-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康
恭喜!
杭州谷禾信息技术有限公司检测实验室
注册号:CNAS L23010
【新突破】
谷禾16S rRNA 扩增子检测项目正式通过中国合格评定国家认可委员会 CNAS 认可,纳入实验室认可范围。
【深扎根】
此前谷禾获认可的宏基因组项目(详情:杭州谷禾检测实验室获CNAS国家认可,微生物检测迈向更高台阶),顺利通过 CNAS 首次监督评审。
关于CNAS
CNAS 是“中国合格评定国家认可委员会”(China National Accreditation Service for Conformity Assessment)的英文缩写,是由国家认证认可监督管理委员会批准设立并授权的国家认可机构,统一负责对检测实验室、校准实验室、认证机构等合格评定机构进行认可。获得 CNAS 认可,表明该实验室具备按照相关国际和国家标准开展检测活动的技术能力和管理水平,其出具的检测报告具有权威性和公信力。
在我国合格评定体系中,CNAS 认可代表的是对机构能力的第三方评价。对于检测实验室而言,是围绕实验室的管理体系、技术能力、人员能力、设备设施、方法确认、质量控制、结果有效性、持续改进能力等方面进行系统评审。
CNAS 认可体系强调实验室活动的公正性、独立性和结果有效性。检测结果是否客观,检测过程是否受控,人员是否能够避免不当压力和利益干扰,都是实验室质量体系中的重要内容。
因此,实验室通过 CNAS 认可,意味着其在获认可范围内具备按照相应认可准则开展检测活动的能力,也意味着检测过程和结果具备更清晰的规范性、可追溯性和可信基础。
同时,CNAS 与国际认可互认机制相衔接,在相关互认范围内,有助于推动检测、校准、检验等合格评定结果在不同国家和地区之间获得更广泛的接受。
对高通量测序检测实验室来说,CNAS 认可不仅是一项资质成果,更是一种来自国家认可体系的能力确认,也是一份面向科研、产业与真实应用场景的可信承诺。
据目前公开认可信息及相关项目范围比对
谷禾获认可的16S rRNA 扩增子检测项目
为国内高通量测序检测类公司中
首个通过 CNAS 认可的同类项目
从宏基因组检测项目首次获得认可
到如今顺利通过监督评审;
从实验室基础质量体系建设
到16SrRNA扩增子检测项目纳入认可范围;
这是一份属于谷禾团队全体成员的喜报
更是一段长期秉持“工匠精神”
日常工作的阶段性回响
也意味着谷禾检测实验室在微生物组检测
和高通量测序检测能力建设的道路上
再次迈出了坚实且具有行业引领性的一步
16S rRNA 扩增子检测,是一条由多个高精细环节环环相扣组成的完整技术链路。通过对微生物特征基因片段进行富集、高通量测序和深度生物信息学分析,能够帮助洞察样本中微生物群落的物种组成、多度分布、多样性特征及结构演变。
然而,正因为这条技术链路横跨了分子生物学、微生态学与计算机信息学等多个学科,其全生命周期的复杂性与不确定性也呈指数级上升。
系统性鸿沟:标准化不足带来的数据可比性挑战
在当今蓬勃发展的微生物组研究领域,方法学标准化不足所导致的研究间可比性差,已成为横亘在科学探索与真实世界应用之间的一道系统性鸿沟,这不仅是对宝贵科研资源的巨大损耗,更是掣肘了微生态研究成果向产业化与临床端安全转化的核心通道。
这就要求实验室不仅在技术层面知道怎么做,更要在体系管理层面清晰、透明、有据可查。
比如说,数据质控阈值是如何经过数理统计学验证的,异常结果出现时的根本原因有没有相应的调查机制,涉及流程、软件版本或关键参数调整时,是否经过必要的技术确认等。
这些都构成了高通量测序检测项目的质量门槛。
谷禾的选择:以标准化支撑检测长期稳定
面对这一阻碍行业健康发展的底层痛点,谷禾健康团队深知,建立贯穿生命科学检测全周期的标准化操作规范(SOP)和严密质控体系,是让检测数据具备稳定性、可追溯性的关键,也是精准指导下游靶向干预、产品研发、功效评价,以及迈入临床级应用的关键基石。
为此,我们历经多年在底层技术上的持续深耕与无数次真实样本的反复打磨,系统性重构并确立了一套严苛、稳定的微生物高通量测序(涵盖16S rRNA及宏基因组)全流程检测技术规范。
正因如此,CNAS 认可对谷禾而言,不是一次单点项目申报,也不是把既有流程简单整理成文件,而是一次对检测能力、质量体系和长期稳定能力的系统性确认。
对于高通量测序实验室而言,认可体系的核心在于:将国际标准内化为员工工作习惯的能力,并在日复一日的枯燥运行中,始终保持检测能力的在册、受控、不走样。
高通量测序的底层逻辑是极其精密且连续的。为了确保发出的每一组测序数据的真实可靠,谷禾检测实验室严格围绕“公正、科学、守信、高效”的质量管理方针,从人员管理、设备量值、物料方法、环境监控、数据记录五大核心维度,构建了多维一体的立体防线,将标准彻底深化在底层秩序中。
在这套质量方针中,公正始终被放在最前面。对谷禾而言,公正意味着:
“ 在谷禾实验室体系,实验室人员,
从来不是简单完成标准操作的机器,
而是质量体系有血有肉的真正执行者。”
一份样本从接收到报告出具,中间会经过多个岗位。
有人负责核对信息,有人负责样本处理,有人负责核酸提取,有人负责文库构建,有人负责上机测序,有人负责数据分析…每个人手中的动作,可能只是流程中的一小步,但连在一起,就成为一份报告背后看不见的质量支撑。
CNAS 认可要求实验室关注人员能力、培训、监督、授权和持续评价。这些要求最终落到日常,各岗位都要知道:为什么这样做,这样做会影响什么?如果出现偏离,应该如何识别、记录、上报和处理等。
同样重要的,是检测人员在岗位职责内保持独立、客观的技术判断。
样本不会说话,数据也不会主动解释自己。守住每一道关口的,是实验室里那些反复核对编号的人,是每次操作前确认试剂状态的人,是发现异常后停下来追原因的人,是报告发出前逐项检查的人…
将CNAS要求落到谷禾检测的日常里,就是一套动态的人员能力验证模型。
质量管理不是把责任简单压给个人,而是让每个人在清晰的规则、充分的训练和持续的监督中,把事情做对、做稳、做久。
这种能力,不是某一次操作优秀,而是长期稳定;不是把标准挂在墙上,而是让标准融入每一次开盖、每一次移液、每一次复核、每一次签发…
这些看似重复的动作,最终都会落到客户所等待的那份结果里。也正因如此,我们愿意把看似细小的记录留得更清楚,把普通的日常做扎实,让质量体系真正落在每一天的工作里。
宁愿在发现疑点时多停一步,
也不愿把不确定带进样本流程。
高通量测序仪、PCR 仪、荧光定量 PCR 仪、生物安全柜、离心机、微量移液器、纯水仪……许许多多设备构成了一份肠道菌群检测报告背后的技术基础。
然而再精密的仪器,如果状态不可确认,也无法支撑可靠的结果。
一支微量移液器,可能只是实验台上最常见的工具,但它每一次吸取和转移,都关系到微量反应体系的稳定。
一台生物安全柜,不只是设备,更是样本保护、人员保护和污染控制的重要屏障。
设备会通过状态、记录和数据回应实验室的管理水平。因此,在谷禾实验室,设备会贯穿采购、验收、标识、使用、维护、核查、校准、异常处理和记录管理的全过程。
“一份看似普通的粪便样本,
背后都承载着一个人
对了解自身肠道微生态的期待。”
样本是检测的起点,也是客户信任进入实验室的第一站。
从采集、保存、运输,到实验室接收、编号、流转、处理和留存,每一步都需要被认真对待。
对每一份样本保持一致的接收、流转和处置要求,也是公正性在样本端的体现。无论样本来自何种项目、何种合作场景,进入实验室后,都应回到同一套受控流程中被认真对待。
样本要有清晰身份
试剂要有受控状态
耗材要适用于相应实验环节
关键物料要经过必要确认
外部提供的产品和服务也需要被纳入管理
成熟的实验室管理,是要把影响结果的因素尽可能管住。
与样本同等重要的,是各种高精密的试剂与反应耗材。高通量测序对试剂批次间的稳健性要求近乎苛刻。
磁珠吸附力的微量起伏、核酸提取试剂盒的微小原料波动,或者聚合酶的活性晃动,都会在敏感的测序仪中被指数级放大,进而干扰微生物群落的真实丰度。
为此,谷禾对所有生产投入品确立了严格的“状态受控”与“关键物料确认”原则。
“ 能说明的,用严谨的数据讲清楚。
不能过度推断的,我们严格守住边界。
把科学的不确定性放在它该在的位置。”
在菌群检测中,方法贯穿样本处理、核酸提取、扩增建库、测序、数据分析和报告审核等多个环节。每一个环节都可能影响最终结果。因此,方法要落实为“流程被理解、被执行、被记录、被复核、被持续改进”。
谷禾检测对方法的理解,是专业,也是克制。
专业,是因为检测方法必须经过充分确认。在检测项目建立和持续运行过程中,我们会围绕方法适用性、关键性能表现、操作边界和结果稳定性等方面开展系统评估,确保方法能够满足预期用途。
好的方法,不是让实验室显得复杂,而是让复杂工作变得稳定。
对于肠道菌群检测中涉及的分子生物学实验环节,谷禾检测关注的除了能不能检出之外,还有方法在不同样本状态、不同批次运行和不同实验条件下是否依然保持稳定。
哪些步骤必须严格一致,哪些条件不能随意改变,哪些异常信号需要暂停复核等等,都是由方法体系本身来约束。
在微生态检测领域,复杂性不仅来自实验台,也来自数据端。高通量测序产生的是海量数据,形成正式的报告之前,还需要经过数据质控、序列处理、分类注释、结果整合、专业审核等。
每一次涉及分析逻辑、版本状态或关键参数的调整,都需要经过相应评估和确认,确保升级带来的是能力提升,而不是结果波动。
CNAS评审专家们对谷禾检测实验室的实验方案、分析流程及关键参数控制,进行全方位、系统性的现场核查与技术确证。
专业的另一面,是克制,是边界。
报告表达必须基于检测数据本身,不能越过证据范围去制造确定性,也不能为了迎合期待而扩大解释。肠道菌群检测可以帮助客户了解肠道微生态的组成和特征,但它不应被包装成超出检测能力范围的承诺。
一次偶发异常结果背后的多轨复核
一处原始记录细节的补充确认
一项分析流程变更前后的评估
一份报告发出前的层层审查
…
它们隐身于幕后,
却真真切切地决定了
一份报告是否值得被托付信任
“ 好的实验环境,
让正确的事更容易发生,
让错误的事更不易发生。”
在微生物检测和基因扩增相关实验中,环境从来不是背景。环境本身,就是质量的一部分。
肠道菌群检测面对的是复杂微生物群落。样本之间、区域之间、人员操作之间,以及仪器状态、空间条件、输入物料等外部变量,都可能对结果稳定性产生影响。
对高通量测序而言,一些风险并不总是以明显故障的形式出现,它可能是设备运行状态中的细微漂移,也可能是空间流转中的潜在交叉干扰。
在空间管理层面,谷禾检测重视功能区域之间的边界感。
涉及核酸处理、扩增和测序相关的关键区域,需要通过合理分区、标识管理、人员与物料流转控制、清洁消毒和污染防控措施,尽可能降低交叉干扰风险。
该隔离的区域要隔离,该限制的进入要限制,该标识的状态要标识,该记录的条件要记录。环境条件不适合时,检测活动就不能被简单推进;存在潜在风险时,就需要先判断、先控制、先确认。
这些日复一日的环境控制,可以保护样本不被无关因素干扰,保护人员在安全条件下工作,也保护最终结果不被不必要的外部变量影响。
谷禾检测的环境,不仅是一间实验室的装修和空间,而是一种质量氛围:
环境会在每一份稳定的结果里,留下自己的痕迹。
应用十多年的底层技术迭代与平台进化
16S rRNA 扩增子检测,作为谷禾的高频基石技术,是谷禾在微生物组学领域扎根最深、应用场景最广、数据资产最雄厚的底层技术支撑。
在谷禾多年的肠道菌群检测、微生态研究服务和高通量测序的大规模实践中,16S rRNA 测序长期扮演着连接微生态样本与高维度生命科学大数据的关键纽带。
作为谷禾最早推向市场、经历了十余年应用与多轮标准化迭代的经典项目,它完整见证了海量复杂样本的自动化流转、实验反应体系的优化、生物信息学分析算法的迭代升级,数以万计科研与临床报告的交付等。
伴随着这一长周期的技术演进历程,谷禾在微生物组检测领域逐步构建起了严密的方法学验证体系,确立了标准作业程序(SOP)的底层逻辑。
一个高通量测序项目真正的成熟与强大,取决于它能否在大规模、跨批次、长周期的实际运行中,有效对抗各种高风险变量,始终保持极高的多中心稳定性和结果复现性。
纳入全球互认体系的底层逻辑升级
此次,16S rRNA 扩增子检测项目通过 CNAS 认可,是谷禾长期的技术沉淀与质量管理体系深度融合的必然结果,意味着谷禾持续运行的检测,全面正式跨入了全球多边互认的合规轨道。
16S rRNA 扩增子检测项目从企业经典向国际认可的跨越,是检测与质量的全面升级。在 CNAS 质量框架下,16S rRNA 项目的运行实现了多维一体的规范化:
经典,代表它经历过时间和样本的检验
认可,代表它进入了更规范、受控和可评价的质量框架
两者结合,构成了谷禾 16S rRNA 扩增子检测项目今天的底气
深度赋能全产业链伙伴
对于长期携手并进的科研学者、临床专家、生物医药企业以及大健康产业同仁而言,这一升级意味着:谷禾为全产业链合作伙伴的课题研究、队列分析、产品研发、商业化落地、功效评价、微生态应用,提供了坚实的数据底座。
-为科学研究、临床转化与大健康产业筑牢底座-
CNAS 认可是一项阶段性成果,也是一项持续性的要求。
从宏基因组检测项目获得认可,到顺利通过 CNAS 首次监督评审;从 16S rRNA 扩增子检测项目长期规模化运行,到正式纳入 CNAS 认可范围,谷禾走过的,是一条把技术做深、把体系做实、把数据做稳的长期主义之路。
这条路没有捷径。
– 它来自十多年的技术迭代,也来自每一天的样本、记录、复核和改进;
– 它来自实验平台的持续进化,也来自质量体系对每一个细节的长期约束;
– 它来自团队所有人对微生物组检测的专业投入,也来自谷禾对客户信任的持续回应
…
这一次,我们为团队感到骄傲;
同时也更清醒地知道,认可之后,标准只会更高,责任只会更重。
未来,谷禾将继续围绕微生物组检测和高通量测序技术,持续完善检测方法、质量控制、生物信息分析和数据管理流程,在每一次样本处理、每一轮数据分析、每一份报告审核中,保持对标准的敬畏和对结果的审慎。
因为我们深知样本的背后,连接着一个个科研问题、一项项转化探索、一次次产品验证,或者一个人对自身健康状态的认真关注。
样本和数据不会说话,但谷禾的选择,定义了数据的分量。
我们专注检测
坚持检测平台的中立性,不因商业压力或主观判断改变检测结果;
我们尊重数据
坚持科学严谨,不为了迎合期待而扩大解释;
我们重视体系
因为只有把标准融入日常,检测能力才能长期稳定地保持;
我们敬畏样本
坚持职业的同理心,因为每一份样本背后,都是一份真实的托付。
以此为新的起点,谷禾将坚定依托这套获得国家级认可的标准化检测体系,以扎实的技术积累和持续的体系建设,推动微生物组检测服务向更加规范、可靠、可持续的方向发展。
谷禾将用这份穿越周期的确定性,为国内外顶尖科研机构的多中心大队列研究、高分顶级临床文献的发表、精准个性化微生态干预方案的制定,乃至下游创新型产品开发,提供精准、可靠、经得起全球科学界与监管机构反复推敲的高质量底层数据支撑。
把看不见的标准
做进看得见的结果里
谷禾,与前沿科学同频
始终与全产业链伙伴同行
编辑

谷禾健康
准确的饮食评估是营养流行病学、临床营养学及公共卫生计划的基石,对于减轻全球饮食相关慢性疾病(如肥胖、三高)至关重要。
然而,传统的评估工具(如食物频率问卷、24小时回忆法和称重食物记录)长期面临着参与者负担重、数据偏差以及可扩展性有限等问题,这些局限性降低了营养数据的可用性和准确性。
人工智能与大型语言模型的兴起
近年来,人工智能(AI),特别是自然语言处理(NLP)和大型语言模型(LLMs)的飞速发展,为自动化营养评估提供了新的契机。以GPT、Claude、Gemini为代表的先进模型,具备了处理复杂自由文本、进行高通量食品分类及提供个性化营养建议的潜力。
编辑
然而,目前的验证研究主要集中在英语数据集上,遇到像波兰语这样语法复杂的语言,它们还能不能这么灵光? 此外,现实生活远比实验室复杂。比如在养老院里,饮食数据往往包含复杂的菜单外食品,要让AI准确识别这些带有强烈文化特色、又没写在标准食谱上的东西,对它的语言天赋和应变能力绝对是个巨大的考验。
分类框架的整合:NOVA与WHO指南
饮食质量的评估通常依赖于互补的框架:既要看食品是如何加工的(NOVA系统),又要看具体的营养成分是否超标(如糖、脂肪和钠),即参照世界卫生组织(WHO)的指南。
编辑
虽然这两大标准是全球饮食指南的基础,但直接让AI照搬却很困难,因为两者在定义上时有模糊,甚至相互打架。尤其是NOVA系统有时过于简单粗暴(非黑即白),无法完全反映食品的复杂性。因此,我们需要一种能同时考量加工深度和营养密度的混合系统,从而让AI做出更精准的判断。
为了填补这一空白,来自波兰的华沙医科大学Aia等人的研究团队利用一份包含1992种食品的波兰语纵向数据集,对目前市面上三款顶流AI模型——Claude Opus 4.5、Gemini 3 Pro 和 GPT-5.1-chat-latest,设了两场考试:一场是死磕标准的硬核测试(结合NOVA和WHO双重标准),一场是凭直觉的快速测试。
编辑
结果很明确,虽然AI面对复杂的波兰语还不能完全独立行医,但只要有人类专家的把关,AI完全有能力成为临床和公共卫生营养评估中的得力助手,让大规模的健康饮食管理变得既快又好。
本文我们一起来详细了解一下这项研究的主要设计、AI模型的真实表现、专家如何配合AI实现人机协作的最佳效果。无论你是营养师、AI从业者,还是对健康饮食感兴趣的科研工作者,相信都能从中获得新的启发。
该研究基于华沙医科大学2017–2021年开展的长期护理机构(LTCF)纵向项目数据,已获伦理批准(AK-BE/212/2017)。
原队列约1000名居民,收集人体测量、生物阻抗、体力活动、临床诊断与膳食信息。研究聚焦居民个人橱柜中菜单外食品(由本人购买或家属带入,非机构配餐),共纳入1992条食品记录,数据以波兰语原文处理。
选取三种大型语言模型(Claude Opus 4.5、Gemini 3 Pro、GPT-5.1-chat-latest),均通过API调用,统一温度参数为1.0,以提高可比性并贴近默认使用场景。
设置两种提示策略
① 结构化双步提示
1
按NOVA识别超加工食品(UPF),一经判定即归为不健康;
2
仅对第1步判为健康的条目,按WHO阈值评估游离糖(>总能量10%)、饱和脂肪(>10%)或钠(>2 g/日),超标则改判不健康。
模型需输出每个产品的名称、重量(g)、热量与二分类结果,并分别汇总两类总热量。
② 简化单步提示
仅要求对每个产品给出健康/不健康判断及简短理由(两列:evaluation与description)。
人工参照标准由两位专家完成:
统计分析以Pearson卡方检验(α=0.05)进行多组两两比较(模型-专家、模型间、提示策略间及“多模型共识”主导类别)。同时以不健康为阳性,基于混淆矩阵计算Accuracy、Precision、Recall、F1与Specificity,评估不同提示与模型的错误模式。
编辑
文中会出现的一个词:Dominant,指的是多模型共识(consensus response across LLMs),你可以理解为对多个模型的结果采用了投票机制,如果大多数模型对某一项食品都给出了相同的分类,那么这个分类就被认为是“Dominant”分类,即多模型共识的分类结果,有助于减少单个LLM的偏见,同时保持保守的风险评估。
1. 结构化双步提示结果
★ 核心结论
三种模型与专家一致性都很高(约90–91%);同时呈现更保守的倾向,也就是说更擅长抓出不健康(Recall很高),但更容易把健康误判为不健康(Specificity下降)。
双步提示下 LLM vs 专家的一致性分布
(占总样本1992的百分比)
编辑
双步提示下的混淆矩阵指标
(UNHEALTHY为阳性)
编辑
怎么看这组结果?
双步提示把规则写死(先NOVA判UPF直接不健康,再用WHO阈值复筛),所以模型更倾向于宁可判不健康也不漏掉,表现为 Recall极高(0.963–0.982)。
代价是 Specificity偏低(0.798–0.844),也就是说有一部分其实健康的条目被判成不健康(假阳性更多)。
2. 简化单步提示结果
★ 核心结论
简化提示下,总体一致率更高(约92.5–94.2%),并且 Specificity明显提升,Recall仍保持较强,整体更像专家的整体判断风格。
同时,作者指出:在简化提示下,模型整体会把更多条目判为健康(相比双步提示),显示规则约束变少后,模型不再那么保守。
简化提示下 LLM vs 专家的一致性分布
(占总样本1992的百分比)
编辑
简化提示下的混淆矩阵指标
(UNHEALTHY为阳性)
编辑
怎么看这组结果?
相比双步提示,简化提示的 Accuracy更高(0.927–0.942),并且 Specificity显著提高(0.897–0.951):更少把健康误判为不健康。
Recall略有回落但仍高(0.909–0.964),整体更均衡。
Dominant在简化提示下表现最好/接近最好(Accuracy 0.942,F1 0.949)。
这提示简化单步策略在召回率和特异性之间找到了一个更好的平衡点,使得模型的分类结果既能较好地识别不健康食品,也能较好地识别健康食品,减少了误报或漏报的情况。
3. 跨模型、跨提示词的整体差异(卡方检验)
为了评估提示结构对不同LLM及人类专家之间的一致性和差异性,使用Pearson’s Chi-square检验,对所有可能的成对比较(36对)进行统计显著性测试。
关键发现
所有两两比较均显著不同:
χ2=1174.5 –1897.1,p < 0.001
编辑
说明即使总体一致率很高,不同模型/不同提示词仍会导致系统性的分类分布差异(不只是随机波动)。
文中还给了两个极值例子:
各模型与专家总一致率(双步 vs 简化)
编辑
尽管多模型共识(Dominant)在一些指标上接近或略优于最佳单一模型,但与专家的判断仍存在显著差异,提示共识并不能完全替代专家判断,尤其在边缘案例上。这些差异也体现出提示词设计的重要性。
当前人工智能所展示的膳食分类的能力,虽然接近人类专家的水平,但无法完全替代专家,适合做初筛和前处理,可以用不确定性阈值触发强制人工复核。
未来的优化方向在于提示词设计、多语言本地化、多模态数据融合(例如包装、配料表的图片等)、混合工作流开发(AI+人类专家)、纵向验证等。
主要参考文献
Ase, A.; Borowicz, J.; Rakocy, K.; Piekarska, B. Large Language Models for Real-World Nutrition Assessment: Structured Prompts, Multi-Model Validation and Expert Oversight. Nutrients 2026, 18, 23. doi.org/10.3390/nu18010023

谷禾健康
如果你正为宏基因组数据的组装和注释而忙于“拼工具、调环境、转格式”,那么annoSnake或许能让你从繁琐中解放。
它是一个基于Snakemake的自动化工作流程,从clean reads组装到物种分类、功能注释,再到MAGs的装配和注释。
作为开源工具,annoSnake具备良好的可重复性、可扩展性和可移植性,非常适合HPC集群环境。
本文将带你了解它的工作流程、在白蚁肠道宏基因组数据上的验证结果,以及它的优势与局限,帮助你快速判断是否值得上手。
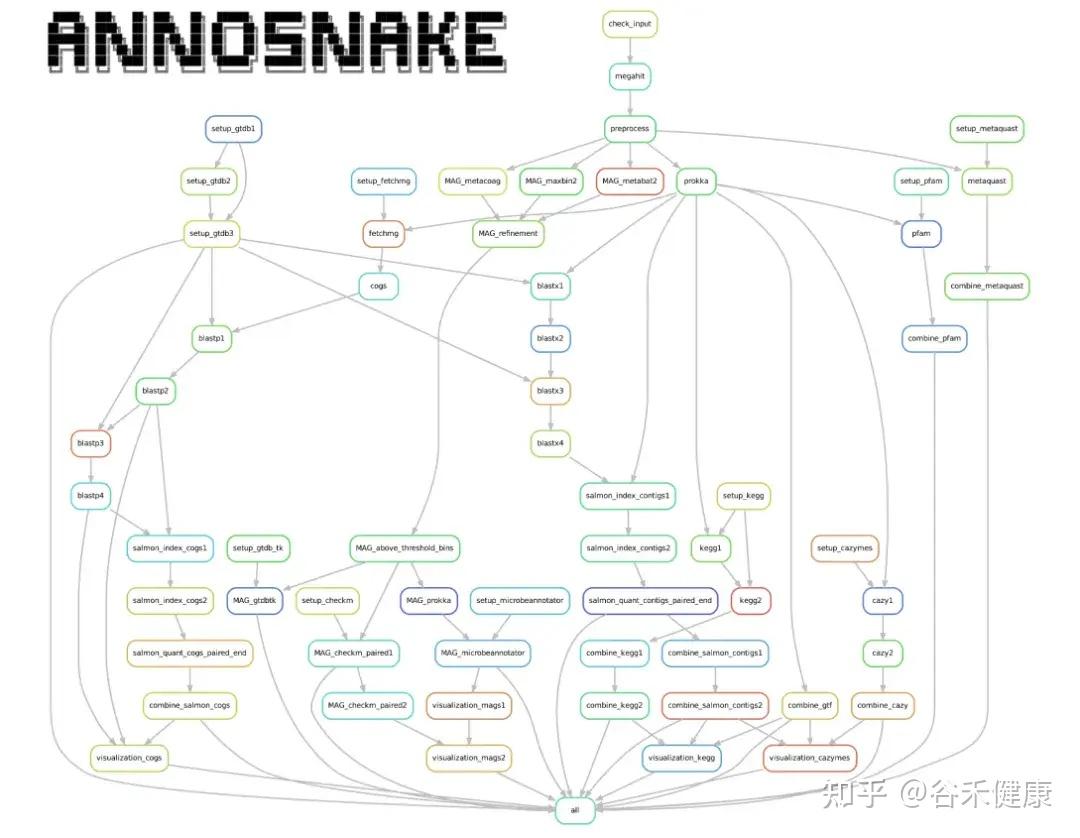
annoSnake以“自动化+模块化”为核心:输入clean reads → 组装 → 注释 → 分箱 → 结果汇总与可视化。
每个标准化步骤里,annoSnake使用的都是主流工具,如果你事先没有任何准备,也无需担心,它会自动创建独立的虚拟环境,并安装所需的分析工具和注释用的数据库。
分析前的准备
1. Mac OS或linux系统设备,磁盘空间推荐>100GB。如果运行单个宏基因组样本,只要有32 GB 内存和 8 核 CPU 基本就能跑完。
若要在集群上批量运行十几个样本或进行MAG分析,最好准备 ≥128 GB 内存和多核服务器。annoSnake 可批量化处理。
2. 安装mamba或conda用于管理环境,然后安装snakemake。
3.克隆Github仓库到本地(git clone https://github.com/bheimbu/annoSnake.git)。
4. 清洗后的测序数据,可以是双端,也可以是交错合并的fastq.gz文件,注意要是gzip格式。
5.编辑./profile/params.yaml和./profile/config.yaml文件。config.yaml 决定“要做什么”与“怎么做”,config.yaml 决定“在哪跑、分配多少资源”。
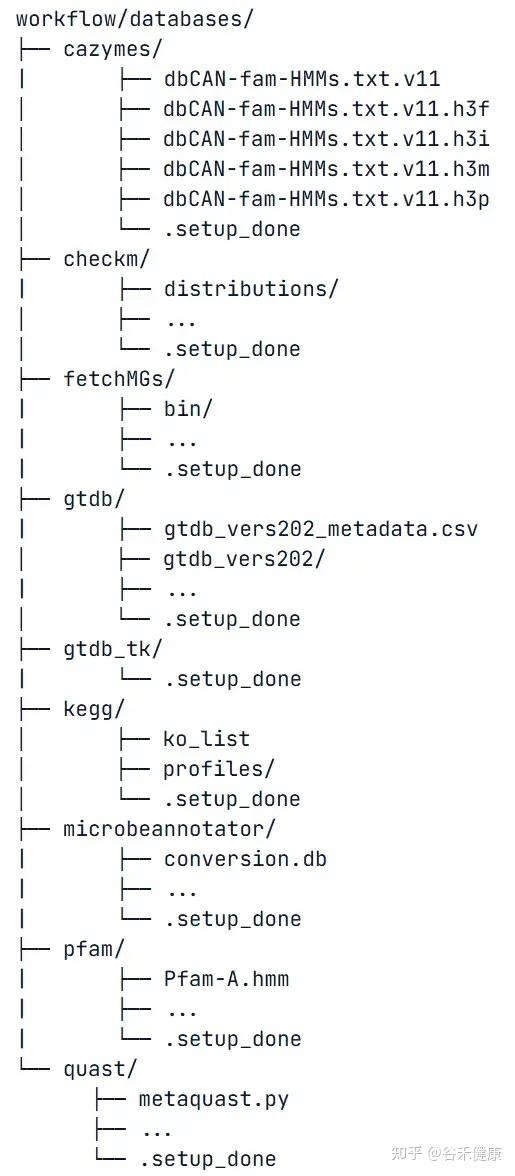
首次分析,annotSnake会自动下载并设置GTDB、dbCAN、Pfam、KEGG等数据库,总量约100GB。
开始分析
1. 组装
MEGAHIT v1.2.9工具进行宏基因组组装,默认–presets meta-sensitive模式组装,保留≥1500 bp的contigs,并以metaQuast评估组装质量。
2. 物种分类注释
Prokka v1.14.6工具识别CDS、rRNA、tRNA;fetchMG v1.2提取40个单拷贝标记基因;结合GTDB(v202, (Parks et al.2022)数据库进行blastp和blastx注释;自定义R脚本gtdb_diamondlca.R进行LCA分类整合。
3. 功能注释
对细菌/古菌的contig执行注释,可以选择的功能数据库有:CAZy(dbCAN version 11)、Pfam(version 35)和KEGG。
针对Pfam搜索结果,可以自行借助在线工具HydDB进一步分类。针对KEGG结果,借助KofamScan工具重建以KEGG为基础的代谢通路。E-values阈值在params.yaml中设定。
4. 基因丰度量化与归一化
Salmon v1.10.2对CDS进行TPM定量,对于TPM>1的,予以保留,然后对剩余TPM做CLR对数转换(默认,log(TPM+0.65))。
5. 分箱与注释(可选)
同是采用三种分箱算法:MetaBAT v2.10.2、MetaCoAG v1.1.1、MaxBin v2.2.7,最后用metaWRAP v1.3的bin_refinement整合最优集合,CheckM 评估MAGs质量,默认阈值是完整性≥50%且污染≤10%。
对优质的MAGs使用GTDB-Tk v2.3.2进行物种分类(数据库v214),Prokka做基因预测,然后用MicrobeAnnotator进行功能注释,该工具使用DIAMOND和KofamScan,并以通路基因存在/缺失评估完整性。
6. 输出与可视化
输出包括CSV表格和ggplot2/plotly生成的PDF/HTML图表。
!
Tips
annoSnake已经内置了“KEGG条目(KO编号)→基因名称/通路名称”的映射表文件。如果你想重点关注某些特定的KEGG基因或通路(比如只看甲烷生成、乙酸生成或硫酸盐还原等),可以在/workflow/rules/scripts目录下,直接编辑这类映射文件,把你关心的KO编号及其基因名、通路名加入或调整。
管道运行时会按你改过的清单去批量检索与汇总这些目标基因/通路的注释与丰度,并在输出图表中优先呈现,从而实现“按课题定制”的结果视图。
作者用来自澳大利亚Amitermes组(AAG)的白蚁肠道宏基因组作为测试数据,与已知发现进行一致性检验。
▸在测序深度不高时仍能识别主要细菌谱系和代谢通路
31个群体,Illumina NextSeq双端,平均每样本约700万条reads。尽管测序深度不高,但annoSnake仍有效识别了主要的细菌谱系,以及大量与木质纤维素消化相关的代谢通路和基因。
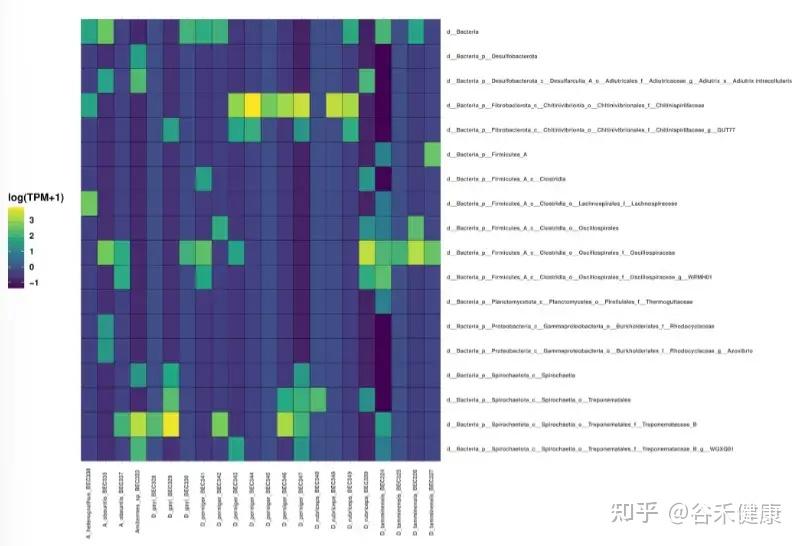
图中是annoSnake识别出的主要菌群,结果显示不同取食类型白蚁肠道的优势类群模式,这与已知发现相符,通常,食草和食木的白蚁其肠道群落以螺旋体为主,而食腐殖质和土壤的白蚁则富含梭菌(clostridia)。
但也有与已知发现不符的结果,D. tamminensis物种的肠道群落以梭菌为主,几乎不见螺旋体或Fibrobacterota,部分D. gayi群体也表现出类似模式,这与“草/木料取食类型的白蚁常以螺旋体占优”的普遍模式不一致,作者解释这是数据特性使然,低覆盖度数据只能恢复高丰度群落成员,而非物种生态学的结论。
▸ 识别出硫酸盐还原等重要通路的基因
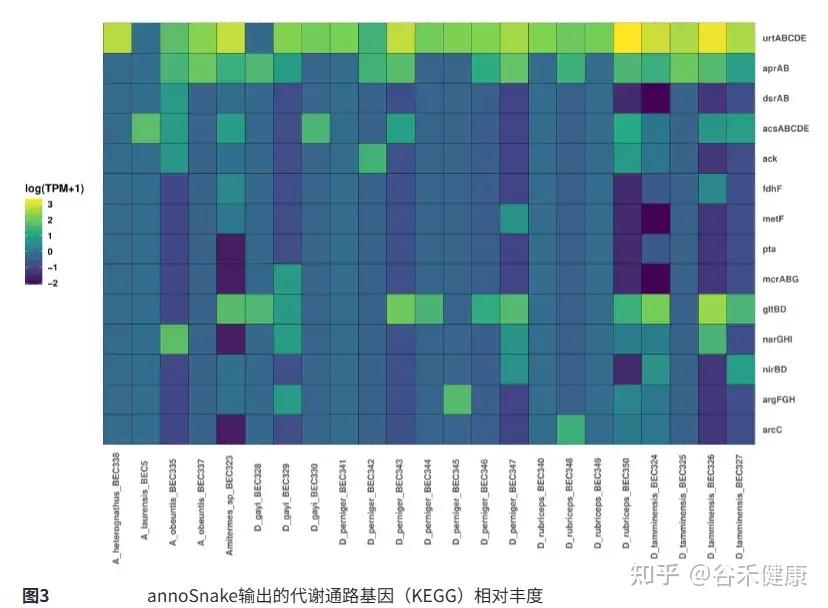
在所有样本中,annoSnake识别出硫酸盐还原通路的关键基因,如aprA、aprB和dsrAB,这符合白蚁肠道微生物组中硫酸盐还原过程的常见模式。还有许多与木质纤维素消化相关的KEGG代谢通路基因,这支持白蚁肠道微生物组在碳循环和能量代谢方面的普遍功能特征。
仅检测到少量与甲烷生成相关的基因,如mcrABG。已知甲烷生成主要局限于厌氧甲烷生成古菌,而本次分析中未有样本被检测到古菌,所以甲烷生成基因稀少是符合预期的。这与低覆盖度数据仅恢复高丰度群落成员的特性一致,古菌可能被低估,而不是KEGG注释的偏差。
识别出fdhF和acsABCDE等基因预测还原性乙酸生成的存在,这一点由分箱得到的15个Bacillota和6个螺旋体MAGs所支持,这两类群包含潜在的乙酸生成菌。这与其他白蚁和千足虫研究中已知的乙酸生成潜力一致。
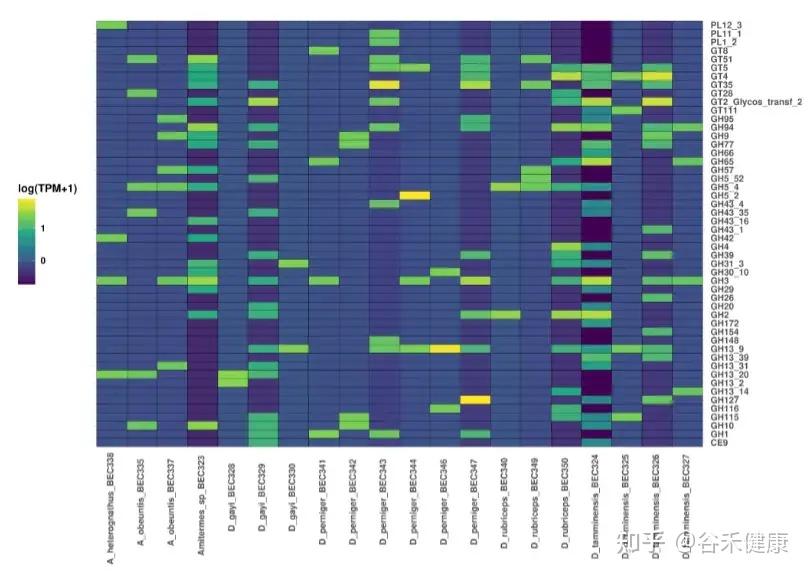
▸ 能够检测到大量碳水化合物活性酶
annoSnake检测到大量CAZymes(碳水化合物活性酶)。D. tamminensis在不同群体间的GHs丰度差异再次暗示饮食灵活性,且部分群体GHs模式与腐殖/土壤取食物种一致;螺旋体主导的D_gayi_BEC329中GHs丰富,符合凋落物取食物种的特性,而在梭菌主导的D. gayi群体中GHs较低。
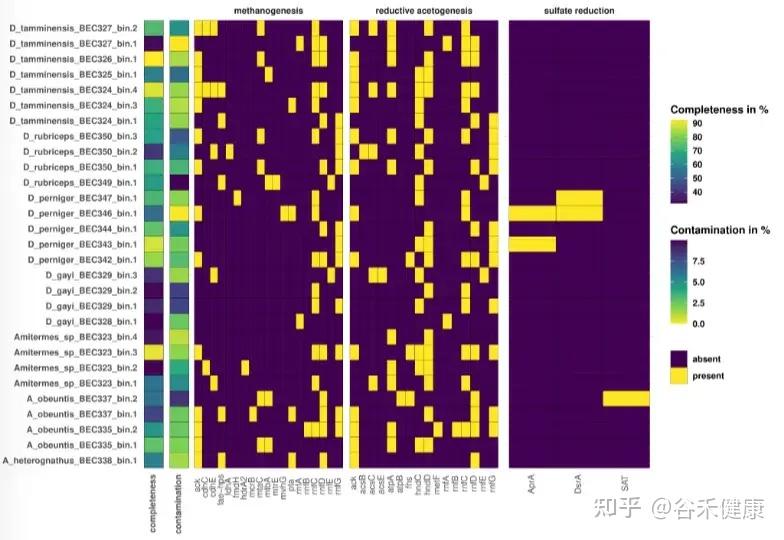
annoSnake从低覆盖度数据中获得30个MAGs,其中包括15个Bacillota(内含大量梭菌纲)、1个Desulfobacterota、7个Fibrobacterota、1个Pseudomonadota、6个Spirochaetota。
图中展示了MAGs中木质纤维素消化相关代谢途径(甲烷生成、还原性乙酸生成、硫酸盐还原)基因的存在/缺失。左侧给出MAG的完整性和污染分值,颜色越浅表示完整性越高,污染越少。 紫色方块表示基因缺失,黄色方块表示基因存在。
优势
• 覆盖全流程的一站式自动化:从输入reads→组装→物种注释→功能注释→丰度定量→分箱(可选)→可视化,节省操作时间。
• 数据库自动下载和配置。
• 兼具一些灵活性,比如可以自定义数据库,也能调整分析参数。
• 可重复、可扩展、可移植。Snakemake内核+HPC优化,可以在不同HPC环境中高效执行。
劣势
• 资源占用较高,数据库体量约100GB,完整流程在大规模数据上更适合HPC环境;本地轻量设备可能受限于存储、内存与时长。
• 需要有一定代码基础,掌握Snakemake、Conda与YAML配置,能调试环境配置时可能出现的错误。
• 范围聚焦细菌/古菌,真核生物未被纳入默认流程,氢化酶精细亚型分类需借助HydDB等外部工具,未在管道集成。
• 低覆盖度数据的固有限制:对稀有类群的恢复能力受限,更偏向于恢复高丰度成员,需要结合研究设计与深度规划权衡。但作者也没有发表对高覆盖度数据的测试结果,所以工具对高覆盖度数据的表现不明确。
• 工具较新,容易出现环境/兼容性问题或边缘情况未覆盖;第三方依赖更新也可能引入不稳定性。数据库管理灵活性受限,版本固定且无更新管道。虽支持自定义数据库,但需自行调整文件格式。
o 输出的图像不够美观,可视化类型单一。
annoSnake适合具备中级生信技能,需快速产出的微生物组学研究者。如样本量大,需批量分析,则需要配备高性能设备。研究范围在细菌/古菌的宏基因组与MAGs。
下面这个网址可访问 annoSnake 文档:
https://annosnake.readthedocs.io/en/latest/index.html
参考文献:
Bastian Heimburger, Rebecca Clement, Tamara R. Hartke
bioRxiv 2025.11.03.686227; doi: https://doi.org/10.1101/2025.11
谷禾健康
报告更新通知
各位合作伙伴,2025-08-25开始出具的报告,以下疾病模型有较大更新,胃病、胆病、肝病、甲状腺疾病、II型糖尿病、肺部疾病,大幅增加了临床病例样本数量,改善了检出率,部分疾病的假阳性也大幅改善了。另外疾病的打分已经过优化,尽可能与之前的打分相匹配。
II型糖尿病:
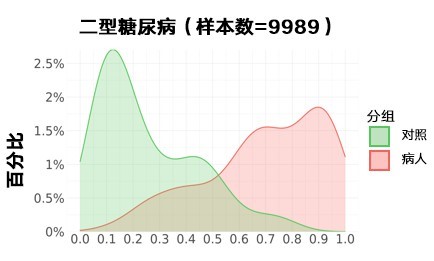
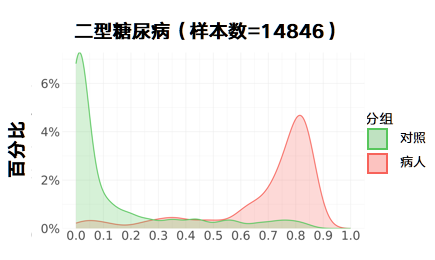
更新后:
胆病:
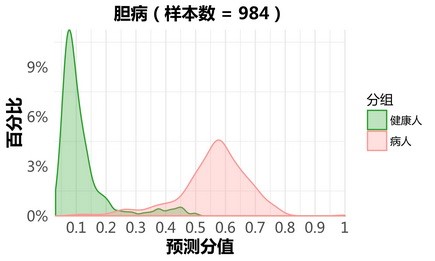
更新后:
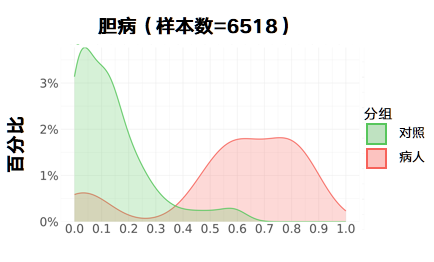
胃病:
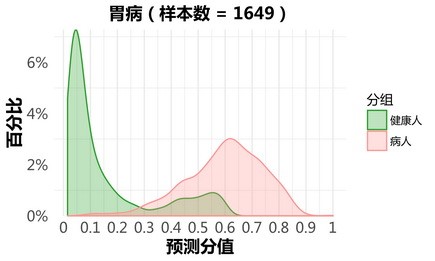
更新后
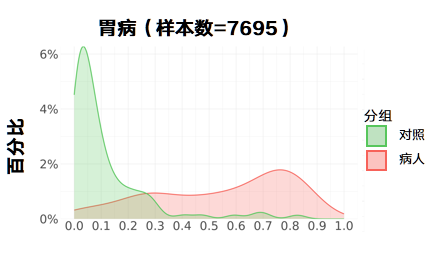
肝病:
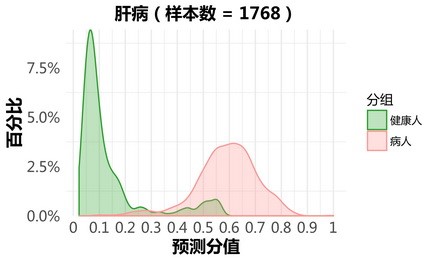
更新后:
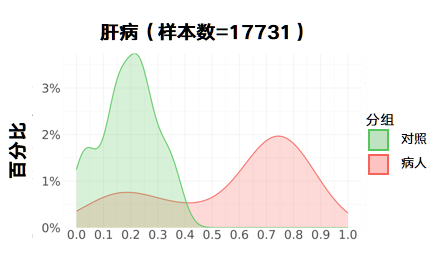
肺部疾病:
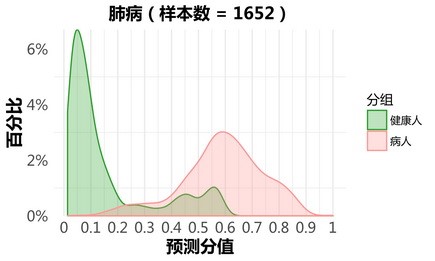
更新后:
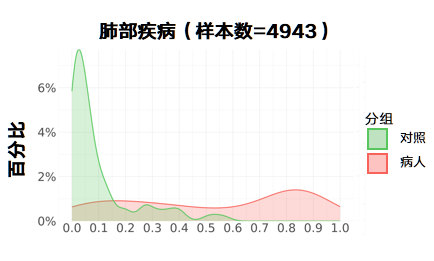
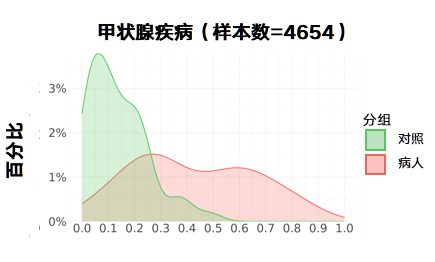
甲状腺疾病:
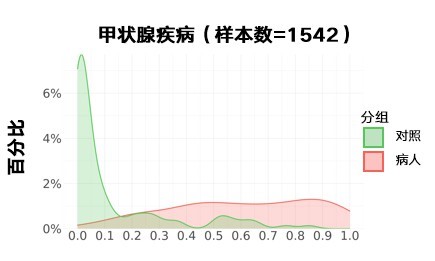
更新后:
杭州谷禾信息技术有限公司
2025-08-25

谷禾健康
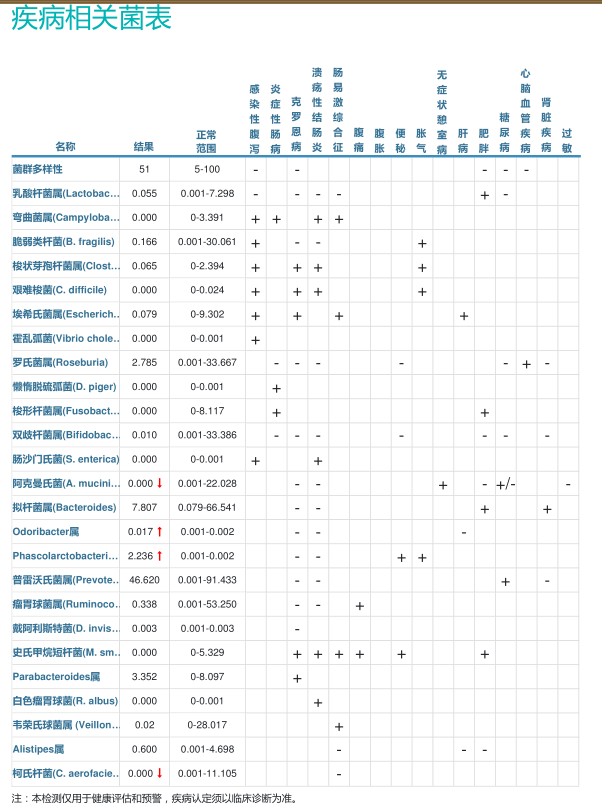
肠道菌群检测临床版(16S rRNA测序),作为谷禾健康最早推向市场、历经十余年打磨的经典产品,凭借其成本效益、高效性与成熟度,为临床端肠道微生态评估以及诸多科研项目基线建立,提供了坚实、可靠且极具价值的数据基础(注:仅用于菌群科学研究和辅助参考,不直接用于临床诊断 )。

临床版对报告版式进行简化,以更符合临床检验的形式呈现分析结果,便于临床医生快速查看和判断异常。
临床版是主要面向临床和医疗机构的版本,主要用于临床科室已有明确症状或诊断,需要对肠道菌群进行进一步分析以为临床提供辅助判断。
该版本减少了基本介绍和文字说明,并对部分指标的异常判定范围和计算方式进行调整,更加适应临床需求。
报告内容截图:


谷禾健康

随着“它经济”的蓬勃发展和“科学养宠”理念的深入人心,宠物已成为家庭的重要成员。然而,面对“毛孩子”们无法言说的病痛,如反复腹泻、顽固皮肤病、食欲不振、呕吐等,传统兽医诊断往往面临挑战。
从宠物医院的实际经营来看,慢性疾病正成为他们面临的核心挑战。慢性肾病、老年痴呆、精神类疾病等病症不仅治疗费用昂贵,而且现有手段往往无法覆盖,特别是小型诊所更是心有余而力不足。
在与许多B端合作伙伴的深度交流中我们发现
宠物腹泻,肾病以及其他疾病等正在增加
后期医疗费用高昂让宠物主人无能为力
异常行为严重影响生活质量和主人养宠体验
情感难舍却不得不放弃…
因此,迫切需要一种更加
科学、经济、精准的健康管理方式
既能降低医疗成本
又能提供个性化的健康方案
还能避免过度医疗
这就需要我们从根本上
重新思考宠物健康管理的方法论
从”治疗导向“转向”预防导向“
✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲
谷禾凭借在人类健康领域积累的深厚微生物组学经验,战略性延伸至动物医学领域,推出宠物菌群精准检测服务。
我们致力于解码宠物肠道微生态的奥秘,为兽医临床、宠物营养和家庭养护提供科学依据,开启宠物健康管理的精准化新时代。

从人类微生态到宠物微生态,不是简单的复制,而是技术能力的升维应用。人类肠道微生态的复杂性研究为我们提供了强大的算法基础和数据分析能力,以及多年来在宠物菌群科研中的持续投入,这些经验在宠物领域的应用中展现出了独特的技术优势。
研究表明,宠物肠道菌群不仅影响消化吸收功能,更与免疫调节、神经系统、皮肤健康等多个生理系统密切相关,成为宠物整体健康状况的重要晴雨表。
谷禾正有序推进构建涵盖不同品种、年龄、健康状况犬猫的肠道菌群数据库,结合最新的机器学习算法,实现对宠物肠道微生态健康状况和营养进行精准评估(注:仅用于菌群科学研究和辅助参考,不直接用于临床诊断 )。
宠物菌群报告展示采用更温馨活泼的配色,通过可视化图表和情感化设计,让复杂数据更直观,帮助主人轻松了解爱宠健康。
谷禾宠物菌群检测报告中包括菌群评估(整体指标)、肠道基础功能评估(屏障功能、炎症水平、代谢状态)、菌群代谢物评估(短链脂肪酸等)、炎症免疫评估(促炎、抗炎等指标)、营养饮食评估(维生素、微量元素)等。
…
…
症状相关菌群分析,包括腹泻、呕吐、过敏等。
…
菌群代谢物评估(短链脂肪酸等)。
…
…
从多维度全面评估宠物肠道健康状况,为宠物主人提供科学的健康管理依据和个性化调理建议。


谷禾健康
在肠道菌群检测系列产品成熟之后,谷禾健康将深耕多年的微生物组学技术平台,延伸至关乎女性全生命周期健康的另一核心领域——阴道微生态。
还包括子宫颈沙眼衣原体、HPV、HSV、EB病毒、巨细胞病毒等。
谷禾阴道菌群检测报告引入科学前沿的菌群状态分型(CST)概念,将复杂的菌群构成归纳为几种易于理解的健康状态类型。
例如,以卷曲乳杆菌为主的CST-I型代表健康的稳定状态,而以加德纳菌等多种厌氧菌为主的CST-IV型则与细菌性阴道病高度相关。这为临床判断和干预效果评估提供了科学支持。
谷禾阴道菌群检测报告中包括阴道菌群总体评估、CST分型、致病菌表(细菌性阴道病,需氧菌性阴道炎,外阴念珠菌病等)、列出异常菌群及相关说明,菌群详细构成等(注:仅用于菌群科学研究和辅助参考,不直接用于临床诊断)。
…
…
…
…
阴道菌群检测让我们能够更全面地了解阴道微生物组的组成及其变化,以及它是如何随着时间的推移或对各种因素(如环境、激素变化、性活动和抗生素使用等)的反应而变化的。
注:本产品可辅助评估和筛查,不用作临床诊断。
05

谷禾健康
谷禾16S+tNGS技术结合了超多重PCR和高通量测序的优势,旨在提供比传统16S rRNA测序和宏基因组测序更优、更全面的病原体及耐药基因检测方案。

传统16S + 病原体精准分型 = 全新升级
它在保留16S报告的基础上
以接近16S的成本和周期
用靶向测序技术在原16S的基础上
增加了125 种消化道病原体的检测
还包括耐药基因和毒力基因等
如幽门螺杆菌、大肠杆菌、艰难梭菌分型
弥补了传统16S无法检测
非细菌/古菌病原体的不足
这是一款突破传统16S检测瓶颈而生的产品
值得一提的是,谷禾在tNGS技术的研发道路上并非一帆风顺。这项看似成熟的技术,在不同应用场景下却面临着截然不同的挑战难度。
tNGS对血液和上呼吸道样本检测较简单,因其主要含病原体。但在消化道特别是粪便样本检测时,技术难度大幅增加。
经过无数次的实验优化和迭代
我们最终突破了这一技术瓶颈
为大家带来真正可靠的
消化道病原体检测解决方案
谷禾16S+tNGS产品特点
弥补16S检测技术边界
实现靶向病原体精准检测
谷禾16S+tNGS报告内容涵盖所有16S报告的内容,即包括健康总分评估、慢病风险预警、肠道屏障及代谢物、神经递质分析、个性化营养评估等。此外还包括常见消化道病原体(注:仅用于菌群科学研究和辅助参考,不直接用于临床诊断)。
例如:
细菌病原体
…
病毒
真菌、寄生虫、其他病原体
…
毒力基因
…
耐药基因
…

也包括相关病原微生物的解释
…
例如,通过检测幽门螺杆菌毒力基因组合,可判断是否需立即治疗,避免对弱毒株患者的不必要抗生素使用。强毒株感染会损伤胃黏膜,增加胃炎和溃疡风险,早期预警能在胃黏膜不可逆损伤前提供治疗窗口期。
注: 由于该技术是检测粪便中的幽门螺杆菌,当浓度低于检测下限(50 copies/mL)时,可能出现假阴性结果。因此,对于临床症状明显但检测结果为阴性的患者,建议结合其他检测方法。

开始写作或按/来选择区块

谷禾健康

目前随着高通量测序,微生物以及代谢、蛋白组学和高精度影像学等快速的发展,有关人体疾病或健康相关的海量数据正在不断产生,依托各种算法尤其AI的算法等,涌现出很多优秀的“模型”用于预测人体疾病以及健康相关的辅助诊断或筛查。
谷禾在人体肠道菌群检测领域深耕多年,积累了大量肠道菌群和个体健康信息数据,并运用建模方法结合人工智能等算法构建了多种疾病和营养预测模型。
对于不是从事专业的人员或大众来说,什么样的预测模型是准确的?如何评价一个模型的好坏?里面涉及比较多的概念和知识,今天谷禾和大家简单聊下这方面的内容。
首先大家评估下:
对于肿瘤筛查,以一个人群发病率1%的肿瘤为例:
模型A的检出率50%,假阳性为1%
模型B的检出率为90%,但是假阳性是10%
请问哪个检测模型作为肿瘤筛查类产品更好 ?
换句话说就是回答:“鉴于测试结果为阳性,该患者患有这种疾病的可能性有多大?
为了回答上面问题,我们需要了解和正确认识以下模型评价参数/指标:
• 该测试的阳性预测值(PPV)是多少?——返回阳性结果的人实际上患有疾病的概率是多少?
• 该测试的阴性预测值(NPV)是多少? ——返回阴性结果的人实际上是健康的概率是多少?
• 测试的灵敏度如何?——它正确地将多少个患病个体识别为患病?
• 测试的特异性是什么?——它正确地将多少个健康个体识别为健康个体?
要回答这个问题,我们需要计算两个模型的阳性预测值(Positive Predictive Value, PPV)。然后一步步分析:
▸ 什么是阳性预测值(PPV)?
阳性预测值(PPV)是样本检测阳性为真阳性的概率值。
发病率较低时的阳性预测值(PPV)
模型A
人群发病率:1%
检出率:50%
假阳性率:1%
模型B
人群发病率:1%
检出率:90%
假阳性率:10%
接下来使用贝叶斯定理来计算每个模型的阳性预测值。
对于模型A:
真阳性率 = 1% × 50% = 0.5%
假阳性率 = 99% × 1% = 0.99%
总阳性结果 = 真阳性 + 假阳性 = 0.5% + 0.99% = 1.49%
阳性预测值(PPV) = 真阳性 ÷ 总阳性结果 = 0.5% ÷ 1.49% ≈ 33.6%
对于模型B:
真阳性率 = 1% × 90% = 0.9%
假阳性率 = 99% × 10% = 9.9%
总阳性结果 = 真阳性 + 假阳性 = 0.9% + 9.9% = 10.8%
阳性预测值(PPV) = 真阳性 ÷ 总阳性结果 = 0.9% ÷ 10.8% ≈ 8.3%
模型A的PPV为33.6%,而模型B的PPV仅为8.3%。
因此,我们可以看到尽管模型B的检出率更高(90%vs50%),但其阳性预测值(PPV)显著低于模型A。
★ 低发病率人群中,高特异性比高敏感性更重要
这意味着:
模型A的阳性结果更可靠
模型A的假阳性风险更低
模型A在这种低发病率的情况下,作为肿瘤筛查产品更好。
这里对于我们应用的启示:在低发病率的人群中,高特异性(低假阳性率)比高敏感性(高检出率)更重要。选择产品时,不仅要看检出率,还要关注假阳性率和阳性预测值。

发病率较高的阳性预测值(PPV)
可能接下来有人会问,这个问题中以肿瘤发病率比较低的例子举例,那对于发病率比较高的疾病,比如心脑血管、糖尿病等如何?
接下来,我们继续举例:
让我们用同样的方法计算发病率为20%时的阳性预测值(PPV)。
模型A
人群发病率:20%
检出率:50%
假阳性率:1%
模型B
人群发病率:20%
检出率:90%
假阳性率:10%
一样的计算
对于模型A:
真阳性率 = 20% × 50% = 10%
假阳性率 = 80% × 1% = 0.8%
总阳性结果 = 真阳性 + 假阳性 = 10% + 0.8% = 10.8%
阳性预测值(PPV) = 真阳性 ÷ 总阳性结果 = 10% ÷ 10.8% ≈ 92.6%
对于模型B:
真阳性率 = 20% × 90% = 18%
假阳性率 = 80% × 10% = 8%
总阳性结果 = 真阳性 + 假阳性 = 18% + 8% = 26%
阳性预测值(PPV) = 真阳性 ÷ 总阳性结果 = 18% ÷ 26% ≈ 69.2%
可以看到当发病率提高到20%时,情况发生了显著变化:
模型A的PPV提高到92.6%
模型B的PPV提高到69.2%
对比之前1%发病率的情况:
模型A:PPV从33.6%提高到92.6%
模型B:PPV从8.3%提高到69.2%
可以发现随着发病率的提高,两个模型的阳性预测值都大幅提升。但是模型A仍然保持更高的PPV,但优势不如低发病率时那么明显。

★ 发病率越高,阳性预测值越高
发病率越高,阳性预测值越高,这是因为真阳性的比例增加。
因此在高发病率的人群中,两种模型的检测价值都显著提高。但模型A仍然是更好的选择,因为其假阳性率更低。
评估不同检测模型和询问检测准确率时,要综合考虑不同疾病的发病率、检出率和假阳性率。
对于要检出阳性结果而言,这个例子就很好地说明了发病率对检测结果解读的重要影响,也体现了贝叶斯定理在医学诊断中的应用。
而阳性预测值(PPV)具有多维意义,对于不同视角的人群可能意义不同:
➤ 政府视角下的PPV
⑴公共卫生决策基础
资源分配效率:高PPV意味着确诊投入的资源浪费更少,每发现一个真实病例的成本更低;
筛查项目评估:决定是否推行大规模筛查项目时,PPV是核心考量因素之一;
卫生经济学分析:假阳性导致的后续检查、治疗和心理干预成本需纳入卫生政策评估。
⑵监管与标准制定
差异化监管:对不同严重程度疾病的筛查产品可设置不同PPV要求;
强制信息披露:要求厂商在不同疾病流行率下公开产品的PPV值;
患者保护:防止低PPV产品导致过度医疗和不必要的医源性伤害。
➤ 受检者视角下的PPV
⑴个人决策与心理影响
决策参考价值:“我的阳性结果有多可靠?”—这是患者最关心的问题;
心理负担差异:假阳性可能导致严重心理压力,尤其对严重疾病筛查;
后续检查意愿:了解PPV有助于患者决定是否及时进行确诊检查。
⑵风险认知与理解
个体化解读:同一PPV值对不同风险人群的意义不同;
检前概率影响:有症状个体的PPV通常高于无症状筛查人群;
教育需求:医生需帮助患者理解PPV与个人情况的关联。
➤ 产品评估的情境差异
⑴临床应用场景分析
筛查工具:可接受相对较低PPV,但应有明确后续路径;
确诊工具:需要更高PPV,减少误诊风险;
监测工具:对症状性疾病监测,PPV要求可能介于两者之间。
⑵疾病特性影响
致命但可治疗疾病:可接受较低PPV,以高灵敏度优先;
慢性管理疾病:需较高PPV避免不必要长期干预;
高耻感疾病:需更高PPV避免标签化伤害。
⑶社会经济环境考量
医疗资源丰富地区:可能更关注灵敏度,接受较低PPV;
资源有限环境:高PPV更为重要,避免资源浪费;
支付方式影响:商业保险vs自费vs政府支付下对PPV的需求不同。
➤ PPV优化与应用建议
⑴预测模型调整
风险分层应用:根据人群特征调整决策阈值;
多指标综合评估:结合阴性预测值(NPV)、阳性/阴性似然比(LR+/LR-)综合评估;
贝叶斯思维应用:根据先验概率(患病率)调整PPV期望。
⑵实际应用最佳实践
透明沟通:向受检者清晰解释结果可靠性;
分级报告:提供风险概率而非简单阳性/阴性结果;
智能决策支持:AI辅助工具结合临床特征提供个性化PPV估计。
那么下面我们看下什么是阴性预测值(NPV),简言之就是把健康人检测为健康的概率。
阳性预测值(PPV)是样本检测阳性为真阳性的概率值,而阴性预测值(NPV)是样本检测真阴性的概率值。
阴性预测值(NPV) =(真阴性数量)/(真阴性数量+假阴性数量)
例如你有一个600人的样本量,根据有效性,假设你知道肯定患有这种疾病的样本(480)或没有这种疾病的个体样本(120)。
测试后,将结果与已知的疾病状态进行比较,发现:
真阳性(测试阳性和正确阳性)= 480
假阳性(试验阳性但实际为阴性)= 15
真阴性(测试阴性和真阴性)= 100
假阴性(试验阴性,但实际为阳性)=5
我们可以计算PPV和NPV如下:
阳性预测值(PPV)=480/(480+15)≈0.97(97%)
阴性预测值(NPV)=100/(100+5)≈0.95(95%)
因此,如果检测结果为阳性,则有97%的机会是正确的,如果结果为阴性,则有95%的机会是正确的。
拓展:什么是假阳性和假阴性?
假阳性:健康人被错误地识别为患病;
假阴性:病人被错误地认为是健康;
真阳性:患病的个体已被正确识别为患有疾病;
真阴性:未患有疾病的个体已被正确诊断为未患有疾病。
或:
真阳性:患者患有疾病且检测呈阳性;
假阳性:患者没有疾病,但检测呈阳性;
真阴性:患者没有疾病,检测结果为阴性;
假阴性:患者患有疾病,但检测结果为阴性。
敏感性
敏感性(Sensitivity)是测试正确识别疾病患者的能力,也称为真阳性率(TPR),即被正确识别为患有疾病的患者的百分比。
敏感性的计算公式为:
敏感性 = 真阳性数 / (真阳性数 + 假阴性数)
或者表示为:
敏感性 = TP / (TP+FN)
其中:
TP (True Positive): 真阳性,即测试正确地将患病者判断为阳性的数量;
FN (False Negative): 假阴性,即测试错误地将患病者判断为阴性的数量。
从另一个角度看,敏感性也可以表示为:
敏感性 = 真阳性率 = 1 – 假阴性率
敏感性的值范围在0到1之间,通常以百分比表示。例如,敏感性为0.95或95%意味着测试能够正确识别95%的实际患病者。
★ 高敏感性特别适用排除诊断
在医学诊断中,高敏感性的测试特别适用于排除诊断(rule-out test),因为如果一个高敏感性测试的结果为阴性,则疾病存在的可能性很小(即”阴性结果可靠”)。
例如:表述中敏感性为100%的检测可正确识别所有患者,敏感性为80%的检测可检出80%的疾病患者(真阳性),但20%的疾病患者未被检测到(假阴性)。
当检测到非常严重的感染类型时,例如前两年进行的COVID-19大流行,高敏感性对于适当的管理和治疗非常重要。COVID-19 IgG/IgM诊断测试是一种筛查COVID-19的测试,据报道敏感性为95%。
当测试用于识别严重但可治疗的疾病(例如宫颈癌)时,高敏感性显然很重要。通过宫颈涂片检测筛查女性人群是一项敏感的检测。然而,它不是很特异,很大一部分宫颈涂片阳性的女性继续进行阴道镜检查,最终被发现没有潜在的病变。

特异性
临床试验的特异性是指检测正确识别无病患者的能力。
特异性的计算公式是:
特异性 = 真阴性数 /(真阴性数 + 假阳性数)
Specificity = TN/(TN + FP)
其中:
TN (True Negative): 真阴性,即正确识别出的健康者数量
FP (False Positive): 假阳性,即错误地将健康者判断为患病的数量
特异性也可以表示为: 特异性 = 正确识别的健康者数/所有实际健康者总数
或: 特异性 = 真阴性率 = 1 – 假阳性率
例如:假设一项检测在100名实际健康者中:
95人被正确判断为阴性(TN = 95)
5人被错误判断为阳性(FP = 5)
特异性 = 95/(95 + 5) = 95/100 = 0.95 或 95%
特异性表明该检测能够正确识别95%的健康者,有5%的健康者被误判为患病。
TIPs:
一种具有高敏感性但低特异性的测试会导致许多没有疾病的患者被告知他们有疾病的可能性,使受检者要接受进一步的测试。理想情况下,测试应该是100%准确的,但这是一个不切实际的场景。或者,对患者进行高敏感性和低特异性的测试,然后进行低敏感性和高特异性的第二次测试,可以识别所有假阳性和假阴性。
在考虑检测对临床医生的价值时,阳性和阴性预测值很有用。它们取决于该疾病在目标人群中的患病率。
但是灵敏度(sensitivity)、特异性(specificity)这两术语对于理解和评估模型的效用也至关重要。敏感性和特异性是用于评估临床试验的术语,它们与测试的目标人群无关。
★ 敏感性和特异性取决于检测阳性的临界值
检测的敏感性和特异性取决于高于或低于检测阳性的临界值。一般来说,敏感性越高,特异性越低,反之亦然。
★ 阳性和阴性预测值取决于被测人群和患病率
与敏感性和特异性不同,阳性预测值(PPV)和阴性预测值(NPV)取决于被测人群,并受疾病患病率的影响。
例如以下示例:使用抗核抗体在普通人群中筛查系统性红斑狼疮(SLE)的PPV较低,因为它产生的假阳性数量很高。然而,如果患者有SLE的迹象(例如 颊部潮红和关节疼痛),测试的PPV会增加,因为患者所在的人群不同(从SLE患病率低的一般人群到患病率高得多的临床可疑人群)。
另一个举例也可以考虑产后出现呼吸困难的女性,其中一种鉴别诊断是肺栓塞。在该患者群体中,D-二聚体检测几乎肯定会升高;因此,该测试的肺栓塞PPV较低。然而,肺栓塞的NPV很高,因为低D-二聚体不太可能与肺栓塞相关。
★ 患病率高时,PPV和NPV准确性更高
阳性预测值(PPV)和阴性预测值(NPV)对疾病患病率的依赖性可以用数字来说明:例如,在一个4000人的人群中,病人和健康人各占一半。该病症筛查试验的灵敏度和特异性均为99%。因此,筛查结果将产生1980个真阳性和1980个真阴性,其中20名健康人被误判为阳性,20名病人被误判为阴性。
该测试的PPV为99%。但是,如果人口中的患病人数只有200人,而健康人数为 3800 人,则假阳性数量从20增加到38人,PPV下降到84%。
本举例实际上想强调这样一个事实,即诊断或筛查疾病的能力既取决于检测的区分价值,也取决于疾病在相关人群中的患病率。
与此同时,要注意:
PPV的互补值是错误发现率(FDR),NPV的互补值是错误遗漏率(FOR),分别等于1减去PPV或 NPV。FDR是错误的结果或 “发现” 的比例。FOR 是被错误拒绝的假阴性的比例。从本质上讲,PPV和NPV越高,FDR和FOR就越低——这对您测试结果的可靠性来说是个好消息。
如果结果是按值的滑动比例给出的,而不是明确的阳性或阴性,则敏感性和特异性值尤为重要。它们允许确定在何处绘制预测结果为阳性或阴性的临界值,甚至可能建议重新测试的灰色区域。
例如:谷禾结直肠癌模型
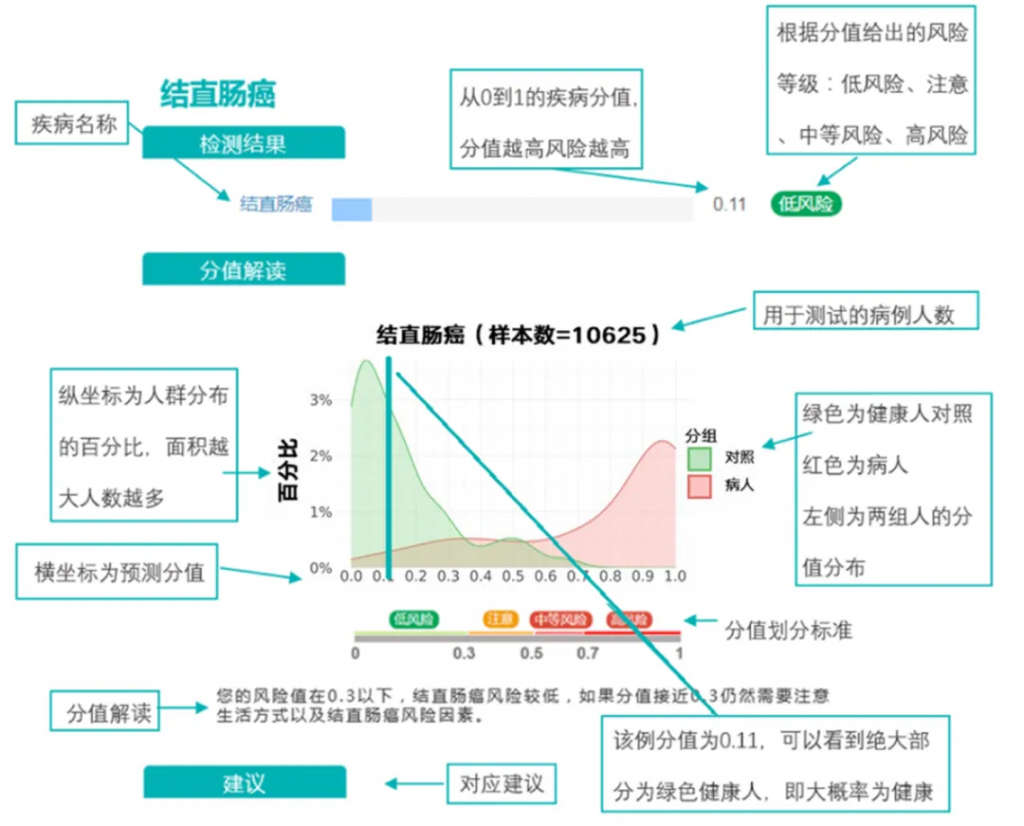
例如,通过将阳性结果的临界值置于非常低的水平,可以捕获所有阳性样品,因此测试非常敏感。但是,这可能意味着许多实际为阴性的样本可能被视为阳性,因此该测试将被视为特异性较差。找到平衡点对于有效和可用的测试至关重要。
★ ROC曲线有助于平衡假阴性和假阳性
在科研项目分析中经常使用受试者工作特征 (ROC)曲线有助于达到最佳平衡点并平衡假阴性和假阳性。但是,对于假阴性是否比假阳性问题更小,或者反之亦然,分析的内容也很重要。例如,在生死攸关的问题上,那么您可能愿意容忍更多的假阳性,以避免遗漏任何真阳性。
▸ 什么是ROC曲线?
ROC曲线是一种图形表示,显示测试的敏感性和特异性如何相互变化。为了构建 ROC 曲线,使用该检验测量已知为阳性或阴性的样本。
对于给定的截止值,将TPR(敏感性)与FPR(1-特异性)作图,得到类似于下图的曲线。理想情况下,选择曲线肩部周围的一个点,这既能限制假阳性,又能最大限度地提高真阳性。
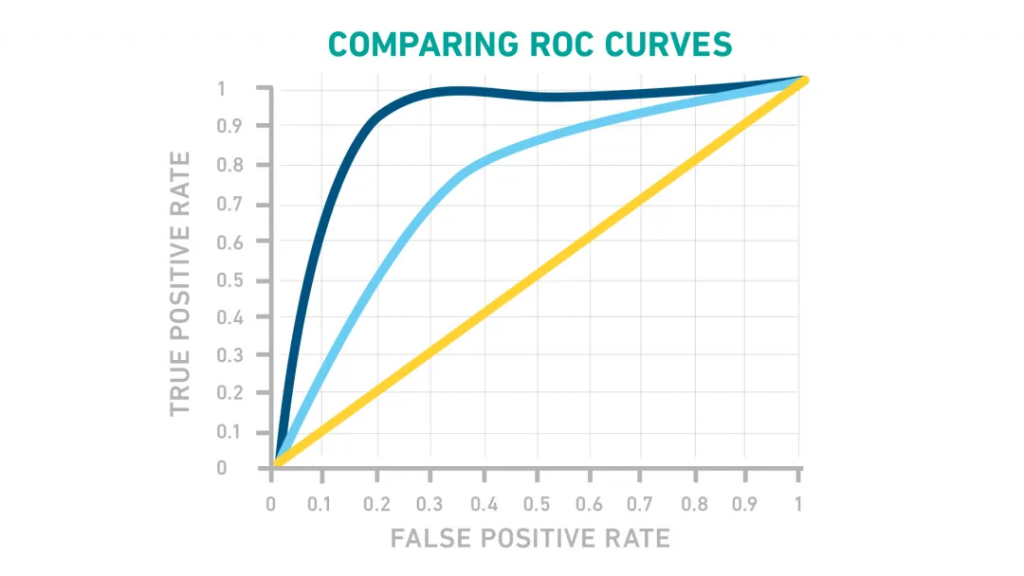
图片来源:Technology Networks
给出ROC曲线(如黄线)的测试并不比随机猜测好,淡蓝色很好,但由深蓝色线表示的测试会更好。这将使临界值确定相对简单,并以非常低的假阳性率产生高真阳性率。
在医疗科技和人工智能快速发展的今天,我们面临着海量的健康相关数据和不断涌现的疾病预测模型。在选择和评估医疗检测产品时,必须综合考虑疾病特性、人群特征和具体应用场景。对于普通大众和医疗从业者而言,理解这些指标不仅是一种专业素养,更是做出明智医疗决策的基础。
我们需要超越简单的”阳性”或”阴性”结果,深入理解检测结果背后的概率和风险。正如本文所强调的,一个优秀的医疗检测模型,应该能够在保证高敏感性的同时,最大限度地降低假阳性和假阴性的风险,为个人健康和公共卫生提供更精准、更有价值的信息。
主要参考文献:
technologynetworks.com/analysis/articles/sensitivity-vs-specificity-318222
Faith Mokobi. What is Sensitivity, Specificity, False positive, False negative? 2021 April 18, Microbenotes.

谷禾健康

近年来,随着微生物组学研究的深入,科学家们发现肠道菌群与人体年龄之间存在着远比想象更密切的关联。一系列突破性研究表明,肠道菌群不仅能够反映个体的实际年龄,更可能是调控生物学年龄的关键因素之一。这一发现正在改变我们对发育、衰老和健康的传统认知。
最新研究揭示,肠道菌群的组成和功能会随年龄发生系统性变化。这种变化表现在多个层面:从婴幼儿期菌群的快速建立与稳定,到成年后菌群多样性的动态平衡,再到老年期特征性菌群的衰退。基于这些规律,研究人员开发出了一系列基于肠道菌群的年龄预测模型,其准确度已经达到前所未有的水平。
更引人注目的是,肠道菌群年龄与多种疾病的发生、发展密切相关。研究发现,肠道菌群年龄的加速老化往往预示着代谢紊乱、免疫功能下降,甚至认知能力衰退。相反,保持年轻和健康的肠道菌群,可能是延缓衰老、预防疾病的重要途径。
本文将重点探讨三个核心问题:
通过梳理最新研究进展,我们将看到肠道菌群不仅是年龄的被动标记物,更可能是一个可调控的”生物时钟”。这一认识正在开启精准医疗和个性化健康管理的新篇章。
doi: 10.14218/ERHM.2024.00008
随着研究的深入和技术的进步,基于肠道菌群的年龄干预策略很可能成为未来医学实践的重要组成部分。
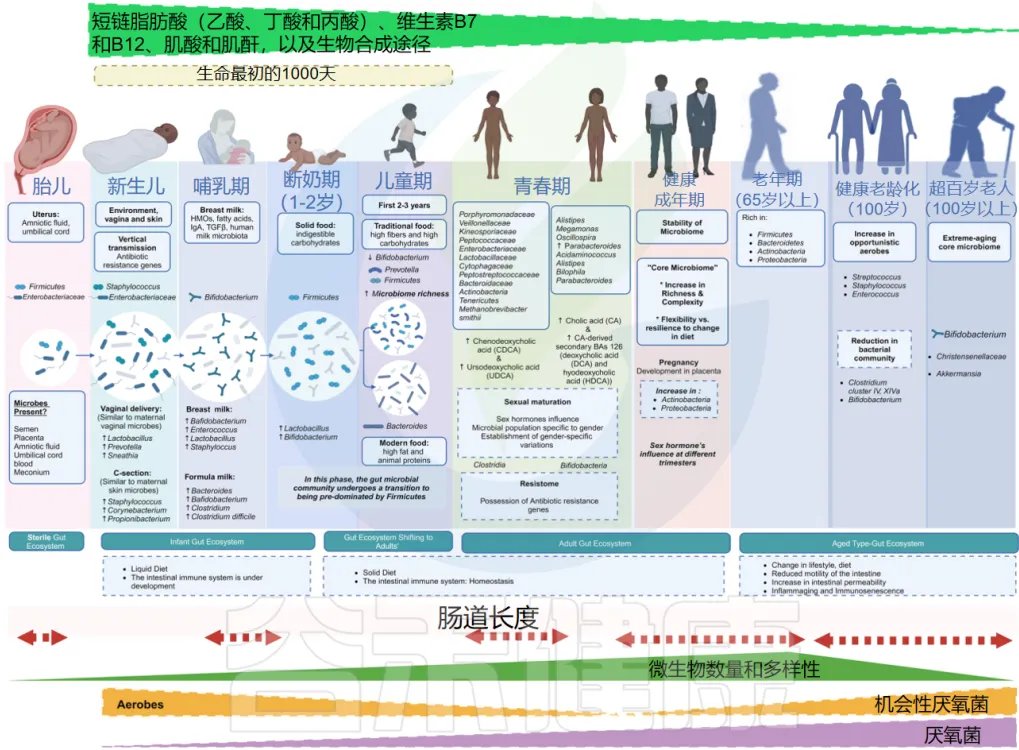
从无菌到共生——婴幼儿肠道菌群的“黄金塑造期”
近年来研究表明,肠道微生物群在婴幼儿早期发育阶段的变化与其发育年龄密切相关。婴幼儿的肠道微生物群呈现出明显的年龄相关性变化特征:在早期阶段主要表现为不稳定的群落结构和较低的微生物成熟度,随着年龄增长,菌群组成从以Firmicutes和Bifidobacterium为主,逐渐过渡到以Bacteroides和Prevotella为主导。这种转变具有确定性和可预测性,为年龄预测模型的建立提供了基础。
肠道微生物群的发育受多种因素影响,包括喂养方式、出生方式、地理位置和环境暴露等。研究发现,母乳喂养与Bifidobacterium的高水平密切相关,而停止母乳喂养则会加速肠道微生物群的成熟。此外,出生方式也显著影响微生物群的发育,如阴道分娩的婴儿肠道中Bacteroides的水平较高。尽管存在这些普遍性特征,但个体间的肠道微生物群发育轨迹仍存在差异,这与特定的生活方式和环境因素密切相关。
doi: 10.3233/NHA-170030
在预测模型研究方面,近年来取得了显著进展。2025年发表在《Nature Communications》上的研究开发出了一个高精度的预测模型,该研究分析了来自12个国家的1,827名婴幼儿的3,154个样本,使用随机森林模型进行预测,预测误差仅为2.56个月。
研究还识别出了关键的分类学预测指标,包括双歧杆菌的减少和粪杆菌的增加。另一项2024年的研究则开发出了“肠道微生物群健康指数”,该指数不仅能够预测婴幼儿在前5年的整体健康状况,还提供了一个可靠的微生物群年龄评估基准。
最新研究进一步证实,婴幼儿肠道微生物组的发育成熟度与年龄高度相关,仅通过肠道微生物群组成就能准确预测婴儿的年龄。研究人员建立了”微生物组年龄”的评估标准,这不仅可以用于年龄预测,还可以作为评估早期肠道发育的重要指标。这些研究成果不仅证实了基于肠道菌群预测婴幼儿年龄的可行性,还为评估婴幼儿的发育状况和健康水平提供了新的工具和方法。
肠道菌群发育滞后会对婴幼儿的多个系统产生深远影响。
在神经系统方面,可能导致认知发育迟缓、语言发育延迟、注意力不集中,甚至增加自闭症谱系障碍(ASD)的风险;
在免疫系统方面,常见表现包括反复呼吸道感染、特应性皮炎、食物过敏,以及哮喘等过敏性疾病的发生率升高;
在代谢系统方面,可能引起生长发育迟缓、体重增长异常、微量元素吸收不良等问题;在消化系统方面,则可能出现腹泻、便秘、肠痉挛等功能性胃肠道疾病。
特别是在出生后最初1000天这个关键期内,肠道菌群的发育状况对婴幼儿的长期健康具有决定性影响。
在这种情况下,规范的肠道菌群检测在婴幼儿发育评估中具有重要意义。检测内容应包括微生物多样性指数、关键菌群(如双歧杆菌、乳酸菌、粪杆菌等)的丰度,以及潜在致病菌的监测。
建议在以下关键时间点进行检测:出生后1-2个月、辅食添加期(4-6个月)、断奶期(12个月左右)以及2岁左右。对于高危人群,如早产儿、剖宫产婴儿、有过敏家族史或自身免疫性疾病家族史的婴幼儿,需要更频繁的监测。
研究显示,及早发现和干预菌群异常可以显著改善预后。具体干预措施包括:调整饮食结构(如添加低聚糖、膳食纤维等益生元),补充特定益生菌(如婴儿双歧杆菌、鼠李糖乳杆菌等),改善生活习惯(如规律作息、适度运动),以及环境因素的调整(如避免不必要的抗生素使用、增加户外活动时间)。对于特定问题,如食物过敏,可以通过补充特定菌株(如常见的LP299V益生菌)来改善症状;对于免疫功能低下,则可以考虑添加具有免疫调节作用的益生菌。
因此,建议在婴幼儿发育的关键时期进行系统的肠道菌群检测和跟踪监测。这种检测不仅能够及早发现发育风险(如自闭症倾向、过敏风险、免疫功能异常等),还能为个性化干预方案的制定提供科学依据。特别是对于已经出现发育迟缓、免疫功能低下、消化吸收障碍等问题的婴幼儿,定期的肠道菌群检测对评估干预效果和及时调整治疗方案具有重要的临床指导意义。
通过肠道菌群的视角:解密衰老时钟背后的微生物密码
➤ 老年肠道菌群的三大特征:
多样性下降、促炎菌增加、代谢功能衰退
随着年龄增长,我们的肠道菌群会发生显著变化。研究发现,老年人的肠道菌群具有三个显著特征,这些变化与多种衰老相关的健康问题密切相关。
◆ 首先是菌群多样性的显著下降
与年轻人相比,老年人的肠道菌群种类明显减少,特别是厚壁菌门数量减少,而拟杆菌门的比例相对升高。同时,一些对健康有益的细菌,如双歧杆菌和瘤胃球菌等数量也显著减少。这种多样性的降低会影响肠道微生态的稳定性。
◆ 第二个特征是促炎菌群的增加
老年人肠道中肠杆菌科(Enterobacteriaceae)和肠球菌属(Enterococcus)等促炎菌群数量明显增多,而具有抗炎作用的普拉梭菌(Faecalibacterium prausnitzii)等益生菌则显著减少。这种失衡会导致肠道处于慢性低度炎症状态,不仅影响局部肠道健康,还可能加速整体衰老进程。
◆ 第三个特征是代谢功能的衰退
老年人肠道中产丁酸菌如真杆菌(Eubacterium)、瘤胃球菌等数量减少,这些菌群对维持肠道健康至关重要。它们的减少会影响营养物质的吸收和能量代谢,同时也会降低肠道屏障功能,增加肠道通透性。这种变化可能导致营养吸收不良,并增加有害物质进入血液的风险。
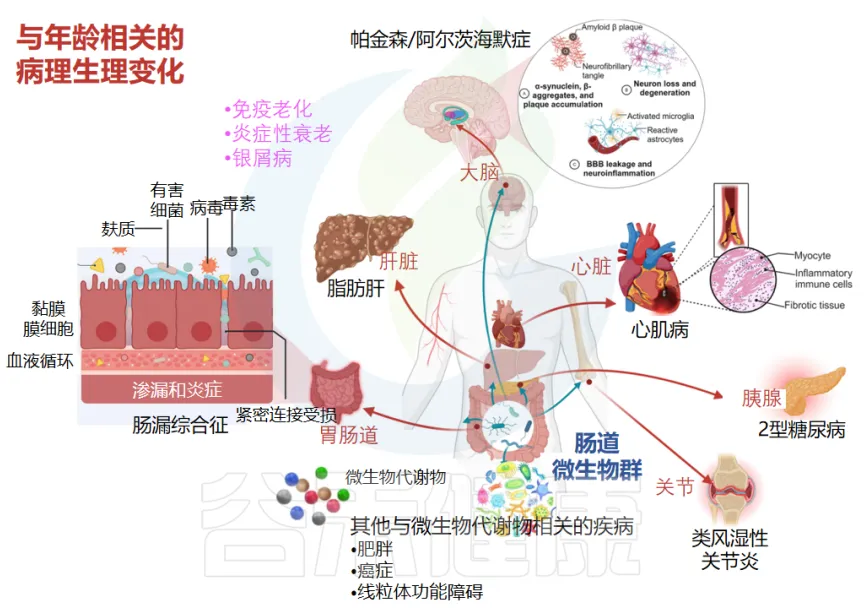
这三个特征之间相互关联,共同构成了一个复杂的失衡网络。例如,菌群多样性的下降会影响代谢功能,促炎菌群的增加又会进一步降低有益菌群的数量。这种恶性循环可能是老年人更容易出现各种健康问题的重要原因之一。了解这些特征对于开发针对性的干预策略,改善老年人健康状况具有重要意义。
➤ 虚弱的菌群特征
虚弱是老年人一种特殊的临床状态,表现为机体对内外压力源的脆弱性增加,往往伴随着食欲下降、肌肉减少、认知功能下降、活动能力减弱以及独立生活能力的丧失。近年来的研究发现,肠道菌群的改变与虚弱程度之间存在着密切的关联,这种关联并不完全依赖于年龄因素。
研究显示,虚弱老年人的肠道菌群具有独特的“虚弱特征”。首先是普氏菌(Prevotella)和普拉梭菌(F. prausnitzii) 的显著减少,这些菌群对维持肠道健康和免疫功能具有重要作用。其次是产丁酸菌,如Eubacterium halii和Eubacterium rectalis的含量降低,这与肌肉蛋白合成的减少密切相关。
研究人员还观察到,在虚弱评分较高的老年人中,乳酸菌(Lactobacilli)、拟杆菌/普氏菌(Bacteroides/Prevotella)等有益菌群显著减少,而瘤胃球菌属(Ruminococcus)、Atopobium和肠杆菌科(Enterobacteriaceae)等菌群则明显增加。
doi: 10.1016/j.advnut.2025.100376
这些菌群变化的意义重大。产丁酸菌的减少会影响能量代谢和肌肉功能,而有益菌的减少则可能导致免疫功能下降和炎症水平升高。相反,在功能状态良好的老年人中,往往可以观察到较高水平的产丁酸菌,特别是普拉梭菌(Faecalibacterium prausnitzii)的含量,这进一步证实了菌群组成对机体功能的重要影响。
这些发现为我们理解和干预老年虚弱提供了新的视角。通过调节肠道菌群,特别是补充特定的益生菌和益生元,可能有助于改善老年人的虚弱状态。这也提示我们,在老年医学实践中,应当将肠道菌群作为评估和干预虚弱的重要靶点之一。
➤ 百岁老人长寿菌群的特殊组成
研究发现,百岁老人的肠道菌群具有独特的“长寿特征”,这种特征不仅区别于普通老年人,更展现出与健康长寿密切相关的微生物组成模式。这一发现为我们理解人类长寿的生物学机制提供了新的视角。
在菌群组成上,百岁老人的肠道中普遍存在较高水平的拟杆菌和罗氏菌属(Roseburia)。这两类菌群在维持肠道健康和免疫平衡方面发挥着重要作用。作为重要的丁酸盐产生菌,能够通过产生短链脂肪酸来调节肠道微环境,增强肠道屏障功能。
更引人注目的是,百岁老人的肠道中还富含Christensenella、双歧杆菌和阿克曼氏菌(Akkermansia)。这些菌群被认为是“长寿菌群”的代表。其中,长寿菌群与人类的遗传因素高度相关,被认为是最具遗传性的菌群之一;双歧杆菌能够维持肠道健康,增强免疫功能;而阿克曼氏菌则在调节代谢、维持肠道屏障完整性方面发挥关键作用。
研究还发现了一些在百岁老人肠道中特有的菌种,如腐生链菌(Alistipes putredinis)和气味杆菌(Odoribacter splanchnicus)。这些特殊菌群的存在可能与长寿个体特有的代谢特征和免疫调节功能有关,为我们理解长寿的微生物学机制提供了重要线索。
这些发现不仅帮助我们理解了百岁老人独特的肠道菌群特征,更为延缓衰老、促进健康长寿提供了新的思路。通过深入研究这些”长寿菌群”的功能和调节机制,有望开发出基于微生物组的健康干预策略,为实现健康老龄化提供新的方案。
值得注意的是,这些长寿相关的菌群特征并非简单的个体差异,而是反映了人体在长期健康状态下的微生态平衡。这种平衡可能是遗传因素、生活方式和环境因素长期共同作用的结果,对于我们理解和促进健康长寿具有重要的指导意义。
➤ 菌群-肠-脑轴在认知衰退中的作用路径
肠道菌群与大脑的关系远比我们想象的要复杂。研究表明,肠道菌群通过复杂的菌群-肠-脑轴与认知功能密切相关。这条轴线不仅包括神经元的直接连接,还涉及神经递质、激素和免疫介质等多重信号分子,构成了一个精密的双向调控网络。
◆ 第一个关键通路是神经递质调节
肠道菌群可以直接产生或调节多种神经递质的合成。研究发现,乳酸菌和双歧杆菌能够将肠道中的谷氨酸转化为GABA(γ-氨基丁酸),这是大脑中主要的抑制性神经递质,对情绪调节和认知功能具有重要作用。
此外,肠道菌群还参与乙酰胆碱和多巴胺等神经递质的产生。动物实验证实,补充Lactobacillus helveticus NS8可以通过降低血浆中皮质醇和促肾上腺皮质激素的水平来改善应激反应,同时提高血清素和去甲肾上腺素的水平。
◆ 第二个重要机制是免疫-炎症通路
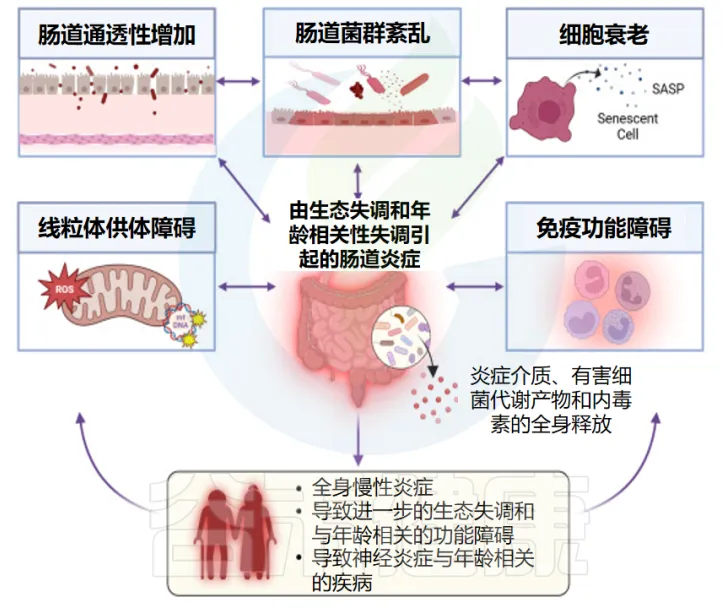
随着年龄增长,肠道菌群失调会导致肠道通透性增加,使脂多糖(LPS)、脂蛋白和细菌RNA等细菌代谢产物进入血液循环。这些物质可以激活免疫系统,导致TNF-α、IL-1β、IL-6等促炎因子的释放,引发系统性炎症反应。
这些炎症因子可以穿过血脑屏障,在大脑中引起神经炎症,加速认知功能的衰退。值得注意的是,小胶质细胞(占中枢神经系统固有免疫细胞的约10%)在这个过程中发挥着重要作用,它们能够快速识别和响应环境变化,参与维持神经系统的稳态。
◆ 第三个关键环节是神经营养因子的调节
肠道菌群对脑源性神经营养因子(BDNF)和神经生长因子(NGF)的表达具有重要影响。这些神经营养因子对神经元的存活、突触可塑性和神经回路的重塑起着关键作用。研究发现,某些益生菌的补充可以提高BDNF的水平,改善认知功能。例如,在小鼠实验中,补充长双岐杆菌(Bifidobacterium Longum)能够显著改善动物的推理能力。
此外,肠道菌群还通过产生短链脂肪酸等代谢产物发挥作用。这些代谢产物可以调节血脑屏障的完整性,影响神经元功能和神经胶质细胞的活化状态。研究还发现,某些菌群数量变化与认知功能直接相关,如Alcaliganeceae和Porphyromonadaceae的数量增加与推理能力下降呈正相关。
在治疗干预方面,益生菌的应用显示出了积极的效果。例如,Lactobacillus、Bifidobacterium、S. thermophilus、Enterococcus、Bacillus等益生菌可以通过增加色氨酸衍生的神经营养因子水平来促进神经递质的释放。
然而,益生菌补充的效果会受到多种因素的影响,如剂量、菌株、补充时间以及患者的年龄、疾病状态、药物使用(尤其是抗生素)、饮食和生活方式等。
需要指出的是,尽管菌群-肠-脑轴的研究取得了显著进展,但仍有许多问题需要进一步探索。目前的研究主要基于动物模型和临床观察,还需要更多的多学科、多系统的研究来阐明相关机制和干预机会。这些研究将有助于开发以肠道菌群为靶点的早期干预策略,为预防和治疗神经退行性疾病提供新的思路。
用AI构建年龄的生物标记——肠道菌群的年龄预测模型
近年来,随着测序技术的进步和机器学习方法的发展,基于肠道菌群的年龄预测模型取得了显著进展。2020年,研究人员通过深度学习方法分析了4000多份来自18-90岁人群的宏基因组数据,构建出预测误差为10.6年的模型。同年,另一项基于随机森林算法的研究分析了4434份样本的16S rRNA测序数据,将预测误差控制在11.5岁。这些早期研究证实了利用肠道菌群特征预测年龄的可行性。
2022年的突破性研究展示了多维度数据整合的优势。通过分析4478份成年人粪便样本,研究发现单独使用分类特征或代谢途径特征的预测模型误差分别为9.5年和10.2年,但将两类特征结合并考虑宿主因素后,预测误差可降至8.3年。这一发现为后续模型开发指明了方向。
2024年一项新研究开发出了基于肠道微生物组多维数据的生物学年龄预测模型(gAge)。该研究通过创新的机器学习方法,成功整合了微生物组的分类学和功能学特征,显著提高了年龄预测的准确性。研究团队识别出164个标志物物种和35个关键代谢通路与宿主年龄变化密切相关,并根据其对衰老的影响将其分类为促进和抑制两类。
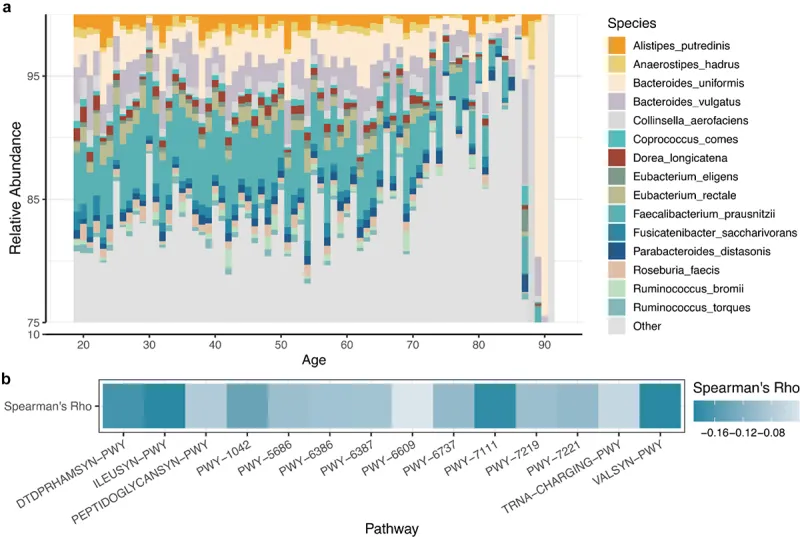
doi: 10.1080/19490976.2023.2297852
研究验证显示,gAge模型及其预测残差与个体健康状况和虚弱程度高度相关。特别是在老年群体中,模型识别的年龄相关标志物与多种疾病和虚弱特征显著重合,为评估和预测衰老风险提供了新的工具。
doi: 10.1080/19490976.2023.2297852
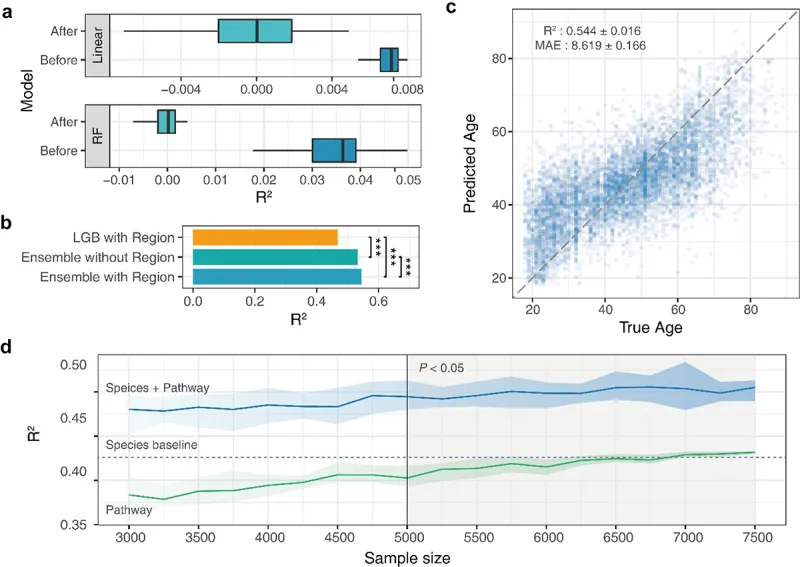
在这些研究基础上,谷禾开发的GUHEAge模型实现了更大的突破。该模型基于60万例不同年龄人群的6万个菌群特征,首次完整分析了从0-120岁的肠道菌群演变过程。通过深度神经网络,我们识别出381个标记物种和1574个标记基因,并根据其对衰老的影响将其分为促进和抑制两类。
在这些标记物中,产短链脂肪酸的有益菌(如双歧杆菌属、嗜粘蛋白阿克曼氏菌和拟杆菌属)表现出抑制衰老的作用。这些菌群可增强肠道屏障功能,调节机体代谢、炎症和免疫反应。相反,某些致病菌(如大肠杆菌和空肠弯曲杆菌)则显示出促进衰老的特征。
更重要的是,GUHEAge模型不仅能准确预测年龄,其预测结果还与个体健康状况和虚弱程度高度相关。验证集表明,模型识别的年龄相关标志物与多种疾病和虚弱特征显著重合,这为精准医疗和个性化健康管理提供了新的思路。
这些进展表明,基于肠道菌群的年龄预测模型已经发展成为一个可靠的生物年龄评估工具。通过整合多维度数据和先进的机器学习方法,为干预衰老提供新的靶点。
<来源:谷禾健康肠道菌群数据库>
可以看到,肠道年龄和实际年龄基本是符合的。健康人的肠道菌群年龄恰恰是最符合真实年龄的,与真实年龄差异大意味着肠道菌群出现偏离。
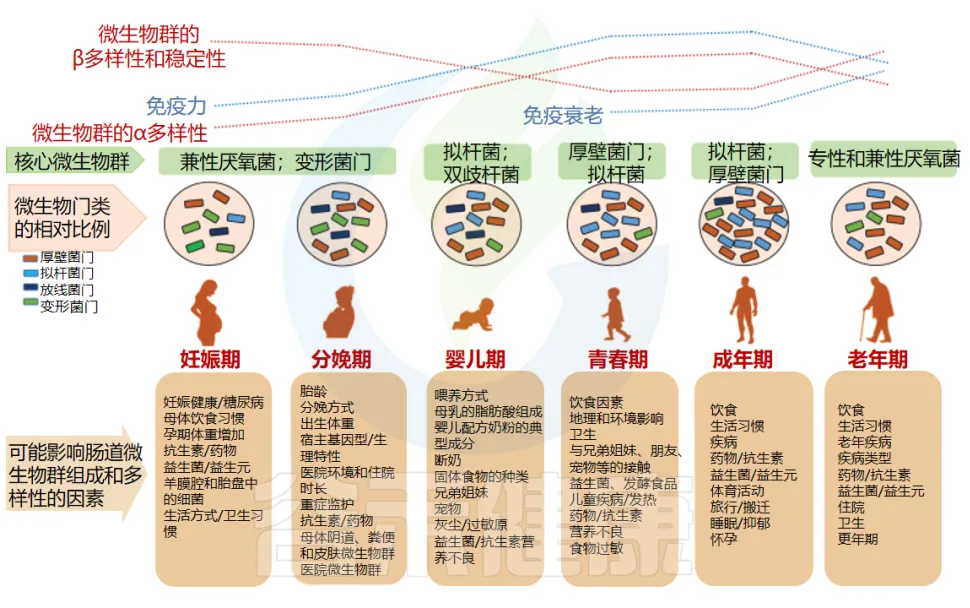
健康的人存在更多样化且平衡的肠道菌群。微生物群中与年龄相关的变化归因于生理,生活方式和健康状况。这些因素中的每一个都与某些菌群的相对丰度变化有关。
例如,饮食、卫生、兄弟姐妹、宠物、过敏、儿童疾病和抗生素是影响儿童微生物组的一些突出因素。到了成年期微生物群相对稳定,而到了老年期,一些有益菌开始逐渐下降,菌群又向另一个阶段过渡。
理解肠道年龄超越数字的生物学意义
➤ 发育迟缓儿童的菌群年龄滞后现象
发育迟缓儿童的肠道菌群呈现出明显的“年龄滞后”特征,这种现象反映在菌群的多样性、组成结构和功能等多个方面。研究表明,这些儿童的肠道菌群发育水平往往落后于其实际年龄,这种滞后可能是导致发育迟缓的重要因素之一。
我们之前发表在《Gut》上的针对自闭症儿童菌群发育的研究显示,自闭症儿童的菌群发育要滞后于健康儿童。
在菌群组成方面,发育迟缓儿童表现出显著的特征性改变。首先是有益菌群的明显减少,特别是双歧杆菌和乳杆菌的含量显著低于同龄健康儿童。这些菌群对维持肠道健康、促进营养物质吸收和调节免疫功能具有重要作用。同时,研究还发现这些儿童的肠道中普氏菌(Prevotella)和粪杆菌(F. prausnitzii)等核心菌菌含量也明显降低。
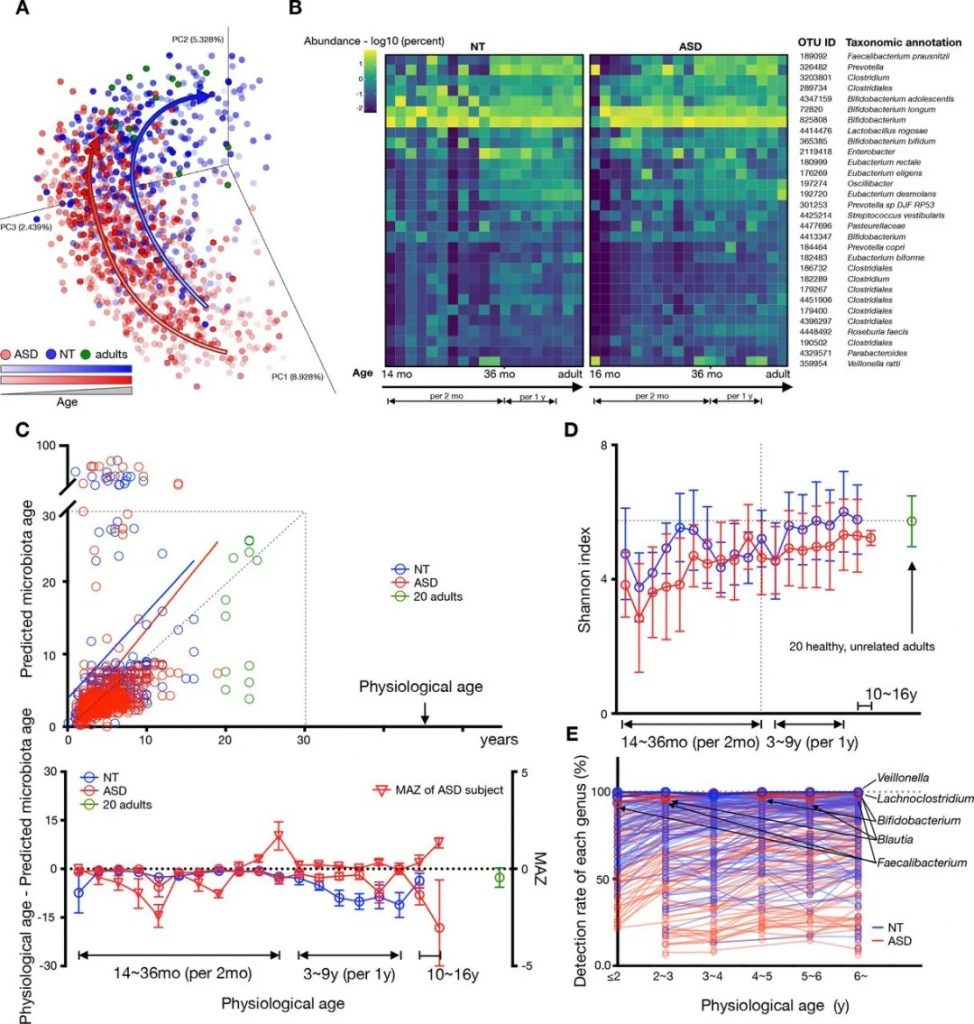
doi.org/10.1136/gutjnl-2021-325115
这种菌群滞后现象可能通过多个机制影响儿童发育:
免疫调节机制:菌群失调导致肠道免疫功能发育不良,增加炎症风险,影响生长发育。研究发现,这些儿童体内往往存在低度慢性炎症状态。
营养代谢影响:关键有益菌群的减少影响营养物质的吸收和利用效率。特别是产丁酸菌的减少,会影响能量代谢和营养物质的吸收。
神经-内分泌调节:菌群失调可能通过菌群-肠-脑轴影响生长激素和其他内分泌激素的分泌,进而影响生长发育。
针对这一问题,目前研究提出了几种潜在干预策略,包括结合营养补充、运动干预和益生菌治疗或全面的菌群置换可以有效改善儿童的生长发育状况。
通过调节肠道菌群,有望开发出更有效的干预策略,帮助发育迟缓儿童实现健康生长。同时,这也提示我们在儿童保健实践中,应当将肠道菌群作为评估和干预的重要指标之一。
➤ 早衰综合征患者的菌群年龄加速特征
早衰综合征患者的肠道菌群呈现出显著的“年龄加速”特征,其菌群组成和功能状态明显超前于实际年龄水平。研究发现,这种菌群年龄加速现象与多种生理功能的提前衰退密切相关。
doi: 10.2147/CIA.S414714
在菌群组成方面,早衰患者表现出显著的特征性改变。有益菌群如粪杆菌(Faecalibacterium prausnitzii)、直肠真杆菌(Eubacterium rectale)和扭链瘤胃球菌(Ruminococcus torques)的含量明显降低。同时,代谢通路也发生了重要变化,包括支链氨基酸代谢通路(如L-缬氨酸和L-异亮氨酸合成)的显著下降,丙酮酸发酵(PWY-7111)能力减弱,以及NAD合成相关代谢通路的异常。
这些改变通过多个机制影响机体功能。首先是炎症反应的加速,表现为促炎因子(TNF-α、IL-6和IL-1)水平升高,导致慢性低度炎症状态持续存在。其次是代谢功能的紊乱,包括氨基酸代谢异常、能量代谢效率下降和营养物质吸收受损。此外,肠道屏障功能受损也是重要特征,导致肠道通透性增加,细菌代谢产物进入血液循环,进一步加重系统性炎症反应。
针对这些问题,研究提出了几种潜在的干预策略:
精准菌群干预:通过补充特定益生菌,如双歧杆菌和乳酸菌,同时添加益生元促进有益菌群生长。研究表明,这种干预方式可以改善肠道菌群组成,延缓衰老进程。
代谢通路调节:包括补充NAD前体物质、调节氨基酸代谢和改善能量代谢。特别是NAD水平的提升,已被证实可以预防衰老和相关疾病。
生活方式干预:优化饮食结构,增加膳食纤维摄入;保持适度运动,提高体能状态;改善睡眠质量,维持昼夜节律。
综合干预方案:结合营养补充、心理支持、运动康复和定期监测菌群状态,全面改善患者状况。
通过对肠道菌群的精准干预,有望延缓早衰进程,改善患者生活质量。
➤ 糖尿病/心血管疾病的菌群老化预警
在代谢性疾病领域,肠道菌群年龄作为一个新兴的生物标志物展现出重要的预警价值。研究发现,糖尿病和心血管疾病患者的肠道菌群往往表现出提前衰老的特征,这种菌群老化现象通常早于临床症状的出现,为疾病的早期预警提供了新的时间窗口。
在菌群组成方面,研究显示代谢性疾病患者的肠道菌群存在显著变化。这些变化与血清生化指标密切相关,特别是与碱性磷酸酶和胰岛素水平显著相关。研究还发现,某些代谢通路的改变,如支链氨基酸代谢通路(BCAA)、NAD合成相关代谢通路的异常,可能是疾病发展的早期信号。
通过肠道年龄预测模型的应用,研究人员发现菌群年龄与多种疾病风险指标具有显著相关性。在一项针对老年人群的研究中,gAge预测残差与个体健康状况和虚弱程度呈现高度相关。特别是在2型糖尿病(T2D)患者中,尽管疾病本身可能不会显著影响预测年龄,但菌群组成的特定改变可以作为疾病进展的预警指标。
在心血管疾病方面,研究发现血清碱性磷酸酶水平与心脑血管疾病风险显著相关,而这一指标与肠道菌群年龄存在明显关联。通过分析不同gAge残差人群的菌群特征,研究者识别出了一系列可能与心血管健康相关的关键菌种和代谢通路,为疾病预防提供了新的干预靶点。
这些发现为代谢性疾病的早期筛查和预防提供了新的思路。通过监测肠道菌群年龄的变化,结合传统的临床指标,可以更早地识别疾病风险,实现更精准的健康管理。随着检测技术的进步和预测模型的完善,基于肠道菌群的疾病预警系统有望成为临床实践中的重要工具。
实现从被动治疗到主动预防的转变
随着人口老龄化加剧,深入理解和干预衰老过程变得愈发重要。近年来,肠道微生物组研究为我们提供了全新的视角,从发育到衰老的全程监测和干预成为可能。
肠道年龄概念的提出,标志着生物学年龄评估领域的一次重要范式转变。这一转变不仅打破了传统chronological age的局限,更是将微生物组这一动态指标引入健康评估体系,为个体化健康管理提供了新的思维框架。
在发育领域,研究发现肠道菌群年龄可以有效反映儿童发育状况。发育迟缓儿童往往表现出明显的”菌群年龄滞后”现象,这为早期干预提供了新的靶点。同时,在早衰综合征患者中观察到的菌群加速衰老特征,以及在糖尿病、心血管疾病患者中发现的菌群老化预警信号,都展示了微生物组在疾病预防和健康管理中的重要价值。
谷禾团队通过分析60万例不同年龄人群的6万个菌群特征,这种基于海量数据的个体化健康监测方法,为微生物组研究开辟了新的范式。通过持续监测个体肠道菌群的动态变化,有助于更早地发现健康隐患,实现疾病的早期预警和干预。
<来源:谷禾健康肠道菌群检测数据库>
从精准医学的角度来看,肠道年龄的概念为个体化健康管理提供了新的工具。通过整合微生物组数据与传统临床指标,我们可以构建更全面的健康评估体系。这种多维度的评估方法不仅能够反映个体当前的健康状态,还能预测未来的健康轨迹,为精准医疗干预提供决策依据。
尽管目前研究已取得显著进展,但仍面临诸多挑战:微生物组与衰老的因果关系有待厘清,个体差异和环境因素的影响需要进一步研究,预测模型在特定人群中的适用性也需要验证。此外,如何将研究成果有效转化为临床应用,实现精准干预,也是未来需要重点解决的问题。
展望未来,随着高通量测序技术的进步和人工智能算法的优化,结合多组学数据的整合分析将帮助我们更全面地理解肠道微生物组在人体发育和衰老过程中的作用。精准医学的未来将更加依赖于这种整合的、动态的健康评估体系,结合人工智能分析,可以更好为每个个体制定个性化的健康管理方案,实现从被动治疗到主动预防的转变。
主要参考文献:
Min M, Egli C, Sivamani RK. The Gut and Skin Microbiome and Its Association with Aging Clocks. Int J Mol Sci. 2024 Jul 8;25(13):7471.
Hohman LS, Osborne LC. A gut-centric view of aging: Do intestinal epithelial cells contribute to age-associated microbiota changes, inflammaging, and immunosenescence? Aging Cell. 2022 Sep;21(9):e13700.
Tao X, Zhu Z, Wang L, Li C, Sun L, Wang W, Gong W. Biomarkers of Aging and Relevant Evaluation Techniques: A Comprehensive Review. Aging Dis. 2024 May 7;15(3):977-1005.
Wang H, Chen Y, Feng L, Lu S, Zhu J, Zhao J, Zhang H, Chen W, Lu W. A gut aging clock using microbiome multi-view profiles is associated with health and frail risk. Gut Microbes. 2024 Jan-Dec;16(1):2297852.