-

 CNAS L23010
CNAS L23010

 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室

谷禾健康

最近有客户在拿到科研报告分析结果的时候问:
“在同样的数据分析流程情况下,为什么我拿到的分析结果提取不出什么有价值的结果,而别人有着类似的项目课题,样本类型也相似,却可以拿到全面的差异分析结果,想要提取的信息一目了然?”
★项目设计和入组标准以及采样至关重要
微生物组研究涉及到生活的方方面面,从环境微生物检测,到人体疾病与微生物群的关系,再到药物与食品营养干预等,以及菌群的发酵产物等。一项好的研究一定离不开前期的项目主题优化以及合理的采样方案。
项目分析主要是依托高通量测序获取的数据和特定数据库对样本数据进行归类,识别,统计检验分析,以及差异化寻找和可视化展示,来获取全面的数据信息。
最终拿到科研分析报告结果,PDF结果报告也好,在线或平台呈现展示也好,是对结果的排列,展示和解读说明等。
个别研究者拿到结果报告后,发现结果不理想,分组没有代表性,组内差异较大,当这个时候发现前期研究不明确,或者取样不具有代表性等问题,想通过后期科研分析的手段进行补救,根据我们多年的分析经验来看,在这种情况下只能进行一定范围内的微调,差异结果大局已定。
如果想得到优质的差异分析结果靠后期数据分析筛选修改很难达到要求。这个时候如果想再次优化课题,重新取样,项目周期会延长,因此,本文就前期实验方案设计和取样环节进行详细描述和讨论,便于科研工作者获得更优质的实验结果。
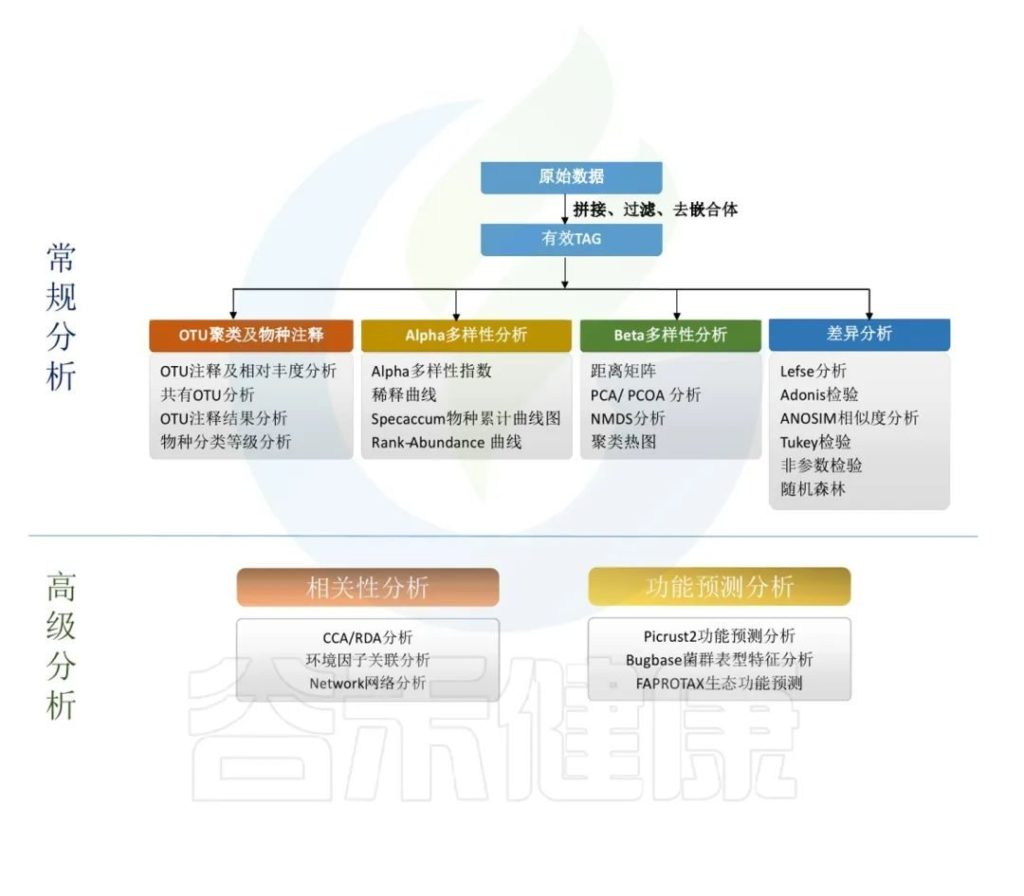
一套完整的扩增子和宏基因组测序流程包括:
1. 确定实验设计与研究目的、样本规模和时间
2. 样本的采集,归纳,保存和运输
3. 预期实验结果计划,分析统计方法选择
4. 样本提取,实验,上机测序的选择
5. 数据下机,分析,与结果的呈现
一般测序公司拿到样本以后先进行样本状态查验以及样本信息核对,核对没问题后会尽快开展实验环节,包括提取,建库,上机,可以通过标准化流程减少实验误差,统一规范操作步骤;而数据分析也是通过标准的SOP分析流程拿到完整的数据分析结果,分析中分析人员可能会根据数据尝试寻找最适配的分析方法,或者增加图表展示的内容和呈现形式,以辅助客户拿到最能反映研究结果的图表。
唯有前两项主题设计与采样环节的不确定性因素较大,需要客户提前设计准备充分,同时也决定了未来数据分析结果的好坏。
实验设计就是为整个课题设定一个总的大纲,确立总的研究方向和目的;取样环节是未来能否将实验方案具体实现的关键。客户提前做好科研课题开题前这两项工作,更有利于后续测序分析的开展。
微生物组最常见的研究方法之一是高通量测序,其原理是基于微生物基因组的特异性序列(细菌16S rDNA,真菌的ITS或宏基因组DNA),进行测序和分类,分析在特定的环境下微生物群落的物种组成和功能,微生物的多样性与丰度。
根据主题的不同可以将微生物组研究主要应用到以下几个方面:
1. 将肠道微生物与疾病进行关联以探讨疾病与健康的个体之间微生物的差异性;
2. 在特定环境下找出特异性微生物,发现耐受菌群及与特定环境具有关联性的代谢通路,通过不同样本之间的比较,研究微生物群内部、微生物与环境、微生物与宿主之间的关系;
3. 通过单一因素直接作用于特定环境或宿主(例如补充维生素、给土壤施加不同的类型的肥料)研究对应的微生物群在不同时间段的变化规律。
依照不同的课题类型提前规划好实验设计方案。
▼
例如可以研究不同用药组菌群构成差异;在不同的地理环境下土壤菌群构成的变化;还可以研究不同疾病类型的病人与健康对照组的菌群做两两分组之间的差异分析。
•保证组内样本的均匀性和一致性
为了尽可能多的从不同分组当中找出差异结果,需要保证组内样本的均匀性和一致性,排除组内样本差异性过大等干扰因素。
例如研究某一种疾病的肠道菌群构成时,需要排除多种因素对菌群构成的影响:
所以在前期实验设计的时候这些细节都需要考虑到位,避免个别的干扰因素影响整体的实验效果。
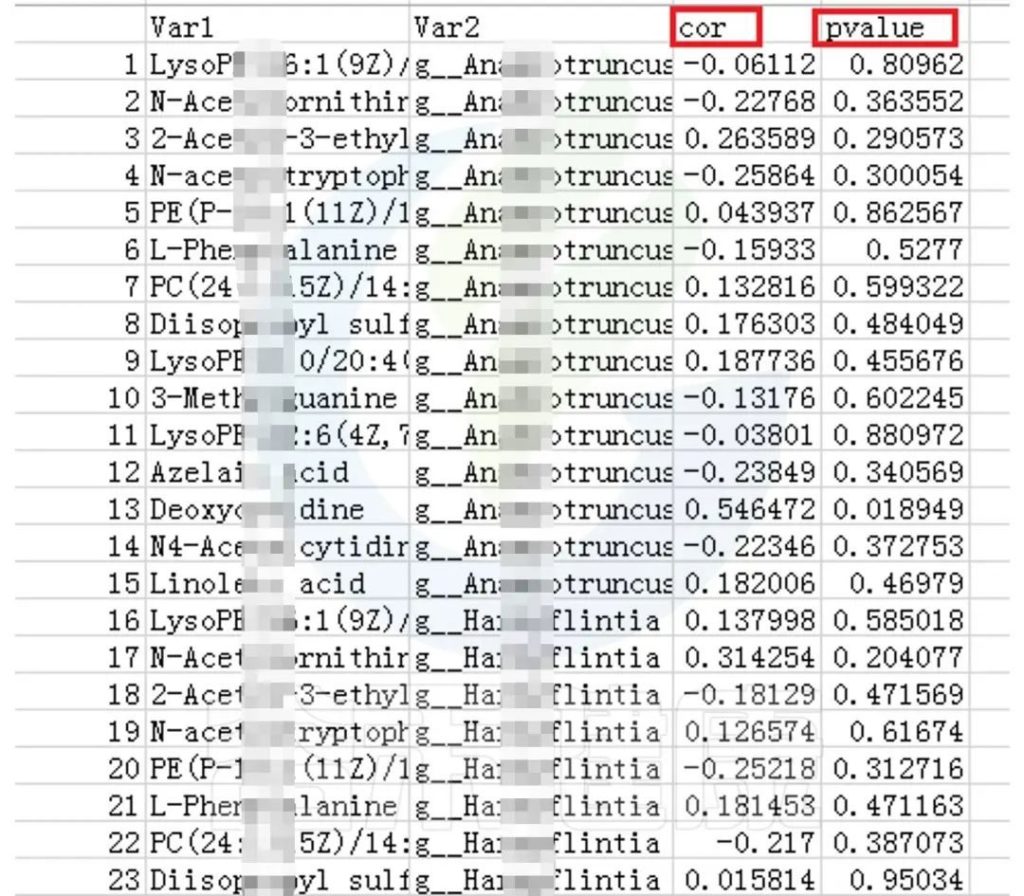
不同分组直接菌属差异比较
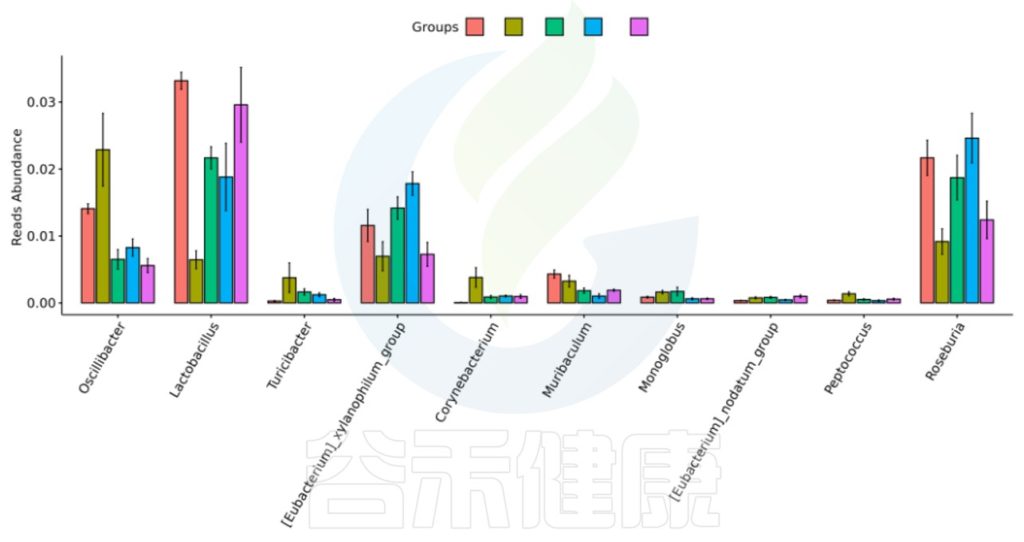
上图表明:尽管是用药组,但是不同的细分用药对菌群的差异影响不同,甚至差异还比较大。
下图可以看下每两组之间是有显著性差异的。
两两分组之间菌属差异比较
这刚好反馈在分组的时候,一定要按照研究方案不同或处理效应尽可能细分分组,或严格入组样本标准,或设置细分的干预浓度等。
合理细分分组,确保实验结果的科学性和有效性,从而推出更可靠的结论。
▼
相关性分析用于研究微生物群落内部,微生物与环境之间,微生物与宿主之间的关系。例如最常见的就是临床病人拿到的各项生理生化指标与微生物群之间做关联分析;或是环境、土壤、发酵样本的各项理化指标与微生物之间做相关性分析等。
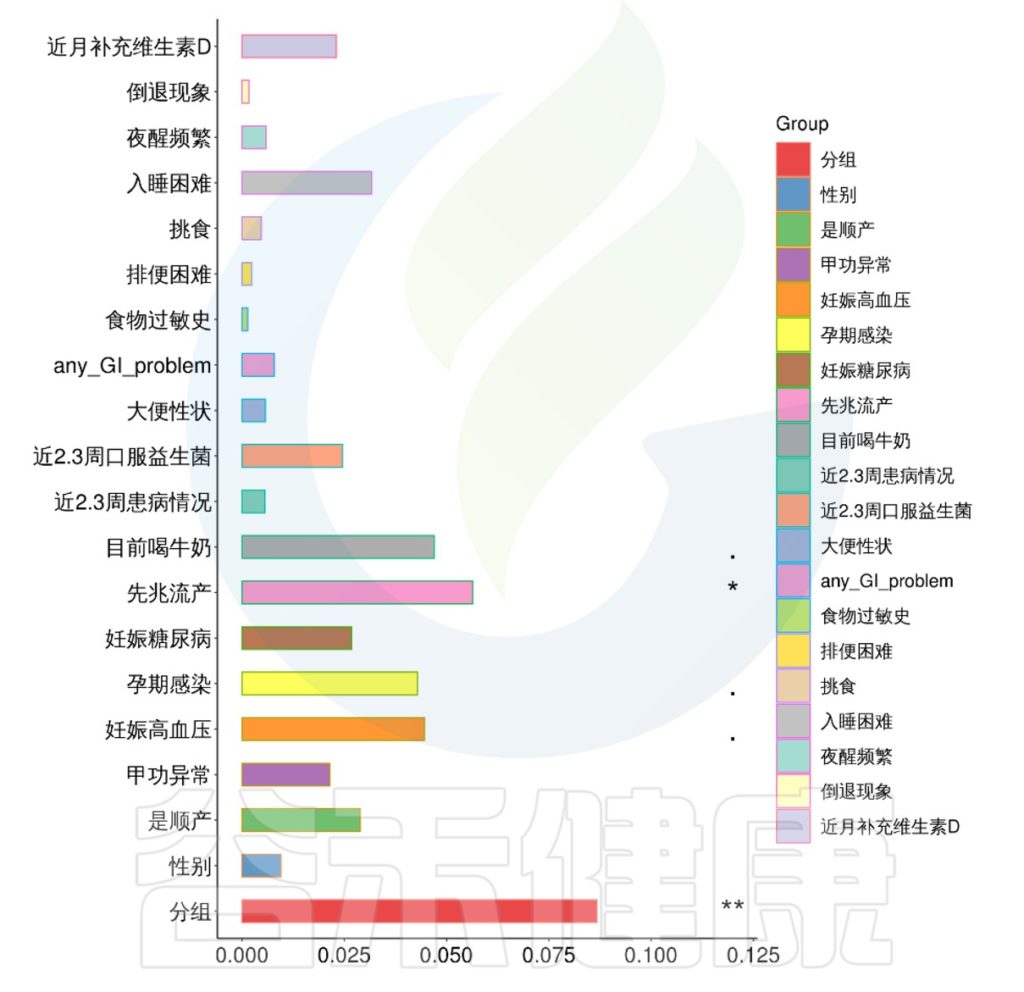
该项目是比较自闭症儿童与正常儿童的菌群差异的项目。客户在样本信息单里详细描述了孕期各种理化指标,根据这些理化指标与菌群数据做envfit相关性分析。
从结果可以看出,自闭症谱系障碍与正常儿童之间的分组与菌群之间相关性极显著**,其次是否有先兆流产的分组与菌群之间有显著相关性*,其他的包括是否喝牛奶、孕期是否感染、妊娠高血压都与菌群有相关性。
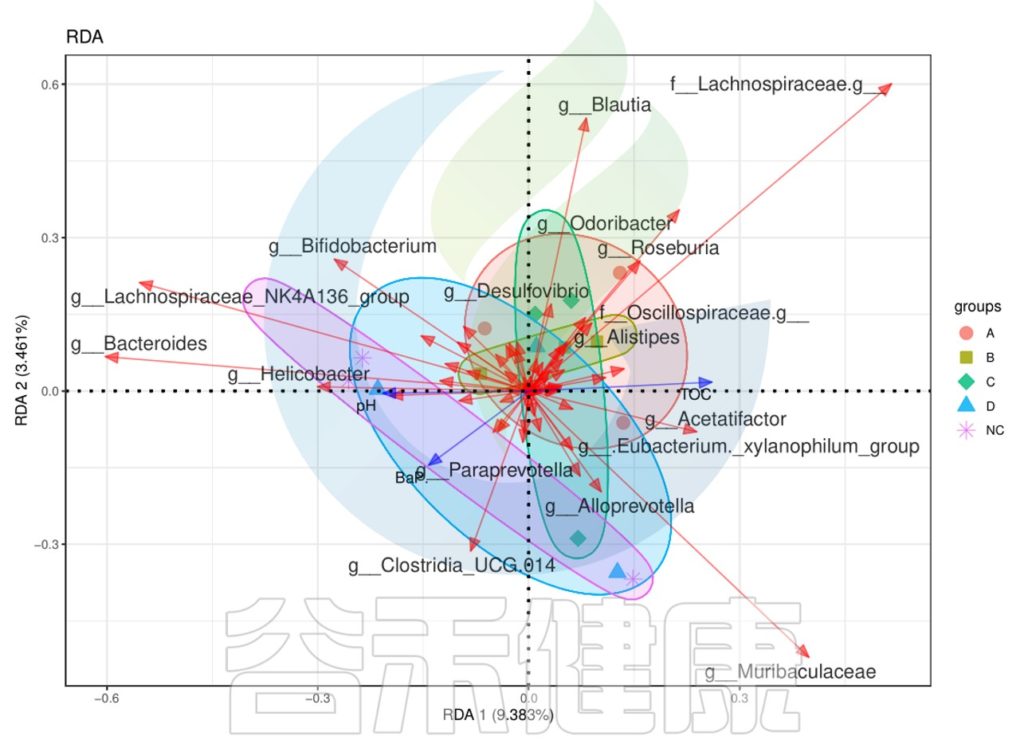
下列CCA图、RDA图可以做环境因子与菌属、样本之间的关联分析。
图中使用点代表不同的样本,从原点发出的箭头代表不同的环境因子。箭头的长度越长,表示环境因子的影响越大。夹角越小,代表相关性越高。样本点与箭头距离越近,该环境因子对样本的作用越强。
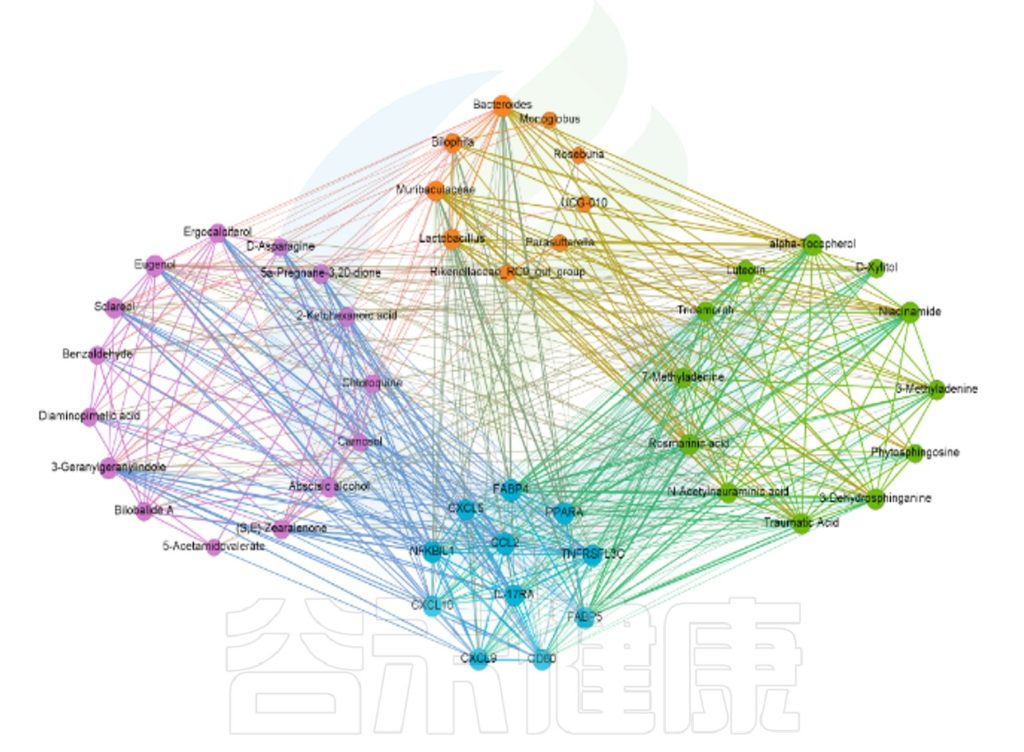
微生物相关性网络分析,可发现物种间的相互关系。图中每个节点代表属水平的物种,节点的颜色代表对应的门水平,节点越大,与之相关的属就越多,物种间的相关性用线条连接,相关性越强,线条越粗。线条红色正相关,绿色负相关。
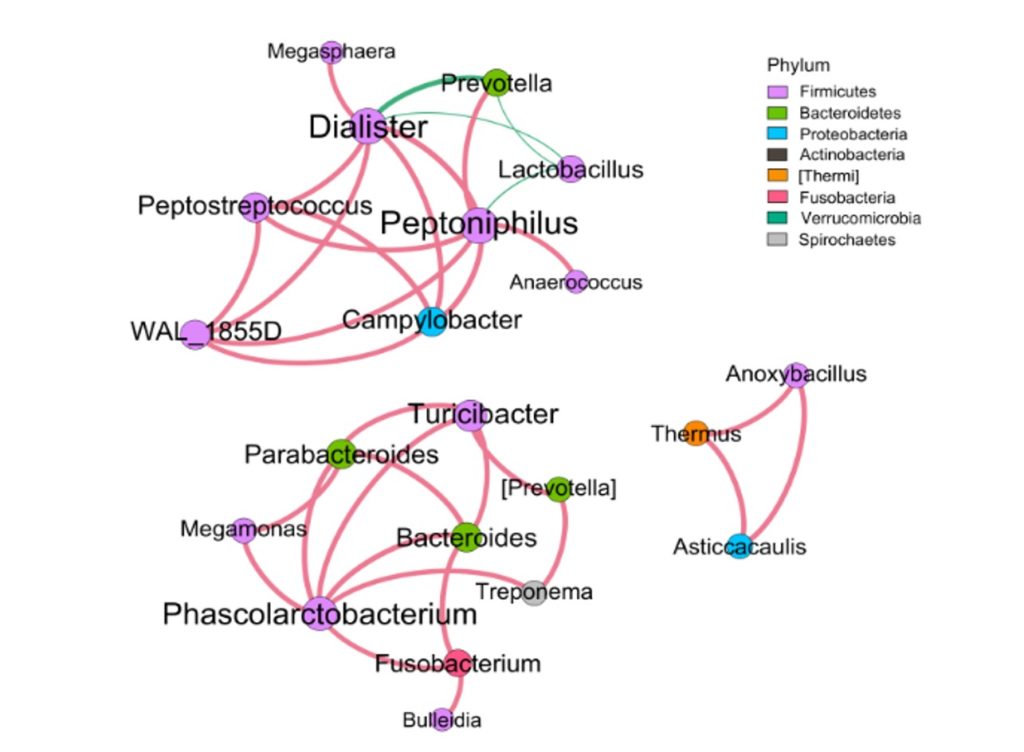
微生物与理化指标的相关性网络图
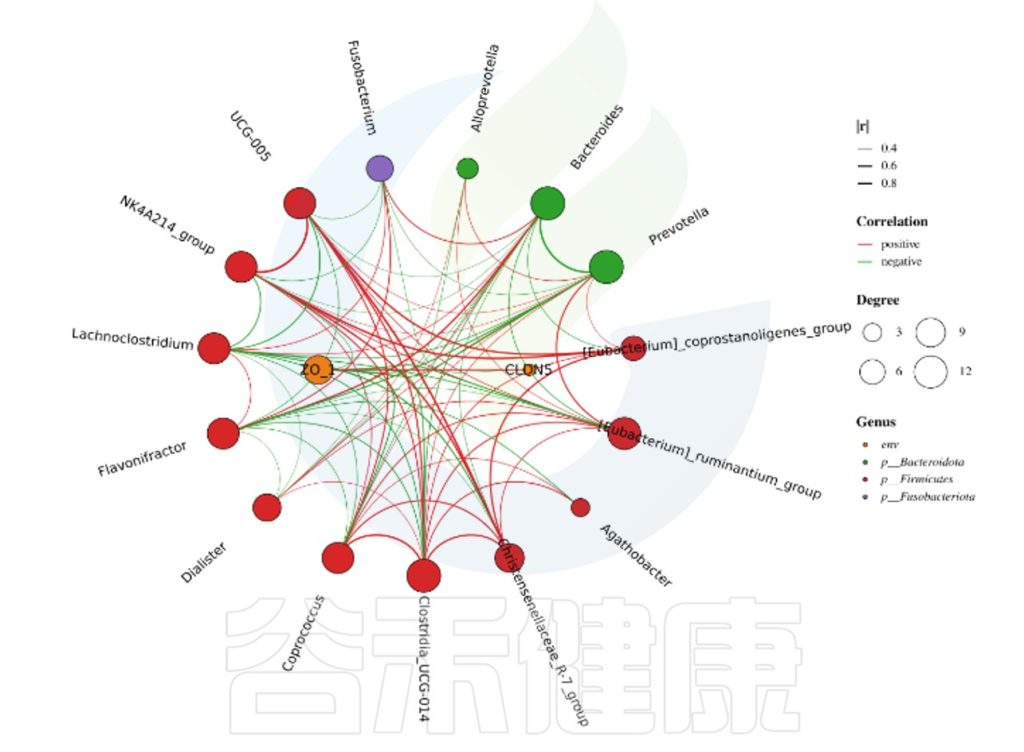
上调代谢物、下调代谢物与菌群和基因的互作关系
理化指标与菌群的相关性热图分析
理化指标与物种的线性回归分析
•实验设计时规划好哪些指标与菌群数据之间做关联分析
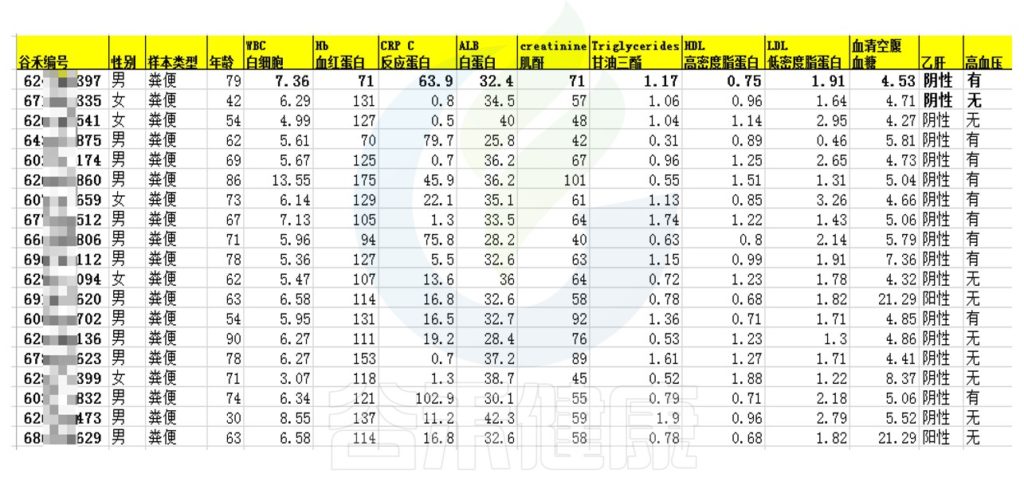
在实验方案设计阶段先规划好哪些生理生化、临床指标可以跟菌群数据之间做关联分析,在取样的同时搜集好样本相关的理化指标质量和范围,有利于后期相关性分析,增强分析结果的说服力。
将详细数据填写在样本信息单里,例如:
▼
微生物群是一个相对较为稳定的组合状态,在受到外部环境变化的影响下,菌群构成也会随之发生改变。在一定的时间范围内,菌群构成呈现出一定的变化规律。
•外界因素影响下肠道菌群在一定时间内变化
例如研究在抗生素药物治疗的情况下,一定时间内病人的肠道菌群变化情况。抗生素会直接影响肠道菌群的丰富度和多样性。
营养饮食的影响相对缓慢
对于益生菌,或者营养饮食等方面的摄入对肠道菌群的干预是缓慢的,被动的,需要长时间的补充摄入才能对整体的菌落构成起到影响作用,直到菌群构成趋于稳定。所以这里时间上要从几周到几个月的时间段内进行选取和采样,形成一个完整的采样周期。
婴儿的肠道菌群变化较为显著,取样间隔更短
又比如婴儿的肠道菌群在头几年变化较为显著,前期取样的时候就需要间隔更短一点,例如几天到几周采一次样,才能更细致的捕捉到婴儿肠道菌群的变化规律。
时间间隔的长短会影响微生物的时间序列分析,依据不同的样本类型,选取恰当的时间间隔周期,能观察短期或长期的菌群变化趋势,可以做个体之间的线性分析,配对T检验。
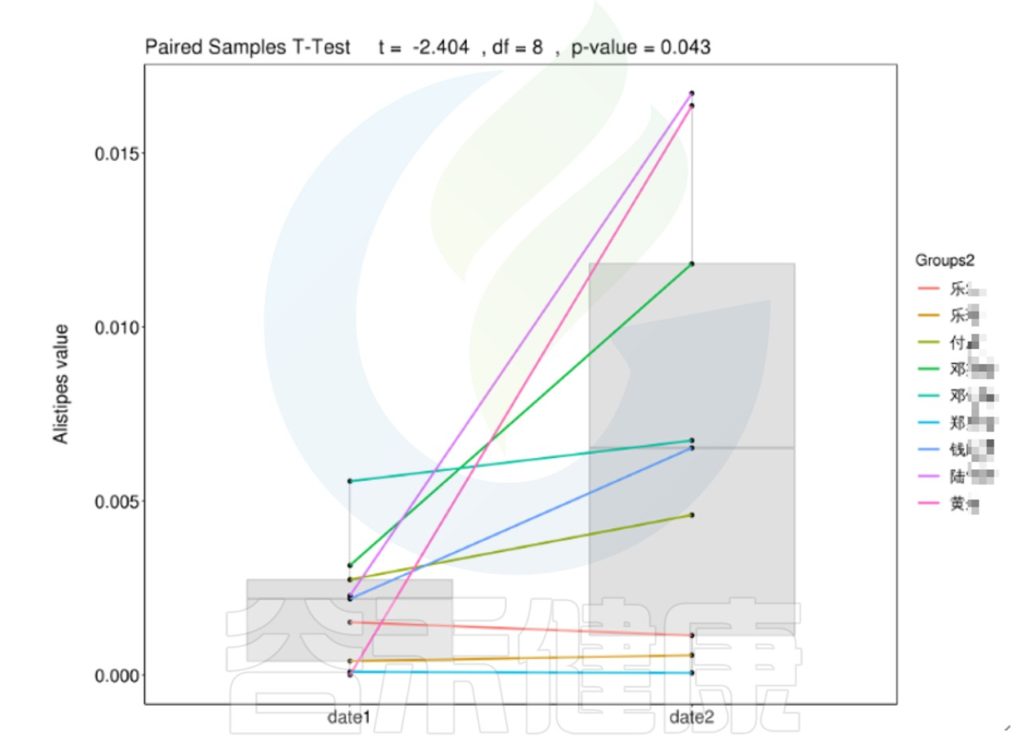
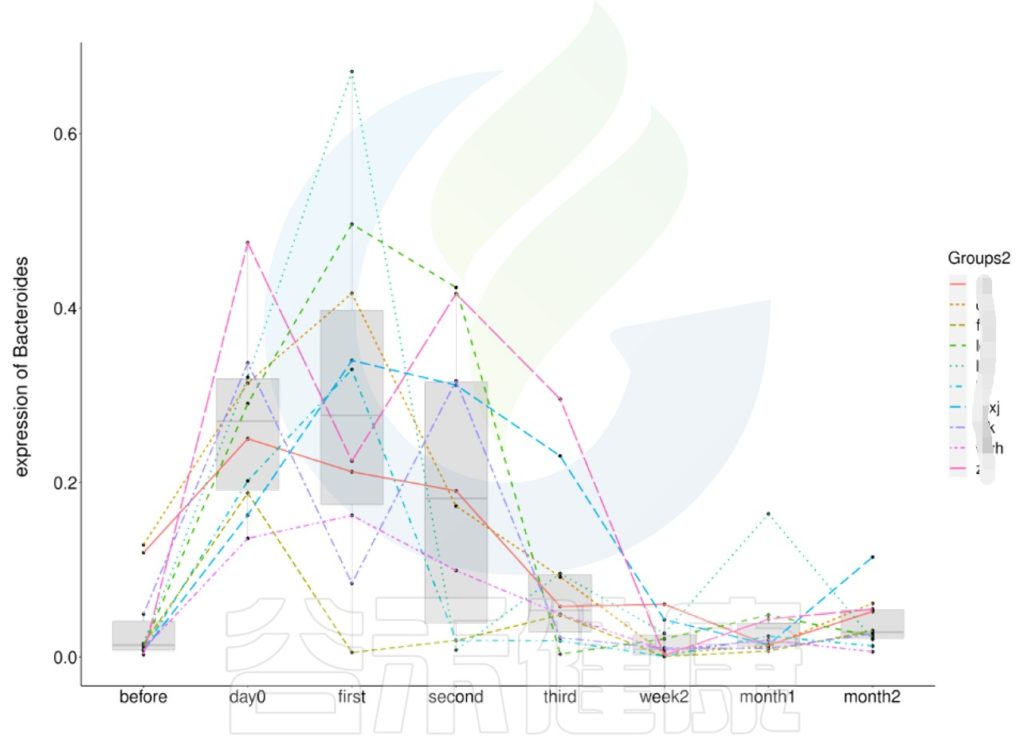
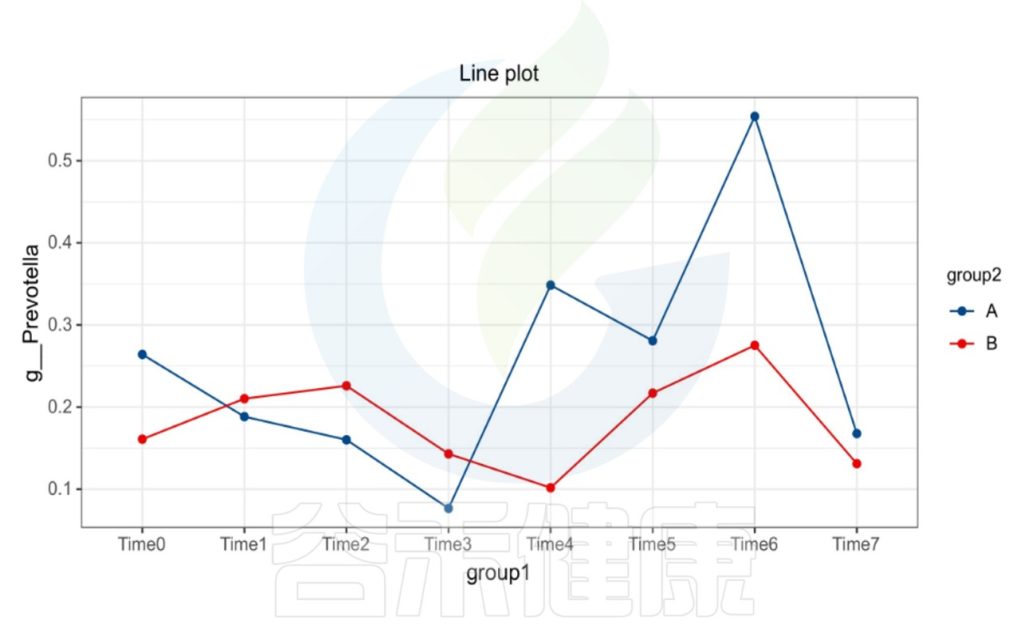
编辑
恰当的取样方法对于微生物组组学研究尤为重要,样本的收集和处理决定了后续微生物组鉴定的准确性。
前期的实验方案设计规划好以后,下一步就是落实,基于实验设计选取符合实验方案要求的高质量样本。在取样时需要注意以下几个点:
重复取样时,确定地理位置,或取样部位、取样深度、体积等都能实现标准化,以保证各个环节的一致性和可重复性。
影响肠道菌群样本的干扰因素有:年龄、地域、感染、疾病,药物治疗如抗生素、化疗、营养饮食干预等。
•收集样本时其他条件尽量保持一致
收集疾病组样本或者健康对照组样本时,尽量选择具有相同地理环境,饮食习惯,年龄段,药物治疗相关的病例作为重复样本。
采集过程的污染物也会被检测到变成样本组成的一部分,所以采样的过程中重复使用的工具,也要进行适当的消毒处理。取样的标准化和可重复性也可以避免由于不同的批次处理带来的样本菌群构成的差异性。
为了能合理的评估不同分组菌群之间的差异性或相似程度,必须要有一定的数据量来评估差异或者效应的大小。
一般来说,数据量规模越小,一组样本内的变异性越大。因此需要有更多的样本量来获得足够的统计能力。
每组至少3个重复,才能满足最低的组间差异统计检验条件,一般实验我们建议每组至少6个样本重复,临床样我们建议每组至少30个样重复。
不同的样本类型,如环境、植物、动物到人类,微生物组的来源差别较大,取样方法也有所差异。
•不同取样部位具有各自的生理特征
关于肠道菌群取样部位问题,该取粪便样本还是取肠道内容物?
根据研究发现不同的取样部位(如:粪便、粘膜活检,肠液等样本)具有各自的生理特征,肠道菌群的研究结果也有所差异。
•肠道内容物具有侵入性,用于某些特定疾病
肠道内容物由于采样方法较为复杂且具有一定的侵入性,所以在日常的应用相对较少。但是如对于一些特定的疾病或者研究问题,需要区分肠道不同部位微生物的组成或特性时,肠道组织样本或肠道内容物样本可能更加适合。
•一般情况下选择粪便样本
从实用的角度考虑,粪便样本可以自然采集、无创,且可以重复采样成为最常用的检测工具,是肠道菌群检测的最优选择。
注意
传统的粪便样本取样方法繁琐,对样本量和保存运输方式条件较为苛刻,例如单次的取样量要达到200mg以上,保存和运输过程中必须冷冻保存。一般临床上病例和健康人收集样本的配合度和接受度也不高,这也大大增加了收集样本的难度和成本。
★谷禾取样管便于采样以及储存运输
为了解决临床和实验过程中对粪便样本的取样遇到的这些实际问题和痛点,谷禾经过多年的肠道菌群检测实践,进一步对粪便样本的取样流程进行优化,开发出了专门用于肠道菌群取样的谷禾肠道菌群取样储存盒,实现了粪便样本的日常常规取样和常温储存运输。
该取样盒操作简单,取样方便,常温可以有效保存7天,日常在家就能实现常规粪便样本取样,缩短了实验人员收集样本的效率,节省了人力成本,所以该取样盒在市面上一经推出,就受到各大院校和临床机构的欢迎。同时也为谷禾做大健康方向前期收集样本阶段,提供了有力的前期技术支持。
谷禾取样管具有以下特征:
1、在家中就可轻松自行采集高质量样品
2、起始样品需要量低至0.01g左右,快速且稳定
3、样本取样7天内常温下储存和运输可保障稳定的群落结构
4、标准样品适合高通量标准化处理
5、能有效获得适用于16S 、宏基因组和qPCR等的高质量DNA
6、条形码化全样本可追溯性

取样方法也比较简单,仅需使用棉签从厕纸上沾取粪便,然后洗脱到取样管的保存液中即可,使保存液可见粪便颜色即表示取样量足够。
谷禾取样管不仅可用于人体粪便样本的采集,动物的粪便也同样适用,例如:大鼠样本用无菌棉签沾取少量粪便样洗脱到样本管的裂解液中;小鼠样本只需要放入1~2粒的粪便粒到裂解液即可。
1.如3天内使用过抗生素类,质子泵类胃药,阿片类精神药物请停药3天后进行检测;
2.感冒、腹泻或其他症状期间不影响取样,拉稀或稀便可以用棉签反复沾取粪便至取样管;
3.如长期便秘无排便可使用开塞露等辅助手段获取粪便样本;
4.取样无时间和饮食限制,取样前按照日常的用餐习惯进食即可;
5.完成取样后样本可常温有效存储一周,为保证检测时效请完成取样后尽快送检。如需长期储存,可将样本冷藏保存,-20℃冷藏最多可保存半年,-80℃可保存一年;
6.样本保存于取样管后需要拧紧瓶盖,防止取样管漏液,或者受到环境污染。

其他样本类型则需要用到传统常规的取样方法严格按照标准取样,之前我们的文章有对不同样本类型的常见取样方法作详细介绍:
微生物取样方法大全 (点击查看)
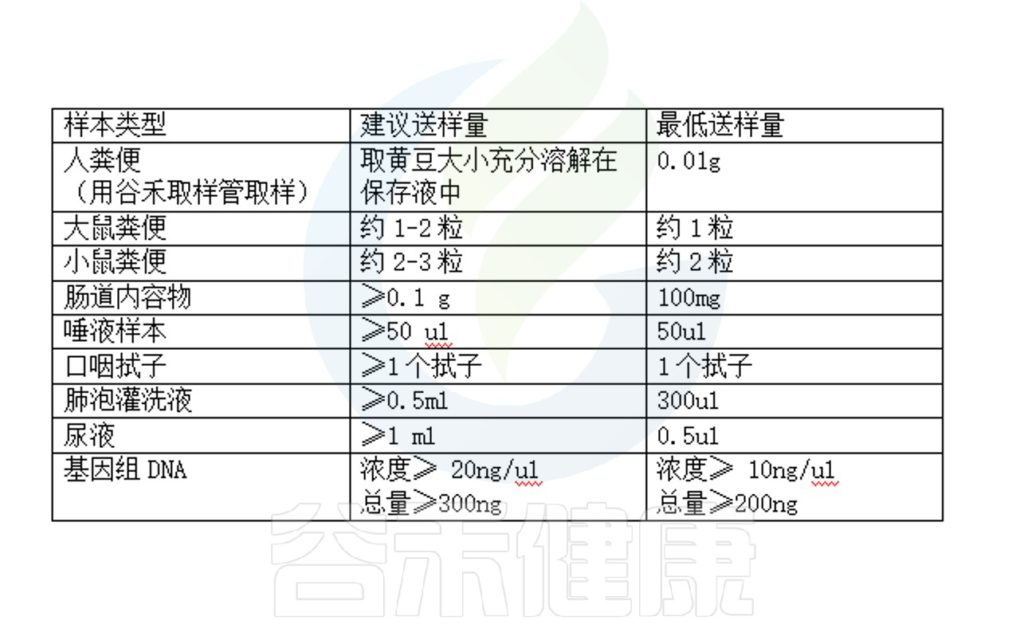
注:以上是16s测序项目送样需求,如果是宏基因组测序,需要达到常规标准,而且高宿主样本例如灌洗液,尿液,组织等样本,需要提前处理。
送样前要做好以下这些准备:
•样本信息单
样本信息单里填写好相关资料,包括谷禾编号,对应的科研分析名称,分组情况,有多少种分组情况就填几列,样本名称和分组名称由英文字母、数字、”.”组成,并以字母开头,不能以数字开头,不支持字符“- ”“_ ”、运算符、中文、空格等特殊字符。
注:名称长度最好不要超过8个字符。
如果要做相关性分析需要提前准备好样本的临床指标、生理生化指标,并将每个样本的具体测得的指标数值填写到样本信息单里。
如果对科研分析不太熟悉,不知道还能提取出哪些有用的分组信息,可以将具体的研究目的、实验方案详细写在备注里,分析人员会根据您的信息给出相应的指导意见和分析方案建议。可以提供一个纸质和电子版的样本信息单。
•样本检查
样本送样前要对样本和运输条件做检查,取样管是否拧紧,是否有漏液情况,样本不受环境污染和干扰,检查样本个数是否能核对的上,在寄样前要在快递箱上附带一张送样信息的纸条,包含以下内容:
寄样单位或联系人:xxx
寄样日期:xxx
样本个数:xxx
实验室接收到样本后会跟送样信息单进行核对,如果样本个数和信息能核对上,则继续对样本进行实验上机,如果样本个数或信息核对不上,会跟客户沟通,核对信息。
小 结
客户在做好实验方案设计和样本收集的前期准备工作以后,后续的实验和测序分析工作就交给我们来完成吧,从收到样本到科研分析报告的出具,周期一般在6~10天左右(16S v4); 15-20天(16S V3V4);15-20天(宏基因组)。
实验室在收到样本以后会先核对信息,然后进行样本DNA提取,质检,建库,测序上机。等测序数据下机以后,对原始数据拆分,质控,然后就进行科研数据分析阶段。
分析思路:
1.先了解不同分组样本都由哪些微生物构成,并计算丰富度,做α多样性分析;
2.比较不同分组微生物群落整体是否有差异,做β多样性分析;
3.进一步找出不同分组之间微生物群落的差异菌属,关键菌属;
4.根据关键菌属可以作为biomarker用于疾病诊断模型构建;
5.找出菌属与临床理化指标的相关性;
6.根据物种构成预测菌属代谢途径,基因功能预测,找出组间差异功能预测,查看菌属是怎么运行宿主的
7.将数据结果整合起来,找出不同疾病环境分组条件下,菌属的差异是否跟疾病相关,并影响宿主。
最终根据科研分析报告的研究方向和侧重科研选择不同的分析方法,并做图像化展示。
差异分析方法:
•Tukey检验
•非参数检验
•基于矩阵的Anosim检验、Adonis检验
•LEfSe分析
•随机森林和ROC曲线
相关性分析方法:
pearson相关系数
spearman相关系数
相关性网络图、相关性热图、线性回归图
功能分析:
PICRUSt2、FAPROTAX、BugBase
PCA、PCoA、NMDS
ROC、RDA
Venn图、花瓣图、Upset图
物种构成柱状图、热图、差异箱型图
GraPhlan物种进化树
metagenomeSeq
Random Forest随机森林
ROC曲线图
graphlan图
Gephi复杂网络图
三元相图
谷禾近期合作发表示例文章:
Hu Y, Li J, Ni F, et al. CAR-T cell therapy-related cytokine release syndrome and therapeutic response is modulated by the gut microbiome in hematologic malignancies[J]. Nature communications, 2022, 13(1): 1-14.
Lou M, Cao A, Jin C, et al. Deviated and early unsustainable stunted development of gut microbiota in children with autism spectrum disorder [J]. Gut, 2021 Dec 20:gutjnl-2021-325115.
Li R, Liu R, Chen L, et al. Microbiota from Exercise Mice Counteracts High-Fat High-Cholesterol Diet-Induced Cognitive Impairment in C57BL/6 Mice[J]. Oxid Med Cell Longev. 2023, 20:2766250.
Xiao W, Zhang Q, Zhao S, et al. Citric acid secretion from rice roots contributes to reduction and immobilization of Cr(VI) by driving microbial sulfur and iron cycle in paddy soil [J]. Sci Total Environ. 2023, 16:158832.
Wang X, Weng Y, Geng S, et al. Maternal procymidone exposure has lasting effects on murine gut-liver axis and glucolipid metabolism in offspring [J]. Food Chem Toxicol. 2023, 174:113657.
Liao J, Dou Y, Yang X, et al. Soil microbial community and their functional genes during grassland restoration [J]. J Environ Manage. 2023, Jan 1;325(Pt A):116488.

谷禾健康

16S科研项目是一个完整的闭环,前期的课题项目设计方案、取样和重复实验设置决定了后期分析报告的数据完整性和项目类型。
想要拿到一手有利用价值的科研报告和项目数据,前期的实验方案设计和后续的分析都起着关键性的作用。
然而有时候拿到报告不知道如何去解读,这里为大家梳理一下16s科研项目的全过程,帮助大家更好的了解报告内容,快速获取关键信息。


实验方案设计就像一个总工程的设计图纸,决定了未来科研分析报告的类型走向,并且前期的分组设计的越详细,各种理化指标、生化指标、代谢物等信息准备越充分,后续报告的完整度越高。


明确项目课题类型
第一步要做的就是明确项目课题类型:
最常见的就是多分组之间差异分析比较:例如,要比较对照组、模型组、实验组,之间的差异结果。
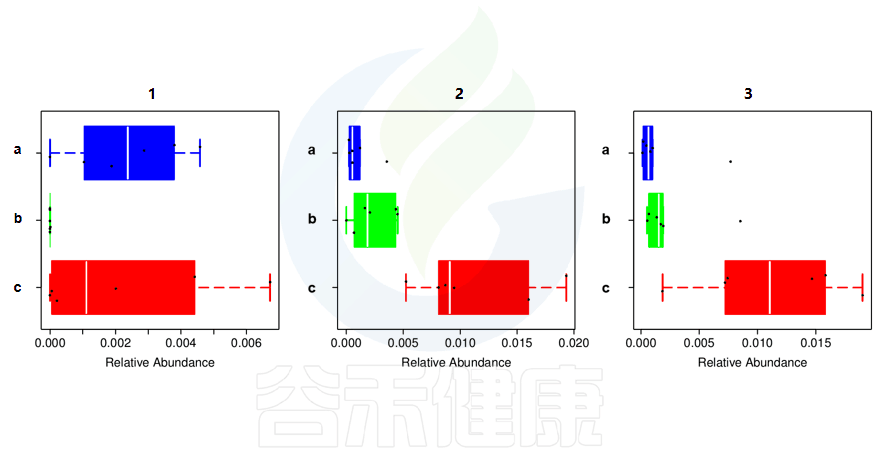

还有多分组中,任意两组之间比较:例如某实验设计了正常组、疾病组、用药组服用奥氮平、阿立哌唑、氨磺必利、利培酮,像比较不同的用药组和疾病组之间的菌群的差异结果,就用到了分组之间两两差异比较。
✦举个例子
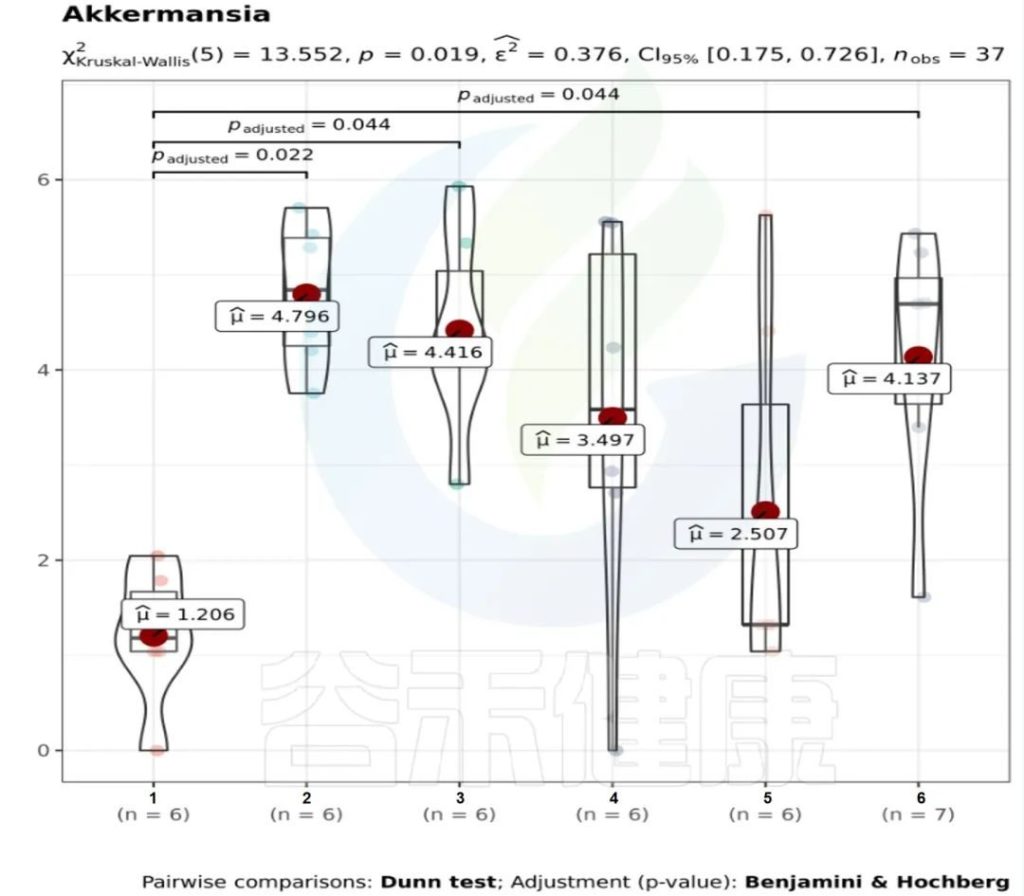
图中1组与3组、4组、6组 组间差异显著
还有随时间的变化比较菌群之间的变化规律:例如在用药不同时间段包括3天,5天,2周,1个月,2个月,观察菌群的变化情况。如下图所示:
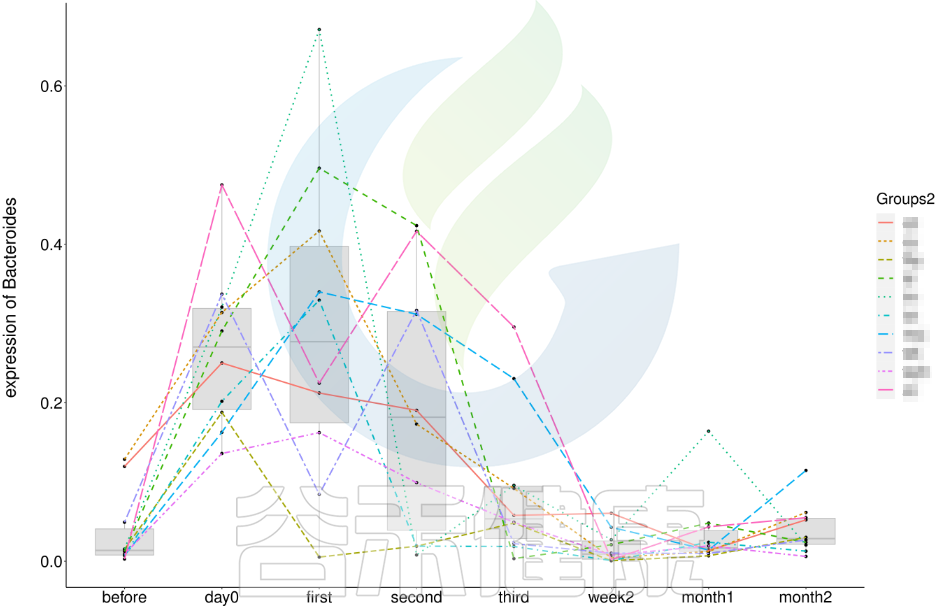
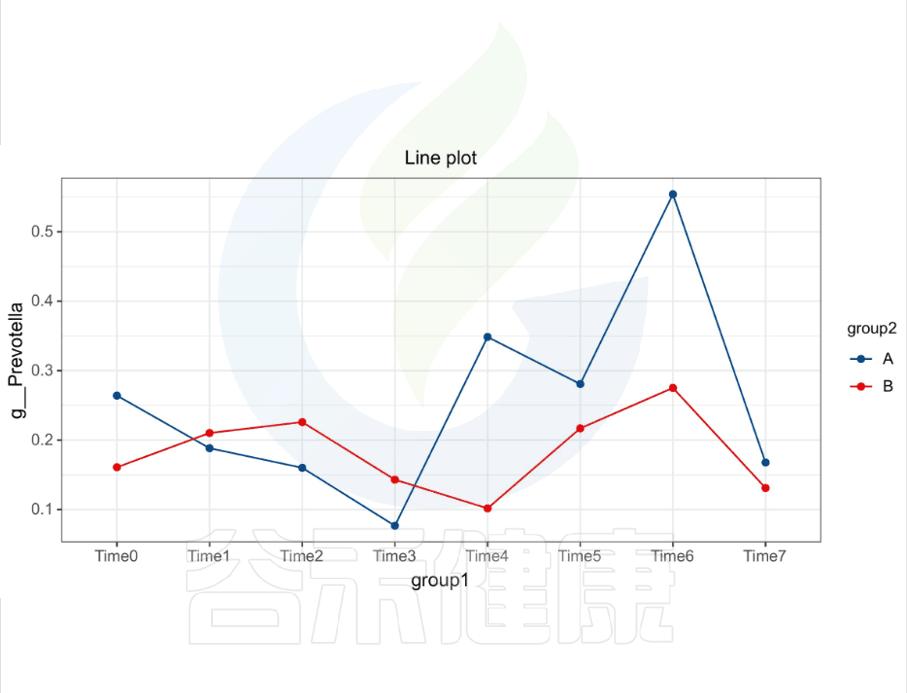
收集理化指标非常重要
如果前期搜集好每个样本的相关理化指标,还可以计算这些指标与菌群之间是否具有相关性。
✦举个例子
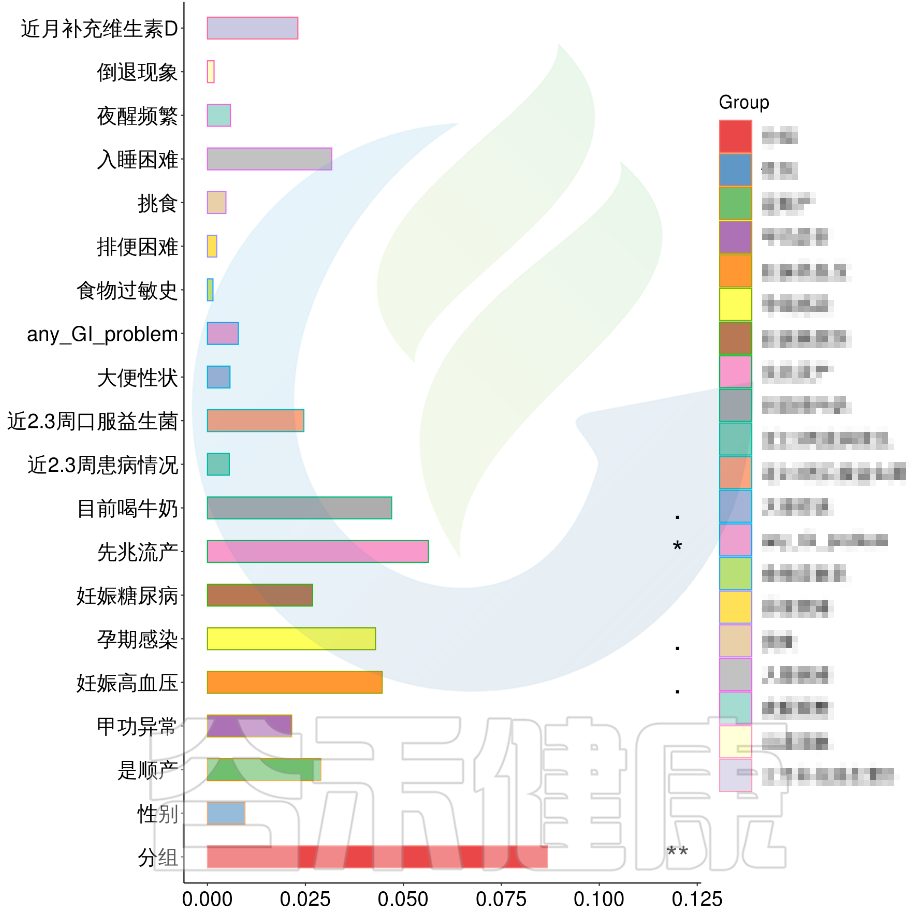
例如该项目比较自闭症儿童与正常儿童的菌群差异。客户在样本信息单里还详细搜集了母孕期的各种详细指标,例如孕期天数、出生体重、白细胞介素6、肿瘤坏死因子a、五羟色氨等数值型理化指标。
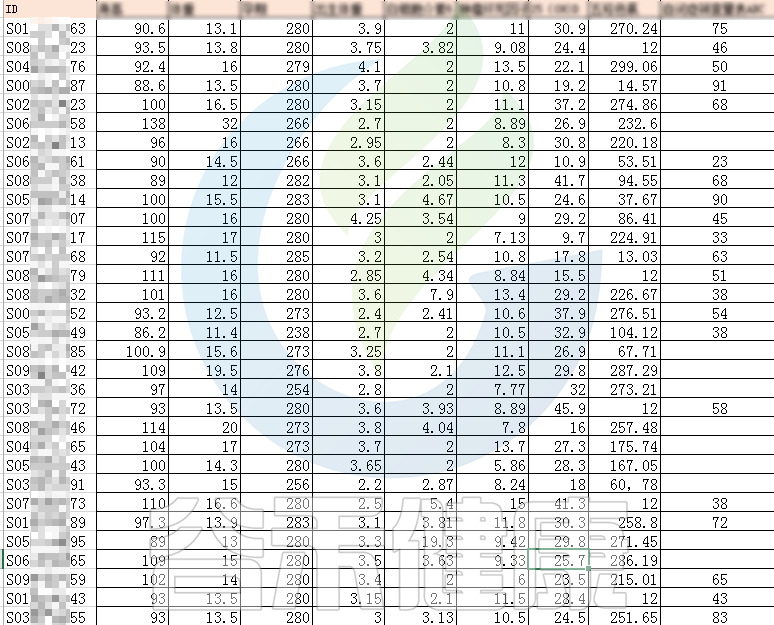
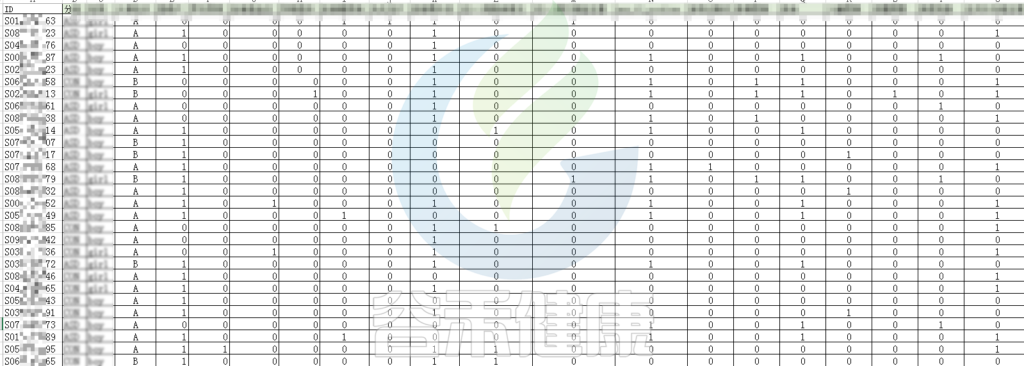
还搜集了是否顺产、是否妊娠高血压、是否孕期感染、是否妊娠糖尿病、是否先兆流产等因子型理化指标。其中0代表否,1代表是:
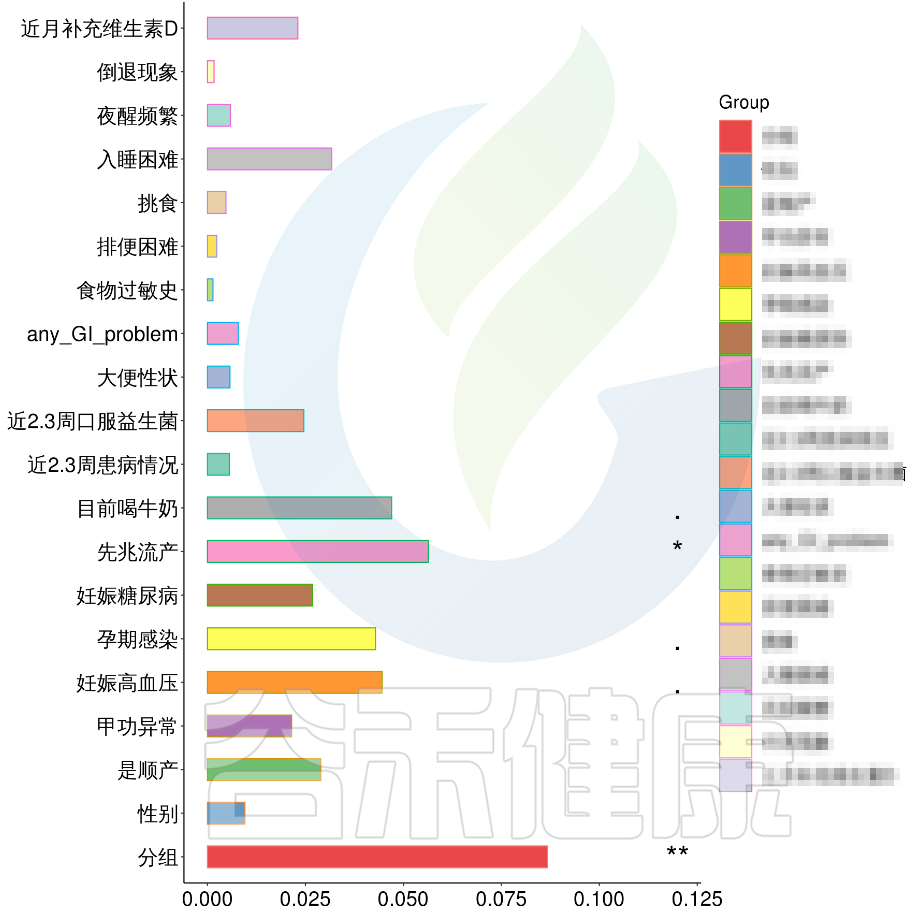
根据这些理化指标与菌群数据做相关性分析,从因子型的结果可以看出,自闭症(ASD)与正常儿童之间的分组与菌群之间相关性极显著**,其次是否有先兆流产的分组与菌群之间有显著相关性*,其他的包括是否喝牛奶、孕期是否感染、妊娠高血压都与菌群有相关性。
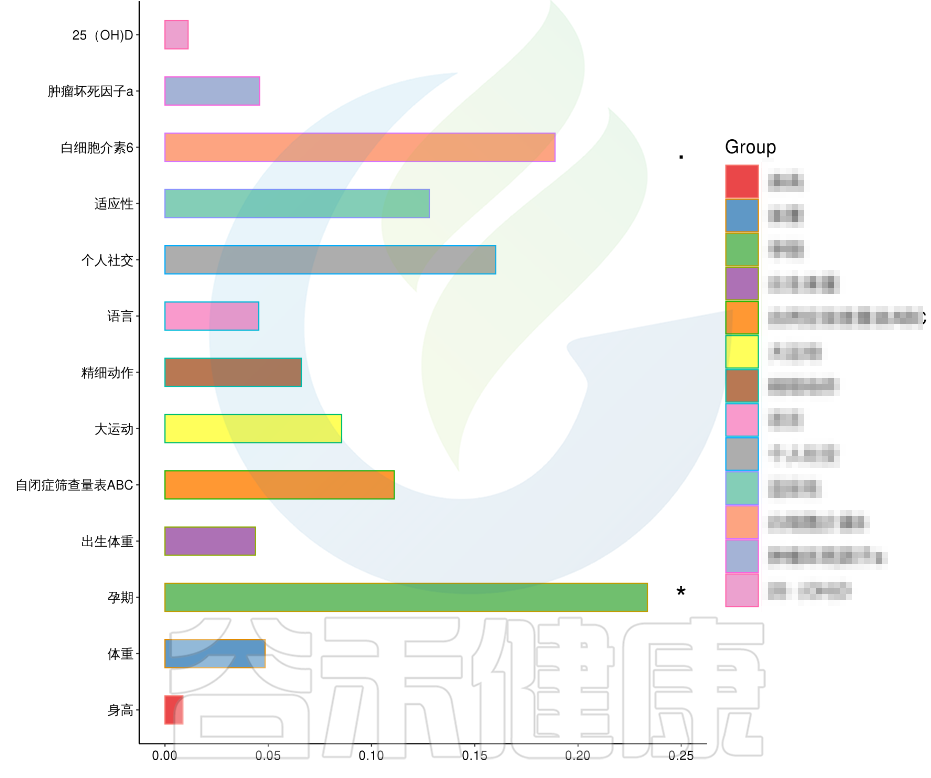
在数值型理化指标中,孕期的天数与菌群之间相关性显著*,其次是白细胞介素6与菌群之间有相关性。
小结
因此,前期搜集相关资料越详细充分,对分析报告的完整性也会有帮助,分析人员也会根据您的样本信息单提供的相关内容,做出个性化的分析和售后指导建议。
首先基于样本类型,最常见的环境样本来源是人体、动物、土壤、水体等。而人体中的肠道菌群样本是目前研究最广泛,可鉴定的物种也最为丰富,谷禾在肠道菌群与人体健康方面有深入研究,目前已完成超20万例临床肠道菌群样本检测,并构建了超过60万各类人群粪便样本数据库。
其他样本类型还包括人体/动物唾液样本、组织样本、尿液样本等。
▸ 粪便样本
目前粪便样本从采样到提取数据分析技术较为成熟、应用较为广泛,谷禾最早在15年就开发了针对粪便样本的取样管,也是最早致力于研发粪菌取样盒的公司,方便实验室、个人日常取样需求,实现了粪菌样本的常温运输。
谷禾取样管常温保存,取样也较为方便卫生,在家就可以轻松完成,相较于传统取样方法都有所升级。并且该取样管也有专利证书。该取样方法被大量客户采用并接纳,大大降低了采集粪便样本的难度,缩短了搜集样本的时间周期。
取样示意图

▸ 其他样本
土壤样本也相对较为容易提取出DNA,但需要注意的是土壤样本的菌群特征容易受植物腐殖质基因的影响和干扰,所以提取时要进行纯化。
而口腔、组织、尿液等样本,由于DNA含量较少,在实验阶段提取相对较为困难,所以提前准备样本时,尽量多取一些,并且可以多取几个重复,尽量避免扩增不出来的情况。
并且这些样还很容易受到环境样的污染,所以在实验阶段,可以取空白样本,和阳性样本ST做对照,数据分析时可以用来纯化样本,排除来自环境的干扰序列。
✦组间差异分析需重复取样
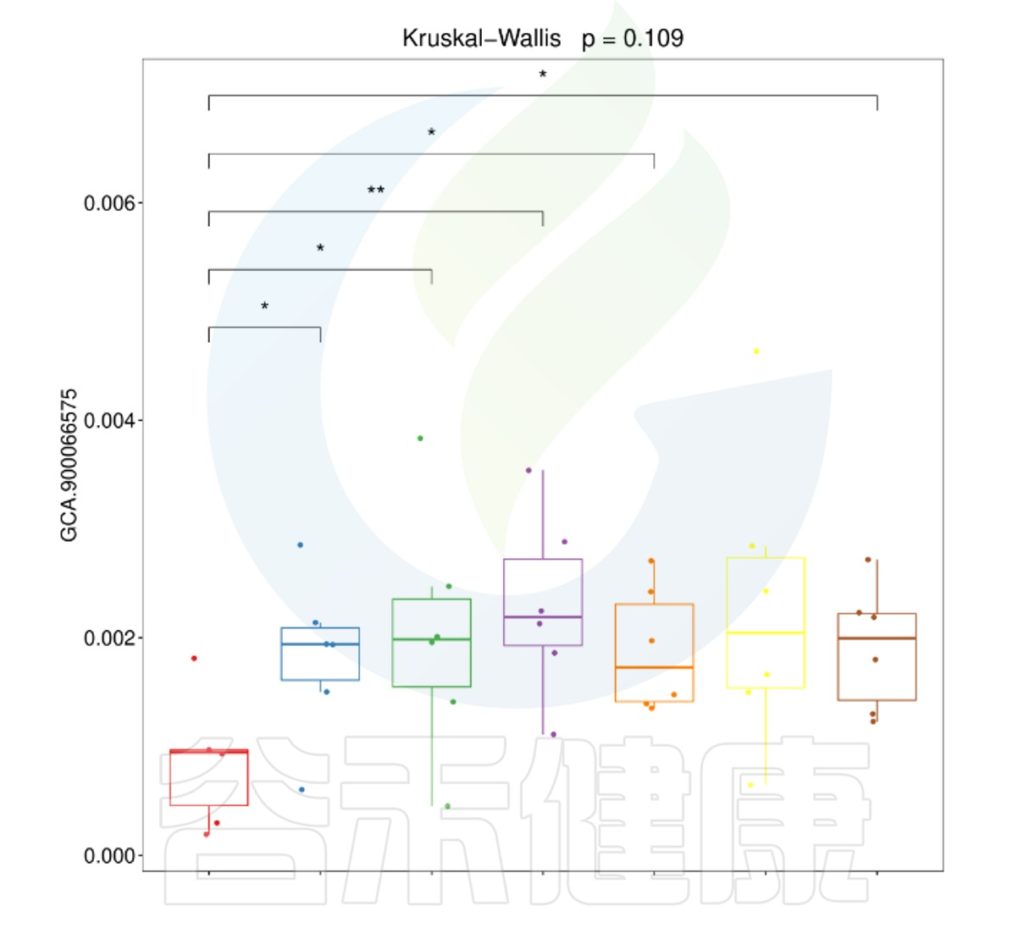
要做组间差异分析时,每组要重复取样,才能做组与组之间的统计检验。理论上,每个组至少3个样就满足基本的统计差异分析需求。所以在重复取样时,每个分组至少取3个样。取样时要保证每个分组内部的样本一致性,如果组内样本之间的个体差异性较大,则会影响后期组间差异结果分析。
✦举个例子
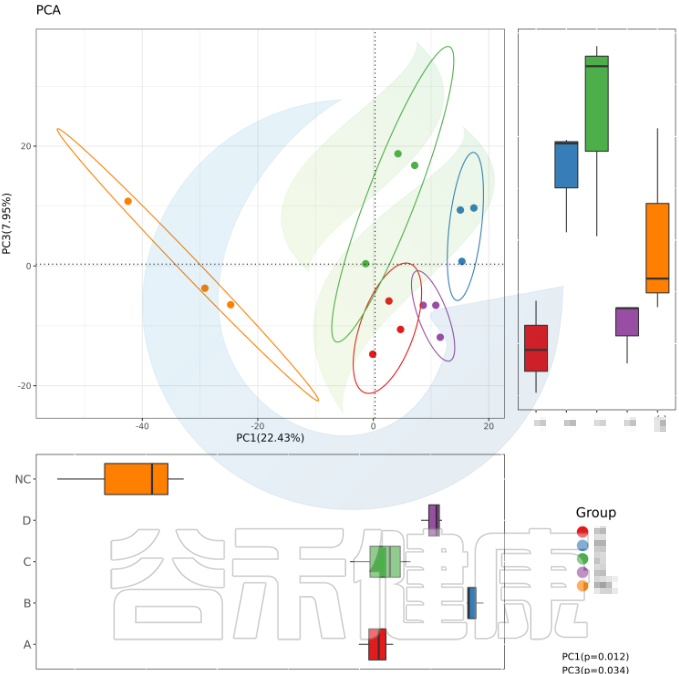
例如从该图可以看出,分组之间组间差异较大,并且组内的样本之间较为接近和相似。
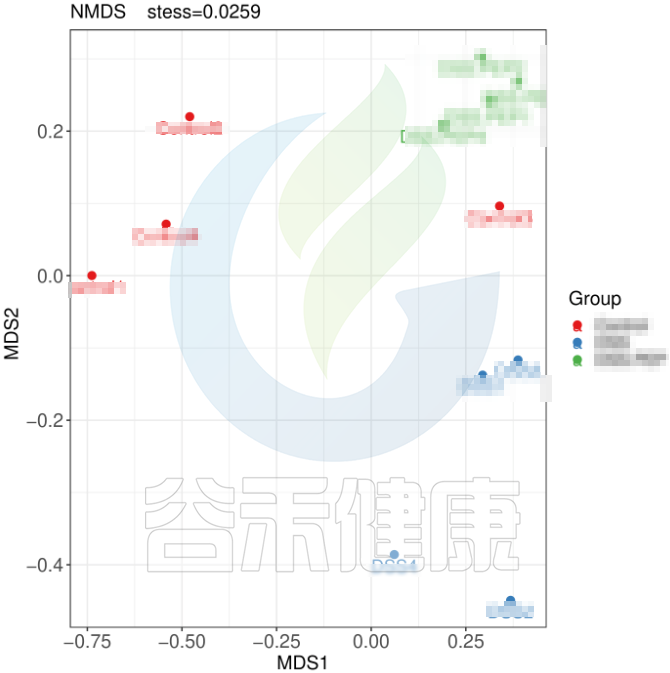
但从该图可以看出,Control组中Control3样本明显与组内的其他样本差异较大,与DSS组内的样本较为相近,这样就对后期组间差异分析的时候会产生影响,需要将该样本去除。
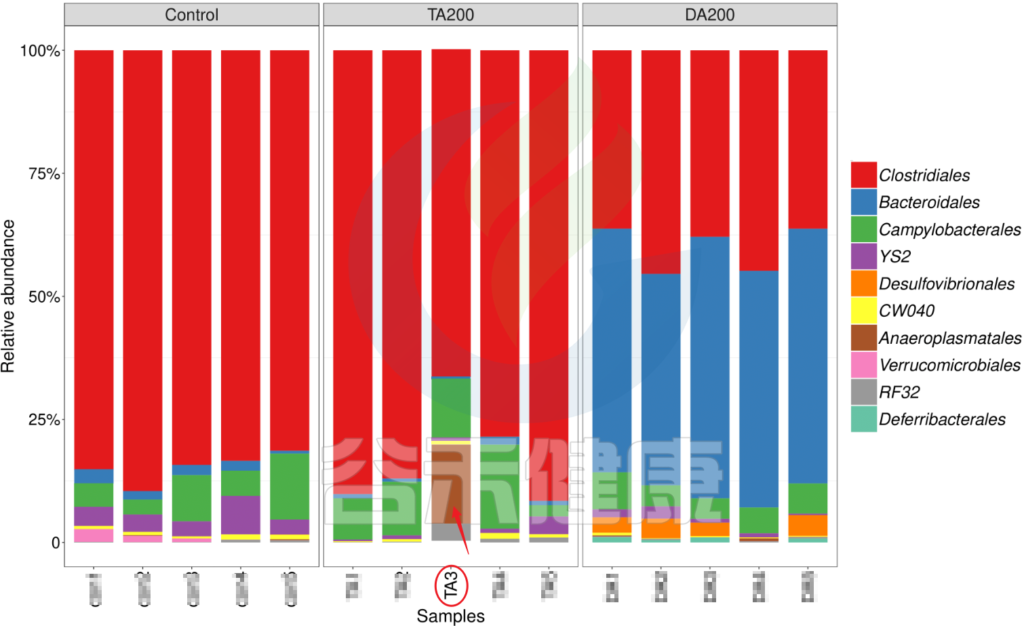
又例如在该图中,TA200组中的TA3样本的Anaeroplasmatales物种丰度含量非常高,该样本与组内的其他样本明显差异较大,该样本可能受到环境污染等其他因素干扰,这样就没有办法保证组内样本的均一性,也会影响分组之间的差异分析统计结果,再后期分析的时候建议把该样本去掉重新分析。
建议
为了便于后期数据整理修改,每个分组需要保留一定量的重复样本,假如每个分组只取了3次重复,假如其中有一个样本质量不好需要去除,该分组只剩2个样本,则不满足每组至少3个样的分组条件,整体就没有办法做组间差异分析统计。
所以这里建议每个分组至少取5个样做重复,一般6到10个样就能分析出比较完善的结果。具体分组和组内的重复取样数量视具体的实验设计方案而定。
在经费允许的情况下,建议多取一些重复。假设每组取50到100个重复或者以上,得到的分析结果就基本可以涵盖该分组情况所有的菌群构成情况,可以较为全面的研究分组之间的菌群构成差异情况。
当拿到16S科研分析报告以后,面对纷繁复杂,各式各样的图表分析结果犯了难,不知道如何从这么多的图表中入手,快速找到报告中需要的图表结果。
这里对16S科研分析结果抽丝剥茧,概括出报告中的主要几大内容板块。
•16S科研分析究竟是在做什么?
16S rDNA 是一种对特定环境样品中所有的细菌进行高通量测序,以研究环境样品中微生物群体的组成,解读微生物群体的多样性、丰富度及群体结构,探究微生物与环境或宿主之间的关系的技术。
主要是对原始数据进行拼接过滤得到的优化序列,降噪方法得到ASV,再对ASV进行物种注释,注释到门、纲、目、科、属、种各层次上的分类结果。
通过ASV表计算Alpha多样性,样本内的多样性指数,Beta多样性,样本间相似性的指标。
对ASV表进行功能预测,例如Picrust2功能预测分析、Bugbase菌群表型特征分析,FAPROTAX生态功能预测等。
得到的每个样的数据结果,根据客户提供的分组情况和理化指标,进一步做组间差异分析,以及和环境理化指标之间做关联分析,相关性分析,比较分组之间是否有差异,差异是否显著,来验证分组是否合理,和环境宿主之间是否有关联性。
原始数据处理
Illumina NovaSeq测序平台测序得到的双端数据Raw PE,经过拼接和质控,根据一定的标准过滤掉低质量数据、接头或PCR错误,得到Raw Tags。再经过去重复序列,去singleton序列,过滤嵌合体,得到可用于后续分析的有效数据 Effective Tags。
OTU(ASV) 表生成
微生物多样性分析中最重要的就是OTU特征表,一切后续分析都围绕OTU表来进行。生成OTU除了传统的聚类的方法(一般按照97%的相似度进行聚类),现在最新用到的技术的是降噪的方法得到ASV。
简单来讲ASV就是在去除了错误序列之后,将Identity的标准设为100%进行聚类,常见的有DADA2、Deblur、Unoise三种降噪方法。项目里用到的是UNOISE2降噪方法获得ASV数据。
物种的分类与注释
采用QIIME2训练分类器方法对ASVs代表序列进行分类学注释,默认选用SILVA138数据库进行物种注释。并在各个分类水平上:domain(域),phylum(门),class(纲),order (目),family(科),genus(属),species(种)对每个样本的群落组成统计。
alpha多样性
Alpha多样性主要反映样本内多样性。对ASV表进行计算可以获得每个样本的simpson,ace,shannon,chao1以及goods_coverage等指数,alpha多样性指数用来来评估样本菌群物种的丰富度(richness)和多样性(diversity)
beta多样性
Beta多样性反映的是样本间多样性,Beta多样性是衡量个体间微生物组成相似性的一个指标。通过计算样本间距离可以获得β多样性矩阵,基于OTU的群落比较方法报告中给出了,欧式距离、bray curtis距离、Unweighted UniFrac距离和Weighted UniFrac距离等。
功能预测
得到群落的微生物组成之后,也可以对群落功能组成进行预测,常用的16S功能预测的相关软件有PICRUSt2、FAPROTAX、BugBase。
PICRUSt2用来预测功能,通常指的是基因家族,PICRUSt2支持基于多个基因家族数据库的预测,报告中包括了KEGG同源基因,KO直系同源物,EC酶分类编号,MetaCyc途径的丰度,CAZy碳水化合物活性酶数据库,GMM是肠道代谢模块和GBM是肠脑模块。
FAPROTAX是原核的微生物注释代谢或其他生态相关的功能(例如硝化,反硝化,发酵)的一个数据库和软件。FAPROTAX预测的功能主要集中在海洋、湖泊等环境样本微生物的功能,特别是硫、碳、氢、氮的循环功能。
BugBase能进行表型预测,其中表型类型包括革兰氏阳性(Gram Positive)、革兰氏阴性(Gram Negative)、生物膜形成(Biofilm Forming)、致病性(Pathogenic)、移动元件(Mobile Element Containing)、氧需求(Oxygen Utilizing,包括Aerobic、Anaerobic、facultatively anaerobic)及氧化胁迫耐受(Oxidative Stress Tolerant)等7类。
以上这些部分,我们通过数据处理分析,得到了每个样本相关的大量数据结果,包括每个样本的序列统计、ASVs表格、物种分类注释统计、alpha多样性指数、beta多样性指数、功能预测等。这些数据主要集中在报告里的这些内容:
▸ 科研分析报告结果文件夹
01_pick_otu/ 文件夹主要是对样本ASV表格统计
02_sequence_statistic/ 文件夹是对样本序列数据的统计
03_diversity-metrics / 文件夹是对样本的alpha多样性指数、beta多样性指数的统计
04_Taxonomic/ 文件夹是对物种分类注释的统计(门到种水平)
Picurst2/ 文件夹是Picrust2功能预测得到的每个样本的相关功能预测数据
Groups/ 文件夹下是对组间差异分析结果

红框是样本个体的相关数据统计,Group是分组比较
根据以上常规分析得到的相关数据进行作图,其路径也在对应文件夹下,可以打开 分析报告.html 有相关分析的图表和对应文件的详细介绍和路径说明。
★拿到样本后需要进行统计分析
当我们拿到这些样本大量的数据结果,之后关键的一步就是做对这些数据进行处理,做统计分析,比较分组之间的差异结果,找出菌群和环境之间的关联性等,对数据进一步做研究,找出课题方案对应的结果。
不同的数据用到的统计检验方法也不太一样,接下来我们对报告中的不同的分析结果对应的统计差异分析方法进行介绍说明。
▸ alpha多样性
alpha多样性指数组间差异统计分析用到的检验方法是:单因素方差分析(如果只有两个分组,用Wilcoxon秩和检验,3个及以上的分组用Kruskal-Wallis 检验),图上方显示P值
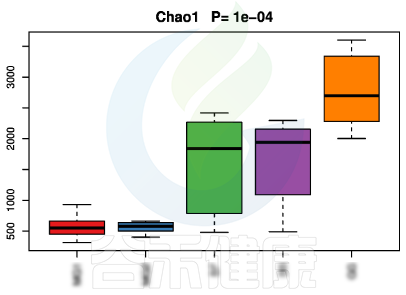
▸ beta多样性
beta多样性指数的统计检验方法有ANOSIM相似性分析和Adonis多元方差分析,这两种都是基于距离矩阵的检验方法。
✦Anosim相似性分析
Anosim分析是一种非参数检验,用来检验组间的差异是否显著大于组内差异,从而判断分组是否有意义。
报告中给出了加权距离和非加权距离的Anosim结果图,图中给出了R值和P值。
R值用于比较不同组间是否存在差异,R-value 介于(-1,1)之间,R-value > 0,说明组间差异大于组内差异。R-value < 0,说明组间差异小于组内差异。R只是组间是否有差异的数值表示,并不提供显著性说明。
统计分析的可信度用 P-value 表示,P< 0.05 表示统计具有显著性。
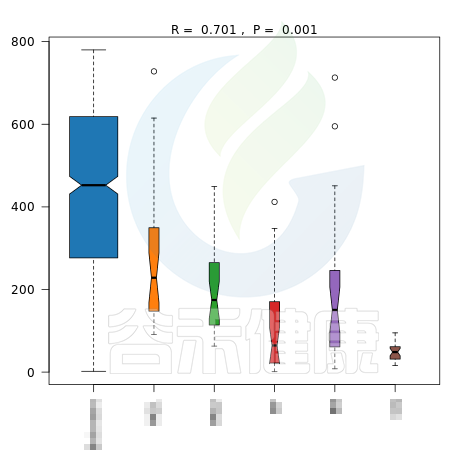
图中能看出R>0,说明组间差异大于组内差异,P<0.05 ,说明差异显著,证明该分组情况效果较好。
✦Adonis多元方差分析
Adonis多元方差分析,其实就是PERMANOVA,亦可称为非参数多元方差分析。
其原理是利用距离矩阵(比如基于Bray-Curtis距离、Euclidean距离)对总方差进行分解,分析不同分组因素对样品差异的解释度,并使用置换检验对其统计学意义进行显著性分析。
它与Anosim的用途相似,也能够给出不同分组因素对样品差异的解释度(R值)与分组显著性(P值)。
报告中PCoA bray距离、PCoA weighted_unifrac距离、PCoA unweighted_unifrac距离的图片右下角有给出PERMANOVA检验的P值和R值。
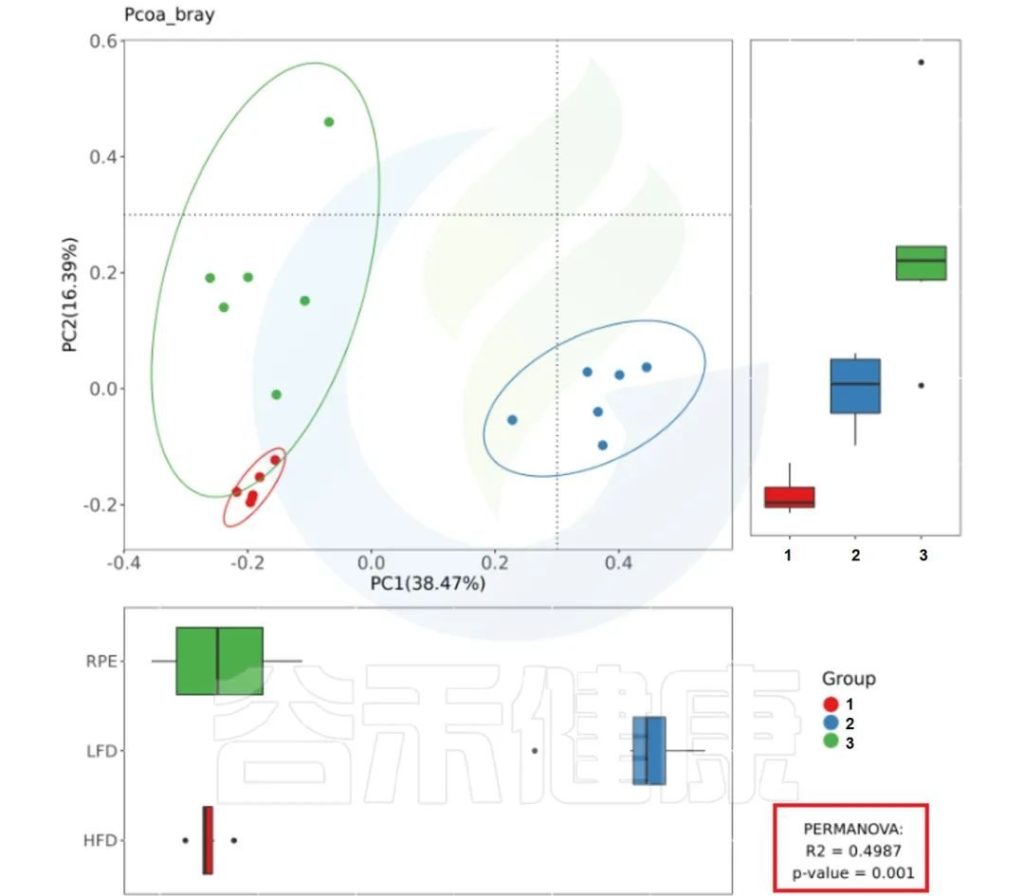
图中看出PCoa bray距离得到的检验P<0.05 组间差异显著,并且分组之间区分较为明显。
PCoa bray距离的PERMANOVA检验结果路径:
多组间检验结果:
Groups/betadiv/pcoa_bray_analysis/PERMANOVA.result_all.csv
两组间检验结果:
Groups/betadiv/pcoa_bray_analysis/ PERMANOVA_paired_result.csv

不同分类水平下的检验方法
在很多分析报告当中,例如在不同疾病的肠道菌群分组中,本身样本个体之间肠道菌群的物种多样性,丰富度差异并不大,alpha多样性组间差异并不显著,beta多样性分组间区分不是很明显,这样就需要进一步找出分组之间的差异物种或者差异功能来进行分析。
对于不同分类水平的物种和功能预测结果用到以下几种检验方法:
Tukey检验
Tukey主要应用于3组或以上的多重比较,适合于各组例数相等的每两两分组之间比较。
Tukey检验的一个重要的优点是非常简单,而且所需实验样本相对较少。
其检验结果的可信度达到95%的置信水平时,最少的情况下只需6个样本进行验证(改善前3个样本、改善后3个样本)。
•举个例子
图中的字母代表显著性差异的字母表示法,只要含有相同的字母,就表明两组之间没有显著性差异。
例如a和ab含有相同字母“a”,表示两组之间没有显著性差异。ab中的“b”表示这一组和其他含有字母b的组(比如bc)没有显著性差异,但是a和bc就有显著性差异了。
图中只展示Tukey检验差异显著的物种或功能,如果数量较多,则只展示前10个。
路径:Groups/diff_analysis/TukeyHSD/
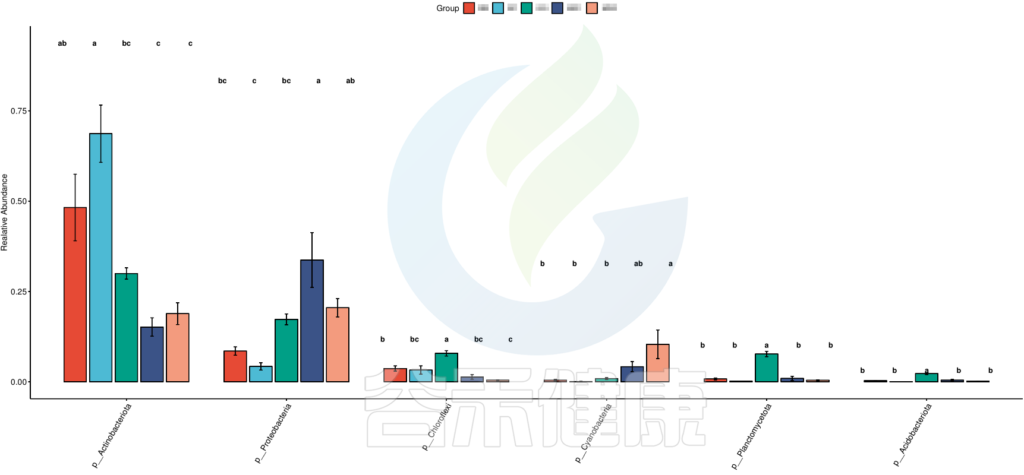
图中显示的都是Tukey检验组间差异显著的物种,依次按照丰度从高到底排列,如果差异结果较大,则显示前10个物种。例如在该图中,Tukey检验结果,门水平物种Actinobacteriota在BB与MG1组、BB与MG2、BF与GG组、BF与MG1组、BF与MG2组,这些分组之间组间差异显著。
组间差异箱型图
组间差异箱型图用到的检验方法是通过单因素方差检验(只有两个分组,用的是Wilcoxon秩和检验,3个及以上的分组用的是Kruskal-Wallis 检验),Var检验和one-way相结合,筛选出组间差异性物种。
路径:Groups/diff_analysis/TaxaMarkers
图中每一个箱型图代表一个组间差异显著的物种
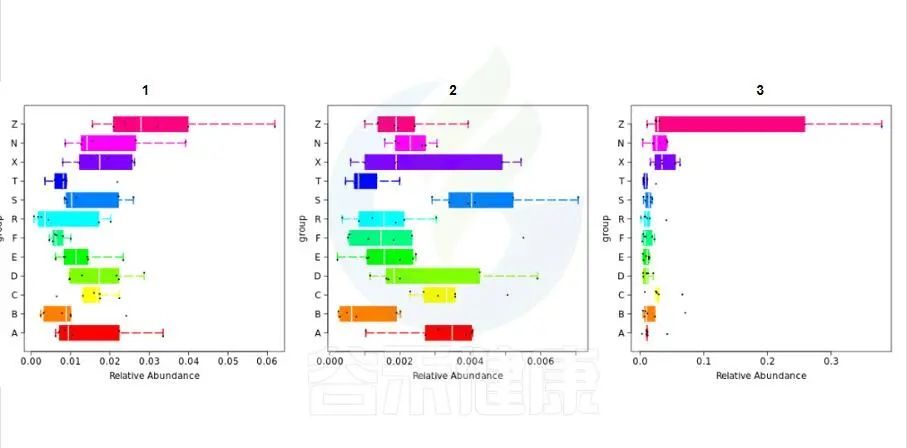
图中显示的都是统计方法得到的差异显著的物种,图中能看出这3个物种分组之间差异显著。
命名格式是,例如:Cen_Nitrosopumilus 指的是,当前分类水平(属水平)的名字 g__Nitrosopu 加上一级分类水平(科水平)的名字 f__Cenarchaeaceae 的前 3 个字母简写Cen,如果当前水平没有注释到名字则以全称的名字表示。
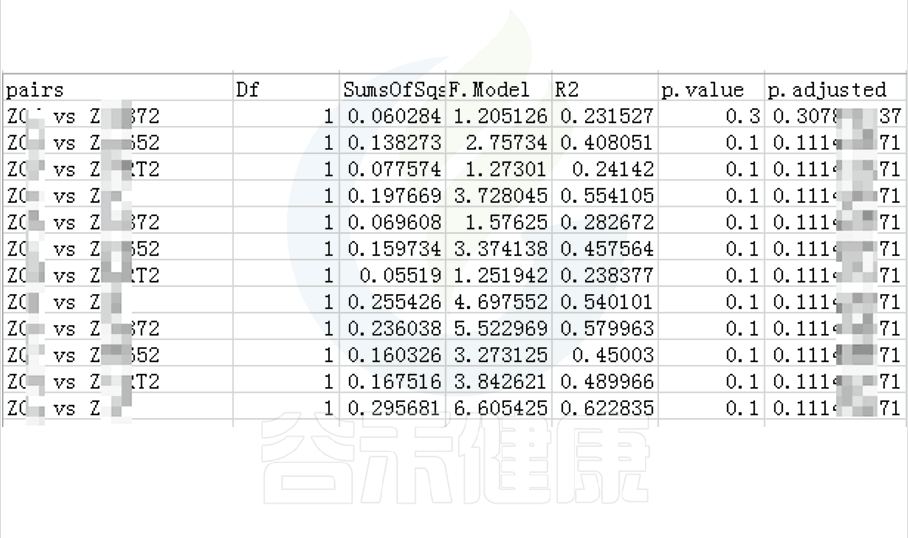
统计结果表:Groups/diff_analysis/TaxaMarkers/ xxx.Groups.sig.meanTests.csv
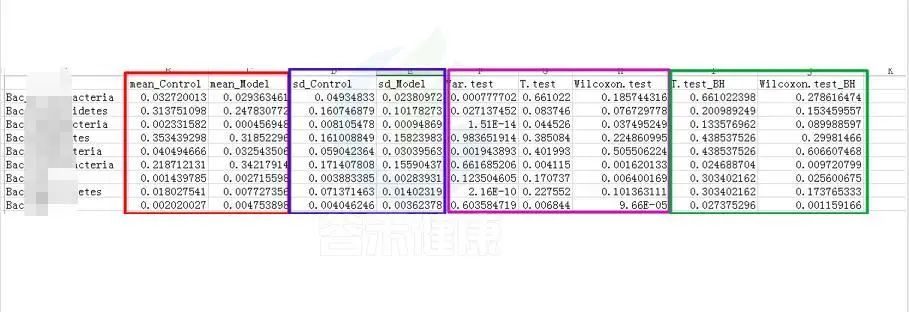
例如这是一个表格的截图
红框 mean_ 是分组组间的平均值
蓝框 sd_ 代表组间的标准差
粉色 .test 代表不同统计检验结果的P-value P值,这里有var检验 T 检验 Wilcoxon检验(或Kruskal-Wallis 检验)
绿色 _BH 例如Wilcoxon.test_BH代表Wilcoxon.test检验BH矫正的Q-value,Q值
UnivarTest检验(单因素方差分析)
单因素方差分析是指如果只有两个分组,用Wilcoxon秩和检验,3个及以上的分组用Kruskal-Wallis 检验。
路径:Groups/diff_analysis/UnivarTestXXX
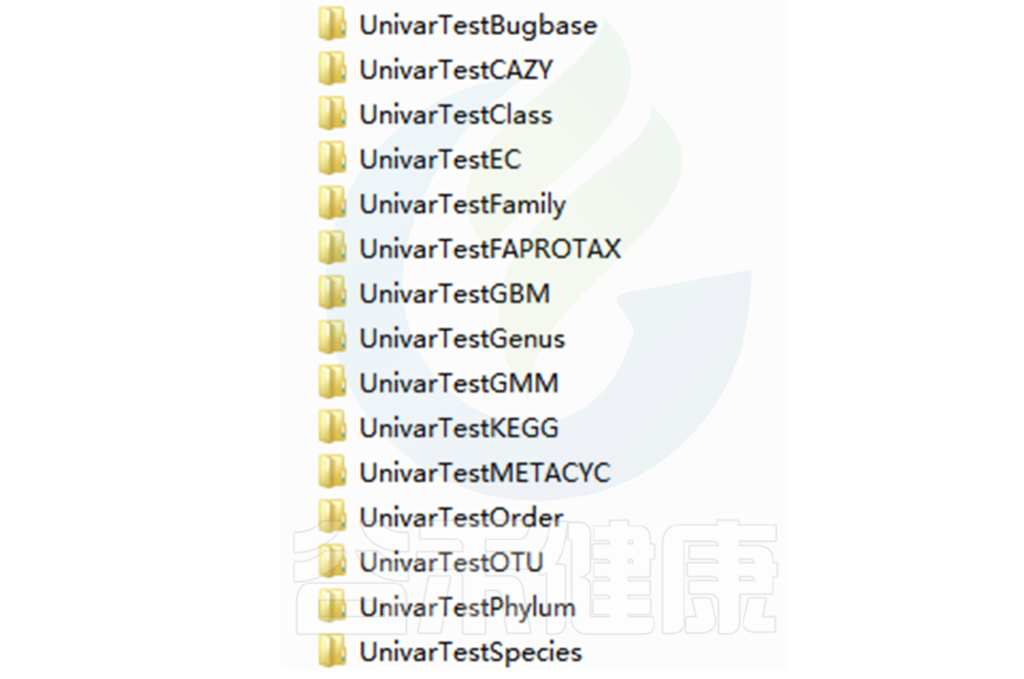
Groups\diff_analysis\UnivarTestKEGG\figure 文件夹下有做成柱状图、箱型图和单个物种之间的图,其中有横着排列和竖着排列的,有用原始值计算的,还有对原始值取log后进行统计的。图中只展示Univar 检验组间差异显著的物种/功能。
统计结果表:Groups/diff_analysis/UnivarTestXXX/ UnivarTest_sign.txt
•举个例子
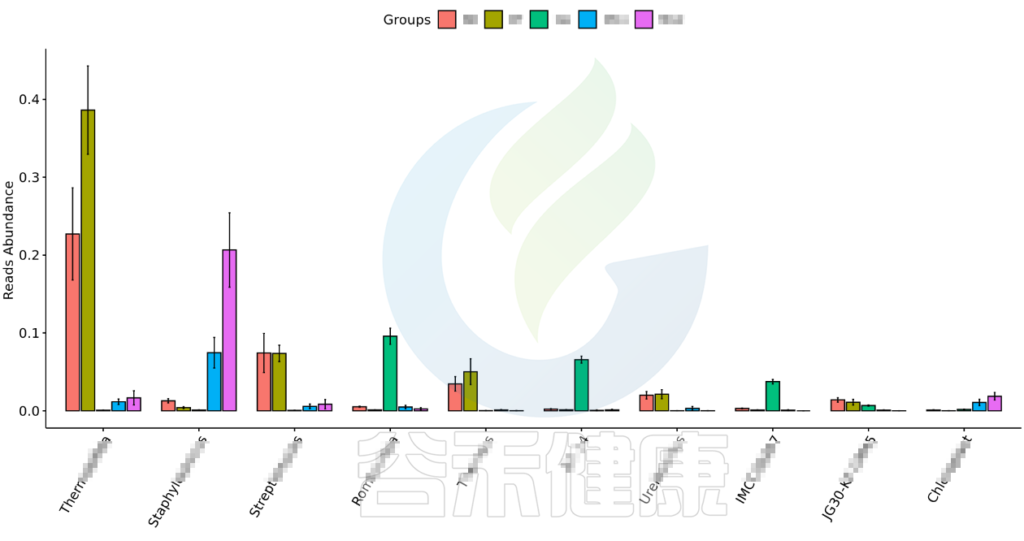
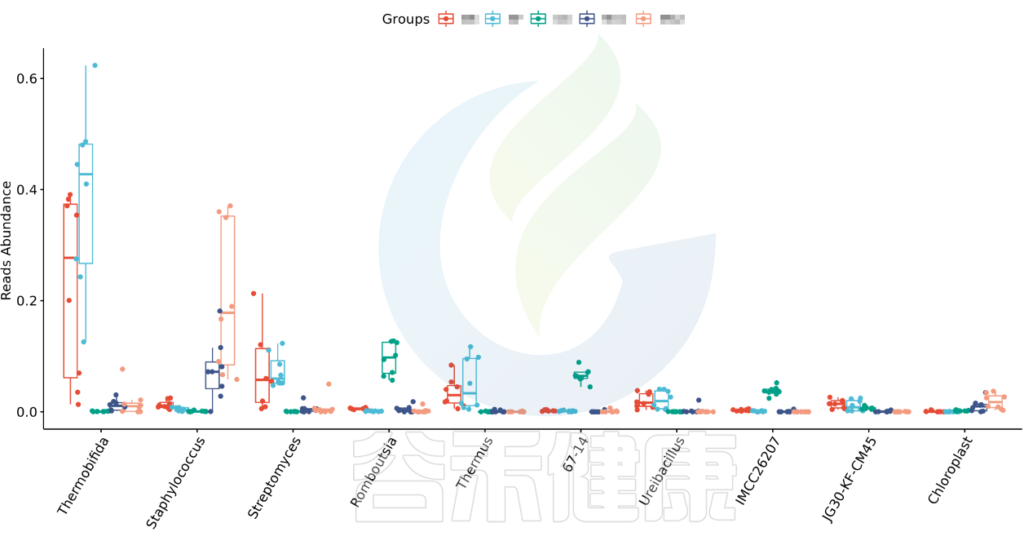
图中显示的是该统计检验差异显著的物种的柱状图或箱型图,按照丰度从高到低排列,如果差异物种/功能较大,则只显示前10个。例如该图中Therobifida、Staphylococcus、Streptomyces等物种用Kruskal-Wallis 检验得到的组间显著差异物种。
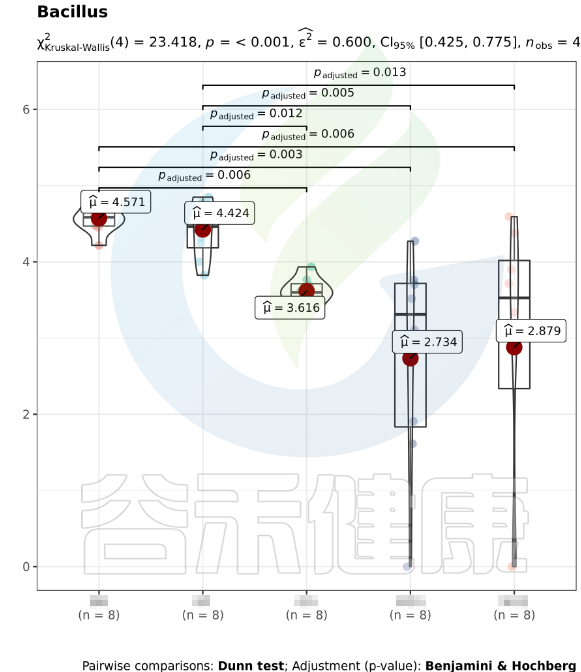
该图展示了Bacillus物种Kruskal-Wallis 检验差异结果,所有分组中P<0,001 多组间差异显著,两组间BB与GG、BB与MG1、BB与MG2、BF与GG、BF与MG1、BF与MG2,组间差异显著。
LEfse分析
LEfse分析即LDA Effect Size分析,是一种用于发现和解释高维度数据生物标识(基因、通路和分类单元等)的分析工具,可以进行两个或多个分组的比较,它强调统计意义和生物相关性,能够在组与组之间寻找具有统计学差异的生物标识(Biomarker)。
LEfSe用到的统计分析方法是将线性判别分析与非参数的Kruskal-Wallis以及Wilcoxon秩和检验相结合。
LEfse分析结果中一般会出现两个图,一张表( LDA值分布柱状图、进化分支图以及特征表)。
LDA值分布柱状图
这个条形图主要为我们展示了LDA score大于预设值的显著差异物种,即具有统计学差异的Biomaker,默认值为2.0(看横坐标,只有LDA值的绝对值大于2才会显示在图中);柱状图的颜色代表各自的分组,长短代表的是LDA score,即不同组间显著差异物种的影响程度。
路径:
Group/Lefse_Analysis/out_formant.cladogram.png
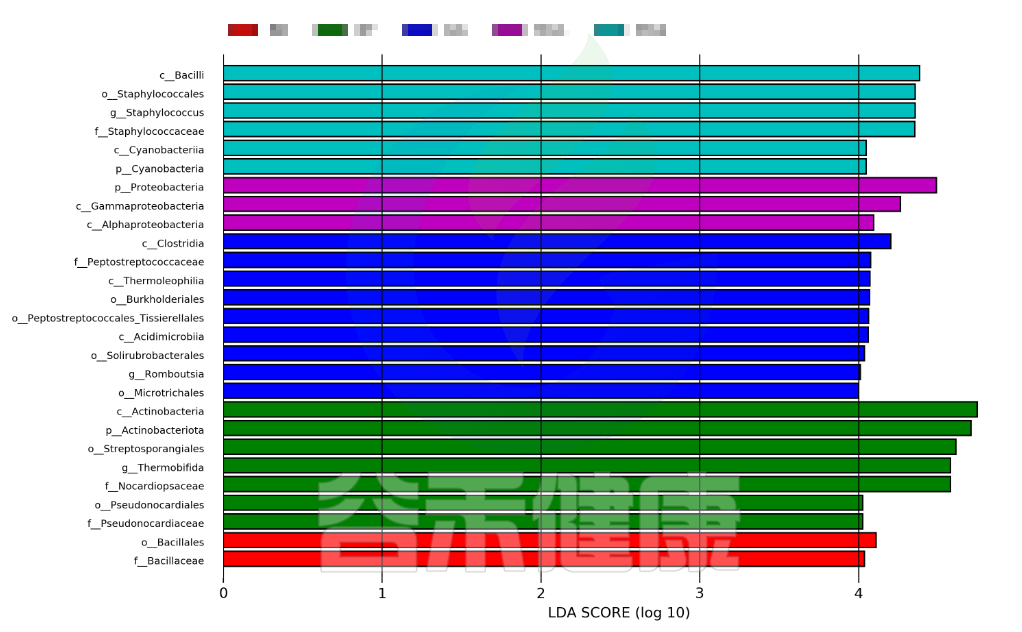
图中展示了不同分组特有的Lefse组间差异标记物,例如BB组的标记物是目水平的Bacillales和科水平的Bacillaceae,不同的分组标记物也不同,图中如果只展示了部分分组,则代表只有部分分组通过Lefse分析筛选出组间差异标记物。
进化分支图
小圆圈: 图中由内至外辐射的圆圈代表了由门至属的分类级别(最里面的那个黄圈圈是界)。不同分类级别上的每一个小圆圈代表该水平下的一个分类,小圆圈的直径大小代表了相对丰度的大小。
颜色: 无显著差异的物种统一着色为黄色,差异显著的物种Biomarker跟随组别进行着色,红色节点表示在红色组别中起到重要作用的微生物类群,蓝色节点表示在蓝色组别中起到重要作用的微生物类群。
未能在图中显示的Biomarker对应的物种名会展示在右侧,字母编号与图中对应(为了美观,右侧默认只显示门到科的差异物种)。
路径:Group/Lefse_Analysis/out_formant.png
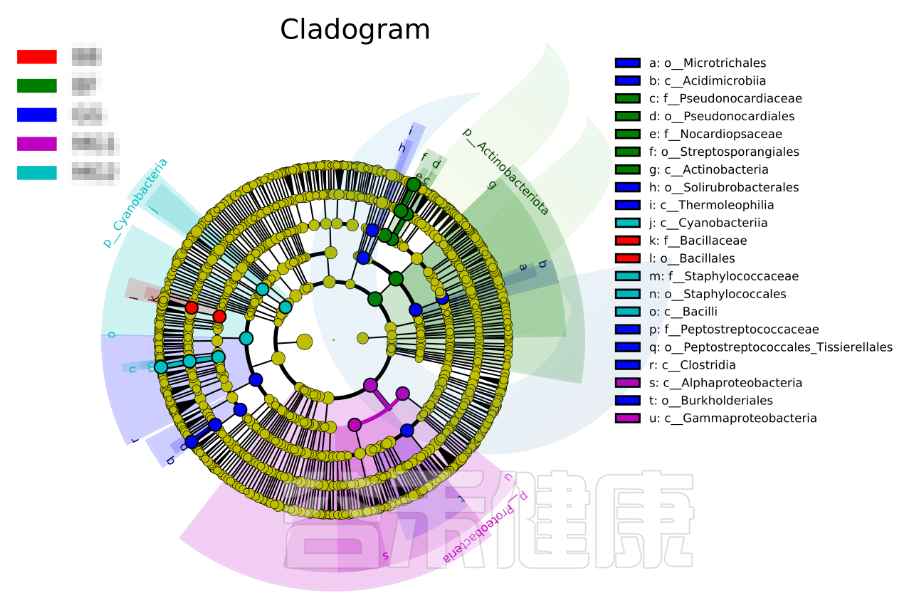
图中右侧展示了分支图中的字母对应的物种信息,例如a 代表GG组的标记物目水平的Microtrichales ,b代表GG组的标记物刚水平的Acidimicrobiia。在分支图的最外层显示的是各分组门水平物种的标记物,例如BF组的是Actinobacteriota、MG1组的是Proteobacteria、
MG2组的是Cyanobacteria
特征表
路径:Group/Lefse_Analysis/out_formant.res.csv
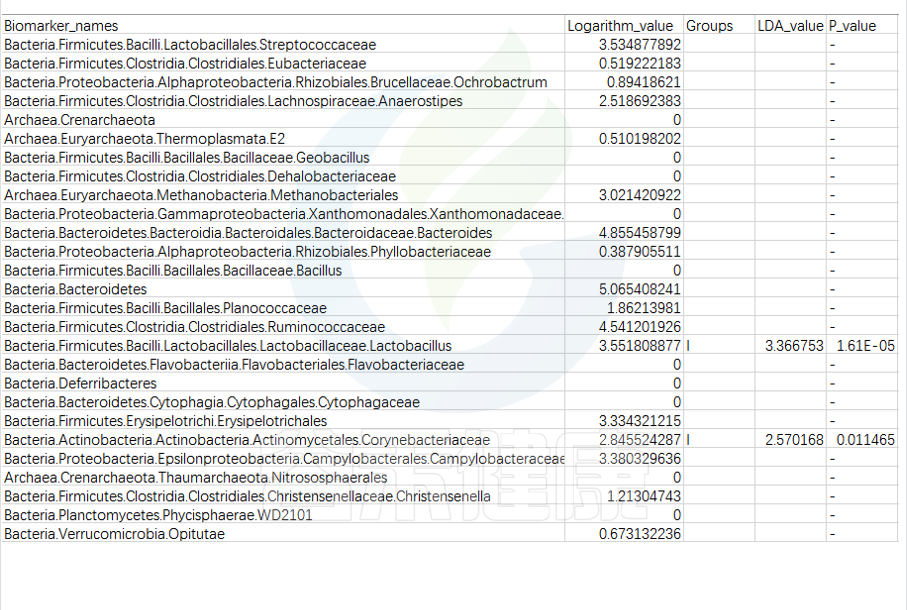
第一列是样本中从门到属水平所有分类单位的列表
Lefse会逐一判断这些分类单位的在分组之间是否具有统计学显著性差异。
第二列:各组分丰度平均值中最大值的log10,如果平均丰度小于10的按照10来计算;如果该分类单位未体现出显著组间差异,则后三列为空。
对于具有统计学差异的分类单位:
第三列:差异基因或物种富集平均丰度最高的分组组名;
第四列:LDA差异分析的对数得分值;
第五列:Kruskal-Wallis秩和检验的p值,若不是Biomarker用“-”表示。
默认LDA>2,P<0.05
通常根据第4列的LDA差异分析对数得分值和第五列的P值,可以描述组间具有显著差异的分类单位统计学效力强弱。
metagenomeSeq
metagenomeSeq是用R开发的一个包,metagenomeSeq的基本思想,用normalization实现分类注释时的biases处理,同时用零膨胀高斯分布(zero-flated Gaussian distribution)处理了测序深度所带来的影响,在此基础上,利用线性模型找到存在的差异所在。
路径:Groups/diff_analysis/ metagenomeRXXX
metagenomeSeq 差异显著物种/功能 热图
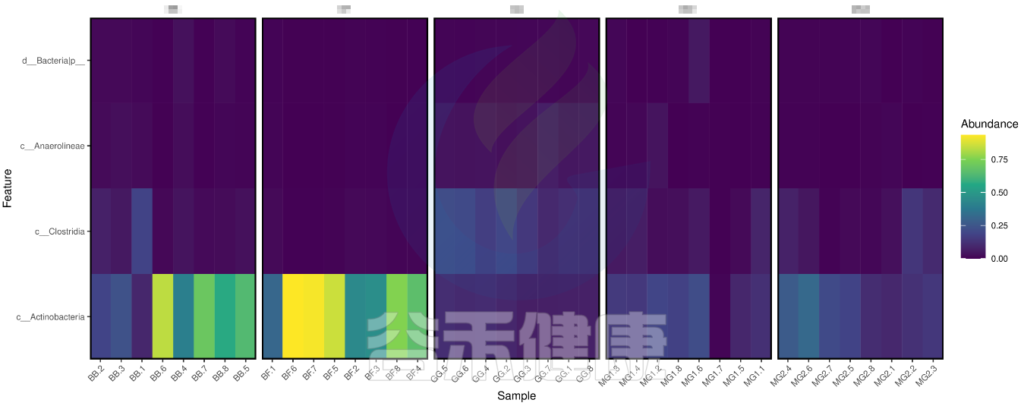
图中颜色越深相关性越小,颜色越接近黄色相关性越大,从图中能看出Actinobacteria物种与BB组和BF组相关性较大。
metagenomeSeq差异菌属于功能代谢关联分析
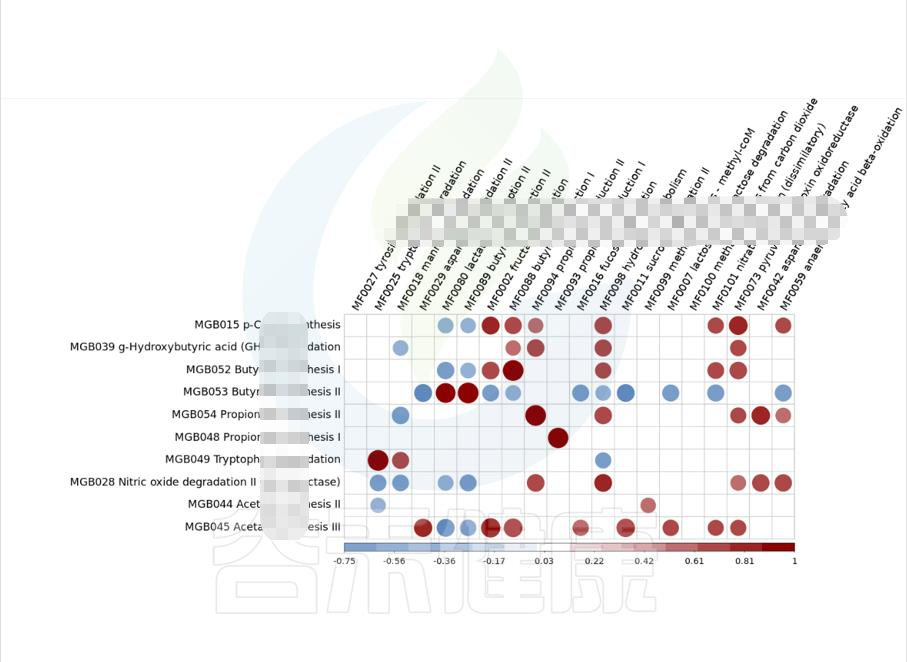
图中红色代表正相关,蓝色代表负相关,颜色越深,圆圈越大,相关性也越大,例如图中能看出MGB049余MF0025 之间成正相关,且相关性较大。
随机森林模型
一种非线性分类器,随机森林属于集成类型的机器学习算法,挖掘变量之间复杂的非线性的相互依赖关系。通过随机森林重要性点图,可以找出分组间差异的关键物种/功能。
反映了分类器中对分类效果起主要作用的特征,按重要性从大到小排列。
Error rate:表示使用下方的特征进行随机森林方法预测分类的错误率,数值越高表示基于特征分类准确度不高,可能分组之间特征不明显。分值越低证明分组效果比较好。
•举个例子
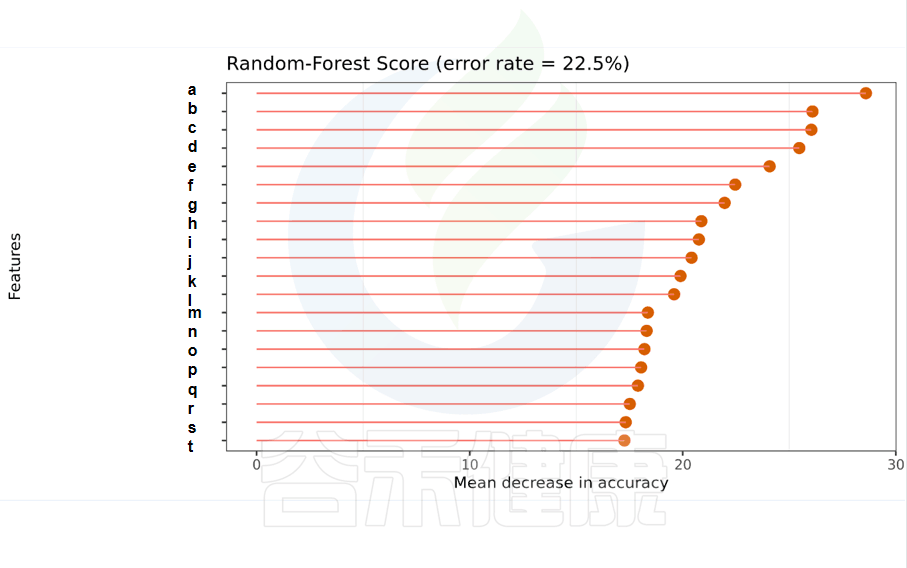
图中按照随机森林模型效果筛选出的对分组效果有重要性作用的物种,按照重要性从高到低进行排列,例如图中最终要的是a,依次往下是b、c等。错误率较小,表明该分组效果较好。
ROC曲线
ROC曲线分析是一种常用的统计学分析方法,在医学研究中主要用于评价诊断试验的效能。在16S测序报告中,我们通过绘制ROC曲线,并计算ROC曲线下面积(AUC),来确定分组对于菌群是否有诊断价值。
ROC曲线图是反映敏感性与特异性之间关系的曲线。ROC曲线下的面积值在1.0和0.5之间。在 AUC>0.5的情况下,AUC越接近于1,说明诊断效果越好。
AUC在0.5~0.7时有较低准确性,AUC在0.7~0.9时有一定准确性,AUC在0.9以上时有较高准确性。AUC=0.5时,说明诊断方法完全不起作用,无诊断价值。AUC<0.5不符合真实情况,在实际中极少出现。
•举个例子
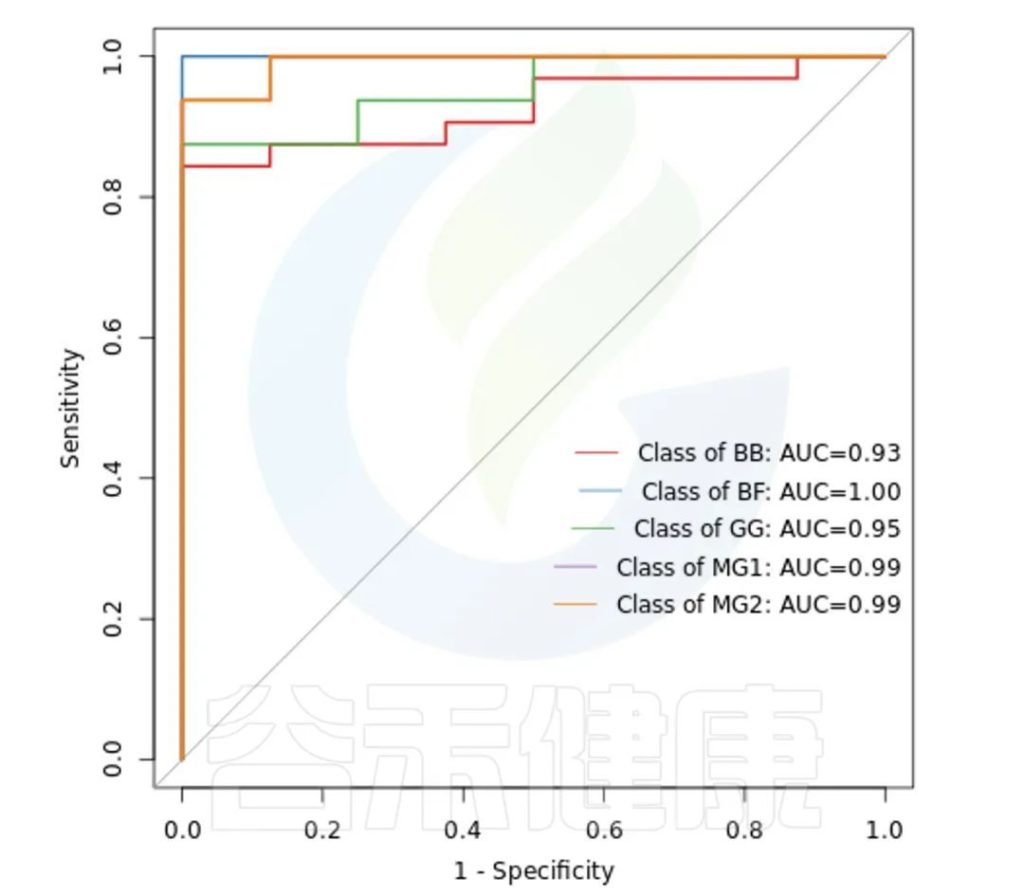
从图中能看出各分组的AUC都大于大于0.9,各分组的分组效果较好,BF组AUC等于1,该分组效果最好,可能样本之间较为相近,并且跟其他分组组间差异也比较大。
以上是组间统计差异的方法介绍,其他的还包括关联分析。
例如客户提供了每个样的相关理化指标数据,想计算这些指标与均属之间有什么相关性,就可以做一下分析。
关联性分析
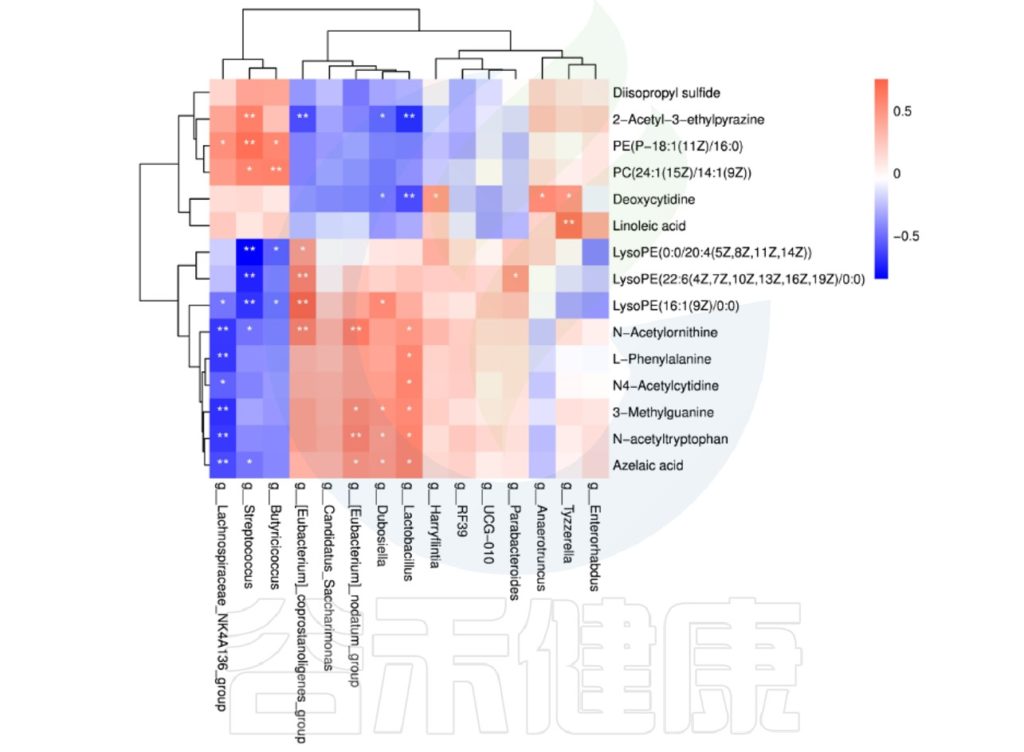
✦相关性热图
图中X轴代表属水平物种,Y轴代表代谢指标,红色代表正相关,蓝色代表负相关,**代表相关极显著P<0.01,* 代表相关性显著P<0.05相关性具有统计学意义。
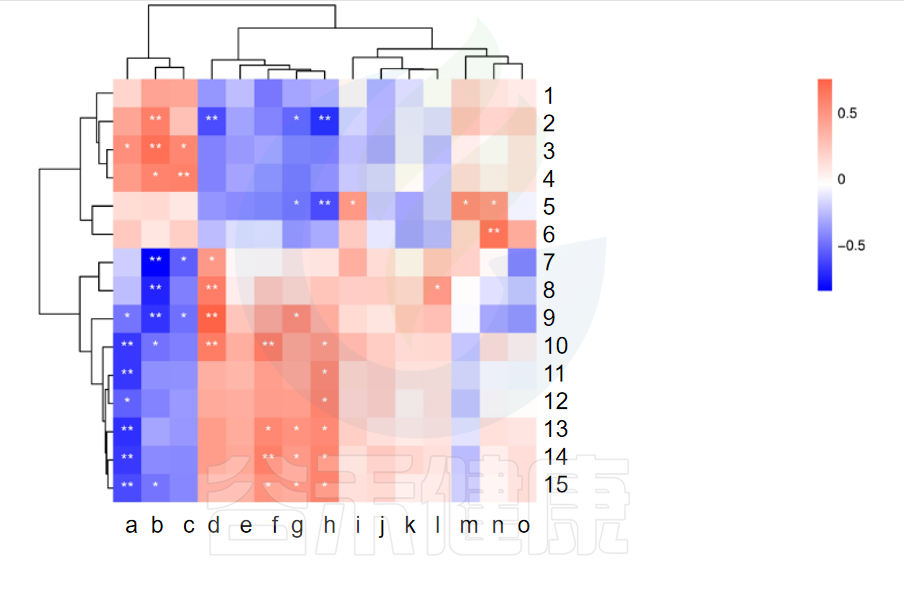
例如从该图中能看出6与n物种成正相关,并且相关性极显著**,7与b物种成负相关,并且相关性极显著**。
可以得到表格:任意菌属和代谢的相关性的值和P值
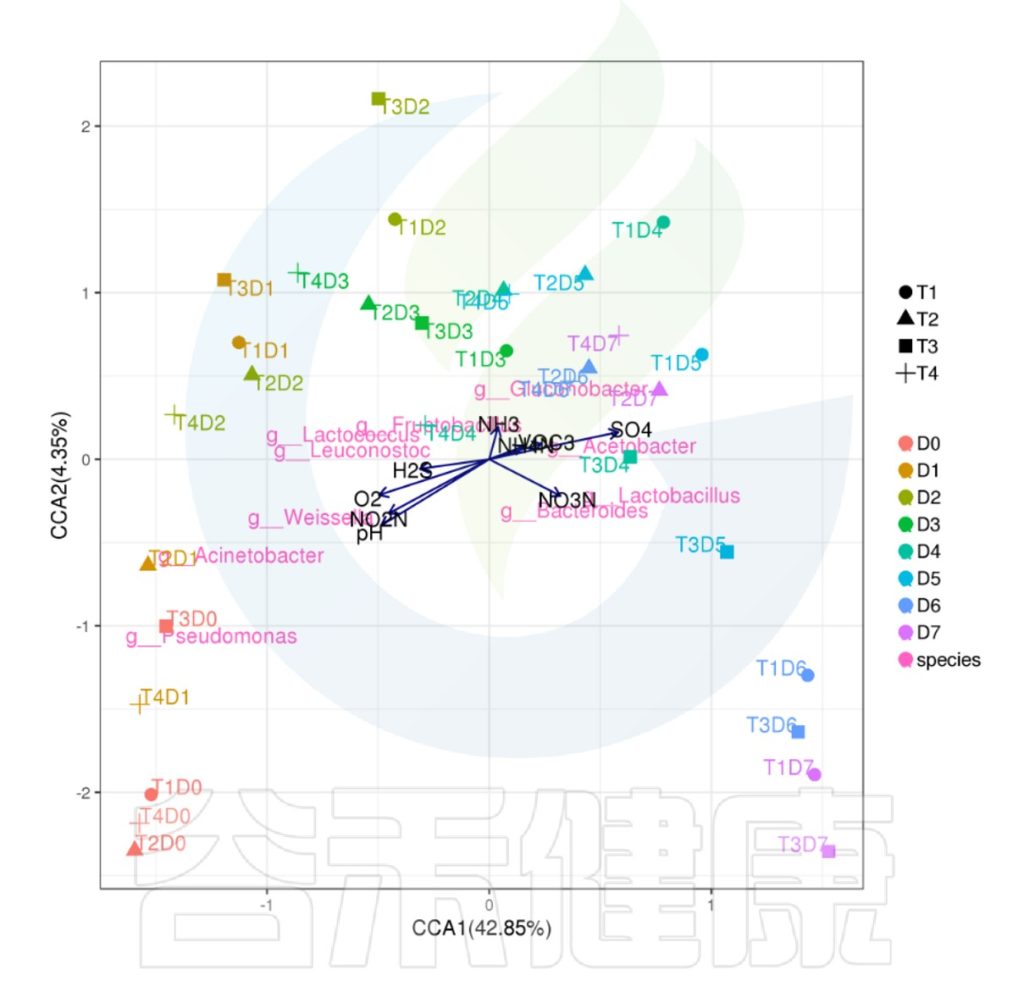
✦CCA图
可以分析样本、菌群、理化指标之间的关联关系。图中使用点代表不同的样本,从原点发出的箭头代表不同的环境因子。
箭头的长度越长,表示环境因子的影响越大;夹角越小,代表相关性越高。样本点与箭头距离越近,该环境因子对样本的作用越强。
图像中坐标轴标签中的数值,代表了坐标轴所代表的环境因子组合对物种群落变化的解释比例。
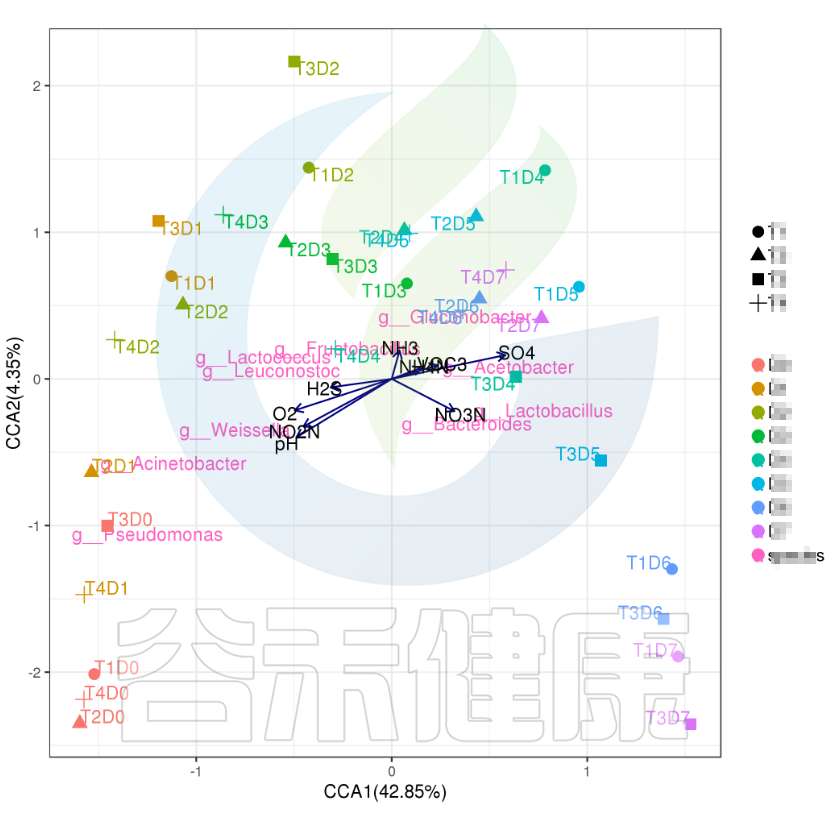
例如从图中能看出pH 、NO2N、02与 Acinetobacter、Weissella等物种成正相关,与T3D0、T1D0、T4D0等D0组的样本成正相关。
✦RDA 冗余分析

例如从图中能看出pH与Helicobacer物种成正相关,相关性较大,pH与NC组有一定的相关性。
✦Envfit分析
回归拟合分析结果:
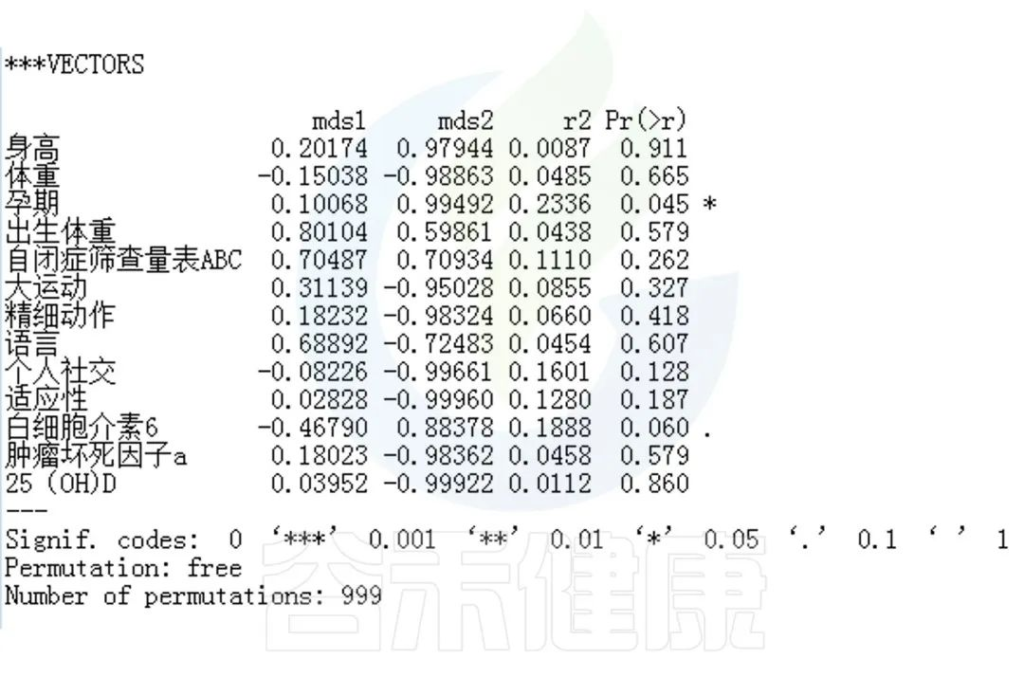
图中能看出ASD与正常儿童之间的分组与菌群之间相关性极显著**,其次是否有先兆流产的分组与菌群之间有显著相关性*,其他的包括是否喝牛奶、孕期是否感染、妊娠高血压都与菌群有相关性。
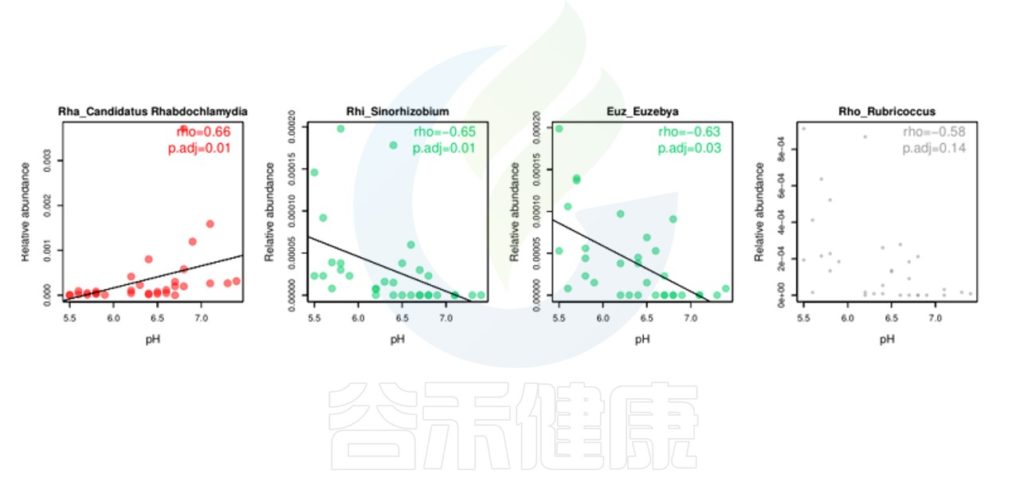
环境因子与功能/物种的相关性线形图P<0.05显著,图中红色点代表正相关,绿色点代表负相关,灰色相关性不显著。
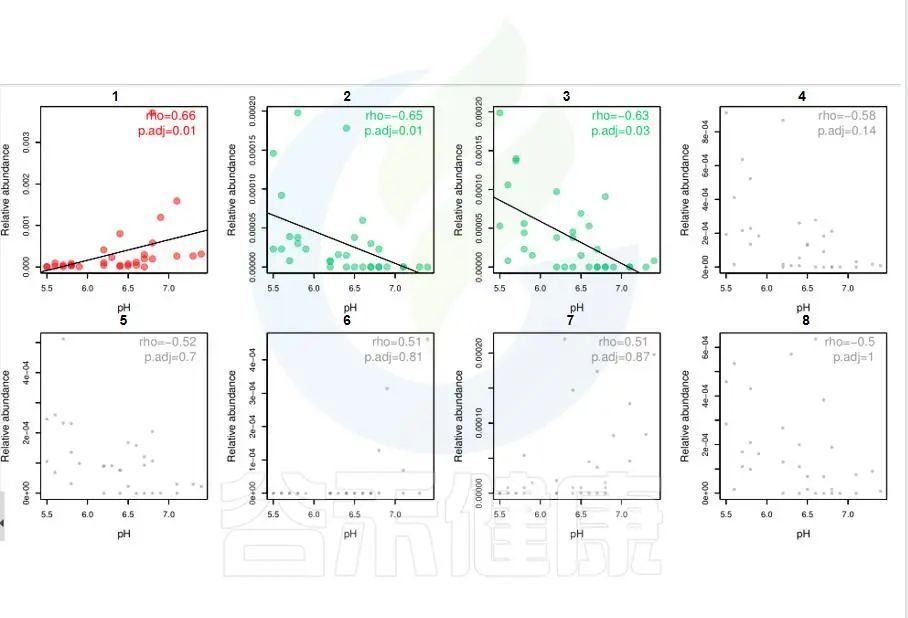
图中能看出pH 与Candidatus Rhabdochlamydia 之间成正相关,且相关性显著,pH 与Sinorhizobium、Euzebya 之间成负相关,切相关性显著。
✦Network网络分析
还可以做菌属之间的网络分析关联图,共发生网络图为研究复杂微生物环境的群落结构和功能提供了新的视角。
由于不同环境下微生物的共发生关系截然不同,通过物种共发生网络图,可以直观看出不同环境因素对微生物适应性的影响,以及某个环境下占互作主导地位的优势物种、互作紧密的物种群,这些优势物种以及物种群往往对维持该环境的微生物群落结构和功能稳定发挥着独特以及重要的作用。
•举个例子
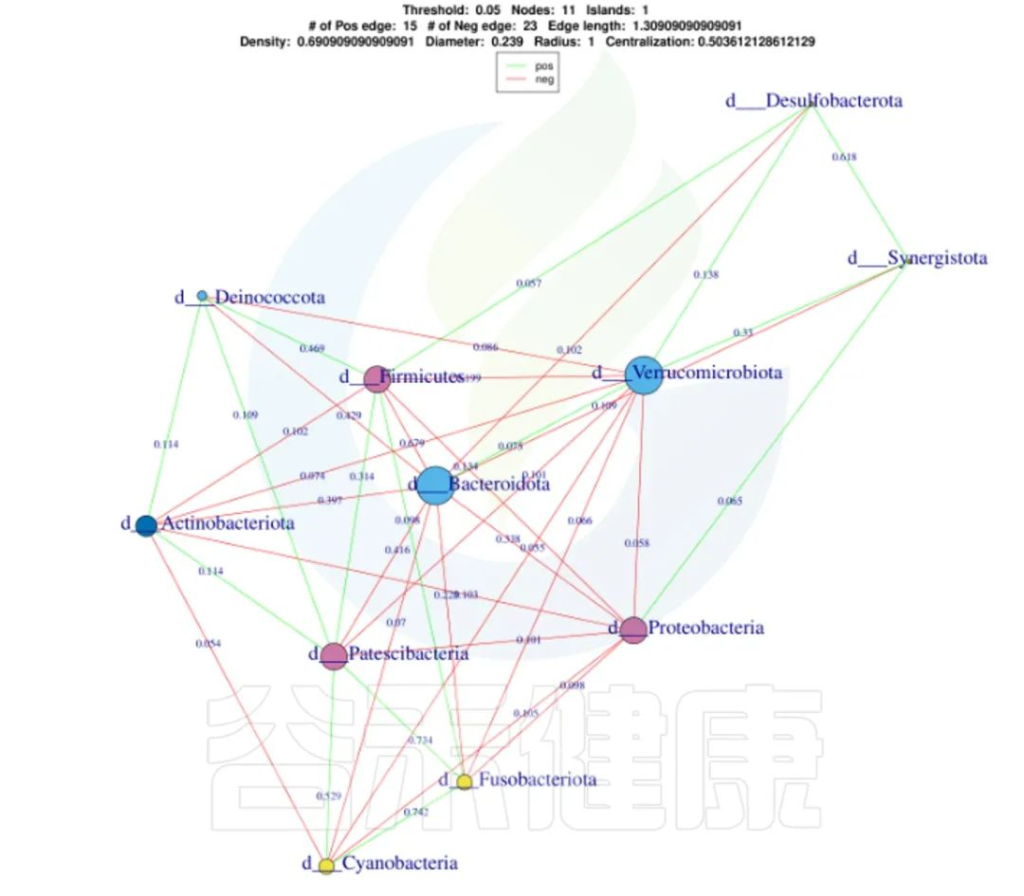
图中展示了相关性的物种,例如Bacteroidota、Actinobacteriota、Proteobacteria 这些物种与其他物种相关较大,图中这些物种与其他物种连线较多,字体比较大也代表相关性较强,例如Actinobacteriota与Deinococcota连线是绿色的代表这两个物种是负相关。
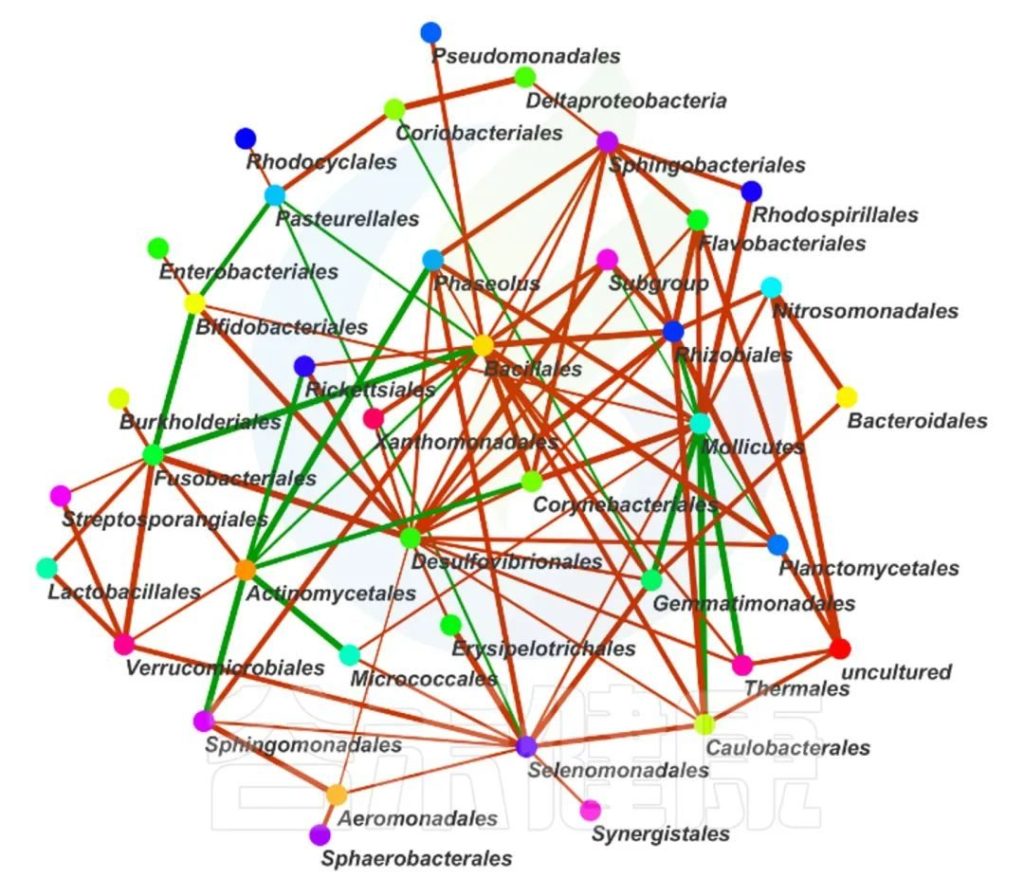
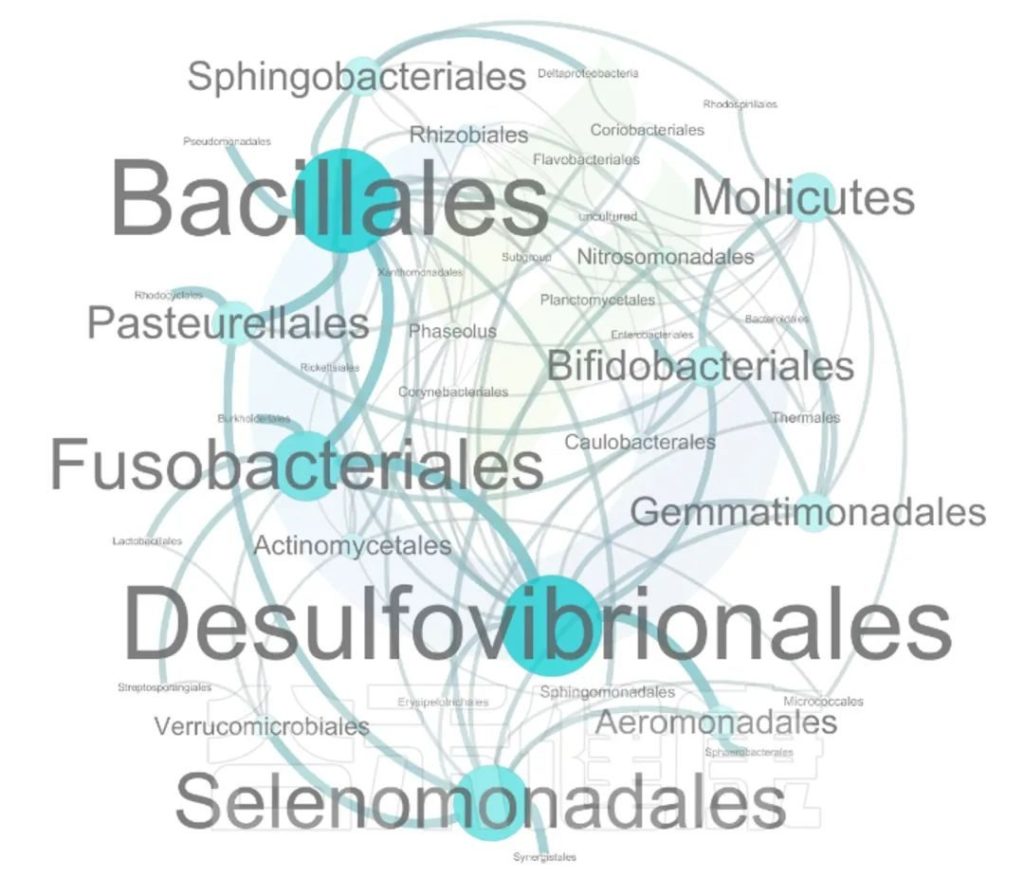
这两个图类似的物种相关性的图,用同一个数据做出来的,图中能看出Bacillales、Desulfovibrionales、Selenomonadales与其他物种相关性较强。
报告中已经基本都涵盖了16S科研数据分析所需要的图表、差异统计,以及相关性分析结果。如果在几种不同类型的统计方法对比之下有略微的差异结果,选取其中一组差异结果即可。
报告里涵盖了大部分16S所需要的图片,不过也有个别个性化的图需要单独用到软件去做,可以单独完成个性化图表生成。
随着16s分析报告的不断升级,报告中的图表以及相应的解读也会越来越精细完善,谷禾也将尽可能为大家的科研之路带来更多便利。


谷禾健康
生物系统——组成
生物系统很复杂,具有许多调节功能,例如DNA,mRNA,蛋白质,代谢物,以及表观遗传功能(例如DNA甲基化和组蛋白翻译后修饰(PTM))。 这些特征中的每一个都可能受到疾病的影响,并引起细胞信号传导级联和表型的改变。 除了宿主对疾病的反应调节机制外,微生物组还可以改变宿主特征的表达,例如它们的基因,蛋白质和/或PTM。
生物系统——疾病
为了深入了解疾病的机制,我们需要研究这些特征及其相互作用。例如,黑色素瘤、肺癌和甲状腺癌等癌症是由BRAF癌基因驱动的。然而,当患者接受抑制BRAF的治疗时,往往会产生耐药性。最近的多组学研究揭示了肿瘤特征的异质性和复杂性,如基因突变、转录组、蛋白质和信号通路。现在人们认识到肿瘤可以绕开治疗而产生耐药性。
生物系统——技术
随着下一代测序和质谱技术的发展,人们越来越需要融合生物特征的能力来研究整个系统。转录组、甲基组、蛋白质组、组蛋白翻译后修饰和微生物组等特征都影响宿主对各种疾病和癌症的反应。由于样品制备步骤、测序所需的材料量和测序深度要求,每个平台都有技术限制。近年来,数据集成方法的发展受到了推动。每种方法都使用诸如概念整合、统计整合、基于模型的整合、网络和路径数据整合等方法来具体整合组学数据的子集。
生物系统——多组学
多组学方法的整合使得对疾病病因学有了更深入的了解,例如:揭示微生物组在减轻或增加疾病风险方面发挥作用的各种方式。双酚A(BPA)是一种大规模生产的化学品,广泛应用于食品包装、塑料和树脂中,双酚A的不完全分解就是一个例子。由于双酚a是一种内分泌干扰物,双酚A已成为日益增长的公共卫生问题。因此,利用微生物手段快速、完全降解双酚A等化合物的研究具有重要意义。
本文讨论每个数据特征的研究设计考虑,基因和蛋白质丰度及其表达率的限制,当前的数据整合方法,以及微生物对基因和蛋白质表达的影响。在开发整合多组学数据的新算法时应考虑的因素。
不同生物基因数量
生物系统是具有多种调控功能的复杂生物。 例如,人类基因组由大约32亿个核苷酸组成,可产生20 000至25 000个蛋白质编码基因,并且通过选择性剪接事件可产生超过100万种蛋白质(下图)。
不同的生物有不同数量的基因和蛋白质。例如,在大肠杆菌、酿酒酵母和智人基因组中分别有大约4300、6000和25 000个基因。这导致大肠杆菌、酿酒酵母和智人的每个细胞中分别有大约2400到7800、15 000和300 000个mRNA分子。线粒体转录物约占多聚腺苷酸化RNA的20%。其他高丰度的转录物包括编码核糖体蛋白质和参与能量代谢的蛋白质的转录物。下图概述了人类DNA、DNA甲基化、组蛋白翻译后修饰、mRNA和蛋白质的复杂性。

Graw et al., 2020 Molecular Omics
染色质结构和基因/蛋白质调控的概述。 DNA通路受DNA甲基化和组蛋白翻译后修饰(PTM)的调控。调节的每一层也可以通过环境和宿主生物中存在的微生物进行修饰。 可以通过使用各种核苷酸和蛋白质/肽测序技术对生物调节的每个水平进行测序。
细胞中蛋白质含量
一个细胞中蛋白质的估计数量约为2.36×106(在大肠杆菌中),约为2.3×109(在晚期智人细胞中)。在一个细胞的全部蛋白质总数中,最丰富的蛋白质可占蛋白质含量的5-10%,由核糖体蛋白、酰基载体蛋白(ACP)(在脂肪酸生物合成中的功能)组成,分子伴侣和折叠催化剂、糖酵解蛋白质(能量和碳代谢的主干)和肌动蛋白等结构蛋白质。
转录因子是一种低丰度的蛋白质,在细菌中每个细胞的拷贝数为1-103,在哺乳动物细胞中为103-106。
最丰富的蛋白质通常在细菌中有数千个拷贝,在哺乳动物细胞中有数百万个拷贝。由转录因子调控的基因数量取决于其浓度。蛋白质含量取决于生长条件和基因诱导。最后,考虑到微生物与宿主细胞数量的比例(取决于宿主细胞类型)和其他因素,这可能会变得更加复杂
mRNA 和蛋白质寿命以及差异
由于仪器检测,动态范围和分子寿命表达的限制,用于各种组学平台的测序技术只能捕获某一时刻某个细胞群体中发生的情况的快照。 例如,mRNA转录本和蛋白质的终生表达差异很大。 在大肠杆菌中,mRNA的中位寿命为5分钟,在发芽酵母中为20分钟,而对于人参则为600分钟。然而,蛋白质的寿命约为1-2天。
转录和翻译的速率因生物体的不同而不同(大肠杆菌:每秒10-100个核苷酸(nt)和10-20个氨基酸(aa)/ s。智人:6-70 nt / s和2 aa per s;分别为转录和翻译速率)。
对于大肠杆菌来说,一个单一的mRNA转录本在被降解之前可以产生10-100个蛋白质。鉴于这一信息,我们可以看到,将我们对组学平台的选择和由此产生的对细胞过程的解释相结合,检测具有更长寿命的蛋白质的机会将增加。
在考虑数据整合研究设计、开发新算法和解释结果时,认识生物体的生物复杂性、分子的动态范围、测序限制以及这些分子的表达寿命非常重要。
近年来,微生物组学在宿主健康中的重要性已得到公认。全生物和全基因组的概念对我们如何看待微生物组有着深远的影响,尤其是在治疗方面。这种微生物-宿主相互作用的密切关系可以更明确地称为“微生物群-营养代谢-宿主表观遗传轴”。微生物与宿主相互作用的紧密关系可以更明确地称为“微生物群-营养代谢-宿主表观遗传轴”。
微生物群及其代谢产物可以通过直接修饰组蛋白,改变DNA甲基化谱图和影响而影响宿主表观遗传。 非编码RNA的性质(上图)。 例如,可以通过改变组蛋白修饰酶的活性和酶底物的水平,通过微生物群来修饰组蛋白。
微生物影响药效
微生物群也可以影响药物的治疗性质。 许多前药,即必须进行代谢转化才能在药理上有用的药物,可能会保持无活性(即不存在介导前药向其活性形式转化的微生物群),或者该药物/前药可能无法生物利用。此外,服用NSAIDs(非甾体类抗炎药)的患者可能会促进抗生素耐药菌的优势,因为24%的非处方非处方NSAIDs被抑制。
这些代谢组学效应引起人们对旨在用于人类和农业系统的治疗药物或其他饮食和治疗方案的潜在副作用的担忧。 例如,抗生素可以消除产生组蛋白脱乙酰基酶(HDAC)抑制剂的微生物。 这些微生物(如果存在)可以增强调节性T(Treg)细胞,从而有助于抗炎过程。
微生物代谢途径的多样性及其对药物药代动力学和药效学的影响可能部分解释了个体和人群之间药物反应的变化。 因此,涉及微生物组的治疗方法可能必须因地制宜。组蛋白可以同时进行变体置换和翻译后修饰(PTM),这些共同构成了“组蛋白密码”。 这些局部排列可以影响染色质结构,从而导致转录活性的激活或抑制。
通过饮食,微生物有能力改变宿主的甲基化和PTM谱,并且还可以通过膳食碳水化合物的发酵影响短链脂肪酸(SCFA)的生成。丁酸盐和乙酸盐等SCFAs可抑制脱乙酰酶水平。这意味着由于乙酰化促进转录活性的增加,染色质结构变得越来越松弛。事实上,已经证明微生物可以以位点特异性和组合方式影响宿主组织乙酰化和甲基化染色质状态,甚至影响宿主发育和代谢表型。
微生物参与干预
未来关注健康医疗策略时,越来越多地考虑对微生物组的发展及其相应的宿主个体发育变化进行建模。考虑到宿主免疫系统不仅必须能够识别“自身”抗原,而且还必须能够识别共生微生物的抗原,这些变化可以通过宿主免疫成熟来证明。
微生物如何影响主要组织相容性复合体(MHC)的表达,或者宿主杂合度如何通过MHC影响微生物群的多样性,这在很大程度上是未知的,也是一个活跃的研究领域。微生物在癌症和免疫治疗中的作用正日益成为治疗策略发展的目标。蛋白质组学与其他组学策略相结合已被用于研究疾病过程。如果我们不考虑微生物群的影响,那么我们可能会错过开发潜在治疗方法的有意义的见解。尤其是那些与代谢紊乱(如肥胖)或代谢物(如胆汁酸)对器官系统的全身影响有关的疾病。
微生物生态学的历史围绕着适当的系统发生标记基因的测序和比对。 WoSes and Fox(1977)首先将16S rRNA基因用作标记基因,是迄今为止最常用的标记基因,其大规模数据库包含从环境和培养来源(例如SILVA,RDP,Greengenes)获得的全长基因分离株 )(表1)。 新的微生物分类数据库,例如基因组分类数据库(GTDB),不仅建立了16S rRNA基因参考数据库,而且还利用系统基因组学信息提供了一个一致的框架,用于确定从元基因组获得的系统发育背景部分或完整基因组 。
大数据集的可用资源列表

选择合适的引物和平台
除了选择标记基因和合适的数据库外,研究人员还可以在测序方法和平台之间进行选择。 由于Illumina和Ion Torrent等短读平台的局限性,研究人员必须在〜1500 bp的16S rRNA基因的可变区之间进行选择。 取决于微生物群落组成,每个可变区提供不同水平的敏感性和特异性。 然后选择在研究中最能区分普通分类群的引物组和扩增子区域的组合。
目前针对扩增子测序可选择的测序平台和方案很多,不同平台的读长和适用的测序区段以及优势各有不同。16s测序主要的测序区段包括v4、v3v4,v1v2,v6,此外还有全长等不同的区段选择,不同可变区或全长由于引物的不同以及不同种属相应区段内的变异多样性差异,对菌属的丰度评估会有一定的差异。
从长度来看,全长16s长度为1.5kb左右,单菌落的16s全长sanger一代测序仍然是菌种鉴定的主要手段,纳米孔和pacbio的三代测序可以高通量的获得全长序列,对于希望更高分辨率的分析菌种的研究有一定优势。三代的测序准确度目前逐渐改进,直接测序准确度可以在90%以上,纠错后可以提高到97~99%以上,已足够提供高精度的分类。三代目前主要问题在于建库成本相对较高,通过使用barcode可以降低部分但仍然偏高,此外普遍测序深度相对于二代测序要低许多。
目前最主要的可变区选择是v4区和v3v4区,v4区长度为256bp左右,加上两侧引物长度为290bp左右,使用双端2x250bp或2x150bp可以测通,此外如454、life、illumina的测序平台读长也可以主要涵盖该区段读长。例如采用illumina Novaseq测序平台对该项目进行双端测序(paired-end),测序得到了fastq格式的原始数据(样本对应一对序列s_1.fastq和s_2.fastq)。再配对拼接成单条序列。其引物通用性相对是所有可变区中最高的,大量的大规模菌群调查研究都采用v4区作为检测区域,包括人体菌群研究如:hmp,肠道菌群如美国肠道计划agp,欧洲的fgfp等,以及全球土壤菌群调查,目前仍然是国际研究中使用最广泛和认可的检测区域。
illumina的miseq提供了长达2x300bp以及hiseq2500和最近的novoseq提供有2x250bp的测序方案,为进一步利用读长,目前有相当一部分研究选择v3v4区,该区段长度在460bp左右,相较于v4度多出了v3区段约100bp左右的片段,在少部分菌属中可以增加一定分辨率。经过对比,v3v4区的检测结果和v4区在绝大部分菌属中的丰度一致,但由于引物不同,在少量菌属中丰度会有不同偏向,v3v4从otu层面上并未发现较v4区有明显增加。引物的选择和提取、储存方法是影响菌群检测丰度构成的主要因素,不同研究之间的比较需要考虑到实验方案的一致,相同的方案可以直接比较。
当前的宏基因组分析技术已使研究人员能够从环境/宿主来源的样品中获得足够的序列覆盖率,从而获得部分和完整的基因组草图。 该覆盖因子高度取决于物种的均匀度和丰富度。 还可以通过拼接组装元基因组组。 但是,由于难以组装和正确分装高度保守的基因(如核糖体亚基基因),因此它们通常必须使用浓缩的通用蛋白将这些基因组置于系统发育背景中。
组合的通用标记基因被用来构建由环境和寄主衍生序列组成的基因组以及来自培养物收集的少数常见微生物基因组的系统发育。微生物基因组测序的热潮使得有必要构建易于使用的软件包以及分析工具,以帮助生物学家学习如何对其全部或部分的元基因组数据进行分析。这类工具的例子如,QIIME 2、metaWRAP、 Sunbeam、SqueezeMeta、metAMOS、 mg RAST、IMG/M、 Anvi’o、MicrobiomeAnalyst、以及biobakery集合中的各种工具(例如MetaPhlan2、PhyloPhlan、HUMAnN、LEfSe)等。
此外,如果深入研究,还需要望整合疾病指标、宿主蛋白质组学和微生物多样性多组学的联合分析。
根据生物学问题的不同,有许多类型的组学技术,针对DNA、总RNA、mRNA、miRNA、DNA甲基化、蛋白质、蛋白质修饰、组蛋白翻译后修饰、宏基因组学、宏蛋白质组学,测序平台经过多年的改进,现在可以在几天内从少量材料中对大型复杂人体样本进行测序(表2)。已经开发了几种工作流程来对整个基因组、整个外显子组(DNA的蛋白质编码部分)和转录组(mRNA)进行排序,并对特定的癌症或免疫相关基因进行排列。此外,还可以利用亚硫酸氢盐全基因组测序或Illumina的甲基化珠芯片阵列分析修饰,如DNA甲基化。 还可以通过牛津纳米孔技术(ONT)MinION平台和PacBio仪器对长读的DNA和RNA进行直接测序来确定此类修饰的检测。
推荐覆盖率和读数

基因组测序
DNA测序技术的错误率和读取长度各不相同。Illumina短读测序(即Hiseq、Miniseq等)通常具有非常低的错误率,约为每碱基0.25%,但对低多样性文库敏感,如16S宏基因组学和靶向基因方法等应用。长读取技术的错误率较高,PacBio为13–15%,Oxford Nanopore instruments为5–20%。Illumina平台的读取长度最大为600个碱基,但长读取技术通常一次读取可达到10–30 kb。最佳读取长度也取决于应用程序。
大多数测序实验可以收集150-300碱基对读取长度的合适信息,但也有例外。对于全基因组测序(WGS),最长的读取可能是最佳的,但是对于长读取技术,错误率随着长度的增加而增加。有许多研究者把“短读”和“长读”结合起来。由于最近长读取排序技术的出现,关于WGS以外应用程序的最佳长读取长度的信息非常缺乏,但Illumina short read sequencing提供了丰富的最佳读取长度建议。
蛋白质测序
在过去5-10年中,质谱仪通过增加测序深度能力也得到了改进。这项技术已经从使用旧的LTQ质谱仪在细胞系实验中对大约3000个蛋白质进行测序,发展到使用新的Orbitrap Lumos和Orbitrap Eclipse质谱仪对8000-10000个蛋白质进行常规测序。大多数蛋白质组学实验都是采用数据相关采集(DDA)模式进行的。在该方法中,选择从液相色谱(LC)柱洗脱的MS1扫描中最丰富的前20个肽在orbitrap中进行裂解,以产生肽序列MS2扫描。样品混合物的复杂性极大地影响了测序深度和将鉴定多少蛋白质。了解样品的蛋白质丰度和组成是至关重要的。如果转录因子是目标分子,那么在质谱分析之前去除高丰度蛋白质的方法可能是必要的。这对于含有大量分子(如白蛋白和血红蛋白)的血清和血浆样品尤其重要。否则,质谱仪将测序数千个白蛋白分子,并错过最有趣的低丰度蛋白质。
最新的质谱技术利用数据独立采集(DIA)来对MS1扫描中所有肽从LC色谱柱洗脱时的序列进行测序,这与仅对最丰富的峰进行测序的DDA方法相反。
对于复杂的混合物,例如上面的血清示例,DIA方法优于DDA。 这种方法有助于克服受高丰度蛋白质高度影响的复杂混合物。
除了对宿主基因和/或蛋白质进行鸟枪法测序外,我们还可以对微生物组利用鸟枪法测序。
当测序深度很浅时,弹枪宏基因组学/元代谢组学只能采样优势菌群。shot弹枪对微生物组测序的主要挑战是由于采样不足而难以组装基因组片段,将肽组装在一起以进行可靠的蛋白质和生物分类鉴定也同样困难。
尽管存在这些潜在问题,但从各种人体部位和疾病(如唾液、肠道/粪便、颈阴道疾病或慢性肾脏疾病)中对微生物蛋白质组进行深度取样是可能的。然而,每个研究必须考虑的研究/取样设计和分析方法可能有很大差异。
从差速离心到双过滤差速分离,几种样品制备方法已被证明能富集微生物生物量。这些方法通常遵循各种优化的微生物裂解方案,通常涉及机械破坏(如打珠、超声波),辅以酶(如胰蛋白酶)和洗涤剂。在成功溶解后,同样重要的是去除残留的酶、洗涤剂和盐。
元蛋白质组学实验的另一个复杂性是由于同一生物体内的蛋白质具有共享的肽序列这一事实。 为了对蛋白质鉴定有信心,应以高可信度鉴定蛋白质的独特肽段匹配。 当将肽序列映射到数百个具有保守蛋白序列的不同物种时,这变得更加复杂。 质谱法不对蛋白质进行测序,而是测量肽的电荷,并依靠与蛋白质序列数据库匹配的质谱进行蛋白质鉴定。
精心挑选的数据库对于正确分析从这些各种测序平台生成的核苷酸和蛋白质测序数据至关重要。 使读数与参考基因组比对的能力仅与参考基因组中存在的序列和注释信息一样好。 有几种资源可以不断地整理和更新核苷酸序列信息和注释,包括加利福尼亚大学圣克鲁斯分校(UCSC)基因组学研究所基因组,美国国家生物技术信息中心(NCBI)GenBank和RefSeq,DNA元素百科全书(ENCODE)和Ensembl 仅举几例。 通用蛋白质资源(UniProt)包含Swiss-Prot(手动注释和审阅)和TrEMBL(自动注释且未审阅)数据库,以获取蛋白质序列信息。
已经开发了几种数据集成方法来集成某些类型的组学数据。 另外,已经创建了大数据存储库来存储来自各种疾病的测序实验的数据。 这些资源提供了有价值的构建基块和大量生物样本,可用于推动数据集成方法的发展。 当前,数据集成工具实现了多种方法,但通常分为两类:多阶段分析和元维度分析。
多阶段集成模型仅使用数据的两个数字或分类特征构建。 例如,将来自RNA-seq实验的基因计数与来自质谱运行的蛋白质信息相结合。元维度分析试图通过级联或转换将所有感兴趣的数据类型合并到可以同时分析的同时矩阵或“元数据”集中。
后一种方法具有更大的统计能力,但在尝试合并来自不同类型数据集的数据时可能会具有挑战性。 但是,研究人员如何确定最合适的工具或方法?
如上所述,生物学问题是选择的分析方法类型的驱动力,诸如采样,平台类型和数据质量等因素很重要。 样品如何收集和准备?
如果测序深度或质量较低,是否可以有效分析数据? 数据类型兼容吗?
归一化和滤波后损失了多少信号?
这些都是在选择适当工具之前应考虑的所有问题。
不幸的是,数据集成和分析非常复杂,并且对于具有有限生物信息学背景的研究人员而言,目前还没有许多用户友好的工具。 许多工具使用统计语言R,除了强大的生物统计知识外,它还需要专业编程知识。 例如,将蛋白质组学,转录组学和途径分析结合到两个数据集上的R包积分学使用了相关分析和偏最小二乘回归。R包mixOmics使用多元分析进行数据探索,降维和可视化。 通过途径分析,iClusterplus和LRACluster进行的miRNA和基因表达使用聚类来整合甲基化和基因表达数据。
多组学数据集成工具



对于多状态和多维方法,都使用了许多不同的算法,但最常见的算法是聚类,网络分析,数据约简(PCA)和贝叶斯分析。Ray等2014年使用贝叶斯分析,使用从癌症基因组图谱项目收集的数据分析卵巢癌中的基因表达和甲基化数据,并检测到一个基因SPON1,该基因似乎受其CpG位点的甲基化调控。当缺乏生化相互作用的先验知识时,基于相关性的分析是有用的。无论采用何种方法,适当的规范化和数据过滤是非常重要的,因为数据来自多个来源。
还有一些基于网络的工具,如Paintomics,试图使数据分析更容易,但对于缺乏经验的用户来说仍然很困难,研究人员必须对他们的数据有很好的工作知识。此外,还有一些数据库常用于综合组学分析,如癌症细胞系百科全书(CCLE)、癌症基因组图谱计划(TCGA)、与基因组学驱动治疗相关的肿瘤改变(TARGET)和组学发现指数(OmicsDI)。CCLE和TCGA已经描述了数以千计的癌症数据集,可以用于数据挖掘和可视化。TARGET利用临床信息并在其网站上提供分析工具的资源。omicdi提供了一个平台,用于搜索各种生物的公共和受保护数据。
对于任何高质量的研究,进行多组学研究应该首先确定研究的范围和限制。仔细的计划和执行将提高研究的稳健性和可重复性,在多组学研究中尤其重要,因为它们涉及大量的比较、定制的统计分析、大量的财力,时间和精力。一旦研究假设被明确定义,选择一个合适的研究设计,最好地解决研究假设。因此,有几个问题需要评估,
例如:是否有一个或多个干预组与对照组(或其自身)进行比较,或者是否在干预前后对同一样本的效果进行评估?
干预效应是在一段时间内产生的,还是在几个不同的时间点测量样本?
生物样本是否会被单独收集或分析?它的科学依据是什么?
哪些类型的组学平台将提供最有价值的以及如何整合多组学数据?
来自同一生物来源的样本是否可用于所有感兴趣的多组学平台?
理想情况下,所有omic平台的样本将从同一来源收集。
然而,由于样品的特殊限制或材料的可及性和数量,这并不总是可能的,从福尔马林固定石蜡包埋(FFPE)组织生成多组学数据对于某些组学平台可能是不可能的。虽然在选择实验设计时有许多问题需要考虑,但选择研究设计的决定因素通常是其可行性和经费限制。
样品和数据的收集应以数据分析为指导,以减少混淆和技术因素,例如批量效应。这些效应可以在样品收集,制备和存储的步骤中引入。
由于与多组学研究相关的数据的复杂性和大量数据,因此针对特定的研究项目量身定制统计分析至关重要。已提出了多种集成多组学数据的方法,并将其归类为受监督的, 半监督或非监督; 以及基于概念,统计,相关性,网络和模型的集成。
一项研究的统计能力取决于几个因素(下图),其中一些因素可以控制,而另一些因素由于研究及其设计而固定。首先,选择了用于分析的统计方法。虽然有些测试比其他测试更强大,但重要的是验证和满足他们的假设。
影响研究统计能力的另一个因素是单个组学平台测量的变量数量,通常由组学平台决定。例如,基因组学通常测量数百万个变体,转录组学量化了成千上万个分子,和蛋白质组学和代谢组学分析了数千个分子。此外,统计效力受表型或处理效应的大小和差异程度(效应大小)的影响。效果有多明显?组间的信号差异有多大?有多少被测变量受到影响?关于效应大小的信息可以从以前的文献或专家知识中获得,但通常是未知的。
这种情况下,初步研究可以帮助估计效应大小,但由于不稳定,这些估计需要谨慎处理。另一个效力影响因素是测量值的均匀性,描述了样品的自然方差、测量仪器的精度和检测限。随着方差的增大,统计效力将减小。样本的方差可能是多方面的结果,例如样本群体的选择、组织类型的选择或混杂因素。
除了样本方差膨胀外,混杂因素也会在数据中引入偏差,因此,收集样本元数据以减轻某些混淆的影响是很重要的。由于影响研究统计能力的大多数因素是固定的或由研究设计决定的,因此最常用于调整研究统计能力的因素是样本量。

多组学研究中影响统计功效的因素
Graw et al., 2020 Molecular Omics
研究的首要考虑正在调查的疾病或研究问题的背景,以及整合在一起时,什么类型的数据将提供有价值的见解。根据生物学问题、材料类型(新鲜组织、FFPE组织、血清/血浆和细胞系)、DNA/RNA/蛋白质的数量、生物复制的数量以及研究中混杂效应的数量,这些因素将决定数据采集所需的最佳样品制备和测序方法。
样品制备方法,包括每个样品制备的日期、提取的DNA、RNA和/或蛋白质的类型、基因组学的文库生成、质谱分析的蛋白质消化和肽标记方法以及测序平台/仪器,都是研究设计和最终结果解释的关键因素结果。
如果一个样本是在不同的日期制备的,而不是其他生物复制品,这将引入方差和/或偏差,并降低分析的统计能力。如果蛋白质组样品使用多个TMT-10plex批次进行复合,这将在整个测序过程中引入批次效应。这些因素应在样品制备前进行讨论。
同样重要的是要知道什么样的调控特征被捕获用于测序和整合。例如,如果在进行质谱分析之前在样品制备过程中膜蛋白没有溶解,那么膜结合蛋白就不能与基因表达数据整合。质谱数据的一个警告是,缺失值并不一定意味着蛋白质没有表达,只是蛋白质低于质谱仪的检测限。生物学问题应该成为多组学数据整合方法的驱动力。
在大多数情况下,当前的工具利用聚类、网络、数据简化和贝叶斯分析。随着数据获取量的不断增加,产生了大量的数据集,使得机器学习对于有效的分析和数据挖掘变得越来越必要。有必要使用易于获取和记录良好的方法、工具和算法。
机器学习在允许科学家集成多组学数据集方面发挥了越来越重要的作用。通过利用机器在大量生物数据中比较和识别模式的能力,可以用更加准确和有效的方法来阐明复杂的细胞机制,在某些情况下还可以预测临床结果。这是通过计算机独特的能力来实现的,它可以同时观察多个层次的组学数据,从而提供一个更全面的系统视图。
尽管多组学数据集可以为个体提供更深入的理解,但这并非没有成本。组学研究通常依赖于大量的比较、正确的数据类型、适当的统计分析以及大量的时间、技术人员和金钱投入。在构建一个实验时,人们必须清楚什么类型的组学数据可以而且应该被整合以获得对所研究系统的最大理解。
高通量的组学平台并不总是回答研究问题所必需的。传统技术:如酶联免疫吸附试验(ELISA)、免疫组织化学(IHC)和定量聚合酶链反应(qPCR),也是验证特定生物学机制所必需的。事实上,为了验证从组学数据中鉴定出的重要分子是一个真正的阳性结果,通常需要这些技术来验证一个更大的组学研究的结果。
但是每种方法都受到其统计能力、样本量、技术变量、批次效应、测序深度、样本制备和许多其他因素的限制。在设计、进行和分析研究以及解释研究结果时,必须牢记这些因素。因此,如果允许,建议研究设计一开始就让生物统计学家/生物信息学家参与进来。
参考文献:
Graw S, Chappell K, Washam CL, Gies A, Bird J, Robeson MS 2nd, Byrum SD. Multi-omics data integration considerations and study design for biological systems and disease. Mol Omics. 2020 Dec 21. doi: 10.1039/d0mo00041h. Epub ahead of print. PMID: 33347526.
A. Zaman , W. Wu and T. G. Bivona , Targeting Oncogenic BRAF: Past, Present, and Future, Cancers, 2019, 11 , 1197
A. Alvarez-Arenas et al., Interplay of Darwinian Selection, Lamarckian Induction and Microvesicle Transfer on Drug Resistance in Cancer, Sci. Rep., 2019, 9 , 9332 .
K. Yu et al., An integrated meta-omics approach reveals substrates involved in synergistic interactions in a bisphenol A (BPA)-degrading microbial community, Microbiome, 2019, 7 , 16.
G. D. Poore et al., Microbiome analyses of blood and tissues suggest cancer diagnostic approach, Nature, 2020, 579 , 567 —574 .
A. Gonzalez et al., Characterizing microbial communities through space and time, Curr. Opin. Biotechnol., 2012, 23 , 431 —436 Search PubMed .
D. Gurwitz The Gut Microbiome: Insights for Personalized Medicine, Drug Dev. Res., 2013, 74 , 341 —343 .
N. Issa Isaac et al., Metaproteomics of the human gut microbiota: Challenges and contributions to other OMICS, Clin. Mass Spectrom., 2019, 14 , 18 —30