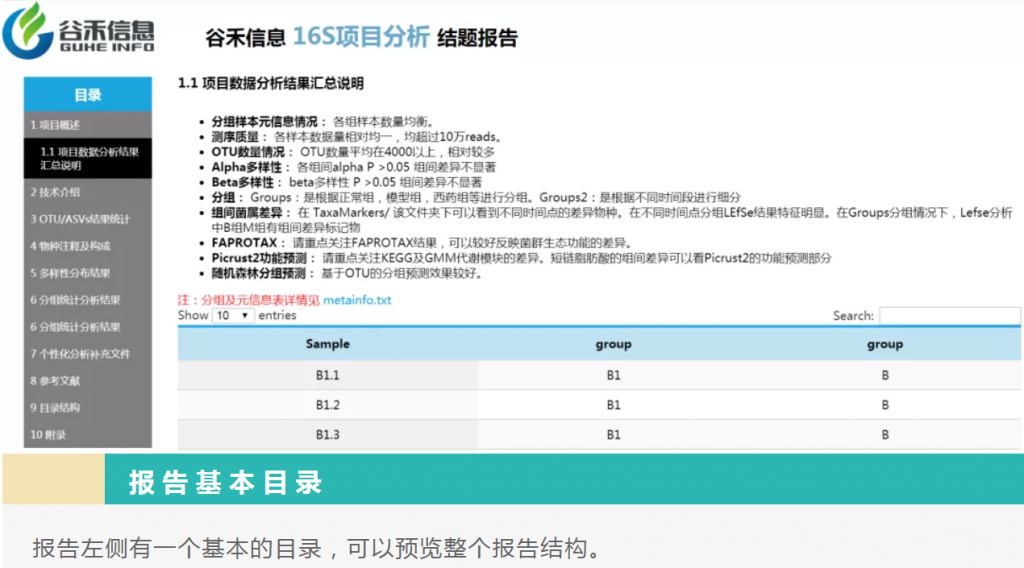
-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康

16S科研项目是一个完整的闭环,前期的课题项目设计方案、取样和重复实验设置决定了后期分析报告的数据完整性和项目类型。
想要拿到一手有利用价值的科研报告和项目数据,前期的实验方案设计和后续的分析都起着关键性的作用。
然而有时候拿到报告不知道如何去解读,这里为大家梳理一下16s科研项目的全过程,帮助大家更好的了解报告内容,快速获取关键信息。


实验方案设计就像一个总工程的设计图纸,决定了未来科研分析报告的类型走向,并且前期的分组设计的越详细,各种理化指标、生化指标、代谢物等信息准备越充分,后续报告的完整度越高。


明确项目课题类型
第一步要做的就是明确项目课题类型:
最常见的就是多分组之间差异分析比较:例如,要比较对照组、模型组、实验组,之间的差异结果。
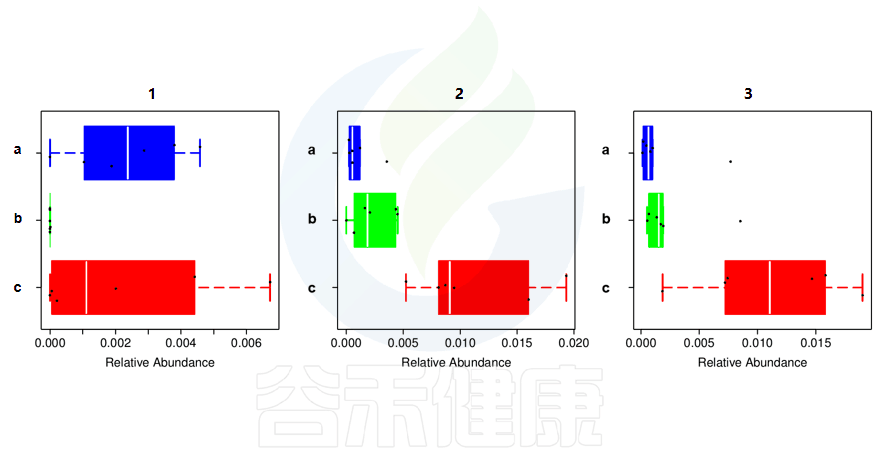

还有多分组中,任意两组之间比较:例如某实验设计了正常组、疾病组、用药组服用奥氮平、阿立哌唑、氨磺必利、利培酮,像比较不同的用药组和疾病组之间的菌群的差异结果,就用到了分组之间两两差异比较。
✦举个例子
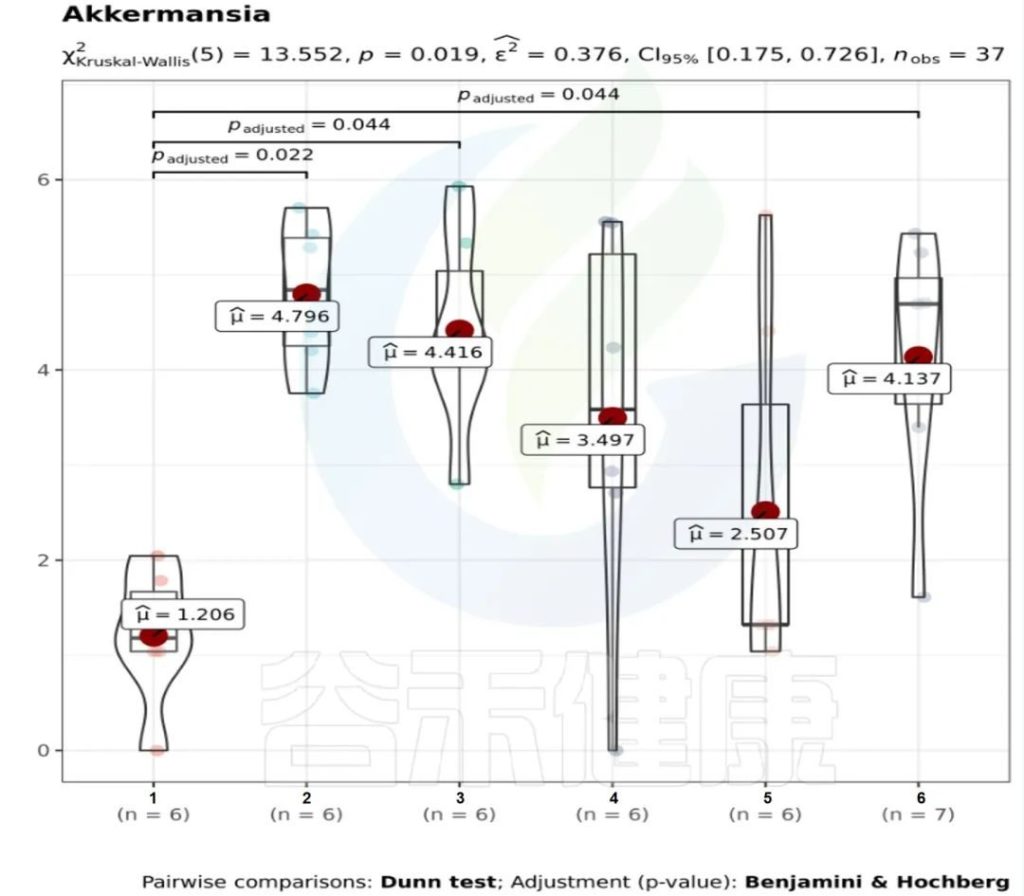
图中1组与3组、4组、6组 组间差异显著
还有随时间的变化比较菌群之间的变化规律:例如在用药不同时间段包括3天,5天,2周,1个月,2个月,观察菌群的变化情况。如下图所示:
收集理化指标非常重要
如果前期搜集好每个样本的相关理化指标,还可以计算这些指标与菌群之间是否具有相关性。
✦举个例子
例如该项目比较自闭症儿童与正常儿童的菌群差异。客户在样本信息单里还详细搜集了母孕期的各种详细指标,例如孕期天数、出生体重、白细胞介素6、肿瘤坏死因子a、五羟色氨等数值型理化指标。
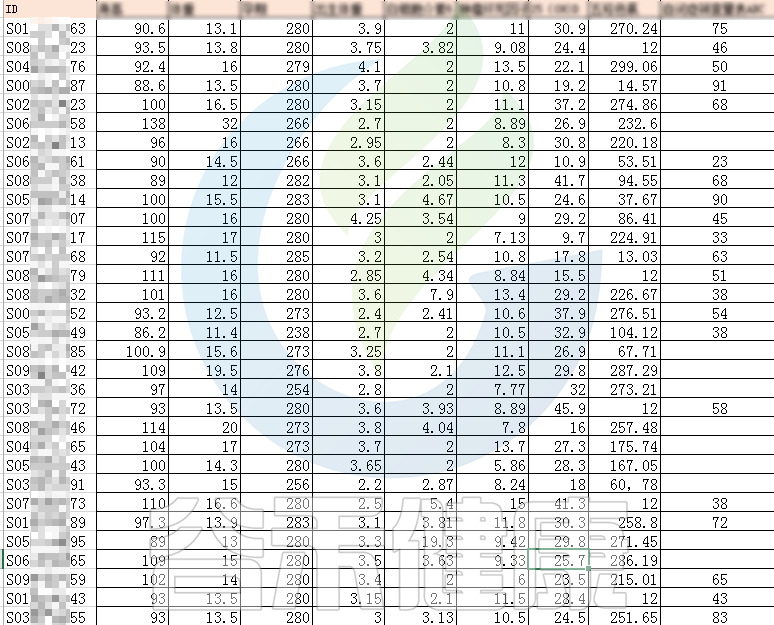
还搜集了是否顺产、是否妊娠高血压、是否孕期感染、是否妊娠糖尿病、是否先兆流产等因子型理化指标。其中0代表否,1代表是:
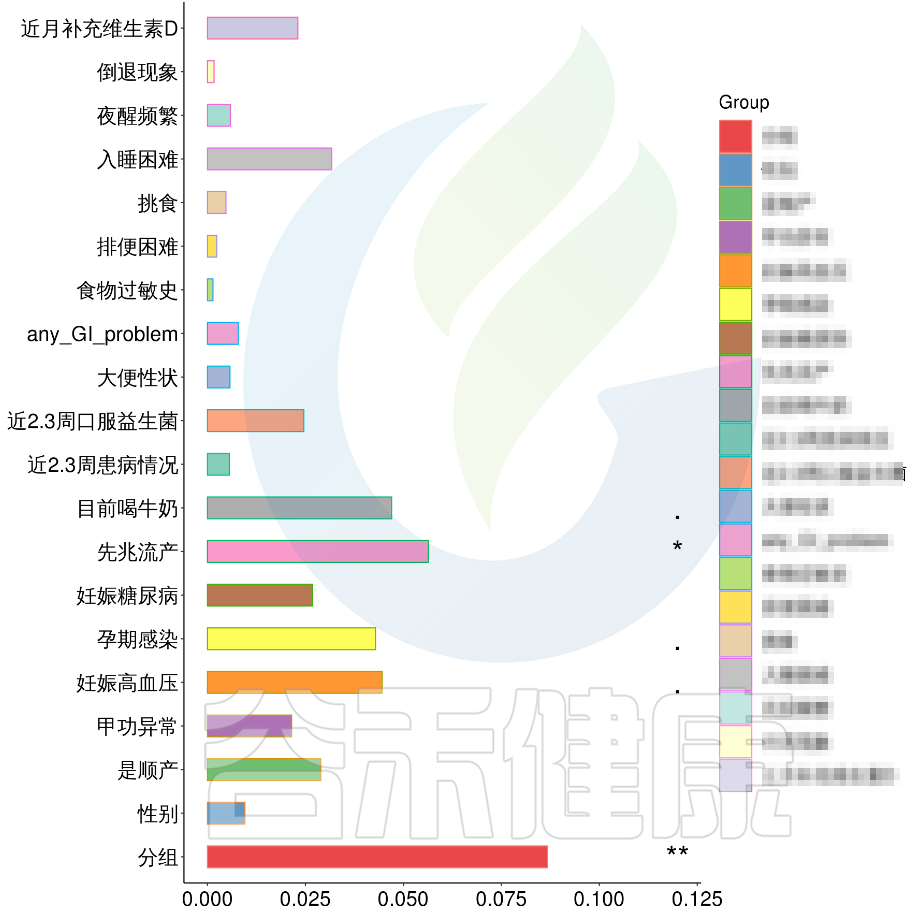
根据这些理化指标与菌群数据做相关性分析,从因子型的结果可以看出,自闭症(ASD)与正常儿童之间的分组与菌群之间相关性极显著**,其次是否有先兆流产的分组与菌群之间有显著相关性*,其他的包括是否喝牛奶、孕期是否感染、妊娠高血压都与菌群有相关性。
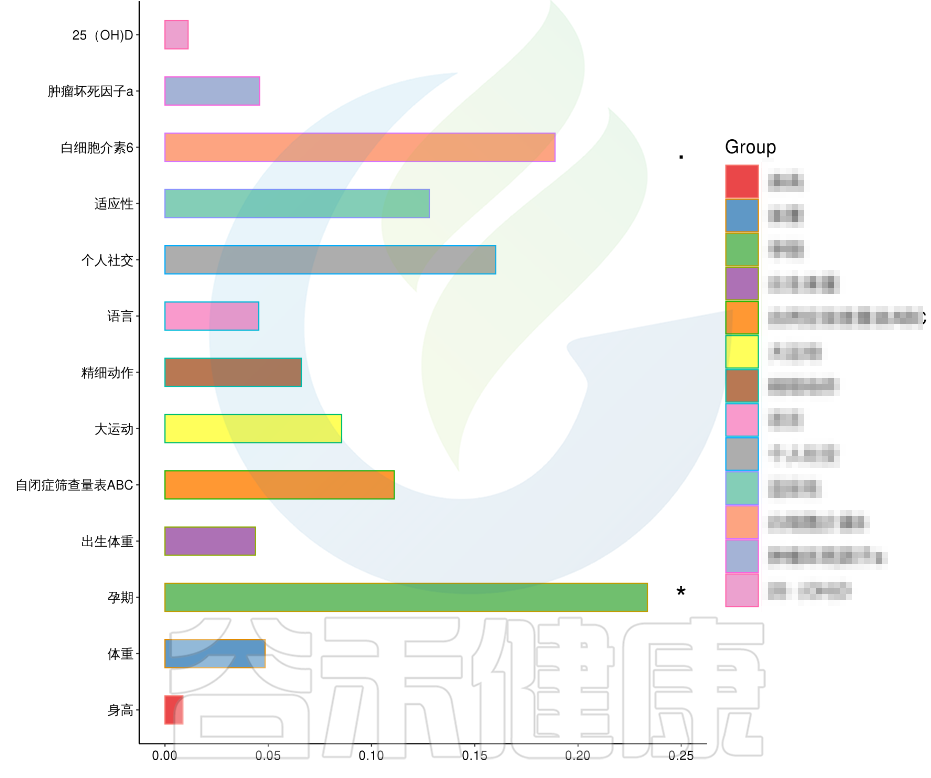
在数值型理化指标中,孕期的天数与菌群之间相关性显著*,其次是白细胞介素6与菌群之间有相关性。
小结
因此,前期搜集相关资料越详细充分,对分析报告的完整性也会有帮助,分析人员也会根据您的样本信息单提供的相关内容,做出个性化的分析和售后指导建议。
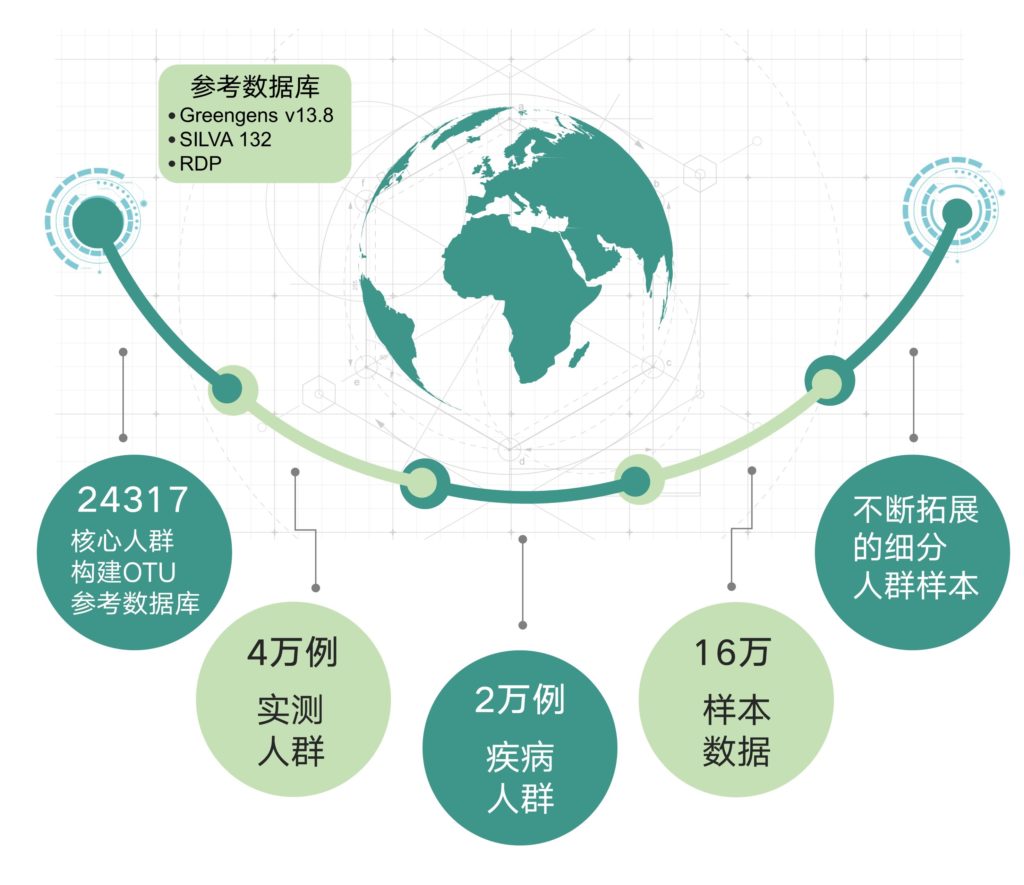
首先基于样本类型,最常见的环境样本来源是人体、动物、土壤、水体等。而人体中的肠道菌群样本是目前研究最广泛,可鉴定的物种也最为丰富,谷禾在肠道菌群与人体健康方面有深入研究,目前已完成超20万例临床肠道菌群样本检测,并构建了超过60万各类人群粪便样本数据库。
其他样本类型还包括人体/动物唾液样本、组织样本、尿液样本等。
▸ 粪便样本
目前粪便样本从采样到提取数据分析技术较为成熟、应用较为广泛,谷禾最早在15年就开发了针对粪便样本的取样管,也是最早致力于研发粪菌取样盒的公司,方便实验室、个人日常取样需求,实现了粪菌样本的常温运输。
谷禾取样管常温保存,取样也较为方便卫生,在家就可以轻松完成,相较于传统取样方法都有所升级。并且该取样管也有专利证书。该取样方法被大量客户采用并接纳,大大降低了采集粪便样本的难度,缩短了搜集样本的时间周期。
取样示意图

▸ 其他样本
土壤样本也相对较为容易提取出DNA,但需要注意的是土壤样本的菌群特征容易受植物腐殖质基因的影响和干扰,所以提取时要进行纯化。
而口腔、组织、尿液等样本,由于DNA含量较少,在实验阶段提取相对较为困难,所以提前准备样本时,尽量多取一些,并且可以多取几个重复,尽量避免扩增不出来的情况。
并且这些样还很容易受到环境样的污染,所以在实验阶段,可以取空白样本,和阳性样本ST做对照,数据分析时可以用来纯化样本,排除来自环境的干扰序列。
✦组间差异分析需重复取样
要做组间差异分析时,每组要重复取样,才能做组与组之间的统计检验。理论上,每个组至少3个样就满足基本的统计差异分析需求。所以在重复取样时,每个分组至少取3个样。取样时要保证每个分组内部的样本一致性,如果组内样本之间的个体差异性较大,则会影响后期组间差异结果分析。
✦举个例子
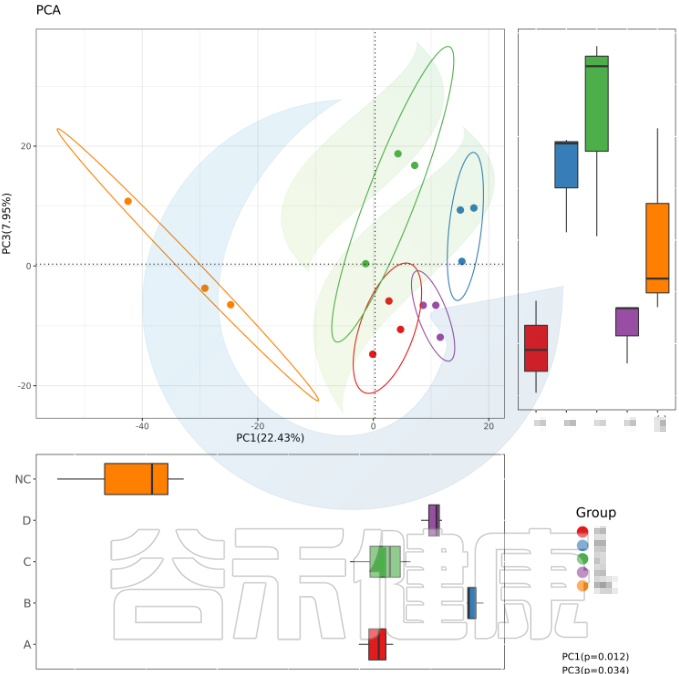
例如从该图可以看出,分组之间组间差异较大,并且组内的样本之间较为接近和相似。
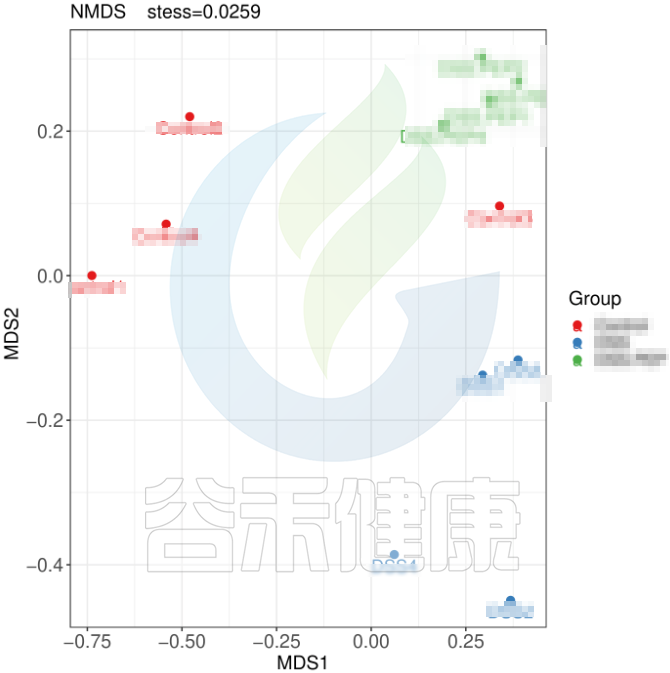
但从该图可以看出,Control组中Control3样本明显与组内的其他样本差异较大,与DSS组内的样本较为相近,这样就对后期组间差异分析的时候会产生影响,需要将该样本去除。
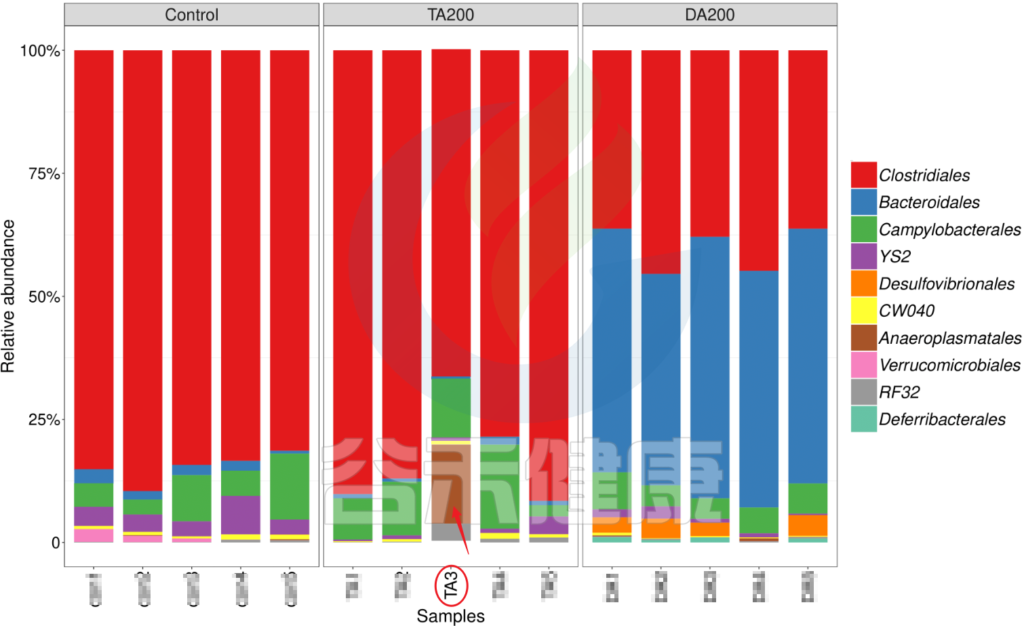
又例如在该图中,TA200组中的TA3样本的Anaeroplasmatales物种丰度含量非常高,该样本与组内的其他样本明显差异较大,该样本可能受到环境污染等其他因素干扰,这样就没有办法保证组内样本的均一性,也会影响分组之间的差异分析统计结果,再后期分析的时候建议把该样本去掉重新分析。
建议
为了便于后期数据整理修改,每个分组需要保留一定量的重复样本,假如每个分组只取了3次重复,假如其中有一个样本质量不好需要去除,该分组只剩2个样本,则不满足每组至少3个样的分组条件,整体就没有办法做组间差异分析统计。
所以这里建议每个分组至少取5个样做重复,一般6到10个样就能分析出比较完善的结果。具体分组和组内的重复取样数量视具体的实验设计方案而定。
在经费允许的情况下,建议多取一些重复。假设每组取50到100个重复或者以上,得到的分析结果就基本可以涵盖该分组情况所有的菌群构成情况,可以较为全面的研究分组之间的菌群构成差异情况。
当拿到16S科研分析报告以后,面对纷繁复杂,各式各样的图表分析结果犯了难,不知道如何从这么多的图表中入手,快速找到报告中需要的图表结果。
这里对16S科研分析结果抽丝剥茧,概括出报告中的主要几大内容板块。
•16S科研分析究竟是在做什么?
16S rDNA 是一种对特定环境样品中所有的细菌进行高通量测序,以研究环境样品中微生物群体的组成,解读微生物群体的多样性、丰富度及群体结构,探究微生物与环境或宿主之间的关系的技术。
主要是对原始数据进行拼接过滤得到的优化序列,降噪方法得到ASV,再对ASV进行物种注释,注释到门、纲、目、科、属、种各层次上的分类结果。
通过ASV表计算Alpha多样性,样本内的多样性指数,Beta多样性,样本间相似性的指标。
对ASV表进行功能预测,例如Picrust2功能预测分析、Bugbase菌群表型特征分析,FAPROTAX生态功能预测等。
得到的每个样的数据结果,根据客户提供的分组情况和理化指标,进一步做组间差异分析,以及和环境理化指标之间做关联分析,相关性分析,比较分组之间是否有差异,差异是否显著,来验证分组是否合理,和环境宿主之间是否有关联性。
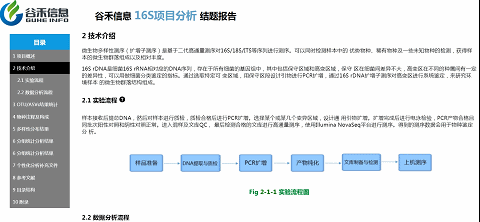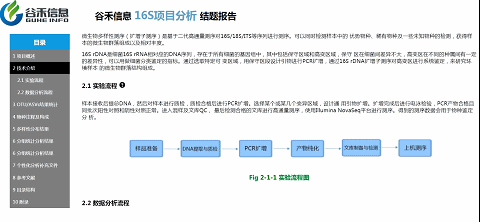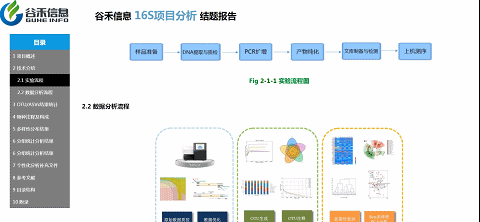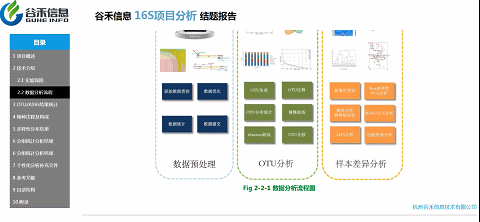
原始数据处理
Illumina NovaSeq测序平台测序得到的双端数据Raw PE,经过拼接和质控,根据一定的标准过滤掉低质量数据、接头或PCR错误,得到Raw Tags。再经过去重复序列,去singleton序列,过滤嵌合体,得到可用于后续分析的有效数据 Effective Tags。
OTU(ASV) 表生成
微生物多样性分析中最重要的就是OTU特征表,一切后续分析都围绕OTU表来进行。生成OTU除了传统的聚类的方法(一般按照97%的相似度进行聚类),现在最新用到的技术的是降噪的方法得到ASV。
简单来讲ASV就是在去除了错误序列之后,将Identity的标准设为100%进行聚类,常见的有DADA2、Deblur、Unoise三种降噪方法。项目里用到的是UNOISE2降噪方法获得ASV数据。
物种的分类与注释
采用QIIME2训练分类器方法对ASVs代表序列进行分类学注释,默认选用SILVA138数据库进行物种注释。并在各个分类水平上:domain(域),phylum(门),class(纲),order (目),family(科),genus(属),species(种)对每个样本的群落组成统计。
alpha多样性
Alpha多样性主要反映样本内多样性。对ASV表进行计算可以获得每个样本的simpson,ace,shannon,chao1以及goods_coverage等指数,alpha多样性指数用来来评估样本菌群物种的丰富度(richness)和多样性(diversity)
beta多样性
Beta多样性反映的是样本间多样性,Beta多样性是衡量个体间微生物组成相似性的一个指标。通过计算样本间距离可以获得β多样性矩阵,基于OTU的群落比较方法报告中给出了,欧式距离、bray curtis距离、Unweighted UniFrac距离和Weighted UniFrac距离等。
功能预测
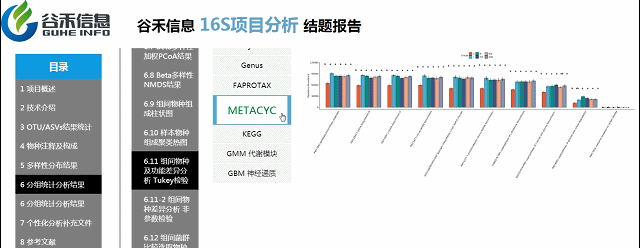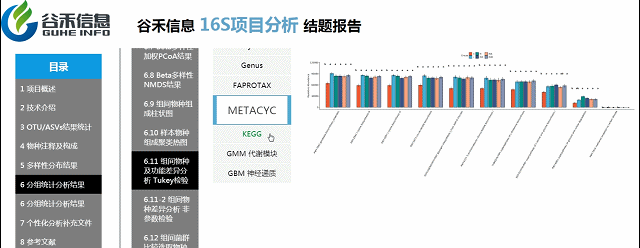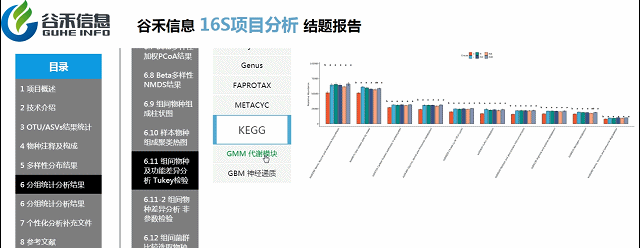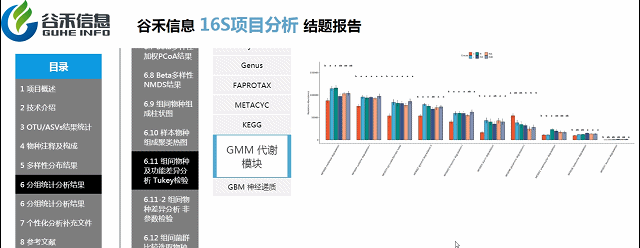
得到群落的微生物组成之后,也可以对群落功能组成进行预测,常用的16S功能预测的相关软件有PICRUSt2、FAPROTAX、BugBase。
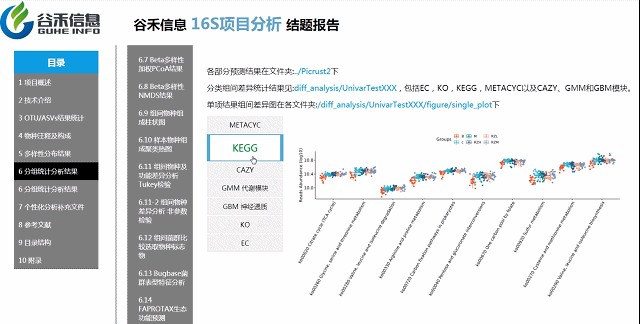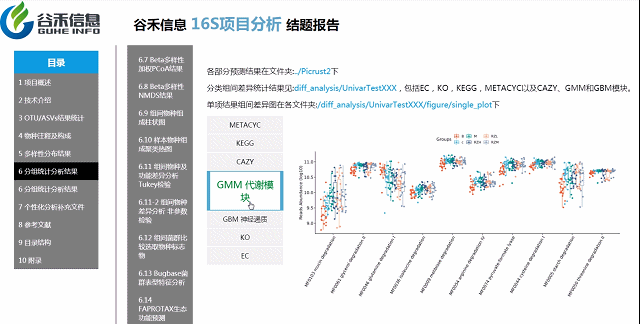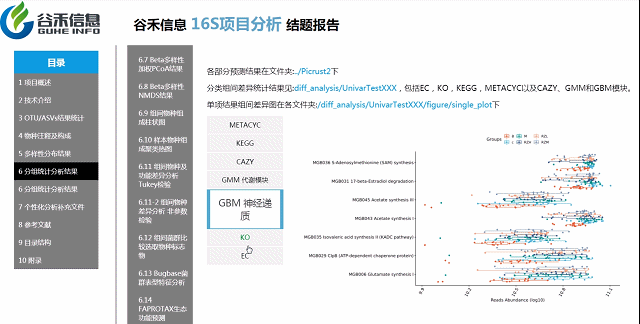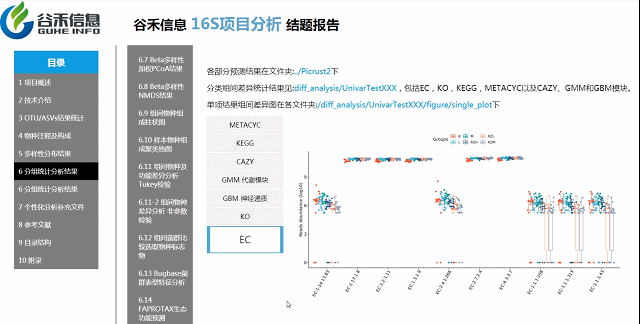
PICRUSt2用来预测功能,通常指的是基因家族,PICRUSt2支持基于多个基因家族数据库的预测,报告中包括了KEGG同源基因,KO直系同源物,EC酶分类编号,MetaCyc途径的丰度,CAZy碳水化合物活性酶数据库,GMM是肠道代谢模块和GBM是肠脑模块。
FAPROTAX是原核的微生物注释代谢或其他生态相关的功能(例如硝化,反硝化,发酵)的一个数据库和软件。FAPROTAX预测的功能主要集中在海洋、湖泊等环境样本微生物的功能,特别是硫、碳、氢、氮的循环功能。
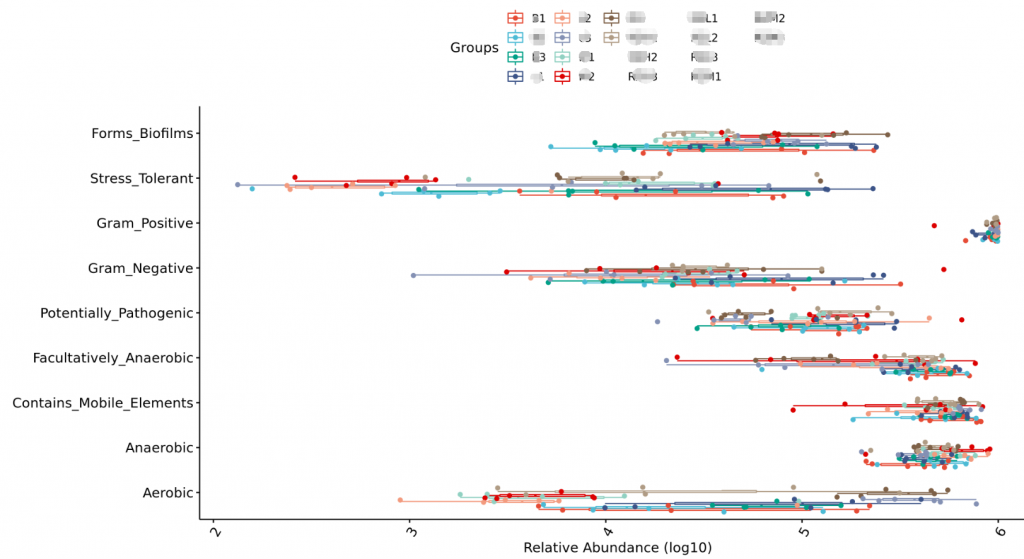
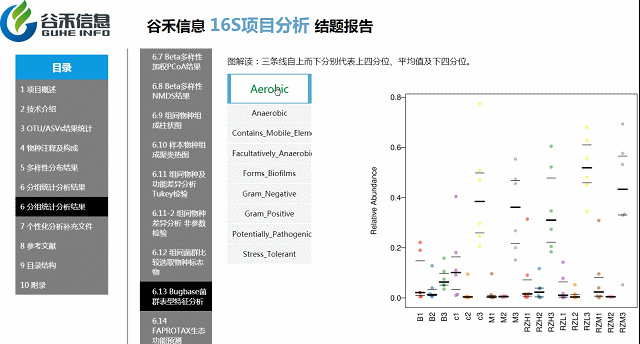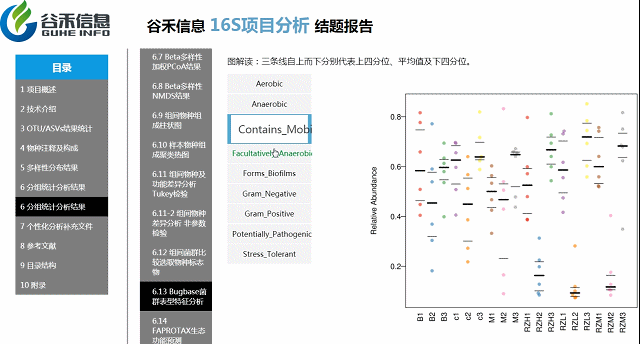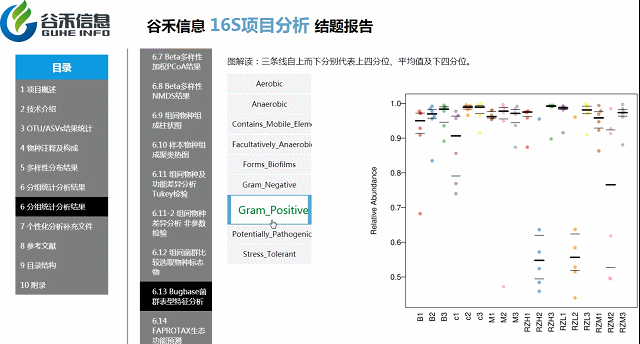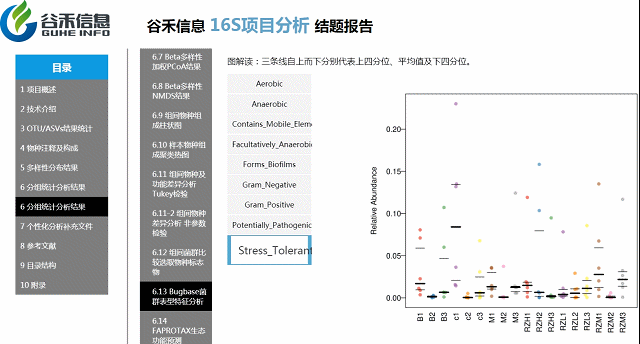
BugBase能进行表型预测,其中表型类型包括革兰氏阳性(Gram Positive)、革兰氏阴性(Gram Negative)、生物膜形成(Biofilm Forming)、致病性(Pathogenic)、移动元件(Mobile Element Containing)、氧需求(Oxygen Utilizing,包括Aerobic、Anaerobic、facultatively anaerobic)及氧化胁迫耐受(Oxidative Stress Tolerant)等7类。
以上这些部分,我们通过数据处理分析,得到了每个样本相关的大量数据结果,包括每个样本的序列统计、ASVs表格、物种分类注释统计、alpha多样性指数、beta多样性指数、功能预测等。这些数据主要集中在报告里的这些内容:
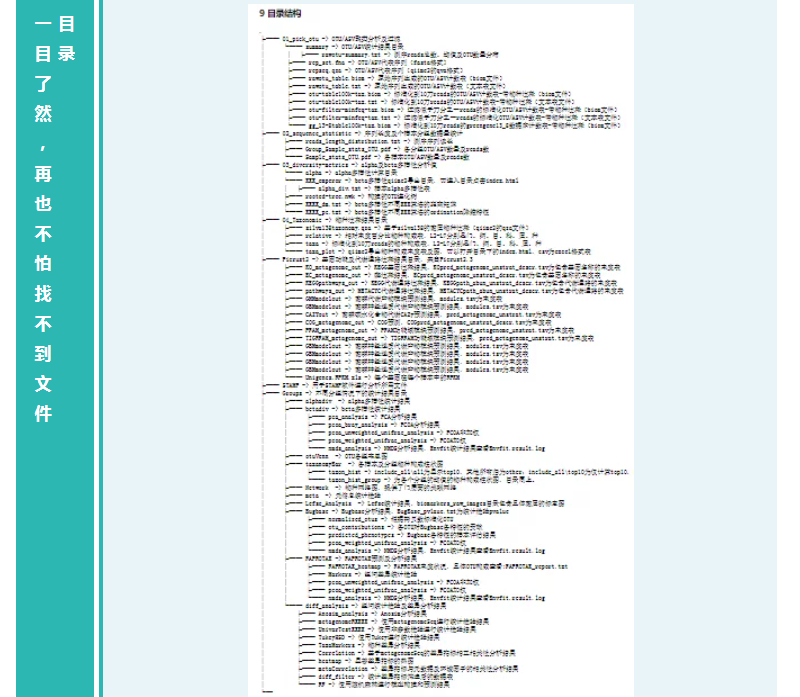
▸ 科研分析报告结果文件夹
01_pick_otu/ 文件夹主要是对样本ASV表格统计
02_sequence_statistic/ 文件夹是对样本序列数据的统计
03_diversity-metrics / 文件夹是对样本的alpha多样性指数、beta多样性指数的统计
04_Taxonomic/ 文件夹是对物种分类注释的统计(门到种水平)
Picurst2/ 文件夹是Picrust2功能预测得到的每个样本的相关功能预测数据
Groups/ 文件夹下是对组间差异分析结果

红框是样本个体的相关数据统计,Group是分组比较
根据以上常规分析得到的相关数据进行作图,其路径也在对应文件夹下,可以打开 分析报告.html 有相关分析的图表和对应文件的详细介绍和路径说明。
★拿到样本后需要进行统计分析
当我们拿到这些样本大量的数据结果,之后关键的一步就是做对这些数据进行处理,做统计分析,比较分组之间的差异结果,找出菌群和环境之间的关联性等,对数据进一步做研究,找出课题方案对应的结果。
不同的数据用到的统计检验方法也不太一样,接下来我们对报告中的不同的分析结果对应的统计差异分析方法进行介绍说明。
▸ alpha多样性
alpha多样性指数组间差异统计分析用到的检验方法是:单因素方差分析(如果只有两个分组,用Wilcoxon秩和检验,3个及以上的分组用Kruskal-Wallis 检验),图上方显示P值
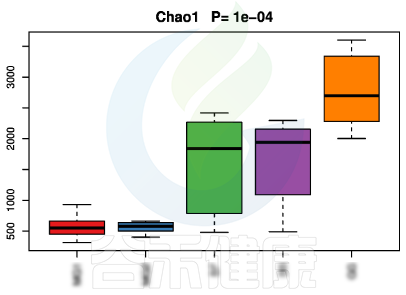
▸ beta多样性
beta多样性指数的统计检验方法有ANOSIM相似性分析和Adonis多元方差分析,这两种都是基于距离矩阵的检验方法。
✦Anosim相似性分析
Anosim分析是一种非参数检验,用来检验组间的差异是否显著大于组内差异,从而判断分组是否有意义。
报告中给出了加权距离和非加权距离的Anosim结果图,图中给出了R值和P值。
R值用于比较不同组间是否存在差异,R-value 介于(-1,1)之间,R-value > 0,说明组间差异大于组内差异。R-value < 0,说明组间差异小于组内差异。R只是组间是否有差异的数值表示,并不提供显著性说明。
统计分析的可信度用 P-value 表示,P< 0.05 表示统计具有显著性。
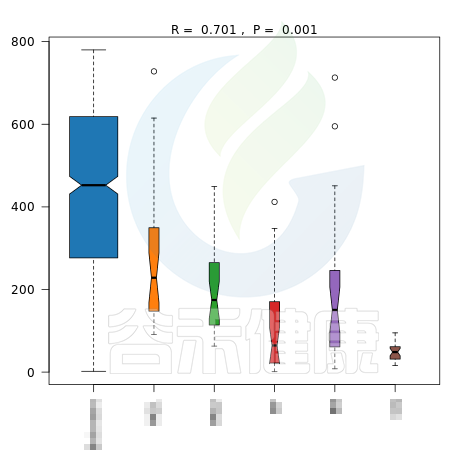
图中能看出R>0,说明组间差异大于组内差异,P<0.05 ,说明差异显著,证明该分组情况效果较好。
✦Adonis多元方差分析
Adonis多元方差分析,其实就是PERMANOVA,亦可称为非参数多元方差分析。
其原理是利用距离矩阵(比如基于Bray-Curtis距离、Euclidean距离)对总方差进行分解,分析不同分组因素对样品差异的解释度,并使用置换检验对其统计学意义进行显著性分析。
它与Anosim的用途相似,也能够给出不同分组因素对样品差异的解释度(R值)与分组显著性(P值)。
报告中PCoA bray距离、PCoA weighted_unifrac距离、PCoA unweighted_unifrac距离的图片右下角有给出PERMANOVA检验的P值和R值。
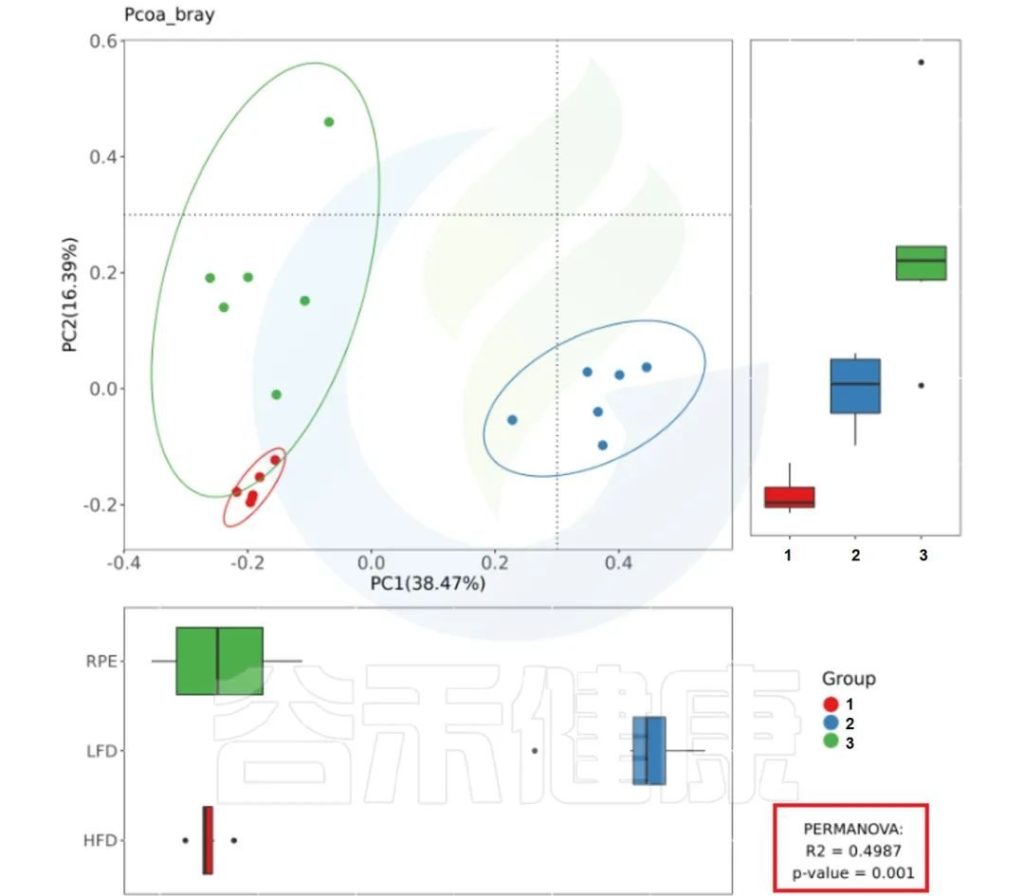
图中看出PCoa bray距离得到的检验P<0.05 组间差异显著,并且分组之间区分较为明显。
PCoa bray距离的PERMANOVA检验结果路径:
多组间检验结果:
Groups/betadiv/pcoa_bray_analysis/PERMANOVA.result_all.csv
两组间检验结果:
Groups/betadiv/pcoa_bray_analysis/ PERMANOVA_paired_result.csv

不同分类水平下的检验方法
在很多分析报告当中,例如在不同疾病的肠道菌群分组中,本身样本个体之间肠道菌群的物种多样性,丰富度差异并不大,alpha多样性组间差异并不显著,beta多样性分组间区分不是很明显,这样就需要进一步找出分组之间的差异物种或者差异功能来进行分析。
对于不同分类水平的物种和功能预测结果用到以下几种检验方法:
Tukey检验
Tukey主要应用于3组或以上的多重比较,适合于各组例数相等的每两两分组之间比较。
Tukey检验的一个重要的优点是非常简单,而且所需实验样本相对较少。
其检验结果的可信度达到95%的置信水平时,最少的情况下只需6个样本进行验证(改善前3个样本、改善后3个样本)。
•举个例子
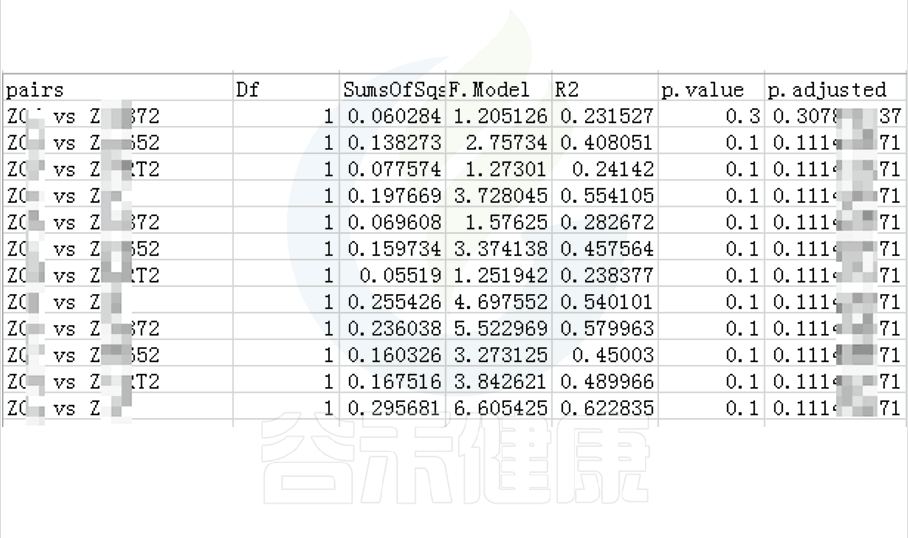
图中的字母代表显著性差异的字母表示法,只要含有相同的字母,就表明两组之间没有显著性差异。
例如a和ab含有相同字母“a”,表示两组之间没有显著性差异。ab中的“b”表示这一组和其他含有字母b的组(比如bc)没有显著性差异,但是a和bc就有显著性差异了。
图中只展示Tukey检验差异显著的物种或功能,如果数量较多,则只展示前10个。
路径:Groups/diff_analysis/TukeyHSD/
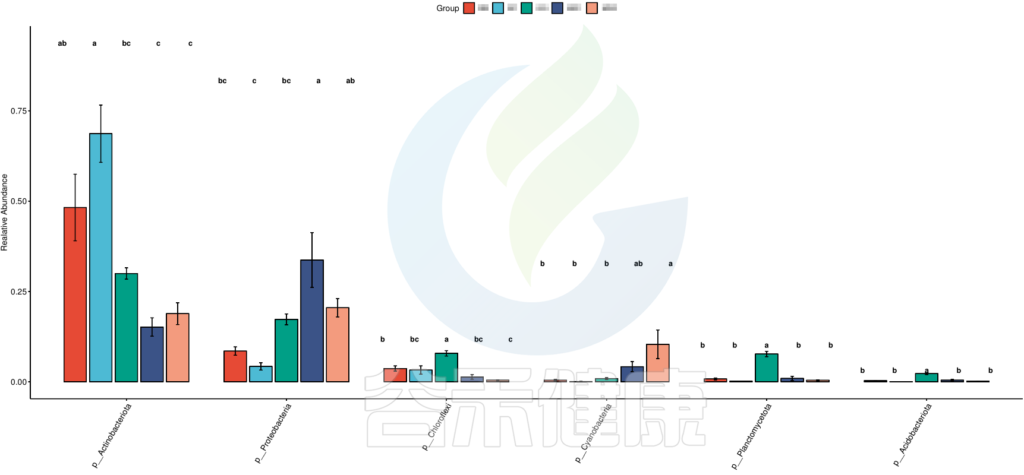
图中显示的都是Tukey检验组间差异显著的物种,依次按照丰度从高到底排列,如果差异结果较大,则显示前10个物种。例如在该图中,Tukey检验结果,门水平物种Actinobacteriota在BB与MG1组、BB与MG2、BF与GG组、BF与MG1组、BF与MG2组,这些分组之间组间差异显著。
组间差异箱型图
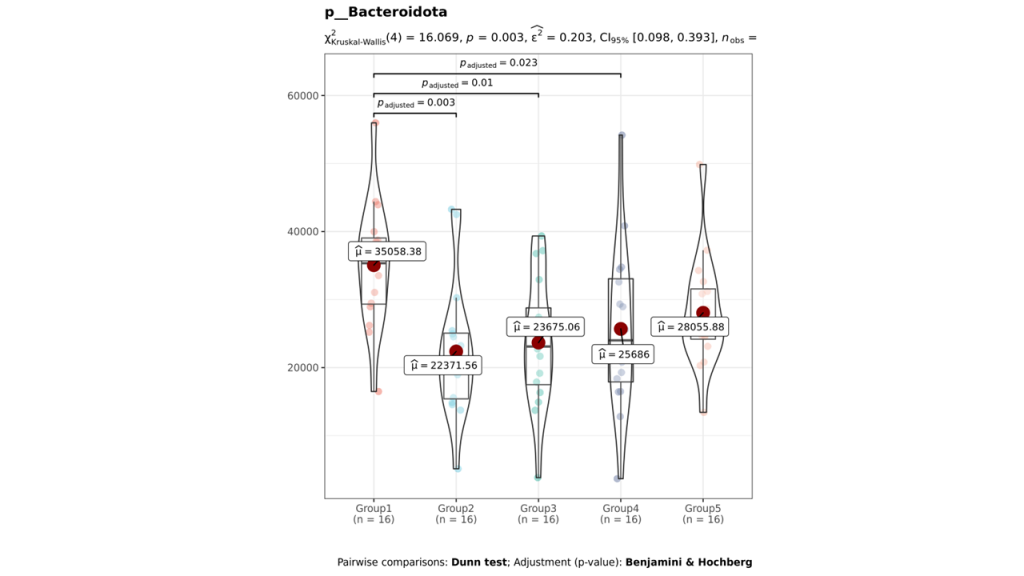
组间差异箱型图用到的检验方法是通过单因素方差检验(只有两个分组,用的是Wilcoxon秩和检验,3个及以上的分组用的是Kruskal-Wallis 检验),Var检验和one-way相结合,筛选出组间差异性物种。
路径:Groups/diff_analysis/TaxaMarkers
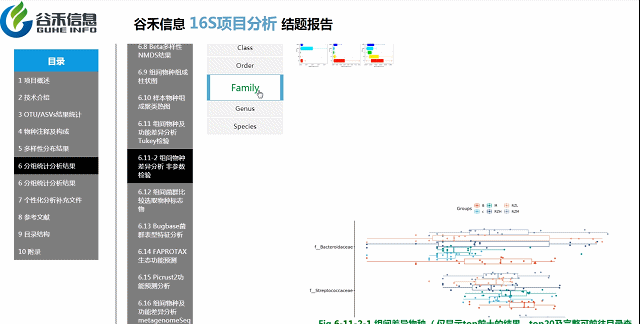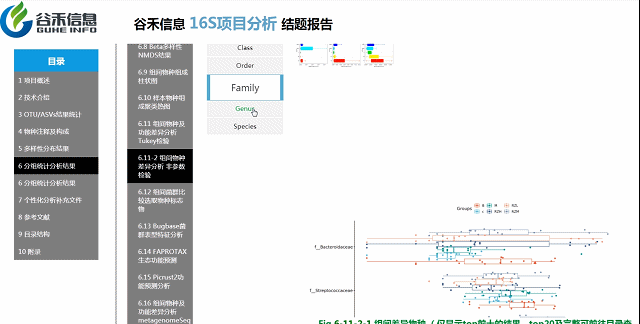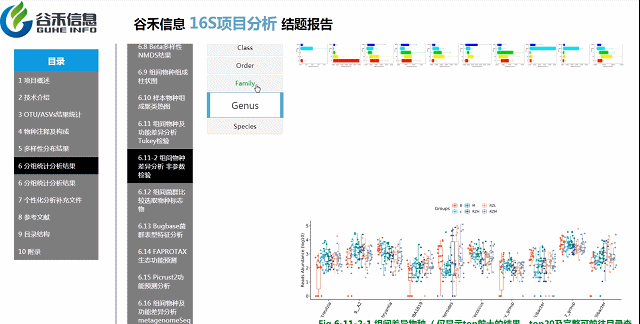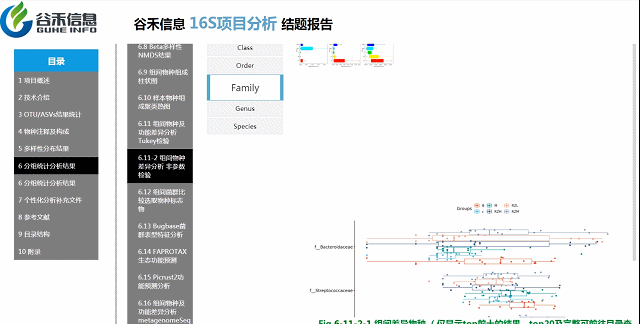
图中每一个箱型图代表一个组间差异显著的物种
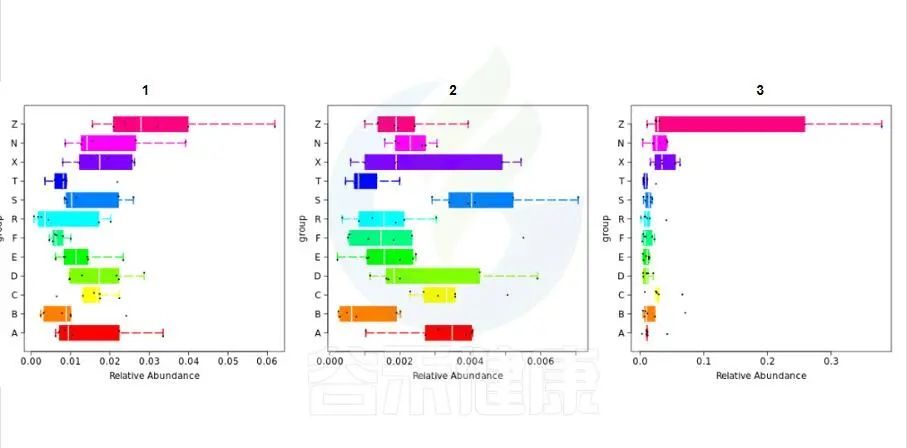
图中显示的都是统计方法得到的差异显著的物种,图中能看出这3个物种分组之间差异显著。
命名格式是,例如:Cen_Nitrosopumilus 指的是,当前分类水平(属水平)的名字 g__Nitrosopu 加上一级分类水平(科水平)的名字 f__Cenarchaeaceae 的前 3 个字母简写Cen,如果当前水平没有注释到名字则以全称的名字表示。
统计结果表:Groups/diff_analysis/TaxaMarkers/ xxx.Groups.sig.meanTests.csv
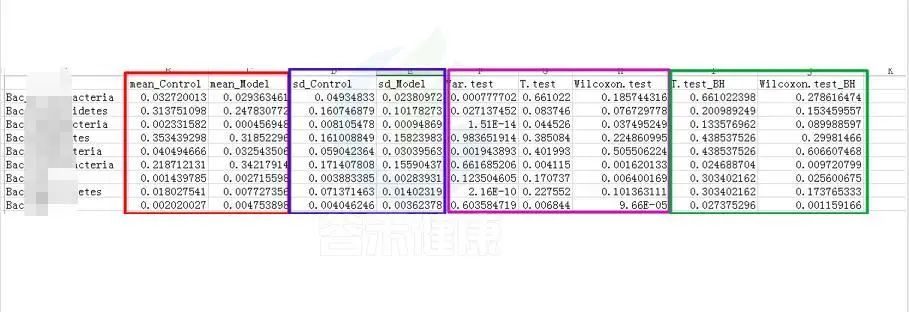
例如这是一个表格的截图
红框 mean_ 是分组组间的平均值
蓝框 sd_ 代表组间的标准差
粉色 .test 代表不同统计检验结果的P-value P值,这里有var检验 T 检验 Wilcoxon检验(或Kruskal-Wallis 检验)
绿色 _BH 例如Wilcoxon.test_BH代表Wilcoxon.test检验BH矫正的Q-value,Q值
UnivarTest检验(单因素方差分析)
单因素方差分析是指如果只有两个分组,用Wilcoxon秩和检验,3个及以上的分组用Kruskal-Wallis 检验。
路径:Groups/diff_analysis/UnivarTestXXX
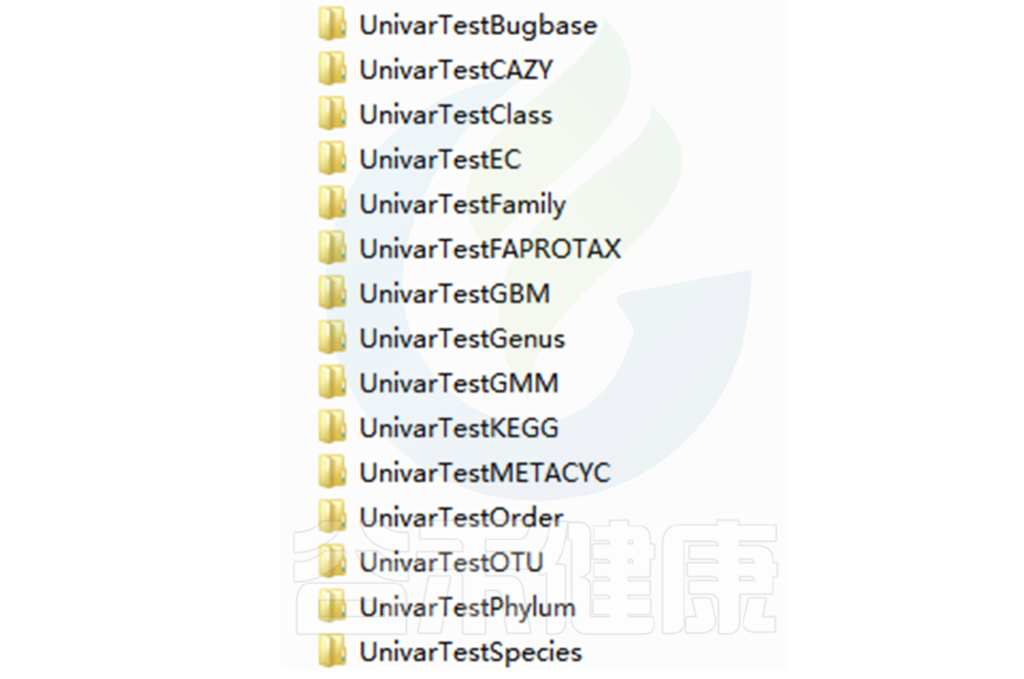
Groups\diff_analysis\UnivarTestKEGG\figure 文件夹下有做成柱状图、箱型图和单个物种之间的图,其中有横着排列和竖着排列的,有用原始值计算的,还有对原始值取log后进行统计的。图中只展示Univar 检验组间差异显著的物种/功能。
统计结果表:Groups/diff_analysis/UnivarTestXXX/ UnivarTest_sign.txt
•举个例子
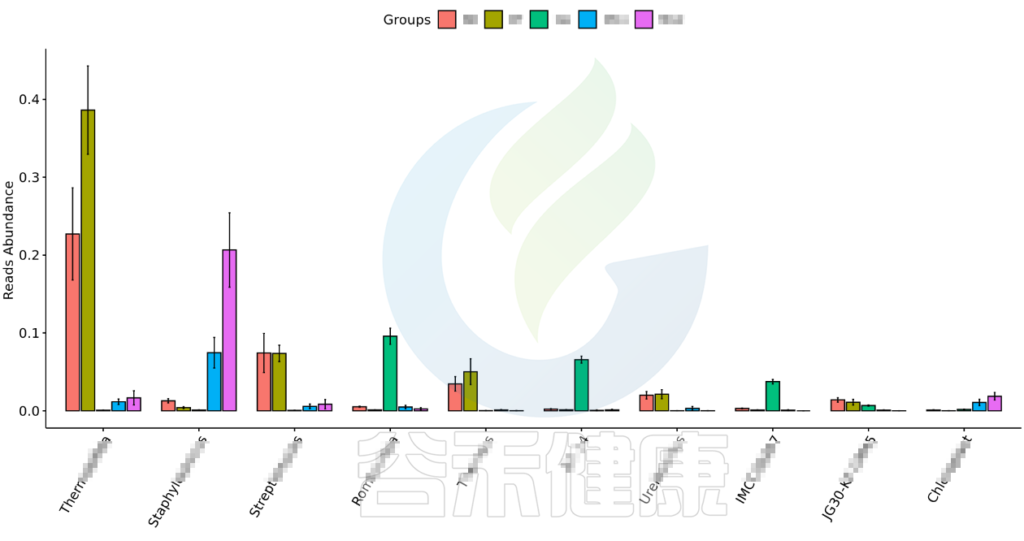
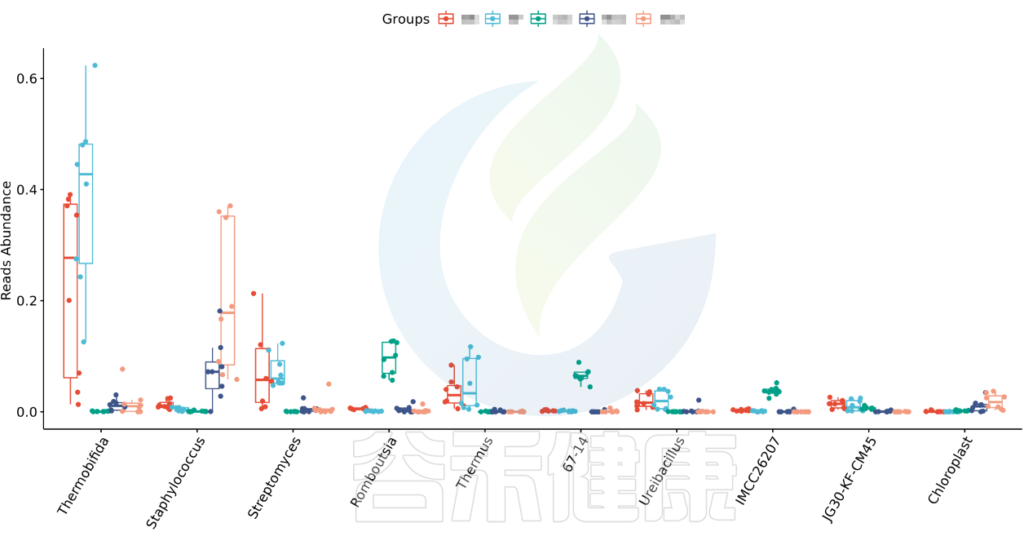
图中显示的是该统计检验差异显著的物种的柱状图或箱型图,按照丰度从高到低排列,如果差异物种/功能较大,则只显示前10个。例如该图中Therobifida、Staphylococcus、Streptomyces等物种用Kruskal-Wallis 检验得到的组间显著差异物种。
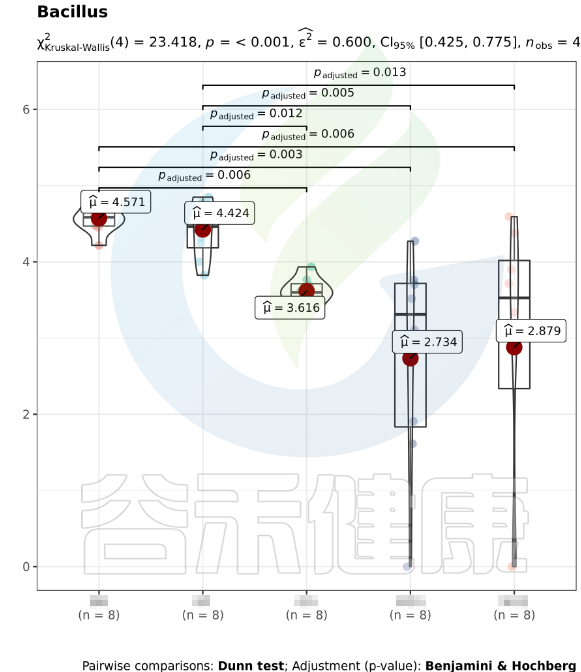
该图展示了Bacillus物种Kruskal-Wallis 检验差异结果,所有分组中P<0,001 多组间差异显著,两组间BB与GG、BB与MG1、BB与MG2、BF与GG、BF与MG1、BF与MG2,组间差异显著。
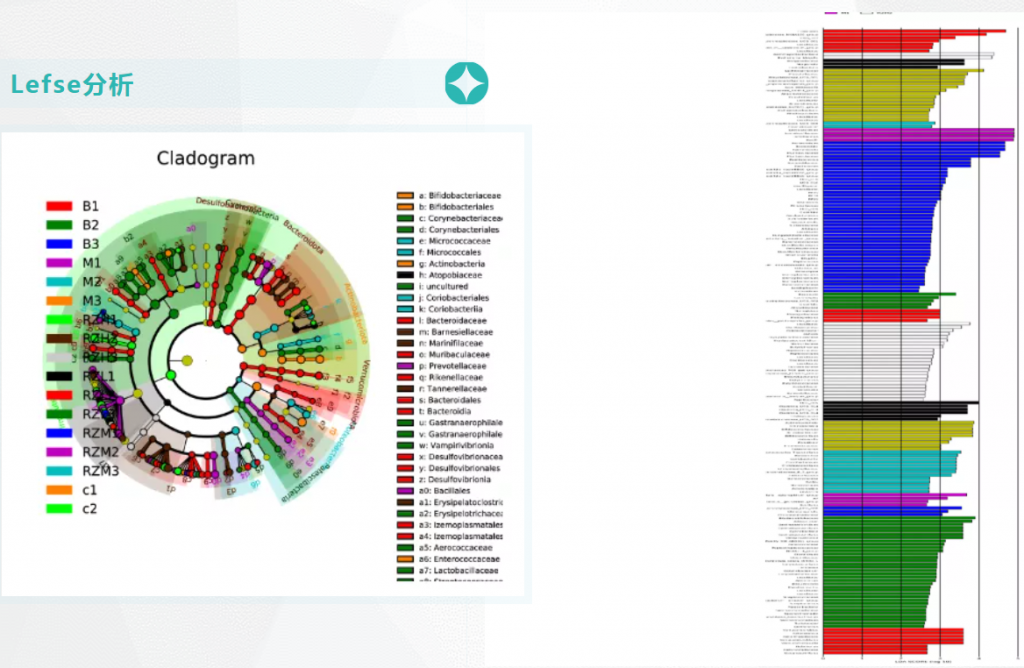
LEfse分析
LEfse分析即LDA Effect Size分析,是一种用于发现和解释高维度数据生物标识(基因、通路和分类单元等)的分析工具,可以进行两个或多个分组的比较,它强调统计意义和生物相关性,能够在组与组之间寻找具有统计学差异的生物标识(Biomarker)。
LEfSe用到的统计分析方法是将线性判别分析与非参数的Kruskal-Wallis以及Wilcoxon秩和检验相结合。
LEfse分析结果中一般会出现两个图,一张表( LDA值分布柱状图、进化分支图以及特征表)。
LDA值分布柱状图
这个条形图主要为我们展示了LDA score大于预设值的显著差异物种,即具有统计学差异的Biomaker,默认值为2.0(看横坐标,只有LDA值的绝对值大于2才会显示在图中);柱状图的颜色代表各自的分组,长短代表的是LDA score,即不同组间显著差异物种的影响程度。
路径:
Group/Lefse_Analysis/out_formant.cladogram.png
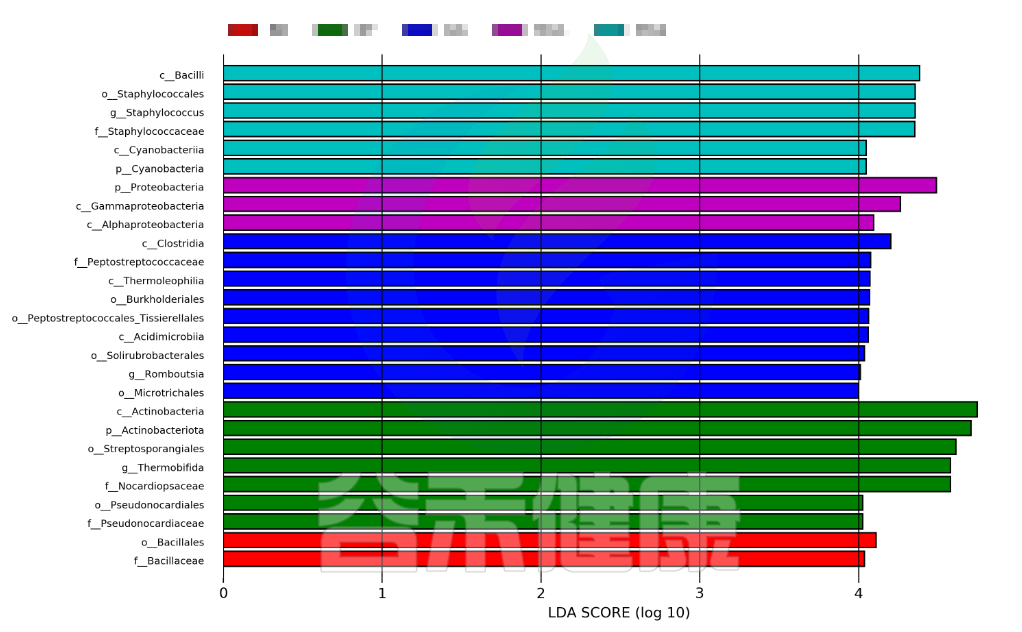
图中展示了不同分组特有的Lefse组间差异标记物,例如BB组的标记物是目水平的Bacillales和科水平的Bacillaceae,不同的分组标记物也不同,图中如果只展示了部分分组,则代表只有部分分组通过Lefse分析筛选出组间差异标记物。
进化分支图
小圆圈: 图中由内至外辐射的圆圈代表了由门至属的分类级别(最里面的那个黄圈圈是界)。不同分类级别上的每一个小圆圈代表该水平下的一个分类,小圆圈的直径大小代表了相对丰度的大小。
颜色: 无显著差异的物种统一着色为黄色,差异显著的物种Biomarker跟随组别进行着色,红色节点表示在红色组别中起到重要作用的微生物类群,蓝色节点表示在蓝色组别中起到重要作用的微生物类群。
未能在图中显示的Biomarker对应的物种名会展示在右侧,字母编号与图中对应(为了美观,右侧默认只显示门到科的差异物种)。
路径:Group/Lefse_Analysis/out_formant.png
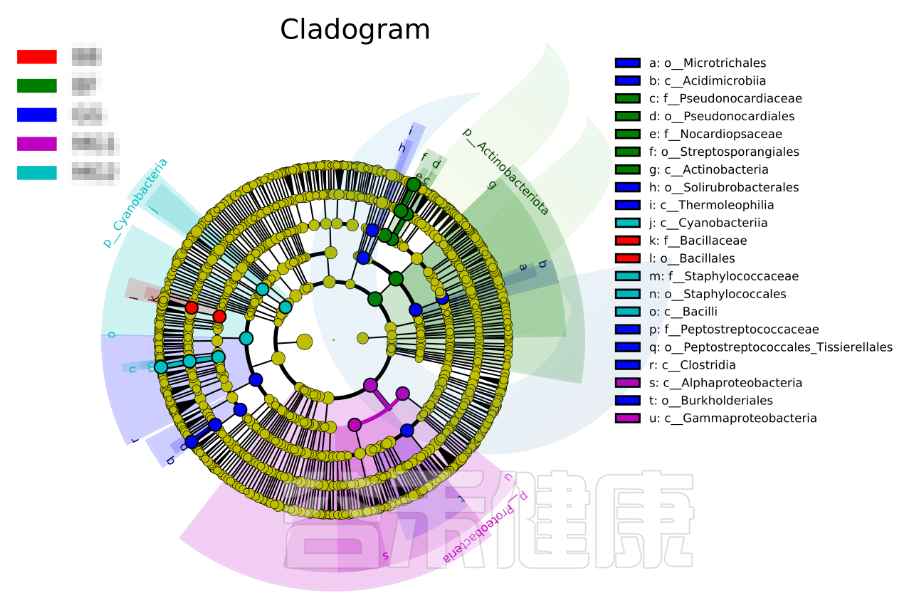
图中右侧展示了分支图中的字母对应的物种信息,例如a 代表GG组的标记物目水平的Microtrichales ,b代表GG组的标记物刚水平的Acidimicrobiia。在分支图的最外层显示的是各分组门水平物种的标记物,例如BF组的是Actinobacteriota、MG1组的是Proteobacteria、
MG2组的是Cyanobacteria
特征表
路径:Group/Lefse_Analysis/out_formant.res.csv
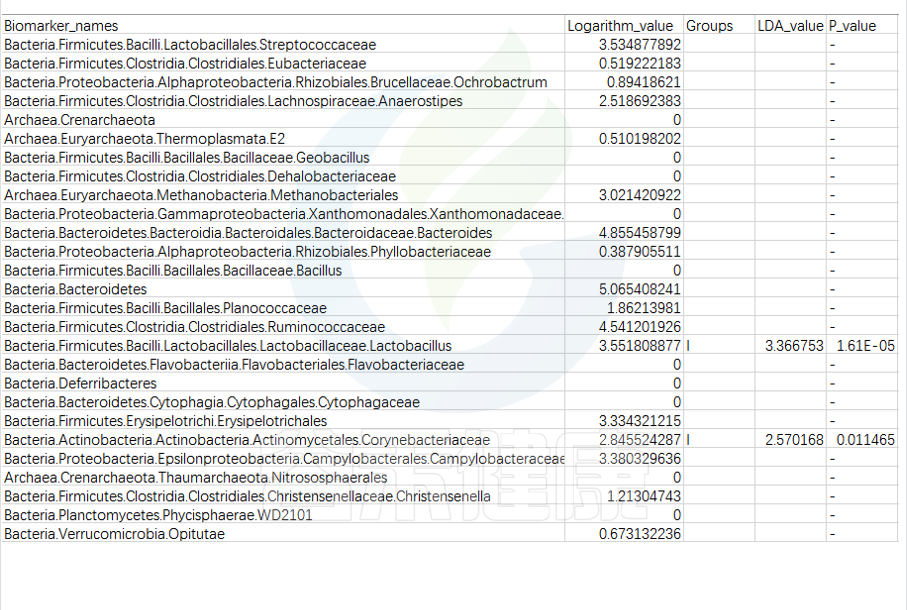
第一列是样本中从门到属水平所有分类单位的列表
Lefse会逐一判断这些分类单位的在分组之间是否具有统计学显著性差异。
第二列:各组分丰度平均值中最大值的log10,如果平均丰度小于10的按照10来计算;如果该分类单位未体现出显著组间差异,则后三列为空。
对于具有统计学差异的分类单位:
第三列:差异基因或物种富集平均丰度最高的分组组名;
第四列:LDA差异分析的对数得分值;
第五列:Kruskal-Wallis秩和检验的p值,若不是Biomarker用“-”表示。
默认LDA>2,P<0.05
通常根据第4列的LDA差异分析对数得分值和第五列的P值,可以描述组间具有显著差异的分类单位统计学效力强弱。
metagenomeSeq
metagenomeSeq是用R开发的一个包,metagenomeSeq的基本思想,用normalization实现分类注释时的biases处理,同时用零膨胀高斯分布(zero-flated Gaussian distribution)处理了测序深度所带来的影响,在此基础上,利用线性模型找到存在的差异所在。
路径:Groups/diff_analysis/ metagenomeRXXX
metagenomeSeq 差异显著物种/功能 热图
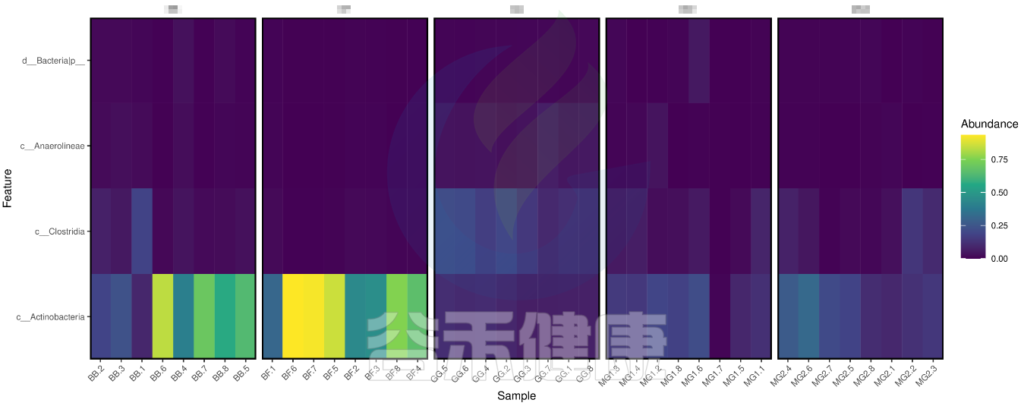
图中颜色越深相关性越小,颜色越接近黄色相关性越大,从图中能看出Actinobacteria物种与BB组和BF组相关性较大。
metagenomeSeq差异菌属于功能代谢关联分析
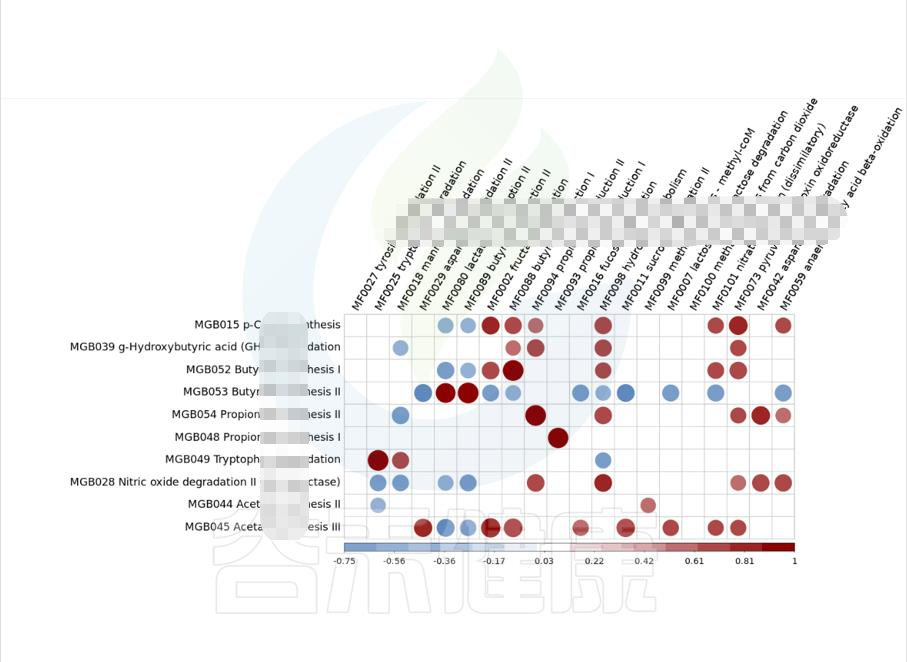
图中红色代表正相关,蓝色代表负相关,颜色越深,圆圈越大,相关性也越大,例如图中能看出MGB049余MF0025 之间成正相关,且相关性较大。
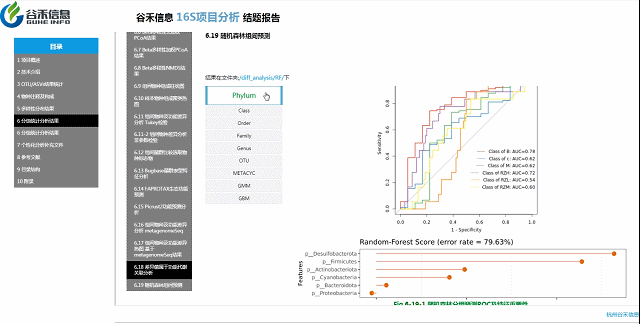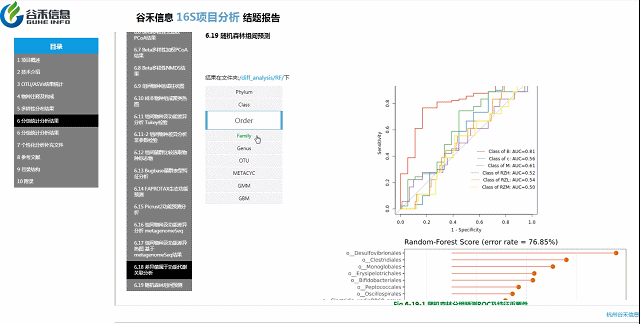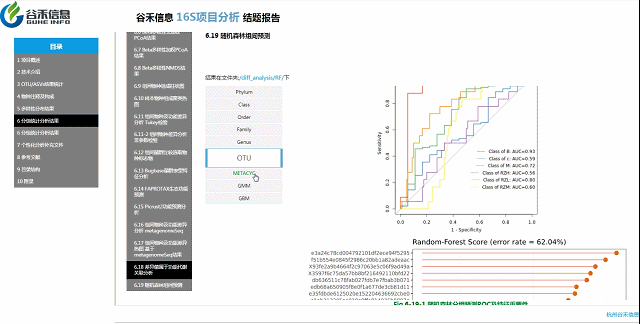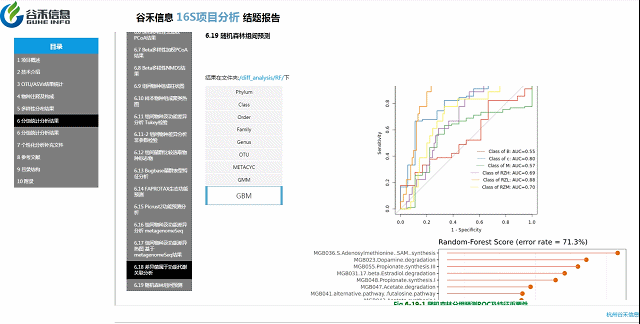
随机森林模型
一种非线性分类器,随机森林属于集成类型的机器学习算法,挖掘变量之间复杂的非线性的相互依赖关系。通过随机森林重要性点图,可以找出分组间差异的关键物种/功能。
反映了分类器中对分类效果起主要作用的特征,按重要性从大到小排列。
Error rate:表示使用下方的特征进行随机森林方法预测分类的错误率,数值越高表示基于特征分类准确度不高,可能分组之间特征不明显。分值越低证明分组效果比较好。
•举个例子
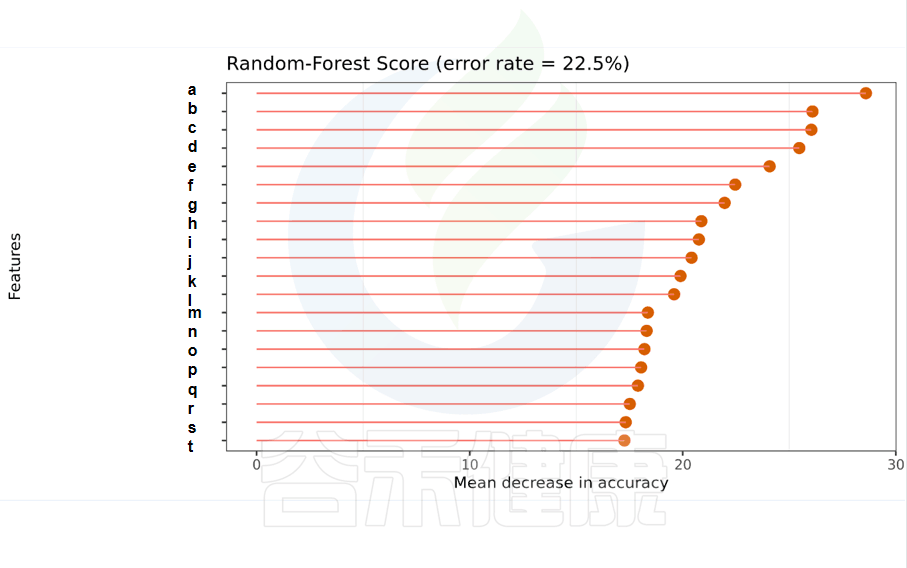
图中按照随机森林模型效果筛选出的对分组效果有重要性作用的物种,按照重要性从高到低进行排列,例如图中最终要的是a,依次往下是b、c等。错误率较小,表明该分组效果较好。
ROC曲线
ROC曲线分析是一种常用的统计学分析方法,在医学研究中主要用于评价诊断试验的效能。在16S测序报告中,我们通过绘制ROC曲线,并计算ROC曲线下面积(AUC),来确定分组对于菌群是否有诊断价值。
ROC曲线图是反映敏感性与特异性之间关系的曲线。ROC曲线下的面积值在1.0和0.5之间。在 AUC>0.5的情况下,AUC越接近于1,说明诊断效果越好。
AUC在0.5~0.7时有较低准确性,AUC在0.7~0.9时有一定准确性,AUC在0.9以上时有较高准确性。AUC=0.5时,说明诊断方法完全不起作用,无诊断价值。AUC<0.5不符合真实情况,在实际中极少出现。
•举个例子
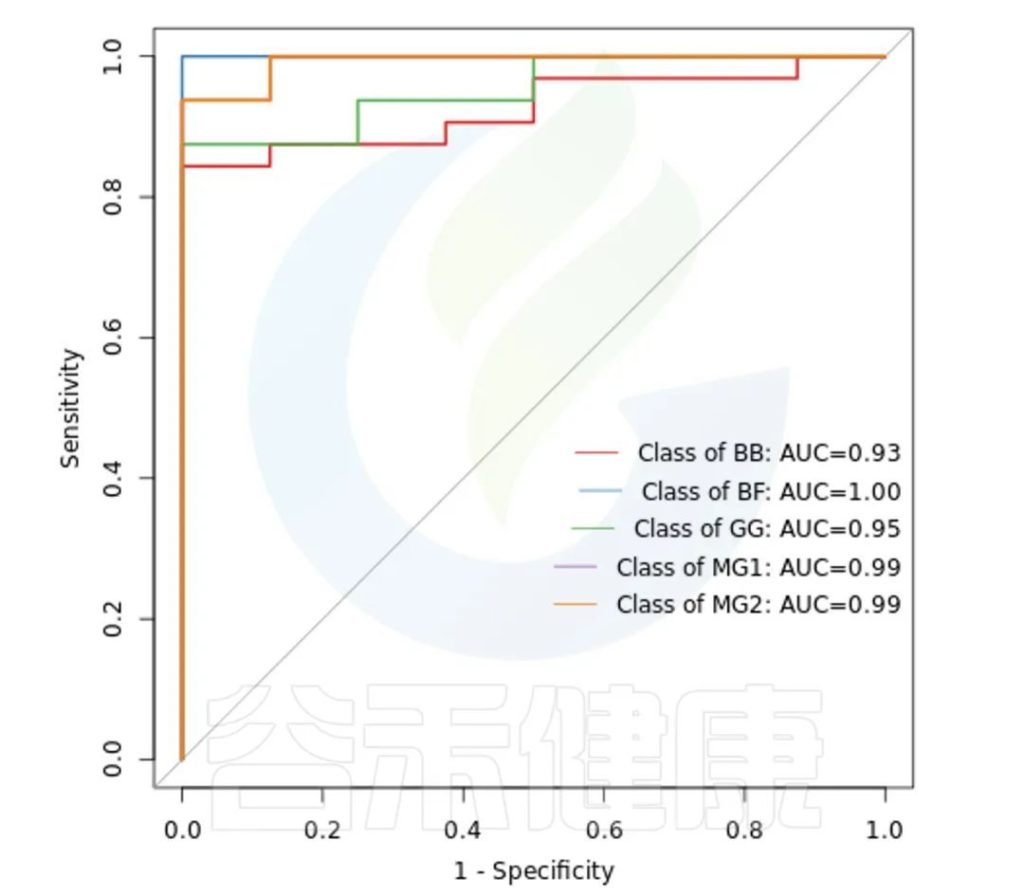
从图中能看出各分组的AUC都大于大于0.9,各分组的分组效果较好,BF组AUC等于1,该分组效果最好,可能样本之间较为相近,并且跟其他分组组间差异也比较大。
以上是组间统计差异的方法介绍,其他的还包括关联分析。
例如客户提供了每个样的相关理化指标数据,想计算这些指标与均属之间有什么相关性,就可以做一下分析。
关联性分析
✦相关性热图
图中X轴代表属水平物种,Y轴代表代谢指标,红色代表正相关,蓝色代表负相关,**代表相关极显著P<0.01,* 代表相关性显著P<0.05相关性具有统计学意义。
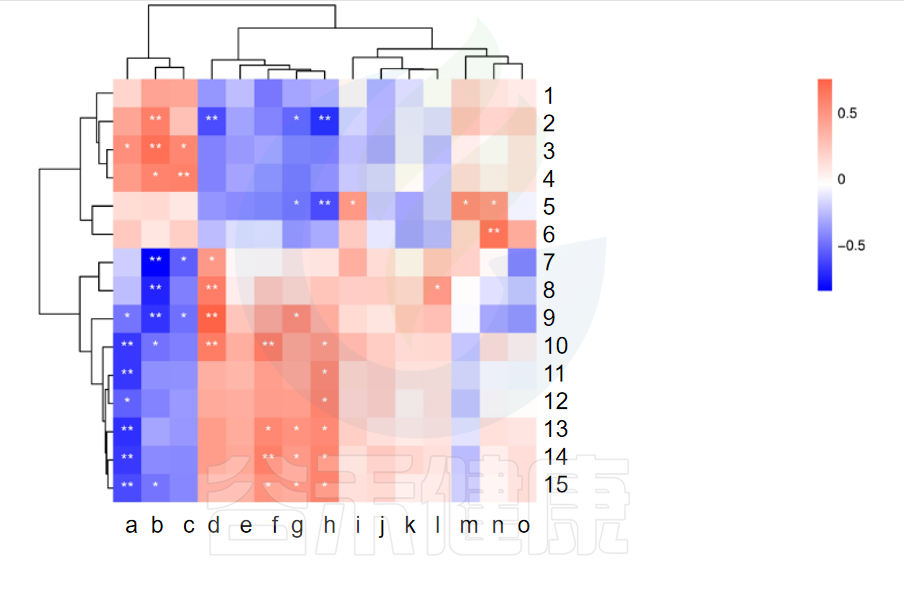
例如从该图中能看出6与n物种成正相关,并且相关性极显著**,7与b物种成负相关,并且相关性极显著**。
可以得到表格:任意菌属和代谢的相关性的值和P值
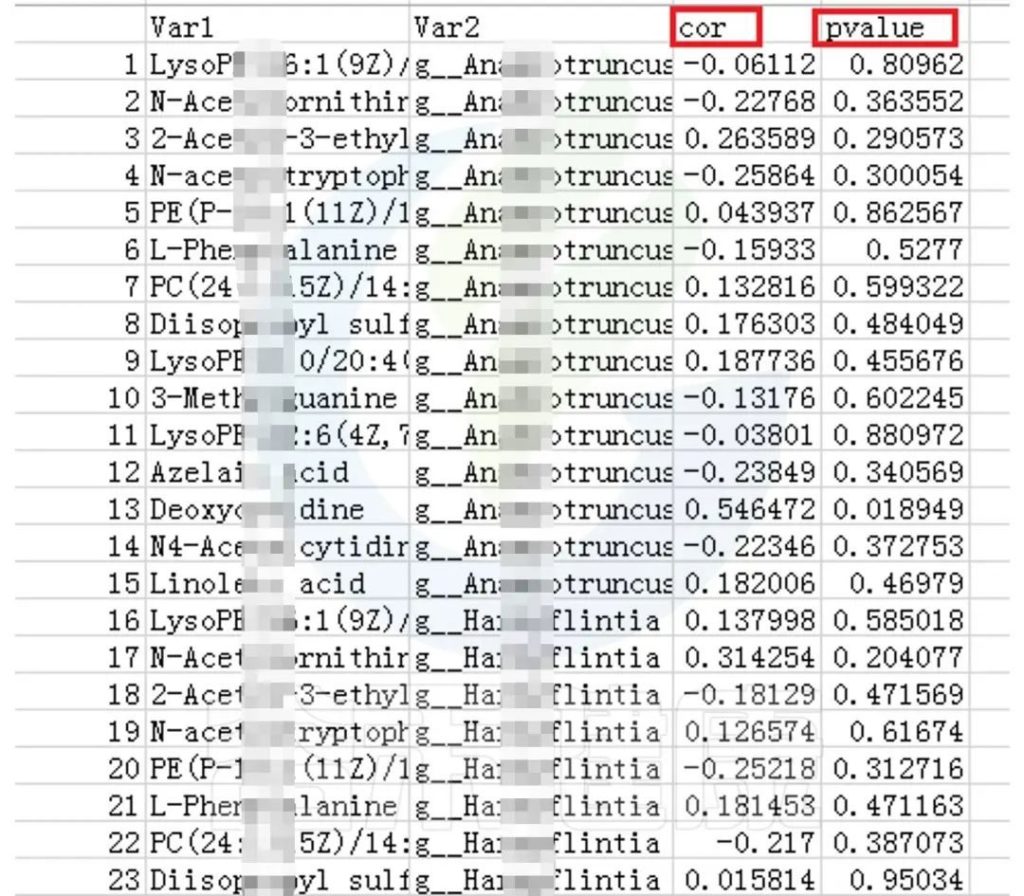
✦CCA图
可以分析样本、菌群、理化指标之间的关联关系。图中使用点代表不同的样本,从原点发出的箭头代表不同的环境因子。
箭头的长度越长,表示环境因子的影响越大;夹角越小,代表相关性越高。样本点与箭头距离越近,该环境因子对样本的作用越强。
图像中坐标轴标签中的数值,代表了坐标轴所代表的环境因子组合对物种群落变化的解释比例。
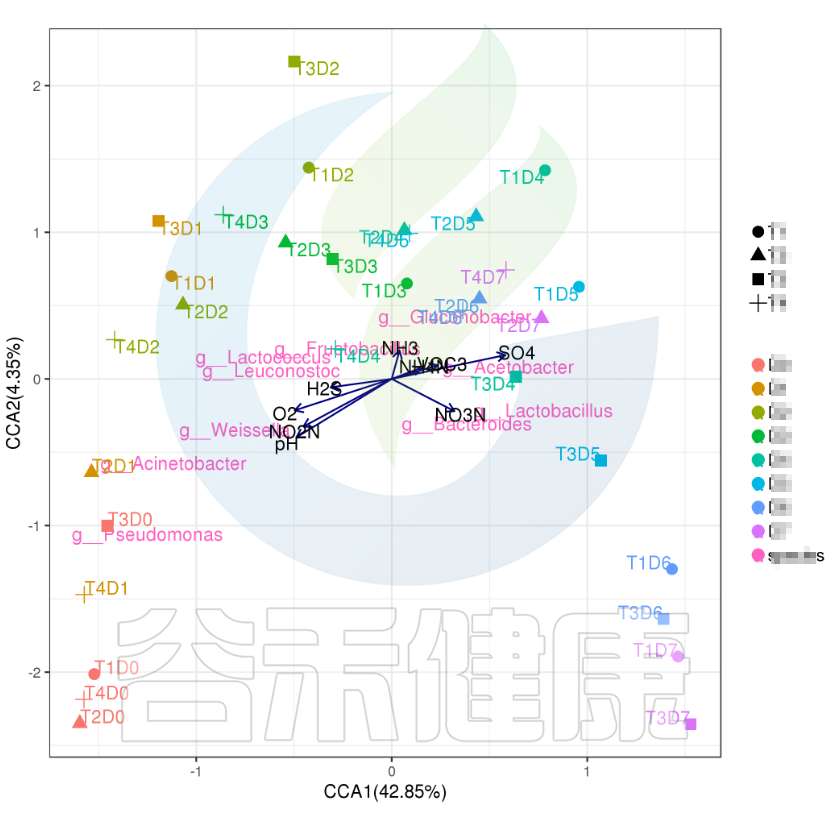
例如从图中能看出pH 、NO2N、02与 Acinetobacter、Weissella等物种成正相关,与T3D0、T1D0、T4D0等D0组的样本成正相关。
✦RDA 冗余分析

例如从图中能看出pH与Helicobacer物种成正相关,相关性较大,pH与NC组有一定的相关性。
✦Envfit分析
回归拟合分析结果:
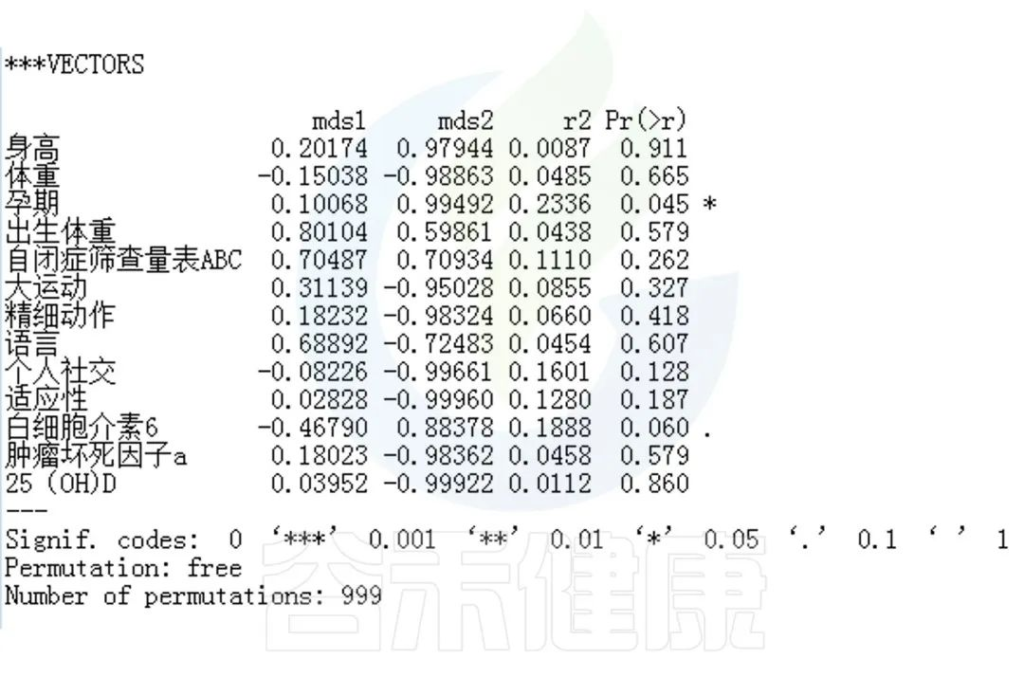
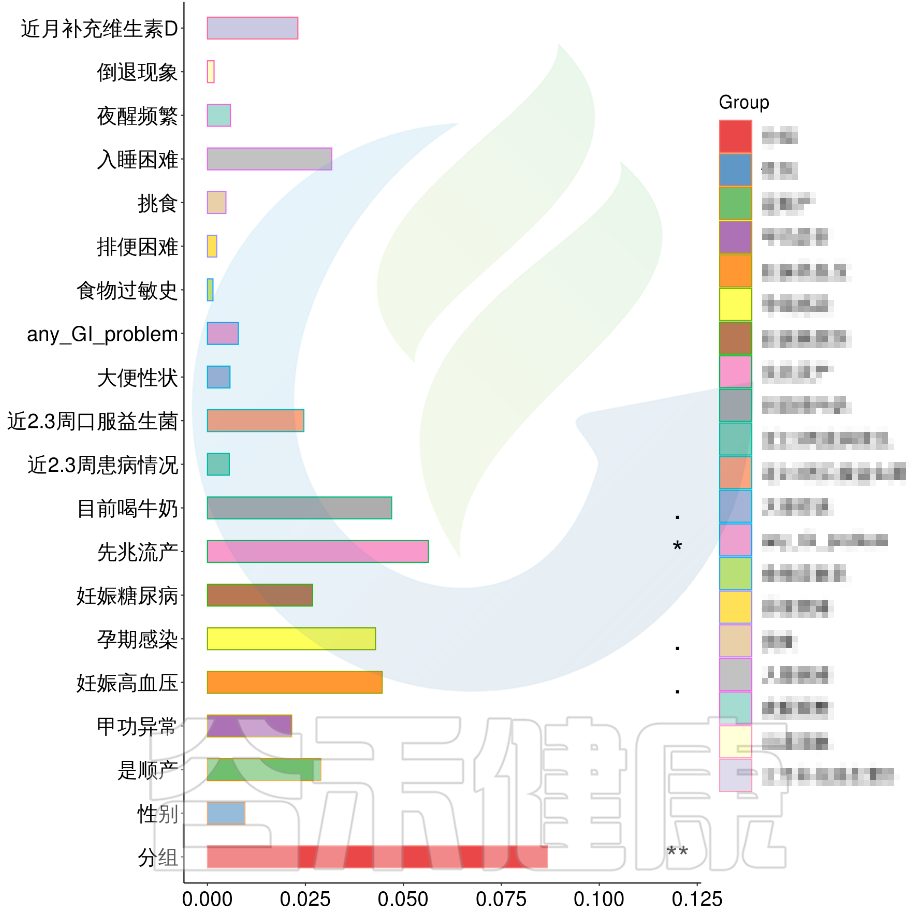
图中能看出ASD与正常儿童之间的分组与菌群之间相关性极显著**,其次是否有先兆流产的分组与菌群之间有显著相关性*,其他的包括是否喝牛奶、孕期是否感染、妊娠高血压都与菌群有相关性。
环境因子与功能/物种的相关性线形图P<0.05显著,图中红色点代表正相关,绿色点代表负相关,灰色相关性不显著。
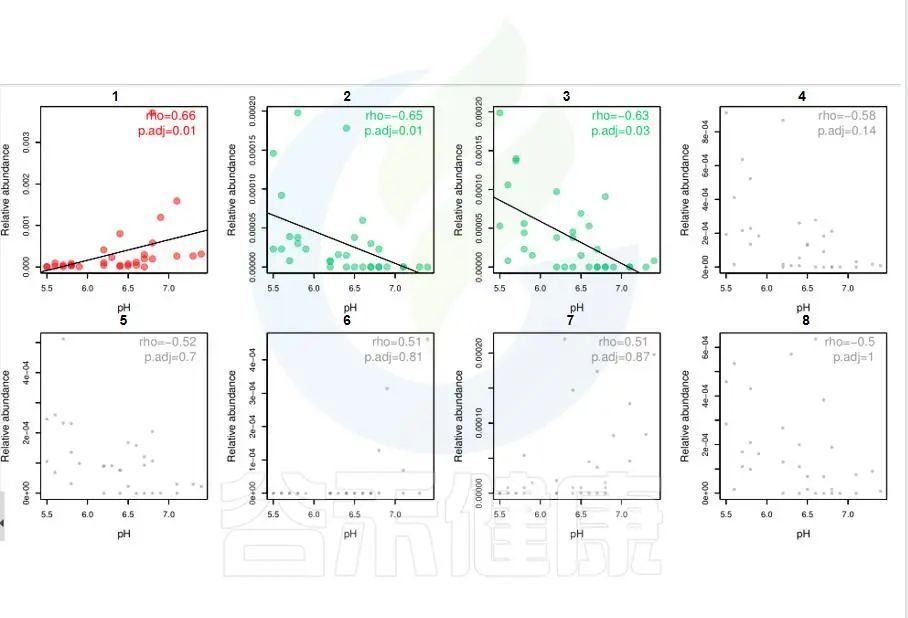
图中能看出pH 与Candidatus Rhabdochlamydia 之间成正相关,且相关性显著,pH 与Sinorhizobium、Euzebya 之间成负相关,切相关性显著。
✦Network网络分析
还可以做菌属之间的网络分析关联图,共发生网络图为研究复杂微生物环境的群落结构和功能提供了新的视角。
由于不同环境下微生物的共发生关系截然不同,通过物种共发生网络图,可以直观看出不同环境因素对微生物适应性的影响,以及某个环境下占互作主导地位的优势物种、互作紧密的物种群,这些优势物种以及物种群往往对维持该环境的微生物群落结构和功能稳定发挥着独特以及重要的作用。
•举个例子
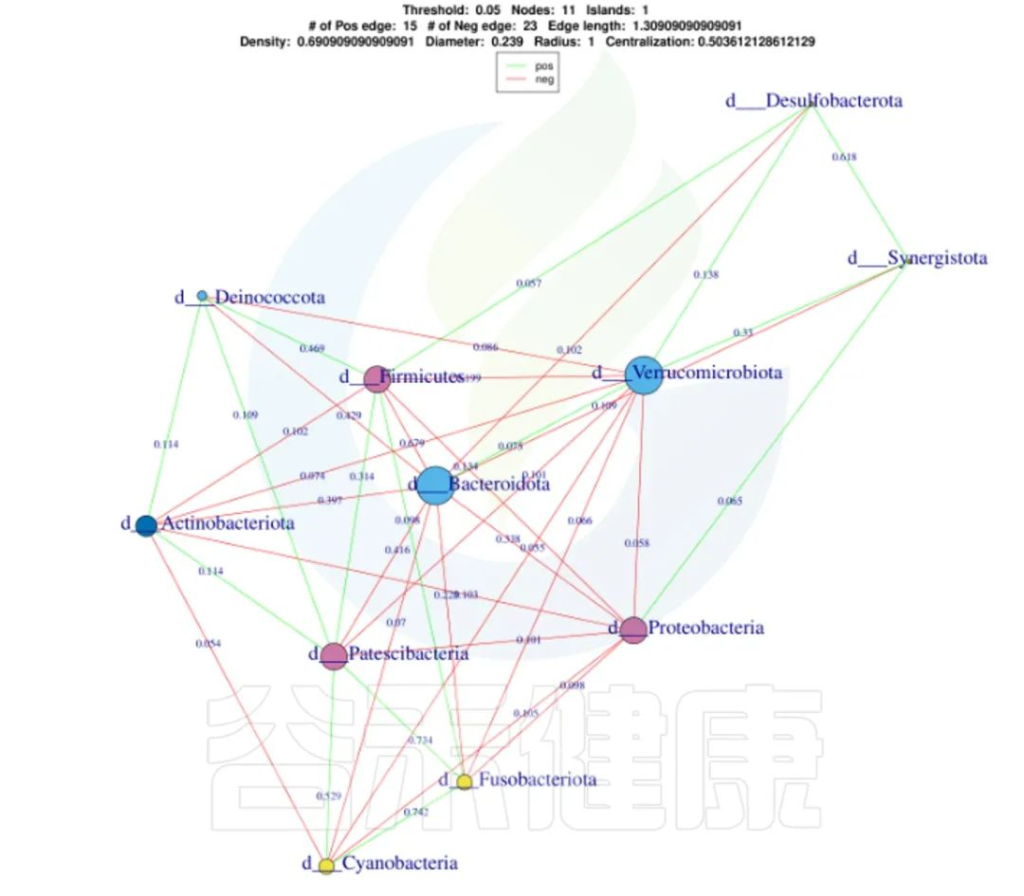
图中展示了相关性的物种,例如Bacteroidota、Actinobacteriota、Proteobacteria 这些物种与其他物种相关较大,图中这些物种与其他物种连线较多,字体比较大也代表相关性较强,例如Actinobacteriota与Deinococcota连线是绿色的代表这两个物种是负相关。
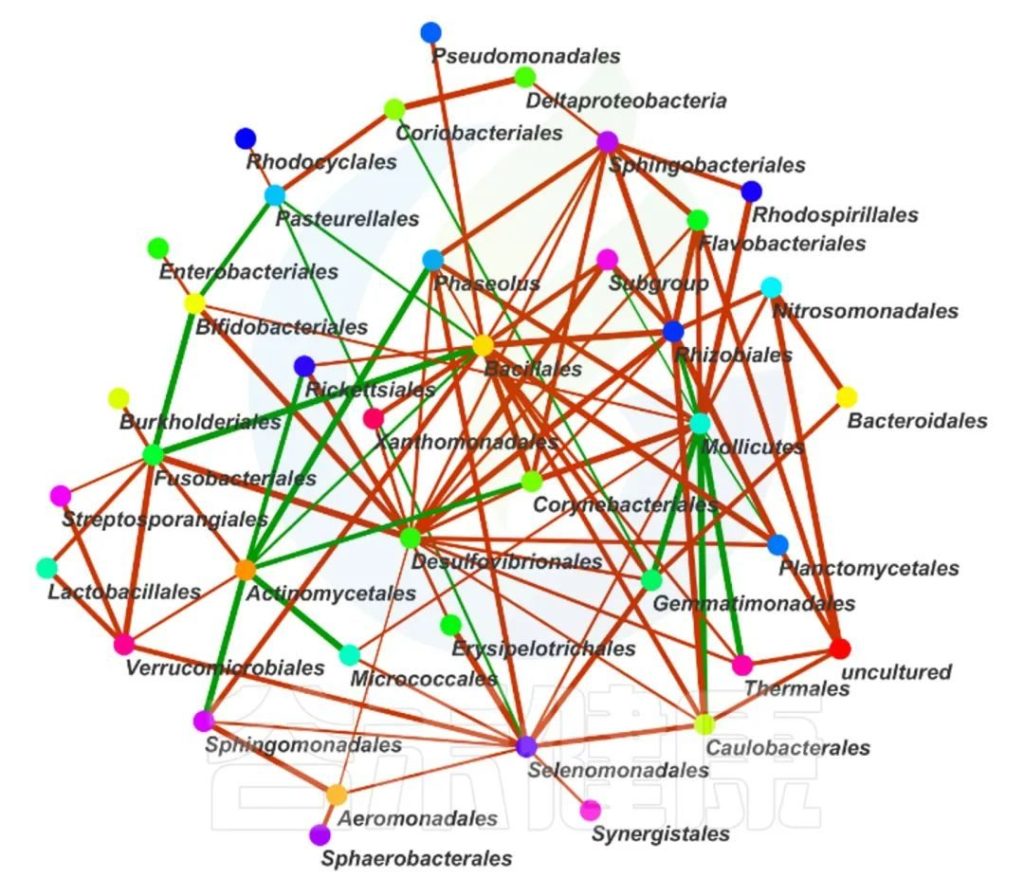
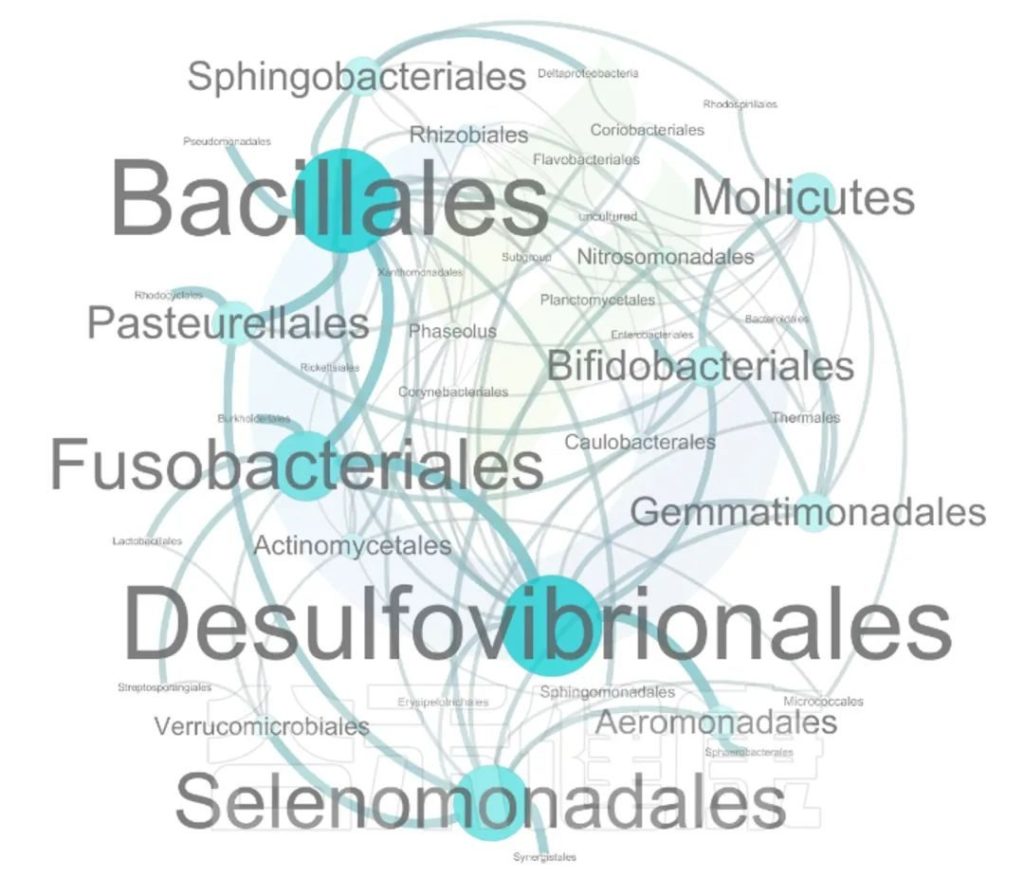
这两个图类似的物种相关性的图,用同一个数据做出来的,图中能看出Bacillales、Desulfovibrionales、Selenomonadales与其他物种相关性较强。
报告中已经基本都涵盖了16S科研数据分析所需要的图表、差异统计,以及相关性分析结果。如果在几种不同类型的统计方法对比之下有略微的差异结果,选取其中一组差异结果即可。
报告里涵盖了大部分16S所需要的图片,不过也有个别个性化的图需要单独用到软件去做,可以单独完成个性化图表生成。
随着16s分析报告的不断升级,报告中的图表以及相应的解读也会越来越精细完善,谷禾也将尽可能为大家的科研之路带来更多便利。


谷禾健康


微生物多样性测序(扩增子测序)是基于二代高通量测序对16S/18S/ITS等序列进行测序。可以同时检测样本中的优势物种、稀有物种及一些未知物种的检测,获得样本的微生物群落组成以及相对丰度。
相信关注我们的小伙伴对此并不陌生。
这次我们整合了大家平时会遇到的一些问题,在原有的基础上对报告进一步完善。



重要指数 :★★★★★
这部分内容必看。
主要是汇总信息,包括样本数据量,测序质量,重复性效果评估,分组信息,组间差异评估,代谢途径上差异,功能预测等。
这里会给出本项目中的一些重要提示,帮你从众多的报告信息中获取关键的部分。


重要指数 :★★★
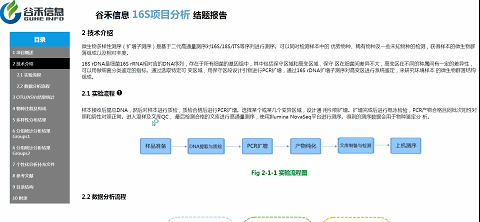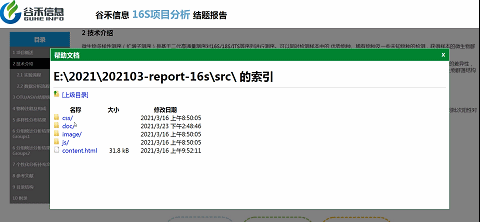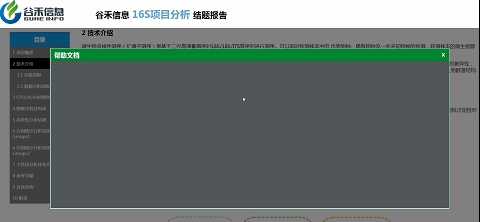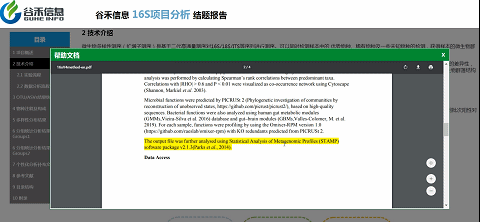
技术介绍这部分内容,就是说我们基于是怎么样一个测序平台、什么方法来获得的最后的数据。

如果你担心
这么直观的报告,
会不会不够详细?
小问号里有宝藏!



如上图,点击实验流程旁边的小问号,弹出的文件夹里就有详细的英文版方法介绍。
重要指数 :★★★★
这部分内容主要是数据统计的图表:
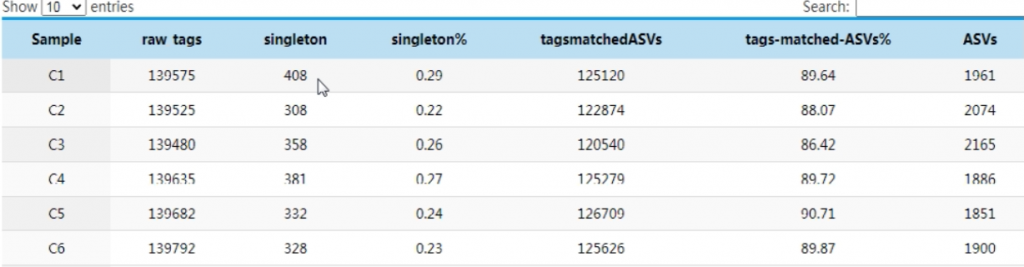

Raw-tags: 样本的原始序列数据
Singleton: 无完全匹配的单条序列数量
tagsmatchedASVs: 比对到最终ASVs的序列数据
ASVs:以及ASVs的种类个数

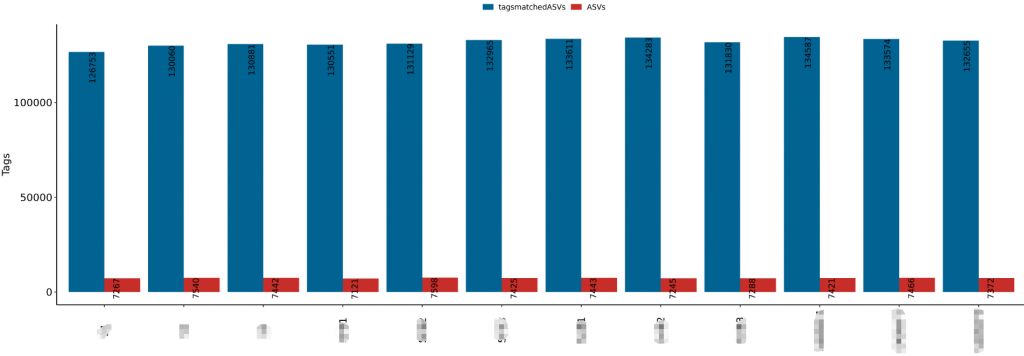


重要指数 :★★★★
经过SILVA138数据库的注释,得到ASVs的物种注释结果。
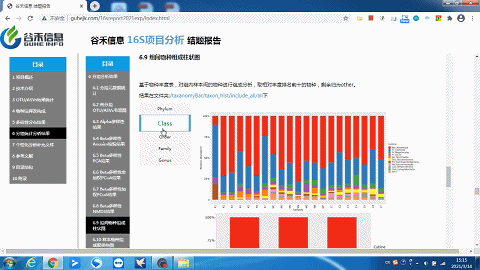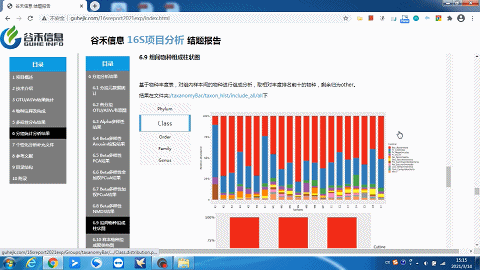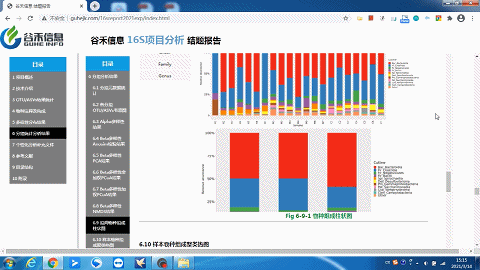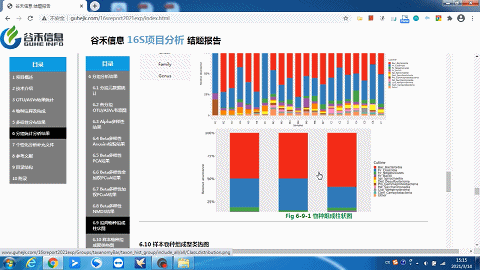
这一部分可以看到每个样本的物种构成比例,Taxonomic Level 可以选择Level1 ~ Level7 界门纲目科属种,不同分类水平下的物种构成。
这里选择level2就是“界”层级(可根据需求自选),另外比如选一个groups分组,如下:
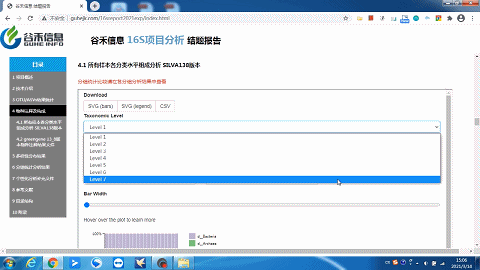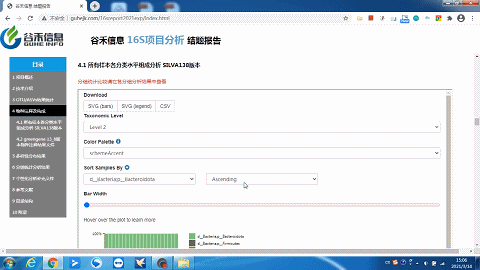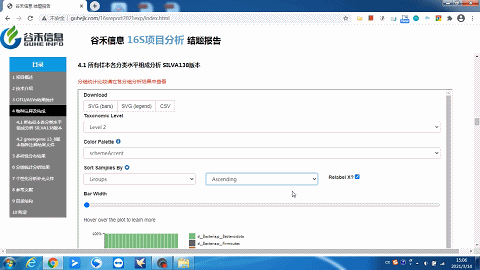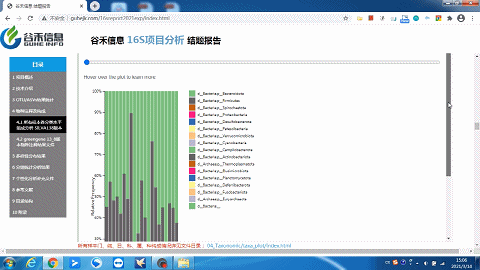

柱状图太宽?太窄?
一拉即可调整!
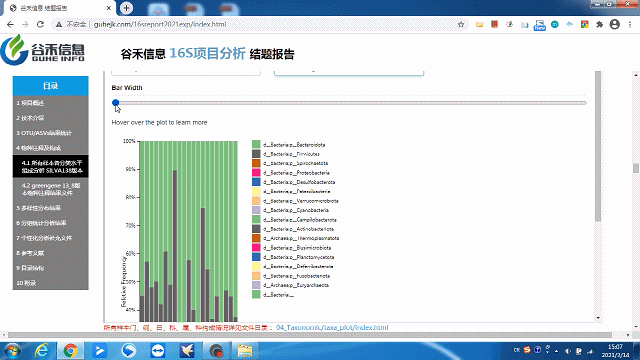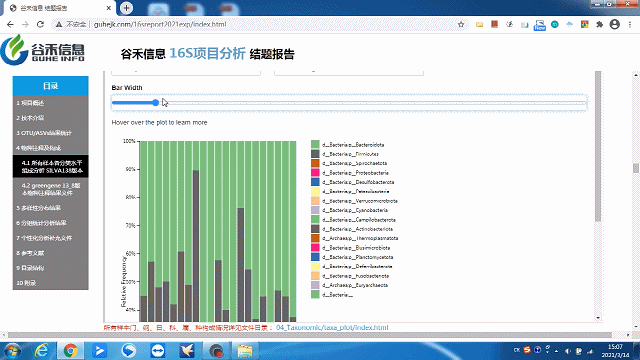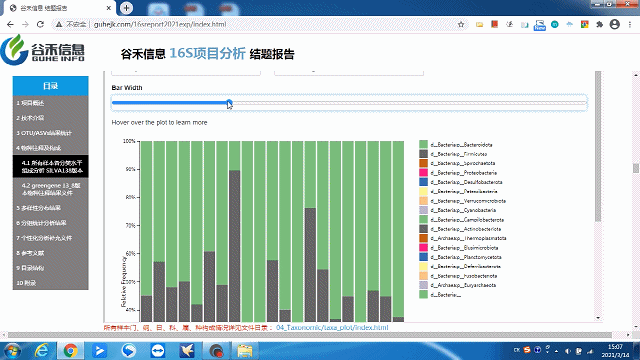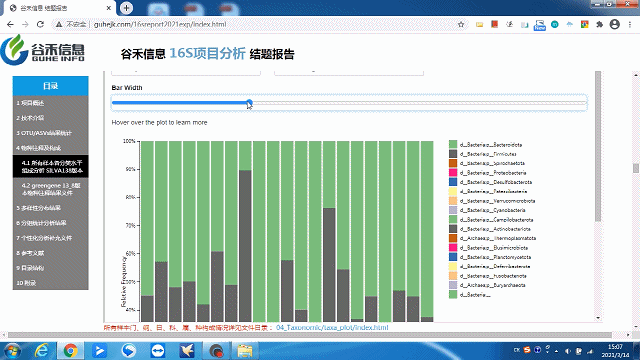

同时给出了各分类水平的相关原始数据,可以到对应路径进行查看。


重要指数 :★★★★
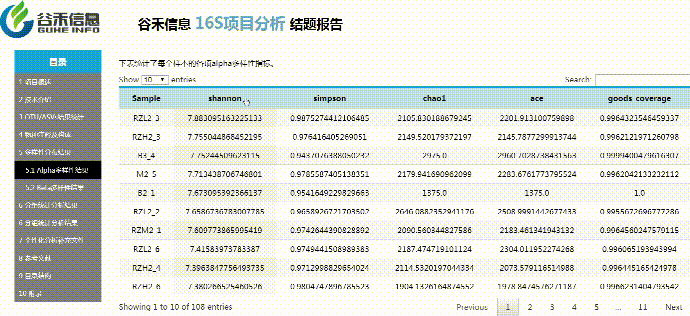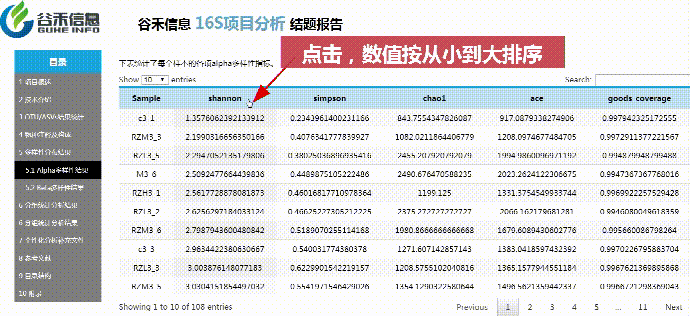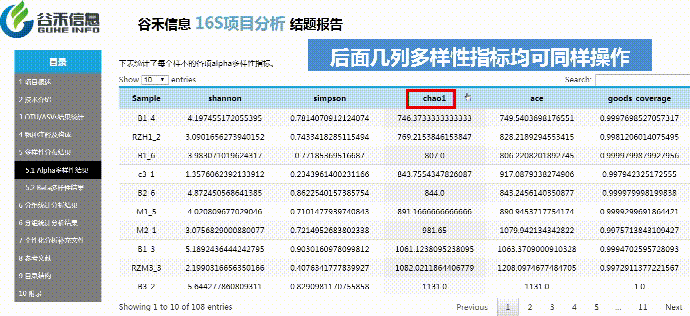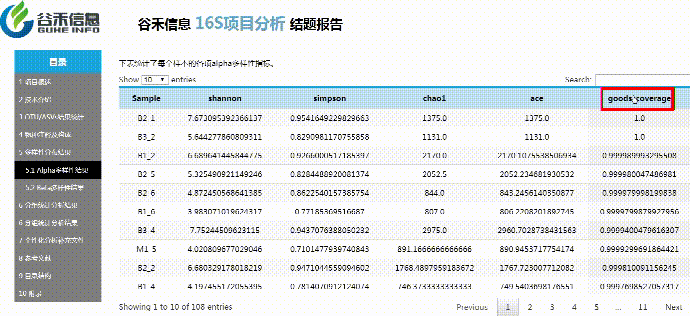
α多样性
评估单个样本内的物种构成的丰度情况
使用Qiime2进行α多样性分析,分别计算获得simpson,ace,shannon,chao1以及goods_coverage数据统计结果。

β多样性
通过降维的方法来考察样本与样本之间的相似度和关系,种属构成特征。
三种聚类方式:
Beta多样性PCA、非加权距离的PcoA、加权距离的PcoA的3D图。
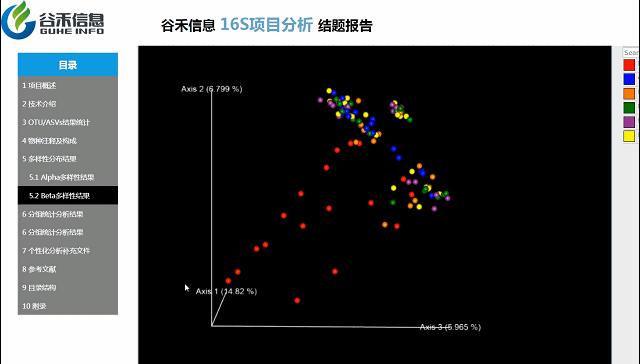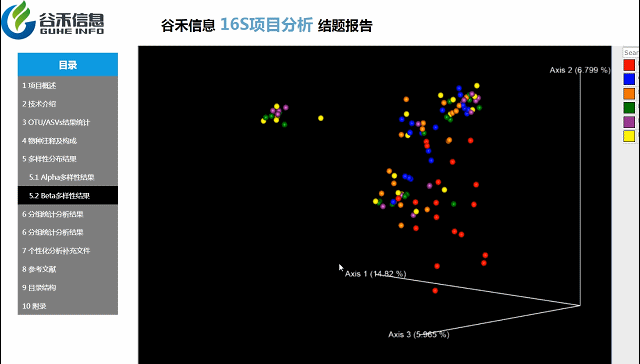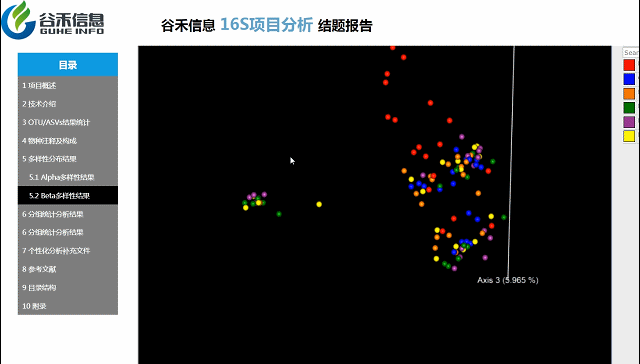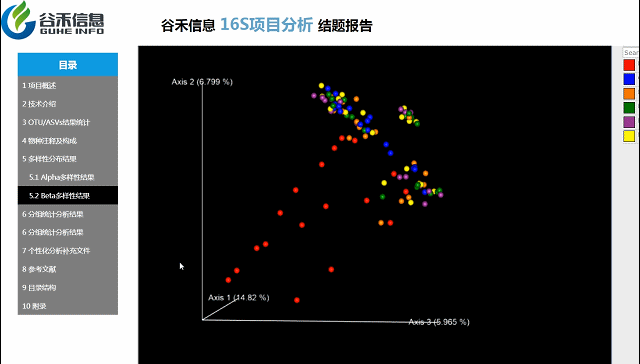

按住鼠标随意拖动,可以看到任意角度的三维坐标自由变换。
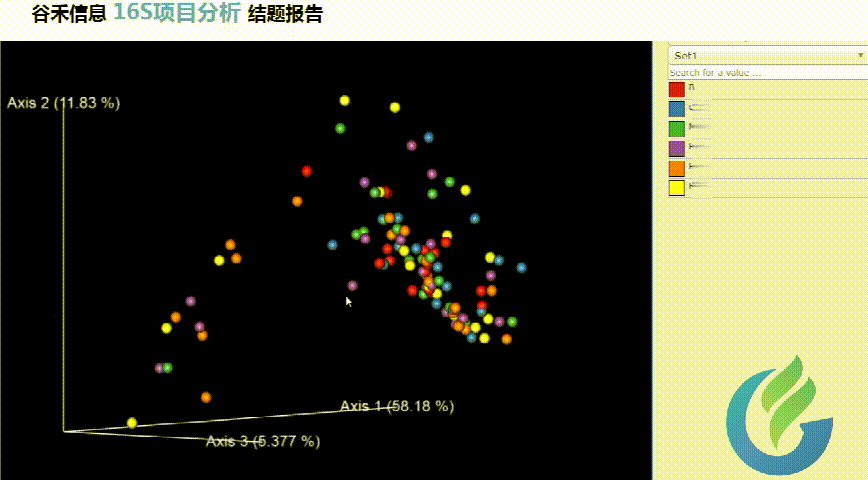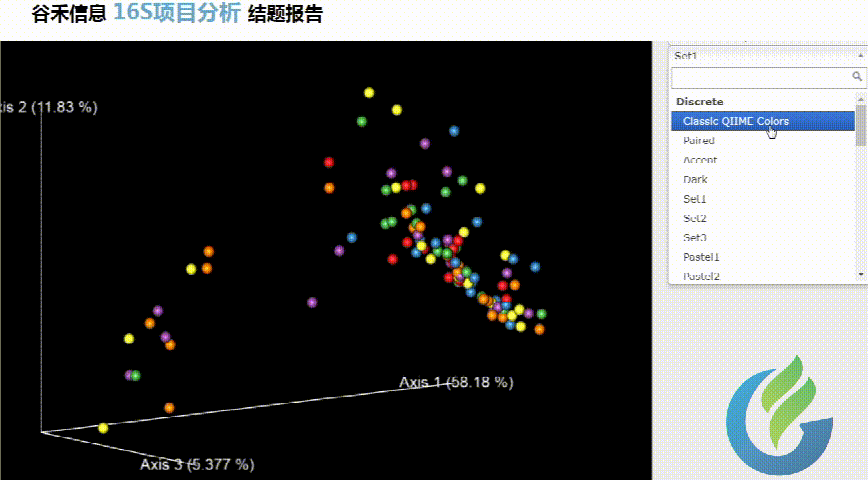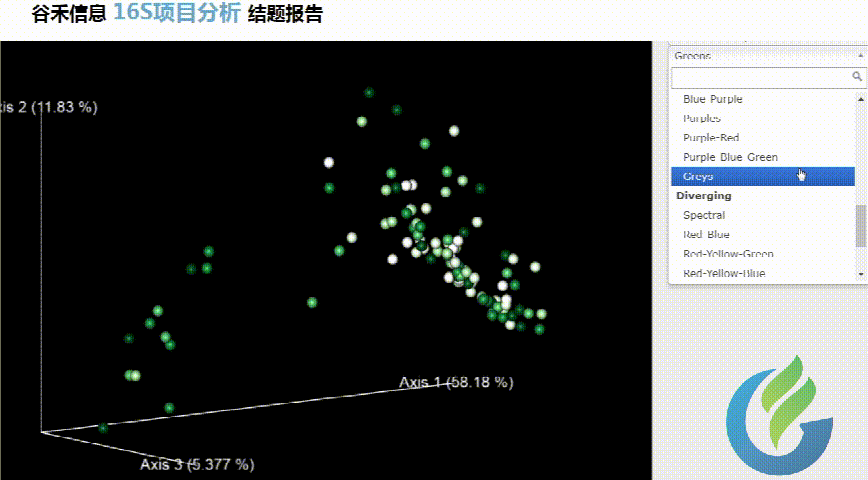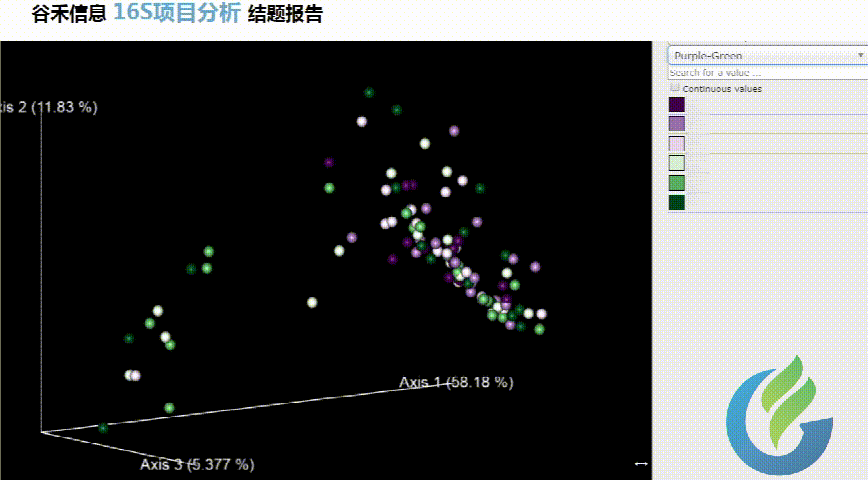

大小可自行调整
多色系任你挑选
总有你想要的图
重要指数 :★★★★★
按照你填写的样本信息单,对各分组情况,进行统计学差异分析。
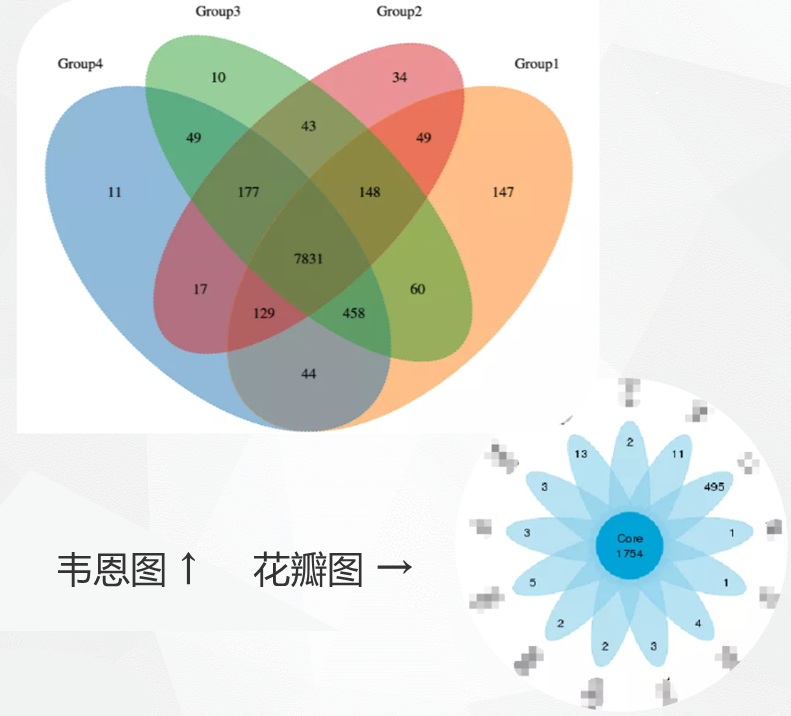
分组Venn图
OTU/ASVs比较韦恩图(样本数/分组数<=5个样本,若分组数大于5出花瓣图)

分组元信息统计
对分组样本及其元数据进行统计
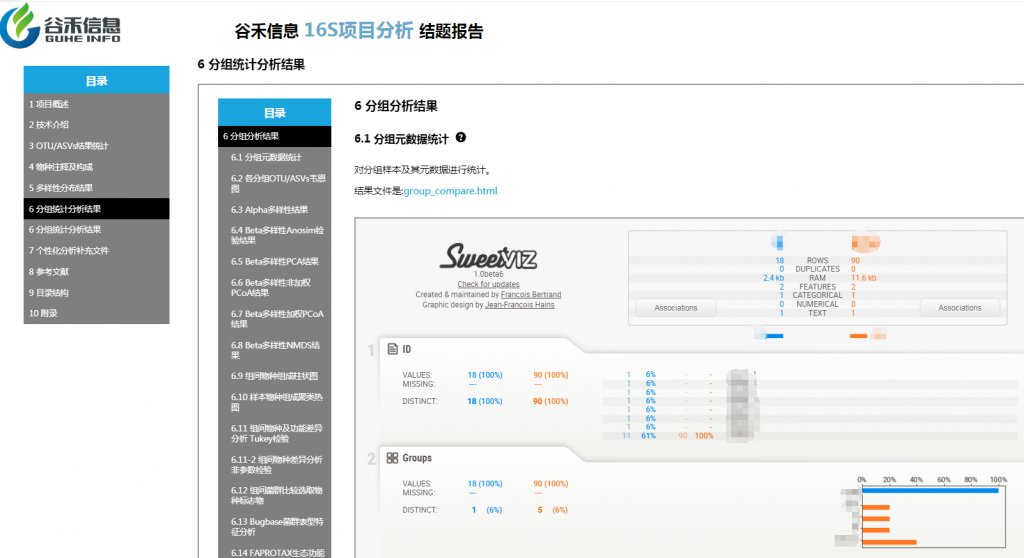

α多样性
分组之间alpha多样性指数使用非参数统计检验
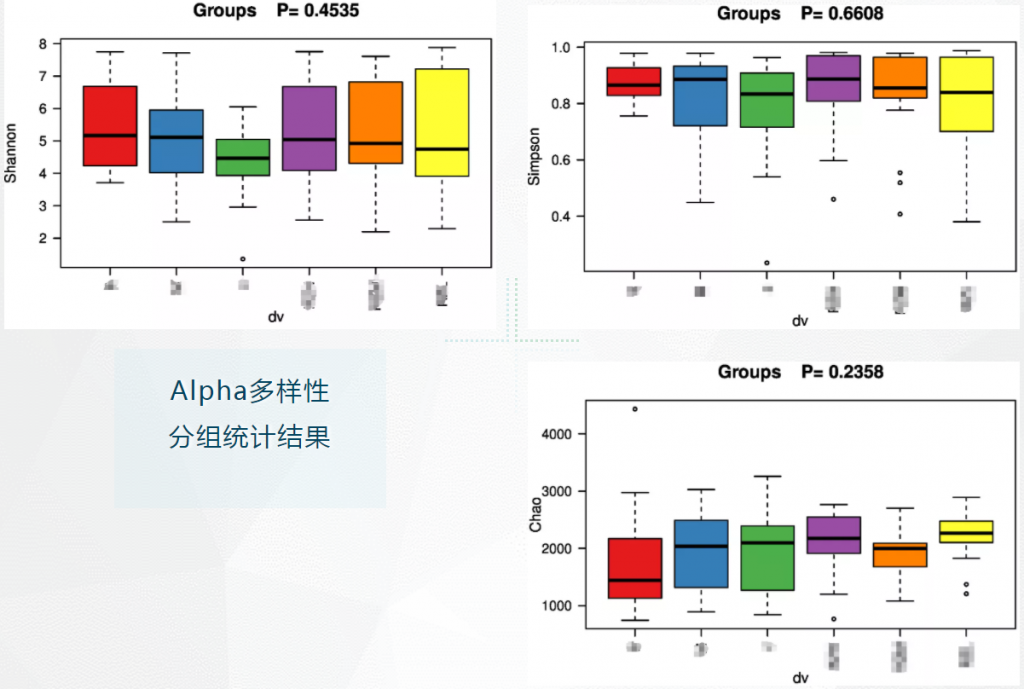

分组是否有意义?——β多样性
Beta多样性分组Anosim检验结果
Anosim分析是一种非参数检验,用来检验组间的差异是否显著大于组内差异,从而判断分组是否有意义。
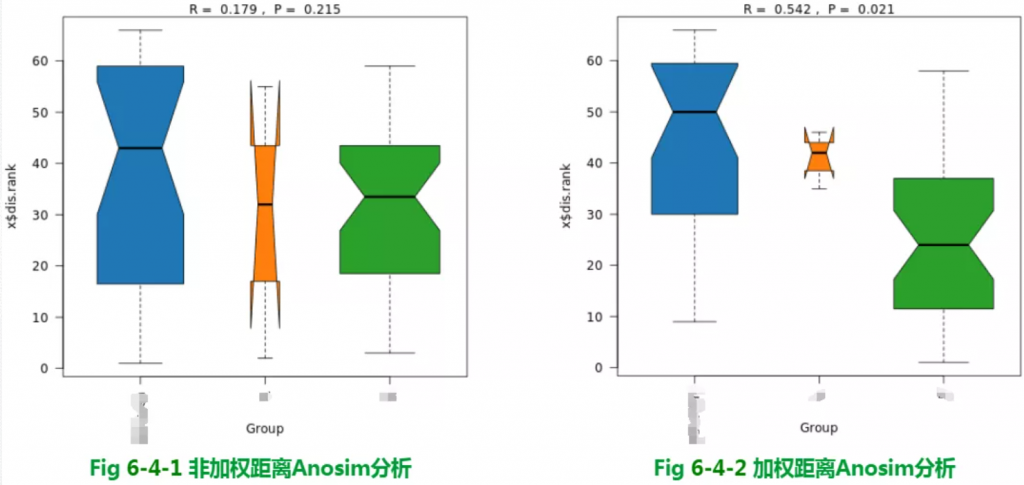

要PCA结果图?
要PCoA结果图?
要NMDS结果图?
要加权?非加权?
… …
全部都有
Beta多样性PCA结果
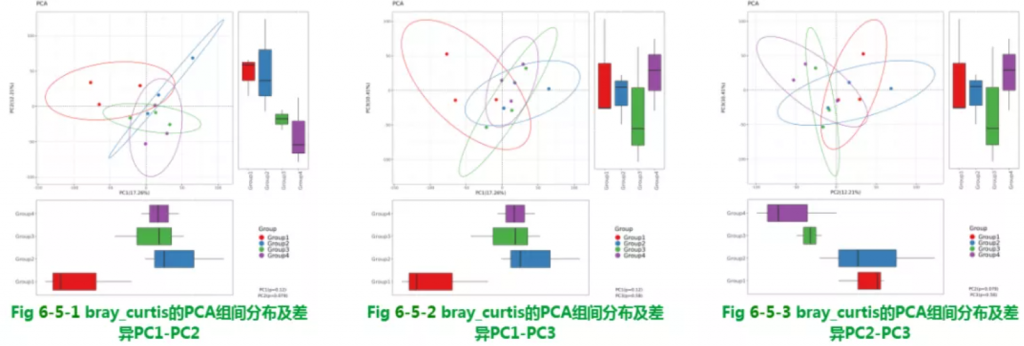

使用bray_curtis的PCA组间分布及差异
Beta多样性非加权PCoA结果
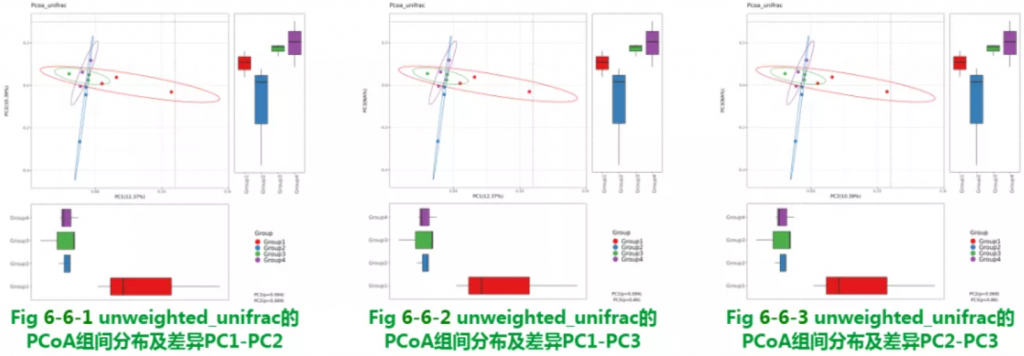

使用unweighted_unifrac的PCoA组间分布及差异
Beta多样性加权PCoA结果
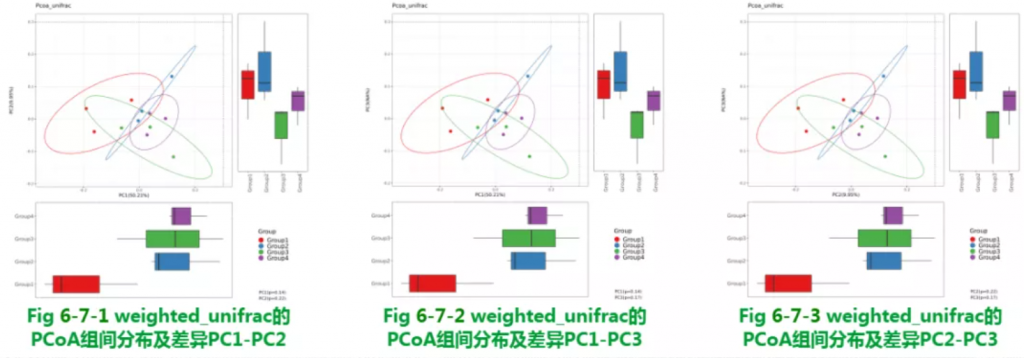

使用weighted_unifrac的PCoA组间分布及差异
Beta多样性NMDS结果
非度量多维尺度分析 NMDS 分析与 PCoA 类似,也是一种基于样本距离矩阵的分析方法,通过降维处理展现样本特定的距离分布。
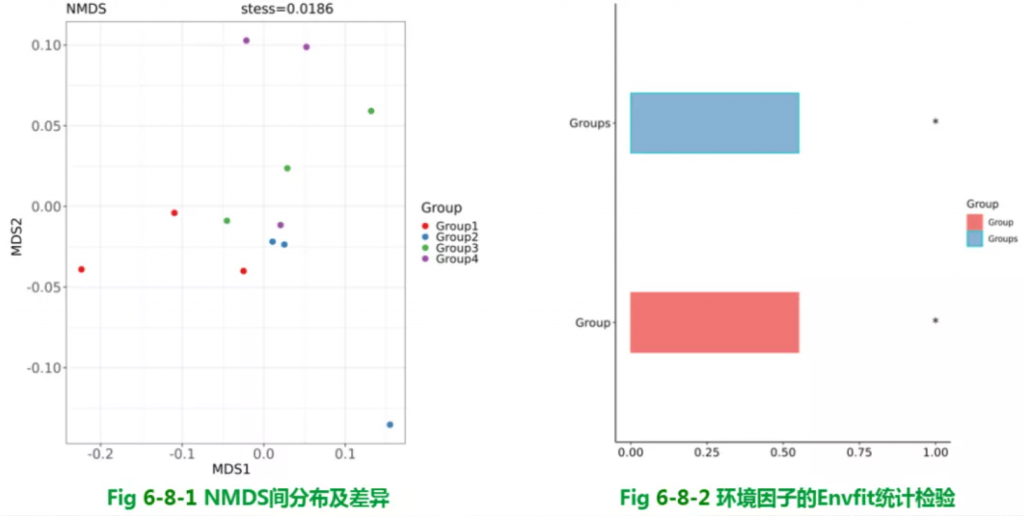

通过对样本距离进行等级排序,使样本在低维空间中的排序尽可能符合彼此之间的距离远近关系(而非确切距离数值)。因此,NMDS 分析不受样本距离的数值影响,对于结构复杂的数据排序结果可能更稳定。


你想要的层级或分组都有——组间物种构成柱状图



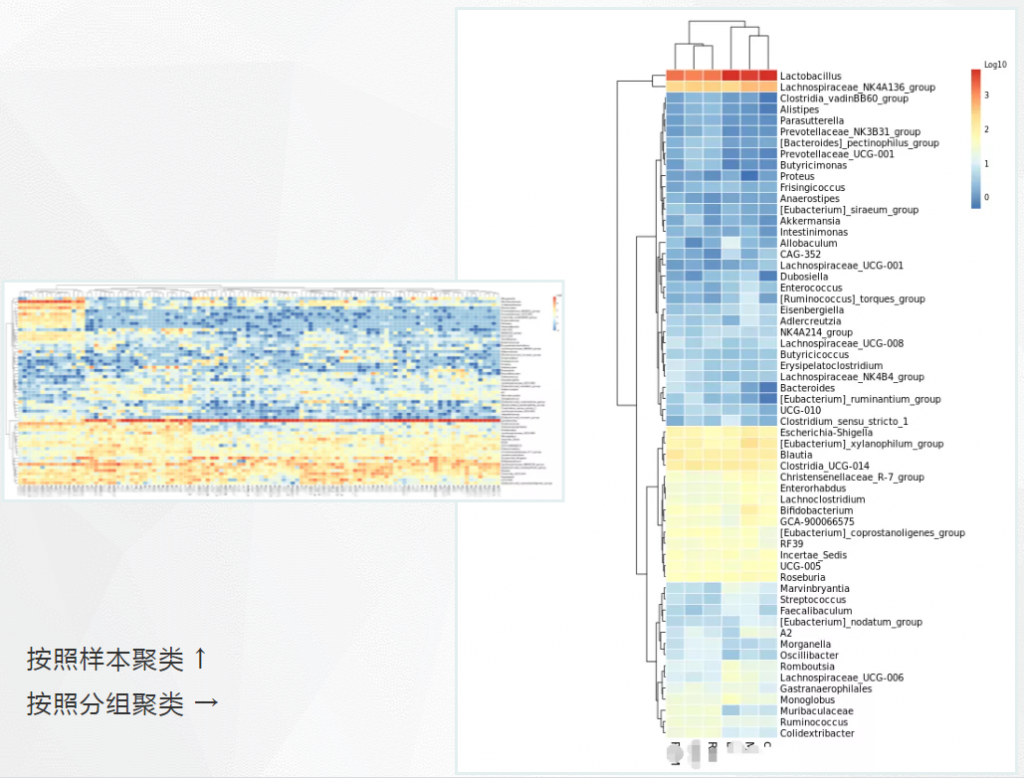
样本及分组之间聚类热图
了解样品之间的相似性以及属水平上的群落构成相似性。

组间各物种分类水平及功能差异
Tukey检验
如果样本每个分组是完全均等的情况(比如说每个组各有10个样),适合用Tukey检验。

优势:
可以快速在图中表现出多个分组之间,哪两个之间存在显著差异。
组间各物种分类水平
非参数检验

各个层级均有相对应的图展示。
组间菌群比较选取物种标志物
Lefse分析

基于线性判定的方式,筛选组与组间的生物标记物——也就是说找到组间存在特别显著的高丰度的菌属。
Bugbase菌群表型特征功能预测分析
基于文献的一些分类,对菌属进行菌群表征,包括对厌氧/好氧,革兰氏阴性/阳性,生物膜形成等分类。


环境样本工具?——FAPROTAX生态功能预测
整合文献原核功能数据库,偏向于代谢和生物学功能的注释。比较适合环境样本,比如说碳、氢、氧、氮、硫等元素的代谢循环的能力。



基因功能预测?——Picrust2功能预测分析
随着研究的不断深入,很多菌的基因组数据有了,基于基因组数据一旦能确定其物种来源,可以推测它具有的基因的拷贝数、代谢通路的构成特征。

2万多的物种,基因覆盖更完整
还包括了CAZY,GMM,GBM等模块
具体差异的意义要结合你的实际研究目标解释
组间各物种分类水平及功能差异
MetagenomeSeq分析
更保守,结果可靠性更高

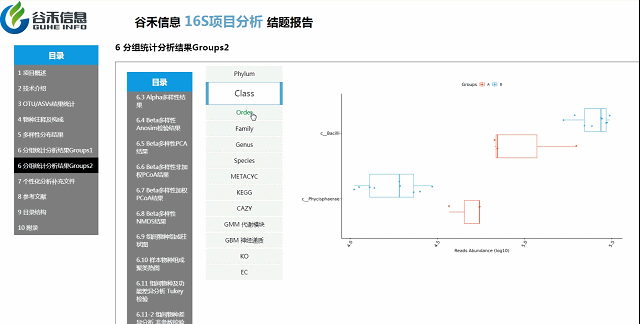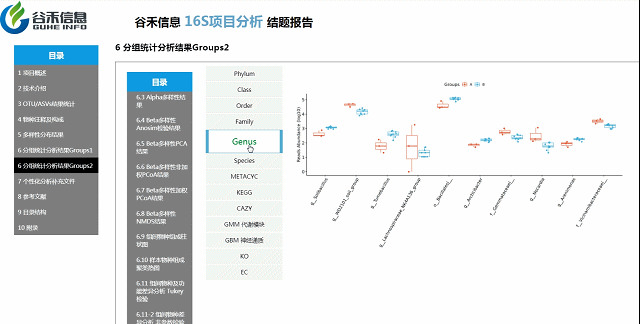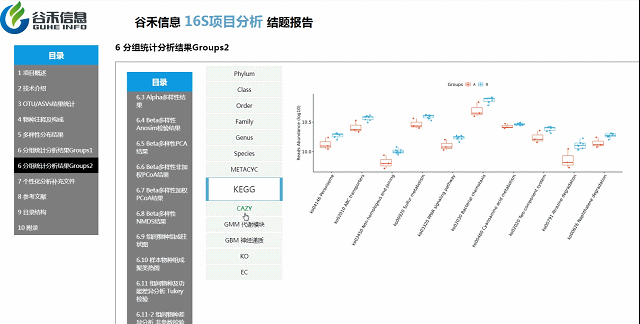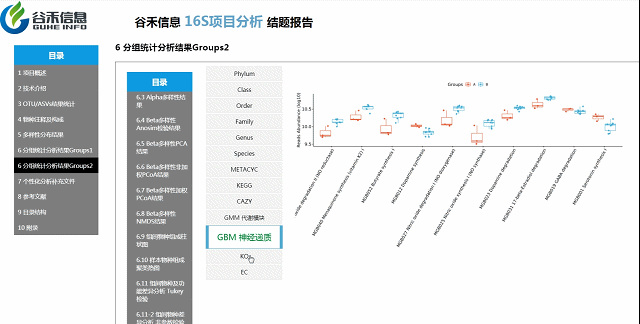

组间物种及功能差异热图
基于上面MetagenomeSeq的结果中,找到差异的物种种属和代谢通路做的热图。

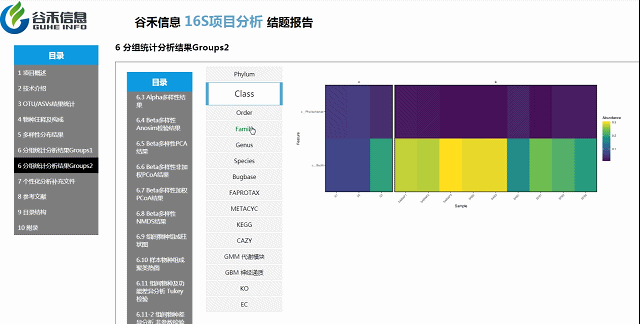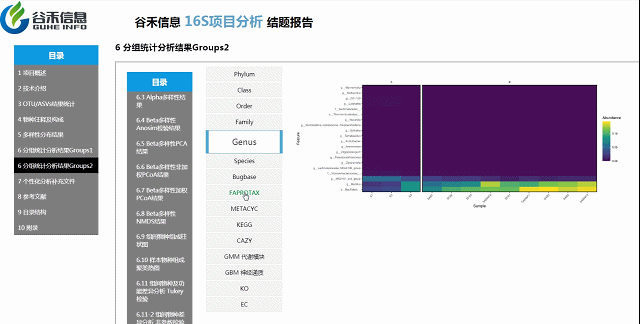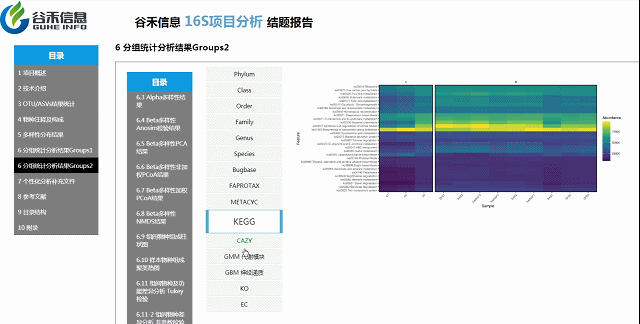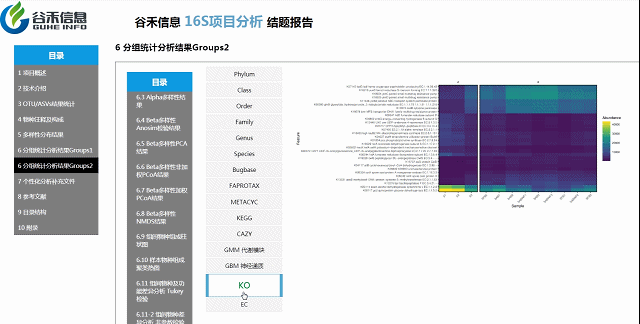
差异菌属与代谢通路之间有什么关系?
差异菌属和功能代谢关联分析

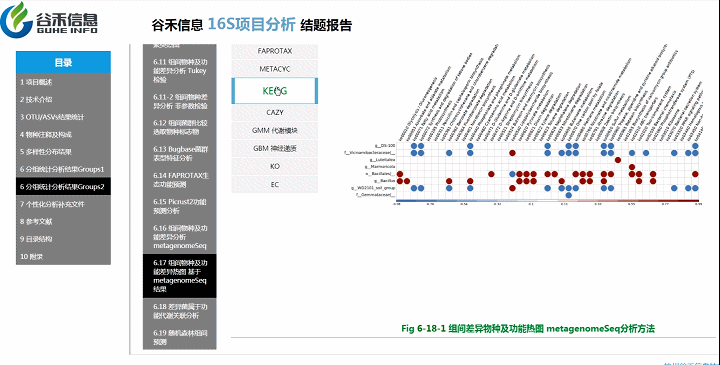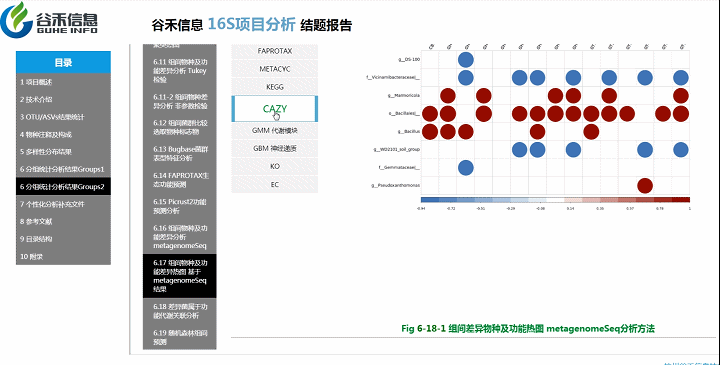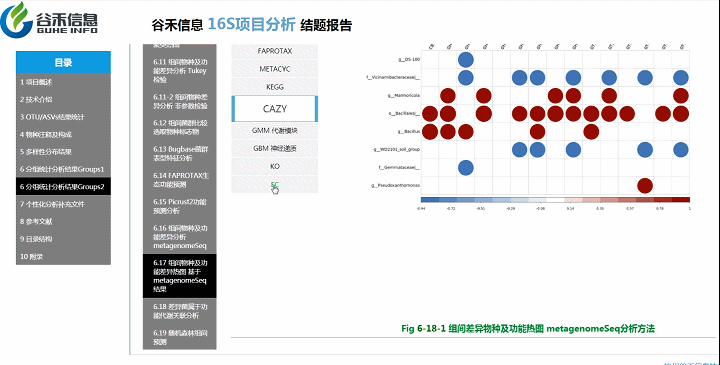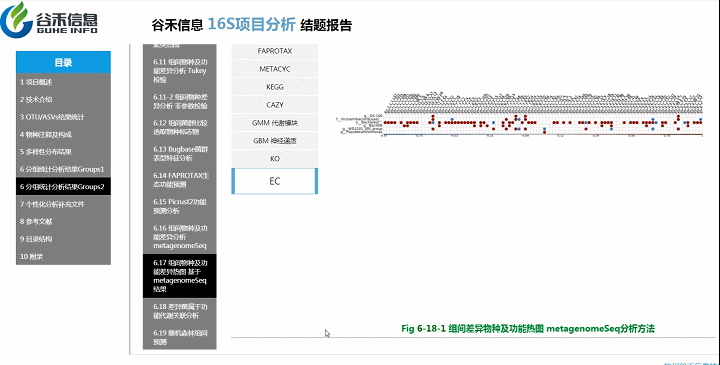
从菌属上的差异,代谢通路的差异等来看,到底是如何关联,是什么类的菌或代谢通路作出贡献。
不同分组之间相对明确区别的模型?
随机森林预测

判断是哪个层面上的数据能最大程度作为分组样本的区分,以及区分效果。



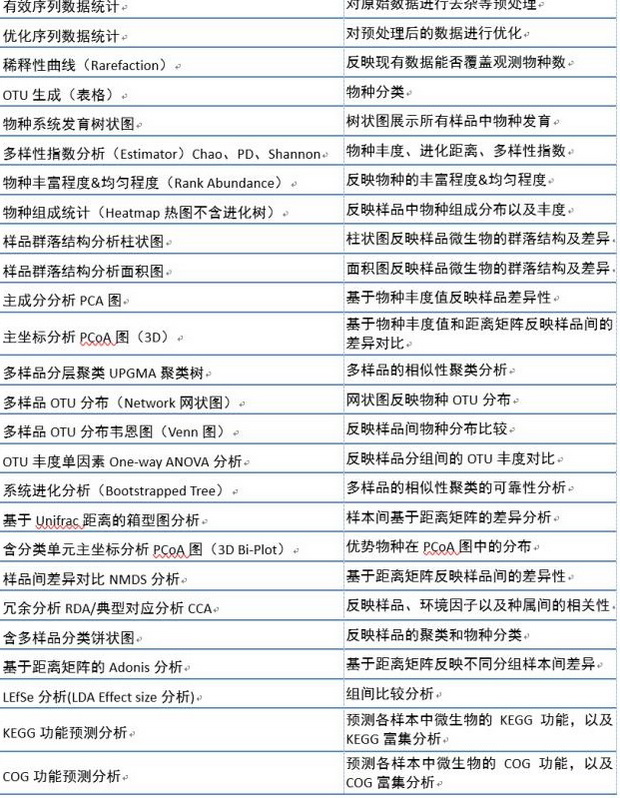
我们提供的基础分析包括以下所有内容:


相关阅读:

针对肠道菌群研究,谷禾进行了一系列的研发和优化,为菌群研究人员提供全套的技术服务。
目前,与谷禾合作的老师们已在各自研究的领域不断发表文章。下面挑选近期几篇文章和大家分享,涵盖了水体,小鼠,土壤多个领域。
最近合作发表文章示例展示:
一 水体样本



01 研究背景
迅速的工业化、城市化和人口增长导致河流污染和水生生态系统的退化。几十年来,许多研究评估了水生生态系统的健康状况,并监测和恢复了河流和湖泊的功能。然而传统的生物指示物种,例如无脊椎动物和硅藻,在严重污染的河流中面临更高的灭绝风险。因此,需要新的敏感和可靠的指标来识别和监测污染严重的河流,如黑臭河的变化。
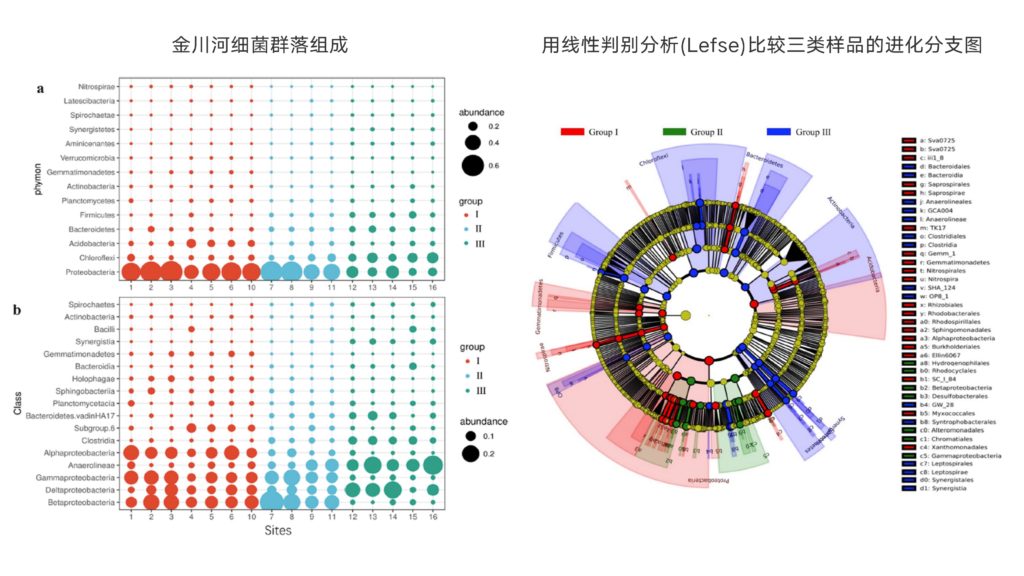
02 主要图表
03 主要结论
研究揭示了不同污染水平的城市河流细菌群落的多样性、组成、共生模式和功能的变化。不同黑臭水平的细菌群落组成和共生关系明显不同,但细菌群落多样性的变化在黑臭水平上没有明显差异。此外,在严重污染的河流中,细菌群落功能受到抑制。与能量代谢以及异生物降解和代谢相关的基因丰度显著降低。
二 小鼠肠道样本



01 研究背景
抑郁症是一种精神疾病,导致显著和持续的情绪和兴趣下降。不健康的饮食或生活方式会导致抑郁。抑郁症的发病率逐年上升,预计到2020年将成为仅次于心脏病的第二大人类疾病。研究发现抑郁症患者表现为脑、内分泌、免疫和肠脑功能异常,脑-肠轴向功能障碍可能是抑郁症的主要病理机制。越来越多的证据表明,膳食补充益生菌可改善重度抑郁症患者或面临社会压力的人的压力引起的抑郁和抑郁行为或情绪。
02 主要图表
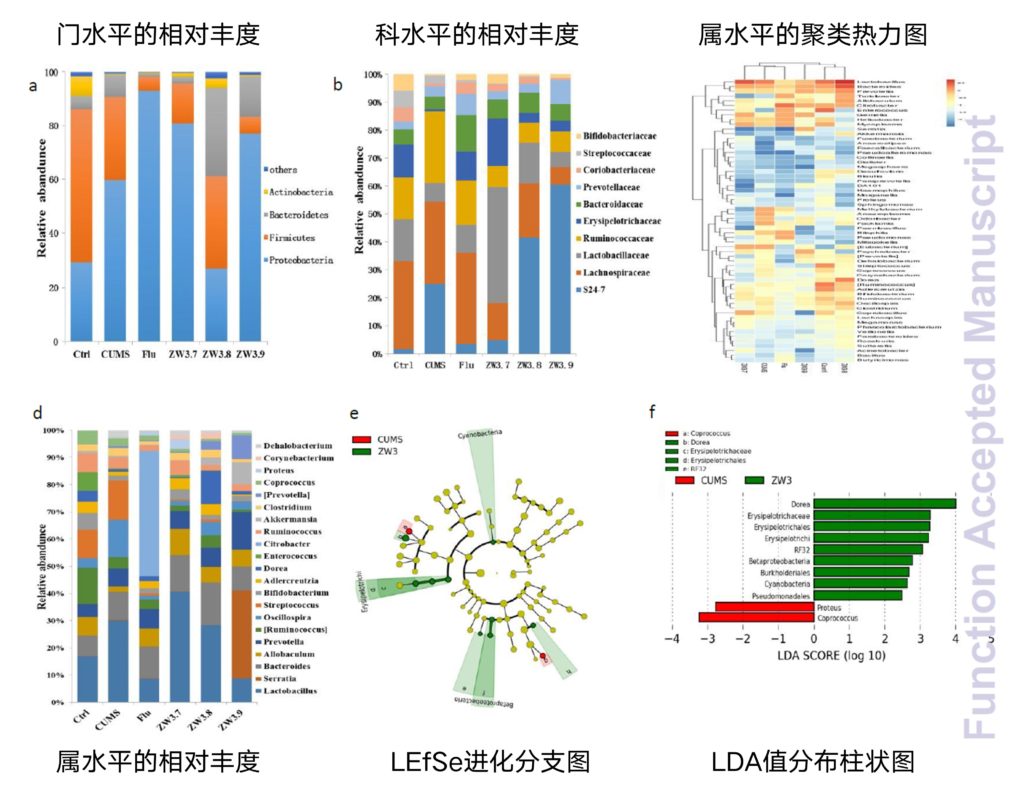
03 主要结论
结果表明马乳酒样乳杆菌ZW3可以通过调节色氨酸代谢紊乱,保护下丘脑-垂体-肾上腺轴,抑制由慢性轻度不可预见性应激引起的炎症,从而改善抑郁症。此外,饲料中添加马乳酒样乳杆菌ZW3可使慢性轻度不可预见性应激抑郁小鼠肠道微生物区系更加平衡。ZW3增加了小鼠粪便中的抗炎和抗应激微生物丰度,如放线菌、拟杆菌、毛螺菌科、红蝽菌科、双歧杆菌科和阿克曼氏菌等,降低了与疾病和应激呈正相关的微生物,如小鼠粪便中的变形菌。
三 土壤样本



01 研究背景
多环芳烃(PAHs)是广泛存在于土壤中一类持久性有机污染物,具有毒性、致突变性和致癌性,可通过食物链进行生物积累,威胁人类健康。
表面活性剂强化植物修复技术(SEPR)是一种高效、经济的有机污染土壤修复技术。植物促进微生物降解是去除污染土壤中多环芳烃的主要贡献。PAHs的去除是由于污染土壤中微生物群落组成改变和PAHs降解微生物数量的增加所致。此外,微生物群落中PAHs的降解是土壤中PAHs消散的主要因素。因此,有必要研究土壤中残留多环芳烃的生物可利用部分和其它组分,以便更好的了解污染土壤中多环芳烃组分的动态变化。
02 主要图表
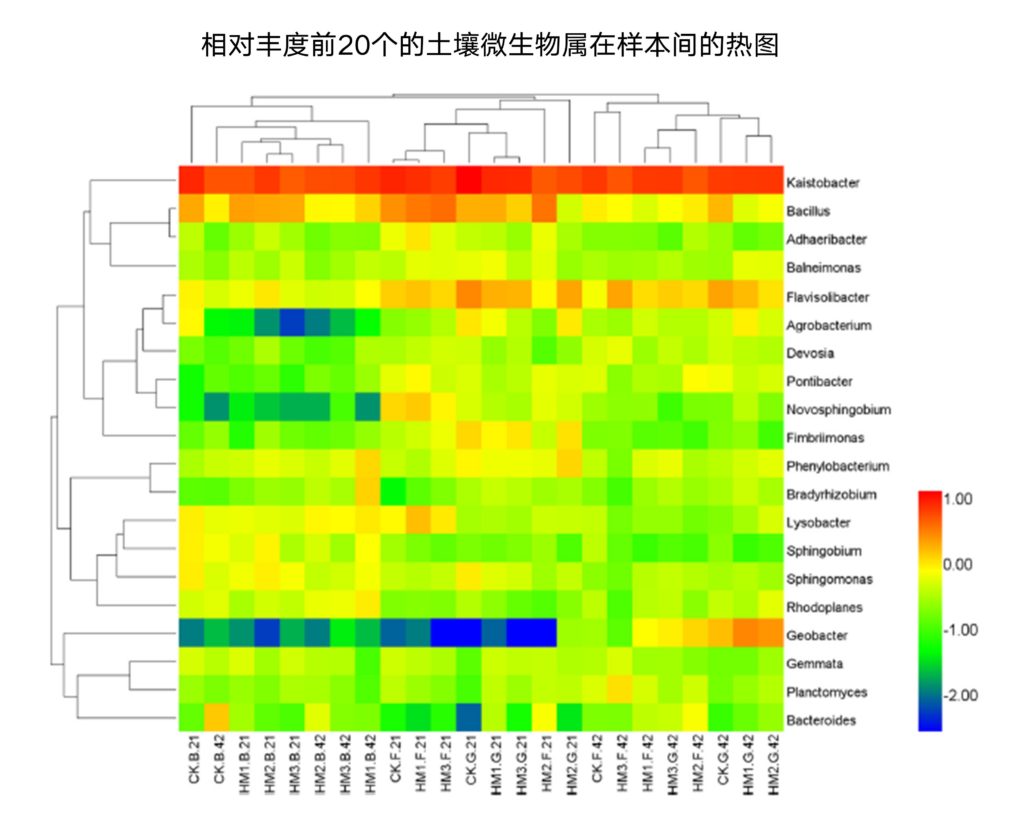
03 主要结论
1∶1的混合比例SDBS-Tween 80表面活性剂增强了菲和芘的修复。DHO活性分析表明,DHO是一种重要的酶,生物有效分数的变化是菲和芘消散的主要指标。混合表面活性剂促进多环芳烃从结合态、残余组分向生物有效组分的转化。此外,混合表面活性剂增加了多环芳烃降解细菌和降解相关基因的丰度,从而促进了多环芳烃的生物有效组分的降解。
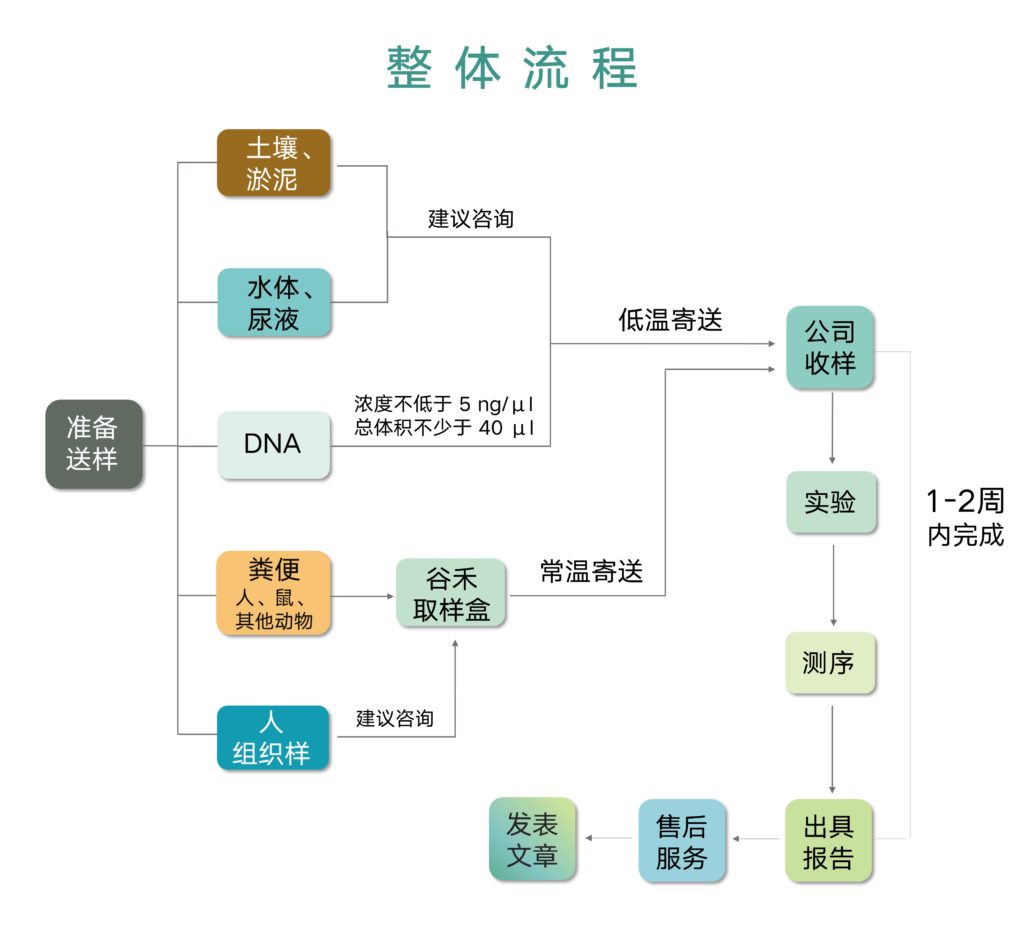
以上几篇文章从建库、测序到分析均由谷禾提供完整的技术服务。
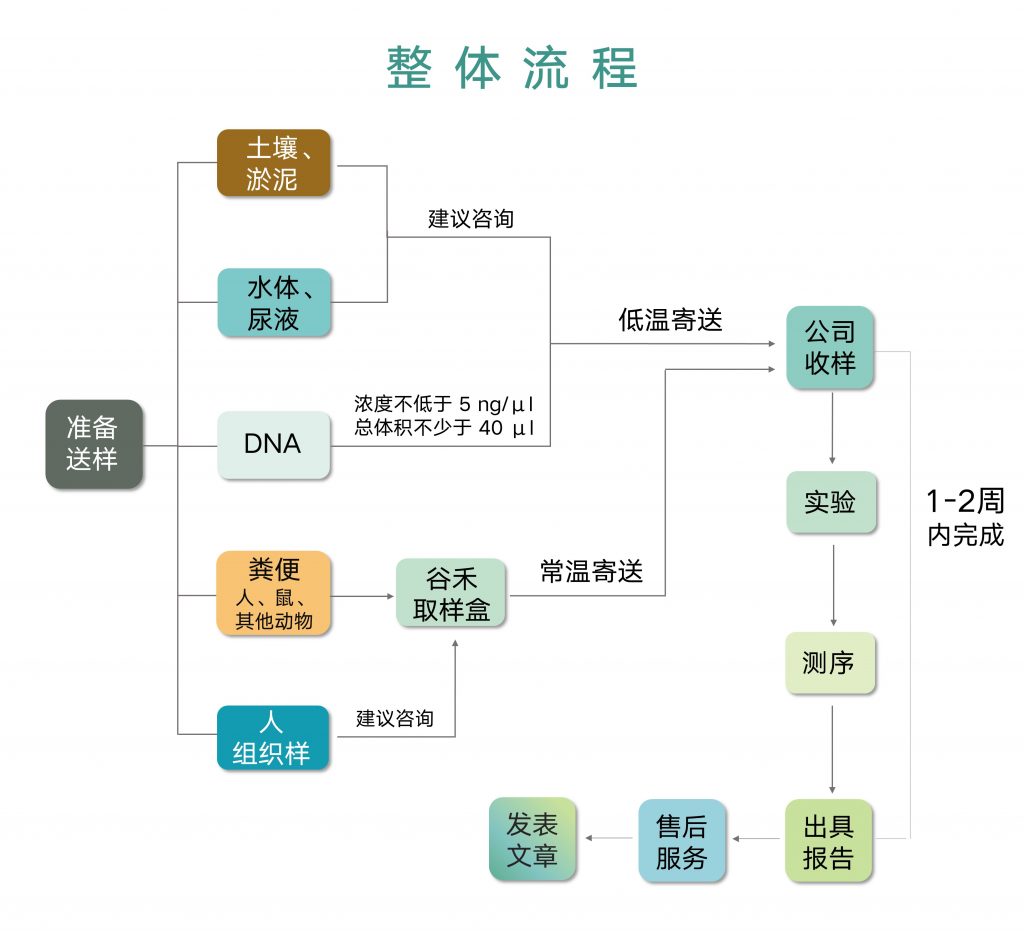
合作服务整体流程如下:

部分分析内容一览表 :

以上所有包括前面文章中的图表都可以做,包括在科研服务内,不额外收费



谷禾的优势
一体化服务
我们在高通量测序行业服务长达8年,无论是科研设计的思路,实验技术支持还是数据分析甚至整个流程的把控都有了非常丰富的经验。
专门面向肠道菌群相关科研实验的完整肠道菌群检测方案

专注于解决肠道菌群实际研究中的各种问题并进行全方位针对性优化。
为研究者提供从项目系统、取样、DNA提取、质控、扩增、测序、科研数据分析、肠道菌群参考数据集、人工智能模型分析并给出全面的科研分析报告。
取样便捷稳定
取样:检测方案为客户提供肠道菌群专用取样盒,没有经过训练的普通人也可以完成稳定可靠的取样。稳定可靠的保存液可在室温下有效完整保存样品DNA至少60天。
[WPGP gif_id=”276″ width=”600″]
简便的样品取样,助力肠道菌群研究
稳定存储样品,提供常温运输能力
运输条件:全程常温运输。
取样管内稳定液经过海量样本测试,并通过大量样本的极端条件重复测试。测试结果表明君验的样品保存可在常温下有效保存超过1个月,4度保存6个月以上。
专利提取技术
实验过程用到严格的提取,建库技术(包括发明专利提取技术、高保真酶、循环数控制、空白和对照试验、独立barcode控制数据切分、凝胶电泳+荧光定量双重质检)。发明专利号:ZL201511009389.7。
DNA提取:用户仅需将取样管通过普通快递寄回,实验室按照严格测试和优化的粪便DNA提取试剂盒进行DNA提取。
质控:因粪便样品易受环境因素和污染干扰,实验室对每一批次样品设置空白和阳性对照,从提取过程到扩增完成均进行全面对照质控。
扩增:经过多年数万例样本的测试,君验将PCR扩增循环数控制在24个循环之内,尽最大可能减少偏差。
测序平台和技术
测序:使用Illumina高通量测序平台,提供高达10万reads的高质量测序,提供达远超饱和的测序深度
Illumina Hiseq / Novaseq 高通量测序
2*150 bp 或者 2*250
Q30 质量大于93%
平均10万reads,最低5万reads
肠道菌群样本70%到种
致病菌95%特异性
扩增引物
V4区:
515F:GTGCCAGCMGCCGCGGTAA
806R:GGACTACHVGGGTWTCTAAT
V3-V4区:
341F5′ -ACTCCTACGGGAGGCAGCA-3′
806R5′ -GGACTACHVGGGTWTCTAAT-3′
注:一般情况下默认是测V4区,如果特殊要求测V3-V4区也可以做,周期会相应延长至一个月。
护
数据分析全覆盖
科研数据分析:提供完整全面的16s菌群分析流程和图表内容,基因功能和代谢途径预测以及个性化图表和自助分析平台帮助。
肠道菌群参考数据集:近20万例肠道菌群样本数据针对性构建的肠道菌群参考数据库远超Greengene或SILVA的覆盖度,提供史无前例的分辨精度。
人工智能分析模型:肠道菌群的复杂程度以及不同样品之间的巨大差异已无法使用简单的统计模型来解析,专业的人工智能专家对数万例样本和40多种疾病深度模型分析之后的结晶,精细化区分各种环境及影响因素的干扰,数量化评估每个特征的贡献。
微生物多样性测序结果如何看?
做过16s测序的小伙伴们都知道
测完之后会拿到一份结果报告
但这并不代表可以开始写文章了
看似一大堆数据图表却不知如何下手
这是很多人头疼的地方
那么怎样给报告中的数据赋予灵魂
让它真正成为对你有帮助的分析呢?
一文扫除困惑
首先什么是16S rRNA?
16S rRNA 基因是编码原核生物核糖体小亚基的基因,长度约为1542bp,其分子大小适中,突变率小,是细菌系统分类学研究中最常用和最有用的标志。
16S rRNA基因序列包括9个可变区和10个保守区,保守区序列反映了物种间的亲缘关系, 而可变区序列则能体现物种间的差异。
16S rRNA基因测序以细菌16S rRNA基因测序为主,核心是研究样品中的物种分类、物种丰度以及系统进化。
二代高通量测序原理
目前二代测序是一个边合成边测序的过程,使用的是荧光可逆终止子。每个可逆终止子的碱基3’端都有一个阻断基团,而在侧边带有一种荧光。由于有4种不同的碱基(ATCG),因此也会有对应4种不同颜色的荧光。开始扩增每次结合上一个碱基,DNA的扩增便会停止,此时能收到一种荧光信号。然后放试剂除去阻断基团,进行下一个碱基的结合,以此类推得到一连串的荧光信号组合序列。而根据荧光的颜色我们便可以确定每一个位点的基因型,即可以得到这一段DNA片段的序列。

环境样品高通量分析需要重复么?
在进行实验设计前,这是有些小伙伴面临的一个问题。环境样本由于来源和条件不完全可控,每个样品之间会存在很大的差异,即便是相同样本的不同取样时间和部位也会存在一定的差异。
基于高通量测序主要是为了了解样品的菌群构成和功能分析,以及寻找不同环境之间的差异,包括菌和功能基因以及代谢。如果仅做单一样本,很可能结论只能代表这个单一取样样本的信息,无法排除不同样本重复之间的差异,也就可能得不到真正代表环境差异的结果。
所以环境样品不仅要重复而且还应该以分组方式取尽量多的样本以全面的代表一个环境条件下的各种变异情况。
测序区段如何选择
确定做重复后,又面临该怎么选择测序区段的问题。目前市面上有v1-v3区/v3-v4区/v4区等可供选择。
16S rRNA编码基因序列共有9个保守区和9个高可变区。其中,V4区其特异性好,数据库信息全,我们通过大量的测序试验证明用v4区扩增出菌群结果的可以很好的反应样本的菌群结构用于后续的数据建模分析,是细菌多样性分析注释的最佳选择。

基本确定好后,就要着手开始实验,实验完送样又是个问题,以往给测序公司送样往往是低温运输,且不说麻烦,还要提心吊胆怕运输过程会不会有什么问题。为此我们免费提供常温保存取样盒,就不用有这样的顾虑,取样及运输全程都只需要常温即可。
16s分析结果详解
很多小伙伴有过这样的经历,在拿到公司出具的报告之后,仍然一头雾水,几十页的报告内容看着丰富却不知该怎么运用。我们一起来理一下关键图表的含义。
OTU是我们要搞清的一个重要概念,可以说是后续分析的基石。
OTU(operational taxonomic units) 是在系统发生学研究或群体遗传学研究中,为了便于进行分析,人为给某一个分类单元(品系,种,属,分组等)设置的同一标志。通常按照 97% 的相似性阈值将序列划分为不同的 OTU,每一个 OTU 通常被视为一个微生物物种。相似性小于97%就可以认为属于不同的种,相似性小于93%-95%,可以认为属于不同的属。样品中的微生物多样性和不同微生物的丰度都是基于对OTU的分析。
有了OTU这个概念之后,就不难理解下表。对每个样本的测序数量和OTU数目进行统计,并且在表栺中列出了测序覆盖的完整度。

其中SampleName表示样本名称;SampleSize表示样本序列总数;OTUsNumber表示注释上的OTU数目;OTUsSeq表示注释上OTU的样本序列总数。
Coverage是指各样品文库的覆盖率,其数值越高,则样本中序列没有被测出的概率越低。该指数实际反映了本次测序结果是否代表样本的真实情况。计算公式为:C=1-n1/N 其中n1 = 只含有一条序列的OTU的数目;N = 抽样中出现的总的序列数目。
下表是对每个样本在分类字水平上的数量进行统计,并且在表栺中列出了在每个分类字水平上的物种数目

其中SampleName表示样本名称;Phylum表示分类到门的OTU数量;Class表示分类到纲的OTU数量;Order表示分类到目的OTU数量;Family表示分类到科的OTU数量;Genus表示分类到属的OTU数量;Species表示分类到种的OTU数量。
我们可以看到绝大部分的OTU都分类到了属(Genus),也有很多分类到了种(Species)。但是仍然有很多无法完全分类到种一级,这是由于环境微生物本身存在非常丰富的多样性,还有大量的菌仍然没有被测序和发现。
当然,对这些种属的构成还可以进行柱状图展示:

横坐标中每一个条形图代表一个样本,纵坐标代表该分类层级的序列数目或比例。同一种颜色代表相同的分类级别。图中的每根柱子中的颜色表示该样本在不同级别(门、纲、目等)的序列数目,序列数目只计算级别最低的分类,例如在属中计算过了,则在科中则不重复计算。
我们还需要对样本之间或分组之间的OTU进行比较获得韦恩图:

样品构成丰度
稀释曲线
微生物多样性分析中如何验证测序数据量是否足以反映样品中的物种多样性?
稀释曲线(丰富度曲线)可以派上用场。它是用来评价测序量是否足以覆盖所有类群,并间接反映样品中物种的丰富程度。
不免有同学有疑惑,稀释曲线怎么来的?
它是利用已测得16S rDNA序列中已知的各种OTU的相对比例,来计算抽取n个(n小于测得reads序列总数)reads时出现OTU数量的期望值,然后根据一组n值(一般为一组小于总序列数的等差数列)与其相对应的OTU数量的期望值做出曲线来。
至此,我们虽然知道了稀释曲线的由来,那么这个五彩缤纷的稀释曲线该怎么看呢?
当曲线趋于平缓或者达到平台期时也就可以认为测序深度已经基本覆盖到样品中所有的物种,增加测序数据无法再找到更多的OTU;
反之,则表示样品中物种多样性较高,还存在较多未被测序检测到的物种。

横坐标代表随机抽取的序列数量;纵坐标代表观测到的OTU数量。样本曲线的延伸终点的横坐标位置为该样本的测序数量。
Shannon-Winner曲线
Shannon-Wiener 曲线,是利用shannon指数来进行绘制的,反映样品中微生物多样性的指数,利用各样品的测序量在不同测序深度时的微生物多样性指数构建曲线,以此反映各样本在不同测序数量时的微生物多样性。
当曲线趋向平坦时,说明测序数据量足够大,可以反映样品中绝大多数的微生物物种信息。

横坐标代表随机抽取的序列数量;纵坐标代表的是反映物种多样性的Shannon指数,样本曲线的延伸终点的横坐标位置为该样本的测序数量。
其中曲线的最高点也就是该样本的Shannon指数,指数越高表明样品的物种多样性越高。
好奇的同学又有疑问,Shannon指数怎么算的?
这里有Shannon指数的公式:

其中,Sobs= 实际测量出的OTU数目;
ni= 含有i 条序列的OTU数目;N = 所有的序列数。
Rank-Abundance曲线
该曲线用于同时解释样品多样性的两个方面,即样品所含物种的丰富程度和均匀程度。

横坐标代表物种排序的数量;纵坐标代表观测到的相对丰度。
样本曲线的延伸终点的横坐标位置为该样本的物种数量
物种的丰富程度由曲线在横轴上的长度来反映,曲线越宽,表示物种的组成越丰富;
物种组成的均匀程度由曲线的形状来反映,曲线越平坦,表示物种组成的均匀程度越高。
如果曲线越平滑下降表明样本的物种多样性越高,而曲线快速陡然下降表明样本中的优势菌群所占比例很高,多样性较低。
但一般超过20个样本图就会变得非常复杂而且不美观!所以假如没超过20个样可以考虑该图哦~
Alpha多样性(样本内多样性)
Alpha多样性是指一个特定区域或者生态系统内的多样性,常用的度量指标有Chao1 丰富度估计量(Chao1 richness estimator) 、香农 – 威纳多样性指数(Shannon-wiener diversity index)、辛普森多样性指数(Simpson diversity index)等。
计算菌群丰度:Chao、ace;
计算菌群多样性:Shannon、Simpson。
Simpson指数值越大,说明群落多样性越高;Shannon指数越大,说明群落多样性越高。

看了那么多指数,可能觉得有点晕,到底每个指数是什么意思呢?
当然要解释下咯:
Chao1:是用chao1 算法计算群落中只检测到1次和2次的OTU数估计群落中实际存在的物种数。Chao1 在生态学中常用来估计物种总数,由Chao (1984) 最早提出。Chao1值越大代表物种总数越多。
Schao1=Sobs+n1(n1-1)/2(n2+1)
其中Schao1为估计的OTU数,Sobs为观测到的OTU数,n1为只有一条序列的OTU数目,n2为只有两条序列的OTU数目。
Shannon:用来估算样品中微生物的多样性指数之一。它与 Simpson 多样性指数均为常用的反映 alpha 多样性的指数。Shannon值越大,说明群落多样性越高。
Ace:用来估计群落中含有OTU 数目的指数,由Chao 提出,是生态学中估计物种总数的常用指数之一,与Chao1 的算法不同。
Simpson:用来估算样品中微生物的多样性指数之一,由Edward Hugh Simpson ( 1949) 提出,在生态学中常用来定量的描述一个区域的生物多样性。Simpson 指数值越大,说明群落多样性越高。

Alpha多样性指数差异箱形图
分别对 Alpha diversity 的各个指数进行秩和检验分析(若两组样品比较则使用 R 中的wilcox.test 函数,若两组以上的样品比较则使用 R 中的 kruskal.test 函数),通过秩和检验筛选不同条件下的显著差异的 Alpha Diversity指数。
Beta多样性分析(样品间差异分析)
也许我们有听说Beta多样性在最近10年间成为生物多样性研究的热点问题之一。具体解释下:
Beta多样性度量时空尺度上物种组成的变化, 是生物多样性的重要组成部分, 与许多生态学和进化生物学问题密切相关!
PCoA分析
PCoA(principal co-ordinates analysis)是一种研究数据相似性或差异性的可视化方法,通过一系列的特征值和特征向量进行排序后,选择主要排在前几位的特征值,PCoA 可以找到距离矩阵中最主要的坐标,结果是数据矩阵的一个旋转,它没有改变样品点之间的相互位置关系,只是改变了坐标系统。

每一个点代表一个样本,相同颜色的点来自同一个分组,两点之间距离越近表明两者的群落构成差异越小。
另一种相似的是PCA分析
主成分分析(Principal component analysis)PCA 是一种研究数据相似性或差异性的可视化方法,通过一系列的特征值和特征向量进行排序后,选择主要的前几位特征值,采取降维的思想,PCA 可以找到距离矩阵中最主要的坐标,结果是数据矩阵的一个旋转,它没有改变样品点之间的相互位置关系,只是改变了坐标系统。

详细关于主成分分析的解释推荐大家看一篇文章,http://blog.csdn.net/aywhehe/article/details/5736659
一起来看看包含PCoA研究的文章
案例解析



研究背景:全球塑料产量飞速增长,而且呈持续上升的趋势,因此导致大量塑料废物排放到环境中,从沿海河口到大洋环流,从东大西洋到南太平洋海域。塑料废弃物具有化学稳定性和生物利用率低的特点,可长期存在于海洋中,从而影响海洋环境包括海洋生物的生存。
作为一个独特的底物,塑料碎片可以吸附海洋中的微生物并形成个“塑性球”。以生物膜形式存在于塑料碎片上的微生物群落。许多研究表明,无论是在海洋还是淡水生态系统中,附着在塑料碎片上微生物群落的组成明显不同于周围环境(水和沉积物),而且易受位置、时间和塑料类型的影响。
主要图表
两两群落差异指数的PCoA图

PCoA 图可以清楚地看到,SW区细菌群落的置信椭圆与pd和sd的置信椭圆有显著的偏差(p<0.05),而sd上细菌群落的置信椭圆几乎覆盖了pd的置信椭圆(p>0.05),这表明pd和sd上的细菌群落有相似之处。
不同样本和处理下的细菌群落( 前 10 位)丰度分布

底物(SW、SD和Pd)上的主要属为细菌和假互斥单胞菌,暴露两周后,这些菌可能是分布广泛和适应性强的三种底物(SW、SD和PD)。暴露4周后,弧菌相对丰度增加.此外,暴露6周后,自养细菌(如扁平菌和硝酸菌)的数量增加。这三种底物上个细菌群落的生长模式也与3.2的结果一致。图5还显示,在6个星期内,在429个原位点中,假单胞菌在pd上的相对丰度高于sw和sd(anova,p<0.05)。

研究结论:首先,营养物质 (TN 和 TP) 与生物膜的平均生长速率呈正相关,而盐度与生物膜的平均生长速率呈负相关。盐度是影响PD的个细菌多样性的主要因素,而温度、溶解氧和养分(TN和TP)在类似的盐度条件下可能具有二次效应。尽管种聚合物类型对PD上的细菌群落的多样性具有较少的影响,但是在细菌群落中的一些属显示对PD的聚合物类型的选择性,并且倾向于将其优选的基质定殖。大的相对丰度SW、PD、SD间属显著差异。盐度是改变河口地区Pd条件致病菌富集的主要因素。另外,在种病原物种丰富的基础上,PD具有较高的致病性。
NMDS分析(非度量多维尺度分析)
NMDS(Nonmetric Multidimensional Scaling)常用于比对样本组之间的差异,可以基于进化关系或数量距离矩阵。

横轴和纵轴:表示基于进化或者数量距离矩阵的数值在二维表中成图。与PCA分析的主要差异在于考量了进化上的信息。
每一个点代表一个样本,相同颜色的点来自同一个分组,两点之间距离越近表明两者的群落构成差异越小。
排序分析
PCA,PcoA,NMDS分析都属于排序分析(Ordination analysis)。
排序(ordination)的过程就是在一个可视化的低维空间或平面重新排列这些样本。
目的:使得样本之间的距离最大程度地反映出平面散点图内样本之间的关系信息。
排序又分两种:非限制性排序和限制性排序。
1、非限制性排序(unconstrained ordination)
——只使用物种组成数据的排序
(1) 主成分分析(principal components analysis,PCA)
(2) 对应分析(correspondence analysis, CA)
(3) 去趋势对应分析(Detrended correspondence analysis, DCA)
(4) 主坐标分析(principal coordinate analysis, PCoA)
(5) 非度量多维尺度分析(non-metric multi-dimensional scaling, NMDS)
2、限制性排序(constrained ordination)
——同时使用物种和环境因子组成数据的排序
(1) 冗余分析(redundancy analysis,RDA)
(2) 典范对应分析(canonical correspondence analysis, CCA)
比较PCA和PCoA
在非限制性排序中,16S和宏基因组数据分析通常用到的是PCA分析和PCoA分析,两者的区别在于:
PCA分析是基于原始的物种组成矩阵所做的排序分析,而PCoA分析则是基于由物种组成计算得到的距离矩阵得出的。
在PCoA分析中,计算距离矩阵的方法有很多种,包括如:Euclidean, Bray-Curtis, and Jaccard,以及(un)weighted Unifrac (利用各样品序列间的进化信息来计算样品间距离,其中weighted考虑物种的丰度,unweighted没有对物种丰度进行加权处理)。
组间菌群比较选取物种标志物
(属水平)样本-物种丰度关联circos弦装图
样本与物种的共线性关系circus 图是一种描述样本与物种之间对应关系的可视化圈图,该图不仅反映了每个样本的优势物种组成比例,同时也反映了各优势物种在不同样本之间的分布比例。
图解读:样本与物种的共线性关系图,左半边表示样本属物种丰度情况。右半边表示属水平在不同样本中的分布比例情况。在最内一圈:左边不同颜色代表不同物种,宽度表示物种丰度,圈外数值表示物种丰度刻度值。一端连接右边的样本,不同颜色代表不同样本,条带端点宽度表示该样本中对应物种的比例分布。最外两圈:左边不同颜色表示不同样本在某一物种的比例,右边不同颜色表示不同物种在某一样本中的比例。
Ternary三元相图
三元相图是重心图的一种,它有三个变量,在一个等边三角形坐标系中,图中某一点的位置代表三个变量间的比例关系。这里表示三组样本之间优势物种的差异,通过三元图可以展示出不同物种在分组中的比重关系。

图解读:三角分别代表三个或三组样本,图中的圆分别代表排名最高哦的属水平的物种,三种颜色分别代表三组不同分组的优势物种,圆圈大小代表物种的相对丰度,圆圈理哪个顶点接近,表示此物种在这个分组中的含量较高。该分析仅限三个样本或三组样本之间分析比较。
相关系数图
通过R 软件的corrplot 包绘制spearman 相关性热图,并通过该热图可以发现优势物种/样本之间重要的模式与关系。

图解读:蓝色系的为正相关,红色系的为负相关,×表示检验水平下无意义。越靠近颜色条两头,相关系数越大。所以说,我们可以通过实心圆的颜色和大小判断相关的方向和相关系数的大小。
LDA差异贡献分析
如果说 PCA,它所作的只是将整组数据整体映射到最方便表示这组数据的坐标轴上,映射时没有利用任何数据内部的分类信息,是无监督的。
那么LDA是有监督的,增加了种属之间的信息关系后,结合显著性差异标准测试(克鲁斯卡尔-沃利斯检验和两两Wilcoxon测试)和线性判别分析的方法进行特征选择。
两者相同点:
都可以对数据进行降维。
降维时都采用了矩阵特征分解的思想。
差异:
1)LDA是有监督学习的降维方法,而PCA是无监督的降维方法。(注:监督学习是从标记的训练数据来推断一个功能的机器学习任务。)
2)LDA选择分类性能最好的投影方向,而PCA选择样本点投影具有最大方差的方向。
除了可以检测重要特征,他还可以根据效应值进行功能特性排序,这些功能特性可以解释大部分生物学差异。这部分希望能详细了解的同学可以参考这篇文章http://blog.csdn.net/sunmenggmail/article/details/8071502 。


不同颜色代表不同样本或组之间的显著差异物种。
使用LefSe软件分析获得,其中显著差异的logarithmic LDA score设为2。
LDA分析究竟能做什么
组间差异显著物种又可以称作生物标记物(biomarkers),这个LDA分析主要是想找到组间在丰度上有显著差异的物种。
案例解析

研究背景:研究表明遗传和环境影响都在I型糖尿病的发展中起作用,增加的遗传风险不足以引起疾病,环境因素也是需要的,而且起着至关重要的作用。肠道菌群也许就是这个重要的环境因素,肠道菌群在免疫系统的成熟中起重要作用,此外还影响自身免疫疾病发展。

不同遗传风险儿童的LDA差异菌群

不同遗传风险分组中包含的常见菌属,部分存在特定分组中

PCoA分析揭示不同遗传风险儿童肠道菌群的在不同地域样本中均存在显著差异

点评:针对I型糖尿病疾病发生过程中遗传HLA分型风险和对应肠道菌群菌的关联分析,揭示了特定肠道菌群与宿主特定遗传风险共同作用推进疾病发生。某些特定菌属可能无法在遗传高风险儿童肠道内定植,可能对疾病发生存在特定作用。此外对于其他遗传风险的自身免疫疾病也具有重要提示意义,例如乳糜泻和类风湿性关节炎。
物种进化树的样本群落分布图
这是另一款和LDA长得有点像的图,当然功能可完全不一样。它是将不同样本的群落构成及分布以物种分类树的形式在一个环图中展示。数据经过分析后,将物种分类树和分类丰度信息通过这款软件GraPhlAn进行绘制
(http://huttenhower.sph.harvard.edu/GraPhlAn )。
其目的是将物种之间的进化关系以及不同样本的物种分布丰度和最高分布样本的信息在一个视觉集中的环图中一次展示,其提供的信息量较其他图最为丰富。

中间为物种进化分类树
不同颜色的分支代表不同的纲(具体的代表颜色见右上角的图例),
接着的外圈的灰色标示字母的环表示的是本次研究中比例最高的15个科(字母代表的科参见左上角的图例)。
之后的外圈提供的是热力图,如果样本数<=10个则绘制样本,如果样本数超过10个则按照分组绘制,每一环为一个样本,根据其丰度绘制的热力图。
最外圈为柱状图,绘制的是该属所占比例最高的样本的丰度和样本颜色(样本颜色见环最下方的样本名字的颜色)。其中热力图和柱状图取值均为原比例值x10000后进行log2转换后的值。
物种相关性分析
根据各个物种在各个样品中的丰度以及变化情况,计算物种之间的相关性,包括正相关和负相关。
相关性分析使用CCREPE算法
怎么画的?
首先对原始16s测序数据的种属数量进行标准化,然后进行Spearman和Pearson秩相关分析并进行统计检验,计算出各个物种之间的相关性,之后在所有物种中根据simscore绝对值的大小,挑选出相关性最高的前100组数据,基于Cytoscap绘制共表达分析网络图。
网络图采用两种不同的形式表现出来。
物种相关性网络图A

○ 图中每一个点代表一个物种,存在相关性的物种用连线连接。
○ 红色的连线代表负相关,绿色的先代表正相关。
○ 连线颜色的深浅代表相关性的高低。
物种相关性网络图B

○ 图中每一个点代表一个物种
○点的大小表示与其他物种的关联关系的多少
○ 其中与之有相关性的物种数越多,点的半径和字体越大
○ 连线的粗细代表两物种之间相关性的大小
连线越粗,相关性越高。
案例解析


研究背景:气候变化导致美国中部草原的降水模式发生变化,对土壤微生物群落构成及代谢影响很大。
研究希望明确土壤微生物群落对土壤水分变化的反应,并确定响应的特定代谢特征。
主要图表
同一样本在不同水分含量孵化处理下土壤菌群的变化

受到水分条件影响的土壤菌群代谢途径和网络分布

研究结论:土壤干燥导致土壤微生物组的组成和功能发生显着变化。相反,润湿后几乎没有变化。由于干旱导致的土壤水分减少对土壤碳循环和土壤微生物组进行的其他关键生物地球化学循环的影响很大。导致渗透保护剂化合物产生的代谢途径受到较大影响。
点评:
相对简单的样本和实验设计,但是从多个维度探寻支持土壤微生物群落对湿润和干燥表型的反应。
与常见的环境采样检测不同,针对同一样本在对照环境下进行环境控制孵化,然后比较菌群变化可以更为有效的控制背景差异。
聚类分析
根据OTU数据进行标准化处理(1wlog10)之后,选取数目最多的前60个物种,基于R heatmap进行作图

○ 热图中的每一个色块代表一个样品的一个属的丰度
○ 样品横向排列,属纵向排列
○ 差异是是否对样品进行聚类,从聚类中可以了解样品之间的相似性以及属水平上的群落构成相似性。
Tips:
如果聚类结果中出现大面积的白或黑是因为大量的菌含量非常低,导致都没有数值,可以在绘制之前进行标准化操作,对每一类菌单独自身进行Z标准化。
案例解析



研究背景:妊娠期糖尿病(GDM)的患病率在全球范围内迅速增加,构成一个重要的健康问题和产科实践的重大挑战(Ferrara,2007)。高脂血症是妊娠常见的合并症。在GDM患者中,血脂的生理变化可能导致怀孕期间潜在的代谢紊乱。肠道失调在宿主代谢异常中起着至关重要的作用,最近关于2型糖尿病(T2D)和肥胖的研究就证明了这一点。这些研究表明,妊娠期间肠道微生物ME的主要变化可能在GDM的发展中起着至关重要的作用。

GDM加高脂血症(M队列)妊娠期间与显著改变的脂质相关的肠道微生物群(属)


研究结论:我们的结果表明,血脂水平可能反映了GDM发展过程中的一些异常变化。所鉴定的多种生物标志物对GDM合并高脂血症的防治有一定的参考价值。

组间物种差异性箱形图
组间物种差异性盒形图描述在不同分组之间具有差异显著的某一物种做盒形图,图中以属水平为例做物种差异性盒形图,展示如下:

○ 图中不同颜色代表不同的分组,更直观显示组间物种差异
○ 每一个盒形图代表一个物种,图上方是物种名。
Anosim检验
Anosim分析是一种非参数检验,用来检验组间的差异是否显著大于组内差异,从而判断分组是否有意义
展示如下:


R-value介于(-1,1)之间,R-value大于0,说明组间差异显著。
R-value小于0,说明组内差异大于组间差异。
统计分析的可信度用 P-value 表示,P< 0.05 表示统计具有显著性。
对Anosim的分析结果,基于两两样本之间的距离值排序获得的秩(组间的为between,组内的为within),这样任一两两组的比较可以获得三个分类的数据,并进行箱线图的展示(若两个箱的凹槽互不重叠,则表明它们的中位数有显著差异)
随机森林分类树属分类效果
随机森林是机器学习算法的一种,它可以被看作是一个包含多个决策树的分类器。
其输出的分类结果是由每棵决策树“投票”的结果。由于每棵树在构建过程中都采用了随机变量和随机抽样的方法,因此随机森林的分类结果具有较高的准确度,并且不需要“减枝”来减少过拟合现象。
随机森林可以有效的对分组样品进行分类和预测。

物种重要性点图。横坐标为重要性水平,纵坐标为按照重要性排序后的物种名称。上图反映了分类器中对分类效果起主要作用的菌属,按作用从大到小排列。
Error rate: 表示使用下方的特征进行随机森林方法预测分类的错误率,越高表示基于菌属特征分类准确度不高,可能分组之间菌属特征不明显。图中以所有水平为例,取前60个作图。
ROC曲线图
ROC 曲线指受试者工作特征曲线(receiver operating characteristic curve), 是反映敏感性和特异性连续变量的综合指标,通过构图法揭示敏感性和特异性的相互关系。
ROC 曲线将连续变量设定出多个不同的临界值,从而计算出一系列敏感性和特异性,再以敏感性为纵坐标、(1-特异性)为横坐标绘制成曲线。
曲线下面积越大,诊断准确性越高。展示如下:
FAPROTAX生态功能预测
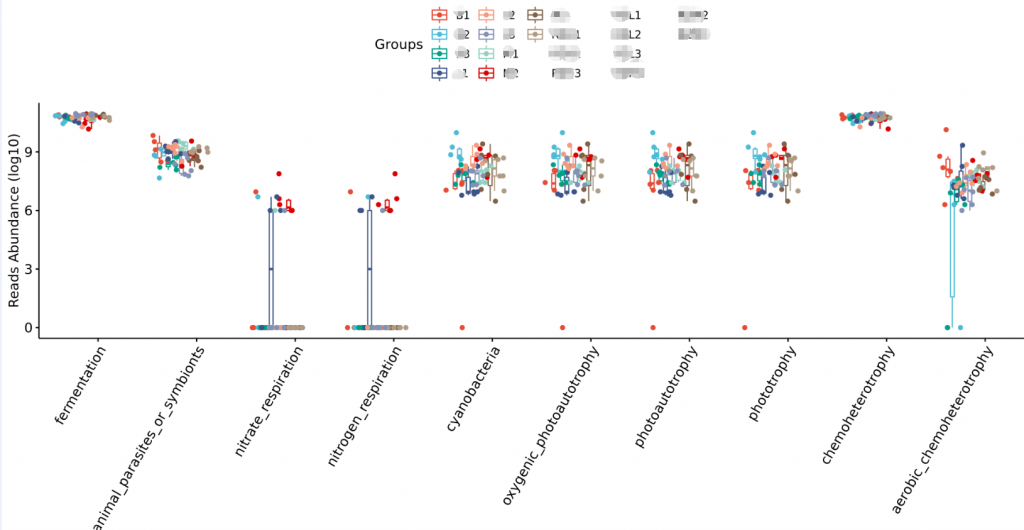
FAPROTAX是一款在2016年发表在SCIENCE上的较新的基于16S测序的功能预测软件。它整合了多个已发表的可培养菌文章的手动整理的原核功能数据库,数据库包含超过4600个物种的7600多个功能注释信息,这些信息共分为80多个功能分组,其中包括如硝酸盐呼吸、产甲烷、发酵、植物病原等。
FAPROTAX对环境样本更友好
如果说PICRUSt(后续会介绍)在肠道微生物研究更为适合,那么FAPROTAX尤其适用于生态环境研究,特别是地球化学物质循环分析。
FAPROTAX适用于对环境样本(如海洋、湖泊等)的生物地球化学循环过程(特别是碳、氢、氮、磷、硫等元素循环)进行功能注释预测。因其基于已发表验证的可培养菌文献,其预测准确度可能较好,但相比于上述PICRUSt和Tax4Fun来说预测的覆盖度可能会降低。
FAPROTAX可根据16S序列的分类注释结果对微生物群落功能(特别是生物地化循环相关)进行注释预测。

图中横坐标代表样本,纵坐标表示包括碳、氢、氮、硫等元素循环相关及其他诸多功能分组。可快速用于评估样品来源或特征。
基于BugBase的表型分类比较
Bugbase也是16年所提供服务的一款免费在线16S功能预测工具,到今年才发表文章公布其软件原理。该工具主要进行表型预测,其中表型类型包括革兰氏阳性、革兰氏阴性、生物膜形成、致病性、移动元件、氧需求,包括厌氧菌、好氧菌、兼性菌)及氧化胁迫耐受等7类。
Gram Negative 革兰氏阴性菌
Picrust群落功能差异分析
通过对已有测序微生物基因组的基因功能的构成进行分析后,我们可以通过16s测序获得的物种构成推测样本中的功能基因的构成,从而分析不同样本和分组之间在功能上的差异(PICRUSt Nature Biotechnology, 1-10. 8 2013)。
Picrust对肠道菌群样本更友好
通过对宏基因组测序数据功能分析和对应16s预测功能分析结果的比较发现,此方法的准确性在84%-95%,对肠道微生物菌群和土壤菌群的功能分析接近95%,能非常好的反映样品中的功能基因构成。
怎么做出来的?
为了能够通过16s测序数据来准确的预测出功能构成,首先需要对原始16s测序数据的种属数量进行标准化,因为不同的种属菌包含的16s拷贝数不相同。
然后将16s的种属构成信息通过构建好的已测序基因组的种属功能基因构成表映射获得预测的功能结果。(根据属这个水平,对不同样本间的物种丰度进行显著性差异两两检验,我们这里的检验方法使用STAMP中的two-sample中T-TEST方法,Pvalue值过滤为0.05,作Extent error bar图。)

此处提供COG,KO基因预测以及KEGG代谢途径预测。当然,跃跃欲试的小伙伴也可自行使用我们提供的文件和软件(STAMP)对不同层级以及不同分组之间进行统计分析和制图,以及选择不同的统计方法和显著性水平。
这里提到的STAMP有些小伙伴说不太了解,别急,后面会有更多介绍。
COG构成差异分析图
图中不同颜色代表不同的分组,列出了COG构成在组间存在显著差异的功能分类以及在各组的比例,此外右侧还给出了差异的比例和置信区间以及P-value。

KEGG代谢途径差异分析图
通过KEGG代谢途径的预测差异分析,我们可以了解到不同分组的样品之间在微生物群落的功能基因在代谢途径上的差异,以及变化的高低。为我们了解群落样本的环境适应变化的代谢过程提供一种简便快捷的方法。
本例图所显示的是第三层级的KEGG代谢途径的差异分析,也可以针对第二或第一层的分级进行分析。

图中不同颜色代表不同的分组,列出了在第三层级的构成在组间存在显著差异的KEGG代谢途径第三层分类以及在各组的比例,此外右侧还给出了差异的比例和置信区间以及P-value。
案例解析


研究背景:尽管普遍认为肠道微生物组的生态多样性和分类组成在肥胖和T2D中发生改变,但与单个微生物或微生物产物的关联在研究之间不一致。缺乏大样本群体研究,从而确定肠道微生物组,血浆代谢组,肥胖和糖尿病表型以及环境因素之间的几种关联。
主要图表:
按照肥胖和糖尿病对人群分为三组,同时进行了16S,代谢和宏基因组的检测。

与肥胖相关的菌属以及代谢途径


研究结论:确定了肠道微生物组,血浆代谢组,肥胖和糖尿病表型以及环境因素之间的几种关联。与肠道微生物组变异相关的主要是肥胖,不是2型糖尿病。存在与肠道微生物组变异相关的药物和膳食补充剂。高铁摄入量影响小鼠的肠道微生物组成。微生物组变异也反映在血清代谢物谱中。
点评:
相对大人群的队列研究,同时涵盖了菌群、代谢和疾病表型以及膳食补充调查的数据。
从结果看菌属和血浆代谢存在关联,但是贡献度都较低,如果样本数量不足很可能找不到显著的联系,这也是这类大样本队列研究的意义。
本研究在人群分组时针对性的研究了肥胖-II型糖尿病和菌群的关联,因而构建了三个主要分组人群,结果显示肥胖与菌群的关联度更大,解释了大部分的菌群差异,而糖尿病的菌群变化较小。
本研究其中较为重要的是发现了不同膳食补充对菌群的影响,并在小鼠实验中得到证实。

基因的差异分析图
除了能对大的基因功能分类和代谢途径进行预测外,我们还能提供精细的功能基因的数量和构成的预测,以及进行样本间以及组间的差异分析,并给出具有统计意义和置信区间的分析结果。
这一分析将我们对于样本群落的差异进一步深入到了每一类基因的层面。

图中不同颜色代表不同的分组,列出了在组间/样本间存在显著差异的每一个功能基因(酶)以及在各组的比例,此外右侧还给出了差异的比例和置信区间以及P-value。
很多小伙伴总希望能亲自上手做点分析,机会来了!
在获得标准报告后如果希望单独修改分组或对某些组之间进行显著性差异分析,可以使用STAMP软件在自己的电脑上进行数据分析。STAMP提供了丰富的统计检验方法和图形化结果的输出。
在使用STAMP之前需要首先准备需要的spf格式文件和样品分组信息表,但是如果数据不会处理,那也很不便。
而在我们的报告中已经将KEGG和KO以及COG的结果文件后经过转换生成了适用于STAMP软件打开的spf格式文件,还有对应的分组信息表文件groupfile.txt。
使用STAMP时的一些相关问题
1、STAMP作图用的原始数据的来源?
STAMP 可以直接使用来自QIIME的biom文件和PICUST的KEGG和ko 文件,groupfile.txt文件的格式为tab-saperated value (tab键隔开的数据)
2、分组问题?
导入数据之后,viewàgroup legend ,在窗口右侧会出现分组栏,根据需要进行分组。
3、Unclassiffied选项中,remain Unclassiffied reads、remove Unclassiffied reads、和use only for calculating frequency profiles 方法的区别?
remain Unclassiffied reads和use only for calculating frequency profiles方法会保留所有的数据,而remove Unclassiffied reads仅仅保留有确定分组信息的数据。
4、Statistical test 中,Welch’s t-test、t-test、white’s non-parametric t-test的区别,各自优缺点?
为了确保统计学意义和准确度和精确性,需要足够多的样本数目,t-test检验可以在最少样本数为4的时候确保高的准确度和精确性。
当两个样本之间具有相同方差的时候,用t-test更为准确,当两个样本没有相同方差,Welch’s t-test更为准确。
当样本数目少于8的时候,可以使用white’s non-parametric t-test,该计算时间较长,当样本数目过多的时候不宜使用该方法。
5、Two-group 中 type: one side和two side的区别?
One side只会显示前一个group与后一个group差异的比例,而two side两者之间的比例均会显示。
6、STAMP在使用时首先打开了一个分析文件,如果新打开一个可能会导致显示错误?
目前版本的STAMP存在一些小问题,一次分析只能使用一个数据文件,如果要打开新的需要关闭软件后再打开。
详细的STAMP使用教程可以参考我们提供的STAMP使用教程。
环境因子分析
冗余分析(redundancy analysis, RDA)或者
典范对应分析(canonical correspondence analysis, CCA)都是基于对应分析发展的一种排序方法,将对应分析与多元回归分析相结合,每一步计算均与环境因子进行回归,又称多元直接梯度分析。主要用来反映菌群与环境因子之间的关系。
RDA 是基于线性模型,CCA是基于单峰模型。分析可以检测环境因子、样品、菌群三者之间的关系或者两两之间的关系。

○ 冗余分析可以基于所有样品的OTU作图,也可以基于样品中优势物种作图;
○ 箭头射线:箭头分别代表不同的环境因子;
○ 夹角:环境因子之间的夹角为锐角时表示两个环境因子之间呈正相关关系,钝角时呈负相关关系。环境因子的射线越长,说明该影响因子的影响程度越大;
○ 不同颜色的点表示不同组别的样品或者同一组别不同时期的样品,图中的拉丁文代表物种名称,可以将关注的优势物种也纳入图中;
○ 环境因子数量要少于样本数量,同时在分析时,需要提供环境因子的数据,比如 pH值,测定的温度值等。
21. 贡献图
我们通过计算每个变量正常计数中值,进一步确定每个被选择的OTU的特征。如果某一变量的中位数数高于任何其他变量,则OTU被定义为对变量有贡献。其中每个OTU条长度对应于多元模型中特征的重要性(对于每个组件上的特定特征,具有正号或负号的多元回归系数)通过从底部开始降低重要性进行排序,并且颜色与贡献变量相匹配。贡献图可以显示任意指定级别的细菌分类。


图解读:加载在comp1组件和comp2组件上贡献最大的OTU图。颜色代表不同分组。条形图越长说明对应OTU在此分组中贡献最大。
25. spls(稀疏偏最小二乘)回归分析
sPLS回归允许整合微生物群落数据矩阵和临床变量矩阵以进行多元回归。它可以处理数据中的共线性和噪声,并且适合对多个响应变量进行建模。
这需要有大量的meta信息,例如一个样本有几十个临床信息,你想知道这些信息与肠道菌群的相关性是怎样的,我们将这些临床信息利用adonis2检验它们与肠道菌群间是否有统计学意义。然后将具有统计学意义的信息利用spls按照它们之间的相关性从大到小排列。数据间的相关性越强越能很好的使用此分析。
| a |

| b |

| c |

图解读:
a图. 前两个sPLS维度的相关圆图显示了> 0.2/< – 0.2的相关性。两个灰色圆圈表示相关系数为0.5和1.0。OUT显示为较小的圆点,根据所属的cluster进行着色。表示变量的圆点附带了标签。距离较近的变量之间呈正相关,投影方向相反的变量之间呈负相关。彼此垂直放置的变量不相关。OTU解释的方差在Component 1上为2.94%,在Component 2为8.77%.
b图. 前两个sPLS维度的聚类图像映射,显示了OTUs(右侧)和临床变量(底部)之间的两两相关。红色和蓝色分别表示正相关和负相关。在基于sPLS回归模型的mixOmics cim()函数内进行层次聚类(聚类方法: complete linkage,距离法:Pearson相关)。
c图. 分别在Component 1和Component 2上贡献最大的OTU的荷载图。长方形条状是根据它们所属的簇而着色的。各OTU的分类信息根据颜色着色(图例见b图)
看完以上内容,也许还有不明白的地方,没关系,我们罗列了一些常见的问题。看看有没有你想问的。
答疑小课堂
Q1
原始数据形式以及数据如何上传?
原始fastq格式是一个文本格式用于存储生物序列(通常是核酸序列)和其测序对应的质量值。这些序列以及质量信息用ASCII字符标识。通常fastq文件中一个序列有4行信息:如

第一行:序列标识,以 @开头。格式自由,允许添加描述信息,描述信息以空格分开。
第二行:序列信息,不允许出现空格或制表符。一般是明确的DNA或RNA字符,通常大写
第三行:用于将序列信息和质量值分隔开。以 +开头,后边是描述信息或者不加。
第四行:质量值, 每个字符与第二行的碱基一一对应,按照一定规则转换为碱基质量得分。进而反映该碱基的错误率,因此字符数必须和第二行保持一致。
Fasta格式
fasta是一种基于文本用于表示核苷酸序列或氨基酸序列的格式。在这种格式中碱基对或氨基酸用单个字母来编码,且允许在序列前添加序列名及注释。由两部分信息组成:如

第一行:序列标记,以 >开头,接序列的标识符,序列标识符以空格结束,后接描述信息。为保证分析软件能区分每条序列,每个序列的标识必须具有唯一性。
第二行:序列信息,使用既定的核苷酸或氨基酸编码符号。
数据提交
原始数据(Raw data),常见的是illumina机器产生的fastq文件,这一类文件需要向NCBI的SRA数据库进行提交,SRA是NCBI为了并行测序的高通量数据(massively parallel sequencing)提供的存储平台。完整提交SRA需要一些独立项目的分步提交,包括BioProject、BioSample、Experiment、Run等,每一部分用以描述数据的不同属性。
Q2
如何判断测序质量是否合格?
原始的Tags数据会经过质控、过滤、去嵌合体,最终得到有效数据(Effective Tags)。所以在判断测序质量是否合格时应该从几个方面去判断。
打开文件01_sequence_statistic/sumOTUPerSample.txt


报告里所有的txt打开如果格式不对的话,可以用excel表打开。
其中tags为经质量过滤后能正确overlap包含正确barcode和高质量序列的数据。
Singleton为非完全相同的序列,只要有1个碱基的差异即为不同序列,该值的高低与OUT数量并无直接关系,OTU是以97%的相似度聚类,测序质量较低导致的碱基错误、PCR扩增过程中的碱基错误、菌种内部的多样性以及OTU数量均会影响该数量。
Chimeras为通过与RDP等标准数据库比对分析判断可能由于PCR过程错误扩增导致的嵌合体比例,chimeras%为百分比,一般低于1。
首先判断下机数据tags和有效数据 clean tags 的数据量是否满足测序要求,一般下机数据量达到3万条reads以上满足测序需要,谷禾16s样本的测序深度可以达到10万条reads左右。如果数据量不够则需要重新补测样本。通过观察嵌合体数chimras 和嵌合体所占百分比chimeras%,可以反应出有效序列的转化率,嵌合体的比例越小序列的利用转化率就越高。
根据稀释曲线可以判断测序深度是否达到饱和,如图中曲线都逐渐趋于平缓,就证明样本的测序深度较好,测序深度基本覆盖能测到的该样本所有的物种,测序深度比较好。同时曲线趋于水平纵坐标的高低也能够反映各样本的微生物多样性情况,曲线越高,证明测到的物种种类越多,样本的微生物多样性就越高。

而从该图可以看出,个别样本的曲线未趋于平缓,证明该样本测序深度不够,测序深度未能很好的反映出该样本的完整菌群构成。如果测序数据量更大的的话会检测到更多物种。
Q3
如何了解分组内部的多个样本的重复性以及多样性情况?
观察分组内部多个样本的重复性如何可以从以下几个方面考虑。
首先在各分类水平的柱状图的菌属构成来看

从构成图来看,Flu组和ZW3.7组,组内样本重复性较好。Ctrl组中Ctrl.2明显区别于组内另外两个样本,可以去掉该样本。而ZW3.8组内样本间差异性较大。
比如人体肠道或小鼠肠道样本本身个体差异性较大,菌群结构组成复杂,即便通过不同疾病的分类的样本,但营养饮食、代谢以及环境的影响都会改变肠道菌群的构成,所以有可能组内样本间差异性会比较大。而经过单因素处理的样本组内差异会比较小。
所以在前期实验设计时,尽量选择同一批次相同处理的小鼠或其他样本,避免组内差异的影响。并且要预留好多余的样本,比如组内只有3个样本,如果去掉一个差异性较大的样本,一个分组内只有2个样本,会影响后续组间差异比较,组间差异性比较分析每组要至少要3个样本。

通过beta多样性分析PCA,PCoA,MNDS 也可以大致观察组内样本重复性情况,左图组内样本重复性较好,右图组内样本间差异性较大,两组间的区割不是很明显。

在加圈图的beta多样性分析中,右下角有给出PC1和PC2的P值,小于0.05则差异显著。
Alpha多样性是针对单个样品中物种多样性的分析,包括chao1指数、ace指数,shannon指数以及simpson指数等。前面4个指数越大,最后一个指数越小,说明样品中的物种越丰富。

其中chao指数和ACE指数反映样品中群落的丰富度(species richness),即简单指群落中物种的数量,而不考虑群落中每个物种的丰度情况。指数对应的稀释曲线还可以反映样品测序量是否足够。如果曲线趋于平缓或者达到平台期时也就可以认为测序深度已经基本覆盖到样品中所有的物种;反之,则表示样品中物种多样性较高,还存在较多未被测序检测到的物种。
而shannon指数以及simpson指数反映群落的多样性(species diversity),受样品群落中物种丰富度(species richness)和物种均匀度(species evenness)的影响。相同物种丰富度的情况下,群落中各物种具有越大的均匀度,则认为群落具有越大的多样性。
稀释曲线是利用已测得序列中已知的各种OTU的相对比例,来计算抽取n个(n小于测得Reads序列总数)Tags时各Alpha指数的期望值,然后根据一组n值(一般为一组小于总序列数的等差数列,本项目公差为500 )与其相对应的Alpha指数的期望值绘制曲线。
Q4
不同的样本之间差异大吗?不同分组之间能否用菌群差异来区分?
观察不同分组间差异的大小可以观察随机森林分类效果图。
路径在07_diff_analysis/RF
图中以该分类水平下选取用于区分不同分组间的差异性起到关键性影响因素的物种作为标志物作图。标志物按重要性从大到小排列,图中随机森林值error rate 表示用随机森林方法预测分组之间的错误率,分值越高代表所选取的标志物准确度不高,并不能很好的用于区分各分组,分组差异不显著。分值越低证明分组效果比较好。

上图中的随机森林按照门和属以及代谢途径分别进行分析作图,各自都有单独文件,报告中仅给出了一个图,其他文件需要到目录中查看。可能存在门或属区分效果不佳,但是代谢途径区分效果较好。
随机森林筛选出来的物种是用于区分所有分组的重要标志。分值越高代表该物种用于区分所有组之间的重要性越大。
Q5
二代测序16s 能用普通酶扩增吗?
16s测序主要为了鉴定菌种,通常在做鉴定的时候区分标准是97%,区分亚种和菌株的时候相似度更高。
普通TAQ酶的复制错误率较高,可能在扩增过程中引入错误,这些错配可能导致相似度下降从而分类错误。
一般我们不建议使用普通TAQ酶进行扩增,都选择高保真酶。
Q6
利用16s rRNA鉴定细菌能确定到种上吗?
16s rRNA长度为1.5k多,作为菌种鉴定一般选择相似度97%的标准,相似度超过97%一般定义为同一种菌。
如果是sanger测序获得16s全长的都可以鉴定到种,甚至能区分亚种。有些细菌并不只有1个16s序列,会包含有1-15拷贝的16s序列,所以单一的16s序列鉴定可能会出现偏差。
利用高通量如454或miseq测序一般由于读长的缘故,通常只有300-500多个碱基被测序,所以在物种鉴定上一般比较可靠的是能分类到属,部分能分类到种。
根据我们的经验,不同的样品会有大约10-50的菌能分类到种。利用新的分析方法,我们现在也可以利用16s rRNA的群落多样性高通测序数据进行亚种级别的分析。主要是利用16s中共同变化的SNP位点进行分型。这样可以大大提高菌种的分类精度,尤其是在有些菌株之间表型差异巨大的时候。
Q7
听说光测16s就可能预测基因和功能,是真的吗?
16s序列能够区分菌的种属,但是并不包含这些菌的基因和代谢功能的信息。不过由于我们已经对大量的细菌基因组进行了测序,所以可以根据16s的菌种信息,利用这个菌属已经测序的细菌基因组的基因信息和代谢功能信息来估计每类基因的上限和下限。
所以答案是可以利用16s序列测序来预测菌群的功能基因分布和代谢途径分布情况。
目前主要使用的软件是PICRUSt和新发表的Tax4Fun。
从我们实际分析和实验结果来看,预测的准确性还是很高的,不过和样品有很大关系。像肠道菌群和土壤以及一些致病菌的测序较多,所以预测的准确度较高可以到85-90%以上。一些海洋的菌由于测序的菌较少,预测准确性要差一些。目前发表的文献基本都是用PICRUSt,新的软件还有待验证。
Q8
测16s rRNA能分到亚种吗?不同菌株都有致病性差异光到种不解决问题啊!
16s rRNA如果是使用sanger测序可以细分到亚种甚至有些可以精确区分菌株,但是要看菌种。
如果是高通量测序,目前的常见分析一般以97%为标准,大部分情况只能到属,少部分能区分到种。如果要进一步细分到亚种甚至更小的区分目前是有可能的,我们在使用oligotype一类的方法时可以将相同变化模式的SNP归类,并对原来的OTU进行进一步细分,理论上可以区分到菌株。
不过这种区分不同菌属差异很大,有些可以很理想的区分,主要用来了解在更细分化尺度上菌株构成的地理和时间变化。
仅通过16s高通量测序恐怕不能完全解决菌株致病性差异这种问题,但是通过对常见OTU的进一步深入分析可以提供可能的解释或方向。如果明确了某一特定类型菌株的变化有关,可以采用比如毒力基因或菌株特异性标记等方法详细了解不同菌株的比例和差异。
可变区和测序选择
目前针对扩增子测序可选择的测序平台和方案很多,不同平台的读长和适用的测序区段以及优势各有不同。16s测序主要的测序区段包括V4、V3V4,V1V2,V6,此外还有全长等不同的区段选择,不同可变区或全长由于引物的不同以及不同种属相应区段内的变异多样性差异,对菌属的丰度评估会有一定的差异。
从长度来看,全长16S长度为1.5kb左右,单菌落的16S全长sanger一代测序仍然是菌种鉴定的主要手段,纳米孔和Pacbio的三代测序可以高通量的获得全长序列,对于希望更高分辨率的分析菌种的研究有一定优势。三代的测序准确度目前逐渐改进,直接测序准确度可以在90%以上,纠错后可以提高到97~99%以上,已足够提供高精度的分类。三代目前主要问题在于建库成本相对较高,通过使用barcode可以降低部分但仍然偏高,此外普遍测序深度相对于二代测序要低许多。
目前最主要的可变区选择是V4区和V3V4区,V4区长度为256bp左右,加上两侧引物长度为290bp左右,使用双端2x250bp或2x150bp可以测通,此外如454、life、Illumina
Hiseq 4000的测序平台读长也可以主要涵盖该区段读长。例如采用Illumina Hiseq测序平台对该项目进行双端测序(Paired-end),测序得到了fastq格式的原始数据(样本对应一对序列S_1.fastq和S_2.fastq)。再配对拼接成单条序列。其引物通用性相对是所有可变区中最高的,大量的大规模菌群调查研究都采用V4区作为检测区域,包括人体菌群研究如:HMP,肠道菌群如美国肠道计划AGP,欧洲的FGFP等,以及全球土壤菌群调查,目前仍然是国际研究中使用最广泛和认可的检测区域。
Illumina的Miseq提供了长达2x300bp以及Hiseq2500和最近的NovoSeq提供有2x250bp的测序方案,为进一步利用读长,目前有相当一部分研究选择V3V4区,该区段长度在460bp左右,相较于V4度多出了V3区段约100bp左右的片段,在少部分菌属中可以增加一定分辨率。经过对比,V3V4区的检测结果和V4区在绝大部分菌属中的丰度一致,但由于引物不同,在少量菌属中丰度会有不同偏向,V3V4从OTU层面上并未发现较V4区有明显增加。引物的选择和提取、储存方法是影响菌群检测丰度构成的主要因素,不同研究之间的比较需要考虑到实验方案的一致,相同的方案可以直接比较。
目前的高通量测序平台可以较低成本的进行大规模的测序,从测序深度角度,土壤菌群的多样性最高,一般需要5万条以上序列可以达到饱和,肠道样本在3万条以上,水体和尿液等1万条以上基本可以到达饱和。
同一批小鼠粪便样本v4(10万 clean reads)和 v3v4(5万clean reads)测序数据比较:
原始序列数据:
V4
V3V4
以上两表是对原始序列数据进行统计,表中可以看出有效序列tags、高质量序列clean_tags、otus数量
V4区都远高于v3v4区。V4区测序获得下机数据在13万条左右,v4区测序获得的下机数据在5万条左右。
Alpha多样性指数比较:
V4
V3V4
以上两个表分别是对Alpha多样性指数计算的结果比较
Chao1 指数和ACE指数是用来评估样本中所含OTU数目的指数,从Chao1 指数和ACE指数可以看出,用 v4测序获得的结果要明显大于v3v4的结果。这是因为v4测序通量更高,测序深度更好,每个样下机的测序数据可以到10万条以上,一般在13万条左右,所以经过序列比对获得的OTU数目更多,相比较用v3v4测序每个样下机的数据大约在4到5万条左右,经过序列比对获得的OTU相对少一点。
Shannon指数和Simpson指数是用来评估菌群的丰富度和均一度 的。从Shannon指数和Simpson指数,用v4和v3v4测序指数相差不大,或v4比v3v4略高一点,证明两种测序之间菌群的丰富度多样性和均一度叫接近。
物种主要构成比较:
V4

V3V4

V3v4
属水平前10个物种构成:Lactobacillus、Adlercreutzia、Flexispira、Allobaculum、Desulfovibrio、Prevotella、Odoribater、Oscillospira、[Prevotella]、Bacteroides
V4
属水平前10个物种构成:Lactobacillus、Akkermansia、Helicobacter、Allobaculum、Desulfovibrio、Adlercreutzia、Odoribacter、Bacteroides、Prevotella、[Prevotella]
从前10个物种构成来看,有8个是相同的,物种的主要构成基本一致,测序的稳定性较好。从种类来看,v3v4测到的属水平个数较多。
各分类水平鉴定到的物种种类比较:
V4

V3v4

以上两张表代表了每个样本在各分类水平上鉴定到的物种种类数。从整体上来看,分别用v4和v3v4测序得到的数据,在各分类水平上鉴定到的物种个数相对比较稳定和接近,(尤其在目水平和科水平上)用v3v4测序获得的物种数比v4相对较多一点,单相差不大,在属水平和种水平则不一定是这种规律,最终鉴定到的物种个数也跟该样本的测序质量有关。
在线系统沟通和下载报告非常方便
项目系统:检测方案将为每一位合作者建立项目系统,全程了解样品和项目情况,并可直接与相应人员沟通。
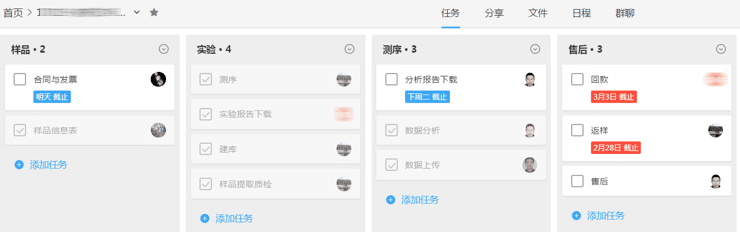
项目进度管理系统为项目提供从项目需求到样品接收以及实验过程和测序分析、售后全程管理和人员责任。
不用担心测完之后的售后问题,项目系统是永久登录的。售后全部都是服务到发文章为止~
多项合作成果发表于Nature communications、PNAS、Plant biotechnology journal、DNA Research、Environmental Science & Technology、Plant、cell & environment、Science of The Total Environment 、Gut Microbes 、Frontiers in microbiologyt、Journal of environmental management 等国际著名学术期刊。
近期发表文章目录
最后附几篇顶级杂志发表的16s V4区的文章
Poyet, M., et al. “A library of human gut bacterial isolates paired with longitudinal multiomics data enables mechanistic microbiome research.” Nature medicine 25.9 (2019): 1442-1452.
(16S library preparation and sequencing. 16S rRNA gene libraries targeting the V4 region of the 16S rRNA gene were prepared by first normalizing template concentrations and determining optimal cycle number by way of qPCR. Two 25 µL reactions for each sample were amplified with 0.5 units of Phusion with 1X High Fidelity buffer, 200 μM of each dNTP, 0.3 μM of 515 F( 5′- AATGATACGGCGACCACCGAGATCTACACTATGGTAATTGTGTGCCAGCMGCCGCGGTAA-3′) and 806rcbc0 (5′- CAAGCAGAAGACGGCATACGAGATTCCCTTGTCTCCAGTCAGTCAGCCGGACTACHVGGGTWTCTAAT-3′).
Tito, Raul Y., et al. “Population-level analysis of Blastocystis subtype prevalence and variation in the human gut microbiota.” Gut 68.7 (2019): 1180-1189.
(We profiled stool samples from 616 healthy individuals from the FGFP cohort as well as 107 patients with IBD using amplicon sequencing targeting the V4 variable region of the 16S rRNA and 18S rRNA genes).
Call, Lee, et al. “Metabolomic signatures distinguish the impact of formula carbohydrates on disease outcome in a preterm piglet model of NEC.” Microbiome 6.1 (2018): 111.
(Gut contents and mucosal samples were collected and analyzed for microbial profiles by sequencing the V4 region of the 16S rRNA gene. Metabolomic profiles of cecal contents and plasma were analyzed by LC/GC mass spectrometry).
Wang, Chao, et al. “High-salt diet has a certain impact on protein digestion and gut microbiota: a sequencing and proteome combined study.” Frontiers in Microbiology 8 (2017): 1838.
(In this study, C57BL/6J mice were fed low- or high-salt diets (0.25 vs. 3.15% NaCl) for 8 weeks, and then gut contents and feces were collected. Fecal microbiota was identified by sequencing the V4 region of 16S ribosomal RNA gene).
Bai, J., Y. Hu, and D. W. Bruner. “Composition of gut microbiota and its association with body mass index and lifestyle factors in a cohort of 7–18 years old children from the American Gut Project.” Pediatric obesity 14.4 (2019): e12480.
(AGP sequenced the V4 region of 16S rRNA gene).
Luthold, Renata V., et al. “Gut microbiota interactions with the immunomodulatory role of vitamin D in normal individuals.” Metabolism 69 (2017): 76-86.
(The association between 25(OH)D and fecal microbiota (16S rRNA sequencing, V4 region) was tested by multiple linear regression).
Iszatt, Nina, et al. “Environmental toxicants in breast milk of Norwegian mothers and gut bacteria composition and metabolites in their infants at 1 month.” Microbiome 7.1 (2019): 34.
(Child fecal samples were characterized by 16S rRNA gene amplicon sequencing of the V4 region. We used Deblur, a novel sub-operational taxonomic-unit (sub-OTU) approach that provides a higher resolution than OTU-based analyses).
Vangay, Pajau, et al. “US immigration westernizes the human gut microbiome.” Cell 175.4 (2018): 962-972.
(We performed amplicon-based sequencing of the 16S rRNA gene V4 region on 550 stool samples (one sample per participant).
Suez, Jotham, et al. “Post-antibiotic gut mucosal microbiome reconstitution is impaired by probiotics and improved by autologous FMT.” Cell 174.6 (2018): 1406-1423.
(For 16S amplicon pyrosequencing, PCR amplification was performed spanning the V4 region using the primers 515F/806R of the 16S rRNA gene and subsequently sequenced using 2X250 bp paired-end sequencing (Illumina MiSeq).
Zmora, Niv, et al. “Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features.” Cell 174.6 (2018): 1388-1405.
(For 16S amplicon pyrosequencing, PCR amplification was performed spanning the V4 region using the primers 515F/806R of the 16S rRNA gene and subsequently sequenced using 2 × 250 bp paired-end sequencing (Illumina MiSeq).
Riquelme, Erick, et al. “Tumor microbiome diversity and composition influence pancreatic cancer outcomes.” Cell 178.4 (2019): 795-806.
(The 16S rDNA V4 region was amplified by PCR and sequenced in the MiSeq platform (Illumina) using the 2×250 bp paired-end protocol yielding pair-end reads that overlap almost completely. The primers used for amplification contain adapters for MiSeq sequencing and single-index barcodes so that the PCR products may be pooled and sequenced directly (Caporaso et al., 2012), targeting at least 10,000 reads per sample. 16S (variable region 4 [v4]) rRNA gene pipeline data incorporated phylogenetic and alignment based approaches to maximize data resolution).
Matson, Vyara, et al. “The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients.” Science 359.6371 (2018): 104-108.
(Specifically, the V4 region of the 16S rRNA gene (515F-806R) was PCR-amplified with region-specific primers that include sequencer adapter sequences used in the Illumina flowcell).
Raman, Arjun S., et al. “A sparse covarying unit that describes healthy and impaired human gut microbiota development.” Science 365.6449 (2019): eaau4735.
(Amplicons generated from variable region 4 (V4) of bacterial 16S rRNA genes present in these 2455 fecal samples were sequenced, and the resulting reads were assigned to operational taxonomic units with ≥97% nucleotide sequence identity (97%ID OTUs).
Gehrig, Jeanette L., et al. “Effects of microbiota-directed foods in gnotobiotic animals and undernourished children.” Science365.6449 (2019): eaau4732.
(Characterizing human fecal microbial communities Methods for V4-16S rRNA gene sequencing and data analysis, calculation of MAZ scores and functional microbiome maturity, and quantification of enteropathogen burden by means of multiplex quantitative polymerase chain reaction (qPCR) are described in the supplementary materials).
Lloyd-Price, Jason, et al. “Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases.” Nature 569.7758 (2019): 655.
(In brief, bacterial genomic DNA was extracted from the total mass of the biopsied specimens using the MoBIO PowerLyzer Tissue and Cells DNA isolation kit and sterile spatulas for tissue transfer. The 16S rDNA V4 region was amplified from the extracted DNA by PCR and sequenced in the MiSeq platform (Illumina) using the 2 × 250 bp paired-end protocol, yielding pair-end reads that overlapped almost completely).
Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019
(In brief, bacterial genomic DNA was extracted from the total mass of the biopsied specimens using the MoBIO PowerLyzer Tissue and Cells DNA isolation kit and sterile spatulas for tissue transfer. The 16S rDNA V4 region was amplified from the extracted DNA by PCR and sequenced in the MiSeq platform (Illumina) using the 2 × 250 bp paired-end protocol, yielding pair-end reads that overlapped almost completely).
emporal development of the gut microbiome in early childhood from the TEDDY study. Nature. 2019
(Bacterial DNA was extracted using the PowerMag Microbiome DNA isolation kit following the manufacturer’s instructions. The V4 region of the 16S rRNA gene was amplified by PCR and sequenced on the MiSeq platform (Illumina) using the 2 × 250 bp paired-end read protocol).
A communal catalogue reveals Earth’s multiscale microbial diversity. Nature. 2018
(We surveyed bacterial and archaeal diversity using amplicon sequencing of the 16S rRNA gene, a common taxonomic marker for bacteria and archaea12 that remains a valuable tool for microbial ecology despite the introduction of whole-genome methods (e.g., metagenomics) that capture gene-level functional diversity13. We amplified the 16S rRNA gene (V4 region) using primers14 shown to recover sequences from most bacterial taxa and many archaea).
Root microbiota drive direct integration of phosphate stress and immunity. Nature. 2017.
(For wild soil experiment 16S sequencing, we processed libraries according to Caporaso, et al.28. Three sets of index primers were used to amplify the V4 (515F-806R) region of the 16S rRNA gene of each sample. In each case, the reverse primer had a unique molecular barcode for each sample).