-

 CNAS L23010
CNAS L23010

 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室

饮用水(DW)中含有多种微生物物种和化学特性。水构成了我们日常饮食的最大部分,既可单独饮用,也用于食物制备。
饮用水是我们主要的液体来源,对维持体内平衡至关重要,还可以提供必需矿物质。
有限的证据表明,饮用水在塑造肠道微生物群方面发挥作用,这意味着它可能影响人类健康。尽管饮用水对饮食有重要贡献,但在研究饮食对肠道微生物群的影响时,饮用水常被忽视。
本文探讨饮用水与肠道微生物群之间联系的理解——这是人类微生物组科学中研究不足的领域。
对饮用水与肠道微生物群关系的深入理解将有助于理解肠道微生物群结构及其与人类健康的关系。这可能导致针对失调状态的具体建议,优化饮用水处理过程,并为研究人员制定具有定制益生菌或化学特征的饮用水铺平道路。
干预措施可能包含或排除特定化学物质甚至益生菌,到优化例如氯离子含量,调节pH值,来促进健康的肠道微生物群。
人们普遍认为人类肠道微生物群对人类健康具有重要影响。饮食因素在决定肠道微生物组成中的作用已明确,各个因素都会影响原核生物的相对丰度和绝对丰度,以及它们的生长动态。
在对肠道微生物群的影响方面,一个被人们忽视的关键饮食因素是饮用水 (DW)。饮用水主要来自地下水和地表水,每种水源都有不同的物理和化学属性及相关微生物群。根据水质,源水通常会经过各种处理和消毒过程,以去除有害化学物质和微生物,生产出可饮用水。许多源水中的微生物能够耐受处理过程,处理后的饮用水中存在多样化的微生物群,细菌浓度约为106–108个细胞/升。
不同源水的化学和微生物学特性因源环境的生物地球化学而显著不同,这些因素与所应用的处理过程相结合,影响了最终的饮用水微生物群。
饮用水是我们饮食的核心组成部分。鉴于饮食模式和因素在塑造肠道微生物群结构中起关键作用。饮用水可以通过直接(即通过驻留的饮用水微生物群)或间接(即由于其化学成分)方式与肠道微生物群相互作用。然而,在研究饮食因素对肠道微生物群影响的队列研究中,饮用水很少被考虑。在少数考虑的几项研究中,发现其具有显著影响。
研究发现,饮用水的化学、物理和生物特性在塑造肠道微生物群结构和功能中起着关键作用,并对人类健康产生连锁效应。
在此,我们总结了从各个领域和不同设计的研究中获得的关于饮用水对肠道微生物群直接和间接影响的有限知识。
个体之间以及饮用水之间的高度变异性将使未来的研究工作复杂化,但这些挑战已经在肠道微生物群研究领域得到解决。
肠道对水的吸收发生在小肠和大肠中。渗透梯度驱动水的吸收,这通常与离子和营养物质的运输有关。在小肠中,葡萄糖和半乳糖转运蛋白 SGLT1 以及 Na+/H +反向转运蛋白负责从肠腔中运输大部分水。
水分吸收和体内平衡是大肠的主要功能之一,而大肠也是肠道微生物的主要聚集地。水分与离子(主要是钠)一起穿过肠上皮,钠离子的运输产生渗透梯度,推动水分吸收。
早在1998年,有一项研究就证实大肠中存在的水量通过改变肠道中的 pH 值和运输时间来影响肠道微生物组的组成。
饮用水中其他成分的吸收通过多种机制并在肠道的特定位置发生。复杂的有机化学物质可能被肠道微生物群发酵并转化为脂肪酸,或者在没有吸收或转化的情况下被排除。肠道中微生物细胞的最高丰度是在大肠中,近端大肠被认为是最高微生物增殖的地方,因为发酵底物的可用性很高。
我们预计饮用水中的化学物质和生物与肠道的相互作用方式与其他摄入的化学物质和生物类似。由于益生元和益生菌在这方面的研究相对较多,我们使用来自该领域的数据来描述潜在的相互作用。
然而值得注意的是,饮用水中的化学物质和生物可能并非都具有有益影响,区分对宿主产生有益和有害影响的相互作用是制定优化饮用水成分以支持公共健康的策略的关键。
A) 饮用水微生物群和肠道微生物群之间的潜在相互作用
B) 识别饮用水和肠道微生物群之间联系的建议方法
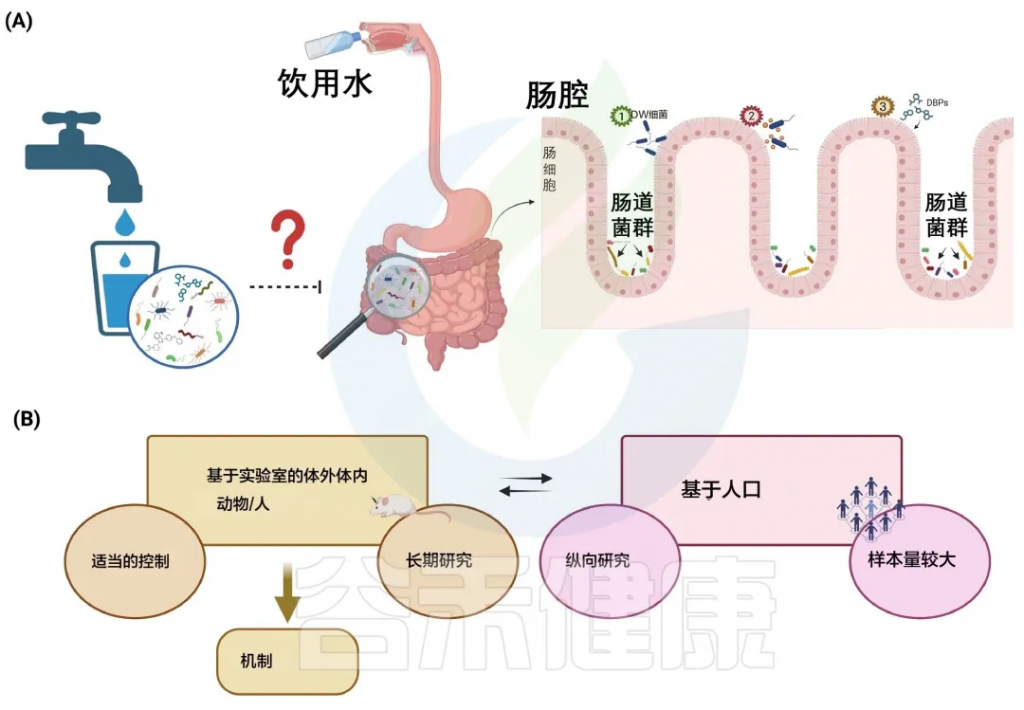
Moghaddam HS et al., Water Res. 2024
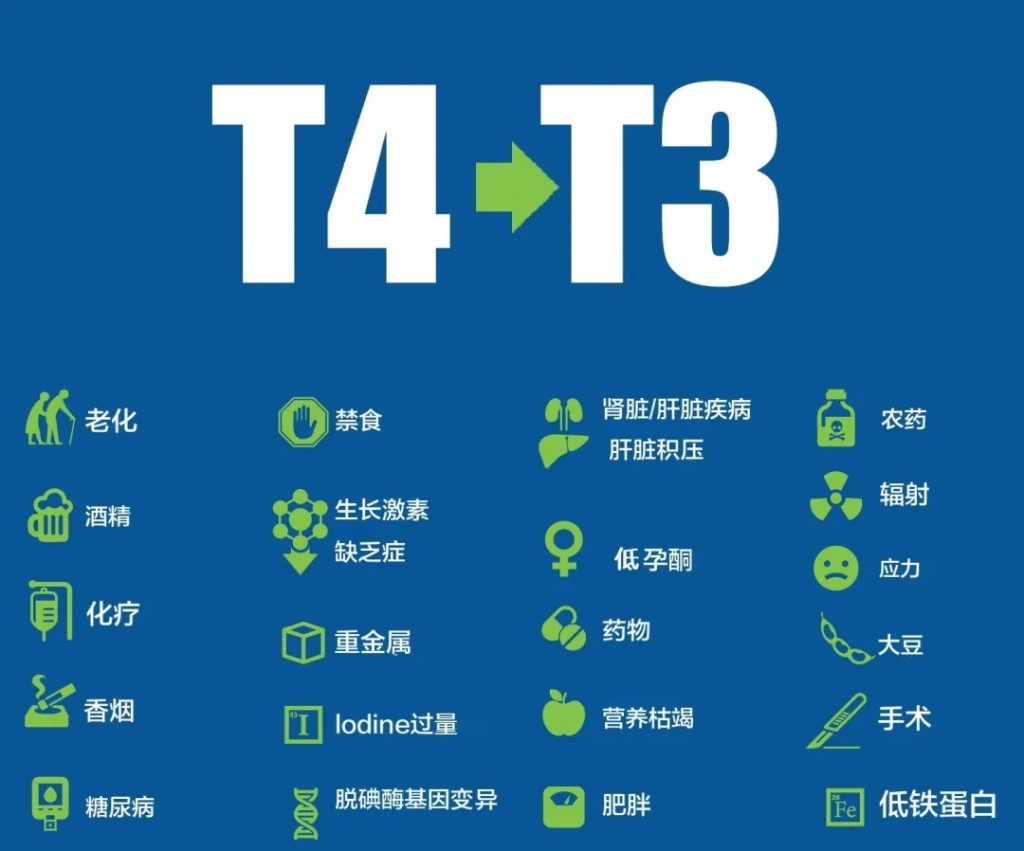
饮用水对肠道微生物群的影响途径:
饮食是塑造肠道微生物群落的主要驱动因素之一。成人每日建议饮水量约为2.0-3.7升。
两项研究使用不同的方法检查了肠道微生物群和饮用水微生物群之间的重叠。使用 16S rRNA 基因扩增子数据鉴定出 35 个属,它们属于 5 个门(假单胞菌门、放线菌门、芽孢杆菌门、拟杆菌门和蓝藻门),存在于经过处理的饮用水和肠道微生物群中。
来自 131 个饮用水样本宏基因组的组装重叠群,并将它们与来自同一国家的 196 个粪便样本的宏基因组进行了比较。通过关注饮用水中大量存在的属,他们确定了与肠道微生物群共享的 6 个关键属:
肠道菌群和肠道菌群重叠程度最大的是假单胞菌门(Pseudomonadota),两项研究都在肠道和饮用水群落中发现了鞘氨醇单胞菌属(Sphingomonas)、假单胞菌属(Pseudomonas)和伯克霍尔德菌属(Burkholderia)。
然而,当应用类似的宏基因组学方法来评估一个人的粪便菌群和饮用水菌群之间的直接联系时,他们只发现了一种常见的生物,即Curvibacter(弯钩菌),这表明,如果饮用水微生物能够在人体肠道中定植,那么存在一些重要的决定因素,这些因素是生物体固有的,也是肠道中生物和非生物条件的共同决定因素。
显然,饮用水和肠道之间存在共同的属并不意味着饮用水是这些微生物的来源。假单胞菌属、鞘氨醇单胞菌属等属广泛存在于各种自由生活和宿主相关环境中。
饮用水质量与肠道微生物群的长期影响
在考虑饮用水对肠道微生物群的长期影响时,也值得考虑劣质饮用水的影响,因为全球超过27%的人口无法获得安全管理的水源。

为了研究水资源不安全和肠道病原体暴露如何影响肠道微生物群的发展,Piperata等人2020年的研究分析了尼加拉瓜儿童的家庭饮用水和粪便样本。在水和粪便样本中观察到了高浓度的沙门氏菌(Salmonella)、弯曲杆菌属(Campylobacter)和大肠菌群(coliforms)。
沙门氏菌(Salmonella)是世界范围内肠道感染(食物中毒)的最常见原因之一。尤其在卫生条件差的地方。沙门氏菌是能动的生物,它们利用鞭毛将自身导向肠细胞,有时候也依赖入侵基因来穿透宿主肠细胞。入侵基因在宿主细胞中介导广泛的动作重排,导致细胞膜变形并使生物体能够入侵。
研究根据饮用水中的大肠菌群含量将家庭分为高含量和低含量两类(高含量为每毫升≥29个菌落形成单位)。虽然这项研究没有直接比较饮用水和粪便样本的微生物组成,但他们发现,饮用高大肠菌群饮用水家庭的儿童,其肠道微生物群的多样性低于饮用低大肠菌群家庭的儿童。
简单来说,肠道微生物群的多样性可能与抵抗肠道病原体的能力有关。也就是说,肠道内的微生物种类越丰富,可能越能抵抗有害细菌的侵袭。不过,某些特定微生物的存在似乎比整体的多样性更为重要。
这强调了确保全球安全和良好管理的饮用水供应的重要性。
除了建立饮用水和肠道微生物群之间的机制联系的重要性之外,抗菌素耐药性基因 (ARG) 从饮用水到肠道的传播也同样重要。饮用水中存在各种携带 ARG 的微生物和可能含有 ARG 的 eDNA。
Tips:这里的eDNA也可能是提取了环境中的微生物内的DNA,很难直接判断:即使没有活的微生物,ARG也可能通过环境DNA的形式存在于水中。
各种水处理工艺都可用于改变耐药基因的丰度。耐药基因从饮用水传播到肠道微生物群的速度尚未得到很好的研究。

2020的如上项目研究了上海和北京饮用水中耐药基因 blaNDM and mcr-1的丰度。他们随后证实,免疫缺陷 (BALB/c) 小鼠能够从饮用水中分离出的微生物中获得这些耐药基因,而这些微生物的浓度与饮用水中相当。这就引发了一个问题:免疫功能低下和/或健康的人是否也可能从饮用水中获得抗菌素耐药性微生物或基因。
饮水来源与肠道微生物群多样性
虽然饮用水和肠道中同一属的成员之间的重叠并不意味着存在直接联系,但两项人群水平的研究已将饮用水来源确定为解释肠道微生物组多样性变化的最重要因素之一。这两项研究都调查了分布在全球不同地区的多个人群,并使用了粪便微生物的16S rRNA基因测序数据。

2022年,Vanhaecke 等人调查了来自美国和英国 3413 个体肠道菌群组成的主要影响因素。微生物群、生活方式和饮食数据来自美国肠道微生物组计划。使用线性和逻辑模型分析了肠道菌群与不同饮用水来源 (瓶装、过滤、自来水和井水) 之间的关联。
饮用水来源是肠道菌群 α 和 β 多样性的关键因素,其效应大小与酒精摄入量和饮食类型相似。
参与者的地理位置多样,这可能导致饮用水的物理化学和生物参数存在较大差异。然而,研究发现市政自来水饮用者与其他类型饮用者之间,以及私人井水饮用者与瓶装水饮用者之间的肠道微生物群存在显著差异。
不同组之间观察到几种物种的丰度差异,包括瓶装水和市政自来水饮用者中链球菌属(Streptococcus)、拟杆菌、Odoribacter的丰度较高,而井水饮用者中Dorea属的丰度较高。
每日饮水量也影响了β多样性(组间差异性),每日饮水量少于1升的人群中弯曲杆菌属(Campylobacter)的丰度较高。
Dorea菌属于厚壁菌门毛螺菌科,广泛存在于人体肠道内,谷禾数据显示该菌在人群的检出率超89%。该菌最早也是从人体粪便中分离出来。
该菌是一类革兰氏阳性厌氧菌,主要存在于人类和动物的肠道中,可以利用多种底物进行发酵代谢,包括葡萄糖、果糖、乳糖和芳香族化合物等。它可能通过诱导Treg并抑制Th17细胞的分化和功能,从而调节肠道免疫反应,维持肠道黏膜屏障的完整性和稳定性。
在多发性硬化症、炎症性肠病患者,甚至结直肠癌、自闭症谱系障碍以及肥胖人群中的Dorea菌高丰度富集,被认为具有促炎作用。多数研究证实Dorea与体重指数 (BMI)、腰围和舒张压呈正相关。基线肠道内富含高丰度的Dorea菌的人群,在减重方面更困难。
然而Dorea菌在抑郁患者和患有食物过敏人群中减少,研究还表明Dorea菌可以预防或治疗过敏性鼻炎。

美国加利福尼亚州斯坦福市斯坦福大学人类微生物组研究中心团队2018年通过研究喜马拉雅山脉 4 个具有不同觅食程度和采用农耕方式时间的人群,研究了区分农耕和觅食生活方式的肠道微生物群的因素。这些人群分为觅食者、过渡到自给性农业的觅食社区和过去两个世纪完全过渡到农耕的人群。
从 56 个人采集了粪便样本。对应分析 (CA) 和微生物多样性分析表明,饮用水是与肠道微生物组成显着相关的两个因素之一。
各组之间没有观察到 α 多样性的显著差异,但几个β多样性指标表明与饮用水来源相关的组成变化。
在将这项研究扩展到坦桑尼亚的猎人采集者人群——哈扎人(Hadza)的季节性肠道微生物群数据时,研究发现饮用水对肠道微生物群组成也有类似的显著影响。这表明饮用水来源在不同生活方式的人群中对肠道微生物群有重要作用。

饮水中的化学成分与口腔及肠道微生物群
随后,英国研究团队于2020年开展的一项研究,他们调查了自来水中溶质浓度(钠、氯和硫酸盐)与肠道微生物群之间的关联。研究对象为英国的36对单卵双胞胎。结果表明,自来水中钠的日均摄入量(ADD)增加显著降低了肠道微生物群的丰富度,而微生物群的组成在硫酸盐和氯的日均摄入量的影响下有所不同。然而,这些关联较弱,并且研究中没有量化一些可能影响肠道微生物群的其他变量。
与饮用水和肠道微生物群之间的相互作用类似,关于饮用水对口腔微生物群影响的研究也很少。一项研究发现,在西班牙青少年的口腔微生物群中,自来水是所有调查因素中影响最大的。
饮用水的几个特征,包括碱度、硬度,以及氟化物和几种离子的存在,对特定菌属的丰度有显著影响。另一项研究也证实了饮用水中的氟化物对口腔微生物群的影响,该研究在小鼠中得出了类似的结果。除了饮用水对口腔微生物群的影响可能间接影响肠道微生物群外,饮水还可能将口腔中的微生物引入肠道。
总之,这些研究表明,饮用水属性对肠道菌群的组成有重要影响。有必要进一步研究这种关系是如何介导的,以及它如何影响人类健康。
关于饮用水(DW)与肠道微生物群关系的研究大多集中在饮用水的物理化学特性上,通常是在动物模型中进行,少数情况下涉及人类。需要注意的是,饮用水的物理化学性质,包括化学成分和处理过程,直接影响其微生物群,这反过来可能直接或间接影响肠道微生物群。一项荟萃分析表明,饮用水的化学性质在动物模型的微生物群研究中起到混杂作用,因此研究时需要注意。特别是,饮用水的 pH 值似乎会改变实验动物的肠道菌群。
■ 灭菌与否

2018年,一项实验动物研究了四种水源对小鼠肠道微生物群的影响:
他们观察到高压灭菌水和非高压灭菌水的肠道菌群存在差异,喂食高压灭菌水的小鼠中,属于芽孢杆菌属(Bacillota)和不动杆菌属(Acinetobacter)的操作分类单元 (OTU) 的丰度降低。
在喂食未高压灭菌水的小鼠中,没有观察到显着差异。这些差异的原因尚不清楚,并且该研究没有评估饮用水微生物群。
■ PH值
实验室研究中使用的动物通常被喂食酸化水,以防止动物护理设施中的微生物污染。酸化水导致 1 型糖尿病发病率增加,并导致小鼠肠道微生物群发生变化。
研究饮用水的pH值对健康成年男性血糖调节和肠道微生物群的影响,参与者在两个为期两周的周期内分别饮用中性(pH7)或碱性(pH9)水。研究未发现血糖调节或肠道微生物群的显著变化,尽管这种差异可能是由于所饮用水的pH值不同所致。
先前的人体研究表明,饮用酸性水与1型糖尿病之间存在关联,但尚未研究其对肠道微生物群的影响。
此外,已知肠道微生物群会影响葡萄糖稳态,而酸性pH值会不同程度地影响肠道常驻微生物的生长速率,这提示了可能的关联。从实际角度来看,市政供水中暴露于酸性饮用水是不常见的,因为饮用水通常以中性到微碱性的条件分配,以防止管道腐蚀。
■ 富含碳酸氢盐/硅酸盐
饮用富含碳酸氢盐的矿泉水或偏硅酸盐碱性矿泉水可能会通过改变肠道微生物群来影响宿主的生理机能(下表)。
饮用水微生物群和化学物质对肠道微生物群的影响

Moghaddam HS et al., Water Res. 2024

上面两篇研究分别在人类和猪仔和发现,饮用富含重碳酸盐的矿泉水或以偏硅酸为基础的碱性矿泉水可能通过改变肠道微生物群影响宿主生理。其中一些观察到的益处包括减少仔猪腹泻发生率,降低与胰岛素抵抗相关的某些标志物。
因此,值得进行样本量更大且具有适当对照组的综合研究将为矿泉水对肠道微生物群和宿主生理的影响提供更有力的证据。
■ 氯化物
自 20 世纪 50 年代以来,氟化物就被引入到饮用水和牙科用品中,如今大多数牙科产品和许多市政水源都已含氟。尽管氟化物使用范围很广,但很少有研究调查其对宿主相关微生物群落的影响。
最近一项关于水氟化对小鼠微生物组影响的研究表明,氟化水对口腔有选择性影响,但对肠道微生物组没有影响(上表)。
流行病学研究已将饮用水氯化和/或接触消毒副产物 与结直肠癌和膀胱癌联系起来。在小鼠结肠直肠癌模型中,高浓度的水氯化(10 mg/L)增加了肿瘤发生。
肠道微生物群中产气荚膜梭菌(C.perfringens)和肠杆菌科(Enterobacteriaceae)等专性厌氧菌同时减少。同样,由于未评估饮用水微生物群,因此无法建立饮用水与肠道微生物群之间的直接联系。
此外,这些发现与实际饮用水系统的相关性尚不清楚,因为这远远超过了正常的氯浓度。美国可接受的最大氯浓度为 4 mg/L。
根据世界卫生组织(WHO)的有关数据,人体对氯的耐受量为每天每公斤体重0.15毫克,假设这些氯全部来自于饮用水,得到自来水氯的允许含量为每升5毫克。根据我国的相关规定,出厂水的余氯含量最低不能低于0.05毫克/升,最高上限为4毫克/升。

在如上一项关于水氯化对 6-61 个月儿童肠道微生物群影响的研究中,未发现氯化对肠道菌群有显著的长期影响,但氯化处理组的腹泻发作和抗生素使用次数较少。氯化水处理组中某些微生物物种的丰度略有增加,抗生素耐药基因 (ARG) 的丰度也有所增加。
耐药基因丰度的增加归因于肠杆菌科细菌相对丰度的增加,这些细菌含有可移动的耐药基因,但被认为最终是有益的肠道居民。
由于本研究再次未测量水中的微生物群,因此这些耐药基因的来源尚不清楚。先前的研究表明,饮用水中存在的大多数耐药基因表现出垂直传播模式,而不是水平传播模式。这些耐药基因可能来自饮用水,或者可能有其他来源,但氯的存在会在肠道中创造一种环境,从而有利于这些肠杆菌科细菌的繁殖。
迄今为止,已有少数研究考察了水氯化对模型生物肠道菌群的影响。几种水氯化会导致肠道菌群发生变化,但这些变化仅在高于饮用水中通常测量的浓度时才会观察到。这些研究中的暴露时间通常也很短。对水中氯离子相关浓度的慢性暴露进行进一步研究将有助于得出与现实世界相关的结论。
众所周知,自来水会影响人体健康,但其影响机制尚不明确。
随着技术的发展,我们现在有很多工具可以帮助研究饮用水对人类肠道微生物群的影响。现代的DNA测序技术已经变得更加经济实惠,使我们能够分析饮用水中的微生物群落。这些技术的应用可以显著提升我们对这一领域的理解。
此外,各种可用的组学技术,包括代谢组学、蛋白质组学和转录组学,以及成像技术,提供了可用于动物模型和人类的工具,以研究饮用水微生物组与水特性和肠道微生物组之间的关系。
综合饮用水对肠道菌群及进一步身体健康的影响,还需要以下研究:
1) 长期研究
大多数现有研究侧重于持续 1 至 3 个月的短期治疗。纵向方法将考虑肠道微生物群和水质与微生物群的时间变化,并有助于了解微生物群和化学性质之间的长期关系。增强我们当前知识的一个简单方法是将饮用水属性(如水源、化学性质和消费模式)纳入肠道微生物群的纵向队列研究中。这还将改善样本量限制,因为许多队列包括数百名参与者。
2) 饮用水微生物群的影响
以前的研究主要集中在水的特性上,例如 pH 值、温度、消毒剂和溶质。然而,了解饮用水微生物群与肠道微生物群之间的直接关系对于理解其对人类健康的潜在影响至关重要。因此,未来的研究工作应该同时考虑饮用水微生物群及其化学属性。这项工作还应考虑耐药基因从饮用水到肠道微生物群的潜在传播。
3) 以人为本的研究
使用动物模型进行的研究有助于在严格控制的环境中检验假设和开发机械联系。需要进行更多研究来直接检查人类和类似人类的状况,因为这些状况可能与动物模型中的状况不同。
最近的创新如可复制的肠道微观生态系统,可能在可控环境中进行类似人类的研究。这些系统减少了对活体动物研究的需求,可用于调查多种因素、肠道发育的不同阶段及其他条件。
在体外系统中获得的重要发现可以通过人类试验并结合运用基因组学、转录组学/蛋白质组学和代谢组学来验证。这种以人为重点的研究将显著有助于理解饮用水微生物群与人类肠道微生物群之间的联系,以及在现实环境中如何体现,为公共卫生考量提供相关见解。
主要参考文献:
Moghaddam HS, Abkar L, Fowler SJ. Making waves: From tap to gut- exploring the impact of drinking water on gut microbiota. Water Res. 2024 Sep 21;267:122503.
Piperata BA, Lee S, Mayta Apaza AC, Cary A, Vilchez S, Oruganti P, Garabed R, Wilson W, Lee J. Characterization of the gut microbiota of Nicaraguan children in a water insecure context. Am J Hum Biol. 2020 Jan;32(1):e23371.
Vanhaecke T, Bretin O, Poirel M, Tap J. Drinking Water Source and Intake Are Associated with Distinct Gut Microbiota Signatures in US and UK Populations. J Nutr. 2022 Jan 11;152(1):171-182.
Khan H, Miao X, Liu M, Ahmad S, Bai X. Behavior of last resort antibiotic resistance genes (mcr-1 and blaNDM-1) in a drinking water supply system and their possible acquisition by the mouse gut flora. Environ Pollut. 2020 Apr;259:113818.
Jha AR, Davenport ER, Gautam Y, Bhandari D, Tandukar S, Ng KM, Fragiadakis GK, Holmes S, Gautam GP, Leach J, Sherchand JB, Bustamante CD, Sonnenburg JL. Gut microbiome transition across a lifestyle gradient in Himalaya. PLoS Biol. 2018 Nov 15;16(11):e2005396.
Bowyer RCE, Schillereff DN, Jackson MA, Le Roy C, Wells PM, Spector TD, Steves CJ. Associations between UK tap water and gut microbiota composition suggest the gut microbiome as a potential mediator of health differences linked to water quality. Sci Total Environ. 2020 Oct 15;739:139697.
Murakami S, Goto Y, Ito K, Hayasaka S, Kurihara S, Soga T, Tomita M, Fukuda S. The Consumption of Bicarbonate-Rich Mineral Water Improves Glycemic Control. Evid Based Complement Alternat Med. 2015;2015:824395.
Chen J, Xu XW, Kang JX, Zhao BC, Xu YR, Li JL. Metasilicate-based alkaline mineral water confers diarrhea resistance in maternally separated piglets via the microbiota-gut interaction. Pharmacol Res. 2023 Jan;187:106580.
Murakami S, Goto Y, Ito K, Hayasaka S, Kurihara S, Soga T, Tomita M, Fukuda S. The Consumption of Bicarbonate-Rich Mineral Water Improves Glycemic Control. Evid Based Complement Alternat Med. 2015;2015:824395.

谷禾健康

慢性子宫内膜炎(Chronic endometritis, CE)是一种慢性非特异性的子宫内膜炎症性疾病,常表现为无症状或非特异性临床症状,慢性子宫内膜炎经常被患者和妇科医生忽略。因此,寻找新的诊断标记对于改善慢性子宫内膜炎的预后至关重要。
针对上述问题,来自武汉大学人民医院生殖医学中心杨静团队的科研人员在《OBSTETRICS AND GYNECOLOGY》上发表了研究论文。
该研究针对慢性子宫内膜炎女性的阴道微生物特征进行深入研究,比较了98名接受子宫内膜活检进行不孕症常规临床检查的女性(49名诊断为CE的女性和49名非CE的女性)的阴道微生物组特征,并采用杭州谷禾核酸提取试剂盒及测序平台,使用16S rRNA基因扩增子测序分析阴道微生物组,对微生物标记物进行了鉴定。
该项研究确定了四个慢性子宫内膜炎的微生物标记物(肠杆菌,普雷沃氏菌,粪杆菌,Phascolarctobacterium),开发了针对慢性子宫内膜炎预测诊断分类器,并进一步探讨了阴道微生物组特征作为慢性子宫内膜炎诊断新工具的潜力。

英文题目:Vaginal microbiome dysbiosis as a novel noninvasive biomarker for detection of chronic endometritis in infertile women
中文题目:阴道微生物群失调作为检测不孕女性慢性子宫内膜炎的新型非侵入性生物标志物
期刊名:OBSTETRICS AND GYNECOLOGY
发表时间:2024年7月10日
▼ 什么是慢性子宫内膜炎?
慢性子宫内膜炎是发生在子宫内膜间质区的持续性细微炎症性疾病,其特征为子宫内膜间质区的浆细胞浸润,通常无症状或仅出现轻微的症状,如异常子宫出血、盆腔疼痛、白带增多等。
▼ 慢性子宫内膜炎有哪些不良后果?
大量研究表明,慢性子宫内膜炎与女性不孕症、反复流产、反复着床失败、子宫内粘连和辅助生殖技术的不良后果密切相关,此外慢性子宫内膜炎还是影响活产和足月分娩的客观因素之一。
▼ 慢性子宫内膜炎的发病率
慢性子宫内膜炎的发病率在不同人群中不尽相同:
其病理改变与内膜中微生物群的定性、定量改变有关。
近年来,人类微生物组学的研究表明,人体不同系统内微生物群与人类健康和疾病发病机制存在密切关系,刺激炎症并增加癌症等疾病发生的风险。作为人体主要的微生物组,阴道菌群对于女性生理和生殖健康起着重要作用。
◆阴道病原体感染是慢性子宫内膜炎的病因之一
Moreno等人通过微生物培养在慢性子宫内膜炎女性子宫内膜中发现了包括无乳链球菌和淋病奈瑟菌在内的细菌,并开发了一种诊断慢性子宫内膜炎的分子方法——逆转录聚合酶链反应试验,从这些病原体中识别DNA,用于慢性子宫内膜炎的诊断。
◆慢性子宫内膜炎:宫腔内致病菌增加
随着微生物组学技术的发展,传统观点认为宫腔内是无菌环境的观念也逐渐被推翻。16S和宏基因组测序等技术均证实宫腔内存在微生物的定植,健康女性以乳杆菌属占绝对优势,且宫腔内微生物与慢性子宫内膜炎的发病存在关联,慢性子宫内膜炎患者宫腔非乳酸杆菌为优势菌群且致病菌如加德纳菌、葡萄球菌、链球菌检出率增加。
在研究中也指出,诊断患有慢性子宫内膜炎的女性阴道微生物群生态失调。与子宫内膜冲洗样品相比,阴道分泌物可以作为诊断慢性子宫内膜炎的更无创和更经济有效的生物标志物。阴道宫颈黏液的免疫学指标已被认为是子宫内膜炎的潜在诊断生物标志物。然而,需要进一步评估阴道微生物组在慢性子宫内膜炎中的诊断潜力。
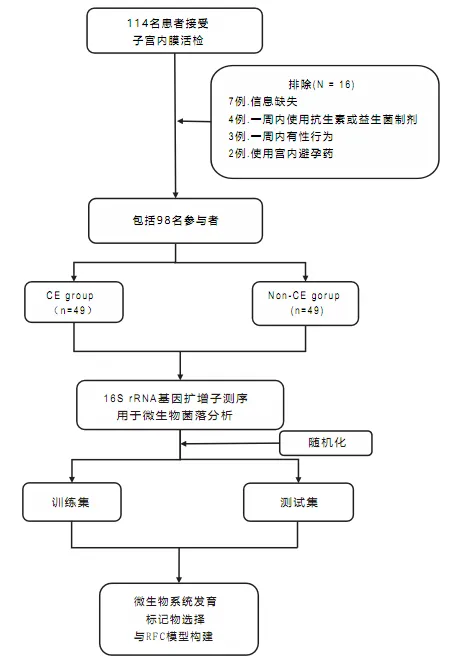
本研究于2023年6月至2023年11月在武汉大学人民医院生殖医学中心完成。研究集中于常规不孕症检查中接受子宫内膜活检的患者。最终共纳入49名患有慢性子宫内膜炎的女性,并与49名非慢性子宫内膜炎对照组相匹配。
慢性子宫内膜炎患者的选择标准如下:
排除标准为:
注:所有受试者均获得书面同意,并经武汉大学人民医院伦理委员会批准(批准通知号:WDRY2023- K090)。
该研究的设计和流程图如图所示:
编辑
在本研究中,所有样本均在子宫内膜活检前采集。用无菌生理盐水擦拭外阴。随后,使用无润滑剂的一次性无菌阴道窥镜完全暴露宫颈。使用两个无菌拭子从后穹窿收集阴道分泌物:一个使用Nugent评分法进行评估,另一个储存在−80°C下用于后续16S rRNA基因测序分析。
项目采用杭州谷禾核酸提取试剂盒(GHFDE100)提取阴道冲洗液样本的基因组DNA。用引物515F(5′- GTGCCAGCMGCCGCGGTAA- 3′)和806R(5′- GGACTACHVGGGTWTCTAAT- 3′)扩增16S rRNA的V4区。为了实现多重测序,将样品特异性对端6bp条形码集成到TrueSeq适配器中。纯化和定量后,整个DNA池在Illumina NovaSeq6000平台(Illumina)上按照协议进行测序。
使用Vsearch v2.22.1组装拼接双端序列,并以100%的相似性阈值将序列聚类为相同的扩增子序列变体(amplicon sequence variant, ASV)。利用QIIME2加权分类器获得ASV的分类信息。基于R包“VennDiagram”生成维恩图来说明分组间共有的和独有的ASV。
α-多样性是指群落内的物种多样性,以物种丰富度和归一化香农熵(均匀度)为其显著组成部分。物种丰富度采用Chao1指数进行量化,而Shannon多样性指数同时评估丰富度和均匀度。通过主坐标分析(PCoA)可视化两个组之间样本的微生物菌落结构。使用R的vegan包进行相似性分析(ANOSIM)评估微生物群落的差异。
此外,为了预测微生物的代谢功能,使用京都基因与基因组百科全书(KEGG)数据库与PICRUSt2 (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States)进行代谢途径富集分析。组间的统计差异使用Kruskal-Wallis检验和Wilcoxon秩和检验进行评估,显著性水平定义为P < 0.05。
为了评估阴道微生物群组成对慢性子宫内膜炎(CE)的诊断潜力,应用随机森林算法,这是一种稳健的监督学习算法。使用R的pROC软件包生成的受试者工作特征(ROC)曲线评估模型的整体性能。曲线下的面积(AUC)量化了ROC曲线的判别能力。此外,还计算模型的特异性、敏感性和准确性。
连续变量的临床特征差异使用Student’s t检验进行评估,而分类变量则需要使用χ²检验或Fisher精确检验进行评估。统计分析使用SPSS版本27(IBM)进行,P < 0.05被认为具有统计学意义。
在98名参与者中,49名被诊断为CE,其标准是在30个随机选择的高倍视野中至少有一个视野中观察到超过5个MUM-1+/CD138+细胞。基线特征包括年龄、体重指数、抗穆勒激素水平和流产史,在两组之间显示了可比性(见表1)。
等级丰度曲线体现了物种丰富度和均匀度
等级丰度曲线描述了两组微生物群落的物种丰富度和均匀度。曲线范围越宽表明物种丰度越高,曲线越光滑表明物种分布越均匀。
从曲线上看,CE组的物种丰富度和均匀度高于非CE组(图2a)。维恩图显示,两组共有1180个ASV中的664个,其中CE组特有的ASV为354个(图2b)。利用Chao1丰富度估计器和Shannon多样性指数量化的α-多样性结果显示,两组间差异不显著(Chao1的P = 0.1733, Shannon的P = 0.9043,图2c,d)。
为了说明样品之间的微生物组分布空间,进行了PCoA,如图2e所示。结果显示,两组样本的分布是对称的。
值得注意的是,ANOSIM结果显示组间差异大于组内差异,尽管没有达到统计学意义(R = 0.022, P = 0.085)(图2f)。
阴道微生物群的细菌多样性
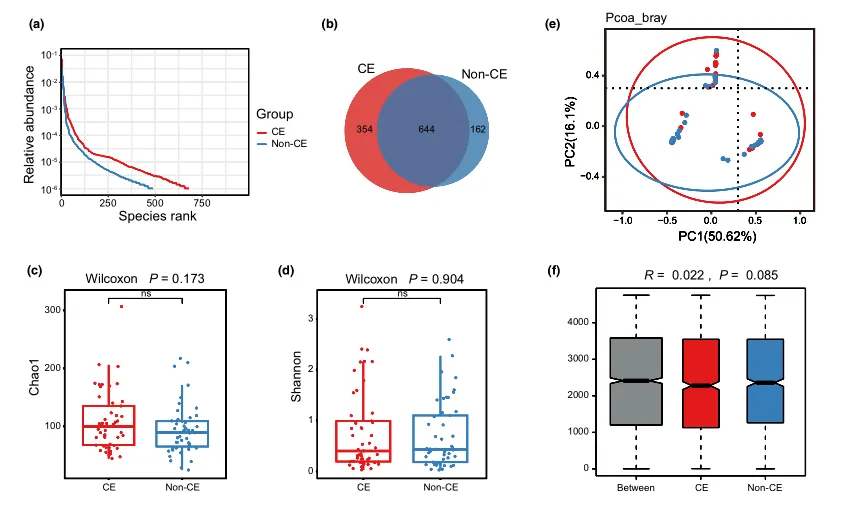
(a)用秩丰度曲线评价细菌的相对均匀度;
(b)显示组间重叠的维恩图;
(c)采用Chao1指数;
(d)Simpson指数估算口腔微生物多样性;
(e)使用Bray-Curtis通过主坐标分析计算β多样性;
(f)通过相似性分析计算了组间微生物群落的差异。
阴道微生物群的组成
文中分析了与CE相关的阴道微生物组的分类组成和变化。门、科和属水平的平均细菌群落组成如图3a-c所示。值得注意的是,与非CE组相比,CE组有10个属的细菌显著富集,包括双歧杆菌、普雷沃氏菌和加德纳菌(均P < 0.05)(图3d)。
慢性子宫内膜炎患者阴道微生物群落的系统发育概况
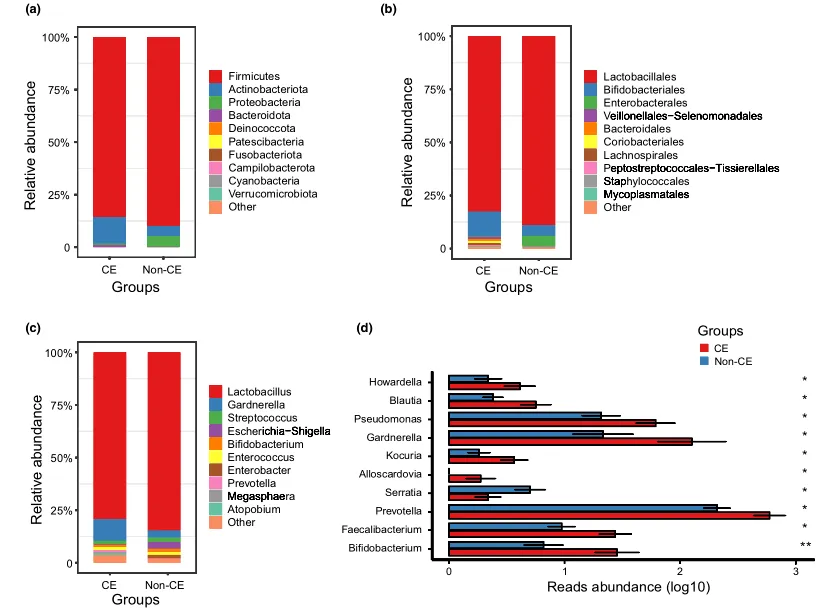
门(a)、科(b)、属(c)水平上细菌群落的平均组成。CE组与非CE组间差异有统计学意义(均 P < 0.05)。0.01 <矫正 P ≤ 0.05 ,标记为*。
代谢途径
通过KEGG途径富集的功能分析揭示了CE组中几种上调的途径,如图4所示。具体而言,参与多糖生物合成和代谢的途径(包括鞘糖脂生物合成、糖胺聚糖降解和其他多糖降解过程)上调。
此外,萜类和聚酮类代谢、II型聚酮类生物合成、鞘脂质代谢、次生代谢物生物合成、链霉素生物合成、外源生物降解、氯烷烃和氯烯烃降解以及凋亡相关途径也上调。总共有8个KEGG模块在CE组显著富集,重点是代谢相关途径(均P < 0.05)。
KEGG代谢通路的丰度差异
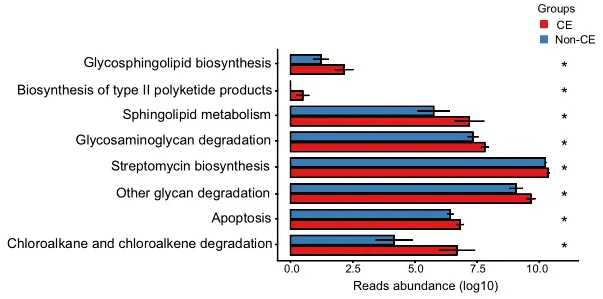
0.01< 校正P≤0.05,标记为*;0.001 <校正P≤0.01,标记为**。CE,慢性子宫内膜炎。
为测试阴道微生物组对慢性子宫内膜炎(CE)的诊断潜力,这里构建了一个随机森林分类器模型,用以区分CE样本和非CE样本。训练集用于模型训练,测试集用于评估模型性能。通过在发现阶段重复五次的10倍交叉验证,我们识别出四个最优标记 ASV 作为CE的标记集(图5a)。
这些选定ASV的对应细菌属包括:
随机森林模型在训练集队列上的AUC值为85.68%(95%置信区间[CI], 75.62%-95.73%),在测试集队列上的AUC值为83.26% (95% CI, 68.67%-97.85%)(图5b)。模型的特异性为82.76%,灵敏度为79.49%,准确率为80.88%。
这些发现表明,基于阴道微生物ASV标记的随机森林模型在区分CE和非CE队列方面表现出强大的诊断潜力。
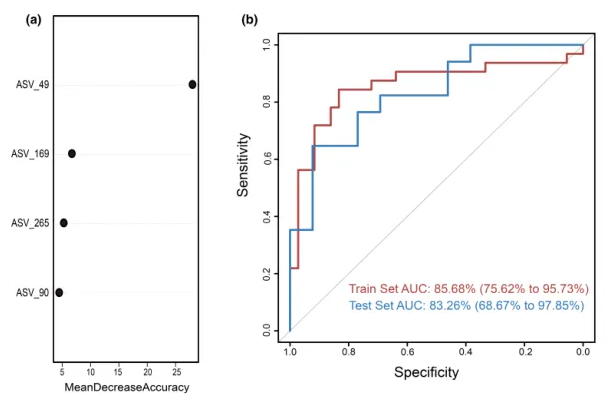
图5.通过随机森林模型鉴定用于临床诊断的微生物标志物。
(a)使用阴道样品中四种选定生物标志物的概况,来自随机森林模型的变量的平均降低准确度。
(b)接收器工作特征曲线基于组织样本的训练集和测试集。ASV,扩增子序列变体; AUC,曲线下面积。
◆阴道菌群的变化可引起宫腔的菌群失调
女性阴道中的微生物组是一个完整的连续体观点已被广泛接受。阴道细菌可以异位定植并改变上生殖道中的微生物组。局部微生物组的生态失调会导致微环境发生复杂的病理生理变化,最终扰乱与免疫反应、炎症和凋亡相关的各种过程,从而促进病理状况。
阴道和宫腔内的菌群变化有一定的同步性,阴道菌群的变化可引起宫腔内的菌群失调,从而影响女性生殖健康。
本研究旨在对有或没有慢性子宫内膜炎(CE)的女性阴道微生物组的群落结构和功能进行初步探索。此外,文章中还提出了基于阴道微生物ASV的新型非侵入性CE诊断标志物。
◆特定的菌群可作为新型非侵入性CE诊断标志物
与先前的研究结果一致,该研究中没有发现慢性子宫内膜炎组和非慢性子宫内膜炎组阴道微生物组之间的α-多样性或β-多样性有显著差异。
值得注意的是,微生物组的低组内变异情况表明其作为潜在诊断标记具有一定的稳健性。与非CE组相比,慢性子宫内膜炎组中几个低丰度属的富集程度不同,包括双歧杆菌,普雷沃氏菌和加德纳菌。
双歧杆菌
双歧杆菌以其保护作用而闻名,其高丰度在CE组中令人惊讶。双歧杆菌菌株作为益生菌用于肠道炎症、严重疾病和抑郁症患者的肠道微生物组调节已有很长的历史。然而,其他研究报告称,在一些不健康的情况下,包括子宫内膜异位症、高危人乳头瘤病毒和不孕症,阴道中双歧杆菌含量很高。这种不一致的潜在机制尚不清楚,需要进一步研究。
加德纳菌和普氏菌
加德纳菌和普氏菌是公认的阴道生态失调的指标。这些属的定植改变了免疫系统,并诱导了促炎反应。在宿主-微生物共培养模型中,加德纳菌与宫颈阴道屏障的破坏有关,同时促进小鼠中普雷沃菌属对上生殖道的侵入性感染。
总的来说,该研究结果表明,两组之间的阴道微生物组发生了实质性变化,突显了阴道冲洗液样本作为检测慢性子宫内膜炎的诊断工具的潜力。
◆阴道菌群的变化可引起宫腔的菌群失调
KEGG途径富集分析强调了关键代谢过程的上调,特别是CE组的聚糖生物合成和代谢。覆盖阴道上皮细胞的聚糖涂层是保护屏障的重要组成部分。乳杆菌与阴道上皮的粘附被认为是具有竞争性地抑制病原体,而上皮的糖胺聚糖在这些过程中起着至关重要的作用。
值得注意的是,糖胺聚糖的消耗与宫颈上皮细胞屏障功能的破坏有关,这可能会增加对局部细菌和上升感染的易感性。这表明CE患者的微生物组失调可能会对微生物侵入阴道的主要门户产生负面影响,并引发宫内炎症。
关于慢性子宫内膜炎(CE)的标准化定义和诊断指南仍存在争议,最被接受的诊断依赖于子宫内膜活检。基于选定的阴道微生物群构建了CE诊断模型。本文研究中,引入了一种新的随机森林模型,该模型可以根据ASV以更高的灵敏度和准确性将CE患者与使用阴道分泌物的健康个体区分开来。该研究结果表明,阴道微生物组的变化可以作为诊断CE的指标,为慢性子宫内膜炎提供了一种更容易获得和更方便患者的诊断选择。
然而,该研究也存在一定的局限性。首先,它主要侧重于检查有慢性子宫内膜炎和没有慢性子宫内膜炎的女性之间的成分差异,而不是研究与慢性炎症相关的免疫失调的潜在机制。其次,阴道微生物样本是在分泌中期收集的,这可能无法解释整个月经周期中微生物群组成的变化。第三,作为一项单中心调查,发现的普遍性可能有限。考虑到饮食和运动等因素,多中心方法可以提供更全面的解读。
最后,多种组学方法的整合(包括培养组学、元转录组学和代谢组学),可以提供对微生物群落功能的全面了解。这种更广泛的视角对于阐明阴道微生态系统内复杂的网络和阴道微环境的潜在变化至关重要。
阴道微生物生态失调是慢性子宫内膜炎的重要指标。该研究确定了与慢性子宫内膜炎发展相关的潜在候选细菌,并提出了潜在的机制。基于阴道微生物群的生物标志物有望成为检测慢性子宫内膜炎的非侵入性工具。需要进一步的研究来发现特定阴道微生物组在慢性子宫内膜炎中的功能,并确定具有诊断能力的强大阴道微生物标记物,以区分患有慢性子宫内膜炎的个体。
谷禾健康
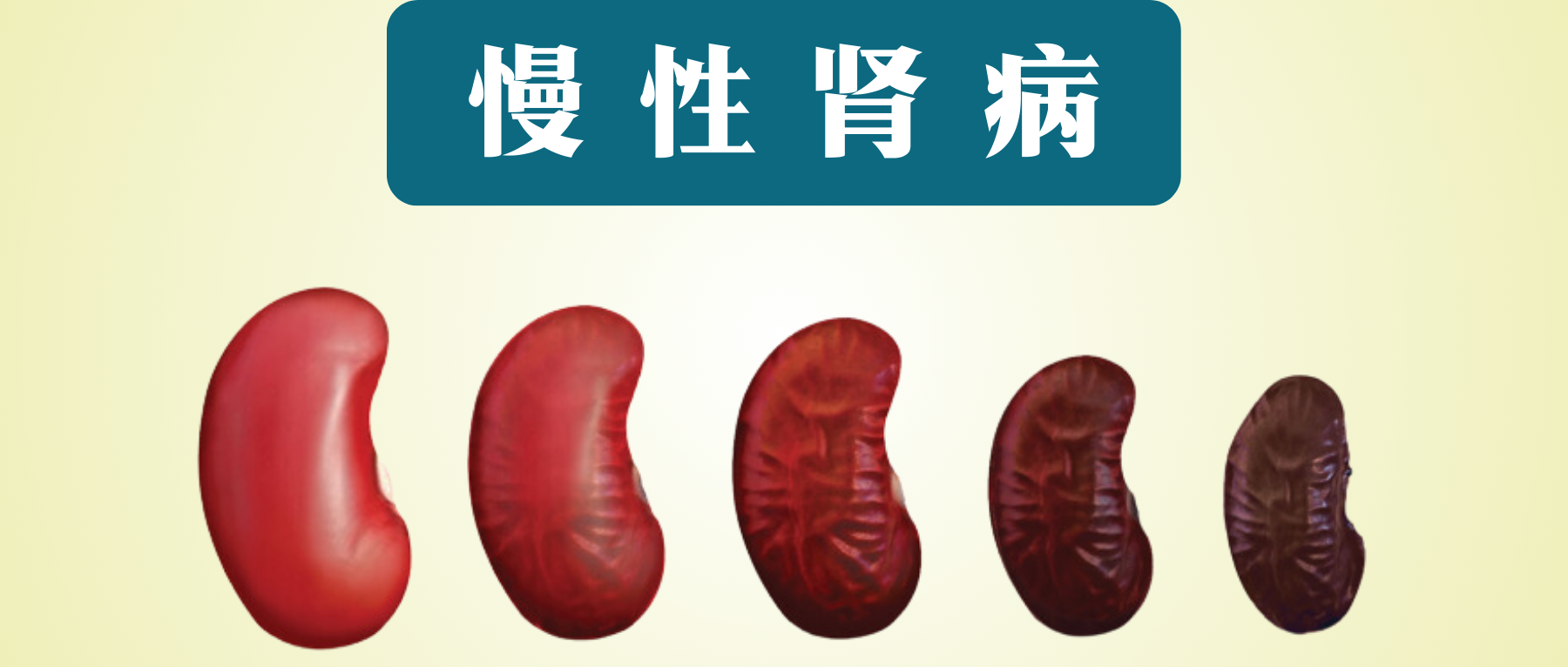
慢性肾病(CKD)被称为“隐形的杀手”,“无形的病”,因为该病在早期大多数人没有明显的症状,直到后期肾脏失去过滤血液中废物和多余液体能力时,已经可能导致肾功能衰竭,需要透析或肾移植来维持生命。
慢性肾病特征是肾脏结构异常或因肾脏损伤导致的肾功能逐渐下降,持续时间至少为 3 个月,并伴有相关的健康后果。全球慢性肾病患病率不断上升,影响全球 10% 以上的人口,全球有超过 8.436 亿人患有慢性肾病。
根据美国疾病控制与预防中心 (CDC) 的数据,美国约有 3700 万人(约占成年人的 15%)患慢性肾病。
慢性肾病的发生通常是一个非常缓慢的过程,最初症状很少。根据 CDC 的数据,90% 患有慢性肾病的成年人并不知道自己患有慢性肾病,而每 2 名肾功能极低且未接受透析治疗的人中,就有 1 名不知道自己患有慢性肾病。
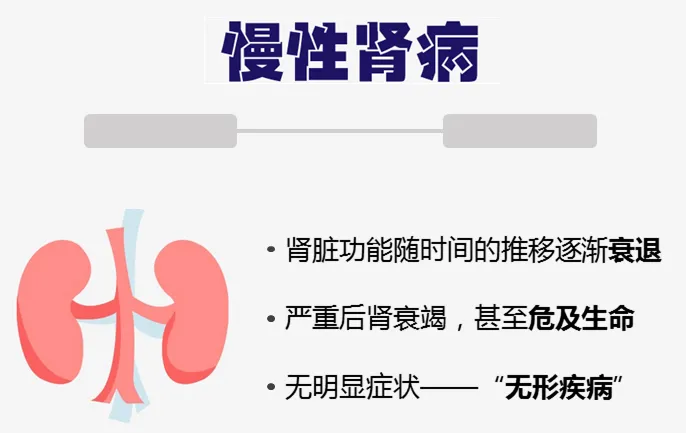
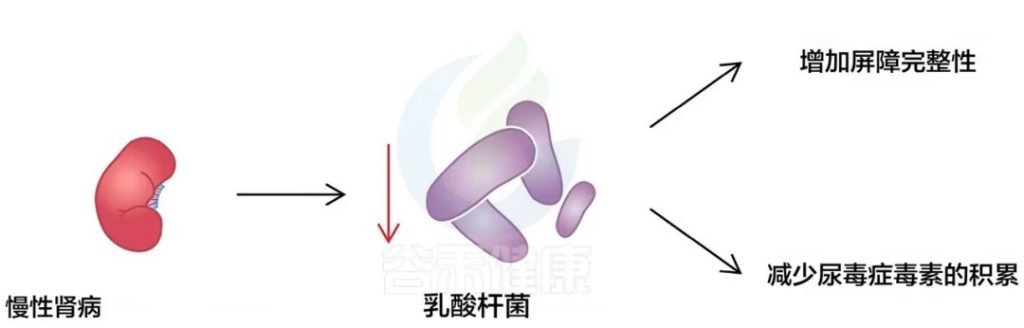
慢性肾病的发生与高血压、糖尿病和肥胖等多种风险因素的持续暴露密切相关,导致肾脏排泄功能不可逆地逐渐衰退。尿毒症毒素在血液循环中的积累,对肾脏造成巨大损害,还会影响其他器官和组织,增加心力衰竭和死亡的风险。
遗憾的是,慢性肾病目前尚无法治愈,现有的治疗方法如生活方式的改变、药物治疗和透析只能缓解症状,延缓病情进展。肾移植虽然是终末期肾病患者的治疗选择,但受限于供体数量和漫长的等待期,其可及性有限。因此,迫切需要新的、有效的干预策略来改善慢性肾病患者的预后。
近年来,研究者们将目光投向了肠道微生物群。肠道菌群失调已被证实与慢性肾病的发生和进展密切相关。肠道微生物的多样性和平衡对维持宿主的代谢、免疫和炎症等功能至关重要。在慢性肾病患者中,肠道屏障功能受损,导致肠黏膜通透性增加,有害物质如内毒素和尿毒症毒素更容易进入血液循环,加重肾脏负担。
具体而言,慢性肾病患者的肠道菌群中有害菌如变形菌门和放线菌门比例增加,而有益菌如拟杆菌门和厚壁菌门减少。这种菌群失调导致短链脂肪酸等有益代谢物的产生减少,而有害代谢物如三甲胺和吲哚硫酸盐的产生增加。这些有害代谢物进入血液循环后,可诱发炎症和氧化应激,加速肾脏纤维化过程。
本文我们来了解一下慢性肾病的发生发展,症状,发病率,病因等,随着对肠-肾轴的深入了解,有助于我们制定优化的干预策略,改善慢性肾病患者的临床结局。
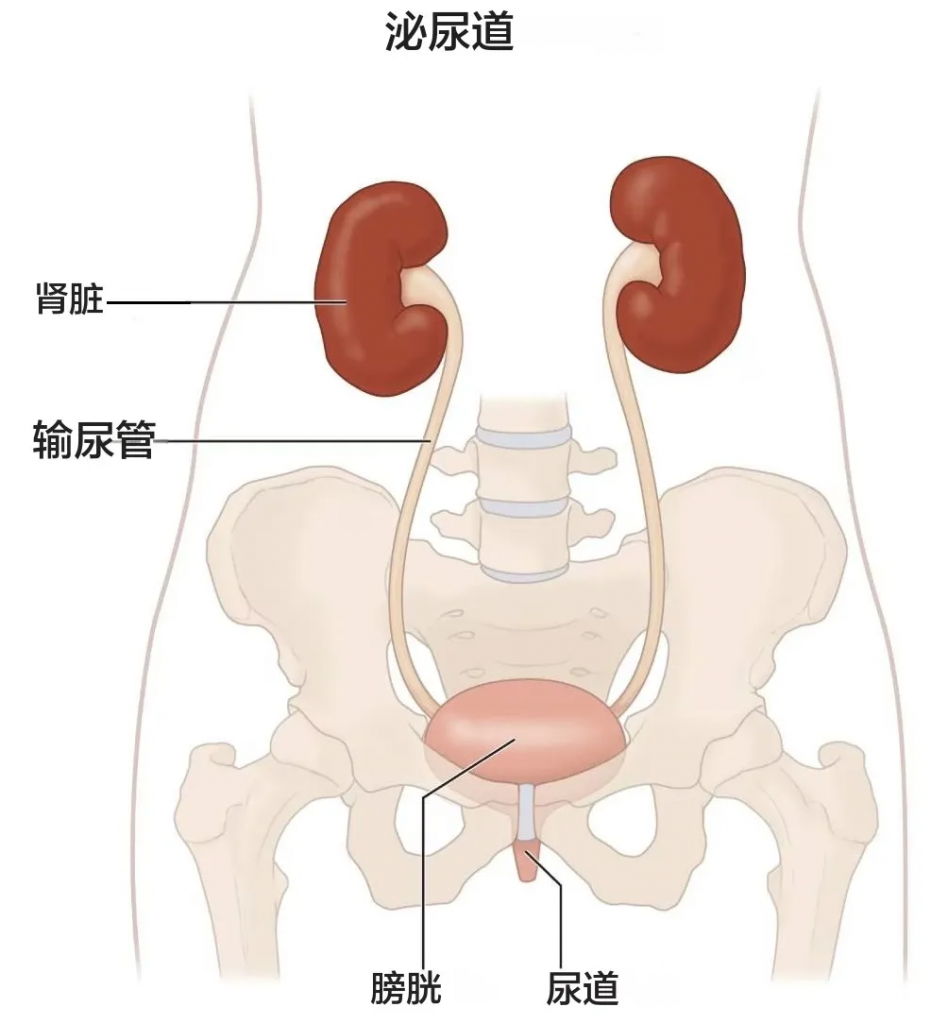
我们的肾脏主要功能是过滤血液中的多余水分和废物,产生尿液排出体外。为了保持身体正常运转,肾脏会平衡血液中循环的盐分和矿物质(如钙、磷、钠和钾)。肾脏还会分泌激素,帮助控制血压、制造红细胞并强健骨骼。
晚期慢性肾病可能导致液体、电解质和废物在体内积聚至危险水平。
图源:NIDDK
慢性肾病的发展取决于个人的年龄、合并症、反复急性肾损伤、蛋白尿水平等。肾功能下降超过三个月称为慢性。
大多数患有慢性肾病的人会出现高滤过、肾肥大、小管间质纤维化、肾素-血管紧张素-醛固酮系统激活和内皮屏障破坏,导致肾小球滤过率和肾脏排泄效率降低。
具体如何定义慢性肾病?
根据“改善全球肾脏病预后”组织 (KDIGO) 的定义,慢性肾病目前定义为肾小球滤过率 (GFR) 降低,<60 mL/min/1.73 m 2(GFR 分类 G3a–G5),或以肾脏损害形式出现的其他肾脏结构或功能异常,对健康有影响,持续至少 3 个月。
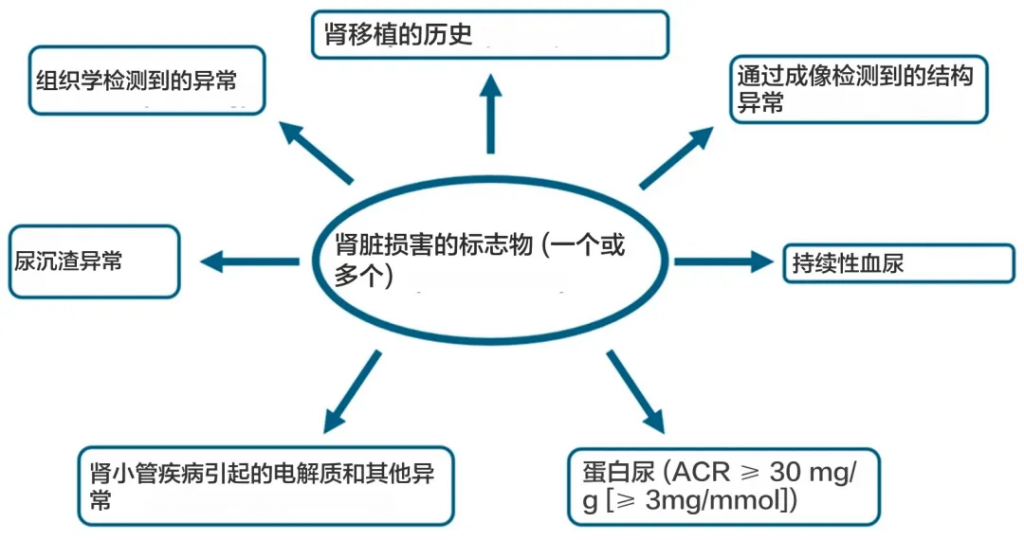
doi.org/10.3390/ijms251910429
慢性肾病的特点是病情进展缓慢,不可逆,发病相对隐匿。该病的显著特点是并发症风险增加,死亡主要与血管疾病有关。
◑ 根据肾功能分级分类
按照肾小球滤过率(GFR)进行分级,慢性肾病通常分为五个阶段,阶段越高,肾功能越差,对患者生活质量和预期寿命的影响越大:
1
GFR ≥ 90 mL/min
通常肾功能正常,可能存在其他肾脏病理改变。
2
GFR 60-89 mL/min
轻微肾功能减退,肾功能异常的早期迹象。
3
GFR 30-59 mL/min
中度肾功能减退,有可能出现明显的临床症状。
4
GFR 15-29 mL/min
重度肾功能减退,需进行透析或等待肾移植。
5
GFR < 15 mL/min
终末期肾病(ESRD),需进行透析或肾脏移植。
随着肾功能的降低,患者面临更高的并发症风险,包括心血管疾病、贫血和电解质紊乱等。
◑ 根据病因分类
慢性肾病还可以依据病因进行分类:
◑ 依据临床表现分类
根据临床症状和生化指标的不同,慢性肾病可以进一步分为:
维持肾脏健康的策略,尤其是在早期阶段,将是预防慢性肾病发展的关键因素。
根据统计,慢性肾病的全球流行率约为13%。
对于高危人群,如糖尿病患者,其慢性肾病的发病率可高达35%以上。
据欧洲肾脏健康联盟统计,慢性肾病的发病率在老年人群中较高:
其中大多数 (10.6%) 处于疾病晚期(3-5 期),0.1% 处于 5 期。当然这个百分比可能不准确,因为1-2 期慢性肾病的人群可能并不知道自己的病情,这可能导致上述数据被低估和扭曲。
5 期患者数量较少,是因为慢性肾病患者过早死亡的可能性比患肾衰竭的可能性高出五到十倍。
慢性肾病(CKD)是一种长期进行性肾脏疾病,随着病情的进展,患者可能会出现多种临床症状和体征。以下是慢性肾病的一些主要表现:
早期无症状阶段
慢性肾病的早期阶段,患者常常无明显症状,可能仅在常规体检时通过血液或尿液检查发现肾功能异常。因此定期监测高危人群(如糖尿病或高血压患者)是至关重要的。
水钠潴留
随着肾功能的逐渐下降,患者可能会出现水钠潴留,这可能导致:
尿液变化
代谢性酸中毒
慢性肾病患者由于肾脏排除酸的能力下降,会出现代谢性酸中毒,其典型症状包括:
疲劳与虚弱
随着肾功能下降,患者常常感到持续的疲乏和虚弱。部分患者可能出现贫血(通常与肾脏无法生成足够的红细胞生成素有关),导致:
肾性骨病
随着肾功能的下降,钙、磷代谢失衡可能会导致肾性骨病。相应的临床表现包括:
神经系统症状
心血管相关症状
皮肤和其他系统表现
任何人任何年龄都可能患上慢性肾病。但有些人的风险比其他人更高。最常见的风险因素是:
高血压和糖尿病:高血压和糖尿病是慢性肾病最常见的病因之一。这两种疾病通过引起肾脏血管的损伤,导致肾小球硬化和肾功能下降。
脱水:脱水,尤其是与高温相关的反复脱水,可能导致永久性的肾脏损伤。这可能通过激活血管加压素、醛固酮-果糖激酶途径和慢性高尿酸血症等机制实现。
年龄:随着年龄的增长,肾脏的质量和功能会逐渐下降,这是慢性肾病的一个重要风险因素。衰老过程中细胞衰老,特别是肾小管上皮细胞的衰老,会导致慢性炎症,进而推动慢性肾病的进展。
遗传因素:某些单基因疾病也是成人慢性肾病的原因之一。
肾小球疾病和肾小管疾病:肾小球疾病(如糖尿病肾病和肾淀粉样变性)和肾小管疾病(如急性肾衰竭后发展为慢性状态)是导致慢性肾病的重要原因。
缺氧:肾脏组织的缺氧是推动慢性肾病进展的一个重要因素。缺氧条件下,肾脏组织的持续炎症攻击是通过诱导白细胞获得粘附表型来实现的。
氧化应激和免疫系统的作用:氧化应激、免疫系统的异常活动、中性粒细胞弹性蛋白酶相关脂ocalin和基质金属蛋白酶的活动在慢性肾病的发展和进展中起着重要作用。
肠道菌群失调:这也在正常稳态的维持和慢性肾病中的失调中起着关键作用。这在接下来的章节我们会详细阐述。
共病条件:除了高血压和糖尿病外,其他共病条件,如肥胖、吸烟和性别(在男性中更为显著),也与慢性肾病的发展有关。
环境毒素和农药暴露:在低收入和中等收入国家或地区,环境毒素(如重金属暴露、农药使用、真菌毒素、水污染和蛇咬)被认为是慢性肾病未知病因的潜在原因。
其他因素:包括但不限于心血管疾病、HIV感染、代谢综合征、药物引起的肾病(如非甾体抗炎药引起的肾病)等也是慢性肾病的潜在病因。
这种进行性的疾病不仅影响患者的肾功能,而且与多种并发症有关,包括心血管疾病、贫血、骨代谢紊乱等。随着病情的发展,慢性肾病患者的症状可能包括尿量改变、水肿、疲劳、恶心和认知功能下降等。
随着对慢性肾病的理解不断深入,研究人员开始关注肠道微生物群在慢性肾病进展中的作用。肠道微生物群的组成和功能在慢性肾病患者中可能会发生显著变化,这些变化可能会通过多种机制影响慢性肾病的进展,包括影响宿主的免疫系统、代谢途径和炎症反应等。接下来我们具体来了解这其中的机制,这对于开发新的治疗策略和管理慢性肾病患者的病情具有重要意义。
近年来,随着医学研究的深入,人们逐渐认识到肠道菌群与慢性肾病之间存在着密切的联系。
一方面,慢性肾病患者的肾功能下降导致体内毒素积累,这些毒素可通过血液循环影响肠道环境,进而破坏肠道菌群的平衡;另一方面,肠道菌群失调会产生更多的有害代谢物,尿毒症毒素如吲哚硫酸盐和对甲苯磺酸盐,这些物质通过肠-肾轴加重肾脏的损伤。此外,肠道菌群失调还可能导致肠道屏障功能下降,使得细菌和内毒素更易进入血液,引发全身性炎症反应,进一步加剧慢性肾病的进展。
下面我们来详细了解一下慢性肾病患者的具体肠道菌群变化特征:
慢性肾病患者的肠道菌群组成有何特点?与健康人群相比有何不同?
慢性肾病肠道菌群基本特征
慢性肾病患者肠道菌群失调,主要表现为:
有害菌增多,有益菌减少。
有害菌增多:
有益菌减少:
肠道菌群失调如何影响慢性肾病进展?
这种菌群失调会导致:
因此,调节肠道菌群、增加有益菌、减少有害菌是改善慢性肾病患者预后的一个重要策略。
肠道菌群生物标志物
肠道菌群有潜力成为慢性肾病早期诊断和预后监测的生物标志物。
早期诊断标志物:
这些细菌属可以较准确地区分慢性肾病患者和健康对照。
疾病进展标志物:
Escherichia-Shigella和Prevotella9(AUC = 0.86)可以准确区分糖尿病肾病患者和年龄/性别匹配的糖尿病患者。
随着慢性肾病的进展,一些特定菌种的变化:
增加的菌种:
减少的菌种:
特定慢性肾病阶段的标志菌:
编辑
肾移植患者的特征菌群:
与健康对照相比,肾移植患者的肠道菌群特征类似于慢性肾病3-4期患者,表现为:
这些菌群变化与慢性肾病进展相关,可能通过以下机制影响疾病:
首先,失调的肠道菌群导致肠道屏障功能受损,使得内源性和外源性毒素(如脂多糖,LPS)进入血液。这些毒素通过激活全身性炎症反应,增强肾脏的负担,进而导致肾功能损害。脂多糖可以与Toll样受体4(TLR4)结合,诱导炎症介质的释放,如肿瘤坏死因子α(TNF-α)和白细胞介素-6(IL-6),这些介质加重肾脏的炎症反应。
其次,肠道微生物产生的代谢物,尤其是短链脂肪酸,在维持肾脏健康中发挥着关键作用。短链脂肪酸生成减少与肠道菌群失调相关,导致的肠道屏障功能进一步减弱,使肾脏暴露于更高的毒素水平和炎症状态。
慢性肾病与肠道菌群失调之间的恶性循环
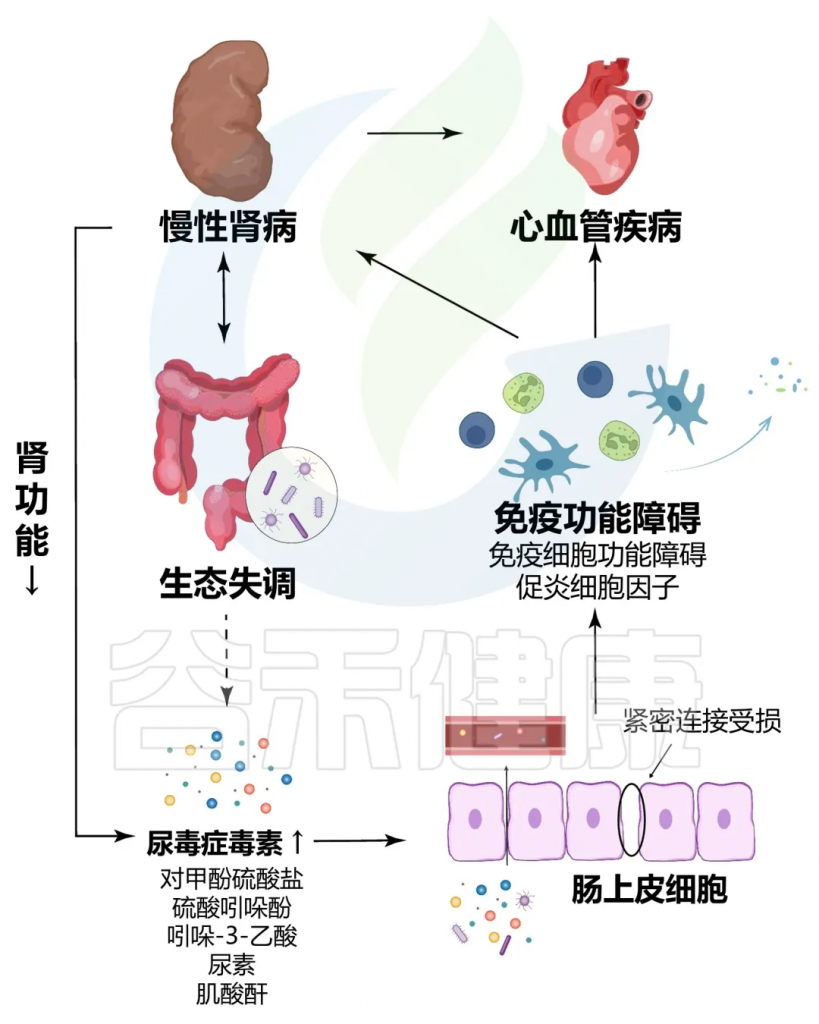
doi.org/10.1186/s12967-023-04455-2
菌群代谢物
短链脂肪酸
SCFAs(短链脂肪酸)主要通过以下几个方面影响慢性肾病:
• 通过抑制NF-κB活性和MAPK信号通路来减少促炎因子的产生
• 激活Nrf2通路,增强抗氧化防御能力。
• 通过AMPK/mTOR信号通路调节自噬,延缓CKD进展
• 改善CKD患者胰岛素敏感性和葡萄糖稳态。
• 调节T细胞和B细胞分化,参与免疫调节。
吲哚硫酸盐(IS)
•IS是一种强效的尿毒症毒素,对肾脏和血管系统有负面影响。
•IS可以促进内皮功能障碍和炎症。
•它增加氧化应激,上调转录因子NF-κB的表达(与炎症有关),并下调转录因子Nrf2(与抗氧化防御有关)。
•IS可以通过影响调节骨化血管平滑肌细胞(VSMCs)转分化的microRNAs来促进血管钙化。
•它可以下调miR-29b,这是一种血管钙化的抑制因子。
•IS可以激活肾素-血管紧张素-醛固酮系统,上调血管紧张素II 1型(AT1)受体,下调2型(AT2)受体。
•在慢性肾病(CKD)患者中观察到的浓度下,IS可以增强血管紧张素II对VSMCs的有害作用。
对甲酚硫酸盐(PCS)
• PCS与心血管损伤有关,随着肾小球滤过率(GFR)下降而在血清中累积。
•它可以促进VSMCs的迁移和增殖,这是血管钙化发展的关键细胞事件。
•PCS可以触发内皮细胞和VSMCs中氧化应激。
•它可以诱导主动脉壁平滑肌收缩,导致主动脉壁向内的共性重塑。
•PCS与动脉僵硬度、血管钙化和颈-股脉搏波速度有关。
•它可以促进主动脉炎症和钙化,通过急性期反应和凝血信号通路。
•PCS与内皮功能障碍、动脉僵硬度、血管钙化、心血管事件和全因死亡率有关。
三甲胺N-氧化物(TMAO)
•TMAO可以剂量依赖性地增加VSMCs中的钙含量,促进血管钙化。
•它刺激与VSMCs骨化分化相关的基因(如Runx2和BMP2)的表达。
•TMAO可以增加矿物质含量,上调负责VSMCs向骨样细胞转分化的基因。
•它可以激活NLRP3炎症小体和上调NF-κB,这些都与IL-1β的转录有关。
•TMAO与主要不良心血管事件的发生有关。
•它可以加速肾功能障碍的进展,影响肾小管间质纤维化和胶原沉积的发展。
•TMAO与高血压风险增加、不良心血管事件和全因死亡率有关。
铁死亡
铁死亡是一种新型的铁依赖性、脂质过氧化驱动的细胞死亡形式。
肠道菌群与多种器官、组织和疾病中的铁死亡有关
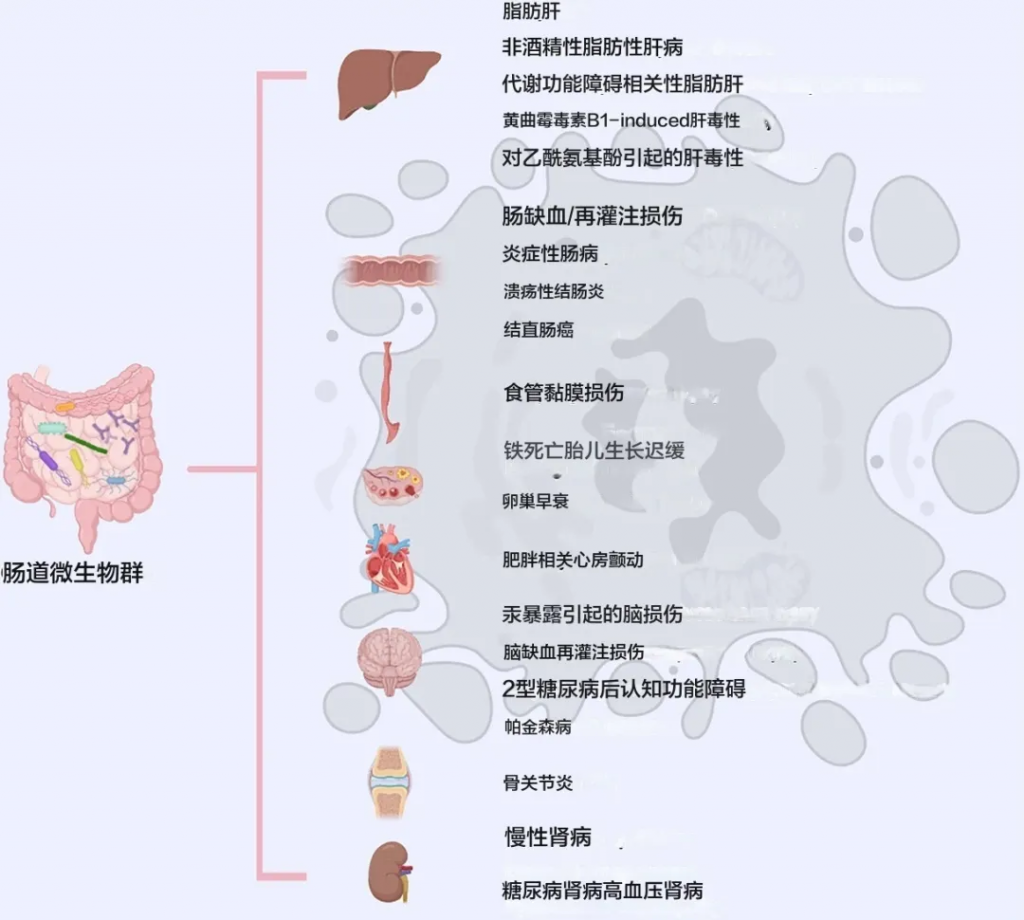
doi.org/10.1038/s41420-024-02000-8
铁死亡影响慢性肾病的方式主要如下:
○ 氧化应激和炎症反应:
铁死亡导致活性氧(ROS)积累,这会加剧氧化应激,进而引发炎症反应,对肾脏细胞造成损伤。
注:谷氨酰胺和支链氨基酸的代谢与铁死亡密切相关,它们可以通过不同机制影响谷胱甘肽合成、ROS生成和铁死亡过程。
○ 脂质代谢紊乱和脂质积聚:
铁死亡涉及多不饱和脂肪酸(PUFAs)的过氧化,这些物质在脂质代谢中起着重要作用。慢性肾病与脂质代谢紊乱和脂质积聚有关。脂质的积聚可激活先天免疫系统,促进炎症纤维化,引发线粒体和肾细胞损伤,并驱动慢性肾病进展。
○ 肾脏细胞损伤:
铁死亡导致的细胞死亡,如果发生在肾脏,将直接导致肾脏细胞的丧失,影响肾脏功能。
肠道菌群与铁死亡的关系主要表现在:
○ 代谢物的影响:
肠道菌群通过其代谢产物,如短链脂肪酸、胆汁酸、色氨酸代谢物等,影响宿主的代谢平衡和免疫状态,进而可能影响铁死亡的过程。
肠道菌群代谢物,如丁酸盐通过Nrf2/GPX4信号通路改善溃疡性结肠炎中的铁死亡,保护肠道粘膜屏障的完整性。
○ 免疫调节:
肠道菌群在维持宿主免疫平衡中起着关键作用。肠道菌群失衡可能导致系统性炎症,影响铁死亡的调节。
○ 铁代谢调节:
肠道菌群参与宿主肠道中铁的吸收和储存,影响铁代谢,进而可能影响铁死亡。
与铁死亡相关的肠道菌群有:
通过球形红杆菌(Rhodobacter sphaeroides)发酵获得的富含辅酶Q10的南瓜汁不仅具有抗氧化能力,尤其是铁离子还原抗氧化能力,而且还能调节哺乳动物的肠道菌群,保护肠道屏障。
总的来说,铁死亡在慢性肾病中的作用和肠道菌群的联系是一个复杂的过程,涉及多种代谢途径和细胞信号传导机制。慢性肾病患者应多食用富含膳食纤维的食物,避免食用高胆碱食物,如 L-肉碱和磷脂酰胆碱,因为肠道菌群代谢胆碱产生的三甲胺-N-氧化物会促进慢性肾病进展和死亡风险。
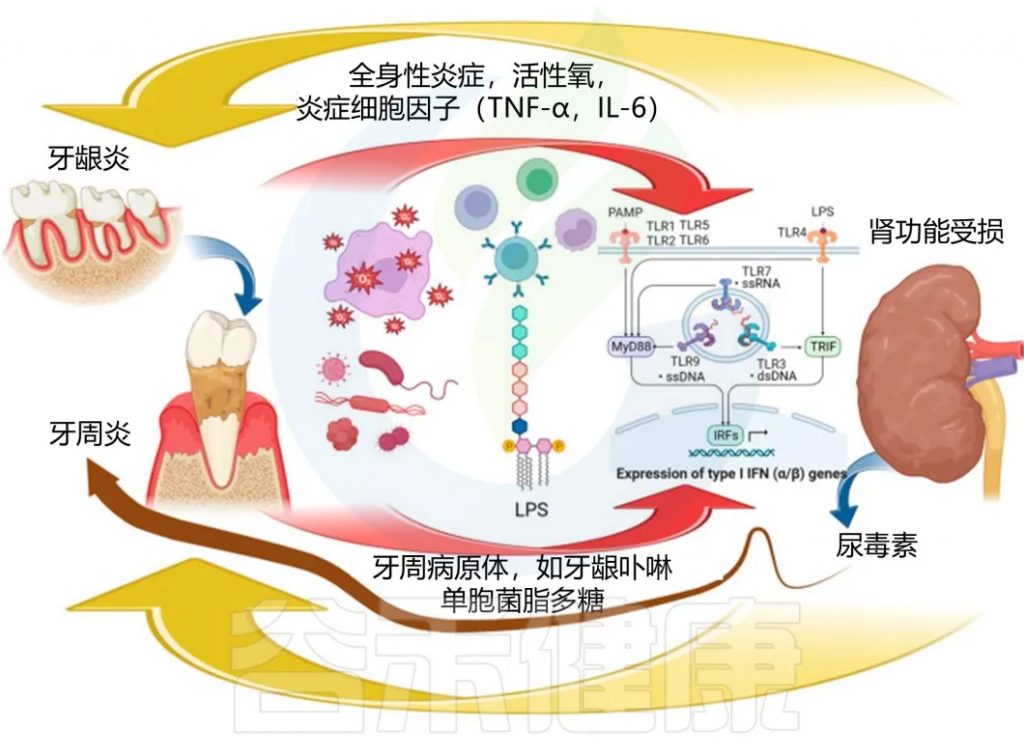
流行病学研究表明,慢性肾病患者很容易患上多种口腔疾病。
肾功能障碍会导致血清和唾液中尿素浓度升高,导致患者(尤其是晚期慢性肾病患者)因尿毒症而反复出现口臭。在口腔中,过量的尿素会被尿素酶阳性的口腔微生物群转化为氨,从而减少唾液流量并导致口干,这通常在终末期肾衰竭患者中常见。
慢性肾病患者在摄入食物后常常会感觉到不舒服的金属味道,这会导致食欲下降、营养摄入不良和蛋白质能量消耗综合征。
除了这些机制之外,其他因素,包括唾液 pH 值改变、口腔卫生不良、菌群失调、使用多种药物以及免疫反应改变,都可能大大增加患牙周病的风险。
某些因素同时增加了慢性肾病和牙周疾病的风险,如糖尿病、高血压、吸烟等。这些共同的风险因素进一步强化了两种疾病之间的联系。
两者之间也存在很大关联,慢性肾病患者的全身状况可能会加剧牙周炎症,而牙周炎症通过促进全身性炎症反应,又可能进一步损害肾脏健康。
慢性肾病对牙周疾病的影响
慢性肾病的营养不良状况、代谢性酸中毒、氧化应激、低度炎症会对口腔健康和牙周产生影响。
a) 免疫功能下降:
慢性肾病患者的免疫系统通常受到抑制,这使得他们更容易受到口腔感染和牙周疾病的影响。
b) 尿素浓度升高:
慢性肾病晚期患者体内尿素浓度升高,导致唾液中尿素浓度也随之升高。口腔中的细菌将尿素转化为氨,这可能改变口腔pH值,影响口腔微生态平衡。
c) 矿物质代谢紊乱:
慢性肾病患者常见钙磷代谢紊乱,这可能影响牙齿和牙槽骨的健康,增加牙周疾病的风险。
d) 药物副作用:
一些用于治疗慢性肾病的药物可能有口腔副作用,如口干,增加了口腔感染的风险。
e) 营养不良:
慢性肾病患者常见的营养不良状态可能影响口腔组织的修复能力,加重牙周疾病。
慢性肾病与牙周炎的直接关联机制
doi:10.3390/biomedicines11113033
牙周疾病对慢性肾病的影响
a) 系统性炎症:
牙周疾病是一种慢性炎症性疾病,可导致口腔内细菌和炎症因子进入血液循环。这些炎症因子(如IL-6、TNF-α、CRP、IL-8、IL-1β)可以引起全身性的低度炎症反应,进而影响肾脏功能。
b) 内皮功能障碍:
牙周病原体可以附着并侵入冠状动脉内皮细胞,导致动脉粥样硬化和血管功能障碍。这种机制同样可能影响肾脏血管,导致肾功能下降。
c) 氧化应激:
牙周疾病会增加体内的氧化应激水平,这可能导致肾脏组织的损伤和纤维化。
d)基质金属蛋白酶
基质金属蛋白酶(MMP)是一组参与组织修复和细胞凋亡的酶,它们在牙周炎症期间上调,并在肾脏中参与调节炎症反应和慢性纤维化,牙周病诱导的全身性MMP过度表达可能导致肾脏损害。
e) 微生物群相关:
菌群失调:如牙龈卟啉单胞菌、T. denticola, S. noxia, A.actinomycetemcomitans, V. parvula,导致IgG水平升高,这与肾功能受损有关。
菌群易位:口腔中的致病菌可能通过血液循环到达肾脏,直接或间接地影响肾脏功能。
口腔-肠道-肾脏轴的概念为我们提供了一个全新的视角,来理解慢性肾病与牙周病之间的联系。口腔中的微生物可以通过血液循环到达肠道,影响肠道微生物群的平衡,进而通过肠道-肾脏轴影响肾脏健康。
口腔-肠道-肾脏轴是一个复杂的动态系统,在慢性肾脏病中发挥着重要作用。这个系统的各个组成部分之间存在着密切的相互作用,形成了一个复杂的网络。以下是这个系统如何动态互作的详细解释:
牙周病与慢性肾病之间的间接关联机制
口腔-肠-肾轴
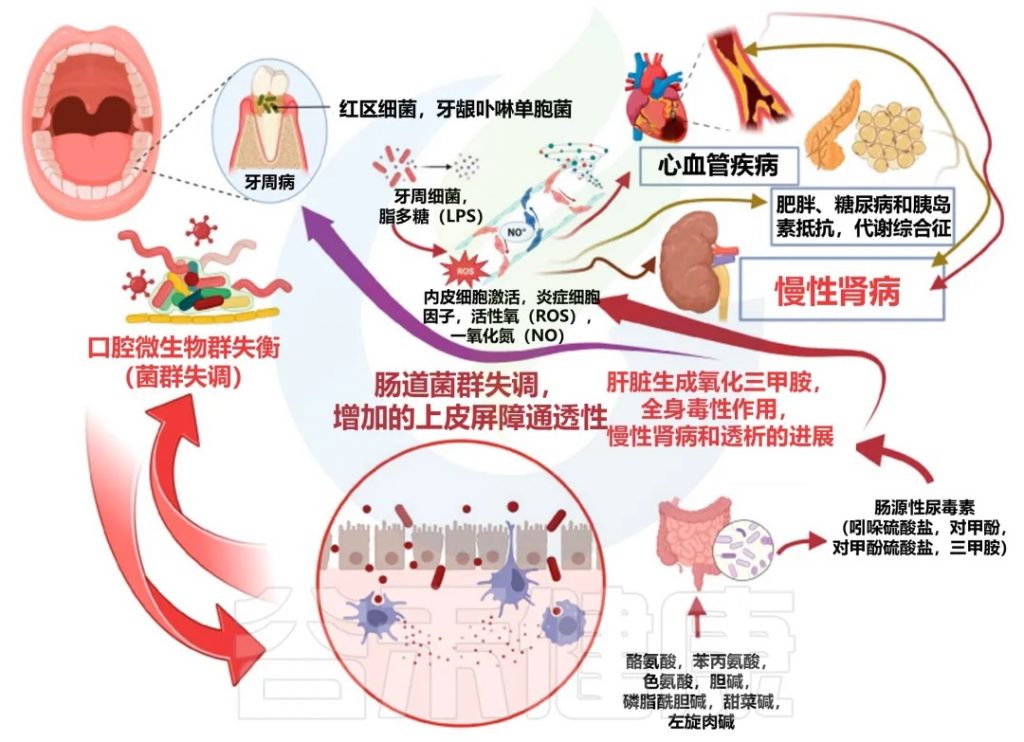
doi:10.3390/biomedicines11113033
口腔 ⇋ 肠道
● 口腔微生物群落影响肠道微生物组成
口腔中的细菌可以随唾液进入消化道,影响肠道微生物的组成。特别是在牙周病患者中,口腔病原体可能会定植在肠道中。
● 共同的炎症通路
口腔和肠道炎症可能共享一些共同的炎症通路,如NF-κB和NLRP3炎症小体的激活。
● 细菌转位
口腔病原体可能通过肠道进入血液循环,影响全身健康。
肠道 ⇋ 肾脏
● 肠道微生物代谢物影响肾功能
肠道微生物产生的代谢物,如短链脂肪酸、三甲胺N-氧化物(TMAO)等,可直接影响肾功能。
● 尿毒症毒素的产生和吸收
肠道微生物参与尿毒症毒素的产生,而这些毒素会进一步损害肾功能。
● 肠道通透性增加
慢性肾病可导致肠道屏障功能受损,增加细菌和内毒素的转位,加重全身炎症。
肾脏 ⇋ 口腔
● 尿毒症对口腔健康的影响
慢性肾病患者体内尿素浓度升高,可导致口腔pH值改变,影响口腔微生物环境。
● 免疫功能改变
慢性肾病导致的免疫功能障碍可能增加口腔感染的风险。
● 矿物质代谢紊乱
慢性肾病患者常见的钙磷代谢紊乱可能影响牙齿和骨骼健康。
牙周治疗
牙周治疗通过改善 eGFR 和肌酐水平,并降低炎症标志物(如 IL-6、CRP、ROS)对终末期肾病患者的肾功能产生积极影响。
强化牙周治疗还与接受腹膜治疗和血液透析患者的营养参数和铁利用率改善有关。3 个月牙周治疗显著降低了慢性肾病患者的全身 TNF-α 水平和其他炎症参数(IL-6、hs-CRP 和正五聚蛋白-3)。
牙周治疗可改善 ESRD 腹膜透析患者的全身炎症、营养状况和促红细胞生成素反应性。
一项为期 6 个月的随机对照临床试验旨在评估非手术牙周治疗对ESRD患者临床反应和全身状态的影响,结果表明,牙周临床参数以及 IL-6、铁蛋白、白蛋白、肌酐、血尿素氮和转铁蛋白水平均有显著改善。
一项全国性队列研究报告称,非手术性牙周治疗(尤其洁牙)效果与慢性肾病患者进展至ESRD、重大不良心血管事件、感染和全因死亡风险降低显著相关。
口腔预防措施
预防措施中,改善口腔卫生与慢性肾病发病率下降相关。
一项回顾性纵向研究报告称,刷牙频率对eGFR下降或透析需求有积极影响。
一项初步研究作者指出,强化牙科预防可能是减少全身炎症并随后降低慢性肾病儿科患者过早发生心血管疾病的一种有前途的方法。
多项研究的结果,提醒临床医生和患者注意口腔健康在控制肾功能方面的关键作用。
糖尿病肾病 (DKD) 是 1 型和 2 型糖尿病的严重并发症,是终末期肾病的主要原因。
糖尿病肾病的患病率和发病率逐年增加,大约30%~40%糖尿病患者会患上糖尿病肾病。
这种慢性肾病的标志是:
除了传统代谢因素,即高血糖和高血压外,肠道菌群的变化被认为是新型的重要影响因素之一。
肾功能下降导致尿素、尿酸等代谢废物在血液中积聚。这些代谢废物通过肠道分泌入肠腔,改变肠道环境。肠道环境改变引起肠道菌群失调。
糖尿病肾病患者的肠道菌群与健康人有明显差异,双歧杆菌、乳酸杆菌等有益菌减少,肠杆菌等致病菌增加。
关于糖尿病肾病肠道菌群变化的研究

doi.org/10.3389/fcimb.2024.1359432
肠道菌群:辅助判别的生物标志物
糖尿病肾病患者肠道菌群组成发生显著变化,可作为临床鉴别诊断或活检确诊糖尿病肾病的生物标志物。对于有肾活检禁忌症的患者,肠道菌群检测可能是一个重要的替代方案。
在四川省经活检确诊的14例糖尿病肾病患者中,Prevotella_9属可准确区分糖尿病患者与健康对照,受试者AUC为 0.900。
大肠杆菌-志贺氏菌和Prevotella_9也可准确区分经活检确诊的糖尿病肾病患者与糖尿病患者,AUC为0.860,有助于诊断糖尿病肾病。
山西地区 35 例经穿刺活检确诊的糖尿病肾病患者研究发现结果存在差异,其中Flavonifractor(AUC=0.909)或Eisenbergiella(AUC=0.886)可准确鉴别糖尿病肾病与糖尿病患者,可能与南北地区及饮食习惯差异有关。
下列可有效区分糖尿病肾病患者与健康对照组:
• Colatridium sp. CAG_768 (AUC=0.941)
• Bacteroides propionicifaciens (AUC= 0.905)
• Colatridium sp. CAG_715(AUC=0.908)
多元线性回归分析显示,Fusobacterium varium、Pseudomonadales、Prevotella sp. 3 联合检测对 糖尿病肾病患者具有较高的鉴别价值。
这些结果提示,肠道菌群可能是诊断糖尿病肾病的有效方式。但目前研究表明,用于诊断糖尿病肾病的肠道菌群生物标志物在不同地区和种族之间存在差异。因此,还需要更多临床研究来探讨其应用价值。
氨基酸代谢:
N-乙酰天门冬氨酸、L-缬氨酸、甜菜碱、异亮氨酸、天门冬酰胺和L-蛋氨酸水平升高
在糖尿病肾病中,肾脏对氨基酸的重吸收和代谢能力下降,导致这些物质在血液中积累。
L-亮氨酸和异亮氨酸水平与肾小球滤过率快速下降显著相关
这两种分支链氨基酸的水平升高可能是肾功能下降的早期标志。它们可能参与胰岛素抵抗和炎症过程,加速肾功能恶化。
谷氨酰胺水平升高
谷氨酰胺是一种重要的氨基酸,参与多种代谢过程。其水平升高可能反映了氮代谢的紊乱和肾脏对氨的处理能力下降。
脂肪酸代谢
硬脂酸水平升高
硬脂酸是一种饱和脂肪酸,其水平升高可能与脂质代谢紊乱和胰岛素抵抗有关。
亚油酸水平降低
亚油酸是一种必需脂肪酸,具有抗炎作用。其水平降低可能加剧糖尿病肾病相关的炎症反应。
其他代谢物:
短链脂肪酸(SCFAs):
尿毒症毒素:
苯硫酸盐可以直接损害肾脏,增加氧化应激,促进肾脏纤维化,从而加速糖尿病肾病的进展。
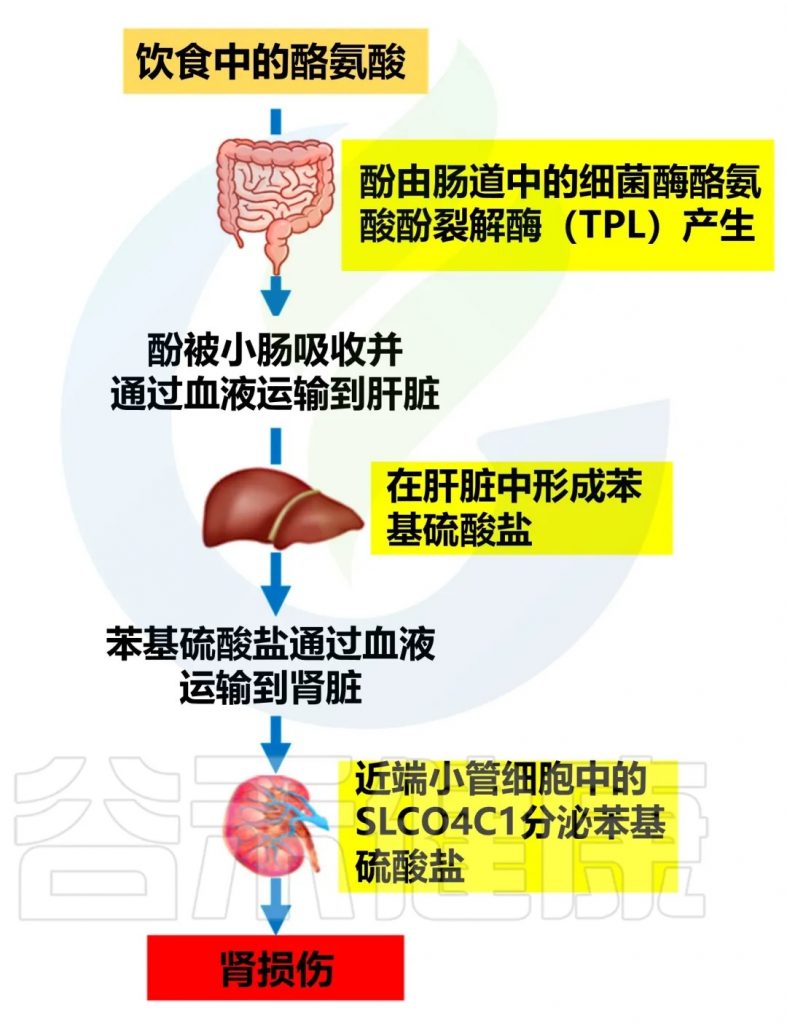
doi.org/10.3390/biom14091153
减轻苯硫酸盐对肾脏有害影响的策略:
慢性肾病相关认知障碍和抑郁风险
慢性肾病患者中16-38%存在认知障碍,肾功能与认知障碍和痴呆的发展相关。
肠道微生物失调和尿毒症毒素积累确实会影响血脑屏障(BBB)完整性,可能导致认知功能障碍。同时,慢性肾病患者更容易出现抑郁和认知障碍,这与肠道微生物改变和炎症有关。
肠道微生物失调和尿毒症毒素对血脑屏障的影响
认知功能障碍与肠道微生物和尿毒症毒素的关系
慢性肾病患者抑郁和认知障碍与肠道微生物和炎症的关系
总之,肠道微生物失调和尿毒症毒素积累通过影响血脑屏障完整性、改变神经递质和神经营养因子水平、促进炎症和氧化应激等多种机制,增加了慢性肾病患者出现认知功能障碍和抑郁的风险。这强调了维持健康肠道菌群和控制尿毒症毒素水平,对慢性肾病患者神经健康的重要性。
慢性肾脏病患者中肌肉减少症的发病机制是多方面的。
慢性炎症
首先,慢性炎症状态是导致肌肉减少症的一个重要因素。在慢性肾病患者中,系统性炎症反应指数(SIRI)与疾病的发生呈正相关,炎症因子如TNF-α、IL-6、IL-8等水平升高,这些炎症因子可以促进肌肉蛋白的降解并抑制肌肉生长。
炎症介质参与了导致肌肉减少症发生的多种机制
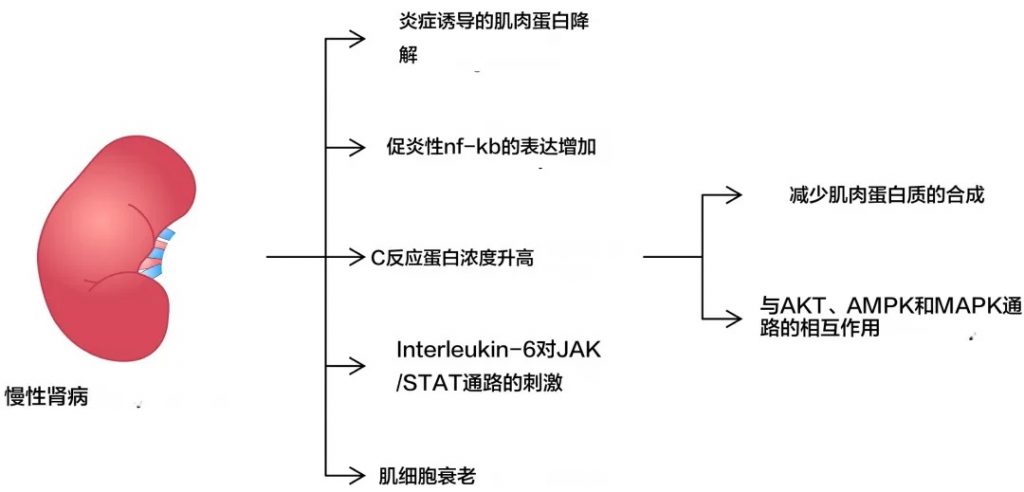
doi.org/10.3390/ijms25158474
代谢毒素积累
其次,代谢和激素调节障碍也与肌肉减少症的发展有关。慢性肾病患者常伴有代谢性酸中毒和尿毒症毒素的积累,这些因素可以导致胰岛素抵抗、线粒体功能下降,进而影响肌肉功能和肌肉质量的维持。
肠道菌群失调
慢性肾病患者的肠道菌群失调也是一个不可忽视的因素。研究发现,慢性肾病患者的肠道菌群组成发生了显著变化,如拟杆菌门和变形菌门的增加,厚壁菌门的减少。
肠道失调与肌少症之间潜在联系
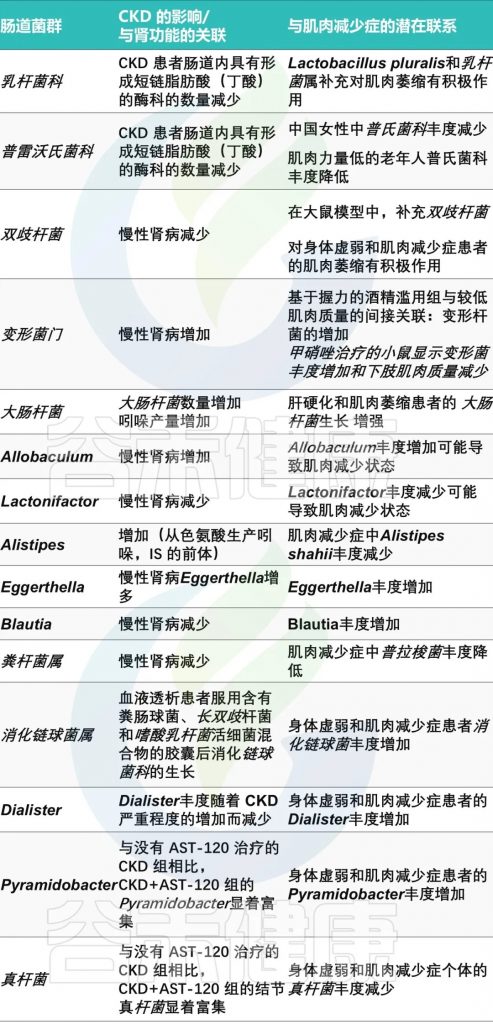
doi.org/10.3390/ijms25158474
慢性肾病状态减少了乳酸杆菌的存在,这与抑制其与肌肉健康相关的有益作用有关:
doi.org/10.3390/ijms25158474
代谢产物:硫酸吲哚酚和色氨酸
肠道菌群代谢产物硫酸吲哚酚(IS)在慢性肾病和肌肉减少症中起重要作用。
IS通过以下机制影响健康:
IS抑制肠上皮细胞的线粒体自噬,损害肠道屏障,可能通过IRF1-DRP1轴影响线粒体功能。
IS作为AhR的激动剂,激活AhR/NF-κB通路,促进炎症和纤维化,这与慢性肾病和肌肉减少症的发展有关。
IS在肌肉中激活NRF2,过度激活抗氧化反应,导致TCA循环减缓和ATP产生不足,进而引起肌肉无力和萎缩。
肠道微生物群产生的另一种代谢物是色氨酸,一种具有吲哚结构的氨基酸,色氨酸可以作为 AhR 的配体,AhR激活增加会导致线粒体损伤,从而导致肌肉损伤和萎缩。
miRNA表达异常
微小RNA(miRNA)的异常表达也参与了慢性肾病相关肌肉减少症的发病机制。例如,miR-29a和miR-29b的表达降低与肌肉减少症有关,它们通过影响YY1蛋白的表达来调节肌肉细胞的分化和凋亡。
总的来说,慢性肾脏病相关肌肉减少症的发病机制涉及炎症、代谢紊乱、肠道菌群失调和miRNA表达异常等多个方面。针对这些机制的治疗策略,如抗炎治疗、调节代谢、改善肠道菌群和miRNA治疗等,可能是未来治疗慢性肾病相关肌肉减少症的潜在方法。
泌尿系统结石是全球主要的健康问题,全球约 10-15% 的人一生中至少会经历一次。
泌尿系统结石的起因复杂且多方面,涉及遗传倾向、饮食习惯、液体摄入以及肥胖、糖尿病和高血压等潜在健康状况。结石的形成主要是由于尿液中含有钙、草酸盐、磷酸盐、尿酸盐、胱氨酸等矿物质过饱和,这个过程受尿液 pH 值以及结晶抑制剂和促进剂之间的平衡影响。
肠源性高草酸尿症
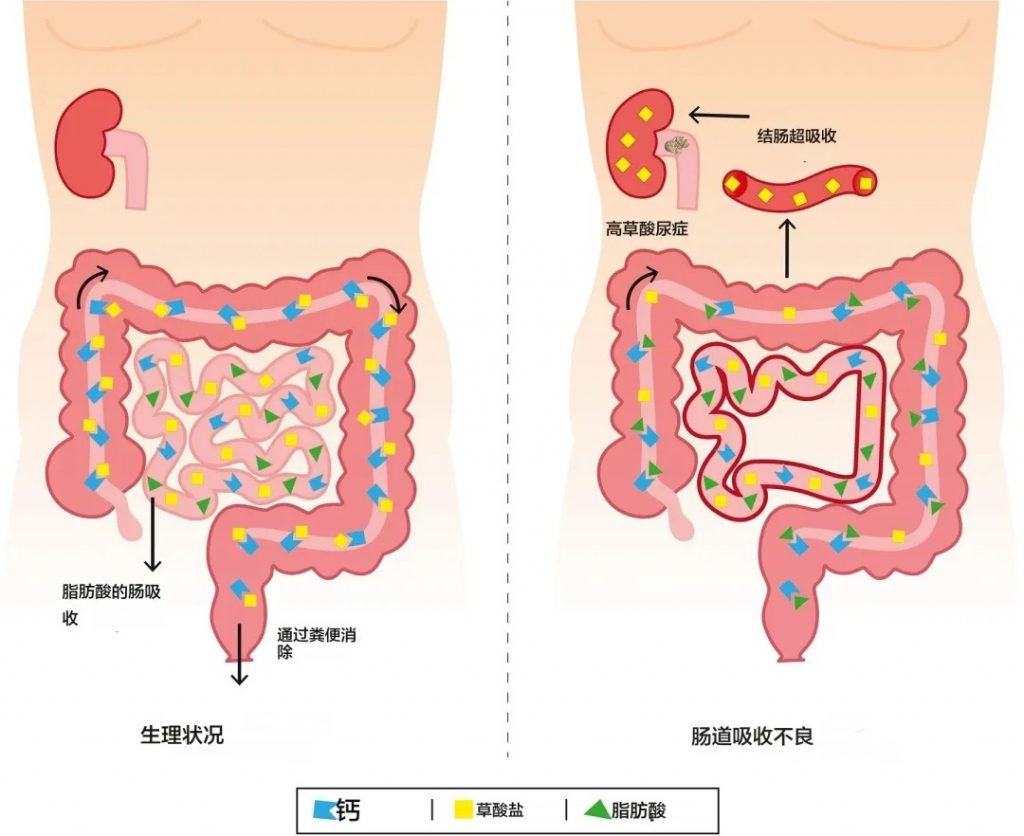
doi.org/10.1016/j.clinre.2024.102322
在生理条件下(左图),草酸的吸收受到限制,因为草酸钙复合物在结肠中不被吸收。在吸收不良状态下,例如短肠综合征或其他引起吸收不良的疾病(右图),结肠中的脂肪酸会导致钙螯合(皂化)。草酸不再与钙结合,可能会被结肠吸收,导致高草酸尿症。
新兴研究表明,肠道菌群失调可能通过调节尿液代谢物、肠道草酸代谢变化和全身炎症反应等机制,显著影响肾结石的风险和发展。
肠道菌群可降解草酸
特别是草酸杆菌属Oxalobacter等细菌,尤其是O. formigenes,以其降解草酸的能力而闻名,这种细菌通过表达草酸脱羧酶等与草酸代谢相关的蛋白来降解草酸,将其转化为甲酸盐和二氧化碳,从而减少肠道对草酸盐的吸收,并降低结石形成的风险。
Klimesova等(2015)和Mogna等(2014)的研究比较了不同菌株降解草酸的效率。
Suryavanshi等(2016)发现肾结石患者肠道微生物群中的O. formigenes 减少,但其他草酸代谢酶表达增加。
动物实验显示给予草酸降解菌可降低血草酸水平和肾钙化风险,但对尿草酸无影响。
肠道菌群调节草酸转运蛋白表达
此外,肠道微生物群可以通过调节草酸转运蛋白的表达来影响草酸的吸收和分泌。例如,SLC26A3和SLC26A6等转运蛋白在肠道草酸的吸收和分泌中起着关键作用。肠道微生物群的组成变化可能会影响这些转运蛋白的表达和活性,进而影响草酸的代谢。
菌群产生的短链脂肪酸影响草酸转运蛋白表达
微生物产生的短链脂肪酸也可以影响草酸转运蛋白的表达,这可能是通过改变肠道环境的酸碱度或通过直接与宿主细胞的信号传导途径相互作用来实现的。
肠道菌群降低胆固醇
多项研究证实了脂肪吸收不良与高草酸尿症之间的关联。
Moreland等(2017)的研究发现,胃旁路手术后患者出现脂肪吸收不良和高草酸尿症。
Chambers等(2022)和de Martines等(2019)的研究表明,慢性胰腺炎和胰腺功能不全患者也存在脂肪吸收不良、高草酸尿症和肾脏草酸钙结晶沉积。
Agrawal等(2014)报告,减肥手术后患者在术后两个月就可能出现显著的尿液草酸水平升高。
肠道微生物群的其他作用也不容忽视。例如,某些微生物如双歧杆菌、乳杆菌属等可降低胆固醇,这可能间接降低草酸的吸收。
一项研究发现,肾结石患者的乳杆菌和双歧杆菌的丰度显著低于健康个体,这与肾结石的风险增加相关。
Bordoni等(2013)和Zanotti等(2015)发现口服双歧杆菌可降低小鼠血清LDL和总胆固醇水平。
Costabile等(2017)报告Lactiplantibacillus plantarum治疗可显著降低轻度高胆固醇血症患者的血清LDL和总胆固醇水平。
益生菌治疗肾结石的潜力与挑战
在治疗肾结石方面,益生菌显示出了潜力,但也面临着定植和存活的挑战。例如,Oxalobacter formigenes是一种有潜力的益生菌,它能够降低原发性高草酸尿症患者的血浆和尿液草酸水平,但这种效果在停止治疗后会消失。乳杆菌属和双歧杆菌属的某些菌株也显示出降低尿液草酸水平的能力。然而,这些益生菌在定植和存活方面面临挑战,需要进一步研究来解决这些问题。
其他方法可以参考:
系统性红斑狼疮 (SLE) 是一种众所周知的系统性自身免疫性疾病,以产生致病性自身抗体和免疫复合物为特征,从而导致多种器官和组织的损害。大约 50% 的系统性红斑狼疮患者会出现肾脏损害,其特征是血尿、蛋白尿、水肿或肾功能减退等症状,称为狼疮性肾炎 (LN) 。在亚洲,系统性红斑狼疮患者的狼疮性肾炎患病率为 33%-82%。
狼疮性肾炎的治疗主要依赖类固醇和非选择性免疫抑制药物,但仅有50%~70%的狼疮性肾炎患者可获得临床缓解,10%~20%的患者在初次诊断后5年内进展为终末期肾病,预后不佳提示狼疮性肾炎的发病机制和治疗仍是一个尚未解决的难题。
越来越多的证据表明,肠道菌群失调可能与狼疮性肾炎相关,因为它会导致免疫失调,而这是其潜在发病机制之一。
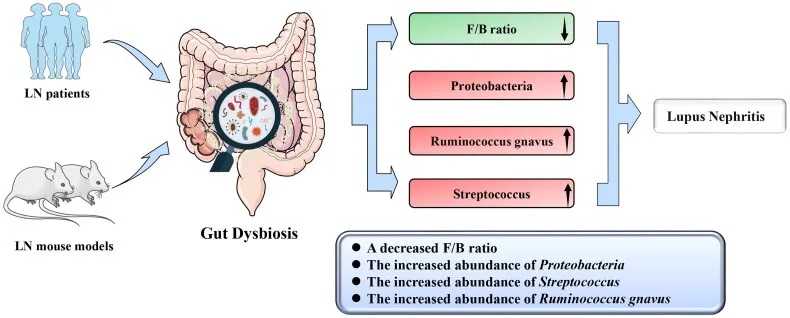
狼疮性肾炎患者肠道菌群特征
菌群多样性降低:
临床和动物研究中狼疮性肾炎肠道菌群变化
doi : 10.1080/0886022X.2023.2285877
菌群组成改变:
Firmicutes/Bacteroidetes (F/B)比例降低
这被认为是病理状态的一个标志。F/B 比率降低可导致Treg细胞和Th17细胞失衡,这可能会加剧先前存在的肠道炎症。狼疮患者 F/B 比率降低引起的菌群失调会导致 ILC3 功能障碍,而 ILC3 是屏障免疫的关键因素。
此外,F/B 比率失衡与系统性红斑狼疮患者血浆脂多糖水平升高相一致;从而增强 B 细胞的活化并促进小鼠模型中的系统性红斑狼疮进展。研究人员认为,多形拟杆菌可能是导致狼疮性肾炎患者 F/B 比降低的关键肠道菌群。
变形菌门、链球菌属、乳杆菌属↑↑
肠道菌群中变形菌增多可能是狼疮性肾炎发病机制中的一个重要因素,或许是基于自身免疫和炎症反应。
变形菌可能在以下两个方面对狼疮性肾炎的发病机制产生重大影响:
首先,从变形菌门成员大肠杆菌中获得的LPS可通过 TLR4–NF-κB和TLR4–p38MAPK通路刺激IL-6的产生,从而引发炎症反应。巧合的是,动物研究表明 IL-6在促进狼疮性肾炎中起着重要作用。
其次,变形菌门成员肠杆菌科的丰度与T细胞有关,而 T 细胞在免疫反应中至关重要。这表明,肠杆菌科丰度的变化可能促使狼疮性肾炎的发病机制和进展。
因此,变形菌的增加可作为肠道菌群改变的特异性标志,并可能通过调节IL-6和T细胞的水平促进狼疮性肾炎的发展。
链球菌属(Streptococcus)丰度增加,可能通过增强自身免疫反应促进狼疮性肾炎发展。
3项临床研究发现,狼疮性肾炎患者的链球菌属丰度高于健康对照组。
链球菌与韦荣球菌(Veillonella)的组合可增加IL-6、IL-10、IL-8和TNF-α的产生,同时抑制IL-12p70的产生。
肺炎链球菌的多糖与抗dsDNA抗体中的五肽具有相同的表位。
某些链球菌种可能通过抗原呈递触发特定CD4+ T细胞和初始B细胞刺激。
细菌同源物与人类自身抗原的分子模拟可能触发T和B淋巴细胞的交叉反应,激活自身免疫。
Ruminococcus gnavus、Lactobacillus reuteri ↑↑
Ruminococcus gnavus(简称RG)的丰度与狼疮疾病活动性和狼疮性肾炎相关,并与C3和C4补体水平呈负相关。
RG2菌株细胞壁脂聚糖具有抗原性,可诱导抗dsDNA抗体的产生。
这些抗体可与狼疮抗dsDNA抗体发生交叉反应,导致不适当的免疫反应。
活动性狼疮性肾炎患者血清中IgG抗RG2抗体浓度较高。
乳杆菌属和鼠李糖乳杆菌(L. reuteri)在狼疮性肾炎发病中存在争议
正面作用:
乳酸杆菌治疗可以促进无炎症的肠道环境,增加血清IL-10水平,恢复肾脏内Treg和Th17细胞的平衡。
负面作用:
罗伊氏乳杆菌灌胃增加了脾脏肿大和浆细胞样树突状细胞(pDCs)在脾脏和派氏结积累,加剧了白细胞向肾脏募集。
罗伊氏乳杆菌单菌定植的小鼠显示pDCs在脾脏和肠系膜淋巴结中积累增加,狼疮性肾炎症状恶化。
这些矛盾的发现表明乳酸杆菌属和鼠李糖乳杆菌在狼疮性肾炎中可能扮演复杂的角色,其作用可能取决于具体的遗传或环境条件。
特定菌群链的富集:
从系统发育图中观察到,肠道分类链拟杆菌-拟杆菌-多形拟杆菌(Bacteroides thetaiotaomicron)在狼疮性肾炎中显著富集。
肠道菌群失调与狼疮性肾炎的可能机制
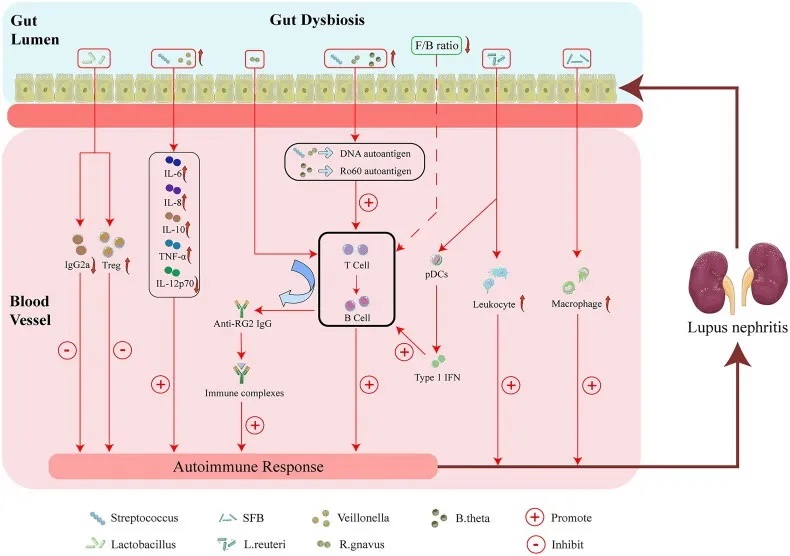
doi : 10.1080/0886022X.2023.2285877
特定微生物类群的改变可能通过以下四个因素促进狼疮性肾炎的发病和进展:
首先,特定微生物类群的改变可通过促进肾脏 M2 样巨噬细胞浸润和白细胞募集而诱发狼疮性肾炎。
其次,肠道菌群可能通过增强自身免疫反应导致狼疮性肾炎。
第三,链球菌与韦荣球菌结合可增强自身免疫反应,包括通过增加IL-6、IL-8、IL-10、TNF-α水平,而 IL-12p70 降低可能会诱发狼疮性肾炎。
第四,特定微生物类群的改变可以增加Treg的丰度,而IgG2a沉积减少可能会减轻狼疮性肾炎。
细菌代谢物:短链脂肪酸的作用
短链脂肪酸是最常被提及的细菌代谢物,对许多肾病具有有益作用。短链脂肪酸产生的变化与 系统性红斑狼疮患者的肠道菌群失调有关,而粪便 短链脂肪酸水平的升高与 F/B 比降低有关。SLE 动物模型显示,补充 SCFA 可以减轻狼疮表型。
此外,短链脂肪酸可能抑制B细胞活化诱导的胞苷脱氨酶和 Blimp1 的表达,限制浆细胞和系统性类型转换自身抗体的分化,并防止 IgG1/IgG2a 在肾脏中的沉积。
基于菌群对狼疮性肾炎的干预措施
通过饮食、益生菌和粪菌移植 (FMT) 成功治疗狼疮性肾炎可能为理解狼疮性肾炎与肠道菌群之间的关系提供新的证据。
首先,营养干预可以改善小鼠模型中的狼疮性肾炎。
其次,益生菌疗法是狼疮性肾炎的另一种治疗选择。口服脆弱拟杆菌可有效缓解狼疮性肾炎。该机制包括降低狼疮性肾炎患者的自身抗体水平、更新 B 淋巴细胞的免疫反应、缓解肠道炎症以及恢复狼疮性肾炎小鼠模型中 Treg 和 Th17 细胞的平衡。
第三,目前,FMT在治疗系统性红斑狼疮和 CKD 方面的安全性和有效性已得到证实。在系统性红斑狼疮治疗中,经过 12 周的 FMT 干预后,血清抗 dsDNA 抗体水平、炎症相关肠道微生物群和外周血中 IL-6 水平降低,同时产生 SCFA 的菌群和 SCFA 的产量增加。
总的来说,F/B 比值降低可能是狼疮性肾炎患者肠道菌群的一个特殊标志,这与 IFN-γ 水平降低、Treg 和 Th17 细胞失衡以及 B 细胞激活有关。变形菌门丰度增加,这可能与 IL-6 和 T 细胞水平有关,链球菌属和R. gnavus的富集也可能通过增强自身免疫和分子模拟在狼疮性肾炎的发病机制中发挥作用。这些发现有助于更好地了解肠道菌群如何影响狼疮性肾炎及其在预防和治疗狼疮性肾炎中未被认识到的作用。
慢性肾病患者更易发生抗菌药物相关神经毒性,主要表现为抗生素相关性脑(AAE)。
慢性肾病是抗生素相关性脑的已知风险因素。AAE被认为是慢性肾病患者中最常见的抗生素引起的不良反应之一。
在接受静脉抗生素治疗的住院终末期肾病患者中,AAE的总体患病率估计为4.4%,但可能被低估。
慢性肾病患者更容易发生抗生素相关神经毒性的原因包括:
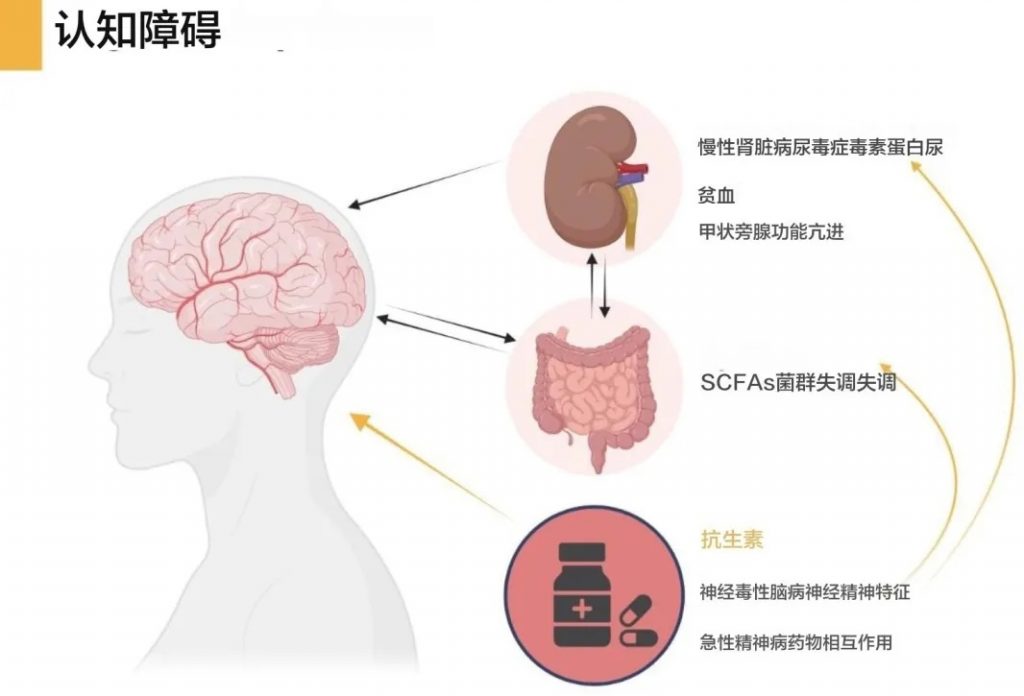
doi.org/10.1093/ckj/sfae174
以下列举的是一些CKD患者可能会用到的抗生素药物及其带来的影响。
◆β-内酰胺类抗生素(如青霉素、头孢菌素和碳青霉烯类)
◆ 氟喹诺酮类(如环丙沙星、左氧氟沙星、莫西沙星)
◆ 氨基糖苷类(如庆大霉素和妥布霉素)
氨基糖苷类抗生素以其强大的杀菌作用著称,但其肾毒性是导致CKD患者使用时的主要顾虑,其毒性作用与药物的血药浓度和使用时间密切相关。
◆ 大环内酯类(如红霉素和阿奇霉素)
大环内酯类抗生素的肾毒性相对较低,但部分药物如红霉素可能通过与肾脏药物转运体相互作用,影响其他药物的排泄,从而间接影响肾功能。
◆ 磺胺类(如磺胺甲噁唑、磺胺嘧啶)
磺胺类抗生素在慢性肾病患者中使用需谨慎,这类药物可能引起晶体性肾尿,特别在尿液酸性时更易形成药物结晶,导致肾小管堵塞和急性肾损伤。
◆ 硝基咪唑类(如甲硝唑和替硝唑):
磺胺类抗生素在CKD患者中使用需谨慎,这类药物可能引起晶体性肾尿,特别在尿液酸性时更易形成药物结晶,导致肾小管堵塞和急性肾损伤。
◆ 糖肽类(如万古霉素)
◆ 多粘菌素类(如多粘菌素B和粘菌素)
◆ 抗结核药物(如异烟肼、利福平、吡嗪酰胺和乙胺丁醇)
症状通常在开始治疗后数天内出现,停药后可逆。但某些抗生素(如甲硝唑)症状出现和恢复可能需要更长时间。
对于血液透析患者,有报道显示血液透析可能有助于清除某些抗生素并改善症状。
CKD患者使用抗菌药物时应进行治疗药物监测,调整剂量
对于慢性肾病(CKD)患者使用抗菌药物时,进行治疗药物监测(TDM)和剂量调整是非常重要的。
剂量调整原则:
监测频率:
总之,对慢性肾病患者使用抗菌药物时,进行治疗药物监测和适当的剂量调整是至关重要的。这不仅可以确保治疗的有效性,还可以显著降低药物不良反应的风险,特别是神经毒性。医生应熟悉治疗药物监测的原则和方法,并根据患者个体情况制定个性化的给药方案。
饮 食
选择健康的饮食方式
✘ 红肉摄入增加和人工甜味剂使用改变了肠道微生物组成,增加了尿毒症毒素的产生。
✘ 富含胆碱和 L-claritin(TAMO 的前体)的食物(如蛋黄、肾脏、肝脏、牛奶和肉类)可能会增加尿毒症毒素并降低肾小球滤过率,尽量避免。
✔ 一系列饮食指南已被研究作为 CKD 的潜在治疗策略,包括地中海饮食(富含蔬菜、坚果、豆类、水果和全谷物)、植物性饮食和低蛋白饮食(LPD;减少饮食蛋白质摄入但避免完全不摄入蛋白质)。
这些饮食旨在通过减少与西方饮食相关的高蛋白质消耗来减少细菌蛋白水解发酵。因此,预计炎症反应和尿毒症毒素的产生会减少,肾功能衰退可能会减缓,心血管风险可能会降低。
选择含盐和钠较少的食物
关键词:
无钠、无盐;或低盐、低钠;无盐或微咸
经常购买新鲜食物。超市或餐厅出售的许多预制或包装食品中都添加了钠(盐的一部分)。
自己烹饪食物,而不是吃预制食品、“快餐”食品、冷冻食品和钠含量较高的罐头食品。当你自己做饭时,你可以控制食物的成分。
使用香料、草药和不含钠的调味料代替盐。
检查食品包装上的营养成分标签上是否有钠。每日摄入量为 20% 或更高意味着食物中钠含量高。
尝试低钠版本的冷冻晚餐和其他方便食品。
食用前请用水冲洗罐装蔬菜、豆类、肉类和鱼类。
摄入适量、合适类型的食物
✔ 摄入超过需要的蛋白质可能会使你的肾脏更加辛苦地工作,因此,吃少量的蛋白质食物更合适。
✔ 防止脂肪在血管、心脏和肾脏中堆积:
烧烤、炙烤、烘培、烘烤或炒制食物,而不是油炸。
使用不粘烹饪喷雾或少量橄榄油代替黄油来烹饪。
食用前,应去掉肉中的脂肪,去掉家禽的皮。
尽量限制饱和脂肪和反式脂肪的摄入。阅读食品标签。
少吃这些食物
饱和脂肪、黄油、猪油、红肉、全脂牛奶、反式脂肪、商业烘焙食品,例如饼干和蛋糕、甜甜圈、炸薯条、氢化植物油、人造黄油等。
可以选择这些食物
单不饱和脂肪和多不饱和脂肪、菜籽油、坚果、麦片、橄榄油、三文鱼、香油。
✔ 随着肾功能下降,可能需要食用磷和钾含量较低的食物。
患有慢性肾病时,磷会在血液中积聚。血液中过多的磷会从骨骼中吸收钙,使骨骼变薄、变弱,更容易断裂。血液中磷含量过高还会导致皮肤瘙痒以及骨骼和关节疼痛。

来源:NIDDK
✔ 选择含适量钾的食物
当血钾水平过高或过低时,就会出现问题。受损的肾脏会导致血液中钾积聚,从而导致严重的心脏问题。
盐替代品的钾含量可能非常高,阅读成分标签。
食用前将罐装水果和蔬菜沥干。

来源:NIDDK
多酚和omega-3脂肪酸等植物成分具有抗氧化和抗炎作用,可能有助于缓解CKD。
✔ 多酚
多酚是植物中广泛存在的次生代谢物,主要包括黄酮类和酚酸类化合物。它们具有以下作用:
✔ Omega-3脂肪酸
Omega-3脂肪酸,特别是α-亚麻酸(ALA)、二十碳五烯酸(EPA)和二十二碳六烯酸(DHA),可以抗血栓形成、降低甘油三酯、还有抗炎作用。
研究表明,CKD患者体内Omega-3脂肪酸水平较低。补充Omega-3脂肪酸可能通过以下机制缓解CKD:
其他植物成分
这些植物成分通过抗氧化、抗炎、调节肠道菌群等多种机制,可能有助于减轻氧化应激和炎症,从而缓解慢性肾病的进展。然而,还需要更多的研究来确定这些成分在慢性肾病患者中的具体作用和适当剂量。
更多抗炎食物可以参考我们之前的文章:
增加维生素D的摄入量
维生素 D对肾脏的正常功能起着重要作用,
维生素D在肌肉生长、调节肌肉收缩和舒张周期、提供能量、维持葡萄糖平衡以及修复肌肉损伤方面起重要作用。
缺乏维生素 D 会导致进一步的肾功能障碍。对CKD患者而言,PTH水平升高和维生素D缺乏共同影响钙磷代谢,可能导致骨矿物质代谢紊乱。这些变化可能影响肌肉功能和生长,参与肌肉减少症的发生。
可以适当吃鲑鱼、鲭鱼、金枪鱼,乳制品和谷物产品等。多晒太阳补充。
其他饮食小Tips:
选择新鲜或冷冻的蔬菜而不是罐装的。
选择糙米或大麦而不是速食米饭或预先包装的调味谷物。
选择吃鸡胸肉等瘦肉蛋白质来源,而不是红肉。
在你的饮食中加入更多鱼。
用低盐鹰嘴豆泥蔬菜条代替薯条作为零食。
吃新鲜水果而不是烘焙食品。
中草药提取物
五层龙(Salacia chinensis)
研究:30名稳定的糖尿病CKD患者,每天两次服1000mg
结果:降低同型半胱氨酸和IL-6水平
西藏苦草(Hygrophila spinosa)
研究:分析植物化学成分,对多重耐药Pandoraea sputorum的杀菌活性,以及对HepG2和HEK 293细胞系的肝肾保护作用
结果:甲醇提取物对CCl4和顺铂诱导的细胞毒性显示肝肾保护作用
蕨麻 (Potentilla anserine L.)
研究:调查rosamultin对顺铂诱导的肾毒性的保护作用
结果:
降低血尿素氮(BUN)
提高HEK293细胞的体外活力
抑制顺铂诱导的细胞凋亡
改善肾功能障碍
减少肾小管损伤
洋甘草 (Glycyrrhiza glabra L.)
研究:分析植物化学成分,研究甘草根提取物对顺铂诱导的体内外肾毒性的保护作用
结果:通过抗氧化、抗炎和抗凋亡活性发挥肾脏保护作用
长梗黄花棯 (Sida cordata)
研究:评估乙酸乙酯提取物对CCL4诱导的肾毒性的抗氧化活性
结果:显示对CCl4诱导的大鼠肾毒性有保护作用,与其含有的抗氧化化合物有关
菝葜 (Smilax cordifolia) 和刺芹属植物(Eryngium carlinae)
研究:评估两种植物煎剂对大鼠肾功能障碍的影响
结果:
降低血清尿酸、白蛋白和尿素浓度
减少与肾小球硬化和肾小管纤维化相关蛋白的积累
增加肌酐清除率
增加促炎和保护性蛋白的浓度
中药大黄 (Rheum L.)
大黄是一种具有泻下作用的传统中药,常用于治疗CKD。
大黄与血管紧张素转换酶抑制剂(ACEI)或血管紧张素受体阻滞剂(ARB)具有协同作用。
大黄灌肠可以调节肠道菌群的丰度和组成,增加短链脂肪酸(SCFA)水平,改善肠道屏障损伤,减少炎症水平,改善肾脏病理,降低血肌酐水平。
这些研究结果表明,这些中草药提取物通过不同的机制(如抗氧化、抗炎、抗凋亡等)展现出肾脏保护作用,有望用于CKD的辅助治疗。然而,还需要进行更多的研究来确定这些植物提取物在CKD患者中的具体作用和适当剂量。
益生菌 和 益生元
给8名血液透析患者口服嗜酸乳杆菌,结果显示血清二甲胺(DMA)减少。
注:DMA这种物质会损害器官血管。
口服胶囊形式的长双歧杆菌可以降低血清IS水平。
干酪乳杆菌L. casei Zhang通过提高血清短链脂肪酸水平改善局部巨噬细胞和肾小管上皮细胞的炎症反应。研究还观察到在服用张氏乳杆菌后,CKD3-5期患者的肾功能下降速度变慢。
约翰逊乳杆菌(Lactobacillus johnsonii)治疗通过增加血清IAld水平来抑制AHR信号通路,从而减轻肾脏病变。
Lactiplantibacillus plantarum N-1在乙二醇诱导的肾结石大鼠模型中,降低了尿液草酸水平和肾脏草酸钙结晶沉积。
初步证据表明,富含益生元低聚果糖的菊粉(p-菊粉) 可促进双歧杆菌的生长、介导减肥、减轻炎症并改善代谢功能。
高膳食纤维摄入量与 CKD 患者炎症风险降低和死亡率降低相关。
益生菌对CKD患者的影响研究



doi: 10.1021/acs.jafc.4c00263
益生菌调节肠道菌群对 CKD 的缓解作用包括:
通过肠肾轴探索益生菌防治慢性肾脏病的机制
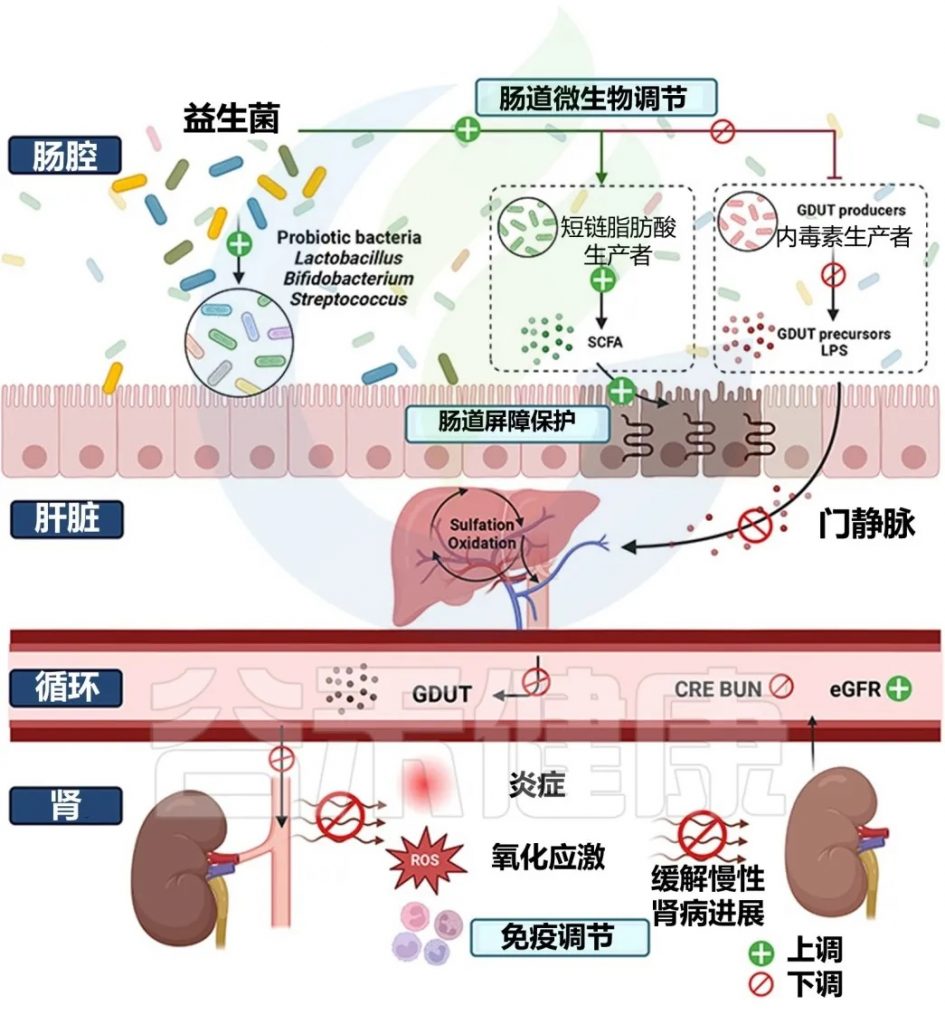
doi: 10.1021/acs.jafc.4c00263
由于饮食、疾病状况和个体年龄的广泛差异,需要充分考虑益生菌的治疗精确性。
粪 菌 移 植
多项 FMT 研究已证实,FMT 已成为一种有用工具,有助于证明肠道菌群的调节可能是 CKD 治疗干预措施发挥肾脏保护作用的关键机制之一。FMT 也已在临床病例研究中用于改善肾功能:
在一份临床病例报告中,从一名健康男性合格供体中提取的 FMT 通过内镜应用于治疗一名膜性肾病患者。经过两次治疗后,FMT 改善了相关的肾病综合征,改善了肾功能,总血清蛋白和白蛋白水平增加,血清肌酐和 24 小时尿蛋白降低。
两例 IgAN 患者通过内镜肠内插管定期接受 FMT 6-7 个月。FMT 治疗降低了两名患者的 24 小时尿蛋白,增加了血清白蛋白,并恢复了肠道菌群。
FMT 通过调节肠道菌群失调来改善肾功能并防止肾损伤,其机制包括恢复宿主的免疫力、调节肠道菌群代谢物、肾素-血管紧张素系统和改善肠道上皮屏障完整性。
FMT 治疗慢性肾病的潜在机制
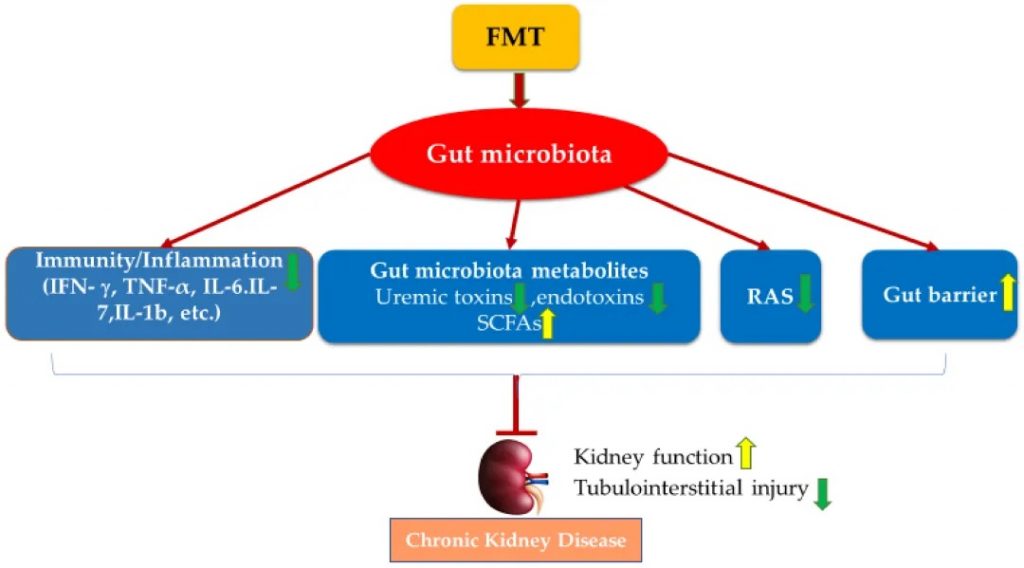
doi.org/10.3390/nu14122528
“一刀切”并不能解决所有问题。因此,特定疾病的供体选择和给药方法是 FMT 成功率的主要决定因素。
FMT 策略需要依靠人工智能、先进的生物信息学技术和机器学习算法的优势,例如多组学相关性分析。需要将肠道菌群检测、血浆、尿毒症毒素、粪便代谢物和肾功能的一组参数联系起来并进行系统分析,这将有力地支持研究人员了解FMT。
生 活 方 式
管理压力水平
重大生活事件带来的巨大压力,会增加急性肾损伤和慢性肾病进展的风险。可以通过听音乐,冥想,瑜伽等方式让身体放松。
定期锻炼
运动锻炼有助于降低血压和改善心脏健康,这两者都是肾脏疾病的风险因素。运动还有助于控制血糖水平和体重,这对糖尿病和肾病患者来说都是重要的因素。CDC建议,每周进行150分钟中等强度的锻炼。两天的肌肉强化活动。
慢性运动对CKD可能的治疗效果
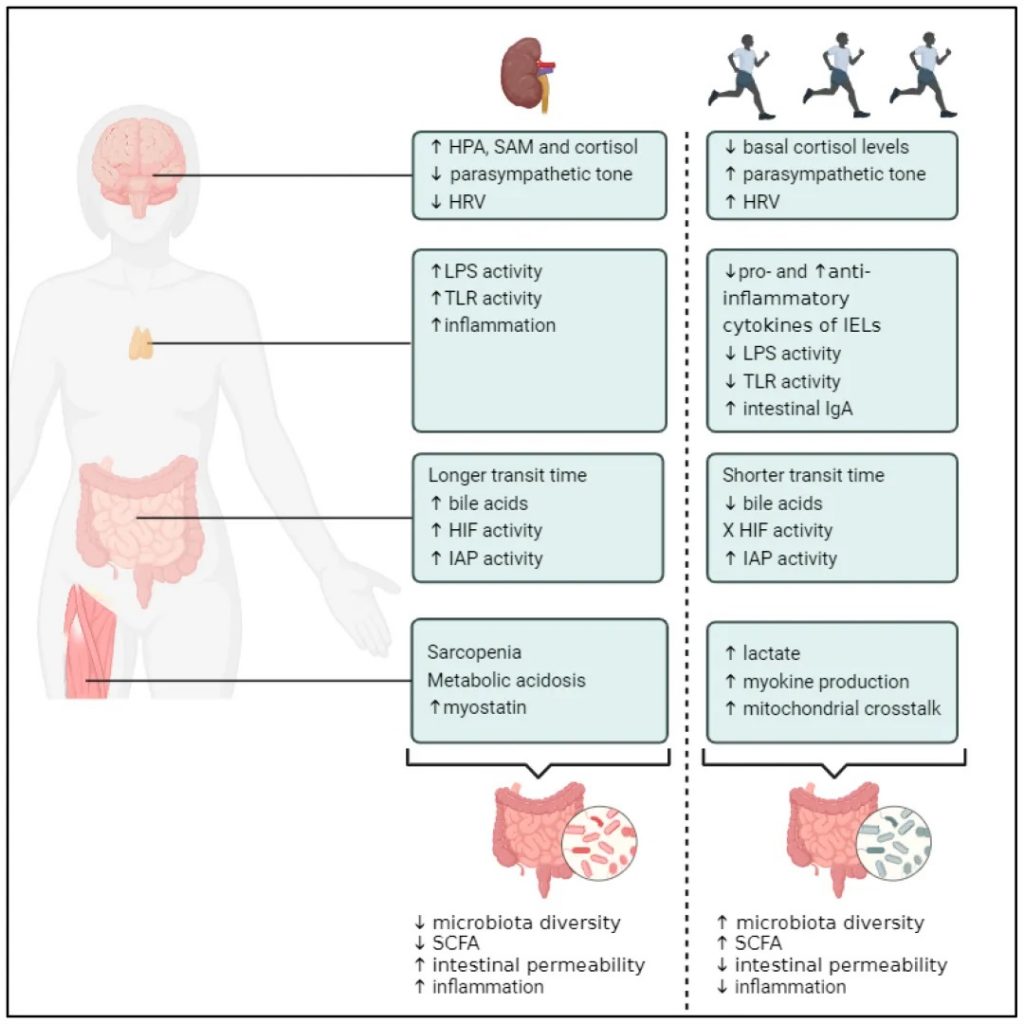
doi.org/10.3390/toxins16060242
保证充足的睡眠
一项研究发现,肾功能正常的中老年人如果每晚睡眠时间不足6小时,患慢性肾病的风险更高。
建立规律的睡眠时间表:每天在同一时间上床睡觉和起床,以调节身体的生物钟。
营造宁静的环境:让卧室安静、黑暗、凉爽。考虑使用遮光窗帘、耳塞或白噪音机。
睡前限制屏幕时间:睡前至少一小时关闭电子设备。
练习放松的睡前习惯:从事一些令人平静的活动,例如阅读、洗个温水澡或完成放松的伸展运动。
限制酒精摄入量
一些研究表明,少量至适量的酒精摄入可能对肾功能有益。
然而,他们并不建议不喝酒的人为了获得潜在的健康益处而开始饮酒。
其他研究警告称,饮酒可能会增加其他疾病的风险,例如通常与肾脏疾病有关的心血管疾病。
避免吸烟
香烟烟雾中含有有害物质,会减少流向肾脏的血液,从而降低肾脏正常工作的能力。
吸烟也是导致高血压和2 型糖尿病的风险因素,这两者都是导致肾脏疾病的主要原因。
据统计,慢性肾病(CKD)的发病率高达10%,其致残和死亡率在所有慢性疾病中增幅最快,防治面临严峻挑战。尽管通过尿液检查、血液检测、核素显像和肾脏超声等方法可以早期发现肾脏损害,但由于缺乏明显症状,许多人忽视定期检查。此外,这些检测的费用较高,增加了筛查的难度。
肾脏病种类繁多,病理类型复杂,临床症状相似,单靠临床判断诊断慢性肾病具有挑战性。患者最关心的是疾病是否会进展到尿毒症阶段。肾活检虽然是诊断的“金标准”,但在病情严重时,其适用性受到限制。治疗方案如激素和免疫抑制剂也存在副作用和风险。
近年来,研究发现慢性肾病与肠道菌群密切相关,调节肠道菌群有助于延缓疾病进展并改善生活质量。然而,个体差异、饮食模式和补充剂等因素使得研究结论难以统一。
为解决这些问题,建议扩大研究范围,利用人工智能和生物信息学技术评估肠道菌群在慢性肾病中的作用。通过肠道菌群检测技术评估干预效果,将更多基于菌群的干预手段推向临床实践。
随着研究的深入,肠道菌群有望成为慢性肾病治疗的重要方向,为患者带来新的希望。
主要参考文献
Mao ZH, Gao ZX, Pan SK, Liu DW, Liu ZS, Wu P. Ferroptosis: a potential bridge linking gut microbiota and chronic kidney disease. Cell Death Discov. 2024 May 15;10(1):234.
Krukowski H, Valkenburg S, Madella AM, Garssen J, van Bergenhenegouwen J, Overbeek SA, Huys GRB, Raes J, Glorieux G. Gut microbiome studies in CKD: opportunities, pitfalls and therapeutic potential. Nat Rev Nephrol. 2023 Feb;19(2):87-101.
Liu, H.; Diep, T.N.; Wang, Y.; Wang, Y.; Yan, L.-J. Diabetic Kidney Disease: Contribution of Phenyl Sulfate Derived from Dietary Tyrosine upon Gut Microbiota Catabolism. Biomolecules 2024, 14, 1153.
Bian, J.; Liebert, A.; Bicknell, B.; Chen, X.-M.; Huang, C.; Pollock, C.A. Faecal Microbiota Transplantation and Chronic Kidney Disease. Nutrients 2022, 14, 2528.
Młynarska, E.; Budny, E.; Saar, M.; Wojtanowska, E.; Jankowska, J.; Marciszuk, S.; Mazur, M.; Rysz, J.; Franczyk, B. Does the Composition of Gut Microbiota Affect Chronic Kidney Disease? Molecular Mechanisms Contributed to Decreasing Glomerular Filtration Rate. Int. J. Mol. Sci. 2024, 25, 10429.
Ramezani A, Raj DS. The gut microbiome, kidney disease, and targeted interventions. J Am Soc Nephrol. 2014 Apr;25(4):657-70.
Vandecruys, M.; De Smet, S.; De Beir, J.; Renier, M.; Leunis, S.; Van Criekinge, H.; Glorieux, G.; Raes, J.; Vanden Wyngaert, K.; Nagler, E.; et al. Revitalizing the Gut Microbiome in Chronic Kidney Disease: A Comprehensive Exploration of the Therapeutic Potential of Physical Activity. Toxins 2024, 16, 242.
Bakinowska E, Olejnik-Wojciechowska J, Kiełbowski K, Skoryk A, Pawlik A. Pathogenesis of Sarcopenia in Chronic Kidney Disease-The Role of Inflammation, Metabolic Dysregulation, Gut Dysbiosis, and microRNA. Int J Mol Sci. 2024 Aug 3;25(15):8474.
Joly PF. Pathophysiology and management of enteric hyperoxaluria. Clin Res Hepatol Gastroenterol. 2024 Jun;48(6):102359.
Tang Z, Yu S, Pan Y. The gut microbiome tango in the progression of chronic kidney disease and potential therapeutic strategies. J Transl Med. 2023 Oct 3;21(1):689.
Wilson S, Mone P, Jankauskas SS, Gambardella J, Santulli G. Chronic kidney disease: Definition, updated epidemiology, staging, and mechanisms of increased cardiovascular risk. J Clin Hypertens (Greenwich). 2021 Apr;23(4):831-834.
Huang HW, Chen MJ. Exploring the Preventive and Therapeutic Mechanisms of Probiotics in Chronic Kidney Disease through the Gut-Kidney Axis. J Agric Food Chem. 2024 Apr 17;72(15):8347-8364.
Liabeuf S, Hafez G, Pešić V, Spasovski G, Bobot M, Mačiulaitis R, Bumblyte IA, Ferreira AC, Farinha A, Malyszko J, Pépin M, Massy ZA, Unwin R, Capasso G, Mani LY; CONNECT Action (Cognitive Decline in Nephro-Neurology European Cooperative Target). Drugs with a negative impact on cognitive functions (part 3): antibacterial agents in patients with chronic kidney disease. Clin Kidney J. 2024 Jun 14;17(8):sfae174.
Liu X, Mo J, Yang X, Peng L, Zeng Y, Zheng Y, Song G. Causal relationship between gut microbiota and chronic renal failure: a two-sample Mendelian randomization study. Front Microbiol. 2024 Apr 3;15:1356478.
Tang Z, Yu S, Pan Y. The gut microbiome tango in the progression of chronic kidney disease and potential therapeutic strategies. J Transl Med. 2023 Oct 3;21(1):689.
Khan MA, Kassianos AJ, Hoy WE, Alam AK, Healy HG, Gobe GC. Promoting Plant-Based Therapies for Chronic Kidney Disease. J Evid Based Integr Med. 2022 Jan-Dec;27:2515690X221079688.

谷禾健康

编辑
大肠杆菌(E.coli)是埃希氏菌属的一个菌种,属于肠杆菌科。生活中经常有人将大肠杆菌和肠杆菌属或肠杆菌科混淆。
大肠杆菌(E.coli)是种,也叫大肠埃希氏菌,肠杆菌属细菌是肠道的正常菌群,有阴沟肠杆菌、产气肠杆菌、阪崎肠杆菌等。
肠杆菌科包括好几种重要的菌属,如:
埃希氏菌属
沙门氏菌属
志贺氏菌属
克雷伯氏菌属
变形杆菌属
耶尔森氏菌属
肠杆菌属
在分类学上,大肠杆菌、肠杆菌属和肠杆菌科都属于变形菌门的γ亚纲。大肠杆菌最为人所知的是,它是人类、其他温血动物和爬行动物正常肠道菌群中普遍存在的成员。
正常情况下,大肠杆菌作为无害的共生菌存在于盲肠和结肠的粘膜层中。这种革兰氏阴性、能运动的细菌已经非常成功地使其新陈代谢适应了这种营养生态位,在上千种其他细菌物种中脱颖而出。
大肠杆菌在出生后数小时内便在婴儿肠道中定殖,并成为人类肠道菌群中最丰富的兼性厌氧菌之一,它具备在不断变化的肠道环境中生长和应对哺乳动物宿主相互作用的能力。
大肠杆菌惊人的代谢和调节能力促进了其在不同生态位中的定植,以及在长期非生长条件下的生存。大肠杆菌的已知栖息地包括土壤、水、沉积物和食物。一些大肠杆菌菌株已经进化并适应了致病生活方式,并可导致不同的疾病病理。
多数大肠杆菌是无害的,可与人体共存,产生人体所必须的维生素B和K,但少数的大肠杆菌具有致病性。
根据感染部位,致病性大肠杆菌菌株可分为肠致病性大肠杆菌(IPEC)和肠外致病性大肠杆菌(ExPEC)。两者进一步细分为不同的致病型,定义为具有某些致病性状的单一物种的一组菌株。
致病型分类基于疾病的临床表现、所涉及的毒力因子(VF)和系统发育背景。
最突出的IPEC致病型是:
肠聚集性大肠杆菌(EAEC)
肠出血性大肠杆菌(EHEC)
肠侵袭性大肠杆菌(EIEC)
肠致病性大肠杆菌(EPEC)
肠产毒性大肠杆菌(ETEC)
弥漫性粘附性大肠杆菌(DAEC)
粘附性侵袭性大肠杆菌(AIEC)
最常见的ExPEC致病型:
泌尿道致病性大肠杆菌(UPEC)
脑膜炎相关大肠杆菌(MNEC)
败血症相关大肠杆菌(SEPEC)
禽类致病性大肠杆菌(APEC)
不同的生活方式使大肠杆菌成为研究宿主和细菌之间相互作用以及互利共生和致病性之间关系的良好候选对象。与此同时,基因和表型的多样性也妨碍了风险评估和菌株分型。
然而,明确区分肠外致病性大肠杆菌和共生性大肠杆菌并不容易,因为具有引起肠外感染能力的菌株是兼性病原体,属于许多健康个体的正常菌群。
比较其共生性和致病性大肠杆菌的系统发育、基因和表型特征。我们大量的检测实践和相关论文发现该菌肠外毒力和肠道适应性之间的界限可能变得模糊,因为适应性和竞争力的提高可能会促进大肠杆菌的肠道定植以及肠外感染。
本文,我们介绍大肠杆菌的生态属性,多样性和遗传变异以及致病性,风险因素感染症状和预防治疗。
▷ 革兰氏阴性、兼性厌氧菌“代表”
大肠杆菌是一种特别典型的革兰氏阴性菌,结构十分简单、有代表性。
作为兼性厌氧菌,大肠杆菌可以同时在有氧和无氧环境中生存。这种细菌还可以在许多不同的营养物质上迅速生长,并且几乎可以从任何人身上分离出来。
大肠杆菌通常只有一条染色体,比高等生物的基因组要小得多,并且基因密度高,没有内含子,很少有重复DNA,易于寻找和分析基因。
大肠杆菌是十分理想的生物遗传信息表达的宿主系统。在生命科学研究中,经常利用大肠杆菌繁殖快、结构简单等特点大量复制DNA、蛋白质等生物大分子以供研究或用于工业生产,比如我们熟知的胰岛素就是通过大肠杆菌发酵实现大批量生产的。
另外,大肠杆菌是单倍体,这意味着即使是隐性突变,也能够表现出突变的表型,同时细菌之间可以方便地进行遗传物质的交换,意味着可以很容易地向大肠杆菌内引入外源基因。大肠杆菌的这些特征便于对其进行遗传学研究。
▷ 亦正亦邪
大肠杆菌(E.coli) 是一类正常生活在健康人和动物肠道中的细菌。生活在胃肠道中的大肠杆菌通常不会伤害您,甚至可以帮助您消化食物。
大肠杆菌最初分离时被命名为芽孢杆菌,这个拉丁名词描述了它作为一种“常见的结肠细菌”的突出特性,这种细菌可以在多种基质中容易地培养。
大肠杆菌在肠道内起着什么作用呢?
-可以分解食物,帮助消化,提高人体的代谢能力;
-在适宜的温度酸度情况下,大肠杆菌能合成B族及K族维生素,为人体提供营养;
-还可以充当卫士,抑制其它致病菌的生长。
▷ 但是以下六种可导致肠道疾病
1.产肠毒素大肠杆菌(ETEC)
这种细菌会引起水样腹泻,在卫生条件较差的地区,食物和水中经常发现这种细菌。这种细菌是旅行者腹泻的最主要诱因。
2.肠致病性大肠杆菌(EPEC)
这种细菌主要引起儿童和婴儿水样腹泻,在卫生条件较差的地区,食物和水中经常发现这种细菌。这种细菌可能会在托儿所或日托中心引发疫情。
3.肠聚集性大肠杆菌(EAEC)
这种病毒会引起持续性急性腹泻,不伴有发烧和呕吐。发展中国家和发达国家均有发现。它也是旅行者腹泻的来源之一。
4.
肠侵袭性大肠杆菌(EIEC)
这与志贺氏菌有关,通常是由于食用受污染的蔬菜、未煮熟的肉类或饮用未经高温消毒的(生)牛奶而引起的。它会导致便血和带粘液的大便、腹部绞痛、呕吐、发烧和发冷。
5. 弥漫粘附大肠杆菌(DAEC)
这是一种鲜为人知的大肠杆菌菌株。它似乎主要影响学龄前儿童,并导致呕吐和腹泻。
6.肠出血性大肠杆菌(EHEC)
这也被称为产志贺毒素大肠杆菌(STEC)。它会产生一种名为志贺毒素的毒素,使您生病。这种毒素会损害您的肠道内壁。它通常存在于肉(加工过程中受到污染且未充分煮熟)、未经高温消毒的牛奶以及使用含有 EHEC/STEC 的粪肥施肥的蔬菜中。
一种特别严重的肠出血性大肠杆菌菌株,称为 O157:H7,会让人病得很重。它会引起腹部绞痛、呕吐和血性腹泻。
它是儿童急性肾衰竭的主要原因。它还会引起危及生命的症状,例如:成人肾衰竭、发烧、出血、困惑、癫痫。
如果你有任何这些症状,应该寻求紧急帮助。
▷ 科研界的“明星”
大肠杆菌是微生物界顶流,货真价实。
大肠杆菌(Escherichia coli),作为微生物界的“顶流”,在科研领域中占据着举足轻重的地位。其重要性不仅体现在数量庞大的研究文献中,也在于它在多个科学突破中的核心作用。
在全球知名的学术搜索引擎Google Scholar中,关于大肠杆菌的论文数量高达200多万篇,而在中国知网(CNKI)中,也有超过13万篇论文提到这一微生物。这些数据无疑证明了大肠杆菌在科研圈中的明星地位。
大肠杆菌曾是多项诺贝尔奖获奖研究的关键对象
乔舒亚·莱德伯格(Joshua Lederberg)通过研究大肠杆菌,揭示了基因如何携带和交流信息,并证明了遗传因子的重组现象,这一研究为他赢得了1958年的诺贝尔生理学或医学奖。
约翰·凯恩斯(John Cairns)则利用放射自显影技术,首次直接观察到大肠杆菌环状DNA的半保留复制机制,为DNA复制研究提供了重要的实验证据。
此外,方斯华·贾克柏(Francois Jacob)和贾克·莫诺(Jacques Monod)以大肠杆菌为研究对象,提出了操纵子学说,揭示了基因表达调控的机制,并因此获得1965年诺贝尔生理学或医学奖。
石野良纯(Yoshizumi Ishino)在分析大肠杆菌基因时发现了CRISPR序列,这一发现为后来的CRISPR/Cas9基因编辑技术奠定了基础。埃玛纽埃勒·沙尔庞捷(Emmanuelle Charpentier)和詹妮弗·A·杜德纳(Jennifer A. Doudna)在此基础上,完整阐释了CRISPR/Cas9技术,并因此被授予2020年诺贝尔化学奖。
如今,大肠杆菌的应用已超越传统的微生物学研究,广泛渗透到材料科学、能源开发、环境保护等多个领域。在材料科学中,大肠杆菌被用于验证新型抗菌材料的性能;在能源领域,它被用于开发生物能源新材料,推动可持续能源的发展。
此外,在上百年的生命健康科学研究中,大肠杆菌也可以说是居功至伟。大肠杆菌在人类微生物菌群和疾病研究中发挥重要作用,这些也扩大了其作为模式生物的价值。
▷ 适应性强,代谢广泛
作为兼性厌氧菌,大肠杆菌可以同时在有氧和无氧环境中生存。这种细菌还可以在许多不同的营养物质上迅速生长,并且几乎可以从任何人身上分离出来。
在有氧条件下,它通过有氧呼吸高效地生成能量,而在无氧条件下,它则通过发酵或厌氧呼吸继续生存。这种灵活性使得大肠杆菌能够在多变的环境中保持生存优势。
大肠杆菌的代谢广泛性还表现在其对多种营养物质的利用能力上。它能够利用葡萄糖、乳糖、甘露糖、果糖等多种糖类作为碳源进行生长。此外,大肠杆菌还可以代谢氨基酸、有机酸和脂肪酸等多种化合物。
这种代谢多样性也使大肠杆菌成为研究代谢途径和生物化学反应的理想模型生物。例如,科学家们通过研究大肠杆菌的代谢途径,揭示了糖酵解、三羧酸循环和氧化磷酸化等基本生物化学过程的机制。
大肠杆菌的适应能力还体现在其对环境压力的耐受性上。它能够在不同的温度、pH值和渗透压条件下生存,并对抗生素和重金属等环境毒素表现出一定的耐受性。这种适应能力使得大肠杆菌能够在自然界中广泛分布,并在人体肠道中稳定存在。
▷ 特点
耐热脂多糖(LPS)是大肠杆菌的主要细胞壁抗原。
大肠杆菌有4种抗原:H、O、K 和 F。
鞭毛抗原
·热和酒精不稳定蛋白
·存在于鞭毛上
·属特异性
·呈现单相
·已确认75种“H”抗原
O或体细胞抗原
·耐热,可耐煮沸2小时30分钟
·发生在外膜表面
·细胞壁的组成部分
·已确认173种“O”抗原
K或荚膜抗原
·热不稳定
·包膜中存在酸性多糖抗原
·煮沸可去除 K 抗原
·抑制吞噬作用
·已识别出103种“K”抗原
菌毛抗原
·热不稳定蛋白
·存在于菌毛中
·K88、K99抗原
营养琼脂上的大肠杆菌,其外形大、圆形、凸度低、呈灰色、白色、湿润、光滑且不透明。其有两种形态:光滑(S)形态和粗糙(R)形态。
生长温度:大肠杆菌为最适生长温度为37℃,生长范围为10℃~45℃左右。
代谢能力:大肠杆菌的生化代谢非常活跃。大肠杆菌可以发酵葡萄糖产酸、产气,还能发酵多种碳水化合物,也可以利用多种有机酸盐。
最值得注意的是,大肠杆菌对乳糖、过氧化氢酶和吲哚呈阳性,对氧化酶、尿素酶和柠檬酸呈阴性,可以产生醛和酸。尽管这些特性中的许多都具有低水平的多态性。
阳性反应:大肠杆菌能够分解乳糖(糖的一种),产生过氧化氢酶(帮助分解过氧化氢),以及产生吲哚(由色氨酸分解产生的一种化合物)。
阴性反应:大肠杆菌不产生氧化酶(参与氧化反应的酶),不产生尿素酶(分解尿素的酶),也不利用柠檬酸作为碳源。
多态性:虽然这些特性是大肠杆菌的典型特征,但在不同的大肠杆菌菌株中,这些特性可能会有一些小的变化。总之这些特性有助于科学家识别和区分大肠杆菌与其他细菌。
一个多世纪以来,大肠杆菌一直是重要的模式生物,用于阐明遗传学、进化、分子生物学和发病机制等关键方面。然而,由于区分细菌种类的特征和标准不断变化,定义哪些菌株真正属于这一物种并不容易,也不稳定。
此外,许多被指定为大肠杆菌的分离株在基因上与志贺氏菌菌株的关系比与其他大肠杆菌的关系更密切,这就造成了整个志贺氏菌属及其四个种都包含在大肠杆菌一个种中的情况。
▷ 大肠杆菌主要分为六个进化枝
对被认为涵盖整个物种多样性的大肠杆菌菌株进行系统发育分析,定义了六个主要进化枝(A、B1、B2、D、E和 F)和几个较稀有的进化枝。
然而,将集合扩大到包括来自其他动物和环境来源的菌株,产生了五个“隐秘”进化枝(称为CI至CV),它们与大肠杆菌的关系都比与其姊妹种埃希氏菌 (Escherichia fergusonii)的关系更密切。这五个未分类的进化枝的分类地位仍然不确定:它们无法基于表型特征与大肠杆菌区分开来,但它们在基因上存在差异,这导致有人提出,这些进化枝中至少有一些(例如,进化枝III+IV和进化枝V)可能代表不同的物种。
▷ 基因特征有助于区分致病性或共生性
显著的基因组可塑性是该物种表现出巨大变异性的关键。通过水平基因转移、基因丢失以及其他基因组修饰(如 DNA 重排和点突变)获取遗传信息可以不断改变基因组内容,从而改变某些生态位中个体变异的适应性和竞争力。
特定的基因亚群和特征与大肠杆菌菌株引起肠道或肠外疾病的可能性增加有关。根据基因组含量和表型特征,可以可靠地将肠道致病性大肠杆菌菌株与非致病性、共生性或肠外大肠杆菌病原体区分开来。
随着更多全基因组被整合到分析中,大肠杆菌的系统发育结构和进化关系变得更加精细,人们认识到越来越多的亚种群,并有迹象表明其中一些可能代表实际或初期物种。为了适应所有分类单元中不断增长的测序菌株数量,基因组分类数据库 (GTDB;gtdb.ecogenomic.org/) 建议应用全基因组身份阈值(类似于 ANI)来定义细菌物种。根据其衡量标准,目前归类为大肠杆菌的菌株将分为六个种:
E.coli
E.coli_E
Escherichia ruysiae
Escherichia marmotae
Escherichia sp001660175
Escherichia sp005843885
▷ 大肠杆菌对多种抗菌药物具有耐药性
大肠埃希氏菌是医院内感染监测中的重要病原菌之一,对多种抗菌药物如克林霉素、达托霉素、夫西地酸、利奈唑胺、利福平、大环内酯类、糖肽类和脂肽类具有天然耐药性。
在中国临床分离的菌株中,对氨苄西林的耐药率超过80%,对头孢菌素的耐药率在20%到60%之间不等,而对喹诺酮类抗生素(如左氧氟沙星和环丙沙星)的耐药率为50%到65%。
然而,值得注意的是,对碳青霉烯类抗生素的耐药率仍保持在极低水平(1%到2%)。
▷ 耐碳青霉烯大肠杆菌的风险因素
耐碳青霉烯的大肠埃希菌通常与医院获得性感染相关,尽管在社区中也偶有发生。其风险因素包括:既往多次或长期住院、入住ICU、接受过侵入性检查或治疗、近期手术史、血液肿瘤等免疫力低下、严重基础疾病以及多种抗菌药物的应用(如喹诺酮类、第三代或第四代头孢菌素和碳青霉烯类)。
这些菌株的耐药机制包括产生分解抗菌药物的酶、外膜孔蛋白变异影响药物作用、以及产生甲基化酶和修饰酶等。
▷ 异质性耐药现象
大肠埃希菌的异质性耐药现象也引起了关注。异质性耐药指的是细菌中的同源亚群对某种抗菌药物表现出不同的敏感性,这是敏感菌进化为耐药菌的中间阶段,临床上难以及时检出,常导致患者反复感染或抗菌药物治疗失败。
近年来,研究发现大肠埃希菌对甲硝唑、碳青霉烯类、粘菌素、磷霉素、替加环素和哌拉西林/他唑巴坦等药物表现出异质性耐药。尽管其机制尚未完全阐明,可能与耐药基因、外排泵基因的表达水平增加或活性增强、以及生物膜的形成有关。
即使是少量大肠杆菌,也有可能被感染。
▷ 感染原因包括:
1.肉
肉没有煮熟到足以杀死细菌。加工肉类时,有时动物肠道中的细菌会进入肉中。碎肉比其他类型的肉更容易出现这种情况,因为碎肉通常来自不止一只动物。
2.未经处理的牛奶
你喝的是未经巴氏消毒的牛奶,这种牛奶没有经过加热杀菌。大肠杆菌可能通过奶牛的乳房或挤奶设备进入牛奶中。
3.蔬菜和水果
你吃的新鲜蔬菜或水果被含有细菌的水污染了。这种情况最常发生在附近动物的粪便与水混合时。生菜和菠菜特别容易引发大肠杆菌。
4.其他食品和饮料
您还可能从未经巴氏消毒的果汁以及用生牛奶制成的酸奶和奶酪中感染大肠杆菌。
如果您让接触过未煮熟的肉(如鸡肉)的刀或砧板接触生吃的食物(如沙拉),那么厨房里的食物也会被污染。
5.共同场所
可能在游泳池、湖泊或池塘游泳时吞下含有大肠杆菌的水。动物粪便可能会感染池塘或河流,而人类粪便可能会感染游泳池。
研究表明,即使经过氯处理,某些大肠杆菌也可能重新生长。还可能从私人水井中获取含有大肠杆菌的饮用水,因为这些水在使用前可能没有经过消毒。
6.他人
您可能会从感染大肠杆菌的人那里感染大肠杆菌,例如,孩子在触摸您的嘴之前没有彻底洗手。如果在感染者之后清理,并且在触摸嘴之前没有彻底洗手,细菌也可能传染给你。
7.动物
大肠杆菌O157天然存在于健康农场动物(如牛、羊和马)的肠道中。如果触摸它们,它会传播到它们的皮肤、毛发和它们漫游的区域,并传播到你的手上。因此,如果前往过动物园或农场,请彻底洗手。
8.受污染的土壤
将新鲜甚至陈年的粪肥作为肥料施用于花园的土壤中,可能会使大肠杆菌与正在种植的粮食作物接触。受污染的水也可能渗入作物土壤。
9.尿液中的大肠杆菌
据美国国家肾脏基金会表示, 80%至90%的尿路感染(UTI) 是由大肠杆菌引起的。女性比男性更容易患尿路感染,因为女性的尿道(将尿液从膀胱排出体外的管道)较短,如果没有正确擦拭自己,细菌就更容易从您的屁股传播到膀胱。
▷ 大肠杆菌感染的风险因素
有些人比其他人更容易感染大肠杆菌,包括:
-65岁以上的人
-新生儿和幼儿
-免疫系统较弱的人(服用免疫抑制药物或患有癌症或艾滋病毒等疾病)
-溃疡性结肠炎或糖尿病患者
-吃过未煮熟的汉堡包或喝过生牛奶、未经高温消毒
-因服用减少胃酸的药物而导致胃酸水平下降的人
-季节(六月至九月)
感染大肠杆菌后2至5天内,您可能会开始感到不适。
最常见的症状是:
-腹部绞痛
-腹泻,可能带血
-恶心
-持续疲劳
您可能不会发烧。如果发烧,也可能只是轻微发烧。
大肠杆菌的严重症状:
-腹泻持续超过3天
-腹泻带血
-腹泻并伴有发烧
-严重呕吐
如果有这些症状,请立即联系就医。
▷ 胃肠炎
产肠毒素大肠杆菌(ETEC)可导致婴儿出现旅行者腹泻或婴儿腹泻。发病机制涉及质粒介导的耐热(ST)和耐热(LT)肠毒素,这些毒素会刺激液体和电解质分泌过多。
EPEC导致发展中国家的婴儿腹泻。发病机制涉及质粒介导的 A/E 组织病理学,破坏正常微绒毛结构,导致吸收不良和腹泻。
EAEC导致发展中国家和发达国家的婴儿腹泻以及旅行者腹泻。发病机制包括质粒介导的杆状体聚集性粘附(“堆叠砖块”),导致微绒毛缩短、单核细胞浸润和出血;液体吸收减少。
STEC会导致出血性结肠炎。STEC由EPEC进化而来;A/E病变会破坏肠微绒毛,导致吸收减少;病理学由细胞毒性志贺毒素 (Stx1、Stx2) 介导,会破坏蛋白质合成。
EIEC引起的疾病在发展中国家和发达国家中都很罕见。发病机制涉及质粒介导的结肠上皮细胞的侵袭和破坏。
▷ 泌尿道感染
最常见的引起尿路感染的细菌是大肠杆菌(E.coli)。其他细菌也会引起尿路感染,但大约90%的罪魁祸首是大肠杆菌。
感染的主要表现包括:
-强烈而持续的尿意
-排尿时有灼热感
-骨盆压力
-下腹不适
-排尿频繁且疼痛
-尿液中有血
大多数引起尿路感染的革兰氏阴性杆菌起源于结肠,污染尿道,上升进入膀胱,并可能迁移到肾脏或前列腺。
▷ 脓毒症
当正常宿主防御能力不足时,大肠杆菌可能会进入血液并引起败血症(脓毒症)。
新生儿可能极易感染大肠杆菌败血症,因为他们缺乏IgM抗体。
败血症可能因泌尿道感染而继发发生。
▷ 急性细菌性脑膜炎
大肠杆菌和B组链球菌是导致婴儿脑膜炎的主要原因。
大约75%的脑膜炎病例中的大肠杆菌具有K1抗原,该抗原与脑膜炎奈瑟菌的 B 组荚膜多糖发生交叉反应。而与K1抗原相关的毒力机制尚不清楚。
患有大肠杆菌脑膜炎的新生儿会出现发烧、发育迟缓或神经系统体征异常。
新生儿的其他发现包括黄疸,喂养减少,呼吸暂停和精神萎靡。1个月以下的患者出现烦躁、嗜睡、呕吐、食欲不振和癫痫发作。
▷ 并发症
感染大肠杆菌的健康人通常在一周内会感觉好些。但有些人会出现一种严重的并发症,称为溶血性尿毒症综合征,这种综合征会影响肾脏。
老年人和儿童更容易发生这种情况。症状包括:
-呕吐
-血性腹泻
-胃痛
-发烧和发冷
随着感染恶化,您可能会出现:
-疲劳和虚弱
-昏厥
-瘀伤
-皮肤苍白
并发症可能导致高血压、肾病、癫痫、血液凝固问题、中风或昏迷。大肠杆菌还会导致营养不良(因慢性腹泻导致营养吸收不足)。
大肠杆菌传统的鉴定方法包括显色实验、乳糖发酵实验、IMViC生化实验,另外也可以通过显色平板、全自动微生物鉴定系统等方法进行鉴定,除此之外,采用分子检测的方法,可直接检测样本中的病原菌DNA,提供快速而准确的检测结果,为临床用药提供科学依据。
医生如何诊断大肠杆菌取决于你的症状。如果您有腹泻或其他消化系统症状,医生会检测粪便样本中的大肠杆菌。如果你有其他症状,医生可能会检测您的尿液、粪便、血液或脑脊液(CSF)。
需要进行哪些测试来检查大肠杆菌?
大肠杆菌的具体检测包括:
-大便检测
-尿液分析或尿液培养
-血培养。
-脊椎穿刺(腰椎穿刺)
肠道内大肠杆菌
可以利用16S或宏基因组技术,测定大肠杆菌的丰度和毒株,判别其丰度是否超标已经对菌群的紊乱影响。
尿路感染中的大肠杆菌
大多数尿液样本是通过清洁中段尿液采集技术从成年患者身上获取的。可以通过对未离心尿液样本进行革兰氏染色、对离心样本进行革兰氏染色或直接观察尿液样本中的细菌来在显微镜下检测菌尿。
染色后,大肠杆菌呈现为无芽孢形成、革兰氏阴性杆状细菌;常规尿液培养物应使用校准环进行半定量法培养。
!
注意
定义严重菌尿的最常用标准是每毫升尿液中存在⩾105CFU。
常规培养所用的培养基类型应仅限于血琼脂和麦康凯琼脂。尿液培养物应在35°C–37°C的环境空气中孵育过夜后再读取。
大肠杆菌通过粪口途径传播,这意味着含有大肠杆菌的粪便细小颗粒被人摄入(通常是通过食物或水),然后人就会生病。虽然这听起来很恶心,但这很常见,而且大多数食源性疾病都是通过粪口途径传播的。
▷ 大肠杆菌的预防
1.勤洗手
为了防止大肠杆菌的传播,洗手非常重要。上完厕所、换完尿布、准备食物(尤其是生肉)前后以及接触动物后洗手是防止疾病传播的最佳方法。
2.不食用生肉或未经消毒的乳制品
未经高温消毒的牛奶、“生”或未经高温消毒的奶酪以及经过绞碎或嫩化的生肉具有传播大肠杆菌感染的高风险。生肉应煮至安全温度以确保细菌被杀死,并且完全不应食用生的或未经高温消毒的乳制品,以降低感染大肠杆菌和其他食源性疾病的风险。
3.不吞咽生水
尽量避免在可能被大肠杆菌污染的地方吞咽水,例如游泳池、婴儿池、湖泊、溪流和池塘。公共水上公园也曾发生过大肠杆菌爆发病例,因此请尽量避免在这类水中人数众多且卫生习惯可能存在问题的地方吞咽水。
尽管大肠杆菌可能很严重,许多人担心感染这种疾病,但大多数情况下,这种疾病会在几天内自行消退,不需要额外治疗。如果有担心的症状,最好咨询医疗保健提供者或做检测。
目前尚无疫苗或药物可以保护你免受大肠杆菌(E.coli)相关疾病的侵害,但研究人员正在研究可能的疫苗。为减少您接触E.coli的几率,请避免饮用湖泊或水池中的水、勤洗手、避免风险食物并预防交叉污染。
▷ 肠杆菌感染的治疗
–磺胺类、氨苄西林、头孢菌素、氟喹诺酮类、氨基糖苷类药物对肠道细菌有明显抗菌作用,但敏感性变异很大,需进行实验室药敏试验。
–大肠杆菌脑膜炎需要使用抗生素,例如第三代头孢菌素(例如头孢曲松)。
–大肠杆菌肺炎需要呼吸支持、充足氧疗和抗生素,如第三代头孢菌素或氟喹诺酮类药物。
-大多数腹泻病患者不需要服用抗生素。治疗大肠杆菌感染的最佳方法是多喝水,避免脱水,并尽可能多休息。但是,患者应避免食用乳制品,因为这些产品可能会引起暂时的乳糖不耐症,从而使腹泻恶化。
▷ 大肠杆菌泌尿道感染的治疗
一些大肠杆菌菌株是肠道微生物群落的正常组成部分,但如果它们进入泌尿系统就会引起尿道感染 (UTI) 。
医生通常使用多种抗生素治疗尿路感染。具体开哪种抗生素取决于尿液中检测到的细菌类型。
用于治疗大肠杆菌相关尿路感染的一些抗生素包括:
-磺胺甲恶唑和甲氧苄啶(复方新诺明)
-奥沙霉素(Monurol)
-呋喃妥因(Macrobid)
-头孢氨苄(Keflex)
对于大多数人来说,三天或五天的抗生素疗程可以成功治疗大多数尿路感染,疼痛和持续的尿意冲动在服用几剂后就会消退。
一些大肠杆菌菌株,称为超广谱β-内酰胺酶(ESBL)大肠杆菌,对许多抗生素治疗具有耐药性。
注:风险最高的人群包括使用导尿管、有复发性尿路感染病史或近期使用抗生素的人。
对于这些人,通常建议采取以下措施:
-卡巴培南类,此类抗生素包括亚胺培南(Primaxin IV)、美罗培南(Merrem)、多利培南(Doribax)和厄他培南(Invanz)。
-其他抗生素呋喃妥因、磷霉素和头孢吡肟(Maxipeme)有时也是可行的治疗选择。
▷ 预防泌尿道感染的方法
有很多方法可以帮助预防尿路感染。一些居家措施包括:
-经常排尿。 大约每两到三个小时排空一次膀胱,有助于在感染开始之前将大肠杆菌从尿道中冲洗出来。(尿液在膀胱中停留的时间越长,细菌繁殖的可能性就越大)
-从前向后擦拭。 这有助于防止细菌从肛门区域传播到尿道。
-性交后排尿。 性交和接触会将肛门中的细菌通过尿道带入膀胱。但性交后排尿有助于清除体内的细菌。
-多喝水。 喝水(尤其是性交后)有助于稀释尿液,刺激排尿次数增加,从而促进排出来自泌尿道的大肠杆菌。
-避免使用隔膜或杀精子剂。 这些物质会促进细菌生长,杀死有助于预防尿路感染的有益细菌。
▷ 如何治疗产志贺毒素大肠杆菌感染
根据前面,我们知道大肠杆菌作为肠道正常细菌中普遍存在的成员,存在于大多数人肠道中,检出时不要太紧张,一般该菌超标才会导致菌群紊乱。
除此之外如果大肠杆菌致病毒株感染,患者治疗症状时不采取的措施与患者采取的措施同样重要。例如,由产志贺毒素大肠杆菌(STEC)引起的肠道大肠杆菌感染不需要抗生素治疗,该病每年存在大量的食源性感染。
事实上,根据《毒素》杂志发表的一份报告,用抗生素治疗这些病例可能会使患溶血性尿毒症综合征 (HUS) 的风险增加三倍,这是一种并发症,毒素会破坏红细胞,破坏肾脏的过滤系统,并可能导致肾衰竭。
同样重要的是,不要使用非处方止泻药治疗STEC感染。根据《临床传染病》发表的一项研究,这些药物也会增加患溶血性尿毒症综合征(HUS)的风险。止泻药会减缓消化系统,从而阻止身体迅速排出毒素。但这并不意味着人们无能为力,无法缓解症状并感觉更好。
根据参阅文献和专家建议在家中进行以下支持疗法,以帮助从STEC感染中恢复:
-多休息。让你的身体休息一下,这样它才能尽力抵抗感染。
-保持水分充足。多喝清澈的液体,包括水和肉汤,有助于避免脱水和疲劳。
-避免食用会加重症状的食物。这些食物包括苹果汁和梨汁、咖啡因、酒精、辛辣食物、乳制品、油腻食物和高纤维食物。
-逐渐在饮食中添加清淡食物。从苏打饼干、烤面包、鸡蛋和米饭等食物开始。
大多数健康成年人感染 STEC 后大约一周即可完全康复,无需任何医疗护理。但如果腹泻持续三天以上,并伴有高烧、便血或剧烈呕吐导致脱水,则必须联系专业医疗人员。
大肠杆菌感染对人体的影响差异很大,取决于细菌的菌株和它们引起的症状。这意味着治疗方法也各不相同。
肠道大肠杆菌感染主要需要休息、多饮水和避免食用可能导致进一步不适的某些食物。如果症状包括腹泻超过三天、便血、发烧和剧烈呕吐,请去看医生,因为可能需要药物治疗。
在某些情况下, 大肠杆菌感染可导致溶血性尿毒症综合征。如果不治疗,HUS会导致肾衰竭,然后需要透析、输血和补液等治疗。保持良好的卫生习惯和食品安全是预防大肠杆菌感染和任何潜在并发症的关键。
大肠杆菌(E.coli)是一种存在于我们周围环境、动物和人类体内的细菌。许多大肠杆菌菌株是无害的,但有些菌株可引起从轻微到严重的疾病,所以我们需要正确认识大肠杆菌并提供更精准检测。
精准的检测手段可以帮助我们及时发现潜在的大肠杆菌感染风险。在食品行业,应采用高效的检测方法对各类食品原材料、加工过程以及成品进行严格筛查,确保食品安全。在医疗卫生领域,临床实验室需要不断提升检测技术水平,以便在患者出现症状时迅速确定是否为大肠杆菌感染,并区分不同菌株的致病性,为制定个性化的治疗方案提供依据。
只有通过不断提高对大肠杆菌的认识和检测水平,我们才能更好地保障公众健康,降低大肠杆菌带来的疾病风险。
主要参考文献
Cobo-Simón M, Hart R, Ochman H. Escherichia Coli: What Is and Which Are? Mol Biol Evol. 2023 Jan 4;40(1):msac273.
Ananthanarayan and Paniker. Textbook of Microbiology.
Bailey and Scott’s Diagnostic Microbiology. Part 3. Section 7. Chapter 22. Enterobacteriaceae, 323.
Mackie and McCartney Practical Medical Microbiology. Section B. Bacteria and Related Organisms. Chapter 20. Escherichia, 361.
Murray, P. R., Rosenthal, K. S., & Pfaller, M. A. (2013). Medical microbiology. Philadelphia: Elsevier/Saunders
Sastry A.S. & Bhat S.K. (2016). Essentials of Medical Microbiology. New Delhi: Jaypee Brothers Medical Publishers.
Scaletsky, I. C., Fabbricotti, S. H., Carvalho, R. L., Nunes, C. R., Maranhão, H. S., Morais, M. B., & Fagundes-Neto, U. (2002). Diffusely adherent Escherichia coli as a cause of acute diarrhea in young children in Northeast Brazil: a case-control study. Journal of clinical microbiology, 40(2), 645-8.
www.ncbi.nlm.nih.gov/books/NBK564298/
www.cdc.gov/ecoli/about/index.html
microbenotes.com/escherichia-coli-e-coli/
CHINET中国细菌耐药性监测结果(2021年)
2020 年全国细菌耐药监测报告
临床微生物检验标准化操作(第三版)

谷禾健康

在现代医学的发展历程中,口服药物因其便捷性和经济效益一直占据着重要地位。然而,随着科学研究的深入,我们逐渐认识到一个复杂现象:药物与人体肠道微生物群之间存在着密切而微妙的相互作用。
这种相互作用有时可能带来积极的效果,例如,二甲双胍这种常用的糖尿病药物能有益地调节肠道菌群。然而,更多情况下,药物可能会导致肠道菌群的失衡,引发一系列健康问题。
对于大多数药物而言,我们仍然缺乏全面深入的了解。这导致在药物的研发和使用过程中,往往没有充分考虑到它们对肠道微生物组的潜在影响。一些药物可能直接表现出抗菌作用,而一些则可能通过改变胃肠道的微环境间接影响微生物平衡。例如,广泛使用的抗精神病药物奥氮平就被发现具有内在的抗菌特性,其使用与肠道微生物群组成的改变以及肥胖等副作用密切相关。
近期,科学界提出了一个概念——”肠道中性“。什么是肠道中性呢?通俗来讲就是,设计和使用药物和配方时,尽量减少与肠道微生物群的不良相互作用。实现这一目标的策略多种多样,包括开发”肠道友好型”药物、采用特殊的药物包装技术,以及结合益生菌和粪便微生物群移植等辅助疗法。
本文将深入探讨药物与肠道微生物组之间复杂的相互作用,分析当前研究面临的挑战,并展望未来可能的解决方案。希望为优化药物治疗效果、减少不良反应,以及促进个体化医疗的发展提供新的思路和方向。
肠道菌群参与药物代谢可能直接导致毒性反应
就比如说:抗焦虑药物硝西泮在梭菌、拟杆菌、真细菌的作用下发生硝基还原,产生7-氨基硝西泮,这是一种致畸代谢物,会干扰妊娠期间胎儿的正常发育。
多重用药也会加剧潜在的肠道微生物组毒性
同时服用索利夫定(一种抗病毒药物)和 5-氟尿嘧啶 (5-FU)(一种抗肿瘤药物)会导致拟杆菌属B. vulgatus、 B. thetaiotaomicron、B. fragilis、B. uniformis、B. eggerthii将索利夫定代谢为 (E)-5-(2-溴乙烯基) 尿嘧啶,从而抑制 5-FU 代谢。这会导致未代谢的 5-FU 在生物体内蓄积,从而增加出血、感染、胃肠道紊乱、呼吸困难、心律失常的风险。
类似地,肠道微生物代谢地高辛可导致多达 10% 的患者药物失活。地高辛用于治疗心律失常,被Eggerthella lenta所含的糖苷还原酶灭活。
药物引起的微生物群失调会破坏肠道微生物群与各种健康系统的联系,从而破坏药物的治疗目的。
例如,用于治疗精神分裂症和躁郁症的精神药物如氯氮平、利培酮、奥氮平,会使微生物群失调从而破坏肠-脑轴,导致神经能量学(神经细胞系统的能量代谢和需求过程)改变和认知障碍。
奥氮平是一种多受体作用药物,主要通过阻断多巴胺D2受体和5-羟色胺5-HT2A受体发挥抗精神病作用。主要用于治疗精神分裂症、双相情感障碍、难治性抑郁症等。常见的副作用可能有体重增加和代谢综合征风险增加、嗜睡、口干等。
与对照组相比,奥氮平在 21 天内使 Sprague-Dawley 大鼠的体重增加了 30%,同时 TNF-α 和血糖增加了 3 倍,血液甘油三酯增加了 2 倍。
这些变化可能会破坏神经能量、认知和情绪稳定,从而导致治疗效果恶化。
此外,代谢功能障碍带来的社会和心理负担,即体重增加,可能导致不依从治疗和进一步恶化心理健康。
药物引起的菌群失调也会阻碍联合用药的治疗
在抗菌耐药性的体外模型中,接触抗抑郁药(尤其是舍曲林)会增加细菌对氯霉素、四环素、环丙沙星等抗生素的耐药性。这是由于肠道代谢组的变化,例如细胞内活性氧化物质的产生增加、外排泵的表达、持留细胞的形成和接合质粒的转移。
抗抑郁药可诱导多种抗生素耐药性并增强抗生素持久性
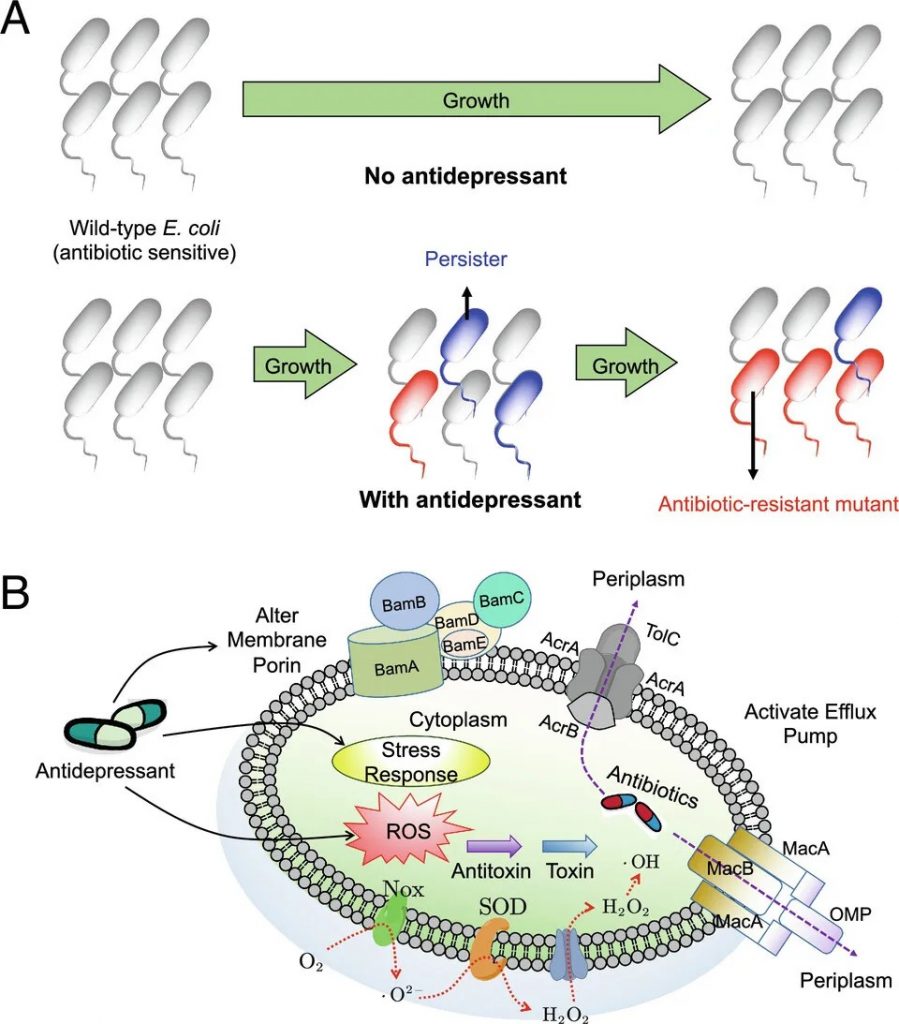
doi.org/10.1073/pnas.2208344120
随着抗抑郁药的广泛使用和对抗生素耐药性的担忧,对药物微生物组学的关注至关重要。
有必要采取策略来克服药物与肠道的不良相互作用,减轻肠道介导的代谢和毒性,同时有利于全面了解药物微生物组学,总结如下图。
克服药物-肠道不良相互作用的策略
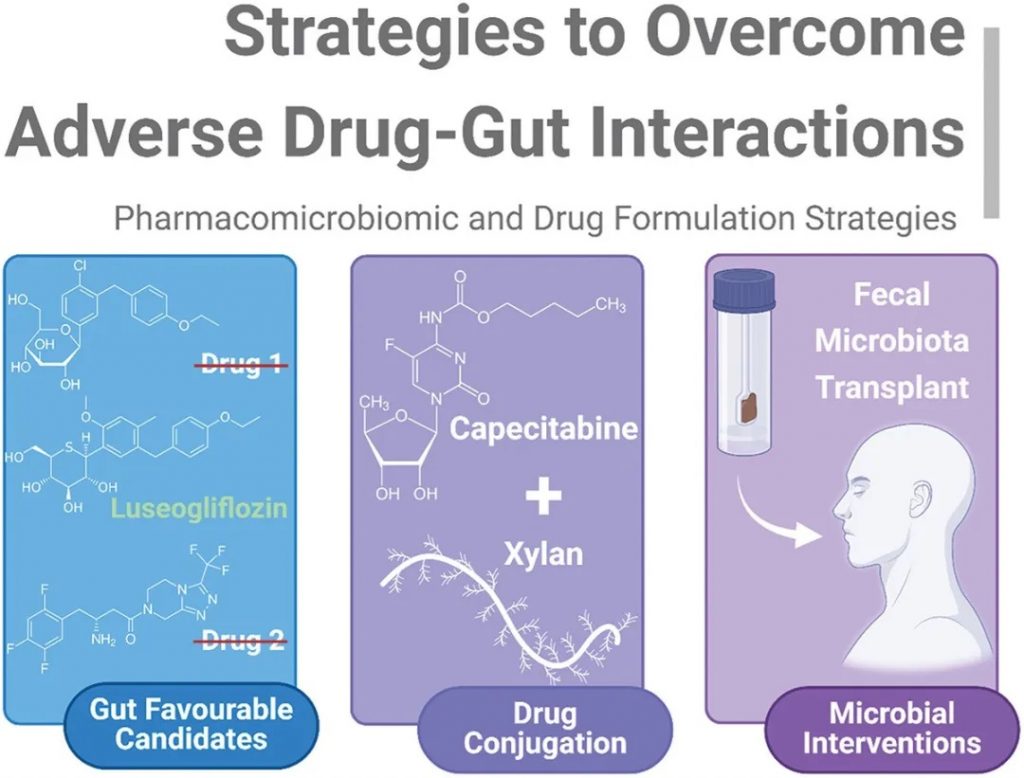
doi.org/10.1080/17425255.2024.2407616
可归纳为不同的子类:
1)药物的内在特性:应选择对肠道中性或阳性的候选药物以确保最大程度的治疗效果。
2)药物结合:益生元纤维与药物的直接结合,如培西他滨-木聚糖-硬脂酸结合物,可减少药物固有的任何非预期代谢或抗菌作用。
3)微生物干预:粪便微生物群移植允许采用非药物方法来减轻非预期的药物-肠道毒性。
药物微生物组学是一个新兴领域,大多数药物-肠道相互作用仍不为人所知。“肠道中性”的概念和基因组测序的进步可能会重振未充分利用的疗法。
★ 鲁拉西酮可能比奥氮平更适合某些患者
与奥氮平不同,非典型抗精神病药物鲁拉西酮在动物研究中对体重、葡萄糖代谢、炎症或肠道微生物群多样性没有显著影响。
注:尽管疗效相当,但鲁拉西酮的处方量仍然不足,这可以归因于该药物的监管和市场批准时间相对较短(与其他抗精神病药物相比)、生物利用度较差以及药物的食物效应,使得患者必须随餐服药,从而给患者的依从性带来了挑战。
与抗精神病药物相比,增加其使用可以减轻微生物组介导的毒性,强调药物微生物组学在药物选择中的重要性。
这些发现强调了鲁拉西酮在维持肠道微生物群平衡方面的优势,可能使其成为代谢敏感患者的更好选择。
★ 卢塞格列净:增加短链脂肪酸,积极调节肠道菌群
类似地,用于治疗糖尿病的钠-葡萄糖协同转运蛋白 2 抑制剂卢塞格列净 (luseogliflozin) 通过增加短链脂肪酸 (SCFA) 来积极调节肠道微生物群。这些短链脂肪酸阻碍了小糖的吸收,从而降低血糖并有助于糖尿病管理。
因此,制药行业必须在候选药物过程中考虑药物对肠道微生物群的影响,通过在早期临床前研究中筛选微生物组介导的毒性。
洛拉米星(lolamicin)证明了这一点。
注:一种针对脂蛋白转运系统的革兰氏阴性细菌特异性抗生素。
★ 洛拉米星:保护肠道菌群,防止艰难梭菌定植
研究发现,洛拉米星对 130 多种多重耐药临床分离菌有活性,在多种急性肺炎和败血症感染小鼠模型中显示出疗效,并且不损害小鼠肠道菌群,防止艰难梭菌的继发感染。与对照组相比,C57BL/6小鼠在服用洛拉米星后,其 α 或 β 多样性没有显著下降。
注:洛拉米星对致病革兰氏阴性菌的选择性杀死是由于致病菌与共生菌中靶标的序列同源性低;这种双重选择策略可以作为开发其他微生物组保护抗生素的蓝图。
洛拉米星保护肠道菌群并防止艰难梭菌定植
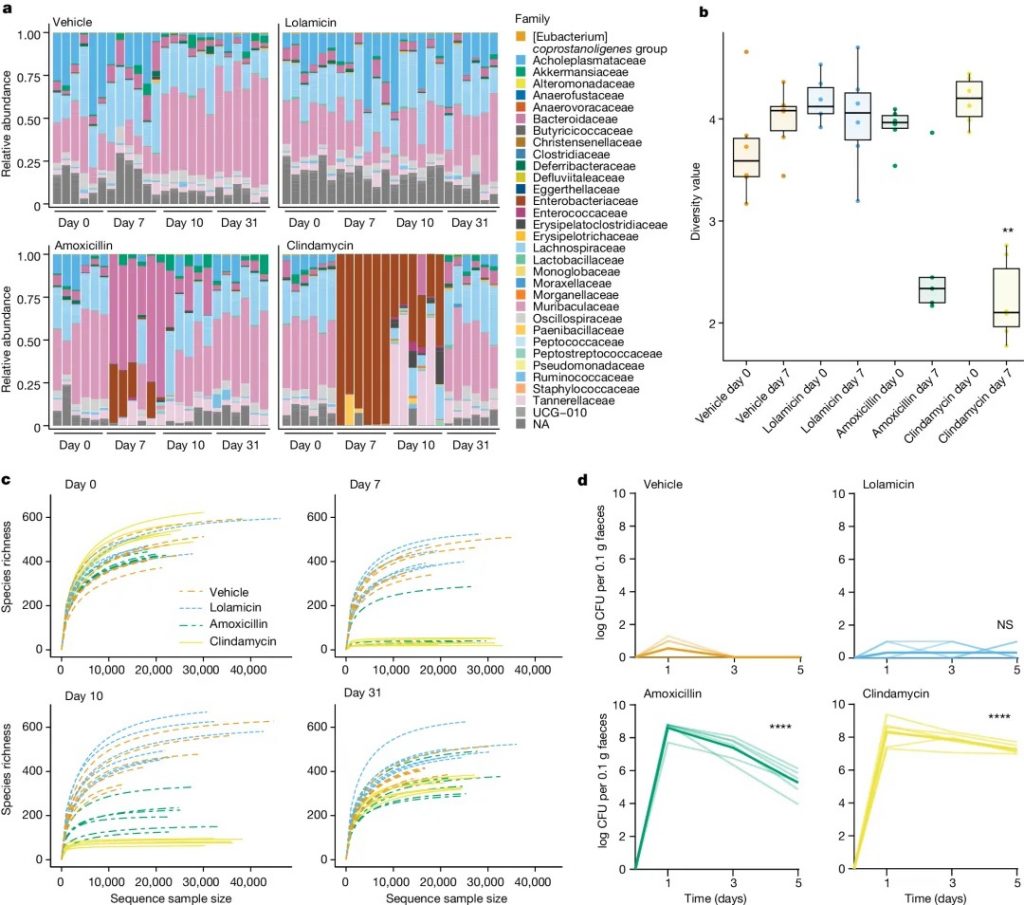
doi.org/10.1038/s41586-024-07502-0
a 在施用抗生素前(第 0 天)和施用抗生素后(第 7 天、第 10 天和第 31 天)31 天内科水平上的菌群变化。
b 用 Shannon 指数测量的施用抗生素前(第 0 天)和后(第 7 天)的多样性分析。
c 在施用抗生素之前(第 0 天)和之后(第 7 天、第 10 天和第 31 天)通过 alpha 稀疏度测量的物种丰富度。
d 从用 1.2 × 104 个艰难梭菌菌株 630孢子进行攻击(第 0 天)开始直至感染后 5 天,用载体、洛拉霉素、阿莫西林或克林霉素治疗的小鼠粪便样本中艰难梭菌的每日计数。
这些观察结果表明,共生双歧杆菌的损失减少,短链脂肪酸的产生增加,消除了机会性致病菌的生长(如艰难梭菌),并维持了适当的肠道屏障功能。
丰富肠道微生物群的策略性药物配方和赋形剂可以减轻微生物群介导的代谢和毒性。
★ 季铵化壳聚糖对姜黄素的影响
硫酸软骨素功能化的棕榈酸和半胱氨酸共接枝的季铵化壳聚糖,将姜黄素的生物利用度提高了 3 倍,同时改善了肠道微生物丰富度,并调节了炎症巨噬细胞和中性粒细胞。
壳聚糖是一种天然多糖,具有良好的生物相容性、生物降解性和黏膜粘附性,在药物递送领域有着广泛的应用。
半胱氨酸能与黏膜层中的糖蛋白形成二硫键,因此将其与壳聚糖偶联可显著提高壳聚糖的黏膜粘附能力,因其能粘附于黏膜层并延长肠道停留时间,是一种很有前途的结肠定位药物递送策略。
棕榈酸是一种十六烷饱和脂肪酸,可作为疏水链供体接枝亲水聚合物,提高疏水性药物的包封率。
doi: 10.1016/j.mtbio.2023.100617
★ 槲皮素可用于缓解 5-FU 引起的粘膜炎
槲皮素也有类似的改善作用,槲皮素是一种五羟基黄酮类化合物,在饮食和作为食品补充剂的重要性已众所周知,但由于其不稳定性、溶解性差和生物利用度低,其口服利用受到限制。
研究人员制备了载槲皮素的壳聚糖衍生物纳米粒子,显著提高了生物利用度。
主要通过增加粘膜粘附性和增加胃滞留时间来提高槲皮素的生物利用度。
该方式显著减轻了 5-Fu 诱导的空肠绒毛和隐窝破坏。有效地减弱了空肠中促炎因子的表达并增强了claudin-1的表达,从而改善了肠道屏障功能。
同时,通过降低拟杆菌的丰度部分逆转了 5-Fu 诱发的粘膜炎小鼠肠道菌群的改变。
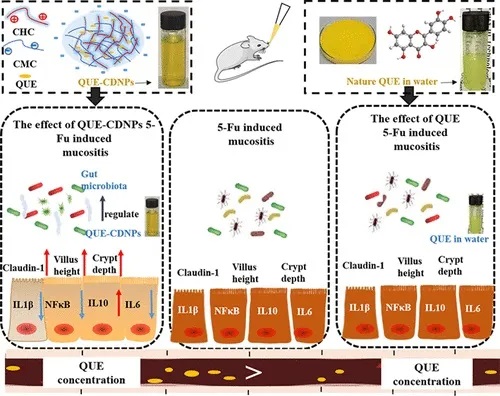
doi.org/10.1021/acsfoodscitech.0c00121
★ 菊粉-脂质核壳微胶囊对鲁拉西酮的影响
菊粉是一种天然存在的可溶性膳食纤维,属于益生元,微胶囊采用脂质作为外壳,能够保护内部包裹的药物,避免其在胃酸和上消化道中的降解。这种核壳结构确保了药物能够稳定地到达肠道。
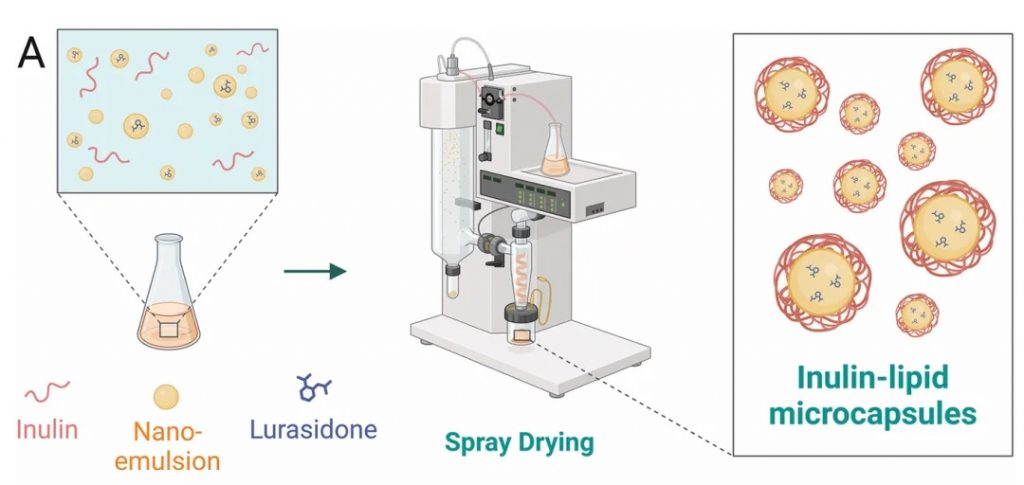
DOI: 10.1002/adfm.202403914
使用菊粉-脂质核壳微胶囊(ILM)靶向肠道微生物群,模拟鲁拉西酮的药物食物效应,获得了类似的益处,同时增加肠道微生物群的丰度和多样性,使口服生物利用度提高了 8.7 倍。
提高溶解度和生物利用度
菊粉-脂质核壳微胶囊(以下简称ILM)显著提高了鲁拉西酮的溶解度和生物利用度。在体外模拟肠道条件下,ILM在空腹和进食状态下都显示出较高的药物溶解度,其空腹/进食状态溶解度比接近1,有效缓解了食物效应。体内药代动力学研究进一步证实,ILM使鲁拉西酮的口服生物利用度提高了8倍以上。
改善肠道微生物组
ILM显著增加了肠道微生物的丰度和多样性(Shannon指数)。研究发现,ILM促进了多种共生菌的生长,而抑制了少数菌种。这种微生物组的改善可能与药物的增强吸收有关。
DOI: 10.1002/adfm.202403914
减轻肠道炎症
ILM降低了肠道组织中促炎细胞因子(IL-1β、IL-6和TNF-α)的水平,有效缓解了肠道炎症。
增加血浆和粪便中的5-HT水平
ILM显著提高了血浆和粪便中的5-羟色胺(5-HT)浓度。研究发现,5-HT水平与肠道微生物的丰度和多样性呈现正相关趋势,这暗示ILM通过调节肠道微生物可能增强了鲁拉西酮的药效学反应。
★ 直接药物-益生元膳食纤维结合也可改善肠道菌群
在一项临床前研究中探索了卡培他滨-木聚糖-硬脂酸结合物 (SCXN)。卡培他滨是一种用于治疗结直肠癌的抗代谢药物,通常会导致严重的肠道微生物失调。
SCXN对肠道微生物群的影响:
肠道屏障功能:
药物耐受性:
抗肿瘤效果:
SCXN 调节肠道菌群
编辑
doi: 10.1038/s41467-023-40439-y
但值得注意的是,这些研究并未考虑先前存在的严重疾病以及无法裂解药物-益生元结合物的影响。
饮食干预/改变提供了一种管理肠道微生物组介导的代谢和毒性的简单方法。
★ 半乳寡糖对奥氮平引起的菌群失调的影响
补充细菌可发酵的半乳寡糖可减轻奥氮平引起的菌群失调、体重增加、甘油三酯和血糖水平。然而,通常需要高剂量的益生元才能发挥治疗作用,这可能会限制其作为辅助疗法的使用。
★ 工程化乳酸乳球菌对抗生素引起的菌群失调的影响
抗生素的使用对肠道菌群的干扰最强烈,但也是可预测的。由于临床用抗生素的药代动力学特性已得到很好的表征,因此可以设计适时的预防措施,以最大限度地减少抗生素在肠道中存在的影响。一项研究开发了一种工程活生物治疗药物(eLBP),用于预防抗生素引起的肠道菌群失调。
研究人员利用合成生物学方法,设计了一种分泌β-内酰胺酶的乳酸乳球菌,可以在小鼠肠道中降解广谱抗生素β-内酰胺类药物,从而保护肠道微生物群落不受抗生素的影响。
该eLBP可以维持肠道微生物多样性,防止抗生素耐药基因的富集,并保持对艰难梭菌的定植抵抗力。这种方法为减少抗生素治疗相关并发症提供了新的策略。
当给 C57BL/6 小鼠服用时,氨苄青霉素引起的菌群失调得到缓解,且不影响血清药物浓度。
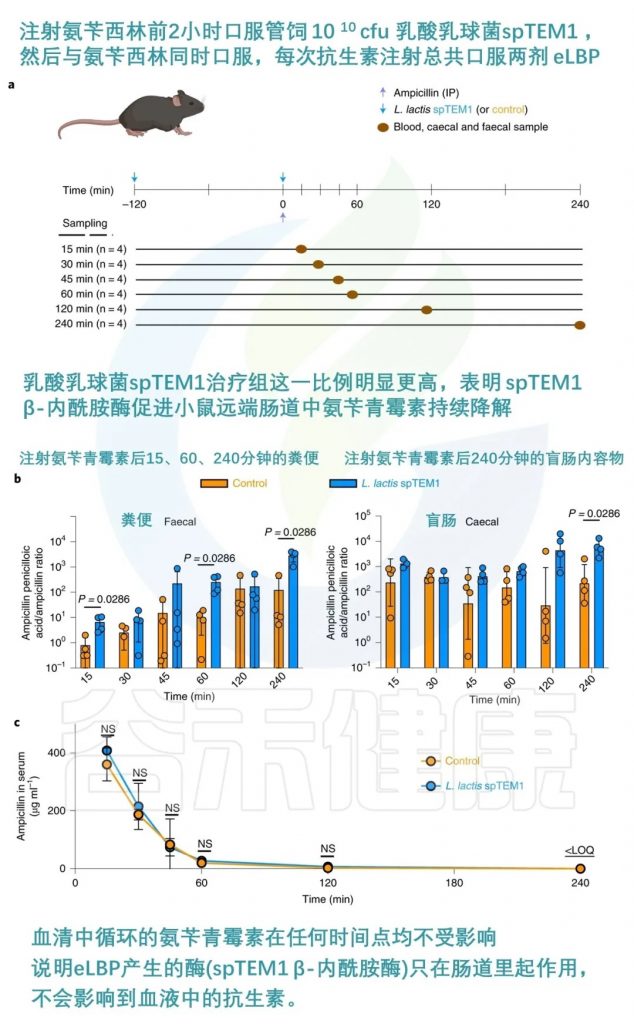
doi.org/10.1038/s41551-022-00871-9
★ 植物乳杆菌IS-10506→氨氯地平吸收率提升2倍
同样,补充植物乳杆菌IS-10506 可通过增加 ATP 结合盒转运蛋白的存在使氨氯地平的吸收率提高 2 倍。
并非所有益生菌都能有效缓解药物引起的菌群失调,有些菌株可能会损害抗生素治疗后的微生物组恢复。因此,选择具有恢复正常生化状态功效的益生菌至关重要。
其他补充疗法(例如 FMT)可解决微生物介导的毒性。接受 FMT 作为抑郁症辅助疗法的患者抑郁症状有较大改善,肠道微生物状况也有所改善。
药物微生物组学领域处于起步阶段,完整的药物微生物组图谱尚未绘制完成,但仍表现出巨大的潜力,将药物微生物组学整合到药物开发和治疗实践中,可以为临床医生和患者提供替代性的疗法,增强治疗效果和最大限度减少不良反应。
鲁拉西酮有益于肠道的特性就是一个典型的例子。作为一种非典型抗精神病药物,鲁拉西酮长期以来处方不足。它的疗效与奥氮平、氯氮平等竞争性抗精神病药物相当,但不会引发肠道菌群失调或影响代谢功能。因此,鲁拉西酮可能更适合代谢敏感的患者,避免了与治疗目标相冲突的风险,降低了加重精神疾病的可能性。
这些例子提醒我们,在药物设计的早期阶段以及开具现有药物时,必须考虑药物微生物组学,以确保实现本文所说的“肠道中性”。通过优先考虑“肠道中性”,可以开发和开出维持肠道微生物组平衡的药物,从而降低与菌群失调和微生物介导的毒性相关的风险。
需要各学科跨领域合作研究,以充分了解药物-微生物组相互作用,并实施战略配方和补充疗法,为更安全、更有效的药物治疗铺平道路,最终有益于患者的护理和健康。
过去二十年对药物-微生物组相互作用的理解已经取得了显著进展,发现了克服不利相互作用的关键策略。
例如,为了提高药物的生物利用度,研究人员设计了基于益生元的递送载体,用于封装或结合药物,既提升了药物疗效,又减少了对肠道微生物组的负面影响。在这种情况下,实现“肠道中性”旨在提高药物的耐受性和安全性。
不过这在临床环境中的研究仍然相对缺乏。这凸显了迫切需要开展针对人类的缓解策略研究,这样有希望将新的发现转化为临床实践,最终改善患者的护理和治疗结果。
控制药物-微生物组相互作用面临诸多挑战,需要有针对性的研究工作。这些挑战包括微生物采样、测序的技术限制,以及获取和分析大数据所需的成本等。为了克服这些障碍,肠道菌群检测技术的进步和标准化,构建预测大数据模型和临床前模型的完善显得尤为重要。通过精确的检测和分析,我们可以更好地理解药物对肠道微生物组的影响,以及微生物组如何影响药物的代谢和疗效。
尽管面临挑战,“肠道中性”的药物和制剂的前景依然很不错。这为那些曾因肠道代谢问题或毒性反应而被搁置的治疗方法重新带来了希望。在药物开发的早期阶段考虑药物微生物组学,是减少后期意外毒性的有效途径。对于现有的疗法,应用肠道中性的制剂可能带来突破性的改善。无论采取何种策略,追求“肠道中性”和调节药物微生物组学,都为我们站在口服药物递送和精准医疗领域的前沿提供了有力的支持。
总的来说,肠道菌群检测在药物微生物组学的研究和临床应用中起着至关重要的作用。这不仅有助于开发更安全、高效的治疗方案,提升患者的护理和治疗效果,也将推动药物研发和个性化医疗的进步,最终促进人类健康水平的提升。
主要参考文献
Kamath, S., & Joyce, P. (2024). A critical need for ‘gut neutrality’: mitigating adverse drug–microbiome interactions. Expert Opinion on Drug Metabolism & Toxicology, 1–4.
Muñoz, K.A., Ulrich, R.J., Vasan, A.K. et al. A Gram-negative-selective antibiotic that spares the gut microbiome. Nature 630, 429–436 (2024).
Wang Y, Yu Z, Ding P, Lu J, Mao L, Ngiam L, Yuan Z, Engelstädter J, Schembri MA, Guo J. Antidepressants can induce mutation and enhance persistence toward multiple antibiotics. Proc Natl Acad Sci U S A. 2023 Jan 31;120(5):e2208344120.
Doll JPK, Vázquez-Castellanos JF, Schaub AC, Schweinfurth N, Kettelhack C, Schneider E, Yamanbaeva G, Mählmann L, Brand S, Beglinger C, Borgwardt S, Raes J, Schmidt A, Lang UE. Fecal Microbiota Transplantation (FMT) as an Adjunctive Therapy for Depression-Case Report. Front Psychiatry. 2022 Feb 17;13:815422.
Xie Y, Xu W, Jin Z, Zhao K. Chondroitin sulfate functionalized palmitic acid and cysteine cografted-quaternized chitosan for CD44 and gut microbiota dual-targeted delivery of curcumin. Mater Today Bio. 2023 Mar 24;20:100617.
Lang T, Zhu R, Zhu X, Yan W, Li Y, Zhai Y, Wu T, Huang X, Yin Q, Li Y. Combining gut microbiota modulation and chemotherapy by capecitabine-loaded prebiotic nanoparticle improves colorectal cancer therapy. Nat Commun. 2023 Aug 7;14(1):4746.
谷禾健康
桥本甲状腺炎(HT)是一种慢性自身免疫性甲状腺炎。其发病率因地区和人群而异,30~50岁人群发病率高,女性比男性患病率高很多。
近几十年来,HT发病率逐步升高,已成为全球公共卫生问题。此外,在碘充足地区,HT是成人甲状腺功能减退症最常见的原因。一项关于中国碘摄入量低、充足和过量地区人群甲状腺疾病发病率的研究显示,HT的累计发病率分别为0.2%、1%和1.3%。
桥本甲状腺炎的病理学特点为淋巴浆细胞浸润、组织纤维化、淋巴滤泡形成、实质萎缩、滤泡细胞嗜酸性改变。
最常见的临床表现是甲状腺肿大,可伴有或不伴有甲状腺功能减退症。肿大的甲状腺压迫颈部,可导致发音困难、呼吸困难和吞咽困难。由于甲状腺功能丧失和原发性甲状腺功能减退症,HT患者常有累及全身多个系统的其他症状,尤其是疲劳和便秘。
在患桥本甲状腺炎的早期,许多人经历了消化不适症状,其中大部分最初并没有将其与甲状腺功能减退或自身免疫性疾病联系起来。直到现在包括有的医生也不明白肠道健康与甲状腺和免疫系统健康有多么密切的关系!
甲状腺疾病或甲状腺功能障碍,例如桥本甲状腺炎(HT)和格雷夫斯病(GD)或甲状腺功能减退症,通常的认知是与不孕、荷尔蒙失调、体重增加、疲劳或焦虑等症状有关。
然而,鲜为人知的是,甲状腺功能障碍的胃肠道症状有很多,包括吞咽困难、胃灼热、消化不良、胃酸分泌减少、恶心或呕吐、胆囊不适、腹部不适、腹胀、腹泻、便秘以及包括肠易激综合征(IBS)在内的一般消化系统不适。胃肠道症状常由血清甲状腺激素改变引起,其中便秘是最常见的临床症状。HT患者常因甲状腺激素降低,肠道蠕动明显减弱,严重者可出现假性肠梗阻或肠梗阻。这是因为甲状腺激素如甲状腺素(T4)和三碘甲状腺原氨酸(T3)决定了整个身体,特别是肠道和内脏器官的基础代谢(完成生物任务/功能所消耗的能量)。
三碘甲状腺原氨酸(T3)还被认为是肠粘膜上皮细胞发育和分化的最重要调节剂之一 。临床上,甲状腺激素血液浓度的变化是胃肠道症状的原因,证据是腺体功能低下和亢进时经常出现的胃肠道紊乱。这种影响可能是由于胃肠道神经运动功能的改变,表现为肠道肌肉收缩传播速度的不同以及由于局部糖胺聚糖浸润导致的肌肉层水肿。
反过来,肠道屏障的破坏以及肠道菌群的失调对桥本甲状腺等自身免疫性疾病的影响非常重要。研究人员已将肠道通透性与每一种自身免疫性疾病联系起来,包括桥本甲状腺炎。
肠道菌群失调一方面导致肠道屏障受损,细菌易位,通过分子模拟、旁观者激活、表位扩散等一系列机制破坏甲状腺自身免疫中的免疫耐受性。
另一方面,它可以通过自身的脱碘酶活性和 促甲状腺激素(TSH)抑制直接影响甲状腺激素水平。肠道微生物群还影响对甲状腺很重要的矿物质的吸收,包括碘、硒、锌和铁。所有这些都是甲状腺功能所必需的,甲状腺功能障碍与这些矿物质水平的改变之间存在明显的联系。例如,碘缺乏可能导致甲状腺肿,可能是甲状腺结节,甚至滤泡性甲状腺癌。高碘摄入量可诱发易感患者的甲状腺功能减退或甲状腺功能亢进。铁是细菌生长所必需的,铁的利用率会影响微生物群的组成,同时,微生物群也会影响铁的利用率。铁对于有效利用碘和甲状腺激素合成至关重要,碘可能导致甲状腺疾病,包括甲状腺激素合成、储存和分泌受损。
目前传统医学对桥本氏病(自身免疫性甲状腺炎)的检查不够及时和全面。导致许多患者在未经充分评估的情况下就开始服用甲状腺药物(一线药物为左旋甲状腺素钠)。但是自身免疫性甲状腺炎患者常对常规药物治疗反应不佳。在精准医学时代,对不同细菌菌株所具有的酶活性的了解越来越多。研究证实肠道菌群组成会影响口服左旋甲状腺素的疗效,即通过改变肠道吸收表面或直接与细菌结合,如大肠杆菌所证明的那样。与肠道菌群正常的小鼠相比,无菌小鼠的促甲状腺激素增加了25%。此外,肠道菌群还在甲状腺激素的肠肝循环和代谢中发挥作用 :事实上,在小鼠和人类粪便内容物中都检测到了β-葡萄糖醛酸酶和硫酸酯酶活性 ,可能导致小肠水平上甲状腺激素的肠道解离和重吸收,可防止甲状腺素随粪便流失,从而使激素在肠肝内循环。
由于HT通常伴随患者终身,且难以康复,往往会严重影响患者的正常工作和生活质量。目前,HT尚缺乏有效的治疗方法,亟待寻找一种有效的,适应个体的干预手段来治愈疾病。
本文将就上述机制进行讨论,在本文中,您将了解有关桥本甲状腺炎和肠道的许多事实。您还将了解:
•桥本氏病的常见症状
•甲状腺和自身免疫与菌群失调和肠漏症的关系
•肠道细菌失衡的常见原因
•桥本应该接受实验室检查
•为什么许多人患甲状腺疾病的根本原因都是相同的
•为什么桥本氏病常常被误诊
•桥本甲状腺炎是可以逆转的
•饮食可以改变桥本甲状腺炎的病情
•桥本氏病患者可能缺乏的常见营养素
如果你服用 Synthroid/左旋甲状腺素或患有甲状腺功能减退症、甲状腺水平低或甲状腺功能低下,则很可能患有桥本甲状腺炎。
★ 注意:桥本甲状腺炎和甲状腺功能减退症不同
桥本甲状腺炎和甲状腺功能减退症并不总是一回事。虽然桥本甲状腺炎是甲状腺功能减退症的主要原因,但桥本甲状腺炎和甲状腺功能减退症之间还是有区别的。一个人可能患有其中一种,而不患有另一种。
大多数单纯性甲状腺功能减退症可通过服用甲状腺激素来治愈。然而,约95%的甲状腺功能减退症病例是由晚期桥本甲状腺炎(自身免疫性甲状腺炎)引起的,即使通过药物恢复了适当的甲状腺激素水平,桥本甲状腺炎和自身免疫攻击仍会持续存在(除非找到并治疗根本原因)。
✦ 甲状腺功能减退症
甲状腺功能减退症的定义是甲状腺无法为身体产生足够的甲状腺激素,通常表现为促甲状腺激素(TSH)水平高或游离T4和游离T3水平低。一些医生将其称为“甲状腺功能低下”。
甲状腺功能减退症是一种临床状态,可能由于多种因素而发生,例如碘缺乏、甲状腺手术切除、放射性碘治疗、甲状腺激素摄入不足、服用甲状腺抑制药物,或者由于感染、事故或桥本甲状腺炎等疾病导致甲状腺受损。
✦ 桥本甲状腺炎
但是,桥本甲状腺炎是一种渐进性自身免疫性疾病,会导致身体攻击自己的甲状腺,最终导致甲状腺功能减退。
桥本甲状腺炎早期患者可能还没有甲状腺功能减退,只是攻击了甲状腺。这种对甲状腺的自身免疫攻击会引起症状,由各种因素引发,并对生活方式的改变作出反应。将在下文进一步讨论这一点。
甲状腺的自身免疫攻击在一个人患上甲状腺功能减退症之前几年就已经出现了,因此及早发现这种疾病可以防止其发展。一个人可能在最终被诊断出患有甲状腺功能减退症并接受甲状腺药物治疗之前,已经患有桥本甲状腺炎多年。
▸ 桥本甲状腺炎的症状
常见症状包括:焦虑、抑郁、健忘、体重增加、情绪波动、疲劳、脑雾、皮肤干燥、便秘、肌肉痉挛、心跳缓慢、指甲易碎、关节炎、手脚冰冷和胃肠道问题。
此外一些症状包括:对多种食物敏感,尤其是对麸质、乳制品和大豆敏感,这可能不会在标准血液测试中显示出来,但可能会在食用这些食物后表现为延迟症状。
另一方面,当我们的身体处于甲状腺功能亢进状态时,症状包括:体重减轻、心悸、焦虑、眼睛凸出、易怒、月经不调、疲劳、不耐热、食欲增加、脱发、甲状腺肿大、易出汗、排便频繁、不孕不育、软指甲、手指震颤、失眠、肌肉无力。
正如您在下图中看到的,人们可能会同时出现甲状腺功能减退和甲状腺功能亢进的症状。
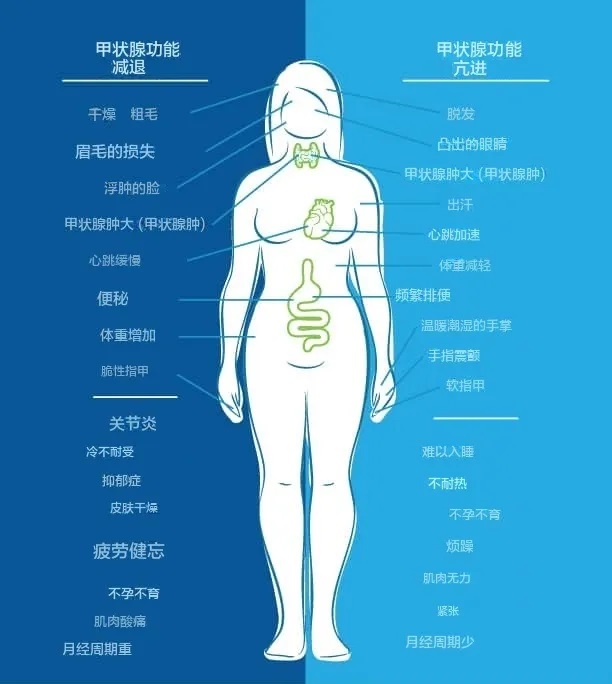
图片来源:thyroidpharmacist
以上种种证明,桥本甲状腺炎症状可能会影响一个人,即使他表面看起来很好!
桥本甲状腺炎是一种自身免疫性疾病,但研究表明多种因素可能共同作用导致该病的发生。以下是一些可能的诱因和关键触发因素:
1
毒素
已发现多种环境毒素可引发桥本氏病和甲状腺抗体。减少接触这些毒素保护肝脏将是减少抗体的关键。需要注意的毒素包括:
·碘过量
·铜中毒
·汞,如牙科填充物(汞合金)中常见的汞
·砷
·氟化物
·其他重金属
·药物,包括锂、避孕药、PPI、干扰素、异维甲酸
·个人护理产品和家用清洁剂中发现内分泌干扰物
·辐射
·镍毒性
·乳房植入物
·网片植入手术
2
感染
感染是桥本甲状腺炎的常见诱因,如果饮食干预不能让你感觉好转,感染往往就是问题所在。这些是口腔和肠道感染,以及多种细菌和病毒感染,在桥本甲状腺炎中很常见。
可能引发桥本甲状腺炎的感染包括:
·口腔感染,如牙脓肿、根管感染和牙周炎
·念珠菌感染
·幽门螺杆菌感染
·细菌感染
·EB病毒
·丙型肝炎
·巨细胞病毒 (CMV)
·莱姆病
·链球菌感染
3
情绪和身体压力
各种压力源都可能引发自身免疫问题。情绪和身体压力源可能包括:
·生活事件(离婚、创伤)
·身体虐待、情感虐待
·社会排斥
·血糖异常
·过度运动
·睡眠呼吸暂停
·外科手术
·睡眠不足
4
营养缺乏
低热量饮食和西式饮食会导致营养缺乏,各种消化酶缺乏也会导致营养缺乏。一些常见的可能引发桥本甲状腺炎的营养素缺乏包括:
·硒
·维生素D
·B族维生素
·铁/铁蛋白
·碘
·锌
5
食物敏感
食物敏感可能是桥本甲状腺炎和自身免疫性疾病的主要诱因。
桥本甲状腺炎最常见的敏感食物有:
·麸质
·奶制品
·大豆
·谷物
·鸡蛋
·坚果
·种子
·茄科植物
6
激素变化
雌激素占主导地位、催乳素升高和孕酮缺乏都可能导致或加剧自身免疫性甲状腺问题。女性生活中最大的激素变化是:
·青春期
·怀孕
·围绝经期
这是女性被诊断患有桥本甲状腺炎的最常见时间。
研究人员过去认为怀孕是诱因,因为胎儿细胞会进入母体的甲状腺;然而,进一步的研究表明,母亲甲状腺内的婴儿细胞实际上可能具有保护作用。
7
输血
我们知道,当一个人接受输血时,他们会接触到捐献者血液中的抗原。输血被发现会增加自身免疫性疾病的发病率,包括桥本氏病。
8
菌群失调为桥本氏病埋下隐患
肠道菌群失调是肠道通透性的潜在原因,我们知道肠道通透性是桥本氏病发展的三个必要因素之一。
厚壁菌门/拟杆菌门比例更高
2017年的一项研究发现,与健康对照组相比,桥本氏病患者的细菌组成(多样性和丰度)存在显著差异。其中一个例子就是厚壁菌门/拟杆菌门的比例,桥本氏病患者和肠易激综合征等其他疾病患者的厚壁菌门/拟杆菌门比例明显更高。比例较高通常是健康状况不佳的征兆,与微生物失衡有关。
注:厚壁菌门与拟杆菌门比例较高也与肥胖有关。减肥后,这一比例会有所改善。
抗炎细菌的水平较低
桥本氏病患者的肠道菌群中,对肠道屏障完整性和调节炎症至关重要的细菌水平较低。例如,拟杆菌属会产生短链脂肪酸 (SCFA),粪杆菌属会产生丁酸并具有抗炎作用,而普氏菌属会产生抗炎代谢物。
最有趣的发现是,某些细菌含量较高与甲状腺抗体存在之间存在相关性。这些细菌包括Blautia、Roseburia、Ruminococcus twists、Romboutsia、Dorea、Fusicatenibacter和Eubacterium hallii。
另外的研究采用16S rRNA基因测序对桥本甲状腺炎(HT)患者肠道菌群进行分析,结果显示在门控水平上,变形菌门和蓝藻门的丰度较高,放线菌门水平提高,而厚壁菌门和拟杆菌门的丰度降低;在科和属水平上,肠杆菌科、产碱菌科和Parasutterella属增加,而普雷沃氏菌科、瘤胃球菌科、韦荣氏球菌科和小杆菌科减少。
同时,实时PCR数据显示,双歧杆菌和乳酸杆菌在桥本甲状腺炎(HT)患者中显著减少。有趣的是,在另一项研究中,发现HT患者肠道中双歧杆菌的丰度随着病情的发展而增加。此外,有研究结果显示, HT患者的物种丰富度指数Chao1显著升高,这可能表明肠道细菌过度生长。虽然高微生物多样性通常与更好的健康结果相关,但也可能带来破坏性的影响,如蛋白质分解增加,多酚转化、黏液分泌和上皮周转减少。
因此,虽然HT患者肠道菌群发生了变化,但还需要更大的样本人群库来确定桥本甲状腺炎的菌群特征。
检测甲状腺激素水平是诊断甲状腺疾病和确定适当治疗方案的第一步。然而,实践中也许医生并不检测桥本甲状腺炎,尽管病人出现了甲状腺疾病的症状。
根据传统指南,大多数初级保健医生仅通过检测患者的TSH(促甲状腺激素)和T4水平(血液中循环的T4甲状腺激素量)来筛查甲状腺功能减退症。然而,这些测试并不总区分自身免疫性甲状腺问题和桥本甲状腺炎,并且经常会忽略桥本甲状腺炎和甲状腺功能减退症早期阶段出现的甲状腺激素细微波动和甲状腺抗体升高。这些阶段通常有症状,可能持续十年,之后TSH或T4水平才会被标记为异常。
经常看到患有桥本甲状腺炎早期阶段的人被告知是“甲状腺测试正常,没有问题”,即使表现出明显甲状腺功能障碍的症状,而这些症状可以通过其他测试明确证实。
因此,进行全面的甲状腺检查非常重要,不仅包括TSH和T4,还应包括T3,以及两种最常见的桥本抗体、TPO和TG抗体(当它们升高时,表明甲状腺内存在自身免疫过程,其水平可以指示对甲状腺的自身免疫攻击的强度,并有助于预测甲状腺完全破坏的时间表)。
此外,超声波检查可以帮助诊断桥本甲状腺炎,并揭示甲状腺的情况并查看是否存在结节。
1
甲状腺筛查测试
促甲状腺激素(TSH)测试用于筛查甲状腺疾病,以及监测个人所需正确药物剂量的测试。该测试很重要,如果可以建议每次检查甲状腺功能时都进行这项测试。
注:TSH是一种脑垂体激素,对循环中甲状腺激素的低/高水平有反应。TSH升高意味着体内没有足够的甲状腺激素,并且可能患有甲状腺功能减退症。这是因为TSH激素可以感知甲状腺水平低下,并在缺乏甲状腺激素时释放,以向身体发出信号以产生更多的甲状腺激素。
在患有晚期桥本甲状腺炎和原发性甲状腺功能减退症的患者中,该测试结果会升高。在患有格雷夫斯病和甲状腺功能亢进症的患者中,TSH 水平会降低。患有桥本甲状腺炎和轻度或中枢性甲状腺功能减退症的患者在该测试中的读数可能为“正常”。
测试前注意事项:建议在清晨测试TSH,并将甲状腺激素测试推迟到测试后,以确保结果准确,特别是如果正在服用含T3的药物。测试前服用药物会抑制TSH,这会是医生判别用药过量,但实际上并非如此。此外,常用于治疗脱发的补充剂生物素也会抑制TSH水平,也会使看起来用药过量或甲状腺机能亢进。
2
甲状腺激素水平测试
已确定有四种主要的甲状腺激素:T1、T2、T3 和 T4。
T4(甲状腺素)和T3(三碘甲状腺原氨酸)是两种主要的甲状腺激素。T4被称为激素原,其生物活性比T3低 300%。T3是主要的生物活性甲状腺激素,它能让我们有美丽的头发、补充能量并促进新陈代谢。
大多数常用的甲状腺药物(如左甲状腺素钠)仅含有T4,因此它们需要在体内转化为活性T3形式。
从理论上讲,T4到T3的转换很顺利,但在现实世界中,在真实的人体中,T4转换为T3会受到很多因素的影响,如下图所示。
图片来源:thyroidpharmacist
通过测试游离T4和游离T3水平,我们可以了解T4与T3的比例,并测量体内可发挥作用的激素。
游离T3和游离T4测试可测量体内循环的活性甲状腺激素水平。当这些水平较低,但同时 TSH 处于正常范围内时,这可能会导致医生怀疑你患有一种罕见的甲状腺功能减退症,即中枢性甲状腺功能减退症。
有些临床医生可能只检测T4,但检测T3也很重要,因为有些人可能无法顺利将T4转化为活性T3。因此,有些人的T4水平可能正常,但T3水平较低。
3
甲状腺抗体
甲状腺疾病患者体内可检测到多种类型的抗甲状腺抗体。甲状腺抗体的存在表明甲状腺已被免疫系统识别为外来入侵者,甲状腺正在遭受攻击。
甲状腺抗体是甲状腺问题的第一个迹象
在桥本甲状腺炎中,触发因素会导致身体出现所谓的“缺乏自我耐受性”。这时身体不再能够将自己的组织识别为自身的一部分,而是开始将其组织视为外来入侵者。它不再“容忍”自己,这会导致自身免疫疾病。当身体开始破坏其免疫耐受性时,我们最初会看到甲状腺抗体升高。
桥本甲状腺炎患者中约有80%至95%有甲状腺抗体。 在许多情况下,甲状腺抗体是甲状腺问题的第一个迹象。在检测到TSH变化之前,它们可能会升高5年、10年,有时甚至15年。即使 TSH 水平“正常”,甲状腺抗体升高也意味着甲状腺被破坏到无法再产生足够激素的程度只是时间问题。
有些临床医生会说,一旦你有甲状腺抗体,你就会一直有甲状腺抗体,所以实际数字并不重要,因为抗体会随机波动。小编相信抗体会因触发因素(如压力等常见因素)而波动,而且它们可以成为追踪疾病进展的极其有用的标记。
桥本氏病中最常见的抗体是甲状腺过氧化物酶抗体(TPO抗体)和甲状腺球蛋白抗体(TG抗体)。大多数桥本氏病患者会出现其中一种或两种抗体升高的情况,而TPO抗体是最常见的。
甲状腺抗体表明甲状腺正在受到主动破坏,因此可用于诊断目的,并可进行监测以追踪缓解情况。
4
甲状腺超声检查
有些人可能患有甲状腺疾病,但血液检查结果可能没有发现任何变化。事实上,研究表明,10%至50%的桥本甲状腺炎患者可能没有抗体检测阳性。 在这些情况下,患者可能患有一种侵袭性较小的桥本甲状腺炎,称为抗体阴性或血清阴性桥本甲状腺炎。
在这种情况下,可以使用甲状腺超声检查来发现指向桥本甲状腺炎的甲状腺物理变化。甲状腺超声检查将帮助医生确定你是否患有与桥本甲状腺炎一致的变化(例如甲状腺弹性变大、甲状腺萎缩、甲状腺肿大或甲状腺异常生长)。
一些生长可能表明存在自身免疫过程或存在良性结节,而其他生长可能表明存在癌性结节。
注:我们了解到,大约10%的桥本甲状腺炎患者是通过超声波诊断出来的,建议每个桥本甲状腺炎或甲状腺疾病患者,尤其是育龄妇女,一生中至少做一次超声波检查。如果发现甲状腺结节,建议每年做一次超声波检查。
5
你的症状
俗话说,自己身体是疾病最好的显示器。我们经常忙于日常生活工作,忽视身体发出的微小信号。对于桥本甲状腺炎来说,你的症状应该作为重要的甲状腺测试。因为有些微妙的信息可能只有你知道。
身体症状为我们体内发生的状况提供了重要线索,并且这些症状会随着甲状腺激素水平的变化而变化。
您是否有甲状腺功能减退或甲状腺激素缺乏的症状。前文我们给出了桥本对应的症状。
!
其他注意事项
这里还是要分享下有些重要因素,它们可能会影响甲状腺测试结果和激素水平。
可能干扰甲状腺检查结果的补充剂和药物
药物相互作用是指甲状腺激素与各种药物、补充剂和食物之间的相互作用,它会导致甲状腺激素水平发生实际变化,一旦停止使用相互作用的药物,这种变化是可逆的。
一些例子包括:钙补充剂、咖啡和非处方 PPI,如奥美拉唑。如果您在服用甲状腺药片后几小时内服用这些药物,这些往往是最相关的。
影响甲状腺功能的物质
虽然有些补充剂会导致甲状腺水平测试误差,但补充剂补充后的测试水平会反映甲状腺水平的真实变化——这种变化可以用于监测实际改善或恶化,这个和任何一项测试一样。
导致甲状腺功能长期改善的物质:例如芦荟、冬虫夏草、维生素A、肌醇在某些情况下可以降低/使 TSH 正常化,这意味着你只需要更少的药物。
导致甲状腺功能长期恶化的物质:锂、胺碘酮和高剂量碘等药物会导致甲状腺损伤,从而需要更多的甲状腺激素。
甲状腺药物需要以适当的剂量使用
用药过量的症状包括但不限于:心跳加快或不规律、紧张、易怒或情绪波动、肌肉无力或震颤、腹泻、月经不调、脱发、体重减轻、失眠、胸痛和过度出汗。
未经咨询医生,请勿擅自开始、更改、增加、减少或停止用药。
总而言之,有些药物会干扰甲状腺检查结果,因为它们实际上会干扰甲状腺激素的功能——这意味着它们会导致甲状腺激素水平发生真正的变化。其中一些变化具有临床意义,当同时服用这两种药物时,可能需要调整甲状腺药物的剂量。
▸ 桥本甲状腺炎与消化道功能的关系
小学生物课上就讲过,消化从口腔开始,口腔会产生富含酶的唾液,从而分解食物,特别是淀粉。因此,分泌足够的唾液对正确启动消化过程非常重要。
✦ 桥本甲状腺炎唾液分泌减少
然而,研究表明,相当一部分患有自身免疫性甲状腺疾病(如桥本甲状腺炎)的人缺乏足够的唾液,并出现“口干”的症状。这在一定程度上是由于促炎细胞因子的过量产生阻碍了唾液的正常产生。
✦ 桥本甲状腺炎胃食管运动能力降低
研究还表明,甲状腺功能减退会显著降低胃食管运动能力,从而导致胃肠功能障碍,因此建议消化不良患者检查甲状腺功能。
既往研究也已证实了甲状腺功能在胃肠道系统中的作用。严重的甲状腺功能减退症可导致食管蠕动。当近端受累时黏液水肿引起吞咽困难,远端可出现食管炎、食管裂孔疝等。
✦ 甲状腺功能减退与消化不良呈正相关
胃肌电研究显示,消化不良与甲状腺功能减退评分呈正相关。甲状腺功能减退症患者由于肌肉水肿、肌电节律减退,常出现胃动力障碍,导致胃排空延迟、胃酸缺乏。
另一方面,甲状腺功能也会影响肠道菌群,研究表明,甲状腺功能明显减退的患者更容易出现肠道细菌过度生长。
▸ 桥本甲状腺炎与消化系统疾病
早在20世纪50年代,胃肠道与甲状腺的关系就被提出,并被称为“甲状腺胃综合征”。这种一致性可以通过共同的胚层起源和微生物群的特定组成来解释。近年来,桥本甲状腺炎与消化道疾病之间的共同机制引起了人们的关注。
✦ 桥本甲状腺炎和乳糜泻(CD)
乳糜泻是一种具有自身免疫特征的小肠炎症性疾病,由摄入小麦、大麦和黑麦中的储存蛋白(麸质)引发和维持。由于麦谷蛋白的分子结构与甲状腺组织相似,可导致多种自身免疫性甲状腺疾病。
一项单中心回顾性病例对照研究表明,桥本是乳糜泻患者中最常见的自身免疫性疾病。加之它们的症状常有重叠,因此我们有理由相信它们可能通过肠-甲状腺轴相互联系。
非乳糜泻小麦敏感性(NCWS)是一种非过敏性、非自身免疫性疾病,它还可以通过Toll样受体(如TLR2和TLR4)增加 TNF-α 的表达和活化。
✦ 桥本甲状腺炎和幽门螺杆菌感染性胃炎
近年来,大量研究已证明幽门螺杆菌(Hp)感染与消化性溃疡、胃癌的发病密切相关。在过去的20年中,Hp与其他非胃肠道疾病的相关性被揭示。甲状腺自身免疫与Hp的关系可以用分子模拟来解释。
首先,CD4+T细胞识别与甲状腺上H/K/ATPase结构相似的Hp表位,激活Th1诱导细胞凋亡。
其次,树突状细胞将Hp表位呈递给幼稚T细胞,由于缺乏外周免疫耐受,Th1会被激活。
最后,INF-γ可以刺激甲状腺滤泡细胞中MHCII的表达。
此外,Hp能产生细胞毒素相关基因A(CagA),研究发现,cag-A阳性的Hp菌株显示与甲状腺过氧化物酶(TPO)序列相似的核苷酸序列,说明血清CagA阳性增加了患自身免疫性甲状腺疾病的风险。
✦ 桥本甲状腺炎和自身免疫性萎缩性胃炎
桥本甲状腺炎(HT)见于近40%的自身免疫性萎缩性胃炎(AAG)患者,其血清中存在大量抗壁细胞抗体。由于AAG时胃酸分泌大大减少,铁吸收不良,无法促进T3和T4的合成。
此外,研究发现,血清胃泌素、嗜铬粒蛋白A水平及肠嗜铬样细胞(ECL)增殖与自身免疫性疾病的共存有显著相关性。
上述研究结果从多个角度提供了理论支持,通过探讨HT与其他胃肠道疾病的关系,可以进一步理解肠-甲状腺的作用和机制,进而拓宽HT及其并发症的临床治疗思路,开发新的治疗方法和药物,进一步提高HT的诊治水平。
▸ 甲状腺对其他消化器官的影响
其他器官在宏量营养素和微量营养素的分解和吸收中发挥着至关重要的作用,而甲状腺健康会影响这些器官。
✦ 肝脏受甲状腺功能影响巨大
肝脏会产生消化和吸收蛋白质、糖和脂肪所必需的酶和胆汁。肝脏还会努力代谢毒素(如杀虫剂或重金属)、酒精,并控制释放到血液中的葡萄糖量。换句话说:如果你的肝脏功能不正常,你的消化功能就会受到影响。
然而,如果没有健康的甲状腺,您的肝脏就无法发挥最佳功能,研究表明,肝脏是受甲状腺功能亢进和减退影响最大的器官。
因此,不健康的甲状腺会导致不健康的肝脏,从而影响胆囊并妨碍最佳消化、营养吸收和 T4/T3 转换。
✦ 胰腺与甲状腺功能也有联系
此外,胰腺和甲状腺之间存在联系,甲状腺对胰腺酶的产生和整体功能完整性有影响。
最后,甲状腺与肠道功能有许多联系,例如运动能力改变是便秘的诱因,关于甲状腺和肠道菌群的下面内容我们详细讲…..
▸ 肠道微生物群失调和桥本氏病
健康的肠道微生物群会影响免疫系统,也对甲状腺功能有重要影响,特别是对桥本甲状腺炎和格雷夫斯病等自身免疫性甲状腺疾病。
研究表明,肠道菌群的变化(包括菌群失调)会影响您的整体甲状腺健康,甚至导致和加剧包括桥本甲状腺炎在内的自身免疫性甲状腺疾病。
✦ 甲状腺功能减退的患者存在小肠细菌过度生长
2007年的一项研究发现,在有自身免疫性甲状腺功能减退症病史的人群中,54%的人小肠细菌过度生长(SIBO)呼气测试呈阳性,而对照组中这一比例为 5%。
✦ 肠道菌群会影响T4向T3的转化
虽然还未完全了解,但肠道微生物群也会影响T4向T3的转化——T3是甲状腺激素的活性形式。碘甲状腺原氨酸脱碘酶在T4向其活性形式T3的转化中起着核心作用。据推测,肠道和肠道微生物群会影响这些酶的活性。
这至少在一定程度上归因于肠道微生物群对将T4转化为T3所需的微量营养素(如硒和锌)的可用性的影响。
但事实可能远不止于此,因为人们在肠壁中发现了脱碘酶活性,而且至少有一项动物研究表明肠道细菌具有结合甲状腺激素的能力。
有趣的是:这个过程的成功在很大程度上取决于胆囊中初级胆汁酸的产生。这些初级胆汁酸在消耗脂肪后从胆囊分泌到小肠中,肠道细菌将它们代谢成所谓的“次级胆汁酸”,从而增加脱碘酶的活性。
而且,这些胆汁酸依赖于矿物质硒,而肠道和甲状腺健康问题患者通常会缺乏硒。而且,甲状腺功能减退症还会阻碍胆汁从胆囊中流出,从而进一步影响 T4/T3 的转换。这种胆汁也具有天然的抗菌作用,这可能进一步解释甲状腺疾病与特定细菌性肠道感染(如小肠细菌过度生长)之间的联系。
建议
如果你患有桥本氏病等自身免疫性甲状腺疾病,情况也是如此。可能认为自己的肠道健康很好。但是,鉴于肠道和甲状腺问题同时普遍存在,最好检查一下你的肠道健康。
▸ 甲状腺和菌群失调和肠漏症的联系
当谈到菌群失调、肠漏、甲状腺功能障碍和自身免疫之间的联系时,需要牢记以下关键点:
所有自身免疫性疾病(包括桥本氏病)都会出现肠道通透性一定程度的增加(导致肠漏)。
✦ 桥本甲状腺炎(HT)的肠道屏障通透性增加
正常情况下,肠黏膜屏障能有效阻止病原体进入循环系统。然而,当肠屏障完整性受损时,黏膜下免疫细胞暴露于细菌、饮食抗原和自身抗原,可导致不良免疫激活或耐受反应失败,从而导致自身免疫性疾病。如今,越来越多的证据表明肠道菌群在维持肠屏障完整性方面起着至关重要的作用。
在极端情况下,无菌动物研究证实,肠道菌群缺乏会导致肠黏膜屏障功能退化,具体表现为肠道整体表面积减少、肠绒毛变短、肠隐窝减少、肠道通透性增加、黏膜层变薄及不稳定。
大量研究表明,肠道菌群失调、细菌过度生长和肠道通透性增加(肠漏)会导致桥本甲状腺炎的发生。与IBD(炎症性肠病)患者的改变类似,桥本甲状腺炎患者也有肠道屏障通透性异常。因此,炎症存在时肠道屏障功能受损,肠道通透性增高导致无法穿过肠道屏障的抗原进入体循环激活免疫系统,产生的抗体攻击甲状腺组织是桥本甲状腺炎发生的重要原因之一。
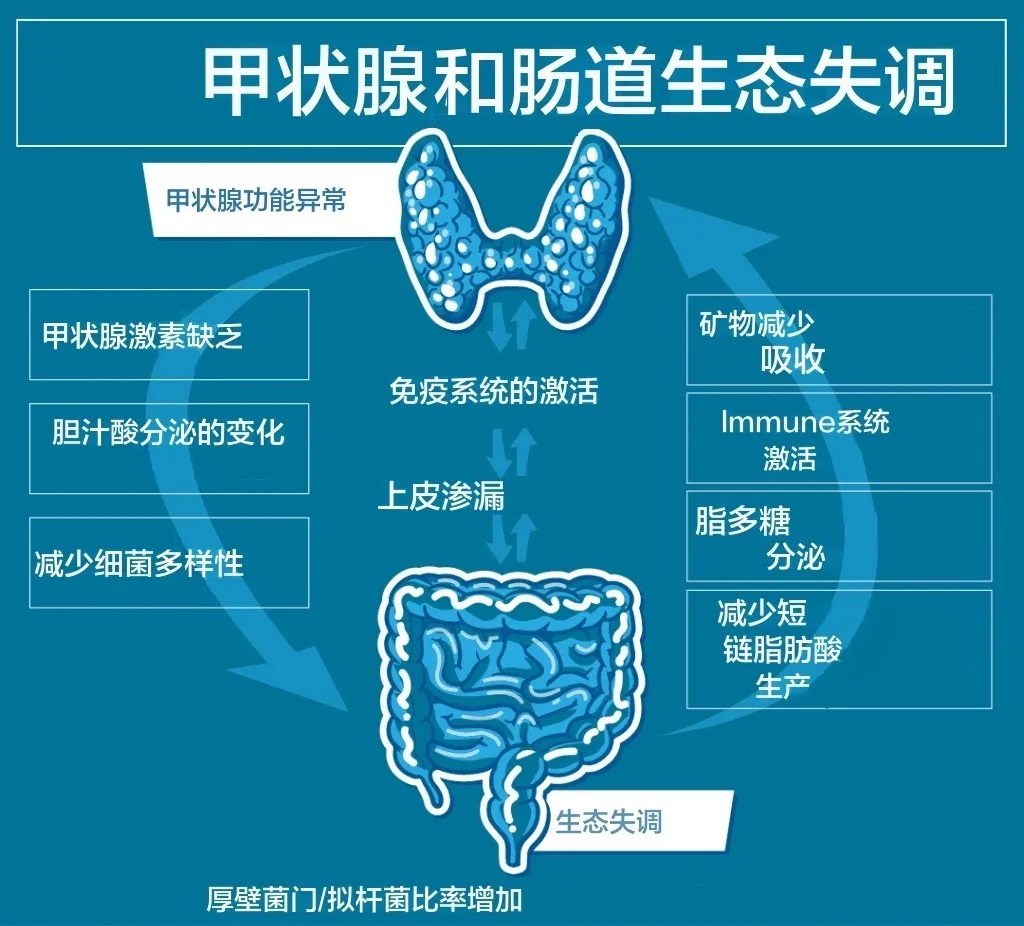
✦ 肠道菌群自噬与甲状腺
肠道菌群与自噬也存在相互作用,与甲状腺自身免疫性疾病的发病机制密切相关。自噬是一个高度保守的生理过程,细胞内的成分经过溶酶体介导的自我消化和循环,受损或老化的生物大分子和细胞器被从细胞质中清除。
一方面,肠道菌群及其代谢产物可以通过mTOR通路调控自噬。同时,自噬缺陷会改变肠黏膜中紧密连接蛋白claudin-2的表达水平。与大多数紧密连接(如occludin、claudin-1、ZO-1)的功能不同,claudin-2蛋白的表达增加了细胞旁路通透性,导致肠上皮屏障破坏,细菌易位和传输增加,进而导致肠道菌群失调。
自噬除了介导炎症反应和免疫外,也是清除过量活性氧(ROS)、防止细胞损伤和死亡的重要机制。自噬缺陷导致去极化的线粒体和蛋白质积聚,并诱导炎症小体激活物(ROS或线粒体DNA)的释放。作为重要的发病机制,炎症因子和ROS也参与了桥本甲状腺炎(HT)的炎症过程。正常情况下,ROS对甲状腺激素的合成至关重要,但过量的氧化应激可诱导甲状腺滤泡细胞(TFC)损伤,引起甲状腺炎症,促进HT的发生。
HT患者血清中可检测到IL-23浓度升高,这是因为IL-23通过促进Th17细胞分化和IL-17的分泌来促进HT的发展。近期有证据揭示了一种新的机制,即 IL-23 作为 AKT/mTOR 信号通路的强诱导剂,能够抑制 TFCs 的自噬活性,导致 ROS 过度积累 。此外,还发现Caveolin-1缺陷可抑制TFCs的自噬活性,诱导AKT/mTOR活化,这可能参与桥本甲状腺炎(HT)的发病过程。而过量的ROS又促进炎症反应和促炎细胞因子及IL-23的产生,形成正反馈循环,加重病情的严重程度。
✦ 菌群失调和甲状腺功能减退症
研究表明,甲状腺功能减退症患者的肠道细菌组成发生改变,产生有益短链脂肪酸(SCFA)的细菌数量减少,而产生炎症驱动脂多糖 (LPS) 的细菌数量增加。
2020年的一项研究涉及52名甲状腺功能减退症患者和40名健康对照者。他们用较新的16SrRNA测序技术分析了他们的肠道菌群,发现两组之间的肠道细菌存在显著差异。研究人员发现,四种特定细菌(Veillonella, Neisseria, Paraprevotella和Rheinheimera)的水平可以初步识别受试者是患有甲状腺功能减退症还是来自健康对照组。此外,甲状腺功能减退症患者的SCFA产生能力显著下降,LPS水平升高。
在物种水平上,荟萃分析最重要的结果是脆弱拟杆菌(Bacteroides fragilis)的相对丰度增加,这种细菌能够激活NLR 家族含吡啶结构域 3(NLRP3)的表达,NLRP3 是炎症小体成分,在 HT 患者的甲状腺组织中过表达。经Spearman相关性分析,一些门、科和属,如拟杆菌、瘤胃球菌科、肠杆菌科、韦荣球菌、链球菌和乳酸杆菌,与抗甲状腺过氧化物酶抗体(TPO)呈正相关,与 TSH 水平呈负相关;此外,链球菌属与抗甲状腺球蛋白抗体水平呈正相关。
谷禾健康经过多年检测实践和与临床的合作,已经积累了超过2000例的甲状腺患者肠道菌群样本,我们还与保定市第一中心医院席永昌医生合作开展“131-碘治疗对甲状腺疾病患者肠道菌群影响的分析的临床研究”,从初步的研究结果看,甲状腺功能减退症和甲状腺癌患者有明显的肠道菌群特征。
研究人员还进行了粪便微生物群移植(FMT),从两组人类身上提取菌群,并将其移植到小鼠体内。结果非常有趣,因为接受甲状腺功能减退组细菌移植的小鼠在移植后显示出总甲状腺激素水平下降。
有研究还证明脂多糖激活 TLR4 可在 NOD H2h4 小鼠中引发甲状腺炎。在对患有此病的甲状腺功能正常的患者进行形态学和功能研究时,检测到了肠道屏障的渗漏,这为肠道微生物群在桥本甲状腺炎 (HT) 发病机制中发挥作用提供了其他线索。
✦ 甲状腺激素药物的吸收和微生物群
研究发现,当发生细菌过度生长时,会导致微生物组成和药物吸收发生变化,并且可能需要更高剂量的甲状腺药物。
对于常见的引发甲状腺疾病的幽门螺杆菌(H. pylori)也发现了同样的结果。
有趣的是,一项较新的研究表明,有益细菌也会影响甲状腺激素的剂量。一项研究发现,与未使用益生菌的对照组相比,使用几种益生菌(三种双歧杆菌属、四种乳酸杆菌属和嗜热链球菌)可减少左旋甲状腺素剂量调整量。 另一项研究表明,罗伊氏乳杆菌可增加甲状腺激素。
▸ 肠道细菌失衡和肠漏的常见原因
许多因素会影响微生物群的组成和肠道通透性的水平,其中一些因素也是桥本甲状腺炎的常见诱因,这并非巧合。
•遗传易感性——某些基因变异会影响人体对细菌的识别和反应,并导致微生物群的变化。
•分娩过程中母体微生物群的定植——当婴儿通过阴道时,他们会接种来自母亲阴道微生物群的细菌(剖腹产出生的婴儿细菌多样性较低)。
•饮食——我们所吃的食物会影响我们微生物群的组成和肠道的通透性水平。例如,某些食品添加剂会改变肠道中的细菌。研究表明,麸质尤其会导致敏感人群的肠道通透性增加。
•食物敏感——食用我们敏感的食物会导致肠道发炎,从而导致肠道通透性改变。这会影响微生物群中的细菌。
•毒素——多种毒素会影响肠道通透性和肠道内各种细菌的水平。这包括环境毒素(如污染)以及内源性毒素(如脂多糖)。毒素超载导致肝功能障碍也是桥本氏病的常见诱因。
•压力——研究发现,社会压力会降低具有抗炎活性的微生物水平,并导致更高程度的炎症。慢性压力也会导致肠道通透性增加。
•肠道感染和寄生虫——幽门螺杆菌、小肠细菌过度生长、人芽囊原虫、贾第鞭毛虫和念珠菌等感染可导致肠道细菌失衡。例如,幽门螺杆菌感染(以及用于治疗幽门螺杆菌的抗生素)可导致细菌多样性降低。
•减肥手术——最近的研究发现,减肥手术对肠道细菌有很大影响(尽管可能通常是有益的)。
•抗生素——广谱抗生素如阿莫西林、四环素和氟喹诺酮类药物是严重的罪魁祸首,它们会降低厚壁菌门和拟杆菌门的多样性和丰富度。
•药物——质子泵抑制剂和避孕药(也有甲状腺毒性) 等常见药物会影响微生物群的组成。
•营养缺乏——现代农业实践(通常导致土壤和食物中的营养成分减少)是造成营养缺乏的部分原因,但消化酶含量低和胃酸含量低也可能是因素。 例如,谷氨酰胺和锌(重建肠道内壁所需的两种物质)含量低会导致肠道通透性增加。
•激素——性激素对微生物群组成和丰度有显著影响。雌性和雄性实际上具有不同的微生物特征。雌性激素占主导地位可能是桥本氏病的诱因。
•其他疾病状态——其他疾病状态,尤其是肥胖等代谢/能量相关疾病,可能是造成微生物组失衡的因素。研究表明,肥胖会增加产生内毒素脂多糖 (LPS) 的细菌数量,并减少抑制 LPS 的细菌数量。肥胖也可能损害肠道屏障的完整性。
★ 甲状腺→肠道菌群→营养素吸收
碘、硒、铁和锌是重要的甲状腺支持营养素,肠道细菌的组成会影响这些营养素的吸收方式。甲状腺功能障碍本身可以改变肠道微生物群的组成,并影响这些矿物质的吸收。
碘和铁对甲状腺激素合成至关重要,而硒和锌对T4到T3的转化和免疫健康很重要。 肠道屏障是一种半透性壁,允许从肠道吸收营养物质,其中一些营养物质对甲状腺的正常功能也至关重要。
碘
碘是甲状腺激素结构的必需成分,人体内的大部分碘都储存在甲状腺中。甲状腺对碘的吸收是一个主动过程,是甲状腺激素合成的限制步骤之一。NIS 是一种广泛弥散的碘转运蛋白,它利用细胞内H+与细胞外Na+交换获得的通量,逆电化学梯度共同转运碘。
此外,碘的肠道吸收是由位于上皮胃肠道质膜顶端的NIS介导的。负责肠道碘吸收的其他转运蛋白包括 Na+/多种维生素转运蛋白和囊性纤维化转运蛋白,但作用程度较小。
✦ 肠道菌群会影响碘的吸收
1972年,一篇论文展示了肠道菌群在调节大鼠碘吸收中的作用:与传统饲养的大鼠相比,用卡那霉素(一种可降低大鼠体内细菌总数的抗生素)治疗的动物在治疗 3 小时以及治疗 42 和 72 天后放射性碘吸收量降低。
然而,在人体研究中,结果更加矛盾。事实上,在短肠综合征和肠外营养患者中,尽管两组之间的微生物组成不同,但碘排泄量与健康对照组并没有显著差异。在减肥后患者的尿碘排泄量中也观察到了类似的结果。然而,在患有炎症性肠病(通常伴有菌群失调)的人中,观察到碘吸收不良的情况。此外,研究还发现,产生丁酸的肠道微生物群的减少与碘吸收减少有关,这一证据与高危甲状腺结节的发病机制有关。
事实上,短链脂肪酸,尤其是丁酸,通过抑制组蛋白去乙酰化酶,可能激活甲状腺癌细胞中NIS的重新表达。
✦ 碘会改变肠道菌群的组成
另一方面,由于碘具有内在的抗菌活性,它可能会改变肠道菌群的组成,影响常驻菌和致病菌。事实上,已经清楚地证明碘可能通过抑制需氧菌的ATP产生来干扰电子链传递;碘破坏微生物细胞壁结构的能力也已被描述。
一些动物模型分析了碘补充的影响,表明碘对微生物群调节的总体影响取决于宿主微生物组成的个体状况。
硒
甲状腺中的硒浓度高于任何其他器官。它是硒蛋白的重要组成部分,硒蛋白参与多种过程,其中最重要的是抗氧化和抗炎作用以及甲状腺激素的代谢活性。
自然界中的硒以无机形式存在,即硒酸盐和亚硒酸盐,以及有机形式,即含硫氨基酸类似物、硒代蛋氨酸和硒代半胱氨酸。这些化合物的吸收发生在十二指肠、盲肠和结肠中,有机形式的吸收比无机形式的吸收更快。
✦ 肠道菌群将硒转化为有机形式更利于吸收
硒和肠道菌群组成相互作用。事实上,大约25%的细菌拥有编码硒蛋白的基因:例如,大肠杆菌的结构中就含有三种硒蛋白。一些乳酸杆菌属能够将细胞内的亚硒酸盐转化为有机形式,促进人体对硒的吸收。
一项针对无菌小鼠和正常饲养小鼠的研究表明,肠道菌群对硒有部分封存作用。这种与宿主对硒吸收的竞争在硒供应有限的情况下最为明显。
✦ 硒摄入量能够调整肠道微生物组成
此外,不同的硒摄入量能够调节肠道微生物环境的组成:与饲喂缺硒饮食的小鼠相比,补充硒的动物的多雷亚菌相对丰度较低,而Turicibacter(在肠道中发挥抗炎活性)和阿克曼氏菌(以保护肠道屏障完整性而闻名)的丰度较高。
事实上,硒对免疫系统调节具有有益作用,还能促进Tregs细胞因子的分泌。桥本甲状腺炎患者经常出现硒缺乏的情况,而补充硒似乎能够降低甲状腺自身抗体水平。
铁
铁在甲状腺功能中起着关键作用,因为TPO酶的活性中心含有铁,并且参与甲状腺激素的储存和分泌。缺铁对甲状腺代谢有很大影响,因为当发生贫血时,它可能会降低氧气运输,诱发类似于甲状腺缺氧障碍的状况。
特别是,缺铁的动物表现出甲状腺功能障碍。所有形式的铁(无机铁、血红素和铁蛋白)的吸收主要发生在十二指肠和空肠上部。铁以还原形式的Fe(II)被吸收,与十二指肠相比,结肠铁的吸收效率仅为15%左右。
✦ 菌群能够调节铁的生物利用度
然而,该百分比可能会受到结肠pH值变化的调节,而结肠pH值变化可能是由 SCFAs 的产生引起的 。此外,已证明发酵乳杆菌具有铁还原活性,可将 Fe(III) 还原为 Fe(II),从而促进铁的吸收。
事实上,细菌能够调节铁的生物利用度,这要归功于几种促进铁吸收的高亲和力蛋白质。因此,与硒的观察结果一样,肠道菌群和宿主竞争铁的吸收。
✦ 缺铁或过量铁都会影响细菌的正常组成
动物研究发现,缺铁饮食会干扰细菌的生长,而高铁饮食会降低肠道菌群的生物多样性。在人类中,补铁会增加肠杆菌科和拟杆菌,同时减少乳杆菌科和双歧杆菌。
值得注意的是,后者不需要铁来生长。对未吸收铁的竞争也会调节菌群组成,对共生菌产生不利影响。富含血红素的肠道环境为能够代谢这种化合物的细菌种类的增殖提供了营养。过量摄入铁会增加致病性肠道细菌(沙门氏菌、志贺氏菌_、致病性大肠杆菌)的数量,这些细菌需要铁来定植和发挥毒性。同样,在摄入过量铁饮食的人类中,可以观察到 SCFA 生成的变化。
锌
锌在甲状腺病理生理中的作用是由于它参与脱碘酶和超氧化物歧化酶的活性。此外,它还是甲状腺激素结合转录因子的组成部分。事实上,锌参与了整个甲状腺机制主要成分的合成 [促甲状腺激素释放激素 (TRH)、TSH 和甲状腺激素]。除此之外,它还可能影响三碘甲状腺原氨酸与其核受体的结合。
✦ 补充锌可以抑制一些病原体的生长
在人类中,已经观察到甲状腺疾病和锌代谢之间的相互关系,因为甲状腺功能减退症患者通常表现为锌水平降低,而锌缺乏与游离甲状腺激素水平低有关。
一些研究表明,肠道微生物组成与锌之间存在相关性。人类证据支持补锌可抑制病原体(即腹泻致病性大肠杆菌相关)的生长,促进乳酸杆菌等有益细菌的生长。此外,在患有甲状腺自身免疫性疾病的人群中,乳酸杆菌和双歧杆菌的相对丰度与锌水平呈正相关。
贫血,桥本,营养素
许多人认为缺铁是贫血的原因。但根据多年查阅文献和检测经验,实际上有三种营养素缺乏会导致贫血,并且常见于桥本氏病患者:铁、维生素B12和叶酸。
铁蛋白
铁蛋白是人体铁储备蛋白的名称。女性的“正常”铁蛋白水平在12至150ng/mL之间。然而,观察到甲状腺功能的最佳铁蛋白水平在90至110ng/mL之间。
铁蛋白是在桥本甲状腺炎患者中发现的最常见的缺乏症之一,除了疲劳之外,铁蛋白缺乏还会导致呼吸短促、不安腿综合症、脱发、失眠、奇怪的食物渴望、反向T3水平升高和情绪波动。
但除非您经检测缺乏铁蛋白,否则不建议补充。因为铁/铁蛋白过多也会有问题。
维生素B12
B12是蛋白质合成、细胞繁殖和正常生长所必需的必需维生素。它还有助于褪黑激素和血清素的产生,从而促进安稳的睡眠和积极的情绪。
一项关于甲状腺功能障碍患者中维生素B12缺乏症患病率的研究发现,自身免疫性甲状腺疾病也与自身免疫性疾病恶性贫血有关,这可能导致维生素B12吸收不良。
由于某些人患有恶性贫血、小肠细菌过度生长或幽门螺杆菌感染等疾病,口服维生素B12可能难以吸收,因此医生可能会选择口服大剂量维生素B12或注射维生素B12以确保适当吸收。
叶酸
维生素B9是一种必需的营养素,又称为叶酸,在 DNA 的形成和细胞生长中起着至关重要的作用。在某些情况下,您的叶酸测试结果可能正常或偏高,但您仍然可能缺乏适合您身体的叶酸。
一些患有桥本氏病的人,有MTHFR基因变异,使他们无法正确处理可能存在于补充剂和加工食品中的叶酸。一些专业人士声称,这种类型的叶酸甚至可能导致体内积聚,从而导致毒性。
那些可能患有肾上腺疲劳、经常感到压力的人以及具有MTHFR基因变异和高同型半胱氨酸水平的人,可能会从活性或甲基化叶酸中受益,主要是因为单靠食物无法提供足够的叶酸。
降低甲状腺抗体的机制主要有以下几种:
•减少触发
•减少氧化应激
•免疫调节
•免疫抑制
•目标移除
1
去除麸质
2001年,意大利的一项多中心前瞻性研究结论强调了无麸质饮食对患有乳糜泻和桥本氏病的人的益处。这项研究中的受试者在开始无麸质饮食之前患有亚临床甲状腺功能减退症,甲状腺抗体升高。
研究显示,71%的受试者恢复了正常的甲状腺功能;19% 的受试者能够在一年内将甲状腺抗体恢复正常。

编辑
无麸质饮食也帮助了大多数患有桥本氏病的患者,尽管他们中的大多数人并没有乳糜泻。
关于麸质的另一个需要注意的是,它通常是导致肠道通透性的原因。(您具备患上桥本氏病的三个必要条件中的两个:遗传易感性、肠道通透性和诱因。)
2
去除乳制品
戒掉乳制品可能不再有肠易激综合征、腹胀、反流等。在该甲状腺资深专家的调查中,79%的桥本氏病患者在停止食用乳制品后感觉好多了,而且抗体减少了20%。
但是查阅文献发现关于乳制品和桥本氏病的研究有限,后续值得在本地人群中开展相关研究。
3
解决幽门螺杆菌感染
多种细菌感染都与引发自身免疫性甲状腺炎有关,包括幽门螺杆菌(一种导致胃炎胃癌的细菌)、伯氏疏螺旋体(与莱姆病有关)和小肠结肠炎耶尔森氏菌。
根据桥本研究专家 IZABELLA WENTZ博士的分享,在其饮食中去除麸质和乳制品时,大部分症状也消失了。但检测甲状腺抗体检测结果仍然呈阳性,所以WENTZ博士不得不深入研究其他根本原因。最终发现是肠道感染。
她还给其80%咨询客户中的人营养干预达到瓶颈的时候推荐了肠道检测,结果检测出至少一种肠道感染。
4
解决小肠细菌过度生长 (SIBO)
小肠细菌过度生长是指小肠内细菌过度生长。虽然从技术上讲这不是一种感染,但小肠细菌过度生长确实有细菌成分,可能是桥本氏病的诱因、后果或加剧因素。
编辑
根据2014年发表的一项研究,多达54%的甲状腺功能减退症患者会出现小肠细菌过度生长(SIBO)。
桥本氏病患者通过治疗小肠细菌过度生长缓解了肠易激综合征、不安腿综合症、低铁蛋白和低维生素 B12 等症状;在某些情况下,甲状腺抗体通过小肠细菌过度生长治疗而降低,甚至消失。
5
肠道治疗方法
肠道在桥本甲状腺炎中起着重要作用——事实上,肠漏是自身免疫性疾病发展的三个必需因素之一(另外两个是遗传因素和诱因)。
虽然并非所有患有桥本氏病或肠漏症的人都会出现腹胀、胃痛、肠易激综合征和胃酸反流等症状,但大多数桥本氏病患者确实存在一定程度的肠道通透性。对于任何患有自身免疫性疾病的人来说,治愈肠道可以大大减轻症状,甚至可能缓解病情。
通过查阅文献了解到2018年意大利的一项研究,该研究特别关注了芦荟对桥本甲状腺炎的益处。有趣的是,发起这项研究的一位研究人员发现,在她开始每天喝芦荟以促进消化并用作治疗便秘的泻药后,她自己的桥本甲状腺炎标志物得到了改善。
编辑
通过优化肠道健康并恢复肠道微生物平衡,可以从内部开始治愈。为此,可以去除反应性食物,补充酶,平衡肠道菌群,滋养肠道。感兴趣可以关注谷禾以往的专题文章。
6
广谱抗菌
肠道感染,往往是自身免疫性疾病发展的诱因。其他感染,如Epstein-Barr virus(EB病毒)和莱姆病,也可能引发体内的自身免疫反应。
EB 病毒会在体内产生潜伏感染,潜伏到合适的时间才会被重新激活并苏醒。重新激活的病毒有可能诱导甲状腺抗体的产生,并可能导致许多使人衰弱的自身免疫症状。
2015 年波兰的一项研究发现,80%的桥本甲状腺炎患者和62.5%的格雷夫斯甲状腺炎患者的甲状腺细胞中存在EB病毒,而对照组的甲状腺细胞中没有 EBV。
一些研究发现黑种草籽油(Black seed oil)是抗感染草药之一,因为它具有广泛证实的抗菌特性,对桥本甲状腺炎患者来说是一种有用的药物。它之所以特别受人关注,是因为最近的研究表明它也能降低甲状腺抗体。
7
血糖平衡
当血糖水平不断波动时,身体会将这些变化视为慢性压力。当肾上腺(负责释放压力激素的腺体)受到压力时,它们会释放过量的皮质醇激素,这也会导致与免疫反应增强相关的炎症蛋白的产生增加。这种模式最终会导致皮质醇释放改变,从而导致多种症状,包括慢性疲劳、情绪波动和甲状腺抗体增加。
建议从食物开始——在用餐时优先考虑蛋白质和健康脂肪,并尽量减少碳水化合物的摄入,尤其是加工食品。锻炼和充足的睡眠也有助于维持健康的血糖水平。
二甲双胍也有助于降低甲状腺抗体
二甲双胍是一种支持血糖代谢和胰岛素敏感性的药物,通常用于治疗糖尿病患者,最近在桥本氏病患者中进行了研究,结果显示它具有降低甲状腺抗体的能力。
小檗碱是一种植物碱,已被证明对血糖有类似的有益作用。研究表明,每天服用一克小檗碱可使糖尿病患者的空腹血糖降低 20%,这与二甲双胍相当。如果您难以平衡血糖,除了改变生活方式外,小檗碱也是一种不错的选择。
8
减肥
近年来,自身免疫性疾病和肥胖症的发病率显著上升,许多研究都探讨了两者之间的关联。一种可能的机制是,由于肥胖症中的脂肪组织发挥着类似内分泌器官的功能,它会释放细胞因子,通过激活辅助T细胞和抑制调节T细胞来引起促炎作用。这可能导致自身免疫反应,尤其是在有遗传倾向的人身上。
2021年的一项研究调查了减肥是否会影响桥本氏病肥胖患者的甲状腺抗体水平。这项研究包括 BMI 超过 30 且 TPO 抗体水平超过 5.6 IU/mL 的个体。在六个月的研究结束时,研究人员发现减肥的人的 TPO 抗体水平也降低了。减掉多余的体重可能对桥本氏病患者有益。
9
减少氧化应激
谷禾以前专门写过氧化应激对健康的威胁的文章(点击查看 大脑退行疾病的两个重要诱因:氧化应激和肠道失衡)。下面将介绍几种减少氧化应激的方法。桥本甲状腺炎患者的氧化应激增加,因此这可能是一种减少甲状腺抗体和感觉较好的有益策略。
✦ 限制碘以减少氧化应激
限制碘摄入已被证明可以减少氧化应激,根据多项研究,有些人通过限制碘摄入量可以降低甲状腺抗体。但请注意,其他人报告说,增加碘摄入量会降低抗体。这恰恰强调了为什么寻找根本原因是因人而异的。
在一项调查中,有356人尝试了高剂量碘,其中 25% 的人表示服用后感觉好些,28% 的人表示服用后感觉更糟。46%的人表示没有发现任何变化,但这并不意味着他们的甲状腺抗体没有受到某种影响。
另一方面,碘限制使31.7%的人感觉好些,7% 的人感觉更糟。
碘是一个非常有争议的话题,但我还是把它放在这里,希望它能帮到你。话虽如此,在考虑改变碘摄入量之前,我会先消除常见的诱因。你也可以采取其他措施来减少氧化应激,效果会更好,比如服用硒。
✦ 硒作为抗氧化剂制造商
桥本甲状腺炎患者常见营养缺乏,通常是由于肠道通透性问题造成的。
桥本氏病中一种特别缺乏的营养物质是硒。硒是一种微量矿物质,参与制造谷胱甘肽过氧化物酶等抗氧化剂。谷胱甘肽过氧化物酶有助于清除受氧化损伤影响的细胞。
临床试验发现,每天200微克的剂量可以降低甲状腺抗体。在一些研究中,硒在六个月内显著降低了抗体。
在一项的调查中,服用硒补充剂的人中有 63% 感觉更好。34%的人感觉没有变化,而3.5%的人感觉更糟。
✦ 肌醇可减轻氧化应激(以及焦虑)
研究表明,焦虑、抑郁和总体感觉“不适”是甲状腺受到自身免疫攻击的早期迹象,并且可能与甲状腺抗体有关,即使 TSH 数值仍然正常。
据报道,甲状腺抗体阳性人群中最常见的焦虑症类型是强迫症 (OCD)。强迫症在A型人格中更为常见,所以强迫症倾向肯定会随着甲状腺抗体的上升而爆发,然后随着抗体的下降而减轻。
研究发现,肌醇有助于治疗多囊卵巢综合征 (PCOS)、强迫症和焦虑症。另外,肌醇被发现可以降低TPO抗体、降低TSH,还能平衡血糖。
在一项将肌醇和硒结合起来治疗桥本甲状腺炎患者的研究中,研究人员发现,这种组合可以降低 TSH、TPO 和 TG 抗体,并改善甲状腺激素水平和个人幸福感。
10
减少甲状腺抗体的免疫调节策略
✦ 系统酶
系统性酶作为天然免疫调节剂,帮助我们的免疫系统保持平衡。
欧洲对系统性酶进行了大量研究。在那里,酶是治疗关节炎和炎症(如桥本氏病)的一种流行止痛药替代品。
系统性酶可以分解自身免疫疾病中形成的循环免疫复合物,从而减少针对食物和甲状腺的抗体。它们还可以通过减少炎症来帮助组织修复。
✦ 阿纳他滨
阿那他滨是一种合成物质,由茄科植物(奇怪的是包括烟草)中发现的一种天然生物碱制成。早在 2012-2014 年,由于临床试验结果良好,它就作为桥本氏病的补充剂上市销售。它有助于减少炎症和抗体,同时寻找免疫系统失衡的根本原因。
一项针对患有桥本甲状腺炎且甲状腺功能正常的患者的研究结果显示,使用阿那他滨后,甲状腺球蛋白抗体显著降低。
✦ 维生素 D – 轻松增强免疫力
如果每天在户外的时间不多,那么可能面临维生素 D 缺乏的风险。维生素 D 缺乏(由于缺乏阳光照射、使用防晒霜或饮食不良等因素)与免疫功能不正常有关。
维生素 D 缺乏症在桥本甲状腺炎患者中也更为常见。一项研究中,桥本甲状腺炎患者中,68% 的人报告自己也患有维生素 D 缺乏症,这种缺乏症与抗甲状腺抗体的存在有关。
另一项研究发现,92% 的桥本甲状腺炎患者缺乏维生素 D,而 2013 年的一项研究发现,维生素 D 水平低与甲状腺抗体较高和疾病预后较差有关。
最近一项研究表明,补充维生素D可以降低自身免疫性甲状腺疾病患者的甲状腺过氧化物酶抗体水平。
✦ 羟氯喹 – 用于治疗多种自身免疫性疾病
羟氯喹(Plaquenil)是一种最初用于预防或治疗疟疾的药物。然而,在第二次世界大战期间,人们发现这种药物对治疗狼疮、类风湿性关节炎和其他自身免疫性疾病的症状也有效。
甲状腺功能障碍和抗体常与系统性红斑狼疮 (SLE) 和类风湿性关节炎 (RA) 有关。
羟氯喹可减少淋巴细胞、自身抗体的产生、免疫介质、细胞因子和 NK 细胞活性;并抑制向 B 细胞、树突状细胞和单核细胞呈递的抗原。
一项研究表明,使用羟氯喹治疗可显著降低甲状腺抗体的 DNA 水解活性。甲状腺功能减退症患者的 DNA 水解 IgG 抗体水平较高。使用羟氯喹可减少这些抗体,并改善甲状腺激素的产生并提高甲状腺的功能活动。羟氯喹还改善了患者的临床状况。
注意,这种药物确实有很多潜在副作用,包括:心律不齐、精神/情绪变化(焦虑、抑郁、幻觉)、呼吸短促、视力模糊、手臂/腿部/背部疼痛、疲劳和胸部不适。
11
靶向去除是降低甲状腺抗体的策略
✦ 甲状腺切除术
通过手术切除甲状腺可以消除甲状腺抗体,因为没有任何东西可以攻击。但是,切除甲状腺并不能消除自身免疫性疾病。无数人因为甲状腺疾病而切除甲状腺(最常见的是格雷夫斯病、结节和甲状腺癌,但偶尔也有桥本甲状腺炎)。在某些情况下,同时患有桥本甲状腺炎和甲状腺癌的患者可能希望采用这种方法。
一般不建议桥本甲状腺炎患者手术切除。有上述很多其他方法可以改善,防止自身免疫的进展。
✦ 新兴解决方案
干细胞最近已成为自身免疫性疾病患者的潜在治疗选择。在干细胞治疗中,患者将接受可修复受损器官的细胞输注。在桥本甲状腺炎病例中,有人报告称甲状腺抗体减少,甲状腺组织再生。请注意,使用捐赠者的干细胞实际上可能会增加自身免疫性疾病的风险,而据报道,使用自己的干细胞有助于治疗自身免疫性疾病。
Tips 降低抗体需要多长时间?
✦
大多数人在开始干预后一个月内就会开始看到抗体减少的趋势,也可能需要三个月到九个月甚至更长时间才能看到全部效果。
对于患有乳糜泻和麸质过敏症的患者,抗体水平的变化似乎与肠道愈合一致。对于患有幽门螺杆菌(H. pylori)的患者,在根除幽门螺杆菌后,肠道内壁可能需要数月才能愈合。
建议在实施干预措施时每 1-3 个月进行一次测试。这可以了解抗体的趋势,并清楚改变正在产生效果。
桥本氏病和其他自身免疫性疾病是由三种因素引起的。遗传易感性就是其中之一。
另外也存在两个因素,即,肠道通透性和自身免疫触发因素,幸运的是,人们可以通过生活方式干预来解决和改善这些问题。
桥本氏病和其他自身免疫性疾病的共同点是,触发因素会导致身体产生一种称为“缺乏自我耐受”的状态。这时身体不再能够将自己的组织识别为自身的一部分,而是开始将其组织视为外来入侵者。它不再对自己“宽容”,这就是导致自身免疫性疾病的原因。这是怎么发生的?
可以说是一个误认的情况。
一些微生物触发因素或感染产生的细菌细胞确实会触发人体免疫系统攻击入侵细胞。但由于所谓的分子模拟,人体可能会瞄准与这些入侵者相似的其他细胞。这可能导致免疫系统攻击自己的身体。
触发因素包括食物敏感、营养素缺乏、毒素暴露、慢性感染、压力反应不佳以及许多其他因素。一些触发因素实际上也会导致肠道通透性(肠漏)问题,例如麸质敏感。
因此,大多数甲状腺疾病都是由于免疫系统失衡而攻击甲状腺造成的。即使通过手术切除甲状腺或用放射性碘治疗,在大多数情况下,自身免疫问题仍然存在。许多人会切除甲状腺,并可能会患上新的自身免疫性疾病,如狼疮、类风湿性关节炎等。好像免疫系统只是找到了不同的目标。
在大多数桥本甲状腺炎病例中,这种自我耐受能力的缺乏在甲状腺功能受到影响之前就开始出现(通常甚至在症状出现之前)。当身体的免疫耐受能力开始崩溃时,就像文中详细提到的首先会看到甲状腺抗体升高。
在桥本氏病中,抗体针对甲状腺球蛋白(80%)和甲状腺过氧化物酶 (TPO) (95% 的人)。因此有很多方法可以检测甲状腺抗体,一旦发现有抗体,就可以采取纠正措施:
•减少触发
•减少氧化应激
•免疫调节
•免疫抑制
•目标移除
从而防止甲状腺疾病出现或发展。
与此同时,很多抗体的半衰期比检察官,可在免疫细胞中停留约两到三个月。它们需要以抗原的形式不断“提醒”,以便继续产生。因此如果去除抗原,抗体也会消失。大部分抗体完全忘记抗原并消失所需的时间是九到十二个月。意味通过降低或靶向去除抗体,是可以扭转桥本甲状腺炎的。
为了让抗体忘记甲状腺,需要做到以下几点:
而甲状腺会因两个原因停止表达TPO:
在自身免疫性疾病的情况下,传统和替代医学从业者可能会专注于重新平衡免疫系统(即传统医学中使用的类固醇和免疫调节药物,或者替代医学中使用的草药、补充剂或针灸)。
虽然这种方法可能有助于在短期内驯服免疫系统或克服自身免疫爆发,但它通常是一种临时解决方案,如果不解决导致免疫系统失衡的根本问题,一旦停止使用药物、针灸、草药和补充剂,免疫系统可能会再次失衡。因此,我们可以说免疫调节只能治疗症状,而不是根本原因。
由于我们无法改变基因,我们解决桥本甲状腺炎根本原因的方法有三方面:
小编寄语
希望今天这些分享能对桥本患者有所帮助!面对桥本甲状腺炎,保持积极的心态和健康的生活方式是非常重要的。记住,你并不孤单,很多人都和你一样,正在经历相同的挑战。
治愈是一个循序渐进的过程,定期就医进行必要的检查,遵医嘱定时服药,合理饮食,适当锻炼,坚持健康的生活方式,关注肠道健康,恢复肠道微生物群平衡,都有助于改善病情。希望大家都能在这个过程中找到适合自己的方法,收获健康与快乐。
免责声明:本文所含信息仅供参考,不应视为医疗建议。如有任何健康问题,以及在进行任何医疗或生活方式改变之前,请咨询您的医生。
主要参考文献
Lzabella Wentz, PharmD, 2018.Thyroid Antibodies Part 1: An Early Warning for Thyroid Disease. Thyroidpharmacist.
Lzabella Wentz, PharmD, 2024.Thyroid Antibodies Part 2: Mechanisms to Reduce Thyroid Antibodies. Thyroidpharmacist.
Hutfless S, Matos P, Talor MV, Caturegli P, Rose NR. Significance of prediagnostic thyroid antibodies in women with autoimmune thyroid disease. J Clin Endocrinol Metab. 2011;96(9):E1466-E1471.
Müssig K, Künle A, Säuberlich AL, et al. Thyroid peroxidase antibody positivity is associated with symptomatic distress in patients with Hashimoto’s thyroiditis. Brain Behav Immun. 2012;26(4):559-563.
Bucci I, Giuliani C, Di Dalmazi G, Formoso G, Napolitano G. Thyroid Autoimmunity in Female Infertility and Assisted Reproductive Technology Outcome. Front Endocrinol (Lausanne). 2022;13:768363.
Geracioti TD Jr, Kling MA, Post RM, Gold PW. Antithyroid antibody-linked symptoms in borderline personality disorder. Endocrine. 2003;21(2):153-158.
Wang N, Sun Y, Yang H, et al. Hashimoto’s Thyroiditis Induces Hippocampus-Dependent Cognitive Alterations by Impairing Astrocytes in Euthyroid Mice. Thyroid. 2021;31(3):482-493.
Geracioti TD Jr, Kling MA, Post RM, Gold PW. Antithyroid antibody-linked symptoms in borderline personality disorder. Endocrine. 2003;21(2):153-158.
Wang N, Sun Y, Yang H, et al. Hashimoto’s Thyroiditis Induces Hippocampus-Dependent Cognitive Alterations by Impairing Astrocytes in Euthyroid Mice. Thyroid. 2021;31(3):482-493.
Mazaheri T, Sharifi F, Kamali K. Insulin resistance in hypothyroid patients under Levothyroxine therapy: a comparison between those with and without thyroid autoimmunity. J Diabetes Metab Disord. 2014;13(1):103. Published 2014 Oct 30.
Brent GA. Environmental exposures and autoimmune thyroid disease. Thyroid. 2010;20(7):755-761. doi:10.1089/thy.2010.1636
Edwards C. Surgical mesh implants may cause autoimmune disorders. Medical Device Network. August 1, 2018. Accessed June 28, 2023.
Fadaee N, Huynh D, Khanmohammed Z, Mazer L, Capati I, Towfigh S. Patients with systemic reaction to their hernia mesh: An introduction to mesh implant illness. Frontiers. January 30, 2023.
Tomer Y, Davies TF. Infection, thyroid disease, and autoimmunity. Endocr Rev. 1993;14(1):107-120.
Mizokami T, Wu Li A, El-Kaissi S, Wall JR. Stress and thyroid autoimmunity. Thyroid. 2004;14(12):1047-1055.
Hu S, Rayman MP. Multiple Nutritional Factors and the Risk of Hashimoto’s Thyroiditis. Thyroid. 2017;27(5):597-610.
Coucke F. Food intolerance in patients with manifest autoimmunity. Observational study. Autoimmun Rev. 2018;17(11):1078-1080.
Rogers MA, Levine DA, Blumberg N, Fisher GG, Kabeto M, Langa KM. Antigenic challenge in the etiology of autoimmune disease in women. J Autoimmun. 2012;38(2-3):J97-J102.
Desai MK, Brinton RD. Autoimmune Disease in Women: Endocrine Transition and Risk Across the Lifespan. Front Endocrinol (Lausanne). 2019;10:265. Published 2019 Apr 29.
Sategna-Guidetti C, Volta U, Ciacci C, et al. Prevalence of thyroid disorders in untreated adult celiac disease patients and effect of gluten withdrawal: an Italian multicenter study. Am J Gastroenterol.
Hollon J, Puppa EL, Greenwald B, Goldberg E, Guerrerio A, Fasano A. Effect of gliadin on permeability of intestinal biopsy explants from celiac disease patients and patients with non-celiac gluten sensitivity. Nutrients. 2015;7(3):1565-1576. Published 2015 Feb 27.
Bell DS, Ovalle F. Use of soy protein supplement and resultant need for increased dose of levothyroxine. Endocr Pract. 2001;7(3):193-4.
Fruzza AG, Demeterco-Berggren C, Jones KL. Unawareness of the effects of soy intake on the management of congenital hypothyroidism. Pediatrics. 2012;130(3):e699-702.
Eschler DC, Hasham A, Tomer Y. Cutting edge: the etiology of autoimmune thyroid diseases. Clin Rev Allergy Immunol. 2011;41(2):190-197.
Aghili R, Jafarzadeh F, Bhorbani R, Khamseh ME, Salami MA, Malek M. The association of Helicobacter pylori infection with Hashimoto’s thyroiditis. Acta Med Iran. 2013;51(5):293-296.
Yason JA, Liang YR, Png CW, Zhang Y, Tan KSW. Interactions between a pathogenic Blastocystis subtype and gut microbiota: in vitro and in vivo studies. Microbiome. 2019;7(1):30. Published 2019 Mar 11.
Lepczyńska M, Białkowska J, Dzika E, Piskorz-Ogórek K, Korycińska J. Blastocystis: how do specific diets and human gut microbiota affect its development and pathogenicity?. Eur J Clin Microbiol Infect Dis. 2017;36(9):1531-1540.
Diagnosis | Blastocystis Research Foundation. Blastocystis Research Foundation. http://www.bhomcenter.org/info/diagnosis.htm.
Rajič B, Arapović J, Raguž K, Bošković M, Babić SM, Maslać S. Eradication of Blastocystis hominis prevents the development of symptomatic Hashimoto’s thyroiditis: a case report. J Infect Dev Ctries. 2015;9(7):788-791. Published 2015 Jul 30.
Patil AD. Link between hypothyroidism and small intestinal bacterial overgrowth. Indian J Endocrinol Metab. 2014;18(3):307-309.
Shenkman L, Bottone EJ. Antibodies to Yersinia enterocoliticain Thyroid Disease. Ann Intern Med. 1976;85:735–739.
Chatzipanagiotou S et al. Prevalence of Yersinia plasmid-encoded outer protein (Yop) class-specific antibodies in patients with Hashimoto’s thyroiditis. Clinical Microbiology and Infection. 2001;7(3):138–143.
Pamphlett R, Doble PA, Bishop DP. Mercury in the human thyroid gland: Potential implications for thyroid cancer, autoimmune thyroiditis, and hypothyroidism. PLoS One. 2021;16(2):e0246748. Published 2021 Feb 9.
Sterzl, Ivan I. Removal of dental amalgam decreases anti-TPO and anti-Tg autoantibodies in patients with autoimmune thyroiditis. Neuro-endocrinology letters. 27(25):0172-780X.

谷禾健康

2024 年美国胃肠病学会 (AGA) 和欧洲神经胃肠病学和运动学会 (ESNM) ,在华盛顿特区组织了第 12 届肠道微生物组健康世界峰会。
通过为期 2 天的全体会议、研讨会、海报展示和现场讨论,学界泰斗、医生、研究人员以及食品药品管理局和制药行业的代表参与其中,会议重点讨论了开发基于微生物组的疗法,预防和治疗人类疾病的策略和挑战。
本文介绍下该会议期间讨论的主要内容,包括:
1)如何更好的识别微生物及其可以调节的靶向目标?
2)哪些措施可以操控干预微生物组?
3)未来的基于微生物的疗法如何与现实需求结合走出临床范式?
doi.org/10.1080/19490976.2024.2400579
在开发基于微生物组的治疗方法时,我们首先需要明确针对特定疾病的目标。这些目标可能是引发疾病的有害微生物,也可能是能够预防或改善疾病的有益微生物。通过对比患者和健康人群的微生物组,我们有望发现这些关键目标。
然而,在疾病状态下找出高度可重复的微生物组特征仍面临诸多挑战。
影响微生物组研究的复杂因素
确定疾病状态下高度可重复的微生物组特征仍然具有挑战性。
部分原因可能是由于影响队列微生物组的复杂混杂因素,包括:
宿主特异性因素
研究特异性因素
不同研究中心的宏基因组测序技术的差异可能会在多个环节引入偏差,包括:
在 MOSAIC 标准挑战赛中,44 家实验室提交了 7 项共享参考标准的结果,以评估微生物组研究方法设计的影响。
16s 测序显示厚壁菌门与拟杆菌门的比例升高,鸟枪法宏基因组学却强调了相反的结果,引发了人们对两种测序策略结果可比性的担忧。
更深入地描述患者群体
尽管存在上述挑战,宿主微生物组研究仍然是确定治疗性微生物靶点的基础。
精心设计队列:有助于克服潜在混杂因素的影响。
选择不同地理位置的患者:有助于区分真正的致病物种和与疾病无关的过客菌株。
改进数据收集方法,深入描述患者表型:
是因还是果?——前瞻性队列设计
使用临床前前瞻性队列设计,例如:调查炎症性肠病 (IBD) 患者一级亲属微生物组的 GEM 研究,可以识别出驱动早期亚临床发病机制的微生物,从而有可能解决“先有鸡还是先有蛋”的难题,即混淆差异丰富的微生物是疾病的原因还是结果。
从干预研究中收集样本可能有助于纳入更好的对照,并更深入地了解治疗效果和潜在机制。
最后,使用面向微生物组输入的人工智能模型,例如微生物组可区分可解释时间规则引擎 (MDITRE),可以更好地利用纵向时间序列数据来揭示与宿主疾病表型相关的微生物靶标。
微生物靶标的可重复性和有效性
可重复性一直是微生物组研究的一大挑战。虽然使用报告指南和一致的测序和分析流程方法可以提高队列的可复制性,美国国家标准与技术研究所即将推出的人类粪便参考材料可能有助于跨研究中心对测序结果进行有意义的比较。
该人类粪便参考材料是通过收集健康捐赠者粪便样本,在-80°C下均质化和储存,然后详细表征,包括宏基因组测序、流式细胞术、培养组学、代谢组学。
在未来的微生物组研究中,实验室可以使用该参考标准来对其协议进行基准测试并描述其流程中的偏差。
在群体中确定疾病特异性微生物靶点后,可以在临床前动物模型中测试微生物的效果。
这种方法已用于验证临床 IBD 相关肺炎克雷伯菌菌株在易患结肠炎的无菌小鼠模型中的致病作用。
类似地,在瘦人中发现Akkermansia muciniphila菌株可以逆转肥胖小鼠模型中高脂饮食引起的代谢紊乱,包括胰岛素抵抗。
通过对患者群体进行大规模数据挖掘,随后在适当的临床前小鼠模型中进行验证,可以概括出一种针对每种疾病识别微生物组治疗核心的方法。
一旦确定了致病或有益微生物目标,有效消除致病菌或将有益微生物引入人体肠道的策略仍然具有挑战性。例如:
抗生素治疗挑战:
粪便微生物移植(FMT)优点:
局限性:
传统益生菌挑战:
然而,开发新型微生物组导向治疗方法还需要克服监管障碍,比如:
针 对 病 原 体
通过使用生态导向治疗方法,可以消除人体内的致病微生物,而不会影响周围的微生物群。
噬菌体是一种很有前途的操控细菌的工具,因为这些病毒可以高度特异性地感染细菌,而不会伤害宿主。
优势
概念验证研究
在一项概念验证研究中,Elinav 及其同事表明,5 种噬菌体鸡尾酒疗法可以特异性地根除肺炎克雷伯氏菌,并改善易患 IBD 的无菌小鼠的结肠炎症。
研究意义:
提供了一个完整的研究框架:
替代方法是改造噬菌体或细菌以携带 CRISPR-Cas9 系统,或利用后生元或微生物产生的分子(如细菌素),可以选择性地消除病原体。
将有益微生物纳入肠道微生物群
下一代益生菌
为了突破传统益生菌或 FMT 的局限性,新方法包括开发下一代益生菌或以人为中心的微生物菌株,以预防或治疗疾病。
特点:
案例研究:Akkermansia muciniphila
在 32 名超重和肥胖志愿者中,这种菌株的给药安全且耐受性良好,改善了胰岛素敏感性、总胆固醇,并减轻了体重和肝功能障碍。
其他进展:
Faecalibacterium prausnitzii在IBD治疗中的临床试验
挑战
潜在解决方案
单菌株益生菌的效果可能有限,因为单菌株可能难以融入复杂的微生物群落并发挥治疗作用。
相比之下,由 8-12 个菌株组成的简单定义的微生物群落可以克服此类挑战。
案例研究
VE303(8个共生梭菌菌株)
GUT-108(11种细菌菌株)
其他应用
局限性
改进方向
复杂的联合体包含 100 多种微生物菌株,可以提供支架和功能潜力,以有意义地改变受体微生物组。
案例研究:Hcom2
优势
潜在应用
挑战
开发基于 FMT 的产品或下一代益生菌需要向 FDA 提交新药临床试验 (IND) 申请。
IND 通常需要评估产品中的微生物菌株、抗生素耐药性概况、相关菌株的基因组序列、制造工艺细节、稳定性数据、效力测试。
一旦研究小组或公司准备将产品投入临床试验,他们可以要求与 FDA 举行 IND 前会议,讨论最佳准备 IND 的方法。FDA 最近已经批准了基于微生物组的产品。
虽然在识别与疾病相关的微生物以及随后开发微生物组导向疗法方面已经取得了概念验证成功,但此类疗法能否在实际应用中提供有意义的临床益处仍是一个挑战。
粪便微生物移植(FMT)的局限性
优点:
局限性:
效果不一致:
肥胖和代谢综合征研究结果:
荟萃分析(6项随机对照试验)显示:
溃疡性结肠炎治疗中的挑战:
虽然 FMT 以外的微生物组导向疗法在治疗复杂疾病方面越来越受到关注,但有几个因素使这些治疗的成功变得复杂,包括:
个性化治疗计划:
挑战:如何根据个体微生物组特征定制治疗方案
需要:更精准的微生物组分析和个体化治疗策略
饮食的补充作用:
挑战:饮食对微生物组的影响如何与治疗协同
需要:整合饮食干预和微生物组治疗
治疗定位:
挑战:如何有效将微生物组疗法与现有治疗结合
需要:考虑疾病阶段和其他治疗方法的影响
高分辨率、纵向微生物组测序纳入多中心临床试验
将高分辨率、纵向微生物组测序研究纳入微生物组导向产品的多中心临床试验,可能提供更多可行的见解,了解接受者资料如何更好地预测微生物组导向治疗的临床意义终点。
通过这种深入的研究,我们可以更准确地评估微生物组导向治疗的临床价值,为个性化治疗方案的制定提供坚实基础。
机器学习方法在预测供体-接受者互补性中的应用
定制的机器学习建模方法也可能更好地预测供体和接受体的互补性。这种创新方法可能带来以下优势:
功能性缺陷纠正作为个性化治疗的新趋势
个性化治疗的新趋势正在从关注分类学变化转向纠正功能性缺陷,也可能被证明是一个更具临床意义的目标。
开发能够快速筛查个体肠道主要代谢物的技术至关重要。这些关键代谢物包括:
通过快速、准确地识别这些代谢物的变化,可以为每位患者量身定制最适合的微生物治疗方案。
应用案例:IBD患者的个性化治疗
以炎症性肠病(IBD)患者为例:
问题识别:
创新解决方案:
治疗目标:
这种方法开辟了微生物组治疗的新途径。
将基于微生物组的疗法与患者饮食相结合
饮食习惯能显著影响宿主的微生物群结构。这种影响主要表现在以下方面:
全植物饮食的影响
纤维干预的效果
饮食干预与微生物疗法的协同作用
虽然健康控制饮食的试验表明可以减轻体重、改善胰岛素敏感性和延长寿命,饮食是否会影响微生物疗法的疗效尚不清楚。
在一项试验中,与标准药物疗法相比,将 FMT 与抗炎饮食相结合能够更好地维持溃疡性结肠炎患者 1 年以上的缓解状态。
然而,特定饮食是否有助于微生物导向疗法在人体中的应用仍是一个未知数。近期,NIH 制定的《精准营养 2020-2030》营养研究战略计划可能为饮食-微生物群相互作用提供基础证据,为个性化饮食设计提供参考,从而提高基于微生物组的疗法的疗效。
在当前治疗模式中定位微生物组导向疗法
如何以及何时将基于微生物组的疗法与传统疗法同时使用,目前在很大程度上仍是未知数。疾病的阶段可能会影响治疗结果。
例如,已知给予 IBD 患者免疫抑制疗法(如肿瘤坏死因子抑制剂)可改善肠道菌群失调,这表明缓解期结肠更有可能支持健康细菌生长。
利用这一概念,使用传统免疫抑制剂治疗宿主,然后利用基于微生物组的疗法来维持健康的疾病状态,可能被证明是一种有用的范例,尽管尚未进行阐明这一概念的研究。
2024 年肠道微生物组健康世界峰会强调,基于微生物组的干预措施在改善人类健康方面取得了进展。
在过去几年中,FDA 批准了 2 种新的基于 FMT 的产品,用于治疗复发性艰难梭菌相关疾病。
大量的下一代益生菌正在开发和测试,以改善慢性人类疾病的结果,而开发联盟的新方法正在扩大定制治疗方案的前景。
测序技术、下游分析和预测模型的快速发展正在推动新一波数据驱动的微生物组研究,为更普遍的基于微生物组的治疗方法的发展提供信息。
然而,推进这些疗法的知识差距仍然存在。围绕这些差距的未来研究可能有助于更好地实现微生物组导向疗法的临床意义,为难以治疗的慢性病患者提供新的、潜在安全和有效的选择。
编辑
doi.org/10.1080/19490976.2024.2400579
会议强调了基于微生物组的个性化治疗方案的重要性,特别是在预防和治疗慢性疾病方面。这与我们肠道菌群检测高度契合。通过深入分析个体的微生物特征,为用户提供量身定制健康建议,优化治疗方案,提高健康管理的效果。
当然,全面准确地表征微生物组是推进微生物组研究和临床应用的关键一步。为了准确表征微生物组,建立一个大规模的数据库是必要的,这个数据库应当包括足够数量的样本,涵盖不同年龄,性别,地域等人群,确保数据的代表性和可靠性。
这种准确表征为后续的改善和干预措施奠定了基础。通过识别与疾病相关的微生物特征,可以用于预测疾病风险,指导预防措施的制定,开发更有针对性的干预策略。
科学技术的不断进步,我们有理由相信,肠道菌群检测和微生物组疗法的应用,将为我们的健康带来革命性的变化。
参考文献:
Jangi, S., & Hecht, G. (2024). Microbiome 2.0: lessons from the 2024 Gut Microbiota for Health World Summit. Gut Microbes, 16(1).

谷禾健康

霍尔德曼氏菌属(Holdemania)是一种棒状或杆状的革兰氏阳性细菌,属于厚壁菌门下的丹毒丝菌科。严格厌氧,不形成芽孢。
其表现出对糖类的代谢能力,能够发酵葡萄糖和其他糖类如阿拉伯糖、纤维二糖、赤藓糖醇等。并产生乙酸,乳酸和琥珀酸,有时还会产生少量甲酸。还发现Holdemania与色氨酸代谢呈正相关,这可能是其影响神经系统疾病的一个机制。
Holdemania通常被认为是人体肠道正常菌群的一部分,在谷禾健康人群检出率为29.33%,在维持肠道微生态平衡中起一定作用,大多数情况下,Holdemania属菌不会引起疾病,是人体共生的细菌。但研究发现其丰度过高与脂质代谢紊乱的严重程度相关。之前的报告还表明,肠道中高丰度的霍尔德曼氏菌会诱导炎症反应。
谷禾查找并整理了相关文献资料,发现霍尔德曼氏菌属(Holdemania)与脂质和葡萄糖代谢受损有关,其在II型糖尿病和肥胖等代谢性疾病中具有潜在危害作用。
此外研究发现在肝炎、抑郁症、肠易激综合征、肺动脉高压、心房颤动、痛风、过敏性哮喘和非酒精性脂肪肝等病症中,Holdemania的相对丰度明显增加。而在胰腺炎、肾结石中Holdemania的丰度减少。此外,一些研究表明霍尔德曼氏菌属可能对巴雷特食管和癌症治疗具有一定的积极作用。
鸭肉、羊肉、乳制品和藻类蔬菜等饮食因素以及利福昔明和伊匹单抗+纳武单抗等药物会增加Holdemanella属的相对丰度。锌过载时Holdemania的丰度也会增加。而使用二陈汤、夏桑菊、针刺疗法等有助于减少Holdemania的丰度。
关于霍尔德曼氏菌属(Holdemania)的研究资料相对有限,本文将根据该菌相关的研究结果以及检测实践数据来综合分享该菌的相关信息和健康特性及其干预措施。
1
细胞形态
霍尔德曼氏菌属(Holdemania)是一种棒状或杆状的革兰氏阳性细菌,最早于1997年被发现,大小约0.5-1.2μm×1.5-6μm,更长的细胞有中央到末端的肿胀。无鞭毛,成对或短链出现,不产生孢子。
该菌的细胞壁含有B组胞壁质类型(B1δ(L-Ala)-D-Glu-L-Asp-L-Lys),这种类型在任何其他细菌物种中均未观察到。
在琼脂上生长的菌落直径为1.0毫米。它们是圆形、完整的、半透明的,具有颗粒状外观。通过葡萄糖肉汤培养浑浊并含有白色沉淀物。
2
生长条件
该菌属通常在37°C下生长最佳,但也能在25°C至45°C的温度范围内生长。它们是专性厌氧菌,不能在有氧条件下生长。适宜生长的pH范围是6.5-7.5,最佳pH是7.0。人体大肠的环境非常适宜其生长。
3
代谢特征
Holdemania有严格的无氧代谢,通常表现出对糖类的代谢能力,而不是蛋白质分解。能够发酵葡萄糖和其他糖类。例如阿拉伯糖、纤维二糖、赤藓糖醇、甘露糖、麦芽糖、甘露糖、棉子糖、鼠李糖、核糖、山梨醇、淀粉、海藻糖或木糖等。
通过代谢葡萄糖的最终产物是乙酸,乳酸和琥珀酸,有时还会产生少量甲酸。不产生过氧化氢酶和吲哚。
此外还发现Holdemania与色氨酸代谢呈正相关,与异丁酸呈负相关。
4
抗生素敏感性
霍尔德曼氏菌属(Holdemania)对万古霉素具有耐药性,但对特定的抗生素敏感,如在一些研究中观察到的对青霉素类和大环内酯类抗生素的敏感性。
5
霍尔德曼氏菌属下的物种
Holdemania filiformis是该菌属的模式种,此外还可以对其他物种进行细分。
Holdemania filiformis
–Holdemania filiformis DSM 12042
Holdemania massiliensis
–Holdemania massiliensis AP2
未分类的 Holdemania
–Holdemania sp.
–Holdemania sp. 1001095H_141210_F2
–Holdemania sp. 1001302B_160321_E10
–Holdemania sp. cp07.24
–Holdemania sp. Marseille-P2844
–Holdemania sp. P15wH7
–Holdemania sp. P23wH1
具有致病性的物种:
Holdemania filiformis
Holdemania sp AP2
6
基因组结构
霍尔德曼氏菌属(Holdemania)的DNA G+C 含量(mol%):38
霍尔德曼氏菌属的模式菌种Holdemania filiformis有一条环状染色体,由3932923个碱基对组成,该基因组采用宏基因组测序法进行测序,已识别出4223个假定的蛋白质编码区。
7
人群中的分布特征
研究表明,霍尔德曼氏菌属(Holdemania)在不同人群中的分布特征存在差异。
儿童和老年人群中Holdemania的丰度较低,而成年人群中丰度较高。这可能与年龄相关的生理变化和免疫系统成熟度有关。性别方面,目前的数据并不充分,但初步研究指出Holdemania在女性中的丰度较高,而在男性中的丰度较低。
Holdemania属的分布特征与地理区域密切相关。不同地区的环境条件、饮食习惯及生活方式等因素都可能对其在人群中的存在产生影响。一项针对亚洲和西方国家的微生物群落研究显示,Holdemania属 在亚洲人群中更为常见。
8
与其他菌群的互作
Holdemania与其他微生物之间存在复杂的互作关系。这些互作可以是共生的、促进的或拮抗的,具体取决于环境条件和微生物群的组成。以下是一些可能的互作方式。
能够增强下列菌属的生长:
•Bacteroidales
•Bacteroides
•Odoribacter
•Peptococcaceae
能够抑制下列菌属的生长:
•Bifidobacterium
•Coriobacteriales
•Adlercreutzia
•Collinsella
•Porphyromonas
•Prevotella
•Clostridium
•Clostridiales incertae sedis
•Clostridiales Family XIII. Incertae Sedis
•Blautia
•Coprococcus
•Dorea
•Lachnospiraceae
•Ruminococcaceae
•Ruminococcus
•Dialister
•Campylobacteraceae
•Erysipelotrichaceae
谷禾查找并整理了相关文献资料,发现霍尔德曼氏菌属(Holdemania)的丰度变化与健康状态或一些疾病存在关联。
研究显示,在肝炎、抑郁症、肠易激综合征、肺动脉高压、心房颤动、痛风、过敏性哮喘和非酒精性脂肪肝等病症中,Holdemania的相对丰度明显增加。
Part 1
Holdemania高丰度相关
前列腺炎、肝炎

霍尔德曼氏菌属(Holdemania)属于丹毒丝菌科,是一个相对不典型的属。它存在于各种环境中,包括动物的胃肠道。一些观察性研究表明,在患有肝炎、肺炎和神经炎症等各种炎症性疾病的患者肠道中,Holdemania的丰度显著增加。
推测其丰度的变化可能影响肠道屏障的完整性,促进微生物产物易位到血液中,诱导IL-8的产生,这被认为是一种潜在的促炎机制,从而引发炎症反应。此外发现Holdemania数量增加还会带来前列腺炎风险,可能与前列腺炎风险呈正相关。
抑郁症

研究发现,抑郁症患者的肠道菌群发生改变,其中Butyricicoccus、Coprococcus、Faecalibacterium、Fusicatenibacter、Romboutsia的含量持续下降,而Eggerthella、Enterococcus、Flavonifractor、Holdemania 和 Streptococcus的含量增加。
这些肠道菌群可能通过影响色氨酸代谢来调节大脑功能。一些编码色氨酸合酶基因的细菌,如肠球菌、乳杆菌和Oscillibacter,被发现在研究中增加。Trp和5-HTP(5-HT的前体)可以通过血脑屏障(BBB),成为脑内5-HT的前体。
同时在患有沮丧的个体中观察到Holdemania的增多,这表明这种细菌与抑郁症状之间可能存在联系。

抑郁症性高血压(DEP-HTN)患者肠道细菌主要富集的菌落包括Eubacterium siraeum、Alistipes obesi、Holdemania filiformis、Lachnospiraceae bacterium 1.1.57FAA和Streptococcus salivariu,这5种细菌的丰度较高。
肠易激综合征

在属水平上,霍尔德曼氏菌属(Holdemania)在肠易激综合征(IBS)患者中显著高于对照组。IBS 粪便样本中有9个属显著丰富,包括Akkermansia、Blautia、Coprococcus、Granulicatella、Holdemania、Oribacterium、Oscillospira、Parabacteroides和Sutterella。
食物蛋白诱导的小肠结肠炎综合征(FPIES)患儿的粪便菌群失调,表现为:青春双歧杆菌显著减少,拟杆菌属(尤其是脆弱拟杆菌)、Holdemania、Lachnobacterium、鲁氏不动杆菌等菌增加,而双歧杆菌利用寡糖产生短链脂肪酸的代谢通路显著减少。
主观记忆障碍

来自大型前瞻性队列的老年女性的研究中,报告有2个或更多主观记忆障碍(SMC)的女性与报告仅有1个或没有的女性相比,霍尔德曼氏菌属(Holdemania)和脱硫弧菌科的相对丰度更高。
与主观记忆障碍(SMC)少于2个的女性相比,SMC 有2个以上女性的Holdemania和脱硫弧菌科的相对丰度分别高2.09倍和2.10倍。
肺动脉高压
肺动脉高压(PAH)患者可能导致丁酸球菌 (Butyricicoccus)和Holdemania的水平升高,同时降低无害梭菌(Clostridium innocuum)、Defluviitaleaceae UCG011、Eisenbergiella和Ruminiclostridium 5的水平 。
心房颤动
Holdemania与心房颤动(AF)风险相关,霍尔德曼氏菌属(Holdemania)与BMI显著相关,而霍尔德曼氏菌属通过BMI对心房颤动风险的中介作用比例为12.01%。
口腔溃疡

三种肠道微生物群与口腔溃疡呈正相关:Holdemania(比值比 [OR] = 1.005,95% 置信区间 [CI]:1.001-1.009,P = .019)、草酸杆菌(OR = 1.004,95% CI:1.000-1.007,P = .032)和瘤胃球菌科 UCG011(OR = 1.006,95% CI:1.001-1.011,P = .029)。
其他疾病
痛风
基于细菌 16sRNA 的测序结果表明,拟杆菌、Holdemania、Anaerotruncus 和其他几种细菌与痛风病呈正相关。
非酒精性脂肪肝
非酒精性脂肪肝(NAFLD)会导致Holdemania和瘤胃球菌丰度增加。
过敏性哮喘
霍尔德曼氏菌属(Holdemania)(P=0.046)与过敏性哮喘(AA)存在正相关关系。
慢性自发性荨麻疹
慢性自发性荨麻疹(CSU)和对照组肠道菌群Holdemania菌属相对丰度分别为0.04%、0.01%,两组间差异有统计学意义(P = 0.025)。
创伤性脊髓损伤
还分析了23名创伤性脊髓损伤(SCI)患者与23名健康对照,并报道了在创伤性脊髓损伤(SCI)患者中Parabacteroides、aliistipes、Phascolarctobacterium、Christensenella、Barnesiella、Holdemania、Eggerthella、nestiinimonas、Gordonibacter、Bilophila和Coprobacillus的丰度高于健康个体。
乙型肝炎肝硬化
研究发现,乙型肝炎肝硬化无腹水(HBLC-WOA)患者中:阿克曼菌属(Akkermansia)、另枝菌属(Alistipes)、肠杆菌属(Enterobacter)、霍尔德曼菌属(Holdemania)的丰度明显升高。
重症肌无力
此外,瘤胃球菌科UCG005属、 Holdemania、 Lachnoclostridium和真细菌属(反刍动物)与重症肌无力(MG)发生概率较大相关。
Part 2
Holdemania低丰度相关
霍尔德曼氏菌属(Holdemania)在一些疾病中的丰度较高,与此同时,某些疾病中霍尔德曼氏菌属(Holdemania)的丰度可能会降低。
胰腺炎

已确定七种微生物群与胰腺炎的发展显著相关。宿主基因驱动的拟杆菌目和拟杆菌纲与胰腺炎风险增加有关。粪球菌属和裂解真杆菌组(Eubacterium fissicatena)也对胰腺炎的发展表现出促进作用。同时,急性胰腺炎与变形菌门和毛螺菌属呈正相关,与Holdemania属呈负相关。
认知障碍

一项对西班牙裔为主的认知障碍(CI)和不认知障碍(NC)个体中口腔和肠道微生物群的测序研究发现:认知障碍(CI)个体表现出一些肠道菌属的丰度下降,包括Shuttleworthia、Holdemania和Subdoligranulum。
肾结石
肾结石患者中显著降低的菌群:霍氏真杆菌、多尔氏菌属、啮齿杆菌、厌氧棒菌属、Ruminiclostridium_5、霍尔德曼氏菌属(Holdemania)、Fusicatenibacter、罕见小球菌属(Subdoligranulum)、戴阿利斯特菌属、Parasutterella、嗜胆菌属。
Part 3
可能存在的益处
尽管霍尔德曼氏菌属(Holdemania)被发现在一些疾病中存在丰度的变化,但是也有一小部分研究发现霍尔德曼氏菌属(Holdemania)可能对一些疾病存在保护作用。
对巴雷特食管可能具有保护作用

研究发现,Alistipes、Lactobacillus、Prevotella 7和Ruminococcaceae UCG004是Barrett食管(BE)的危险因素,而Flavonifractor和Ruminococcaceae UCG004是食管腺癌(EAC)的危险因素。
另一方面,某些肠道微生物群似乎对BE和EAC都有保护作用。这些菌群包括Eubacterium、 Holdemania、Lactococcus和Actinomyces。
预防3-4期子宫内膜异位症

研究了肠道菌群与子宫内膜异位症不同阶段之间的因果关系。霍尔德曼氏菌属(Holdemania)和瘤胃球菌科UCG002可能在子宫内膜异位症的发展中起着至关重要的作用。
Holdemania和瘤胃球菌科UCG002参与肠道丁酸的产生。值得注意的是,瘤胃球菌科UCG002与调节性T细胞(Treg)呈正相关,其丰度与促炎细胞因子水平密切相关。
目前推测霍尔德曼氏菌属(Holdemania)是预防3-4期子宫内膜异位症的保护因素。相反,较高水平的瘤胃球菌科UCG002会增加1-2期子宫内膜异位症的风险。
在肿瘤治疗中的积极作用
霍尔德曼氏菌属(Holdemania)、多形拟杆菌和普鲁士尼茨菌在对肿瘤免疫治疗响应的患者中显著富集。
此外,肝细胞癌(HCC)恢复正常的群体有几个微生物属相对于延迟恢复(DR)患者显著富集,包括双歧杆菌、丹毒菌科、霍尔德曼氏菌属(Holdemania)。
某些食物和药物的摄入可能对Holdemania的丰度产生影响,导致其水平升高或降低。这种影响取决于食物的种类、药物的成分以及个体的具体健康状况。通过调整饮食和药物使用,可以间接调节Holdemania的丰度。
✦ 果胶
果胶的添加富集了厚壁菌门的微生物成员,包括梭菌纲下的Mogibacteriaceae科、Lachnospiraceae(毛螺菌科)以及产芽胞菌科。
其中,毛螺菌科的Butyrivibrio(丁酸弧菌属)、Coprococcus(粪球菌属)、Moryella、Holdemania(霍尔德曼氏菌属)和Bulleidia属对果胶表现出显著的特异性。
✦ 纳米硒
代谢组学和微生物组学分析结果表明,纳米硒SeNPs能够参与调节机体脂质代谢途径,提高霍尔德曼氏菌的丰度及其代谢产物短链脂肪酸(乙酸、丙酸)的含量。
✦ 利福昔明
利福昔明干预后丰度升高的菌属包括粪杆菌属(Faecalibacterium)、嗜胆菌属(Bilophila)、副萨特氏菌属(Parasutterella)、粪芽孢菌属(Coprobacillus)、霍尔德曼菌(Holdemania)、双歧杆菌属、真杆菌属(Eubacterium)。
✦ 伊匹单抗+纳武单抗
使用伊匹单抗结合纳武单抗治疗的患者肠道内厚壁菌门的Faecalibacterium prausnitzii和Holdemania filiformis菌以及拟杆菌门的Bacteroides thetaiotamicron增多。
✦ 针刺疗法
2型糖尿病患者,通过针刺疗法,在细菌属、种水平上,针刺组的埃希菌属(Escherichia)、肠球菌属(Enterococcus)、明串珠菌属(Leuconostoc)、霍尔德曼氏菌属(Holdemania)、罗氏菌属(Rothia)、普雷沃氏菌属(Prevotella)、罗斯氏菌属(Roseburia)、弯曲杆菌属(Campylobacter)、厌氧棍状菌属(Anaerotruncus)、厌氧消化链球菌(Peptostreptococcus anaerobius)和Shuttleworthia satelles的丰度下降。
✦ 低FODMAP黑麦面包
一项随机临床试验比较低FODMAP黑麦面包与普通黑麦面包对肠易激综合征患者破坏菌群的影响,结果显示食用低FODMAP黑麦面包减少了拟杆菌、Flavonifractor、Holdemania、Parasutterella 和克雷伯菌的丰度。
✦ 夏桑菊

夏桑菊是一种上市药物和膳食补充剂,用于治疗具有炎症的代谢疾病。夏桑菊可以引起肠道菌群组成的变化,颤杆菌丰度增加,胆菌属、Candidatus Stoquefichus、Holdemania、Parasutterella和Rothia减少。
✦ 二陈汤
二陈汤(ECD):使用二陈汤主要减少了Prevotella、Blautia和Holdemania菌属的含量,这些菌属及丙酸水平与宿主肥胖表型(特别是脂质代谢紊乱)存在正相关关系。
✦ 开菲尔
使用开菲尔干预后Holdemania属的显著减少可能通过炎症产生了积极的微生物群组成变化。
主要参考文献
Liu H, Li J, Guan C, Gao W, Li Y, Wang J, Yang Y, Du Y. Endometriosis is a disease of immune dysfunction, which could be linked to microbiota. Front Genet. 2024 Jun 21;15:1386411.
Gao M, Wang J, Liu P, Tu H, Zhang R, Zhang Y, Sun N, Zhang K. Gut microbiota composition in depressive disorder: a systematic review, meta-analysis, and meta-regression. Transl Psychiatry. 2023 Dec 8;13(1):379.
Wu F, Davey S, Clendenen TV, Koenig KL, Afanasyeva Y, Zhou B, Bedi S, Li H, Zeleniuch-Jacquotte A, Chen Y. Gut Microbiota and Subjective Memory Complaints in Older Women. J Alzheimers Dis. 2022;88(1):251-262.
Zhou F, Liu Y, Shi Y, Wu N, Xie Y, Zhou X. Association between gut microbiota and acute pancreatitis: a bidirectional Mendelian randomization study. J Gastroenterol Hepatol. 2024 Jun 18.
Wadop YN, Vasquez EL, Mathews JJ, Muhammad JAS, Mavarez RP, Satizabal C, Gonzales MM, Tanner J, Maestre G, Fonteh AN, Seshadri S, Kautz TF, Fongang B. Differential Patterns of Gut and Oral Microbiomes in Hispanic Individuals with Cognitive Impairment. bioRxiv [Preprint]. 2024 Jul 29:2024.07.27.605455.
Mishra AK, Lagier JC, Pfleiderer A, Nguyen TT, Caputo A, Raoult D, Fournier PE. Non-contiguous finished genome sequence and description of Holdemania massiliensis sp. nov. Stand Genomic Sci. 2013 Dec 15;9(2):395-409.
Zhang M, Fang J, Zheng C, Lin Q, Zhang J. Gut microbiota and autoimmune neurologic disorders: a two-sample bidirectional Mendelian randomization study. Front Microbiol. 2024 Apr 24;15:1337632.

谷禾健康

“民以食为天”,人的一生都离不开“吃”,并且“病从口入”,饮食直接或间接影响着我们的健康。不健康的饮食不仅导致多种疾病发生,甚至还可能减少寿命!
2019年,著名医学期刊《柳叶刀》发表了一篇大规模研究,分析了195个国家和地区饮食结构造成的死亡率和疾病负担。研究发现不良饮食造成的死亡人数超过吸烟等任何其他风险,改善饮食习惯可以预防全球五分之一的死亡。

并且可能颠覆以往认知的是,我国因为饮食结构而导致的高死亡率和疾病发生率,最大“杀手”可能不是糖和脂肪,而是钠摄入量高,全谷物摄入量低,水果摄入量低。换句话可以理解为:我们吃的食物偏咸、全谷物类食物摄入不足、蔬菜水果摄入太少,甚至疾病风险比高糖高油饮食的欧美国家还要高出了许多。
终止高血压饮食(DASH饮食) 和地中海饮食等健康饮食模式已被验证有益心血管健康,但考虑到西方饮食模式不易被中国人广泛接受,以及中国膳食文化、习惯的复杂性,今年,由北京大学临床研究所,复旦大学附属华东医院临床营养科、广东省食品营养与健康重点实验室、四川大学公共卫生学院营养系牵头共同发表在《美国临床营养杂志》,研究指出,中国心脏健康(CHH)饮食有效降低了血清总胆固醇(TC)和10年心血管疾病风险,并降低轻度高血压中国成年人的血糖水平。DASH饮食可将10年心血管疾病(CVD)风险降低10.3%。该研究中采用CHH饮食的影响更显著(降低27%)。

该研究的CHH饮食在开发阶段借鉴了健康膳食的主要食物特点,如低盐、富含蔬菜水果、增加全谷物和豆类等,以及主要营养素含量,包括脂肪、蛋白质、碳水化合物的比例、膳食纤维、钾、钙,并降低钠的摄入等。
CHH膳食方案经过严格的多中心随机对照试验证明了,只要食物搭配合理,符合中国人口味的健康中餐也能有效降低血压水平。
与此,同时《美国新闻与世界报道》今年发布了2024年度最佳饮食榜单。在这份最佳饮食排行榜中,“地中海饮食”仍牢牢稳居榜首,这也是该饮食模式7年蝉联榜首。“地中海饮食”确实在减重、减少“三高”、降低心血管疾病风险、防癌等方面有突出优势。
这些权威饮食研究表明只要将不健康的膳食转变为健康的膳食,就能够大幅度降低血压,并改善血脂和血糖,炎症且有望显著减少疾病事件。这一研究结果促使人们认识到通过健康膳食能够预防疾病、促进健康的巨大作用。
比起DASH饮食能够将10年内心血管疾病风险降低10.3%,中国心脏健康(CHH)饮食的效果更为显著,能降低27%,造成这一差异的可能原因是什么?
“一方水土养育一方菌”。
众多研究证明,饮食是塑造肠道微生物群的主要和关键因素。例如:
◆ 精制谷物、乳制品和茶或咖啡的摄入与颤螺菌目 (Oscillospirales)相对丰度增加有关;
◆ 甜饮料和甜点、动物脂肪和土豆与未分类的普雷沃氏菌种的相对丰度增加有关;
◆ 蔬菜和全谷物的摄入量与 Subdoligranulum丰度相关。
因此,肠道微生物组的独特性可能是CHH饮食显著降低心血管风险的重要原因。
一个人在日常生活中选择的食物的种类、数量、进食频率和时间安排等方面构成了他的饮食模式。不同的饮食模式具有不同的特点和健康益处或疾病风险。
不同人群可能需要符合自身需求的饮食模式。以下总结了一些主流饮食模式,帮助您了解其特点、益处和潜在不足,以便选择更适合自己的健康饮食。

首先要介绍一种典型的不健康饮食模式,也是现在很多年轻人都喜爱的西方饮食模式。
西方饮食是一种现代饮食模式,在西方社会中普遍存在。近年来,随着生活节奏加快,这种饮食在我国也越来越受欢迎。
“
西方饮食有什么特点?
•高糖和高碳水化合物: 西式饮食中的高糖和高碳水化合物主要来自于精制食品,如白面包、糕点、甜点和含糖饮料。
•高饱和脂肪酸: 这类饮食含有大量的饱和脂肪酸,主要来源于红肉、全脂奶制品和某些植物油(如棕榈油和椰子油)。
•高动物蛋白: 西式饮食中动物蛋白的摄入量较高,主要来自于牛肉、猪肉、鸡肉和奶制品。
•低膳食纤维: 膳食纤维摄入量低,因为这类饮食中新鲜水果和蔬菜的比例较低。
“
为什么这么多人选择西方饮食?
•口感和风味诱人:西式饮食的多样化和丰富的口味可以给人们带来饮食上的享受。
•方便快捷:许多西式食品,如快餐和即食食品,准备起来非常方便和快速,适合忙碌的现代生活节奏。
“
西方饮食对健康有哪些危害?
西方饮食在带来味觉上享受的同时,也伴随着许多健康隐患。
•易导致炎症:西方饮食与全身性慢性炎症和脂多糖易位有关,高脂饮食的摄入增加了促炎细胞因子的产生,导致全身性慢性炎症和脂多糖易位。这可能增加患心血管疾病、糖尿病和其他炎症性疾病的风险。
•增加慢性病风险: 长期摄入西方饮食与多种慢性疾病的风险增加有关,包括心血管疾病、某些类型的癌症、代谢综合征和认知能力下降。
•影响免疫系统: 西方饮食可能会影响免疫系统的功能,增加自身免疫性疾病和感染性疾病的风险。
“
对肠道微生物群有什么影响?
西方饮食中易于消化的非细胞营养素增加,可能会改变肠道pH值、微生物群组成和新陈代谢,影响肠道微生物群稳态的调节和维持。
与其他饮食相比,西方饮食与肠道微生物组多样性的显著降低有关,其肠道特征转向以拟杆菌属为主的肠道特征。其他丰富的物种属于Ruminococcus、Faecalibacterium、双歧杆菌属、Alistipes、Blautia、Bilophila。
并且由于纤维摄入较少和不同的微生物组成,相关的微生物群产生的短链脂肪酸也较少。
小结
综上所述,西方饮食虽然在某些方面为人们提供了便利,但其对健康的负面影响不容忽视。
为了改善健康状况,建议采取更加均衡和多样化的饮食模式,如地中海饮食,同时限制加工食品和高糖食品的摄入。

地中海饮食已经七年位于饮食榜单总体排名第一。
“
什么是地中海饮食?
地中海饮食是一种以蔬菜水果、鱼类、五谷杂粮、豆类和橄榄油为主的饮食风格,源自希腊、西班牙、法国和意大利南部等地中海沿岸的南欧国家。
这种饮食风格强调食物的天然、简单和清淡,富含营养,有助于减少患心脏病的风险,保护大脑免受血管损伤,降低中风和记忆力减退的风险。
“
地中海饮食有什么特点?
•植物性食物的丰富性:地中海饮食模式强调大量摄入水果、蔬菜、豆类、全谷物、坚果和种子。
•健康的脂肪来源:以橄榄油为主的不饱和脂肪,特别是单不饱和脂肪酸,作为主要的脂肪来源。
•适量的动物蛋白:适量摄入鱼类、海鲜、鸡蛋、家禽和低脂或脱脂乳制品。
•限制红肉和加工肉类:减少红肉和加工肉类的摄入,这些食品通常含有较多的饱和脂肪。
•适度饮酒:适量饮用红酒,尤其是晚餐时。
•香料的使用:使用香料和草药代替盐来增加食物的风味。
•高膳食纤维:通过食用全谷物、豆类和蔬菜来增加膳食纤维的摄入。
地中海饮食推荐的食物
特级初榨橄榄油;
新鲜水果,如蓝莓、草莓、无花果、桃子、芒果、梨和苹果;
新鲜蔬菜,如菠菜、羽衣甘蓝、芝麻菜、洋蓟、茄子、西葫芦、红薯、球芽甘蓝、芹菜、洋葱和胡萝卜;
藜麦、燕麦、鹰嘴豆、扁豆、杏仁、核桃、亚麻籽、奇亚籽;
植物奶,如杏仁奶或燕麦奶、希腊酸奶;
肉类如鸡胸肉、火鸡肉末、三文鱼、金枪鱼;
鸡蛋;
香草(新鲜或干燥)和香料,包括罗勒、牛至、迷迭香、百里香和大蒜
“
地中海饮食应该避免吃哪些食物?
地中海饮食的优点之一是限制性不强。这种饮食方法不禁止食用任何食物或食物种类,但会鼓励您限制食用某些食物的量。
•红肉:可以将红肉作为配菜。
•甜食:将甜食视为偶尔的庆祝食物,而不是日常的放纵。
•酒精:虽然适量饮用红酒是可以的,但过量饮酒可能会损害健康。不建议目前不喝酒的人饮酒。
•黄油:用更健康的替代品来代替黄油,比如橄榄油。
•全脂乳制品:减少食用冰淇淋和其他全脂乳制品。
•含糖饮料:不建议饮用含糖饮料,包括果汁。
“
地中海饮食的健康益处有哪些?
大量研究发现,地中海饮食存在以下的健康益处:
•降低患心脏病和中风的风险;
•预防认知能力下降和痴呆症;
•可能预防2型糖尿病;
•可能减轻炎症和自身免疫性疾病(如类风湿性关节炎)的症状和进展;
•可能有助于缓解抑郁症
改善心血管健康
地中海饮食已得到广泛研究,并一直被证明对心脏健康有益。
在希腊、意大利和日本等七个国家的一项大规模研究调查了13000名男性的饮食与心脏病之间的关系。研究表明,摄入的膳食脂肪类型(特别是不饱和脂肪)比脂肪总量更有益于心脏健康。
此后,包括2019年的PREDIMED研究和2022年的随机临床试验在内的众多研究发现,坚持地中海饮食与降低血压、胆固醇和体重有关,从而降低心血管疾病、死亡率、冠心病和中风的发病率。
降低癌症死亡率及糖尿病风险
一项对美国25,315名女性的前瞻性研究显示,那些坚持地中海饮食模式的人在25年的随访期间全因死亡率降低了23%。
这项研究还显示,较高的地中海饮食摄入量与20年随访期间未来2型糖尿病风险降低30%相关。地中海饮食模式可能还对癌症有保护作用。实际上,高度遵守这种饮食与普通人群中的癌症死亡率降低、癌症幸存者的全因死亡率降低,以及降低发展结直肠癌、头颈癌、呼吸、胃、肝和膀胱癌风险有关。
改善大脑健康
地中海饮食以海鲜、坚果、种子、特级初榨橄榄油、豆类、绿叶蔬菜和全谷物为主,对大脑健康有诸多益处。
多项研究表明,地中海饮食可以减缓人们大脑的衰老迹象。此外,越来越多的研究表明,这种均衡的饮食可以降低患痴呆症和阿尔茨海默病的风险。
事实上,一项2023年的研究和2018 年的研究表明,坚持地中海饮食的人患痴呆症的风险降低了 23%,并且对阿尔茨海默病进展的保护期为1.5至 3.5 年。地中海饮食不仅可以改善认知功能和预防神经退行性疾病,还可以改善心理健康。
一项2019年的研究显示,补充了鱼油的地中海式饮食改善了抑郁症患者的心理健康。多吃蔬菜、水果、全谷物、坚果和豆类,同时限制不健康食品,可以显著改善包括抑郁症在内的心理健康状况。
“
地中海饮食可以帮助减肥吗?
地中海饮食有助于减肥,特别是如果你从高糖、高钠和高饱和脂肪的饮食转变过来,但如果你想快速减肥,地中海饮食可能不适合你。
虽然地中海饮食不是为减肥而设计的,但这种饮食强调高纤维食物,如水果、蔬菜和全谷物,可以帮助你更长时间保持饱腹感。此外,用不饱和脂肪代替不健康的饱和脂肪已被证明可以改善代谢健康并有助于减肥。
地中海饮食已被证明可以促进长期减肥
多项研究表明,地中海饮食以植物为主的全食健康饮食方式对想要减肥的人有益。例如,2015 年的一项研究发现,与低脂饮食的人相比,遵循地中海饮食一年多的人减掉的体重更多。
利于体重维持和管理
2018年的一项大型纵向研究对32000多名参与者进行了评估,结果发现,对于基线体重正常的参与者而言,遵循地中海饮食可降低6至20年后体重增加和肥胖的风险,并降低腹部脂肪的风险。
“
对肠道微生物群有什么影响?
这些因饮食而导致的微生物组变化与短链脂肪酸产量的增加和代谢副产物(如乙醇、对甲酚和二氧化碳)产量的减少有关。
地中海饮食中富含的膳食纤维能够促进有益细菌的生长,如双歧杆菌、粪杆菌、Tenericutes、Dorea等。
膳食纤维还能增加产丁酸菌的丰度,如Roseburia hominis、Agathobaculum butyriciproducens、Faecalibacterium prausnitzii和厌氧菌Anaerostipes hadrus,这些细菌能够产生短链脂肪酸,对维持肠道屏障功能和抗炎作用至关重要。
两项干预研究将地中海饮食与特定分类特征联系起来,增加Faecalibacterium prausnitzii、Roseburia丰度,减少Ruminococcus gnavus、Collinsella aerofaciens、Ruminococcus torques丰度。
此外,地中海饮食中的植物化学物质,包括多酚、硫代葡萄糖苷、萜类等,也是肠道微生物群的重要底物,可以促进有益菌的生长,并通过微生物群依赖的生理效应,如短链脂肪酸介导的胰岛素抵抗衰减,对宿主健康产生积极影响。
并且地中海饮食中推荐的鱼类和海鲜是长链ω-3脂肪酸EPA和DHA的主要来源,这些脂肪酸对心脏健康有益,并且可能通过调节肠道微生物群的组成和功能来发挥作用。
这些因饮食而导致的微生物组变化与短链脂肪酸产量的增加和代谢副产物(如乙醇、对甲酚和二氧化碳)产量的减少有关。
“
地中海饮食是否存在不足?
•文化和可获得性:地中海饮食可能不适合所有人或每种文化,且某些地区可能难以获得地中海饮食中推荐的食物。
•成本:一些地中海饮食推荐的食物,如特级初榨橄榄油、新鲜海鲜和坚果,可能成本较高。
•个体差异:每个人的营养需求和健康状况都不同,地中海饮食可能需要根据个人情况进行调整。
•饮酒问题:虽然适量饮酒是地中海饮食的一部分,但并不适合所有人,特别是那些有酒精依赖或其他健康问题的人。
“
哪些人不适合地中海饮食?
地中海饮食通常对所有人都是安全的,包括老年人、儿童和孕妇。
小结
地中海饮食因其丰富的蔬菜、水果、全谷物和健康脂肪来源,被视为一种健康的饮食模式,有助于降低多种慢性疾病的风险并改善肠道健康。
但也存在一些局限性和不足,在采纳这种饮食模式时,应考虑个人的健康状况、文化背景和经济能力。

被评为最佳快速减肥饮食第一名的是生酮饮食。
“
什么是生酮饮食?
生酮饮食(keto)是一种高脂肪、低碳水化合物的饮食,旨在让你的身体进入酮症状态,燃烧脂肪以帮助人们减肥。这种饮食的目的是快速减肥,而不会感到饥饿或渴望。
“
生酮饮食为什么有助于快速减肥?
碳水化合物是人体首选的能量来源。当碳水化合物被摄入时,肝脏会将其转化为葡萄糖来为身体提供能量。然而,在生酮饮食中,你摄入的碳水化合物会比你平时少得多。
当你剥夺身体的碳水化合物,就会欺骗它,让它相信自己正在挨饿,迫使它几乎完全依赖脂肪,你的身体就会开始分解储存的脂肪来获取能量,这会导致你减肥。
研究表明,生酮饮食在短期内可以有效减肥,但从长远来看,它可能并不一定比摄入更多碳水化合物的低脂饮食更好。
“
生酮饮食有什么特点?
•高脂肪:生酮饮食中,脂肪通常占总热量摄入的70%至80%,这包括了饱和脂肪、单不饱和脂肪和多不饱和脂肪。
•适量蛋白质:蛋白质的摄入量需要控制,以避免身体将过多的蛋白质转化为葡萄糖,从而影响生酮状态。
•极低碳水化合物:碳水化合物的摄入量通常限制在每天20至50克,远低于一般饮食的摄入量。
•生酮比例:生酮饮食的生酮能力定义为脂肪克数与碳水化合物和蛋白质克数之和的比值,常见的生酮比为4:1或3:1。
•包括特定食物:饮食中鼓励摄入的食物包括肉类、鱼类、蛋类、健康脂肪(如橄榄油、椰子油)、非淀粉性蔬菜等。
“
生酮饮食有哪些健康益处?
生酮饮食对一些人的健康有益,包括:
•短期内减轻体重:生酮饮食通过限制碳水化合物的摄入,迫使身体燃烧脂肪来获取能量,有助于体重减轻。
•减少糖尿病患者对胰岛素的需求:生酮饮食有助于降低血糖水平,对糖尿病患者可能有积极影响。
•改善认知功能:一些研究表明,生酮饮食可能有助于提高认知功能和记忆力。
•减少炎症:生酮饮食中富含的抗炎食物,如浆果等,有助于减少身体的炎症反应。
•可能的神经保护作用:生酮饮食可能有助于改善线粒体功能,对某些神经退行性疾病有潜在的保护作用,并被发现可以减少癫痫患者的发作。
•癌症治疗的潜在辅助:一些研究表明,生酮饮食可能有助于减少某些癌症治疗的副作用,并可能对癌细胞的生长有一定的抑制作用。
生酮饮食已被证明可以帮助糖尿病患者降低糖化血红蛋白水平并减少胰岛素剂量。在一项临床试验中,采用极低碳水化合物生酮饮食的参与者将胰岛素剂量减少了一半,并在24周内达到正常血糖水平,比采用低热量饮食的另一组更快。
研究表明,生酮饮食可能对患有某些神经系统疾病的成年人有治疗作用,包括癫痫、阿尔茨海默病、偏头痛和神经胶质瘤,这些疾病是由人体代谢营养物质的方式发生紊乱引起的。
“
生酮饮食可能存在的健康风险?
虽然生酮饮食可以在短期内快速减轻体重,但同时也存在一些健康风险。包括以下几点:
•生酮饮食可能增加心血管疾病风险,如增加低密度脂蛋白(“坏”胆固醇)水平较高。
•水果和蔬菜摄入量减少可能导致营养缺乏,如维生素和矿物质摄入不足。
•可能影响肌肉消耗和运动表现。
•高蛋白质摄入可能增加代谢疾病的风险。
•肉类和乳制品的增加可能促进某些有害肠道细菌代谢产物产生,如氧化三甲胺和硫化氢。
•此外生酮饮食还可能导致肾结石和其他肾脏问题,脂肪肝和其他肝脏问题或是心脏病。
“
对肠道微生物群有什么影响?
临床前研究也表明,肠道微生物组的组成在响应生酮饮食时发生了显著变化,最明显的是:
•Akkermansia,乳杆菌属、Roseburia、副拟杆菌属(Parabacteroides)增加
•Turicibacter、Desulfovibrio、大肠杆菌和志贺菌属物种大幅减少。
——超重成年人
在涉及17名超重成年人的研究中,为期4周的生酮饮食显示在人肠道中放线菌门(Actinobacteria)和厚壁菌门的大量减少。具体来说,有益的双歧杆菌的19种物种减少了,而拟杆菌门丰度增加。这些变化部分是通过宿主产生酮体诱导的。
——癫痫儿童
在涉及12名严重癫痫儿童的为期3个月的研究中,遵循生酮饮食的儿童显示健康促进和消耗纤维的双歧杆菌属、直肠真杆菌(E.rectale)和Dialister属的丰度大幅减少。相反,儿童显示拟杆菌属和大肠杆菌属的丰度增加,后者部分归因于大肠杆菌(Escherichia coli)的增加。
“
哪些人不适合遵循生酮饮食?
不建议以下人群食用生酮饮食:
•患有胰腺疾病的人;
•患有肝脏疾病的人;
•甲状腺有问题;
•脂肪代谢紊乱;
•饮食失调或有饮食失调史;
•胆囊疾病或已切除胆囊的人
此外,孕妇、未接受过减肥医学建议的儿童、患有某些类型癌症的人、患有心脏病的人和高水平运动员都不应尝试这种饮食。
这种饮食还会对胰岛素和生殖激素产生巨大影响。糖尿病患者采用生酮饮食是有争议的,尤其是接受胰岛素治疗的人,至少需要仔细的医疗监测。
小结
总的来说,生酮饮食是一种特殊的饮食模式,它最大的优势在于能够快速减肥,并可能具有一定的神经保护作用,减轻炎症、改善认知功能,但同时也存在潜在的风险和挑战。
在开始生酮饮食之前,建议咨询医生或营养专家,以确保这种饮食模式适合个人的健康状况和生活方式。并且由于生酮饮食具有严格的限制性,不建议长期使用。

DASH饮食是一种预防及控制高血压的饮食模式,也是除地中海饮食外,最利于心脏健康的饮食模式。
“
什么是DASH饮食?
DASH饮食是推荐给想要预防或控制高血压的人的饮食计划。它通过增加纤维、水果、低脂(或脱脂)奶,和有益心脏健康的矿物质(包括钙、钾和镁)的摄入量,同时减少钠和不健康脂肪的摄入量来实现这一目标。
特点
•多不饱和脂肪:DASH饮食强调摄入多不饱和脂肪,如橄榄油、亚麻籽油、山茶油等,这些脂肪有助于改善心血管健康。
•全谷物:饮食中包括丰富的全谷物,如糙米、全麦面包和全麦意大利面,它们提供膳食纤维和必要的维生素和矿物质。
•蔬菜和水果:鼓励大量摄入各种蔬菜和水果,以提供抗氧化剂、维生素和矿物质。
•低脂乳制品:推荐食用低脂或无脂牛奶、酸奶和奶酪,这些乳制品是钙和维生素D的良好来源。
•限制饱和脂肪和胆固醇:减少黄油、起酥油、人造黄油、奶酪和熏肉等饱和脂肪和胆固醇含量高的食物的摄入。
•减少钠的摄入:DASH饮食建议减少钠的摄入,将钠的每日摄入量限制在2300毫克,以帮助降低血压。
“
如何开始DASH饮食?
您无需对饮食做出重大改变,只需从饮食习惯的小变化开始 DASH 饮食即可。
例如:
•每餐添加一份蔬菜或水果;
•每周吃两顿或两顿以上的无肉餐;
•使用香草和香料可以使食物更美味,而无需加盐;
•吃杏仁、山核桃或其他坚果代替薯片;
•尽可能将白面粉换成全麦面粉;
•午餐或晚餐后(或两者皆有)散步15分钟;
“
DASH饮食有哪些健康益处?
•降低血压:DASH饮食能够有效降低高血压,减少心血管疾病的风险。
•改善心脏健康:通过减少饱和脂肪和胆固醇的摄入,以及增加多不饱和脂肪和膳食纤维的摄入,有助于改善心脏健康。
•促进消化系统健康:富含膳食纤维的食物有助于维持肠道健康,预防便秘。
•控制体重:DASH饮食由于其高纤维和低脂肪的特点,有助于控制体重。
•预防糖尿病:研究表明,DASH饮食有助于预防2型糖尿病。
•抗炎作用:由于富含抗氧化剂和抗炎成分,DASH饮食可能有助于减少慢性炎症。
“
对肠道微生物群有什么影响?
DASH饮食与地中海饮食有许多相似之处,包括对全谷物、蔬菜、水果和低脂乳制品的摄入。因此,我们可以合理推测,DASH饮食也可能对肠道微生群产生积极影响,包括:
•增加有益菌群:DASH饮食中丰富的纤维和植物性食物可能促进有益菌群的增长,如双歧杆菌和乳酸杆菌、部分产短链脂肪酸菌,这些菌群与健康状态相关。
•改善肠道环境:DASH饮食可能通过增加肠道中的短链脂肪酸(SCFA)产生,改善肠道环境。SCFA有助于维持肠道屏障的完整性,调节免疫反应,并提供能量。
•减少炎症:DASH饮食中较低的饱和脂肪和反式脂肪含量可能有助于减少肠道炎症,改善肠道健康。
•促进肠道蠕动:DASH饮食中丰富的纤维摄入有助于促进肠道蠕动,预防便秘,并为肠道微生物提供营养。
“
可以通过DASH饮食减肥吗?
可以通过DASH饮食安全且可持续地减肥。但是,诀窍是留意份量并考虑您选择食物的卡路里含量。
通常,当人们从正常的饮食模式过渡到DASH饮食方式时,他们可能会自动减少卡路里,因为他们在饮食中添加了更多的蔬菜和均衡的膳食。
短期减肥
虽然DASH饮食并不是专门为减肥而设计的,但它可以帮助人们减肥。临床试验的荟萃分析表明,与采用对照饮食方案的患者相比,DASH 饮食方案的患者在8至24周内额外减轻了3.1磅体重,在24周内腰围减少了1.05厘米。
在另一项随机临床试验中,与对照饮食相比,DASH 饮食八周后体重和BMI显著下降。
长期减肥
如果您像 DASH 饮食一样从饮食中去除加工、含糖、含盐和高脂肪的食物,并定期锻炼,您很可能会继续减肥。
体重维持和管理
一旦达到目标体重,就应该能够通过DASH饮食来维持体重。纤维和蛋白质会让人有饱腹感,而卡路里含量足以让人精力充沛。
“
DASH饮食有什么优缺点?
优点
✔营养丰富;
✔不用计算碳水化合物或卡路里;
✔能够带来饱腹感——富含高纤维食物;
✔一份包含食谱的明确计划;
✔多样的食物和口味;
✔具有已证实的健康益处。
缺点
•成本:DASH饮食中推荐的食物,如新鲜水果、蔬菜和全谷物,可能比加工食品和快餐更昂贵;
•口味适应:一些人可能需要时间适应DASH饮食中推荐的健康食物,特别是如果他们习惯于高盐、高脂肪的食物;
•便利性:在外就餐时,遵循DASH饮食可能较为困难,因为餐馆的食物往往含有较多的钠和不健康脂肪。
•食物选择限制:需要避免或减少某些食物的摄入,如高饱和脂肪和高胆固醇的食物,这可能限制了食物的选择。
“
哪些人不适合尝试DASH饮食?
DASH饮食对大多数成年人来说都是健康的选择,除非医生建议限制某种营养素。例如,患有肾病的人可能需要低钾饮食。
小结
总的来说,DASH饮食是一种对大多数人来说健康且有益的饮食模式,特别是对于那些需要控制血压的人。
然而,DASH饮食可能不适合所有人。例如,严格素食者可能会发现在不摄入动物产品的情况下很难遵循DASH饮食的建议,因为它包括了低脂奶制品。
此外,对某些食物成分有过敏或不耐受的人,如乳糖不耐受者,可能需要调整DASH饮食以适应他们的特定需求。肾脏疾病患者也可能需要在遵循DASH饮食时调整蛋白质的摄入量。

亚洲饮食,通常指的是东亚、东南亚和南亚等地区的食物习惯,如中餐、日本料理和韩国料理,具有其独特的特点和对肠道微生物群及整体健康的影响。
亚洲饮食强调整体健康和平衡,注重天然食材的使用,富含蔬菜、水果、全谷物、豆类、鱼类和适量的肉类。以下是亚洲饮食的主要特点以及它们对肠道微生物群和健康的潜在影响。
“
亚洲饮食的特点
•蔬菜和水果的高摄入量:亚洲饮食通常包含大量的新鲜蔬菜和水果,它们是膳食纤维和各种植物化学物质的重要来源。
•全谷物:亚洲饮食中的主食经常是全谷物,如糙米、燕麦和小米,富含B族维生素和矿物质。
•豆类和豆制品:豆类,包括大豆、绿豆和红豆,以及豆制品如豆腐和味噌,是亚洲饮食中蛋白质的主要来源。
•鱼类和海鲜:亚洲饮食中常见的鱼类和海鲜,如三文鱼、金枪鱼和鳕鱼,是优质蛋白质和omega-3脂肪酸的丰富来源。
•适量的肉类:与西方饮食相比,亚洲饮食中的肉类摄入量通常较少,更倾向于使用瘦猪肉、鸡肉和偶尔的红肉。
•传统发酵食品:亚洲饮食中常见的发酵食品,如泡菜、味噌、纳豆和酸奶,含有益生菌,有助于肠道健康。
“
亚洲饮食对健康有什么影响?
•降低慢性疾病风险:亚洲饮食通常包含大量的蔬菜、水果和全谷物,这些食物富含纤维、维生素和矿物质,有助于维持消化系统的健康,预防慢性疾病,与较低的心血管疾病、2型糖尿病和某些癌症风险相关。
•改善消化健康:亚洲饮食中的高纤维和发酵食品有助于改善消化健康,减少便秘和肠易激综合征的症状。
•抗炎抗氧化:亚洲饮食中的茶、香料和某些蔬菜(如姜、大蒜)富含多酚和抗氧化剂,这些成分具有抗炎和抗氧化作用,能够减少氧化应激和炎症反应。
•促进体重管理:亚洲饮食中的食物通常热量较低,有助于控制体重和减少肥胖的风险。
•增强免疫力:亚洲饮食中的抗氧化剂和免疫调节成分有助于增强免疫力,减少感染的风险。
•改善认知功能:一些研究表明,亚洲饮食中的成分,如omega-3脂肪酸和多酚,可能有助于改善认知功能和预防神经退行性疾病。
“
亚洲饮食存在哪些不足?
•营养不均衡:在某些亚洲饮食中,可能缺乏足够的蛋白质和某些维生素(如维生素D和B12),特别是在素食主义者中。
•高盐摄入:一些亚洲饮食,特别是中国和韩国的饮食,可能含有较高的盐分,这与高血压和心血管疾病的风险增加有关。
•饮酒文化:在某些亚洲文化中,饮酒被视为社交活动的一部分,过量饮酒可能导致肝脏疾病和其他健康问题。
•加工食品的增加:随着全球化的影响,亚洲饮食中加工食品和快餐的摄入量有所增加,这可能导致不健康脂肪和糖分的摄入增加。
•传统烹饪方法:某些传统的烹饪方法,如油炸和烧烤,可能增加不健康脂肪的摄入。
•食物过敏和不耐受风险:亚洲饮食中可能含有一些常见的过敏原,如花生、海鲜等,可能会引起一些人的食物过敏或不耐受,影响其健康。
小结
亚洲饮食的健康益处主要体现在其丰富的植物性食物、低饱和脂肪和高纤维摄入。然而,高盐、高糖和加工食品的摄入也是亚洲饮食中需要注意的潜在健康隐患。
为了最大化亚洲饮食的健康益处并减少潜在风险,建议采取均衡的饮食方式,减少加工食品和高盐、高糖食品的摄入,同时增加蔬菜、全谷物和健康蛋白质的摄入。

素食饮食包括纯素饮食(Vegan Diet)和现在新出现的一种弹性素食。
纯素饮食是指一种不包含任何动物产品的饮食方式,包括肉类、鱼类、奶制品、鸡蛋、蜂蜜等。纯素饮食者通常会食用植物性食品,如蔬菜、水果、全谷物、豆类、坚果和种子等。
而弹性素食饮食又称为半素食饮食,不像传统素食饮食那样严格,而是多吃植物性食物少吃肉。不用完全遵循无肉生活方式又可获得素食主义带来的健康益处。可以大多数时候选择无肉餐,但在特殊场合仍可享用汉堡或牛排等肉类。
“
素食饮食的特点
•植物性食品的高摄入量:纯素饮食强调食用大量的植物性食品,包括全谷物、豆类、坚果、种子和蔬菜。
•更高的纤维摄入:由于植物性食品通常富含纤维,纯素饮食者可能会摄入更多的纤维。
•低饱和脂肪:由于不食用动物产品,纯素饮食通常含有较低的饱和脂肪。
•可能的高抗氧化剂和植物化学物质摄入:植物性食品富含抗氧化剂和植物化学物质,这些对健康有多种益处。
“
素食饮食对健康有什么影响?
•降低心脏病风险:由于纯素饮食中饱和脂肪和胆固醇的含量较低,可以降低心脏病的风险。
•改善血糖控制:纯素饮食有助于改善血糖控制,对于预防和控制2型糖尿病有益。
•促进健康的体重:由于富含纤维和低热量密度,纯素饮食有助于维持健康的体重。
•可能降低某些癌症的风险:一些研究表明,植物性食品中的抗氧化剂和其他植物化学物质可能有助于降低某些类型癌症的风险。
•改善肠道健康:高纤维饮食有助于维持肠道健康,促进良好的肠道菌群平衡。
“
对肠道微生物群有什么影响?
首先,素食饮食通常富含碳水化合物和纤维,增加了肠道微生物的多样性和丰富性。这种饮食模式特征是拟杆菌门、普雷沃氏菌属的数量增加,而厚壁菌门与拟杆菌门的比例降低。
此外,纯素饮食中的纤维成分,是结肠微生物的主要营养来源。膳食纤维在结肠中经细菌酵解可以形成短链脂肪酸,这对维持结肠细胞的营养和功能完整是必需的。短链脂肪酸如丁酸和丙酸对维持肠道屏障功能、调节免疫反应和降低炎症具有重要作用。因此,纯素饮食通过提供丰富的膳食纤维,有助于促进短链脂肪酸的生产,进而有益于肠道健康。
另一方面,纯素饮食中也包含多种植物化学物质,如多酚、类胡萝卜素、植物甾醇、木脂素和生物碱,这些物质已被证明对肠道微生物群具有调节作用。例如类胡萝卜素,有助于维持肠道免疫稳态,可能通过诱导IgA产生来提高抗肿瘤效率。
同时,纯素饮食中可能包含的益生元,如低聚半乳糖,对肠道微生物群也有积极影响。纯素饮食中的益生元成分可能有助于改善肠道微生物群的组成,促进有益菌的生长。
“
素食饮食存在哪些潜在不足?
•营养缺乏的风险:纯素饮食者可能面临某些营养素缺乏的风险,特别是维生素B12、铁、钙、锌和ω-3脂肪酸。
•蛋白质摄入可能不足:虽然植物性食品也含有蛋白质,但纯素饮食者需要更加注意蛋白质的来源和质量,以确保足够的必需氨基酸摄入。
•社会和文化挑战:在某些文化和社会环境中,纯素饮食可能会遇到社会接受度和可获得性方面的挑战。
•需要更多的计划和教育:为了确保营养均衡,纯素饮食者需要更多的计划和教育,以确保他们能够从植物性食品中获得所有必需的营养素。
小结
纯素饮食可以是一种健康的饮食方式,特别是当它被精心规划以确保足够的营养摄入时。然而,它也需要对食物选择和营养需求有更深入的了解,以避免潜在的营养不足。
对于那些选择纯素饮食的人,定期咨询营养专家或评估营养状况并提供个性化建议是非常重要的。

过去50年中,一种受到极大关注的饮食疗法是日常热量限制(CR),它被定义为在保持充足营养的同时,将饮食摄入量减少至低于维持体重所需的能量水平。观察性、临床前和临床试验的发现表明,CR可能将寿命延长1-5年,同时改善生活质量。
“
热量限制饮食的健康益处
最严格的CR随机试验来自国家老龄化研究所资助的CALERIE(减少能量摄入长期效应综合评估)联盟。CALERIE研究的发现显示,短期和长期CR都可以减少体重、皮下脂肪、内脏脂肪和肝内脂肪含量。
•这些改善与多种代谢益处相关,包括增加胰岛素敏感性、增强胰岛β细胞功能和降低空腹胰岛素。
•改善了代谢灵活性,降低了血压、低密度脂蛋白胆固醇和甘油三酯水平,并提高了高密度脂蛋白胆固醇水平。
•心血管疾病的10年风险降低了29%,这归因于氧化应激和炎症的减少,以及CR过程中维持的内皮一氧化氮功能的保护。
热量限制饮食减少了微生物表达的酶
这些酶能够使脂多糖A生物合成,从而限制了脂多糖(LPS)的产生,并以药理学上已知能刺激脂肪细胞褐化和减少内脏脂肪的方式抑制了LPS-TLR4途径。
“
热量限制饮食可能存在的不足?
•营养不足:如果热量限制不当,可能会导致某些营养素的摄入不足,影响身体健康。
•难以持续:长期的热量限制可能会导致饥饿感和食欲增加,使得饮食计划难以持续。
•可能的肌肉损失:如果热量限制过于严格,可能会导致肌肉质量的损失,影响身体的代谢率和整体健康。
•社会和心理压力:严格的饮食限制可能会影响社交活动和心理健康,造成压力和焦虑。
小结
综上所述,热量限制饮食在实施时需要综合考虑个人的营养需求、生活方式和健康状况,以确保既能达到减肥目标,又能维持健康。
思考
传统饮食与新兴饮食个性化饮食的兴起

地中海饮食长期以来一直被广泛认为是一种有效且均衡的减肥方法,可改善肥胖、2 型糖尿病和非酒精性脂肪肝等代谢疾病患者的代谢状况。
个性化饮食结合各种因素和不同人群,提高预测准确性
最近的研究表明,地中海饮食在人际间存在差异,即使个体食用同质饮食,依赖微生物组的代谢物的变异系数也不会显著下降。因此,营养学领域——与医学的许多其他领域一样——正朝着采用“个性化方法”的方向发展,以满足不同患者群体的特殊需求。因此,在 100 名糖尿病前期患者中,将地中海饮食的有效性与机器学习系统开发的个性化餐后目标饮食 (PPT) 进行了比较。
PPT 饮食基于一种人工智能算法,该算法整合了临床和微生物组特征来预测个人餐后血糖反应。
PPT 饮食的饮食建议以菜单的形式提供,餐点从研究生成的餐食库中选择。菜单设计有各种食物和餐食选项,以实现多样性,确保均衡饮食,并满足参与者的个人口味和喜好。
PPT 饮食平均总能量摄入量为 1881.0 kcal/天,碳水化合物、蛋白质和脂肪平均摄入量分别为 182.7、85.9 和 84.9 克/天。
结果表明,经过 6 个月的营养干预后,与地中海饮食相比,PPT 饮食导致 alpha 多样性显著增加,肠道微生物组成的变化更为显著。关于心脏代谢结果,作者发现坚持 PPT 饮食与 HbA1c、HDL 胆固醇和甘油三酯值的改善相关,并且通过因果中介分析,他们表明这种关联部分由属于拟杆菌目、毛螺菌科、颤螺菌目的三种细菌介导。
这些研究结果表明,虽然像地中海饮食这样的传统饮食评分依赖于将微量营养素、食物和慢性病风险联系起来的研究,在预测一般人群的结果方面相当有效。但较新的评分系统正在出现,它们考虑到各种因素和不同人群的独特特征,从而提高预测准确性。
其他的例子如:
黑色素瘤患者研究
对115名接受免疫检查点抑制剂治疗的黑色素瘤患者进行的分析表明,肠道微生物特征与治疗效果和免疫相关不良事件相关。
以瘤胃球菌科为主的微生物群患者相比拟杆菌科为主的患者反应更佳。纤维和omega-3脂肪酸减少摄入可能导致免疫疗法反应不佳。
溃疡性结肠炎管理
一项研究将标准治疗与从多名农村捐赠者获得的新鲜粪便微生物群移植(FMT)以及抗炎饮食相结合,发现这种组合能显著提高轻度至中度溃疡性结肠炎患者的临床反应和缓解率。
这些发现强调了个性化方法在增强肠道微生物的多样性和变化组成方面的潜在益处,这反过来可以在影响心血管健康标志物、癌症治疗反应和炎症性肠病缓解方面发挥关键作用。

个性化饮食离不开好的营养筛查工具
20多年来,营养风险筛查一直被认为是营养护理过程中的关键步骤。在临床营养领域,营养风险筛查被定义为“一种快速识别营养风险个体并分类进行营养干预的过程”。
在过去的几十年里,全球范围内为将营养风险筛查纳入议程付出了巨大的努力。这些努力取得了显著成效,最终促使许多国家的医院将营养风险筛查作为常规实践。然而,国际指南对于如何选择筛查工具缺乏明确规定。目前,不同人群和医疗保健环境中使用了多种筛查工具。
人工智能与机器学习在临床营养决策中的应用前景
近年来,医学领域见证了人工智能和机器学习的兴起。机器学习是人工智能的一个分支,涉及计算机(“机器”)从数据中学习的方式。这些技术不依赖于预先编程的规则,而是通过接触实例进行学习和改进,旨在帮助临床决策并提高护理质量和效率。随着患者病情和医疗技术的日益复杂,机器学习在医学中的重要性日益凸显。
机器学习可以通过广泛筛查、促进诊断、个性化治疗、预测临床结果、提供早期预警和评估患者对治疗的反应性等多种方式改善临床决策。
谷禾健康多年来一直在积累和构建不同人群样本数据库,利用机器学习技术,结合大量的肠道菌群数据和粪便代谢组学数据,以及人体测量、生化和临床数据和饮食习惯,获得了数十万独特的数据特征。这些数据被用来创建机器学习模型,实现预测不同的营养需求。根据肠道菌群和代谢数据,使用机器学习算法可以给出个性化的饮食建议,干预后的临床结果表现出了显著的改善。
通过对训练队列进行子采样,发现随着队列规模的增加,预测准确性也随之提高。模型方法旨在对出现特定可诊断症状之前检测出整体健康的潜在不利变化。这种检测可以指导饮食或生活方式的改变,以防止轻微问题升级为严重的健康问题,或促使进一步的诊断测试。
与现有的疾病特定指数不同,我们的指数涵盖多种疾病和多种营养,强调了泛疾病(或者说,一般健康)肠道微生物组特征。这种广泛的适用性在临床场景中可能特别有用,例如在选择粪菌移植(FMT)捐赠者时,肠道健康可以作为整体健康状况的反映。
主要参考文献
Best Diabetes Diets 2024,U.S.NEWs World Report
GBD 2017 Diet Collaborators. Health effects of dietary risks in 195 countries, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2019 May 11;393(10184):1958-1972.
Willett WC, Stampfer MJ. Current evidence on healthy eating. Annu Rev Public Health. 2013;34:77–95.
World Cancer Research Fund/American Institute for Cancer Research Diet, nutrition, physical activity and cancer: a global perspective. Continuous Update Project Expert Report. 2018.
Lloyd-Jones DM, Hong Y, Labarthe D. Defining and setting national goals for cardiovascular health promotion and disease reduction: the American Heart Association’s strategic Impact Goal through 2020 and beyond. Circulation. 2010;121:586–613.
GBD 2015 Risk Factors Collaborators Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1659–1724
谷禾健康
糖尿病是一种复杂的多系统代谢紊乱,其特征是高血糖,它还会导致并发症,降低生活质量并增加死亡率。糖尿病病理生理学包括β细胞、脂肪组织、骨骼肌和肝脏功能障碍。
1型糖尿病(T1D)是由免疫介导的β细胞破坏引起的。更常见的2型糖尿病(T2D)是一种异质性疾病,其特征是不同程度的β细胞功能障碍与胰岛素抵抗共同作用。
肥胖和2型糖尿病之间的密切关联涉及由中枢神经系统调节的通路,这些通路控制食物摄入和能量消耗,并整合来自外周器官和环境的输入。
患糖尿病或其并发症的风险代表了遗传易感性和环境因素之间的相互作用,包括营养食品的可用性和其他健康的社会决定因素。
糖尿病已经困扰人类数千年。从对这一疾病的“早期”描述到现代,目前人们对全球日益增多的糖尿病病人的流行病学、病理生理学、并发症和治疗选择的理解已经大大加深。
在过去的 50 年里,我们共同目睹了有关1型糖尿病和2型糖尿病病理生理学的知识爆炸式增长,而这种知识正在彻底改变糖尿病的治疗和预防方法。因此,任何关于糖尿病病理生理学和治疗进展的观点都不可能详尽无遗,或完全统一。
本文根据顶级期刊《Cell》近期发布的关于近50年有关糖尿病研究进展总结内容,与大家分享。
◮ 关于糖尿病的基础知识
糖尿病是由于胰岛素不足以刺激生理性葡萄糖处理,从而促进能量在脂肪组织、肌肉和肝脏中的储存而发展起来的。
糖尿病的表现范围从几乎完全的胰岛素缺乏症,如1型糖尿病(T1D)中出现的情况,到在胰岛素抵抗的情况下胰岛素相对缺乏的情况——2型糖尿病(T2D)的特征。
虽然糖尿病的诊断是基于测量血糖或糖化血红蛋白,但这种疾病应该被视为一个与多种合并症相关的多系统疾病。
◮ 糖尿病的大致分类
•1型糖尿病:由免疫介导的胰岛细胞破坏引起;
•2型糖尿病:与胰岛素抵抗和相对胰岛细胞功能不全相关;
•特定单基因疾病、药物毒性或胰腺功能不全引起的糖尿病综合征;
•妊娠期糖尿病
◮ 糖尿病的患病情况
2型糖尿病患者人数最多,1型糖尿病则不到所有病例的5%。2021年,全球糖尿病的患病率估计为6.1%,相当于5.29亿人,某些地区的患病率甚至高达12.3%。2型糖尿病占96%的病例,其中超过50%与肥胖有关。
糖尿病流行的趋势令人担忧,预计到2050年,将有13.1亿人患糖尿病。其他分析指出,2021年全球患病率已经超过10%。此外,2021年估计还有4.64亿人有糖耐量受损,2.98亿人有空腹血糖受损,总体上代表了糖尿病前期。
◮ 糖尿病大大增加了患其他病的风险
糖尿病大大增加了由心血管疾病和肾脏病造成的全因死亡率,且导致多种其他病症,包括失明、肢体丧失、慢性疼痛和残疾。
尽管升高的循环葡萄糖是任何原因引起的糖尿病的特征,但2型糖尿病是一种异质性疾病,不同人群亚群的结果存在差异。
鉴于肥胖与2型糖尿病之间的关联,有人认为,只要更加注重改善营养、增加身体活动和减少肥胖的政策,这种负担的大部分是可以预防的。然而,糖尿病的异质性表明,预防和治疗策略最好量身定制,以最大限度地发挥其在特定人群中的功效。
◮ 1型糖尿病概况
1型糖尿病(T1D)占所有糖尿病病例的5%–10%,由自身免疫介导的胰腺β细胞破坏导致。
胰岛素的发现使1型糖尿病从曾经的致命诊断变为可管理的慢性病。尽管取得了管理进步和新疗法的问世,只有约20%的患者能实现最佳血糖控制,且预期寿命较短。
◮ 遗传研究和疾病进展
全基因组关联研究(GWAS)鉴定了60多个与1型糖尿病相关的基因位点,显示出该病的高度遗传性。
对自然史的研究通过新生儿筛查和自身抗体筛查提供了对环境因素、疾病触发因素、胰岛自身免疫轨迹及代谢和免疫表型的见解。
重要观察之一是综合分析显示,有两种或更多胰岛自身抗体的人在15年内发展为临床1型糖尿病的风险超过80%。
◮ 疾病分期
1期T1D:存在两种或两种以上自身抗体
2期T1D:多种自身抗体阳性和血糖紊乱
3期T1D:明显的高血糖症
历史背景
在上世纪末,关于2型糖尿病的遗传学理解主要集中在少数几个基因座上,这些基因座与环境因素共同影响个体患病风险。
现代研究进展
随着全基因组关联研究(GWAS)的发展,现在已发现数百种与2型糖尿病相关的遗传变异。这些变异大多对患病风险的影响很小。
变异的作用机制
大多数与2型糖尿病相关的变异并不直接改变蛋白质功能,而是通过影响非编码基因组序列中的调控元件来改变基因表达的丰度。这些调控元件在特定的细胞类型和发育时间点发挥作用。
挑战与机遇
当前的挑战是将这些调控信号与具体的效应基因(转录本)关联起来,以了解其对糖尿病风险的影响。识别这些基因有助于理解糖尿病的机制,并可能发现新的治疗靶点。
单细胞多组学数据的应用
单细胞分辨率多组学数据集的出现为研究人员提供了工具,以更好地将遗传变异与其效应基因关联起来。这些数据结合高通量细胞表型分析,有助于评估变异相关的基因表达改变与疾病的关系。
临床应用的局限性
尽管这些研究提供了对糖尿病潜在病理生理学的生物学洞察,但目前尚无直接的精准诊断或治疗方法。
多样性研究的需求
大多数遗传学研究是在欧洲人群中进行的,因此需要在更广泛和多样化的人群中开展类似研究,以避免因缺乏多样性导致的健康差异,尤其是在糖尿病护理方面。
未来研究方向
研究人员对将心脏代谢和血糖特征的基因数据与2型糖尿病风险的基因数据结合起来很感兴趣。这些共享信号可能揭示潜在的组织和机制,通过这些机制,变异体会影响糖尿病或其并发症的风险。
聚类分析
通过不同聚类方法,研究人员正在识别2型糖尿病中常见的缺陷过程(如胰岛素作用、β细胞功能、血脂异常),并评估其临床效用。
20世纪90年代初,通过识别单基因糖尿病的突变,揭示了葡萄糖代谢与胰岛素分泌偶联机制的重要性。这些研究表明,关键的糖酵解酶如葡萄糖激酶的功能丧失突变影响胰岛素分泌。
转录因子HNF1A/HNF4A对内分泌胰腺的发育和维持至关重要,其发现为进一步研究胰腺和内分泌细胞发育的重要步骤奠定了基础。
基因变异的影响
罕见的完全渗透性突变在生命早期表现为糖尿病,而影响较小的等位基因也会增加2型糖尿病的风险。
胰腺功能的复杂性
研究发现外分泌和内分泌胰腺之间存在意想不到的联系。罕见的消化酶基因突变和与糖尿病相关的常见变异揭示了胰腺疾病(如胰腺炎和囊性纤维化)与内分泌细胞功能障碍之间的联系。
对β细胞功能受损的各种方式的机制理解得益于人类基因发现(下图)。
胰腺β细胞因环境和遗传因素受损的潜在方式示意图
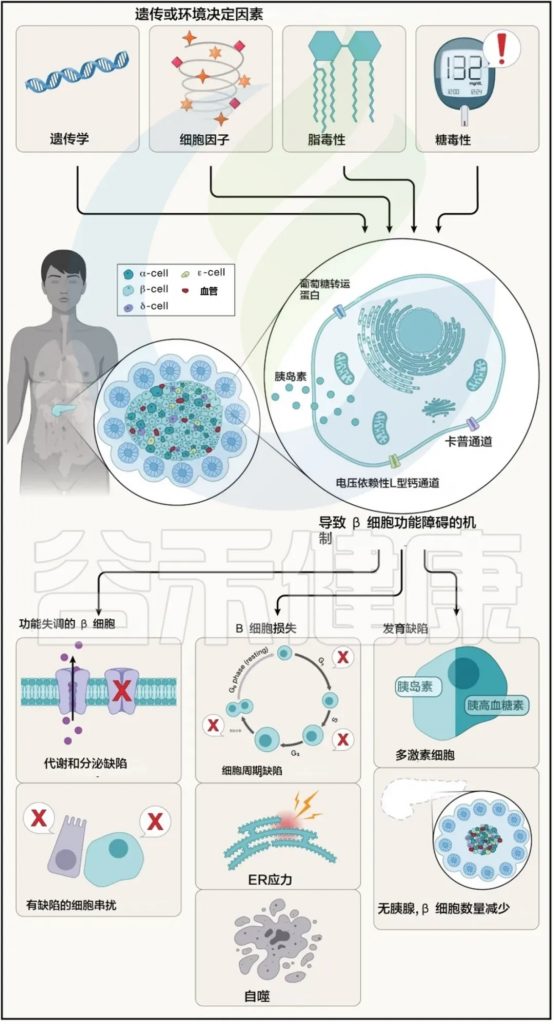
Abel ED,et al.Cell.2024
提升β细胞功能的兴趣
由于β细胞在维持正常葡萄糖耐受性中的关键作用,提高“功能性β细胞质量”被视为治疗1型和2型糖尿病的重要策略。
潜在治疗靶点
人类遗传学研究支持K-ATP通道和葡萄糖激酶为改善胰岛素分泌的潜在靶点。
SLC30A8基因中的特定蛋白质变体(ZnT8的锌转运蛋白)主要在胰腺β细胞中表达,并被证明可保护个体免受2型糖尿病的侵袭,因此相关药物的开发在进行中。
GLP-1受体(GLP-1R)
虽然在GWAS证实其作为治疗靶点的有效性之前人们就已经对它进行了广泛研究,但人类遗传学为其在降低血糖和改善心脏代谢健康方面的作用提供了有力证据。
GLP-1R激动剂不仅有效控制血糖,还具有减肥和降低心血管死亡率的积极作用。
开发新疗法的挑战
GLP-1类药物的多重成功显示了从多个角度治疗疾病生物学的巨大潜力,但也增加了专注于改善β细胞功能新疗法开发的难度。
社会健康决定因素(SDoH)指的是影响健康的社会条件,包括经济、教育、食品安全、医疗保健等因素。它们对包括2型糖尿病在内的多种疾病的发病和进展有深远影响。
SDoH通过复杂的途径影响2型糖尿病的发展,从生活方式行为到分子机制(例如HPA轴激活、微生物群失调)。粮食不安全和空气污染等具体因素已经被证明与糖尿病有关,表明需要多层次的干预策略。
粮食和营养不安全
粮食安全被定义为“所有人在任何时候都能获得足够的食物以过上积极和健康的生活”。
营养安全进一步扩展为“能够公平、稳定地获取、负担得起和利用促进健康、预防和治疗疾病的食品和饮料的状态”,包括饮食质量和社会健康决定因素(SDoH)。
粮食不安全的影响:粮食不安全导致水果和蔬菜摄入减少,精制碳水化合物、饱和脂肪和添加糖摄入增加,整体饮食质量下降。长期不良饮食习惯导致氧化应激、炎症和脂肪组织扩张。
粮食不安全与增加的炎症、生物荷负和肠道微生物多样性降低有关。社会经济压力则加剧了这种状态,影响补偿行为,并激活炎症途径。
空气污染
空气污染是重大环境健康风险,社会经济地位较低的人群受影响更大。关键污染物包括臭氧 (O₃)、二氧化氮 (NO₂) 和各种颗粒物 (PM),如PM2.5。
对糖尿病的影响:空气污染与胰岛素抵抗、血糖紊乱、高脂血症和2型糖尿病的发病率增加有关。观察性研究与孟德尔随机化研究支持其因果关系。
机制:初级启动途径:包括氧化应激、淋巴细胞活化、组织内DAMP产生及直接影响胰腺β细胞等。
次级效应途径:涉及全身炎症、神经内分泌失调、肝脂肪变性以及肠道微生物群失调等。
共同的病理生理影响:无论是营养不良还是空气污染,均通过氧化应激、炎症反应、HPA轴激活等机制影响糖尿病的发展。
粮食不安全和空气污染等因素可能在从胚胎到婴儿期影响后代未来的糖尿病风险。饥荒和空气污染对母体的营养和污染暴露都有证据表明会影响后代的代谢健康。
研究和政策发展:需跨学科团队的合作来深度研究SDoH的病理生理影响,指导新的干预策略和政策制定,以提高糖尿病预防和管理的公平性。
总之,粮食不安全和空气污染通过复杂的生物和环境机制显著影响2型糖尿病的进程。理解这些机制将有助于开发更有效的公共卫生政策和临床干预措施。
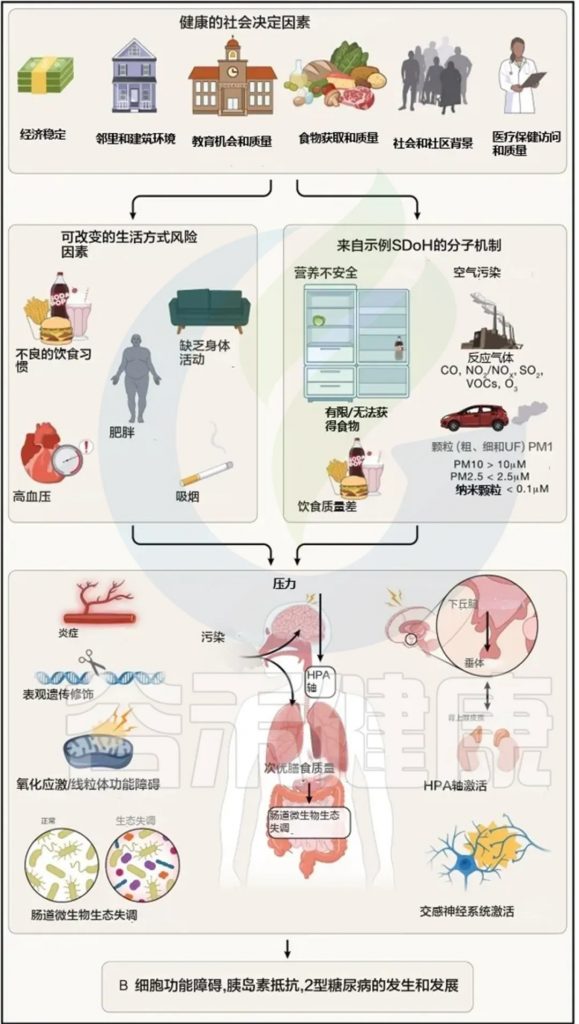
Abel ED,et al.Cell.2024
大量临床前数据强调了大脑在控制体重以及易感人群中胰岛素抵抗和肥胖发展中的重要作用。尤其中枢神经系统(CNS)在控制血糖和2型糖尿病病理生理学中的重要性。
◮ 重要性与作用
大量临床前数据显示,大脑在体重控制、胰岛素抵抗(IR)、肥胖及2型糖尿病发病机制中扮演关键角色。其中肥胖是2型糖尿病的重要风险因素。
食欲和能量消耗的调节对于肥胖的发生至关重要,受中枢系统全面调控。
肠道激素如生长素释放肽和瘦素,以及肠道微生物群的变化,在调节过程中起重要作用。
已在下丘脑和脑干中对这些调控神经回路的了解取得显著进展。
临床研究
人类研究重点关注中枢及自主神经系统如何整合体重调节及葡萄糖代谢,尤其在维持循环葡萄糖浓度中的作用。
动物模型研究
动物模型研究提供了许多见解,显示迷走神经及中枢神经系统回路对营养感知的重要性。
研究表明脂肪或糖摄入与多巴胺释放及暴饮暴食行为之间存在关联。
大脑与外周之间相互作用的示意图
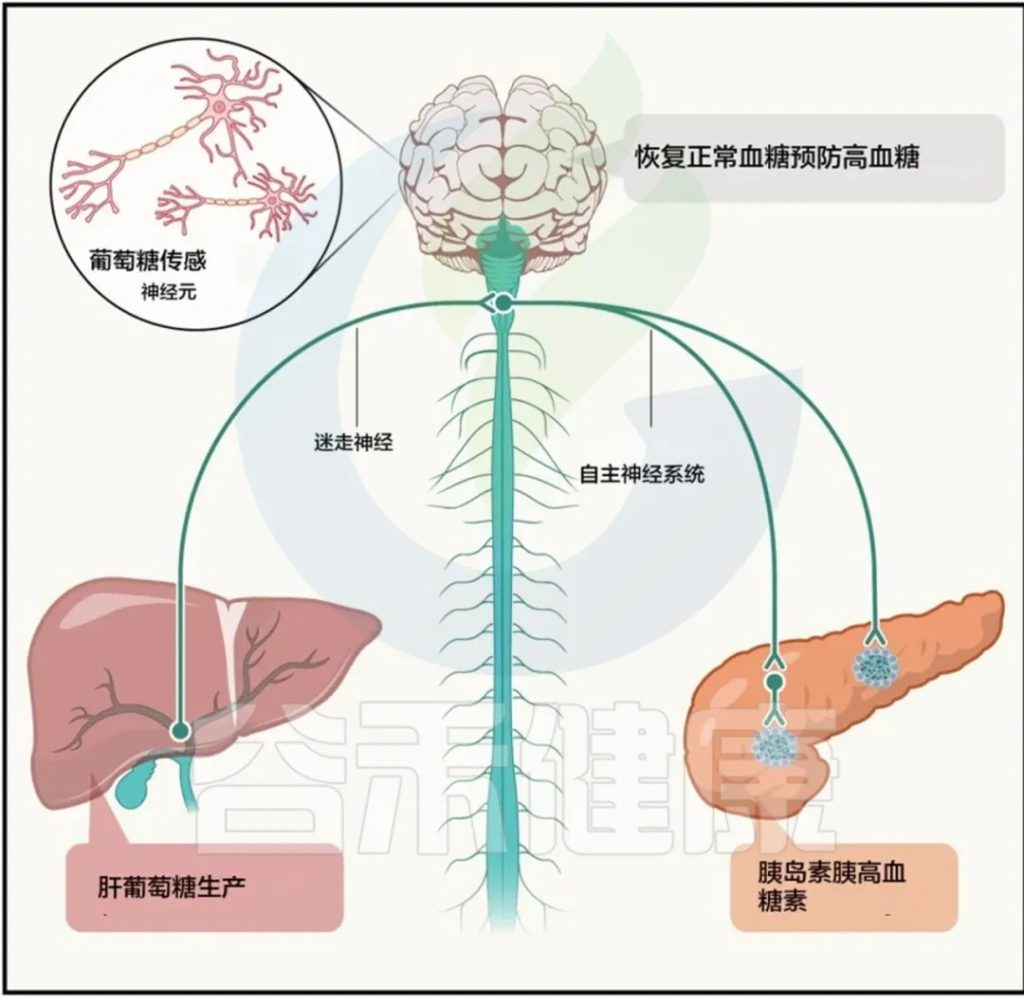
Abel ED,et al.Cell.2024
注:这些见解大多来自动物模型研究
为了更好地科普中枢神经系统(CNS)在糖尿病和代谢调节中的作用,可以从以下几个方面进一步理解:
基本概念
中枢神经系统(CNS)不仅仅是控制思维和行为的中心,它还在调节身体的代谢功能中发挥重要作用。特别是在血糖调节方面,CNS通过调控肝脏的葡萄糖生成和分解来维持血糖的稳定。
机制
下丘脑是CNS中一个关键区域,它通过神经信号影响胰腺的功能,调节胰岛素的分泌。胰岛素是降低血糖的主要激素,因此下丘脑的调控对于维持正常血糖水平至关重要。
胰岛素的作用
胰岛素不仅在外周组织中发挥作用,它在大脑中也有重要功能。研究表明,胰岛素可以通过鼻内给药直接作用于大脑,影响胰岛素的分泌和全身的胰岛素敏感性。
个体差异
不同个体对大脑胰岛素的反应存在差异,这种差异可能影响到胰岛素的分泌和代谢功能。这种差异也与肥胖和胰岛素抵抗(IR)有关。
FGF的潜力
成纤维细胞生长因子(FGF)在动物实验中显示出通过作用于大脑来缓解糖尿病的潜力。这种方法不依赖于体重减轻,而是直接改善葡萄糖代谢。
药物的CNS作用
一些用于治疗2型糖尿病的药物,如GLP-1受体激动剂,通过中枢神经系统发挥作用,影响食欲、体重和胰岛素分泌。
关联性
2型糖尿病与认知功能受损和神经退行性疾病(如阿尔茨海默病)之间存在关联。研究发现,糖尿病患者的大脑胰岛素反应性可能受损,这与认知功能下降有关。
潜在治疗
鼻内胰岛素和GLP-1受体激动剂在临床研究中显示出神经保护作用,可能降低痴呆症的发病率。
更具脑渗透性的药物
开发能够更好地穿透血脑屏障的药物,可能增强其神经保护作用,同时减少不良反应。
共同途径的探索
由于糖尿病和神经退行性疾病的发病率随着年龄增长而增加,研究共同的病理机制可能带来同时治疗这两种疾病的新方法。
胰岛素抵抗(IR)
当胰岛素无法有效地降低血糖时,称为胰岛素抵抗。这种情况不仅影响糖代谢,还导致脂肪组织释放更多的游离脂肪酸(FFA)进入血液。
脂解作用增加
在IR条件下,脂肪组织经历脂肪分解增加,产生更多的FFA。这些游离脂肪酸流入血液并扩散到其他组织,如肝脏和骨骼肌,进一步导致这些组织的发展胰岛素抵抗。
二酰甘油的作用
二酰甘油(DAG)是一种重要的信号分子。研究表明,在胰岛素抵抗的状态下,血浆中DAG水平升高。DAG可以与细胞膜上的蛋白激酶Cε (PKCε)结合,从而使胰岛素受体发生抑制性磷酸化。这一过程会限制胰岛素受体的激酶活性,削弱胰岛素的信号传导,导致细胞对胰岛素的反应降低。
特异性结合:DAG的不同立体异构体对作用靶点的结合亲和力不同,其中sn-1,2 DAG与PKCε结合最强,定位在质膜上,而其他异构体如sn-1,3 DAG和sn-2,3 DAG倾向于定位在脂滴和内质网(ER)。
神经酰胺的作用
神经酰胺是另一种导致胰岛素抵抗的脂质。高FFA水平促进长链神经酰胺(如C16和C18)的产生。神经酰胺通过激活特定的激酶,如蛋白激酶C ζ (PKCζ)和蛋白磷酸酶2A (PP2A),抑制关键信号分子Akt的磷酸化。这种抑制作用破坏了正常的胰岛素信号传导。
选择性影响:神经酰胺特异性地促进胰岛素抵抗,这是因为其他类似脂类(如二氢神经酰胺和鞘磷脂)未能在实验模型中表现出同样的效果。
异位脂质沉积
骨骼肌和肝脏中DAG和神经酰胺的积累会加剧胰岛素抵抗。这些组织的『选择性胰岛素抵抗』意味着通常应该增加葡萄糖吸收的组织变得不再敏感,反而可能继续将葡萄糖转化为脂质储存。
加速糖尿病的进展
当骨骼肌和肝脏对胰岛素的敏感性降低时,会加速糖尿病的进展。这些脂质代谢产物的积累不仅影响胰岛素信号传导,还可能引发脂质毒性,损伤细胞功能并加重代谢紊乱。
以上细节描述展示了脂质介质如何在分子水平上影响胰岛素信号通路,从而推动全身性胰岛素抵抗的发展。这些见解为开发针对性更强的干预措施和治疗策略提供了依据,以改善代谢健康并减缓或逆转糖尿病。
下面是肥胖和脂肪组织通过多种机制驱动胰岛素抵抗(IR)的详细总结:
1.脂肪酸羟基脂肪酸酯与胰岛素敏感性
脂肪酸羟基脂肪酸酯(FAHFA)的角色:FAHFA是一类由两个脂肪酸通过酯键结合形成的复合脂质。研究表明,FAHFA可以改善胰岛素敏感性,其水平在肥胖状态下和2型糖尿病中会降低。
机制:FAHFA通过与某些代谢重要组织中的G蛋白偶联受体结合,起到调节小鼠胰岛素敏感性、脂肪生成和能量消耗的作用。此作用途径表明FAHFA可能通过影响受体来调控广泛的代谢过程。
2.脂肪甘油三酯脂肪酶与FAHFA
脂肪甘油三酯脂肪酶(ATGL)的双重角色:ATGL是一种负责脂肪分解的酶,被认为可能在FAHFA代谢中也发挥作用。
ATGL可能充当FAHFA的合酶,通过改变ATGL功能,提供了FAHFA与2型糖尿病中的脂肪分解之间的潜在联系。调节ATGL功能的变化可能影响FAHFA的合成和分解。
3.前馈循环与全身性胰岛素抵抗
前馈循环:肥胖状态下,脂肪组织中多种信号分子的变化(包括FAHFA的减少)会驱动一种前馈的代谢循环。这种循环会进一步加剧全身胰岛素抵抗。
注释:代谢生理学中,”前馈循环” (feed-forward loop) 指的是一种生物学过程,其中某一信号或变化会增强或放大某一响应,而这种响应又会使初始信号或变化更加显著。上文中前馈循环是指肥胖状态下脂肪组织中多种信号分子的变化(例如 FAHFA 的减少)导致的一系列代谢事件。这些变化不仅直接影响机体的代谢状况,还通过促进某些过程或信号的放大,进一步加剧全身胰岛素抵抗。
具体来说,脂肪组织中信号分子的变化作为初始刺激,会引发代谢反应或变化,这些代谢反应可能通过各自的信号机制,反过来进一步促进初始信号或加大初始变化。如果这种循环不被打断,就可能导致长期的代谢失调,比如全身性的胰岛素抵抗,从而增加糖尿病等代谢疾病的风险。
4. 脂肪组织在全身性胰岛素抵抗中的作用
脂肪组织重塑:肥胖引起的脂肪组织重塑是全身性胰岛素抵抗、炎症以及葡萄糖和脂质代谢异常的关键驱动因素。
5. 高血糖与胰岛素抵抗的恶性循环
高血糖的影响:高血糖本身会通过增加己糖胺生物合成途径的通量,加剧胰岛素抵抗。这一途径生成尿苷二磷酸-N-乙酰葡萄糖胺 (UDP-GlcNAc),是N和O连接糖基化的前体。
O连接糖基化:包括Akt在内的胰岛素信号蛋白的O连接糖基化会进一步诱导胰岛素抵抗,形成恶性循环。
在胰岛素抵抗状态下,脂肪细胞的葡萄糖摄取功能受损。这种情况与脂肪细胞释放的胰岛素敏感因子和复合脂质的变化有关,这些变化会影响其他组织对胰岛素的响应。
肝脏和骨骼肌是胰岛素作用的主要目标组织,在胰岛素抵抗状态下,其糖代谢功能受损,尤其是肝脏的葡萄糖输出增加和骨骼肌的葡萄糖摄取减少。这些机制是糖尿病患者血糖水平升高的主要驱动因素。
糖原合成减少
在胰岛素抵抗的早期阶段,骨骼肌中胰岛素介导的糖原合成减少。这是因为胰岛素信号传导受损,影响了糖原合成酶的活性。
GLUT4易位受损
胰岛素通常促进葡萄糖转运蛋白4(GLUT4)从胞内储存池转移到质膜,以增加葡萄糖摄取。然而,在胰岛素抵抗状态下,这一过程受损,导致葡萄糖摄取减少。
葡萄糖氧化减少
由于胰岛素信号传导的缺陷,骨骼肌中葡萄糖的氧化能力降低,这进一步影响能量代谢。
长期影响
随着时间推移,骨骼肌胰岛素抵抗会导致肌肉萎缩、运动能力下降以及线粒体功能和质量的下降。
肝糖输出(HGP)增加
肝脏胰岛素抵抗的主要表现是肝糖输出增加。这是因为胰岛素对糖异生基因的抑制作用受损,导致肝脏中糖异生增加。
脂质积累
胰岛素抵抗还促进肝脏中脂质的积累,进一步影响肝脏功能。
组织间通讯的理解
未来的研究需要深入了解不同组织间的通讯机制,包括外泌体等新型介质在内的作用。
脂质动员机制
研究控制组织和细胞器之间脂质动员的机制,以及未表征脂质的功能,将有助于揭示2型糖尿病中的脂质失调。
复杂病理生理学的探索
进一步探索这些途径在持续胰岛素抵抗中的复杂病理生理学影响,将有助于开发新的治疗策略,改善代谢稳态。
综上所述,理解骨骼肌和肝脏的胰岛素抵抗机制及其与脂肪组织的相互作用,对于揭示2型糖尿病的发病机制和寻找新的治疗靶点至关重要。
近年来,越来越多的研究揭示了2型糖尿病与代谢功能障碍相关脂肪肝病(MASLD)之间的紧密联系,除了影响血糖稳态外,还对心血管并发症以及其他健康问题有显著的影响。
病理生理学背景
糖尿病患者常伴有肝脏代谢功能障碍,这是血糖稳态受损和糖尿病心血管并发症的主要因素之一。肥胖引发这些患者肝脏葡萄糖和脂质的异常增加。
MASLD的演变
MASLD从单纯脂肪变性(最初是可逆且普遍的状态)到代谢功能障碍相关脂肪性肝炎(MASH),可以进展至纤维化,这在严重情况下是主要的死亡原因。
下面是对2型糖尿病和MASLD相互作用的总结:
1.多重打击假说
这一理论解释不同患者发展严重并发症的原因,重点在于脂质蓄积后非实质细胞(NPC)的异常激活。对于早期疾病,GLP-1 基药物疗法可能有效,但对晚期纤维化无明显效果。
2.过量从头脂肪生成(DNL)
过量从头脂肪生成(DNL)在2型糖尿病患者中明显增加。健康人群中,胰岛素通过Akt信号通路抑制肝糖输出(HGP),同时激活脂肪生成的促进元素,如SREBP-1c。但在胰岛素抵抗状态下,FoxO1抑制失效增加葡萄糖生成,然而胰岛素仍然能推动DNL。
尽管胰岛素信号转导的某些功能失效,DNL却仍然活跃。这可能由于糖尿病患者CHREBP诱导Akt磷酸化调控机制存在异常,尤其是PHLPP2磷酸酶水平降低,导致了过量DNL。
3.胰岛素信号转导的动力学变化
在慢性高胰岛素血症背景下,正常的胰岛素信号分叉模型可能发生变化,影响胰岛素在早期和晚期阶段的作用。
早期阶段:胰岛素的快速作用通过抑制FoxO1来减少葡萄糖生成。
晚期阶段:延长的Akt活性则促进了DNL和脂质合成,潜在机制包括细胞自主信号、自主脂肪信号以及肠道介导的信号。
4.双向风险与相互促进
2型糖尿病和MASLD不仅拥有共同的风险因素,还可能通过如上机制双向增加各自的风险。研究显示,肝脏脂肪过多与2型糖尿病高度相关,即便在调整了BMI和其他潜在混杂因素后,二者之间的联系依然显著。
5.临床与研究启示
由于代谢功能障碍相关脂肪肝病(MASLD)在肥胖率较高的人群中表现为一种日益增长的健康挑战,当前的研究重点在于理清组织间通讯的复杂网络、揭示潜在的细胞机制、探索治疗新策略以及更好地管理代谢性肝病。
通过全面的机制研究和临床分析,我们可以更好地理解2型糖尿病与MASLD的复杂交互机制,为治疗和预防这些相关病理状态带来新的希望。
在2型糖尿病与代谢功能障碍相关脂肪肝病(MASLD)的研究中,理解肝脏脂质堆积及其进展至肝脏病变的过程尤为重要。以下是详细汇总和对当前研究状况的总结:
1.肝脂质过量与疾病进展
影像学发现:许多2型糖尿病患者在影像学检查中展示了肝脂质过量。然而,哪些患者将发展为有临床意义的肝脏疾病,以及进展的时间和诱导因素,仍不明确。
遗传因素:基因组广泛关联研究(GWAS)揭示了一些与体重和肝脏脂质积累相关的常见风险等位基因(如FTO、PNPLA3、TM6SF2和APOB)。然而,这些基因标记在预测疾病进展为代谢功能障碍相关脂肪性肝炎(MASH)方面的效能有限。
2.脂质管理障碍
2型糖尿病和MASLD患者的肝细胞可能有储存脂质的倾向,但难以在不损伤细胞的情况下管理过量的脂质。这导致了细胞的炎症反应和纤维化。
3.肝细胞的信号贡献
肝细胞在疾病中的作用:研究表明肝细胞不仅在血糖调节中起作用,还通过分泌促进NPC浸润和活化的趋化性和纤维化细胞因子,直接参与肝脏炎症和纤维化过程。
Notch信号的再激活:这是2型糖尿病和MASLD患者肝细胞炎症的一部分,影响HGP和DNL的增加。
4.临床研究和治疗前景
治疗性干预:使用如GalNAc修饰的反义寡核苷酸或siRNA,可以靶向性调制肝细胞信号通路,有可能改善病情。
5.当前研究的主要方向和挑战
高胰岛素血症的作用:2型糖尿病患者IR导致的高胰岛素血症,与肝脏DNL正相关,可能是MASLD病理机制之一。现有研究正在探讨抑制胰岛素分泌的策略,但其在糖尿病患者中的适用性仍需验证。
其他激素和营养因素:胰高血糖素、果糖和胆固醇也可能对肝脏脂质生成有影响。此外,肝交感神经系统的参与是否在脂质生成功能中有重要贡献,需要进一步探索。
通过这些研究,肝脏的代谢调节能力、遗传和环境因素的交互作用构成了2型糖尿病和MASLD在病理进程中的复杂图景。理解这些机制对于开发有效的治疗方案和风险评估方法至关重要。
尽管最近取得了进展,但许多问题依然存在,包括一下几点。
1.高胰岛素血症和非激素因素的作用
2型糖尿病的胰岛素抵抗(IR)显示出代偿性高胰岛素血症。人类数据表明血浆胰岛素水平与肝脏DNL正相关,动物研究证实胰岛素作用时机不当可能导致MASLD。有趣的是,奥曲肽通过阻断胰岛素分泌降低了大鼠的DNL标记物和肝脏甘油三酯。
目前,这一概念正在非糖尿病个体中使用二氮嗪(NCT05729282)进行测试,其结果是否适用于T2D患者尚不明确。其他激素如胰高血糖素也通过促进糖原分解和降低肝脏脂质起作用。
此外,果糖和胆固醇可能影响肝脏脂质生产。最后,非营养和非激素的肝糖输出(HGP)决定因素,如交感神经对肝脏的影响及其对脂质生产的共调节作用,仍不清楚。
2.脂肪性肝炎的空间决定因素
组织异质性:肝脏作为一种异质性组织,由于不同区域的氧张力和营养状态,产生了肝内糖异生和脂肪生成功能的分区。这种分区影响了MASH的区域特异性,表现为中枢周围或门管周围类型,后者尤为常见于儿童患者。
区域性亚型:代谢综合征和2型糖尿病患者倾向于发生门管周围类型的疾病。这种区域亚型与代谢和肝脏病理的发展程度相关,但其机制尚不清楚。针对肝脏区域特定通路(例如Notch、法呢醇X受体[FXR]、甲状腺激素受体[TR]和过氧化物酶体增殖激活受体[PPAR])的治疗可能提供新的干预策略。
3.纤维化消退的途径
尽管对肝脏促炎和纤维化途径的理解有所增加,但对纤维化清除及肝细胞途径影响纤维化消退的研究较少。我们推测,所有肝细胞决定的“纤维化开启”信号都有相应的“纤维化关闭”信号。
系统研究回归途径对肝纤维化的影响,类似于动脉粥样硬化病变的解决,可能带来重要的影响。对于部分对肠促胰岛素治疗抵抗的T2D患者,新的治疗靶点可能特别有价值。
4.双向肝细胞-非实质细胞串扰
虽然肝细胞在脂质过载中的作用被强调,但非实质细胞(NPC)对肝细胞功能也有显著影响。例如,肝星状细胞不仅是肝细胞再生的决定因素,还是一个潜在的调节因子。在2型糖尿病中,免疫细胞增加从而改变胰岛素敏感性。
6.对脂质超载的遗传适应
近来的研究揭示了MASLD/MASH患者肝细胞中FOX01的体细胞突变,这些突变可能是一种保护机制,减轻脂质超载的损伤。这种基因适应可能说明MASLD与2型糖尿病的关联。
7.与心血管疾病的关系
肝脏脂质过多增加了肝病相关死亡率的风险,但与2型糖尿病患者类似,MASLD/MASH患者的主要死因是心血管疾病(CVD)。鉴于直接加速CVD的常见并发症,需在临床前动物模型和人类中进行进一步机制研究以揭示其潜在机制。
糖尿病是一种影响全身的慢性疾病,会对多种系统造成广泛且复杂的影响,显著增加患病率和死亡率。以下是对糖尿病相关代谢异常和其主要并发症的详细分析。
增高的风险:糖尿病患者患动脉粥样硬化性心血管疾病(ASCVD)的风险增加2至5倍。
其他风险因素:包括高血压、血脂异常、肥胖和代谢综合征。
糖尿病性心肌病:糖尿病导致特有的心脏结构和功能异常,不完全依赖于冠状动脉疾病(CAD),而与异常代谢环境直接相关。
综合风险:没有单一机制可以解释CVD风险的增加,糖尿病引起的多种风险因素复杂相互作用导致CVD。
机制:与内皮细胞(EC)和血管平滑肌细胞(VSMC)功能障碍有关。
糖尿病影响:特征是内皮功能障碍,包括一氧化氮合酶功能下降和NO缺乏。
动脉粥样硬化驱动因素:糖尿病通过激活髓系细胞和炎症细胞(如单核细胞、巨噬细胞)的生成,直接加剧动脉粥样硬化。
风险因子和生物标志物:LDL胆固醇、血脂异常、HbA1c、肾功能和身体活动为疾病预测因子。新型生物标志物(如线粒体代谢物)可能用于风险分层。
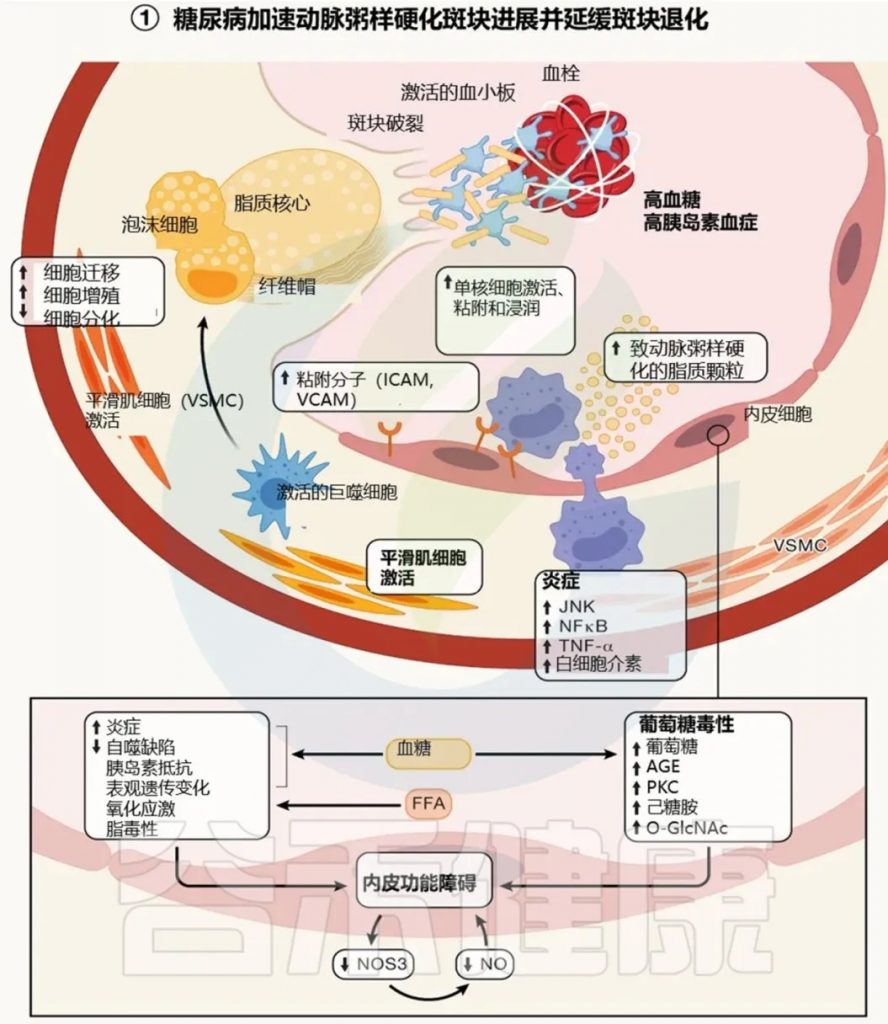
Abel ED,et al.Cell.2024
风险增加:糖尿病增加心力衰竭风险,这与冠状动脉疾病(CAD)风险增加无关。
机制:碳毒性(脂毒性和糖毒性)
氧化应激
线粒体生物能量学受损
线粒体解偶联
心肌兴奋-收缩偶联受损
促纤维化途径激活
选择性胰岛素抵抗引起的肥大性信号通路激活
人类研究证据:心脏移植研究显示,正常供体心脏在移植到糖尿病患者体内后数月内出现代谢异常。
观察到甘油三酯超载、神经酰胺等有毒脂质积聚、线粒体呼吸功能不全、氧化应激和炎症。
注:前驱糖尿病患者也观察到线粒体氧化缺陷,二甲双胍治疗可能减轻这些变化。
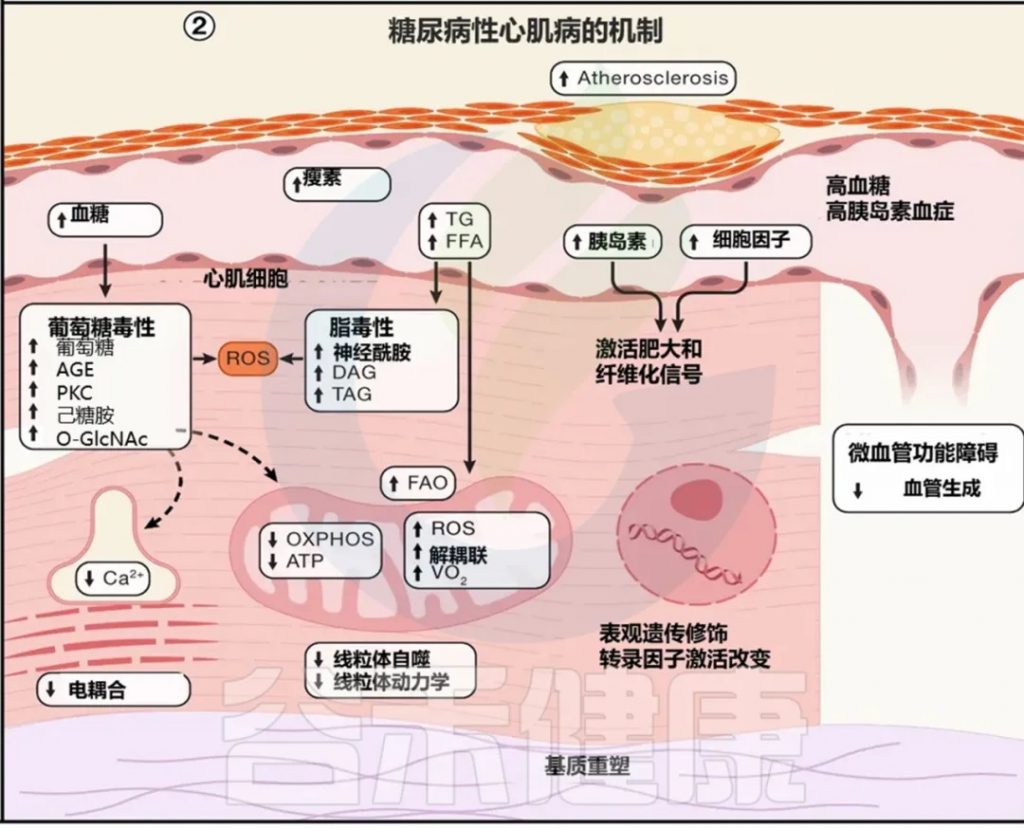
Abel ED,et al.Cell.2024
特征:糖尿病降低多种促血管生成因子的表达,导致血管生成信号通路紊乱。
影响:增加外周血管疾病和严重肢体缺血的风险。
机制:VEGF抵抗
一氧化氮信号受损
血管生成干细胞前体水平降低
周细胞丢失
微小RNA和长链非编码RNA的失调
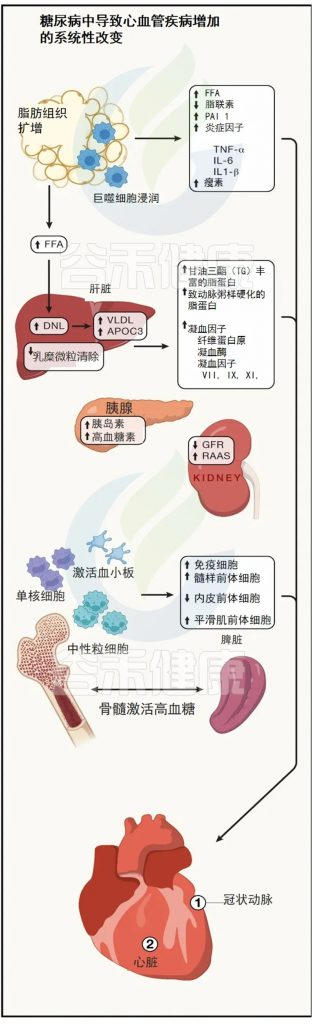
Abel ED,et al.Cell.2024
特征:白蛋白尿和估计肾小球滤过率(eGFR)降低。
患病率:约40%的糖尿病患者会发展为DKD。
肾血管功能障碍:肾小球高滤过是早期特征之一;
肾小管肾小球反馈机制导致肾小球传出动脉收缩。
肾小球和肾小管上皮细胞变化:
足细胞代谢改变,氧化应激增加。
RhoA/Rac1通路导致足突消失。
mTOR和生长因子信号改变导致足细胞增大。
足细胞丢失是疾病进展的关键步骤。
近端小管(PT)细胞:
高代谢活性,负责大量水和盐的重吸收。
初期细胞大小和数量增加,后期出现能量耗竭和细胞身份丧失。
线粒体损伤激活炎症途径。
小结
总的来说,糖尿病通过多种复杂的机制影响心血管系统和肾脏,涉及代谢异常、血管功能障碍、炎症反应和表观遗传变化等多个方面。
这些发现为理解糖尿病并发症的发生机制和开发新的治疗策略提供了重要的科学基础。
传统疗法的局限:早期治疗通常在T1D的3期开始,对于改变疾病进展的效果有限。
新进展:创新的分期系统为早期干预提供了框架,报告聚合抗CD3单克隆抗体teplizumab治疗延迟T1D进展的结果。这一研究促使FDA批准teplizumab作为首个T1D疾病改良疗法。
病毒传播与诊断挑战:目前缺乏统一的筛查指导方针,正测试多种自身抗体筛查策略。
胰岛素管理技术(如优化的药物动力学胰岛素、胰岛素泵和持续血糖监测设施)不断革新。
β细胞替代疗法:胰腺或供体胰岛移植显示出β细胞替代的可行性,但供体资源有限。
干细胞技术:通过多能干细胞分化得到的β细胞有望克服移植局限性,Vertex制药公司正在测试相关疗法(VX-880)。
异质性挑战:T2D患者的疾病表现由于遗传和环境因素而异,例如,非洲-加勒比和亚洲人群的T2D表现差异明显。
◮ 精准医疗的方法
临床分层工具:如BMI和eGFR用于HbA1c反应的预测,最新的研究表明此类因子可以指导选择更有效的治疗方法。
复杂数据整合:利用机器学习分类法如集群分析,识别糖尿病亚型,与特定并发症风险相关。
◮ 临床实施与研究挑战
标准化的必要性:研究需要一致的方法来评估精准治疗的优劣及其经济性。
证据差距:精准糖尿病治疗需要更多随机对照试验来证明其临床优势和成本效益。
精准医疗成功的例子:例如葡萄糖激酶突变的单基因糖尿病无需药物治疗,而HNF1A突变可用磺酰脲类药物有效管理。
综合来看,1型糖尿病的治疗正在向提前预防和细胞治疗方向发展,而2型糖尿病的精准医疗方法强调对不同人群亚型的特定管理。
随着研究的深入和技术的进步,这些策略有望显著改善糖尿病患者的治疗效果和生活质量。
在2型糖尿病的精准医疗和全球健康公平领域,取得了许多令人瞩目的成就,但仍面临资源可及性不均与种族多样性不足等挑战,并且新型疗法不断涌现,进一步推动病情管理和并发症预防。
◮ 资源限制与成本效益
2型糖尿病精准医疗是否能在不同资源环境中实施仍不明确,当前诊断和治疗的费用较高,尤其在中低收入国家。
如果精准医疗能提高治疗精度,减少资源浪费,则可能长期节约成本,但这些长期获益难以精准量化。
◮ 全球资源差异
许多中低收入国家患者难以获得基本药物,更复杂的精准医疗诊断尚难普及。简便的分层方法和决策支持工具可能在这些地区更具实用性。
糖尿病护理的差距往往影响社会经济地位低、少数民族群体。未经过周密计划和执行的精准医疗可能导致不平等加剧。
◮ 种群多样性的影响
研究多样性不足:大多数精准医疗研究在欧洲白人群体中进行,跨种族的应用性受限。需在不同种族背景下进行更多研究,以开发具有全球代表性的解决方案,适应不同遗传机制。
基因组研究局限:多样性缺乏的基因组研究可能导致偏见。例如,某些族群的2型糖尿病特征存在重大差异。开发跨祖先适用的基因风险评分,不同族群的参与和持续研究至关重要。
总体来看,精准医疗已经在2型糖尿病领域取得长足进步,但其广泛和公平实施受到资源、技术和种族多样性等多个因素的限制。未来的研究应致力于开发合适的工具和管理策略,以改善全球范围内的健康结果。
药物创新
GLP-1受体激动剂、DPP-4抑制剂和SGLT-2抑制剂在改善血糖及心血管、肾脏健康方面展现无与伦比的效果。
GLP-1RA的增强版本显示出非凡的减肥和心血管保护作用。SGLT-2抑制剂同样有助于不同心肾健康目标。
开发多激动剂如Tirzepatide(GIP受体-GLP-1R共激动剂),有效性超越单一疗法。此类疗法在减重和改善心血管风险方面效果显著,开创了治疗新方向。
基于GLP-1的治疗心脏代谢和神经退行性疾病
Abel ED,et al.Cell.2024
针对不同疾病机制的新疗法和联合疗法的临床试验持续进行,探索能够提供更高效、个性化处理方案的创新疗法。
多种基于GLP-1的联合疗法目前正处于临床后期开发阶段,期待它们在提高血糖控制和体重减轻效果的同时,继续展现心血管益处。这些治疗选项包括:
合成及联合应用疗法
-卡格列肽与索马鲁肽每周一次联合使用;
-胰高血糖素受体GLP-1R共激动剂如survodutide;
-GIP受体拮抗剂—GLP-1R激动剂抗体;
-三联胰高血糖素-GIP-GLP-1R多激动剂。
注:卡格列肽是一种肠促胰岛素激素,用于治疗2型糖尿病。
索马鲁肽,英文名称 “Semeglutide”,是一种新型的,长效的,每周皮下注射一次的 GLP-1 类似物,是基于利拉鲁肽基本结构而开发的长效剂型,其治疗2型糖尿病的效果更好。
新兴口服疗法
-包括danuglipron和orforglipron在内的小分子口服GLP-1R激动剂,以及ECC5004和GSBR-1290等其他小分子GLP-1RA。
-高剂量口服索马鲁肽(每天高达50毫克)与新配方吸收促进剂的应用。
这些创新药物不仅可能增强降糖和减重效果,还可能在外周动脉疾病、代谢性肝病、神经退行性疾病等领域拓展GLP-1药物的应用。
此外,对成瘾相关行为、遗传性肥胖、多囊卵巢综合征、1型糖尿病等多种适应症的研究也在进行之中。未来十年有望出现更为便捷和有效的GLP-1药物,这些药物的广泛适应症及安全性将通过更大规模临床试验的验证,为更多患者提供治疗选项。
随着糖尿病大流行的发展,我们对病理生理学以及治疗和预防方法的理解也呈指数级增长。目前的知识为提高识别增加β细胞功能障碍易感性标志物的特异性奠定了基础,特别是在肥胖环境中。
我们对大脑在体重调节中的作用以及脂肪组织分泌的新因子的理解可能有助于改进治疗或预防肥胖的方法。增加对肝功能障碍对胰岛素抵抗(IR)贡献的理解以及增加对代谢功能障碍相关肝病的理解代表了进一步研究的重要领域,以避免可能成为日益流行的肝功能衰竭。
心血管和肾脏疾病仍是糖尿病的主要死亡和发病因素,复杂的病理生理学涉及多器官系统和全身环境的交互作用。
糖尿病大流行由环境和社会因素推动,需综合管理。治疗进展为预防或治愈1型糖尿病和治疗2型糖尿病带来希望,不仅改善代谢稳态,还降低心肾疾病风险。
最后,随着我们了解和开发出识别导致糖尿病及其并发症的潜在异质性的工具,我们将为有针对性的治疗和预防策略奠定基础,以优化其在不同人群和医疗资源中的应用。
参考文献:
Abel ED, Gloyn AL, Evans-Molina C, Joseph JJ, Misra S, Pajvani UB, Simcox J, Susztak K, Drucker DJ. Diabetes mellitus-Progress and opportunities in the evolving epidemic. Cell. 2024 Jul 25;187(15):3789-3820.