-

 CNAS L23010
CNAS L23010

 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室

谷禾健康
迄今为止,已经有了许多对呼吸道微生物组通过16S rRNA高通量测序的研究。这其中所有基于扩增子的研究的共同之处就是PCR的应用:
一是扩增待测序的目标标记基因,
二是为多样本的混合测序添加必要的索引序列。
这些步骤可以通过一步PCR或两步PCR完成,但没有研究说明两步PCR方案相关的实验室处理步骤是否会使样品比一步PCR方案更容易受到来自实验室的细菌DNA污染的影响。
本文
试图确定对16S rRNA V3V4与V4基因区域的一步或两步PCR的建库方案对上呼吸道和下呼吸道微生物组的影响
对收集的样本进行了三个设置下的lllumina MiSeq测序
设置1(两步PCR,V3V4区域)
设置2(两步PCR,V4区域)
设置3(一步PCR,V4区域)
分别对这三个设置产生的测序数据进行分析
结论
PCR步骤数量的差异会影响对呼吸道微生物群落的物种组成分析,且对上呼吸道(高细菌载量)的影响小于下呼吸道(低细菌载量),这表明PCR设置的偏差与样本生物量有关。
通过三个实验,即对模拟群落样品HM-783D、NCS样品、呼吸道样品采用三种PCR方案进行建库后的测序结果分析,研究这三种建库方案对其菌群描述的影响。
模拟群落样品HM-783D,来自20种不同细菌物种(17个属)的基因组DNA。
阴性对照样本NCS
呼吸道样品,从Bergen COPD微生物组研究中选择了23名研究对象,其中9名健康,4名患哮喘,10名患COPD(慢阻肺)。上呼吸道样本以漱口水(OW)为代表,下呼吸道样本以标本刷(PSB)和支气管肺泡灌洗液(PBAL)为代表。
三种PCR方案:
细菌DNA提取后通过三种不同的建库设置进行MiSeq测序,分别为
Setup1(两步PCR,V3V4区域);
Setup2(两步PCR,V4区域);
Setup3(一步PCR,V4区域)
下图完整的展示了三个PCR设置下的使用呼吸道样本的生物信息学过滤步骤:
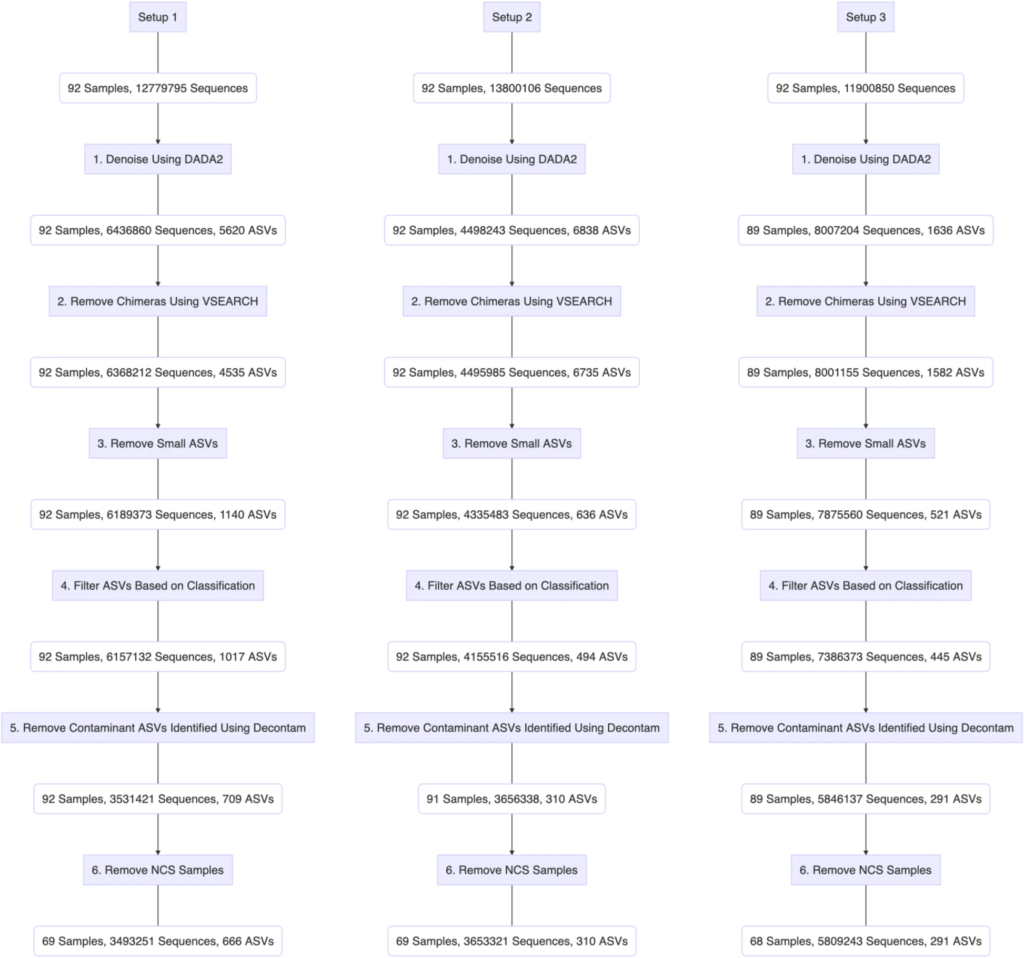
最终:
设置1:得到了666个ASVs
设置2:得到了310个ASVs
设置3:得到了291个ASVs
1. 对模拟群落样品HM-783D的分析
在设置1中进行了四次测序,设置2和设置3分别进行了一次。
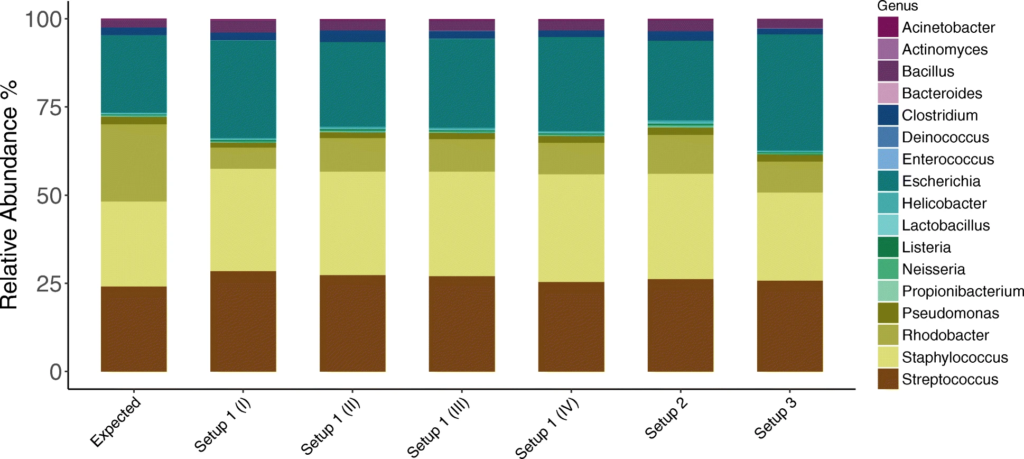
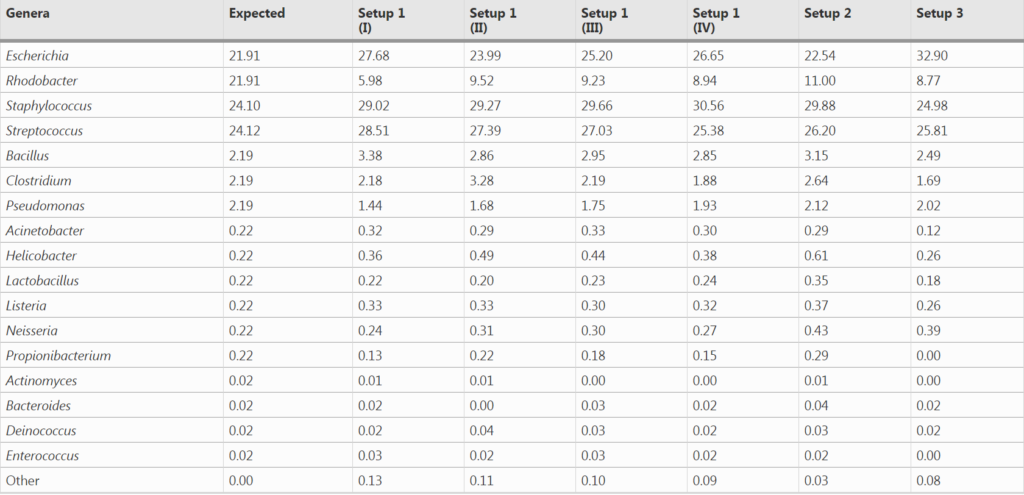
与预期丰度(Expected)相比,柱状图中观察到三种PCR设置下的各菌属的相对丰度与预期丰度相差不大,表中数据显示,三种PCR设置都在恢复高丰度物种方面具有最高的效率,但设置3在回收低丰度物种时的效率最低。
2. 对阴性对照样品的分析
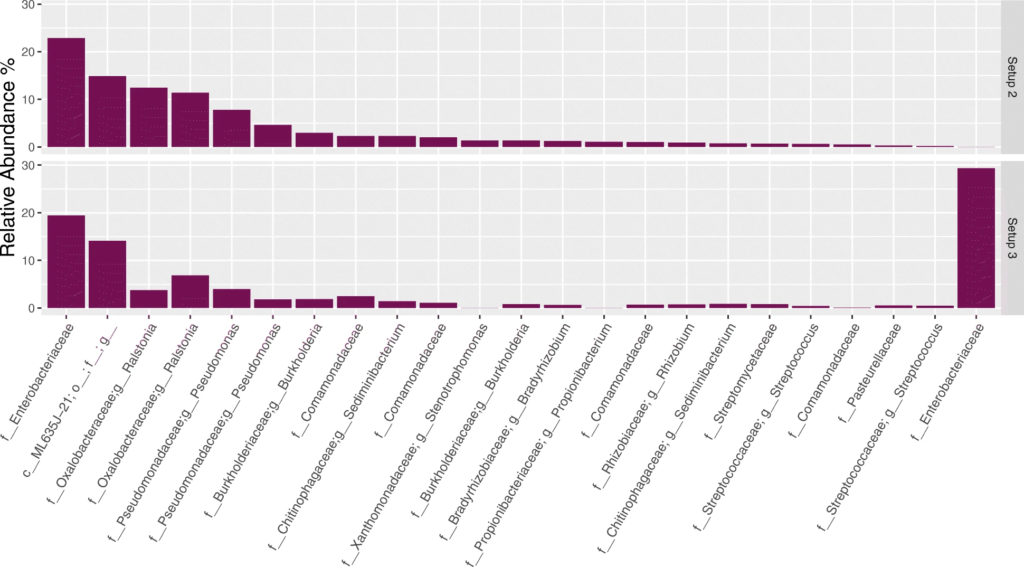
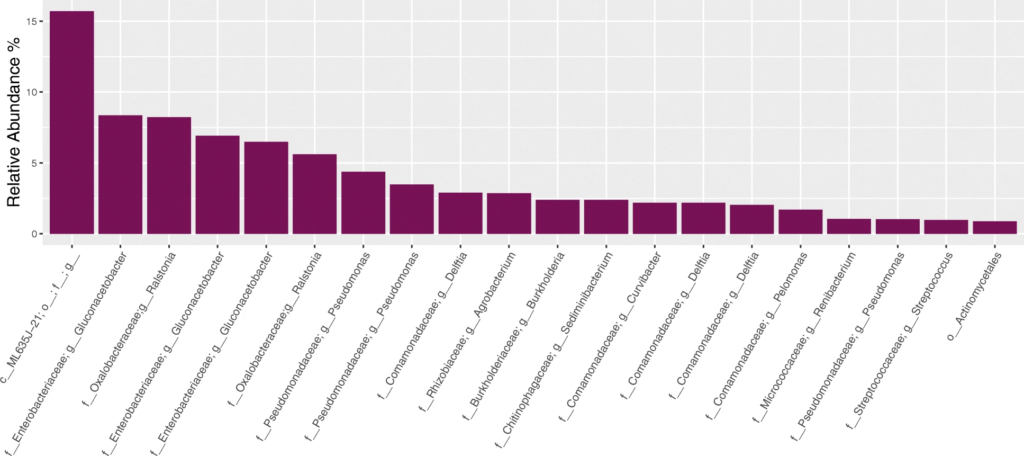
从上至下分别为设置123测序后,在NCS样品中观察到的20种最丰富的ASV。通过R包Decontam去除污染物,在设置23之间差异最大的是属于肠杆菌科的ASV,与后续的对水样品进行设置23下的测序分析结果相比较,发现大肠杆菌ASV就是在建库步骤中使用设置3的试剂时引入的污染物。
3. 对采集的呼吸道样品的分析
在去除污染物前后,代表为呼吸道菌群的链球菌,普雷伏氏菌,Veillonella和Rothia属的相对丰度变化不大,而去除污染物后,预测作为污染物代表的数量较少的物种被滤出。基于主坐标分析,发现高细菌载量的OW样品聚集在一起,低细菌载量PBAL,PSB一句设置23分离开。
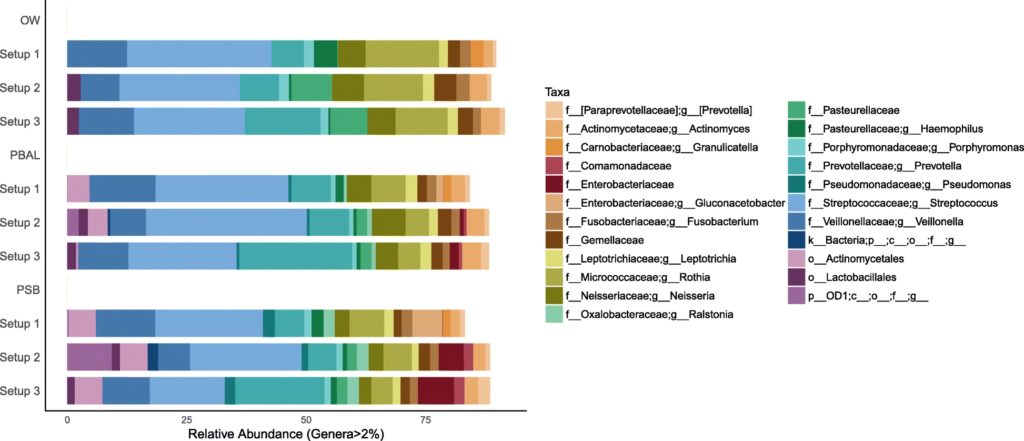
去除污染物之前的三种类型样品的三种PCR设置下的物种分类
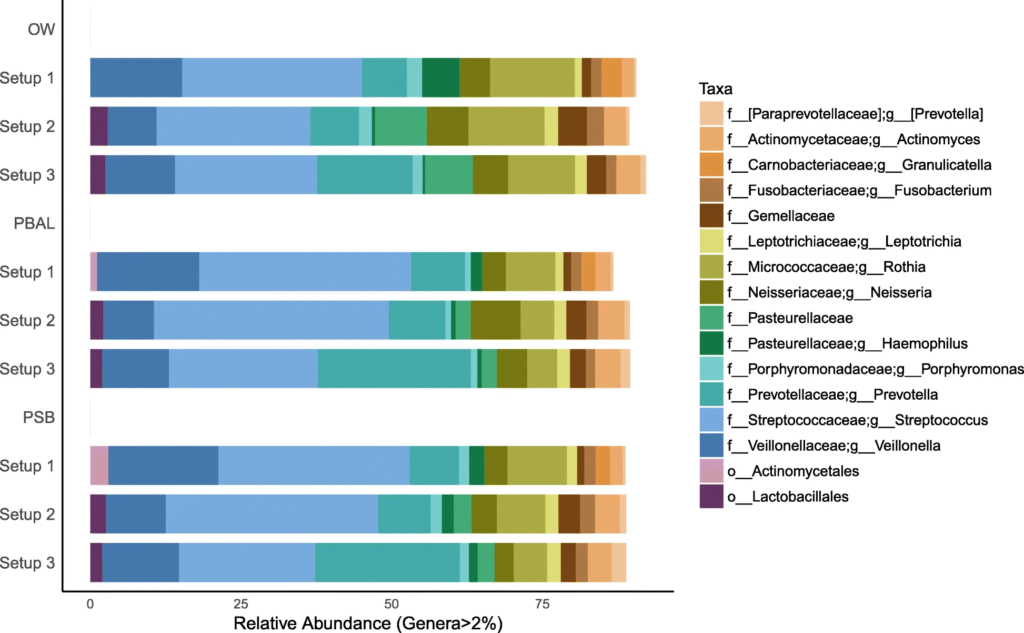
去除污染物后的
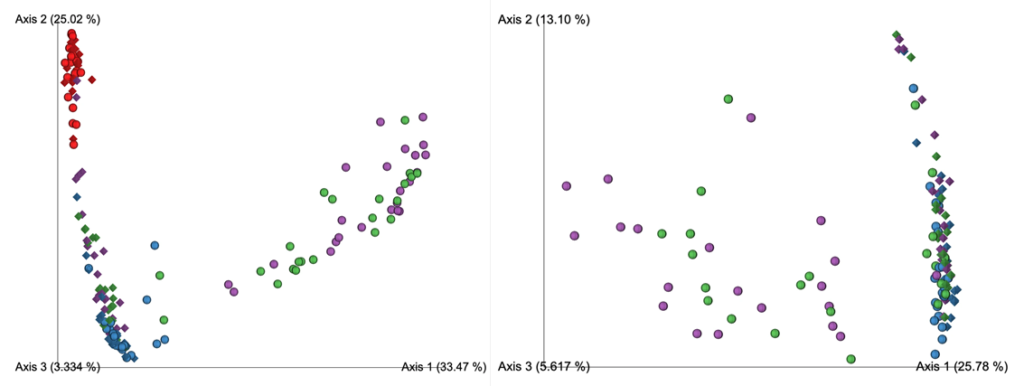
从左至右分别为去除污染物前后的未加权UniFrac距离的主坐标分析
OW:蓝色; PBAL:绿色;PSB:紫色;NCS:红色。
设置2(球形),3(菱形)
文章作者给出的结论是文库制备和测序方法的选择会对呼吸道微生物组的分析产生影响,且对上呼吸道的影响小于下呼吸道。靶向扩增子区域的差异(16S rRNA基因V3 V4与V4)并未表现出对细菌群落描述的重大影响。对于整篇研究存在的主要的局限性在于仅研究了DNA提取后的PCR步骤,污染或影响也可能来自于更前期的处理。
编者按
在使用测序技术进行的微生物研究中,测序偏差和污染物是一直存在的问题,也因此诞生了许多工具和计算方法用于尽可能的消除或降低这方面的影响。这篇研究也提醒了我们,在呼吸道微生物组的研究中,要注意上呼吸道与下呼吸道的菌群差异或相似可能不仅仅来源于样本自身,还可能掺杂着PCR方法选择上的影响。
参考文献:
Drengenes C, Eagan TML, Haaland I, Wiker HG, Nielsen R. Exploring protocol bias in airway microbiome studies: one versus two PCR steps and 16S rRNA gene region V3 V4 versus V4. BMC Genomics. 2021 Jan 4;22(1):3.
相关阅读:
谷禾健康
宏基因组中短序列的注释是理解测序微生物群落潜在功能的重要步骤之一。单纯利用局部匹配的注释容易混淆那些蛋白同源性且局部序列非常相似的序列,进而不能真实准确反映复杂蛋白质家族中多变的结构和功能域。

今天我们介绍一种新方法MetaGeneHunt,该方法可以识别特定的蛋白质结构域,并根据结构域的长度对hit-counts进行标准化。使用MetaGeneHunt对MG-RAST对公开获取的宏基因组进行分析,包括哺乳动物微生物群和Twin Gut肠道菌群研究,以评估短序列中含GH蛋白的频率和位于GH区域的匹配频率。
在对糖苷水解酶(GHs)的研究,发现在所有样本中4726,023条含有GH区域蛋白匹配的短读序列中,有58.3%的序列位于目标区域之外。接下来,在比较样本之前,将匹配到目标区域的hit-counts标准化,以说明对应的域长度。肠道和盲肠中的菌群显示出与不同微生物组合相匹配的GH谱特征。
相反,胃和结肠的菌群在结构和功能上显示出更多样性和多变性。在样本中,尽管有波动,但碳水化合物处理的潜在功能变化与群落组成的变化相关。这表示,在利用MG-RAST平台处理宏基因组测序序列时,MetaGeneHunt是一种能快速准确地识别短序列宏基因组中离散蛋白结构域的新方法。
在过去的几十年里,宏基因组DNA的高通量测序已经产生了大量的序列,这些序列的特征为我们了解微生物群落的结构和功能提供了许多认知。例如,截至2019年12月,MG-RAST托管了约40万个可公开访问的带注释的数据集。在数据处理过程中,不考虑目标区域(或蛋白质)的长度会导致两个主要的系统偏差。
首先,目标区域越长,他们的频率就越容易被高估。其次,如果数据处理涉及稀疏性,较短的、不太丰富的域,尽管重要,也可能被丢弃。为了解决这些问题,研究人员设计了MetaGeneHunt来精确注释从MG-RAST检索到的短序列宏基因组中的蛋白质结构域。MetaGeneHunt将MG-RAST提供的短序列局部比对与M5nr数据库中精确的基于PFam的蛋白质结构域识别相结合,以在公共可访问数据集中识别蛋白质结构域。
MetaGeneHunt简要说明:
MetaGeneHunt的设计基于MG-RAST平台注释的数据集的。在使用GeneHunt创建的M5nr数据库中,MetaGeneHunt使用了糖苷水解酶和辅助结构域(如CBMs)的精确的特定结构域注释(PFam)作为参考注释表(RAT)。
首先,MetaGeneHunt使用MG-RAST应用程序接口从MG-RAST(“330”和“650”文件)检索M5nr注释的宏基因组。接下来,使用来自RAT的注释命中的MD5id,在文件“650”中识别与潜在的GHs匹配的序列。
接下来,对于这些局部匹配,将精确对齐位置与RAT中特定于域的注释进行比较。如果查询中的>20AAs与特定的蛋白质结构域(考虑到RAT中的HMM-envelope位置)对齐,则该结构域注释被转移到查询中。
相反,如果查询的>20AAs匹配在目标区域之外(例如,在连接域、辅助域、信号肽中),则该注释被认为是否定的。用户可以随意修改重叠(overlapping)的阈值。接下来,从序列聚集文件( “330”文件)中检索每个识别出的命中的实际序列计数。最后,在后续的数据处理和标准化过程中,根据Pfam数据库中蛋白质结构域的大小,对每个蛋白质结构域的命中计数进行标准化。
方法验证:
文中使用的原始数据和预处理数据可在MG-RAST服务器上公开访问。在mgp20861项目中可获得对应于〜555百万个100 bp序列的小鼠微生物组数据。使用MG-RAST API 检索了哺乳动物微生物组数据(mgp116)和双肠肠道菌群研究(mgp10)其他数据集。哺乳动物微生物组研究糖苷水解酶(GHs)和相关酶的附加注释表是从Brian Muegge(直接对应)获得的。使用MG-RAST API检索了预处理的数据,包括从门到属水平的读物分类注释。数据分析和统计使用R统计语言。
1. 糖苷水解酶的识别,识别蛋白质结构域并考虑其长度产生了一个健壮的功能注释系统,对hit-count的标准化反应了目标区域的实际分布。

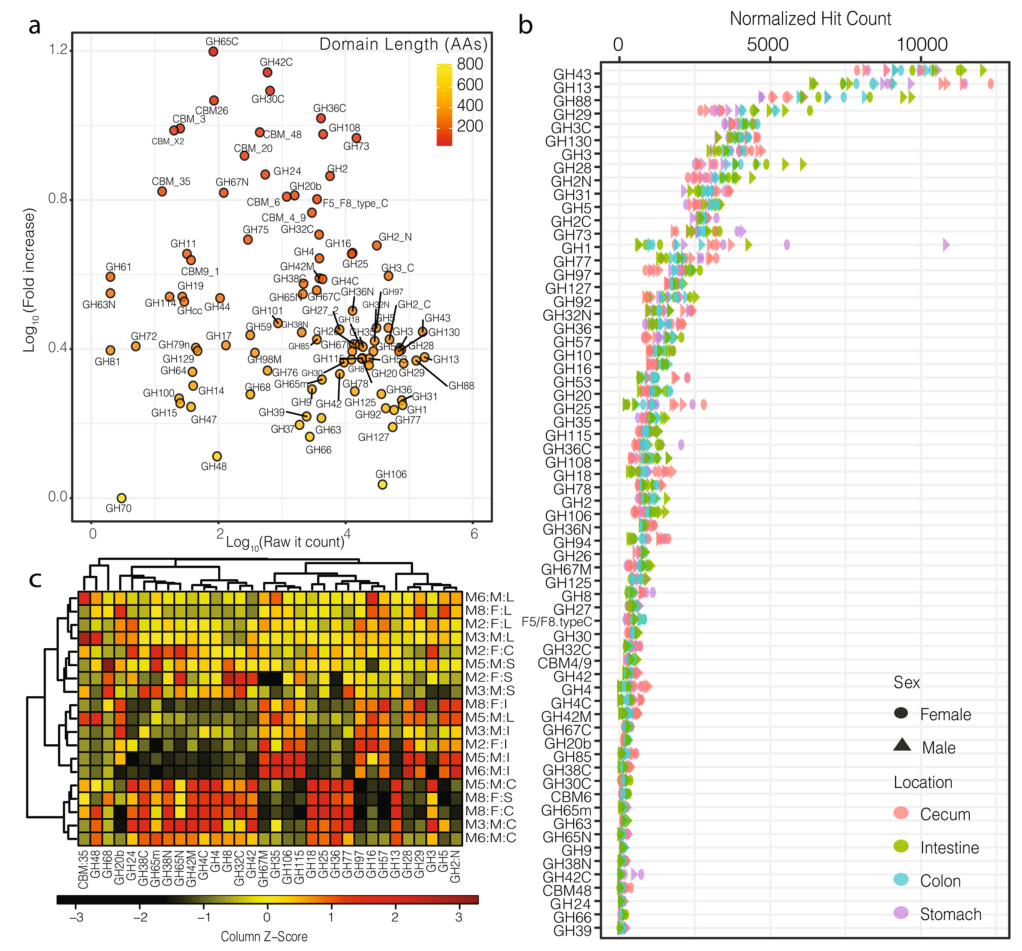
a).横轴为目标区域的原始hit-count,纵轴为标准化后的hit-count,图中的颜色阶梯表示目标区域的长度。这种标准化主要影响长度短的域(例如,GH78、GH25)、小的亚域(例如,GH31N、GH36C)和目标区域的附属域(例如,CMB5_12)。
b).小鼠胃肠道中目标区域的标准化后的hit-count(仅显示大于100的hit-count的区域),可见,标准化后的hit-count与结构域长度无关(附加文件中有对两者做相关分析,结果分别为P.pearson=0.38,P.spearman=0.33)
c).热图显示了小鼠胃肠道中最受样本来源影响的被稀疏标准化的GH区域的分布(two-way方差分析)。纵轴的注释列Mx:F/M:S/I/C/L分别表示小鼠(样本号):雌性/雄性:胃/肠/盲肠/结肠
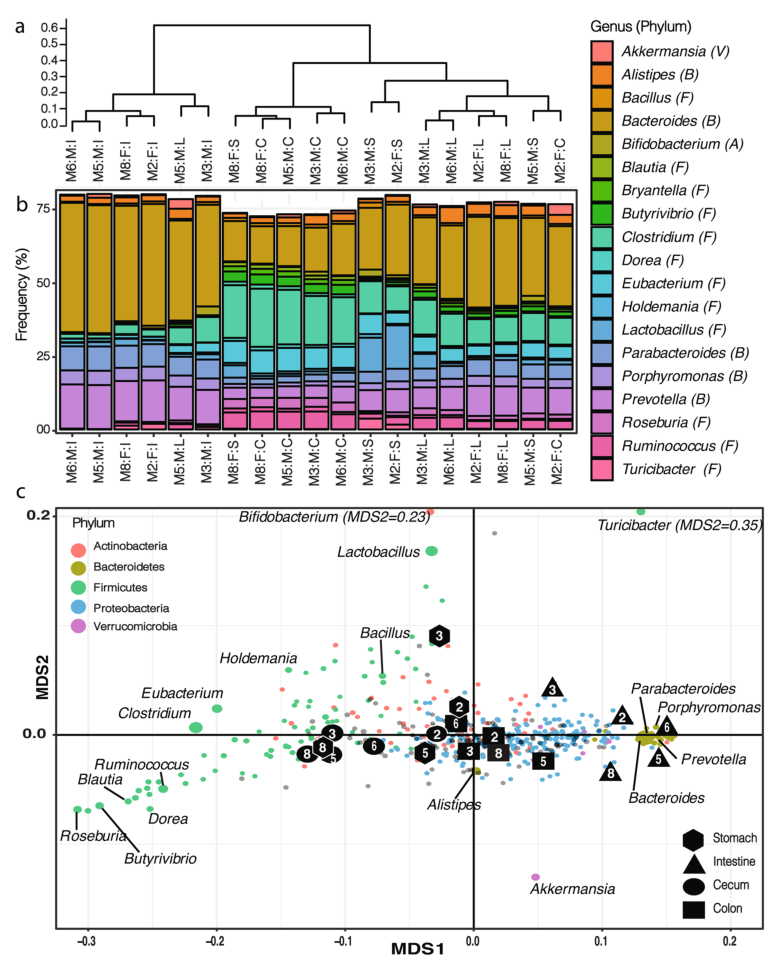
2. 小鼠肠道菌群的结构,与盲肠中的微生物群落相比,结肠与肠道中的微生物群落结构更相似,结肠和胃中的微生物群落有较高的相似性。

a).对受样本来源影响较大的样本根据属水平进行样本聚类(Bray-Curtis距离指数,complete linkage)。
b).样本间的微生物群落组成,只展示了相对丰度至少占群落中1%的属水平物种(V:疣微菌门,B:拟杆菌门,A:放线菌门,F:厚壁菌门)。
c).NMDS分析(2D stress=0.020),展示了在样本聚类中都存在的这些菌属,在b)中的主要类群用标签指示,不同门水平按颜色区分,点的大小反映该属在样本中的最大频率。
微生物组中的结构-功能关系,多样性仍然与潜在功能高度相关。胃和盲肠的群落在结构和功能上是最多样化的。其次,肠道中的群落组成和功能大多是保守的,而与保守的微生物群落相关的大肠则显示出可变功能潜力。
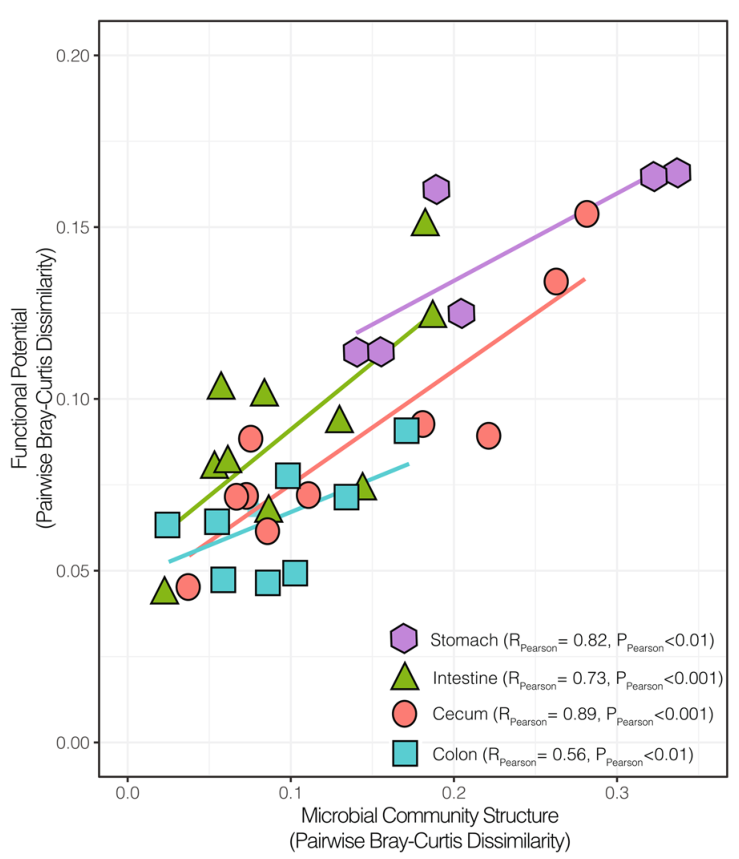

对同一位置的样本的微生物群落结构和功能差异进行成对比较(Bray-Curtis),线条为线性回归的结果。在胃,肠,盲肠和结肠中,属水平群落结构的变化与多糖解构功能的相关性分析结果表示除大肠外,其余的P.pearson的值都在0.001以下。胃和盲肠的群落在结构和功能上是最多样化的,尽管多样性仍然与功能潜力高度相关。其次,肠道中的群落组成和功能大多是保守的,而与保守的微生物群落相关的大肠则显示出可变的功能潜力。
MetaGeneHune提供了一种新的方法来识别短序列宏基因组中的GHs及其相关结构域。识别结构域而不是蛋白质是至关重要的,因为GH结构域与许多可变结构域相关。这种新方法基于GeneHunt注释方法,并对其进行补充,旨在分析MG-RAST中的短序列宏基因组。因此,它不需要大型计算机基础设施。
通过这种新方法对小鼠胃肠道菌群的GHs研究发现,在胃中,虽然富含碳水化合物处理的酶,但相对于胃肠道的其他部分,胃中没有特定酶可供选择;在肠道中,出现了更保守的菌群,最为富集的是拟杆菌门,它们的潜在功能主要在多糖处理上;来自结肠和胃的菌群虽然是距离最远的,但在结构和功能上却表现出高度的相似性。
在未来,利用GeneHunt和MetaGeneHunt相结合创建新的专用参考注释表将为研究宏基因组的潜在功能提供新的更有效的途径。
MetaGeneHunt和GH的RAT可在GitHub上公开访问。(https://github.com/renober/MetaGeneHunt)
参 考 文 献
Berlemont R, Winans N, Talamantes D, Dang H, Tsai HW.MetaGeneHunt for protein domain annotation in short-read metagenomes. Sci Rep.2020 May 7;10(1):7712. doi: 10.1038/s41598-020-63775-1. PMID: 32382098; PMCID:PMC7205989.
Muegge BD, et al. Diet drives convergence in gut microbiomefunctions across mammalian phylogeny and within humans. Science.2011;332:970–4. doi: 10.1126/science.1198719
Turnbaugh PJ, et al. A core gut microbiome in obese and leantwins. Nature. 2009;457:480–484. doi: 10.1038/nature07540.
Berlemont R, Martiny AC. Glycoside Hydrolases acrossEnvironmental Microbial Communities. PLOS Comput. Biol. 2016;12:e1005300. doi:10.1371/journal.pcbi.1005300.
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity,stability and resilience of the human gut microbiota. Nature. 2012;489:220–30.doi: 10.1038/nature11550.
Sharpton TJ. An introduction to the analysis of shotgunmetagenomic data. Front. Plant Sci. 2014;5:209. doi: 10.3389/fpls.2014.00209.