-

 CNAS L23010
CNAS L23010

 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室
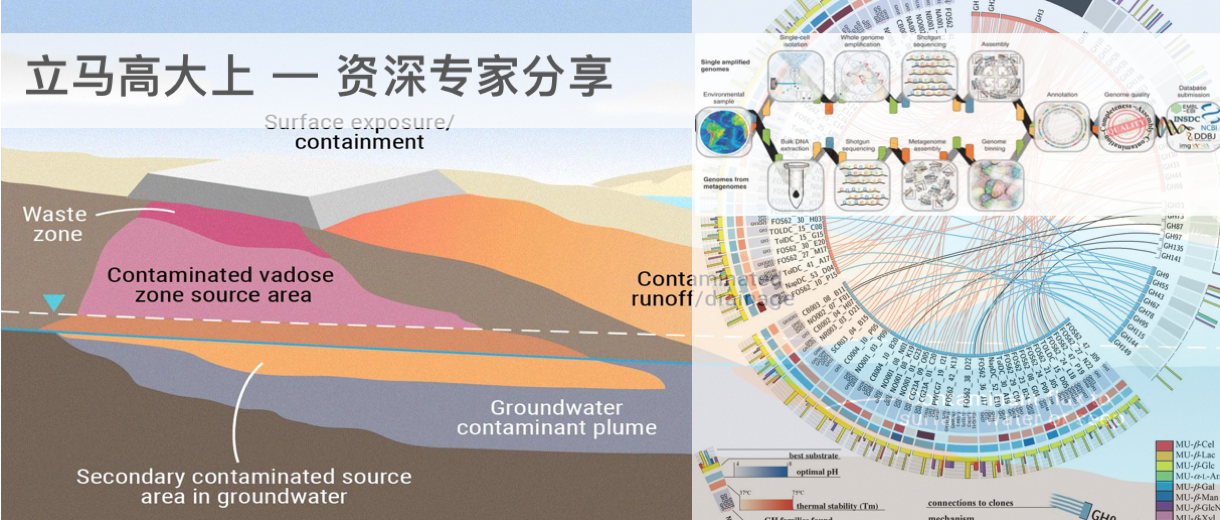
近年的研究热点集中于环境和生物体相互作用的微生物群体,而大量复杂的微生物群体存在培养困难,构成复杂(包括细菌、古菌、真菌、原生生物、病毒甚至小型真核生物)。 为了克服传统纯培养技术的不足,充分挖掘此类未培养微生物所蕴涵的巨大潜能,发展了 宏基因组学的研究方法,该方法绕过纯培养技术来研究微生物的多样性及功能。
宏基因组测序也就是shotgun测序, 以环境中所有微生物基因组为研究对象,通过对环境样品中的全基因组DNA进行高通量测序,获得单个样品的饱和数据量,基于denovo组装进行微生物群落结构多样性, 深度全面的了解微生物群体的构成,甚至获得单个菌株的完整基因组,在这些高质量基因组序列的基础上可以更加精细化开展其基因构成、分布,次生代谢合成,抗生素耐药基因及其演化, 微生物群体基因组成及功能,特定环境相关的代谢通路等分析 甚至菌株的生态演变和水平转移事件等研究。 进一步发掘和研究具有应用价值的基因及环境中微生物群落内部、微生物与环境间的相互关系。
此外对于缺乏深度研究和高质量参考基因组的样本,如土壤和特殊环境下的样本,宏基因组获得的较为完整的基因组不仅可以丰富参考基因组数据库,同时可以提供更加准确的物种分类。
建库流程
1) 将检测合格的基因组DNA样品用酶切随机打断成400bp-500bp的片段并末端修复加A;
2) 将测序引物连接到DNA片段上;
3) 进行PCR扩增引入测序index
4) 对构建的文库进行质量检测;
5) 将质量检测合格的文库上机测序。
粪便、动物肠道内容物、皮肤、组织、痰液、血液、唾液、牙菌斑、尿液,阴道分泌物、发酵物,瘤胃,废水,火山灰,冻土层、病害组织、淤泥、土壤、堆肥、污染河流,养殖水体、空气等有微生物存在的样本都可以用于宏基因组测序。
测序平台:Illumina Novaseq,PE150,默认:5-6G/样,每增加一个G加100元。
周期:2-3周
粪便、动物肠道内容物、皮肤、组织、痰液、血液、唾液、牙菌斑、尿液,阴道分泌物、发酵物,瘤胃,废水,火山灰,冻土层、病害组织、淤泥、土壤、堆肥、污染河流,养殖水体、空气等有微生物存在的样本都可以用于宏基因组测序。




第一项研究是关于肥胖患者减肥手术后的宏基因组和代谢数据的分析研究。
研究纳入了61名严重肥胖的受试者,他们是可调节胃束带术(AGB,n = 20)或Roux-en-Y胃旁路术(RYGB,n = 41)的候选人。减肥手术后1、3和12个月随访24名受试者。使用宏基因组学测序和LC-MS分析肠道菌群和血清代谢组。另外纳入了10人和147人分别作为宏基因组和代谢检测的验证集。
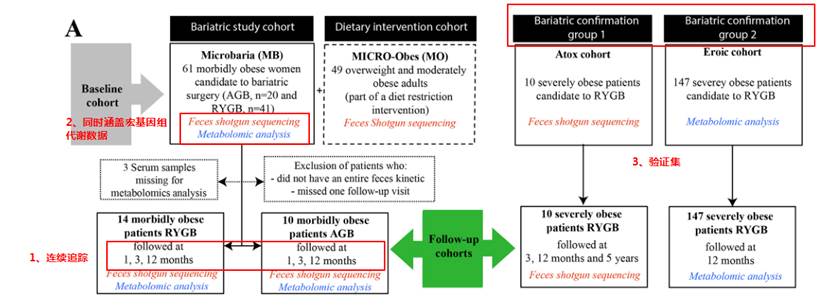
研究思路

这样的设计分别有什么作用?
第一点持续的动态采样可以获得持续变化情况,尤其是在一个特定变化后(减肥手术),持续的最终采样有助于确认菌群的变化出现和特定事件或生理病理变化的前后,尤其是在确定因果中有重要帮助。
第二点获得多维的数据有助于帮助我们全方位的了解菌群变化背后的带来的生理和代谢变化以及之间的关联。
第三点独立验证集的存在将大大增强研究的可信度,尤其是该研究纳入的样本量并不多,无法全面有效的控制无关因素,使得很多统计检验的效力无法显现。这也导致该研究仅在基因总量和多样性上获得较好的重复效果,而更多的菌群精细特征以及具体基因和代谢通路没有得到深入分析。但是独立验证集保证了核心结论的可靠性和重复性,这点在宏基因组研究中非常重要。
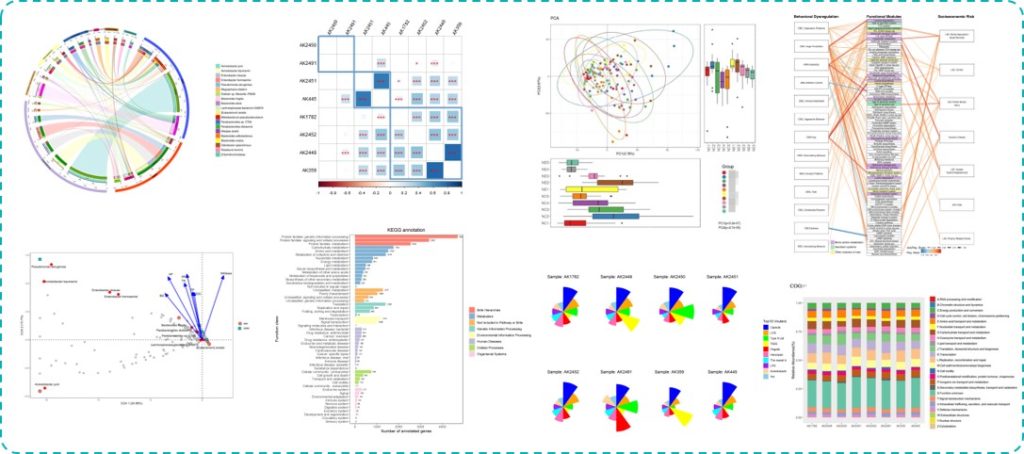
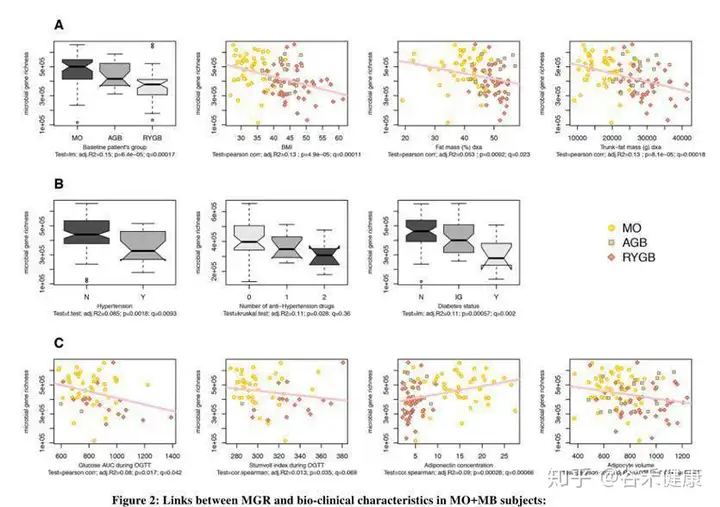
从下图可以看到研究针对样本的总基因多样性水平与生理指标和疾病状态进行相关性分析和组间差异分析,图中给出了显著相关和差异的指标。
使用的统计检验方法是pearson和sperman相关和t-test以及Kruskal-Wallis检验。
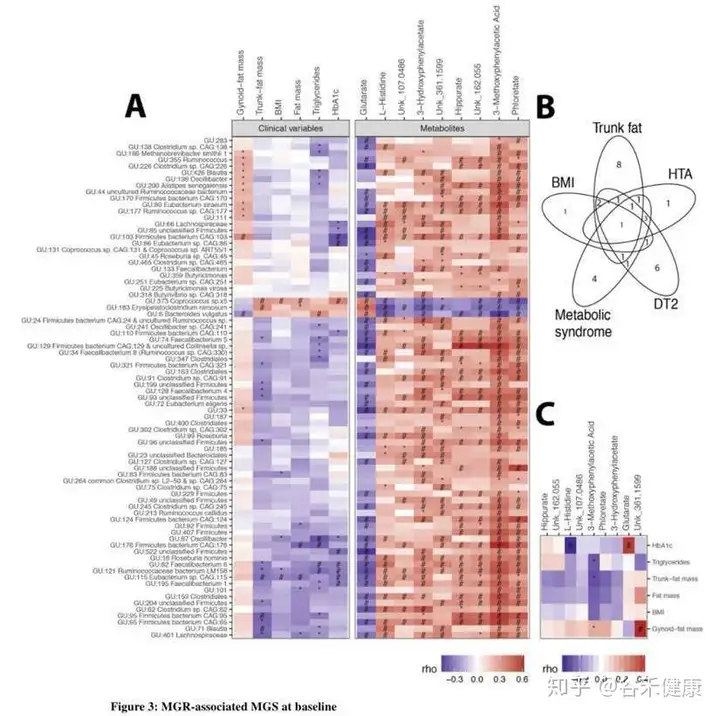
下图是研究将MAGs与各项生理和代谢值进行相关性分析后的热力图。该研究由于测序较早,并未独立拼接,而是直接使用了之前一项人类肠道菌群研究获得组装基因组参考序列。
进一步研究分析了术后特定变化模式的MAGs以及它们与代谢生理指标的相关性,见下图:
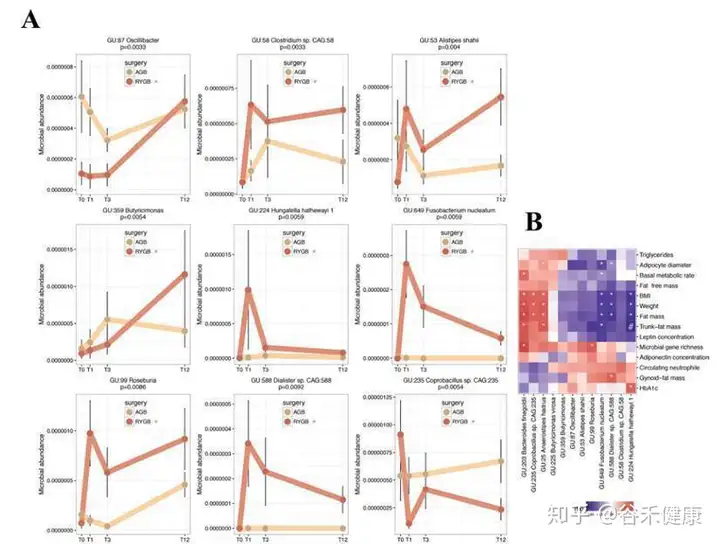
上图的研究可以通过pattern search的方法寻找特定变化模式的菌种。
研究的主要结论发现是低基因丰富度(LGC)存在于75%的患者中,并且与躯干脂肪质量和合并症(2型糖尿病,高血压和严重程度)增加相关。LGC改变了78种宏基因组种(MGS),其中50%与不良的身体成分和代谢表型有关。九种血清代谢产物(包括谷氨酸盐,3-甲氧基苯基乙酸和L-组氨酸)和含有参与其代谢的蛋白质家族的功能模块与低MGR密切相关。术后一年,BS会增加MGR,但尽管RYGB患者的代谢改善比AGB患者大,但术后一年的MGR仍然很低。
总体而言该项研究可以使用浅宏基因组来完成所有测序和分析,进一步扩大样本数量,如果能同时获得人的转录组数据甚至能更加明确的找到菌群变化与特定代谢通路的关联关系。
< 案例二 > 食物与人类肠道微生物组
第二项研究是Dan Knights实验室发表在Cell Host & Microbe,2019的一篇针对34个人17天每日饮食和菌群变化的相关研究,试图揭示日常食物选择与人类肠道微生物组组成之间的精细关系。
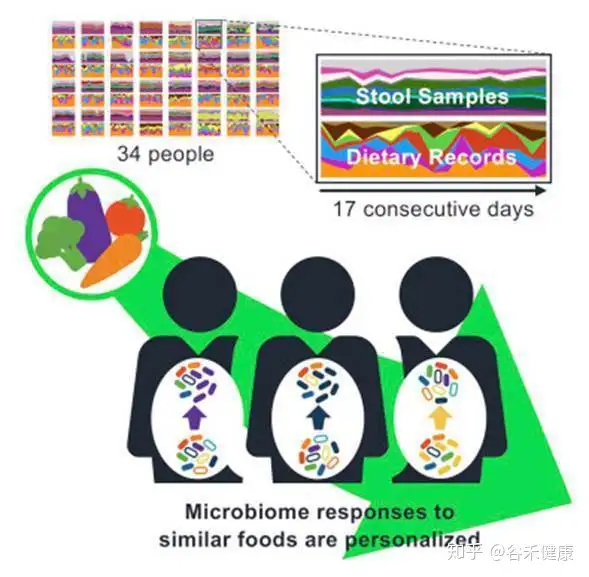
可以看到,研究同时记录了粪便样本的菌群宏基因组和每日的饮食记录。研究的核心在于将每日饮食的食物通过营养构成进行量化,并构建类似物种进化树的食物物候树。
此外由于有每日的数据,可以通过前一日的食物与第二日的菌群数据进行时间序列分析,构建食物与菌之间的关联以及时间相关性。
最后基于菌群数据和前一日饮食来构建模型预测判断后一日的菌群状态,帮助我们了解食物对于个体菌群的影响因素并实现定量和预测。
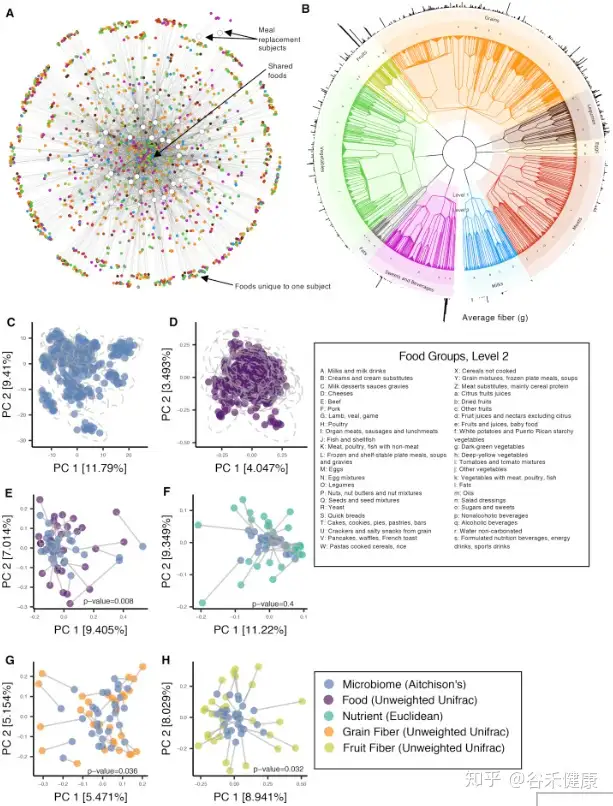
研究中对数据的处理过滤标准如下:删除所有具有低读取计数(每个样品<23,500个读取)的样品。物种级别的分类表仅限于研究对象中至少存在25%的研究日,且在10%的研究样本对象中发现的那些物种。
最后,相对丰度<0.01%的稀有物种被丢弃,将物种数量限制为290个注释。将得到的分类表汇总到较高的分类级别(即属,科,门等),以进行下游分析。
菌群和饮食以及营养构成的堆叠图很好展现了变化和对应。
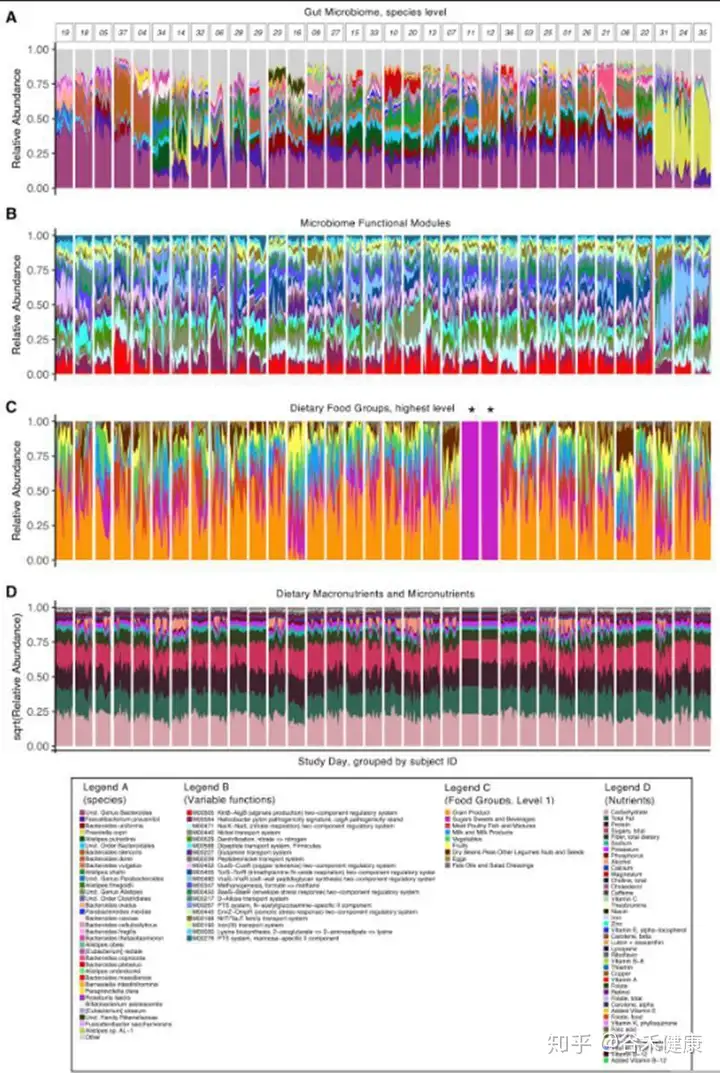
下面这张图很好的显示了饮食食物的变化与菌群变化之间的时间变化关系:
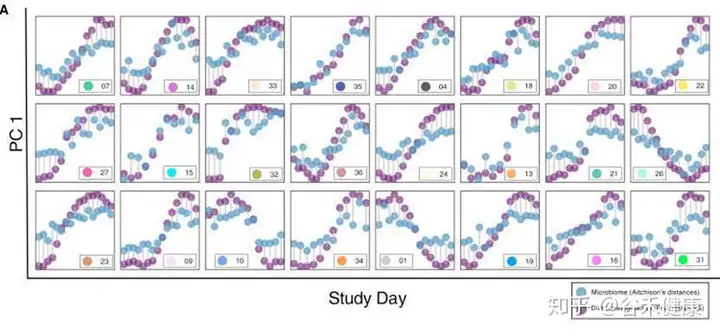
下面这张图通过对每个人单独进行菌属与食物的Spearman相关,展现了菌与食物之间的关联的个体化差异,在特定菌属对应相同食物不同人会出现完全不同方向的变化,这也正是这项研究所揭示的,这种关联关系的复杂性。
本研究虽然有大量样本,但并未进行组装,而是直接使用了Refseq的细菌完成基因组序列作为参考。研究由于样本数量众多,测序深度也很有限,类似研究也可以使用浅宏基因组方式完成。
接下来的一个研究是比较典型的宏基因组组装并与疾病进行关联分析的案例,研究的是类风湿关节炎人群的肠道微生物组的全基因组关联研究。
研究使用较高深度的宏基因组shotgun测序(每个样本平均13 Gb)对日本人群(病例 = 82,对照 = 42)进行了RA肠道微生物组的MWAS分析。MWAS由三个主要的生物信息学分析渠道(系统发育分析、功能基因分析、途径分析)组成。
使用了之前研究中6139个完成拼接日本人肠道宏基因组作为参考序列以及其他几项研究的参考基因组,在过滤部分种过多的基因组之后,最后一共使用了7881个参考基因组。
将QC后的序列直接比对到参考基因组,并根据基因组长度计算对应物种的相对丰度。
基因方面选择denovo组装,使用MegaHIT,然后再contig上完成ORF预测和CD-HIT聚类去冗余,最后与UniRef和KEGG数据库比对。
最后使用bowtie2将测序序列比对到注释后的unigene序列上获得基因丰度,经过KEGG注释得到代谢途径的丰度。研究的数据处理流程图如下:
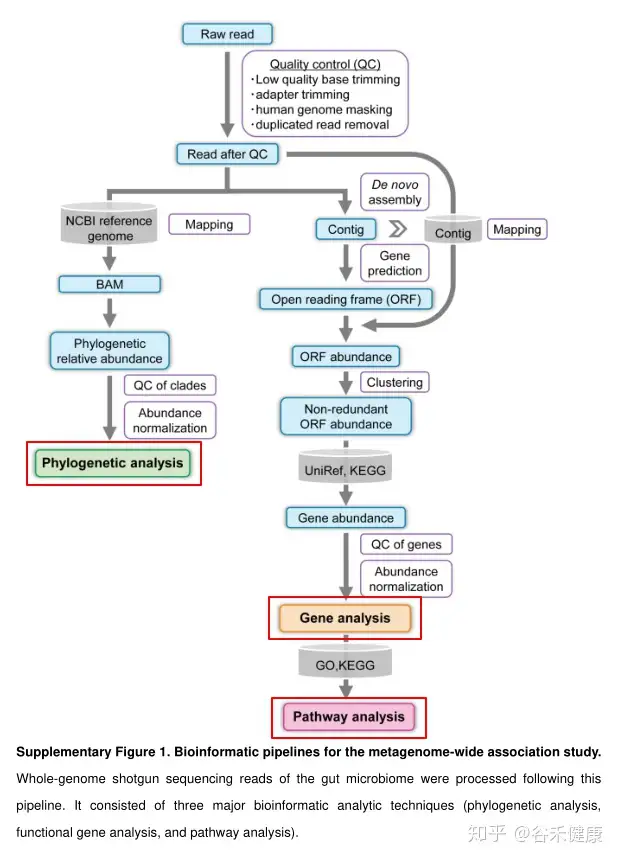
在数据分析流程和方案选择上人体肠道菌群由于研究众多,以及有多个深度测序拼接完成的Binning参考基因组数据集,确实可以直接使用参考基因组直接比对。
对于其他一些环境或来源的样本这个深度的数据量可以考虑独立拼接和分箱。该研究中使用已有参考基因组,大概88%的序列能比对到参考基因组,如果直接组装这个比例应该可以更高一些。另外在获得基因丰度是可以考虑使用Salmon,比对获得基因丰度更为方便。
获得相应数据后对相对丰度,该研究使用Box-Cox transformation对数据进行标准化,并过滤了一些低丰度的菌属。Case-control的相关性分析使用的R的glm2模块,将年龄、性别和测序上机分组作为协变量。
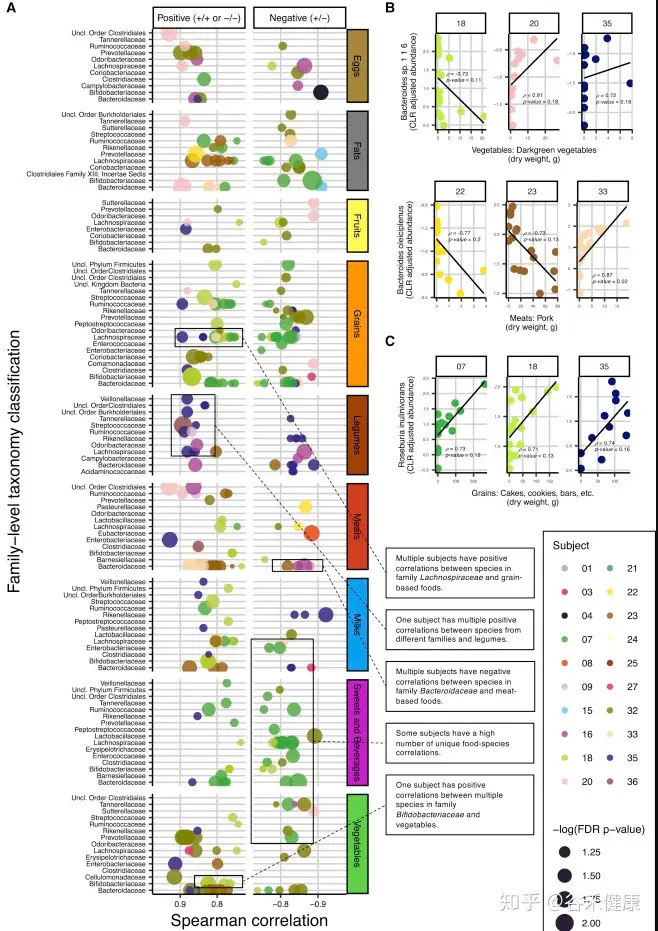
对于菌属的关联分析,最终将显著性结果以火山图和GraPhlAn图的形式展现如下:
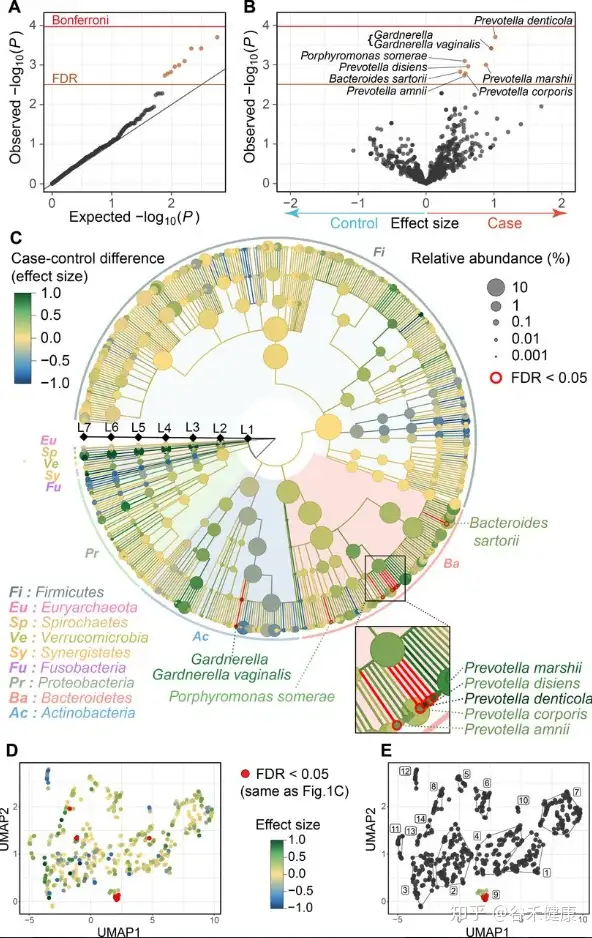
上面其中D图使用每个菌的丰度进行UMAP分析,并映射关联效应的展示比较有意思。
不过在基因层面上并未找到相应的关联,可以看到下图UniRef90的NMDS分布图两组之间无法有效区分,多样性也没有显著差异。
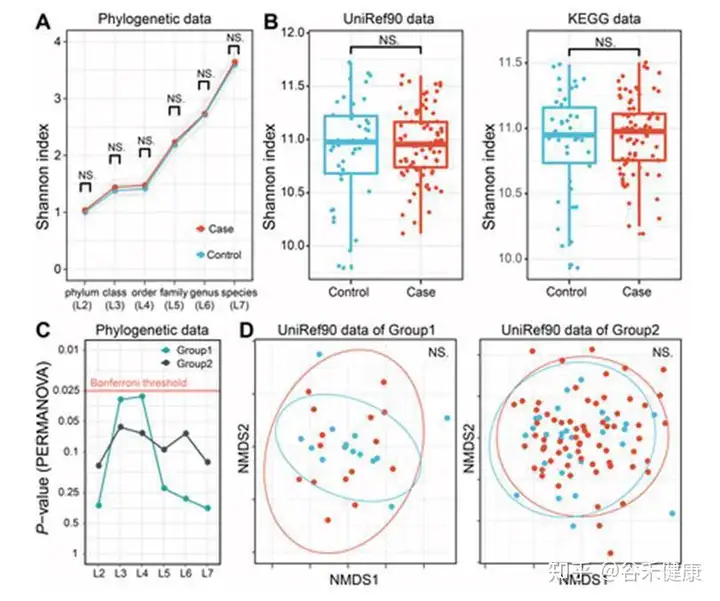
这项研究在菌层面发现了多个普雷沃氏菌属的菌在日本人群中与类风湿性关节炎存在关联,不过除此之外其他方面的发现并不多,仅找到一个基因存在显著关联,涉及的临床调查也相对有限,且人群队列数量不算多,并无独立验证集,因此亮点并不多。如果能纳入免疫相应指标可能能研究的更细致一些。
最后这项研究是对来自永冻土融化梯度的214个样品的宏基因组测序组装了1,529个基因组,揭示了参与有机物降解的关键种群,包括其基因组编码先前未描述的木糖降解真菌途径的细菌。
通过宏基因组denovo组装和分箱Binning,最终获得了1529个永冻土菌群基因组。基于这些数据描绘了永冻土融化梯度下的菌群构成特征,如下图。
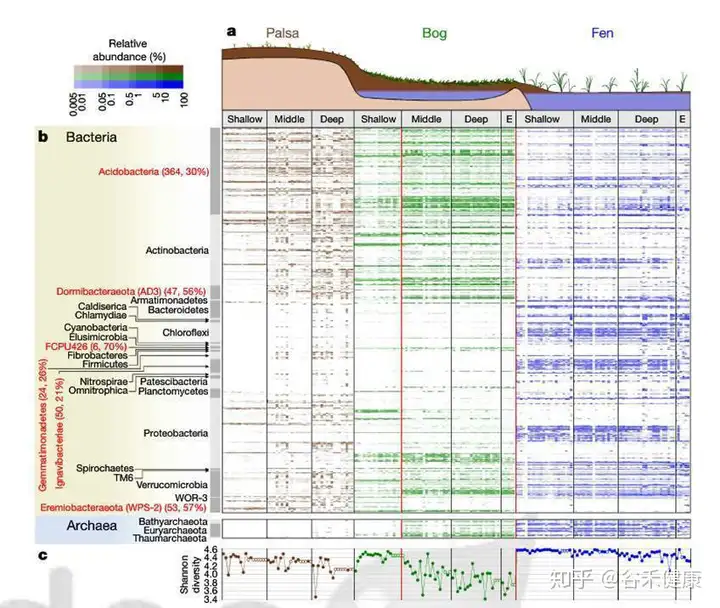
论文是2018年发表的,测序是在2011和12年测的,使用的是CLC Genomics Workbench 较早的4.4版分单样本组装,然后使用MetaBAT进行分箱,最后的标准是70%完成度和低于10%的污染。
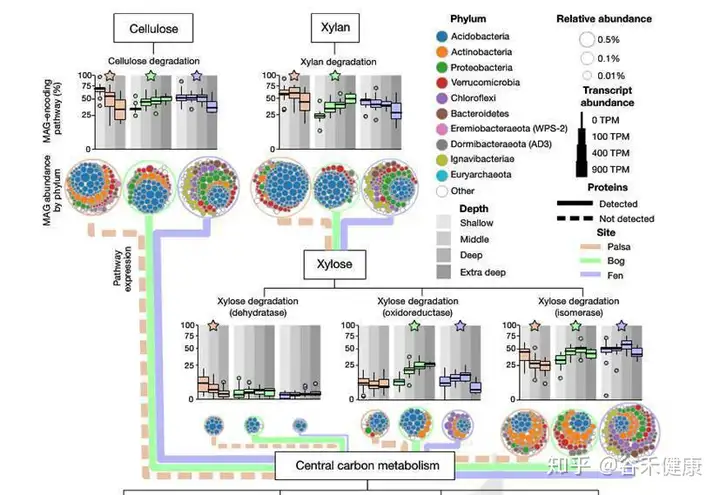
其中糖苷水解酶基因使用dbCAN数据库的HMM进行预测,碳代谢使用KEGG数据。
研究还同时选择了部分样本进行了宏转录组和宏蛋白组的测序,对碳代谢同时结合转录组和蛋白组的数据,展现了特定通路下不同永冻土的菌群构成和表达丰度差异。
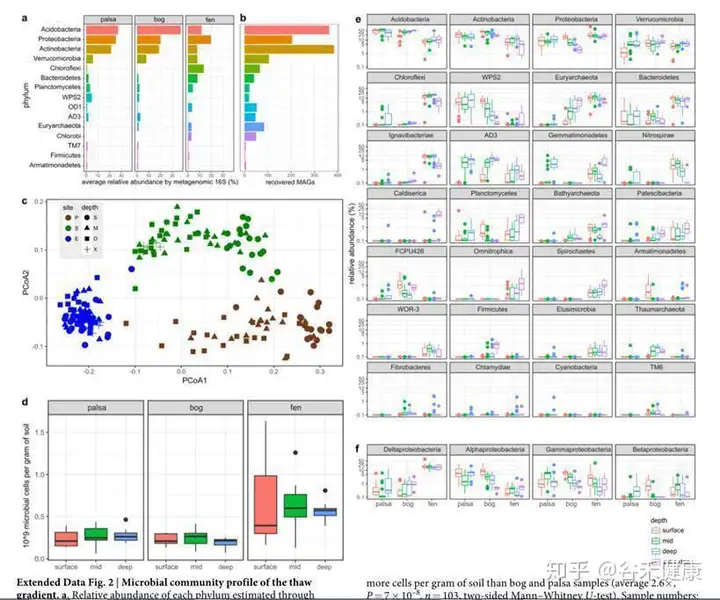
基因组拼接的分布情况,以及不同地域样本分布和菌属丰度情况如下:
木糖降解途径在每个样本中的分布和维恩图,另外详细的展现了主要门对每个代谢途径的贡献和基因表达丰度,如下图:
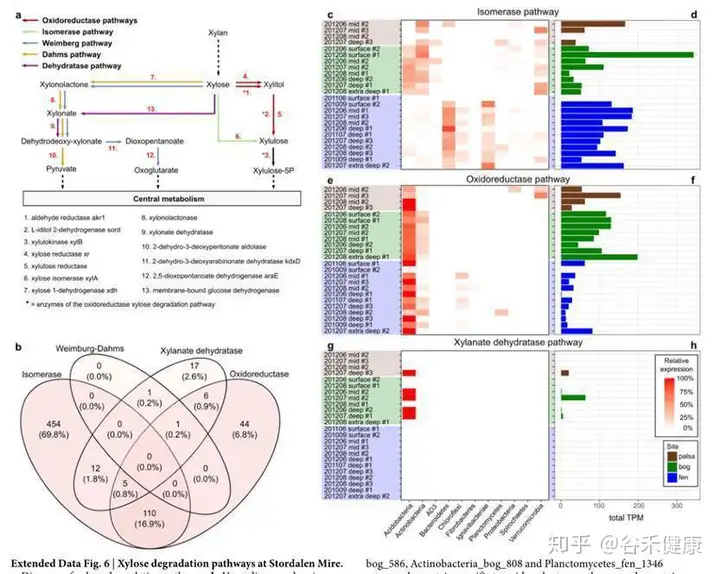
这张图分析了特定菌与地理位置和CO2以及甲烷的浓度的关联性,如下图:
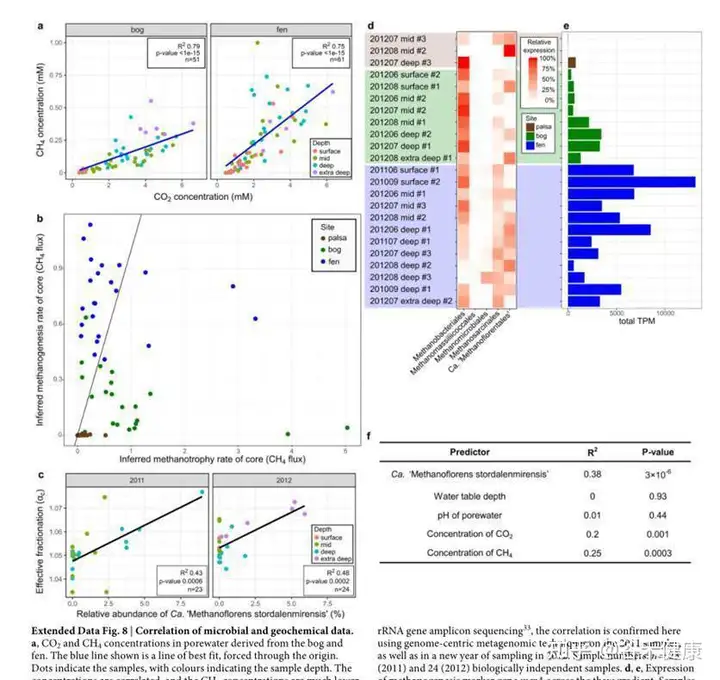
对关键物种的CH 4 :CO2浓度比相关代谢途径的重建,以及相应基因的基因家族分析。
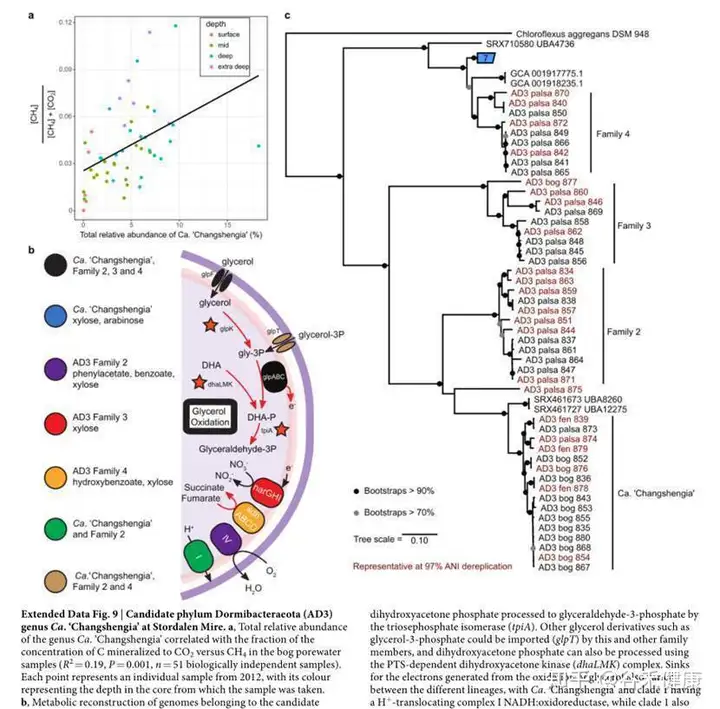
总结一下这项研究,永冻土的菌群参考基因组数据缺乏,该研究从大量地点采集样本重建了1500多个参考基因组。
首先从物种构成特征上与永冻土融化阶段特征进行分析,并与重要环境因子进行分析,锁定重要的特征菌。
然后针对重要的代谢途径和关键基因结合宏转录组和宏蛋白组全面解析代谢途径的分布和差异变化。对关键的物种重建了相关代谢途径并对其相关基因家族进行分析。
研究基本上从头构建了一个生态环境下的菌群结构数据,并利用获得的基因组深度解析特定代谢途径和基因的构成和表达变化,应该说既宽又深。很多样本采集和测序是2011年和 2012年就开展的,虽然测序技术远不如现在成本低和成熟,但是其独特的研究对象和全面深入的分析仍然使整项研究和目前的一些研究相比完成的更加出色。



样本需求量低:常规宏基因组建库建议样本量在500ng以上,公司研发实现了低当量微生物样本提取和建库,保证提取丰度以及片段完整性同时,样本量需求低于同行其他公司要求;对于样本获取困难的样本,也可以选择微量建库,样本量可低至10ng。
免费取样盒和针对性取样建议:粪便及环境样本提供取样盒助力临床/科研取样,人体口腔、痰液、腹水、脑脊液、尿液、皮肤等高寄主细胞含量样本可根据我们的处理方案简单处理后大幅降低宿主DNA比例。
严格标准的实验流程:自动化样品处理平台辅助,每轮设置阳性对照,上轮检测样本对照,阴性对照。评估污染,轮次比对,最大化减少误差,保证样本重复性和稳定性
Illumina测序平台:宏基因组测序(PE150)采用先进的Illumina Novaseq测序平台,快速、高效地读取高质量的测序数据、结合样品特点和数据的产出,充分挖掘环境样品中的微生物菌群和功能基因
大数据分析流程质量流程控制严格:优化的数据质量控制,包括过滤比对质量低、非特异性扩增、覆盖度低、低复杂度的序列,从而快速准确获得样本中微生物信息及其丰度信息,最大化提高质量数据
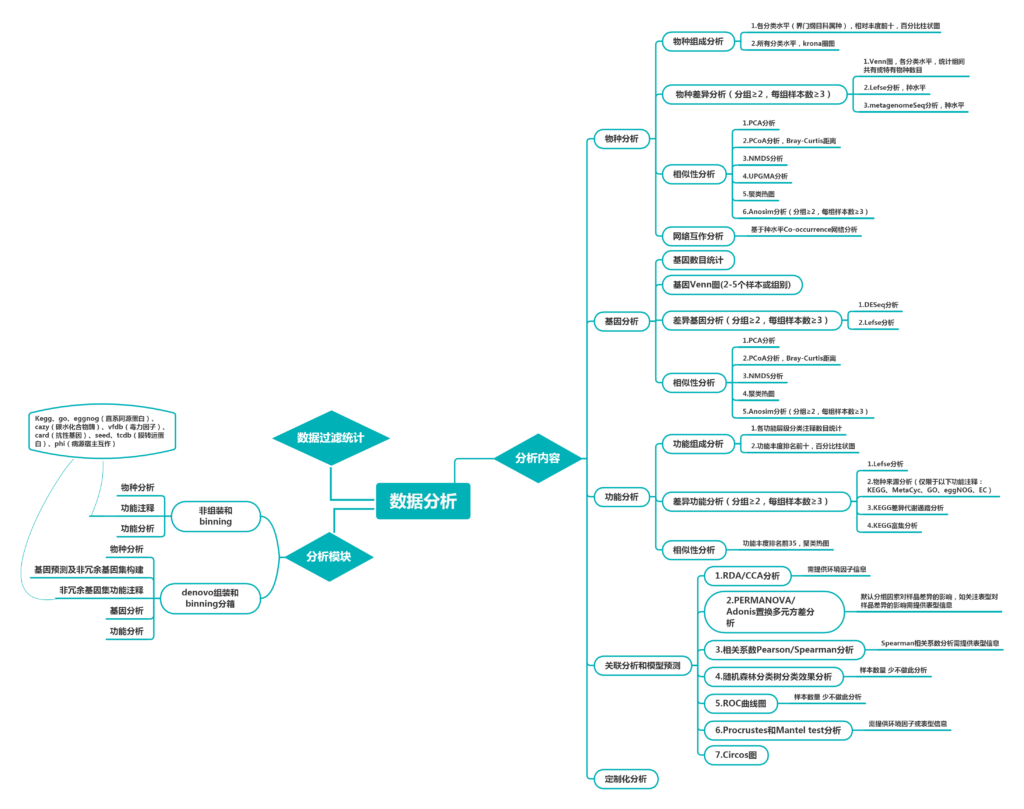
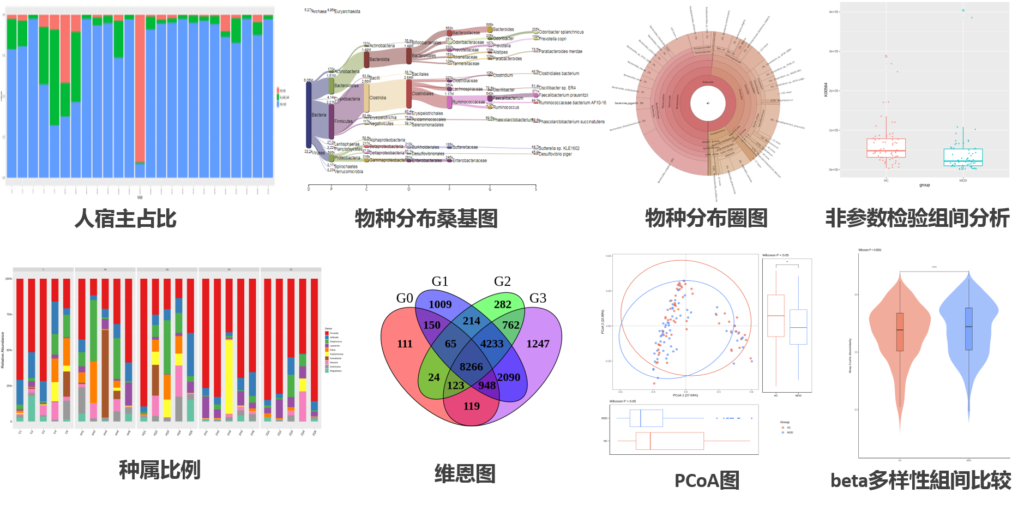
分析内容丰富全面:物种分析,基因预测与分析,多样性和相似性分析,功能分析,网路互作分析,代谢网络,关联分析等
完整详细的报告:提供质检实验报告,分析统计报告,分析报告解读,原始数据
高效个性化服务:在线项目系统方便您及时查看项目动态和下载报告以及与分析人员高效交流,免费支持个性化图表修改以及重新分组出报告。
价格低,周期快:包括提取,测序到分析,最快2周出报告。
大数据分析团队和多中心大项目分析经验(团队主要源自浙江大学,包括生物信息学,计算机,微生物以及统计分析等专业,积累了多年的大健康项目多中心项目分析经验,有助于宏基因组大数据,多样本,多表型,多组学联合分析
兼容性强的合作模式:有专门团队负责,提供切实可行的项目方案,兼顾临床和科研双需求模式。
3 comments so far
张琦Posted on4:08 下午 - 5月 7, 2021
土壤微生物多样性宏基因组
谷禾健康Posted on10:44 上午 - 5月 21, 2021
您好,可以做的,具体可以联系牛博13336028502
谷禾健康Posted on11:08 上午 - 5月 25, 2021
可以做的,周期是2-3周,价格咨询13336028502