 国家高新企业 | ISO9001认证
国家高新企业 | ISO9001认证 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 二级病原微生物安全实验室

谷禾健康

随着一日三餐米面肉蛋菜等一些列食物的食用,数百种化学成分会进入我们的消化道。在那里,它们被肠道微生物组进一步代谢,这是数千种微生物物种的独特集合。
因此,肠道微生物组在决定营养如何影响健康方面发挥着重要作用。然而到目前为止,微生物组中的许多微生物的代谢能力仍然是未知的。这意味着我们不知道它们以什么物质为食,以及它们是如何处理这些物质的。
近期,来自普林斯顿大学的研究人员在《CELL》期刊上发表了最新的文章:
“Gut bacterial nutrient preferences quantified in vivo”,研究人员使用同位素追踪定量研究了小鼠肠道微生物群的输入和输出。

微生物碳水化合物发酵的主要输入是膳食纤维,支链脂肪酸和芳香代谢物的主要输入为膳食蛋白质。此外,循环宿主乳酸、3-羟基丁酸和尿素(但不是葡萄糖或氨基酸)为肠道微生物群提供食物。
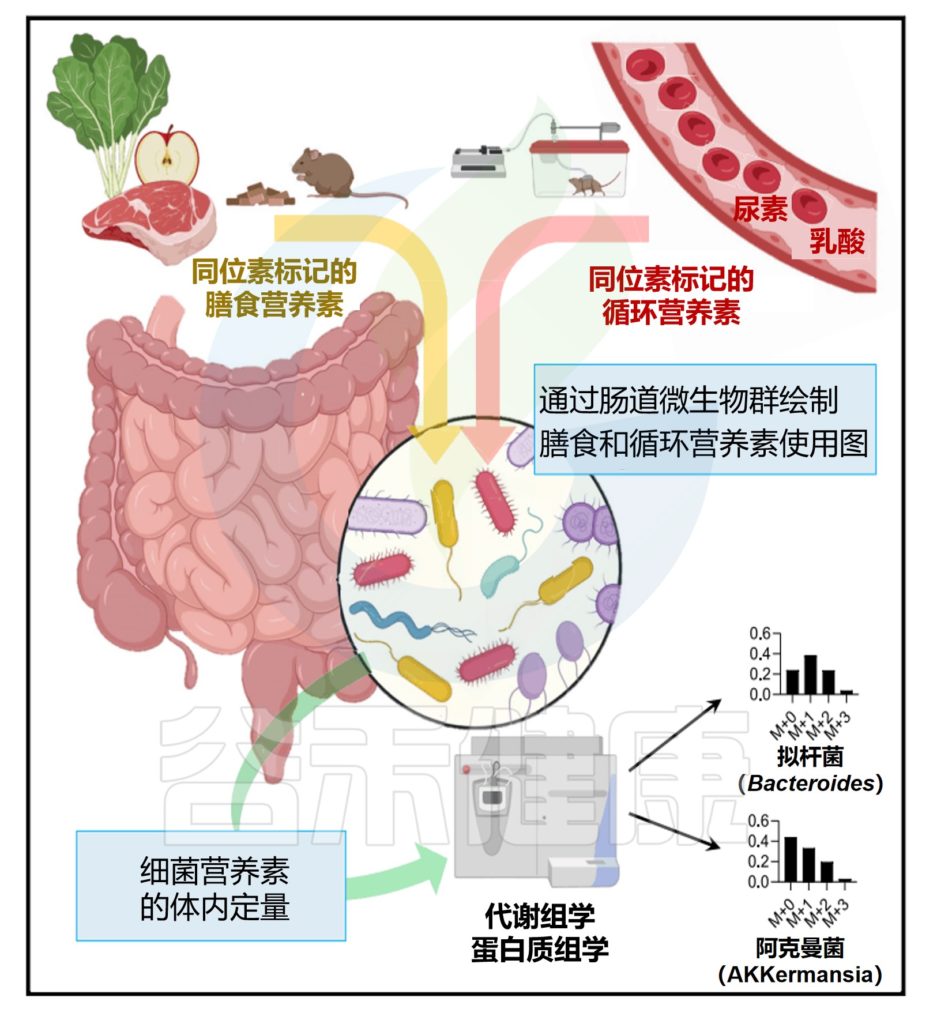
肠道菌群拥有巨大的酶多样性,超过哺乳动物基因组的数量100多倍。这些酶的能力能使摄入的膳食营养物质加工成一些列微生物代谢物。
为了复制自身和释放代谢产物,肠道细菌需要营养输入。这些形式包括摄入的食物、宿主合成的肠道粘液和宿主循环代谢物。
//
在本文中,研究人员通过对肠道菌群及其进入宿主循环系统的代谢物进行了大规模的定量评估。
研究了膳食淀粉、纤维和蛋白质的贡献以及宿主粘液的贡献,也研究了大多数主要的循环宿主营养素,发现乳酸、3-羟基丁酸和尿素在从宿主传递到肠道微生物群中表现突出。基于对细菌特异性肽序列的测量,评估了不同细菌属的营养偏好,并表明这些偏好与响应改变饮食的微生物组分变化一致。
同位素追踪能够定量测量代谢物和生物量的输入。与质谱检测相结合的稳定同位素示踪剂,使得能够测量特定下游产物的标记。通过注入氮标记的苏氨酸来标记宿主粘液,研究人员能够比较饮食和粘液蛋白对肠道微生物群的贡献,并观察到喂食低蛋白饮食的小鼠中粘液贡献的变化。
从小鼠尾部静脉抽取血样;
使用注射器从小鼠膀胱采集尿液;
所有血清样品在没有抗凝剂的情况下置于冰上 15 分钟,并在 4°C 下以 16,000 x g 离心 15 分钟。
用预冷的Wollenberger钳在液氮中快速分离并快速冷冻(< 5秒)获得组织;夹紧前取出肠内容物;盲肠内容物取样时,先将小鼠盲肠取出并在表面切开,然后用镊子将盲肠内容物挤出。
取新鲜粪便,轻揉小鼠腹部诱导排便。将血清、组织和粪便样本保存在 -80 ºC 直至进一步分析。
为了测定血清和组织样本中的代谢物浓度,进行了同位素标配(isotope spike-in)或标准标配(standard spike-in )。
对于前者将已知浓度的同位素标记标准品加入血清或组织提取液中,通过标记与未标记代谢物的比值计算浓度。
当没有同位素标准品时,加入连续稀释的非标记标准品,测量的总离子计数与加入的标准品浓度之间产生线性拟合。然后通过拟合线的x截距确定内源代谢物的浓度;蛋白质氨基酸组成采用酸水解法测定。
首先,使用13C同位素标记的不同营养物质,通过口服管饲法对小鼠进行灌胃采集小鼠的血清、组织和粪便样本。对粪便和肠内容物进行16S rRNA测序获得细菌分类。
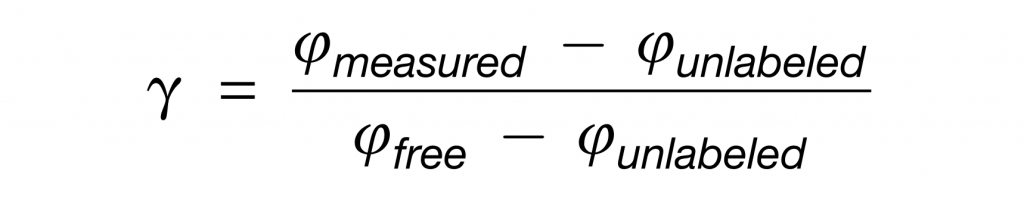
首先使用代谢组学方法测定盲肠内容物中游离氨基酸13C-或15N标记。
然后,对于每个肽,模拟了未标记(Iunlabeled)和由游离盲肠氨基酸(Ifree)合成的肽的同位素包膜模式。标量γ可以通过将测量的肽同位素分布(Imeasured)与Iunlabeled和Ifree的线性组合拟合来确定。
注意,当一个菌属使用的特定营养素超过该营养素对盲肠游离氨基酸的贡献时,γ将大于1。
具体来说,测量的每个肽的γ如下:

对于细菌属水平的原料贡献程度的测量,分析中只保留测量超过3个肽的属,多肽的中位数为γ-genus。
对于细菌科水平,仅分析在蛋白质组学中始终检测到的属,以及在 16S rRNA 基因扩增子测序中检测到 (> 0.5%) 的属的上一级科。
每种营养物质对菌属的贡献程度的定量公式如下:
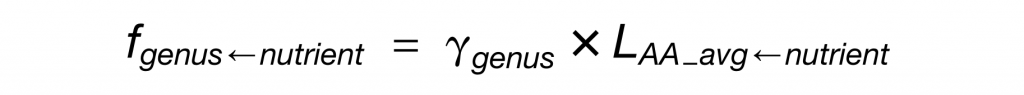

LAA_avg-nutrient为各营养物质对细菌蛋白质的贡献程度,其计算公式如下:
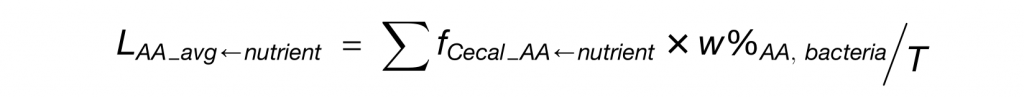

1 微生物组消耗较少的可消化膳食成分
微生物群影响宿主生理学的主要机制是通过分泌代谢产物。研究人员在门静脉和体循环以及盲肠内容物中测量了微生物衍生的50多种代谢产物的绝对浓度。
微生物群相关代谢物的绝对浓度和来源
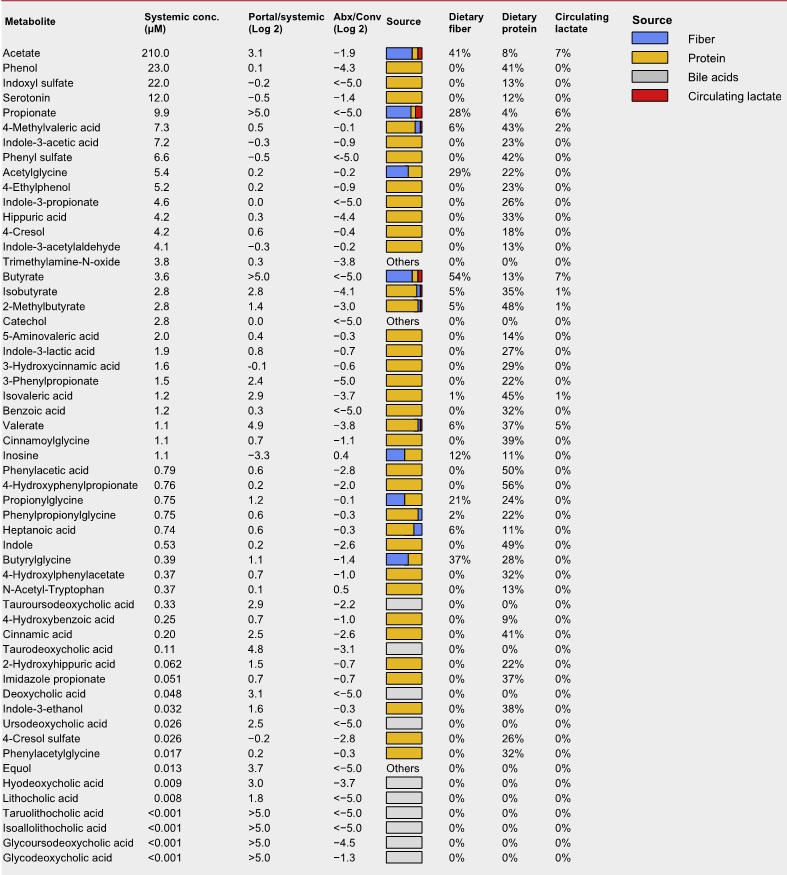
上表可以看到,与全身血液相比,大多数在门静脉循环中升高,除两种(肌苷和N-乙酰色氨酸主要来源于宿主)外,其余均被抗生素治疗耗尽。
门静脉血中主要排泄产物是短链脂肪酸。
其他相对丰富的微生物群产物是芳香族氨基酸发酵产物(苯酚、吲哚硫酸盐和3-苯丙酸盐)和支链脂肪酸(戊酸盐、异戊酸盐,4-甲基戊酸、异丁酸盐和2-甲基丁酸盐)。
探索肠道微生物产物的膳食输入:淀粉、菊粉
研究人员通过口服管饲法、淀粉(易消化葡萄糖聚合物)和菊粉(易消化果糖聚合物,即可溶性纤维)喂养小鼠:
13C淀粉灌胃后,标记的葡萄糖、乳酸和丙氨酸迅速出现在门脉循环中,并占大多数淀粉碳(约75%)。
13C菊粉和13C淀粉有什么不同?
13C菊粉灌胃后,没有观察到大量标记的果糖、葡萄糖、乳酸和丙氨酸,取而代之的是标记的门静脉代谢产物以短链脂肪酸的形式缓慢出现,约40%的菊粉碳成为短链脂肪酸,其余未消化并随粪便排出。
膳食菊粉,而不是淀粉,在盲肠内容物中广泛标记糖酵解和TCA中间体和氨基酸。
藻类蛋白大量标记了微生物群衍生的门静脉代谢物:短链脂肪酸、支链脂肪酸和芳烃(吲哚、吲哚-3-丙酸盐和3-苯丙酸盐)。
“难以消化的碳水化合物和蛋白质直接为微生物组提供营养,并通过微生物产物间接为宿主提供营养。”
研究中发现宿主循环系统中的乳酸,3-羟基丁酸以及尿素能为肠道细菌提供营养。
如图A,将同位素标记的营养物质通过静脉输注到小鼠的全身血液循环中。 2.5 小时后收集血清和粪便以量化每种营养物质对相应菌群代谢物的碳贡献。
图BCD表示了13C标记的各种营养物质在小鼠的血液和粪便中的含量,可见乳酸和 3-羟基丁酸有进入肠道菌群中,而其余大部分营养物质如柠檬酸盐、葡萄糖、氨基酸等都没有进入到肠道菌群中。
图F为15N标记的营养物质,可见尿素也同样被菌群大量利用。
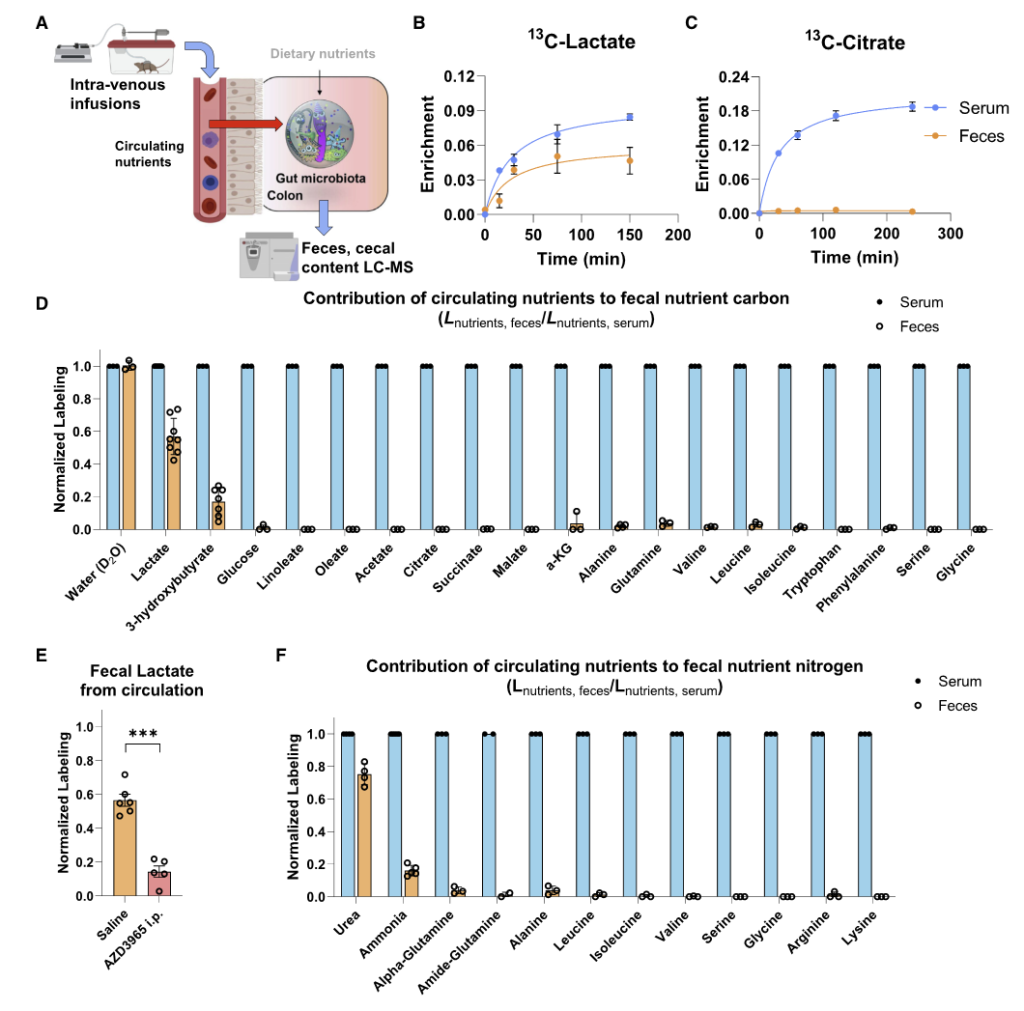
为了定量确定微生物代谢物的来源,研究人员给小鼠喂食部分纤维、脂肪或蛋白质13C标记的标准食物,盲肠标记在12小时内达到稳定状态。
为了说明循环营养输入,研究人员还注入了13C乳酸或3-羟基丁酸。
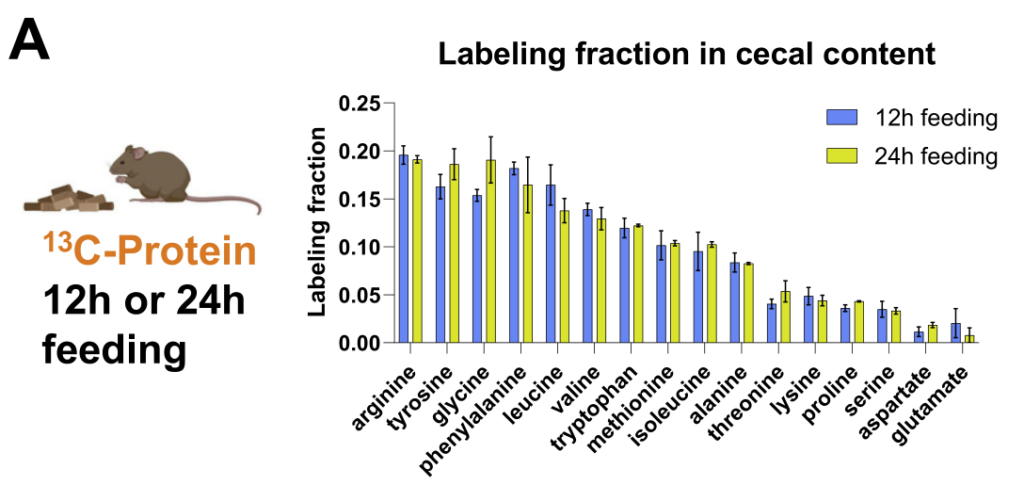
这些研究确定了大多数微生物群中心代谢物中的碳供给:
接下来,研究人员检查了微生物组游离氨基酸的输入,并用15N标记的膳食蛋白和注入的尿素进行追踪。
与哺乳动物不同,大多数肠道细菌具有合成所有20种蛋白质氨基酸的生物合成能力。
然而,研究人员观察到“必需氨基酸”主要来源于膳食蛋白质,哺乳动物无法制造,需要在细菌中表达广泛的生物合成途径。
“非必需氨基酸”主要在肠道微生物群中合成,使用膳食菊粉和循环乳酸作为碳源。
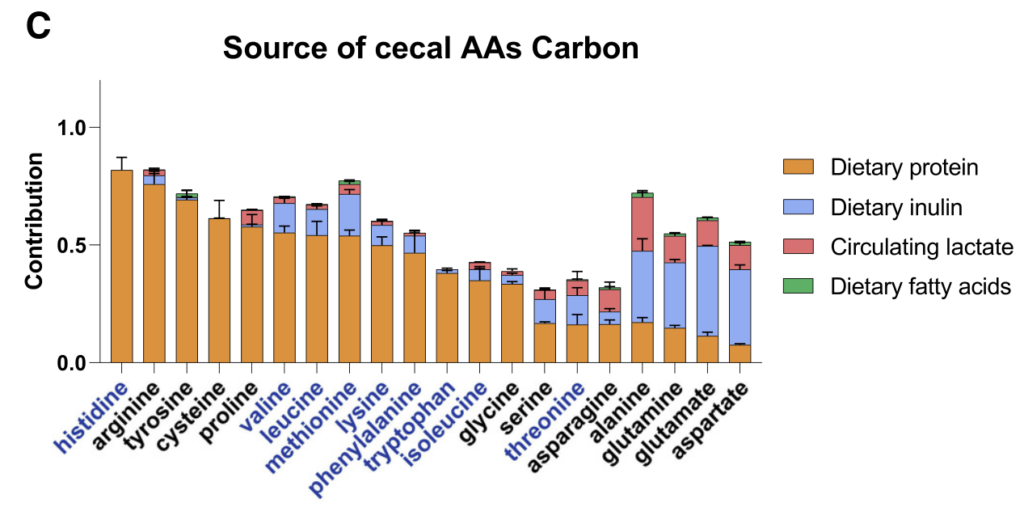
抗生素或无菌小鼠中的微生物群消耗有利于盲肠中氨基酸的积累(基于同位素追踪研究),这些氨基酸主要来自膳食蛋白质和微生物合成的氨基酸的消耗。
膳食蛋白质是必需氨基酸和非必需氨基酸的主要氮源,宿主尿素对非必需氨基酸也有很大贡献。
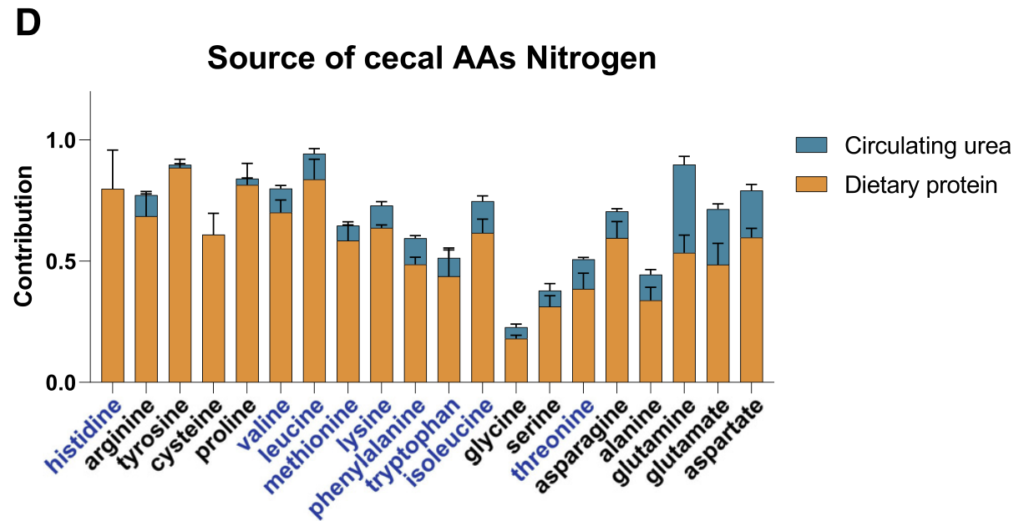
研究人员的发现如下:
【1】必需氨基酸,尽管能够由微生物群合成,但主要来自饮食,不经历任何碳重排;
【2】与TCA连接最紧密的非必需氨基酸基本上由微生物群合成,使用来自纤维的碳,通过中心代谢反应与其他碳争夺;
【3】转氨反应部分地将来自饮食衍生氨基酸的氮与来自宿主尿素的氮混合。
研究人员发现,许多微生物来源的代谢物来源于到达结肠的未吸收膳食蛋白。假设这些代谢物的循环水平将取决于膳食蛋白质到达结肠微生物群的程度。
为了控制这一点,研究人员给小鼠喂食的食物中,一部分蛋白质(酪蛋白,部分到达结肠微生物群)被游离氨基酸(基本上在小肠中完全吸收)取代。
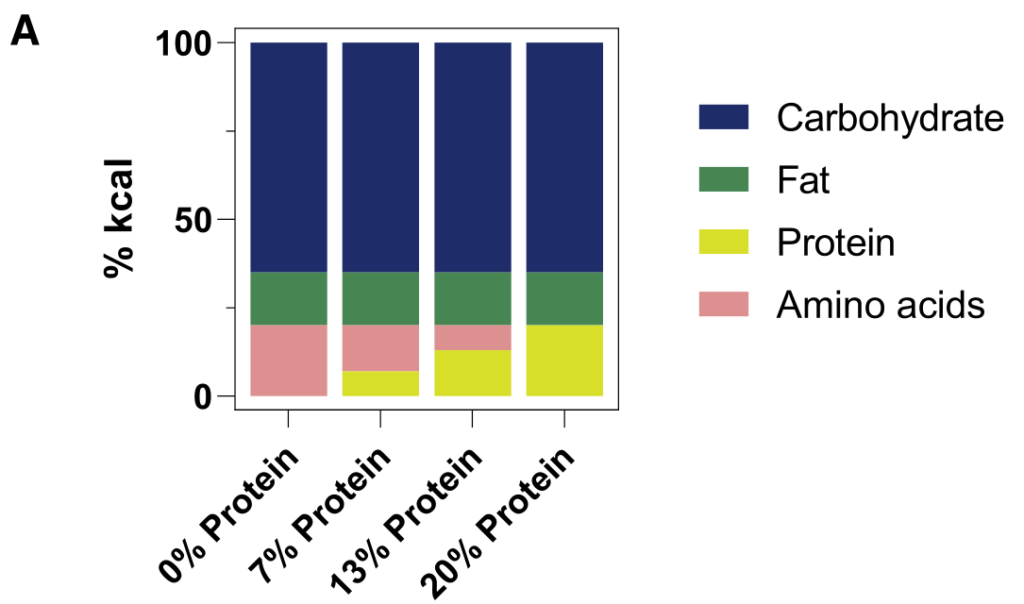
2周后对全身血液进行代谢组学研究。含有较少完整蛋白质和更多游离氨基酸的饮食往往会增加循环氨基酸水平。
重要的是,蛋白质衍生的循环微生物代谢物(酚类、吲哚类和酰基甘氨酸)串联下降。
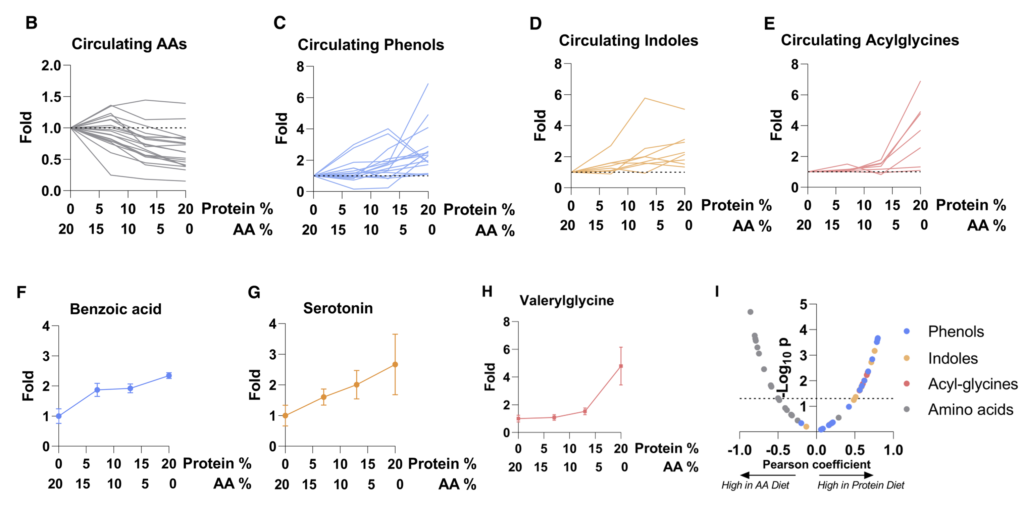
“微生物代谢物营养来源的知识可用于操纵其系统水平。”
研究人员通过结合13C营养标记和蛋白质组学来定量不同微生物的碳原料。
每种13C标记的营养素(膳食菊粉、膳食藻蛋白或循环乳酸)提供24小时,这足以在肠道细菌中实现稳态标记。
如同B-D,分别计算了在膳食中使用的菊粉和蛋白质以及乳酸在各细菌内的喜好程度,这个喜好程度也就是将在细菌特异性肽上被同位素标记的程度进行了量化。
结果可见:
拟杆菌属和梭状芽胞杆菌利用菊粉的程度是 Akkermansia、Muribaculum 或 Alistipes 的 4 倍多。
总体而言,厚壁菌门下的菌属比拟杆菌门的使用膳食中的蛋白质(厚壁菌0.237±0.052;拟杆菌0.175±0.031,p=0.02)。
Akkermansia通常被认为是一种促进健康的肠道微生物,使用的菊粉和蛋白质最少。相比之下,它使用了来自宿主的循环乳酸最多。
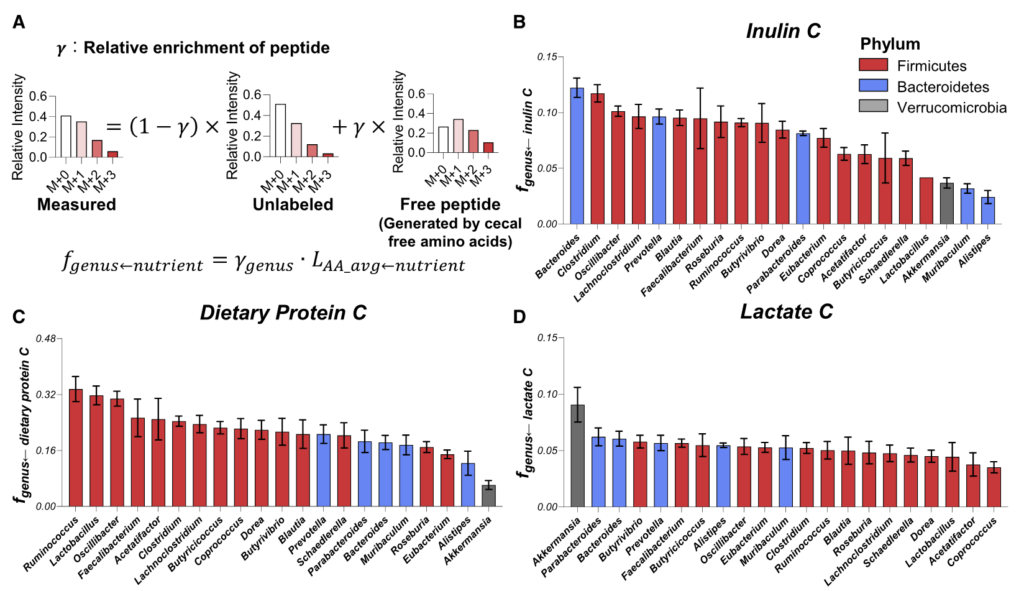
为了知晓这些细菌的营养偏好是否能预测饮食变化后的肠道菌群的组成变化。研究人员给小鼠喂食富含菊粉或藻类蛋白的饮食 2 天,并通过 16S rRNA 测序测量微生物组的组成。
结果如图F和I:
利用最多菊粉的拟杆菌属在高菊粉饮食后增加了4倍;
另一种利用较多菊粉的梭状芽胞杆菌也增加了2倍;
利用较少菊粉的菌属要么没有变化,要么略有下降;
富含藻类蛋白饮食的实验结果同理。
图G和J计算了这两种营养物与对其利用程度最高的前两名菌属相对丰度的相关性,p<0.05呈显著相关。
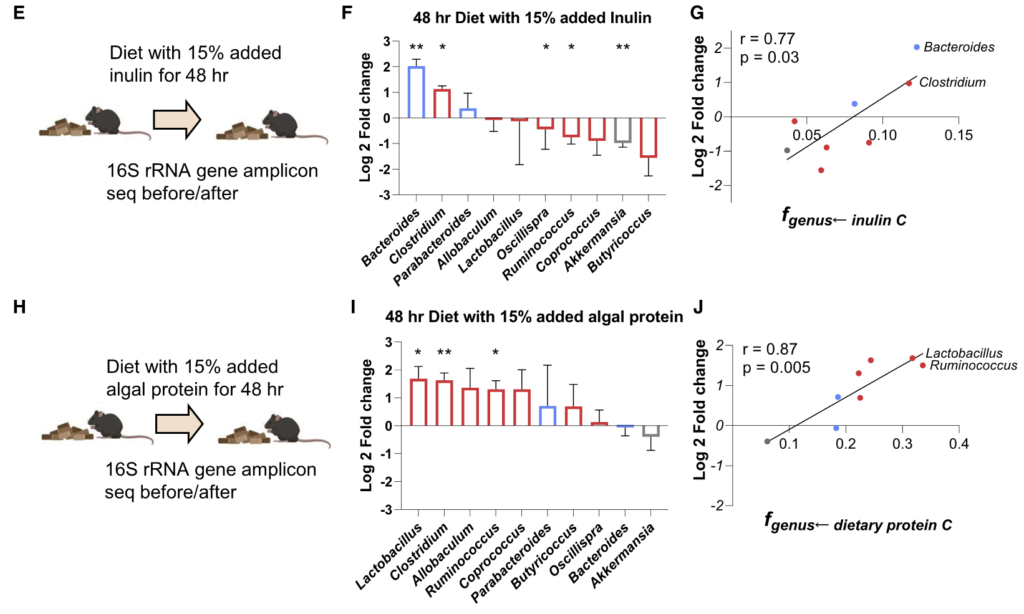
“不同肠道细菌的营养偏好有助于解释饮食操作后微生物组分的变化。”
最后,研究人员转向不同肠道细菌的氮源偏好,比较15N标记的膳食蛋白喂养和15N尿素输注。
高度利用膳食蛋白质中碳的细菌属也高度利用膳食蛋白中的氮,这与细菌蛋白质组中完整吸收的膳食蛋白质中的氨基酸一致。
厚壁菌喜欢从膳食蛋白质获取氮
在厚壁菌门成员中,偏好尿素氮的属往往是菊粉的疯狂使用者,即使用菊粉和尿素合成自己的氨基酸。这包括一些脲酶阴性菌属,它们可能通过交叉喂养获得尿素氮。
此外,在厚壁菌中也看到了一些属更喜欢从膳食蛋白质中获得氮,而其他属更喜欢循环尿素。
静脉注射尿素以提高循环尿素浓度后,偏好尿素的厚壁菌以及阿克曼菌的丰度大幅增加。
拟杆菌喜欢从宿主分泌的蛋白质中获取氮
与厚壁菌相比,拟杆菌对膳食蛋白质和循环尿素氮的利用率较低,这提出了一个关键问题:
拟杆菌如何获得氮?
肠道微生物群的一些成员(如拟杆菌和阿克曼菌)能够消化宿主分泌的蛋白质,如粘蛋白。
假设宿主分泌的蛋白质是拟杆菌氮的关键来源。为了探索这种可能性,研究人员进行了长期15N标记的赖氨酸和精氨酸输注(12、18和36小时),以标记结肠中的宿主蛋白。
尽管没有直接给微生物组喂食,但在36小时输注后,赖氨酸和精氨酸确实起作用,这与通过宿主蛋白进行的标记一致。这种标记优先发生在拟杆菌和阿克曼菌中。
膳食和分泌宿主蛋白的氮贡献呈负相关,与某些肠道细菌优先消耗膳食蛋白和其他宿主蛋白一致。
“膳食蛋白质和循环尿素是厚壁菌的主要氮原料,而分泌的宿主蛋白质为拟杆菌提供氮。”
研究人员开发了定量同位素追踪方法来测量肠道细菌的营养偏好。除了膳食纤维和分泌的宿主蛋白外,还将膳食蛋白和循环宿主乳酸、3-羟基丁酸和尿素确定为喂养肠道细菌的重要营养素。排除了其他循环宿主营养素(如葡萄糖和氨基酸)对结肠微生物群的直接贡献。
一项关键技术成就是能够从不同碳源和氮源追踪到细菌特异性肽,从而揭示复杂和竞争性肠腔环境中不同细菌的营养偏好。
厚壁菌门倾向于从膳食蛋白质获得氨基酸,而拟杆菌门更多地依赖宿主分泌蛋白。同样,一些厚壁菌门(如梭菌属)大量利用纤维(菊粉),而其他厚壁菌门则不利用纤维。
动物饮食干预实验发现,拟杆菌属和梭菌属是转化纤维最活跃的菌属。宿主循环代谢物水平也可能影响微生物组的营养获取和最终组成。
本文提供了关于哪些营养素喂养肠道微生物群以及哪些细菌更喜欢哪些营养素的基础知识。
文中所开发的方法具有广泛的应用前景,最终将有助于全面和定量地了解饮食-微生物-健康的关系。
参考文献:Zeng X, Xing X, Gupta M, Keber FC, Lopez JG, Lee YJ, Roichman A, Wang L, Neinast MD, Donia MS, Wühr M, Jang C, Rabinowitz JD. Gut bacterial nutrient preferences quantified in vivo. Cell. 2022 Sep 1;185(18):3441-3456.e19. doi: 10.1016/j.cell.2022.07.020. PMID: 36055202; PMCID: PMC9450212.

谷禾健康

随着高通量多组学技术的快速创新推动,微生物群,尤其是肠道菌群失调已被明确与许多人类疾病有关,包括 2 型糖尿病和炎症性肠病。
多组学数据的综合分析,包括宏基因组学和代谢组学以及宿主指标的检测和细菌物种的分类,已经确定了许多与疾病相关的细菌和细菌产物。然而,深入了解微生物影响肠道健康的机制需要从关联拓展到因果关系。
目前对肠道微生物群对疾病因果关系的贡献的理解仍然有限,这主要是由于微生物群落结构的异质性、疾病进化的个体差异以及对将微生物群衍生信号整合到宿主信号通路中的机制的不完全理解。
最近,德国慕尼黑工业大学从事肠道菌群和营养研究的Haller 教授团队在《Nature reviews gastroenterology & hepatology》 (自然评论胃肠病学和肝病学)发表评论文章,系统讨论了目前已知的炎症和代谢紊乱相关微生物组的特征和认知,并讨论提高对其作用机制理解的困难所在。
在这里我们将文章整理与大家分享。


关键信息
1、肠道菌群组成的改变和细菌衍生代谢物经宿主加工后的变化与 IBD 和 T2DM 相关,并提供了共同的潜在致病机制。
2、益生菌与 IBD 或 T2DM 之间的因果关系已通过无菌小鼠实验和综合多组学研究明确。
3、对于疾病特异性生物标志物发现的挑战,包括确定观察到的变化的因果关系,了解它们在疾病机制中的功能以及肠道微生物群的地理和种族差异。
4、特定细菌菌株、其编码基因和代谢副产物的大数据细化、测试和验证对于识别疾病生物标志物是必要的。
文章内容
人体消化道含有一系列复杂的微生物,包括细菌、古细菌、病毒和真菌。由于消化道及其微生物组被认为位于免疫和代谢过程的交叉点,本文重点关注炎症性肠病 (IBD) 和 2 型糖尿病 (T2DM) 作为微生物群相关疾病的范例。
IBD 和 T2DM 都被认为是多因素疾病,随着工业化的进展其发病率在全球范围内呈上升趋势。病因涉及遗传易感性、环境诱因和城市生活方式相关因素的复杂相互作用。
在这种共同的背景下,代谢疾病(如 T2DM)的另外特征是肝脏、脂肪组织、肌肉、胰腺和肠道的慢性亚临床炎症,而炎症性胃肠道疾病,如克罗恩病(CD)和溃疡性结肠炎(UC) ,也与炎症驱动的代谢改变有关。
环境触发因素的重要性(肠道菌群)
全基因组关联研究已经确定了大量的遗传变异与 T2DM (143 位点) 或 IBD (>240 位点)的易感性增加相关。然而,这些变异共同解释了这些疾病的一小部分遗传性:T2DM < 10 %,UC < 15 % 和 CD < 50 % 。这种情况表明环境触发因素的重要性,特别是肠道微生物组,作为这些疾病病因的主要贡献者。对大型人群研究和 IBD 或 T2DM 患者队列的多项分析已经确定了与特定疾病表型、复发风险和治疗反应相关的微生物组特征。
IBD 和 T2DM 都与特征性微生物改变有关,特别是随着有益微生物的减少和病原菌的增加而降低群落多样性。尽管它们的病理学不同,IBD 和 T2DM 有几个共同的机制特征。T2DM 的代谢特征伴随着慢性低度炎症和肠道屏障的破坏,IBD 患者的复发性炎症发作与细胞和全身水平的代谢改变共同发生。
这些复杂疾病的治疗仍然具有挑战性,但粪便微生物群移植(FMT) 的对照试验已显示出对T2DM和IBD的临床疗效,包括UC以及较轻的 CD。
FMT对炎症、免疫和代谢疾病有效果但存在差异
FMT 的临床试验还提供了肠道菌群与其他炎症、免疫或代谢疾病之间存在因果关系的证据。例如,FMT 在治疗大约 90% 的艰难梭菌(以前的艰难梭菌)感染患者方面非常有效并已被评估为治疗肥胖和移植物抗宿主病。
在四项随机临床试验中,FMT 在 28% 的 UC 患者中诱导了临床缓解。但很少有临床试验检查过 FMT 在 CD 患者中的疗效,而且结果相当不同。
在一项对 174 名接受 FMT 治疗的 CD 患者进行的研究中,20% 的患者获得了临床缓解,总体而言,43% 的患者获得了临床缓解。
一项单独的随机对照试验发现 FMT 对 CD 患者的临床缓解率没有影响,但供体微生物群的植入增加与维持缓解有关。相反(尽管大量研究表明,特定的菌群失调或特定的微生物群谱与代谢紊乱有关),FMT 对代谢性疾病患者有益的证据尚不明确。具有里程碑意义的研究表明,从较瘦、健康的捐赠者那里接受 FMT 的代谢综合征患者的代谢改善以及肠道微生物组的有益变化。然而,这些影响是不一致和短暂的,这可以通过供体微生物群的有限移植或基线时供体粪便微生物多样性的变化来解释。
有趣的是,口服 FMT 后补充低发酵性纤维可改善肥胖和代谢综合征患者的胰岛素敏感性、增加微生物多样性,并延长供体微生物定植。这些数据强调了微生物调节疗法在逆转代谢功能障碍中的价值。
与这些发现一致,来自代谢受损的肥胖供体的 FMT 会暂时恶化代谢综合征受体的胰岛素敏感性,而胃旁路术后健康供体的 FMT 会导致代谢综合征受体的胰岛素敏感性略有增加。
几项大型队列研究(表 1、表2)通过分析 IBD 患者的肠腔和黏膜微生物群落,研究了肠道微生物群的改变。
总体而言,活跃期IBD 与某些菌群的数量过多有关,如:
肠杆菌科Enterobacteriaceae
梭杆菌属Fusobacterium
咽峡炎链球菌Streptococcus anginosus
肠球菌Enterococcus
巨球菌Megasphaera
弯曲杆菌Campylobacter
Gammaproteobacteria
Deltaproteobacteria
相反,IBD 与有益菌群的缺失有关,例如:
Faecalibacterium prausnitzii
Christensenellaceae
Collinsella
Roseburia
Ruminococcus
其他产丁酸盐的细菌
在我们检测的炎症性肠病的菌群报告中也发现,炎症性肠病风险高的人群中,炎症水平很高,肠杆菌科Enterobacteriaceae,梭杆菌属Ruminococcus gnavus偏高,而Faecalibacterium prausnitzii和Roseburia丰度普遍降低或者缺乏。


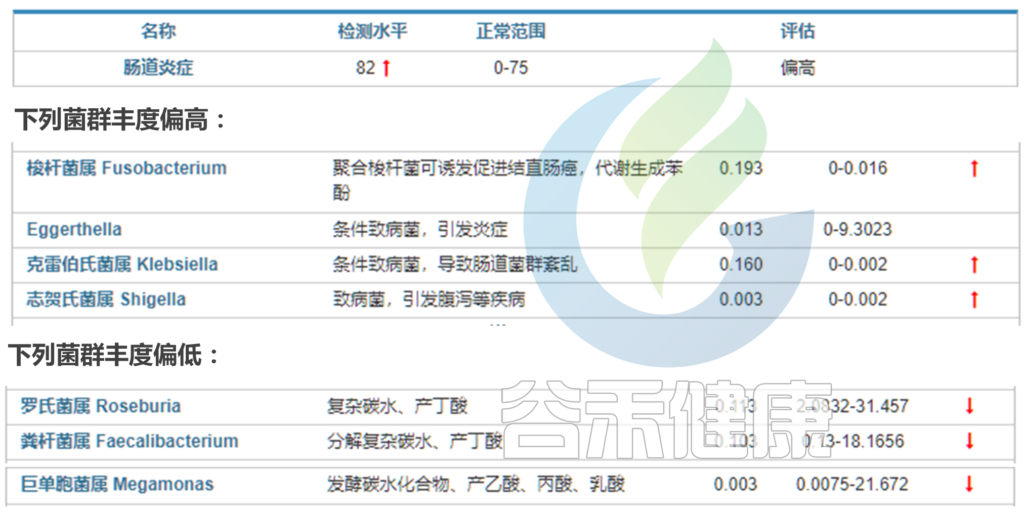
< 选自:谷禾肠道菌群健康检测报告 >
菌群代谢功能
粪便样本的宏基因组学为 IBD 中发生的功能失调和代谢途径的扰动提供了更全面的观点。这些研究表明,参与含硫氨基酸合成、核黄素代谢、谷胱甘肽转运蛋白、氧化应激和营养转运的代谢途径均被上调。
一项能够将微生物群落分解到物种内单个菌株水平的研究显示,与健康对照相比,IBD 或肠易激综合征 (IBS) 患者粪便样本中致病菌的菌株多样性增加,而有益微生物的菌株多样性降低。深入分析表明,219个类群(包括152种)与CD相关,102个类群(包括93种)与UC相关。
CD的主要特征是属于毛螺菌科和瘤胃球菌科的分类群减少和属于肠杆菌科的分类群增加,而对于UC观察到属于拟杆菌科的分类群减少和属于毛螺菌科的分类群增加。与这种异质性一致,在不同的 IBD 研究中仅存在少数物种的共同变化,这表明尽管疾病表型和病程相似,但在 CD 患者群体中仍存在个体间差异。
IBD和肠道菌群的因果关系探究
肠道微生物群在 IBD 中的致病作用的首批临床证据之一源于实验表明,从 CD 患者的小肠发炎段转移粪便流可改善疾病症状。粪便流的恢复和新末端回肠术后暴露于肠腔内容物会诱发炎症,这表明肠道微生物群会引发 CD的术后复发。此外,抗生素治疗对活动性 CD 患者亚群的疗效强调了肠道细菌和 IBD 之间的因果关系。
急性和慢性肠道炎症小鼠模型的机制研究为微生物失调与 IBD 之间因果关系的提供了进一步证据。例如,将IBD 患者的菌群移植到无菌受体小鼠,会将IBD 表型转移到无菌小鼠中。而具有 IBD 遗传易感性的小鼠在无菌条件下不会发生自发性炎症。
免疫响应
IBD 发病的遗传易感小鼠的失调菌群,能够将这种疾病症状转移到无菌受体小鼠。将 IBD 微生物群转移到无菌的野生型小鼠体内会导致肠道 T 细胞反应失衡,肠道 T 辅助 17 (TH17) 细胞和 TH2 细胞数量增加,RORγt +数量减少。同样,肠道微生物群的人类共生细菌脆弱拟杆菌对无菌小鼠的定植诱导CD4 + T 细胞转化为产生IL-10的FOXP3 + T reg细胞,这表明微生物群驱动的存在IBD 的发病机制。
在 T2DM 中也发现了几种细菌类群的丰度变化很大。
例如,据报道,2型糖尿病患者中下列菌相对丰度增加:
大肠杆菌E. coli、
韦荣氏菌属Veillonella、
布劳氏菌属Blautia、
厌氧菌属Anaerostipes、
乳杆菌属Lactobacillus、
粪杆菌属Faecalibacterium、
梭状芽胞杆菌属Clostridiales(等)
相反,下列菌丰度降低:
拟杆菌属Bacteroides
双歧杆菌属Bifidobacterium、
副拟杆菌属Parabacteroides、
颤螺菌属Oscillospira
可降解粘蛋白的阿克曼菌Akkermansia muciniphila
2019 年发表的一项宏基因组和宏蛋白质组学研究分析了来自 254 名中国个体的粪便样本中的肠道微生物群组成和功能,其中包括 77 名未接受治疗的 T2DM 患者、80 名糖尿病前期患者和 97 名葡萄糖耐量正常的对照个体。与代谢健康的对照组相比,T2DM 患者和前驱糖尿病患者的梭菌目Clostridiales细菌丰度较低,而埃氏巨球形菌Megasphaera elsdenii的丰度较高。
菌群代谢功能
在 T2DM 患者和糖尿病前期患者的微生物组中观察到功能差异。与对照个体相比,糖尿病前期个体的肠道微生物群显示出与糖磷酸转移酶系统、细菌分泌系统和氨基酸的 ATP 结合盒 (ABC) 转运蛋白有关的途径富集。这些发现表明,在糖尿病前期患者转变为 T2DM 之前,可以检测到肠道微生物组的疾病特异性变化。
环境因素影响
细菌种类和代谢基因簇谱的差异已被用于确定一组具有正常葡萄糖耐量或 T2DM 的个体的糖尿病状态。然而,包括地理位置、种族、健康状况和用药史在内的混杂因素导致在识别与 T2DM 相关的微生物变化方面不同研究存在不一致。
因果关系研究
几项研究提供了肠道微生物群特定成员与 T2DM 发病机制之间因果关系的证据。例如,A. muciniphila属于在人类和小鼠研究中显示对代谢紊乱具有保护作用的分类群。有趣的是,补充益生元使A. muciniphila的丰度正常化并改善了人类的代谢健康。同样,对喂食高脂肪饮食的小鼠施用A. muciniphila可逆转其增加的脂肪量、代谢性内毒素血症、脂肪组织炎症和胰岛素抵抗。
此外,产生丁酸盐的细菌Anaerobutyricum soehngenii(以前称为Eubacterium hallii菌株 L2-7)显示出丰度增加,这与来自瘦供体的FMT受体的外周胰岛素敏感性改善相关。
对T2DM 患者A. soehngenii菌水平进行管理,在治疗 4 周后改善了外周胰岛素敏感性,这些益处伴随着微生物群组成的改变和胆汁酸代谢的变化。
奇怪的是,特定的细菌分类群在 IBD 和 T2DM 中表现出相似的变化,这表明免疫介导和代谢疾病的共同特征导致微生物群的相似适应。
下列菌丰度下降:
梭状芽孢杆菌属 Clostridium spp.
粪杆菌属 Faecalibacterium
瘤胃球菌属 Ruminococcus
阿克曼氏菌属 Akkermansia
柯林斯氏菌属 Collinsella
罗斯氏菌属 Roseburia
下列菌丰度增加:
肠杆菌科 Enterobacteriaceae
大肠杆菌 E. coli
具核梭杆菌 Fusobacterium nucleatum spp.
这为定义疾病特异性标志物提出了挑战(下图)


Metwaly et al.,Nat Rev Gastroenterol Hepatol. 2022.
例如,一项针对 2,045 名 IBD 患者的研究的作者确定了 8 个分类群的特征,包括克里斯滕森菌科Christensenellaceae和梭杆菌属Fusobacterium的未知成员,它们可以区分 CD 患者和健康个体。
然而,Christensenellaceae 的丰度增加与低 BMI 和体重减轻有关,这是一种在 IBD 患者中经常观察到的分解代谢状况。同样,梭杆菌的富集被认为是转移性结直肠癌患者预后不良的标志。鉴于 IBD 患者患结直肠癌的风险增加,这一提议的微生物组特征可能是一种附带现象,与潜在的疾病机制没有因果关系。
对来自 132 名个体的微生物组、代谢组和转录组数据集的综合网络分析确定了连接关键细菌分类群(即F. prausnitzii 、未分类的Subdoligranulum、Alistipes、大肠杆菌、Roseburia)的某些代谢物(短链脂肪酸、辛酰肉碱和几种脂质)。有趣的是,有和没有 IBD 的研究参与者之间的差异在粪便代谢组中,比在粪便宏基因组、粪便宏转录组或粪便蛋白质组中更明显。
在综合个人组学分析研究 (iPOP) 中,血浆代谢物与来自 106 名个体的纵向样本中的胰岛素抵抗密切相关,这表明宿主代谢组和肠道微生物组之间的相互作用在胰岛素抵抗个体中受到干扰。
许多研究调查了微生物改变作为疾病生物标志物的效用,特别是在 CD 或 UC 患者中。最初试图定义可以作为疾病活动指标的单一细菌分类群。
例如,F. prausnitzii (一种产生丁酸盐的厚壁菌)在 CD患者中被耗尽。CD 患者回肠黏膜活检样本中这种细菌丰度的降低与回肠切除术后内镜下复发风险的增加密切相关。相反,粘附的侵袭性大肠杆菌丰度增加与回肠 CD相关。
然而,由于大多数细菌物种由许多个体菌株组成,这些菌株可以表现出相当大的基因组和蛋白质组变异,因此菌株多样性具有重要的功能,特别是在确定致病性方面。
例如,R. gnavus和大肠杆菌的亚种都与 IBD 的严重程度增加有关。此外, R. gnavus的一个特定亚种表明,来自 IBD 患者的粪便样本中丰度增加含有菌株特异性基因(与改善的细菌定植有关)。这些基因涉及诸如氧化应激反应、细菌粘附、铁获取和宿主粘液利用等功能。同样,不同的脆弱拟杆菌菌株表现出功能差异,导致 IBD 相关小鼠模型中 IgA 诱导水平不同。这些遗传上不同的脆弱拟杆菌菌株在接种到受体小鼠时也表现出不同的致结肠和免疫调节作用。在一项旨在定义用于监测 IBD 患者疾病活动的关键菌群失调的研究中,使用定量 PCR 计算了F. prausnitzii和大肠杆菌的绝对丰度比(也称为 F-E 指数)。F-E 指数的使用提高了 UC 和 IBS 患者与 CD 患者的区分,并有助于区分结直肠癌与其他肠道疾病。然而, F-E 指数无法区分 IBD亚型,这表明单一分类群指标在分类疾病亚型方面的效用有限。
大规模生物标志物分析
基于微生物特征的判别模型
几项研究已经使用机器学习算法来验证横截面和纵向患者队列中复杂的微生物组特征。
例如,2017 年发表的一项研究使用 16S rRNA 微生物群来分析来自大量 IBD 患者和没有 IBD 的对照个体的粪便样本。研究人员使用序列聚类算法根据八种细菌类群的丰度来识别 CD 特异性微生物特征。
此外,另一组研究表明,基线肠道微生物组组成的特征可以预测 IBD 患者在治疗开始后14周对抗整合素治疗的反应。由深度神经网络生成的微生物组特征的受试者工作特征曲线 (AUC) 下面积为 0.87,而基于临床协变量的模型的 AUC 为 0.62。研究小组还评估了微生物组特征作为 IBD 和 T2DM 生物标志物的效用。
在一项研究中,检查了 29 名接受过自体造血干细胞移植的 CD 患者的独特队列中的疾病活动性和对治疗的反应。来自人类供体和人源化小鼠的微生物组和代谢组风险概况的整合将疾病结果的预测模型的性能从 AUC 0.79 提高到 0.96,并确定了与硫代谢相关的疾病相关细菌和代谢物网络。
这些发现听起来很有前景,但重要的是要承认微生物组风险概况是基于来自前瞻性队列研究中的人群或患者组的预测模型,因此比起对于个人的预测结果,对于相似患者组(人群或队列)可能更准确。重要的是记住,预测的风险可能不会直接转化为个体患者,这可能是由于在异质环境中风险概况的普遍性有限。
不忽略混杂变量
另一项研究调查了以德国人群为基础的 1,976 人队列中的代谢健康和肠道微生物群的昼夜节律性。粪便微生物群分析确定了 13 个微生物分类群的风险特征,这些分类群显示 T2DM 患者的昼夜节律性受到破坏。基于这种心律失常风险特征的预测模型成功识别出有患 T2DM 风险的个体,当模型中包含 BMI 时,AUC 为 0.78。
这些例子为微生物组特征在用于诊断和治疗目的的生物标志物发现中的作用提供了证据。然而要注意,生态失调指数不是独立的测量值,需要整合到额外的宿主衍生数据和临床数据中。这些指标的标准化和验证需要大规模究研,包括对潜在生物标志物的纵向评估,并考虑可能的混杂变量,例如饮食、年龄、种族、病史和最后的排便时间,所有这些因素都会影响微生物组的改变。
在寻找疾病生物标志物时,代谢物作为疾病活动的最接近指标,并且与作为疾病机制基础和调节疾病机制的调节信号密切相关。事实上,代谢组和微生物组都随着饮食、环境、衰老和整体健康状况等内源性和外源性因素而波动。
许多研究报告了 IBD或 T2DM患者的肠道代谢物谱的显着变化。
例如,已在 IBD 患者的粪便代谢组中发现中链脂肪酸(如戊酸和己酸水平降低)和 B 族维生素水平降低。相反,据报道,成人和儿童 IBD 患者的粪便和血清中氨基酸、胺和肉碱的含量分别增加。
一项具有里程碑意义的研究结果表明,代谢物分析可以区分 IBD 患者和健康个体。该报告之后有许多其他人一致表明 IBD 患者的代谢物表型与健康个体的不同。有趣的是,代谢物分析还可以区分不同形式的 IBD,例如 CD 和 UC,并且可以进一步将 CD 患者分类为患有回肠或结肠炎症。同样,T2DM 患者的代谢物分析表明代谢途径的活性发生了改变。
在多份报告中,支链和芳香族氨基酸(如亮氨酸、异亮氨酸、缬氨酸、苯丙氨酸、酪氨酸和色氨酸)的血清水平与胰岛素抵抗、肥胖和发生 T2DM的风险相关。
T2DM 患者的代谢物分析还揭示了特定细菌代谢物水平与疾病发作之间的强关联。
例如,色氨酸代谢途径包括几个候选代谢物生物标志物,这些生物标志物由于与人类和小鼠研究中炎症和代谢疾病的发展相关而引起了研究关注。色氨酸是一种从饮食中获得的必需氨基酸,主要在小肠中吸收,尽管一小部分在结肠中分解代谢为吲哚代谢物。
一项纵向队列研究证实,在 213 名中国个体(包括 51 名继续发展为 2 型糖尿病和 162 名保持代谢健康的个体)中,高基线空腹血清色氨酸浓度与患 T2DM 的风险增加有关。此外,色氨酸水平作为生物标志物的预测能力与五种现有氨基酸在区分患有和未患 T2DM 的个体方面的预测能力相当 。
值得注意的是,先前的报告表明,几种氨基酸的血清水平可以以不同的准确性识别来自不同人群的 T2DM 患者。例如,苯丙氨酸和缬氨酸在美国人群中表现最好,而酪氨酸在南亚人群中最准确。这些发现指出了识别区域特异性生物标志物在实现最佳诊断准确性方面的重要性。
有价值的生物标志物必须为从临床信息中获得的分类能力提供额外的分类能力。因此,粪便生物标志物是粘膜疾病诊断标志物的明显来源,因为粪便流与肠粘膜直接接触。
钙卫蛋白
粪便钙卫蛋白是一种可在粪便中检测到的粒细胞衍生的细胞溶质蛋白,由于炎症严重程度与粪便钙卫蛋白水平之间的相关性强,它是最广泛使用的用于炎症性疾病的粪便生物标志物。
两份报告证实了粪便钙卫蛋白水平检测内窥镜炎症的能力,报告的敏感性为70-100% ,特异性为44-100%。这些值的广泛范围可以通过每项研究中应用的截止阈值的变化来解释。
然而,粪便钙卫蛋白水平升高并不是 IBD 特有的,而是反映了肠道炎症状况,这也与其他肠道和代谢疾病(包括 IBS、胃肠道恶性肿瘤、肥胖和 T2DM)有关。
例如,对来自 1,792 个人的粪便样本中肠道微生物群的鸟枪宏基因组分析能够区分 IBD 和 IBS,与单独的粪便钙卫蛋白水平 (AUC) 相比,机器学习算法显着提高了这些预测模型的准确性 (AUC 0.91>0.80)。
重要的是,具有最高预测准确度 (AUC 0.93) 的模型包括粪便钙卫蛋白水平以及前 20 个选定分类群的宏基因组分析。这些结果表明,临床和微生物生物标志物的整合提高了诊断的准确性。这种综合方法已被用于预测 IBD 患者对治疗的反应。
在这项研究中,基线临床数据(包括血清学、内窥镜和临床生物标志物)不足以预测缓解(AUC 0.62),而添加分类学和代谢谱将诊断能力分别提高到 AUC 0.72 和 AUC 0.74。
此外,仅粪便钙卫蛋白水平就能够区分储袋炎患者和无储袋炎患者 (AUC 0.63)。相比之下,微生物组物种模型(有或没有粪便钙卫蛋白水平作为额外的预测因子)实现了 0.78 的 AUC,证实微生物分析在识别储袋炎方面具有优于仅粪便钙卫蛋白水平的诊断性能。
用于诊断 T2DM 的葡萄糖代谢受损的血清学生物标志物包括空腹血糖水平、75g口服葡萄糖激发后 2 小时血糖水平(口服葡萄糖耐量试验)和糖化血红蛋白水平。
一项使用来自两个瑞典队列136数据的研究确定了预测 T2DM 进展的血清学和微生物组生物标志物的组合。
在这项研究中,多变量分析表明胰岛素抵抗程度与微生物组变异之间存在很强的相关性。
有趣的是,使用基于微生物组的机器学习模型来区分验证队列中胰岛素抵抗程度最低和最高的个体的 AUC 为 0.78,这表明肠道微生物群是 T2DM 进展的重要调节因子。
事实上,尽管已经为 T2DM 提出了广泛的诊断生物标志物,但它们中的大多数未能捕捉到这种疾病的复杂性或掌握微生物和代谢的变化。在这方面,已将代谢物生物标志物与已确定的临床风险因素结合使用,以显着改善疾病分类。
微生物特征
了解单个细菌类群(病原菌)和/或复杂微生物群落(生态失调)变化的功能作用和特异性对于解决IBD或T2DM中微生物-宿主相互作用的发病机制至关重要。在这种情况下,肠道微生物组的功能改变可能代表宿主适应的结果。

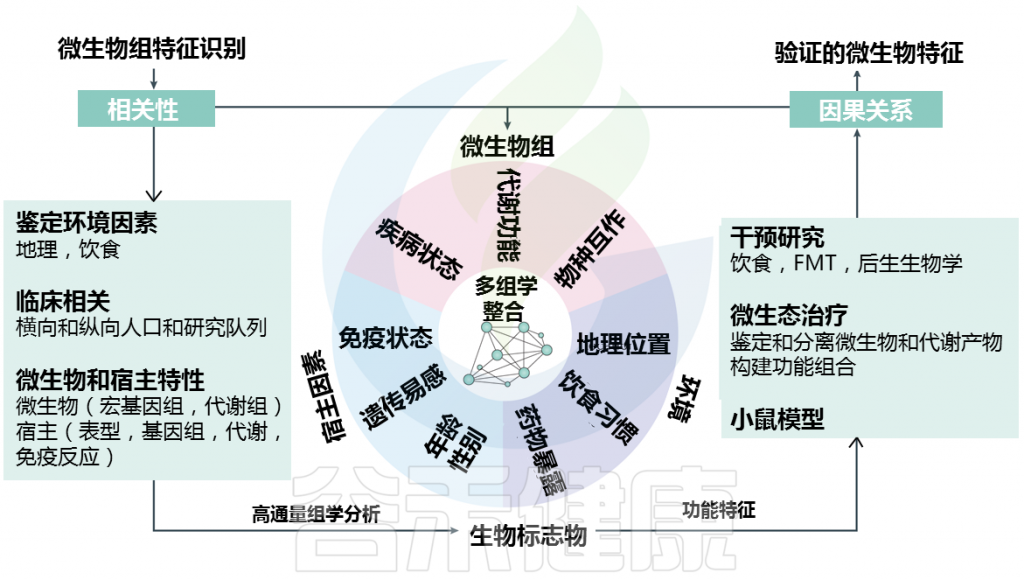
Metwaly et al.,Nat Rev Gastroenterol Hepatol. 2022.
肠道微生物群与多种疾病之间存在因果关系,已在小鼠实验中得到证实。无菌小鼠模型可以选择性地用单一细菌菌株、最小细菌聚生体或来自人类粪便或其他供体材料的定义复杂的肠道微生物生态系统进行定植,以研究它们对宿主表型的影响。在 IBD 中,无菌小鼠模型的单菌株定植有多种共生细菌,包括大肠杆菌、粪肠球菌、普通拟杆菌和Bilophila wadsworthia使我们能够了解疾病引发或保护的一些潜在机制。已有的研究工作表明,肠道菌群是驱动结肠炎小鼠模型炎症所必需的,而这些细菌与 CD患者的复发风险相关。肠道微生物群移植到无菌小鼠体内导致了几种疾病状态,从而揭示了与炎症有关的共享功能代谢途径。同样,以前的工作表明葡萄糖耐量和胰岛素抵抗受肠道微生物组组成的影响,已通过一系列 FMT 研究得到证实。
在过去的二十年里,人类和小鼠研究的证据揭示了肠道微生物组在炎症和代谢疾病(如 IBD 和 T2DM)的发病机制中的基本作用。肠道微生物生态系统结构和功能的变化(失调),与这些疾病患者的疾病活动、复发风险或对治疗的反应有关。然而,大多数这些疾病的复杂性和多因素发病机制,以及人类研究中存在多种混杂因素,依然对微生物组特征在诊断、预测预后和治疗决策中的临床应用提出重大挑战。
当前的微生物组研究不仅仅局限于描述微生物群落结构和疾病关联,还在了解肠道菌群在复杂慢性病发病机制中的致病作用方面取得进展。预计这些努力将增强微生物组建模,并推进可用于临床环境的基于菌群特征和/或疾病风险的模型开发。
参考文献:
Metwaly A, Reitmeier S, Haller D. Microbiome risk profiles as biomarkers for inflammatory and metabolic disorders. Nat Rev Gastroenterol Hepatol. 2022 Feb 21. doi: 10.1038/s41575-022-00581-2. Epub ahead of print. PMID: 35190727.