 国家高新企业 | ISO9001认证
国家高新企业 | ISO9001认证 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 二级病原微生物安全实验室

谷禾健康


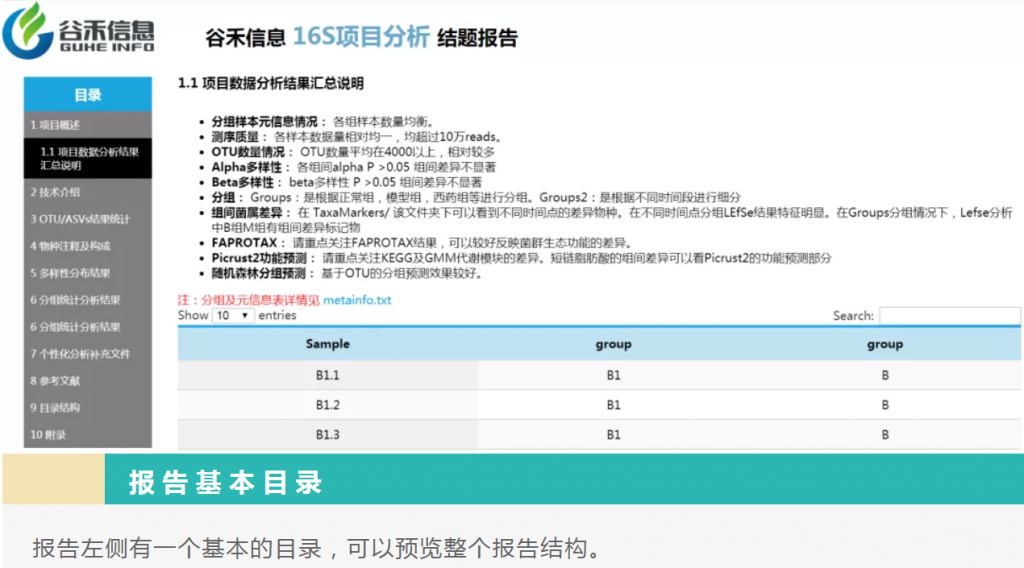
微生物多样性测序(扩增子测序)是基于二代高通量测序对16S/18S/ITS等序列进行测序。可以同时检测样本中的优势物种、稀有物种及一些未知物种的检测,获得样本的微生物群落组成以及相对丰度。
相信关注我们的小伙伴对此并不陌生。
这次我们整合了大家平时会遇到的一些问题,在原有的基础上对报告进一步完善。



重要指数 :★★★★★
这部分内容必看。
主要是汇总信息,包括样本数据量,测序质量,重复性效果评估,分组信息,组间差异评估,代谢途径上差异,功能预测等。
这里会给出本项目中的一些重要提示,帮你从众多的报告信息中获取关键的部分。


重要指数 :★★★
技术介绍这部分内容,就是说我们基于是怎么样一个测序平台、什么方法来获得的最后的数据。

如果你担心
这么直观的报告,
会不会不够详细?
小问号里有宝藏!


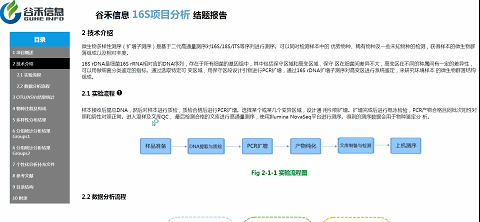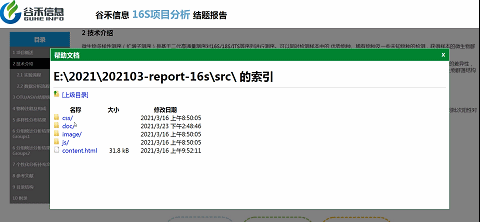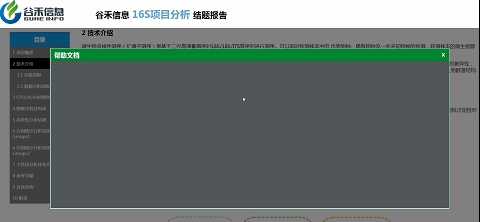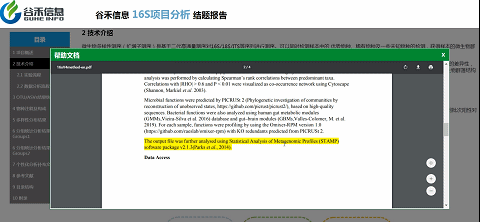

如上图,点击实验流程旁边的小问号,弹出的文件夹里就有详细的英文版方法介绍。
重要指数 :★★★★
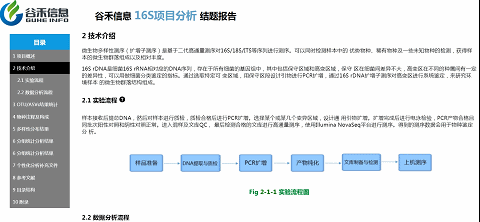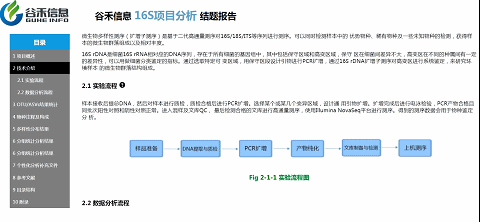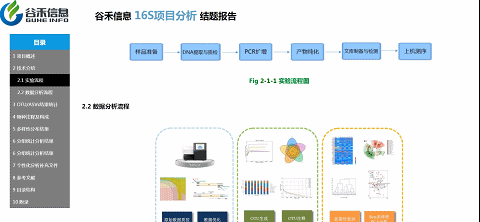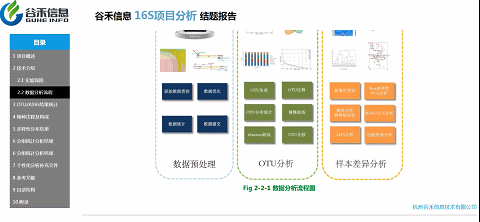
这部分内容主要是数据统计的图表:
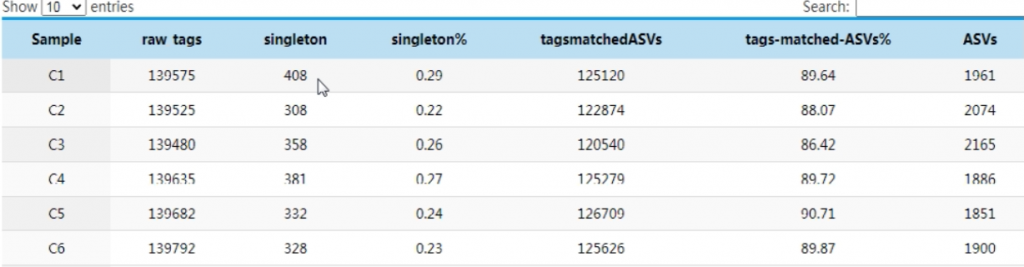

Raw-tags: 样本的原始序列数据
Singleton: 无完全匹配的单条序列数量
tagsmatchedASVs: 比对到最终ASVs的序列数据
ASVs:以及ASVs的种类个数



重要指数 :★★★★
经过SILVA138数据库的注释,得到ASVs的物种注释结果。
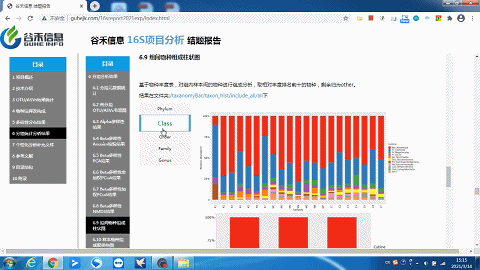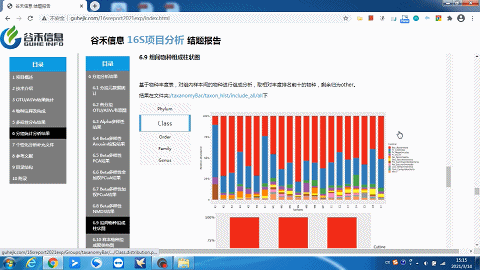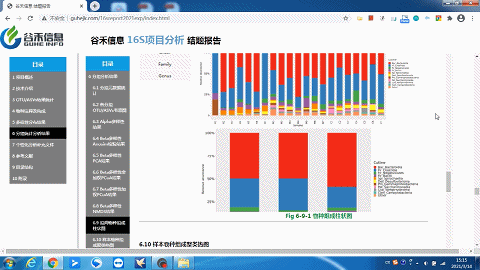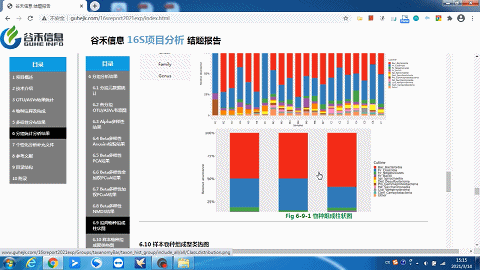
这一部分可以看到每个样本的物种构成比例,Taxonomic Level 可以选择Level1 ~ Level7 界门纲目科属种,不同分类水平下的物种构成。
这里选择level2就是“界”层级(可根据需求自选),另外比如选一个groups分组,如下:
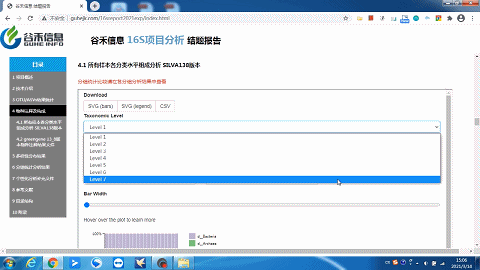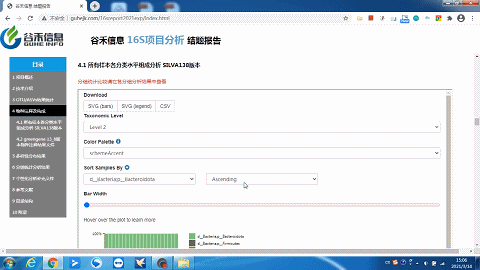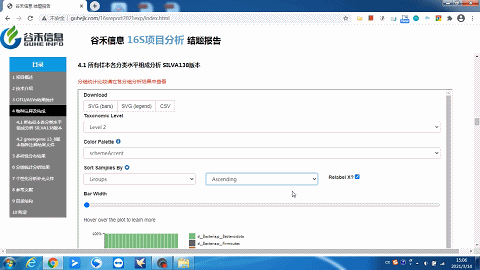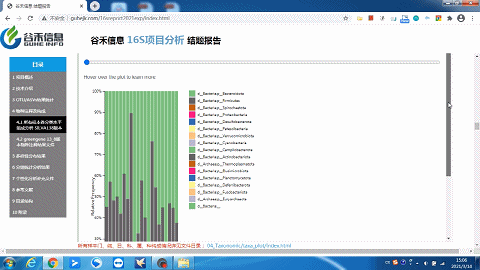

柱状图太宽?太窄?
一拉即可调整!
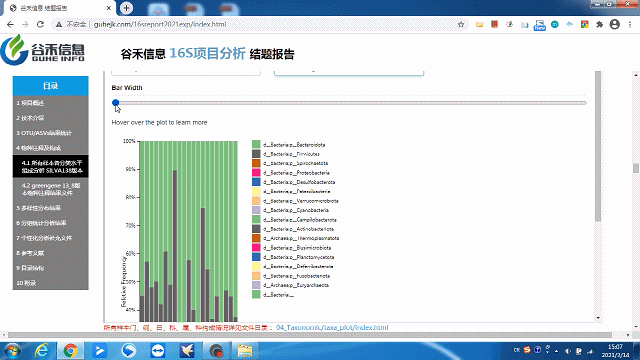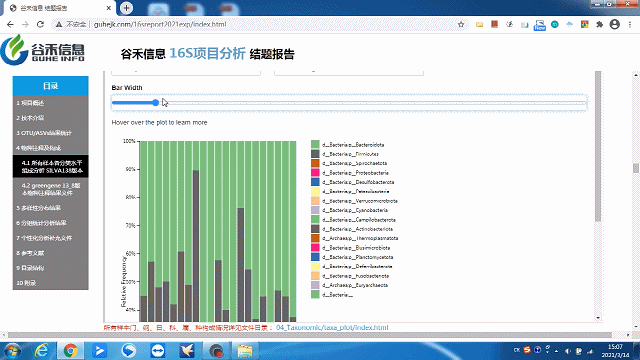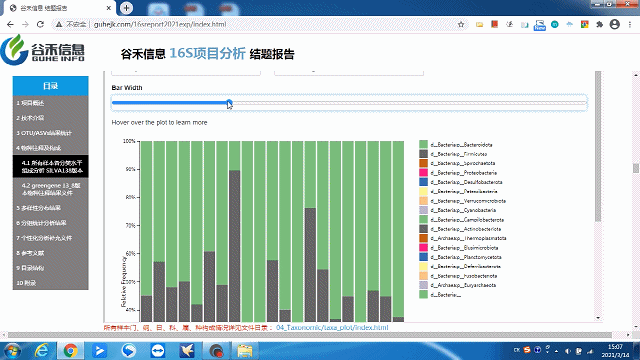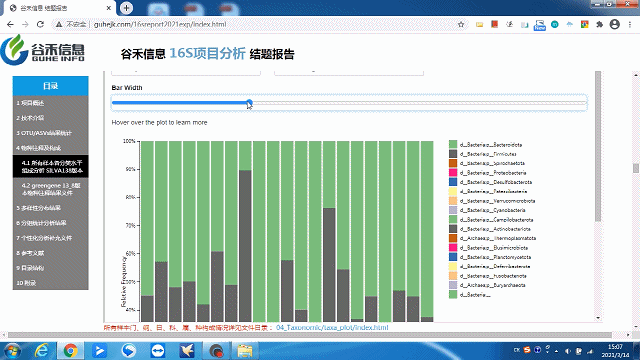

同时给出了各分类水平的相关原始数据,可以到对应路径进行查看。


重要指数 :★★★★
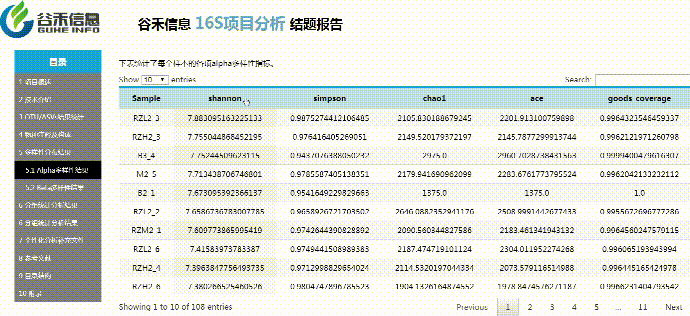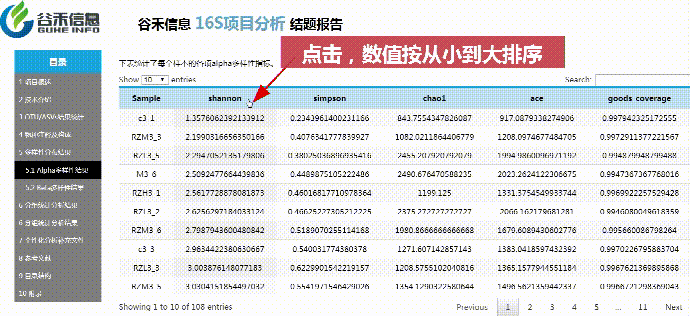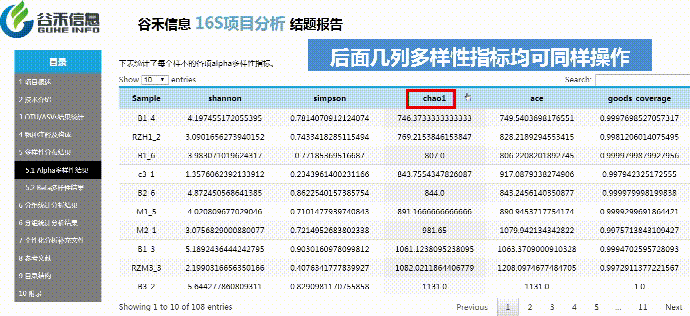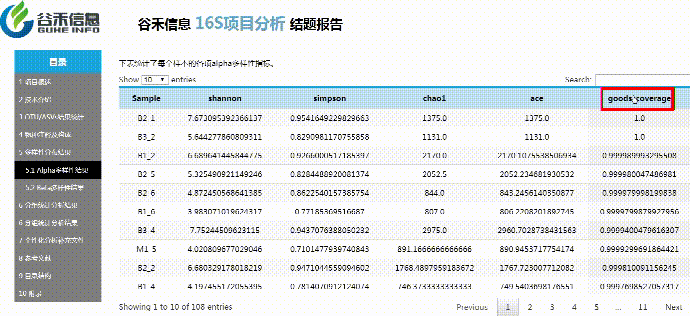
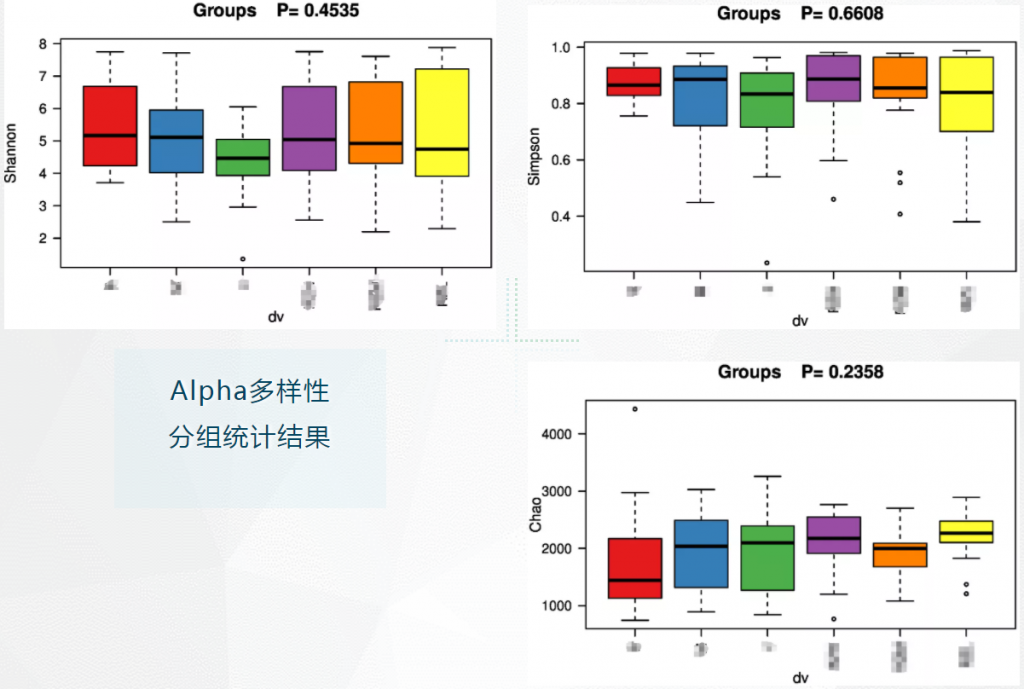
α多样性
评估单个样本内的物种构成的丰度情况
使用Qiime2进行α多样性分析,分别计算获得simpson,ace,shannon,chao1以及goods_coverage数据统计结果。

β多样性
通过降维的方法来考察样本与样本之间的相似度和关系,种属构成特征。
三种聚类方式:
Beta多样性PCA、非加权距离的PcoA、加权距离的PcoA的3D图。
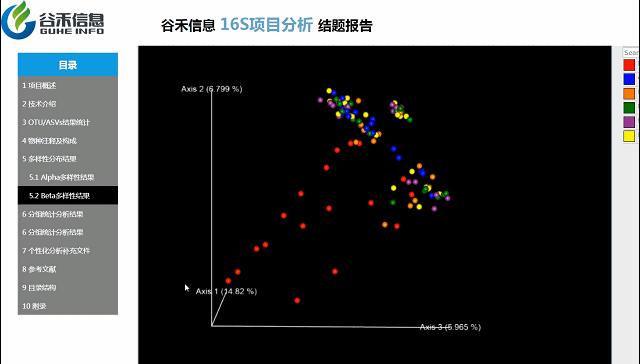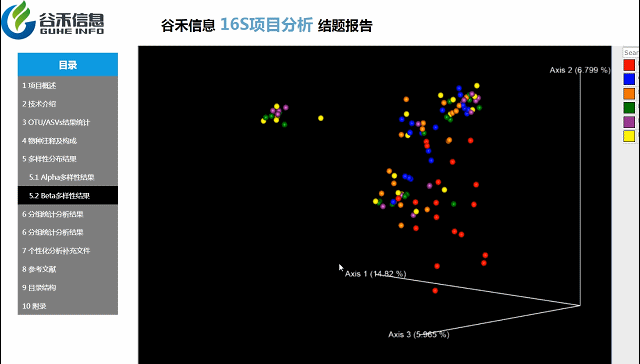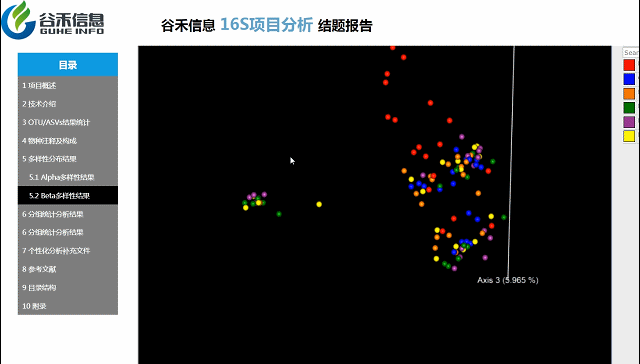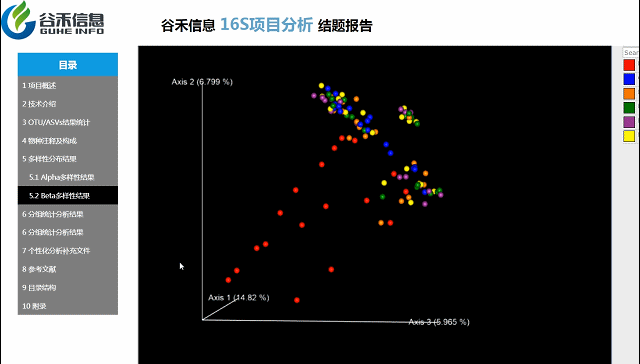

按住鼠标随意拖动,可以看到任意角度的三维坐标自由变换。

大小可自行调整
多色系任你挑选
总有你想要的图
重要指数 :★★★★★
按照你填写的样本信息单,对各分组情况,进行统计学差异分析。
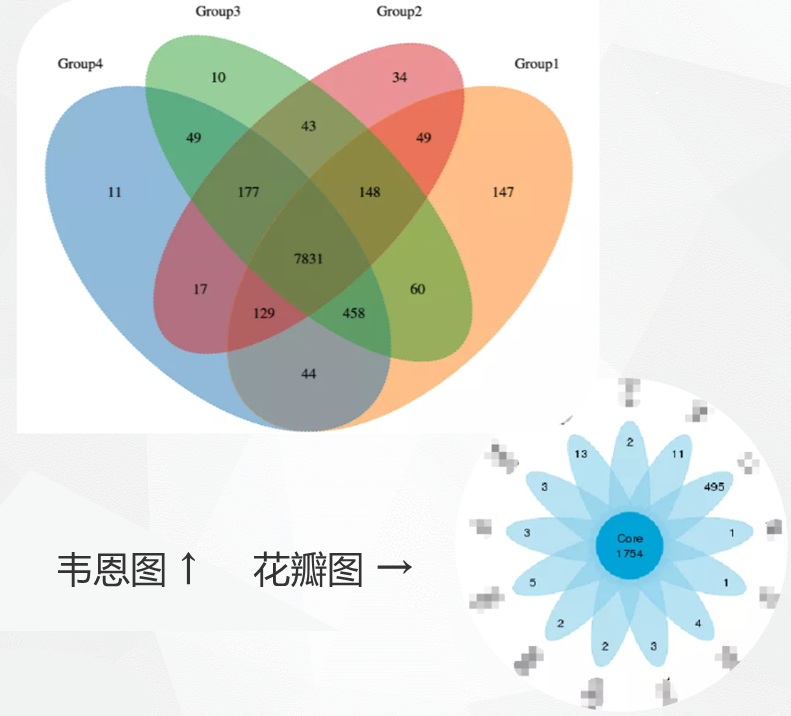
分组Venn图
OTU/ASVs比较韦恩图(样本数/分组数<=5个样本,若分组数大于5出花瓣图)

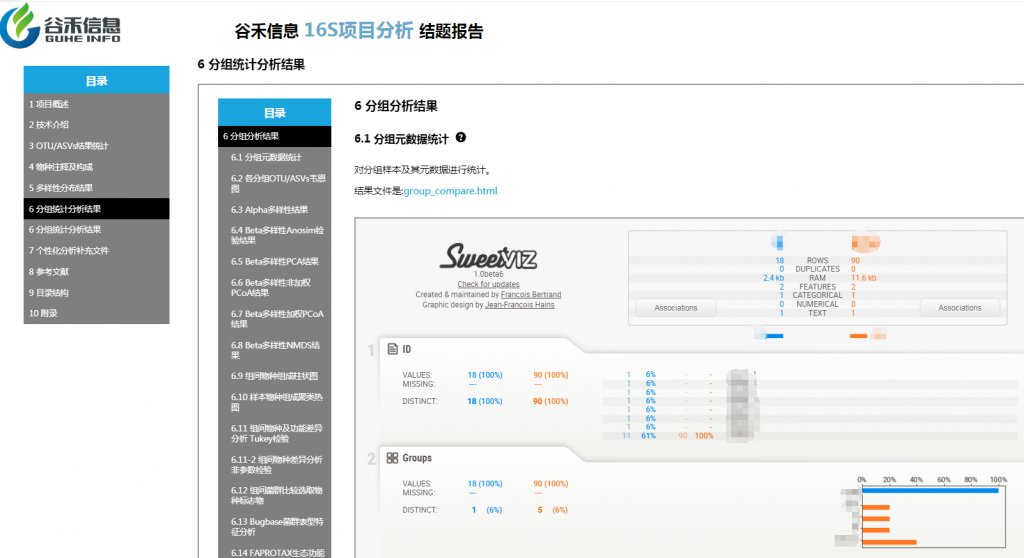
分组元信息统计
对分组样本及其元数据进行统计

α多样性
分组之间alpha多样性指数使用非参数统计检验

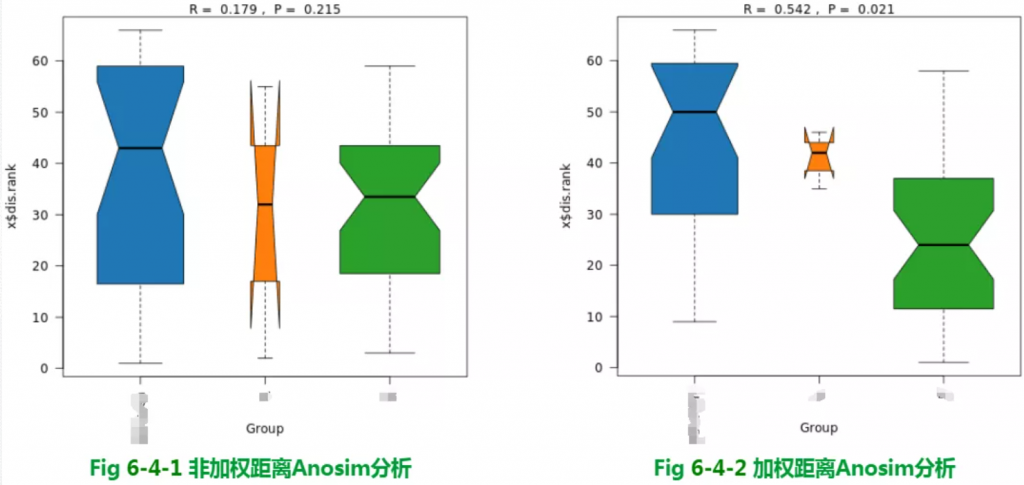
分组是否有意义?——β多样性
Beta多样性分组Anosim检验结果
Anosim分析是一种非参数检验,用来检验组间的差异是否显著大于组内差异,从而判断分组是否有意义。

要PCA结果图?
要PCoA结果图?
要NMDS结果图?
要加权?非加权?
… …
全部都有
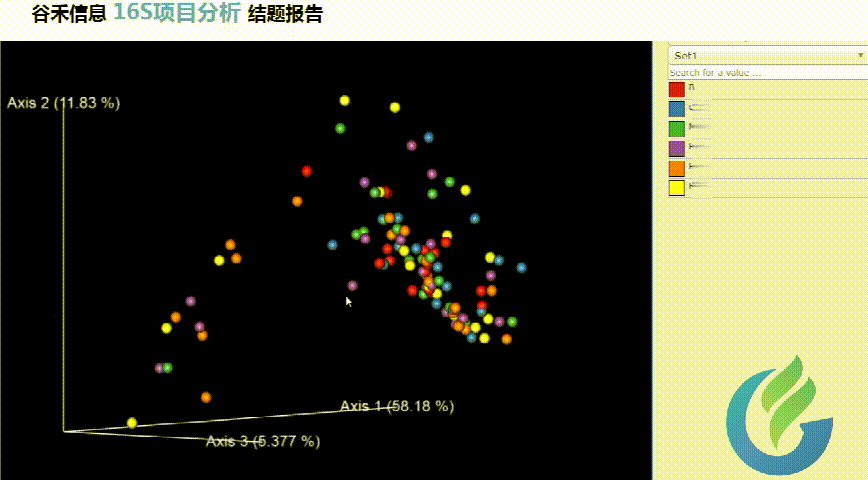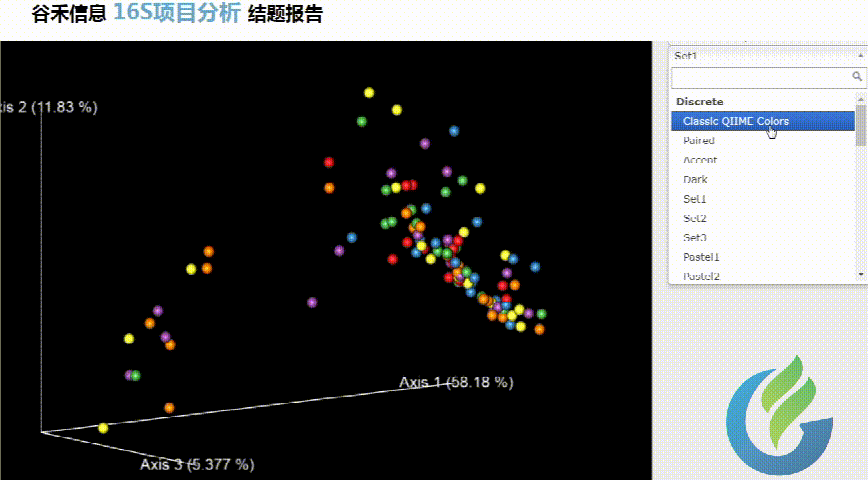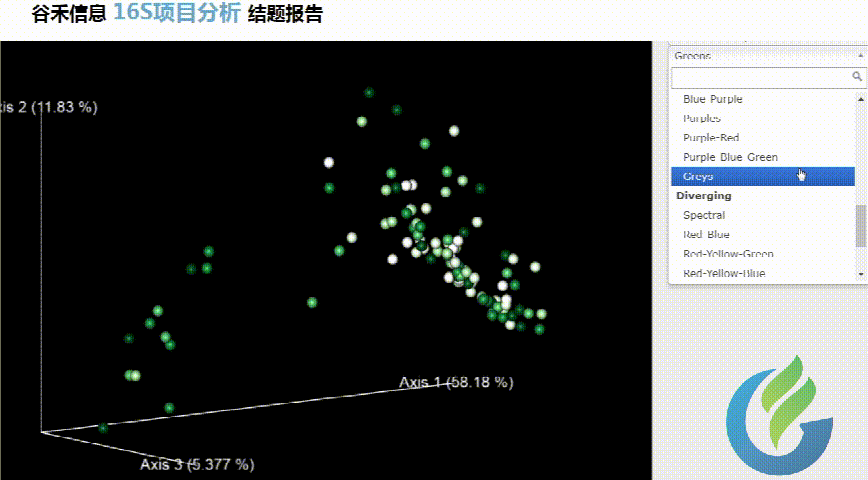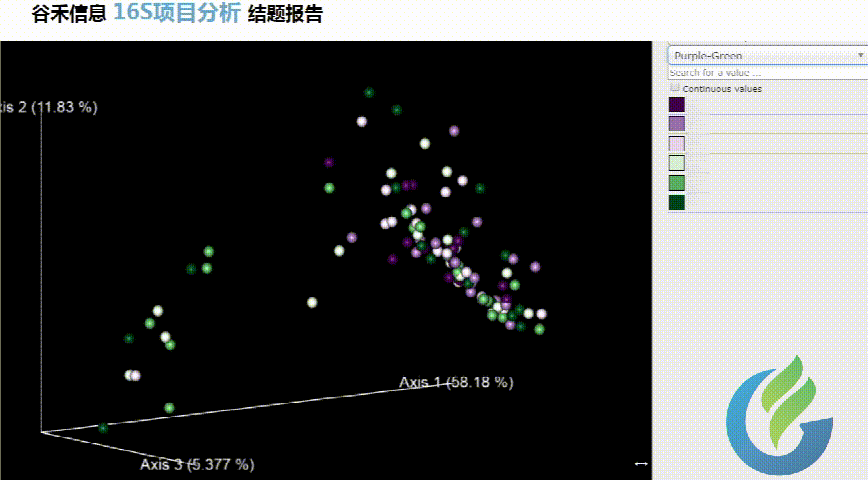
Beta多样性PCA结果
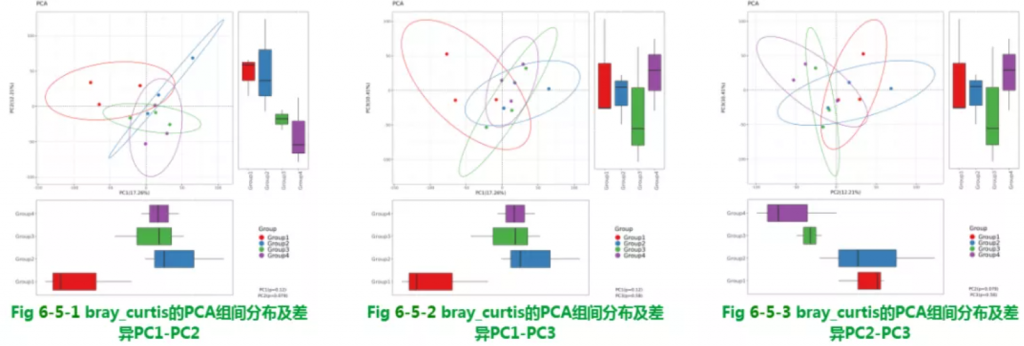

使用bray_curtis的PCA组间分布及差异
Beta多样性非加权PCoA结果
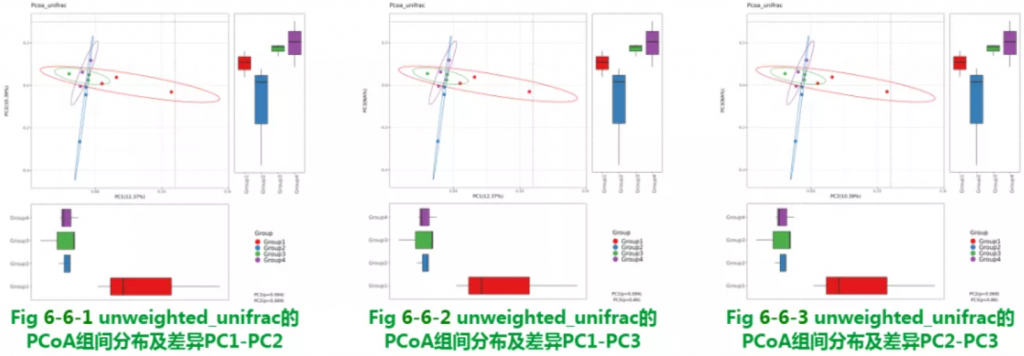

使用unweighted_unifrac的PCoA组间分布及差异
Beta多样性加权PCoA结果
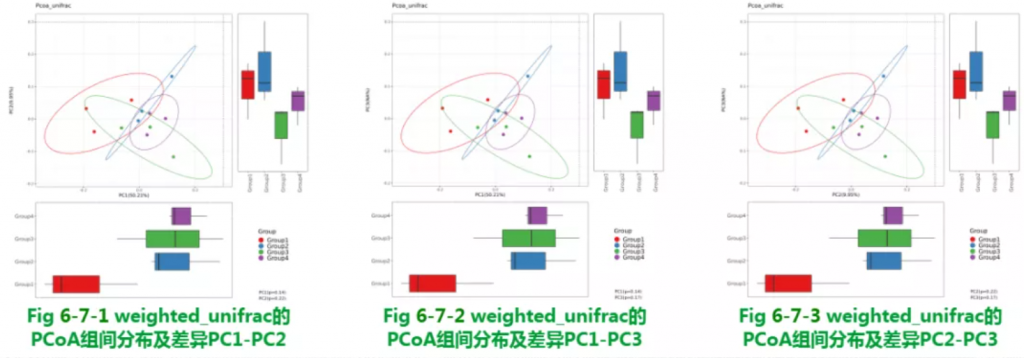

使用weighted_unifrac的PCoA组间分布及差异
Beta多样性NMDS结果
非度量多维尺度分析 NMDS 分析与 PCoA 类似,也是一种基于样本距离矩阵的分析方法,通过降维处理展现样本特定的距离分布。
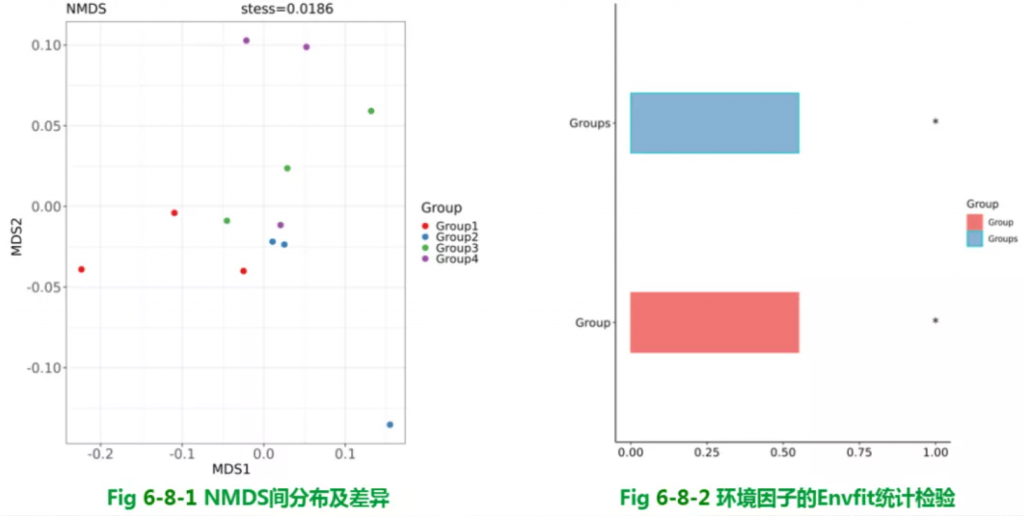

通过对样本距离进行等级排序,使样本在低维空间中的排序尽可能符合彼此之间的距离远近关系(而非确切距离数值)。因此,NMDS 分析不受样本距离的数值影响,对于结构复杂的数据排序结果可能更稳定。


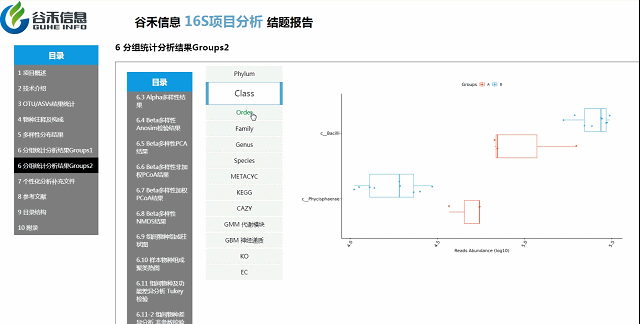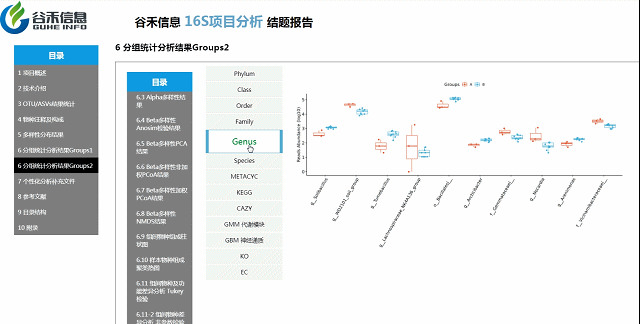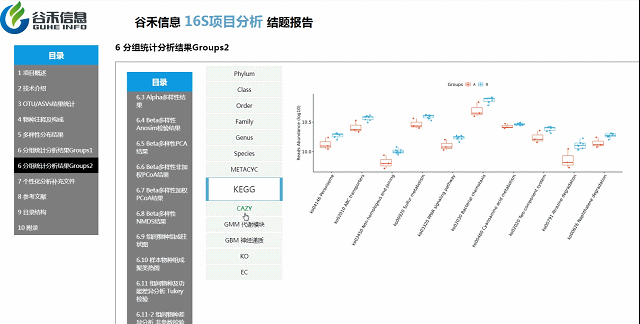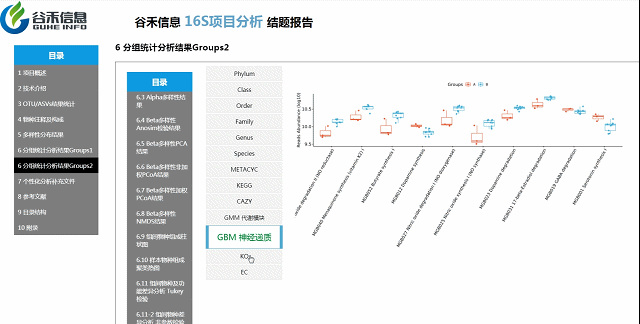
你想要的层级或分组都有——组间物种构成柱状图



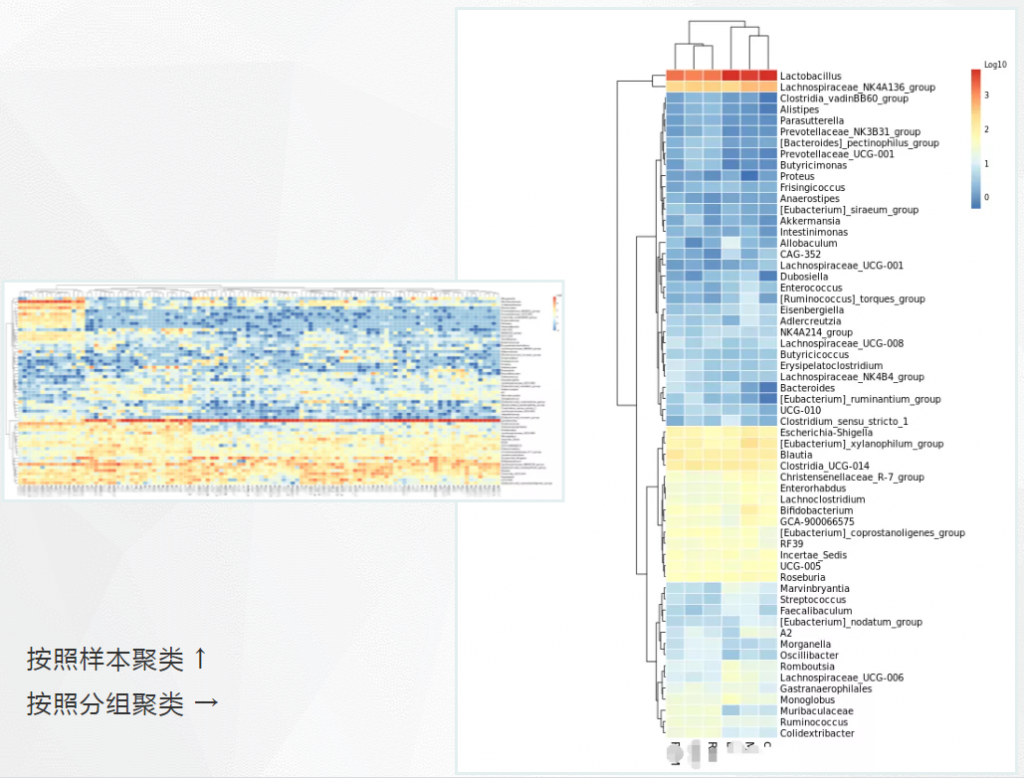
样本及分组之间聚类热图
了解样品之间的相似性以及属水平上的群落构成相似性。

组间各物种分类水平及功能差异
Tukey检验
如果样本每个分组是完全均等的情况(比如说每个组各有10个样),适合用Tukey检验。

优势:
可以快速在图中表现出多个分组之间,哪两个之间存在显著差异。
组间各物种分类水平
非参数检验
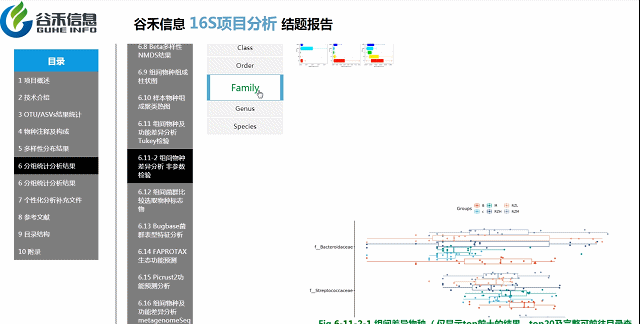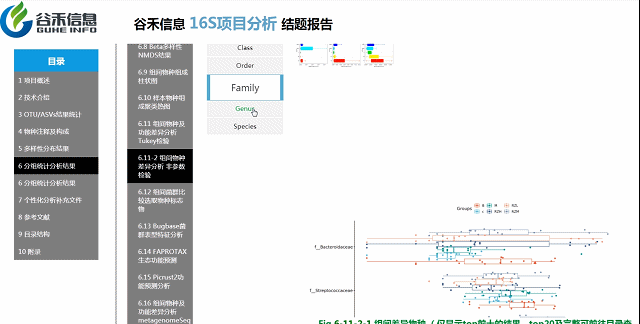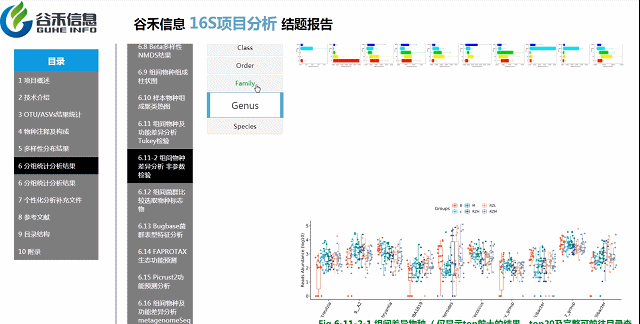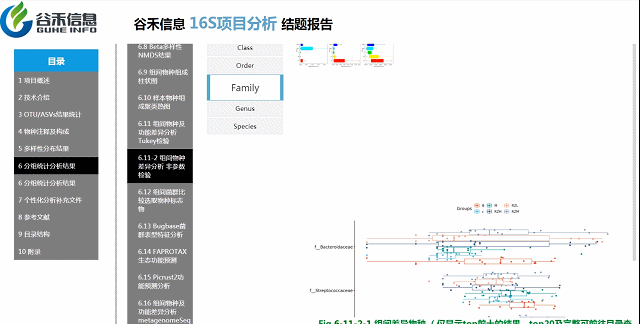

各个层级均有相对应的图展示。
组间菌群比较选取物种标志物
Lefse分析
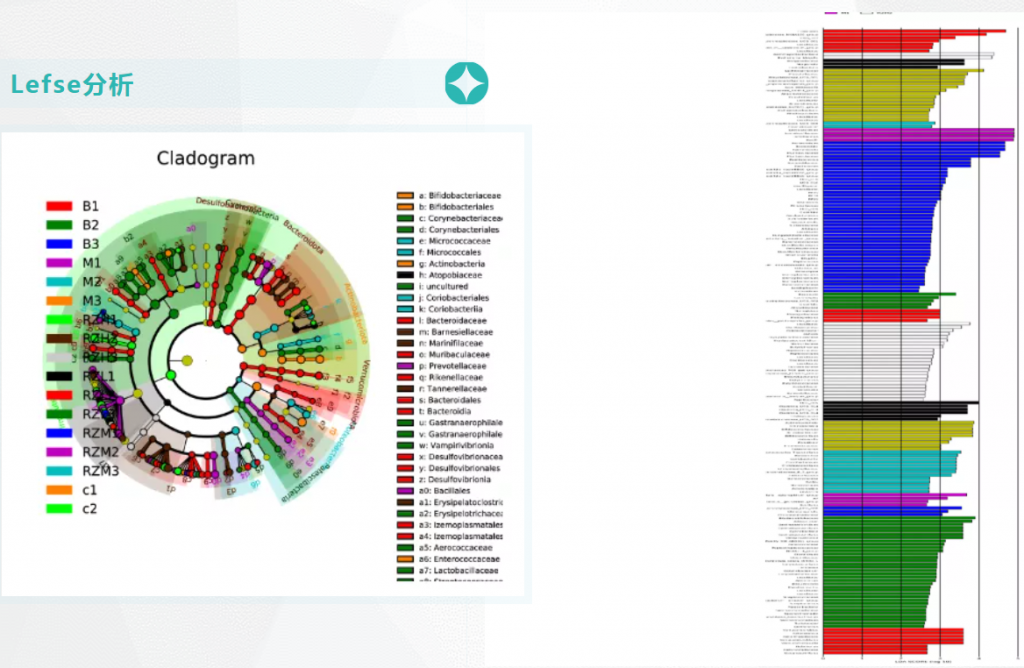

基于线性判定的方式,筛选组与组间的生物标记物——也就是说找到组间存在特别显著的高丰度的菌属。
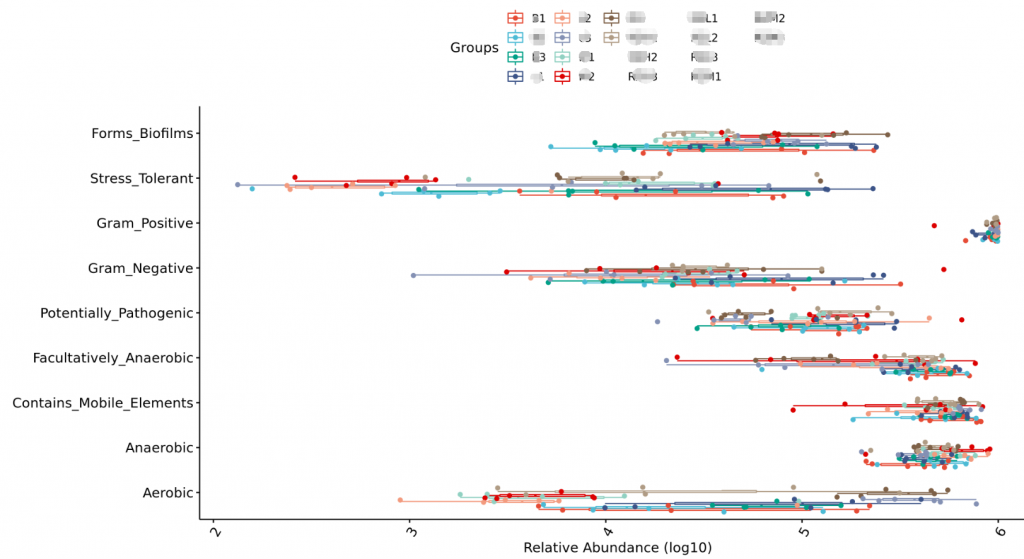
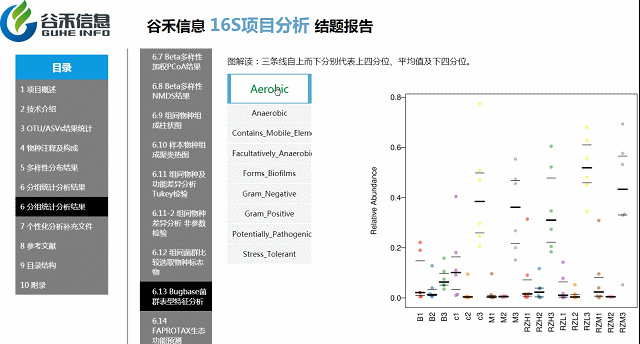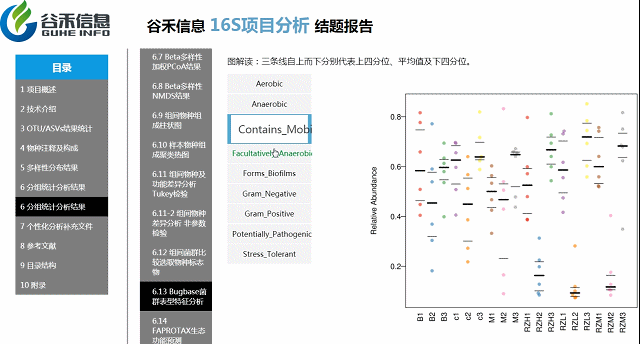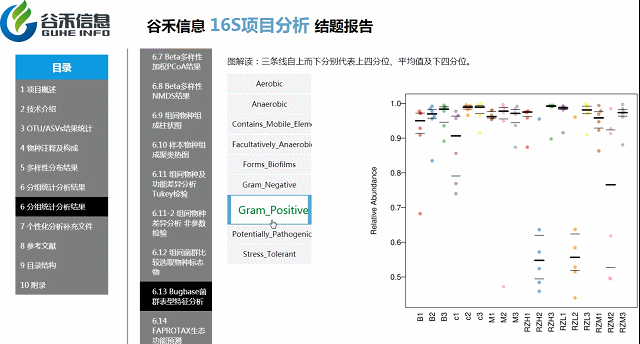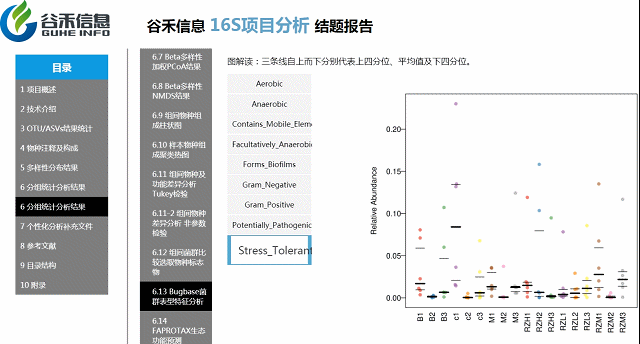
Bugbase菌群表型特征功能预测分析
基于文献的一些分类,对菌属进行菌群表征,包括对厌氧/好氧,革兰氏阴性/阳性,生物膜形成等分类。


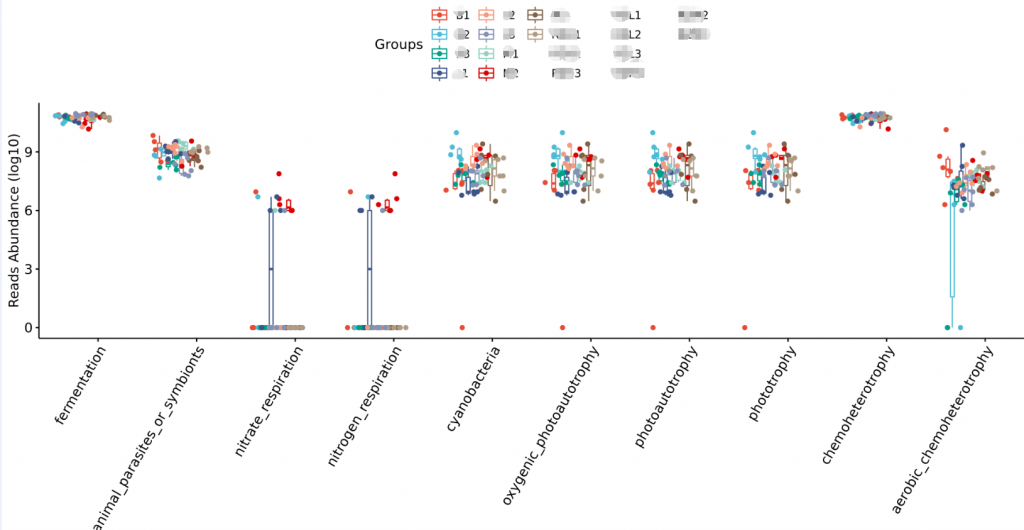
环境样本工具?——FAPROTAX生态功能预测
整合文献原核功能数据库,偏向于代谢和生物学功能的注释。比较适合环境样本,比如说碳、氢、氧、氮、硫等元素的代谢循环的能力。



基因功能预测?——Picrust2功能预测分析
随着研究的不断深入,很多菌的基因组数据有了,基于基因组数据一旦能确定其物种来源,可以推测它具有的基因的拷贝数、代谢通路的构成特征。

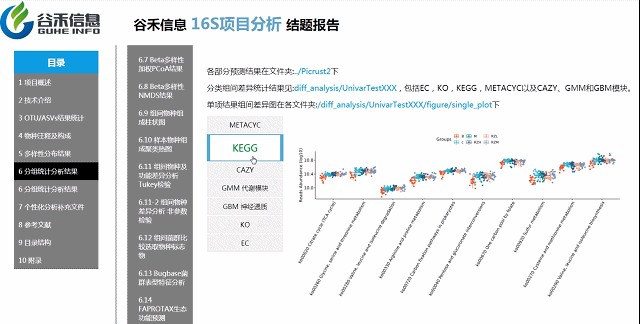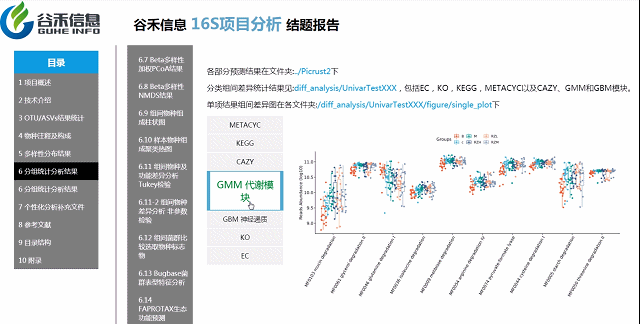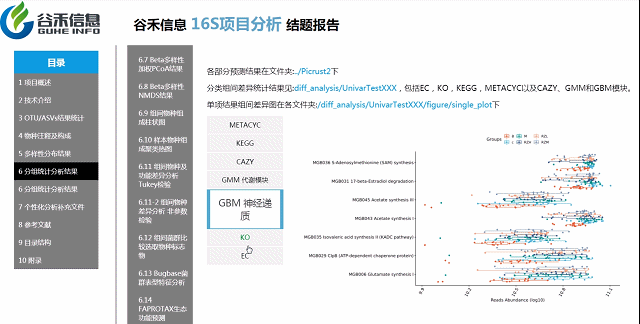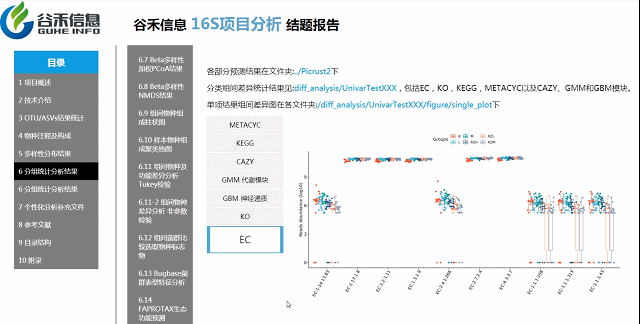
2万多的物种,基因覆盖更完整
还包括了CAZY,GMM,GBM等模块
具体差异的意义要结合你的实际研究目标解释
组间各物种分类水平及功能差异
MetagenomeSeq分析
更保守,结果可靠性更高
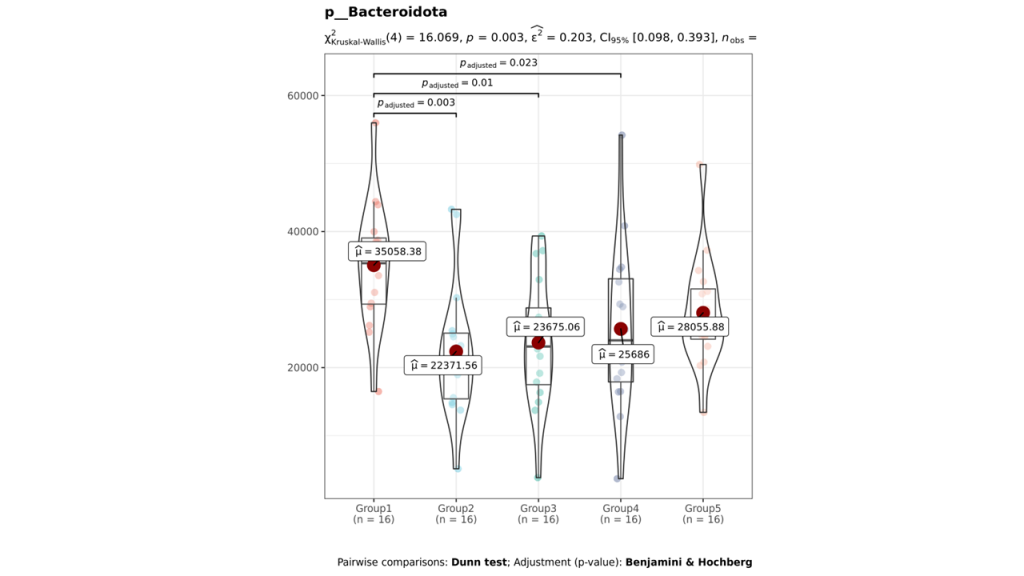


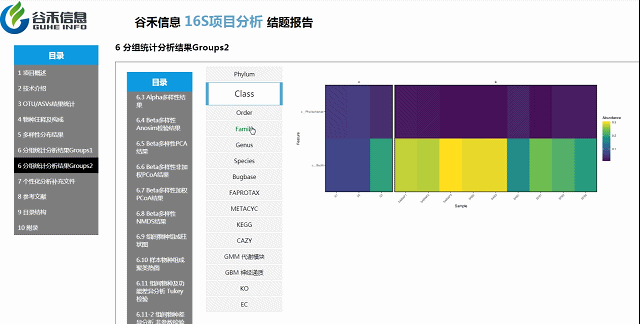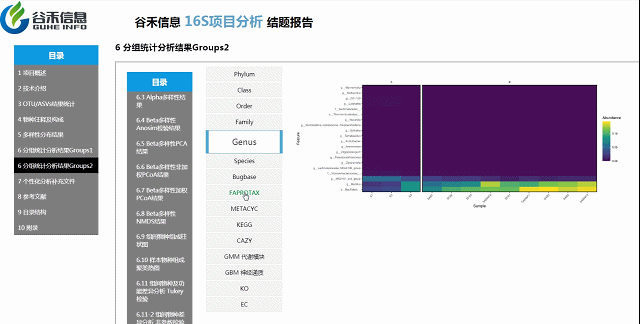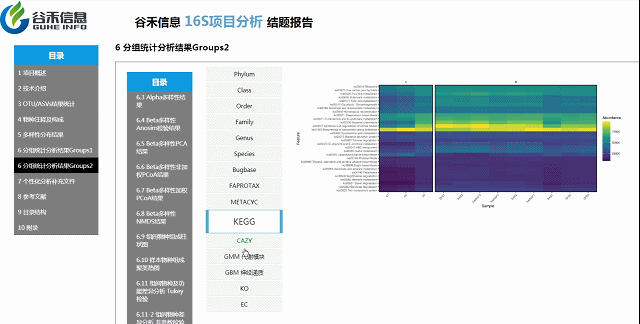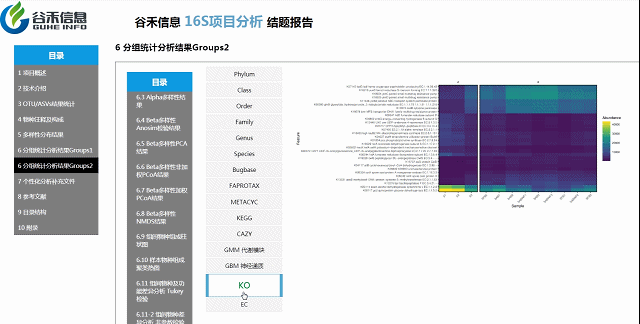
组间物种及功能差异热图
基于上面MetagenomeSeq的结果中,找到差异的物种种属和代谢通路做的热图。

差异菌属与代谢通路之间有什么关系?
差异菌属和功能代谢关联分析

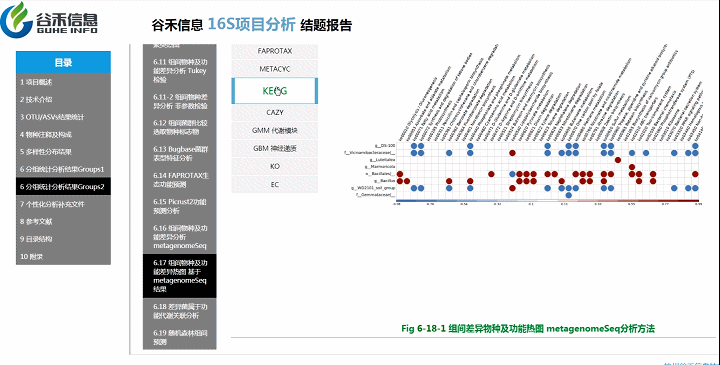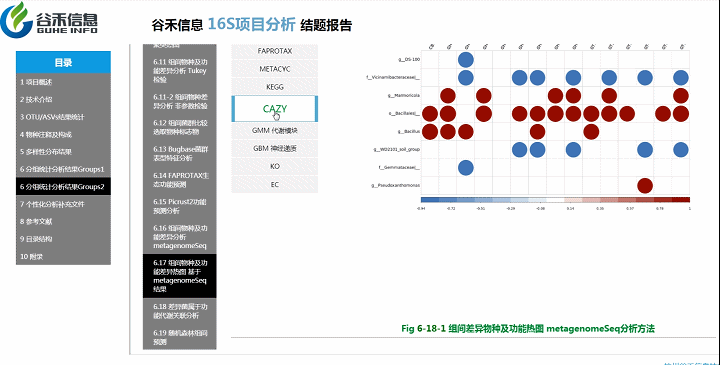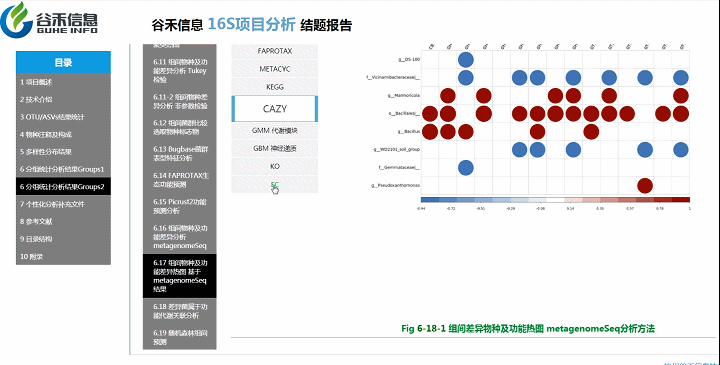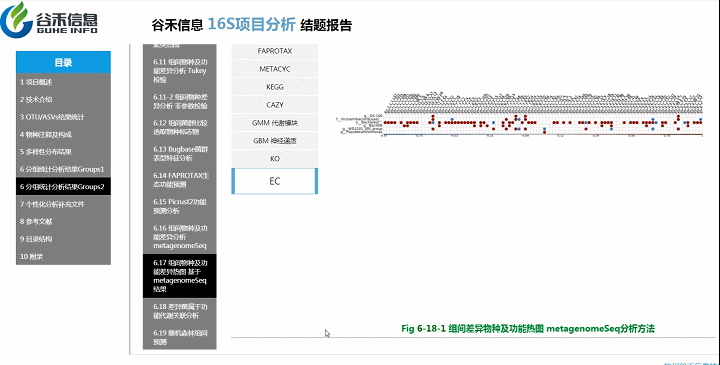
从菌属上的差异,代谢通路的差异等来看,到底是如何关联,是什么类的菌或代谢通路作出贡献。
不同分组之间相对明确区别的模型?
随机森林预测
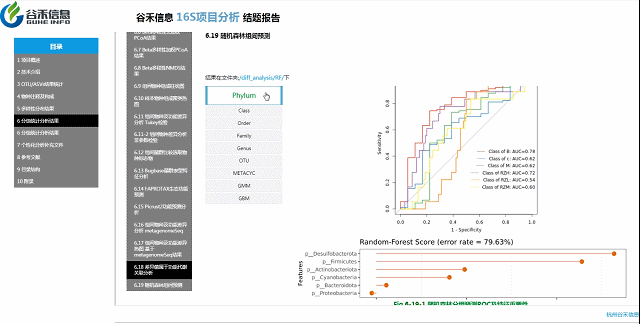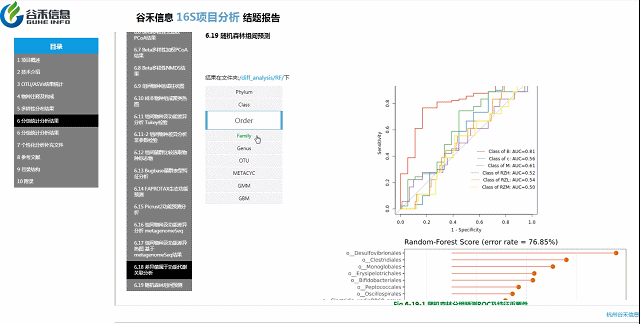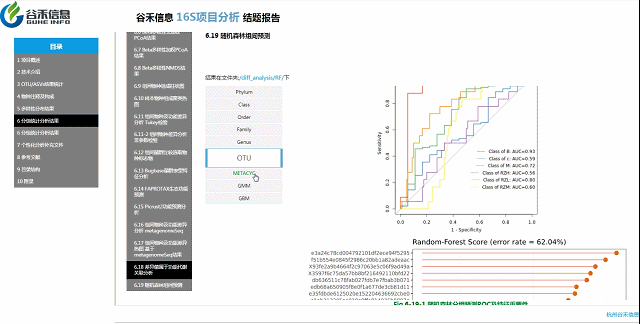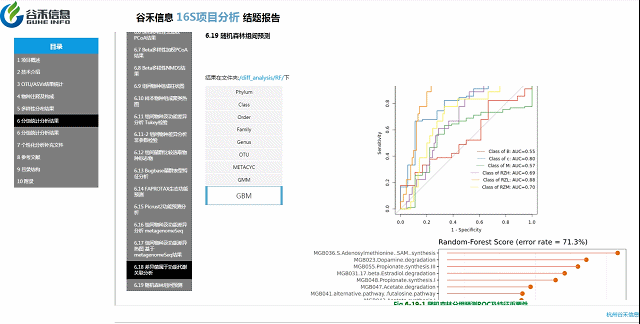

判断是哪个层面上的数据能最大程度作为分组样本的区分,以及区分效果。
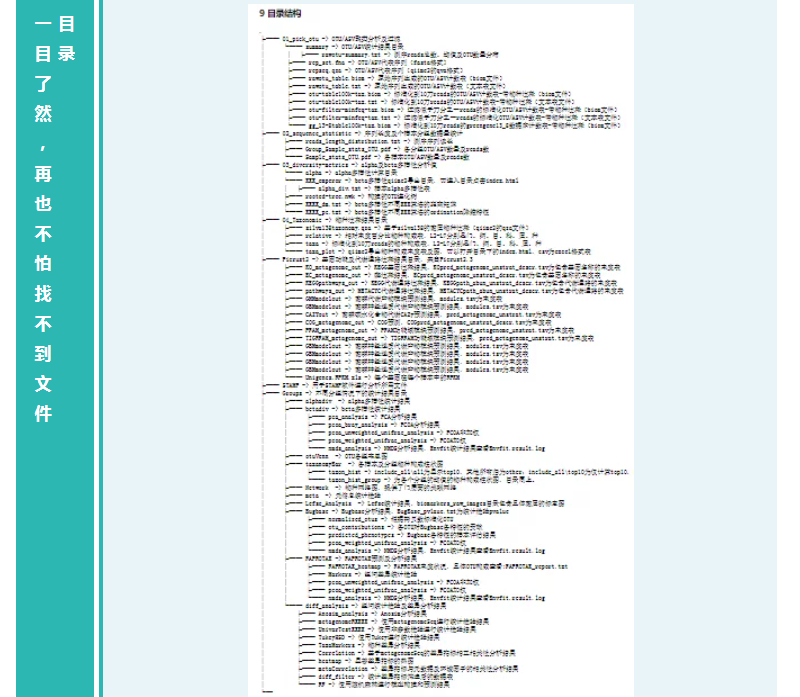



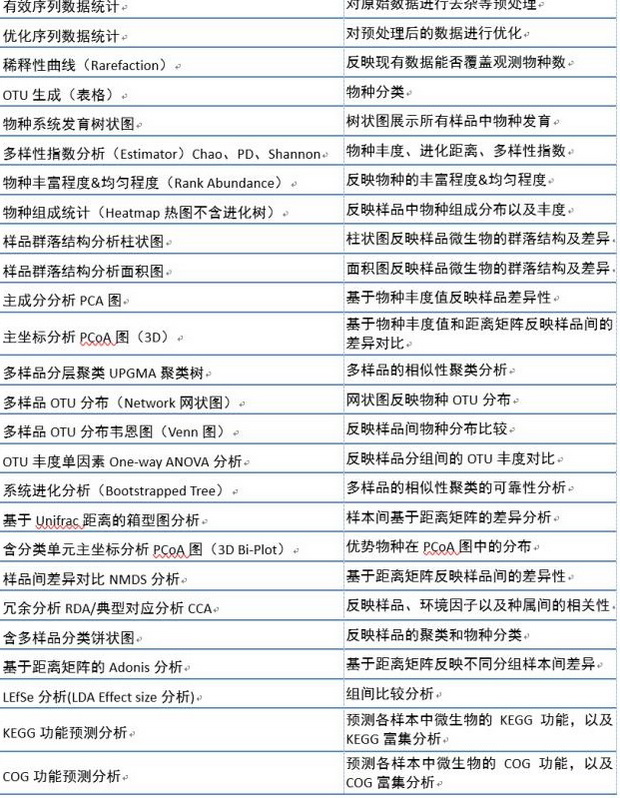
我们提供的基础分析包括以下所有内容:


相关阅读:

谷禾健康
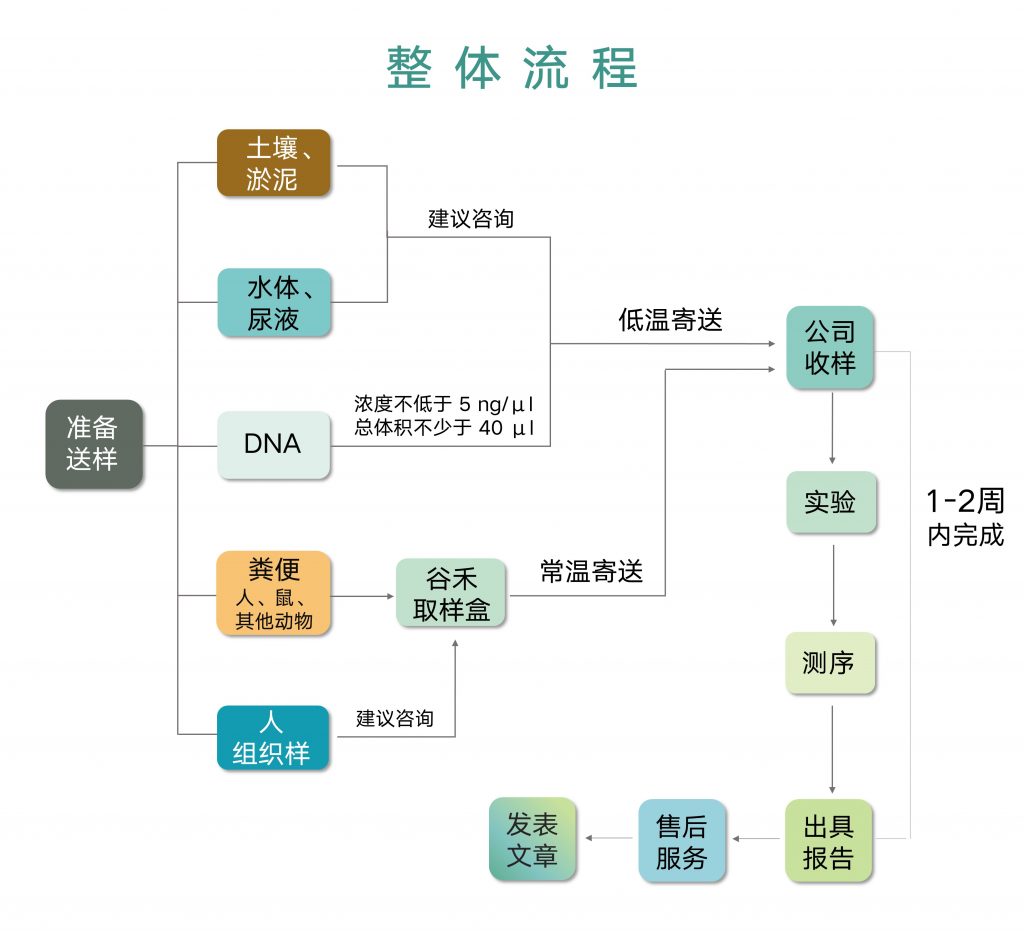
肠道菌群检测的第一步是取样,取样的重要程度不言而喻,实验人员只有在拿到合格样本后才能开展后续实验。
如果储存和运输不当菌群结构就会发生变化,进而导致菌群测序不准确,因此,便捷可靠是关键。
谷禾经过多年肠道菌群检测实践和研发,开发出适用于肠道菌群取样和常温储存的取样管,可以采集并稳定DNA,用于定量肠道菌群组成分析。

整个取样盒包括:
取样管(内含裂解液和稳定液);
无菌棉签;
回寄袋;
每个取样管上均有唯一条码。
主要特点:
在家中轻松自行采样高质量样品
起始样品需要量低至0.01g,快速且稳定
常温保存运输
标准样品适合手动或高通量自动处理
获得适用于16S ,qPCR,宏基因组的高质量DNA
条形码化全样本可追溯性
谷禾取样管的独特特点使得取样变的异常简便,下面是取样演示:
仅需使用棉签从厕纸上沾取粪便,然后洗脱到取样管的保存液中即可,使保存液可见粪便颜色即表示取样量足够。

适用于-20°C至65°C下保持DNA完整性
室温下有效存储长达60天
与新鲜样本一致的菌群构成特征
低成本
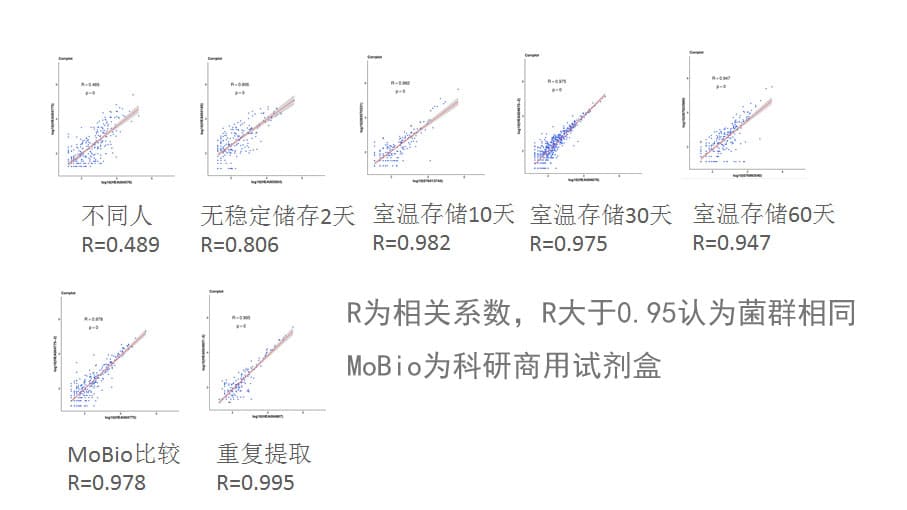
下面来看一下取样管在不同条件下的保存效果,我们使用凝胶电泳来检测不同保存处理条件下提取菌群DNA的状态:
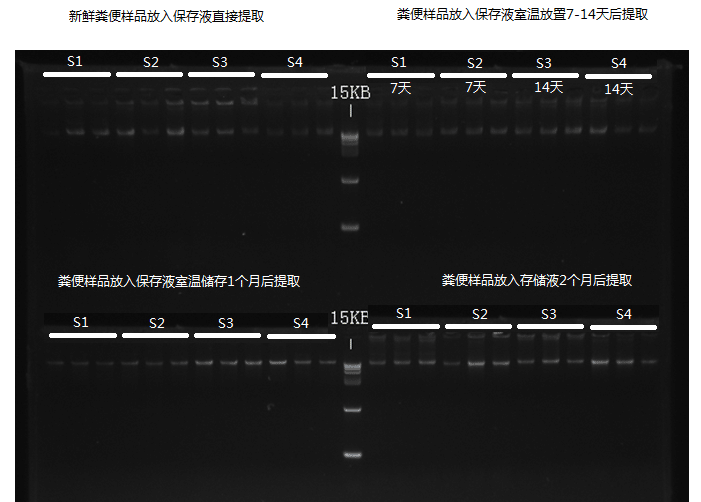
可以看到,使用谷禾保存管的DNA样品即便在存储至60天仍然没有出现明显的DNA降解情况。
专利号:ZL201511009389.7
配合谷禾肠道菌群取样保存管
适用于提取极低当量菌群DNA
具备以下特点:
磁珠法-适用于自动化高通量提取
起始量限制低
与MoBio试剂盒一致性高
现有样本处理量450例/天

下图可以看到我们使用谷禾提取方法与MoBio试剂盒比较以及重复提取的菌群相关性。另外同时比较了使用谷禾取样管保存不同天数后的提取菌群结果。
专业的实验环境
让整个实验操作
得以高效可靠运行
二级生物安全实验室

注:生物安全实验室的分级

生物安全实验室一般实施两级隔离。一级隔离通过生物安全柜、负压隔离器、正压防护服、手套、眼罩等实现;二级隔离通过实验室的建筑、空调净化和电气控制系统来实现。

谷禾健康
基因组规模代谢网络模型(Genome-scale metabolic model,GEM),是一种包含了某种特定生物或者是细胞基因组范围代谢反应,及其酶及基因关联的数学模型。
这里,我们基于文章的描述,介绍一款新软件——MetaGEM。
研究者认为,目前代谢建模的工作流程仍然是倾向于依赖参考基因组作为重建和模拟GEMs的起点,这忽略了微生物群落中存在的物种内和物种之间的多样性。也限制了对已知参考基因组空间中的代谢网络的分析和解释。
可能导致假阳性(即在参考基因组中存在但在群落中的变量中缺失的通路)或假阴性(即在参考基因组中缺失但在群落变量中存在的通路)结果,最终导致对个别物种代谢通路以及交互营养共生(cross-feeding)相互作用的不准确预测。
也就是说当前的代谢建模方法很可能无法捕捉特定物种在不同环境中的特定代谢特征,例如具有不同疾病状况的个体的微生物群。为了克服这一局限,研究者们开发了MetaGEM。
MetaGEM可以不依赖参考基因组,直接从短读的宏基因组数据中重建样本特定的代谢模型。
下图是该软件的流程图,图中蓝底白字的部分是该流程中所使用到的软件,都是已经由他人开发完成的。
研究者们自己开发的部分有两个:
一是end-to-end的框架,能够进行群落水平的代谢交互模拟;
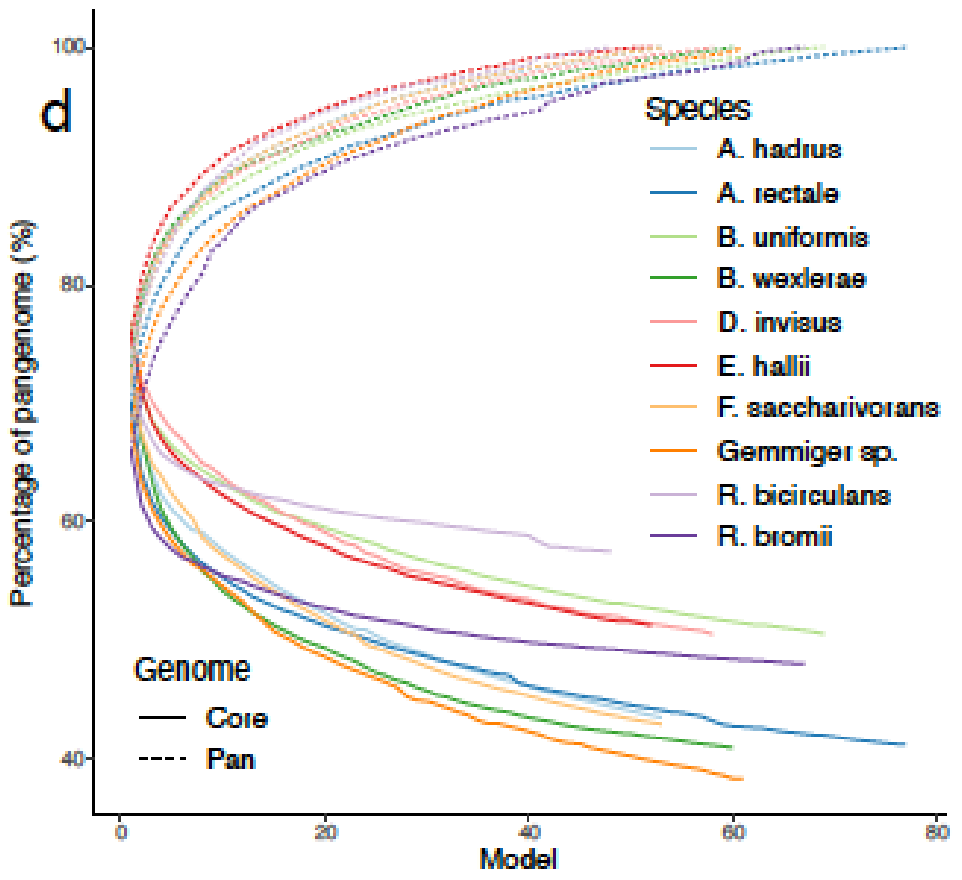
二是一个来自宏基因组生物群落的14,000多个MAGs,包括3750份高质量的MAGs,以及来自人类肠道微生物组研究和全球微生物组项目的相应的随时可用的GEMs。
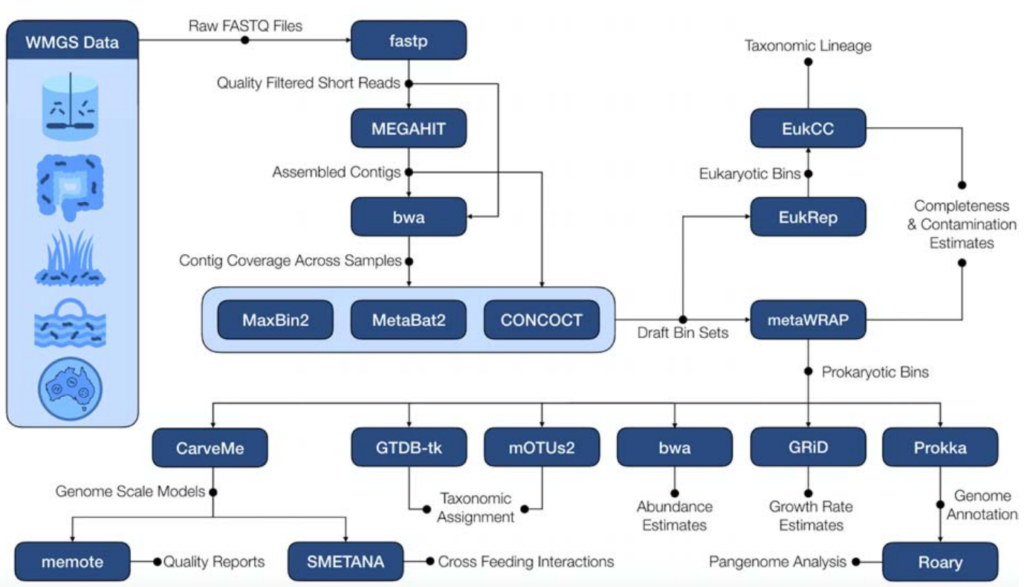
整个流程使用Snakemake实现,从原始的宏基因组的fastq文件开始,质控、组装、估计contig覆盖率、binning、Bin的改进和重组、MAG丰度定量和物种分类、CarveMe进行基因组规模代谢模型重建及质量报告,Smetana模拟重建的基因组规模代谢模型的肠道微生物群落。
(这里只简单介绍了处理步骤,文章中的“Methods”部分有给出使用的参数)
除了以上的必备选项,该流程还有一些附加功能可供用户选择。可以使用GRID估计中和高覆盖率的MAGs的增长率。
Prokka可以对MAGs做功能注释,并且其结果可以提供给Roary,获得一组MAGs的核心MAG和泛基因组的可视化结果。
EukRep可以用于寻找真核生物的MAGs。
EukCC可以对真核生物的bins做后续的分析。
MetaGEM流程具有两个特点:
一是直接从宏基因组获得高质量的代谢重建;
二是可以为个性化的人类肠道群落建模,研究者通过两个实验进行了描述:
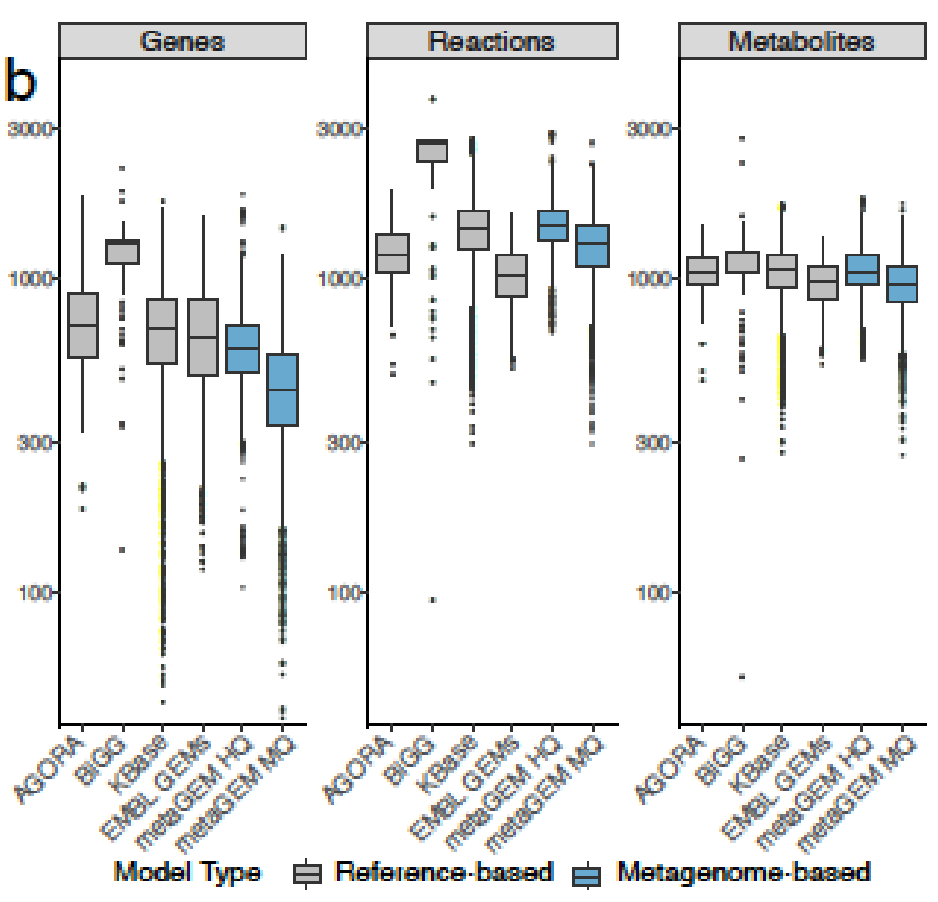
MetaGEM模型与EMBL、AGORA、KBase和Bigg模型相比较
用MetaGEM基于宏基因组短读序列构建MAGs,分为HQ(高质量的),MQ(中等质量的),并以此进行代谢重建,总共获得14087个GEMs,然后将它们与高度精选的基于参考基因组的BIGG模型、AGORA、EMBL和KBase模型进行了比较。
利用基于定位的方法(方法)生成的丰度估计值与基于标记基因的丰度估计值完全相关;
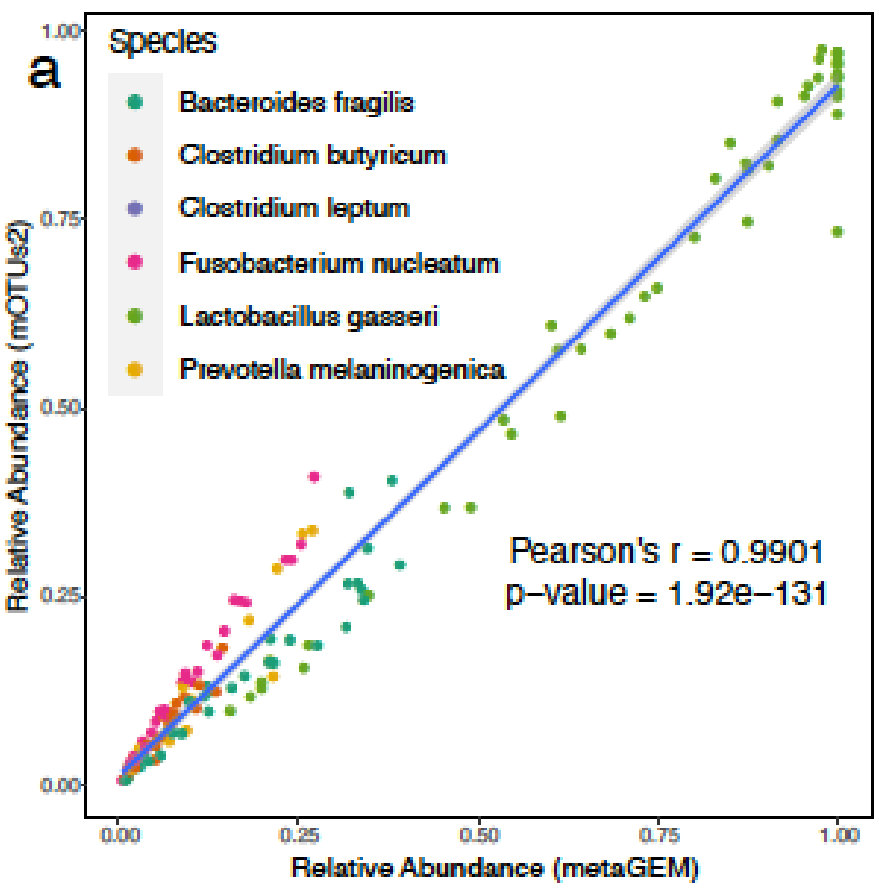
MetaGEM和其他模型都具有类似数量的反应和代谢物,但基因数量相比较少;
通过计算模型之间成对的代谢之间的距离,发现MetaGEM具有相似的酶多样性分布;
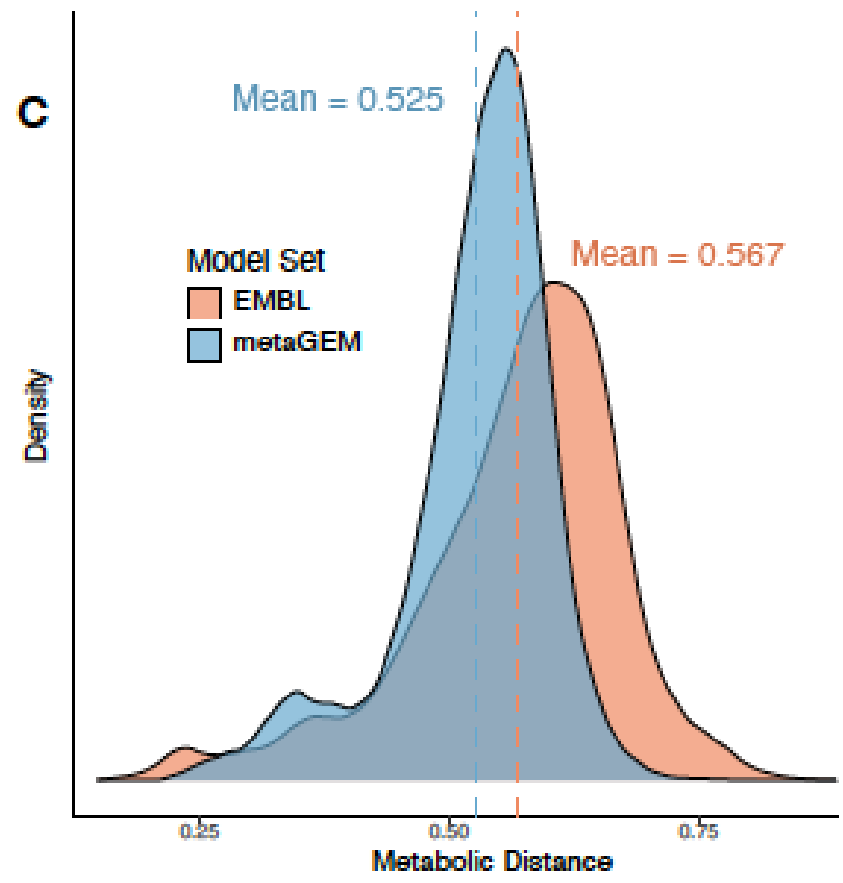
可以捕捉到种水平物种间的显著的代谢差异。高达60%的代谢多样性存在于物种泛基因组中,metaGEM模型捕获的物种内代谢变异程度显著
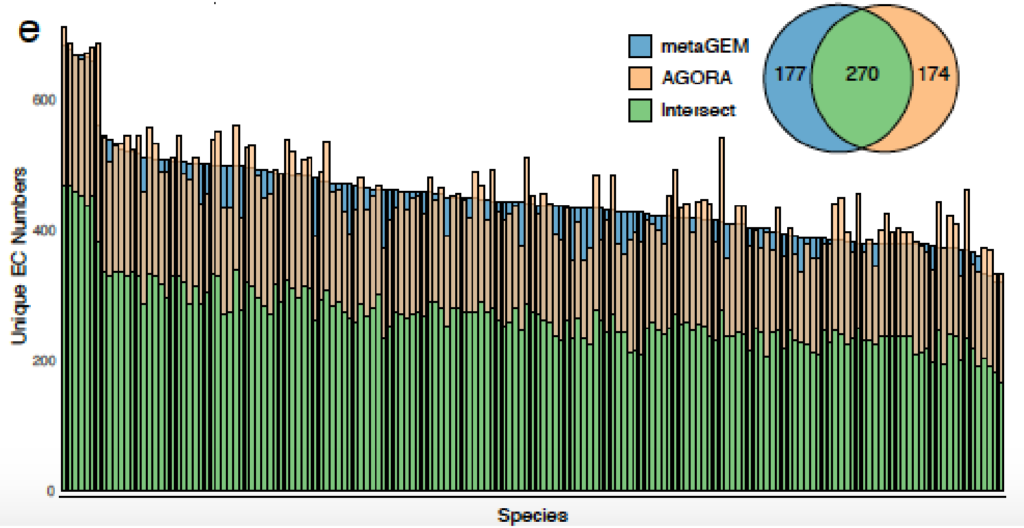
与基于参考基因组的肠道物种代谢模型AGORA比较,发现基于参考的模型引入的代谢反应不一定存在于每个宏基因组环境中,而MetaGEM模型是完全基于实际的宏基因组在特定环境下重建的代谢模型。
AGORA和MetaGEM模型的EC数的交集在48.9%到69%之间,其中53.9%的情况下MetaGEM模型比相应的AGORA模型包含更多的EC数。
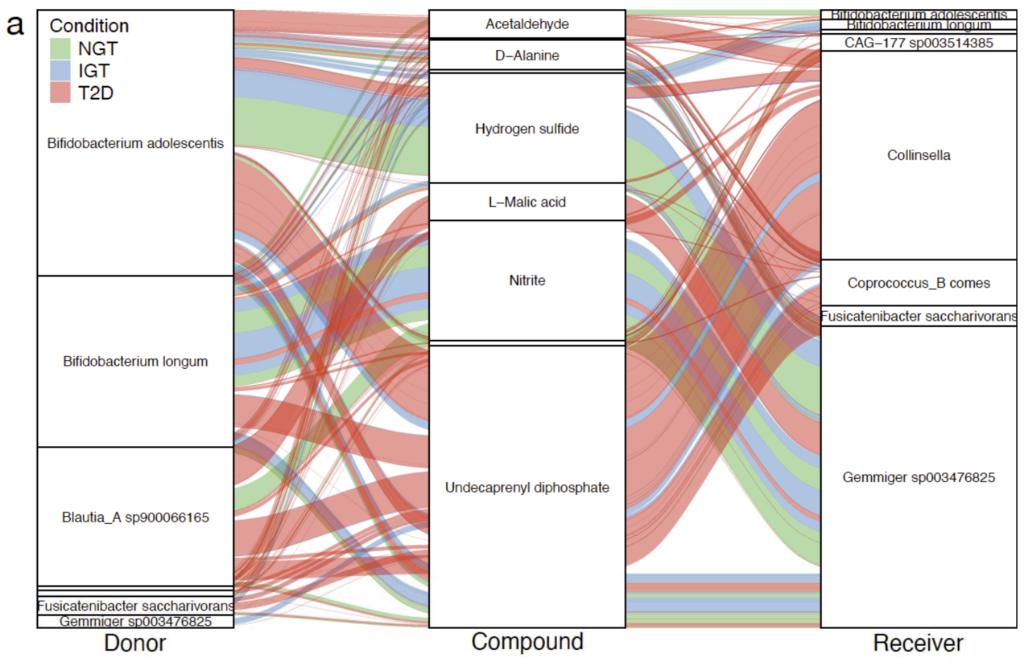
研究健康和代谢受损的2型糖尿病患者肠道微生物群落中潜在的微生物代谢相互作用。
使用metaGEMs通过137个宏基因组数据重建了4127个个性化的GEMs。
根据疾病状况分类,即正常糖耐量(NGT,n=42)、糖耐量受损(IGT,n=42)、 2型糖尿病(T2D,n=53),然后应用Smetana软件模拟微生物群落中的物种间依赖关系,Smetana为每个群落输出一个分数表,对应于在给定条件下为支持群落成员的成长而应发生的交叉喂养相互作用强度的度量,即物种A生长的可能性取决于物种B的代谢物X。
不同的2型糖尿病疾病组(NGT、IGT、T2D)相对应的肠道代谢基因组产生具有不同代谢结构的群落。
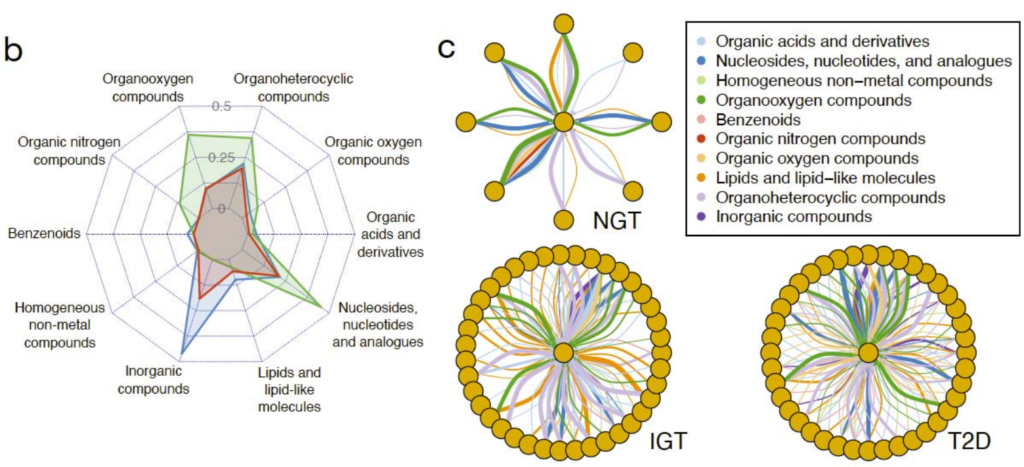
MetaGEM具有完善的流程,搭载的工具也是生物信息分析中常用的处理工具,下载很方便,用conda就能完成。无需参考基因组,这也意味着不需要下载动辄几十Gb的文件。使用Snakemake做流程的自动化管理,运行命令简单,也可以分步骤运行。
总体而言,MetaGEM可以直接从宏基因组数据中研究复杂微生物群落中特定样本(sample-specific)的新陈代谢。
【附录】
关于文中MetaGEM流程搭建所应用到的宏基因组分析软件,这其中也有我们常用的软件,比如fastp、MEGAHIT、bwa、SAMtools、metaWRAP,它们在处理数据时非常的方便也易于上手。
参考文献:
Zorrilla F, Patil K R, Zelezniak A. metaGEM: reconstruction of genome scale metabolic models directly from metagenomes[J]. bioRxiv, 2021: 2020.12. 31.424982.
相关阅读:

谷禾健康
迄今为止,已经有了许多对呼吸道微生物组通过16S rRNA高通量测序的研究。这其中所有基于扩增子的研究的共同之处就是PCR的应用:
一是扩增待测序的目标标记基因,
二是为多样本的混合测序添加必要的索引序列。
这些步骤可以通过一步PCR或两步PCR完成,但没有研究说明两步PCR方案相关的实验室处理步骤是否会使样品比一步PCR方案更容易受到来自实验室的细菌DNA污染的影响。
本文
试图确定对16S rRNA V3V4与V4基因区域的一步或两步PCR的建库方案对上呼吸道和下呼吸道微生物组的影响
对收集的样本进行了三个设置下的lllumina MiSeq测序
设置1(两步PCR,V3V4区域)
设置2(两步PCR,V4区域)
设置3(一步PCR,V4区域)
分别对这三个设置产生的测序数据进行分析
结论
PCR步骤数量的差异会影响对呼吸道微生物群落的物种组成分析,且对上呼吸道(高细菌载量)的影响小于下呼吸道(低细菌载量),这表明PCR设置的偏差与样本生物量有关。
通过三个实验,即对模拟群落样品HM-783D、NCS样品、呼吸道样品采用三种PCR方案进行建库后的测序结果分析,研究这三种建库方案对其菌群描述的影响。
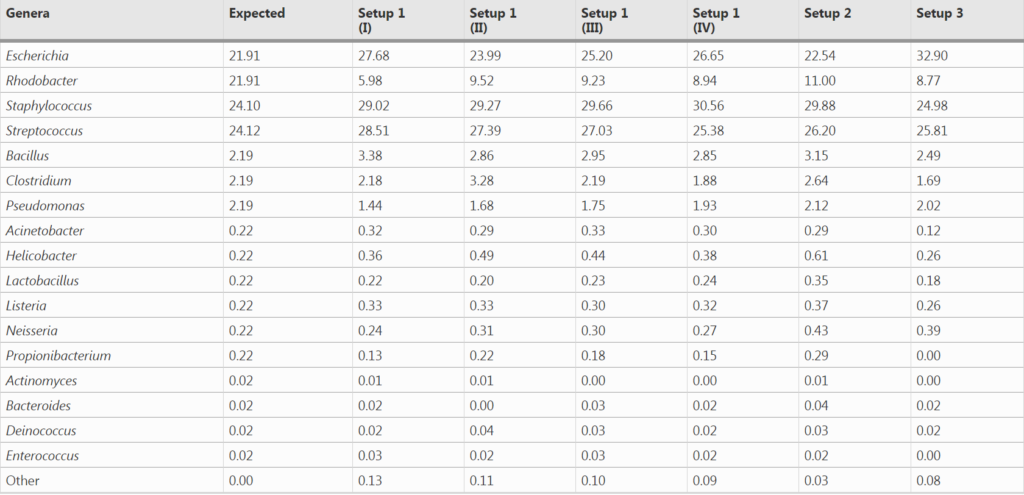
模拟群落样品HM-783D,来自20种不同细菌物种(17个属)的基因组DNA。
阴性对照样本NCS
呼吸道样品,从Bergen COPD微生物组研究中选择了23名研究对象,其中9名健康,4名患哮喘,10名患COPD(慢阻肺)。上呼吸道样本以漱口水(OW)为代表,下呼吸道样本以标本刷(PSB)和支气管肺泡灌洗液(PBAL)为代表。
三种PCR方案:
细菌DNA提取后通过三种不同的建库设置进行MiSeq测序,分别为
Setup1(两步PCR,V3V4区域);
Setup2(两步PCR,V4区域);
Setup3(一步PCR,V4区域)
下图完整的展示了三个PCR设置下的使用呼吸道样本的生物信息学过滤步骤:
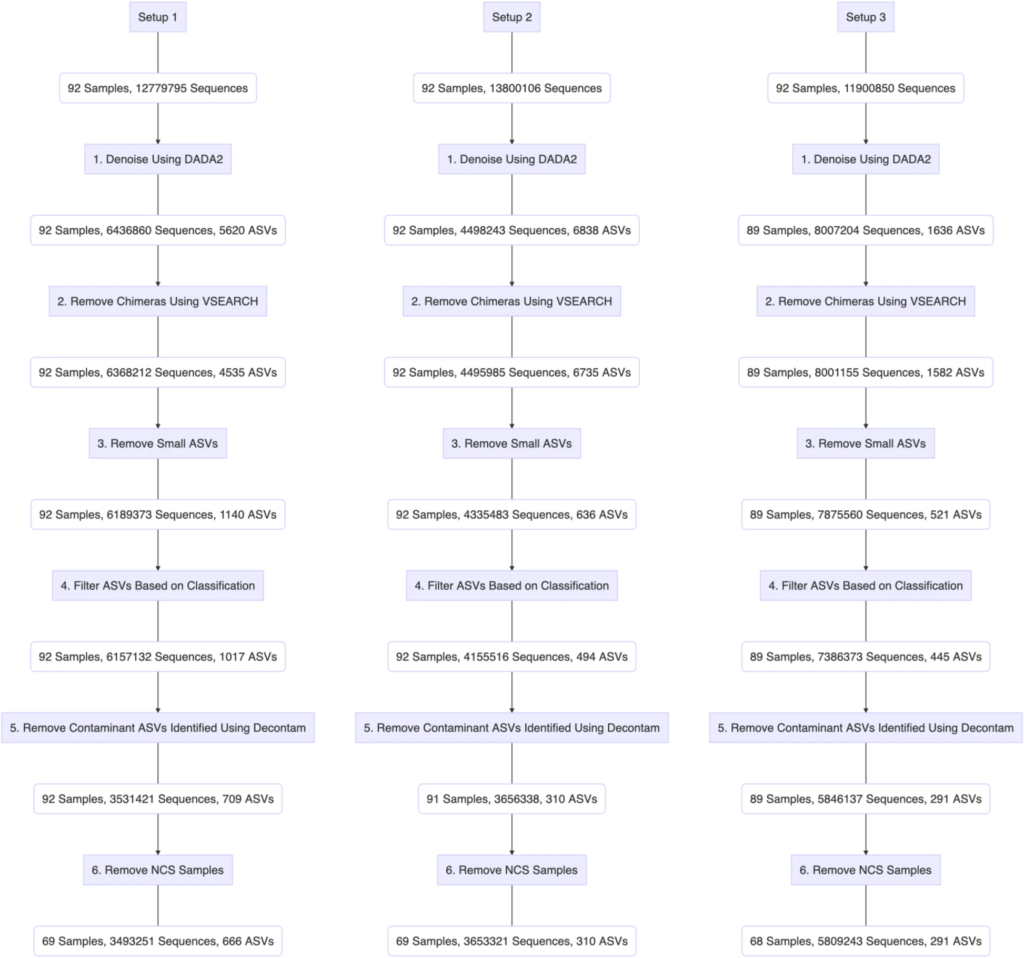
最终:
设置1:得到了666个ASVs
设置2:得到了310个ASVs
设置3:得到了291个ASVs
1. 对模拟群落样品HM-783D的分析
在设置1中进行了四次测序,设置2和设置3分别进行了一次。
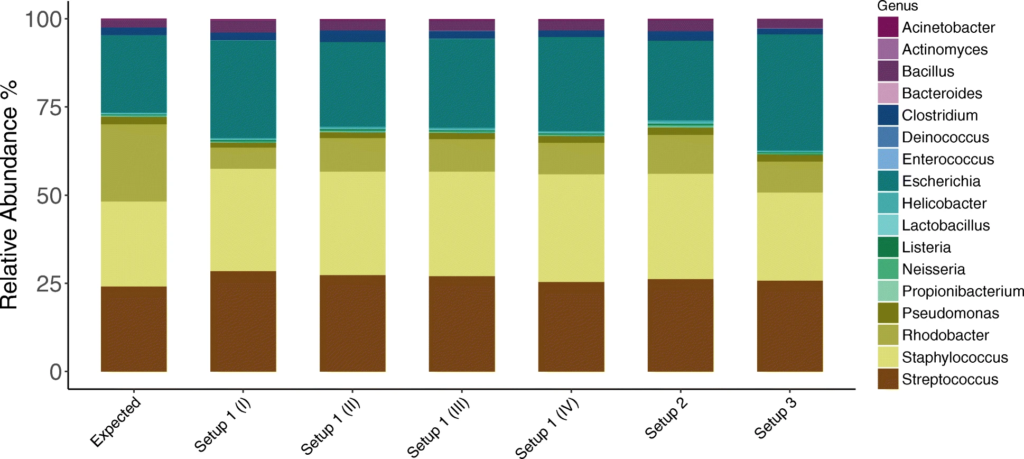
与预期丰度(Expected)相比,柱状图中观察到三种PCR设置下的各菌属的相对丰度与预期丰度相差不大,表中数据显示,三种PCR设置都在恢复高丰度物种方面具有最高的效率,但设置3在回收低丰度物种时的效率最低。
2. 对阴性对照样品的分析
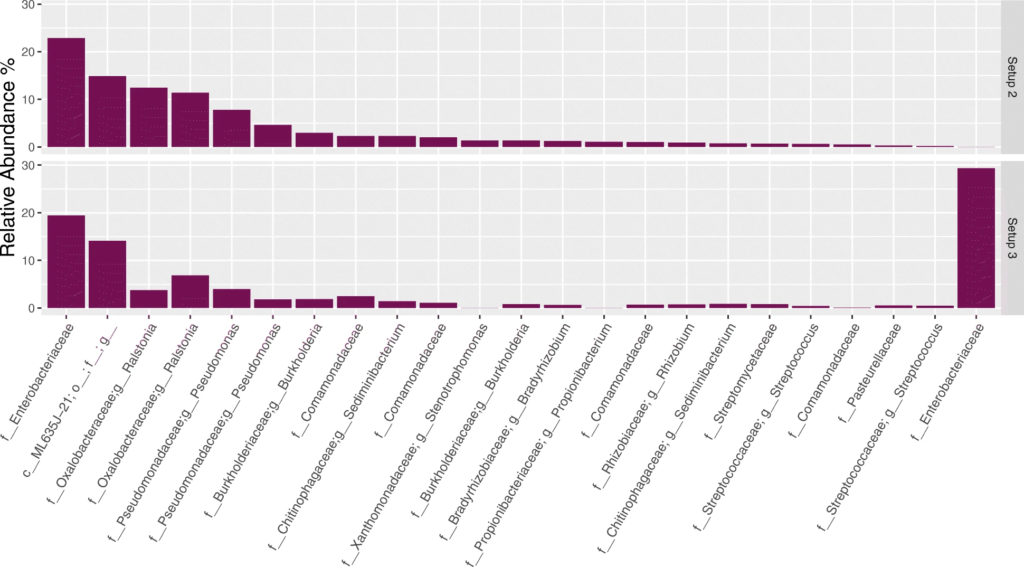
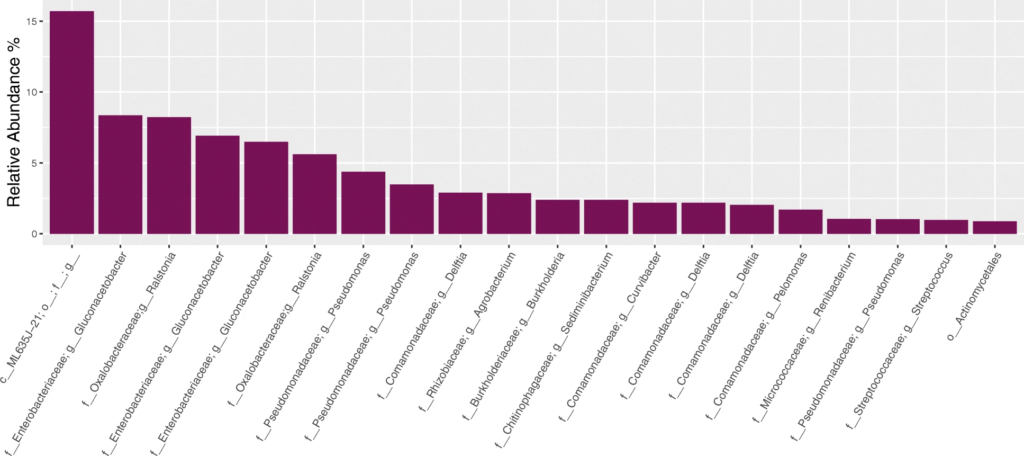
从上至下分别为设置123测序后,在NCS样品中观察到的20种最丰富的ASV。通过R包Decontam去除污染物,在设置23之间差异最大的是属于肠杆菌科的ASV,与后续的对水样品进行设置23下的测序分析结果相比较,发现大肠杆菌ASV就是在建库步骤中使用设置3的试剂时引入的污染物。
3. 对采集的呼吸道样品的分析
在去除污染物前后,代表为呼吸道菌群的链球菌,普雷伏氏菌,Veillonella和Rothia属的相对丰度变化不大,而去除污染物后,预测作为污染物代表的数量较少的物种被滤出。基于主坐标分析,发现高细菌载量的OW样品聚集在一起,低细菌载量PBAL,PSB一句设置23分离开。
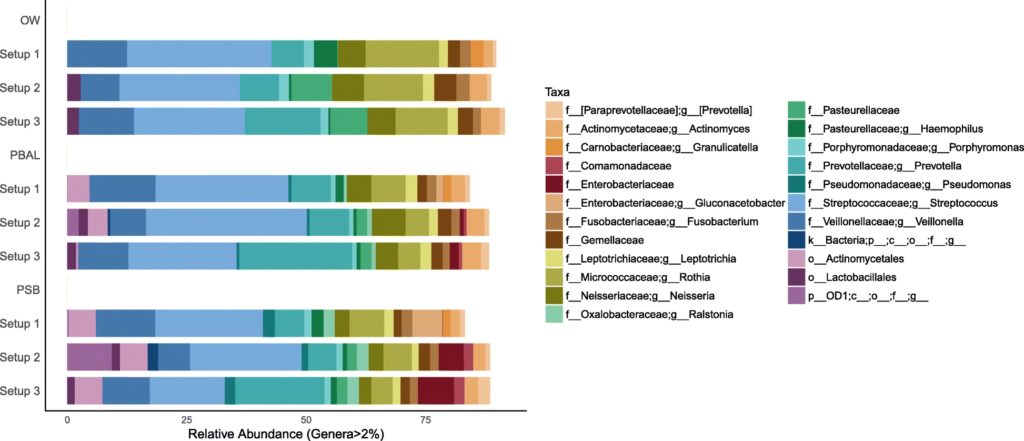
去除污染物之前的三种类型样品的三种PCR设置下的物种分类
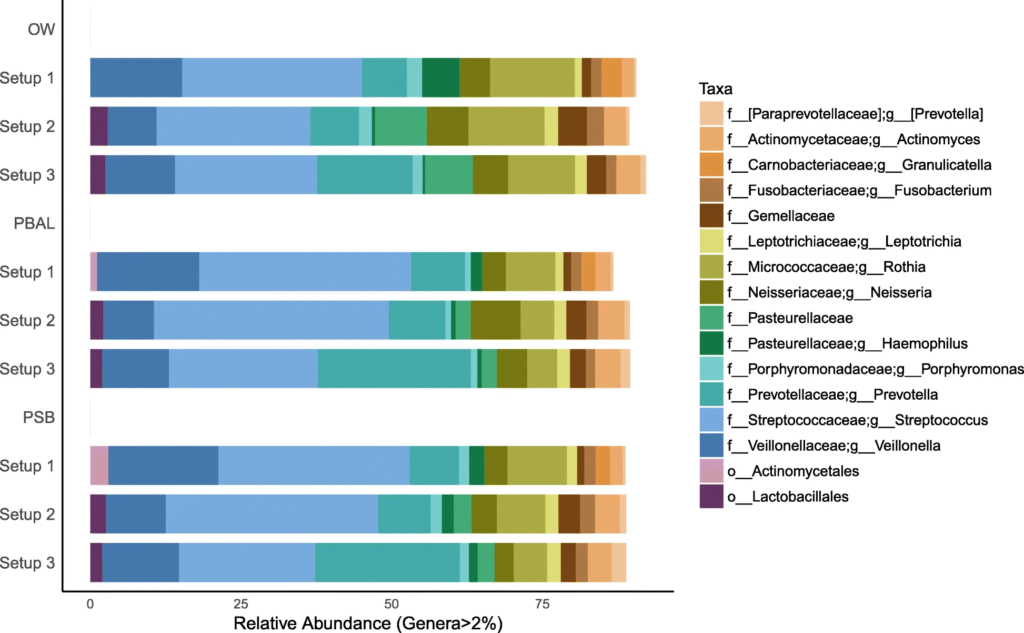
去除污染物后的
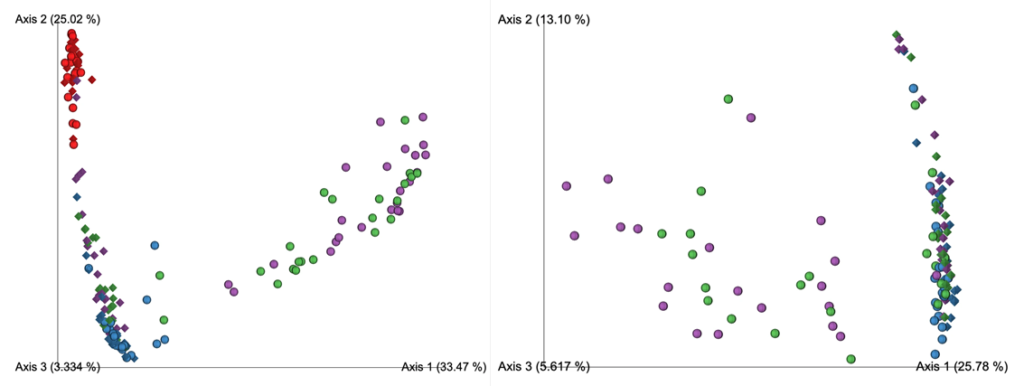
从左至右分别为去除污染物前后的未加权UniFrac距离的主坐标分析
OW:蓝色; PBAL:绿色;PSB:紫色;NCS:红色。
设置2(球形),3(菱形)
文章作者给出的结论是文库制备和测序方法的选择会对呼吸道微生物组的分析产生影响,且对上呼吸道的影响小于下呼吸道。靶向扩增子区域的差异(16S rRNA基因V3 V4与V4)并未表现出对细菌群落描述的重大影响。对于整篇研究存在的主要的局限性在于仅研究了DNA提取后的PCR步骤,污染或影响也可能来自于更前期的处理。
编者按
在使用测序技术进行的微生物研究中,测序偏差和污染物是一直存在的问题,也因此诞生了许多工具和计算方法用于尽可能的消除或降低这方面的影响。这篇研究也提醒了我们,在呼吸道微生物组的研究中,要注意上呼吸道与下呼吸道的菌群差异或相似可能不仅仅来源于样本自身,还可能掺杂着PCR方法选择上的影响。
参考文献:
Drengenes C, Eagan TML, Haaland I, Wiker HG, Nielsen R. Exploring protocol bias in airway microbiome studies: one versus two PCR steps and 16S rRNA gene region V3 V4 versus V4. BMC Genomics. 2021 Jan 4;22(1):3.
相关阅读:
谷禾健康
宏基因组中短序列的注释是理解测序微生物群落潜在功能的重要步骤之一。单纯利用局部匹配的注释容易混淆那些蛋白同源性且局部序列非常相似的序列,进而不能真实准确反映复杂蛋白质家族中多变的结构和功能域。

今天我们介绍一种新方法MetaGeneHunt,该方法可以识别特定的蛋白质结构域,并根据结构域的长度对hit-counts进行标准化。使用MetaGeneHunt对MG-RAST对公开获取的宏基因组进行分析,包括哺乳动物微生物群和Twin Gut肠道菌群研究,以评估短序列中含GH蛋白的频率和位于GH区域的匹配频率。
在对糖苷水解酶(GHs)的研究,发现在所有样本中4726,023条含有GH区域蛋白匹配的短读序列中,有58.3%的序列位于目标区域之外。接下来,在比较样本之前,将匹配到目标区域的hit-counts标准化,以说明对应的域长度。肠道和盲肠中的菌群显示出与不同微生物组合相匹配的GH谱特征。
相反,胃和结肠的菌群在结构和功能上显示出更多样性和多变性。在样本中,尽管有波动,但碳水化合物处理的潜在功能变化与群落组成的变化相关。这表示,在利用MG-RAST平台处理宏基因组测序序列时,MetaGeneHunt是一种能快速准确地识别短序列宏基因组中离散蛋白结构域的新方法。
在过去的几十年里,宏基因组DNA的高通量测序已经产生了大量的序列,这些序列的特征为我们了解微生物群落的结构和功能提供了许多认知。例如,截至2019年12月,MG-RAST托管了约40万个可公开访问的带注释的数据集。在数据处理过程中,不考虑目标区域(或蛋白质)的长度会导致两个主要的系统偏差。
首先,目标区域越长,他们的频率就越容易被高估。其次,如果数据处理涉及稀疏性,较短的、不太丰富的域,尽管重要,也可能被丢弃。为了解决这些问题,研究人员设计了MetaGeneHunt来精确注释从MG-RAST检索到的短序列宏基因组中的蛋白质结构域。MetaGeneHunt将MG-RAST提供的短序列局部比对与M5nr数据库中精确的基于PFam的蛋白质结构域识别相结合,以在公共可访问数据集中识别蛋白质结构域。
MetaGeneHunt简要说明:
MetaGeneHunt的设计基于MG-RAST平台注释的数据集的。在使用GeneHunt创建的M5nr数据库中,MetaGeneHunt使用了糖苷水解酶和辅助结构域(如CBMs)的精确的特定结构域注释(PFam)作为参考注释表(RAT)。
首先,MetaGeneHunt使用MG-RAST应用程序接口从MG-RAST(“330”和“650”文件)检索M5nr注释的宏基因组。接下来,使用来自RAT的注释命中的MD5id,在文件“650”中识别与潜在的GHs匹配的序列。
接下来,对于这些局部匹配,将精确对齐位置与RAT中特定于域的注释进行比较。如果查询中的>20AAs与特定的蛋白质结构域(考虑到RAT中的HMM-envelope位置)对齐,则该结构域注释被转移到查询中。
相反,如果查询的>20AAs匹配在目标区域之外(例如,在连接域、辅助域、信号肽中),则该注释被认为是否定的。用户可以随意修改重叠(overlapping)的阈值。接下来,从序列聚集文件( “330”文件)中检索每个识别出的命中的实际序列计数。最后,在后续的数据处理和标准化过程中,根据Pfam数据库中蛋白质结构域的大小,对每个蛋白质结构域的命中计数进行标准化。
方法验证:
文中使用的原始数据和预处理数据可在MG-RAST服务器上公开访问。在mgp20861项目中可获得对应于〜555百万个100 bp序列的小鼠微生物组数据。使用MG-RAST API 检索了哺乳动物微生物组数据(mgp116)和双肠肠道菌群研究(mgp10)其他数据集。哺乳动物微生物组研究糖苷水解酶(GHs)和相关酶的附加注释表是从Brian Muegge(直接对应)获得的。使用MG-RAST API检索了预处理的数据,包括从门到属水平的读物分类注释。数据分析和统计使用R统计语言。
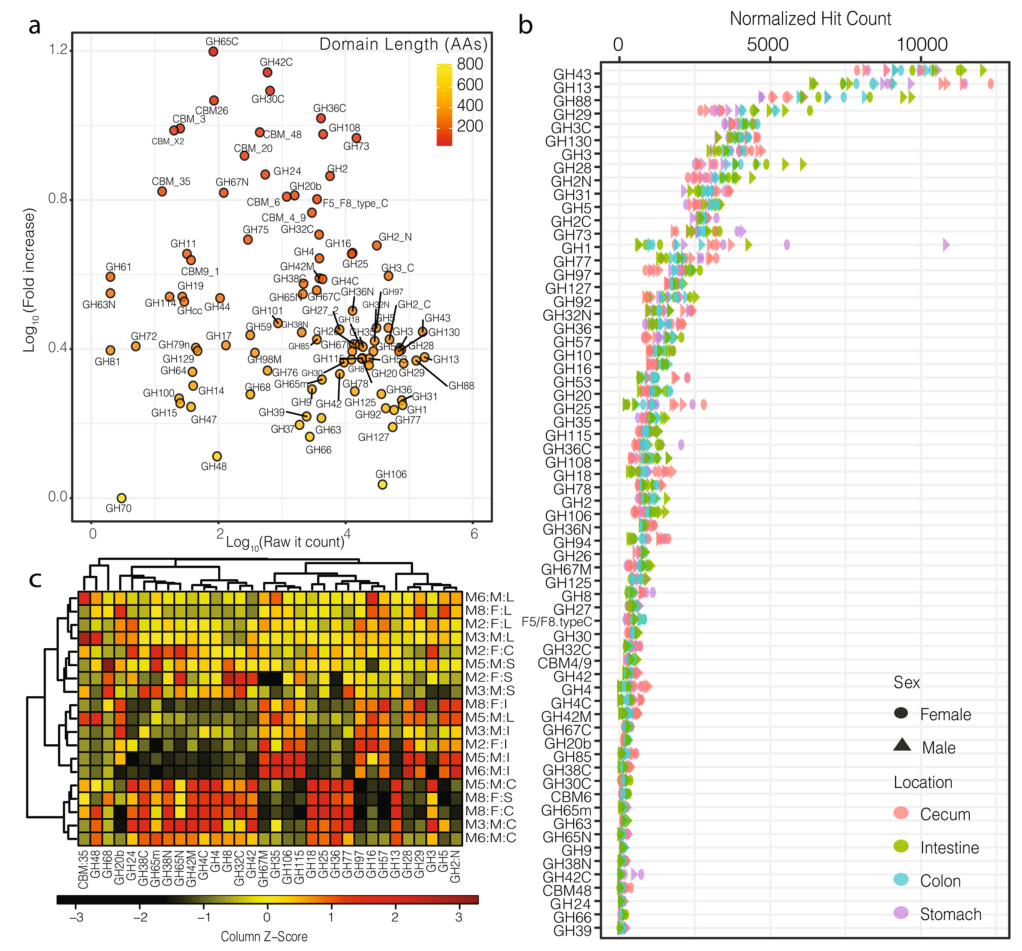
1. 糖苷水解酶的识别,识别蛋白质结构域并考虑其长度产生了一个健壮的功能注释系统,对hit-count的标准化反应了目标区域的实际分布。

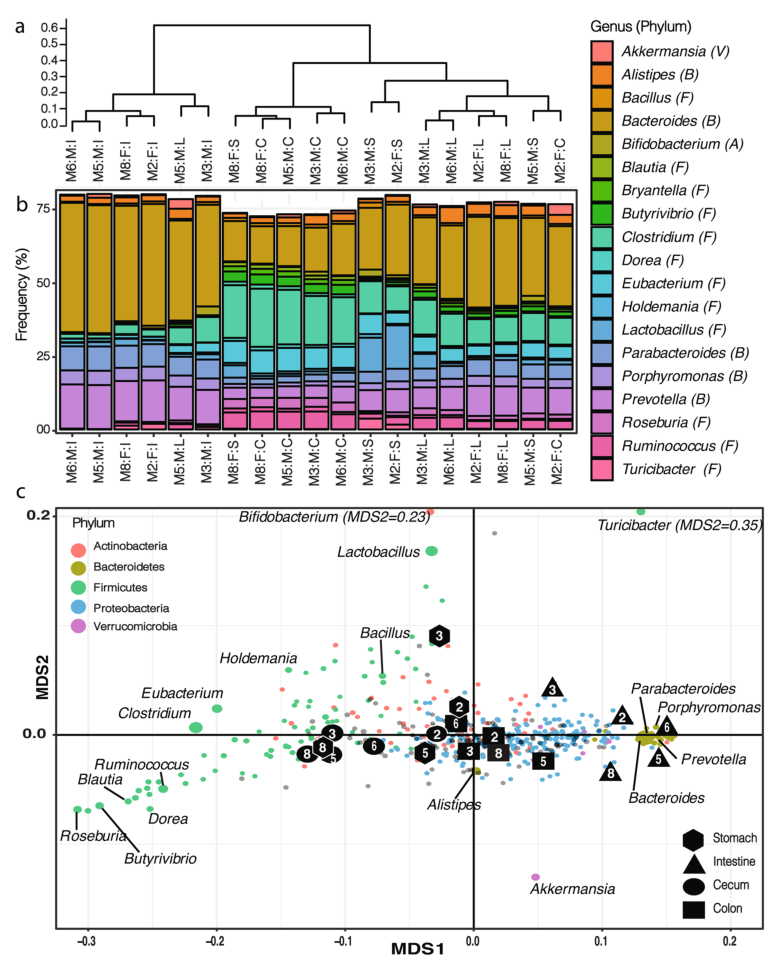
a).横轴为目标区域的原始hit-count,纵轴为标准化后的hit-count,图中的颜色阶梯表示目标区域的长度。这种标准化主要影响长度短的域(例如,GH78、GH25)、小的亚域(例如,GH31N、GH36C)和目标区域的附属域(例如,CMB5_12)。
b).小鼠胃肠道中目标区域的标准化后的hit-count(仅显示大于100的hit-count的区域),可见,标准化后的hit-count与结构域长度无关(附加文件中有对两者做相关分析,结果分别为P.pearson=0.38,P.spearman=0.33)
c).热图显示了小鼠胃肠道中最受样本来源影响的被稀疏标准化的GH区域的分布(two-way方差分析)。纵轴的注释列Mx:F/M:S/I/C/L分别表示小鼠(样本号):雌性/雄性:胃/肠/盲肠/结肠
2. 小鼠肠道菌群的结构,与盲肠中的微生物群落相比,结肠与肠道中的微生物群落结构更相似,结肠和胃中的微生物群落有较高的相似性。

a).对受样本来源影响较大的样本根据属水平进行样本聚类(Bray-Curtis距离指数,complete linkage)。
b).样本间的微生物群落组成,只展示了相对丰度至少占群落中1%的属水平物种(V:疣微菌门,B:拟杆菌门,A:放线菌门,F:厚壁菌门)。
c).NMDS分析(2D stress=0.020),展示了在样本聚类中都存在的这些菌属,在b)中的主要类群用标签指示,不同门水平按颜色区分,点的大小反映该属在样本中的最大频率。
微生物组中的结构-功能关系,多样性仍然与潜在功能高度相关。胃和盲肠的群落在结构和功能上是最多样化的。其次,肠道中的群落组成和功能大多是保守的,而与保守的微生物群落相关的大肠则显示出可变功能潜力。
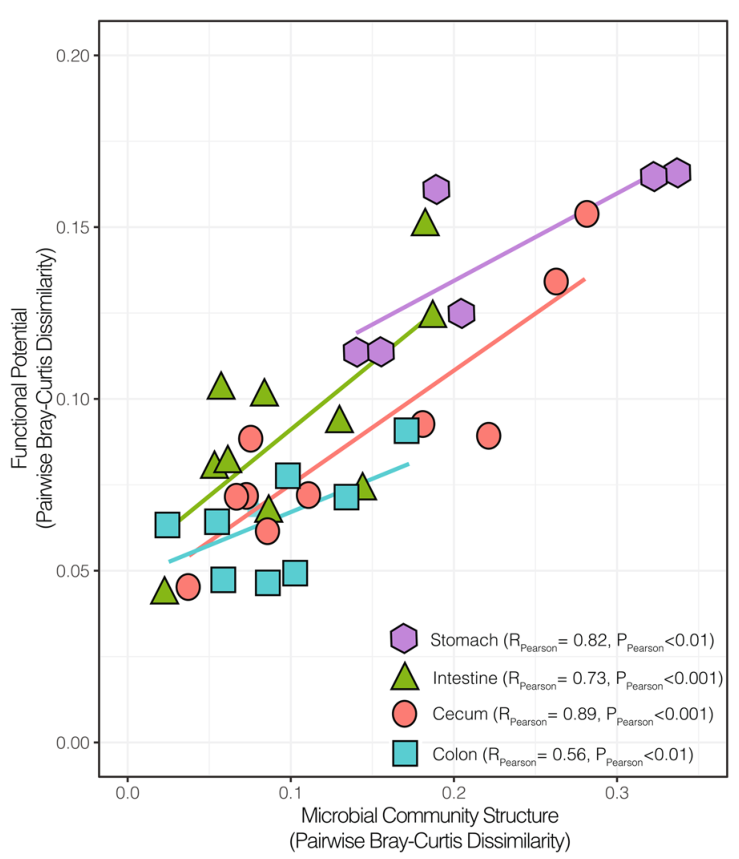

对同一位置的样本的微生物群落结构和功能差异进行成对比较(Bray-Curtis),线条为线性回归的结果。在胃,肠,盲肠和结肠中,属水平群落结构的变化与多糖解构功能的相关性分析结果表示除大肠外,其余的P.pearson的值都在0.001以下。胃和盲肠的群落在结构和功能上是最多样化的,尽管多样性仍然与功能潜力高度相关。其次,肠道中的群落组成和功能大多是保守的,而与保守的微生物群落相关的大肠则显示出可变的功能潜力。
MetaGeneHune提供了一种新的方法来识别短序列宏基因组中的GHs及其相关结构域。识别结构域而不是蛋白质是至关重要的,因为GH结构域与许多可变结构域相关。这种新方法基于GeneHunt注释方法,并对其进行补充,旨在分析MG-RAST中的短序列宏基因组。因此,它不需要大型计算机基础设施。
通过这种新方法对小鼠胃肠道菌群的GHs研究发现,在胃中,虽然富含碳水化合物处理的酶,但相对于胃肠道的其他部分,胃中没有特定酶可供选择;在肠道中,出现了更保守的菌群,最为富集的是拟杆菌门,它们的潜在功能主要在多糖处理上;来自结肠和胃的菌群虽然是距离最远的,但在结构和功能上却表现出高度的相似性。
在未来,利用GeneHunt和MetaGeneHunt相结合创建新的专用参考注释表将为研究宏基因组的潜在功能提供新的更有效的途径。
MetaGeneHunt和GH的RAT可在GitHub上公开访问。(https://github.com/renober/MetaGeneHunt)
参 考 文 献
Berlemont R, Winans N, Talamantes D, Dang H, Tsai HW.MetaGeneHunt for protein domain annotation in short-read metagenomes. Sci Rep.2020 May 7;10(1):7712. doi: 10.1038/s41598-020-63775-1. PMID: 32382098; PMCID:PMC7205989.
Muegge BD, et al. Diet drives convergence in gut microbiomefunctions across mammalian phylogeny and within humans. Science.2011;332:970–4. doi: 10.1126/science.1198719
Turnbaugh PJ, et al. A core gut microbiome in obese and leantwins. Nature. 2009;457:480–484. doi: 10.1038/nature07540.
Berlemont R, Martiny AC. Glycoside Hydrolases acrossEnvironmental Microbial Communities. PLOS Comput. Biol. 2016;12:e1005300. doi:10.1371/journal.pcbi.1005300.
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity,stability and resilience of the human gut microbiota. Nature. 2012;489:220–30.doi: 10.1038/nature11550.
Sharpton TJ. An introduction to the analysis of shotgunmetagenomic data. Front. Plant Sci. 2014;5:209. doi: 10.3389/fpls.2014.00209.

谷禾健康
链读测序(Linked-read sequencing)通过将相同的barcode与长DNA片段(10-100kb)的序列连接在一起,能够消除其中的一些错读,从而改进宏基因组组装。但目前还不清楚在使用链读测序时参数的选择对组装的质量的影响如何。
近日,香港浸会大学研究人员发表文章 “通过链读测序对宏基因组组装全面研究”。


模拟数据和模拟菌群中的分析结果表明,模拟数据(simulated data)中读取深度(C)与组装序列的长度呈正相关,但对组装序列的质量影响不大,模拟菌群的研究中读取深度(C) 对组装序列的质量以及被注释为基因组草图的bin的比例有轻微影响。
另一方面,宏基因组组装质量受CR(每个短读长片段的平均深度)和CF(由长DNA片段计算的基因组的平均物理深度)的影响。对于相同的读取深度,较深的CR 会产生更多的基因组草图,而较深的CF 会提高基因组草图的质量。
还发现μFL(未加权的DNA片段的平均长度)对组装有边际效应,而NF/P(每个分区的片段数)对局部组装涉及到的偏离目标读数(off-target reads)有影响,即较低的NF/P值会通过减少off-target序列的错读而有更好的组装效果。
总体而言,与Illumina的短读长相比,使用链读改善了组装中重叠群的N50,但与PacBio CCS的长读长相比则没有改善。
人体微生物群是一个复杂的系统,在生理活动和疾病中起着重要的作用。对微生物群中的微生物基因组进行测序可以帮助我们研究其功能。
然而,微生物基因组序列很难获得,微生物群中的绝大多数微生物不能被分离出来进行单个测序。目前的宏基因组项目中使用短读长测序对混合的微生物基因组进行测序。
这些结果在基因组组装过程中是有错读的,导致微生物基因组的完整性和重叠群的连续性结果不理想。长读长测序已经被用来尝试减轻这些问题,如Nicholls等人和Sevim等人的研究。特别是Moss等人的研究,其成果优化了纳米孔测序的长读长文库制备方案,并获得了更完整的细菌基因组。
但实际应用中,长读长测序是昂贵的。虽然链读序列(linked-reads)的基因组组装的质量无法与PacBio CCS的长读长相提并论,但其低成本和高碱基质量的优点是值得去使用的。
01 三组链读序列数据集的来源及构成:
模拟数据(simulated data):
从MBARC-26数据集中下载了23个细菌和3个古细菌菌株,按丰度分类,L-sim,低丰度微生物,摩尔浓度<10-15;M-sim,中等丰度微生物,10-15 < 摩尔浓度 < 10-14;H-sim,高丰度微生物,摩尔浓度 > 10-14
模拟菌群(mock community):
(ATCC MSA-1003)是一个由20个菌株组成的池,同样按丰度分类,L-mock,低丰度微生物;M-mock,中等丰度微生物;H-mock,高丰度微生物;UH-mock,超高丰度微生物。
人类肠道菌群:
一份来自健康的中国人粪便样本
02 DNA提取、文库制备和测序:
对于模拟菌群,从ATCC 20菌株交错的混合基因组材料中提取DNA,不进行大小选择。
对于人类肠道菌群,用Qiagen QiAaMP粪便迷你试剂盒提取DNA,去掉5kb以下的DNA片段。
脉冲场凝胶电泳后,按照厂商的说明制备10x Chromium文库。使用Illumina XTen双端2x150bp测序。人类肠道微生物组的DNA也被用于标准的Illumina XTen短序列测序。
03 DNA长片段重建和链读序列二次抽样:
Long Ranger v2.2.1用于纠正barcode碱基错误,计算PCR重复率,并完成barcode感知的链读序列比对。
使用BWA-MEM v0.7.17比对短序列和没有barcode的链读序列。根据映射得到的具有共同的barcode的短序列的坐标重建DNA长片段。
链接序列首先按barcode排序,然后按它们的映射坐标排序。如果最近的barcode序列大于50kb,则终止延伸长DNA片段。每个片段必须包括至少两个具有共同barcode的成对序列,并且最小长度为2kb。
04 宏基因组组装:
对于链读序列的组装,没有 barcode 的链读序列首先由 metaSPAdes v3.11.1使用默认参数组装为“seed”重叠群,并通过BWA-MEM v0.7.17与重叠群比对。
最后使用 Athena-meta v1.3 通过汇集在 scaffold 中的两个“seed”重叠群里共享相同 barcode 的序列进行局部组装。
05 组装效果评估:
MaxBin v2.2.4将长于1kb的重叠群分组到bins中,并通过CheckM v1.0.12评估其完整性和污染率。
Quast v5.0.0统计了基础信息,如重叠群的N50、NG50、NGA50、总比对长度(total aligned length)和基因组覆盖率(genomic coverage)。
Kraken v0.10.6基于内置数据库MiniKrakenDB为bins做物种注释。每个bins都作为一个基因组草图,被分类为高质量的(完整性>90%,污染率<5%),中等质量的(完整性≥50%,污染率<10%),低质量的(完整性<50%,污染率<10%)
来自人类肠道菌群和Illumina短序列链读序列二次抽样的组装效果统计
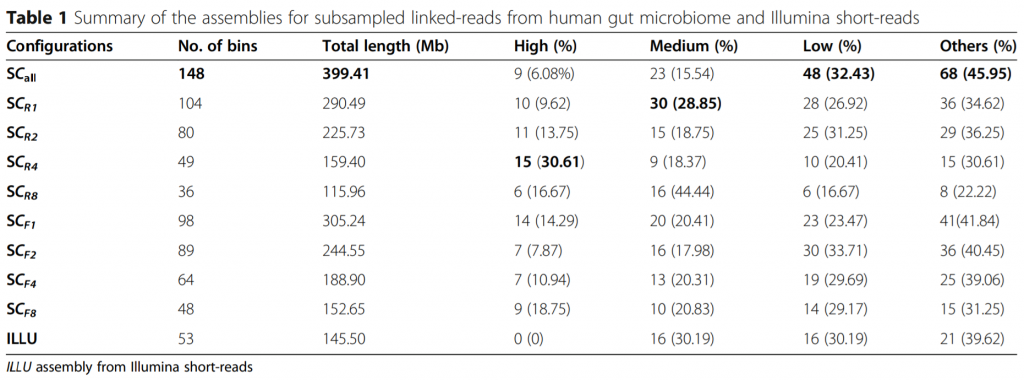

ILLU,Illumina短序列的组装
SC-all,模拟菌群和人类肠道菌群总共的两个测序lane链读序列
在链读测序中,有四个关键参数可能会影响宏基因组组装,如下图。
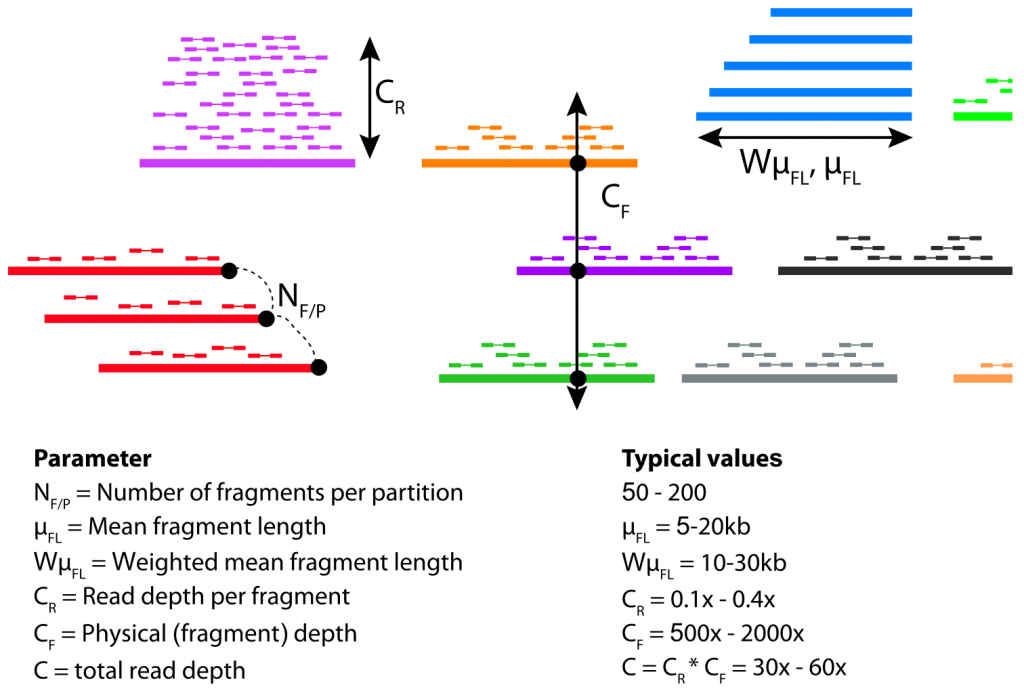

这些参数中有几个是相互依赖的。例如,输入DNA的量越大,CF和NF/P都会增加,CR就会降低;CF和CR的绝对值是由总读取深度(C)增加多少来设置的,因为CR×CF=C。
L-sim,模拟数据中的低丰度微生物,青色
M-sim,模拟数据中的中等丰度微生物,蓝色
H-sim,模拟数据中的高丰度微生物,红色
L-mock,模拟菌群中的低丰度微生物
M-mock,模拟菌群中的中等丰度微生物
H-mock,模拟菌群中的高丰度微生物
UH-mock,模拟菌群中的超高丰度微生物
“-”表示测序lane的倒数,例如MSCR4/MSCF4表示四分之一测序lane的序列被二次采样
MSCR-,模拟菌群中的短序列
MSCF-,模拟菌群中的长DNA片段
MSC-1,模拟菌群和人类肠道菌群总共的一个测序lane链读序列
SC-all,模拟菌群和人类肠道菌群总共的两个测序lane链读序列
相关阅读:
参考文献:
Zhang L, Fang X, Liao H, Zhang Z, Zhou X, Han L, Chen Y, Qiu Q, Li SC. A comprehensive investigation of metagenome assembly by linked-read sequencing. Microbiome. 2020 Nov 11;8(1):156. doi: 10.1186/s40168-020-00929-3. PMID: 33176883; PMCID: PMC7659138.
He S, Chandler M, Varani AM, Hickman AB, Dekker JP, Dyda F: Mechanisms of evolution in high-consequence drug resistance plasmids. MBio 2016;7(6): e01987–16.
Peng Y, Leung HC, Yiu SM, Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth.Bioinformatics. 2012;28(11):1420–8.
Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast singlenode solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31(10):1674–6.
Nurk S, Meleshko D, Korobeynikov A. Pevzner PA: metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27(5):824–34.
Nicholls SM, Quick JC, Tang S, Loman NJ. Ultra-deep, long-read nanopore sequencing of mock microbial community standards. Gigascience. 2019;8(5): 1–9.
Sevim V, Lee J, Egan R, Clum A, Hundley H, Lee J, Everroad RC, Detweiler AM, Bebout BM, Pett-Ridge J, et al. Shotgun metagenome data of a defined mock community using Oxford Nanopore, PacBio and Illumina technologies. Sci Data. 2019;6(1):285.

谷禾健康
用于生物数据可视化的方法不断改进,但是在一些可视化图形的着色方面仍然存在根本性的挑战。
生物学数据的视觉不应淹没,掩盖或偏倚结果,而应使其更易于理解。这是对于在创建可视化效果时如何有效使用颜色的挑战。
生物数据的可视化处理是计算机图形学,科学可视化和信息可视化在生命科学各个领域的应用。

本文将介绍10条简单的规则来对生物数据进行可视化着色。
总览
规则1:确定数据的性质
规则2:选择色彩空间
规则3:根据所选颜色空间创建调色板
规则4:将调色板应用于数据集以进行可视化
规则5:套用调色板后,检查数据中的颜色上下文
规则6:在数据可视化中评估颜色的交互作用
规则7:要了解特定学科的颜色约定和定义
规则8:评估色差
规则9:考虑网络内容的可访问性和打印实际情况
规则10:黑白分明
数据是有价值的信息记录。可视化数据是将这些数据中包含的想法、经历和故事联系起来的一种重要而有力的方式。
图形和数据可视化促进了生物信息在不同背景下的表达和交流,形成叙述、想法和经验。要使数据中包含的信息具有形状,了解数据的性质是重要的。借用描述性统计的领域知识,数据如性别、年龄、身高、体重和眼睛颜色等被称为变量。变量的类型与数据的性质有关。
区分变量类型的一种方法是依赖于分配给变量的值中的信息的性质。这个被称为测量的水平或尺度,将观察到的变量分为4个级别:名义、序数、区间和比率。这些数据也可以分为两种不同的数据类型:定性或分类(名义、序数)和定量(区间、比率)。
下面我们分别用一个例子来描述和解释:
名义描述了一个变量的属性,只通过名称(类别)来区分,没有顺序(等级、方向或位置)。
例如:性别、生物种类、眼睛颜色,血型(A、B、AB、O),细菌类型(球菌、芽孢杆菌、螺旋菌等)。它们是一个多值变量,没有明确的尺度来适应不同的值。
序数层次描述了按顺序(等级、规模或位置)区分的变量的分类属性,但没有关于它们之间差异相对程度的信息。要注意这种变量可能会用数字编码。
举例:热度(低、中、高);疾病的严重程度(轻度、中度、重度);一致量表,如李克特量表,(强烈不同意、不同意、无意见、同意或强烈同意)。
注:李克特量表是一种心理反应量表。
区间级别描述变量的属性,通过差异程度来区分,没有绝对零度,并且属性之间没有已知的比率。通常,该变量的数值为正、负或零。
例如:公制摄氏温标,温差(摄氏度和开尔文),1年的间隔。20℃和30℃之间的差异与25℃和35℃间的差异相同.。
比率级别描述变量的属性,这些属性通过它们之间的差异程度来区分,绝对为零,并且属性之间的比率是已知的。具有负值是不典型的。
例如:年龄、身高、体重、持续时间、开尔文温标。此外,假设数值的定量数据(区间或比率)可以进一步分为离散或连续。
离散(可计算的)变量仅假设整数和某种计数。
例如:年龄和日期是离散的。年龄在1年内保持不变,而日期在24小时内保持不变。它们都以“1”跳跃或增加。
连续(定义范围内的任何值)变量可以取某个值范围内的任何值。对这种测量的观察会受到测量仪器的限制。
例如:身高(厘米,英寸),体重(公斤,磅),温度(摄氏度,华氏度),时间(小时,分,秒)。温度逐渐升高,时间不断流逝。
当只有两个可能值时,二进制或二分变量类型是一种特殊类型。示例:是或否调查问卷和二进制数字(0或1)。
表1根据4个不同的测量相关类别介绍了4个测量级别,包括从最低到最高的测量分辨率。
表1 四个层次的测量


使用4个与测量相关的类别来比较等级:分辨率、属性、数学运算符和中心趋势。
颜色空间指的是颜色转化为数字的颜色模型。基于一组原色,颜色模型创建许多颜色。每个模型都有其可以产生的特定颜色范围,该范围定义了色彩空间。
通常,红、绿、蓝(RGB)和青色、洋红、黄色和黑色(CMYK)是最常见的系统,当然还有其他系统。例如,色调、饱和度和亮度/值(HSB/HSV)颜色空间是RGB颜色模型或标准红绿蓝(sRGB)颜色空间的替代表示。
注:关于这些维度的更多信息,大卫·布里格斯的网站名为《颜色的维度》是一个关于颜色理论和使用的信息宝库。
传统的颜色工具,如色轮,鼓励艺术/手工颜色选择。颜色或代码的数值是不同的,将颜色视为特定颜色空间中的数字。此外,由于我们选择的数字和输出颜色之间可能会出现差异,颜色空间应该在感知上是一致的。
在颜色科学领域,已经努力建立独立于特定颜色显示或复制设备的颜色空间。人们努力创造出感觉上统一的色彩空间。这些颜色空间背后的动机是使空间与人类视觉感知颜色属性的方式紧密一致。
下表是常用的颜色空间(表2),接下来将要讨论的是解决感知一致性问题的颜色空间。
表2 常用色彩空间的优缺点
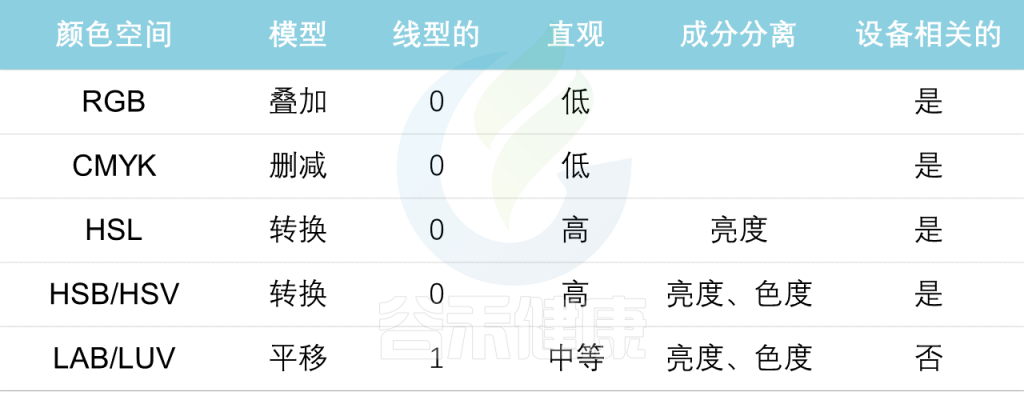

由于复杂的颜色转换,光线混合的维度反映了人类视觉的工作方式。下面几种是我们需要考虑的各种特征:模型,线性,直观,组件分离以及设备相关。
模型
一个有序的系统,用于从一小组原色中创建一个完整的颜色范围
线性
颜色值相同的变化应该会产生视觉重要性大致相同的变化
直观
指颜色维度易于重新映射到不同的颜色模型
组件分离
指相对于其他维度分离1个颜色维度。
例如,色调、饱和度和亮度(HSL)分离亮度分量(明度),在图像处理的领域知识中特别有用。
设备相关
颜色空间依赖于所使用的设备来设置、制作和渲染的情况。
亮度是光的可见能量或根据人类视觉系统的逐波长响应加权的物理光能。
色度是一个区域的颜色,它被判断为一个相似的被照亮的区域的亮度的一个比例,这个区域看起来是白色的或高度透射的。虽然色度描述了光的心理物理颜色,但它与光的强度(亮度)无关。
所列出的感知一致颜色空间优于RGB和CMYK颜色空间。RGB用来表示颜色,但它不足以进行颜色处理,并且不是行业标准。
由于CMYK主要用于印刷,它有许多缺点,将在下一篇规则9中进一步讨论。然而,它们并非没有混淆的效果,例如亮度随色调而急剧变化。
LUV和LAB都追求感知一致性。虽然两者都已被CIE(国际照明委员会)采用,但通过依靠三个组成部分并计算相邻颜色之间的椭圆距离,可以观察到空间中不同颜色的相对概念差异。
因为它们是独立的,我们建议使用它的颜色空间。如果选择了,就需要为数据创建一个合适的调色板。
创建一个调色板很像选择一套衣服。重要的是要了解允许选择颜色来给数据可视化着色的规则。
为了根据特定的颜色空间选择调色板,通常使用色轮。它是一种围绕一个圆圈组织不同颜色以显示颜色之间关系的工具。通常,色轮包含12种颜色。
创建色彩和谐是一个选择在图像合成中协同工作的色彩的过程。基于色轮上的颜色组合,有助于为色彩如何协同工作提供共同的指导方针。
我们可以区分有助于使用色轮创建配色方案的软件和/或网络工具,即Adobe color和配色方案设计器Paletton。
除了创造美学上令人愉悦的颜色组合,调和性还可以用来指导调色板的创建。它们包括单色、模拟和互补。
下图描绘了青色调中的三个调和示例。应该注意颜色的小点,以描绘出特定的色调排列(单色、相似和互补)。
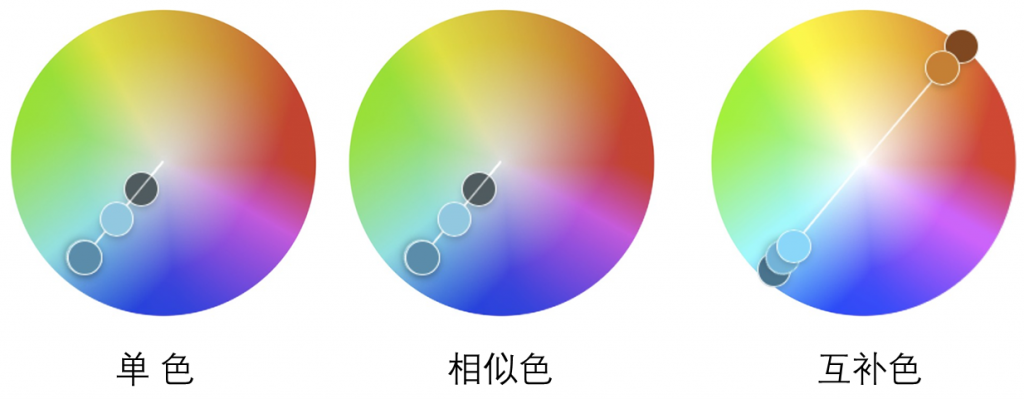

单色或单调色度
是一种单一色调,它在色调、色度和饱和度方面有所变化。一个特别的例子是单调方案,但非彩色(没有色调),仅由从黑色到白色的灰度值组成,即灰度。
相似色
是那些位于任何给定颜色的两边或被一个颜色分开的颜色。这些通常是自然界中的配色方案。
互补色
是色轮上彼此直接相对的颜色。他们经常形成对比,相互突出。当用作数据中的高亮颜色时,它们非常有用。
为了更好地将颜色的使用与数据类型联系起来,信息设计师和数据科学家将上述数据类型(规则1)简化为三种主要类型:连续的、发散的和定性的。
这些分类是在ColorBrewer工具中开发的,最初旨在为制图提供颜色建议。这一概念已经被数据可视化社区所采用,反映在蒙兹纳的可视化分析和设计教科书中。该网络工具可以在colorbrewer2.org找到。
下图展示出了每个数据类型的调色板的例子。
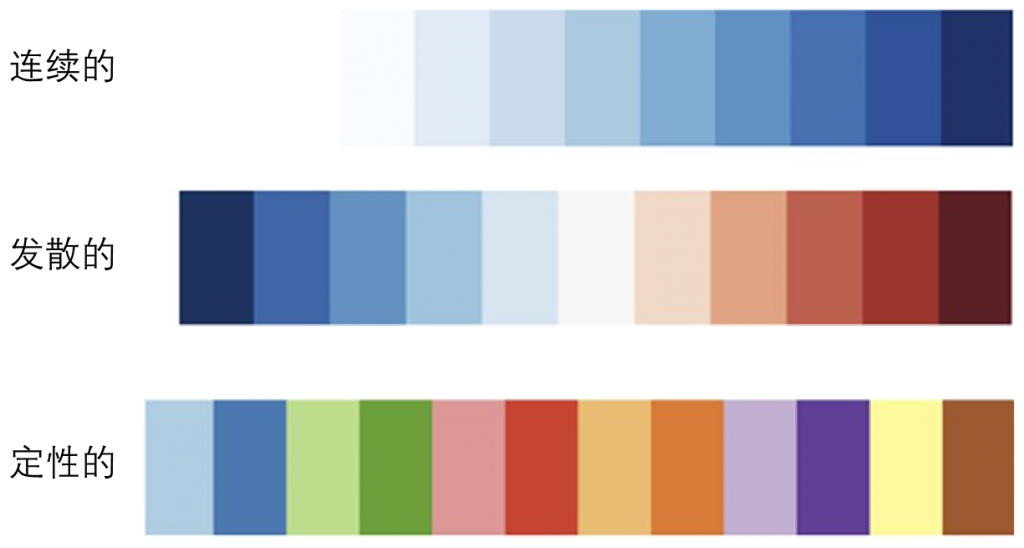

顺序调色板
适用于从低到高变化的有序数据。视哪一方对观察者来说最重要,视觉编码是两种颜色之间的变化,分别从白色或较亮的颜色到黑色或较暗的颜色。这种颜色使用是明度逐步变化,通常重要的数据值具有较暗的颜色。这些调色板对应于包含1种颜色变化的单色调色板。
发散调色板
显示两个方向的视觉变化。主要用于在区间数据范围的两端同等强调中间值和极值,它们通常是对称的。颜色在黑暗中增加,以表示断点(如零变化或平均值)周围与数据中特定有意义的中间值之间的差异。
定性调色板
不依赖或暗示类别之间的数量差异。通常,色调以一致的亮度来表示名义和分类数据。还有另外两种变体:成对和强调。处理无序数据时,成对调色板通过视觉关联类来处理成对数据,但强调调色板通过更饱和的颜色来强调相关类。
除了前面提到的工具ColorBrewer,还有两个调色板:一个用于连续数据,另一个用于定性数据。
对于连续数据,推荐 viridis调色板。它在感觉上是均匀的,并以多种色调显示单调增加的亮度。多亏viridis调色板和其他调色板,一个连续数据集的所有数据点都具有同等的视觉重要性。此外,我们将在下一篇的规则8中看到,这些调色板对色弱和色盲是友好的。
对于定性数据,Tableau 10调色板可以推荐给大家。它包含几个非常不同的色调,亮度值范围很广。虽然它是用10种颜色设计的,而且很适合三色异常,但它所有颜色的使用对其他颜色缺陷是一个挑战。我们将在规则4和下一篇规则8中讨论定性数据的颜色限制。
另外还有一个创建调色板的网络工具,即Colorgorical( http://vrl.cs.brown.edu/color)。
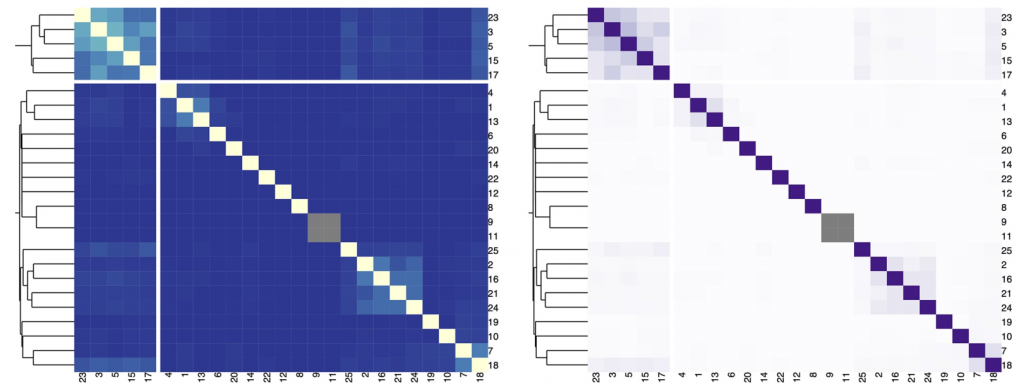
要应用选定的调色板,需要考虑将颜色映射到数据点的过程。基于规则3,我们考虑了3种不同的颜色映射调色板:连续的、发散的和定性的。
对于连续调色板,色调应该受到限制,只有亮度或饱和度应该变化。根据背景颜色、手头的任务和数据的性质,将较高的值映射到较暗或较亮的颜色非常重要。
在下图中,我们展示了一个热图,描述了不同字符串之间的Jaccard索引和由分层聚类提供的背景信息。

对于发散的调色板,当数据有有意义的或关键的中断时很重要。通常,关键断点应该采用中性颜色,如灰色,端点应该采用饱和颜色。一般来说是对称的,临界断点可以是平均值、中间值或零变化值。
在平均值或中位数的情况下,通常有低终点和高终点。在负值和正值有零值中断的情况下,端点应使用不同的色调。为了突出分歧,中断可以去饱和,端点可以饱和。
对于定性调色板,建议仅使用5 –6 种颜色,如果绝对需要,也可以使用更多颜色。实际上,当使用ColorBrewer时,限制被设置在3到9的范围内。
如果有理由的话,我们认为颜色是不变的,即使它们在不同的光线下。事实上,颜色恒常性是感知物体颜色的能力,不受光源颜色的影响。这主要是因为颜色是一种相对的媒介。
举个例子,比如我们可以看到一根香蕉在阳光充足的中午或光线微弱的黑暗房间里呈黄色。然而,在某些情况下,相邻的颜色会改变我们的感知和区分某种颜色影响的能力。
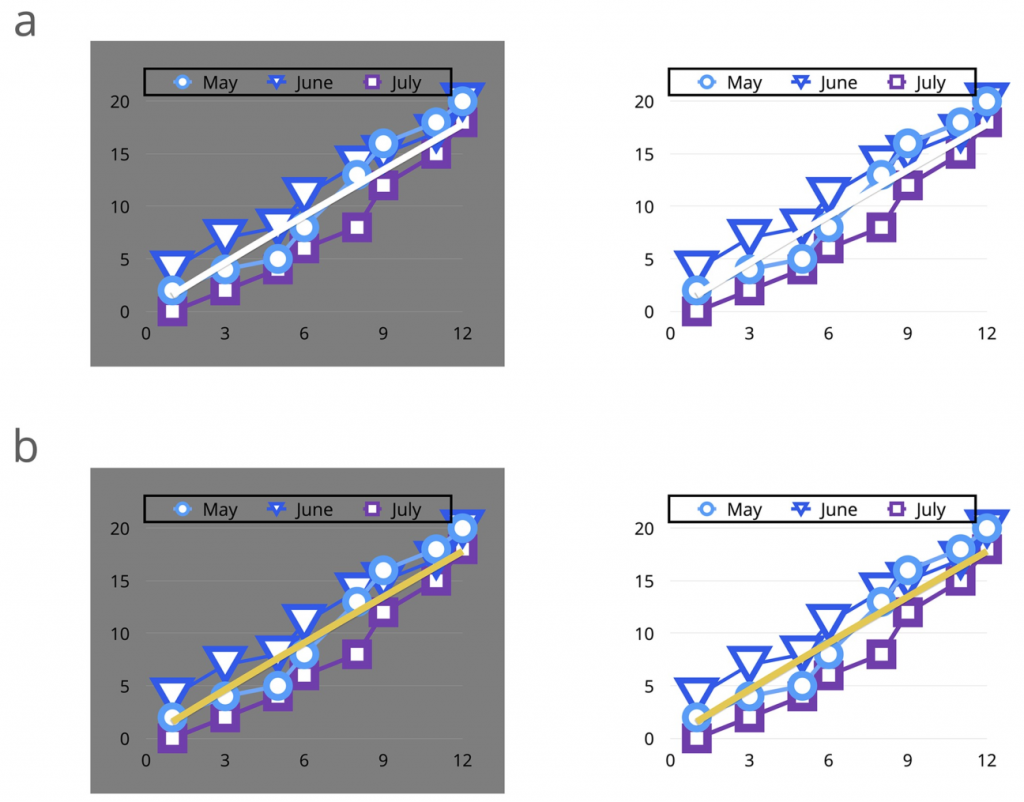
下图显示了一个数据视觉的例子,其中白色可以与灰色背景区分开来,比如在你的电脑屏幕上。然而,同样的白线在白色背景下很难区分,也许当打印在白纸上时。当白线变成黄线时,情况就解决了。

有个“Interaction of color”app可以进一步教会你如何意识到颜色的背景,它是约瑟夫·阿尔伯斯50年前写的《色彩交互》一书的数字化延伸。它为在不同的显示背景中学习颜色提供了练习。该应用还允许在interactionofcolor.com创建个性化的色彩研究和调色板。
在生物数据可视化中,通常会看到红色/蓝色的数据可视化。由于同时对比,对红/蓝颜色组合的偏好是可以解释的。下图就是这种情况。
-1024x454.png)
-1-1024x454.png)
左:红色/蓝色组合。右图:绿色/紫色补色组合改善了数据可视性。
同时对比是指两种不同的颜色相互影响的方式。这也是蓝色背景下很难阅读红色文字的原因。理论是,当两种颜色并排放置时,一种颜色可以改变我们对另一种颜色的色调感知。实际的颜色本身不会改变,但我们认为它们已经改变了。
法国化学家米歇尔·欧仁·切夫勒发展了这种同时对比的规则。它坚持认为,如果两种颜色靠得很近,每种颜色都将呈现相邻颜色补色的色调。类似的结果也可以发生在数据可视化中。然而,同时使用对比色可能难以评估数据趋势的变化。
颜色的使用取决于大量数据和介质特性。除了某些颜色的不良相互作用之外,我们会看到颜色可能带有某种意义。
对于交互,存在红/蓝颜色或文本内容的交互。首先,文本看起来模糊不清,伤害眼睛。这是称为色差的现象的结果,色差对应于不能同时聚焦在两种颜色上。
对于下图中提供的例子,还需要解决色彩不足问题的补色组合。绿色/紫色方案提供了两个数据变量之间的转换组合,同时允许有色觉障碍的个人区分这两个变量。绿色和紫色是这种特殊情况下的最佳组合。
-2-1024x454.png)
-1-1024x454.png)
在某些情况下,感知的一致性至关重要。一个简单的例子是,在不规则的颜色空间中,选择一种随机的颜色以便在黑暗的背景下可读,这是很困难的,因为相同亮度或光度的颜色看起来非常不同(蓝色和黄色在HSV中都有100%的亮度,但是蓝色比黄色暗得多)。
为了解决这个问题,需要考虑所选色调的复杂计算,以使随机颜色看起来同样明亮。有一个更简单的方法,即选择更好的颜色空间。
基于jet或彩虹的调色板是最常用的调色板,因为它是在软件工具中作为标准提供的。它具有很高的对比度,这使得它能够突出手头数据的特征。
然而,当查看颜色图表时,色带或色块尤其出现在青色和黄色区域。这个看似不错的调色板在应用于描述同等重要的顺序数据时会导致急剧的转换,尽管底层数据变化均匀。事实上,由于非恒定的感知颜色变化,这是误导,甚至对色弱的个人更是如此。
下图描述了这些部分。
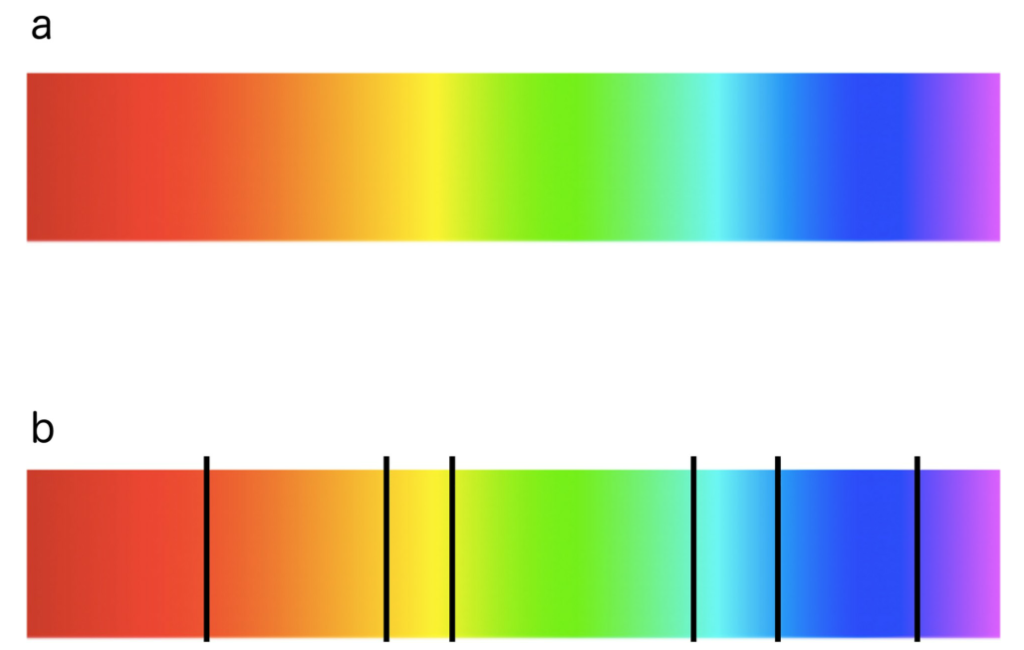
虽然许多研究人员都在抱怨它的误导,但在实际许多应用中,基于彩虹的调色板仍然在被使用,并且有可能对解决任务的准确性有潜在的负面影响。
不幸的是,由于人类通常将颜色分类,彩虹调色板的使用会给数据的解释带来偏差。此外,由于色调的自然顺序,这甚至可能被放大。然而,不同的方面可以被智能地集成。例如,不同的亮度强调某些标量值,而低亮度颜色(例如,蓝色)可能隐藏高频。
生物学描述了生物组织的不同层次(从分子到细胞、有机体到生态系统),整合了多种领域,如生物化学和生物物理学。这涉及到大量不同风格的数据,这些数据可能受特定领域惯例的约束。
我们简要讨论4个与生物化学、生物物理学、解剖学和细菌学相关的显著例子。
生物化学
在化学中,一个分子中不同原子的颜色遵循标准的科里·鲍林·科尔顿(CPK)规则。最重要的颜色是氢的白色(H),碳的黑色(C),氮的蓝色(N),氧的红色(O),硫的深黄色(S),磷的紫色(P)。其余的原子呈现出亮、中、暗、卤素组为深绿色,金属组为银色。生物化学遵循这些惯例,例如,给20种蛋白质氨基酸的生化结构着色。
生物物理学
在过去的几年里,已经开发了广泛的荧光蛋白遗传变异体,其特征是荧光发射光谱分布几乎跨越了整个可见光谱。借助这种分子和显微镜技术,科学家可以看到特定的细胞反应甚至亚细胞机制。例如,这种特定的分子可以在不同的光谱范围内(例如青色、绿色、黄色或红色)发出荧光。当然,最著名的分子是绿色荧光蛋白。如果数据集涉及荧光图谱,或包括光谱范围的信息,惯例是根据它们给数据着色。
解剖学
解剖学上,颜色约定从第一张解剖草图就已经存在。虽然第一幅彩色印刷的医学插图显示了文字颜色的用法,但现代颜色的用法是相当象征性的。事实上,颜色通常用于肤色、内脏、循环和神经系统,甚至是选定的身体组织(如肌肉或脂肪)。尽管在体内动脉和神经呈白色,静脉呈淡蓝色,但既定的颜色惯例是动脉为红色,静脉为蓝色,神经为黄色。
细菌学
在细菌学中,科学家对许多细菌特性和机制感兴趣,例如革兰氏染色、形态学、遗传学和抗生素抗性。前者通过细胞壁的化学和物理特性来区分细菌(革兰氏阳性细菌有厚厚的肽聚糖细胞壁,保留了结晶紫的主要染色)。后者发生在细菌和真菌等细菌有能力击败用来杀死它们的抗生素(如青霉素)时。
基于包含3种最流行的抗生素对16种细菌的性能的数据,我们在下图中展示了青霉素与新霉素的有效性的2个实例数据。
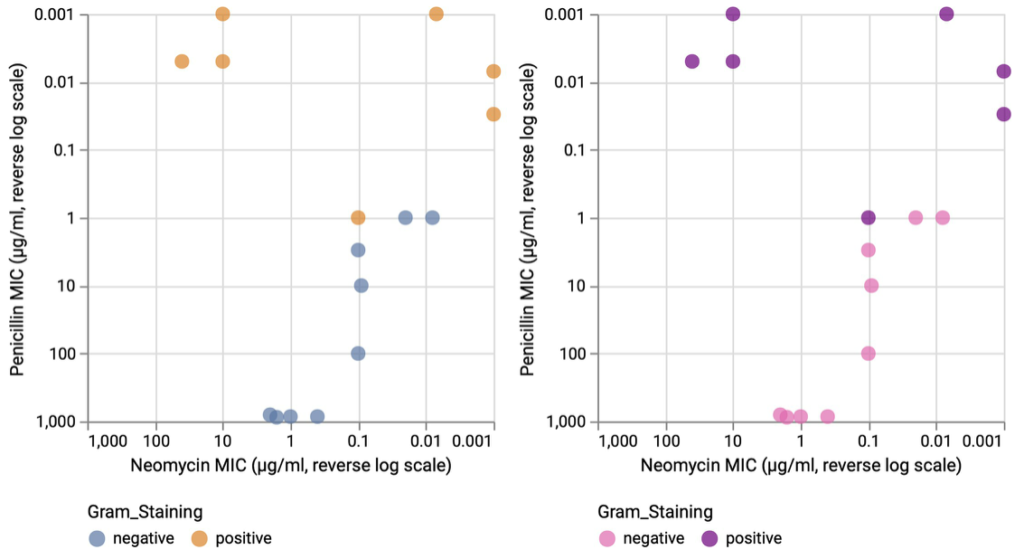
虽然选择蓝色/橙色的配色方案是为了为名义比较提供可感知的可分辨颜色(左),但采用革兰氏染色颜色惯例呈现出更适合具体问题的颜色用法(右)。
其他生物领域
其他实践存在于特定的生物研究领域。例如,在分子和进化生物学中,基因表达水平和基因保护的视觉编码依赖于红/蓝发散调色板。然而,我们不能谈论颜色惯例,因为在红/绿、红/蓝以及断点不是白色而是黄色的其他情况下,这个值会有很大差异。
值得一提的是要注意文化习俗。事实上,在不同的国家或文化中,颜色可能具有非常不同的象征意义,如果不是相反的话。一个很好的例子是红色,它在西方社会象征着危险和激情,在东方社会象征着幸福和繁荣。
人类有3种感光细胞或视锥细胞,每一种都对视觉光谱的不同部分敏感,以促进丰富的彩色视觉。我们需要尊重一些人的颜色感知是不同的,并评估所选择的调色板是否适合有色弱或色盲的人。
如果一个或多个视锥细胞不能正常工作,就会导致色觉缺失。红色锥细胞缺乏症被归类为红色盲。绿色锥细胞缺陷被归类为绿色盲。蓝锥细胞缺陷被归类为蓝色盲。
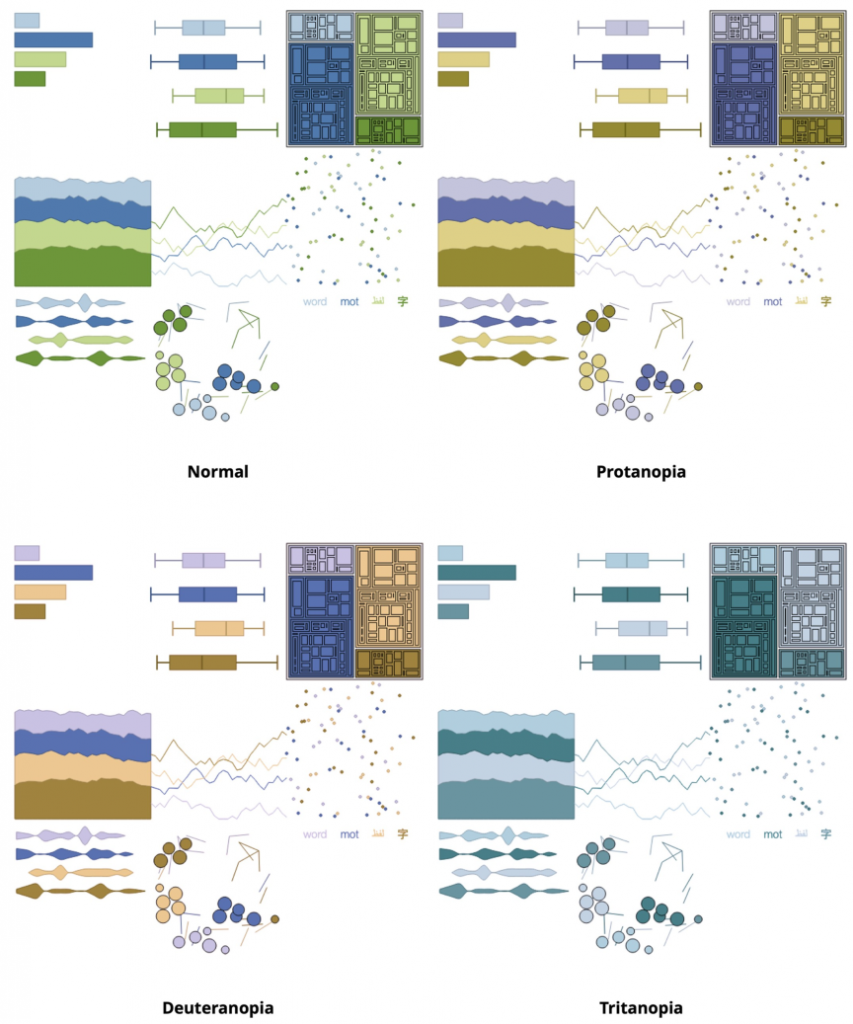
当创建或选择调色板时,不同的网络工具允许测试色弱和色盲。一方面,可以使用Adobe Color web工具(color.adobe.com)或Paletton–配色方案设计师(paletton.com)来测试调色板的颜色缺陷。另一方面,网络工具Coblis (Matthew Wickline和人机交互资源网络)使我们能够评估数据可视化是否对更大的受众可用,包括颜色缺陷。
另一个值得注意的工具是Viz调色板。它允许通过模拟选定的信息可视化示例来测试特定调色板的颜色缺陷。下图结合了3个工具,ColorBrewer, Viz Palette和 Coblis,提供了一个用例的例子。

在许多情况下,生物数据可视化超越了研究工作,成为一般在线(如网站)和印刷(如期刊论文)出版物的一部分。对于这些情况,我们简要讨论了网络内容的可访问性和打印现实。
对于基于网络或桌面和移动设备,我们建议遵循万维网联盟(W3C)制定的网络内容可访问性指南(WCAG)。网站必须是可感知的、可操作的、可理解的和健壮的,根据4个原则组织有12个指南。
虽然有一些技术可以帮助作者满足指导方针和成功标准,但是这些技术会随着时间的推移而发展和适应。在列出的技术中,有8种与颜色有关。我们将范围限制在非交互式数据可视化,并借用有利于数据可视化可访问性的技术:
· 确保通过颜色差异传达的信息在文本中也可用
· 使用颜色和图案
·使用颜色提示时使用语义标记
·当使用文本颜色差异来传达信息时,确保附加的视觉提示可用
·对周围的文本使用3:1的对比度,并在链接或控件的焦点上提供额外的视觉提示,仅用颜色来识别它们
·包括用于彩色表单控件标签的文本提示
事实上,其中大部分是为了网页颜色的使用,但我们认为它们是相关的。所报道的技术解决了如何提高看不见颜色的用户的可访问性,因此可以寻找或倾听文本线索;使用盲文显示器或其他触觉界面的人可以通过触摸来检测文本提示。
此外,一些技术解决了实现文本信息和内容的更好对比度的问题。即G17:确保文本(和文本的图像)和文本后面的背景之间存在至少7:1的对比度。
事实上,这个想法是为了确保阅读文本时亮度有对比,而不是色调有对比。
网络工具 Colorable 允许使用十六进制格式的web十六进制代码测试两种颜色,并提供滑块来控制色调、饱和度和亮度。
观看和阅读生物数据可视化取决于目标受众使用的媒介。一方面,使用桌面和移动设备,其中光源用于在RGB颜色空间中混合不同强度的红色、绿色和蓝色。当所有颜色混合时,出现白色。
另一方面,使用纸张打印件,其中打印机将不同程度的CMYK颜色与物理墨水颜色(青色、品红色、黄色和黑色)相结合。当所有颜色混合在一起时,就会产生黑色。为了方便起见,我们可在以下工作时提出一项易于遵守的入围名单要求:
台式机和移动设备,最适合的色彩空间是RGB。关于在网页上应用颜色的指导性文件可以在这里找到:w3.org/TR/css-color-3/#rgb-color
小印刷件,如小册子,或期刊纸图,我们鼓励在300 DPI分辨率的CMYK色彩空间的图像。
非常大的图形并不总是控制它们的质量,我们建议从灰度、位图或RGB颜色空间转换到打印机友好的CMYK颜色空间。
在某些需要考虑印刷成本的情况下,黑白配色方案可能是首选。此外,黑白会增加那些色盲者以与您相同的方式看到和阅读数据可视化的机会。
在不同的领域,如图形和渲染,甚至摄影,这条规则通常被表述为“检查它在黑白和彩色中是否工作良好”。在数据可视化中,这通常与测试所呈现的故事是否仍然可见或可辨别有关。
黑白分明意味着两件事。当不确定调色板时,尝试用灰度显示数据,或者比较两个彩色版本的数据,当不确定哪个更易读时,用黑白打印出来。大多数情况下,后者是找到对比度更好的可视化。
此外,一个建议是关于影印友好的调色板。为了对抗影印过程的损耗,单色或连续调色板是最有弹性和最合适的。
着色并不容易。如果对颜色有需求,可以选择适合并使用少量的颜色,避免饱和的颜色,并且要符合阅读者的期望。


以上两图详细说明了根据示例任务的黑白数据可视化的变化。安全选择的一个例子是选择一种颜色和几种灰色阴影。
最后,总结一下这十种规则。

【参考文献】
Hattab G, Rhyne T-M, Heider D (2020) Ten simple rules to colorize biological data visualization. PLoS Comput Biol 16(10): e1008259.
Rhyne T. M. (2017). Applying Color Theory to Digital Media and Visualization., CRC Press, Boca Raton, Florida, ISBN 9781498765497
Smith N, van der Walt S. A better default colormap for Matplotlib. SciPy2015. 2015
Stokes D., Matthen M., & Biggs S. (Eds.). (2015). Perception and its modalities. Oxford University Press, USA
Gramazio CC, Laidlaw DH, Schloss KB. Colorgorical: Creating discriminable and preferable color palettes for information visualization. IEEE Trans Vis Comput Graph. 2016; 23(1):521–530.

谷禾健康


在现代测序技术的帮助下,微生物组研究的范围被扩大,通过16S rRNA测序或鸟枪法宏基因组测序可以生成大量的微生物组数据。而微生物群落研究中的一个重要问题是对这些微生物的归类,模拟和分析人类微生物群。
通常使用16S rRNA技术量化微生物群落的组成,但量化后的数据是偏斜的,带有过多的0。目前还缺乏对复杂的微生物群落测序数据的标准化的聚类分析方法。
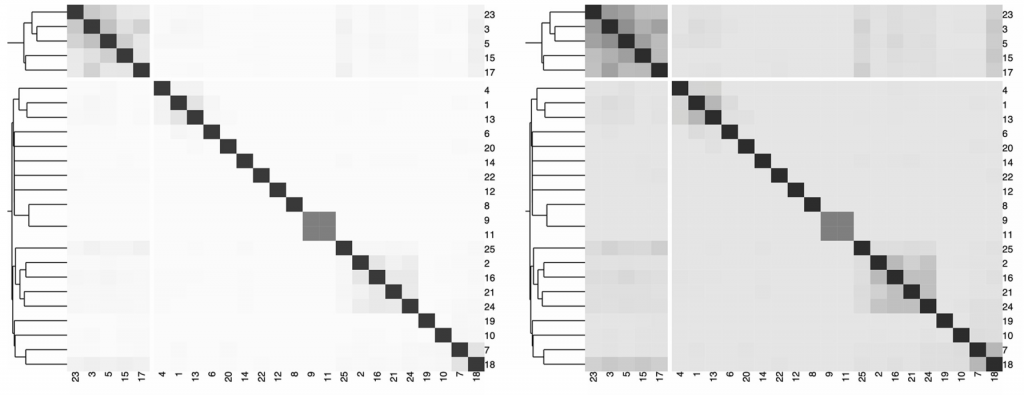
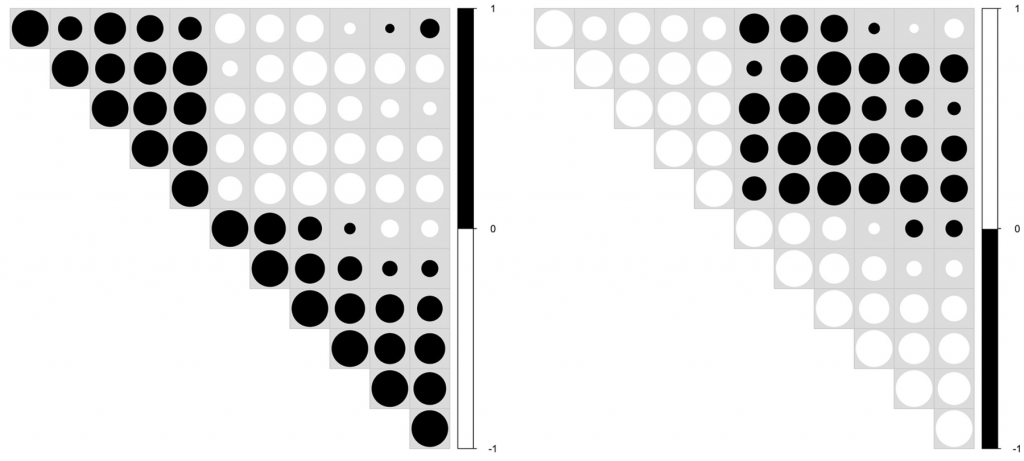
近日,加拿大多伦多大学研究人员在《Microorganisms》上发表的一篇研究,针对上述问题构建了一个参数化的混合模型用于计算聚类分析的距离度量,模型根据观察到的OTU计数和估计的混合权重产生sample-specific的分布。这个方法可以准确的估计真实的0比例,从而构建一个精确的beta多样性度量。
大量的模拟研究表明,与一些被广泛使用的距离度量方法相比,当存在较大比例的0时,该方法取得了较好的聚类效果。
该研究人员提出了一种具有特定beta多样性度量的聚类算法,该算法可以解决稀疏计数数据遇到的有无偏差问题且能有效的度量样本距离,达到分层的目的。
微生物群落研究中的一个重要问题是对这些微生物的归类,它们是否能被划分为亚群。如果有,有多少组亚群,如何解释这个亚群。例如,这种分类是否区分了治疗方法、疾病或遗传类型。
为了回答这些问题,需要测量两个微生物群落之间的相似性。beta多样性是为了适应不同的目的而提出的,在评估群落之间的差异时提供不同的结果。对于微生物组成,beta多样性根据测量丰度来衡量群落之间的距离,丰度可以是观察到的计数,也可以是相对丰度,这些丰度是根据不同或距离度量计算出来的,以量化样本之间的相似性。
现如今,已经有许多非参数统计方法来量化距离度量。例如Euclidean和Manhattan距离是最常用的。其它beta多样性指标,例如Bray-Curtis距离、Jensen-Shannon距离、Jaccard指数、UniFrac距离(未加权的、加权的和广义的)也经常用于微生物组研究。
除了距离度量之外,还引入了用于生态关联推理的稀疏逆协方差估计(SPICE-EASI)的图形网络模型。然而这些方法都会有一定的局限性,例如SPIEC-EASI方法依赖于单一的方差-协方差矩阵,由于微生物群落结构复杂,可能无法完全恢复底层OTU网络。
于是,研究人员开发了一种创新的聚类方法,以混合模型而不是beta多样性度量作为距离度量,并将聚类算法应用于微生物群落数据来表征亚群。该算法还包括根据选择的内部指标选择最优聚类数,并将结果在几种距离度量和不同评估方法之间进行比较。通过全面的模拟研究和一个真实的帕金森病肠道微生物群数据集对该算法的性能进行了评估。
1. 构建混合模型
混合模型是一种概率模型,用于表示在无监督学习中经常使用的总体内的子群体。该模型关注单个OTU在种群中的分布,可以解决样本间的稀疏性问题。它参数化地模拟了计数的潜在分布,包括低计数OTU和极高计数。对于个体样本之间的成对距离,在L2范数距离中使用公式化的混合概率。
2. 距离度量
在确定混合模型分布后,使用概率分布通过样本之间的两两距离计算距离度量。为了进行比较,考虑了基于L2范数的三种距离度量(L2-PDF、L2CDF、L2-DCDF、L2-CCDF)。
除此之外还选择了一些其他的距离度量进行比较,即Manhattan距离和Euclidean距离,以及微生物组分析中特有的三个距离度量:Bray-Curtis距离、加权UniFrac距离和广义UniFrac距离。本研究不考虑未加权的UniFrac距离,因为它不包含类群丰度信息。
3. 聚类分析验证指数
这些指数用于衡量集群在集群内部和集群之间的可分离性表现很好。验证指标可以分为内部指标和外部评估,许多内部验证指标被用来选择最优聚类数。外部评估分数是在假设标签在建模阶段没有使用时,通过直接将划分结果与之前的标签进行比较来计算的。
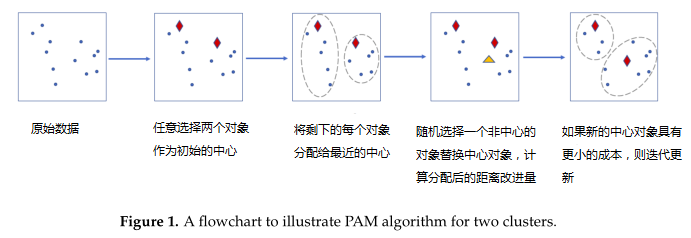
4. 用于聚类的分区算法(PAM)

使用混合分布的聚类过程的详细步骤:
为了测试该方法在聚类中的表现如何,研究人员推导了其准确性和Jaccard指数。
准确性是指聚类结果与真实的聚类指数的接近程度。它被定义为正确聚集的受试者所占的比例。
Jaccard指数衡量聚类结果与原始聚类标签之间的相似性,原始聚类标签定义为正确分类的主题数量(预测集与真实集的交集 )与两组总样本量(两集的并集)之比。
研究人员用类标签模拟数据来模拟OTU计数及其复杂的结构。研究人员考虑两个有两个子类和三个子类的场景,每个子类包含200个样本,总样本量分别为400和600。所有的结果被重复了100次。
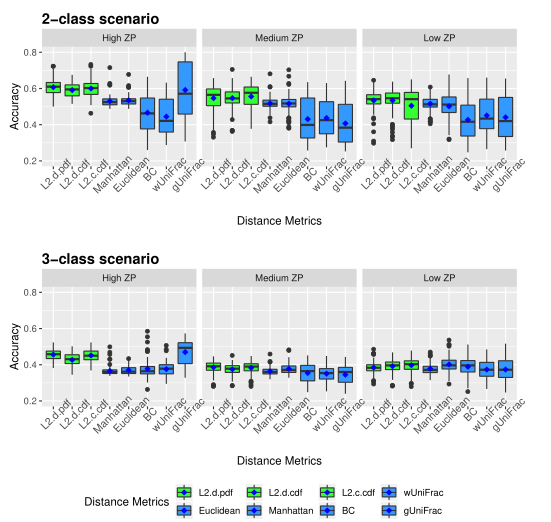
下图展示了模拟数据的准确性。评估了三种不同的0的比例(ZP)情况,左中右分别为高ZP、中等ZP、低ZP。
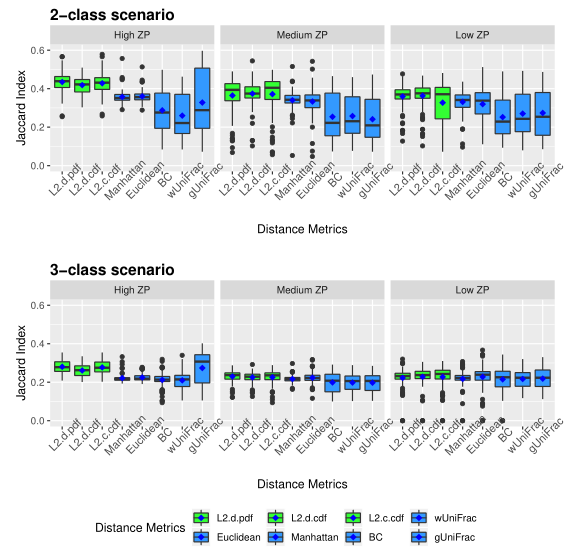
下图展示了模拟数据的Jaccard指数。同上图一样评估了三种不同的0的比例。
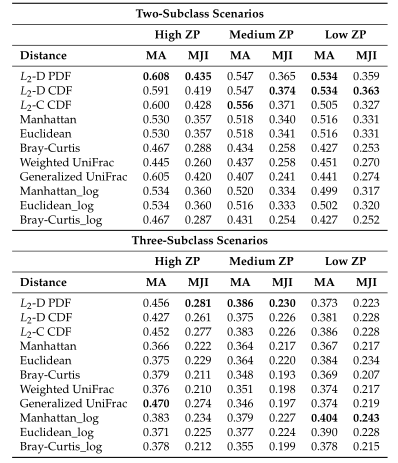
以上两图显示了具有不同ZP和子类数量的不同场景下模拟数据集的聚类结果。通过准确率和Jaccard指数对基于距离的算法性能进行了评估。填充颜色为绿色的箱形图为研究人员建议使用的距离度量。所有的距离都是根据相对丰度计算的。
Table1平均准确率(MA)和平均Jaccard指数(MJI)估计。粗体表示每个方案下的最佳情况。Log表示对输入的模拟数据进行了对数变换。
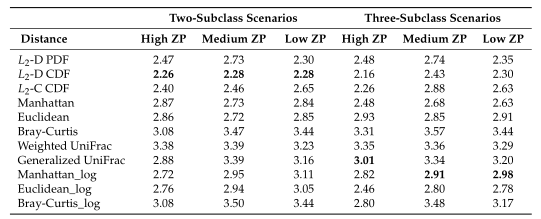
Table2所有模拟场景的平均集群数。根据Dunn内部指数计算出每次重复的最优聚类数。
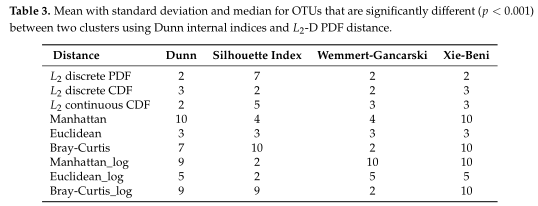
Table1 的结果是通过对每个场景中的100个重复结果求平均值计算得出的。观察得到在聚类算法中实现的距离度量(即绿色标识的箱形图)的准确率和Jaccard指数都优于其他距离度量,特别是在数据集中包含大量0时(高ZP)。在MA和MJI方面,L2范数也显示了其优势,在基于100次重复计算的L2范数在两个子类场景下的;平均准确率约为0.6秒,在三个子类场景下的平均准确率为0.45。而广义Unifrac距离(gUniFrac)在准确性估计中有很大的波动变化。
数据集为197名PD患者和130名对照的粪便样本的16S rRNA测序数据。首先过滤掉在80%的OTU中相对丰度都为0的OTU,因此,此次分析中共使用280个OTUs。将基于相对丰度计算的L2范数与其他三个距离度量进行了比较,包括对数变换和不变换的比较。
如Table3所示,距离度量采用各种内部验证指标(列名)进行灵敏度分析。对于Dunn和Xie-Beni指数,l2范数倾向于将数据聚类为两到三个亚群,而在有和没有对数变换的情况下,除了未变换的欧氏距离外,Manhattan、Euclidean和Bray-Curtis更倾向于聚类更多的亚群。(设置了最多聚类数目为10)
接着选择L2-D PDF范数作为进一步分析的例子。
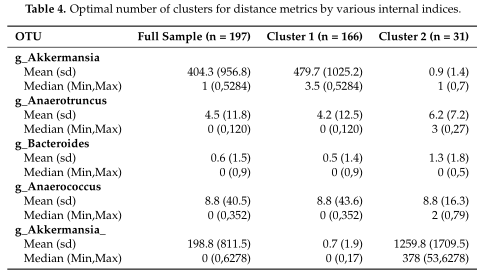
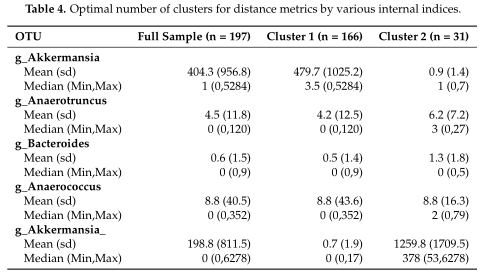
结果在Table4中展示,对数据集中的两个集群之间的OTUs进行了探索,得到了聚类之间差异最大的前5个OTU。
研究认为该聚类算法在高ZP和中等ZP的情况下表现最好,因此,当数据中出现大量的0时,建议使用。并且,在PAM框架下,文章中列出的那些距离度量都可以用作聚类的输入。
如模拟研究中显示的那样,在各种场景下,由混合模型计算的成对距离比其他距离度量表现的更好。但是与所有聚类方法一样,都有一个局限性,就是很难为每个新数据选择合适的内部指标,因此很难获得最优和最稳健的集群数。
此外,对于L2范数距离,在聚类中无法进行变量选择。但不可否认,该聚类算法结合了微生物测序数据的特殊距离,所介绍的聚类算法除了目前使用的方法之外,还可以被看作是分析微生物数据的一个很好的辅助工具。
研究人员指出,下一步会基于Dirichlet-Polyomial等模型进行分区,与文章中的方法进行比较,并努力将该方法扩展到其他微生物群落和疾病相关的数据上。
【参考文献】
Yang D, Xu W. Clustering on Human Microbiome Sequencing Data: A Distance-Based Unsupervised Learning Model. Microorganisms. 2020 Oct 20;8(10):E1612.