-

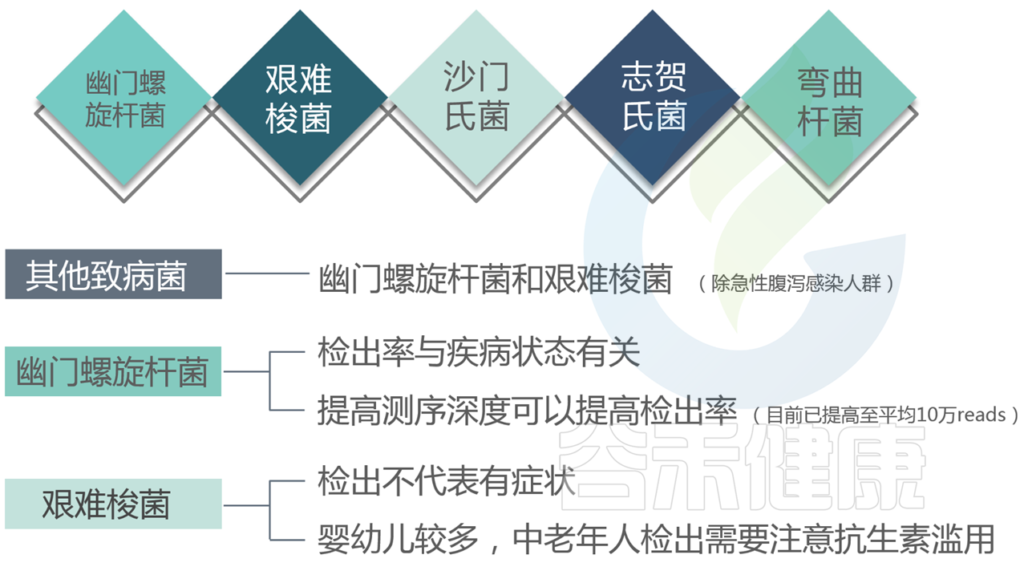
 CNAS L23010
CNAS L23010

 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室
谷禾健康
从 2024 年 8 月 24 日后,报告指标及排版更新说明如下:
◼ 增加了【核心菌属】这个指标。
◼ 增加了【肠道炎症】和【肠道产气】两个指标。
◼ 原有指标的值和报告顺序均没有改变。
◼ 版式调整:页面宽度增加,指标异常的配色和背景提示颜色进行了改变。
◼ 首页基本信息部分:表格排列形式修正,避免出现姓名不显示的错误,备注可以支持 更长的文字。
◼ 代谢物和神经递质指标的说明均改为单页。 ◼ 部分神经递质名字进行了调整:一氧化氮、维生素 K2。
◼ 修正了部分标点符号和语句描述。
◼ 肠道年龄的描述单独成页。
◼ 菌的描述和异常菌的字体大小进行了修改。
◼ 短链脂肪酸的缺乏异常和描述增加。
◼ 页码从 130 多变为:儿童 142 页~成年人 156 页之间。
◼ 页码部分增加的总页数,便于判断文档是否完整。
◼ pdf 生成大小变为 2.8M 左右,生成速度和可靠性大大提高。

谷禾健康

5月29日,是世界肠道健康日。肠道是人体最重要的消化系统之一,与人体健康紧密相关。而肠道菌群作为肠道重要组成部分,在肠道健康中发挥着重要的作用。
编辑
由于基因、环境、饮食、药物等因素的影响,每个人的肠道菌群都有所不同,因此我们常常说每个人的肠道菌群都是独一无二的。
然而在不同年龄段不同人群中,有一类菌群却普遍存在,它们在人体的健康和稳定性方面发挥着重要的作用,这就是“肠道核心菌属”。
在这个世界肠道健康日,我们一起来聊聊关于“肠道核心菌属”。结合肠道菌群检测报告的部分案例,我们可以更深入直观地了解肠道核心菌属,这对于我们保持肠道健康和整体健康有重要意义。
谷禾在多年的肠道菌群检测中,发现大批量的肠道菌群检测报告中有那么一些菌群在大多数人群中普通存在,这些菌群的存及其丰度构成在肠道健康中起着至关重要的作用。一些科研文献中也有关于肠道核心菌属的相关研究。
一项大规模研究,利用微生物扩增子变异体精确作图 (RExMap)对来自全球 16 个地区的 29,349 名个体的现有 16S 数据进行重新分析,RExMap 的重新分析来自 10 项人口规模研究的现有 16S 微生物组数据绘制了来自世界各地 29,349 名个体的 17,786 个 OSU。
在此荟萃分析中,平均 122 个 OSU 占单个人微生物组丰度的 99%,平均 2000 个 OSU 占一个地理区域内 95% 的微生物组丰度。揭示了不同人群和地理区域的肠道微生物物种的详细景观。表明人类确实存在共享肠道核心微生物组。
这些核心菌属在不同的人群中,核心微生物之间的相对比例高度保守,这可能表明存在稳定的生态或功能关系。
我们的数据和这篇大规模荟萃分析表明这些核心微生物群对于其它菌群的定植,营养代谢,体重指数以及整个微生态健康起非常关键的作用。
目前,对于核心菌属的定义还没有统一的标准,但一般认为,核心菌属应该具备以下特征:
1. 在不同人群中普遍存在:核心菌属应该在不同人群中都能够被检测到,而不是仅存在于某些特定人群中。
2. 数量相对较高:核心菌属应该在肠道菌群中占据较高的比例,而不是仅占据极小的比例。
3. 对人体健康具有重要作用:核心菌属应该对人体健康具有重要作用,如参与食物消化、维持肠道屏障功能、调节免疫系统等。
谷禾健康基于超过60万人肠道菌群检测样本数据,这些样本包括了来自全球各大洲不同人种、地区饮食习惯和所有年龄段人群,给出了人体肠道菌群核心菌属的标准:
在90%人群检出,人群平均丰度1%以上的菌属为核心菌属(双歧杆菌和乳杆菌作为益生菌相对丰度低于1%但是重要性很高)。
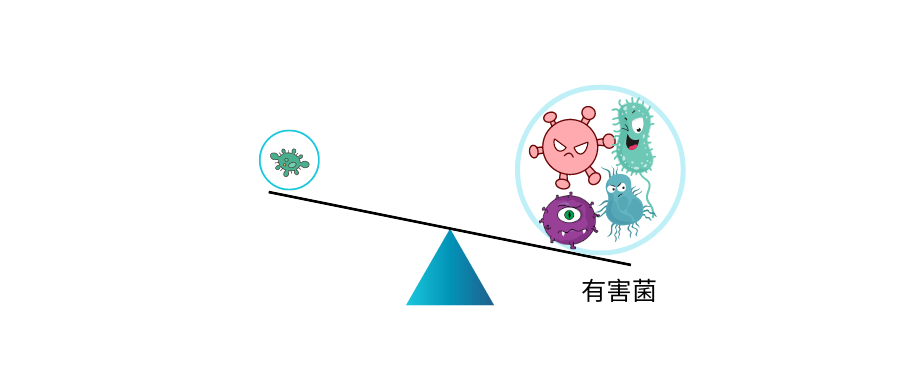
核心菌属及有益菌累加占总肠道菌群比例低于60%就可能出现肠道菌群紊乱,菌群是一种生态系统,健康的核心菌属占据菌群构成的绝大部分后,其他可能导致感染、免疫反应以及无法有效代谢食物的异常菌的比例就会被抑制。
核心菌属缺乏就给了病原菌或非肠道菌群生存空间,导致肠道健康问题。
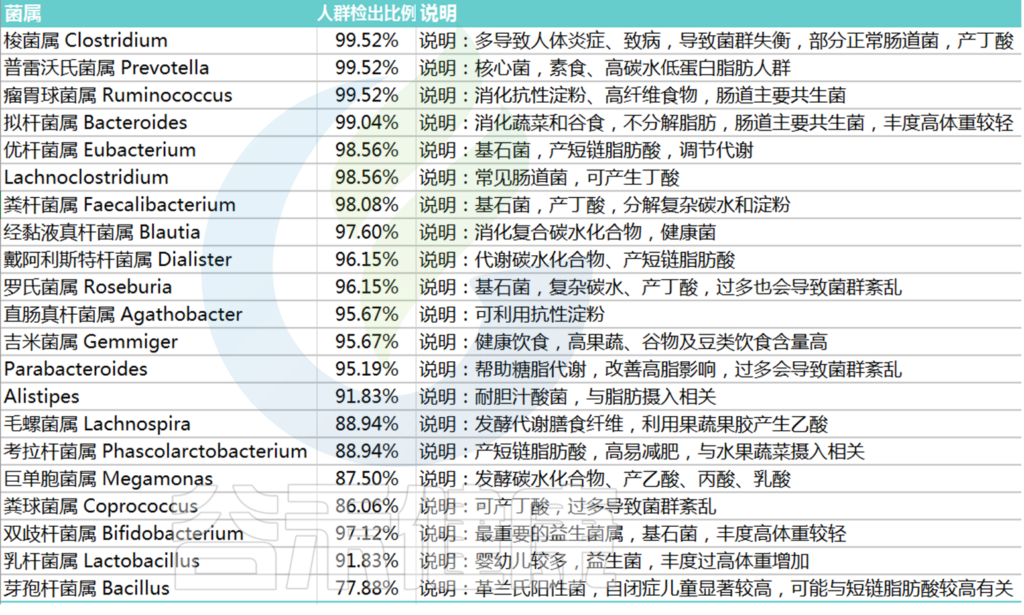
<来源:谷禾肠道菌群检测数据库>
上述这些核心菌属比如:
1. 拟杆菌 Bacteroides
Bacteroides是人体肠道中最常见的菌属之一,能够分解多糖类物质,产生短链脂肪酸,维持肠道屏障功能,调节免疫系统等。
2. 普雷沃氏菌属 Prevotella
Prevotella是一种厌氧菌,能够分解多糖类物质,产生短链脂肪酸,参与肠道免疫调节等。
3. 粪杆菌属 Faecalibacterium
Faecalibacterium是一种产生丰富的丁酸和丙酸的梭菌,能够维持肠道屏障功能,调节免疫系统等。
4.瘤胃球菌属Ruminococcus
Ruminococcus是一种产生丰富的丙酸和丁酸的菌属,能够参与食物消化、维持肠道屏障功能等。
虽然都属于核心菌属,且广泛存在于各类人群的肠道中,但每个人各个菌属的相对丰度却存在很大差异,不同的饮食习惯、母体传递以及生活地区都会导致这些核心菌属构成的偏好。
由于使用相对丰度来评估,其中一类菌丰度占比更高通常会导致其他菌属的相对丰度占比下降,因此早期肠道菌群研究中根据占据最主要丰度的菌属来将肠道菌群构成划分为多个肠型,常见的有拟杆菌、普氏菌、罗氏菌型三种。
不过肠型的划分相对比较粗放,也忽略了其他菌属的构成,那么:
这么多菌属怎么样的比例才是最健康的呢?
是不是都在范围内就都正常了,可是这些范围看着都好大,有的是从0.001~36%,都一样吗?
针对这些问题,我们给出了另一个指标:
该菌属丰度处于人群%水平
对于核心菌属我们评估较为:
因此评估菌群总体状况时较简单的方法,就是看过高和过低的核心菌属数量,超标越多菌群问题越大。当绝大部分菌属都处于正常范围之后我们就可以优化我们的核心菌属构成,使其尽量处于50~70%的水平。
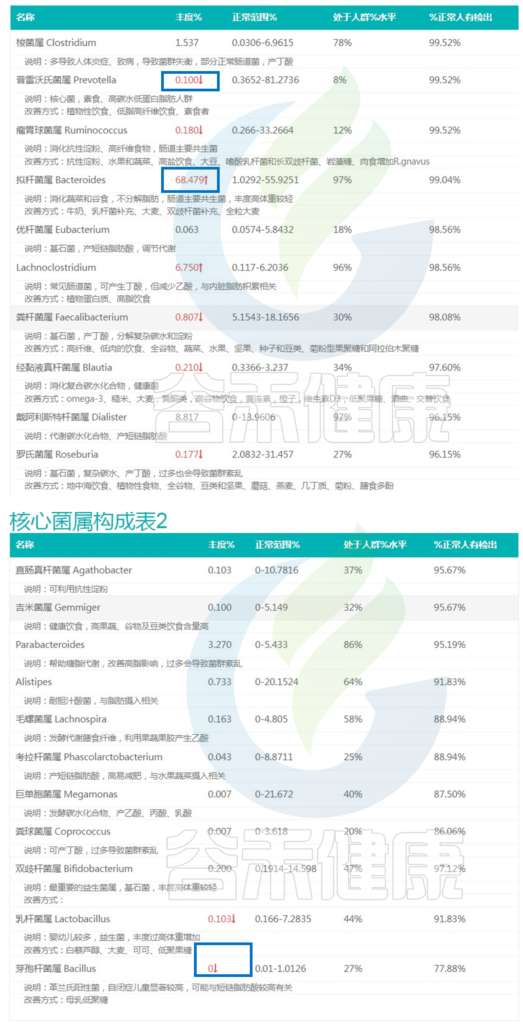
下图其中第4列就给出了这个指标,意思是这个核心菌属现有丰度超过了人群中%的人。
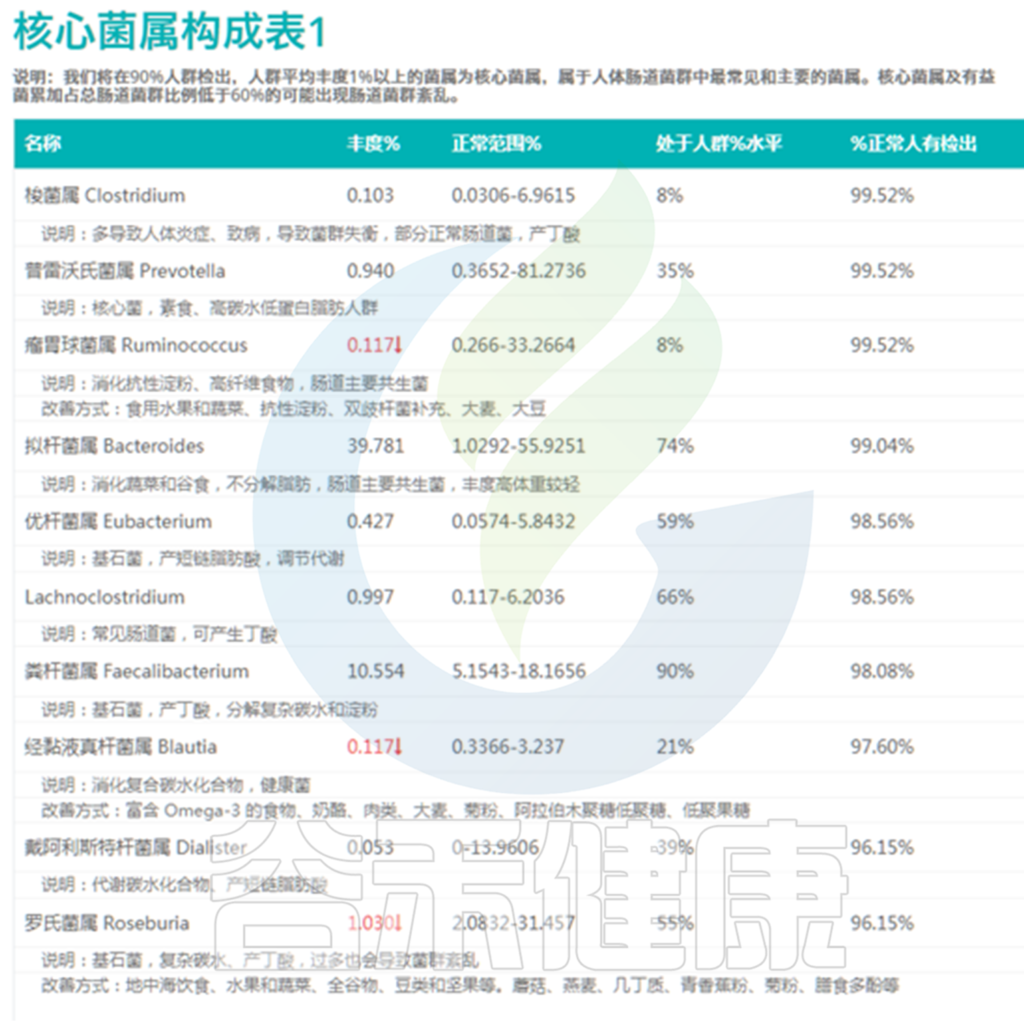
<来源:谷禾肠道菌群检测数据库>
其中瘤胃球菌属的丰度水平是0.117%,并给出了红色向下箭头,表明其丰度水平过低,超标了,对应的人群水平是8%,也就是这个丰度仅比8%的人要高。
除了箭头标示的过低菌属之外,其实像普雷沃氏菌属的35%人群水平也属于偏低,而粪杆菌属的90%人群水平就偏高了。
这样通过饮食和补剂定向干预可以使菌群中占比最主要的核心菌属达到更加稳定和合理的水平,整体肠道菌群的健康状况也会得到极大改善。
作为核心菌属,它们对于人体肠道菌群的作用主要包括:
1
参与食物消化
核心菌属能够分解多糖类物质,产生短链脂肪酸等,帮助人体消化食物
2
维持肠道屏障功能
核心菌属能够维持肠道黏膜屏障的完整性,防止有害物质进入血液循环。
3
调节免疫系统
核心菌属能够调节肠道免疫系统的功能,维持肠道微生物群落的稳定性。
4
构建稳定的肠道菌群生态
经过长期的共同进化和适应,核心菌属中的大部分能够有效代谢和利用人类饮食并消化进入结直肠部位的物质。这些菌属通过代谢途径的互补相互之间构建起一个菌群代谢网络,并维持稳定的菌群结构。
类似于我们常见的生态系统,一次抗生素的使用或某次感染后免疫系统的清理重建,相当于大草原或森林的一场大火,会摧毁大部分的生态成员,但一旦雨季来临生态又会重新开始建立。最底层的食物和能量来源就相当于雨水,最早能利用这些食物和能量来源的菌群构成了肠道菌群的基石。
研究发现上述核心菌属中的大部分都属于能代谢特定碳水化合物的降解和能量产生途径,包括拟杆菌(Bacteroides thetaiotaomicron)和青春双歧杆菌(Bifidobacterium adolescentis),以及Ruminococcus torques、Bacteroides coprocola、Faecalibacterium prausnitzii、Coprococcus catus、Parabacteroides johnsonii、Alistipes shahii等。
当上述这些核心菌属含量较高且构成合理时,肠道菌群的整体生态环境就会非常稳定,从最初代谢碳水化合物产生短链脂肪酸,进一步维持肠道内下游菌群的生长,同时调节肠道屏障,供给肠道黏膜细胞养分,维持免疫系统的均衡。
总之,核心菌属在人体肠道菌群中具有重要作用,对于人体健康具有重要意义。
谷禾肠道菌群检测数据库中显示,肠道核心菌属的异常与以下66种疾病相关:
以下是与每种核心菌属相关的疾病数量:
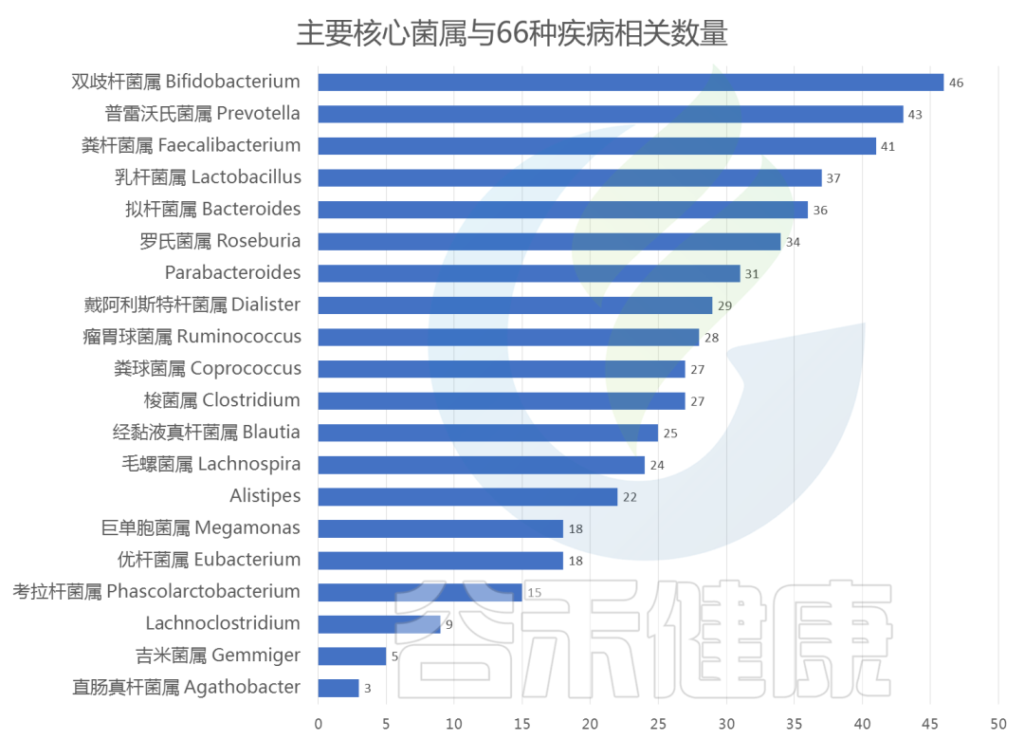
<来源:谷禾肠道菌群检测数据库>
★
结合谷禾肠道菌群检测报告案例,我们来看看一些慢性疾病患者的肠道核心菌属构成情况。
一般来说,他们的核心菌属异常分为两种情况:
这里我们来看关于慢性腹泻和便秘患者的两个案例。
➤➤ 慢性腹泻患者:
该慢性腹泻患者的肠道菌群检测报告中,许多核心菌属均处于低丰度的情况。
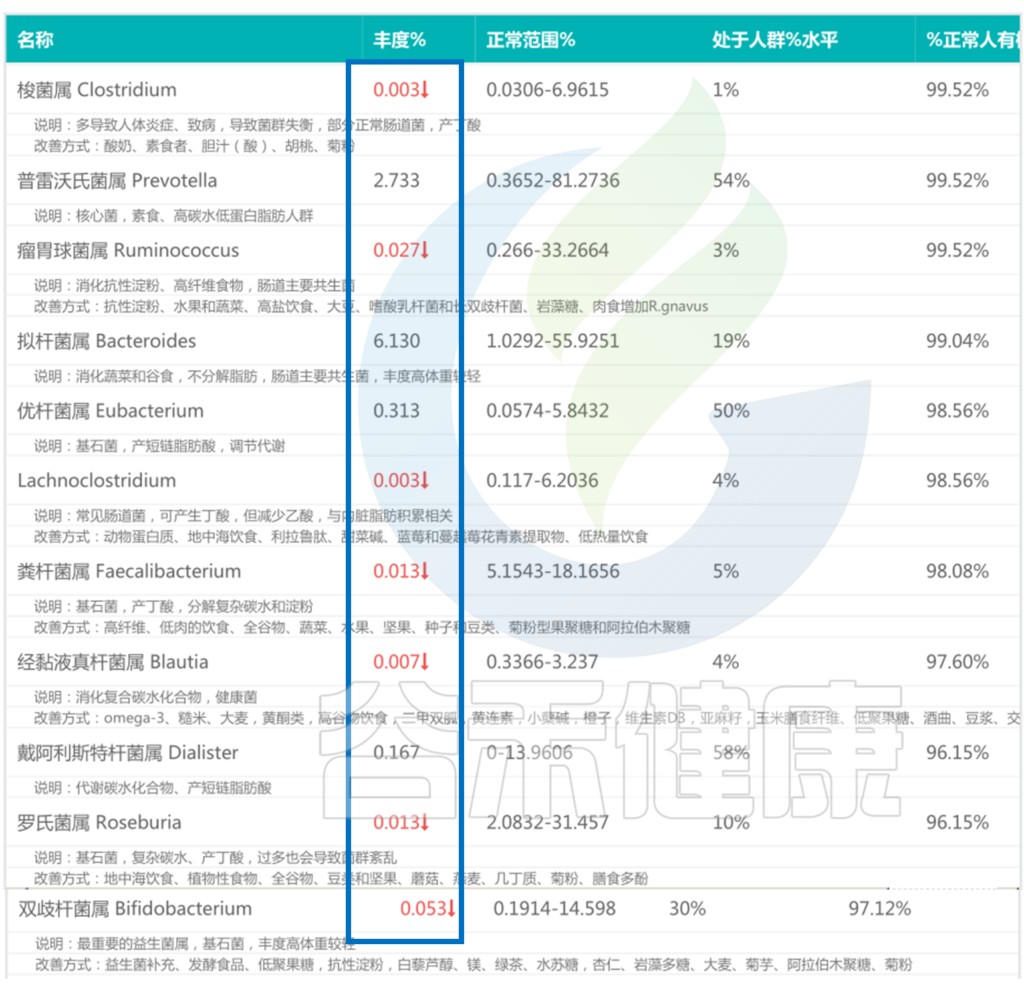
<来源:谷禾肠道菌群检测数据库>
当多数核心菌属丰度较低处于劣势的情况下,肠道有害菌逐渐开始占上风:
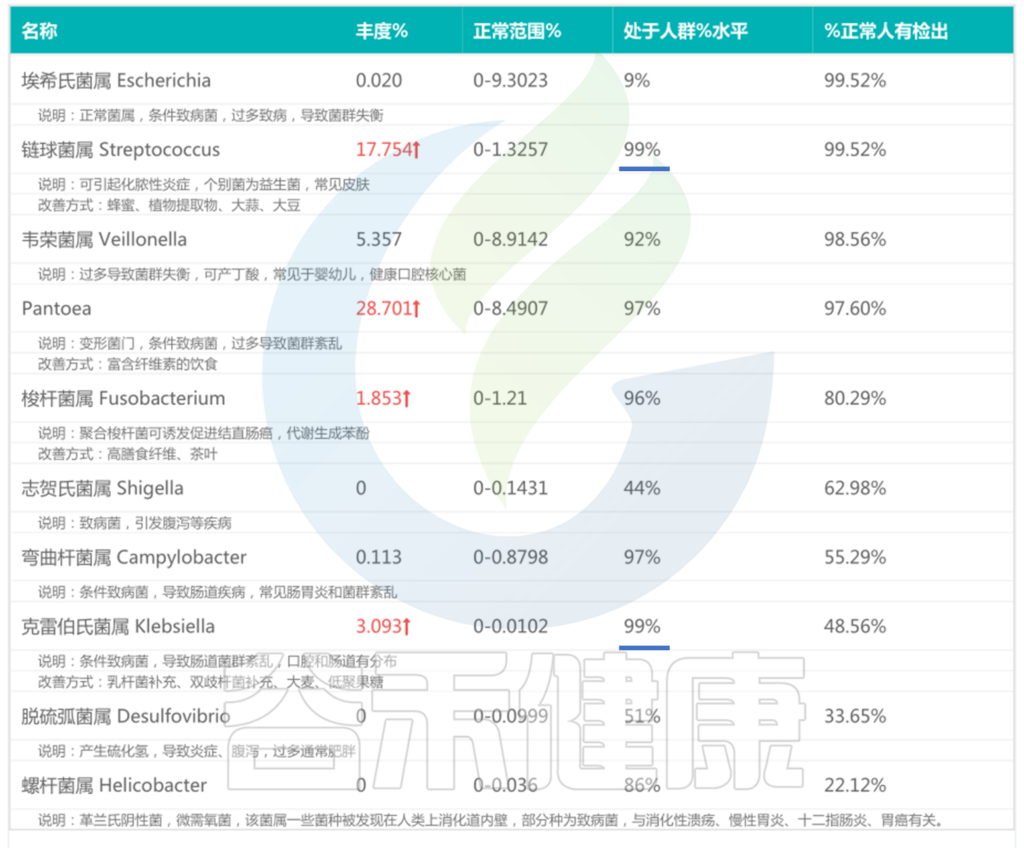
<来源:谷禾肠道菌群检测数据库>
可以看到,链球菌属、Pantoea、克雷伯氏菌属等有害菌远高于正常水平。
链球菌通常是存在于人体正常的口腔、鼻咽、肠道等部位的常见菌群,一些链球菌感染可以引起喉咙痛、扁桃体肿胀等症状。
肠道中检出的过量链球菌,可能是由呼吸道感染后通过体循环进入肠道,链球菌到了肠道可能会影响肠道的健康和功能,导致一些症状,包括腹泻、腹痛、消化不良等。
链球菌在自身免疫性疾病中也会富集,和免疫相关疾病中自身抗体的产生或Th17细胞的激活有关。
Pantoea是肠杆菌科内一个高度多样化和多变的人类机会性人类病原菌,一般由伤口或医院获得性感染引起,主要发生在免疫功能低下的个体中。在我们的肠道菌群检测实践中也发现许多健康状况不良的人群,尤其幼儿和老年患者中检出高丰度的Pantoea菌。
只有大约 20% 的病例能够在培养中分离出泛菌属物种。大多数关于Pantoea 物种可以通过适当的抗生素和支持性管理进行治疗。
克雷伯氏菌可以自然存在于健康个体的肠道和呼吸道中,具有健康免疫系统的人很少发生感染疾病。一旦克雷伯氏菌属进入体内并在免疫系统的防御中幸存下来,它们就会影响各种器官。疾病的症状通常取决于感染开始的位置。
当克雷伯氏菌属感染肠道后,由于克雷伯氏菌通过肠道分泌毒素,导致肠道蠕动加快,从而引起腹泻,同时患者可能出现腹部不适的症状。有时会伴随发热等全身反应,这可能是由于克雷伯氏菌在肠道内繁殖并释放炎症介质所致。具体症状和严重程度因人而异。
而拟杆菌的存在可以阻止肺炎克雷伯菌的肠道定殖。上述报告中拟杆菌丰度并不高,对克雷伯菌的抑制程度有限。
易感因素包括营养不良,接触程度,不卫生的环境、酒精、年龄(衰老)、遗传易感性、药物、慢性肝病、糖尿病、大型手术等。
以上我们可以看到仅这三种有害菌的丰度加起来已经占一半了,那么核心菌属自然就没有生长空间,肠道菌群整体趋于失调。
有害菌过多会影响肠道内环境,如pH值,含氧量,肠道内毒素等,可能会导致出现一些机会感染和机会致病菌入侵,进而诱发肠道炎症和相关疾病。

➤➤ 便秘患者:
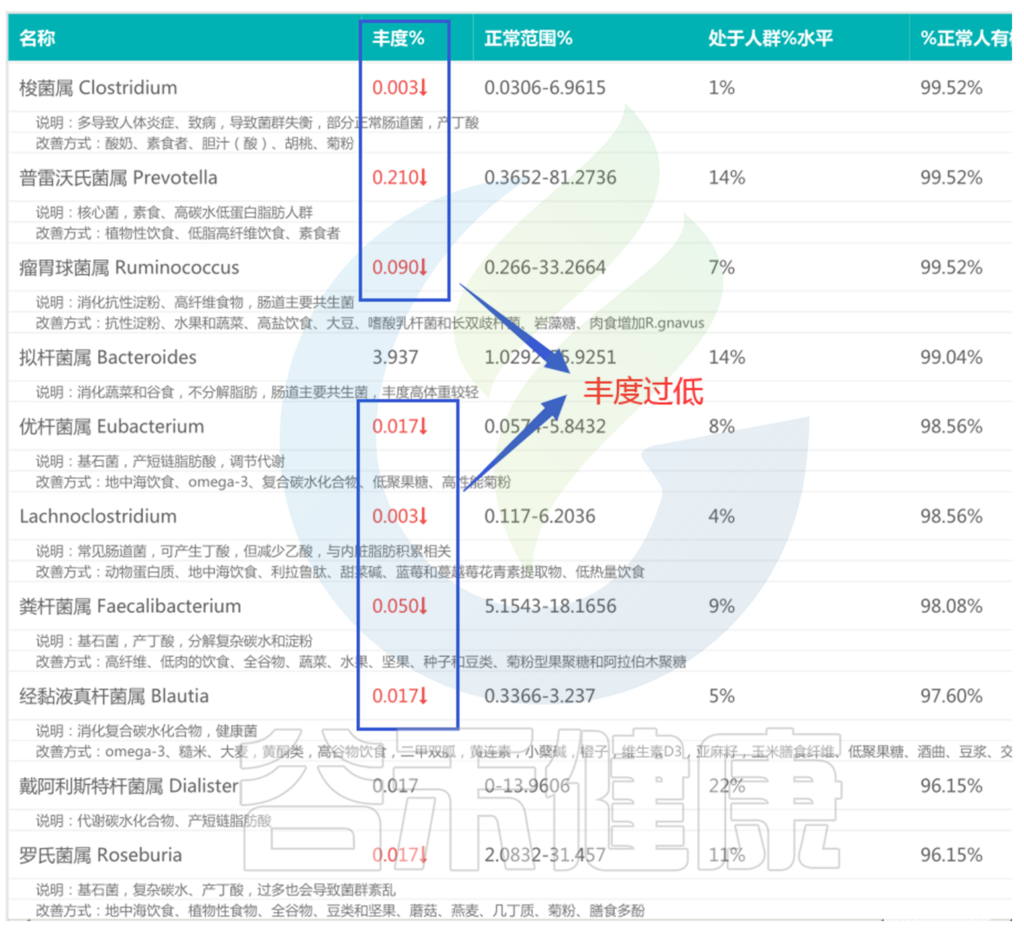
<来源:谷禾肠道菌群检测数据库>
同样,在便秘患者的肠道菌群检测报告中,部分核心菌属丰度是过低的。
上述报告中,与产丁酸相关的核心菌属丰度较低。有研究表明,便秘患者可能出现肠道菌群和丁酸代谢失调。
核心菌属丰度低,伴随着部分有害菌的丰度大幅上升,肠道菌群多样性下降。
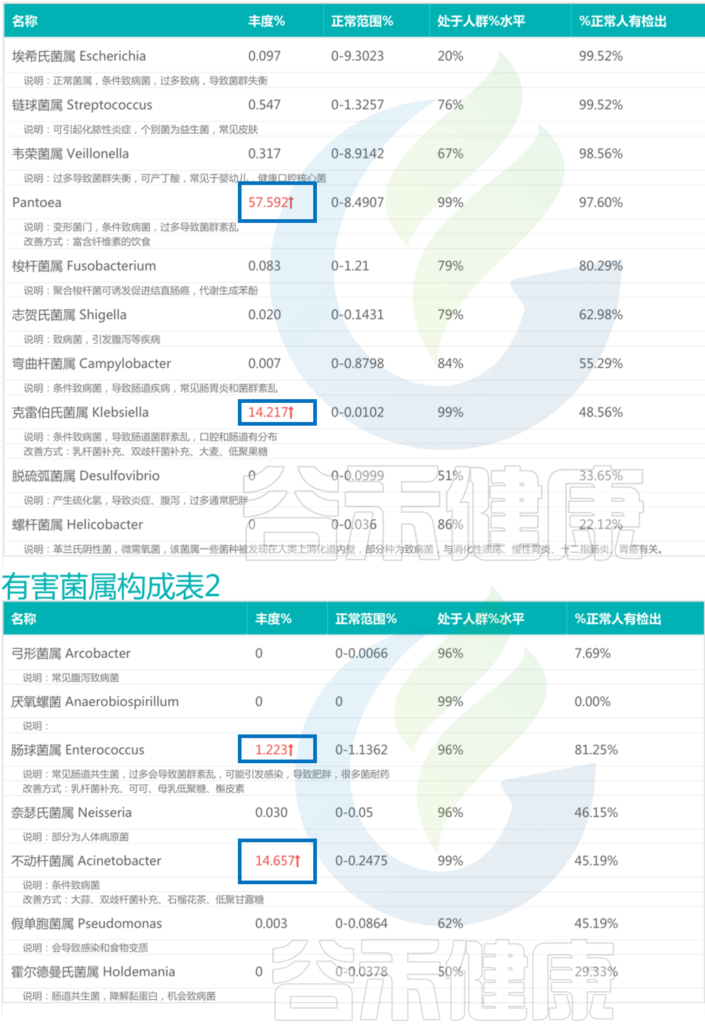
<来源:谷禾肠道菌群检测数据库>
不动杆菌属Acinetobacter是一类革兰氏阴性菌,该菌属于常见的医院感染病原体之一。不动杆菌属感染常见的病症包括呼吸道感染、泌尿道感染、创伤感染等,严重情况下还可能导致败血症。
不动杆菌属对常见抗生素经常会产生抗药性。这里不动杆菌属丰度较高可能与抗生素使用较多相关。
泛菌属Pantoea:57.592%
克雷伯氏菌属:14.217%
肠球菌属:1.223%
不动杆菌属:14.657%
以上四种有害菌加起来已经超过85%,核心菌属的生存空间受到严重影响,整体丰度很低,肠道多样性下降,这可能与便秘相关。
以上是核心菌属低,有害菌大量入侵的情况,那么如果核心菌属丰度整体很高,是不是意味着健康的肠道菌群?
不一定。也有可能出现核心菌属内部不平衡,个别核心菌属疯长的情况。
肠易激综合征、消化不良患者:
<来源:谷禾肠道菌群检测数据库>
可以看到虽然核心菌属整体占比较高,但是个别核心菌属丰度过高,例如:拟杆菌、Lachnoclostridium菌群丰度属于过高,那么其他核心菌属的生存空间就会受到影响,这就导致菌群单一化,多样性下降,肠道菌群稳定性较差。
多项研究表明拟杆菌、普拉梭菌、瘤胃球菌属和双歧杆菌与肠易激综合征相关。其中拟杆菌的过度生长可能与肠易激综合征的症状加重相关。
研究表明,肠易激综合征患者中短链脂肪酸水平降低,尤其是丁酸盐,上述报告中与产丁酸相关的核心菌属如粪杆菌属,罗氏菌属等都有所下降。
同时,与消化复合碳水化合物相关的经黏液真杆菌属丰度也较低,可能与患者的消化不良有关。
以上是核心菌属异常的情况,健康人的肠道菌群检测报告中核心菌属具体如何?
这里从谷禾肠道菌群检测数据库选取一例健康人的报告,健康总分在89分。
注:在谷禾肠道菌群检测报告评分机制中,超过80分已经算很健康的状态。

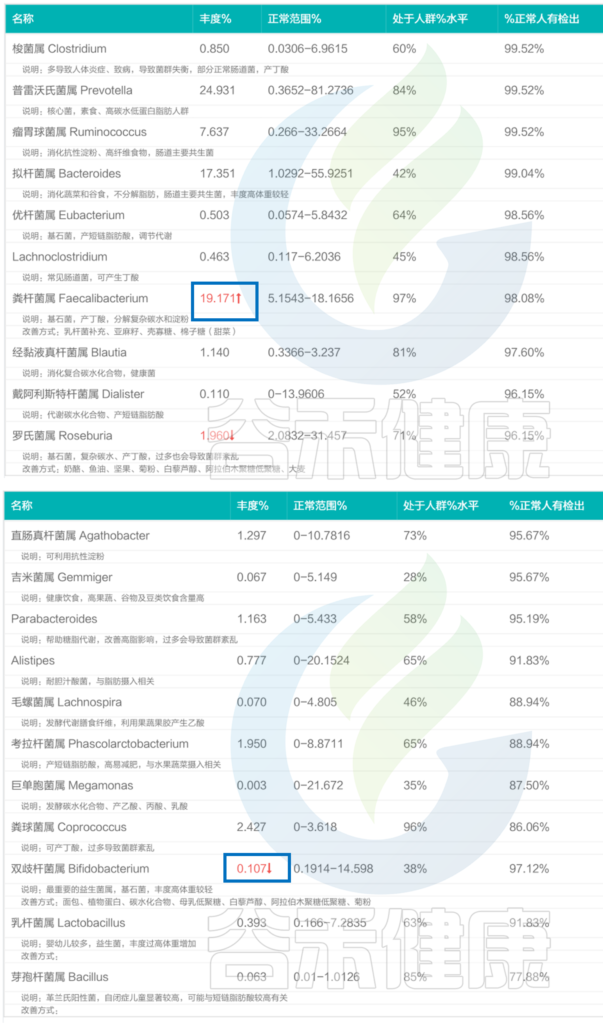
其核心菌属丰度如下:
<来源:谷禾肠道菌群检测数据库>
可以看到,健康人的核心菌属绝大部分都在正常范围内,其肠道菌群结构非常稳定。
当然也有个别菌群超标。其中粪杆菌属丰度为19.171%,比正常范围的上限略高一点点,而粪杆菌属,可以帮助分解膳食纤维,产短链脂肪酸,有助于维持肠黏膜健康、增强免疫系统功能,维持肠道内长期的稳定状态。
其他像罗氏菌属的丰度在1.960%,双歧杆菌属丰度在0.107%,略微低于正常水平,对整体健康基本不构成威胁。
还有像芽孢杆菌属在一些亚健康人群中有很多未检出或者极低,而该报告中芽孢杆菌属能检测到且丰度属于正常水平,有助于提升其整体肠道菌群结构的稳定性。
我们需要关注肠道菌群中的核心菌属,因为这些菌属在整个人体肠道菌群中扮演着网络节点或基础菌群的角色。核心菌属及其重要菌属的紊乱会直接影响营养物质的吸收、代谢产物的合理构成以及维持肠道正常生理环境的稳定性等。
如何保持这些核心菌属的健康水平呢?
一方面要坚持通过健康的饮食和生活状态来实现。尽量选择多样化新鲜卫生的食物、富含含欧米伽-3的不饱和脂肪酸,平衡动物和植物蛋白,更多的十字花科蔬菜、富含多酚的水果、根茎类蔬菜以及合理的脂肪。
另外一方面,了解肠道健康和菌群状态,针对性的调整异常菌属和菌群结构,保持核心菌属的平衡和喂养,才能构筑一个强大的菌群网络结构,维护正常的免疫状态和身体健康。
世界肠道健康日,希望每个人都拥有更健康的肠道和更好的身体~

谷禾健康

如果你是一名科研人员,在研究的过程中需要用到代码,那么你可能不需要像专业码农那样从头到尾一句一句去写完整的,而是可以将网上的一段符合应用场景的现成代码拿过来直接用。
这听起来是不是很简单?然而实际上…
目前,虽然学术界有资源可以用来编写更好的代码,但很少关注使用他人的代码可能遇到的问题,比如:有时候看着别人的代码过于繁琐、晦涩难懂;又或者绕进去,反而忘了自己一开始要干什么;或者添油加醋最后一团乱麻;甚至即便是费了九牛二虎之力搞懂了,deadline却已经在眼前了…
学会使用他人的代码是现代学术生活的重要组成部分。
本文主要在将行业、现有文献和一些科研人员自己的经验结合起来,为学术研究人员提供相对实用的十条规则。
这10条规则可以分为计划、理解、更改和发布类别。
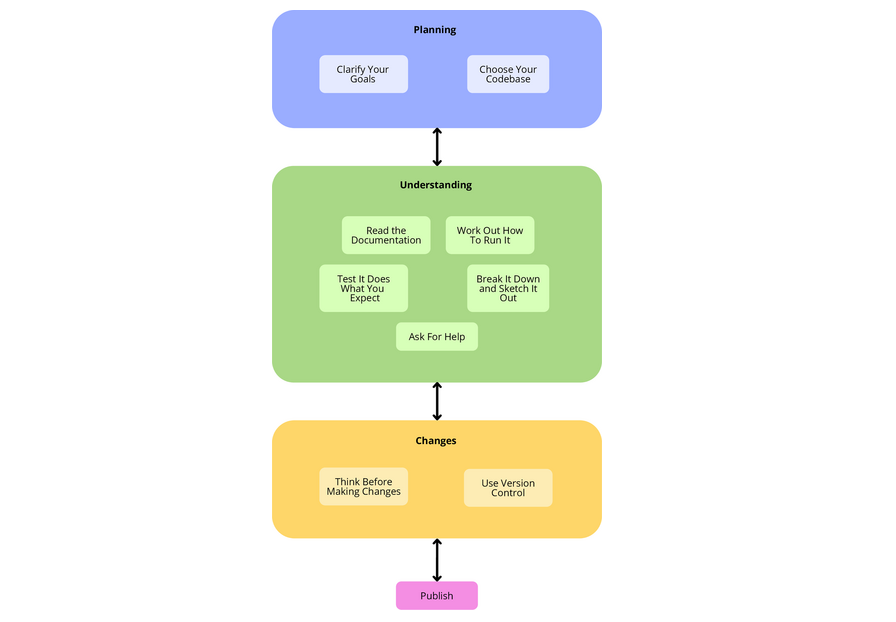
doi.org/10.1371/journal.pcbi.1011031.g002
下面我们就来详细看一下。
在正式开始动手写之前,先想一想,你真正想要实现的目标是什么?
是需要计算一些东西吗?
还是需要添加一些代码中当前不存在的功能?

不同的目标需要对应不同的方法。
如果需要计算,那么关键的要求是确信计算是正确的;而如果要添加某些功能,那么可能需要更多地理解代码。
试想一下,是不是每次都需要通过浏览整个代码库,理解每一行代码来实现呢?
明确目标会防止你做无用功,提升效率。这是一个有用的练习。
当你建立对代码的理解时,你的目标可能会改变,这会影响到你如何改变以及是否做出改变。
在学习或者写代码的过程中,要记住这个一开始就已明确的目标,一旦发现行动走偏的时候不断把自己拉回来,不断回到问题,从而以轻松高效的方式完成。
如果要使用别人写好的程序,你需要在网上搜索,选择一个合适的代码库。那么问题来了,所谓的“合适的代码库”怎么选呢?
几个要点:
当然,其他还可以考虑:
代码库的可访问性、代码的可读性。
也可以考虑选择其他熟悉的编程语言。
有时候别人的代码看不懂,自己的又跑不通,很容易把它全部扔掉,然后重新开始。
到底是否需要重新写一遍代码?
重新写一遍代码也是一种选择。
➪ 好处是可以让你更深入地理解代码的功能和运作方式,也就是说可以学到深度知识,这些知识在使用他人代码时很难挖掘到。
一位计算机科学博士说,他们更喜欢编写自己版本的算法来完全理解它们,一旦完成,他们就会使用现有的库。
➪ 主要缺点是复制已经完成的工作,这可能很耗时,也不必要。
当然,可以阅读现有的代码和文档,了解如何构建自己的版本。也可以使用旧代码来测试新代码,并检查是否正常。
同时也别忘了第一条,明确目标,从而确定是不是要重新写一遍。
►►
使用哪种代码库以及是否重新开始可能都是艰难的选择。重要的是,不要急于做决定,花时间了解别人的代码,再考虑该怎么做。
阅读文档有什么用?
文档,就像是代码的翻译官,可以帮你理解它们。虽然不需要像看小说一样读完整个文档,但是拿来当参考还是蛮重要的。刚开始接触他人的代码时,简单地浏览一下文档可以帮助你大概了解它们的结构。
文档里有哪些内容?
如果卡在了某个问题上,也可以去文档里找答案。文档通常会包含一些重要、难懂的部分,这些部分我们读了以后会更清楚。即使文档不太好或者太少,也应该去看一看。如果文档实在少得可怜,那么这说明代码可能不太靠谱,不是遵循最佳实践编写的。
文档不仅包括README文件或在线API,还包括代码注释、文档字符串、变量和函数名,以及任何写下来的测试。好的项目通常会有在线文档,里面可能包含“入门教程”,对你上手很有帮助。
如果文档不存在(或不正确),自己加文档
这不仅对你自己很有帮助,而且还能帮助未来其他使用代码的人。文档不一定要多么正式或完美,它就是你对代码的注释。在探索代码时,你可能会记录很多笔记,把它们保存成文档,过个几周或几个月后可以更容易回到代码中学习或修改。
如何加文档?
加文档可以简单地创建一个README文件,该文件通常从较高的层次解释功能,但也可能包括安装步骤或强调如何通过API调用特定函数。另外,你也可以在代码本身中添加或更改注释,解释特定部分是如何工作的。
一个很好的建议是:
想象你下一年回到这段代码时已经忘记了所有内容,这时候你添加文档就相当于在帮助未来的自己。
使用他人代码时的一个基本问题是首先得让它跑的通。这看起来很简单,但实际上却是非常困难的一步。
很多因素导致别人的代码不一定能跑通
不同的代码需要不同的操作系统、编程语言、数据库甚至是硬件,在一台计算机上跑通的代码在另一台计算机可能不行。如果环境不匹配,代码就无法正常运行。
对于旧代码来说,这尤其是个问题——《Nature》最近的一篇文章向研究人员提出了一个问题,即他们是否可以运行10年后的研究代码,发现了与过时的环境以及不完整的文档相关的问题。
通常,运行他人代码的主要问题是使用正确版本的编程语言和代码依赖关系。编程语言和单个软件包经常更新,有时这些更改是在考虑向后兼容性的情况下进行的,因此旧代码在新版本中仍然可以工作,但有时情况并非如此。
除了编程语言和依赖性之外,使用特定的操作系统、安装特定的数据库或图形卡、传感器等硬件,甚至需要软盘驱动器等。
发现跑不通的时候应该怎么办?
——看error提示
为了弄清楚如何运行代码,最好的第一步是简单地尝试在当前环境中按原样运行它。如果不行,计算机可能会发出一个error ,让你知道出了什么问题。
——查看文档
好的文档会清楚地说明预期的环境,包括所需的编程语言和软件包版本。不幸的是,并不是所有的文档都包括这些。
——使用互联网搜索
找出编写代码时软件的最新版本,并从那里往回推。
——创建虚拟环境来运行
当你确定代码需要运行的特定环境时,可以开始在计算机上复制该环境。如果不想更改计算机上的环境,我们可以创建一个虚拟环境来运行代码。虚拟环境允许使用特定版本的编程语言和程序包运行代码。
除此之外,虚拟机可以模拟整个计算机系统(例如,在Mac上运行虚拟Windows实例)。虚拟化有很多选项,最佳选项取决于当前的计算机和要虚拟化的系统。
——目前已有的配置虚拟环境工具
可能是其他人已经制定了运行代码的虚拟化设置。例如:
Conda和DockerHub都是社区驱动的平台,你也可以贡献自己的虚拟化设置来帮助他人(以及未来的自己)。
——利用调试工具深入挖掘代码
可能你遇到的问题不仅仅是寻找合适的环境。例如,可能不清楚需要什么样的输入。当代码的其他部分出现错误时,某些功能可能会正常工作。混淆这些问题,错误信息本身可能无法为问题提供很好的线索。在这些情况下,你可能需要更深入地挖掘代码的各个部分,并尝试弄清楚它是如何工作的。这可能很耗时,需要耐心和毅力。
调试工具可以在这里提供帮助,一步一步地跟踪代码的执行,并查看正在使用的资源和数据类型。当然,代码的某些部分是有可能被破坏的,可能需要进行更严格的测试或进行更改。
为什么要做测试?
一个警示故事:
2006年,Geoffrey Chang因软件bug撤回了《科学》杂志上的几篇引人注目的论文。在这些论文中,Chang使用了一些代码来计算蛋白质的结构。
不幸的是,这个从另一个实验室继承的代码包含了一个小错误,这意味着由此产生的蛋白质结构是不正确的。如果进行更好的软件测试,这个问题可能会更早发现。
当使用他人的代码时,测试是一个好方法,可以检查代码做了什么(或没做什么),并验证它是否真的有效。
运行现有的测试
通常,一个项目已经有了测试,找到并运行这些测试应该是起点。这些通常可以在一个叫“tests”或类似的文件夹中找到,理想情况下,文档将包括运行这些测试的说明。
运行测试你会进一步熟悉代码库,也是一种衡量他人代码质量的方法。你甚至可能会发现测试失败,在这种情况下,可以考虑进行更改来修复代码。
编写自己的测试
除了现有的测试之外,你可能还想编写自己的测试。一个好的起点是健全性检查,这可以检验代码是否给出了期待的正确答案。
注:健全性检查是一种数据完整性检查方法,用于确保数据的准确性和一致性,是数据管理的重要组成部分。
健全性检查是值得的,不过自动化测试更彻底。这可以简单到通过编写测试来检查函数的输出是否正确,从而自动进行健全性检查。
——单元测试
通常,这是通过使用“assert”语句来实现的,如果输出不是预期的,则该语句会引发错误。这是一种单元测试的形式,它检查软件的特定单元,在这种情况下是一个功能,是否按预期工作。大多数编程语言都包括单元测试功能或库,其中包括断言语句以及快速轻松地运行许多测试的方法。
——功能测试
另一种常见的测试类型是功能测试,可以检查整个软件是否符合期望。
例如,你可能打算从论文中复制一个图形,这将作为对用于生成图形分析的软件的一种测试。功能测试可以是一种很好的方法,可以检查代码是否做了它应该做的事情,而不必太了解代码是如何工作的。
对于单元测试和功能测试,需要一些数据来检查结果
代码库可能已经包括了一些演示数据,这是一个很好的起点,也是代码所需数据格式的一个好例子。你可能需要添加自己的测试数据,这些数据可能是真实数据,例如在复制图形的情况下。
或者可以模拟数据,例如,可以模拟具有已知参数的数据,以检查拟合算法是否给出合理的结果。
对于真实或模拟的数据集,你可能不知道“正确”的答案,因此在可能的情况下,通常使用另一种现有工具来验证结果。
►►
总的来说,需要的测试的深度和类型将取决于你的具体目标。在许多情况下,一些简单的健全性检查就足以验证代码是否按预期工作。当对代码的质量有疑问时,以及当代码的正确性能至关重要时,需要进行更严格的测试。
一种很好的策略:
将问题分解为较小的单元或模块,然后将这些单元放在一起解决复杂的问题。
同样的事情也适用于使用其他人的代码。拆解代码并弄清楚代码的每一部分做什么,这通常比一次完成要容易得多。
这种方法的具体操作之一是:
绘制代码是如何工作的以及各部分是如何交互的。
这不需要完美,可以简单地用纸笔或黑板快速绘制草图。你可能想可视化代码的结构、数据的传递方式、哪些函数调用其他函数等。只要把一些东西写在纸上就可以帮助可视化系统。
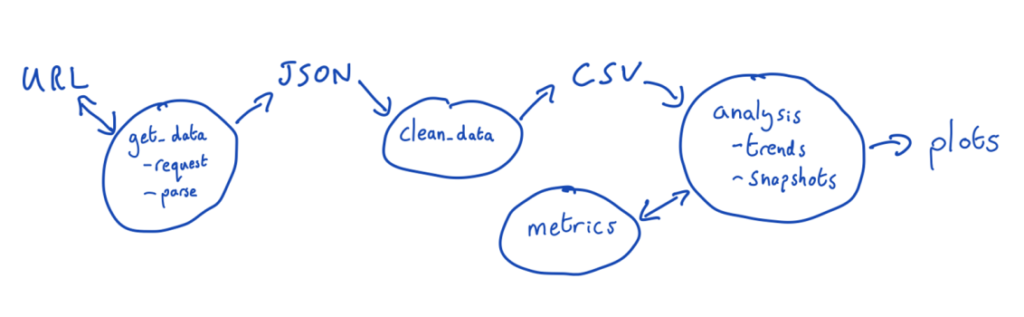
doi.org/10.1371/journal.pcbi.1011031.g002
根据项目和目标,你可能会决定通过使用可视化软件设计图形来正式化这些图纸。
可以将其包含在项目的编写中,也可以将其添加到代码库的文档中。对于那些感兴趣的人,你甚至可以使用统一建模语言,这是一种可视化软件系统的通用方法。
研究有时候也是需要合作努力。协作在处理代码时尤其有用,因为每个人在各种编程语言和范式中具有不同的知识和技能水平。利用现有知识是提高自己研究效率的一个很好的策略。
获得帮助的途径有很多:
➤ 谷歌(或其他搜索引擎)
可以依靠搜索引擎来帮助写代码,一项研究发现,开发人员花费了大约20%的时间搜索网络。当涉及到使用他人的代码时,包括项目名称在内的谷歌搜索可以显示在线资源,如代码库、教程、论坛等。谷歌是一个查找代码片段、简短解释和诊断错误的好资源。
-小tips:
一个好的搜索策略是:直接把错误消息复制粘贴到谷歌中。
➤ StackOverflow
StackOverflow是一个有用的资源,经常出现在谷歌搜索结果中。该在线门户允许用户询问和回答特定的编码问题。
一般来说,你的问题已经有人问过并回答过问题。如果你无法通过搜索找到解决方案,那么也可以自己问,基本上会很快得到答案。
也有可能你正在处理的代码库过于模糊,无法直接引用。就算这样,也可以发布代码块或提出更一般的问题并获得帮助。
其他有用的门户网站包括W3Schools和Quora,还有专门研究特定研究领域的论坛,如生物信息学的BioStars。
➤ github
许多项目都会在GitHub上托管代码,这是托管和共享代码库的行业和学术标准。
如果你对代码有问题,比如bug、建议的新功能,或者只是一个问题,那么你可以在GitHub Issues中提出这个问题,项目开发人员会回答或解决这个问题。这是最佳实践,也是许多开发人员提出问题的首选方式。
在提出问题之前,可以浏览你过去的问题,看看是否得到了解决。
➤ 其他项目门户网站
正在进行的软件项目通常有各种在线资源来获得帮助。这可能包括Slack、Gitter、Discord和论坛等平台。这些可以从GitHub页面链接,也可以通过谷歌搜索找到。
➤ 合作者
如果你有合作者,可以问他们。他们可能愿意提供帮助,甚至可能在过去使用过该代码。
➤ 其他研究人员
很有可能其他人以前也使用过该代码。你可以查看引用该软件或相关研究文章的论文。
如果代码托管在GitHub上,可以在存储库页面上检查问题,或者查看它是否已被分叉。或者在GitHub中搜索存储库名称并查找其他版本。
如果运气好的话,可能已经有人熟悉了这个代码库,他们可能会愿意为你提供资源,甚至成为你的合作伙伴。
➤ 原作者
值得与原作者联系,他们应该愿意回答特定的问题,甚至提供一些在网上找不到的文档,或者他们可能也想合作。你用他们的代码,他们也会高兴。如果你们合作起来,说不定他们会为你增加一些新功能或修复一些bug呢。
➤ 研究软件工程师
根据你所在的机构,你可能会获得研究软件工程师团队的帮助。这可以是一般性建议或支持的形式,也可以是他们愿意承担与代码库相关的项目并跟你合作。

当我们想要使用别人的代码时,总是想着要去写代码,改进代码。但是有没有想过:
我们真的需要去改变它吗?
其实,完成任务的最有效方法是:意识到你根本不需要去做它。当然,有时候改变是必要的,但在动手之前,先花点时间思考一下。

改变代码可能会带来很多意想不到的麻烦,特别是当你不太了解代码的运作方式或者理解不到位时,这种后果就更加可能发生。有些你看起来垃圾的代码可能是出于某种重要的原因而写成这样的。一旦你改变了代码,你就改变了代码的本质,不能确定它是按预期的,还是因为你的改变引起的。
关于修改代码,有本书叫《代码修改的艺术(Working Effectively with Legacy Code)》

改代码的目的不是要破坏原有功能,而是尽可能有效地完成改动。所以,我们需要明确改动的目的,只做必要的改动。
比如,你在修复bug时发现一个循环可以优化,但是要忍住不要掉入这种陷阱!这个循环的效率可能并不重要,每一次改动都可能引发严重问题。就像计算机科学家Donald Knuth说的,在大多数情况下,我们不应该过于关注代码的小细节,过早地优化是所有问题的根源。
改代码的最佳实践应该是什么样的?
在你做任何改动之前,最好先写一些单元测试,覆盖现有的功能,以及覆盖你想要添加的新功能。一旦做出改变,你可以运行这些测试来检查是否破坏了任何东西,更改是否生效。这个过程相对简单。
比如,如果你要重构一个特定的函数,你可以先写一些自动化测试,基于当前函数的输出(也就是特征测试)。改了代码后,你可以运行这些测试,确保重构没有改变功能。另外,记得把改过的地方记录在文档中,包括更改的内容,为什么要更改等。
版本控制是备份、共享、协作和跟踪代码的最佳方式。
很多人会通过电子邮件、共享文件夹来传输代码,或者根本不备份,如果你不经常使用版本控制,那么这条很重要。
当我们谈到版本控制时,通常会想到Git和GitHub。GitHub提供免费账户,其中包括无限的代码存储库,可以是私有的,也可以是公共的。
还有其他选择,比如BitBucket。如果代码(或数据)是敏感的,不能发送到GitHub的服务器上,那么还有像GitLab这样的选择,可以让你运行自己的私人的Git服务器。
学习使用Git难不难?有哪些资源推荐?
学习使用Git实际上入门很简单。你不需要成为专家,只要能够推动和拉动更改就足够了。在这里,我们不会重复介绍如何使用Git的指南,已经有很多现成的资源了。
对于初学者建议阅读PLOS文章《使用Git和GitHub进行版本控制的快速介绍》。
对于更感兴趣的人,Pro Git书籍是一个有用的资源,还有许多其他的在线指南和教程。
如果你拿来的代码没有存储在版本控制的存储库中怎么办?
遇到这样的情况,你可以开始将现有代码移动到一个Git存储库中,并创建一个起始提交,将所有文件添加进去。你自己所做的任何更改都可以像往常一样进行跟踪,使用后续的提交。这样做可以确保始终可以将代码恢复到以前的状态,即使引入了错误,也不用担心继承的原始版本代码丢失。
Git还有什么其他功能?
对于高级用户,Git还提供了一系列有用的工具,例如GitHub Actions,可以自动化工作流程的某些部分。这可以包括自动化测试,检查代码更改是否破坏了任何东西,并且项目中的测试是否仍然能通过。
Git还允许轻松对代码进行版本控制,便于你可以将代码库回溯到特定版本或时间,这对于研究的可重复性非常重要。

根据你的目标,你可能已经对代码和/或文档进行了更改。可以考虑分享这些更改。
为什么要在网上发布代码?有什么好处?
——为研究社区做出贡献
很多人可能和你一样,也有着相同的目标,你的更改可以为他们节省大量时间和困难。
——帮助你的职业发展
在线发布代码将建立你的在线个人资料和声誉,这在申请工作或其他机会时非常有帮助。
——优化代码、书写更加规范
可能有其他人帮你的代码库做出贡献和改进,或者只是发现你忽略的bug。
发布代码是一个好习惯。通常,如果我们知道代码将被发布,那就会有意识地写得更加规范,避免那些可能带来问题的懒惰捷径。
在研究项目的同时发布代码的趋势越来越明显,如果你已经养成了这个习惯,就会变得更加容易。
——增加合作机会
其他研究人员看到你的代码,可能会联系你进行合作。发布代码还将使其他人或者你自己在未来更容易地复制你的工作。
有研究人员讲述了这么一个故事:
在他们发布了几年后有人联系他们合作一个类似的项目,这是一个很好的机会,但他们差点拒绝了——因为他们找不到代码,之前写在一台旧笔记本电脑上的,现在已经无法访问了。幸运的是,他们意识到当时已经在GitHub上发布了代码,因此才有可能达成合作。

GitHub是共享代码的标准平台
在研究和工业领域,共享代码的标准是GitHub。如果代码已经作为现有的GitHub存储库发布,那么你可以“fork”创建一个包含更改的存储库副本。根据这些更改的内容,你甚至可以向现有存储库发起“pull request”,将你的更改合并到原始代码库中。
发布更改前需检查软件许可证
如果没有现有的存储库,那么你可以创建一个新的存储库。但是,在发布更改之前,需要注意一点:最好检查一下软件许可证是否允许这样做。
许可证通常在主项目目录中,命名为“LICENSE.txt”或类似的名称。虽然许多研究软件都是在宽松的开源许可证下发布的,但并不是每一个都这样,需要进行检查。即使是开源许可证也可能有一些条件,例如必须注明原作者。
以上总结了使用他人的代码的一些经验,供大家参考交流。学会用好他人的代码,对于自身代码水平的提升也有帮助。毕竟在接触不同的编码实践、设计模式、工具的过程中,你的编程知识和视野都在拓宽。
希望本文的一些驾驭源代码的技巧和心法,能帮你在科研的路上走得更稳,更加高效地得到想要的结果。当然你也可能有自己的心得体会,欢迎分享。
参考文献
Pilgrim C, Kent P, Hosseini K, Chalstrey E. Ten simple rules for working with other people’s code. PLoS Comput Biol. 2023 Apr 20;19(4):e1011031.

谷禾健康

大多数细菌对我们的生活是有帮助和必要的,某些细菌可以帮助消化,为身体提供能量,分解毒素,保护肠道,增强免疫力等,从而有益健康;也有一些细菌会给我们的健康带来一些危害。
然而有些菌并不是天生注定就是致病菌,也许正常状况下,它只是体内默默地存在着,但可能在某种特定条件下突然变身,彰显其致病威力。
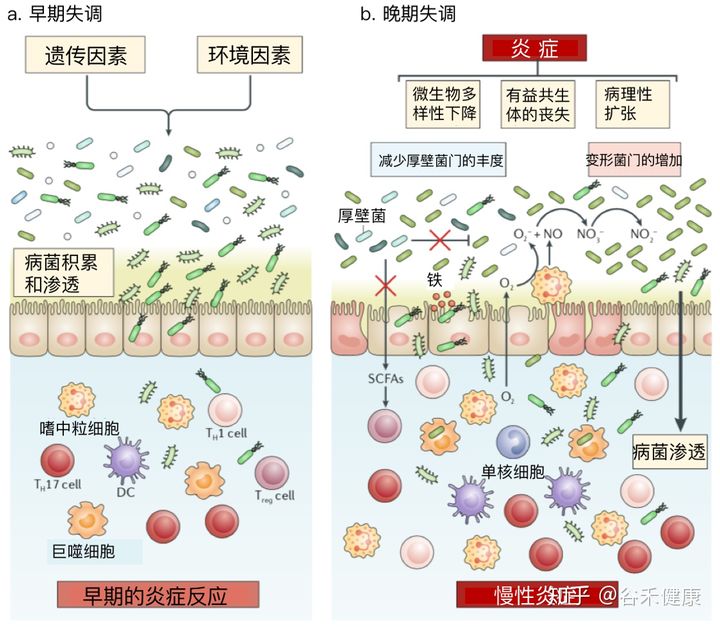
事实上,许多严重的疾病是由健康个体的皮肤、粘膜或肠道中常见的细菌引起的。在这些情况下,致病菌根本不是专性病原体,而是遵循新的生态轨迹的共生体,通常会迁移到与宿主不再和谐相处的侵入性生态位。
细菌从共生到致病的转变在肺炎、脑膜炎、全身感染和医院获得性感染等疾病中发挥重要作用。当然,宿主环境扰动可能会提供感染机会。
因此,我们需要对致病菌的个性、不同环境下的状态有个基本的了解。
在谷禾肠道菌群健康检测报告中,我们会看到关于有益菌,有害菌,致病菌的检测:
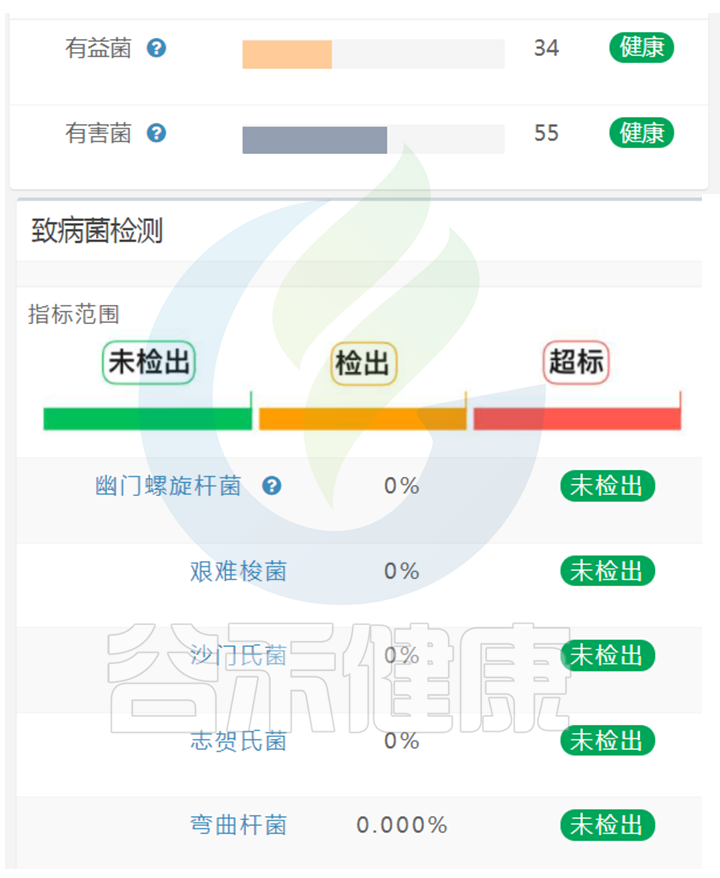
有小伙伴有疑问,这里既显示有害菌又有致病菌,有害菌不是致病菌吗?
携带致病菌就一定会生病吗?
条件致病菌是如何引起感染的?
有害菌很多,该如何改善?
…
本文也将围绕着这些问题,展开一些讨论。
致病菌是引起疾病的细菌,也称为病原菌。当它进入身体时,就会破坏细胞或干扰身体的正常活动,人可能会患上轻微疾病或致命疾病。
✦ +
致病菌通过多种方式导致宿主生病。最明显的方法是在复制过程中直接破坏组织或细胞,通常是通过产生毒素,使病原体到达新组织或离开它复制的细胞。细菌毒素是已知的最致命的毒物之一,包括著名的例子,如破伤风、炭疽等。
然而,对宿主的损害通常是通过强烈的或有时是过度的免疫反应自行造成的,这种免疫反应会不加选择地杀死受感染和未感染的细胞并损害宿主组织。免疫系统过度反应的典型例子包括乙型肝炎中的肝硬化和肝癌。
一些病原体受益于宿主的免疫反应,可以在受感染的宿主内传播或增加它们向未感染宿主的传播。
流感主要通过它引起的打喷嚏和咳嗽产生的气溶胶传播。
霍乱弧菌在肠道粘膜中引发强烈的炎症反应,导致水样腹泻,并确保其在环境中的释放,从而感染更多的宿主。
✦ +
从概念上来讲,致病菌和病原菌意思差不多。需要注意的是,这里我们报告用的两个词分别是:
肠道致病菌,病原菌。
✦ +
正常情况下,肠道内是存在少量的病原菌,但是其丰度相对较低,丰度低于健康人群98%以下,不一定会导致疾病的发生。
这里我们可以了解一下关于“细菌感染”:
细菌感染:
病原菌侵入宿主体内并引起病理变化称为“感染”。
也就是说,如果报告中只是检出极少的病原菌,同时并没有症状,那么可能只是表面有病原菌摄入,需要注意饮食和生活卫生,这并不能称之为病原菌感染,因此不需要过于恐慌。
如果报告中病原菌检出已经超过98%人群,则代表可能存在感染的风险,需要结合相应症状和具体菌群丰度比例综合进行判断。
此外,如果出现多种病原菌或病原菌丰度水平很高,则需要引起注意。
✦ +
需要注意的是,谷禾肠道菌群检测报告中的疾病风险,是综合了多项指标判别的,并不只是考虑了致病菌。
慢病是每一种病单独构建模型,不一定和有害菌或菌群平衡指标直接有关。
下面我们来看一些致病菌,这些致病菌一旦感染可能会给人体带来较大危害。
✦ +
★ 炭疽杆菌 Bacillus anthracis
炭疽杆菌是革兰氏阳性、非运动、兼性厌氧、孢子形成和杆状细菌。
炭疽杆菌是一种人畜共患病的病原体,是一种专性病原体,因为细菌的繁殖周期只发生在合适的宿主中。
编辑
图源:tvmdl.tamu.edu . by Mallory Pfeifer
炭疽杆菌的发病机制主要由两种毒力因子引起:三方外毒素和聚γ-d-谷氨酸(γ-DPGA)。
人类可以通过四种不同的方式感染炭疽:吸入、摄入、通过皮肤和注射。
吸入性炭疽被认为是最致命的炭疽形式。如果不进行治疗,病死率接近95%,而立即干预可以将病死率降至50%。
胃肠道炭疽的发生是由于食用了来自感染炭疽的动物制备不当的肉类或肉制品。死亡率是可变的,通过适当的抗生素治疗可以达到≤40%.
皮肤炭疽通常通过皮肤接触受感染的动物或动物产品而发生。这种形式的炭疽病占全球人类病例的 95%。治疗后病死率<1%。潜伏期为 3-7 天。临床表现从轻度到重度不等。
抗生素疗法用于治疗炭疽感染。青霉素、强力霉素、环丙沙星。抗生素治疗必须在接触后立即开始,因为其有效性会随着毒血症的进展而降低。炭疽疫苗在治疗中也很重要。在确诊或疑似接触炭疽孢子的情况下,建议进行 60 天抗生素治疗。
★ 鼠疫耶尔森菌 Yersinia pestis
鼠疫耶尔森菌是一种小型、非运动的革兰氏阴性细菌,属于肠杆菌科。鼠疫耶尔森氏菌是鼠疫的病原体,鼠疫是一种罕见但高度致命的人畜共患病。
图源:onlinebiologynotes
大多数人类感染是由于与受感染的动物接触或被受感染的跳蚤叮咬造成的。
鼠疫在全球超过 25 个国家流行。尽管有有效的抗生素治疗,流行地区的死亡率仍超过 10%,这主要是由于发病机制的快速发展。
鼠疫主要以三种形式发生:肺鼠疫、腺鼠疫和败血症鼠疫。
鼠疫最严重的表现,发展最快的是肺鼠疫,在没有治疗的情况下死亡率接近100%.
肺鼠疫通过呼吸道飞沫在人与人之间传播。经过 2-4 天的潜伏期后,疾病的各种症状包括发烧、头痛、恶心、不适、呕吐、咳痰带血、呼吸困难和胸痛。如果在症状出现后 24 小时内给予适当的抗生素治疗,死亡率可降低高达 50%。
腺泡形式是最常见的,腺鼠疫是鼠疫的主要形式,占病例的 80-95%。死亡率为 10–20%。腺鼠疫由受感染的跳蚤叮咬引起。腺鼠疫的特点是形成淋巴结(淋巴结肿大)。通常的潜伏期从 2 到 6 天不等,有时更长。
如果腺鼠疫没有在适当的时间得到诊断和治疗,它会通过血液传播细菌而发展成败血性鼠疫。这种形式的瘟疫也可能是由传染性跳蚤叮咬通过受损的皮肤或粘膜直接进入鼠疫耶尔森氏菌引起的。通常的潜伏期为 2-7 天,但这种类型的鼠疫甚至在临床表现出现之前就可能导致死亡。
败血性鼠疫的症状包括腹痛、皮肤和其他器官出血。皮肤和其他组织可能会坏死,尤其是鼻子、手指和脚趾。此外,可以观察到发烧、腹泻、呕吐和虚弱。败血症性鼠疫以高菌血症为特征,并伴有危险的内毒素血症。
★ 土拉弗朗西斯菌 Francisella tularensis
土拉弗朗西斯菌是一种革兰氏阴性、非运动、非产孢球杆菌。它是一种小的细胞内病原体,具有高毒力和低感染剂量(1-10 个细胞)的特点。
细菌可以通过接触受污染的水进一步传播。吸入受感染的气溶胶,或直接接触受感染动物的组织和液体,也可用作细菌传播的途径。
细菌进入体内后,在局部繁殖,引起溃疡和坏死,然后侵入血液和淋巴管,扩散至肝、脾、肺、肾、浆膜、骨髓等淋巴结和器官,引起多发性凝固性坏死灶。
图源:DeviantArt
土拉弗朗西斯菌不仅容易在巨噬细胞中生长,而且还可以感染许多其他细胞类型,如上皮细胞、肝细胞、肌肉细胞和中性粒细胞。
有六种主要形式的土拉菌病,根据症状分类:肺病、腺病、溃疡腺病、口咽病、伤寒病和眼腺病。
该病的潜伏期通常为暴露后3-5天。
溃疡腺形式是最常见的,是节肢动物媒介叮咬或在与受感染动物接触期间通过皮肤获得感染的结果。
细菌通过磨损进入生物体后,可能会发生腺体形式的土拉菌病。
食用未煮熟、受感染的食物或受污染的水后,可能会形成口咽形式,之后细菌会感染咽部。患者常出现发热、咽痛、颈淋巴结肿大伴耳下淋巴结受累。
在疾病的严重形式中,可能会出现由于肠溃疡导致的胃肠道出血。
当细菌进入循环系统并从另一个感染部位扩散到肺部时,就会出现继发性肺炎性土拉菌病。症状包括高烧、干咳、胸痛和肺门淋巴结肿大;也可能出现肺部浸润或胸腔积液。
伤寒形式的主要症状是发烧、发冷和严重疲劳。然后出现呕吐、腹泻、谵妄和腹痛。临床表现还包括全身疲劳、败血症和死亡。伤寒形式是最难诊断的,因为它的一般症状没有明显的外部病变或区域淋巴结肿胀。
★ 肉毒杆菌 Clostridium botulinum
革兰氏阳性菌,厌氧菌,可运动,致病菌。
它会引起一种罕见但严重的疾病。产生一种特殊类型的外毒素,通过抑制神经肌肉连接的活动来影响神经系统。因此,肉毒杆菌毒素表现为一种神经毒素,它会阻止神经递质的释放。
图源:Science Photo Library
肉毒中毒的症状通常始于控制眼睛,面部,嘴巴和喉咙的肌肉无力。
这种无力可能会蔓延到脖子,手臂,躯干和腿部。肉毒杆菌中毒还会削弱呼吸所涉及的肌肉,从而导致呼吸困难甚至死亡。
分为五种传播类型:
预防:
包括减少微生物污染水平,酸化,减少水分水平,以及尽可能破坏食物中所有肉毒杆菌孢子。
易感食物包括罐装芦笋、绿豆、油蒜、玉米、汤、熟橄榄、金枪鱼、香肠、午餐肉、发酵肉、沙拉酱和熏鱼。食用前,考虑将这些食物煮沸10分钟。
以上列举了一些常见的致病菌。致病菌与非致病菌并不是绝对的,是一个动态作用的过程。我们知道肠道中还有大量其他菌群,这些菌群在特定条件下,也可能转为致病菌,我们把这类细菌称为条件致病菌,接下来章节我们来详细了解一下条件致病菌的特点。
正常菌群与宿主、其他菌群之间,通过营养竞争、代谢产物的相互制约等因素,维持着良好的生存平衡。
然而,在一定条件下这种平衡关系被打破,一部分平时看起来正常的细菌就开始“作妖”,变成可以致病的细菌,这就是所谓的 “条件致病菌”。
条件致病菌包括引起肺炎、血流感染、脑膜炎和其他疾病的细菌。它们存在于环境中,可以通过皮肤上的伤口或吸入含有细菌的灰尘进入人体。
健康的免疫系统可以对抗许多病原体,但如果它受损,条件致病菌通常会引起感染。
与免疫系统受损相关的因素有:
遗传易感性;
癌症化疗;
给予免疫抑制药物以防止移植后排斥反应;
艾滋病感染;
严重营养不良;
长期抗生素治疗;
天生免疫系统较弱的婴儿和老年人;
导致白细胞生成减少的骨髓疾病;
怀孕;
正所谓 “橘生淮南则为橘,生于淮北则为枳”。某些细菌在肠道内是乖乖成长的,可一旦冲破束缚,例如在肠漏的情况下,细菌或其代谢物泄露到循环中,可能会变身“有害菌”,可以通过诱导慢性或急性炎症反应,导致疾病发生,包括损害肝脏和胰腺等重要器官,肝癌和胰腺癌可能与细菌易位有关。
如果没有健康的器官和器官系统来适当调节正常的身体功能,条件致病菌就有下手的机会,利用此环境推动疾病的发生。
手术通常涉及切口和伤口,这些切口和伤口为病原体进入身体创造了入口。这也为条件致病菌创造了有利条件。
长期使用会破坏体内正常的微生物群,杀死有益细菌,并导致条件致病菌增殖。
其实细菌(即便是致病菌),它们要生存下来并没有我们想象的容易,生活在复杂的群落中,多个物种和菌株存在相互竞争。
例如,在共生葡萄球菌中,分泌蛋白酶的表皮葡萄球菌菌株,会抑制金黄色葡萄球菌的生物膜形成和鼻腔定植。此外,生物膜破坏介导与其他物种的相互作用,并增加金黄色葡萄球菌对宿主免疫反应成分的易感性。可以看到宿主和共生细菌发出协同反应以排除其他物种。
然而你以为只有宿主和其他菌会带来威胁吗?不,另一种威胁来自与它们同居的同一物种的另一个谱系,同种内部也会互相竞争。这种种内争夺优势的斗争在金黄色葡萄球菌中得到了例证。
关于细菌之间如何交流详见:
金黄色葡萄球菌
Staphylococcus aureus
金黄色葡萄球菌是一种需氧的革兰氏阳性细菌,通常在健康人的鼻腔和皮肤上定植。
但当存在于假体关节和静脉输液管等内部装置中时,可能会导致感染,感染可能导致严重的败血症。
铜绿假单胞菌
Pseudomonas aeruginosa
铜绿假单胞菌是一种需氧、不发酵、高度运动的革兰氏阴性菌。
当感染宿主时,铜绿假单胞菌需要铁。因此铜绿假单胞菌合成了两种铁载体:pyochelin和pyoverdin。铜绿假单胞菌随后将这些嗜铁细胞秘密地存在于细胞的外部,与铁紧密结合并将铁带回细胞。铜绿假单胞菌还可以利用肠杆菌素中的铁来满足其铁需求。
其优先代谢是呼吸,通过将电子从葡萄糖(还原的底物)转移到氧(最终的电子受体)来获得能量。当处于厌氧状态时,铜绿假单胞菌使用硝酸盐作为末端电子受体。
形成生物膜,附着在金属,塑料,医疗植入材料和组织表面。
症状:
肺部感染:发烧和发冷、呼吸困难、胸痛、疲倦、咳嗽
尿路感染:强烈尿频冲动、小便疼痛、尿液中难闻的气味、尿液混浊或带血、骨盆区域疼痛
伤口感染:伤口发炎、漏液
耳部感染:耳痛、听力下降、外耳发红或肿胀、发热
也可能是囊性纤维化患者或机械呼吸机患者呼吸道感染的重要原因。
传播:
当暴露于受该菌污染的水或土壤中时,可以传播到医院患者。还可以在医院中通过被污染的手,设备或表面从一个人传播到另一个人。
治疗:
抗生素治疗
注:铜绿假单胞菌感染通常难以治疗,因为该细菌对许多抗生素具有抗性,并且具有形成生物膜的非凡能力。
易感人群:
免疫力低下人群
使用呼吸机患者、使用导管等装置、有手术或烧伤的伤口患者
预防:
患者和护理人员应保持双手清洁,避免感染。尤其是在护理伤口或触摸医疗设备之前和之后要彻底洗净双手。每天打扫房间。避免共享个人物品。
艰难梭菌
Clostridium difficile
革兰氏阳性,厌氧菌。经过氨基酸发酵,以产生ATP作为能量来源,并且还可以利用糖。
产生两种毒力因子:在70%的菌株中发现肠毒素(毒素A)和在所有菌株中发现的细胞毒素(毒素B)。毒素通过糖基化使Rho-gtpase失活,破坏肠上皮细胞的紧密连接,导致细胞旁通透性增加,从而导致体液分泌(腹泻)、粘膜损伤和炎症。
艰难梭菌存在于整个环境中,包括土壤,空气,水,人类和动物的粪便以及食品(例如加工肉)中。
疾病症状:
艰难梭菌通常影响住院的老年人。
常见症状有:严重腹泻、发热、排便频繁、胃部压痛或疼痛、食欲不振、恶心。
更严重的症状可能包括:
水样腹泻,每天10至15次;
脱水、心跳加快、体温升高、食欲不振、严重的腹部绞痛和疼痛、粪便中有脓液或血、体重下降、腹部肿胀、肾功能衰竭等。
传播:
通过粪-口途径在人与人之间转移。感染通常在医院发生。
该菌可以从粪便传播到食物,然后传播到其他物体表面,如果不洗手或者不正确洗手,就容易感染。
治疗:
抗生素(万古霉素,非达霉素)治疗。患有艰难梭菌感染的人容易脱水,可能需要住院治疗。但是,使用抗生素治疗可能会攻击体内有益细菌。
其他治疗感染的方法,包括:
脆弱拟杆菌
bacteroides fragilis
无芽孢,专性厌氧革兰氏阴性杆菌,人类结肠正常菌群的一部分。
BF毒素引起剧烈的炎症和“细胞间附着物的丧失”,从而引起典型的腹痛和腹泻。
某些脆弱类芽孢杆菌菌株无毒,甚至对其宿主生物有益。
疾病症状:
菌血症、阑尾炎、褥疮、化脓性关节炎、脑膜炎、喉咙发炎、上呼吸道感染、皮肤感染、心内膜炎、软组织感染、心包炎
该菌是正常肠道菌群成员,但是如果转移到周围组织中也会引起腹腔内感染。
这些感染包括产后子宫内膜炎,盆腔脓肿,会阴切开后软组织感染,微管卵巢脓肿和盆腔炎。
菌血症定义为血液中细菌的存在。脆弱拟杆菌是血液培养后最常见的厌氧菌。细菌在腹部,软组织和女性生殖道中的存在是最常见的菌血症来源。
该菌不是人体皮肤微生物菌群的一部分。如果这种细菌进入任何组织,则会引起皮肤感染,例如坏疽和坏死。糖尿病患者还容易受到该菌引起的皮肤感染的影响。
该菌还与引起心包炎,心内膜炎,脑膜炎,阑尾炎和咽喉发炎有关。
传播:
如果细菌被转移到任何其他人体组织,则可能导致该组织的疾病;可能由于外伤,割伤,烧伤,异物侵入或由于不当的外科手术做法造成感染。
治疗:
单一药物治疗或组合治疗
易感人群:
患有腹腔内感染的患者,糖尿病患者易感染
预防:
产气荚膜梭状芽胞杆菌
Clostridium perfringens
革兰氏阳性细菌,是嗜温菌,最适生长温度为37℃,产生内生孢子的非运动性菌。
通过无氧呼吸产生能量,使用硝酸盐作为其电子受体。可能导致许多胃肠道疾病,严重程度从轻微的肠毒血症到致命的气性坏疽。
还具有进行糖酵解和糖原代谢所需的所有酶,利用各种糖酵解酶将糖化合物分解为更简单的形式。
可以在人体肠道,污水和土壤中正常发现。
图源:Food Safety News
疾病症状:
食源性疾病(食物中毒)的最常见原因之一。
关于食源性疾病详见:正值夏季,警惕食源性疾病,常见的食物中毒的病原菌介绍
大多数感染了产气荚膜梭菌的人在食用受污染的食物后6-24小时内会出现腹泻和胃痉挛。这种疾病通常突然发作,持续不到24小时。
这种感染通常不会引起发烧或呕吐。
传播:
营养和食品卫生状况较差的地区人群;
糖尿病和动脉粥样硬化等患者;
产气荚膜梭菌感染也与多发性硬化症有关
危险因素:
常见感染来源包括肉类,禽类,肉汁和其他不安全温度下的食物。
爆发往往发生在医院,学校食堂,监狱和疗养院,以及带有餐饮的活动。最常发生在11月和12月。
治疗:
大多数人未经抗生素治疗即可恢复。
如持续腹泻应多喝水,多休息。
对于源自深层伤口的感染, 必须尽可能清洁该区域,并应使用抗生素。
预防:
将食物煮至安全温度;
食物煮熟后应保持在大于60度或低于4度
可以将热食直接放入冰箱,从冰箱拿出来后加热食用。
结核分枝杆菌
Mycobacterium tuberculosis
革兰氏阳性,专性需氧菌,无运动能力,无孢子形成,细胞内生长的细菌。
通常感染单核吞噬细胞。
在感染的潜伏期,结核分枝杆菌利用一系列效应蛋白将宿主免疫系统弄混,并使其生活方式驻留在肉芽肿中,肉芽肿是宿主为应对持续感染而建立的复杂和有组织的免疫细胞结构。肉芽肿中的结核分枝杆菌通常被限制在具有免疫能力的宿主中,但是当宿主免疫力受损时,它可能导致结核病复发。
常见的症状:
低烧、盗汗、疲劳、厌食(食欲不振)、体重下降。肺结核患者通常会产生咳嗽,并伴有低烧发冷,肌痛(疼痛)和出汗。
潜伏期没有症状。
引发疾病:
传播:
吸入飞沫(咳嗽或打喷嚏)
治疗:
抗生素治疗
预防:
肺炎链球菌
Streptococcus pneumoniae
柳叶刀形,革兰氏阳性,兼性厌氧菌,α-溶血性,条件致病菌。
该菌通过胞外酶系统获得大量的碳和氮,胞外酶系统允许多糖和己糖胺的代谢,并对宿主组织造成损害并使其定植。
荚膜多糖的组成和数量在毒力中起主要作用。产生最大量多糖的菌株可能是最强毒的。
图源:Science Photo Library
疾病症状:
从无症状的咽部定植到粘膜疾病(中耳炎,鼻窦炎,肺炎)再到侵袭性疾病(通常在无菌部位的细菌;菌血症,脑膜炎,脓胸,心内膜炎,关节炎)
传播:
通过呼吸道飞沫直接进行人与人接触,上呼吸道携带细菌的人自动接种。
易感人群:
治疗:
抗生素;静脉注射疗法
预防:
无乳链球菌
Streptococcus agalactiae
革兰氏阳性双球菌,不耐酸,不形成孢子,不易动,兼性厌氧的条件致病菌。
通常称为B组链球菌(GBS),是四种Beta-溶血性链球菌之一,可导致血琼脂上细菌菌落周围宽阔清晰区域显示的血细胞完全破裂。
使用葡萄糖作为能源。该细菌能够通过氧化磷酸化合成ATP。
是一种异养菌,能够导入多种碳源。能够将不同的碳源发酵成多种副产物,如乳酸,乙酸盐,乙醇,甲酸盐或乙酰丙酮。
需要许多氨基酸才能生长,因为它不存在任何TCA循环来合成氨基酸。
毒力因子: 多糖胶囊, β溶血素毒素
属于生殖道的正常菌群。5-20%的女性阴道定植。
疾病症状:
尿路感染, 新生儿和幼儿败血症,脑膜炎
患有基础疾病的成年人:肺炎,心内膜炎,皮肤和软组织感染等。
传播:
GBS阴道或直肠定植的母亲所生的婴儿中,有1%至2%的婴儿发生早发感染。
通过孕妇在怀孕或分娩期间的生殖器官和/或肠道,以及来自其他新生儿或妇产医院的医院工作人员。
治疗:
抗生素治疗
易感人群:
预防:
流感嗜血杆菌
Haemophilus influenzae
革兰氏阴性杆菌。有6种囊化血清型(指定为a至f)具有不同的囊化多糖。
该菌在人的鼻子和喉咙中,通常不会造成伤害。但有时会移动到身体的其他部位并引起感染:
肺炎(肺部感染),菌血症,脑膜炎,喉咙肿胀,蜂窝织炎(皮肤感染);
引起儿童耳部感染和成人支气管炎;
较不常见的感染包括心内膜炎和骨髓炎。

图源:ecdc.europa.eu
易感人群:
5岁以下和65岁以上感染风险增加,免疫力低下的(如艾滋病患者,癌症患者)感染风险增加。
传播方式:
咳嗽或打喷嚏;新生儿可以通过吸入羊水或与含有该菌的生殖道分泌物接触而感染。
治疗:
服用抗生素
预防:
婴儿接种Hib疫苗
衣氏放线菌
Actinomyces israelii
直径为1μm的革兰氏阳性杆菌,厌氧细菌,它是肠道正常菌群的一部分,条件致病菌。
浸润性,组织渗透/破裂;可引起慢性化脓性感染,放线菌病。
通过在各种手术(牙科,胃肠道),抽吸或病理性疾病(例如憩室炎)过程中破坏粘膜屏障来建立感染。
感染部位:
口腔,宫颈,面部疾病是最常见形式,有时感染可能发生在胸部(肺放线菌病),腹部,骨盆或身体其他部位。
当细菌进入人体时,它可以在软组织上形成脓肿。随着脓肿随着时间的流逝而扩大,它会穿透皮肤表面,引起皮肤溃疡。这些脓肿或肿块通常会影响头和颈部,并且会引起肌肉痉挛,阻止下巴正常运动。
其他常见症状包括:
发烧,体重减轻,咳嗽,胸痛和窦腔过度引流。症状可能发展缓慢,但是早期治疗是迅速康复的关键。
易感人群:
治疗:
抗生素;如果与宫内节育器有关,则须卸下
预防:
良好的口腔卫生和定期看牙医可能有助于预防某种形式的放线菌病。
嗜肺军团菌
Legionella pneumophila
具有一定铁含量的细胞内多形革兰氏阴性细菌,条件致病菌。
普遍存在于水环境中,例如饮用水系统、温泉、冷却水,可引起人类感染军团菌肺炎。
通过抽吸或直接吸入到达肺部后,会附着在呼吸道粘膜上。

图源:apotheekteirlinck.be
症状:
高烧(可能到40℃或更高),发冷,咳嗽,肌肉酸痛和头痛。
传播:
可以在人类制造的水系统(例如空调)中繁殖。大多数人吸入含有军团菌细菌的微小水滴时会被感染。这可能来自淋浴,水龙头或漩涡浴池中的喷雾,或者来自大型建筑物中通风系统中的水。
如何避免致病菌感染?
尽快恢复免疫力,减少致病菌感染的发生率。
个人应通过煮熟鸡蛋和肉类来避免传染源,饮用巴氏杀菌乳制品,避免感染个人和医院的潜在感染源,避免与粪便、灰尘或农场动物接触,这些是主要的感染源。
不要饮用未经处理的水,例如直接来自湖泊或河流的水。避免在国外饮用自来水。使用瓶装水或滤水器。
不要共用针头、注射器或其他药物注射设备。
有害菌和肠道内的其他共生菌共同构成菌群微生态,也是大部分人群肠道内常见的菌群。
有害菌是相对而言的。正常肠道菌群也包含许多有害菌,但有害菌比例或个别菌属丰度超标可能预示着肠道菌群的健康状况受到破坏。
有害菌过多会影响肠道内环境,如pH值,含氧量以及肠道内毒素等,可能会导致出现一些机会感染和机会致病菌入侵,进而诱发炎症和疾病。
谷禾肠道菌群检测报告中的有害菌包含了原发致病菌和条件致病菌,以及属内主要菌种为致病菌的属。
为便于统计,我们在计算的时候统一按照属层级进行计算比例。
报告中的有害菌包括了以下的菌属:韦荣氏球菌属、葡萄球菌科、变形菌属、弓形菌属、弯曲菌属、螺杆菌属、厌氧螺菌属以及弧菌属等。
具体每个菌相关介绍详见:
全面认识——肺炎克雷伯菌 (Klebsiella pneumoniae)
慢病是每一种病单独构建模型,不一定和有害菌或菌群平衡指标直接有关。
肠道菌群中的有害菌过多,也就是说肠道菌群趋向于失衡,这会给身体带来诸多麻烦。
当肠道内的有害菌增多时,起初可能会有些症状出现,例如:
随着时间的推移,如果没有采取相应的干预措施,有害菌变得过多,则可能会对身体方方面面产生影响:
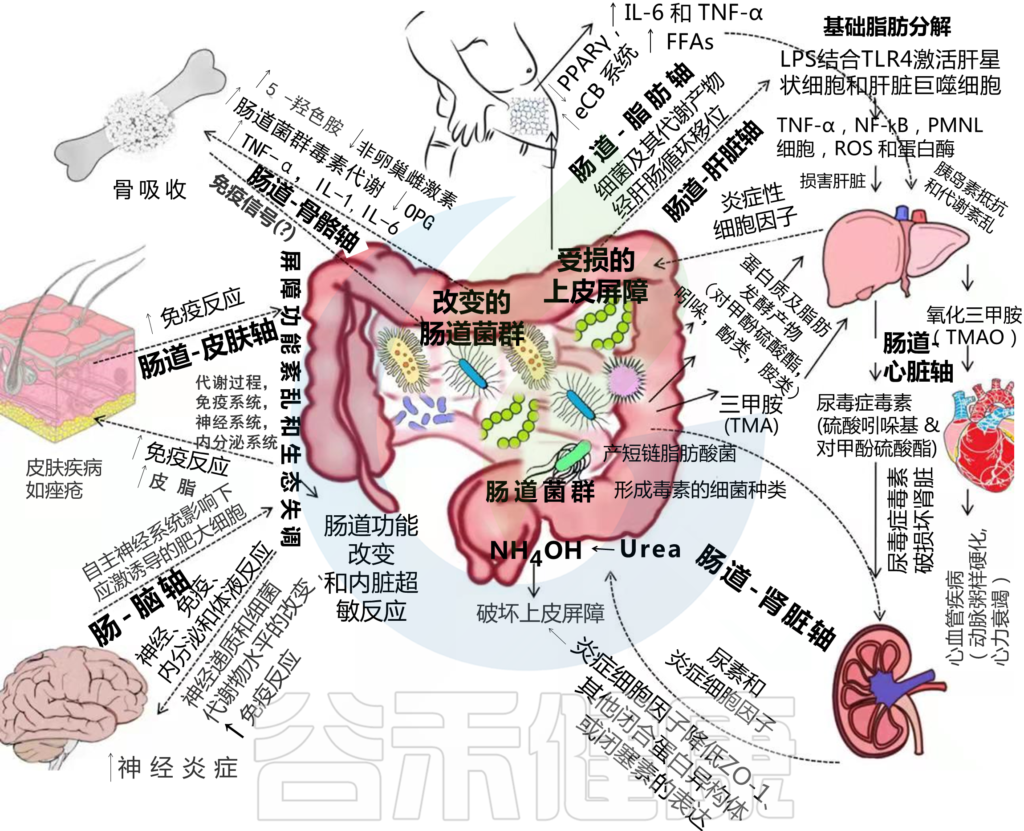
特殊类型的细菌会产生一种化学物质,肝脏会将其转化为三甲胺- n -氧化物(TMAO)。
TMAO产生的增加会导致血管中胆固醇的积聚,从而可能导致心脏病。
详见:
TMAO产生的增加也与慢性肾脏疾病有关。也会导致肾结石的发展。
与健康对照组相比,终末期肾病患者体内的TMAO浓度可高出20倍。
对终末期肾病患者的类似研究表明,从普雷沃氏菌向拟杆菌转变,产丁酸菌减少。

有害细菌将纤维转化为脂肪酸。身体可能会把它们沉积在肝脏中,如果不及时治疗,可能导致代谢综合征的发展。
肝硬化患者中韦荣球菌属和链球菌增多。
肝细胞癌与肠道大肠杆菌过度生长有关,患者微生物群多样性增加,与产丁酸菌属(如Alistipes)减少有关,而致病性产脂多糖菌(如克雷伯氏菌)增加。
研究表明,肠道菌群在宿主的代谢和疾病状态中起着重要的作用。特别是2型糖尿病,其病因复杂,包括肥胖、慢性低度炎症,受肠道微生物群和微生物代谢产物的调节。
在2型糖尿病患者普遍具有相对高丰度的特定属:
Blautia、Coprococcus、Sporobacter、Abiotrophia、Peptostreptococcus、Parasutterella、Collinsella
2型糖尿病患者肠道菌群详见:
谷禾数据库统计发现,在有害菌属的丰度水平分布上,肥胖人群要高于对照人群。进一步对具体菌属进行分析,发现肺炎克雷伯氏菌的丰度水平肥胖人群更高。
肥胖患者肠道菌群详见:
肠道有许多直接与大脑沟通的神经末梢,称为肠脑轴。肠道炎症和菌群失调与心理健康不良有关。过多的有害肠道细菌会导致:
详见:
不良的肠道细菌会影响你的整体健康。它会增加消化问题的风险,这些症状通常伴有体重减轻和腹痛。比如:
肠道上皮、免疫系统和共生细菌之间的串扰是启动全身炎症反应的关键。有益菌和有害菌的失衡,抗炎和促炎细胞因子之间的失衡,包括白细胞介素(IL)-1β、肿瘤坏死因子、干扰素(IFN)-γ、白细胞介素-6、白细胞介素-12和白细胞介素-17,在参与RA发病机制的炎症过程中起着核心作用。
为了支持肠道菌群成分变化在类风湿性关节炎发病和进展中起重要作用的假说,已经提出了肠道菌群与关节炎相关的几种机制。
这些包括调节宿主免疫系统(触发T细胞分化)、通过作用Toll样受体(TLR)或NOD样受体(NLR)激活抗原呈递细胞(APC)、通过酶促作用促进肽的瓜氨酸化、抗原模拟和增加肠粘膜通透性。关于对APCs TLRs表达的影响,这可能导致Th17/Treg细胞比率失衡,这种局部免疫反应可能导致系统性自身免疫。
有害菌过多会导致肠道通透性和肠道屏障破坏。肠黏膜屏障功能障碍可能导致血清脂多糖 (LPS) 水平升高,从而导致代谢性内毒素血症。早期研究表明,LPS 可促进体内股骨的骨质流失和体外破骨细胞的。
与骨骼疾病相关的肠道菌群变化如下:

编辑
详见:
鉴于具有高度稳定性的平衡肠道微生物群与宿主的免疫系统具有共生相互作用,能够抑制有害菌增长。然而不稳定的状态例如肠道紊乱,慢性疾病,由遗传易感性、化学物质或肠道病原体感染引起的肠道炎症会导致有害菌增加。
其他包括饮食、生活方式、环境等因素也会影响肠道微生物群的分类和功能组成。例如,西方饮食、高糖饮食,饮食结构过于单一,加工食品过多摄入等不健康的饮食方式,睡眠不足,作息不规律,不运动等不良生活习惯,压力过大,服用药物等因素都会导致有害菌增多。
有害菌和肠道内的其他共生菌共同构成菌群微生态,如果有害菌过多,通常我们可以通过服用益生菌或益生元的方式首先增加有益菌的比例,相应的有害菌比例就会降低。
双歧杆菌和乳酸杆菌有助于发酵碳水化合物,同时会产短链脂肪酸,有助于维持良好的消化系统。
乳酸杆菌
乳酸杆菌菌株产生乳糖酶。它有助于分解乳制品中的乳糖,有助于维持肠道的酸度水平,对于吸收关键矿物质至关重要。
双歧杆菌
保护肠壁;维持肠道的酸度;限制产生硝酸盐菌的生长;生产 B 族维生素和维生素 K 等。
益生菌抑制其他菌群的生长:
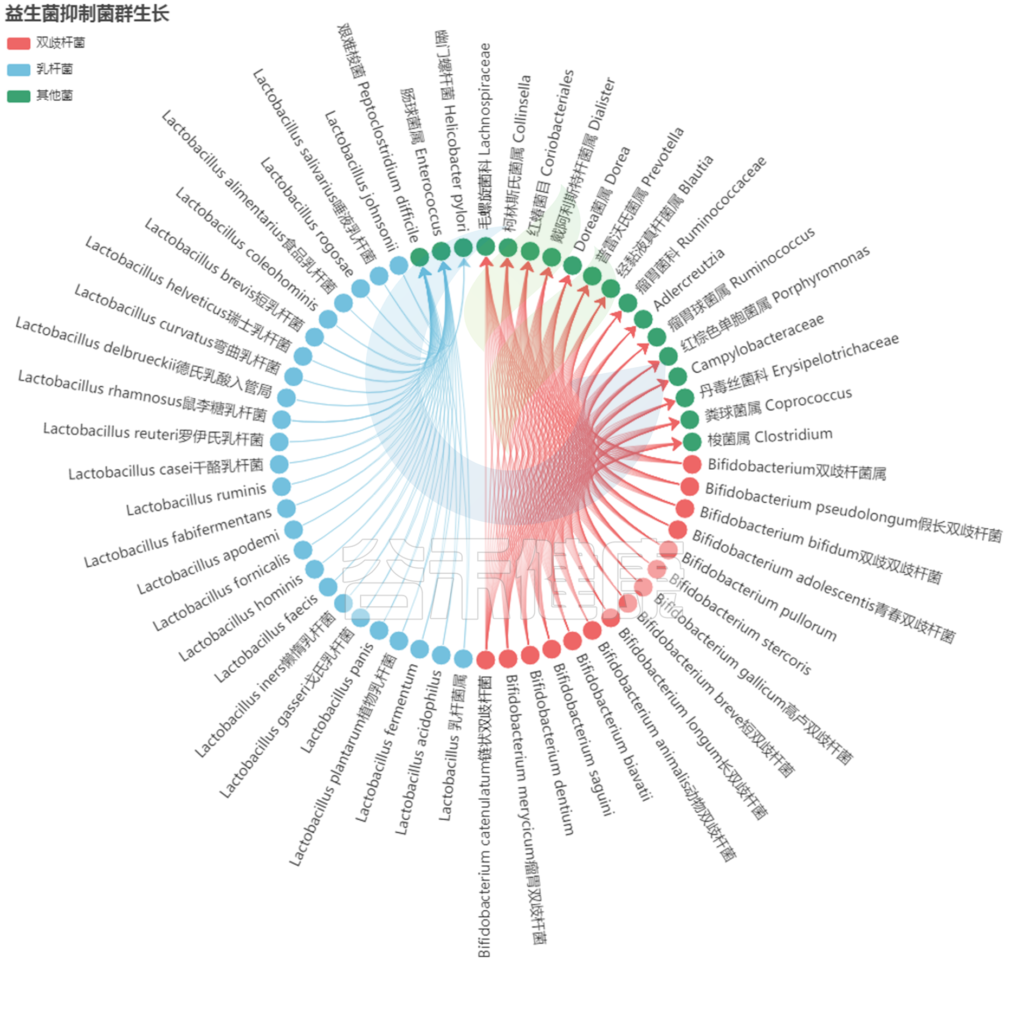
编辑
<来自谷禾健康数据库>
除了直接服用益生菌这种方式之外,有些食物中也富含益生菌,如:
乳制品:酸奶、牛奶、开菲尔等
发酵食品:泡菜、酸菜、味噌汤、豆豉等
益生元是一种可溶于水的可溶性纤维,可以作为益生菌的“食物”。
最广泛认可的益生元包括低聚果糖 (FOS)、菊粉和低聚半乳糖 (GOS) 等。
此外还包括抗性淀粉、果胶寡糖 (POS)、多酚等。
含益生元的食物包括:菊芋、青香蕉或青香蕉粉、大麦和燕麦、魔芋根、菊苣根、牛蒡根、亚麻籽、海藻、苹果、土豆等。
关于益生菌,益生元的补充详见:
想要持久的改善菌群结构降低有害菌水平就需要改善生活方式,适当增加抗性淀粉等膳食纤维并规律饮食和睡眠,增加运动等。
下面介绍一些日常生活中可以自行调整的饮食及生活方式。
尝试食用多种食物,避免每天食物一样
饮食多样性更有利于菌群维持健康平衡。体内的微生物群就好比一群挑剔的孩子,每个孩子都会去吃自己喜欢的食物。当你吃下各种食物时,就相当于喂食了各种微生物。
如果可以的话,一个星期的饮食中可以摄入 40 种及以上不同类型的天然食物,尽可能地提高肠道多样性。
减少西方饮食
西方饮食(其特点是大量摄入脂肪、蛋白质、糖、盐和加工食品),可能增加有害菌,与伴随的导致自身免疫疾病发展的微生物变化之间的联系越来越明显。
尝试地中海饮食
其他可以参考地中海饮食结构摄入,适量食用纤维,多吃各种颜色的水果和蔬菜。将红肉的摄入限制在每月两三次,可以将其视为一种“奢侈品”,每周食用两到三次家禽。它的饱和脂肪和胆固醇比红肉少得多,选择健康的脂肪,每周可以食用两次三文鱼等,做菜选择橄榄油,适量食用坚果(杏仁、巴西坚果、榛子、松子、开心果和核桃都是非常健康的坚果类型),它们是单不饱和脂肪的重要来源。
尝试抗炎饮食
很多种食物均具有抗炎特性,其中包括抗氧化剂和多酚含量高的食物。肠道抗炎饮食推荐的食物包括:浆果类,西兰花,牛油果,辣椒,姜黄,洋葱,大蒜等。
详见:
避免膳食纤维过多或过少
膳食纤维细菌发酵的产物(短链脂肪酸、乙醇和乳酸)过多会破坏细菌。纤维也会让人“上瘾”,其发展方式:随着发酵破坏细菌,需要越来越多的纤维来形成粪便。
但是膳食纤维摄入不足或突然停止所有纤维摄入,也会发生菌群失衡,导致便秘或其他肠道问题,从而导致有害菌增加。
减少摄入加工和包装食品
食用的加工食品越多,饮食越无菌,加工食品会减少我们体内有益细菌的数量,相应的有害菌会逐渐增多,破坏肠道菌群原有的平衡。
具体来说,防腐剂,比如聚山梨酯80和羧甲基纤维素(CMPF),它们是许多加工食品中常见的乳化剂,直接改变了肠道微生物群的组成。
食品添加剂对菌群的影响详见:
你的焦虑可能与食品添加剂有关,警惕食品添加剂引起的微生物群变化
尽量避免含糖饮食
对于我们大多数人来说,糖在我们的饮食中太普遍了。
在现代饮食中,糖无处不在,而且形式多种多样。我们大多数人现在都知道,过度消费“游离糖”的精制糖并不健康。糖也是造成菌群失调的主要因素。然而在忙碌的现代生活中,很难不过度摄入糖分。
高糖食物的常见罪魁祸首包括碳酸饮料、能量饮料、糖果、饼干、甜点、蛋糕、果汁和谷物等,它们都会导致菌群失调。
还有一些食物,含有的糖分更加隐蔽。这些包括:即食食品、腌泡汁、酸辣酱和泡菜、一些酒精饮料、调味酱、白面包、白米饭和土豆。
糖对菌群的影响详见:
吃八分饱
少吃多餐,每三到四个小时吃一顿小餐或吃零食,以补充精力。
间歇性禁食
间歇性禁食是一种越来越流行的健康实践,研究发现间歇性禁食导致肠道菌群结构改变,进食时机和频率可以一定程度上改善生活方式和心血管代谢,防止2型糖尿病和心血管疾病的发生。
关于间歇性禁食详见:
维生素维持肠道菌群稳态和减少肠道炎症以预防癌症的机制;肠道菌群帮助吸收营养,并参与维生素代谢。几项观察表明,微生物群失调和维生素缺乏是相互关联的。
例如:
补充维生素 C 可减少肠杆菌科细菌的数量,增加乳酸杆菌的丰度,抑制有害菌的生长,促进有益菌的增加。
维生素 D 的缺乏会增加拟杆菌门、变形杆菌门和螺杆菌科的丰度。
维生素E对变形菌有抑制作用,而维生素E(和纤维)的摄入量较低与Sutterella水平较高相关。

详见维生素的文章:
当肠道渗漏也就是屏障受损时,大量的有害菌及微生物代谢毒素、食物中的有毒物质逃离肠道,涌入血液循环,这可能会产生炎症并导致组织损伤,器官从感染到炎症再到功能缺失,甚至是到癌症。
关于什么情况会导致肠漏,肠漏带来的危害详见:
即使是很小的压力也会触发体内激素和化合物的释放。压力会慢慢积累,如果你是一个压力大的人,经常烦躁,愤怒,那么会对微生物群产生负面影响,扰乱 HPA 轴。不过这是双向的,肠道中的某些细菌菌株也会影响体内神经递质的方式。
适当给自己减压,可以帮助菌群恢复平衡。减压的方式包括:
农村环境和微生物群与过敏患病率的降低有关。通过暴露于农村室内灰尘来调节肠道微生物群可以改善过敏预防。
城市儿童和小鼠暴露在城市灰尘提取物中,肠道菌群向拟杆菌类的变化是明显的。相比之下,农村儿童和接触农村粉尘提取物的小鼠肠道菌群分别富含普雷沃氏菌属和梭状芽孢杆菌属。
环境对菌群的文章详见:
环境污染物通过肠脑轴影响心理健康,精神益生菌或将发挥重要作用
睡眠质量与肠道菌群组成之间存在双向关系。

编辑
拟杆菌门和厚壁菌门的丰度与睡眠质量呈正相关,而毛螺菌科(Lachnospiraceae)、棒状杆菌(Corynebacterium)、Blautia等几种菌与睡眠质量测量值呈负相关。
睡眠不足或者其他因素如受伤、食物摄入、压力、昼夜节律和运动等,可致肠屏障损伤和细菌移位,增加感染易感性,激活HPA轴从而影响菌群。
详见睡眠对肠道菌群的影响的文章:
抗生素
正常情况下,强大的免疫系统会追捕并消灭病原体,但在系统较弱的情况下,人体的免疫防御系统无法控制病原菌的生长。
服用抗生素会杀死体内的许多好细菌和坏细菌。在瑞典的一项临床试验中,研究人员发现,在服用抗生素仅一周后,一些参与者在一整年后就破坏了微生物组。
抗生素使用对肠道菌群变化的不同影响
Yang L, et al., AMB Express. 2021
我们之前写过抗生素对菌群的影响文章,详见:
其他包括非甾体抗炎药、质子泵抑制剂 (PPI)等也会影响肠道菌群的组成。
限制饮酒量
酒精诱导的胃肠道菌群组成和代谢功能的变化可能有助于建立酒精诱导的氧化应激、肠道对菌群产物的高通透性和随后发展的酒精性肝病和其他疾病之间建立明确联系。如果体内酒精过多,肝酶可能没有足够的能力对其进行处理。过量的酒精会在身体的其余部分循环,产生负面影响。
坚持适量运动
运动锻炼与肠道微生物群组成之间可能存在密切关联。经常中等强度的耐力运动对肠道微生物产生最有益的影响,促进健康和抗炎细菌增加;长期运动的人菌群多样性更高。
运动改善菌群多样性,增加菌群种类,有益于提高菌群稳定性,降低有害菌的相对比例。
运动对菌群的影响详见:
肠道微生物组如何影响运动能力,所谓的“精英肠道微生物组”真的存在吗?
在我们的检测实践中,以上的这些干预调节方式可以有效的定向改善特定有害菌和致病菌的超标,并最终带来整体健康状况的改善。
主要参考文献:
Janik E, Ceremuga M, Niemcewicz M, Bijak M. Dangerous Pathogens as a Potential Problem for Public Health. Medicina (Kaunas). 2020 Nov 6;56(11):591. doi: 10.3390/medicina56110591. PMID: 33172013; PMCID: PMC7694656.
Sheppard SK. Strain wars and the evolution of opportunistic pathogens. Curr Opin Microbiol. 2022 Jun;67:102138. doi: 10.1016/j.mib.2022.01.009. Epub 2022 Feb 12. PMID: 35168173.
Balloux F, van Dorp L. Q&A: What are pathogens, and what have they done to and for us? BMC Biol. 2017 Oct 19;15(1):91. doi: 10.1186/s12915-017-0433-z. PMID: 29052511; PMCID: PMC5648414.
Kinnula H, Mappes J, Sundberg LR. Coinfection outcome in an opportunistic pathogen depends on the inter-strain interactions. BMC Evol Biol. 2017 Mar 14;17(1):77. doi: 10.1186/s12862-017-0922-2. PMID: 28288561; PMCID: PMC5348763.
Engen PA, Green SJ, Voigt RM, Forsyth CB, Keshavarzian A. The Gastrointestinal Microbiome: Alcohol Effects on the Composition of Intestinal Microbiota. Alcohol Res. 2015;37(2):223-36. PMID: 26695747; PMCID: PMC4590619.
Kouzu K, Tsujimoto H, Kishi Y, Ueno H, Shinomiya N. Bacterial Translocation in Gastrointestinal Cancers and Cancer Treatment. Biomedicines. 2022 Feb 4;10(2):380. doi: 10.3390/biomedicines10020380. PMID: 35203589; PMCID: PMC8962358.

谷禾健康
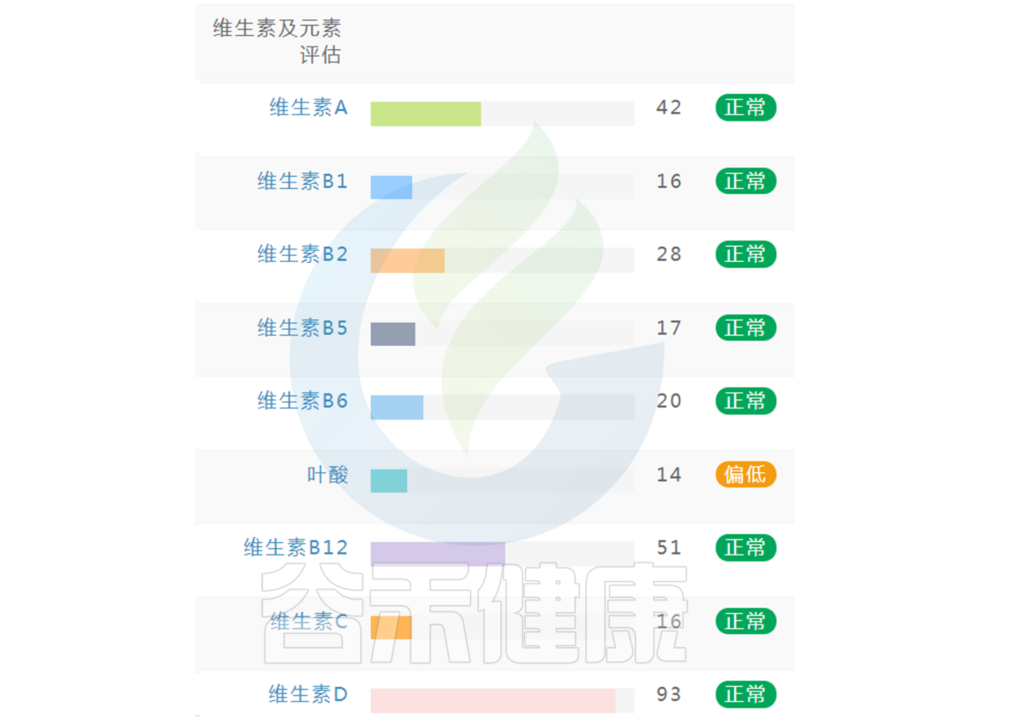
在谷禾肠道菌群健康检测中,我们会看到结果报告中关于维生素的评估如下:

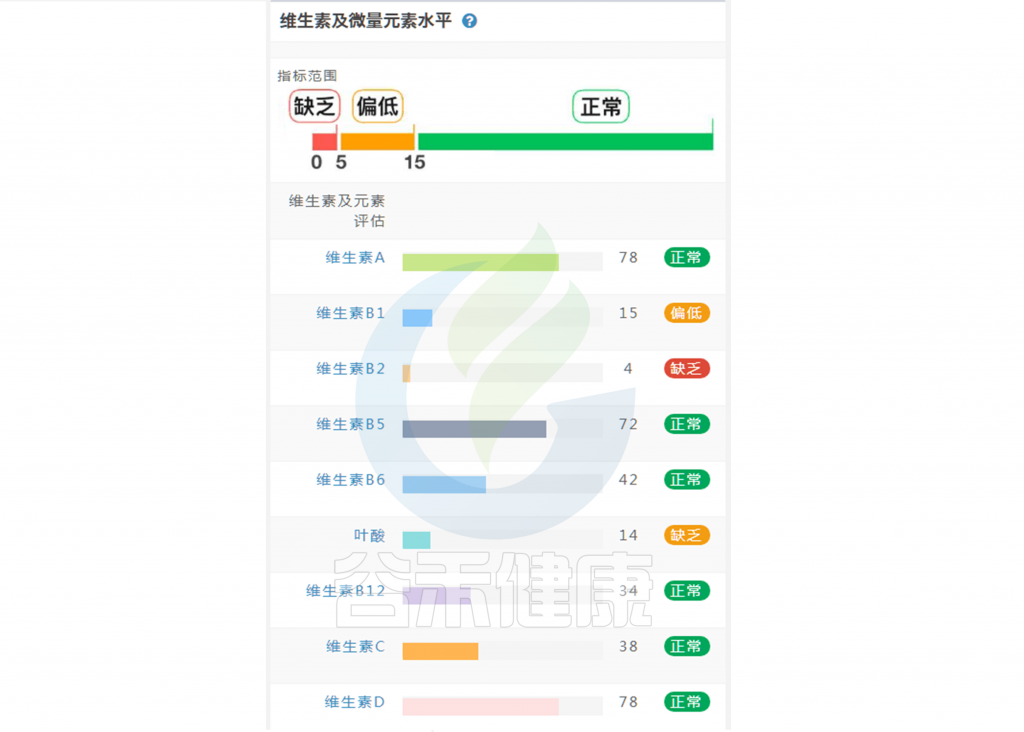
摄入水平建议保持在70-80分之间最佳,如果单项指标低于5表明摄入比例在人群中属于最低的5%,评估为缺乏,如上图中维生素B2;低于15评估为偏低,如上图中叶酸;达到或超过95则表明该项指标可能摄入比例偏高,可适当减少摄入;其余则为正常范围。
上图可以看到,像维生素C这项指标分值在38,虽然正常但相对于最佳来说是偏低的。
一些小伙伴可能会存在这样的疑惑:
为什么肠道菌群检测可以评估维生素?
这些维生素指标的分值代表着什么含义?
肠道菌群和维生素之间有什么样的关联?
它们如何影响人体健康/疾病?
如何判断维生素是否缺乏?
该如何补充?
…
本文就以上问题进行详细解答,同时也包括维生素-微生物群之间的相互作用,维生素维持肠道菌群稳态和减少肠道炎症以预防癌症的机制,产生维生素的益生菌,补充调节维生素的方式包括饮食、益生菌等。
在阅读本文之前,可以先了解一下各类常见的维生素功能,缺乏导致的症状。


每个维生素的详细介绍可以点开以下查看(请在谷禾健康微信公众号找到这篇文章查看)。
维生素B1(硫胺素)
维生素B2(核黄素)
维生素B3(烟酸)
维生素B5(泛酸)
维生素B6(吡哆醇)
维生素B7(生物素)
维生素B9(叶酸)
维生素B12(钴胺素)
以上每个都有关于该维生素的详细介绍,包括:
—正文—
维生素是一种微量营养素,在人体的生长、新陈代谢和发育中起着至关重要的作用。
在谷禾肠道菌群健康检测报告中,维生素分值即代表该维生素的膳食摄入水平和菌群代谢能力(报告中显示的分值是经过一系列计算得到的一个相对值)。
其中B族维生素很多需要通过肠道菌群对初始原料进行代谢之后才会产生,因此肠道菌群相应的基因和代谢途径的丰度水平也会直接反映这些维生素的摄入水平。
我们知道维生素的缺乏可能引起一些不良后果,导致维生素缺乏的原因有很多,摄入不足,吸收不良等都会导致维生素缺乏。
我们日常主要从饮食中获取维生素,肠道是主要吸收部位。例如,维生素 A 主要在近端空肠吸收,维生素 D 在远端空肠吸收最佳,维生素 E 和 K 主要在回肠吸收。因此,肠道功能受损可能会影响维生素的吸收。当然,影响维生素吸收的其他原因还包括年龄,某些疾病,药物等因素。
那么肠道菌群和维生素之间有什么关联?
肠道菌群是人体生理和健康的重要决定因素。肠道菌群帮助吸收营养,并参与维生素代谢。
肠道有益菌:乳酸菌和双歧杆菌,可以重新合成B族和K族维生素,为宿主提供约30%的每日摄入量。与从食物中获得的维生素不同,微生物产生的维生素主要在结肠中吸收。接下来了解一下具体哪些菌群,如何产生维生素。
前一章节我们知道,除了通过饮食提供维生素外,人体肠道中的细菌也可以产生一些维生素,如果吸收得当,可以部分满足人体的需要。
可以把这些细菌微生物想象成小小的维生素工厂。细菌确保为自己和与他们共生的微生物朋友提供维生素,同时也会为人体提供维生素。
合成的B族维生素的菌群较多
研究人员估计了人体肠道细菌可以提供维生素每日参考摄入量的百分比,得出的结论是可以提供:
40-65% 的人体肠道菌群具有合成 B 族维生素的能力。两种最常见的合成维生素是维生素B2和B3,预测分别有 166 和 162 个生产者。
可以合成 B 族维生素的细菌以及B 族维生素缺乏对肠道健康的影响
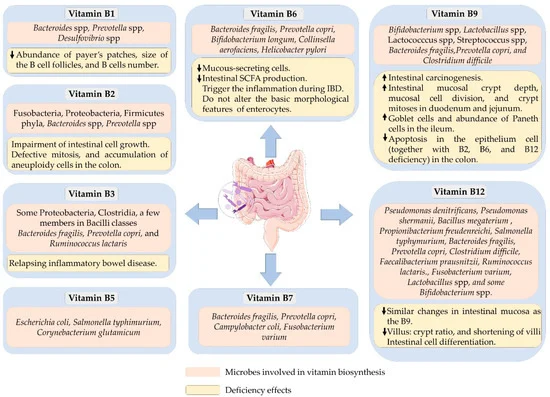
doi.org/10.3390/microorganisms10061168
大部分肠道菌群都参与维生素的合成
随着基因组注释方法的不断完善,研究人员可以预测维生素代谢途径并评估维生素生物合成潜力。通过检索 UniProt 数据库,研究人员发现:
厚壁菌门是维生素的主要代谢相关菌,其次是变形菌门,再然后是拟杆菌和放线菌。这四种菌群是人体肠道菌群的主要组成部分,占总菌群的60%-90%.
下表列出了参与合成B族维生素的肠道菌群,以及相应的代谢机制。


以上是肠道菌群对维生素产生的影响,而维生素和肠道菌群之间的作用是双向的,维生素也会影响肠道菌群,下一章节我们详细了解维生素对肠道菌群的影响。
维生素通过调节免疫力、细菌生长和新陈代谢来改变肠道微生物群的组成。
例如,膳食补充剂中的维生素 B、C、D 和 E 通过有利于双歧杆菌、乳酸杆菌和罗斯氏菌等有益菌属的肠道黏膜扩张和定植,在很大程度上有助于微生物组的组成。
肠道微生物组和宿主之间的微量营养素交换
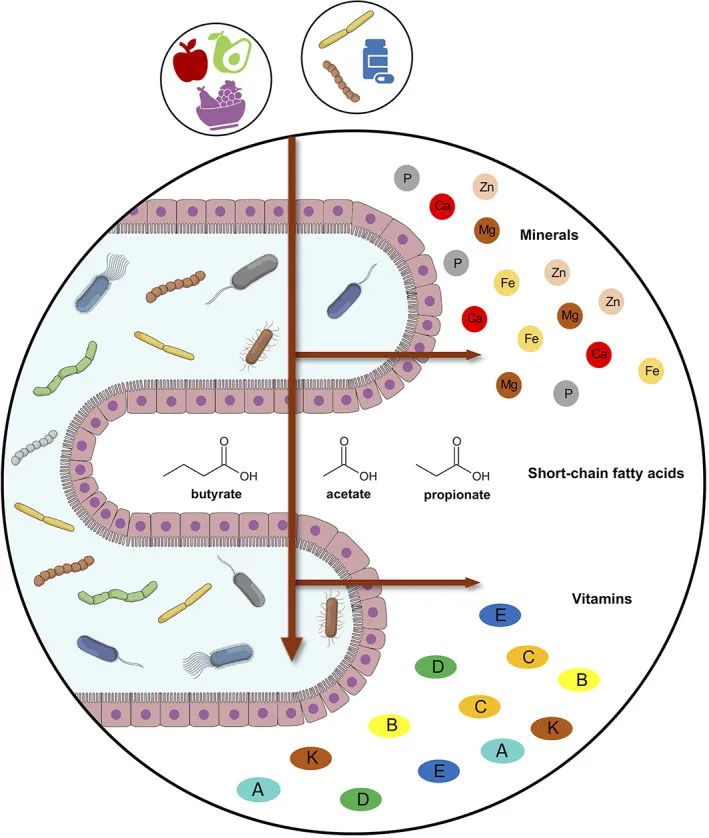
doi: 10.1002/biof.1835
一项研究调查了 96 名健康志愿者,结果表明:
补充维生素 B2 后肠道微生物的种类数量显着增加;联合补充维生素 B2 和 C 导致Sutterella显着减少,但Coprococcus数量增加;
维生素C显着提高肠道微生物的α多样性;
服用维生素D后,促进放线菌的生长和抑制拟杆菌的生长。
补充维生素对人体肠道菌群的影响
doi.org/10.3390/nu14163383
/ 维生素A /
维生素 A 的充足状态可能与微生物多样性增加有关。在小鼠实验中,普通拟杆菌(Bacteroides vulgatus )在维生素 A 缺乏期显着增加。维生素 A 缺乏导致的粘膜反应受损,粘蛋白和防御素 6 表达减少,可能使病原菌更容易穿透肠道屏障。
维生素A缺乏使厚壁菌门中毛螺菌_NK4A136_群、厌氧菌、颤杆菌的数量减少,毛螺菌的含量也降低;然而,Parasutterella呈上升趋势。TLR4 可能参与了维生素 A 调节微生物群的过程。
/ B族维生素 /
研究人员在一个小的成年志愿者群体中进行了一项试点研究,该群体补充了过量核黄素(100mg),持续14天。他们发现,在补充期间,每克粪便中的Faecalibacterium prausnitzii数量增加。作者还注意到厌氧菌Roseburia 增加,大肠杆菌减少。
其他关于B族维生素对肠道菌群的影响详见:
/ 维生素C /
补充维生素 C 可减少肠杆菌科细菌的数量,增加乳酸杆菌的丰度,抑制有害菌的生长,促进有益菌的增加。
也有研究表明,维生素 C 服用4周导致 α 多样性增加,短链脂肪酸浓度增加。

/ 维生素D /
维生素 D 和肠道微生物群的相互作用对免疫稳态至关重要。补充高水平的维生素 D 增加了普氏菌,减少了韦荣氏菌和嗜血杆菌。
婴儿饮食中补充维生素 D 对早期微生物组成的变化有重要影响,而儿童缺乏维生素 D 会导致细菌多样性降低。
最近的一项研究表明,维生素D的活性代谢物1,25-二羟基胆钙化醇,维生素D受体的配体(VDR),影响美国不同地区老年人肠道菌群的α -和β -多样性。
研究人员报告了通过食物频率问卷评估的微量营养素摄入量与孕妇微生物群组成之间的相关性。他们观察到,高脂溶性维生素,特别是维生素D的膳食摄入量与微生物α多样性降低有关(P值<0.001),维生素D和视黄醇与变形菌相对增加有关,变形菌门是一个已知包含多种病原体并具有促炎特性的门。
/ 维生素E /
维生素E对变形菌有抑制作用,而维生素E(和纤维)的摄入量较低与Sutterella水平较高相关,据报道,自闭症和某些胃肠道疾病婴儿的Sutterella水平大量增加。
体外维生素E 可以防止几种人类病原体的生物膜形成,特别是金黄色葡萄球菌和表皮葡萄球菌。
/ 维生素K /
一项动物实验表明,缺乏维生素 K 的小鼠的肠道中,瘤胃球菌、毛螺菌科、Muribaculaceae的含量较多。
关于维生素对人体肠道微生物组直接影响的研究
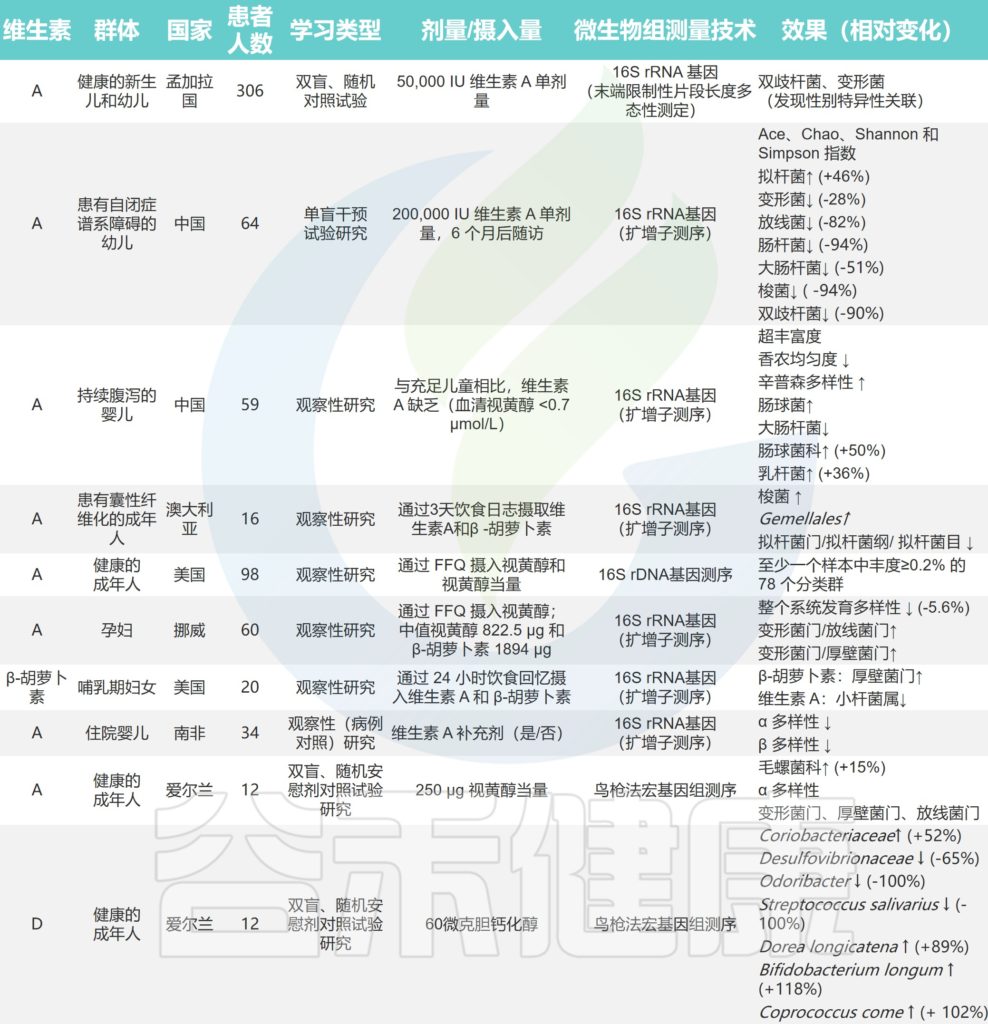
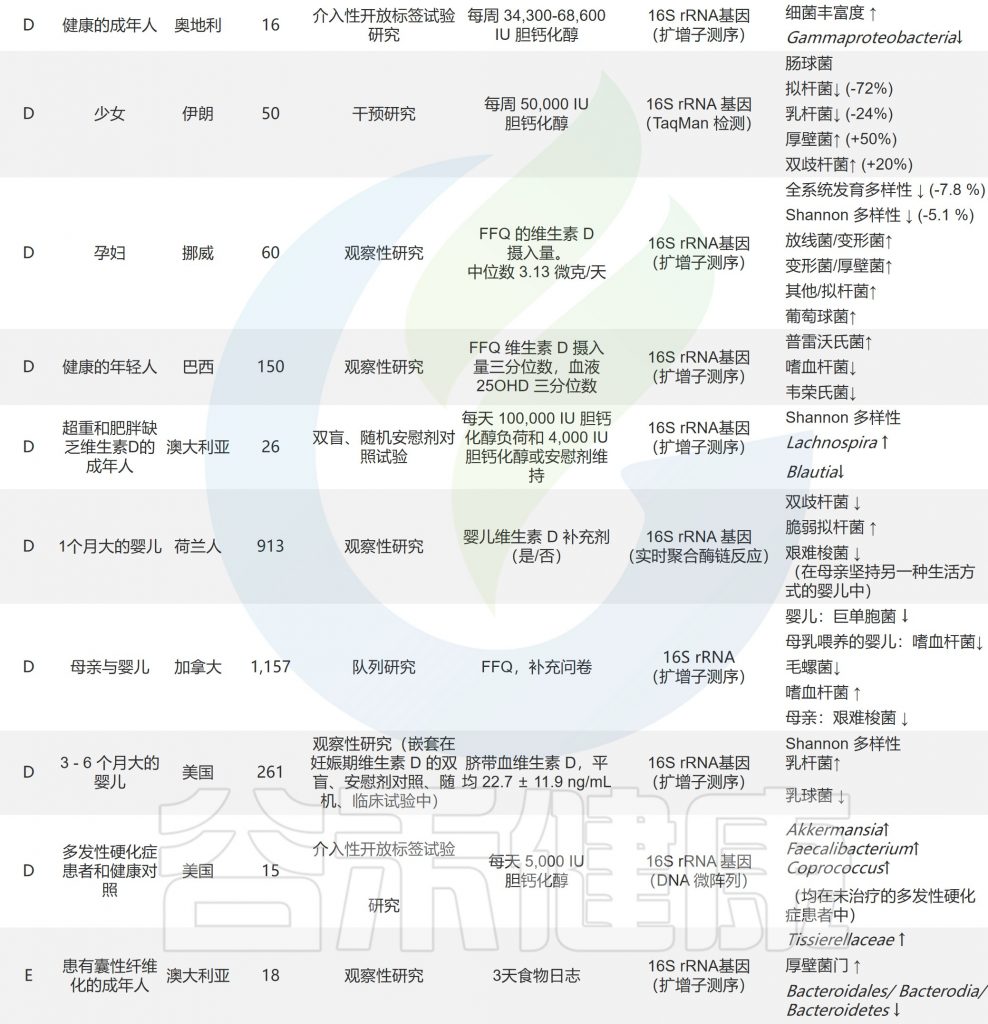
doi.org/10.1016/j.nutres.2021.09.001
饮食是维生素的主要来源,通过饮食补充维生素也会影响菌群。
注:由于测试饮食干预效果所需的随机试验的样本量和持续时间,相关发病率的研究具有挑战性。此外,由于特定的营养素不是孤立地消耗的,而是作为饮食模式的一部分,并且饮食成分之间相互作用,因此饮食带来的实际影响可能只有作为一个整体考虑时才会变得明显。
因此这里我们主要考虑饮食模式,例如地中海饮食等饮食方式。
地中海饮食是营养均衡饮食的典型代表,其特点是大量且频繁地摄入重要的纤维来源(谷物、蔬菜、豆类、水果和坚果)和具有抗氧化特性的化学成分(维生素、类黄酮、植物甾醇、矿物质、萜烯和酚类)。

同时地中海饮食还富含复杂和不溶性纤维含量。我们知道,大量摄入膳食纤维可促进肠道中有益菌群的生长,例如增加拟杆菌、普雷沃氏菌属、罗斯氏菌属、瘤胃球菌属、普拉梭菌等菌属的丰度,从而在肠道中产生高水平的短链脂肪酸,包括丁酸盐。
响应地中海饮食而增殖的细菌可以充当“基石”物种,也就是说它们对于稳定的“肠道生态系统”至关重要。这些变化主要是由于膳食纤维和相关维生素和矿物质的增加,特别是维生素C、B6、B9、铜、钾、铁、锰和镁。
总之,维生素似乎是微生物-宿主间代谢相互作用的重要媒介。
越来越多证据表明,维生素缺乏会导致肠道菌群紊乱,进而引发肠道疾病,甚至促进炎症和肿瘤的发展。下一章节详细讨论,维生素-微生物群相互作用对健康/疾病的影响。
最近的几项观察表明,微生物群失调和维生素缺乏是相互关联的。
维生素对宿主健康的影响

doi.org/10.1016/j.nutres.2021.09.001
这种关系可能直接影响宿主健康:例如,克罗恩病恶化与参与抗炎介质核黄素、硫胺和叶酸生物合成的微生物基因减少有关。
此外,2型糖尿病受试者在与微生物介导的维生素代谢相关的基因丰度谱中显示出显著变化。
营养不良儿童的微生物群显示,参与B族维生素代谢的多种途径(包括烟酸/NADP生物合成)显著减少。
在经历饮食振荡以诱导急性短期维生素A缺乏的灵长类小鼠模型中,Hibberd等人观察到细菌群落结构和宏转录组的调节,其中Bacteroides vulgatus是显著的应答者,在缺乏维生素A的情况下其丰度增加。有趣的是,B.vulgatus是在人类肠道微生物群的灵长类小鼠模型中鉴定的一种生长差异物种。
所有这些观察结果表明,维生素缺乏可能会改变肠道微生物群,从而影响人体健康。
下面我们以肠道疾病和精神类疾病两大类疾病为例,来具体了解维生素-微生物相互作用及其在疾病中的影响。
维生素 A 和 D 分别在近端和远端空肠吸收。维生素E和K主要在回肠吸收;微生物产生的维生素主要在结肠中吸收。维生素缺乏会加重肠道炎症,甚至通过多种机制促进癌症。
肠道菌群->维生素->肠道疾病中的作用
慢性 IBD 发生和发病机制中的关键作用是微生物(尤其是共生菌群)对宿主黏膜免疫功能的影响。同时,肠道微生物群和慢性炎症已被证明与肿瘤发生密切相关。
维生素具有调节肠道菌群和保护肠道的功能。因此,维生素和微生物群的相互作用可能在 IBD 和结直肠癌的治疗中具有巨大的潜力。
维生素A通过促进黏膜愈合、促进产生ASCFA的相关菌增加、降低UC相关菌的水平来达到治疗UC的效果。
费氏丙酸杆菌ET-3 产生维生素 K2 的前体,即 1,4-二羟基-2-萘甲酸 (DHNA),可激活芳烃受体 (AhR) 以改善结肠炎并调节肠道微生物群。
维生素 D 的缺乏会增加拟杆菌门、变形杆菌门和螺杆菌科的丰度,降低厚壁菌门和去铁细菌的丰度门,并且还影响 E-钙粘蛋白表达并减少耐受树突状细胞的数量。
然而,在治疗 IBD 时,维生素 D 与利福昔明的共同给药会影响肠道菌群和利福昔明的疗效。维生素 D 促进A. muciniphila的生长以保护肠粘膜屏障,这些作用对于对抗结直肠癌的发展尤为重要。
研究表明,维生素 E 及其代谢物在调节肠道菌群、减少炎症和抑制癌变方面具有巨大潜力。此外,维生素 Eδ-生育三烯酚 (δTE) 及其代谢物δTE-13′-羧基色原酚 (δTE-13′) 增加了肠道中的乳球菌和拟杆菌,并抑制炎症因子的产生。
维生素->肠道菌群->肠道疾病中的作用
▸维生素在IBD和结直肠癌中的作用不容忽视
大量临床研究表明,缺乏维生素 B 和维生素 D 的人群中结直肠癌的患病率较高。同时,IBD 的长期不愈合使患者面临更高的结直肠癌风险。维生素 D 水平低的 IBD 患者疾病严重程度和预后较差。
▸为什么肠道炎症容易导致癌症高风险?
在炎症背景下,敲除 IKKbetaβ(炎症与癌症之间的联系)可减少由于上皮细胞凋亡增加而导致的癌症发生。在一项关于结肠炎相关癌前癌 (CApC) 的研究中,IL-6 反式信号转导的存在增加了炎症性致癌的风险。如果不及时治疗,由肠道菌群紊乱和维生素缺乏引起的肠道炎症最终可能发展为癌症。
维生素 A 在肠道炎症和癌症中的作用
维生素 A 及其活性代谢物视黄酸 (RA) 在人体免疫系统中发挥着关键作用,并可能对辅助 T 细胞的分化产生影响。
炎症下:视黄酸从保护转变为破坏作用
在非炎症条件下,视黄酸能够抑制 IL-6 受体的表达和 Th1/Th17 的产生。
在炎症条件下,视黄酸从对粘膜的保护作用转变为破坏作用;这反映在活动期 IBD 患者黏膜中视黄酸水平显着升高,伴随着 CD4 和 CD8 分泌的 IL-17 和 IFN-γ 的上调。
维生素A及其代谢物:发挥抗炎作用
维生素 A 及其代谢物通过阻断 Th1 和 Th17 的激活,抑制 IL-17、INF-γ 和 TNF-α 的产生而显示出抗炎作用。同时,它们可以通过与TGF-β协同作用,提高Foxp3的水平,发挥免疫功能,从而促进抗炎因子的发挥。
一项数据显示,低水平的维生素 A 会激活核 NF-kB 并促进胶原蛋白的形成,从而加剧结肠炎的炎症。补充维生素后,肠道炎症明显缓解。
全反式维甲酸 (AtRA) 可降低 UC 和结直肠癌患者结肠黏膜分泌的 TNF-α 和一氧化氮合酶 2 (NOS2) 蛋白的表达。

维生素A保护肠黏膜屏障,其潜在机制是拮抗LPS的肠道破坏作用
在一项关于维生素 A 缺乏对结肠炎和结直肠癌发展的影响的检查中,研究人员使用葡聚糖硫酸钠 (DSS) 诱导小鼠结肠炎;此外,偶氮甲烷 (AOM) 预注射和 DSS 结肠炎的组合诱导了结直肠癌。缺乏维生素的小鼠肠道炎症水平较高,黏膜愈合较慢,免疫反应增强,更容易发生结直肠癌。
AtRA具有抗癌作用,结直肠癌中AtRA 水平降低
在结直肠癌小鼠模型中,肠道细菌引起的炎症影响 AtRA 代谢;这导致其水平下降。在 UC 及其相关结直肠癌的临床样本中发现 AtRA 代谢酶活性降低和 AtRA 水平降低。同时,AtRA通过激活CD8 + T细胞发挥抗癌作用;这为 CAC 的治疗提供了新的见解。
视黄醇和视黄醇结合蛋白(RBP)的结合激活致癌基因STRA6,促进结直肠癌的发生;Holo-RBP/STRA6 通路可通过促进成纤维细胞的致癌作用进一步发挥致癌作用。
在一项关于维生素 A 缺乏对结肠炎和结直肠癌发展影响的动物实验中,当维生素 A 处于低水平时,小鼠体内的维生素 A 脂滴会被降解,免疫反应会增强,结肠炎症会加重,癌变进程将加快。
维生素 B12 和叶酸在肠道炎症和癌症中的作用
IBD 患者缺乏维生素 B12 和叶酸的原因有很多,包括回肠和空肠微生物过度生长、维生素 B12 摄入不足或身体需求增加、维生素肠道破坏增加或吸收能力降低、某些药物(如甲氨蝶呤或柳氮磺胺吡啶)的不良反应、一些病理原因例如蛋白丢失性肠病、肝功能异常、回肠相关病变或手术切除、肠瘘等。
维生素 B12 缺乏不会影响健康的肠道微生物群组成;然而,它会导致实验性结肠炎中肠道菌群失调,并促进条件致病菌的生长。出乎意料的是,维生素 B12 缺乏减少了结肠组织的损伤;这可能与抗炎细胞因子 IL-10 的增加有关。

对甲基缺乏饮食 (MDD) 的潜在作用进行了一项研究,该饮食可降低维生素 B12 和叶酸的血浆浓度,并提高同型半胱氨酸水平,对 DSS 诱导的小鼠结肠炎的影响。喂食 MDD 的 DSS 治疗小鼠比其他治疗组患有更严重的结肠炎。
尽管超氧化物歧化酶和谷胱甘肽过氧化物酶活性保持稳定,但 caspase-3 和 Bax 的水平受到影响。除Bcl-2表达增加外,炎症相关标志物如胞质磷脂酶A2和环氧合酶2的表达也有明显增加趋势;这伴随着金属蛋白酶组织抑制剂(TIMP)3蛋白的表达降低。因此,维生素 B12 缺乏可能会加重实验性 IBD 的炎症程度。
高维生素 B12 水平可通过减少 DNA 甲基化来降低结直肠癌的风险
在结直肠癌患者中,与低血清维生素 B12 组相比,高维生素 B12 组的肿瘤区域和外周血单个核细胞 (PBMC) 中长散布的核元素 1 (LINE1) 甲基化被证明是降低的;肿瘤区域的LINE1甲基化水平也低于周围的非肿瘤区域。
氧化应激是结直肠癌发病机制之一;此外,叶酸和维生素 B12 的水平与体内抗氧化剂谷胱甘肽的水平呈正相关。提高 AOM 诱导的结直肠癌中的叶酸和维生素 B12 水平显示出显着的抗凋亡、抗氧化应激和抗 AOM 细胞毒性。
在对 4517 名 IBD 患者的系统评价和荟萃分析中,补充叶酸被证明可以降低 IBD 患者的结直肠癌风险并防止结直肠癌发展。
有趣的是,有证据表明缺乏甲基供体营养素叶酸、胆碱、蛋氨酸和维生素 B12 会抑制 Apc 突变小鼠的肿瘤发展。总而言之,维生素B12和叶酸在肠道疾病中的作用需要更深入的研究。
维生素 D 在肠道炎症和癌症中的作用
流行病学和动物实验表明,维生素 D 缺乏是 IBD 和 结直肠癌的高危因素。维生素 D 补充剂有助于降低疾病严重程度,可能通过多种机制,包括调节免疫细胞运输和分化,以及抗菌肽合成。
维生素D可以维持肠黏膜屏障的正常功能,提高机体的先天性和适应性免疫
1α,25-二羟基维生素 D3(骨化三醇)是维生素 D 的活性形式,可与 TGF-β 结合,提高 IL-2 水平,调节 T 细胞抑制炎性细胞因子的产生,增强 Foxp3 + Treg 细胞的存活和功能。

维生素D受体(VDR)是维生素D调节免疫和发挥抗炎作用的重要途径
相关资料显示,VDR对肠道有保护作用;它可以通过调节 JAK/STAT 通路来维持肠道稳态并预防癌症。
在 IBD 患者 中,结肠上皮中VDR的含量明显低于正常人。在实验性结肠炎模型中,与缺乏 VDR 的小鼠相比,表达 hVDR 的转基因小鼠的结肠炎症较少。用 hVDR 转基因恢复上皮 VDR 表达可减轻严重结肠炎并降低死亡率。内在机制是 VDR 通过抑制 NF-κB 活化发挥抗凋亡作用,以保护肠道屏障缓解结肠炎。
肠道菌群通过犬尿氨酸通路(合成维生素),在精神健康方面发挥作用
关于肠道细菌在心理健康方面的作用的关键方面,是它们通过犬尿氨酸通路参与调节色氨酸代谢。微生物群能够合成犬尿氨酸途径 (KP) 的酶促辅助因子,如维生素 B2 和 B6。

犬尿氨酸是主要的色氨酸代谢途径,其中 95% 的这种氨基酸被代谢为各种免疫和神经调节犬尿氨酸/色氨酸分解代谢物 (TRYCAT),在大脑中,犬尿氨酸途径主要在神经胶质细胞中分隔。
犬尿氨酸通路在精神、神经退行性和神经系统疾病中的作用是至关重要的,包括重度抑郁症,双相情感障碍,精神分裂症,阿尔茨海默病,亨廷顿病和帕金森病,与 HIV 感染相关的痴呆,手术后认知能力下降,肌萎缩侧索硬化(ALS) 等。
精神病理学和炎症中维生素缺乏与高同型半胱氨酸血症有关
精神病理学和炎症中维生素缺乏的另一个关键机制与高同型半胱氨酸血症(hHcy)有关,这可能是由叶酸、维生素 B6 和 B12 缺乏引起的。
高同型半胱氨酸血症和维生素 B 缺乏在重度抑郁症、精神分裂症、双相情感障碍、自闭症、焦虑症和痴呆症(包括阿尔茨海默病和帕金森病)中起关键作用。
同型半胱氨酸(Hcy)是在蛋白质消化过程中获得的另一种氨基酸蛋氨酸代谢过程中形成的氨基酸和中间体。该反应需要维生素 B12 作为酶促辅因子和叶酸衍生物(5-甲基四氢叶酸)作为甲基供体。
注:Hcy-同型半胱氨酸,是人体内含硫氨基酸的一个重要的代谢中间产物,可能是动脉粥样硬化等心血管疾病发病的一个独立危险因子。
此外,Hcy 可以在需要维生素 B6 作为酶辅因子参与的途径中转化为半胱氨酸。
因此,Hcy 被认为是叶酸和维生素 B12 缺乏的敏感标志物。
高同型半胱氨酸血症导致神经和精神病理学的机制包括:
促进免疫炎症反应、增加肠道和血脑屏障通透性、NMDA受体激动和神经毒性、诱导神经元凋亡、氧化应激、线粒体功能障碍和由于甲基化受损导致的单胺能神经递质合成失调。
目前对体内维生素水平的检测例如:
抽取血液检测其中维生素的含量水平,可以判断是否存在维生素的缺乏情况。
其他,例如通过肠道菌群健康检测,也可以查看近期体内维生素状况。
与通过血液进行维生素检测不同,肠道菌群的评估更加反映一段时间 ( 一般2周左右 ) 的长期状态,如部分B族维生素无法在体内留存,需要每日补充,血液检测波动较大。
注:菌群会受检测前一天饮食的影响,造成15~30%的菌群改变,同样也会反映在营养状况的评估上,因此建议检测前一天尽量保持近期正常饮食 ,这样能更好的反映真实的营养饮食状态。
在了解补充维生素的干预措施之前,我们先从肠道菌群的角度,来了解一下影响维生素合成吸收的因素。
人类基因的变异与肠道结构和微生物组组成有关。人类肠道微生物群中存在不同的维生素 B 生物合成途径支持人类遗传变异影响维生素 B 合成的观点。
维生素的合成吸收不仅需要靠饮食补充,还与吸收相关。而维生素的吸收涉及到相关基因,例如:
MTHFR 基因的突变会影响我们产生加工维生素 B9的酶——亚甲基四氢叶酸还原酶。
亚甲基四氢叶酸还原酶是叶酸代谢通路中的一种重要的辅酶,亚甲基四氢叶酸还原酶基因缺陷,容易造成叶酸在体内的代谢障碍,MTHFR基因最主要的两种突变为C677T、A1298C基因多态性。该两种位点同时突变可显著降低MTHFR活性进而降低叶酸水平。
VDR基因(维生素 D 受体):维生素 D(来自阳光、食物或补充剂)经过转化步骤后,活性形式骨化三醇 (1,25(OH)2D3 ) 可以通过VDR在细胞内发挥作用,是打开或关闭基因的转录因子。该基因突变可能导致维生素D缺乏引起的佝偻病。
维生素缺乏是一个严重的问题,尤其是在老年人中。随着年龄的增长,营养需求会随之变化。
由于食物中的维生素B12 需要胃酸及胃蛋白酶的作用才能释放出来被吸收,而老年人胃酸及胃蛋白酶分泌减少,就会影响维生素B12 的吸收。
患有维生素B12缺乏症的老年人可能出现神经精神或代谢缺陷。
一些药物会改变营养物质的吸收或代谢方式。例如,抗惊厥药也会减少叶酸的吸收。
肠道菌群通过各种代谢途径影响维生素的合成,例如拟杆菌属、肠球菌属和双歧杆菌属等人类肠道共生菌可以从头合成维生素 K 和大多数水溶性 B 族维生素,这在前面第二章节的表已经详细阐述。
在 B 族维生素合成中暴露于抗生素的反应因使用的抗生素类型而异。例如,在饮食中添加青霉素和金霉素会增加雄性大鼠的肝脏维生素 B2 浓度,以及 B2 和 B3 在尿液中的排泄。然而,链霉素和放线菌酮的施用降低了肝脏中维生素 B9 和 B12 的浓度。维生素合成对抗生素暴露的混合反应尚不清楚,但它们可能是由肠道微生物群的选择性改变引起的。
自由基是含有不成对电子的化学物质,可以诱导氧化应激。一个这样的例子是一氧化氮,它与金属离子形成复合物,包括钴,维生素 B12 的一种结构成分,因此使其无法用于细菌维生素 B12 的生物合成。此外,维生素生产者(如脆弱拟杆菌)暴露于过氧化氢等自由基会抑制其生长 ,从而降低维生素的生物合成能力。
维生素主要在小肠中吸收,其生物利用度取决于食物成分,相关相互作用等。
饮食和膳食的组成会通过影响肠道转运时间和/或混合胶束的肠道形成来影响某些维生素的吸收。
饮食中足量的水和膳食脂肪对于分别吸收水溶性和脂溶性维生素至关重要。
*水溶性维生素包括:B族维生素,维生素C;
脂溶性维生素包括:维生素 A、D、E 、K.
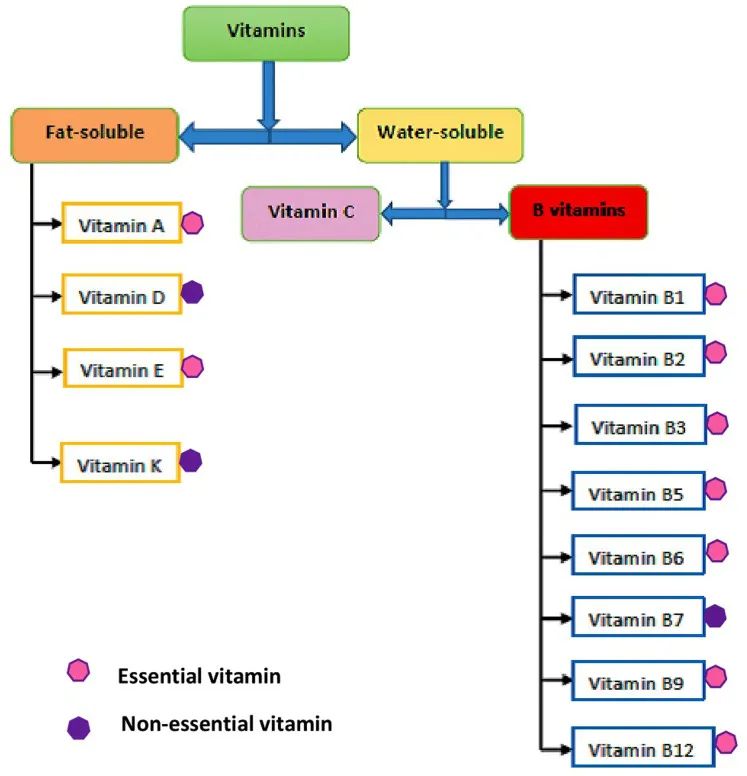
doi: 10.7717/peerj.11940
由于脂溶性维生素可以溶解在脂肪中,因此与膳食脂肪一起食用时最容易被吸收。例如,一种富含维生素 A 的小胡萝卜,如果单独食用,将在食物中获取维生素 A,但如果它是在含有一些膳食脂肪的食物成分中(比如说,橄榄油),将增加体内维生素 A 的吸收。
食物的性质(物理状态)也会影响维生素的吸收效率。例如,存在于可消化性较差的纤维植物材料中的类胡萝卜素已被证明相对于维生素A表现出较低的生物利用度。
当我们看到维生素缺乏的时候,可能希望通过饮食来补充相应缺乏的维生素,下表列出了常见的维生素的食物来源,可供参考。
此外,宿主饮食作为肠道中细菌的底物,其对肠道微生物分布的影响已被广泛研究。含有益生元和其他膳食营养素(如微量营养素和多酚)的饮食可以显着影响有益细菌的生长,包括维生素生产菌。
一些维生素,如核黄素,可作为氧化还原介质并刺激营养缺陷菌(如Faecaibacterium prauznitsii)的生长。
在即将形成共生关系的环境中,限制这些基质会增加微生物和微生物与宿主之间的竞争。
除了通过饮食直接补充之外,我们还可以通过补充益生菌来调节维生素水平,从而改善疾病。
双歧杆菌
在健康成人中补充益生菌菌株青春双歧杆菌DSM 18350、青春双歧杆菌DSM 18352 和假链双歧杆菌DSM 18353,导致粪便中叶酸浓度显着增加。
乳酸菌
乳酸菌通过不同的机制抑制炎症过程,包括调节IBD患者肠道菌群紊乱、保护肠道屏障和黏膜的正常功能、调节人体免疫反应等。乳酸菌通过产生核黄素(维生素 B2)和叶酸发挥抗炎和抗氧化作用。
产维生素的乳酸菌不仅对急性肠炎有抗炎作用,还能有效缓解复发性结肠炎。此外,在与美沙拉秦合用过程中,可有效降低不良反应,提高疗效。
研究人员发现注射产生叶酸的乳酸菌会缓解 5-FU 引起的肠炎小鼠的腹泻,改善结肠组织的结构和功能。这一发现降低了癌症化疗期间发生的肠黏膜炎症的严重程度,并提高了药物有效性;因此,这提高了患者的生活质量。
此外,乳酸菌 和 5-FU 的联合使用可减少 5-FU 引起的血细胞计数减少,并使患者获得完整的治疗周期。
产维生素的益生菌在肠道疾病中的作用

doi.org/10.3390/nu14163383
研究人员从 150 个收集的人类粪便样本中分离出三种产生核黄素和叶酸的益生菌;他们用它们来治疗乙酸引起的大鼠结肠炎。他们发现这些益生菌可以保护结肠黏膜,促进溃疡性病变的愈合;此外,它们具有抗炎和抗氧化应激作用。
一种新分离的具有产生叶酸能力的细菌——清酒乳杆菌LZ217,具有促进丁酸产生和改善肠道菌群组成的作用。
Akkermansia muciniphila 是肠道中的一种常见细菌,可调节 CLT 以保护肠道免受炎症和肿瘤侵袭;它还产生维生素 B12 以缓解 IBD 患者的维生素缺乏症。
研究发现,丙酸杆菌菌株 P. UF1 合成维生素 B12;这对肠道免疫和肠道健康有积极的调节作用。
大肠杆菌通过产生维生素来缓解 IBD. 使用大肠杆菌生产两种产生β-胡萝卜素的菌株来治疗维生素A缺乏症。这些结果显示出巨大的临床潜力。
维生素 A 及其代谢物与短乳杆菌KB290 的组合提高了 CD11c + MP/CD103-DC 比率;因此,这在结肠炎的治疗中起着积极的作用。
此外,肠道中的分段丝状细菌 (SFB) 可以产生 AtRA,以抵消感染对肠道的损害。
益生菌对维生素D及其受体活性有积极作用,如鼠李糖乳杆菌GG(LGG)和植物乳杆菌(LP);同样在沙门氏菌结肠炎模型中,使用 VDR (-/-) 小鼠验证 LGG 对 IBD 的缓解作用是通过 VDR 信号通路。
此外,胆汁盐水解酶 (BSH)活性罗伊氏乳杆菌NCIMB 30,242 可调节血浆中的活性维生素 D 水平。磷虾油 (KO)、益生菌罗伊氏乳杆菌和维生素 D 的混合物显着降低病理评分和炎症因子的释放,促进黏膜愈合并减少机会性感染的发生。
经益生菌 VSL#3 预处理后,VDR 水平显着提高,共同保护肠黏膜,防止损伤;这对预防CRC的发展起到一定的作用。
用从韩国泡菜中分离的乳酸菌条件培养基处理 HCT116 细胞或肠类器官后,其分泌的蛋白质 P40 和 P75 与 VDR 的表达增加有关;它们还增强自噬反应,共同具有抗炎作用。肠道微生物合成的石胆酸 (LCA) 充当连接 VDR 与微生物的桥梁,从而提高维生素 D 水平。
益生菌配方有助于抑郁症患者维生素水平的增加
一项随机对照试验中,重度抑郁症患者接受了多种益生菌配方,其中含有双歧杆菌W23、乳双歧杆菌W51、乳双歧杆菌W52、嗜酸乳杆菌W22、干酪乳杆菌W56、副干酪乳杆菌W20、植物乳杆菌W62、唾液乳杆菌W24、乳酸乳杆菌W19。
此外,益生菌组和安慰剂组的患者接受了相同剂量的维生素 B7。在两组中,抑郁症的临床参数都有所改善,然而,益生菌干预组与安慰剂组相比,仅在微生物 β 多样性方面存在差异,临床结果指标没有差异。有趣的是,尽管两组都接受了相同剂量的生物素,但接受益生菌的那组维生素 B6 和 B7 的合成上调。
多种益生菌相结合通过增加叶酸和维生素 B12血浆水平,改善精神疾病
八周的个性化饮食与含有多种益生菌的菌株相结合:婴儿双歧杆菌DSM 24737、长双歧杆菌DSM 24736、短双歧杆菌DSM 24732、嗜酸乳杆菌DSM 24735 、德氏乳杆菌、保加利亚乳杆菌DSM 24734、副干酪乳杆菌DSM 24733、植物乳杆菌DSM 24730 、嗜热链球菌DSM 24731 (VSL#3),在健康老年人中增加了叶酸和维生素 B12 血浆水平并降低了 Hcy 血浆水平。
此外,益生菌的添加导致粪便双歧杆菌浓度增加,这种变化与叶酸和维生素 B12 水平呈正相关。
在精神病患者中引入高同型半胱氨酸的评估和治疗可能非常有价值,益生菌可能成为治疗工具之一。
维生素是相互关联的、具有协同作用的微量营养素,当它们处于适当的平衡状态时,它们的全部潜力就会得到充分发挥。
因此,在食用益生菌和发酵食品时,应考虑维生素生产者与代谢者之间复杂的相互作用。
除了以上方式干预菌群之外,也可以通过良好的生活方式调理菌群,从而使维生素达到一个相对健康稳定的水平,减少各类疾病风险。
▸在服用维生素的同时可以服用益生菌吗?
可以。在大多数情况下,服用益生菌不会影响其他补充剂的效果。
一项 2021 年对临床试验的系统评价发现,益生菌可以改善健康人群的微量营养素水平,特别是维生素 B12、叶酸(维生素 B9)、钙、铁和锌。
2017 年的一项非随机临床试验发现,服用益生菌和铁补充剂的参与者比不服用益生菌的铁吸收明显更多。
有研究表明,维生素 D 和益生菌之间存在协同关系。
随机对照试验发现,维生素 D 补充剂与益生菌一起可以改善多囊卵巢综合征患者和同时患有冠心病的糖尿病患者的各种心理健康参数、一般健康状况、代谢和炎症标志物。
2019 年对随机对照试验的系统评价和荟萃分析发现,维生素 D 强化酸奶(富含益生菌嗜酸乳杆菌)有助于改善维生素 D 和胆固醇水平、代谢功能和身体测量值。
然而以上研究都没有单独研究维生素 D 和益生菌的作用,因此尚不清楚结果是否与两者的综合影响有关。
研究人员认为,无论有没有维生素,服用益生菌的时间很重要。作为一般规则,服用益生菌的最佳时间是空腹,大约在进食前 30 分钟。
研究人员担心胃酸的存在会影响益生菌的生存能力。在餐前或餐后几个小时服用时,当胃酸自然降低时,益生菌可以进入肠道,从而提高其生存几率。
何时服用维生素取决于维生素的种类。复合维生素通常最好在早上第一时间服用,非常适合搭配早餐前的益生菌。脂溶性维生素,如 A、D、E 和 K 以及一些矿物质,包括铁和镁,最好与食物一起服用。否则可能会导致胃部不适。
▸应该从食物中补充维生素还是通过维生素补充剂?
2020 年的一篇文献综述发现,与浓缩补充剂相比,许多微量营养素在其全食物形式中的生物利用度更高。因此提倡补充方式以食物为先。
一般认为,对于健康人来说,营养均衡的饮食可以提供身体需要的维生素,不需要额外补充,但对于可能存在免疫功能、肠道健康、吸收不良等问题的人群,可以考虑维生素补充剂进行补充,具体补充剂量请遵医嘱。
下表是维生素易缺乏的高风险人群:

▸可以长期服用维生素补充剂吗?补充过量会带来副作用吗?
一般健康人不需要长期服用维生素补充剂。
对于服用复合维生素片,多余的维生素会被排出体外,因此不用过于担心会带来危害。但是如果长期十倍以上的用量,对身体是有危害的。

doi: 10.7717/peerj.11940
在发现维生素缺乏的症状的时候,我们可能希望通过补充相应的维生素补充剂来改善健康。然而服用任何补充剂之前,我们应该寻找其根本原因而不是直接根据症状盲目补充。
通过肠道菌群健康检测可以了解维生素缺乏状况,且可以根据各类菌群丰度来推断维生素的菌群代谢状况。如果是由于菌群的代谢异常,可能直接补充并没有太大效果,这时候优先调节菌群或许是更好的选择。
如果维生素指标都显示正常没有缺乏(如下图),保持常规饮食不需要刻意补充。还想要更健康,指标更接近70的话,可以在数值略小的指标上,针对性地通过饮食进行补充调理。
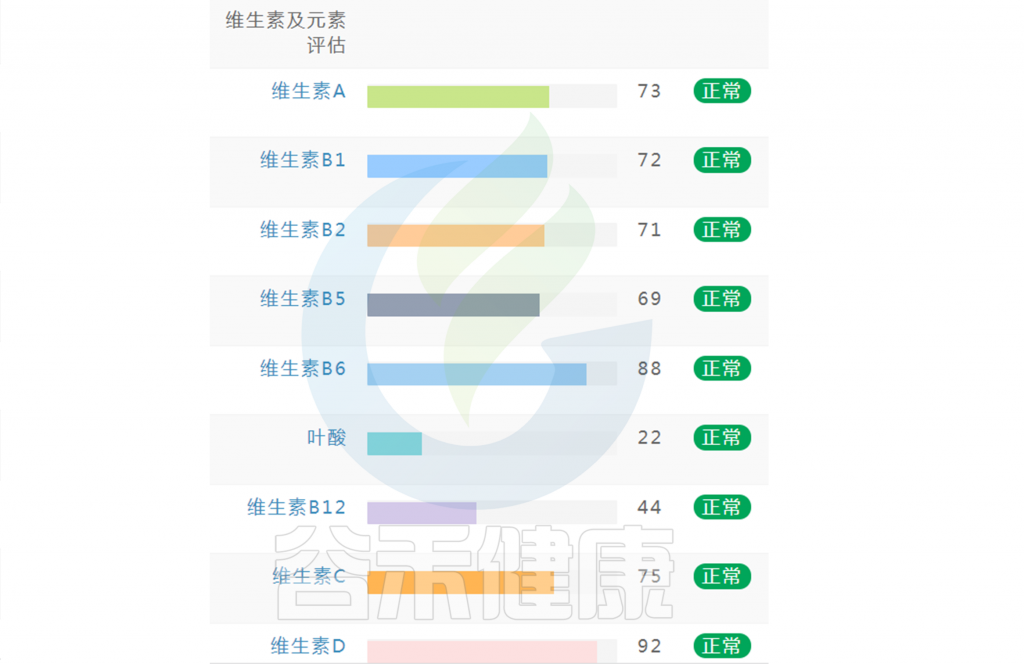

如果维生素指标中出现个别指标缺乏或偏低(如下图),可以通过饮食针对性地进行改善调整,如果已经出现对应症状,例如缺乏维生素A,同时出现干眼症或者夜盲症等相应的症状,可以使用相应的维生素补充剂进行干预,或者根据菌群代谢通路判别,通过菌群调理进行相应干预。
如果维生素指标中出现缺乏或偏低的指标较多,则需要选用复合维生素,各类维生素之间可能存在协作关系,同时配合饮食、菌群进行干预。
选择补充剂,应优先考虑生产规范良好的产品,比如说可以查看是否有“OTC”标志。
注:本账号内容仅作交流参考,不作为诊断及医疗依据。
主要参考文献:
Zhai Z, Dong W, Sun Y, Gu Y, Ma J, Wang B, Cao H. Vitamin-Microbiota Crosstalk in Intestinal Inflammation and Carcinogenesis. Nutrients. 2022 Aug 17;14(16):3383. doi: 10.3390/nu14163383. PMID: 36014889; PMCID: PMC9414212.Zhai Z, Dong W, Sun Y, Gu Y, Ma J, Wang B, Cao H. Vitamin-Microbiota Crosstalk in Intestinal Inflammation and Carcinogenesis. Nutrients. 2022 Aug 17;14(16):3383. doi: 10.3390/nu14163383. PMID: 36014889; PMCID: PMC9414212.
Bellerba F, Muzio V, Gnagnarella P, Facciotti F, Chiocca S, Bossi P, Cortinovis D, Chiaradonna F, Serrano D, Raimondi S, Zerbato B, Palorini R, Canova S, Gaeta A, Gandini S. The Association between Vitamin D and Gut Microbiota: A Systematic Review of Human Studies. Nutrients. 2021 Sep 26;13(10):3378. doi: 10.3390/nu13103378. PMID: 34684379; PMCID: PMC8540279.
Ofoedu CE, Iwouno JO, Ofoedu EO, Ogueke CC, Igwe VS, Agunwah IM, Ofoedum AF, Chacha JS, Muobike OP, Agunbiade AO, Njoku NE, Nwakaudu AA, Odimegwu NE, Ndukauba OE, Ogbonna CU, Naibaho J, Korus M, Okpala COR. Revisiting food-sourced vitamins for consumer diet and health needs: a perspective review, from vitamin classification, metabolic functions, absorption, utilization, to balancing nutritional requirements. PeerJ. 2021 Sep 1;9:e11940. doi: 10.7717/peerj.11940. PMID: 34557342; PMCID: PMC8418216.
Steinert RE, Lee YK, Sybesma W. Vitamins for the Gut Microbiome. Trends Mol Med. 2020 Feb;26(2):137-140. doi: 10.1016/j.molmed.2019.11.005. Epub 2019 Dec 17. PMID: 31862244.
Pham VT, Dold S, Rehman A, Bird JK, Steinert RE. Vitamins, the gut microbiome and gastrointestinal health in humans. Nutr Res. 2021 Nov;95:35-53. doi: 10.1016/j.nutres.2021.09.001. Epub 2021 Oct 21. PMID: 34798467.
Hossain KS, Amarasena S, Mayengbam S. B Vitamins and Their Roles in Gut Health. Microorganisms. 2022 Jun 7;10(6):1168. doi: 10.3390/microorganisms10061168. PMID: 35744686; PMCID: PMC9227236.
Rudzki L, Stone TW, Maes M, Misiak B, Samochowiec J, Szulc A. Gut microbiota-derived vitamins – underrated powers of a multipotent ally in psychiatric health and disease. Prog Neuropsychopharmacol Biol Psychiatry. 2021 Apr 20;107:110240. doi: 10.1016/j.pnpbp.2020.110240. Epub 2021 Jan 9. PMID: 33428888.
Barone M, D’Amico F, Brigidi P, Turroni S. Gut microbiome-micronutrient interaction: The key to controlling the bioavailability of minerals and vitamins? Biofactors. 2022 Mar;48(2):307-314. doi: 10.1002/biof.1835. Epub 2022 Mar 16. PMID: 35294077; PMCID: PMC9311823.

谷禾健康
宏基因组测序可以使我们深度全面地了解微生物群的构成,对于缺乏深度研究和高质量参考基因组的样本,宏基因组获得的较为完整的基因组不仅可以丰富参考基因组数据库,同时可以提供更加准确的物种分类。
关于宏基因组的介绍可见我们之前的文章:

在宏基因组分析过程中,可能遇到的问题,及问题相关解决思路如下:








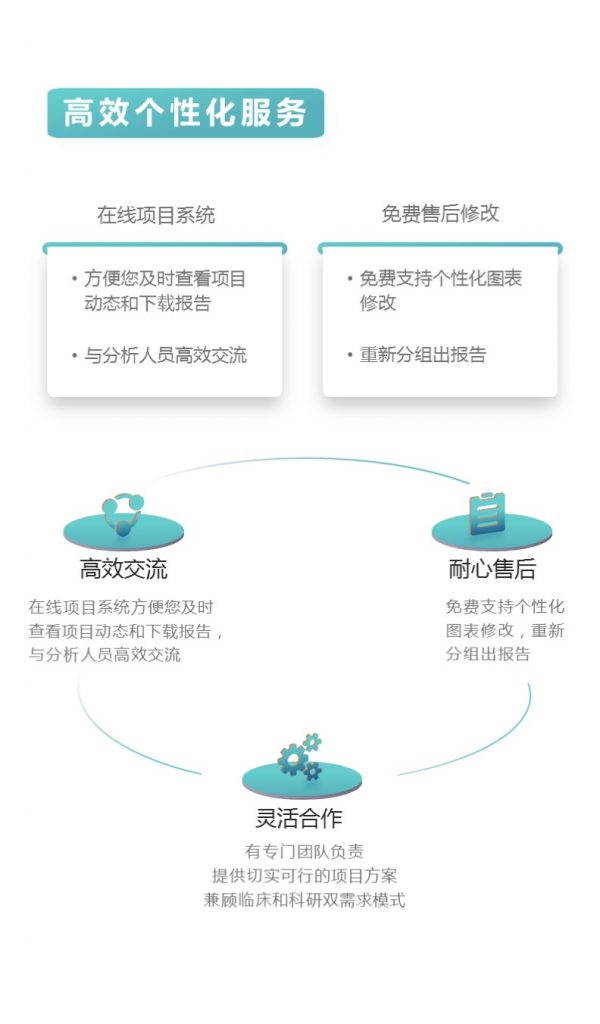






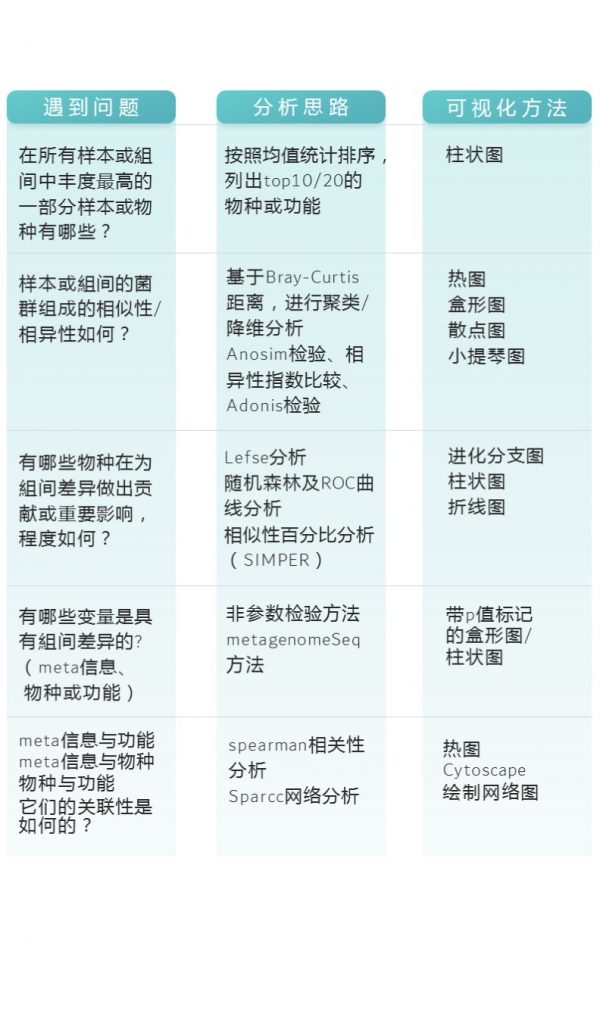


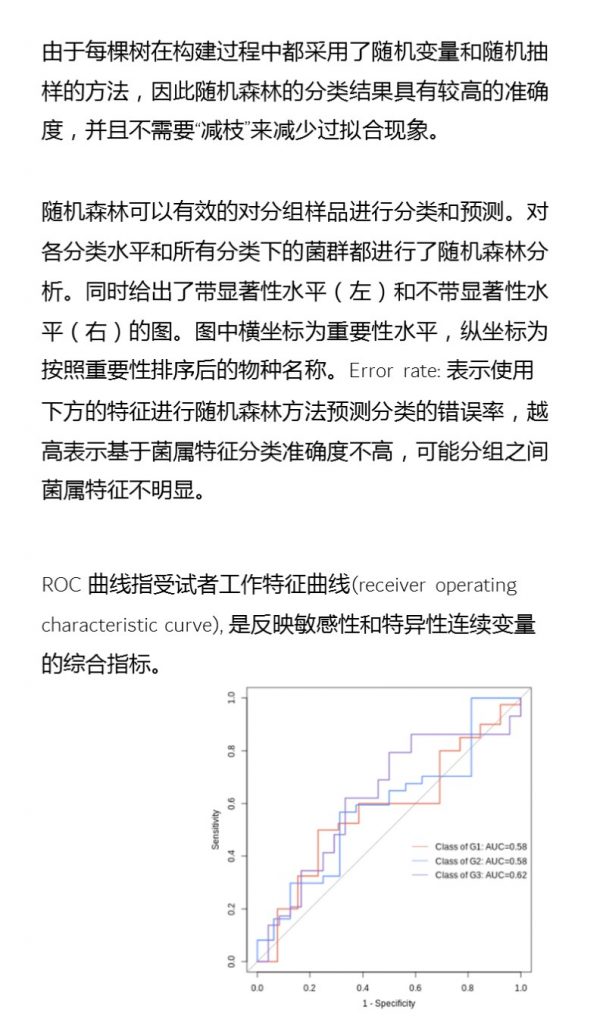

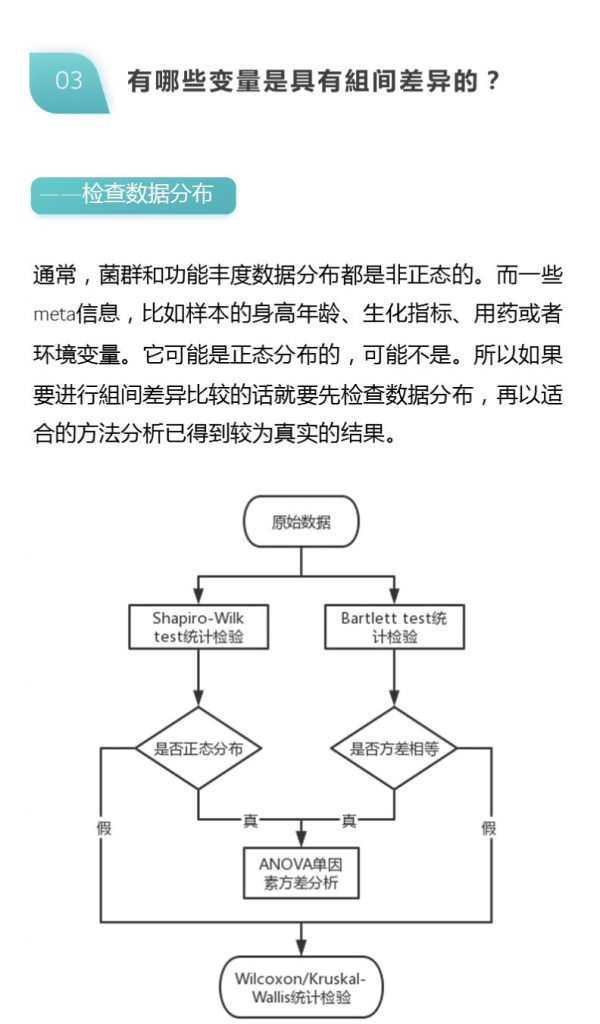

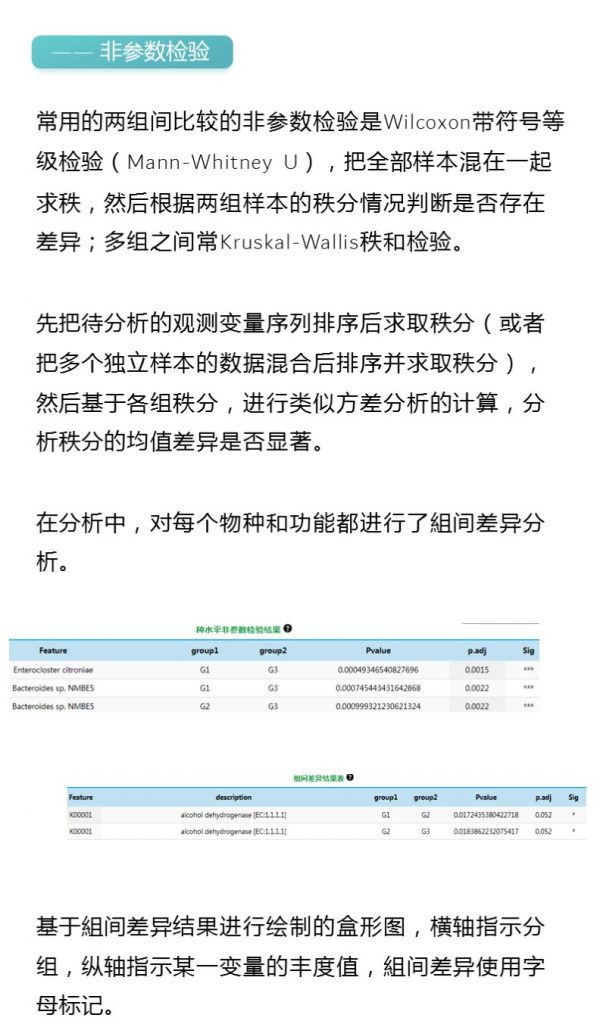

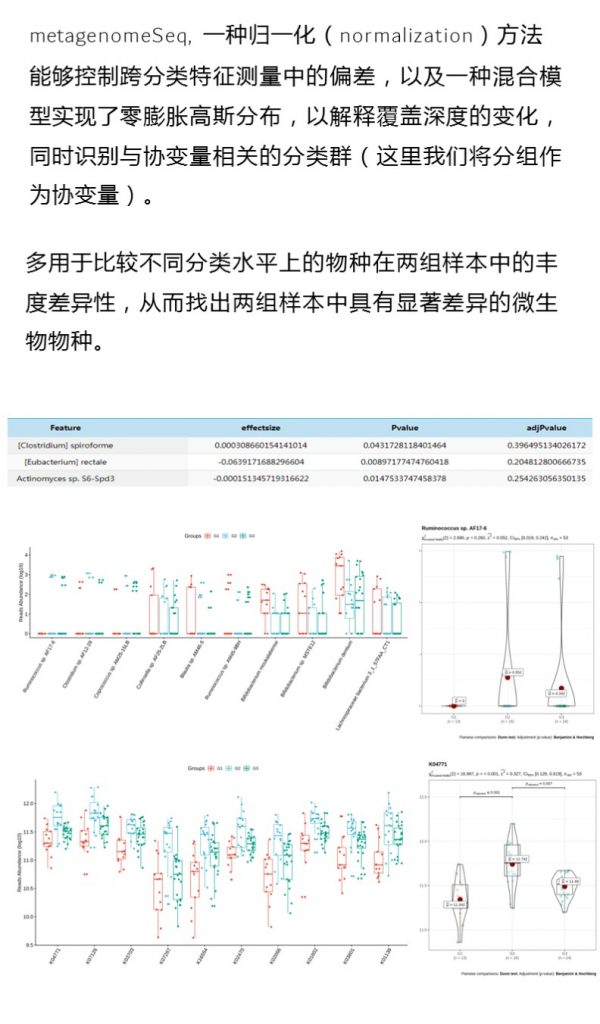

























更多关于宏基因组科研服务详询:
商务经理:13336028502(微信同号)

谷禾健康

在持续的肠道菌群检测实践过程中,我们收到很多新的问题反馈和对肠道菌群检测在具体问题中的疑问。在此谷禾基于长期和大规模样本群的经验以及实验分析,对部分常见问题进行汇总和整理。

一次肠道菌群检测好比一场健康考试,你拿到报告的那一刻,等同于拿到了你考的那张卷子,那么你首先会关心自己考了多少分。
在肠道菌群检测报告中,同样也有基于肠道菌群的健康评估分数,即健康总分。
基于大数据和整体性评估,报告中会给出健康总分这项指标。这个健康总分是如何计算得出的?
还是拿我们最熟悉不过的考试举例,一场语文考试可能包括了拼音词语、阅读理解、写作等模块,所以最后你的总分是综合各个模块的测试之后得到的(比如说拼音写错了扣1分,阅读理解错了一题扣5分……),通过各模块测评后得到的总分反映的是你的综合能力。
健康总分也是一样,综合计算了三个部分:肠道菌群健康状况、疾病风险情况和营养饮食均衡情况综合评估计算。总分100分,采取扣分制,疾病风险和营养不均衡以及菌群失衡都会相应的减分。
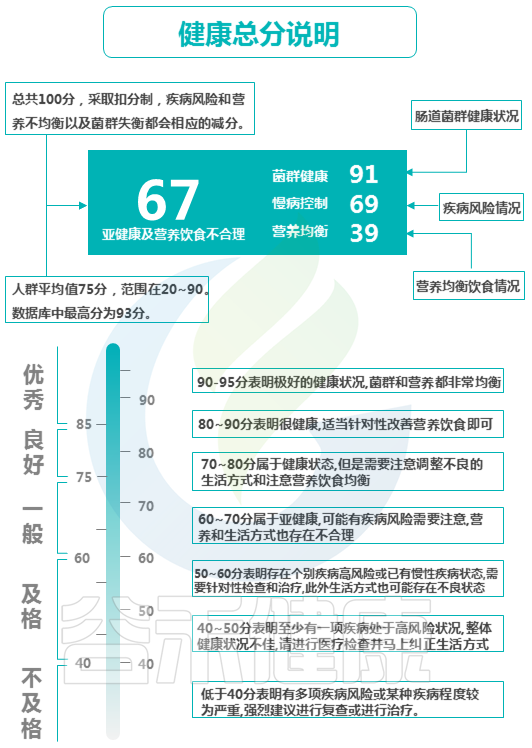
以上是具体的评分标准。
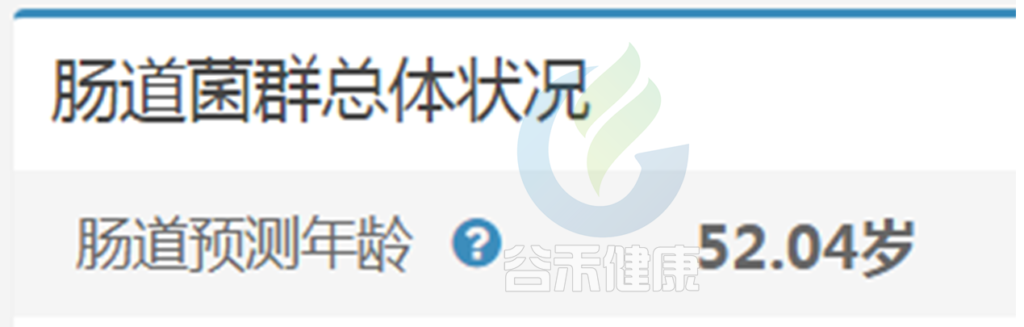
健康总分可以说是非常直观的一个指标,除此之外,整体性评估指标还有一个:肠道预测年龄。
生理年龄是指人达到某一时序年龄时生理和其功能所反映出来的水平,是从医学、生物学角度来衡量的。
谷禾肠道预测年龄是基于超过6万人群队列的深度学习模型构建的,对健康人群的肠道年龄预测与真实生理年龄吻合度很好。
肠道预测年龄和生理年龄就像齿轮运作,井井有条匹配状态,身体这个系统运作起来相对健康轻松。
疾病人群或菌群紊乱人群,肠道年龄会较大偏离真实年龄,也就是这个齿轮系统出现一些偏差问题。
如果肠道菌群多样性下降,且以大肠杆菌为主,可能会被预测为10岁以下儿童,也就是预测年龄远小于真实年龄。
如果存在较多病原菌,则预测年龄会偏向远大于真实年龄。
如果菌群预测年龄和实际生理学年龄相差很大,如何解读?
还是用考试来说,每个年龄段都应具备该年龄段的能力。如果你是一个初中学生,那么就应该答出初中阶段学生该会的题,这时候给你做个测评,发现还停留在幼儿园水平或者已经到了大学生水平,要么太幼稚要么太早熟,都不符合健康的身心发展规律。
肠道预测年龄同样,如果肠道预测年龄偏离实际年龄很大,两种情况,一种是偏大,另一种是偏小。
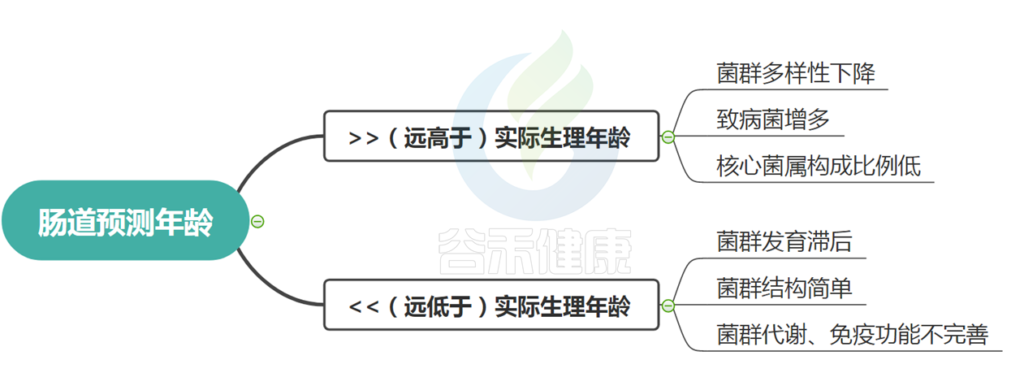
这两种情况均表明菌群发育成熟偏离了实际生长发育,我们均认为其代表菌群状况不太好,存在菌群异常或不健康状况。
如果偏小,即肠道年龄远小于生理学年龄,一般菌群发育滞后或者偏幼龄,菌群构成简单,代谢以及免疫功能不完善。
如果偏大,即肠道年龄远大于生理学年龄,一般菌群多样性下降,变形菌、肠杆菌等致病菌增多,核心菌属构成比例低等。
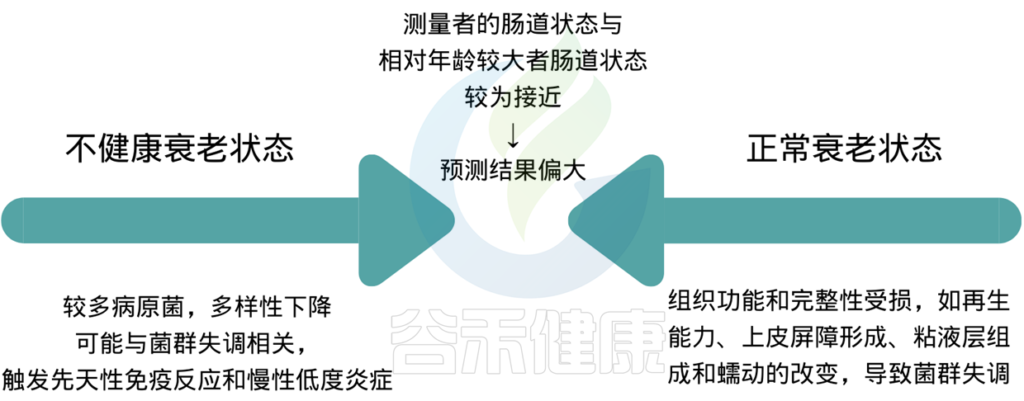
而在正常范围内,肠道预测年龄小于生理学年龄,那么表示菌群发育正常,菌群构成和代谢偏向于更年轻,比较好。那么什么是正常范围呢?
谷禾肠道年龄预测如下范围内表示正常:
0~2岁:偏差小于3个月
3~5岁:偏差在6个月以内
6~15岁:偏差在1岁左右
16~50岁:偏差在3岁以内
50岁以上:偏差在5岁以内
真实年龄与肠道预测年龄在范围内的差异可以反映其肠道菌群的发育和衰老状况。以下情况可能会导致肠道预测年龄完全偏离真实年龄,包括:
▪ 肠道菌群紊乱
▪ 菌群结构过于单一
▪ 近期服用可能严重干扰菌群的药物(如抗生素)
▪ 病原菌感染或者处于疾病状态
▪ 长期补充益生菌
由于肠道年龄考虑了整体的肠道菌群结构,如果肠道年龄严重偏离真实年龄,通过干预调整或去除上述干扰因素肠道年龄是能够恢复正常范围,但该干预周期一般需要1个月以上。
有益菌
有益菌包括益生菌,益生菌主要来自两个菌属:
分别是双歧杆菌属和乳杆菌属,目前已获得批准的有效益生菌菌株均来自这两个细菌属。
其中双歧杆菌可有效改善肠道状况,而特定的乳杆菌菌株可以改善精神健康,包括焦虑和情绪,也能改善肠道健康。双歧杆菌和乳杆菌也是人体肠道菌群中常见的菌。
虽然说是常见菌,却不见得它们数量多。在成年人肠道菌群中,双歧杆菌的比例较低,在1%左右,乳杆菌更是低于1%,甚至很多人(20~40%)的肠道菌群中比例低至万分之一。
下表是谷禾检测的益生菌列表,列出了主要的常见益生菌。
除了上述益生菌,有益菌还包括下列种属,这些菌属是构建肠道菌群的核心菌属,在评估有益菌水平时根据菌属对肠道菌群结构的重要性会给予不同的权重。
Faecalibacterium、Ruminococcus、Roseburia
Phascolarctobacterium、Prevotella、Parabacteroides
Oscillospira、Megamonas、Lachnospira
Lachnoclostridium、Gemmiger、Eubacterium
Coprococcus、Dorea、Dialister
Clostridium、Blautia、Bacteroides
Akkermansia、Alistipes、Agathobacter
通常益生菌的检出率比较低,一般在益生菌补充一周左右在报告中可以体现。从大数据来看,益生菌检出的同时,菌群的相关指标也会有所提升,比如说有害菌降低,改善菌群平衡状况。
有害菌
有害菌和肠道内的其他共生菌共同构成菌群微生态,也是大部分人群肠道内常见的菌群。
有害菌是相对而言的,正常肠道菌群也包含许多这些菌属的菌,但有害菌比例或个别菌属丰度超标可能预示着肠道菌群的健康状况受到破坏。这些菌过多会影响肠道内环境,如pH值,含氧量以及肠道内毒素等,可能会导致出现一些机会感染和机会致病菌入侵,进而诱发炎症和疾病。
我们报告中的有害菌包含了致病菌和条件致病菌,以及属内主要菌种为致病菌的属。为便于统计,我们在计算的时候统一按照属层级进行计算比例。
报告中的有害菌包括了以下的菌属:韦荣氏球菌属、葡萄球菌科、变形菌属、弓形菌属、弯曲菌属、螺杆菌属、厌氧螺菌属以及弧菌属等。
在肠道菌群检测报告中会有对有益菌,有害菌的整体评估。
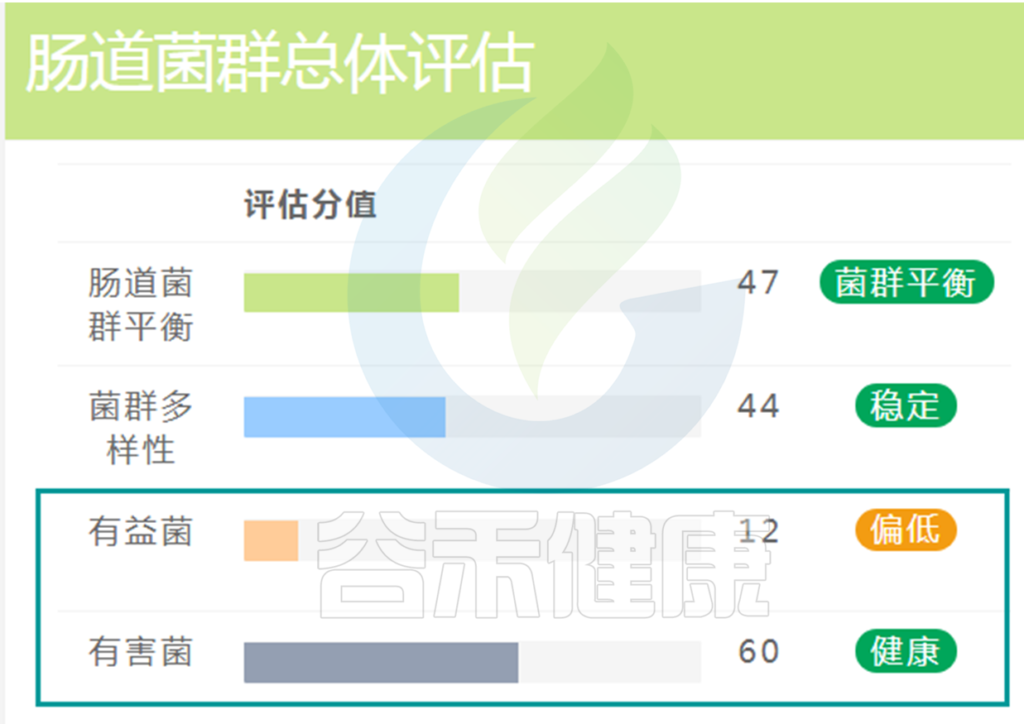
如果有害菌过多,通常建议服用益生菌或益生元的方式首先增加有益菌的比例,相应的有害菌比例就会降低。想要持久的改善菌群结构降低有害菌水平就需要改善生活方式,适当增加抗性淀粉等膳食纤维并规律饮食和睡眠,增加运动等。
整个生态系统平衡对于地球而言十分重要,同理,肠道菌群平衡对于我们人体健康也很重要。健康的肠道菌群丰富且多样性高。
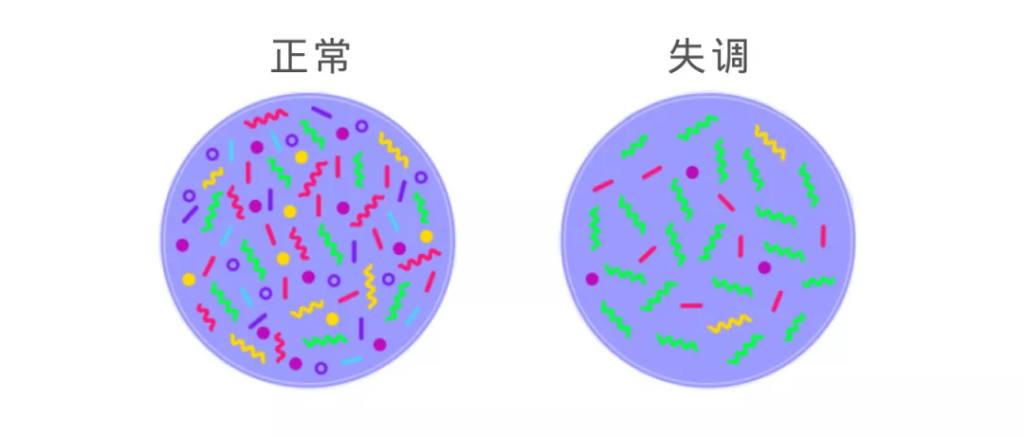
菌群失调是指体内微生物群不平衡,这可以表现为某些细菌的出现率较高,细菌的出现率较低,细菌的多样性不足,有害菌,有益菌比例失调等。
通常临床上采用大便常规检查,通过显微镜下观察统计染色细菌中杆菌和球菌以及革兰氏阴性和阳性菌的比值是否超标来判别的。
其中致病菌多为球菌和革兰氏阴性菌,而肠道有益菌多为杆菌和阳性菌,因而在传统临床上简单比较两者的比值评估是否菌群紊乱,是相对比较粗放的。
谷禾菌群检测报告中的菌群失调:
基于高通量测序可以精准的检测低至万分之一水平的菌,甚至可以分类到种水平,因此可以更加精细化评估菌群是否出现紊乱和异常。
基于谷禾超过30万人群的菌群数据库分析结果,我们将在90%的人群都有检出,且人群平均丰度1%以上的菌属做为核心菌属。这些核心菌属通过长期与人类共生,在帮助消化复杂碳水化合物和产生短链脂肪酸外还影响整个肠道环境,抑制病原微生物的定植生长。因此当这些核心菌属占总肠道菌群比例低于60%时,肠道菌群很可能处于紊乱状态。
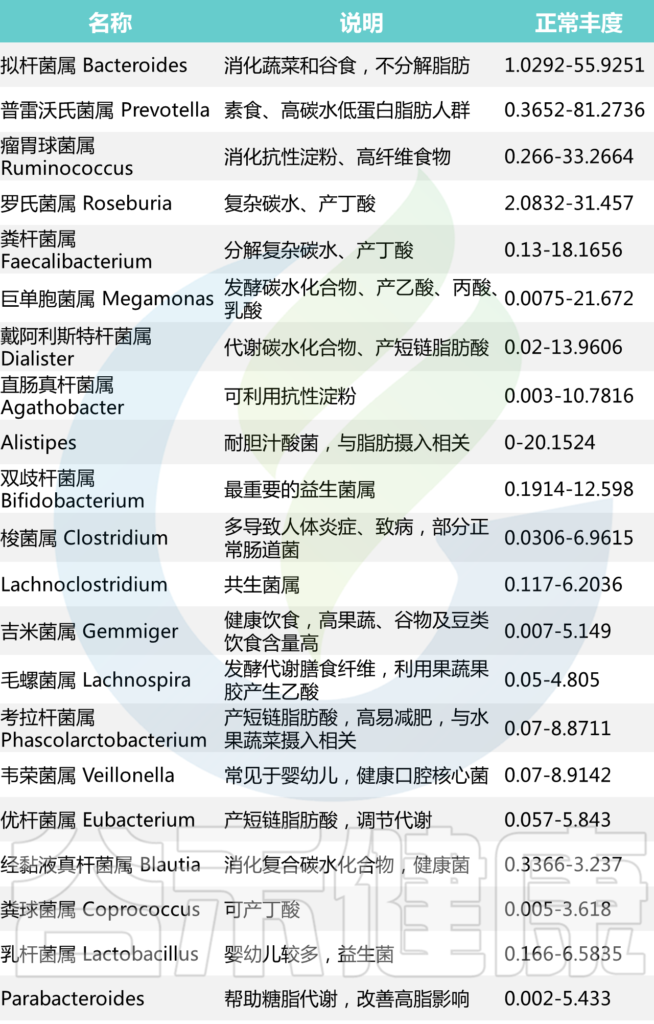
【谷禾健康菌群数据库】
如果出现菌群严重失衡,例如致病菌占了相当大比例,那么首先应考虑针对致病菌使用相应的抗生素治疗,然后再通过益生菌补充及饮食、生活方式的改变进行调理,直到菌群恢复平衡。
多样性包含两个维度。
一个是肠道菌群种类,人群中肠道菌群的种类参考范围在100~2000种,种类数量越多多样性越高。
另一个维度是均匀性,即各个菌种的含量丰度较为均一没有出现单一菌种占据绝大部分的情况。
多样性的评估一般通过一个叫做香农-维纳多样性指数的指标来进行评估,计算公式为:
H=-∑(Pi)(log2Pi)
其中Pi为每个菌的占比例,值越大代表物种种类越多,均匀性也更好相应的多样性也越高。正常人群中香浓指数在2~9之间,一般大于3以上表明具有一定多样性。
换句话说,肠道菌群多样性表现在:微生态系统的稳定性,以及面对外界致病菌等入侵的抵御能力。
在一定范围内,更高的多样性通常代表饮食更加丰富多样,同时也意味着更健康的身体状况。
菌群多样性高可能与下列情况有关:
环境,农村儿童比城市儿童菌群多样性高;
饮食,低脂饮食与菌群多样性较高有关;
年龄,长寿老人的菌群多样性较高;
……
多样性低不代表一定有疾病,但是更容易受到饮食,环境或疾病的影响,包括更易发生水土不服或更容易因饮食不洁导致腹泻等。
多样性低可能与下列情况有关:
分娩方式,剖腹产宝宝菌群多样性较低;
饮食营养,营养不良的孩子菌群多样性会下降;
药物,抗生素的使用会大幅降低菌群多样性,并且需要一段时间才能恢复。其他药物也会降低菌群多样性,如治疗胃溃疡和反酸的质子泵类药物也会导致菌群多样性降低;
环境,医院的ICU病房、更衣室等消毒严格,可能导致环境菌群多样性下降。
此外,神经系统、代谢、免疫等慢性疾病也与多样性下降有关。
你可以通过在饮食中增加纤维素,从高脂饮食逐渐转为低脂饮食来提高菌群多样性,另外规律运动也可增加多样性。
另外,我们在实际检测中会发现有这样一种情况:
多样性指标虽然很高,但是整体看起来健康总分并不理想。甚至还有很多慢性疾病风险,这是为什么呢?
这种情况可能是核心菌群丰度不够,核心菌群在代谢、免疫等方面都发挥重要作用,一旦核心菌群丰度下降,则可能造成外源物质侵入。感染、旅行等可能会出现这种情况。
看过我们检测报告的可能会发现,报告里有包括肠道致病菌和病原菌,分别代表什么?

<篇幅关系,此处仅展示部分>
肠道致病菌列出了最主要和常见的感染类肠道致病菌。(注意这里重点是肠道)

病原菌中给出的包括几十种人体的致病菌,不仅仅是肠道的。<如果没有检出就没有列出>
病原菌和条件致病菌的区别是什么?
病原菌一般极少存在于健康人的肠道菌群,正常范围很小,条件致病菌一般会在正常人群的肠道内存在,丰度较高或菌群结构单一到一定程度会引发疾病。如大肠杆菌和肺炎克雷伯氏菌正常人群中都会有检出,但当丰度较高是就会导致肠道菌群紊乱或疾病。
报告中如果出现病原菌超标的情况,不一定直接认为有病,需要结合症状。
如果出现相应的腹泻等症状,需要考虑是不是因为这些病原菌导致的。单纯超标如果没有症状只是表面有病原菌摄入,注意一下饮食和生活卫生,无须过于担心。
★ 幽门螺杆菌
为什么在医院检查出幽门螺杆菌感染,而报告中并未显示?
注意:本检测未检出并不代表完全不存在该致病菌感染,可能由于比例或其他因素未能达到检测丰度或未检出。
如果肠道菌群检测报告中检出幽门螺杆菌,是否需要去医院进行幽门螺杆菌呼气检测?
如果肠道菌群检测报告显示该项为超标,且同时存在胃部不适或其他胃酸、胃胀等症状,建议前往医院进行幽门螺旋杆菌检测,及早发现治疗。
★ 沙门氏菌
在食物中毒案例中,通常伴随着沙门氏菌,沙门氏菌粘附到肠上皮表面是发病机制中重要的第一步,并且是其在肠道定植的核心。
关于沙门氏菌的治疗及预防详见:食物中毒一文
扩展阅读:细菌大盘点(二) | 葡萄球菌、沙门氏菌、弯曲杆菌
通过以上部分,我们大概了解了菌群的构成及其扮演的角色,那么我们能利用检测到的这些菌的信息,给我们的健康带来什么帮助呢?
很重要的几个点:
第一,也就是前面所述的,菌群的构成本身就可以反映你的肠道内的环境是不是健康菌群,如果紊乱,它会带来很多的问题,比如说儿童菌群紊乱,可能会营养不良,因为菌群紊乱本身会影响营养吸收。
第二,对病原物的抵抗,也就是说身体是不是比较容易出一些状况,比如说腹泻,感染等问题。
第三,它还会诱发一些长期的慢性疾病,比如说糖尿病,实际上当然饮食是一个问题,但是有一些炎症相关的菌群,会诱发慢性的持续的炎症,从而导致慢性疾病的发展。
这就是我们接来下要讲的,疾病风险这块内容。
目前我们疾病风险检测部分包括16类主要疾病,根据疾病检测准确度和稳定性,我们将检测疾病的水平分为三个等级:低风险、中风险和高风险。
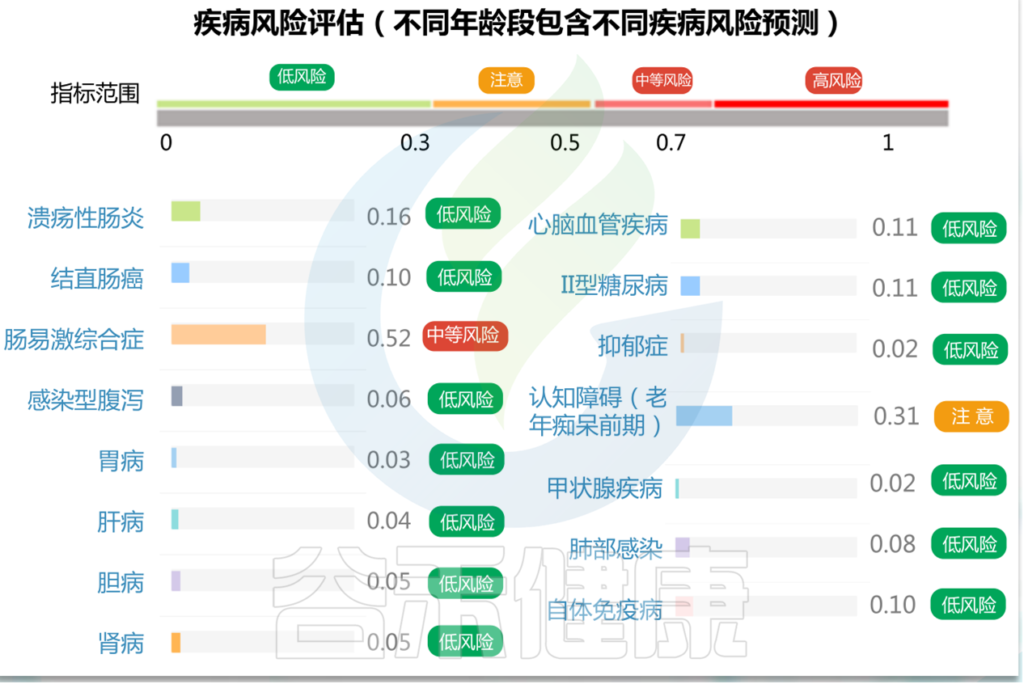
根据每种病的分值,0~0.3归为低风险,0.3~0.5评估为注意,0.5~0.7为中等风险,超过0.7为高风险。
目前报告中提供的疾病均经过大量病例样本检验并且准确率超过90%,虽然不作为疾病的诊断依据,但是其分值的高低仍然具有很强的指示作用。
0-0.3
如果某种疾病的风险值低于0.3以下表明菌群状态提示疾病风险较低,不同身体条件和生活方式下会有0.05的波动。
0.3-0.5
如果某种疾病的风险值位于0.3~0.5之间我们认为属于疾病前期阶段,通过饮食调理和相应的注意就可以降低风险。
0.5-0.7
如果某种疾病的风险值位于0.5~0.7之间表明可能患有该疾病或处于疾病风险阶段,这时候我们建议最好前往医院相关科室进行检查,如果不便前往医院也可根据建议先进行饮食调理和相应的注意,一般一个月后再进行一次检测查看疾病风险是否下降到正常范围,如果仍然较高甚至升高建议最好前往医院复查。
0.7- 1
如果某种疾病的风险值超过0.7表明有很大可能已患有该疾病,且分值越高表明风险越高。因此我们强烈建议去医院进行相应检查并听从医生建议。
注意:本检测目前尚不属于医疗诊断,疾病分值作为提示,低分值不代表完全没有疾病,只表示风险较低,也可能存在一定的未检出。高分值只表示存在很大疾病风险,疾病的确诊和精确诊断需要通过进一步的医疗检查确认。
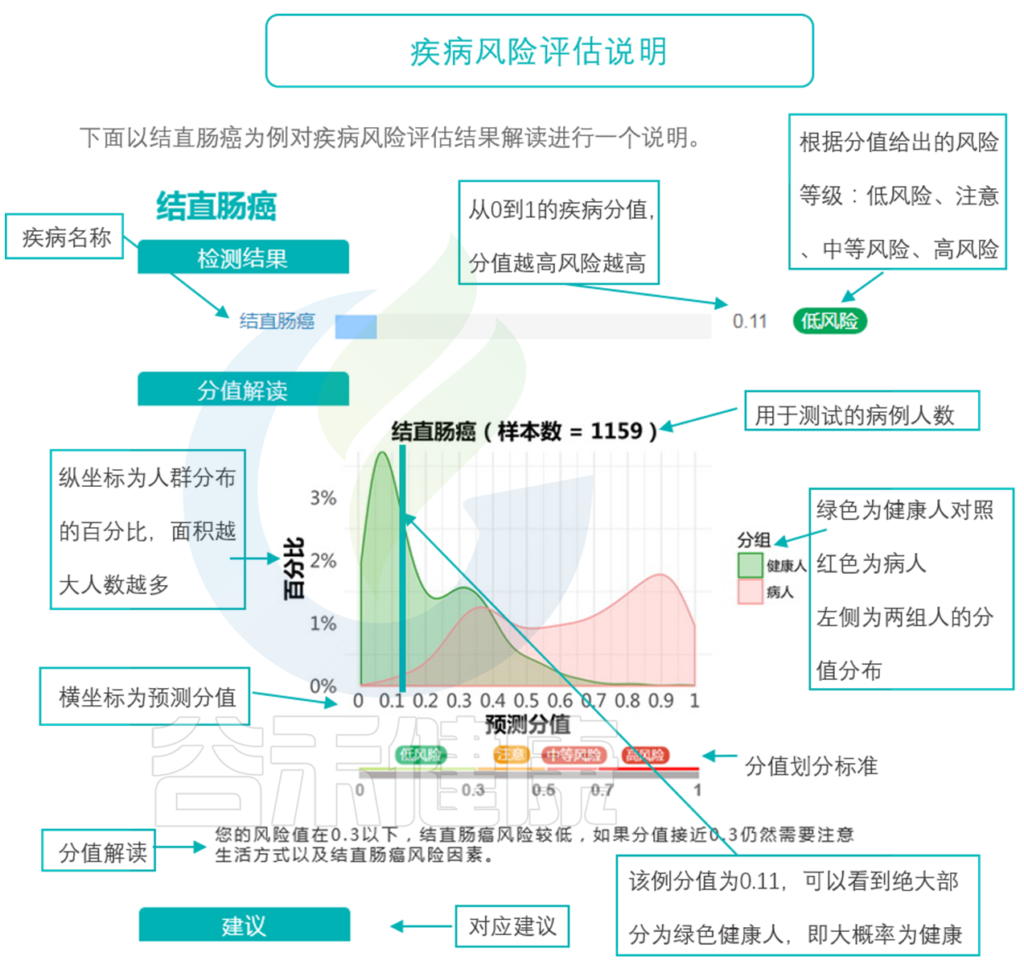
说到这里,可能有人对以上这个0.3,0.5…这些风险值有所不解,风险值是你们自己确定的吗?如何计算得出这个值的呢?有参考依据吗?
这里我们来了解一下风险值的计算。
通过模型的构建和大规模人群队列的测试和学习,现在大概已经有几十种病,我们可以比较好的通过菌的构成,来预测到底有没有这个疾病。虽然现在它还做不到直接确诊,但它可以起到一个很好的提示作用,以及对病程进展的评估。
那么,具体哪些方面的疾病跟菌群有重要的关系,并且能够用菌群来预测和评估呢?
消化系统疾病
首先当然是消化道疾病,这很好理解,因为菌群本身就在消化道环境内。像肠炎,就包括克罗恩病,溃疡性结肠炎之类的,还有消化性的腹痛、腹胀这些问题,可能是由于菌群的特征变化造成。
炎症性肠病中的菌群失调
详见:炎症性肠病一文
还有过敏性腹泻,有人可能对一些食物过敏,吃完之后会导致一些腹泻,菌群特征变化很明显,包括甚至一些肠道病毒的感染,比如说诺如病毒、轮状病毒的感染。它也会体现出非常特定的菌群变化特征。
在肠道菌群检测报告中,这类疾病风险呈现如下:
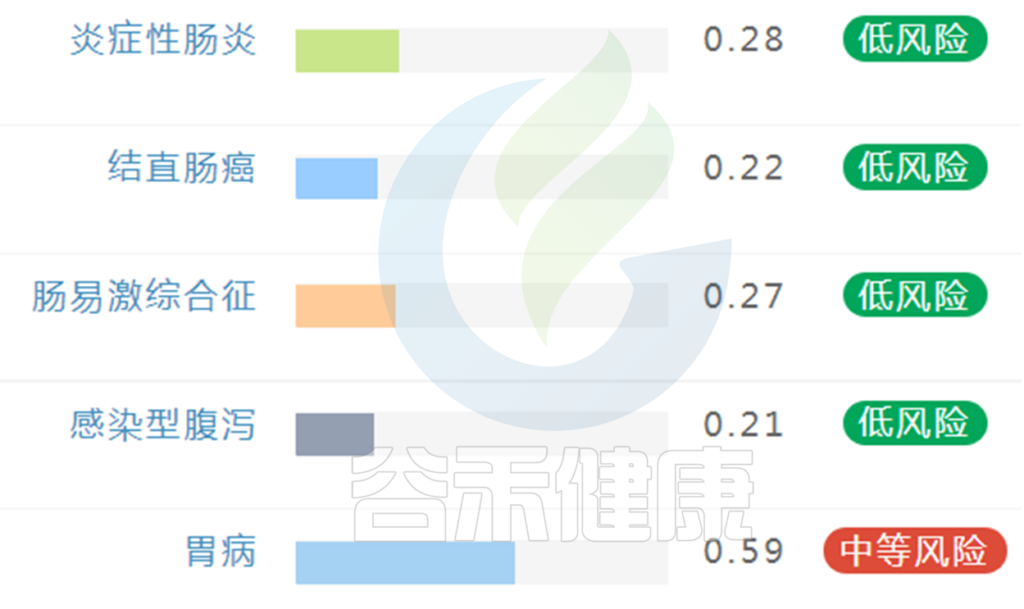
上图样本可以看到胃病有中等发现,其备注信息里有填:胃痛,可能要开始注意这方面的疾病隐患,通过饮食等调理一段时间,或前往医院就诊。
★ 胃癌
胃部更严重一点的疾病就是胃癌,胃癌与肠道菌群之间也有很大关系,最近,在“谷禾开放基金项目”中,也有相关论文也已发表。
肠道菌群在区分胃癌患者和健康人方面具有高度的敏感性和特异性,表明肠道微生物群是胃癌诊断的潜在无创工具。
胃炎与胃癌具有某些微生物群特征,化疗降低了胃癌患者的微生物丰度和多样性。乳酸杆菌Lactobacillus和巨球菌Megasphaera,是胃癌的预测标志物。
★ 结直肠癌
现在已经有多项研究表明,通过菌群可以做一个很好的标志物。虽然做不到所有的结直肠癌患者都能够被检出,但是最终的准确率相对来说还是挺高的,甚至比一些,包括肿瘤标注可能还要更高一些。
我们现在大概能做到70%多的肿瘤患者是能被筛查出来。并且准确度其实能够到90%,作为普筛或者健康评估来说,已经是一个比较有效的标志物了。
化疗与手术会大幅降低风险分值,但仍比健康人高。
此外,结直肠癌会经历从息肉到腺瘤到癌症多个阶段,应结合年龄和家族史判断息肉和结直肠癌。
肝胆类疾病
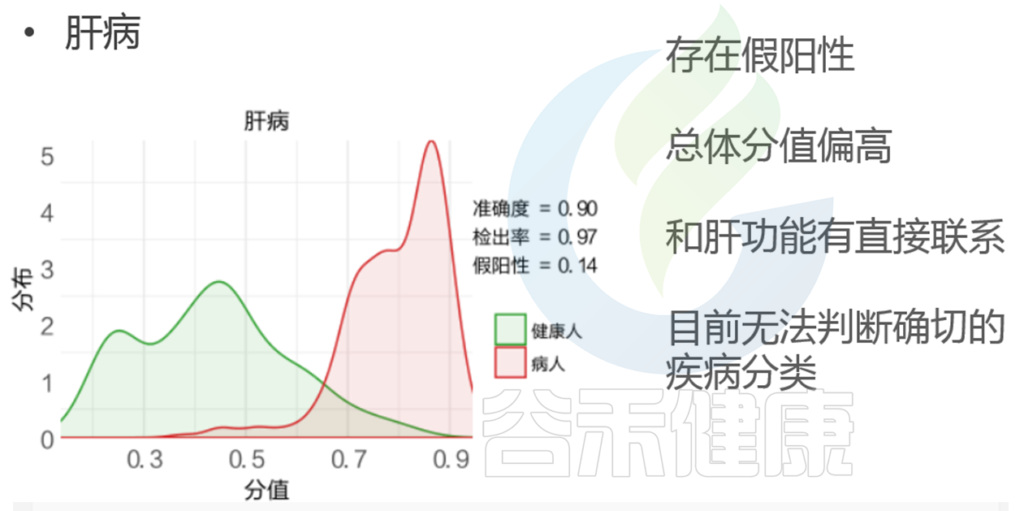
肝脏类疾病,比如说非酒精性脂肪肝跟肠道菌群有相当大的关系。
不同肝病有不同的菌群特征,尤其是脂肪肝的严重程度,肝功能异常的严重程度。
扩展阅读:深度解析 | 肠道菌群与慢性肝病,肝癌
因为菌群会产生大量的刺激代谢物,这些代谢物本身可能会加重肝脏的负担,并且诱发一些肝脏的疾病,但反过来肝脏的代谢能力的减弱和一些慢性肝脏疾病进展又会反映在菌群的构成上,所以它们是相互的。当然也可以用菌群的构成来反映具体肝病的特征。

由于不同阶段肝功能异常,脂肪肝等情况都统一归类在肝病这个大类,因此目前还无法判断确切的疾病分类,后续如果有更多细分疾病的样本用于建模,报告也会随之迭代更新。
代谢类疾病
代谢类疾病,比如糖尿病,肥胖等,都与肠道菌群有密切关联。
★ 2型糖尿病
2型糖尿病的发病率越来越高,也有更多人开始关注菌群与2型糖尿病的关系。很多文献都有报道它们之间的关联性。
2型糖尿病人群中个体微生物群的差异
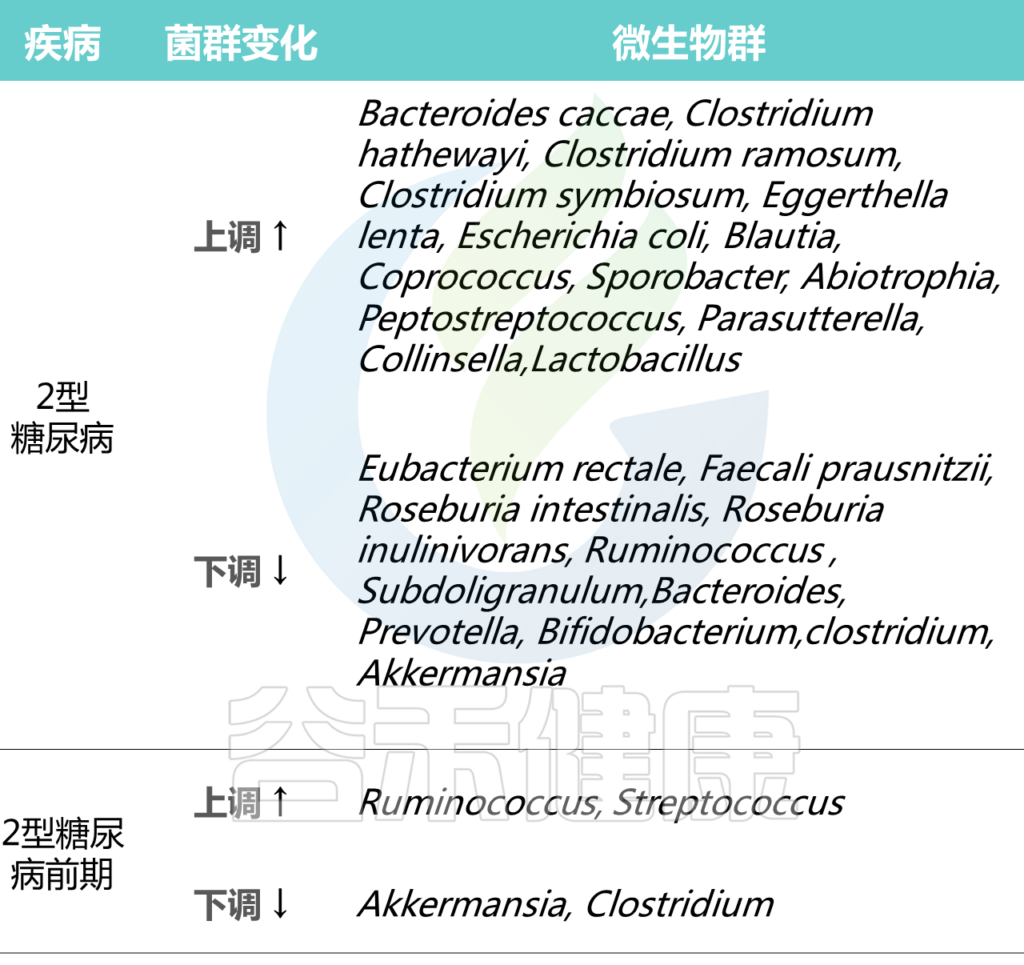
Cunningham A L et al., Gut Pathog, 2021
在2型糖尿病患者普遍具有相对高丰度的特定属:Blautia、Coprococcus、Sporobacter、Abiotrophia、Peptostreptococcus、Parasutterella、Collinsella。
2型糖尿病患者中,产生丁酸菌特别缺乏,特别是梭菌目,包括:
Ruminococcus、Subdoligranulum,Eubacterium rectale、Faecali prausnitzii、Roseburia intestinalis 、
Roseburia inulinivorans
通过肠道菌群检测,一方面健康人群可以查看是否有患病风险,另一方面如果已经患病人群,也可以查看菌群是否异常,推测是否是因菌群显著变化导致的,从而能进行更有针对性的干预。
肠道菌群检测报告中疾病风险预测如下:
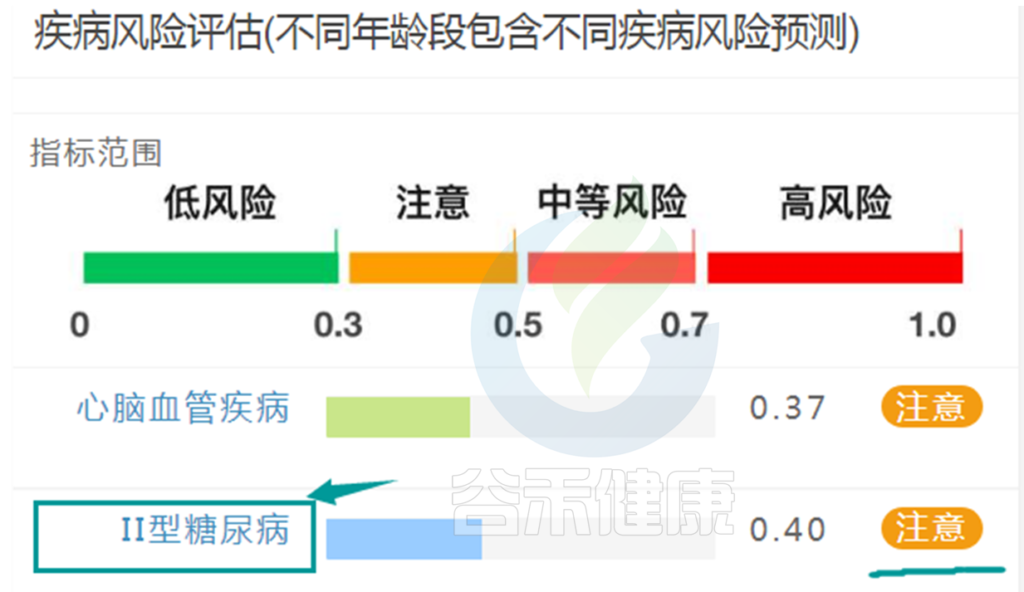
2型糖尿病的检出率相对较高,可以达到95%以上,准确的也较高,可以预测早期糖尿病风险。
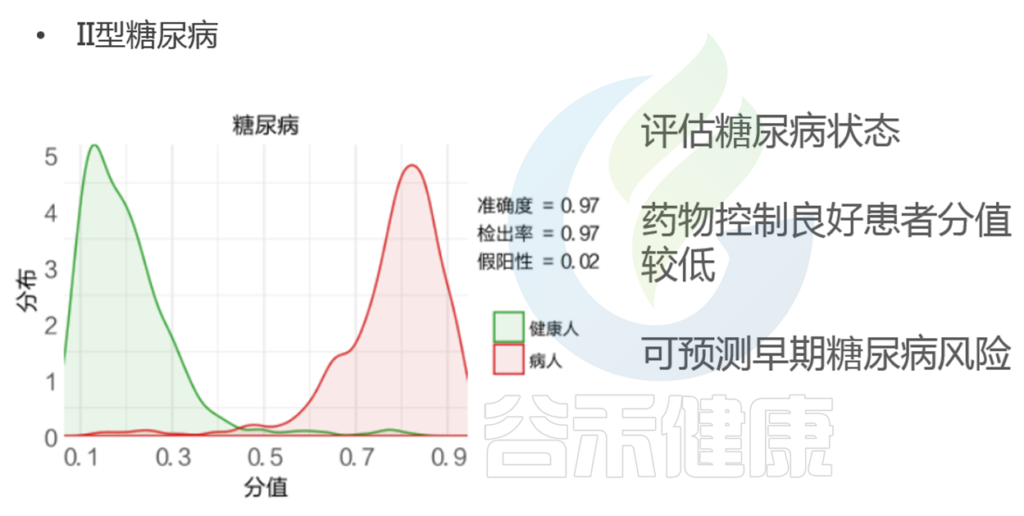
★ 肥胖
目前已有很多关于肠道菌群和肥胖之间关系的研究。
人体摄入大量营养素、肠道菌群与肥胖的关系
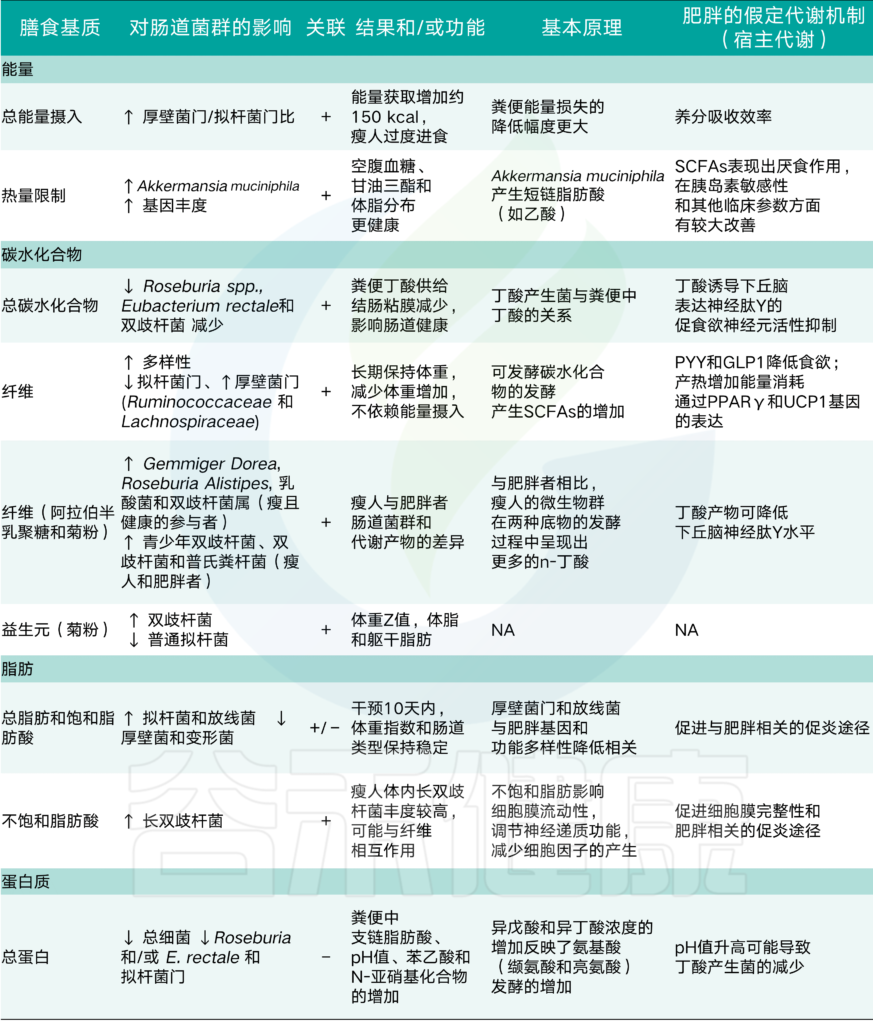
↑, 增加;↓,减少;NA,不可用;第三列:营养物质和/或饮食基质与肠道微生物群之间的关系
有人说,为什么我们的肠道菌群报告没有判别测试者是否肥胖?
首先,肥胖不肥胖这个症状是肉眼可见的,也就是说测试者自身已经了解,这种情况下用模型来判别没有意义。
而我们更希望通过肠道菌群检测来可以告诉你,可能是什么因素造成的肥胖,饮食结构的,还是某些菌属代谢问题。
通过菌群知道营养构成,以及是否存在一些特定代谢菌的异常,比如说Akk菌,它是一种在一定程度上帮助减肥的菌群。
如果在你的肠道内该菌特别少,那么可能同样减肥,控制饮食,别人一个月假设瘦十斤,你就不一定能达到这个效果。这些都是菌群可以提供的一些信息。
在肠道菌群检测报告中,会列出肥胖正负相关菌群,及其是否超标。
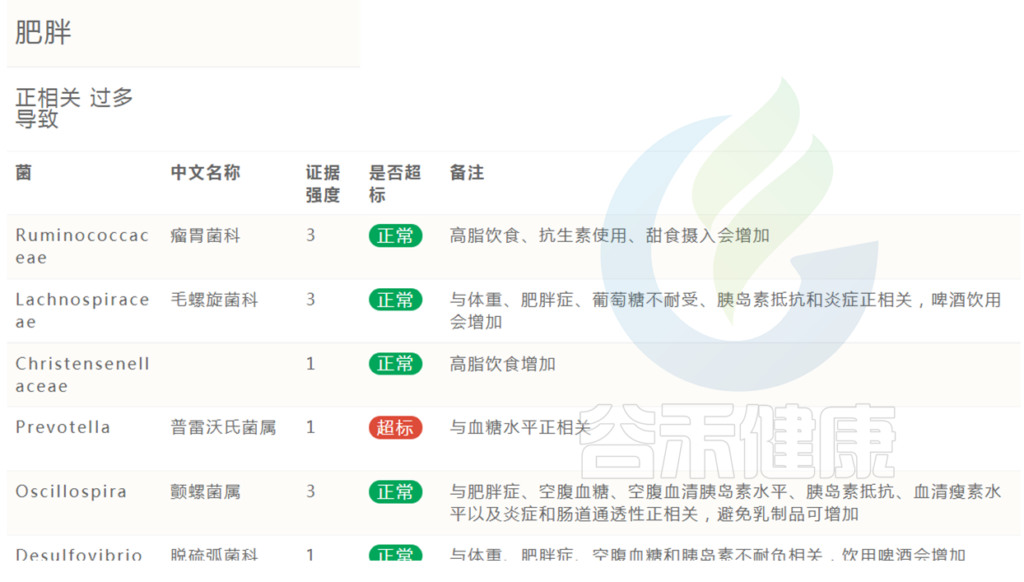
同理,其他各类肉眼可见的症状(包括腹泻、便秘、腹胀、过敏、皮肤状况等)正负相关菌群都会在报告中呈现,此处就不一一列举。
神经系统疾病
听起来神经系统好像没什么关系,但实际上很多肠道菌群能代谢产生大量神经递质及其他代谢产物。
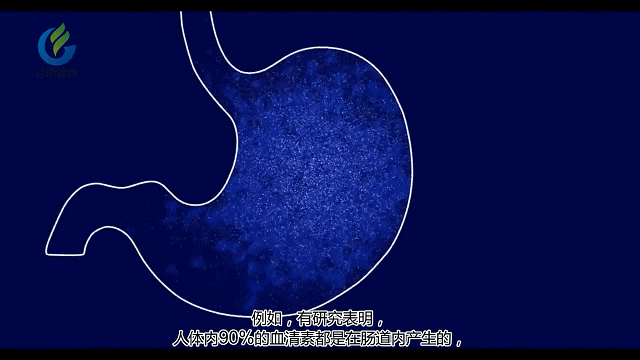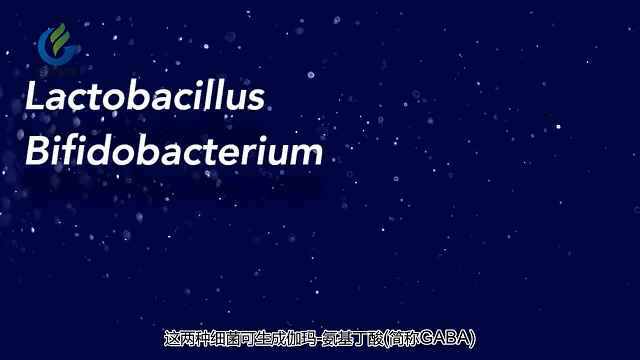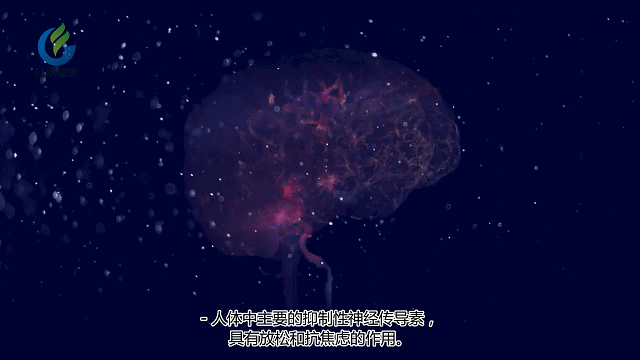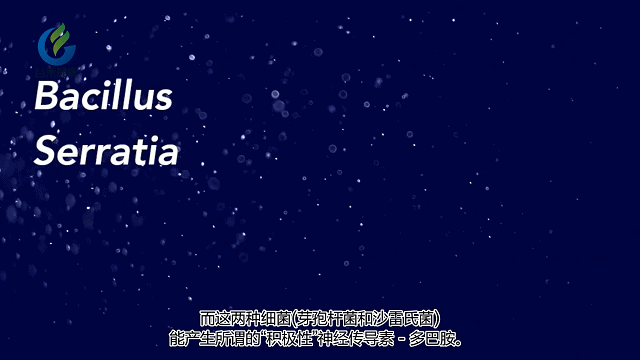
肠道菌群会影响HPA轴的发育,该轴调节压力反应并参与皮质醇的释放。在抑郁和长期处于压力下的人中,HPA轴可能失调,导致过量的皮质醇(一种压力激素)被循环。
肠道菌群的部分代谢物质也会通过免疫系统影响神经系统。促炎性细胞因子的失衡可导致慢性炎症和自身免疫性疾病,通常与抑郁症同时发生。
通过肠道菌群检测,可以了解体内血清素水平及激素水平,同时也可以了解神经系统相关疾病风险,包括自闭症,抑郁症,阿尔兹海默症等。

肺部疾病
宿主,微生物组和环境之间的三重相互作用在健康功能中维持了肺稳态。
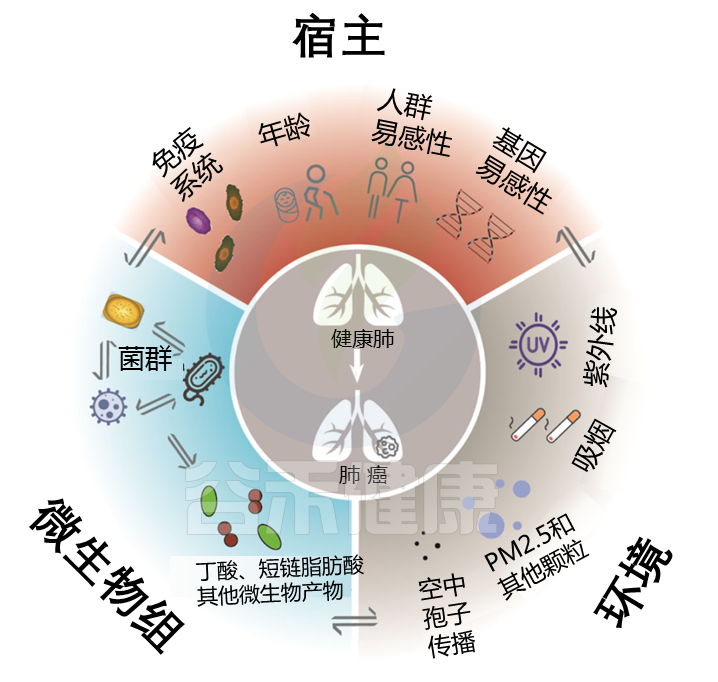
Liu NN, et al., NPJ Precis Oncol. 2020
在大量的临床样本数据当中可以发现,肺部感染,包括社区性肺炎,慢性阻塞性肺疾病,通过血氧浓度和全身的免疫反应,一定程度上是可以反映在肠道菌群上。
另外像肺部的感染,比如说在肺炎链球菌之类的感染中,肺部的病原菌可以通过痰或者是呼吸进入到肠道,所以我们在肠道当中是能检测到这些肺部的感染菌,并且随着其严重程度和感染进程,菌群的丰度会越来越高。
肠道菌群检测报告中也有对肺部相关疾病风险提示。

免疫疾病
肺部感染会出现咳嗽等症状,但咳嗽不一定仅是肺部感染,也可能是哮喘。
★ 哮喘
在哮喘中,微生物群是导致肺和肠道之间相互作用的重要因素。肠道微生物可以影响肺部的免疫反应,而肺部刺激可以导致肠道反应。
在一项研究中,来自加拿大的三个月大婴儿哮喘高风险的粪便样本中观察到 Lachnospira, Veillonella, Faecalibacterium, Rothia显著下降。这种菌群特征在1岁时不再明显,同时伴随着粪便乙酸的减少和肝肠代谢物失调。
肠道微生物对哮喘的影响部分是由细菌代谢物介导的,1岁时粪便中含有大量丁酸和丙酸的儿童,其特应性敏感性明显降低,3至6岁之间哮喘的可能性较小。此外,哮喘患者的粪便中Akkermansia muciniphila 菌水平均有所降低。
★ 过敏
已知的婴儿期与过敏性疾病相关的微生物群改变如下:
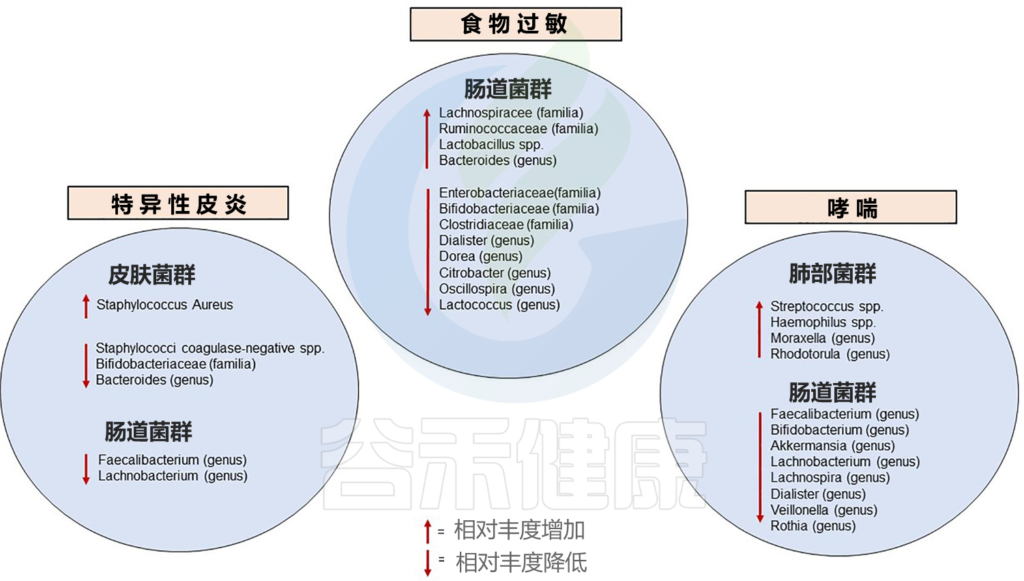
Diego G. Peroni et al, Front.Immunol. 2020
肠道菌群检测报告中有列出与过敏正负相关菌群,及是否超标。
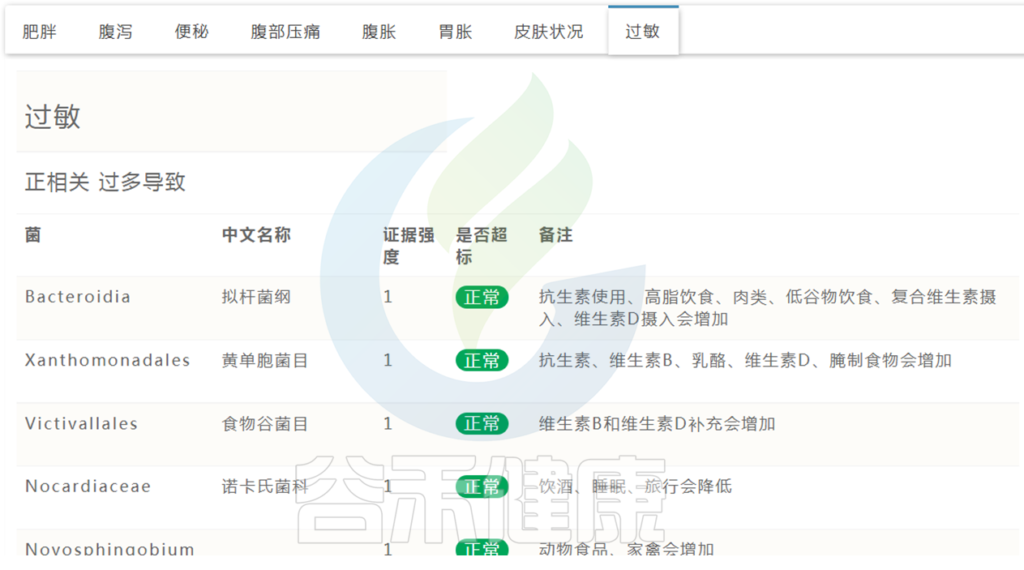
< 篇幅关系,此处仅展示部分 >
菌群生长需要养分,它的食物来源取决于你的肠道,有句话叫:you are what you eat (在我们这篇文章中有详细解释它们之间的关系 深度解读 | 饮食、肠道菌群与健康)。
也就是说,你吃的食物会帮助构建你的专属菌群。有的菌擅长代谢碳水化合物,有些菌擅长代谢脂肪,所以饮食结构不同,也就是食物来源比例不同,最后会塑造不同的菌。
那反过来,如果知道你的菌群的构成,就可以相对数量化的去了解你的饮食构成,包括营养摄入具体是什么样子,所以菌群很大的另外一个作用就可以反映你的营养饮食摄入状况。
这部分内容在我们报告中的呈现如下:
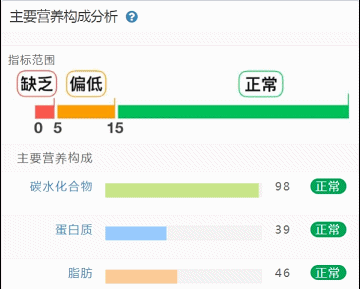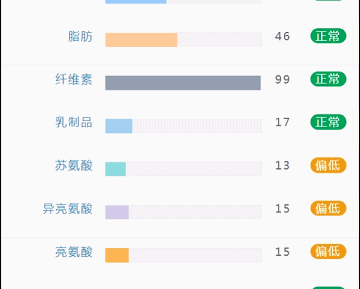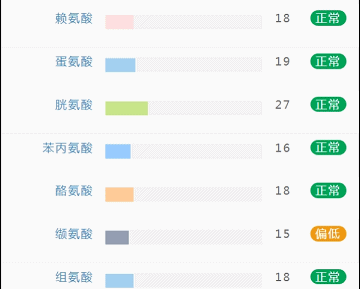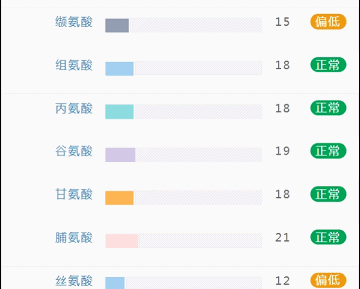
那么这里可能又会有疑惑,以上这些数值是什么意思,如何计算的呢?
不同的细菌有不同的代谢能力,需要不同的营养物质进行繁殖。通过评估特定营养供给下的偏好菌群的比例,即可反映不同营养物质的摄入比例。所以报告中的主要营养代谢分值评估的是主要营养物质摄入的比例在人群中的分布水平。
因此不会出现所有主要营养物质均高或均低的情况,也因此主要营养指标的最佳分值在70,且更关注不同营养物质的均衡性。
单项营养物质的分值低于5,表明摄入比例在人群中属于最低的5%,评估为缺乏,低于15评估为偏低。
而如果某项指标达到或超过95,则表明该项可能摄入比例偏高,通常对应会有其他营养成分较低。只需要针对性的增加缺乏或偏低的营养成分摄入,维持不同营养成分相对一致即达到营养均衡的目标。
为什么会出现所有的营养指标都很低?
这可能是菌群失调引起的。营养指标的评估是基于菌群构成特征和菌群代谢生成特定营养素的途径来评估的,如果菌群结构异常,将导致后续的预测失常,例如大量氨基酸都评估缺乏的情况。
这时候需要先调节菌群,等菌群指标恢复到一定水平后再次检测,评估营养指标。
我们日常摄入的除了上一小节提到的宏量营养素之外,还包括微量元素和维生素等。有些维生素比如说B族维生素中有相当一部分,甚至百分之六七十需要通过肠道菌群对初始原料进行代谢之后才会产生,也就是说有些细菌会代谢我们食物中的一些成分,转换成B族维生素。
而你的菌群构成和代谢B族维生素的能力,会直接决定是否缺乏该类维生素。当然也有部分受基因影响,因此肠道菌群相应的基因和代谢途径的丰度水平也会直接反映这些维生素的摄入水平。
总的来说,菌群在这其中起重要作用。在我们报告中呈现如下:

微量营养元素和维生素的评估分值与主要营养物质不同,是通过调查人群的单项营养成分水平,然后寻找与该项成分异常相关的菌群,并基于这些菌群和代谢途径计算丰度并转换为人群分布后的值。
简单来说,报告中的微量营养元素的分值即代表该营养元素的摄入水平。
菌群检测营养状况与血液检测有什么区别吗?
通过肠道菌群评估的维生素一般反映一段周期内的维生素状况,因为肠道菌群在没有突发疾病的情况下相对稳定,受一段周期的饮食影响为主,一般是2周。B族维生素是水溶性维生素,每日摄入后会通过尿液代谢排出,通过血液检测,不同时间检测波动较大。
菌群评估营养和血液检测营养趋势是一样的,在极端缺乏和极端过量是吻合的,中间档可能在数值上不是完全吻合,血液反映的营养水平比较及时。
★
当了解了体内的营养素和维生素是否缺乏,以及哪方面的缺乏,就可以进行有针对性地补充。菌群也是需要营养物质的,这就离不开我们的日常饮食,那么该如何补充呢?
我们的肠道菌群检测报告中有个体化饮食推荐表。

<篇幅关系,此处仅展示部分>
以上食物推荐表是怎么来的?
这是经过综合考虑疾病风险和营养缺乏状况计算得到的。主要是计算每种食物的营养构成与目前营养状况的匹配度,以及特定疾病需要避免的食物。
该表推荐的食物分数从-100~100,排序为不推荐到强烈推荐,日常饮食可以参考这个推荐表。推荐分值,表示基于目前的菌群和营养状况对食物的推荐指数,正数分值越大,建议优先选择,同时也是对改善最有帮助;负数分值越大,并不表示不能吃,而是目前状况下不优先推荐或尽量少吃。
p.s. 如果有特殊疾病需要忌口的,优先遵医嘱。
该表包括几百种日常食物,如下图。

<个体化饮食推荐,建议用电脑查看,目前手机端展示不太美观>
对于长期调理菌群而言,饮食无疑是最主要的驱动因素之一。
下一步我们将利用更全和详细的菌群结构,食物营养,人群膳食构成以及营养数据库推出个性化膳食营养升级方案,特别会针对个别菌属的异常和失衡状况以及营养元素异常和缺乏问题。
前面章节我们知道,通过菌群可以反映你的饮食状况,那么反过来,如果你吃了一个东西,会对菌群检测造成影响吗?是不是菌群就变了,那检测就不准了?
这也是比较重要的一部分,也就是肠道菌群检测的准确性,它能允许多大范围内的变化?什么因素会影响?
其实,菌群变化算快,也不算快。饮食对菌群是有一定影响没错,但这种影响呢,一般来说是前一天的饮食会影响第二天的菌群结构的百分之十几,也就是说,假设你昨天吃大餐,大量吃肉,蛋白摄入非常高,而你之前是以碳水化合物为主的,那么第二天饮食当中,你的蛋白质相关的这部分菌的比例可能会有15%,最高到20%可能会有,但一般来说是在15%以内,会有一个波动。
然而,总体的核心菌群构成,不会因为你今天一顿大餐,就直接从素食的变成肉食的菌群结构,核心菌是相对稳定的,那么多久会发生变化呢?
一般来说坚持两周,饮食结构的变化,核心菌群就会发生一个迁移改变。但两周只是一个短暂的周期,如果你两周后又换回先前的那种饮食方式,菌群也会随之改变到之前的状态。那要怎么样才能持久改变菌群呢?
这个时间线可能要拉长到两个月。
这是在我们的菌群干预中,很多人会遇到的一个周期性的问题。也就是如果你想有效改善菌群,至少需要两周会见到相对明显的菌群结构变化,那如果把干预延伸到持续两个月的周期,甚至是持续干预周期更长,那效果会更好。
取样前饮食会不会造成影响?
前面我们知道,菌群会受检测前一天饮食的影响,造成15~30%的菌群改变,同样也会反映在营养状况的评估上,因此建议检测前一天尽量保持近期正常饮食,这样能更好的反映真实的营养饮食状态。
此外,如果你是在调理一段时间后再次检测,想要和上次比较的话,最好在检测前保持饮食大体相似(意思是不要突然吃和平时不一样的食物或者吃完大餐后取样)。
取样过少会怎么样?
取样不能太少,如果太少的话,可能会影响DNA提取,另外会导致一些低丰度的菌检测不到。
取样过多会怎么样?
如果说取样太少导致样本不合格可以理解,那么取样过多为什么也会有问题呢?
我们的采样管中有保存液,可以将菌群固定在采样的瞬间,但是如果取样过多的话,可能导致部分粪便无法完全溶解于粪便,这部分样不能正常保存可能会使其中的大肠杆菌等兼性厌氧菌开始在管内繁殖。
正确合格取样量(黄豆大小,约200mg,如果是稀便,反复沾取)

只需棉签沾取少量,混匀于保存液,固体粪便取样不能超过管子1/5体积(右图刻度线)。且保存液带有粪便颜色即可。(右图所示)

详见:肠道菌群取样方法
注 意 事 项
如3天内使用过抗生素类、质子泵类胃药、阿片类精神药物请停药3天后进行检测(如果长期服用某种药物,如降压、降血糖药等,不建议停药,检测反映的是用药控制的菌群和身体状况)。
感冒、腹泻或其他症状期间不影响取样,拉稀或稀便可以用棉签反复沾取粪便至取样管。
★
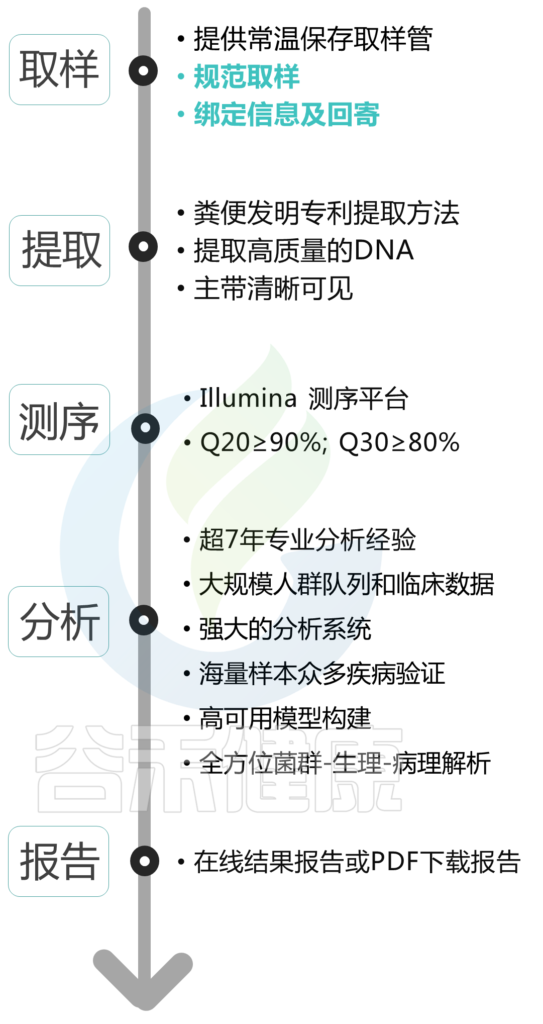
总的来说,取样虽然很重要,但也只是其中一个环节。每一个样本的结果呈现都凝聚了我们与你共同的努力。那么,从取样到结果报告呈现的那一刻,中间经历了什么?
在你取完样之后,把样本用快递寄到我们这里之后,它会经历提取->测序->分析->报告到你手上。下图绿色标注部分是你需要完成的。

近年来,我国将全面健康和预防作为国家重点领域。我们致力于将信息技术(IT)与生物技术(BT)相融合,发展推动肠道菌群基因检测进入成为精准和预防医学时代下的“生命健康新基建”,尽管目前的菌群检测,包括疾病关系,算法,数据库,后端干预均在成长积累阶段,但是菌群检测正在进入大数据时代,菌群基因中蕴藏海量对人体生命和健康的重要数据,我们致力于将这些数据和实际应用相结合,最终转化为疾病预防、改善健康的有效方案。
前沿技术正在不断创新发展,报告也在迭代更新中,谷禾肠道菌群健康检测在辅助判别慢病风险、精准营养、亚健康管理、临床治疗干预中显示出其广泛的社会需求和指导价值。
你问我答
不同部位间的样本(如前段/中段/后段),检测结果差异性有多大?
答:会有不同的,不过主要反映在具体的菌种丰度上,有无这种菌的差异不大。另外慢病的评估也影响不大慢病模型中使用了高维特征,丰度的变化波动对结果的影响没那么大。营养和代谢部分受菌群丰度影响相对大一些,同一个人的前后两天的取样最大可能有15%左右的差异。
肠道菌群在肠道内不同部位以及粪便的不同部分其实都存在差异,含水量、连续几餐的饮食构成和排便周期的长度都会对菌群各个菌种的丰度造成影响。单纯从绝对丰度上来看是一个动态变化的过程,各个菌属在继承之前的构成比例的情况下因各种因素的变化增长或降低。因此并不存在一个绝对的菌群构成以及完全准确的单一指标。肠道菌群检测获取的丰度含量本身信息量很大,但是稳定性和一致性并不很高。
更高层级的菌群相对比例顺序则相对稳定一些,之后具体包含的菌种也相对稳定。目前我们使用的疾病预测模型主要通过高维的菌群结构特征,并不单纯依靠每个菌的绝对丰度来评估,稳定性很高。针对一些特定的病原菌或问题菌,需要通过与人群范围比较,在正常范围内并无问题。
日常多添加有益菌或益生菌的酸奶,可以改善肠道菌群状况吗?
答: 大范围人群调查显示添加益生菌的酸奶可以改善肠道健康,但效应因人和状态而定。总体而言我们支持服用益生菌酸奶有益,但需要注意酸奶饮料可能包含果糖,游离糖等,其作用仍然非常有限。
同一份样本,不同批次的实验环节如上机测序,差异有多大?这种差异率是否有一个范围呢?
答:不同批次上机影响很小,菌群数据相关性不低于98%。我们会在每轮设置一个阳性对照,一个上轮检测样本对照,一个阴性对照。评估污染,轮次比对。理论上不同的实验室,扩增引物,方法都会带来对不同菌丰度的系统误差,我们尽力保证本实验体系下各个轮次之间最小化的实验误差。另外使用的引物是经过大量验证的标准化引物。
实际患者建不建议送检,我们这个产品主要针对健康体检,还是也可以辅助诊断和预后治疗呢?
答:产品主要针对健康体检,如果临床诊断判断可能菌群异常或疾病症状与菌群相关,产品可以通过菌群检测提供临床参考,用于辅助诊断和治疗方案的评估。产品关于疾病和菌群相关指标的评估仅限于菌群相关方面,以临床诊断为准,不适用于单独使用产品进行疾病诊断。
抗生素是如何影响菌群的,菌群的敏感性和抗性基因是什么?
答:广谱抗生素会杀死细菌,但是部分细菌在抗生素选择或滥用的情况下会在抗生素靶点基因产生突变或携带耐药基因,从而对特定抗生素产生耐药。不同菌目前的耐药菌比例以及携带的耐药基因水平不同,对应的抗生素耐药水平和种类也有不同。
有在吃富含某种事物或者相关营养素,为什么报告显示缺乏?
答:营养指标的评估是基于菌群构成特征和菌群代谢生成特定营养素的途径来评估的,直接的营养素补充会反映在相关菌群构成上,但部分营养素因为吸收部位不同以及菌群代谢途径上下游的影响,预测可能有一定差异。另外菌群构成异常的情况也会导致营养指标预测失常,如大量氨基酸都评估缺乏的情况。
有人说长期服用益生菌,会让肠道自己产生的益生菌的能力减弱或者可以说是肠道自主平衡的能力减弱,不能长期服用。这种说法是否有依据?长期服用一种益生菌,也容易产生耐药性,那么是否建议定期更换或者调整益生菌的菌种和数量呢?
答:持续服用单一或特定组合的益生菌确实会存在效力减退的情况,主要是菌群具有适应性,如果不配合生活方式和饮食结构的改变,会较快失效。可以根据菌群检测结果来调整益生菌的方案。
样品的稳定性对于那些数据的影响是比较大的哪些是影响比较小的?
答:越是直接和具体菌相关的指标变化越快越大,和菌群结构相关的指标,比如一些慢病风险还有总体饮食结构一类的变化较稳定。
从波动性排序来看,具体菌丰度>多样性>微量营养(锌 铁 氨基酸 维生素)>消化道疾病风险评估 (受当前状态影响较大)>肠龄>宏量营养素(碳水 蛋白 脂肪 纤维素 乳制品)>抗生素水平 >菌属是否出现>其他慢病风险
中大龄儿童小孩检测到自闭症风险高,如何解读?
答:肠道菌群在1-3岁期间主要是菌群发育滞后会影响神经发育和营养,3~6岁左右菌群参与的神经递质代谢异常会加剧自闭症的程度,但这个年龄段已有的神经发育滞后不光靠菌群改善就能解决了。
所以如果是0~2岁的如果这个风险值较高,不管有没有症状都建议改善菌群。如果是3~6岁甚至6岁以上,如果就风险值高没有相应的神经或行为异常,就问题不大,可能是菌群代谢构成不太好,不会导致自闭症的。如果有症状那改善菌群有助于改善症状。
肠道菌群平衡,为何多样性指数是低的?
答:菌群平衡和多样性指数是2个不同指标;
多样性仅仅评估肠道菌群的种类数量和丰度分布,与具体是有益和有害无关。多样性主要与饮食摄入,药物如抗生素类以及疾病状态有关。
菌群平衡对应的异常称为肠道菌群失调,临床上有I度失调和更严重的II度失调。大便常规检查是通过显微镜下观察统计染色细菌中杆菌和球菌以及革兰氏阴性和阳性菌的比值是否超标来判别的。本报告同时提供了另一评估算法,通过有益菌/有害菌的总体情况来评估菌群平衡状态,低于2为重度失衡,低于5为失衡,同时分值也提示菌群平衡水平,越高越正常。
菌群失衡如何调整?
从菌群失衡的评估角度来看,首先就是快速增加有益菌特别是双歧杆菌的丰度可有效改善该项指标。因此临床上通常提供多联的益生菌制剂来快速补充益生菌,可以短期有效改善菌群平衡比例。
菌群平衡和多样性分值都高的,但是肠道年龄预测比实际大,年龄预测模型是不参考多样性和平衡性参数的?
答:肠道年龄是靠机器学习和人群大队列做的,不是只根据菌群平衡和多样性,每个年龄段都有核心和标致的菌群特征,比如婴儿的双歧杆菌,老年人瘤胃球菌等,这几个指标都是表征菌群的状态和健康的。
END
声明
谷禾专注于提供肠道菌群基因检测和基于此的健康评估咨询,肠道菌群对人体健康的影响和关联性已被广泛研究和认可,但基于对健康的慎重和法规,谷禾重申其提供的肠道菌群基因检测目前不用于临床疾病诊断,仅作为菌群状况构成检测和健康评估以及基于菌群的科研。分析报告中疾病风险和健康相关评估来自于公开研究数据和谷禾构建的大人群队列数据分析的预测评估结果,涉及临床诊断和医疗建议请遵照临床诊断和医生的医嘱。由于技术进步和样本数据不断积累,报告中可能存在尚未完全涵盖的因素或状况,不可避免的存在一定概率部分风险未被完全检出的情况。
做过16s测序的小伙伴们都知道
测完之后会拿到一份结果报告
但这并不代表可以开始写文章了
看似一大堆数据图表却不知如何下手
这是很多人头疼的地方
那么怎样给报告中的数据赋予灵魂
让它真正成为对你有帮助的分析呢?
今天我们来详细解读下。
一文扫除困惑
首先什么是16S rRNA?
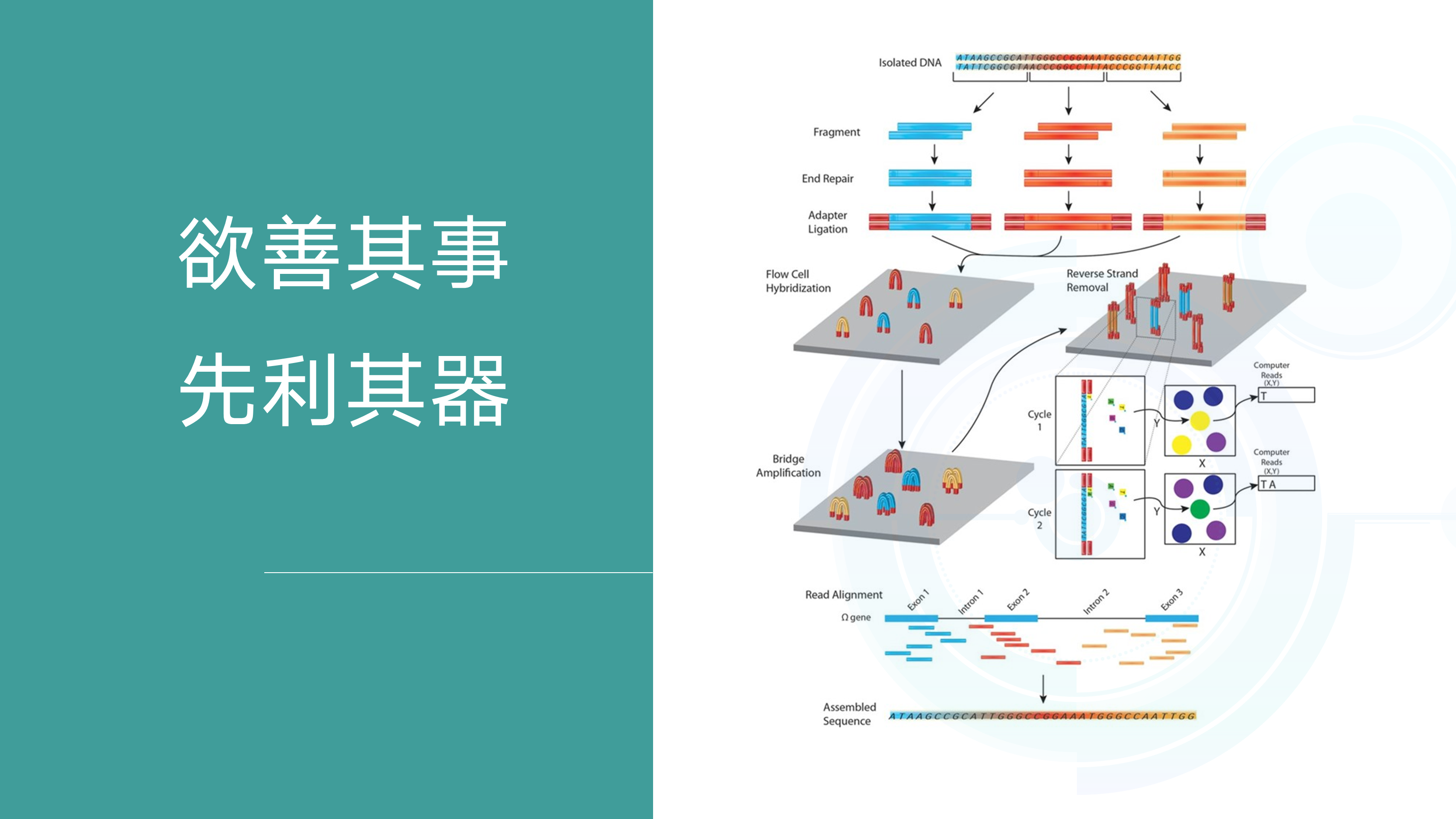
16S rRNA 基因是编码原核生物核糖体小亚基的基因,长度约为1542bp,其分子大小适中,突变率小,是细菌系统分类学研究中最常用和最有用的标志。
16S rRNA基因序列包括9个可变区和10个保守区,保守区序列反映了物种间的亲缘关系, 而可变区序列则能体现物种间的差异。
16S rRNA基因测序以细菌16S rRNA基因测序为主,核心是研究样品中的物种分类、物种丰度以及系统进化。
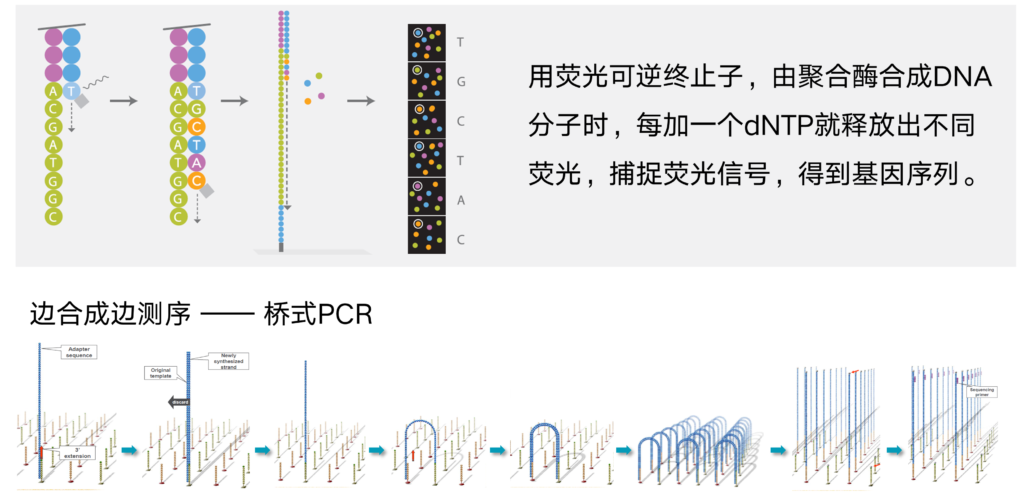
二代高通量测序原理
目前二代测序是一个边合成边测序的过程,使用的是荧光可逆终止子。每个可逆终止子的碱基3’端都有一个阻断基团,而在侧边带有一种荧光。由于有4种不同的碱基(ATCG),因此也会有对应4种不同颜色的荧光。开始扩增每次结合上一个碱基,DNA的扩增便会停止,此时能收到一种荧光信号。然后放试剂除去阻断基团,进行下一个碱基的结合,以此类推得到一连串的荧光信号组合序列。而根据荧光的颜色我们便可以确定每一个位点的基因型,即可以得到这一段DNA片段的序列。
环境样品高通量分析需要重复么?
在进行实验设计前,这是有些小伙伴面临的一个问题。环境样本由于来源和条件不完全可控,每个样品之间会存在很大的差异,即便是相同样本的不同取样时间和部位也会存在一定的差异。
基于高通量测序主要是为了了解样品的菌群构成和功能分析,以及寻找不同环境之间的差异,包括菌和功能基因以及代谢。如果仅做单一样本,很可能结论只能代表这个单一取样样本的信息,无法排除不同样本重复之间的差异,也就可能得不到真正代表环境差异的结果。
所以环境样品不仅要重复而且还应该以分组方式取尽量多的样本以全面的代表一个环境条件下的各种变异情况。
测序区段如何选择
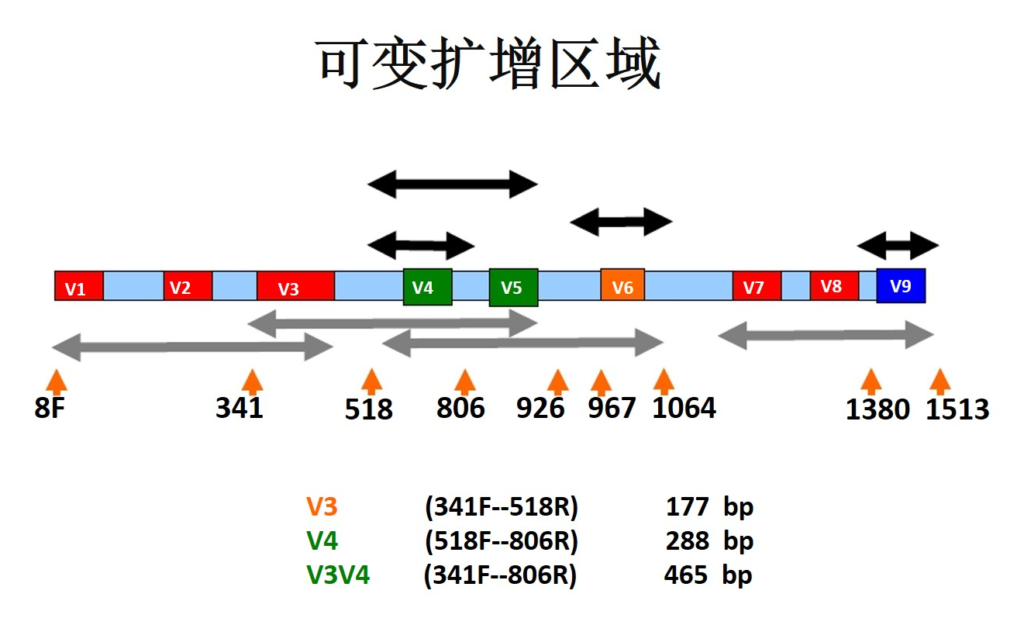
确定做重复后,又面临该怎么选择测序区段的问题。目前市面上有v1-v3区/v3-v4区/v4区等可供选择。
16S rRNA编码基因序列共有9个保守区和9个高可变区。其中,V4区其特异性好,数据库信息全,我们通过大量的测序试验证明用v4区扩增出菌群结果的可以很好的反应样本的菌群结构用于后续的数据建模分析,是细菌多样性分析注释的最佳选择。
基本确定好后,就要着手开始实验,实验完送样又是个问题,以往给测序公司送样往往是低温运输,且不说麻烦,还要提心吊胆怕运输过程会不会有什么问题。为此我们免费提供常温保存取样盒,就不用有这样的顾虑,取样及运输全程都只需要常温即可。
样品到公司之后就更不用操心,全套服务等着呢!

16s分析结果详解
很多小伙伴有过这样的经历,在拿到公司出具的报告之后,仍然一头雾水,几十页的报告内容看着丰富却不知该怎么运用。我们一起来理一下关键图表的含义。
OTU是我们要搞清的一个重要概念,可以说是后续分析的基石。
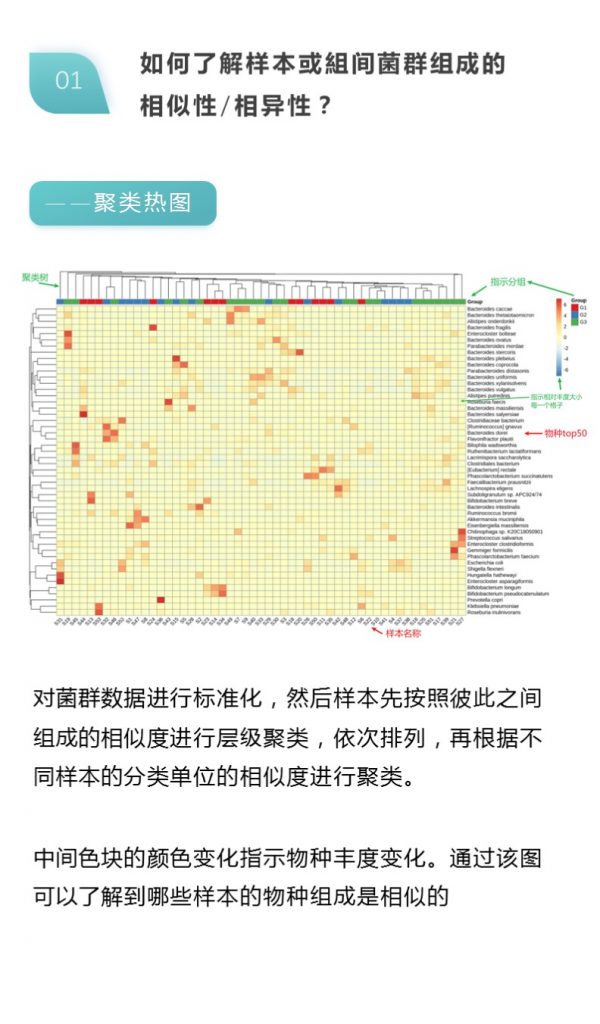
OTU(operational taxonomic units) 是在系统发生学研究或群体遗传学研究中,为了便于进行分析,人为给某一个分类单元(品系,种,属,分组等)设置的同一标志。通常按照 97% 的相似性阈值将序列划分为不同的 OTU,每一个 OTU 通常被视为一个微生物物种。相似性小于97%就可以认为属于不同的种,相似性小于93%-95%,可以认为属于不同的属。样品中的微生物多样性和不同微生物的丰度都是基于对OTU的分析。
有了OTU这个概念之后,就不难理解下表。对每个样本的测序数量和OTU数目进行统计,并且在表栺中列出了测序覆盖的完整度。
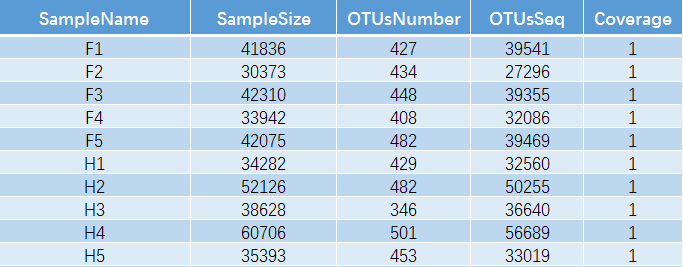
其中 SampleName表示样本名称;SampleSize表示样本序列总数;OTUsNumber表示注释上的OTU数目;OTUsSeq表示注释上OTU的样本序列总数。
Coverage是指各样品文库的覆盖率,其数值越高,则样本中序列没有被测出的概率越低。该指数实际反映了本次测序结果是否代表样本的真实情况。计算公式为:C=1-n1/N 其中n1 = 只含有一条序列的OTU的数目;N = 抽样中出现的总的序列数目。
下表是对每个样本在分类字水平上的数量进行统计,并且在表栺中列出了在每个分类字水平上的物种数目
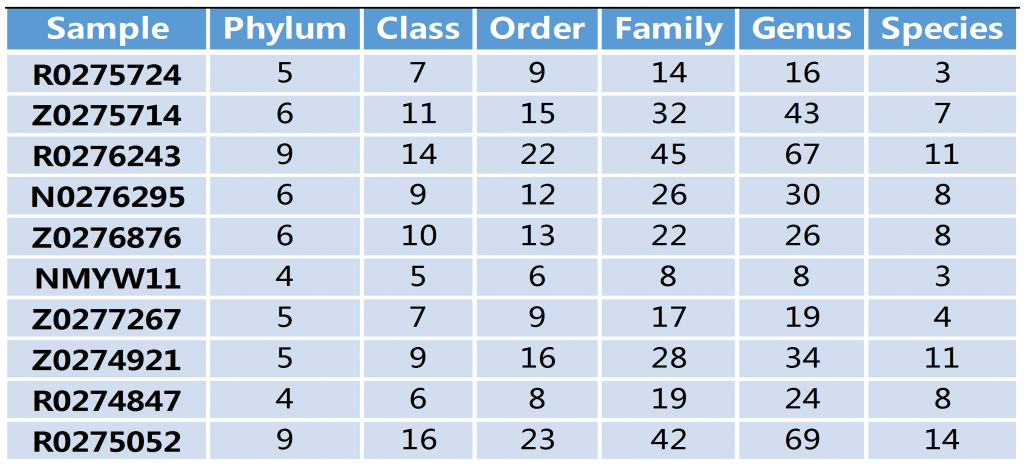
其中SampleName表示样本名称;Phylum表示分类到门的OTU数量;Class表示分类到纲的OTU数量;Order表示分类到目的OTU数量;Family表示分类到科的OTU数量;Genus表示分类到属的OTU数量;Species表示分类到种的OTU数量。
我们可以看到绝大部分的OTU都分类到了属(Genus),也有很多分类到了种(Species)。但是仍然有很多无法完全分类到种一级,这是由于环境微生物本身存在非常丰富的多样性,还有大量的菌仍然没有被测序和发现。
当然,对这些种属的构成还可以进行柱状图展示:
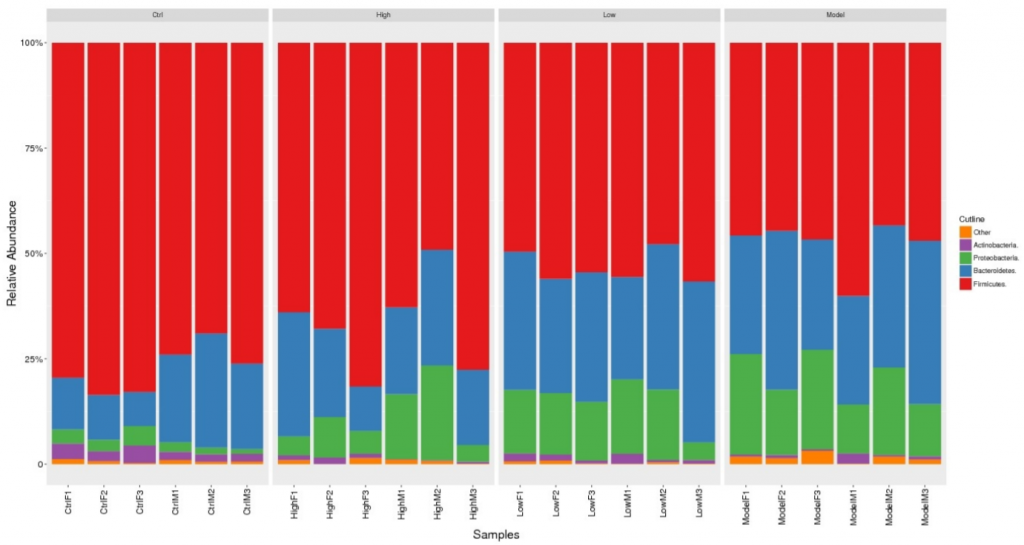
横坐标中每一个条形图代表一个样本,纵坐标代表该分类层级的序列数目或比例。同一种颜色代表相同的分类级别。图中的每根柱子中的颜色表示该样本在不同级别(门、纲、目等)的序列数目,序列数目只计算级别最低的分类,例如在属中计算过了,则在科中则不重复计算。
我们还需要对样本之间或分组之间的OTU进行比较获得韦恩图:
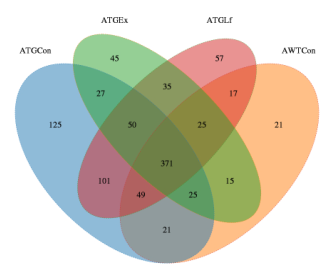
样品构成丰度
稀释曲线
微生物多样性分析中如何验证测序数据量是否足以反映样品中的物种多样性?
稀释曲线(丰富度曲线)可以派上用场。它是用来评价测序量是否足以覆盖所有类群,并间接反映样品中物种的丰富程度。
不免有同学有疑惑,稀释曲线怎么来的?
它是利用已测得16S rDNA序列中已知的各种OTU的相对比例,来计算抽取n个(n小于测得reads序列总数)reads时出现OTU数量的期望值,然后根据一组n值(一般为一组小于总序列数的等差数列)与其相对应的OTU数量的期望值做出曲线来。
至此,我们虽然知道了稀释曲线的由来,那么这个五彩缤纷的稀释曲线该怎么看呢?
当曲线趋于平缓或者达到平台期时也就可以认为测序深度已经基本覆盖到样品中所有的物种,增加测序数据无法再找到更多的OTU;
反之,则表示样品中物种多样性较高,还存在较多未被测序检测到的物种。
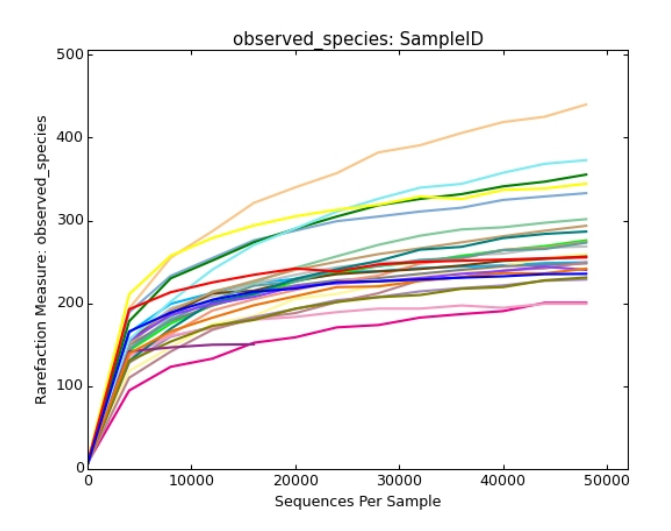
横坐标代表随机抽取的序列数量;纵坐标代表观测到的OTU数量。样本曲线的延伸终点的横坐标位置为该样本的测序数量。
Shannon-Winner曲线
Shannon-Wiener 曲线,是利用shannon指数来进行绘制的,反映样品中微生物多样性的指数,利用各样品的测序量在不同测序深度时的微生物多样性指数构建曲线,以此反映各样本在不同测序数量时的微生物多样性。
当曲线趋向平坦时,说明测序数据量足够大,可以反映样品中绝大多数的微生物物种信息。
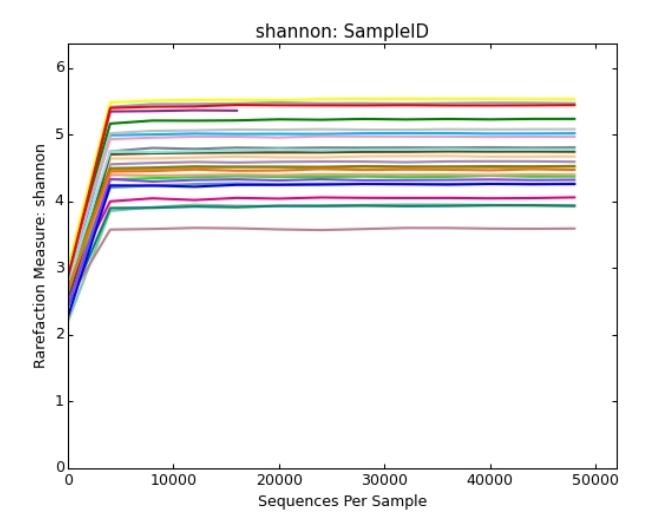
横坐标代表随机抽取的序列数量;纵坐标代表的是反映物种多样性的Shannon指数,样本曲线的延伸终点的横坐标位置为该样本的测序数量。
其中曲线的最高点也就是该样本的Shannon指数,指数越高表明样品的物种多样性越高。
好奇的同学又有疑问,Shannon指数怎么算的?
这里有Shannon指数的公式:
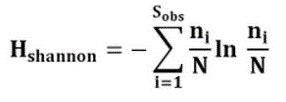
其中,Sobs= 实际测量出的OTU数目;
ni= 含有i 条序列的OTU数目;N = 所有的序列数。
Rank-Abundance曲线
该曲线用于同时解释样品多样性的两个方面,即样品所含物种的丰富程度和均匀程度。
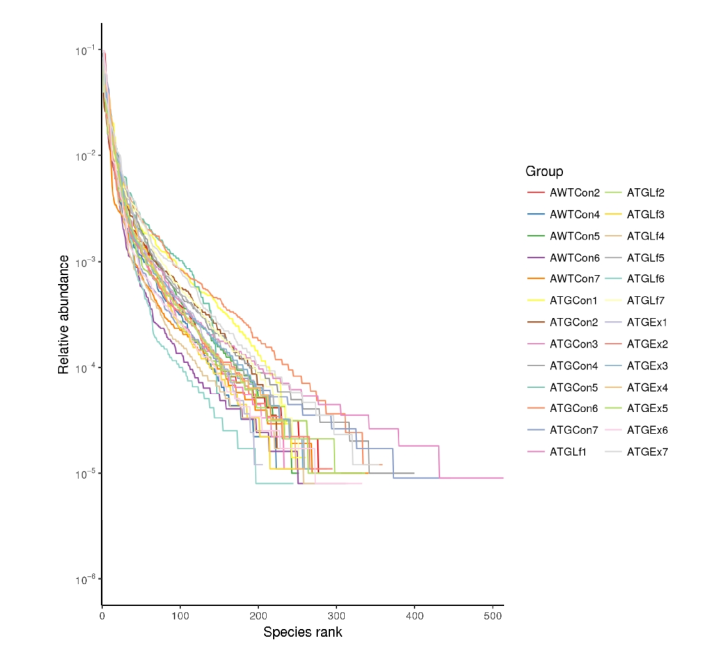
横坐标代表物种排序的数量;纵坐标代表观测到的相对丰度。
样本曲线的延伸终点的横坐标位置为该样本的物种数量
物种的丰富程度由曲线在横轴上的长度来反映,曲线越宽,表示物种的组成越丰富;
物种组成的均匀程度由曲线的形状来反映,曲线越平坦,表示物种组成的均匀程度越高。
如果曲线越平滑下降表明样本的物种多样性越高,而曲线快速陡然下降表明样本中的优势菌群所占比例很高,多样性较低。
但一般超过20个样本图就会变得非常复杂而且不美观!所以假如没超过20个样可以考虑该图哦~
Alpha多样性(样本内多样性)
Alpha多样性是指一个特定区域或者生态系统内的多样性,常用的度量指标有Chao1 丰富度估计量(Chao1 richness estimator) 、香农 – 威纳多样性指数(Shannon-wiener diversity index)、辛普森多样性指数(Simpson diversity index)等。
计算菌群丰度:Chao、ace;
计算菌群多样性:Shannon、Simpson。
Simpson指数值越大,说明群落多样性越高;Shannon指数越大,说明群落多样性越高。
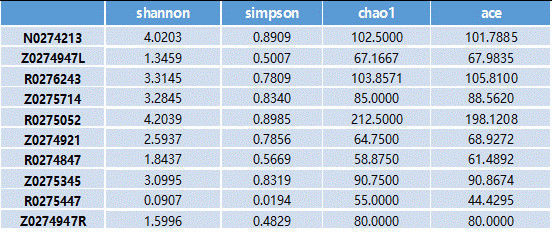
看了那么多指数,可能觉得有点晕,到底每个指数是什么意思呢?
当然要解释下咯:
Chao1:是用chao1 算法计算群落中只检测到1次和2次的OTU数估计群落中实际存在的物种数。Chao1 在生态学中常用来估计物种总数,由Chao (1984) 最早提出。Chao1值越大代表物种总数越多。
Schao1=Sobs+n1(n1-1)/2(n2+1)
其中Schao1为估计的OTU数,Sobs为观测到的OTU数,n1为只有一条序列的OTU数目,n2为只有两条序列的OTU数目。
Shannon:用来估算样品中微生物的多样性指数之一。它与 Simpson 多样性指数均为常用的反映 alpha 多样性的指数。Shannon值越大,说明群落多样性越高。
Ace:用来估计群落中含有OTU 数目的指数,由Chao 提出,是生态学中估计物种总数的常用指数之一,与Chao1 的算法不同。
Simpson:用来估算样品中微生物的多样性指数之一,由Edward Hugh Simpson ( 1949) 提出,在生态学中常用来定量的描述一个区域的生物多样性。Simpson 指数值越大,说明群落多样性越高。

Alpha多样性指数差异箱形图
分别对 Alpha diversity 的各个指数进行秩和检验分析(若两组样品比较则使用 R 中的wilcox.test 函数,若两组以上的样品比较则使用 R 中的 kruskal.test 函数),通过秩和检验筛选不同条件下的显著差异的 Alpha Diversity指数。
Beta多样性分析(样品间差异分析)
也许我们有听说Beta多样性在最近10年间成为生物多样性研究的热点问题之一。具体解释下:
Beta多样性度量时空尺度上物种组成的变化, 是生物多样性的重要组成部分, 与许多生态学和进化生物学问题密切相关!
PCoA分析
PCoA(principal co-ordinates analysis)是一种研究数据相似性或差异性的可视化方法,通过一系列的特征值和特征向量进行排序后,选择主要排在前几位的特征值,PCoA 可以找到距离矩阵中最主要的坐标,结果是数据矩阵的一个旋转,它没有改变样品点之间的相互位置关系,只是改变了坐标系统。
重要的是,它是可以用来观察个体或群体间的差异的。
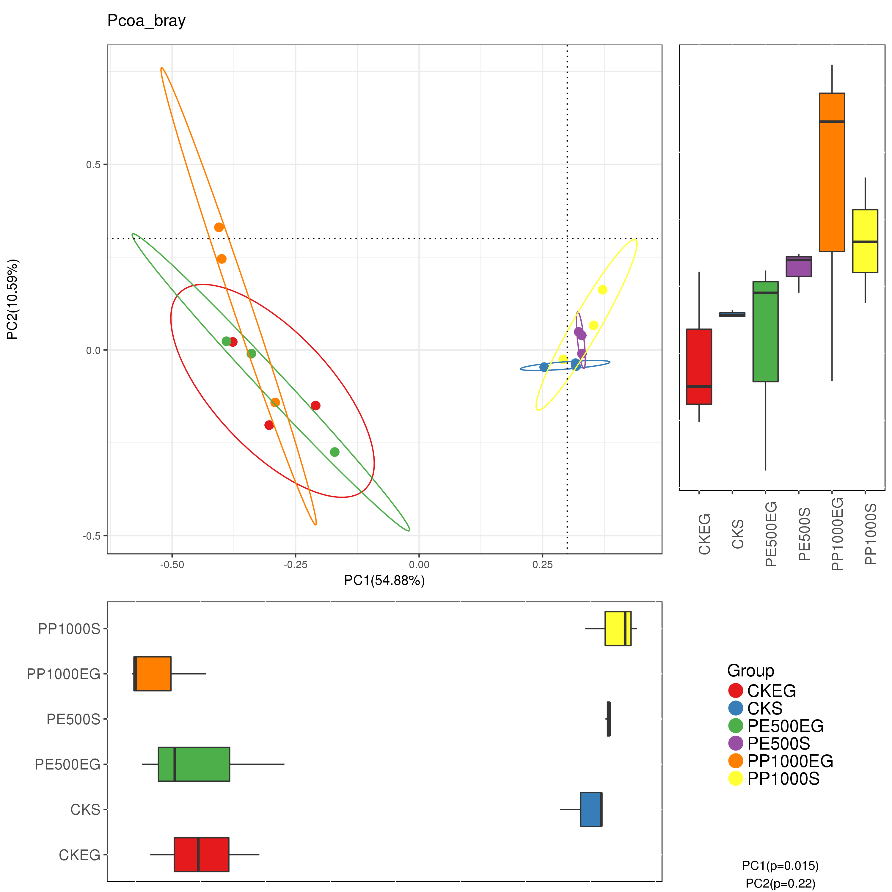
每一个点代表一个样本,相同颜色的点来自同一个分组,两点之间距离越近表明两者的群落构成差异越小。
另一种相似的是PCA分析
主成分分析(Principal component analysis)PCA 是一种研究数据相似性或差异性的可视化方法,通过一系列的特征值和特征向量进行排序后,选择主要的前几位特征值,采取降维的思想,PCA 可以找到距离矩阵中最主要的坐标,结果是数据矩阵的一个旋转,它没有改变样品点之间的相互位置关系,只是改变了坐标系统。
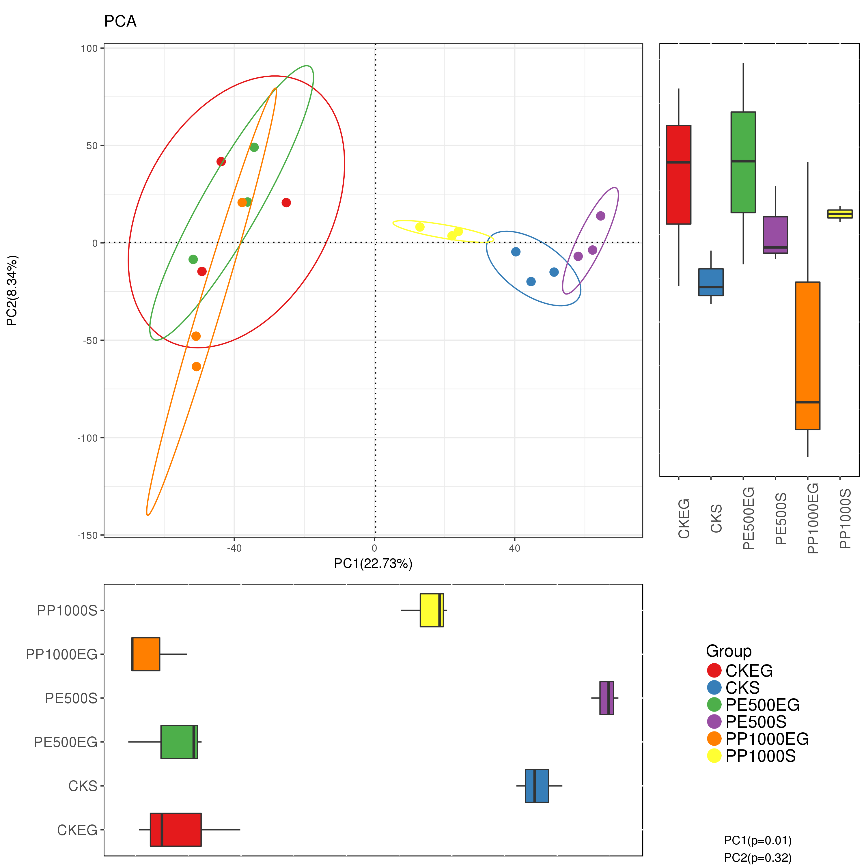
详细关于主成分分析的解释推荐大家看一篇文章,http://blog.csdn.net/aywhehe/article/details/5736659
一起来看看包含PCoA研究的文章
案例解析



研究背景:全球塑料产量飞速增长,而且呈持续上升的趋势,因此导致大量塑料废物排放到环境中,从沿海河口到大洋环流,从东大西洋到南太平洋海域。塑料废弃物具有化学稳定性和生物利用率低的特点,可长期存在于海洋中,从而影响海洋环境包括海洋生物的生存。
作为一个独特的底物,塑料碎片可以吸附海洋中的微生物并形成个“塑性球”。以生物膜形式存在于塑料碎片上的微生物群落。许多研究表明,无论是在海洋还是淡水生态系统中,附着在塑料碎片上微生物群落的组成明显不同于周围环境(水和沉积物),而且易受位置、时间和塑料类型的影响。
主要图表
两两群落差异指数的PCoA图
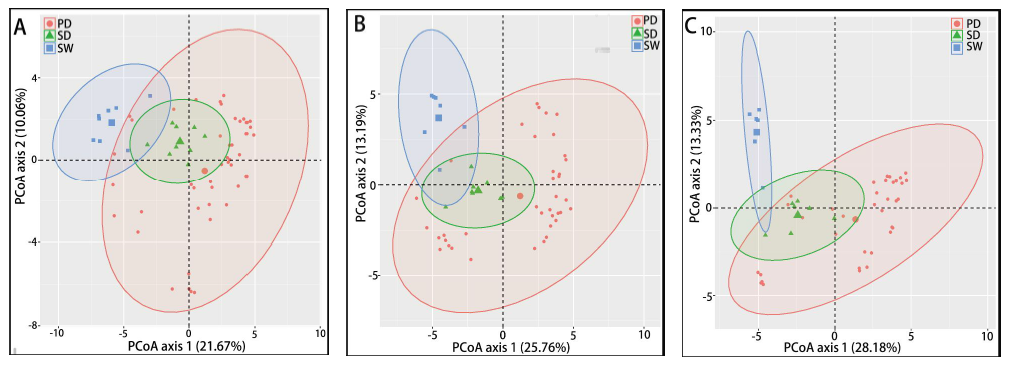
PCoA 图可以清楚地看到,SW区细菌群落的置信椭圆与pd和sd的置信椭圆有显著的偏差(p<0.05),而sd上细菌群落的置信椭圆几乎覆盖了pd的置信椭圆(p>0.05),这表明pd和sd上的细菌群落有相似之处。
不同样本和处理下的细菌群落( 前 10 位)丰度分布
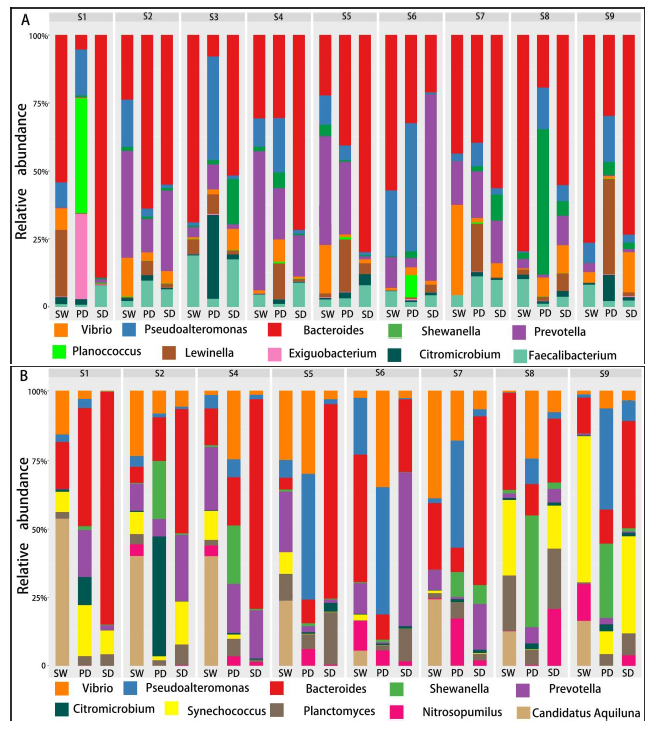
底物(SW、SD和Pd)上的主要属为细菌和假互斥单胞菌,暴露两周后,这些菌可能是分布广泛和适应性强的三种底物(SW、SD和PD)。暴露4周后,弧菌相对丰度增加.此外,暴露6周后,自养细菌(如扁平菌和硝酸菌)的数量增加。这三种底物上个细菌群落的生长模式也与3.2的结果一致。图5还显示,在6个星期内,在429个原位点中,假单胞菌在pd上的相对丰度高于sw和sd(anova,p<0.05)。
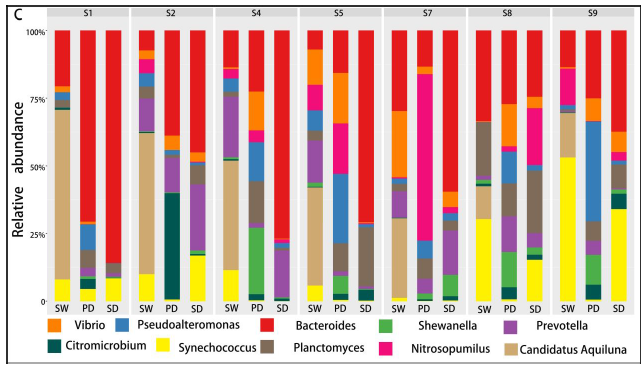
研究结论:首先,营养物质 (TN 和 TP) 与生物膜的平均生长速率呈正相关,而盐度与生物膜的平均生长速率呈负相关。盐度是影响PD的个细菌多样性的主要因素,而温度、溶解氧和养分(TN和TP)在类似的盐度条件下可能具有二次效应。尽管种聚合物类型对PD上的细菌群落的多样性具有较少的影响,但是在细菌群落中的一些属显示对PD的聚合物类型的选择性,并且倾向于将其优选的基质定殖。大的相对丰度SW、PD、SD间属显著差异。盐度是改变河口地区Pd条件致病菌富集的主要因素。另外,在种病原物种丰富的基础上,PD具有较高的致病性。
NMDS分析(非度量多维尺度分析)
NMDS(Nonmetric Multidimensional Scaling)常用于比对样本组之间的差异,可以基于进化关系或数量距离矩阵。
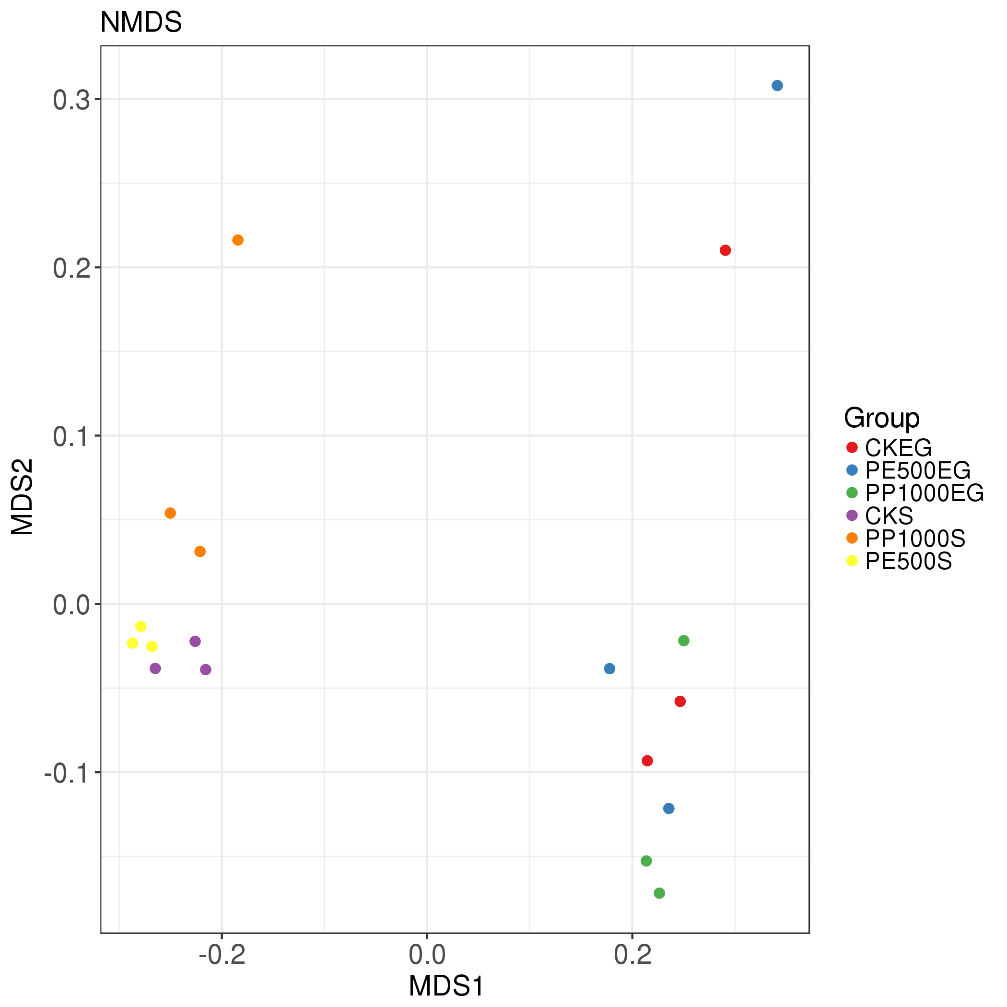
横轴和纵轴:表示基于进化或者数量距离矩阵的数值在二维表中成图。与PCA分析的主要差异在于考量了进化上的信息。
每一个点代表一个样本,相同颜色的点来自同一个分组,两点之间距离越近表明两者的群落构成差异越小。
排序分析
PCA,PcoA,NMDS分析都属于排序分析(Ordination analysis)。
排序(ordination)的过程就是在一个可视化的低维空间或平面重新排列这些样本。
目的:使得样本之间的距离最大程度地反映出平面散点图内样本之间的关系信息。
排序又分两种:非限制性排序和限制性排序。
1、非限制性排序(unconstrained ordination)
——只使用物种组成数据的排序
(1) 主成分分析(principal components analysis,PCA)
(2) 对应分析(correspondence analysis, CA)
(3) 去趋势对应分析(Detrended correspondence analysis, DCA)
(4) 主坐标分析(principal coordinate analysis, PCoA)
(5) 非度量多维尺度分析(non-metric multi-dimensional scaling, NMDS)
2、限制性排序(constrained ordination)
——同时使用物种和环境因子组成数据的排序
(1) 冗余分析(redundancy analysis,RDA)
(2) 典范对应分析(canonical correspondence analysis, CCA)
比较PCA和PCoA
在非限制性排序中,16S和宏基因组数据分析通常用到的是PCA分析和PCoA分析,两者的区别在于:
PCA分析是基于原始的物种组成矩阵所做的排序分析,而PCoA分析则是基于由物种组成计算得到的距离矩阵得出的。
在PCoA分析中,计算距离矩阵的方法有很多种,包括如:Euclidean, Bray-Curtis, and Jaccard,以及(un)weighted Unifrac (利用各样品序列间的进化信息来计算样品间距离,其中weighted考虑物种的丰度,unweighted没有对物种丰度进行加权处理)。
LDA差异贡献分析
如果说 PCA,它所作的只是将整组数据整体映射到最方便表示这组数据的坐标轴上,映射时没有利用任何数据内部的分类信息,是无监督的。
那么LDA是有监督的,增加了种属之间的信息关系后,结合显著性差异标准测试(克鲁斯卡尔-沃利斯检验和两两Wilcoxon测试)和线性判别分析的方法进行特征选择。
两者相同点:
差异:
1)LDA是有监督学习的降维方法,而PCA是无监督的降维方法。(注:监督学习是从标记的训练数据来推断一个功能的机器学习任务。)
2)LDA选择分类性能最好的投影方向,而PCA选择样本点投影具有最大方差的方向。
除了可以检测重要特征,他还可以根据效应值进行功能特性排序,这些功能特性可以解释大部分生物学差异。这部分希望能详细了解的同学可以参考这篇文章http://blog.csdn.net/sunmenggmail/article/details/8071502 。
LDA分析究竟能做什么
组间差异显著物种又可以称作生物标记物(biomarkers),这个LDA分析主要是想找到组间在丰度上有显著差异的物种。
案例解析

研究背景:研究表明遗传和环境影响都在I型糖尿病的发展中起作用,增加的遗传风险不足以引起疾病,环境因素也是需要的,而且起着至关重要的作用。肠道菌群也许就是这个重要的环境因素,肠道菌群在免疫系统的成熟中起重要作用,此外还影响自身免疫疾病发展。

不同遗传风险儿童的LDA差异菌群
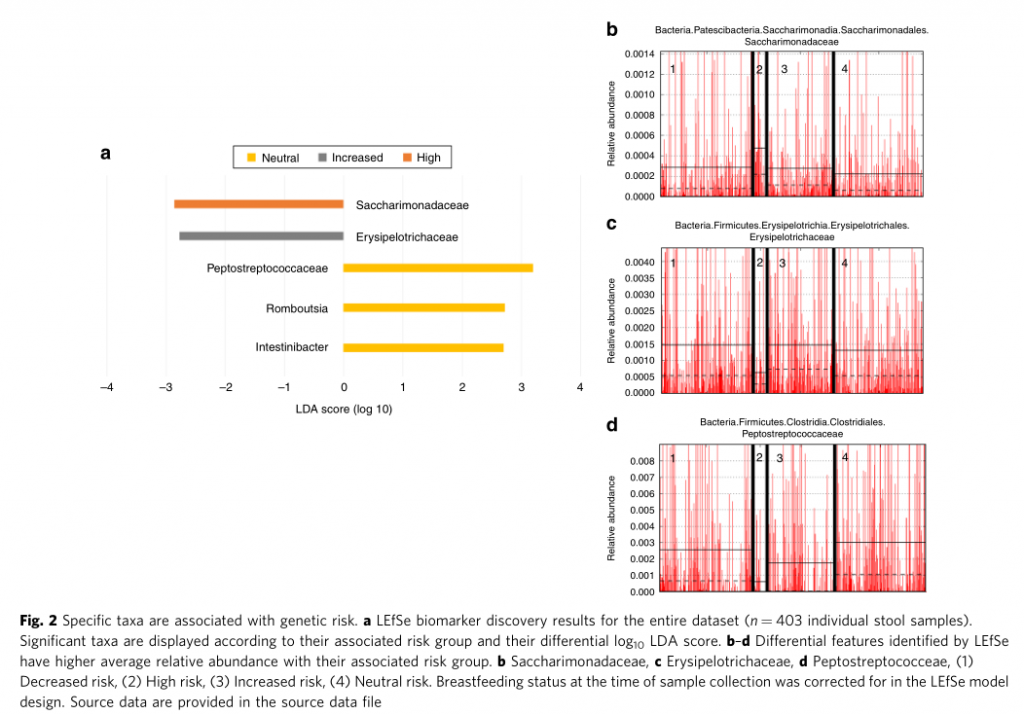
不同遗传风险分组中包含的常见菌属,部分存在特定分组中
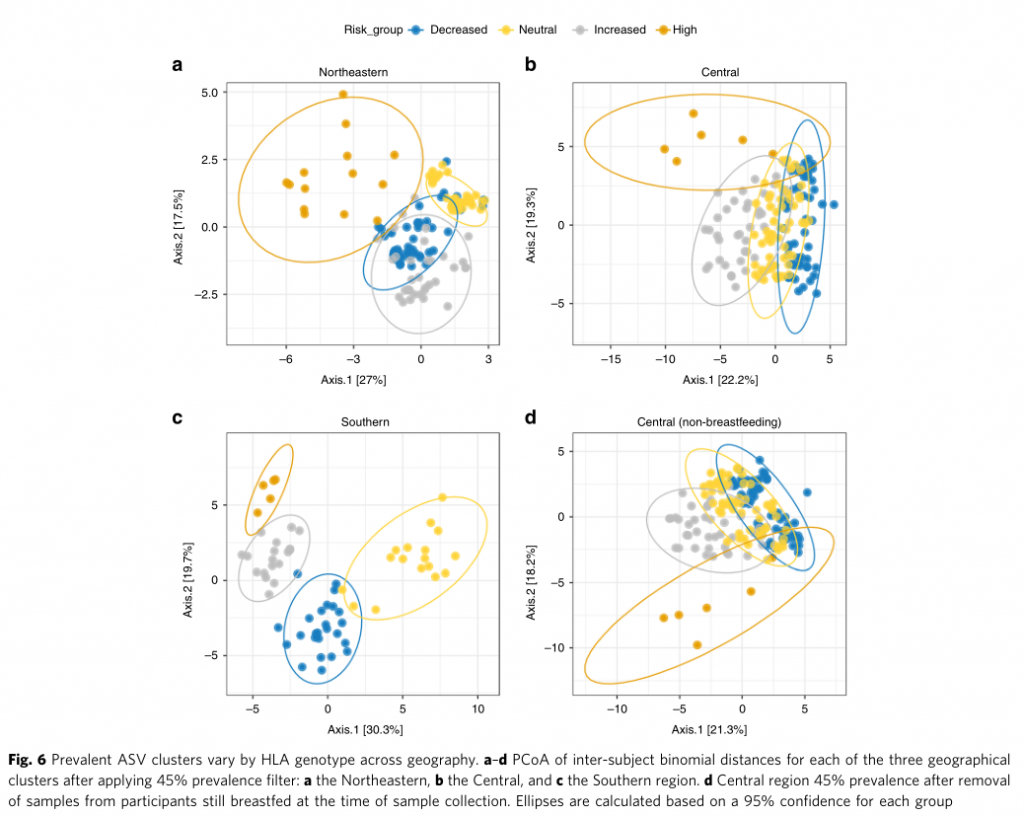
PCoA分析揭示不同遗传风险儿童肠道菌群的在不同地域样本中均存在显著差异
点评:针对I型糖尿病疾病发生过程中遗传HLA分型风险和对应肠道菌群菌的关联分析,揭示了特定肠道菌群与宿主特定遗传风险共同作用推进疾病发生。某些特定菌属可能无法在遗传高风险儿童肠道内定植,可能对疾病发生存在特定作用。此外对于其他遗传风险的自身免疫疾病也具有重要提示意义,例如乳糜泻和类风湿性关节炎。
物种进化树的样本群落分布图
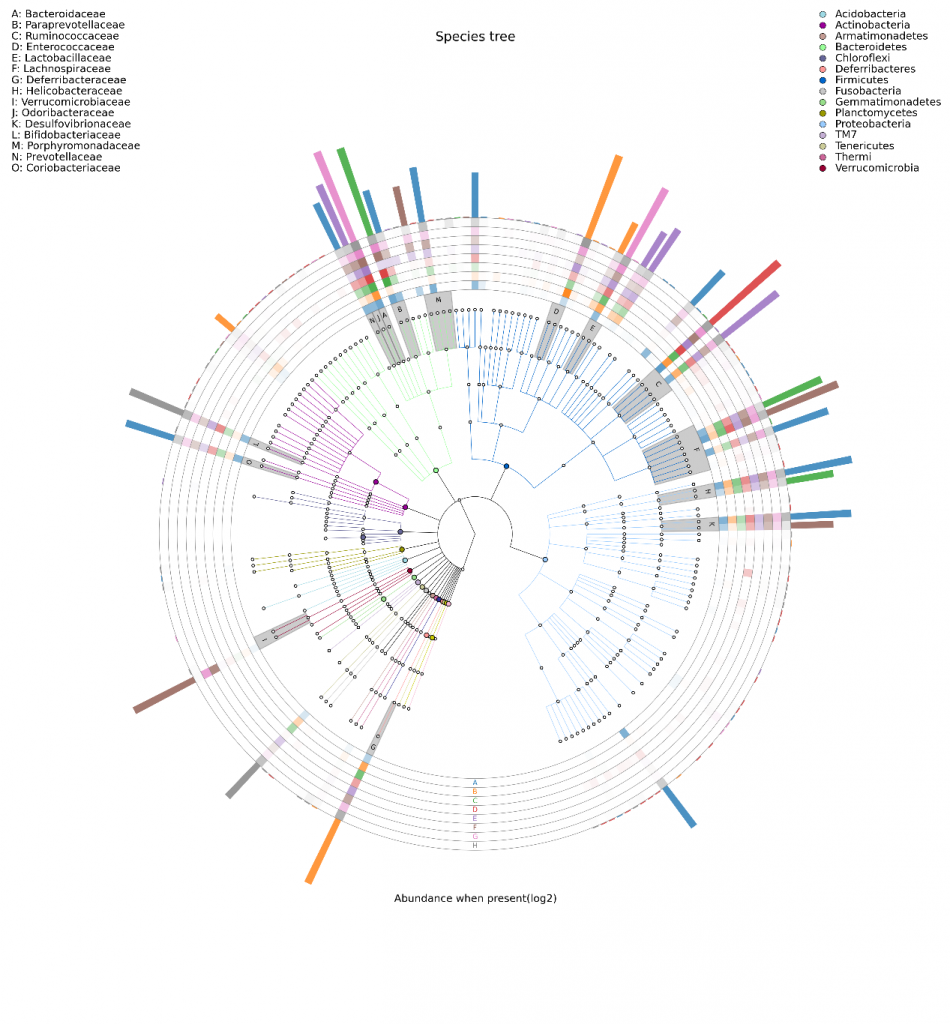
这是另一款和LDA长得有点像的图,当然功能可完全不一样。它是将不同样本的群落构成及分布以物种分类树的形式在一个环图中展示。数据经过分析后,将物种分类树和分类丰度信息通过这款软件GraPhlAn进行绘制
(http://huttenhower.sph.harvard.edu/GraPhlAn )。
其目的是将物种之间的进化关系以及不同样本的物种分布丰度和最高分布样本的信息在一个视觉集中的环图中一次展示,其提供的信息量较其他图最为丰富。
物种相关性分析
根据各个物种在各个样品中的丰度以及变化情况,计算物种之间的相关性,包括正相关和负相关。
相关性分析使用CCREPE算法
怎么画的?
首先对原始16s测序数据的种属数量进行标准化,然后进行Spearman和Pearson秩相关分析并进行统计检验,计算出各个物种之间的相关性,之后在所有物种中根据simscore绝对值的大小,挑选出相关性最高的前100组数据,基于Cytoscap绘制共表达分析网络图。
网络图采用两种不同的形式表现出来。
物种相关性网络图A
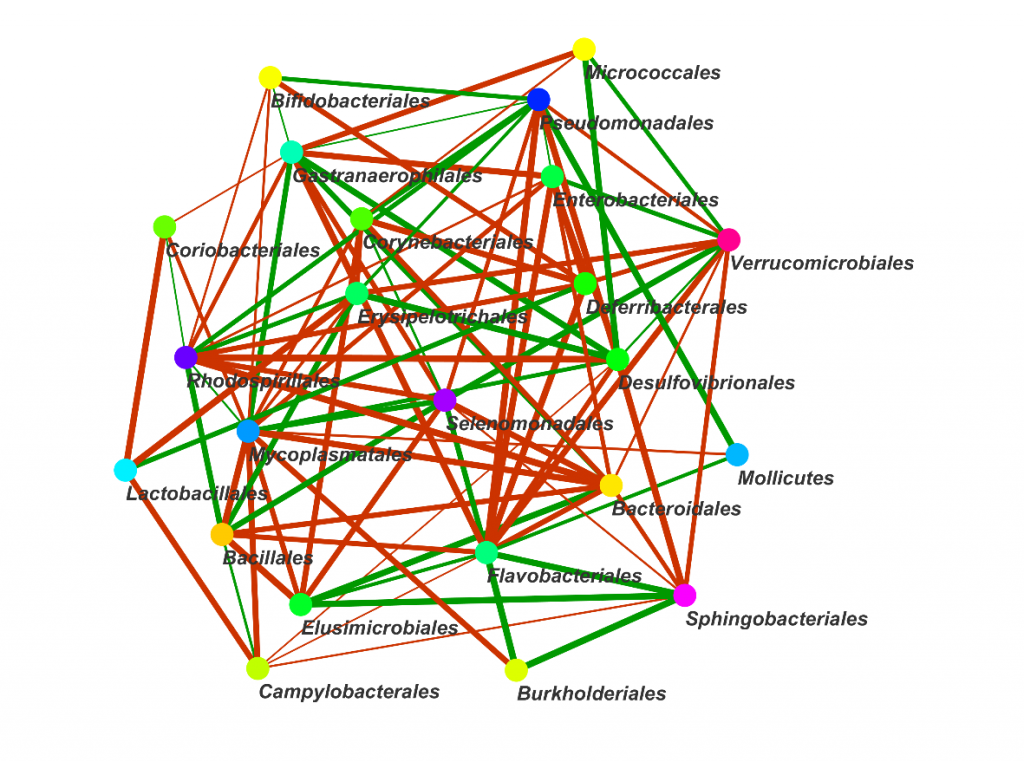
○ 图中每一个点代表一个物种,存在相关性的物种用连线连接。
○ 红色的连线代表负相关,绿色的先代表正相关。
○ 连线颜色的深浅代表相关性的高低。
物种相关性网络图B
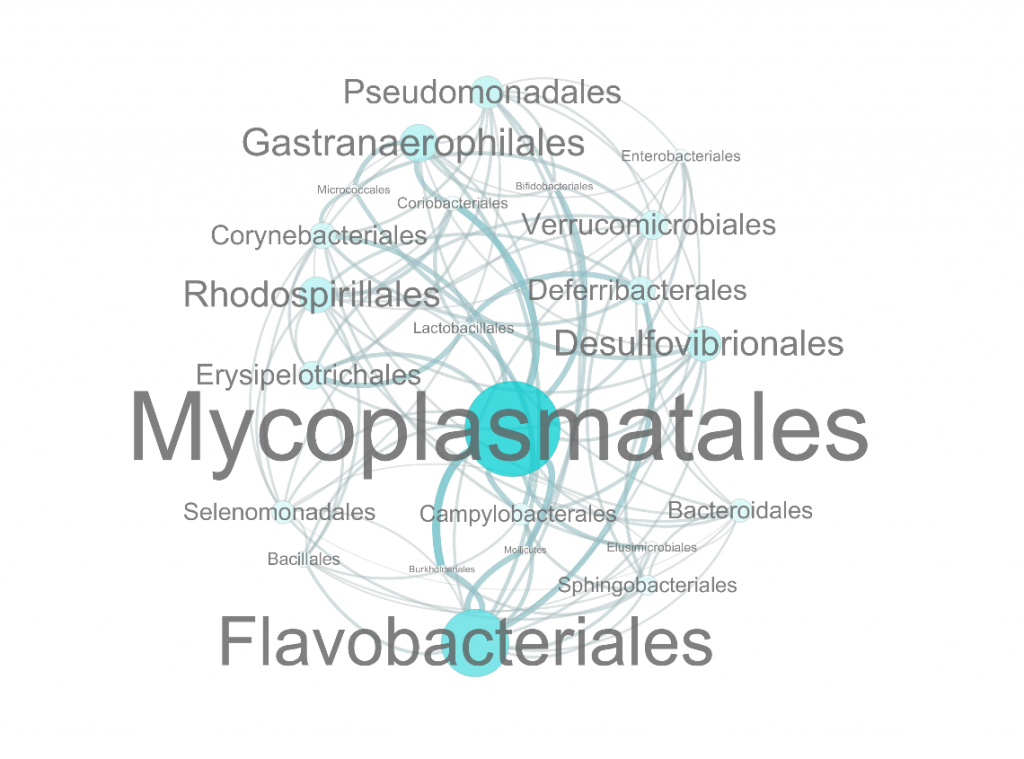
○ 图中每一个点代表一个物种
○ 点的大小表示与其他物种的关联关系的多少
○ 其中与之有相关性的物种数越多,点的半径和字体越大
○ 连线的粗细代表两物种之间相关性的大小
连线越粗,相关性越高。
案例解析


研究背景:气候变化导致美国中部草原的降水模式发生变化,对土壤微生物群落构成及代谢影响很大。
研究希望明确土壤微生物群落对土壤水分变化的反应,并确定响应的特定代谢特征。
主要图表
同一样本在不同水分含量孵化处理下土壤菌群的变化

受到水分条件影响的土壤菌群代谢途径和网络分布
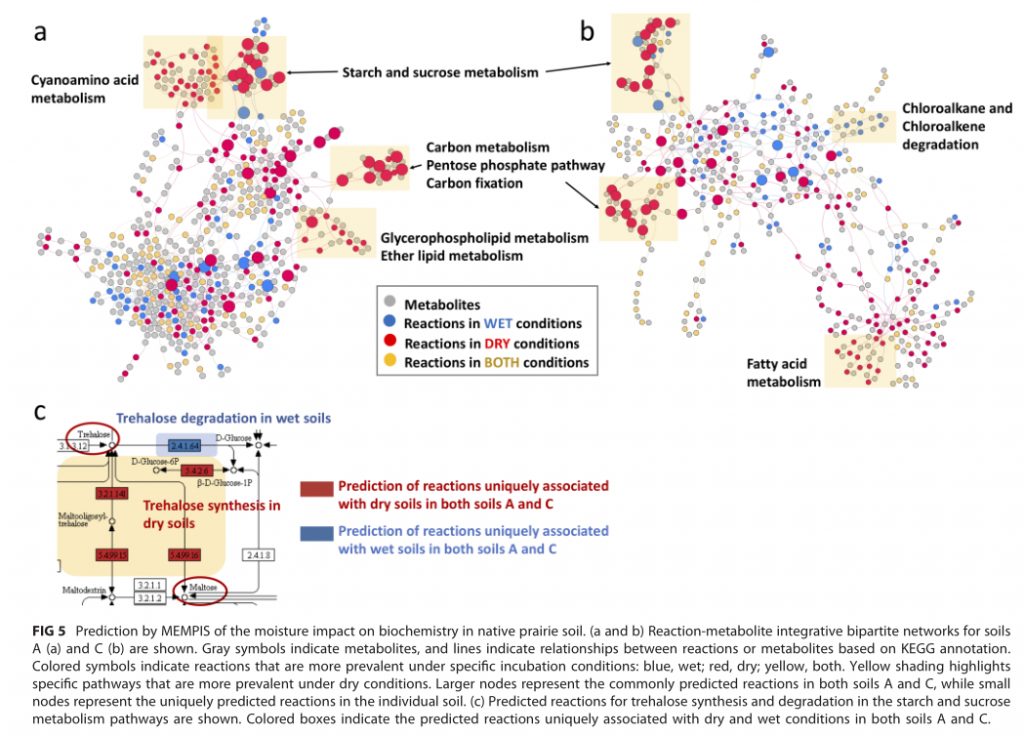

研究结论:土壤干燥导致土壤微生物组的组成和功能发生显着变化。相反,润湿后几乎没有变化。由于干旱导致的土壤水分减少对土壤碳循环和土壤微生物组进行的其他关键生物地球化学循环的影响很大。导致渗透保护剂化合物产生的代谢途径受到较大影响。
点评:
相对简单的样本和实验设计,但是从多个维度探寻支持土壤微生物群落对湿润和干燥表型的反应。
与常见的环境采样检测不同,针对同一样本在对照环境下进行环境控制孵化,然后比较菌群变化可以更为有效的控制背景差异。
聚类分析
根据OTU数据进行标准化处理(1wlog10)之后,选取数目最多的前60个物种,基于R heatmap进行作图
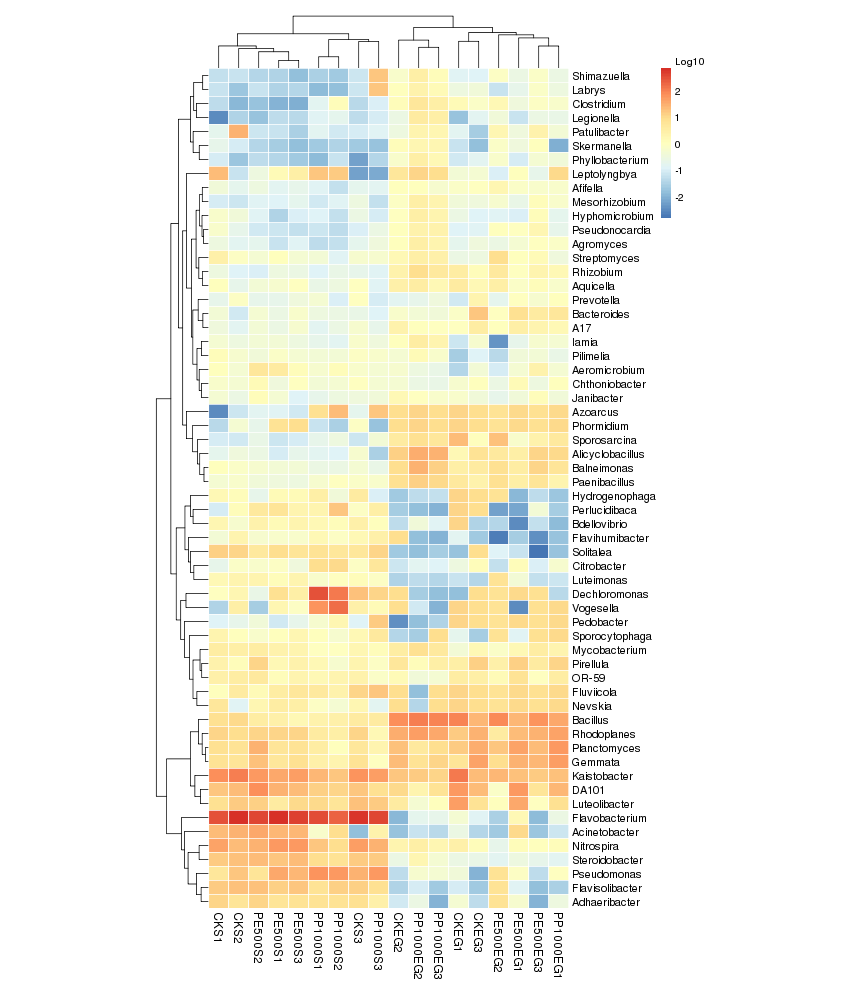

○ 热图中的每一个色块代表一个样品的一个属的丰度
○ 样品横向排列,属纵向排列
○ 差异是是否对样品进行聚类,从聚类中可以了解样品之间的相似性以及属水平上的群落构成相似性。
Tips:
如果聚类结果中出现大面积的白或黑是因为大量的菌含量非常低,导致都没有数值,可以在绘制之前进行标准化操作,对每一类菌单独自身进行Z标准化。
案例解析






研究背景:妊娠期糖尿病(GDM)的患病率在全球范围内迅速增加,构成一个重要的健康问题和产科实践的重大挑战(Ferrara,2007)。高脂血症是妊娠常见的合并症。在GDM患者中,血脂的生理变化可能导致怀孕期间潜在的代谢紊乱。肠道失调在宿主代谢异常中起着至关重要的作用,最近关于2型糖尿病(T2D)和肥胖的研究就证明了这一点。这些研究表明,妊娠期间肠道微生物ME的主要变化可能在GDM的发展中起着至关重要的作用。

GDM加高脂血症(M队列)妊娠期间与显著改变的脂质相关的肠道微生物群(属)
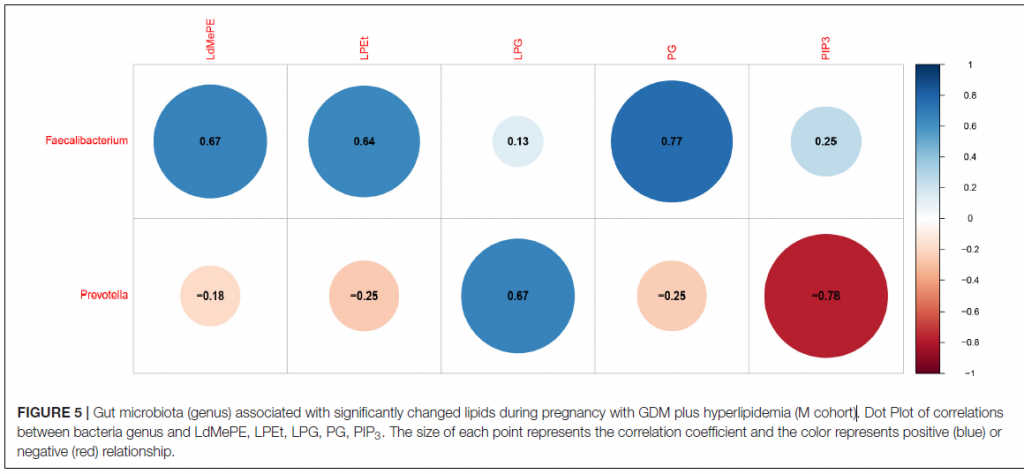


研究结论:我们的结果表明,血脂水平可能反映了GDM发展过程中的一些异常变化。所鉴定的多种生物标志物对GDM合并高脂血症的防治有一定的参考价值。

组间物种差异性箱形图
组间物种差异性盒形图描述在不同分组之间具有差异显著的某一物种做盒形图,图中以属水平为例做物种差异性盒形图,展示如下:

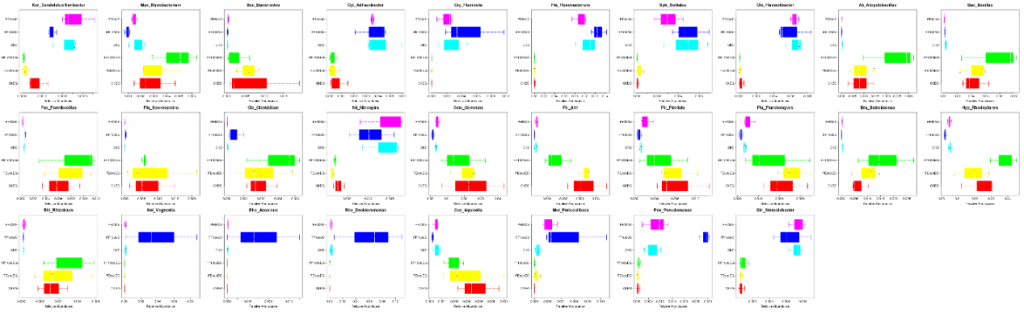

○ 图中不同颜色代表不同的分组,更直观显示组间物种差异
○ 每一个盒形图代表一个物种,图上方是物种名。
Anosim检验
Anosim分析是一种非参数检验,用来检验组间的差异是否显著大于组内差异,从而判断分组是否有意义
展示如下:


R-value介于(-1,1)之间,R-value大于0,说明组间差异显著。
R-value小于0,说明组内差异大于组间差异。
统计分析的可信度用 P-value 表示,P< 0.05 表示统计具有显著性。
对Anosim的分析结果,基于两两样本之间的距离值排序获得的秩(组间的为between,组内的为within),这样任一两两组的比较可以获得三个分类的数据,并进行箱线图的展示(若两个箱的凹槽互不重叠,则表明它们的中位数有显著差异)
随机森林分类树属分类效果
随机森林是机器学习算法的一种,它可以被看作是一个包含多个决策树的分类器。
其输出的分类结果是由每棵决策树“投票”的结果。由于每棵树在构建过程中都采用了随机变量和随机抽样的方法,因此随机森林的分类结果具有较高的准确度,并且不需要“减枝”来减少过拟合现象。
随机森林可以有效的对分组样品进行分类和预测。
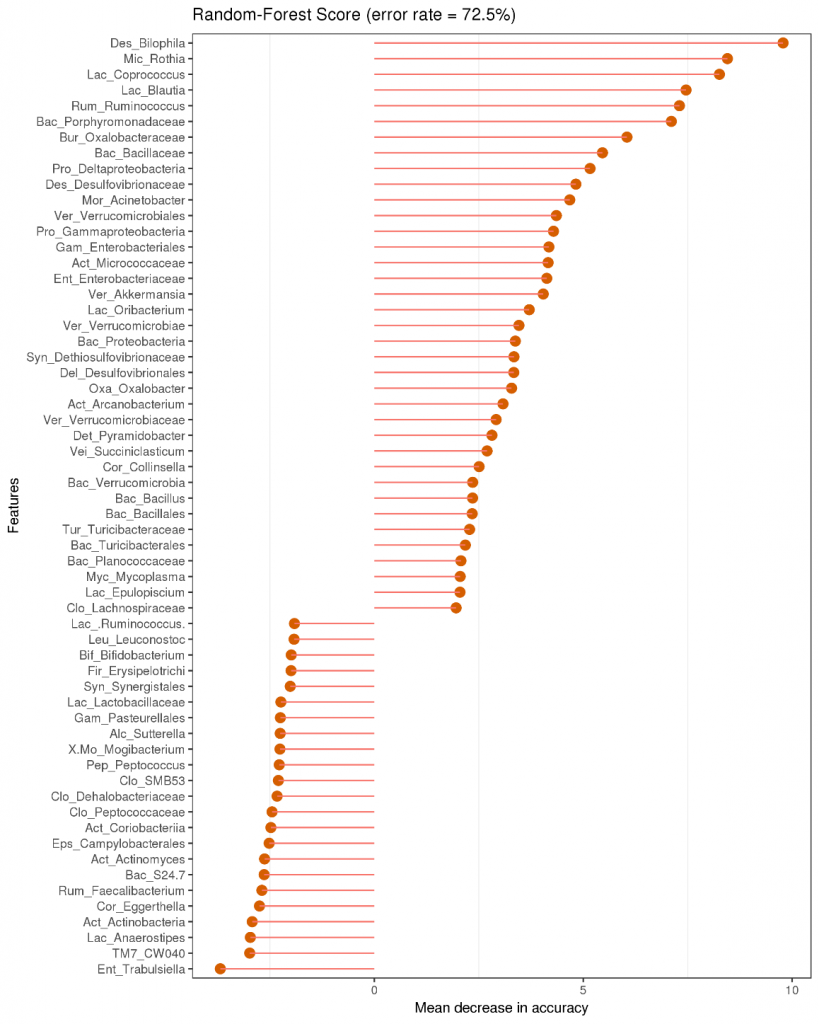

物种重要性点图。横坐标为重要性水平,纵坐标为按照重要性排序后的物种名称。上图反映了分类器中对分类效果起主要作用的菌属,按作用从大到小排列。
Error rate: 表示使用下方的特征进行随机森林方法预测分类的错误率,越高表示基于菌属特征分类准确度不高,可能分组之间菌属特征不明显。图中以所有水平为例,取前60个作图。
ROC曲线图
ROC 曲线指受试者工作特征曲线(receiver operating characteristic curve), 是反映敏感性和特异性连续变量的综合指标,通过构图法揭示敏感性和特异性的相互关系。
ROC 曲线将连续变量设定出多个不同的临界值,从而计算出一系列敏感性和特异性,再以敏感性为纵坐标、(1-特异性)为横坐标绘制成曲线。
曲线下面积越大,诊断准确性越高。展示如下:
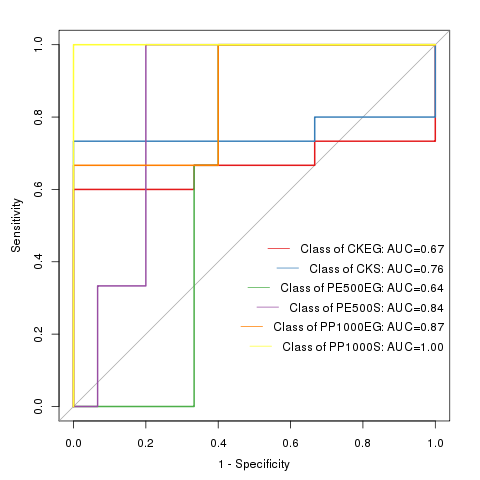

FAPROTAX生态功能预测
FAPROTAX是一款在2016年发表在SCIENCE上的较新的基于16S测序的功能预测软件。它整合了多个已发表的可培养菌文章的手动整理的原核功能数据库,数据库包含超过4600个物种的7600多个功能注释信息,这些信息共分为80多个功能分组,其中包括如硝酸盐呼吸、产甲烷、发酵、植物病原等。
FAPROTAX对环境样本更友好
如果说PICRUSt(后续会介绍)在肠道微生物研究更为适合,那么FAPROTAX尤其适用于生态环境研究,特别是地球化学物质循环分析。
FAPROTAX适用于对环境样本(如海洋、湖泊等)的生物地球化学循环过程(特别是碳、氢、氮、磷、硫等元素循环)进行功能注释预测。因其基于已发表验证的可培养菌文献,其预测准确度可能较好,但相比于上述PICRUSt和Tax4Fun来说预测的覆盖度可能会降低。
FAPROTAX可根据16S序列的分类注释结果对微生物群落功能(特别是生物地化循环相关)进行注释预测。
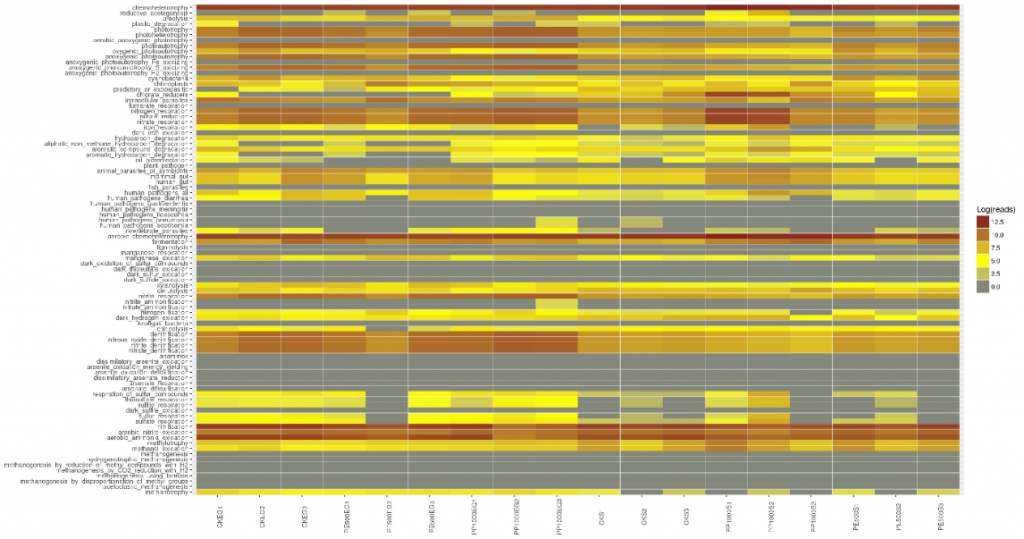

图中横坐标代表样本,纵坐标表示包括碳、氢、氮、硫等元素循环相关及其他诸多功能分组。可快速用于评估样品来源或特征。
基于BugBase的表型分类比较
Bugbase也是16年所提供服务的一款免费在线16S功能预测工具,到今年才发表文章公布其软件原理。该工具主要进行表型预测,其中表型类型包括革兰氏阳性、革兰氏阴性、生物膜形成、致病性、移动元件、氧需求,包括厌氧菌、好氧菌、兼性菌)及氧化胁迫耐受等7类。
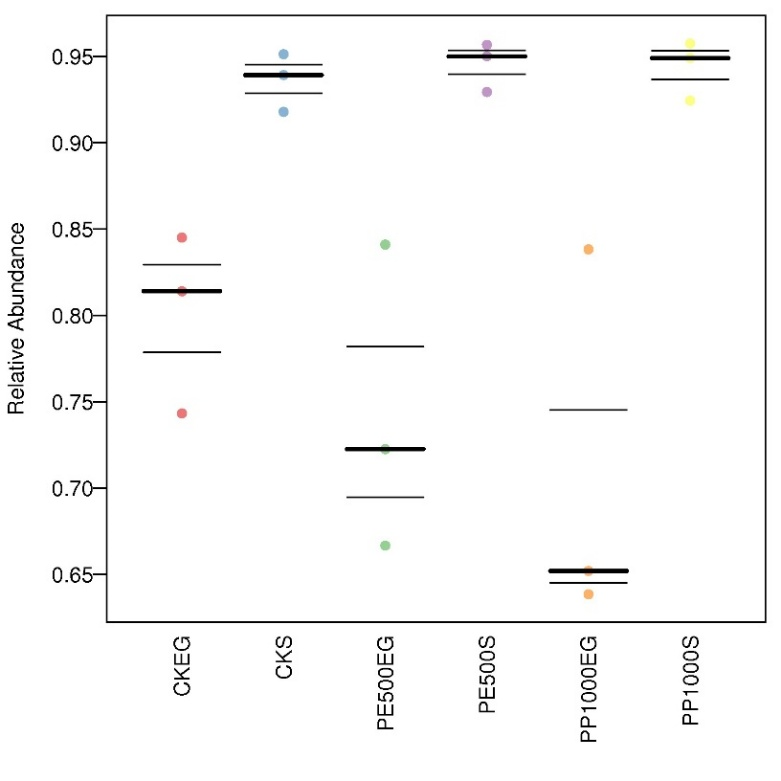

Gram Negative 革兰氏阴性菌
Picrust群落功能差异分析
通过对已有测序微生物基因组的基因功能的构成进行分析后,我们可以通过16s测序获得的物种构成推测样本中的功能基因的构成,从而分析不同样本和分组之间在功能上的差异(PICRUSt Nature Biotechnology, 1-10. 8 2013)。
Picrust对肠道菌群样本更友好
通过对宏基因组测序数据功能分析和对应16s预测功能分析结果的比较发现,此方法的准确性在84%-95%,对肠道微生物菌群和土壤菌群的功能分析接近95%,能非常好的反映样品中的功能基因构成。
怎么做出来的?
为了能够通过16s测序数据来准确的预测出功能构成,首先需要对原始16s测序数据的种属数量进行标准化,因为不同的种属菌包含的16s拷贝数不相同。
然后将16s的种属构成信息通过构建好的已测序基因组的种属功能基因构成表映射获得预测的功能结果。(根据属这个水平,对不同样本间的物种丰度进行显著性差异两两检验,我们这里的检验方法使用STAMP中的two-sample中T-TEST方法,Pvalue值过滤为0.05,作Extent error bar图。)
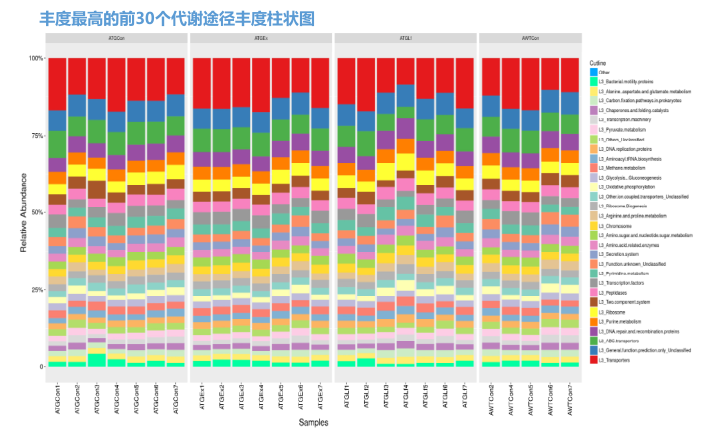

此处提供COG,KO基因预测以及KEGG代谢途径预测。当然,跃跃欲试的小伙伴也可自行使用我们提供的文件和软件(STAMP)对不同层级以及不同分组之间进行统计分析和制图,以及选择不同的统计方法和显著性水平。
这里提到的STAMP有些小伙伴说不太了解,别急,后面会有更多介绍。
COG构成差异分析图
图中不同颜色代表不同的分组,列出了COG构成在组间存在显著差异的功能分类以及在各组的比例,此外右侧还给出了差异的比例和置信区间以及P-value。
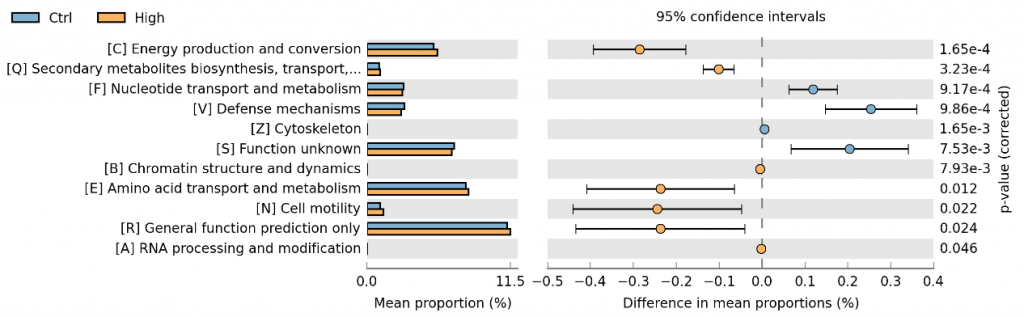

KEGG代谢途径差异分析图
通过KEGG代谢途径的预测差异分析,我们可以了解到不同分组的样品之间在微生物群落的功能基因在代谢途径上的差异,以及变化的高低。为我们了解群落样本的环境适应变化的代谢过程提供一种简便快捷的方法。
本例图所显示的是第三层级的KEGG代谢途径的差异分析,也可以针对第二或第一层的分级进行分析。
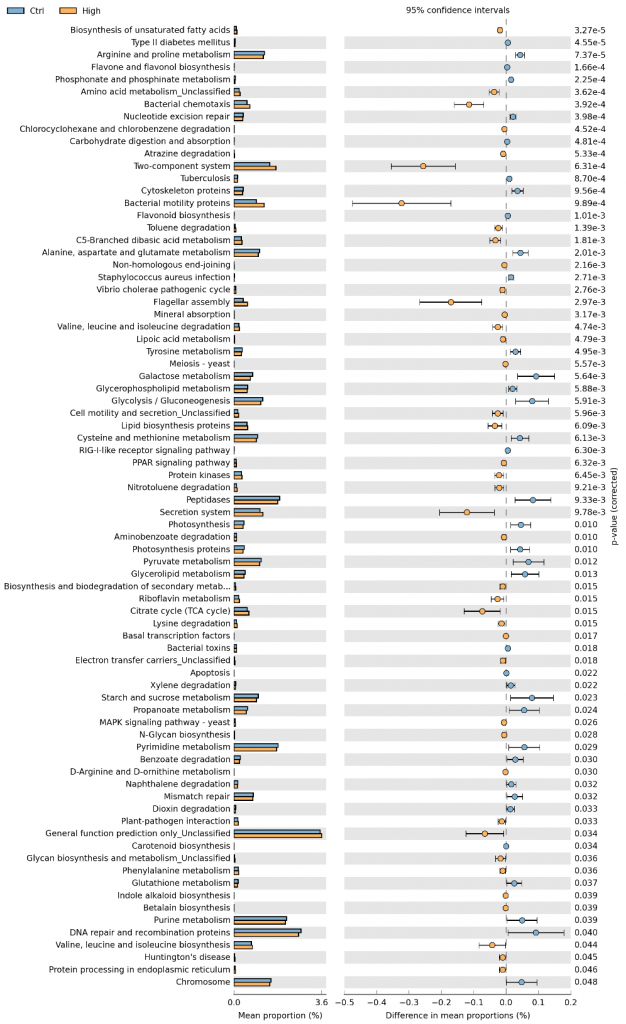

图中不同颜色代表不同的分组,列出了在第三层级的构成在组间存在显著差异的KEGG代谢途径第三层分类以及在各组的比例,此外右侧还给出了差异的比例和置信区间以及P-value。
案例解析




研究背景:尽管普遍认为肠道微生物组的生态多样性和分类组成在肥胖和T2D中发生改变,但与单个微生物或微生物产物的关联在研究之间不一致。缺乏大样本群体研究,从而确定肠道微生物组,血浆代谢组,肥胖和糖尿病表型以及环境因素之间的几种关联。
主要图表:
按照肥胖和糖尿病对人群分为三组,同时进行了16S,代谢和宏基因组的检测。

与肥胖相关的菌属以及代谢途径
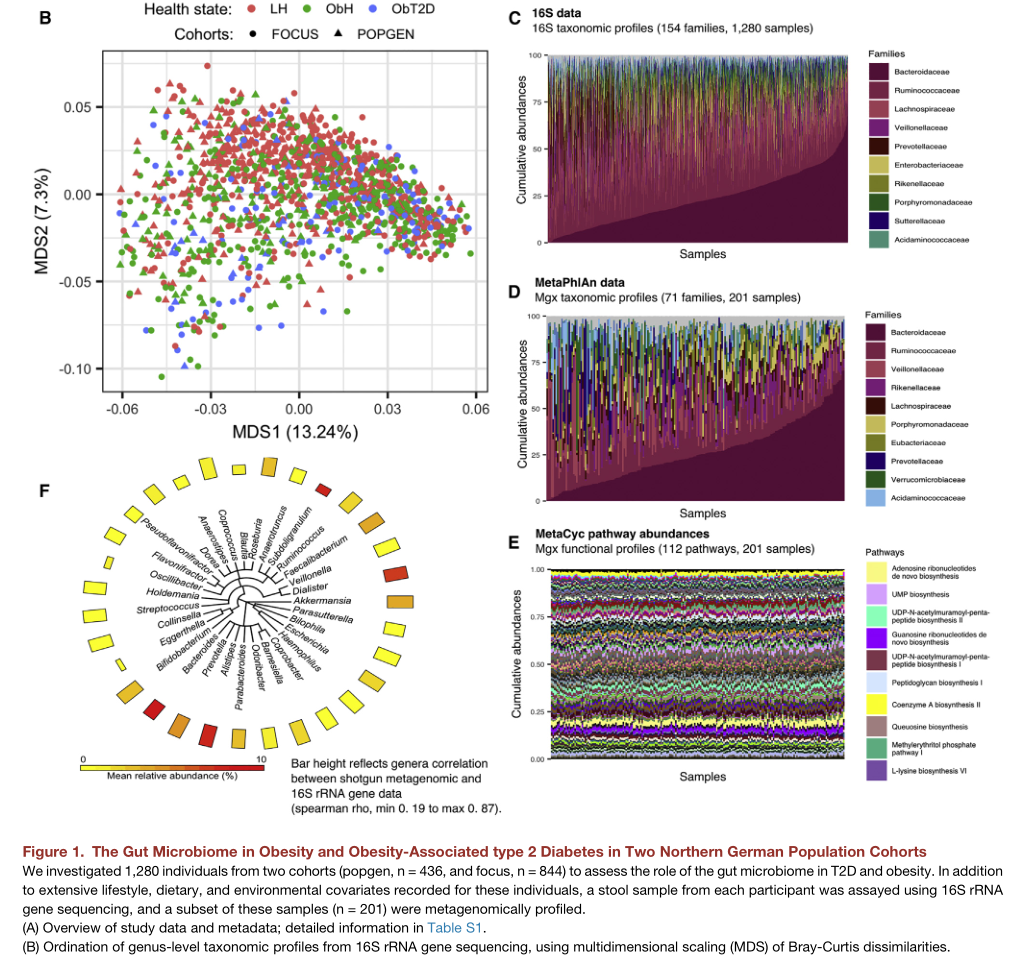

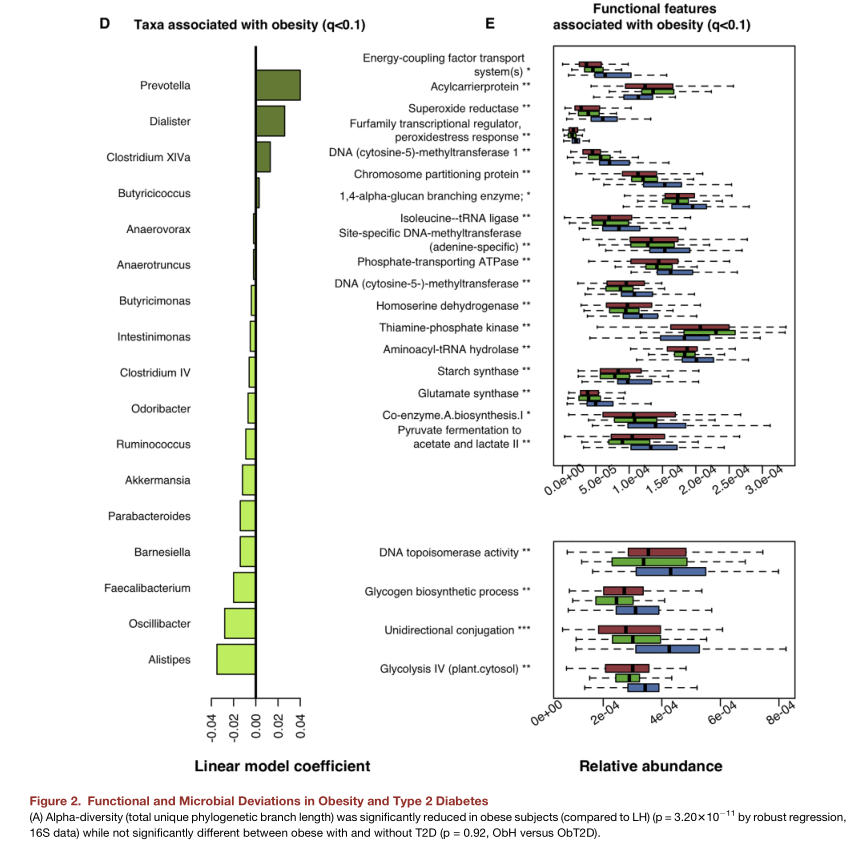

研究结论:确定了肠道微生物组,血浆代谢组,肥胖和糖尿病表型以及环境因素之间的几种关联。与肠道微生物组变异相关的主要是肥胖,不是2型糖尿病。存在与肠道微生物组变异相关的药物和膳食补充剂。高铁摄入量影响小鼠的肠道微生物组成。微生物组变异也反映在血清代谢物谱中。
点评:
相对大人群的队列研究,同时涵盖了菌群、代谢和疾病表型以及膳食补充调查的数据。
从结果看菌属和血浆代谢存在关联,但是贡献度都较低,如果样本数量不足很可能找不到显著的联系,这也是这类大样本队列研究的意义。
本研究在人群分组时针对性的研究了肥胖-II型糖尿病和菌群的关联,因而构建了三个主要分组人群,结果显示肥胖与菌群的关联度更大,解释了大部分的菌群差异,而糖尿病的菌群变化较小。
本研究其中较为重要的是发现了不同膳食补充对菌群的影响,并在小鼠实验中得到证实。

基因的差异分析图
除了能对大的基因功能分类和代谢途径进行预测外,我们还能提供精细的功能基因的数量和构成的预测,以及进行样本间以及组间的差异分析,并给出具有统计意义和置信区间的分析结果。
这一分析将我们对于样本群落的差异进一步深入到了每一类基因的层面。


图中不同颜色代表不同的分组,列出了在组间/样本间存在显著差异的每一个功能基因(酶)以及在各组的比例,此外右侧还给出了差异的比例和置信区间以及P-value。
很多小伙伴总希望能亲自上手做点分析,机会来了!
在获得标准报告后如果希望单独修改分组或对某些组之间进行显著性差异分析,可以使用STAMP软件在自己的电脑上进行数据分析。STAMP提供了丰富的统计检验方法和图形化结果的输出。
在使用STAMP之前需要首先准备需要的spf格式文件和样品分组信息表,但是如果数据不会处理,那也很不便。
而在我们的报告中已经将KEGG和KO以及COG的结果文件后经过转换生成了适用于STAMP软件打开的spf格式文件,还有对应的分组信息表文件groupfile.txt。
使用STAMP时的一些相关问题
1、STAMP作图用的原始数据的来源?
STAMP 可以直接使用来自QIIME的biom文件和PICUST的KEGG和ko 文件,groupfile.txt文件的格式为tab-saperated value (tab键隔开的数据)
2、分组问题?
导入数据之后,viewàgroup legend ,在窗口右侧会出现分组栏,根据需要进行分组。
3、Unclassiffied选项中,remain Unclassiffied reads、remove Unclassiffied reads、和use only for calculating frequency profiles 方法的区别?
remain Unclassiffied reads和use only for calculating frequency profiles方法会保留所有的数据,而remove Unclassiffied reads仅仅保留有确定分组信息的数据。
4、Statistical test 中,Welch’s t-test、t-test、white’s non-parametric t-test的区别,各自优缺点?
为了确保统计学意义和准确度和精确性,需要足够多的样本数目,t-test检验可以在最少样本数为4的时候确保高的准确度和精确性。
当两个样本之间具有相同方差的时候,用t-test更为准确,当两个样本没有相同方差,Welch’s t-test更为准确。
当样本数目少于8的时候,可以使用white’s non-parametric t-test,该计算时间较长,当样本数目过多的时候不宜使用该方法。
5、Two-group 中 type: one side和two side的区别?
One side 只会显示前一个group与后一个group差异的比例,而two side 两者之间的比例均会显示。
6、STAMP在使用时首先打开了一个分析文件,如果新打开一个可能会导致显示错误?
目前版本的STAMP存在一些小问题,一次分析只能使用一个数据文件,如果要打开新的需要关闭软件后再打开。
详细的STAMP使用教程可以参考我们提供的STAMP使用教程。
环境因子分析
冗余分析(redundancy analysis, RDA)或者
典范对应分析(canonical correspondence analysis, CCA)都是基于对应分析发展的一种排序方法,将对应分析与多元回归分析相结合,每一步计算均与环境因子进行回归,又称多元直接梯度分析。主要用来反映菌群与环境因子之间的关系。
RDA 是基于线性模型,CCA是基于单峰模型。分析可以检测环境因子、样品、菌群三者之间的关系或者两两之间的关系。
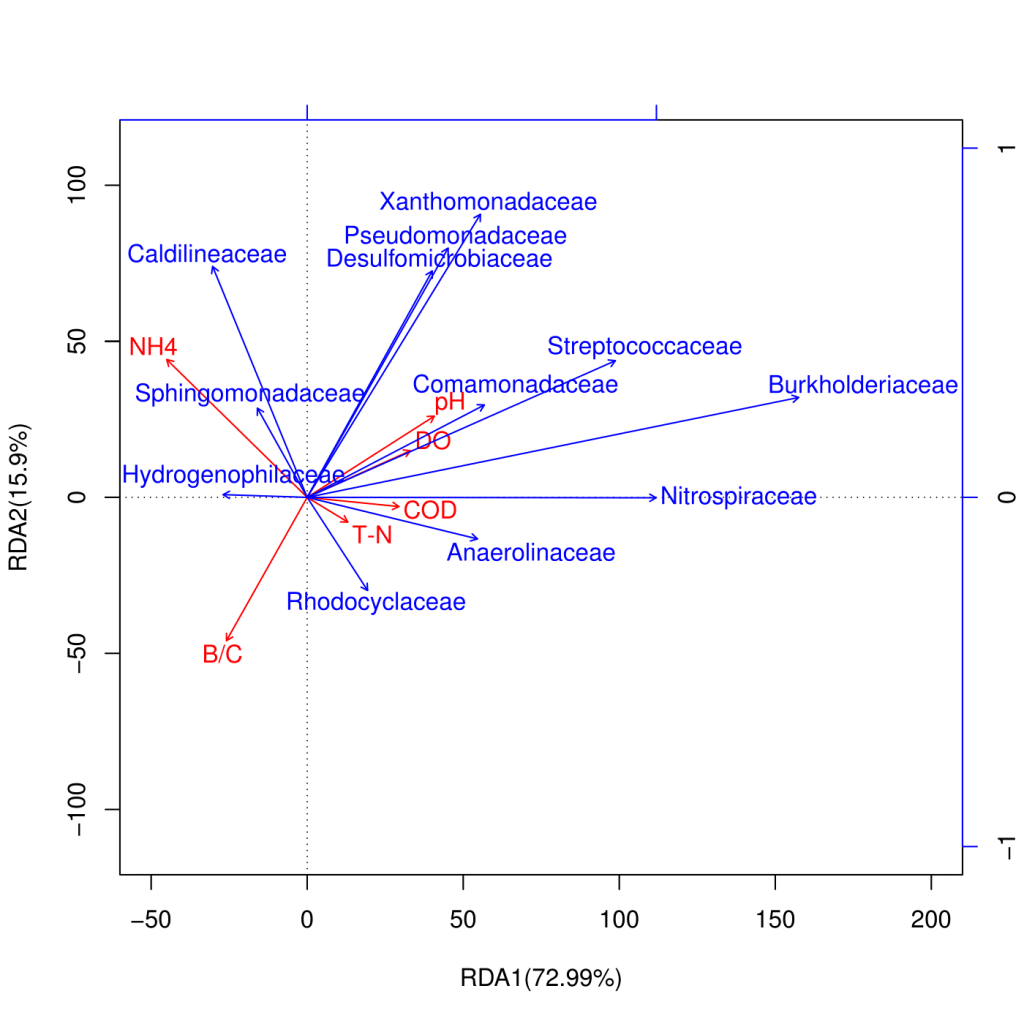

○ 冗余分析可以基于所有样品的OTU作图,也可以基于样品中优势物种作图;
○ 箭头射线:箭头分别代表不同的环境因子;
○ 夹角:环境因子之间的夹角为锐角时表示两个环境因子之间呈正相关关系,钝角时呈负相关关系。环境因子的射线越长,说明该影响因子的影响程度越大;
○ 不同颜色的点表示不同组别的样品或者同一组别不同时期的样品,图中的拉丁文代表物种名称,可以将关注的优势物种也纳入图中;
○ 环境因子数量要少于样本数量,同时在分析时,需要提供环境因子的数据,比如 pH值,测定的温度值等。
个性化图表

除以上部分,还可以进行个性化图表定制,像下面这样:

看完以上内容,也许还有不明白的地方,没关系,我们罗列了一些常见的问题。看看有没有你想问的。
答疑小课堂
Q1
原始数据形式以及数据如何上传?
原始fastq格式是一个文本格式用于存储生物序列(通常是核酸序列)和其测序对应的质量值。这些序列以及质量信息用ASCII字符标识。通常fastq文件中一个序列有4行信息:如
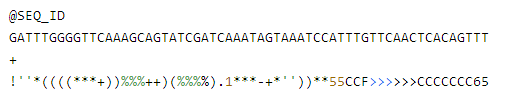

第一行:序列标识,以 @开头。格式自由,允许添加描述信息,描述信息以空格分开。
第二行:序列信息,不允许出现空格或制表符。一般是明确的DNA或RNA字符,通常大写
第三行:用于将序列信息和质量值分隔开。以 +开头,后边是描述信息或者不加。
第四行:质量值, 每个字符与第二行的碱基一一对应,按照一定规则转换为碱基质量得分。进而反映该碱基的错误率,因此字符数必须和第二行保持一致。
Fasta格式
fasta是一种基于文本用于表示核苷酸序列或氨基酸序列的格式。在这种格式中碱基对或氨基酸用单个字母来编码,且允许在序列前添加序列名及注释。由两部分信息组成:如

第一行:序列标记,以 >开头,接序列的标识符,序列标识符以空格结束,后接描述信息。为保证分析软件能区分每条序列,每个序列的标识必须具有唯一性。
第二行:序列信息,使用既定的核苷酸或氨基酸编码符号。
数据提交
原始数据(Raw data),常见的是illumina机器产生的fastq文件,这一类文件需要向NCBI的SRA数据库进行提交,SRA是NCBI为了并行测序的高通量数据(massively parallel sequencing)提供的存储平台。完整提交SRA需要一些独立项目的分步提交,包括BioProject、BioSample、Experiment、Run等,每一部分用以描述数据的不同属性。
Q2
如何判断测序质量是否合格?
原始的Tags数据会经过质控、过滤、去嵌合体,最终得到有效数据(Effective Tags)。所以在判断测序质量是否合格时应该从几个方面去判断。
打开文件01_sequence_statistic/sumOTUPerSample.txt
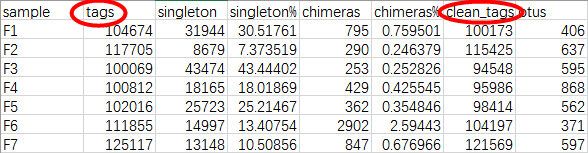


报告里所有的txt打开如果格式不对的话,可以用excel表打开。
其中tags为经质量过滤后能正确overlap包含正确barcode和高质量序列的数据。
Singleton为非完全相同的序列,只要有1个碱基的差异即为不同序列,该值的高低与OUT数量并无直接关系,OTU是以97%的相似度聚类,测序质量较低导致的碱基错误、PCR扩增过程中的碱基错误、菌种内部的多样性以及OTU数量均会影响该数量。
Chimeras为通过与RDP等标准数据库比对分析判断可能由于PCR过程错误扩增导致的嵌合体比例,chimeras%为百分比,一般低于1。
首先判断下机数据tags和有效数据 clean tags 的数据量是否满足测序要求,一般下机数据量达到3万条reads以上满足测序需要,谷禾16s样本的测序深度可以达到10万条reads左右。如果数据量不够则需要重新补测样本。通过观察嵌合体数chimras 和嵌合体所占百分比chimeras%,可以反应出有效序列的转化率,嵌合体的比例越小序列的利用转化率就越高。
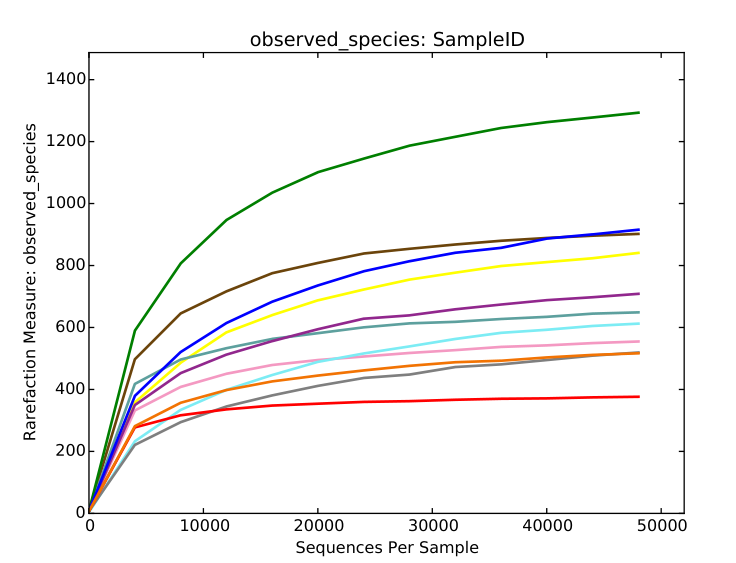

根据稀释曲线可以判断测序深度是否达到饱和,如图中曲线都逐渐趋于平缓,就证明样本的测序深度较好,测序深度基本覆盖能测到的该样本所有的物种,测序深度比较好。同时曲线趋于水平纵坐标的高低也能够反映各样本的微生物多样性情况,曲线越高,证明测到的物种种类越多,样本的微生物多样性就越高。
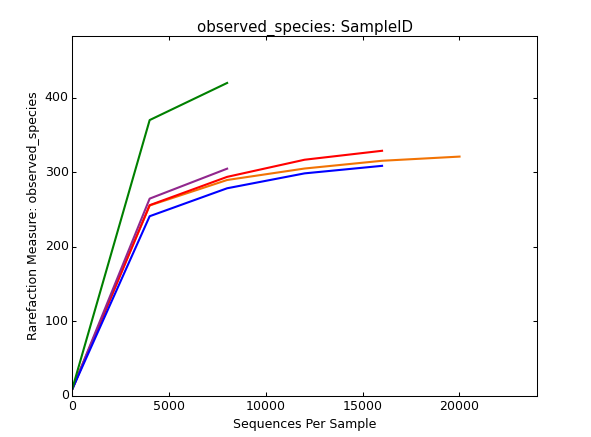

而从该图可以看出,个别样本的曲线未趋于平缓,证明该样本测序深度不够,测序深度未能很好的反映出该样本的完整菌群构成。如果测序数据量更大的的话会检测到更多物种。
Q3
如何了解分组内部的多个样本的重复性以及多样性情况?
观察分组内部多个样本的重复性如何可以从以下几个方面考虑。
首先在各分类水平的柱状图的菌属构成来看
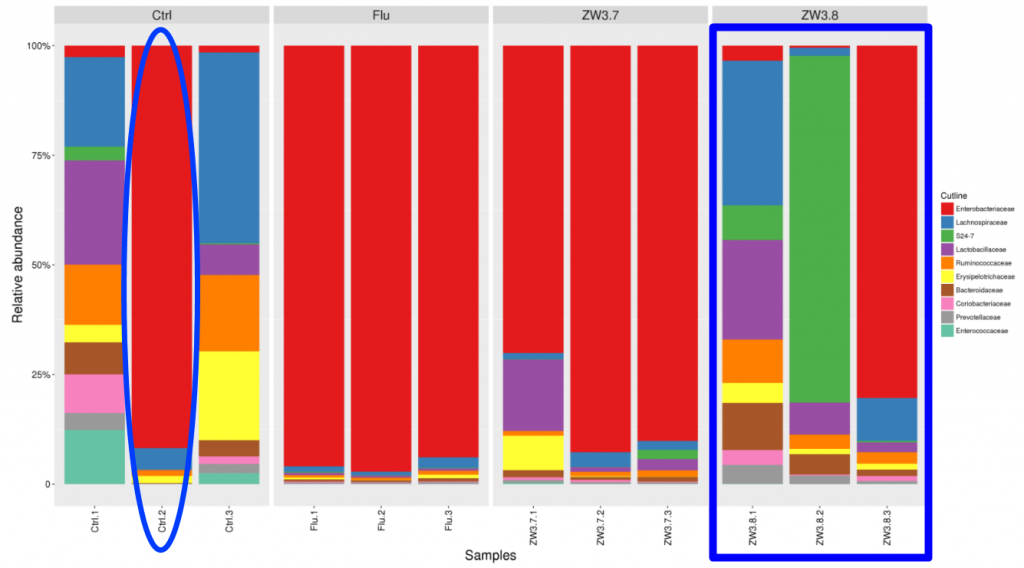

从构成图来看,Flu组和ZW3.7组,组内样本重复性较好。Ctrl组中Ctrl.2明显区别于组内另外两个样本,可以去掉该样本。而ZW3.8组内样本间差异性较大。
比如人体肠道或小鼠肠道样本本身个体差异性较大,菌群结构组成复杂,即便通过不同疾病的分类的样本,但营养饮食、代谢以及环境的影响都会改变肠道菌群的构成,所以有可能组内样本间差异性会比较大。而经过单因素处理的样本组内差异会比较小。
所以在前期实验设计时,尽量选择同一批次相同处理的小鼠或其他样本,避免组内差异的影响。并且要预留好多余的样本,比如组内只有3个样本,如果去掉一个差异性较大的样本,一个分组内只有2个样本,会影响后续组间差异比较,组间差异性比较分析每组要至少要3个样本。

通过beta多样性分析PCA,PCoA,MNDS 也可以大致观察组内样本重复性情况,左图组内样本重复性较好,右图组内样本间差异性较大,两组间的区割不是很明显。
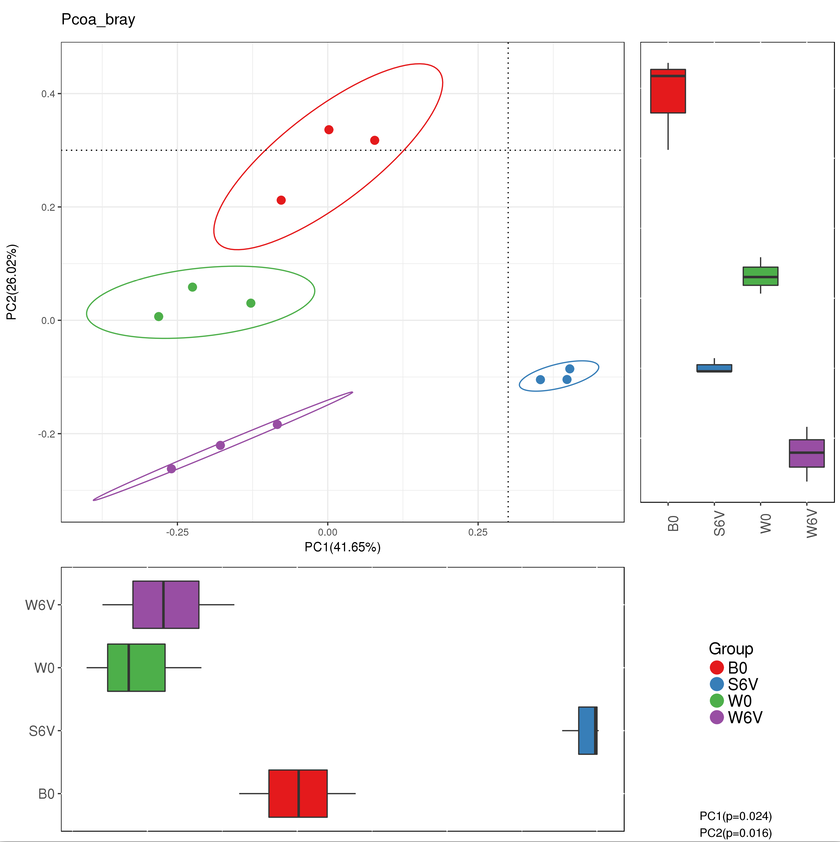


在加圈图的beta多样性分析中,右下角有给出PC1和PC2的P值,小于0.05则差异显著。
Alpha多样性是针对单个样品中物种多样性的分析,包括chao1指数、ace指数,shannon指数以及simpson指数等。前面4个指数越大,最后一个指数越小,说明样品中的物种越丰富。
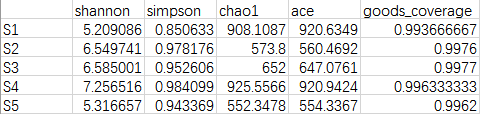

其中chao指数和ACE指数反映样品中群落的丰富度(species richness),即简单指群落中物种的数量,而不考虑群落中每个物种的丰度情况。指数对应的稀释曲线还可以反映样品测序量是否足够。如果曲线趋于平缓或者达到平台期时也就可以认为测序深度已经基本覆盖到样品中所有的物种;反之,则表示样品中物种多样性较高,还存在较多未被测序检测到的物种。
而shannon指数以及simpson指数反映群落的多样性(species diversity),受样品群落中物种丰富度(species richness)和物种均匀度(species evenness)的影响。相同物种丰富度的情况下,群落中各物种具有越大的均匀度,则认为群落具有越大的多样性。
稀释曲线是利用已测得序列中已知的各种OTU的相对比例,来计算抽取n个(n小于测得Reads序列总数)Tags时各Alpha指数的期望值,然后根据一组n值(一般为一组小于总序列数的等差数列,本项目公差为500 )与其相对应的Alpha指数的期望值绘制曲线。
Q4
不同的样本之间差异大吗?不同分组之间能否用菌群差异来区分?
观察不同分组间差异的大小可以观察随机森林分类效果图。
路径在07_diff_analysis/RF
图中以该分类水平下选取用于区分不同分组间的差异性起到关键性影响因素的物种作为标志物作图。标志物按重要性从大到小排列,图中随机森林值error rate 表示用随机森林方法预测分组之间的错误率,分值越高代表所选取的标志物准确度不高,并不能很好的用于区分各分组,分组差异不显著。分值越低证明分组效果比较好。

上图中的随机森林按照门和属以及代谢途径分别进行分析作图,各自都有单独文件,报告中仅给出了一个图,其他文件需要到目录中查看。可能存在门或属区分效果不佳,但是代谢途径区分效果较好。
随机森林筛选出来的物种是用于区分所有分组的重要标志。分值越高代表该物种用于区分所有组之间的重要性越大。
Q5
二代测序16s 能用普通酶扩增吗?
16s测序主要为了鉴定菌种,通常在做鉴定的时候区分标准是97%,区分亚种和菌株的时候相似度更高。
普通TAQ酶的复制错误率较高,可能在扩增过程中引入错误,这些错配可能导致相似度下降从而分类错误。
一般我们不建议使用普通TAQ酶进行扩增,都选择高保真酶。
Q6
利用16s rRNA鉴定细菌能确定到种上吗?
16s rRNA长度为1.5k多,作为菌种鉴定一般选择相似度97%的标准,相似度超过97%一般定义为同一种菌。
如果是sanger测序获得16s全长的都可以鉴定到种,甚至能区分亚种。有些细菌并不只有1个16s序列,会包含有1-15拷贝的16s序列,所以单一的16s序列鉴定可能会出现偏差。
利用高通量如454或miseq测序一般由于读长的缘故,通常只有300-500多个碱基被测序,所以在物种鉴定上一般比较可靠的是能分类到属,部分能分类到种。
根据我们的经验,不同的样品会有大约10-50的菌能分类到种。利用新的分析方法,我们现在也可以利用16s rRNA的群落多样性高通测序数据进行亚种级别的分析。主要是利用16s中共同变化的SNP位点进行分型。这样可以大大提高菌种的分类精度,尤其是在有些菌株之间表型差异巨大的时候。
Q7
听说光测16s就可能预测基因和功能,是真的吗?
16s序列能够区分菌的种属,但是并不包含这些菌的基因和代谢功能的信息。不过由于我们已经对大量的细菌基因组进行了测序,所以可以根据16s的菌种信息,利用这个菌属已经测序的细菌基因组的基因信息和代谢功能信息来估计每类基因的上限和下限。
所以答案是可以利用16s序列测序来预测菌群的功能基因分布和代谢途径分布情况。
目前主要使用的软件是PICRUSt和新发表的Tax4Fun。
从我们实际分析和实验结果来看,预测的准确性还是很高的,不过和样品有很大关系。像肠道菌群和土壤以及一些致病菌的测序较多,所以预测的准确度较高可以到85-90%以上。一些海洋的菌由于测序的菌较少,预测准确性要差一些。目前发表的文献基本都是用PICRUSt,新的软件还有待验证。
Q8
测16s rRNA能分到亚种吗?不同菌株都有致病性差异光到种不解决问题啊!
16s rRNA如果是使用sanger测序可以细分到亚种甚至有些可以精确区分菌株,但是要看菌种。
如果是高通量测序,目前的常见分析一般以97%为标准,大部分情况只能到属,少部分能区分到种。如果要进一步细分到亚种甚至更小的区分目前是有可能的,我们在使用oligotype一类的方法时可以将相同变化模式的SNP归类,并对原来的OTU进行进一步细分,理论上可以区分到菌株。
不过这种区分不同菌属差异很大,有些可以很理想的区分,主要用来了解在更细分化尺度上菌株构成的地理和时间变化。
仅通过16s高通量测序恐怕不能完全解决菌株致病性差异这种问题,但是通过对常见OTU的进一步深入分析可以提供可能的解释或方向。如果明确了某一特定类型菌株的变化有关,可以采用比如毒力基因或菌株特异性标记等方法详细了解不同菌株的比例和差异。

多项合作成果发表于Nature communications、PNAS、Plant biotechnology journal、DNA Research、Environmental Science & Technology、Plant、cell & environment、Science of The Total Environment 、Gut Microbes 、Frontiers in microbiologyt、Journal of environmental management 等国际著名学术期刊。
近期发表文章目录