-

 CNAS L23010
CNAS L23010

 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室

谷禾健康

更年期标志着女性衰老的一个关键里程碑,引发激素,组织学和微生物组的变化。
随着卵巢功能衰退,雌激素与孕激素的断崖式下降,不仅重塑了女性体内的激素图谱,更改变了口腔、肠道、阴道等关键部位的微生物群落平衡。
最新研究揭示,更年期的症状,从反复的腹胀、顽固的牙周炎到阴道感染,其背后与微生物组的“失控”密切相关:肠道菌群的多样性降低可能加剧骨质流失,而口腔微生物的代谢紊乱甚至与心血管风险形成隐秘联动。
激素波动在塑造这些微生物群落方面发挥关键作用,对疾病易感性有影响。雌激素影响微生物群落,而微生物可以代谢并影响雌激素水平。因此,激素与微生物组之间的相互作用是复杂且双向的。
联合国大会已将2021-2030年期间定为”健康老龄化十年“。联合国“健康老龄化十年”计划强调,尽管女性寿命更长,但更年期带来的独特健康挑战,如骨质疏松、心血管疾病和泌尿生殖系统感染,正威胁着她们的晚年生活质量。
社会经济因素和文化规范(如生活方式和营养习惯)也影响她们的健康和老龄化体验。因此,关注性别特定的健康需求并推广定制化的健康老龄化策略,对于提高老年女性的健康和生活质量至关重要。
本文系统阐述了更年期女性激素波动与微生物组(口腔、肠道、阴道)的动态互作机制,揭示了雌激素下降引发的菌群失衡如何加剧骨质疏松、心血管疾病、牙周病等健康风险,这为更年期健康管理从“症状缓解”转向“精准干预”提供了科学依据。
理解更年期转变包括激素变化、环境因素和微生物动态如何影响更年期症状和女性健康。这一见解可能推动精确疗法的开发,以缓解症状并最小化相关健康问题的风险,最终提高更年期女性的生活质量。
更年期是由于卵巢滤泡活动减少导致的连续12个月自然停经的永久现象,通常发生在50岁左右,但自然变化范围在40~59岁之间。
更年期女性有哪些症状?
常见症状
大多数情况下,更年期会随着时间的推移而发生。导致绝经的几个月或几年称为围绝经期或绝经过渡期。
在过渡期间,卵巢产生的激素量会有所不同。围绝经期可持续 2~8 年。平均约为 4 年。
激素变化会引起以下症状:
此外,更年期时口腔健康会因雌激素水平下降和口腔组织老化而受到影响。
不同的人有不同的更年期症状。大多数情况下,月经在结束之前并不规律。
围绝经期,通常,月经周期跳过一个月并返回。或者跳过几个月,然后再次开始每月一次的周期,持续几个月。围绝经期早期的月经周期往往会变短,随着更年期的临近,月经间隔会越来越远,直到结束。
需要注意的是,随着年龄增长,老化过程以及系统性疾病或药物对口腔变化的影响会增加。口干感、颞下颌关节功能障碍和心理生理障碍引起的并发症是一些更年期女性出现饮食障碍的多种原因之一。
另外,由于老化或雌激素下降导致的免疫功能受损可能会显著影响口腔感染的发生。
并发症
绝经后患某些疾病的风险会增加。比如:
更年期与什么因素有关?
30 多岁时,卵巢开始减少控制经期的激素,如雌激素和黄体酮。如果这些激素水平低,较难怀孕。
在 40 多岁时,月经期可能会变长或变短、变重或变轻,并且发生频率更高或更低。随着时间的推移,卵巢会停止释放卵子。那么就没有更多的月经了。这种情况平均发生在 51 岁左右。
卵巢产生控制月经周期的激素,包括雌激素、黄体酮。切除卵巢的手术会导致立即绝经。
月经停止,可能会出现潮热和其他绝经症状。症状可能严重,因为手术会导致激素立即下降,而不是在几年内缓慢下降。
切除子宫但不切除卵巢的手术称为子宫切除术,通常不会立即绝经。卵巢在一段时间内仍然会释放卵子并产生雌激素和黄体酮。
这些癌症疗法可导致更年期。它们会在治疗期间或治疗后不久引起潮热等症状。化疗后月经有时会恢复,仍然可以怀孕。
针对骨盆、腹部和下脊柱的放射疗法可导致绝经。用于干细胞移植的全身放疗也可能导致绝经。对身体其他部位(如乳房组织或头部和颈部)进行放射疗法可能不会影响绝经。
大约 1% 的更年期患者在 40 岁之前过早绝经。过早绝经可能是由于卵巢没有产生通常的激素水平造成的,也就是原发性卵巢功能不全,它可能由基因变化或自身免疫性疾病引起。
更年期期间,激素水平变化会影响肠道菌群组成和功能。研究发现某些肠道菌群能直接代谢雌激素和孕激素,形成双向关系:激素影响菌群,菌群也参与激素代谢。这种微生物变化可能加剧更年期症状。
具体我们将在下一章节详细阐述。
更年期时,体内激素变化会改变口腔、肠道、阴道和皮肤的微生物环境。研究发现某些肠道菌群能直接代谢雌激素和孕激素,当女性进入更年期时,激素水平下降导致的生理变化也会影响微生物群的组成和功能。
因此,探索微生物组与更年期之间的复杂相互关系揭示了缓解更年期症状和改善整体健康的有前景途径。通过调整饮食、使用益生菌和个性化微生物干预等方法,有望改善更年期女性的健康状况。
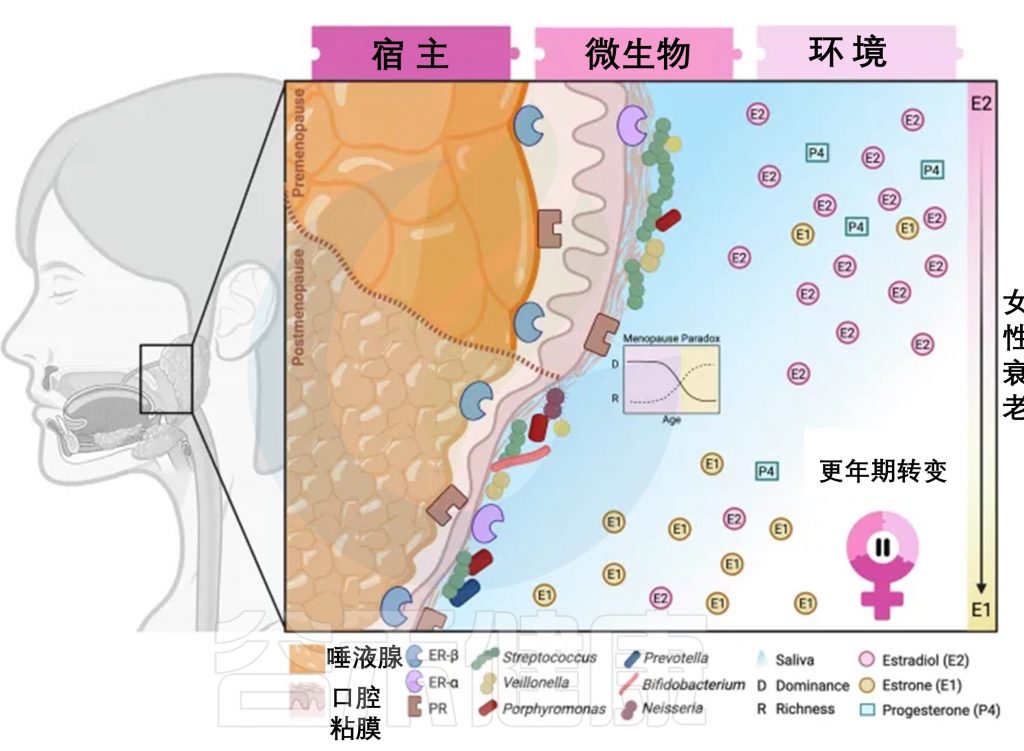
doi.org/10.1038/s44294-024-00050-y
宿主、微生物、环境在更年期的变化
宿主变化
更年期期间,女性的身体会发生一系列改变。例如,唾液腺和口腔黏膜(如牙龈、舌头表面)会逐渐变薄甚至萎缩。类似的变化也可能出现在肠道、阴道等对激素敏感的部位,因为这些地方的组织里存在能接收雌激素、孕激素信号的“接收器”(激素受体)。
微生物变化
更年期期间,微生物组多样性(微生物种类分布)和丰富度(种类数量)会发生变化,这些变化受到宿主组织互动、激素代谢和环境变化的影响。
口腔细菌不仅局限于黏膜生态环境,还存在于牙龈或舌头等其他口腔部位。它们被视为能够代谢性类固醇激素的常驻口腔微生物群的一部分。
更年期悖论表现为阴道环境中微生物优势度降低但丰富度增加,这种现象可能适用于微生物群体中的其他身体部位。从临床角度看,微生物适应能力的变化可能导致感染等健康问题。
环境变化
唾液减少:唾液分泌减少,导致口腔干燥,细菌更容易滋生。
雌激素类型转换:卵巢不再生产强效的雌二醇,转而依赖脂肪组织合成的雌酮(效果较弱)。
孕激素“断崖式下降”:这种激素的减少可能削弱身体对炎症的控制能力。
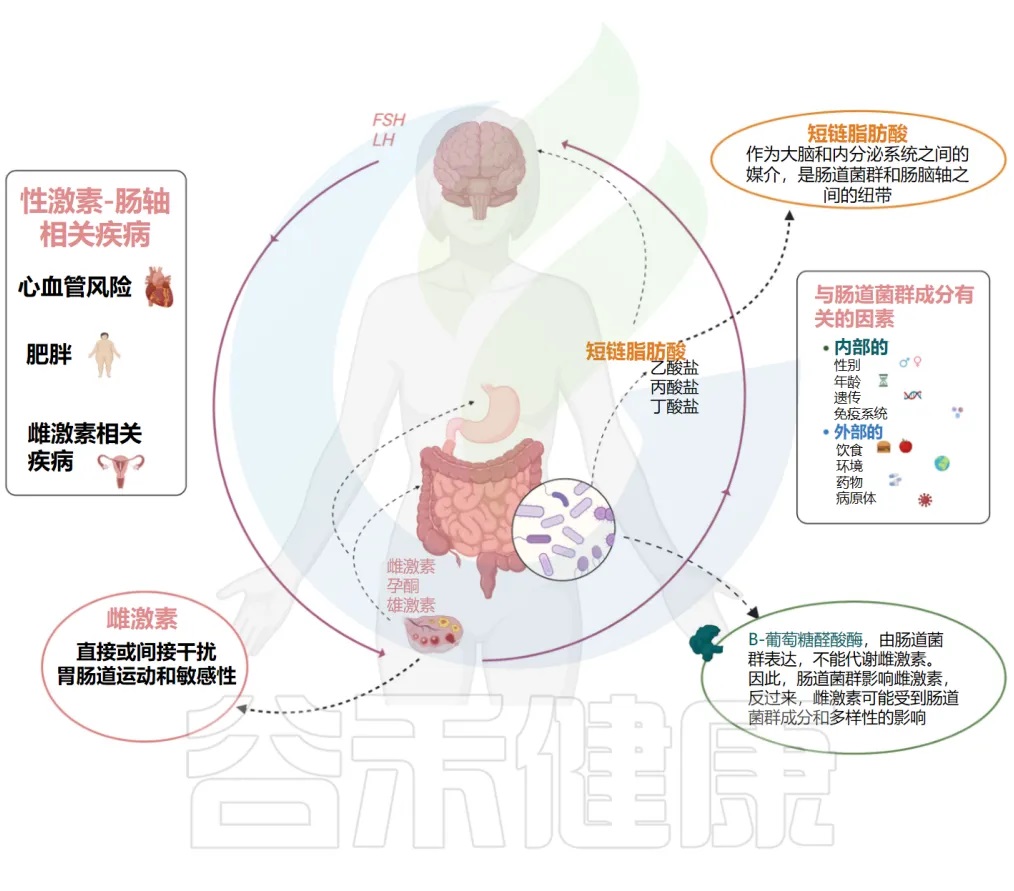
雌激素与微生物组的双向互动
雌激素是与女性内分泌转变相关的主要性激素,包括青春期、妊娠期、更年期。雌激素就像是调节女性身体的总开关,特别在这些重要时期起作用:
在更年期过渡期间,女性的粘膜组织变薄变干,导致阴道和口腔细菌失调,这可以通过更年期激素治疗来缓解。
微生物组和性激素之间存在动态的双向相互作用,随着一些因素如衰老而变化。
▸雌激素存在两种形式:
人体内的雌激素水平通过以上两种形式的平衡来调节。
▸微生物的特殊能力:
肠道里的某些细菌有一种特殊”钥匙”(β-葡萄糖醛酸苷酶),这把”钥匙”能打开储备形式的雌激素,让它变成活跃形式,这些特殊细菌群体被称为”雌激素组”细菌。
游离雌激素可以被运输到许多部位,如阴道,并促进微生物组中乳杆菌的优势地位。因此,代谢性激素的微生物可以改变宿主对可用性的控制,从而影响与激素相关的生理过程。
▸互相影响的循环
雌激素水平影响微生物群落的组成,微生物又能影响体内实际可用的雌激素量;循环关系会随着年龄增长而改变。在更年期,这种平衡被打破。
因此了解这种关系很重要,因为通过调整微生物群(比如益生菌),可能有助于缓解一些更年期症状。
雌激素:从全身健康到口腔微生态的塑造
雌激素变化如何塑造女性一生的健康轨迹
雌激素,主要以雌二醇和雌酮形式存在,在女性从青春期到更年期的不同生命阶段经历显著变化。
在生育年龄,雌二醇是最强效且普遍的雌激素形式,随月经周期呈现周期性波动,在排卵期达到峰值。这种激素在生殖功能中扮演关键角色,影响全身健康,维持骨密度、心血管健康、性健康、情绪调节和认知健康。
然而,随着更年期的到来,雌激素动态发生明显转变。卵巢功能下降导致雌二醇产量减少,使雌酮成为主要雌激素形式。与雌二醇不同,雌酮在脂肪组织中合成,在排卵停止后维持某些雌激素活动。不过,在绝经后,整体雌激素活性降低。
在谷禾的检测实践中,也发现部分的45岁以上女性雌激素水平缺乏或偏低。
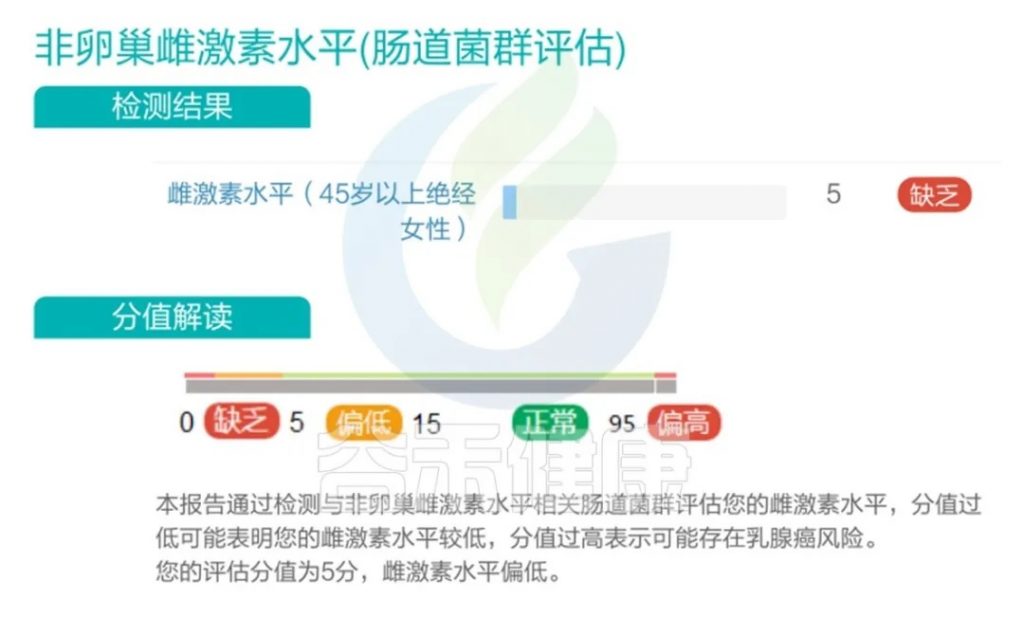
<来源:谷禾健康肠道菌群检测数据库>
多项研究将这种下降与骨质疏松、心血管疾病、认知障碍风险增加相关联。这一时期反映了雌激素在女性一生中的复杂相互作用,雌酮在雌二醇水平降低的情况下提供残留但不足的雌激素效应。这些女性激素水平的波动可能是女性在更年期过渡期间经历的影响其健康的身体和心理症状的原因。
雌激素循环途径
Valeri F,et al.Front Neuroendocrinol.2021
激素如何影响口腔健康?
口腔液体自然含有激素。在口腔中,唾液和牙龈沟液对维持健康完整性起着重要作用。唾液和牙龈沟液共享一些代谢物和途径,但也有各自独特的成分。
唾液主要由水、矿物质、电解质、激素、酶、免疫球蛋白和细胞因子组成。唾液的生理水平对口腔健康至关重要,因为它影响口腔中的各种因素,如防止龋齿、免疫过程、消化。唾液中含有卵巢分泌的高活性天然雌激素17β-雌二醇、孕激素、雌酮。
在唾液腺腺泡和导管细胞中发现雌激素-β受体,表明雌激素缺乏可能是更年期女性唾液分泌和无机成分变化的病因。唾液的成分和流量也可能受某些药物影响,如抗抑郁药、降压药、口腔消毒剂和癌症治疗药物。
更年期唾液变化:酸性环境与口腔健康风险的隐秘关联
激素缺乏可能导致唾液在数量和质量上发生变化,破坏口腔内环境平衡,影响口腔微生物群并导致细菌定植。绝经后女性唾液pH值和流量的变化直接导致口腔疾病增加。
唾液pH值与年龄之间的关系存在争议
患者体内的氢离子浓度随年龄增长而增加,使他们处于更酸性的环境中。有研究报告显示,与绝经前女性相比,绝经后女性的唾液pH值较低,而其他研究则发现不同组别之间的唾液pH值没有显著变化。
2018年发表的一项病例对照研究(n=80)显示,与对照组(n=40)相比,绝经后女性(n=40)的唾液流量和pH值降低。因此,更年期的激素变化可能导致女性体内pH值更酸性,增加口腔组织与老化相关的损伤风险。然而,身体其他部位的pH值却会增加,从而导致细菌感染。
激素波动如何破坏牙龈健康?微生物与炎症
相比之下,牙龈沟含有血清衍生的牙龈沟液。牙龈液中含有雌二醇和孕激素,其波动影响牙龈组织。雌激素降低角质化程度,增加上皮糖原,并影响成纤维细胞增殖和蛋白质生成。
孕激素增强血管通透性,减少糖胺聚糖合成,改变胶原蛋白生成速率和模式,并抑制IL-6生成。女性类固醇激素对牙龈产生促炎作用。
存在牙龈炎等牙龈炎症期,这与微观解剖结构改变以及牙龈和龈下细菌群落增加相关。尽管雌二醇水平在更年期期间急剧下降,但雌酮等其他形式的雌激素可能影响牙龈组织。
由于牙龈环境中有常驻微生物生物膜群落,而唾液中含有暂时性浮游微生物群,这两种口腔环境都能在激素波动期间影响宿主-微生物相互作用。因此,研究口腔液体是否含有各种浓度的激素以及微生物群可获取的形式至关重要。
更年期女性的身体“改造”:从皮肤到骨骼
激素撤退的连锁反应:黏膜屏障弱化与阴道健康警报
卵巢激素受体存在于鼻咽黏膜、胃肠道和女性泌尿生殖系统中。性激素的作用影响口腔黏膜、牙龈和唾液腺组织。更年期发生的激素变化产生血管运动改变,导致血管通透性和炎症介质发生变化。
雌激素和孕激素调节女性生殖道的黏膜屏障和免疫反应,若它们的水平改变可能引起阴道症状。与更年期相关的最常见阴道症状是因雌激素缺乏导致的阴道干燥以及阴道上皮变薄、失去防御元素。
一些最常见的病理状况包括外阴阴道萎缩、复发性尿路感染、细菌性阴道病和阴道念珠菌病。结果,阴道区域出现灼热感、瘙痒和刺痛。
口腔与阴道的”镜像变化”
更年期女性常惊讶于口腔干燥与阴道灼痛的同时出现,这背后暗藏黏膜系统的深层关联。在微观层面,口腔和阴道上皮在超微结构、角蛋白丝分布、水渗透性和化学成分方面表现出相似性。这一点特别值得注意,因为阴道上皮在更年期期间及之后会经历各种变化,这暗示着对口腔上皮可能的影响。
多项研究发现,两种黏膜的上皮细胞层数没有显著差异。同样,角质化模式和细胞间隙的脂质层分布也很相似。所有这些表明,鉴于它们的微观相似性,绝经后女性阴道黏膜因缺乏激素刺激而观察到的变化也可能以相同方式影响口腔上皮。
有研究表明,口腔、阴道和肠道微生物群的组成可能受雌激素水平调节。因此,性激素的减少被发现会引起宿主的炎症反应增加,可能导致口腔微生物平衡失调。这可能导致各种牙龈病理,其中更年期龈口炎尤为突出。
口腔里的激素“接收器”与更年期连锁反应
我们的口腔中藏着一些特殊的“激素接收器”——牙龈和唾液腺里分布着能感应雌激素、孕激素的受体。就像乳腺组织一样,唾液腺里主要存在雌激素受体α(ERα),而牙龈中则富含另一种“接收器”雌激素受体β(ERβ)。这些受体像开关一样,帮助激素调控口腔组织的健康状态。
此外,唾液腺和牙龈细胞里还发现了孕激素受体(PR),协同调节激素对口腔的影响。
更年期为何容易“口干”?
唾液腺中的腺泡和导管细胞(尤其是腮腺、颌下腺)密布激素受体。当更年期雌激素断崖式下降,这些“接收器”失去信号,唾液分泌可能减少,导致口干、黏膜干燥等问题。
激素撤退的全身风暴
更年期后性激素水平的急剧下降,尤其是雌激素水平,对神经系统、心血管系统、风湿性疾病、内分泌系统、胃肠道和泌尿生殖道有众多影响。在绝经前女性中,性类固醇激素通过其受体展示直接血管扩张作用,表明其心血管益处。在年轻女性中,雌激素有助于心脏保护,这一功能在更年期后减弱。
骨质疏松症是更年期另一种常见疾病,尽管其病因复杂。在这一阶段,激素波动和钙代谢改变可能导致骨吸收水平增加,使该疾病在更年期后更为普遍。
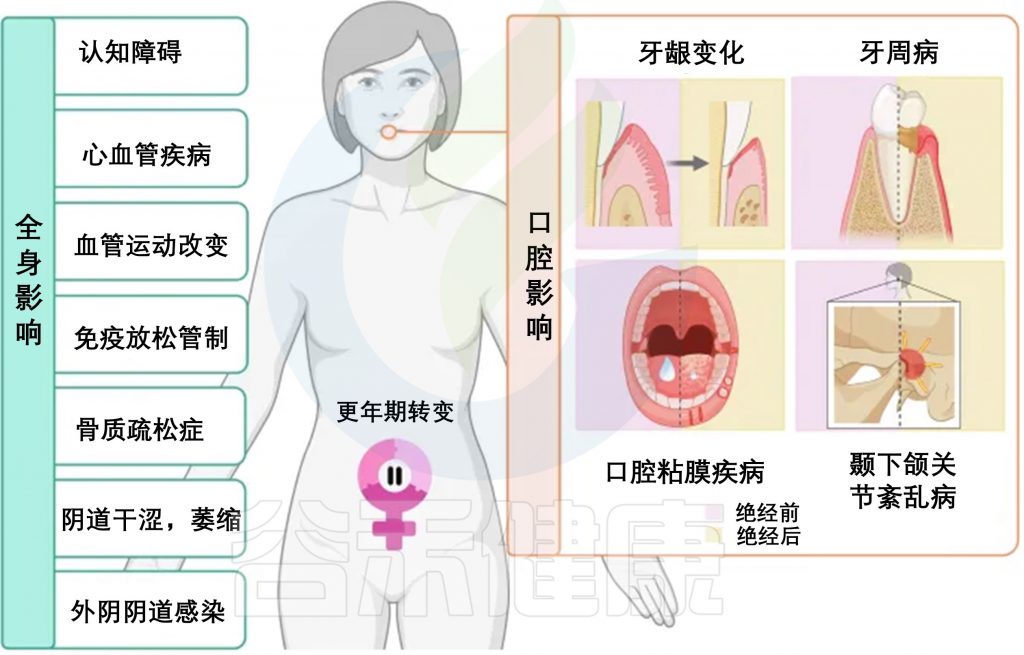
doi.org/10.1038/s44294-024-00050-y
更年期转变对口腔的影响
在更年期,由于女性生殖衰老导致的雌激素下降,会发生一系列生理变化。这种明显的低雌激素状态影响口腔颌面系统和其他系统,产生各种全身和口腔临床表现,影响女性的健康。
多项研究探索了更年期口腔状况与全身表现之间的关联,发现了积极的相关性。
更年期如何悄然影响牙周健康?
牙周病是一种慢性细菌源性炎症疾病,破坏牙齿支持组织,最终导致牙齿脱落。这是一种多因素病因学病理,由必要但不充分的主要病因因素、易感宿主以及影响两者的环境因素相互作用引起。
性激素被认为是重要的修饰因素,可以增加宿主对牙周病原体的易感性,因此影响牙周病的患病率、进展和严重程度。性激素水平的显著波动以及更年期特有的骨质疏松症已导致多项研究将更年期与牙周病联系起来。
骨质疏松症可能导致单位体积内牙槽嵴骨密度降低,这会加速牙周感染引起的骨吸收刺激下的骨质流失。基于这一假设,一项针对绝经后女性的研究未能将较低的系统性矿物质密度与对口腔感染反应中更大的牙周附着丧失相关联,尽管观察到随着患者年龄增长,这种关联变得显著。
更年期激素治疗:牙周健康的潜在保护因素
后续研究报告显示,绝经后女性的牙周病患病率高于绝经前女性,而绝经前女性与接受更年期激素治疗(MHT)的绝经后女性的牙周状况相似。一些研究甚至指出MHT是减轻牙痛、改善牙齿活动度和降低牙周袋探测深度的保护因素。
最近有研究探讨了绝经后女性牙齿脱落的主要原因是口腔卫生不良和骨矿物质密度低,低雌激素水平-骨质疏松症与牙齿脱落呈正相关,但需要更多研究来明确更年期与牙周病之间的关系,考虑到与两种状况相关的混杂因素,如高龄、教育程度、慢性疾病、烟草和酒精消费,特别是口腔卫生和饮食。
颞下颌关节紊乱与更年期的关系
颞下颌关节紊乱(TMD)是一组影响咀嚼肌和颞下颌关节(TMJ)的肌肉骨骼疾病。女性TMD发病率高于男性,以及在TMJ盘中检测到雌激素和孕激素受体,使研究者考虑女性性激素在这种关节紊乱的多因素病因学中的作用。
一些作者观察到绝经后女性的TMD患病率和严重程度高于绝经前女性。相关研究中,对不同月经周期状态下患有TMD的女性进行的研究得出结论,当雌激素水平较低时,TMD相关慢性疼痛程度、咀嚼功能障碍、抑郁症状和躯体化症状更为严重。
然而,其他研究者评估了绝经后女性TMD的存在及其与疼痛和更年期激素治疗(MHT)使用的关系,发现TMD与绝经后期之间没有关系,MHT使用与TMD疼痛之间也没有关系。这些相互矛盾的结果需要进一步研究,以了解更年期对TMJ的影响。
更年期唾液危机:口腔干燥与健康变化的激素关联
雌激素缺乏可能与更年期女性唾液分泌变化有关,这是由于唾液组织改变所致。唾液减少与口腔变化相关,如口腔黏膜光泽丧失、黏膜干燥、舌背裂纹、口角炎、唾液变稠、口腔感染频率增加、非典型位置出现龋齿以及主要唾液腺体积增大。
绝经后女性常见口腔症状包括干燥、灼热感、口腔疼痛、味觉改变、牙齿脱落,通常伴有吞咽困难、发音问题和口臭。
口干或口腔干燥症是围绝经期和绝经后期的主要口腔症状,大多数患者报告唾液流量减少。唾液中孕激素水平与更年期口腔干燥感直接相关。
然而,目前尚不清楚干燥是否仅与激素下降有关,因为药物或衰老等其他因素也会影响唾液分泌率。
更年期口腔干燥与相关综合征
口腔干燥感通常伴随着口腔灼烧综合征(BMS),这也与绝经后期的真菌感染如口腔念珠菌病有关。情绪不稳定,特别在表现为口腔黏膜疼痛和灼热的BMS患者中明显,与抑郁或焦虑等心理障碍相关,突显了更年期口腔健康问题的多方面性质。
此外,面部、牙齿和颞下颌关节紊乱以及溃疡也有所增加。总体而言,唾液流量减少会导致其他症状,如口味差或异常、吞咽困难、唾液粘稠、口腔黏膜炎、扁平苔藓、口炎和类天疱疮。
因此,通过制定预防性口腔护理计划和针对更年期的特定治疗干预措施,解决老龄女性的口腔疾病非常重要。
口腔微生物组是由宿主-微生物互动及环境条件共同塑造的复杂生态系统,分布于口腔软硬组织中。激素波动可直接影响该系统的平衡,例如改变唾液成分(如流量、pH值及抗菌物质含量),从而调控微生物群落的结构与功能。
研究表明,激素水平变化(如青春期、妊娠期及更年期)会通过唾液特性改变,促进特定致病菌增殖或抑制有益菌定植。以更年期为例,雌激素锐减引发的唾液酸化与黏稠度增加,可显著改变微生物群落的组成,加剧龋齿及牙周病风险。此外,分析口腔微生物组的动态变化,可能成为监测系统性激素波动的潜在生物标志物。
口腔微生物组与激素变化的关系
更年期时,女性体内的雌激素水平会随着卵巢功能衰退而逐渐降低。但有意思的是,在更年期初期(过渡期),某些性激素可能会暂时升高,这种波动会刺激口腔组织发炎,并扰乱牙龈部位的微生物平衡,导致有害细菌过度繁殖。与此同时,肠道内的雌激素水平变化也会打破人体和肠道微生物之间的平衡,影响消化和免疫健康。
口腔-肠道-雌激素组轴提示全身雌激素和口腔健康之间存在联系。比如,口腔里的某些细菌能分解雌激素、孕激素等激素,甚至反过来调节它们的水平。这意味着,如果雌激素代谢出了问题,最早可能在口腔菌群失衡中表现出来,比如牙龈出血、口臭等症状,都可能成为身体发出的预警信号。
具体来说:
孕激素能“压制”有害菌:研究发现,像奈瑟菌(Neisseria)、葡萄球菌这类容易引发口腔感染的有害菌,遇到孕激素时生长会被抑制,这种效果和激素浓度有关。
“吃激素”的细菌更危险:密螺旋体等细菌会“偷吃”人体产生的类固醇(如雌激素)作为营养,这可能让它们变得更活跃,甚至加重牙周疾病。
维生素K的替代来源:
一些口腔细菌如Prevotella intermedia、Prevotella nigrecens、 Capnocytophaga等需要维生素K才能存活,而雌激素和孕激素可以替代这种营养。这些细菌在牙龈组织中随着雌二醇和孕酮周期而同步增加。
最近的一项研究调查了雌二醇、雌三醇、孕激素或睾酮对体外口腔生物膜诱导毒力因子表达的影响。研究显示对生物膜形成、微生物组成和蛋白水解活性的影响很小。在口腔牙龈组织中,牙龈含有能够特异性结合雌激素的受体,其微生物组具有β-葡萄糖醛酸苷酶(GUS)作为雌激素代谢酶。
源自人类口腔微生物组的GUS图谱与53种独特的GUS酶相关。许多这些酶在常与牙周病相关的属中被鉴定,如Tannerella, Treponema、普雷沃菌属、梭杆菌属。特别是,口腔微生物组中发现的GUS蛋白与胃肠道中发现的不同。性激素影响口腔生态位的确切机制仍在持续探索中。
内源性类固醇与口腔微生物代谢的关联
内源性类固醇能够直接触发正常微生物群改变的能力的发现,为类固醇水平波动标记的口腔和一般健康之间的联系提供了新见解。类固醇脱氢酶已在多个细菌属中被鉴定,包括梭菌属、棒状杆菌属(Corynebacterium)、芽胞杆菌属(Bacillus)、分枝杆菌属(Mycobacterium)、Nocardia、假单胞菌属(Pseudomonas)和链霉菌属(Streptomyces)。这突显了口腔微生物细胞的居民可以通过微生物酶代谢孕激素和睾酮。
变形链球菌(Streptococcus mutans)同时拥有5α-和5β-类固醇还原酶,以及3α-、17β-和20α-羟类固醇脱氢酶,促进孕激素和睾酮的代谢。
此外,牙龈沟中发现的其他口腔细菌已知含有参与类固醇转化的细菌酶。例如,密螺旋体通过5α-还原酶、3β-和17β-羟类固醇脱氢酶代谢胆固醇、孕激素和睾酮。
因此,关注更年期过渡期间微生物在激素代谢中的作用,强调了有针对性干预的潜力,并为更年期症状管理的先前未探索的途径提供了启示。
唾液皮质醇如何影响微生物与更年期相关疾病
唾液皮质醇是用于检查人类应激反应的生物标志物。
心身性头颈部疾病如口疮性口炎、非典型面部疼痛、口腔扁平苔藓、口腔灼烧综合征(BMS)和口干症与更年期阶段相关。
在一项包括200名绝经后女性的临床试验中,唾液皮质醇水平显示出统计学显著性,证明患有心身疾病的绝经后女性具有更高水平的皮质醇。
最近一项关于应激相关皮质醇对口腔微生物组影响的宏转录组功能分析发现,在皮质醇存在的情况下,梭菌门成员变得更加活跃。有趣的是,之前与牙龈炎相关的Leptotrichia goodfellowii活性显著增强。总体而言,口腔微生物组暴露于皮质醇可改变整个细菌群落的活动。
这些变化包括宿主对口腔细菌的免疫应答过度表达,以及细菌方面蛋白质水解、寡肽转运、铁代谢和鞭毛组装的增加。这些活动先前已与功能性菌群失调和口腔疾病(如牙周病)的进展相关。这提出了一个有趣的可能性,即口腔微生物可能直接响应应激激素的存在。
更年期对唾液与口腔微生物的隐秘影响:争议与新发现
唾液流量和成分与更年期的关联存在争议。
AMICA项目比较了20名绝经后女性与对照组(19名生育年龄女性)的唾液口腔微生物组。该研究未发现更年期女性与有规律月经周期女性之间唾液口腔微生物组成分存在显著差异。
最丰富的细菌属是已知在健康个体中占主导地位的,如链球菌、奈瑟菌、卟啉单胞菌、普雷沃菌和韦荣球菌。
相比之下,研究人群中占主导地位的细菌科是普雷沃菌科和链球菌科。具体而言,绝经后普雷沃氏菌显著增加,而Veillonella tobetsuensis减少。此外,经历严重唾液分泌减少的女性与唾液流量正常的女性相比,细菌谱相似。
研究在绝经后女性唾液中发现了显著的代谢物变化,雌二醇水平与非刺激性唾液流量呈正相关。
另一项研究确定了老年女性唾液分泌减少相关的唾液成分差异,特别关注口腔微生物组。研究结果表明,由于唾液流量减少导致的雌二醇水平下降可能导致更年期口腔问题并改变某些口腔细菌。
进一步的大规模人群和纵向研究将阐明更年期转变对唾液环境的影响。
更年期女性的牙周疾病与口腔微生物组
生物膜相关牙周疾病是老年和更年期女性常见的口腔疾病。绝经后女性的龈下微生物组与牙周疾病的存在和严重程度相关。与这种疾病相关的一些最具特征性的病原体属于拟杆菌属、牙龈卟啉单胞菌、螺旋体、牙形螺旋体、拟杆菌目和梭杆菌属。
厚壁菌门和拟杆菌属之间的相互作用可以作为老年患者微生物栖息地的良好指标。研究描述了更年期女性厚壁菌门与拟杆菌属的比例趋向更高。
特定物种在更年期女性的龈下微生物组中存活,不受牙周疾病存在或严重程度的影响。特别是,这些包括Veillonella dispar、Veillonella parvula、口腔链球菌(Streptococcus oralis)和齿双歧杆菌(Bifidobacterium dentium)。
研究表明,齿双歧杆菌的存在可抑制牙周疾病中的显著病原体牙龈卟啉单胞菌的增殖。这一观察可能解释了为什么在没有牙周疾病的老年女性群体中检测到的牙龈卟啉单胞菌水平相对较低。
更年期期间口腔微生物组发生变化,特定病原体促进了老年女性的牙周疾病。尽管如此,某些物种在更年期女性的龈下微生物组中持续存在,可能影响牙周健康。
龈沟液与激素变化对牙周健康的影响
龈沟液是一种牙周渗出物,由含有各种与牙周定植微生物群相互作用的代谢物的血清组成。从临床上看,牙周炎症增加与激素调节失调的情况如妊娠期和雌激素依赖性疾病如子宫内膜异位症相关。
因此,老年女性牙周疾病相关的许多龈下微生物与年轻牙周病患者研究中观察到的龈下微生物群相似。一个显著的例外是缺乏伴随侵袭性牙周炎的聚集性放线杆菌(A. actinomycetemcomitans),这在老年人中很少见。
这些发现表明口腔微生物组与更年期之间存在复杂的动态关系,对研究和治疗具有重要意义。
更年期激素失衡与真菌失调
虽然先前的研究主要关注细菌物种,但认识共生真菌群落在口腔微生物复杂性中的作用至关重要。在更年期,由于衰老和激素失衡,真菌繁殖增加,导致菌群失调和机会性物种如白色念珠菌(Candida albicans)的增殖,特别是在绝经后阶段。
以下因素会促进念珠菌病的发展:
更年期慢性念珠菌病与口腔灼烧综合征
慢性念珠菌病可引起口腔灼烧感,这是口腔灼烧综合征(BMS)的特征症状,这两种情况都与长期服药和佩戴活动假牙有关。
虽然在45.16%患有BMS的绝经后女性中检测到白色念珠菌(C. albicans),但其与该综合征病因的关联尚无定论。
口腔环境的酸碱度和唾液分泌量对白色念珠菌的存在似乎没有明显影响,这表明念珠菌在口腔中的定植受到多种因素的复杂影响。虽然更年期的生理变化可能有利于真菌生长,但研究表明更年期本身并不直接增加口腔念珠菌感染的风险。
虽然在绝经后女性中未观察到唾液流量与白色念珠菌侵袭之间的关联,但以往文献表明可能存在潜在联系,特别是在那些使用质子泵抑制剂(可能影响口腔微生物平衡)的女性中。
最近研究已确定光滑念珠菌(Candida glabrata)是一种机会性病原体,负责黏膜和全身感染,通常见于老年人、免疫功能低下者和医疗机构环境中。
性激素对多部位微生物组的系统性调节
卵巢激素受体已在脑部、口腔、鼻咽部、胃肠道和女性泌尿生殖系统的黏膜中被发现。这表明神经系统、口腔、肠道、阴道和膀胱微生物组的组成可能受到性激素水平的系统性调节。
尽管我们体内的不同部位(如口腔、肠道和生殖系统)拥有各自独特的微生物环境,但研究发现某些对激素变化敏感的细菌能同时在多个区域生存。例如,某些乳杆菌不仅能在口腔中找到,也能在阴道和直肠中繁衍,有时甚至能通过腹部组织从肠道迁移到阴道。
当女性进入更年期后,体内激素水平的变化会对不同身体部位的微生物群落产生不同影响,改变人体与这些微生物之间的相互关系。这种变化可能是更年期女性在不同身体系统中出现各种症状的原因之一。
性类固醇激素影响的微生物-宿主相互作用
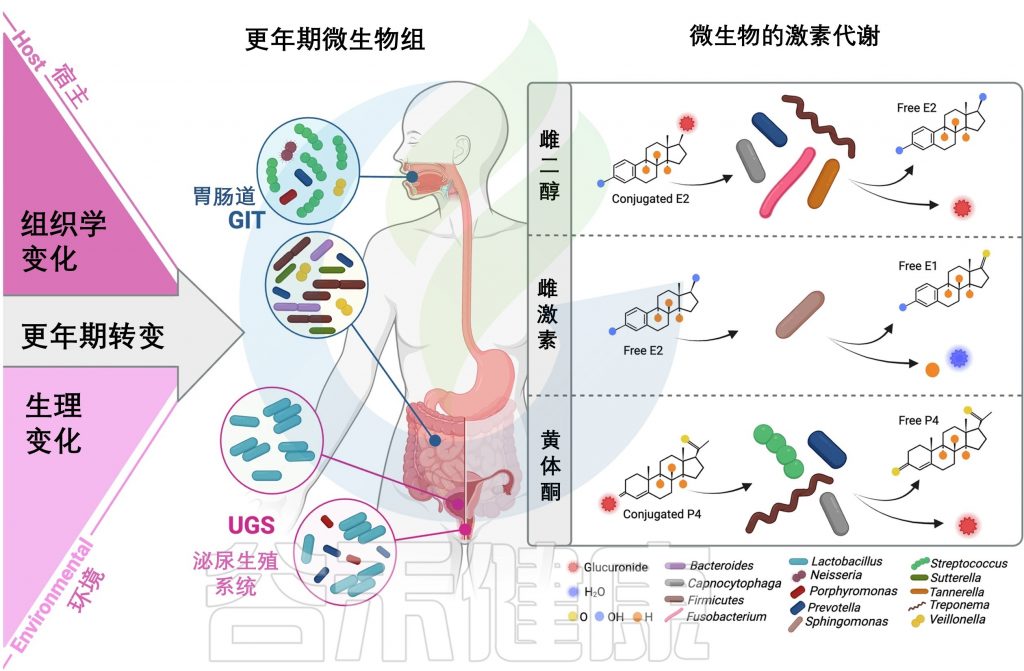
doi.org/10.1038/s44294-024-00050-y
最新研究显示,肠道中的微生物群落可能在女性衰老过程中扮演重要角色,特别是对更年期期间体内激素变化有显著影响。由于从口腔到肛门的整个消化系统被视为一个连续的环境,肠道中的细菌可能与口腔中的微生物有关联,反之亦然。
性激素和肠道菌群之间的相互关系
Calcaterra V, et al., Front Endocrinol (Lausanne). 2022
微生物群的α多样性由样本内微生物分布决定,包括存在的分类群的数量和相对丰度。研究表明,由于激素水平下降,更年期与胃肠道微生物群多样性的改变相关。
不过,科学界对此并未达成共识。一些相反的研究结果表明,绝经前和绝经后女性之间没有明显差异。
这些矛盾的发现表明更年期对肠道微生物组的影响可能比预期的更为复杂,受到多种因素影响。
更年期女性肠道微生物组的变化
研究者们正日益关注女性衰老过程中肠道微生物组所经历的变化。一项纵向研究发现,绝经后女性(n=1027)的肠道微生物组比绝经前女性(n=295)的多样性更低。
绝经后女性体内以下菌群丰度更高:
绝经后女性以下微生物丰度降低:
Prevotella和Sutterella 在其他研究中曾与肥胖相关。同样,肠道拟杆菌可能产生有益或有害影响,这取决于它与其他微生物组和宿主因素的关系。能在口腔中找到适宜条件的微生物,如普氏菌属、拟杆菌属和厚壁菌门,在胃肠道水平产生不同类型的群落。
更年期女性 Odoribacter 的增加导致短链脂肪酸、氢和硫化氢水平升高。短链脂肪酸增加脂肪酸氧化和能量代谢,参与血清素的合成和稳定神经元,并增加刺激成骨的循环胰岛素样生长因子-1。因此,更年期女性中短链脂肪酸 的Odoribacter 相关增加可能会降低肥胖、高脂血症、抑郁症和骨质疏松症的风险。相反,硫化氢产生增加会导致炎症反应。因此,Odoribacter 既有积极影响,也有不良反应(类似于绝经后综合征的影响)。
Biophila 产生的硫化氢可以放松回肠平滑肌,增加胃肠黏膜的血液供应。更年期妇女体内嗜胆汁细胞增多会导致硫化氢产生增加,引发局部炎症和粘膜损伤,血清内毒素浓度增加,以及几种组织的炎症反应。炎症因子引起的细胞内炎症反应通过干扰胰岛素信号转导导致胰岛素抵抗。在骨骼中,炎症因子(如肿瘤坏死因子-α、白细胞介素(IL-1和IL-6)增强破骨细胞的功能,导致骨质减少。外周血中的炎症因子穿过血脑屏障,激活中枢神经系统的小胶质细胞,导致神经元炎症,加剧神经纤维缠结和β-淀粉样蛋白的聚集和积累,从而导致阿尔茨海默病。
目前尚不清楚更年期过渡期间激素水平的变化是否会影响肠道微生物群的平衡,可能导致菌群失调。
性别&肠道菌群:从青春期差异到绝经后趋同
肠道微生物组的性别二态性指男性和女性在微生物组成和多样性方面的差异。研究发现:
值得注意的是,绝经后女性的肠道微生物群与男性的相似度高于绝经前女性。
2022 年 4 月发表在《mSystems》上的一项研究研究了近 1,000 名西班牙裔男性、1,000 名绝经后的西班牙裔女性和近 300 名绝经前西班牙裔女性的肠道微生物。研究发现,绝经后女性的微生物组与男性相比,与男性更相似。
这表明性激素水平可能在调节肠道微生物组成方面发挥重要作用。
然而,普氏菌属对人类健康的影响存在矛盾,其效果因涉及的特定菌株而异。有些菌株可能有益,而其他菌株可能与某些健康问题相关,这取决于具体条件和宿主因素。
肠道微生物群功能与更年期健康关系
居住在消化道中的微生物群体执行多种功能:
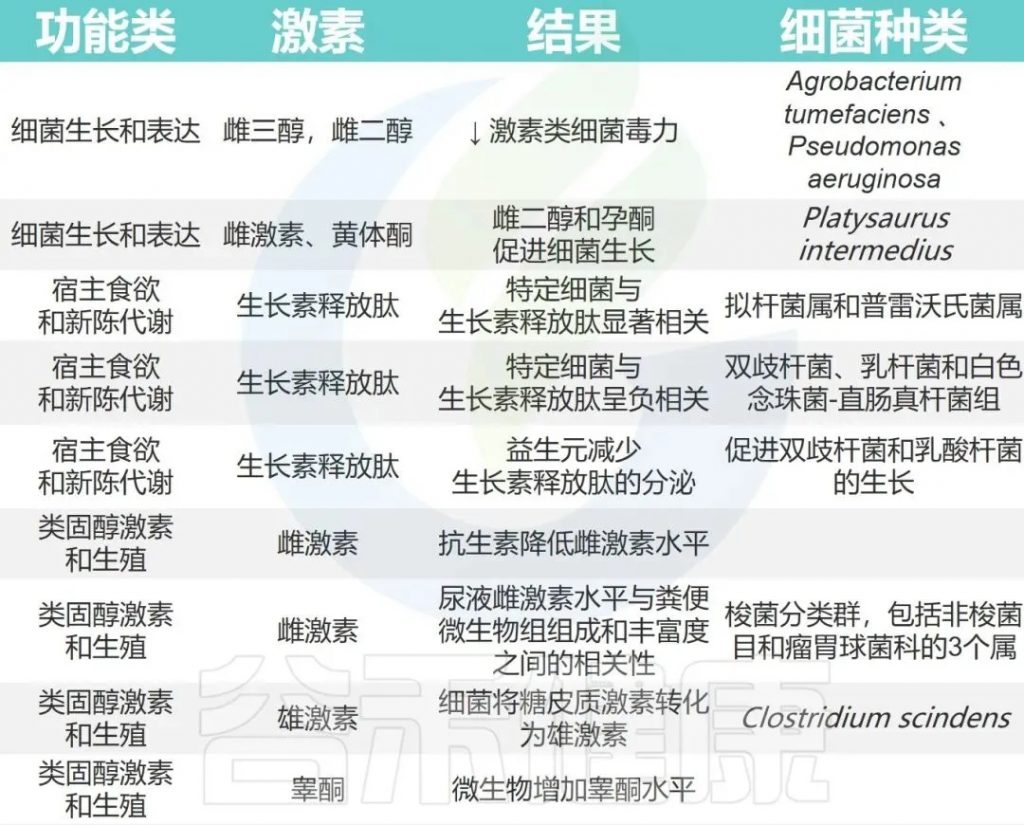
一些激素与肠道菌群的已知关系
Hussain T,et al.Anim Nutr.2021
研究发现,绝经后自身免疫疾病的发病率增加。这可能与肠道微生物组的变化有关。
瘤胃球菌属(Ruminococci),属于梭菌目,是短链脂肪酸的生产者。这些短链脂肪酸具有神经活性特性,能够促进脑-肠轴的通信,这是一种有益功能。在克罗恩病和系统性红斑狼疮患者中,已观察到某些瘤胃球菌种类的丰度降低。
值得强调的是,与绝经前女性相比,绝经后女性体内瘤胃球菌的丰度较低。这一发现表明,更年期激素变化可能通过影响这些有益菌的丰度,对免疫功能和炎症过程产生影响。
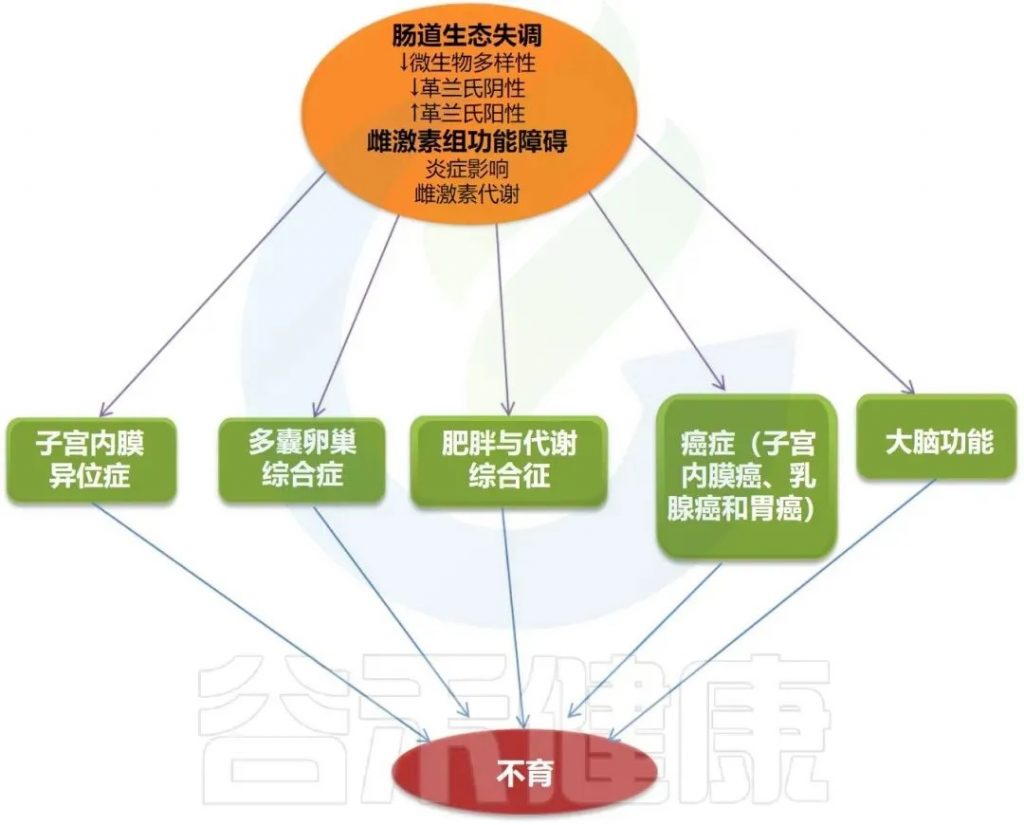
肠道微生物组与雌激素及相关疾病的关系
Hussain T,et al.Anim Nutr.2021
骨健康状态
关于通常影响更年期妇女的骨质疏松症,研究报告了与骨质疏松症严重程度相关的肠道微生物组的显着差异。
那些没有骨质疏松症的人有大量的 Romboutsia、unclassified_Mollicutes 、Weissella。
相比之下,骨质疏松症患者下列菌群丰富:
在骨质疏松症的不同严重程度中,肠道微生物组的变化比阴道微生物组的变化更明显,这表明女性的肠道微生物组可能会影响骨骼代谢。
更年期心血管健康
由于心血管疾病是老年女性的主要疾病之一,近期研究评估了肠道微生物组在激素相关心血管保护中的作用。
一项大型绝经后女性队列研究收集了粪便样本和15种性激素的血清水平,发现雌激素与以下微生物的多样性和丰度增加相关:
有趣的是,研究者提出肠道微生物组可能影响雌酮与颈动脉斑块之间的联系。然而,该研究中81%的女性患有艾滋病毒(HIV),这可能干扰性激素与微生物组之间的直接相关性。
另一方面,孕酮降低免疫系统活性,导致对病原体的易感性增加。绝经后,孕酮水平与雌激素同时下降。研究发现,绝经后女性的血浆孕酮浓度可根据肠道微生物组的组成预测循环孕酮水平。
由于缺乏机制研究,确定这些细菌在肠道微生物组中的具体作用及其与更年期的关系具有挑战性。因此,肠道微生物组在女性衰老中的具体健康影响尚未被充分理解。
更年期阴道微生物组变化及健康影响
阴道微生物组在更年期经历深刻变化,影响女性健康和对各种感染的易感性。
具体而言,在阴道水平,微生物失衡可触发慢性炎症,这可能导致某些感染性疾病风险升高,包括:
持续的炎症状态和相关感染还可能增加恶性转化的可能性,从而提高致癌风险。
这强调了更年期激素变化对阴道健康和相关风险的潜在影响。激素水平的下降改变了阴道环境,影响了保护性微生物的生存条件,可能导致更易感染和炎症反应增强,形成一个可能危及女性晚年健康的复杂互动网络。
更年期生殖微生物组变化与子宫内膜癌风险
近期研究表明,更年期期间生殖微生物组的变化与子宫内膜癌(EC)风险增加有关。
Walsh等人的研究发现,绝经后女性的微生物组多样性增加,这可能提高疾病风险。具体而言:
这表明卟啉单胞菌属(Porphyromonas)可能在更年期疾病发展中发挥作用。这些发现突显了女性更年期期间生殖道微生物组变化与癌症风险之间的重要联系,为未来预防和治疗策略提供了潜在目标。
微生物组健康与女性生殖道健康
微生物组健康度指特定环境(如肠道或口腔)中微生物群落的健康状态和恢复力。健康的微生物组具有多样性,由支持宿主健康的有益微生物组成,而失衡的微生物组可能导致疾病。
在健康的阴道微生物组中,乳杆菌属(Lactobacillus)通常是占主导地位的细菌,乳杆菌属可以:
乳杆菌的这种优势对维持阴道健康和预防感染至关重要。
它们形成一种保护性屏障,通过多种机制(包括pH调节、抗菌物质产生和竞争性排斥)抵抗病原体。
在更年期过渡期间,激素水平的变化可能会影响这种微生物平衡,导致乳杆菌减少和有害菌增加,从而增加感染和其他健康问题的风险。
更年期悖论:阴道微生物组变化的双面性
更年期悖论描述了绝经前和绝经后女性阴道微生物组之间观察到的相互矛盾趋势。这一现象的特征是:
因此,绝经后女性的阴道微生物组可能变得更加多样化,适应生态位内更广泛的微生物物种谱。这似乎违反了通常认为的微生物多样性对健康有益的观点。
导致更年期女性优势度和丰富度之间这种悖论关系的因素包括:
从临床角度看,这种悖论可能导致健康问题,如由于微生物适应性变化引起的感染,强调了在更年期女性医疗保健中解决这些微生物组变化的重要性。
这表明阴道微生物组的最佳状态在生命不同阶段可能有所不同,而不仅仅是追求最大的多样性。
更年期荷尔蒙变化对阴道微生物生态的影响
首先,与更年期相关的荷尔蒙变化会改变阴道环境,从而影响微生物群落。绝经期间雌激素水平的下降导致阴道pH值和湿度水平发生变化,创造了一个不太有利于某些微生物物种生长的环境,特别是在绝经前女性中占主导地位的乳杆菌。
阴道微生物组被分为五种群落状态类型(CST),其分类取决于乳杆菌物种的存在和丰度:
编辑
随着绝经的发生,许多女性从乳杆菌占优势的状态转变为CST IV状态,这种变化可能增加对某些感染和健康问题的易感性。这种转变反映了荷尔蒙调节对维持健康阴道微生物组的重要性。
绝经后阴道微生物组变化及健康影响
绝经后女性经历乳杆菌水平降低,导致微生物多样性增加。然而,这可能增加厌氧菌定植的易感性,与多种感染相关。
当细菌多样性增加时,检测到的物种包括:
其中一些与特定阴道感染相关,如细菌性阴道病(BV)。临床研究表明,BV的特征性厌氧菌过度生长与绝经后女性的微生物组相关,与绝经前女性相比有明显差异。
此外,研究已发现非乳杆菌占优势的阴道菌群与阴道干燥之间存在相关性。阴道干燥、性交疼痛和阴道疼痛症状更严重的女性往往具有更大的阴道微生物多样性,且不以乳杆菌为主。
普雷沃氏菌菌和卟啉单胞菌是女性生殖道和口腔的组成部分,它们参与细菌性阴道病和口腔牙周炎等多微生物感染。这表明微生物组变化可能在多个身体系统中产生广泛影响。
绝经前后免疫功能与阴道健康差异
绝经前后女性在免疫功能和阴道健康方面的差异也可能导致微生物多样性与丰富度的悖论。免疫反应和阴道上皮完整性的变化能够影响微生物定植模式和群落结构。
乳杆菌的保护作用
乳杆菌通过乳酸发酵保护女性免受侵入性病原体的侵害,促进阴道和膀胱健康。绝经前后最显著的差异是乳杆菌水平的降低。乳杆菌的主要代谢途径是乳酸和糖原。乳杆菌通过乳酸(一种主要的抗菌剂)消灭失调的微生物和各种病原体,从而维持微生物平衡。
乳酸水平变化
绝经前:乳酸含量占总量的98%
绝经后:乳酸浓度显著降至94.2%,阴道液pH值升高
雌激素与糖原关系
较高水平的雌激素促进阴道上皮中糖原的积累,有利于乳杆菌的优势地位。增加的游离糖原水平促进更厚的复层鳞状上皮和保护性粘液层形成,这也与较高的乳杆菌水平相关。
绝经前:阴道粘膜中游离糖原水平显著高于绝经后
绝经后:雌激素水平急剧下降,阴道微生物组和上皮受到影响
绝经后女性乳杆菌水平较低,可能是由于雌激素依赖性糖原可及性降低。此外,在患有阴道萎缩的女性中,细菌微生物组缺失。
这些变化说明了激素、上皮环境和微生物组之间复杂的相互作用,解释了为什么绝经可能导致微生物多样性增加但并不总是有益健康。
阴道与泌尿生殖系统微生物组的相互关系
泌尿生殖系统与阴道密切相关。阴道微生物组与泌尿系统和胃肠道系统中的其他微生物群落相互作用。阴道乳杆菌可能对泌尿道起保护作用。此外,泌尿道可能作为阴道乳杆菌的储存库,并可能帮助在因更年期相关代谢变化或病理引起的菌群失调后重新定植。
在乳杆菌物种中,詹森乳杆菌(L. jensenii)也常见于尿道,与惰性乳杆菌(L. iners)和卷曲乳杆菌(L. crispatus)一起,是阴道中最常分离出的菌种。阴道中乳杆菌丰度与其在尿道中存在显著的相关性。因此,促进乳杆菌在阴道中的定植可以积极影响其在泌尿系统中的存在,从而在女性健康中发挥重要作用,尤其是在绝经后。
这一发现具有重要的临床意义,表明针对阴道微生物组的干预可能会对整个泌尿生殖系统健康产生连锁效应。绝经后这种微生物生态系统的变化可能解释了为什么许多女性在更年期后容易出现泌尿道感染和其他泌尿系统问题,强调了在更年期医疗保健中考虑整个泌尿生殖轴的重要性。
更年期泌尿生殖系统并发症影响50岁或以上女性的三分之一
泌尿系统也可能因粘膜干燥而受到影响。可能出现的泌尿生殖系统症状包括性交疼痛、排尿困难和复发性尿路感染(UTI)。存在一种称为更年期泌尿生殖综合征(GSM)的病理状态,影响约50%的更年期女性,同样影响女性的性健康和功能健康。更年期引起阴道微生物组的变化,导致阴道症状。
2021年的一项研究确定普雷沃氏菌和卟啉单胞菌(经典的牙周病原体)是与接受抗生素治疗的尿路感染相关的微生物。
2013年的一项横断面研究(n=87)表明,轻度或中度外阴阴道萎缩表现出更大的微生物组多样性,缺乏乳杆菌,而没有外阴阴道萎缩的女性则表现出以卷曲乳杆菌为主的微生物组。因此,阴道微生物组动态的复杂性需要多方面的方法来阐明潜在机制。
未来研究应采用纵向研究和先进的组学技术,揭示宿主生理、微生物组成和环境因素在更年期阴道微生物组塑造中的复杂相互作用。通过更多地了解这些动态,可以开发有针对性的干预措施,促进阴道健康并减轻更年期女性感染的风险。
更年期常见的肠道健康问题
随着更年期荷尔蒙的变化,消化通常会变得更加不可预测。许多女性会出现腹胀、便秘、胃酸倒流和食物敏感,这些在过去从来都不是问题。这些问题与消化缓慢、胃酸降低和肠道细菌的变化密切相关。了解这些症状发生的原因以及如何管理它们可以帮助更好地控制自己的肠道健康。
腹胀和胀气
随着消化速度减慢,食物在肠道中停留的时间会更长,从而导致发酵和气体积聚。激素波动会进一步影响食物在消化系统中移动的效率,从而引起不适。此外,肠道细菌的变化会破坏正常的消化,使腹胀更加频繁和明显。
通过肠道菌群检测可识别甲烷菌、硫化氢代谢菌等特定菌群的丰度,从而制定针对性策略。若检测j结果显示产甲烷菌丰度高,需减少豆类、十字花科蔬菜摄入;若硫化氢菌为主,则限制红肉和含硫食物。
如何干预?
便秘和消化迟缓
当雌激素水平下降时,消化系统会减慢,导致食物在肠道中停留的时间更长。随着胃酸和酶的产生降低,分解食物变得更加困难,导致消化缓慢。此外,肠道肌肉收缩减弱会使排便不那么规律,从而导致不适。
肠道菌群检测报告中,一些产丁酸菌可能辅助判断菌群代谢能力,若丁酸菌不足,补充菊粉或抗性淀粉(如青香蕉);若普氏菌属丰度高,也可以改用其他例如低聚半乳糖。
如何干预?
食物敏感和炎症增加
肠道通透性增加引起的肠漏,使未消化的食物颗粒进入血液,引发免疫反应。肠道细菌的变化会改变身体处理某些食物的方式,使其更容易产生敏感性。较高的炎症水平也会使身体对乳制品、麸质和加工食品更敏感,导致消化不适和不耐受。
如何干预?
支持更年期肠道健康的食物
吃合适的食物可以在平衡肠道菌群、减少腹胀、改善消化和支持更年期的整体健康方面产生巨大影响。由于激素变化会影响肠道功能,因此专注于富含纤维、益生菌、抗炎和营养丰富的食物有助于维持消化平衡。
减少肠道炎症的健康脂肪
Omega-3 脂肪酸和健康脂肪支持肠道内壁的完整性并减少消化道的炎症。
《营养生物化学杂志》发表的一项研究发现,Omega-3 脂肪酸补充可以改善绝经后妇女的肠道微生物群健康并减少炎症。
建议每天摄入约250-500毫克Omega-3脂肪酸。
健康脂肪的最佳来源:
如果经历持续的肠道炎症,健康的脂肪会有所帮助,但它们本身并不总是足够的。有针对性的抗炎营养计划可以进一步减轻更年期与肠道相关的症状。
舒缓肠道的抗炎食物
慢性炎症会削弱消化并导致食物敏感。抗炎食物有助于修复肠道内壁并减少腹胀。
最好的抗炎食物:
如果炎症导致持续腹胀或肠道不适,可以考虑进行功能医学肠道健康咨询,确定食物触发因素和解决方案。
《营养学杂志》发表的一项研究发现,大豆补充可以改善绝经后妇女的雌激素水平。
骨汤和胶原蛋白用于肠道内壁修复
更年期会削弱肠道内壁,增加肠漏和炎症的风险。骨汤和胶原蛋白含有谷氨酰胺等氨基酸,有助于增强肠壁。
最佳来源:
如果怀疑肠漏导致了食物敏感性,可以考虑优先解决肠漏方案来帮助治愈和恢复消化。
多补充水分
保持水分可以保持消化顺畅并防止便秘。除了喝水,吃补水食物也会有所帮助。
最佳保湿食物:
益生菌
益生菌是发酵食品中的活性微生物,存在于酸奶、酸菜和酸面包,也可以在膳食补充剂中找到。摄入益生菌可以改善消化、增强免疫系统并调节雌激素水平。
改善肠道健康
它们被认为通过恢复有益菌和抑制肠道有害菌的生长来改善肠道健康。肠道微生物平衡的转变可以导致改善雌激素代谢,有助于维持健康的雌激素水平。
一项研究发现,连续六周每天食用益生菌补充剂的女性肠道健康显著改善。
改善雌激素水平
研究表明,益生菌对雌激素水平有积极影响,通过增加β-葡萄糖醛酸酶的产生,这是一种促进体内雌激素排泄的酶。
根据《临床胃肠病学杂志》发表的一项研究,益生菌补充可以改善代谢综合征的绝经后妇女的雌激素水平。
益生元
益生元是人体无法消化但肠道微生物组可以消耗的纤维素类型。
益生元可以在许多食物中找到,最佳益生元来源:
改善肠道健康、调节雌激素水平
摄入益生元促进肠道中有益菌的生长,减少有害菌的数量,有助于改善肠道健康并调节雌激素水平。
研究表明,益生元对雌激素水平有积极影响,通过增加短链脂肪酸的产生,这可以帮助调节雌激素水平。
《营养学杂志》发表的一项研究发现,益生元补充可以改善绝经后妇女的雌激素水平。
此外,益生菌和益生元还可能对与激素失衡有关的疾病,如多囊卵巢综合症产生积极影响。
《功能性食品杂志》发表的一项研究发现,连续12周食用益生菌补充剂的多囊卵巢综合症女性,睾酮水平下降,胰岛素敏感性改善,这两个指标与多囊卵巢综合症密切相关。
总之,益生菌和益生元对肠道健康和激素平衡,特别是在女性中有积极影响。将这些补充剂纳入饮食中可能对与激素失衡有关的疾病有益,如多囊卵巢综合症。
然而,需要进行更多的研究来全面了解它们的影响机制,也可以借助肠道菌群检测了解自身需求,确定最佳的剂量和补充时间。
维生素D
这种维生素在维持肠道健康和免疫功能方面起着至关重要的作用。一些富含维生素D的食物包括富含脂肪的鱼类、蛋黄和强化的乳制品。
《妇女健康杂志》发表的一项研究发现,维生素D补充可以改善绝经后妇女的肠道微生物群组成并减少炎症。
缓解压力
压力是我们日常生活中常见的现象,它可以对肠道微生物组产生深远的影响。
压力改变肠道菌群组成
几项研究表明,慢性压力可以改变肠道细菌的组成,导致有益菌的减少和有害菌的增加。
例如,发表在《大脑、行为和免疫》杂志上的一项研究发现,压力诱导的肠道微生物组变化与炎症性肠病的风险增加有关。
压力影响激素的产生和代谢
此外,压力还可以影响激素的产生和代谢,包括雌激素。下丘脑-垂体-肾上腺(HPA)轴是压力反应系统的关键组成部分,已知与下丘脑-垂体-性腺(HPG)轴相互作用,后者调节雌激素的产生。
研究表明,慢性压力可以破坏这些轴的调节,导致雌激素水平的改变。
因此,减少压力水平可以是改善肠道健康和恢复雌激素水平的有效策略。一些减压的方式包括冥想、深呼吸练习、瑜伽、定期运动。
一项研究发现,定期运动可以改善绝经后妇女的雌激素水平,运动还可以降低患乳腺癌等雌激素相关疾病的风险。
– 每天练习10-15分钟的正念冥想或深呼吸练习
– 每天进行至少30分钟的有氧运动,如快走、慢跑或骑车
– 将力量训练融入每周的运动计划中
– 减少暴露于压力因素,如过度的工作负担或负面关系
– 参与有趣的活动,如兴趣爱好或与亲人共度时光,以减轻压力水平
然而,肠道健康并不是一刀切的。每个女性的身体对激素变化的反应都不同,对一个人有效的方法可能对另一个人不起作用。这种个体差异与肠道菌群的独特组成密切相关。例如,部分女性因产甲烷菌丰度过高而对豆类敏感,而另一些人可能因丁酸菌不足而需针对性补充抗性淀粉。
通过肠道菌群健康检测可通过解析菌群结构,明确优势菌群(如普氏菌、双歧杆菌)及致病菌定植状态,指导膳食调整方向。了解功能基因及代谢物水平(如短链脂肪酸、硫化氢等),判断炎症风险与营养吸收能力,从而制定个性化干预方案;通过定期复测评估益生菌定植率或益生元/饮食干预后的菌群重塑效果,避免盲目补充。
更年期作为女性生命周期的关键转折点,其健康管理已从传统的症状缓解转向基于微生物组学的精准干预。肠道菌群与雌激素代谢的双向调控机制、菌群失衡与更年期综合征的关联性研究,为健康老龄化提供了全新的科学视角。
然而,现有研究过度依赖西方人群样本,地理与生活方式差异(如饮食结构、环境暴露)导致微生物组数据缺乏多样性,限制结论的普适性。例如,非洲或亚洲女性因饮食差异可能具有独特的菌群特征。
解决这一差距至关重要。包括来自非洲、亚洲和拉丁美洲等更多样化人群的研究,可以提供对更年期如何全球性影响微生物组的更全面理解。更具地理包容性的方法将有助于揭示微生物群落的变异,从而开发更个性化的干预措施,更好地满足不同群体更年期女性的需求。
随着研究的深入,未来将围绕以下方向展开:
检测技术的革新与普及
精准医疗的核心在于“检测先行”。宏基因组测序、代谢组学及AI驱动的数据分析技术将推动肠道菌群检测的标准化。
唾液、阴道分泌物等多部位微生物组联合检测技术的开发,将揭示“口腔-肠道-生殖道”微生物轴的联动机制,为系统性干预提供依据。
个性化益生菌/益生元产品的迭代
基于菌群检测结果的靶向菌株筛选与定制化产品研发将成为主流。例如,针对绝经后女性双歧杆菌丰度下降、产甲烷菌过度增殖等问题,开发含特定菌株的益生菌组合,或设计差异化的益生元配方。
同时,微生物代谢工程技术的应用,可人工合成具有抗炎、调节雌激素功能的代谢产物,为药物开发开辟新路径。
数据驱动的健康管理平台
整合肠道菌群数据、激素水平、临床表型及生活方式的多组学数据库将构建更年期健康管理的智能中枢。AI算法可通过分析大规模队列数据(如中国更年期女性队列),预测骨质疏松、心血管疾病等风险,并推荐个性化干预方案,同时联动医疗机构进行动态跟踪。
跨学科研究与产业协同
微生物组学与内分泌学、免疫学、营养学的交叉融合,将催生“微生物-激素”共调控疗法。例如,通过靶向调控肠道菌群的β-葡萄糖醛酸酶活性,促进雌激素再循环以缓解阴道萎缩;或利用益生菌代谢产物调节骨吸收相关通路,降低骨质疏松风险。此外,中医药与微生物组研究的结合有望挖掘传统疗法的现代科学价值。
未来,随着技术的突破与产业链协同,女性将不再被动应对更年期挑战,而是通过“检测-干预-监测”的全周期管理,实现从激素平衡到全身健康的精准调控。通过整合肠道菌群检测、激素水平分析与临床表型数据,可构建预测模型,为女性提供从症状缓解到疾病预防的全周期方案。
注:本账号内容仅作交流参考,不作为诊断及医疗依据。
主要参考文献
Nieto M R, Rus M J, Areal-Quecuty V, et al. Menopausal shift on women’s health and microbial niches[J]. npj Women’s Health, 2025, 3(1): 3.
Tatullo M, Nor J, Orrù G, Piattelli A, Cascardi E, Spagnuolo G. Oral-Gut-Estrobolome Axis May Exert a Selective Impact on Oral Cancer. J Dent Res. 2024 May;103(5):461-466.
Park MG, Cho S, Oh MM. Menopausal Changes in the Microbiome-A Review Focused on the Genitourinary Microbiome. Diagnostics (Basel). 2023 Mar 21;13(6):1193.
Shen X, Wang C, Zhou X, Zhou W, Hornburg D, Wu S, Snyder MP. Nonlinear dynamics of multi-omics profiles during human aging. Nat Aging. 2024 Nov;4(11):1619-1634.
Hu S, Ding Q, Zhang W, Kang M, Ma J, Zhao L. Gut microbial beta-glucuronidase: a vital regulator in female estrogen metabolism. Gut Microbes. 2023 Jan-Dec;15(1):2236749.
Peters BA, Santoro N, Kaplan RC, Qi Q. Spotlight on the Gut Microbiome in Menopause: Current Insights. Int J Womens Health. 2022 Aug 10;14:1059-1072.
Łaniewski P, Herbst-Kralovetz MM. Connecting microbiome and menopause for healthy ageing. Nat Microbiol. 2022 Mar;7(3):354-358.

谷禾健康

双歧杆菌属是人类肠道微生物群的关键成员,已包括婴儿双歧杆菌、两歧双歧杆菌、长双歧杆菌、青春双歧杆菌、短双歧杆菌和假小链双歧杆菌等几十个物种。
两歧双歧杆菌(Bifidobacterium bifidum)是其中一个具有许多促进健康益处的双歧杆菌,革兰氏阳性菌、厌氧、不具备运动性,且不形成芽孢。能够通过代谢乳糖、低聚糖等碳水化合物生成乙酸和乳酸。
这种益生菌是最早定植于婴儿肠道的菌株之一,在新生儿和婴幼儿体内丰度高,但随年龄增长逐渐减少。B.bifidum作为益生菌被用于改善消化问题,研究发现其具有改善腹痛、腹泻、便秘、胃酸相关消化不良等益处。
除此之外,还有一些研究发现其在治疗或改善其他疾病方面的作用。如:减轻炎症性肠病症状、改善肠易激综合征、减轻幽门螺杆菌感染症状、改善血脂状况,减少不健康胆固醇、减轻了乙酰对氨基苯酚引起的急性肝损伤、还有助于抗氧化、预防和改善婴儿湿疹等。
两歧双歧杆菌(Bifidobacterium bifidum)作为一种肠道有益菌。它是如何工作的,它还有哪些其他好处?本文将带您了解更多。
两歧双歧杆菌(Bifidobacterium bifidum)是人类肠道内的一种重要共生细菌,尤其在新生儿和婴幼儿肠道中占据重要地位,成年后丰度有所降低。它是典型的益生菌,广泛应用于食品、药品和肠道调节剂中。
细胞形态:B.bifidum是一种革兰氏阳性厌氧菌,形态呈分叉状或杆状,常见为Y形或V形,无鞭毛,不具备运动性,且不形成芽孢。
两歧双歧杆菌BGN4粘附在上皮Caco-2细胞上

doi: 10.3390/ijms17091544.
(a)光学(放大倍数1000×);(b)扫描电子显微镜(放大倍数为20000×)
厌氧性:B.bifidum是严格厌氧菌,不需要氧气,氧气可能抑制其生长。
大小:细胞长度约为1.0–3.0µm。
B.bifidum是母乳喂养婴儿中的常见菌种,通过代谢碳水化合物生成短链脂肪酸(如乙酸和乳酸),可以抑制有害菌增殖,维持肠道微生态平衡,具有减少肠易激综合征、腹泻和病原体感染等健康益处。
Bifidobacterium bifidum主要分布于人类肠道,特别是婴幼儿肠道,也存在于部分温血动物(哺乳类)的消化道中。
此外,它还存在于女性泌尿生殖道、母乳和一些发酵乳制品中。
该菌最适生长温度为36℃-38℃,pH适宜范围为5.0-8.0,仅在严格厌氧环境下高效增殖。pH值低于4.5-5.0或高于8.0-8.5时不会发生生长。
发酵型代谢:特有的双歧杆菌途径(异葡糖酸旁路)代替糖酵解途径,代谢六碳糖(如乳糖、寡糖等)生产2分子乳酸和3分子乙酸作为主要代谢产物。
营养需求:营养要求较高,主要利用乳糖、低聚糖(如人乳寡糖)和葡萄糖作为能量来源。
饮食来源的糖类利用:B.bifidum也能代谢饮食来源的碳水化合物,如淀粉类食物,这得益于其多样的碳水化合物活性酶系统。
宿主来源的糖类利用:B.bifidum还能够利用宿主来源的糖类,如婴儿期的母乳低聚糖和成年期的粘蛋白中的糖类。这种能力是通过其基因组中丰富的糖代谢相关基因实现的,这些基因包括糖苷水解酶、糖基转移酶和碳水化合物结合模块等。
注:一项研究比较了牛奶配方奶粉或母乳喂养的婴儿的粪便微生物群组成,结果显示,母乳喂养婴儿粪便中的两歧双歧杆菌丰度高于配方奶粉喂养的婴儿,这可能得益于母乳低聚糖可以被其利用。而在成人肠道中的存在可能得益于聚糖底物(如粘蛋白)的支持。
重要代谢产物:乙酸、乳酸、维生素(如硫胺素(维生素B1)、核黄素(维生素B2)、维生素B6和维生素K、叶酸、烟酸(维生素B3)和吡哆醇(维生素B6))。
其他代谢物:如γ-氨基丁酸(GABA)和生物素。
一些研究表明,两歧双歧杆菌(B.bifidum)能够通过多种途径与宿主进行相互作用,这些相互作用机制为B.bifidum对宿主的有益作用提供了重要基础。
两歧双歧杆菌在人体肠道中发挥的主要特性

doi: 10.3390/microorganisms7110544.
• 调节免疫蛋白和炎症因子的产生

-通过增加IgA、IgM和IgG,同时降低IgE。B.bifidum可能有助于增强免疫系统和减少过敏。
-B.bifidum还可以通过降低促炎细胞因子IL-4和IL-5的产生,平衡IFN-γ的水平,增加抗炎细胞因子IL-10和特定免疫细胞类型(CD25 和 Foxp3)的产生来帮助治疗过敏。
-在衰老小鼠模型中,B.bifidum通过显著增加细胞因子IL-2和IFN-γ同时降低IL-6和TNF-α水平来刺激免疫系统。
• 影响免疫细胞活性和免疫反应

-在患有炎症性肠病(IBD)的小鼠中,B.bifidum通过抑制Th1反应和减少促炎细胞因子(IL-1β、IL-6和角质形成细胞衍生的趋化因子)、趋化因子(MCP-1)和酶(COX-2、MPO)来减少炎症。它还可以通过增加Toll样受体2(TLR2)和前列腺素E2(PGE2)的产生来减少细胞死亡。
-在幽门螺杆菌感染的小鼠中,B.bifidum抑制促炎性IL-8和NF-κB信号通路。
B.bifidum还增加了外周CD8+细胞的细胞毒活性,而不会降低淋巴细胞存活率。一些菌株可能会刺激Th17的产生。
B.bifidum是一种具有重要作用的细菌,它能够有效抑制一些致病性有害细菌的生长和繁殖,从而维护肠道微生态的平衡。此外,已有研究表明,B.bifidum在下列疾病中发现了潜在健康益处,其表现出有助于改善疾病以恢复宿主健康。
1)改善腹痛、腹泻等胃肠道不适

在一项针对37人的临床试验中,持续食用含有两歧双歧杆菌YIT 10347 的发酵乳可改善功能性胃肠道疾病患者的胃肠道和心理症状。如腹痛、腹泻、便秘、胃酸相关消化不良、愤怒和敌意。
在另一项对300多名未服用药物的人进行的临床试验中,摄入两歧双歧杆菌可显著降低胃和下腹部症状的患病率。
含两歧双歧杆菌(B.bifidum)、动物双歧杆菌(B.animalis)、嗜酸乳杆菌(Lactobacillus acidophilus)的益生菌配方,在24名马拉松运动员的临床试验中降低了胃肠道症状的频率。
还发现B.bifidum通过增加大鼠粘蛋白的产生来减轻急性胃损伤。
2)调整肠道菌群

在一项针对27名健康志愿者的临床试验中,摄入两歧双歧杆菌(B.bifidum)减少了Prevotellaceae和 Prevotella,增加了瘤胃球菌科和Rikenellaceae。
在另一项针对18名老年人的试验中,与两歧双歧杆菌和乳双歧杆菌共生可增加有益双歧杆菌和乳酸杆菌的粪便计数。
在一项针对53名慢性肝病患者的临床试验表明,两歧双歧杆菌是可有效防止小肠细菌过度生长的三种益生菌之一。此外,在66名酒精性肝损伤患者的试验中,它与植物乳杆菌联合改善了肠道菌群。
在另一项针对30例患者的试验中,两歧双歧杆菌(联合嗜酸乳杆菌)在抗生素治疗后也恢复了肠道菌群。
摄入两歧双歧杆菌可以通过增加益生菌(乳酸杆菌)的数量同时减少不需要的细菌种群(肠杆菌、大肠杆菌)来改善肠道的微生物群。
3)减轻炎症性肠病症状

一项试验显示,抗炎药和益生菌包括两歧双歧杆菌和其他菌株的组合改善了溃疡性结肠炎,试验对象为60名服用2年的患者。
在一项针对15名老年人的小型试验中,一种含有两歧双歧杆菌和嗜酸乳杆菌的益生菌减少了肠道内壁的炎症。

坏死性小肠结肠炎是一种危及早产儿生命的疾病,由细菌侵入肠道引发严重炎症并损害肠道壁。一项针对400多名婴儿的临床试验显示,含两歧双歧杆菌(B.bifidum)和嗜酸乳杆菌的益生菌有助于预防该病。
在小鼠研究中发现,两歧双歧杆菌减轻了小鼠炎症性肠病(IBD)的症状,例如肠壁增厚、炎性细胞浸润和高水平的炎性细胞因子。
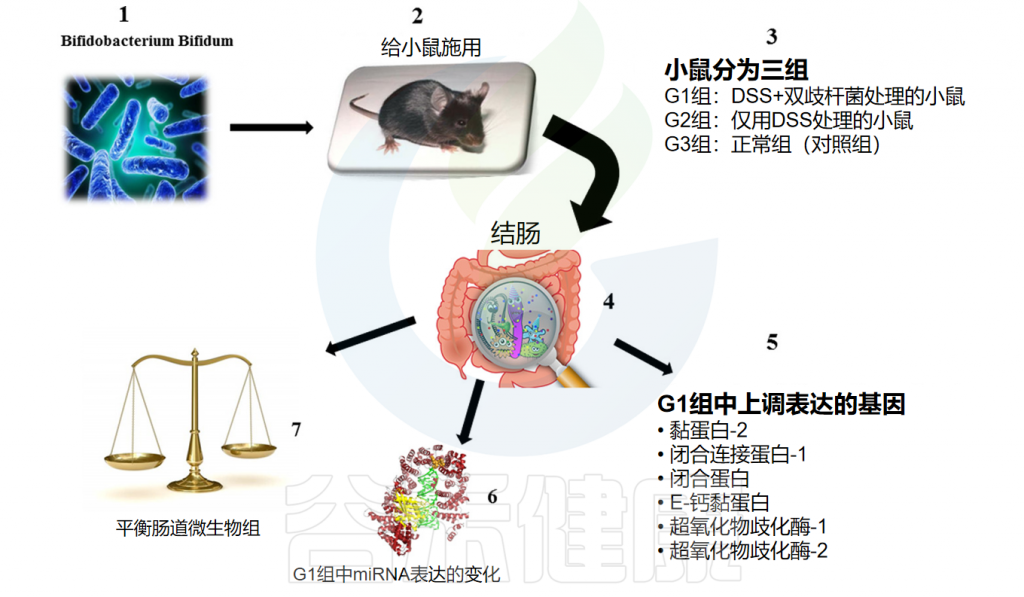
doi: 10.1016/j.jnutbio.2020.108353.
4)改善肠易激综合征(IBS)

在一项针对122名肠易激综合征(IBS)患者的临床试验中,两歧双歧杆菌有效缓解了IBS症状(疼痛/不适、腹胀/腹胀、尿急和消化系统疾病)并改善了生活质量。
注:大约47%服用两歧双歧杆菌的受试者报告症状明显缓解,而服用安慰剂的受试者中只有11%出现症状缓解。
但在另一项针对52名IBS患者的试验中,一种含有乳双歧杆菌和嗜酸乳杆菌的多菌种益生菌同样有效。
5)预防或减轻急性腹泻

在一项针对4400多名婴儿的临床试验中,添加到婴儿配方奶粉中的两歧双歧杆菌和嗜热链球菌益生菌有助于预防急性腹泻。
同样,在一项针对63名患有癌症的女性的临床试验中,两歧双歧杆菌和嗜酸乳杆菌的联合用药预防了放疗引起的腹泻。

在另一项针对67名已经患有这种疾病的婴儿的试验中,两歧双歧杆菌和嗜酸乳杆菌的组合缩短了腹泻的持续时间,并减少了排便次数。
另外两种混合物(两歧双歧杆菌与德氏乳杆菌保加利亚亚种、嗜酸乳杆菌和嗜热链球菌的混合物,以及两歧双歧杆菌与长双歧杆菌、嗜嗜酸乳杆菌、鼠李糖乳杆菌和屎肠球菌的混合物)在2项针对近700名急性腹泻儿童的试验中也有效。
6)减轻幽门螺杆菌感染症状

在一项针对79名幽门螺杆菌感染者的临床试验中,两歧双歧杆菌(B.bifidum)改善了上消化道和总症状的发生率。
在另一项针对89名儿童的试验中,两歧双歧杆菌和嗜酸乳杆菌增强了幽门螺杆菌标准三联疗法(克拉霉素、阿莫西林和质子泵抑制剂)的有效性。
在小鼠模型中发现,两歧双歧杆菌(B.bifidum)减轻了幽门螺杆菌对小鼠胃组织的损伤。
总而言之,上述这些证据表明两歧双歧杆菌(B.bifidum)可能有助于解决消化问题,如肠道生态失调、炎症性肠病、肠易激综合征和幽门螺杆菌感染。
但需要与医生讨论,是否可以将其作为您当前治疗方案的补充。重要的是,严格按照医生的建议服用这种益生菌,永远不要用它来代替批准的疗法。
两歧双歧杆菌(B.bifidum)不仅在改善多种胃肠道疾病方面表现出显著效果,还被发现对一些肠外疾病的治疗具有辅助作用。
7)控制血糖、减轻糖尿病危害

在4项针对200多名2型糖尿病患者的临床试验中,两歧双歧杆菌与其他细菌菌株联合使用,降低了空腹血糖、胰岛素水平,改善了血糖控制和抗氧化状态。
益生菌与两歧双歧杆菌(B.bifidum)的混合物在4项针对250名妊娠期糖尿病女性的临床试验中同样有效。
两歧双歧杆菌降低了糖尿病大鼠和健康小鼠的空腹血糖、糖化血红蛋白(HbA1c)、总胆固醇、甘油三酯、低密度脂蛋白(LDL)和极低密度脂蛋白(VLDL),增加了高密度脂蛋白(HDL)和胰岛素。
它还通过减少脂质过氧化和增加谷胱甘肽、超氧化物歧化酶、过氧化氢酶、谷胱甘肽过氧化物酶、谷胱甘肽还原酶和谷胱甘肽-S-转移酶的水平来改善糖尿病大鼠的氧化应激。
证据表明两歧双歧杆菌(B.bifidum)可能有助于控制糖尿病患者的血糖。如果您的医生推荐,您可以将它们用作补充方法。但切勿用两歧双歧杆菌(B.bifidum)代替医生开的抗糖尿病药物。
8)减轻了乙酰对氨基苯酚引起的急性肝损伤

研究发现,两歧双歧杆菌(B.bifidum)可以减少氧化应激并改变肠道菌群,以减轻N-乙酰对氨基苯酚引起的急性肝损伤。B.bifidum给药显著减轻了N-乙酰对氨基苯酚(APAP)暴露诱导的肝酶升高——特别是ALT、AST和ALP。
注:血浆ALT和AST水平可作为指示急性肝损伤和不同程度肝坏死的生化标志物。
我们对文献总结得出两歧双歧杆菌可以通过以下几种方式来减轻急性肝损伤:
(1)B.bifidum减少急性肝损伤(AILI)中活性氧的产生;
(2)B.bifidum激活AILI中的肝脏KEAP1-NRF2通路;
(3)B.bifidum通过降低血浆脂多糖和减少结肠炎症来减轻急性肝损伤中APAP诱导的肝细胞凋亡;
(4)B.bifidum通过调节结肠微生物群来改善急性肝损伤。
B.bifidum抑制急性肝损伤中的氧化应激和细胞凋亡
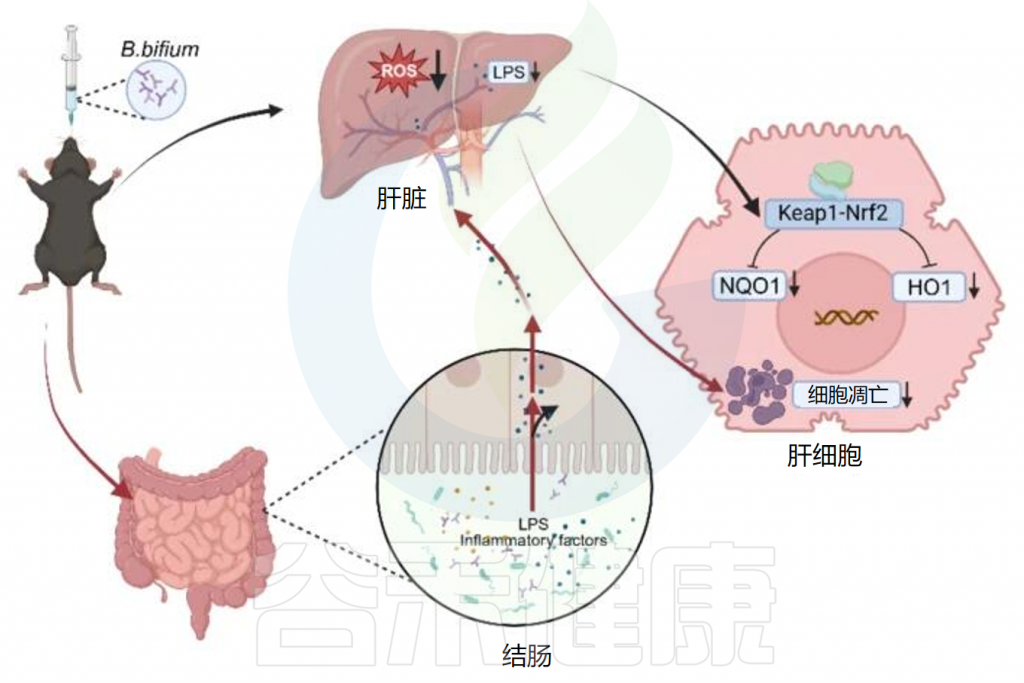
Yin J,et al.BMC Microbiol.2025
9)改善血脂状况,减少不健康胆固醇
在2项针对80名2型糖尿病患者的临床试验中,B.bifidum和乳杆菌益生菌(单独或与干酪乳杆菌联合使用)改善了血脂状况,尤其是“好”胆固醇(HDL)
同样,在两项针对130名妊娠期糖尿病女性的临床试验中,这些组合降低了甘油三酯和“坏”胆固醇(低密度脂蛋白(LDL))。
在一项针对64名患有非酒精性脂肪肝的肥胖儿童的临床试验中,两歧双歧杆菌、乳双歧杆菌、嗜酸乳杆菌和鼠李糖乳杆菌的组合降低了血液甘油三酯、总胆固醇和“坏”胆固醇(LDL)。
在动物模型中也发现了相似的结果,两歧双歧杆菌(B.bifidum)降低了糖尿病大鼠的总胆固醇、“坏”胆固醇(LDL和VLDL)和甘油三酯,同时增加了“好”胆固醇(HDL)。
10)增强免疫力、调节免疫因子水平

在一项针对近600名压力大的学生的临床试验中,两歧双歧杆菌增加了每个参与者的健康天数比例,并降低了干预期间报告感冒/流感的学生百分比。
另一项针对55名老年人的研究表明,两歧双歧杆菌补充能降低促炎细胞因子TNF-α和调节细胞因子IL-10的水平,同时提高抗炎细胞因子TGF-β1的水平。
在一项针对77名感染艾滋病毒(HIV)的儿童临床试验中,一种含有两歧双歧杆菌和嗜热链球菌的益生菌配方增加了CD4+细胞的计数,可能有助于改善免疫系统。
在一项针对32名老年人的临床试验中,一种含有两歧双歧杆菌、长双歧杆菌和加氏乳杆菌的益生菌混合物维持了CD4+淋巴细胞的产生,同时减少了促炎细胞因子。
在一项针对33名训练有素的运动员的临床试验中,一种含有两歧双歧杆菌的益生菌混合物预防了上呼吸道感染。
两歧双歧杆菌(B.bifidum)延迟小鼠轮状病毒相关性腹泻的发作;由两歧双歧杆菌产生的低聚半乳糖可减少鼠伤寒沙门氏菌及其相关病理的定植。
两歧双歧杆菌(B.bifidum)通过增强胸腺和脾脏的抗氧化活性和改善免疫功能来延缓小鼠的免疫衰老。
总而言之,证据表明,两歧双歧杆菌(单独使用和与其他益生菌联合使用)可以增强免疫系统,从而降低传染病的发病率。如果您的医生确定它可能有帮助,您可以服用它来增强您的免疫系统。
11)抗氧化作用
在针对40名重度抑郁症患者、60名健康孕妇、60名糖尿病孕妇、120名糖尿病患者和43名超重妇女的6项临床试验中,将两歧双歧杆菌与其他细菌菌株(如动物双歧杆菌、嗜酸乳杆菌和干酪乳杆菌)结合的益生菌干预措施改善了抗氧化状态(以谷胱甘肽水平升高和MDA水平降低衡量)。
两歧双歧杆菌(B.bifidum)在小鼠中也表现出抗氧化活性。虽然研究有限,但证据表明两歧双歧杆菌益生菌可能会改善抗氧化状态。进一步的研究应该确定如何在治疗上使用它们。
12)预防和改善婴儿湿疹
在一项针对40名婴儿的临床试验中,两歧双歧杆菌(B.bifidum)预防和改善了湿疹。

在对200多名孕妇进行的2项临床试验中,孕妇及其新生儿补充两种益生菌混合物(一种含有两歧双歧杆菌、乳双歧杆菌和嗜酸乳杆菌,另一种含有两歧双歧杆菌、乳双歧杆菌和乳酸乳球菌)可以预防湿疹的发展。
然而,在另一项针对400多名孕妇的试验中,另一种益生菌(含有两歧双歧杆菌、动物双歧杆菌、唾液乳杆菌和副干酪乳杆菌)并没有帮助预防婴儿湿疹,但确实预防了皮肤对常见过敏原的致敏。
尽管证据有限,但表明两歧双歧杆菌可能有助于预防婴儿湿疹。
两歧双歧杆菌(B.bifidum)在其他健康方面也有潜在益处,但证据尚不充分,包括以下几点:
1)减轻压力
在一项针对近600名学业压力大的本科生的临床试验中,两歧双歧杆菌减少了自我报告的压力和与压力相关的腹泻/消化问题。
尽管结果很有希望,但单一的临床试验不能被视为支持双歧杆菌使用的充分证据。需要更多的临床试验。
2)减少上呼吸道感染
2015年进行并发表在《英国营养学杂志》上的一项双盲研究表明,与安慰剂治疗对照组相比,服用两歧双歧杆菌可显著降低上呼吸道感染的风险。
3)改善过敏
在一项针对173名季节性过敏症患者的临床试验中,两歧双歧杆菌、长双歧杆菌和加氏乳杆菌的组合改善了呼吸道和眼部(鼻结膜炎)症状和生活质量。
两歧双歧杆菌还显著降低了小鼠的气道高反应性、肺部炎症和Th2反应;口服两歧双歧杆菌显著抑制了抗原诱导的豚鼠过敏性鼻反应,如打喷嚏和鼻塞。
结果很有希望,但只进行了一项临床试验。需要进一步的临床研究来调查两歧双歧杆菌对过敏的影响。
4)可能有助于狼疮恢复
肠道菌群失调,其特征是厚壁菌门/拟杆菌门比率降低,已在系统性红斑狼疮患者中报道。此外,补充两歧双歧杆菌可防止CD4+ 淋巴细胞过度激活,并可能有助于恢复狼疮中发现的Treg/Th17/Th1失衡。
5)可能对抗癌症
两歧双歧杆菌(B.bifidum)提取物在实验室中抑制结直肠癌细胞的生长。
1)饮食因素
高脂饮食: 长期摄入高脂肪食物会抑制B.bifidum生长;
低纤维饮食: 缺乏膳食纤维(尤其是菊粉、低聚果糖等益生元)导致B.bifidum缺少能量来源;
精制糖和加工食品: 大量摄入会抑制B.bifidum生长;
蛋白质摄入过量: 可增加肠道内蛋白质发酵产物,不利于双歧杆菌生长。
2)药物因素
抗生素使用: 广谱抗生素会非选择性地杀死肠道菌群,包括两歧双歧杆菌;
非甾体抗炎药(NSAIDs): 长期使用可能损害肠黏膜,影响微生物组成;
质子泵抑制剂: 降低胃酸可能改变上消化道微生物组成,间接影响两歧双歧杆菌;
泻药: 频繁使用可冲洗肠道菌群,减少两歧双歧杆菌。
3)生理因素
年龄增长: 随着年龄增长,双歧杆菌自然减少;
长期慢性压力: 压力激素可直接影响肠道菌群组成;
睡眠不足: 破坏昼夜节律,影响肠道微生物多样性;
缺乏运动: 久坐不动会降低肠道菌群多样性;
吸烟: 香烟中的毒素会导致肠道菌群失调
过度饮酒: 酒精对肠道菌群有直接毒性作用。
4)疾病状态
肠道炎症性疾病: 如炎症性肠病(IBD)、肠易激综合征(IBS);
代谢性疾病: 如糖尿病、肥胖症;
自身免疫性疾病: 如类风湿性关节炎、系统性红斑狼疮;
慢性肝病: 肝脏功能障碍会影响肠-肝轴;
胃肠道感染: 病原体感染可能改变菌群平衡。
以上这些因素都可能导致两歧双歧杆菌(B.bifidum)减少。
主要参考文献
Yin J, Chen L, Lin Y, Qiu J, Liu F, Wang Y, Dou X. Bifidobacterium bifidum reduces oxidative stress and alters gut flora to mitigate acute liver injury caused by N-acetyl-p-aminophenol. BMC Microbiol. 2025 Feb 25;25(1):87.
Turroni F, Duranti S, Milani C, Lugli GA, van Sinderen D, Ventura M. Bifidobacterium bifidum: A Key Member of the Early Human Gut Microbiota. Microorganisms. 2019 Nov 9;7(11):544.
Turroni F, Duranti S, Bottacini F, Guglielmetti S, Van Sinderen D, Ventura M. Bifidobacterium bifidum as an example of a specialized human gut commensal. Front Microbiol. 2014 Aug 21;5:437.
Din AU, Hassan A, Zhu Y, Zhang K, Wang Y, Li T, Wang Y, Wang G. Inhibitory effect of Bifidobacterium bifidum ATCC 29521 on colitis and its mechanism. J Nutr Biochem. 2020 May;79:108353.
Turroni F, Bottacini F, Foroni E, Mulder I, Kim JH, Zomer A, Sánchez B, Bidossi A, Ferrarini A, Giubellini V, Delledonne M, Henrissat B, Coutinho P, Oggioni M, Fitzgerald GF, Mills D, Margolles A, Kelly D, van Sinderen D, Ventura M. Genome analysis of Bifidobacterium bifidum PRL2010 reveals metabolic pathways for host-derived glycan foraging. Proc Natl Acad Sci U S A. 2010 Nov 9;107(45):19514-9.
Arboleya S, Watkins C, Stanton C, Ross RP. Gut Bifidobacteria Populations in Human Health and Aging. Front Microbiol. 2016 Aug 19;7:1204.
Devika NT, Raman K. Deciphering the metabolic capabilities of Bifidobacteria using genome-scale metabolic models. Sci Rep. 2019 Dec 3;9(1):18222.
Ku S, Park MS, Ji GE, You HJ. Review on Bifidobacterium bifidum BGN4: Functionality and Nutraceutical Applications as a Probiotic Microorganism. Int J Mol Sci. 2016 Sep 14;17(9):1544.
Abdelhamid AG, El-Dougdoug NK. Comparative genomics of the gut commensal Bifidobacterium bifidum reveals adaptation to carbohydrate utilization. Biochem Biophys Res Commun. 2021 Apr 2;547:155-161.
Hidalgo-Cantabrana C, Delgado S, Ruiz L, Ruas-Madiedo P, Sánchez B, Margolles A. Bifidobacteria and Their Health-Promoting Effects. Microbiol Spectr. 2017 Jun;5(3):10.1128/microbiolspec.bad-0010-2016.
谷禾健康
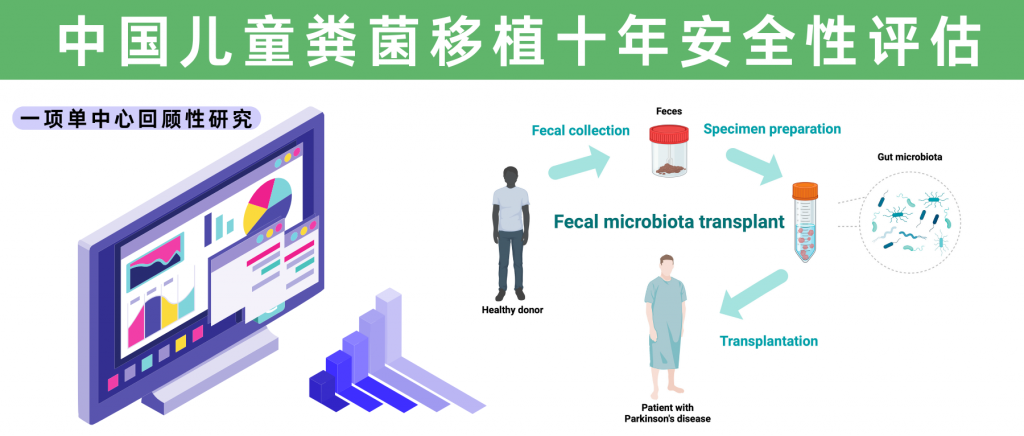
随着现代医学的不断进步,人类对于肠道微生态的研究正逐渐深入,粪菌移植(FMT)作为一种新兴的、生物学意义深远的治疗手段,在临床实践中展现出了革命性潜力。FMT广泛用于治疗艰难梭菌感染等复杂疾病,并逐渐延伸至免疫功能调节、代谢紊乱等领域,尤其是在儿童人群中的应用引发了广泛关注。
然而,在这项技术日益普及的同时,其在特殊人群中的安全性问题始终是临床医生和产业从业者关注的焦点。大多数关于FMT安全性的研究主要集中在成人人群中,而对儿科人群的了解较少,目前所有儿科FMT数据均来自小型病例系列和病例报告。
近日,一项来自上海儿童医院的大规模单中心回顾性研究,揭示了FMT在儿童人群中的长期安全性数据。这项跨越十年(2013-2023年)、涵盖813名儿童患者的研究,不仅是目前样本量最大的儿科FMT安全性评估,更是首次系统性地呈现了中国儿童接受FMT后的长期随访结果。

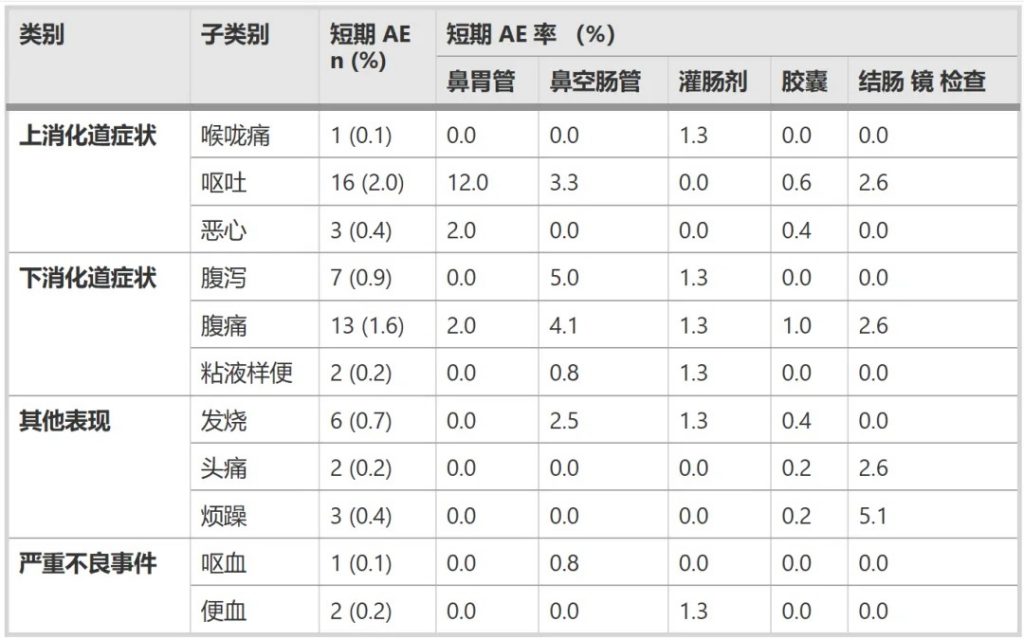
研究发现,FMT在儿科人群中总体表现安全,短期不良事件(AE)发生率仅为5.8%,且大多为轻微自限性症状;在长达122个月的随访期内未观察到长期不良事件。
这些珍贵的临床数据将为技术优化、产品研发和临床应用提供重要参考,并为拓展儿科FMT市场提供有力支撑。
本文我们来详细了解一下这项研究。

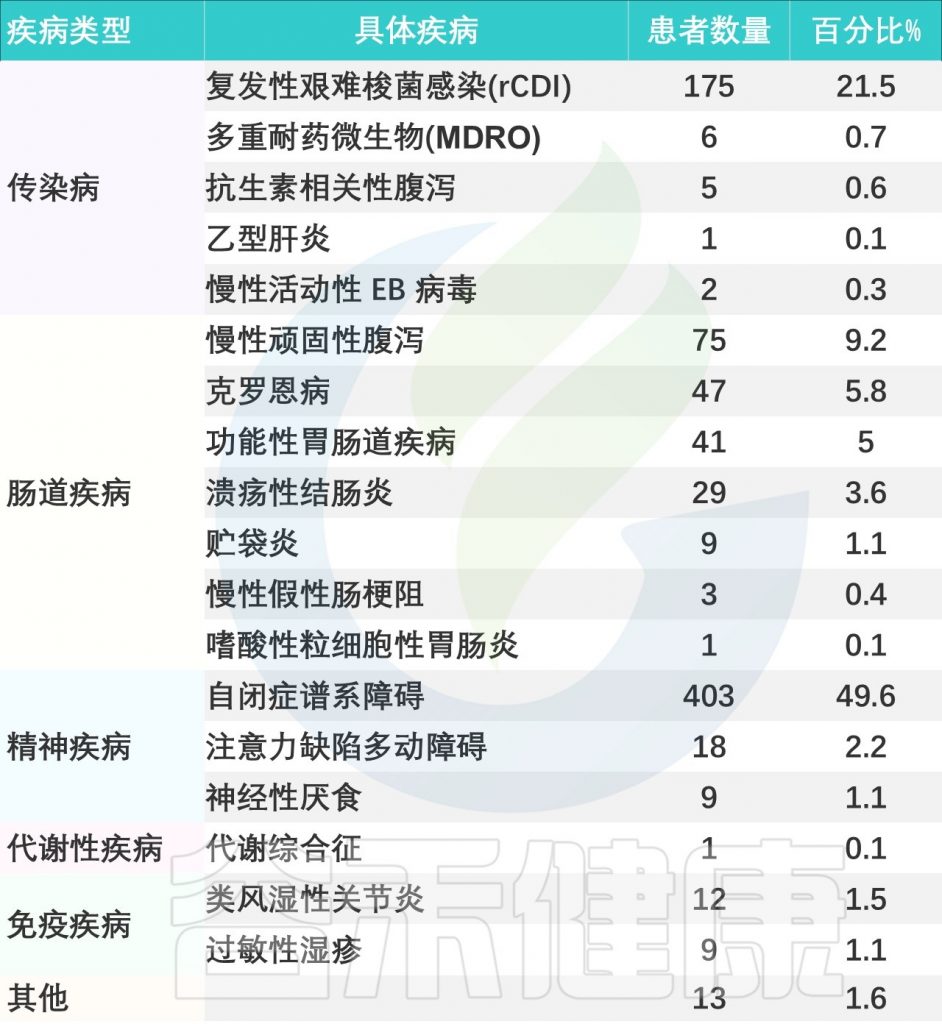
该研究回顾性分析了2013年12月~2023年12月在上海儿童医院接受 FMT 治疗的儿科患者,例如复发性艰难梭菌感染、抗生素腹泻、乙肝、慢性顽固性腹泻、炎症性、慢性肠假性梗阻、自闭症、多动症、神经性厌食症等,详见下表。
doi.org/10.1186/s12866-025-03858-z
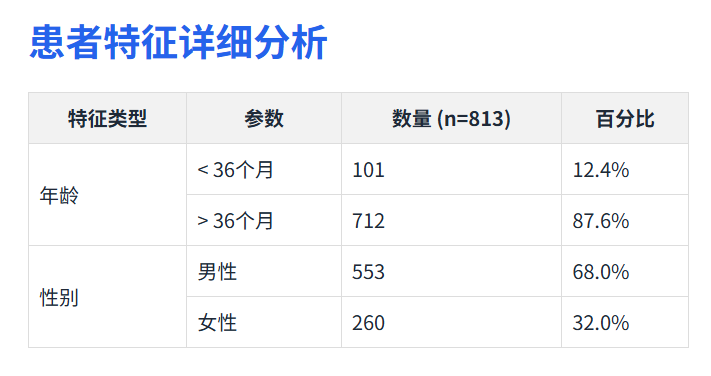
患者特征详细分析

1. FMT相关不良事件
短期不良事件总发生率为5.8%(47/813例)
最常见的短期不良反应包括:
doi.org/10.1186/s12866-025-03858-z
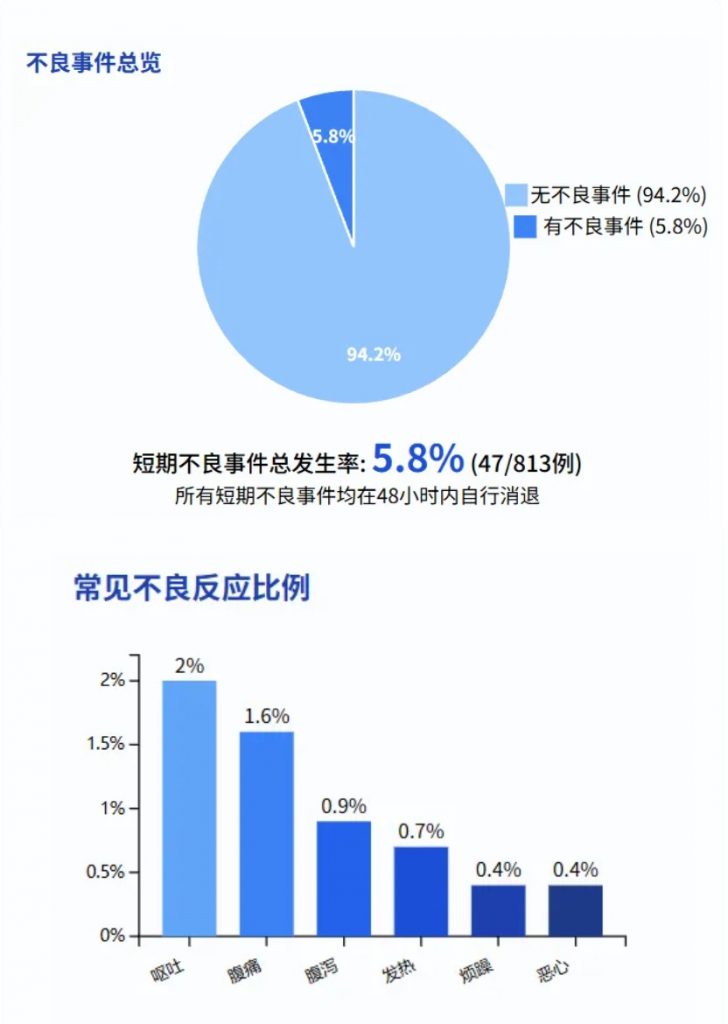
2. 给药途径与不良事件关联
FMT 的给药途径在短期不良事件的发生中起着重要作用。
给药选择包括上消化道(UGI),例如使用鼻胃管或鼻空肠管、内窥镜检查、胶囊,以及下消化道途径(LGI),如保留灌肠、乙状结肠镜检查或结肠镜检查。
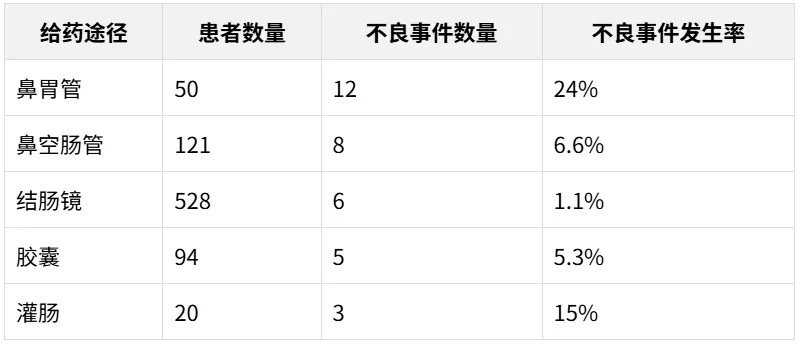
与其他途径相比,胶囊的短期不良事件发生率较低;鼻胃管(16.0%,8/50) 和鼻空肠管(14.0%,17/121) 的短期不良事件发生率更高。
FMT不同给药方式下的短期不良事件发生率(%)
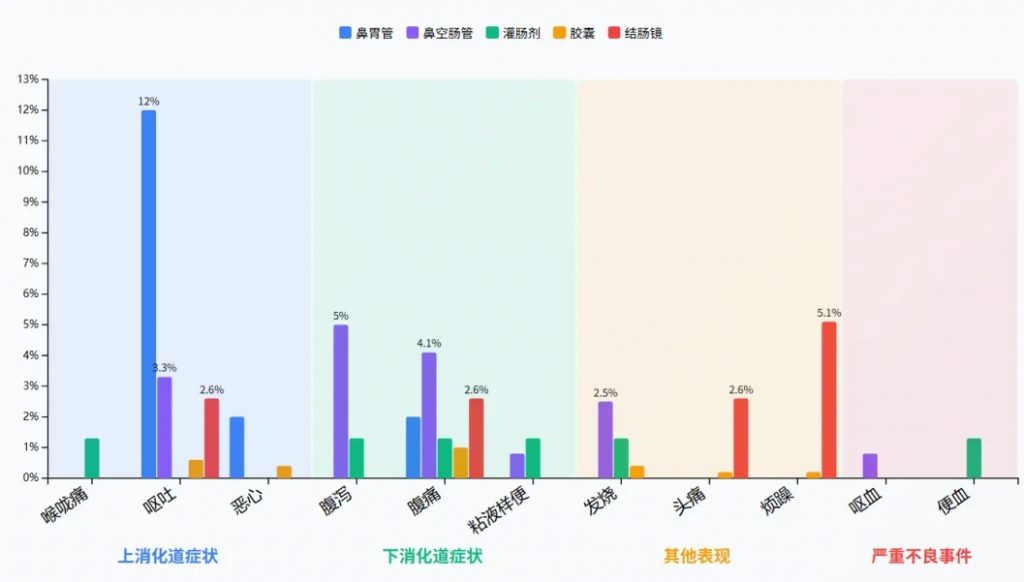
鼻胃管给药方式下,呕吐的发生率最高(12.0%),是所有给药方式中单一不良事件最高的发生率。
鼻空肠管给药方式下,腹痛(4.1%)和腹泻(5.0%)的发生率较高。
严重不良事件(呕血、便血)总体发生率极低(0.1%-0.2%)。

胶囊形式安全也更易于标准化生产、储存和运输,有利于FMT治疗的普及和推广。
3. 疾病类型与不良事件关联
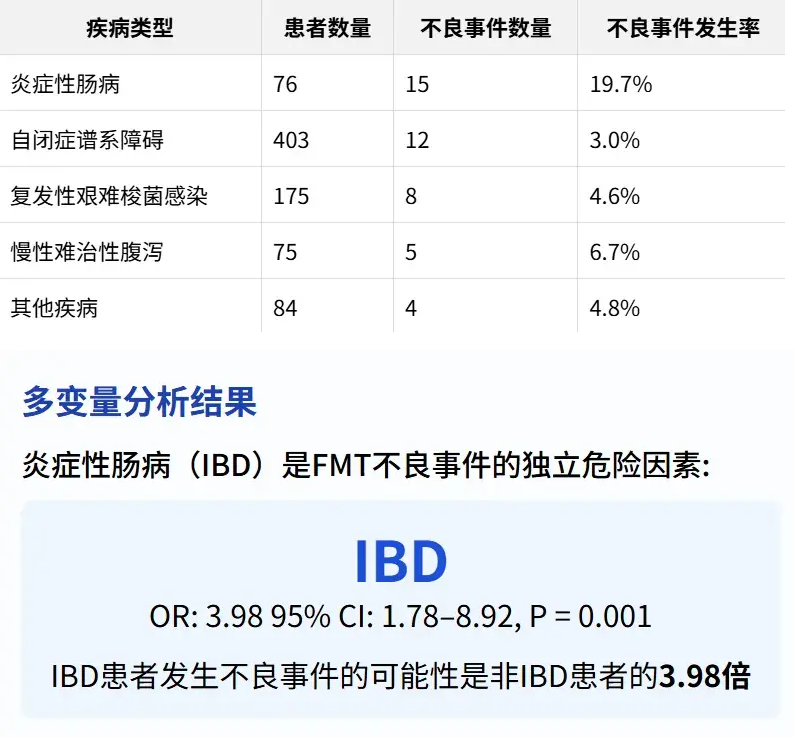
提示在产品开发中需针对IBD患者群体设计更安全的给药方案。
4. 长期随访结果
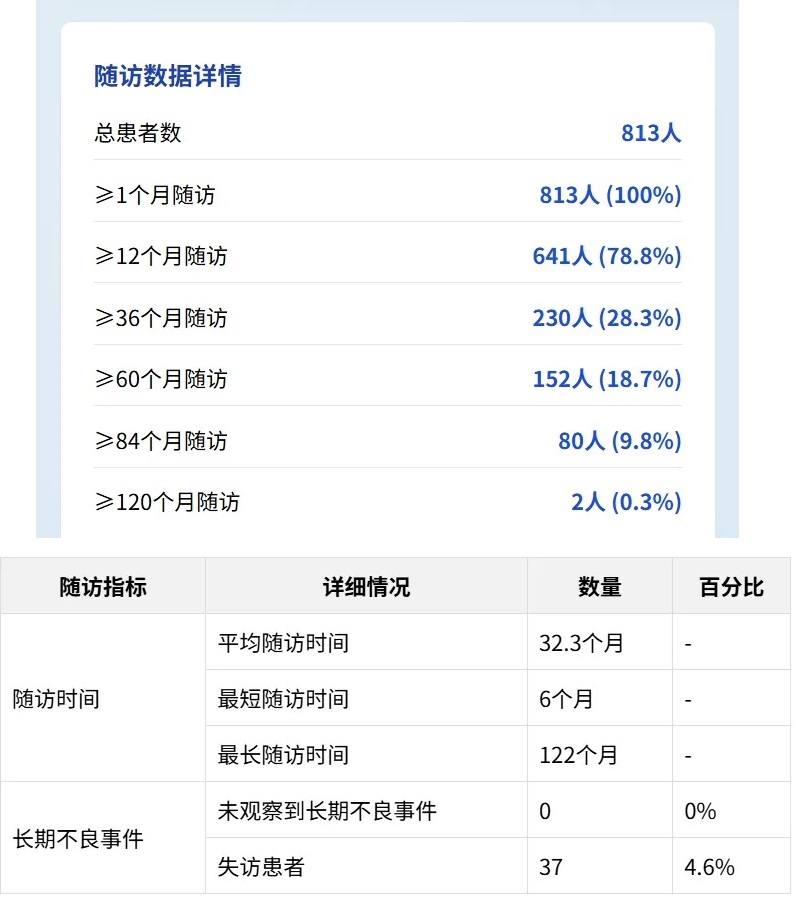
研究纳入813名患者,平均随访时间达32.3个月,最长随访期达122个月,形成了一个极具代表性的儿科FMT安全性数据库。
该研究作为迄今为止中国儿科领域样本量最大、随访时间最长的粪菌移植(FMT)安全性评估,具有较大的科学价值和临床指导意义。
这种大样本、长随访的队列设计克服了以往研究样本量小、随访时间短的局限性,为FMT安全性评估提供了更为可靠的证据。
且该研究覆盖了多种疾病类型和给药途径,包括感染性疾病、肠道疾病、精神疾病、代谢性疾病等多种适应症,以及鼻胃管、鼻空肠管、灌肠剂、胶囊和结肠镜等多种给药方式,能够指导不同疾病和不同情况下FMT的应用策略。
该研究提供的长期安全性数据将增强医患对FMT的接受度,促进其临床应用的规范化,也为微生物组治疗领域的产业化发展奠定了基础。未来,FMT产业有望通过以下方向发展:
未来需要加强微生物组移植后的菌群定植与功能研究,深入探索宿主-菌群互作机制,开发更加精准的微生物组干预策略。
随着高通量测序技术和人工智能技术的发展,FMT治疗前后的连续性菌群数据采集成为可能。这种基于海量微生物组动态变化数据的安全性评估,将远超传统的症状观察方法,能够在潜在安全问题出现前就发出预警,大幅提升FMT的安全性保障水平

谷禾健康
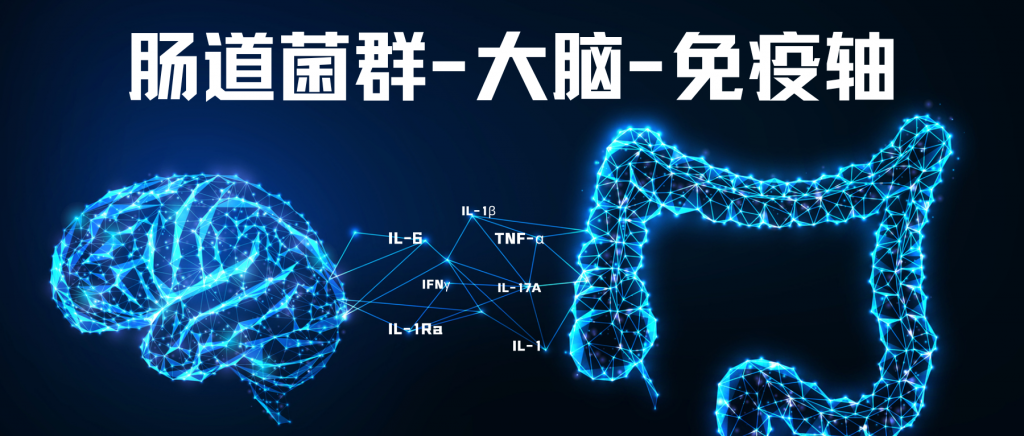
近年来,微生物组研究颠覆了传统神经科学对大脑疾病的理解,微生物群-肠-脑轴对人类健康有重大影响,包括胃肠道生理学、大脑功能和行为。免疫系统代表了沿该轴线与与健康和疾病中的神经炎症有关的微生物组进行通信的关键途径。
肠道菌群通过免疫、代谢和神经通路与中枢神经系统的互作机制。在这个交流网络中,先天免疫系统作为第一道防线快速响应微生物信号,而适应性免疫系统则提供特异性和记忆性反应,两者共同构成了肠脑交流的核心通路。
这种“肠-脑-免疫轴”的交互网络,通过短链脂肪酸、色氨酸代谢物和神经递质(如5-羟色胺)等分子信使,动态调节小胶质细胞功能、血脑屏障通透性及HPA轴活性,成为神经炎症和认知障碍的核心驱动因素。
从抑郁症到阿尔茨海默病,从自闭症到精神分裂症,许多研究证实,肠道微生物群的紊乱与多种神经精神疾病密切相关。靶向这一轴线的治疗策略正在兴起:精神益生菌(如乳杆菌和双歧杆菌)可改善情绪和减轻焦虑;高纤维饮食增加SCFA产生,提高认知能力;粪菌移植通过减少全身炎症标志物,为抑郁和焦虑症治疗开辟新途径。
本文主要分享讨论肠道微生物群如何与大脑相互作用的机制,重点关注在肠脑轴疾病中经常被破坏的先天免疫和适应性免疫。
还详细解释了这些观察结果的含义,以及如何通过跨学科研究来推进它们。利用对这些相互作用如何调节免疫力的加深理解,助力神经发育和神经系统疾病精准临床干预更前一步。
•
微生物在人类健康和疾病各个方面的重要性越来越得到认可,包括大脑健康。当前越来越重视微生物组-肠-脑轴这一轴线的沟通机制。肠道微生物群,尤其是细菌组,因其对免疫成熟、神经炎症和神经行为的影响而备受关注,而不仅局限于多样性指标。
这两大免疫分支共同构成肠脑轴通信的核心,介导肠道微生物群对中枢神经系统的多重影响,涉及多种神经和心理过程,影响着我们的思考、感受和行为方式。
免疫系统和肠脑轴的相互作用
肠道微生物群和大脑通过各种途径进行交流,包括神经元连接和化学信息传递,但促进这些相互作用机制的确切细节仍有待完全阐明。在以下部分中,我们重点介绍肠道和大脑之间通信的主要免疫调节机制,然后关注参与微生物组-肠道-免疫-大脑轴通信的先天性和适应性免疫系统的特定方面。
肠道微生物群-免疫-脑轴通讯机制
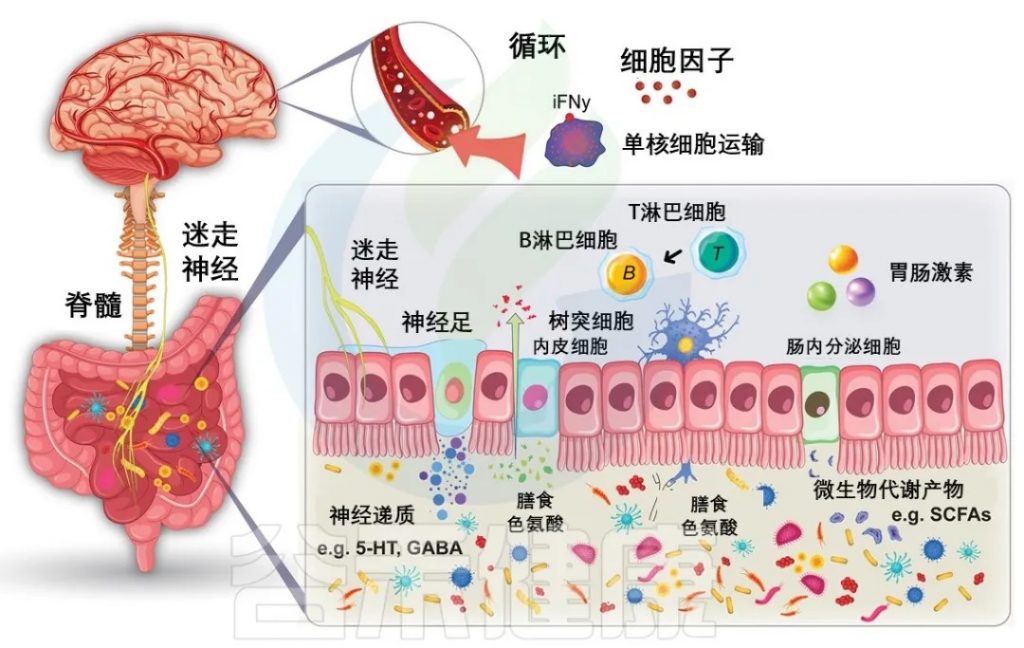
doi: 10.1016/j.xcrm.2025.101982
自 主 神 经 系 统
自主神经系统(ANS)通过交感神经和副交感神经分支与免疫系统相连,在调节生理功能和维持体内平衡方面发挥着关键作用。它促进微生物组-肠道-大脑轴内的双向通信,影响关键的胃肠道功能并通过反馈回路对环境刺激做出反应。
▸ 影响情绪和行为反应
迷走神经和骨盆传入神经是这个通信网络的关键部分,在肠道和大脑之间提供直接联系。 它们通过庞大的传入纤维网络从肠道收集信息,并通过传出的正交感神经/内脏神经和副交感神经系统调节胃肠道和免疫功能,这些神经被传递到大脑,影响情绪和行为反应。
迷走神经和盆腔神经支持体内平衡,而内脏神经支配主要传递伤害性信号。然而,双侧盆腔神经切片可减轻大鼠有害结直肠扩张期间的疼痛行为,表明它们在急性疼痛中的作用。
▸ 迷走神经是炎症反射的关键部分
涉及结肠组织化学刺激物的研究进一步表明,炎症期间盆腔纤维敏感,暗示它们参与伤害感受。 迷走神经是炎症反射的关键组成部分:炎症反射是一种神经反射通路,可调节先天免疫反应和炎症,以应对病原体入侵和组织损伤。
迷走神经传入神经可以检测到来自肠道的一系列信号,包括来自微生物群的更迭、张力和化学信号。研究表明,通过迷走神经切断术或迷走神经刺激等方法改变迷走神经信号传导,可以影响情绪调节、肠道功能和免疫反应,这强调了这种游荡神经在肠脑交流中的重要性。
虽然迷走神经和骨盆通路主要向大脑传递非痛苦刺激,如饱腹感、膨胀和运动,但脊髓内脏神经支配在将复杂的感觉信息(包括疼痛)从肠道传递到中枢神经系统方面起着关键作用。这些信号通过脊髓、脊髓脑、脊髓下丘脑和脊髓丘脑等途径传播,投射到脑干、丘脑和下丘脑区域,影响对肠道来源刺激的情绪和自主神经反应。
肠 神 经 系 统
肠道神经系统(ENS)通常被认为是自主神经系统(ANS)的第三臂, 在胃肠道内充当神经网络,与肠道的免疫细胞群(包括巨噬细胞、T细胞和先天淋巴细胞)接口。这种相互作用使 ENS 能够将环境化学信号解释为神经反应,这对于管理肠道与其饮食、病原体和微生物组的相互作用至关重要,从而影响整体健康。
▸ 微生物成分会影响肠道神经系统的发育、活性
肠道神经系统的发育和功能会受到肠道微生物群的影响,研究表明,微生物成分会影响ENS的发育、活性和肠道免疫反应。 在无菌临床前模型中,肠道微生物群的缺失导致明显的ENS不成熟和免疫失调,突出了肠道微生物群在ENS发展和免疫功能中的重要作用。
抗生素和饮食会改变 ENS 结构和免疫功能,影响肠道蠕动和肠道分泌。这种相互作用表明有可能靶向微生物群或其代谢产物,用于神经精神和神经系统疾病的治疗干预。
▸ 肠道神经系统与免疫细胞双向通讯影响免疫作用
ENS 神经元可以通过神经递质相互作用调节免疫细胞活性,特别是涉及儿茶酚胺,这会影响粘膜内巨噬细胞的功能。
最近的研究表明,肠道神经系统(ENS)活性不仅响应免疫信号传导,而且积极塑造免疫信号传导,在维持胃肠道稳态和响应微生物刺激方面起关键作用。
ENS和免疫细胞之间的这种双向通讯强调了肠道中神经免疫相互作用的复杂性。此外,新出现的证据强调了肠道水平微生物群与宿主相互作用的重大影响,这导致细胞因子、趋化因子、神经递质、神经肽、内分泌信使和微生物副产物的释放。这些分子可以渗透血液和淋巴系统或调节迷走神经和脊髓传入神经元携带的神经信息。通过这些机制,肠道不断与大脑交流,更新其健康状况并调节大脑功能和行为。
内 分 泌 途 径
微生物内分泌学强调宿主和微生物之间共享的神经化学语言,细菌产生并响应血清素(5-羟色胺)、γ-氨基丁酸、儿茶酚胺和吲哚衍生物等神经化学物质,影响宿主的情绪、认知和免疫反应。
▸ 细菌来源的代谢产物会影响炎症、代谢健康等
哺乳动物和细菌共有色氨酸代谢,产生5-HT和犬尿氨酸,影响胃肠道血清素能系统、免疫调节和心理健康。 细菌色氨酸衍生的代谢物(如吲哚)会影响肠道屏障的完整性、炎症和代谢健康,而它们的产生是应激反应性的和昼夜性的。
细菌来源的组胺会诱导内脏痛觉过敏,而β-葡萄糖醛酸酶(由宿主和微生物产生的酶)会影响解毒、炎症和疾病。
肠内分泌细胞通过感应微生物代谢物并释放胰高血糖素样肽-1(GLP-1)和肽YY(PYY)等激素,影响饱腹感、免疫反应和食物摄入量,在肠脑通讯中发挥关键作用。值得注意的是,L细胞通过神经足与 ENS 形成直接突触连接,从而实现快速的肠到脑信号传导。
注:在消化系统中L细胞是一种特殊类型的肠内分泌细胞,主要分布在肠道中。这些细胞呈锥形,底部位于基底膜上。
▸ 细菌通过血清素影响宿主大脑、免疫等功能
产孢梭菌(Clostridia) 通过增加结肠中5-HT(5-羟色胺)的合成,间接影响大脑功能,这种作用通过迷走神经活动和免疫反应实现。此外,新生儿肠道富含特定细菌产生的5-HT,这些细菌还下调单胺氧化酶A以提高5-HT的可用性。在新生儿中,肠道细菌来源的5-HT通过促进调节性T细胞分化来支持免疫耐受,强调了其在早期免疫发育中的关键作用。
肠道和大脑之间的微生物代谢物信号传导
短链脂肪酸(SCFA)和次级胆汁酸等微生物代谢物可增强肠道肽、GLP-1和PYY的分泌,尤其是在远端肠道中。 脱氧胆酸和石胆酸多胺等次级胆汁酸会破坏上皮屏障,使脂多糖、肽聚糖和鞭毛蛋白等细菌成分易位到血液或其他组织中,从而触发免疫反应和全身炎症。
细菌素是小核糖体合成的抗菌肽或蛋白质,也由细菌产生,这些细菌对其他细菌具有窄谱或广谱活性,通常针对密切相关的物种或特定菌株。
下丘脑-垂体-肾上腺轴
下丘脑-垂体-肾上腺(HPA)轴是应激反应协调的核心,也是微生物组-肠-脑轴内的关键通信途径。
▸ 压力会影响激素分泌及微生物群活性
通过微生物易位和炎性细胞因子激活证明的免疫-HPA 相互作用强调了肠道微生物群在HPA信号传导中的作用。压力会激活下丘脑释放促肾上腺皮质激素释放因子,促使垂体前叶分泌促肾上腺皮质激素,刺激糖皮质激素从肾上腺皮质释放。这些糖皮质激素使身体为“战斗或逃跑”反应做好准备,激发免疫系统反应,并向下丘脑和垂体提供负面反馈。
压力不仅影响肠道微生物群的组成和活性,还会促进微生物易位,增强炎性细胞因子激活(例如,肿瘤坏死因子α)并增加肠道通透性,加剧压力相关症状。
细胞因子介导的炎症使微生物产物能够影响全身和神经功能,从而影响HPA轴活性,如在表现出高度应激反应的无菌小鼠中所见。益生菌等干预措施可以调节细胞因子水平,减轻压力对HPA轴的不利影响并恢复肠道稳态。这些相互作用突出了使其成为管理压力相关疾病的潜在治疗靶点。
HPA轴与其他微生物群-脑通讯通路(包括迷走神经刺激和免疫相互作用)之间的对话突出了影响压力和炎症反应的复杂相互作用。 最近,人们越来越强调微生物群在整合整个昼夜节律周期中的HPA轴反应中的作用。
肠道微生物群-免疫-脑轴:关注先天性和适应性免疫系统
免疫系统最初被认为防御病原微生物;现在人们认识到,我们的免疫系统与肠道微生物群广泛相互作用,这有助于宿主健康(下图)。
先天免疫与适应性免疫和肠道微生物组
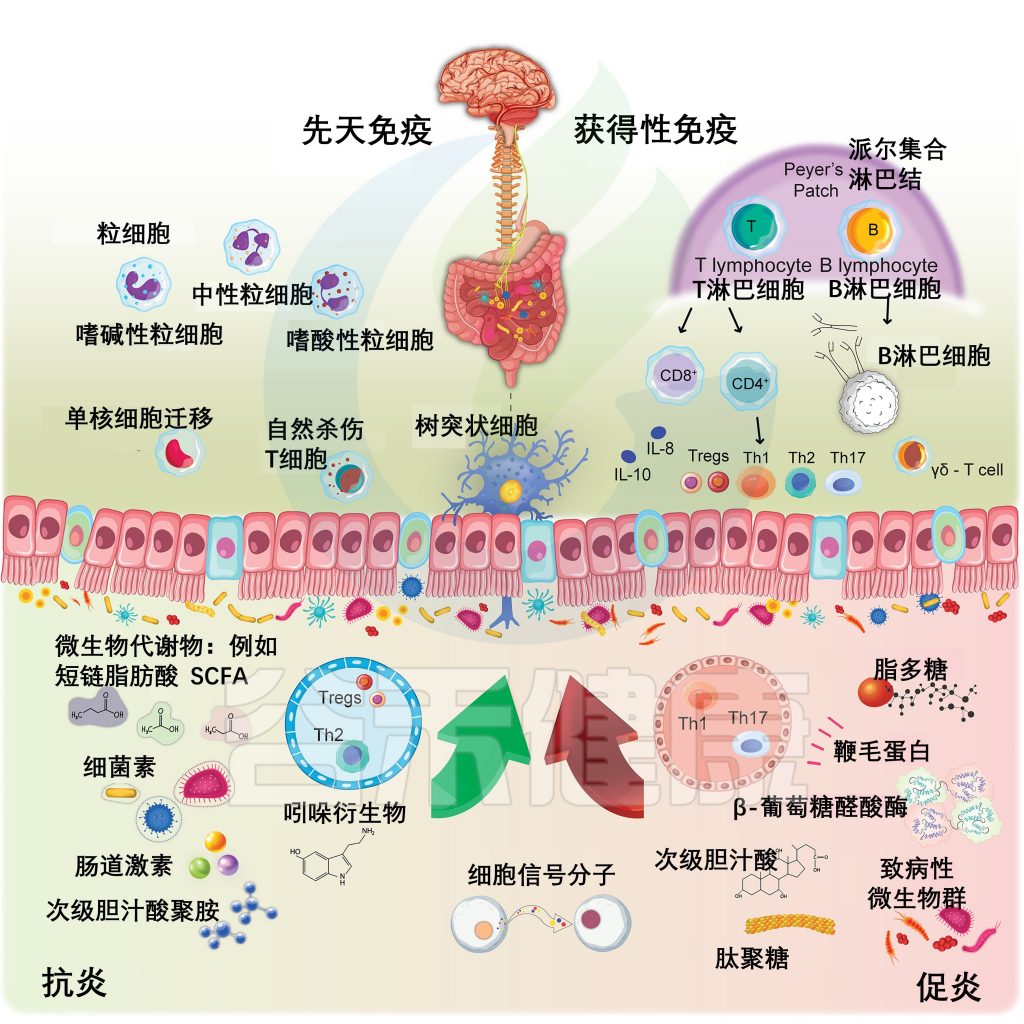
doi: 10.1016/j.xcrm.2025.101982
宿主识别通过前面描述的各种途径(即SCFA、色氨酸代谢物和胆汁酸)发生。人们也越来越关注免疫系统细胞如何影响行为和认知,而免疫功能也会影响关键的大脑过程,如对感染、损伤或自身免疫的反应。
▸ 免疫系统会影响行为和认知
免疫细胞可以浸润大脑,引发炎症反应。神经炎症可导致大脑功能和结构发生变化,从而影响认知、情绪和行为。细胞因子和趋化因子可以穿过血脑屏障(BBB),在那里它们影响神经元活动、突触传递和神经发生。
此外,皮肤和粘膜表面含有多种微生物。随着时间的推移,对共生细菌的免疫反应形成了先天免疫和适应性免疫(包括Peyer斑块内的 B 和 T 淋巴细胞、浆细胞和分化的细胞因子),在所有三个系统之间形成了密切的联系。然而,这些联系背后的机制尚不完全清楚。
肠道微生物组、压力和免疫系统:对脑部疾病的影响
人们越来越关注促进免疫细胞迁移到大脑的细胞过程,特别是肠道微生物群在这些动力学中的作用。虽然历史上认为中枢神经系统(CNS)与外周免疫系统隔离,但现在人们了解循环细胞因子会影响大脑功能和行为。 外周白细胞,包括单核细胞、T细胞和B细胞以及自然杀伤T细胞,可以进入脑脊液、脑膜、脉络丛和大脑。
在中枢神经系统内,脉络丛、脑膜和血管周围巨噬细胞、肥大细胞和小胶质细胞(大脑的巨噬细胞)检测病原体或组织损伤并启动免疫反应。
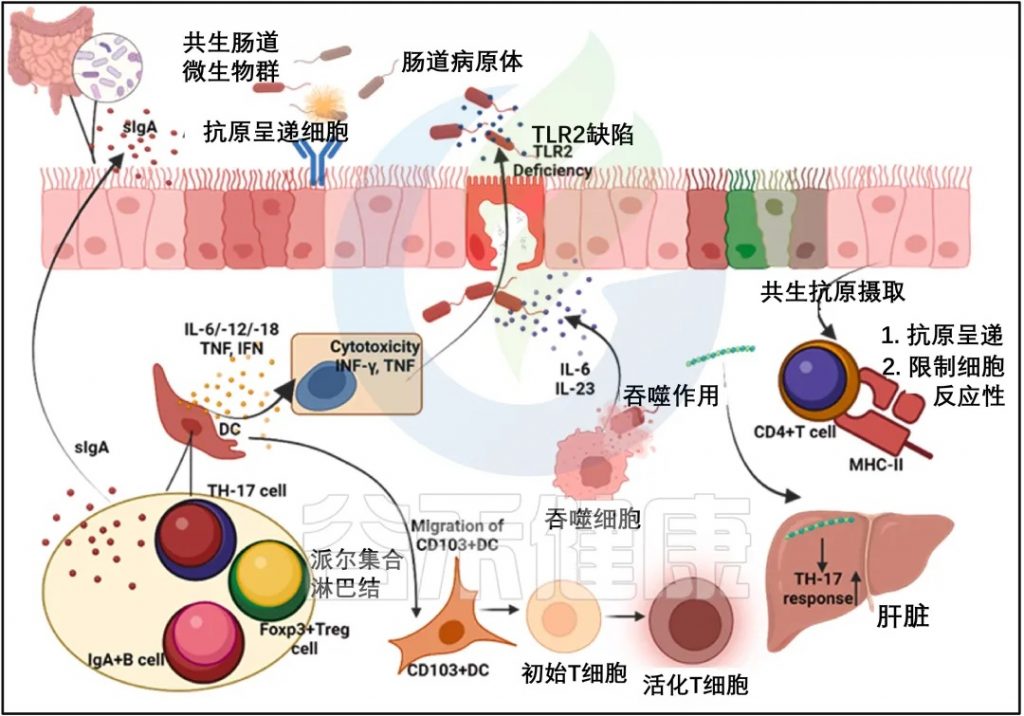
DOI: 10.1016/j.heliyon.2024.e34092
▸ 细胞因子的水平受到肠道微生物群的影响
趋化因子驱动的淋巴细胞募集到血管周围间隙进一步支持中枢神经系统免疫。细胞因子水平的失衡和单核细胞迁移增加可能导致神经炎症,可能受肠道微生物群的影响。
免疫状态的这种变化也可能受到肠道微生物群的影响,可能会对神经炎症反应产生深远影响,从而可能加剧神经精神和神经系统疾病。小胶质细胞体现了肠道微生物群如何通过先天免疫机制影响大脑。事实上,无菌小鼠表现出小胶质细胞缺陷,包括数量、成熟、形态和代谢功能的改变,这与对感染的反应受损有关。 这些过程似乎受到微生物来源的短链脂肪酸,特别是乙酸盐的调节。细菌来源的乙酸盐在稳态下调节小胶质细胞的关键代谢过程,并且可以挽救无菌小鼠中受损的小胶质细胞成熟。
N6-羧甲基赖氨酸是一种微生物代谢物,也诱导衰老小胶质细胞的线粒体功能障碍。中性粒细胞影响肠道微生物群,反之亦然,代谢物调节中性粒细胞的产生和功能。
此外,中性粒细胞驱动的肠道炎症与自闭症谱系障碍(ASD)、帕金森病(PD)和阿尔茨海默病(AD)有关。
在阿尔茨海默病中,中性粒细胞积累在β-淀粉样蛋白(Aβ)沉积物附近,它们在疾病早期阶段的耗竭改善了小鼠模型的记忆力。
最近的一项研究表明,急性肠道炎症通过中性粒细胞外渗加速Aβ积累,这可以通过中性粒细胞耗竭来缓解。 这些发现强调了通过肠脑轴靶向中性粒细胞的潜在治疗作用,尽管需要进一步研究。
压 力 暴 露
肠道微生物组也受到压力的影响。研究已在多种细菌菌株、生物体和压力模型中验证这一发现。压力不仅改变肠道微生物组的组成,还影响胃肠道的生理与功能。
一项研究表明,将慢性不可预测轻度应激(CUMS) 小鼠的肠道微生物群移植至无特定病原体(SPF) 小鼠,可诱导抑郁样行为。微生物组转移还导致SPF小鼠出现补体C3激活和小胶质细胞介导的突触修剪,这与CUMS小鼠的抑郁样行为相关联。
▸ 一些有益肠道细菌能够减轻焦虑和抑郁行为
树突状细胞(DC)激活也与压力暴露和随之而来的焦虑的临床前模型有关。与载体处理的动物相比,用鼠李糖乳杆菌(JB-1)治疗的雄性小鼠应激诱导的焦虑样行为降低。该JB-1菌株还被证明可以减弱与应激相关的 DC 激活,同时增加白细胞介素(IL-10)和调节性T淋巴细胞。
此类研究表明,微生物群可能通过与树突状细胞的交流影响某些神经和行为结果。产丁酸盐的细菌(如普拉梭菌)已被证明在结肠炎中发挥抗炎作用,并减少焦虑和抑郁样行为,从而影响活化淋巴细胞的 Th17/Treg 比率。
值得注意的是,IL-17A是连接适应性免疫系统与肠道微生物组的重要分子。研究表明,分泌IL-17A的共生特异性T细胞通过IL-17A受体A向感觉神经元传递信号,促进神经元修复。在粘膜表面,适应性免疫系统与微生物组的细胞协同作用,影响中枢神经系统的修复。
•
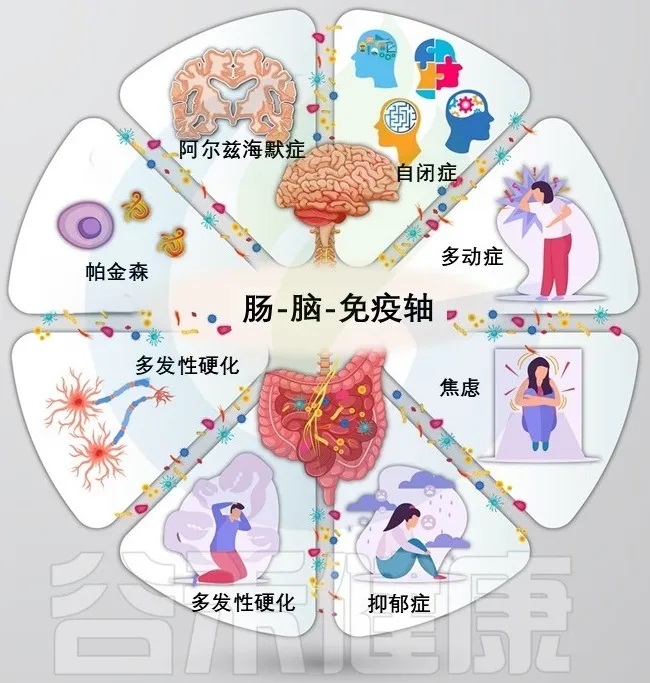
doi: 10.1016/j.xcrm.2025.101982
神 经 精 神 疾 病
重 度 抑 郁 症
重度抑郁症(MDD) 等情绪障碍是受炎症和肠道微生物组影响比较复杂。HPA 轴的破坏、免疫激活的改变和肠道微生物群紊乱是导致重度抑郁症病理的原因。
免疫系统“罢工”引发抑郁——炎症因子成幕后黑手
慢性低度炎症被认为是抑郁症发展的关键因素,在重度抑郁症患者中,循环免疫细胞(例如单核细胞和粒细胞)水平升高;抑制这些细胞因子可降低动物模型中的抑郁样行为。此外,肥大细胞是关键的先天免疫调节因子,通过与色氨酸代谢和神经炎症相关的机制与抑郁症有关。
此外,T 细胞有助于重度抑郁症病理学,荟萃分析揭示了免疫功能障碍,包括 CD4+ 辅助细胞和活化 T 细胞计数的改变。越来越多的证据表明C反应蛋白(CRP) 等炎症标志物与抑郁、焦虑和认知缺陷有关。
孟德尔随机化暗示了 CRP 和焦虑之间的因果关系,支持免疫细胞谱作为患者分层的潜在重度抑郁症生物标志物。总的来说,这些研究强调了免疫细胞谱作为生物标志物的潜在用途,用于识别重度抑郁症的亚型并指导未来试验中的患者分层。
肠道菌群紊乱影响情绪——基于菌群的治疗
同样,此类大规模人群研究还发现, 生活质量较高者体内富含Coprococcus和Dialister菌属的菌株,而未经治疗的抑郁症患者体内这些菌群含量较低。丁酸球菌属(Butyricicoccus) 在抗抑郁治疗反应机制中同样扮演着重要角色。
来自重度抑郁症个体的粪便移植(FMT)在受体动物中诱导了抑郁样行为,表明肠道微生物组与抑郁症的病理生理学有关。
益生菌干预(例如瑞士乳杆菌 、长双歧杆菌菌株、丁酸梭菌和植物乳杆菌 )已证明对抑郁评分、认知功能和治疗反应性有益。肠道微生物组在适应性免疫中的作用,影响 T 和 B 淋巴细胞功能,表明微生物群靶向治疗或可以解决重度抑郁症。了解微生物组、免疫系统和抑郁症之间的相互作用可能会为压力相关疾病的新治疗方法提供信息。
社 交 焦 虑 障 碍
社交焦虑症 (SAD)(又名社交恐惧症)是一种心理健康状况,其特征是对在社交场合被审视、评判或羞辱的深刻而持续的恐惧。这种极度恐惧导致患者回避社交或在社交场合中备受煎熬。
最近的研究表明,在社交焦虑症的情况下,肠道微生物群、免疫功能和焦虑之间存在显著联系。社交焦虑症个体对小鼠的 FMT 显示,肠道微生物群可诱导对社交恐惧的高度敏感,这与社交恐惧症的症状相似。这种反应伴随着中枢和外周免疫功能的显著变化以及终纹床核内催产素表达的降低。改变的免疫反应包括 IL-17A 产生减少和肠道相关淋巴组织中 T 细胞谱改变,表明免疫信号中断,这与社交恐惧增加的行为表型相关。
社交焦虑症的肠道菌群组成和功能与健康对照不同,Anaeromassillibacillus 和Gordonibacter水平升高,对照组的 Parasutterella 患者水平升高。需要更大规模的纵向研究来证实这些发现并探索其临床相关性。
神 经 发 育 障 碍
精 神 分 裂 症
虽然精神分裂症是一种神经精神疾病,但也被认为是一种神经发育障碍,是由于遗传易感性与产前和产后环境压力源之间的相互作用引起的。
精神分裂症表现为阳性(幻觉)和阴性症状(情感麻木),以及认知缺陷。基因与环境之间的相互作用使微生物组和免疫系统处于治疗精神分裂症的多系统方法的最前沿。
肠道菌群紊乱,免疫系统拉警报
精神分裂症的几个危险因素突出了胃肠道是一个关键的调查领域。这些包括炎症、麸质敏感性和暴露于刚地弓形虫(Toxoplasma gondii)。
强有力的证据支持微生物组和免疫系统在精神分裂症中的综合作用。细菌易位和肠道通透性的生物标志物,例如,可溶性CD14和脂多糖结合蛋白,与精神分裂症患者的CRP水平相关,并受抗精神病药物使用的影响。
精神分裂症个体表现出肠道微生物组β多样性的改变,代谢途径与炎性细胞因子有关。同样,口腔微生物组显示链球菌增加和普雷沃氏菌减少,链球菌与 TNF-α 和 IL-9 升高、慢性炎症和血脑屏障破坏有关。这些促炎细胞因子与慢性低度炎症和血脑屏障中断有关。肠道微生物群的变化,例如Eggerthella 增加和拟杆菌减少,与连蛋白和 CRP 等炎症标志物相关。
此外,神经认知障碍个体中,血浆IgM 和IgA对共生菌株,特别是 Hafnia alvei, Pseudomonas aeruginosa, Morganella morganii, Pseudomonas putida ,肺炎克雷伯菌的反应增强,将微生物组与先天免疫和适应性免疫联系起来。
自 闭 症 谱 系 障 碍
自闭症谱系障碍 (ASDs)也是多方面的神经发育障碍,其特征是沟通和社交技能缺陷以及重复的刻板行为。儿童自闭症和胃肠道合并症的高患病率激发了人们对肠道微生物组在自闭症发病机制中的作用的兴趣。
来自自闭症的粪便样本显示拟杆菌门增加和双歧杆菌、乳酸杆菌、普雷沃氏菌和瘤胃球菌属定植的改变,并与炎症和免疫激活增加有关。事实上,双歧杆菌通常与保护性抗炎活性有关,因此它在自闭症中的水平降低被认为是有害的而不是保护性的。
自闭症的慢性炎症与炎症性肠病转录组重叠,而 自闭症患儿的外周血单核细胞产生升高的粘膜相关细胞因子(IL-5、IL-15 和 IL-17)和连蛋白,表明肠道通透性。
母亲的炎症,孩子的风险
由于感染、自身免疫性疾病或妊娠期炎症引起的母体免疫激活(MIA)会增加后代患自闭症的风险。
关键的炎性细胞因子,包括 IL-6 和 IL-17A,会破坏胎儿大脑发育,导致神经发育异常。MIA 小鼠模型表明,母体炎症会改变后代的大脑连接、行为和免疫启动,其中 IL-17A 是关键介质。
益生菌
新出现的证据强调了母体肠道微生物组在调节 MIA 效应中的作用,表明了潜在的干预目标。自闭症中肠道微生物组与免疫系统联系的进一步证据来自在丙戊酸(VPA)大鼠模型中使用益生菌和益生元的研究。 多菌株益生菌(VSL#3) 治疗与改善社交能力、社交互动、焦虑样行为相关,以及挽救 VPA 诱导的 IL-6 增加和前额叶皮层 5-HT 水平降低。
益生元
此外,特定的益生元饮食(3% 低聚半乳糖 [GOS]/低聚果糖 [FOS])已被证明可以通过恢复微生物群落、肠道通透性和减少与小脑相关的神经炎症来减轻 VPA 效应。它还增强了 VPA 暴露小鼠的 Foxp3 + Tregs,表明免疫平衡的调节。
上所述,这些研究强调了自闭症中肠道微生物组和免疫系统之间的复杂相互作用,并表明可以通过靶向该轴来缓解症状。
注 意 缺 陷 多 动 障 碍
注意力缺陷多动障碍(ADHD) 是一种神经发育疾病,其特征是注意力不集中、多动和冲动,人们越来越关注微生物组在其发育中的作用。研究表明,饮食因素,尤其是西式饮食和消除饮食,可能通过影响肠道微生物群来影响多动症症状。
少吃反而更专注?
多动症个体双歧杆菌属水平升高可能与肠道来源的多巴胺前体的调节有关。少食饮食法(Few-Foods Diet)饮食对多动症症状的影响发现,63% 的参与者表现出症状显著减轻。
注:少食饮食法(Few-Foods Diet)
少食饮食法是一种针对注意力缺陷多动障碍(ADHD)症状的饮食干预方法。这是一种消除饮食的形式,其基本原理是:
限制食物摄入种类,只食用少量特定食物。
从神经科学角度看,少食饮食法可能通过增加大脑楔前叶区域的激活来发挥作用,这与多动症症状的减少有关联。这提示了该饮食方法可能通过特定的神经认知机制来改善多动症症状。
尽管与抑制控制相关区域的大脑激活变化与症状改善无关,但楔前区激活的增加与多动症症状的减少有关,这表明少数食物饮食可能通过一种神经认知机制发挥其益处。
基于肠道菌群的干预
最近一项关于益生元、益生菌和合生元对多动症有益影响的系统评价发现,特定菌株如鼠李糖乳杆菌和两歧双歧杆菌,可能会对神经认知和行为结果产生积极影响。
另一项系统评价确定了与多动症相关的肠道微生物组特征的差异,强调了与多巴胺代谢相关的 Odoribacter和 Eggerthella 等属的丰度增加,以及与炎症相关的Faecalibacterium丰度减少降低。虽然肠道微生物群特征与多动症症状之间存在一些相关性,但该评论强调需要进一步研究来探索这些关系,考虑可能影响多动症肠脑联系的因素,例如年龄和饮食习惯。
自 身 免 疫 性 疾 病
多 发 性 硬 化 症
微生物群-免疫轴失调驱动中枢神经炎症
微生物群免疫通讯与自身免疫性疾病(如多发性硬化症)之间的联系已得到充分证实。多发性硬化症(MS)是一种以中枢神经系统髓鞘破坏为特征的慢性自身免疫性疾病,与单核细胞中炎症通路表达增加相关的微生物组改变有关。
微生物干预:重塑免疫平衡的新策略
DC受肠道微生物组调节,短链脂肪酸(SCFA)通过Fms相关酪氨酸激酶3配体表达促进DC造血。在临床前多发性硬化模型(实验性自身免疫性脑脊髓炎[EAE])中进行,益生菌如Lactibiane Iki 通过减少脱髓鞘和诱导致耐受性DC表型来改善临床结果。类似地,酿酒酵母及其衍生物Selemax增加了CD103+ DC并减少了肠道炎症。
脑膜在免疫监视中的作用
依赖于肠道微生物组的脑膜 IgA + 细胞对肠道炎症的反应增加并抑制实验性自身免疫性脑脊髓炎相关炎症。
微生物-饮食-免疫平衡
饮食影响 B 细胞功能,对神经元发育有影响,尤其是参与髓鞘形成的 B1a 亚型。如前所述,越来越多的证据表明肠道微生物组与多发性硬化的发生和进展有关,研究人员观察到多发性硬化患者某些共生体组成的变化。
这些包括细菌细胞壁成分的低水平易位、血脑屏障的破坏或参与髓鞘形成的基因表达的改变。
抗生素预处理增强了调节性 T 细胞和 B 细胞的数量,通过改变细胞因子谱从而降低了实验性自身免疫性脑脊髓炎的严重程度,这表明微生物群的调节可能会影响多发性硬化自身免疫。
神 经 退 行 性 疾 病
帕 金 森
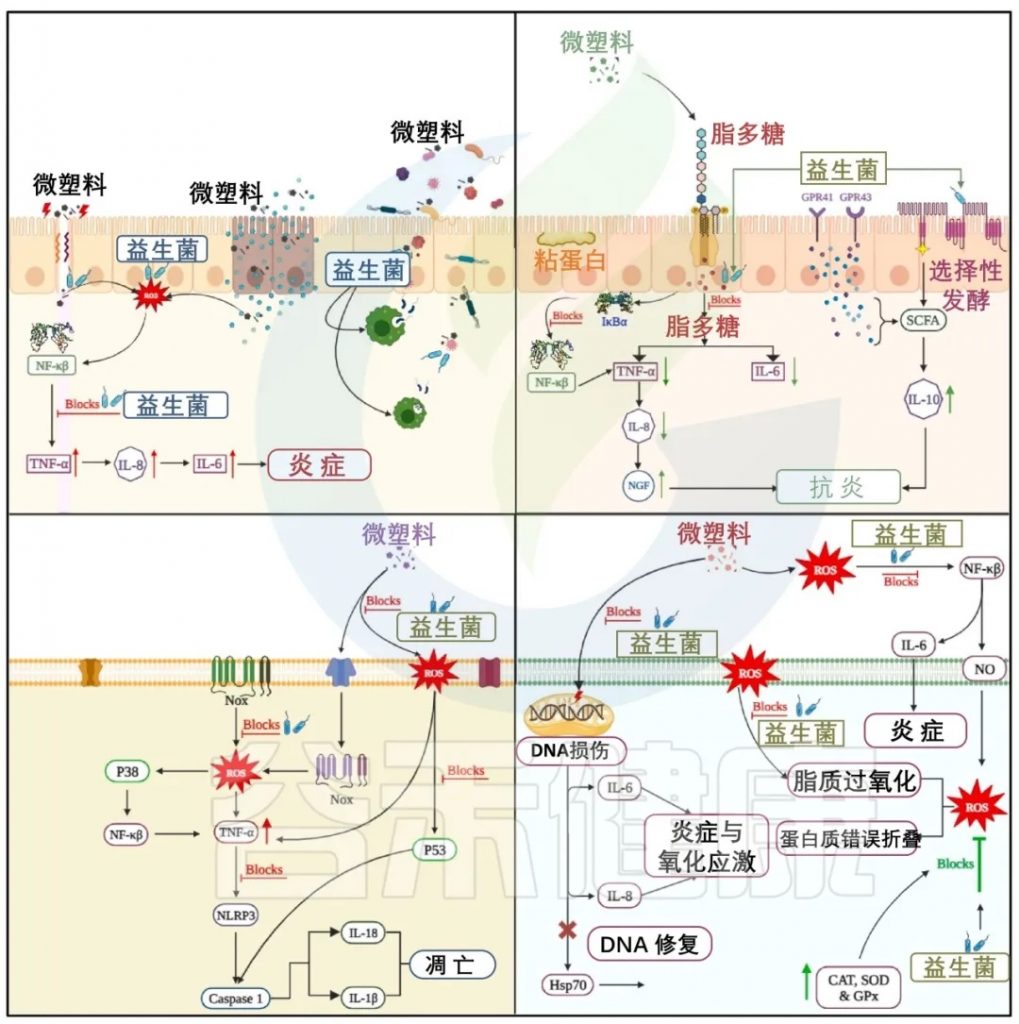
二十多年前,一项开创性研究提出了震惊医学界的假设:帕金森病可能起源于肠道,而非大脑。这种与年龄相关的神经退行性疾病,以多巴胺能神经元丢失、α-突触核蛋白积累和神经炎症为特征,通常在诊断前数年出现便秘。
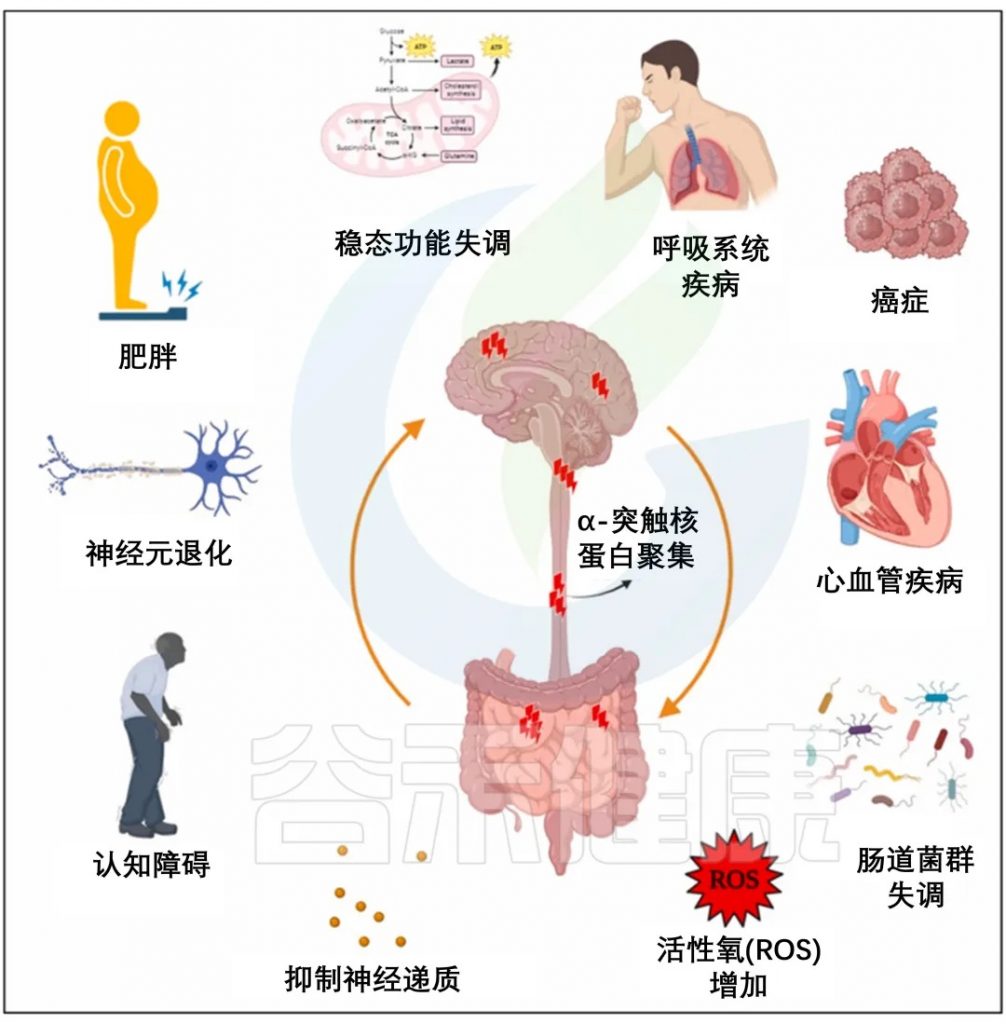
DOI: 10.1016/j.heliyon.2024.e32004
帕金森病的”肠道起源”
已在肠道神经纤维和神经节中鉴定出 α-突触核蛋白,小鼠模型显示肠道微生物群影响 α-突触核蛋白聚集和蛋白质清除。
此外,迷走神经可能介导肠到脑的 α-突触核蛋白运输,这得到了躯干迷走神经切断术降低小鼠帕金森风险并阻止小鼠 α-突触核蛋白病进展的研究结果的支持。
然而,对于是否存在帕金森特有的独特微生物模式尚未达成共识,部分原因是纵向研究有限和样本量小。
与帕金森的免疫学联系有据可查,调节免疫活性的基因和细胞因子信号转导与帕金森风险相关。
新兴研究表明,源自肠道的炎症在帕金森中起病理作用,推动了针对 α-突触核蛋白和免疫介质的免疫疗法的发展。帕金森脑组织的尸检分析显示,补体、细胞因子和趋化因子的产生以及炎性小体的激活增加,所有这些都与小胶质细胞激活相结合,表明免疫系统在整个疾病进展过程中参与。
帕金森患者的独特肠道菌群特征
微生物组研究揭示了不同人群的一致发现。在德国、芬兰、俄罗斯和日本的队列中报道了普雷沃氏菌丰度降低,这表明全球模式与种族或饮食无关。Akkermansia muciniphila 增加与帕金森患者的便秘相关,而改变的产短链脂肪酸细菌是帕金森的标志,可能与 SCFA 失调与神经炎症有关。
稳态破坏
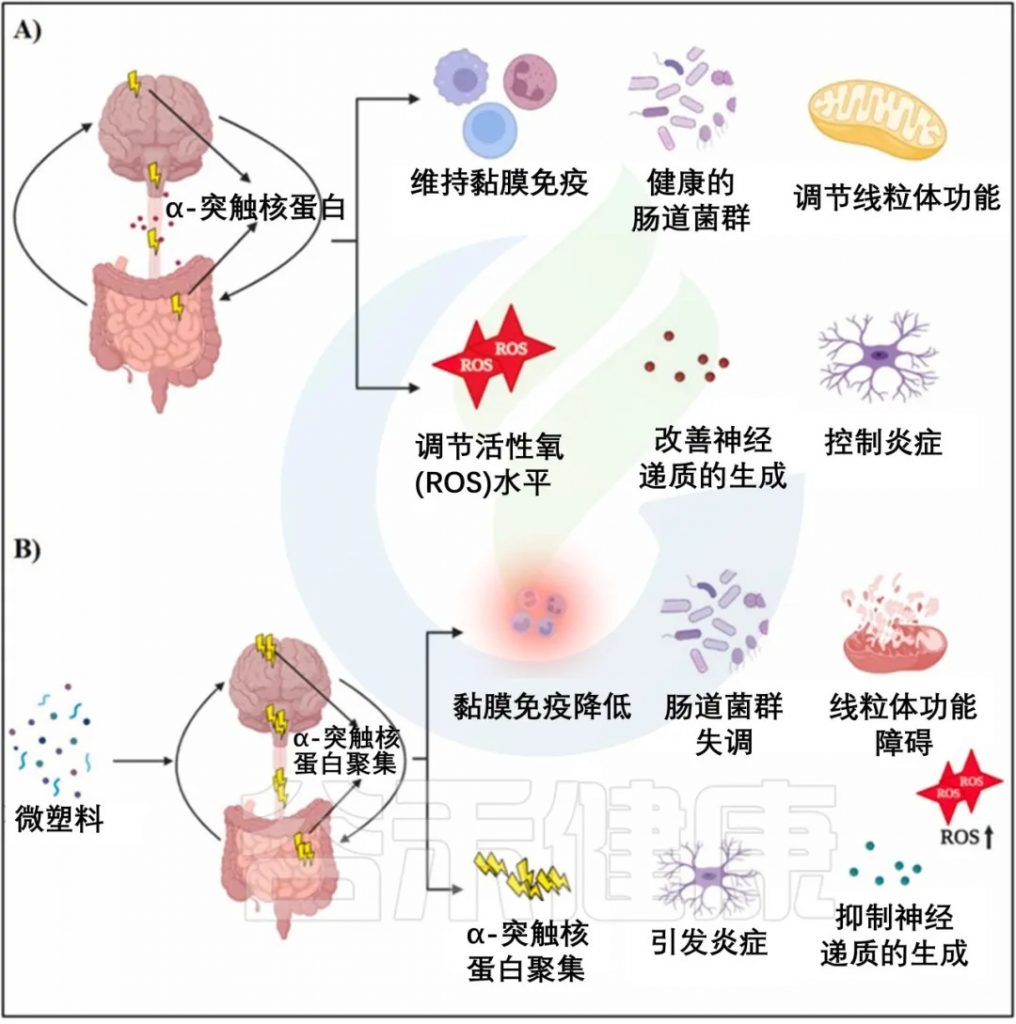
DOI: 10.1016/j.heliyon.2024.e32004
越来越清楚的是,微生物组是帕金森进展所必需的,未来的工作应该确定疾病发展过程中不同时间点的微生物组组成,包括前驱期。
阿 尔 茨 海 默 病
阿尔茨海默病(AD) 是最普遍的神经退行性疾病,可导致痴呆,其特征是形成 Aβ 斑块、过度磷酸化的 tau 蛋白和神经原纤维缠结、神经元丢失和神经炎症。疾病从跨鼻皮层发展到海马体和皮质区域。尽管 Aβ 积累是阿尔茨海默的核心,但神经炎症会加速认知能力下降,Aβ 激活 Toll 样受体会触发炎性小体复合物和小胶质细胞驱动的炎症。
错误折叠的 tau 会破坏突触处的蛋白质周转,进一步导致神经元功能障碍。然而,Aβ 积累是免疫反应失调还是阿尔茨海默的直接驱动因素仍然是一个悬而未决的问题,需要进一步研究。
肠道菌群失衡:有益菌减少、致病菌增加
新出现的证据将病原微生物与阿尔茨海默发病机制联系起来。阿尔茨海默小鼠中拟杆菌属水平升高与小胶质细胞吞噬活性降低相关,促进 Aβ 积累。
阿尔茨海默患者通常表现出肠道微生物群失衡,微生物多样性减少,直肠真杆菌、双歧杆菌和腹泻菌等有益细菌减少,以及埃希氏菌/志贺氏菌、拟杆菌属和瘤胃球菌等致病菌增加。
致病菌与促炎细胞因子存在显著相关性
据报道,阿尔茨海默个体血清中埃希菌/志贺氏菌水平升高与促炎细胞因子(IL-1β 和趋化因子 CXCL2)增加之间存在显著相关性,表明肠道菌群改变与阿尔茨海默外周炎症之间存在联系。 假说表明 Aβ 作为一种抗菌肽,将病原体捕获在纤维聚集体中。病毒受累,尤其是单纯疱疹病毒 1(HSV-1),与阿尔茨海默有关,因为 HSV-1 与大脑中的 Aβ 斑块和 tau 缠结共定位。
抗 HSV-1 IgM 抗体增加了阿尔茨海默风险,表明再激活而不是持续存在可能会驱动病理。此外,将阿尔茨海默匀浆从人类接种到灵长类动物和小鼠中诱导了阿尔茨海默样病理的传播性。
微生物组靶向治疗的潜力——粪菌移植
肠道微生物群的改变与阿尔茨海默相关。抗生素诱导的微生物组变化调节神经炎症和 Aβ 沉积。无菌淀粉样蛋白前体蛋白/早老素 1 小鼠的 Aβ 病理降低,而来自健康供体的 FMT 减少了阿尔茨海默小鼠模型中的 Aβ 和 tau 异常。
益生菌调控
从治疗的角度来看,在阿尔茨海默小鼠模型中,植物乳杆菌的给药通过减少 Aβ 斑块形成和 tau 过度磷酸化来防止认知能力下降。另一方面,动物研究的因果证据表明,阿尔茨海默患者的微生物群可以诱发认知缺陷。
临床试验显示结果喜忧参半,一些益生菌改善了轻度阿尔茨海默的认知能力,但对晚期病例的影响有限。虽然阿尔茨海默的微生物组靶向疗法前景广阔,但需要进一步研究来阐明机制,开发用于早期检测的生物标志物,并改进干预措施。基于微生物群的治疗具有作为辅助疗法的潜力,可减缓或阻止阿尔茨海默进展,为管理这种复杂疾病提供一种变革性的方法。
•
新出现的证据通过微生物组-肠道-免疫脑轴将肠道微生物群与精神和神经系统疾病联系起来,导致了针对该轴的治疗方法的发展。这些疗法的重点是调节肠道微生物群以影响免疫反应和大脑功能,其中益生菌、益生元、合生素、益生菌和FMT,结合生活方式选择和饮食建议的指导,是目前正在研究的主要策略。

doi: 10.1016/j.xcrm.2025.101982
益 生 菌
益生菌是赋予宿主健康益处的活微生物,已在微生物组-肠道-大脑轴的背景下进行了广泛的研究。精神益生菌,最初定义为通过肠脑相互作用专门针对心理健康的益生菌,可改善情绪、认知和压力反应。该定义已扩展到包括针对可能增强大脑过程的微生物组的其他方法。
乳杆菌和双歧杆菌:改善情绪
推定益生菌的好处似乎与菌株有关;特别是,乳杆菌和双歧杆菌在临床前模型和人体研究中都显示出改善情绪、焦虑、抑郁评分的前景。
干酪乳杆菌改善了老年参与者的情绪评分,尤其是那些基线情绪较低的参与者。嗜酸乳杆菌、L. casei 和 B. bifidum 显示重度抑郁患者血清高敏 CRP 和抑郁症状显著降低, 而乳杆菌和双歧杆菌补充剂可降低妊娠糖尿病患者的 CRP 和 TNF-α 水平。
乳杆菌和肠球菌:抗炎
临床前模型中,特定菌株减少了炎症,例如高脂肪饮食诱导的炎症模型,其中乳杆菌和肠球菌菌株降低了 IL-6、TNF-α 和 IL-1β。菌株特异性至关重要;例如,长双歧杆菌缓解了肠易激综合征患者的抑郁,但没有缓解焦虑。
同样,L. plantarum减轻了应激成人的压力和犬尿氨酸,但对 TNF-α、IL-6 或 IL-1β 水平没有影响。此类工作表明,在临床应用中需要仔细选择益生菌菌株,因为并非所有菌株都具有相同的治疗潜力。
双歧杆菌与乳杆菌的行为调节作用
一些益生菌(例如双歧杆菌和乳酸杆菌)在自闭症的临床前模型中显示出改善行为和神经化学障碍的潜力,其中双歧杆菌和乳杆菌可改善行为和神经化学障碍。
人体研究是初步的,研究表明嗜酸乳杆菌 、鼠李糖乳杆菌、长双歧杆菌可能会改善行为;但同样,在一项研究中测试的任何组中,均未观察到 TNF-α、IL-6 和 IL-1b 或皮质醇的显著变化。
外周免疫异常与精神分裂症有关,但益生菌显示出有限的精神益处。补充鼠李糖乳杆菌和动物双歧杆菌可降低血管性血友病因子水平并调节单核细胞趋化蛋白-1、脑源性神经营养蛋白 (BDNF)、趋化因子配体 5 和巨噬细胞炎症蛋白-1 β。
通路分析表明,这些变化与通过 IL-17 细胞因子对免疫细胞和肠上皮细胞的调节有关,表明益生菌可能有助于控制胃肠道通透性精神分裂症。使用短双歧杆菌的开放标签研究还报告了对精神分裂症焦虑和抑郁的积极影响,以及反应者中 TNF 相关激活诱导的细胞因子和 IL-22 水平的增加,尽管缺乏安慰剂组限制了这些发现。
DOI: 10.1016/j.heliyon.2024.e32004
益 生 元
益生元是选择性选择的不易消化的底物,用于靶向宿主微生物,使有益微生物能够茁壮成长,代表了调节微生物组-肠-脑轴的另一种方法。益生元存在于多种食物中,包括水果、蔬菜、全谷物和母乳。
通过促进双歧杆菌等有益细菌的生长,益生元可以增强肠道健康,并可能改善心理健康。FOS 和 GOS 是研究最多的益生元之一,研究表明它们能够调节肠道微生物组成和减少压力反应。
GOS三周:压力减轻,减轻IBS焦虑
补充 GOS 三周降低了健康志愿者的皮质醇觉醒反应,表明压力减轻,尽管没有改变压力和炎症或心理健康的生物标志物。
另一项研究发现,GOS 降低了 IBS 患者的焦虑评分,突出了益生元缓解肠道和情绪相关症状的潜力。
最近的一项随机对照研究表明,为期 14 天的高剂量益生元干预减少了超重成人在食物决策过程中与奖励相关的大脑激活,肠道微生物群同时发生变化,包括产短链脂肪酸的双歧杆菌科的增加,这表明益生元与与食物选择相关的大脑功能的调节之间存在潜在联系。
FOS/GOS 混合物:抗抑郁
临床前研究还证明了益生元的抗抑郁作用。FOS/GOS 混合物用于减轻子宫内 VPA 诱导的 自闭症小鼠模型的改变。益生元饮食使关键微生物分类群正常化,改善肠道通透性,恢复免疫平衡,减少神经炎症(特别是降低 CD68 表达),并增强 VPA 暴露后代的社会行为和认知。
给予小鼠的 FOS/GOS 混合物还能够降低皮质酮水平并增加海马体中的 BDNF 表达,这对情绪调节至关重要。这些发现表明,益生元不仅可以影响肠道微生物群,还可以影响与神经精神疾病有关的关键神经生物学途径。然而,关于益生元和心理健康的人类研究仍然有限,需要进一步的研究来充分了解它们的治疗潜力。
DOI: 10.1016/j.heliyon.2024.e34092
合生元:结合益生菌和益生元以增强效果
合生元结合了益生菌和益生元, 旨在通过为益生菌提供可发酵的底物来提高益生菌的生存和功效。这种方法在改善肠道健康和心理健康方面显示出前景。
瑞士乳杆菌和长双歧杆菌+GOS
一项针对轻度至中度抑郁症患者的研究表明,含有瑞士乳杆菌和长双歧杆菌 的合生元与 GOS 相结合,可降低抑郁评分并改善色氨酸代谢,色氨酸代谢是参与血清素产生的关键途径。
长双歧杆菌+母乳低聚糖(HMO)
在另一项研究中,由多菌株益生菌与益生元结合的合生元改善了帕金森患者的功能性胃肠道症状,表明肠道健康与神经退行性疾病的大脑相关结果之间存在联系。最近的一种合生元方法使用长双歧杆菌(婴儿)与母乳低聚糖(HMO)作为益生元,在成人肠道微生物组中实现了可预测的植入。
婴儿双歧杆菌,通常在成人中不存在,以 HMO 依赖性方式成功移植,无需任何抗生素预处理,覆盖高达 25% 的细菌种群并促进有益代谢物的变化。合生元还提高了丁酸盐水平并抑制了肠道病原体的生长,提供了一种潜在的新型治疗策略。虽然合生元在调节微生物组-肠-脑轴方面显示出潜力,但很少有研究采用这种策略,需要更大规模的临床试验来确定它们在各种精神和神经系统疾病中的疗效。
后生元:具有潜在益处的微生物的非活性制剂
后生元是无生命微生物和/或其成分的无生命制剂(例如,热灭活),可为宿主带来健康益处,作为一种调节肠脑轴的新方法而受到关注。
热灭活 L. gasseri :减轻学生考试压力
为期 12 周的热灭活 L. gasseri 干预减轻了学生与考试相关的压力,提高了焦虑分数、皮质醇水平和睡眠质量。
热灭活的副干酪乳杆菌:抗焦虑抗抑郁
临床前研究还表明,热灭活的 副干酪乳杆菌可以通过逆转与抑郁症相关的大脑区域降低的多巴胺水平来发挥抗抑郁和抗焦虑作用。
乳酸菌 LB 是一种源自发酵乳杆菌和德氏乳 杆菌发酵的后生元,已被证明会影响回肠离子转运和运动,可能有助于其对急性腹泻和 IBS 的治疗效果。与活益生菌相比,后生元具有许多优势,包括更长的保质期和可能增强的安全性。
微 生 物 代 谢 物
微生物代谢物是肠道微生物在新陈代谢过程中产生的有机化合物,通过免疫调节、炎症调节和肠道屏障维持影响宿主的生理和健康。
这些包括各种生化产品,如 SCFA、氨基酸和维生素。微生物代谢物在各种生物学功能中起着至关重要的作用,包括调节免疫反应、调节炎症和维持肠道屏障完整性。
补充SCFA可能有益于治疗神经退行性疾病
微生物衍生的 SCFA 对小胶质细胞活化的调节已被证明可以增强帕金森的病理生理学。
然而,另一项在帕金森背景下的研究表明,增加 SCFA 产生的高纤维饮食改善了运动缺陷,减少了 α-突触核蛋白聚集,并促进了帕金森模型中的保护性巨噬细胞亚群,这种影响被小胶质细胞耗竭消除了,突出了小胶质细胞依赖性肠-脑相互作用。
同样,在阿尔茨海默模型中,SCFA 治疗减少了小胶质细胞活化,改善了斑块负担,并在几项不同的研究中挽救了记忆障碍。综上所述,这表明 SCFA 治疗可能有益于治疗神经退行性疾病;但是,这也可能取决于这种情况发生在疾病的哪个阶段。
产丁酸盐细菌、SCFA补充改善神经炎症
对帕金森患者的进一步研究确定了 Blautia 和丁酸盐的缺陷;在帕金森模型中补充产丁酸盐细菌 B. producta 可减轻小胶质细胞介导的神经炎症,改善运动功能障碍并抑制小胶质细胞活化。
纤维缺乏的饮食也被证明与小胶质细胞、肠脑轴和认知障碍有关。在这里,长期纤维缺乏的小鼠表现出物体位置记忆、时间顺序记忆的缺陷和日常活动能力的降低。这些缺陷与海马炎症增加和小胶质细胞对突触的吞噬增加有关。
进一步的临床前研究表明,补充 SCFA 可以减少动物模型中的焦虑和压力相关行为,这可能是通过调节免疫和炎症途径。
粪菌移植:恢复肠道微生物平衡
FMT 涉及将粪便从人类或啮齿动物供体转移到个体或啮齿动物受体,以便移植指示供体的肠道微生物特征,转移肠道微生物组特征和/或表型以进行治疗或进一步研究。虽然 FMT 已成功用于治疗复发性艰难梭菌感染和溃疡性结肠炎等疾病,但其在精神疾病中的应用仍处于起步阶段。
然而,鉴于肠道疾病、炎症和心理健康之间的密切联系,FMT 正在被探索作为抑郁症和焦虑症的潜在治疗方法。
临床前研究表明,将肠道微生物群从抑郁个体转移到健康动物可以诱导类似抑郁的行为, 从而支持肠道微生物群在情绪调节中的作用。事实上,虽然一项研究报告在先天免疫细胞群或适应性免疫细胞群中没有观察到差异,但另一项研究发现 IL-1β 和 TNF-α 的产生减少,并且 Iba1 阳性小胶质细胞和 NLRP3 炎性小体的激活存在脑区域特异性抑制。
肠易激综合征与情绪障碍:FMT的双重治疗潜力
在人类中,小型临床试验表明,FMT 可能会缓解 IBS 患者的症状,IBS 患者通常与焦虑和抑郁共病。来自临床和动物研究的证据表明,FMT 可能通过减少全身炎症来发挥作用。在接受葡聚糖硫酸钠诱导的溃疡性结肠炎的小鼠中,来自健康供体的 FMT 改善了结肠炎症并恢复了肠道微生物群组成,同时降低了结肠促炎标志物 IL-1 和 IFNγ 的 mRNA 表达。
对溃疡性结肠炎患者的临床研究还表明,FMT 后血清 CRP 和炎性细胞因子(如 IL-6 和 IL-1Ra)以及炎性趋化因子 IP-10 和 ENA-78 显著降低,表明存在潜在的免疫系统调节。但需要更大规模的对照试验来充分评估 FMT 作为精神疾病治疗选择的潜力。
•
动物模型的局限与多组学人类队列研究的前景
动物模型为解析微生物组与宿主互作的生物学机制提供了重要工具,但其对人类复杂生理病理环境的还原度有限,尤其在神经免疫领域存在显著物种差异。因此,基于动物实验的发现需通过人类队列研究(如整合基因组、代谢组与免疫表型的多组学纵向研究)进行验证,并进一步探索遗传背景、社会环境及认知行为等混杂因素的调节效应。
此外,将研究扩展到包括跨物种研究,例如涉及斑马鱼的研究,可以提供对宿主-微生物组相互作用的进化见解,可以充分了解这些复杂的相互作用如何影响健康和疾病。
饮食调节微生物组:从日常饮食到功能性营养的转化医学路径
探索饮食干预,包括益生元和益生菌,以及评估不同的极端饮食,如纯素食与生酮饮食, 仍然至关重要。饮食是改变肠道微生物组的一种快速、安全和重要的途径,可能会影响免疫系统和大脑。最近的研究表明,增加膳食纤维摄入量可以提高认知能力,而精神益生菌饮食已被证明可以稳定肠道微生物组并改善健康个体的感知压力。肠道菌群检测可通过识别关键功能菌群(如产丁酸菌Faecalibacterium)的丰度,指导饮食方案的个性化定制。
个体化微生物干预:从健康到疾病的精准医学
必须区分这些干预措施如何影响健康个体和处于疾病状态的个体的免疫系统和肠道微生物组,从而能够制定个性化的治疗策略。关于 FMT 调节大脑和免疫系统相互作用的能力,尤其是在免疫介导的疾病中,仍然存在许多悬而未决的问题。了解其在不同健康环境中的影响可以开辟新的治疗途径。
肠道菌群检测结合宿主基因组(如APOE ε4)、血浆炎症标志物(如CRP)及脑脊液生物标志物(如Aβ42/tau),可构建多维治疗响应预测模型。
技术创新与数据革命
正在开发新技术,这些技术使用一种高通量、不依赖培养物的方法,测量外周血中针对肠道共生细菌的全身 IgG,能够突出炎症性疾病中微生物组与免疫系统之间的相互作用;未来可能会使这项技术适应神经系统疾病。
在数据分析中使用人工智能可能会彻底改变我们对微生物组-肠道-免疫-大脑相互作用的理解,并增强疾病预测、治疗策略和个性化医疗。
鉴于处理大量生物数据的能力不断扩大,将有机会更全面地整合免疫-微生物相互作用,肯定会产生更有效的治疗方法。机器学习和深度学习可以通过处理生活方式因素、免疫标志物读数、先天信息和宿主数据来设计量身定制的医疗干预措施,从而提高诊断的准确性和治疗的有效性。
主要参考文献:
Pan I, Umapathy S. Probiotics an emerging therapeutic approach towards gut-brain-axis oriented chronic health issues induced by microplastics: A comprehensive review. Heliyon. 2024 May 28;10(11):e32004.
O’Riordan KJ, Moloney GM, Keane L, Clarke G, Cryan JF. The gut microbiota-immune-brain axis: Therapeutic implications. Cell Rep Med. 2025 Mar 18;6(3):101982.
Ashique S, Mohanto S, Ahmed MG, Mishra N, Garg A, Chellappan DK, Omara T, Iqbal S, Kahwa I. Gut-brain axis: A cutting-edge approach to target neurological disorders and potential synbiotic application. Heliyon. 2024 Jul 4;10(13):e34092.

谷禾健康

抗生素可有效治疗细菌感染,但其广泛使用引发了抗生素耐药性,抗生素耐药性是个全球性的大问题,这与人类疾病密切相关。这里存在着一个被我们长期忽视的关键因素:肠道耐药组。
当我们在医学上使用抗生素时,肠道里的微生物数量和基因组成可能会发生变化,这样就增加了抗生素耐药性在肠道中积累的风险。
肠道耐药组(gut resistome)是指肠道微生物群中存在的所有抗生素耐药基因(ARG)的集合,可以从自然环境和食物摄入中获得,并且可以随着食物中抗生素和其他抗菌物质诱导的选择压力进一步积累。
这些耐药基因不只是让细菌对抗生素产生抵抗力,更可能通过影响肠道微生物平衡、改变代谢功能、触发炎症反应,与多种慢性疾病建立起令人担忧的联系,包括代谢紊乱、心血管疾病、肝病、神经系统疾病等。
当我们服用口服抗生素时,药物直接穿过消化系统,破坏肠道微生物的平衡,促进抗生素耐药基因的出现和扩散。更令人忧虑的是,这些耐药基因可以通过食物链、水源和环境污染从自然环境进入人体,并在肠道微生物间通过水平基因转移快速传播。
肠道耐药组的富集不仅加剧了抗生素耐药性,还导致了多重耐药性感染的出现。这些感染构成了重大的公共卫生挑战,因为它们更难治疗,通常需要更昂贵的疗法,这些疗法可能具有更大的副作用且效果较差。
肠道耐药组已成为连接抗生素使用、耐药危机和慢性疾病的关键环节。
已知膳食成分会影响肠道微生物组的发生率、活性和多样性,例如,研究表明,地中海饮食(大量摄入完整的、未加工的植物性食物、橄榄油和乳制品;适量的家禽和鱼类;以及最少食用红肉)与粪杆菌属和短链脂肪酸的增加有关,它们具有抗炎作用。相比之下,西方饮食(高热量,富含动物蛋白、饱和脂肪、单糖和超加工食品,而纤维、水果和蔬菜含量低)与 Blautia、拟杆菌属、瘤胃球菌属物种的丰度较高有关,这与代谢紊乱和慢性炎症的风险增加有关。
在此基础上,饮食不仅会影响有益细菌,而且还有可能改变肠道中抗生素耐药细菌的存在。到目前为止,已发现食品中的生物活性常量营养素、植物化学物质和益生菌可以改变肠道中的抗生素耐药细菌,为调节肠道耐药组的功能性食品打开了新的研究窗口。
随着科学进步,肠道菌群检测技术已能够识别耐药基因的存在和丰度,使个体ARG状况的评估成为可能,为个性化的肠道耐药组管理提供客观依据。
本文强调了肠道耐药组与人类疾病(如代谢紊乱、心血管疾病、肝病和神经系统疾病)之间的联系,包括抗生素作用机制和耐药性的发展,还讨论了饮食习惯和饮食成分(包括生物活性宏量营养素、植物化学物质和益生菌)通过增强抗生素疗效和潜在降低耐药性,对肠道抗药组的组成产生影响。
强调了对靶向肠道耐药组的功能性食品的新兴趋势,以及越来越关注具有调节抗生素耐药性潜力的生物活性植物化学物质。
靶向肠道耐药组的功能性食品,正从概念走向临床,未来,通过“膳食-菌群-肠道耐药组”三位一体的精准干预,有望将耐药危机防控窗口前移至日常饮食,从功能性食品的创新到临床营养的精准化应用,为全球公共卫生提供可持续解决方案。
★
随着抗生素在现代医学和农业中的广泛应用,一个隐形的健康危机正在悄然形成——肠道抗生素耐药基因组(ARG)的扩张。
ARG 可以从亲本细菌(垂直基因转移)遗传,也可以通过水平基因转移从其他微生物获得,水平基因转移是遗传物质在细菌之间移动的过程,甚至跨物种移动。
这些耐药基因不仅可以使细菌对抗生素产生抵抗力,更可能通过影响肠道微生物平衡、改变代谢功能、触发炎症反应,与多种慢性疾病建立起令人担忧的联系。
给药方法会显著影响肠道微生物组和耐药组
静脉注射(注射到血液中)或局部(直接涂抹在皮肤上)的抗生素通常与肠道微生物组的直接接触最少,因此对肠道耐药组的影响较小。
然而,口服抗生素是最常见的治疗形式,它通过消化系统,直接影响肠道微生物群。这种相互作用会破坏肠道微生物的平衡,并促进抗生素耐药基因(ARG)的出现。
Zhang等人强调了这一差异,表明小鼠口服四环素或阿莫西林会迅速增加肠道中ARG的存在,而静脉注射则会延迟或减少这种作用。这种差异与药物的排泄途径有关:氨苄西林主要通过肾脏排泄,限制了静脉注射后的肠道暴露,而四环素通过肾脏和胆汁排出,即使在静脉注射后也会暴露肠道。这些发现强调了口服抗生素在塑造肠道抗药性中的重要作用。
此外,抗生素的广泛使用,无论是直接在人类身上还是间接在动物身上,都会导致抗生素耐药性在自然环境和生物中的积累,从而促进人类肠道耐药性的扩大。
严重的健康问题与肠道耐药组有关,包括代谢紊乱、心血管疾病、肝病、神经系统疾病(下图)。
肠道耐药组对人体健康的影响
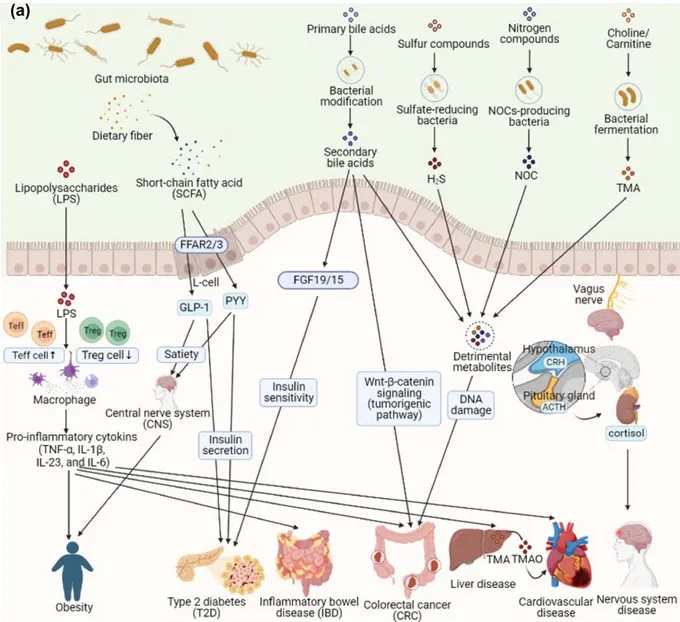
doi:10.1111/1541-4337.70143
a) 肠道微生物群在健康和疾病中的机制。短链脂肪酸(SCFA)来源于膳食游离脂肪酸受体(FFAR2/3),通过释放肽YY(PYY)和胰高血糖素样肽1(GLP-1)等肠道激素来促进饱腹感,同时增强胰岛素分泌和敏感性,导致肥胖和2型糖尿病(T2D)。来自革兰氏阴性菌的脂多糖(LPSs)通过激活巨噬细胞和释放促炎细胞因子(TNF-α、IL-1β、IL-6)触发全身炎症,导致肥胖、2型糖尿病、炎症性肠病(IBD)、癌症(CRC)、心血管疾病和肝脏疾病。次生胆汁酸、三甲胺-N-氧化物(TMAO)、硫化氢(H2S)和含氮有机化合物(NOC)会导致胆汁酸信号失调、氧化应激和DNA损伤,从而导致CRC等慢性疾病。此外,肠道和中枢神经系统(通过迷走神经)以及肠道衍生代谢物(如三甲胺(TMA)和TMAO)之间的相互作用会影响神经和心血管健康。
下面我们来看具体与肠道耐药组相关的疾病。
抗生素耐药性通过破坏肠道微生物群及其相关代谢途径,在包括肥胖、2型糖尿病(T2D)、炎症性肠病(IBD)和结直肠癌(CRC) 在内的代谢紊乱中发挥着至关重要的作用。肠道耐药组是肠道微生物组的一部分,也与这些疾病的发展有关。
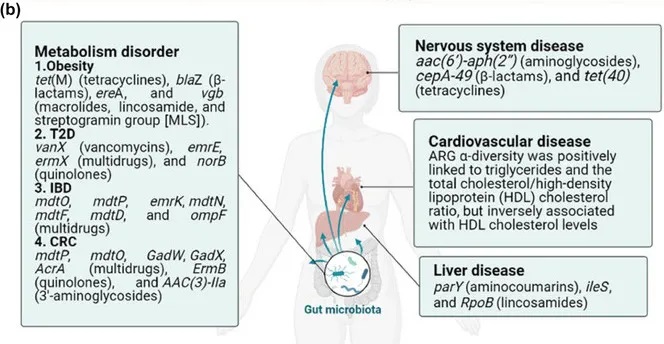
doi:10.1111/1541-4337.70143
b) 抗生素抗性基因对疾病的影响。特定的抗生素抗性基因(ARG)与代谢紊乱、神经系统疾病和肝病有关。ARGα多样性与心脏代谢风险增加有关。
肥胖
在肥胖人群中,肠道生态失调通过改变短链脂肪酸和脂多糖(LPS)影响能量代谢和慢性炎症(图a)。短链脂肪酸通过刺激GLP-1和PYY等激素的释放来调节饱腹感,这些激素作用于下丘脑。此外,肠道细菌的不平衡,特别是革兰氏阴性菌如变形杆菌的丰度增加,会导致LPS水平升高,并通过增加促炎细胞因子(TNF-α、IL-1β、IL-6)的产生引发全身炎症。
一项研究中,与富营养化参与者相比,肥胖个体中梭杆菌、肠球菌、大肠杆菌的丰度更高,同时ARG水平升高,如tet(M)(四环素类)、blaZ(β-内酰胺类)、ereA和vgb(大环内酯类、林可酰胺类、链菌素组[MLS])。这表明ARG与革兰氏阴性菌之间的关系,是由拟杆菌门和变形杆菌门之间的不平衡所驱动的。
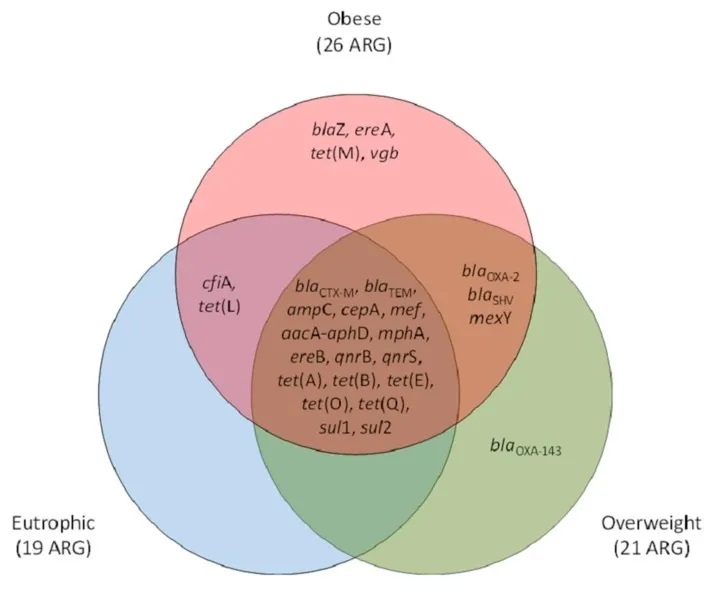
doi: 10.3390/genes10050349
2型糖尿病
在2型糖尿病中,肠道微生物失调,包括抵抗体的变化,导致SCFA产生减少,损害PYY和GLP-1的分泌,这对胰岛素调节和胰高血糖素水平至关重要(图a)。
胆汁酸代谢的改变刺激成纤维细胞生长因子19/15(FGF19/15)的释放,进一步促进胰岛素敏感性和葡萄糖耐量。
ARG丰度的增加,包括Vancomycin_vanX、Multidrug_emrE、MLS_ermX、喹诺酮_norB,与2型糖尿病风险的增加有关,耐药组的变化发生在肠道微生物群变化之前(图b)。
在IBD中,能够产生短链脂肪酸的细菌数量减少,这会削弱短链脂肪酸的抗炎作用。短链脂肪酸通常有助于Treg和效应性T细胞(Teff)的分化。由于这种减少,炎症和肠道上皮细胞损伤会加剧,从而为有害细菌的滋生创造条件,并导致慢性炎症的持续。大肠杆菌中携带的抗生素抗性基因,如mdtO、mdtP、emrK、mdtN、mdtF、mdtD、ompF,变得更加普遍。这些ARGs通过破坏肠道屏障功能,增加肠道的通透性,进而促进炎症反应(如图b所示)。
此外,ARGs和细菌的毒力因子可以通过水平基因转移在微生物群落中传播,进一步加重IBD中的炎症状况。
在结直肠癌中,肠道菌群失衡会通过减少粘液层厚度和增加肠道通透性来促进癌症的发生和发展(图a)。
有害细菌过度生长会导致脂多糖(LPS)和其他有毒代谢物的释放,比如次生胆汁酸、三甲胺-N-氧化物(TMAO)、硫化氢(H₂S)和含氮有机化合物,这些物质会引发低度炎症并激活致癌信号。

上述研究发现,结直肠癌患者体内大肠杆菌、柠檬酸杆菌、不动杆菌水平升高,同时存在耐药基因如mdtP、mdtO、GadW、GadX、AcrA(多药耐药)、ErmB(喹诺酮类耐药)和AAC(3)‐IIa(3′-氨基糖苷类耐药)等耐药基因水平升高(图b)。这些抗生素抗性基因通过加剧肠道菌群失衡、炎症反应和肠道屏障功能障碍,进一步推动CRC的发展,突出了肠道耐药组在代谢性疾病发病机制中的作用。
总体而言,新出现的证据表明,肠道耐药组不仅反映了微生物失衡,而且通过抗生素耐药性的传播积极推动代谢紊乱。ARG在肠道微生物群中的存在和传播会破坏关键的生理过程,如能量代谢、免疫调节和肠道屏障的完整性。通过营造一个有利于致病菌及其毒力因子的环境,耐药组会放大炎症,改变代谢信号通路,并促进生态失调。
肠道微生物组通过炎症反应和TMAO代谢机制参与心血管疾病的发病机制,包括心力衰竭和冠状动脉疾病(图a)。
心血管疾病患者体内产丁酸菌减少,这会引发局部炎症,加剧肠道菌群失衡,并导致肠道屏障功能受损。这些患者体内TMAO水平的升高,会干扰胆固醇运输、促进泡沫细胞形成和诱导血小板聚集,从而可能导致急性冠状动脉综合征,引发动脉粥样硬化的发展。
此外,较高的肠道抗生素抗性基因α多样性与心脏代谢风险增加有关。
一项研究发现,较高的肠道ARGα多样性指数与甘油三酯和总胆固醇/高密度脂蛋白胆固醇比值(TC/HDL)呈正相关,但与高密度脂蛋白胆固醇(HDL)水平呈负相关。
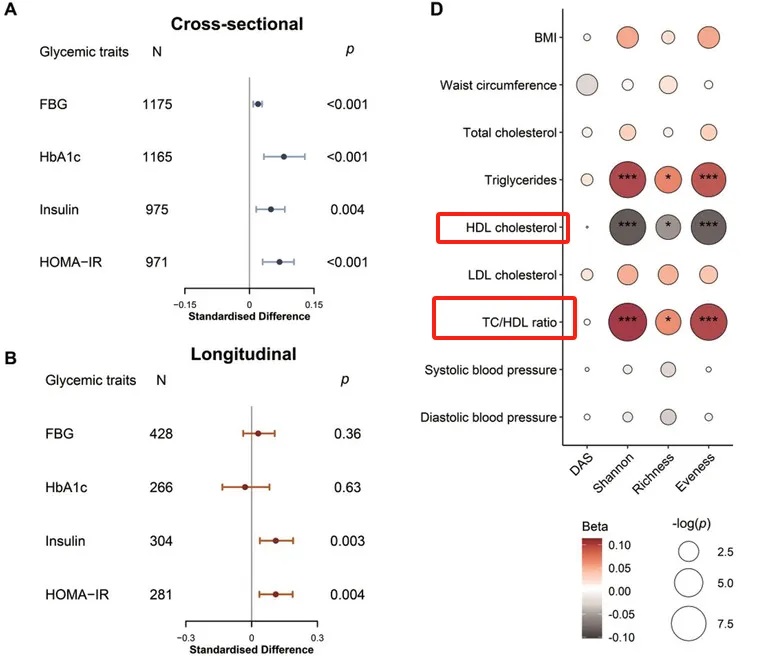
doi: 10.1002/advs.202104965
这表明ARG在扰乱脂质和葡萄糖代谢方面发挥作用,并可能通过血脂异常和胰岛素抵抗增加心血管疾病的风险。
肠道微生物群通过引发炎症反应,显著影响肝脏的正常功能(图a)。
当肠道微生物组因抗生素耐药性发生变化时,肠道的通透性会增加,使细菌及其产物(如脂多糖LPS)能够通过肝肠循环进入肝脏。LPS进入肝脏后,会引发急性炎症反应,导致肝细胞受损,进而可能引发多种肝脏疾病,如非酒精性脂肪肝、脂肪性肝炎、酒精性肝病、肝癌,甚至肝性脑病。
肠道耐药组的变化与肝脏疾病的发生密切相关。研究发现,肝硬化患者的肠道耐药组中,与肠杆菌科、链球菌、肠球菌和不动杆菌属等病原菌相关的耐药基因丰度更高。
此外,这些患者还表现出对β-内酰胺酶、大环内酯类、喹诺酮类、糖肽类、磷霉素、四环素类抗生素的更大耐药性。肝性脑病患者则表现出更高水平的氨基香豆素耐药基因parY和林可酰胺耐药基因(ileS和RpoB)。这些抗性组的变化会破坏肠道与肝脏之间的正常相互作用,以及胃肠道的免疫反应,随着疾病的进展,这种破坏会进一步恶化。
肠道微生物组在通过肠道-大脑轴塑造神经系统的发育和功能方面起着关键作用。迷走神经传递神经元、内分泌和免疫信号,而下丘脑通过释放促肾上腺皮质激素释放激素(CRH)来调节应激反应,导致皮质醇的产生,从而影响肠道的上皮屏障和免疫系统。
肠道耐药组的改变也与神经发育障碍有关。
一项研究发现,患有自闭症的儿童具有更高丰度的屎肠球菌aac(6′)-aph(2′)基因、Megasphaera elsdenii 的cepA‐49和tet(40)基因以及脆弱拟杆菌的cepA-49基因。

研究还发现,健康个体和自闭症患者在对大环内酯类抗生素耐药性的基因方面存在显著差异,包括肠球菌和未培养细菌的氨基糖苷类耐药基因、拟杆菌属和Acidaminococcus的β-内酰胺抗性基因,以及来自Megasphaera和Alistipes的四环素抗性基因。因此,肠道微生物群平衡和抗性基因的破坏可能会降低神经代谢潜力,影响大脑功能所需的神经递质和代谢物的产生。
通常用抗生素治疗的疾病与扩大的肠道耐药性有关,这表明曾经有接触抗生素史对疾病相关菌株的ARG获得产生了相当大的选择压力。
未来的研究需要更好地了解肠道耐药性与疾病之间的相互作用及其机制。将多基因风险评分与肠道宏基因组风险模型相结合可以提高对常见慢性病的预测能力,为疾病预测和预防提供更全面的方法。
通过上述研究,我们看到肠道耐药组对人体健康的多层面影响,是如何超越简单的感染问题而演变成一个系统性的健康风险。然而,要有效应对这一挑战,我们需要深入了解:这些耐药基因是如何产生的?它们又是如何从环境进入人体并在肠道微生物间传播的?…
★
抗生素耐药性并非凭空出现,而是有其明确的生物学机制和传播途径,下面章节我们来详细了解这一过程中的关键环节,包括垂直遗传、水平基因转移等核心机制,以及食物链、水源和环境污染在耐药基因传播中的角色。
在这里重点介绍针对细菌的抗生素,介绍它们的作用机制,并讨论抗生素耐药性如何在肠道中发展和积累。
将治疗细菌性疾病的抗生素引入临床应用是 20 世纪最大的医学突破。抗生素的抗菌机制可细分为五类:
抗生素作用机制和抗生素耐药性
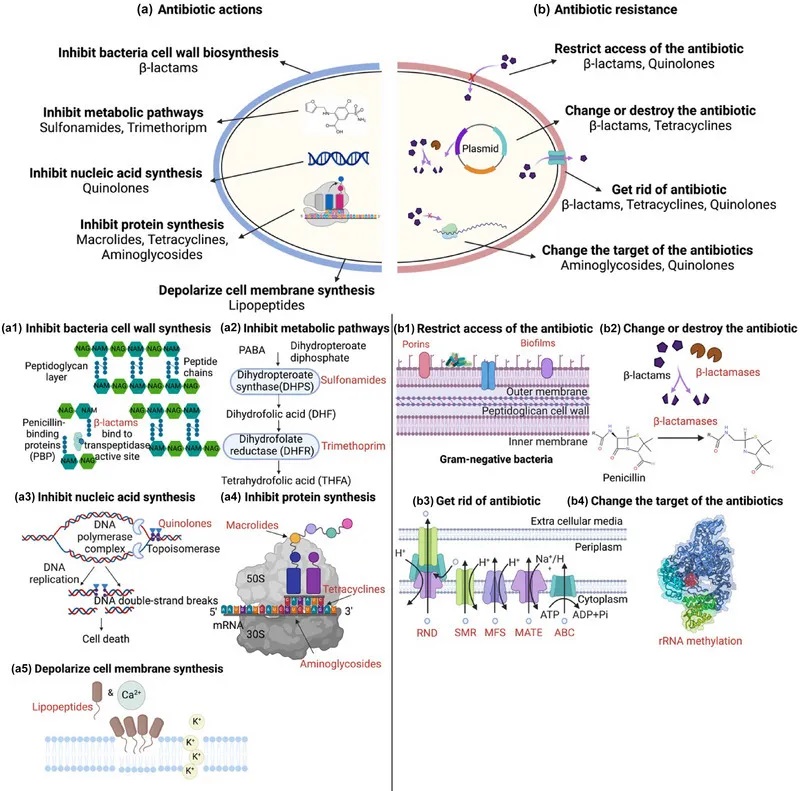
doi:10.1111/1541-4337.70143
a)抗生素作用:抗生素针对关键的细菌过程来抑制生长和存活。
b)抗生素耐药性:细菌通过各种策略产生耐药性。
下面我们先来看一下抗生素的作用。
阻碍细菌细胞壁的合成——结构性破坏
β-内酰胺类抗生素,可以伪装成细菌细胞壁合成所需的关键分子。这类抗生素会:
• 模仿细菌细胞壁合成的天然底物(d-丙氨酸)
• 结构相似性使抗生素能够与DD转肽酶结合(d-丙氨酸是DD转肽酶的底物),防止DD转肽酶与其天然底物进一步结合
• 破坏肽聚糖层的交联,肽聚糖框架减弱
• 最终导致细胞壁结构损坏,细胞死亡
典型的β-内酰胺类抗生素包括:青霉素类、头孢菌素类、碳青霉烯类、单巴坦类。
抑制细菌代谢途径——叶酸合成阻断
磺胺类药物抑制二氢蝶酸酯合酶,这是叶酸生产中必不可少的酶。这种抑制阻断了DNA和RNA生产所必需的核苷酸的合成,阻止了细菌复制并导致细胞死亡。
最常用的磺胺类抗生素:磺胺甲氧基嘧啶、磺胺二甲嘧啶、磺胺甲恶唑、磺胺嘧啶。
协同用药:甲氧苄啶(靶向二氢叶酸还原酶,DHFR)与磺胺类联用,双重阻断叶酸代谢链。
抑制核酸生物合成所需的细菌RNA聚合酶
作用靶点:细菌RNA聚合酶β亚基
机制:阻断转录起始→抑制mRNA合成→细胞分裂停滞
代表药物:利福霉素类(如利福平)
作用靶点:DNA旋转酶(拓扑异构酶II/IV)
机制:
与 DNA 和旋转酶结合→阻止DNA超螺旋/松弛
捕获DNA上的拓扑异构酶→抑制核酸合成→DNA双链断裂→抑制细菌的生长
代表药物:喹诺酮类(诺氟沙星、环丙沙星、氧氟沙星)
阻断蛋白质合成
大环内酯类(红霉素、克拉霉素、阿奇霉素)
与50S核糖体亚基上的23S rRNA产生的结合肽基转移酶中心结合→抑制核糖体沿mRNA的运动→多肽链延伸终止→细菌蛋白质合成的抑制→细菌细胞死亡
四环素类(四环素、土霉素、多西环素)
靶点:与30S核糖体亚基结合
阻断氨酰-tRNA与A位点结合→抑制氨基酸掺入生长的多肽链
氨基糖苷类(卡那霉素、链霉素、庆大霉素)
靶点:30S核糖体16S rRNA上的 A位点
诱导mRNA误读→多肽链提前终止→破坏蛋白质合成完整性→细胞死亡
破坏细胞膜完整性
脂肽类抗生素(达托霉素)
结构特征:含脂尾的肽核
钙离子依赖型寡聚→达托霉素分子嵌入细胞膜形成孔道→膜电位去极化→胞内成分泄漏→细胞裂解
不同种类抗生素作用的方式

Uddin TM, et al., J Infect Public Health. 2021
细菌抗生素耐药性是微生物数千年进化的自然现象。这种固有的抗生素耐药性也被称为固有耐药性。
除了固有耐药性外,过度使用抗生素还会导致抗生素耐药性的积累。细菌采用各种策略来抵抗抗生素,例如减少抗生素的摄取,修饰或灭活抗生素,主动将其从细胞中排出,或改变抗生素的靶标以阻止其作用。
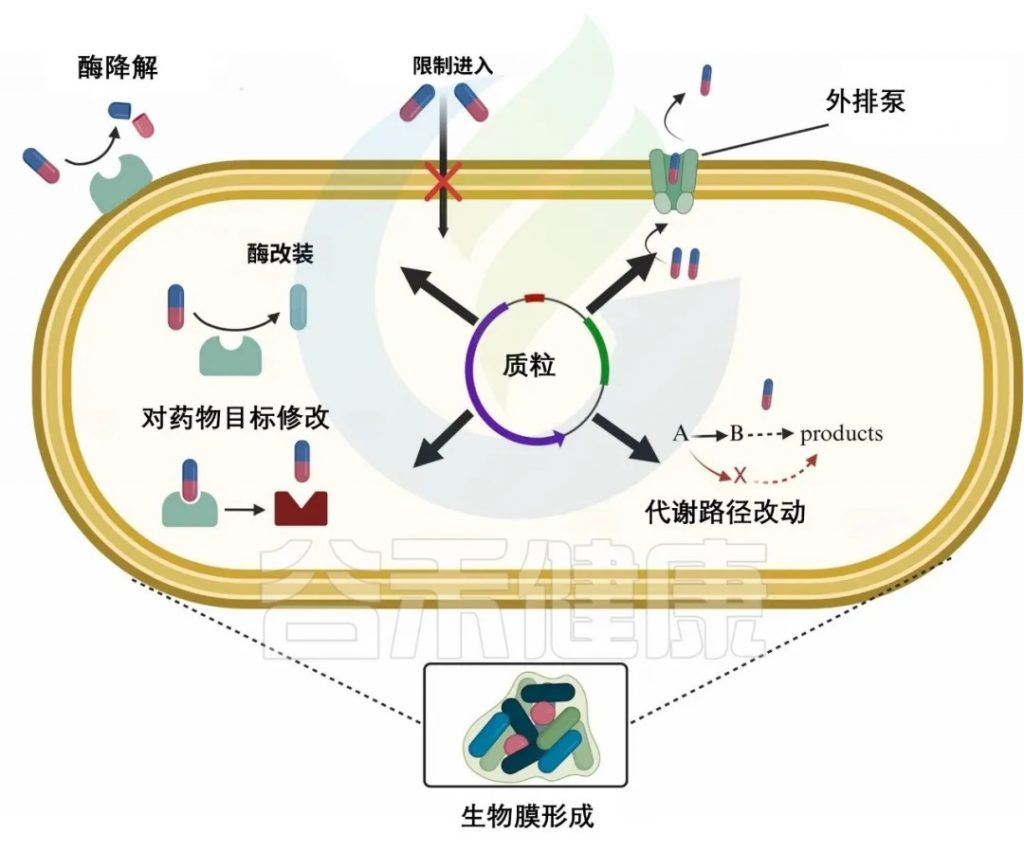
doi.org/10.1016/j.glmedi.2024.100081
细菌耐药核心机制如下:
1. 细菌改变外膜疏水性、孔蛋白突变或形成生物膜,阻止抗生素渗透,导致耐药性
革兰氏阴性菌有一层外膜,可以作为屏障,阻止抗生素(β-内酰胺类、喹诺酮类)进入细胞。大多数抗生素需要穿透外脂膜才能到达目标。疏水性药物如大环内酯类(红霉素)可以通过扩散穿过膜,而亲水性抗生素如β-内酰胺类需要通过孔蛋白(图b1)。
外膜的变化,如疏水性的改变或孔蛋白的突变,可能导致革兰氏阴性菌产生抗生素耐药性。一些细菌可以产生大量的细胞外聚合物,包括胞外多糖、蛋白质、细胞外DNA和脂质。这些物质可以在生物膜内的细菌细胞周围形成物理屏障,防止抗生素直接到达细菌细胞。这减少了抗生素的直接暴露,导致抗生素疗效明显降低。
例如,金黄色葡萄球菌菌株产生的生物膜可以显著降低苯唑西林、万古霉素和头孢噻肟的渗透性。
2. 细菌产生分解抗生素分子的酶,对抗生素修饰或降解,使其无效
β-内酰胺酶
细菌产生β内酰胺酶来降解β内酰胺类,这些酶通过不可逆地打开β-内酰胺环使其失活,从而使其无法与青霉素结合蛋白结合,使其无效。
氨基糖苷修饰酶
如AAC(3)乙酰转移酶修饰庆大霉素,降低其与核糖体结合能力,从而降低其疗效。
3. 细菌外排泵,以能量依赖性方式从细菌细胞中去除抗生素,使细菌在高浓度抗生素中存活
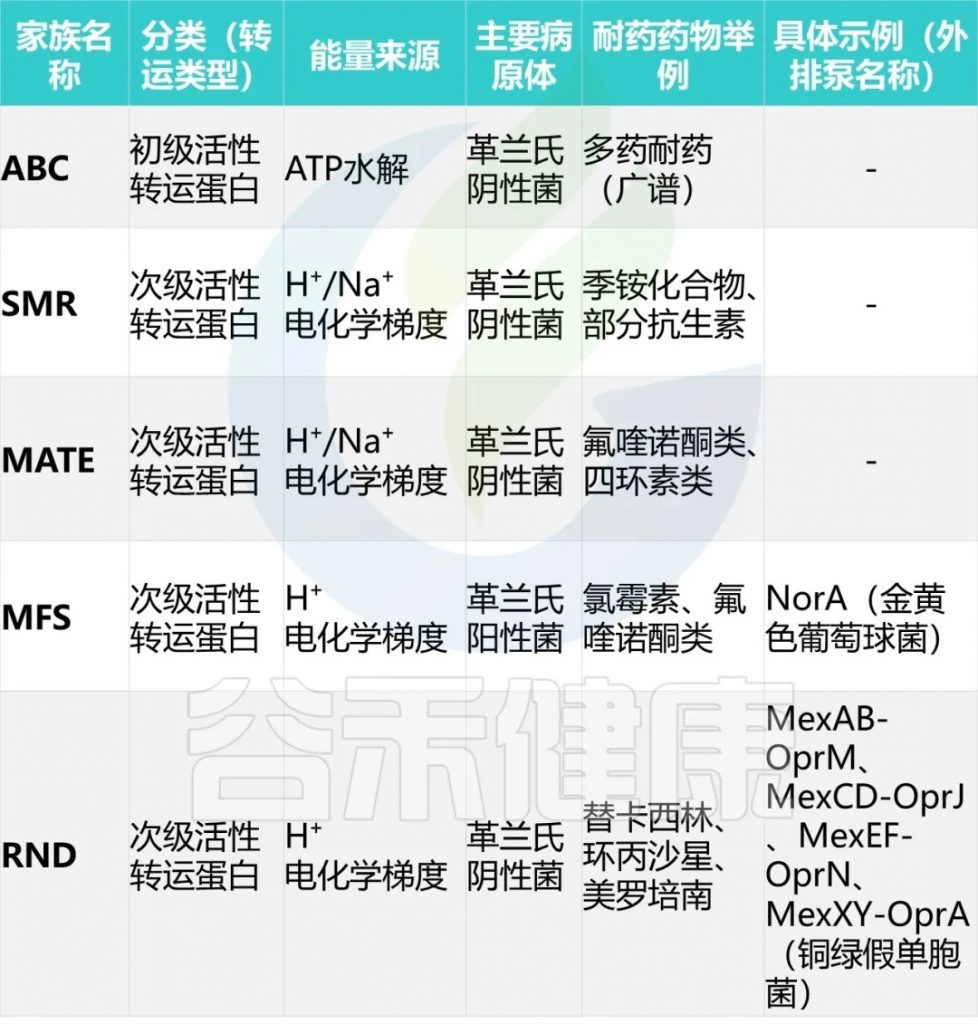
细菌药物外排泵可分为五个家族——ABC、SMR、MATE、MFS、RND。
4. 改变抗生素的靶点结合位点,降低结合效率
16S rRNA甲基转移酶改变氨基糖苷类核糖体结合位点的构象,从而降低氨基糖苷类对核糖体的亲和力,使抗生素更难有效结合。
氨基糖苷修饰酶可分为两类,甲基化G1405和A1408。
G1405甲基化:改变了核糖体RNA在这些4,6-二取代2-脱氧链霉素(DOS)氨基糖苷类关键结合位点的结构,如卡那霉素、庆大霉素、妥布霉素、阿米卡星。
A1408甲基化(NpmA酶):可对4,5和4,6二取代的2-DOS氨基糖苷类产生耐药性。
随着分子技术的进步,了解抗生素耐药性背后的机制变得越来越复杂。其中,基因测序已成为一种强大的工具,能够根据遗传数据预测抗性表型。这允许更精确地跟踪耐药性演变,并为有针对性的干预措施提供信息,以防止耐药菌株的传播。
此外,细菌采用多种策略来确保其存活,即使是少量耐药菌株也可能在胃肠道的复杂微生物环境中持续存在,从而导致耐药细菌种群的选择和存活。
了解分子耐药机制对于减轻抗生素耐药性和揭示人体肠道中耐药性积累的动态是必要的。
抗生素耐药性发生在自然环境,如土壤、水生态系统、动物粪便,在社区环境中也有,如医院污水、农业实践中。当人类食用食物中的抗生素耐药细菌时,抗生素耐药性可以通过食物链从自然环境传播给人类,比如,通过受污染的食物和直接的公共接触传播给人类。
环境中抗生素耐药性的累积
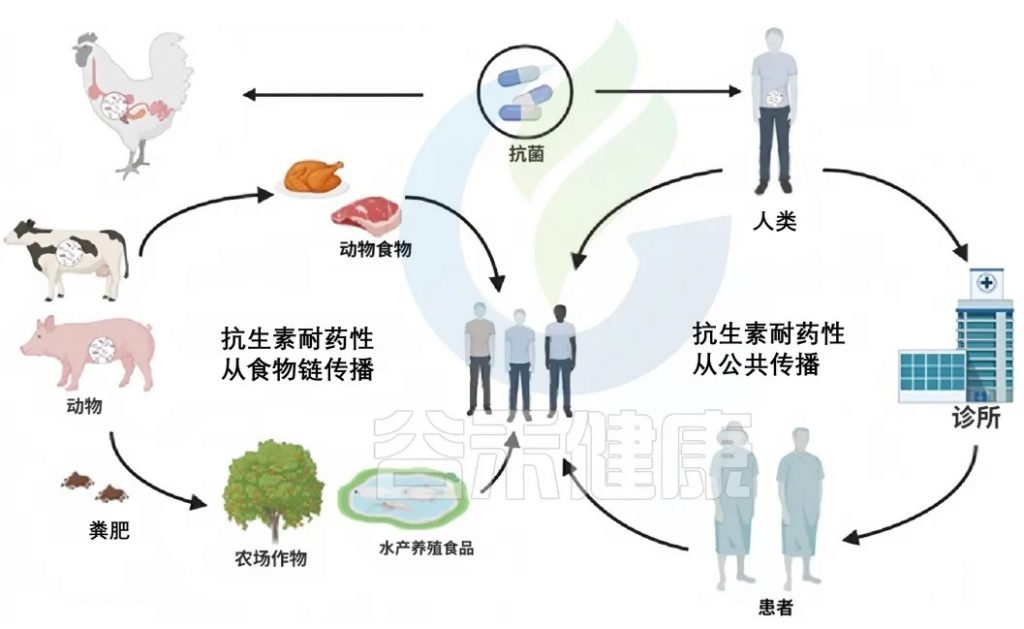
doi:10.1111/1541-4337.70143
在自然环境中,不同的生物和基质(如动物、土壤和水)之间存在联系。这种相互联系促进了抗生素耐药性在各种环境因素中的传播。土壤、水和沙子是抗性细菌和遗传元素可以持续存在并相互作用的水库。
整个自然界就像一张巨大的“互联网络”——动物、土壤、水甚至沙子之间都有看不见的“网线”连接。这些“网线”让抗药细菌和它们的耐药基因能在环境中四处“串门”。
抗生素耐药性从环境传播到植物、动物和昆虫涉及多种机制。农业实践,包括在畜牧业中使用抗生素和将处理过的动物粪便施用到田地里,将抗性基因引入土壤。水源可能通过农田径流,特别是那些用牲畜粪便施肥的农田径流,被抗生素抗性细菌和遗传元素污染。水生生态系统中的昆虫和其他生物可以成为抗性基因的载体,进一步在环境中传播抗生素抗性。
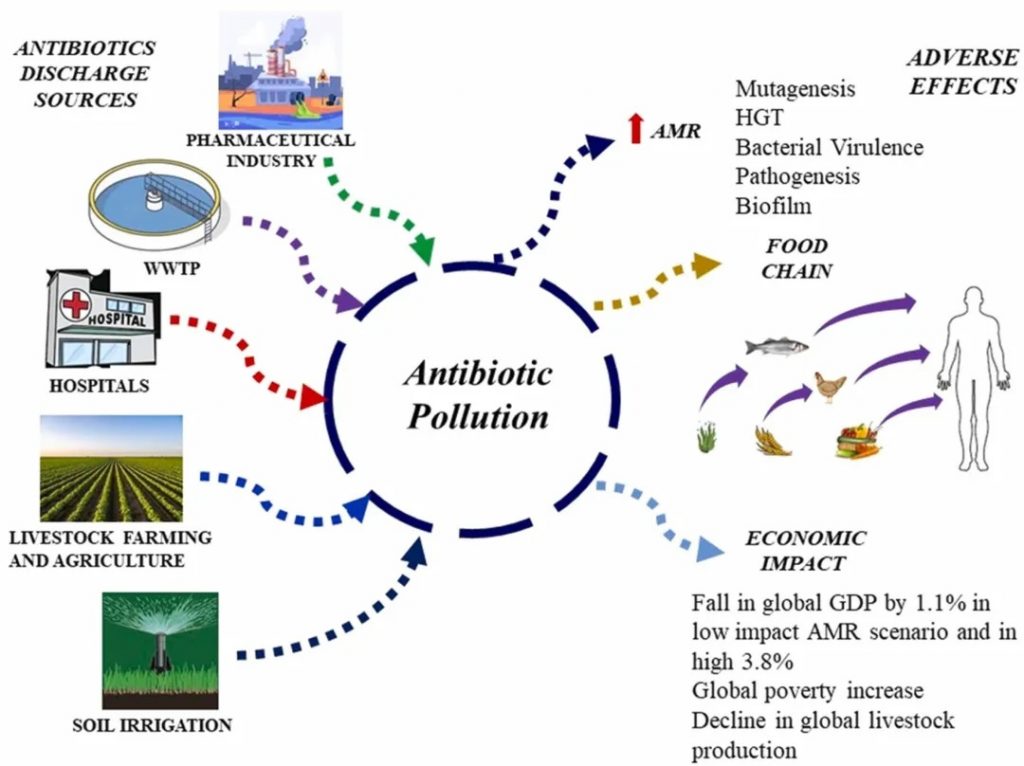
doi.org/10.1016/j.hazl.2024.100105
人类活动对抗生素排放到环境中起着重要作用。这通过多种途径发生,包括城市和医院废物、工业制造、农业径流和垃圾填埋场渗滤液。因此,抗生素耐药性可能通过各种途径进入人体肠道。人类可能通过受污染的食物、水和与环境的直接接触(如家庭、社区或医疗保健互动)接触到抗生素抗性细菌和遗传因素。
宠物狗和猫可携带多种多重耐药细菌,包括耐甲氧西林金黄色葡萄球菌和耐β-内酰胺类抗生素的肠杆菌科细菌。
这些耐抗生素微生物可能通过直接接触、体外寄生虫和气溶胶从宠物传给人类。特别是弱势群体,包括免疫功能低下的老年人和婴儿,可能面临更大的风险。
详见我们之前的文章:宠物猫可能塑造人类肠道中的抗生素耐药性和益生菌
ARG在自然环境、动物和人类中的发生、积累和传播依赖于垂直遗传和水平基因转移(下图)。在细菌的生长和繁殖过程中,基因突变使它们对特定抗生素产生耐药性,它们的后代将继承突变基因(下图a)。携带突变基因的细菌,更有可能存活,并且可以世代垂直传播。随着时间的推移,细菌种群将由具有耐药基因的细菌主导,这种选择性优势使耐药细菌得以增殖,有助于抗生素耐药性的扩大。
ARG具有高流动性,可以通过水平基因转移传播,包括四种机制:基因转化、基因转导、基因接合、膜囊泡融合(下图b)。
通过垂直遗传、水平基因转移以及影响这些机制的因素在肠道中积累抗生素耐药性
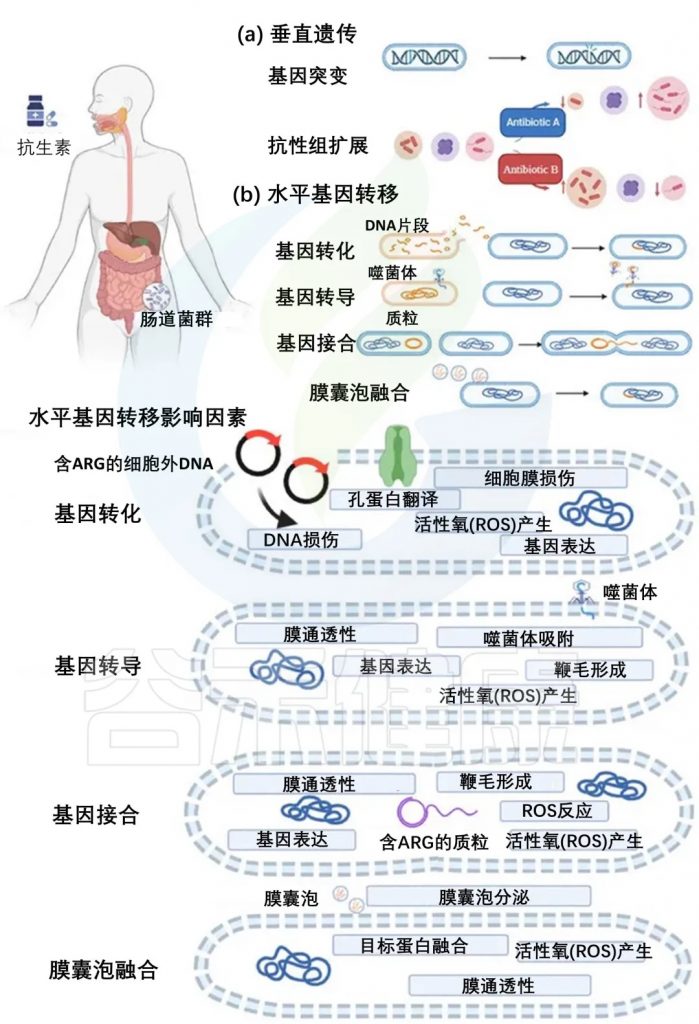
doi:10.1111/1541-4337.70143
a) 垂直遗传:抗生素耐药性可以通过基因突变和耐药体扩增进行垂直遗传。
b) 水平基因转移及其影响因素:
水平基因转移发生在四种途径中:
抗生素可以影响细菌中的水平基因转移,特别是在ARG的情况下。它们创造了一个有利于携带抗性基因的细菌存活和增殖的选择性环境。除了抗生素选择压力外,各种非抗生素因素,包括抗菌剂和环境条件,都会影响ARG的水平转移。
在大多数细菌中,获取抗性基因的主要和最有效的方法涉及使用可移动的遗传元件,如质粒作为基因交换的载体。在多种细菌中观察到水平基因转移,如大肠杆菌和肺炎克雷伯氏菌,使耐药基因能够在细菌种群中快速有效地传播。一般来说,ARG的水平转移会加剧肠道中抗生素耐药性的传播。
总体而言,垂直遗传和水平基因转移是导致ARG在自然环境、农场动物和最终人类肠道中发生和传播的两条主要途径。值得注意的是,ARGs的水平基因转移是决定其传播的关键因素。
饮食干预在抑制ARGs的水平基因转移中起着重要作用。已经发现,一些膳食植物化学物质,如酚类化合物、硫化物、萜类化合物、生物碱,有可能诱导ROS的产生,导致细菌细胞膜不可逆的损伤,生物膜活性降低,毒力基因表达下调,从而抑制水平基因转移。下面章节将进一步讨论食物成分在控制ARG中的作用。
★
通过饮食成分改变肠道耐药性是一个新兴的研究领域,人们越来越关注食物选择如何影响肠道中的抗生素抗性细菌。例如,发酵食品中含有大量微生物,乳酸菌和凝固酶阴性葡萄球菌被确定为四环素、青霉素、氯霉素和大环内酯类ARG的携带者。这表明食用发酵食品可能会直接影响肠道耐药性。
食品中存在的许多天然分子,如酚类、萜类和酚酰胺,具有抗菌能力,能够调节肠道微生物群。然而,关于食物成分对肠道抗生素耐药细菌影响的研究有限,这是一个新兴的研究领域。
这里总结了对大量人群的营养研究,以了解饮食习惯对肠道耐药性的潜在影响,并强调了选定类别的饮食成分对抗生素耐药细菌的调节作用。
长期的饮食习惯会影响肠道微生物群和肠道抵抗力。
Stege等人(2022)选择了149名荷兰个体,他们的饮食习惯分为四个不同的群体:杂食者、鱼素食者、素食者、纯素食者。研究发现长期的饮食习惯并没有显著影响肠道微生物组的关键组成和多样性。在前10个最丰富的ARG中没有观察到显著差异。然而,两个ARG[lsa(C)和tet(L)]在杂食者和鱼食者之间显示出不同的丰度。
此外,宏基因组分析显示,与鱼素者相比,tet(X)在杂食动物的抗性体中更为丰富。
在来自三种不同饮食习惯(杂食性、蛋乳素食主义、严格素食主义)的58名志愿者的肠道抵抗力中观察到了类似的结果。
在所有习惯组中,最常见的耐药基因是针对四环素类抗生素[tet(A)、tet(B)、tet(M)、tets(O)、tet(Q)]、β-内酰胺类(blaTEM、blaSHV、mef)和MLS耐药基因[erm(B)、erm(C)],其次是磺胺类(sul1、sul2)和氨基糖苷类耐药基因(aacA-aphD)。
blaCTX‐M仅在卵乳素食组的样本中发现,而tet(E) 仅在杂食组的样本中检测到。有趣的是,这些经常检测到的ARG也常见于农场动物粪便中,这表明饮食习惯和环境ARG之间存在潜在联系。
虽然将饮食与肠道抗药性联系起来的确切机制尚不清楚,但饮食干预可能提供一种有前景的方法来减轻抗生素耐药性的负担。
Oliver等人(2022)研究了290名健康成年人,以探索饮食、肠道微生物组和抗生素耐药性之间的关系,并观察到高膳食纤维摄入量的个体ARG水平较低。
低ARG人群在肠道中显示出更多梭菌科的专性厌氧菌,而高ARG人群的链球菌科和肠杆菌科水平更高。此外,低ARG个体摄入的蛋白质较少,尤其是来自牛肉和猪肉的蛋白质。
该研究表明,增加纤维摄入量可能会促进肠道微生物组环境,有利于专性厌氧菌的生长,同时限制兼性厌氧菌,从而可能降低肠道中抗生素耐药性的发生率。
这是因为由专性厌氧菌发酵的膳食纤维会产生短链脂肪酸,降低肠道环境的pH值,使其对专性厌氧杆菌有利,但对兼性厌氧菌不利。
特定的饮食成分通过各种机制在调节肠道微生物群方面发挥着至关重要的作用。生物活性常量营养素,如凝集素、乳铁蛋白和多不饱和脂肪酸(PUFA),来源于碳水化合物、蛋白质和脂质,主要因其营养作用而被认可,但它们也具有生物活性,通过破坏营养可用性和细菌膜完整性来抑制抗生素细菌。
多酚等植物化学物质通过破坏细胞功能、干扰代谢过程或抑制酶活性表现出抗菌特性。
益生菌,如乳杆菌和双歧杆菌,通过竞争性排斥、分泌抗菌化合物和免疫调节来抑制病原体。
有趣的是,其中一些化合物还具有选择性抑制抗生素耐药细菌的能力,通过靶向其脆弱性,如损害生物膜形成、抑制耐药酶和破坏外排泵。
研究这些化合物如何特异性靶向细菌,包括抗生素耐药菌株,对于制定控制或减少肠道致病菌群的饮食策略至关重要。这有助于提供了一种非抗生素方法来管理肠道健康,并有助于遏制抗生素耐药性的传播。
生物活性常量营养素
常量营养素包括碳水化合物、蛋白质、脂肪,是人类饮食中支持生命和调节各种生理功能的重要组成部分。虽然它们的主要作用是为身体提供能量和构建块,但某些生物活性常量营养素也具有固有的抗菌特性。
这些特性可能有助于控制抗生素耐药细菌的生长,减轻其对肠道微生物组的影响,从而可能提供一种对抗抗生素耐药性的替代策略。
碳水化合物
不仅是能量的来源,而且具有生物活性,可以提高抗生素的有效性。
来自紫色鞘豆(Dioclea violacea)种子的甘露糖结合凝集素已被证明可以提高抗生素的疗效。关于什么是凝集素可以详见我们之前的文章:什么是凝集素,食物中的凝集素如何影响肠漏和自身免疫
Santos等人(2023)证明,从紫色鞘豆中提取的凝集素作为佐剂,通过碳水化合物识别增加细菌膜附近氨基糖苷类的生物利用度,并促进抗生素进入细菌细胞质,从而增强新霉素对多药耐药金黄色葡萄球菌和铜绿假单胞菌的活性。
蛋白质
如母乳和牛乳中的乳铁蛋白,也表现出抗菌和抗菌膜特性。
Avery等人(2021)报告称,人和牛乳铁蛋白都可以显著抑制测试的多重耐药鲍曼不动杆菌菌株中生物膜的形成,而人乳铁蛋白的最低抑菌浓度(MIC)低于牛乳铁蛋白,表明其效力更高。除了乳铁蛋白,来自植物的抗菌肽(AMP)在对抗抗生素耐药细菌方面也显示出了希望。
Heymich等人(2021)从鹰嘴豆中鉴定出21种AMP候选物,其中两种对耐甲氧西林的金黄色葡萄球菌ATCC 3300具有杀菌活性。这些肽与细菌膜相互作用,通过膜破裂导致细胞裂解,这与它们的杀菌作用直接相关。
膳食脂肪
特别是PUDA,也可以提高某些抗生素的有效性。
Zang等人(2021)发现,花生四烯酸(AA)和二十二碳六烯酸(DHA)等PUFA可增强氨基糖苷类药物对多重耐药鲍曼不动杆菌的疗效。
AA和DHA都通过细胞外隔离减轻了粘菌素的抗菌作用,使其难以穿透细菌的亲水性包膜和脂多糖屏障,并上调了外排系统基因表达(adeABC和adeIJK),从而通过将抗生素排出细胞来增加细菌的抗药性。
上述研究强调了生物活性常量营养素在增强抗生素有效性和影响耐药机制方面的作用。
果胶等碳水化合物可以通过与抗生素相互作用来提高抗生素的疗效,而乳铁蛋白等蛋白质则具有强大的抗菌和抗菌膜特性。然而,AA和DHA等膳食脂肪的作用更为微妙,因为它们都可以通过激活外排泵来增强抗生素作用并上调细菌耐药性机制。
总之,生物活性常量营养素不仅可以作为能量来源,还可以在对抗抗生素耐药细菌方面提供巨大的潜力。虽然它们在调节细菌耐药机制方面的作用很复杂,但这些发现强调了进一步研究饮食成分补充或增强传统抗生素治疗潜力的重要性。通过利用这些天然化合物,可能能够开发出新的策略,来缓解抗生素耐药性日益增长的挑战。

doi:10.1111/1541-4337.70143
植物化学物质
植物化学物质来源于植物性食品,主要包括多酚、含硫化合物、萜类、生物碱。这些化合物对抗生素耐药细菌具有抗菌活性,其中一些会破坏细菌膜结构,另一些则通过抑制外排泵来增强抗生素的有效性,从而防止耐药性的发展。
★ ■ 多酚类
柿子单宁——直接抗菌作用
一种经过充分研究的多酚,即来自青涩柿子的柿子单宁,已显示出对耐甲氧西林金黄色葡萄球菌的抑制作用。柿子单宁抑制细胞增殖,降低膜电位和细胞内ATP浓度,破坏全细胞蛋白质完整性,并诱导细胞S期细胞周期阻滞。
EGCG、原花青素——协同作用、增强抗生素疗效
Parvez等人(2019)报道了表没食子儿茶素没食子酸酯(EGCG)与庆大霉素联合使用时具有明显的协同作用,增强了对金黄色葡萄球菌和大肠杆菌多药耐药菌株的抗生素疗效。这是因为EGCG可以破坏细菌细胞膜,阻碍DNA超螺旋,增加细胞通透性,最终导致细菌死亡,并减轻耐药性的发展。
同样,从美国蔓越莓(Vaccinium macrocarpon L.)中提取的原花青素与β-内酰胺类抗生素显示出显著的协同作用。
Gallique等人(2021)报告称,原花青素抑制β-内酰胺酶,并选择性增强苯唑西林和羧苄青霉素对耐甲氧西林葡萄球菌的有效性,可能是通过调节葡萄球菌中变异转肽酶PBP2a的表达。PBP2a的低表达降低了细菌抵抗β-内酰胺类抗生素的能力,使细菌更容易受到抗生素的影响。
扩展阅读:
★ ■ 含硫化合物
大蒜素衍生物——释放硫化物破坏致病生物膜
例如,Xu等人(2018)将从大蒜鳞茎中分离出的大蒜素转化为纳米级硫化铁。这些纳米颗粒可以释放杀菌的有机硫化物(Polysulfanes),有效抑制生物膜形成和耐药的金黄色葡萄球菌。
异硫氰酸盐——抑制生物膜代谢活性
同样,来源于旱金莲和辣根的异硫氰酸酯对铜绿假单胞菌等抗生素耐药细菌具有抗菌潜力,特别是在生物膜抑制方面。当与美罗培南联合使用时,异硫氰酸酯通过抑制铜绿假单胞菌生物膜的代谢活性来增强抗生素疗效。
皂苷——破坏细胞壁结构,改变膜通透性
此外,da Silva等人(2024年)从Sarcomphalus joazeiro中提取了六种皂苷——红枣皂苷B、红枣皂苷III、酸枣仁苷、红枣皂苷IV、红枣皂苷II和红枣皂苷I,以及三种皂苷衍生物。当与庆大霉素和诺氟沙星联合使用时,富含皂苷的组分显示出增强作用,增加了它们对多药耐药铜绿假单胞菌和金黄色葡萄球菌的活性。皂苷与细菌细胞壁相互作用,导致结构损伤,改变膜通透性,并诱导膜破裂。
★ ■ 萜类药物
1,8-桉叶醇——增加膜通透性、诱导氧化应激导致成分泄漏
Moo等人(2021)发现,1,8-桉叶醇对碳青霉烯类抗生素耐药肺炎克雷伯菌具有抗菌作用。它增加了细菌表面电荷,增强了外膜通透性,并诱导了氧化应激,导致膜损伤、细胞内成分(如核酸、蛋白质和脂质)泄漏,最终导致细胞死亡。
芳樟酯——产生ROS、引发脂质过氧化
另一种萜烯,邻氨基苯甲酸芳樟酯(LNA),存在于薰衣草和百里香等植物中,对碳青霉烯类耐药肺炎克雷伯菌具有杀菌活性。
Yang等人(2021)证明,当LNA与美罗培南联合使用时,会降低MIC,使细菌更容易感染。LNA的抗菌作用归因于其通过产生ROS诱导氧化应激的能力,ROS可能进一步引发脂质过氧化并对细菌膜造成损伤。
萜类化合物——细菌外排泵的重要抑制剂
Oluwatuyi等人(2004)在迷迭香中鉴定出几种萜类化合物,如鼠尾草酸、鼠尾草酚和12-甲氧基-反式鼠尾草酸。这些化合物已被证明可以通过抑制NorA外排泵来增强红霉素对多药耐药金黄色葡萄球菌的活性,从而降低细菌排出抗生素的能力并增加其细胞内浓度。
★ ■ 生物碱
原小檗碱类生物碱——抑制H+-ATP酶泵,阻断ATP合成
Guefack等人(2022)从绿花恩南番茄(Enantia chlorantha)中鉴定出的原小蘖碱类生物碱(protoberberine alkaloids)(哥伦比亚胺、假哥伦比亚胺、药根碱、巴马汀、4,13-二羟基-3,9,10-三甲氧基原小檗碱和13-羟基-2,3,9,10-四甲氧基原黄连素)可以有效抑制多药耐药细菌的生长,包括大肠杆菌、产气肠杆菌、肺炎克雷伯菌、普罗维登斯氏菌(Providencia stuartii)、金黄色葡萄球菌,MIC值低于100µg/mL。重要的是,其中一种名为哥伦比亚胺的生物碱抑制了H+-ATP泵,阻碍了细胞ATP的产生。
苦参碱——抑制Mex泵,增强氟喹诺酮敏感性
来自苦豆子种子的苦参碱与环丙沙星对铜绿假单胞菌的抗生素耐药菌株显示出协同作用。总生物碱显示出抑制生物膜形成和降低Mex泵活性的作用,从而增强细菌对氟喹诺酮类药物的敏感性。
研究人员从Erythroxylum revolutum Mart.中分离出生物碱,包括6-(2′-甲基丁酰氧基)-3-羟基托烷和6-丁酰氧基-3-羟基托巴烷,可增强诺氟沙星和红霉素对多药耐药金黄色葡萄球菌的疗效。这种作用可能与托烷生物碱削弱耐多药金黄色葡萄球菌中ABC多药转运蛋白的能力有关。
总之,植物化学物质通过破坏细菌膜、抑制外排泵和阻碍细菌生物膜的形成来发挥抗菌能力。当与抗生素结合时,一些植物化学物质表现出协同作用,可以提高抗生素的有效性并改变抗生素的耐药性,表明有可能管理肠道中的抗生素耐药细菌并影响肠道耐药组。
未来,有必要研究这些植物化学物质在人体肠道中的生物利用度、剂量和长期影响,这可能会导致更有针对性的饮食干预。深化我们对特定植物化学物质如何选择性调节肠道抗药性的理解,对于开发创新方法来减轻抗药性基因的传播和改善人类健康结果至关重要。

doi:10.1111/1541-4337.70143
益生菌
益生菌是具有健康益处的活微生物,通常被掺入酸奶和奶酪等发酵食品中。然而,这些产品通常只有在用未经巴氏消毒的牛奶制成时才含有益生菌,因为巴氏消毒可以显著减少或消除活的益生菌菌株。益生菌可以通过调节肠道微环境、增强免疫功能和抑制过敏反应来影响肠道中的抗生素耐药细菌。
★ ■ 直接抗菌效应
某些益生菌菌株已显示出对耐药病原体的抗菌活性。
乳杆菌类——破坏细胞膜完整性,与抗生素协同抑制有害菌
例如,嗜酸乳杆菌NCFM和鼠李糖乳杆菌GG通过破坏细菌细胞膜的完整性对耐甲氧西林金黄色葡萄球菌表现出抗菌作用。
益生菌胶囊(嗜酸乳杆菌CL1285、干酪乳酸杆菌LBC80R、鼠李糖乳杆菌CLR2)与妥布霉素联合使用时,通过限制病原体定植和阻碍生物膜的形成,对耐甲氧西林的金黄色葡萄球菌和铜绿假单胞菌表现出协同作用。
双歧杆菌——促进四环素进入,破坏蛋白质合成
双歧杆菌会产生生物表面活性剂和乙酸盐,这有助于分离粘附的大肠杆菌并改变细胞内的阴离子组成。这可以增强四环素进入大肠杆菌,破坏蛋白质合成,最终导致协同杀死大肠杆菌。这些研究为益生菌作为功能性食品调节肠道抗菌药物耐药性的潜在应用铺平了道路。
★ ■ 发酵食品中的益生菌活性
除了这些单独的益生菌菌株外,酸奶和泡菜等发酵食品中发现的益生菌已被证明可以抑制抗生素耐药细菌的生长。
酸奶来源菌株抗菌物质:类细菌素肽或蛋白
从商业酸奶中分离出的石蜡乳杆菌对14种多药耐药细菌具有抗菌活性,包括大肠杆菌、铜绿假单胞菌、鲍曼不动杆菌、产气杆菌、奇异变形杆菌、肺炎克雷伯菌。该菌株产生的抗菌物质可能是一种细菌素样肽或蛋白质,对密切相关的细菌菌株具有活性,表明其在抑制致病性多药耐药细菌生长方面具有潜在作用。
泡菜来源菌株——抑制耐药菌、生物膜
Yi和Kim(2023)从泡菜中分离出益生菌乳酸菌(Pediococcus inopinatus K35),该菌能有效抑制多药耐药铜绿假单胞菌的生长和生物膜形成。这些研究共同强调了益生菌的巨大潜力,特别是那些来自发酵食品的益生菌,作为能够对抗抗生素耐药细菌的天然药物。

doi:10.1111/1541-4337.70143
然而,一个值得关注的问题是益生菌菌株可能携带ARG,ARG可以转移到致病菌,从而导致耐药性的传播。
例如,Selvin等人(2020)从膳食补充剂中分离出益生菌菌株,发现粪肠球菌和肠系膜芽孢杆菌对青霉素G有耐药性,嗜酸乳杆菌对氨苄青霉素有耐药性。
Montassier等人(2021)的一项研究表明,摄入市售益生菌补充剂(含有11种常见的益生菌菌株:嗜酸乳杆菌、干酪乳杆菌、副干酪乳酸杆菌、鼠李糖乳杆菌、植物乳杆菌、双歧杆菌、短双歧杆菌、长双歧杆菌亚种、婴儿长双歧杆菌、乳链球菌和嗜热链球菌)导致ARG丰度降低。
然而,益生菌也可以作为肠道耐药组扩展的储库,因为益生菌的摄入可以通过水平基因转移将携带ARG的菌株扩展到病原体,因为移动遗传元件含量(转座酶和整合酶)的检测与ARG丰度相关。
这些发现强调了在将益生菌纳入饮食时需要谨慎,特别是在商业益生菌补充剂的情况下。虽然益生菌可以在减少致病菌定植和增强抗菌活性方面发挥作用,但必须仔细评估它们作为ARG载体的作用。为了降低潜在风险,需要监测补充剂和功能性食品中使用的益生菌菌株中的ARG含量,确保它们不会导致耐药性的进一步传播。
总之,虽然益生菌作为功能性食品具有调节肠道耐药性和对抗抗生素耐药性的潜力,但它们储存和转移ARG的潜力需要更彻底的研究。未来的研究应侧重于鉴定ARG转移风险最小的益生菌菌株,并优化益生菌食品,以提高其治疗效果,同时限制对肠道耐药的不利影响。
以上我们已经了解了抗生素耐药性的发生机制、传播途径、饮食干预的方法和机制。在实施上述饮食干预策略的同时,如何客观评估其对肠道耐药组的影响至关重要。
随着测序技术的快速发展,肠道菌群检测已经能够识别肠道中的抗生素耐药基因,例如,谷禾宏基因组肠道菌群检测报告中,专门设有抗生素耐药基因分析板块,可检测β-内酰胺酶、氨基糖苷类、大环内酯类、喹诺酮类、四环素耐药基因等多种耐药机制的存在和丰度,这为评估饮食干预效果提供了客观依据。
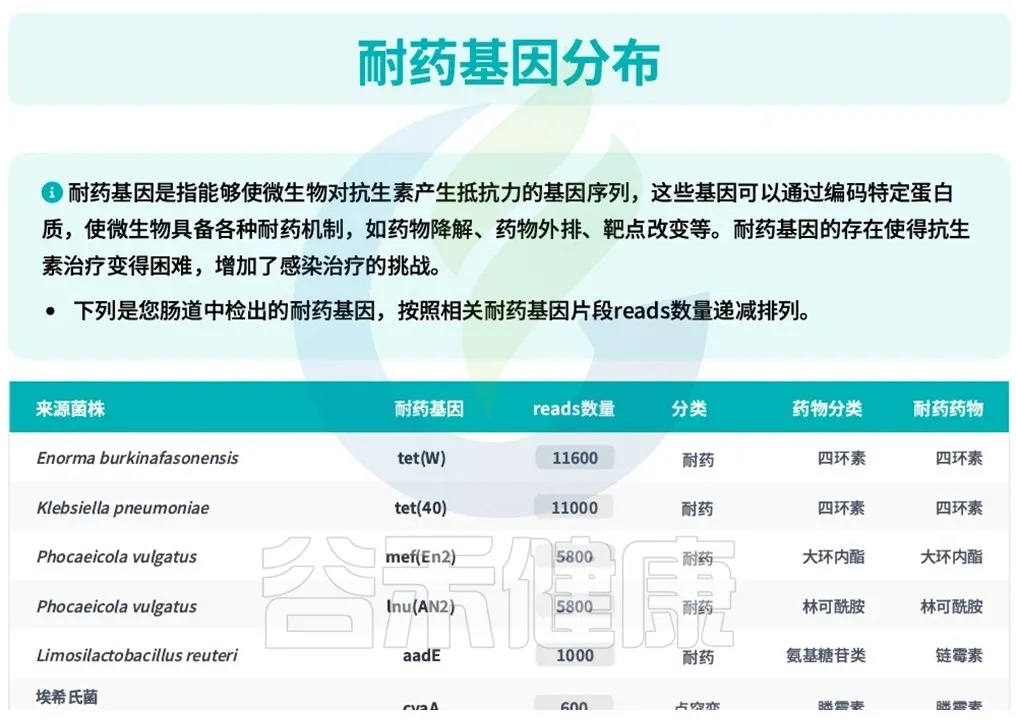
<来源:谷禾宏基因组肠道菌群检测报告>
通过干预前后的对比检测,我们可以了解特定饮食成分对耐药基因表达的影响,从而不断优化干预方案,实现个性化的肠道耐药组管理。
例如,患者A因肺炎需要使用抗生素治疗,在用药前安排了肠道菌群检测。报告显示其肠道中存在高水平的β-内酰胺酶耐药基因,那么根据报告调整相关抗生素方案,避开容易产生耐药的β-内酰胺类抗生素,减少治疗期间耐药基因的扩增风险。
通过识别高丰度ARGs,预测治疗失败风险,避免可能造成更加严重抗生素耐药。
一名反复发生尿路感染的患者B,常规抗生素治疗无效。通过肠道菌群宏基因组检测,发现其肠道中携带高丰度的 blaCTX-M(广谱β-内酰胺酶基因) 和 mecA(甲氧西林耐药基因),提示对β-内酰胺类及甲氧西林类药物耐药。
根据检测结果,避免使用头孢类(靶向β-内酰胺酶敏感菌),改用磷霉素(对blaCTX-M无交叉耐药)或联合多黏菌素(针对mecA阳性菌)。补充蔓越莓原花青素(抑制β-内酰胺酶活性)或大蒜素纳米颗粒(破坏生物膜),增强抗生素渗透性。
识别关键耐药基因,指导临床选择非交叉耐药抗生素并辅以天然抗菌成分,从源头减少抗生素滥用导致的耐药性扩增。
一名2型糖尿病患者宏基因组检测显示,其肠道中 Vancomycin_vanX(万古霉素耐药基因) 和 Multidrug_emrE(多药耐药基因) 丰度升高,且与胰岛素抵抗指数呈正相关。
ARG丰度高的糖尿病患者血糖控制失败率相对较高,因此可以从这方面入手,增加苦参碱摄入(抑制Mex泵活性),联合二甲双胍改善胰岛素敏感性。
通过识别耐药基因丰度,可预测糖尿病血糖控制失败的风险,针对性干预,减少耐药基因对慢性疾病的协同恶化作用。
抗生素耐药性已经从单纯的医学挑战演变为一个多维度的健康问题,而肠道耐药组的研究为我们提供了新的思路应对这一挑战。
肠道菌群检测技术已能够识别耐药基因的存在和丰度,使个体ARG状况的评估成为可能,这不仅可以帮助临床医生了解患者的耐药风险,也为饮食干预提供了科学依据。
日常饮食的预防性策略
加富含多酚的食物(如绿茶、浆果、深色蔬菜)、含硫食物(如十字花科蔬菜、大蒜)及发酵食品(如酸奶、泡菜),通过天然成分抑制耐药基因传播。
控制高脂高糖饮食(如红肉、加工食品),避免促进肠杆菌科等耐药菌增殖。地中海饮食模式(富含纤维、橄榄油)可提升短链脂肪酸水平,抑制耐药菌生长。
通过将这些科学发现转化为具体的饮食策略和产品,结合肠道菌群检测技术,我们有望在不依赖新抗生素开发的情况下,通过日常饮食管理减轻抗生素耐药性的负担,为公共健康问题提供可持续的补充解决方案。
功能性食品的开发创新
开发含高浓度EGCG的绿茶提取物饮品,或添加原花青素的蔓越莓咀嚼片,辅助治疗泌尿系统耐药菌等相关感染。
利用大蒜素纳米颗粒或异硫氰酸盐制成肠道缓释胶囊,抑制生物膜形成并减少水平基因转移。
基因编辑乳杆菌(如表达抗菌肽的菌株),靶向清除肠道内携带ARGs的致病菌。
通过微胶囊化或脂质体包裹提高多酚类成分的肠道吸收率;结合AI预测模型,从天然产物库中筛选新型抗菌分子,加速功能性配方的迭代。
建立功能性食品中ARGs迁移风险的动态监测体系,避免益生菌载体成为耐药基因传播媒介。
功能性食品作为“可食用疗法”,通过精准调控肠道耐药组,为临床耐药危机提供了“非抗生素”解决方案。未来需融合合成生物学、纳米技术与营养学,推动第三代功能性食品的落地。
临床营养的精准化应用
本文中列举的一些研究表明,植物多酚(如柿子单宁、表没食子儿茶素没食子酸酯)、含硫化合物(如大蒜素)及萜类(如1,8-桉叶油醇)可通过破坏细菌生物膜、抑制外排泵或增强抗生素渗透性,降低耐药菌存活率。临床可开发含此类成分的功能性营养剂,辅助治疗耐药菌感染(如MRSA、铜绿假单胞菌),或用于术后耐药菌感染预防。
特定益生菌(如乳酸杆菌、双歧杆菌)可通过竞争排斥或分泌抗菌肽抑制耐药菌定植。临床中可针对术后感染或抗生素滥用患者,定制含益生菌的肠内营养配方,重建肠道菌群平衡。
乳铁蛋白、植物抗菌肽等可增强抗生素疗效。例如,乳铁蛋白与氨基糖苷类联用可显著抑制多重耐药鲍曼不动杆菌,适合用于重症患者的营养支持方案。
2型糖尿病患者中,苦参碱与二甲双胍联用可抑制肠道内产气荚膜梭菌的Mex泵活性,改善胰岛素敏感性。
结直肠癌患者化疗期间,添加大蒜素纳米颗粒的营养配方可减少肠道内携带mdtP基因的多重耐药大肠杆菌丰度,缓解化疗相关性腹泻。
然而,也需要建立植物化学物质与抗生素的相互作用数据库,避免协同毒性(如EGCG与环丙沙星联用可能加重肝损伤)。
同时可以开展剂量梯度临床试验,明确如多酚类成分的最佳治疗窗口。
联合微生物学、药理学与食品科学,开发兼具营养支持与耐药调控功能的特医食品。通过跨学科协作,特医食品不仅可成为感染患者的营养支持方案,也能作为耐药危机的一线防控工具。
未来,跨领域合作结合人工智能,有望加速功能性食品的迭代,在降低抗生素依赖的同时,重塑肠道健康生态,为应对耐药危机提供可持续路径,最终实现“以食为药”的精准健康管理。
主要参考文献:
Abbas A, Barkhouse A, Hackenberger D, Wright GD. Antibiotic resistance: A key microbial survival mechanism that threatens public health. Cell Host Microbe. 2024 Jun 12;32(6):837-851.
Liang Z, Liang Z, Hu HW, Howell K, Fang Z, Zhang P. Food substances alter gut resistome: Mechanisms, health impacts, and food components. Compr Rev Food Sci Food Saf. 2025 Mar;24(2):e70143.
Pramod Barathe, Kawaljeet Kaur, Sagar Reddy, Varsha Shriram, Vinay Kumar, Antibiotic pollution and associated antimicrobial resistance in the environment,Journal of Hazardous Materials Letters, Volume 5,2024.
Langford BJ, Soucy JR, Leung V, So M, Kwan ATH, Portnoff JS, Bertagnolio S, Raybardhan S, MacFadden DR, Daneman N. Antibiotic resistance associated with the COVID-19 pandemic: a systematic review and meta-analysis. Clin Microbiol Infect. 2023 Mar;29(3):302-309.
Laxminarayan R. The overlooked pandemic of antimicrobial resistance. Lancet. 2022 Feb 12;399(10325):606-607.
Sarmiento MRA, de Paula TO, Borges FM, Ferreira-Machado AB, Resende JA, Moreira APB, Dutra Luquetti SCP, Cesar DE, da Silva VL, Diniz CG. Obesity, Xenobiotic Intake and Antimicrobial-Resistance Genes in the Human Gastrointestinal Tract: A Comparative Study of Eutrophic, Overweight and Obese Individuals. Genes (Basel). 2019 May 7;10(5):349.
谷禾健康
艰难梭菌是一种革兰氏阳性、形成孢子的厌氧芽孢杆菌,是艰难梭菌感染(CDI)的病原体。该菌于1935年首次分离得到,1977年报道了第一例确诊的CDI病例。从那时起,CDI的发病率逐年上升,艰难梭菌感染现在是全世界医院腹泻的最常见原因。
艰难梭菌在我国健康成年人结肠的定植率约为4%-7%,而腹泻患者的检出率高达15%-40%。但为什么一些人群肠道中存在艰难梭菌,却没有出现任何症状?而另一些则会出现腹泻、腹痛,甚至发展为假膜性结肠炎?
这是因为艰难梭菌可分为产毒型和非产毒型,通常只有产毒型会引发临床症状,其感染的表现主要受多种毒力因子影响,这些毒力因子可能比艰难梭菌的存在更为重要。并且健康的肠道微生物组对艰难梭菌感染具有保护作用。平衡的微生物和宿主因子可抑制艰难梭菌的发芽和生长,同时微生物群与宿主免疫系统的相互作用调节免疫反应,刺激抗菌肽和分泌型IgA的产生,维持菌群平衡,通过营养竞争、生态竞争和生态位排斥等机制抵抗艰难梭菌的定植和感染。
例如一些有益菌通过产生短链脂肪酸降低管腔pH值(不利于艰难梭菌),并刺激粘蛋白和抗菌肽的生成以增强防御屏障。丁酸盐还能稳定缺氧诱导因子-1(HIF-1)、增强紧密连接,保护肠道上皮免受艰难梭菌毒素损害。
艰难梭菌的孢子和生长依赖特定胆汁酸,肠道共生细菌通过调节胆汁酸代谢产物,抑制其萌发和定植。例如,Clostridium scindens可催化胆汁酸7α-脱羟基化,生成次级胆汁酸,从而增强对艰难梭菌感染的抵抗力。
当肠道微生物组的平衡状态受到干扰或破坏时,艰难梭菌感染(CDI)的易感性会显著增加。例如由于使用抗生素,年龄增长、其他胃肠道疾病,营养状态不佳、肥胖、癌症化疗这些因素都会增加艰难梭菌感染的风险。
由于艰难梭菌的危害由毒力因子决定,检测产毒菌株或基因显得尤为重要。目前,一些检测方法只能识别艰难梭菌菌株,无法区分是否为产毒菌株,可能导致误诊或过度治疗。为提高诊断准确性,近年来开发了多种技术,包括检测毒素基因的分子诊断技术和直接检测毒素蛋白的免疫学方法。16S测序仅能分辨到物种层面,宏基因组测序(包括一些靶向的测序)则可识别毒力基因。这些技术能够较快速、准确地识别产毒菌株,帮助临床医生制定更有效的治疗方案,减少不必要的抗生素使用及相关并发症。
艰难梭菌感染的治疗和预防也是人们所关心的,其治疗方法包括针对细菌(抗生素)、针对毒素(抗体、结合剂)以及微生物群(保护或恢复)的方法,在暴发性病例或非手术治疗失败时,还可选择结肠切除术或其他微创手术。通过疫苗和益生菌预防艰难梭菌感染的研究逐渐显示出一定的临床效果。
希望通过本文的内容,能够帮助人们更加全面和清晰地了解艰难梭菌的相关知识,同时提高对其危害的认识,从而采取更加科学和有针对性的措施来预防和应对艰难梭菌感染。
艰难梭菌是一种革兰氏阳性、形成孢子的厌氧芽孢杆菌,近年来,由于抗生素的滥用导致肠道菌群失调,艰难梭菌感染(CDI)的发病率在中国和全球范围内不断上升,显示出这一公共卫生问题的严重性。
2022年,中国艰难梭菌感染治疗市场规模达34.14亿元,全球艰难梭菌感染治疗市场规模达到78.74亿元,预计全球艰难梭菌感染治疗市场规模将在2028年达到147.95亿元。
艰难梭菌的感染率不断上升,很多人可能想知道自己体内是否存在这种细菌,以及是否艰难梭菌存在于人体就会致病及造成危害?随着谷禾的视角一起往下看。
1
毒力因子影响艰难梭菌的致病性
首先要强调的一点是,艰难梭菌感染的临床表现受到多种毒力因子的影响,这些毒力因子可能比艰难梭菌的存在更为重要。艰难梭菌分为产毒型和非产毒型,通常只有产毒型会引发临床症状。
产毒艰难梭菌主要产生毒素A(肠毒素)和毒素B(细胞毒素),少部分仅产生毒素B。高毒力菌株(如027型)除了产生这两种毒素外,还产生二元毒素。
据国外综合医院统计,A(+)B(+)占艰难梭菌的57%,A(-)B(+)占34%,A(-)B(-)占9%。毒素A阳性,毒素B阴性的菌株尚未被发现,毒素B可以单独导致艰难梭菌致病。
◮ 艰难梭菌的毒力因子
毒素A:主要作用于肠道,导致肠道上皮细胞的损伤和炎症反应。它通过结合肠道细胞表面的受体,诱导细胞内信号转导,导致细胞凋亡和肠道通透性增加,从而引发腹泻。
毒素B:具有更强的细胞毒性,能够直接破坏细胞骨架,导致细胞死亡,破坏紧密连接并丧失肠道屏障功能。它在致病过程中,尤其是在高毒力菌株中,起着关键作用。
二元毒素:一种ADP-核糖基转移酶,导致肌动蛋白细胞骨架解聚(导致屏障功能丧失和紧密连接破坏)和微管突起(导致艰难梭菌粘附增加)。
致病性基因和毒素作用方式
Buddle JE,et al.Virulence.2023
◮ 毒素诱导引发的免疫反应导致各种症状
这些毒素在致病性位点(PaLoc)内编码,能够与肠上皮细胞的受体结合并被内化。在细胞内,它们使小的Rho蛋白葡萄糖化,导致紧密连接破裂、上皮完整性降低,并增加细菌对宿主上皮的粘附。
毒素诱导的肠道屏障损伤会引发免疫反应,其特征是分泌促炎细胞因子和趋化因子,导致中性粒细胞、肥大细胞、单核细胞和先天性淋巴细胞的募集和激活;以及花生四烯酸代谢物的分泌、P物质和活性氧中间体的生产。这些细胞因子和免疫细胞的作用会引发艰难梭菌感染的临床症状。例如,肥大细胞脱颗粒刺激组胺释放,增加肠道屏障的通透性,导致大量液体流失到管腔中,从而引发严重的腹泻、痉挛、脱水和伪膜性肠炎等症状。
艰难梭菌的各种毒力因子

Buddle JE,et al.Virulence.2023
2
产毒艰难梭菌及我国的流行情况
目前已鉴定出几十种艰难梭菌菌株,其相对比例在过去几年中迅速变化,且毒力和感染能力可能存在差异。我们这里简单介绍几种高毒性艰难梭菌菌株以及我国的主要流行情况。
◮ 毒力较高的艰难梭菌
⑴027菌株
027菌株是近年来备受关注的高毒力菌株,部分027菌株在体外能够产生更多毒素,并且更容易与人类肠道上皮细胞结合,通常与严重腹泻和高死亡率相关。该菌株在北美和欧洲的医院中广泛传播,成为医院获得性腹泻的主要原因。
⑵017菌株
这种菌株在1990年代首次被识别,主要在亚洲地区流行。017菌株通常被认为是毒素A阴性、毒素B阳性的菌株。由于毒素A检测通常用于识别粪便样本中的艰难梭菌,因此可能会遗漏这种菌株。
⑶078菌株
078介导的艰难梭菌感染患者更常见于靠近农场的地区,078菌株是食用动物中最常见的类型,在牛和猪中都有发现,人可能通过食用动物导致感染。078菌株的毒力特征与027菌株相似,所有027和078菌株似乎都具有完整的肌动蛋白特异性ADP核糖基化毒素。
⑷其他菌株
除了027、017和078菌株,还有其他核糖型的艰难梭菌菌株,如001、106和053等。然而每种核糖型的相对比例在过去几年中迅速变化。核糖型001的比例从25.1%下降到7.8%,核糖型106的比例从26.2%下降到20.2%,且106几乎只在英国发现。
◮ 我国的艰难梭菌流行情况

一项荟萃分析统计了我国大陆部分省份的艰难梭菌感染发病率及主要流行和耐药的艰难梭菌。
艰难梭菌在健康成年人结肠的定植率约为4%-7%,而国内腹泻患者的检出率为15%-40%(共分析了15,313个样本)。在中国大陆,ST-37(017)和ST-3是最普遍的菌株;幸运的是,ST-1(027)和ST-11(078)等高毒力菌株迄今很少出现。
并且不同省份的阳性率存在差异显著:
湖北:23%
河北:19%
安徽:19%
四川:17%
宁夏:4%
河南:3%
doi: 10.1038/srep37865.
中国临床艰难梭菌的分子特征
Wu Y,et al.Anaerobe.2022
中国大陆的流行艰难梭菌株并非高毒力类型,这可能是近年来没有艰难梭菌感染爆发的原因。此外,关于毒素A阴性和毒素B阳性菌株的报道逐渐增加,许多研究也发现了这一现象。在亚洲,毒素A阴性和毒素B阳性菌株的数量明显高于欧美国家。
◮ 我国的艰难梭菌耐药情况
我国的艰难梭菌对环丙沙星、克林霉素和红霉素的耐药率高于其他地区;然而,报道的艰难梭菌分离株均未对甲硝唑、万古霉素、替加环素或哌拉西林/他唑巴坦耐药。
3
非产毒艰难梭菌
◮ 菌株特性不同,不会致病
非产毒性艰难梭菌(NTCD)是指那些不产生致病毒素的艰难梭菌菌株。这些菌株能够在肠道中定殖,并且通常从无症状个体中分离出来。
非产毒性艰难梭菌菌株表现出与产毒性菌株不同的表型特征,如更高的甲硝唑耐药性和更高的孢子形成效率。
◮ 非产毒菌株定植可能具有保护作用
非产毒性艰难梭菌能够在肠道中定殖而不引起症状,并有研究发现其可能有助于降低由产毒性艰难梭菌引起的感染风险。研究表明,NTCD的定殖可以通过竞争性抑制机制,防止产毒性菌株的定殖和毒素产生。
临床应用与研究:在临床试验中,NTCD-M3等菌株已被用于预防艰难梭菌感染,通过诱导肠道和系统性免疫反应,从而减少产毒性艰难梭菌的附着和感染。
了解艰难梭菌感染的发病机制对于制定有效的疾病治疗和预防措施至关重要。让我们一起来看下人体是如何感染艰难梭菌以及感染后会出现的症状。
◮ 艰难梭菌主要通过孢子传播感染
艰难梭菌(C.difficile)作为一种专性厌氧菌,通常无法在大气中存活,那么它是如何传播到人体的呢?这是通过形成孢子来实现的,这些孢子即使在恶劣的环境条件下也能存活。除了提供对氧的抵抗力外,孢子还对紫外线、干燥、热、许多消毒剂和抗生素具有抵抗力。
注:艰难梭菌孢子已在各种环境来源中检测到,包括家畜、水源和土壤。
艰难梭菌感染的发病机制
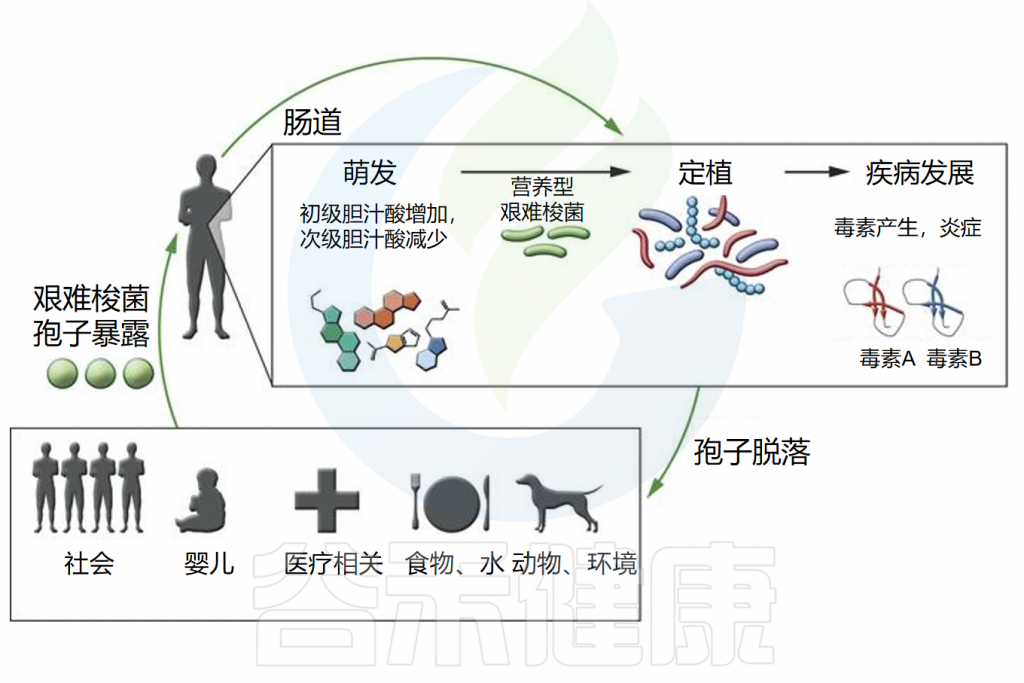
doi.org/10.1172/JCI72336.
1
感染过程
◮ 孢子生长必须依赖特定的胆汁酸
通过粪口途径摄入艰难梭菌孢子后,孢子需要发芽并生长为定植于胃肠道的营养细胞。然而,摄入孢子并不总是会导致定植,因为胃肠道环境必须适宜这一过程的发生。
体外研究表明,发芽和生长成营养形式取决于特定初级胆汁酸(如牛磺胆酸)的存在。相反,其他胆汁酸,如鹅去氧胆酸,可能会抑制艰难梭菌孢子的发芽。
胃肠道内的微生物在胆汁酸代谢中起关键作用,微生物群落的调节会影响代谢物的可用性。来自抗生素处理小鼠的盲肠提取物含有高水平的胆盐并促进孢子萌发,而来自未处理小鼠的盲肠提取物则没有。我们推测:抗生素的使用改变了肠道微生物群的代谢,导致胆汁酸种类更易促进艰难梭菌孢子的生长。
◮ 定植后毒素介导炎症和疾病
一旦定植,艰难梭菌会引发毒素介导的炎症和疾病。它产生两种主要毒素,即艰难梭菌毒素A和B(TcdA和TcdB),这些毒素在营养生长的静止期产生,主要导致粘膜上皮损伤和炎症反应。
另一种毒素,艰难梭菌二元毒素(CDT),会破坏肌动蛋白细胞骨架,研究表明其存在可能增加菌株的毒力。
由于孢子暴露和艰难梭菌定植不一定导致临床疾病,胃肠道微生物群和宿主在艰难梭菌的疾病发展中可能发挥重要作用。
2
艰难梭菌感染后的临床症状
产毒艰难梭菌菌株感染的临床表现通常从无症状携带到轻度或中度腹泻,或是暴发性,有时是致命的。症状在定植后不久开始,中位发病时间为2至3天。
◮ 常见的症状
腹泻:这是艰难梭菌感染最常见的症状,通常表现为水样腹泻,频率可达每天多次。腹泻可能伴有粘液或脓性分泌物。
腹痛和腹部不适:患者常感到腹部绞痛或不适,疼痛可能是间歇性的,且通常与腹泻发作相关。
发热:部分患者可能出现低热,体温通常在37.5°C至38.5°C之间。
恶心和食欲减退:患者可能会感到恶心,伴随食欲减退,进而导致体重下降。
脱水:由于频繁腹泻,患者可能出现脱水症状,如口干、尿量减少、皮肤弹性下降等。
腹胀:部分患者可能会感到腹部胀气或腹部膨胀。
◮ 严重并发症
在一些情况下,艰难梭菌感染可能导致更严重的并发症,包括:
伪膜性结肠炎:表现为严重的腹痛、腹泻和发热,肠道内形成伪膜。
肠穿孔:极少数情况下,感染可能导致肠道穿孔,表现为剧烈腹痛和急性腹膜炎。
中毒性巨结肠:具有全身中毒症状及全结肠或节段性结肠扩张的临床表现。是一种危及生命的并发症,表现为腹痛、腹胀、腹泻和全身症状加重,可能需要手术干预。
◮ 肠外表现
艰难梭菌感染不仅会导致肠道相关的症状,还可能引发一些肠道之外的表现,包括脱水、电解质紊乱、低白蛋白血症、低血压、肾功能衰竭、全身炎症反应综合征、关节炎、菌血症、败血症甚至死亡。
感染的核心是肠道菌群平衡被破坏
艰难梭菌感染发病机制的核心是微生物群的破坏。健康的肠道微生物群对于防止病原体定植(称为定植抗性)至关重要 。
未被破坏的微生物群能够抵抗病原体的定植,健康平衡的微生物和宿主因子均可抑制艰难梭菌的发芽和生长。同时微生物群和宿主免疫系统之间的串扰导致调节免疫反应。此外,微生物群可以刺激抗菌肽和分泌型IgA的产生,从而维持微生物群的组成。并且已经提出了多种机制来解释为什么微生物群被破坏会导致定植抗性丧失,包括营养竞争、生态竞争和生态位排斥。
而由于抗生素使用、药物、年龄变化、饮食或炎症等因素导致微生物群的破坏,可导致艰难梭菌感染的发展。由于结构或代谢环境的变化,菌群失调会导致定植抗性丧失。特定群落成员的损失可能会影响微生物和宿主产生的代谢物的水平,从而促进孢子萌发和艰难梭菌生长。菌群失调也可能通过免疫调节的丧失和促炎状态导致免疫反应失衡,这两者都会影响疾病的发展。营养型艰难梭菌产生的毒素可刺激炎性细胞因子、中性粒细胞和抗毒素抗体的生成。
微生物群对CDI期间病原体抗性和宿主的机制
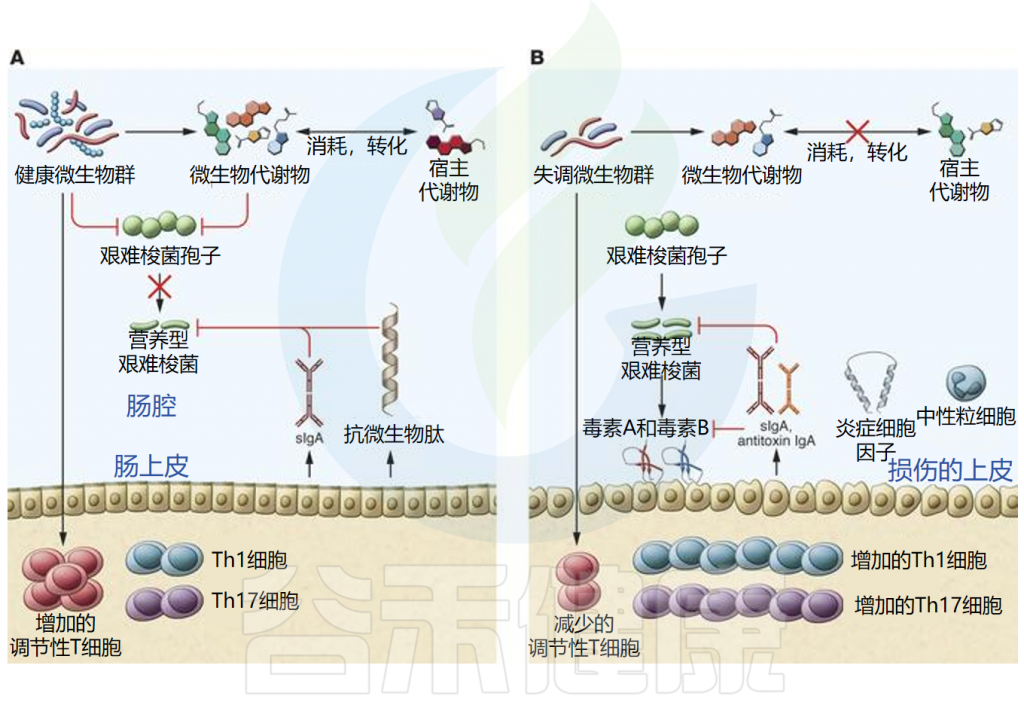
doi.org/10.1172/JCI72336.
据研究文献报道,以下这些因素会增加获得CDI的风险。
1
使用抗生素
◮ 使用过抗生素的人患病率更高
使用抗生素是艰难梭菌感染中最常见的诱发因素。一项大型回顾性研究分析了10154例艰难梭菌感染患者的数据,发现78%的患者在感染前3个月内使用过抗生素。
大多数抗生素都与艰难梭菌感染(CDI)发展相关,但最常见的药物包括青霉素、头孢菌素和氟喹诺酮类药物。在一项关于社区获得性CDI中抗生素使用情况的荟萃分析中,克林霉素发生社区获得性CDI的风险最高,然后依次是氟喹诺酮类药物、头孢菌素类、青霉素类、大环内酯类和磺胺类/甲氧苄啶类。
◮ 使用抗生素导致肠道微生物平衡被破坏增加艰难梭菌感染的风险
抗生素的使用,尤其是广谱抗生素,会显著改变肠道微生物群的组成和多样性,导致微生物群失调。这种失调主要表现为有益菌群(如双歧杆菌、乳酸菌等)数量减少,抑制艰难梭菌的生长和毒素产生的能力减弱,导致更易感染艰难梭菌。
艰难梭菌感染的发病机制
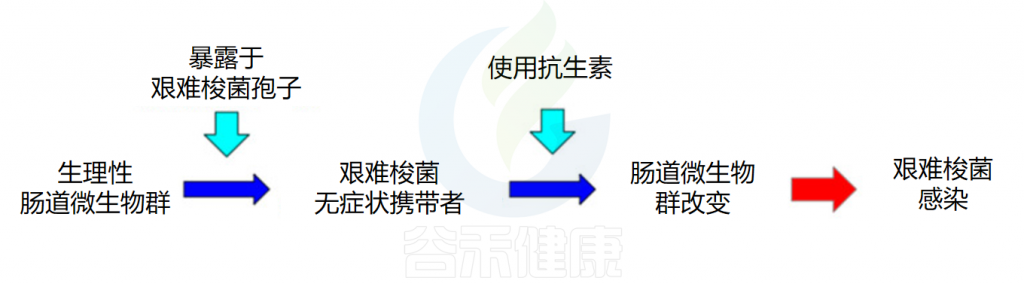
Piccioni A,et al.Int J Mol Sci.2022
2
高龄
◮ 高龄人群CDI的患病率和死亡率更高
正如多项研究所记录的那样,高龄是CDI的重要风险因素。65岁及以上的老人患病人数显著增加,人口发病率比其他年龄组高5倍以上。
并且还发现CDI的死亡率随着年龄的增长而显著升高。在2011年对美国CDI负担的最新研究中,发现65岁及以上的人大约占CDI病例总数的57%,但该年龄组的CDI死亡占CDI死亡总数的83%。
◮ 年龄增长免疫退化、微生物多样性下降
年龄增长会影响肠道微生物组结构。人类肠道微生物组在一生中经历显著变化,老年人群的微生物组多样性较低且不断变化。研究发现,老年人保护性物种(如双歧杆菌和部分厚壁菌门成员)减少,有害物种(如变形菌门)增加。这些变化与免疫系统退化(即免疫衰老)有关。
尽管年龄是CDI的独立危险因素,但其与抗生素使用增加、更频繁的医院就诊及疾病发展密切相关,这些因素共同提高了艰难梭菌的易感性。
新生儿与成人艰难梭菌感染的比较
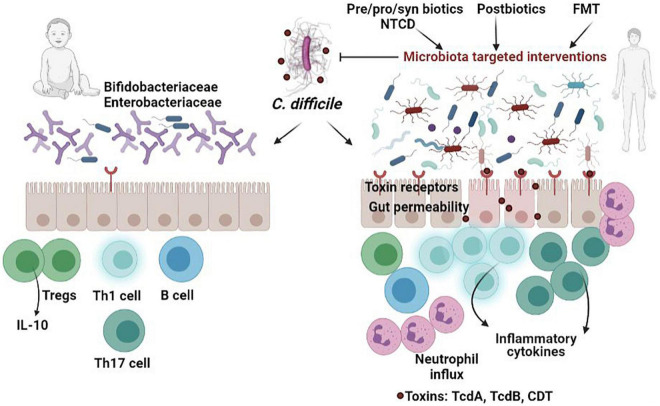
Vasilescu IM,et al.Front Microbiol.2022
3
其他胃肠道疾病
患有其他胃肠道疾病的患者也可能更容易感染艰难梭菌。炎症性肠病(IBD)已被证实是CDI的危险因素,并与更严重的疾病结果相关。IBD患者肠道菌群呈现多样性降低,同时存在以变形菌门为主的多种潜在致病菌。然而,这些微生物群落如何影响艰难梭菌易感性的具体机制较为复杂。
◮ 肠道炎症性疾病会促进艰难梭菌感染
宿主免疫反应能调节微生物群,而IBD加重CDI病情表明炎症可促进CDI发展。抗菌肽脂质运载蛋白-2和钙卫蛋白等炎症产物限制肠道环境中营养物质可用性,可能为艰难梭菌创造有利条件。肠道菌群类型影响粘膜IgA库,复发性CDI患者结肠活检中IgA产生细胞减少。
还观察到各种微生物会影响T细胞的亚群,例如梭菌属诱导Treg物种和分段丝状细菌诱导Th17细胞分化。这些微生物种群的调节,例如抗生素后,可能会影响艰难梭菌的定植。
4
其他风险因素
除此之外,还有一些因素也会增加艰难梭菌感染的风险。如:
医疗机构暴露:住院患者,特别是长期住院者,长期护理机构居住者容易艰难梭菌感染;
胃酸抑制剂使用:质子泵抑制剂(PPIs)和H2受体拮抗剂,降低胃酸可能导致艰难梭菌孢子存活率增加,长期使用与CDI风险增加相关。
手术和医疗操作:胃肠道手术、鼻胃管和胃肠营养管的使用、结肠镜检查会导致艰难梭菌感染风险升高;
营养状态不佳:低蛋白血症、营养不良的人群易感染艰难梭菌;
肥胖、癌症化疗也可能会增加艰难梭菌感染的风险。
肠道微生物群被破坏是艰难梭菌感染发病的关键机制。健康的肠道菌群对防止艰难梭菌定植和感染至关重要。那么,微生物群究竟如何在这一过程中发挥作用?那我们一起来看看。
艰难梭菌感染中的微生物群改变
正常的肠道菌群通过对艰难梭菌的定植抗性在预防 艰难梭菌感染中起着核心作用。这导致了一个问题,即是否存在一种微生物群紊乱模式,这种模式易导致艰难梭菌定植和感染。
◮ 菌群多样性降低,厚壁菌门增加
多项研究一致显示,与健康人群相比,艰难梭菌感染(CDI)患者的菌群复杂性和丰富度明显降低。这种多样性降低被认为是CDI发病和复发的关键因素。
厚壁菌门(Firmicutes)减少:CDI患者中厚壁菌门的丰度和多样性显著降低,尤其是瘤胃球菌科(Ruminococcaceae)和毛螺菌科(Lachnospiraceae)等保护性菌群。
拟杆菌门(Bacteroidetes)改变:CDI患者通常表现为拟杆菌门多样性降低,但有研究显示有症状患者中某些拟杆菌属(Bacteroides)可能增加。
变形菌门(Proteobacteria)增加:CDI患者体内变形菌门(特别是肠杆菌科)的比例明显增高。
◮ 产丁酸细菌减少,机会性病原体增加
接受多轮抗生素治疗的复发性CDI患者肠道微生物群组成遭受严重破坏。对比CDI患者、艰难梭菌阴性院内腹泻患者和健康对照受试者的远端肠道微生物群发现,艰难梭菌感染可能导致功能性菌群发生以下变化:
产丁酸盐细菌减少:包括罗氏菌属(Roseburia)、普拉梭菌(Faecalibacterium Prausnitzii)、假丁酸弧菌属等。
产乳酸细菌增加:特别是肠球菌属(Enterococcus)。
机会性病原体增加:研究发现,与健康者相比,白色念珠菌和光滑念珠菌在艰难梭菌阳性样本中更常见。
在艰难梭菌感染患者的肠道微生物群中,肠球菌、乳酸菌、大肠杆菌、肠杆菌、副拟杆菌、嗜粘蛋白阿克曼菌的相对丰度增加,以及粪杆菌、Roseburia、Blautia、Prevotella、链球菌的水平降低。
艰难梭菌感染相关的肠道微生物群失调

Vasilescu IM,et al.Front Microbiol.2022
生化和免疫紊乱影响感染
肠道菌群失调还引发多种生化和免疫紊乱:如短链脂肪酸(SCFA)水平降低、初级胆汁酸增多、碳水化合物可利用性提高、免疫功能受抑及竞争微生物缺乏。这些变化共同促进艰难梭菌的定植、孢子萌发和生长繁殖。
◮ 丁酸盐有助于减轻艰难梭菌毒素的损害
短链脂肪酸可以通过降低管腔pH值(对艰难梭菌不利)并通过产生粘蛋白和抗菌肽来刺激防御屏障。丁酸盐还可以通过稳定缺氧诱导因子-1(HIF-1)和增加紧密连接来保护肠道上皮免受艰难梭菌毒素的损害,抑制肠道炎症和细菌易位。
在小鼠的饮用水中添加丁酸盐,施用丁酸盐的前药、三丁酸甘油酯或富含菊粉的饮食(菊粉可以被肠道共生细菌发酵,产生短链脂肪酸,主要是乙酸盐、丙酸盐和丁酸盐)可使小鼠免受CDI的侵害。
◮ 胆汁酸会影响艰难梭菌的定植和生长
艰难梭菌孢子萌发由蛋白酶CspC和CspA感知胆汁盐和氨基酸复合物而调控。某些胆酸盐衍生物和甘氨酸可促进孢子萌发,而脱氧胆酸盐抑制艰难梭菌生长,鹅去氧胆酸盐则阻断牛磺胆酸盐介导的萌发过程。
共生肠道梭菌通过调节胆汁酸代谢产物,创造不利于艰难梭菌萌发和定植的环境。例如,Clostridium scindens能催化胆汁酸7α-脱羟基化,产生次级胆汁酸,增强对艰难梭菌感染的抵抗力。失去这类能将初级胆汁酸转化为具抗菌活性次级胆汁酸的微生物,将显著增加CDI风险。
◮ 艰难梭菌会刺激其他细菌产生吲哚造成不利肠道环境
最近的研究发现,CDI患者肠道腔中的吲哚水平增加(色氨酸代谢物参与微生物生长、毒力诱导、抗酸性、生物膜形成),艰难梭菌本身不能产生这种代谢物,但会刺激其他细菌产生吲哚,以阻止和抑制吲哚敏感菌株的生长和发展,包括保护性肠道微生物群代表, 从而确保有利于艰难梭菌生存的肠道环境。
微生物群介导艰难梭菌的定植和感染
一些人群中虽然有艰难梭菌定植,但并不会出现致病症状,这可能与肠道微生物群的保护作用相关。
◮ 婴儿艰难梭菌的定植率较高,但很少出现症状
由于婴儿肠道的不成熟和肠道微生物群的不稳定,其特别容易受到艰难梭菌定植的影响,但并不会出现症状和发展为疾病。
在<1个月大的婴儿中,艰难梭菌的平均定植率为 37%,范围在0到61%之间。在1到6个月大之间,定植率仍然很高,为30%,出生后第一年结束时下降到约10%。不同研究报告的12个月以下儿童的定植率从14%到71%不等,这个年龄组最常被艰难梭菌定植,并且他们没有症状。
无症状携带率在3岁左右下降至0-3%,接近成人水平。同时,出生至24个月间血清针对毒素A和B的IgG抗体浓度逐渐升高。3岁左右,儿童肠道微生物群趋于稳定并具备成人特征,这可能导致症状性CDI从该年龄开始增加。
注:新生儿微生物群以革兰氏阳性球菌、肠杆菌科或双歧杆菌科为主,逐渐过渡到以双歧杆菌科为主。双歧杆菌可通过上调肠道树突状细胞产生IL-10,解释了艰难梭菌定植婴儿无症状的原因。在剖宫产新生儿中,T细胞和CD4+辅助性T细胞水平降低,可能因未成熟的免疫系统无法激活炎症反应所致。
研究发现,配方奶喂养婴儿的艰难梭菌定植率高于母乳喂养婴儿,且母乳喂养婴儿的菌落计数明显更低,这可能与母乳中含有毒素A和B的抗体有关。
总之,新生儿艰难梭菌定植的高携带率可以用新生儿肠道的不成熟和肠道微生物群的存在来解释。然而,出生后的母乳喂养带有毒素抗体,加上艰难梭菌毒素受体的缺乏,可以帮助婴儿免受艰难梭菌毒素的有害影响。
◮ 肠道微生物多样性降低的人群和老年人更易受艰难梭菌影响
几项研究还比较了老年人群的肠道微生物样本,这些人群更容易受到艰难梭菌影响。
老年艰难梭菌感染患者的肠杆菌科、肠球菌属和乳酸杆菌的数量较高,而健康的老年人携带更多样化的拟杆菌属菌株。并且与任一老年人群相比,健康成年人也更有可能拥有更多的双歧杆菌和拟杆菌。最近使用16S rRNA 基因高通量测序的研究更深入地研究了艰难梭菌阳性人群的群落结构。观察到,与健康患者相比,活动性CDI患者的肠道微生物群多样性较低。
类似研究发现,与健康成人相比,CDI腹泻患者的肠道微生物群多样性显著降低,尤其是厚壁菌门的多样性较低。健康人群中以Lachnospiraceae、Ruminococcaceae和Bacteroidaceae为主导,而CDI和非CDI腹泻患者的微生物群落高度相似,表明腹泻或炎症可能与特定微生物群落相关。
在小鼠模型中进行了类似观察,与人类相似,抗生素降低了小鼠肠道微生物群多样性,使其更易患肠道疾病,包括CDI。研究发现,头孢哌酮、克林霉素或多种抗生素处理后,易感小鼠感染前以乳酸菌科和肠杆菌科为主,而对CDI具有保持抗性的动物以Lachnospiraceae为主。后续研究表明,被Lachnospiraceae定植的小鼠比被大肠杆菌定植的小鼠艰难梭菌定植减少且病情较轻。小鼠模型为识别CDI保护性成分提供了可测试的方法。
◮ 复发性CDI:微生物群恢复不完全
艰难梭菌感染(CDI)最常见的并发症是恢复不完全和反复感染。初次感染后复发率约为20-30%,3次感染后高达60%。研究发现,复发患者的微生物群多样性低于单次CDI患者,这表明可能可以根据感染期间存在的微生物群落预测复发。尽管一些研究在分析中包含了复发样本,但尚未确定复发性CDI患者特有的微生物特征。
另外有研究发现,复发性CDI患者体内初级胆汁酸浓度较高。粪菌移植后,次级胆汁酸浓度增加,接近健康供体水平,而这些酸在FMT前样本中未检测到。该结果与体外和小鼠研究一致,表明次级胆汁酸(如石胆酸和脱氧胆酸)可抑制艰难梭菌生长。尽管细菌群落决定代谢环境,但不同细菌群落可能通过相似功能实现相同代谢结果,仅靠群落结构可能不足以预测复发风险。
如何定义艰难梭菌感染病例?
只有产生毒素的产毒艰难梭菌菌株才具有致病性。根据欧洲临床微生物学和传染病学会(ESCMID)指南,CDI定义为:(i)符合CDI的临床表现,且粪便中检测到毒素A或毒素B的艰难梭菌,且无其他腹泻原因;或(ii)伪膜性结肠炎(PMC)患者。
美国医疗保健流行病学学会和美国传染病学会的定义类似:CDI病例需具备症状(通常为腹泻),且粪便检测产毒艰难梭菌或其毒素阳性,或结肠镜或组织病理学显示PMC。
那我们该如何检测艰难梭菌感染(CDI)?艰难梭菌感染检测通常包括以下方法:
1
临床评估
•症状:难梭菌感染病的主要症状包括腹泻(每天3次或以上稀便,持续2天以上)、腹痛、发热、恶心和脱水等。严重病例可能出现伪膜性结肠炎(PMC)、中毒性巨结肠或感染性休克。
•病史:重点关注患者的抗生素使用史(尤其是第三代头孢菌素、氟喹诺酮类等)、住院史、免疫抑制状态以及质子泵抑制剂(PPI)的使用。
2
实验室检测方法
◮ 细菌培养
用环丝氨酸头孢西丁果糖琼脂培养基(CCFA)等进行厌氧培养,需时72小时,灵敏度高且可获得菌株,但不能检测毒素不能区分非产毒株。
但分离菌株后,可以通过直接从菌落悬浮液或细菌生长的肉汤上清液中检测其体外毒素的产生来确定其致病潜力。
◮ 高通量测序
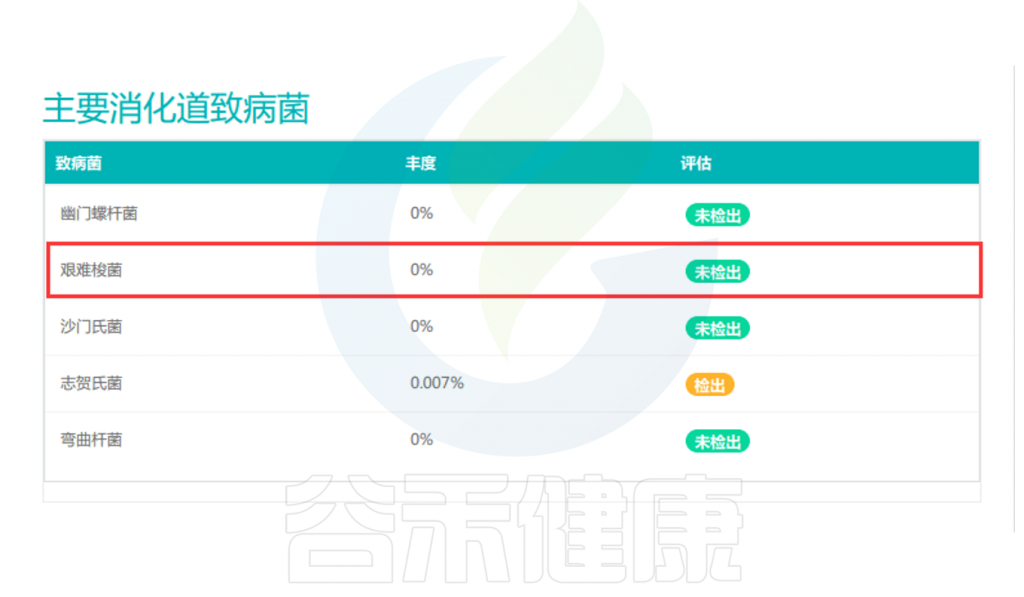
高通量测序可检测艰难梭菌菌株或毒力基因,16S测序仅能分辨到物种层面,可通过谷禾16S健康报告的解读,并结合自身有没有腹泻症状。然后进一步去判别是否存在产毒菌株。
与此同时,宏基因组测序(包括一些靶向的测序)则可识别毒力基因。
◮ 谷氨酸脱氢酶测定
谷氨酸脱氢酶(GDH)是所有艰难梭菌菌株表达的代谢酶,可通过ELISA或免疫层析法检测。阳性结果仅表明存在艰难梭菌,无法判断其是否产毒。由于其高阴性预测值(NPV,80.0%-100%),GDH阴性结果通常可排除感染,常用于初筛。
◮ 毒素检测
细胞毒性测定被认为是检测艰难梭菌的游离毒素(主要是毒素 B)的参考方法。
酶联免疫吸附试验(EIA):检测艰难梭菌毒素A或毒素B,快速但敏感性较低。
细胞毒素中和试验:检测毒素活性,敏感性和特异性较高,但耗时较长。
◮ 核酸扩增检测(NAATs)
实时PCR或环介导等温扩增法(LAMP) :检测艰难梭菌毒素基因,耗时短,敏感性高,但可能检测到无症状定植者,且成本高。
3
综合算法
根据欧洲临床微生物学和传染病学会(ESCMID)指南,推荐使用两步或三步算法:
初筛:谷氨酸脱氢酶(GDH)抗原检测或核酸扩增试验(NAAT)。
确认:毒素检测(EIA或细胞毒素试验)。
4
其他辅助检查
血常规:轻、中度感染者外周血白细胞可正常,严重感染者白细胞升高。
降钙素原(PCT):对诊断CDI意义不大,但PCT>0.2 ng/ml时,提示CDI有重症化趋势。
腹部CT:对重症CDI患者有辅助诊断意义,即结肠壁增厚、结节状结肠袋增厚、水肿厚度>4 cm,特别是炎症部位在升结肠。
内镜检查:内镜检查可作为辅助诊断,典型征象包括黏膜充血、水肿、糜烂、溃疡、直肠乙状结肠有多发性隆起的斑片或融合为大片的灰绿色、黄褐色伪膜覆盖黏膜表面。部分患者表现可不典型,尤其炎症性肠病合并艰难梭菌感染时多无特征性病变。伪膜性改变可能表明感染了产毒艰难梭菌。
艰难梭菌感染(CDI)的治疗包括针对细菌(抗生素)、针对毒素(抗体、结合剂)、宿主反应(调节炎症)或微生物群(保护或恢复)的方法,以及在暴发性病例或非手术方法失败的情况下,结肠切除术或其他侵入性较小的手术。选择取决于疾病严重程度、病史和宿主因素。
1
针对细菌——抗生素
停用有问题的抗生素以恢复正常肠道菌群是治疗CDI的理想方法。但在感染负担高或需持续抗生素治疗其他感染时,通常需要使用针对艰难梭菌的抗菌药物。
然而,几乎所有抗生素,包括针对艰难梭菌的药物,都可能进一步破坏肠道微生物群,延迟耐药性菌群的恢复,并增加再次感染的风险。目前用于治疗CDI的主要抗菌药物包括:
•万古霉素:万古霉素是美国食品药品监督管理局(FDA)批准的第一种CDI药物。最近的临床试验数据表明,万古霉素在治疗重症时优于甲硝唑。
•甲硝唑:与万古霉素的疗效相当,成本优势以及对万古霉素耐药肠球菌传播的担忧,甲硝唑成为CDI的推荐药物。但随着艰难梭菌流行菌株 BI/NAP1/027/III 的出现,甲硝唑治疗失败的报道越来越多。
•非达霉素:非达霉素也是获得FDA批准的其他治疗CDI药物。
•新型小分子抗菌剂:乳酸菌素3147是一种由乳酸乳球菌产生的双组分抗生素,它靶向细胞壁前体脂质II,抑制肽聚糖的生物合成,并在细胞膜上形成孔,实现细胞死亡。粪便发酵模型证明了乳酸素 3147 对一系列艰难梭菌分离株的强效细胞杀伤活性,在 30 分钟内完全消除艰难梭菌。
与传统药物相比,这种窄谱抗菌药物可能具有多种优势,包括减少对微生物组的影响、降低复发率、优于常规治疗以及改善药代动力学特征。然而,在保持非孢子形成厌氧菌和总革兰氏阴性厌氧菌完好无损的同时,这种抗菌剂对乳酸杆菌和双歧杆菌产生了负面影响。
2
针对毒素——阻断、中和毒素
CDI的发病机制以毒素介导为主,因此中和或阻断毒素是合理策略。但尽管抗毒素在动物模型中可改善疾病,但在人类中仅表现为减少复发性CDI。
•单克隆抗体:一项随机双盲安慰剂对照研究评估了针对艰难梭菌毒素A和B的两种中和单克隆抗体。在200名参与者中,治疗组的CDI复发率显著低于安慰剂组。然而,单克隆抗体未能缩短腹泻持续时间、降低严重程度或减少住院时间。住院、年龄较大、有严重潜在疾病或重症CDI的患者对治疗反应较差。
•Tolevamer:Tolevamer 是一种高分子量苯乙烯磺酸盐聚合物,可在体外中和艰难梭菌毒素。一项针对轻中度疾病的研究显示,6克剂量的Tolevamer与每日500毫克万古霉素疗效相当。
注:然而,Tolevamer的临床成功率低于甲硝唑和万古霉素。汇总分析显示,Tolevamer组的临床成功率为44.2%,显著低于甲硝唑组的72.7%和万古霉素组的81%。随着疾病严重程度增加,Tolevamer的疗效进一步下降。
•免疫球蛋白(IG):在人体研究中,针对毒素A和B的血清IgG抗体与保护相关,静脉注射IG治疗复发性或重症CDI的效果尚无系统研究支持,仅有少量病例报告和系列报道提供个案证据。剂量、给药次数和间隔存在较大差异。静脉注射IG对临床效果的差异可能与人群中抗毒素抗体水平的不可预测性有关。
3
微生物疗法
肠道微生物群的扰动是艰难梭菌感染(CDI)发生的关键因素。一些抗生素治疗无症状感染会导致艰难梭菌持续脱落和复发风险增加。通过特定微生物群恢复受损的肠道微生物群,已成为打破CDI-抗生素-CDI循环的策略。
• 粪菌移植的治疗效果优于仅用抗生素
粪菌移植(FMT)或微生物群替代疗法的原理是通过正常供体的粪便恢复CDI患者受损的肠道微生物群。系统评价显示,CDI患者接受FMT的腹泻消退率约为77%-90%。
一项随机对照试验比较了粪菌移植(FMT)与两种对照方案。FMT组在口服万古霉素(500mg,每天4次)4-5天后接受移植,对照组则接受14天相同剂量的万古霉素治疗。结果显示,FMT组3个月症状消退率为81%,显著高于万古霉素组(31%)和万古霉素加灌肠组(23%)。
尽管FMT已使用数十年,但其对严重复杂CDI的疗效、免疫功能低下患者的安全性及不明供体粪便长期安全性仍存疑。据报道,28.5%的患者出现FMT相关不良事件,最常见为腹部不适和排便异常,具体取决于给药途径。
• 口服微生物组疗法—SER-109
SER-109是一种由活性厚壁菌门细菌孢子组成的口服微生物组疗法,用于降低艰难梭菌感染复发风险。其机制可能通过与艰难梭菌竞争必需营养物质或调节胆汁酸谱来重建定植抵抗力,或两者兼具。
进行了一项3期、双盲、随机、安慰剂对照试验,其中艰难梭菌感染发作3次或以上(包括符合条件的急性发作)的患者在标准护理抗生素治疗后接受SER-109或安慰剂(每天4粒胶囊,持续3天)。
结果显示,在接受标准护理抗生素治疗后艰难梭菌感染症状消退的患者中,口服SER-109在降低复发感染风险方面优于安慰剂。
以微生物群为中心的治疗方法

Vasilescu IM,et al.Front Microbiol.2022
4
手术干预
艰难梭菌感染(CDI)的手术治疗通常用于严重或复杂病例,尤其是当药物治疗(如抗生素或粪便微生物群移植)无效,或患者出现危及生命的并发症时。以下是艰难梭菌感染的主要手术治疗方法:
• 全结肠切除术
适应症:严重的中毒性结肠炎、肠穿孔或肠坏死;严重的腹腔感染(腹膜炎),药物治疗无效且病情迅速恶化。
优点:快速去除感染源,降低死亡风险。
缺点:创伤较大,术后恢复时间长,可能影响生活质量。
• 分段结肠切除术
适应症:感染局限于结肠的某一部分。
优点:保留更多的肠道功能。
缺点:感染可能扩散,手术效果不如全结肠切除术稳定。
• 回肠造口术联合结肠灌洗
适应症:不适合全结肠切除的高危患者(如老年人或伴有多种合并症)。
优点:创伤较小,适合高危患者。
缺点:疗效可能不如全结肠切除术。
• 腹腔引流术
适应症:腹腔内脓肿或局部感染。
优点:辅助治疗,减轻感染负担。
缺点:仅适用于局部并发症,不能根治感染。
5
噬菌体疗法
噬菌体疗法利用天然噬菌体感染并裂解病原菌,是应对抗菌药物耐药性危机的潜在治疗方法。与抗生素不同,噬菌体具有进化能力,可持续克服细菌的抗性机制,从而避免疗法过时。
• 噬菌体可以降低艰难梭菌的生长和毒素水平
一些噬菌体在治疗艰难梭菌感染中表现出潜力。例如,ФCD27在CDI分批发酵模型中可显著降低艰难梭菌的生长和毒素水平。一种噬菌体组合已在体外完全裂解艰难梭菌,并在小鼠模型中减轻疾病症状和细菌定植。该组合进一步优化为4种噬菌体,成功在发酵容器中彻底根除艰难梭菌,显示出作为治疗选择的可行性。
总体而言,特异性以及对抗细菌耐药性的能力表明噬菌体治疗作为艰难梭菌治疗剂的巨大前景。
6
通过疫苗预防艰难梭菌感染
随着艰难梭菌感染(CDI)的发病率、死亡率和医疗成本不断上升,预防疾病的免疫接种成为理想选择。人类研究表明,对艰难梭菌毒素的强体液反应可减少复发和无症状定植。目前,多种候选疫苗正在开发中,包括基于类毒素、重组毒素肽、DNA和表面蛋白抗原的疫苗,但大多处于临床前阶段。
• 疫苗有助于减少复发和无症状定植
已发现部分纯化的类毒素A和B疫苗在30名健康成人中具有安全性和免疫原性,≥90%的受试者对两种毒素产生血清抗体反应。在一项试点研究中,3名多次复发CDI患者在第0、7、28和56天接受类毒素肌肉注射,其中2人对毒素A和B的IgG水平显著升高,且3人均在疫苗接种后停用万古霉素,随访6个月无复发。
两项2期试验已完成,测试疫苗在高危中老年人和首发CDI患者中的效果。一项3期试验正在17个国家中进行,计划评估疫苗在多达15,000名参与者中预防首发CDI的效果。其他候选疫苗包括基因和化学修饰的全长TcdA和B及重组融合蛋白IC84,其1期研究已完成,但结果尚未公布。
7
通过益生菌预防艰难梭菌感染
• 益生菌有助于预防艰难梭菌定植及相关腹泻
多项小型研究及荟萃分析表明,益生菌的使用可能与预防艰难梭菌相关腹泻有关。益生菌通常通过抑制艰难梭菌定植、调节肠道微生物群和胆汁酸代谢、破坏细胞壁和细胞膜、下调炎症反应、改善肠道屏障功能以及缓解病原性结肠炎来预防和治疗CDI。
最常研究的益生菌为嗜酸乳杆菌和双歧杆菌属。最近,一项多中心、随机、双盲、安慰剂对照研究测试了含嗜酸乳杆菌和双歧杆菌(双歧双歧杆菌和乳双歧杆菌)的高剂量益生菌制剂对≥65岁接受抗生素治疗患者的疗效。1,493名受试者接受益生菌,1,488名接受安慰剂。分析显示,益生菌组发生抗生素相关性腹泻和艰难梭菌感染的数量少于安慰剂组。
• 益生菌发挥抗菌活性抑制艰难梭菌定植
短双歧杆菌(YH68)通过抑制生长、孢子形成、毒力发生及毒力基因表达,展现出抗艰难梭菌活性,并增强抗艰难梭菌抗生素的体外效果或预防体内临床表现。
克劳氏芽孢杆菌和罗伊氏乳杆菌可分泌直接抑制艰难梭菌的可溶性化合物,而Clostridium scindens等产生次生胆汁酸的菌体可增强艰难梭菌的定植抗性。
此外,研究发现了一些可能有效的新型益生菌和制剂,如:
Bacteroides thetaiotaomicron,
E.thailandicus strain d5B,
B.amyloliquefaciens C-1,
B.longum JDM301,
Pediococcus pentosaceus LI05,
B.breve (YH68)
未来需要更大样本量的多中心双盲研究,以明确益生菌在CDI中的作用,同时考虑菌株和抗菌剂类型等因素。
艰难梭菌感染作为一种全球性公共卫生挑战,其发病率和医疗负担正不断攀升。通过本文的系统介绍,我们可以了解到,艰难梭菌感染的致病性主要取决于其产生的毒素,而非仅仅是菌株的存在。
健康的肠道微生物群在预防艰难梭菌感染中扮演着至关重要的角色,肠道微生物平衡的破坏是导致感染风险增加的关键因素。随着科学研究的深入,我们对艰难梭菌的检测、诊断、治疗和预防手段也在不断完善,从传统抗生素到粪菌移植、益生菌干预和疫苗开发等多元化策略均显示出积极成效。
了解艰难梭菌感染的本质和发病机制,对于临床医生制定合理治疗方案、减少不必要的抗生素使用以及患者接受适当预防措施至关重要。希望在未来研究的推动下,我们能够进一步降低艰难梭菌感染的发病率和复发率,减轻其对公共健康的威胁。
注:本账号内容仅供学习和交流,不构成任何形式的医疗建议。
主要参考文献
Buddle JE, Fagan RP. Pathogenicity and virulence of Clostridioides difficile. Virulence. 2023 Dec;14(1):2150452.
Piccioni A, Rosa F, Manca F, Pignataro G, Zanza C, Savioli G, Covino M, Ojetti V, Gasbarrini A, Franceschi F, Candelli M. Gut Microbiota and Clostridium difficile: What We Know and the New Frontiers. Int J Mol Sci. 2022 Nov 1;23(21):13323.
Wu Y, Wang YY, Bai LL, Zhang WZ, Li GW, Lu JX. A narrative review of Clostridioides difficile infection in China. Anaerobe. 2022 Apr;74:102540.
Arcay R, Barceló-Nicolau M, Suárez L, Martín L, Reigada R, Höring M, Liebisch G, Garrido C, Cabot G, Vílchez H, Cortés-Lara S, González de Herrero E, López-Causapé C, Oliver A, Barceló-Coblijn G, Mena A. Gut microbiome and plasma lipidome analysis reveals a specific impact of Clostridioides difficile infection on intestinal bacterial communities and sterol metabolism. mBio. 2024 Oct 16;15(10):e0134724.
Vasilescu IM, Chifiriuc MC, Pircalabioru GG, Filip R, Bolocan A, Lazăr V, Diţu LM, Bleotu C. Gut Dysbiosis and Clostridioides difficile Infection in Neonates and Adults. Front Microbiol. 2022 Jan 20;12:651081.
Maslanka JR, Gu CH, Zarin I, Denny JE, Broadaway S, Fett B, Mattei LM, Walk ST, Abt MC. Detection and elimination of a novel non-toxigenic Clostridioides difficile strain from the microbiota of a mouse colony. Gut Microbes. 2020 Nov 9;12(1):1-15.
Nibbering B, Gerding DN, Kuijper EJ, Zwittink RD, Smits WK. Host Immune Responses to Clostridioides difficile: Toxins and Beyond. Front Microbiol. 2021 Dec 21;12:804949.
Samarkos M, Mastrogianni E, Kampouropoulou O. The role of gut microbiota in Clostridium difficile infection. Eur J Intern Med. 2018 Apr;50:28-32.
Kelly CR, Fischer M, Allegretti JR, LaPlante K, Stewart DB, Limketkai BN, Stollman NH. ACG Clinical Guidelines: Prevention, Diagnosis, and Treatment of Clostridioides difficile Infections. Am J Gastroenterol. 2021 Jun 1;116(6):1124-1147.
Dawson LF, Valiente E, Wren BW. Clostridium difficile–a continually evolving and problematic pathogen. Infect Genet Evol. 2009 Dec;9(6):1410-7.

谷禾健康

目前随着高通量测序,微生物以及代谢、蛋白组学和高精度影像学等快速的发展,有关人体疾病或健康相关的海量数据正在不断产生,依托各种算法尤其AI的算法等,涌现出很多优秀的“模型”用于预测人体疾病以及健康相关的辅助诊断或筛查。
谷禾在人体肠道菌群检测领域深耕多年,积累了大量肠道菌群和个体健康信息数据,并运用建模方法结合人工智能等算法构建了多种疾病和营养预测模型。
对于不是从事专业的人员或大众来说,什么样的预测模型是准确的?如何评价一个模型的好坏?里面涉及比较多的概念和知识,今天谷禾和大家简单聊下这方面的内容。
首先大家评估下:
对于肿瘤筛查,以一个人群发病率1%的肿瘤为例:
模型A的检出率50%,假阳性为1%
模型B的检出率为90%,但是假阳性是10%
请问哪个检测模型作为肿瘤筛查类产品更好 ?
换句话说就是回答:“鉴于测试结果为阳性,该患者患有这种疾病的可能性有多大?
为了回答上面问题,我们需要了解和正确认识以下模型评价参数/指标:
• 该测试的阳性预测值(PPV)是多少?——返回阳性结果的人实际上患有疾病的概率是多少?
• 该测试的阴性预测值(NPV)是多少? ——返回阴性结果的人实际上是健康的概率是多少?
• 测试的灵敏度如何?——它正确地将多少个患病个体识别为患病?
• 测试的特异性是什么?——它正确地将多少个健康个体识别为健康个体?
要回答这个问题,我们需要计算两个模型的阳性预测值(Positive Predictive Value, PPV)。然后一步步分析:
▸ 什么是阳性预测值(PPV)?
阳性预测值(PPV)是样本检测阳性为真阳性的概率值。
发病率较低时的阳性预测值(PPV)
模型A
人群发病率:1%
检出率:50%
假阳性率:1%
模型B
人群发病率:1%
检出率:90%
假阳性率:10%
接下来使用贝叶斯定理来计算每个模型的阳性预测值。
对于模型A:
真阳性率 = 1% × 50% = 0.5%
假阳性率 = 99% × 1% = 0.99%
总阳性结果 = 真阳性 + 假阳性 = 0.5% + 0.99% = 1.49%
阳性预测值(PPV) = 真阳性 ÷ 总阳性结果 = 0.5% ÷ 1.49% ≈ 33.6%
对于模型B:
真阳性率 = 1% × 90% = 0.9%
假阳性率 = 99% × 10% = 9.9%
总阳性结果 = 真阳性 + 假阳性 = 0.9% + 9.9% = 10.8%
阳性预测值(PPV) = 真阳性 ÷ 总阳性结果 = 0.9% ÷ 10.8% ≈ 8.3%
模型A的PPV为33.6%,而模型B的PPV仅为8.3%。
因此,我们可以看到尽管模型B的检出率更高(90%vs50%),但其阳性预测值(PPV)显著低于模型A。
★ 低发病率人群中,高特异性比高敏感性更重要
这意味着:
模型A的阳性结果更可靠
模型A的假阳性风险更低
模型A在这种低发病率的情况下,作为肿瘤筛查产品更好。
这里对于我们应用的启示:在低发病率的人群中,高特异性(低假阳性率)比高敏感性(高检出率)更重要。选择产品时,不仅要看检出率,还要关注假阳性率和阳性预测值。

发病率较高的阳性预测值(PPV)
可能接下来有人会问,这个问题中以肿瘤发病率比较低的例子举例,那对于发病率比较高的疾病,比如心脑血管、糖尿病等如何?
接下来,我们继续举例:
让我们用同样的方法计算发病率为20%时的阳性预测值(PPV)。
模型A
人群发病率:20%
检出率:50%
假阳性率:1%
模型B
人群发病率:20%
检出率:90%
假阳性率:10%
一样的计算
对于模型A:
真阳性率 = 20% × 50% = 10%
假阳性率 = 80% × 1% = 0.8%
总阳性结果 = 真阳性 + 假阳性 = 10% + 0.8% = 10.8%
阳性预测值(PPV) = 真阳性 ÷ 总阳性结果 = 10% ÷ 10.8% ≈ 92.6%
对于模型B:
真阳性率 = 20% × 90% = 18%
假阳性率 = 80% × 10% = 8%
总阳性结果 = 真阳性 + 假阳性 = 18% + 8% = 26%
阳性预测值(PPV) = 真阳性 ÷ 总阳性结果 = 18% ÷ 26% ≈ 69.2%
可以看到当发病率提高到20%时,情况发生了显著变化:
模型A的PPV提高到92.6%
模型B的PPV提高到69.2%
对比之前1%发病率的情况:
模型A:PPV从33.6%提高到92.6%
模型B:PPV从8.3%提高到69.2%
可以发现随着发病率的提高,两个模型的阳性预测值都大幅提升。但是模型A仍然保持更高的PPV,但优势不如低发病率时那么明显。

★ 发病率越高,阳性预测值越高
发病率越高,阳性预测值越高,这是因为真阳性的比例增加。
因此在高发病率的人群中,两种模型的检测价值都显著提高。但模型A仍然是更好的选择,因为其假阳性率更低。
评估不同检测模型和询问检测准确率时,要综合考虑不同疾病的发病率、检出率和假阳性率。
对于要检出阳性结果而言,这个例子就很好地说明了发病率对检测结果解读的重要影响,也体现了贝叶斯定理在医学诊断中的应用。
而阳性预测值(PPV)具有多维意义,对于不同视角的人群可能意义不同:
➤ 政府视角下的PPV
⑴公共卫生决策基础
资源分配效率:高PPV意味着确诊投入的资源浪费更少,每发现一个真实病例的成本更低;
筛查项目评估:决定是否推行大规模筛查项目时,PPV是核心考量因素之一;
卫生经济学分析:假阳性导致的后续检查、治疗和心理干预成本需纳入卫生政策评估。
⑵监管与标准制定
差异化监管:对不同严重程度疾病的筛查产品可设置不同PPV要求;
强制信息披露:要求厂商在不同疾病流行率下公开产品的PPV值;
患者保护:防止低PPV产品导致过度医疗和不必要的医源性伤害。
➤ 受检者视角下的PPV
⑴个人决策与心理影响
决策参考价值:“我的阳性结果有多可靠?”—这是患者最关心的问题;
心理负担差异:假阳性可能导致严重心理压力,尤其对严重疾病筛查;
后续检查意愿:了解PPV有助于患者决定是否及时进行确诊检查。
⑵风险认知与理解
个体化解读:同一PPV值对不同风险人群的意义不同;
检前概率影响:有症状个体的PPV通常高于无症状筛查人群;
教育需求:医生需帮助患者理解PPV与个人情况的关联。
➤ 产品评估的情境差异
⑴临床应用场景分析
筛查工具:可接受相对较低PPV,但应有明确后续路径;
确诊工具:需要更高PPV,减少误诊风险;
监测工具:对症状性疾病监测,PPV要求可能介于两者之间。
⑵疾病特性影响
致命但可治疗疾病:可接受较低PPV,以高灵敏度优先;
慢性管理疾病:需较高PPV避免不必要长期干预;
高耻感疾病:需更高PPV避免标签化伤害。
⑶社会经济环境考量
医疗资源丰富地区:可能更关注灵敏度,接受较低PPV;
资源有限环境:高PPV更为重要,避免资源浪费;
支付方式影响:商业保险vs自费vs政府支付下对PPV的需求不同。
➤ PPV优化与应用建议
⑴预测模型调整
风险分层应用:根据人群特征调整决策阈值;
多指标综合评估:结合阴性预测值(NPV)、阳性/阴性似然比(LR+/LR-)综合评估;
贝叶斯思维应用:根据先验概率(患病率)调整PPV期望。
⑵实际应用最佳实践
透明沟通:向受检者清晰解释结果可靠性;
分级报告:提供风险概率而非简单阳性/阴性结果;
智能决策支持:AI辅助工具结合临床特征提供个性化PPV估计。
那么下面我们看下什么是阴性预测值(NPV),简言之就是把健康人检测为健康的概率。
阳性预测值(PPV)是样本检测阳性为真阳性的概率值,而阴性预测值(NPV)是样本检测真阴性的概率值。
阴性预测值(NPV) =(真阴性数量)/(真阴性数量+假阴性数量)
例如你有一个600人的样本量,根据有效性,假设你知道肯定患有这种疾病的样本(480)或没有这种疾病的个体样本(120)。
测试后,将结果与已知的疾病状态进行比较,发现:
真阳性(测试阳性和正确阳性)= 480
假阳性(试验阳性但实际为阴性)= 15
真阴性(测试阴性和真阴性)= 100
假阴性(试验阴性,但实际为阳性)=5
我们可以计算PPV和NPV如下:
阳性预测值(PPV)=480/(480+15)≈0.97(97%)
阴性预测值(NPV)=100/(100+5)≈0.95(95%)
因此,如果检测结果为阳性,则有97%的机会是正确的,如果结果为阴性,则有95%的机会是正确的。
拓展:什么是假阳性和假阴性?
假阳性:健康人被错误地识别为患病;
假阴性:病人被错误地认为是健康;
真阳性:患病的个体已被正确识别为患有疾病;
真阴性:未患有疾病的个体已被正确诊断为未患有疾病。
或:
真阳性:患者患有疾病且检测呈阳性;
假阳性:患者没有疾病,但检测呈阳性;
真阴性:患者没有疾病,检测结果为阴性;
假阴性:患者患有疾病,但检测结果为阴性。
敏感性
敏感性(Sensitivity)是测试正确识别疾病患者的能力,也称为真阳性率(TPR),即被正确识别为患有疾病的患者的百分比。
敏感性的计算公式为:
敏感性 = 真阳性数 / (真阳性数 + 假阴性数)
或者表示为:
敏感性 = TP / (TP+FN)
其中:
TP (True Positive): 真阳性,即测试正确地将患病者判断为阳性的数量;
FN (False Negative): 假阴性,即测试错误地将患病者判断为阴性的数量。
从另一个角度看,敏感性也可以表示为:
敏感性 = 真阳性率 = 1 – 假阴性率
敏感性的值范围在0到1之间,通常以百分比表示。例如,敏感性为0.95或95%意味着测试能够正确识别95%的实际患病者。
★ 高敏感性特别适用排除诊断
在医学诊断中,高敏感性的测试特别适用于排除诊断(rule-out test),因为如果一个高敏感性测试的结果为阴性,则疾病存在的可能性很小(即”阴性结果可靠”)。
例如:表述中敏感性为100%的检测可正确识别所有患者,敏感性为80%的检测可检出80%的疾病患者(真阳性),但20%的疾病患者未被检测到(假阴性)。
当检测到非常严重的感染类型时,例如前两年进行的COVID-19大流行,高敏感性对于适当的管理和治疗非常重要。COVID-19 IgG/IgM诊断测试是一种筛查COVID-19的测试,据报道敏感性为95%。
当测试用于识别严重但可治疗的疾病(例如宫颈癌)时,高敏感性显然很重要。通过宫颈涂片检测筛查女性人群是一项敏感的检测。然而,它不是很特异,很大一部分宫颈涂片阳性的女性继续进行阴道镜检查,最终被发现没有潜在的病变。

特异性
临床试验的特异性是指检测正确识别无病患者的能力。
特异性的计算公式是:
特异性 = 真阴性数 /(真阴性数 + 假阳性数)
Specificity = TN/(TN + FP)
其中:
TN (True Negative): 真阴性,即正确识别出的健康者数量
FP (False Positive): 假阳性,即错误地将健康者判断为患病的数量
特异性也可以表示为: 特异性 = 正确识别的健康者数/所有实际健康者总数
或: 特异性 = 真阴性率 = 1 – 假阳性率
例如:假设一项检测在100名实际健康者中:
95人被正确判断为阴性(TN = 95)
5人被错误判断为阳性(FP = 5)
特异性 = 95/(95 + 5) = 95/100 = 0.95 或 95%
特异性表明该检测能够正确识别95%的健康者,有5%的健康者被误判为患病。
TIPs:
一种具有高敏感性但低特异性的测试会导致许多没有疾病的患者被告知他们有疾病的可能性,使受检者要接受进一步的测试。理想情况下,测试应该是100%准确的,但这是一个不切实际的场景。或者,对患者进行高敏感性和低特异性的测试,然后进行低敏感性和高特异性的第二次测试,可以识别所有假阳性和假阴性。
在考虑检测对临床医生的价值时,阳性和阴性预测值很有用。它们取决于该疾病在目标人群中的患病率。
但是灵敏度(sensitivity)、特异性(specificity)这两术语对于理解和评估模型的效用也至关重要。敏感性和特异性是用于评估临床试验的术语,它们与测试的目标人群无关。
★ 敏感性和特异性取决于检测阳性的临界值
检测的敏感性和特异性取决于高于或低于检测阳性的临界值。一般来说,敏感性越高,特异性越低,反之亦然。
★ 阳性和阴性预测值取决于被测人群和患病率
与敏感性和特异性不同,阳性预测值(PPV)和阴性预测值(NPV)取决于被测人群,并受疾病患病率的影响。
例如以下示例:使用抗核抗体在普通人群中筛查系统性红斑狼疮(SLE)的PPV较低,因为它产生的假阳性数量很高。然而,如果患者有SLE的迹象(例如 颊部潮红和关节疼痛),测试的PPV会增加,因为患者所在的人群不同(从SLE患病率低的一般人群到患病率高得多的临床可疑人群)。
另一个举例也可以考虑产后出现呼吸困难的女性,其中一种鉴别诊断是肺栓塞。在该患者群体中,D-二聚体检测几乎肯定会升高;因此,该测试的肺栓塞PPV较低。然而,肺栓塞的NPV很高,因为低D-二聚体不太可能与肺栓塞相关。
★ 患病率高时,PPV和NPV准确性更高
阳性预测值(PPV)和阴性预测值(NPV)对疾病患病率的依赖性可以用数字来说明:例如,在一个4000人的人群中,病人和健康人各占一半。该病症筛查试验的灵敏度和特异性均为99%。因此,筛查结果将产生1980个真阳性和1980个真阴性,其中20名健康人被误判为阳性,20名病人被误判为阴性。
该测试的PPV为99%。但是,如果人口中的患病人数只有200人,而健康人数为 3800 人,则假阳性数量从20增加到38人,PPV下降到84%。
本举例实际上想强调这样一个事实,即诊断或筛查疾病的能力既取决于检测的区分价值,也取决于疾病在相关人群中的患病率。
与此同时,要注意:
PPV的互补值是错误发现率(FDR),NPV的互补值是错误遗漏率(FOR),分别等于1减去PPV或 NPV。FDR是错误的结果或 “发现” 的比例。FOR 是被错误拒绝的假阴性的比例。从本质上讲,PPV和NPV越高,FDR和FOR就越低——这对您测试结果的可靠性来说是个好消息。
如果结果是按值的滑动比例给出的,而不是明确的阳性或阴性,则敏感性和特异性值尤为重要。它们允许确定在何处绘制预测结果为阳性或阴性的临界值,甚至可能建议重新测试的灰色区域。
例如:谷禾结直肠癌模型
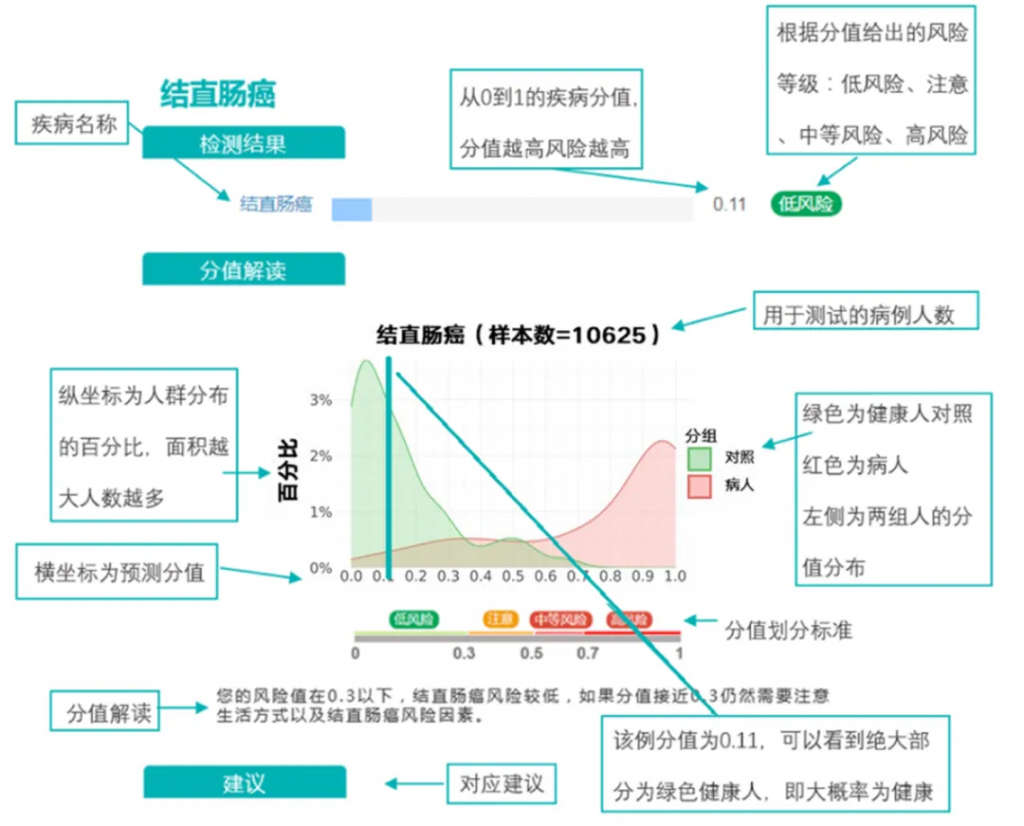
例如,通过将阳性结果的临界值置于非常低的水平,可以捕获所有阳性样品,因此测试非常敏感。但是,这可能意味着许多实际为阴性的样本可能被视为阳性,因此该测试将被视为特异性较差。找到平衡点对于有效和可用的测试至关重要。
★ ROC曲线有助于平衡假阴性和假阳性
在科研项目分析中经常使用受试者工作特征 (ROC)曲线有助于达到最佳平衡点并平衡假阴性和假阳性。但是,对于假阴性是否比假阳性问题更小,或者反之亦然,分析的内容也很重要。例如,在生死攸关的问题上,那么您可能愿意容忍更多的假阳性,以避免遗漏任何真阳性。
▸ 什么是ROC曲线?
ROC曲线是一种图形表示,显示测试的敏感性和特异性如何相互变化。为了构建 ROC 曲线,使用该检验测量已知为阳性或阴性的样本。
对于给定的截止值,将TPR(敏感性)与FPR(1-特异性)作图,得到类似于下图的曲线。理想情况下,选择曲线肩部周围的一个点,这既能限制假阳性,又能最大限度地提高真阳性。
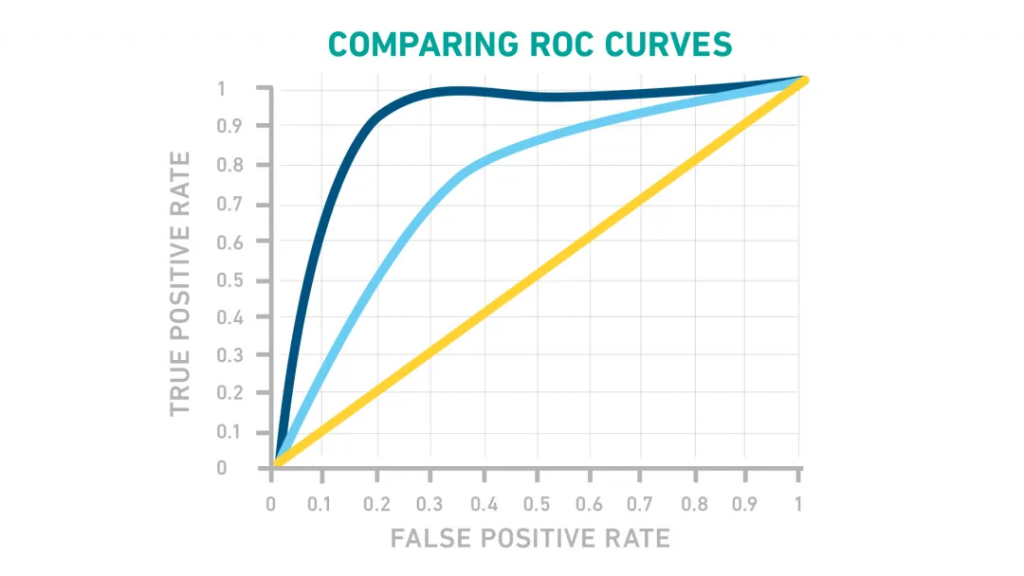
图片来源:Technology Networks
给出ROC曲线(如黄线)的测试并不比随机猜测好,淡蓝色很好,但由深蓝色线表示的测试会更好。这将使临界值确定相对简单,并以非常低的假阳性率产生高真阳性率。
在医疗科技和人工智能快速发展的今天,我们面临着海量的健康相关数据和不断涌现的疾病预测模型。在选择和评估医疗检测产品时,必须综合考虑疾病特性、人群特征和具体应用场景。对于普通大众和医疗从业者而言,理解这些指标不仅是一种专业素养,更是做出明智医疗决策的基础。
我们需要超越简单的”阳性”或”阴性”结果,深入理解检测结果背后的概率和风险。正如本文所强调的,一个优秀的医疗检测模型,应该能够在保证高敏感性的同时,最大限度地降低假阳性和假阴性的风险,为个人健康和公共卫生提供更精准、更有价值的信息。
主要参考文献:
technologynetworks.com/analysis/articles/sensitivity-vs-specificity-318222
Faith Mokobi. What is Sensitivity, Specificity, False positive, False negative? 2021 April 18, Microbenotes.

谷禾健康
扭链瘤胃球菌(Ruminococcus torques)是一种革兰氏阳性厌氧细菌,杆状、不形成孢子,不运动。根据环境条件可呈现链状或簇状排列,最适生长温度为37°C。
R.torques含有多种编码降解复杂碳水化合物的酶系,是一种纤维素分解细菌,这意味着它可以降解纤维素,将纤维素分解为葡萄糖和其他糖。在单糖中可以利用葡萄糖、果糖、半乳糖、N-乙酰半乳糖胺和N-乙酰葡萄糖胺生长,还能参与胆汁酸的代谢。通过代谢能够产生短链脂肪酸(主要是乙酸和丙酸,以及少量丁酸),是一种产丁酸盐的细菌。
R.torques最显著的特征是其强大的粘蛋白降解能力,可以利用MUC2(人肠道中主要分泌的粘蛋白)。其拥有一系列专门的糖苷酶和蛋白酶,能够有效分解肠道粘蛋白的复杂糖链结构。研究发现,R.torques、A.muciniphila、B.bifidum和R.gnavus均能降解人类结肠黏蛋白,但R.torques降解能力是最强的。粘蛋白是构成肠道粘液层的主要成分,对维持肠道屏障功能至关重要。
R.torques是人体肠道微生物组的常见成员,在健康人群中普遍存在,但其丰度通常维持在相对较低的水平。它与肠道粘液层的动态平衡密切相关,在正常情况下参与粘液更新和营养物质循环。
但多项研究表明,R.torques异常增殖与多种疾病相关,包括克罗恩病、肥胖、糖尿病相关疾病、桥本甲状腺炎及多囊卵巢综合征等患者体内均发现其丰度显著升高。
R.torques丰度过高可导致粘蛋白过度降解,使粘液层变薄并增加肠道通透性,从而引发细菌移位、低度慢性炎症,并干扰免疫调节与代谢功能。深入研究R.torques生物学特性及其与宿主的相互作用,对开发基于微生物组的肠道屏障相关诊断和治疗策略具有重要意义。
1
Ruminococcus torques在炎症性肠病中显著增加
许多研究和谷禾的检测数据发现,即使在没有明显肠道炎症的情况下,炎症性肠病(IBD)患者的肠道微生物群都会发生显著变化,尤其是粘膜降解相关细菌丰度有所增加。这表明在IBD中,粘膜降解相关细菌处于一个更有利的生态位。
结肠杯状细胞持续分泌的粘液层在结肠细菌与上皮细胞间形成屏障,这一屏障具有免疫调节和保护作用。而粘膜降解细菌可以降解这一防御屏障。

研究人员发现,在非炎症性克罗恩病和溃疡性结肠炎的肠道中,总粘膜相关细菌分别平均增加了1.9倍和1.3倍。但活泼瘤胃球菌(Ruminococcus gnavus)和扭链瘤胃球菌(Ruminococcus torques)在IBD患者中增加的数量比总粘膜相关细菌高出许多倍。
与控制对照组相比,在克罗恩病和溃疡性结肠炎的肠道中,R.gnavus分别平均增加了12.3倍和4.4倍。在控制对照样本中,仅有10%检测到Ruminococcus torques,但在克罗恩病和溃疡性结肠炎患者中分别有38%和45%检测到,其总体丰度约为对照组的几十倍。
炎症性肠病患者与对照组的细菌相对丰度
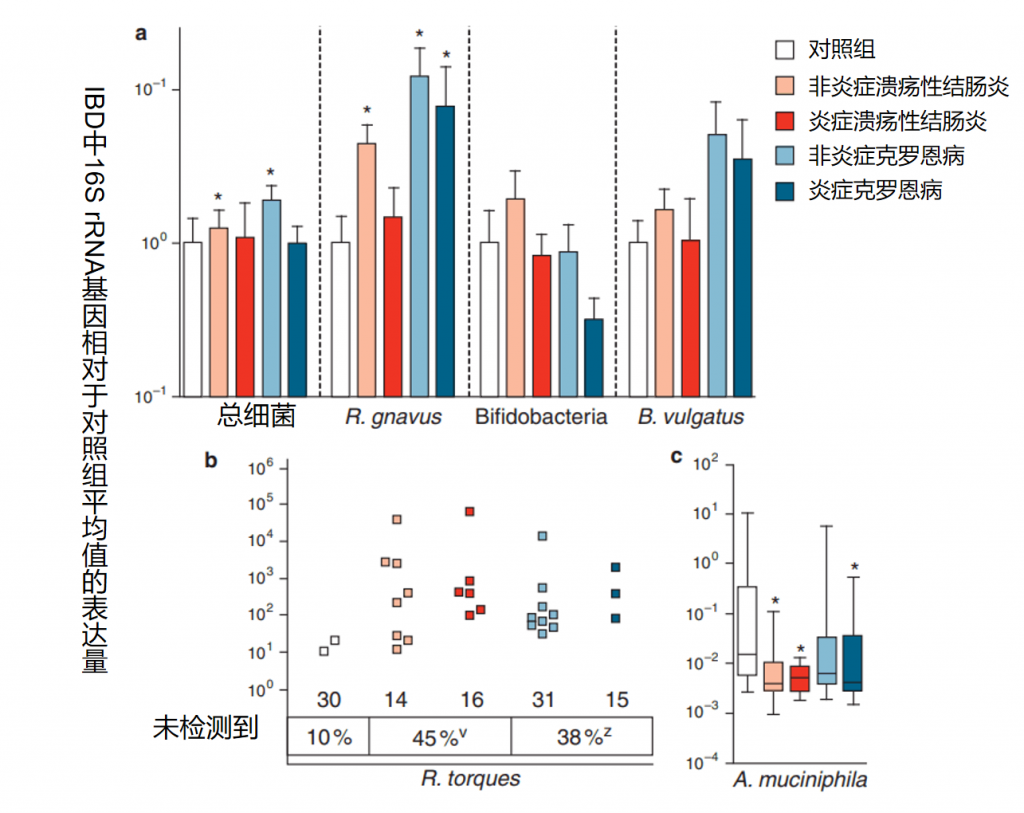
doi: 10.1038/ajg.2010.281.
值得注意的是,我们之前介绍过的下一代益生菌嗜黏蛋白阿克曼菌(A.muciniphila)在炎症性肠病患者中含量降低。
2
高丰度R.torques过度降解黏蛋白破坏肠道屏障
为何同为黏液降解相关菌,A.muciniphila被视为益生菌,而R.torques在炎症性肠病中富集并具有致病性?
研究表明,R.torques、A.muciniphila、B.bifidum和R.gnavus均能降解人类结肠黏蛋白(MUC2),但部分降解菌仅能利用特定粘蛋白成分,而R.torques降解能力是最强的。
黏蛋白降解细菌丰度过高或过度活跃会破坏肠道黏蛋白屏障,增加肠道通透性,促使管腔抗原和有害物质渗入肠壁,从而诱发炎症反应和免疫失调——这被认为是克罗恩病的关键病理特征之一。研究表明,黏蛋白降解菌丰度与肠道炎症程度相关,即使被视为有益的A.muciniphila,过量存在同样有害。
R.torques过多导致代谢通路受损影响屏障功能
研究基于KEGG数据库功能注释分析发现,克罗恩病上消化道受累患者中两条重要保护性通路显著减少:L-蛋氨酸生物合成III通路和棕榈酸酯生物合成II通路,这两条通路与R.torques活性呈负相关。
注:消化道受累患者是指消化系统的部分或全部结构因疾病而受到影响或损害的病人。
蛋氨酸能增强小肠粘膜完整性、屏障功能及绒毛形态,而棕榈酸酯促进肠道杯状细胞产生MUC2,形成厚粘液层,在维持肠道屏障功能中起关键作用。
3
R.torques增强了其他细菌的底物利用和生长
Ruminococcus torques生长过程中降解的特定粘蛋白聚糖结构或顽固降解的结构,这些降解产物可能被其他微生物群成员利用。
例如R.torques分泌细胞外的α-和β-糖苷酶,这些酶能够去除这些末端糖分以降解粘蛋白。一旦末端糖分被去除,暴露出寡糖链和蛋白质核心,粘蛋白基质就变得容易降解。可以被许多其他结肠细菌单独或作为微生物群落的一部分所降解。

上面这项研究发现,R.torques通过分泌酶降解粘蛋白糖蛋白结构域和胃肠O-聚糖,并从完整的粘蛋白中释放出产物,这些产物可供B.thetaiotaomicron利用。R.torques将完整粘蛋白糖蛋白释放的产物交叉喂养到B.thetaiotaomicron的能力支持R.torques能促进其他细菌生长的假设。
当肠道中的微生物过度降解黏蛋白并且数量异常增殖时,会打破原本稳定的肠道菌群平衡状态。这种失衡不仅影响正常共生菌的生存环境,还会显著扰乱整个肠道微生态系统的稳态。随着黏蛋白屏障的持续被破坏,肠道上皮细胞的保护层变得薄弱,导致肠道通透性增加,有害物质更容易渗透进入肠壁组织。这一系列连锁反应最终会损害肠道的正常生理功能,降低营养物质的吸收效率,并可能引发局部炎症反应,最终导致克罗恩病症状。
4
临床意义:与克罗恩病的辅助诊断相关
抗酿酒酵母抗体(ASCA)是酿酒酵母菌抗原蛋白的血清反应性抗体,是一种免疫球蛋白,常见于炎症性肠病,尤其是克罗恩病。对成人和儿童克罗恩病的诊断有辅助作用,具有高度特异性但敏感度较差。
注:ASCA在CD患者中的出现率为30%到60%,在一般人群中仅为5%。ASCA与小肠受累、狭窄和穿透性克罗恩病形式有关。

研究发现,Ruminococcus torques与克罗恩病患者中抗酿酒酵母抗体(ASCA)阳性状态有显著关联。具体来说,R.torques在ASCA阳性患者中的存在频率较高,而在ASCA阴性患者中则较低。这可能反映了这些患者在免疫反应和微生物群落组成上的差异。
ASCA阳性患者与Ruminococcus torques和Yersinia enterocolitica显著相关,而ASCA阴性患者与Enterobacter cloacae和Faecalibacterium prausnitzii显著相关。
在ASCA阳性患者中,R.torques的平均丰度显著高于ASCA阴性患者。这表明R.torques的丰度可能与ASCA的产生或存在直接相关。
通过影响免疫反应
Ruminococcus torques可能通过其代谢产物或结构成分侵袭肠道黏膜,触发宿主免疫反应。这种肠道细菌的存在增加了宿主对微生物抗原的敏感性,进而可能促进抗酿酒酵母抗体(ASCA)的产生,因为ASCA通常源于对肠道微生物抗原的免疫应答。
肠道微生态失衡
R.torques的存在可能反映了肠道微生态的失衡,这种失衡可能与ASCA阳性患者的免疫系统异常有关。ASCA阳性患者可能具有特定的免疫特征,使其更容易对某些肠道细菌产生免疫反应。
疾病严重性
由于R.torques与ASCA阳性状态的关联,这种细菌可能与克罗恩病的严重程度有关。ASCA阳性患者通常表现出更复杂的疾病表型,包括更高的手术风险和更频繁的疾病复发。
R.torques在克罗恩病中的临床意义
总体而言,Ruminococcus torques与抗酿酒酵母抗体的关联及其丰度变化表明,该细菌可能作为克罗恩病的潜在生物标志物,在患者免疫炎症反应和疾病进程中发挥重要作用。
未来的研究可以进一步探讨R.torques在克罗恩病发病机制中的作用,并探索其在临床诊断和治疗中的应用价值,特别是在风险评估、患者筛查和预防上消化道受累方面的潜力。
Ruminococcus torques不仅与克罗恩病存在明显的关联性,其他科学研究已经证实,在多种代谢、免疫、甚至神经系统相关疾病的患者样本中,同样能够观察到Ruminococcus torques菌群丰度的显著增加,表明这种微生物可能在多种疾病的发病机制中扮演着重要角色。
1
R.torques在肥胖患者中增加

越来越多的证据表明,肠道微生物群通过代谢物-宿主相互作用调节肥胖。上述研究发现肠道缺氧诱导因子2α(HIF-2α)通过调控肠道乳酸脱氢酶A(LDHA)表达正向调节乳酸水平。
小鼠肠道特异性HIF-2α消融导致乳酸水平降低,普通拟杆菌(Bacteroides vulgatus)和扭链瘤胃球菌(Ruminococcus torques)丰度增加。这些变化共同导致牛磺酸结合胆酸(TCA)和脱氧胆酸(DCA)水平升高,并激活脂肪G蛋白偶联胆汁酸受体GPBAR1(TGR5)。
这种激活上调了解偶联蛋白(UCP)1和线粒体肌酸激酶(CKMT)2的表达,导致白色脂肪组织产热升高。TCA和DCA的施用反映了这些表型,B.vulgatus和R.torques的定植分别抑制和诱导产热。
2
自闭症中R.torques丰度增加

患有自闭症谱系障碍(ASD)、便秘和腹泻的儿童粪便中Sutterella wadswarensis以及扭链瘤胃球菌(Ruminococcus torques)和活泼瘤胃球菌(Ruminococcus gnavus)的丰度增加。
而Akkermansia muciniphila数量减少,这一模式与炎症性肠病(IBD)患者的肠道微生物特征相似。并且研究提到自闭症谱系障碍儿童中有一个亚群存在肠道通透性增加的问题,R.torques的增加可能与此相关。它可能具有作为自闭症谱系障碍(ASD)特定亚群的潜在生物标志物。
3
糖尿病周围神经病变患者R.torques增加
南京医科大学第二附属医院的一个研究团队将80名受试者分为三组:糖尿病周围神经病变(DPN)患者45名、无DPN的2型糖尿病患者21名,及健康对照组14名。研究比较了三组间肠道菌群构成差异,并分析了肠道菌群与临床指标的相关性。
研究发现,在属水平上,糖尿病周围神经病变(DPN)组拟杆菌属和粪杆菌属的丰富度显著降低,而埃希氏菌-志贺氏菌(Escherichia-Shigella)、Lachnoclostridium、经黏液真杆菌属(Blautia)、巨球菌属(Megasphaera)和扭链瘤胃球菌(Ruminococcus torques)的丰度增加。
稳态模型评估的胰岛素抵抗指数与巨球菌属(Megasphaera)丰富度呈正相关。甘氨酸熊去氧胆酸与瘤胃球菌群和气囊杆菌丰富度正相关,而牛磺熊去氧胆酸则与瘤胃球菌和副拟杆菌属丰富度正相关。
4
妊娠糖尿病患者R.torques增加

一项亚洲研究发现,妊娠糖尿病(GDM)患者的肠道菌群组成与正常孕妇存在显著差异。GDM患者体内柯林斯菌属(Collinsella)、经黏液真杆菌属(Blautia)、瘤胃球菌属(Ruminococcus)、活泼瘤胃球菌(Ruminococcus gnavus)和扭链瘤胃球菌(Ruminococcus torques)以及真杆菌(Eubacterium hallii)的丰度明显增加(p< 0.05)。
5
多囊卵巢综合征患者R.torques增加
据研究报道,多囊卵巢综合征患者体内梭状芽孢杆菌属(Fusobacterium)、大肠杆菌-志贺菌属(Escherichia-Shigella)、扭链瘤胃球菌(Ruminococcus torques)的水平升高,而粪杆菌属(Faecalibacterium)、另枝菌属(Alistipes)、Subdoligranulum和Clostridium IV的水平降低。
6
桥本甲状腺炎患者R.torques增加
研究表明桥本氏甲状腺炎(HT)中肠道微生物群和微生物代谢物谱紊乱。
详细的粪便菌群 Mann-Whitney U 检验显示: 桥本甲状腺炎患者Blautia、Roseburia、Ruminococcus torques、Romboutsia、Dorea、Fusicatenibacter和Eubacterium hallii丰度增加,而粪杆菌属(Faecalibacterium)、拟杆菌属(Bacteroides)、普雷沃菌属(Prevotella_9)和Lachnoclostridium丰度降低。
Spearman相关分析证实7个临床参数间存在相关性。线性判别分析效应量法进一步显示两组间27个菌属有显著差异,这些菌属与临床参数密切相关。
瘤胃球菌属(Ruminococcus)是厚壁菌门中很重要的革兰阳性菌,也是人体肠道菌群的核心菌菌属,在分解碳水化合物和消化抗性淀粉方面发挥关键的作用,瘤胃球菌能有效地分解坚硬的植物物质,如细胞壁,这样消化蔬菜不太可能引起胃痛。除此之外还对健康有很多好处,包括:
稳定肠道屏障
逆转腹泻
降低结直肠癌风险
减少肾结石
增加能量
目前其底下的几个物种被越来越多的研究发现,与人体的疾病发展密切相关,甚至可以通过监测其变化作为疾病的辅助诊断生物标志物。例如前不久我们科普过的活泼瘤胃球菌(Ruminococcus gnavus)——多种疾病风险的潜在标志物,在肥胖、抑郁症、炎症性肠病、2型糖尿病等疾病患者中丰度偏高。
研究发现R.gnavus能够产生一种炎性多糖,该分子诱导树突状细胞产生炎性细胞因子,如TNFα(肿瘤坏死因子),可能导致人体的炎症反应。并且越来越多的证据表明,R.gnavus的过度生长可能与某些肠道疾病有关,包括炎症性肠病(IBD)、肠易激综合征(IBS)和结直肠癌。
本文分享的是瘤胃球菌的另外一个重要物种:扭链瘤胃球菌(Ruminococcus torques),该菌可以分解纤维素,利用胆汁,同时可以产生丁酸。但是该菌最显著的特征是其强大的粘蛋白降解能力,可以利用MUC2(人肠道中主要分泌的粘蛋白)。其拥有一系列专门的糖苷酶和蛋白酶,能够有效分解肠道粘蛋白的复杂糖链结构。
高丰度R.torques过度降解黏蛋白破坏肠道屏障。R.torques异常增殖与多种疾病相关,包括克罗恩病、肥胖、糖尿病相关疾病、桥本甲状腺炎及多囊卵巢综合征等患者体内均发现其丰度显著升高。
Ruminococcus torques与抗酿酒酵母抗体的关联及其丰度变化表明,该细菌可能作为克罗恩病的潜在生物标志物,在患者免疫炎症反应和疾病进程中发挥重要作用。未来的研究可以进一步探讨R.torques在克罗恩病发病机制中的作用,并探索其在临床诊断和治疗中的应用价值,特别是在风险评估、患者筛查和预防上消化道受累方面的潜力。
主要参考文献
Schaus SR, Vasconcelos Pereira G, Luis AS, Madlambayan E, Terrapon N, Ostrowski MP, Jin C, Henrissat B, Hansson GC, Martens EC. Ruminococcus torques is a keystone degrader of intestinal mucin glycoprotein, releasing oligosaccharides used by Bacteroides thetaiotaomicron. mBio. 2024 Aug 14;15(8):e0003924.
Kwak MS, Cha JM, Shin HP, Jeon JW, Yoon JY. Development of a Novel Metagenomic Biomarker for Prediction of Upper Gastrointestinal Tract Involvement in Patients With Crohn’s Disease. Front Microbiol. 2020 Jun 3;11:1162.
Wu Q, Liang X, Wang K, Lin J, Wang X, Wang P, Zhang Y, Nie Q, Liu H, Zhang Z, Liu J, Pang Y, Jiang C. Intestinal hypoxia-inducible factor 2α regulates lactate levels to shape the gut microbiome and alter thermogenesis. Cell Metab. 2021 Oct 5;33(10):1988-2003.e7.
Wang Y, Ye X, Ding D, Lu Y. Characteristics of the intestinal flora in patients with peripheral neuropathy associated with type 2 diabetes. J Int Med Res. 2020 Sep;48(9):300060520936806.
Gupta A, Chan SY, Toh R, Low JM, Liu IMZ, Lim SL, Lee LY, Swarup S. Gestational diabetes-related gut microbiome dysbiosis is not influenced by different Asian ethnicities and dietary interventions: a pilot study. Sci Rep. 2024 Apr 29;14(1):9855.
Yang Y, Cheng J, Liu C, Zhang X, Ma N, Zhou Z, Lu W, Wu C. Gut microbiota in women with polycystic ovary syndrome: an individual based analysis of publicly available data. EClinicalMedicine. 2024 Oct 18;77:102884.
Zhao F, Feng J, Li J, Zhao L, Liu Y, Chen H, Jin Y, Zhu B, Wei Y. Alterations of the Gut Microbiota in Hashimoto’s Thyroiditis Patients. Thyroid. 2018 Feb;28(2):175-186.
谷禾健康
过去二十年,微生物组研究取得显著进展,从代谢性疾病、免疫调节到神经退行性疾病,微生物组是宿主生理与病理状态的重要调控者。然而,将研究结果转化为临床应用方面仍然面临挑战。微生物组的动态异质性、宿主-微生物互作的因果性争议,以及动物模型与人类生理的系统性差异,共同构成了从基础研究到临床应用的转化鸿沟。
当前微生物组研究面临的核心挑战在于:相关性研究难以区分因果。例如,结直肠癌患者的肠道菌群中特定共生菌的减少可能是疾病进展的“乘客”而非“驱动者”;而动物模型(如小鼠)的肠道解剖结构、免疫细胞分布和代谢速率与人类存在显著差异,导致机制外推受限。
因此,需要系统性策略,能够整合大数据驱动的假设生成与多层次实验验证,贯通关联到因果、实验室到临床的转化研究。
近日发表于《Cell》的文章“从大数据和实验模型到临床试验:微生物组研究中的迭代策略”,正是针对这一需求提出的解决方案,创新性地提出“迭代研究框架”——通过计算模型(如多组学方法)、实验模型(如体外、体内和离体模型)和临床验证的循环反馈,逐步解析宿主-微生物互作机制,并优化干预策略。
多组学整合与因果验证
通过宏基因组、代谢组和单细胞转录组数据的交叉分析,识别关键菌群-宿主互作通路(如丁酸代谢与免疫调控),并利用器官芯片(如模拟肠脑轴的neuroHuMiX)和“野生化”小鼠模型验证机制,减少对传统动物模型的依赖。
人工微生物疗法的理性设计
基于机制研究筛选功能互补菌株(如治疗IBD的VE202菌群联盟),或通过基因编辑构建产丁酸工程菌,结合结肠靶向递送技术(如丁酸微胶囊)突破代谢物应用的局限。
临床试验的适应性优化
利用机器学习筛选患者分层标志物(如双歧杆菌丰度预测免疫治疗响应),结合实时菌群监测动态调整干预方案,提升个性化治疗精准性。
这为微生物组研究提供了一个系统的框架,有助于推动从基础研究到临床应用的转化。
具体内容如下:


转化微生物组研究的迭代实验方法

小鼠模型和人类之间的生理、环境和生物学差异
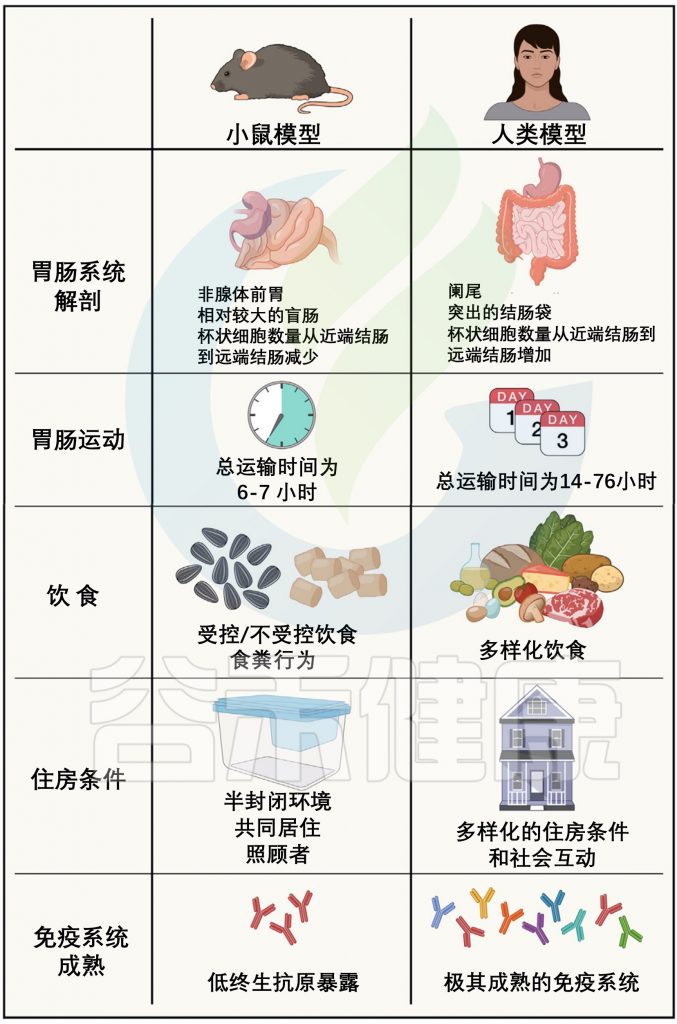
编辑



谷禾健康

编辑
双歧杆菌是人类肠道微生物组的重要成员,在整个宿主生命周期中表现出优越的定植和糖苷利用能力。
从出生开始,双歧杆菌在母乳喂养婴儿的肠道中迅速建立优势地位,并随着宿主年龄的增长和饮食结构的变化,不断调整自身的代谢策略,始终保持在肠道微生物群落中的关键地位。这种随年龄和饮食变化而演变的适应性,使双歧杆菌成为连接人类早期生命到老年期肠道微生态平衡的关键菌群。
我们在之前的文章也详细阐述过双歧杆菌的基本特性、不同种类、代谢特点、在人体各年龄段的分布变化、健康益处以及对疾病的潜在治疗作用等,点此详见:肠道核心菌属——双歧杆菌,你最好拥有它
然而,这里还有一些重要的内容需要补充,关于双歧杆菌在肠道微生态系统中的一个关键功能——”交叉喂养”机制及其在微生物网络中的特殊生态位选择策略。
今天要给大家科普的是,双歧杆菌如何通过精妙的代谢网络与其他微生物建立互惠关系,以及这种关系对肠道健康的深远影响。
什么是微生物的”交叉喂养”?
“交叉喂养”是指一种菌分解复杂糖分子后,释放中间产物到肠道环境中,而这些产物被其他无法直接分解原始糖分子的菌所利用的现象。这形成了一种资源共享的微妙生态平衡。
交叉喂养是肠道微生态系统稳定性和弹性的关键基础。通过交叉喂养,不同微生物贡献各自的酶系统,形成”流水线作业”,显著提高了整个微生物群落对复杂碳水化合物的利用效率。同时也能使微生物群落能够适应饮食变化和宿主生理状态的转变。
这种从“单一微生物”到“微生物网络”的研究视角转变,正引领我们进入微生物干预的新时代。
本文根据江南大学2024年发表在《Npj Biofilms and Microbiomes》的一篇综述性资料分享,探讨双歧杆菌的糖苷利用策略及其与其他微生物之间的交叉喂养关系,以及这些关系对宿主健康的潜在益处。
核心问题
了解双歧杆菌的糖苷利用和交叉喂养策略,不仅有助于揭示肠道微生态系统的运作机制,更为开发针对性的益生菌组合提供了科学基础,有助于改善肠道稳态和缓解相关疾病,包括由剖腹产和抗生素使用引起的菌群失调、代谢综合征和免疫缺陷等现代文明病。
通过深入理解这些微观世界的精妙协作,有望开发更精准的微生物干预措施,实现从”群落微生物学”向”分子营养学”的转型,为个性化健康管理和精准医学提供新思路。
定植早期肠道,随年龄增长发生选择和变化
双歧杆菌在生命很早期就开始在肠道中定植,可以通过母亲传递给后代,发现母乳喂养的幼儿肠道双歧杆菌含量很高。
随着年龄增长,人类肠道中双歧杆菌的数量和多样性会发生变化,随年龄的增长,双歧杆菌的数量会下降。
在婴儿期,粪便中常见的是:
在成年期下列是优势菌种:
青春双歧杆菌的相对丰度随年龄增长而减少,而短双歧杆菌(B. breve)和长双歧杆菌(B. longum) 在老年人肠道中占主导地位。关于长双歧杆菌,我们在以前的文章写过,详见:双歧杆菌:长双歧杆菌
这些变化反映了双歧杆菌与人体生命周期的密切关联,对理解肠道健康与衰老的关系具有重要意义。
双歧杆菌的定植优势以及交叉喂养潜力
肠道中的富含且复杂的碳源为肠道微生物群提供了一个营养生态位。糖类的差异利用影响了肠道生态位中的双歧杆菌的形成,这可能解释了为什么某些种类的双歧杆菌在婴儿或成人的肠道中更为常见。
婴儿的母乳和成人的饮食中含有大量复杂结构的糖类和寡糖,这些物质无法被人体消化,因此作为肠道微生物群的底物经过大肠。
双歧杆菌的”口味偏好”
双歧杆菌代谢单糖、双糖和寡糖;然而,不同种类的双歧杆菌有着各自的”口味偏好”:有些喜欢植物来源的菊粉型果聚糖(ITF);有些专门消化抗性淀粉(RS);有些则偏爱半乳糖聚糖、木聚糖、阿拉伯糖。还有一些”挑剔”的种类专门利用来自宿主的糖类,比如母乳中的人乳寡糖(HMO)或肠道粘液中的粘蛋白。
那么,这些微生物是如何分解这些复杂的糖分子的呢?
人类肠道微生物群通过碳水化合物活性酶(CAZymes) 介导复杂糖苷的代谢。
研究估计,双歧杆菌利用约14.64%的CAZyme基因,包括糖苷水解酶(GHs) 和糖苷转移酶(GTs),用于糖苷的运输、降解和调节,这一比例仅略低于拟杆菌,显示了它们在糖分代谢中的专业性。
拟杆菌是利用糖的多面手,谷禾以往的文章有专门写过,详见:
肠道核心菌——副拟杆菌属(Parabacteroides),是否是改善代谢减轻炎症的黑马?
拟杆菌属重要菌种——单形拟杆菌 (Bacteroides uniformis),控制好其稳态很重要
人群检出率较高的粪便拟杆菌(Bacteroides stercoris),你了解它吗?
肠道微生物群采用类似的策略来维持和分解复杂糖苷。
双歧杆菌编码一系列模块化的葡聚糖酶复合体,通过跨膜结构锚定在细胞表面。这些复合体包含编码GHs的基因,用于在细胞外消化长肽糖或糖苷,以及腺苷三磷酸(ATP)结合盒(ABC)运输系统的底物结合蛋白(SBPs),用于捕获在进入细胞前消化的寡糖。目前尚未发现能利用所有糖苷的菌株;然而,特定类型的糖苷是被利用的。
不同双歧杆菌与其他肠道微生物在可用糖苷的范围、种类及利用策略方面存在代谢特异性,没有任何一种双歧杆菌能够利用所有类型的糖分子,这促使它们发展出了一种巧妙的生存策略——”交叉喂养“,促进双歧杆菌在肠道的适应性。
在这种关系中,一种微生物分解复杂糖分子后,会释放一些中间产物到肠道环境中,而这些产物恰好可以被其他无法直接分解原始糖分子的微生物所利用。这是一种资源共享的微妙平衡。
例如,在婴儿肠道中,长双歧杆菌和双歧双歧杆菌能够分解母乳寡糖(HMO),释放出岩藻糖和短链脂肪酸等中间产物,这些物质随后被其他微生物(如嗜酸乳杆菌)所利用。这种交叉喂养策略不仅增强了整个肠道微生物对糖类的利用效率,也通过代谢产物的交换建立了微生物间的互惠关系,直接影响宿主的肠道稳态和健康。
如人乳寡糖和植物糖苷
双歧杆菌的定植优势依赖于特定营养资源的可用性、需求和消耗率。双歧杆菌在不同阶段的碳水化合物代谢方式影响其对不同肠道环境的选择性适应。
由于营养来源的差异,婴儿的肠道环境与成人不同,特征是以双歧杆菌为主,它们以母乳低聚糖为主要碳源,包括:
然而,在断奶后引入复杂饮食时,具有更强植物糖苷分解能力的双歧杆菌,如下列菌更普遍:
双歧杆菌对糖苷的分解依赖于分布于细胞外或细胞壁内的大量的糖苷水解酶。双歧杆菌进化出几种具有重叠但不同底物特异性的同源酶。不同的物种对不同的糖苷结构具有不同的肠道适应性。
复杂糖苷首先由细胞外的糖苷水解酶降解为单糖或低聚糖,其中一些通过转运蛋白直接运输到细胞质中,而另一些被磷酸化。这些单糖、低聚糖以及其他简单碳水化合物进入双歧代谢途径(果糖-6-磷酸磷酸酮酶中心发酵途径)进行代谢,并用于ATP的生产。
双歧杆菌的代谢途径与肠道中细胞外糖的类型和浓度紧密相关。双歧杆菌通常根据可用糖的数量产生各种代谢物,包括乙酸、乙醇和甲酸;
当细胞外糖丰盛时,会生成乙酸和乳酸。因此,双歧杆菌采取不同策略进行定植,双歧杆菌对糖苷的捕获、降解和代谢能够决定它们在肠道中的营养生态位。
双歧杆菌的主要聚糖利用策略
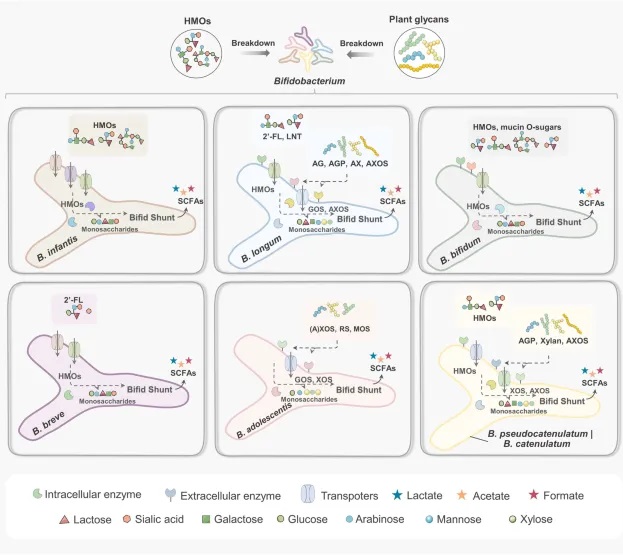
doi: 10.1038/s41522-024-00524-6
总结如下:
双歧杆菌的糖苷利用策略
到达结肠的复杂糖苷(如人乳寡糖和植物糖苷)被双歧杆菌通过两种主要方式利用:
不同种类的双歧杆菌具有不同的糖苷利用能力和策略。
主要双歧杆菌种类的糖苷利用特点
B.infantis
可通过转运系统内化大多数人乳寡糖
B.bifidum
通过丰富的胞外酶降解人乳寡糖(包括岩藻糖化糖苷)进行进一步内化和吸收
B.breve
利用有限的人乳寡糖(主要是2′-FL)
B.longum
擅长利用含有阿拉伯糖结构的植物糖苷
B.adolescentis
抗性淀粉的优秀利用者
B.pseudocatenulatum和B.catenulatum
可消耗一些人乳寡糖和植物糖苷
1
婴儿双歧杆菌 (B. infantis)
婴儿双歧杆菌主要利用人乳寡糖(HMOs)
婴儿双歧杆菌(B. infantis)在细胞内利用大多数母乳寡糖(HMOs)的能力使其相较于其他利用HMO物种(如B. breve)具有竞争优势,从而在母乳喂养期间在婴儿肠道中占据主导地位。婴儿双歧杆菌对2ʼ-富糖乳糖(2ʼ-FL) 的加工依赖于ABC转运蛋白进行细胞内消化。
婴儿双歧杆菌包含几种细胞内酶,包括富糖酶、β-半乳糖苷酶、LNB磷酸化酶、N-乙酰-β-氨基己糖苷酶和唾液酸酶,以降解富糖化和唾液酸化的HMOs。
婴儿双歧杆菌的特异性转运蛋白
婴儿双歧杆菌有专门的”接收器”(转运体)来吸收母乳中的特殊糖分。研究人员发现,这种细菌有两种特殊的转运体,可以吸收母乳中的两种岩藻糖基乳糖(2′-FL和3′-FL)。细菌体内有酶能将这些复杂糖分解为岩藻糖,然后进一步转化为其他物质,如1,2-丙二醇、乙酸和乳酸。
除了含岩藻糖的成分外,母乳中其他一些复杂糖分,如LNT、LNnT、LNB和N-乙酰葡萄糖胺,也能促进婴儿双歧杆菌的生长。

如上研究发现,一种特定的婴儿双歧杆菌株(EVC001)能利用LNT和LNnT这两种母乳糖分,这与它拥有一个名为Blon2175-2177的特殊转运体系统有关。
这个系统位于一个称为H5的基因簇中,包含了利用母乳寡糖所需的所有基因。没有这个H5基因簇的菌株在以这些母乳糖分为食物源时,生长就会受到限制。
这些发现说明了婴儿双歧杆菌如何专门适应于婴儿肠道环境,能高效利用母乳中的独特成分作为营养来源。
2
长双歧杆菌 (B. longum)
长双歧杆菌可以代谢阿拉伯木聚糖
长双歧杆菌(B. longum)是一种能在人体肠道中长期存在的有益菌,从出生到老年,它都能在我们体内稳定生存。这种菌的数量多少与我们在不同年龄段吃的食物有很大关系。当我们的饮食发生变化时,这种菌会相应调整自己的基因,改变获取和分解复杂碳水化合物的能力。
长双歧杆菌之所以能如此成功地在人体内定居,是因为它有两个特殊本领:
长双歧杆菌有些种类能分解母乳中特定类型的复杂糖分,特别是名为LNT和2′-FL的成分,但不能分解带有唾液酸的母乳糖分。它们还能利用肠道黏液中的某些糖链。这使得这些细菌在婴儿早期就能在肠道中找到自己的”生态位”(即生存空间)。

日本东京大学2017年的一项研究发现,一种特定的长双歧杆菌(JCM1217)能产生一种特殊的酶(LnbX),这种酶可以把母乳中的LNT分解成更小的糖分子(LNB和乳糖)供细菌使用。另外,研究人员还发现另一种长双歧杆菌(M12)含有一种叫做α-1-2-L-岩藻糖苷酶的酶,使它能够利用2′-FL和LNT这两种母乳糖分生长。
长双歧杆菌——植物纤维分解专家
长双歧杆菌有一套专门的工具(酶和转运蛋白)来分解植物中的复杂碳水化合物,特别是那些含有阿拉伯糖的复合物。这种能力让它在成人和老年人的肠道中具有生存优势。
这种细菌拥有多种GH43家族的酶,就像一套精密的”分子剪刀”,可以协同工作来切断植物纤维中的化学键。不同的长双歧杆菌菌株对不同类型的植物纤维有不同的偏好,这取决于纤维的具体结构、长度和复杂程度。
有些特别复杂的植物纤维(如阿拉伯半乳聚糖蛋白)对大多数肠道细菌来说太”难啃”了,但某些长双歧杆菌菌株进化出了特殊的酶,可以至少部分地分解这些复杂物质。科学家们发现了一种名为GAfase的新型酶,它能去除阻碍其他酶工作的部分,就像是先”清理障碍“然后再进行主要的分解工作。
不过,即使是长双歧杆菌也不能单独完全分解所有的复杂植物纤维,它需要与肠道中的其他细菌合作才能彻底利用这些物质。此外,长双歧杆菌还能分解一些含氮的糖类结构,通过多种酶的协同作用释放出甘露糖和N-乙酰葡萄糖胺等更简单的糖分子。
这些能力使长双歧杆菌成为肠道中一个重要的”食物分解专家“,能够适应人类饮食中各种复杂的植物纤维。
3
短双歧杆菌 (B. breve)
短双歧杆菌在复杂的人乳寡糖上生长有限
短双歧杆菌处理母乳中复杂糖分的方式与婴儿双歧杆菌很相似。它们都使用一种名为”ABC转运系统”的机制将完整的糖分子吸收进细胞内部,然后在细胞内进行分解。
不过,短双歧杆菌的能力有一定局限性。它不像婴儿双歧杆菌那样拥有处理所有母乳糖分的完整工具箱。短双歧杆菌主要能利用一些特定类型的母乳糖分,如LNT、LNnT和LNB,但对含有岩藻糖或唾液酸的更复杂糖分的处理能力有限。
短双歧杆菌的基因组中主要包含用于分解乳糖、岩藻糖、唾液酸等物质的基因。它有一种特殊的酶(外部唾液酸酶)可以从肠道粘液中释放唾液酸并利用它生长。另外,它还能分解某些含硫的糖分子。
菌株多样性与交叉喂养:依赖微生物网络的代谢策略

如上爱尔兰和荷兰团队研究了20种不同的短双歧杆菌菌株,发现它们都有各种类型的酶来分解不同的糖类物质,如麦芽糖、纤维二糖和半乳糖。
有趣的是,大多数短双歧杆菌不能直接利用母乳中含岩藻糖的成分(如2′-FL或3′-FL),但它们能利用其他细菌分解这些物质后产生的岩藻糖。这说明肠道菌群中的细菌会相互协作,共同分解利用复杂的食物成分。
4
两歧双歧杆菌(B. bifidum)
两歧双歧杆菌在细胞外降解HMOs和粘蛋白O-糖
两歧双歧杆菌与其他双歧杆菌有个显著不同:它有一套完整的”外部工具”(胞外酶)来处理母乳中的复杂糖分。这些酶可以在细菌细胞外直接分解复杂的母乳寡糖,将它们切成更小的部分,如乳糖、岩藻糖和唾液酸。
研究发现两歧双歧杆菌有一种特殊的酶(SiaBb2),它不仅能分解糖分,还能帮助细菌附着在肠道内壁上,这对细菌在肠道中的定居非常重要。
避免“内卷”,找到自己的“生存路”
有趣的是,两歧双歧杆菌似乎专门”选择”了一条与众不同的生存之路。它不太擅长处理一类叫做N-糖苷的物质,而是专注于另一类叫做O-糖苷的分子(主要存在于肠道粘液中) 。这种专业化让它避免了与其他双歧杆菌直接竞争,找到了自己的生态位。
两歧双歧杆菌拥有特殊的酶来分解肠道粘液中的糖类结构,还有一些特殊的”抓手“(碳水化合物结合模块)能帮助这些酶更好地接触到目标物质。研究还发现,它能分解粘液中特定的化学键以及含硫的糖分子,这些能力可能不仅影响它自己的生长,还能影响其他肠道细菌的代谢活动。
这些特性使两歧双歧杆菌成为肠道生态系统中一个独特而重要的成员,它以自己的方式与其他细菌和宿主建立互利关系。
5
青春双歧杆菌(B. adolescentis)
青春双歧杆菌更喜欢降解淀粉
青春双歧杆菌的竞争优势主要体现在其对植物糖甘露聚糖(如抗性淀粉、甘露寡糖和菊粉)的利用上。
抗性淀粉:一种通常不被我们消化系统完全分解的淀粉,存在于冷却的煮土豆、生香蕉等食物中
菊粉:一种存在于菊芋、洋葱等植物中的天然糖类
甘露寡糖:存在于豆类和某些坚果中的复杂糖类
青春双歧杆菌含有α-淀粉酶、糖原支链酶、聚糖酶和特定的淀粉结合模块,这些酶促进了抗性淀粉的完全降解。
编码其他糖甘露聚糖的酶,包括半乳糖苷酶、甘露糖苷酶、β-木糖苷酶和阿拉伯呋喃糖苷酶,也同样反映了青春双歧杆菌在降解膳食糖甘露聚糖方面的偏好。
青春双歧杆菌的寡糖代谢与短链脂肪酸产生能力
Mulualem等人从青春双歧杆菌ATCC15703中鉴定了一种α-半乳糖苷酶(BgaC),它通过转糖基作用从乳糖中产生半乳寡糖(GOS)。值得注意的是,青春双歧杆菌消耗寡糖,并且更倾向于在甘露寡糖(DP ≤ 4)和甘露糖上生长,使用β-葡萄糖苷酶(GH1)和β-果糖呋喃糖苷酶(GH32),而不是复杂的β-甘露聚糖。
Salas-Veizaga等人报道,青春双歧杆菌代谢葡萄糖醛酸化的木寡糖(GXOs)和木寡糖(XOs)(DP 2-6)以产生短链脂肪酸。青春双歧杆菌更喜欢(A)XOS而不是阿拉伯糖或木糖,并编码一种能够在小麦AX上生长的内切1,4-β-木糖酶基因。

6
假小链双歧杆菌(B. pseudocatenulatum)
假小链双歧杆菌和B. catenulatum消耗宿主和植物糖元
假小链双歧杆菌和B.catenulatum存在于婴儿和成人的肠道中。它们的CAZyme基因在不同年龄的宿主中有所不同,这表明这两种物种对宿主和植物糖类都有相对发达的降解系统。
消化母乳中的特殊糖分
假小双歧杆菌(B. pseudocatenulatum) MP80菌株有α-岩藻糖苷酶(GH29和GH95)这两种特殊酶,能够专门消化母乳中含岩藻糖的低分子量人乳寡糖,比如乳二岩藻四糖。这让它能在婴儿肠道中获取独特的营养来源。
分解植物纤维的能力
这种细菌还拥有内切-1,4-β-木聚糖酶(GH10),能够利用来自植物细胞壁的长链木聚糖衍生物(聚合度2-4的木寡糖)。这是很特别的能力,因为大多数双歧杆菌分解木聚糖的能力都很有限。
这种酶像”剪刀”一样切断木聚糖主链,产生木寡糖和木糖。有趣的是,这种细菌不会独占所有养分——部分产物自己吸收利用,其余则释放到肠道环境中供其他细菌使用,形成一种”共享经济”。
其他消化能力
在MCC10289菌株中,科学家们发现了β-L-阿拉伯糖苷酶,这种酶帮助细菌消化阿拉伯半乳聚糖蛋白侧链和L-阿拉伯糖,这些都是植物中常见的成分。

研究者Hosaka等人还发现JCM 1200菌株能产生蔗糖磷酸化酶和β-果糖苷酶,这些酶能分解蔗糖(普通食糖)和类似的二糖N-乙酰蔗糖胺。
总的来说,B. pseudocatenulatum拥有多种特殊的”消化工具“,使它能在肠道中高效利用不同来源的碳水化合物,无论是母乳中的特殊糖分还是植物纤维,这可能是它对人体健康有益的原因之一。
7
链状双歧杆菌 (B. catenulatum)
B. catenulatum的基因组中包含多种参与消化人乳寡糖、木聚糖、淀粉及其衍生低聚糖的糖苷水解酶基因,这让它能够适应宿主饮食的变化。这种细菌能将人乳寡糖(包括2′-岩藻糖乳糖和乳-N-二岩藻己糖)转运到细胞内,然后通过岩藻糖苷酶、β-半乳糖苷酶和乳-N-二糖苷酶的协同作用,将它们分解成N-乙酰葡萄糖胺和单糖。
此外,B. catenulatum的细胞外木聚糖酶能将阿拉伯木聚糖切割成木寡糖和阿拉伯木寡糖,这些产物随后被细胞内的阿拉伯糖苷酶和木糖苷酶进一步分解成阿拉伯糖和木糖。
简单总结说,链状双歧杆菌和前面提到的假小链双歧杆菌类似,都具有强大的消化能力。它能消化婴儿母乳中的特殊糖分,也能分解植物中的复杂纤维,将这些难以吸收的物质转化为简单的糖分。这种多样化的消化能力使它成为肠道中非常有价值的益生菌,能随着人从婴儿期到成人期饮食的变化而调整自己的代谢方式。
双歧杆菌的主要聚糖利用策略

doi: 10.1038/s41522-024-00524-6
肠道微生物的代谢网络
肠道微生物群大多是辅助营养型的,不同物种对糖复合物的同化可能相似(代谢冗余)。因此,它们仍然需要与其他物种竞争或从肠道环境中获取营养。依赖于释放代谢物到环境中的其他微生物,竞争物种可以在平衡中共存;这些无成本的代谢物促进了微生物之间的相互作用。
不同代谢物之间存在交叉喂养,特别是那些利用特定复杂化合物以释放代谢副产物的物种,这些副产物进一步被那些无法单独在这些化合物上生长的物种同化。双歧杆菌依靠其酶降解和寡糖运输系统从宿主中消耗糖复合物,导致肠道的稳定定植,并促进双歧杆菌种群内或与其他细菌(如拟杆菌和丁酸盐生产者)之间的交叉喂养关系。
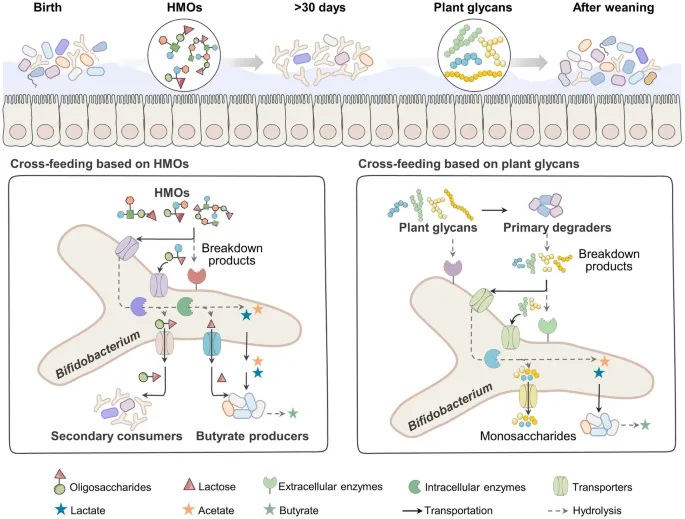
doi: 10.1038/s41522-024-00524-6
双歧杆菌在整个生命周期中都存在交叉喂养。主要类别是基于母乳低聚糖(HMO)和基于植物多糖的交叉喂养。具体而言,当同时培养于HMO上时,双歧杆菌更可能将HMO降解产物交叉喂养给其他非HMO降解主导细菌和丁酸盐生产者。当在植物多糖上进行交叉喂养时,双歧杆菌更可能依赖于其他主导降解细菌的细胞外降解产物而繁荣生长。
基于HMO的双歧杆菌交叉喂养使其在婴儿肠道中占主导地位
基于人乳寡糖的双歧杆菌与产丁酸菌的交叉喂养
B. infantis 代谢人乳寡糖,产生寡糖和代谢物(如 1, 2-丙二醇和乙酸),这些物质通过与其他细菌(如 Eubacterium hallii 和 Anaerostipes caccae)的交叉喂养,在母乳喂养婴儿的肠道中发挥重要作用。
Cheng 等人发现,B. infantis 利用 6ʼ-唾液糖乳糖(6ʼ-SL)进行乙酸的生产,并与 Faecalibacterium prausnitzii 进行交叉喂养以转换为丁酸,这导致 B. infantis 增殖。这可能与 F. prausnitzii 分泌细胞外唾液酶以促使 B. infantis 中唾液酶的表达,从与增加乙酸的生产有关,并且这种交叉喂养活动的发生依赖于人乳寡糖的分子结构。
两种双歧杆菌物种之间的合作性HMO降解
在细胞外降解人源低聚糖(HMOs)时,双歧杆菌(包括B. bifidum和B. longum)产生的产物部分释放到公共环境中,诱导与其他利用HMOs能力较弱的双歧杆菌种类进行交叉喂养。
例如,B. longum降解LNnT产生的产物可以被B. pseudocatenulatum交叉喂养,而B. pseudocatenulatum降解2ʼ-FL产生的低聚糖支持B. longum的生长。
B. bifidum并不是婴儿肠道中的主要物种;然而,作为HMOs的细胞外降解者,它通过释放HMO衍生物与其他物种进行交叉喂养,从而影响肠道微生物组的组成。B. bifidum与B. breve之间的人源低聚糖代谢是互补的,这也促进了这两种菌株之间的交叉喂养。B. bifidum利用岩藻糖酶和FL转运蛋白在细胞外释放乳糖和岩藻糖,这些产物交叉喂养给B. breve。这表明B. bifidum容易与缺乏细胞外岩藻糖酶的其他岩藻糖消费者交叉喂养。
然而,B. bifidum更倾向于在乳糖上生长而不是岩藻糖,这表明无私是其主要功能。除了合作外,B. breve可以与B. bifidum竞争乳糖;共存的关键在于B. bifidum通过上调相关酶和转运蛋白的基因表达释放更多乳糖。类似的现象也在粘蛋白和唇糖的共培养中观察到了。

研究人员证明B. bifidum从唾液酸化聚糖中释放唾液酸和乳糖,这支持了B. infantis和B. breve的生长,作为交叉喂养的次级代谢产物。
B. bifidum不吸收岩藻糖、半乳糖、唾液酸和N-乙酰氨基葡萄糖-6-磺酸,因此,粘蛋白O-糖链的降解可能与其他物种合作完成,例如B. breve,后者利用这些源自粘蛋白的碳水化合物片段。
HMO基础上双歧杆菌与其他肠道细菌的互喂养
建立在双歧杆菌与其他肠道细菌之间的交叉喂养关系可以帮助完善早期生活中肠道微生物群内的相互作用网络。程等人表明,通过发酵阿拉伯糖产生的代谢物1,2-PD可以促进重组肠道无菌小鼠中Limosilactobacillus reuteri的定植。

Nogacka等人研究了B. bifidum和Lactobacillus gasseri之间基于2-ʼFL的代谢相互作用,发现B. bifidum IPLA20048通过交叉喂养细胞外降解产物(半乳糖、阿拉伯糖和乳糖)促进了L. gasseri IPLA20136的增殖,并且在B. bifidum中与碳水化合物运输相关的α-阿拉伯糖苷酶编码基因上调,从而增加了碳水化合物的生产。
母乳喂养婴儿的肠道微生物群在断奶前后会因饮食而发生显著变化,从而促进更复杂的微生物相互作用,并诱导肠道微生物群的结构和成分逐渐稳固和成熟。
基于聚糖利用的双歧杆菌交叉饲养策略

doi: 10.1038/s41522-024-00524-6
基于植物糖苷的双歧杆菌与产丁酸菌的交叉喂养
乙酸和乳酸是双歧杆菌对寡糖代谢的最终产物,是双歧杆菌与丁酸菌之间交叉喂养的重要底物。当在AXOS上生长时,除了将乙酸转化为丁酸外,产丁酸菌消耗AXOS以生产阿拉伯糖和木糖,这些糖可以作为B. longum的底物。
Bhattacharya等人调查了基于半乳糖取代的β-甘露聚糖寡糖的B. adolescentis和Roseburia之间的交叉喂养。

Moens等人证实双歧杆菌可以轻易地在FOS和菊粉型果聚糖上进行交叉喂养,以产生双歧杆菌的生长与丁酸产生效应,并且乙酸转化为丁酸的程度与双歧杆菌种之间的糖苷酶降解能力差异密切相关。具体来说,当与F. prausnitzii共同培养时,B. breve只能利用果糖,不能消耗FOS和ITF,并依赖F. prausnitzii产生的单糖降解果聚糖以进行生长。
B. adolescentis更喜欢短链FOS而非ITF,使其能够将乙酸交叉喂养给F. prausnitzii,并利用其FOS继续生长。相反,B. longum或B. angulatum在ITF上与F. prausnitzii竞争,导致共培养系统中丁酸合成效率最低。
基于植物多糖的双歧杆菌物种之间的交叉喂养扩大了碳源的可用性
当共培养时,无法降解植物糖苷的双歧杆菌种类依赖于能够降解的其他种类。例如,Centanni等人报告称,当在高玉米淀粉培养时,B. pseudolongum通过产生1型外源性蒲白酶和α-淀粉酶释放葡萄糖、麦芽糖、麦三糖,这些产物交叉馈送给B. animalis。更重要的是,具有相似营养代谢的双歧杆菌种类可以通过交叉馈送相同的底物来扩展其营养生态位,而不是竞争。
例如:B. bifidum PRL2010缺乏利用植物糖苷的降解系统,如淀粉和木聚糖;然而,当与其他菌共培养时,如B. adolescentis 22 L、B. breve 12 L、B. thermophilum JCM1207,它可以利用其他双歧杆菌释放出的简单碳水化合物生长,并促进其糖利用基因的表达。
当与其他双歧杆菌共培养时,B. adolescentis在参与木糖代谢的基因表现出最显著的变化,反映了这些菌株的遗传适应性增强,这可能有助于宿主在特定饮食变化期间扩展碳水化合物的利用潜力。
双歧杆菌与其他肠道细菌协作降解复杂的植物糖苷
复杂糖类的降解产物在肠道微生物群中的交叉喂养取决于目标糖类的复杂性。由于缺乏完整的降解和转运系统,双歧杆菌对难以降解的植物糖类(包括木聚糖、长链菊粉和甘露聚糖)具有有限的能力;因此,它们依靠其他细菌将可溶性短糖类物质作为交叉喂养底物(如AXOS、XOS、FOS和甘露糖)在细胞外释放。一些拟杆菌属和乳酸菌属的成员是主要的糖类降解细菌,能够与双歧杆菌交叉喂养,包括B.longum、B.animalis、B. adolescentis。
当在简单的木聚糖(如葡萄糖醛酸木聚糖和小麦阿拉伯木聚糖)上培养时,B. adolescentis ATCC 15703依赖于来自Bacteroides ovatus ATCC 8483产生的AXOS,在缺乏木聚糖酶(GH98)降解系统的情况下生长。
果聚糖的交叉喂养与糖链的取代基和长度密切相关,复杂的果聚糖(即含有蔗糖单元的果聚糖)被主要降解者降解以产生短链化合物,供次级消费者利用。B. longum倾向于使用短链菊粉,促进与Lactobacillus paracasei的合作,完全降解长链菊粉并产生果糖、乳酸和乙酸。
B. longum与拟杆菌之间基于木聚糖、II型各糖苷和AGP存在交叉喂养关系。Wang等报告称,当与Bacteroides caccae ATCC 43185共同培养时,B. longum NCC 2705依靠B. caccae降解的落叶松木AG的碳水化合物片段生长良好。

Vega-Sagardía等报告称,B. ovatus HM222降解木聚糖并通过释放XOS和细胞外酶促进B. longum PT4的生长,并且B. longum PT4中α-L-阿拉伯糖苷酶、L-阿拉伯糖异构酶和木酮糖异构酶的分泌量增加。然而,由于拟杆菌在pH <5.5时生长不良,B. longum发酵产生乙酸和乳酸,导致拟杆菌基质转化(从琥珀酸转化为丙酸)的效率降低,进而导致琥珀酸积累。因此,交叉喂养拟杆菌受到pH限制,用于降解复杂AG或木聚糖的联菌种需要其他物种的参与,例如Prevotella、Roseburia intestinalis。
值得注意的是,B. longum与拟杆菌之间的交叉喂养关系是互惠的,B. longum的植物糖苷代谢的次级产品可以交叉喂养某些糖苷代谢系统较差的拟杆菌。例如,B. longum降解的与AGP相关的低聚糖可以与Bacteroides vulgatus交叉喂养,这促进了后者菌株中木糖苷酶的表达。
B. longum在与植物糖苷共同培养时促进了Bacteroides thetaiotaomicron中甘露糖苷酶和木糖苷酶的功能。
考虑到数据有限,双歧杆菌的交叉喂养互动是否对整个肠道群落内碳水化合物利用的平衡产生积极贡献仍有待确定。
协同组合
双歧杆菌产生的交叉喂养活动凸显了它们在获取或提供其他细菌的底物方面的关键生态作用。这种相互依赖的互动也可以增强参与者的代谢功能,从而扩展它们原有的营养生态位。补充含有双歧杆菌的多菌株共生菌可以帮助肠道微生物群在早期生活中成熟和稳定,并增加有益代谢物(如丁酸)的分泌,最终预防与肠道微生物群失衡相关的慢性疾病。
这种菌株的协同组合协同调节肠道微生物群和健康稳态,并且比单一菌株更不易受到内源性微生物的影响。然而,目前对多菌株共生菌调节健康的研究仅停留在现象描述层面,对组合的基本原因、配方基础和有效机制的明确解释仍然稀缺。
因此,应基于益生菌对糖苷的偏好及其交叉喂养关系探索这种多菌株共生菌的可能性和必要性。肠道微生物群常常受到年龄、饮食习惯和抗生素的影响,因此需要根据不同宿主的需求,使用适当和个性化的策略来调节菌群稳态。
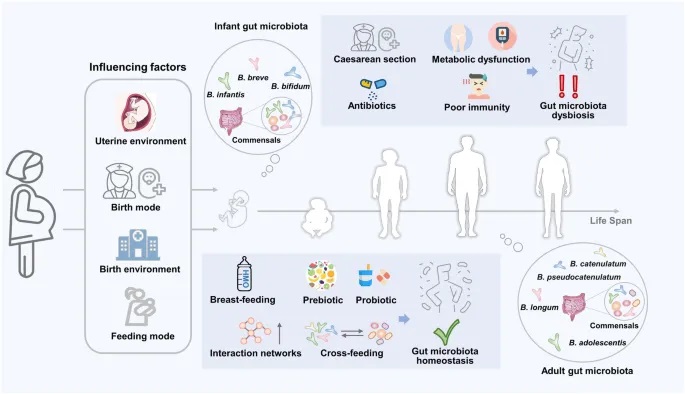
doi: 10.1038/s41522-024-00524-6
宿主的双歧杆菌从出生起受多种因素影响。母乳喂养以及益生菌和益生元的使用有助于建立肠道微生物群的互动网络,包括交叉喂养。这种交叉喂养关系可以促进多菌株合生元的配制,这对改善肠道稳态和缓解因剖腹产和抗生素使用而导致的相关疾病,包括菌群失调、代谢综合征和免疫缺陷,是有效的。
婴儿早期生命中交叉喂养的益处
○ 促进婴儿肠道微生物组的早期定植和多样性
○ 增强免疫系统发育和功能
○ 减少致病菌定植的风险
○ 缓解剖腹产和抗生素使用导致的微生物组变化
成人生命中交叉喂养的益处
○ 维持肠道微生物组的多样性和稳定性
○ 改善肠屏障功能
○ 增加短链脂肪酸的产生
○ 减轻代谢性疾病的风险
○ 调节免疫反应
老年期交叉喂养的益处
○ 延缓肠道微生物组多样性的年龄相关下降
○ 减轻炎症性疾病
○ 改善认知功能
○ 增强免疫系统功能
生命早期——双歧杆菌交叉喂养的有益效应
改善剖腹产和抗生素使用导致的微生物定植的不良后果
出生方式(剖腹产)和抗生素治疗对婴儿期肠道微生物群产生负面影响,这表现为致病菌的积累,双歧杆菌和拟杆菌的减少,以及增加婴儿代谢、炎症和免疫紊乱的风险。
补充具有广泛人乳寡糖代谢能力的双歧杆菌菌株(如婴儿双歧杆菌),益生菌和能够刺激双歧杆菌生长的益生元,已被用来逆转由分娩方式引起的肠道微生物群失调。
——补充R. gnavus可以促进B. breve的增殖
B. breve利用前者从2′-FL中释放的乳糖,并有助于将早产儿微生物组转变为富含双歧杆菌的群落。
——由Lactobacillaceae rhamnoides、B.breve 、Propionibacterium freundenreichii与FOS结合的混合物
该混合物促进了双歧杆菌的定植,并减少了肠球菌科、梭菌科和韦荣球菌科的丰度,最终纠正了由剖腹产和抗生素诱导导致的肠道微生物组的不良变化。
一项宏蛋白质组分析显示,双歧杆菌中用于HMO降解的酶(包括β-半乳糖苷酶和β-半乳糖基N-乙酰己糖胺磷酸化酶)表达显著,而鼠李糖乳杆菌可能从HMO降解释放的单糖中受益。这些研究表明,与肠道居住菌种(特别是双歧杆菌)作为盟友一起施用益生菌和/或益生元在恢复婴儿肠道微生物群失调方面是有效的;这种具有协同作用的多菌株和益生元组合可作为婴儿配方食品的补充。
促进双歧杆菌的优势地位和完善肠道菌群构建
基于HMO的代谢相互作用可以塑造婴儿肠道微生物群的组成。双歧杆菌(尤其是B. infantis)在母乳喂养婴儿的肠道定植中被优先考虑,这取决于它们吸收HMO的优越能力。
双歧杆菌在出生后不久基于HMO代谢发挥优先效应,消耗肠道中大部分碳源,从而阻止后来物种的定植,促进婴儿肠道稳态的建立,并抑制病原体(如艰难梭菌和沙门氏菌)的生长。
B. breve的肠道定植是优先效应的一个例子。
考虑到B. breve对HMO的有限利用能力,其优势地位的形成不仅仅与HMO消耗的表型相关,而是依赖于与其他双歧杆菌的交叉喂养。
双歧杆菌中涉及的交叉喂养活动在肠道微生物群的组装中起着关键作用。
B.infantis Bg2D9促进了Prevotella copri Bg131在营养不良小鼠肠道中的定植,并促进了含有阿拉伯聚糖的饮食中阿拉伯糖的释放,这通过交叉喂养促进了肠道中其他物种的增殖,包括B. catenulatum、Blautia obeum。这可用作严重急性营养不良儿童的饮食干预。
总之,肠道微生物群在出生后具有高度可塑性,可以通过引入含有适当协同生物的饮食来促进肠道稳态。确定这些相互作用的过程和结果可用于饮食干预,以实现个性化营养。
成年期间——双歧杆菌交叉喂养的有益效应
重建肠道微生物群结构和肠道稳态
内源性双歧杆菌的普遍存在程度在成年期低于早期生命阶段;然而,由于饮食多样性的增加,双歧杆菌参与的交叉喂养活动,促进了与肠道微生物群和宿主之间的更多相互作用。
多菌株合生元的膳食补充有助于促进双歧杆菌的增殖并重建稳定的肠道菌群。抗生素暴露可能破坏肠道微生物群的丰度和多样性,而引入已删除的核心微生物群可以通过互营关系帮助重建肠道免疫稳态。
一种人乳寡糖和婴儿型双歧杆菌的合生元,通过促进乙酸盐和丁酸盐的产生,来改善肠道微生物群的不平衡并影响肠杆菌科细菌的生长。
后续研究发现,这种依赖于人乳寡糖的双歧杆菌策略,可以增加乳酸摄取者(韦荣氏菌属)的丰度,并通过减少促炎代谢物对甲酚硫酸盐来缓解抗生素诱导的肠道微生物群失调。
对甲酚详见:对甲酚——自闭症辅助诊断和干预的关键指标
刺激有益代谢物分泌,改善2型糖尿病、肥胖等
由于与产丁酸菌的交叉喂养关系,双歧杆菌丰度的变化与粪便中乙酸盐和丁酸盐浓度密切相关,它们的代谢相互作用可以调节如2型糖尿病(T2D)和肥胖等代谢综合征。
丁酸盐在调节血糖水平方面发挥着重要作用。一种含有Akkermansia muciniphila、婴儿双歧杆菌(B.infantis)、三种产丁酸菌和菊粉的合生元,以改善2型糖尿病,并确定粘液噬菌菌和婴儿双歧杆菌与丁酸盐生产菌的交叉喂养活动促进了丁酸盐的产生,这需要体内验证。
扩展阅读:AKK菌——下一代有益菌
多菌株合生元也被用于改善肥胖个体的能量代谢、免疫功能和肠道微生物群。
引入了一种由GOS、长双歧杆菌(B. longum)和B. bifidum组成的合生元来调节肥胖,并确定瘤胃球菌(Ruminococcus)的增加可用于丁酸盐的生产。
Nguyen等人148通过共现网络分析使用阿拉伯木聚糖治疗肥胖,发现长双歧杆菌通过降解阿拉伯木聚糖释放寡糖,与拟杆菌属、考拉杆菌属(Phascolarctobacterium)和Subdoligranulum进行协同和相互作用,促进乙酸盐的产生并缓解肥胖。
扩展阅读:肠道核心菌属——考拉杆菌属(Phascolarctobacterium),与减肥相关?
饮食改变和合生元干预可用于增加肠道中双歧杆菌的丰度并促进有益代谢物的分泌,从而缓解代谢紊乱;然而,其临床意义仍有待确定。
不同阶段营养——驱动双歧杆菌的适应性演化
肠道中双歧杆菌的种群动态与宿主的营养阶段密切相关。从婴儿期的母乳(富含人乳寡糖等简单糖苷)到成年期的复杂植物糖苷(如阿拉伯木聚糖、β-葡聚糖),饮食结构的转变直接塑造了双歧杆菌的糖苷代谢偏好。
这种代谢适应性使其能够通过调整糖苷水解酶(GHs)和转运系统,在不同年龄段维持肠道定殖优势。然而,相比拟杆菌等肠道菌群,目前对双歧杆菌胞内外GHs酶系及其底物转运机制的认知仍存在显著空白,尤其是底物识别能力如何影响其与其他菌群的互作,亟待深入研究。
交叉喂养机制:协同互作与科学挑战
双歧杆菌通过糖苷偏好性与其他肠道微生物(如拟杆菌、乳酸杆菌)建立交叉喂养网络。例如,其分解阿拉伯木聚糖(AX)或阿拉伯半乳聚糖(AG)的代谢产物可被拟杆菌二次利用,而β-葡聚糖、果聚糖等糖苷的协同代谢机制尚未明确。
种间协作的未知领域:现有研究多聚焦单一菌种,而双歧杆菌属内(如长双歧杆菌与假链状双歧杆菌)及跨属(如与乳酸杆菌)的代谢分工机制亟待阐明。
研究范式局限:当前对多菌株协同效应的研究偏重健康干预的终点指标(如菌群丰度变化),却忽视对互作过程的动态解析(如底物传递路径、酶系统互作),导致协同组合设计缺乏理论依据。
未来研究方向与转化应用策略
机制层面:阐明双歧杆菌(如长双歧杆菌与假链双歧杆菌)间的种内协同机制,以及其与乳酸杆菌等菌的种间互作规律;
技术层面:结合多组学手段,揭示GHs酶系与转运系统的底物特异性,明确关键代谢节点;
应用层面:基于年龄或营养阶段特征,开发靶向双歧杆菌交叉喂养网络的个性化菌群配方,并优化干预时机。
个性化精准营养与功能性食品开发
基于双歧杆菌的糖苷偏好设计靶向性膳食补充剂或功能性食品。
可能的应用方向:
□ 开发含特定糖苷(如LNT、阿拉伯木聚糖)的“益生元+益生菌”组合产品,定向激活双歧杆菌的代谢通路;
□ 例如将B. infantis与丁酸产生菌和特定益生元(如人乳寡糖或阿拉伯木聚糖)结合,形成基于交叉喂养原理的协同生物产品;
□ 开发基于肠道菌群特征的“糖苷-菌株匹配算法”,优化益生元/益生菌组合方案;
□ 针对老年人肠道菌群特征,设计富含双歧杆菌偏好糖苷的老年营养配方(如抗性淀粉微胶囊);
□ 利用双歧杆菌的唾液酸代谢缺陷,设计不含唾液酸化糖苷的婴幼儿配方奶粉,抑制致病菌定殖。
□ 基于不同双歧杆菌菌株之间以及与其他微生物的交叉喂养关系,开发新型发酵食品,优化营养价值和健康效益。
代谢疾病干预与菌群疗法
通过调控双歧杆菌的交叉喂养网络治疗肥胖、糖尿病等代谢疾病。
可能的应用方向:
□ 筛选能高效分解膳食纤维(如β-葡聚糖)的双歧杆菌工程菌株,用于改善胰岛素抵抗;
□ 构建“双歧杆菌-乳酸杆菌”共生菌群胶囊,通过协同降解果聚糖缓解肠道炎症;
□ 开发靶向双歧杆菌GH43酶的小分子激活剂,增强植物纤维代谢能力以控制血糖。
跨学科健康管理工具开发
整合双歧杆菌研究数据构建健康预测与干预平台。
可能的应用方向:
□ 建立“肠道糖苷代谢图谱”数据库,结合机器学习预测更精细化的个体菌群失衡风险;
□ 整合肠道菌群多组学数据与饮食记录,训练机器学习模型预测特定双歧杆菌菌株对宿主代谢的影响阈值;
□ 开发便携式肠道糖苷检测设备(如生物传感器试纸),实时指导饮食调整。
□ 建立母乳低聚糖(HMO)与婴儿粪便中双歧杆菌丰度的关联数据库,指导母乳喂养不足时的精准营养补充。
通过将菌群营养研究提升至分子互作水平,可推动微生物组治疗向“分子营养学”转型,例如设计特定糖苷底物激活双歧杆菌的靶向代谢通路,或利用协同菌群组合重塑宿主-菌群互惠关系,从而为代谢疾病防治提供新策略,是微生物组治疗的未来趋势。
主要参考文献
Xiao M, Zhang C, Duan H, Narbad A, Zhao J, Chen W, Zhai Q, Yu L, Tian F. Cross-feeding of bifidobacteria promotes intestinal homeostasis: a lifelong perspective on the host health. NPJ Biofilms Microbiomes. 2024 Jun 19;10(1):47.
heng L, Kiewiet MBG, Logtenberg MJ, Groeneveld A, Nauta A, Schols HA, Walvoort MTC, Harmsen HJM, de Vos P. Effects of Different Human Milk Oligosaccharides on Growth of Bifidobacteria in Monoculture and Co-culture With Faecalibacterium prausnitzii. Front Microbiol. 2020 Oct 30;11:569700.
Nguyen NK, Deehan EC, et al., Gut microbiota modulation with long-chain corn bran arabinoxylan in adults with overweight and obesity is linked to an individualized temporal increase in fecal propionate. Microbiome. 2020 Aug 19;8(1):118.
Vega-Sagardía M, Delgado J, Ruiz-Moyano S, Garrido D. Proteomic analyses of Bacteroides ovatus and Bifidobacterium longum in xylan bidirectional culture shows sugar cross-feeding interactions. Food Res Int. 2023 Aug;170:113025.
Moens F, Weckx S, De Vuyst L. Bifidobacterial inulin-type fructan degradation capacity determines cross-feeding interactions between bifidobacteria and Faecalibacterium prausnitzii. Int J Food Microbiol. 2016 Aug 16;231:76-85.
Nogacka AM, Cuesta I, Gueimonde M, de Los Reyes-Gavilán CG. 2-Fucosyllactose Metabolism by Bifidobacteria Promotes Lactobacilli Growth in Co-Culture. Microorganisms. 2023 Oct 29;11(11):2659.
Hosaka H, Kawamura M, Hirano T, Hakamata W, Nishio T. Utilization of sucrose and analog disaccharides by human intestinal bifidobacteria and lactobacilli: Search of the bifidobacteria enzymes involved in the degradation of these disaccharides. Microbiol Res. 2020 Nov;240:126558.