-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
谷禾健康
噬菌体(bacteriophages,phages)是专一感染细菌的病毒,它曾长期被视为抗生素时代之前的“旧疗法”,但在多重耐药(multidrug resistance, MDR)日益严峻和微生物组医学兴起的背景下,噬菌体疗法被重新定位为一种“可编程的微生物群编辑工具”。
噬菌体这一名称源于“吃细菌”之意。它广泛存在于一切有细菌的环境中,包括土壤、地壳深处、动植物体内以及海洋等,其中海洋是全球噬菌体最密集的天然库之一。
本文系统梳理噬菌体的生物学基础、肠道噬菌体组的生态学规律,以及噬菌体疗法在胃肠疾病与肝病中的证据链与关键瓶颈。
同时顺便讨论了噬菌体相较抗生素的优势:高度靶向特异性、可在靶点原位扩增、具备穿透和降解生物膜的潜力,以及与抗生素耐药机制的不交叉性;并分析其临床转化面临的核心挑战,如宿主谱狭窄、耐噬菌体进化、免疫反应与微生物组外溢效应、疗效评价体系与监管标准的不完善,以及高质量公共噬菌体库的匮乏。
更重要的是,肠道与肝病领域正在形成“从感染到菌群失衡相关疾病”的范式迁移:噬菌体不再仅是“杀菌药”,而是用于选择性削减致病共栖菌以恢复肠道稳态、影响免疫与代谢表型的精准干预手段。
噬菌体是一种感染细菌细胞并在其内部繁殖的病毒,它们形态和遗传物质类型多样。噬菌体由称为衣壳的蛋白质外壳包裹内部的DNA或RNA。噬菌体通常只识别特定细菌物种或菌株,在多种环境中对细菌种群的调控起着关键作用。
噬菌体疗法最大的优点之一是不会攻击其他细菌,只攻击特定类型或群体,我们的身体习惯了它们,因为我们被它们包围着。
噬菌体的发现历史
噬菌体实际上存在的时间比我们还长。
1915-1940年——发现与发展时期
1910年:噬菌体的发现;
1920年:噬菌体用于细菌感染治疗。
1940-2000年——停滞时期
抗生素的使用压制了西方国家的噬菌体疗法研究,但在苏联及其盟友中蓬勃发展,特别是在冷战期间。
1940年:难以重现,缺乏基本科学数据;
1970年:噬菌体与抗生素治疗比较。
2000年-至今——复兴时期
抗生素耐药性威胁加剧,结合现代生物技术和基因工程的进步,重新激发了对噬菌体疗法的兴趣。相关研究论文、临床试验及成功临床应用案例的数量均不断增加。
2005年:首次安全性测试;
2007年:人类微生物组计划;
2014年:CRISPR-Cas系统武装的噬菌体;
2019年:噬菌体可在肠道中特异性靶向细胞溶解性E.faecalis;
2020年:肠道微生物基因水平的原位调节;
2021年:口服噬菌体疗法BX002的I期药代动力学研究取得积极成果;
2023年:首个CRISPR噬菌体药物候选者进入临床试验。
2024年:小鼠肠道中细菌的原位靶向编辑。

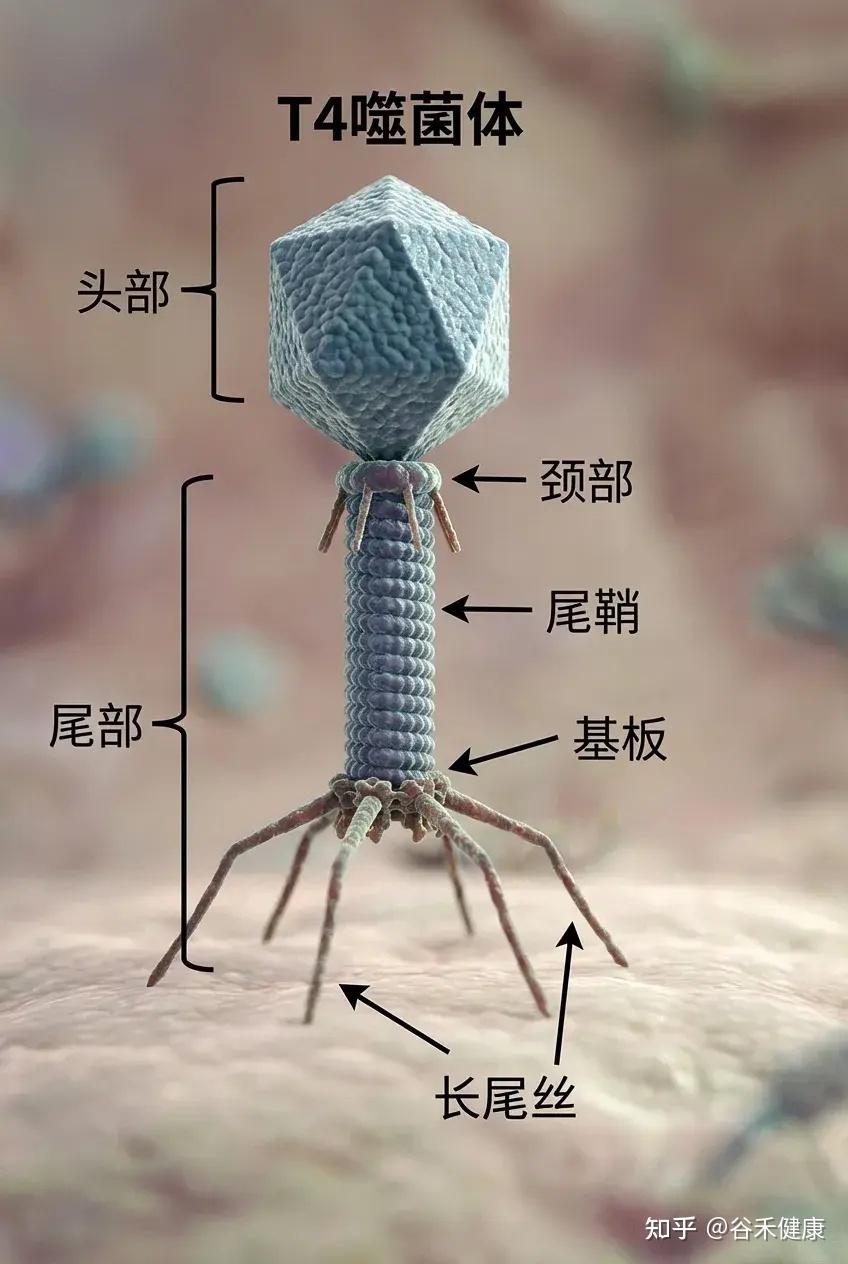
噬菌体结构
噬菌体由三部分构成:一个多面体的头部,一个短短的领口,以及一个螺旋形的尾部。
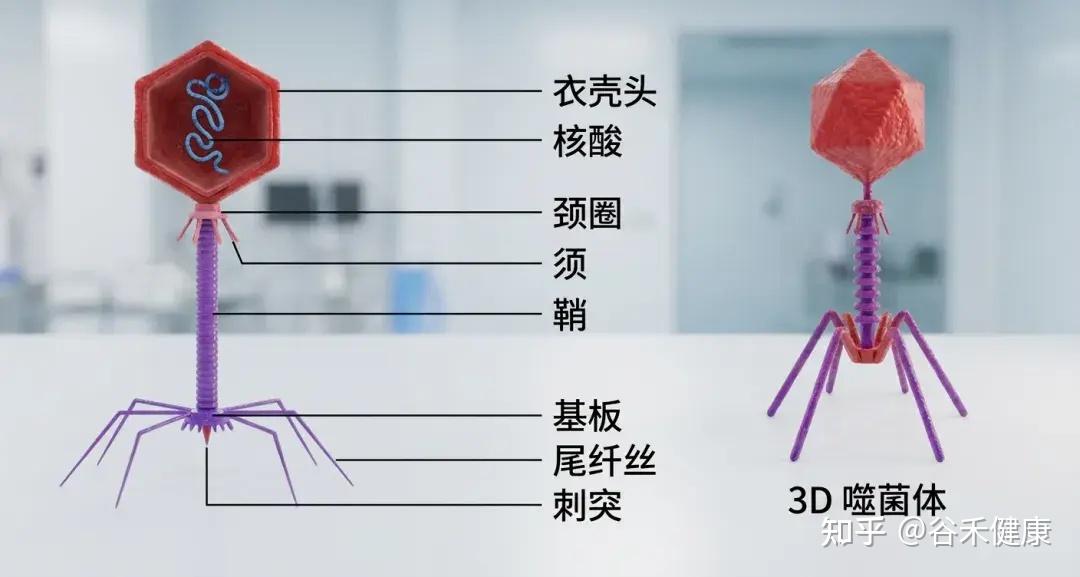
①头部
形状:多面体(就像一个有很多面的几何体)。
结构:由大约2000个囊聚体(capsomere)组成,囊聚体是构成头部外壳的基本单元。
内部:包含双链DNA,也就是噬菌体的遗传物质。
简单来说: 头部的作用就是保护噬菌体的DNA。
②项圈
位置位于头部和尾部之间,提供结构支撑,促进头部与尾部的连接。
③尾部
尾鞘:一种收缩结构,见于某些噬菌体(如T4噬菌体),有助于将遗传物质注入宿主体内。感染期间,鞘收缩并推动尾管穿过细菌细胞壁。
尾管:一种中空的管道,遗传物质通过它进入宿主细菌体内。
底板位于尾部末端,是连接细菌表面的关键。它通常包含受体结合蛋白、分解细菌细胞壁的酶以及其他有助于附着和穿透的特征。
尾纤维丝:长而灵活的延伸部分,使噬菌体能够识别并连接细菌细胞表面的特定受体。这些纤维非常特殊,决定噬菌体的宿主范围。
简单来说: 尾部的作用是像一根“注射器”,将噬菌体的DNA注入细菌体内。尾丝用来“抓住”细菌。
例如,T4噬菌体(如下图)是一种感染大肠杆菌的病毒,大肠杆菌是分子生物学领域研究最为深入的细菌类型之一。由于T4噬菌体感染大肠杆菌,科研人员得以结合对细菌的既有认知,借助T4噬菌体研究病毒在细胞内的感染与复制机制。
噬菌体的生命周期
噬菌体是感染细菌的病毒,它们有两种主要的生命周期:裂解循环和溶原循环。
•裂解循环或致病循环
裂解循环是指噬菌体感染细菌后,在细菌体内复制自身,最终导致细菌破裂(裂解)并释放出大量新的噬菌体,从而感染更多细菌。可以理解为“快速杀灭”的模式。这个过程包含以下几个步骤:
吸附 (Adsorption): 噬菌体利用其尾部的纤维,结合到细菌表面的特定受体上,就像钥匙插入锁一样。
穿透 (Penetration): 噬菌体的尾鞘收缩,并利用一种酶削弱细菌的细胞壁。然后,噬菌体将自身的DNA注入到细菌细胞内。
噬菌体组分的合成: 噬菌体的DNA进入细菌后,会“劫持”细菌的代谢系统,开始合成新的噬菌体所需的各种部件,例如头部、尾部以及各种蛋白质。这些蛋白质分为早期蛋白和晚期蛋白,早期蛋白负责复制DNA和合成其他蛋白质,晚期蛋白主要用于组装病毒颗粒。
成熟与组装: 噬菌体的头部、尾部等各个部件组装在一起,形成完整的病毒颗粒,称为病毒体。 噬菌体DNA会被装入头部中。
释放:最终,大量新形成的噬菌体会产生一些酶来破坏细菌的细胞壁,导致细菌裂解。 细菌破裂后,这些噬菌体就被释放出来,可以去感染其他细菌。
•溶原循环
噬菌体的生命周期要么是裂解性,要么是溶原性。
溶原循环是指噬菌体感染细菌后,并不立即导致细菌裂解,而是将自身的DNA整合到细菌的染色体中,成为前噬菌体。前噬菌体随着细菌的复制而复制,并传递给细菌的后代。可以理解为“潜伏”的模式。
当细菌分裂繁殖时,携带前噬菌体的细菌(称为溶原细菌)也随之分裂,将前噬菌体传递给所有后代。这样,噬菌体就可以在不杀死细菌的情况下进行繁殖。
但是,前噬菌体并非永久地潜伏在细菌体内。在某些条件下(例如紫外线照射),前噬菌体可能会从细菌的染色体上切除下来,并进入裂解循环,导致细菌裂解。
溶原细菌在繁殖过程中,也有可能因为切除作用而失去前噬菌体。
•裂解循环与溶原循环的区别
总而言之,裂解循环相当于“快攻”:迅速杀死宿主并大量产生子代,方式激进;溶原循环则类似“潜伏”:先藏入宿主基因组,随宿主繁殖扩增,在需要时再转入裂解。噬菌体选择哪种模式取决于细菌的生理状态和环境条件等多种因素。
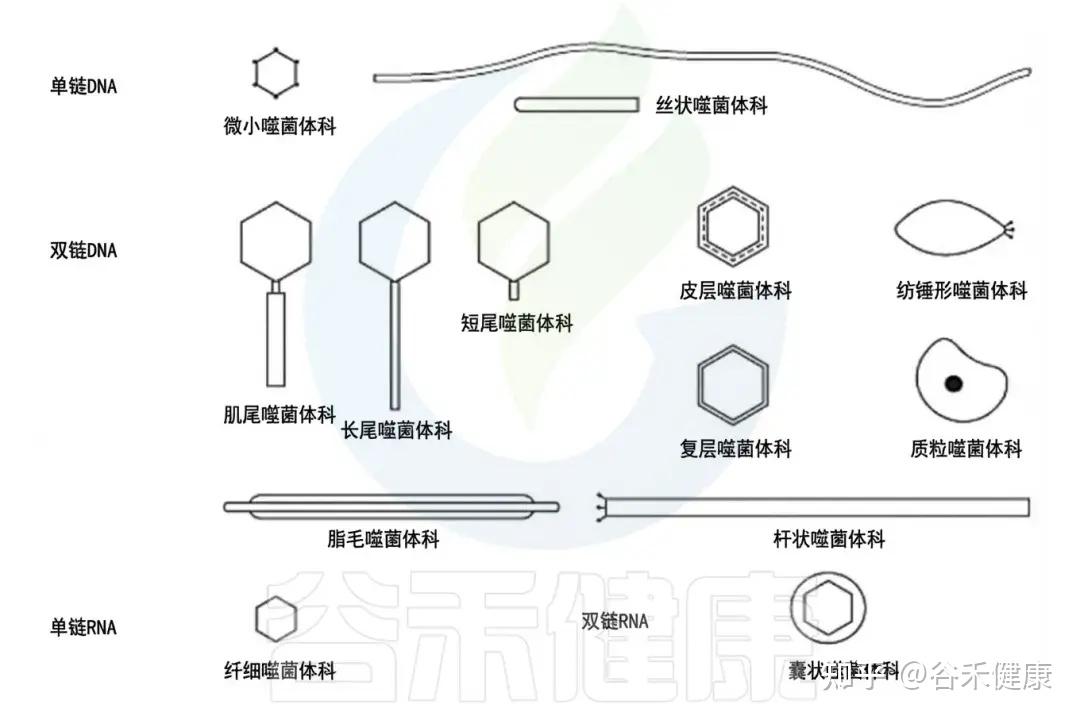
噬菌体的类型
•按基因组分:
根据基因组,噬菌体可分为以下四种类型:
1.dsDNA 噬菌体(双链 DNA);
2.ssDNA 噬菌体(单链 DNA);
3.dsRNA 噬菌体(双链 RNA);
4.ssRNA 噬菌体(单链 RNA)。
噬菌体主要类群的示意图
•按照生活史/复制策略分:
裂解性噬菌体: 这些噬菌体感染细菌细胞并在体内繁殖,最终使细胞裂解(破裂)并释放新形成的噬菌体。T4噬菌体是一种常见的裂解性噬菌体。
溶原噬菌体: 这些噬菌体将 DNA 整合进细菌染色体,并能长时间保持潜伏状态。在某些情况下,噬菌体 DNA 可以在复制过程中传递给子细胞,从而实现稳定感染。λ噬菌体是一种著名的溶原噬菌体。
•按形态结构分:
有尾噬菌体: 这些噬菌体具有独特的尾部结构,有助于附着在宿主细胞细菌表面 。偶T噬菌体(T2、T4、T6)是有尾噬菌体的例子。
丝状噬菌体: 这些噬菌体具有细长的丝状结构,有助于感染细菌细胞。它们通常感染革兰阴性菌,且没有尾巴。M13是一种著名的丝状噬菌体。
长尾噬菌体科(Siphoviridae):是国际病毒分类委员会确认的噬菌体主要科系,具有长而无收缩性的尾部结构,头部呈正多面体或扁形衣壳。其基因组为线型双链DNA,通过尾部吸附宿主细菌细胞壁实现感染,主要侵染大肠杆菌等革兰氏阴性菌。
该科成员以λ噬菌体和T5噬菌体为典型代表,能够在宿主基因组中整合并进行温和性复制。作为天然抗菌剂,其裂解酶特性被应用于绿脓杆菌感染治疗及工业发酵污染防控。
短尾噬菌体科(Podoviridae):这类噬菌体具有短尾、一般不可收缩;宿主范围因种而异,很多感染革兰氏阴性菌,代表如T7噬菌体。
噬菌体的分布
噬菌体无处不在;水、空气、土壤,几乎任何地方都能找到细菌。有人甚至认为,正因为噬菌体的存在和独特功能,我们的环境才没有被细菌完全占据。他们认为噬菌体是被允许控制自然界细菌水平的生物体。
噬菌体疗法已有近百年历史,但在抗生素广泛应用后迅速被边缘化。随着抗生素效果下降以及测序和微生物组研究的推进,噬菌体疗法重新受到关注,并被视为在缺乏有效手段时“恢复肠道微生物群”的潜在临床策略,其依据包括对靶菌的杀灭作用和免疫调节功能。
在胃肠—肝病领域,噬菌体疗法的应用已从霍乱、腹泻等感染扩展至多种与特定菌群相关的非传染性疾病,如克罗恩病中的黏附侵袭性大肠杆菌(AIEC)、溃疡性结肠炎中的艰难梭菌、结直肠癌相关的聚合梭杆菌、NAFLD 中的“产酒精”肺炎克雷伯菌以及酒精相关肝炎中的粪肠球菌等。
另一方面,从微生物组视角出发,噬菌体是人体最小且广泛存在的微生物之一,对细菌群体施加选择压力并塑造微生物组;噬菌体可与黏膜表面相互作用并跨越上皮层,同时还能调节免疫系统。这种“生态—免疫—代谢”的三重效应,使噬菌体疗法天然适配“精准编辑肠道菌群”的医学目标。
噬菌体的潜在应用
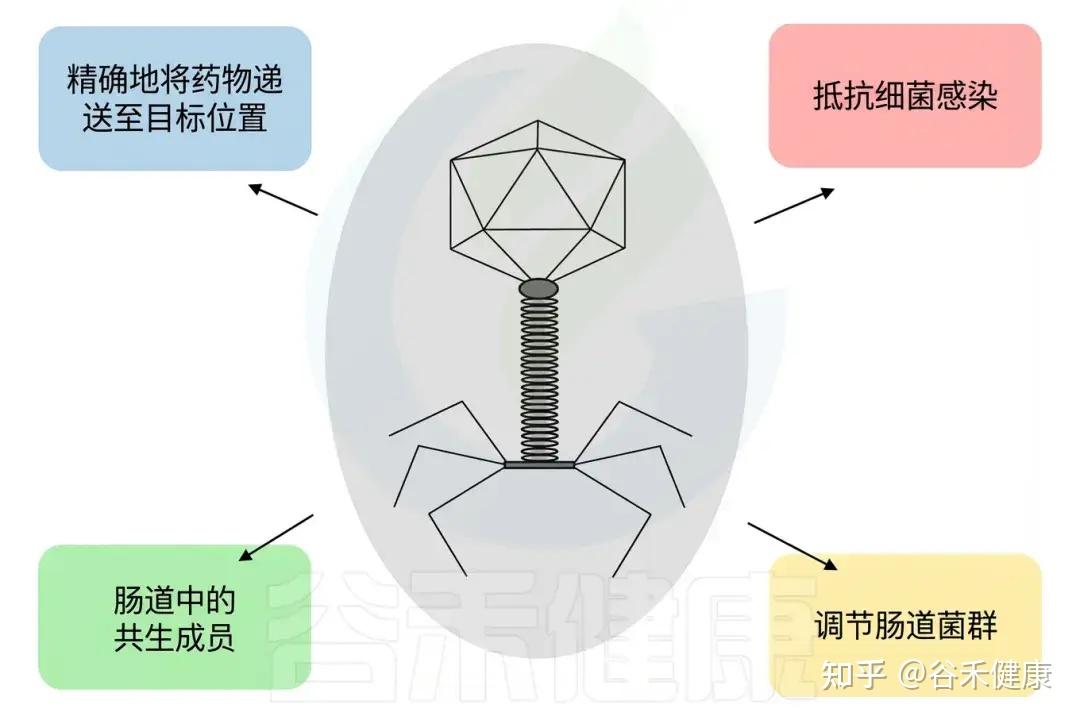
治疗为何偏好裂解噬菌体?
噬菌体的复制策略包括裂解型、温带型(溶原/可裂解)和慢性型。临床应用时,出于监管和安全性考虑,通常优先选择“严格裂解型噬菌体”。
温带噬菌体可整合为前噬菌体并介导基因转移,因而监管接受度较低;但在人类微生物群中却普遍存在,其受饮食、生活方式、化疗等因素诱导激活,可能导致群落扰动和菌群失调。
因此,“理解并可控诱导前噬菌体”正被视为新一代微生物群调控的重要方向。
机制性优势:为何噬菌体常被视为抗生素的互补/替代
•特异性靶向:噬菌体仅攻击被识别的细菌,避免广谱抗生素造成的微生物群破坏和继发感染风险;抗生素易诱发菌群失调,而噬菌体可选择性清除致病共栖菌。
•原位增殖与“自放大药代学”:在存在靶菌时,噬菌体可在局部扩增,呈现近似“单次击中”的动力学特征,其剂量—时间关系不同于需反复给药的抗生素。
•生物膜清除潜力:噬菌体及其脱聚合酶、裂解酶可穿透或降解生物膜胞外基质,提高对耐药生物膜相关感染的控制能力。
•耐药机制不交叉:细菌对抗生素和噬菌体的耐受机制不同,抗生素耐药菌仍可作为噬菌体靶标,反之亦然,因此噬菌体与抗生素的联合和协同应用成为可行策略。
•适应度权衡与表型重塑:噬菌体受体突变可在“噬菌体感染—细菌毒力—抗生素敏感性”之间形成适应度权衡。例如,当外排泵为噬菌体受体时,其突变虽可带来噬菌体耐受,却可能削弱外排泵功能,从而恢复对抗生素的敏感性。
★肠道噬菌体组是菌群稳态的重要调控层
肠道微生物组除细菌外还包括以噬菌体为主的病毒组(virome)。肠道病毒组中约90%为原核病毒、10%为真核病毒,其组成受饮食、生活方式和年龄等因素影响,呈现显著个体差异。
在生态学层面,噬菌体通过对细菌施加选择压力重塑菌群结构,可与黏膜表面互作并跨越上皮屏障,同时调节免疫系统。因此,噬菌体并非单纯的“外源抗菌剂”,而是长期参与“微生物—黏膜—免疫”互作的常驻调控因子。
在生命周期层面,噬菌体可表现为裂解、溶原(前噬菌体)或慢性感染。溶原噬菌体可整合入细菌染色体并在特定触发因素下被诱导进入裂解周期;伪溶原性则允许噬菌体DNA在不利环境中以外染色体形式“待机”,在条件适宜时再进入裂解或溶原通路。
这些机制表明:肠道噬菌体组对菌群稳态的作用既可能体现为“抑制性捕食”(裂解),也可能表现为“功能性重塑”(溶原带来的基因与适应度效应)。这为噬菌体疗法用于肠道疾病提供了理论基础:通过引入特定裂解噬菌体,对关键病原体或致病共栖菌施加定点压力,从而实现菌群结构与功能的可控回调。

噬菌体疗法的“靶向层级”
将噬菌体疗法用于“肠道菌群精准编辑”时,靶点的定义通常可分为三个层级:物种—菌株—功能表型。
•物种层级(species-level):对已知致病或与疾病密切相关的物种进行清除或抑制;
•菌株/群体层级(strain-/pathotype-level):针对同一物种内部与疾病相关的特定致病群体(例如具黏附侵袭表型的菌株群);
•功能表型层级(function-level):以“代谢/毒力功能”定义靶点(例如“产酒精”或“细胞溶解型”表型),强调并非该物种的所有成员均为治疗对象。
★噬菌体选择性靶向并清除特定菌株
通过选择性靶向并清除特定菌株来调节肠道菌群平衡的思路是一致的,也更有助于解释胃肠及肠—肝轴相关疾病机制。
噬菌体的核心优势在于高度特异性,但这也带来宿主谱受限的问题。其宿主范围受感染过程和受体识别等多重因素限制,在临床应用中,宿主谱过窄是公认的主要挑战之一。多数感染或菌群失衡涉及多菌株甚至多菌种,因此往往需要多噬菌体组合(鸡尾酒或联合)以扩大覆盖范围。在治疗设计上,可区分三类“覆盖策略”:
窄谱噬菌体(多为菌株水平):适用于“精准编辑”,最大限度减少对非靶向共栖菌的影响;
相对宽谱噬菌体(同种多株或同属多成员):可降低匹配难度,但并非“广谱抗生素式”全面覆盖;
噬菌体鸡尾酒/联盟(phage cocktails/consortia):通过组合实现更大覆盖并降低耐受风险,实践中常用于应对肠道靶菌的菌株多样性及耐噬菌体进化。
需强调的是,在“微生物群治疗”的语境下,真正意义上的“广谱杀菌”通常并非目标;更契合这一领域价值主张的,是“定点削减关键致病共栖菌、保留有益群落”,以减少菌群失调和继发问题的风险。
基于噬菌体的肠道微生物组生态与基因工程

从感染性腹泻到IBD与CRC相关菌
噬菌体疗法在胃肠领域的研究正由传统感染治疗拓展至与慢病相关的菌群失衡。早期研究主要针对霍乱等感染,而现阶段已开始在溃疡性结肠炎(UC)的艰难梭菌相关结肠炎,以及克罗恩病(CD)中黏附侵袭性大肠杆菌(AIEC)的清除方面开展探索。
•噬菌体定点清除靶菌而不破坏整体菌群
该方向的共同逻辑是:以裂解噬菌体定点清除加重炎症或与复发相关的关键靶菌,从而在不显著破坏整体菌群的前提下恢复微生态平衡。
此外,胃肠领域的综述也将噬菌体疗法延伸至肿瘤相关菌,如将结直肠癌(CRC)相关的具核梭杆菌(Fusobacterium nucleatum)视为潜在靶点。在“菌群—肿瘤微环境”框架下,噬菌体的特异性为“削减疾病相关菌而非整体抑菌”提供了方法学基础,但目前人体临床试验仍然有限。
炎症性肠病和肝病被视为“精准编辑”应用的代表性场景,在这些疾病中通过精确调控微生物或相关因子实现有针对性的干预尤为重要。
炎症性肠病(IBD)的治疗
原则:选择性抑制病原体/致病共栖菌,尽量保留有益菌群。
IBD(包括CD和UC)与菌群失调密切相关。当前调节菌群的手段主要是饮食干预和抗生素治疗,但多重耐药的上升削弱了抗生素的效果,并突显出对新策略的需求。
在此背景下,噬菌体疗法被视为可行替代方案:通过选择性靶向并清除致病细菌来重塑肠道菌群,从而影响炎症进程及临床结局。
(1)UC相关:艰难梭菌(Clostridioides difficile)
艰难梭菌过度生长常由抗生素诱发,可加重UC症状;裂解噬菌体治疗被认为可能帮助恢复微生物平衡。其临床意义在于:在“抗生素引发或加重菌群失调”的情境下,噬菌体的定点作用有潜力降低“治疗—再失衡”的循环风险。
(2)CD相关:黏附侵袭性大肠杆菌(AIEC)
AIEC在CD中被视为重要致病相关群体。综述明确指出,裂解噬菌体疗法正用于探索根除CD中的AIEC,并可能通过清除该致病群体减少炎症与复发。
这一靶点体现了“菌株/致病表型层级靶向”的典型特征:同为E.coli,但治疗目标锁定为与疾病表型相关的特定群体而非全部大肠杆菌。
肝病的治疗
肠道菌群改变与多种肝病密切相关,肠道病毒组的变化也被报道与疾病发生有关。在此背景下,噬菌体疗法的定位正由单纯“抗感染”转向“通过定点清除关键靶菌来编辑肠道菌群”,为肝病干预提供新的路径。
•靶向Enterococcus在治疗肝硬化中有效
近期一项RCT显示,靶向特定Enterococcus物种的噬菌体治疗在肝硬化患者中“非常有效”。研究指出,噬菌体可特异性靶向细胞溶解型(cytolytic)E.faecalis,从而实现对肠道微生物群的精确编辑。其重要意义在于:治疗靶点从“属/种”进一步下沉至“功能/毒力表型”(如细胞溶解型),与前文所述的“功能表型层级靶向”概念相契合。
在肠—肝轴相关肝病中,目前明确的代表性靶点包括:
酒精相关肝炎/肝硬化:肠球菌属(Enterococcus)及E.faecalis(尤其细胞溶解型);
非酒精性脂肪性肝病:Klebsiella pneumoniae(产酒精克雷伯氏菌),是以“功能表型”界定治疗对象的典型例子。
这些靶点共同体现了肝病领域噬菌体治疗的主线逻辑:并非追求“广谱清菌”,而是针对与病理表型高度相关、且具可解释机制链的关键菌群成员实施定点干预,并观察肠道生态与肝脏结局能否实现同步改善。
噬菌体疗法开发的核心在于确保所用噬菌体的安全性与有效性,当前研究主要依赖体内动物模型或合适的体外系统进行评估。同时,宿主谱狭窄与噬菌体耐受进化是公认难题,实践中往往需采用鸡尾酒策略以扩大覆盖并降低耐受风险。
因此,在炎症性肠病(IBD)情境下,研究重点不仅是“找到能裂解靶菌的噬菌体”,还包括:如何应对患者间菌株差异、如何设计组合以抑制耐受出现,以及如何量化其对整体菌群结构与功能的外溢影响。
转化层面上,由于人体试验数量有限,标准化方案和可比终点仍明显不足。尽管噬菌体在胃肠领域已展现出“较为理想的安全性特征”并进入临床评估,但用于胃肠疾病治疗的人体试验仍偏少。
因此,要在肠—肝轴方向开展“可发表、可转化”的研究,需要:
1.明确靶点定义(物种/菌株/功能表型)、患者分层及菌株匹配策略;
2.采用可比较的疗效终点(如靶菌负荷、菌群结构与功能指标、炎症与肝功能指标等);
3.系统评估耐噬菌体出现及菌群外溢效应风险,并据此论证鸡尾酒策略的必要性。
监管与安全性关注点
多个综述一致强调:西方主流监管机构尚未全面批准噬菌体用于常规人用治疗,主要担忧包括噬菌体耐药、对细菌基因组进化的影响、对人体微生物群的影响等;目前更多是在“同情用药”框架下开展个案或探索。
2019年人类微生物群焦点综述进一步把监管审批的科学问题具象化为三问:
•外源噬菌体治疗是否影响微生物群?
•是否影响非靶向共生菌?
•是否出现耐噬菌体突变体,其对微生物群平衡的影响是什么?
并指出现有文献稀缺且结论不一致。
•要注意肠道环境带来的“药物递送”问题
肠道环境的酸度与离子条件可显著影响噬菌体稳定性与滴度;例如空腹人胃pH中位数约1.7,而消化道部分区域pH可>6。因此口服噬菌体疗法常需抗酸策略(抗酸剂、包埋/包衣保护基质)。
这一点对“肠道微生物群编辑”的临床可重复性尤为关键:疗效的不确定性可能来自递送失败而非生物学无效。
主要争议与关键科学问题
需要从“仅仅证明治疗有效”进一步走向“疗效可预测、过程可监管”,实现从单点结果到全程可控的升级。
•宿主谱狭窄与鸡尾酒策略
噬菌体宿主谱狭窄既是优势也是临床障碍;多数感染或失衡为多菌种/多亚型并存,需多噬菌体组合扩大谱系。但鸡尾酒带来配方复杂性、生产一致性、批间稳定性与监管评估难度。
•耐噬菌体进化与共进化外溢
噬菌体—细菌呈捕食—猎物系统并持续共进化,细菌可快速产生耐受突变,噬菌体也可反适应。
鸡尾酒可通过叠加选择压力限制细菌对单一噬菌体的耐受进化。然而,在肠道这样高度异质且多物种互作的生态位中,耐受突变体与原菌株、其他共栖菌的竞争关系可能改变群落结构与功能,导致结果偏离体外培养预测。
•免疫反应与炎症风险
裂解噬菌体可能造成快速大量细菌裂解释放细胞壁成分与噬菌体蛋白,可能触发宿主免疫反应并诱导抗体,影响安全性与重复给药有效性。这也是需要系统研究“噬菌体—免疫”相互作用与合理剂量方案的原因。
•噬菌体选择标准不清与公共库缺乏
几篇权威研究一致指出:治疗性噬菌体选择不能只看宿主谱,还应考虑感染静止期细菌能力、酶活性、血清失活稳定性、突变率等指标;同时公共噬菌体库匮乏导致研发重复、共享不足、难以快速匹配患者菌株。这直接限制了“个体化噬菌体疗法”的规模化与规范化。
★ 什么是噬菌体?
噬菌体是一种感染细菌的病毒。
★ 噬菌体是如何工作的?
噬菌体附着在细菌细胞表面,并将其遗传物质注入细胞内。这些物质随后接管细胞,利用其资源制造更多病毒拷贝。
★ 噬菌体是如何感染细菌的?
噬菌体基因组接管宿主的代谢机制,利用它来生产新的噬菌体。最终,宿主细胞被裂解(破裂)以释放新的噬菌体,这些噬菌体随后可以感染其他细菌。
★ 噬菌体在生物学中有什么重要意义?
噬菌体在控制细菌数量方面发挥着关键作用,防止细菌过度繁殖并对其他生物造成危害。
★ 噬菌体在医学中是如何被利用的?
为抗生素耐药感染的替代治疗方法。它们可以外用或口服,在某些情况下已被证明能有效减少感染中的细菌数量。
将噬菌体送达胃肠道环境
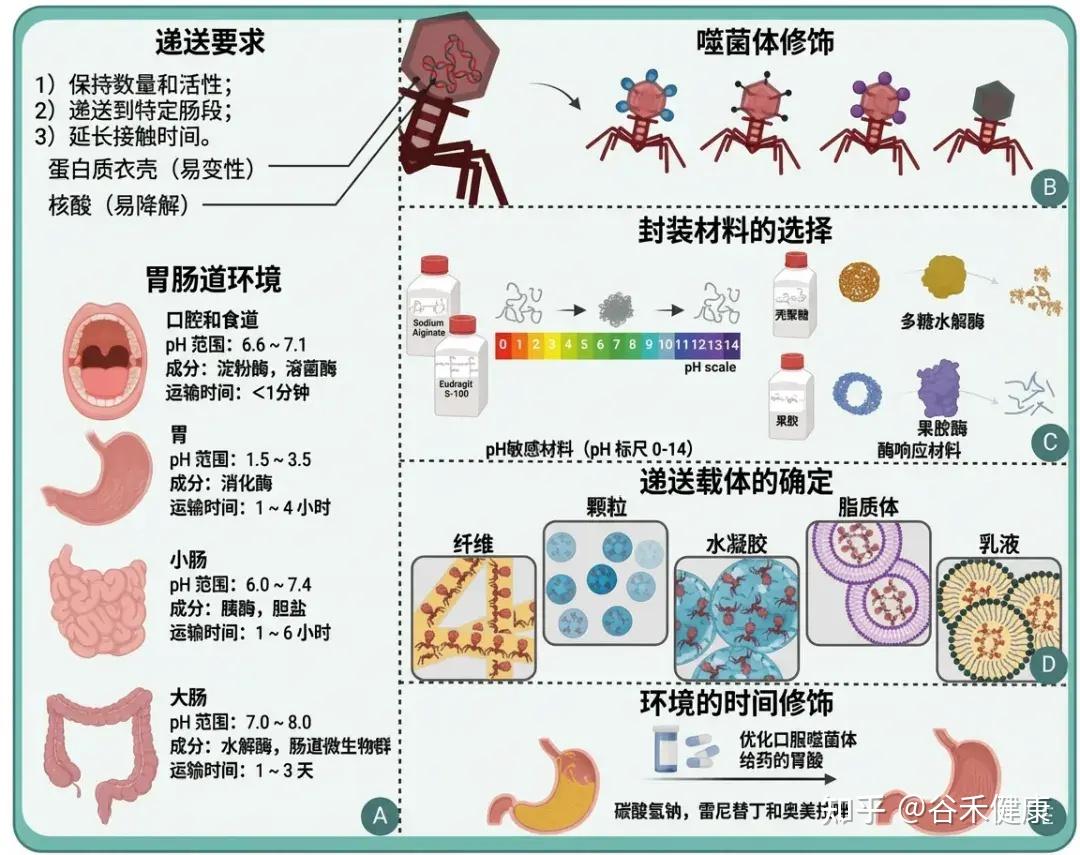
(A)噬菌体给药背景;(B)噬菌体的修饰;(C)封装材料的选择;(D)确定投递承运人;(E)环境的时间性变化。
★ 噬菌体是抗生素更好的替代品吗?
在某些情况下,噬菌体可能是抗生素的有用替代品,尤其是在导致感染的细菌具有抗生素耐药性时。
★ 使用噬菌体有哪些好处?
噬菌体可用于对抗细菌感染,因为它们具有高度特异性,能够针对特定类型的细菌。这常被用作抗生素的替代方案,因为它更具针对性,且对患者产生不良反应的可能性较低。
★ 使用噬菌体会有风险吗?
一个潜在风险是细菌耐药性的发展,因为病毒可能变异并对噬菌体产生抗药性。负责任地使用噬菌体并监测任何抗药性迹象非常重要。
主要参考文献:
Kuang X, Shen J, Zheng L, Duan Y, Ma Y, Leung EL, Dai L. Applications of bacteriophages in precision engineering of the human gut microbiome. Eng Microbiol. 2025 Jan 6;5(1):100189.
Sheraz, M., Shi, H. & Banerjee, S. Human Microbiome and Bacteriophages: Impacts on Health and Disease. Curr Clin Micro Rpt 12, 7 (2025). https://doi.org/10.1007/s40588-025-00243-2
Divya Ganeshan S, Hosseinidoust Z. Phage Therapy with a Focus on the Human Microbiota. Antibiotics (Basel). 2019 Aug 27;8(3):131.
De P, Bhat VG, Kamath V, Kolathur KK, Mazumder N. Insight into Bacteriophage Therapy for Bacterial Infections and Cancer. Mol Biotechnol. 2025 Jul 9.
Bozidis P, Markou E, Gouni A, Gartzonika K. Does Phage Therapy Need a Pan-Phage? Pathogens. 2024 Jun 20;13(6):522. doi: 10.3390/pathogens13060522.
Chen X, Mendes BG, Alves BS, Duan Y. Phage therapy in gut microbiome. Prog Mol Biol Transl Sci. 2023;201:93-118.doi: 10.1016/bs.pmbts.2023.04.005.

谷禾健康
夜幕落下,一场疲惫的战斗即将开始
翻来覆去,时间在无情地流逝
盯着天花板,期待天亮的时刻
却又害怕那一刻的真正到来
这意味着又一个疲惫的早晨
从整个歌单的催眠音乐到数了一万只羊
最终都只是让自己在煎熬中更加清醒

在快节奏的现代生活中,良好的睡眠已成为许多人难以实现的奢望。随着压力的增加,睡眠障碍,如失眠、睡眠呼吸暂停和不宁腿综合症等,成为越来越普遍问题。这些睡眠障碍不仅影响人的生活质量,还常伴随其他共病,如焦虑症、抑郁症、心血管疾病甚至癌症等。
当前,失眠的治疗方法主要有药物治疗和心理疗法。例如,苯二氮䓬类药物虽然能快速缓解失眠症状,但常常伴随神经毒性、成瘾和耐受性等副作用。而认知行为疗法作为一种有效的心理治疗方式,依然缺乏熟练的治疗师,还有高昂的费用,令人望而却步。
当药物的副作用让人却步,心理疗法的门槛又难以触及,我们是否还有更安全、更普适的选择?
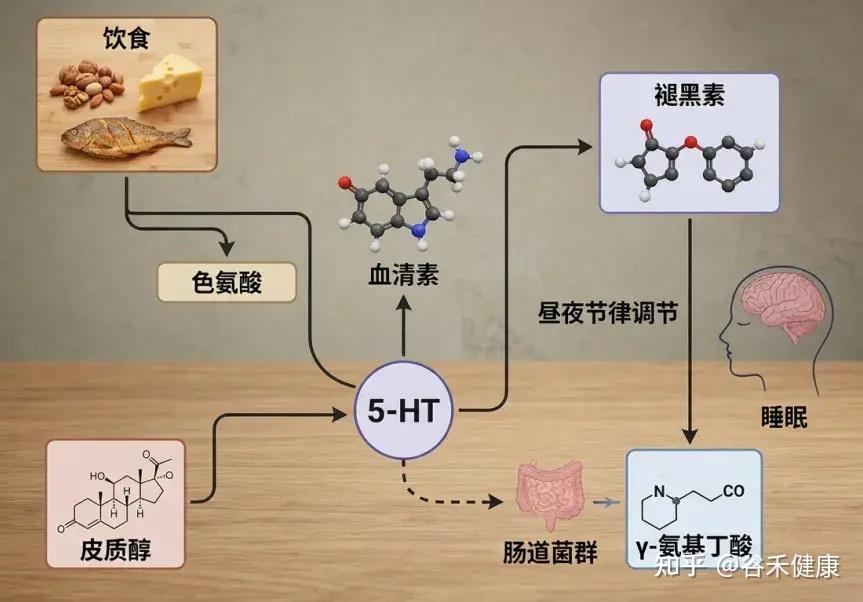
答案或许就藏在我们日常的饮食中。饮食不仅能通过营养成分直接作用于神经系统,还能通过调节肠道菌群这个第二大脑,间接影响我们的睡眠质量和昼夜节律。
例如,富含色氨酸的食物(如坚果和牛奶)有助于促进血清素及褪黑素的合成,而神经递质和激素对调节睡眠周期至关重要。
健康的肠道菌群能够通过调节神经递质和激素,促进更好的睡眠。例如,乳杆菌和双歧杆菌,能够产生GABA等神经递质;进而帮助改善睡眠深度和质量。
然而,饮食与睡眠的关联并不是千篇一律的,因为我们每个人体内都有一套独特的生物钟。对于习惯早起的晨型人来说,他们的进食节奏通常与肠道菌群的活跃周期保持一致,而“夜猫子”们往往习惯晚睡晚起,甚至在深夜进食。
那么,晨型人与夜猫子不同的进食时间和作息习惯,会对肠道菌群的昼夜节律产生怎样的影响?
饮食作为连接两者的关键桥梁,怎样才能更好地帮助我们协调自身节律与肠道菌群的作息,从而改善睡眠呢?
本文我们将探讨饮食中关键的食物成分与睡眠之间的关系,分析饮食如何通过调节昼夜节律和肠道菌群来影响我们的睡眠状态,列举各类改善睡眠质量的营养干预措施,为大家提供切实可行的饮食建议,为您的轻松入睡增添更多选择。
本文目录
01 睡眠调节中的关键营养素及生物活性化合物
氨基酸:色氨酸、甘氨酸
微量元素:镁、锌、铁
维生素:维生素 D、B 族维生素
多酚:芹菜素、槲皮素、白藜芦醇等
02 饮食模式对睡眠质量的影响
地中海饮食、生酮饮食、植物性饮食
饮食时间
03 肠道菌群与睡眠的关系
肠道菌群如何影响睡眠
神经递质合成(GABA等)
昼夜节律与肠道菌群的双向关系
早鸟型or夜猫子型?不同睡眠类型的肠道菌群
饮食与微生物干预措施
04 增强睡眠的功能性食品
酸樱桃汁和褪黑素
猕猴桃和血清素
草本茶与镇静植物化合物
甘氨酸、L-茶氨酸和CBD
05 营养睡眠干预的实际应用与个性化营养方法
睡眠健康的一般饮食建议
场景化食谱与搭配建议
饮水与睡眠(补水节奏、起夜、微醺)
饮食与生活方式的协同作用(正念饮食、光照)
睡眠障碍:个性化营养方案

氨基酸:色氨酸、甘氨酸
✦ 色 氨 酸
色氨酸是一种必需氨基酸,意味着我们的身体无法自行合成它,而必须通过食物获得。色氨酸是合成血清素和褪黑素的重要前体,这两种神经递质对于调节我们的睡眠周期至关重要。特别是,褪黑素帮助信号传递,让我们的身体准备入睡。
无论是通过富含蛋白质的食物还是补充剂摄入色氨酸,都与缩短睡眠潜伏期和改善主观睡眠质量有关。
然而,色氨酸在经过血脑屏障进入大脑时,会与其他大型中性氨基酸(LNAAs)争夺运输通道。因此,膳食中碳水化合物与蛋白质的比例也会影响色氨酸在大脑中的可用性。如果你的饮食中碳水化合物的比例适当,就能更好地促进色氨酸的有效运输,有助于改善睡眠。
注:目前的共识倾向于高碳水、低蛋白的组合最有利于色氨酸进入大脑。研究显示,高升糖指数(高Gl)碳水化合物可能比低Gl碳水化合物更能显著提升Trp/LNAA比率。
来源
小贴士
早餐搭配全麦面包或燕麦粥食用富含色氨酸的食物(如鸡蛋、牛奶)。
下午茶喝一杯牛奶或吃一片全麦饼干,配合少量坚果。
睡前仪式一杯温牛奶,不仅含色氨酸,更重要的是其舒缓的温度也有助于褪黑素分泌。
在摄入色氨酸补充剂或富含色氨酸的食物时,避免吃入大量肉类或乳制品。
✦ 甘 氨 酸
甘氨酸是一种非必需氨基酸,通过在中枢神经系统中作为抑制性神经递质发挥促进睡眠的作用。
研究表明,口服甘氨酸补充剂可以改善主观睡眠质量并缩短入睡潜伏期。这种效果可能与甘氨酸降低核心体温有关,而降低体温是身体准备入睡的重要生理信号。
来源
小贴士
避免空腹大剂量:甘氨酸在高浓度下可能刺激胃黏膜,建议随餐或餐后服用。
搭配富含维C食物:维生素C能促进胶原蛋白合成,间接支持甘氨酸的代谢循环。
适量饮水:甘氨酸是水溶性的,充足的水分有助于维持体内代谢平衡。
在严重失眠时,甘氨酸(负责降温/放松)与褪黑素(负责启动睡眠)联合使用可能效果更佳。
微量元素:镁、锌、铁
✦ 镁
镁在睡眠调节中发挥着关键作用,它通过调节N-甲基-D-天冬氨酸(NMDA)受体和GABA系统的活性帮助我们入睡,这两个系统对催眠至关重要。
研究发现,镁缺乏与夜间觉醒次数增加和慢波睡眠减少相关,因此,确保摄入足够的镁对获得高质量的睡眠至关重要。
✦ 锌
锌是一种微量矿物质,具有抗氧化和神经调节特性,参与调节睡眠结构。
针对婴儿和学龄前儿童的研究表明,血清锌水平与睡眠时长之间存在正相关关系。这意味着锌的摄入可能与更长的睡眠时间有关。
锌可能通过影响突触可塑性和调节谷氨酸能神经递质来改善睡眠。
✦ 铁
铁是大脑健康的另一重要元素,尤其是在调节睡眠-清醒周期时起着关键作用。
铁缺乏与不宁腿综合症(RLS)有关,这是一种在休息时会感觉到不适并有强烈想抖腿的冲动的疾病。补充铁可以帮助缓解这些症状,同时改善睡眠质量。因此,获得足够的铁对维持良好的睡眠也是非常重要的。
来源

小贴士
吃富含铁/锌的植物食物(如菠菜、豆类)时,搭配富含维C的食物(如橙子、番茄),可显著提高吸收率。
避免过量咖啡/茶:咖啡因和鞣酸会抑制铁和锌的吸收,建议在餐后1小时再饮用。
动物性食物中的血红素铁(来自肉类)比植物性非血红素铁更容易被人体直接吸收。
维生素:维生素D、B族维生素
✦ 维生素D
维生素D在多种与睡眠调节相关的脑区中存在受体,包括下丘脑。
观察性研究发现,维生素D水平低与睡眠障碍的风险增加有关,特别是短睡时间和睡眠效率差。这可能与维生素D在调节炎症反应和促进褪黑素合成中的作用有关,褪黑素是调节睡眠的重要激素。
来源
维生素 D 是唯一一种人体可以通过皮肤在阳光下自行合成的脂溶性维生素。虽然食物是重要补充,比如蘑菇、香菇、三文鱼、燕麦奶、杏仁露等,但更重要的维生素 D 来源于晒太阳。
小贴士
晒背:每天中午前后(10:00-14:00)暴露手臂和腿部皮肤 15-30分钟,无需涂抹防晒霜。
注意吸收:维生素D是脂溶性的,建议随餐食用以提高吸收率。
避免过量:长期过量摄入可能中毒,每日上限通常为4000 lU。
✦ B族维生素
B族维生素,尤其是B6(吡哆醇)、B9(叶酸)和B12(钴胺素),在神经递质代谢中发挥着重要作用。维生素B6对于将色氨酸转化为血清素至关重要,而叶酸和维生素B12则参与支持褪黑素合成的甲基化过程。缺乏这些维生素可能导致睡眠问题,如失眠和睡眠片段化。
来源

小贴士
B12 是唯一一种几乎只存在于动物性食物中的 B 族维生素。素食者需通过强化食品或补充剂获取。
B族维生素属于水溶性,容易随尿液排出,因此需要每日通过饮食规律补充。
多酚:芹菜素、槲皮素、白藜芦醇…
多酚,尤其是存在于水果、蔬菜、茶和可可中的类黄酮,因其神经保护和促进睡眠的特性而备受关注。
类黄酮如芹菜素(在洋甘菊中发现)和槲皮素通过与GABA_A受体的相互作用,展现出抗焦虑和镇静效果。
其他多酚化合物,如白藜芦醇(存在于葡萄和浆果中)和表没食子酸没食子酸酯(EGCG)(来自绿茶),具有抗炎和抗氧化效果。
这些化合物可能通过降低全身性炎症和氧化应激间接促进睡眠。这些作用至关重要,因为慢性炎症和氧化应激的升高与失眠以及其他睡眠障碍密切相关。
来源
芹菜素:芹菜(尤其西芹含量极高)、洋葱、大蒜、欧芹、香菜、罗勒;苹果、梨、葡萄等。
槲皮素:苹果(果皮含量极高)、覆盆子、桑葚、樱桃、葡萄;洋葱、菠菜、羽衣甘蓝、番茄;小麦、大麦、燕麦等。
白藜芦醇:葡萄(果皮)、红酒(发酵过程产生)、桑葚、蓝莓;花生、核桃等。
小贴士
芹菜素和槲皮素属于脂溶性或半脂溶性物质,与橄榄油、坚果或牛油果同食可促进吸收。
槲皮素主要集中在苹果的果皮中,吃苹果记得连皮一起吃。
肠道菌群能将多酚转化为活性代谢物(如阿魏酸),因此富含膳食纤维的食物有助于提高利用率。
褪黑素和L-茶氨酸
褪黑素是主要由松果体分泌的内源性激素,是调节昼夜睡眠-清醒节律的关键因素。而外源性褪黑素(可以通过补充剂或一些食品如酸樱桃获取)越来越多地被用于改善睡眠,特别是对那些有昼夜节律障碍的人群。研究表明,褪黑素补充剂可以有效缩短入睡所需时间,增加总睡眠时间,并改善整体睡眠质量。
L-茶氨酸是一种主要存在于绿茶中的氨基酸,它展现出促进放松而不引起镇静的潜力。临床试验证实,L-茶氨酸补充可以改善睡眠质量,减少睡眠干扰,并通过调节大脑α波活动和GABA水平增强睡眠的恢复阶段。

地中海饮食
地中海饮食(MD) 特点
研究表明,遵循这种饮食模式与更好的睡眠效果密切相关。许多研究发现,地中海饮食能够提高睡眠持续时间、效率和主观睡眠质量,这可能与其抗炎和抗氧化特性有关。
关键营养成分
这种饮食富含多酚、ω-3脂肪酸和复杂碳水化合物,这些成分有助于合成褪黑素,减少氧化压力,并增强血清素的传递,这些都是调节睡眠所需的关键途径。此外,地中海饮食中的镁丰富食物,也可能通过调节NMDA和GABA受体来促进睡眠。
Li Rang等人的一项大型横断面研究报告称,对地中海饮食的更高依从性与老年人失眠症状风险的降低相关。地中海饮食中普遍存在的复合碳水化合物通过增加色氨酸在大脑中的可用性,可能有助于睡眠的开始和持续。
地中海饮食对睡眠的影响
生酮饮食
生酮饮食的特点
生酮饮食(KD)是一种高脂肪、极低碳水化合物的饮食模式,因其在治疗癫痫、肥胖和代谢疾病方面的疗效而受到关注。近期研究表明,生酮饮食可能还会影响睡眠结构。
对睡眠结构的影响
研究发现,处于生酮状态时,慢波睡眠(SWS)的比例增加,而快速眼动睡眠(REM)减少。这种变化可能与腺苷信号传导和能量代谢的改变有关。
一项关于癫痫儿童的临床试验显示,生酮饮食能改善睡眠质量,通过减少夜间觉醒和提高睡眠效率。同时,酮体本身可能具有神经保护和抗炎作用,有助于稳定与睡眠调节相关的神经网络。
复杂的影响
然而,生酮饮食对睡眠的影响并非一成不变。饮食的初期阶段常伴随生酮适应症状,这可能暂时干扰睡眠,因此饮食的坚持时间和个人的代谢反应是影响睡眠的重要因素。
植物性饮食
植物性饮食的特点
植物性饮食,强调以水果、蔬菜、豆类、全谷物、坚果和种子等植物来源的食物为主,同时尽量减少或不摄入动物产品。流行病学数据表明,植物性饮食者往往报告的睡眠质量优于杂食者。
对睡眠的积极影响
植物性饮食中的高纤维含量有助于改善肠道微生物组成,增强肠-脑轴信号,这可能有助于改善睡眠。此外,植物性饮食丰富的植物营养素和抗氧化剂形成的抗炎特性,可能有助于降低全身性炎症,而炎症与睡眠质量差有关。
营养素的关注
然而,植物性饮食需要仔细规划,以防止维生素B12、铁和ω-3脂肪酸等营养素的缺乏,这些营养素对良好的睡眠调节至关重要。有研究表明,这些微量营养素的低水平可能会影响睡眠质量。
饮食时间
新兴研究表明,在饮食中,不仅食物的选择很重要,饮食的时间也对睡眠健康有显著影响。不规律的就餐时间、夜间进食、跳过早餐与昼夜节律失调、入睡延迟和睡眠质量降低相关。
饮食时间对睡眠的影响
晚间食用高脂肪或高能量餐食可能会通过干扰昼夜节律激素的分泌(包括褪黑激素)来影响入睡和睡眠深度。相反,建议在睡前约4小时摄入高升糖指数的食物,这可能有助于减少入睡的延迟,表明餐食的合理时间安排能够改善睡眠。
限时饮食的益处
限时饮食(TRE)要求在一天中的特定时间窗口内进食。这种饮食方式已经显示出增强昼夜节律,从而提高睡眠效率和持续时间的潜力。

肠道菌群如何影响睡眠
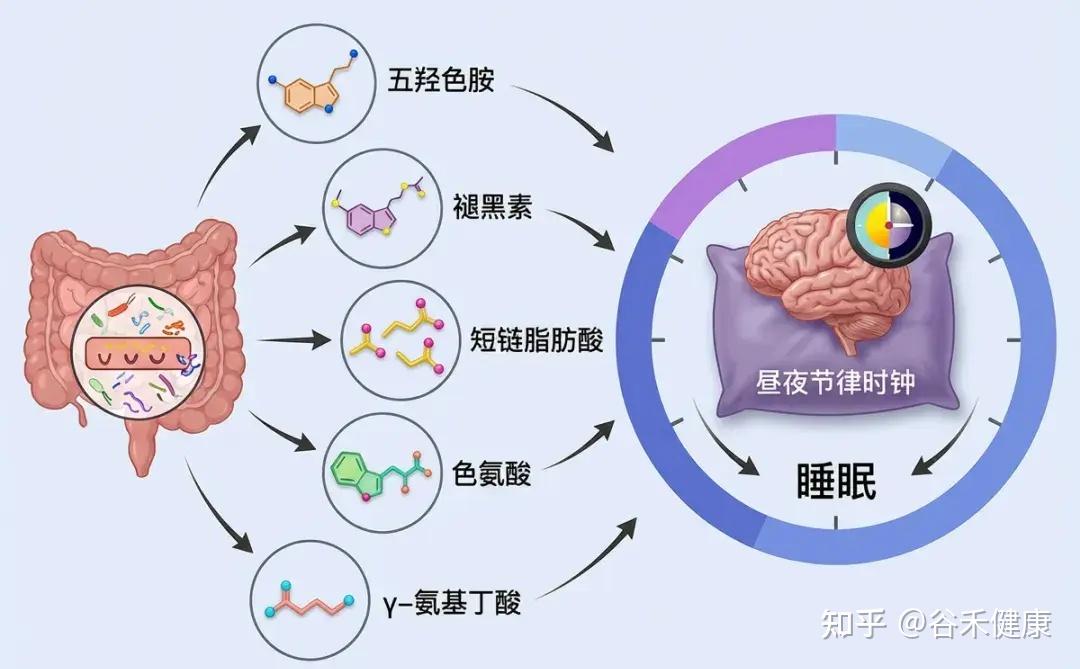
新兴研究表明,肠道微生物群可以通过多种机制影响睡眠,包括调节免疫系统、调节代谢途径、产生神经递质以及与昼夜节律系统的相互作用。
神经递质合成
部分肠道细菌能够合成关键的神经递质,如GABA(γ-氨基丁酸)、血清素和多巴胺。人体约90%的血清素(睡眠激素褪黑素的前体)产生于肠道。这些神经递质的水平直接影响睡眠-觉醒周期的调节。
GABA
GABA(γ-氨基丁酸)是一种非蛋白质氨基酸,是大脑中主要的抑制性神经递质,主要负责抑制神经活动。它在缓解压力和调节睡眠方面起着重要作用。GABA通过引导突触后神经元的超极化过程,帮助产生抑制性的电信号。这导致细胞内负电荷增加,使神经细胞更难以被激活,从而减少神经冲动的产生。
一些研究表明GABA补充可能有助于睡眠质量。
一项研究显示,每天给失眠患者提供300毫克GABA,连续四周,与提高睡眠效率和降低睡眠潜伏期相关。
另一项研究,用含GABA和L-茶氨酸的混合物可减少睡眠潜伏期,并增加快速眼动和非快速眼动期。
GABA的饮食来源:
普通豆类和豌豆芽、大米、燕麦、小麦、菠菜、土豆以及许多蔬菜。
通过饮食摄入GABA有助于提高体内的水平,但食物中含量相对还是较少。研究表明,特定的肠道微生物能够有效地合成GABA,增加其生物利用度。
乳杆菌和双歧杆菌是GABA产生的关键成员
双歧杆菌在肠道粘液层定植:具有将谷氨酸、谷氨酰胺和琥珀酸转化为GABA的酶机制。
乳杆菌 :多个菌株都具备GABA合成能力
– Limosilactobacillus fermentum L18:
高效分泌GABA的专业户,通过增加连接蛋白的浓度和有益肠道细菌的丰度来增强肠道屏障。
– 鼠李糖乳杆菌GG:
除了保护肠道屏障外,还可以通过调节肠脑轴来提高海马和杏仁核中的脑源性神经营养因子(BDNF)和GABA受体水平。
– 短乳杆菌:
通过发酵产生高剂量GABA,可以提高肠道中有益细菌的相对丰度和SCFA的水平,进而可以上调GABA能和5-羟色胺能神经递质的mRNA和蛋白质表达水平。这将导致θ和δ波以及非快速眼动睡眠的显著增加。
免疫系统调节
肠道微生物通过调节炎症细胞因子(如IL-6和TNF-α)来影响睡眠结构。健康的肠道菌群能促进短链脂肪酸(SCFA)等抗炎代谢物的产生,从而改善睡眠连续性;反之,慢性低度炎症通常与肠道菌群失调有关,这也是导致失眠和睡眠碎片化的重要因素。
代谢途径与屏障功能
微生物发酵产生的短链脂肪酸(乙酸盐、丙酸盐、丁酸盐)已被证实能穿过血脑屏障,通过调节神经炎症和神经可塑性来优化睡眠调节。
注意:慢性或周期性纤维缺乏可能导致黏膜糖蛋白作为营养来源分泌,进而侵蚀结肠黏膜屏障。这反过来可能导致肠道屏障功能障碍,进而升级为致命的结肠炎。
研究表明,某些细菌菌株,包括Lachnospiraceae UCG004 ,Odoribacter,参与短链脂肪酸的产生,有助于延长睡眠时间。短链脂肪酸可能通过调节GABA和血清素的合成来影响睡眠。GABA是神经系统中的主要抑制性神经递质,通过抑制唤醒通路在促进睡眠中发挥重要作用。
昼夜节律与肠道菌群的双向关系
光是负责人类昼夜节律最佳功能和精准调节的主要因素,该时钟位于下丘脑的视交叉上核。然而,许多其他刺激和环境信号也可能显著影响昼夜节律,包括用餐时间、食物类型、运动、体温,甚至社交互动。
肠-脑轴是一个复杂的生物系统,促进大脑与肠道之间的双向通信。肠道微生物群在调节这种相互作用中发挥了关键作用,影响着多种信号通路。
微生物群的多样性和组成会发生变化,这些变化受到一天中的时间、所摄入食物的类型和禁食习惯等影响。因此,异常或单一的饮食习惯可能对生物钟的正常功能产生负面影响。此外,肠道微生物群与昼夜节律之间是一种双向关系,彼此的干扰和紊乱会相互影响。
一方面,宿主昼夜节律的紊乱(如轮班工作、时差或不规律饮食)会导致微生物生态失调,进而加剧睡眠问题。
昼夜节律紊乱对肠道菌群的具体表现:α‑多样性显著降低;有害菌如 Ruminococcus torques、Streptococcus、Alloprevotella增多,益生菌如 Lactobacillus johnsonii、Lactobacillus、Bifidobacterium减少。
菌群失调与睡眠障碍
肠道菌群失调与多种睡眠障碍的病理生理学密切相关,包括失眠、阻塞性睡眠呼吸暂停(OSA)和不宁腿综合征(RLS)。
例如,阻塞性睡眠呼吸暂停患者常表现出特征性的肠道菌群改变,具体为微生物多样性降低及促炎细菌丰度增加。这种失调引发的全身性炎症可能通过肠-脑轴反馈至中枢神经系统,进一步恶化睡眠病理。
另一方面,通过规律的作息维持同步的昼夜节律,可促进健康的微生物群,进而反哺睡眠质量。
固定进食窗口产生 “餐后高峰‑空腹低谷” 的碳源波动。
产短链脂肪酸的菌(如Faecalibacterium, Roseburia)在餐后 2‑4 h 达到丰度峰值;胆汁酸代谢菌(如Clostridium spp., Bacteroides)在胆汁分泌高峰时活跃。
规律作息降低全身 IL‑6、TNF‑α,使肠道屏障更完整(ZO‑1、occludin 表达↑),阻止致炎菌(如Enterobacteriaceae)越过黏膜进入血液。低炎症环境进一步促进产短链脂肪酸的菌代谢活性,形成正反馈。
规律作息通过同步中枢与外周时钟、固定进食窗口、调节激素与免疫状态,形成一个“时间‑营养‑免疫”三位一体的生态网络,从而塑造更为多样、功能更完整的肠道菌群。
然而,这种双向调节在每个个体身上的表现并非千篇一律。要真正理解微生物组与生物钟的深层联系,我们不仅要看作息是否规律,还要看每个人独特的生物钟设定,你是早鸟型还是夜猫子型,本身就不一样。
早鸟型or夜猫子型?不同睡眠类型的肠道菌群
什么是睡眠类型,有哪几种常见的?
所谓睡眠类型,指的是一个人的昼夜节律表型,通俗来说,就是你在何时醒来、何时活跃以及何时入睡的天然偏好。
根据人体内部的昼夜节律,我们通常将人分为“晨型人”和“夜型人”(这两类又可细分为极端型和温和型)。此外,研究还确认了第三种类型:中间型(Neither-types)。
有趣的是,只有约40%的成年人属于典型的晨型或夜型,其余约60%都属于中间型。这类人群的作息往往更具弹性,能够根据具体情况或个人喜好灵活调整睡眠和活动时间。
早起鸟 和 夜猫子 在日常生活中有何不同?
这两类人的表现截然不同。晨型人倾向于早睡早起,他们的作息与日出日落的时间紧密同步,通常在一天中的早些时候达到身心状态的巅峰。
相反,夜型人倾向于晚睡晚起,他们的身心最佳状态通常出现在一天的后半段。然而,这也带来了一个问题:如果在不得不早起的情况下,他们更有可能面临精神不振的困难。
当夜型人被迫在他们生物钟认为的“深夜”(比如早上 7 点)进食早餐时,肠道代谢系统尚未完全苏醒(胰岛素敏感性低、消化酶分泌少)。
我们的肠道细菌与这种睡眠生物钟有关吗?
早起时的不适感可能不仅仅是大脑的问题,还可能与肠道有关。
研究发现,不同睡眠类型的人群拥有不同的肠道微生物群。例如,与晨型人相比,夜猫子人体内的 肠杆菌目(Enterobacteriales) 和 肠杆菌科(Enterobacteriaceae) 数量往往更高。
可能与延长睡眠时间相关的菌群包括:
微生物群-肠道-大脑轴如何影响昼夜节律
例如,硒单胞菌目(Selenomonadales)和Negativicutes 丰度较高的个体可能面临更高的失眠风险。相反,像Anaerofilum和肠杆菌目这样的细菌可以影响肠道上皮的时钟基因,从而增加成为夜猫子的概率,而夜猫子人与较高的体重相关。
给晨型人(早鸟型)的建议
利用早餐优势: 你的胰岛素敏感性在早晨极高。把一天中大部分的碳水化合物(全谷物、水果)放在早餐吃,不仅代谢快,还能为一整天提供能量。
警惕晚上能量透支: 晨型人容易在傍晚感到能量崩溃。
晚餐摄入适量的复合碳水化合物(如红薯、糙米),这有助于血清素的合成,不仅能维持情绪稳定,还能为入睡提供充足的褪黑素原料,防止“睡太早、半夜醒”的情况。
给夜型人(夜猫子)的策略
“时间限制进食法(TRE)”是关键:对夜型人来说,最危险的不是晚睡,而是晚吃。
在睡前 3-4小时 坚决停止进食。这给肠道留出了宝贵的清洗时间,防止细菌在不该繁殖的小肠内过度生长。
既然体内时钟慢,如果觉得需要,可以靠光来校准。早起第一件事接触阳光,这是告诉肠道菌群,新的一天开始了。
补给策略: 夜型人往往缺乏维生素D(晒太阳少)和B族维生素。结合肠道菌群检测相关的营养指标,如有缺乏则针对性补充这些营养素,有助于稳定生物钟基因的表达。
饮食与微生物干预策略
基于“饮食-微生物-睡眠”的调节路径,利用益生菌、益生元及膳食纤维进行干预,为管理睡眠障碍提供了有前景的新途径。
膳食纤维与益生元
膳食纤维(富含于水果、蔬菜、全谷物)作为微生物发酵的底物,能显著促进SCFA的产生。
对照喂养研究发现,较高的纤维摄入量与更长的慢波睡眠(深睡眠)时间相关,而高饱和脂肪摄入则导致睡眠质量下降。
益生元(如菊粉、FOS、GOS)被证明能增加非快速眼动睡眠(NREM),并提高机体对压力诱导的睡眠中断的抵抗力。
益生菌(精神益生菌)
益生菌在改善睡眠、缓解压力方面的潜力已在多项临床及临床前研究中得到证实。特定的益生菌菌株(有时被称为“精神益生菌”)通过调节神经递质途径和下丘脑-垂体-肾上腺(HPA)轴发挥作用。
临床证据
Nishida等人的随机对照试验表明,每日摄入
加氏乳杆菌CP2305 可显著提高轻度睡眠障碍成人的睡眠效率并缩短入睡潜伏期,这归因于其降低唾液皮质醇水平的能力。
机制研究
最近的研究进一步明确了特定菌株的神经化学机制。例如,鼠李糖乳杆菌GG被证明能增强海马和杏仁核中GABA受体的表达,通过迷走神经信号传导发挥抗焦虑和促睡眠作用。
代谢调节
长双歧杆菌1714和植物乳杆菌P8显示出降低皮质醇水平、改善主观睡眠质量的效果。这些菌株通过增加色氨酸的可用性并调节犬尿氨酸途径,增强中枢血清素的合成,从而巩固睡眠结构。
-只改睡眠时间而不管饮食,菌群会恢复吗?
部分恢复(尤其是夜间褪黑素对 Akkermansia 有正向作用),但若进食时间仍散乱,底物供给的节律仍缺失,恢复幅度有限。
-是否有推荐的作息模式?
目前证据最稳妥的模式是 “早起早睡 + 8‑10 h 进食窗口 + 早晨强光+ 低GI/高纤维饮食”,这套组合在多项研究中均能提升 α‑多样性、短链脂肪酸产生和睡眠质量。
注:作息干预的效果往往在 2‑4 周 开始出现可检测的菌群变化(α‑多样性、特定属丰度),完整的 功能恢复(SCFA 产量、炎症标记下降) 通常需要 8‑12 周 的持续坚持。
小 结
肠道微生物组通过产生神经活性代谢物(如短链脂肪酸、血清素前体)、调节炎症及与昼夜节律的交互,成为睡眠调节的关键一环。未来仍需更多临床试验来确立具体的因果关系及主要效果。
近年来,功能性食品和(提供基本营养以外的健康益处的食品)在睡眠健康方面的作用引起了人们的关注。几种富含生物活性化合物的天然产物已被证明可以通过多种生物机制调节睡眠质量、潜伏期和持续时间,包括抗氧化活性、抗炎作用和神经递质调节。
酸樱桃汁和褪黑素
酸樱桃(如蒙特莫伦西品种)是褪黑素的自然来源,这种荷尔蒙对调节我们的睡眠-觉醒周期至关重要。研究表明,喝酸樱桃汁可以改善睡眠的持续时间和质量,这主要得益于其中的褪黑素含量和抗氧化特性。
在一项随机、双盲、安慰剂对照的研究中,研究人员发现,患有慢性失眠的成年人每天喝两次酸樱桃汁后,睡眠持续性有所改善。这一结果表明,酸樱桃汁可能有助于提升睡眠的稳定性。
另一项研究发现,健康成年人饮用蒙特莫伦西酸樱桃浓缩汁后,平均睡眠时间增加了约34分钟,且睡眠效率也有所提高。这种改善可能与酸樱桃中增加的褪黑素有关。

猕猴桃和血清素
猕猴桃富含抗氧化剂、维生素(特别是维生素C和E)、叶酸以及血清素前体,这使其成为促进睡眠的理想食物。
临床研究显示,睡前1小时食用两个猕猴桃能够显著改善睡眠的开始时间、持续时间和效率,尤其对有睡眠问题的人群效果明显。
注:老年人缺乏直接的临床试验数据;基于生理差异,推测同样的摄入方案可能仍有助于改善睡眠(通过抗氧化、纤维促进肠道健康、提供血清素前体),但效果幅度可能低于年轻人。
这种积极的效果可能与猕猴桃中丰富的血清素含量有关,因为血清素能促进褪黑素的合成,从而调节睡眠周期。此外,猕猴桃的抗氧化特性可能有助于减少氧化应激,而氧化压力与睡眠障碍的发生有一定关系。
草本茶与镇静植物化合物
多种草本茶因其镇静和抗焦虑的效果而被传统使用,其中洋甘菊、缬草、薰衣草、百香果花是较常研究的几种。
洋甘菊茶富含一种叫芹菜素的类黄酮,它能与大脑中的苯二氮卓受体结合,有助于促进困倦感并减少夜间醒来的次数。
缬草根中含有缬草酸,这种成分可以调节GABA活性,从而产生镇静效果。
有系统评估的研究发现,缬草制剂与安慰剂相比显著改善了睡眠质量,尽管不同研究之间的结果存在一定的差异。
薰衣草和百香果花茶(西番莲茶)也具有抗焦虑特性,可能通过调节中枢神经系统中与压力和警觉性相关的通路来促进睡眠。
Omega-3脂肪酸与睡眠
Omega-3多不饱和脂肪酸,尤其是二十碳五烯酸(EPA)和二十二碳六烯酸(DHA),因其抗炎和神经保护特性而受到关注。近期的研究表明,Omega-3的补充可能与改善睡眠模式有关。
在一项随机对照试验中,研究发现儿童体内DHA水平较高与睡眠干扰显著减少相关。此外,补充富含DHA的鱼油也显示出能延长睡眠时间,并减少夜间觉醒的次数。
注意:儿童研究中常使用的剂量(如 600mg DHA/天)在成人身上可能产生不同的代谢反应。此外,成人的睡眠问题往往由压力、焦虑或环境因素主导,单纯补充 DHA 的效果可能不如在发育期大脑中显著。
这些潜在机制包括调节褪黑素的分泌、减少促炎细胞因子的水平,以及稳定神经元膜,而这些都是维持健康睡眠结构所必需的因素。

EPA 和 DHA 在改善睡眠中的作用是否存在协同或拮抗效应?
多项研究同时使用 DHA 与 EPA(常以鱼油形式)进行干预,均报告了睡眠质量的改善。例如,针对 2 型糖尿病患者的 14 个月随机对照试验显示,鱼油(DHA + EPA)显著降低睡眠障碍风险、提升睡眠时长,并与血浆 DHA + EPA 水平呈正相关。
日本的研究把 DHA + EPA(约 860 mg/天)与单独 EPA(约 1260 mg/天)或单独 DHA(约 1170 mg/天)进行比较,结果显示,高剂量的 DHA + EPA 组合在提升睡眠效率和主观评价方面优于单一成分,提示两者可能产生加和或协同效应。
现有证据倾向于 EPA 与 DHA 共同补充对睡眠具有协同或至少加和的正向作用,而非拮抗。两者在调节褪黑素合成、炎症抑制以及生物钟分子(如 RORα、BMAL1)表达方面可能互补,从而共同改善睡眠质量。
甘氨酸、L-茶氨酸和CBD
新兴的营养药物作为改善睡眠的辅助疗法越来越受到重视:
甘氨酸是一种非必需氨基酸,研究表明,在睡前服用甘氨酸可以改善睡眠的主观质量并减少疲劳。甘氨酸可能通过降低体温和增强血管扩张来发挥作用,这些生理变化对于入睡至关重要。
L-茶氨酸是一种独特的氨基酸,存在于绿茶中,能够促进放松而不引起嗜睡,主要是通过增加GABA、多巴胺和血清素的水平。临床研究报告显示,L-茶氨酸补充能够提高有广泛性焦虑症的个体的睡眠质量。
CBD(大麻二酚)是一种来源于大麻植物的非精神活性成分,显示出通过抗焦虑和镇痛的作用改善睡眠的潜力。尽管初步证据支持CBD在减轻特别是与焦虑相关的失眠中的作用,但仍需进行更为严谨的长期临床试验来验证其效果。

将科学研究成果转化为可操作的饮食策略,对改善睡眠健康至关重要。随着关于特定营养素、饮食模式和生物活性化合物影响睡眠的证据日益增强,临床实践和个体健康管理中应用这些见解的机会也在增加。
睡眠健康的一般饮食建议
研究表明了一些可以推荐给大众的饮食策略,以促进更好的睡眠。
增加有助于睡眠的营养素摄入
通过均衡饮食确保摄入如镁、锌、Omega-3脂肪酸和色氨酸等关键营养素,有助于神经递质合成和昼夜节律调节。
采用抗炎饮食模式
强调地中海饮食或植物性饮食,这些饮食富含抗氧化剂、纤维和抗炎化合物,能通过减少系统性炎症和氧化应激来提高睡眠质量。
注意用餐时间
调整饮食模式与昼夜节律相吻合,例如在白天吃大餐,避免临睡前吃重餐,以优化入睡效率。
限制干扰睡眠的食物
减少晚间咖啡因、酒精和高升糖指数食物的摄入,有助于预防睡眠碎片化和干扰。
这些一般建议作为基础,但“放之四海而皆准”的方法可能无法满足个体在睡眠需求和饮食干预反应上的差异。
场景化食谱与搭配建议(仅供参考)


饮水与睡眠
在饮食干预中,液体摄入同样扮演着重要角色。水分的补给不仅影响身体的整体健康,也对睡眠质量有显著的影响。确保身体在白天的充分水分补给,对于身体性能和睡眠质量都至关重要。
补水节奏
白天补水:成年人通常需要每天摄入大约2-3升水(包括食物中的液体)。通过均匀分配全日饮水量,确保在工作和活动期间补充足够的水分。
睡前限制:在睡前2小时限制摄入液体,尤其是咖啡因和酒精饮料,以减少夜间起夜的可能性,从而保持更好的睡眠连续性。
平衡口渴与起夜的矛盾
饮水的时机和量的把控也很关键。为了平衡口渴与夜间起床的矛盾,可以采取以下策略:
-早上喝水
在一天开始时,补充70%甚至更多的日需水分,有助于唤醒身体,并提高白天的精神状态。
-定时小口喝水
可以考虑将白天的饮水分成多个小量,避免一次大量饮水。
-适当的液体选择
在喝水的同时,适当选择含电解质的液体(如椰子水、运动饮料)来补水,尤其是在炎热的天气或剧烈活动后,有助于更快速恢复水分和电解质平衡。
酒精的微醺更助眠?
很多人习惯,睡前喝上几杯更容易入眠。
这里要说明的是,虽然酒精确实能缩短入睡时间,但它可能会严重破坏快速眼动睡眠,也许并不是优质睡眠。
取而代之,睡前选择温和的茶类(如洋甘菊茶、洛神花茶、薄荷茶等无咖啡因饮品)将更有利于放松和准备入睡。
饮食与生活方式的协同作用
正念饮食
正念饮食是一种基于“正念”理念的饮食方式,强调在吃饭时保持全神贯注,关注食物的色香味,聆听身体的饥饿和饱腹信号。研究表明,进食时的情绪和心理状态不仅影响用餐体验,还会影响消化、吸收,最终影响睡眠质量。
饮食与情绪的关系
情绪状态不佳,比如焦虑、压力大或分心时,会导致以下影响:
建议
在用餐时,尽量保持放松的状态,可以采取以下措施促进正念饮食:
光照与饮食的配合
研究表明,光照与饮食的结合对维生素D的合成及身体生物钟的调节都有显著影响。
“早餐 + 晨光”的最佳组合
在早晨接触自然光可以帮助重置身体的昼夜节律,促进褪黑素和皮质醇的分泌,增强清醒状态,进而有助于晚上入睡的质量。

在早餐时,尽早接触自然光,可以进行以下实践:
睡眠障碍:个性化营养方案
个性化营养的概念是将饮食建议根据个体的生物学、遗传和生活方式因素进行调整,这为管理睡眠障碍提供了很大潜力。
遗传因素
参与昼夜节律调节(如CLOCK和PER3基因)、褪黑素生成(如MTNR1B基因)和营养代谢(如FADS1基因与Omega-3脂肪酸合成)等基因的多态性可能影响睡眠模式及对饮食干预的反应。营养基因检测可帮助识别适合特定营养素或时钟营养策略的个体。
代谢健康状况
肥胖、胰岛素抵抗和代谢综合症与睡眠呼吸暂停等睡眠障碍密切相关。针对减重和血糖控制的个性化干预可显著改善睡眠结果。
肠道菌群分析
越来越多的研究表明,肠道菌群的组成与睡眠质量之间存在显著关联。健康的肠道菌群不仅能促进消化,还通过调节神经递质的合成,影响情绪和睡眠模式。肠道菌群检测可以帮助识别个体特有的微生物特征,从而为优化干预方案提供依据。基于检测结果,个性化的益生菌或益生元治疗可以针对特定菌群进行设计,旨在改善整体睡眠质量与健康。因此,加强对肠道菌群的关注,可能是提升睡眠质量的重要策略。
生活方式和行为因素
睡眠习惯(如规律的作息、优化睡眠环境和减少临睡前的刺激性活动)、身体活动水平、压力管理和工作时间(如轮班工作)与饮食模式相互影响。个性化计划应整合这些行为因素以最大化效果。
生物钟类型(晨型人与夜猫子型)也会改变对用餐时间的代谢反应。个性化方法结合遗传、肠道菌群、代谢分析和生活方式因素,可以为特定睡眠障碍(如失眠、睡眠呼吸暂停和延迟睡眠相位综合征)提供精准的饮食推荐。
未来个性化工具:可穿戴设备和数字健康
技术进步使得可穿戴睡眠追踪器、数字饮食日志和基于机器学习的预测模型得以应用于高度个性化和动态的营养-睡眠程序。可穿戴设备能够实时监测睡眠指标,为饮食调整提供即时反馈。数字健康平台也可以整合遗传、代谢和生活方式数据,生成个性化、适应性的营养推荐。
在改善睡眠质量的过程中,饮食调整是一种有效的干预方式。同时,借助肠道菌群检测,可以更深入地了解自身状态,从而在营养干预上做出更加明智的决策。
将个性化营养与肠道菌群的研究相结合应用于睡眠医学,不仅有助于提高个体效果,也有助于通过尽早识别高风险人群并积极干预,为广泛人群制定有效策略。
本文内容主要集中在睡眠问题的营养干预措施,此外,还有中草药方面的介入措施,详细信息请参见我们之前撰写的文章。
主要参考文献
Abou-Khalil R. Nutritional Interventions for Enhancing Sleep Quality: The Role of Diet and Key Nutrients in Regulating Sleep Patterns and Disorders. Food Sci Nutr. 2025 Dec 4;13(12):e71309.
Sejbuk M, Siebieszuk A, Witkowska AM. The Role of Gut Microbiome in Sleep Quality and Health: Dietary Strategies for Microbiota Support. Nutrients. 2024 Jul 13;16(14):2259.
Bautista J, Hidalgo-Tinoco C, Di Capua Delgado M, Viteri-Recalde J, Guerra-Guerrero A, López-Cortés A. The gut-brain-circadian axis in anxiety and depression: a critical review. Front Psychiatry. 2025 Oct 30;16:1697200.
Minari TP, Pisani LP. Unraveling the connection between the Mediterranean diet and sleep health: from biological mechanisms to clinical implications. Sleep Breath. 2025 Nov 28;29(6):369.
Zhao D, Zou B, Do QL, Wu SK, Shen Y, Yang Y, Kang JX, Su KP, Wang B. Circadian rhythms and gut microbiota Dysbiosis: emerging gut-brain axis pathways in insomnia pathophysiology and Therapeutics. Brain Behav Immun. 2025 Nov 30;132:106203.
BaHammam AS, Pirzada A. Timing Matters: The Interplay between Early Mealtime, Circadian Rhythms, Gene Expression, Circadian Hormones, and Metabolism-A Narrative Review. Clocks Sleep. 2023 Sep 6;5(3):507-535.
Chaput JP, McHill AW, Cox RC, Broussard JL, Dutil C, da Costa BGG, Sampasa-Kanyinga H, Wright KP Jr. The role of insufficient sleep and circadian misalignment in obesity. Nat Rev Endocrinol. 2023 Feb;19(2):82-97.

谷禾健康
阴道是一个复杂的动态环境,会随着女性生命阶段的变化而不断调整,对于青春期、育龄期(包括未怀孕、产前和产后)及更年期的健康至关重要。
阴道微生态具有独特的特征,如糖原和黏蛋白含量高、pH值低、细胞免疫活跃,以及影响复杂微生物群的激素信号波动。例如,雌激素影响阴道上皮的屏障功能和糖原可用性,而糖原分解在塑造阴道微生物群中发挥关键作用。同时,激素调节粘液分泌,增强对病原体的抵抗力,而黏蛋白又为阴道微生物提供附着基质和营养来源。
尽管传统上认为阴道微生物群主要由乳杆菌主导的五种典型群落状态(CST I–V)构成,但新兴研究突出了微生物群的多样性,包括共生微生物和潜在致病微生物,同时揭示了阴道微生物群的时空动态。最近提出的27种宏基因组CST(mgCST)为潜在群落功能提供了关键线索,使研究重点从“有哪些微生物”转向“它们在做什么”。
本文采用生态系统视角,探讨阴道黏膜、微生物群、宿主及环境因素之间的多维关系。重点分析激素如何影响阴道微生物群及环境变化,强调月经周期中微生物群落的动态特征,以及阴道环境变化对定植抵抗力的影响。不同的阴道生态失调状态与乳杆菌丰度及微生物多样性变化密切相关。
此外,我们还将研究阴道中微生物与免疫细胞的相互作用,探讨免疫系统如何调节局部环境。通过宿主与驻留微生物之间的反馈循环,提升对阴道健康与疾病的理解。这些复杂相互作用的理解对于推动女性阴道健康至关重要。
众所周知,生殖激素信号传导可以驱动生理和环境变化,从而在塑造阴道微生物组方面发挥关键作用。
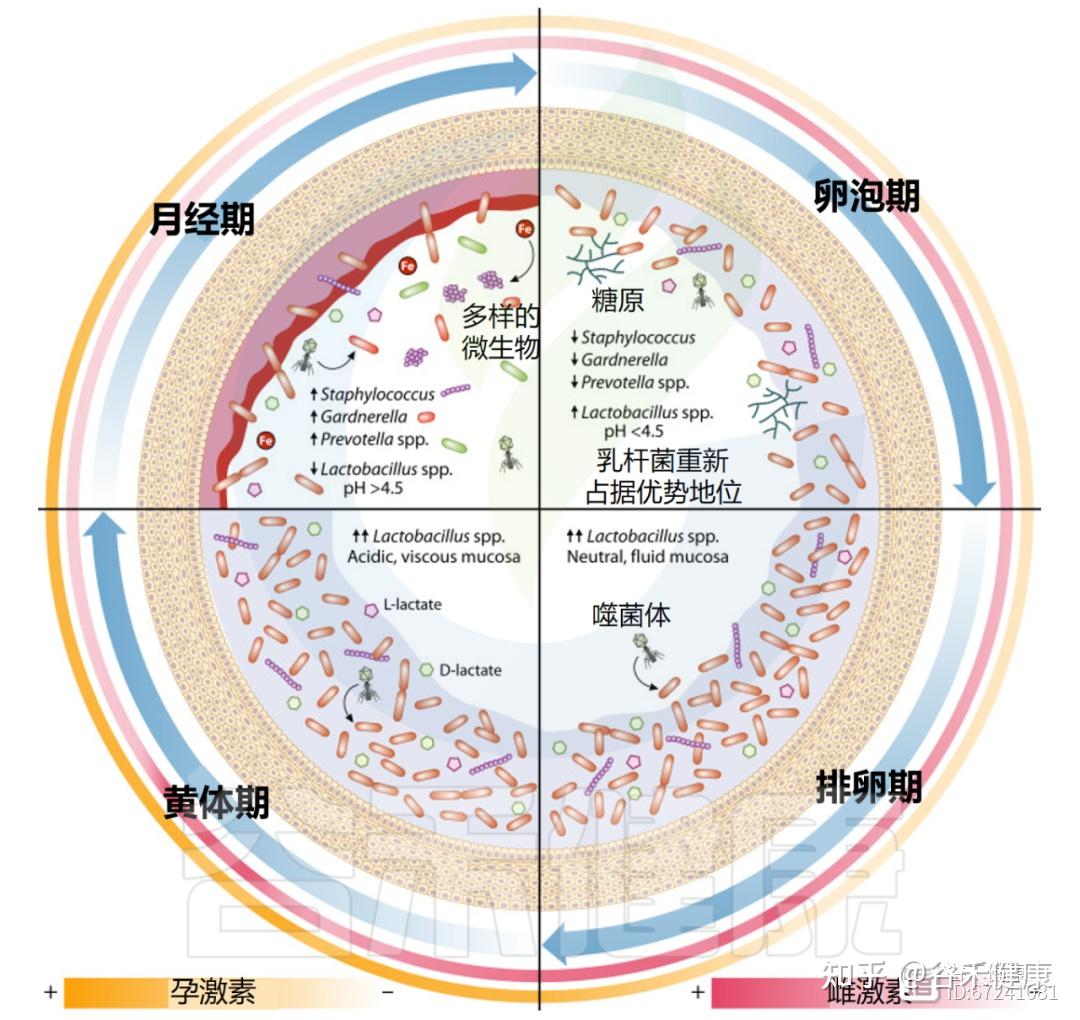
女性一生中阴道微生物群组成的变化
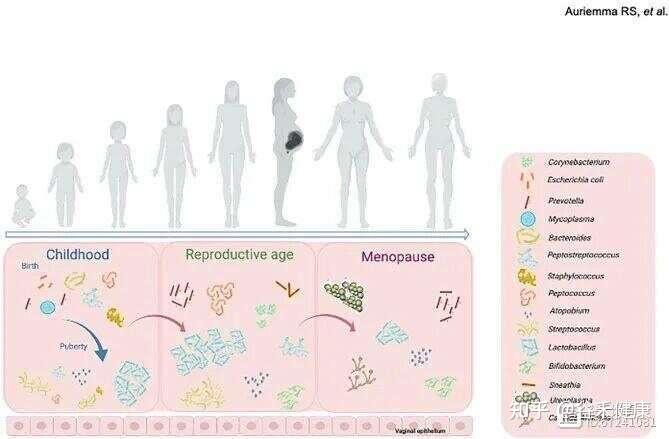
月经周期包括月经期、卵泡期(增殖期)、排卵期和黄体期,其特征是雌激素、孕激素、黄体生成素及促卵泡激素的波动,这些激素共同调控子宫内膜增厚及卵子释放。
阴道环境对月经周期及整个生殖生理过程中的荷尔蒙变化高度敏感,激素水平的波动会影响泌尿生殖系统对感染的抵抗力或易感性。宿主激素与微生物的相互作用构成了“微生物内分泌学”的研究基础,用以揭示激素对细菌生长与毒力的影响。本节将探讨激素分泌背景下阴道黏膜的变化,并分析其对营养供给、屏障功能及微生物群落结构的影响。
月经影响营养供应和阴道微生态稳定
月经期间,阴道微生物群发生显著变化,细菌多样性和丰富度增加。虽然这些变化源于月经期的生理波动,但个体差异明显。
据此,研究提出了四种阴道群落动力学(VCD),用于描述月经周期中微生物群的时间变化。VCD基于“稳定共生”与“持续失调”的特征,其中超过80%的日常样本分别由卷曲乳杆菌(Lactobacillus crispatus)、詹氏乳杆菌(Lactobacillus jensenii)、惰性乳杆菌(Lactobacillus iners)或高多样性群落占主导。低于80%生态阈值的个体则归为“不稳定”群落亚组。
另有约12%个体形成“月经相关失调”亚组,其益生性群落在增殖期、排卵期和黄体期占主导,而月经期间出现可检测的生态失调。
◮ 月经期乳杆菌减少,微生物稳定性较差
其他研究亦观察到月经期乳杆菌减少、菌群多样性上升。在一项为期16周、涉及32名育龄女性的纵向研究中,月经被认为与最低的微生物群稳定性相关,部分个体出现由卷曲乳杆菌主导向惰性乳杆菌或链球菌主导转变的现象。
这些变化可能源于糖原沉积减少及经血流入的影响。血液中的血红素铁可促进L.iners生长,从而取代L.crispatus优势;与此同时,碱性血液流入使阴道pH升至7.2–7.4,进一步改变微生物生态。这种环境转变促使金黄色葡萄球菌、链球菌等机会致病菌的短暂定植,以及细菌性阴道病(BV)相关菌如加德纳菌、普雷沃氏菌、Fannyhessea vaginae、Sneathia amnii、微小脲原体、Veillonella montpellierensis与消化链球菌等的增长。
目前尚无证据表明微生物多样性的变化仅由单一因素——如铁、糖原或pH变化——直接驱动。更可能的是,激素波动、营养变化及血流剪切力共同作用,塑造了月经期阴道微生态的动态特征。
注:血流剪切力是指血液流动时对血管内皮细胞产生的摩擦力,是影响血管功能和结构的重要机械力。
月经激素信号影响阴道微生态
增殖期:影响上皮屏障与糖原可用性
月经期普雷沃氏菌和加德纳菌的增多可延续至增殖期初,此时雌激素水平仍较低;而在卵泡晚期至排卵前,雌激素达到峰值,与乳杆菌重新占优势相吻合。
◮ 雌激素影响阴道上皮屏障与糖原可用性
雌激素的作用超越月经周期,在青春期其分泌上升,促使阴道上皮成熟增厚,形成由富含糖原的多层细胞构成的屏障。顶层细胞周期性脱落,释放糖原至阴道腔内。卵泡晚期乳杆菌优势的恢复与糖原可用性增加密切相关,并在整个黄体期保持稳定。
雌激素和乳杆菌属
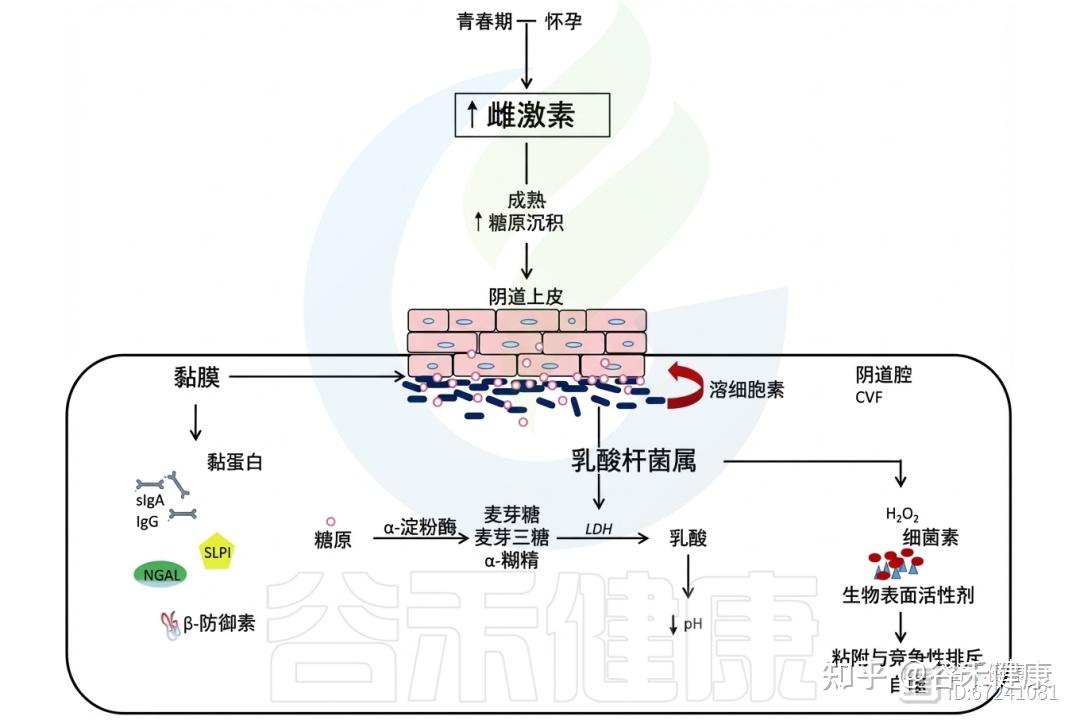
糖原是一种由α−1,4和α−1,6糖苷键组成的复杂分子,其在阴道中的浓度随月经周期波动且个体差异明显。有学者提出阴道乳杆菌可能将糖原作为碳源。然而,由于糖原结构复杂,微生物无法直接吸收,需经细胞外酶分解为糊精、麦芽糖、麦芽三糖等可利用的小分子糖,形成供微生物共享的“公共资源”。
阴道液中含有宿主和微生物来源的糖原水解酶,如α-淀粉酶和支链淀粉酶,分别裂解α−1,4和α−1,6糖苷键,其中宿主α-淀粉酶在阴道腔与宫颈内膜中含量丰富。
◮ α-淀粉酶有助于维持乳杆菌主导的环境
糖原的微生物利用最初被认为主要由保护性的乳杆菌完成,多项研究发现糖原水平、乳杆菌丰度及低阴道pH值之间存在正相关。但并非所有乳杆菌株都能利用糖原。
最新研究表明,各类阴道群落状态(CST)样本中均存在编码糖原降解酶的细菌基因,显示糖原可作为共生菌和病原菌在定植或感染期间的能量来源。研究发现,乳杆菌丰富的阴道样本中α-淀粉酶水平较高,而在乳杆菌减少或患有细菌性阴道病(BV)的个体中则显著降低,提示α-淀粉酶的产生有助于维持乳杆菌主导的环境。
相反,另一研究发现,当L.iners取代L.crispatus成为主要菌时,孕早期阴道分泌物中α-淀粉酶水平提高约5.4倍,可能反映宿主为恢复L. crispatus优势和稳定微生物群所作的代偿性反应。
◮ 糖原分解在塑造阴道微生物群中发挥关键作用
除乳杆菌外,阴道中的其他共生或致病微生物,如加德纳菌、无乳链球菌、白色念珠菌和阴道毛滴虫,也能表达糖原降解酶,这表明糖原分解对多种菌群的代谢至关重要。
值得注意的是,不同微生物的糖原酶在不同pH条件下活性最强:部分在低pH下最优,适应乳杆菌占优势的环境;而另一些则在pH 6–7的较中性环境中活跃。这意味着不同菌种可在月经周期或微生态失调等条件变化时,灵活利用糖原。
糖原降解酶的分泌还促进了“营养交叉喂养”,使无法直接分解糖原的菌种(如粪肠球菌)能利用分解产物生存。
尽管糖原可用性显然在塑造阴道微生物群结构与功能中发挥了关键作用,但其具体机制仍存在重要知识空白。进一步研究糖原供给与群落动态的因果关系,以及探明糖原是促进乳杆菌生长的驱动力,还是乳杆菌优势反过来维持糖原丰富的上皮屏障,将有助于全面理解糖原代谢对阴道健康的深远影响。
排卵期和黄体期:影响阴道粘膜屏障
排卵发生在增殖期雌激素激增之后,标志着黄体期的开始,其特点是黄体酮升高和雌激素的低次级峰值。
此阶段的激素变化显著影响阴道黏膜屏障的物理特性。该屏障由富含无机离子、防御素和免疫球蛋白的黏蛋白糖蛋白水凝胶组成,统称宫颈阴道黏液(CVM)。黏蛋白分为表面锚定型和分泌型,两者均高度O‑糖基化,形成带负电的大分子屏障。阴道及宫颈外上皮细胞主要表达MUC1和MUC16,而宫颈内皮与杯状细胞分泌MUC5B、MUC5AC及MUC6。
尽管MUC4在宫颈内高表达,近期蛋白质组学研究仅检测到MUC1和MUC16,未发现MUC4肽,其在CVM中的作用仍不明确。
阴道微生物组对女性性激素的反应
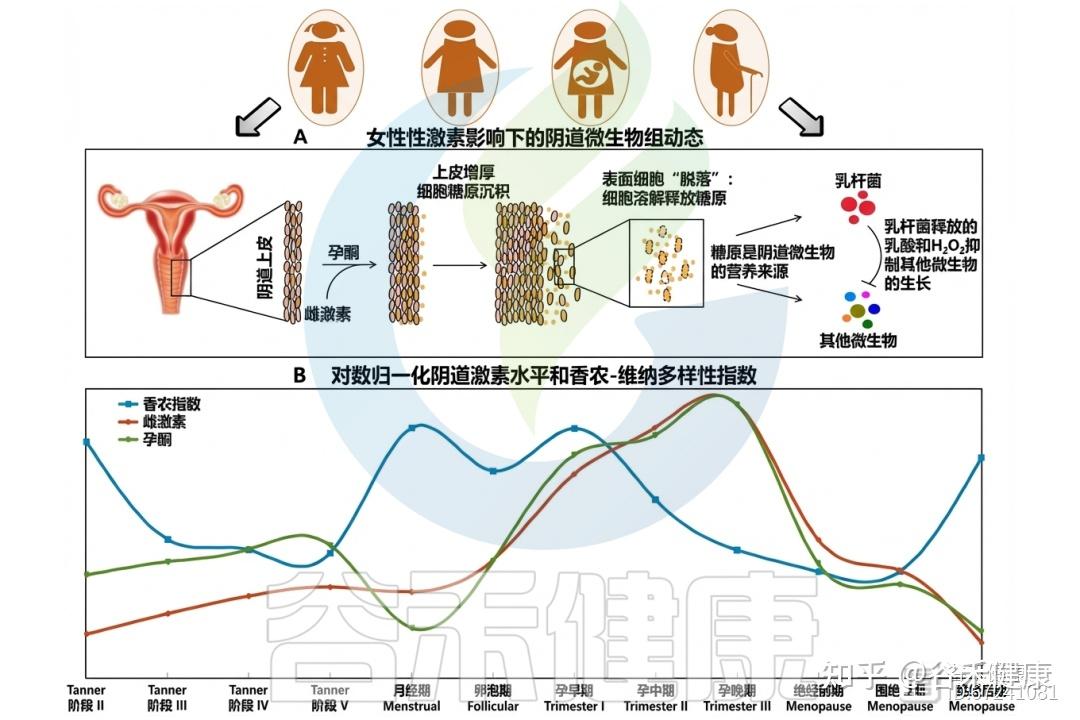
◮ 激素影响粘液的分泌量调节抵抗病原体能力
在排卵期,雌激素促使MUC5B分泌增加,使黏液稀薄、水样且pH更中性,有利于精子穿透。至黄体期,黄体酮上升使黏液减少,质地变黏稠、带负电,形成“黄体酮效应”,强化屏障功能,防御病原体入侵。CVM中含IgG,可抑制HIV‑1运动,其中以L.crispatus为主的群落较L. iners或失调型更具抗病毒力。
此外,噬菌体被发现可附着于黏膜表面,进一步阻止病原体定植。妊娠期,MUC5B与MUC5AC形成宫颈黏液栓,将富菌阴道环境与无菌子宫隔离,并富含抗菌肽(乳铁蛋白、溶菌酶、钙卫蛋白)、免疫球蛋白及具防御性的黏弹性结构。在小鼠模型中,缺失Muc5B会削弱屏障功能,增加感染与早产风险。
◮ 黏蛋白为阴道微生物提供附着基质和营养来源
黏液除了充当感染屏障外,还为阴道微生物提供附着基质和营养来源。无乳链球菌(S.agalactiae)和粪肠球菌(E.faecalis)通过表达可直接结合黏蛋白的菌毛结构,增强在阴道及肠道的定植能力。
同样,许多葡萄球菌和乳杆菌也能结合黏液,乳酸菌(LAB)中特有的黏蛋白结合结构域被认为参与黏液的附着或降解。感染后,黏蛋白及杯状细胞分泌通常上调,依赖NF‑κB信号通路,是维持黏膜屏障的先天免疫反应。
然而,部分微生物在长期适应过程中进化出可降解黏蛋白糖链、用于营养获取的糖苷酶。在阴道环境中,此类酶(如葡萄糖苷酶)的活性主要见于细菌性阴道病相关病原体,它们会破坏黏膜屏障,降低其黏度并增加感染风险。
◮ 细菌性阴道炎或菌群失调会影响酶活性
研究一致发现细菌性阴道炎女性中糖苷酶和唾液酸酶活性升高,而宏转录组分析显示CST IV群落患者的唾液酸酶表达最强。这种与细菌性阴道炎或菌群失调相关的酶活性增加,会导致N和O连接糖链耗竭,削弱黏膜屏障完整性并加剧炎症。
唾液酸酶主要由Gardnerella和Prevotella属产生,但近期研究发现其转录本在所有阴道CST类型中均较高,提示其作用可能超越病原入侵,参与群落代谢调控。支持这一观点的证据包括唾液酸酶促进微生物群共生与营养交互,如梭杆菌的唾液酸酶产物促进G. vaginalis生长与阴道定植。
类似的黏蛋白分解产物交叉喂养机制在肠道中更为常见,如Akkermansia muciniphila以黏液为主要营养源,分泌多种唾液酸酶和岩藻糖苷酶,助长与产丁酸梭菌的共生;双歧杆菌产生的唾液酸酶同样推动其与其他肠道双歧杆菌的共代谢。
阴道中,A.muciniphila与S.agalactiae的共栖可增强后者的持久性,代谢模型显示两者存在潜在的营养交换,但这种互作究竟源于免疫调节、物种交流或代谢互馈仍需进一步研究。
产后与绝经激素波动影响微生物群
◮ 产后和绝经后雌激素变化影响阴道微生物群结构
产后和绝经后阶段虽是关键的生理时期,却仍是生殖健康研究的薄弱环节。产后数天内雌激素水平急剧下降,常伴随出血、子宫收缩和疲劳等症状。更年期通常发生于45岁后,表现为雌激素分泌减少、月经不规律、阴道干燥及感染易感性增加。
这两个阶段雌激素下降均会削弱乳杆菌的优势。产后阴道微生物群常向多样化状态转变,L. iners、F. vaginae、G. vaginalis、Finegoldia magna和Prevotella spp. 等菌种显著富集,部分个体在分娩后一年的微生物组仍保持高多样性。同样,在绝经后,雌激素与黄体酮减少导致乳杆菌丰度下降,而无乳链球菌、表皮葡萄球菌及丙酮棒状杆菌等细菌性阴道病(BV)相关菌增多。
激素替代疗法是绝经后常用的治疗手段,可促使上皮成熟、提高碳水化合物供应以支持天然微生物群,并降低微生物多样性。然而,仍然需要更多研究扩大我们对这些人群阴道健康的理解。
阴道菌群CST分型最初源于对来自育龄女性进行16S rRNA测序的分析。结果确定了五种典型群落状态(CST I–V):CST I以L.crispatus为主,II、III和V分别以L. gasseri、L.iners和L.jensenii为主;CST IV则呈高度多样性,以Prevotella、Dialister、Fannyhessea、Gardnerella、Megasphaera、Peptoniphilus、Sneathia、Eggerthella、Aerococcus、Finegoldia和Mobiluncus等厌氧菌为特征。

乳杆菌优势群落通常具保护作用,惟CST III(以L.iners为主)被认为是一种过渡状态,反映阴道微生物群在健康、亚健康及细菌性阴道病(BV)状态间的动态变化。
阴道微生物群落在不断变化
随着新技术的应用与研究视角的拓展,CST分类体系不断完善。VALENCIA工具通过样本相似性对群落进行分层,将原有五类CST扩展为多级结构,提升分辨率,涵盖高、低丰度的特征物种及复杂混合群落的细节。
另一项研究采用混合成员主题模型分析群落结构,结合纵向变化探讨妊娠影响并识别亚群落。基于孕期与非孕期的大规模阴道拭子样本,共确定九种亚群落,其中四种以乳杆菌(L. crispatus、L. jensenii、L. iners、L. gasseri)为主,其余五种为非乳杆菌群落,富含链球菌、Prevotella、Gardnerella、Corynebacterium、Fannyhessea vaginae和Finegoldia等。
◮ 阴道菌群分型有助于动态平衡与功能多样性
宏基因组学和宏转录组学的进展促进了物种水平的功能解析,更深入揭示了阴道微生物群的时空动态。最新提出的27种宏基因组CST(mgCST)为潜在群落功能提供了关键线索,使研究焦点从“有哪些微生物”转向“它们在做什么”。
尽管CST方法在特定时点揭示了群落结构特征,但其静态视角限制了对动态变化和多样性的理解。为此,提出基于CST并结合纵向采样的VCD系统,以反映月经周期中细菌与噬菌体群落的协同变化。研究发现,“持续稳态”多与CST I和V(>80%检出率)相关,而“持续失调”则常见于CST III和IV。
阴道微生物组与健康及疾病状态关系
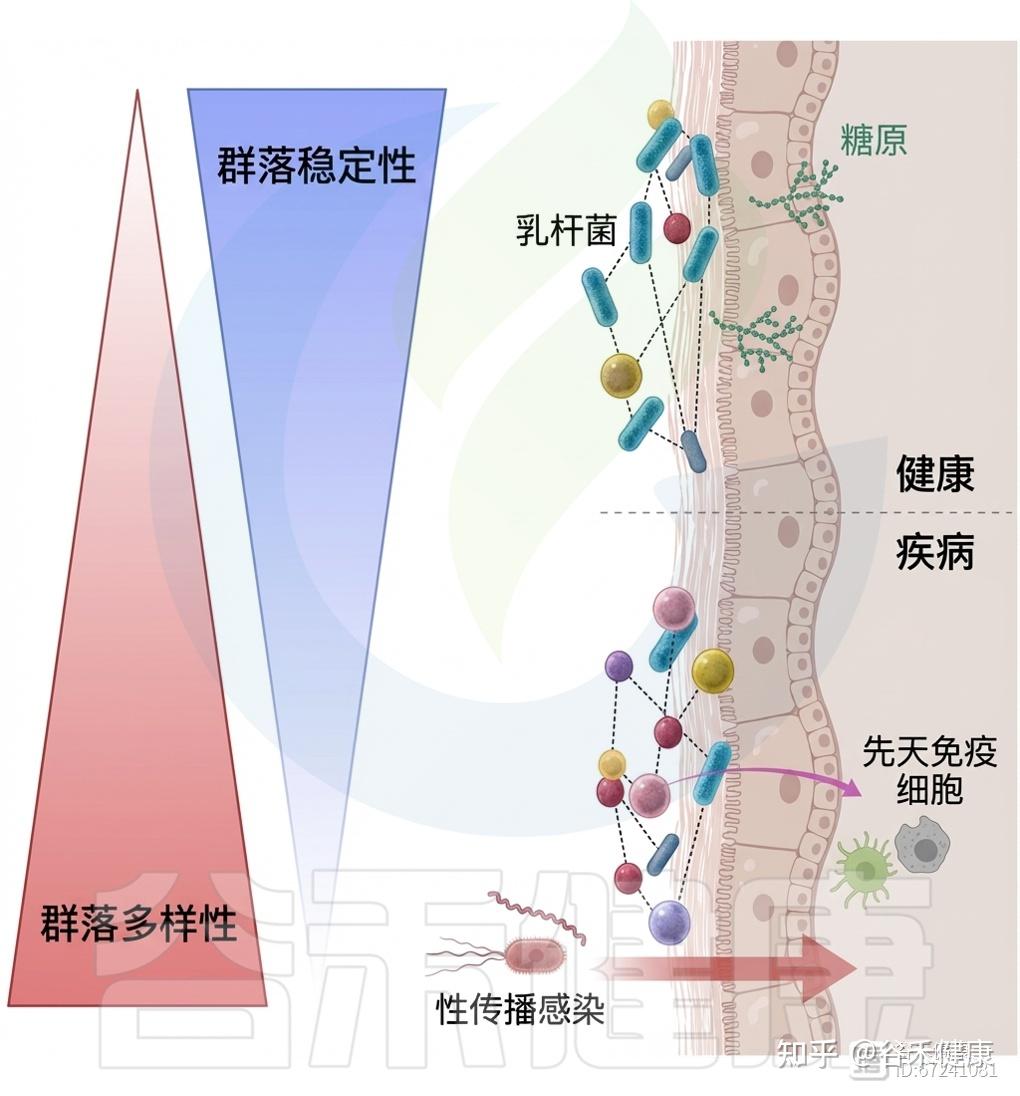
VCD分析进一步显示,不稳定或月经期失调状态下大肠杆菌丰度为稳态群落的两倍,且S. agalactiae、L. iners和U. parvum在这些状态中更为丰富。噬菌体鉴定显示,“稳态”个体的噬菌体丰度较其他类型高出约10倍,尽管群落多样性较低。这些研究为理解阴道微生态的动态平衡与功能多样性提供了新视角。
定植抗性:乳杆菌的保护作用
阴道低pH主要源于宿主与微生物共同将葡萄糖和糖原发酵为乳酸,从而使环境酸化。除上皮细胞产生的L-乳酸外,研究表明D-乳酸占阴道乳酸的大部分。
◮ 乳杆菌降低pH并增强黏膜屏障,形成保护环境
乳酸是乳酸菌(LAB)糖发酵的主要代谢产物,其中L. crispatus、L. gasseri和L. jensenii可产生高水平D-乳酸,而L. iners因缺乏D-乳酸脱氢酶而不具此能力。
微生物来源的乳酸通过降低pH并增强黏膜屏障,形成有利于乳杆菌定植、抑制其他细菌生长的保护性环境,是定殖抵抗的重要机制之一。然而,有研究发现,当pH升高时,乳酸对致病性厌氧菌不具显著抑制作用,提示乳酸产量与环境酸化对维持阴道定殖抵抗至关重要。
◮ 阴道pH变化可能是微生物群落失调的重要指标
阴道pH变化被视为细菌群落失调的重要指标之一,pH升高(>4.5)常与加德纳菌、普雷沃菌属和Sneathia等厌氧菌的增殖相关。能降解阴道带负电黏膜屏障的细菌可能削弱黏液层,从而导致pH升高。
此外,月经周期同样影响阴道pH,因糖原可用性、黏膜完整性及碱性经血的共同作用,短暂的pH升高可能促进潜在致病菌生长。通常,乳杆菌占优势时阴道pH < 4.5,但近期研究发现相当比例的乳杆菌型mgCST个体出现酸化减弱。
具体而言,L. crispatus的宏基因组亚型mgSs2与pH > 4.5相关,推测与第二种D-乳酸脱氢酶缺失有关。在包含1890个样本的研究中,16/27 mgCST以乳杆菌为主,但仅31%的样本pH < 4.5,69%的样本pH > 4.5,提示临床上存在不同程度的菌群失调。
部分乳杆菌可产生细菌素和过氧化氢,有助于定殖抵抗,但最新证据表明阴道中过氧化氢水平有限,其抗菌作用可能主要来自酸性环境或其他尚未明确的机制。
阴道生态系统在健康状态下维持着以乳杆菌为主的动态平衡,其低pH和代谢环境可有效抑制致病菌生长。然而,当这种平衡受到破坏时,便会出现不同类型的生态失调状态。常见的失调形式包括细菌性阴道炎(BV)、需氧性阴道炎(AV)、外阴阴道念珠菌症(VVC)、细胞溶解性阴道炎(CV)等。
这些状态常伴随乳杆菌减少、病原菌或真菌过度生长、阴道pH升高及黏膜防御功能减弱。阴道生态失调不仅影响局部微生物群结构和生理功能,还与性传播感染、高危妊娠及宫颈病变等一系列不良健康结局密切相关。因此,识别并理解不同类型的阴道生态失调及其微生态机制,对女性生殖健康的维护具有重要意义。
细菌性阴道炎(BV)
细菌性阴道炎(BV)是生育年龄女性中最常见的阴道炎类型,全球患病率为23%–29%。细菌性阴道炎与乳杆菌属数量偏低以及厌氧细菌过度繁殖有关,这些细菌来自阴道加德纳菌(Gardnerella)、普雷沃菌属(Prevotella)、Sneathia、动弯杆菌属(Mobiluncus)、Fannyhessea和Finegoldia等属。
携带细菌性阴道炎相关微生物的个体感染性传播感染(STI)和艾滋病病毒(HIV)、不良妊娠结局及宫颈发育不良的风险增加。虽然不建议孕期常规筛查细菌性阴道炎,但荟萃分析显示,阳性患者早产风险是正常孕妇的两倍,自然流产风险高出九倍。
◮ 短链脂肪酸在细菌性阴道炎中增加可能有害
细菌性阴道炎的特征是管腔内乳酸减少,短链脂肪酸(SCFAs)丰度增加,这些脂肪酸通常由微生物群中的厌氧成员通过发酵和氨基酸分解产生。肠道中的SCFAs已被证明具有抗炎作用;然而,近期研究表明,SCFA暴露(代表BV相关代谢产物)会诱导阴道上皮中促炎细胞因子的产生,并降低上皮屏障完整性。
研究还表明,肠道中的SCFA增加了罗伊氏乳杆菌(Lactobacillus reuteri)对原噬菌体元件的诱导。长链脂肪酸(LCFAs)在哺乳动物黏膜表面很常见,并已在从阴道腔采集的拭子样本中检测到。体外研究表明,LCFAs,特别是油酸,可以抑制L.iners及其他BV相关细菌的生长,并可能促进L.crispatus及其他有益乳杆菌的生长。
这些数据表明,长链脂肪酸和脂肪酸代谢可能作为阴道群落结构的潜在驱动因素发挥重要作用。
需氧性阴道炎(AV)
与细菌性阴道炎(BV)不同,阴道群落也可能被需氧性机会性病原体主导,这些病原体导致促炎标志物如白介素(IL)-6的增加,这种情况称为需氧性阴道炎(AV)。
◮ 大肠杆菌、无乳链球菌等可以成为需氧性阴道炎的病原体
需氧性阴道炎(AV)的患病率低于BV,仅占总人口的7%至12%。与Nugent评分用于BV诊断类似,AV诊断采用湿式相位对比显微镜评分系统。诊断标准依据乳杆菌的存在、炎症细胞的数量和上皮的形态。
AV最常见的病原体包括大肠杆菌(E.coli)、无乳链球菌(S.agalactiae)、粪肠球菌(E.faecalis)、肺炎克雷伯菌(Klebsiella pneumoniae)、凝血酶阴性葡萄球菌(如表皮葡萄球菌)和金黄色葡萄球菌。
外阴阴道念珠菌症(VVC)
念珠菌属,主要是白色念珠菌,是阴道微生物群中最常见的真菌之一,像许多细菌一样,可以作为共生菌在基础层面存活。念珠菌的过度生长可能引发外阴阴道念珠菌症(VVC),其症状包括阴道酸痛或不适、阴道瘙痒及异常分泌物。
◮ 阴道微生态失衡引起念珠菌过度生长
与阴道微生物群的其他成员一样,环境因素和群落动态会影响念珠菌的生长,其相互作用往往由乳杆菌及宿主因素驱动。例如,乳杆菌形成的生物膜有助于维持念珠菌的共生酵母状态,而非致病的菌丝形式。
尽管VVC及念珠菌对免疫系统的影响已有充分研究,但共生真菌在阴道微生物组中的具体作用仍需进一步探索。
细胞溶解性阴道炎(CV)
◮ 乳杆菌过度生长和乳酸水平过高也不健康
细胞溶解性阴道病(CV)是由乳杆菌过度生长和乳酸高于正常水平引起的,主要由卷曲乳杆菌(L.crispatus)引起。CV表现出类似的症状,经常被误诊为VVC,导致治疗不当,往往导致持续的症状。
细胞溶解性阴道病是一种有趣的病理学,表明群落平衡是生态平衡的一个更重要的因素,而不是纯粹的乳杆菌优势。
治疗阴道炎的方法
对有症状的细菌性阴道炎(BV)和好氧性阴道炎(AV)患者,推荐的治疗方法是使用处方抗生素。虽然抗生素通常是首选治疗,但滥用可能破坏阴道内细菌的自然平衡,影响致病菌和有益菌,增加对继发感染(如外阴阴道念珠菌症)的易感性。
◮ 通过微生物干预也有助于治疗阴道疾病
因此,已提出使用鼠李糖乳杆菌(Lactobacillus rhamnosus)或卷曲乳杆菌(L.crispatus)菌株的益生菌作为BV的替代治疗方案,以及选择性靶向BV相关微生物的重组噬菌体衍生内溶素。
对于细菌性阴道炎、好氧性阴道炎和细胞溶解性阴道炎是否应视为传染病,或其细菌群落状态是否是正常波动,仍存在不同意见。一些讨论建议对根据Amsel标准或Nugent评分被诊断为临床BV的患者,尤其是那些表现出可变或不一致症状的患者,进行更多关注,甚至对无症状但可检测到的细菌物种(如G. vaginalis)进行干预。
随着研究进展,应继续探讨这些问题,特别是在无症状个体的“生态失调或非最佳”微生物群落中引入抗生素时。
拓展:阴道环境中未充分研究的微生物
我们目前对阴道微生物群落的理解主要由优势物种定义。然而,在大型数据集中,稀有丰度物种的代表性通常较差,或被归为“其他”类别。关键物种生态的概念是阴道微生物组领域的一个新兴而未充分研究的方向,表明低丰度物种可能对微生物群落产生与其丰度不成比例的影响。
◮ 一些低丰度物种可能在阴道生态中起重要作用
近期的研究开始关注这些低丰度物种对阴道生态系统的重要性。例如,A. muciniphila促进无乳链球菌的定植并可能影响出生结果;奇异菌科(atopobiaceae)及Fannyhessea vaginalis在宫颈癌患者中常见,并与癌症免疫生物标志物增加相关;Sneathia与西班牙裔个体的宫颈癌和HPV发病率升高有关;Mobiluncus mulieris的细胞外囊泡刺激阴道和宫颈细胞中促炎细胞因子的产生;F.magna则被证明能激活中性粒细胞并触发中性粒细胞外陷阱(NET)释放等。
注:除了在性传播感染之外的阴道中发现的低丰度细菌种类外,栖息在这种环境中的病毒经常被忽视,尽管它们可能在阴道中发挥着重要作用。
◮ 噬菌体可能是阴道微生态的重要组成部分
研究评估阴道病毒组表明,只有4%-6%的总读数属于真核病毒,其余的94%-96%与噬菌体对齐。这些临床研究揭示了细菌与其在阴道中各自噬菌体之间的关联,表明噬菌体群落的组成是细菌性阴道炎(BV)的重要预测因子,并将特定噬菌体(如芽孢杆菌病毒Camphawk和Pony)与BV诊断联系起来。
噬菌体的分类与环境中宿主细菌的存在密切相关,已发现低噬菌体多样性与乳杆菌优势群落相关,而高噬菌体多样性则与非乳杆菌优势相关。
另一项研究指出,妊娠中期阴道中的噬菌体来自乳杆菌、链球菌、葡萄球菌和大肠杆菌,而非BV相关微生物,但特定噬菌体家族与CST之间并无联系。尽管噬菌体与阴道细菌群落之间存在某种关联,但尚不清楚噬菌体是否驱动了细菌群落的变化,或者特定噬菌体的存在是否仅是变化的伪影。
未来的研究,包括对阴道微生物组的纵向采样,将有助于阐明病毒与细菌群落之间的动态关系。
阴道具备一个强大的免疫细胞库,这些免疫细胞在维持阴道内环境的平衡和健康方面发挥着显著作用。这个多样化的细胞库不仅能够识别和应对潜在的威胁,还能够有效地影响炎症反应和免疫功能。
宿主免疫细胞与阴道炎症反应
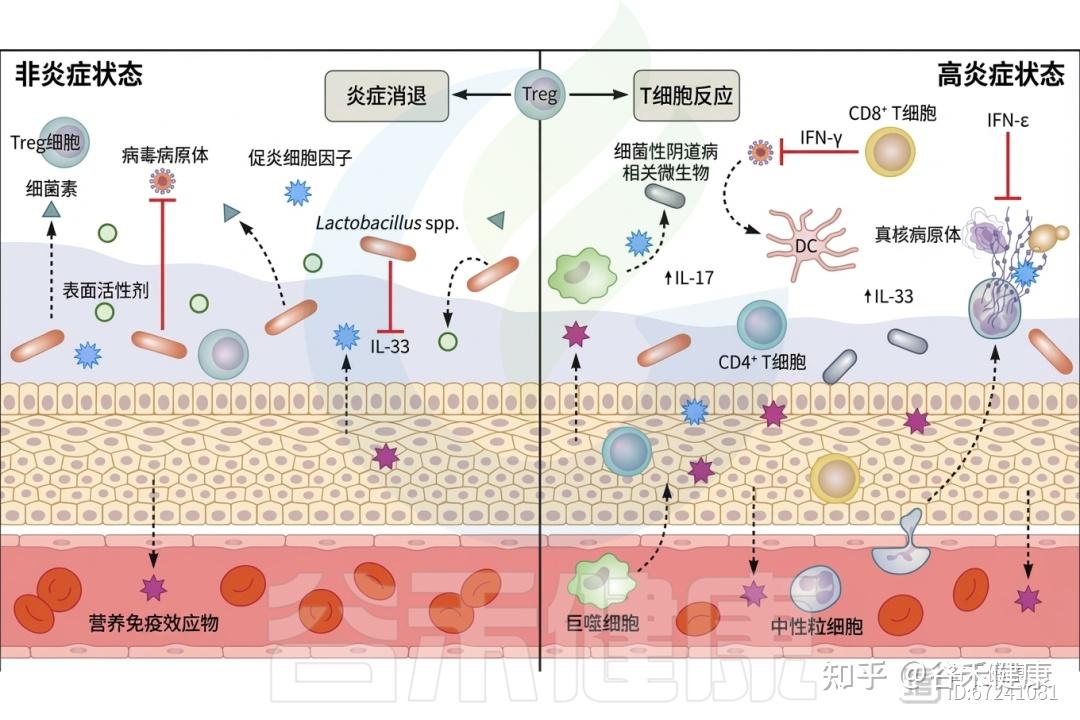
T细胞占比较高
其中CD8+ T细胞和CD4+ T细胞占阴道白细胞的50%。T细胞主要在性传播感染的研究中受到关注。CD8+组织驻留记忆淋巴细胞负责监测病原体入侵,并在识别后释放干扰素γ(IFN-γ),启动免疫细胞的招募。
◮ 调节性T细胞是阴道免疫稳态的关键参与者
调节性T细胞(Treg)是参与适当免疫反应启动的重要细胞,能够成功招募T细胞、自然杀伤细胞和树突状细胞(DC)。Treg在监测阴道炎症中发挥关键作用,激活后可抑制炎症细胞因子的产生、分泌抗炎性IL-10,并产生颗粒酶B,这是一种丝氨酸蛋白酶,有助于控制免疫相关的组织损伤和促进炎症消退。HIV阳性个体的Treg数量减少,伴随炎症粘膜(Tim)CD8+ T细胞的扩张,从而加剧阴道环境中的炎症。
此外,细菌性阴道炎(BV)阳性个体的Th17细胞中检测到了与BV阴性个体相比的T细胞功能变化和促炎标志物表达增加。这些慢性炎症状态会增加对性传播感染和BV等继发感染的易感性,以及感染HIV的风险。
其他免疫细胞
其他白细胞,如中性粒细胞、单核细胞、巨噬细胞和树突状细胞(DC),也在调节阴道免疫中发挥协同作用。
◮ 中性粒细胞具备多种杀菌机制
在阴道病原体入侵过程中,中性粒细胞通常是首批被招募的免疫细胞,具备多种杀菌机制,包括脱颗粒、吞噬作用和NETs的形成。NETs可以捕获病原体,其形成(NETosis)是针对病毒、真菌和寄生虫阴道感染的反应。
从非乳杆菌主导的阴道微生物群个体采集的宫颈阴道拭子和灌洗液中发现,促炎细胞因子和中性粒细胞相关标志物丰度增加,而闭留蛋白和去雾化素-1表达减少,反映出上皮屏障的完整性降低。
在直接实验中,感染BV相关微生物M.mulieris和G.vaginalis的小鼠中,观察到中性粒细胞招募和激活增加。
◮ 阴道微生物群影响抗原呈递细胞的活化
抗原呈递细胞(APC),如巨噬细胞、树突状细胞(DCs)和朗格汉斯细胞(LCs),在调控阴道免疫应对病原体方面发挥着重要作用。
阴道微生物群落影响巨噬细胞的极化, 厌氧消化链球菌(Peptostreptococcus anaerobius)促进抗炎的M2极化;而在孕小鼠中,白色念珠菌则驱动胎盘巨噬细胞的促炎M1极化,可能增加与念珠菌病相关的不良妊娠结局风险。
树突状细胞(DCs)的成熟是通过模式识别受体的信号激活而实现的,暴露于细菌性阴道病(BV)相关细菌或从BV患者采集的宫颈阴道灌洗后,DC的激活明显高于健康个体。然而,DC的活化并不针对所有阴道微生物,暴露于L.crispatus并未能诱导活化。
DC对病毒感染的反应同样重要;黏膜下DC负责向CD4+ T细胞呈递抗原并诱导IFN-γ产生,从而促使针对单纯疱疹病毒2型(HSV-2)的保护性Th1反应。尽管DC对HSV-2感染无直接反应,与细菌相关的Prevotella timonensis却使LCs更易摄取HIV-1病毒颗粒。
◮ 阴道乳杆菌丰度低且多样性高的个体,促炎细胞因子水平更高
乳杆菌丰度较低、微生物多样性较高的个体促炎细胞因子丰度增加。尽管高阴道多样性与乳杆菌主导微生物群体的DC、单核细胞和巨噬细胞丰度无显著差异,但这些抗原呈递细胞(APC)群体的转录组却存在显著差异。
该信号的潜在来源可能与细胞包膜相关,因为转录组分析中的目标基因与脂多糖刺激APC反应密切相关。然而,许多与高多样性相关的生物并不产生脂多糖,因此该领域需要进一步研究。
免疫分泌因子
除了细胞免疫反应,抗菌肽、趋化因子和细胞因子等分泌因子也有助于增强宿主防御。
◮ 白细胞介素-33生成受阴道菌群影响
一个重要的分泌因子是由阴道上皮产生的警报素IL-33,其生成通常受到阴道菌群的抑制。然而,在生态失调时,上皮细胞分泌IL-33作为危险信号,抑制干扰素γ的产生,从而影响宿主对HSV-2(单纯疱疹病毒)的易感性。
◮ 干扰素ε与黄体酮受体的表达相关
另一种在阴道中特殊表达且关键于防止病原体入侵的分泌因子是干扰素ε(IFN-ε)。IFN-ε在阴道和宫颈内皮细胞中以组成型方式表达,但在子宫内膜中受到激素调节,其表达与黄体酮受体的表达相关,卵泡期IFN-ε水平较低,而黄体期水平较高。IFN-ε已被证明对衣原体感染及HIV、寨卡病毒和HSV-2的病毒感染具有保护作用。
◮ 细菌性阴道炎个体IL-17及抗菌肽水平不同
此外,在活动性性传播感染个体的宫颈阴道灌洗中观察到IL-17水平增加,且这一现象与Th17细胞数量无关。需要注意的是,IL-17可以由多种细胞类型产生,包括中性粒细胞、粘膜相关不变T细胞(MAIT)和巨噬细胞等。
在乳杆菌占主导的微生物群体与BV相关微生物相比,阳离子抗菌肽(CAMP)也存在差异表达。最近的阴道宏转录组荟萃分析显示,与非BV微生物组样本相比,从BV相关微生物组样本中涉及CAMP耐药性的基因谱有所增加。
◮ 不同微生物产生的表面活性剂抑制病原菌定植
在阴道中,微生物和宿主细胞均能分泌调节环境的表面活性剂,这影响微生物定植和宿主对感染的反应。宿主表面活性剂通过与真菌细胞的碳水化合物部分相互作用,促进其吞噬。
近期研究发现,L.crispatus产生的细菌表面活性剂可以减少念珠菌对上皮细胞的粘附,并抑制宿主细胞的沙眼衣原体感染。同样,L.gasseri产生的表面活性剂也能阻止耐甲氧西林金黄色葡萄球菌的生物膜形成,表明表面活性剂在阴道微生物群落的建立与形成中可能发挥直接作用。
小结
阴道微生物群与免疫细胞之间的相互作用在维持阴道健康中发挥着重要作用。阴道内的免疫细胞库,特别是T细胞,能够有效识别和应对病原体,同时调节炎症反应。CD8+ T细胞监测病原体,并释放干扰素γ以招募其他免疫细胞。
其他免疫细胞,包括中性粒细胞和抗原呈递细胞(APC),共同参与阴道免疫的调节。研究表明,低丰度乳杆菌且高多样性的微生物群体与促炎细胞因子水平升高相关,提示其在炎症反应中可能发挥关键角色。
此外,分泌因子如IL-33和干扰素ε等对于防止病原体入侵及调节免疫反应也至关重要。不同微生物产生的表面活性剂可有效抑制病原菌的定植,彰显了微生物群在维持阴道微环境中的显著影响。
总的来看,阴道微生物群和免疫细胞的动态平衡对于宿主健康至关重要,但还需进一步研究以深入理解其机制与临床意义。
营养免疫是宿主的一种防御机制,主要涉及抗菌肽,这些肽通过封存可用的营养金属离子,旨在抑制入侵病原体的生长。
营养免疫效应分子对阴道健康的影响
◮ 钙卫蛋白有效捕捉锌、铁等重要金属离子
例如,效应蛋白如S100家族蛋白、脂素和乳铁蛋白能够螯合金属并在宿主的防御机制中发挥重要作用。具体而言,钙卫蛋白是一种由S100A8和S100A9四异二聚体构成的蛋白质,能够有效捕捉锌、锰和铁等金属离子。
另一个例子是S100A7,这种蛋白质表现出对锌的高亲和力;而S100A12,即钙颗粒素C,则表现出对锌和铜的双重亲和力。此外,脂质运载白蛋(lipocalin),亦称lipocalin-2或中性粒细胞相关明胶酶脂素,以及乳铁蛋白,分别是结合铁载体和铁离子的有效铁螯合分子。
◮ 健康阴道中钙卫蛋白丰富,有助于抵抗病原体
值得注意的是,许多营养免疫效应分子由宿主的角质细胞、上皮细胞和先天免疫细胞表达,尤其是中性粒细胞,在炎症期间这些效应分子会大量存在。
在以惰性乳杆菌(L. iners)为主的群落中,研究发现lipocalin-2和钙卫蛋白的浓度明显增加,这指向其在维持阴道健康中的潜在作用。相对而言,有研究表明,与细菌性阴道炎患者相比,健康对照人群中的阴道脂肪钙蛋白更为丰富,这可能与细菌性阴道炎相关微生物的免疫抑制潜力有直接关系。在观察到的动态变化中,阴道乳铁蛋白水平最初在月经后立即上升,并且与阴道微生物多样性的增加相关联。
然而,近来的证据表明,在某些个体中,月经期间及之后的时间窗口与微生物多样性的增加有本质的联系,这可能成为连接这两者的重要线索。这些分子共同作用,可以限制生物利用性的养分金属,同时帮助减少病原微生物的入侵或扩散,包括白色念珠菌、淋病奈瑟菌,以及机会性病原体无乳链球菌(S.agalactiae)等。
效应分子还能影响先天免疫
除了在营养免疫中的重要作用外,S100家族的蛋白质还可以作为警报蛋白或损伤相关分子模式,起到激活和增强先天免疫反应的作用。
◮ S100蛋白影响免疫及炎症反应
例如,钙卫蛋白已被证明能够通过抑制Toll样受体4的活性促进炎症的发生,多种S100家族蛋白结合该受体进行高级糖基化终产物,进而激活转录因子NF-кB。这些数据表明,S100蛋白不仅能够作为金属螯合剂,还可能在功能上促进炎症的发生。
需要指出的是,炎症在阴道环境中是一个研究较少的重要领域,对阴道健康有着显著影响,但这方面的知识仍然存在巨大的空白。因此,未来的研究需要着重深入探讨一些关键问题,例如理解饮食对金属供应的影响、金属摄入对于宿主易感性或感染韧性的作用、在塑造微生物群落形成中的重要性,以及探究在复杂微生物群落中如何使营养金属的获取受到宿主的主动抑制。
阴道微生态是一个动态且错综复杂的生态系统,由微生物群落与宿主之间的相互作用塑造,这些相互作用会随着激素信号传导、屏障完整性和营养可用性的变化而波动。
阴道微生物群的早期研究基于相关数据,但现在的研究已开始深入探索群落互动及其变化的后果。关键研究领域包括:
(1)群落变化的驱动因素:宿主是否通过调节营养选择微生物群落,或微生物代谢导致环境变化?
(2)明确“阴道健康”的定义:健康是以微生物为主还是个性化的标准?应评估临床症状而非仅用Amsel和Nugent评分;
(3)群落优势的概念:微生物优势定义为>50%或>30%?不同群落的保护等级是否不同?
(4)低丰度物种的贡献:阴道并非乳杆菌的单一构成;低丰度微生物在其中起什么作用?
(5)研究真核微生物、古菌和噬菌体的贡献,因为这些在阴道中仍被忽视。
在这些研究领域的基础上,我们还需进一步探讨微生物群落与宿主之间的相互关系及其如何受到环境因素的影响。例如,激素波动可能改变阴道内的pH值,从而影响微生物的组成,进而影响免疫反应。
此外,研究需要关注细菌与宿主免疫系统之间的相互作用。微生物不仅在维持阴道生态平衡中发挥作用,还通过其代谢产物调节宿主的免疫状态。我们必须深入理解这些微生物代谢产物如短链脂肪酸、共生抗菌肽等如何影响免疫反应的强度与方向。
对于不同个体,微生物群的组成及其功能可能有所不同,这也提示我们必须考虑遗传因素、生活习惯和饮食在阴道微生物组发展中的作用。这些差异可能会导致对相同微生物群体的不同免疫响应或营养利用,进而影响女性的健康状况。
最后,随着对阴道微生物群的了解不断深化,跨学科的合作将变得愈发重要。微生物学、免疫学和营养学等领域的相互结合,能够为我们提供更全面的视角,帮助我们理解更复杂的生物系统。这种综合的研究方法将有助于填补当前知识空白,并推动我们在改善女性生殖健康方面取得新的进展。
主要参考文献
Landolt EF, da Conceição Mendonça J, Behler AE, Lumsdaine SW, Jafar T, Burcham LR. Exploring the vaginal ecosystem: insights into host-microbe interactions and microbial community dynamics. Infect Immun. 2025 Sep 9;93(9):e0049924.
Greenbaum S, Greenbaum G, Moran-Gilad J, Weintraub AY. Ecological dynamics of the vaginal microbiome in relation to health and disease. Am J Obstet Gynecol. 2019 Apr;220(4):324-335.
Amabebe E, Anumba DOC. The Vaginal Microenvironment: The Physiologic Role of Lactobacilli. Front Med (Lausanne). 2018 Jun 13;5:181.
Shen L, Zhang W, Yuan Y, Zhu W, Shang A. Vaginal microecological characteristics of women in different physiological and pathological period. Front Cell Infect Microbiol. 2022 Jul 22;12:959793.
Holm JB, France MT, Gajer P, Ma B, Brotman RM, Shardell M, Forney L, Ravel J. Integrating compositional and functional content to describe vaginal microbiomes in health and disease. Microbiome. 2023 Nov 30;11(1):259. doi: 10.1186/s40168-023-01692-x. Erratum in: Microbiome. 2024 Feb 6;12(1):21.
Ma B, Forney LJ, Ravel J. Vaginal microbiome: rethinking health and disease. Annu Rev Microbiol. 2012;66:371-89.
Torcia MG. Interplay among Vaginal Microbiome, Immune Response and Sexually Transmitted Viral Infections. Int J Mol Sci. 2019 Jan 11;20(2):266.
Kaur H, Merchant M, Haque MM, Mande SS. Crosstalk Between Female Gonadal Hormones and Vaginal Microbiota Across Various Phases of Women’s Gynecological Lifecycle. Front Microbiol. 2020 Mar 31;11:551.

谷禾健康
肠道屏障是将宿主与外界隔离的主要防御,具有多项关键生理功能,包括营养消化、吸收以及防止潜在有害的膳食抗原和致病微生物的侵害。然而,饮食、药物、昼夜节律紊乱、年龄、肠道微生物群、微生物代谢物和遗传易感性等多种因素都可能破坏肠道屏障。这种破坏可能导致细菌易位,进而引发肠肝和全身性炎症。
目前,肠道屏障受损已被认为与多种疾病的发病机制有关,包括炎症性肠病(克罗恩病和溃疡性结肠炎)、肠易激综合征和结直肠癌等肠道疾病。此外,肝病(如代谢功能障碍相关脂肪性肝病、酒精性肝病和原发性硬化性胆管炎)和全身性代谢疾病(如糖尿病和肥胖)也与肠道屏障受损有关。
然而,目前大多数临床数据仅显示相关性,尚无法明确肠道屏障损伤是这些疾病的原因还是结果。目前,全世界药监督管理局尚未批准专门用于修复肠道屏障损伤的药物。现有疗法主要侧重于疾病的预防和管理,并严重依赖免疫抑制剂来控制炎症。
不幸的是,持续的屏障损伤和延迟愈合会降低这些治疗的疗效,并可能导致治疗耐受甚至复发。因此,开发直接靶向肠道上皮屏障的治疗策略至关重要。
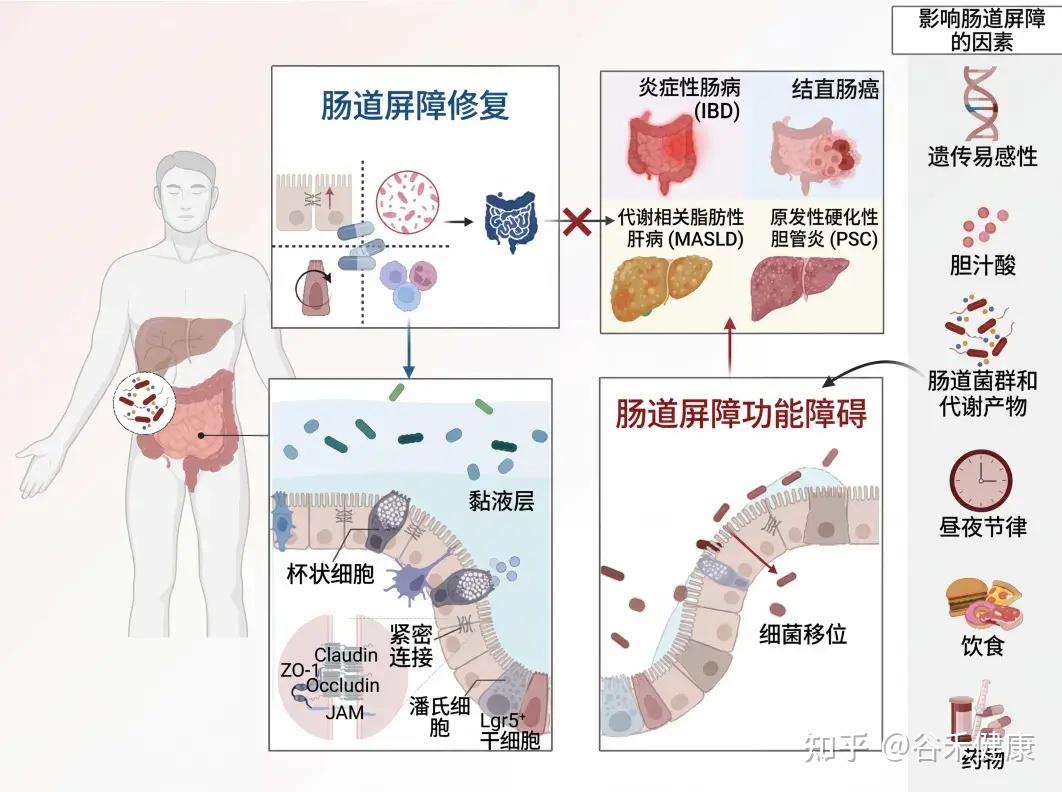
本文介绍肠道屏障的基本功能,生理结构和功能、影响其完整性的内外因素,重点介绍一些潜在的治疗策略,旨在恢复屏障完整性,改善和优化疾病管理。
肠道是一个独特的器官,在食物消化、营养吸收、动态的宿主与环境互动以及维持身体稳态方面发挥着关键作用。
为了保护宿主免受外部威胁,例如长期暴露于膳食抗原和病原微生物,肠道上皮细胞(IECs)形成了多种类型的屏障,包括机械屏障、富含共生微生物的粘液层,以及由免疫细胞及其活性物质组成的免疫屏障(下图)。

机械屏障:由紧密连接(TJs)形成的紧密排列的IECs层构成,确保肠道的结构完整性,调节肠道通透性,并控制水和大分子的运输。
粘液层:由杯状细胞分泌的粘蛋白组成,为共生细菌提供栖息地和营养。其独特的结构特征限制了病原体的渗透,进一步增强了TJs的物理隔离功能。
干细胞微环境:位于肠腺隐窝底部,由隐窝驻留肠道干细胞(ISCs)、间充质细胞、免疫细胞以及肠道分泌细胞(如杯状细胞和潘氏细胞)组成。这个微环境具有高度增殖性,因此负责组织更新和肠道屏障修复。它还介导抗原吞噬并释放抗菌肽(AMPs),通过清除潜在病原体来维持内部稳态。
这些元素共同构建了物理和生化屏障,保护宿主并调节内部和外部环境之间的交流。这种复杂的平衡对于胃肠系统的正常运作和维持身体整体平衡至关重要。
1
粘液层组成与免疫监测
粘液层的物理与生化屏障
粘液层是胃肠道的第一道防线,主要由90%-95%的水、1%-2%的脂质和1%-5%的粘蛋白组成。粘蛋白通过糖基转移酶高度糖基化,含有50%-80%的碳水化合物(w/w)。
粘蛋白聚糖多样而复杂的结构,为粘液相关细菌(如 R.torques、A.muciniphila、B.bifidum和R.gnavus)提供了理想的定植位点和营养来源。
动态防御:细菌即是住客,也是建筑师
MUC2是胃肠道中表达的主要粘蛋白。在细菌暴露后,杯状细胞通过meprin β介导的裂解机制分泌MUC2,形成保护性粘蛋白层。该结构允许共生微生物在外粘液层定植,并利用其多糖降解酶从粘蛋白O-聚糖中获取营养能量。这种宿主与微生物的相互作用,有助于调节近端结肠微生物群的结构和转录。
别让细菌吃肠壁:低纤维饮食的代价
研究表明,低纤维饮食会促进微生物群降解宿主粘蛋白,导致粘液层变薄,从而削弱屏障功能。Muc2缺陷小鼠表现出结肠组织学损伤增加、细菌易位至肝脏增多以及肠道紧密连接蛋白显著减少。此外,粘蛋白O-糖基化紊乱导致的粘液屏障完整性和功能受损与代谢疾病的发病机制有关。
粘液层的水龙头:谁在掌控肠道保护液的释放?
最近的研究发现,Gasdermin D (GSDMD) 是一种参与细胞凋亡的成孔效应蛋白,它通过scinderin介导的F-肌动蛋白解聚,促进钙依赖性胞吐作用,从而调节杯状细胞的粘蛋白分泌。GSDMD缺陷会破坏粘液屏障,使病原体粘附到上皮细胞,导致肠道疾病的发生。
严防细菌偷渡:一套保护肝脏的精密免疫系统
粘液层凭借其独特的粘弹性,能够有效滞留并扩散来自潘氏细胞和杯状细胞的抗菌物质及免疫细胞因子,形成化学屏障。
-抗菌肽 (AMPs) 与IgA的协同作用
潘氏细胞产生隐匿防御素、抗菌素和溶菌酶等AMPs。这些AMPs大量存在于肠道上皮表面,能够直接清除有害微生物。在新生非肥胖糖尿病小鼠模型中,生态失调导致的结肠AMPs缺乏会导致1型糖尿病中的胰腺自身免疫。
AMPs与微生物特异性免疫球蛋白IgA协调作用,在维持屏障稳定性和抑制炎症中发挥关键作用。
-IL-17和IL-22的调控
它们的调节受T辅助17(Th17)细胞和III型固有淋巴细胞(ILC3)产生的IL-17和IL-22的影响。
ILC3依赖于树突状细胞(DC)相关的Mincle信号通路,该通路与酪氨酸激酶偶联的C型凝集素受体有关。在Mincle缺失或酪氨酸激酶受损的情况下,肠道再生胰岛衍生III-γ(RegIIIγ)和IgA的合成会减少,从而导致肠道微生物群移位,进而引发肝脏炎症和脂质代谢失调。
这些发现表明粘液层完整性在维持肠肝稳态中的关键作用。
2
上皮连接:构筑肠壁防线的灰浆与砖块
微绒毛:不仅仅是吸收养分的地毯
功能:肠道上皮细胞顶端的微绒毛密集排列成刷状缘。它们既是营养吸收的高效界面(扩大表面积),又是阻止细菌附着的第一道物理防线。
脆弱性:在克罗恩病中,这些绒毛会变短、基因表达混乱。
破坏机制:就像拆除帐篷的支柱一样,肠出血性大肠杆菌的毒力因子通过CDK1-Formin信号轴,攻击支撑微绒毛的骨架蛋白(ACT-5),导致微绒毛坍塌消失,引发严重腹泻、出血性结肠炎等。
紧密连接(TJs):细胞间的拉链
在微绒毛下方,紧密连接(TJs)像拉链一样把相邻细胞的细胞膜紧紧锁死。
核心作用:这种吻合结构封堵了细胞间的空隙,相当于门控功能——只允许特定的物质通过,严防细菌和有害大分子渗透。
关键零件:谁在控制拉链的松紧?
病菌如何撬开防御?
紧密连接的稳定性高度依赖于细胞骨架的支撑,这成为了病原体的攻击目标:
3
动态防御机制:从干细胞再生到免疫感知
隐窝深处的生命源泉:肠道干细胞 (ISCs)
肠道屏障之所以能抵御消化道内持续的磨损与危害,归功于其惊人的自我更新能力。
核心机制: 位于肠道隐窝底部的肠道干细胞是这一过程的“总工程师”。它们通过持续增殖,不断分化补充受损的肠道上皮细胞(IECs),维持着组织的修复与动态平衡。
谁在调控修复?信号通路、压力与衰老
ISCs 的功能受到微环境信号的精密调控,同时也易受外部因素干扰:
-修复的加速器
当肠道受损时,IL-1R1 信号 和 Wnt 激动剂 RSPO3 会协同作用,强力促进 ISCs 的修复功能,加速伤口愈合。
-心理压力:让干细胞电量耗尽
心理压力不仅仅是情绪问题,它能产生实实在在的生理毒性。
心理压力会导致 ISCs 内部的 线粒体能量代谢 受损,进而干扰细胞分化。这种微观层面的能量危机削弱了宏观的肠道屏障,这科学地解释了为何精神疾病患者常伴有肠道问题。
-衰老的阻滞剂
随着年龄增长,ISCs 的数量和活性会下降,导致屏障完整性受损及菌群失衡。
关键原因:维持 ISCs 活性的关键信号——Wnt 信号通路随衰老而减弱,导致干细胞枯竭。
簇状细胞:不仅是免疫哨兵
簇状细胞是肠道屏障中一种重要的分泌型肠上皮细胞,与潘氏细胞和杯状细胞共同发挥作用。它们通过分泌IL-25来抵御病原体感染,激活2型免疫并清除病原体。簇状细胞还能感知病原体代谢物,并通过G蛋白偶联信号通路产生PGD2,从而刺激杯状细胞分泌粘液并促进自身增殖,进一步增强抗菌防御。此外,在肠道损伤时,簇状细胞可以充当储备肠道干细胞,协助屏障修复。
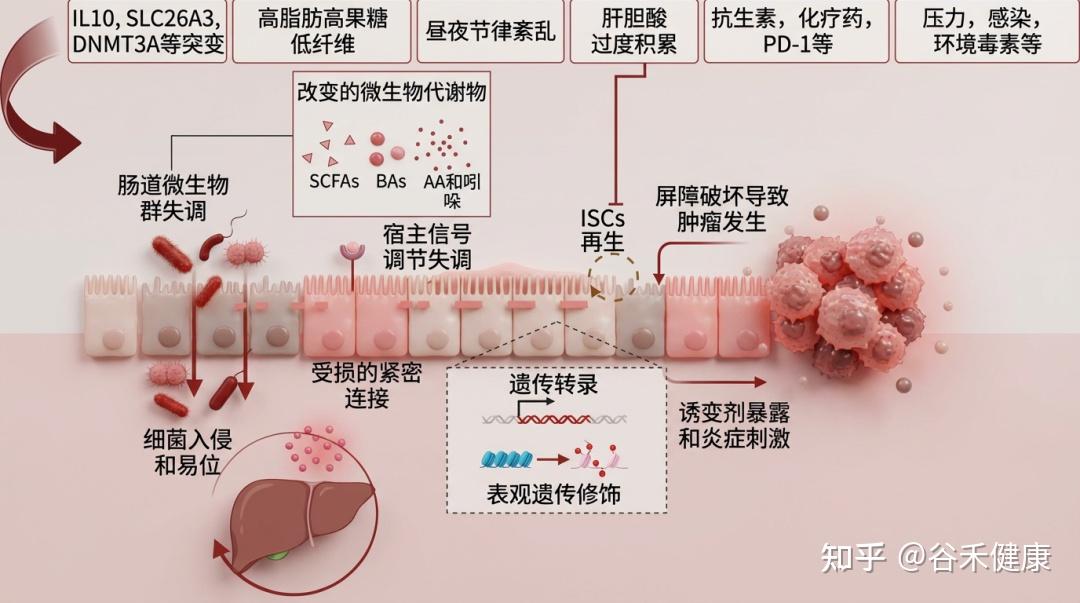
肠道屏障的健康受多种因素影响,包括遗传、饮食(如西式饮食)、药物(抗生素等)、疾病、生活习惯(昼夜节律)、心理状态(压力)和生理过程(衰老)。这些因素共同作用,可导致肠道微生物失衡,进而引发全身性代谢紊乱。这些代谢紊乱又会加剧炎症反应,进一步损害肠道屏障,形成一个恶性循环,持续破坏肠道健康。
下表总结了影响肠道和肝脏疾病的因素及机制与肠道屏障功能障碍相关的研究

1
遗传易感性
基因组关联研究(GWAS)的进展显著提升了疾病易感基因的识别和相关生物学通路的理解,这对临床转化具有重要价值。这些研究分析基因组中的遗传变异,以探索基因型和表型之间的关系。
免疫刹车失灵:IL-10 信号通路的遗传缺陷
尽管基因突变与肠道屏障功能障碍之间的直接联系有限,但GWAS已识别出许多与IBD(炎症性肠病)发展相关的基因座。例如,IL10基因突变是首批被发现能诱发IBD的突变之一。IL10基因敲除小鼠会自发发展结肠炎并增加肠道微生物易位。IL10受体(IL10R)的突变(由IL10RA和IL10RB基因编码的IL10R1和IL10R2蛋白组成)已与早发性小肠结肠炎相关联。这些突变损害了IL10诱导的信号传导,这可能增加TNF-α和其他促炎细胞因子的分泌,从而加剧炎症并削弱肠道耐受性。
离子转运障碍:SLC26A3 与物理屏障的松动
GWAS还显示SLC26A3基因(编码DRA蛋白,一种肠道氯离子转运蛋白)的显著下调。该基因在人类基因组中有害IBD变异中排名在前1%。SLC26A3表达的降低显著增加结肠旁细胞通透性,降低紧密连接(TJ)和黏附连接(AJ)蛋白的表达,从而增加对IBD的易感性。
黏液防线的溃败:ST6GALNAC1 与糖基化异常
糖蛋白组学分析揭示,先天性IBD(炎症性肠病)患者可能携带ST6GALNAC1(ST6)基因突变,该基因编码一种对维持黏液屏障稳态至关重要的唾液酸转移酶。该基因的突变会导致肠道黏膜层厚度减少,并破坏其保护功能。阐明这些调控机制对于理解先天性IBD的发病机制至关重要。此外,屏障功能还受到遗传易感性和环境因素相互作用的影响。DNA甲基转移酶编码基因DNMT3A的突变与IBD风险增加相关,已被证明能减少杯状细胞数量,缩短黏附连接(AJ)复合体,并增加肠道通透性。这些改变增加了对结肠炎的易感性,并阻碍了上皮再生和修复过程。
2
胆汁酸
胆汁酸(BAs)不仅是消化和脂质吸收的关键,还扮演着调节全身代谢和免疫的激素角色。它们通过激活FXR和TGR5等受体发挥作用。
修复的动力(次级胆汁酸)
最近研究发现,胆汁酸在维持肠道上皮屏障方面至关重要,能刺激Lgr5+肠道干细胞(ISCs)的自我更新,从而促进肠道修复。
修复的阻力(过量初级胆汁酸)
然而,过量的初级胆汁酸(如胆酸)会通过抑制脂肪酸氧化来减缓ISC的增殖,进而影响屏障修复。另一方面,TGR5在肠道代谢稳态中作用显著,其被次级胆汁酸(脱氧胆酸)激活后,会促进ISCs中YAP1和SRC等因子的转录,从而有效驱动肠道上皮的再生。
孕烷X受体 (PXR):抗炎与修复的化学感应器
孕烷X受体(PXR)是调节肠道上皮屏障稳态的关键核受体,它通过调控外源性物质代谢和先天免疫发挥作用。
溃疡性结肠炎患者中PXR及其靶基因的下调表明,PXR功能受损可能导致肠道损伤后屏障修复缺陷。PXR缺陷会加剧肠道上皮功能障碍,并增加对肠道损伤的易感性。
PXR的激活(例如通过石胆酸LCA)通过抑制NF-κB通路和炎症因子释放,以及维持紧密连接的完整性来发挥保护作用。
维生素 D 受体 (VDR):从抗菌防御到防癌屏障
胆汁酸也是维生素D受体(VDR)的内源性配体,对肠道稳态具有保护作用。VDR通过调节上皮分化和增强紧密连接表达来维护肠道屏障完整性,并支持潘氏细胞的抗菌功能。然而,在炎症性肠病(IBD)和结直肠癌(CRC)中,VDR的保护机制受损,其信号通路的失调与CRC的加速进展和不良预后密切相关。VDR激活不仅直接抑制肿瘤细胞增殖,还能通过增强黏膜屏障完整性来限制肿瘤进展,从而抑制CRC。
3
肠道微生物群及衍生代谢物
肠道微生物群通常与宿主共生,并被肠道上皮屏障限制在黏液层外。但有些微生物能突破屏障,损害胃肠道健康。抗生素虽看似能保护肠道,但实际上可能削弱屏障功能,增加新发炎症性肠病(IBD)的风险,这已在大量的研究中得到证实。与此相反,良好的肠道微生物结构对肠道屏障修复有积极作用。研究发现,肠道微生物群能通过激活巨噬细胞信号促进结肠上皮前体细胞生长,从而帮助修复受损的肠道上皮屏障。
色氨酸代谢物:PXR 与 AhR
肠道微生物群通过产生多种代谢物(如次级胆汁酸和色氨酸衍生物)来维护肠道屏障的完整性和整体健康。在IBD患者中,色氨酸水平显著降低,其代谢改变对疾病进程和预后有重要影响。
路径一:芳烃受体 (AhR) 的激活
色氨酸衍生物中的吲哚类物质是芳烃受体(AhR)的天然配体,AhR的激活能调节免疫细胞(如Tregs和Th17)及其细胞因子(特别是IL-22)的产生,从而促进肠道屏障的保护、修复和稳态。
路径二:孕烷 X 受体 (PXR) 的增强
除了AhR途径,这些代谢物还能通过孕烷X受体(PXR)增强屏障。例如,吲哚-3-丙酸(IPA)作为内源性PXR激动剂,通过TLR4信号通路减轻肠道通透性和炎症。PXR激活通过稳定紧密连接蛋白ZO-1、抑制MLCK表达和JNK1/2磷酸化来保护肠道屏障。
综上所述,色氨酸衍生的吲哚代谢物通过同时激活PXR和AhR信号通路,对肠道上皮发挥双重保护作用。
反面:犬尿氨酸 (Kyn) 途径
色氨酸代谢的犬尿氨酸(Kyn)途径与IBD进展密切相关,其限速酶IDO在炎症下促进Kyn途径,IDO的抑制或缺陷可减轻肠道炎症并增强屏障功能。
短链脂肪酸:能源与信号的结合
短链脂肪酸(SCFAs),包括丁酸、乙酸和丙酸,是肠道微生物分解膳食纤维等产生的关键代谢物,对肠道屏障功能至关重要。它们通过激活G蛋白偶联受体(如GPR43或FFAR2)来增强肠道屏障,并保护免疫细胞免受损伤,从而有助于预防结直肠癌(CRC)。
丁酸抑制组蛋白脱乙酰酶(HDACs)以调节基因转录,并通过调控紧密连接处的蛋白来促进屏障修复。GPCR信号和HDAC共同抑制维护上皮屏障完整性。此外,SCFAs还能刺激杯状细胞分泌黏蛋白,进而促进黏蛋白降解菌生长,通过消耗黏蛋白促进肠道干细胞分化,确保肠道上皮的再生能力。
4
昼夜节律
昼夜节律,常被称为生物钟,是代谢稳态的内部控制系统,旨在同步诸如光暗周期等周期性环境信号。该系统通过转录、转录后和翻译后修饰,在特定时间协调基因表达,以实现最佳代谢适应。
“什么时候吃”可能比“吃什么”更重要
多项研究表明,昼夜节律紊乱与代谢疾病之间存在密切关联。具体而言,不规律的进食时间会增加小肠暴露于膳食抗原和微生物刺激的风险,从而加重肠上皮细胞(IECs)主要组织相容性复合体II(MHC II)的负担。这种紊乱会削弱肠道微生态的调节功能,并减少IL-10的分泌。
神经免疫回路的失调:VIP 与 ILC3 的博弈
昼夜节律还会影响肠道中的神经免疫回路,这些回路受进食行为激活,并在饮食不规律时可能导致病理变化。食物摄入会触发肠道神经元分泌血管活性肠肽(VIP),该肽会上调与脂质吸收和转运相关的蛋白质。同时,VIP会降低IECs中的AMP水平,并减少ILC3产生的IL22。这种饮食节律的紊乱有助于病原体的肠道定植,尤其是在神经免疫回路的屏障功能受损时。
微生物振荡器:细菌也有生物钟
肠道微生物群自身也表现出丰度和功能的昼夜波动,被称为微生物振荡器,它们通过微生物代谢物或自身抗原影响宿主昼夜节律。肠道微生物群与宿主生物钟之间的这种相互作用显著影响屏障完整性和先天免疫反应。例如,短链脂肪酸通过抑制组蛋白脱乙酰酶(HDAC)活性,有助于调节小肠的昼夜节律相移。
肠道中的分节丝状细菌(SFB)驱动着与宿主节律同步的节律性ILC3回路振荡,从而通过时间依赖性地表达抗菌肽(AMPs)来介导感染抵抗力的昼夜变化。
5
饮食
高脂与快餐:胆汁酸的黑化与致癌风险
高脂肪和高糖、高加工饮食,与现代社会代谢疾病患病率的增加密切相关。饮食成分通过改变肠道微生物群以及主动参与宿主生理过程的次级代谢产物的产生,显著影响肠道微环境。
研究表明,现代快餐和西方饮食引起的肠道微生物群变化会提高脱氧胆酸(DCA)水平,这通过激活肠道FXR和I型干扰素(IFN)信号通路损害潘氏细胞。
动物研究进一步揭示,高脂肪饮食(HFD)在不同的结肠癌模型中,包括偶氮甲烷(AOM)-葡聚糖硫酸钠(DSS)诱导模型以及Apc突变诱导的自发模型,都会加剧肠道屏障损伤。HFD受损的肠道屏障允许更多病原体和衍生代谢物穿透上皮,从而加速结直肠癌(CRC)的发展。
糖衣炮弹:果糖与高血糖的双重打击
除了高脂肪摄入,过量的膳食果糖摄入也会损害上皮屏障。果糖水平升高会增加循环内毒素,从而激活巨噬细胞上的Toll样受体4(TLR4),引发全身性炎症反应。同时,葡萄糖代谢既对代谢综合征具有治疗潜力,也是肠道屏障功能的关键协调者。
在瘦素缺乏(ob/ob)和瘦素受体(LepR)缺乏(db/db)的2型糖尿病(T2DM)小鼠模型中,高血糖通过诱导肠上皮细胞(IECs)中葡萄糖转运蛋白2(GLUT2)依赖性转录重编程来破坏肠道屏障,这随后损害了紧密连接(TJ)和黏着连接(AJ)结构的完整性。
富含果糖的食物

6
药物
抗生素:精准打击与地毯式搜捕
抗菌素的使用对肠道微生物群落的丰度和结构有着深远的影响,进而影响肠道屏障的完整性。
例如,利福昔明-α (Rifaximin-α)常用来治疗小肠细菌过度生长:这种抗生素可以减少破坏粘膜的细菌,增加肠道内TNF-α和IL-17的水平,从而增强对病原体的抵抗力。它还能通过增加回肠中的乳杆菌水平来改善应激引起的肠道屏障功能障碍。
广谱抗生素会降低肠道微生物多样性,导致免疫失调,并增加感染的易感性。在健康成年小鼠中,广谱抗生素治疗会导致菌群失调,肠道上皮紧密连接(TJ)的完整性受损,表现为ZO-1表达减少和NLRP3炎症小体激活。
非抗生素药物:阿司匹林的隐形副作用
非抗生素药物也可能损害肠道屏障,例如阿司匹林,广泛用作消炎镇痛药,但会引起胃肠道损伤。
它会激活肠道FXR信号,并减少戈氏副拟杆菌(Parabacteroides goldsteinii)的数量。
戈氏副拟杆菌产生7-酮-LCA,这种物质能抑制肠道干细胞(ISC)的干性,从而减缓肠道屏障的修复。
化疗药物:再生能力的丧失
化疗药物:化疗是导致肠道屏障损伤的另一个主要原因。例如,5-氟尿嘧啶会加速黏膜细胞的死亡,而非再生。因此,超过40%的化疗患者会出现胃肠道损伤,表现为腹泻、便秘和消化不良等症状。
免疫检查点抑制剂:免疫激活的附带损伤
免疫检查点抑制剂(ICIs):靶向PD-1的ICIs彻底改变了抗肿瘤治疗。尽管这些抗体通过阻断PD-1通路来重建正常的免疫反应,但它们经常引起胃肠道毒性。
PD-1信号被发现对调节结肠淋巴组织诱导(LTi)细胞(ILC3的一个特定亚群,对维持免疫稳态至关重要)至关重要。
PD-1信号的缺失会导致LTi细胞中脂肪酸氧化过度激活,并反馈抑制LTi细胞的激活和IL-22的产生。这种失衡导致生态失调、肠道屏障损伤,并增加肠炎的易感性。
功能性测试:探针分子与 LMR 比值
乳果糖/甘露醇比值 (LMR) 是评估肠道通透性的重要生物标志物。
注:乳果糖(大分子)的尿液排泄量反映“肠漏”程度和上皮损伤。甘露醇(小分子)主要通过跨细胞途径吸收,代表非特异性上皮转运。
通过口服探针分子后检测尿液排泄来评估肠道通透性,这是最常用和直接的方法。
常用的探针分子包括不易代谢、吸收差的糖类(如甘露醇、鼠李糖、三氯蔗糖、乳果糖)或低分子量聚乙二醇(PEG)和乙二胺四乙酸(EDTA)。
为了避免饮食干扰,这些探针常使用同位素标记。
乳果糖-甘露醇测试 (LMT) 是临床广泛使用的评估方法。
在动物模型中,常用FITC标记的葡聚糖或铬-51标记的EDTA (51Cr-EDTA) 吸收到体循环的量来评估肠道屏障完整性。
其他方法:生物标志物的间接分析和多组学分析平台也是新兴的评估手段。
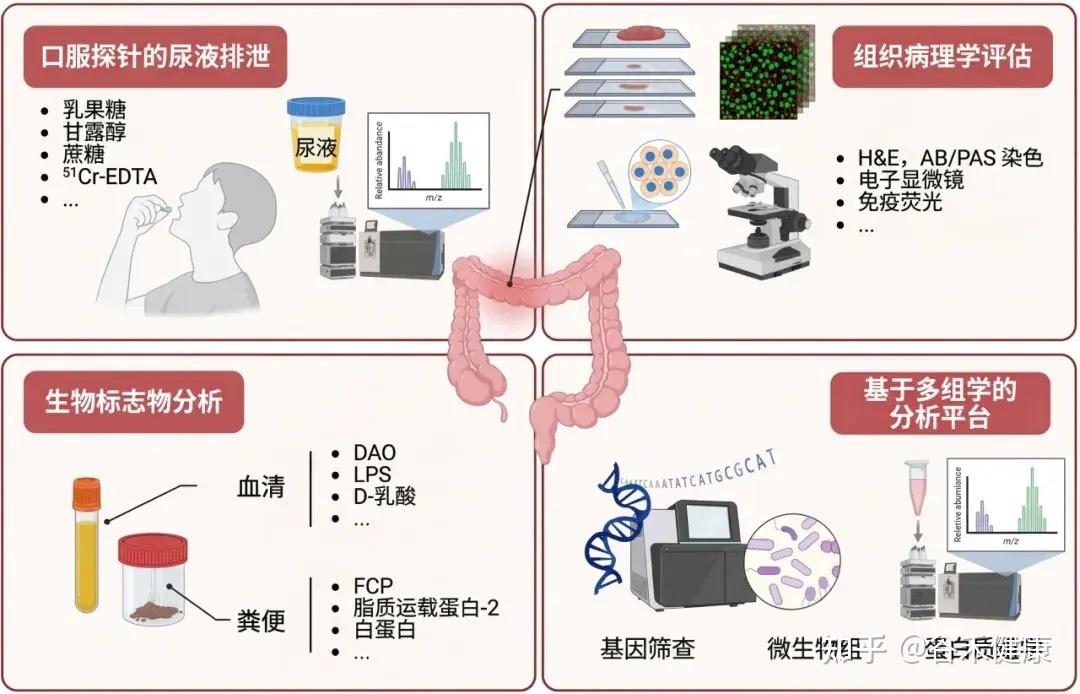
生物标志物:血液与粪便的线索
血清蛋白质组学和基因筛查技术:用于早期检测屏障功能障碍,并支持对其潜在原因的机制研究。
这些间接方法通过检测血液、粪便或分子水平上的特定物质,提供肠道屏障健康状况的重要信息。
组织病理学:屏障损伤的可视证据
内窥镜活检标本的组织病理学评估可以详细检查上皮绒毛形态和评估肠道屏障完整性。可以使用黏蛋白特异性阿尔辛蓝/过碘酸-希夫 (AB/PAS) 染色来评估黏液层,使用电子显微镜进行超微结构分析,以及使用免疫荧光技术精确定位和量化紧密连接 (TJ) 蛋白的表达,从而系统地分析黏膜成分的结构改变。
这些方法共同为评估肠道屏障完整性提供了全面的视角。
肠道上皮屏障的失调对多种肠道和肝脏疾病的发展和进展具有重要意义。肠道上皮的紧密连接功能障碍增加了通透性,促进细菌及其致病因子穿越屏障进入全身循环及其他组织。这种入侵会引发全身性炎症,破坏代谢稳态,而代谢稳态是肠道和肝脏疾病发生和进展的关键事件。虽然尚不确定肠道屏障的损伤是主要驱动因素还是偶然的病理特征,但越来越多的证据表明,肠道屏障的完整性在这些疾病的病因中起着关键作用。
1
炎症性肠病与结直肠癌
IBD 的病理核心:炎症因子的破坏机制
炎症性肠病 (IBD),包括克罗恩病 (CD) 和溃疡性结肠炎 (UC) 的特征是慢性胃肠道炎症。上皮屏障功能障碍是导致 IBD 发生和发展的关键因素。
临床研究表明,UC 和 CD 患者的肠道紧密连接 (TJ) 结构发生改变,导致肠道通透性增加。此外,黏膜免疫激活会引起TNF-α、IL-13、IL-17等细胞因子的产生。
屏障损伤是“因”而非“果”
进一步研究表明,由于 TJ 功能障碍和细胞骨架变化,细胞旁通透性增加可能在疾病发生前就已出现,例如 CD 患者亲属中早期肠屏障异常以及肠易激综合征 (IBS) 的较高患病率。同样,大规模队列研究已经发现 CD 诊断前三年内的屏障障碍标志物。这些发现支持屏障功能障碍是疾病进展的主要原因,而不是次要效应。
致癌风险:暴露于诱变环境
上皮屏障缺陷会使肠道内壁暴露于诱变化合物或长期炎症刺激,这可能通过氧化应激等机制启动并促进结直肠肿瘤的发生。在早期和晚期结直肠癌中,各种屏障相关蛋白的表达显著降低,这促进了微生物移位,引发炎症,并进一步加速肿瘤生长。然而,结直肠癌发生发展过程中上皮屏障成分异常表达的确切机制仍不清楚。
关键分子机制:GOLM1 与 NDRG2
事实上,实验模型表明,上皮屏障受损的小鼠更容易患结肠炎和炎症驱动的肿瘤。例如:
-GOLM1 缺失(细胞分化紊乱)
高尔基体膜蛋白 1 (GOLM1) 的缺失会过度激活 Notch 通路,从而破坏肠道稳态,改变肠上皮细胞 (IEC) 的分化和分泌细胞谱,并减少杯状细胞的数量,所有这些都会增加肠道通透性和促肿瘤炎症。机械屏障在预防结直肠癌发展中也至关重要。
-NDRG2(肿瘤抑制基因)
肿瘤抑制基因 N-myc 下游调节基因 2 (NDRG2) 已被证明可以增强 E3 连接酶 FBXO11 和 E-钙粘蛋白抑制剂 Snail 之间的相互作用,促进 Snail 泛素化。E-钙粘蛋白的这种稳定作用增强了黏附连接,从而限制了结肠炎相关肿瘤的发展。
2
慢性肝病
慢性肝病,包括 MASLD、ALD 和 PSC,常伴有肠道屏障受损和肠肝轴的失调。胃肠道物质,如营养素、分泌因子和微生物代谢物, 通过门静脉进入肝脏,并被代谢用于全身利用。这一过程使肝脏对病原体相关分子模式(PAMPs)极为敏感,成为肠道物质的主要靶点。
代谢功能障碍相关脂肪性肝病
代谢功能障碍相关脂肪性肝病 (MASLD, Metabolic dysfunction-associated steatotic liver disease):以前被称为NAFLD(非酒精性脂肪性肝病)。它是一种与代谢功能障碍相关的肝脏疾病,通常与肥胖、2型糖尿病、高血压和血脂异常有关。主要特征是肝脏中脂肪过度积聚,但与大量饮酒无关。
MASLD中,内毒素血症常见,提示肠屏障与肝炎症相关。DSS诱导小鼠肠屏障损伤加剧肝损伤。Il-10敲除鼠中,西方饮食+DSS处理降低BA水平,抑制肝脏FXR信号,加重MASLD。
肠道菌群失调是MASH早期驱动因素,宏基因组研究表明MASLD患者肝病发作前已存在肠道菌群失调。MASH患者粪外囊泡可能通过nmMLCK机制降低TJ表达,加剧肠屏障功能障碍。恢复肠屏障可逆转结肠炎引起的肝脂代谢失衡,特别是通过调节次级BA-TGR5/mTOR/氧化磷酸化通路。
酒精性肝病
酒精性肝病 (ALD, Alcoholic Liver Disease):是由于长期过度饮酒引起的肝脏损伤。 它可以表现为脂肪肝、酒精性肝炎和肝硬化等不同阶段。
酒精暴露破坏上皮连接,诱导肠屏障分解。ALDH2缺乏加剧酒精引起的肠道TJ/AJ蛋白降解,肠道ALDH2可能是酒精诱导的肠-肝轴损伤靶点。慢性酒精摄入减少肠道cDCs,导致AMP产量下降,保护性A. muciniphila丰度下降,AJs破坏,最终通过IL-12-IFNγ信号通路引起肝损伤。
原发性硬化性胆管炎 (PSC)
原发性硬化性胆管炎 (PSC, Primary Sclerosing Cholangitis):是一种慢性胆汁淤积性肝病,其特征是肝内外胆管的炎症和瘢痕形成。 最终可能导致胆管狭窄、肝硬化和肝功能衰竭。
PSC常与溃疡性结肠炎共病。PSC患者肠道微生物群与健康个体不同。门静脉中存在微生物提示肠屏障破坏,可能导致细菌易位。肺炎克雷伯菌等致病菌可通过接触依赖性细胞凋亡诱导上皮孔形成,加剧炎症和肝胆损伤。BDL诱导胆汁肝病小鼠模型中,肠道屏障破坏独立于菌群失调发生。敲除CHOP可缓解BDL小鼠肠屏障损伤、ISC干性丧失以及肝脏炎症和纤维化。
肠道屏障受损可刺激PSC中的保护性负反馈回路,LPS激活NF-κB通路抑制肝细胞BA代谢,减缓胆汁性肝病进展。抗生素或泛半胱天冬酶抑制剂能减弱菌群失调诱导的内毒素易位或NLRP3炎症小体激活,有望治疗PSC。
免疫靶向疗法,包括抗炎药、免疫抑制剂和生物制剂,已被证明在控制多种肠道疾病(如炎症性肠病)炎症方面有效。然而,这些治疗并不能直接解决肠道屏障的根本损伤。不幸的是,长期使用这些药物甚至可能增加感染、耐药性和疾病复发的风险,以及恶性肿瘤和死亡等严重不良反应。
确实,虽然减少炎症至关重要,但促进上皮屏障的愈合和恢复对于肠道疾病,尤其是炎症性肠病(IBD)的长期缓解至关重要。目前,没有临床批准的疗法专门针对肠道屏障的修复和维护。鉴于屏障功能障碍与肠道和肝脏疾病发病机制的密切联系,及时修复和恢复上皮屏障是一种有前景的治疗策略。与其仅关注症状管理,提升上皮愈合和健康完整性的治疗方法,可以减缓肠道和肝脏疾病的进展。
通过加强屏障,这类疗法还可能减少细菌易位、炎症以及肝损伤的加重。

1
紧密连接蛋白调节
靶向 MLCK:紧密连接稳态的核心调控
肠道屏障的维持在很大程度上依赖于多种 TJ 蛋白。肌球蛋白轻链激酶(MLCK)被视为开发炎症性肠病障碍疗法的有力候选目标,其在调节 TJs 及其分解中发挥核心作用,通过调节紧密连接(TJs)及其分解来保护屏障功能免受免疫诱导损伤。
对 MLCK 的缺失或抑制可以有效保护屏障功能免受免疫诱导损伤,但这些干预无法预防晚期结肠炎,后者涉及细胞凋亡和黏膜损伤,这些过程与 MLCK 介导的 TJ 分解无关。这些发现反映了 MLCK 抑制剂的治疗局限性。目前的 MLCK 抑制剂对上皮和平滑肌的催化域缺乏特异性,因此可能出现毒性副作用。
新机制发现:TCPTP 的双重保护
紧密连接 (TJ) 通过调节闭合蛋白 (occludin) 和封闭蛋白 (claudin) 来维持肠道完整性,而闭合蛋白和封闭蛋白是两种关键蛋白。
一项研究发现,上皮细胞中的蛋白酪氨酸磷酸酶 (TCPTP) 通过以下两种方式保护肠道屏障:
后者可以防止闭合蛋白 (occludin) 错位并降低上皮细胞的通透性。
目前尚无专门针对 TJ 蛋白的药物进入临床试验阶段,因此,需要在此领域进行深入研究和开发。
2
肠道微生物群的生态调控
益生菌疗法:从天然菌株到工程改造
益生菌已被发现通过多种机制调节屏障功能。例如,罗伊氏乳杆菌(Lactobacillus reuteri)激活Wnt/β-catenin信号通路,诱导Paneth细胞分化和AMP(抗菌肽)分泌,同时刺激Lgr5+肠道干细胞(ISCs)增殖以促进上皮修复。
此外,罗氏菌属(Roseburia)的鞭毛蛋白与Toll样受体5(TLR5)结合,可以上调occludin和MUC2,从而改善肠道屏障完整性,并增加IL-22和REG3γ水平,进一步调节肠道生态。
嗜黏蛋白阿克曼氏菌(AKK菌,Akkermansia muciniphila)也通过调节ISC程序来支持肠道上皮的修复、增殖、分化和稳定。因此,靶向肠道微生物失调成为恢复受损屏障功能的一种有前景的治疗策略。
最新的进展包括使用生物纳米材料包裹过表达人工酶的基因工程益生菌,这是一种用于黏膜修复和炎症治疗的新方法。长双歧杆菌(Bifidobacterium longum)经过修饰以表达过氧化氢酶和超氧化物歧化酶,表现出增强的肠道定植能力、强大的抗炎活性,促进肠道屏障重塑,并调节微生物平衡。这些进展有望减少传统抗炎药物的不良反应。
胆汁酸信号:FXR 激动剂的多重获益
胆汁酸在宿主与肠道菌群的交流以及肠道菌群的构成调节中起着关键作用。胆汁酸合成失调与多种疾病的发生密切相关。激活法尼酯X受体(FXR)能有效缓解胆汁酸过量带来的危害,它通过抑制胆固醇代谢、促进肝细胞将胆汁酸转运出去,从而减少胆汁淤积性肝损伤。
研究显示,FXR激动剂奥贝胆酸(OCA)能够:
此外,OCA治疗还能通过稳定内皮细胞内的β-连环蛋白来防止肠道血管屏障受损。
色氨酸代谢:AHR 通路与 IDO 的平衡
-AHR:屏障完整性的总开关
AHR(芳香烃受体)是一种色氨酸代谢物的受体,对维持肠道屏障的完整性至关重要。研究发现,Ahr−/−小鼠(即缺乏AHR的小鼠)的上皮屏障功能明显丧失,表现为肠道机械屏障受损以及细胞无法正常分化。缺乏AHR配体的小鼠也会出现类似的症状。
进一步研究表明,来自食物的AHR配体可以通过促进细胞内的锌离子(Zn2+)信号传导,从而提高紧密连接(TJ)蛋白的表达,有效预防损害肠道屏障功能的疾病。
-AHR 激动剂的治疗潜力:以尿石素 A 为例
其他AHR激动剂,例如微生物代谢产物尿石素A(UroA),也显示出保护肠道屏障功能的潜力。UroA具有抗炎作用,并且可以通过激活AhR–NrF2依赖性通路来上调TJ蛋白的表达,从而促进肠道屏障的修复。
这些研究表明,激活肠道AHR通路对于治疗酒精性肝病(ALD)具有重要意义,因为ALD与TJ屏障的丧失密切相关。因此,AHR可能成为修复肠道屏障的一个有价值的治疗靶点,尤其是在与肠道屏障功能障碍相关的慢性疾病中。
-IDO 抑制剂:代谢平衡与潜在毒性
正向:抑制 IDO -> 阻断色氨酸向犬尿氨酸转化 -> 迫使色氨酸转化为吲哚
抑制IDO(一种将色氨酸转化为犬尿氨酸的酶)可能有助于治疗肠道屏障功能障碍。IDO抑制剂通过促进色氨酸转化为吲哚,增加AHR配体的生成,从而改善肠道完整性。
虽然IDO抑制剂在癌症治疗中显示出潜力,但它们在治疗肠道屏障损伤方面的应用仍需更多研究。Indoximod是一种IDO抑制剂,已被证明能有效维持细胞间的紧密连接,并显著减轻DSS诱导的结肠损伤。
反向:过度抑制 IDO -> 减少犬尿酸生成 -> GPR35 失去激活 -> 削弱屏障保护
需要注意的是,IDO抑制也可能导致不良的肠道毒性反应。完全阻断犬尿氨酸通路可能会适得其反,损害上皮屏障的完整性。
研究表明,化疗药物引起的肠道损伤会激活色氨酸-犬尿氨酸-犬尿酸通路,增加肠道内犬尿酸的生成。犬尿酸随后激活GPR35,从而增强肠道完整性。这意味着,过度抑制犬尿氨酸通路可能会降低犬尿酸水平,削弱GPR35介导的保护作用,并可能延缓屏障修复。
综上所述,色氨酸代谢在肠道健康中扮演着复杂的角色。因此,在开发治疗肠道屏障功能障碍的新方法时,需要进行更深入的研究。
临床挑战:个体化差异
虽然大量研究表明直接调节肠道微生物群可以改善肠道屏障功能并缓解肠肝疾病,但基于微生物的疗法在临床应用上仍然受到限制。
肠道微生物群产生的有益代谢物,如吲哚-3-乳酸(ILA),其增强肠道屏障的特性取决于宿主特异性的微生物群调节。临床疗效主要取决于患者的初始肠道微生物群组成,这会影响微生物的定植情况。
此外,宿主的遗传变异也是治疗结果的关键因素。例如,携带CARD9风险基因的炎症性肠病(IBD)患者,其膳食色氨酸向芳香烃受体(AHR)激活代谢物的微生物转化能力受损。这些发现解释了微生物靶向疗法中个体差异显著的分子机制。
益生菌作为疾病状态下耗尽的共生微生物,需要稳定的宿主微环境才能定植和发挥作用。如果事先不恢复受损的肠道生态系统,微生物干预疗法往往无法建立持久的微生物平衡或实现有意义的临床结果。因此,未来的治疗策略应采用结合微生物调节和微环境恢复的双靶点方法。
3
肠道干细胞再生
Lgr5+ 肠道干细胞 (ISCs):修复的原动力
肠上皮细胞的更新、修复和再生在很大程度上依赖于位于隐窝底部的Lgr5+肠道干细胞(ISCs)。
Lgr5作为ISCs的特异性标记,编码一种受体,该受体能够响应Wnt等信号,从而触发ISCs重编程为上皮细胞谱系。ISC的活性受到细胞外信号和旁分泌信号的精密调控,以维持肠道稳态,并在损伤发生时启动适应性分化,从而保障肠道的基本生理功能。
调控胆汁酸水平,恢复干细胞活力
胆汁酸在介导肠肝轴通讯中扮演着关键角色,并整合了调控ISC功能的饮食和代谢信号。近期研究提示,通过减少病理条件下肠道内过量的胆汁酸累积,或可为治疗炎症性肠病(IBD)相关的肠道损伤提供新的干预策略。
例如,FXR激动剂治疗通过抑制肝脏中CYP8B1的表达,进而降低肠肝胆汁酸水平,从而减轻胆汁酸对Lgr5+ ISCs的相关毒性。上述发现提示,基于跨器官代谢调节靶向ISCs有望成为治疗IBD的新型目标。
4
免疫调节
传统抗炎疗法的局限性
传统的抗炎疗法,包括皮质类固醇、5-氨基水杨酸制剂以及新型TNF-α单克隆抗体,一直是炎症性肠病(IBD)和其他免疫介导疾病的标准治疗方法。
虽然这些疗法能有效减轻炎症并缓解症状,但它们在实现长期愈合方面往往力有不逮,尤其是在肠道屏障方面。许多患者随着时间推移出现疗效丧失和疾病复发。例如,尽管抗IL-6抗体疗法在临床试验中对中度至重度克罗恩病(CD)或溃疡性结肠炎(UC)有效,但在一些患者中仍持续发生脓肿和肠穿孔等严重不良反应。
这些疗法通常未能解决根本的上皮功能障碍问题,而这正是维持慢性肠道炎症的关键因素。
新兴策略:直接靶向屏障修复
为了解决这些局限性,人们对靶向更直接参与上皮屏障修复的免疫通路越来越感兴趣。IL-10是一种有效的抗炎和组织再生细胞因子,最近开发的IL-10制剂旨在通过靶向黏膜屏障来增强治疗效果。
另一个有前景的靶点是IL-22,其受体IL-22R在上皮细胞上表达,这使得IL-22和IL-22R成为旨在恢复上皮完整性和增强黏膜愈合疗法的潜在候选者。
IL-22通过刺激AMPs(抗菌肽)和粘蛋白的产生以及ISC(肠道干细胞)的再生,在维持肠道稳态中发挥关键作用。因此,IL-22被认为是屏障修复的潜在治疗方法。
IL-22 激动剂的临床与代谢获益
IL-22融合蛋白激动剂Efmarodocokin alfa (UTTR1147A) 目前正在研究中,用于活动性溃疡性结肠炎(UC)和克罗恩病(CD)的治疗(NCT02749630)。
临床试验表明,UTTR1147A在UC患者和健康个体中都能激活IL-22R信号通路,并改善UC相关的菌群失调。
此外,肠道中IL-22信号的特异性激活可以在代谢紊乱模型中以微生物依赖的方式增强肝脏和全身葡萄糖和脂质代谢稳态。此外,IL-22还对MASLD(代谢功能障碍相关脂肪性肝病)、ALD(酒精性肝病)和饮食诱导的肥胖表现出积极作用。
当外源性给药时,IL-22通过其在肠上皮细胞(IECs)而非肝细胞上的受体发挥治疗作用,然后激活STAT3并抑制WNT–β-catenin信号传导以减少吸收性肠上皮细胞的数量。
然而,IL-22在肠道屏障维持中的作用仍存在争议,人们担心可能产生致病性的免疫调节作用,例如在结肠组织中介导CXCR2+中性粒细胞的趋化作用,以及增加对IL-23单克隆抗体Ustekinumab的抵抗力。这些观察结果表明,IL-22靶向疗法可能并非总能达到预期的疗效,需要进一步研究以更好地了解IL-22激活的全部影响和潜在副作用。
5
肠道屏障功能增强相关的临床试验进展
前面介绍了目前针对肠道屏障完整性的多种创新疗法,包括药物、微生物疗法、吸附剂、饮食干预和工程益生菌,并同时也探讨了这些疗法在临床验证中的进展和未来面临的挑战。
其实核心要点如下:
创新疗法:多项创新疗法正在进行临床验证,以靶向肠道屏障完整性。
小分子创新 / 微生物疗法
ISM5411:一种新型肠道限制性选择性脯氨酰羟化酶结构域(PHD)抑制剂,通过AI平台开发,已完成I期临床试验。其在肠黏膜修复和免疫调节方面具有双重机制,在IBD模型中显示出显著疗效。
利福昔明-α (Rifaximin-α):通过上调粪便中的TNF-α和IL-17E来调节肠道微环境,增强抗菌防御,有效促进肠道屏屏障修复。
ZED1227:在乳糜泻中,作为转谷氨酰胺酶2抑制剂,显著改善十二指肠黏膜结构,减少上皮内淋巴细胞浸润,通过抑制免疫原性谷蛋白肽中谷氨酰胺残基的脱酰胺化来防止T细胞活化和黏膜损伤。
粪便菌群移植(FMT):健康供体FMT在恢复糖尿病远端对称性多发性神经病变(DSPN)患者肠道屏障功能和减轻全身炎症方面显示出治疗潜力。
工程益生菌:在恢复肠道屏障完整性和维持黏膜稳态方面具有显著治疗潜力,目前研究重点是结合其屏障增强和免疫调节作用,但仍在临床前阶段。
吸附剂:非吸收性、肠道限制性工程化碳珠吸附剂Yaq-001通过改善肠道屏障功能障碍和减少全身内毒素负荷,在肝硬化中显示出临床疗效。

饮食干预
临床研究证实,膳食干预在多种胃肠道疾病中有效。
谷氨酰胺补充剂:显著恢复肠道通透性并缓解感染后肠易激综合征的腹泻 (NCT01414244)。
在肠易激综合征-腹泻、代谢紊乱和轻中度克罗恩病中均显示出益处。

未来挑战
尽管这些进展显示了靶向屏障修复策略的广阔前景,但仍需要在人体试验中全面评估长期安全性和有效性,以推进临床应用。
肠道屏障功能障碍是多种胃肠和肠外疾病(如IBD、MASLD)的关键因素。传统免疫抑制疗法虽能改善症状,但无法根治屏障问题,且副作用明显。新型疗法应结合屏障修复策略,如干细胞再生、微生物疗法、胆汁酸调节、TJ调节剂等。
肠道屏障功能障碍与疾病互为因果,受遗传、环境等因素影响。屏障破坏可引发炎症和多器官功能障碍,导致MASH、IBD、CRC等。同时,疾病微环境反过来又损害屏障,形成恶性循环。
免疫抑制疗法可能抑制黏膜愈合,加剧微生物失衡。需深入研究肠道细胞间通讯,以确定有效治疗靶点。
独立于免疫抑制的屏障防御和修复是治疗肝肠疾病的重要目标。个性化治疗策略可能更有效。增强肠道屏障完整性在MASLD预防和逆转中潜力巨大。未来人体研究至关重要。
恢复肠道屏障是治疗肝肠疾病的重要机遇。新生物技术、再生医学和微生物组研究有望重塑胃肠道治疗格局。
主要参考文献:
Zhang Y, Liu Y, Liang X, Wen Y, Zhao J, He Y, Xie Q, Xie C. Intestinal barrier in chronic gut and liver diseases: Pathogenesis and therapeutic targets. Acta Pharm Sin B. 2025 Nov;15(11):5515-5536.
Macura B, Kiecka A, Szczepanik M. Intestinal permeability disturbances: causes, diseases and therapy. Clin Exp Med. 2024 Sep 28;24(1):232.
Farré R, Vicario M. Abnormal Barrier Function in Gastrointestinal Disorders. Handb Exp Pharmacol. 2017;239:193-217.
Brandl C, Bucci L, Schett G, Zaiss MM. Crossing the barriers: Revisiting the gut feeling in rheumatoid arthritis. Eur J Immunol. 2021;51(4):798–810.
Ramakrishna BS. Role of the gut microbiota in human nutrition and metabolism. J Gastroenterol Hepatol. 2013;28(Suppl 4):9–17.
Bäumler AJ, Sperandio V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature. 2016;535(7610):85–93
谷禾健康
在人类与微生物的共生世界中,新的发现正在不断刷新我们对健康与疾病的认知。肠道、呼吸道、生殖道——这些看似熟悉的环境中,栖息着数量庞大却性质各异的细菌,它们有的温和共生,有的在特定条件下摇身一变成为病原菌。
本次谷禾报告更新,新增了多种具有代表性的潜在致病菌和机会性感染菌——从龋齿放线菌到医院相关耐药菌,从新生儿高危感染的GBS到隐秘的粘质沙雷氏菌——这些微生物不仅展示了复杂的生态角色,也折射出人体微环境的脆弱平衡。
我们希望通过科学、系统的解读,帮助读者理解这些菌群在健康和疾病之中的真实意义:哪些只是短暂停留的“旁观者”,哪些则可能成为引发炎症、感染乃至系统性疾病的“潜在危险信号”。这既是一次对微生物多样性的探索,也是一场关于精准健康管理的知识升级。
龋齿放线菌(Actinomyces odontolyticus)是一种革兰氏阳性、厌氧、非产孢、非运动性杆菌,最早于1958年从口腔龋齿中分离。它通常作为口腔、上消化道和泌尿生殖道的正常菌群存在,但在特定条件下可成为机会致病菌,导致多种感染。
1
基本信息
•形态特征:革兰氏阳性,细长分枝杆菌样,呈丝状。
•需氧情况:严格或微需氧厌氧菌。
•人体分布:人类口腔、咽部、胃肠道和泌尿生殖道的常驻菌群之一。
•特点:与其他放线菌(如A. israelii)类似,可形成肉芽肿样、慢性脓肿,有“硫磺颗粒”。当黏膜屏障受损(如牙科操作、外伤、免疫抑制)时,易由定植转为致病。
2
可能导致的疾病
我们在下表中系统整理了由Actinomyces odontolyticus可能引起的各类疾病:

3
典型症状
•全身症状:患者可出现发热、乏力、体重减轻。局部出现慢性硬结,逐渐形成波动性脓肿,并可伴多发瘘管。脓液中常见黄白色“硫磺样颗粒”,实为菌落团。起病缓慢,虽少见明显全身中毒症状,但可长期低热、消耗,局部愈合不良。
•口腔/颈面部:牙痛、牙龈红肿、口腔脓肿、颈部肿胀。
•胸部/肺部:咳嗽、咳痰、呼吸困难、胸痛、胸腔积液。
•腹部/盆腔:腹痛、肝区压痛、腹部包块、女性可见盆腔炎症。
•中枢神经系统:头痛、意识障碍、神经功能缺损。
•皮肤/导管:脓性分泌物。
4
致病机理要点
•机会致病菌:Actinomyces odontolyticus通常在宿主免疫力下降或局部组织受损时,通过黏附并侵入受损的黏膜或牙龈组织,借此获得感染机会并逐步定植。
•在缺氧或厌氧条件下,该菌可迅速繁殖,形成致密的丝状菌丝团块。这些菌团被机体的肉芽组织所包绕,逐渐构成类似肉芽肿的慢性炎性病灶,使局部组织结构被破坏并持续扩展。
•随着感染进展,病原体可沿组织间隙缓慢向周围扩散,通过连续性扩展和瘘道形成进一步蔓延。由于这种蔓延方式不遵循正常的解剖间隙,病灶范围常远超出皮肤或黏膜表面肉眼可见的受累区域,导致诊断和治疗更加复杂。
5
在肠道里的意义
•这类放线菌本身就可以是口腔和消化道的共生菌之一,在粪便或肠道菌群中检出,并不罕见。多数情况下,仅仅检出不代表有病。
•还有部分研究显示,A.odontolyticus具备硝酸盐还原能力,可能影响口腔及肠道的亚硝酸盐水平,间接参与宿主的抗菌和血管舒张等生理过程 。
6
可能提示或潜在问题
•若在有腹腔感染、慢性脓肿、术后持续不愈的病例中,从腹腔脓液或病灶同时检出放线菌,才需要考虑:是否存在腹部放线菌病(如阑尾穿孔、肠穿孔后形成的慢性肉芽肿性炎症、瘘管等)。
•单纯在“肠道菌群报告”中出现,多半只是共生/或短暂存在,不必单独处理。
总结
A.odontolyticus虽为常见口腔定植菌,但在免疫低下或黏膜损伤时可引发多系统感染,临床表现多样,需高度警惕并结合多种检测手段早期诊断与干预。
摩氏摩根氏菌(Morganella morganii)是一种革兰氏阴性菌,兼性厌氧、杆状细菌,属于肠杆菌科,广泛存在于人类、哺乳动物、爬行动物肠道及环境中。
它通常作为肠道正常菌群存在,但在免疫力低下或特定条件下可成为机会致病菌,引发多种感染,且近年来耐药性和毒力增强,临床关注度显著提升。
1
基本信息
•分类:肠杆菌科,Morganella属,主要有M.morganii subsp. morganii和M.morganii subsp. sibonii 两个亚种。
•生物学特点:无芽孢、运动性强、杆状、兼性厌氧,具强尿素酶活性,能分解尿素。
•生态分布:广泛分布于水体、土壤、动物及人类肠道。
•重要性:常为医院获得性感染的机会致病菌。
2
可能导致的疾病
M. morganii作为机会致病菌,能引发多种感染,常见于以下疾病:
•尿路感染(UTI):常见于留置导尿、结构异常者或免疫低下、老年人、儿童中。
•伤口及软组织感染:包括手术后伤口感染、蜂窝织炎、坏死性筋膜炎等。
•败血症/菌血症:可导致高死亡率,尤其在老年及有基础疾病患者中。
•腹腔及肝胆系统感染:如腹膜炎、肝脓肿、胆道感染。
•其他:脑脓肿、心内膜炎、骨髓炎、关节炎、绒毛膜羊膜炎、食物中毒(组胺中毒)等。
3
典型症状
M. morganii感染的临床表现多样,取决于感染部位和宿主状况,常见症状包括:
•尿路感染:尿频、尿急、尿痛、血尿、发热。
•伤口/软组织感染:局部红肿、疼痛、化脓、坏死、发热,严重时可进展为坏死性筋膜炎。
•败血症:高热、寒战、低血压、意识障碍,严重时可致多器官功能衰竭。
•腹腔感染:腹痛、腹胀、发热、黄疸等。
•其他部位感染:如脑脓肿可表现为头痛、神经功能障碍,心内膜炎可有心脏杂音、心衰表现。
4
致病机理要点
•多重耐药性:M.morganii具备多种耐药基因(如 blaNDM-1、qnrD1、blaKPC-2),对多类抗生素(包括碳青霉烯类、氨基糖苷类等)表现耐药,治疗难度大。
•毒力因子:包括溶血素、尿素酶、IgA 蛋白酶、鞭毛、黏附素等,增强其致病性和组织侵袭能力。
•高危人群:老年人、免疫抑制者、慢性基础病患者、住院及手术后患者感染风险更高,且病死率较高
5
健康风险
•基因毒性与肿瘤风险:
M. morganii 可产生新型基因毒素“indolimines”,能直接损伤肠上皮细胞DNA,增加肠道通透性,并在动物模型中加重结肠肿瘤负担,提示其可能促进结直肠癌的发生与进展。
•炎症与免疫失调
该菌与炎症因子(如IL-6、IL-8、TNF-α)表达升高相关,且与短链脂肪酸水平降低有关,可能加剧肠道炎症和免疫功能障碍。在HIV感染者中,M.morganii丰度升高与抗逆转录病毒治疗后免疫重建不全密切相关。
•精神健康影响
研究发现M. morganii产生的特殊磷脂可激活免疫反应,诱导炎症,且其丰度与抑郁症发病风险增加有关。
6
在肠道里的意义
•属于肠杆菌科,本来就可以是肠道正常菌群的一部分。
•但它也是典型的机会致病菌+容易耐药的医院相关菌。
7
可能提示或潜在问题
•菌群失衡/抗生素压力下(特别是住院、用广谱抗生素的人):Morganella 可能扩增,成为潜在的感染源,例如尿路感染(尤其有导尿管者)、伤口感染、腹腔感染、菌血症、败血症(免疫抑制或重症患者)。
•从菌群学角度:此类条件致病菌大量增多,常提示肠道菌群多样性下降、共生菌被压制。
•什么时候要警惕:在ICU、肿瘤或老年患者中,若粪便或直肠拭子多次检出且丰度较高,并伴发热而无明确感染灶,应警惕其作为潜在的“肠源性血流或尿路感染”病原库,对经验性抗生素选择具有重要参考意义。
总结
M. morganii 虽为肠道常见菌,但其基因毒性、炎症促进、耐药性及与多种疾病的关联,使其成为值得高度警惕的潜在健康威胁。
无乳链球菌(Streptococcus agalactiae)(又称B群链球菌,GBS)是一种革兰氏阳性球菌,广泛存在于人类肠道和泌尿生殖道的正常菌群中,但在特定条件下可转变为重要的人类和动物病原体,导致多种严重感染。
1
基本信息
•革兰染色:革兰阳性球菌,成链状排列。
•溶血性:多为β溶血。
•分类:Lancefield B 群 → 常称“B 族链球菌(GBS)”目前已鉴定出10种血清型(Ia、Ib、II-IX),不同地区和宿主的流行型别有所差异。
•自然栖息地:成人肠道和女性阴道、下生殖道常见定植菌。
2
可能导致的疾病
•新生儿感染(核心重点):
-早发型:新生儿早期败血症、肺炎;
-晚发型:脑膜炎、败血症。
•妇产科感染:
产褥感染、绒毛膜羊膜炎、尿路感染。
•成人侵袭性感染:
皮肤软组织感染、尿路感染、菌血症、肺炎、骨髓炎等,尤其是糖尿病、老年或免疫低下者。
3
典型症状
•新生儿早发型:出生后数小时~数天内出现:呼吸急促、发绀、喂养困难、发热或体温不稳、嗜睡,可进展为呼吸窘迫、休克。
•新生儿脑膜炎:惊厥、反应差、长期可遗留神经系统后遗症。
•妇女:发热、下腹痛、恶露异常、尿频尿急尿痛等。
•成人/老年人:发热、意识障碍、关节炎、皮肤软组织感染、尿路感染、心内膜炎、脑膜炎等,重症可致死亡。
4
致病机理要点
GBS定植依赖多种黏附因子(如Fbs、Lmb、SfbA、HvgA等)和生物膜形成能力,这些机制增强其在肠道黏膜的持久性和侵袭性。
•荚膜多糖(CPS):抗吞噬作用,是主要毒力因子。
•表面蛋白(如C蛋白):有助黏附与免疫逃逸。
•妊娠晚期母体阴道/直肠定植 → 围产期经产道传播给新生儿。
•新生儿免疫系统未成熟,补体及吞噬功能不足,易进展为侵袭性败血症和脑膜炎。
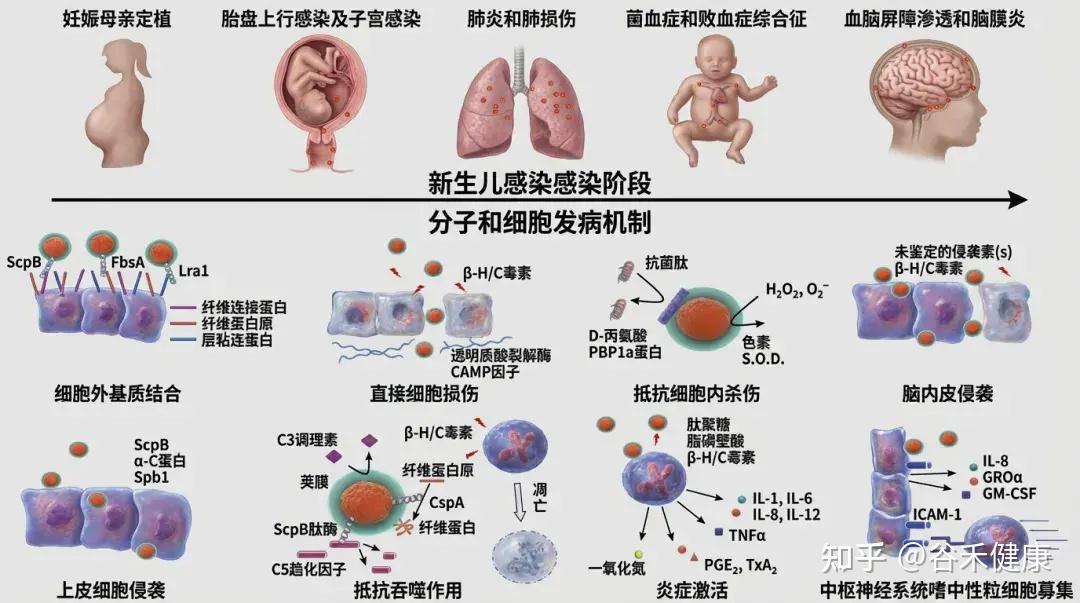
5
在肠道里的意义
•GBS 本身就可定植在肠道和阴道,成人特别是健康人群中检出并不少见。
•对普通非孕成人,大多是良性定植。
6
可能提示或潜在问题
•孕妇:
肠道是阴道定植的“菌库”,肠道检出 GBS,说明机体有GBS 定植风险。
阴道/直肠 GBS 定植 → 分娩时可传给新生儿 → 新生儿早发型败血症/肺炎/脑膜炎。
实务上,真正决定产时预防用药的,还是孕晚期阴道+直肠拭子筛查结果,而不是单纯一份“肠道菌群报告”。
•老年/慢病/免疫抑制人群:
肠道是 GBS 侵袭性感染的潜在来源(菌血症、软组织感染),但总体风险仍属相对低,要结合临床症状。
总结
GBS作为肠道定植菌,既是正常微生态的一部分,也具有转化为致病菌的潜力。其在肠道的存在与新生儿、孕妇及免疫低下人群的感染密切相关。
粘质沙雷氏菌(Serratia marcescens)是一种广泛分布于环境中的革兰氏阴性杆菌,近年来作为机会致病菌在医院获得性感染中日益受到关注,尤其威胁免疫功能低下者和新生儿。
1
基本信息
•革兰染色:革兰阴性杆菌。
•特点:部分菌株产生红色色素,肉眼可见“红色菌落”但临床分离株中无色素菌株更为常见。
具备多种耐药机制,对多类抗生素(如青霉素、头孢菌素、四环素等)有固有或获得性耐药。
•自然栖息地:水、土壤、植物以及医院潮湿环境(洗手池、呼吸机管路等)。
•重要性:典型的医院获得性感染和器械相关感染病原菌。
2
可能导致的疾病
S. marcescens可引发多种感染,尤其在医院环境和免疫低下人群中高发:
•血流感染(菌血症):常见于中心静脉导管相关感染。
•肺炎:包括医院获得性肺炎和重症肺炎。
•尿路感染:尤其在留置导尿管患者中。
•脑膜炎:多见于新生儿或免疫低下者。
•伤口和手术部位感染。
•罕见病例:可见于皮肤软组织感染、心内膜炎、骨髓炎等、角膜炎、结膜炎。
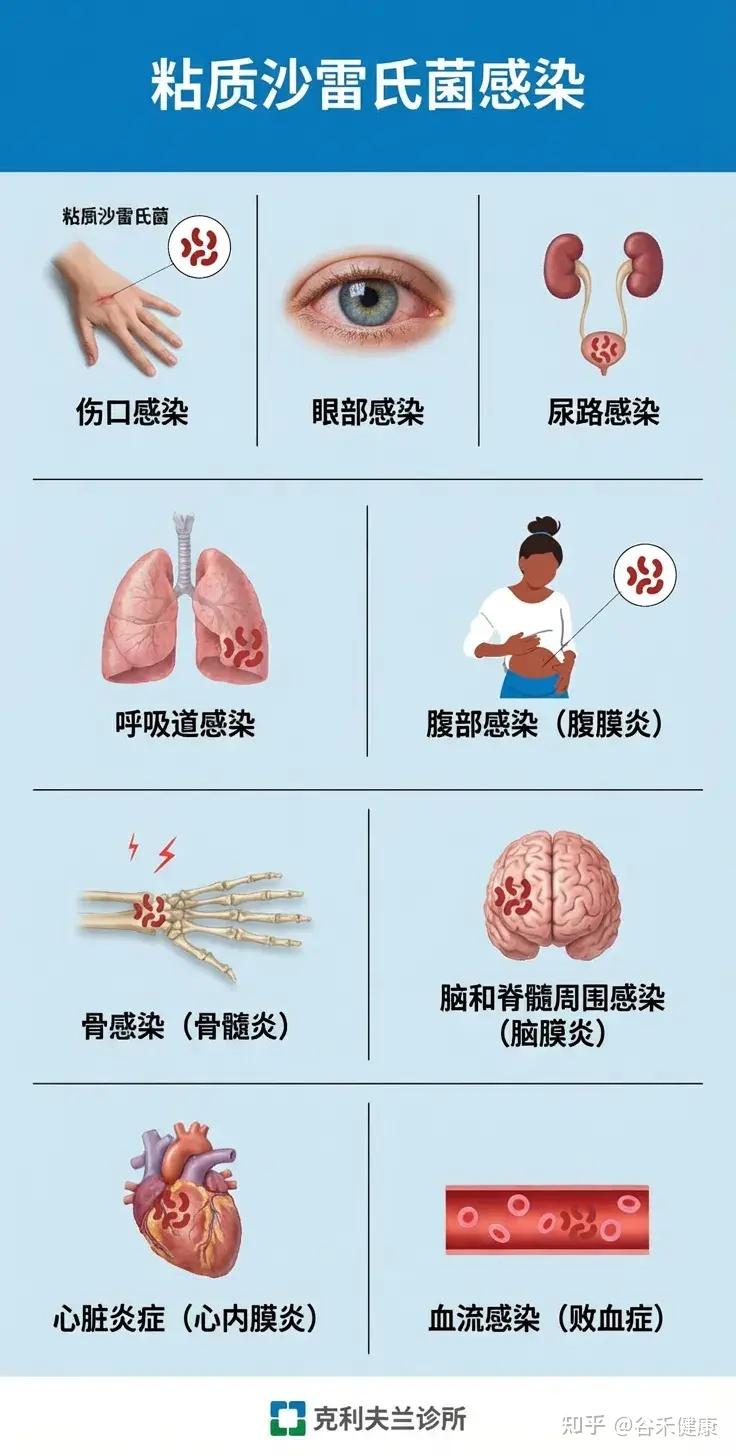
3
典型症状
S.marcescens与其他革兰阴性杆菌感染类似:发热、局部红肿热痛、脓性分泌物,重症可出现败血症、脓毒性休克。具体取决于感染部位和宿主状况:

4
致病机理要点
•黏附与生物被膜形成:在导尿管、静脉导管、呼吸机管路表面形成生物膜,增加持久定植与耐药。
•产生多种水解酶和毒力因子:如蛋白酶、脂酶等,这些物质能够分解宿主组织成分,破坏细胞结构,削弱局部屏障功能,从而进一步导致组织损伤和感染扩散。
•常携带产β-内酰胺酶和多药外排泵:导致多重耐药 → 治疗困难。
•医院潮湿环境的储存与反复污染:是暴发院感的重要原因。
5
在肠道里的意义
•本身不是典型稳定的“常驻共生菌”,更常见为医院环境相关的机会致病菌。
•健康人群肠道中较少见,但在住院患者,尤其是ICU或长期使用抗生素者中检出率显著升高。
•新生儿与免疫低下:肠道定植可导致院内感染暴发,易扩散至血流、呼吸道等部位,致病率和死亡率高。
•抗生素耐药:具多重耐药性,易获得广谱抗生素耐药基因,增加治疗难度。
6
可能提示或潜在问题
•表明患者已被多重耐药医院菌群定植:肠道成为储存库,增加导尿管相关尿路感染、呼吸机相关肺炎及静脉导管相关菌血症的风险。
•肠道菌群失衡:定植后抑制益菌生长,降低菌群多样性,影响宿主免疫与发育。
•公共卫生与院感防控角度:若大量患者肠道定植 Serratia,病区院感暴发风险显著上升。并且可经医院环境、医护人员手部及医疗器械传播,且难以根除。
7
需要关注的人群
•ICU、血液肿瘤科、器官移植患者:如果粪便/直肠拭子筛查提示有 Serratia marcescens(尤其是已知多重耐药株):通常会在经验性抗生素选择、隔离防护上额外注意。
总结
Serratia marcescens是重要的机会致病菌,在肠道中不仅可引发局部和全身性感染,还会扰乱肠道微生态,促进耐药性传播,尤其对新生儿和免疫低下人群威胁极大。加强监测、感染控制和合理用药,是降低其危害的核心措施。
索氏志贺氏菌(Shigella sonnei),也叫宋内志贺菌,是全球重要的肠道致病菌,近年来在发达国家和部分发展中国家发病率持续上升,且耐药性问题日益突出。
1
基本信息
•革兰染色:革兰阴性短杆菌。
•生物学:不运动、不产H2S,极少发酵糖类。
•分类与流行病学:Shigella sonnei属于肠杆菌科、志贺菌属的四个主要种之一,在发达地区更常见(与 S. flexneri 相对),但随着全球卫生条件改善,其在中低收入国家的比例也在上升。
•传播途径:主要通过粪-口途径传播,包括被污染的水、食物、接触受污染物品等。人是其主要宿主,极低的感染剂量(1-100个细胞)即可引发疾病,极易造成暴发流行。
•耐药性:S.sonnei对多种抗生素(如氟喹诺酮、头孢菌素、阿奇霉素等)耐药性显著增强,已成为全球公共卫生关注的多重耐药菌株。
2
可能导致的疾病
•主要疾病:S.sonnei是细菌性痢疾(志贺菌病,shigellosis)的主要致病菌之一,尤其在发达国家占主导地位。
•特殊人群风险:儿童、免疫力低下者、男男性行为者(MSM)、流浪者等为高风险人群,部分病例可出现菌血症、重症结肠炎甚至败血症。
•暴发场景:可引起食源性、饮水型、性传播及机构内(如学校、收容所)暴发。
3
典型症状
•常见症状:水样腹泻、腹痛、发热、恶心、呕吐。部分病例可出现血便(即“痢疾”),严重者可有脱水、休克等并发症。
•潜伏期:通常为1-4天,部分个体可无症状但仍具传染性。
•重症:脱水、电解质紊乱、毒血症,少数可出现中毒性肠麻痹或中毒性脑病(儿童)。
•特殊表现:免疫力低下者、儿童及老年人更易发展为菌血症和全身感染。
4
致病机理要点
•极低的感染剂量(10~100个菌即可致病),因此高度传染性。
•S.sonnei通过多种毒力因子(如III型和VI型分泌系统、O抗原胶囊、群体感应信号4-HBA等)侵袭肠上皮细胞,破坏肠黏膜屏障,诱发炎症反应。
S.sonnei的毒力因子
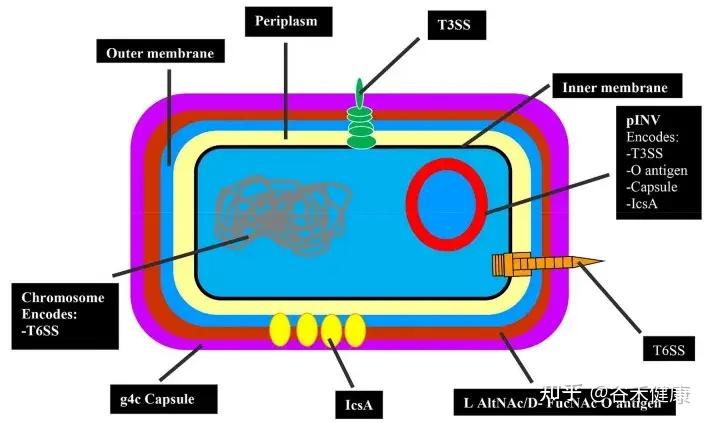
•其VI型分泌系统和细菌素(colicins)赋予其在肠道内与其他菌群竞争、占据生态位的能力,促进其流行。
•通过M细胞入侵肠上皮 → 细胞内增殖 → 从细胞侧面传播,造成上皮细胞坏死、溃疡。
•产生志贺毒素(Shiga toxin,一些菌株):抑制蛋白合成,导致黏膜细胞死亡,也可引起全身毒性反应
•粘膜受损导致渗出性炎症和溃疡 → 典型的黏液脓血便与里急后重。
•感染可显著扰乱肠道菌群多样性,部分患者即使临床恢复后,肠道微生态仍长期异常。
5
在肠道里的意义
•志贺菌是典型肠道致病菌,不是共生菌。
在粪便或肠道菌群中检出,尤其丰度不低时,通常意味着:
•正在感染期;
•或刚经历过感染,处于带菌/恢复期排菌状态。
6
可能提示或潜在问题
•急性期:细菌性痢疾 → 腹痛、里急后重、黏液脓血便、发热。
•恢复/带菌期:人本身症状可能已好转或无症状,但仍有:继续向他人传播的风险(粪–口),公共场所/托幼机构尤其要警惕。
•少数患者可发展为:
脱水、电解质紊乱;
中毒性肠炎/中毒性脑病(多见儿童);
感染后肠易激综合征等后遗症。
•耐药基因发展
S.sonnei对多种一线和二线抗生素(包括氟喹诺酮、头孢菌素、阿奇霉素等)耐药株迅速扩散,导致治疗选择受限。耐药基因可通过质粒在肠道内水平转移,增加耐药性在肠道微生物群中的传播风险。
7
检出后的处理思路
•若伴随腹泻、发热等症状 → 需要按细菌性痢疾评估和管理。
•即使暂时无症状,但粪便志贺菌阳性,尤其在托幼机构、食品从业人员等,应当从传染病防控角度处理。
总结
Shigella sonnei在肠道中不仅引发急性腹泻,还因其强竞争力、复杂致病机制和多重耐药性,带来持续的公共卫生威胁。加强监测、合理用药、疫苗研发及肠道微生态修复是未来防控的关键方向。
化脓性链球菌(Streptococcus pyogenes,简称GAS)是A群β溶血性链球菌。是全球重要的人体病原体,能引发从轻微到危及生命的多种疾病。
1
基本信息
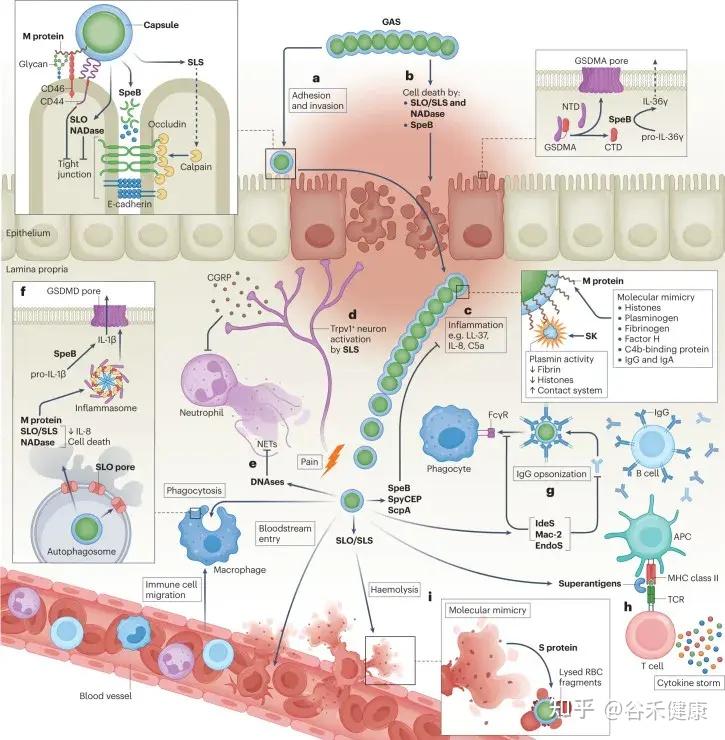
•分类与特性:S. pyogenes 属于A群β溶血性链球菌,革兰阳性球菌,常呈链状排列。该菌能在富血培养基上生长并产生完全溶血。
•重要特点:多种毒力因子,可致急性感染及免疫后遗症。
GAS的毒力因子
•传播途径:主要通过呼吸道飞沫、密切接触传播。上呼吸道和皮肤是主要的感染部位。
•流行病学:全球每年导致约50万例死亡,尤其在卫生条件较差地区危害更大。
2
可能导致的疾病
Streptococcus pyogenes感染后可能引发多种类型的疾病,如下:

3
典型症状
•咽炎/扁桃体炎:喉咙痛、发热、咽部充血、扁桃体肿大、化脓、软腭点状出血、颈部淋巴结肿大。
•猩红热:皮疹(常为沙粒状)、“草莓舌”、高热、咽痛、腺体肿大。
•皮肤感染:局部红肿、疼痛、脓疱、溃烂,严重时可发展为坏死性筋膜炎。
•侵袭性疾病:高热、休克、意识障碍、器官功能障碍(如肺炎、脑膜炎、败血症等)。
•免疫并发症:风湿热表现为关节炎、心脏炎、皮下结节等;急性肾小球肾炎表现为血尿、水肿、高血压,多在皮肤或咽部感染后1–3周出现。
4
致病机理要点
•M蛋白:抗吞噬、促进黏附,是关键毒力因子,也与风湿热的分子模拟相关。
•外毒素(SPE:streptococcal pyrogenic exotoxins):可作为超抗原 → 引起大量细胞因子释放 → 毒性休克、猩红热皮疹。
•溶血素(SLO、SLS):溶血、损伤组织。
•透明质酸荚膜:抗吞噬作用。
•免疫后遗症(风湿热、肾炎):多是免疫交叉反应与免疫复合物沉积所致,而非细菌直接侵袭。
5
在肠道里的意义
•主要定植于咽部和皮肤,并非典型肠道菌。
•在肠道菌群检测中检出,多见几种情况:
1.咽部分泌物被吞咽→通过粪便短暂排出;
2.可能存在上呼吸道感染;
3.极少数真正的肠道/腹腔感染(如坏死性小肠结肠炎等极罕见情形)。
6
可能提示或潜在问题
一般来说,单在菌群检测里偶然检出意义有限。真正需要警惕A组链球菌的,是:
•侵袭性疾病风险
GAS可通过肠道引发罕见但严重的腹膜炎,常被误诊为阑尾炎,延误治疗可能危及生命。女性、免疫抑制者及有水痘史者为高危人群,早期识别和抗生素治疗至关重要。
•免疫相关并发症
反复或持续感染可诱发免疫介导的疾病,如急性风湿热、风湿性心脏病和急性肾小球肾炎,尤其在发展中国家致死率高。
•功能性消化不良与肠道功能障碍
最新动物研究显示,GAS定植可通过皮肤-肠道轴影响胃肠动力,抑制RhoA/ROCK1信号通路,导致食欲下降、体重减轻和胃肠蠕动障碍,提示其可能加重功能性消化不良。
•抗生素耐药性与公共卫生挑战
GAS对青霉素的敏感性下降及大环内酯类耐药的增加,威胁一线治疗手段,需密切监测和合理用药。
总结
S.pyogenes是一种高度多样且致病力强的细菌,既可引发常见的咽炎、皮肤感染,也可导致危及生命的侵袭性疾病和免疫相关并发症。典型症状因疾病类型而异,需临床高度警惕并及时治疗。
解脲支原体(U.urealyticum,Uu),又称为溶脲脲原体。主要寄居于泌尿生殖道,在特定条件下可致病。
1
基本信息
•归类于支原体类,无细胞壁(对β-内酰胺类天然耐药)
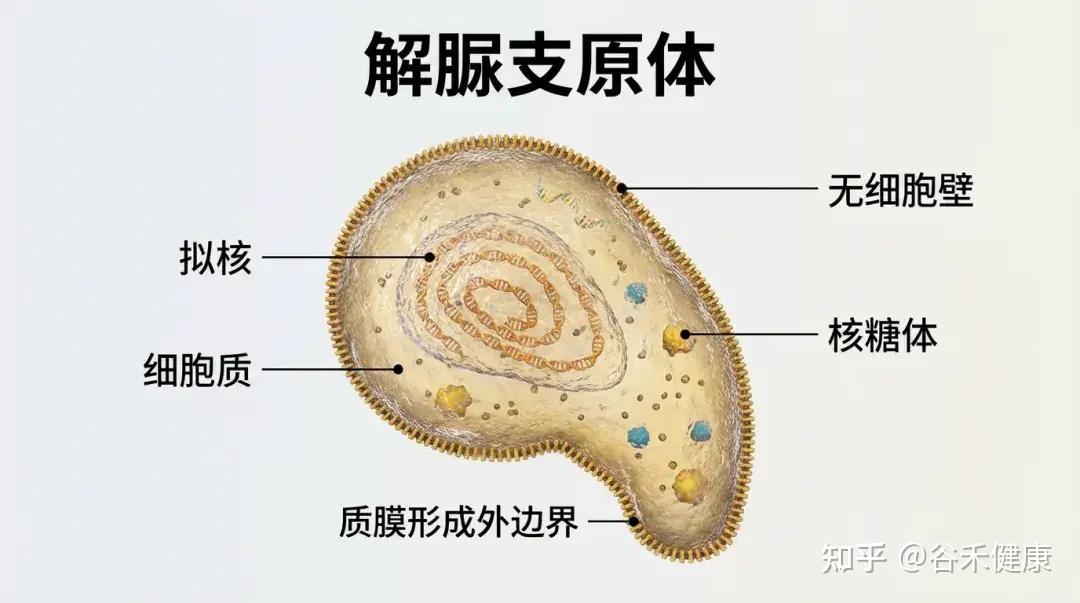
•极小型(“类原核生物”),通过固着于黏膜表面生存。
•自然栖息地:男女泌尿生殖道黏膜正常菌群之一,常为性传播。
2
可能导致的疾病
•男性:非淋菌性尿道炎(NGU),精囊炎、附睾炎等。
•女性:宫颈炎、盆腔炎、输卵管炎、不孕风险增加。
•妊娠相关:绒毛膜羊膜炎、羊水感染、早产、胎膜早破。
•新生儿:肺炎、菌血症、脑膜炎,尤其极低出生体重儿。
•免疫低下者:机会性感染。
3
典型症状
•男性非淋菌性尿道炎:尿道刺痛、瘙痒、轻度分泌物,症状可较轻、反复发作。
•女性宫颈炎/盆腔炎:下腹痛、白带增多、性交痛、不规则出血。
•妊娠并发症:发热、下腹痛、胎动异常、早产征象。
•新生儿:呼吸窘迫、发热或体温不稳、反应差等。
4
致病机理要点
•黏附因子:附着于泌尿生殖道上皮细胞表面,干扰纤毛功能。
•尿素酶:分解尿素→生成氨和二氧化碳:
过量氨对上皮细胞有毒性作用,引发局部炎症;
改变局部pH,影响黏膜屏障。
•诱导宿主产生多种炎症因子(如IL-6、IL-8),使局部出现慢性炎症与微环境改变,影响孕囊稳定和胎膜完整性。
5
在肠道里的意义
•其典型定植部位主要位于泌尿生殖道黏膜,包括尿道、宫颈及阴道等区域,在这些部位最易形成稳定的定植状态并长期存在。
•在粪便或肠道菌群中也可检出该菌,但多数情况下为采样过程中会阴部污染所致,尤其在女性样本中这一情况更为常见。
6
可能提示或潜在问题
•对肠道本身的影响:目前尚缺乏充分而有力的科学证据,尚不能明确证明其在肠道内具有直接的致病作用,更多情况下可能仅为暂时定植或伴随现象。
•需要重点关注的情况是:如果在生殖道相关样本(如尿道拭子、宫颈分泌物)中检测到该菌丰度较高,同时伴随出现尿道炎、不孕或妊娠相关并发症等临床表现,此时检测结果才真正具有临床意义和诊断参考价值。
注:若仅在肠道菌群中偶然检出该菌,而未见相应的临床症状或体征,通常不建议仅依据此结果进行疾病诊断或启动抗菌治疗。
Mycoplasma pneumoniae(肺炎支原体)是一种无细胞壁、体积极小的专性寄生细菌,是全球社区获得性肺炎(CAP)尤其是儿童和青少年中最常见的病原体之一。其感染可导致多种呼吸道及肺外疾病,临床表现多样,部分病例可进展为重症甚至危及生命。
1
基本信息
•肺炎支原体为最小的自由生活原核生物之一,缺乏细胞壁,对β-内酰胺类抗生素天然耐药。
•主要通过飞沫传播,潜伏期为2-3周。
•该菌主要寄生于呼吸道上皮细胞,通过特殊的终末附着器实现黏附、侵袭和免疫逃逸。
•近年来,耐大环内酯类(如红霉素)菌株比例显著上升,尤其在中国和亚洲地区。
2
可能导致的疾病

3
典型症状
•呼吸道症状:持续性干咳(最常见,85-96%)、发热(86-96%)、咽痛、头痛、乏力、气促、胸痛、喘息等。
•肺外症状:皮疹、恶心/呕吐、关节痛、神经系统症状(如头痛、意识障碍)、心肌损伤、肝肾功能异常等,约1/4患者可见。
•儿童表现:咳嗽、哮喘样症状、低热,部分伴有皮肤表现(25%)、消化道症状(33%)。
•影像学:单侧或多叶浸润、实变、胸腔积液等,部分病例可见肺不张或大面积病变。
4
致病机理要点
•P1黏附蛋白:结合呼吸道上皮细胞表面的纤毛,牢固黏附 → 破坏纤毛运动,干扰黏液清除。
•产生CARDS toxin(Community-Acquired Respiratory Distress Syndrome toxin)等毒素,损伤上皮细胞,引发炎症。
•诱发宿主细胞免疫和体液免疫反应,其中部分交叉反应导致肺外免疫并发症(如溶血性贫血、皮疹、神经系统损害等)。
•由于细胞壁缺失,细菌形态柔软、易穿行于黏膜表面间隙,且对多种抗生素类别不敏感(需用大环内酯、四环素或氟喹诺酮类)。
5
在肠道里的意义
•是典型的呼吸道病原体(气道黏附),不属于肠道定植菌。
•在肠道菌群测序中检出,通常是吞咽气道分泌物中的菌体/DNA,短暂通过消化道。
6
可能提示或潜在问题
•一般不直接导致肠道疾病,更多表明可能存在呼吸道感染。但M. pneumoniae感染可显著改变肠道菌群结构,降低有益菌如乳酸杆菌和双歧杆菌的丰度。这种菌群失衡与炎症因子升高密切相关,尤其在伴喘息的患儿中更为明显。同时肠道菌群的变化会加剧免疫失调,促进炎症反应,增加哮喘等并发症风险。
•肺炎支原体真正的危害仍在:非典型肺炎、持续干咳、肺外免疫并发症(皮疹、溶血性贫血、中枢神经系统累及等)。
总结
M.pneumoniae不仅影响呼吸系统,还可通过肠道微生态紊乱和免疫调节,导致肠道损伤和多系统炎症反应。关注其肠道影响有助于早期识别非典型病例,并为综合治疗提供新思路。
Clostridium symbiosum(共生梭菌)是一种严格厌氧、具鞭毛、可运动、不产毒素的芽孢杆菌,属于厚壁菌门,在人体肠道中可作为正常菌群存在,但近年来被认为是新兴的致病菌,临床分离较为罕见。
1
基本信息
•形态与生理特征:C. symbiosum为芽孢形成、鞭毛运动、革兰氏阳性杆菌,但因其对氧的敏感性,常表现为革兰氏阴性染色。它不产毒素,能发酵多种糖类。
•生态分布:主要存在于人类肠道正常菌群中,也可在动物肠道及环境中发现。
•致病性:与其他梭菌如 C.perfringens、C.difficile 相比,其致病性报道较少。多在严重基础病或免疫抑制、肠道屏障破坏时成为机会致病菌。
2
可能导致的疾病
目前临床报道不多,可见于:
•腹腔内感染:如穿孔性腹膜炎、腹腔脓肿(与肠道穿孔/手术有关)。
•菌血症/败血症:多为重症患者、肿瘤患者、长期住院或使用广谱抗生素者。
•结直肠癌:C. symbiosum在结直肠癌患者肿瘤组织中显著富集,并与腺瘤复发风险增加相关。动物实验显示其可促进肠道干细胞增殖和癌变,机制涉及支链氨基酸代谢和胆固醇合成通路激活。
•可能参与某些肠道菌群失衡相关疾病(如代谢性疾病、肠炎)中的“共病菌”,但具体因果尚在研究中。
3
典型症状
•无特异性,通常与原发病及感染部位相关:
-腹痛、发热、白细胞升高、腹膜刺激征(腹腔感染);
–寒战、高热、低血压、脓毒性休克表现(菌血症)。
•其他:如合并腹腔脓肿、手术部位感染等,表现为局部红肿、疼痛、化脓等。
•多数为混合感染:与其他肠道厌氧和需氧菌同在,单独分离报道较少。
4
致病机理要点
•作为梭菌,可形成芽孢,在不利环境中长期存活;进入厌氧环境(如坏死组织、闭合脓腔)后即可迅速繁殖。
•当肠道屏障受损(如穿孔、手术、肿瘤浸润)时,可随肠内容物移位至无菌体腔或血流,引发机会性感染。
•可能分泌组织降解酶和毒力因子,但其毒力谱较艰难梭菌(C. difficile )等梭菌研究尚不充分。
5
在肠道里的意义
这是典型的肠道厌氧共生菌之一,在健康人粪便中检出是常见且正常的。
•能量代谢与交互喂养:C. symbiosum 能分解膳食抗氧化剂(如麦角硫因),产生可被其他菌(如 Bacteroides xylanisolvens)利用的电子受体,促进厌氧能量代谢和菌群协同生长。这种交互喂养机制有助于维持肠道微生态稳定,并可能影响个体疾病风险。
•短链脂肪酸(SCFA)合成:C. symbiosum 参与乙酸等 SCFA 的合成,SCFA 对维持肠道屏障、调节免疫和抑制肿瘤细胞增殖具有重要作用。
6
可能提示或潜在问题
•菌群失衡的“标志菌”之一(研究层面):
一些研究发现,在某些疾病(如结直肠癌、代谢性疾病、肠道炎症状态)中,某些梭菌(包括C.symbiosum在内)丰度升高或降低,可作为潜在“生物标志物”。但这些更多是统计学相关性,并不能简单说“检出/增多 = 一定有某种病”。
•机会致病菌:
当肠道屏障严重破坏(穿孔、手术、肿瘤浸润)或极度免疫抑制时,共生梭菌可随肠内容物进入腹腔或血流,引起腹腔感染或厌氧菌败血症。真正发生时,往往是与其他肠道菌“混合感染”,而不是它单独“作恶”。
•抗药性:
C. symbiosum中携带多种环境来源的抗生素抗性基因,提示其在抗药性传播中的重要角色。
总结
Clostridium symbiosum 在肠道生态、能量代谢、疾病发生及抗药性传播中扮演着双刃剑角色。其作为结直肠癌等疾病的潜在生物标志物和微生态调节靶点,具有重要研究和临床应用前景,但也需警惕其条件致病性和抗药性扩散风险。

谷禾健康
在当代营养流行病学的视野中,我们长期致力于寻找一种最佳饮食模式。无论是地中海饮食、生酮饮食还是植物基饮食,焦点往往集中在吃什么。然而,这种静态的视角忽略了人类饮食行为中最显著的一个特征:时间上的波动性。
举个例子,工作日与周末的割裂,很多人在周一至周五严格控制热量,吃高纤维的健康沙拉;而一旦到了周五晚上或周末,各种吃大餐,高脂、高糖食物轮流上,或者饮食时间变得极不规律。

如果我们将时间跨度拉长,计算其营养摄入的平均值,这些人的数据可能依然显示为健康。这就带来了一个问题:
如果平均营养摄入达标,为什么代谢紊乱和肠道微生态失调的问题依然普遍存在?
近期《Nature》几个子刊的研究,揭示了被长期忽视的真相:
饮食的规律性与营养供给的精准度,可能比单一的追求饮食质量,更能影响肠道微生态的健康。

Singh 等人发表的最新研究(Nat Commun, 2025),通过分析“Food & You”数字队列中近1000名参与者长达两周的高分辨率饮食记录与微生物组数据,饮食质量的时间波动性是破坏肠道菌群多样性的独立风险因素。
Estrela等人的研究(Nat Micro, 2025)深入微观层面,告诉我们肠道菌群挨饿的时候到底会发生什么。
随机休眠-唤醒,可能会让细菌本应用于繁殖的能量储备耗尽,共生菌因无法快速复苏而被淘汰。细菌一旦应激,可能啃食肠壁,使屏障功能变薄;若饥荒持续,则启动终极生存策略——同类相食。这种层层升级的生存博弈,最终将肠道生态系统推向从共生到对抗的危险边缘。
那么,如何才能让肠道菌群真正吃饱吃好,避免生存博弈演变成对宿主的反噬?
膳食纤维与脂肪、蛋白质之间又存在怎样的微妙平衡,才能既满足人体需求又不伤害有益菌?
那些看似不起眼的微量元素,又是如何在这场微观战争中决定菌群的站队?
…
本文将综合这些前沿成果,详细了解菌群喂养的底层逻辑,从关注宏观层面饮食营养的平均达标,转向整合多维度包括时间层面的规律饮食与微观层面的精准喂养。我们将探讨如何通过消除隐性饥饿、维持微量元素平衡以及建立稳定的饮食节律,从而为预防代谢性疾病和构建深层免疫防线提供可落地的精准营养行动指南。
在既往的营养学研究中,我们习惯于通过平均值来评估一个人的饮食质量。例如,如果一个人在一个月内摄入了足够总量的膳食纤维,无论他是每天均匀摄入,还是集中在几天内突击摄入,他在传统的营养评估中得到的得分往往是相似的。
然而,Singh等人的研究通过高分辨率的时间序列分析,彻底打破了这一平均值的说法,揭示了饮食规律性作为独立健康维度的决定性作用。

突破传统的方法论:从回忆问卷到动态观测
该研究依托于“Food & You”数字队列(http://clinicaltrials.gov NCT03848299),这是一个极具前瞻性的研究项目。不同于传统流行病学依赖参与者回忆过去一年饮食习惯的食物频率问卷,这种方法类似一张模糊的静态快照,Singh 等人利用智能手机应用程序辅助的实时追踪技术,记录了近 1000 名参与者在两周内每一餐的详细构成。
这种方法论的革新使得研究者能够计算出一个全新的指标:每日健康饮食指数(Daily HEI)。传统的平均健康饮食指数掩盖了每日的波动,而每日健康饮食指数则像一部动态电影,捕捉了饮食质量在时间轴上的每一次震荡。
研究人员进一步引入了个体内变异系数(Intra-individual Coefficient of Variation, CV),专门用于量化这种震荡的幅度。通过将微生物组测序数据(16S rRNA gene sequencing)与这些高频饮食数据进行关联分析,一个隐藏在平均值背后的规律浮出水面。
每天吃沙拉,健康饮食的人,菌群多样性反而不如饮食普通但规律的人?
数据的统计分析揭示了一个单调且稳健的负相关关系:
饮食质量的变异性越高,肠道微生物群的 α-多样性越低(尤其是 Shannon 指数)。
不同生理和饮食因素与香农多样性之间的相关性
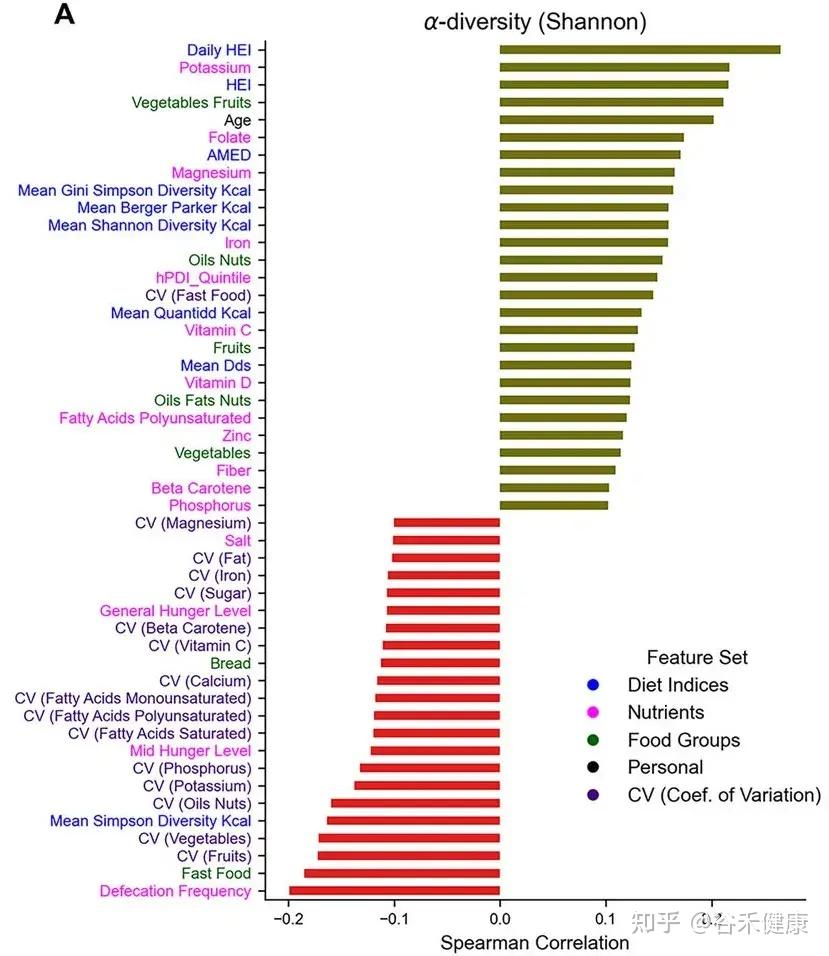
这一发现具有极高的临床意义。Shannon 指数反映了群落中物种的丰富度和均匀度,通常被视为生态系统健康和韧性的标志。
研究数据显示,即使在那些平均饮食质量较高的参与者中,如果他们的饮食习惯呈现出高波动性(例如:周一吃素,周二高脂,周三断食),其微生物多样性依然显著低于那些饮食质量中等但非常规律的个体。
这意味着,肠道微生态系统对于“不确定性”忍耐度有限。 剧烈的饮食波动相当于在肠道内制造了不稳定的生态位,导致只有少数菌能够快速适应环境变化的,能够存活,从而降低了整体生态系统的复杂度和稳定性。
具体菌群的响应
Singh 等人的研究并未止步于宏观的多样性指标,而是深入到了分类学(Taxonomy)层面,明确指出了哪些细菌是饮食规律性的受益者,哪些则是波动的获益者。
规律饮食滋养:毛螺菌科、粪球菌属
数据表明,属于厚壁菌门下的毛螺菌科(Lachnospiraceae),尤其是粪球菌属(Coprococcus),与饮食的规律性呈现最强的正相关。
毛螺菌科,如真杆菌属(Eubacterium)、毛螺菌属(Lachnospira)和丁酸杆菌属(Butyribacter),均是预测高健康饮食指数、蔬菜、水果、坚果及膳食纤维摄入的关键特征菌。
粪球菌属是肠道内重要的产丁酸者。丁酸盐不仅是结肠上皮细胞的主要能量来源,还具有强大的抗炎功能。
研究发现,粪球菌属的丰度对于蔬菜、水果和全谷物摄入的稳定性高度敏感。当这些富含纤维的食物来源在几天内忽高忽低时,粪球菌属的种群数量会迅速崩塌。
这表明这类有益菌需要持续、稳定的底物供应,来维持其在竞争激烈的肠道环境中的生态位。
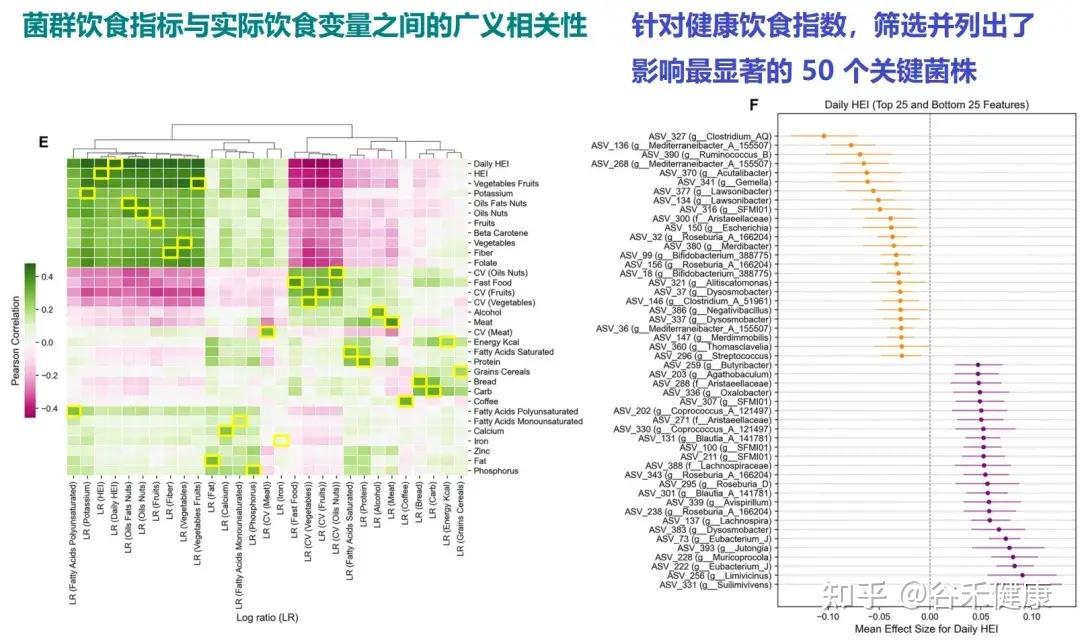
趁乱反噬:活泼瘤胃球菌
与之形成鲜明对比的是,饮食质量的高变异性与活泼瘤胃球菌(Ruminococcus gnavus)的富集显著相关。
研究还发现,Mediterraneibacter、Lawsonibacter、Dysosmobacter等,与肉类摄入、快餐消费以及蔬菜、水果、坚果摄入的不规律性(高 CV)呈正相关,并与腹泻比例增加显著相关。
活泼瘤胃球菌是一种典型的病原共生菌。在健康肠道中,它可能以低丰度存在,但在炎症性肠病、克罗恩病患者体内,常观察到该菌的过度生长。
该菌具有分解含有唾液酸的宿主黏蛋白的能力。当饮食中的外源性营养(如膳食纤维)波动剧烈导致供应中断时,活泼瘤胃球菌能够迅速切换代谢模式,利用宿主的黏液层作为备用能源。
数据暗示,饮食的不规律性实际上是在为活泼瘤胃球菌、Mediterraneibacter、Lawsonibacter这类具有高度适应性的机会主义者创造竞争优势,从而增加了肠道促炎环境的风险。
蔬果摄入波动大,是菌群失衡的关键推手
研究进一步细化了不同食物类别的波动影响。结果显示,蔬菜、水果、坚果摄入量的变异性,即吃得忽多忽少,对微生物组的影响最为显著。相比之下,精制谷物或乳制品的摄入波动对菌群多样性的冲击较小。
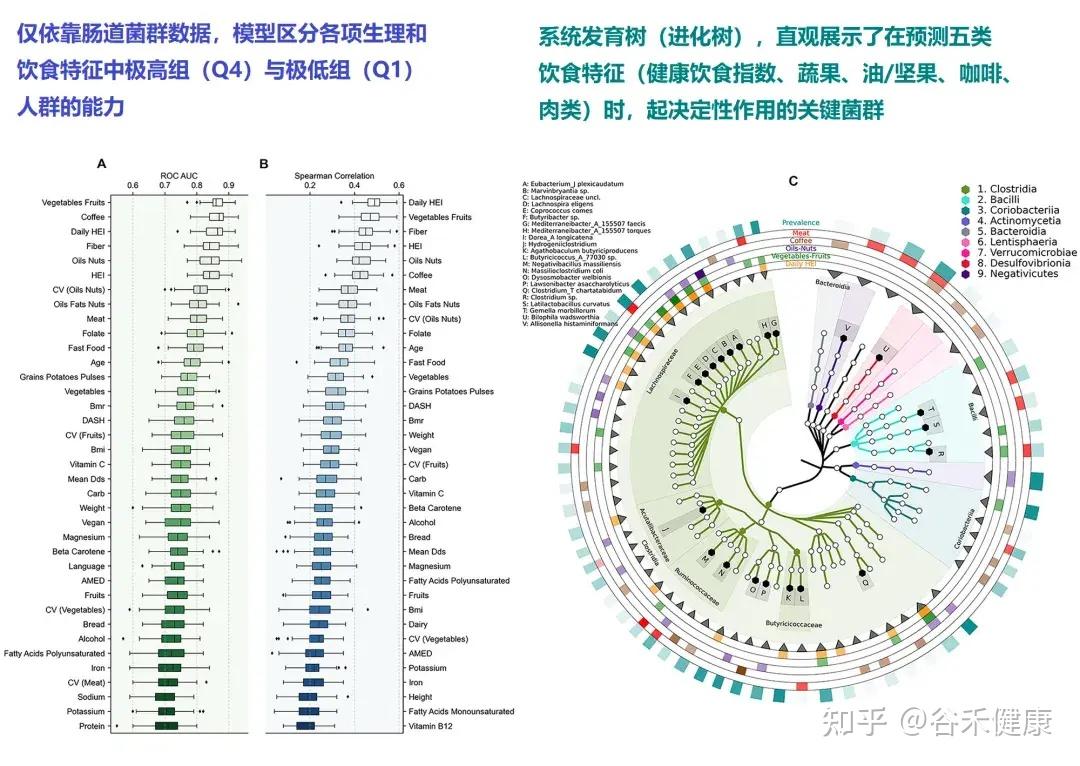
这背后的逻辑在于:蔬菜和水果是膳食纤维的主要来源,对于肠道细菌而言,这些纤维是赖以生存的“口粮”。
如果这些纤维的供应不稳定(高变异性),那些专性依赖纤维发酵的细菌(如前述的毛螺菌科)就会面临周期性的饥饿压力,无法建立稳定的种群;而那些代谢灵活、能吃“杂食”甚至能“吃人”(降解黏液)的细菌则会占据主导地位。
小结:规律性是独立的健康参数
上述研究确立了一个新的营养学范式:规律性本身就是一个独立的、关键的健康参数。
我们不能再单纯地建议人们多吃蔬菜,而应该建议每天稳定地吃蔬菜。因为从微生态的视角来看,三天吃一次大量的沙拉,其生态效益远不如每天吃一小份沙拉。
不规律的饮食习惯,实际上是在肠道内进行着一场持续的、负面的自然选择,逐渐淘汰了那些维护健康的共生菌,而筛选出了潜在的致病菌。
那么,为什么细菌对波动如此敏感?在微观层面上,当营养供应中断的那几个小时或几天里,细菌究竟经历了什么?
下一章节,我们接着来看Estrela等人的一篇文章,有详细的机制解析,深入到细菌的细胞内部,去洞察微观世界的隐形饥荒。
如果说上述研究从宏观层面来告诉我们,饮食波动会导致多样性下降和机会病原菌滋生。但要真正理解这场危机的根源,我们需要深入到细菌的视角,去体验它们在面对“断粮”时的生死抉择。
Estrela等人发表在《Nature Microbiology》的文章为我们揭开了这场微观“饥饿游戏”的规则。

研究表明,细菌并不只是被动地等待死亡,它们演化出了一套极其复杂的策略来应对营养匮乏。对于肠道细菌而言,宿主少吃一顿,或者饮食结构的一次剧烈跳变,就意味着它们必须在毫秒级的时间内启动应急预案。
肠道里的“盛宴与饥荒”循环
尽管现代人往往处于营养过剩的状态,但这并不意味着肠道细菌总能吃饱。相反,细菌生活在一种“盛宴与饥荒”交替的极端环境中。
盛宴期
当宿主进食后,膳食成分进入结肠,营养物质瞬间激增。此时,那些生长速度快、擅长降解复杂膳食多糖的厚壁菌门细菌占据绝对优势。例如,罗氏菌属(Roseburia)、真杆菌属(Eubacterium)和瘤胃球菌属(Ruminococcus),它们在有食物时不仅自身快速增殖,还通过交叉喂养滋养其他菌群。
饥荒期
在宿主睡眠期间、餐间空隙,或者当宿主摄入缺乏纤维的精细饮食时,肠道内的可发酵膳食纤维会迅速耗尽。此时,群落结构发生变化:厚壁菌门的丰度下降,而拟杆菌门、变形菌门以及疣微菌门开始主导局面。
研究人员指出,正是这种不可预测的饥荒期长度,决定了群落的最终结构。不规律的饮食习惯,本质上是在人为地延长或随机化“饥荒期”,迫使细菌长期处于应激状态。
策略一:蛰伏以求生——休眠与生长停滞
面对饥饿,细菌的第一反应往往是节能。
许多细菌会进入一种被称为生长停滞或休眠的状态。它们关闭大部分代谢活动,停止细胞分裂,将原本用于繁殖的能量,转用于维持细胞结构的完整性。
断臂求生:细菌的饥饿应对机制
细菌会通过复杂的分子机制(如 (p)ppGpp 介导的严谨反应)下调核糖体的合成,这是细胞内最耗能的过程,所以立刻叫停这个耗能的事。
对于大肠杆菌和其他变形菌来说,它们会依赖 Sigma因子 RpoS 来重新编程基因表达,将原本用于繁殖的能量转用于维持细胞结构的完整性,甚至不惜降解自身的核糖体,以回收氨基酸和核苷酸作为应急储备。
持久细胞:极度不活跃,却能保生存
文章还提到了一种“两面下注策略”(bet-hedging),即细菌分化出一小部分代谢极度不活跃的持久细胞。它们虽然不生长,但能耐受极端的压力(包括抗生素),确保存活。
内生孢子的形成
某些厚壁菌门的成员,如艰难梭菌或枯草芽孢杆菌,能够形成高度耐受的内生孢子。这就像是把自己锁进一个时间胶囊里,以此度过长达数天甚至数年的饥荒。
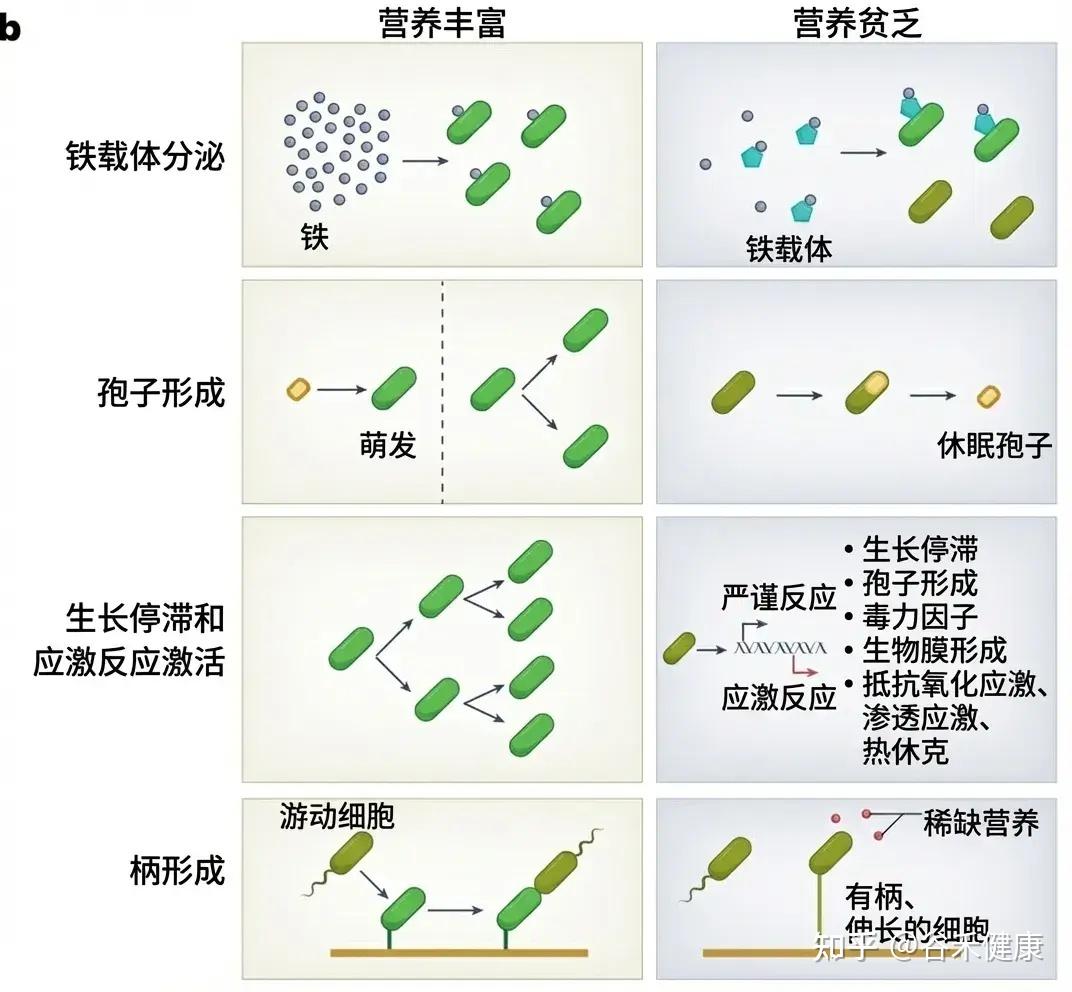
然而,这种策略是有代价的。进入休眠意味着放弃了繁殖的机会。如果饮食波动过于频繁,细菌反复在“唤醒”和“休眠”之间切换,会消耗巨大的能量储备。那些无法快速从休眠中复苏的菌种,就会在下一轮盛宴来临时因为起步太慢而被竞争对手淘汰。这也解释了前面的研究提到的饮食高变异性会导致微生物多样性的丧失——许多脆弱的共生菌因为经不起反复折腾而灭绝了。
策略二:吃宿主——营养毒力与黏液降解
如果说休眠是一种被动的防御,那么另一种策略则显得极具侵略性:当外源性食物(膳食纤维)断供时,去找内源性的替代品——宿主自己。
这就是研究人员强调的“营养毒力”概念。
我们的肠道内壁覆盖着一层厚厚的黏液层,主要由高度糖基化的黏蛋白组成。对于细菌来说,这层黏液不仅是屏障,更是一个巨大的、全天候供应的备用糖库。
黏液降解菌的崛起
正常情况下,像AKK菌(Akkermansia muciniphila)和多形拟杆菌(Bacteroides thetaiotaomicron)这样的细菌,会以受控的方式降解少量陈旧的黏液,这有助于黏液的更新。
但在饥饿状态下,当膳食纤维严重缺乏或供应中断时,多形拟杆菌会通过转录调控,迅速上调分解宿主聚糖的酶系表达。而AKK菌更依赖宿主黏蛋白上的糖链作为能量来源,在别的菌饿得不行的时候还能活得不错,数量甚至上升。
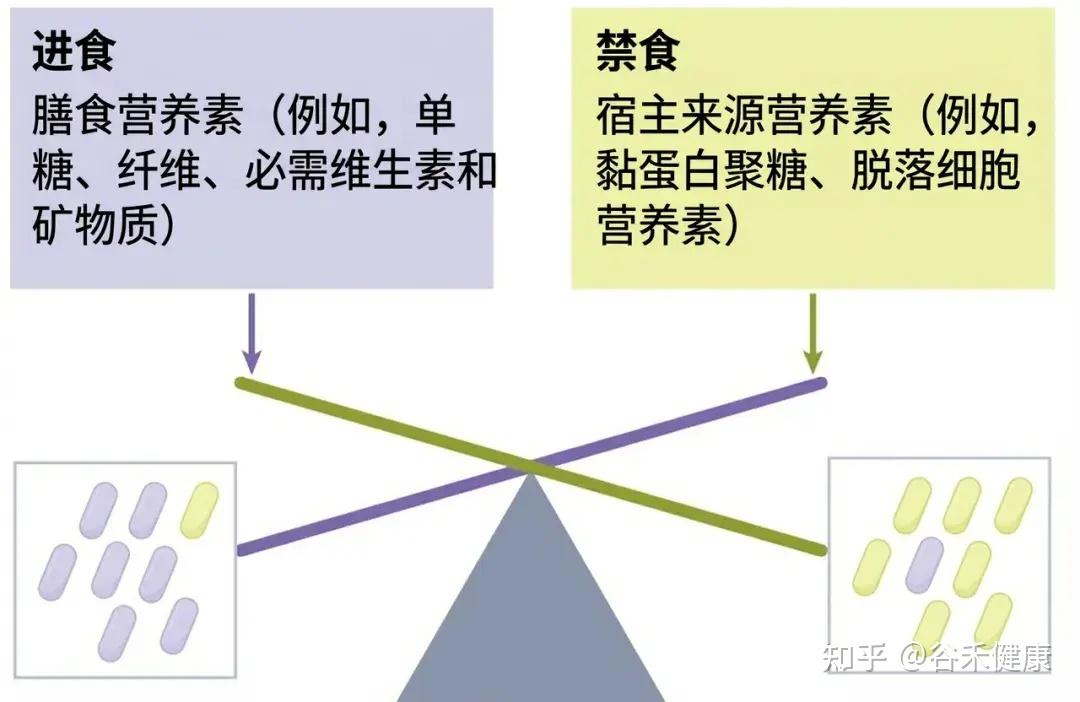
这种代谢切换使它们能够生存下来,但代价是过度啃食保护肠壁的黏液层。
黏液层变薄:病原菌的入侵
更危险的是,这种黏液层的变薄为病原菌打开了缺口。例如,肠出血性大肠杆菌(EHEC)在饥饿状态下会感知这种环境信号,上调其毒力因子的表达,利用这一机会粘附并侵袭肠上皮细胞。
这就是为什么饮食不规律(尤其是纤维摄入的波动)不仅影响菌群结构,更直接威胁肠道屏障功能。你的细菌饿急了,是真的会“吃”你的。
游离糖:病原菌的扩张燃料
此外,拟杆菌在降解黏液时,会释放出岩藻糖、唾液酸等游离糖。在盛宴期,这些糖滋养共生菌;但在饥荒期,它们成了病原菌的“救命粮”。
鼠伤寒沙门氏菌(Salmonella Typhimurium)和艰难梭菌能够利用这些游离的唾液酸和岩藻糖迅速扩增。
策略三:GASP表型——长期饥饿,同类相食
如果饥荒持续的时间足够长(例如极端的节食或长期的饮食紊乱),细菌就不会仅仅满足于生理调节,而是会开启基因层面的进化。
强者通吃:残酷的 GASP 现象
文中提到了一个经典的微生物学现象:
在长期的营养限制下,细菌会发生基因突变,产生“长期静止期生长优势”表型(简称GASP)。
如果将已经饿了10天的大肠杆菌培养物,与仅培养了1天(刚进入静止期)的同种细菌混合,那些饿了10天的老细菌反而会竞争过并取代那些年轻的细菌。
激进策略:弃守为攻与同类相食
老细菌并不是单纯地在等死,而是通过 RpoS 基因突变采取了激进策略。它们主动放弃了原本用于防御(如耐热、耐酸)的盾牌以节省能量,转而全力强化进食能力。这让它们能掠夺环境中仅存的资源,比如说,大肠杆菌被证明能够利用死细胞释放的天冬氨酸、谷氨酸、甘氨酸、丝氨酸、苏氨酸来维持生存,从而在同类相食的竞争中,把那些还在老实防守的同伴彻底淘汰。
生态隐患
虽然这保证了某个物种的延续,但这种突变往往伴随着功能的丧失或毒力的增强。这意味着,经过长期饥饿筛选后的肠道菌群,可能在遗传上已经发生了漂变,肠道菌群变成了一个由一群“生存狂”组成的群落:它们为了活命可能丧失了原有的有益功能,甚至增强了毒力,对宿主不再那么友好。
人类不同的饮食习惯(如长期断食、热量限制、饮食结构改变)对菌群产生的共同影响
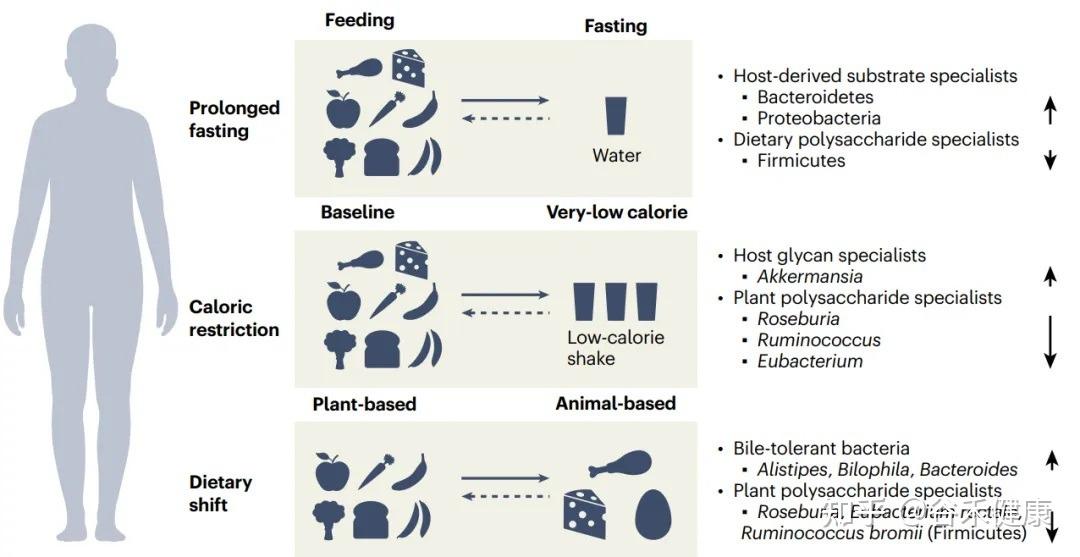
例如,当饮食结构从植物基转向动物基(素食转向肉食)时,会导致耐胆汁细菌的增加,同时那些专门负责分解植物多糖的细菌则会减少。
小结:微观机制的宏观映射
多样性下降是因为许多菌种无法承受反复的“休眠-复苏”能量损耗,或在竞争中落败。
病原菌(如沙门氏菌)的泛滥,是因为它们具备更灵活的代谢切换能力,能在纤维断供时迅速利用宿主黏液糖、琥珀酸等替代营养源。
饮食的规律性,本质上就是为肠道微生物提供一份安全感。消除了对迫近饥荒的恐惧,那些对宿主有益但相对脆弱的专性共生菌才能安心定植,繁衍生息,而不是被迫进入生存模式去啃食宿主的肠壁。
在解决了‘不饿着它们’这一基础问题后,我们需要深入探讨更深层的问题:如何通过特定的饮食成分,将肠道菌群从一个仅仅是活着的群体,引导为一个能为宿主提供健康的共生群落。
一旦细菌脱离了饥饿造成的极端环境,它们的功能重点就会从生存转向代谢。最新的综述研究指出,此时底物的类型(即你吃了什么)将决定菌群的代谢产出。

饮食习惯是肠道微生物群变异的主要决定因素,其影响力远超宿主遗传背景。换句话说,你每天吃进嘴里的食物,才是你肠道微生态的“总设计师”。
★ 一般性饮食模式
我们先来说一些的日常饮食习惯。
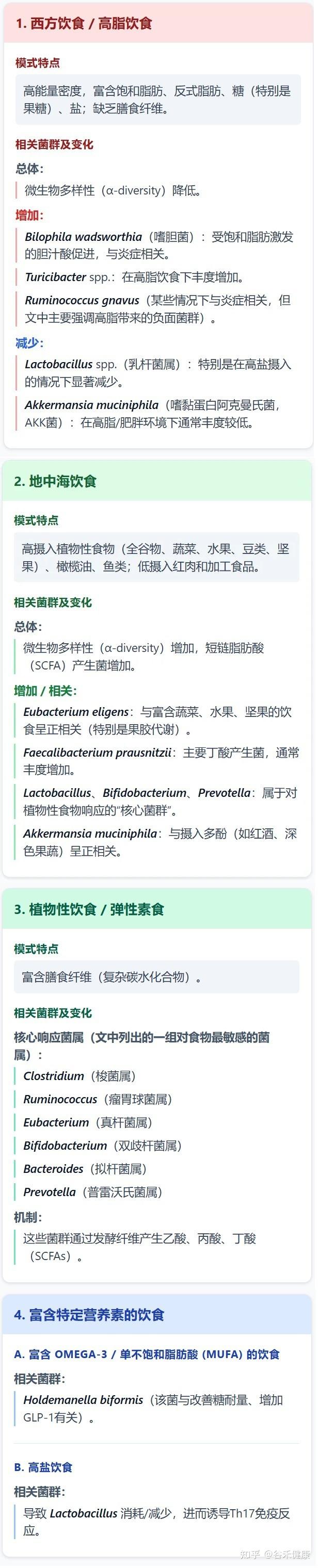
★ 治疗性/医学饮食模式
接下来我们来说一些饮食,通常用于特定疾病的干预。
膳 食 纤 维
在传统的营养学认知中,膳食纤维往往被简化为帮助通便的工具。但在微生物组的视角下,它的定义远不止于此。这是肠道共生菌最主要、甚至是赖以生存的能量来源。
从纤维到短链脂肪酸:点石成金的转化
膳食纤维进入结肠后,并不会像在小肠里那样被简单吸收,未被消化的膳食纤维被细菌发酵后,会产生一系列具有强大生物活性的代谢产物——短链脂肪酸(SCFAs),乙酸、丙酸和丁酸。
膳食纤维代谢产物在维护肠道屏障中的核心作用
-加固屏障,紧密连接
细菌产生的丁酸具有组蛋白去乙酰化酶(HDAC)抑制剂的功能。它能直接作用于肠上皮细胞的基因表达,上调紧密连接蛋白(如 Occludin 和 Claudin)的合成,好比给肠道细胞之间的缝隙抹上了强力胶水,防止细菌内毒素渗入血液。
-筑牢免疫防线(Treg 和 IL-22)
纤维发酵产物能与免疫细胞表面的G蛋白偶联受体(如 GPR43/GPR109A)结合。这种结合会向免疫系统发送和平信号,促进调节性T细胞(Treg)的分化,并刺激IL-22的分泌。IL-22 被称为屏障守护因子,它能加速上皮细胞的修复,并诱导细胞分泌抗菌肽(AMPs),将病原体拒之门外。
-降低环境 pH 值
发酵产生的酸性环境(低 pH)能有效抑制许多对酸敏感的致病菌(如沙门氏菌、大肠杆菌)的生长,这是一种最原始但有效的生态位竞争策略。
吃多少才够?
上一章节我们已经了解到,膳食纤维如果喂不饱菌群,那么菌群就会吃你的粘液层,肠道屏障就会随之薄弱,可能出现肠漏,低度慢性炎症等情况,那么吃多少才够呢?
目前的膳食指南通常建议每日摄入 25-30g 的膳食纤维。然而,Sanz 等人在综述中提出一个观点:这个标准可能仅仅是维持基本功能的底线,而非优化健康的理想值。
研究人员指出,为了达到预防非传染性慢性病(如心血管疾病、代谢综合征)的最佳效果,膳食纤维的摄入量可能需要提高到 35-50g/日。这个数字更接近人类祖先(狩猎采集时期)的摄入水平。
但这里有一个关键的注意事项:
对于肠道已经处于高度敏感或炎症状态(如 IBD 或 IBS 患者)的人群,突然摄入大量粗纤维可能会导致耐受不良。这也引出了精准营养的概念——不同人的菌群对纤维的处理能力是不同的。
因此,策略不是盲目地吃草,而是循序渐进,并注重纤维种类的多样性(可溶性与不可溶性纤维的搭配),以喂养更广泛的共生菌群。

蛋 白 质
在健身房里,蛋白质是增肌的必需品;但在结肠深处,蛋白质的命运却充满了变数。
与膳食纤维的健康益处不同,蛋白质对肠道微生物组的影响是双向的。这取决于两个关键因素:蛋白质的来源,以及你是否同时摄入了足够的碳水化合物。
当有益菌遇到色氨酸:免疫系统的调停者
首先,我们来看看蛋白质天使的一面。
蛋白质由氨基酸组成,比如说,色氨酸(Tryptophan),是连接饮食、微生物和宿主免疫的关键桥梁。
肠道细菌(如乳杆菌属和部分梭菌属)能够将未消化的色氨酸代谢为吲哚(Indole)及其衍生物(如吲哚-3-丙酸 IPA、吲哚-3-乙醛等)。这些吲哚类物质并非废料,而是高级的生化信使,它们可以:
从这个角度看,适量的蛋白质摄入,特别是富含色氨酸的食物,是维持肠道免疫稳态的重要一环。

黑暗面:当发酵转向“腐败”
当膳食纤维摄入不足,或者蛋白质摄入过量导致大量未消化的蛋白质进入结肠(尤其是远端结肠)时,微生物的代谢模式会发生危险的逆转。
在这个过程中,细菌会产生一系列具有细胞毒性的代谢产物:
这种“腐败发酵”主要发生在远端结肠,那里是碳水化合物最先被耗尽的地方,也是结直肠癌的高发部位。这非巧合。
关键在于“碳氮比”:别让细菌只吃肉
那么,我们该如何避免蛋白质的“黑化”?
研究人员认为,碳水化合物(纤维)的存在可以抑制蛋白质的腐败发酵。
这是一种名为分解代谢抑制的生存策略:只要有容易发酵的碳水化合物存在,细菌就会优先利用它们,而此时产生的酸性环境会进一步抑制蛋白水解酶的活性。
因此,问题的核心往往不在于肉吃多了,而在于菜吃少了。
植物蛋白 vs 动物蛋白
相比于红肉(富含血红素铁,可能催化致癌物 N-亚硝基化合物的形成),植物蛋白通常自带膳食纤维。这种自带解药的属性,使得植物蛋白在被细菌分解时,往往伴随着更有益的代谢环境。

肠道菌群与蛋白质代谢14 赞同 · 3 评论 文章

动物蛋白与植物蛋白对肠道微生态的不同“改造”与健康风险”6 赞同 · 2 评论 文章
脂 肪
与纤维(作为食物)不同,脂肪更多是通过改变肠道环境(特别是胆汁酸代谢)来间接筛选微生物。
长期以来,我们对脂肪的恐惧主要集中在变胖和心血管堵塞。但在微观视角下,高脂饮食对健康的打击是从肠道屏障的崩塌开始的。
这其中牵涉到两个核心机制:
胆汁酸的改变 与 内毒素的易位。
高脂饮食的代价:当助消化的胆汁变成致癌毒素
当你吃下一块肥美的五花肉,肝脏会立刻分泌胆汁酸(Bile Acids)来乳化这些油脂,帮助消化吸收。这本是正常的生理过程。
然而,问题在于量。
长期摄入高脂肪饮食,会迫使肝脏源源不断地向肠道泵入大量的初级胆汁酸。当这些胆汁酸流经小肠未被完全回收,进入结肠后,它们就落入了微生物的手中。

肠道细菌(如梭菌属)通过7α-脱羟基作用会将初级胆汁酸进行二次加工,转化为次级胆汁酸,如脱氧胆酸(DCA)和石胆酸(LCA)。
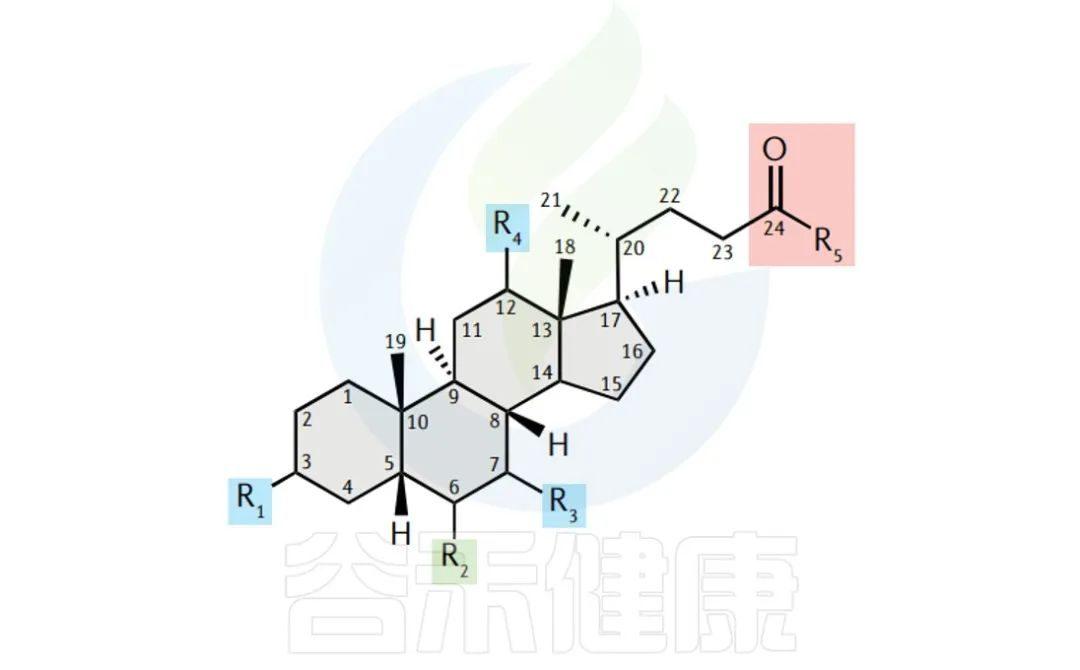
什么是胆汁酸,其与肠道微生物互作如何影响人体健康18 赞同 · 0 评论 文章
次级胆汁酸的蓄积,是一个危险的信号:
当然,次级胆汁酸也有另一面——某些特定的衍生物(如 isoalloLCA)能特异性地抑制艰难梭菌等病原体。
但在典型的高脂“西方饮食”模式下,负面效应往往占据了主导地位。
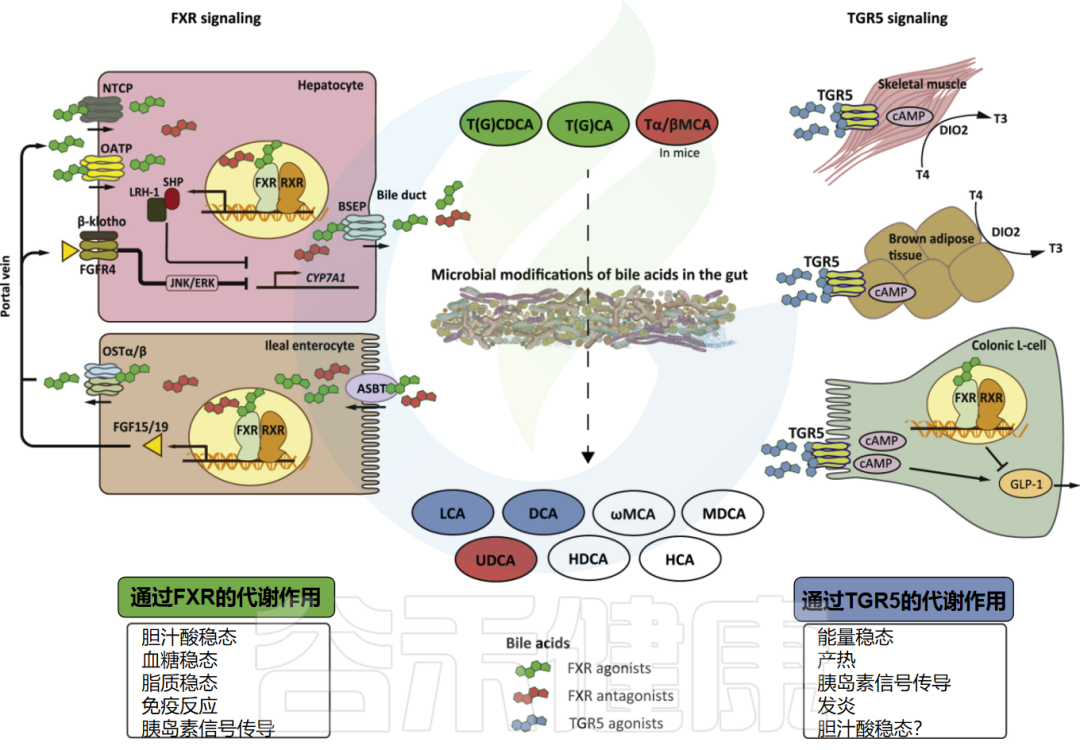
肠道微生物群对胆汁酸代谢和信号传导影响的最新研究成果20 赞同 · 1 评论 文章
脂肪的类型(质量)远比数量重要
研究对比了饱和脂肪酸(如猪油、棕榈油)与多不饱和脂肪酸(PUFAs,如鱼油、亚麻籽油):
-饱和脂肪:代谢性内毒素血症
摄入过多的饱和脂肪,不仅会降低微生物的多样性,更关键的是它充当了细菌毒素的载体。
革兰氏阴性菌的细胞壁成分——脂多糖(LPS,即内毒素),极易溶解在脂肪微粒(乳糜微粒)中。当饱和脂肪穿过肠壁进入血液时,它会顺手把 LPS 也带进去。
这就是著名的代谢性内毒素血症。血液中微量的 LPS 升高,会触发全身性的低度慢性炎症,诱导胰岛素抵抗,成为肥胖和糖尿病的隐形推手。
-Omega-3
相反,富含 Omega-3 的多不饱和脂肪酸,被证明能够增加产丁酸菌丰度,逆转由饱和脂肪引起的菌群失调,并有助于维护肠道屏障的完整性。
探索Omega-3脂肪酸:健康益处与营养补充12 赞同 · 0 评论 文章
维 生 素
我们常以为维生素只是为了身体好,其实肠道里的细菌也是维生素大户。
许多有益菌(如双歧杆菌)虽然能为我们合成维生素K和部分B族维生素,但它们自身往往无法从头合成生长所需的全部维生素(尤其是B族),必须依赖我们的饮食摄入或原本的交叉喂养网络。
B2(核黄素)
当饮食中缺乏维生素B2(核黄素)时,肠道中产丁酸菌(如普拉梭菌)就会因缺乏口粮而减少,而这可能间接给大肠杆菌等潜在有害菌腾出生存空间。
B9(叶酸)与 B12(钴胺素)
乳杆菌和双歧杆菌通常缺乏从头合成叶酸和钴胺素的完整酶系,属于维生素营养缺陷型微生物。必须依赖宿主摄入。如果叶酸摄入不足,这些益生菌的生长就会受限,进而削弱其对肠道微生态的调节功能。
B3(烟酸)
烟酸不仅是细胞能量代谢(NAD+合成)的关键前体,还是结肠上皮及免疫细胞表面 GPR109A 受体的特异性配体。烟酸通过激活该受体发挥抗炎作用,抑制促炎因子的释放。
缺乏烟酸会导致肠道免疫耐受性降低,增加结肠炎易感性,并伴随微生物群落Alpha 多样性的显著下降及微生态结构的崩塌。严重缺乏会导致糙皮病。
B7(生物素)
生物素是细菌羧化酶不可或缺的辅助因子,在菌群的种间竞争中起着关键作用。研究显示,在生物素缺乏的环境下,某些机会性致病菌(如鼠乳杆菌Lactobacillus murinus)会利用代谢优势发生异常增殖,导致严重的菌群失调;而恢复生物素水平则能有效遏制这种过度扩张,通过生态位竞争维持肠道微生态的稳态。

B族维生素与肠道菌群互作52 赞同 · 1 评论 文章
维生素A
维生素 A 的代谢产物(视黄酸)是调节肠道黏膜免疫的关键信号。它能直接促进 B 细胞分化,上调分泌型免疫球蛋白 A (sIgA) 的合成与分泌。sIgA 能够特异性地包裹细菌,在阻止致病菌黏附上皮组织的同时,协助乳杆菌(Lactobacillus) 等共生菌在黏膜表面的稳定定植,防止菌群易位。
维生素D
维生素 D 通过激活肠上皮细胞中的维生素 D 受体 (VDR),上调紧密连接蛋白(如 Occludin 和 Zonulin)的表达,从而维持肠道屏障的物理完整性。
此外,维生素D还能发起主动进攻,它能诱导宿主分泌防御素和组织蛋白酶等抗菌肽。这种选择性压力有助于重塑菌群结构,研究显示其可显著促进Akkermansia muciniphila的生长,并抑制变形菌门等潜在致病菌。
维生素 C & E:调节氧化还原电位
肠道核心菌群(如厚壁菌门和拟杆菌门)多为专性厌氧菌,对氧气极为敏感。维生素 C 和 E 具有显著的抗氧化作用,能够清除肠腔内的活性氧 (ROS),维持肠道较低的氧化还原电位。这种厌氧环境有利于核心有益菌的代谢与生存,同时限制肠杆菌科(多为兼性厌氧菌)等机会性致病菌的过度增殖。
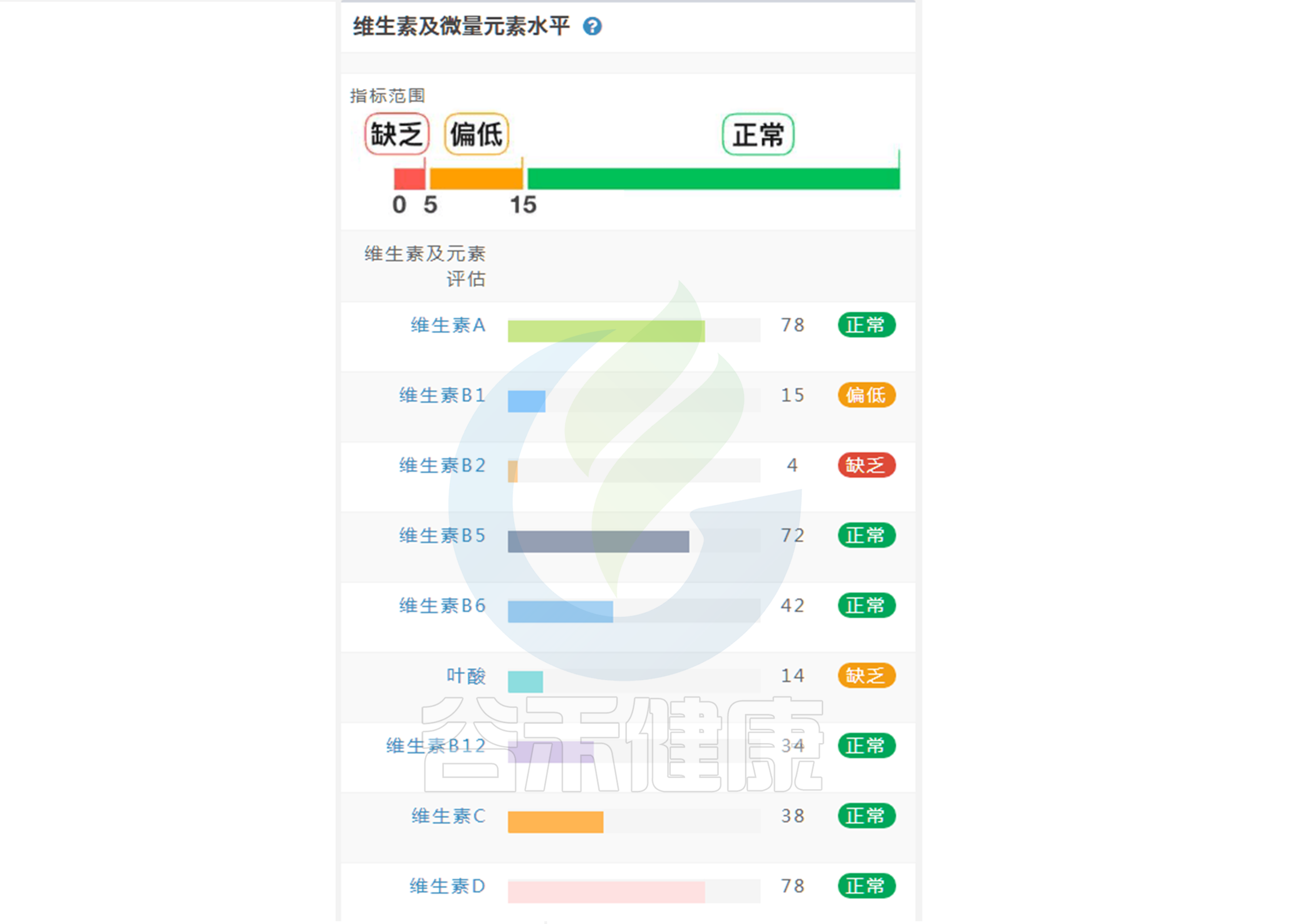
维生素摄入水平肠道菌群可以告诉你14 赞同 · 0 评论 文章
下表列出了常见的维生素的食物来源,可供参考。

矿 物 质
铁
铁是细菌界的稀缺资源。正常情况下,人体会限制肠道内的游离铁(营养免疫),防止细菌疯长。
但问题在于,沙门氏菌和致病性大肠杆菌等肠杆菌科细菌,进化出了极强的“抢铁工具”——铁载体(Siderophores)。相比之下,乳杆菌等有益菌抢夺铁的能力很弱。
人与菌对铁的竞争吸收 | 塑造并控制肠道潜在病原菌的生长15 赞同 · 0 评论 文章
因此,一旦结肠中出现未被吸收的游离铁(比如吃太多红肉或过量补铁),这些铁就会优先被致病菌抢走,助长它们爆发性繁殖,从而压制有益菌并引发炎症。

锌
锌不仅能加固肠道屏障,更是维持菌群结构稳定的关键。
研究发现,缺锌会直接导致肠道中的主力军——厚壁菌门的数量大幅下降。这种多样性的丧失,意味着肠道的定植抵抗力被削弱了。

膳食锌缺乏或过量对人体肠道菌群及健康的影响11 赞同 · 1 评论 文章
这就好比城墙上的守卫变少了,原本被压制的艰难梭菌就会趁虚而入,利用空出来的生态位大量繁殖并分泌毒素,引发生严重的肠道感染。

这种微观层面的营养不良,往往是导致菌群失调和系统性炎症的隐蔽开端,也进一步凸显了通过高质量饮食纠正营养短板的紧迫性。
前面关注了“吃什么”,“吃多少”,接下来我们来看第三个维度:进食时机。
人体内存在两套主要的生物钟系统:
微生物也会“倒时差”
你可能不知道,肠道菌群也是分早班和晚班的。肠道微生物的组成和功能会呈现出显著的昼夜震荡。
这种节律性波动是如此重要,以至于产生的代谢物(如我们前面提到的短链脂肪酸、胆汁酸衍生物)在一天中的浓度也是波动的。这种波动指导着宿主的肝脏何时储存脂肪、何时释放葡萄糖。
现代生活方式:“光”与“食”错位
当我们在深夜(大脑认为是休息时间)吃一顿宵夜(肠道接收到工作信号)时,中枢生物钟和外周生物钟就发生了脱钩。
这种时钟错位会导致肠道微生物节律的丧失。原本应该在休息期清理毒素的细菌被迫加班处理食物,不仅效率低下,还会引发代谢混乱。这被认为是倒班工作者更容易患肥胖、糖尿病和肠道炎症的核心机制之一。
限时进食(TRE):重启肠道节律
限时进食(Time-Restricted Eating, TRE)——即把每天的进食窗口限制在 8-10 小时内,其余时间禁食。
这不仅仅是减少热量摄入的手段,更是一种微生物重塑策略。
–恢复昼夜节律
TRE 能够强力恢复微生物群落的昼夜节律震荡。让不同的细菌在正确的时间做正确的事,重建宿主代谢通路与微生物活动的同步性。
-特定功能菌群的富集
延长的禁食窗口为特定共生菌提供了生态位优势。
研究显示,禁食期间,以AKK菌(Akkermansia muciniphila)为代表的黏液降解菌丰度显著增加。这类菌群利用内源性黏蛋白进行增殖,反向刺激杯状细胞分泌黏液,从而强化肠道屏障功能,减少内毒素血症风险。
-代谢保护
动物模型和人类研究,表明 TRE 可以预防由高脂饮食引起的肥胖和代谢综合征。哪怕摄入的总热量没有改变,仅仅是改变进食的时间窗口,就能显著改善血糖稳态。
对于现代人来说,最糟糕的饮食习惯可能是进食时间的不规律。今天不吃早饭,明天半夜撸串,这种行为让肠道微生物始终处于“倒时差”的应激状态中。

因此,为了维护肠道健康,除了多吃膳食纤维、少吃加工食品外,我们还需要给微生物一个明确的作息表:在白天的活跃期进食,在夜晚的休息期禁食。这不仅是对身体的尊重,也是对居住在你体内的万亿微生物生命的尊重。
小结:从填饱肚子到精准滋养微生态
热量达标不代表菌群吃饱。必须保证膳食纤维的充足供应。这是防止菌群因底物匮乏而倒戈吞噬肠道黏膜的第一道防线。
蛋白质与脂肪对菌群的影响,可能取决于纤维的在场。控制饱和脂肪摄入,选择优质脂肪,能减少次级胆汁酸对有益菌的杀伤,避免内毒素血症。
均衡的微量营养素摄入,比如维生素和矿物质,能精准强化共生菌的竞争优势,助其在生态位争夺战中抢占先机,从而有效压制机会致病菌的野蛮生长。
当我们站在微生物生态学的前沿回望,传统的“金字塔式”饮食建议显得过于粗糙且缺乏维度。Singh等人的数据警示了“饮食大波动有风险”,Estrela等人的机制揭示了“细菌怕长期挨饿”,而Sanz等人的综述则描绘了通过饮食调节菌群的多维度健康蓝图。
这就要求我们必须升级我们的营养系统,从单纯关注成分转向关注生态稳定性与精准化。
新时代的营养观
对于普通大众而言,建立一个基线饮食节奏至关重要。
建立核心菜单
不要让每一顿饭都成为随机事件。尝试确立每天固定的核心食物,例如早餐固定的全谷物燕麦,或晚餐固定的深色蔬菜。这能确保护肠道内的核心共生菌每天都能获得稳定的底物供应,避免其因“断粮”而进入休眠或衰退。
平滑过渡
当你试图改变饮食习惯(比如从高碳水转向低碳水,或者开始吃地中海饮食)时,切忌急刹车或急转弯。根据 Estrela等人的理论,剧烈变换的底物会诱发细菌的饥饿应激和黏液降解。更合适的做法是循序渐进,给微生物群落留出数周的适应期,让代谢网络慢慢重塑。
精准营养 2.0:整合微生物组
每个人的肠道菌群构成都是独一无二的,这意味着同样的食物在不同人体内会引发截然不同的微生物反应(例如,不同人吃同样的香蕉,血糖反应可能截然不同)。未来的营养干预将不再是千篇一律的指南,而是根据人群的代谢表型或微生物特征,提供分层建议。
肠道菌群监测
比如说核心菌群,如厚壁菌门/拟杆菌门比值、产丁酸菌比例等指标,建立个人化菌群基线。
长期的饮食习惯会在菌群上留下印记。例如,长期缺乏膳食纤维的人,其肠道内负责降解植物多糖的酶系可能已经退化。此时若盲目大量补充纤维,不仅无法被利用,反而可能导致腹胀不适。这时肠道菌群数据能提示我们,可能需要采用缓慢饮食过渡方案,给菌群以适应和重建的时间。
靶向性补充
针对特定的功能缺失进行干预。例如,如果发现黏液降解菌(如 Akkermansia)异常升高,这往往是肠道缺乏膳食纤维、菌群正在吃内源性黏液的信号。可能不仅要补充益生菌,更需精准增加抗性淀粉、菊粉等摄入,以提供替代碳源,保护肠道屏障。
拥抱时间营养学
我们不仅要问吃什么,还要问什么时候吃。微生物组具有自身的昼夜节律,这种节律受宿主进食时间的强烈驯化。
保持规律的进食时间,避免深夜进食
这能让微生物群的代谢活动与宿主的生理时钟同步,优化代谢产物的分泌时机,使其在宿主最需要的时候(如日间活动时)发挥作用。
避免长期的随机断食
间歇性断食在某些情况下有益,但无序的、饥一顿饱一顿的“随机断食”是细菌的噩梦。如果你选择断食,请保持断食窗口的规律性,让细菌能够建立起预期的代谢循环。
保持饮食的节奏,不仅仅是一种自律的生活态度,更是一种对生命的敬畏。维护肠道内的和平与稳定,我们实际上是在守护自身健康的根基。正如研究所示,每日稳定摄入蔬菜、水果等富含纤维的食物,远比间断性大量食用更能维持菌群多样性。
在这个充满不确定性的世界里,科学的节奏与精准的滋养,或许就是你能给身体最好的确定性。
主要参考文献
Estrela S, Long JZ, Huang KC. How nutrient starvation impacts the gut microbiome. Nat Microbiol. 2025 Nov;10(11):2663-2672. doi: 10.1038/s41564-025-02139-9. Epub 2025 Sep 30. PMID: 41028229.
Singh R, McDonald D, Hernandez AR, Song SJ, Bartko A, Knight R, Salathé M. Temporal nutrition analysis associates dietary regularity and quality with gut microbiome diversity: insights from the Food & You digital cohort. Nat Commun. 2025 Sep 30;16(1):8635. doi: 10.1038/s41467-025-63799-z. PMID: 41028733; PMCID: PMC12484809.
Sanz Y, Cryan JF, Deschasaux-Tanguy M, Elinav E, Lambrecht R, Veiga P. The gut microbiome connects nutrition and human health. Nat Rev Gastroenterol Hepatol. 2025 Aug;22(8):534-555. doi: 10.1038/s41575-025-01077-5. Epub 2025 Jun 4. PMID: 40468006.

谷禾健康
氨基酸(AA)稳态对人体健康至关重要,其紊乱与多种疾病的发生和进展密切相关,如2型糖尿病和炎症性肠病(IBD),并且常常是治疗结果的决定性因素。
肠道微生物群可能通过多种机制调节宿主氨基酸的可用性,例如影响消化酶(如胰蛋白酶)的活性。肠道微生物群定殖还可能改变肠道通透性,从而影响胃肠道中游离氨基酸的运输和吸收。此外,肠道微生物可能直接利用或代谢肠道中的氨基酸,或合成并向宿主提供氨基酸。以往研究表明,肠道微生物组的变化(或其存在)可能影响肠道氨基酸谱。然而,参与调控宿氨基酸稳态的关键菌株及代谢基因仍未完全明确。
今天分享的这一篇发表在《Cell Host & Microbe》期刊上的研究论文”Microbiota metabolism of intestinal amino acids impacts host nutrient homeostasis and physiology”,通过比较Met无菌(GF)小鼠与无特异病原体(SPF)小鼠的分解物谱已证明,肠道细菌定殖会影响胃肠道(GI)中游离氨基酸谱,还通过高效代谢肠道氨基酸来重塑宿主氨基酸的格局。为确定责任微生物/基因,该研究开发了基于代谢组学的检测法,筛查了104个共生菌,并识别出能高效利用氨基酸的候选基因,并发现这些基因调控了肠道和循环氨基酸的可用性。
结果显示,不同氨基酸具有特定的细菌消费者,消耗效率差异明显。例如,天冬酰胺和谷氨酸被拟杆菌和部分梭菌高效利用,而芳香族氨基酸(色氨酸、酪氨酸)及支链氨基酸(BCAAs)仅为部分厚壁菌门成员所代谢,效率较低。值得注意的是,支链氨基酸与色氨酸代谢相关的微生物基因还能通过调节外周血清素间接影响宿主葡萄糖平衡。
总体而言,该研究首次系统揭示了肠道微生物群编码的氨基酸代谢活动对宿主营养稳态的深远影响,为理解微生物介导的氨基酸利用及其对宿主代谢调控机制提供了关键分子依据。
氨基酸稳态对人类健康至关重要,其紊乱与2型糖尿病和炎症性肠病(IBD)等多种疾病的进展密切相关。虽然传统上认为肠道和肝脏是调控营养代谢的主要器官,但最新研究发现,肠道微生物群通过代谢氨基酸能够深刻重塑宿主的氨基酸谱。
★该研究的核心意义在于:
•揭示了肠道微生物群通过代谢肠道氨基酸影响宿主氨基酸稳态的分子机制;
•鉴定了高效消耗氨基酸的特定肠道微生物及其代谢基因;
•阐明了微生物群氨基酸代谢基因如何通过外周血清素调节宿主葡萄糖稳态;
•为通过调节肠道微生物群代谢活性改善人类健康提供了新的靶点和策略。
▸ 肠道微生物群对宿主氨基酸水平的影响
研究首先通过比较无菌(GF)小鼠、无特定病原体(SPF)小鼠以及接受SPF微生物群移植(FMT)的GF小鼠,证实了肠道微生物群定植显著降低了肠道和循环氨基酸水平。
靶向代谢组学分析显示,SPF小鼠或接受SPF微生物群的GF小鼠肠道和循环氨基酸浓度普遍降低,这与之前的研究结果一致,表明微生物对肠道氨基酸的利用可能是影响宿主氨基酸稳态的主要途径之一。
▸ 高效代谢氨基酸的肠道微生物筛选
为识别高效代谢肠道氨基酸的微生物,研究者建立了一种基于活细胞的高通量代谢组学筛选方法,对104种肠道共生菌进行了系统分析。该方法综合考虑多个因素:
•生长阶段差异:绘制各菌株生长曲线并进行参数拟合,确保在可比生长阶段进行筛选;
•时间动态分析:分别在15、30和60分钟测定氨基酸消耗量,以精确评估不同菌株对特定氨基酸的利用速率;
•营养条件优化:在测定缓冲液中除氨基酸外,补充无机氮源、微量矿物质、维生素及额外碳源(如葡萄糖),以避免因营养限制导致的氨基酸非特异性快速消耗。
研究流程图展示了从识别消耗氨基酸的肠道微生物、鉴定负责的代谢基因、单基因水平的体内调节到对葡萄糖稳态影响的完整研究路径。
▸ 微生物代谢基因的鉴定与功能验证
研究利用CRISPR-Cas9和ClosTron等基因编辑技术,在系统发育多样的肠道微生物中开展大规模基因缺失分析,以鉴定氨基酸代谢的关键基因。筛选标准包括:
1.代谢基因编码的酶以氨基酸为直接底物;
2.这些基因存在于高效代谢氨基酸的肠道菌株中;
3.携带相关基因的菌株能稳定定植于无菌(GF)小鼠肠道,并降低宿主体内相应氨基酸水平。
通过构建基因敲除和互补菌株并结合代谢组学分析,研究识别出多种氨基酸代谢相关基因,并在单菌定植的GF小鼠模型中验证了其对肠道及血清氨基酸水平的调控作用。
▸ 高效代谢氨基酸的肠道微生物鉴定
通过大规模筛选,研究发现不同肠道微生物对氨基酸的代谢效率和偏好存在显著差异:
鉴定高效利用氨基酸的肠道微生物候选者
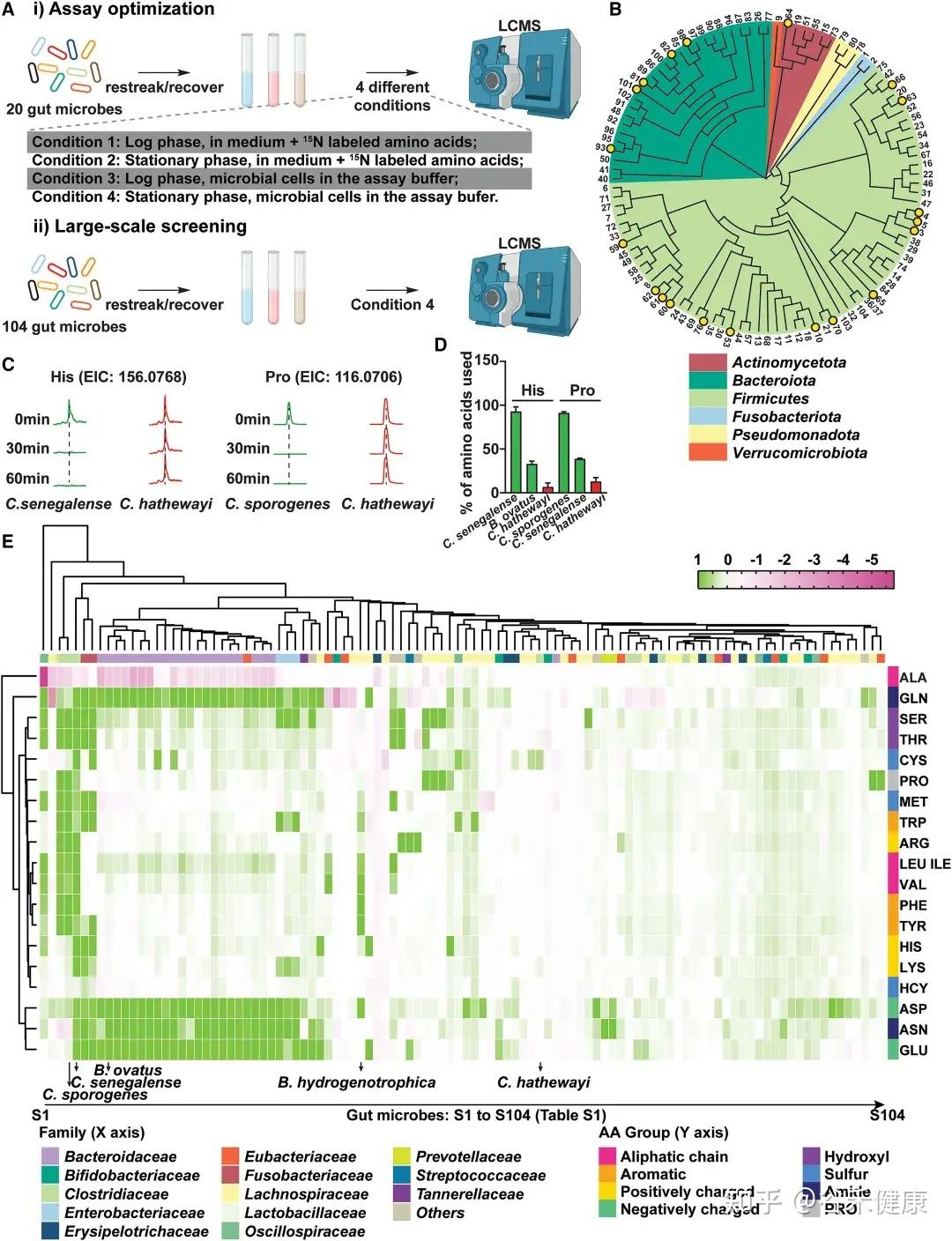
热图展示了104种肠道微生物对不同氨基酸的代谢情况,揭示了微生物系统发育与氨基酸代谢效率之间的复杂关系。
★ 关键发现包括:
•梭菌属微生物:如Clostridium sporogenes ATCC15579和Clostridium senegalense DSM25507,可高效代谢多种氨基酸,包括精氨酸、芳香族氨基酸、支链氨基酸、组氨酸、赖氨酸和脯氨酸等。
•拟杆菌属共生菌:作为肠道多糖的主要利用者,也能有效代谢天冬酰胺(Asn)、天冬氨酸(Asp)和谷氨酰胺(Gln)等氨基酸。
•氨基酸特异性代谢:不同氨基酸具有特定的细菌“消费者”,消耗效率差异明显。天冬酰胺和谷氨酸通常被拟杆菌和部分梭菌快速利用,而芳香族氨基酸(色氨酸、酪氨酸)及BCAA仅被部分厚壁菌门或梭菌较低效地代谢。
▸ 氨基酸代谢相关微生物基因的鉴定
通过对高效代谢氨基酸的肠道微生物进行大规模基因缺失分析,研究鉴定出多种关键氨基酸代谢基因。这些基因编码的酶包括转氨酶、脱羧酶、消旋酶和氨裂解酶等,具有以下特征:
•功能冗余性:单株细菌常含多个具有相似功能的基因。例如,C. sporogenes 携带两种精氨酸脱亚胺酶基因(adiA 和 adiB),单基因敲除影响较小,而双敲除可完全消除其精氨酸代谢能力。
•底物特异性:代谢酶展现高度底物选择性。例如,C. sporogenes 的转氨酶 ArtA 特异作用于色氨酸和酪氨酸,但对苯丙氨酸代谢影响极小。
•催化多样性:不同类型的酶参与多条代谢途径。支链氨基酸和色氨酸经转氨途径代谢,脯氨酸通过还原反应被 C. sporogenes 利用,而谷氨酸、甲硫氨酸、丝氨酸和苏氨酸则由氨裂解酶快速分解。
•功能可转移性:这些酶的催化功能具有可转移性,在其他肠道共生菌中表达其编码基因可赋予相应氨基酸的消耗能力。
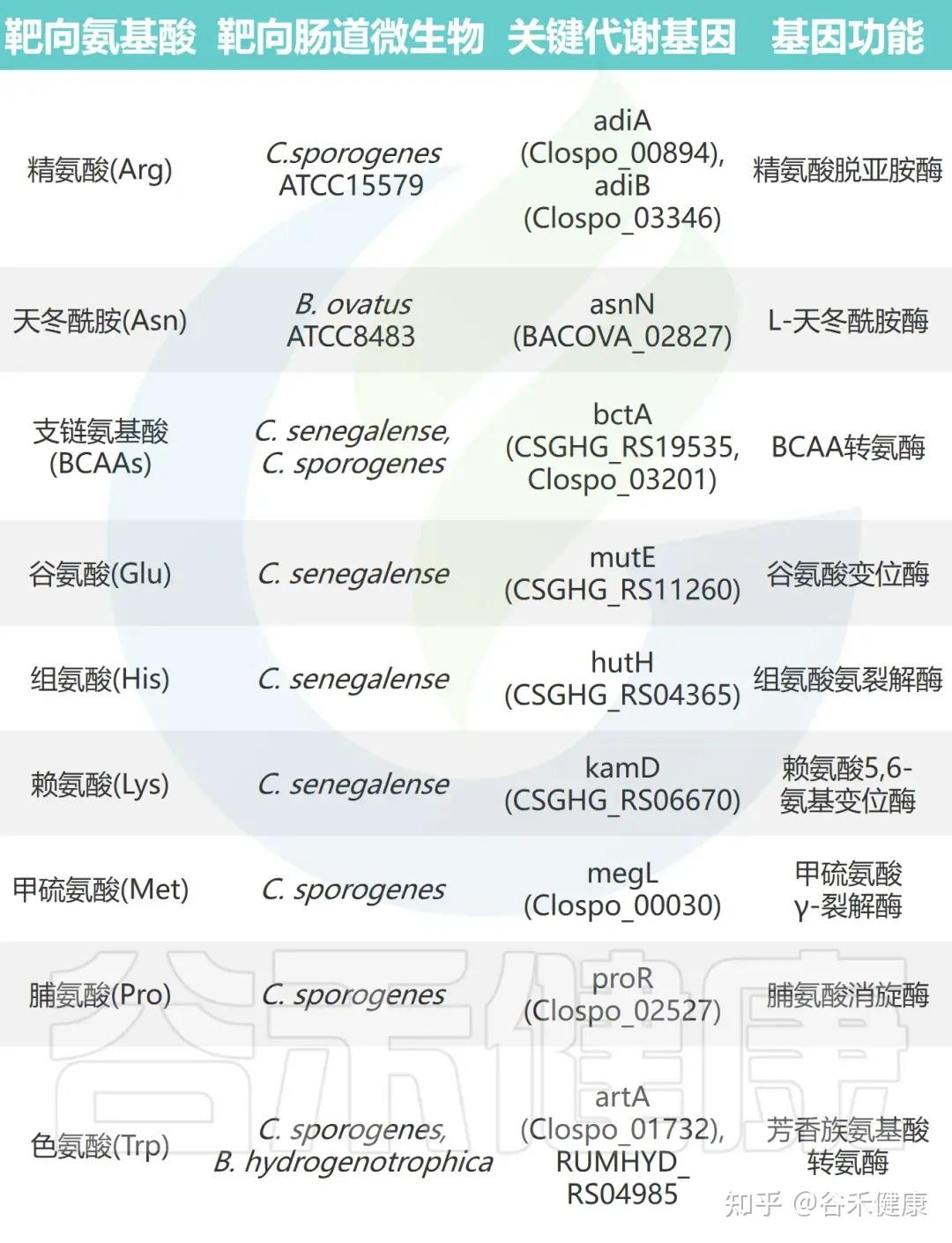
▸ 微生物代谢基因调控宿主氨基酸稳态
通过将野生型菌株及其代谢基因缺失突变体分别单菌定植于GF小鼠,研究发现这些基因显著影响宿主肠道及循环系统中的氨基酸水平:
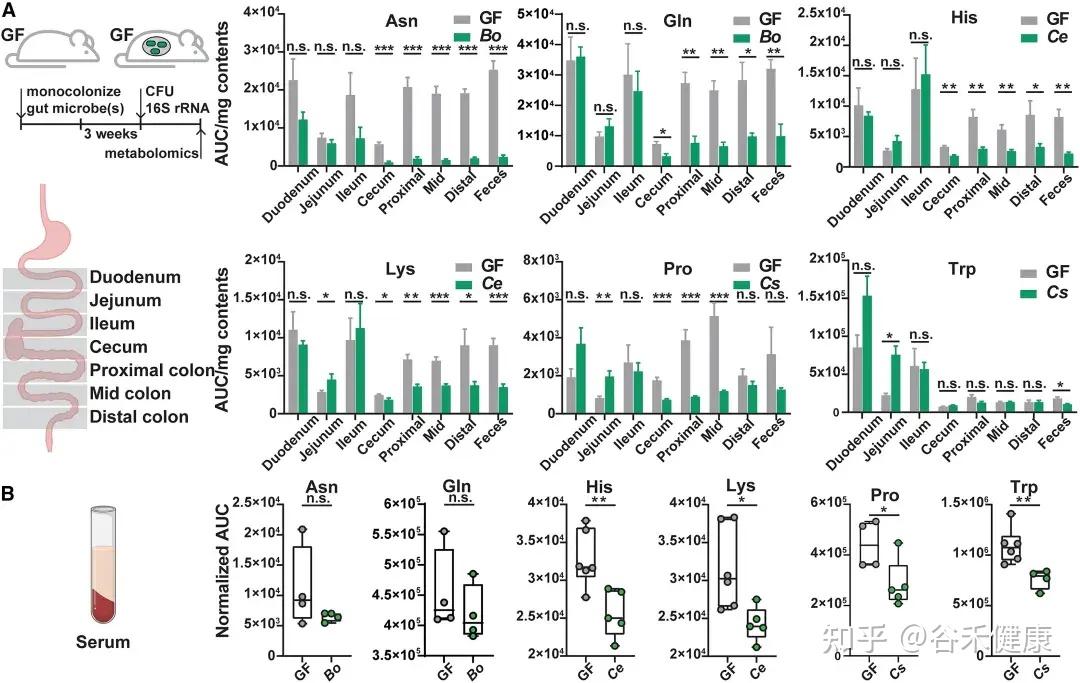
无菌小鼠单一定植B. ovatus、C. senegalense和C.sporogenes野生型菌株后,肠道和血清中特定氨基酸浓度显著降低。
★ 主要发现包括:
•肠道氨基酸稳态调控:多数鉴定出的微生物代谢基因或酶影响肠道氨基酸平衡。由于突变体的相应代谢途径被阻断,肠道内容物和粪便中相关氨基酸显著积累。
•循环氨基酸谱变化:部分代谢基因还影响宿主血清氨基酸水平。定植 Asn、Leu/Ile、Pro 和 His 利用缺陷突变体的无菌小鼠,其血清中对应氨基酸明显升高。
•系统性调节的复杂性:循环氨基酸水平的变化并不总与微生物在肠道中的代谢能力一致。例如,定植 C. sporogenes 精氨酸利用缺陷突变体可显著提高肠道精氨酸水平,但对循环精氨酸影响有限,提示肠上皮或肝脏代谢等额外机制亦参与精氨酸的全身稳态调控。
单基因水平的微生物群氨基酸代谢调控
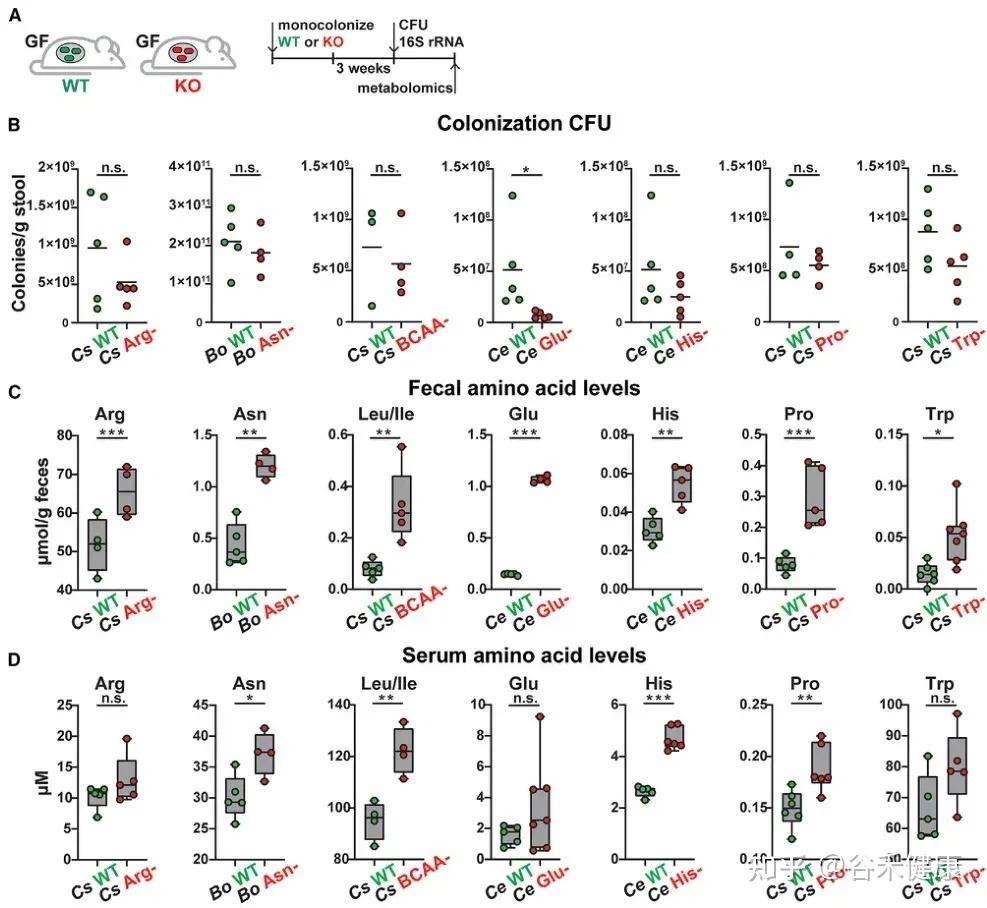
单基因水平的体内调节实验显示,氨基酸代谢基因缺陷突变体定植的无菌小鼠粪便和血清中相应氨基酸水平显著升高。
▸ 微生物氨基酸代谢通过外周血清素调控宿主葡萄糖稳态
研究发现,微生物中支链氨基酸(BCAAs)和色氨酸代谢相关基因可通过外周血清素间接调节宿主葡萄糖稳态。将 C. sporogenes 野生型、BCAA 缺陷型及色氨酸缺陷型突变体分别单菌定植于 GF 小鼠,并进行口服葡萄糖耐量试验(OGTT),结果显示以下关键发现:
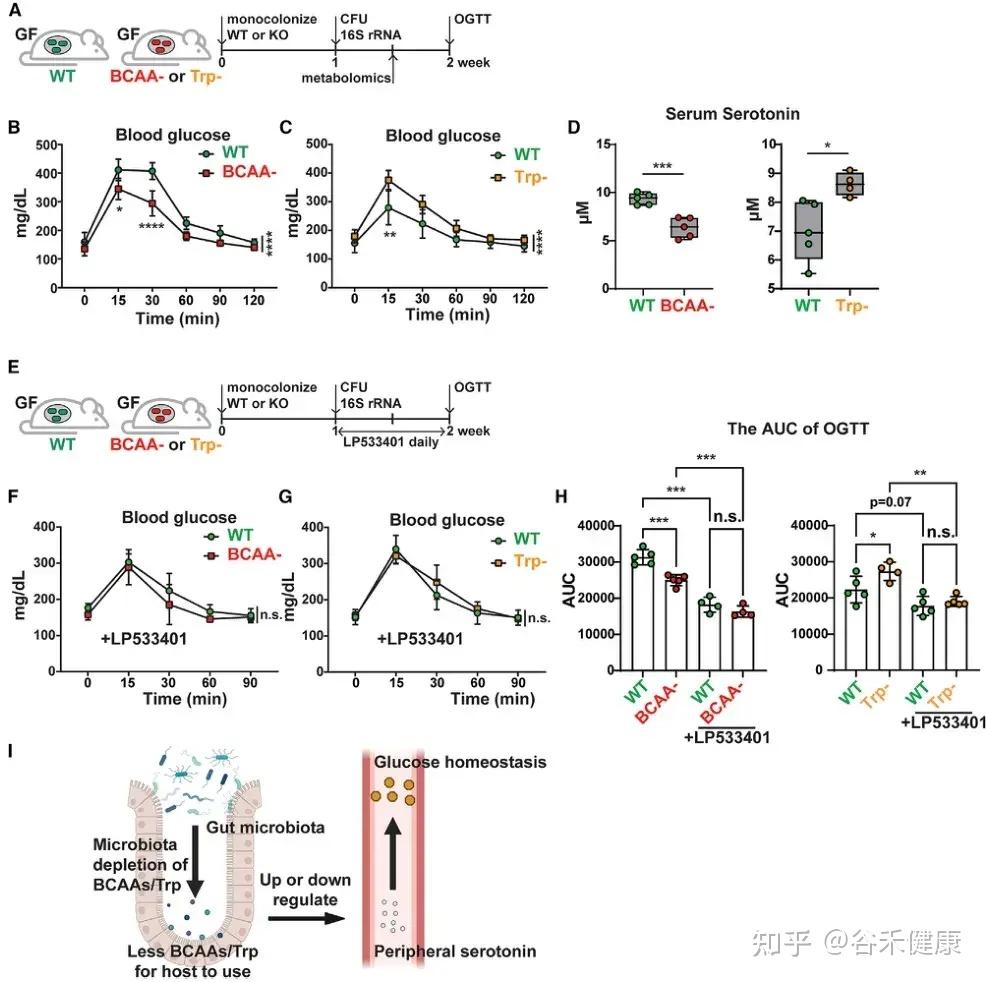
微生物支链氨基酸和色氨酸代谢通过调节外周血清素水平影响宿主葡萄糖稳态的机制示意图及实验验证。
•BCAA代谢与葡萄糖耐受性:定植 BCAA 缺陷突变体的小鼠葡萄糖耐受性显著增强,伴随血清素水平下降。
•色氨酸代谢与葡萄糖耐受性:定植色氨酸代谢缺陷突变体的小鼠表现相反,葡萄糖耐受性下降,血清素水平升高。
•血清素介导机制:应用 Tph1 抑制剂 LP533401 阻断外周血清素合成后,野生型与突变体定植小鼠间的葡萄糖处理差异显著减小,证实微生物氨基酸代谢通过调控外周血清素影响宿主葡萄糖稳态。
这一发现揭示了肠道微生物群、氨基酸代谢、外周血清素及葡萄糖稳态间的复杂关联,为理解代谢性疾病的机制提供了新思路。
!
研究局限性
尽管该研究取得了重要发现,但仍存在一些局限性:
1.微生物氨基酸代谢的多样性:研究主要关注肠道微生物对氨基酸的消耗,而其合成、转运等途径也可能影响宿主氨基酸稳态。体外测定仅反映特定条件下的代谢效率,难以全面代表其在宿主体内的作用。
2.模型简化的局限性:研究采用单菌定植的 GF 小鼠模型,未来应在更接近生理状态的模型(如定植简化微生物群的 GF 小鼠)中验证这些机制。
3.复杂微生物群中基因操作的挑战:虽然本研究成功在宿主定植背景下操纵单一氨基酸代谢途径,但在复杂微生物群中实现精确基因操作仍需进一步技术突破。
4.宿主响应机制研究不足:研究揭示了野生型与突变体共生菌定植小鼠间的生理差异,但尚需深入解析相关宿主细胞类型及信号通路的具体调控机制。
未来的研究在单基因分辨率下揭示了肠道微生物群如何通过代谢肠道氨基酸影响宿主氨基酸稳态。研究鉴定出多种积极代谢肠道氨基酸的细菌,并通过大规模基因缺失分析确定了其关键代谢基因。功能实验显示,这些代谢途径通过直接调控宿主肠道和循环氨基酸的生物利用度,或间接影响外周血清素合成,从而调节宿主的葡萄糖代谢和整体营养稳态。
研究表明,定植微生物群可能以“竞争”模式与宿主共同调控营养平衡,为理解其分子层面的调节机制提供了新视角。更值得关注的是它揭示了一个新的通路:微生物氨基酸利用 → 改变宿主血中氨基酸/代谢产物 → 改变激素/神经递质(如外周 5‑HT)→ 改变葡萄糖代谢等生理功能,所以这些菌在肠道里的存在,更可能是:微调宿主营养和代谢状态,而不是简单的“好/坏”。
参考文献:
Li TT, Chen X, Huo D, Arifuzzaman M, Qiao S, Jin WB, Shi H, Li XV; JRI Live Cell Bank Consortium; Iliev ID, Artis D, Guo CJ. Microbiota metabolism of intestinal amino acids impacts host nutrient homeostasis and physiology. Cell Host Microbe. 2024 May 8;32(5):661-675.e10.

谷禾健康
镁是人体中调节多种生理和病理过程的必需营养素。作为几百种酶促反应的辅因子,它参与能量合成、蛋白质与DNA稳定、神经传导、骨骼健康、肌肉收缩以及心律调节等多种生理过程。镁不仅是细胞能量代谢的核心元素,还在维持离子稳态、抗氧化防御和炎症调节中扮演着关键角色,被誉为“细胞代谢的守护元素”。
然而,现代社会中镁缺乏的现象正在快速上升,已成为全球范围内被低估的公共健康问题。许多国家人群的平均膳食镁摄入量远低于推荐标准。饮食结构改变和食物精制化是导致这一问题的主要原因。随着加工食品和高能量低营养饮食的普及,天然富镁食物(如全谷物、坚果、绿叶蔬菜)的摄入显著减少。此外,慢性压力、睡眠不足、过量咖啡因与酒精摄入,以及某些药物(如利尿剂和质子泵抑制剂)都会加速体内镁的流失。
镁缺乏对健康具有广泛而深远的影响。低镁状态会破坏细胞能量供应,削弱神经与肌肉系统的稳定性,易引发神经兴奋过度和心律失常。研究显示,血镁水平偏低与高血压、动脉硬化、心力衰竭、2型糖尿病、骨质疏松、抑郁症及认知障碍等多种慢性疾病密切相关。同时,镁不足还会增加炎症反应和氧化应激,成为多系统慢性损伤的重要激发因素。
改善镁缺乏除了常规的依赖膳食或补充剂外,更应从调节肠道微生态入手。研究表明,肠道微生物群在镁的吸收与代谢中发挥关键作用:肠道菌群通过发酵膳食纤维产生短链脂肪酸,可降低肠腔pH,促进镁盐溶解,并激活肠上皮细胞镁转运通道(TRPM6与TRPM7)的表达,从而增强镁吸收。健康菌群还能维护肠黏膜屏障,防止炎症性渗漏导致的镁流失;部分菌株则通过代谢产物调节肾脏镁重吸收,维持体内镁稳态。相反,菌群失衡会削弱这些机制,降低镁吸收效率并加剧缺镁,形成“低镁—炎症—菌群紊乱”的恶性循环。
镁缺乏的普遍化反映出现代生活方式与饮食结构下的系统性营养失衡。鉴于镁在多种疾病中的关键作用,深入探索肠道菌群对镁稳态的影响,并将其纳入营养干预策略,有望实现更精准、可持续的镁补充方式,从而在慢性疾病预防与代谢健康维护中发挥更积极的作用。

▸ 镁在人体的含量
镁(Mg)是人体中调控多项生理功能的一种重要矿物质。是人体内第四丰富的金属元素(Ca²+ > K+ > Na+ > Mg²+),也是仅次于钾的细胞内第二丰富的阳离子。
人体镁含量约为24–29克,其中约2/3储存在骨骼,1/3分布于细胞内和肌肉组织,少于1%存在于细胞外其他组织及血液中(约100毫克)。血清镁浓度维持在0.75–0.95 mmol/L之间。
人体中镁的分布
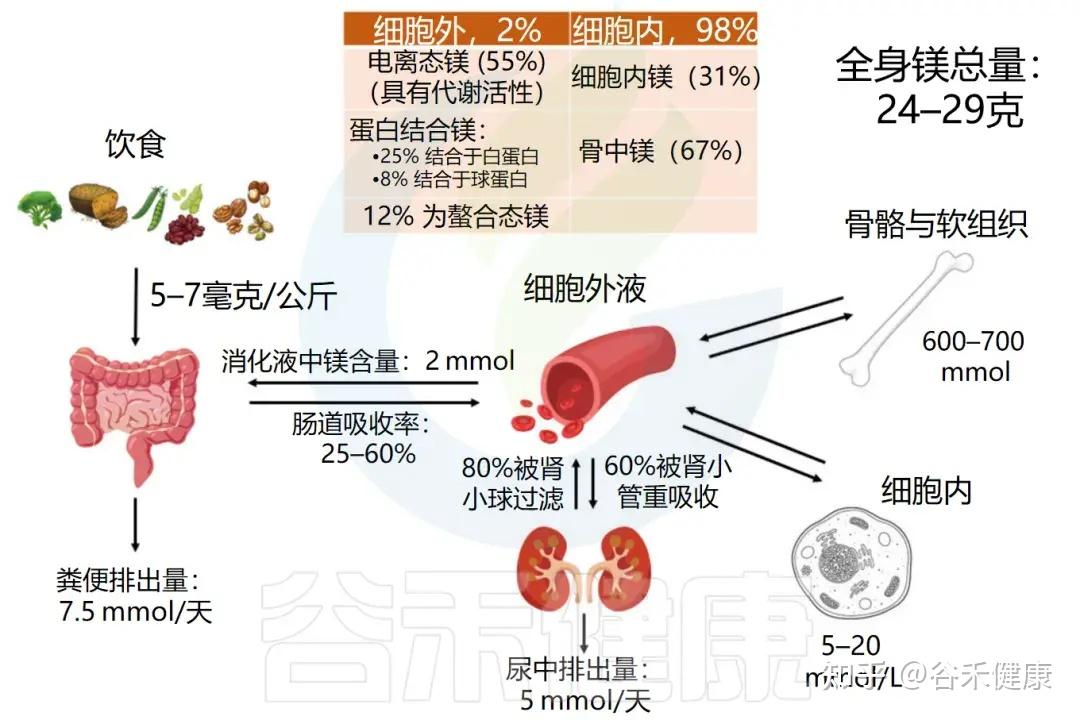
▸ 镁的生理功能
镁(Mg)是维持人体健康的必需矿物质,参与了许多对身体正常运作至关重要的生理过程。其重要性涵盖多个器官系统,包括细胞功能、心血管系统、肌肉骨骼系统和神经系统,突显了其在维持整体健康中不可或缺的作用。
镁在多个细胞过程中的参与
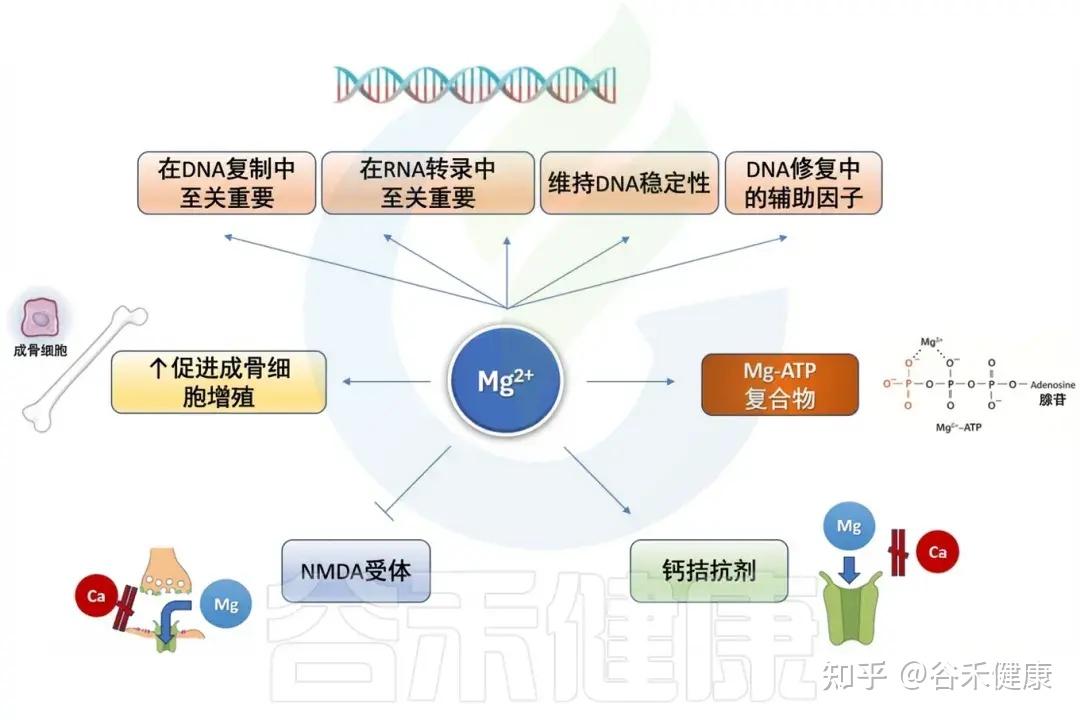
⑴影响能量代谢—是酶促反应核心因子
•镁是人体内超过600种酶的必需辅因子,参与能量代谢(在细胞中,ATP 以 MgATP²⁻ 复合物形式存在);
•促进糖酵解关键酶(己糖激酶、磷酸果糖激酶)的活性;
•调节线粒体氧化还原反应与电子传递链稳定性。
注:辅因子是与酶结合且在催化反应中必要的非蛋白质化合物。
镁离子和MgATP的化学结构
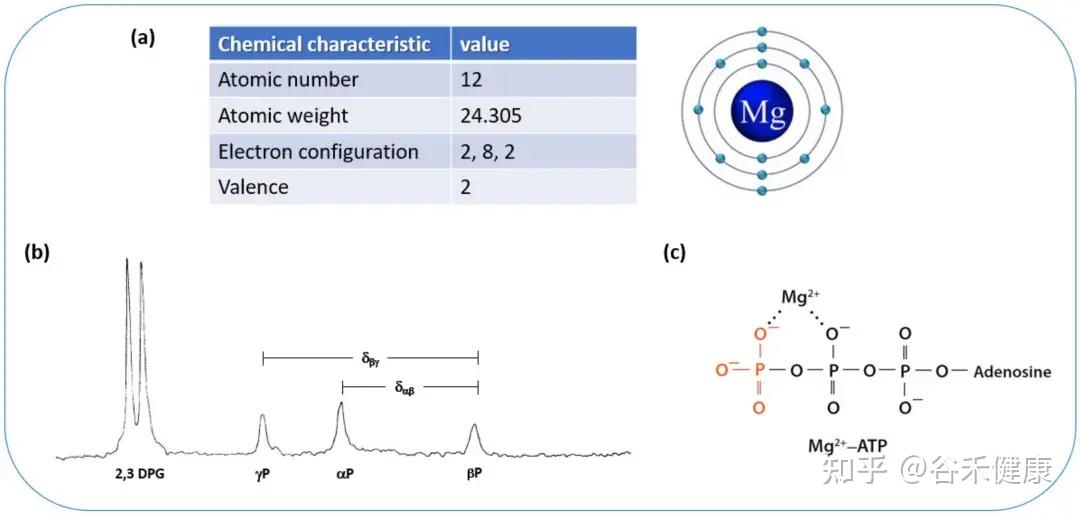
⑵稳定核酸与蛋白质等结构
镁参与骨骼(约储存体内60%镁)、线粒体、核酸及膜结构的稳定,对DNA修复、染色体完整性和细胞衰老的延迟具有重要作用。
⑶免疫调控与炎症反应
镁是免疫细胞(T/B淋巴细胞、巨噬细胞)正常功能所必需,调控细胞因子分泌、吞噬作用及抗原应答。
镁不足可促发慢性低度炎症,增加IL-6、TNF-α等炎性标志物。
⑷神经传导与兴奋调节
•镁稳定神经膜,抑制NMDA受体的过度激活 → 防止兴奋性毒性;
•促进GABA受体敏感性,具有一定镇静与抗焦虑作用;
•调节神经递质(乙酰胆碱、谷氨酸、血清素)的合成与释放;
•缺镁与中枢神经兴奋、偏头痛、抑郁和神经退行性疾病风险升高相关。
⑸调节肌肉兴奋与收缩
•与钙协同控制骨骼肌与心肌的收缩和舒张;
•Mg²⁺ 参与肌动蛋白‑肌球蛋白相互作用的能量供应;
•缺镁可导致肌肉痉挛、震颤、血管痉挛及心律失常。
⑹参与维持心血管功能
•镁可以调节血管平滑肌张力,维持血压稳定;
•抑制血小板聚集与血栓形成;
•增强心肌顺应性并降低心律失常发生;
•低镁水平与高血压、动脉粥样硬化、心律失常、猝死密切相关。
⑺骨骼健康与矿物代谢
•镁与钙、磷共同参与骨基质形成与矿化;
•是维生素D活化(25‑和1α‑羟化)过程中关键辅因子;
•缺镁会影响甲状旁腺激素(PTH)分泌,导致低钙血症与骨密度下降;
•充足镁水平有助于降低骨质疏松与骨折风险。
镁在骨代谢中的作用
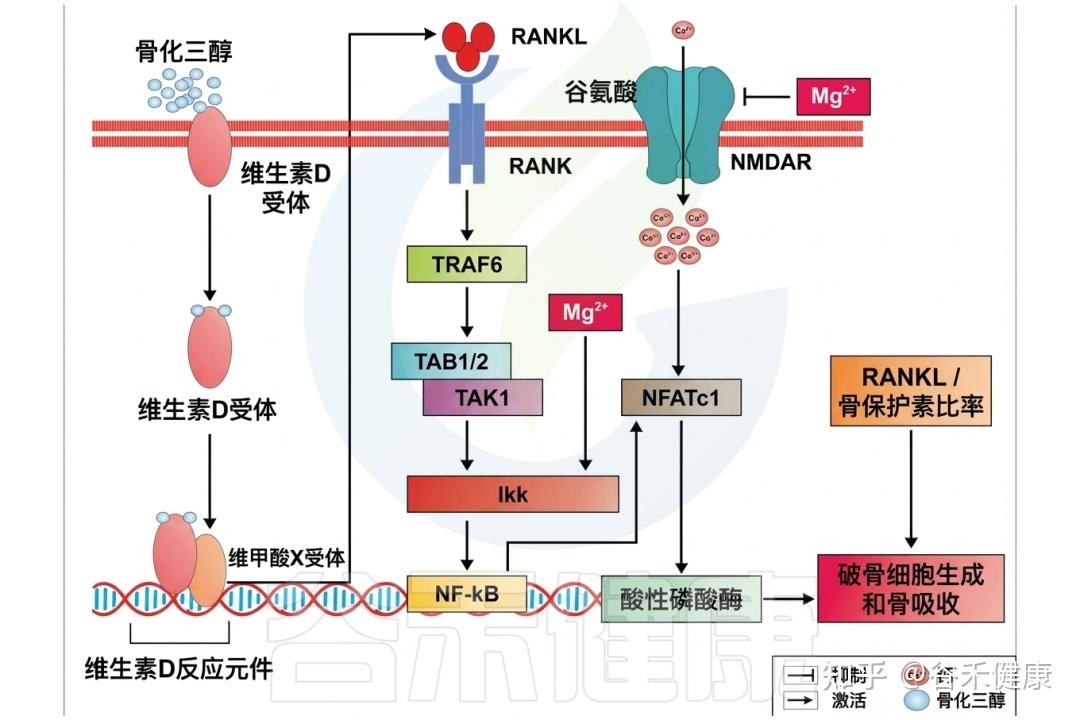
⑻抗氧化、抗衰老
•镁有助于减少活性氧(ROS)生成,增强谷胱甘肽和抗氧化酶系统活性;
•稳定线粒体功能,降低氧化应激造成的细胞损伤;
•维持DNA修复与细胞代谢平衡,延缓组织功能退化与衰老进程;
•抑制炎症因子表达,减轻慢性炎症性衰老。
⑼调节内分泌与代谢稳态
•镁是胰岛素信号转导的关键因子:激活胰岛素受体酪氨酸激酶,增强葡萄糖摄取;低镁状态与胰岛素抵抗及2型糖尿病风险增加相关。
•还参与脂质合成、氧化及甲状腺激素代谢。
镁影响胰岛素信号传导
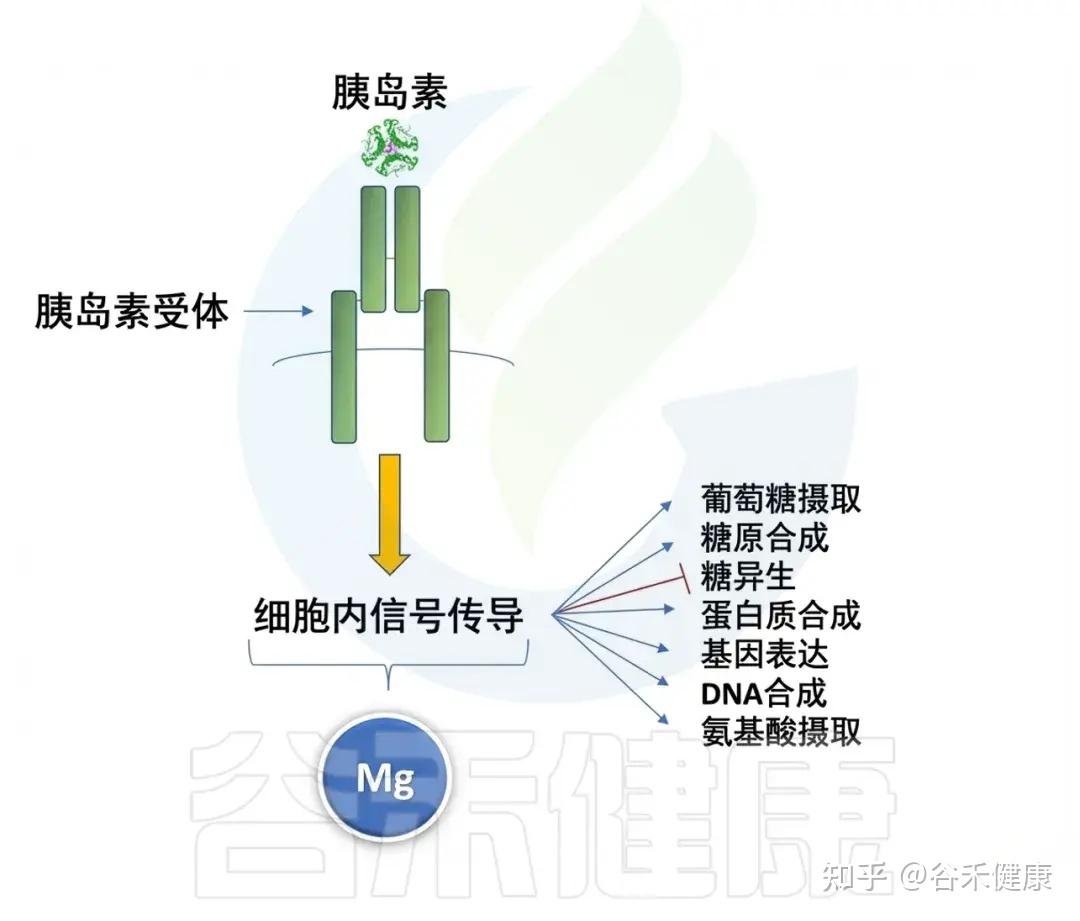
★ 不同人群对镁的需求存在差异
根据2015–2020年美国膳食指南,女性每日镁推荐摄入量为320 毫克,男性为420 毫克。在怀孕、衰老、运动或感染、2型糖尿病等情况下,机体对镁的需求可能进一步增加。
然而,许多因素可能改变Mg的平衡:饮食中高含量的Na、Ca、蛋白质、酒精或咖啡因,或某些药物的使用(利尿剂,如呋塞米;质子泵抑制剂,如奥美拉唑等)。
影响镁平衡的因素:
•胃肠道吸收能力;
•肾脏排泄;
•健康人群的饮食建议每天约5–7毫克/千克体重来保持平衡;
•细胞外的Mg与储存中的Mg处于平衡状态;
•骨头是Mg的主要储存点。
▸ 镁的吸收与代谢过程
我们每天大约从食物和饮水中摄入约300–400毫克的镁,主要来源包括:
•深绿色蔬菜:如菠菜、羽衣甘蓝、甜菜叶等。
例如,100克煮熟的菠菜含约80毫克镁(占成人每日推荐量的20%)。
•坚果与种子:南瓜籽、杏仁、腰果、巴西坚果是镁的优质来源。
30克南瓜籽约含150毫克镁,接近成人每日需求量的37%。
•全谷物:糙米、燕麦、藜麦、荞麦等未经精制的谷物中,镁含量较高。
100克煮熟的藜麦约含65毫克镁。
•鱼类和豆制品:黑豆、鹰嘴豆等豆类每半杯煮熟的豆子含60-80毫克镁。三文鱼、鲭鱼等深海鱼也含镁(100克约含30-50毫克),同时提供Omega-3脂肪酸,建议每周食用2-3次。
•黑巧克力:可可含量70%以上的黑巧克力每30克约含65毫克镁,同时含抗氧化物质。
•硬水:(自来水或矿泉水中也含少量镁)。
①肠道吸收——“通往血液的第一道关口”
当镁随食物进入小肠后,约有30%~50%能被吸收进入血液。吸收主要发生在小肠(尤其是回肠和空肠),有两种途径:
被动扩散途径:当饮食中镁含量较高时,它通过肠壁间隙“自然渗透”进入血液。
主动转运途径:在镁摄入较低时,肠细胞会动用“镁专用通道”——TRPM6和TRPM7蛋白,把镁主动吸收入细胞。
影响吸收效率的因素包括:
促进吸收:维生素D、益生菌、乳酸盐、中等脂肪摄入。
抑制吸收:过多的钙、磷酸盐、酒精、咖啡因或高脂饮食。
维生素D:促进肠道镁吸收,而镁又是维生素D活化所必需。
甲状旁腺激素(PTH):促进肾脏与肠道吸收镁,调节骨中镁释放。
雌激素:通过上调TRPM6通道增强镁再吸收。
细胞内转运系统:TRPM6/7、MagT1以及SLC41家族是调节镁跨膜转运和细胞内稳态的关键通道;其功能异常与代谢紊乱、免疫及神经疾病均有关。
②进入血液——“镁流通的高速公路”
吸收后的镁进入血液循环。成人血浆中正常镁浓度约为 0.75–0.95 mmol/L;其中约1%的镁在血液中自由流动,剩下的99%存储在骨骼(约2/3)和肌肉、软组织(约1/3)中。
镁在血浆中有三种存在形式:
•离子态 Mg²⁺(有生理活性的形式)
•与蛋白结合的镁(主要结合白蛋白)
•与阴离子结合的复合物(如磷酸盐、柠檬酸盐)
血中的镁浓度受到肠道吸收、骨骼储备和肾脏排泄三方面的精密平衡控制。
③肾脏的守护机制——排泄与调节
肾脏是镁平衡的核心调节器,肾小球对血浆反复滤过,每日滤过量可达约2.4g进入尿液;其中约95%(2300 毫克)在肾小管被重新吸收;实际仅约100 毫克以尿液形式排出体外。
肾脏可以调节排泄量,具体取决于血清镁的水平:当镁不足时,肾脏会提高重吸收率;当摄入过多时,则相应增加排泄量。
人体中的镁平衡
▸ 可能导致镁缺乏的因素
虽然日常饮食可提供一定量的镁,但现代饮食结构与生活方式的变化使许多人处于轻中度缺镁状态。长期摄入不足、吸收不良或排泄过多都可能扰乱体内镁平衡。以下列举了可能导致镁缺乏的因素:
导致低镁状态的原因及影响
⑴摄入不足
饮食结构失衡
•过度精制(精米、精面、糖类食品)导致镁含量锐减 > 60%;
•现代“西式饮食”(高脂、高糖、高加工)普遍镁摄入不足;
•素食或高钙高磷饮食中钙/镁比例失衡(Ca:Mg > 3:1)抑制镁吸收;
•减肥、禁食及流质饮食者镁摄入低于推荐摄入量。
饮水镁含量降低
•现代净化/反渗透水镁浓度低于 10 mg/L(天然水约 20–50 mg/L);
•长期饮用低矿物质水可能导致血镁水平降低,有研究提示可达约10–15%,但不同研究差异较大。
食物生长环境贫镁
•现代农业中过度使用化肥、酸性土壤、除草剂(尤其草甘膦)导致土壤镁枯竭。植物镁含量相比1950年代平均下降 ≈ 25–30%。
⑵吸收障碍
胃肠疾病
•慢性腹泻、吸收不良综合征、克罗恩病、乳糜泻导致镁大量丢失;
•胆汁酸吸收障碍降低肠道对镁的电解质吸收能力;
•小肠细菌过度生长(SIBO)或肠黏膜萎缩破坏吸收通道(TRPM6 / TRPM7);
•胰腺外分泌障碍及脂肪泻减少脂溶性螯合离子的吸收。
手术相关
胃旁路手术/小肠切除术使吸收面积显著减少,短肠综合征患者吸收率下降 50% 以上。
药物相关吸收受阻
质子泵抑制剂(PPI,如奥美拉唑、泮托拉唑)抑制胃酸分泌,减少Mg²⁺离子溶解及TRPM6通道活性。
长期使用PPI > 1年可使低镁血症风险增加10–12 倍。
⑶肾性镁丢失
肾脏每日滤过≈2400 mg 镁,其中95% 被重吸收。任何影响肾小管Mg²⁺重吸收的因素都会导致缺镁。
药物诱导
某些药物的使用可能促进排泄而导致镁流失。具体作用机制和影响方式如下所示:

内分泌疾病
•高甲状旁腺功能 → 胃肠Ca²⁺升高→ 钙竞争性抑制Mg再吸收;
•抗利尿激素分泌异常会伴随低钠低镁症;
•糖尿病性肾病 → 高血糖引起渗透性利尿与电解质丢失。
酒精性肾损伤
•乙醇抑制肾小管再吸收并引发代谢性酸中毒,促使镁随尿排出。
•慢性酗酒者血镁常降低 20–30%。
⑷代谢与激素调控缺陷
甲状旁腺及维生素D轴异常
•Mg 是PTH(甲状旁腺激素)分泌与受体敏感性所必需的辅因子;
•缺镁→ PTH 分泌减少 + 靶器官抵抗 → 继发性低钙血症;
•维生素D活化需要Mg依赖性羟化酶(25‑和 1α‑羟化酶)。
遗传性离子通道缺陷
•TRPM6、CNNM2、SLC41A1 突变可致原发性家族性低镁血症;多合并癫痫、智力低下、低钙。
•Gitelman、Bartter 综合征:肾小管Mg‑Ca转运障碍。
⑸其他因素
应激与慢性炎症
•慢性应激激活下丘脑-垂体-肾上腺轴,升高皮质醇 → 增强镁排泄;
•炎性细胞因子(IL‑6、TNF‑α)会下调Mg转运体表达。
酒精与咖啡因
•酒精会促进尿镁排泄,抑制胃肠吸收;
•咖啡因增加肾小管滤过率,加速镁流失。
高钙补充剂及磷酸盐过量
•过高钙/磷摄入形成不溶性Mg盐,减少吸收。
⑹老龄化
吸收能力下降
随着年龄增长,小肠对镁的吸收效率降低。
肾功能减退
老年人肾脏重吸收镁的能力下降,排泄量增加。
饮食摄入不足
食欲减退、饮食单一或牙口问题常导致镁摄入不足。
药物干扰
老年人常用的利尿剂、质子泵抑制剂等药物会促进镁流失。
慢性疾病影响
糖尿病、胃肠疾病等常见老年病可进一步加重镁缺乏。
多项研究与临床证据表明,镁缺乏在全球范围内普遍存在且常被低估。
▸ 镁缺乏的患病率
大约10–25%的人群血镁浓度低于正常参考值(<0.75 mmol/L),并且不同人群镁缺乏的发生率不同。我们总结在下表中:

▸ 镁缺乏的主要症状
▸ 早期阶段(轻度缺乏)
镁水平轻度下降时,症状往往隐蔽而非特异性。常见表现包括:
•精力不足、易疲劳、注意力不集中;
•失眠或睡眠浅、焦虑、易怒、情绪波动;
•偶尔出现肌肉轻微抽搐、眼皮跳、手脚麻感;
•食欲减退或轻微便秘。
此阶段多数人并未意识到与镁有关,但代谢系统和神经功能已开始受到影响。
▸ 加重阶段(中至重度缺乏)
当缺镁持续或加重时,细胞功能紊乱导致全身性症状明显加剧:
•持续肌肉痉挛、抽筋或手足震颤;
•心悸、脉搏不齐、胸闷等心律异常表现;
•明显的神经兴奋性亢进,可出现焦虑、抑郁、甚至轻度意识障碍;
•持续疲劳、虚弱无力,运动耐力下降;
•骨骼变脆、骨痛或骨密度下降;
•代谢紊乱,如胰岛素抵抗、血糖波动、钙钾失衡。
▸ 严重阶段(临床性缺镁)
若缺乏未得到纠正,体内镁储备枯竭,可引发严重后果:
•明显心律失常、心肌功能障碍;
•痉挛性抽搐或癫痫样发作;
•电解质紊乱同时出现(低钙、低钾);
•严重者可能出现意识混乱、昏迷或猝死。
镁缺乏对人体健康的影响是全身性和多层面的。作为重要矿物质,镁在能量代谢、神经传导、心肌收缩、骨骼形成及免疫调节中均发挥关键作用。
当体内镁水平下降时,不仅会引起疲乏、肌肉痉挛、情绪不稳等早期症状,还会干扰糖代谢、增加心血管风险、促进慢性炎症及加速细胞老化。长期缺镁更与高血压、糖尿病、动脉硬化、骨质疏松以及神经退行性疾病等多种慢性病密切相关,显示出镁在维持机体整体稳态中的核心意义。
1
对神经系统与心理健康的影响
√神经兴奋性增强:
镁是调节神经膜电位和 NMDA 受体活性的重要离子,缺乏镁会使神经细胞过度兴奋,易导致焦虑、易怒、失眠及肌肉抽搐等症状。
√抑郁与焦虑关联:
镁缺乏可影响血清素和 GABA 的合成与受体功能,是抑郁症和焦虑症的重要生化基础。多项研究显示,低镁个体中抑郁风险增加20–40%。
√认知功能退化与神经退行性疾病:
•镁缺乏可促进 β‑淀粉样蛋白沉积、线粒体功能下降和突触丢失,与阿尔茨海默病病理密切相关。
•多项长期随访研究发现,血镁水平较低者认知下降速度更快,发生痴呆的风险增加约 20–40%。
注意:不同研究之间结果存在一定差异,上述数值多基于观测性研究,这里相关性而非严格因果。
2
对心血管系统的影响
√心律失常与电生理紊乱:
镁是维持心肌膜稳定性和钾钙平衡的关键元素。缺镁会导致室性早搏、房颤等心律失常发生率上升。
√高血压与动脉硬化:
镁具有血管舒张作用,可抑制钙离子内流、降低血管张力。缺镁使血管平滑肌收缩增强,从而促进高血压和动脉硬化的形成。
√心血管疾病事件风险上升:
大规模临床研究表明,血镁水平低于 0.75 mmol/L 的人群,其冠心病和突然心源性死亡风险增加 30–50%。
3
对骨骼和肌肉系统的影响
√骨质疏松与骨代谢障碍:
镁与钙、维生素 D 协同调控骨骼矿化。长期缺镁会降低成骨细胞活性并促进破骨。文献显示,低镁摄入可使骨密度下降约 5–8%。
√肌肉功能障碍:
缺镁常导致肌肉过度兴奋,表现为抽筋、痉挛、震颤、肌无力等症。严重时可引发横纹肌溶解。
4
对代谢与内分泌系统的影响
√胰岛素抵抗与糖尿病:
•镁是葡萄糖代谢关键酶的辅因子,缺镁会削弱胰岛素信号传导。
•部分研究显示,在低镁个体中,胰岛素敏感性可下降约 25–30%;2型糖尿病患者中约 1/3 存在血镁或细胞内镁下降。
√脂质代谢异常:
镁不足会增加甘油三酯与低密度脂蛋白(LDL)水平,降低高密度脂蛋白(HDL)水平,促进脂肪肝和动脉粥样硬化进程。
√内分泌紊乱:
镁参与皮质醇、甲状腺素及性激素的合成,其缺乏可导致应激反应过度、月经紊乱、骨质丢失加剧。
5
对免疫系统与炎症反应的影响
√慢性低度炎症:
•低镁状态会激活免疫细胞,升高 CRP、IL‑6、TNF‑α 等炎症标志物。
•长期炎症反应被认为是多种代谢性疾病和衰老过程的共同基础。
镁缺乏通过多种信号通路引发炎症
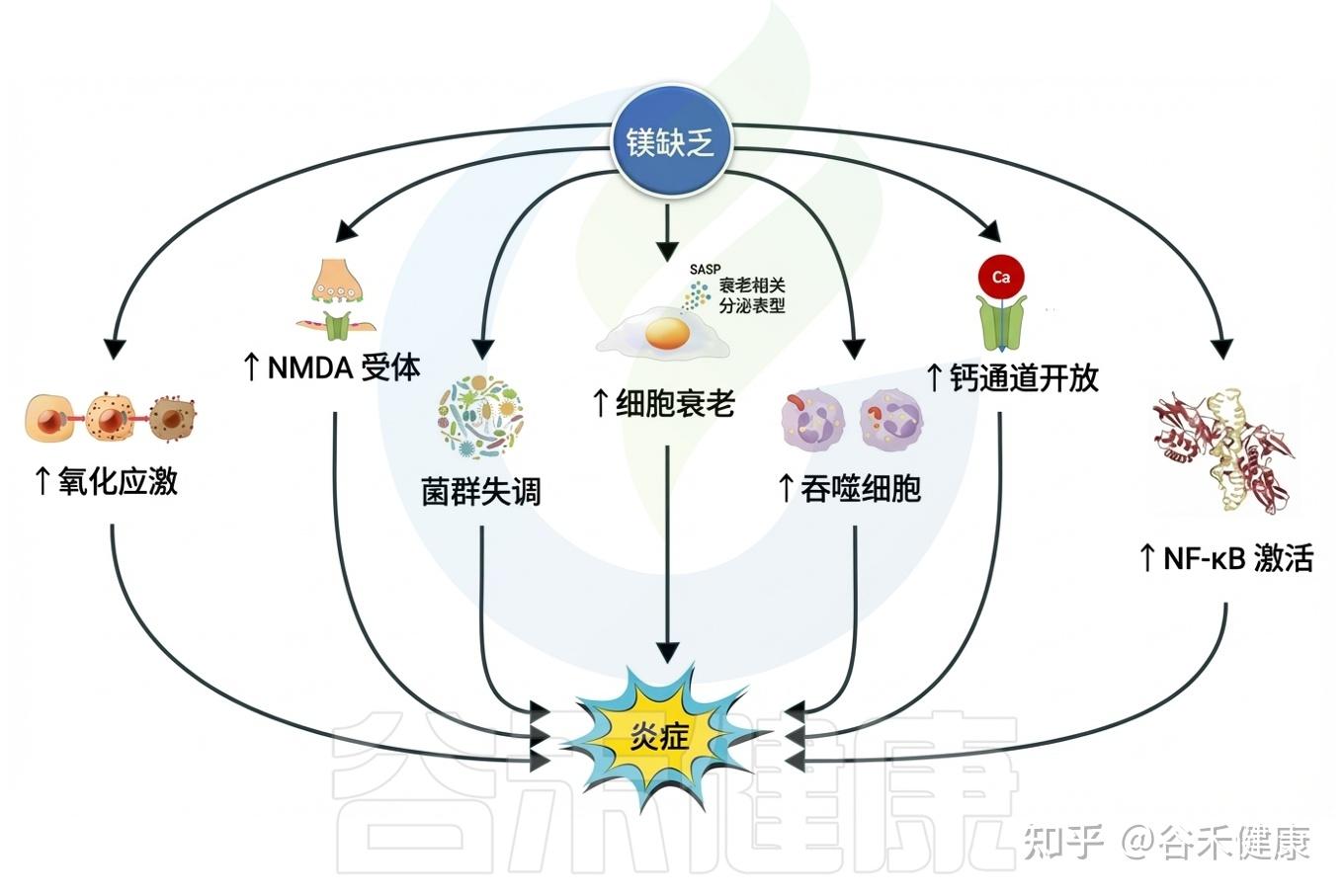
√免疫失衡与感染易感性:
镁缺乏削弱先天及获得性免疫反应,老年群体更易发生感染。
6
对呼吸系统的影响
√平滑肌功能紊乱与支气管高反应性:
镁能抑制钙离子进入平滑肌细胞,促进支气管舒张。缺镁时支气管平滑肌紧张度上升,气道收缩反应增强,易诱发或加重哮喘和慢性阻塞性肺疾病(COPD)。
多项研究发现,哮喘患者的血镁水平显著低于正常人群;在部分严重急性哮喘、对常规支气管扩张剂疗效欠佳的患者中,静脉硫酸镁可作为辅助治疗,有研究提示可改善肺功能与症状。
√呼吸衰竭与重症结局风险:
在重症监护人群中,低血镁与呼吸衰竭发生率及机械通气时间延长显著相关。有研究强调,老年COPD(慢性阻塞性肺疾病)患者中缺镁率可达 45–65%。
7
对衰老与细胞层面的影响
√线粒体功能下降:
镁是 ATP 合成复合物的重要组成部分,缺乏镁会直接影响能量代谢效率并促进氧化应激。
√DNA 稳定性与端粒退化:
镁是 DNA 聚合酶与修复酶的关键辅因子。缺乏镁导致 DNA 损伤积累与端粒缩短,加速细胞老化。
√细胞衰老信号激活:
镁缺乏可上调 p53、NF‑κB、MAPK 信号通路,促进细胞衰老及炎症反应。
8
对其他系统的影响
√电解质紊乱:
缺镁常伴随低钾血症和低钙血症,因为镁调节钙钾通道活性。若未及时纠正镁缺乏,补钾或补钙疗效有限。
√胃肠道与消化功能减退:
低镁可导致胃肠蠕动缓慢、食欲低下和长期便秘。
√妊娠期并发症风险增加:
妊娠缺镁与先兆子痫、胎儿生长受限及早产风险上升有关。
除了对神经、心血管、骨骼和代谢等系统具有广泛影响外,镁还在维持肠道微生态平衡中发挥关键作用。研究表明,镁不仅调节宿主代谢和免疫功能,还直接影响肠道微生物的组成与活性。
肠道微生态是人体健康的重要调控因素,参与营养吸收、代谢调节、免疫稳态及神经—肠轴信号传导,对整体生理平衡至关重要。
镁对肠道微生态的影响可分为两种情况:既包括镁缺乏状态下的不利变化,也涵盖过量摄入时的微环境干扰。但在实际人群中,镁缺乏更为常见且影响更显著。
肠道微生物与镁之间的联系

★ 镁主要通过三大途径调控肠道微生物
•直接离子效应:影响细菌膜电位、ATP合成酶及多种金属依赖酶的活性。
•间接宿主效应:通过上皮屏障、黏液层厚度、免疫细胞激活及pH变化改变菌群生态位。
•代谢共生效应:与短链脂肪酸、胆汁酸、氨基酸代谢互作,调节菌群能量网络结构。
▸ 镁缺乏对肠道微生态的影响
我们通过查阅和整理多篇相关研究文献,对镁缺乏与肠道微生态之间的关系进行了系统分析与比较,总结出镁缺乏对肠道微生态的主要影响如下:
▸ 总体特征
•α-多样性下降(Shannon 与 Chao1 指数显著降低);
•β-多样性结构偏移;
•微生物群表现出“致炎生态型”特征;
•Firmicutes/Bacteroidetes(F/B)比例上升,典型菌群表现为条件致病菌扩张。
▸ 主要菌属变化

▸ 代谢通路与产物变化
短链脂肪酸(SCFAs)水平下降:
•丁酸盐水平下降40–60%;
•丙酸盐水平下降;
•乙酸盐水平略降或不变。
氨基酸与氮代谢紊乱:
•蛋白质/氨基酸发酵菌扩张,支链脂肪酸升高;
•伴随氨、硫化氢(H₂S)和吲哚产物增加,引发局部上皮炎症。
胆汁酸代谢改变:
•Desulfovibrio 扩张促进次级胆汁酸(如脱氧胆酸、石胆酸)升高;
•抑制黏液层再生并促进炎症。
氧化还原平衡破坏:
•低镁降低过氧化氢酶与超氧化物歧化酶活性,造成还原应激环境,偏好肠杆菌科。
宿主相互代谢效应:
•降低肠道TRPM6/7表达(镁吸收能力进一步下降);
•形成“缺镁—菌群紊乱—炎症—吸收障碍”的恶性循环。
▸ 对肠道微生态和整体健康的影响
肠屏障受损与“肠漏”效应:
•缺镁小鼠肠道紧密连接蛋白(ZO‑1、Occludin)显著下降,肠道通透性增加,血浆脂多糖浓度升高;这促发系统性炎症及肝脏TNF‑α、IL‑6表达上升。
慢性炎症与免疫激活:
•镁缺乏促进单核细胞浸润、NF‑κB信号及细胞因子(TNF‑α、IL‑1β)上调,形成“肠—免疫炎症环”。
代谢与系统影响:
•长期缺镁相关菌群变化与胰岛素抵抗、肥胖有高度相关性(增多的 SMB53、Turicibacter 报告于低镁和肥胖模型中)。
•同时,产丁酸菌减少可能加剧代谢综合征风险,损害肠代谢稳态。
▸ 镁过量对肠道微生态的影响
通常,短期补充镁不会造成体内过量,因为机体具有良好的调节与排泄功能。然而,若长期高剂量摄入或肾功能受限,仍可能导致镁积累,进而扰乱肠道渗透压与离子平衡,影响微生物群的结构与代谢活动。
▸ 极高剂量会扰乱生态稳态
轻度镁富集(生理范围上限)可促进有益菌,但极高剂量可扰乱生态稳态。

▸ 高镁环境也改变了微生物的代谢
高镁环境重塑了肠道菌群的代谢谱,使代谢活动偏向能量储存与抗氧化代谢强化。
KEGG 与 PICRUSt2 功能预测显示,高镁饮食促进“糖代谢”(glycolysis)、“戊糖磷酸通路”(PPP)与“脂肪酸β氧化”相关基因丰度上升。与此同时,丙酮酸代谢和丁酸代谢略下降,提示产能途径更侧重于快速代谢而非发酵稳定性。
高镁显著上调谷胱甘肽代谢(glutathione metabolism)与嘌呤代谢,表明抗氧化能力增强。但同时,色氨酸→吲哚代谢途径增强,部分产物可调控免疫及神经—肠轴。
编者小结
总体而言,镁对肠道微生物群的影响取决于剂量与持续时间。
镁缺乏通常导致有益菌如双歧杆菌和产丁酸菌减少,而条件致病菌如肠杆菌科增多,引发短链脂肪酸生成下降、肠屏障受损及慢性炎症反应。
适度高镁摄入(生理上限范围内) 可促进益生菌如 Lactobacillus 和 AKK菌增殖,提升多样性,增强肠屏障与抗氧化防御。
长期或过高剂量则可能干扰渗透平衡,减少部分厌氧产丁酸菌,导致短暂微生态波动和通便反应。
因此,维持肠道内镁水平在适宜范围,是确保微生态稳态与代谢健康的关键条件:不足易致炎,过量则扰稳,唯有平衡方能良性共生。
上文我们提到,镁不仅能够通过多种途径影响肠道微生态的平衡与整体健康状态,但这种关系并不是单向的。实际上,镁与肠道菌群之间存在一种紧密而动态的双向作用关系。肠道菌群的组成和代谢活动同样会反过来调节镁在体内的吸收与利用效率,从而共同维持机体的营养稳态与代谢平衡。
人体镁吸收主要发生在小肠(回肠和空肠)与结肠,包含两条途径:
被动扩散:依赖浓度梯度;约占吸收总量的 80–90%。
主动转运:由肠上皮细胞内的TRPM6、TRPM7通道调控,在低镁环境下尤为重要。
人体肠道矿物质吸收
★ 肠道微生物群是影响镁吸收利用的重要因素
而近十年研究表明,肠道微生物群是第三个决定性因素,通过影响肠环境的理化条件、代谢产物和宿主转运通路,系统地调控镁的生物利用度。
▸ 肠道菌群影响镁吸收的主要机制
⑴调节肠腔环境(pH与溶解度)
•产酸菌(如 Lactobacillus、Bifidobacterium)通过发酵碳水化合物生成短链脂肪酸(SCFAs)——乙酸、丙酸、丁酸。
•SCFAs 降低肠腔 pH,使难溶性镁盐(如Mg(OH)₂、Mg₂PO₄)溶解度升高,从而促进镁离子化,提高吸收效率。
•酸性环境还刺激肠上皮表达钙/镁通道蛋白(TRPM6/7)及螯合肽转运体,从而增强主动摄取能力。
⑵短链脂肪酸促进上皮转运
短链脂肪酸可激活肠上皮 GPR43/GPR109A 受体,增强细胞紧密连接蛋白(ZO‑1、occludin)表达,改善屏障完整性,使镁离子有效跨膜转移。
乙酸与丙酸促进钠依赖型共转运(Na⁺/Mg²⁺ exchang),间接提升镁摄取。
注:有研究指出:丁酸在过高浓度条件下会通过抑制TRPM6通道电流,会降低Mg²⁺跨上皮细胞通量。该效应具有剂量依赖性双向调节特征——低浓度丁酸有利于吸收,而过高浓度则抑制。

因此,短链脂肪酸总体上促进镁吸收,但丁酸对TRPM6的局部抑制提示了菌群平衡对镁代谢的微妙调控关系。
⑶菌群代谢物与转运信号互作
•肠菌产物如多胺(腐胺、精胺)可通过调控AMPK和ERK信号通路促进TRPM6转录。
•粘蛋白降解菌 (Akkermansia muciniphila) 可增强上皮再生,维持适宜的吸收表面积与紧密连接。
•此外,一些细菌通过产生螯合有机酸(柠檬酸、乳酸)可形成可吸收镁螯合物,显著提高生物利用度。
⑷益生菌直接吸附与释放作用
•多种益生菌细胞壁富含带负电的羧基(COO⁻)与磷酸基(PO₄²⁻),能暂时结合Mg²⁺。
•在酸性环境中,结合的镁离子再被逐步释放,有助于维持小肠近端镁浓度梯度。
•Lactobacillus plantarum与Bifidobacterium longum 对镁的吸附容量尤高,故被视为“镁生物富集菌”。
⑸肠道微生物影响肠黏膜的吸收能力
有研究发现莲子抗性淀粉可增加Lactobacillus、Ruminococcus 丰度,SCFAs升高50–60%,并改善绒毛高度与微绒毛密度。
与对照组相比,小鼠肠道镁、钙吸收率分别提高约30%与25%。说明膳食诱导的菌群改善能通过结构性(形态学)重塑增强矿物吸收能力。
⑹肠—菌—宿主信号反馈调节
•微生物群通过信号环路调控肠上皮基因表达:提高 TRPM6、CLDN15(紧密连接蛋白)与 SLC41A1(镁排出转运体)转录水平,使吸收与细胞出口协调。同时通过产生代谢信号(如吲哚衍生物、丁酸)影响上皮AMPK和Nrf2通路,调节能量与氧化还原状态,从而间接优化镁吸收条件。
•微生物组成改变还能影响胆酸代谢与黏膜黏液厚度,这两者共同决定离子交换界面的有效面积与pH稳定性。
▸ 肠道菌群与镁利用的全身效应
⑴肝门循环途径调节
部分短链脂肪酸(SCFAs)被肠道上皮吸收后通过门静脉进入肝脏,在肝细胞内可上调镁稳态相关蛋白,即CNNM家族成员的表达,从而增强肝脏对镁离子的储存与调控功能。
这一过程在维持全身镁平衡中起着重要作用,有助于调节系统性储镁能力并保证体内镁稳态的稳定。
⑵肾脏再吸收作用
目前在动物模型和体外实验中观察到:肠道菌群产生的代谢产物,尤其是丙酸,在被吸收入血后能够激活宿主的PXR受体信号通路,从而促进肾脏中TRPM7通道蛋白的表达水平上升。通过这种分子途径,肠源代谢信号间接改变了肾脏对镁离子的再吸收与排泄能力,使镁的排泄率得到动态调节,进而参与维持全身镁稳态。
⑶免疫效应层面
健康的肠道菌群能够有效维持低炎症的内环境,抑制过度的免疫反应,从而减少由TNF‑α介导的TRPM6通道蛋白内化过程。通过保持TRPM6的稳定表达与功能活性,肠道上皮细胞得以持续高效地吸收镁离子,从整体上提升机体对镁的长期吸收效率与稳态维持能力。
▸ 肠道微生物紊乱与镁吸收障碍
当肠道菌群的平衡被打破时,例如在长期或不当使用抗生素、高脂饮食摄入过多、或发生炎症性肠病等情况下,肠道微生态的稳定性受到显著干扰,进而引发一系列连锁反应。
⑴Mg²⁺ 盐溶解性下降
首先,短链脂肪酸(SCFAs)的生成量明显下降,使得肠道内容物酸度降低,肠腔pH值常升高至7以上,从而导致镁盐溶解度下降并出现沉淀,削弱了可吸收镁离子的供给。
⑵TRPM6/7 下调,吸收效率降低
其次,肠上皮细胞中TRPM6通道的表达受到抑制,导致活性吸收途径效率降低,同时肠屏障的渗透性增强,离子外漏和炎性反应趋于加剧。
⑶长期失衡引发低镁血症
若这种失衡状态长期持续,则可能引发系统性低镁血症,进而影响机体的能量代谢与细胞信号传导,引起继发性代谢紊乱,如胰岛素抵抗的形成及炎症反应的持续增强,最终损害整体代谢健康。
肠道微生物群通过多层次机制深刻调控镁的吸收与利用,是维持机体镁稳态的关键生物学因素。健康的菌群结构可通过发酵产生短链脂肪酸,降低肠腔pH、增强镁盐溶解度,并上调肠上皮镁转运通道(如TRPM6/7)的表达,从而促进镁离子高效吸收。同时,益生菌的代谢活动改善肠屏障功能,抑制炎症介导的通道降解,维持镁吸收持续进行。
相反,当菌群失衡时,SCFA水平下降、肠道环境碱化以及通道表达受抑,会显著降低镁的生物利用度,甚至引发系统性低镁血症及代谢功能障碍。
因此,保持菌群多样性与代谢平衡,对优化镁吸收与全身代谢健康具有重要意义。
当前评估体内镁含量的方法可分为直接测定法与间接评估法两大类。每种方法在反映镁状态的敏感性、特异性及可行性方面各具优缺点。
1
血清(血浆)镁浓度测定
原理: 直接测定外周血清或血浆中的总镁离子含量,是临床常用的基础方法。
常用标准值:0.75–0.95 mmol/L(或1.8–2.3 mg/dL)。
优点:
•操作简便、成本低,可通过常规化学或光谱分析快速获得结果;
•适用于急性临床监测,如电解质紊乱、肾功能异常等情况。
缺点:
•仅反映体内总镁的约 1% ,对镁储备状态敏感性极低;
•血镁浓度受饮食、激素水平、肾功能和血液稀释度影响明显;
•在轻度缺镁或慢性低镁状态下常表现为“假正常”,容易漏诊。
2
红细胞镁(RBC Mg)测定
原理: 测量红细胞内镁离子浓度,更能反映细胞内镁水平。
优点:
•与组织镁含量和代谢活性状态相关性较强;
•适用于慢性缺镁、糖尿病、心血管疾病人群的研究。
缺点:
•样本分离流程复杂,需即时离心处理;
•红细胞寿命较长,难以反映短期动态变化;
•测定标准化程度低,不同实验室间的可比性较差。
3
离子镁(Mg²⁺)检测
原理: 直接测量血液中自由镁离子(生物活性形式)浓度,利用离子选择电极或离子色谱法。
优点:
•能准确反映生理活性镁水平,比总镁更具临床相关性;
•在重症监护、心律失常管理中应用价值高。
缺点:
•仪器昂贵,检测条件严格(需无空气暴露、控制pH和温度);
•样品保存与运输难度高,不适合集大样本研究。
4
尿镁排泄测定
原理: 通过24小时尿液或随机尿镁/肌酐比评估肾脏排镁功能。
优点:
•可判断低镁状态来源:分辨肾性镁丢失与摄入不足;
•利于药物(如利尿剂、铂类药物)诱导的电解质异常评估。
缺点:
•尿镁受肾小管功能、摄水量与饮食镁摄入影响大;
•单次检测波动性强,需与血镁水平联合判断。
5
骨骼或肌肉镁测定
原理:镁的大部分储存在骨骼和肌细胞中,采用骨活检或肌肉组织分析评估体内总储镁。
优点:
•最能代表全身镁储备量,是研究性“金标准”。
缺点:
•操作侵入性强、成本高,不适合常规临床使用;
•检测周期长,临床价值主要局限于科研应用。
6
非侵入性或间接评估方法
包括: 饮食问卷(FFQ)、头发镁、指甲镁或唾液镁检测。
优点:
•采样方便,可用于营养流行病学调查;
•能反映长期摄入趋势。
缺点:
•与生理有效镁缺乏相关性弱;
•易受环境污染、样品处理影响,数据可靠性不足。
综合来看,血清镁是初筛与临床常规监测的首选指标,而红细胞镁与离子化镁更适用于研究或特殊人群诊断。若需明确镁缺乏性质,应结合尿镁排泄、饮食摄入评估及必要的负荷试验,以全面评价机体镁稳态。
未来研究趋势正向多指标综合模型发展,结合代谢组学与离子组学数据,可更真实反映体内镁的动态分布及生理活性状态。
当我们了解到自身存在镁缺乏的情况时,就需要采取一些切实可行的方法来进行有效补充,以帮助身体恢复正常的镁平衡状态。
那么,在我们的日常饮食与生活习惯中,都有哪些常见且实用的途径能够帮助增加镁的摄入量、促进吸收与利用呢?接下来,让我们一起了解这些改善低镁状态的方法。
1
优化饮食结构提高镁摄入与吸收
√富镁食物摄入
•主要来源包括:深绿色叶菜(菠菜、羽衣甘蓝)、全谷物、豆类、坚果(杏仁、腰果)和种子(南瓜籽、亚麻籽)。
•海产品(三文鱼、海藻、贝类)和黑巧克力中亦含有高可溶性镁。
•膳食模式建议:遵循地中海式或DASH饮食可显著提升镁摄入并改善心代谢健康。
√避免阻碍吸收的膳食因素
•过量膳食脂肪、草酸(如菠菜、甜菜)、植酸(谷物外皮)和高磷饮食会与镁形成难溶盐,降低吸收率。
•过量钙、酒精、咖啡因及精制糖也会削弱肠镁吸收或加速排泄。
镁和饮食
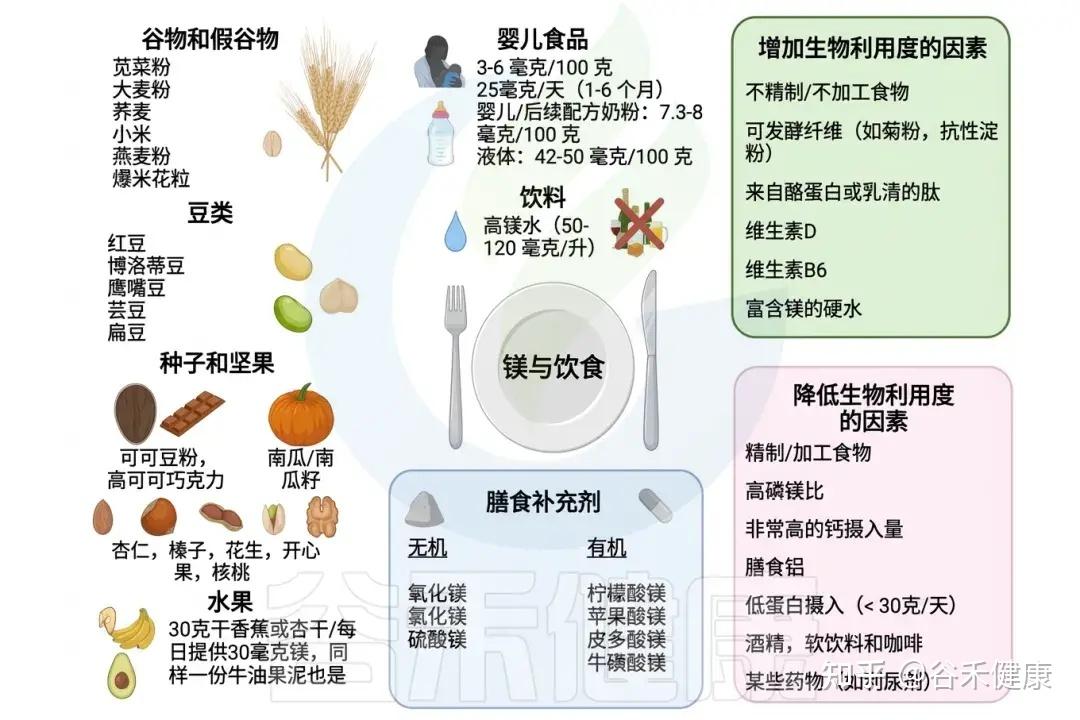
2
调整肠道微生态
√益生菌补充:
部分菌株(如Lactobacillus plantarum、Bifidobacterium longum)具有生物螯合和离子交换能力,能直接结合或转运镁离子,并改善肠环境酸度、增强屏障功能并提高镁吸收。
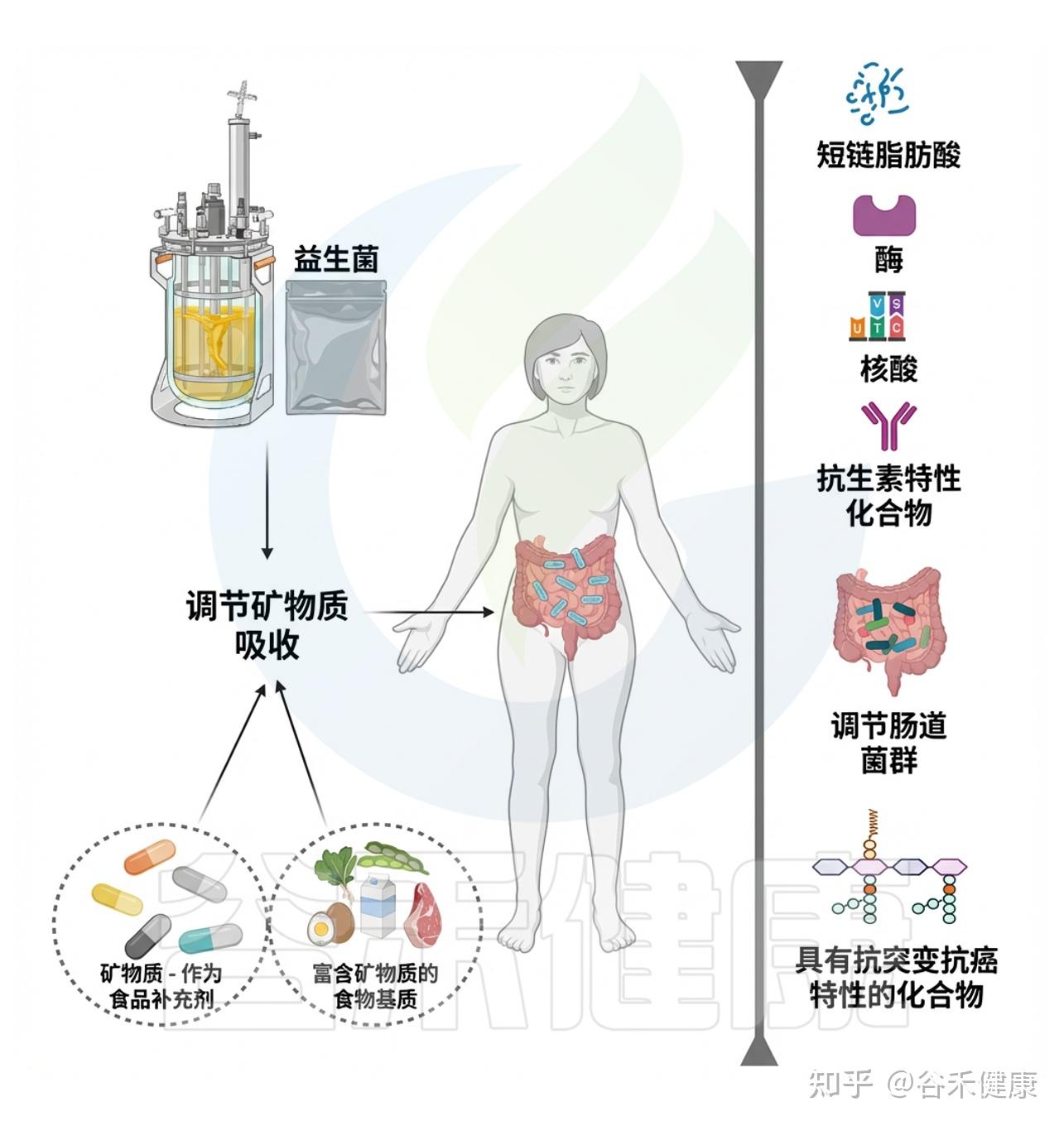
√益生元摄入:
•菊粉、抗性淀粉、低聚半乳糖等促进有益菌生长与SCFA生成;与镁补充剂合用可产生“协同吸收效应”。
•天然食材包括菊苣根、洋葱、香蕉、燕麦、抗性玉米淀粉及豆类。
√合生元策略:
•将益生菌与益生元联合应用,持续重构菌群结构,稳定镁吸收体系。
•常见组合为 Lactobacillus rhamnosus+菊粉,或 Bifidobacterium bifidum+低聚果糖。
3
合理的镁补充剂
常用镁补充剂类型与吸收特征:

√与其他补充剂可能存在竞争或协同作用:
•维生素D与镁存在协同作用:D₃促进TRPM6表达并增强肠吸收。
•避免同时与高钙或铁补充剂同服,以减少竞争性吸收。
√特殊人群调节方案:
•老年人:胃酸分泌低、吸收率下降,应选择有机镁盐并配合益生菌。
•肾功能受损者:慎用高剂量补充,监测血镁。
•糖尿病与代谢综合征患者:镁可改善胰岛素敏感性,推荐持续低剂量补充。
4
促进镁利用减少体内丢失
√控制促镁排泄因素:
•减少长期使用袢利尿剂、质子泵抑制剂(PPI)和胰岛素过量。
•控制高醇饮用与慢性应激,因皮质醇及儿茶酚胺可显著促进镁排泄。
√维持酸碱平衡与肾功能健康:
•酸负荷饮食(高蛋白、高磷)会增加尿镁流失;适当碱化饮食(蔬果丰富)有助保镁。
•保证充足水合状态,稳定肾小管重吸收。
√协同营养素支持:
•维生素B₆:促进镁向细胞内转运。
•维生素D:增强肠镁吸收及骨镁沉积。
•钾水平充足也有助于稳定细胞内镁含量。
5
重视长期个性化健康
√联合评估与动态调整:
•定期监测血清镁、尿镁及相关代谢指标(葡萄糖、CRP、肾功能)。
•使用生物可利用性更高的镁盐并结合饮食优化,可在8–12周显著改善低镁状态。
√持续干预与个体化方案:
•长期追踪应以低剂量持续,而非短期高剂量“冲击补充”;
•根据个体肠道菌群、代谢状态及药物使用情况调整补充途径与剂型。
镁不仅是基础营养素,更是连接营养代谢、肠道微生态与整体健康的关键枢纽。鉴于其在人体代谢和系统调整中的诸多作用,镁有望作为代谢紊乱的潜在标志物。镁稳态的失衡可能预示心血管疾病、糖尿病、神经退行性疾病、胃肠道疾病或肾病、骨质疏松及慢性炎症等多种疾病风险的早期信号。
值得注意的是,近年来肠道微生物群在镁吸收、储存与代谢调控中的重要作用逐渐被揭示。菌群不仅影响了镁的生物利用效率,还通过其代谢产物影响宿主的免疫、炎症及能量代谢网络。
未来的健康干预方向应超越单纯的营养补充,转向以微生态调节为核心的“营养—菌群—代谢”综合模式。通过益生菌、益生元及合生元的个体化应用,可实现镁吸收效率的长期优化,并间接改善血糖、血压及炎症状态,从而强化整体健康管理。
深入研究镁与菌群互作的分子机制,将为精准营养学、代谢疾病防控及功能性食品研发提供新的理论支撑。构建以肠道生态平衡为基础的镁稳态干预体系,或将成为促进健康老龄化、预防慢性病及实现个体化营养治疗的发展方向。镁与菌群的协同作用,不仅代表了一种新的营养理念,更是未来健康科学从“补充”走向“调控”的关键转折点。

谷禾健康
宝宝湿疹反复瘙痒总难根治,季节交替时喷嚏不断,擦不完的鼻涕,整晚咳嗽甚至发展为哮喘,到了学龄期孩子仍无法专注听讲…
如今约40%中国儿童在成长过程中会出现至少一种过敏症状(尤其是过敏性鼻炎),神经发育问题发生率也逐年上升。
多中心流行病学调查和系统综述显示:
神经发育障碍整体患病率呈上升趋势,已从 0.5 %‑1 % 逐步提升至约 5 %‑10 %,包括所有子类的累计比例。
中国儿童常见过敏性疾病的患病率总体呈上升趋势,哮喘约 3%–7%,过敏性鼻炎约 20%–25%,湿疹/特应性皮炎约 8%–20%,食物过敏约 3%–8%,且城市和经济发达地区患病率更高。
而这些看似独立的健康难题,都指向了肠道微生物群。
从出生开始,我们的肠道微生物群便陆续完成定植,在大约三年的时间里,从几乎一片空白,逐步演变为一个多样、稳定且高度复杂的微生态系统。这一阶段被认为是影响儿童健康的关键窗口期,而现代生活方式导致的环境污染、抗生素滥用、高加工食品摄入过多等问题,都在悄悄破坏着孩子娇嫩的肠道环境。
大量研究提示,生命早期一旦出现肠道菌群失衡,不仅会增加炎症性肠病、过敏性疾病(如哮喘、湿疹、过敏性鼻炎等)、自闭症、注意力缺陷多动障碍、儿童肥胖、糖尿病等多种儿科疾病的风险,其影响还可能延续至成年。相反,如果在这一时期通过合理的方式塑造一个平衡的肠道菌群,则有机会改善儿童免疫与代谢反应,甚至为一生的健康打下基础。
基于此,本文将围绕“早期肠道菌群与常见儿科疾病”这一主题,回顾当前研究进展,解析早期肠道菌群在疾病发生发展中的作用机制,并探讨可能的干预思路与临床应用前景。
我们知道肠道菌群不仅仅帮助我们消化食物,它还在免疫、营养吸收、抵御病菌、维护肠道屏障的完整性中扮演着关键角色。
尤其是在生命早期,肠道菌群会建立起连接大脑、肺、皮肤等远处器官的沟通轴线。这个关键发育窗口期建立的菌群平衡,就给未来打下了扎实的基础。如果这时候建立的菌群平衡被打破,就可能对儿童健康产生深远的不利影响,甚至引发多种疾病。
肠道与各大器官的交流
肠道菌群通过复杂的免疫、代谢和神经内分泌途径,与身体多个器官进行着持续的双向沟通。
这是最有名的这一条,大脑和肠道菌群,其实从宝宝还很小的时候就一起长大。
肠道菌群会产生影响情绪和行为的神经递质(如GABA),通过迷走神经和血液循环传递给大脑。
反过来,我们一紧张、焦虑,脑子里的压力信号会改变肠道里的环境,比如让酸碱度、激素水平发生变化,进而影响肠道菌群。这条轴线的失调与自闭症谱系障碍和注意力缺陷多动障碍有关。
再来看肠道和皮肤的关系。很多人都说“脸就是肠道的镜子”,其实还真有点道理。
肠道如果不太健康、菌群有点失衡,就很容易引发一些慢慢的、低度的炎症,还有一些代谢产物。这些东西会跟着血液到皮肤那儿去,影响皮肤表面的菌群平衡和局部免疫状态。
结果就是,像婴儿湿疹之类的问题,可能就会变得更严重或者更难治愈。
听起来好像肠道和肺离得挺远的,但它们之间也在悄悄互相影响。肠道菌群产生的代谢物,还有各种免疫信号,也可以通过血液到达肺部,帮忙调整呼吸道的免疫反应。肠道菌群失衡会增加儿童患哮喘和过敏的风险。
其他重要轴线: 研究还揭示了 肠-胰腺轴、肠-脂肪轴 等通路的存在。
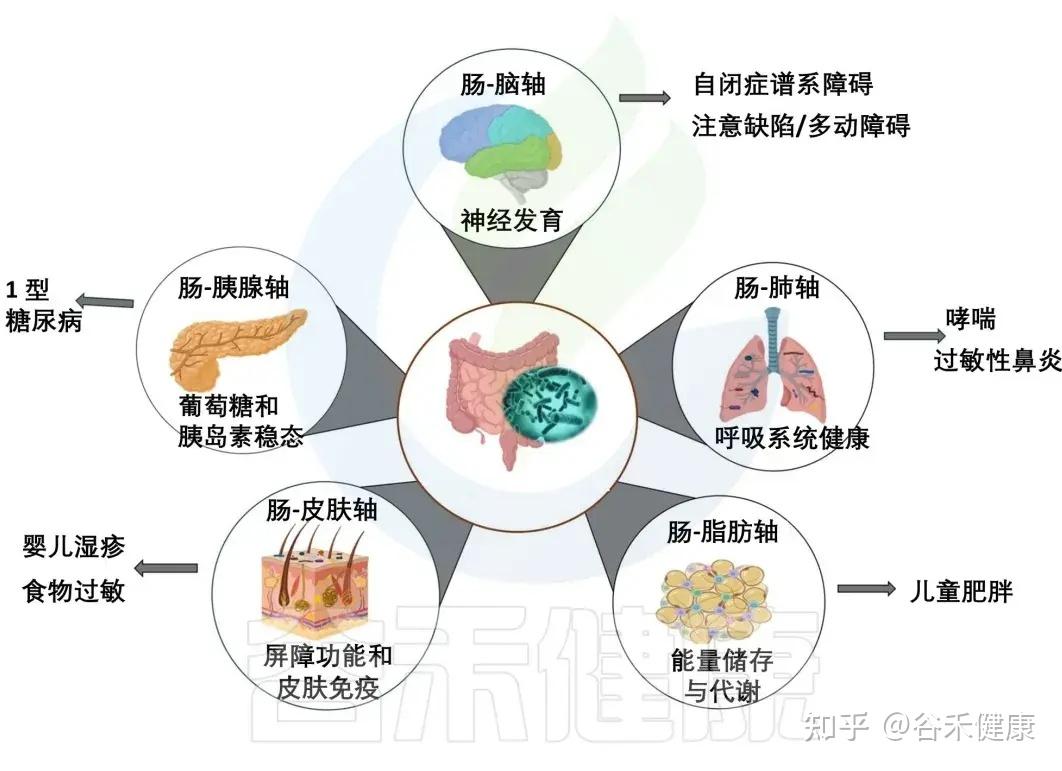
总之,肠道菌群通过这些复杂的代谢和信号通路,构建了一个覆盖全身的通讯网络,成为调节人体从免疫到神经系统等多个方面健康的核心枢纽。
母亲围产期营养状态
婴儿肠道菌群的初始状态,很大程度上继承自母亲。母亲在围产期保持均衡、健康的饮食,是为后代肠道菌群打下良好基础的第一步,也是至关重要的一步。
失衡的风险
如果母亲在孕期偏爱高热量、高脂肪的西式饮食,可能导致自身肠道菌群的失调。这种失调会通过胎盘、产道或母乳传递给婴儿,影响宝宝肠道健康。同样,营养不良也会导致菌群失调,可能增加如γ-变形菌门这类潜在致病菌群的出现。
▸细菌的入住,在出生前就开始了
与传统的“子宫无菌”理论不同,新的证据显示,细菌定植其实早在胎儿的宫内阶段便已开始。研究发现,非致病菌在妊娠期间能从母体肠道转移至胎儿,并存在于胎盘、羊水、脐带、胎儿组织及胎粪中。
而这个宫内微环境的菌群构成,又会受到母亲的健康状况、饮食习惯以及孕期菌群变化的影响,这为婴儿早期肠道菌群形成奠定了最初的基础。
分娩方式:顺产 or 剖腹产?
分娩方式,主要是顺产(即阴道分娩)与剖宫产,是显著影响新生儿肠道菌群组成和发育的关键因素。
▸出生方式决定了宝宝的第一批肠道居民
-顺产
顺产新生儿会接触到母体阴道与肠道来源的多样菌群,包括乳酸杆菌和双歧杆菌等优势菌株,这些菌群构成了初始肠道定植的核心。
顺产宝宝的菌群不仅更稳定、多样性也更高,而且他们的粪便菌群构成与母亲相似。
-剖宫产
与顺产儿相比,剖宫产儿的肠道菌群表现为双歧杆菌和乳酸杆菌的定植延迟、拟杆菌门的多样性较低,而主导菌群则通常是医院环境中常见的肠球菌属。
剖宫产宝宝的肠道里的菌群种类更少,而且这些菌群更像是来自妈妈皮肤和口腔里的。
注:
刚出生那段时间,两种分娩方式带来的菌群差别很大,这个差别会随着时间推移而慢慢变小。
有研究发现剖宫产宝宝的这种菌群特点,可能会让他们以后更容易得一些慢性病(比如哮喘、1型糖尿病、肥胖,但这个说法目前还有争议,不是所有研究结果都一样。
▸如何为剖腹产宝宝补回有益菌?
母体的阴道和粪便菌群是新生儿获得初始微生物的最主要、最丰富的来源。
——第一个办法:阴道菌群移植
为了弥补剖宫产宝宝在这方面的缺失,一些研究探索了“阴道菌群移植”的效果,就是说把母亲的阴道分泌物涂抹给新生儿。
部分研究显示,这种方法有助于部分恢复菌群定植;然而,另一些研究却得出相反结论,认为没什么影响。这种结果上的分歧,很可能是因为阴道菌群难以在婴儿肠道内实现长期定植。
——第二个办法:粪便菌群移植(仅科研探索阶段)
相对来说,母体消化道的菌群多样性要高得多,对宝宝肠道菌群定植的影响可能更深远。
基于这一理念,有些研究认为,母体粪便菌群移植是一种更有效的方法。它能近乎完全地恢复母婴间的微生物垂直传播,有效重塑新生儿的早期肠道菌群。
注:在我们看来,当前有关粪菌移植在儿童中的应用,仍主要停留在科研探索层面,且存在不少限制。成人与儿童的肠道菌群在组成和功能上存在显著差异,而目前临床检测和数据库中可用于精准匹配的儿童菌群数据仍相对有限。因此,现阶段粪便菌群移植更多作为研究工具和小范围、慎重的学术交流内容,并不适合作为常规干预手段。
喂养方式:母乳 or 配方奶?
▸母乳:不仅仅是食物
母乳 (HBM) 不仅为婴儿提供必需的营养,更在生命早期肠道菌群的成熟过程中扮演着至关重要的角色。
母乳富含多种生物活性成分,如免疫细胞、细胞因子、乳铁蛋白、抗菌蛋白和肽、抗体以及母乳低聚糖(HMOs)。这些成分能够对婴儿的肠道菌群进行正向调节,帮助宝宝抵抗感染。
与牛奶相比,母乳含有超过250种不同类型的母乳低聚糖。这些母乳低聚糖对于调节婴儿肠道微生物和影响免疫系统至关重要。
▸菌群差异:母乳 v.s. 配方奶
先前的研究已证实,母乳喂养和配方奶喂养的婴儿,其肠道菌群存在显著差异。
基于母乳喂养带来的整体健康益处,世界卫生组织推荐在婴儿出生后一小时内即开始母乳喂养,纯母乳喂养至六个月,并持续到至少一岁。
▸配方奶粉:添加母乳低聚糖
母乳喂养好处多多,但很多母亲会因乳腺炎、奶水不足或产后抑郁等原因而无法实现。为了弥补这一差距,现代高端婴幼儿配方奶粉开始添加在母乳中发现的关键成分——母乳低聚糖(HMOs),如2-岩藻糖基乳糖和乳糖-N-新四糖,这些有可能促进健康早期肠道微生物群的发展。
有研究发现,添加了HMOs的配方奶粉确实能带来积极效果:
▸生命早期,母婴间菌群如何传递?
母乳里的有益菌从何而来?研究人员认为,存在一个“肠道-乳腺通路”:
母亲肠道中的一些有益菌株可以被自身的免疫细胞(如树突状细胞)捕获。
这些免疫细胞随后通过淋巴系统,将这些细菌穿透肠上皮运送到乳腺。
最终,这些来自母亲肠道的有益菌就进入了母乳,并在喂养时传递给宝宝。
此外,母亲肠道细菌分泌的一些含有生物活性物质的小包裹(即细菌外囊泡,BEV),也可能通过同样的方式进入母乳,影响宝宝的肠道菌群建立。
总而言之,母亲自身的肠道健康状况,会直接影响母乳的微生物构成,进而深刻地影响着宝宝早期的肠道健康。
辅食添加:菌群发展的分水岭
当婴儿长到6个月左右开始添加辅食时,其肠道菌群会经历一次重大的转变。菌群会从以双歧杆菌为主的相对简单的状态,演变为一个相对更多样化的菌群。
-时机很关键
辅食添加太早或太晚,都会干扰菌群的正常成熟过程。因此,推荐在6个月左右开始添加营养丰富的辅食。
营养成分
不同的营养成分对肠道菌群有不同的影响。
▸宏量营养素(碳水、蛋白质等)
-碳水化合物
高纤维和低升糖指数的食物是肠道有益菌的“优质口粮”,有助于促进普雷沃氏菌属、双歧杆菌、毛螺菌属(Lachnoclostridium)和罗斯氏菌属(Roseburia)的生长。
一项研究表明,在婴儿的补充喂养阶段,食用来自各种全谷物、豆类、水果和蔬菜的膳食纤维会导致大量产丁细菌,如粪杆菌、粪球菌、Dorea、Oscillospira。
-蛋白质
相比动物蛋白,植物蛋白如扁豆,豆类,植物性饮食通常富含膳食纤维,含有足够量的多不饱和脂肪酸,通常更有利于结肠细菌的健康。
当然,为了维持肠道稳态,均衡摄入两种蛋白是最佳选择。
▸微量营养素(维生素与矿物质)
-维生素
多数维生素都能促进有益菌的生长,例如双歧杆菌和Akkermansia菌,同时抑制像艰难梭菌这样的致病菌。
例如,维生素A、B2、D、E、β-胡萝卜素可以增加有益共生菌的丰度。
维生素B2、E可以增加有益的SCFA产生菌。
-矿物质
它们的作用比较复杂。镁、钙、硒等有益于菌群。但需要特别注意的是,给婴儿补充铁剂可能是一把双刃剑,过量时可能会抑制双歧杆菌和乳杆菌科等有益菌,反而为一些有害菌的繁殖提供机会。
▸植物营养素(特别是多酚)
这是一类广泛存在于水果、蔬菜中的强大活性成分,能直接调节菌群。例如,生命早期服用葡萄多酚可以促进有益的Akkermansia和乳酸杆菌的生长。
更重要的是,母亲在孕期和哺乳期摄入的植物营养素可以传递益处。例如,母亲饮食中:
生命早期是一个塑造肠道菌群的黄金窗口期。在此期间,通过科学合理的营养干预来优化菌群构成,是预防多种儿童期乃至成年期疾病的有效策略。
运 动
除了我们吃什么,我们的“动与不动”也是影响肠道菌群的另一个关键因素。
▸运动如何优化肠道菌群?
规律的运动被证明可以从多个方面积极地重塑肠道微生态。
-增加有益菌和多样性
运动能显著增加肠道中有益菌的种类和数量,特别是那些能产生短链脂肪酸(SCFAs)的好菌。
宏基因组分析显示,运动量与罗斯氏菌属(Roseburia)和Akkermansia菌丰度呈正相关。
-久坐的危害
相反,久坐不动的生活方式则会破坏这种平衡。研究表明,久坐会导致肠道菌群多样性降低,并使得那些能更高效捕获食物能量的厚壁菌门增多,而放线菌门细菌减少。
▸生命早期的运动
在正确的时间运动,效果可能事半功倍,生命早期或许就是这样一个黄金窗口期。
-早期锻炼效果更佳
一项动物研究发现,如果在还很年幼的时候就开始运动,相比长大成年后才动起来,更能明显、积极地改变肠道菌群。
-具体的菌群变化
研究显示,幼年期的运动能显著增加拟杆菌门的比例,同时降低厚壁菌门的比例,这种菌群构成上的变化与增加瘦体重)直接相关。
基于这些益处,世界卫生组织建议,5岁以下的孩子,每天应累积进行至少180分钟的各类身体活动。
▸母亲的运动:给宝宝的第一份健康礼物
运动的影响力甚至可以追溯到生命开始之前。过去我们认为,父母传给孩子的是基因。现在我们知道母亲还可以传递一个健康的微生态系统。
-孕期运动的益处
母亲在孕前和孕期的规律运动,可以帮助宝宝的肠道减少有害菌,同时富集能产生短链脂肪酸的有益菌,从而改善宝宝整体的代谢健康。
-可能的机制
科学家推测,这其中的一个可能机制是,运动改变了母亲体内的代谢环境,如激素水平、血液循环和能量利用。这些生理变化信号传递到乳腺,影响了母乳的生产过程。
母乳中母乳低聚糖(HMOs)的种类和数量发生了改变。HMOs是母乳中仅次于乳糖和脂肪的第三大固体成分,但宝宝自己无法消化它。它不是给宝宝吃的,而是专门给宝宝肠道里的有益菌吃的。这样一来,宝宝的有益菌在菌群建立的早期就占据主导地位,抑制了有害菌的生长。
均衡的营养加上规律的身体活动,能最大程度地对早期肠道菌群的健康发展产生积极和深远的影响。
在生命早期操纵微生物发育的因素
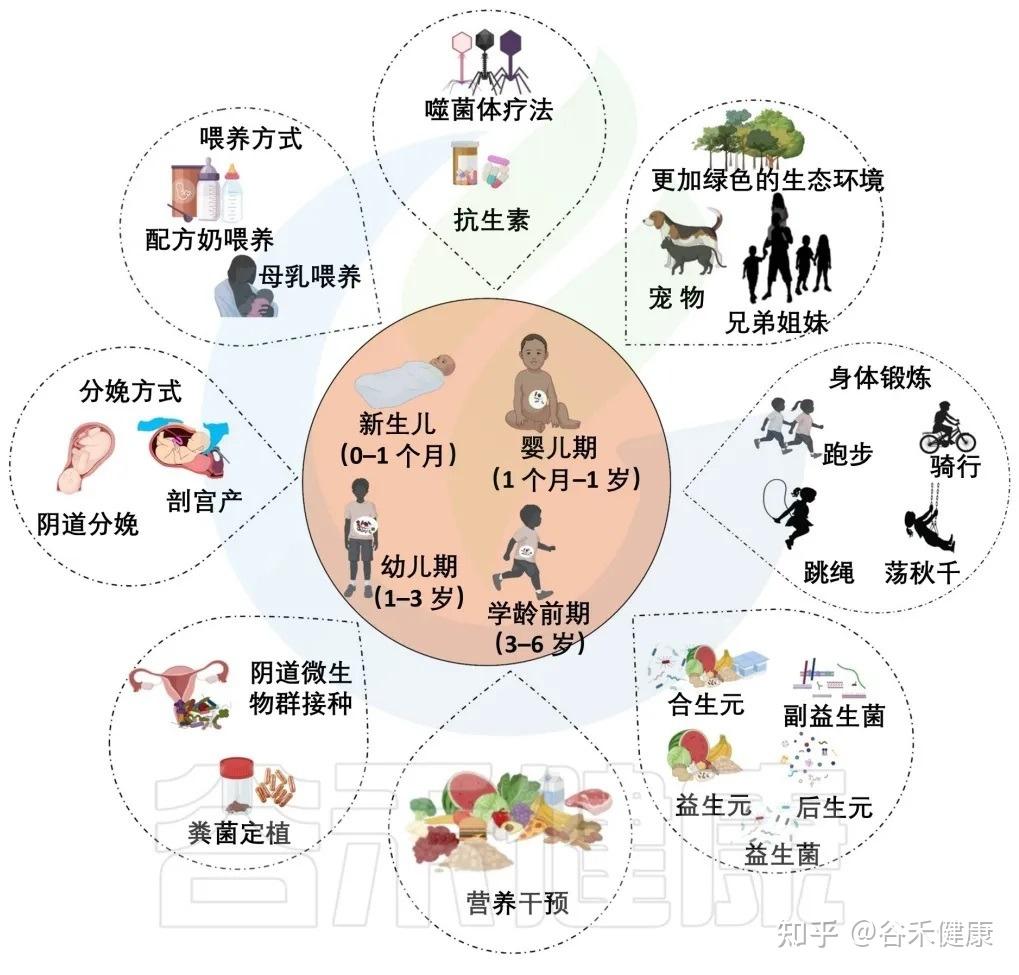
抗生素的使用:有利有弊
有些家长有过这种经历,带孩子看了一次病,用了抗生素,当时觉得病总算好了,松了口气,结果没过几天,新的问题又来了:孩子就开始拉肚子,或者突然吃一些东西开始食物不耐受了,甚至之前控制得好好的湿疹也加重了。这其实也跟肠道菌群有关。
关于抗生素,它确实对付细菌感染特别管用,不过呢,凡事都有两面,尤其是在生命早期,在宝宝身体各种系统都还在搭建的时候使用它,可能需要付出一些看不见的代价。
抗生素最大的问题在于其“不分敌我”的特性,它们不仅杀死了致病菌,也对肠道内大量的有益菌造成了连带伤害,导致肠道菌群失调。这种早期的菌群失调与日后的多种健康问题,如肥胖、哮喘以及抗生素耐药性等都有可能相关联。
婴儿接触到抗生素主要有两种途径:
▸母亲用药:穿越屏障的连锁反应
即使是母亲使用抗生素,其影响也能传递给宝宝。
-影响机制
在分娩期间,抗生素不仅会通过脐带传递给胎儿,还会改变母亲产道和粪便中的菌群,这些都是婴儿在出生时获得初始菌群的来源。
-对婴儿菌群的直接冲击
研究发现,经历过母亲产时抗生素暴露的婴儿,其粪便菌群表现出:
整体多样性降低;
放线菌门和拟杆菌门丰度显著减少;
变形菌门占据了主导地位。
-对母乳的影响
母亲使用的抗生素还会改变母乳中的微生物,这会直接阻碍双歧杆菌和乳酸杆菌等健康菌株在婴儿肠道的正常定植。
▸婴儿用药:艰难梭菌感染风险增高
当婴儿自己直接使用抗生素时,对肠道菌群的影响更为直接。
研究证实,这会导致婴儿肠道菌群构成发生剧变,主要表现为:有益的双歧杆菌数量锐减,而肠球菌属和克雷伯氏菌属等机会性致病菌的数量则会升高。
有些儿童在使用抗生素之后会发现,艰难梭菌(Clostridioides difficile)感染的风险会显著升高,艰难梭菌感染可能会引起严重的腹泻和结肠炎症。
除艰难梭菌外,抗生素使用还可能导致耐药菌株的选择性增长。例如,耐甲氧西林金黄色葡萄球菌(MRSA)和产超广谱β-内酰胺酶(ESBL)的肠杆菌科细菌,这些在抗生素治疗后更容易定植。
长远来看,可能会让宝宝更容易反复出现肠胃不适、拉肚子,或小病不断。因此,在抗生素用药之后,需要格外关注宝宝的饮食,排便,以及肠道菌群状态。
环 境
除了饮食和运动,我们生活于其中的物理和社会环境,也对生命早期的肠道菌群发展有着深刻而直接的影响。从家里的宠物到社区的公园,都在悄无声息地参与塑造这个微小的生态系统。
▸ “不干不净,吃了没病”
过度干净的环境,有时反而不利于建立一个强大的免疫系统和健康的肠道菌群。适度地接触来自外界的微生物,对婴儿来说是一种有益的早期训练。
-宠物带来的益处
宠物猫狗陪伴着孩子长大,像家人一般为孩子带来情感上的安慰与支持,也为孩子性格带来温暖的影响,让孩子感受更多无条件的爱与接纳。
从肠道菌群的角度来说,研究发现,与宠物(如猫狗)一起长大的婴儿,其肠道菌群的丰富度和多样性通常更高。具体来说,他们的肠道中比如双歧杆菌、颤螺菌属(Oscillospira)和瘤胃球菌属(Ruminococcus)这些有益菌的丰度会更丰富。

有趣的是,养猫的人肠道里的双歧杆菌水平也比不养猫的人要高。这就意味着,和猫咪一起生活,可能会通过菌群的交流,悄悄地帮我们补充有益菌,提升肠道健康水平。
注:然而也存在一些需要注意的潜在风险。研究指出,猫肠道里的抗生素耐药基因(ARG)丰度显著高于人类。这些耐药基因可能会通过猫咪在家里的活动(比如它走过地板、沙发),转移到环境中,进而有机会进入人体。简单说,就是猫可能会把它携带的耐药基因传递给主人,这增加了我们体内细菌变得耐药的可能性。
-兄弟姐妹的菌群共享
同样,有哥哥姐姐的孩子,其肠道菌群的多样性也更高。
道理其实很简单。宠物和哥哥姐姐会将更多来自室外(如土壤)或他们自身的微生物带入环境中,让婴儿有机会接触到更多样的细菌,这就像是给婴儿的肠道菌群进行了一次自然接种,有助于其发展得更加健全和有韧性。
▸新冠的意外发现:社交隔离对菌群的影响
新冠为我们提供了一个独特视角,观察环境剧变如何影响肠道菌群。
-菌群多样性下降
研究人员发现,在疫情期间出生的婴儿,其粪便样本中的微生物多样性显著低于疫情之前的同龄婴儿。
-可能的原因
这种变化被认为与疫情期间的特殊生活方式有关,包括:社交活动大幅减少,以及消毒剂、洗手液等清洁产品的使用频率急剧增加。这使得婴儿接触外界微生物的机会大大减少。
▸ 环境的力量:来自大自然的馈赠
我们与自然环境的接触,也能产生意想不到的健康关联。
-母亲接触绿色环境的益处
最近的一项研究指出,如果母亲在孕期更多地接触住宅区周围的公园、绿地等绿色环境,其母乳中母乳低聚糖(HMOs)的多样性和浓度都会更高。

我们已经知道,HMOs是婴儿肠道有益菌的超级食物。因此,通过提升母乳的质量,母亲接触绿色环境的益处最终能够传递给婴儿,积极地影响其肠道菌群的健康发育。
因此,生命早期丰富的环境暴露至关重要。一个不过分无菌、能适度接触来自他人、宠物和大自然的微生物的环境,是帮助孩子建立一个多样、稳定且有韧性的肠道菌群的关键因素之一。
生命从出生到3-6岁的这段时间,是肠道菌群发育的“关键窗口期”。这个阶段的任何干扰,都可能让菌群的健康发展偏离轨道,从而为日后儿童期的各种疾病埋下隐患。
儿童过敏
儿童过敏性疾病,包括湿疹、哮喘、过敏性鼻炎、食物过敏等,是一组以免疫系统对无害物质产生过度反应为特征的常见慢性病。它们的发病,往往与遗传和环境因素共同相关。
从临床分型上看,这些疾病都属于特应性疾病,其核心是身体产生了过度的IgE抗体来对抗那些本应无害的环境物质(如花粉、尘螨、某些食物等)。并且,患有其中一种过敏的孩子,往往更容易患上另一种,这被称为过敏进程。
▸肠道菌群:过敏病程中的关键调控者
这几年,无论是基础研究还是临床队列,都越来越一致地指向一个关键环节,肠道菌群在过敏的发生发展中扮演着关键的调控角色。
在我们对过敏儿童样本的菌群数据分析中,可以看到一种具有代表信的肠道菌群失调。
具体来说,过敏儿童的菌群通常具备几个共性:
更细分到食物过敏的时候会发现,不同食物过敏原(比如牛奶蛋白、鸡蛋蛋白、花生等)相关的菌群变化还可能各有其特异性的模式,不同分型这也是我们正在关注的方向。
▸失控的免疫反应:当前主流的菌群-免疫假说
那肠道菌群究竟是通过什么路径去影响免疫系统,进而推动过敏的呢?目前在学术界相对被广泛接受的,是围绕短链脂肪酸,尤其是丁酸盐的一条核心假说:
丁酸盐的缺失:菌群失调导致关键的代谢产物——丁酸盐浓度下降。
刹车失灵:丁酸盐是促进初始T细胞分化为调节性T细胞(Tregs)的关键信号。Tregs在免疫系统中可以理解为一个刹车装置,负责抑制过度的免疫反应,维持免疫平衡。
免疫过度激活:丁酸盐减少导致Tregs数量不足或功能受损,刹车失灵,免疫系统便会对无害物质反应过度。
典型免疫表型:在这种菌群失调的背景下,我们在过敏儿童身上通常可以看到这样一组特征性变化:
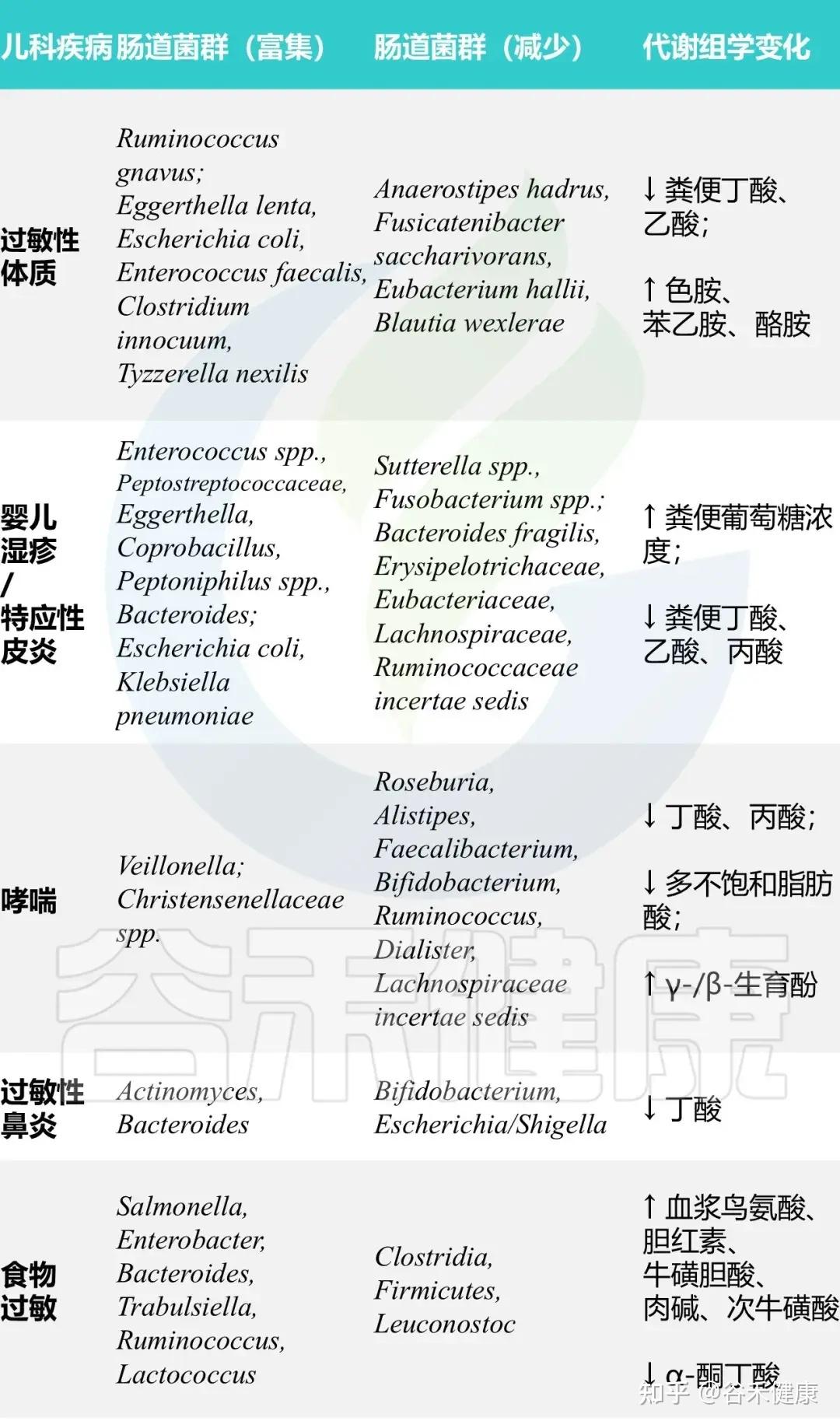
总而言之,生命早期的肠道菌群失调,通过影响免疫系统的正常教育过程,为日后一系列过敏性疾病的发生埋下了伏笔。而具体的菌群和代谢物变化,则在不同的过敏性疾病中呈现出各自的特征。
肠道菌群检测就是力求精准地捕捉到这些早期的菌群和代谢物特征,在实际应用中,我们并不会仅看哪个菌高了或者低了,而是会把菌群特征与已有的临床和免疫学指标放在一起综合判断。
例如,结合谷禾肠道菌群检测报告中的过敏相关菌群模块、肠道屏障评分,以及部分炎症相关因子和免疫调节指标(如 IL‑6 等),我们可以更准确地判断一个孩子的肠道生态,是不是已经处在一个更偏向炎症和过敏反应的状态,尽可能实现对高风险儿童的早期识别,从而为临床的早期预警和干预提供思路。
IBS 与 IBD
在儿童常见的胃肠道疾病中,炎症性肠病(IBD)和肠易激综合征(IBS)是两个重要的类别,它们都与肠道菌群的失衡密切相关。
虽然生命早期的肠道菌群构成与这两种疾病的确切因果关系仍在研究中,但科学家们已经在年龄较大的患病儿童中发现了明确的菌群改变。
▸失衡的菌群
IBS患儿的菌群特征:
IBD患儿的菌群特征:
▸代谢物的变化(化学信号的紊乱)
在IBD患儿体内,色氨酸、琥珀酸盐和3-羟基异丁酸等关键微生物代谢产物的水平较低。
在IBS患儿体内,则观察到葡萄糖、甾醇、乳酸水平升高,而有益的丁酸盐水平下降。
尽管如此,与生命早期相关的特定代谢组学特征,目前仍是一个有待深入探索的领域。
▸失控的免疫反应:炎症
在这两种疾病的背后,一个共同的核心病理机制是:由肠道屏障缺陷和微生物失调共同驱动的、失控的黏膜免疫反应。
IBD:
某些能够降解肠道黏液保护层的致病共生菌,例如黏附侵袭性大肠杆菌(adherent-invasive Escherichia coli),能够诱导巨噬细胞等免疫细胞分泌IL-1β,这会直接促进Th17细胞的分化,点燃肠道炎症。
从口腔跑到肠道的克雷伯氏菌属,也能促进树突状细胞和巨噬细胞产生促炎因子,从而推动初始T细胞分化为Th1和Th17细胞,加剧炎症。
此外,大量能产生IgG抗体的浆细胞被招募到肠道,也参与了IBD的疾病进展。
IBS:
虽然炎症程度不如IBD剧烈,但在IBS患儿的结肠黏膜和血液中,也观察到了先天免疫的过度活跃,特别是来自肥大细胞和单核细胞的活动增强。
适应性免疫同样参与其中,表现为肠道内T细胞数量增加、B细胞活性改变以及抗体的产生。
简而言之,IBD和IBS的发生,是肠道菌群失衡与免疫系统功能紊乱相互作用、相互放大的恶性循环。然而,上述这些复杂的免疫机制,在那些具有高患病风险的婴幼儿身上是如何运作的,仍需要更多未来的研究来进一步验证和阐明。
常见儿科疾病中的早期肠道生态失调
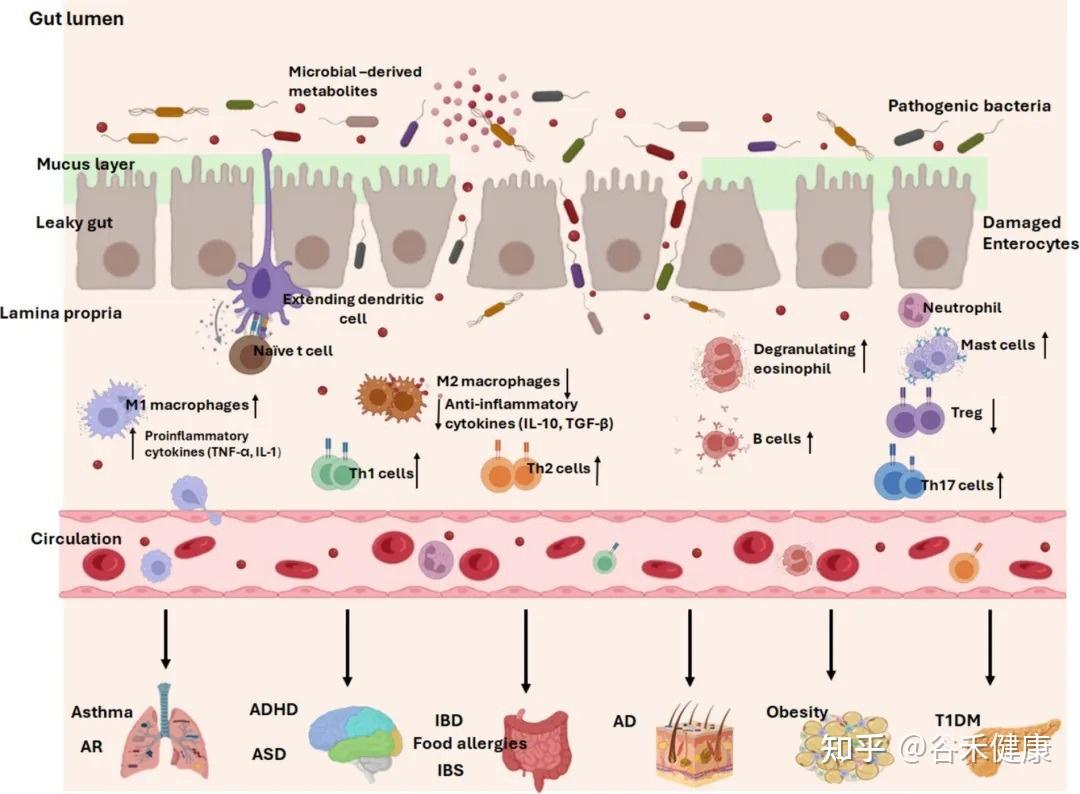
神 经 发 育
生命最初的几年,不仅是肠道菌群建立的关键窗口,也恰好与中枢神经系统发育的关键阶段,如神经突触的形成和髓鞘化高度重叠。这并非巧合。一条被称为肠-脑轴的复杂通讯网络,将肠道和大脑紧密地联系在一起,使得肠道菌群的状况能够深刻影响神经发育,甚至可能参与了自闭症谱系障碍(ASD)和注意力缺陷多动障碍(ADHD)等疾病的发生。
这两种情况有时会同时出现在一个孩子身上。
▸失衡的菌群,失调的化学信号
由于肠道微生物能通过其代谢产物和影响免疫通路来调控神经系统的发育和功能,科学家们将其视为自闭症和多动症发病的一个潜在贡献者。研究发现,与正常儿童相比,具有高患病风险的幼儿,其肠道菌群的构成和产生的生物活性代谢物均存在显著差异。
-菌群失衡
-关键神经递质的缺失
一个尤为引人注目的发现是,这些高风险儿童体内由细菌产生的、具有神经活性的分子——γ-氨基丁酸(GABA)的水平显著降低。
这些发现清晰地揭示了在生命早期,肠道菌群-代谢产物-神经系统之间存在的深刻联系。
▸从肠道炎症到大脑炎症
GABA的减少可能不仅仅是一个孤立的现象,它或许是连接肠道失衡与大脑功能异常的关键一环。GABA是大脑中主要的抑制性神经递质(负责让大脑冷静下来),它还在调节焦虑、改善睡眠、缓解疼痛以及帮助维持神经系统的整体平衡方面发挥着关键作用。
-连锁反应的启动
菌群失调导致GABA等代谢产物水平改变。
GABA水平的改变,加上菌群本身的失调,引发了不正常的免疫反应和全身性炎症(表现为血浆中细胞因子水平的异常)。
这种慢性的全身性炎症会逐渐破坏精密的血脑屏障的完整性。血脑屏障一旦受损,炎症细胞和炎症因子便更容易进入大脑,最终引发神经炎症,从而影响大脑的正常发育和功能。

简而言之,ASD和ADHD的发生,可能部分源于肠道开始,经由免疫系统,最终波及大脑。然而,这其中的具体机制非常复杂。尤其是在菌群发育的关键窗口期,究竟哪些早期的免疫学特征能够预示ASD和ADHD的发生,仍然是未来研究需要迫切解答的重要问题。
肥胖及1型糖尿病
在儿童期,最常见的代谢性疾病莫过于肥胖和1型糖尿病(T1DM),而这两者的发生,都与生命早期的肠道菌群失调有着千丝万缕的联系。
▸肠道菌群:调节新陈代谢
我们的肠道菌群在宿主的新陈代谢中扮演着至关重要的“总调度师”角色,其影响力涵盖了脂质与葡萄糖代谢、能量消耗与脂肪储存,乃至胰岛素信号的传导等多个方面。
更关键的是,T1DM的发病高峰期通常在生命最初的5-6年,这与肠道菌群发育的关键窗口期完全重合,这暗示了两者之间强烈的内在联系。
▸失衡的菌群
与健康儿童相比,患有或高风险患有这两种代谢病的儿童,其肠道菌群构成显示出特定的指纹。
-T1DM高风险儿童的预警信号
研究发现,在T1DM高风险儿童体内出现自身抗体(血清学转换)之前,他们的肠道中多形拟杆菌(Bacteroides dorei)和普通拟杆菌(B. vulgatus)的数量就已经显著增加。值得警惕的是,这两种拟杆菌属细菌均与肠道炎症有关。
-肥胖儿童的菌群
从婴儿早期开始,肥胖儿童的肠道中就表现出脆弱拟杆菌(Bacteroides fragilis)丰度更高,而有益的双歧杆菌属(Bifidobacteria)和柯林斯菌属(Collinsella)数量更少。这种菌群模式与日后过度的体重增加直接相关。
▸代谢紊乱:疾病发生前的信号
在疾病真正显现之前,身体的化学信号——代谢产物,可能已经发出了警报。
-肥胖儿童的代谢特征
他们的体内GABA(一种能调节食欲和体重的神经递质)水平显著较低。
同时,血液中对称性二甲基精氨酸和不对称性二甲基精氨酸的水平升高,这两种物质被认为与全身性炎症有关。
-代谢综合征儿童的发现
另一项研究观察到,在5岁的代谢综合征儿童体内,参与糖异生、氨基酸和脂肪酸代谢的循环代谢产物显著增加,这些代谢通路的变化与他们的BMI、腰围和空腹血糖水平密切相关。

▸免疫与炎症:连接菌群与疾病
早期肠道菌群与免疫系统之间的相互作用,是驱动这些代谢疾病的关键。
-肥胖与炎症
在3-5岁的重度肥胖儿童中,可以观察到C反应蛋白(一种全身性炎症标志物)的水平升高。
失调的肠道细菌及其代谢产物(分解代谢物)会损害脂肪组织的稳态,并诱发慢性低度炎症,从而助长儿童肥胖的发生。
-T1DM与炎症
儿童T1DM患者体内常常同时存在肠道菌群失调和全身性及组织特异性的炎症反应升高,这清晰地表明,肠道微生物在维持宿主代谢健康中扮演着不可或缺的角色。
因此,无论是儿童肥胖还是1型糖尿病,其背后都涉及生命早期的肠道菌群失调,通过扰乱宿主正常的新陈代谢通路和触发不当的免疫炎症反应,为这些慢性代谢疾病的发生和发展铺平了道路。
在生命早期干预和重塑肠道微生物群,是促进长期健康的有效方法。儿童的肠道菌群比成人更容易被环境因素改变,且该时期是免疫系统成熟的关键窗口,而免疫系统又受到肠道菌群的调节。
这里我们来了解一下在常见儿童疾病中,通过操纵早期肠道微生物组进行干预的前沿研究。
儿 童 过 敏
既然我们知道了肠道菌群失调是儿童过敏的关键推手,那么,我们是否能主动出击,通过调整菌群来拨乱反正,预防或缓解过敏呢?
答案是肯定的,现有研究已经为我们指明了几个有希望的方向。
▸抗生素的阴影与警示
在讨论干预之前,必须再次强调预防的重要性。大量证据表明,无论是母亲在孕期还是孩子在生命早期使用抗生素,都会增加儿童患过敏性疾病的风险。
剂量效应: 一项研究发现,母亲孕期使用抗生素的剂量越大,孩子日后患过敏病的风险就越高。
菌群变化: 生命早期使用大环内酯类抗生素,会导致肠道菌群发生剧烈变化,表现为放线菌门数量锐减,而拟杆菌门和变形菌门的数量则会增加。这种菌群的偏移,与哮喘风险和高BMI直接相关。
▸益生菌与益生元
直接补充益生菌及益生元,是最直接的干预手段之一,并且已经展现出不错的效果。
-针对湿疹
一项研究测试了一种含有两歧双歧杆菌(Bifidobacterium bifidum)和短双歧杆菌(B. breve)的特殊婴儿配方。结果显示,使用该配方的湿疹儿童肠道中,短双歧杆菌的丰度显著增加,同时 与乙酸盐合成相关的代谢通路被激活,这与湿疹症状的缓解显著相关。
-预防湿疹
另一项类似的研究也证明,补充益生菌和低聚半乳糖能够丰富肠道中的乳酸杆菌和双歧杆菌,从而有效预防了湿疹的发生。
-针对过敏性鼻炎
在一项芬兰的研究中,研究人员给孕晚期的母亲和她们出生后6个月内的婴儿补充四种益生菌菌株和低聚半乳糖(GOS)。在后来患上鼻炎的孩子组中,这种干预显著提升了双歧杆菌的数量,并减少了拟杆菌的数量。
▸母乳喂养与运动:有争议
-母乳喂养
理论上,母乳是建立婴儿免疫耐受的核心。但关于母乳喂养与预防过敏之间的关系,证据却并不完全一致。一些研究报告了其保护作用,而另一些则认为延长母乳喂养并没有预防效果。这说明其中的机制可能比我们想象的更为复杂。
-体育锻炼
在菌群可塑性最强的童年期,运动也可能扮演着重要角色。有研究显示,3-6岁时体育活动水平低的孩子,在儿童后期患哮喘的风险更高。然而,一个关键的不足是,这些研究并未分析运动究竟是如何影响肠道菌群的。
▸饮食:塑造菌群的日常选择
饮食是影响肠道菌群最基础、也最持久的因素。
PASTURE的一项研究发现,1岁时粪便中丁酸盐和丙酸盐水平高的婴儿,未来患上哮喘和发生过敏性致敏的风险更低。而这些短链脂肪酸的水平,又与婴儿饮食中摄入的酸奶、鱼、水果、蔬菜显著相关。
另一个代谢组学研究则指出,富含油炸和加工肉类的饮食习惯,与哮喘风险以及肠道中克里斯滕森菌科(Christensenellaceae)细菌的增多呈正相关。

「哮喘」最新研究已逐步渗透到更精细层面28 赞同 · 3 评论 文章
IBD 和 IBS
面对由菌群失调驱动的炎症性肠病(IBD)和肠易激综合征(IBS),研究人员正在积极探索一系列干预措施,试图通过调控肠道菌群,来调节肠道内的紊乱。
▸抗生素与噬菌体
越来越多的证据将矛头指向了抗生素。
一项涵盖22项研究的系统性回顾发现,生命最初两年内使用抗生素,与日后患上IBD的风险存在强烈关联。
另一项荟萃分析也指出,母亲在孕期接触抗生素和烟草烟雾,都会增加后代患IBD的风险。
噬菌体疗法:作为抗生素的潜在替代方案,噬菌体疗法(利用病毒去攻击特定的有害细菌)已在人体中显示出良好的安全性。它或许能成为治疗儿童胃肠道疾病的一种新方法,但要真正应用于临床,还需要大量深入的研究。
▸压力
早期生活压力的破坏力:肠道不仅感受生理刺激,也感受心理压力。一项动物研究利用母婴分离模型来模拟早期生活压力,结果发现,这种压力能够诱导小鼠从童年到成年持续出现类似IBS的症状,其肠道菌群也发生了改变,表现为乳酸杆菌、肠杆菌属等有益菌的减少。
▸饮食:塑造肠道的终极力量
在所有干预手段中,饮食调整可能是最基础、也最有效的方法之一。
-需要警惕的饮食
研究表明,高脂肪饮食以及富含可发酵寡糖、双糖、单糖和多元醇(FODMAPs)的饮食,会加剧IBD和IBS的症状。而在一项大型研究(斯堪的纳维亚出生队列的汇总研究数据)中,1岁时大量摄入含糖饮料也与日后更高的IBD风险相关。
-值得推荐的饮食
富含多酚、矿物质和高纤维的饮食则被证实有助于缓解症状。同一项研究发现,1岁时鱼类和蔬菜摄入量高的孩子,未来患IBD的风险更低。
-草本的力量
一些草本植物也显示出潜力。例如,在一项针对103名IBS患儿的随机双盲试验中,服用洋车前子(一种富含纤维的草本)长达6周的儿童,其腹痛发作的次数显著少于服用安慰剂的儿童。
但有趣的是,尽管症状得到了改善,两组儿童的肠道菌群构成却没有观察到显著差异,这提示其作用机制可能不仅仅是通过改变菌群。
肥胖和糖尿病
通过干预和调控生命早期的肠道菌群,我们或许能找到重置儿童新陈代谢的关键,从而预防这些疾病的发生。
▸抗生素
来自母亲的影响:现在的人群数据已经给了比较一致的信号。比如有研究发现,妈妈在备孕阶段频繁使用某些抗生素(像青霉素、喹诺酮类),和孩子日后T1DM 风险上升是相关的。
宫内暴露的后果:胎儿在子宫内接触到抗生素(如青霉素),与宝宝出生后的生长迟缓相关。这种暴露会导致婴儿肠道菌群多样性降低,并引发一系列菌群结构的改变:厚壁菌门和乳杆菌目减少,而变形菌门和拟杆菌门等则相对增多。
▸母乳喂养的黄金标准
在生命最初的几个月里,母乳是主要的营养来源,塑造早期肠道微生物群,促进双歧杆菌主导的肠道,这一点和我们日常检测到的健康婴幼儿菌群特征也是一致的。
明确的保护作用:来自两个基于人群的队列的数据显示,非母乳喂养的婴儿,其日后患T1DM的风险翻了一倍。同时,一项涵盖25项研究的荟萃分析也明确指出,母乳喂养是预防儿童肥胖的显著保护因素。
然而,大多数此类研究未能建立起从“母乳喂养”到“特定菌群改变”再到“代谢改善”之间的完整证据链。
▸益生菌
针对T1DM:研究显示,在生命最初的27天内进行益生菌干预,与T1DM高风险儿童的自身免疫风险降低相关。
针对肥胖:母亲在孕期及产后补充特定的益生菌(如鼠李糖乳杆菌HN001),能有效降低婴儿在2岁时过度肥胖的风险,这提示“母亲补充,婴儿受益”的策略或许能预防儿童肥胖。
▸运动的悖论:明确的效果与未解之谜
在肥胖儿童中的效果:研究证实,在肥胖儿童中,体育锻炼能够降低血糖和促炎通路,同时改善菌群(降低γ-变形杆菌,增加罗斯氏菌属Roseburia等),并提升有益的短链脂肪酸水平。
然而,这些研究几乎都是在年龄较大、菌群已趋于成熟的儿童或青少年中进行的。体育锻炼对于菌群可塑性最强的婴幼儿时期会产生怎样的影响,亟待未来的研究来解答。
▸营养:从母亲到孩子的饮食智慧
-母亲饮食的深远影响
母亲在孕期的饮食可以直接影响其自身的菌群,这种状态又能通过传递来重塑婴儿的早期菌群,从而为孩子未来的代谢健康打下基础。
-植物化学物质
多酚:如葡萄多酚,在动物实验中被证明能促进有益的Akkermansia菌和乳酸杆菌的定植,增加短链脂肪酸的产生。
实验室的研究也表明,无论是来自西兰花的萝卜硫苷,还是来自大豆的染料木黄酮,在生命早期摄入,都能有效减少后代的过度肥胖,改善其整体代谢健康。
这些发现揭示了巨大的潜力,但下一步需要通过转化研究来验证这些营养成分在人类儿童身上是否同样有效,以及它们是如何通过影响肠道菌群来发挥作用的。

膳食纤维对代谢健康和肥胖的影响8 赞同 · 1 评论 文章
儿 童 神 经 发 育
一切始于源头,生命最初的经历深刻地影响着大脑的发育轨迹。
-母乳喂养的保护
一项研究表明,更长时间的纯母乳喂养能够降低儿童患ASD和ADHD的风险。
-需要警惕的风险因素
相反,母亲吸烟、压力大、母乳喂养时间过短(< 4个月)则与更高的ADHD风险显著相关。而孕期感染和母亲患有自身免疫性疾病,也与ASD的发生呈正相关。
-补偿剖腹产的损失
对于剖腹产导致菌群未能正常传递的问题,一种名为阴道菌群移植(VMT)的新技术显示出潜力。研究发现,接受了VMT的新生儿,其肠道菌群和代谢物状况更优(如乳酸杆菌、双歧杆菌增多,克雷伯氏菌减少),并且在6个月大时,其神经发育评分也显著更高。
▸益生菌
直接补充特定的益生菌,为预防神经精神障碍提供了新的思路。
一项长达13年的里程碑式的研究中,科学家们给75名婴儿在生命最初的6个月里补充了鼠李糖乳杆菌GG。结果发现,这一干预引发了长期的、有益的菌群变化。更令人震撼的是,追踪到13岁时,这些接受了益生菌干预的孩子,其患上ADHD的风险显著降低了。
真实世界研究显示,植物乳杆菌PS128 服用者在注意力、沟通技能上有显著改善,且副作用小。
▸体育锻炼
体育锻炼不仅强健身体,更能健脑。
研究证实,让患有ASD的幼儿在进行课堂活动前进行体育锻炼(尤其是有氧运动),能够显著改善他们的学业反应能力。
▸母亲的饮食习惯
母亲通过塑造自身的肠道菌群,能将其影响传递给下一代,深刻影响孩子的行为和大脑功能。
母亲菌群多样性重要:一项大型队列研究发现,母亲孕期肠道菌群的α-多样性越高,其孩子在2岁时表现出的行为问题就越少。
有益菌的传承:在行为正常的孩子组中,他们的母亲肠道中富含能产生丁酸盐的菌科(如毛螺菌科Lachnospiraceae、瘤胃球菌科)。
健康的孕期饮食,与更高的母亲菌群多样性和更少的儿童内化行为问题直接相关,这清晰地表明,母亲的饮食是支持婴儿早期大脑发育的关键一环。
▸营养
研究报告称,ω-3脂肪酸、维生素、锌、镁、植物化学物质可能在管理与自闭症和多动症相关的生态失调方面发挥有益作用。
基于微生物的可改变因素对儿童疾病发展的影响
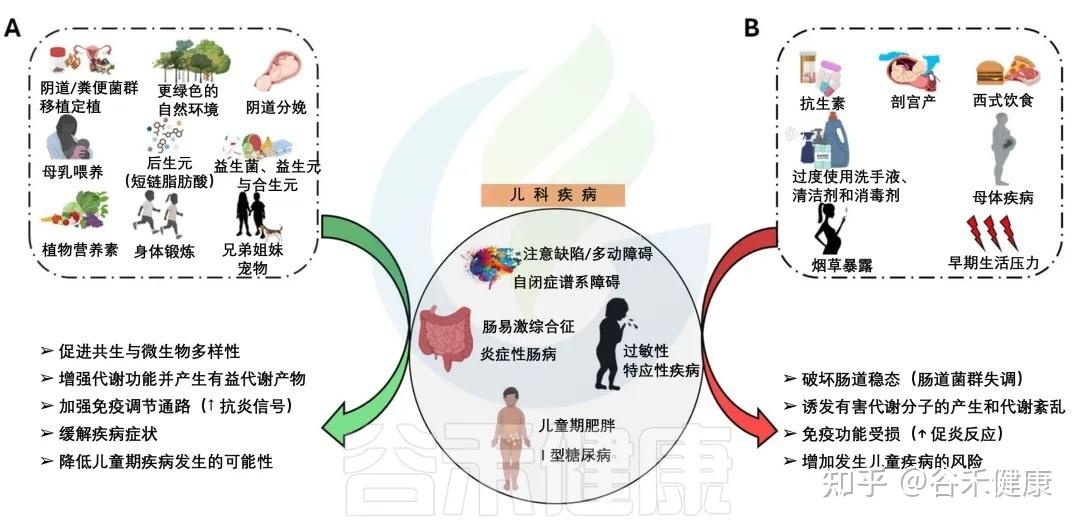

儿童神经发育异常的脑肠轴视角 – 自闭症早期风险判别和干预新路径6 赞同 · 0 评论 文章

探索大脑健康的宝藏:神经营养素、肠道菌群与我们的思维宇宙8 赞同 · 0 评论 文章
甲状腺功能障碍
甲状腺,这个位于颈部的蝴蝶状腺体,是身体新陈代谢的总开关。而它的功能状态与远在腹部的肠道菌群息息相关,形成了一条“肠道-甲状腺轴”。
肠道菌群失调和肠道屏障通透性增加(肠漏),与自身免疫性甲状腺疾病的风险增加有关,这是导致儿童甲状腺功能亢进或减退的主要原因。
▸肠道 ⇌ 甲状腺
肠道对甲状腺的影响:肠道细菌能影响甲状腺合成激素所必需的矿物质(如碘、硒、锌)的吸收和利用。同时,它们也参与甲状腺激素的代谢和激活。甲状腺免疫也受肠道微生物及其代谢产物的调节,从而导致自身免疫性甲状腺疾病的发病机制。
肠道对甲状腺的影响
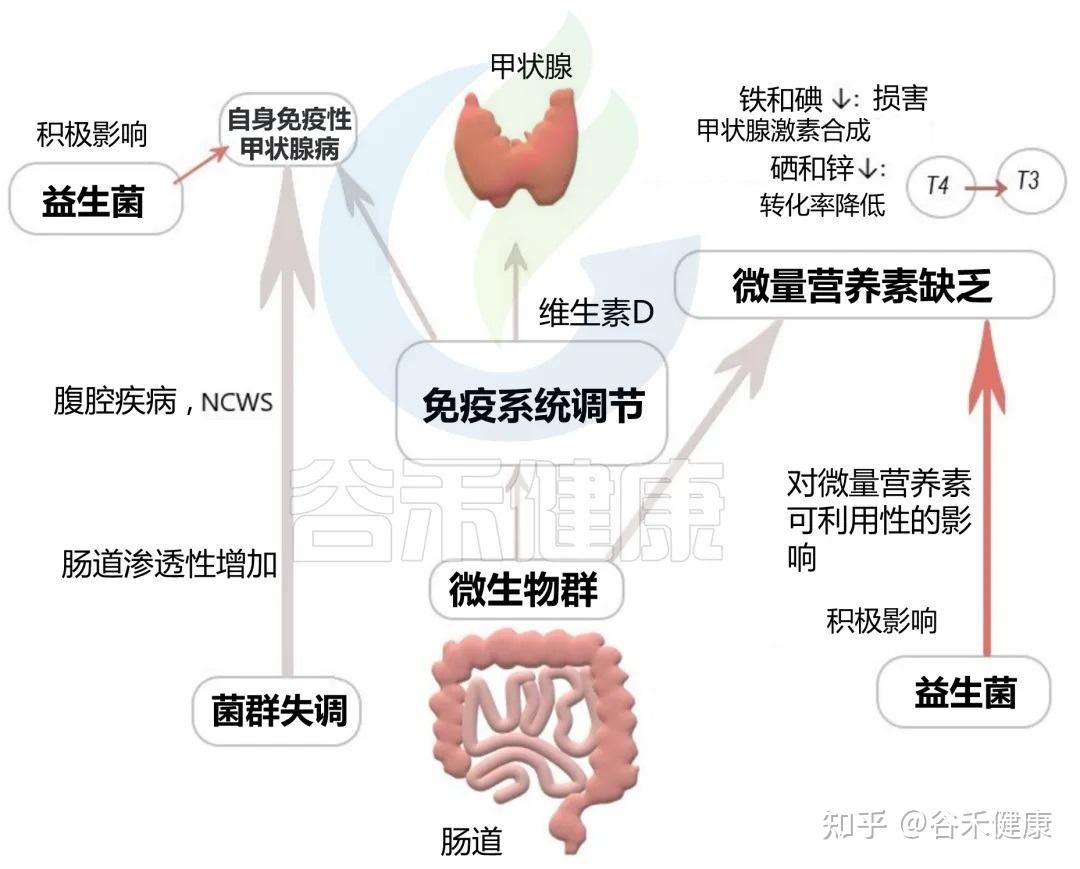
甲状腺对肠道的影响:甲状腺激素反过来也能影响胃肠道的蠕动,并可能直接调控肠道菌群的构成。
▸甲状腺相关疾病患者肠道菌群变化
在成人中:
甲亢患者的双歧杆菌和乳杆菌减少;
而桥本氏甲状腺炎患者则有Akkermansia菌和双歧杆菌的富集。
在儿童中,尤其是在菌群发育的关键窗口期,这方面的研究仍然非常稀少。
▸潜在的干预策略
研究提示,高纤维饮食、补充甲状腺相关的微量营养素、益生菌/益生元、体育锻炼甚至粪菌移植(FMT)都可能对保护甲状腺功能有益。相反,环境中的内分泌干扰物则是一个明确的风险因素。


慢性自身免疫性疾病——桥本甲状腺炎,改变认知和抓住关键16 赞同 · 1 评论 文章
性 早 熟
性早熟(PP),即第二性征过早出现,是儿童中一种日益普遍的内分泌紊乱,尤其在女孩中更为常见。最新的研究将目光投向了肠道菌群,认为它可能在“性激素-肠道菌群轴”上扮演了催化剂的角色。
▸菌群如何催熟?
影响雌激素代谢:某些肠道细菌,如瘤胃球菌属和拟杆菌属,能够通过产生β-葡萄糖醛酸酶来影响雌激素的代谢和循环,从而影响青春期的启动。
通过“肠-脑轴”通讯:菌群产生的短链脂肪酸(SCFAs)和神经递质,也能通过“肠-脑轴”向大脑发送信号,间接调控青春期的启动时机。
一项研究发现,与健康女孩相比,患有中枢性性早熟的女孩肠道中,多种特定细菌显著增加,如:
而这些细菌恰恰与肥胖、短链脂肪酸产生和雌激素代谢有关。
代谢组学分析进一步发现,这些女孩粪便中的代谢物发生了改变,与促进性腺激素分泌的通路(如类固醇生物合成)被激活有关。
性激素和肠道菌群之间的相互关系
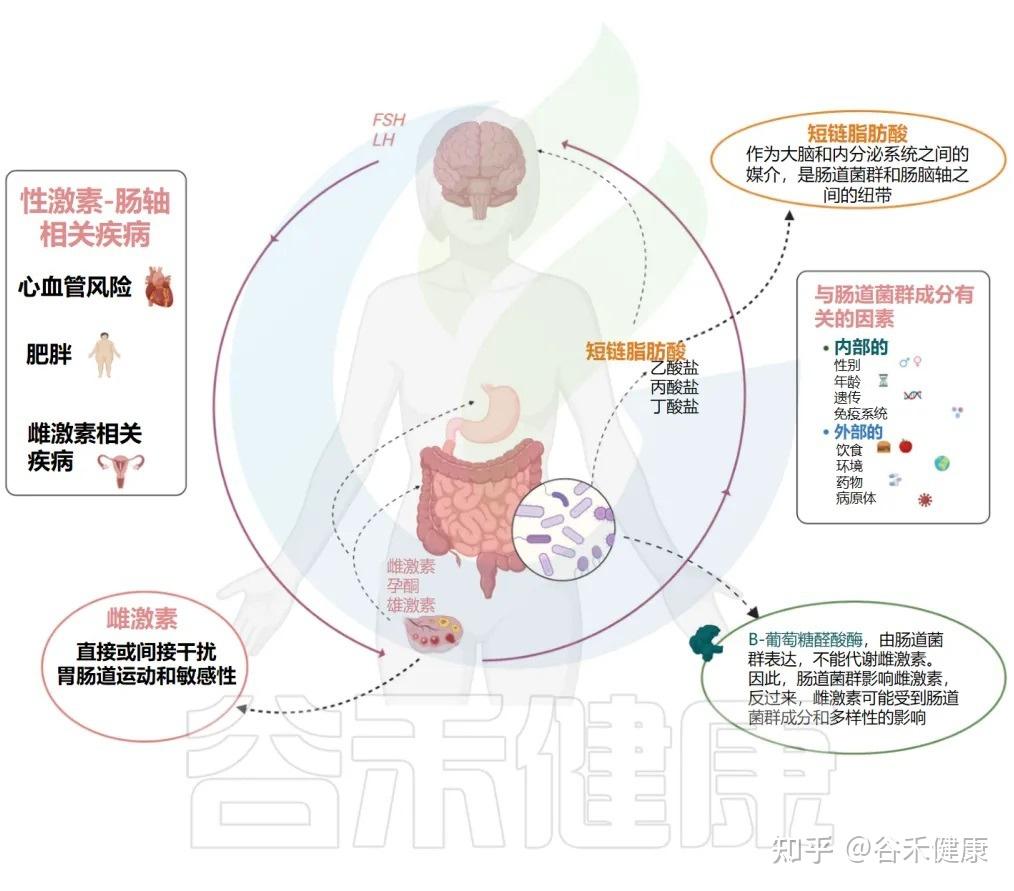
▸干预与警示
有益的干预:动物实验表明,益生菌治疗可以逆转由早期压力或环境物质诱导的性早熟。在人类中,维生素D补充、复杂的碳水化合物和高蛋白饮食(如坚果、蔬菜、海鲜等)被认为可能对预防性早熟有益。
有害的暴露:生命早期接触内分泌干扰化学物质和环境污染物,会扰乱肠道菌群,从而增加性早熟的风险。
无论是甲状腺功能还是青春期的启动,这些看似与肠道无关的生命过程,都可能受到肠道菌群的深刻调控。这为我们理解和干预儿童内分泌疾病打开了一扇全新的大门。

性早熟和微生物群:性激素-肠道菌群轴的作用3 赞同 · 0 评论 文章
综上所述,我们看到许多儿童常见的健康问题,从反复过敏到体重管理,再到消化不适,似乎都与肠道这位“看不见的伙伴”——肠道菌群息息相关。生命早期是肠道菌群蓬勃发展并奠定基础的关键时期,它像一片微型森林,受到出生方式、母乳、饮食、环境等多种因素的影响。
案例一:
肠道整体健康的宝宝,可以看到他的菌群健康、慢病控制、营养均衡三个维度都处在良好水平,有益菌充足、有害菌处于正常低水平,核心菌属完善。
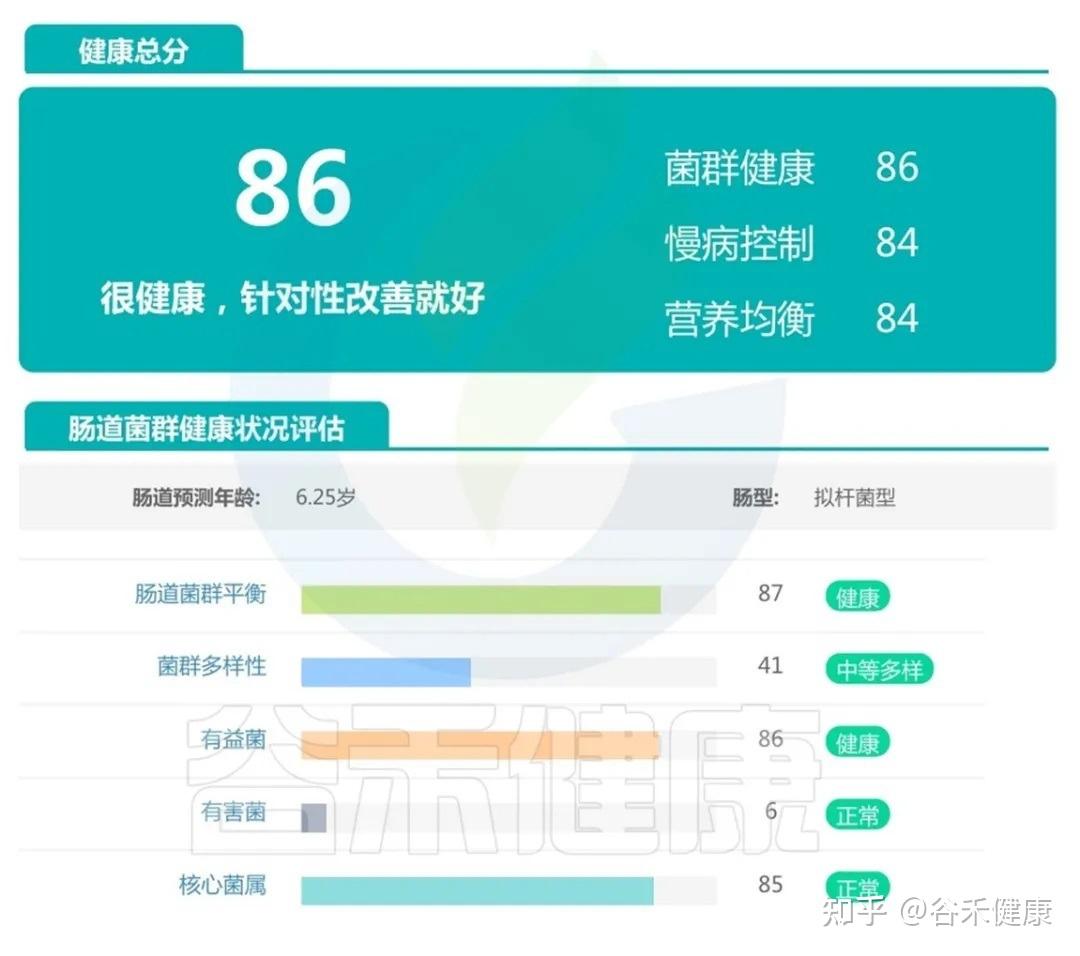
案例二:
这个宝宝处于中等平衡状态,菌群健康指标较前一位儿童有所减弱,核心菌属开始出现缺乏,提示他可能已经携带了某些代谢问题的风险信号。
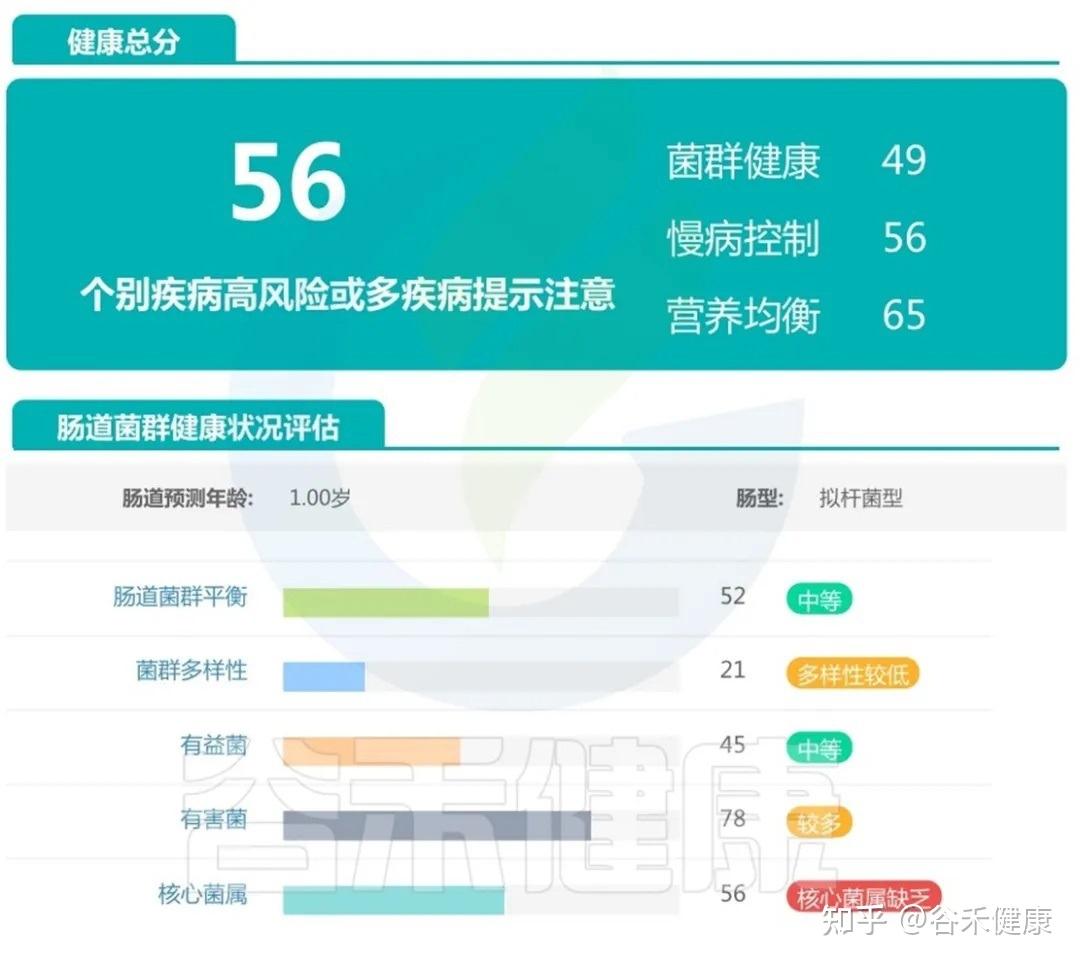
菌群多样性不足,有益菌占比下降,有害菌及产气相关菌群相对偏高。
这种菌群构成,容易让孩子陷入功能性消化不适的恶性循环:肠道动力紊乱、气体产生过多、肠腔内压波动增大,最终就表现为便秘与腹泻交替,夜间睡眠不安等问题。
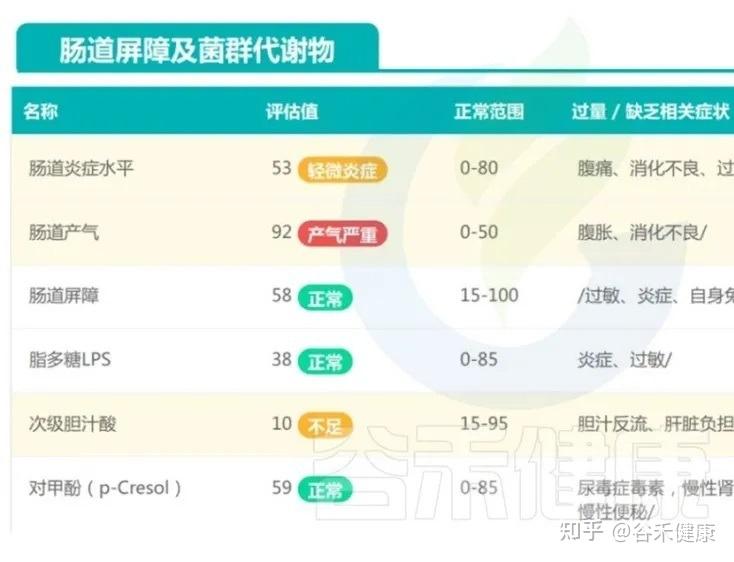
换句话说,这个孩子目前并不是存在明确器质性病变,更符合“肠道微生态已经出现早期失衡、但整体仍具有可逆性”的状态。
在这个阶段,如果能通过规范的饮食结构调整(例如逐步优化配方、增加优质膳食纤维、合理控制脂肪比例)、改善作息和生活方式,适当进行微生态干预(如针对性的益生菌/益生元支持),再配合后续的复查,就有较大机会把他的肠道环境拉回一个更平衡、更稳定的轨道。
案例三:
这个孩子目前看上去发育基本正常,仅社交能力稍不佳,从传统角度看,这样的孩子很容易被归为“现在没什么大问题”。但肠道菌群检测报告给出的信息却更加细腻:
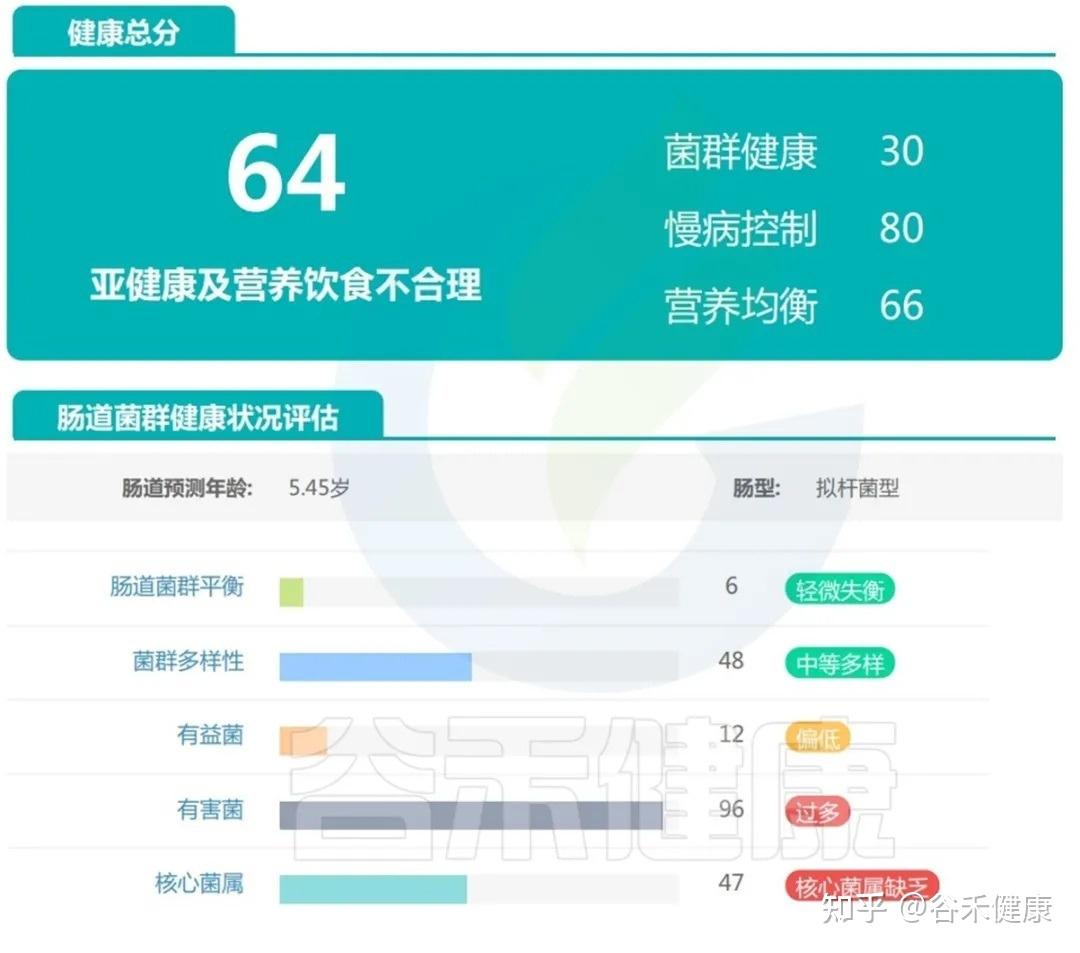
可以看到总体评分落在“轻微失衡”,已偏离理想健康状态。
有益菌丰度不足,有害菌比例偏高,核心菌属缺乏,这意味着肠道微生态的“稳定性”和“恢复力”都打了折扣。
结合该宝宝早期多次抗生素使用,很可能在肠道菌群发育的关键窗口期打断了原本的自然演化轨迹,走向一个“有益菌不足、有害菌偏多”的模式;这样的菌群背景,会让孩子在后续几年中更容易出现:反复上呼吸道感染、皮炎/湿疹、过敏性鼻炎等问题,这也与这与大量关于早期菌群失调和过敏性疾病风险上升的研究结果是吻合的。
同时,在肠–脑轴的影响下,肠道微生态的轻度失衡,过度增殖有害菌及超标代谢物(如对甲酚、硫化氢)会通过“肠脑轴”影响神经递质稳定性(如DOPAC升高提示多巴胺代谢素乱),影响孩子的情绪调节和社交意愿。
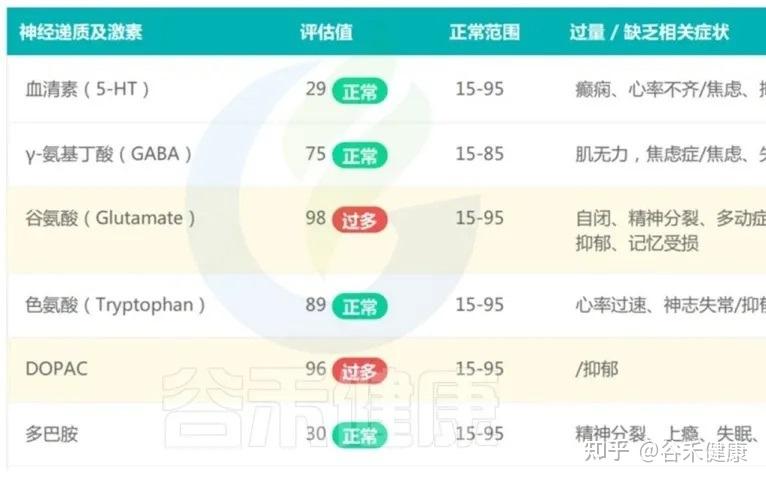
对于这种情况,肠道菌群检测不在于发现一个明确的器质性疾病,而是帮助家长在孩子看起来挺正常的当下,就开始有意识地保护和重建他的肠道微生态——包括减少不必要的抗生素使用、优化饮食结构、增加天然膳食纤维摄入、合理安排户外活动和环境暴露。

通过定期菌群监测,观察肠道菌群、有无新的过敏发作、呼吸道感染频次以及情绪和社交状态的变化,形成一个“早识别—早管理—可追踪”的长期支持路径。
因此,持续关注理解并监测儿童肠道菌群的发育与动态变化,不仅能帮助我们更全面地认识这些健康挑战,还能为未来的干预提供新的思路。通过科学的探索,我们期待能更好地守护儿童肠道微生态的健康成长,为他们一生的健康打下坚实而有力的根基。
主要参考文献
Bankole T, Li Y. The early-life gut microbiome in common pediatric diseases: roles and therapeutic implications. Front Nutr. 2025 May 29;12:1597206.
Shulhai AM, Rotondo R, Petraroli M, Patianna V, Predieri B, Iughetti L, Esposito S, Street ME. The Role of Nutrition on Thyroid Function. Nutrients. 2024 Jul 31;16(15):2496.
Yue M, Zhang L. Exploring the Mechanistic Interplay between Gut Microbiota and Precocious Puberty: A Narrative Review. Microorganisms. 2024 Feb 4;12(2):323.
Stokholm J, Blaser MJ, Thorsen J, Rasmussen MA, Waage J, Vinding RK, Schoos AM, Kunøe A, Fink NR, Chawes BL, Bønnelykke K, Brejnrod AD, Mortensen MS, Al-Soud WA, Sørensen SJ, Bisgaard H. Maturation of the gut microbiome and risk of asthma in childhood. Nat Commun. 2018 Jan 10;9(1):141.
Knezevic J, Starchl C, Tmava Berisha A, Amrein K. Thyroid-Gut-Axis: How Does the Microbiota Influence Thyroid Function? Nutrients. 2020 Jun 12;12(6):1769.
Ma T, Wu Z, Lin J, Shan C, Abasijiang A, Zhao J. Characterization of the oral and gut microbiome in children with obesity aged 3 to 5 years. Front Cell Infect Microbiol. 2023 Mar 29;13:1102650.
Tian X, Liu X, Wang Y, Liu Y, Ma J, Sun H, Li J, Tang X, Guo Z, Sun W, Zhang J, Song W. Urinary Metabolomic Study in a Healthy Children Population and Metabolic Biomarker Discovery of Attention-Deficit/Hyperactivity Disorder (ADHD). Front Psychiatry. 2022 May 20;13:819498.
Agrawal M, Sabino J, Frias-Gomes C, Hillenbrand CM, Soudant C, Axelrad JE, Shah SC, Ribeiro-Mourão F, Lambin T, Peter I, Colombel JF, Narula N, Torres J. Early life exposures and the risk of inflammatory bowel disease: Systematic review and meta-analyses. EClinicalMedicine. 2021 May 15;36:100884.

谷禾健康
宠物 营养 健康
在全球范围内,猫犬早已不再只是传统意义上的“宠物”,而成为许多家庭中重要的“毛孩子”。这种“宠物人性化”的趋势,极大地推动了宠物主人对爱猫爱犬健康的关注,促使他们寻求超越传统温饱的、更精细化、科学化的喂养方案。
如今,延长宠物寿命、提升健康福祉已成为宠物主人与兽医共同的目标。从过去仅满足基本能量与营养需求的狗粮,到如今注重护理、饮食与功能性食品的综合方案,宠物饲养理念正日益进步。
与人类相似,犬猫的健康同样受到饮食与肠道微生物组的深刻影响。肠道微生物组作为宿主体内重要的代谢与免疫调节中心,其稳态或失衡直接决定整体健康水平。
最近,宠物饮食中越来越多地引入功能性食品,以促进肠道微生物组的良性调节。这些食品包括益生菌、益生元、合生元、后生元,以及多不饱和脂肪酸和植物营养素等必需营养成分。
功能性食品虽非药物,却体现了“食疗同源”的理念,通过日常摄入实现主动健康干预。对犬类而言,这类食品可视作“第四餐”,在常规日粮之外帮助优化生理机能、增强免疫力、延缓衰老,并辅助疾病管理。

本文结合多篇核心研究,探讨了功能性营养干预在调节犬猫肠道微生物组及其在营养代谢、免疫成熟、神经行为调控等方面的作用,同时分析了肠道生态失调作为多种疾病(包括胃肠道、皮肤、代谢及神经系统疾病)共同病理基础的分子机制。
在此基础上,系统总结了各类功能性营养成分的关键特性:益生菌的菌株特异性免疫调节;益生元对特定菌群的选择性促进及短链脂肪酸(SCFAs)的生成;合生元的协同增效作用;后生元的创新应用潜力;多不饱和脂肪酸(PUFAs)尤其是EPA和DHA在炎症调控中的核心角色;以及植物营养素的多靶点抗氧化与抗炎效应等。

狗和猫与人类相伴已经超过一万年,作为伴侣动物,它们在人类社会中扮演着非常重要的角色。最近的一份报告估计,全球大约已经有4.77亿只狗和3.77亿只猫被当作宠物饲养。
★ 宠物有助于人身心健康
除了作为人类的伙伴,宠物还有助于个人身心的整体健康。根据一些证据,养宠物有助于减压、缓解抑郁、降低血压、预防心脏病,并提升自尊与幸福感。此外,哮喘、过敏和肥胖等疾病的发生风险也可能随之降低。
并且随着社会经济的发展与人文关怀的提升,犬猫在家庭中的地位发生了根本转变——它们被视为家庭成员,其健康与福祉备受关注。
这种“宠物人性化”趋势推动了宠物健康管理理念的革新。现代兽医学已从单纯的疾病治疗转向以预防、维护健康和提升生命质量为核心的主动健康管理,目标在于实现“健康老龄化”和延长健康寿命。
★ 肠道微生物组:一个多功能“超级器官”
在这一过程中,功能性营养学作为一种非侵入性、可持续的干预手段,其科学与临床价值日益凸显。功能性食品或营养品,是指除提供基础营养外,经科学验证能改善特定生理功能或降低疾病风险的食品或活性成分。其作用机制虽多样,但越来越多的研究表明,关键靶点在于寄居于宿主肠道内的复杂微生物生态系统——肠道微生物组。
作为身体的“隐形器官”,肠道微生物组深刻影响犬猫的营养吸收、免疫防御、新陈代谢及情绪行为。其平衡是健康的基石,而失调则被认为是多种犬猫常见疾病的关键驱动因素。
从急性腹泻、慢性肠病,到特应性皮炎、肥胖、糖尿病、骨关节炎及认知功能障碍,几乎都与菌群失衡密切相关。因此,通过精准营养干预调节肠道微生物组、恢复生态稳定,已成为现代兽医营养学中最具潜力的研究与应用方向之一。
1
营养代谢中心:发酵、合成与转化
犬猫的消化系统无法完全分解复杂碳水化合物,尤其是膳食纤维。这一过程主要由结肠中的厌氧菌群完成,它们通过发酵将纤维转化为短链脂肪酸(SCFAs),为宿主提供关键代谢产物。
• 产生丁酸盐:结肠上皮的重要能量,抗炎物质
丁酸盐(Butyrate)主要由厚壁菌门中的Clostridium簇IV和XIVa(如Faecalibacterium、Roseburia)合成,是结肠上皮细胞超70%的能量来源。
它还可以抑制组蛋白去乙酰化酶(HDACs),发挥抗炎和抗肿瘤作用,并上调紧密连接蛋白(Occludin、Claudin-1、ZO-1)表达,强化肠道屏障。
• 产生丙酸盐:调节脂质合成,影响血糖
丙酸盐(Propionate)主要由拟杆菌门产生,被吸收至肝脏后可抑制糖异生、调节脂质合成,从而改善血糖稳态与胰岛素敏感性。
• 产生乙酸盐:含量最高的短链脂肪酸
乙酸盐(Acetate)是含量最高的SCFA,可由多种菌群生成,既为外周组织供能,也是产丁酸菌合成丁酸的重要前体。
• 代谢胆汁酸,合成一些必需维生素
此外,肠道菌群还负责代谢胆汁酸,将肝脏合成的初级胆汁酸转化为次级胆汁酸(如脱氧胆酸LCA和石胆酸DCA)。这些次级胆汁酸作为信号分子,通过激活法尼醇X受体(FXR)和G蛋白偶联胆汁酸受体1(TGR5),调控宿主的脂质、葡萄糖代谢及炎症反应。
同时,菌群还能合成犬猫必需的维生素K、多种B族维生素(生物素、叶酸、核黄素)及氨基酸等,维持营养与代谢平衡。
2
免疫系统的“首席教官”
肠道相关淋巴组织(GALT)是机体最大的免疫器官,而肠道菌群在免疫系统的发育、成熟和调控中发挥着关键作用。
• 诱导免疫耐受
在新生期,肠道菌群的定植是建立口服耐受的关键,帮助免疫系统对食物抗原和共生菌保持耐受,从而预防过敏和自身免疫疾病。
• 塑造黏膜免疫
菌群及其代谢产物(尤其是短链脂肪酸)可刺激上皮细胞和浆细胞分泌大量sIgA,后者能中和毒素并阻止病原体附着,是黏膜免疫的第一道防线。
• 调节T细胞分化
菌群组成深刻影响辅助性T细胞(Th)亚群平衡。分段丝状菌(SFB)促进促炎性Th17细胞分化,提高对胞外菌的防御力;而产丁酸梭菌则诱导调节性T细胞(Treg)分化,Treg通过分泌IL-10与TGF-β抑制过度免疫反应,维持免疫稳态。肠道菌群失衡常伴随Treg/Th17比例紊乱,导致免疫异常。
3
“肠-脑轴”的核心信使
肠道与大脑之间通过“肠-脑轴”形成复杂的双向通讯网络,而肠道菌群是其中的核心信号调节者。
• 神经递质的合成与调控
肠道菌群可直接生成多种神经递质,如GABA、去甲肾上腺素和多巴胺,或通过代谢膳食前体物质(如色氨酸)影响中枢血清素水平。菌群代谢产物(如丁酸)还能调节肠嗜铬细胞产生血清素,占全身总量的90%以上。
• 迷走神经通路
菌群代谢物可促使肠上皮细胞释放信号分子,这些信号经迷走神经传递至大脑,进而影响情绪与行为。
• 调节HPA轴
肠道菌群稳态对下丘脑-垂体-肾上腺(HPA)轴的发育与应激反应至关重要。无菌动物常表现出过度应激反应,而补充特定益生菌可恢复其正常。研究还发现,具有攻击性或恐惧行为的犬只,其肠道菌群结构与健康犬存在显著差异。
功能性食品在犬猫营养中具有重要意义,它不仅提供基础能量与必需营养素,还能通过特定的生理活性成分,支持机体健康、延缓衰老并预防疾病。
近年来,越来越多研究证实,富含抗氧化剂、ω-3脂肪酸、植物营养素、益生元及功能性蛋白的配方,可改善犬猫的免疫功能、皮肤与毛发状况、肠道健康及认知能力。
1
功能性食品与犬类健康
多项研究探讨了功能性食品在犬中的作用,相关结果见下表。


• 低聚果糖等益生元改善整体健康
例如,成年雄性比格犬摄入富含低聚果糖(FOS)的饲粮后,粪便中氨及梭菌浓度降低,而需氧菌含量上升,改善整体健康。
以肉类为主的犬粮中,单独或联合添加FOS和甘露寡糖(MOS),可提高回肠免疫球蛋白A水平,降低粪便中吲哚和酚含量。
测试了一种含多种成分的维持性饲粮(包括多种植物提取物及抗氧化剂)对12只成年犬的影响。6个月后,犬只氧化应激显著下降,活性氧代谢物减少,而视黄醇及血浆抗氧化力(BAP)升高,提示氧化平衡得以恢复,说明均衡饮食对维持犬的抗氧化状态至关重要。相反,摄入高血糖指数食物后可能导致氧化失衡。
注:进一步研究表明,在测试4种复杂碳水化合物(大麦、玉米、豌豆、大米)及葡萄糖后,豌豆的血糖指数最低(29%),大麦和大米较高(分别为51%和55%),支持它们被纳入平衡膳食。
• 富含抗氧化剂的食物有助于提升代谢和繁殖能力
有研究评估了一种富含抗氧化剂和功能性成分的配方(含水解蛋白、维生素、酵母提取物和植物提取物)对不同年龄雄犬繁殖力的影响。
实验持续4个月,结果显示,在45天内犬的游离甲状腺素和睾酮水平升高,代谢和生育能力改善,特别是2–7岁组的精子活力显著提高,表明富含抗氧化剂的饮食可增强生殖表现与内分泌功能。
• 含抗氧化剂与植物化合物改善认知衰老和行为障碍
此外,犬类模型还用于研究认知衰老、脑源性神经营养因子(BDNF)及饮食之间的关系。研究发现,给予含抗氧化剂与植物化合物(如鱼油、甜菜浆、木瓜、石榴、芦荟、葡萄及迷迭香等)的日粮,可显著提高脑源性神经营养因子(BDNF)水平,降低氧化应激,并改善神经内分泌平衡。
另一系列实验显示,富含功能性植物提取物及色氨酸的饮食能减轻犬的行为障碍,改善相关临床症状。
综上所述,功能性食品通过抗氧化、抗炎及神经保护等多重机制,不仅能优化犬的代谢与免疫状态,还可能成为支持认知健康和行为管理的有效营养干预手段。
• 功能性饮食还有助于改善狗的口臭、皮肤健康
口臭是一种常见于犬及人类的疾病,影响彼此关系。一项双盲、安慰剂对照交叉研究评估了专用功能性饮食对16只不同品种和年龄犬慢性口臭的改善作用。
注:该饮食含米饭、鱼粉、植物脂肪、鱼油、甜菜浆、矿物质、脱水酵母、低聚果糖、丝兰提取物、溶菌酶、生物类黄酮、胸腺提取物与黑肋骨等成分。
经气相色谱测定,犬只口腔中关键挥发性硫化物(甲基硫醇、硫化氢、二甲硫化物)在30天内显著下降,且停用饮食20天后仍维持改善,显示其持久疗效。
有研究进一步发现,一种含水解蛋白、植物提取物(包括茶树、百里香、大蒜、玫瑰果)及矿物质的功能性饮食,可显著缓解犬慢性外耳炎症状,减少药物使用需求。15只患犬接受该饮食与局部药物治疗(前7天使用抗炎滴耳剂OTOMAX,后83天仅靠饮食干预),90天后耳道阻塞、红斑、分泌物与异味均明显减轻,体现出其抗炎与抗氧化双重功效,为抗生素替代方案提供了潜在方向。
此外,研究了免疫调节饮食(IMMD)在犬利什曼病(CL)治疗中的辅助作用。与标准饮食(SD)组相比,IMMD组犬在3、6、12个月内均恢复了因CL降低的调节性T细胞水平,并在后期阶段呈现T辅助细胞数量正常化,表明该饮食可在药物治疗期间调节免疫反应。
另一项研究证实,IMMD还能有效减轻犬角膜结膜炎的典型症状,包括泪液减少、结膜炎症、角膜角化与色素沉积过多等。
综上所述,针对犬的功能性饮食还能在口腔健康、皮肤炎症及免疫调节等方面发挥作用,为临床营养干预提供了有益证据。
2
功能性食品与猫类健康
与犬相比,猫为严格食肉动物,具有独特的营养需求。多项研究已探讨功能性食品在猫营养与健康中的作用(见下表)。
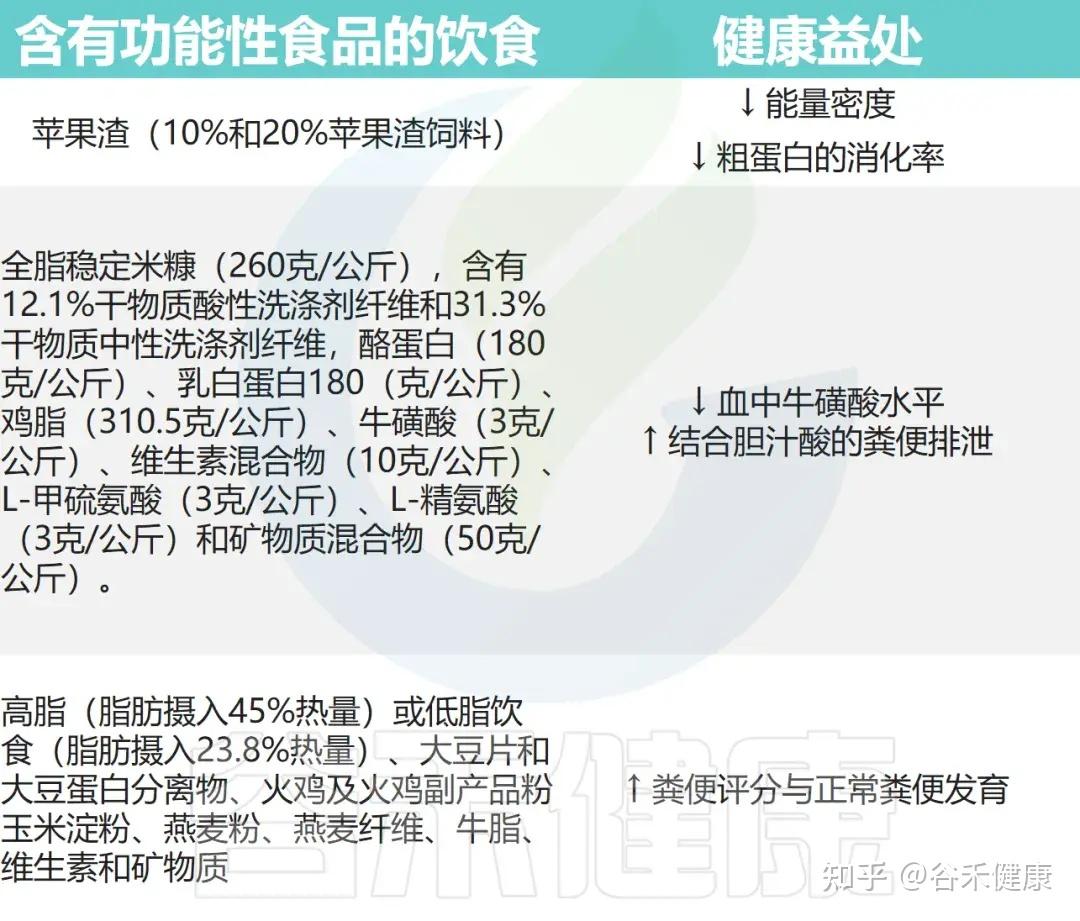
• 功能性食品改善了猫咪的腹泻
一项针对55只患慢性腹泻猫的随机、双盲、对照试验评估了高脂(23%)和低脂(10%)高消化性饮食的效果。该饮食由大豆片、大豆蛋白分离物、火鸡粉、玉米淀粉、燕麦粉、燕麦纤维、牛脂及维生素矿物质组成。结果显示,所有猫在第一周内粪便评分(FSa)均显著提高,3周后达到最佳改善,其中约三分之一的猫恢复正常排便。
• 含有纤维的饮食可能有助于猫咪减肥
苹果渣,即酿造苹果酒的剩余部分,可能是猫咪减肥饮食中的功能性食物。九只成年、绝育、肥胖的猫咪被喂食以肉类为主的饮食,并添加了不同程度的苹果渣。苹果渣分别用10%和20%,并未降低可口性,但确实降低了食物的能量密度。
因此,在肥胖猫的饮食中以有限的比例加入可入口纤维成分,是降低食物能量含量并维持生理食物摄入水平的好方法。
• 一些猫粮应适当提高牛磺酸含量
另一项研究显示,在纯化猫粮中添加26%全脂米糠后,猫的全血牛磺酸浓度显著低于含26%玉米淀粉的对照组。
这种降低与粪便中结合胆酸排泄增加及米糠中不可消化蛋白引发的后肠菌群变化有关,后者可导致牛磺酸降解。为避免缺乏,含米糠配方的猫粮应适当提高牛磺酸含量。
如前所述,调控肠道微生物组以维持或恢复生态平衡,是犬猫功能性食品的核心策略之一。根据作用机制与来源,目前应用最广的功能性成分主要包括:益生菌、益生元、合生元、后生元、多不饱和脂肪酸和植物营养素。它们各具优势,相互协同,共同守护宠物健康。
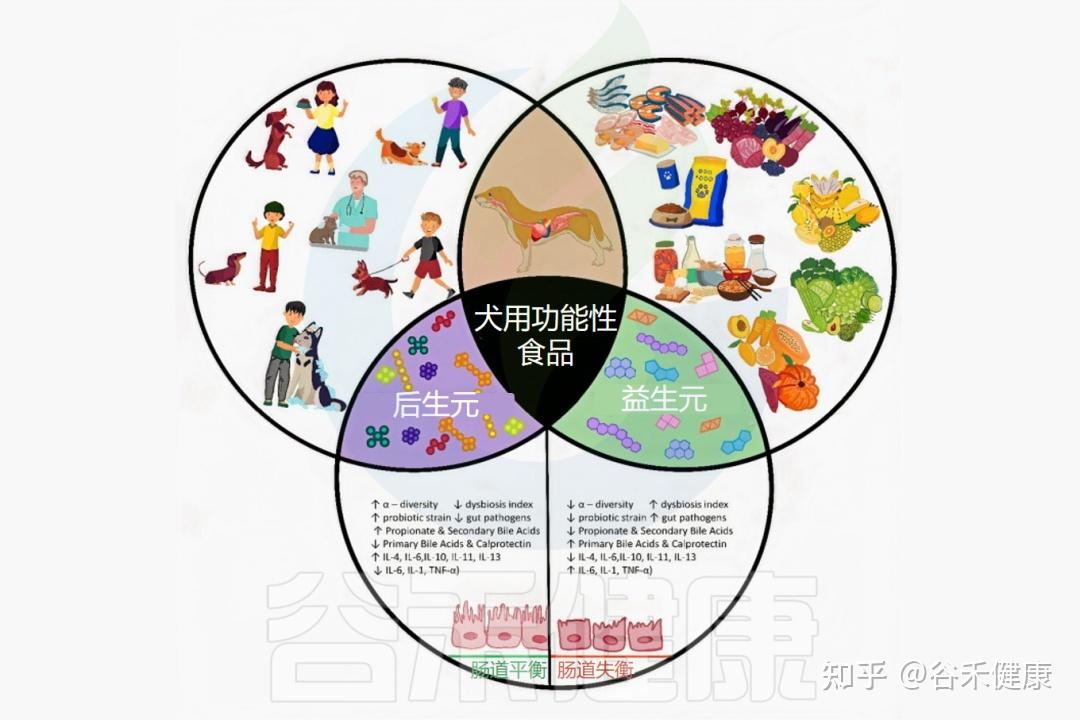
1
益生菌:直接补充“有益菌群
• 益生菌对狗的益处
研究表明,并非所有益生菌都适用于犬类。理想的菌株应具备宿主特异性,通常来源于犬只自身,以更好地适应其肠道环境。常见犬用益生菌包括:
乳杆菌属(Lactobacillus):
如嗜酸乳杆菌(L.acidophilus);
约氏乳杆菌(L.johnsonii);
发酵乳杆菌(L.fermentum);
鼠李糖乳杆菌(L.rhamnosus)。
双歧杆菌属(Bifidobacterium):如动物双歧杆菌(B.animalis)。
肠球菌属(Enterococcus):如屎肠球菌(E.faecium)的特定菌株(如SF68)。
注:但该属的使用需谨慎,因其部分菌株可能携带抗生素抗性基因。
多项研究表明,补充益生菌(如E. faecium SF68、B. animalis AHC7)可有效缩短犬急性腹泻的持续时间并改善粪便稠度。在慢性肠病(CE)中,它们可作为辅助疗法,帮助恢复菌群平衡、减轻炎症。
不同菌株的免疫作用各异。Lactobacillus casei 可诱导Th1型免疫反应(分泌IFN-γ),而Lactobacillus rhamnosus GG 更倾向促进Treg细胞分化。这种差异与细胞壁中不同的微生物相关分子模式(MAMPs)如肽聚糖、脂磷壁酸等有关。
多菌株组合制剂(如VSL#3)基于“菌种协同”理念。在犬炎症性肠病(IBD)研究中,使用VSL#3显著改善临床症状,降低肠道黏膜炎症评分,并增强Treg关键转录因子FoxP3的表达,证明其免疫调节作用。
• 益生菌对猫的益处
研究显示,健康成猫补充L.acidophilus DSM13241的膳食剂后,粪便中乳杆菌和嗜酸乳杆菌数量增加,而粪肠球菌和梭菌减少。
补充L.acidophilus CECT 4529能改善粪便质量。由S. boulardii和Pediococcus acidilactici组成的复合益生菌可提高粪便中丁酸和总短链脂肪酸含量,降低炎症标志物(髓过氧化物酶、钙卫蛋白)水平,并增强抗氧化酶活性(超氧化物歧化酶、谷胱甘肽)。
此外,口服地衣芽孢杆菌发酵产物可缓解腹泻,增加梭菌簇XIVa成员并减少慢性腹泻猫中C.perfringens数量。含八株乳酸菌的复合制剂可改善慢性便秘和特发性巨结肠猫的症状,并增加粪便中乳酸杆菌和链球菌。肠球菌E. faecium SF68在收容所猫中可显著降低腹泻率;另一粪源E. faecium菌株则改善肠道健康。
多种源自健康猫的菌株,如B.adolescentis、L.acidophilus、L.plantarum和L.rhamnosus,表现出对病原菌的益生活性和抗菌潜力。
• 益生菌的应用:
抗生素相关性腹泻:在犬只必须使用抗生素治疗时,同时或之后补充益生菌,有助于减少抗生素对肠道菌群的破坏,预防腹泻等副作用。
免疫调节:研究发现,给健康的犬只补充益生菌可以提高其免疫细胞(如白细胞)的吞噬活性,增强对病原体的防御能力。
压力引起的消化问题:对于寄养、旅行、更换环境等应激情况,提前补充益生菌有助于稳定肠道菌群,预防应激性腹泻。
!
注意事项
不同益生菌菌株的免疫效应存在差异。选择时,应优先考虑针对犬或猫、有随机对照临床试验(RCT)支持并经同行评审的特定产品,同时关注其活菌数量(CFU)及保质期内的稳定性。
2
益生元:为有益菌提供“超级口粮”
益生元是“可被宿主肠道微生物选择性利用、并对宿主健康产生益处的底物”,通俗地说,就是肠道有益菌喜爱的“食物”,可促进其生长和繁殖。
• 常见的益生元
低聚果糖(FOS):存在于菊苣、洋葱、香蕉等植物中。
菊粉(Inulin):FOS的一种长链形式,主要来源是菊苣根。
低聚果糖(FOS)和菊粉主要被双歧杆菌高效利用,因此被称为“双歧因子”。研究表明,在犬猫日粮中添加1–2%的FOS或菊粉,可显著提高粪便中双歧杆菌丰度,同时降低粪便pH值和氨含量。
甘露寡糖(MOS):通常提取自酵母细胞壁。甘露寡糖除具益生元作用外,还能吸附并清除部分病原菌,特别适用于幼年动物及预防应激性腹泻。MOS能与带I型菌毛的革兰氏阴性菌(如沙门氏菌、大肠杆菌)结合,而不影响有益菌。
注:不同类型和剂量的益生元会影响短链脂肪酸(SCFAs)的产量和构成,例如抗性淀粉更易生成丁酸,而果胶则倾向于产生乙酸和丙酸。
• 益生元的应用:
改善粪便质量:补充低聚果糖、菊粉等益生元能显著改善犬的粪便稠度,减少腹泻和便秘。
调节菌群结构:研究一致表明,饲喂益生元能显著提高犬粪便中双歧杆菌和乳杆菌的数量,同时降低梭菌等潜在有害菌的比例。
增强免疫:通过增加短链脂肪酸的产量,益生元能间接调节免疫系统,减轻炎症。有研究显示,给怀孕母犬补充FOS,能提高其初乳中的免疫球蛋白水平,从而使幼犬获得更好的被动免疫。
代谢健康:在超重犬的研究中,补充益生元有助于改善葡萄糖代谢和胰岛素敏感性。
3
合生元、后生元:下一代微生态调节剂
合生元是益生菌与益生元的复合产品,旨在通过益生元为益生菌提供特定营养来源,提高其在肠道内的存活率与定植效率,从而实现协同增效。
后生元则代表微生态干预的进阶方向,将关注点从“活菌”转向其“功能分子”,包括灭活菌体(如热灭活的Lactobacillus acidophilus)、细胞组分(如细胞壁碎片、胞外多糖)及发酵代谢产物(如短链脂肪酸、有机酸、酶和细菌素)。
系统综述与荟萃分析显示,健康犬的短链脂肪酸(SCFA)水平普遍高于患病犬(如IBD个体),说明SCFAs可作为反映健康状态的功能性标志物。其他研究同样发现,慢性肠病犬的粪便SCFA含量下降。
而肽聚糖(PTDG)和磷壁酸(TEIA)是细胞壁的重要组成成分,具有显著的免疫调节作用。TEIA分为脂质型(LTEIA)和壁连型(WTEIA):前者通过糖鞘脂附着在细菌膜上,后者与肽聚糖链形成共价键。乳酸杆菌属的肽聚糖可抑制炎症性细胞因子的释放,其中来自L. rhamnosus的PTDG能增强感染致病链球菌小鼠的先天免疫反应。LTEIA还能通过诱导防御素和猫地霉素的产生,有效治疗皮肤感染并预防细菌及病毒感染。
• 合生元、后生元的应用:
合生元在应对复杂肠道疾病方面展现出显著潜力。研究表明,在犬慢性肠病中,含多种益生菌菌株与益生元(FOS)的合生元产品能迅速缓解腹泻、呕吐等症状,改善肠道组织的炎症评分,并通过上调紧密连接蛋白表达修复受损肠道屏障。
另一项针对顽固性慢性结肠炎犬的研究显示,富含MOS与FOS的高纤维处方粮结合多菌株益生菌混合物,可在约8.5天内显著缓解症状并恢复肠道菌群平衡。
后生元的优势在于其高安全性与稳定性,适合用于免疫低下或危重动物。临床上,可直接补充丁酸钠或丁酸钙以促进急性肠炎和炎症性肠病犬的肠道修复,或利用特定发酵产物实现靶向免疫调节。
4
多不饱和脂肪酸:抗炎与细胞结构的关键
多不饱和脂肪酸(PUFAs)是分子中含有多个双键的脂肪酸。其中,Omega-3(ω-3)和Omega-6(ω-6)两大系列是犬只自身无法合成,必须从食物中获取的必需脂肪酸。
• 主要类型与来源
①ω-6脂肪酸:
亚油酸(LA):广泛存在于玉米油、葵花籽油等植物油中,是维持皮肤和毛发健康的关键营养素。
花生四烯酸(AA):可由犬体内的亚油酸转化而来。
②ω-3脂肪酸:
α-亚麻酸(ALA):来源于亚麻籽油、奇亚籽等植物,但犬体内转化为长链ω-3的效率低。
二十碳五烯酸(EPA)与二十二碳六烯酸(DHA):主要来自鱼油(如三文鱼、沙丁鱼)、磷虾油及海藻,是发挥主要生理作用的活性ω-3。
• 作用机制
抗炎调节:ω-6与ω-3是类花生酸的前体——前者(AA)产生的分子多促炎,后者(EPA)则具抗炎或弱促炎作用。通过提高饮食中ω-3比例(降低ω-6:ω-3比值),可改变细胞膜脂肪酸构成,促进抗炎介质生成,减少过度炎症反应。
炎症消退机制:EPA和DHA还能生成消退素、保护素与脂氧素等“促炎症消退介质”,它们通过抑制中性粒细胞浸润、促进巨噬细胞清除凋亡细胞,加速组织从炎症向修复转变。
结构与功能作用:DHA是脑和视网膜的重要组成成分,对幼犬神经及视觉发育至关重要,也有助于维持老年犬的认知功能。
其他生理效应:可改善血脂、增强心血管功能并调节免疫反应。
• 多不饱和脂肪酸的应用:
骨关节炎(OA):富含EPA+DHA(干物质基础>2.5%或50–100 mg/kg/日鱼油补充)的饮食可显著减轻狗的关节疼痛和跛行,提升活动能力,机制与其抗炎作用相关。
皮肤健康与过敏:提高ω-3摄入可缓解特应性皮炎的炎症、瘙痒与红肿。
心血管健康:ω-3补充可改善犬的心肌功能,降低心律失常风险。
认知与发育:DHA促进幼犬大脑发育,并有助于延缓老年犬的认知衰退,改善学习与记忆。
肾脏健康:ω-3的抗炎特性可能有助于减缓慢性肾病进展。
5
植物营养素:来自大自然的“抗氧化卫士”
植物营养素是植物中具有生物活性的天然化合物,虽非传统维生素或矿物质,但具显著的抗氧化和抗炎作用。
• 主要类别与来源
①多酚类:
类黄酮:如槲皮素(苹果、洋葱)、儿茶素(绿茶)、花青素(蓝莓、蔓越莓)。
其他多酚:如白藜芦醇(葡萄皮)、姜黄素(姜黄)。
②类胡萝卜素:
类胡萝卜素是脂溶性色素,存在于红色、橙色和黄色的蔬菜和水果中。常见于犬类和人类膳食补充剂及食品中的类胡萝卜素有:α-和β-胡萝卜素、番茄红素、玉米黄素、隐黄素和叶黄素。
例如如β-胡萝卜素(胡萝卜)、番茄红素(西红柿)、叶黄素(菠菜)。
③植物甾醇
植物甾醇存在于油、蔬菜、水果、坚果、种子和豆类中。植物甾醇在降低高脂血症犬只的高血浆三酸甘油酯和胆固醇水平中起着重要作用。
这其中尤为重要的两个:
姜黄素(Curcumin)其抗炎机制不仅限于抑制NF-κB,还包括抑制COX-2、LOX、诱导型一氧化氮合酶(iNOS)等多种炎症相关酶的活性和表达。其主要限制在于口服生物利用度低。现代制剂通过将其与磷脂结合(形成植物体)、使用纳米颗粒或与胡椒碱(能抑制其在肝脏的快速代谢)联用,同时调节肠道菌群来提高其吸收率。
白藜芦醇(Resveratrol):作为一种SIRT1激动剂,它能模拟热量限制带来的部分健康效应,如改善线粒体功能、提高胰岛素敏感性、减轻氧化应激。在犬模型中,白藜芦醇显示出心脏保护作用。
注:抗氧化剂“鸡尾酒”疗法:在犬认知功能障碍(CCD)的研究中,单独使用某一种抗氧化剂(如维生素E)的效果往往有限。而将多种抗氧化剂(维生素E, C, L-肉碱, α-硫辛酸)与富含多种植物营养素(来自菠菜、番茄、葡萄、胡萝卜、柑橘)的饮食相结合,则显示出强大的协同效应。这可能是因为不同抗氧化剂在细胞的不同区室(水溶性/脂溶性)起作用,并能相互再生,形成一个抗氧化网络。
• 作用机制
抗氧化:机体在新陈代谢过程中会产生自由基,过多的自由基会攻击细胞,导致氧化损伤,这是衰老和多种慢性疾病的根源。植物营养素能直接中和自由基,或激活机体自身的抗氧化酶系统,从而保护细胞免受损伤。
抗炎:许多植物营养素能抑制关键的炎症通路(如NF-κB),减少促炎细胞因子的产生。
其他:抗癌、调节免疫、保护心血管等。
• 植物营养素的应用:
认知功能与抗衰老:一项针对老年比格犬的著名研究发现,饲喂富含多种抗氧化剂(包括来自菠菜、番茄、葡萄的多酚)的食物,并结合行为丰富化训练,能显著改善犬只的学习和记忆能力,延缓认知衰退。蓝莓和葡萄提取物也被证明能改善老年犬的认知表现。
骨关节炎:姜黄素、鳄梨豆非皂化物(ASU)等被研究用于辅助治疗犬OA,它们通过抗炎和抗氧化作用减轻关节疼痛和炎症。
癌症:一些植物营养素在体外实验中显示出抑制犬肿瘤细胞(如骨肉瘤细胞)生长的能力,如番茄红素、黄芩素等。但这方面的体内研究仍需加强。
心血管保护:葡萄籽提取物、槲皮素等在犬模型中显示出保护血管、抗血小板聚集的作用。
编者小结
以上五类功能性成分从不同的作用途径出发,以各自独特的机制在细胞、组织及系统层面协同发挥效应,共同促进并维持犬猫的整体健康与生理平衡,帮助其保持良好的生命状态和长期活力。
要将上述功能性成分有效地整合到临床实践中,需要充分考虑不同疾病的病理生理学特征,并依据现有的循证研究结果进行科学应用。同时,应结合个体差异和具体健康状况,选择合适的配方与干预方式,以确保营养策略在临床实施中的安全性与有效性。
1
慢性肠病(CE)/炎症性肠病(IBD)
饮食试验:采用水解蛋白或新型蛋白饮食进行6–8周的严格试验,是诊断和治疗食物反应性肠病(FRE)的金标准。
微生态干预:对于确诊的炎症性肠病(IBD)或抗生素反应性肠病(ARE),在饮食管理基础上应联合使用经验证的多菌株、高剂量益生菌或合生元,以恢复肠道菌群平衡并调节免疫反应,从而抑制异常炎症。
抗炎支持:在饲粮中补充富含EPA的鱼油(50–100 mg/kg/天)可有效辅助抗炎治疗。
纤维调整:依据病情(如结肠炎或小肠性腹泻)适当调整可溶性与不可溶性纤维比例,其中低聚果糖、菊粉等益生元纤维为首选。
2
糖尿病与胰岛素敏感性
糖尿病(DM)在犬类中越来越常见,通常是1型糖尿病。自发性1型DM的犬只存在严重的胃肠道菌群失调和粪便中未结合胆汁酸水平的改变。此外,治疗DM的药物也会影响胃肠道微生物群。
个性化微生物治疗:益生菌在糖尿病治疗中具有潜力,可通过减少自身免疫反应、炎症及氧化应激来提高胰岛素敏感性,同时增强黏附蛋白表达以降低肠道通透性。例如,补充双歧杆菌已被证明可改善葡萄糖耐受性。由于个体间微生物组结构和功能差异显著,基于微生物组的糖尿病治疗需采取个体化策略以实现最佳效果。
饮食成分调整:部分植物活性物质具有调节胰岛素和血糖的潜力。例如,高脂饮食中以改性大豆蛋白代替牛肉可显著降低餐后胰岛素水平,可能与其碳水化合物和纤维特性相关。不同谷物、豆类及脂肪酸类型也会影响犬的胰岛素敏感性。
此外,多种植物可影响犬的糖代谢,如辣椒素、迷迭香、罗勒和绿茶提取物。研究表明,它们能降低血糖和胰岛素水平、增强胰岛素分泌,并通过多酚保护胰腺β细胞功能。
功能性食品开发流程
3
骨关节炎(OA)
骨关节炎的改善需要采用多模式的综合管理方法,通过多种干预手段协同作用以获得更好的治疗效果。
体重控制:是所有治疗方案中最重要的一环。减轻体重能直接降低关节负荷。
核心营养干预-高剂量EPA:应使用专门为关节健康设计的处方粮,或在现有日粮基础上添加足量的高品质鱼油,确保EPA+DHA的摄入量达到治疗水平。需告知主人,该干预起效较慢,需坚持8-12周。
辅助营养成分:可联合使用葡萄糖胺/软骨素和ASU作为软骨保护剂。高生物利用度的姜黄素、乳香提取物或新西兰绿唇贻贝提取物可作为额外的抗炎支持。
4
认知功能障碍(CCD)
核心:选择含有复合抗氧化剂(维生素E, C)和多种植物营养素(来自水果蔬菜提取物)的老年犬专用处方粮。
结构支持:DHA:确保日粮中DHA的含量充足。
能量支持:对于已有明显认知衰退症状的犬只,换用含有6-10% 中链脂肪酸(MCTs)的处方粮已被证明能显著改善其认知评分。
肠道支持:通过补充益生菌/益生元来维护老年犬的肠道健康,有助于减轻可能加剧神经炎症的“炎性衰老”。
针对肠道微生物组的功能性营养干预,将在未来犬猫的健康维护与疾病管理中扮演愈发关键的角色。从益生菌的菌株特异性免疫调节,到EPA对炎症通路的分子调控,再到抗氧化剂组合在延缓神经退行性病变中的协同作用,功能性营养已从一种辅助手段,发展成为现代循证兽医学中用于管理多种慢性疾病的、不可或缺的科学工具。
然而,我们必须清醒地认识到当前领域的挑战。最大的挑战来自于显著的个体差异性。不同品种、年龄、遗传背景和生活环境的动物,其基础微生物组构成和对营养干预的反应都可能截然不同。当前基于“群体平均值”的推荐方案,在个体应用中效果可能打折扣。
随着高通量测序成本的降低,多组学技术将使我们能够为每一只动物绘制出其独特的肠道微生态“指纹”。通过将这些个体化的多组学数据与临床表型、生活方式等信息相结合,并利用人工智能(AI)和机器学习算法进行深度分析,我们将能够:
实现疾病的早期预测与诊断:在临床症状出现前,通过识别特定的微生物组失调模式或代谢物谱异常,来预警疾病风险。
制定高度个性化的营养方案:根据动物特定的菌群缺陷(如缺乏某种产丁酸菌)或代谢异常,为其精准匹配最有效的益生菌菌株、益生元种类或后生元组合。
动态监测与方案优化:通过定期检测,评估干预效果,并对营养方案进行动态调整,形成一个“诊断-干预-监测-优化”的闭环管理。
综上所述,功能性营养学正引领着一场宠物健康管理的深刻变革——从“千篇一律”的传统模式迈向“量体裁衣”的精准时代。对临床工作者而言,这意味着需要将前沿的科学工具整合为个体化的诊疗方案。而对宠物主人来说,这更赋予了他们一种全新的能力:通过科学喂养,更主动、更深刻地参与到守护爱宠长期健康的责任中来。
主要参考文献:
https://www.sciencedirect.com/science/article/pii/S1756464623003444
Di Cerbo A, Morales-Medina JC, Palmieri B, Pezzuto F, Cocco R, Flores G, Iannitti T. Functional foods in pet nutrition: Focus on dogs and cats. Res Vet Sci. 2017 Jun;112:161-166.
Rindels JE, Loman BR. Gut microbiome – the key to our pets’ health and happiness? Anim Front. 2024 Jun 20;14(3):46-53.
Kates AE, Jarrett O, Skarlupka JH, Sethi A, Duster M, Watson L, Suen G, Poulsen K, Safdar N. Household Pet Ownership and the Microbial Diversity of the Human Gut Microbiota. Front Cell Infect Microbiol. 2020 Feb 28;10:73.
Yang, Q.; Wu, Z. Gut Probiotics and Health of Dogs and Cats: Benefits, Applications, and Underlying Mechanisms. Microorganisms 2023, 11, 2452. https://doi.org/10.3390/microorganisms11102452
Hernandez J, Rhimi S, Kriaa A, Mariaule V, Boudaya H, Drut A, Jablaoui A, Mkaouar H, Saidi A, Biourge V, Borgi MA, Rhimi M, Maguin E. Domestic Environment and Gut Microbiota: Lessons from Pet Dogs. Microorganisms. 2022 Apr 30;10(5):949.
Atuahene, D.; Mukarram, S.A.; Balouei, F.; Antwi, A. Gut Health Optimization in Canines and Felines: Exploring the Role of Probiotics and Nutraceuticals. Pets 2024, 1, 135-151.
Grześkowiak Ł, Endo A, Beasley S, Salminen S. Microbiota and probiotics in canine and feline welfare. Anaerobe. 2015 Aug;
Guo X, Wang Y, Zhu Z, Li L. The Role of Plant Extracts in Enhancing Nutrition and Health for Dogs and Cats: Safety, Benefits, and Applications. Vet Sci. 2024 Sep 12;11(9):426.
Caterino C, Aragosa F, Della Valle G, Costanza D, Lamagna F, Piscitelli A, Nieddu A, Fatone G. Clinical efficacy of Curcuvet and Boswellic acid combined with conventional nutraceutical product: An aid to canine osteoarthritis. PLoS One. 2021 May 28;16(5):e0252279.
Wernimont SM, Radosevich J, Jackson MI, Ephraim E, Badri DV, MacLeay JM, Jewell DE, Suchodolski JS. The Effects of Nutrition on the Gastrointestinal Microbiome of Cats and Dogs: Impact on Health and Disease. Front Microbiol. 2020 Jun 25;11:1266