1 项目概述
| 项目名称 | 单位 | ||
| 样本数 | 样本类型 | ||
| 测序平台 | Illumina NovaSeq6000 | 测序区段 | 16S V4区 |
- 示例报告详细解读视频(上): http://www.guhejk.com/wordpress/?p=3654
- 示例报告详细解读视频(下): http://www.guhejk.com/wordpress/?p=3660
1.1 项目数据分析结果汇总说明
- 分组样本元信息情况: 各组样本数量均衡。
- 测序质量: 各样本数据量相对均一,均超过10万reads。
- OTU数量情况: OTU数量平均在1600-2000左右,相对较多。
- Alpha多样性: 各组间alpha多样性无差异。
- Beta多样性: beta多样性组间存在显著差异,K组与其他组差异较大,M组组内重复较好。
- 物种构成: 物种构成符合菌群构成特征。
- 组间菌属差异: 组间菌属差异较多,LEfse结果差异较明显。
- Bugbase情况: Bugbase差异不显著。
- FAPROTAX: FAPROTAX结果中K组病原菌较多。
- Picrust2功能预测: 代谢部分CAZY差异较多。
- 随机森林分组预测: 基于OTU的分组预测效果较好可以完全区分各分组。
2 技术介绍
微生物多样性测序(扩增子测序)是基于二代高通量测序对16S/18S/ITS等序列进行测序。可以同时检测样本中的 优势物种、稀有物种及一些未知物种的检测,获得样本的微生物群落组成以及相对丰度。
16S rDNA是细菌16S rRNA相对应的DNA序列,存在于所有细菌的基因组中,其中包括保守区域和高变区域,保守 区在细菌间差异不大,高变区在不同的种属间有一定的差异性,可以用做细菌分类鉴定的指标。通过选取特定可 变区域,用保守区段设计引物进行PCR扩增,通过16S rDNA扩增子测序对高变区进行系统鉴定,来研究环境样本 的微生物群落结构组成。
2.1 实验流程
 |
| Fig 2-1-1 实验流程图 |
2.2 数据分析流程
 |
| Fig 2-2-1 数据分析流程图 |
3 OTU/ASVs结果统计
3.1 样本有效数据及OTU/ASVs统计结果
测序完成之后获得的原始序列(raw reads)在正常情况下会存在一部分低质量数据、接头或PCR错误,根据一定的标准过滤掉低质量数据,本报告中的raw-tags为已经过上述质量过滤的clean reads,然后通过聚类方法获得ASVs,并将原始序列比对回ASVs。
数据过滤的标准如下:
- 低质量测序序列的过滤及切除(去除每4bp平均质量Q≤20的读段和低于50bp的reads);
- 接头序列及无关序列的剔除(Adapter和N);
- 双端overlap小于完全匹配的5bp;
下表统计了每个样本的原始序列数量:raw-tags,无完全匹配的单条序列数量:singleton,及其比例:singleton%,比对到最终ASVs的序列数量:tagsmatchedASVs,及其比例:tags-matched-ASVs%和每个样本的ASVs数量。
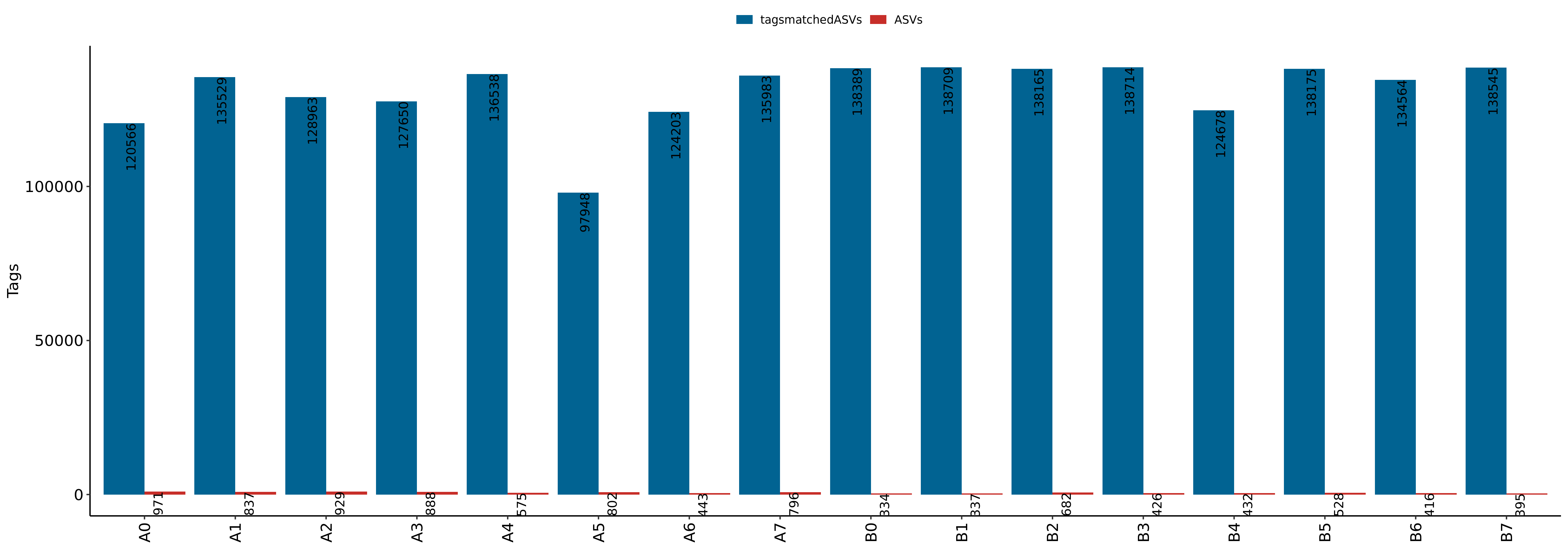 |
| Fig 3-1-1 各样本ASVs及比对到ASV的序列统计 |
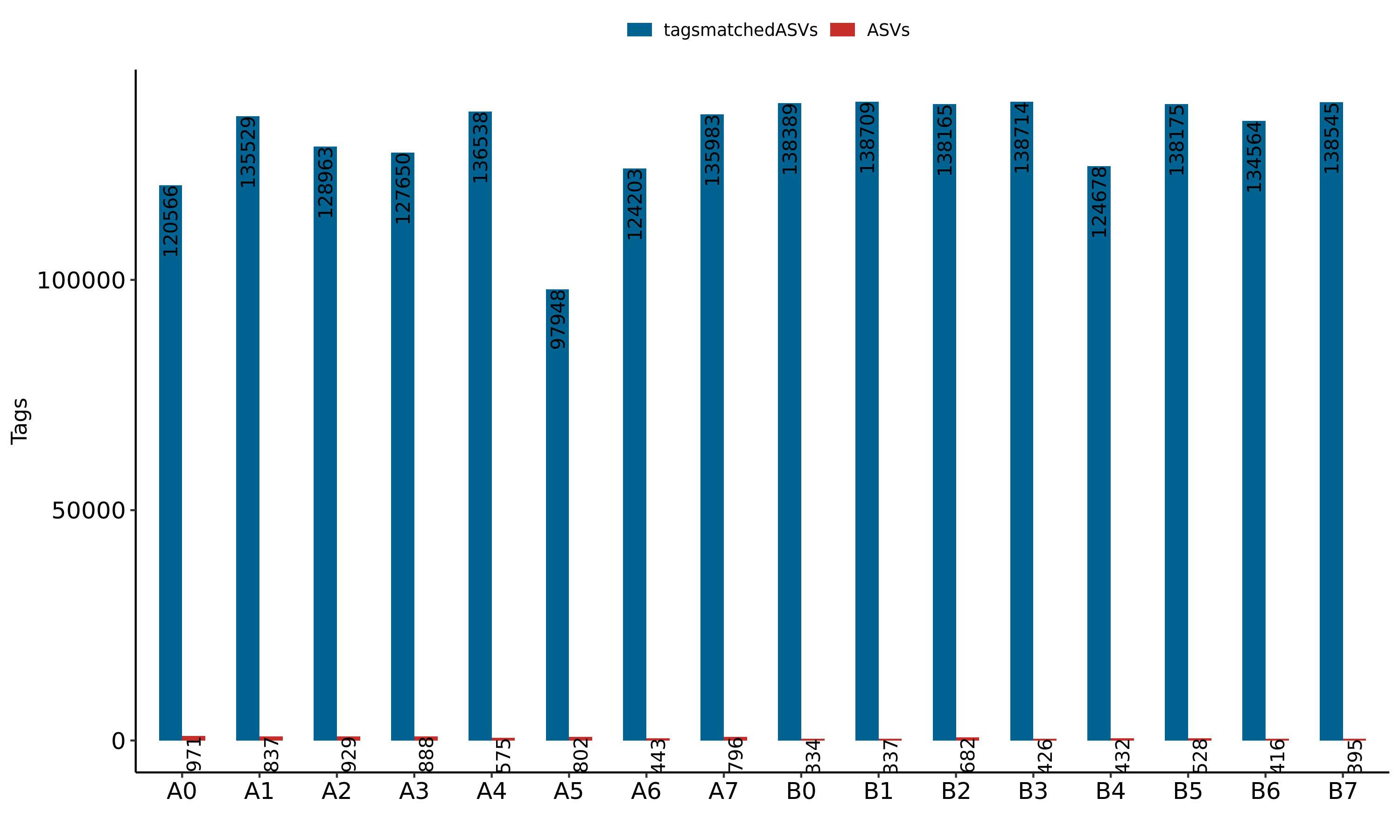 |
| Fig 3-1-2 各分组ASVs及比对到ASV的序列统计 |
注:原始样本reads总数,及总共的ASVs结果统计详情见 rawotu-summary.txt
ASVs的代表序列文件见 rep_set.fna
可以使用记事本等软件打开查看,序列名为hash编码,名称相同表明序列相同。
首先对样本数据进行TSS标准化,并归一到总丰度10万reads,然后对OTU/ASVs结果进行如下过滤:
- 删除平均丰度低于10万分之一的OTU;
4 物种注释及构成
4.1 所有样本各分类水平组成分析 SILVA138版本
分组统计比较请在各分组分析结果中查看所有样本门、纲、目、科、属、种构成情况详见文件目录: 04_Taxonomic/taxa_plot/index.html
以下为标准化到10万reads的物种构成结果文件,可以使用excel打开。
- 门 构成表文件: 04_Taxonomic/taxa_plot/level-2.csv
- 纲 构成表文件: 04_Taxonomic/taxa_plot/level-3.csv
- 目 构成表文件: 04_Taxonomic/taxa_plot/level-4.csv
- 科 构成表文件: 04_Taxonomic/taxa_plot/level-5.csv
- 属 构成表文件: 04_Taxonomic/taxa_plot/level-6.csv
- 种 构成表文件: 04_Taxonomic/taxa_plot/level-7.csv
- OTU/ASVs 表文件: 01_pick_otu/otu-table100k-tax.txt
以下为百分比的物种构成结果文件,可以使用excel打开。
- 门 构成表文件: 04_Taxonomic/relative/feature_table_L2.txt
- 纲 构成表文件: 04_Taxonomic/relative/feature_table_L3.txt
- 目 构成表文件: 04_Taxonomic/relative/feature_table_L4.txt
- 科 构成表文件: 04_Taxonomic/relative/feature_table_L5.txt
- 属 构成表文件: 04_Taxonomic/relative/feature_table_L6.txt
- 种 构成表文件: 04_Taxonomic/relative/feature_table_L7.txt
各分类水平的微生物物种构成汇总统计表: 04_Taxonomic/relative/Otu_all_level/OTU_summary.csv
所有OTU/ASVs对应的silva v138版本物种注释结果文件: 04_Taxonomic/taxonexported/taxonomy.tsv
4.2 greengene 13_8版本物种注释结果文件
- OTU/ASVs 表文件: 01_pick_otu/gg_13-8table100k-tax.txt
5 多样性分布结果
5.1 Alpha多样性结果
分组统计比较请在各分组分析结果中查看使用Qiime2进行alpha多样性分析,命令如下:qiime diversity alpha,分别计算获得simpson,ace,shannon,chao1以及goods_coverage。
α多样性是对单个样本中的物种多样性分析,通过一系列统计学指数来评估菌群物种的丰富度(richness)和多样性(diversity)。其中丰富度是衡量单个样本中物种的种类个数,通过分类单位的个数来衡量,多样性指数是衡量群落的异质性。
计算群落丰富度(Community richness)的指数:
Chao1:用Chao1算法估计样本中所含OTU数目的指数,通过计算群落中只检测到1次和2次的OTU数估计群落中实际存在的物种数。Chao1在生态学中常用来估计物种总数,由Chao(1984)最早提出。
ACE:用来估计群落中含有OTU数目的指数,预设将序列量10以下的OTU都计算在内,从而估计群落中实际存在的物种数。是生态学中估计物种总数的常用指数之一,与Chao1算法不同。
计算群落多样性(Community diversity)的指数:
Shannon:香农-威纳指数综合考虑了群落的丰富度和均匀度。Shannon指数值越高,表明群落的多样性越高。
Simpson:辛普森多样性指数对菌群多样性评估,Simpson指数值越高,表明群落多样性越高。一般而言,Shannon指数侧重对群落的丰富度以及稀有OTU,而Simpson指数侧重均匀度和群落中的优势OTU。
下表统计了每个样本的各项alpha多样性指标。
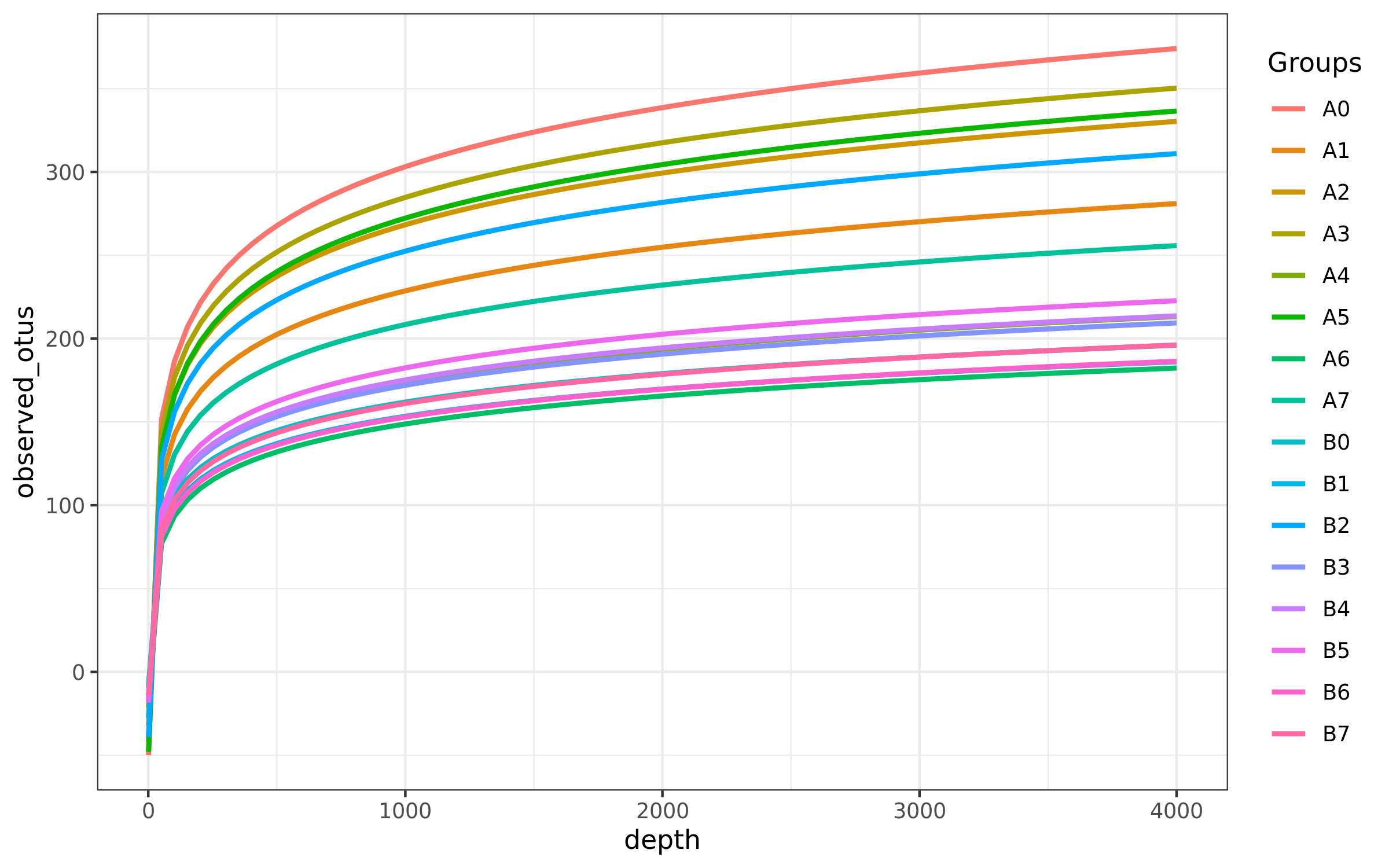
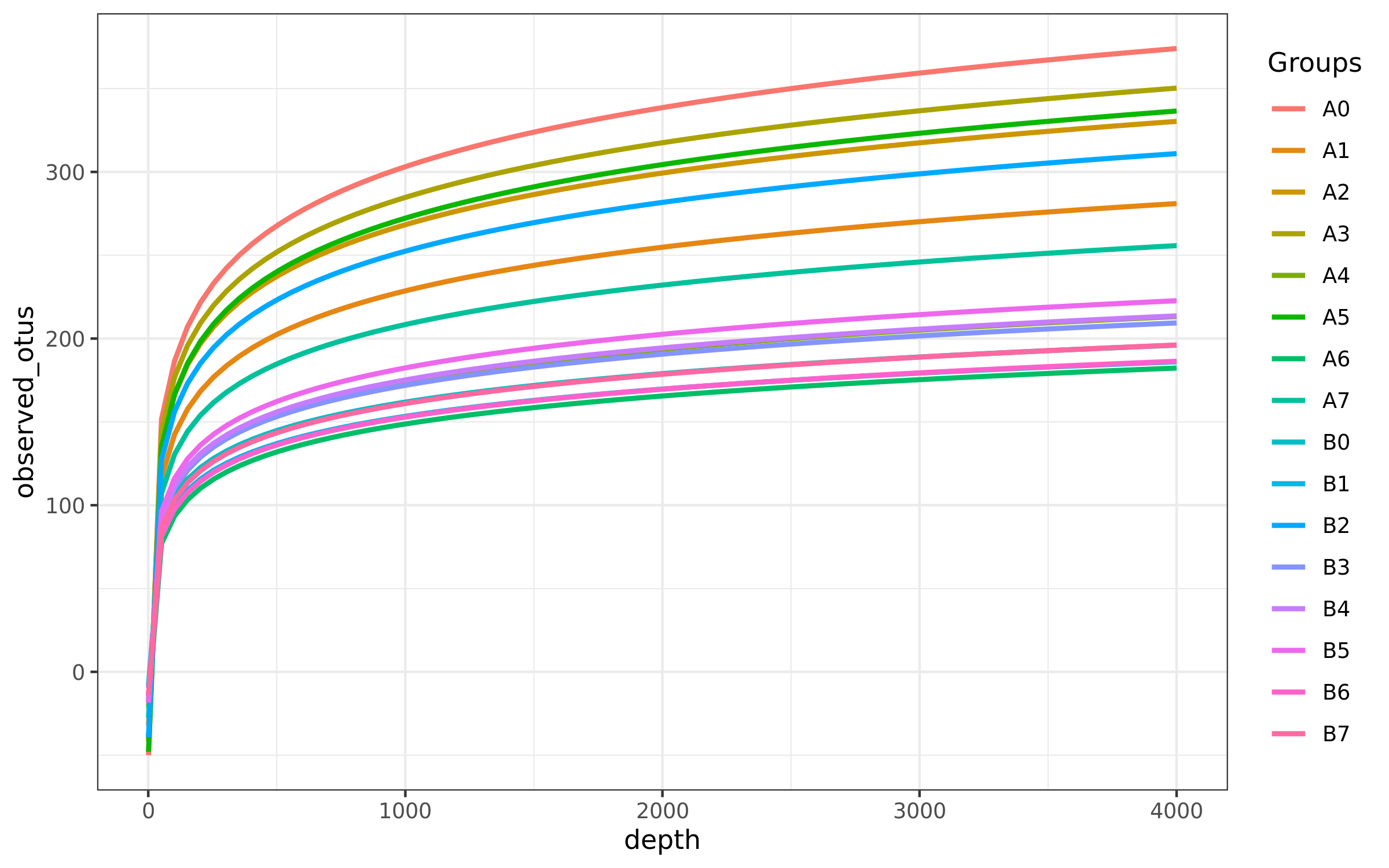
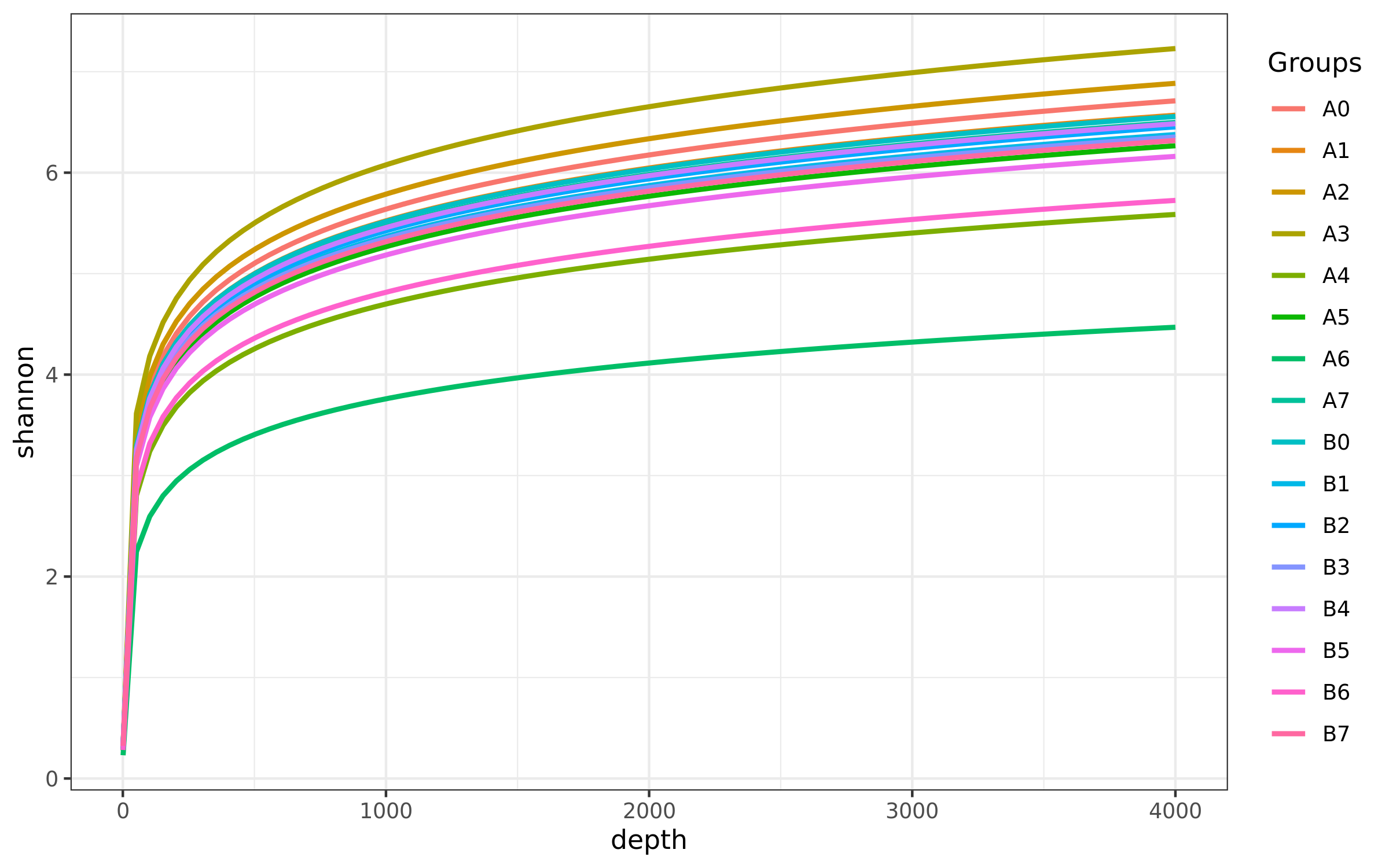
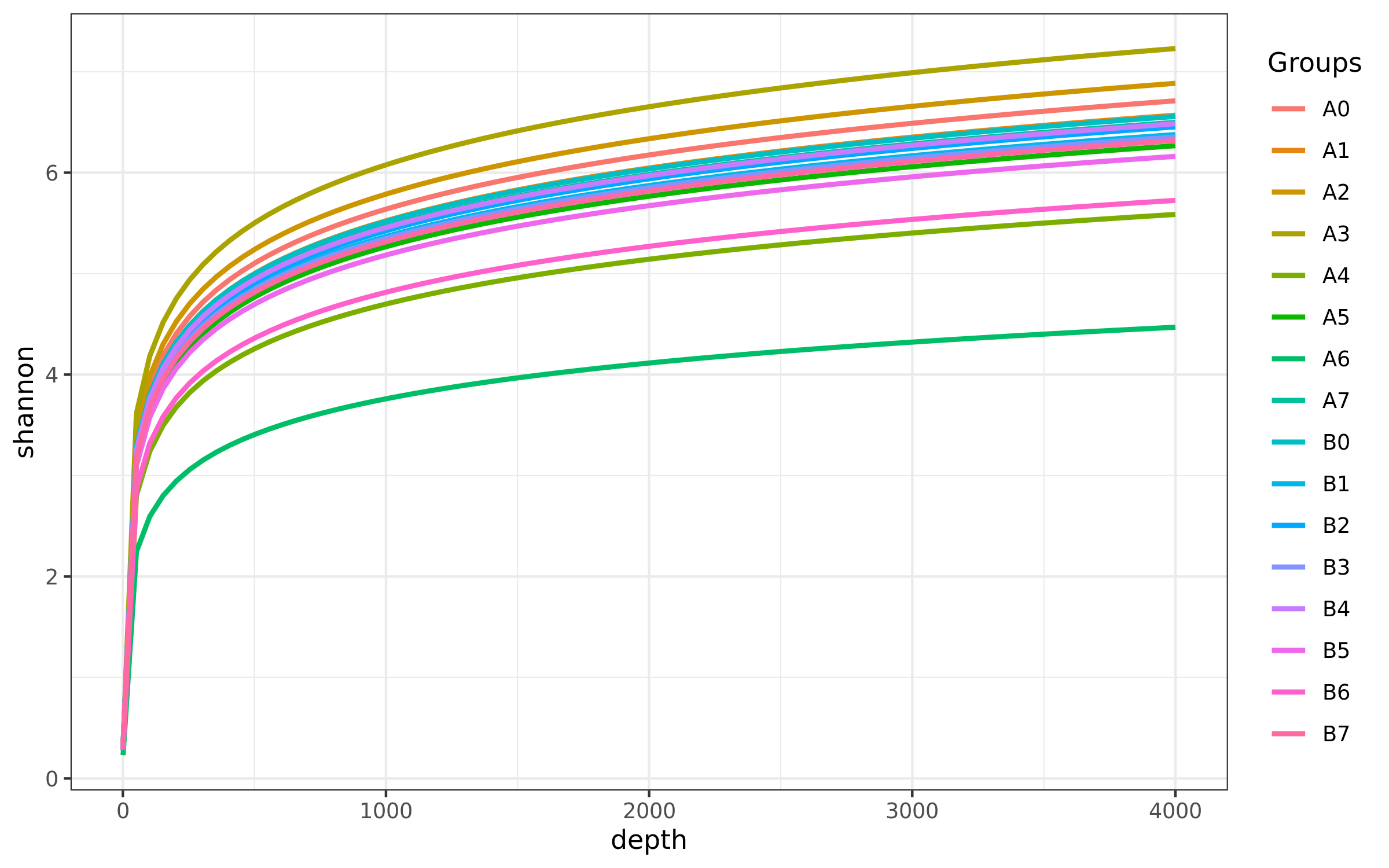
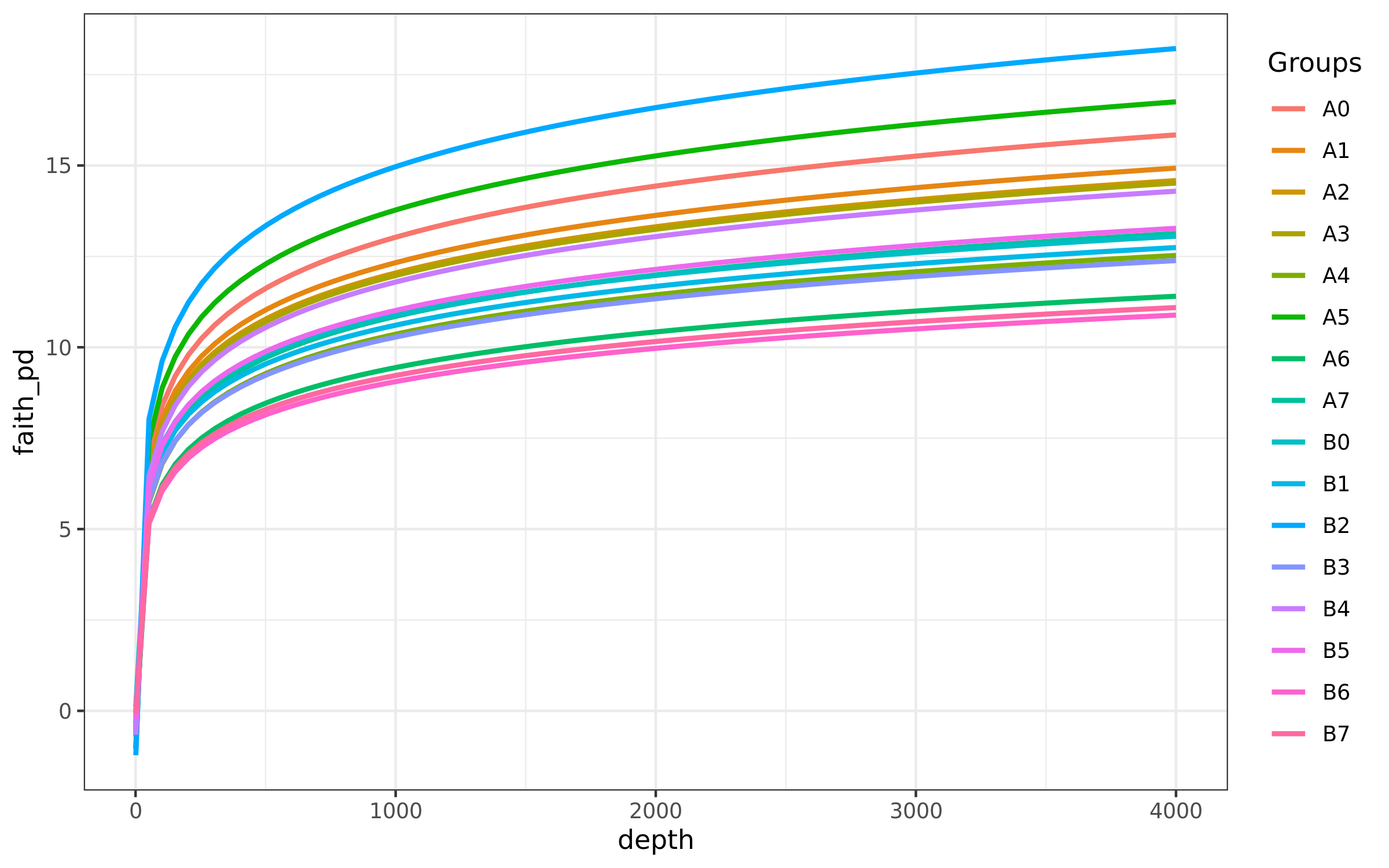
Alpha多样性不同指数的稀释曲线结果
样品物种丰度 Alpha 多样性指数稀释曲线图,横坐标表示抽取 reads 数量,纵坐标表示相应 Alpha 多样性指数的的值,图中一个颜色代表一个样本,或一组。测序条数不能覆盖样本时,曲线呈上升趋势, 当曲线趋向平坦时,说明测序数据量足够大,可以反映样品中绝大多数的微生物物种信息。
结果在文件夹:/03_diversity-metrics/alpha_rarefaction下
- observed_otus
- shannon
- faith_pd
|
|
|

Fig 5-1 稀释曲线图
5.2 Beta多样性结果
Beta 多样性指针是用来比较多组样本之间的差别度量。首先根据所有样品的物种注释结果和 OTUs 的丰度信息,将 相同分类的 OTUs 信息合并处理得到物种丰度信息表(Profiling Table)。同时利用 OTUs 之间的系统发生关系,进 一步计算 Unifrac 距离(Unweighted Unifrac)。Unifrac 距离是一种利用各样品中微生物序列间的进化信息计算样品 间距离,两个以上的样品,则得到一个距离矩阵。然后,利用 OTUs 的丰度信息对 Unifrac 距离(Unweighted Unifrac)进一步构建 Weighted Unifrac 距离。最后,通过多变量统计学方法主成分分析(PCA,Principal Component Analysis),主坐标分析(PCoA,Principal) 等方法,从中发现不同样品(组)间的差异。
 |
使用bray_curtis的PCA
使用unweighted_unifrac_emperor的PCoA
使用weighted_unifrac_emperor的PCoA
6 分组统计分析结果
7 个性化分析补充文件
QIIME2数据文件目录
STAMP数据文件目录
MicrobiomeAnalysis.ca数据文件目录
8 参考文献
- Yongxin Liu, Yuan Qin, Xiaoxuan Guo, Yang Bai. Methods and applications for microbiome data analysis. Hereditas(Beijing)2019/9/2
- Shifu Chen, Yanqing Zhou, Yaru Chen, Jia Gu. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, Volume 34, Issue 17, 01 September 2018, Pages i884–i890
- Steven R. Gill et al. Metagenomic Analysis of the Human Distal Gut Microbiome. Science 02 Jun 2006
- P Ewels, M Magnusson, S Lundin, M Käller. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics [Internet]. 2016; 32: 3047–8
- A Gurevich, V Saveliev, N Vyahhi, G Tesler. QUAST: quality assessment tool for genome assemblies. Bioinformatics, 2013
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Research, 25: 1043–1055
- Nicola Segata et al. Metagenomic biomarker discovery and explanation. Curtis Huttenhower Genome Biology, 12:R60, 2011
- Lu J, Breitwieser FP, Thielen P, Salzberg SL. (2017) Bracken: estimating species abundance in metagenomics data. PeerJ Computer Science 3:e104, doi:10.7717/peerj-cs.104
- Ondov BD, Bergman NH, and Phillippy AM. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics. 2011 Sep 30; 12(1):385
- Li, D., Liu, C-M., Luo, R., Sadakane, K., and Lam, T-W., (2015) MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics, doi: 10.1093/bioinformatics/btv033 [PMID: 25609793]
- Buchfink B., Xie C. and Huson D.H. Fast and sensitive protein alignment using DIAMOND. Nat Methods, 2015;12(1):59-60
- Fu L., et al. CD-HIT: accelerated for clustering the next-generation sequencing data.Bioinformatics, 2012;28(23): 3150-3152
- Langmead B, Salzberg S. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012, 9:357-359
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup, The Sequence alignment/map (SAM) format and SAMtools, Bioinformatics (2009) 25(16) 2078-9 [19505943]
- Doug Hyatt et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics volume 11, Article number: 119 (2010)
- Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., & Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nature Methods
- Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Jaime Huerta-Cepas, Damian Szklarczyk, Lars Juhl Jensen,Christian von Mering and Peer Bork. Submitted (2016)
- Alcock et al. 2019. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Research, gkz935. [Epub ahead of print] [PMID 31665441]
- Liu B, Zheng DD, Jin Q, Chen LH and Yang J, 2019. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47(D1):D687-D692
- Chen LH, Zheng DD, Liu B, Yang J and Jin Q, 2016. VFDB 2016: hierarchical and refined dataset for big data analysis-10 years on. Nucleic Acids Res. 44(D1):D694-D697
- Kanehisa M., et al., The KEGG resource for deciphering the genome. Nucleic acids research,2004;32(suppl 1): D277-D280
- Yuki Moriya et al. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007 Jul; 35(Web Server issue): W182–W185
- Dongwan Kang et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ Preprints
- Gherman V. Uritskiy et al. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome volume 6, Article number: 158 (2018)
- Martin Urban, Alayne Cuzick, James Seager, Valerie Wood, Kim Rutherford,Shilpa Yagwakote Venkatesh et al. PHI-base: the pathogen–host interactions database. Nucleic Acids Research, Volume 48, Issue D1, 08 January 2020, Pages D613–D620
- Milton H. Saier, Jr, Vamsee S. Reddy, Brian V. Tsu, Muhammad Saad Ahmed,Chun Li, Gabriel Moreno-Hagelsieb. The Transporter Classification Database (TCDB): recent advances. Nucleic Acids Research, Volume 44, Issue D1, 4 January 2016, Pages D372–D379
- Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M, de Crecy-Lagard V, Diaz N, Disz T, Edwards R, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:5691–5702
- Caner Bağcı.Sina Beier.Anna Górska.Daniel H. Huson. Introduction to the Analysis of Environmental Sequences: Metagenomics with MEGAN. Evolutionary Genomics pp 591-604
9 目录结构
. ├── 01_pick_otu -> OTU/ASV聚类分析及过滤 │ └── summary -> OTU/ASV统计结果目录 │ │ ├── rawotu-summary.txt -> 测序reads总数、均值及OTU数量分布 │ ├── rep_set.fna -> OTU/ASV代表序列(fasta格式) │ ├── repseq.qza -> OTU/ASV代表序列(qiime2的qva格式) │ ├── rawotu_table.biom -> 原始序列生成的OTU/ASV计数表(biom文件) │ ├── rawotu_table.txt -> 原始序列生成的OTU/ASV计数表(文本表文件) │ ├── otu-table100k-tax.biom -> 标准化到10万reads的OTU/ASV计数表-带物种注释(biom文件) │ ├── otu-table100k-tax.txt -> 标准化到10万reads的OTU/ASV计数表-带物种注释(文本表文件) │ ├── otu-filter-minfeq-tax.biom -> 过滤低于万分之一reads的标准化OTU/ASV计数表-带物种注释(biom文件) │ ├── otu-filter-minfeq-tax.txt -> 过滤低于万分之一reads的标准化OTU/ASV计数表-带物种注释(文本表文件) │ └── gg_13-8table100k-tax.biom -> 标准化到10万reads的greengene13_8数据库计数表-带物种注释(biom文件) ├── 02_sequence_statistic -> 序列长度及个样本分组数据量统计 │ ├── reads_length_distribution.txt -> 测序序列读长 │ ├── Group_Sample_stats_OTU.pdf -> 各分组OTU/ASV数量及reads数 │ └── Sample_stats_OTU.pdf -> 各样本OTU/ASV数量及reads数 ├── 03_diversity-metrics -> alpha及beta多样性分析值 │ └── alpha -> alpha多样性计算目录 │ └── XXX_emperor -> beta多样性qiime2导出目录,可进入目录点击index.html │ │ ├── alpha_div.txt -> 样本alpha多样性表 │ ├── rooted-tree.nwk -> 构建的OTU进化树 │ ├── XXXX_dm.txt -> beta多样性不同XXX算法的距离矩阵 │ └── XXXX_pc.txt -> beta多样性不同XXX算法的ordination降维特征 ├── 04_Taxonomic -> 物种注释结果目录 │ ├── silva138taxonomy.qza -> 基于silva138的菌属物种注释(qiime2的qza文件) │ ├── relative -> 相对丰度百分比物种构成表,L2-L7分别是门、纲、目、科、属、种 │ ├── taxa -> 标准化到10万reads的物种构成表,L2-L7分别是门、纲、目、科、属、种 │ └── taxa_plot -> qiime2导出物种构成丰度表及图,可以打开目录下的index.html,csv为excel格式表 ├── Picrust2 -> 基因功能及代谢通路注释结果目录,来自Picrust2.3 │ ├── KO_metagenome_out -> KEGG基因注释结果,KOpred_metagenome_unstrat_descr.tsv为包含基因名称的丰度表 │ ├── EC_metagenome_out -> 酶注释结果,ECpred_metagenome_unstrat_descr.tsv为包含基因名称的丰度表 │ ├── KEGGpathways_out -> KEGG代谢通路注释结果,KEGGpath_abun_unstrat_descr.tsv为包含代谢通路的丰度表 │ ├── pathways_out -> METACYC代谢通路注释结果,METACYCpath_abun_unstrat_descr.tsv为包含代谢通路的丰度表 │ ├── GMMmodelout -> 菌群代谢产物模块预测结果,modules.tsv为丰度表 │ ├── GBMmodelout -> 菌群神经递质代谢产物模块预测结果,modules.tsv为丰度表 │ ├── CAZYout -> 菌群碳水化合物代谢CAZy预测结果,pred_metagenome_unstrat.tsv为丰度表 │ ├── COG_metagenome_out -> COG预测,COGpred_metagenome_unstrat_descr.tsv为丰度表 │ ├── PFAM_metagenome_out -> PFAM功能域模块预测结果,pred_metagenome_unstrat.tsv为丰度表 │ ├── TIGRFAM_metagenome_out -> TIGRFAM功能域模块预测结果,pred_metagenome_unstrat.tsv为丰度表 │ ├── GBMmodelout -> 菌群神经递质代谢产物模块预测结果,modules.tsv为丰度表 │ ├── GBMmodelout -> 菌群神经递质代谢产物模块预测结果,modules.tsv为丰度表 │ ├── GBMmodelout -> 菌群神经递质代谢产物模块预测结果,modules.tsv为丰度表 │ ├── GBMmodelout -> 菌群神经递质代谢产物模块预测结果,modules.tsv为丰度表 │ └── Unigenes.RPKM.xls -> 每个基因在每个样本中的RPKM ├── STAMP -> 用于STAMP软件进行分析所需文件 ├── Groups -> 不同分组情况下的统计结果目录 │ ├── alphadiv -> alpha多样性统计结果 │ ├── betadiv -> beta多样性统计结果 │ │ ├── pca_analysis -> PCA分析结果 │ │ ├── pcoa_bray_analysis -> PCOA分析结果 │ │ ├── pcoa_unweighted_unifrac_analysis -> PCOA非加权 │ │ ├── pcoa_weighted_unifrac_analysis -> PCOA加权 │ │ └── nmds_analysis -> NMDS分析结果,Envfit统计结果查看Envfit.result.log │ ├── otuVenn -> OTU各组韦恩图 │ ├── taxanomyBar -> 各样本及分组物种构成柱状图 │ │ ├── taxon_hist -> include_all\all为显示top10,其他所有归为other;include_all\top10为仅计算top10,不包含其他;without_unknown为去除了未分类的分类单元 │ │ └── taxon_hist_group -> 为各个分组的均值的物种构成柱状图,目录同上。 │ ├── Network -> 物种网络图,提供了门层面的关联网络 │ ├── meta -> 元信息统计检验 │ ├── Lefse_Analysis -> Lefse统计结果,biomarkers_raw_images目录包含具体菌属的标志图 │ ├── Bugbase -> Bugbase分析结果,BugBase_pvlaue.txt为统计检验pvalue │ │ ├── normalized_otus -> 根据拷贝数标准化OTU │ │ ├── otu_contributions -> 各OTU对Bugbase各特征的贡献 │ │ ├── predicted_phenotypes -> Bugbase各特征的样本评估结果 │ │ ├── pcoa_weighted_unifrac_analysis -> PCOA加权 │ │ └── nmds_analysis -> NMDS分析结果,Envfit统计结果查看Envfit.result.log │ ├── FAPROTAX -> FAPROTAX预测及分析结果 │ │ ├── FAPROTAX_heatmap -> FAPROTAX丰度状况,具体OTU构成查看:FAPROTAX_report.txt │ │ ├── Markers -> 组间差异统计检验 │ │ ├── pcoa_unweighted_unifrac_analysis -> PCOA非加权 │ │ ├── pcoa_weighted_unifrac_analysis -> PCOA加权 │ │ └── nmds_analysis -> NMDS分析结果,Envfit统计结果查看Envfit.result.log │ └── diff_analysis -> 组间统计检验及差异分析结果 │ ├── Anosim_analysis -> Anosim分析结果 │ ├── metagenomeRXXXX -> 使用metagenomeSeq进行统计检验结果 │ ├── UnivarTestXXXX -> 使用非参数检验进行统计检验结果 │ ├── TukeyHSD -> 使用Tukey进行统计检验结果 │ ├── TaxaMarkers -> 物种差异分析结果 │ ├── Correlation -> 基于metagenomeSeq的差异指标相互相关性分析结果 │ ├── heatmap -> 显著差异指标的热图 │ ├── metaCorrelation -> 差异指标与元数据及环境因子的相关性分析结果 │ ├── diff_filter -> 统计差异指标筛选后的数据表 │ └── RF -> 使用随机森林进行模型构建和预测结果 └─
10 附录
10.1 英文Methods
10.2 文章引用与致谢
如果您的研究课题使用了杭州谷禾信息技术有限公司的测序和分析服务,我们期望您在论文发表时,在Method部分或Acknowledgements部分引用或提及杭州谷禾信息技术有限公司。
