-

 CNAS L23010
CNAS L23010

 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室

谷禾健康

目前随着高通量测序,微生物以及代谢、蛋白组学和高精度影像学等快速的发展,有关人体疾病或健康相关的海量数据正在不断产生,依托各种算法尤其AI的算法等,涌现出很多优秀的“模型”用于预测人体疾病以及健康相关的辅助诊断或筛查。
谷禾在人体肠道菌群检测领域深耕多年,积累了大量肠道菌群和个体健康信息数据,并运用建模方法结合人工智能等算法构建了多种疾病和营养预测模型。
对于不是从事专业的人员或大众来说,什么样的预测模型是准确的?如何评价一个模型的好坏?里面涉及比较多的概念和知识,今天谷禾和大家简单聊下这方面的内容。
首先大家评估下:
对于肿瘤筛查,以一个人群发病率1%的肿瘤为例:
模型A的检出率50%,假阳性为1%
模型B的检出率为90%,但是假阳性是10%
请问哪个检测模型作为肿瘤筛查类产品更好 ?
换句话说就是回答:“鉴于测试结果为阳性,该患者患有这种疾病的可能性有多大?
为了回答上面问题,我们需要了解和正确认识以下模型评价参数/指标:
• 该测试的阳性预测值(PPV)是多少?——返回阳性结果的人实际上患有疾病的概率是多少?
• 该测试的阴性预测值(NPV)是多少? ——返回阴性结果的人实际上是健康的概率是多少?
• 测试的灵敏度如何?——它正确地将多少个患病个体识别为患病?
• 测试的特异性是什么?——它正确地将多少个健康个体识别为健康个体?
要回答这个问题,我们需要计算两个模型的阳性预测值(Positive Predictive Value, PPV)。然后一步步分析:
▸ 什么是阳性预测值(PPV)?
阳性预测值(PPV)是样本检测阳性为真阳性的概率值。
发病率较低时的阳性预测值(PPV)
模型A
人群发病率:1%
检出率:50%
假阳性率:1%
模型B
人群发病率:1%
检出率:90%
假阳性率:10%
接下来使用贝叶斯定理来计算每个模型的阳性预测值。
对于模型A:
真阳性率 = 1% × 50% = 0.5%
假阳性率 = 99% × 1% = 0.99%
总阳性结果 = 真阳性 + 假阳性 = 0.5% + 0.99% = 1.49%
阳性预测值(PPV) = 真阳性 ÷ 总阳性结果 = 0.5% ÷ 1.49% ≈ 33.6%
对于模型B:
真阳性率 = 1% × 90% = 0.9%
假阳性率 = 99% × 10% = 9.9%
总阳性结果 = 真阳性 + 假阳性 = 0.9% + 9.9% = 10.8%
阳性预测值(PPV) = 真阳性 ÷ 总阳性结果 = 0.9% ÷ 10.8% ≈ 8.3%
模型A的PPV为33.6%,而模型B的PPV仅为8.3%。
因此,我们可以看到尽管模型B的检出率更高(90%vs50%),但其阳性预测值(PPV)显著低于模型A。
★ 低发病率人群中,高特异性比高敏感性更重要
这意味着:
模型A的阳性结果更可靠
模型A的假阳性风险更低
模型A在这种低发病率的情况下,作为肿瘤筛查产品更好。
这里对于我们应用的启示:在低发病率的人群中,高特异性(低假阳性率)比高敏感性(高检出率)更重要。选择产品时,不仅要看检出率,还要关注假阳性率和阳性预测值。

发病率较高的阳性预测值(PPV)
可能接下来有人会问,这个问题中以肿瘤发病率比较低的例子举例,那对于发病率比较高的疾病,比如心脑血管、糖尿病等如何?
接下来,我们继续举例:
让我们用同样的方法计算发病率为20%时的阳性预测值(PPV)。
模型A
人群发病率:20%
检出率:50%
假阳性率:1%
模型B
人群发病率:20%
检出率:90%
假阳性率:10%
一样的计算
对于模型A:
真阳性率 = 20% × 50% = 10%
假阳性率 = 80% × 1% = 0.8%
总阳性结果 = 真阳性 + 假阳性 = 10% + 0.8% = 10.8%
阳性预测值(PPV) = 真阳性 ÷ 总阳性结果 = 10% ÷ 10.8% ≈ 92.6%
对于模型B:
真阳性率 = 20% × 90% = 18%
假阳性率 = 80% × 10% = 8%
总阳性结果 = 真阳性 + 假阳性 = 18% + 8% = 26%
阳性预测值(PPV) = 真阳性 ÷ 总阳性结果 = 18% ÷ 26% ≈ 69.2%
可以看到当发病率提高到20%时,情况发生了显著变化:
模型A的PPV提高到92.6%
模型B的PPV提高到69.2%
对比之前1%发病率的情况:
模型A:PPV从33.6%提高到92.6%
模型B:PPV从8.3%提高到69.2%
可以发现随着发病率的提高,两个模型的阳性预测值都大幅提升。但是模型A仍然保持更高的PPV,但优势不如低发病率时那么明显。

★ 发病率越高,阳性预测值越高
发病率越高,阳性预测值越高,这是因为真阳性的比例增加。
因此在高发病率的人群中,两种模型的检测价值都显著提高。但模型A仍然是更好的选择,因为其假阳性率更低。
评估不同检测模型和询问检测准确率时,要综合考虑不同疾病的发病率、检出率和假阳性率。
对于要检出阳性结果而言,这个例子就很好地说明了发病率对检测结果解读的重要影响,也体现了贝叶斯定理在医学诊断中的应用。
而阳性预测值(PPV)具有多维意义,对于不同视角的人群可能意义不同:
➤ 政府视角下的PPV
⑴公共卫生决策基础
资源分配效率:高PPV意味着确诊投入的资源浪费更少,每发现一个真实病例的成本更低;
筛查项目评估:决定是否推行大规模筛查项目时,PPV是核心考量因素之一;
卫生经济学分析:假阳性导致的后续检查、治疗和心理干预成本需纳入卫生政策评估。
⑵监管与标准制定
差异化监管:对不同严重程度疾病的筛查产品可设置不同PPV要求;
强制信息披露:要求厂商在不同疾病流行率下公开产品的PPV值;
患者保护:防止低PPV产品导致过度医疗和不必要的医源性伤害。
➤ 受检者视角下的PPV
⑴个人决策与心理影响
决策参考价值:“我的阳性结果有多可靠?”—这是患者最关心的问题;
心理负担差异:假阳性可能导致严重心理压力,尤其对严重疾病筛查;
后续检查意愿:了解PPV有助于患者决定是否及时进行确诊检查。
⑵风险认知与理解
个体化解读:同一PPV值对不同风险人群的意义不同;
检前概率影响:有症状个体的PPV通常高于无症状筛查人群;
教育需求:医生需帮助患者理解PPV与个人情况的关联。
➤ 产品评估的情境差异
⑴临床应用场景分析
筛查工具:可接受相对较低PPV,但应有明确后续路径;
确诊工具:需要更高PPV,减少误诊风险;
监测工具:对症状性疾病监测,PPV要求可能介于两者之间。
⑵疾病特性影响
致命但可治疗疾病:可接受较低PPV,以高灵敏度优先;
慢性管理疾病:需较高PPV避免不必要长期干预;
高耻感疾病:需更高PPV避免标签化伤害。
⑶社会经济环境考量
医疗资源丰富地区:可能更关注灵敏度,接受较低PPV;
资源有限环境:高PPV更为重要,避免资源浪费;
支付方式影响:商业保险vs自费vs政府支付下对PPV的需求不同。
➤ PPV优化与应用建议
⑴预测模型调整
风险分层应用:根据人群特征调整决策阈值;
多指标综合评估:结合阴性预测值(NPV)、阳性/阴性似然比(LR+/LR-)综合评估;
贝叶斯思维应用:根据先验概率(患病率)调整PPV期望。
⑵实际应用最佳实践
透明沟通:向受检者清晰解释结果可靠性;
分级报告:提供风险概率而非简单阳性/阴性结果;
智能决策支持:AI辅助工具结合临床特征提供个性化PPV估计。
那么下面我们看下什么是阴性预测值(NPV),简言之就是把健康人检测为健康的概率。
阳性预测值(PPV)是样本检测阳性为真阳性的概率值,而阴性预测值(NPV)是样本检测真阴性的概率值。
阴性预测值(NPV) =(真阴性数量)/(真阴性数量+假阴性数量)
例如你有一个600人的样本量,根据有效性,假设你知道肯定患有这种疾病的样本(480)或没有这种疾病的个体样本(120)。
测试后,将结果与已知的疾病状态进行比较,发现:
真阳性(测试阳性和正确阳性)= 480
假阳性(试验阳性但实际为阴性)= 15
真阴性(测试阴性和真阴性)= 100
假阴性(试验阴性,但实际为阳性)=5
我们可以计算PPV和NPV如下:
阳性预测值(PPV)=480/(480+15)≈0.97(97%)
阴性预测值(NPV)=100/(100+5)≈0.95(95%)
因此,如果检测结果为阳性,则有97%的机会是正确的,如果结果为阴性,则有95%的机会是正确的。
拓展:什么是假阳性和假阴性?
假阳性:健康人被错误地识别为患病;
假阴性:病人被错误地认为是健康;
真阳性:患病的个体已被正确识别为患有疾病;
真阴性:未患有疾病的个体已被正确诊断为未患有疾病。
或:
真阳性:患者患有疾病且检测呈阳性;
假阳性:患者没有疾病,但检测呈阳性;
真阴性:患者没有疾病,检测结果为阴性;
假阴性:患者患有疾病,但检测结果为阴性。
敏感性
敏感性(Sensitivity)是测试正确识别疾病患者的能力,也称为真阳性率(TPR),即被正确识别为患有疾病的患者的百分比。
敏感性的计算公式为:
敏感性 = 真阳性数 / (真阳性数 + 假阴性数)
或者表示为:
敏感性 = TP / (TP+FN)
其中:
TP (True Positive): 真阳性,即测试正确地将患病者判断为阳性的数量;
FN (False Negative): 假阴性,即测试错误地将患病者判断为阴性的数量。
从另一个角度看,敏感性也可以表示为:
敏感性 = 真阳性率 = 1 – 假阴性率
敏感性的值范围在0到1之间,通常以百分比表示。例如,敏感性为0.95或95%意味着测试能够正确识别95%的实际患病者。
★ 高敏感性特别适用排除诊断
在医学诊断中,高敏感性的测试特别适用于排除诊断(rule-out test),因为如果一个高敏感性测试的结果为阴性,则疾病存在的可能性很小(即”阴性结果可靠”)。
例如:表述中敏感性为100%的检测可正确识别所有患者,敏感性为80%的检测可检出80%的疾病患者(真阳性),但20%的疾病患者未被检测到(假阴性)。
当检测到非常严重的感染类型时,例如前两年进行的COVID-19大流行,高敏感性对于适当的管理和治疗非常重要。COVID-19 IgG/IgM诊断测试是一种筛查COVID-19的测试,据报道敏感性为95%。
当测试用于识别严重但可治疗的疾病(例如宫颈癌)时,高敏感性显然很重要。通过宫颈涂片检测筛查女性人群是一项敏感的检测。然而,它不是很特异,很大一部分宫颈涂片阳性的女性继续进行阴道镜检查,最终被发现没有潜在的病变。

特异性
临床试验的特异性是指检测正确识别无病患者的能力。
特异性的计算公式是:
特异性 = 真阴性数 /(真阴性数 + 假阳性数)
Specificity = TN/(TN + FP)
其中:
TN (True Negative): 真阴性,即正确识别出的健康者数量
FP (False Positive): 假阳性,即错误地将健康者判断为患病的数量
特异性也可以表示为: 特异性 = 正确识别的健康者数/所有实际健康者总数
或: 特异性 = 真阴性率 = 1 – 假阳性率
例如:假设一项检测在100名实际健康者中:
95人被正确判断为阴性(TN = 95)
5人被错误判断为阳性(FP = 5)
特异性 = 95/(95 + 5) = 95/100 = 0.95 或 95%
特异性表明该检测能够正确识别95%的健康者,有5%的健康者被误判为患病。
TIPs:
一种具有高敏感性但低特异性的测试会导致许多没有疾病的患者被告知他们有疾病的可能性,使受检者要接受进一步的测试。理想情况下,测试应该是100%准确的,但这是一个不切实际的场景。或者,对患者进行高敏感性和低特异性的测试,然后进行低敏感性和高特异性的第二次测试,可以识别所有假阳性和假阴性。
在考虑检测对临床医生的价值时,阳性和阴性预测值很有用。它们取决于该疾病在目标人群中的患病率。
但是灵敏度(sensitivity)、特异性(specificity)这两术语对于理解和评估模型的效用也至关重要。敏感性和特异性是用于评估临床试验的术语,它们与测试的目标人群无关。
★ 敏感性和特异性取决于检测阳性的临界值
检测的敏感性和特异性取决于高于或低于检测阳性的临界值。一般来说,敏感性越高,特异性越低,反之亦然。
★ 阳性和阴性预测值取决于被测人群和患病率
与敏感性和特异性不同,阳性预测值(PPV)和阴性预测值(NPV)取决于被测人群,并受疾病患病率的影响。
例如以下示例:使用抗核抗体在普通人群中筛查系统性红斑狼疮(SLE)的PPV较低,因为它产生的假阳性数量很高。然而,如果患者有SLE的迹象(例如 颊部潮红和关节疼痛),测试的PPV会增加,因为患者所在的人群不同(从SLE患病率低的一般人群到患病率高得多的临床可疑人群)。
另一个举例也可以考虑产后出现呼吸困难的女性,其中一种鉴别诊断是肺栓塞。在该患者群体中,D-二聚体检测几乎肯定会升高;因此,该测试的肺栓塞PPV较低。然而,肺栓塞的NPV很高,因为低D-二聚体不太可能与肺栓塞相关。
★ 患病率高时,PPV和NPV准确性更高
阳性预测值(PPV)和阴性预测值(NPV)对疾病患病率的依赖性可以用数字来说明:例如,在一个4000人的人群中,病人和健康人各占一半。该病症筛查试验的灵敏度和特异性均为99%。因此,筛查结果将产生1980个真阳性和1980个真阴性,其中20名健康人被误判为阳性,20名病人被误判为阴性。
该测试的PPV为99%。但是,如果人口中的患病人数只有200人,而健康人数为 3800 人,则假阳性数量从20增加到38人,PPV下降到84%。
本举例实际上想强调这样一个事实,即诊断或筛查疾病的能力既取决于检测的区分价值,也取决于疾病在相关人群中的患病率。
与此同时,要注意:
PPV的互补值是错误发现率(FDR),NPV的互补值是错误遗漏率(FOR),分别等于1减去PPV或 NPV。FDR是错误的结果或 “发现” 的比例。FOR 是被错误拒绝的假阴性的比例。从本质上讲,PPV和NPV越高,FDR和FOR就越低——这对您测试结果的可靠性来说是个好消息。
如果结果是按值的滑动比例给出的,而不是明确的阳性或阴性,则敏感性和特异性值尤为重要。它们允许确定在何处绘制预测结果为阳性或阴性的临界值,甚至可能建议重新测试的灰色区域。
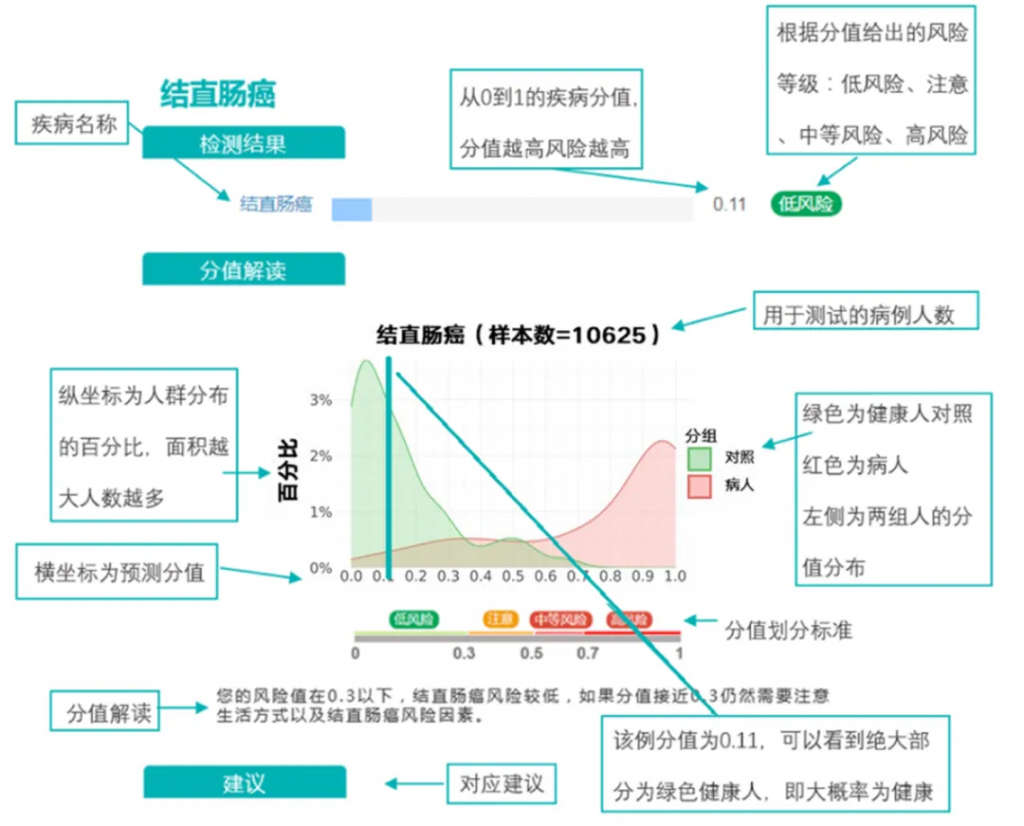
例如:谷禾结直肠癌模型
例如,通过将阳性结果的临界值置于非常低的水平,可以捕获所有阳性样品,因此测试非常敏感。但是,这可能意味着许多实际为阴性的样本可能被视为阳性,因此该测试将被视为特异性较差。找到平衡点对于有效和可用的测试至关重要。
★ ROC曲线有助于平衡假阴性和假阳性
在科研项目分析中经常使用受试者工作特征 (ROC)曲线有助于达到最佳平衡点并平衡假阴性和假阳性。但是,对于假阴性是否比假阳性问题更小,或者反之亦然,分析的内容也很重要。例如,在生死攸关的问题上,那么您可能愿意容忍更多的假阳性,以避免遗漏任何真阳性。
▸ 什么是ROC曲线?
ROC曲线是一种图形表示,显示测试的敏感性和特异性如何相互变化。为了构建 ROC 曲线,使用该检验测量已知为阳性或阴性的样本。
对于给定的截止值,将TPR(敏感性)与FPR(1-特异性)作图,得到类似于下图的曲线。理想情况下,选择曲线肩部周围的一个点,这既能限制假阳性,又能最大限度地提高真阳性。
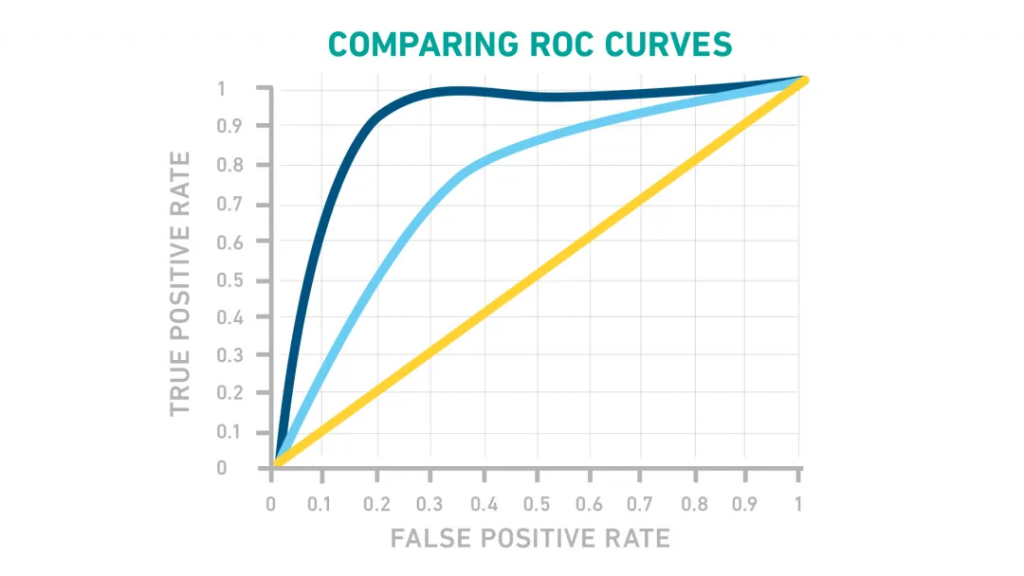
图片来源:Technology Networks
给出ROC曲线(如黄线)的测试并不比随机猜测好,淡蓝色很好,但由深蓝色线表示的测试会更好。这将使临界值确定相对简单,并以非常低的假阳性率产生高真阳性率。
在医疗科技和人工智能快速发展的今天,我们面临着海量的健康相关数据和不断涌现的疾病预测模型。在选择和评估医疗检测产品时,必须综合考虑疾病特性、人群特征和具体应用场景。对于普通大众和医疗从业者而言,理解这些指标不仅是一种专业素养,更是做出明智医疗决策的基础。
我们需要超越简单的”阳性”或”阴性”结果,深入理解检测结果背后的概率和风险。正如本文所强调的,一个优秀的医疗检测模型,应该能够在保证高敏感性的同时,最大限度地降低假阳性和假阴性的风险,为个人健康和公共卫生提供更精准、更有价值的信息。
主要参考文献:
technologynetworks.com/analysis/articles/sensitivity-vs-specificity-318222
Faith Mokobi. What is Sensitivity, Specificity, False positive, False negative? 2021 April 18, Microbenotes.

谷禾健康

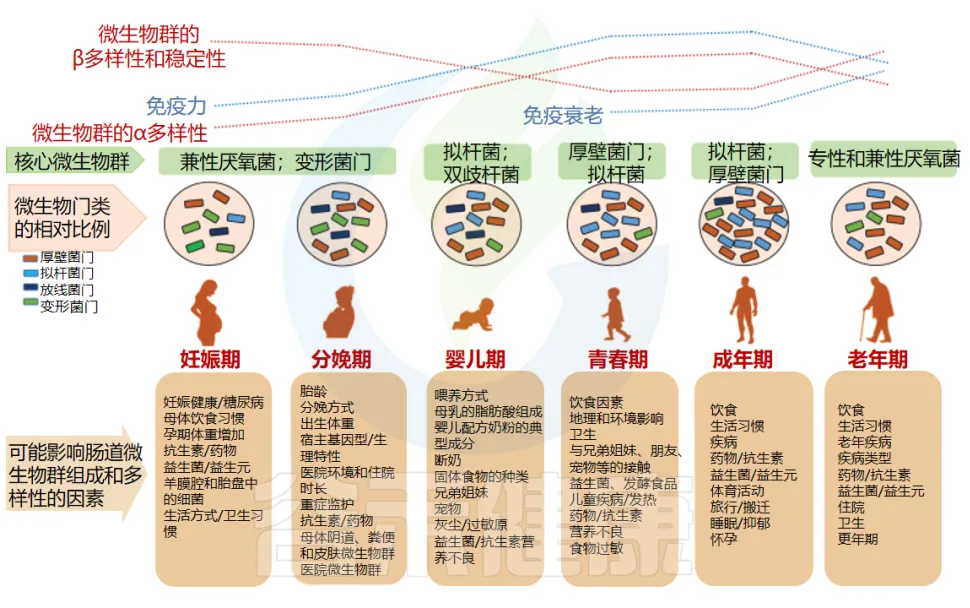
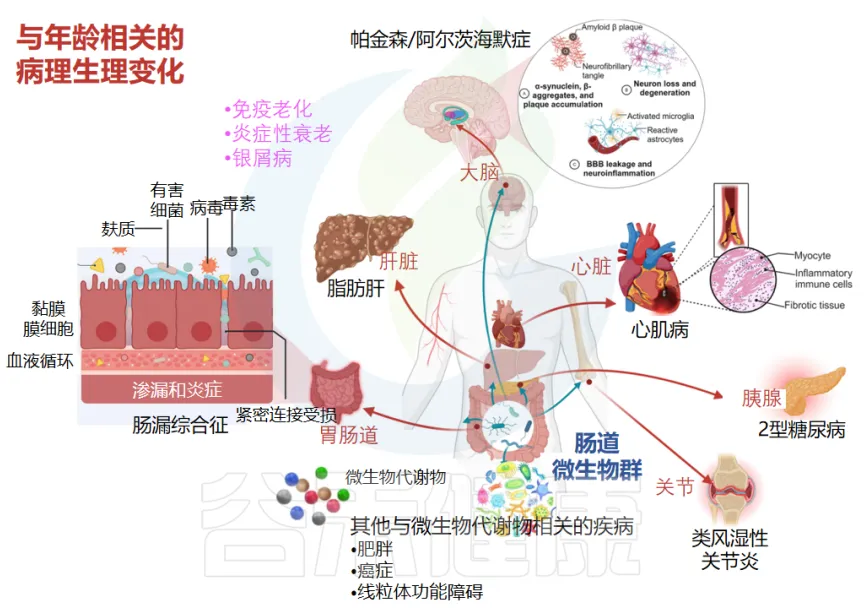
近年来,随着微生物组学研究的深入,科学家们发现肠道菌群与人体年龄之间存在着远比想象更密切的关联。一系列突破性研究表明,肠道菌群不仅能够反映个体的实际年龄,更可能是调控生物学年龄的关键因素之一。这一发现正在改变我们对发育、衰老和健康的传统认知。
最新研究揭示,肠道菌群的组成和功能会随年龄发生系统性变化。这种变化表现在多个层面:从婴幼儿期菌群的快速建立与稳定,到成年后菌群多样性的动态平衡,再到老年期特征性菌群的衰退。基于这些规律,研究人员开发出了一系列基于肠道菌群的年龄预测模型,其准确度已经达到前所未有的水平。
更引人注目的是,肠道菌群年龄与多种疾病的发生、发展密切相关。研究发现,肠道菌群年龄的加速老化往往预示着代谢紊乱、免疫功能下降,甚至认知能力衰退。相反,保持年轻和健康的肠道菌群,可能是延缓衰老、预防疾病的重要途径。
本文将重点探讨三个核心问题:
通过梳理最新研究进展,我们将看到肠道菌群不仅是年龄的被动标记物,更可能是一个可调控的”生物时钟”。这一认识正在开启精准医疗和个性化健康管理的新篇章。
doi: 10.14218/ERHM.2024.00008
随着研究的深入和技术的进步,基于肠道菌群的年龄干预策略很可能成为未来医学实践的重要组成部分。
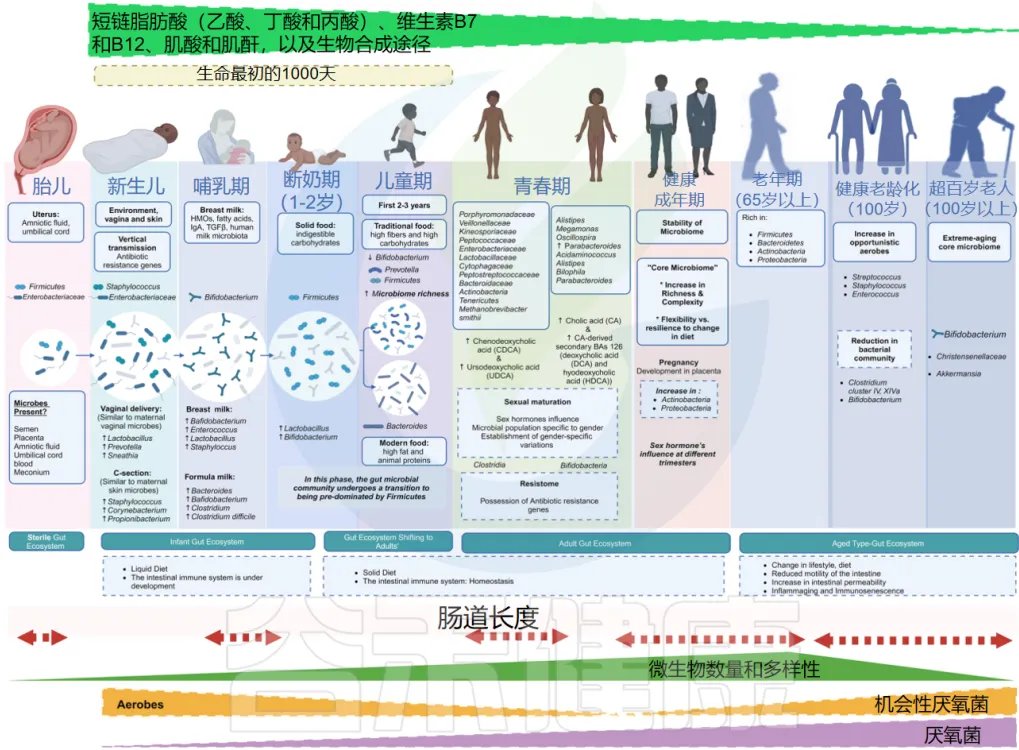
从无菌到共生——婴幼儿肠道菌群的“黄金塑造期”
近年来研究表明,肠道微生物群在婴幼儿早期发育阶段的变化与其发育年龄密切相关。婴幼儿的肠道微生物群呈现出明显的年龄相关性变化特征:在早期阶段主要表现为不稳定的群落结构和较低的微生物成熟度,随着年龄增长,菌群组成从以Firmicutes和Bifidobacterium为主,逐渐过渡到以Bacteroides和Prevotella为主导。这种转变具有确定性和可预测性,为年龄预测模型的建立提供了基础。
肠道微生物群的发育受多种因素影响,包括喂养方式、出生方式、地理位置和环境暴露等。研究发现,母乳喂养与Bifidobacterium的高水平密切相关,而停止母乳喂养则会加速肠道微生物群的成熟。此外,出生方式也显著影响微生物群的发育,如阴道分娩的婴儿肠道中Bacteroides的水平较高。尽管存在这些普遍性特征,但个体间的肠道微生物群发育轨迹仍存在差异,这与特定的生活方式和环境因素密切相关。
doi: 10.3233/NHA-170030
在预测模型研究方面,近年来取得了显著进展。2025年发表在《Nature Communications》上的研究开发出了一个高精度的预测模型,该研究分析了来自12个国家的1,827名婴幼儿的3,154个样本,使用随机森林模型进行预测,预测误差仅为2.56个月。
研究还识别出了关键的分类学预测指标,包括双歧杆菌的减少和粪杆菌的增加。另一项2024年的研究则开发出了“肠道微生物群健康指数”,该指数不仅能够预测婴幼儿在前5年的整体健康状况,还提供了一个可靠的微生物群年龄评估基准。
最新研究进一步证实,婴幼儿肠道微生物组的发育成熟度与年龄高度相关,仅通过肠道微生物群组成就能准确预测婴儿的年龄。研究人员建立了”微生物组年龄”的评估标准,这不仅可以用于年龄预测,还可以作为评估早期肠道发育的重要指标。这些研究成果不仅证实了基于肠道菌群预测婴幼儿年龄的可行性,还为评估婴幼儿的发育状况和健康水平提供了新的工具和方法。
肠道菌群发育滞后会对婴幼儿的多个系统产生深远影响。
在神经系统方面,可能导致认知发育迟缓、语言发育延迟、注意力不集中,甚至增加自闭症谱系障碍(ASD)的风险;
在免疫系统方面,常见表现包括反复呼吸道感染、特应性皮炎、食物过敏,以及哮喘等过敏性疾病的发生率升高;
在代谢系统方面,可能引起生长发育迟缓、体重增长异常、微量元素吸收不良等问题;在消化系统方面,则可能出现腹泻、便秘、肠痉挛等功能性胃肠道疾病。
特别是在出生后最初1000天这个关键期内,肠道菌群的发育状况对婴幼儿的长期健康具有决定性影响。
在这种情况下,规范的肠道菌群检测在婴幼儿发育评估中具有重要意义。检测内容应包括微生物多样性指数、关键菌群(如双歧杆菌、乳酸菌、粪杆菌等)的丰度,以及潜在致病菌的监测。
建议在以下关键时间点进行检测:出生后1-2个月、辅食添加期(4-6个月)、断奶期(12个月左右)以及2岁左右。对于高危人群,如早产儿、剖宫产婴儿、有过敏家族史或自身免疫性疾病家族史的婴幼儿,需要更频繁的监测。
研究显示,及早发现和干预菌群异常可以显著改善预后。具体干预措施包括:调整饮食结构(如添加低聚糖、膳食纤维等益生元),补充特定益生菌(如婴儿双歧杆菌、鼠李糖乳杆菌等),改善生活习惯(如规律作息、适度运动),以及环境因素的调整(如避免不必要的抗生素使用、增加户外活动时间)。对于特定问题,如食物过敏,可以通过补充特定菌株(如常见的LP299V益生菌)来改善症状;对于免疫功能低下,则可以考虑添加具有免疫调节作用的益生菌。
因此,建议在婴幼儿发育的关键时期进行系统的肠道菌群检测和跟踪监测。这种检测不仅能够及早发现发育风险(如自闭症倾向、过敏风险、免疫功能异常等),还能为个性化干预方案的制定提供科学依据。特别是对于已经出现发育迟缓、免疫功能低下、消化吸收障碍等问题的婴幼儿,定期的肠道菌群检测对评估干预效果和及时调整治疗方案具有重要的临床指导意义。
通过肠道菌群的视角:解密衰老时钟背后的微生物密码
➤ 老年肠道菌群的三大特征:
多样性下降、促炎菌增加、代谢功能衰退
随着年龄增长,我们的肠道菌群会发生显著变化。研究发现,老年人的肠道菌群具有三个显著特征,这些变化与多种衰老相关的健康问题密切相关。
◆ 首先是菌群多样性的显著下降
与年轻人相比,老年人的肠道菌群种类明显减少,特别是厚壁菌门数量减少,而拟杆菌门的比例相对升高。同时,一些对健康有益的细菌,如双歧杆菌和瘤胃球菌等数量也显著减少。这种多样性的降低会影响肠道微生态的稳定性。
◆ 第二个特征是促炎菌群的增加
老年人肠道中肠杆菌科(Enterobacteriaceae)和肠球菌属(Enterococcus)等促炎菌群数量明显增多,而具有抗炎作用的普拉梭菌(Faecalibacterium prausnitzii)等益生菌则显著减少。这种失衡会导致肠道处于慢性低度炎症状态,不仅影响局部肠道健康,还可能加速整体衰老进程。
◆ 第三个特征是代谢功能的衰退
老年人肠道中产丁酸菌如真杆菌(Eubacterium)、瘤胃球菌等数量减少,这些菌群对维持肠道健康至关重要。它们的减少会影响营养物质的吸收和能量代谢,同时也会降低肠道屏障功能,增加肠道通透性。这种变化可能导致营养吸收不良,并增加有害物质进入血液的风险。
这三个特征之间相互关联,共同构成了一个复杂的失衡网络。例如,菌群多样性的下降会影响代谢功能,促炎菌群的增加又会进一步降低有益菌群的数量。这种恶性循环可能是老年人更容易出现各种健康问题的重要原因之一。了解这些特征对于开发针对性的干预策略,改善老年人健康状况具有重要意义。
➤ 虚弱的菌群特征
虚弱是老年人一种特殊的临床状态,表现为机体对内外压力源的脆弱性增加,往往伴随着食欲下降、肌肉减少、认知功能下降、活动能力减弱以及独立生活能力的丧失。近年来的研究发现,肠道菌群的改变与虚弱程度之间存在着密切的关联,这种关联并不完全依赖于年龄因素。
研究显示,虚弱老年人的肠道菌群具有独特的“虚弱特征”。首先是普氏菌(Prevotella)和普拉梭菌(F. prausnitzii) 的显著减少,这些菌群对维持肠道健康和免疫功能具有重要作用。其次是产丁酸菌,如Eubacterium halii和Eubacterium rectalis的含量降低,这与肌肉蛋白合成的减少密切相关。
研究人员还观察到,在虚弱评分较高的老年人中,乳酸菌(Lactobacilli)、拟杆菌/普氏菌(Bacteroides/Prevotella)等有益菌群显著减少,而瘤胃球菌属(Ruminococcus)、Atopobium和肠杆菌科(Enterobacteriaceae)等菌群则明显增加。
doi: 10.1016/j.advnut.2025.100376
这些菌群变化的意义重大。产丁酸菌的减少会影响能量代谢和肌肉功能,而有益菌的减少则可能导致免疫功能下降和炎症水平升高。相反,在功能状态良好的老年人中,往往可以观察到较高水平的产丁酸菌,特别是普拉梭菌(Faecalibacterium prausnitzii)的含量,这进一步证实了菌群组成对机体功能的重要影响。
这些发现为我们理解和干预老年虚弱提供了新的视角。通过调节肠道菌群,特别是补充特定的益生菌和益生元,可能有助于改善老年人的虚弱状态。这也提示我们,在老年医学实践中,应当将肠道菌群作为评估和干预虚弱的重要靶点之一。
➤ 百岁老人长寿菌群的特殊组成
研究发现,百岁老人的肠道菌群具有独特的“长寿特征”,这种特征不仅区别于普通老年人,更展现出与健康长寿密切相关的微生物组成模式。这一发现为我们理解人类长寿的生物学机制提供了新的视角。
在菌群组成上,百岁老人的肠道中普遍存在较高水平的拟杆菌和罗氏菌属(Roseburia)。这两类菌群在维持肠道健康和免疫平衡方面发挥着重要作用。作为重要的丁酸盐产生菌,能够通过产生短链脂肪酸来调节肠道微环境,增强肠道屏障功能。
更引人注目的是,百岁老人的肠道中还富含Christensenella、双歧杆菌和阿克曼氏菌(Akkermansia)。这些菌群被认为是“长寿菌群”的代表。其中,长寿菌群与人类的遗传因素高度相关,被认为是最具遗传性的菌群之一;双歧杆菌能够维持肠道健康,增强免疫功能;而阿克曼氏菌则在调节代谢、维持肠道屏障完整性方面发挥关键作用。
研究还发现了一些在百岁老人肠道中特有的菌种,如腐生链菌(Alistipes putredinis)和气味杆菌(Odoribacter splanchnicus)。这些特殊菌群的存在可能与长寿个体特有的代谢特征和免疫调节功能有关,为我们理解长寿的微生物学机制提供了重要线索。
这些发现不仅帮助我们理解了百岁老人独特的肠道菌群特征,更为延缓衰老、促进健康长寿提供了新的思路。通过深入研究这些”长寿菌群”的功能和调节机制,有望开发出基于微生物组的健康干预策略,为实现健康老龄化提供新的方案。
值得注意的是,这些长寿相关的菌群特征并非简单的个体差异,而是反映了人体在长期健康状态下的微生态平衡。这种平衡可能是遗传因素、生活方式和环境因素长期共同作用的结果,对于我们理解和促进健康长寿具有重要的指导意义。
➤ 菌群-肠-脑轴在认知衰退中的作用路径
肠道菌群与大脑的关系远比我们想象的要复杂。研究表明,肠道菌群通过复杂的菌群-肠-脑轴与认知功能密切相关。这条轴线不仅包括神经元的直接连接,还涉及神经递质、激素和免疫介质等多重信号分子,构成了一个精密的双向调控网络。
◆ 第一个关键通路是神经递质调节
肠道菌群可以直接产生或调节多种神经递质的合成。研究发现,乳酸菌和双歧杆菌能够将肠道中的谷氨酸转化为GABA(γ-氨基丁酸),这是大脑中主要的抑制性神经递质,对情绪调节和认知功能具有重要作用。
此外,肠道菌群还参与乙酰胆碱和多巴胺等神经递质的产生。动物实验证实,补充Lactobacillus helveticus NS8可以通过降低血浆中皮质醇和促肾上腺皮质激素的水平来改善应激反应,同时提高血清素和去甲肾上腺素的水平。
◆ 第二个重要机制是免疫-炎症通路
随着年龄增长,肠道菌群失调会导致肠道通透性增加,使脂多糖(LPS)、脂蛋白和细菌RNA等细菌代谢产物进入血液循环。这些物质可以激活免疫系统,导致TNF-α、IL-1β、IL-6等促炎因子的释放,引发系统性炎症反应。
这些炎症因子可以穿过血脑屏障,在大脑中引起神经炎症,加速认知功能的衰退。值得注意的是,小胶质细胞(占中枢神经系统固有免疫细胞的约10%)在这个过程中发挥着重要作用,它们能够快速识别和响应环境变化,参与维持神经系统的稳态。
◆ 第三个关键环节是神经营养因子的调节
肠道菌群对脑源性神经营养因子(BDNF)和神经生长因子(NGF)的表达具有重要影响。这些神经营养因子对神经元的存活、突触可塑性和神经回路的重塑起着关键作用。研究发现,某些益生菌的补充可以提高BDNF的水平,改善认知功能。例如,在小鼠实验中,补充长双岐杆菌(Bifidobacterium Longum)能够显著改善动物的推理能力。
此外,肠道菌群还通过产生短链脂肪酸等代谢产物发挥作用。这些代谢产物可以调节血脑屏障的完整性,影响神经元功能和神经胶质细胞的活化状态。研究还发现,某些菌群数量变化与认知功能直接相关,如Alcaliganeceae和Porphyromonadaceae的数量增加与推理能力下降呈正相关。
在治疗干预方面,益生菌的应用显示出了积极的效果。例如,Lactobacillus、Bifidobacterium、S. thermophilus、Enterococcus、Bacillus等益生菌可以通过增加色氨酸衍生的神经营养因子水平来促进神经递质的释放。
然而,益生菌补充的效果会受到多种因素的影响,如剂量、菌株、补充时间以及患者的年龄、疾病状态、药物使用(尤其是抗生素)、饮食和生活方式等。
需要指出的是,尽管菌群-肠-脑轴的研究取得了显著进展,但仍有许多问题需要进一步探索。目前的研究主要基于动物模型和临床观察,还需要更多的多学科、多系统的研究来阐明相关机制和干预机会。这些研究将有助于开发以肠道菌群为靶点的早期干预策略,为预防和治疗神经退行性疾病提供新的思路。
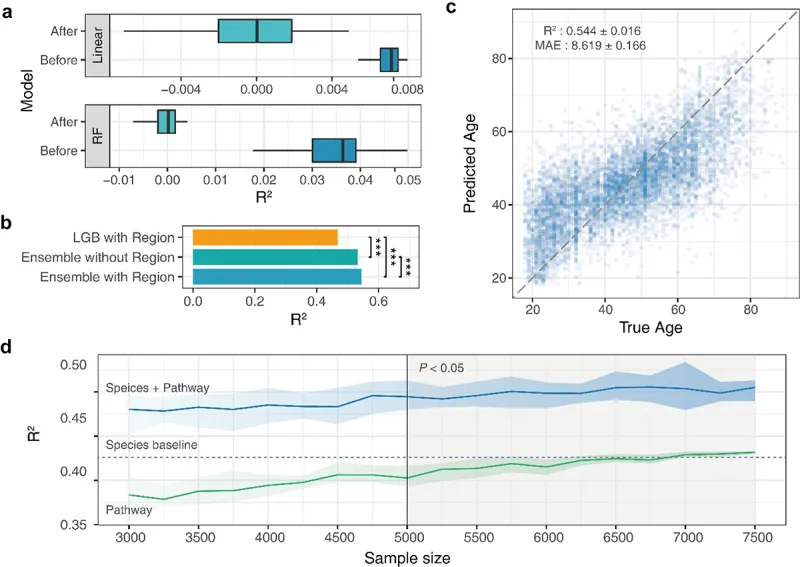
用AI构建年龄的生物标记——肠道菌群的年龄预测模型
近年来,随着测序技术的进步和机器学习方法的发展,基于肠道菌群的年龄预测模型取得了显著进展。2020年,研究人员通过深度学习方法分析了4000多份来自18-90岁人群的宏基因组数据,构建出预测误差为10.6年的模型。同年,另一项基于随机森林算法的研究分析了4434份样本的16S rRNA测序数据,将预测误差控制在11.5岁。这些早期研究证实了利用肠道菌群特征预测年龄的可行性。
2022年的突破性研究展示了多维度数据整合的优势。通过分析4478份成年人粪便样本,研究发现单独使用分类特征或代谢途径特征的预测模型误差分别为9.5年和10.2年,但将两类特征结合并考虑宿主因素后,预测误差可降至8.3年。这一发现为后续模型开发指明了方向。
2024年一项新研究开发出了基于肠道微生物组多维数据的生物学年龄预测模型(gAge)。该研究通过创新的机器学习方法,成功整合了微生物组的分类学和功能学特征,显著提高了年龄预测的准确性。研究团队识别出164个标志物物种和35个关键代谢通路与宿主年龄变化密切相关,并根据其对衰老的影响将其分类为促进和抑制两类。
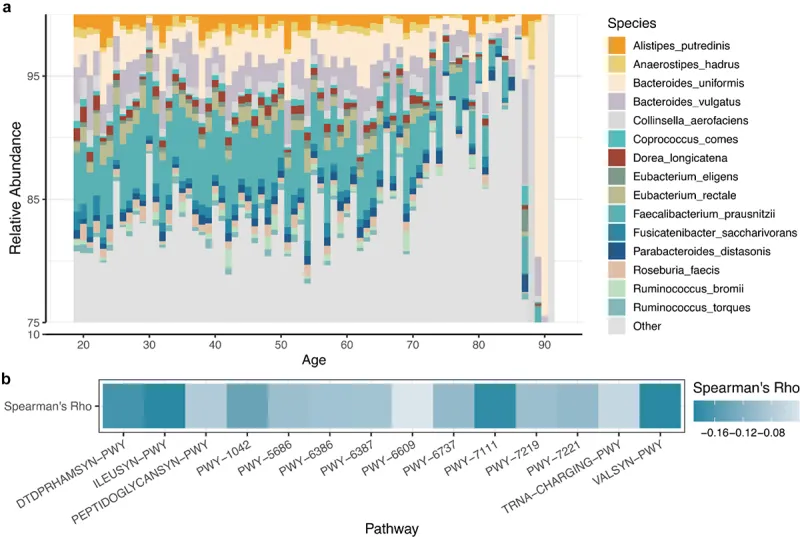
doi: 10.1080/19490976.2023.2297852
研究验证显示,gAge模型及其预测残差与个体健康状况和虚弱程度高度相关。特别是在老年群体中,模型识别的年龄相关标志物与多种疾病和虚弱特征显著重合,为评估和预测衰老风险提供了新的工具。
doi: 10.1080/19490976.2023.2297852
在这些研究基础上,谷禾开发的GUHEAge模型实现了更大的突破。该模型基于60万例不同年龄人群的6万个菌群特征,首次完整分析了从0-120岁的肠道菌群演变过程。通过深度神经网络,我们识别出381个标记物种和1574个标记基因,并根据其对衰老的影响将其分为促进和抑制两类。
在这些标记物中,产短链脂肪酸的有益菌(如双歧杆菌属、嗜粘蛋白阿克曼氏菌和拟杆菌属)表现出抑制衰老的作用。这些菌群可增强肠道屏障功能,调节机体代谢、炎症和免疫反应。相反,某些致病菌(如大肠杆菌和空肠弯曲杆菌)则显示出促进衰老的特征。
更重要的是,GUHEAge模型不仅能准确预测年龄,其预测结果还与个体健康状况和虚弱程度高度相关。验证集表明,模型识别的年龄相关标志物与多种疾病和虚弱特征显著重合,这为精准医疗和个性化健康管理提供了新的思路。
这些进展表明,基于肠道菌群的年龄预测模型已经发展成为一个可靠的生物年龄评估工具。通过整合多维度数据和先进的机器学习方法,为干预衰老提供新的靶点。
<来源:谷禾健康肠道菌群数据库>
可以看到,肠道年龄和实际年龄基本是符合的。健康人的肠道菌群年龄恰恰是最符合真实年龄的,与真实年龄差异大意味着肠道菌群出现偏离。
健康的人存在更多样化且平衡的肠道菌群。微生物群中与年龄相关的变化归因于生理,生活方式和健康状况。这些因素中的每一个都与某些菌群的相对丰度变化有关。
例如,饮食、卫生、兄弟姐妹、宠物、过敏、儿童疾病和抗生素是影响儿童微生物组的一些突出因素。到了成年期微生物群相对稳定,而到了老年期,一些有益菌开始逐渐下降,菌群又向另一个阶段过渡。
理解肠道年龄超越数字的生物学意义
➤ 发育迟缓儿童的菌群年龄滞后现象
发育迟缓儿童的肠道菌群呈现出明显的“年龄滞后”特征,这种现象反映在菌群的多样性、组成结构和功能等多个方面。研究表明,这些儿童的肠道菌群发育水平往往落后于其实际年龄,这种滞后可能是导致发育迟缓的重要因素之一。
我们之前发表在《Gut》上的针对自闭症儿童菌群发育的研究显示,自闭症儿童的菌群发育要滞后于健康儿童。
在菌群组成方面,发育迟缓儿童表现出显著的特征性改变。首先是有益菌群的明显减少,特别是双歧杆菌和乳杆菌的含量显著低于同龄健康儿童。这些菌群对维持肠道健康、促进营养物质吸收和调节免疫功能具有重要作用。同时,研究还发现这些儿童的肠道中普氏菌(Prevotella)和粪杆菌(F. prausnitzii)等核心菌菌含量也明显降低。
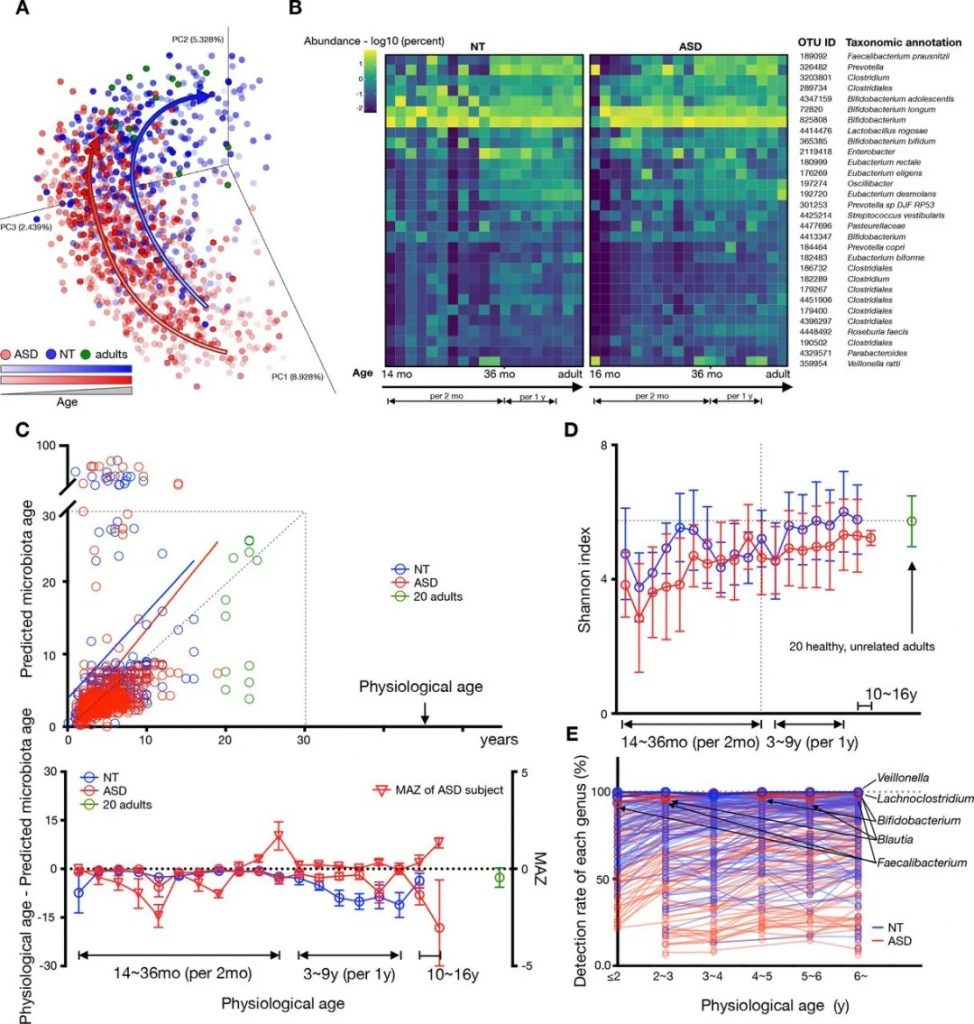
doi.org/10.1136/gutjnl-2021-325115
这种菌群滞后现象可能通过多个机制影响儿童发育:
免疫调节机制:菌群失调导致肠道免疫功能发育不良,增加炎症风险,影响生长发育。研究发现,这些儿童体内往往存在低度慢性炎症状态。
营养代谢影响:关键有益菌群的减少影响营养物质的吸收和利用效率。特别是产丁酸菌的减少,会影响能量代谢和营养物质的吸收。
神经-内分泌调节:菌群失调可能通过菌群-肠-脑轴影响生长激素和其他内分泌激素的分泌,进而影响生长发育。
针对这一问题,目前研究提出了几种潜在干预策略,包括结合营养补充、运动干预和益生菌治疗或全面的菌群置换可以有效改善儿童的生长发育状况。
通过调节肠道菌群,有望开发出更有效的干预策略,帮助发育迟缓儿童实现健康生长。同时,这也提示我们在儿童保健实践中,应当将肠道菌群作为评估和干预的重要指标之一。
➤ 早衰综合征患者的菌群年龄加速特征
早衰综合征患者的肠道菌群呈现出显著的“年龄加速”特征,其菌群组成和功能状态明显超前于实际年龄水平。研究发现,这种菌群年龄加速现象与多种生理功能的提前衰退密切相关。
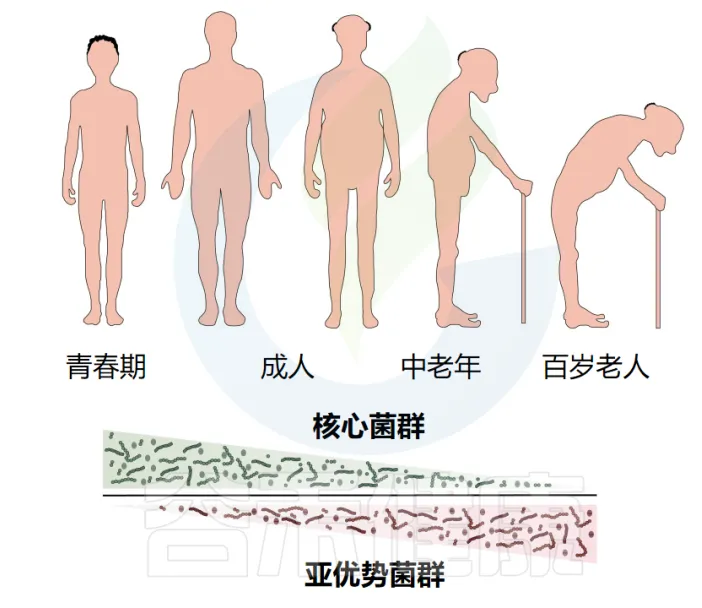
doi: 10.2147/CIA.S414714
在菌群组成方面,早衰患者表现出显著的特征性改变。有益菌群如粪杆菌(Faecalibacterium prausnitzii)、直肠真杆菌(Eubacterium rectale)和扭链瘤胃球菌(Ruminococcus torques)的含量明显降低。同时,代谢通路也发生了重要变化,包括支链氨基酸代谢通路(如L-缬氨酸和L-异亮氨酸合成)的显著下降,丙酮酸发酵(PWY-7111)能力减弱,以及NAD合成相关代谢通路的异常。
这些改变通过多个机制影响机体功能。首先是炎症反应的加速,表现为促炎因子(TNF-α、IL-6和IL-1)水平升高,导致慢性低度炎症状态持续存在。其次是代谢功能的紊乱,包括氨基酸代谢异常、能量代谢效率下降和营养物质吸收受损。此外,肠道屏障功能受损也是重要特征,导致肠道通透性增加,细菌代谢产物进入血液循环,进一步加重系统性炎症反应。
针对这些问题,研究提出了几种潜在的干预策略:
精准菌群干预:通过补充特定益生菌,如双歧杆菌和乳酸菌,同时添加益生元促进有益菌群生长。研究表明,这种干预方式可以改善肠道菌群组成,延缓衰老进程。
代谢通路调节:包括补充NAD前体物质、调节氨基酸代谢和改善能量代谢。特别是NAD水平的提升,已被证实可以预防衰老和相关疾病。
生活方式干预:优化饮食结构,增加膳食纤维摄入;保持适度运动,提高体能状态;改善睡眠质量,维持昼夜节律。
综合干预方案:结合营养补充、心理支持、运动康复和定期监测菌群状态,全面改善患者状况。
通过对肠道菌群的精准干预,有望延缓早衰进程,改善患者生活质量。
➤ 糖尿病/心血管疾病的菌群老化预警
在代谢性疾病领域,肠道菌群年龄作为一个新兴的生物标志物展现出重要的预警价值。研究发现,糖尿病和心血管疾病患者的肠道菌群往往表现出提前衰老的特征,这种菌群老化现象通常早于临床症状的出现,为疾病的早期预警提供了新的时间窗口。
在菌群组成方面,研究显示代谢性疾病患者的肠道菌群存在显著变化。这些变化与血清生化指标密切相关,特别是与碱性磷酸酶和胰岛素水平显著相关。研究还发现,某些代谢通路的改变,如支链氨基酸代谢通路(BCAA)、NAD合成相关代谢通路的异常,可能是疾病发展的早期信号。
通过肠道年龄预测模型的应用,研究人员发现菌群年龄与多种疾病风险指标具有显著相关性。在一项针对老年人群的研究中,gAge预测残差与个体健康状况和虚弱程度呈现高度相关。特别是在2型糖尿病(T2D)患者中,尽管疾病本身可能不会显著影响预测年龄,但菌群组成的特定改变可以作为疾病进展的预警指标。
在心血管疾病方面,研究发现血清碱性磷酸酶水平与心脑血管疾病风险显著相关,而这一指标与肠道菌群年龄存在明显关联。通过分析不同gAge残差人群的菌群特征,研究者识别出了一系列可能与心血管健康相关的关键菌种和代谢通路,为疾病预防提供了新的干预靶点。
这些发现为代谢性疾病的早期筛查和预防提供了新的思路。通过监测肠道菌群年龄的变化,结合传统的临床指标,可以更早地识别疾病风险,实现更精准的健康管理。随着检测技术的进步和预测模型的完善,基于肠道菌群的疾病预警系统有望成为临床实践中的重要工具。
实现从被动治疗到主动预防的转变
随着人口老龄化加剧,深入理解和干预衰老过程变得愈发重要。近年来,肠道微生物组研究为我们提供了全新的视角,从发育到衰老的全程监测和干预成为可能。
肠道年龄概念的提出,标志着生物学年龄评估领域的一次重要范式转变。这一转变不仅打破了传统chronological age的局限,更是将微生物组这一动态指标引入健康评估体系,为个体化健康管理提供了新的思维框架。
在发育领域,研究发现肠道菌群年龄可以有效反映儿童发育状况。发育迟缓儿童往往表现出明显的”菌群年龄滞后”现象,这为早期干预提供了新的靶点。同时,在早衰综合征患者中观察到的菌群加速衰老特征,以及在糖尿病、心血管疾病患者中发现的菌群老化预警信号,都展示了微生物组在疾病预防和健康管理中的重要价值。
谷禾团队通过分析60万例不同年龄人群的6万个菌群特征,这种基于海量数据的个体化健康监测方法,为微生物组研究开辟了新的范式。通过持续监测个体肠道菌群的动态变化,有助于更早地发现健康隐患,实现疾病的早期预警和干预。
<来源:谷禾健康肠道菌群检测数据库>
从精准医学的角度来看,肠道年龄的概念为个体化健康管理提供了新的工具。通过整合微生物组数据与传统临床指标,我们可以构建更全面的健康评估体系。这种多维度的评估方法不仅能够反映个体当前的健康状态,还能预测未来的健康轨迹,为精准医疗干预提供决策依据。
尽管目前研究已取得显著进展,但仍面临诸多挑战:微生物组与衰老的因果关系有待厘清,个体差异和环境因素的影响需要进一步研究,预测模型在特定人群中的适用性也需要验证。此外,如何将研究成果有效转化为临床应用,实现精准干预,也是未来需要重点解决的问题。
展望未来,随着高通量测序技术的进步和人工智能算法的优化,结合多组学数据的整合分析将帮助我们更全面地理解肠道微生物组在人体发育和衰老过程中的作用。精准医学的未来将更加依赖于这种整合的、动态的健康评估体系,结合人工智能分析,可以更好为每个个体制定个性化的健康管理方案,实现从被动治疗到主动预防的转变。
主要参考文献:
Min M, Egli C, Sivamani RK. The Gut and Skin Microbiome and Its Association with Aging Clocks. Int J Mol Sci. 2024 Jul 8;25(13):7471.
Hohman LS, Osborne LC. A gut-centric view of aging: Do intestinal epithelial cells contribute to age-associated microbiota changes, inflammaging, and immunosenescence? Aging Cell. 2022 Sep;21(9):e13700.
Tao X, Zhu Z, Wang L, Li C, Sun L, Wang W, Gong W. Biomarkers of Aging and Relevant Evaluation Techniques: A Comprehensive Review. Aging Dis. 2024 May 7;15(3):977-1005.
Wang H, Chen Y, Feng L, Lu S, Zhu J, Zhao J, Zhang H, Chen W, Lu W. A gut aging clock using microbiome multi-view profiles is associated with health and frail risk. Gut Microbes. 2024 Jan-Dec;16(1):2297852.

谷禾健康

随着技术的发展,高通量测序技术已成为研究微生物群落的重要工具。这种技术使得科学家们能够解析巨量微生物DNA序列,从而获得丰富的微生物组数据,包括16S rRNA基因、ITS序列和宏基因组。然而,这些数据只是迈向揭示微生物群落复杂性的第一步。
通过对环境样本的可变区域如16S、18S、ITS序列进行高通量测序获得的原始序列数据,再对其进行聚类,数据分析,统计学差异比较等得到微生物多样性分析报告。那么,什么是微生物群落多样性?
微生物群落多样性(Microbial Community Diversity)是指在特定环境中存在的微生物种类的数量和分布情况,它不仅包含不同种类微生物的丰度,还包括它们之间的相互关系。多样性可以从不同角度进行评价,主要分为以下几种:
α多样性(Alpha Diversity): 这是衡量某一特定样本内部多样性的一种指标。常用的α多样性指标包括物种丰富度(Species Richness)、香农指数(Shannon Index)和辛普森指数(Simpson Index)。这些指标可以帮助我们了解样本内部的复杂性和均一性。
β多样性(Beta Diversity):不同样本之间的多样性比较被称为β多样性。常用的β多样性指标包括Bray-Curtis距离和Jaccard指数,通过这些指标可以探索样本之间的相似性和差异性,揭示不同环境或条件下微生物群落的变化模式。
γ多样性(Gamma Diversity): 这是指在一个更大尺度、多个样本的总体多样性,通常用以评估一个较大区域的整体多样性水平。
为理解这些多样性指标,我们可以借助一些简单的比喻来形象解释。例如,α多样性就像是在观察一个花园的花卉种类和数量;β多样性则是比较不同的花园之间的相似性或不同之处;而γ多样性则是对一个城市中所有花园的总览评价。
在接下来的部分中,我们将深入探讨这些多样性指标的详细内涵,以及从多个角度展示如何通过高通量测序技术解析微生物群落中的这些多样性规律。
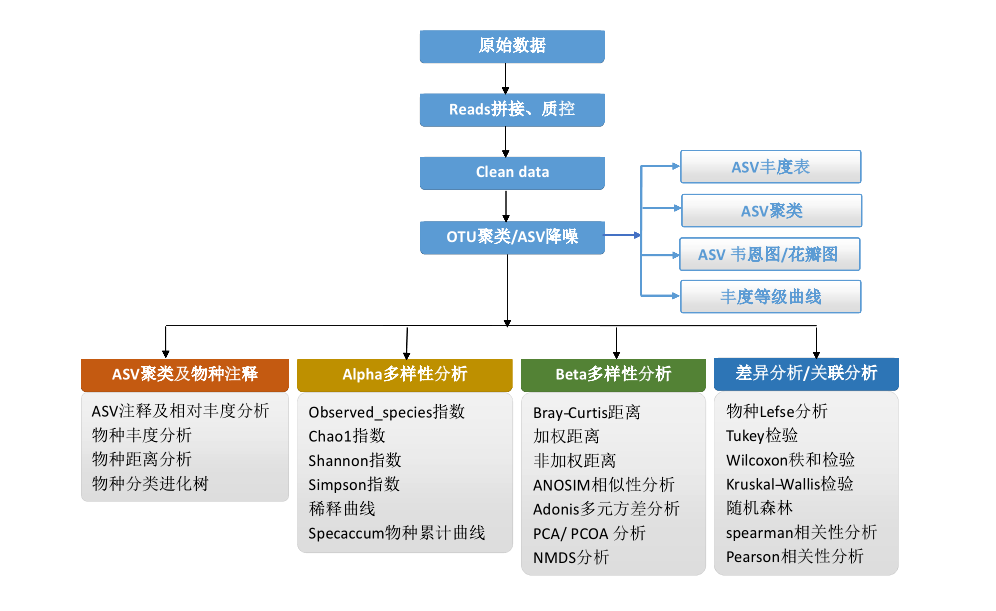
下图是实验上机测序流程,提取的样本总DNA经过质检、PCR扩增、建库等步骤进行高通量测序得到测序原始数据。

原始数据经过Reads拼接、tags过滤、去嵌合体等步骤得到有效数据clean data。在特定的相似度下进行聚类得到OTU/ASV,报告中通过降噪方法得到ASV表,一切后续分析都围绕ASV表来进行。根据ASV表可以继续做物种分类注释、丰度计算、多样性分析、差异分析、功能预测等。所以ASV特征表是微生物多样性分析中关键数据结果。
OTU和ASV的区别
OTU和ASV是微生物组学中用来表示微生物多样性的两个不同概念。两者都是从环境样本中获得的DNA序列数据,通过一定的分析方法分类得到的用于表示微生物种类或种群的单位。它们之间的主要区别在于定义的精确度和建立的方法。
– OTU(Operational Taxonomic Units):
OTU是一种将序列通过相似度聚类的传统方式,来表示相似序列组成的种群。通常,这种聚类方法会将序列之间相似度达到97%(或其他设定的阈值)的序列分到同一个OTU。OTU聚类通常不考虑序列中的单个变异位点,而是基于整体相似度。
由于使用阈值聚类,OTU不能准确反映序列之间的实际差异,可能会将生态学意义上不同的微生物序列归为一个OTU。OTU分析可能过于简化,有时无法捕获低水平的微生物多样性。
– ASV(Amplicon Sequence Variants):
ASV采用较新的降噪方法,可以精确地解析序列中的每一个核苷酸差异,简单来说就是以100%相似度进行聚类,对低质量序列进行去除和校正,这种方法可以生成“零半径OTU”,即互不相同的基于序列的变体。
ASV通常使用误差校正算法来排除测序错误,从而提供更精确的序列变体识别。ASV方法对单一核苷酸变异敏感而能提供更细粒度的微生物多样性解析。ASV为每一种变异提供更一致、可复制的标识符,这在比较不同研究之间的微生物群落组成时非常有用。
简而言之,ASV方法提供了比OTU更高分辨率、更精准的序列变体检测。换句话说,ASV提供了一种微生物组多样性分析的“高清”视角,它更可能捕捉到微生物群落内变化的微妙差异,尤其是在不同环境或时间点间的比较中。
原始序列数据(raw tags)经过质控、过滤、去嵌合体,最终得到有效数据(effective tags)。再对有效数据进行UNOISE降噪处理,得到ASV特征表。数据处理过程中各步骤得到的序列进行途径统计,可以直观的反映每个样本的数据量和物种丰度。
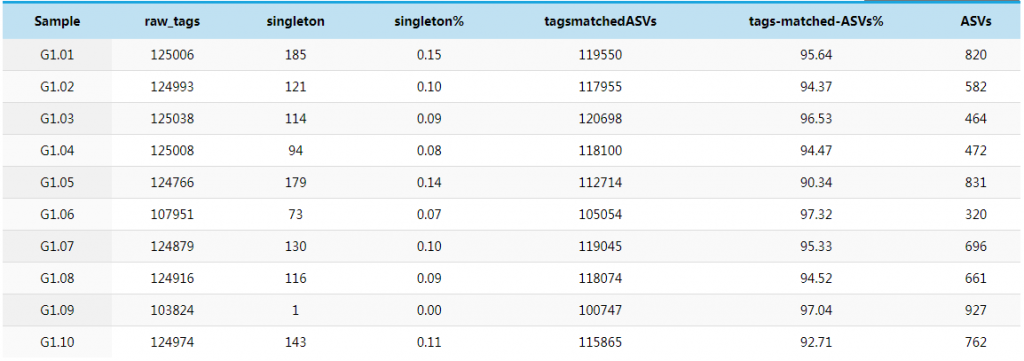
文件目录:
01_pick_otu/summary/sumOTUPerSample.txt
raw-tags:每个样本的原始序列数据;
singleton :每个样本中无完全匹配的单条序列的数量。singleton ASV 是指只有单条代表序列的 ASV,可能由于测序错误,或者是来自于PCR过程中产生的嵌合体;
tagsmatchedASVs: 每个样本中比对到ASVs的最终有效序列数据 及其比例,聚类的同时vsearch会根据UCHIME算法将singleton ASV及嵌合体去除,得到最终的有效序列数据 Effective Tags;
ASVs:每个样本的ASVs数量。
一般文献中的测序原始数据量raw-tags 要求达到3万条以上,可以满足数据分析的基本要求。绝大多数文献数据量平均在5万条左右。世面上不同公司承诺的数据指标有所不同,谷禾测序得到的原始数据一般可以达到10万 reads左右,足够满足当前文章发表要求的参考数据量。
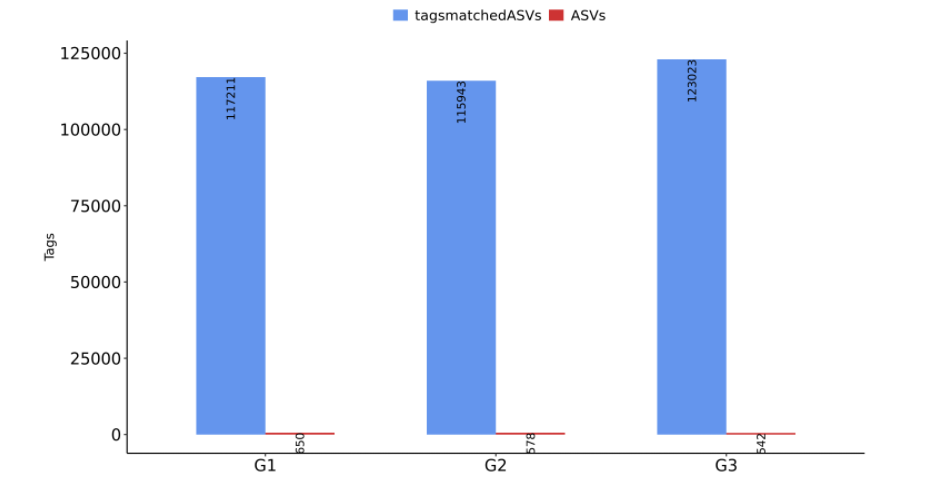
若原始数据量低于1万条,尤其是少于3000条reads以下,则很有可能受环境污染的杂带较多,建议重新上机补测数据。ASVS列可以反映每个样本的物种多样性,一般一个ASVs就代表一个物种。因此可以用ASV数量来代表物种数量。将每个样本的有效原始数量和ASVs数据可视化做成柱状图,可以更直观的观察每个样本/分组数据量的变化。
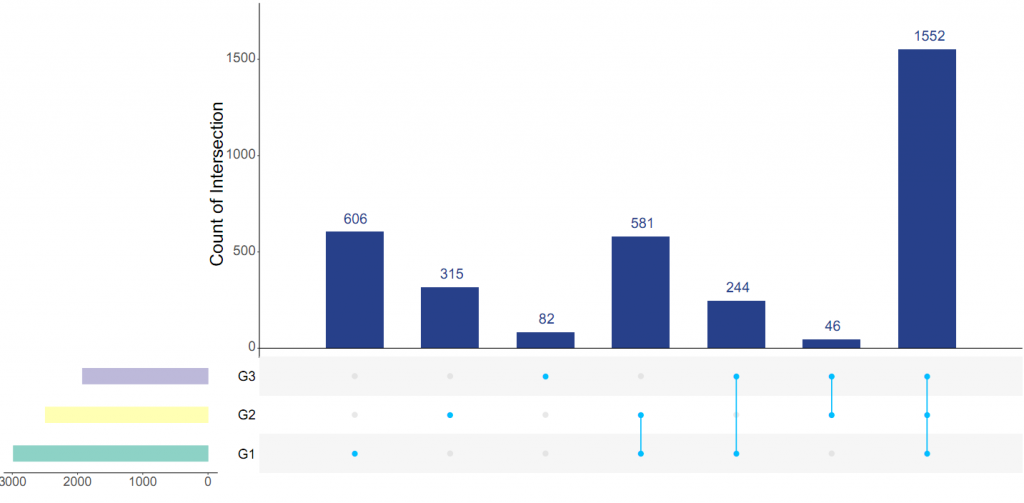
每个样本/分组可能会有一些共有的和独有的ASV,通常用韦恩图或花瓣图表示(样本数/分组数<=5个样本用维恩图,数量大于5出花瓣图)。除了用Venn图将几个数据集之间的交集进行可视化,还可以使用upset图表示。
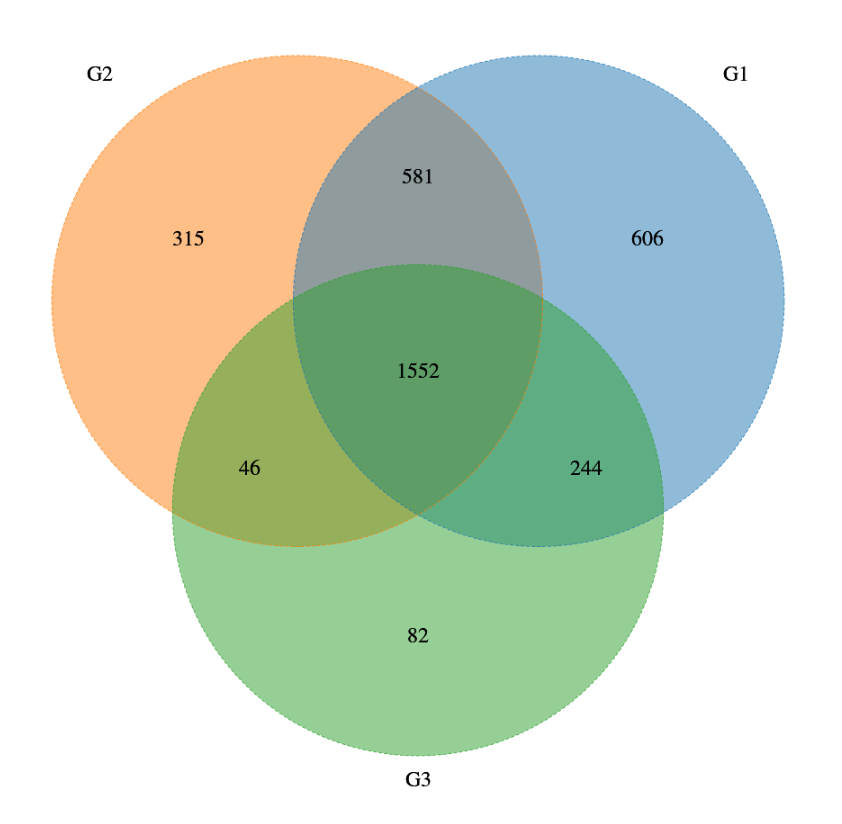
韦恩图中不同颜色的圆圈代表一个样本/分组,圈之间的重迭区域表示样本/分组间共有的ASVs,每个区域的数字大小表示该区域对应的ASVs数目。
UpSet图主要包含三个部分:上部分为各个分组独有和共有的ASV数量,下部分为各个分组独有和共有的分类情况,左部分每一个行代表一个分组。
alpha多样性主要用来衡量单个样本内的菌群多样性,不涉及样本之间的比较。alpha多样性与两个因素相关,分别是:一、丰富度(richness),二、多样性(diversity)。
丰富度指的是单个样本物种的种类数目;而多样性是指菌群在个体中分配的均匀度。样本的丰度高不一定就代表菌群的多样性丰富,丰度高如果是因为里边含有较多低丰度的杂带,这些可能是来源于环境的污染物导致的,这些低丰度的物种并不会使菌群的多样性增加。
alpha多样性有三类相关指数,其中包括菌群丰度指数(Chao1和ACE)、菌群多样性指数(shannon和simpson)和测序深度指数(Goods coverage和Observed spieces)。
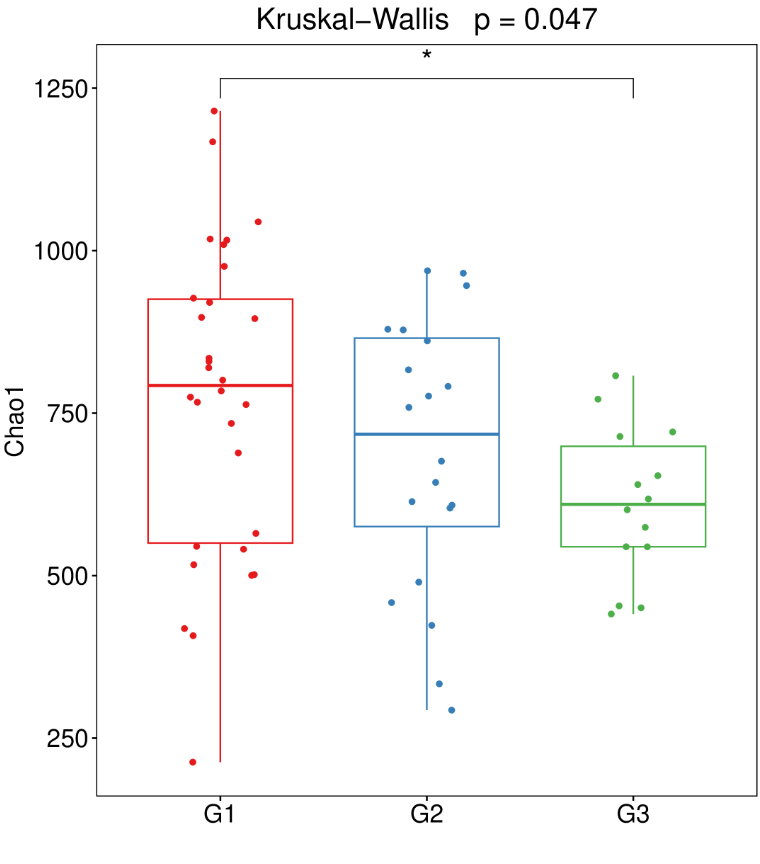
Chao1:Chao1算法用于评估样本中所含ASV数目的指数,Chao1在生态学中常用来估计物种总数,由Chao(1984)最早提出。通过计算群落中只检测到1次和2次的ASV数估计群落中实际存在的物种数。chao1指数可以评估一个样本中的ASV数量,chao指数越大,ASV数目越多,说明该样本物种数越多。
计算公式如下:
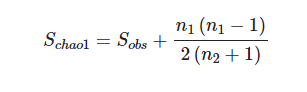
编辑
其中:
Schao1=估计的OTU数;
Sobs=观测到的OTU数;
n1=只有一条序列的OTU数目(如“singletons”);
n2=只有两条序列的OTU数目(如“doubletons”)。
ACE:用来估计群落中含有ASV数目的指数,由Chao提出,是生态学中估计物种总数的常用指数之一,与Chao1的算法不同。预设将序列量10以下的ASV都计算在内,从而估计群落中实际存在的物种数。
计算公式如下:
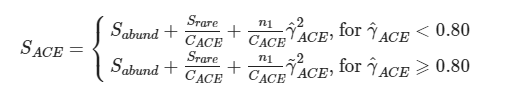
其中
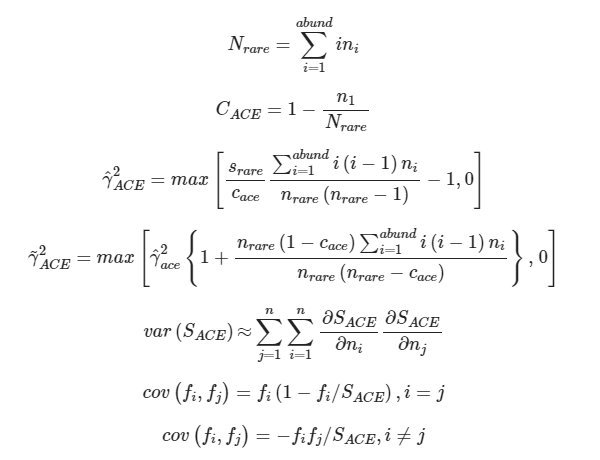
ni=含有i条序列的ASV数目;
Srare=含有“abund”条序列或者少于“abund”的OTU数目;
Sabund=多于“abund”条序列的OTU数目;
abund=被视为“优势”的ASV的阈值,默认为10。
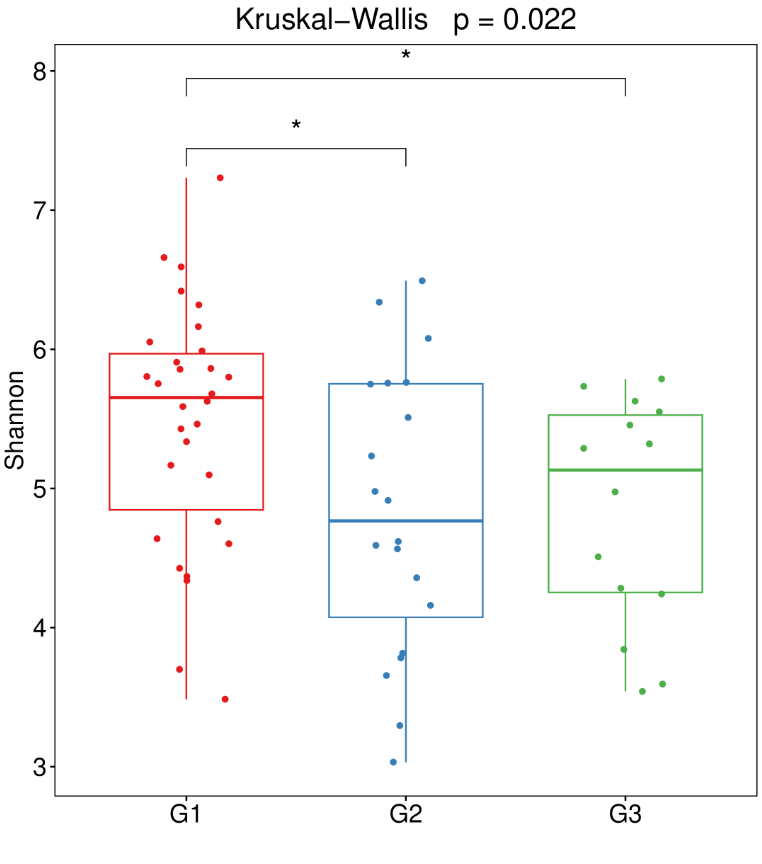
Shannon:香农-威纳指数综合考虑了群落的丰富度和均匀度,是用来评估样本中微生物多样性指数之一。Shannon指数值越高,表明群落的多样性越高。
计算公式如下:
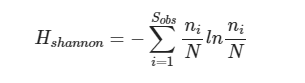
其中:
Sobs=观测到的ASV数目;
ni=含有i条序列的ASV数目;
N=所有的序列数。
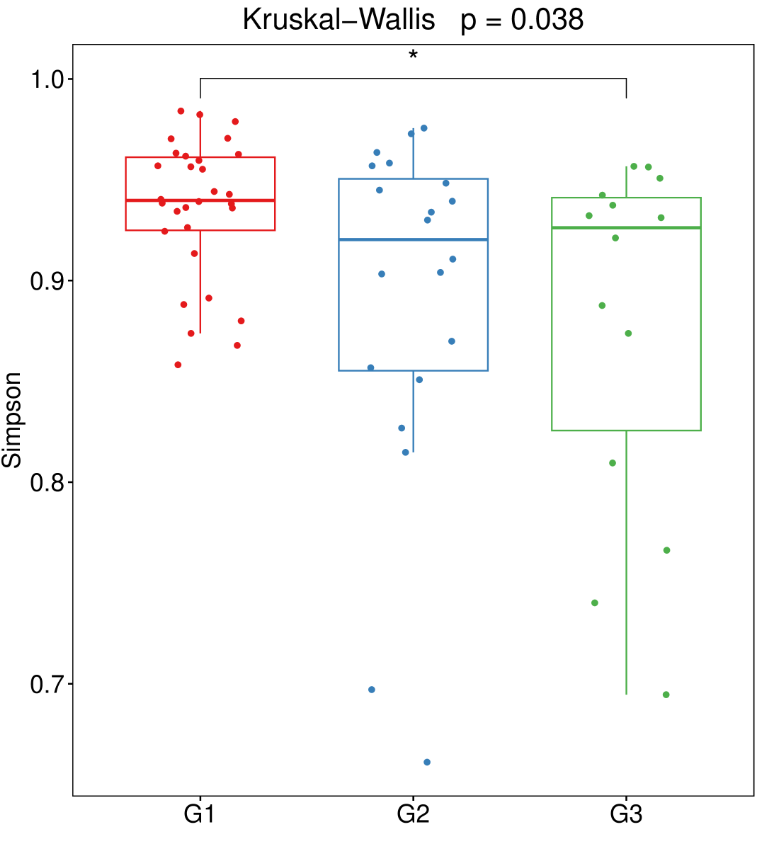
Simpson:辛普森多样性指数对菌群多样性评估,Simpson指数值越高,表明群落多样性越高。由EdwardHugh Simpson(1949)提出,在生态学中常用来定量描述一个区域的生物多样性。一般而言,Shannon指数侧重对群落的丰富度以及稀有ASV,而Simpson指数侧重均匀度和群落中的优势ASV。
计算公式一如下:
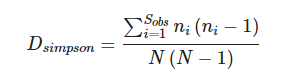
计算公式二如下:
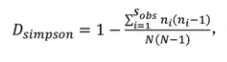
此时,Simpson指数越大,说明群落多样性越大。报告中用到的是计算公式二。
其中:
Sobs=观测到的ASV数目;
ni=含有i条序列的ASV数目;
N=所有的序列数。
Coverage:是指各样品克隆文库的覆盖率,其数值越高,则样品中序列被测出的概率越高,而没有被测出的概率越低。该指数反映本次测序结果是否代表了样品中微生物的真实情况。
计算公式如下:
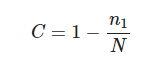
其中:
n1=只含有1条序列的ASV数目;
N=所有的序列数。
下表统计了每个样本的各项alpha多样性指标:
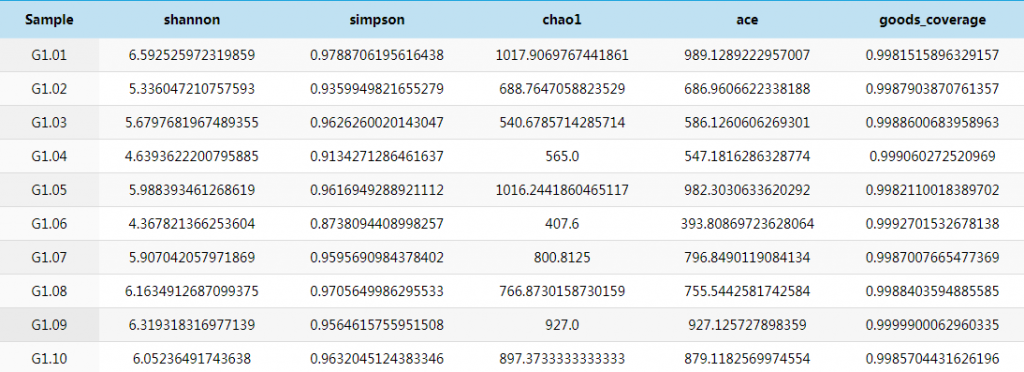
结果目录:
03_diversity-metrics/alpha/alpha_div.txt
可以选择不同的alpha多样性指数进行显著性差异比较,一般常用丰富度指数Chao1,多样性指数Shannon、simpson,比较不同组间指数是否有显著差异。Alpha多样性分析将样本的菌群群整体研究并转换为具体的指数与p值,来说明群落的变化与差异。
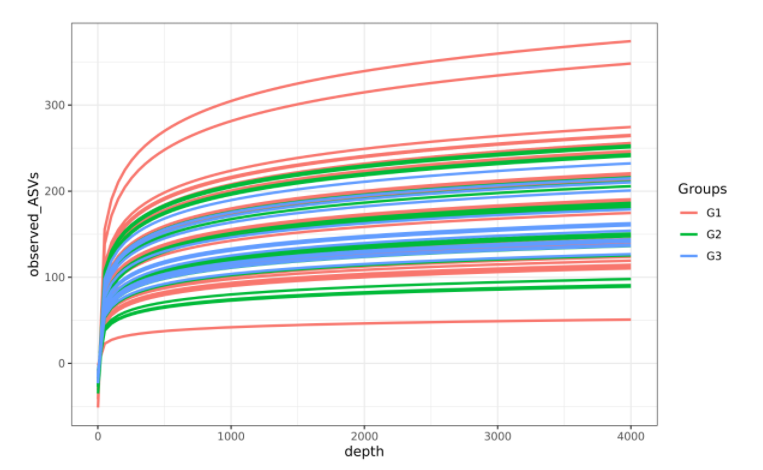
•稀释性曲线(Rarefaction curve)
稀释曲线是从每个样本中随机抽取一定数量的序列,统计这些序列所代表的ASV数目,以随机抽取的序列数与ASV数量来构建曲线。可以用来比较不同样本中的物种多样性,也可以用来说明样本出测序数据量是否足以反映环境中的物种多样性。
•
菌群多样性指数(shannon和simpson)
丰度等级曲线(Rank abundance curve)是分析多样性的一种方式。构建方法是统计单一样品中,每一个OTU所含的序列数,将OTU按丰度(所含有的序列条数)由大到小等级排序,再以OTU等级为横坐标,以每个OTU中所含的序列数(也可用OTU中序列数的相对百分含量)为纵坐标做图。
Rank-abundance曲线可用来解释多样性的两个方面,即物种丰度和物种均匀度。在水平方向,物种的丰度由曲线的宽度来反映,物种的丰度越高,曲线在横轴上的范围越大;曲线的形状(平滑程度)反映了样品中物种的均度,曲线越平缓,物种分布越均匀。
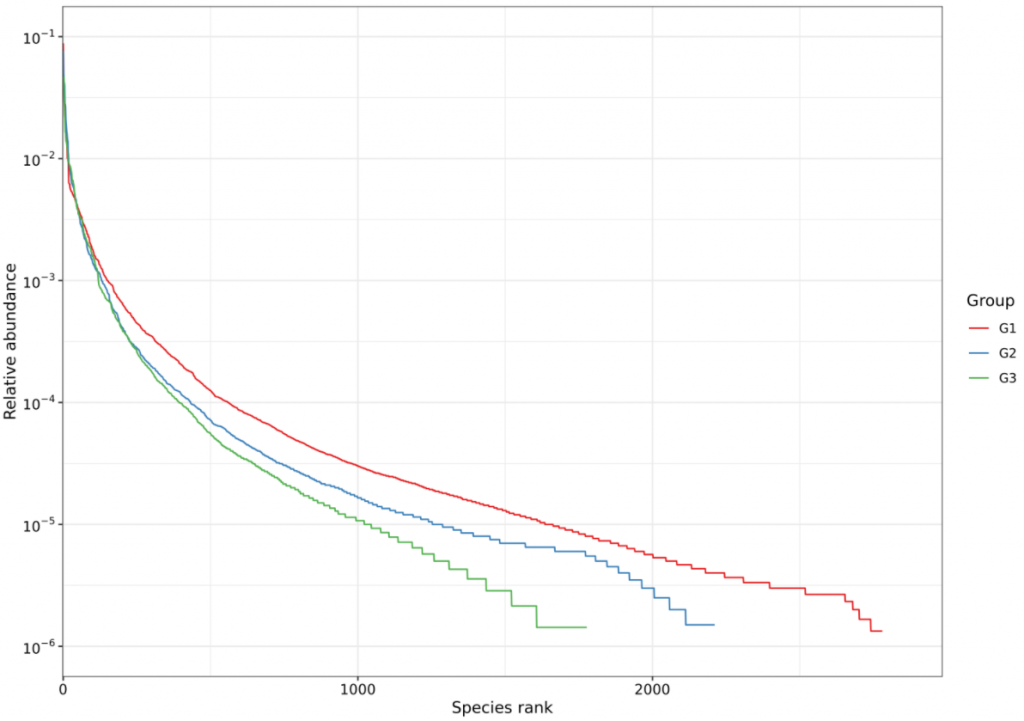
Beta多样性指的是样本间多样性,Beta多样性是衡量个体间菌落构成相似度的一个指标。通过计算样本间距离可以获得beta多样性距离矩阵,Beta多样性计算主要基于OTU的群落比较方法,有欧式距离、bray curtis距离等,这些方法优势在于算法简单,考虑物种丰度(有无)和均度(相对丰度),但其没有考虑OTUs之间的进化关系,认为OTU之间不存在进化上的联系,每个OTU间的关系平等。
另一种算法Unifrac距离法,是根据系统发生树进行比较,并根据16s的序列信息对OTU进行进化树分类,因此不同OTU之间的距离实际上有“远近”之分。而其他距离算法认为OTU之间的关系是平等的。Unifrac距离分为加权距离和非加权距离。
1
欧式距离(Euclidean distance):
欧几里得距离是空间中两点间“普通”(即直线)距离。
2
Bray-Curtis距离:
Bray-Curtis距离是生态学中用来衡量不同样地物种组成差异的测度。由J. Roger Bray and John T. Curtis 提出。其计算基于样本中不同物种组成的数量特征(多度,盖度,重要值等)。
计算公式为:
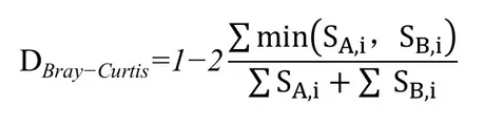
SA,i=表示A样本中第i个OTU所含的序列数;
SB,i=表示B样本中第i个OTU所含的序列数。
3
Unweighted UniFrac距离:
非加权距离包含特征之间的系统发育关系的群落差异定性度量。
4
Weighted UniFrac距离:
加权距离包含特征之间的系统发育关系的群落差异定量度量。
两者的区别在于:Weighted Unifrac 距离是一种同时考虑各样品中微生物的进化关系和物种的相对丰度,计算样品的距离,而Unweighted Unifrac则只考虑物种的有无,忽略物种间的相对丰度差异。
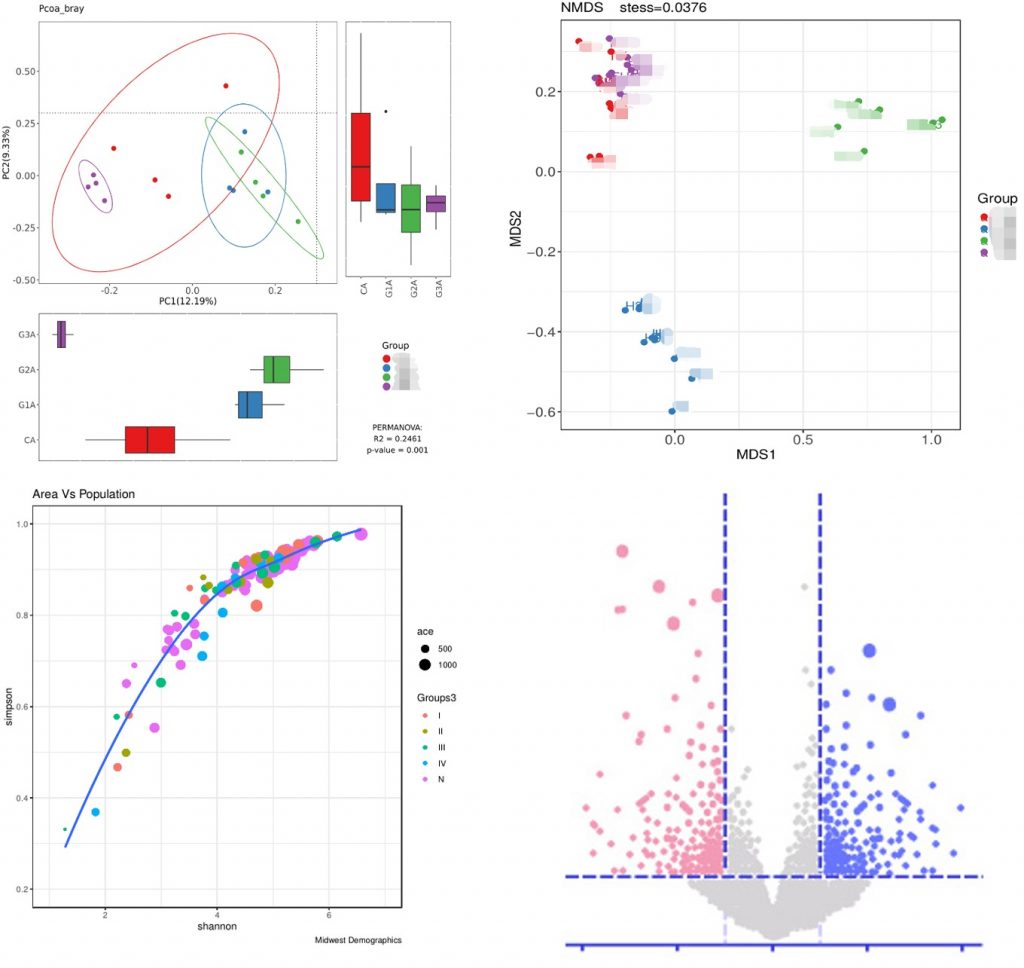
一般采用PCA、PCoA、NMDS等进行图像化展示,区分样本间的菌群组成差异。其原理是利用降维思想把样本平铺到二维平面上,使得相似的样品距离相近,相异的样品距离较远。
PCA图是基于ASV table的欧式距离,PCoA是基于两两样品之间的距离矩阵(有Bray-Curtis距离、加权距离、非加权距离),基于距离矩阵的统计检验方法有ANOSIM相似性分析和Adonis多元方差分析。
Anosim分析是一种非参数检验,用来检验组间差异是否显著大于组内差异,从而判断分组是否有意义。对 Anosim 的分析结果,基于两两样本之间的距离值排序获得的秩(组间的为 between,组内的为 within),这样任一两两组的比较可以获得三个分类的数据,并进行箱线图的展示(若两个箱的凹槽互不重迭,则表明它们的中位数有显著差异)。
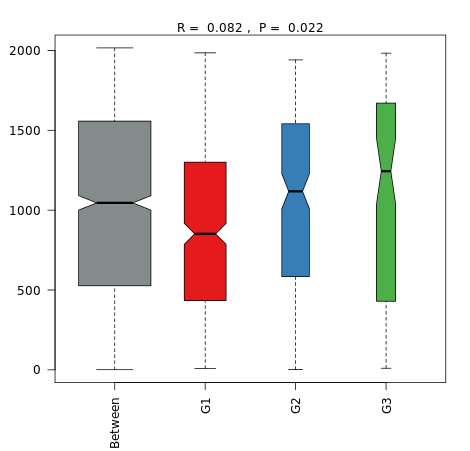
该方法主要有两个数值结果:R值,用于比较不同组间是否存在差异;P值,用于说明是否有显著差异。
R-value 介于(-1,1)之间,R-value > 0,说明组间差异大于组内差异。R-value < 0,说明组间差异小于组内差异, R只是组间是否有差异的数值表示,并不提供显著性说明。统计分析的可信度用 P-value 表示,P< 0.05 表示统计具有显著性。
Adonis检验,多元方差分析,其实就是PERMANOVA,亦可称为非参数多元方差分析。其原理是利用距离矩阵(比如基于Bray-Curtis距离、Unifrac距离)对总方差进行分解,分析不同分组因素对样品差异的解释度,并使用置换检验对其统计学意义进行显著性分析。它与Anosim的用途相似,也能够给出不同分组因素对样品差异的解释度(R值)与分组显著性(P值)。
PCA(Principal Components Analysis)即主成分分析,首先利用线性变换,将数据变换到一个新的坐标系统中;然后再利用降维的思想,使得任何数据投影的第一大方差在第一个坐标(称为第一主成分)上,第二大方差在第二个坐标(第二主成分)上。
这种降维的思想首先减少数据集的维数,同时还保持数据集的对方差贡献最大的特征,最终使数据直观呈现在二维坐标系。经过一系列的特征值和特征向量进行排序后,选取PCA分析得到的前三个主成分(PC1、PC2和PC3)中的任意两个数据作图。通过PCA 可以观察个体或群体间的差异。
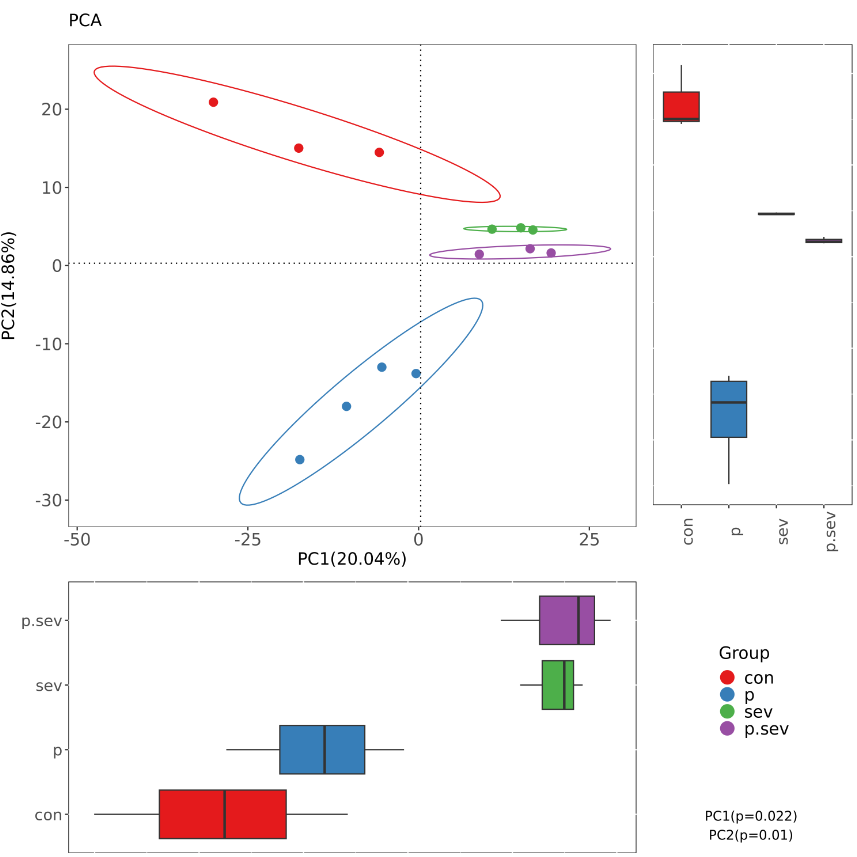
主坐标分析 PCoA (Principal component analysis)是一种非约束性的数据降维分析方法,可用来研究样本群落组成的相似性或相异性。通过 PCoA 可以观察个体>或群体间的差异。
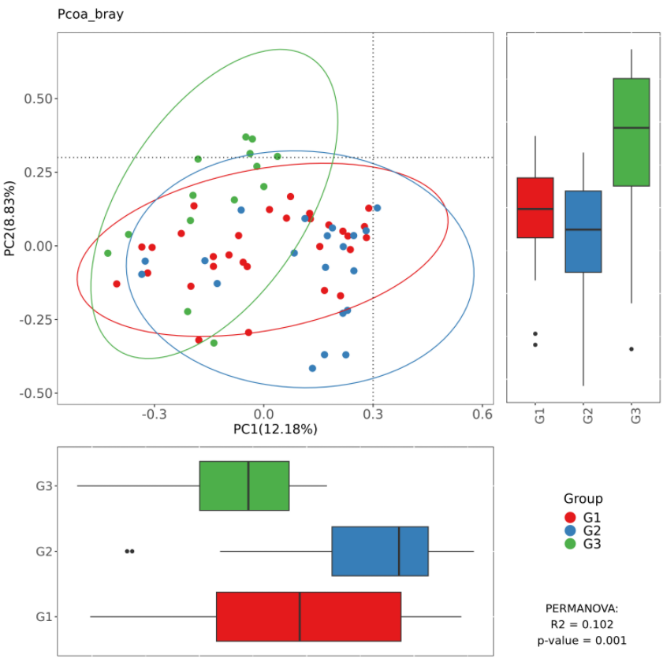
它与PCA类似,两者的区别为PCA是基于样本的相似系数矩阵(如欧式距离)来寻找主成分,而PCoA是基于距离矩阵(欧式距离以外的其他距离)来寻找主坐标。我们基于Bray-Curtis 距离、 Weighted Unifrac 距离和Unweighted Unifrac 距离来进行 PCoA 分析。
该图是基于Bray-Curtis距离做的PCoA图,图中右下角的P值就是基于Adonis检验得到的结果:
编辑
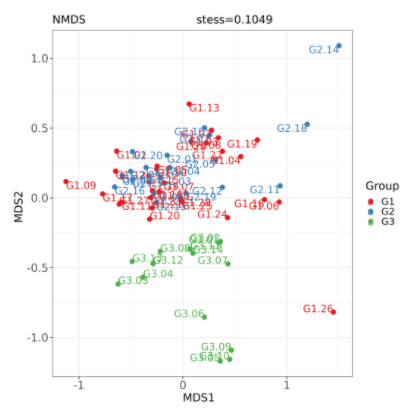
非度量多维尺度分析 NMDS 分析(Nonmetric Multidimensional Scaling)与上述 PcoA 分析类似,也是一种基于样本距离矩阵的分析方法,通过降维处理展现样本特定的距离分布。
与 PcoA 的区别是 NMDS 分析不依赖于特征根和特征向量的计算,而是通过对样本距离进行等级排序,使样本在低维空间中的排序尽可能符合彼此之间的距离远近关系(而非确切距离数值)。因此,NMDS 分析不受样本距离的数值影响,对于结构复杂的数据排序结果可能更稳定。

谷禾健康


DNA 测序是测量各种生命形式主要特性的基础。自20世纪50年代发现DNA双螺旋结构后,全世界科学家们就开始致力于确定不同物种基因组的原始序列。这一任务被称为基因组测序,旨在揭示不同生物的基因组组成和基因的排列顺序。
现代基因组研究标志之一是生成大量原始序列数据。这项工作的重要性在于,基因组序列的破译可以提供关于生物的遗传信息,包括基因功能、遗传变异和进化关系等方面重要线索。
在过去的几十年里,随着测序技术的不断发展和突破,测序的速度和精度都得到了显著提高。早期的测序方法主要依赖于Sanger测序技术,该技术基于DNA链延伸的原理,通过测量DNA链延伸反应中释放的荧光标记物来确定DNA序列。然而,由于其低通量和高成本的限制,Sanger测序逐渐被新一代测序技术(NGS)所取代。
随着新一代测序技术的兴起,如Illumina的高通量测序和454 Life Sciences的Roche测序平台以及华大基因(BGI)DNBSEQ-T7等测序平台,基因组测序进入了一个全新的时代。这些技术利用并行测序的原理,可以同时测序数百万个DNA片段,大大提高了测序的速度和效率。同时,这些技术也在成本和准确性方面取得了显著的突破,使得大规模基因组测序成为可能。
随着测序技术的不断进步,越来越多的原核和真核基因组序列被测序出来,并存储在公共数据库中,目前四个主要的数据库是:
它们目前拥有丰度的实验以及样本核苷酸序列原始数据,此外还有蛋白质序列或大分子结构数据。这些数据库为科学家们提供了宝贵的资源,可以用于研究和比较不同物种的基因组,从而增进对生物多样性、进化和基因功能的理解。
除了基因组序列的分析,还需要开发各种生物信息学工具和数据库,以帮助解释和注释基因组数据。这些工具可以用于预测基因的功能、识别调控元件、比较不同物种之间的基因组差异等。
随着计算机技术的不断进步,测序数据也逐渐借助人工智能和机器学习等技术。这些技术可以辅助人们更快速、准确地分析和解释基因组数据,发现隐藏在数据中的模式和关联。机器学习算法可以用于预测基因的功能、识别基因组中的重要调控区域或精确区分相近物种等。
例如谷禾肠道菌群16S数据库,是谷禾健康从几十万人肠道菌群检测数据中提取的16S序列库,通过宏基因组匹配数据和模型构建,重新完成到种的物种注释。进一步通过基因组数据研究和分析,为您提供深度个性化的健康检测方案。
目前测序技术已经在许多应用领域产生了广泛的影响。例如,基因组测序为研究人类遗传疾病的发病机制提供了重要线索,短读长、高测量通量和低成本为个体化医学和精准治疗奠定了基础。此外,测序还广泛应用于农业、环境科学和生物工程等领域,为改良作物、保护环境和生产高效生物工艺提供了有力支持。
因此本文和大家分享DNA测序相关的知识以及测序技术的发展和测序的注意事项等。
DNA(脱氧核糖核酸)是遗传物质,存在于所有生物的细胞中,以双螺旋结构存在,由核苷酸组成,包括磷酸基团、脱氧核糖和四种氮含基(腺嘌呤A、胸腺嘧啶T、胞嘧啶C和鸟嘌呤G)。DNA负责存储遗传信息,指导蛋白质合成,并在细胞分裂时复制自身,确保遗传信息传递给子代。
不同物种的DNA在结构上非常相似,但在序列和组织上存在差异
人类DNA包含约30亿个碱基对,组成大约2万到2万5千个基因,这些基因分布在23对染色体上。人类DNA中的遗传信息决定了我们的外貌、生理功能和健康状况。人类的遗传多样性虽然存在,但所有人类的DNA序列大致相似,约有99.9%的相似度。个体基因组有 3-400 万个碱基对位置存在差异。这些变异可在单核苷酸多态性(SNP)中捕获 ,但也存在一些称为结构变异(SV) 的较大变异。

大多数病毒基因组有 10000 个 bp;某些植物的基因组长达数千亿个碱基对。细菌,通常具有较小的基因组,可以从几百万到几千万碱基对不等。细菌的DNA通常是单个环状染色体,而不是多条线性染色体。此外,许多细菌还含有质粒,这些是小型的DNA分子,可以在细菌之间进行转移,促进基因的水平传播,这是细菌适应环境和抗药性发展的重要机制。
总之,不同物种的DNA在功能上都是遗传信息的载体,但是在大小、形态和序列上存在差异,这些差异导致了物种之间的多样性。
不同物种个体基因组差异的产生有两个原因:
➼ 随机突变,在进化过程中发生,因为自然选择有利于某些表型。这些主要是由于细胞分裂期间 DNA 复制过程中的“错误”造成的。大多数突变都是有害的,会导致有害的表型变化并导致细胞死亡。有时,自然选择有利于某些突变,而这些突变会保留在种群中。
➼ 重组,发生在哺乳动物等高等生物的繁殖过程中。在重组过程中,亲本生物体传递给子代的遗传物质是来自亲本生物体的遗传物质的混合物。
DNA双链碱基互补
DNA 是双链的,并以双螺旋形式构建,其中核苷酸对作为螺旋的“横档”(因此称为“碱基对”)。腺嘌呤总是与胸腺嘧啶发生化学结合,而胞嘧啶总是与鸟嘌呤发生化学结合。换句话说,A 与 T互补,类似地 C 与 G 互补。AT 和 CG 对称为互补对。
DNA的结构如下所示:
图源:medlineplus
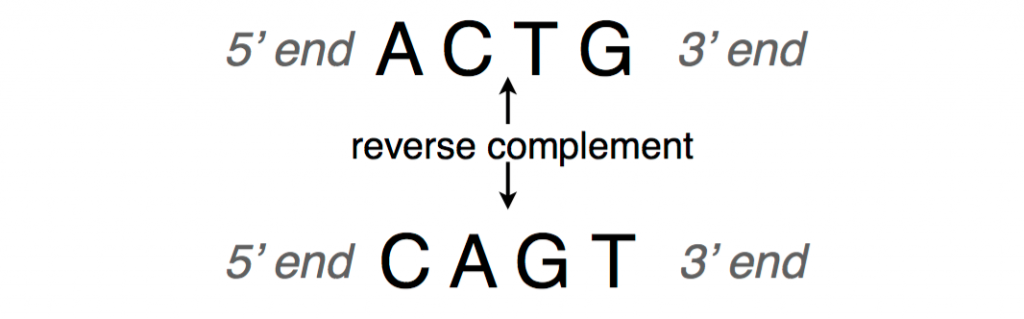
DNA序列通常以5’端(头部)到3’端(尾部)的方向展示或写入。当我们有一条 DNA 链时,由于知道互补对,所以可以推断出另一条链是第一条链的反向互补链。
为了获得反向互补,可以反转原始字符串中核苷酸的顺序,然后互补核苷酸(即,将 A 与 T 互换,将 C 与 G 互换)。
下图显示了 DNA 片段及其反向互补链的示例。
DNA 补体
DNA复制
DNA 是细胞复制的基础。当细胞进行细胞分裂(也称为有丝分裂)时,细胞核中的 DNA 会被复制,并通过下图所示的一系列步骤,一个亲代细胞产生两个相同的子细胞。
有丝分裂的图
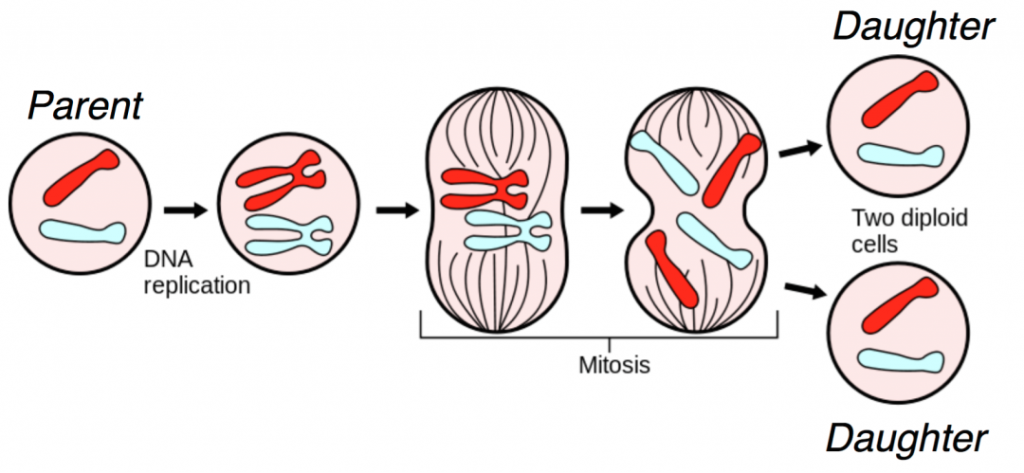
图源:wikipedia
有丝分裂过程中涉及多种生物分子,我们在这里对有丝分裂过程进行了高度简化的解释。
在图中,我们从两条染色体开始:红色和蓝色。
首先,DNA 被复制,产生更熟悉的 X 形染色体。通过生物分子信号的复杂级联和细胞内重组,(现已复制的)染色体在细胞中部排列。对于每条染色体,两半被拉开,两个子细胞中的每一个都会收到原始染色体的副本。这会产生两个与原始亲本细胞在遗传上相同的子细胞。
DNA 复制是这张图中最重要的部分;这是用来进行测序的基础过程。DNA复制如下图所示:
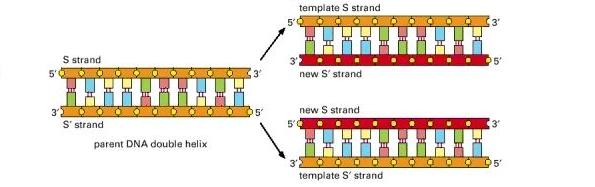
在 DNA 复制过程中,两条 DNA 链首先被解压缩,产生两条单链,每条链都充当复制的模板。然后将短 RNA 引物附着到 DNA 上的特定位点;引物中的碱基与位点中的碱基互补。酶促进(或“催化”)化学反应,而DNA 聚合酶是催化新核苷酸与延伸结合引物的模板 DNA 互补配对的酶。
DNA 聚合酶用来延伸链的核苷酸称为dNTP(脱氧核苷酸三磷酸)。从生物化学角度来看,它们与核苷酸略有不同,因为它们在 DNA 复制过程中更容易使用。对应于A、C、G和T的dNTP分别是dATP、dCTP、dGTP和dTTP。
获取DNA序列主要依赖于测序技术。常用的测序技术有桑格测序法和下一代测序法。下一章节会详细介绍。
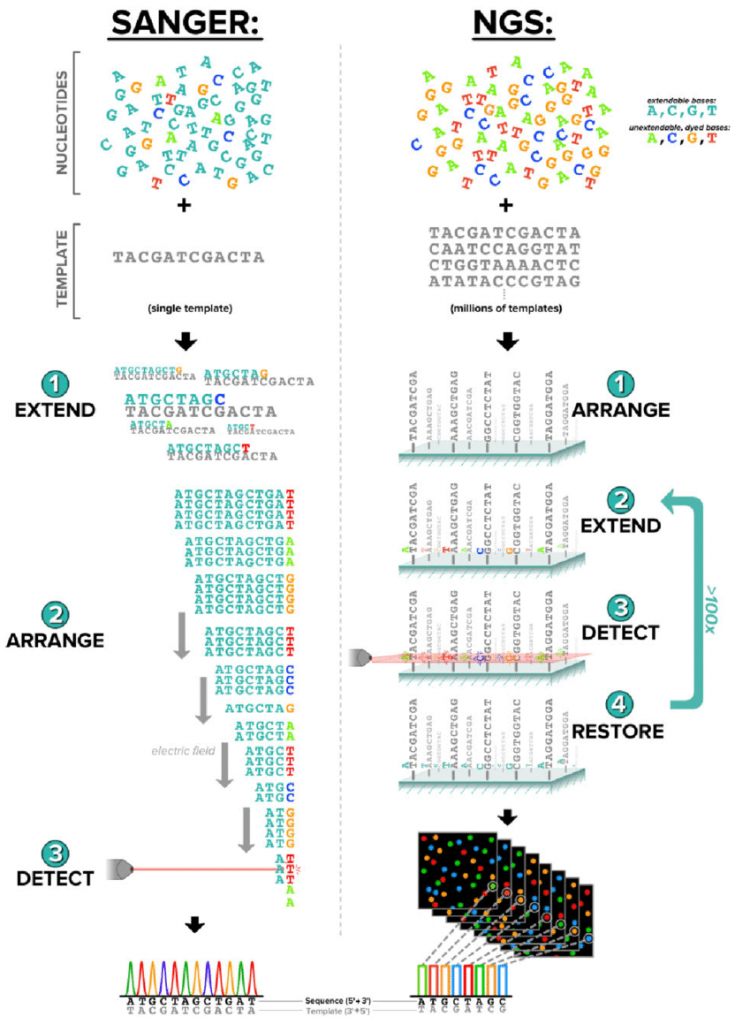
图源:praxilabs
桑格测序法准确度高但通量低。下一代测序法代表了高通量测序技术,它实现了并行化操作,大大提高了序列通量,降低了测序成本和时间,因此适合进行整个基因组或transcript组的测序。这些技术使大规模、高精度的DNA测序分析成为可能。
DNA 测序方法的发展在 2000 年左右达到顶峰,主要基于四位研究人员的贡献。
➛
Allan Maxam 和 Walter Gilbert 在 70 年代开发了一种 DNA 测序的化学方法,其中末端用放射性磷标记的 DNA 片段经过碱基特异性化学切割,并通过凝胶电泳分离反应产物。
➛
1977 年,Frederick Sanger采用另一种方法,通过使用链终止双脱氧核苷酸类似物完善了测序方法,该类似物导致引物DNA合成的碱基特异性终止。在这种方法中,引物通常用放射性磷标记。
➛
Leroy Hood 与他的同事 Michael Hunkapiller 和 Lloyd Smith 在1986年通过使用荧光标记的双脱氧核苷酸将 Sanger 方法修改为更高的通量配置。这种方法可避免使用寿命有限的放射性化合物,而是使用稳定的荧光探针。此外,所有核酸碱基的分析可通过仅读取一个而不是四个电泳泳道来完成,并且读取过程可以自动化。
这种高通量配置用于第一个人类基因组的测序,该测序于 2003 年通过人类基因组计划完成,该计划历时 13 年。

由于方法的改进和自动化,2008 年,另一个人类基因组在 5 个月的时间内完成了测序。第一份人类基因组草图的完成只是现代 DNA 测序时代的开始,它带来了更多的发明和新的、先进的高通量 DNA 测序策略,即所谓的下一代测序 (NGS)。
NGS 策略的发展正在满足我们对测序通量和成本的需求,从而在基因组研究中实现多种当前和未来的应用。这些先进方法需要开发新的生物信息学工具,作为分析过程中产生的大量数据的必要先决条件。
第一代测序——桑格测序
Fred Sanger 及其同事开发了一种基于放射性标记部分消化片段检测的相关技术。
著名的桑格测序起源于 20 世纪 70 年代末,当时桑格开发了一种基于凝胶的方法,将 DNA 聚合酶与标准核苷酸和链终止核苷酸 (ddNTP) 的混合物结合起来。将 dNTPS 与 ddNTP 混合会导致 PCR 期间测序反应随机提前终止。四个反应并行进行,每个反应包含一种版本的链终止核苷酸。使用凝胶电泳可视化该过程使得能够逐个碱基读取序列。在当时,这项技术是革命性的。它能够对 500-1,000bp 片段进行测序。
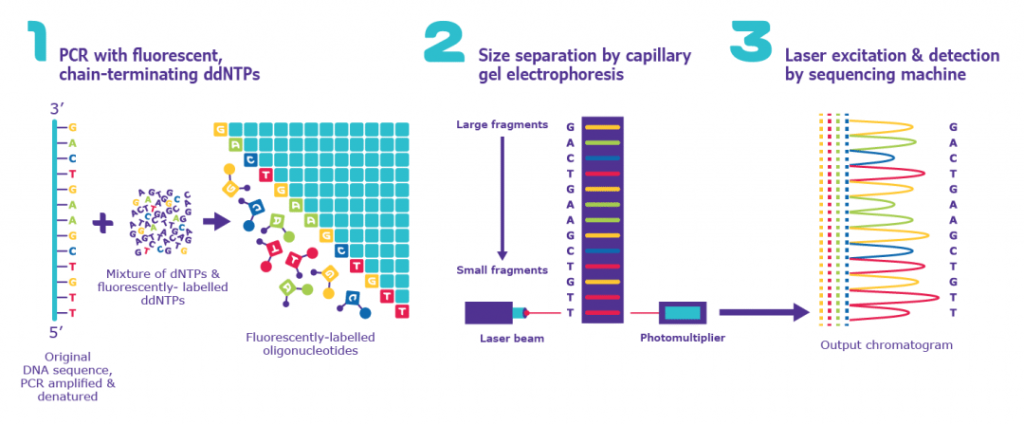
图源:praxilabs
桑格方法的一种变体——加减法,由桑格和艾伦·科尔森开发,于1977年获得了第一个DNA基因组序列,即噬菌体φX174。
图源:pixels
两年后,艾伦·麦克萨姆和沃尔特·吉尔伯特发表了他们的化学裂解技术,该技术成为第一个广泛采用的 DNA 测序方法。

到了 20 世纪 80 年代,桑格最初的方法已经自动化(毛细管电泳)。大块凝胶被更细的丙烯酸毛细管取代,结果可以在电泳图上查看。这项技术对于 2003 年人类基因组计划的完成至关重要。尽管如此,即使在人类基因组计划之后,毛细管电泳的成本仍然过高,无法实现大规模测序项目。
到 2000 年代中期,人们做出了一些努力来降低测序成本。世界各地的实验室正在测试用于更高通量筛选的新方法和技术。
第二代测序技术
第二代测序,也称为下一代测序(NGS)。简单来说,二代测序是依靠PCR文库构建和激光探针荧光信号读取的短读长测序。
目前最常见的平台有Illumina和华大基因(BGI)。
Illumina测序平台
由 illumina 等公司开发的第二代 NGS 技术可分为两大类:杂交测序或合成测序。
华大测序平台
华大基因测序化学方法被称为组合探针锚定合成(cPAS)。它采用Phi 29 DNA聚合酶进行滚动圆环复制,合成一条长的单链DNA,自组装成约300纳米大小的纳米球。然后进行鉴定以确定 DNA 序列。
随着大规模双脱氧测序技术的进步,一项新技术的出现奠定了下一代测序(NGS)技术的基础。这项名为焦磷酸测序的方法,利用DNA合成过程中焦磷酸盐产生的光信号来确定核苷酸序列。在这个过程中,模板DNA被固定在一个固相表面上,随着每个核苷酸的加入,通过检测焦磷酸释放的光信号来推断DNA的序列。此技术后续还引入了珠子,以便更有效地附着DNA分子。
焦磷酸测序技术被454 Life Sciences公司所开发,并最终被罗氏公司收购,成为市场上第一个取得重大成功的商业化NGS平台。
乳液PCR
在这一平台中,DNA文库通过油包水乳液PCR技术附着在微小珠子上。在测序过程中,当较小的珠联酶和dNTPs被引入到反应板上时,便可进行焦磷酸测序。这种高度并行化的方法显著提高了测序的吞吐量,实现了数量级的提升。
桥式放大
继454测序技术取得成功之后,许多新的并行测序技术相继出现。其中最显著的是Solexa测序技术,该技术后来被illumina公司收购。
Illumina测序平台因此成为了第一个实现商业化的高通量并行测序技术。

其他
随着时间的推移,新技术不断涌现,其中包括:
图源:slideserve
这些创新技术已经成为下一代测序(NGS)技术领域的一部分。NGS平台目前是主流的测序技术,它们可以以相对较低的成本进行高通量的测序工作。然而,这些平台的读长通常有限,一般产生的读长在50到500碱基对(bp)之间。
本文我们主要介绍Illumina和华大这两个平台的测序原理。其他的简单说明一下。
Illumina测序平台介绍
Illumina 的首个测序平台是通过收购Solexa公司获得的,被命名为基因组分析仪,并于2007年开始商业化运作。这台设备能够在每个测序通道中对600万个扩增的DNA片段进行测序,最初每个片段的读取长度大约是30个碱基。Illumina 不久后提升了这一读取长度,增至100多个碱基对。同时,流动池中扩增片段的数量也得到了提升,使得基因组分析仪的输出能力达到了80吉字节的碱基信息。
注:吉字节(GigaByte),又称千兆字节,是计算机存储容量单位,简称GB。
2010年,Illumina 推出了其第二代NGS设备——HiSeq。这款设备配备了两个流动池:
紧接着,Illumina 又发布了HiSeq X10,该设备通过使用图案化的流动池凹坑(代替了传统的随机扩增簇),进一步提高了可分析片段的数量。
目前,Illumina 提供了多种测序设备,包括NextSeq 和 NovaSeq 系列,以及适用于不同规模需求的台式测序仪,如iSeq100和 MiniSeq。

NextSeq
NextSeq 500 于 2014 年推出,采用两种染料测序技术,而不是其前代产品使用的四种染料测序技术。仅拍摄红色和绿色图像,从而显着缩短周期和数据处理时间。该仪器能够在大约 30 小时的运行时间内读取4亿个碱基对。
NextSeq 1000 和 2000 机器于2020年发布,旨在通过提供机载信息学和基于云的技术来简化工作流程。P3 流动池扩展了 NextSeq 2000 仪器的范围,在单次测序运行中提供 11 亿次读取。
NovaSeq6000
NovaSeq 6000于2017年发布。它能够运行三种不同的芯片,并且可以生成100 GB的序列输出,价格仅为375美元——这个价格仅适用于测序,不包括DNA分离、文库制备、测序分析或数据贮存。
本质上,该机器每次运行能够对多达 48 个完整人类基因组进行测序,这可能需要长达 44 小时。其他关键应用包括单细胞分析、转录组测序和宏基因组分析。

HiSeq X 系列
HiSeq X Ten 测序仪是一种高性能的测序系统,它能够在单次运行中产生高达16 Tb的序列输出。使用该系统,可以以不到1000美元的价格对人类基因组进行30倍或更高倍数的测序,并且每年可以提供超过18,000个人类基因组的测序数据。每个流动池可以生成多达520亿次的读取,最长运行时间为48小时。
该系统具有超越人类物种的全基因组测序能力,并且还可以用于全外显子组测序、转录组测序、单细胞分析和多组学研究。
华大测序平台(BGI) 介绍
华大基因集团成立于1999年、参与人类基因组计划的中国公司。华大基因于2012年收购了Complete Genomics,其产品由子公司(华大智造)销售。
DNBSEQ-T7
DNBSEQ-T7于2019年推出,旨在支持健康项目和临床研究的一系列大规模测序应用。据报道,与百万基因组整体解决方案软件和硬件一起,DNBSEQ-T7 每年可以对多达 800,000 个样本进行测序。

硬件解决方案包括自动化文库制备系统,这意味着测序机可以24小时运行,无需人工干预,每天可以完成60个人类全基因组测序。其商业化预计将把个人全基因组测序的成本降低至 500 美元以下,从而改变测序格局。
华大基因测序化学
BGI的测序化学方法被称为组合探针锚定合成(cPAS)。它采用Phi 29 DNA聚合酶进行滚动圆环复制,合成一条长的单链DNA,自组装成约300纳米大小的纳米球。荧光探针被结合在其中,纳米球被连接到硅片流动池上,选择性地与带正电的材料高度有序地结合。然后,荧光发射被成像和测量,以记录碱基位置。
与所有短读取测序方法一样,BGI平台主要缺点是无法获得长的DNA序列。然而,基于cPAS的测序的一个重要优势是Phi 29 DNA聚合酶的高准确性,确保了环状模板的准确扩增。此外,由于DNA纳米球在流动池上保持不动,它们不会产生光学重复,并且不会干扰相邻的DNA。
DNBSEQ-G99(G99)
DNBSEQ-G99(以下简称“G99”)基因测序仪采用的是基于聚合酶链式反应(PCR)原理的测序技术。测序过程中,首先使用特定的引物引导 DNA 序列进行体外扩增,然后添加一种含有测序所需的四种不同颜色的 dNTP(脱氧核苷酸)和荧光标记物。当引物与待测序列结合后,聚合酶开始合成新的链,同时荧光标记物被激活并发出不同颜色的荧光。通过记录这些荧光信号,并使用计算机进行数据分析和解码,最终确定每个碱基的序列。

G99突破性地实现了12小时可完成PE150测序,从用户需求出发,在提供高质量的测序数据的同时,做到快速、简单、灵活,能为测序工作带来更好的体验,应用场景大大扩展了。
而且DNBSEQ-G99获批国家药品监督管理局(NMPA)医疗器械注册证(国械注准20233221289)。此次获批意味着,中小通量测序仪中的“速度王者”DNBSEQ-G99被准许在国内市场应用于临床,将能够充分发挥其快速、灵活的优势,服务于临床方向的应用需求。
第三代测序技术
第三代测序技术的原理主要基于单分子测序或合成测序方法,通过直接读取DNA分子的序列来进行测序。
单分子测序:通过将DNA固定在表面上,并使用荧光染料或其它探针进行测序。
单分子实时测序(SMRT):使用PacBio公司的SMRT技术,通过监测DNA聚合酶在DNA模板上的荧光信号来进行测序。
纳米孔测序(Nanopore):使用Oxford Nanopore Technologies(ONT)的纳米孔测序技术,通过将DNA分子通过纳米孔,测量通过纳米孔的电流变化来进行测序。

合成测序:通过在反应体系中逐步合成DNA序列,并使用荧光标记的核苷酸来标记每个碱基。第三代测序技术通常具有较长的读取长度,可以读取数千到数百万个碱基。
第三代测序技术的不断发展和改进,为基因组学研究提供了更多的可能性,可以更好地解析复杂的基因组结构和功能。适用于长片段的测序,如全基因组测序、长读段转录组测序、甲基化测序等。然而,第三代测序技术也面临一些挑战,如测序错误率、数据处理和分析等方面的问题,需要进一步的研究和改进。
市场上的其他三代测序平台:
MinION:MinION设备是一种便携式的纳米孔测序仪器,可以实现实时测序,并且具有较小的体积和较低的成本。
GridION:GridION设备是一种高通量的纳米孔测序仪器,可以同时进行多个样品的测序。
PromethION:PromethION设备是一种高产量的纳米孔测序仪器,可以进行大规模的基因组测序。
此外国内目前也有多家公司已推出或正在开发三代测序仪,包括真迈生物,齐碳科技等。
测序将继续变得越来越高效和经济实惠,彻底改变与基因组学相关的多个领域。目前,所有高通量测序(NGS )方法都需要文库制备。该方案发生在 DNA 片段化之后,其中接头连接到每个片段的末端。接下来通常是 DNA 扩增步骤,以产生一个文库,然后可以通过 NGS 平台进行测序。
➦1
样品制备的本质是将生物样品中的核酸混合物转化为不同类型的文库,以准备进行NGS技术所需的测序步骤。如果未正确遵循方案,测序将会受到影响。每个准备步骤都是基础性的,并且根据样本和NGS平台的类型有不同的考虑因素。因此,在开始实验之前,考虑如何执行最有效的方案以确保最高质量的结果非常重要。
样品制备的一般步骤如下:
步骤1:提取遗传物质
这是每个样品制备方案的第一步。从各种生物样品中提取核酸(DNA 或 RNA)。
步骤2:文库准备
生成文库需要一系列步骤,最终目标是将提取的核酸转换成适合所选测序技术的格式。这是通过将目标序列片段化至所需长度,然后将特定的接头序列连接到这些目标片段的末端来完成的。
适配器还可以包括条形码,识别特定样品并允许多重分析。片段化可以通过物理或酶促方法完成。
步骤3:放大
这是一个可选步骤,但通常也是必需的。这取决于 NGS 的应用和样本量。扩增对于获得足够的覆盖范围以对含有少量起始材料的样品进行可靠测序至关重要。聚合酶链式反应(PCR) 是增加 DNA 量的常用方法。有关可实现小样本核酸检测的 PCR 方法出现的更多信息。
步骤4:纯化和质量控制
此步骤是必要的,以去除可能阻碍测序的任何不需要的材料。一些NGS平台可能对尺寸要求较窄,因此丢弃太大或太小的片段可以提高测序效率。最佳文库大小由测序应用决定。这种“清理”通常通过基于磁珠的清理或琼脂糖凝胶来完成。
质量控制是进行测序之前的最后一个过程。确认 DNA 的质量和数量可以提高测序数据的可信度。后续的实验既耗时又昂贵,因此需要严格的质量控制步骤以确保所有样品都适合其应用。
➦2
挑战 1
许多样本是从有限数量的样本或甚至单个细胞中提取的。它们本身并不能提供足够的遗传物质,因此需要进行 PCR。然而,该扩增步骤很容易给样品引入偏差。PCR 重复是指存在完全相同的 DNA 片段的多个拷贝。太多的 PCR 重复会导致实验的测序覆盖率不均匀。
解决方案 1:消除所有偏差来源有些不可能,但了解偏差发生的位置并采取所有实际步骤将其最小化非常重要。高 PCR 重复率表明文库制备需要进行一些修改,可能需要提高 NGS 文库的复杂性。
许多程序都可以删除 PCR 重复项,最常用的是 Picard MarkDuplicates 和 SAMTools。此外,特定的PCR 酶已被证明可以最大限度地减少扩增偏差。最终,文库制备的目标是最大限度地提高样品的复杂性,并最大限度地减少扩增造成的偏差。
挑战 2
建库效率低下是样品制备过程中面临的问题。具有正确适配器的片段比例较低反映了这一点。其后果是获得的测序数据量减少,嵌合片段数量增加。嵌合读数源自基因组中彼此不相邻的部分,并且是测序期间错误的来源。
解决方案 2:据报道,PCR 产物的有效A 加尾可防止嵌合体形成,该程序是通用的,可应用于多种不同的文库构建技术。此外,链分割伪影读数 (SSAR) 已被建议减少样本中嵌合伪影的数量,并且嵌合体检测程序可用于过滤原始序列,以实现仅 1% 的总体嵌合率。
挑战 3
样品污染是一个固有的问题,因为单独的文库通常是并行制备的。最可能的主要污染源是预扩增,这是一种在 PCR 之前增加核苷酸序列量的方法。
解决方案 3:可以通过质控,阴性对照,设置重复等步骤识别污染,确保在样品制备过程中使用无菌技术和无菌实验条件,以防止外源性污染的引入。
此外使用独特的条码和标签对样品进行标识(谷禾所有样本全程唯一条形码溯源识别管理),以避免混淆和交叉污染。最后,做好定期清洁和消毒:定期清洁和消毒实验室设备和工作区域,以减少污染的积累和传播。
挑战 4
文库制备的巨大成本主要归因于实验室设备、需要经过培训的人员和试剂成本。
解决方案 4:通过优化实验步骤和条件,可以减少试剂的使用量和浪费,从而降低成本。确保实验室人员接受适当的培训和技术支持,以提高实验的效率和准确性。与其他实验室或研究团队合作,共享设备和资源,共同承担成本和实验负担。随着自动化技术变得越来越流行,样品制备的准确性和效率可能会提高。
什么是碱基平衡?
测序中一个不可忽视的原则就是碱基平衡,是指测序过程中,每个循环中A、C、G和T四种碱基,比较均匀地存在。需要兼顾的平衡度与复杂度。在测序过程中,保持碱基平衡是非常重要的,可以确保测序结果的准确性和可靠性。
什么是碱基不平衡文库?
就是扩增子产生的文库,扩增子的特点是有特定的起始位点的。反应到测序图像上,就会呈现一张照片特别亮,光点很多,而其他三张照片就特别暗。这时软件做空间上比对就比较难。结果是判断的可靠性比较差,导致对于碱基的判读就会出现错误,从而导致测序质量值大幅度下降。一般添加诸如基因组DNA文库,或掺入大量的平衡碱基文库,包括phix文库等。同时,也可以尽量多掺入不同类型的扩增子文库。
此外,碱基平衡还涉及到检测和纠正测序过程中的碱基偏差。在测序过程中,可能会出现碱基的插入、缺失或错误,这些错误会影响测序结果的准确性。为了纠正这些错误,开发了各种生物信息学工具和算法,例如质量控制和碱基校正等。
文库长度含两侧测序接头和插入目标片段,整个文库的长度范围不能过宽,一般建议在250bp-450bp之间比较好,超过600bp以上就会造成一些不利影响。
过长的文库长度会降低测序效率
在Illumina测序等高通量测序平台上,测序片段长度会影响测序的质量和效率。过长的文库长度会增加测序过程中的错误率,并且会导致测序片段的读取长度变短。这会降低测序的可靠性和准确性,影响后续的生物信息学分析和数据解读。但是如果文库片段过短的话,该短片段测序到后期,就是要测接头序列了,有的时候连接头序列都测完了,那就没有信号了,后续会读取一些假信号,降低测序质量值。
过长的文库长度会降低簇密度
簇密度是基于单次只测一个碱基的边合成边测序原理,要求对各个分子簇的反应时间要求一致。也就是各个分子簇必须同时进行反应。理想状态当然是如此,但是实际PCR反应过程中,各个分子的反应时间还是不尽相同的(一般体系和酶要控制好)。因此,会产生有的分子簇内的分子反应的快,有的慢的情况过长的文库长度会降低簇密度。在Illumina测序中,DNA片段会被固定在测序芯片上的聚合酶链反应(PCR)产物中,形成簇。过长的文库长度会导致PCR扩增效率降低,从而降低簇密度。低簇密度会减少每个簇中的测序片段数量,进而降低测序的覆盖度和深度,影响后续的数据分析和解读。
过长的文库长度可能导致碱基偏移
在测序过程中,由于DNA聚合酶的滑动等原因,长片段的文库容易出现碱基偏移的情况。
数据量(yield)
数据量指一次测序所获得的PF数据的总量。注意,是PF数据(PF数据是指通过滤波后的有效测序数据,即通过质量控制筛选后的测序片段),而不是原始数据。数据量当然越多越好,实际成绩与测序仪型号有关,不同的机器,产量不一样。
PF数据的总量是衡量测序深度和测序质量的一个重要指标。较高的PF数据总量表示测序过程中得到了更多的有效测序片段,可以提供更高的测序覆盖度和深度,从而提高后续数据分析的可靠性和准确性。
Q30
Q30是指在测序过程中,质量值(Quality Value,QV)大于或等于30的碱基。质量值是根据测序仪器对每个碱基的测量结果和信号峰值计算得出的,用来表示该碱基的质量好坏。Q30值越高,表示测序数据中高质量的碱基比例越高。
by:Alexander William Eastman
需要注意的是,Q30的大小与测序片段(read)的读长有关。如果读长较长,即测序片段包含的碱基数较多,那么要求每个碱基的质量值都达到或超过30就更加困难,因此平均%Q30可能会降低。相反,如果读长较短,即测序片段包含的碱基数较少,那么要求每个碱基的质量值达到或超过30就相对容易,平均%Q30可能会提高。
比对率(mappingrate)
将测序数据与参考序列(reference)进行比对,是测序数据分析中的一项重要步骤。比对率是指在比对过程中,测序数据与参考序列完全一致的碱基占测序数据总碱基数的比例。比对率越高,表示测序数据的准确性和可靠性越高。在细菌16S测序中,可以根据具体的需求选择适合的比对工具。
常用的比对工具是基于Smith-Waterman算法的BLAST(Basic Local Alignment Search Tool)和基于Burrows-Wheeler变换的Bowtie、BWA等工具。高比对率是测序数据质量好的重要指标之一。它表示测序数据的准确性和可靠性较高,能够提供更准确的基因组信息和变异位点等重要信息(在谷禾16s测序中,尤其粪便样本约超过 70%能比对到种)。在后续的数据分析和解读中,高比对率的测序数据更有助于准确地进行变异检测、基因表达分析、功能分析等。
需要注意的是,比对率受到多种因素的影响,包括测序数据质量、参考序列的准确性、数据库以及比对算法的选择等。在进行测序数据分析时,需要综合考虑比对率、测序数据质量和其他相关指标,以获得准确可靠的分析结果。
覆盖度(coverage)
由于测序数据的生成过程中存在一些技术和生物学上的随机性,导致不同区域的测序数据的覆盖深度(coverage depth)是不一样的。
覆盖深度是指在某个特定位置的测序数据的读段数目或测序碱基数目。覆盖深度越高,表示该位置的测序数据越丰富,测序结果的准确性和可靠性越高。
需要注意的是,覆盖深度的均匀性和高低受到多种因素的影响,包括测序深度、测序技术、样本质量等。
重复率(duplicationrate)
在二代测序文库的构建过程中,除了无PCR流程(PCR-free approach),其他方法都需要进行PCR扩增。PCR扩增会导致染色体的不同区域放大程度不一致,有部分序列被过度放大。这是一种人为引入的偏差。重复率与文库构建试剂的质量有关,对于人类全基因组测序来说,通常<10%。
捕获率(capturerate)
杂交捕获建库是通过探针杂交捕获来从基因组文库中富集相应序列的,探针杂交捕获存在着捕获效率高低的问题,因此考察、评价这一步骤成败、好坏的参数就是捕获率,越高越好。捕获率与所用的捕获试剂有关,不同的试剂,捕获率不同。
高通量测序操作包括样品准备、文库构建、PCR扩增、测序仪器运行等。只有按照标准SOP规范操作,才能保证实验的准确性和可重复性。新测序仪平台和技术的不断出现和改进,使得高通量测序无论通量、质量、速度和成本都在快速进步,高通量测序的应用范围也大大得到拓展,不久的将来有望以低成本随时随地的开展高通量测序应用。

谷禾健康

当谈到肠道菌群研究时,16S测序是一种常用的方法,它在了解微生物组成和多样性方面非常重要且实用。
16S rRNA是细菌和古细菌中的一个高度保守的基因片段,同时具有一定的变异性。通过对16S rRNA基因进行测序,可以确定微生物群落中存在的不同菌属和菌种,并评估它们的相对丰度。
针对肠道菌群16s测序概括起来可以了解以下内容:
揭示微生物组成:通过16S rRNA测序,可以获得关于肠道菌群中存在的不同菌属和菌种的信息。这有助于了解菌群的组成,包括有益菌、有害菌和其他微生物的相对丰度。
探索微生物多样性:16S rRNA测序可以评估微生物群落的多样性水平。通过分析不同样本中的菌群组成和多样性指标,可以比较不同个体、不同健康状态或不同治疗干预对微生物多样性的影响。
鉴定潜在病原菌:通过16S rRNA测序,可以鉴定肠道中存在的潜在病原菌。这有助于了解潜在的疾病风险,并为相关疾病的预防和治疗提供指导。
监测治疗效果:在临床研究中,通过定期进行16S rRNA测序,可以监测治疗干预对肠道菌群的影响。这有助于评估治疗效果,并为个体化治疗提供依据。
指导个体化营养干预:通过16S rRNA测序,可以了解个体的肠道菌群状态,进而指导个体化的营养干预。根据菌群组成和功能,可以制定相应的饮食方案,促进肠道健康和营养吸收。
以上总结了16s对于了解肠道菌群的重要价值,今天本文主要分享和介绍16s测序相关的定义和术语,以及菌群命名的规则等。
正确的说法是16S rRNA(不是16s rRNA或16s rDNA)。
16S rRNA 代表16S核糖体核糖核酸 (rRNA),其中S(Svedberg) 是测量单位(沉降率)。该rRNA是原核核糖体小亚基(SSU)以及线粒体和叶绿体的重要组成部分。
下图显示了16S rRNA(简称16S)如何参与原核核糖体。
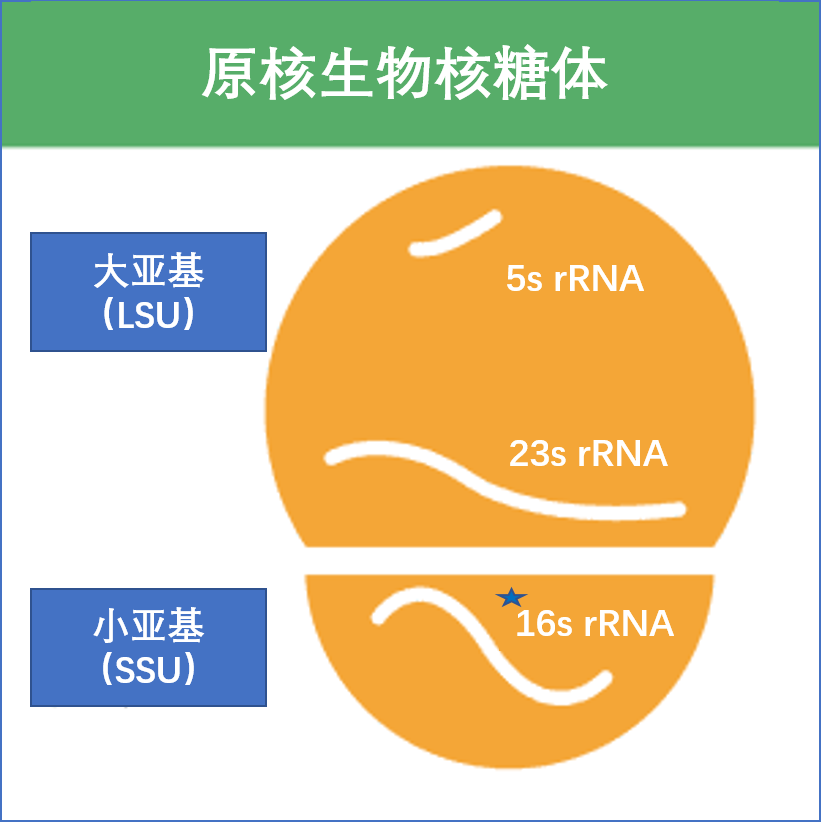
•16S rRNA在原核生物中发挥重要功能
16S rRNA是一种细菌和古细菌中常见的核糖体RNA分子。它在细胞中起着重要的功能,包括参与蛋白质合成的核糖体组装和识别启动子序列的功能。
由于16S rRNA在细菌和古细菌中具有高度保守的序列区域和变异的序列区域,因此它被广泛用于细菌分类和进化研究中。
相比之下,16S rDNA这个术语并不常用。rDNA是指编码核糖体RNA的DNA序列,因此16S rDNA可以理解为指代编码16S rRNA的DNA序列。
•科研中通常使用16S rRNA
在科研文献和学术讨论中,通常更常见地使用16S rRNA测序或16S ribosomal RNA测序。这种测序通常针对编码16S rRNA的DNA序列进行扩增测序,用来研究微生物的多样性和进化关系,特别是细菌和古细菌。
通过对16S rRNA基因进行测序,可以对微生物群落的组成和结构进行分析。这项技术在微生物学、生态学、医学和农业等领域有广泛的应用。
要用作DNA条形码,基因应具有以下特征:
•已知的细菌和古细菌都具有16S rRNA
它应该无处不在。否则,我们不能包括所有生物。已知细菌和古细菌的所有成员都具有16S基因。
它应该包含足够的系统发育信息。16S大约有1500bp长,不算太短也不算太长。
原核生物中发现的16S基因内的遗传变异足以用于广泛分类范围的系统发育分析。它成功地用于推断门之间的系统发育关系,同时也用于同一属的物种之间的比较。
•容易通过PCR扩增
它应该很容易通过PCR扩增。16S基因有多个保守区域可以作为引发位点。这成为基于NGS的短读长测序的一个显著优势。
经过多年的国内外研究和检测积累,我们拥有几乎所有已知细菌和古菌物种的16S序列数据库。通过在这些数据库中搜索16S序列,即使没有严格的分类学知识,也可以识别新分离的菌。
引物用于扩增
已知细菌16S之间的序列变异分布不均匀。在细菌中鉴定出9个高变区,分别命名为V1至V9。
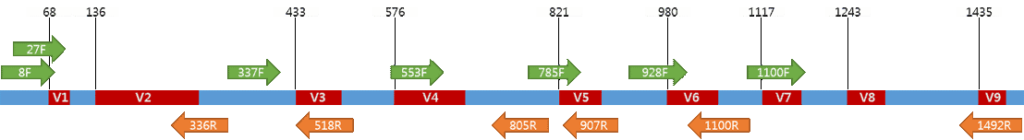
从长度来看,全长16S长度为1.5kb左右,单菌落的16S全长sanger一代测序仍然是菌种鉴定的主要手段,纳米孔和Pacbio的三代测序可以高通量的获得全长序列,对于希望更高分辨率的分析菌种的研究有一定优势。
•三代测序建库成本较高,测序深度较低
三代的测序准确度目前逐渐改进,直接测序准确度可以在90%以上,纠错后可以提高到97~99%以上,已足够提供高精度的分类。三代目前主要问题在于建库成本相对较高,通过使用barcode可以降低部分但仍然偏高,此外普遍测序深度相对于二代测序要低许多。
•二代测序由于读长限制需要引物进行扩增
由于二代测序读长限制,无法使用高通量测序技术对16S rRNA整个基因全长进行测序,因此必须针对基因的某一片段设计引物进行扩增测序。
注:虽然有大量的文献研究不同片段的优缺点,但由于采用的样本类型、区域引物以及分析角度的不同,尚没有针对所有样本的最佳可变区片段的共识。
基于大量项目经验和文献调研, 目前最主要的可变区选择是V4区和V3V4区,V4区长度为256bp左右,加上两侧引物长度为290bp左右,使用双端2x250bp或2x150bp可以测通,此外如454、life、Illumina Hiseq 4000, Illumina Novaseq 6000的测序平台读长也可以主要涵盖该区段读长。
•16S V4 引物通用性是所有可变区中最高的
传统的认知中,普遍认为测序片段越长,测到物种数据就越多,故倾向选择16S V3V4。然而16S V4(515-806)引物通用性相对是所有可变区中最高的。且在大规模菌群调查研究中,如人体菌群研究HMP,地球微生物计划EMP,欧洲的FGFP、美国肠道计划AGP以及全球土壤菌群调查,都采用V4区作为检测区域。16S V4目前仍然是国际研究中使用最广泛和认可的检测区域。
细菌的命名法和命名受《国际原核生物命名法典》 (简称《原核生物法典》)的约束 ,但实际物种的分类则不然。例如,大肠杆菌(Escherichia coli)这个名称受《原核生物守则》的规范,但物种本身的特性和分类学分类不受规范。这意味着没有官方分类法,只有名称受到控制。
因此,“有效物种”、“正式物种”或“官方物种”等术语没有意义。只能验证名称,因此可以使用 “具有有效发布名称的物种”。
物种名称应由二项式系统的两部分组成:
(1)通用名称和(2)特定词缀。
如下示例:
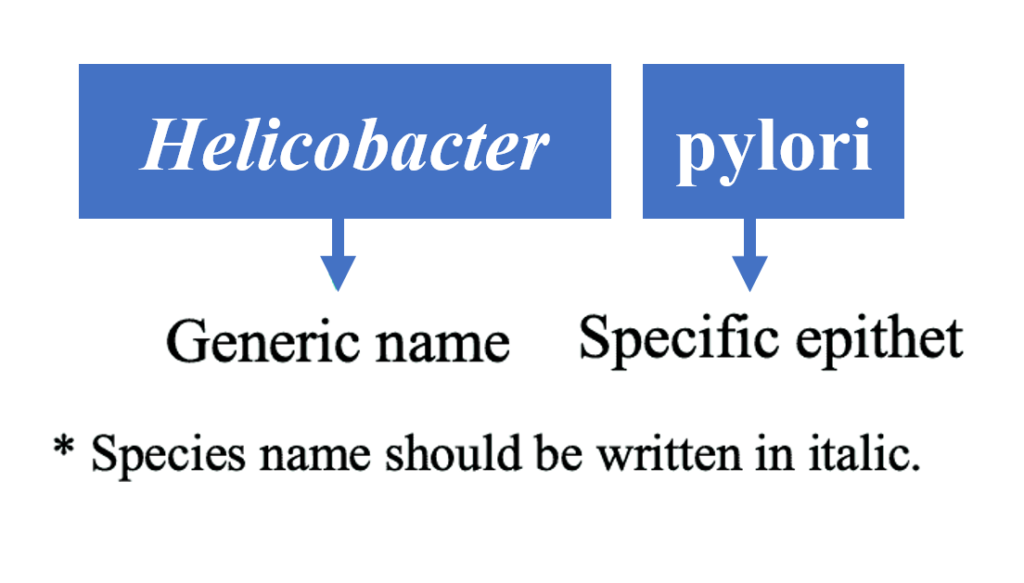
该组合代表“物种名称”,并且在命名系统中应该是唯一的。所有分类群的学名必须被视为拉丁文;物种等级以上的属群名称是单个词。
•生物分类按照不同等级
名称被组织成一个层次系统。
门(Phylum):门是细菌分类的最高级别之一,代表着细菌的大类别。细菌门包括了多个相关的纲和目。
纲(Class):纲是细菌分类的次级别,比门更具体。同一纲的细菌通常具有一些共同的特征和特性。
目(Order):目是细菌分类的更具体级别,比纲更详细。同一目的细菌通常具有更相似的形态、生理特征和生态习性。
科(Family):科是细菌分类的更具体级别,比目更详细。同一科的细菌通常具有更相似的基因组组成和代谢途径。
属(Genus):属是细菌分类的更具体级别,比科更详细。同一属的细菌通常具有相似的形态、生理特征和基因组组成。
种(Species):种是细菌分类的基本单位,代表着具有相似形态和生理特征的细菌群体。
亚种(Subspecies):亚种是种的更具体级别,用于进一步划分具有细微差异的细菌。
目前,门等级不受原核代码控制。提议将“门”纳入守则,但该提案尚未得到控制原核生物守则的机构国际原核生物系统学委员会(ICSP)的讨论和批准。
在细菌分类中,门、纲、目、科、属、种和亚种的命名规则和方法遵循国际细菌命名法。
以下是一些要注意的事项:
命名规则:细菌的命名应该是拉丁化的,以便在全球范围内使用和理解。通常使用斜体字体或以斜体书写。
门、纲、目、科、属、种和亚种的命名:这些分类级别的名称通常是基于拉丁词根、描述性词语、地名、科学家的名字或其他相关特征来命名的。例如,属名可以以科学家的姓氏命名,种名可以以形容词或名词来描述特定的特征。
•每个细菌分类级别的名称是唯一的
命名的唯一性:为了避免混淆,每个细菌分类级别的名称必须是唯一的。这意味着同一级别下的不同分类单元不能有相同的名称。
命名的正式发表:细菌分类的名称需要通过正式的科学出版物发表,以确保其被广泛认可和接受。这有助于维护分类的一致性和准确性。
命名的修订和更新:随着科学研究的进展和新的发现,细菌分类可能需要进行修订和更新。新的分类单元可能会被提出或已有的分类单元可能会被重新归类。这些修订和更新需要经过科学界的讨论和认可。
例如下面是假单胞菌菌名称和分类:
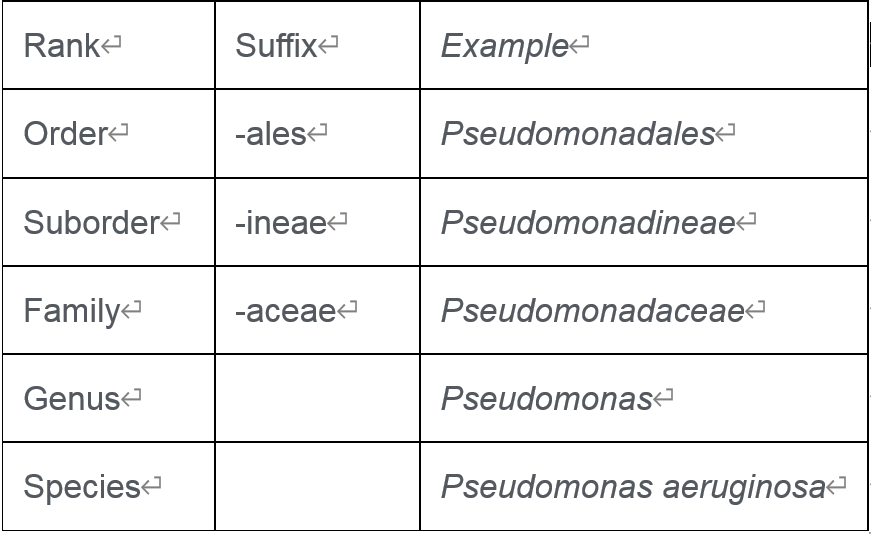
这些名称是由提出物种并将其分配给属的科学家给出的。例如, Thermacanus属已被提出,但并未将其归属于已知科。在NCBI分类学中,它属于芽孢杆菌目的暂定科“ Bacillales Family X. Incertae Sedis ” 。
EzBioCloud数据库将每个物种分配在完整的等级系统下,Thermicanus 被分配在暂定创建的家族“ Thermicanus_f ”下(请注意,这是一个无效名称,并且不是斜体)。当然,任何人都可以通过发表提案来提出Thermacanaceae家族的建议。
如前所述,名称由原核代码控制。要验证名称,该名称应包含在下述列表之一中。
▸ 批准的细菌名称列表
该论文由Skerman、McGowan 和 Sneath于1980年发表 ,标志着原核生物命名法的新开始。
此列表中的任何后续更改均由《国际系统与进化微生物学杂志》(IJSEM)(前身为《国际系统细菌学杂志》(IJSB))发布。
IJSEM中发布了两种类型的更新列表,即“通知列表”和“验证列表”。
▸ 通知列表
IJSEM是国际原核生物系统学委员会(ICSP)和国际微生物学会联盟(IUMS)细菌学和应用微生物学部的官方期刊。这意味着IJSEM是原核生物命名法的官方期刊。但是注意,没有官方的细菌分类法。
任何包含在IJSEM中发布的名称更改(新分类群或名称更改)的分类提案都将自动由IJSEM的列表编辑进行审核。如果论文符合《原核生物守则》的要求,则拟议的更改将被列入“通知列表”并被正式“验证”。
▸ 验证列表
原核代码还允许科学家有效验证在IJSEM以外的期刊上发表的名称。一旦名称被公布,就被称为“有效公布”。
任何关心科学的人都应该确保任何有效发布的名称都得到验证,方法是将出版物PDF发送给 IJSEM,并证明(对于物种和亚种)指定的类型菌株可以不受限制地从位于不同地区的至少两个公共培养物种保藏中心获得。
这可以由包括作者在内的任何人来完成。不这样做的带来的后果可能是有害的,因为该名称“有效发布”但从未“验证”。如果满足原核生物守则的所有要求(对于新物种和亚种,特别是规则27和30),IJSEM 的列表编辑将在验证列表中添加名称。
▸ 无效名称
不幸的是,在某些情况下名称无法验证。例如:
•一个名称在1980年之前被广泛使用,但在1980年没有被列入批准的细菌名称列表。
•一个名字在IJSEM之外发布,但从未被验证。
•没有人将PDF和进一步所需的文档发送给 IJSEM 进行验证。
•该出版物不符合准则的最低要求。
注:未有效公布的名称要用引号括起来,例如“Selenomonas Massiliensis”以区别于有效公布的名称。
•验证新物种的最低且唯一要求
根据原核生物守则, 验证新物种和亚种名称的最低且唯一的要求是:
•必须指定类型菌株。有时,描述新物种的论文不会提及哪个菌株是模式菌株。在这种情况下,名称无法验证。
•模式菌株应保藏于两个不同国家的两个培养物保藏中心。这确保了典型菌株仍然可用,例如当培养物保藏中心停止其活动或丢失菌株时。
•模式菌株必须通过培养物保藏中心可供任何人使用。专利菌株不能作为模式菌株。如果您想为某个菌株申请专利并限制其分布,则它不能作为物种或亚种的命名类型。
小结
以上总结起来如下:
确认命名规范:首先,确保您了解国际微生物学命名规范,如IJSEM是国际原核生物系统学委员会。这些规范包括命名原则、命名要求和规则,以确保新细菌名称的准确性和一致性。
检查已有命名:在确定您的新细菌名称之前,进行一次详细的文献调研,以确保该名称没有被其他研究人员使用或已经被正式命名。搜索相关数据库和文献,如国际细菌命名索引(International Journal of Systematic and Evolutionary Microbiology)和PubMed,以避免重复命名。
提交命名申请:一旦您确认您的新细菌名称符合命名规范并且尚未被使用,您可以准备提交命名申请。通常情况下,命名申请需要包括详细的描述、分类学特征、基因序列数据等相关信息。您可以向国际微生物学协会或相关的命名委员会(前面说的IJSEM)提交命名申请。
合作与审查:在提交命名申请之前,建议与其他研究人员、领域专家或命名委员会进行合作和讨论。这样确保命名申请符合规范。
审核和正式发布:一旦命名申请被提交,相关的命名委员会将对其进行审查。如果您的命名申请符合规范,经过审核后,新细菌名称将被正式发布,并被纳入相应的细菌命名索引和数据库中。
数据库很重要?
细菌名称的设计或命名是为了科学地组织,以提供基于系统发育的生物学见解;使用分层分类系统可以实现这一点。
•正确的名称对于解释微生物与其他数据的关联非常重要
例如,我们认为大肠杆菌与其他大肠杆菌物种比与芽孢杆菌物种有更多的表型特征。因此,使用正确的名称对于解释微生物组和其他类型的数据至关重要。
不幸的是,在当前的命名法中仍然有许多名称位于错误的位置。例如,Clostridium scindens因其在人类肠道菌群胆汁酸代谢中的重要作用而闻名,并且仍然保留着Clostridium的名称。然而,它甚至与梭状芽胞杆菌属并不接近 ,应归类为毛螺菌科的一个新属。
真正的梭菌属(Clostridium)属于梭菌科(Clostridiaceae)。例如Ruminococcus Sp(梭状芽胞杆菌)
梭状芽胞杆菌簇XIVa和IV 是已知在人类微生物组中发挥主要作用的细菌群。这些细菌簇在1994年才进入16S rRNA系统发育。
然而梭菌簇XIVa和IV 不代表正式命名法,也不表示单个分类群,例如属或科。自1994年以来,许多最初指定的属于梭状芽胞杆菌簇XIVa和IV的物种已被重新分类为新属。
•一些细菌存在错误的命名和分类
然而,这些簇中仍然存在错误分类的梭菌属物种,从而保留了旧名称。比如:Faecalibacterium prausnitzii(普拉梭菌),革兰氏阴性,对氧极度敏感,属于梭菌科,厚壁菌门。
该物种就是属于Clostridium clusterIV分组的Clostridium leptum group柔嫩梭菌类群,是该类群的最优势菌种。一般中文翻译柔嫩梭菌指的就是这个类群,其代表物种就是普氏栖粪杆菌,又名普拉梭菌。
以下是当前EzBioCloud数据库中对应于梭状芽胞杆菌簇XIVa和IV的分类群。
“Candidatus“是一个拉丁语词汇,通常用于微生物学中。它表示对一种微生物的命名,表示该微生物是一个新发现的候选物种,尚未被正式分类或命名。在中文中,”Candidatus“通常被翻译为”候选物种”或”候选菌种”。这个术语用于描述那些已经被发现但尚未被完全分类的微生物,通常是由于它们的特殊性质或难以培养。
总结来说,使用正确的细菌名称可以确保科学研究的准确性和一致性。细菌命名规范旨在为每个细菌物种提供唯一的、明确的名称,以避免混淆和误解。通过使用正确的名称,研究人员可以准确地标识和描述细菌物种,使得研究结果具有可比性,并促进科学交流和合作。
使用庞大的菌群的数据库,便于数据整合和共享。通过将新细菌与已知的细菌物种进行比对,可以确定其在分类学上的位置和相关信息。这有助于构建细菌分类树、建立物种间的关系,并为进一步的研究提供基础数据。
此外,正确的细菌命名和细菌样本数据库的比对可以促进研究进展和新的发现。通过对新细菌进行分类和命名,研究人员可以更好地理解细菌的多样性、生态学角色和潜在的应用价值。
人体肠道菌群健康检测需要依托本地化人群菌群样本检测的大数据库,区分菌群的组成和功能在个体间存在差异,并且与本地人体的生理状态、生化代谢、免疫炎症以及疾病风险等建立关联。通过建立大数据库,可以收集大量的菌群样本和相关数据,进行统计学分析和模式识别,从而揭示不同菌群组成与人体健康之间的关联。
总之,使用正确的细菌名称和拥有庞大的细菌样本数据库进行比对是确保科学研究准确性、促进数据整合和共享、推动研究进展和发现的关键步骤。
在某些情况下,即使一个物种是真实的并且具有科学重要性,也无法满足原核生物守则的基本要求。
根据正式命名法,这些物种的名字永远不会得到有效的公布。因此,分类学家创造了“Candidatus”一词来支持暂定命名。
对于Candidatus名称,没有纯培养物或分离株,因此没有典型菌株。由于Candidatus不属于正式命名法,因此不受原核法典管辖。
一个典型的例子是Candidatus Pelagibacter ubique该物种含有来自海洋的未培养或培养的细菌,可能是地球上最丰富的物种,因此它一定非常重要。
它从未得到验证的原因是该物种可以培养,但不能以培养物保藏所可用于长期储存的方式或规模进行培养。因此,无法满足该准则的最低要求。此外,特定的加词(ubique)格式错误。
并非所有Candidatus类群都得到了很好的表征,因为Candidatus的等级不受原核生物守则的规定。
•一般Candidatus的最低标准:
•提供全长高质量16S 序列,因为该基因是细菌和古细菌分类学的框架。
•未受污染且相当完整的基因组序列的可用性(通过域级核心基因的存在进行检查)。
•应提供最低限度的生态和代谢特征。
名称最好能代表该物种的典型特征。例如, Plasticicumulans这个名字的意思是“积累的塑料”。该属因积累生物塑料聚羟基脂肪酸酯而得名。
命名新分类单元的另一种流行方式是以人的名字命名。这个人应该有对科学做出贡献,尤其在微生物方面。著名的埃希氏菌(Escherich)和沙门氏菌(Salmon)是以微生物学家埃舍里奇(Theodor Escherich)和萨蒙(Salmon, D.E.)的名字命名的。
此外,以地理位置命名也很流行,但一般不鼓励。

谷禾健康

微生物群通常由数百个物种组成的群落,这些物种之间存在复杂的相互作用。绘制微生物群落中不同物种之间的相互关系,对于理解和控制其结构和功能非常重要。
微生物群高通量测序的激增导致创建了数千个包含微生物丰度信息的数据集。这些丰度可以转化为共现网络,让我们了解微生物组内的关联。
然而,处理这些数据集以获得共现信息依赖于几个复杂的步骤,每个步骤都涉及大量工具和相应参数的选择。
本文给大家介绍一个标准化流程——MiCoNE,该流程可以从微生物群落的16S序列数据中生成稳健且可重复的共现网络,并使用户能够交互式地探索在每个步骤中使用不同的替代工具和参数时网络会如何变化。
MiCoNE推理出的共现网络结果可以导出为json格式,也可以通过Python包导出为Cytoscape、GML或其他常见的格式。
MiCoNE模块化式的构建使它可以分步骤运行,用户可以随时停止,也可以随时从任一步骤开始。文中使用了真实的实验数据、模拟微生物群数据以及合成的微生物相互作用数据,对MiCoNE的性能表现进行了一些测试和评估。
下图为MiCoNE的工作流程介绍。
该流程主要由五个模块组成,分别为:
每个模块下的方框都告知了该模块下包含的进程,以及执行该进程可选的工具或方法。
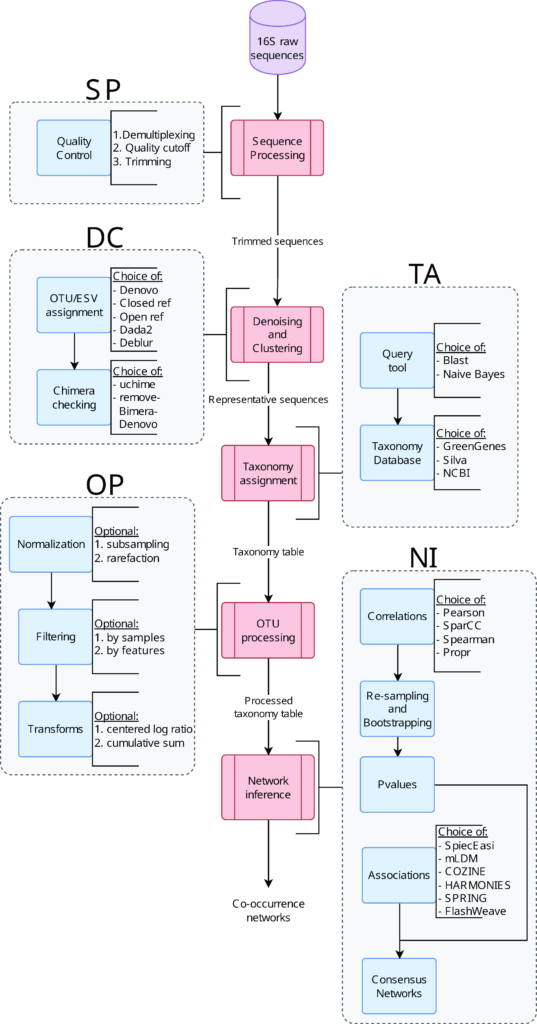
在SP模块中主要执行对序列的质检和修剪,在MiCoNE中提交单个或多个样本混合的序列都可。
DC模块主要执行去噪和聚类,然后检查并移除序列中存在的嵌合体,该模块的输出是一个计数矩阵,它描述了每个样本(矩阵的列)中存在的特定OTU或ESV(矩阵的行)的读取次数。
TA模块主要执行物种分类。可选的参考数据库有:
这些数据库是使用RESERT QIIME2插件下载和构建的。
在分配过程中,代表序列可能会被分配给一个“unknown”属,原因有两个:
一是数据库中与该序列相关联的分类标识符中没有包含给定的属信息
第二个更可能的原因是,数据库包含多个与查询(代表)序列非常相似的序列,而Consensus算法(来自QIIME2)无法以所需的置信度指定一个特定的属信息,也就是说如果数据库中没有该属信息,或者数据库中有多个与查询序列非常相似的序列,那么该代表性序列可能被标记为“unknown”属。
OP模块主要执行OTU或ESV计数矩阵的归一化、过滤和数据转换。默认情况下:
如果一个样本中的总读数少于500,那么过滤掉该样本;
如果特征的相对丰度小于1%,则过滤掉该特征;
如果特征在所有样本中的出现频率(含有该特征的样本百分比)小于5%,并且所有样本中该特征的计数总和小于100,则过滤掉该特征。
换句话说,如果一个样本的数据量太小或者一个特征在样本中的出现频率、丰度都很低,那么它们都将被过滤掉。
NI模块主要执行网络推理,该模块包括四种基于相关性的方法和六种基于直接关联的方法,可以自由组合,对于计算显著性水平P值,根据关联强度和p值过滤后,应用研究人员开发出的共识算法,最终生成共现网络。
▼
使用五种方法处理了FMT研究的16S数据,分别为:
前三种方法来自QIIME2的vsearch插件,OR和CR方法使用的参考数据库为Greengenes。
通过计算所有样本的平均UniFrac距离,并在不同方法中进行比较,发现除了Deblur之外,其它方法产生的代表性序列在按其丰度加权时彼此相似;差异主要在于分配较低丰度序列时。
哪个工具最能准确地概括样本中的参考序列?
使用相同的步骤处理了模拟数据集(mock4、mock12、mock16),并将预测的代表性序列与真实序列及其分布进行比较。
结果如图CD,预测的序列分布与预期的完全不同。数据集之间的差异表明数据集本身在方法性能中有着很大的影响力。
总体而言:
DADA2似乎是最可靠的,如图AB,它的加权UniFrac值在所有模拟数据集上都有更高的表现。
其次是Deblur,因为比较而言,OR和DN方法返回的OTU数量要多很多,如果不执行严格的过滤,将影响NI步骤的准确性。
但如果需要对不同16S区域进行测序的研究进行比较,CR和OR方法可能是更好的选择。
去噪后,要对序列进行嵌合体检查,在MiCoNE中应用了两种检查方法,uchime-Denovo和remove-bimera,经测试这两种方法之间没有显著差异。
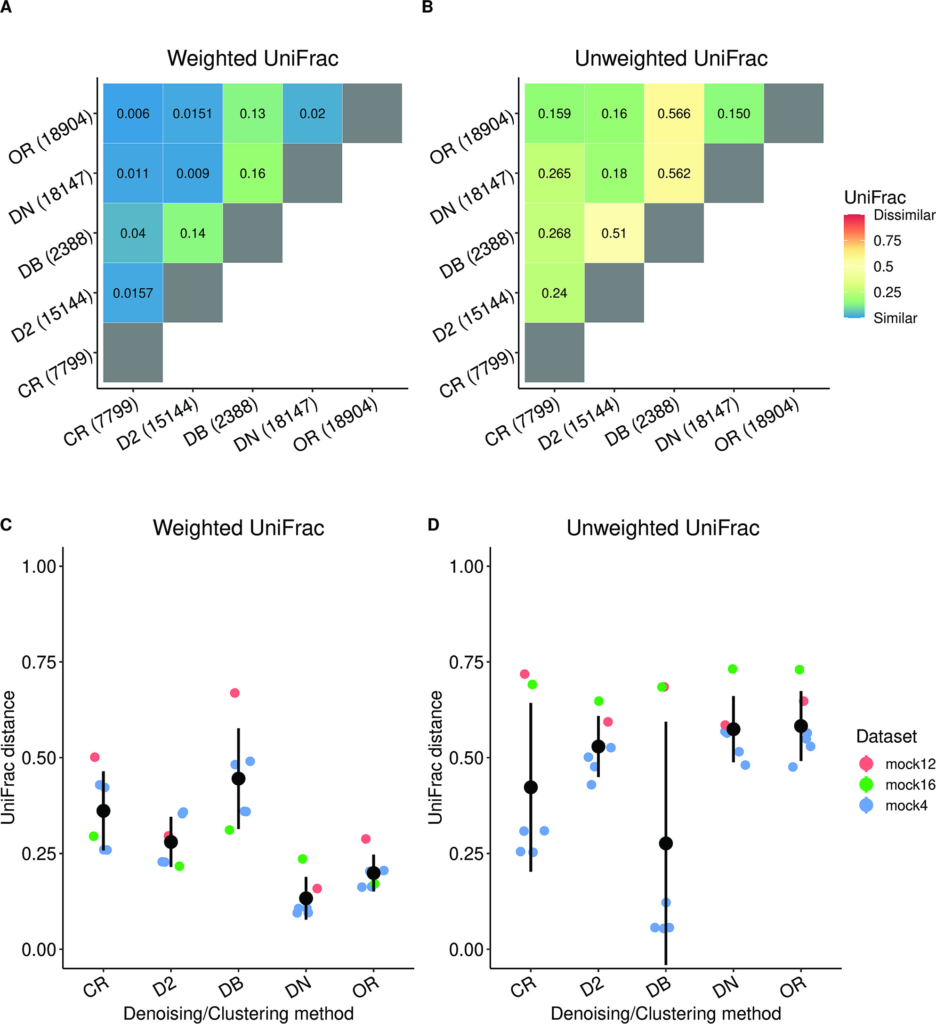
图AB用于分析的数据来自FMT研究,图CD中用于分析的数据来自模拟数据的mock 4、mock 12和mock 16数据集。基于以上结果,DC模块中的默认方法是DADA2+remove-bimera。
▼
MiCoNE使用的16S分类参考数据库分别是:
对于GG和Silva数据库,使用QIIME2的“naive Bayes”分类器,对于NCBI数据库,使用作为QIIME2插件的“BLAST”工具。
这些工具都经过了很好的量化和优化,因此都使用的默认参数。
结果如下图所示:
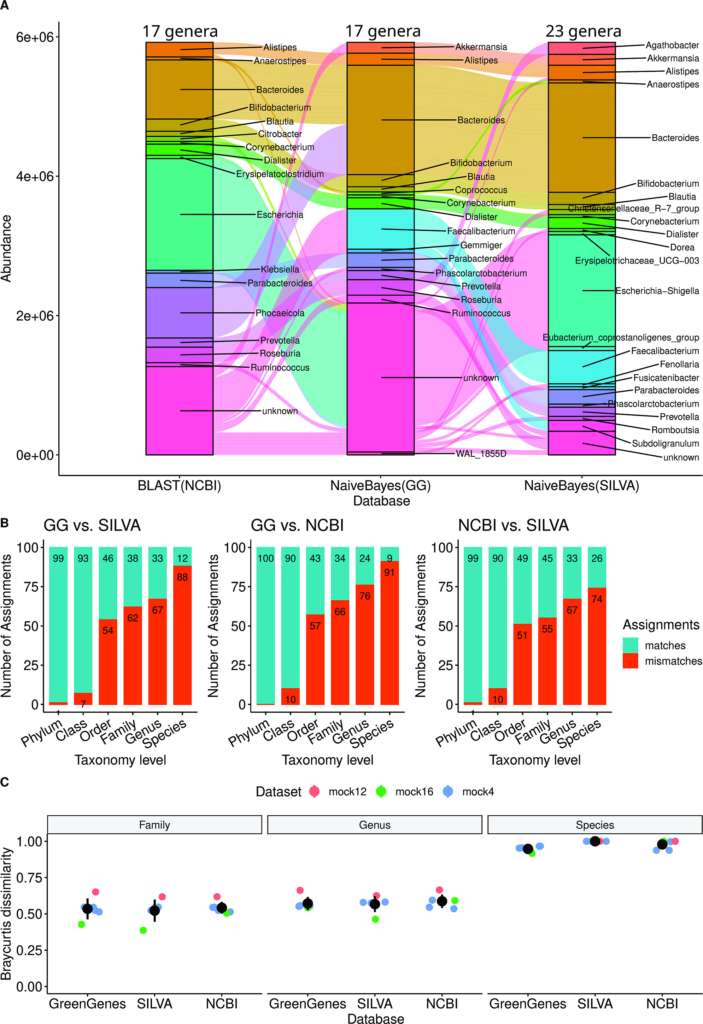
图A展示了三个不同的参考数据库对前50个具有代表性的序列进行的属水平的物种注释,说明了相同的序列是如何被分配到不同数据库中的不同属中的。在Greengenes和NCBI数据库中,代表序列的很大一部分被归入一个“unknown”。
图B比较了不同参考数据库之间分配给同一水平的代表性序列的数目(总和是前100个代表性序列),在较高的分类水平上,不匹配较少,但即使在目水平上,也存在超过51%的不匹配,这表明不同数据库的物种注释结果一致性较差。
图C比较了模拟数据中不同数据库预测的物种和已知物种之间的Bray-Curtis距离,差异结果表明对于每个数据集,不存在唯一的最佳数据库选择,因为所有的数据库都表现出相似的性能。但由于Greengenes数据库的主流性,它被选为MiCoNE的TA步骤的默认参考数据库。
▼
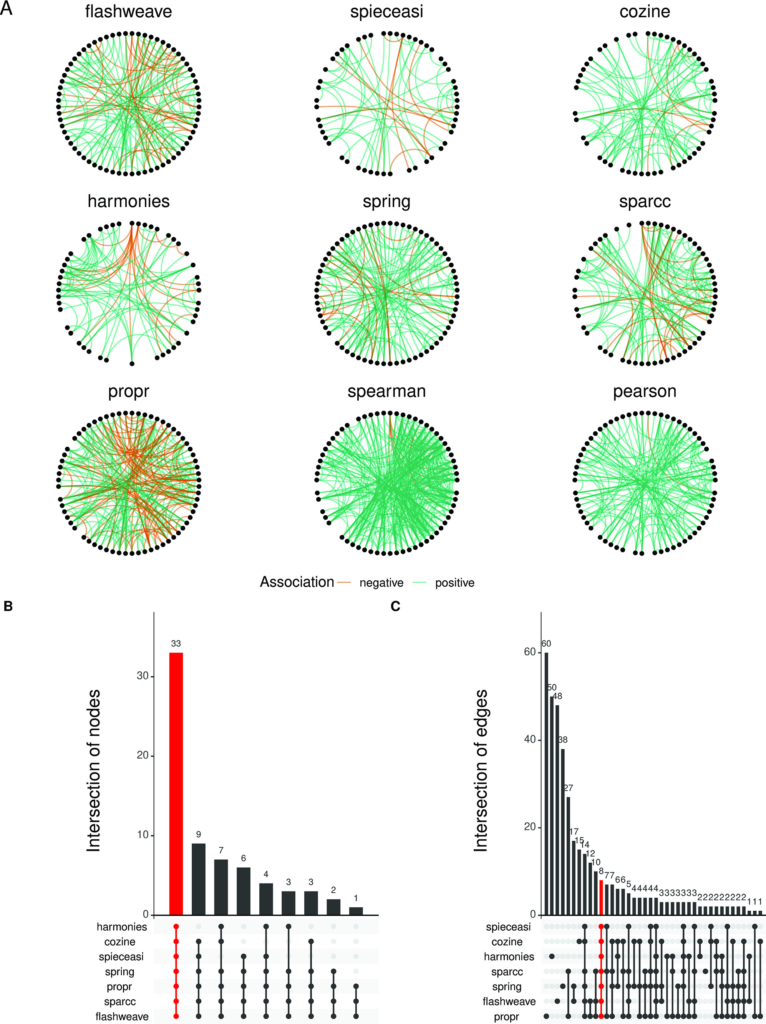
如图A, 对来自FMT研究的健康人群数据使用不同网络推断方法生成了九个网络。每个网络的节点(代表物种)在圆形布局中排列,可以直接可视化和比较它们之间的连接差异。绿色的连线表示正相关,橙色的连线表示负相关。
这些网络看起来不同,并且在连接性方面差异很大,值得注意的是基于相关性的方法通常会产生具有更高边缘密度的网络。
而在本文中基于相关性的方法有sparcc、propr、spearman和pearson,设置了0.3的阈值;基于直接关联的方法有flashweave、spieceasi、cozine、harmonies和spring,设置了0.01的阈值。
为了量化网络之间的差异,如图B,使用Upset图展示所有网络中有很大比例的共享或单一节点的分布(68个里有33个是共享的)。
图C边缘重叠Upset图显示,这些连接中的一小部分实际上是共享的(202条里有8条是共享的)。
▼
研究人员开发了两种方法来生成共识网络(consensus network),分别为:
它们将基于相关性和直接关联方法计算并过滤后的网络进行组合。基准数据集是计算机合成的相互作用数据,用于比较基于MiCoNE流程中的每种关联方法生成的预测关联的精确度和灵敏度。结果如下图:
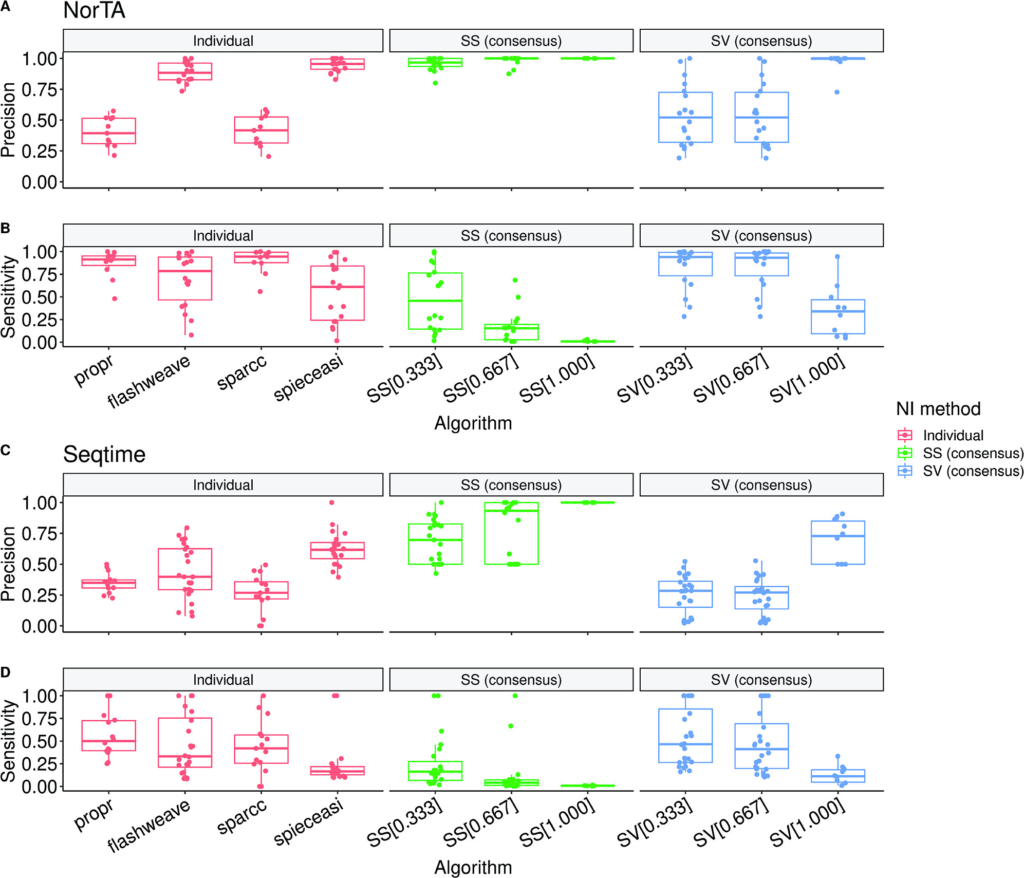
图AC为精确度的结果,图BD为灵敏度的结果。总体而言,θ=0.333的SS方法表现最佳,灵敏度和精确度处在良好的平衡上,因此在MiCoNE工具中默认使用SS方法。
▼
为了分析不同的处理方法对推理的共现网络的影响(在共识估计之前),研究人员使用MiCoNE中所有的方法组合生成网络,并量化每种选择导致的可变性,结果如下图:
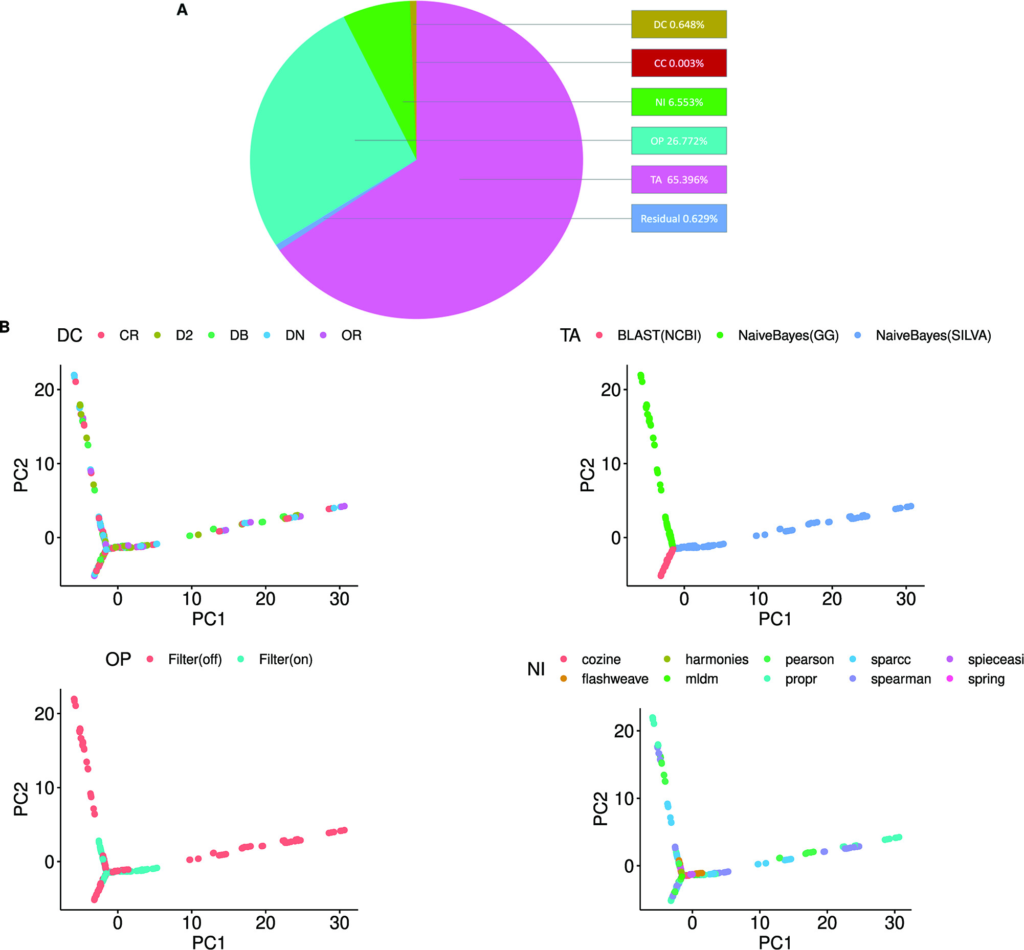
图A为在线性模型上使用方差分析(ANOVA)计算MiCoNE流程中的DC、CC、TA、OP和NI步骤所贡献的网络方差百分比(从FMT数据集生成)。
图B为PCA图,每个点表示使用MiCoNE流程中可用的工具和参数的不同组合推理的网络。点的颜色对应于流程中每一步骤(DC、TA、OP和NI)。
数据显示TA步骤对网络方差的影响最大,这意味着参考数据库的变化将导致截然不同的网络,其次是OP步骤的过滤水平和所使用的NI算法。
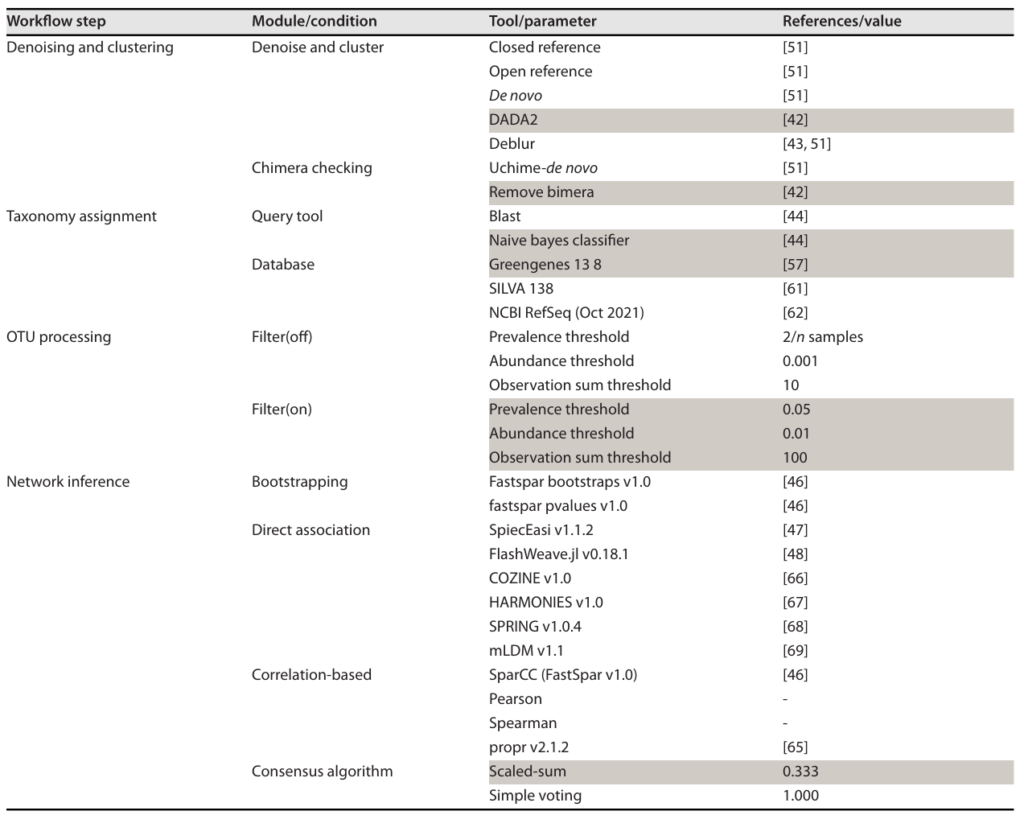
经过上面的测试和分析,研究人员发现工具和参数的选择对最终呈现的网络有很大的影响,因此提供了一组默认设置,如下表,灰色突出显示的工具是MiCoNE的默认工具,这些工具是基于模拟和合成数据集的基准测试推荐的。
使用上面的默认工具和参数从FMT数据集中分别对自闭症人群(ASD)和健康对照(Healthy)生成的网络进行比较,结果如下图,对照样本的网络中有22个独有的连接,自闭症样本的网络中有12个独有的连接,两个网络之间有7个共同的边。
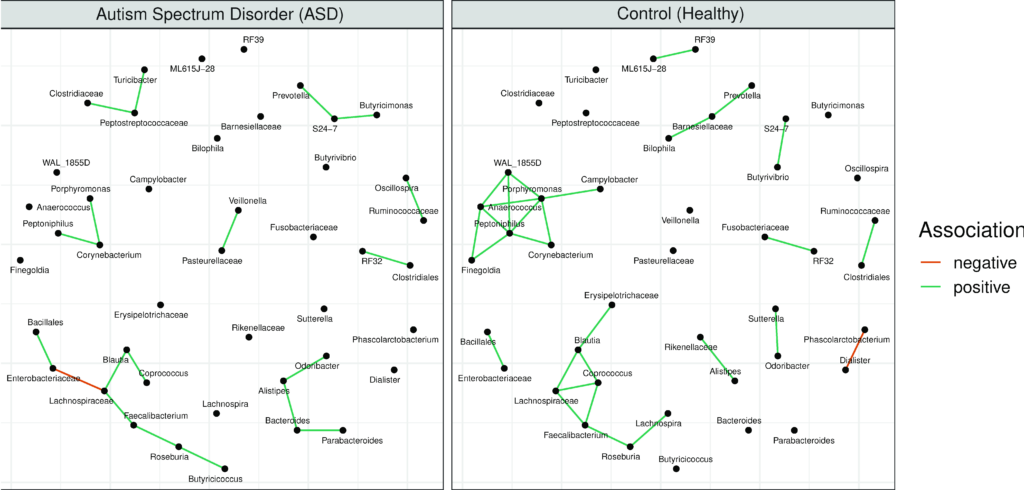
研究人员认为尽管这些独有的关联并不意味着实际的相互作用,但它们仍然可以作为文献调查和进一步探索菌群失调机制的起点。
MiCoNE工作流程提供了一个平台,可以轻松评估任何其他感兴趣的数据集在每个工作流程步骤的准确性、方差和其他属性。虽然MiCoNE内包含的工具方法较多,但研究人员基于测试结果也提供了一套默认参数,公开的测试结果也提高了可信度。
目前而言,MiCoNE的网络分析主要以属水平为基础,节点的最低分辨率是属水平,如果无法确定到属水平,就会使用上一层分类级别(例如,科水平)。不过,研究人员表示会持续更新和扩大MiCoNE的工作范围。
该项目的github地址:
GitHub – segrelab/MiCoNE: The Microbial Co-occurrence Network Explorer
参考文献:
Kishore D, Birzu G, Hu Z, DeLisi C, Korolev KS, Segrè D. Inferring microbial co-occurrence networks from amplicon data: a systematic evaluation. mSystems. 2023 Jun 20:e0096122. doi: 10.1128/msystems.00961-22. Epub ahead of print. PMID: 37338270.

谷禾健康

感染是人类面临的健康威胁之一。各种病原体,如细菌、病毒、真菌、寄生虫等,存在于我们日常接触的环境、物品、食物等中。一些常见的感染病例包括感冒、流感、腹泻、组织器官或血液感染等,在全球范围内广泛传播。这些疾病的传播方式多样,可能通过空气、水源、接触和食物等途径传播。
感染的症状包括发热、头痛、咳嗽、呕吐、腹泻等,严重感染可能导致器官衰竭、败血症等并发症。感染的原因包括接触感染源、免疫力下降、不良生活习惯等,易感人群有免疫力低下者、老年人、慢性疾病患者等。
研究发现,肠道菌群与感染也有密切关系。肠道菌群有助于消化、吸收营养物质,同时还调节人体免疫系统的功能。但如果肠道菌群失衡,会导致多种疾病,包括感染。
本文我们来了解一下关于感染的种类,症状,致病途径,易感因素,免疫系统应对病原体的三大防线,肠道菌群失衡与免疫系统的关联,以及肠道菌群检测在临床感染案例中的应用。
▼
感染可以按多种不同方式分类。
按影响部位分为局部和全身的:
按影响持续时间分为急性和慢性的:
按病原体类型划分:
病毒、细菌、真菌、寄生虫等都是导致感染的不同类型的病原体。
► 细菌感染
细菌感染是由体内过量的有害细菌引起的。正常情况下,人体的免疫系统能够通过白细胞和吞噬细胞等机制对抗细菌感染。但如果人体的免疫系统出现异常或受到外界因素干扰,细菌就可能趁虚而入,导致细菌感染的发生。
常见的细菌感染包括:肺炎、腹泻、皮肤感染、脑膜炎、膀胱炎、中耳炎等。
► 真菌感染
真菌感染多为继发性感染,由机会致病性真菌引起,特别是深部真菌感染多是由于各种诱因使机体免疫功能显著下降所致。
真菌是一类与细菌和病毒不同的微生物,它们包括霉菌、酵母菌等,常见的真菌感染有白色念珠菌感染、曲霉菌感染等。
► 病毒感染
病毒感染与细菌感染不同,因为病毒本身并不能独立存在,只有当它侵入人体细胞后才能产生病毒性病变。当病毒进入细胞时,它会留下迫使细胞复制的遗传物质。
当细胞死亡时,它会释放新的病毒感染其他细胞。并非所有的病毒都会破坏细胞——有些病毒会改变细胞的功能。
病毒会导致多种疾病,包括普通感冒、流感、登革热、水痘、艾滋病等。
► 寄生虫感染
寄生虫是需要以其他生物为食才能生存的生物。有些寄生虫不会对人产生明显影响,而有些寄生虫会生长、复制并侵入器官系统。
常见的寄生虫感染有阿米巴病、血吸虫病、肠道蠕虫感染等。
▼
无论何时发生感染,身体的一线反应都是炎症性的。
炎症是身体抵御疾病的方式,同时促进受影响组织的愈合。炎症的主要症状:红、肿、热、痛、组织功能的暂时丧失。
在感染期间,由于潜在的炎症反应,人们通常会出现非特异性的全身症状。症状和严重程度可能因受影响的器官系统而异,但可能包括:
▼
► 病史采集
询问患者或病人家属发病情况、既往病史、服药史、过敏史、流行病学史等。
► 体格检查
体温、呼吸、心率、血压、身体部位肿胀、红肿、局域性压痛、淋巴结肿大等都是感染体征,可以作为诊断和判断感染病情严重程度的参考指标。
► 实验室检查(抽血、尿检等)
通过血液、尿液、呼吸道分泌物、伤口分泌物等样本的化验检查可以更直接地确定病原体的种类和数量,以及感染程度的严重程度。
► 影像学检查(做CT等)
如X射线、CT、MRI等技术,可以帮助医生了解感染的程度以及可能受影响的部位,从而确定感染的类型和严重程度。影像学检查为临床医生在明确诊断之前的经验性治疗提供线索。
► 快速诊断测试(做核酸等)
如血清学检测、ELISA试剂盒、PCR技术等,可以在短时间内快速准确地检测出患者血清中的病原体抗体、抗原以及核酸等,提供对感染病原体的敏感性和特异性的快速诊断。
▼
呼 吸 道
普通感冒的另一个名称是上呼吸道感染。鼻病毒是引起感冒的最常见病毒。其他通过空气传播的传染病也以这种方式感染。
感冒和流感通常不会直接影响肺部,但它们可能会导致继发性细菌感染——肺炎。
消 化 道
如果不小心吃了被细菌或病毒感染的食物、饮料或水、或是农产品在种植或运输过程中接触了污染物,或者过期变质的食物等,病原体也一并被吞入,感染胃部或肠道,有时表现为腹泻和/或呕吐。一个常见的例子是细菌性肠胃炎,也称为食物中毒。
泌尿生殖系统
病原体也可以通过泌尿系统进入人体,如尿路感染,或生殖系统,如性传播疾病。传染源可能停留在局部,也可能进入血液。例如,性传播疾病最常感染生殖器,而艾滋病毒通过体液携带,也可以通过唾液、精液或血液传播。
皮 肤 接 触
皮肤的功能之一是充当抵御感染的屏障。但如果有割伤、抓伤、虫咬或任何类型的开放性伤口,皮肤阻挡不住,病原体可能会进入血液。如:蜂窝组织炎、脓疱疮等都是通过皮肤接触开始的常见感染。
▼
无论来源如何,任何人都可能感染。但有些人更容易感染,或感染并发症的风险增加,例如:
以上我们可以看到,那些免疫系统功能低下的人更容易发生感染。那么当病原体入侵时,免疫系统正常运作会发生什么呢?
下一章节我们来了解一下,默默帮助我们抵抗病原体的——免疫系统。
前面章节我们知道感染是微生物进入人体并造成伤害的结果。引起感染的生物体很多,包括病毒、细菌、真菌、寄生虫等。
当它们试图攻击的时候,免疫系统是如何运作的?
第一道防线——物理屏障
正常情况下,皮肤、呼吸道等组成的物理屏障在保护你,一般只有水分、气体等小分子物质进出,细菌、病毒难以入侵。
然而,也许你只是摔了一跤擦破了皮,也许只是水果刀不小心划伤了…皮肤作为免疫系统的第一道防线破了。
这时候细菌就开始争先恐后进入体内,不断繁殖,增强武力值。
第二道防线——先天性免疫防线
(广泛执行任务)
巨噬细胞——以吞制敌
巨噬细胞开始介入这场搏斗,它往往通过吞噬的方式以一敌百,不管三七二十一先吞下将其困在膜泡里,利用酶等物质分解杀敌,如果细菌还来不及生长繁殖就被吞完了,这场搏斗也就随之终止了。该过程可能引发局部的炎症,表现为红,肿,痛,热等。

但如果一直持续下去,巨噬细胞也可能累了,它开始发出紧急信号,召集后援团…
中性粒细胞——不分敌我直接干
中性粒细胞出现了,它离开了原本在血液中巡逻的路线,第一时间冲过来加入,武力值开始飙升,健康细胞也会被它误杀,它也会给细菌设置障碍,从而困住细菌将其干掉。
它们也会“适可而止、功成身退”,几天后自杀,不带来更多伤害。
如果这还不足以击退,发出的求救信号越来越多,当中枢神经检测到,下丘脑便会开始调节体温上升,也就是发烧。较高的体温可以抑制病原体的活性与繁殖。
以上就是人体免疫系统的第二道防线,也就是先天性免疫(非特异性免疫)。
如果第二道防线没有阻止入侵者呢?
最终对抗会发生在第三道防线,也就是后天性免疫。
第三道防线——后天性免疫防线
(形成记忆,长期保护)
后天性免疫系统是一个成长、学习和经验的累积,学习和记忆着病原体的模式,进而识别并产生特异性的免疫应答。具有极强的打击和防守能力,可以持续有效地保护我们的身体。

这时候,树突状细胞登场了。
树突状细胞——先天性免疫和后天性免疫之间的桥梁
树突状细胞开始手撕敌军,把病原体碎片暴露在自己的细胞膜上,带着病原体的信息去淋巴结,这里有很多T细胞待激活,当T细胞出生时会经过一个训练,之后会配备一个装备。
树突状细胞要寻找的正是可以和病原体表面特征蛋白结合的那个T细胞。
T细胞——等待激活,加入战斗
找到后该T细胞会被瞬间激活,然后开始分裂复制,并分化出三种T细胞,分别为:
B细胞——产生抗体,继续奋斗
当辅助T细胞过去找到B细胞识别结合之后,B细胞瞬间激活,大量复制,产生抗体。
抗体的出现意味着胜利的曙光,抗体表面有多个结合点,可以把多个病原体结合在一起,病原体捆在一起行动不便,失去活力。其他免疫细胞过来可以轻松把它们干掉。
抗体还可以拉着病原体连接杀伤细胞,将其消灭。
记忆T细胞和B细胞——学会对付,默默守护
在清除病原体后,分化出来的大量免疫细胞完成任务,会程序性自杀,防止继续占用资源。会有一小部分留下来,也就是记忆T细胞和记忆B细胞,等待病原体下次来的时候直接杀敌。
以上就是免疫系统的三大防线,它们的存在为人体提供了多重保护。正是因为它们的不懈努力,在感染发生后我们也会挺过来。
研究发现肠道菌群与人体免疫功能以及许多疾病的发生密切相关。
肠道菌群失衡如何与感染相关?肠道菌群与免疫系统之间存在怎样的关联?
下一章节我们继续来看。
肠道菌群中某些微生物和宿主的免疫系统之间存在相互作用,肠道菌群能够影响和调节上述第三大防线中B细胞和T细胞的活性,通过调节免疫细胞的行为,促进抗原递呈细胞的活化和作用,影响细胞因子及其受体的表达等。
肠道微生物代谢产物能够影响免疫平衡。例如,肠道菌群可以通过代谢产生短链脂肪酸、色氨酸胆汁酸等代谢产物,这些代谢产物能够通过激活特定的神经元和免疫细胞来调节免疫反应,从而维持肠道黏膜的免疫稳态。
短链脂肪酸可能具有抗炎和免疫调节作用,当然短链脂肪酸只是一部分,因为除了产生的酶和其他代谢物之外,细菌本身的成分,包括脂多糖、细胞囊碳水化合物和其他内毒素,也可能被释放并对宿主产生二次影响。
肠道微生物群衍生的代谢物丙酸盐(一种短链脂肪酸)水平降低可能导致 Th17/Treg 细胞分化的病理失衡。
一些菌群代谢物,如 4-甲基苯甲酸、3,4-二甲基苯甲酸、己酸和庚酸等,已被证明会加剧结肠上皮细胞的损伤,从而影响免疫。
在肠道菌群失调的情况下,某些有益菌可能会缺乏消化酶等必要的代谢产物和物质,使机体免疫系统无法及时有效地对病原体进行攻击。
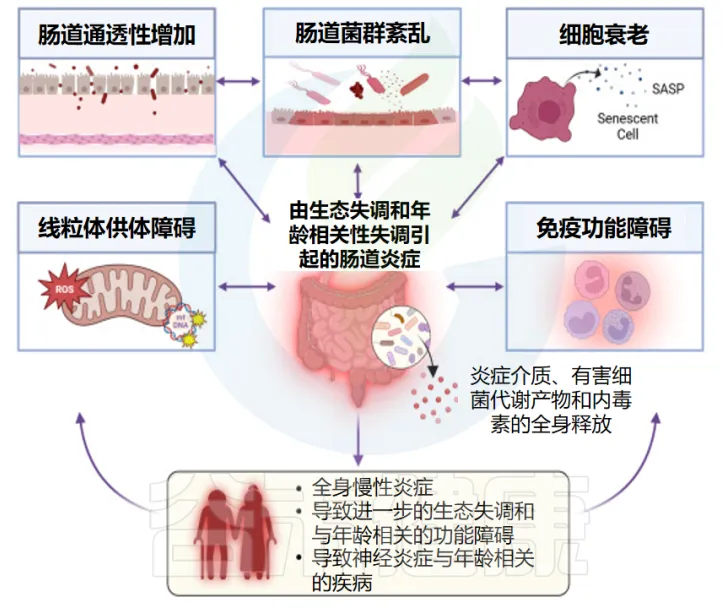
许多致病菌与肠道屏障的特定元素相互作用。一些有益的菌群可以激活上皮细胞,加强它们的黏附能力和抗病原性,从而提高肠黏膜的免疫防御能力。
致病菌和抗生素的使用可能会通过增强黏液降解,或抑制正常的共生黏液生成触发器来扰乱肠道黏液层,从而破坏肠道屏障,也就是我们常说的“肠漏”。
肠道屏障的破坏也会诱发体内炎症:与微生物相关的代谢物离开肠道并进入血液。作为回应,身体会产生细胞因子和其他介质,有效地启动炎症反应。
肠道上皮组织内的细胞将细菌代谢物传递给免疫细胞,从而在局部和全身范围内促进炎症。这种情况的持续存在可能导致亚急性或慢性炎症,随后可能会导致炎症性肠病、糖尿病或心血管疾病等疾病的发展。
doi.org/10.3390/biomedicines11020294
C 反应蛋白 (CRP) 是一种下游炎症标志物,可以通过特定肠道微生物的抗炎代谢产物的作用下调。当巨噬细胞和 T 细胞分泌白细胞介素 IL-6 时,该分子随后以高水平释放到循环中。我们在肠道菌群检测报告中也可以用CRP来辅助判断炎症状况。
炎症标志物是复杂的双向调节机制的组成部分,并且参与多个重叠过程,与感染和体内稳态的维持有关。
由此,肠道菌群变化对于免疫功能强弱具有重要指示作用,那么我们可以结合肠道菌群检测中的实际案例,来了解更多关于肠道菌群和感染的关联。
在谷禾肠道菌群健康检测中,有许多关于感染的病例,我们选取一例来详细看一下。
年初,一名7岁女孩入院,经诊断后发现患有:
菌血症是一种严重的感染症状,通常由细菌或真菌等微生物感染引起。菌血症分为原发性和继发性。
只有大约20%病例是原发性菌血症,也就是说病原体最初进入血液系统并引起血流感染,例如创伤、手术、注射等导致血管受损直接感染。
其余大多数都是继发性菌血症,也就是在其他部位先感染,未能及时处理,致使病原菌进入血液系统引起继发性菌血症。
常见的菌血症类型包括葡萄球菌菌血症,肺炎链球菌菌血症,鲍曼不动杆菌菌血症,克雷伯菌菌血症,真菌菌血症等,都是相应的致病菌感染引起的。
入ICU后,出现腹胀问题,且愈发严重 ⇊
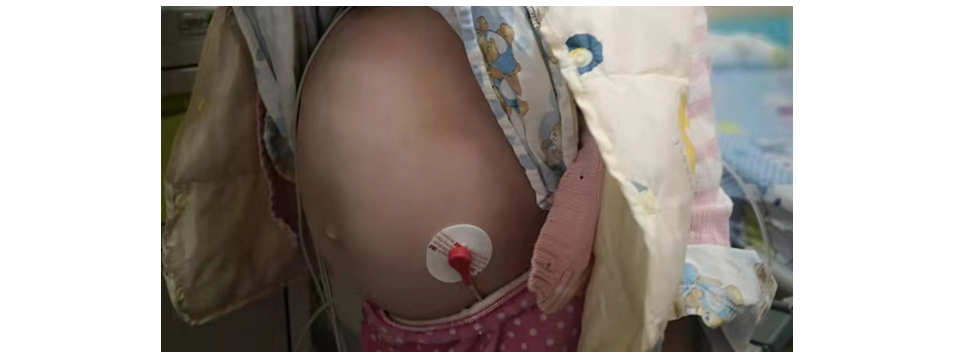
肠道菌群检测结果显示:
1. 患儿肠道菌群严重失调
<来源:谷禾健康肠道菌群检测数据库>
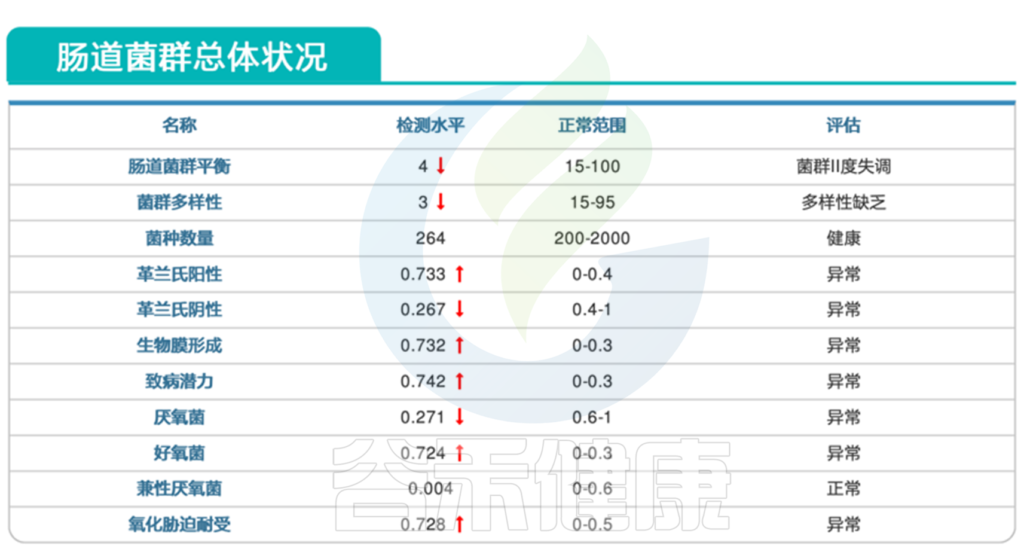
显然,该患者的肠道菌群失调,多项指标均显示异常。
从报告的肠道菌群总体状况这部分,我们可以看到,菌群多样性下降,其中革兰氏阳性菌明显偏高,革兰氏阳性菌包括葡萄球菌,链球菌,梭菌等,可能与多种感染相关。同时报告中致病潜力这项指标也是大幅上升。
2. 存在艰难梭菌携带超标、酵母菌感染、不动杆菌超标严重(感染)情况
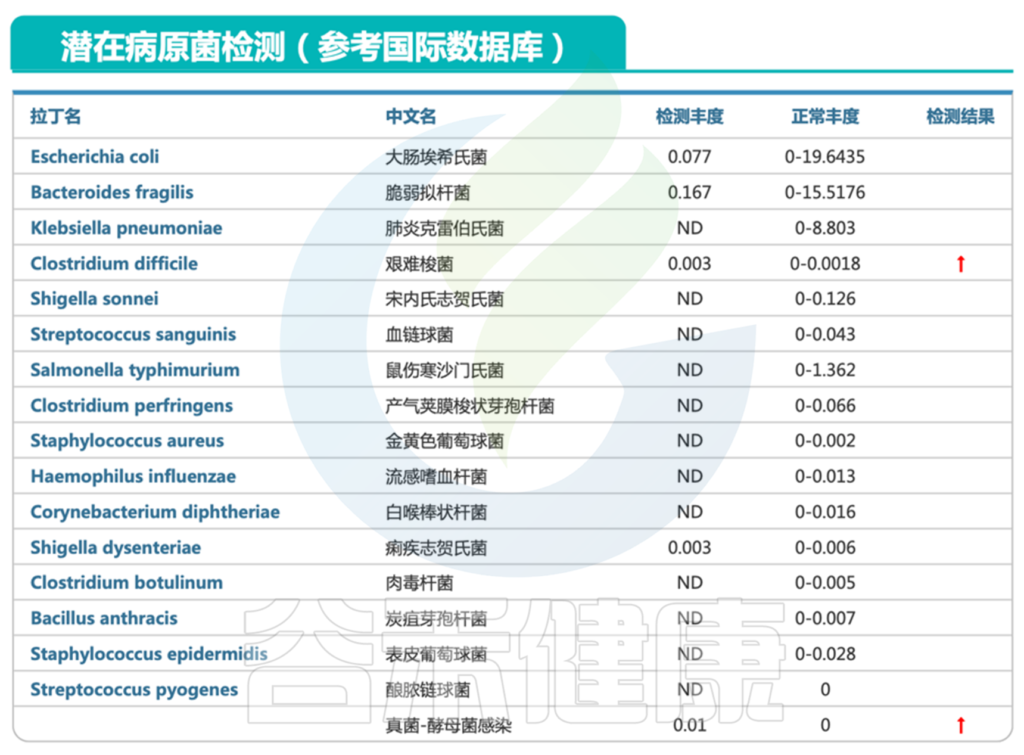
<来源:谷禾健康肠道菌群检测数据库>
艰难梭菌超标 ↑
艰难梭菌是一种常见的肠道病原菌。它可以引起广泛感染,症状最常见的是腹泻,伴随着腹部疼痛,刺痛,腹胀等不适,有些人还可能会出现发热,发冷,胃部压痛或疼痛,恶心呕吐的症状。
更严重的可能出现水样腹泻,心跳加快、体温升高、腹部绞痛、体重下降、腹部肿胀、肾功能衰竭等。
这与患儿的某些症状相符,从肠道菌群检测中可以发现艰难梭菌感染。
艰难梭菌感染与免疫功能有怎样的关联?
艰难梭菌毒素通过炎症小体和TLR4、TLR5和含有核苷酸结合寡聚结构域的蛋白1(NOD1)信号通路刺激先天免疫细胞,随后产生大量促炎细胞因子(如IL-12、IL-1β、IL-18、IFN-γ和TNFα)和趋化因子(MIP-1a、MIP-2、IL-8),导致粘膜通透性增加、肥大细胞脱颗粒、上皮细胞死亡和中性粒细胞浸润,引发免疫功能低下。
这与患者一开始诊断中中性粒细胞功能低下相符。
艰难梭菌感染通常与抗生素使用相关
通过某些抗生素,特别是克林霉素,可能会去除大量肠道中的有益细菌,使艰难梭菌得以繁殖,导致结肠炎和随后的腹泻。
真菌——酵母菌感染 ↑
报告中可以看到酵母菌超标,存在感染。这与入院时的粪便培养中的真菌感染相吻合。
真菌感染可能通过影响肠道微生物群和免疫反应,促进病原体的定殖和发展。
<来源:谷禾健康肠道菌群检测数据库>
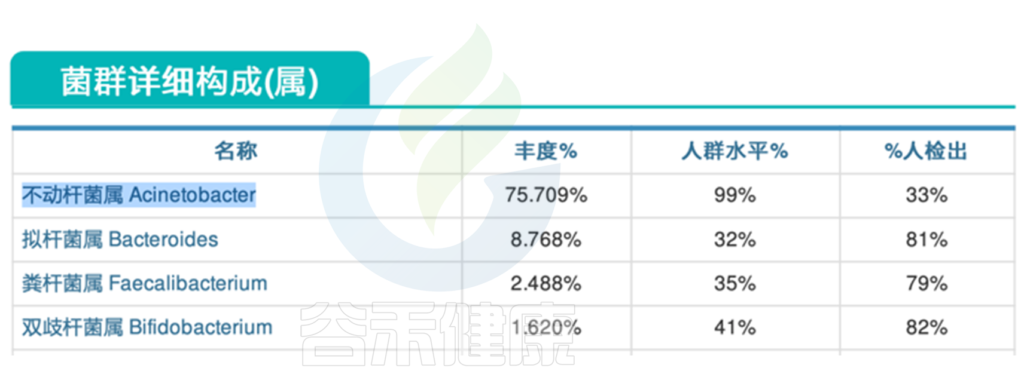
不动杆菌属(75.709%)占比过多 ↑
从报告中的菌群详细构成这部分可以看到,不动杆菌属的丰度竟然超过75%,严重超标。
这种情况下,不动杆菌属严重挤压了其他肠道核心菌属的生存空间,有益菌生长受限,菌群多样性下降,肠道菌群整体失调。
不动杆菌属Acinetobacter是一类革兰氏阴性菌,该菌属于常见的医院感染病原体之一。不动杆菌属感染常见的病症包括呼吸道感染、泌尿道感染、创伤感染等,严重情况下还可能导致败血症。
不动杆菌属对常见抗生素经常会产生抗药性。这里不动杆菌属丰度较高可能与抗生素使用较多相关。
结合该患者的情况,一开始入院时的组合抗生素一周未见好转,可能与不动杆菌属占比过多相关,对许多常见的抗生素已经产生耐药性,且免疫功能低下,因此难以好转。
针对上述情况,临床考虑口服抗生素,针对消化道微生物进行抗感染治疗。
初步选定:口服利福昔明 + 甲硝唑治疗。2天未见明显改善。
后改为:口服万古霉素 + 阿莫西林治疗。当天腹胀症状明显缓解,3天治疗腹胀明显改善。
同时辅助康复新液+B族维生素+谷氨酰胺治疗。
用药两周后,患儿整体情况平稳,仍存在轻微腹胀⇊

肠道菌群复查提示菌群结构基本恢复正常。
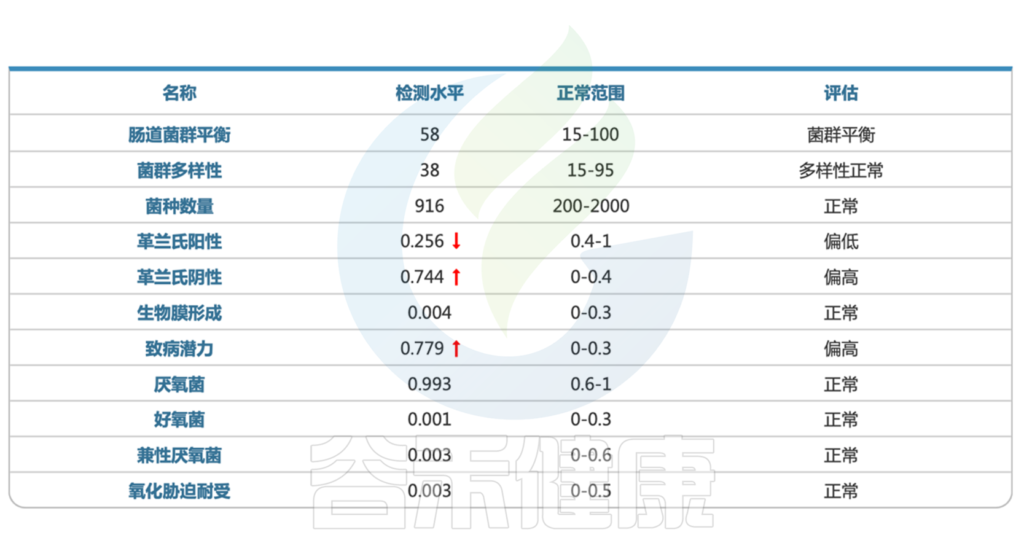
<来源:谷禾健康肠道菌群检测数据库>
一年后随访,该儿童身体已经好转,生活正常。
备注:临床中还有其它治疗药物等没有全部分享,主要分享了与菌群相关的治疗,仅做交流,不能作为治疗参考。
宿主感染与肠道菌群之间存在复杂的相互作用,免疫系统有两个分支,即先天性和适应性,它们协同工作以保护身体免受外部和内部威胁。这两个免疫系统分支都受到严格调节,肠道微生物群是其中的一个重要因素。
要揭开这其中的机制关联可能需要依靠营养学、流行病学、医学、免疫学和微生物学等多学科研究人员的共同努力。
不过从以上案例我们可以看到,肠道菌群健康检测在感染治疗中发挥重要价值。
辅助判别
在传统诊断中得知患者存在菌血症,真菌感染,但通过肠道菌群健康检测可以了解更多具体的感染信息,比如说艰难梭菌感染可能引起的腹胀等问题,不动杆菌属占比过大形成的抗生素耐药性。
除了致病菌超标等直观的信息之外,我们还可以判断整个肠道微生态的健康状况。肠道微生物群的失调可能导致肠道易感性增加,使得易感性疾病如艰难梭菌感染更容易发生。
辅助用药
针对这些具体信息可以使用药更加精确,而不是直接使用常见的组合抗生素。
用对抗生素对于恢复速度有很大提升,同时也避免了患者因使用不必要的抗生素产生耐药性。

谷禾健康

微生物组的纵向研究是一种长期跟踪微生物组变化的研究方法。在这类研究中,样本从同一人群或个人中多次采集,通过检测样本中微生物群落丰度的变化(如不同菌群的比例和种类),来了解微生物组随时间的变化趋势和特征。
根据微生物组不同菌群的变化,可以发现一些与健康或疾病相关的特征,从而为疾病预防和治疗提供参考。
但是,由于不同研究对象在纵向时间轴上的时间点分布不均匀且数量不同,难以进行有效的综合分析,一些时间点上的样本缺失导致大量数据无法利用。
为此,研究人员提出了一种名为DeepMicroGen的双向循环神经网络-生成对抗网络(GAN)模型,该模型根据观测值之间的时间关系进行训练,从而在时间序列数据中填补缺失值。
本文介绍了DeepMicroGen模型的算法、并在性能评估上使用模拟和真实数据集与几种标准基线方法进行了比较,结果都表示DeepMicroGen模型无疑是优秀的。

如图中所示,DeepMicroGen模型主要分为两部分,生成器(Generator)和判别器(Discriminator)。
•生成器
生成器的目标是产生更真实的伪造数据,用于填补缺失值。
•辨别器
判别器的目标是辨别真实的数据和伪造的数据,促使生成器不断地生成更加真实的虚假数据。
两者通过反复训练来相互对抗和优化,最终达到生成器生成的数据足以欺骗判别器的效果,从而提高模型的生成和预测能力。
在进入模型训练前,要对输入的微生物组数据进行预处理,使用clr方法进行了归一化,同时,为了避免某些物种在所有样本中完全缺失的情况,即数据中不会出现零值,计算了伪计数(最小相对丰度除以2)。
▷DeepMicroGen模型中的生成器(Generator)训练
先判断每个时间点上是否存在该物种,0表示数据缺失,1表示数据存在。

然后计算出时间差δf,一个向量,用于表示前一个观测值与当前时间点之间的时间差。δb,与δf定义类似,但计算方向相反,这反映了下一个观测值的时间与当前时间点之间的差异。
这些变量的计算有助于更准确地捕捉时间序列数据中的时间相关性。
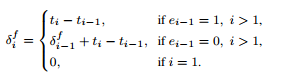
接着将x(观测值)、e、δf和δb变量值输入至DeepMicroGen模型的生成器中,使用CNN模块提取捕捉系统发育关系的特征。
计算门水平的OTUs之间的Spearman相关性,得到相关性系数矩阵(一个p×p矩阵,其中p个OTUs在一个聚类中),利用下面的公式将矩阵中的每一行简化为相关系数的几何平均值,其中ρOTUjp是OTUj和OTUp之间的Spearman相关性。
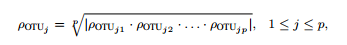
然后OTUs按照几何平均值从大到小进行排序,进一步强化门水平OTU之间的关联。之后,将CNN模块分别应用于每个聚类并提取特征,该模块由两个1D-CNN层组成,卷积核大小为3,分别具有16和8个滤波器,每个滤波器后接一个Leaky ReLU激活函数和一个max-pooling层。
从每个聚类中提取的特征被连接并传递到biRNN模块。通过这种方式,DeepMicroGen模型可以同时利用门聚类和时间序列数据的相关性,提高对微生物组数据中缺失值的填充精度。
为了训练模型,需要捕捉时间序列之间的关系,以便填充缺失值。因此,在模型中增加了一层具有tanh激活函数的单层biRNN模块,用于捕捉正向和反向的时间关系。
RNN(循环神经网络)单元的定义如下:其中,Wh是权重矩阵,bh是bias,hi−1是前一个时间点的隐藏状态(hidden state)
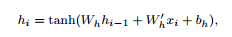
在biRNN 模块中的生成器应用中,前向(forward)和反向(backward)RNN单元都会产生一个生成的输出,分别用 x˜f和x˜b指示。
然后,每个生成的输出将与其对应的权重系数λf和λb相乘,λf和λb是根据时间差分计算出来的,bias向量bλf和bλb是模型通过训练学习得到的参数,最终的插补值 ˜x是由输入数据和生成数据按一定比例加权组合而成,如果输入数据中存在真实值,那么生成值就会被真实值替代。
其中,1是一个全1矩阵,xe是一个矩阵,其中第i列是x乘以一个单位向量ei(即只有第i个位置是x,其他位置均为0)。

▷ DeepMicroGen模型中的判别器与生成器之间的对抗训练
判别器由一个带有LSTM单元的一层RNN模块构成,判别器的训练过程旨在最小化两个不同的损失函数lossD和lossT,以此优化判别器的性能,从而提供准确的反馈信息给生成器,改进生成器的生成结果。
其中,lossD用于评估判别器的鉴别能力,即判断一个样本是真实值还是生成值的能力;lossT用于评估判别器对时间点的预测准确性。

在生成器的训练过程中,会分别计算lossG、lossR和lossC。使得生成器产生更准确、一致且真实的填充值。
lossG表示生成器的损失函数(Generator Loss),表示生成器在生成伪造数据过程中的误差;lossR表示真实数据损失函数(Real Data Loss),表示判别器对真实数据分类的误差;lossC表示伪造数据损失函数(Fake Data Loss),表示判别器对伪造数据分类的误差。
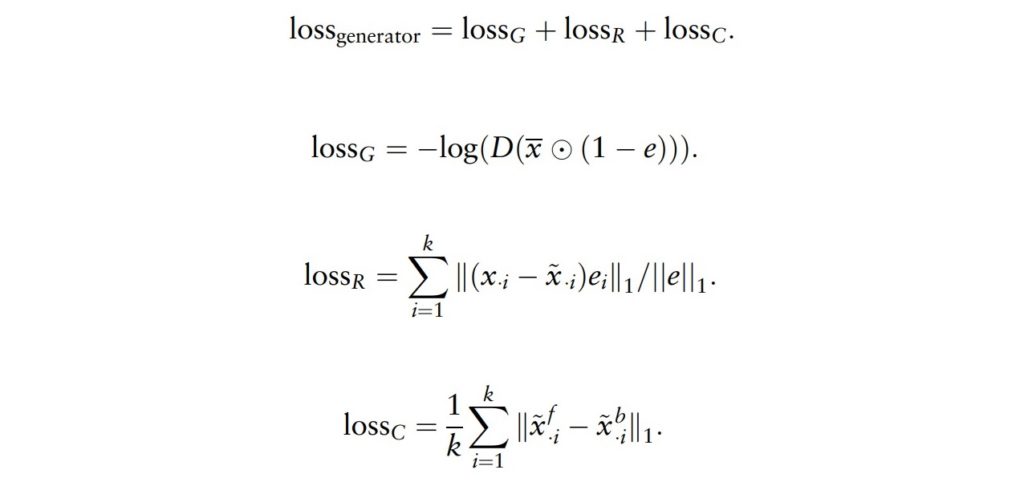
最终,通过相互对抗和共同优化,判别器和生成器可以同时得到优化,并且生成器可以生成具有真实样本类似但又与数据集不完全相同的新数据。
DeepMicroGen是基于Tensorflow库(v1.8.0)构建的。在训练中使用了Adam优化算法,Adam的学习率被设置为0.001。
每一轮训练,生成器会在判别器经过5次迭代之后进行一次训练。当生成器的损失函数连续1000轮训练没有降低时,训练就会被结束。同时使用了Dropout(CNN层中的0.7比率)来缓解过拟合的问题。
下图为训练过程的损失函数曲线:

实验设计
使用的测试数据集包括1个模拟数据集和2个真实数据集(DIABIMMUNE和BONUS-CF数据集)。
DIABIMMUNE数据集内含从三个国家(芬兰、爱沙尼亚和俄罗斯)招募的婴儿粪便样本,这些国家在1型糖尿病和过敏的发病率方面存在很大差异。
根据16s rRNA扩增子测序,选择了133名受试者的1064份粪便样本(时间点为第4、7、10、13、16、19、22和28个月),其中包含115个种水平的OTUs。BONUS-CF数据集内含231个婴儿的粪便样本,这些婴儿中,有一部分存在囊性纤维化(CF)或高风险发展成囊性纤维化的情况。
研究人员选择了113名受试者的452份在5个月、6个月、8个月和10个月时的粪便样本,使用WGS测序。模拟数据是基于DIABIMMUNE数据集模拟了200名受试者。
使用了三种不同类型的缺失值填补方法进行性能评估。采用简单的方法(平均值和中位数)进行插补,然后比较了线性曲线拟合、三次曲线拟合和基于移动窗口(window-size = 3)的缺失值填补方法。
还使用了广泛用于纵向数据集的缺失值填补方法:基于链式方程的多重插补(MICE)和最后一次观测值向前插补(LOCF)。使用平均绝对误差(MAE)来衡量不同缺失值填补方法的性能。
★ 实验结果表明DeepMicroGen模型优于其他方法
DeepMicroGen模型在模拟数据集上的MAE为1.866,优于其他方法,其中MICE的表现次之,为1.942。其他方法的MAE分别为:三次曲线拟合(1.943)、线性曲线拟合(1.990)、Median(2.277)、Mean(2.298)、Moving window(2.452)、LOCF(2.698)。DeepMicroGen在真实数据集上也取得了最好的成绩,MAE分别为1.609和0.486。
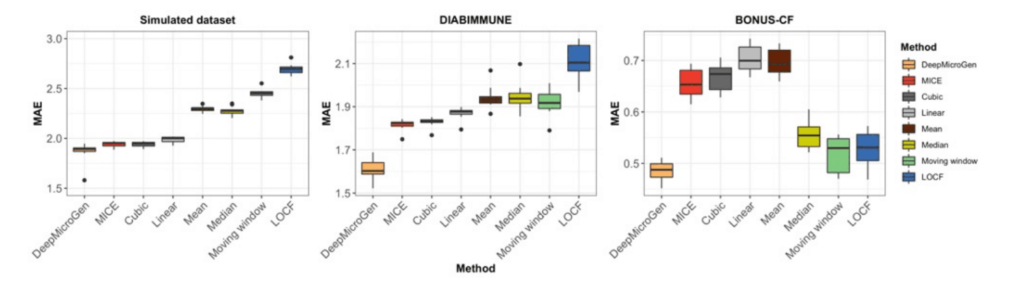
DeepMicroGen模型利用GAN与CNN结合提取OTU特征来推算微生物组数据。研究人员为了观察不同组件对imputation性能的影响,通过移除或改变模型中的组件设计了四种变体DeepMicroGen模型。然后分别使用DIABIMMUNE数据集进行10倍交叉验证,并计算MAE进行性能评估。
如下图所示,与所有四种变体模型相比,DeepMicroGen表现出最佳性能。

评估DeepMicroGen模型在不同数据缺失率下的表现。随机(MAR)或非随机(MNAR)删除10%到80%的数据,使用DeepMicroGen模型进行缺失值填补,实验重复5次,计算MAE。结果表示除MNAR缺失率为40%外,DeepMicroGen模型在大多数情况下的MAE仍然是最低的。
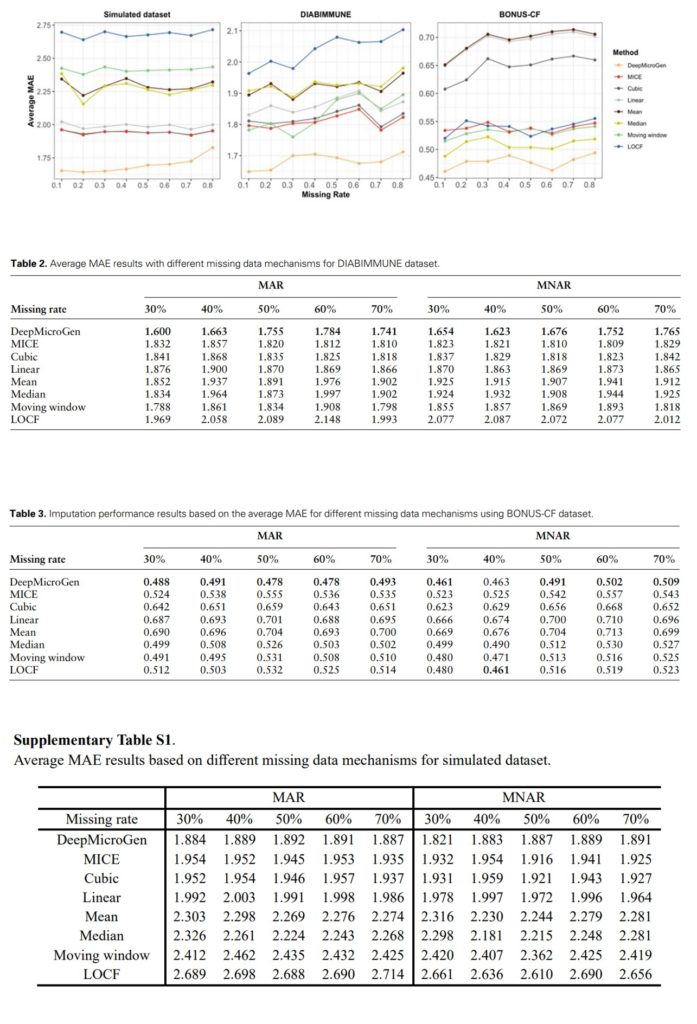
评估DeepMicroGen模型生成的插补值是否能够与原始数据保持相似性。研究人员随机丢弃10%-80%的数据,并使用DeepMicroGen和其他方法进行填补。
对于每种方法,分别比较了Shannon指数结果,NMDS分析结果。还计算量真实值和插补值分别与这两者间的pearson相关系数。
结果表示mean、median、MICE、linear和cubic方法的插补值与真实值的alpha多样性存在显著差异(P<0.05)。
MW和LOCF方法进行插补得到的数据,其alpha多样性与真实数据较为接近,但它们与真实数据的相关性较低。
★ DeepMicroGen模型的到的插补值与真实数据相似,相关性最高
而通过DeepMicroGen模型得到的插补值,其alpha多样性与真实数据相似,并且相关性最高。在比较beta多样性时,还可以看到DeepMicroGen模型生成的插补值与真实数据之间有重叠,而其他方法却几乎没有重叠。
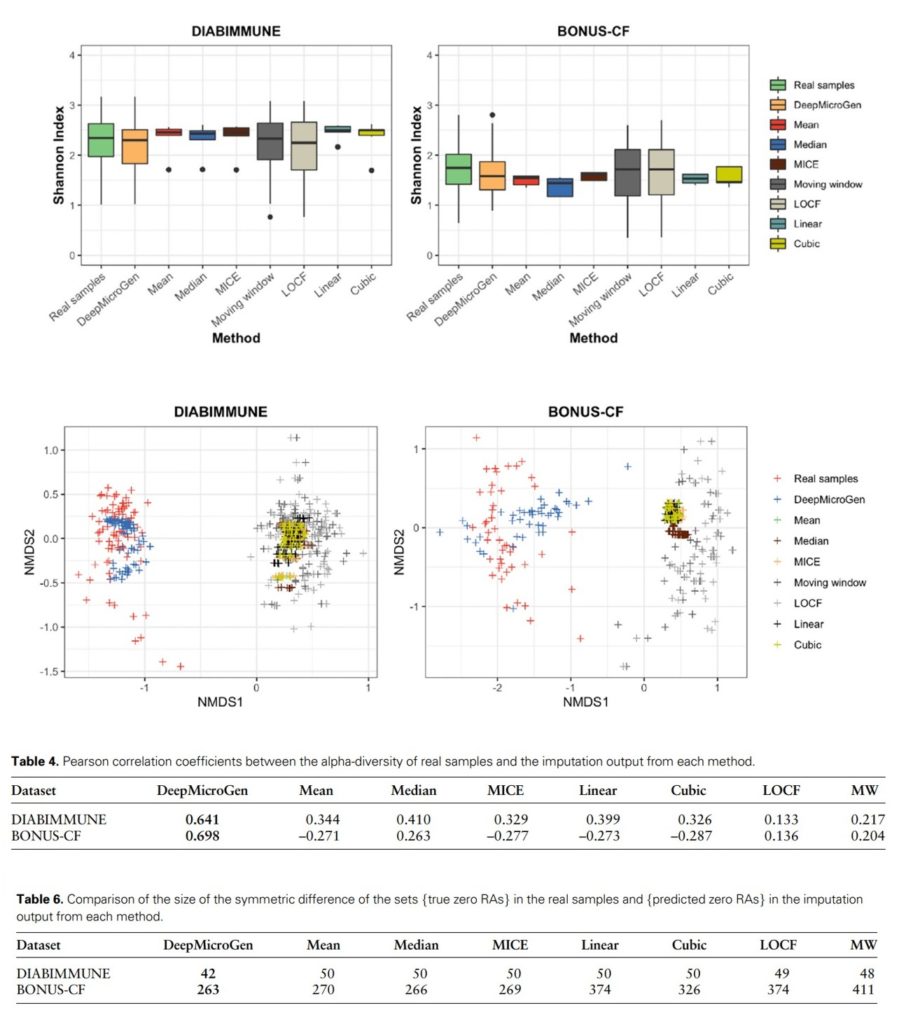
此外,研究人员还测试了是否能够识别真实数据中相对丰度值(RA)为0的,并准确预测。准备了两个数据集合,分别是{true zero RAs}和{predicting zero RAs},分别表示真实数据集中RA=0的OTU集合和生成插补值数据集中RA=0的OTU集合。依次比较这两个集合之间的对称差,预测准确率和MAE值。
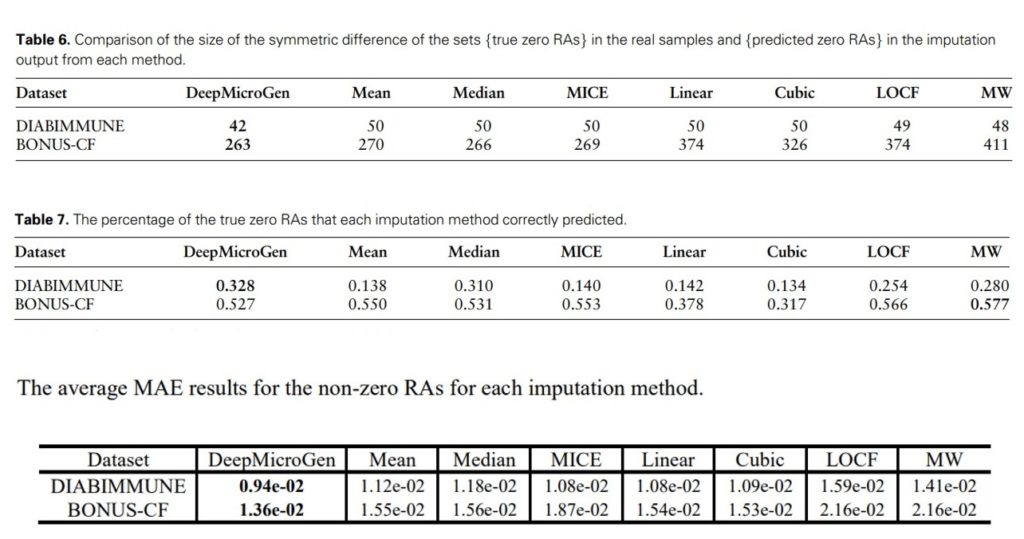
★ 能够有效保留原始数据特征,利于下游分析
这些结果都表明DeepMicroGen模型生成的插补值能够有效地保留原始数据的特征,更有助于下游分析。
评估DeepMicroGen模型是否可以提高疾病预测能力。在DIABIMMUNE数据集中,141名婴儿有鸡蛋、牛奶和花生过敏结果的临床信息,其中117名婴儿有8个时间点的所有样本,而其他25名受试者没有样本。
研究人员利用LSTM神经网络分类器对蛋、牛奶和花生过敏等疾病进行了预测,使用clr转换后的相对丰度数据作为DeepMicroGen模型数据输入,比较经重复5次5-fold交叉验证计算 的平均AUC值,与其他插补值生成方法相比,DeepMicroGen模型在所有过敏疾病的预测表现中实现了最高的改善效果,其中花生过敏的预测表现提高了19.5%。
编辑
由于DeepMicroGen模型较好的结果表现是假定样本是在相同的时间间隔下生成的,如果样本的时间间隔不规律,那么DeepMicroGen模型的表现可能不太好。
因此,最好有一定数量的样本在每个时间点上。当然,对于这类数据,DeepMicroGen模型也能训练并生成插补值,只是精确度可能会降低。
DeepMicroGen 模型为微生物组数据缺失值填补提供了一种有效的方法,并且在实验中表现出了良好的性能,在疾病预测方面的潜力也不逊色。
据文中介绍,基于深度学习的纵向数据填补方法已经被用于特定的研究领域,如用于阿尔茨海默病进展的MRI特征插补,患者管理的电子健康记录插补,以及肺癌风险评估的CT图像插补。
这为利用不完整的纵向数据集进行微生物组研究的科学家们提供了支持。
参考文献
Choi JM, Ji M, Watson LT, Zhang L. DeepMicroGen: a generative adversarial network based method for longitudinal microbiome data imputation. Bioinformatics. 2023 Apr 26:btad286.

谷禾健康
微生物组经由二代测序分析得到庞大数据结果,其中包括OTU/ASV表,物种丰度表,alpha多样性、beta多样性指数,代谢功能预测丰度表等,这些数据构成了微生物组的变量,大量数据构成了高纬度数据信息。
针对不同的微生物数据,和研究目的的不同,数据分析时需要用到不同的统计分析方法,例如降维、聚类、机器学习等。
本文将介绍和比较微生物测序数据统计方法和分析工具,以及微生物组数据分析结果展示原则。旨在帮助谷禾合作方或客户更好地理解和解读微生物组数据分析结果。
假设检验是根据一定的假设条件,由样本推断总体的一种方法。
假设检验的基本思想是小概率反证法思想,小概率思想认为小概率事件在一次试验中基本上不可能发生,在这个方法下,我们首先对总体作出一个假设,这个假设大概率会成立,如果在一次试验中,试验结果和原假设相背离,也就是小概率事件竟然发生了,那我们就有理由怀疑原假设的真实性,从而拒绝这一假设。
根据微生物组研究目的的不同,可以将数据检验方法分为:
1)组间差异检验
2)微生物组与临床或实验数据之间的相关性检验
组间差异分析或者叫组间差异显著性检验,差异显著性检验是微生物数据分析时用到的最多的统计学方法。
通常我们需要比较两组或多组数据之间是否有显著差异性,同时还要根据显著性检验识别出不同分组之间的差异变量。这需要用到统计学上的假设检验方法,检验分组之间是否有差异性,并且计算其差异程度。
注:所有的差异检验都基于一个假设:组间没有差异,及变量之间没有关系(即原假设,H0)。
在统计学上,假设检验方法可分为两大类,参数检验和非参数检验。
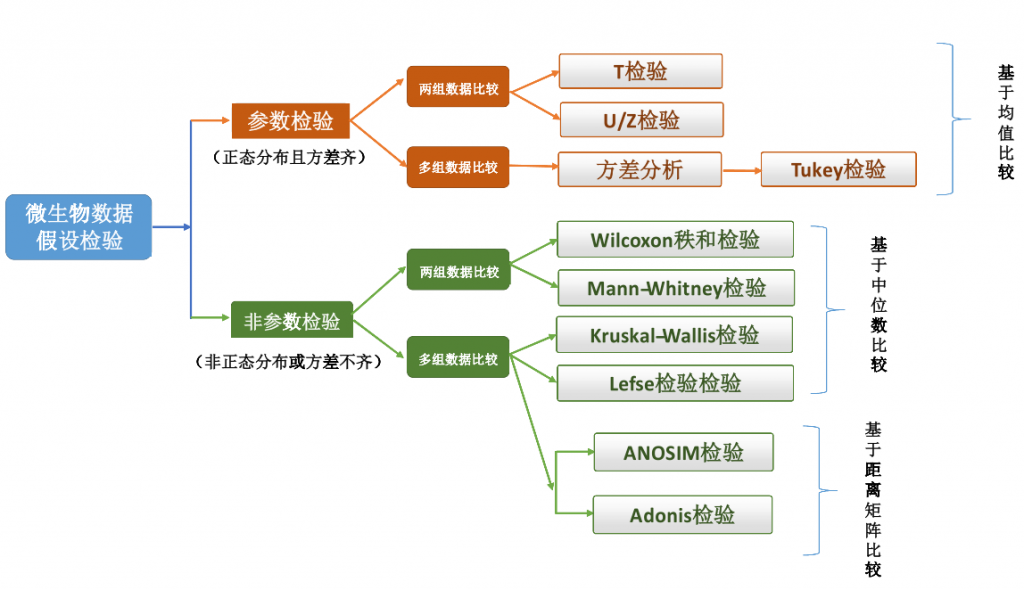

参数检验(Parameter test)是基于样本的观测数据对总体参数(比如总体均值、方差)及总体参数差异性的检验。
•参数检验需要具备一定的条件
参数检验要求总体具备一定条件才能适用,例如总体需呈正态分布、涉及两个总体时两总体必须满足方差同质性(即方差相等)。t检验、F检验(方差分析),都属于参数检验方法。
在实际分析过程中,数据会经常遇到不满足参数检验前提条件的情况,比如总体不服从正态分布,或者总体分布未知,这时就不能使用参数检验方法,不然得到的检验结果会不准确。在参数检验方法不适用的情况,就需要采用非参数检验方法进行统计检验。
•总体分布情况未知可使用非参数检验
非参数检验(Non-parametric test)是在总体分布未知或者知之甚少的情况下,通过样本数据对总体分布形态等特征进行推断的统计检验方法。
由于此种检验方法在由样本数据推断总体的过程中不涉及总体分布的参数,不要求总体分布满足某些条件,使用条件较为宽松,因此被称为非参数检验。
参数检验与非参数检验的一些区别


微生物组数据具有以下特点:
1.多样性高:微生物组数据通常包含大量的微生物种类,且种类之间的相对丰度差异较大。
2.维度高:微生物组数据通常包含大量的特征,例如16S rRNA基因序列或基因组序列,每个特征都可以看作是一个维度。
3.数据稀疏:微生物组数据通常是稀疏的,即大部分微生物种类的相对丰度很低,只有少数微生物种类的相对丰度较高。
4.非正态分布:微生物组数据通常不满足正态分布假设,且方差通常与均值相关。
5.数据复杂性高:微生物组数据中存在许多复杂的相互作用和关系,例如共生、拮抗和协同作用等。
考虑到微生物数据的以上特点,我们更推荐采用非参数假设检验的算法,在报告中我们大部分也采用的是非参数检验方法对组间差异进行检验。
差异显著性检验的方法有很多种,需要根据不同的情况,选择适合自身实验情况的方法进行分析。接下来对几种常见的统计检验方法进行介绍。
1
T 检验
Student t检验(Student’s t test),亦称T检验,是用t分布理论来推论差异发生的概率,从而比较小样本量两组间的差异是否显著,主要用于样本含量较小(例如n<30),总体标准差σ未知的正态分布数据。
注:Welch T检验(Welch’s t test)在两组数目方差不相等时可选择该检验。
•T检验的步骤:
第一步:建立虚无假设H0:μ1=μ2,即先假定两个总体平均数之间没有显著差异;
第二步:计算统计量T值,对于不同类型的问题选用不同的统计量计算方法。
2
U 检验
U检验又称Z检验,一般用于大样本(即样本容量大于30)平均值差异性检验的方法(总体的方差已知)。
它是用标准正态分布的理论来推断差异发生的概率,从而比较打样板量两组间的差异是否显著。
•U检验的步骤:
第一步:建立虚无假设H0:μ1=μ2,即先假定两个平均数之间没有显著差异;
第二步:计算统计量Z值,对于不同类型的问题选用不同的统计量计算方法。
3
方差检验
方差分析(ANOVA)又称“变异数分析”或“F检验”。用于正态分布、方差齐性的多组间(两个及两个以上样本均数)差别的显著性检验。
当自变项的因子中包含等于或超过三个类别情况下,检定其各类别间平均数是否相等的统计模式。方差分析首先是比较的多组间整体是否有差异性。再进行组间两两比较。
•方差检验的步骤:
第一步:建立检验假设;
H0:多个样本总体均值相等;
H1:多个样本总体均值不相等或不全等。
注:检验水准为0.05。
第二步:计算检验统计量F值;
第三步:确定P值并作出推断结果。
4
Tukey检验
Tukey检验也称为Tukey HSD(Honestly Significant Difference)检验,用于比较多个样本均值的两组之间的差异性。
Tukey检验的假设条件是各组数据的方差相等且服从正态分布。Tukey检验的原理是计算每两组之间的平均差异,然后根据方差和样本量的大小来计算显著性水平。Tukey检验通常用于方差分析的后续多重比较。
1
Wilcoxon秩和检验
Wilcoxon秩和检验又叫Mann-Whitney U 检验,Wilcoxon秩和检验,是两组独立样本非参数检验的一种方法。
其原假设为两组独立样本来自的两总体分布无显著差异,通过对两组样本平均秩的研究来实现判断两总体的分布是否存在差异,该分析可以对两组样品的物种进行显著性差异分析,并对p值计算假发现率(FDR)q值。
2
Kruskal-Wallis H检验
K-W检验的全称为,Kruskal-Wallis检验,它是用于正态分布条件不满足情况下,多组独立样本方差分析的替代。我们可以把它理解为“非参数检验的方差分析”。
微生物高纬度数据的多个变量根据不同的距离算法求取距离矩阵,然后根据距离矩阵进行相应的检验。所谓基于距离也就是检验的是群落差异而不是具体的某个物种。
以上这些检验方法,可以得到分组之间是否有显著差异(即简单的理解为差异的有无),如果想进一步知道这些差异的程度(可以理解为多少),就需要用到Anosim检验和Adonis检验。
这些方法不但可以输出检验显著性结果(p值),还有程度结果(R值),R值可以用来判断分组贡献度大小。Anosim、Adonis这些可用于多元统计检验的模型就非常适合了。
例如肠道微生物受到A、B等多种因素影响,如果一个项目里有3个分组分别是A处理、B处理和对照组,如果A处理和B处理相对于对照组都显著差异(P<0.05),想知道A处理、B处理哪个更为重要,就需要比较R值的大小了。
1
Anosim检验
Anosim(Analysis of similarities)是一种非参数检验方法。它首先通过变量计算样本间关系(或者说相似性),然后计算关系排名,最后通过排名进行置换检验判断组间差异是否显著不同于组内差异。
相似性分析ANOSIM检验用来检验组间(两组或多组)的差异是否显著大于组内差异,从而判断分组是否有意义。
这个检验有两个重要的数值,一个是p值,可以判断这种组间与组内的比较是否显著;一个是R值,可以得出组间与组内比较的差异程度。
R值范围实际范围是(-1,1),但一般介于(0,1)之间,R>0,说明组间存在差异,一般R>0.75:大差异;R>0.5:中等差异,R>0.25:小差异。R等于0或在0附近,说明组间没有差异。R偶尔也会<0,这种情况是组内差异显著大于组间差异,这就说明我们的采样或者分组出现大问题,可以认为是无效数据。
2
Adonis检验
Adonis,其实就是PERMANOVA。它与Anosim的用途其实差不多,也能够给出不同分组因素对样品差异的解释度(R值)与分组显著性(P值)。
Adonis又称置换多因素方差分析(permutationalMANOSVA)或非参数多因素方差分析(nonparametricMANOVA)。它利用半度量(如Bray-Curtis)或度量距离矩阵(如Euclidean)对总方差进行分解,分析不同分组因素对样品差异的解释度,并使用置换检验对划分的统计学意义进行显著性分析。
不同点是应用的检验模型不同,ADONIS本质是基于F统计量的方差分析,所以很多细节与上述方差分析类似。
图表解释
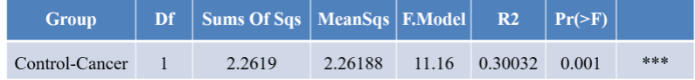

Group:表示分组;
Df:表示自由度;
SumsOfSqs:总方差,又称离差平方和;
MeanSqs:平均方差,即SumsOfSqs/Df;
F.Model:F检验值;
R2:表示不同分组对样品差异的解释度,即分组方差与总方差的比值,即分组所能解释的原始数据中差异的比例,R2越大表示分组对差异的解释度越高;
Pr(>F):通过置换检验获得的P值,P值越小,表明组间差异显著性越强。
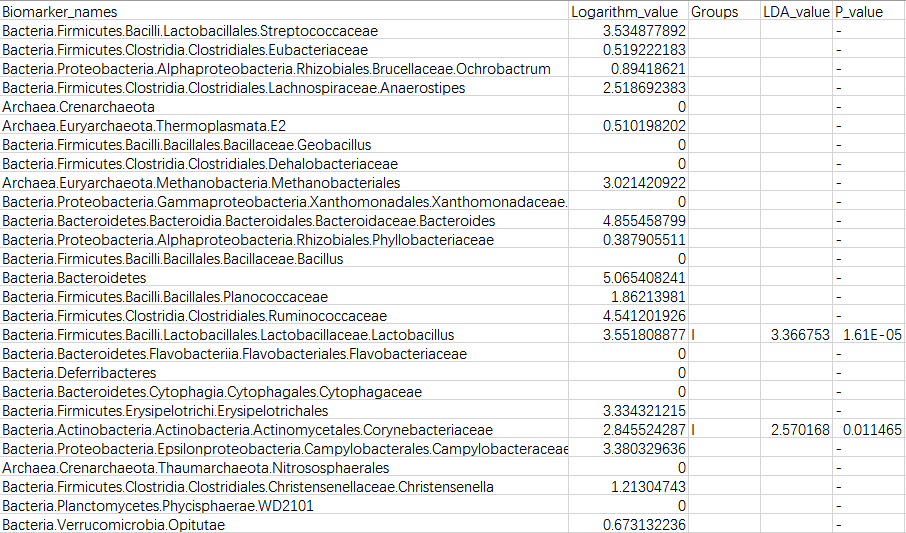
LEfSe(LDA EffectSize)是用于发现生物标识和揭示基因组特征的软件。以上分析我们找出了分组的差异显著性。
而Lefse的作用就是找出各分组的微生物标记物,即biomarker的筛选,以及这些特征对组间差异的影响程度,用于发现不同分组样本中最能解释组间差异的特征。
√ 分析步骤
A)软件首先使用Kruskal-Wallis (KW) (非参数因子克鲁斯卡尔—沃利斯秩和验检)检测多分组间具有显著丰度差异特征。
B)找出与丰度有显著性差异的物种后进一步使用Wilcoxon 秩和检验进行组间差异分析。
C)最后Lefse采用线性判别分析(LDA)估计每个分组的差异特征对差异效果的影响大小(即LDA score值)。
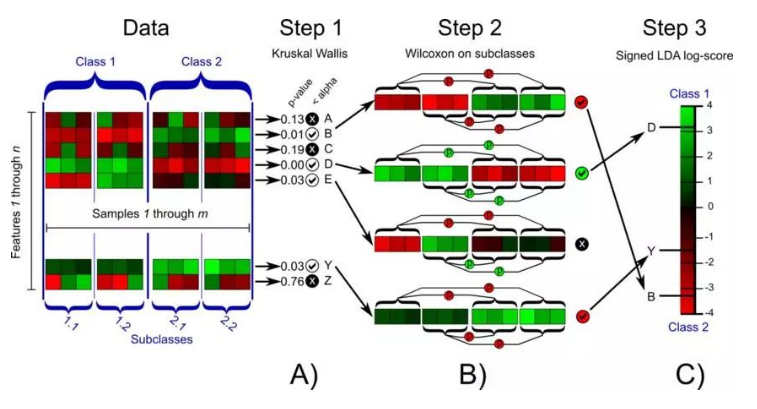

LEfSe结果包含两张图一张表,即(LDA值分布柱状图、 进化分支图及特征表)。
✦ LDA值分布柱状图
LDA值分布柱状图中展示了LDA Score大于设定值(默认设置为2)的差异物种,即组间具有统计学差异的Biomarker。
展示了不同组中丰度差异显著的物种,柱状图的颜色代表各自的组别,柱状图的长度代表差异物种的影响大小(即为 LDA Score),即不同组间显著差异物种的影响程度。
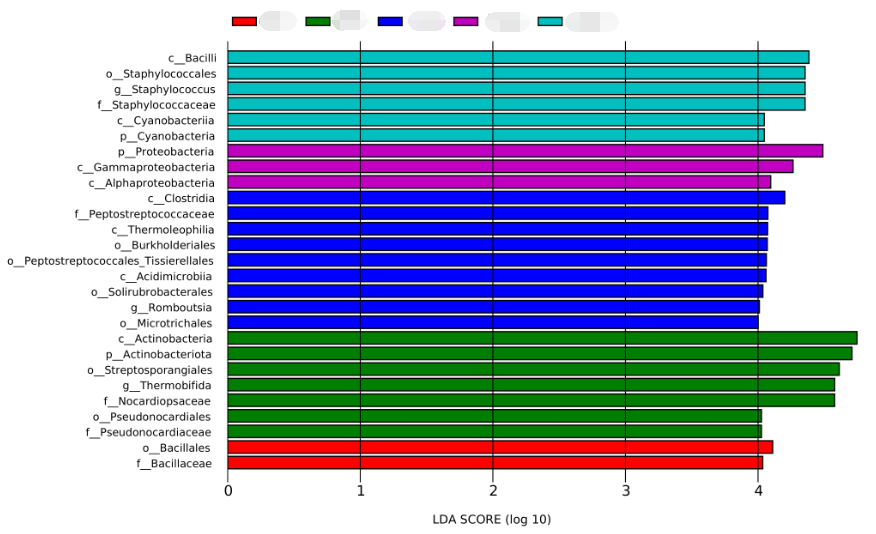

✦ 进化分支图
进化分支图中,由内至外辐射的圆圈代表了由门至属的分类级别。在不同分类级别上的每一个小圆圈代表该水平下的一个分类,小圆圈直径大小与相对丰度大小呈正比。
着色原则:无显著差异的物种统一着色为黄色,差异物种Biomarker跟随组进行着色,红色节点表示在红色组别中起到重要作用的微生物类群,绿色节点表示在绿色组别中起到重要作用的微生物类群。
注:图中英文字母表示的物种名称在右侧图例中进行展示。(为了美观,右侧默认只显示门到科的差异物种)。
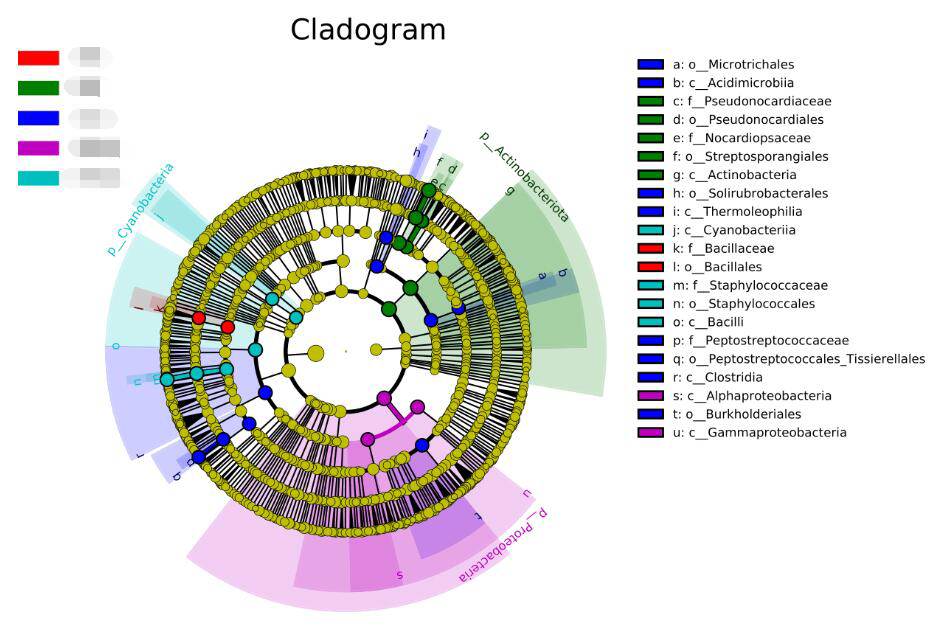

✦ 特征表解读:
第一列:Biomarker名称;
第二列:各组分丰度平均值中最大值的log10,如果平均丰度小于10的按照10来计算;
第三列:差异基因或物种富集的组名;
第四列:LDA值;
第五列:P值,若不是Biomarker用“-”表示。

基于微生物组数据的高维度、稀疏性,传统的统计方法不一定能很好的适用于微生物数据统计分析,机器学习面对复杂的数据结构逐渐成为更优的选择。
机器学习(machine learning,ML)就是计算机能够自主“学习” (拟合训练数据),通过神经网络、决策树等算法,从数据中自动分析寻找规律,从而生成经验模型,用于新数据的预测或分类。
随机森林(Random Forest)是一种基于决策树(Decisiontree)的机器学习算法。属于非线性分类器。
通过对对象和变量进行抽样构建预测模型,即生成多个决策树,并依次对对象进行分类。最后将各决策树的分类结果汇总,得出随机森林最终预测的分类结果。
•随机森林可以对样本进行分类和预测
随机森林可以对样本进行分类和预测。16S测序数据通过RandomForest随机森林可以找出与分组有关的关键物种/功能。
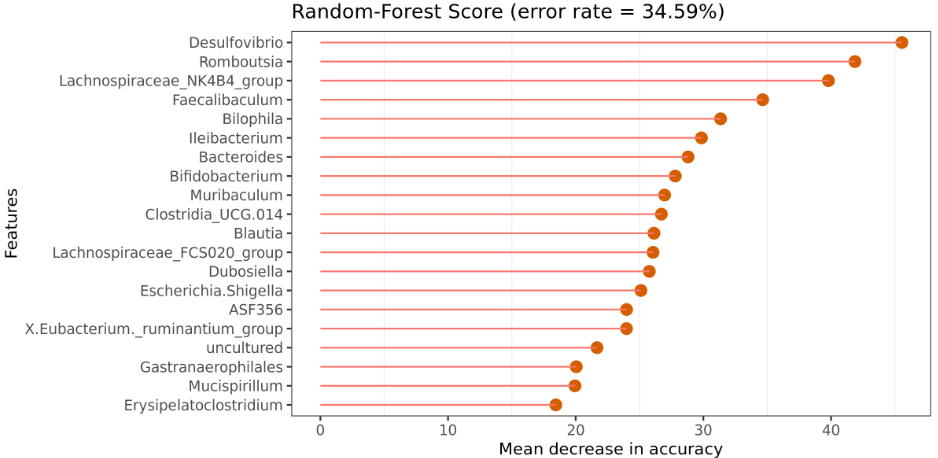

微生物数据通过RandomForest随机森林可以找出与分组有关的关键物种/功能。
图中展示了分类器中对分类效果起主要作用的特征,按重要性从大到小排列。Error rate :表示使用下方的特征进行随机森林方法预测分类的错误率,数值越高表示基于特征分类准确度不高,可能分组之间特征不明显。分值越低证明分组效果比较好。
根据随机森林方法筛选出的最佳模型,绘制ROC曲线,ROC是一种常用的统计学分析方法,在医学研究中主要用于评价诊断试验的效能,用来评判分类、检测结果的好坏。
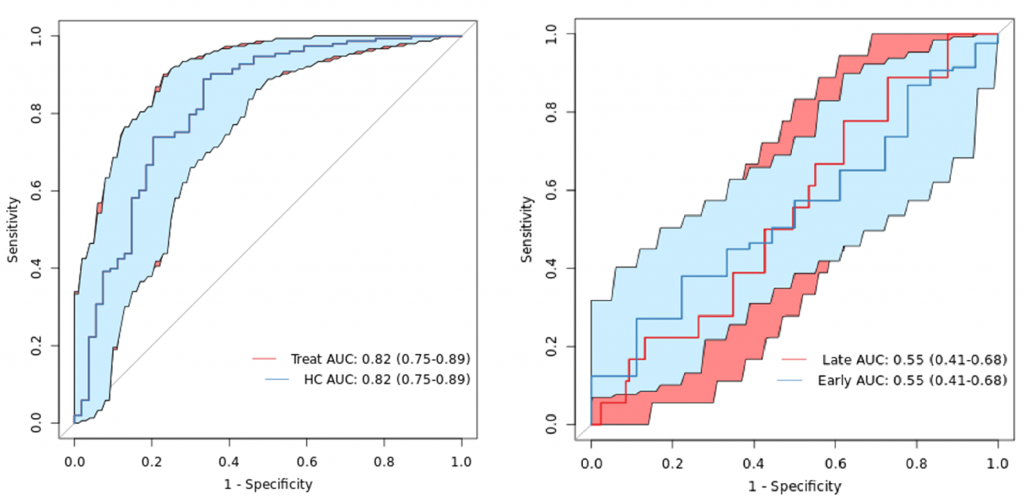

根据曲线位置,把整个图划分成了两部分,曲线下方部分的面积被称为AUC,用来表示预测准确性。
AUC值越高,也就是曲线下方面积越大,说明预测准确率越高。曲线越接近左上角(X越小,Y越大),预测准确率越高。
ROC曲线下的面积值在1.0和0.5之间。在 AUC>0.5 的情况下,AUC越接近于1,说明诊断效果越好。
•AUC值不同时的准确性:
AUC在0.5~0.7时有较低准确性,AUC在0.7~0.9时有一定准确性,AUC 在0.9以上时有较高准确性。AUC=0.5 时,说明诊断方法完全不起作用,无诊断价值。AUC<0.5 不符合真实情况,在实际中极少出现。
微生物高维度数据信息进过统计检验得到庞大的检验结果。如何将这些结果进行可视化展示,也是微生物数据分析过程中的重要组成部分,常见的可视化方法有箱型图、柱状图、散点图等。接下来是对这些图的详细介绍。
▷箱型图
箱型图(boxplot)又叫盒图,箱型图由5点组成:最小值(min),下四分位数(Q1),中位数(median),上四分位数(Q3),最大值(max)。
•便于多组间形态比较
箱型图可以直观的识别出异常数据,判断数据的侧重方法,方便多组间比较形态比较。其他类似变形的还有柱状图,小提琴图等就不做详细赘述。
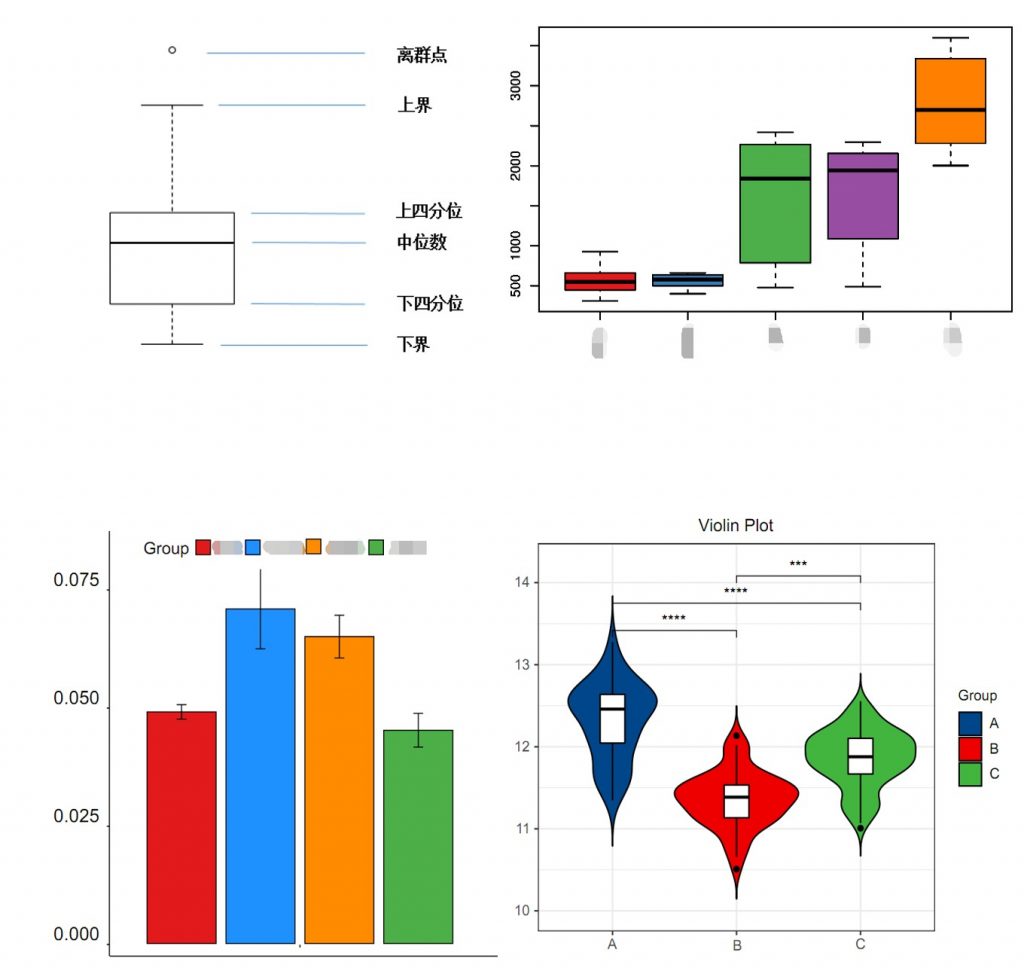

▷散点图
散点图(Scatter plot)是数据点在直角坐标系平面上的分布图,散点图表示因变量随自变量而变化的大致趋势,据此可以选择合适的函数对数据点进行拟合。比如PCA、PCoA、NMDS、回归分析、火山图等。

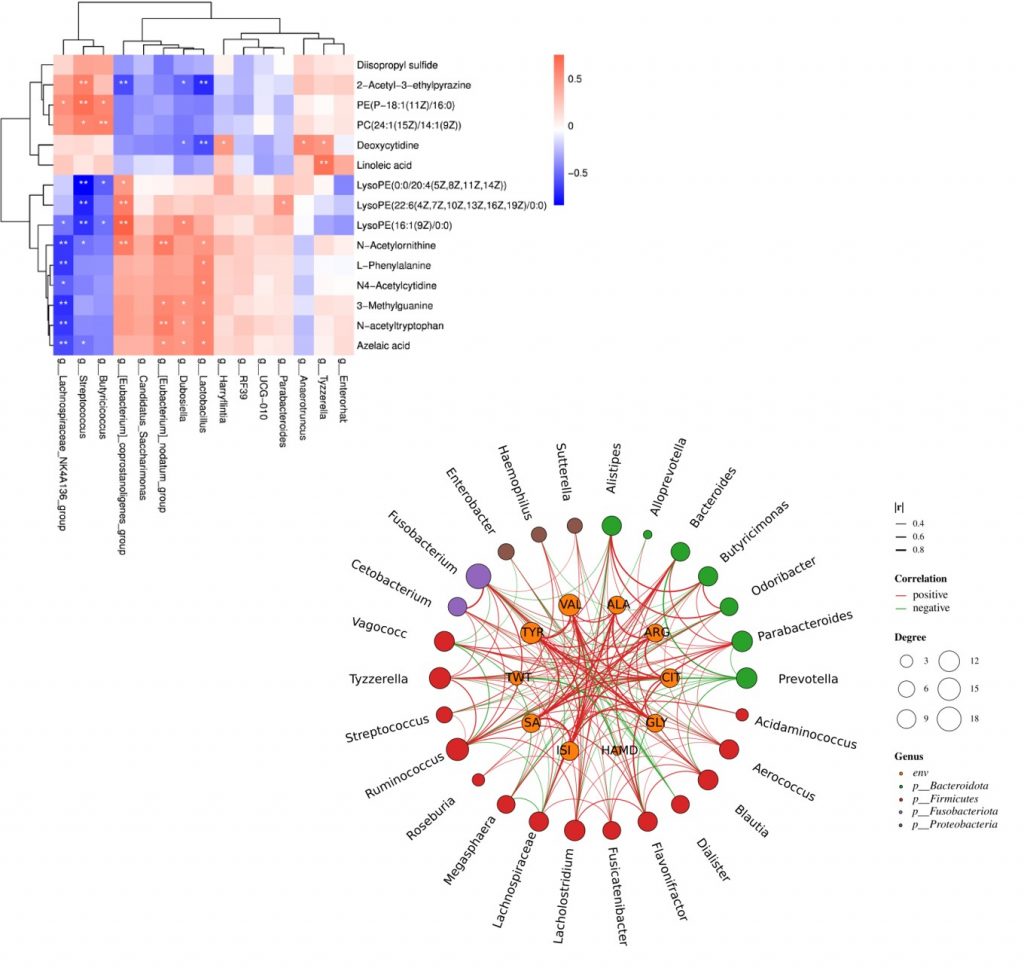
相关性是两个变量之间统计学相关关系的度量,可以预测变量间的相关关系。相关性的原始之间存在一定的联系或者相关性。
用相关系数来描述两个变量之间的相关程度。相关性分析常用的分析方法:
(1)pearson相关系数
(2)spearman相关系数
• pearson相关系数
皮尔逊相关系数(r),是衡量两个变量之间线性相关性的统计量。皮尔逊相关性只能评估两个连续变量之间的线性相关(仅当一个变量的变化与另一个变量的比例变化相关时,关系才是线性的)。
当两个变量都是正态连续变量,且两者之间呈线性关系时,则可以用Pearson来计算相关系数。取值范围[-1,1]。
• spearman相关系数
Spearman相关系数是排序相关性(ranked values)的非参数度量。
使用单调函数描述两个变量之间的关系的程度。Spearman相关性可以评估两个变量之间的单调关系。

接下来是对微生物统计检验分析中经常会遇到的一些问题进行汇总和解答。
➤ 参数检验和非参数检验有什么区别?
参数检验是对参数做假设检验,非参数检验是对总体分布情况做的假设。
•是否利于总体的信息
参数检验利用总体的信息(总体分布、总体的参数特征如方差等),以总体分布和样本信息对总体参数作出推断;非参数检验不需要利用总体的信息(总体分布、总体的一些参数特征如方差),以样本信息对总体分布作出推断。
➤ T检验和U检验有什么区别?
T检验和U检验都是常用的参数检验方法,都是用于检验两个样本的均值是否存在显著差异。
•样本量的大小不同
它们的区别在于,T检验主要用于小样本量(例如n<30)的正态分布数据,而U检验主要用于大样本量(即样本容量大于30)平均值差异性检验的方法。T检验的检验统计量是t值,U检验的检验统计量是U值。
➤ 什么是T值,什么是F值?为什么在报告中没有找到?
T值和F值都是统计学中常用的指标,用于检验假设是否成立。T值是T检验的统计量,用于检验两个样本均值是否有显著差异的指标。
•T值的计算公式
它的计算公式为:T = (x1 – x2) / (s * sqrt(1/n1 + 1/n2)),其中x1和x2分别是两个样本的均值,s是两个样本的标准差的合并估计值,n1和n2分别是两个样本的样本量。
T值越大,说明两个样本的均值差异越显著。
•F值的计算公式
F值F检验,即方差分析的检验量。是用于检验多个样本均值是否有显著差异的指标。
它的计算公式为:F = (SSB / (k – 1)) / (SSE / (n – k)),其中SSB是组间平方和,SSE是组内平方和,k是组数,n是总样本量。F值越大,说明组间差异越显著,即均值之间的差异不是由于随机误差造成的。在方差分析中,F值用于检验多组间平均数是否相等。
这两个指标均来自参数检验的统计量,由于微生物数据具有高纬度,多样性高,数据稀释复杂等特点,而参数检验对数据分布要求比较严格,微生物数据一般都呈现非正态分布,报告中大部分都采用非参数的检验方法进行检验计算。所以报告中没有直接给出T值和F值。
➤ Pearson相关系数和Spearman 相关系数有什么区别?
Pearson适用于两个变量之间呈线性关系,而Spearman适用于单调关系。
Pearson分析要求数据符合正态分布;线性相关;并且数据为连续值。如果不满足,则使用Spearman相关系数。
Pearson 处理变量的数据原始值,而Spearman处理数据排序值(需要先做变换,transform)。
➤ 报告中给出了几种检验方法得出的差异结果,该如何选择?
报告中分别使用了非参数检验、Tukey检验、Lefse分析、metagenomeSeq 差异分析。
•不同统计方法的结果略有不同
不同的统计方法对数据的处理方式和假设条件不同,所以不同的统计方法可能给出的差异结果略有不同。
•统计方法可能存在一定的偏差和误差
此外,不同的统计方法还可能存在偏差和误差,例如样本量不足、数据分布不符合假设条件、测量误差等因素都可能影响统计结果的准确性。
因此,在进行统计分析时,需要根据实际情况选择合适的统计方法,并结合实际情况进行综合分析和判断。这里建议选择其他一种方法的检验结果就可以。或者用到不同的检验方法的时候,在文章中加以说明。
➤ 报告中给出的多组间差异的P值只有一个吗?是哪两组之间的P值呢?
非参数检验多组间差异分析时就用到的是Kruskal-Wallis H检验,检验多组间是否有差异,所以只有1个P值。
例如该图是属水平多组间非参数检验的差异结果:

路径:
Groups\diff_analysis\UnivarTestGenus\figure\barplot
对应P值:
Groups\diff_analysis\UnivarTestGenus\ UnivarTest_sign.txt
如果想看在多个分组的统计检验过程中,具体哪两组间之间有差异,可以看:
Groups\diff_analysis\UnivarTestGenus\figure\single_plot
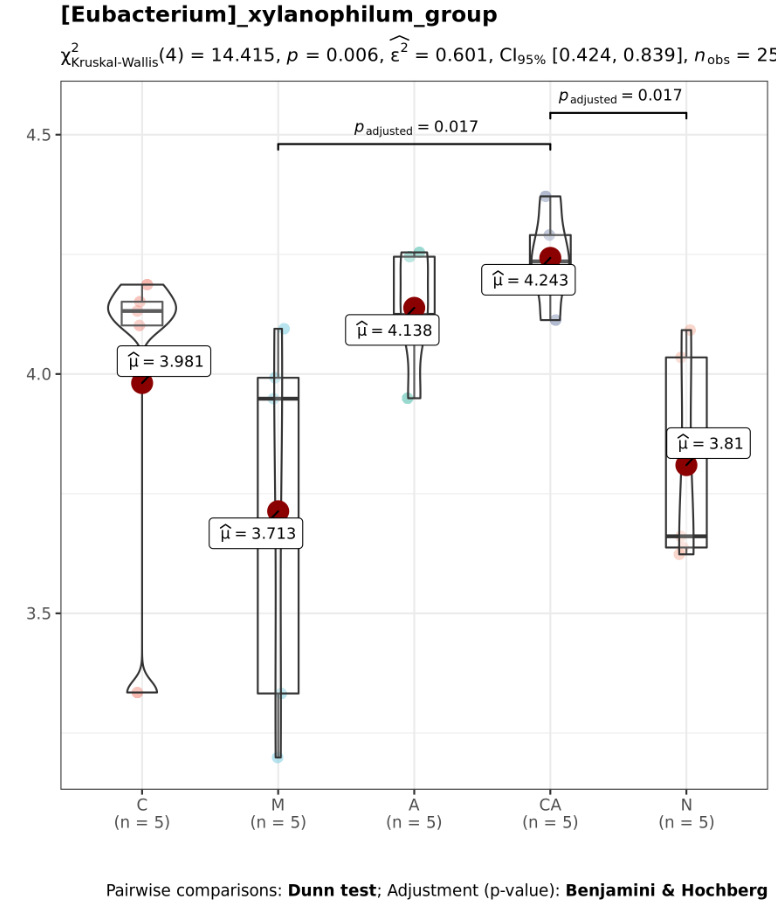
图上方的P值0.006是多组间非参数检验的P值,图中展示的是任意两组间差异显著的结果,例如P=0.017是M组和CA组之间的结果。P=0.017是CA组和N组之间的P值。如果任意两组间有差异,则在图中显示对应P值。

PCoa基于Bray距离的Adonis检验得到的P值=0.001右下角,也是多组间统计检验的结果。
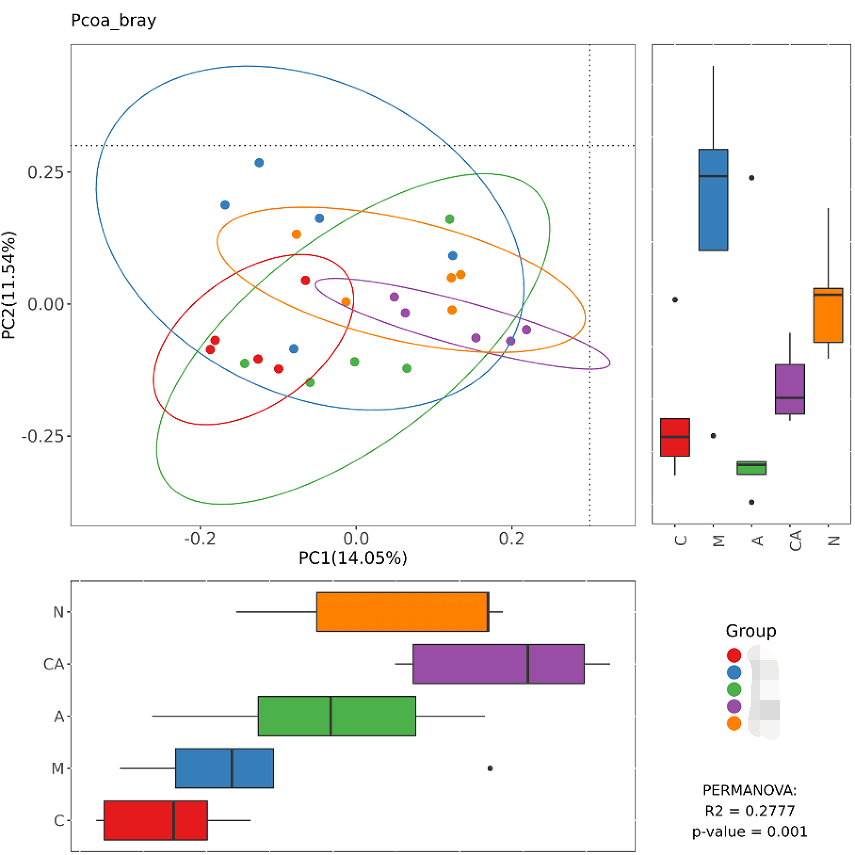

想看任意两组间Adonis检验是否差异显著,可以看下表:
Groups\betadiv\pcoa_bray_analysis\ PERMANOVA_paired_result.csv
表中展示了任意两组间Adonis检验的P值和R值。
编辑
✦
附 录
1
热 图
热图是一种常用的展示方法,可以将微生物组数据转化为颜色矩阵,从而直观地显示不同样本或物种之间的相似性和差异性。可以使用热图来展示微生物组数据的OTU/ASV表、物种丰度表等。

2
柱 状 图
柱状图是一种常用的展示方法,可以将微生物组数据转化为柱状图,从而直观地显示不同样本或物种的相对丰度。可以使用柱状图来展示微生物组数据的物种丰度表等。
3
散 点 图
散点图是一种常用的展示方法,可以将微生物组数据转化为散点图,从而直观地显示不同样本或OTU/ASV之间的关系。可以使用散点图来展示微生物组数据的beta多样性、代谢功能预测丰度表等。
4
网 络 图
网络图是一种常用的展示方法,可以将微生物组数据转化为网络图,从而直观地显示不同微生物组物种之间的相互作用关系。可以使用网络图来展示微生物组数据的共生关系、代谢通路等。
5
生 态 图
生态图是一种常用的展示方法,可以将微生物组数据转化为生态图,从而直观地显示不同微生物组物种之间的生态关系和生态功能。可以使用生态图来展示微生物组数据的生态功能、物种丰度表等。
6
积 柱 状 图
积柱状图是一种常用的展示方法,可以将微生物组数据转化为堆积柱状图,从而直观地显示不同样本或物种的相对丰度,并且可以分别显示不同分类水平的物种。可以使用堆积柱状图来展示微生物组数据的物种丰度表等。
7
饼 图
饼图是一种常用的展示方法,可以将微生物组数据转化为饼图,从而直观地显示不同物种的相对丰度。可以使用饼图来展示微生物组数据的物种丰度表等。
8
箱 线 图
箱线图是一种常用的展示方法,可以将微生物组数据转化为箱线图,从而直观地显示不同样本或物种的分布情况。可以使用箱线图来展示微生物组数据的alpha多样性、代谢物浓度等。
9
简 单 线 图
简单线图是一种常用的展示方法,可以将微生物组数据转化为简单线图,从而直观地显示不同样本或物种的变化趋势。可以使用简单线图来展示微生物组数据的时间序列数据、代谢物浓度等。
总的来说,展示微生物组数据分析结果需要选择合适的展示方法,并且需要根据研究目的和数据类型进行选择。以上展示方法是常见的方法,并不是唯一的方法,实际应用中需要根据具体情况选择合适的方法。

谷禾健康
什么是CAZymes?
CAZymes(碳水化合物活性EnZymes)是一种酶,主要以糖苷键为目标,降解、合成或修饰地球上的所有碳水化合物。CAZymes在植物和植物相关微生物中非常丰富。
例如,人体肠道是富含碳水化合物的环境,碳水化合物降解细菌的多样性非常高。肠道微生物组的CAZyme组合组成了数以万计的附加基因。
CAZymes对人类健康、营养、肠道微生物组、生物能源、植物疾病和全球碳循环的研究极为重要。
在过去的5年里,大量的微生物组测序,来自各种生态环境的数十万个宏基因组组装基因组 (MAGs) 现在可以在公共数据库中获得。
例如欧洲生物信息学研究所的MGnify数据库和 IMG/M联合基因组研究所数据库。目前,没有数据库从微生物组MAG中收集CAZymes和CAZyme基因簇(CGC)并在网络上提供它们。
✦CAZyme研究对人体健康非常重要
与此同时,CAZyme生物信息学领域继续发展。现在可以推断CAZymes和CGCs的碳水化合物底物,这对应用微生物组非常有兴趣。例如,基于微生物组的个性化营养旨在使用个性化饮食干预策略来调节人体肠道微生物组,以改善人体健康。
预测患者肠道微生物组可能对哪些益生元聚糖做出反应的能力将对营养师和营养学家提出个性化饮食建议非常有用。
✦最新更新了dbCAN-seq数据库
最近在内布拉斯加大学的团队在Nucleic Acids Research发表的题为:“dbCAN-seq update: CAZyme gene clusters and substrates in microbiomes”的文章,更新了dbCAN-seq数据库 ( https://bcb.unl.edu/dbCAN_seq)


包括以下新数据和特征:
(i)来自四种生态(人类肠道、人类口腔、牛瘤胃和海洋)环境的9421个MAG~498000个CAZyme和~169000个CAZyme基因簇(CGC);
(ii) 通过两种新方法(dbCAN-PUL 同源搜索和 eCAMI 亚家族多数表决)推断的41447(24.54%) 个CGC的聚糖底物(这两种方法就4183个CGC的底物分配达成一致);
(iii) 重新设计的CGC页面,包括CGC基因组成的图形显示、查询 CGC 和 dbCAN-PUL 的主题PUL(多糖利用位点)的比对,以及支持预测底物的 eCAMI 亚家族表;
(iv) 一个统计页面,用于根据底物和分类门组织所有数据,以便于CGC访问;
(v) 批量下载页面
总之,这个更新的dbCAN-seq数据库突出显示了预测来自微生物组的CGC的聚糖底物。
dbCAN-seq数据库于2018年发布,提供了5349株细菌分离株基因组的CAZyme和CGC (CAZyme基因簇)序列及注释数据。
CGC是研究人员定义的一个术语,用于描述微生物基因组中含有CAZyme的基因簇。
PUL指多糖利用位点,是一个更流行的术语,描述利用复杂碳水化合物底物的基因簇,例如XUL(木聚糖利用位点),ChiUL(几丁质利用位点)和XyGUL(木聚糖利用位点)。随着CAZyme在生物信息学领域的发展,dbCAN-seq也紧跟步伐,推出了新的版本。
在dbCAN-seq数据库的更新中,主要取得了两个重要的进展:
(i) dbCAN-seq现在提供了四个生态环境(人类肠道,人类口腔,牛瘤胃,海洋)的微生物组的全面的CAZyme和CGC目录。
(ii) dbCAN-seq使用了两种新的方法来预测微生物组CGCs的底物,并提供了底物查阅功能,允许搜索针对不同微生物组中预测的特定底物的CGCs。
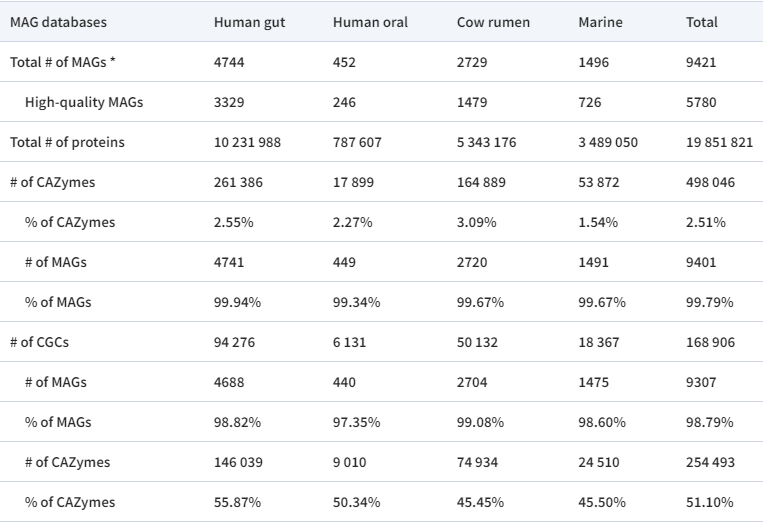
对EBI MGnify数据库中的4个MAG数据集,选取其中质量最好的代表性基因组,如下表,一共9421个基因组,使用dbCAN2的run_dbcan程序进行注释,可见有近50万个蛋白质被注释为CAZymes。
人类肠道MAG中CGCs的CAZymes比例最高(55.87%),而牛瘤胃中CGCs的CAZymes比例最低(45.45%)。
在预测出CGCs之后,研究人员开发了两种计算方法来推断它们的糖基底物,如下图。
第一种方法
使用BLAST比对CGCs的蛋白质序列(图A)与dbCAN-PUL的612个PULs的蛋白质序列(图B),选择BLAST总评分最高的最佳命中且至少有一个CAZyme与至少一个来自其它特征基因类别,如TC、TF等相匹配(例如图C),然后通过表S1,PUL→底物的映射文件,获知对应的底物;
第二种方法
使用eCAMI工具对CAZyme亚家族进行注释(图D),确定了CAZyme蛋白的eCAMI亚家族和EC数量后,根据eCAMI亚家族和EC编号建立的CAZyme亚家族→EC→底物的映射文件(表S2),使用简单的多数投票规则来推断CGC中的底物分配。
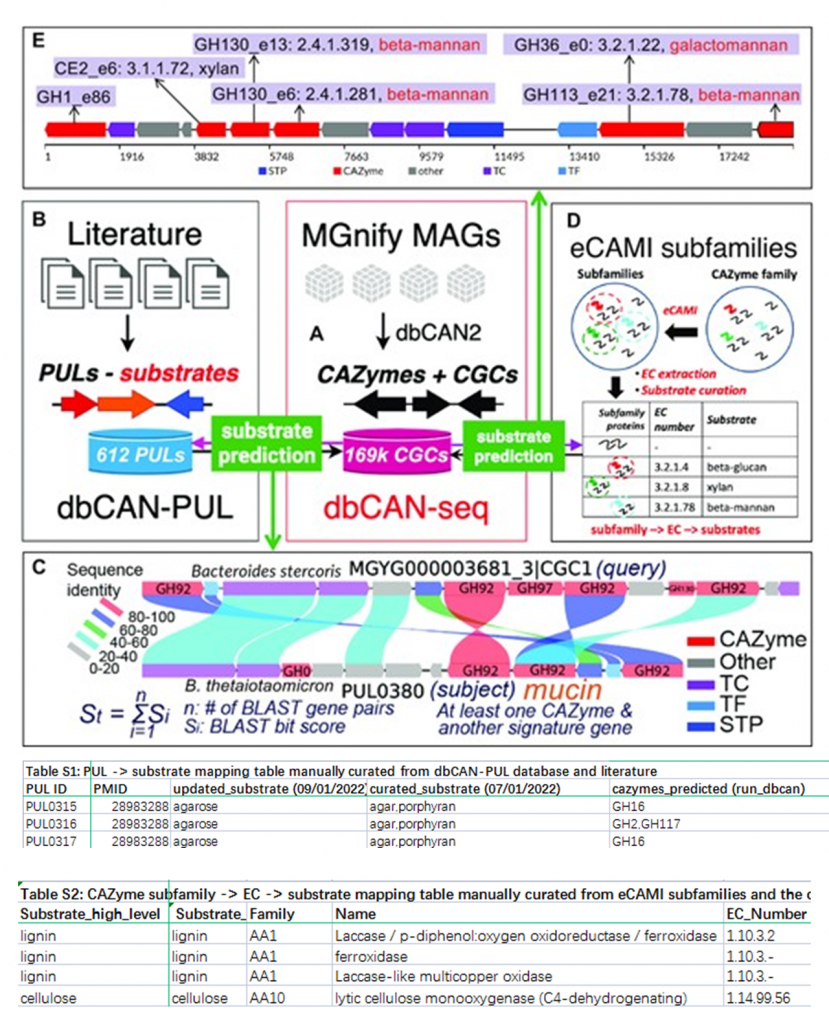

通过以上两种方法,目前dbCAN-seq数据库中新增的预测底物的CGC计数如下图:
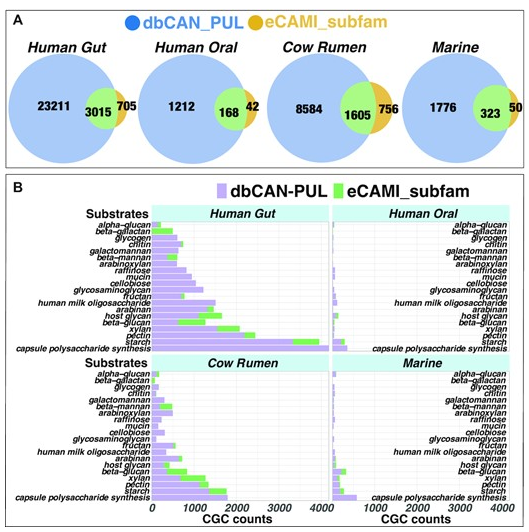
图A为用dbCAN-PUL方法和eCAMI亚族方法注释CGCs的韦恩图。
图B各数据集中丰度top20的底物。
dbCAN-seq的官网为:
如下图,网站也新增了版块:
图A可选五个数据集。
图B可按目录浏览。
图C和D分别为支持通过eCAMI或dbCAN-PUL预测的底物查询对应的CGCs
研究人员计划每年更新dbCAN-seq数据库,以包含更多生态环境的MAG数据集,例如来自小鼠、猪、山羊的肠道MAG,以及来自土壤和地球微生物组项目的MAG。
也会继续探索用于底物预测的方法,比如无监督机器学习方法,以预测自然界中未知的底物的新型CGCs。