-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康
精神分裂症(SCZ)是一种严重的持续性精神障碍,表现为阳性症状(如妄想和幻觉)、负性症状(如动力丧失和社交退缩)以及认知症状(包括工作记忆和认知灵活性缺失)。此外,大多数精神分裂症患者伴有显著的睡眠障碍(SD),常出现入睡困难、睡眠维持问题、睡眠结构紊乱和昼夜节律失调。
睡眠障碍这种共病会加重精神症状,导致更频繁的偏执意念、幻觉和思维紊乱,以及更高程度的抑郁和焦虑。还显著影响治疗依从性和长期预后。
精神分裂症的发病机制涉及遗传、环境、免疫和神经发育等多种因素的相互作用。与此同时,现有研究表明,肠道微生物群通过微生物群-肠道-大脑轴调节大脑功能,影响神经递质的代谢和免疫炎症反应,因此可能在精神分裂症和睡眠障碍的发生和发展中发挥重要作用。
精神分裂症患者常表现出产丁酸细菌减少、产乳酸菌增加,以及与谷氨酸和γ-氨基丁酸(GABA)代谢相关的细菌增加。特别是,在精神分裂症患者中,这种肠道微生物群破坏可能通过影响神经递质平衡、促进神经炎症和干扰昼夜节律等机制,促成精神症状和睡眠问题的发展。
本文整合了现有研究,首先介绍了精神分裂症(SCZ)和睡眠障碍(SD)患者的肠道微生物群特征。探讨了SCZ和SD中关键微生物群及其代谢物的共同影响,以及肠道微生物群通过神经免疫、内分泌和神经递质通路的作用机制。最后,提出了针对肠道微生物群的干预策略在改善精神分裂症伴随SD中的潜在应用。
精神分裂症(SCZ)是一种严重的精神障碍,表现为阳性症状(超出正常范围的行为和思维)如妄想和幻觉;负性症状(缺乏正常的情感和行为)如动力丧失和社交退缩;以及认知症状,包括工作记忆和认知灵活性的缺失。
★ 许多精神分裂症患者伴有睡眠障碍
除了典型的精神症状外,睡眠障碍(SD)是精神分裂症最常见的共病之一,表现为失眠、睡眠片段化、慢波睡眠减少和昼夜节律紊乱。
研究显示,约80%的精神病患者至少患有一种类型的SD。睡眠质量的恶化会加重精神症状,导致更频繁的偏执意念、幻觉和思维紊乱,以及更高程度的抑郁和焦虑。研究发现,睡眠质量受损和昼夜节律紊乱的精神分裂症(SCZ)患者在负性症状评估上得分更高,认知功能障碍更为明显。
此外,SD不仅加重精神分裂症的精神病症状,精神分裂症的核心症状也会破坏睡眠生理。多导睡眠图研究表明,更严重的阳性症状与短的快速眼动(REM)潜伏期、长的入睡潜伏期和低睡眠效率相关,而明显的负性症状与非快速眼动(NREM)睡眠中的慢波振幅降低和REM起始潜伏期缩短有关。
这些发现表明精神分裂症与睡眠障碍之间存在双向病理循环,睡眠障碍不仅是精神分裂症的常见共病,也是影响其症状表达和疾病进展的关键因素。
1
流行病学特征
研究表明,精神分裂症(SCZ)患者的睡眠障碍(SD)患病率显著高于普通人群,影响其生活质量和疾病预后。
精神分裂症患者快速眼动睡眠时间缩短
通过睡眠脑电图(EEG)观察精神分裂症患者与健康对照组的睡眠结构差异发现,精神分裂症患者的快速眼动(REM)潜伏期缩短,REM密度增加,并且REM睡眠比例与症状严重程度密切相关。在非快速眼动(NREM)睡眠期间,精神分裂症患者呈现慢波睡眠减少、纺锤体波密度和振幅下降,以及持续时间缩短。
不同精神分裂症患者睡眠障碍也表现出差异
同时,不同精神分裂症患者组间的睡眠障碍表现也存在显著差异,例如,住院患者在睡眠时间、活动水平和生物钟稳定性方面较门诊患者表现更加严重。这表明精神分裂症临床症状的严重程度不仅取决于病理机制,还受到患者生活环境和治疗状态等多种因素的影响。
2
睡眠障碍对精神分裂症的影响
夜间睡眠不足会进一步导致白天功能障碍和生活质量下降。大量临床研究证实,睡眠障碍与精神分裂症患者核心症状组之间存在显著且特异的关联模式。
睡眠障碍会加重精神分裂症症状并影响治疗效果
睡眠障碍不仅加重精神分裂症的阳性症状(如幻觉和妄想),还会加重负性症状(如情感冷漠和社交退缩),进而影响患者的认知功能。此外,患有睡眠障碍的精神分裂症患者表现出治疗依从性差、静坐不能和攻击性言语。
睡眠障碍的精神分裂症患者自杀行为风险增加
在一项为期八年的纵向研究中,结合Kaplan–Meier生存分析和对数秩检验,发现患有精神分裂症谱系障碍且频繁失眠的患者,其累计自杀行为风险显著增加,证实睡眠障碍会增加这些患者的自杀风险。这些结果显示睡眠问题对精神分裂症患者产生负面影响,强调了加强临床关注精神分裂症患者睡眠的必要性。
3
现有治疗的局限性
目前,治疗合并睡眠障碍(SD)的精神分裂症(SCZ)患者主要依赖传统抗精神病药物,特别是非典型抗精神病药物如氯氮平、奥氮平、喹硫平、利培酮、齐拉西酮和帕利立酮。
这些药物有助于提高精神分裂症患者的总睡眠时间和睡眠效率。然而,它们在改善SD方面的疗效有限,并伴有如体重增加和代谢综合征等副作用,部分患者在接受治疗后仍出现睡眠质量差和睡眠结构障碍。
药物疗法存在副作用,生物行为疗法逐渐兴起
尽管药物治疗是主要干预手段,但患者对药物依赖和副作用的担忧使得部分人不愿长期使用。此外,认知行为疗法(CBT)在改善持续妄想或幻觉患者的睡眠上已显示一定效果,但其临床应用仍需更多研究支持。这表明当前治疗精神分裂症合并睡眠障碍的方法面临显著局限,需探索新的治疗靶点和方法。
近年来,褪黑素的研究显示其在调节睡眠-觉醒节律、改善睡眠质量和缓解精神分裂症症状方面有效。同时,生物反馈疗法及其他非药物干预(如光疗和睡眠卫生教育)也被认为是有效的辅助治疗方式,可以改善患者睡眠质量。
总体而言,现有精神分裂症与睡眠障碍的联合治疗方法在疗效和耐受性上存在明显局限,迫切需要通过进一步研究和临床试验来探索新的治疗方案,以为患者提供更有效和安全的治疗选择。
近年来,研究发现肠道微生物群在调节精神健康和睡眠质量方面扮演着重要角色。数据显示,精神分裂症患者的肠道微生物群与健康个体存在显著差异,表现为多样性降低和特定微生物群落的失衡。与此同时,睡眠障碍的发生也与肠道微生物群的组成变化密切相关,这种相互作用可能通过微生物群-肠道-大脑轴影响患者的情绪、认知功能和睡眠模式。
1
精神分裂症患者的肠道菌群失调
多项研究发现,肠道微生物群与精神分裂症(SCZ)的病理机制、症状和认知功能密切相关。
关于精神分裂症患者肠道微生物群变化的研究
编辑
编辑
变形菌增加,拟杆菌减少,这会影响代谢健康
高通量测序结果表明,与健康对照组相比,精神分裂症患者的肠道微生物群丰富度指数(Chao)和多样性指数(Shannon)均较低,且特定微生物群的组成和丰度存在显著差异。曼-惠特尼U检验显示,精神分裂症患者的拟杆菌门(Bacteroidetes)显著减少,而变形菌显著增加。
Faecalibacterium等产生丁酸盐的抗炎菌减少
在属层面,与抗炎和神经保护作用相关的丁酸产生菌Faecalibacterium、Coprococcus和Bacteroides的丰度下降,而Prevotella和Collinsella丰度增加。短链脂肪酸(SCFAs)产生菌减少和促炎细菌增加可能与精神分裂症(SCZ)患者的病情严重程度有关。
微生物群的差异可能有助于判断疾病不同阶段
在不同疾病阶段的研究中,急性精神分裂症患者的嗜血杆菌属(Haemophilus)和Faecalibacterium数量减少,而缓解期患者则表现出巨单胞菌属(Megamonas)和Megasphaera的增加。
注:在这里小编推测嗜血杆菌属(Haemophilus)在肠道里变少可能更像“口源菌/黏膜相关菌”的信号,急性期患者的黏膜生态位与免疫/炎症微环境发生了系统性改变,而不是单纯少了一种好菌或坏菌,也可能是研究存在饮食,治疗等的混杂因素。
此外,口腔常驻细菌如Veillonella atypica、唾液链球菌(Streptococcus salivarius)和Bifidobacterium dentium在精神分裂症患者肠道微生物群中显著富集,这种病理状态可能削弱肠道屏障和免疫功能,为口腔细菌的肠道定殖提供有利条件。
一项涵盖细菌、真菌、古菌和病毒的综合多界微生物组分析发现,精神分裂症患者肠道中链球菌、脱硫弧菌(利用氨基酸或脂肪酸作为碳源)和Methanobrevibacter smithii等微生物数量增加,而丁酸盐产生菌的数量减少。
精神分裂症人群中吸烟率、牙周状况差异常见;如果口腔来源菌输入或口腔生态改变,使得口腔菌更容易通过吞咽等进入肠道并在那里存活、甚至黏附定殖肠道菌群组成更像“口腔来源的组合”,常见于炎症、屏障受损或环境改变的肠道。可以推测病源菌增加 + 产丁酸菌(如 Faecalibacterium)下降+ 屏障/炎症指标异常(如 LPS/DAO/zonulin、粪便钙卫蛋白、炎症因子),可能是未来辅助判别急性精神分裂症的重要标志组合。
2
与睡眠障碍相关的微生物群变化
在具有睡眠障碍(SD)的人类和动物模型中也观察到了微生物群变化。
关于睡眠障碍患者肠道微生物群变化的研究
编辑
微生物群的多样性与睡眠效率和睡眠时间正相关
对健康成年人睡眠的研究发现,微生物群的多样性与睡眠效率和总睡眠时间呈正相关,而与睡眠碎片化呈负相关。简而言之,肠道微生物群的多样性促进了更健康的睡眠。
此外,针对学龄前儿童的研究表明,夜间睡眠时间较长的孩子肠道微生物群结构与睡眠时间较短的孩子不同,且微生物群中的双歧杆菌和拟杆菌与睡眠时间延长、睡眠效率提高和夜间清醒时间缩短相关。
睡眠紊乱导致肠道菌群失调,普雷沃氏菌等增加
研究还表明,睡眠和昼夜节律紊乱会导致人类和动物模型中的肠道微生物群失调,表现为致病菌增加和有益菌减少。在人类中,睡眠不足和质量差常与普雷沃氏菌科(Prevotellaceae)和丹毒丝菌科(Erysipelotrichaceae)的增加及瘤胃球菌属(Ruminococcus)的减少有关。
在动物模型中,睡眠剥夺和破碎会导致毛螺菌科(Lachnospiraceae)和丹毒丝菌科(Erysipelotrichaceae)增加,而乳杆菌科(Lactobacillaceae)和双歧杆菌科(Bifidobacteriaceae)减少。
人类和小鼠的平行实验显示,不同物种的肠道微生物群存在昼夜节律振荡。此外,将人类在经历时差反应前后的肠道微生物群移植到无菌小鼠中,会导致微生物群结构改变,并出现代谢异常,如体重增加、血糖升高和体脂肪堆积。
不同类型的睡眠障碍患者显示出微生物群的特征差异
不同类型的睡眠障碍(SD)患者似乎表现出不同的肠道微生物群特征。慢性失眠患者主要表现为放线菌门数量增加,抗炎菌属Faecalibacterium含量减少,这一变化可能与长期低度炎症状态相关。
相比之下,急性失眠患者的肠道微生物群中,厚壁菌门增加而拟杆菌门减少,这一模式与急性24小时睡眠剥夺实验的发现高度相似,表明这种短期微生物失衡可能是对睡眠模式突然变化的压力反应。
此外,厚壁菌门数量增加,而拟杆菌门减少,导致(F/B)比例升高。关于急性睡眠时间表延迟的研究进一步证实,睡眠与觉醒周期的改变会增加F/B比。研究还报告指出,急性昼夜节律紊乱促进疾病相关的嘌呤代谢和丁酸生成乙酰辅酶A发酵途径,这些通路与宿主能量代谢和炎症反应相关。
注:这些代谢变化在恢复期内往往会逆转。
3
精神分裂症与睡眠障碍的微生物共性
基于研究发现,精神分裂症(SCZ)患者和睡眠障碍(SD)患者的肠道微生物多样性均降低,且特定微生物群组成发生类似变化。这种多样性的缺乏可能削弱肠道微生态系统的稳定性,增加疾病风险。
能代谢膳食纤维的菌种减少,而促炎菌增加
在微生物群组成方面,抗炎菌减少、促炎菌增加,这与疾病易感性密切相关。此外,结肠中的微生物如拟核菌、双歧杆菌和Faecalibacterium能发酵不易消化的碳水化合物和寡糖,从而合成短链脂肪酸(SCFAs)。这些属的减少显著影响丁酸盐、丙酸酯和乙酸盐的生产。
微生物群通过肠脑轴影响精神分裂症和睡眠障碍
与睡眠密切相关的特定微生物群在精神分裂症患者中数量减少。例如,双歧杆菌的减少与睡眠效率降低和睡眠碎片化有关,而Prevotella的增加可能干扰睡眠结构。
在关于睡眠与肠道微生物组组成的研究中,曼-惠特尼U测试显示,Ellagibacter isourolithinifaciens和Senegalimassilia faecalis在睡眠质量良好的患者中显著富集。在属层面,Senegalimassilia与患者的较好睡眠质量呈正相关。
这些微生物改变的特征表明,精神分裂症和睡眠障碍通过微生物群-肠道-大脑轴(MGBA)相互作用。这一过程涉及短链脂肪酸生成减少、全身炎症加剧和昼夜节律紊乱等机制,这些因素共同促进并加重两种疾病的共存和进展。
在肠道微生物群影响精神疾病的复杂病理网络中,宿主的遗传背景和环境因素共同构成了疾病易感性的基础。全基因组多效性分析揭示了胃肠疾病与精神疾病之间的广泛遗传相关性、共同的致病基因和通路,以及它们与肠道微生物群的遗传关联,证实了微生物群-肠道-大脑轴(MGBA)在这两类疾病的共同遗传基础中的关键作用。
多种环境因素,如地理位置、饮食和生活方式,动态调节肠道微生物群的丰富性和多样性,同时影响MGBA的功能稳态。MGBA作为宿主与微生物相互作用的调控枢纽,通过神经、内分泌和免疫系统之间的多层次双向通信网络,整合肠道微生态系统与中枢神经系统的功能耦合。
具体而言,肠道微生物群通过迷走神经直接调节中枢神经系统的活动,影响血脑屏障的通透性、神经炎症及通过细胞因子介导的免疫调节通路促进神经递质的合成和代谢。MGBA功能障碍可能导致病理变化,如下丘脑-垂体-肾上腺(HPA)轴异常激活及小胶质细胞持续激活,这些与精神疾病如精神分裂症(SCZ)和睡眠障碍(SD)的共病机制密切相关。
肠道微生物群及其代谢物通过肠-脑轴相互作用
编辑
1
神经递质调控
肠道菌群通过神经递质影响精神状态和睡眠
在精神分裂症(SCZ)和睡眠障碍(SD)研究中,神经递质的异常代谢被认为是影响症状的重要因素,而肠道微生物群及其代谢物在神经递质的产生和功能调控中发挥关键作用。如血清素(5-HT)、GABA和多巴胺(DA)的代谢异常可能妨碍SCZ的神经传导及SD的睡眠调节。
研究显示,肠道微生物群通过调控色氨酸代谢直接影响5-HT的生物合成。在松果体中,5-HT经历一系列酶转换,通过N-乙酰化转化为N-乙酰丝氨酸,并进一步O-甲基化合成褪黑素。该生物合成途径的完整性直接影响内源性褪黑素的生成,进而影响睡眠的开始和维持。
因此,肠道微生物群可以通过影响色氨酸的可用性来调控该代谢途径,从而通过特定微生物群增加色氨酸转化为5-HT而非犬尿氨酸途径,从而提高5-HT和褪黑素的水平。
乳杆菌和双歧杆菌通过GABA影响精神分裂症
研究表明,特定微生物群如乳杆菌和双歧杆菌不仅能促进色氨酸转化为血清素(5-HT),还可以直接合成GABA。作为中枢神经系统的主要抑制性神经递质,GABA与精神分裂症(SCZ)患者的焦虑和抑郁症状密切相关,其代谢异常对症状产生影响。通过与GABA受体结合,它有效降低神经兴奋性,延长慢波睡眠时间。
总之,神经递质的代谢在精神分裂症和睡眠障碍的病理机制中扮演重要角色。肠道微生物群通过影响神经递质的水平,不仅影响精神症状,还通过改变睡眠加重病情。因此,针对神经递质代谢的干预措施为治疗SCZ合并SD患者提供了新的思路与策略。
2
免疫炎症通路
肠道微生物群组成的变化会促进神经炎症,而这种炎症被认为是多种精神疾病(包括精神分裂症(SCZ)和睡眠障碍(SD))的重要病理机制。
IL-6和TNF-α等促炎细胞因子增加易导致精神分裂症恶化
肠道微生物群及其代谢物是调控小胶质细胞的成熟、形态和功能的关键分子。微生物群的变化可激活胶质细胞,促使IL-6和TNF-α等促炎细胞因子的释放,这影响神经元的存活与功能,破坏血-脑屏障的完整性,使外周炎症细胞因子和神经活性代谢物渗透中枢神经系统,最终诱发神经炎症反应。
针对SCZ患者的纵向研究显示,IL-6和TNF-α水平较高与脑源性神经营养因子(BDNF)水平下降及认知障碍相关。这些炎症标志物水平较高的SCZ患者更可能出现症状恶化。免疫系统在特定症状中也发挥作用,炎症刺激能改变健康个体腹侧纹状体区域的神经活动,导致动机减弱和奖赏处理缺陷,表明炎症与负性和认知症状之间的病理生理关系。
急性炎症升高也影响睡眠时间和深度
此外,睡眠障碍(SD)与炎症标志物如C反应蛋白(CRP)和IL-6的增加有关,急性炎症升高可改变睡眠的时间和深度。使用曼-惠特尼大学测试的统计分析发现,与对照组相比,接受72小时快速眼动(REM)睡眠剥夺的小鼠血浆内脂多糖浓度升高和TNF-α显著上调。
限制大鼠睡眠后,血-脑屏障的选择性过滤功能减弱,降低了阻断有害物质的能力,增加了神经炎症的风险。
通过调节肠道菌群的抗炎治疗有助于改善精神分裂症合并睡眠障碍
在临床干预中指出,使用认知行为疗法(CBT)治疗慢性和原发性失眠的老年患者后,炎症风险标志物减少,显示免疫炎症通路与睡眠障碍存在显著关联。
抗炎治疗可能是改善SCZ合并SD患者的重要策略。通过降低炎症通路则可减少小胶质细胞的激活和促炎细胞因子的产生,从而改善患者的整体症状和生活质量。
未来的研究应集中于探索特定的抗炎干预方法(如细胞因子拮抗剂或微生物组调控)对精神分裂症(SCZ)合并睡眠障碍(SD)患者的治疗效果及潜在神经保护机制。
3
神经内分泌调节
下丘脑-垂体-肾上腺轴(HPA)是神经内分泌系统的核心部分,在调节应激反应中发挥关键作用,其功能障碍已成为肠道微生物群介导睡眠障碍(SD)的重要机制之一。
压力下的高糖皮质激素导致认知障碍和焦虑增加
在压力条件下,下丘脑分泌的促肾上腺皮质激素释放因子(CRF)激活垂体前叶,释放促肾上腺皮质激素(ACTH),随后刺激肾上腺皮层合成糖皮质激素,如皮质醇。
长期过度暴露于糖皮质激素会影响大脑不同区域的神经元可塑性。这些结构重塑与认知功能障碍和动物实验中焦虑类行为增加密切相关,表明糖皮质激素介导的异常神经回路可能是慢性压力和精神障碍中认知缺陷和情感症状的重要病理基础。大量临床证据表明,HPA轴功能障碍在精神分裂症中普遍存在。
具体而言,精神分裂症患者的皮质醇觉醒反应(CAR)呈现明显异常模式,包括过度多动或钝化。这些患者的晨间基础皮质醇水平显著高于健康人群,而这种HPA轴功能障碍的模式具有疾病特异性,在高风险精神病人群中未观察到类似变化。
精神分裂症患者松果体受损影响褪黑素合成分泌
神经影像学研究显示精神分裂症(SCZ)患者的松果体体积减少、钙化加重,导致褪黑素合成和分泌功能受损。这种多层次内分泌障碍与SCZ临床症状的严重程度相关,并扰乱睡眠与觉醒周期,增加睡眠起始潜伏期和睡眠连续性受损情况。
可见,神经内分泌异常与睡眠障碍(SD)在SCZ中形成复杂的双向调控网络。睡眠不足激活下丘脑CRF神经元,导致HPA轴过度激活和皮质醇增加,而高皮质醇水平则抑制褪黑素合成,干扰睡眠调节,进一步加重SD。这一过程形成“内分泌障碍-睡眠问题-疾病恶化”的病理闭环,促进SCZ的进展和症状维持。
4
代谢物介导的调控
肠道微生物群生成的代谢物,如短链脂肪酸(SCFA)、色氨酸衍生物和胆汁酸,直接或间接参与中枢神经系统的调控,影响神经递质的平衡、维持免疫稳态,并调节昼夜节律。
丁酸盐有助于改善精神分裂症状和睡眠质量
菌群失调可破坏肠道屏障的完整性,增加肠道通透性,损害血-脑屏障的功能。这些双重屏障的破坏使得细菌代谢产物和促炎细胞因子等有害物质更易进入中枢神经系统,引发神经炎症反应。
丁酸盐作为关键代谢物,不仅对维持肠道黏膜的完整性至关重要,还能穿越血-脑屏障,通过激活迷走神经调节睡眠和清醒周期。动物研究发现,静脉注射丁酸盐显著延长了大鼠的非快速眼动睡眠(NREMS)时间。
此外,血清丁酸盐水平的升高与精神分裂症(SCZ)患者阳性和阴性综合征量表(PANSS)的阳性症状分数下降相关,暗示其潜在的神经保护作用。
血清素和胆汁酸水平下降可能是影响精神分裂症和睡眠障碍的重要机制
精神分裂症(SCZ)患者还表现出显著的色氨酸代谢障碍,研究发现其血浆色氨酸水平通常较低,这可能由于色氨酸向犬尿氨酸的转化增强和5-羟色胺(5-HT)合成途径的减少。这种转变导致神经递质5-HT和褪黑素的合成减少,由于这些物质在睡眠调节中起关键作用,其缺乏会加重精神分裂症患者的睡眠障碍(SD)。
代谢物分析显示,精神分裂症患者胆酸通路中的代谢物如糖果酸、牛磺酸脱氧胆酸和牛磺鹅脱氧胆酸的浓度显著降低,胆汁酸水平的下降通过影响抗炎信号通路,促进慢性低度炎症微环境的形成,从而增加神经退行性疾病和精神障碍的风险。研究还发现慢性失眠与特定胆酸结构和组成之间存在关联。
因此,代谢轴的异常不仅是精神分裂症的典型生物标志物,也是精神分裂症中睡眠障碍发病的关键机制之一。针对肠道微生物群及其代谢物的干预措施可为改善精神分裂症合并睡眠障碍患者提供新的治疗靶点。
5
昼夜节律紊乱
肠道微生物群与昼夜节律密切相关,其组成会随宿主生物钟的变化而波动,反之肠道微生物群的紊乱也会影响生物钟、肠道免疫功能和营养代谢。
肠道微生物群影响人体生物钟在精神疾病中发挥重要作用
肠道微生物群的组成和代谢物对宿主时钟基因(如CLOCK和Bmal1)的表达具有独特的调控作用。研究发现,微生物代谢物可以调节中枢神经系统和肝脏的生物节律,比如,短链脂肪酸(SCFAs)直接调控肝细胞中的时钟基因表达,影响宿主生物节律的稳定性。
肠道微生物群失衡在精神分裂症(SCZ)患者中较常见,这种失衡可能通过肠道-大脑轴影响宿主时钟基因的表达。分析首次发作SCZ患者的单核血细胞发现,与健康对照相比,患者的CLOCK、PER2和CRY1基因表达显著降低。而在慢性SCZ患者的成纤维细胞中,时钟基因CRY1和PER2的节律表达丧失。这些研究揭示了肠道微生物群通过代谢物与时钟基因的相互作用,对SCZ患者昼夜节律的稳定性至关重要。
时钟基因表达的变化不仅影响肠道微生物群的结构,还进一步影响核心昼夜节律输出通路的效率。研究表明,昼夜节律紊乱与肠道-大脑轴(MGBA)之间存在显著相互作用,MGBA在精神疾病的整个过程中持续影响患者的健康状况和临床症状。
昼夜节律调节神经内分泌活动影响精神分裂患者临床症状
结合本文前面的内容,昼夜节律通过调节神经内分泌活动和自主神经系统活动,调节睡眠、新陈代谢和免疫等生理过程。该输出通路的失效可能导致睡眠结构紊乱、压力激素节律失调,以及代谢和免疫反应的周期性紊乱,这些共同构成了精神分裂症患者常见的临床症状基础。与此同时,时钟基因表达的变化反过来会影响肠道微生物群的组成,显示出丰富度和多样性的减少,形成复杂的相互作用网络。
例如,研究显示宿主昼夜节律的核心激活基因(Bmal1)和抑制基因(Per1, Per2)共同调节肠道微生物群组成的昼夜振荡。这些时钟成分的基因敲除消除了这些节律波动,改变了小鼠的微生物群落结构。
深入研究肠道微生物群与其代谢物及时钟基因之间的关系,不仅有助于理解精神分裂症的发病机制,还将为基于肠道微生物群干预的未来治疗策略提供新思路。例如,通过调整饮食或补充益生菌以改善肠道微生物群的组成,调控时钟基因的表达以优化昼夜节律信号的传递效率,改善精神分裂症患者的临床症状。
小编总结
这组证据总体在说明:睡眠/昼夜节律紊乱本身就是一个能“驱动”肠道微生态改变的上游因素,而这种改变不仅是“相关”,还可能通过菌群把代谢风险传递出去。菌群不是静态的,它会随进食时间、胆汁酸分泌、肠蠕动、激素(如皮质醇/褪黑素)等出现日内波动,群落结构不稳定和功能(例如短链脂肪酸、胆汁酸转化、内毒素负荷)也会随之波动。
如果精神分裂症患者同时存在常见的睡眠/昼夜节律紊乱,那么观察到的某些肠道菌群特征(如特定门、科/属的增减,口源菌富集,以及产丁酸菌减少等)可能并非完全由疾病本身导致,而是共同受到多条路径的影响,包括:疾病相关应激、药物与生活方式,以及睡眠—生物钟失调这一通路。
与此同时,膳食纤维代谢菌减少提示菌群功能发生改变:尤其是短链脂肪酸(SCFAs)生成能力下降,可能削弱其对肠道屏障维护与免疫调节的作用;而促炎菌增加、抗炎菌减少则意味着机体更偏向促炎状态与肠道高通透,进而可能通过“肠—免疫—脑”轴影响中枢免疫激活与神经炎症。
在监测精神分裂症菌群时,睡眠状态可能是重要的混杂因素,也可能是可干预的上游靶点(改善睡眠可能间接改善菌群与代谢/炎症表型)。
尽管睡眠障碍与多种精神疾病的发生和发展密切相关,但针对精神分裂症(SCZ)合并发病的干预研究仍然不足。目前大多数治疗策略集中于症状控制,如通过抗精神病药物改善核心精神症状,而较少关注肠道微生物群失调机制。
因此,以下讨论提出针对肠道微生物生态的新治疗策略,包括特定益生菌、益生元、饮食干预、粪便微生物移植(FMT)和靶向代谢物治疗,旨在调节肠道-脑轴功能,以改善精神分裂症患者的睡眠问题及相关精神症状。
针对精神分裂症患者的微生物群干预方法
编辑
1
个性化补充益生菌
个体间肠道微生物群的差异会影响药物的疗效和毒性,因此个性化的肠道微生物群调控有助于改善药物反应。
通过检测精神分裂症(SCZ)患者的肠道微生物群,可以制定个性化益生菌方案,有效调节肠道微生物组成,从而改善症状。
长双歧杆菌等益生菌能改善精神状态
益生菌的疗效因菌株而异,不同菌株在调节炎症和压力方面表现出不同作用机制。例如,婴儿双歧杆菌(Bifidobacterium infantis 35624)已被证明能逆转HPA轴功能障碍,并与抗炎和促炎细胞因子比例的正常化相关,具有临床意义。
鼠李糖乳杆菌(Lactobacillus rhamnosus,JB-1)能降低压力诱发的皮质酮水平,缓解焦虑和抑郁行为。长双歧杆菌(Bifidobacterium longum NCC3001)通过MGBA内的迷走神经通路传递信号,使低度肠道炎症小鼠的焦虑行为和海马BDNF水平恢复正常。该菌株在人类试验中也显示抗抑郁效果,与多个情绪处理脑区活动变化相关。
益生菌有助于改善炎症相关、情绪状态及睡眠
在一项针对失眠患者的双盲研究中,参与者接受了植物乳杆菌(Lactobacillus plantarum PS128)或安慰剂。结果显示,PS128组在疲劳水平、脑电波活动和深度睡眠期间醒来次数方面有所改善,同时也缓解了焦虑和抑郁症状。这表明特定益生菌菌株可能通过微生物群-肠道-脑轴的机制(如GABA能系统调控)影响睡眠结构和情绪状态。
一项针对双相情感障碍和精神分裂症谱系障碍患者的双盲随机安慰剂对照试验表明,补充多株益生菌配方能改善与肠道通透性和炎症相关的生物标志物,并对认知功能产生积极影响。
这些研究结果揭示了益生菌补充剂在调节神经功能方面的潜在治疗价值,为进一步探索益生菌的干预机制提供了重要理论基础。
2
膳食益生元补充
益生元是一类能够抵抗宿主消化酶的功能性膳食成分,能选择性促进有益肠道微生物群的代谢活动,从而发挥生理调节功能。
菊粉等益生元改善精神分裂症的精神和行为症状
多项研究发现,半乳糖、菊糖型果聚糖及其合成制剂能够降低促炎因子(如高敏感性CRP、IL-6和TNF-α)的水平,表明特定的益生元干预可以调控炎症相关生物标志物的表达。菊粉作为可溶性膳食纤维,已被证实改善精神分裂症(SCZ)模型小鼠的精神和行为症状,同时增加有益菌数量并改善肠道通透性。
服用低聚果糖和低聚半乳糖的复合制剂也改善了SCZ模型小鼠的肠道功能。这些变化可能通过重塑肠道微生物群、降低促炎细胞因子水平以及增强肠道通透性,从而减少病原体与肠道黏液层的接触,为改善多种精神疾病的核心症状提供重要的病理生理基础。
益生元调节神经递质平衡改善精神状态及睡眠
另一项研究表明,短链半乳糖与长链果糖的联合干预显著促进了短链脂肪酸中乙酸和丁酸的生成。这些益生元成分还有效调控下丘脑和海马体中核心时钟基因BMAL1和CLOCK的表达。
这表明益生元通过多重靶点的协同效应发挥神经调节功能。特别是,益生元对时钟基因的调节可能通过恢复昼夜节律改善睡眠问题,并通过调节神经递质平衡潜在改善精神分裂症核心症状。
为促进益生元疗法在精神疾病临床实践中的应用,后续研究应明确不同益生元的应用价值、个体化剂量策略以及治疗的安全性和有效性评估。
3
饮食调整干预
特定饮食成分通过与肠道菌群互作影响睡眠质量
饮食调整是个性化治疗的重要部分,已有大量科学证据证明饮食与睡眠之间存在关系。健康饮食有助于改善睡眠质量,而加工食品和高糖食物则与睡眠质量较差相关。
特定饮食成分(如脂肪和蛋白质)及习惯(如牛肉、咖啡和干果摄入)通过与肠道微生物群相互作用影响睡眠。
生酮和高纤维饮食有助于改善睡眠质量和免疫
近年来,生酮饮食受到广泛关注,已被证明是改善伴有代谢异常的精神疾病患者心理和代谢健康的一种可行辅助治疗方法。根据匹兹堡睡眠质量指数(PSQI)评估,接受生酮饮食干预后,双相情感障碍和精神分裂症(SCZ)患者的主观睡眠质量有所改善。这表明,生酮饮食可以纳入精神疾病患者的综合治疗策略,有望在控制精神症状、调节代谢和改善睡眠方面带来多重益处。
此外,从高纤维饮食的角度来看,短链脂肪酸(SCFAs)是通过肠道共生细菌发酵膳食纤维产生的。当膳食纤维摄入不足时,SCFAs的生成水平会降低,这将对宿主的各种生理功能产生不利影响。增加膳食纤维摄入以促进SCFA的产生,不仅能增强血液和脑屏障的保护功能,维持肠道黏膜的完整,还能调节肠道微生物群的组成,从而协同促进免疫稳态的建立。
基于这一机制,这种饮食干预策略为改善精神障碍患者的睡眠提供了新思路。通过缓解肠道炎症状态并调节神经递质水平,它可以改善患者的精神症状和睡眠质量。
4
粪菌移植
粪菌移植(FMT)是一种以重建微生物群落为核心的生物治疗策略,通过将健康供体的粪便移植到患者的胃肠道,直接重组受体的肠道微生物群,促进微生物生态系统的平衡,从而实现临床治疗目标。
粪菌移植有助于调节情绪、行为和睡眠质量
作为一种突破性干预方法,粪菌移植(FMT)在动物模型和初步临床研究中展现出独特优势。研究发现,通过移植精神分裂症(SCZ)患者的肠道微生物群,被移植小鼠出现了类似SCZ的行为,包括多动、焦虑增加、社交互动受损和记忆缺陷。
注:这些小鼠还显示出周围和中枢神经系统中色氨酸代谢犬尿氨酸-犬尿酸通路的显著激活,前额叶皮层基底细胞的多巴胺能神经递质释放增强,以及海马体5-HT水平升高,同时谷氨酸能神经递质浓度下降,谷氨酰胺和GABA水平上调。
FMT在人体临床研究中展现出重要价值。比较接受健康供体FMT前后的粪便样本显示,α多样性显著增加,柯林斯氏菌属(Collinsella)和双歧杆菌的丰度也有所上升。FMT治疗还显著降低了患者的血液皮质醇水平,同时改善了睡眠和情绪相关评估指标。
作为潜在治疗干预,FMT通过调节肠道微生物群的组成和功能,促进神经递质及其前体物质的合成,从而发挥治疗作用。这些研究表明,FMT能够有效调节情绪和行为,促进多种精神疾病患者的症状缓解。
!
注意事项
尽管粪菌移植(FMT)展现了广泛的治疗前景,但其临床应用面临标准化不足、疗效不一及长期安全性验证的挑战。高通量测序技术可帮助识别健康稳定的供体。口服FMT胶囊相比传统灌肠方法更方便,患者依从性更高。多样化的给药途径,如结肠镜输注、鼻肠管插管或口服胶囊,有助于平衡疗效与患者依从性。建立安全监测系统可预防和控制感染风险,加强安全监管。
此外,在进行FMT治疗时,必须密切关注精神障碍患者的知情同意能力,确保他们能够在充分了解潜在风险和益处的基础上做出自主决策,并通过伦理审查委员会的批准进行标准化申请。尽管仍需更多研究验证FMT在精神分裂症(SCZ)和睡眠障碍(SD)治疗中的长期效果,但作为个性化治疗的一部分,FMT显示出广泛的应用前景。
5
靶向代谢物药物治疗
在精准医疗领域,靶向代谢物药物治疗正成为新研究方向。尽管传统微生物群移植在临床应用中取得了一定成效,但个体肠道微生物群的差异严重限制了其治疗效果的稳定性和重复性。因此,直接补充微生物代谢物或前体物质为解决这一问题提供了新思路。
靶向代谢物能够避免个体菌群差异,更精准治疗
这种策略能够绕过微生物群移植中的个体差异,直接作用于人体代谢网络,实现更精确的调控。短链脂肪酸(SCFAs)和色氨酸代谢在生理和病理过程中起着关键作用。
以吲哚胺2,3-二氧加氧酶(IDO)抑制剂为例,IDO是色氨酸代谢中的限速酶,其过度活化可导致色氨酸耗竭和一系列神经毒性代谢物的产生,进而引发异常免疫反应和神经功能障碍。IDO抑制剂通过抑制其活性,可以有效调节免疫反应,减少神经毒性代谢物的产生,展现出治疗多种疾病的潜力。
SCFAs和色氨酸代谢物与精神分裂症(SCZ)发病机制及睡眠调节密切相关。未来研究应进一步探讨代谢物的具体作用机制,推动其临床应用的发展,为合并睡眠障碍的SCZ患者提供更安全、更有效的治疗选择。
治疗策略总结
并非所有精神分裂症(SCZ)患者都适合微生物组靶向干预。潜在候选者的识别应基于具体特征,例如明显的胃肠道症状、抗精神病药物反应不良或显著副作用、异常睡眠结构与疾病活动的强相关性,以及肠道微生物组分析结果显示与健康对照有显著偏差。
根据临床表型和微生物特征进行对应治疗选择
目标群体应根据临床表型和微生物特征的组合进行选择。在推进微生态干预时,应明确不同策略的优先级,优先考虑低风险、非侵入性的方法。例如,补充特定益生菌和益生元可作为基础干预,而饮食模式调整可作为长期管理策略。
相比之下,高风险且难以预测的治疗如粪便微生物移植(FMT)应仅考虑用于难治症状、对传统疗法反应不足及严重肠道菌群失调的患者,并在充分知情同意和严格伦理监督下使用。
此外,实施时需认真考虑个体差异和潜在风险。益生菌和益生元干预应考虑菌株特异性效应和个体耐受性,饮食干预需关注患者的依从性和营养平衡。对于FMT,严格的供体筛查至关重要,并需密切监测感染、免疫和代谢不良反应,以及长期精神病学结局,同时建立动态的疗效-安全性评估体系。
本文分析了精神分裂症(SCZ)及其相关睡眠障碍患者的典型肠道微生物群特征。研究发现,这两种状况均表现出肠道微生物群α多样性下降、短链脂肪酸产生细菌减少以及促炎微生物群比例增加。由此可以推测,SCZ患者的肠道菌群失调可能是导致睡眠质量下降的重要因素,而睡眠质量恶化又通过反馈机制加重精神症状,形成恶性循环。
肠道菌群失调、中枢神经系统功能障碍和睡眠稳态紊乱在宿主体内形成自我延续的动态循环,导致精神病理表现和生理节律紊乱的周期性恶化。这些发现为理解精神分裂症患者共病睡眠障碍的机制提供了重要的理论框架。针对肠道微生物群以打破这一恶性循环,为这类患者提供了有前景的新颖治疗视角。
现有研究表明,治疗应采取综合策略。虽然非典型抗精神病药物能改善精神病症状,但对睡眠结构的复杂影响仍需深入评估。认知行为疗法同样对失眠有效,但需根据精神病症状进行调整。因此,针对肠道微生物群的干预策略正在向多层次和精准化方向发展,具体包括补充特定益生菌和益生元,结合饮食调整,通过多途径共同改善精神症状和睡眠质量。尽管粪便微生物移植(FMT)作为高级干预措施展现出潜力,但其效果仍需通过标准化研究来验证。实施时应重点关注胃肠道症状、药物反应不足、精神症状严重程度与睡眠节律紊乱以及肠道微生物群特征显著偏离健康标准的患者。整合临床表型与微生物特征的模型将为精准应用干预策略提供基础,最终推动微生物群靶向治疗的系统化及个性化发展。
我们每个人都在不断追求身心健康的道路上前行,理解肠道微生物群在我们的心理与生理健康中所扮演的角色,不仅能增进我们对自身健康的认识,更能激励我们在日常生活中关注饮食、生活方式等易被忽视的细节。希望未来的研究能够持续启发我们,推动科学领域的进步,为那些受到精神障碍困扰的人们带来新的希望和解决方案。通过这样的努力,我们将共同迈向一个更加健康和明亮的未来。
注:本账号发表的内容仅是用于信息的分享,在采取任何预防、治疗措施之前,请先咨询临床医生。
主要参考文献:
Huang, Z., Huang, Z., Du, Z., Gao, X., Jiang, Y., Zhou, Z., & Zhu, H. (2026). Role and mechanism of gut microbiota and metabolites in schizophrenia complicated with sleep disorder. Gut Microbes, 18(1). https://doi.org/10.1080/19490976.2025.2607817.
McCutcheon RA, Reis Marques T, Howes OD. Schizophrenia-An Overview. JAMA Psychiatry. 2020 Feb 1;77(2):201-210.
Meyer N, Faulkner SM, McCutcheon RA, Pillinger T, Dijk DJ, MacCabe JH. Sleep and Circadian Rhythm Disturbance in Remitted Schizophrenia and Bipolar Disorder: A Systematic Review and Meta-analysis. Schizophr Bull. 2020 Sep 21;46(5):1126-1143.
Reeve S, Sheaves B, Freeman D. Sleep Disorders in Early Psychosis: Incidence, Severity, and Association With Clinical Symptoms. Schizophr Bull. 2019 Mar 7;45(2):287-295.
McGuinness AJ, Davis JA, Dawson SL, Loughman A, Collier F, O’Hely M, Simpson CA, Green J, Marx W, Hair C, Guest G, Mohebbi M, Berk M, Stupart D, Watters D, Jacka FN. A systematic review of gut microbiota composition in observational studies of major depressive disorder, bipolar disorder and schizophrenia. Mol Psychiatry. 2022 Apr;27(4):1920-1935.
Mayeli A, LaGoy AD, Smagula SF, Wilson JD, Zarbo C, Rocchetti M, Starace F, Zamparini M, Casiraghi L, Calza S, Rota M, D’Agostino A, de Girolamo G; DiAPAson Consortium; Ferrarelli F. Shared and distinct abnormalities in sleep-wake patterns and their relationship with the negative symptoms of Schizophrenia Spectrum Disorder patients. Mol Psychiatry. 2023 May;28(5):2049-2057.
Penninx BWJH, Lange SMM. Metabolic syndrome in psychiatric patients: overview, mechanisms, and implications. Dialogues Clin Neurosci. 2018 Mar;20(1):63-73.
Zhu C, Zheng M, Ali U, Xia Q, Wang Z, Chenlong, Yao L, Chen Y, Yan J, Wang K, Chen J, Zhang X. Association Between Abundance of Haemophilus in the Gut Microbiota and Negative Symptoms of Schizophrenia. Front Psychiatry. 2021 Jul 30;12:685910

谷禾健康
植物性食物 Plant-based Diet
食物,尤其是植物性饮食,具有复杂的化学多样性。然而,肠道细菌大规模代谢植物营养素的活动在很大程度上仍是不完全清楚的。
近日,来自德国耶拿莱布尼茨天然产物研究与感染生物学研究所(Leibniz-HKI)微生物组的 Gianni Panagiotou团队联合上海交通大学医学院于发表在《Nature microbiology》整合并系统分析了包含酶反应和食物健康益处信息的多个数据库,以及3,068个全球公开的人类微生物组数据和来自可食用植物的775种植物营养素,发现植物营养素的转化与多种肠道微生物编码的酶相关。
通过结构预测与体外实验证实关键菌株(如 Eubacterium ramulus)对黄酮类化合物的特异代谢作用及其关键酶的催化功能。植物营养素的转化能力存在显著个体差异和地域特异性,五大洲人群共享630种核心转化通路,且疾病,年龄,肥胖等都会改变我们的代谢能力。
最终,这项研究告诉我们:我们吃下的植物性食物中含有数千种化学物质,其中许多需要经过你肠道中数万亿微生物的“加工处理”才能被人体有效利用。
同时该研究创新性地建立了“膳食化学—微生物酶—宿主健康”多层耦合框架,暗示在未来,营养师和医生可能会常规分析你的肠道微生物组,以推荐最适合你个人“肠道化工厂”的饮食方案,让我们真正迈向个性化营养的新时代。
“饮食决定健康”这一简单概念正被更复杂的理解所取代:我们的健康不仅取决于吃什么,还取决于我们的肠道微生物如何处理我们所吃的食物。”
就如刚发表在《Nature Microbiology》上的这项开创性研究,系统地探索了肠道微生物如何代谢植物性食物中的化学物质(称为植物营养素),以及这一过程如何影响我们的健康。来自德国、丹麦、中国等国家的科学家团队整合了全球3,068个人类肠道微生物组数据和1,300多种植物营养素信息,揭示了一个肠道惊人的”代谢宇宙”:我们肠道中约70%的微生物酶参与植物营养素的生物转化。
这意味着,当你咬下一口新鲜的草莓或清脆的西兰花时,你肠道中的微生物正在加班加点地将这些复杂化合物转化为对你身体有益的物质——但前提是拥有理想的微生物团队。
黄酮类化合物是植物性食物重要的次级代谢产物,隶属多酚类物质,广泛存在于多种膳食植物来源中,如水果、蔬菜、草药、种子、谷物及饮料等。其基本骨架通常由芳香环 A 与杂环 C 相连,并与另一芳香环相结合形成特征性结构。依据化学结构差异,黄酮类可进一步分为黄酮、黄酮醇、黄烷酮、黄烷醇、异黄酮和花青素等主要类型。
摄入后,部分黄酮类化合物可在小肠中经上皮酶 O-脱糖基化后被吸收;进入肠上皮细胞与肝脏后,吸收的黄酮类苷元进一步发生 I 相与 II 相代谢转化。然而,多数膳食黄酮类化合物(及其 I/II 相代谢产物)会到达大肠,并在肠道菌群作用下被进一步改造。大肠细菌酶可介导多种结构修饰反应,如脱羧、开环、还原、异构化和去甲基化等。
由于膳食黄酮多以糖苷形式存在,肠道细菌的 α-L-鼠李糖苷酶 可去除末端 α-L-鼠李糖,将其转化为葡萄糖苷;随后 β-葡萄糖苷酶进一步脱糖生成可吸收的苷元,苷元既可在大肠被吸收,也可继续代谢为多种开环产物。
肠道菌群介导的黄酮类代谢中,雌马酚(equol)生物合成途径是研究最为深入的代表之一,其核心在于一系列关键酶将异黄酮苷元逐步转化为雌马酚。
另一条较为清晰的途径是黄酮/黄酮醇的降解代谢,其中 Flavonifractor plautii 与 Eubacterium ramulus被认为是参与转化的活跃菌种;在这些菌种中已鉴定出多种相关酶,包括黄酮/黄酮醇还原酶(Flr)、查尔酮异构酶(Chi)、黄烷酮/黄烷醇裂解还原酶(Fcr)以及根皮苷水解酶(Phy)。
黄酮类化合物及其微生物代谢产物因具备抗炎、抗氧化与抗癌等生物活性而与多种健康获益相关,其保护作用已在结直肠癌(CRC)和炎症性肠病(IBD)等疾病研究中得到支持。
植物营养素与微生物酶的完美匹配
当想研究这个复杂关系时,首先面临的挑战是如何将植物性食物中的化学物质与肠道微生物产生的酶联系起来。研究团队人员从NutriChem 2.0数据库中获取了7,825种与可食用植物相关的低分子量植物营养素,并功将超过1,500种化合物与具有独特酶库编号的酶联系起来。经过精细筛选,最终确定了1,388种植物营养素与4,678种酶相关联。
通过分析全球3,068个健康人的肠道微生物组数据,研究团队发现这些植物营养素中有775种可被肠道微生物酶转化,涉及1,118种可食用植物。令人惊讶的是,约64%(1,226种)的这些酶仅存在于肠道微生物物种中,而在人类基因组中不存在。这意味着,如果没有肠道微生物的帮助,我们的身体将无法代谢这些植物性化合物。
广泛的代谢能力
约67%的肠道微生物酶可能参与植物营养素的生物转化
独特的微生物贡献
约64%的植物营养素转化酶仅存在于肠道微生物中,人类基因组中不存在
主要参与者
拟杆菌门和变形菌门,是最常见的植物营养素转化菌门
核心化学类别
萜类、黄酮类和生物碱是主要被转化的植物营养素类别
主要酶类别
氧化还原酶、转移酶和水解酶是参与转化的主要酶类
值得注意的是,达人群中肠道细菌物种的丰度和流行度与其基因组转化植物营养素的能力呈正相关。这意味着那些在我们肠道中更常见、数量更多的细菌,往往具有更强的代谢植物性化合物的能力。
研究团队特别关注了21种被认为是健康饮食选择的常见食物,发现它们中超过一半的已识别植物营养素与肠道微生物酶相关联。例如,黄酮类化合物根皮素的糖基化(由EC 2.4.1.4催化)和根皮素向3-(4-羟苯基)丙酸的转化(由查尔酮异构酶EC 5.5.1.6催化)都是已知的与健康相关的生物转化过程。
注:Parsnips 是英文里对 “欧洲防风草 的称呼,一种根茎类蔬菜,外形有点像白色胡萝卜。
这些发现表明,我们日常食用的许多健康食物,如蓝莓、绿茶、坚果等,其健康益处可能在很大程度上依赖于我们肠道微生物的代谢能力。换句话说,如果你肠道中缺乏特定的微生物或其产生的酶,即使食用”超级食物”也可能无法获得其全部健康益处。
随着益生菌产品的普及,许多人可能认为服用益生菌补充剂可以获得与健康肠道微生物相同的益处。但这项研究揭示了一个重要事实:常见益生菌在代谢植物营养素方面的能力远不如我们肠道中自然存在的某些细菌。
益生菌的短板 与 肠道细菌的特长
研究人员注释了59种益生菌的基因组,发现这些菌株中的酶可以潜在地转化775种与肠道微生物酶相关的植物营养素中的525种。然而,在186种与次级代谢相关途径的植物营养素中,只有116种可被益生菌和肠道微生物酶共同修饰,而70种只能被肠道微生物酶修饰。
更重要的是,能够修饰这些植物营养素的肠道细菌种类平均为158种,而能够做到这一点的益生菌种类平均仅为8种。这意味着,许多植物性食物中的生物活性化合物需要我们肠道中特定的”本土”细菌才能被有效代谢,而常见的益生菌补充剂可能无法提供这种能力。
研究还发现,共享和肠道限制性植物营养素在结构特征上存在差异。含有羧酸和羰基的植物营养素(如反式肉桂酸和柚皮素)特别富集在共享化学空间中,而肠道限制性植物营养素则富含苯环和双环基团。
注:
原文中 shared phytonutrients(共享):既能被肠道常驻菌也能被常见益生菌,预测/注释到可代谢的植物营养素。
gut‑restricted phytonutrients(肠道限制性):只被肠道常驻菌预测/注释到可代谢,而未在益生菌物种中预测到可代谢的植物营养素。
所以,肠道限制性在这里就是一个相对概念:相对于他们纳入分析的那批益生菌而言,这些化合物的代谢能力更“限制在肠道本土菌里”。
体外实验验证:Eubacterium ramulus的明星表现
为了验证这些发现,研究团队在实验室中测试了六种具有高生物转化潜力的肠道菌株对36种植物营养素的代谢能力。结果令人瞩目:Eubacterium ramulus(一种常见的肠道细菌)表现出最强的生物转化活性,能显著代谢12种底物中的11种。
黄色的代表Eubacterium ramulus 菌种
进一步研究发现,Eubacterium ramulus含有一种名为EC 5.5.1.6的酶,能够催化两种黄酮类化合物——butein和isoliquiritigenin的生物转化。当存在活的E. ramulus时,这些化合物的转化速度显著加快,并且只在活菌存在的情况下才能检测到下游产物。这一发现不仅验证了计算预测,还为理解特定细菌如何影响植物营养素的代谢提供了机制性见解。
研究结果对益生菌行业提出了挑战。许多商业益生菌可能无法提供我们肠道中自然存在的某些细菌所具有的代谢能力。同时也意味着保护我们原生肠道微生物组多样性的重要性。
如果你曾经疑惑为什么某些食物对你的朋友或家人有效却对你似乎没什么作用,这项研究可能给了答案。研究发现,植物营养素的生物转化存在显著的个体间和地理变异性,这解释了为什么相同的饮食对不同人会产生不同的健康效果。
个体差异:与α多样性(群落丰富度/均匀度)的关系
分析显示,在个体微生物组中注释的酶中,平均70%与植物营养素生物转化相关,而在次级代谢相关途径中这一比例高达90%。然而,植物营养素相关酶的比例与微生物组α多样性呈负相关,这表明植物营养素生物转化是肠道微生物群的普遍特性。但对于参与次级代谢的酶来说,这种相关性是正的,表明肠道微生物转化次级代谢相关植物营养素的能力存在差异。
个体差异很大,但洲际总体很相似
个体间植物营养素转化范围差异明显:每人可被转化的植物营养素数量在264–620之间(覆盖5个地理区域)。
植物营养素相关的微生物组独特性在不同个体之间显著高于同一个体内部。
但把人按“洲/地区”汇总后,整体“植物营养素与食物生物转化相关的酶学机器”在各地区高度相似:5个地区共同关联到的植物营养素有630种;
只有少数显示强地域特异性(如亚洲10种、大洋洲2种)。
例如黄烷醇类(taxifolin)及其相关酶 EC 1.1.1.219 只在亚洲个体中被注释到,可能与当地食物/药用植物相关。
地理差异:祖籍可能影响你的代谢能力
地理差异的有趣发现所有地区的肠道微生物组都能代谢630种常见植物营养素;然而,也有一些植物营养素表现出高度的地理特异性。
例如,黄酮类化合物taxifolin(存在于荔枝和柘树等食物中,后者是东亚的传统药物)及其相关酶EC 1.1.1.219仅在亚洲个体中被注释到。这种地理特异性可能反映了长期饮食传统与肠道微生物组之间的共同进化。
某些植物营养素的代谢能力具有地理特异性;且不同洲的酶谱仍显著不同,反映饮食差异。非洲和亚洲与其他地区的差异尤为显著,这反映了区域饮食的巨大差异。酶的变化与特定化学类别(包括黄酮类和脂肪酸相关化合物)显著相关。
为了探索饮食如何塑造这些模式,研究人员使用了已有的连续饮食信息和配对的宏基因组数据。Procrustes分析显示,可食用植物饮食记录与这些可食用植物1,665种植物营养素相关的酶丰度之间存在显著一致性,表明食用植物的摄入与微生物组的植物营养素生物转化潜力相关。
更有趣的是,对美国出生的个体和居住在泰国、刚移民到美国或已在美国生活至少20年的泰国人的数据集进行的相似性分析发现,尽管饮食习惯的适应相对温和,但泰国参与者的食用植物相关酶组成随着时间推移变得与美国出生的参与者显著相似。这表明,我们的肠道微生物组及其代谢能力可以通过改变饮食来调节,尽管这可能需要相当长的时间。
研究还发现,植物营养素相关酶的比例与年龄呈负相关,而植物营养素相关微生物组的独特性与年龄呈正相关。这表明,随着年龄增长,我们的肠道微生物组可能失去一些代谢植物营养素的能力,同时变得更加个性化。此外,511种酶与BMI显著相关,随着BMI增加,植物营养素相关酶的数量减少,这可能部分解释了为什么肥胖人群对某些植物性食物的反应不同。
研发发现
疾病状态会显著改变肠道微生物群对“具有健康益处的植物食物/其植物营养素”的生物转化潜力;这些与植物营养素相关的微生物酶特征还能用于区分健康人与患者。
研究设计
对象/数据:公共肠道宏基因组病例-对照队列,覆盖三类疾病:
方法主线
把肠道细菌酶(EC/酶学分类,且做了species-stratified分层归因到物种)与其可作用的具有健康益处的植物营养素空间关联,评估不同疾病中这些酶的丰度变化,并进一步用机器学习做判别。
主要发现1:与植物营养素相关的酶在疾病中大量失衡
IBD:炎症性肠病(Inflammatory Bowel Disease)
CRC:结直肠癌(Colorectal Cancer)
NAFLD:非酒精性脂肪性肝病(Non-Alcoholic Fatty Liver Disease)
识别到的植物营养素相关酶数量:
IBD:608
CRC:1,038
NAFLD:517
其中在患者 vs 对照中显著差异丰度
IBD:59.7%(363/608)
CRC:49.9%(518/1,038)
NAFLD:22.2%(115/517)
说明IBD 与 CRC 中,这类能处理健康植物营养素的微生物酶谱改变更广泛;NAFLD 相对较少但仍存在系统性改变。
主要发现 2:对具体健康食物的生物转化潜力改变程度不一
按每种有益食物对应的植物营养素相关 EC来看,平均有显著改变的比例:
IBD:63.0%
CRC:33.6%
NAFLD:25.3%
并且存在显著异质性:不同疾病之间差异很大;
同一疾病内部,不同食物差异也很大;
有些食物对应的酶中超过 50%都显著改变。
意味着不是“整体代谢能力统一下降/上升”,而是呈现“食物/营养素通路特异”的重塑。
主要发现 3:饮食相关酶特征可用于疾病判别,且优于非饮食相关酶
随机森林判别模型使用的特征数与内部表现:
IBD:18 个酶,auROC = 0.892
CRC:28 个酶,auROC = 0.763
NAFLD:22 个酶,auROC = 0.95
外部队列验证:三病均保持较高准确度(auROC 约 0.72–0.79)。
对照实验:若改用非饮食相关 EC(不与植物营养素关联)做 species-stratified 特征,验证集 auROC 降低:
IBD:0.779
CRC:0.698
NAFLD:0.584
这些可以推测与植物营养素生物转化直接相关的酶信号更贴近疾病相关的微生物功能改变,具有更强的可迁移判别力(尤其 NAFLD 的差距最大)。
总之,这些说明疾病状态不仅改变菌群有哪些菌,也改变其处理健康植物食物成分的功能酶库;这种功能改变具有食物/营养素通路特异性。
动物实验验证:草莓的抗炎作用为何依赖肠道微生物?
编辑
为了在体内测试参与植物营养素生物转化的肠道微生物酶是否对健康饮食的保护作用至关重要,研究人员在特定无病原体(SPF)和无菌(GF)条件下的结肠炎小鼠模型中研究了抗炎食物草莓的作用。
草莓的保护作用依赖肠道微生物
编辑
草莓抗炎作用的微生物机制
关键酶的作用:EC 4.1.1.11(天冬氨酸→β-丙氨酸和色氨酸→色胺)与减轻炎症相关
基因表达的重要性:不仅酶的存在(DNA水平)重要,其表达(RNA水平)也至关重要
治疗潜力:特定酶(如EC 6.3.1.1)可能成为炎症性肠病的治疗靶点
这些发现有力地证明,抗炎植物性食物(如草莓)的全部益处取决于特定肠道微生物酶的存在和转录活性。这解释了为什么同一食物可能对某些人有显著的健康益处,而对另一些人则效果有限——这完全取决于他们肠道中是否存在并表达这些关键酶。
这项研究通过整合全球微生物组数据和植物营养素信息,为我们理解饮食-微生物组-健康之间的联系提供了前所未有的深度和广度。研究结果不仅揭示了肠道微生物如何代谢植物性食物中的数千种化合物,还为个性化营养和精准医学开辟了新的可能性。
对益生菌和功能性食品开发的启示
这项研究对益生菌行业提出了重要挑战。虽然益生菌在各种疾病背景下已显示出疗效,但研究发现常见益生菌在代谢植物营养素方面的能力远不及我们肠道中自然存在的某些细菌。这表明,超越乳酸杆菌和双歧杆菌的益生菌开发对于更广泛地应用于人类健康至关重要。
研究人员发现,一小部分肠道细菌物种(如11种细菌的组合)可以潜在地修饰43种次级代谢相关代谢物(常见益生菌缺乏这种遗传潜力)。这为通过选择具有特定代谢能力的细菌进行发酵来开发功能性食品提供了新方向。
重新定义”健康饮食”
研究结果强调了重新审视”健康饮食”概念的重要性。饮食的有效性可能在很大程度上取决于健康微生物组的存在。对于肠道微生物组失衡的人来说,食用具有已证实健康益处的可食用植物可能不足以获得全部益处。因此,个性化营养可能需要特定食物和有益微生物的组合,或食物的离体发酵,以实现植物性饮食的全部营养潜力。
特别值得注意的是,植物营养素相关酶的比例与年龄呈负相关。这表明,老年人可能失去代谢某些植物营养素的能力,这为开发针对老年人的特定益生菌或发酵食品提供了依据,以弥补这种与年龄相关的代谢能力下降。

谷禾健康
维生素
B族维生素和维生素K是微生物和人类生理学中多种细胞过程所必需的微量营养素。传统上,这些维生素被认为主要来自饮食,但近年来的研究显示,人类肠道微生物群具备合成这些维生素的能力,提供了重要的内源性来源,这一贡献过去常被低估。
值得注意的是,微生物对宿主维生素储备的贡献正逐渐被视为维生素稳态的重要功能。在结肠中,微生物合成的维生素能够通过特定的运输机制被吸收,以发挥其生物学作用。
什么是维生素?
维生素是一类化学结构各异的有机化合物,人体只需要很少的量,但它们对维持细胞和全身的生理功能至关重要。
与提供结构或能量的宏量营养素(如碳水化合物、蛋白质和脂肪)不同,维生素主要通过作为辅因子或辅酶前体的形式发挥作用,帮助酶完成各种反应,这些反应涉及能量代谢、物质合成、氧化还原调节和细胞信号传导。
即使是轻微的维生素缺乏,也可能损害代谢的完整性,并增加患贫血、神经退行性疾病和免疫缺陷等多种疾病的风险。
分类
根据溶解性和生理特性,维生素通常分为水溶性和脂溶性两类。
• 水溶性维生素
水溶性维生素包括B族维生素(维生素B1、B2、B3、B5、B6、B7、B9和B12)和维生素C。它们通常作为辅基或辅酶参与酶促反应,尤其是在能量代谢、核苷酸合成和一碳单位转移反应中。由于其亲水性,水溶性维生素不易在体内储存。
• 脂溶性维生素
脂溶性维生素(维生素A、D、E和K)通过脂类介导的途径被吸收,并储存在脂肪组织和肝脏中,形成一个相对稳定的储备。
脂溶性维生素的功能更偏向于调节而非催化,但对于维持正常的视力、免疫调节、抗氧化防御以及钙磷平衡至关重要。比如,维生素K是凝血因子中谷氨酸残基γ-羧化反应的辅因子。
来源
• 大部分维生素依赖饮食获取或微生物产生
值得注意的是,由于人体无法合成大多数维生素,因此它们是必需的营养素。只有少数例外,如维生素D,它可以在紫外线照射下由皮肤中的7-脱氢胆固醇合成。对于其他所有维生素,人体主要依赖饮食和微生物的产生来获取。
• 肠道菌群合成维生素的能力对人体代谢非常重要
与高等真核生物不同,许多细菌物种具备从头合成必需辅因子的遗传机制,包括维生素B1、B2、B3、B5、B6、B7、叶酸(B9)、B12和甲萘醌(K2)。
其中,6种B族维生素(B1、B2、B3、B5、B6和B9)是关键的辅酶前体,这些辅酶在细菌和哺乳动物中发挥作用,如硫胺素焦磷酸(TPP)、黄素单核苷酸/黄素腺嘌呤二核苷酸(FMN/FAD)、烟酰胺腺嘌呤二核苷酸(NAD/NADP)、辅酶A(CoA)、磷酸吡哆醛(PLP)和四氢叶酸(THF)。
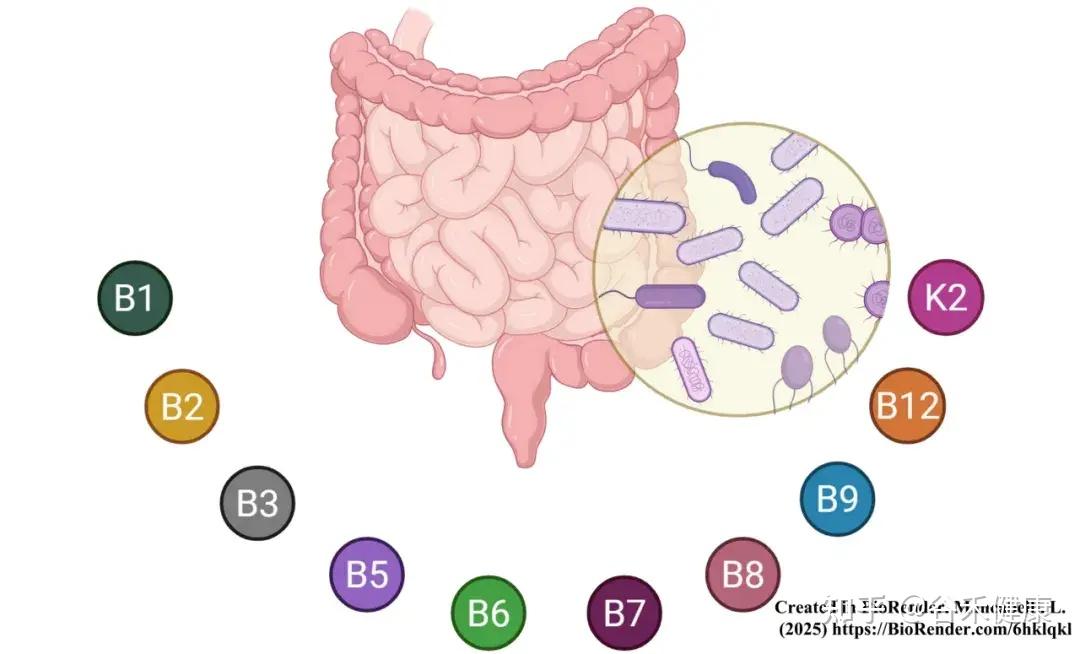
生物素则是一种重要的羧化/脱羧辅因子,在脂肪生成、碳水化合物和氨基酸代谢中发挥重要作用。钴胺素是B12辅酶家族(包括氰钴胺素、甲基钴胺素和腺苷钴胺素)的前体,对所有动物和许多(但并非所有)细菌物种都是必需的。
因此,人们越来越关注人体肠道菌群的合成能力,将其视为B族维生素和维生素K的潜在内源性来源,并逐渐认识到它对宿主维生素稳态的功能性贡献。
这种观念的转变得益于肠道菌群检测方法学的改进和多样化,使得谷禾能够以更高更大规模的分辨率检测和功能性地解释微生物维生素的生物合成。
本文主要关注和讨论目前对人体肠道相关细菌合成B族维生素和维生素K的理解。重点关注用于研究肠道菌群产生维生素潜力的各种方法,包括基因组学、宏基因组学等,以更好地了解微生物与宿主之间的维生素相互作用,从而改善人类健康。
人类肠道菌群是一个庞大且代谢活跃的微生物生态系统,包含丰富的基因功能库。随着新物种和功能的不断发现,我们对它的认识也在不断加深。
哪些证据说明肠道菌群合成维生素
(1)无菌大鼠 vs. 普通大鼠的对比实验
为了证明细菌生产的维生素,我们以一个关于维生素K的经典例子:
实验对象A(无菌大鼠): 肠道里没有任何细菌。
给它们吃不含维生素K的食物,它们就会生病(凝血酶原水平降低,容易出血),因为它们既没从食物中吃到维生素K,肠道里也没有细菌帮忙制造。
实验对象B(普通大鼠): 肠道里有正常的细菌。
结果: 即使吃完全一样的不含维生素K的食物,它们却很健康,凝血功能正常。
结论: 这说明普通大鼠肠道里的细菌“生产”了足够的维生素K,弥补了食物的缺失。进一步推测细菌的产量足甚至以满足生理需求。
因此以上实验表明细菌不仅仅是“产生”了维生素,而且产生的量很大,可能足够达到维持宿主健康(如正常凝血)的生理标准。
(2)人体研究
同样,在人类研究中,即使低维生素K饮食持续3-4周,也不会导致明显的维生素缺乏,除非同时使用广谱抗生素抑制肠道菌群。在这种情况下,会观察到血浆凝血酶原的明显下降,这清楚地表明了肠道菌群在维持维生素K稳态方面的重要作用。
(3)体外研究
一些计算机模拟研究也探讨了肠道菌群对宿主相关微量营养素摄入的潜在贡献,特别是关于B族维生素的膳食参考摄入量(DRI)。这些计算方法发现了能够产生多种B族维生素的微生物生物合成途径,表明肠道菌群可能对宿主的微量营养素摄入产生影响。
具体来说,尽管这仍然是推测性的,但有目前研究普遍认为肠道微生物合成的维生素B3、B6、B9和B12可能占人类相应DRI的约27%-86%。
(4)结肠转运系统
此外,在人类结肠中检测到B族维生素转运系统,进一步加强了微生物和膳食维生素可能协同作用,维持全身维生素稳态的观点。然而,由于微生物产生的维生素在结肠环境中的实际生物利用度仍未得到充分表征,需要进一步明确的研究。
!
注意局部和全身
与小肠中吸收效率明确的膳食维生素不同,微生物合成的维生素吸收取决于其化学形式和宿主转运蛋白的能力。例如,结肠中存在大量微生物合成的类咕啉素(包括维生素B12的变体),其结构常与具有生物活性的钴胺素不同,导致人类宿主的生物利用度降低。
此外,微生物合成的B族维生素主要满足自身的代谢需求,因此,与膳食来源相比,剩余部分(无论是主动分泌还是通过细菌细胞自发裂解释放)所占比例可能较小。
同时,微生物产生的维生素在塑造局部肠道环境(特别是在肠腔内和黏膜界面)中具有重要的生物学意义。已知这些维生素可以支持结肠上皮细胞的代谢需求,影响屏障完整性,调节局部免疫反应,并发挥抗氧化和保护作用。
谷禾数据库整理了大样本人群微生物基因组参考集合的基因组学研究,初步细致地刻画了肠道微生物群中各个成员合成维生素的潜力。
▸ 谁能自己造维生素?
把这些信息汇总到“门”(phylum)这一分类层级时,拟杆菌门(Bacteroidota)和假单胞菌门(Pseudomonadota)中“维生素原养型”(prototrophic,能从头合成某种化合物)的物种比例更高。
这两个门里包含能够合成几乎所有B族维生素的物种,但维生素B12(钴胺素)是个例外:大约只有30%的拟杆菌门基因组和约42%的变形菌门基因组显示出合成B12的潜力。
• 拟杆菌属和大肠杆菌是重要的B族维生素生产者
Bacteroides(拟杆菌属): 一些拟杆菌属的成员可以合成全部八种B族维生素。
E.coli(大肠杆菌), Phocaeicola dorei, Bifidobacterium(双歧杆菌属), Segatella copri(以前叫Prevotella copri): 这些也是肠道中B族维生素的重要生产者。
• 放线菌和芽孢杆菌中能产生维生素的较少
相对地,放线菌门(Actinomycetota)和芽孢杆菌门(Bacillota)中维生素“营养缺陷型/需外源型”(依赖外部来源满足维生素需求)的成员比例更高。
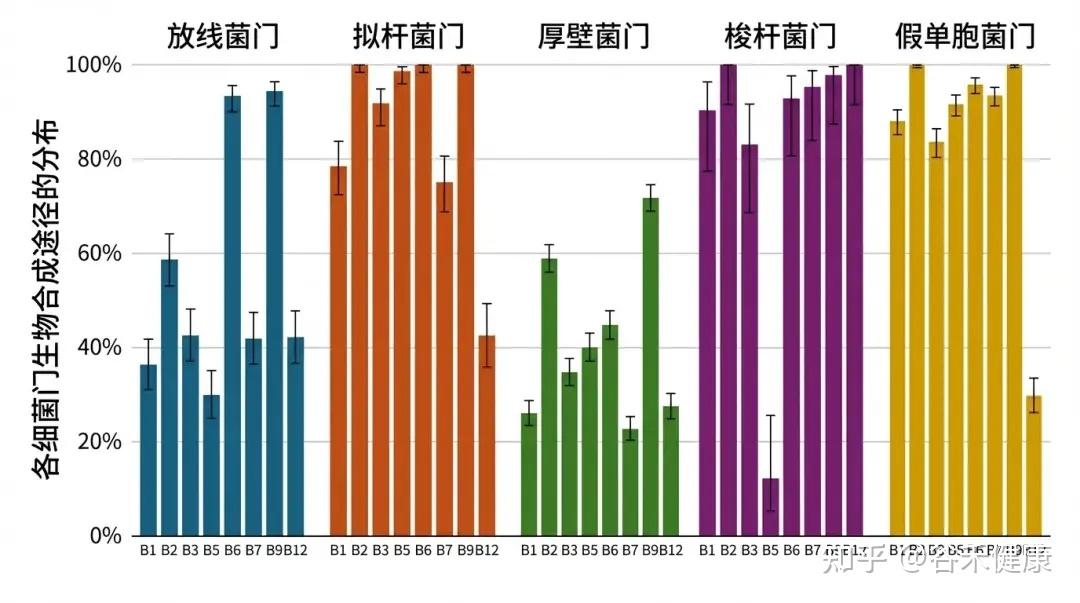
注:研究数量有限,仅供参考
▸ 肠型可能影响维生素的合成能力
• 不同肠型擅长合成的维生素不同(仅供参考)
肠道菌群的组成(肠型)会影响维生素的合成能力。
肠型1(以Bacteroides为主): 更擅长合成生物素、核黄素和泛酸。
肠型2(以Prevotella copri为主): 更擅长合成硫胺素和叶酸。
• 谁是维生素“消费者”?
Faecalibacterium prausnitzii(普拉梭菌): 它会消耗多种B族维生素,尤其是核黄素,核黄素能帮助它在肠道内抵抗氧化。
尽管基因组研究已阐明单个菌株或基因组的生物合成能力,现有方法仍较少将这一视角扩展至整个微生物群落,因此尚难在人群层面绘制维生素生物合成潜力图谱。
现有研究和谷禾的研发表明,特定微生物类群通过其生物合成能力模式和维生素利用策略,在塑造肠道维生素“生态”中起着关键作用。深入理解不同类群对维生素代谢的特异性贡献对于阐明微生物群与宿主营养状态的功能性相互作用至关重要,这些认识还可能指导基于微生物组的干预策略设计,以优化维生素可获得性并解决营养缺乏问题。
在此背景下,比如用能分泌叶酸的乳酸菌做发酵剂,生产叶酸强化乳制品;也在筛选肠道来源的产叶酸菌作为潜在益生菌,用于改善维生素供应。这些例子说明:关于微生物维生素代谢的知识可以转化为食品和治疗策略,最终为利用肠道微生物群有益代谢潜力的营养学方法铺平道路。
尽管人们越来越认识到人类肠道菌群具备合成维生素的能力,但最近的研究强调,肠道菌群中维生素的可用性并非仅仅由生物合成能力决定,还受到微生物群落内部以及宿主处复杂的代谢相互作用的影响。
这些动态过程包括微生物之间的交叉代谢、维生素摄取的竞争以及宿主介导的吸收,所有这些因素共同塑造了最终的营养产出。
▸ 微生物—微生物互作
在微生物层面,维生素的交叉喂养代谢源于不同分类群之间生物合成能力的不均匀分布,且这种差异不仅存在于亲缘关系较远的类群间,也体现在同一属内的不同物种之间。
最近的研究中,研究人员利用 SEED 平台分析了一个包含2000多个经过人工校对的人类肠道细菌基因组的参考数据集。该平台整合了所有已知和推断的生物合成及补救途径组件,包括酶和转运蛋白。这一方法通过预测不同分类等级中原养型(可自身合成)和营养缺陷型(需外部获取)菌株的比例,实现了对维生素生物合成潜力变异性的定量评估。这些跨越不同维生素和分类级别的保守性与变异性模式,为微生物群落中的潜在代谢相互作用奠定了基础。
• 原养型微生物的产物可能支持缺陷型的生长
当原养型和营养缺陷型微生物共存于同一生态位时,这种差异可能导致代谢相互依赖,即原养型微生物释放的代谢产物或中间体能够支持营养缺陷型微生物的生长和代谢功能。
一个明显的例子是维生素B12,这一结构复杂且代谢成本高昂的辅助因子。由于完整生物合成途径需高昂的代谢成本和遗传负担,只有少数厌氧肠道细菌(主要属于厚壁菌门和放线菌门)能够自主生产维生素B12。
然而,这种辅助因子对许多群落成员至关重要,众多成员已进化出高亲和力的转运系统以从环境中获取。例如,Anaerobutyricum hallii(以前称E.hallii)是一种特征明确的丁酸产生菌,能够合成维生素B12,而嗜黏蛋白阿克曼氏菌(Akkermansia)则通过共生利用该维生素进行丙酸的合成。
同样,在肠道微生物群落模型中,缺乏叶酸合成能力的Roseburia intestinalis M50/1生长受到经体外验证的叶酸原养型菌株双歧杆菌Bifidobacterium bifidum CNCM I-3650的益处。这些发现表明,原养型细菌可以通过主动交换代谢物和微量营养素,或通过被动机制(如细胞裂解并释放胞内维生素),来缓解共存菌株的营养缺陷。
这些以维生素为媒介的相互作用不仅是合作性的,竞争也在塑造人类肠道维生素可用性方面起着关键作用。
维生素的可用性受微生物间相互作用等的影响
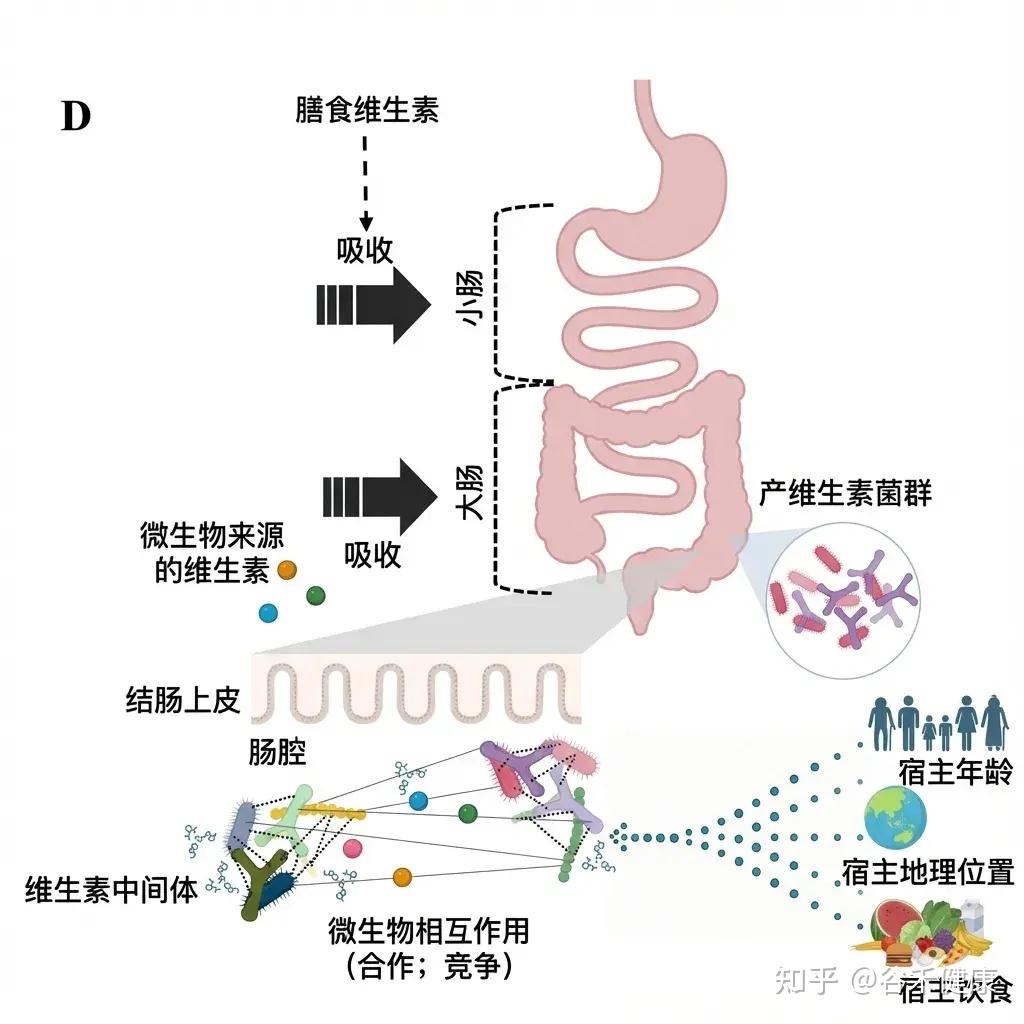
• 肠道微生物群会竞争有限的维生素资源
微生物通过产生维生素结合蛋白和利用特定转运蛋白,与其他群落成员竞争有限的维生素资源。因此,肠道环境可视为一个严格调控的微量营养素市场,其可用性由生物合成能力和摄取效率共同决定。
例如,多形拟杆菌(Bacteroides thetaiotaomicron)表达三种功能性、同源的维生素B12转运系统以获取钴胺素。这些转运蛋白对不同类咕啉类似物表现出独特的特异性,这可能为其在竞争激烈的人类肠道环境中提供选择性优势,同时也体现了肠道微生物为确保获得必需辅助因子所采用的复杂策略和巨大努力。
▸ 微生物—人体互作
宿主与微生物之间的相互作用使这一网络愈加复杂。实际上,宿主通过免疫调节、结肠吸收、粘液分泌和饮食等方式,间接影响维生素的动态变化,从而影响微生物的组成和功能。
• 不同年龄的肠道菌群组成影响维生素合成能力
最近的研究显示,年龄和地理起源等宿主因素显著影响人类肠道菌群的维生素生物合成潜力。具体而言,肠道菌群的测序分析表明,婴儿期维生素B9(叶酸)和K2的生物合成途径非常普遍,这与能合成叶酸的双歧杆菌属(Bifidobacterium spp.)的主导地位以及能够产生维生素K2的大肠杆菌(E.coli)的存在相一致,这些都是早期肠道微生物组的特征。
另一项研究将人群按年龄分组(从几个月大到80岁以上),并将微生物特征与年龄进行关联分析。结果显示,新生儿组(0-4岁)的微生物与参与硫胺素(维生素B1)和烟酸(维生素B3)生物合成的酶呈正相关,表明这些维生素路径与生命早期相关,这与老年组形成对比。
成人的肠道菌群则富含Bacteroides和Prevotella copri,展现出更广泛的代谢能力,因为这些类群通常是维生素B的原养型生物,具备多种B族维生素的完整生物合成途径。
• 不同地区的饮食和生活方式导致维生素合成能力差异
此外,新兴证据表明,地理起源导致全球不同人群肠道菌群在B族维生素生物合成能力上的差异,可能与饮食和生活方式相关因素影响肠道菌群的组成和功能有关。
在这个复杂的生态和代谢网络中,结合本地大数据库明确哪些微生物类群在结肠中参与维生素生物合成、这些维生素是否被宿主吸收以及哪些因素影响其产生,对于理解微生物群对宿主的营养贡献至关重要。这也有助于设计针对微生物群的策略(如益生元、益生菌或工程菌群),以改善维生素状况。
1
维生素B1(硫胺素)
▸ 生物学功能与重要性
硫胺素(维生素B1)是一种水溶性维生素,作为辅酶参与多个核心代谢过程。在活性磷酸化形式TPP(焦磷酸硫胺素)下,维生素B1是碳水化合物代谢中多种酶的关键辅因子,包括丙酮酸脱氢酶、α-酮戊二酸脱氢酶和转酮醇酶,这些酶是三羧酸循环(TCA)和磷酸戊糖途径的关键参与者。
通过这些途径,硫胺素有助于能量生成、氧化还原平衡、核苷酸和脂质的生物合成,并间接参与神经递质的代谢。
▸ 微生物合成与已知微生物贡献者
细菌中的硫胺素生物合成遵循模块化途径,涉及两个部分的分别合成:4-氨基-5-羟甲基-2-甲基嘧啶磷酸(HMP-P)和4-甲基-5-(β-羟乙基)噻唑磷酸 (THZ-P)。随后,这些部分由 ThiE 酶偶联生成磷酸硫胺素,最后转化为活性的 TPP 形式。基因组调查显示,相当大比例的肠道相关细菌基因组,包括拟杆菌属(Bacteroides)、普雷沃氏菌属(Prevotella)、肠球菌属(Enterococcus)和双歧杆菌属(Bifidobacterium)等主要菌属的代表,都拥有从头合成磷酸硫胺素所需的完整基因库。
然而,许多肠道共生菌对硫胺素是营养缺陷型的,依赖于外源性硫胺素或诸如 HMP-P 之类的中间体,这些物质可以从环境中回收或在微生物之间交换。在这些维生素B1营养缺陷型细菌中,已鉴定出不同的补救策略:有些菌株可以回收两种硫胺素中间体,而另一些则缺乏其中一条生物合成支路,依赖外部来源提供相应的缺失化合物,还有一部分完全依赖于摄取预先形成的硫胺素。
值得注意的是,所有营养缺陷型变体都保留了一定的补救能力,反映了对不完整生物合成潜力的多样化进化适应。
有趣的是,普拉梭菌(F. prausnitzii)在将丙酮酸转化为乙酰辅酶A的代谢过程中需要这种维生素,这是丁酸生物合成途径的一部分,突显了维生素B1在肠道菌群成员生产短链脂肪酸(SCFA)中的关键作用。
// 小结
总结来说,维生素B1(硫胺素)不仅是你身体的“能量充电宝”,也是将食物(碳水化合物)转化为能量所必需的“点火器”。没有它,我们的细胞无法有效产生能量,也无法制造DNA和脂肪,甚至会影响神经系统的信号传递。
肠道里住着很多细菌,其中一些是“生产者”。比如拟杆菌和双歧杆菌等常见菌,可以自己从头制造维生素B1,不仅满足自己,可能还能分给别人。还有很多细菌是“消费者”(营养缺陷型),它们自己造不出维生素B1,或者只能造出一半。
有的细菌完全依赖别人,必须从环境里捡现成的维生素 B1 吃。有的细菌只能造一半零件,需要从邻居那里借另一半零件来组装。说明为了生存,细菌逐步进化出复杂的“借用”和“互助”关系。
2
维生素B2(核黄素)
▸ 生物学功能与重要性
核黄素(维生素B2)是两种主要黄素辅酶——黄素单核苷酸(FMN)和黄素腺嘌呤二核苷酸(FAD)的前体。这两种辅酶在核心代谢中广泛参与氧化还原反应,包括电子传递链、脂肪酸氧化、氨基酸分解代谢,以及维生素B6和B9的激活反应。
此外,核黄素通过支持谷胱甘肽还原酶的活性和调节氧化应激,有助于维持抗氧化防御系统。
▸ 微生物合成与已知贡献者
细菌中的核黄素生物合成涉及一组保守基因(ribA、ribB、ribC、ribD和ribE),这些基因将三磷酸鸟苷(GTP)和5-磷酸核酮糖转化为核黄素,随后核黄素被磷酸化为FMN,再腺苷酸化为FAD。
最近的基因组重构研究显示,完整的操纵子在肠道共生细菌中广泛存在,包括拟杆菌属(Bacteroides)、肠球菌属(Enterococcus)和多种乳杆菌。尤为重要的是,乳酸乳球菌(Lactococcus lactis)、植物乳杆菌(Lactiplantibacillus plantarum)和罗伊氏乳杆菌(Limosilactobacillus reuteri)已被提议作为天然生物强化剂,用于提高酸奶、奶酪和发酵乳制品中的核黄素浓度。
然而,基因组分析显示,芽孢杆菌门(Bacillota)和放线菌门(Actinomycetota)中的一些成员缺乏完整的核黄素生物合成操纵子。有趣的是,许多这些核黄素缺乏型细菌编码高亲和力的核黄素转运系统,如RibU(ECF家族)和RibXY(ABC超家族),显示出它们对核黄素衍生辅助因子(如FMN和FAD)的强烈需求。
除了营养作用外,微生物生成的核黄素还参与调节宿主粘膜表面的免疫反应,其关键机制涉及粘膜相关恒定T(MAIT)细胞,这些细胞在识别由非多态性MHC I类相关分子MR1呈递的核黄素生物合成中间体后被激活。该途径建立了微生物维生素B2代谢与粘膜免疫监视之间的直接联系,强调了微生物核黄素生成在宿主-微生物相互作用中的更广泛生理相关性。
// 小结
维生素B2(核黄素)不仅是身体必需的营养素,也是肠道细菌与免疫系统之间的沟通桥梁。进入体内后,维生素B2转化为两种“超级助手”(FMN和FAD),参与无数化学反应,尤其是在将脂肪和蛋白质转化为能量,以及协助产生其他维生素(如B6和叶酸)。它还能帮助抵抗“氧化压力”,保护身体免受损伤。
肠道细菌也能合成B2,许多细菌(如某些拟杆菌和乳酸菌)具备特定基因,能够自己生产维生素B2。然而,并非所有细菌都能合成B2;一些细菌缺乏这种能力,但进化出强大的“抢夺”能力(高亲和力转运系统),能够从环境中获取B2以维持生存,显示出B2对细菌生存的重要性。
意外发现是,细菌制造B2的过程中还会产生一些中间产物,这些产物能够激活免疫系统中的特殊巡逻兵“MAIT细胞”。当MAIT细胞探测到细菌生成B2时,这些细胞会被激活,像是细菌发送的信号,通知免疫系统“我在这里”。这不仅帮助身体识别细菌,还启动免疫监视,保护肠道粘膜的健康。
3
维生素B3(烟酸)
▸ 生物学功能与重要性
维生素B3,即烟酸,包含两种生物活性形式:烟酸(NA)和烟酰胺(NAM)。这两者都是烟酰胺腺嘌呤二核苷酸(NAD+)和烟酰胺腺嘌呤二核苷酸磷酸(NADP+)的前体。这两种二核苷酸是参与氧化还原反应、能量代谢、DNA 修复和细胞信号传导的重要辅助因子。
NAD+是糖酵解、三羧酸循环和氧化磷酸化中的关键辅酶,而NADP+对合成代谢途径和抗氧化防御至关重要。
▸ 微生物生物合成与已知贡献者
与大多数其他B族维生素不同,烟酸可以在体内(主要在肝脏)内源性合成,通过犬尿氨酸途径将色氨酸转化为喹啉酸,后者是NAD+生物合成的关键中间体。然而,这一途径通常不足以满足日常需求,因此饮食摄入或微生物的贡献显得尤为重要。
在肠道中,许多共生细菌拥有一条替代的烟酸从头合成途径,从天冬氨酸开始生成喹啉酸,再转化为烟酸单核苷酸,最终合成NAD+。这一生物合成途径广泛分布于假单胞菌门(Pseudomonadota)和拟杆菌门(Bacteroidota)中,尽管在属和种的水平上存在显著差异。大肠杆菌(E. coli)、脆弱拟杆菌(B.fragilis)和多种双歧杆菌(Bifidobacterium)被确认是肠道环境中的烟酸生成者。
然而,并非所有菌种都编码完整的生物合成基因,许多细菌依赖烟酸补救途径,利用宿主或微生物衍生的前体。最新证据强调了宿主与其肠道菌群之间维生素B3前体的双向交换,支持共享的NAD+代谢。
尽管饮食中的烟酸前体大多在上消化道/小肠中被吸收,但结肠中的微生物可以利用可发酵纤维(如菊粉)或宿主细胞代谢释放的NAM来合成NAD+。微生物来源的NAD+随后转化为NAM和NA,这些物质能够被宿主肠道组织吸收,并在宿主细胞内用于再生NAD+。这突显了一个动态循环,即微生物活动与宿主来源底物共同维持循环中NA水平,即使在缺乏直接饮食摄入的情况下。
// 小结
维生素B3是身体细胞正常运转不可或缺的成分。你可以把它想象成制造“电池”的原料。它主要有两种形式(烟酸和烟酰胺),在体内会变成两种超级重要的物质(NAD+和NADP+)。这两种物质负责帮我们把食物变成能量、修复受损的 DNA,以及保护细胞不“生锈”(抗氧化)。我们的肝脏可以利用蛋白质中的某种成分(色氨酸)自己合成一点维生素 B3,但这通常不够用,大部分需要通过食物摄入。如果你吃不够,你肚子里的细菌能帮大忙。
在你肠道里的许多细菌(比如大肠杆菌和双歧杆菌)拥有特殊的“生产线”,可以用完全不同的原料(天冬氨酸)制造出维生素 B3。
细菌还可以利用我们吃下去的膳食纤维(比如菊粉)或者我们身体代谢剩下的废料,制造出 NAD+。然后它们把这些成品分解成我们可以吸收的形式(烟酸或烟酰胺),这就相当于细菌在结肠里给我们“供货”。我们身体产生的某些前体物质也能被细菌利用。
4
维生素B5(泛酸)
▸ 生物学功能与重要性
维生素B5(泛酸)是辅酶A(CoA)的生化前体,辅酶A作为核心代谢辅助因子,参与广泛的生物过程,包括三羧酸循环(TCA)、脂肪酸合成与β-氧化以及氨基酸代谢,主要在能量生成和合成代谢中充当酰基载体。此外,胆固醇、类固醇激素、血红素和乙酰胆碱的生物合成也需要辅酶A。
维生素B5对于维持细胞能量平衡和代谢功能至关重要。
▸ 微生物生物合成与已知贡献者
细菌可以通过一条从头合成途径来制造泛酸,该途径将泛解酸与β-丙氨酸结合在一起。这条途径由一组高度保守的基因簇(panB、panC、panD、panE)编码,合成出的维生素随后会被转化为辅酶 A。
在人类肠道菌群中,基因组分析显示,几乎所有的假单胞菌门(Pseudomonadota)和拟杆菌门(Bacteroidota)都具备合成泛酸的遗传能力,而受评估的厚壁菌门(Bacillota)和放线菌门(Actinomycetota)成员中,只有不到一半编码了完整的泛酸合成途径。不过,许多无法自身合成泛酸的菌株(营养缺陷型)仍然保留了将外源性泛酸转化为辅酶A的酶机制,这使它们即使无法从头合成,也能完成基本的代谢功能。
正因为假单胞菌门和拟杆菌门普遍具备这种合成能力,所以在成年人的肠道菌群中,尤其是典型的以拟杆菌为主的肠型中,维生素B5的生物合成途径是最常见的代谢途径之一。
// 小结
维生素 B5 之所以重要,是因为它是我们体内一种叫“辅酶 A”的关键物质的原材料。你可以把辅酶 A 想象成细胞工厂里的“搬运工”和“点火器”。 它是身体燃烧脂肪、分解蛋白质和碳水化合物产生能量的过程中不可或缺的一环。身体制造胆固醇、性激素、血红蛋白(血液中携带氧气的成分)以及神经传导物质时,都离不开它。
5
维生素B6(吡哆醇)
▸ 生物学功能与重要性
维生素B6是由六种可相互转化的形式组成,包括吡哆醇(PN)、吡哆醛(PL)、吡哆胺(PM)及其磷酸化形式,其中5′-磷酸吡哆醛(PLP)是最活跃的形式。PLP作为辅酶参与了约140种酶促反应,使维生素B6成为功能多样且复杂的微量营养素之一。除了糖原磷酸化酶外,几乎所有依赖PLP的酶都涉及氨基化合物的生化过程,尤其是氨基酸代谢。
▸ 微生物生物合成与已知贡献者
在人类肠道中,PLP通过两条截然不同的途径合成:较长的依赖脱氧木酮糖5-磷酸(DXP)的途径,这在拟杆菌门(Bacteroidota)和假单胞菌门(Pseudomonadota)中普遍存在;以及较短的不依赖DXP的途径,这在放线菌门(Actinomycetota)中更为常见。
基因组分析表明,PLP的产生在肠道细菌中分布不均。尽管预测双歧杆菌属(Bifidobacterium)、拟杆菌属(Bacteroides)和普雷沃氏菌属(Prevotella)能合成PLP,但许多著名的肠道共生菌,如韦荣氏球菌(Veillonella spp.)、粪肠球菌(Enterococcus faecalis)、普拉梭菌(F.prausnitzii)和Roseburia inulinivorans则被认为是营养缺陷型(无法自行合成)。这些生物体通常携带补救途径的遗传特征,使它们能够将环境中的B6维生素形式转化为可用的PLP。
6
维生素B7(生物素)
▸ 生物学功能与重要性
生物素,也被称为维生素B7,是一种含硫的水溶性维生素。它作为羧化酶的辅酶,参与关键的代谢途径,包括脂肪酸代谢、氨基酸代谢、碳水化合物代谢、聚酮化合物生物合成以及尿素利用。生物素通过赖氨酸残基与靶酶共价结合,促进二氧化碳转移反应,这对维持能量稳态和脂质代谢至关重要。
▸ 微生物生物合成与已知贡献者
细菌中的生物素合成通过两条截然不同的途径进行。在大肠杆菌(Escherichia coli)中,生物素是通过一种改良的脂肪酸合成途径合成的,该途径依赖于bioC和bioH基因,这是一条规范的且在生物素合成的肠道细菌中广泛存在的路径。相比之下,枯草芽孢杆菌(Bacillus subtilis)则利用以bioW基因为中心的独立途径,这条依赖bioW的路线在Bacillota成员中更为常见。
根据最近的基因组调查,所有分析的放线菌门(Actinomycetota)基因组均缺乏从头合成生物素所需的必需基因,这种特征在厚壁菌门也很少见。与其营养缺陷特征相符,大多数放线菌门细菌编码高亲和力的生物素摄取系统,强调了它们对环境生物素来源的依赖,并表明存在选择性机制以确保高效清除这种必需辅因子。
7
维生素B9(叶酸)
▸ 生物学功能与重要性
维生素B9包括一类与四氢叶酸(THF)结构相关的水溶性化合物,THF在“一碳代谢”中作为辅酶发挥作用。THF及其衍生物对于许多反应中转移一碳单位至关重要,这些反应涉及嘌呤、胸苷酸和多种氨基酸的合成,以及核苷酸的合成、DNA的复制、修复和甲基化。
因此,叶酸在核苷酸合成、表观遗传调控和细胞分裂中发挥关键作用,尤其在妊娠和婴儿期等快速生长阶段更为重要。
▸ 微生物生物合成与已知贡献者
细菌从头合成叶酸需要先生成并缩合两种关键中间体:二氢蝶呤三磷酸(DHPPP)和对氨基苯甲酸(pABA)。这些前体通过不同的代谢途径生成,最终汇合形成二氢叶酸(DHF),随后被还原为具有生物活性的四氢叶酸(THF)。
在肠道微生物中,适应婴儿肠道的物种(如B.bifidum、B.longum和B.breve)被认为是重要的叶酸生产者。这些菌株已能在培养基中积累叶酸,表明它们可能为婴儿肠道的叶酸库作出贡献。然而,双歧杆菌通常需要外源补充pABA才能合成叶酸,表明它们在该前体上存在一定程度的“营养缺陷”。
乳杆菌也是能产叶酸的细菌。例如,德氏乳杆菌(Lactobacillus delbrueckii)、瑞士乳杆菌(Lactobacillus helveticus)、罗伊氏乳杆菌(Limosilactobacillus reuteri)、清酒乳杆菌(Latilactobacillus sakei)和植物乳杆菌(Lactiplantibacillus plantarum)的菌株在pABA存在的生长环境中能合成叶酸。
值得注意的是,植物乳杆菌表现出无需补充pABA就能稳定产生叶酸的能力。许多研究评估了肠道微生物对动物宿主叶酸摄入的贡献,已证实肠道细菌合成的叶酸可以在结肠被吸收并被宿主利用。基于这一发现,研究人员探讨了来自各种发酵食品的乳杆菌作为发酵剂生产叶酸强化乳制品,同时也探索将人体肠道来源的乳杆菌作为产叶酸益生菌。
8
维生素B12(钴胺素)
▸ 生物学功能与重要性
维生素B12(钴胺素)是一种含钴的辅因子,也是自然界中体积最大、结构最复杂的非聚合生物分子之一。在人体内,它作为辅酶参与两项关键的线粒体反应:
1)甲基丙二酰辅酶A变位酶(methylmalonyl‑CoA mutase):参与丙酸代谢与能量产生;
2)蛋氨酸合成酶(methionine synthase):在叶酸循环中把同型半胱氨酸再生为蛋氨酸,起关键作用。
在B12的多种形式中,氰钴胺(cyanocobalamin)化学性质最稳定;它被吸收后会经过酶促加工,转化为具有活性的辅因子形式:甲基钴胺(Me‑Cbl)和腺苷钴胺(Ado‑Cbl)。
▸ 微生物生物合成与已知贡献者
钴胺素的生物合成过程非常复杂且耗能,涉及30多个基因,可走需氧或厌氧两条途径,最终都汇合到咕啉环(corrin ring)形成以及钴离子螯合的步骤。
该合成能力主要局限于某些细菌类群,许多肠道微生物并不具备。大规模基因组分析显示,约60%–80%的人类肠道共生菌缺乏合成B12所需的完整基因组合,例如双歧杆菌属(Bifidobacterium)、普雷沃菌属(Prevotella)以及Bacteroides thetaiotaomicron等。
相反,Propionibacterium freudenreichii、梭菌属(Clostridium)、脆弱拟杆菌(Bacteroides fragilis)、阿克曼菌(Akkermansia muciniphila)以及部分Bacillota和放线菌门(Actinomycetota)细菌,编码了完整或近乎完整的B12合成操纵子。
但由于合成B12非常耗能,很多细菌进化出了替代策略来获得这种辅因子及其类似物(咕啉类/钴胺素类似物,corrinoids):例如回收前体或直接从环境/其他菌那里摄取完整分子,从而在肠道微生物群中形成合作与资源共享。
不过在人类体内,结肠产生的B12吸收效率很低,因此微生物来源B12对全身B12水平的直接贡献仍不明确。
// 小结
B12对人体很关键:它参与能量代谢的一条关键反应,以及“叶酸循环/同型半胱氨酸→蛋氨酸”的关键步骤。补剂里常见的氰钴胺只是稳定形态:进入人体后会被转换成真正起作用的两种活性形式(甲基钴胺、腺苷钴胺)。
肠道菌并非都能制造B12:多数常见肠道共生菌缺乏完整“生产线”,只有部分细菌具备较完整的合成基因。很多细菌选择“省钱模式”:不自己合成,而是从环境或其他细菌那里“捡现成/回收零件”。未来还需要继续加强研究。
9
维生素K2(甲萘醌,Menaquinones)
▸ 生物学功能与重要性
维生素K主要有两种形式:一种是叶绿醌(K1),存在于绿色叶菜中;另一种是甲萘醌(K2),它是一类多样的异戊二烯衍生物,主要由细菌产生。甲萘醌(MK-n)的差异在于其异戊二烯侧链长度不同,这会影响其生物利用度和在组织中的分布。
在细菌细胞中,MK-n在细胞质膜的电子传递中起核心作用,支持形成芽孢的过程,并与致病菌的毒力有关。在人类中,维生素K2不足与骨质疏松、心血管疾病以及胰岛素敏感性下降有关。
▸ 微生物生物合成与已知贡献者
细菌中的甲萘醌合成途径涉及两个关键部分的汇合:萘醌环结构与异戊二烯侧链。这两部分首先通过不同代谢途径分别生成,随后再结合形成醌的核心结构。之后,异戊二烯侧链通过逐步添加异戊二烯单位而延长,产生侧链长度不同的甲萘醌变体。这种结构多样性带来功能上的分工,也可能有助于不同细菌适应环境。
目前,若干能产生甲萘醌的细菌菌株已作为工业食品发酵的发酵剂使用,并显示出可提高培养基中维生素K含量的能力。例如,将乳酸乳球菌(Lactococcus cremoris、L.lactis)和乳明串珠菌(Leuconostoc lactis)在复原奶粉或豆奶中培养时,可产生长链MK,并且水平几乎接近成人每日适宜摄入量。这些发现支持:发酵食品可以成为维生素K2的重要膳食来源。
// 小结
总结来说,维生素K有两大类:K1主要来自绿叶菜;K2主要由细菌合成。K2不是单一物质,而是一系列“MK-n”家族成员,侧链长短不同,会影响人体吸收与在体内去向。
对细菌来说,K2是能量代谢(电子传递链)的关键分子,还与芽孢形成和致病性相关。对人来说,K2不足可能与骨质疏松、心血管问题以及胰岛素敏感性变差有关。有些用于发酵的乳酸菌在牛奶或豆奶里能产出一定量的K2,说明发酵食品可能是补充K2的一个重要来源。
▸ 分子测序方法
近一二十年来,高通量测序、多组学与计算建模的重大进展,极大提升了我们对人类肠道微生物组的理解,包括其参与维生素生物合成的作用。
通过测序的基因组与生物信息学绘制“群落层面”的维生素合成潜力。
分享一些有趣关联:高纤维饮食人群的肠道微生物组更富集叶酸与核黄素“潜在生产者”,而西式饮食与更高的“维生素合成缺失”(更依赖外源获取)相关。不同年龄也有差异:成人整体合成潜力更强;婴儿相对更富集维生素K2相关的甲萘醌与叶酸合成潜力。
在临床队列中,2型糖尿病患者的维生素K2及多种B族维生素合成基因丰度下降,提示微生物维生素代谢可能与健康状态相关。
有研究发现炎症性肠病(IBD)患者的钴胺素(维生素B12)与硫胺素(维生素B1)合成相关基因表达显著降低,说明疾病本身会影响微生物维生素代谢的“实际运行”。
!
不足之处
但需要强调的是:无论是16S rRNA 测序还是宏基因组测序,目前主要评估的是菌群的潜在功能能力”——其推断结果高度依赖检测机构所采用的整合菌群基因与代谢通路数据库。现有数据库仍可能存在注释不全或偏倚:替代通路、新发现酶及非模式菌的功能注释缺口更为常见,从而带来功能推断的不确定性。
现阶段,基因组学层面的“检出”并不能直接证明相关基因已在体内表达、对应酶活性确实发生并产生了可观的生理效应;这类问题往往需要结合转录组、蛋白组(必要时包括代谢组)等证据链来支撑。
因而,更理想的路径是将菌群组学结果与人群膳食摄入、临床指标与其他组学数据联合分析,才能更清晰地反映人体维生素“合成—转化—吸收—利用”的真实状态;这也与谷禾健康正在持续研发与构建的综合模型思路一致。
基于上述限制,在谨慎选择菌群检测机构的同时,也应对菌群评估报告中的营养相关指标保持审慎解读:建议结合受试者的症状体征、既往史与用药/补充剂使用、饮食结构及实验室检查结果,进行综合判断与个体化干预,避免将“功能预测”直接等同于“临床结论”。
总体而言,基因组与生物信息学为绘制肠道微生物合成、改造或获取维生素的遗传潜力提供了强有力的入口。它们无法单独给出真实产量与功能输出,但能指导饮食和了解评估肠道菌群代谢维生素的合成和利用潜力,并在群落尺度上建模预测代谢。
▸ 培养方法
测序等基于序列的方法极大拓展了我们对微生物代谢的认识,但要在实验上“坐实”某个微生物是否真的能合成维生素,培养(culture-based)方法仍然不可或缺。通过培养,我们可以直接测试代谢能力、分离出能产维生素的菌或需要外源维生素才能生长的菌(营养缺陷/依赖型),并在营养成分严格可控的条件下设计实验来研究微生物之间的相互作用。
在实验室条件下评估微生物维生素生物合成潜力的一种经典且至今仍广泛使用的方法,是利用“营养缺陷指示菌株”(auxotrophic indicator strains)。这类菌的生长依赖某一种特定B族维生素的外源供给,因此可以通过其生长反应来定量推断样品中该维生素的可用量。这种测定已大量用于研究乳酸菌和双歧杆菌产生叶酸、硫胺素(维生素B1)和钴胺素(维生素B12)的能力。
研究者也会用体外体系直接观察微生物之间的“维生素互喂/交叉供养”(cross-feeding):把一种需要维生素的菌和一种能合成维生素的菌配对共培养。比如在缺乏叶酸的培养基中,R. intestinalis 与 B. bifidum 共培养时能够生长,因为后者会被动向环境释放叶酸合成的中间产物。类似地,在没有外源硫胺素的情况下,Roseburia faecis 能维持对硫胺素依赖的F.prausnitzii 的生长,说明物种间确实发生了有效的营养共享。
为了更接近肠道微生物生态系统的复杂性并延长观察时间,更复杂的体外培养平台(如基于生物反应器的发酵系统)也被用于相关研究。这类封闭、动态系统可以精确调控环境参数(如pH、滞留时间、底物输入),并能长期维持稳定、可重复的“合成”微生物群落。尽管它们已被广泛用于模拟结肠条件下的微生物生态与代谢产物研究,但直接用于研究肠道菌B族维生素的合成与交换目前仍相对有限。不过,这些系统为未来在更接近生理条件的环境中捕捉微生物营养互作提供了很有前景的框架。
!
局限性
但培养方法也有先天局限:相当一部分肠道微生物在标准实验室条件下仍难以培养,限制了我们对一些关键群落成员维生素代谢能力的评估。尽管如此,培养仍是功能微生物学的基石,因为它能对维生素的合成、转运以及物种间交换进行直接的实验验证。
▸ 分析化学方法与代谢组学
代谢组学在阐明微生物代谢方面居于补充重要地位:它通过检测和定量代谢产物,直接提供“微生物实际产生了什么”的证据,因此适合用来研究宿主—微生物以及微生物—微生物之间的代谢互作,包括微量营养素(如维生素)的动态变化。
代谢组学方法大体分为靶向和非靶向两类。靶向代谢组学使用已知标准品,对特定代谢物进行精确定量,例如B族维生素及其衍生物/不同活性形式(vitamers)。
非靶向代谢组学则试图给出更全面的代谢物“全景图”,用于发现意料之外或新的代谢变化。
目前,对代谢物的检测、鉴定与定量能力依赖于分析化学平台的快速发展,包括:
-气相色谱(GC)
-液相色谱(LC)
-高/超高效液相色谱(HPLC、UPLC)。
其中,LC尤其是与质谱联用(LC‑MS)已成为最通用、应用最广的代谢组学工具之一。LC‑MS通常配合电喷雾电离(ESI),可以在食品、牛奶、药物制剂、婴儿配方奶粉、血液和人类粪便等复杂样品中,同时分析多种B族维生素和甲萘醌(维生素K2的一类化合物)。
LC‑MS的一种更高级形式是液相色谱‑串联质谱(LC‑MS/MS)。它通过两级连续的质量分析显著提高灵敏度和特异性,不仅能检测痕量化合物,还能区分结构非常相近的分子,比如同一种维生素的不同同分异构体/不同活性形式。例子是:近期已有研究用LC‑MS/MS实现了维生素B12三种活性形式的同时精准定量,体现了该技术在解析微量营养素“精细多样性”方面的价值。
!
注意
尽管这些方法已成功用于从复杂食物和生物样品中区分并定量多种B族维生素的活性形式,但它们用于研究人体肠道中“微生物自身合成维生素”的工作仍相对有限。不过,由于其高特异性和高灵敏度,这类技术很有潜力成为未来研究的重要工具。
▸ 临床前模型
要真正理解肠道菌群的维生素代谢是否真的有功能意义、能否影响维生素稳态,并为调控菌群来改善健康提供依据。因此,需要把“微生物活动”和“宿主反应”放在同一个整体里研究的宿主相关模型。
▸ 为什么常用鼠类模型?
研究肠道菌群与宿主互作最常用的是动物模型,尤其是小鼠和大鼠。它们的优势是环境和变量可控,能系统地观察“菌群—维生素—宿主”的因果关系。
例如:在叶酸缺乏的大鼠中,给予能大量产叶酸的双歧杆菌(如 Bifidobacterium adolescentis、B.pseudocatenulatum)可以帮助恢复叶酸水平。这类实验说明:微生物合成的维生素有可能在体内发挥营养补充作用。
此外,由于B族维生素与多种人类疾病相关,鼠类也常用来模拟疾病状态,以研究菌群维生素代谢对病理过程的影响。
▸ 维生素合成益生菌:对炎症性疾病的潜力
能合成B族维生素的益生菌在治疗炎症性疾病(尤其是肠道炎症)方面显示出潜力:产核黄素(维生素B2)的 Lactiplantibacillus plantarum CRL2130:在小鼠溃疡性结肠炎和肠黏膜炎模型中,可降低炎症细胞因子、改善症状。
产叶酸的 Streptococcus thermophilus(CRL808、CRL415):在化学诱导的小鼠模型中,可减轻肠黏膜炎症。
若联合使用“产叶酸 + 产核黄素”菌株:在乳腺癌化疗动物模型中,既能提高化疗效果,又能降低如黏膜炎等副作用。
▸ 对神经系统也可能有帮助(动物证据)
一些产B族维生素的菌株在神经保护方面也有动物实验支持:
在帕金森病小鼠模型中,口服产核黄素的L.plantarum CRL2130 或产硫胺素(维生素B1)的 L. plantarum CRL1905,可改善运动协调能力,并减少多巴胺能神经元损失。
这些效果与直接补充商业维生素相当,提示“微生物在体内合成的维生素”确实影响人体维生素的浓度和利用,并能在功能上支持宿主的营养与生理过程。
!
局限性
鼠类模型很有价值,但也要谨慎外推到人:小鼠/大鼠在人类的解剖结构、免疫系统、代谢方式上都有差异,所以动物结果未必等同于人体效果。即便如此,它们仍然是连接体外机制研究与人体真实生理的重要桥梁,用于在“整体生物体”层面验证假设。
人类肠道中微生物维生素生物合成形成的复杂网络,正在成为理解“微生物组—宿主”相互作用的核心内容之一。肠道微生物合成、共享并调节维生素可获得性的能力,并不只是简单的代谢功能,而是一条涉及生态协作、支持宿主以及进化适应的关键轴线。
在过去十年里,高通量测序、功能基因组学以及整合组学的发展显示:许多肠道细菌并不具备合成必需维生素的完整生物合成途径。相反,它们通过代谢交叉喂养形成合作网络,在群落成员之间交换代谢中间体或完整维生素。这些网络有力地说明:肠道微生物群并非一群彼此独立的个体,而更像一个协同运作的“代谢共同体”,并且与宿主健康紧密相关。
要在生态系统层面全面理解这种代谢,需要多学科的方法组合,用于绘制维生素合成的潜力,培养与实验验证能够提供因果证据与机制洞见;动物模型则用于在生理环境中验证微生物贡献。
只有整合这些方法,我们才能把握微生物维生素代谢的全部深度与细微差别。
首先,需要提升组学数据的分辨率与功能解读能力,尤其是针对非模式微生物类群以及非典型维生素变体。这要求建立更完整、更高质量的数据库,并使用更高分辨率,更大人群的组学技术来完善预测,揭示维生素代谢的调控与生态动态。
同样重要的是,将基础发现转化为可行干预手段的需求正在增长。通过使用具有明确维生素产能谱的益生菌、调整饮食以促进微生物合成,或开发个性化的微生物组营养治疗,用于营养支持、疾病预防与个体化健康管理。