-

 CNAS L23010
CNAS L23010

 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室

谷禾健康

微生物物种的遗传变异研究通常包括单核苷酸多态性(SNPs)、结构变异(structural variants ,SV)和可移动遗传元件(mobile genetic elements,MGEs)。
在宏基因组中,SNP被用来量化种群结构、追踪菌株和鉴定微生物表型的遗传决定因素。然而,现有的基于比对的宏基因组SNP检测方法需要高性能的计算和足够的覆盖深度来区分SNP和测序错误。
为了解决这些问题,美国加利福尼亚大学研究人员使用高质量基因组,构建了 909 个人类肠道物种中 1.04 亿个 SNPs 的目录,并使用针对该目录的独特 k-mers 表征来自 7,459 个样本的肠道菌群的全球种群结构,开发了GenoTyper for Prokaryotes(GT-Pro),可以对宏基因组的这些 SNPs进行快速基因分型的方法。该研究成果近日公开在《Nature Biotechnology》发表。
该方法与使用读长对齐的方法相比,GT-Pro 更准确,速度快两个数量级,作者构建了一个GT-Pro数据库,基于大约25,000个宏基因组样本,并展示了GT-Pro如何用于数千种菌群的菌株水平探索,可以实现在个人电脑上快速高效地对数百万个SNP进行宏基因组分型。

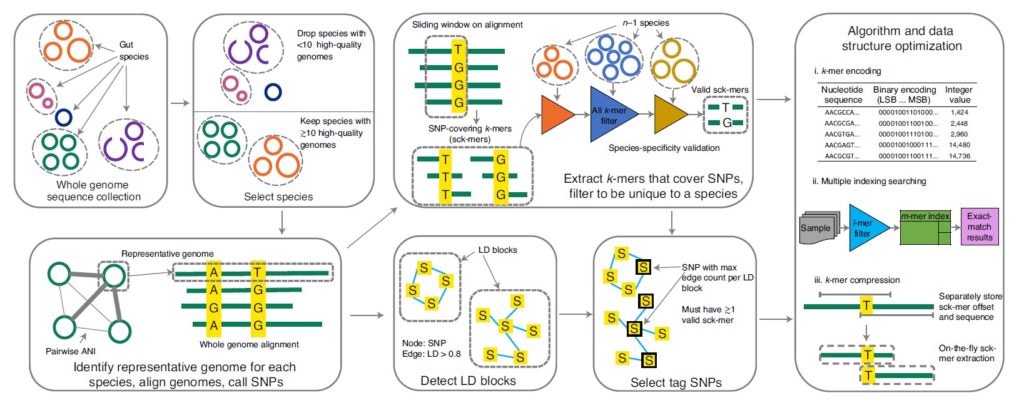
如图,按箭头方向所示。
首先从全基因组序列中识别高质量基因组的物种(去除<10 个高质量基因组的物种,高质量基因组:≥90% 的完整性和≤5% 的污染),对于每个物种,一个有代表性的基因组是根据平均核苷酸一致性(Average Nucleotide Identity,ANI)和组装质量指标选择的,确定代表性基因组后,对每个物种,通过MUMmer软件将每个同种基因组(conspecific genome )与代表性基因组比对,确定SNP,在这些SNP中选择常见的双等位基因SNP用于分型(site prevalence ≥90% and minor allele frequency >1%)。
接下来提取覆盖SNPs的k-mers(sck-mers),过滤出独有的物种,同时检测LD块,并选择具有物种特异性的sck-mers的SNPs和该块中其它SNP的最高LD。LD块为基于跨基因组的共现模式将 SNP 聚类成linkage disequilibrium block。检测LD块使用R2 阈值 (0.81) 。具有物种特异性的sck-mers即删除了两个或多个物种共有的任何sck-mer。
最右边的方框里简要是GT-Pro的算法和数据结构的优化方法。也是该研究的主要目标之一,正是利用了该方法构建的SNP索引才能实现快速地分型。
首先是k-mers编码,选择了k=31,以便使用64位整数编码,通过这一步骤,GT-Pro 数据库缩小了四倍。
其次是多索引检索和进一步压缩SNP数据结构。
优化后的GT-Pro数据库由两个表组成:
(1)10.6 GB的sck-mers表,包含每个k-mer的4字节条目;
(2)2.4 GB的sc-span表,包含每个等位基因的24字节条目。
所需的总存储空间为13 GB,是原始sck-mer表的bzip2压缩的两倍。也使得GT-Pro可以在个人计算机中高效运行。
1.从模拟宏基因组中准确识别SNP
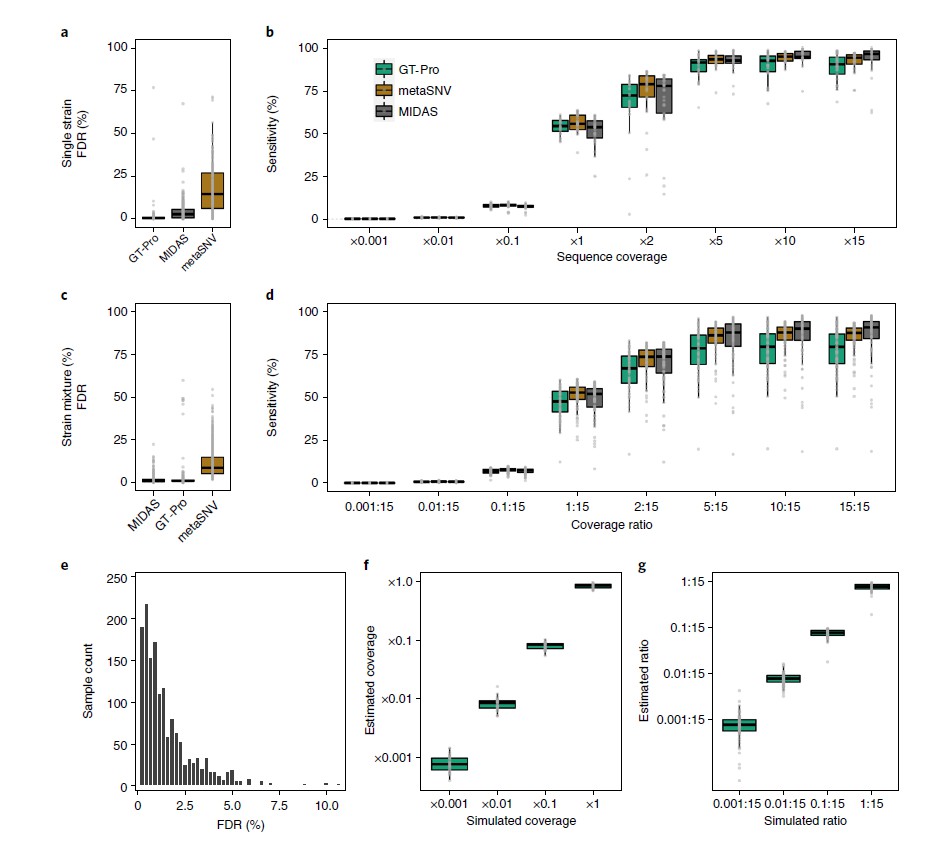
比较GT-Pro、MIDAS和metaSNV宏基因分型的准确性,使用232个未用于开发这些方法的人类肠道分离株的模拟宏基因组(大约2600万次reads)。
图a为FDR比较,假阳性指不正确的基因型,是由测序错误和读数映射到错误位点导致的。假阴性指缺失的基因型,在没有读数映射时产生。在宏基因组中,FDR最低的是GT-Pro(中位数,0.4%),而 metaSNV 最高(中位数,14.5%)。
图b为对图a的灵敏度调查,用于直接比较不同方法。敏感性是指在GT-Pro数据库中检测到分离株基因组(参考和非参考等位基因)中存在基因型的概率。结果表示,随着覆盖度的加深,GT-Pro的灵敏度损失较小。
图c为比较三个工具在一对同种分离株但不同覆盖率下的FDR,目的是检查宏基因组分型方法对菌株混合物的表现。其中一个菌株始终为15倍的覆盖率,另一个菌株的覆盖率从 0.001 到 15 倍不等。FDR包括纯合位点和杂合位点。
总体而言,GT-Pro的 FDR与 MIDAS 相似但低于 metaSNV。
图d为对图c的灵敏度调查,敏感性是指正确判断reads所模拟的基因组的基因型(纯合位点和杂合位点)的概率。GT-Pro 的灵敏度低于基于比对的方法,基于比对的宏基因分型通常使用覆盖率和等位基因频率过滤来减少错误的杂合性调用。
图e为基于图a中模拟的等位基因,从tag SNPs推算的基因型的FDR。结果表示大多数物种的 FDR 较高但仍低于 5%。
图f和图g,为了探索 GT-Pro 是否能用于定量估计物种丰度,使用从单个分离株和对同种分离株中模拟的宏基因组,比较了sck-mer匹配reads的平均数量和已知的基因组覆盖率。结果表示GT-Pro等位基因的调用和计数可以用一个小的校正因子来估计物种和菌株的相对丰度。
所有的结果表示,在模拟宏基因组的测试中,metaSNV 和 MIDAS 对于丰富的物种(>5×覆盖度)和保守位点表现良好,但 GT-Pro 对典型覆盖率值、非参考和杂合位点更准确和敏感,同时对错配和测序错误更为稳健。只是,与 metaSNV 和 MIDAS 相比,GT-Pro 无法检测其数据库中缺少的新 SNP。
结论是,在保守的基因组区域仔细选择sck-mers能使 GT-Pro 能对来自鸟枪法宏基因组数据的已知 SNPs 进行敏感和特异性的基因分型。
2.从模拟宏基因组中准确识别SNP
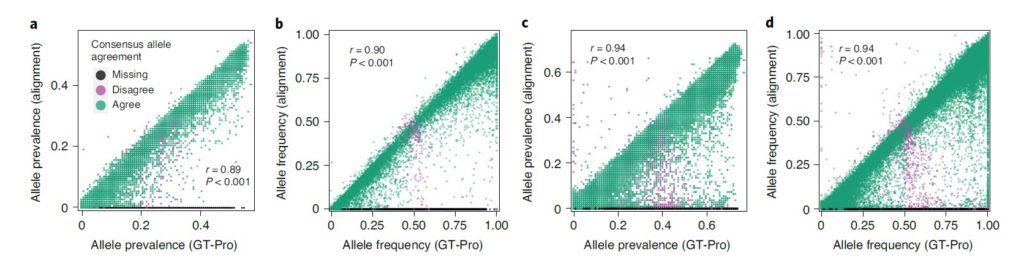
使用GT-Pro对肠道微生物组样本进行宏基因组分型,结果与基于比对的MIDAS宏基因组分型比较。
图a和b分别为流行率(prevalence)、平均等位基因频率(Average allele frequency)
图c和d类似图a和b,只是物种不同。
每个点代表一个 SNP,颜色表示两种方法的共有等位基因(即样品中最常见的)是否相同(绿色),两种方法都返回某些样品的基因型,但共有等位基因不同(紫色)或仅GT-Pro 返回基因型(黑色)。
结果表示对于高覆盖率物种,基于比对的方法能检测到GT-Pro数据库中没有的SNP,而GT-Pro 在中低覆盖率物种中检测到更多SNP位点。这部分结果也与模拟宏基因组测试时的结论一致。
1.使用GT-Pro的SNP估算结构变异
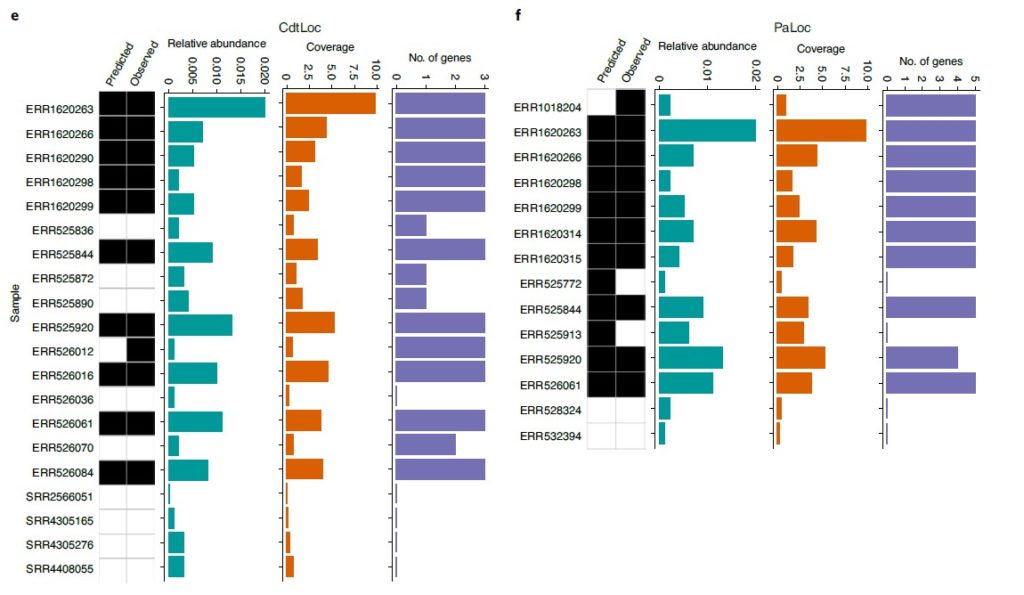
研究人员试图使用GT-Pro的SNP推断附近基因或操纵子的存在,从而作为结构变异的生物标志物。
首先对艰难梭菌的毒性控制位点CdtLoc和PaLoc的侧翼区域使用GT-Pro检索SNP。
接着用艰难梭菌的参考基因组训练了一个随机森林分类器,用于预测来自混合群组(n = 7,459)的人类肠道宏基因组中存在/不存在艰难梭菌毒素基因位点。
图e和f分别代表CdtLoc基因和PaLoc基因,对每个样本,最左边的热图,第一列为预测的,第二列为基于比对方法得到的,黑色表示存在,白色表示不存在。
从左到右的条形图分别指艰难梭菌的相对丰度、全基因组序列覆盖率,从毒素位点检测到的基因数目,所有这些都是通过比对到艰难梭菌的代表性基因组来估计的。结果表示预测到艰难梭菌毒素位点的概率>0.6。
对CdtLoc的几个预测与宿主的表型相关(P < 0.001),包括5名艰难梭菌阳性和CdtLoc(+)的克罗恩病患者,这与该人群对艰难梭菌病理的高易感性相一致。与此相反,CdtLoc基因座在大多数可检测到艰难梭菌的健康婴儿中没有被预测,这与婴儿期艰难梭菌常见的无症状定殖一致。这些结果表明,GT-Pro可以预测具有临床相关性的linked structural variants。
2. 使用 GT-Pro 捕获新的种内遗传结构
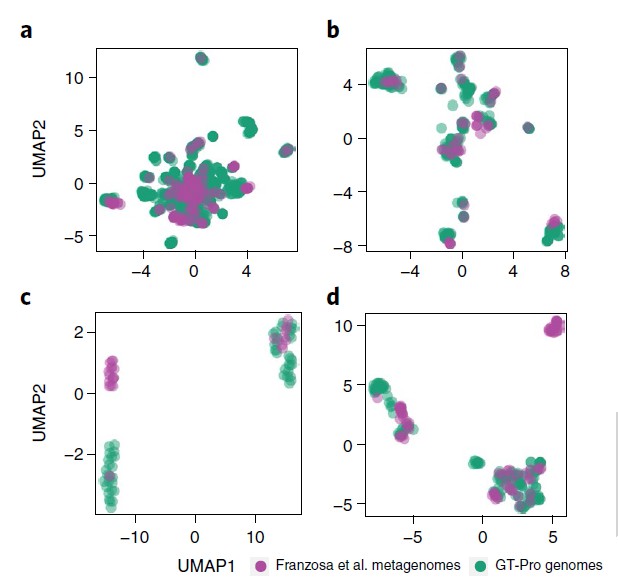
GT-Pro 可以对从参考基因组中鉴定的已知 SNP 进行宏基因组分型分析。但研究人员认为GT-Pro还有更广阔的发展,假设GT-Pro可以基于 SNP 等位基因的不同组合检测新的菌株变异。
为了验证该假设,研究人员使用GT-Pro 对最近发表的北美炎症性肠病 (IBD) 队列 的 220 个粪便宏基因组中发现的物种进行基因分型。使用UMAP降维分析,每个图都是将UMAP应用于一个物种GT-Pro SNPs基因型矩阵的结果。每个点代表该物种的一个菌株(杂合宏基因组的主等位基因)。紫色为队列样本,绿色为GT-Pro基因组。
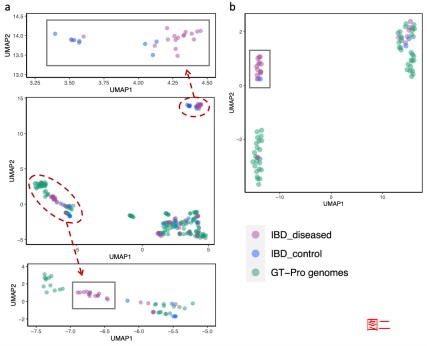
结果表明 GT-Pro 的数据库代表了这些个体的常见菌株多样性,对于大多数物种,如图一的a和b,粪便样本组与参考基因组聚集在一起,相比之下,对于少数物种。
如图二的c和d,分别是新的亚种,观察到基因型与数据库中任何参考基因组不同的粪便样本群,包括一些富含IBD患者的样本。这说明可以使用 GT-Pro 常见 SNP 发现新的亚种遗传结构。
3. GT-Pro 探索全球人类肠道微生物组遗传变异
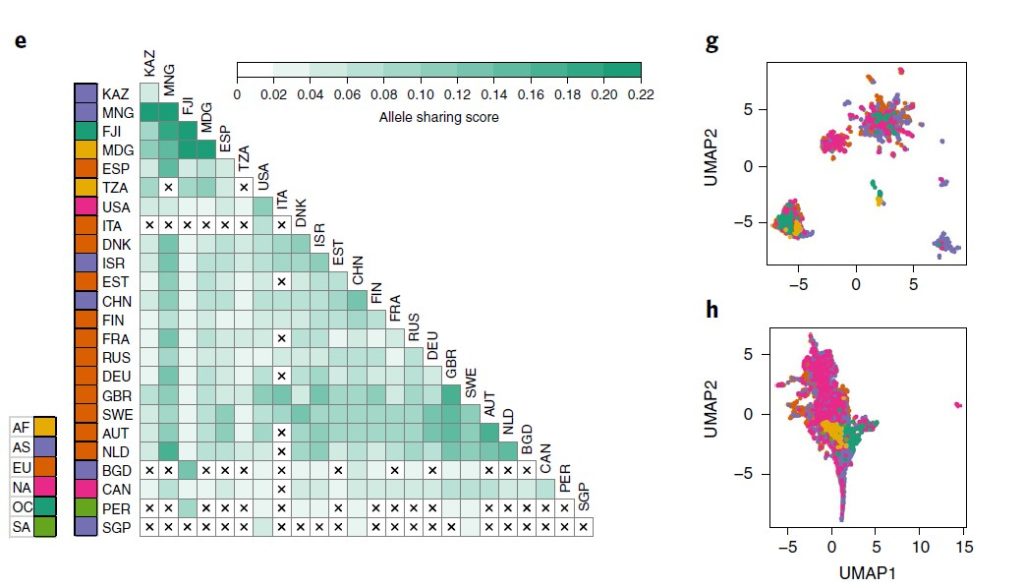
来自六大洲 31 个地点的 7,459 个肠道样本中发现的 881 个物种的 5180 万个 SNP的多个物种的种内遗传变异荟萃分析。
图e来自不同国家的宏基因组间的等位基因平均共享分数的热图。打叉单元格表示由于样本对不足(<5,000)而导致分数缺失。
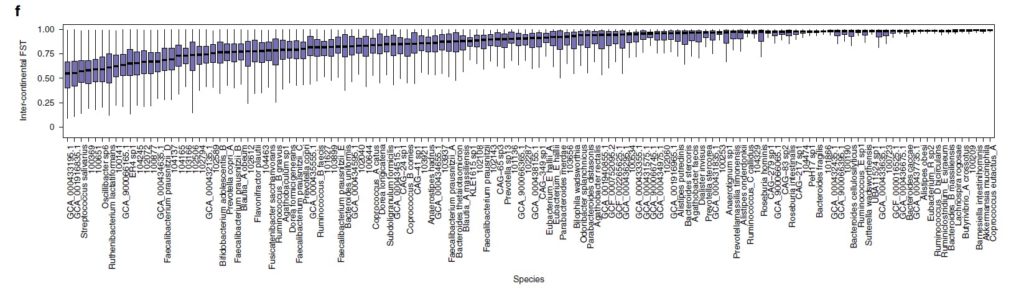
图f为78 个常见物种的洲际种群分化分析(大陆内部与大陆之间的遗传相似性,用 F 统计检验测量亚种群 (FST) 中捕获的总遗传变异的比例)。
每个箱线图代表一个物种的洲际 FST 分布,按中位数排序。图g为通过直肠Agathobacter rectalis(物种ID 102492)的GT-Pro宏基因组基因型中的种内遗传变异捕获的地理模式的示例。
图h为为基于图g中相同样本的物种相对丰度的UMAP 分析。每个点都是一个宏基因组样本。颜色与图e示意一致。
结果表示,等位基因共享与工业化程度以及宿主关系明显关联;洲际种群间的分化程度有巨大差异,具有高FST的物种显示出明显的宿主集群,但不是所有宿主集群都与地理相关。
这与菌株在宿主中殖民的生活方式和环境的作用相一致。相比之下,在基于物种相对丰度的UMAP分析中,宿主间并没有明显集群,这表明宏基因组基因型可能揭示了在丰度分析中缺失的微生物生态学和微生物群落-宿主关系。
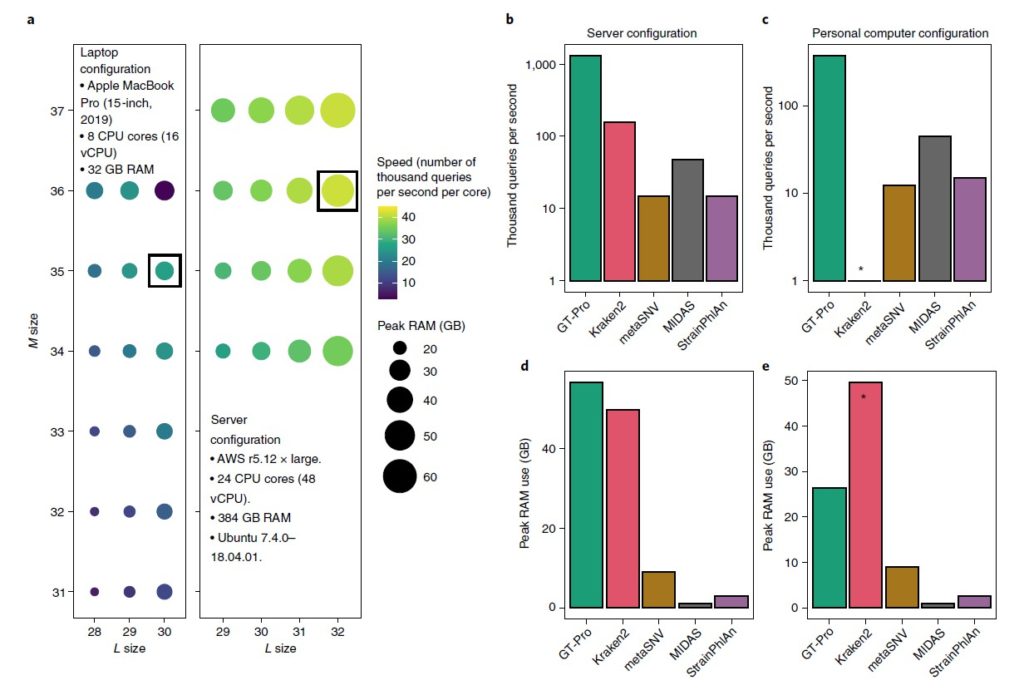
图a评估GT-Pro在笔记本电脑(左)和服务器环境(右)中的计算性能,以bits为单位。颜色表示处理速度,圆圈大小为RAM使用峰值。黑色方框表示最优状况。
图b-c为GT-Pro与metaSNV、MIDAS、StrainPhlAn、 Kraken2之间的速度比较,分别在服务器环境和笔记本电脑下比较。
图d-e为RAM使用峰值的比较,分别在服务器环境和笔记本电脑下比较。* 由于超出可用 RAM,Kraken2 无法在笔记本电脑中运行。
这些分析表明,与其他方法相比,GT-Pro 在服务器上大约快 8.5-570 倍,在笔记本电脑上快 8.3-163.6 倍。平均而言,处理每个宏基因组只需要在服务器上不到 4 秒,在笔记本电脑上大约需要 13 秒(平均为 497 万次读取)。虽然 GT-Pro 比其他方法更快,但它在服务器上需要 1.1-53.7 倍的 RAM和笔记本电脑上的 2.9-29.2 倍的 RAM(不包括内存不足的 Kraken2)。因此,只要计算机具有足够的 RAM,GT-Pro 数据结构和算法就可以极大地加速宏基因组分型。
研究人员在该文章中使用GT-Pro大约分析了2.5万个宏基因组,展示了GT-Pro是如何快速准确的识别SNP以及探索结构变异、种内遗传变异等。GT-Pro不使用基于比对的方法,而是类似于Kraken2,通过编码k-mers来快速检索,并适用于个人计算机或服务器环境。
但是它也不是完美的,目前GT-Pro存在的不足和如何应对:
第一,GT-Pro 数据库并未捕获所有人类肠道微生物多样性:但是通过基因组测序,会持续扩大SNPs的数量和涵盖的物种。
第二,GT-Pro 类似于基因分型阵列,因此不能识别新的 SNP,这需要其他方法,例如基于比对的宏基因组分型或单细胞基因组测序。
第三,由于基因组集合中存在高度相关的物种,少数物种缺乏物种特异性的 sck-mers。替代策略,例如使用更长的 k-mer 或不太常见的 SNP,可以对这些物种使用 GT-Pro 。
第四,尽管非常严格的挑选了用于构建GT-Pro的基因组和SNPs,但不可能完全排除错误(例如,不完整、污染和物种错误分类)。
最后,GT-Pro 不直接对结构变异进行基因分型。
“
考虑几个GT-Pro的未来发展方向,比如:
将GT-Pro与下游算法结合起来,以识别代表新微生物菌株的SNPs簇,或准确标记参考数据库中已知菌株的SNPs;
将GT-Pro的计算框架扩展到其他微生物环境中;为短插入缺失和结构变异开发无比对宏基因组分型;
将微生物组应用于精准医学,综合识别与疾病或其他特征(如致病性、抗菌耐药性、药物降解)相关的SNPs;
将GT-Pro用于检测污染、重组和跟踪变化,比如变异或菌株随时间、宿主生活方式和地理位置的变化。
主要参考文献
Shi ZJ, Dimitrov B, Zhao C, Nayfach S, Pollard KS. Fast and accurate metagenotyping of the human gut microbiome with GT-Pro. Nat Biotechnol. 2021 Dec 23. doi: 10.1038/s41587-021-01102-3. Epub ahead of print. PMID: 34949778.