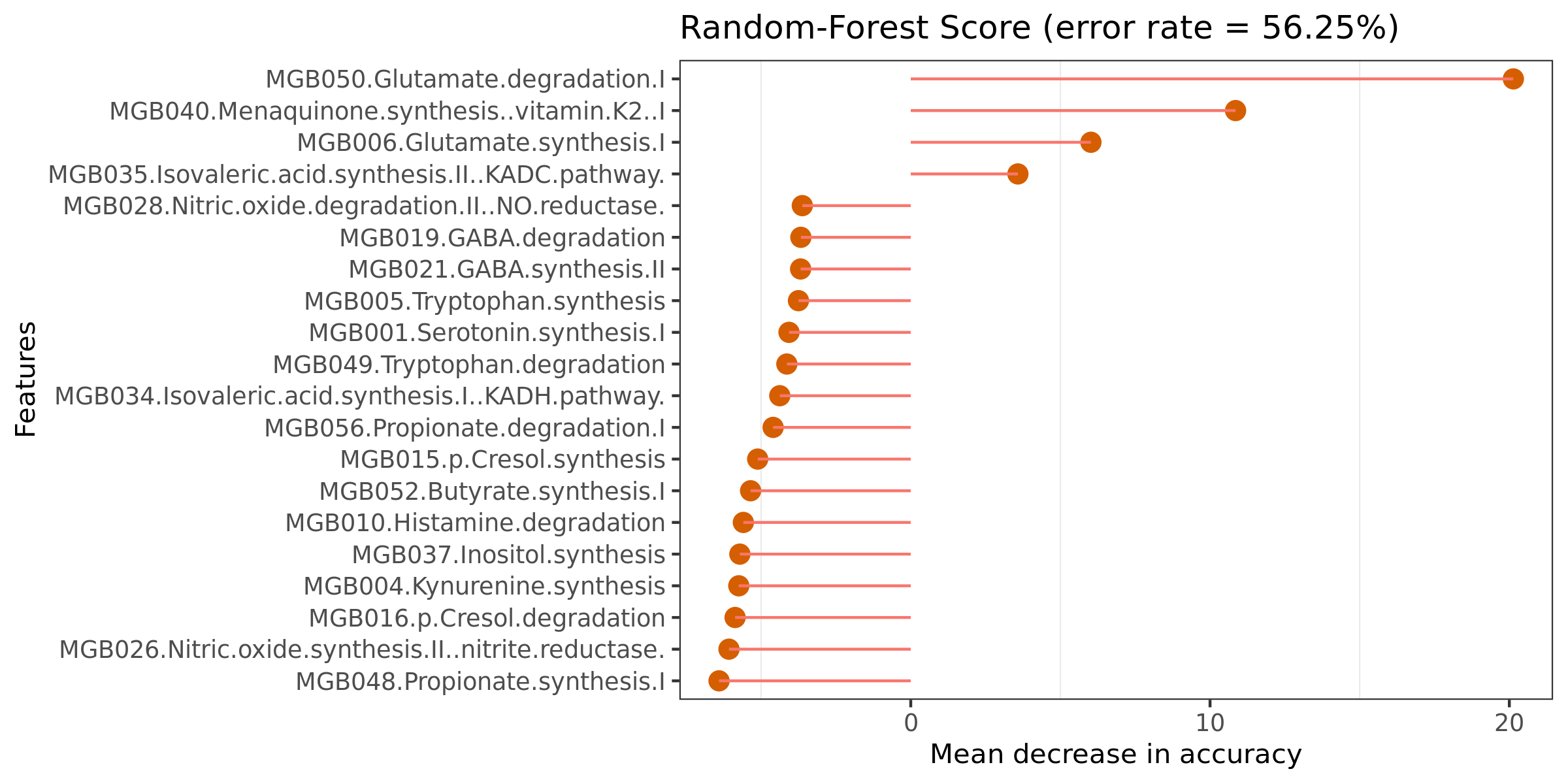
特征重要性点图。横坐标为重要性水平,纵坐标为按照重要性排序后的物种名称。上图反映了分类器中对分类效果起主要作用的特征,按重要性从大到小排列。Error rate :表示使用下方的特征进行随机森林方法预测分类的错误率,数值越高表示基于特征分类准确度不高,可能分组之间特征不明显。分值越低证明分组效果比较好。
结果在文件夹:/diff_analysis/RF/下
- Phylum
- Class
- Order
- Family
- Genus
- OTU
- METACYC
- GMM
- GBM
Fig 6-19-1 随机森林分组预测ROC及特征重要性
杭州谷禾信息技术有限公司