-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
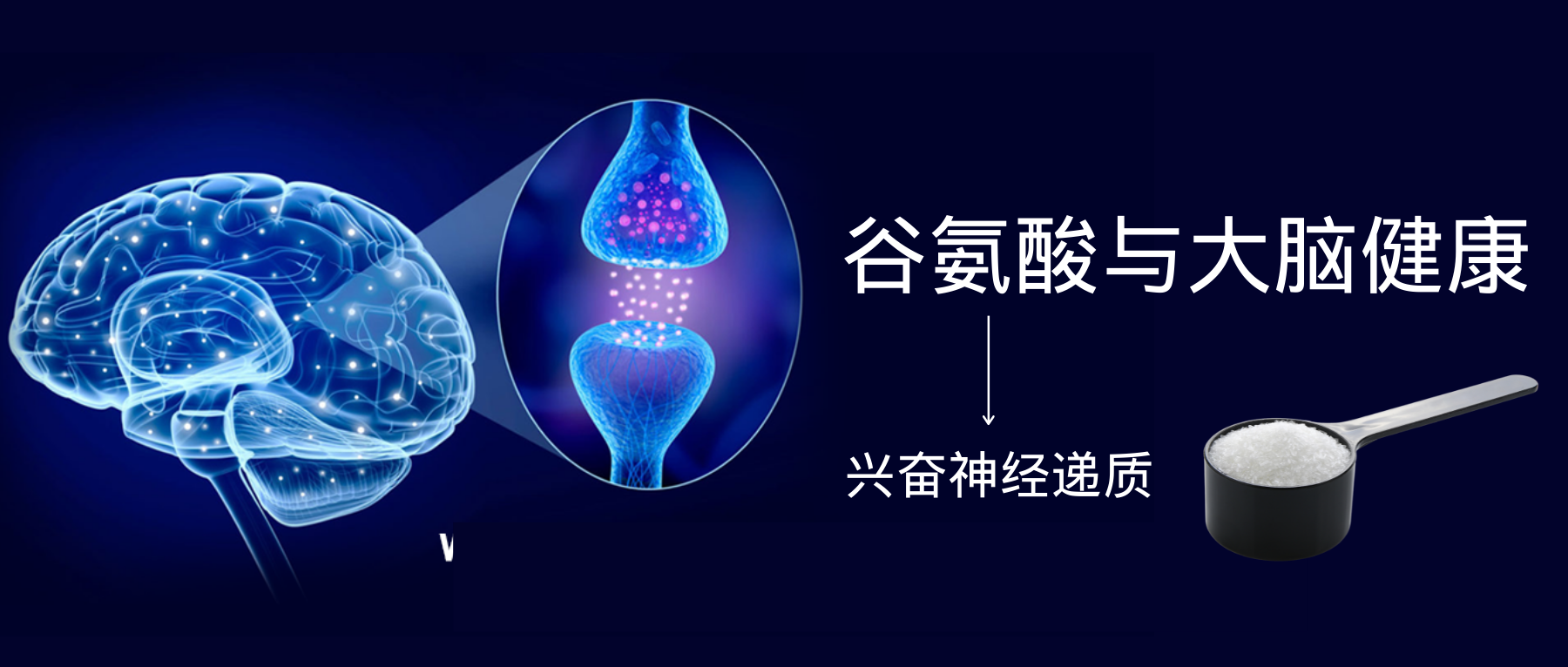
谷禾健康
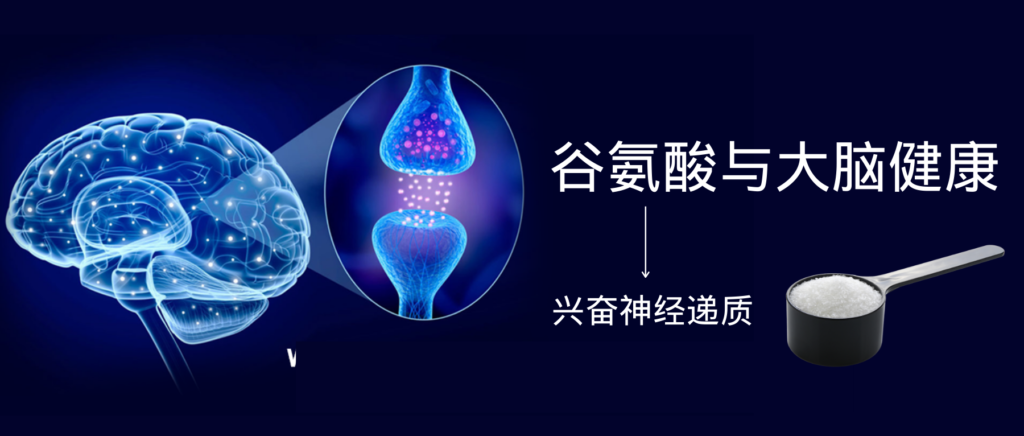
关于谷氨酸
谷氨酸是一种多功能氨基酸,它处于多种代谢途径的十字路口,不仅参与消化系统和免疫系统,还与大脑健康密切相关。
谷氨酸是大脑中最丰富的游离氨基酸,也是大脑主要的兴奋性神经递质。它可以帮助我们说话、处理信息、思考、运动、学习新事物、存储新知识和集中注意力学习等。
学习和记忆是脑的高级功能,是一个相当复杂的生理过程,其神经生物学基础是突触可塑性。突触的传递是谷氨酸及其受体实现的。
谷氨酸也是γ-氨基丁酸(GABA)的前体,γ-氨基丁酸是脑内普遍存在的抑制性神经递质,这二者之间的平衡对于我们大脑健康非常重要(下次我们专门讲)。
很早以前人们就已经知道,谷氨酸广泛存在于人体各组织和食物以及母乳,这凸显了谷氨酸对人类成长中脑和躯体发育的重要性;但直至20世纪80年代,谷氨酸在中枢神经系统(CNS)和大脑内的作用才得到认识。在过去的30年中,越来越多的研究进展已揭示了谷氨酸及其受体在神经退行性疾病疾病(阿尔茨海默氏症、肌萎缩侧索硬化症、多发性硬化症、癫痫、帕金森等)以及肠道疾病如克罗恩病 (CD) 和溃疡性结肠炎 (UC)的病因中起核心作用。
根据区域的不同,每公斤脑组织含有 5-15 mmol 谷氨酸,比其他任何氨基酸都多。因此,谷氨酸应该在正确的时间、正确的位置以正确的浓度存在。
细胞应该对谷氨酸有正确的敏感性,并且有足够的能量来承受正常的刺激,并且应该以适当的速率从正确的位置去除谷氨酸。谷氨酸过多和谷氨酸过少都是有害的。
海马中靠近血管的突触周围的谷氨酸转运蛋白分布示意图
Zhou Y and Danbolt NC. J Neural Transm. 2014
四个谷氨酸能神经末梢 ( T ) 显示在树突棘 ( S ) 上形成突触。指示星形胶质细胞分支 ( G )。请注意星形胶质细胞的 EAAT2(红点)和 EAAT1(蓝点)的密度非常高。
随着工业水平的发展和人类口味的发展,越来越多的谷氨酸衍生物添加到许多食物中,以赋予“鲜味”味道。鲜味感知分布在人类口腔和胃肠道中的多个受体系统,这些系统激活大脑中涉及不同功能的多个区域,从食物识别到与特定食物相关的情感价值的形成,进而影响食欲和情感等。
加工食品是游离谷氨酸的最大来源。因此目前谷氨酸过量带来的神经毒性不容忽视。一些研究将味精以及谷氨酸衍生添加剂与体重增加、麻木/刺痛、虚弱、高血压、哮喘发作、胃肠道问题、代谢综合征和敏感人群的短期副作用联系起来。
现在强有力的证据表明肠道微生物产生神经活性分子,如神经递质(即去甲肾上腺素、多巴胺、血清素、GABA 和谷氨酸)和代谢物(即,色氨酸代谢物,短链脂肪酸等)维持宿主和细菌之间跨界跨区域交流。谷氨酸代表了在这种跨界交流中活跃的众多神经活性分子之一。
在我们的检测实践中,也发现在精神科病人或存在精神症状的人群中,谷氨酸指标异常。

这些表明谷氨酸以及肠道菌群的异常与大脑健康问题存在关联,它们之间的因果及其发病机制还需更大和更精细的研究和临床探索,但是至少我们可以看到谷氨酸及其与肠道菌群和精神健康方面不可忽视的关联。
本文主要讨论谷氨酸是什么,对人体的健康益处和影响,通过食物或生活方式如何改善谷氨酸缺乏或中毒症,以及共同探讨通过传统药理学方法以及使用产生神经活性分子的益生菌作为治疗神经胃肠道和/ 或精神疾病以及相关的中枢神经系统疾病,如焦虑和抑郁。
谷氨酸(Glutamic acid), 化学构成为2-氨基-5羟基戊酸,作为一种非必需氨基酸,但却是人体最丰富的氨基酸,广泛存在与大脑和肌肉中,是谷氨酰胺,脯氨酸以及精氨酸的前体。
谷氨酸也被定义为功能性氨基酸,意味着其可以调节关键代谢途径以改善动物和人类的健康、生长、发育和繁殖。这个将我们的视野扩展到营养必需或非必需氨基酸的营养范式之外。
在哺乳动物的中枢神经系统中,由于与特定受体的相互作用,谷氨酸充当重要的兴奋性神经递质。此外,谷氨酸还在细胞能量产生和蛋白质合成,免疫功能中发挥重要作用 。
化学信使:谷氨酸将信息从一个神经细胞传递到另一个神经细胞。
脑细胞的能量来源:当细胞的主要能量来源葡萄糖储备低时,可以使用谷氨酸。
学习和记忆的调节:谷氨酸有助于随着时间的推移增强或减弱神经元之间的信号,以塑造学习和记忆。
疼痛传递器:较高水平的谷氨酸与增加的疼痛感有关。
睡眠和清醒介质:大鼠模型研究表明,当我们清醒或快速眼动 (REM) 睡眠期间,谷氨酸水平最高。丘脑是个例外,在非快速眼动睡眠期间谷氨酸水平最高。
谷氨酸是所谓的兴奋性神经递质。它的作用很像兴奋剂,就像咖啡一样。太多会导致问题,但太少也不好。
如果谷氨酸太少,我们无法对进入大脑的刺激做出快速反应,无法很好地记住事物,难以集中注意力,学习会更加困难。
过多的谷氨酸会导致兴奋性毒性,从而破坏神经元。由于谷氨酸是神经元的兴奋剂,过多会导致神经元过度激活并死亡。
如果高水平不受控制,这种神经递质会过度刺激细胞,一直到它们采取剧烈行动自杀以保护周围的细胞。
我们体内的细胞一直在死亡,其中大部分是可以被替换的。然而,谷氨酸导致自杀的神经元,大脑无法制造新的来代替,因此保持它们的健康和安全很重要。
谷氨酸作为兴奋性毒素的作用,与多种神经退行性疾病有关,例如多发性硬化症、 阿尔茨海默病和肌萎缩性侧索硬化症(ALS 或 Lou Gherig 病)。谷氨酸失调也被认为是纤维肌痛和慢性疲劳综合征的原因之一。
两种形式的谷氨酸,结合的和游离的,是显着不同的。
谷氨酸的结合形式存在于完整的蛋白质来源中。结合谷氨酸是天然存在于未加工食品中的氨基酸形式,尤其是蛋白质含量高的食品。它与其他氨基酸结合,当你吃它时,它通常被缓慢消化和吸收,并能够精确调节你摄入的量。这种形式的谷氨酸很少有任何敏感性。因为多余的量可以简单地通过废物排出,以防止毒性。
在蛋白质中,谷氨酸提供负电荷,这可能对稳定蛋白质结构很重要。例如,涉及谷氨酰残基的离子对于稳定转录因子 GCN4 的亮氨酸拉链结构很重要。带电荷的残基,例如谷氨酸,经常出现在球状蛋白质的外表面。表面上极性残基的重要性,很容易通过血红蛋白 (HbS) β 链 6 位上的谷氨酸 → 缬氨酸突变的破坏性影响来证明,从而在具有这种突变的杂合子和镰状细胞中产生镰状细胞性状纯合子个体的疾病。
脱氧血红蛋白在 EF 连接处有一个疏水口袋(phe B85,leu B88)。表面上允许谷氨酸,因为它在能量上不利于它与疏水袋相互作用,因此脱氧 HbA 保持可溶性。然而,在 HbS 中取代它的缬氨酰残基从表面突出并很容易嵌入疏水袋中,导致 HbS 分子粘在一起,导致长而僵硬的纤维扭曲红细胞。
谷氨酸不仅在合成蛋白质时掺入蛋白质中,而且可以在合成后作为翻译后修饰以聚谷氨酰尾的形式添加。例如,微管蛋白的多谷氨酰化被认为会影响其与其他蛋白质的相互作用,例如微管相关蛋白 (MAP) 和分子马达。
在蛋白质中,谷氨酸与阳离子的结合相当弱,但它对钙的亲和力可以通过维生素 K 依赖性 (VKD) 羧化作用大大增加,这种羧化作用在翻译后将 γ-羧化谷氨酰残基 (gla) 引入蛋白质中。
所有 VKD 蛋白都含有一个同源氨基酸序列,该序列将蛋白靶向羧化酶。羧化发生在“gla 结构域”内的多个谷氨酸残基上。VKD 蛋白包括许多参与止血的蛋白:凝血酶原和因子 VII、IX 和 X。其他 VKD 蛋白参与骨形态发生(骨 gla 蛋白和基质 gla 蛋白)。
VKD 羧化酶功能的抑制对于基于香豆素的抗凝治疗至关重要,因为 4-OH 香豆素类似物会抑制维生素 K 环氧化物还原酶 (VKOR),该酶将维生素 K 环氧化物重新转化为还原的维生素 K.
这些研究清楚地说明了谷氨酸残基在蛋白质中所起的关键作用之一,包括它们的翻译后修饰。
另一方面,游离谷氨酸是吸收更快的修饰形式。谷氨酸敏感性在游离形式中更为常见。游离谷氨酸不与其他氨基酸结合,更快地被吸收到您的系统中。这种快速吸收率会导致血液中谷氨酸水平的峰值。
一些天然食物来源含有游离谷氨酸,但最成问题的来源之一是加工和包装食品。谷氨酸钠 (MSG) 形式的谷氨酸在这些产品中用作防腐剂和增味剂。这种形式存在于一些完整/未加工的食品中,但在许多超加工和包装食品中更为常见。
多年来,味精一直被用来给食物调味,尤其是汤、薯条和某些类型的亚洲食物。一些吃很多这些食物的人在进食后会出现症状。
谷氨酸被归类为非必需氨基酸,这意味着它可以在体内以足够的数量合成。事实上,它必须在体内合成。这是对各种动物肠道进行仔细平衡研究的结果,结果表明膳食谷氨酸几乎在肠道内定量代谢,主要是通过肠细胞。
这首先由 Windmueller 和 Spaeth 在1975 -1980年间使用灌注的大鼠肠道以及在大鼠体内进行了展示。随后,对仔猪、早产儿和成年人的研究表明,膳食谷氨酸被肠道广泛代谢。
1993年后,Matthews 和Battezzati 等在人群中做了临床实验,表明大部分的肠内谷氨酸可以通过代谢排出。事实上,膳食谷氨酸是一种重要的代谢燃料,其中大部分被完全氧化为 CO2。对仔猪的详细研究表明,只有小部分的肠内谷氨酸出现在门静脉血中。
在肠内喂养的早产人类婴儿中,大约 74% 的谷氨酸在第一次通过肠道时被去除。肠道谷氨酸代谢的后果之一是血浆谷氨酸水平不受膳食谷氨酸的特别影响。事实上,健康人循环中的谷氨酸盐严格维持在相当低的浓度。
谷氨酸的肠道代谢有一个非常重要的后果:身体的大部分谷氨酸需要内源性合成。谷氨酸可以以两种不同的方式合成。
天然富含谷氨酸的食物通常(但不总是)富含蛋白质,包括:
肉、家禽、奶酪、蛋、蘑菇、酱油、番茄、葡萄、鱼露、西兰花、豌豆、核桃、骨汤…
谷氨酸和谷氨酰胺的区别
谷氨酸很容易与谷氨酰胺混淆,谷氨酰胺是体内富含的另一种重要氨基酸。
两者的化学结构略有不同;谷氨酰胺具有氨 (-NH3) 基团,而不是羟基 (-OH) 基团。
谷氨酰胺
是一种非必需氨基酸(蛋白质的组成部分)。它存在于植物和动物蛋白中。一般在肌肉中大量制造,它是体内最丰富的氨基酸。谷氨酰胺有助于肠道功能、大脑功能、免疫系统、氨基酸产生和压力。
压力和某些药物会消耗它,肌肉萎缩是常见的结果。在身体需要氮的地方(例如,在伤口修复中),其中三分之一来自谷氨酰胺。
谷氨酰胺有助于“治愈”肠道。它可以修复肠壁中受损的细胞,也是肠道内免疫球蛋白产生和免疫系统的重要氨基酸来源。
谷氨酸
谷氨酸也是一种非必需氨基酸。它可以通过多种方式进入人体,例如,以蛋白质或味精、谷氨酸钠的形式。它可以完整或以结合形式提供。
但在体内,它也可以作为多种化合物的分解产物——比如来自谷氨酰胺,也来自叶酸和葡萄糖。谷氨酸广泛存在于肌肉中所有的蛋白质储存中。
谷氨酸对大脑和神经功能也是必不可少的。它是神经中的主要传递器。具有更多谷氨酸受体的人往往具有更高的智商。然而,大脑通过血脑屏障摄取的谷氨酸非常低。绝大多数谷氨酸是谷氨酰胺转化的结果,因为神经元不能从大脑内的葡萄糖中产生谷氨酸。
谷氨酰胺是谷氨酸的来源,它是由谷氨酰胺酶产生的。
谷氨酰胺-谷氨酸循环
星形胶质细胞摄取的谷氨酸可通过三羧酸循环代谢,用于蛋白质合成或转化为谷氨酰胺。谷氨酸向谷氨酰胺的转化由谷氨酰胺合成酶(GLUL) 以 ATP 依赖性方式催化。但是由于谷氨酰胺转运蛋白迄今尚未在脑组织的末端中得到阳性鉴定。因此谷氨酰胺-谷氨酸循环已经研究和争论了很多年,至今没有明确答案。
1) 支持大脑功能
谷氨酸是脑功能正常的重要神经递质
大脑和脊髓(中枢神经系统)中几乎所有的兴奋性神经元都是谷氨酸能神经元。作为主要的兴奋性神经递质,它向大脑和全身发送信号。它有助于认知功能、记忆、学习和其他大脑功能 。
人脑的质子-MRS 研究显示谷氨酸浓度非常高(10-12 mM),但这平均了不同亚细胞区室中非常不同的谷氨酸浓度,从脑脊液中的约 1 μM 到分泌颗粒中 100 mM。
谷氨酸通过两种主要类型的受体在中枢神经系统中发挥作用:
每种受体有许多亚型。对大脑中谷氨酸信号传导的详细说明不在这详细展开,只要了解其在突触可塑性中的作用,这是其在学习和记忆等认知功能中的作用的基础。
有限的研究将大脑谷氨酸水平低与神经和精神疾病联系起来。例如,在一项研究中,精神分裂症成人的谷氨酸水平低于健康成人。
代谢型谷氨酸受体5型(mGluR5)含量低表明癫痫患者大脑发育不良。
在小鼠中,低谷氨酸释放已被用于模拟自闭症谱系障碍。
在大鼠中,亮氨酸会增加谷氨酸进入大脑,这有助于在脑损伤后恢复大脑功能 。
2) 血脑屏障和谷氨酸
血脑屏障位于血液和大脑间质液之间。它是在哺乳动物中受脑细胞影响后由内皮细胞形成的。另一个屏障位于分泌脑脊液 (CSF) 的脉络丛上皮中。
这些屏障很重要,从生理学的角度来看,维持大脑稳态,从药理学的角度来看,可以防止药物进入脑组织。
神经系统通过屏障将自身与血液隔离。谷氨酸是一种非必需氨基酸,不能穿过血脑屏障,必须在脑细胞内局部由谷氨酰胺和其他前体产生 。
然而,如果血脑屏障“渗漏”(由于血脑屏障损伤或功能障碍导致通透性增加),血液中的谷氨酸可能会进入大脑,这会使身体增加炎症。
更高水平的炎症也会导致血脑屏障发生渗漏。这放松了对进入大脑的内容的控制。在这种情况下,反应性代谢物可以进入大脑。
如果你已经对谷氨酸敏感,并且还在吃富含游离谷氨酸的食物,这些情况可能会使症状加重。
3) GABA 的前体(与GABA保持平衡)
身体使用谷氨酸来产生神经递质 GABA(γ-氨基丁酸),这是一种在学习和肌肉收缩中起重要作用的抑制性神经递质。
在大脑中,GABA 作用于下丘脑,帮助调节睡眠、温度、HPA 轴和自主神经系统。下丘脑的主要作用是使身体保持体内平衡,而没有 GABA 则无法做到这一点。
GABA—谷氨酸:
“相互作用,相辅相成,适量才和谐”
正如谷氨酸是一种兴奋性神经递质一样,GABA(γ-氨基丁酸)是一种抑制性神经递质。它的目的是让大脑和神经系统慢下来。这两种神经递质在大脑中平衡工作。
GABA 和谷氨酸之间复杂而相互关联,这有助于保持身体平衡,任何一种过量都会导致严重的疾病状况。GABA 和谷氨酸有助于保持交感神经和副交感神经系统之间的平衡。如果没有这两种神经递质,我们会不断地发现自己受到这些系统中的一个或另一个的刺激。我们要么对外部刺激反应过度,要么反应不足。
“一个太少,另一个就会负重前行”
如果 GABA太少,就会过分强调谷氨酸的作用,从而在肾上腺疲劳、慢性疲劳、惊恐发作和化学敏感性的发展中发挥重要作用。体内的 GABA 过少可能会导致所谓的“超负荷现象”,即由于谷氨酸含量高而对神经元产生过多的刺激。由于过多的刺激,这最终导致神经元死亡。
据报道,代谢型 Glu 和 GABA B受体,以及细菌周质氨基酸结合蛋白,可能是从一个共同祖先进化而来的 。
Mazzoli R and Pessione E. Front Microbiol. 2016
谷氨酸能/GABA能神经网络的表示,包括兴奋性(谷氨酸能)锥体神经元(红色)、抑制性(GABA能)神经元(蓝色)和相关连接。左侧的抑制神经元集成到反馈 (FB) 电路中,而右侧的抑制神经元集成到前馈 (FF) 环路中。针对锥体细胞体细胞或树突的抑制性突触被浅蓝色染色包围,这表明背景 GABA 浓度有助于整体抑制。
4) 在免疫中发挥作用
谷氨酸受体存在于免疫细胞(T 细胞、B 细胞、巨噬细胞和树突状细胞)上,这表明谷氨酸在先天免疫系统和适应性免疫系统中均发挥作用。
谷氨酸还参与肺、肾、肝、心脏、胃、骨骼和免疫系统组织的功能。
科学家们正在研究谷氨酸对调节性 T 细胞(Treg)、B 细胞和炎症性神经退行性疾病的影响。
一项研究得出结论,谷氨酸对正常以及癌症和自身免疫病理性 T 细胞具有有效作用。这意味着它可能能够增强对抗癌症和感染的有益 T 细胞功能 。
根据至少一项针对多发性硬化症小鼠模型的研究,这些受体还具有抑制自身免疫发展和保护中枢神经系统免受炎症影响的潜力。
不过,大多数情况下,科学家发现谷氨酸对免疫系统有益还是有害,都取决于体内的浓度。
谷氨酸诱导的神经元细胞损伤的关键机制之一是小胶质细胞释放谷氨酸。小胶质细胞本质上是大脑和神经系统的常驻免疫细胞。了解神经系统和免疫系统之间的联系至关重要。小胶质细胞释放谷氨酸是一个关键环节。
一旦谷氨酸的游离池增加,谷氨酸受体,如 NMDA 就会被过度刺激。然后,这会导致大量钙离子流入细胞,导致神经元细胞损伤和细胞死亡。
许多因素可以作为小胶质细胞激活的诱导剂。汞毒性已显示可迅速诱导小胶质细胞活化,并且汞毒性已显示可改变小鼠大脑中谷氨酸的转运和摄取。有毒金属镉也显示出强大的诱导小胶质细胞活化的能力。
基本上任何类型的“非自身”分子都能够引发大脑和神经元组织中的小胶质细胞活化。这包括各种病原体和抗原、病毒、疫苗、佐剂以及任何穿过血脑屏障的外源物质。
5)支持肠道
我们从食物中获得的谷氨酸为肠道细胞提供能量,并有助于激活消化系统。
在胃肠道上皮细胞的顶膜中发现了多种介导谷氨酸吸收的转运蛋白,主要存在于小肠中,但也存在于胃中,而氨基酸从管腔到门户的转运很少或没有血液出现在结肠中。它是营养物质吸收和代谢的主要能量来源。
谷氨酸是肠细胞的主要营养素之一。对不同动物模型(包括早产儿和成人)的几项研究一致认为,存在于 GI 腔中的大多数谷氨酸被氧化为 CO2,或者其次被肠粘膜转化为其他氨基酸。
只有一小部分(5% 到 17%,取决于研究)摄入的 谷氨酸被转运到门静脉循环,但这通常不会在很大程度上影响血浆中的谷氨酸浓度。此外,血浆谷氨酸浓度增加(摄入谷氨酸后)并不一定会影响脑组织中的谷氨酸浓度,因为人们普遍认为谷氨酸不能通过血脑屏障。
发现啮齿动物的脑组织需要血浆谷氨酸浓度增加 20 倍(或更多)。膳食摄入(或肠道微生物群的谷氨酸生物合成)后血浆中达到如此高的谷氨酸浓度似乎不太可能。
谷氨酸还可以通过帮助产生抗氧化剂谷胱甘肽来保护肠壁。
一项动物研究发现,补充 L-谷氨酸有助于改善仔猪的肠道完整性,这有利于营养物质的消化和吸收。
谷氨酸还可以预防由于幽门螺杆菌 和长期使用阿司匹林等非甾体抗炎药(非甾体抗炎药)引起的胃肠道损伤。
α-gustducin,一种味觉特异性 G 蛋白,在胃和肠中的作用导致人们认识到味觉细胞存在于肠道中。现在很清楚,胃和小肠中都存在类似味觉的谷氨酸受体和细胞。在胃中发现了离子型和代谢型谷氨酸受体很明显,谷氨酸是唯一能刺激传入胃迷走神经的氨基酸。
谷氨酸的胃内输注刺激特定的前脑区域,包括边缘系统和下丘脑。它还刺激胃收缩活动。谷氨酸与 IMP 一起刺激大鼠十二指肠中的碳酸氢盐分泌,这可能是中和胃蛋白消化过程中产生的胃酸的保护作用。
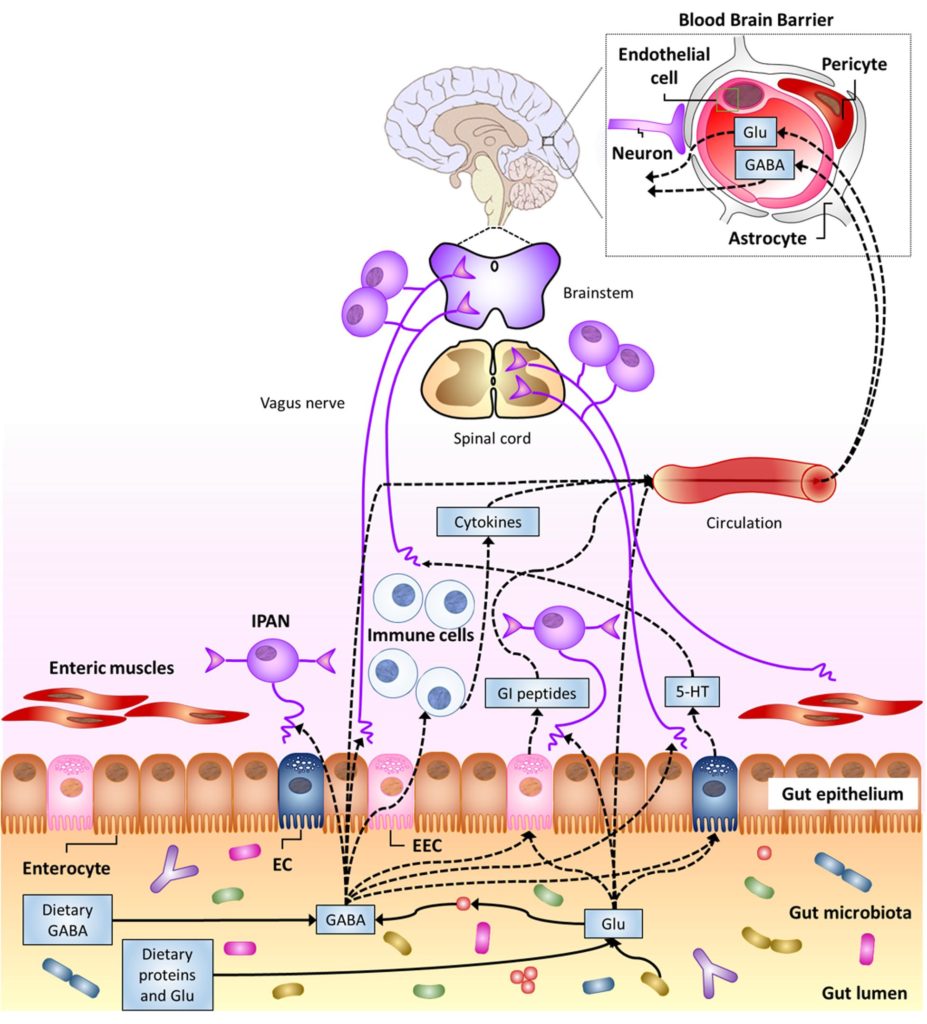
Cryan, Dinan,2012
肠道中 Luminal Glu/GABA(饮食或肠道微生物群来源)影响神经系统的潜在途径。5-羟色胺,血清素;EC肠嗜铬细胞;EEC,肠内分泌细胞;GI,胃肠道;网;内在初级传入神经元
此外,包括普通拟杆菌和空肠弯曲杆菌在内的肠道微生物群会影响谷氨酸代谢,并减少谷氨酸代谢物 2-酮基谷氨酸。同时,具有谷氨酸消旋酶的肠道细菌,包括谷氨酸棒杆菌Corynebacterium glutamicum、乳发酵短杆菌 Brevibacterium lactofermentum和Brevibacterium avium,可以将 L-谷氨酸转化为 D-谷氨酸。由肠道细菌代谢的 D-谷氨酸可能会影响痴呆患者的谷氨酸 NMDAR 和认知功能。
6) 支持肌肉功能
谷氨酸可能在肌肉功能中发挥重要作用。
在运动过程中,谷氨酸在提供能量和支持谷胱甘肽生产方面起着核心作用。
然而,在动物模型中,谷氨酸可能会延缓缺乏维生素 D 的动物的肌肉萎缩症。进一步的研究应该探索谷氨酸、肌肉功能和肌肉萎缩疾病之间的联系。
7)对骨骼很重要
谷氨酸也适用于骨骼和肌肉,用于神经和非神经信号。神经递质对骨骼的健康很重要,并且可能在多种骨骼疾病的潜在治疗中发挥作用。
8)谷氨酸与味觉
鲜味,特别是谷氨酸,被认为可以调节对富含蛋白质的食物的食欲反应。因此,它们在评估食物的营养价值方面发挥着重要作用。
谷氨酸通过激活鲜味受体发挥关键的信号传导作用。这些味觉受体的激活还需要作为共配体的 5′-核糖核苷酸,例如单磷酸肌苷 (IMP) 或单磷酸鸟苷。
对于完全表达鲜味所需的受体多样性仍然存在一些不确定性。当然,涉及 G 蛋白偶联受体 T1R1 和 T1R3 的异二聚体。此外,最近利用 T1R3 敲除小鼠的研究表明,这些动物继续区分鲜味和其他促味剂,这表明其他受体的作用。特别是,两种代谢型谷氨酸受体 mGluR1 和 mGluR4 可能很重要。
一个促成因素是食用加工食品,由改良的游离形式谷氨酸制成的。例如,谷氨酸用于制造味精(或谷氨酸钠),这是一种合成化学物质,会添加到许多食品中,增加咸味和吸引力。

但要注意的是,有些时候这些高水平可能不是由于饮食中的谷氨酸,而是身体的其他缺陷或功能障碍。
过量水平可能是由压力或对谷氨酸脱羧酶 (GAD) 的自身免疫反应引起的,谷氨酸脱羧酶 (GAD) 旨在将谷氨酸转化为其镇静伴侣 GABA。
谷氨酸是大脑中必不可少的(也是主要的兴奋性)神经递质。然而,谷氨酸在某些情况下会变得有毒——这一过程称为谷氨酸兴奋性毒性 (GE):如果大脑中有过量的谷氨酸或谷氨酸受体被过度刺激。GE在神经退行性变中起关键作用。
兴奋性氨基酸过度激活谷氨酸受体会导致许多负面影响,即细胞钙稳态受损、自由基的产生、线粒体通透性转变的激活和继发性 GE。
GE 还与抑郁症、焦虑症、自闭症、多动症、慢性疼痛、中风和脑肿瘤以及许多其他疾病有关。
过多的谷氨酸可能在多发性硬化症 (MS)、阿尔茨海默病、亨廷顿病、癫痫和 ALS(肌萎缩性侧索硬化症或 Lou Gehrig 病)中起作用。
在亨廷顿舞蹈病中,谷氨酸受体的敏感性是主要特征。已经发现,降低谷氨酸的活性可以产生治疗作用。
ALS 被认为是由谷氨酸诱导的兴奋性毒性引起的,利鲁唑等药物试图控制谷氨酸水平。
涉及谷氨酸的通路功能障碍也可能导致多发性硬化症和阿尔茨海默病的发展。
研究人员正在研究专注于改善谷氨酸途径以治疗退行性脑病的药物和疗法。
谷氨酸过多(或过少)也与抑郁症和精神分裂症等心理健康障碍有关。
大量研究发现,在患有重度抑郁症的人中经常发现高水平的谷氨酸或过度活跃的谷氨酸受体。
还有一个普遍的假设是,过量(或不足)的谷氨酸活性可能导致精神分裂症症状。当然也可能没有那么简单,该领域的相互矛盾的研究表明,症状可能是由神经递质异常(包括谷氨酸)以及可能的基因突变和其他发育问题共同引起的。
谷氨酸在痛觉和传递中也起着举足轻重的作用。
这意味着,可以通过靶向谷氨酸受体和降低谷氨酸的影响来缓解慢性疼痛。
高浓度的谷氨酸盐和偏头痛之间也有密切的联系。一项研究发现,偏头痛患者的血浆谷氨酸水平显着升高。另一项研究得出结论,GABA 能药物(那些改变 GABA 作用的药物)可能有助于治疗偏头痛。
同时,阻断谷氨酸受体的药物也可能有效治疗偏头痛。
有一些证据表明,随着时间的推移,高水平的谷氨酸会导致 1 型和2 型糖尿病的发生。一项研究发现,谷氨酸水平对两种类型的糖尿病中 β 细胞的损失都有直接和间接的影响。
如果对谷氨酸敏感并怀疑自己的谷氨酸含量很高,最实际的方法是消除添加的游离谷氨酸的来源。尤其经过改良以改善口味的加工食品和包装食品,限制味精,酱油等摄入量。选择完整的、未经加工的食物,这是让你的水平恢复到正常、健康范围内的最佳方法。
总体而言,最好限制或避免使用富含谷氨酸的食物包括:酱油、硬奶酪、腌肉、谷物(尤其是含有麸质的)、骨汤,土豆片、快餐、方便面、沙拉酱等
除了监测提供这种氨基酸的食物的摄入量外,增加抗炎食物的摄入量也是有益的,因为这些食物可能在一定程度上有助于抵消过量谷氨酸的影响。
一些抗炎食物有:
关于抗炎饮食可详见之前的这篇文章:
深度解析 | 炎症,肠道菌群以及抗炎饮食
PPAR -γ激活剂可能是对抗 GE(谷氨酸兴奋性毒性)的最佳方法之一。
PPAR(过氧化物酶体增殖物激活受体)是对身体具有深远影响的特定蛋白质。它们属于所谓的核受体超家族(该类的其他成员包括维生素 A 和 D、雌激素甲状腺和糖皮质激素受体)。
许多食物和草药具有降低谷氨酸兴奋性毒性的能力。它们有多种作用机制,起作用的一个关键原因是 PPAR- γ激活剂。对文献的回顾发现,以下许多天然草药具有激活 PPAR 的潜力(括号中的是 PPAR -γ – 活化成分):
注意在多发性硬化或其他自身免疫性疾病的情况下,应谨慎使用或干脆避免使用过度刺激免疫系统的补充剂,如紫锥花、黄芪、人参甚至绿茶。
维生素 B6 有助于减少谷氨酸过量,因为参与将谷氨酸转化为 GABA。维生素 B6 缺乏可能是谷氨酸过量积累并且不能正确转化为 GABA 的一个原因。B6 缺乏症不会单独发生,通常与 B12 和叶酸一起发生。
维生素 B2(核黄素)本身具有神经保护作用,可以通过几种不同的方式抵消谷氨酸过量:
核黄素依赖性酶在吡哆醇活化、色氨酸-犬尿氨酸途径和同型半胱氨酸代谢中具有重要作用。
还发现维生素 B12(甲基钴胺素)对 GE 具有保护作用(可能通过 SAM 介导的甲基化改变膜特性起作用)。
维生素 B9(叶酸)补充剂:仅限低剂量。谷氨酸在结构上与叶酸相似,它可能会竞争神经元上的结合位点并可能导致问题。
维生素 D – 单独使用或与处方药联合使用
辅酶Q10 – 改善谷氨酸兴奋性毒性、线粒体功能和氧化应激
镁是健康神经信号传输所必需的矿物质。分子和动物研究表明,健康的镁水平可以防止神经元过度兴奋引起的细胞死亡。
从理论上讲,意味着增加镁含量可能有助于预防与细胞死亡有关的疾病,包括:
一项针对 60 名患有纤维肌痛的女性的小型研究发现,每天服用 300 毫克柠檬酸镁超过 8 周,可以降低压痛点的数量和报告的疼痛强度水平。然而,还需要进行更大规模的研究。
除了服用镁补充剂外,还可以尝试食用更多富含镁的食物,包括:
谷氨酸是大脑里需求量比较大的一种酸性氨基酸,主要是参与脑内蛋白质或者是脂肪酸等的合成和代谢,过低可能影响人的精神状态,也可能诱发神经衰弱。
谷氨酸在情绪障碍中的作用:
正在研究的一种此类情绪障碍是重度抑郁症(MDD),其症状包括空间记忆受损和快感缺失(无法感受到快乐)。研究人员发现,阻止大鼠吸收谷氨酸会导致类似抑郁的效果,这可能反映了快感缺失。
谷氨酸在慢性疲劳综合征中的作用:
关于谷氨酸失调是否在慢性疲劳综合征中起作用的研究存在分歧,这种情况还涉及感觉超负荷、焦虑和运动/平衡问题。
也有一些证据表明慢性疲劳综合征可能涉及与谷氨酸失调相关的基因。
有谷氨酸补充剂或增加谷氨酸的处方。如果想尝试提高谷氨酸水平,可能需要考虑在饮食或生活方式中加入其前体,前体是身体制造其他物质所需的物质。
提高谷氨酸水平/途径可能是一项非常复杂的任务。促进线粒体健康、减少氧化应激和炎症、平衡谷氨酸与大脑中其他神经递质以及改善糖和脂肪代谢的补充剂、疗法和生活方式的改变都是有益的。
运动
运动实际上可以帮助你的身体产生更多的谷氨酸。研究人员研究了近 40 名健康人类志愿者的谷氨酸和 GABA 水平。他们在三个持续 8 到 20 分钟的剧烈运动之前和之后立即测量了两个不同大脑区域的这些神经递质水平。
锻炼的参与者的谷氨酸或 GABA 水平增加。即使在停止运动后效果仍然存在,这表明谷氨酸水平会随着运动而发生更持久的变化。
在服用任何膳食补充剂之前,请先咨询医生。如果患有其他疾病,包括慢性病或怀孕,这一点尤其重要。
有助于提高谷氨酸水平的补充剂包括:
5-HTP:身体将 5-HTP 转化为血清素,血清素可以增强 GABA 活性,这可能会影响谷氨酸的活性。谷氨酸是 GABA 的前体。
一些前体包括:
GABA:理论上,由于 GABA 镇静和谷氨酸刺激,两者是对应的,一种不平衡会影响另一种。然而,研究尚未证实 GABA 是否可以纠正谷氨酸的失衡。
谷氨酰胺:身体将谷氨酰胺转化为谷氨酸。谷氨酰胺可以作为补充剂使用,也可以在肉类、鱼类、鸡蛋、奶制品、小麦和一些蔬菜中找到。
牛磺酸:对啮齿动物的研究表明,这种氨基酸可以改变谷氨酸的水平。牛磺酸的天然来源是肉类和海鲜。它也可作为补充剂使用,并存在于一些能量饮料中。
茶氨酸:这种谷氨酸前体可以通过阻断受体降低大脑中的谷氨酸活性,同时提高 GABA 水平。天然存在于茶中,也可作为补充剂使用。
不建议大多数人使用谷氨酸补充剂,一般人都可以从饮食中能获得足够的谷氨酸,而且人体会自己制造一些。
如上所述,经过改良以改善口味的加工食品是游离谷氨酸的最大来源。
天然高谷氨酸食物包括:
发酵、陈化、腌制、压力烹制的食品。其中包括:
陈年奶酪、慢煮肉类和家禽、蛋、酱油、大豆蛋白、鱼露,某些蔬菜,如蘑菇、成熟的西红柿、西兰花和豌豆、核桃、大麦麦芽。
多运动,保持足够睡眠。
本账号内容仅作交流参考,不作为诊断及医疗依据。
主要参考文献
Albarracin SL, Baldeon ME, Sangronis E, Petruschina AC, Reyes FGR. L-glutamate: a key amino acid for senory and metabolic functions. Arch Latinoam Nutr. 2016 Jun;66(2):101-112. PMID: 29737666.
Purves D, Augustine GJ, Fitzpatrick D. Glutamate. Neuroscience 2nd edition.
Huntington’s Outreach Project for Education, Stanford. About glutamate toxicity.
Dana Foundation. Protecting the brain from the glutamate storm.
Sheng P, Zhang Y, Zhang J, Wang H, Ren B. Glutamate receptors and signal transduction in learning and memory. Mol Biol Rep. 2011 Jan;38(1):453-60. doi: 10.1007/s11033-010-0128-9
Watson CJ, Lydic R, Baghdoyan HA. Sleep duration varies as a function of glutamate and GABA in rat pontine reticular formation. J Neurochem. 2011 Aug;118(4):571-580. doi:10.1111/j.1471-4159.2011.07350.x
Bechtholt-Gompf AJ, Walther HV, Adams MA, Carlezon WA Jr, Ongür D, Cohen BM. Blockade of astrocytic glutamate uptake in rats induces signs of anhedonia and impaired spatial memory. Neuropsychopharmacology. 2010;35(10):2049-2059.
Maddock RJ, Casazza GA, Fernandez DH, Maddock MI. Acute Modulation of Cortical Glutamate and GABA Content by Physical Activity. J Neurosci. 2016 Feb 24;36(8):2449-57. doi:10.1523/JNEUROSCI.3455-15.2016
Byun J, Shin YY, Chung S, Shin WC. Safety and efficacy of gamma-aminobutyric acid from fermented rice germ in patients with insomnia symptoms: a randomized, double-blind trial. J Clin Neurol. 2018;14(3):291-. doi:10.3988/jcn.2018.14.3.291
Cruzat V, Macedo Rogero M, Noel Keane K, Curi R, Newsholme P. Glutamine: metabolism and immune function, supplementation and clinical translation. Nutrients. 2018;10(11):1564-. doi:10.3390/nu10111564
Bulley S, Shen W. Reciprocal regulation between taurine and glutamate response via Ca2+- dependent pathways in retinal third-order neurons. J Biomed Sci. 2010;17(Suppl 1):S5-. doi:10.1186/1423-0127-17-S1-S5
White D, de Klerk S, Woods W, Gondalia S, Noonan C, Scholey A. Anti-stress, behavioural and magnetoencephalography effects of an l-theanine-based nutrient drink: a randomised, double-blind, placebo-controlled, crossover. Trial Nutrients. 2016;8(1):53-. doi:10.3390/nu8010053
Holton KF, Taren DL, Thomson CA, Bennett RM, Jones KD. The effect of dietary glutamate on fibromyalgia and irritable bowel symptoms. Clin Exp Rheumatol. 2012 Dec 14;30(6 Suppl 74):10-17. PMID:22766026
Bagis S, Karabiber M, As I, Tamer L, Erdogan C, Atalay A. Is magnesium citrate treatment effective on pain, clinical parameters and functional status in patients with fibromyalgia?. Rheumatol Int. 2013 Jan 22; 33(1):167-72. doi:10.1007/s00296-011-2334-8

谷禾健康

嗜黏蛋白阿克曼菌(Akkermansia muciniphila, 简称A. muciniphila, Akk菌)的缺乏或减少与多种疾病(如肥胖、糖尿病、肝脂肪变性、炎症和对癌症免疫治疗的反应)有关。
关于AKK菌,我们之前的一篇文章也有详细介绍过,点击详见:
肠道重要菌属——Akkermansia Muciniphila,它如何保护肠道健康
谷禾肠道样本大数据库显示A. muciniphila缺乏或未检出情况在人群中很常见,尤其是那些有肠道问题的人。在健康个体中其约占肠道微生物群总数的0.5%–3%。
现如今,关于AKK菌的研究正在从动物模型转向人类验证试验,AKK菌与疾病之间的研究不仅仅停留在相关性,更是开始向因果性及具体产生作用的机制方面深入探讨。
– A. muciniphila –
本文我们将依次介绍A. muciniphila的起源,主要特性,以及它与不同疾病间的联系,并解释产生有益作用的主要机制。
一所在瓦格宁根(Wageningen)的微生物实验室的研究人员使用一种专门用于分离优势细菌的策略,即稀释到消亡(即基于连续稀释的分离),从一名健康成人身上分离出一株高度丰富的粘液降解菌株。
该菌株似乎是疣微菌门(Verrucomicrobiota)中一个新属的新种,被命名为嗜黏蛋白-阿克曼菌(Akkermansia muciniphila, A. muciniphila, Akk菌),并以典型菌株MucT为代表。
粘液主要由粘蛋白组成,粘蛋白是一种由粘多糖组成的保护肠道细胞的糖蛋白。
长期以来,粘液的降解被认为是一种可能导致宿主紊乱的不良特性。然而,对于结肠微生物群来说,粘蛋白在肠道中大量分泌,因此提供了持续的宿主产生的碳、能量和氮源。粘蛋白的代谢转换需要一组酶,如唾液酸酶和硫酸酯酶,这些酶参与顺序降解。
粘蛋白降解“专家”
A. muciniphila 是人类早期生命中存在的独特的粘蛋白降解“专家”。此外,对无菌小鼠的单体型关联研究表明,A. muciniphila MucT不会损害宿主,并在结肠中显示出特异性的代谢和免疫信号。其2.7Mb的基因组预测了粘蛋白降解的酶机制,比较生长分析表明,A. muciniphila是体外利用粘蛋白最有活性的菌株。
A. muciniphila MucT也可以利用人乳寡糖作为能量来源。值得注意的是,在纯培养中,只有少数其他底物被发现能刺激其生长,包括二甲双胍、甜菜碱和色氨酸。
由于A. muciniphila是人类肠道中疣微菌门的唯一代表,许多报道这一门的16S rRNA基因序列调查往往代表A. muciniphila 。最近的一项研究分析了2,000多个Akkermansia基因组,表明A. muciniphila是迄今为止具有高度相似(超过98%同一性)16S rRNA 序列的优势物种。
非工业化人群A. muciniphila下降
在这个大型宏基因组数据库中对四个已确定的A. muciniphila亚种的详细分析表明,它们的基因组通常存在于西方和中国人群中。对哈扎部落和其他非工业化人群的肠道微生物群与工业化世界个体的肠道微生物群进行分析比较,发现拟杆菌纲和疣微菌门微生物群的丰度减少。这些结果表明,非工业化种群的A. muciniphila丰度水平有所下降,但这一观察结果是否与粪便样本保存、DNA提取或测序深度的技术问题有关仍有待调查。
值得注意的是,A. muciniphila宏基因组已在非人类灵长类动物中发现,包括野生和圈养。类似地,与模式菌株MucT具有高度基因组同源性的A. muciniphila已从同样生活在野外的各种动物中分离出来。
部分A. muciniphila菌株产维生素B12
长期以来,A. muciniphila型菌株MucT是唯一可用的人类分离株然而,在过去的 5 年中,从不同的肠道环境中分离到了其他A. muciniphila菌株,包括野生哺乳动物和圈养哺乳动物,特别是人类。在临床前试验中报告了人类相关 A. muciniphila菌株之间的一些变异性,但尚不清楚观察到的差异的稳定性和可重复性如何。
迄今为止,观察到的菌株之间最显著的代谢差异是产生维生素B12的能力,而维生素B12是产生丙酸盐所必需的。大约三分之一的A. muciniphila分离株能产生类似于A. glycaniphila的维生素B12。多种结肠微生物可以产生可供A. muciniphila利用的维生素B12,正如霍氏真杆菌(Eubacterium hallii)(重命名为Anaerobutyricum soehn genii)所显示的那样,在共培养实验中,发现该细菌与菌株MucT形成微生物网络,导致从粘液中产生丙酸盐和丁酸盐。
代谢紊乱
由于观察到益生元补充剂(即低聚果糖)对肥胖发育的保护作用与啮齿动物疣微菌门的主要繁殖相吻合,因此引起了人们对A. muciniphila和代谢紊乱的兴趣。随后在人类和啮齿动物上进行的观察研究都表明,与瘦小的对应物相比,患有肥胖症的个体肠道中的A. muciniphila反复出现表达不足。代谢紊乱,包括肥胖、T2DM、非酒精性脂肪性肝病(NAFLD)和心血管疾病,都与Akkermansia spp.丰度减少有关。
研究发现,11名超重者和38名肥胖者体内A. muciniphila的丰度与更健康的代谢状态以及热量限制后较好的临床结果呈正相关。通过在小鼠和人类身上注射该模式菌株,研究了A. muciniphila在肥胖过程中的因果作用及其潜在的有益影响。首次证明,小鼠每天服用活的A. muciniphila MucT可以逆转高脂饮食引起的代谢紊乱,包括脂肪增加、代谢内毒素、脂肪组织炎症和胰岛素抵抗。
在合成培养基中生长的A. muciniphila MucT的巴氏杀菌增强了它减少小鼠脂肪团发育、胰岛素抵抗和血脂异常的能力。事实上,A. muciniphila MucT可以降低小鼠的胆固醇水平和血清甘油三酯水平。
代谢健康背景下黏质阿克曼菌的代谢效应及主要影响因素
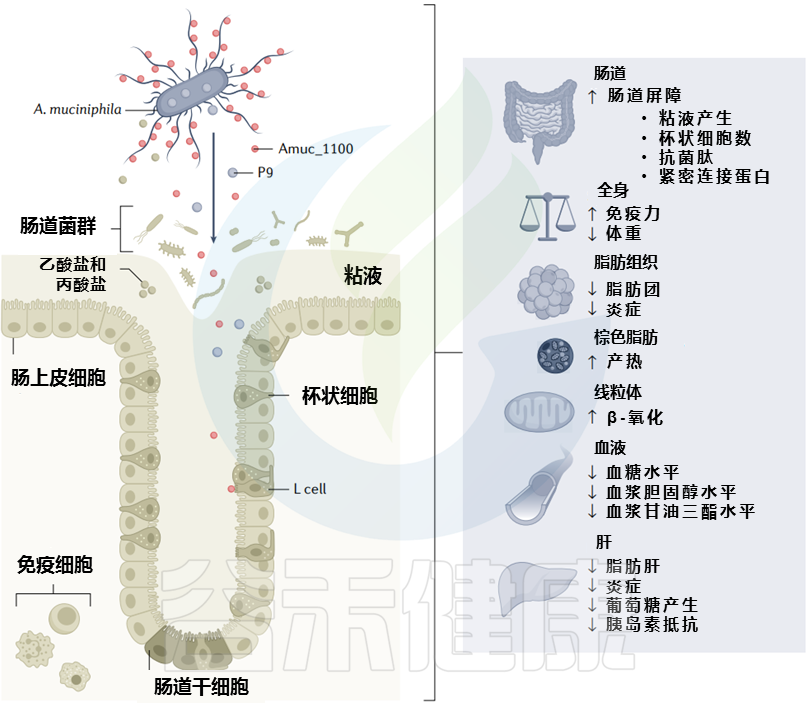
Cani PD, et al. Nat Rev Gastro Hepat. 2022
影响肝脏脂肪代谢
研究发现A. muciniphila MucT补充剂通过调节与脂肪合成有关的基因的表达(例如,降低肝脏中固醇调节元件结合蛋白的表达)和不同的炎症标志物(例如,降低IL-1β和IL-6的表达,ALT和髓过氧化物酶活性),可以积极地影响小鼠的肝脏脂肪代谢,预防非酒精性脂肪性肝病。
减轻动脉粥样硬化
在动脉粥样硬化发展的小鼠模型(载脂蛋白E缺陷(ApoE−/−)小鼠)中,A. muciniphila MucT的注射似乎减轻了动脉粥样硬化的损害。
降低糖尿病发病率
有趣的是,除了T2DM,在啮齿类动物和人类中,A. muciniphila通过降低肠道通透性、减少炎症和胰岛炎症的机制减少了T1DM的发病,从而有助于胰岛的保存。早期应用万古霉素可增加非肥胖型糖尿病小鼠(T1DM小鼠模型)的A. muciniphila丰度,降低糖尿病发病率。
神经退行性疾病
2016年至2020年间发表的几份报告表明,A. muciniphila在帕金森病和多发性硬化症中的作用,因为其相对丰度与受影响患者的疾病严重程度呈正相关。然而,尽管将患者的粪便微生物移植到无菌小鼠体内会导致疾病某些方面的发展,但这些研究都没有检测到受体小鼠粪便中的A. muciniphila,从而表明其他细菌可能参与了该病的发生表型。
多发性硬化症
对于多发性硬化症,在一项纳入62名复发缓解性疾病患者的研究中,作者发现脑脊液中的抗-Akkermansia IgG水平高于健康同行,并且局部脑脊髓特征与残疾评分呈正相关,而在同一个人的血液中检测到IgG水平没有明显的改变。
虽然在这些研究中观察到的抗-Akkermansia IgG水平的增加与肠道中嗜粘蛋白A. muciniphila丰度的改变无关,但另一项研究提供了支持 A. muciniphila在多发性硬化症中的积极和有益作用的机制解释。
这项研究表明,在多发性硬化症实验性自身免疫性脑脊髓炎小鼠模型的粪便和未治疗的多发性硬化症高峰期患者的粪便中发现的A. muciniphila的大量繁殖与miR-30d的富集有关。
有人发现,从这些小鼠或人类身上收集的粪便的转移导致A. muciniphila的丰度增加,并有利于调节性T (Treg)细胞的扩增,进而控制效应T细胞以抑制疾病症状。研究人员表明在患有多发性硬化症的小鼠中接种A. muciniphila可降低疾病评分、减少轴突损失的脱髓鞘和增加Treg细胞群。
相反,A. muciniphila衍生肽被确定为环境因素,与多发性硬化症(即HLA-DR15单倍体型)最强的遗传风险易感因素相结合,可以介导免疫调节介导的患者自身反应性T细胞的激活。
更具体地说,从患者体内分离出的 HLA-DR-SP反应性 CD4+ T细胞可以被某些外来因子(例如Epstein-Barr病毒和从A. muciniphila中分离出的一些肽)激活,然后对血液中的潜在致病多肽或自身抗原(如髓鞘碱性蛋白)产生反应,可能还会在脑脊液和/或脑中发生交叉反应,从而针对脑组织。
帕金森病
文献中关于阿尔茨海默病和A. muciniphila的发病机制存在差异,相关研究显示阿尔茨海默病患者的A. muciniphila含量较高,而干预研究清楚地报告了这种细菌在病理学中的有益作用。阿尔茨海默病是一种疾病,其最强的病理标志之一是淀粉样蛋白β肽42 (Aβ42)在老年斑中的积累。
由于肥胖和T2DM是痴呆的重要危险因素,一项机制研究探讨了在喂食高脂肪饮食的阿尔茨海默病小鼠模型中施用A. muciniphila的影响。除了抗肥胖作用外,无论饮食如何,接种都能显着减少大脑中的 Aβ并改善认知测试的表现。A. muciniphila给药还恢复了生命早期(生命3周时)暴露于高脂肪饮食的小鼠的认知能力下降和海马发育障碍。
癌症免疫治疗反应
免疫疗法是一种利用免疫系统对抗肿瘤形成细胞的癌症疗法,并已发展成为治疗各种癌症的成功方法。
肠道微生物组的相关性是增强对检查点抑制剂治疗的临床反应的标志物和辅助剂。
该疗法通过阻断程序性死亡受体1(PD1)等免疫检查点来恢复对抗癌细胞的活性。对100名对抗-PD1抗体反应良好的非小细胞肺癌和肾细胞癌患者的微生物组进行了详细的表征,结果显示A. muciniphila富集。此外,在疾病进展迅速的个体和后来死亡的个体中,A. muciniphila的患病率最低(34%)。
A. muciniphila菌株在用抗-PD1 药物治疗后,在移植无应答者粪便的无菌小鼠和预先暴露于抗生素的小鼠中改善了抗肿瘤活性。一项具有统计学意义的研究旨在专门评估338例非小细胞肺癌患者的A. muciniphila 基线检测与临床反应之间的关联。证实了A. muciniphila的存在与临床反应之间的密切关联。
在另一项研究中,42名主要使用抗-PD1药物治疗的转移性黑色素瘤患者的肠道微生物群也被证实富含多种分类群,包括Akkermansia。2020 年发表的一项研究确定肌苷是抗癌细胞活性的潜在因素,尽管这种核苷是由许多其他细菌产生的。
总之,这些研究表明,A. muciniphila是一种有望提高对检查点抑制剂免疫疗法的临床反应的候选菌。
肠道屏障和肠道炎症
最初发现A. muciniphila MucT 可以通过恢复小鼠的粘液层厚度以及抗微生物肽Reg3g的肠道表达来改善肠道屏障功能,这在肥胖和代谢紊乱期间都会发生改变。后来,在A. muciniphila MucT补充剂中观察到的粘液层厚度增加与小鼠粘液产生细胞数量的增加有关。
此外,来自A. muciniphila MucT (AmEVs)的细胞外囊泡,也显示通过调节小鼠的紧密连接来降低肠道通透性。
肠道炎症的情况下保护的肠道屏障
已有研究表明,与健康人相比,克罗恩病 (n = 26)和溃疡性结肠炎(n = 20和n = 15 ) 患者相比,A. muciniphila显著减少。这种相关性已在临床前模型中进行了因果研究。
改善结肠炎
已发现A. muciniphila MucT在结肠炎中的有益作用,AmEVs可保护葡聚糖硫酸钠 (DSS) 诱导小鼠结肠炎的进展。
随后,几项研究观察到A. muciniphila MucT细胞恢复肠道屏障功能并改善了:
改善衰老引起的变化
衰老是另一种肠道屏障减少和炎症增加的情况。各种人类研究报告称,与年轻成人(<50岁)和百岁老人相比,老年人(>65岁)的A. muciniphila丰度较低。
这一发现导致了旨在评估A. muciniphila MucT给药对不同衰老小鼠模型中几个年龄相关参数的影响的研究。
总之,这些研究表明,施用A. muciniphila MucT改善了一些与年龄相关的变化,包括炎症、屏障完整性和行为。
代谢综合征
在啮齿动物中,A. muciniphila被认为是健康的生物标记物。在啮齿类动物中,许多对A. muciniphila种群具有生长促进作用的膳食补充剂因其促进健康的作用而受到广泛关注。
在患有代谢综合征的人类中,给药被证实是可行的、安全的和有良好的耐受性。事实表明,与炎症、血液学、肾脏、肝脏和肌肉功能相关的任何标志物的变化,都可以用任何A. muciniphila MucT制剂来观察。这一结果是在不考虑所使用的形式以及短期(即2周)和长期(即3个月暴露)的情况下观察到的。
巴氏灭菌Akk菌比活菌效果好
以前的结果表明,高压灭菌灭活A. muciniphila可消除其对小鼠代谢综合征的保护作用。对植物乳杆菌或干酪乳杆菌菌株进行的研究表明,不剧烈的热诱导灭活,如巴氏杀菌,可以使细菌在稳定它们的同时保留部分有益的特性。
因此,有人在相同的饮食诱导肥胖小鼠模型上,通过比较活菌和巴氏灭菌菌的给药效果,检验A. muciniphila巴氏灭菌的效果。结果是,接受巴氏灭菌菌株的小鼠在体重、脂肪质量增加、血脂和胰岛素抵抗标志物方面的降幅甚至比接受活细菌的小鼠更大。除了提高A. muciniphila粘液的有效性,巴氏杀菌还具有提高菌稳定性和延长其保质期的好处,从而便于给人服用。
在未经治疗的患者中,给予巴氏灭菌A. muciniphila MucT成功地防止了与代谢综合征相关的参数的自然恶化。与补充安慰剂的志愿者相比,补充已过活的A. muciniphila MucT显著改善了胰岛素敏感性,降低了胰岛素血症和血浆总胆固醇水平。
为了进一步评估巴氏灭菌A. muciniphila MucT的安全性,对大鼠进行了一项强有力的毒理学长期评估。结果表明,口服90天,即使在测试的最高剂量(每公斤体重9.6×1010A. muciniphila MucT细胞,没有观察到不良反应的水平)下,也没有转化为亚慢性毒性,而体外遗传毒性试验显示阴性结果。
如前面所述,A. muciniphila可能有多种作用模式,所有这些都已用 MucT菌株进行了研究。当使用巴氏杀菌细菌时,可以观察到A. muciniphila MucT的几种有益效果和令人惊讶的功效增加。
产短链脂肪酸,作用于宿主代谢
最明显的答案是短链脂肪酸(如丙酸盐)的一般作用,因为A. muciniphila MucT在维生素B12存在下被称为丙酸盐生产者。虽然A. muciniphila对肠道中丙酸盐总水平的贡献可能相对于其他主要的丙酸盐生产者(例如拟杆菌)相对较低,尤其是在禁食条件下。但 Akkermansia似乎在动物和人类中都上调,并且可能对生产这种短链脂肪酸做出重大贡献。
在这种情况下,某些影响可能与肠道上皮细胞和肠道黏膜中存在的不同类型免疫细胞上表达的FFAR3和FFAR2受体(也分别称为GPR41和GPR43)有关。然而,值得注意的是,巴氏杀菌的 A. muciniphila MucT也可以作用于宿主代谢,不会改变丙酸盐的产生。
通过外膜上的蛋白质Amuc_1100发挥作用
2017年,有人确定了A. muciniphila MucT可以在宿主健康中发挥作用的特定和独特的分子机制。他们发现存在于A. muciniphila外膜中的一种特定蛋白质,称为Amuc_1100,概括了这种细菌的有益作用。A. muciniphila的所有有益作用是否都可归因于Amuc_1100目前尚不清楚,但其每个细胞的生产水平足以解释临床前模型中的作用。
重要的是,他们发现这种蛋白质在用于巴氏杀菌的温度下仍保持其活性构象,从而解释了为什么巴氏杀菌的A. muciniphila MucT在小鼠和人体实验中保持活性。
此外,发现Amuc_1100可激活Toll样受体2 (TLR2),单独给药可复制A. muciniphila MucT细胞的大部分有益作用,包括在肠道炎症和结肠癌的特定疾病模型中的作用。
这一发现表明,即使死亡(即巴氏杀菌后),A. muciniphila MucT仍然可以改善宿主健康,并反对需要潜在的分泌代谢物来观察细菌的有益作用。
外膜上的蛋白质 P9,刺激GLP1水平增加
2021年的一项研究确定了A. muciniphila MucT产生的另一种蛋白质。发现A. muciniphila处理的饮食诱导的肥胖小鼠在口服葡萄糖挑战期间表现出循环GLP1水平略有增加。
通过体外实验,作者鉴定了一种84kDa的蛋白质,命名为蛋白 9(P9;由早期鉴定的 Amuc_1831 基因编码),它负责体外刺激GLP1。
在体内,他们测试了 8周以非常高的剂量口服P9的效果,发现体重增加和脂肪量增加都有所减少,胰高血糖素前体(即GLP1的前体)的肠道表达增加。
这些影响怎么与较高的循环 GLP1 水平联系起来?
研究发现P9的药理作用激活 ICAM2,这些作用与可能的GLP1分泌有关,因为抗-ICAM2抗体在体外部分消除了P9对GLP1分泌的影响。
此外,A. muciniphila MucT增加了小鼠回肠中IL-6的表达。除了证实IL-6是一种促炎细胞因子外,这种细胞因子还在体外剂量依赖性地增加GLP1的分泌。
因此,有人假设A. muciniphila MucT也可以通过 IL-6-GLP1信号传导发挥作用。为了进一步支持他们的发现,研究人员使用了IL-6敲除小鼠,发现这些小鼠对P9诱导的GLP1分泌没有反应,而阻断GLP1受体也消除了P9对产热的影响。尽管这些发现是相关的,但应评估其他因素,例如P9的生理剂量和位置(可能被分泌)。
与Akkermansia或相关分子在疾病中的作用相关的主要机制
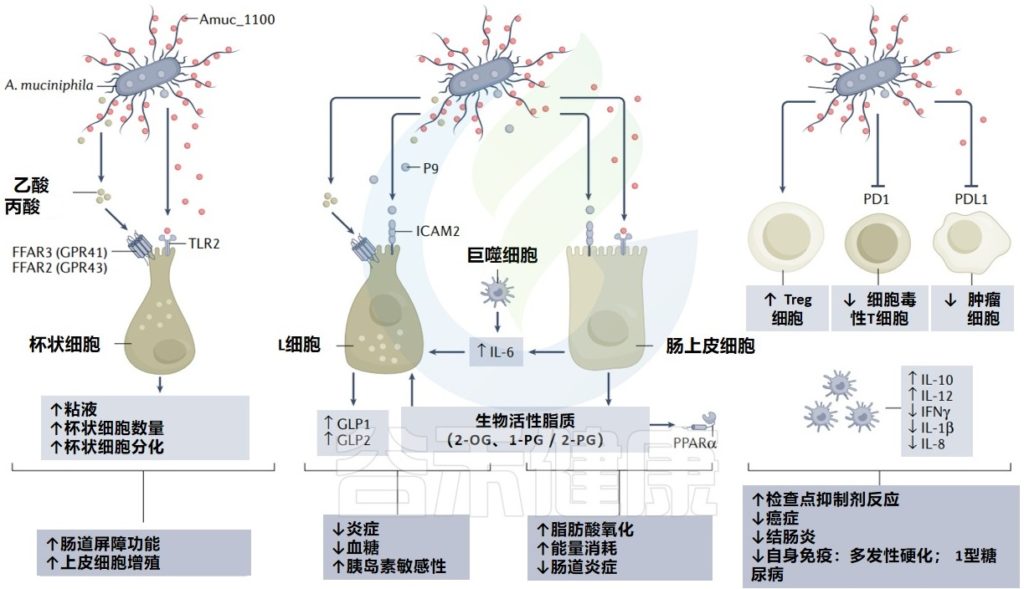
Cani PD, et al. Nat Rev Gastro Hepat. 2022
A. muciniphila MucT表达的酶潜在益处
一项研究证明天冬氨酸蛋白酶Amuc_1434*通过TRAIL介导的细胞凋亡途径抑制LS174T细胞活力。然而,没有进一步的位置、热稳定性或体内数据报告,因此尚无法评估这一发现的潜在和生理相关性。
第二项研究表明,A. muciniphila MucT表达的β-乙酰氨基己糖苷酶Amuc_2109*保护小鼠免受DSS诱导的结肠炎;然而,尚未报告热稳定性数据。
此外,已在人类身上表明,活的或巴氏杀菌的A. muciniphila MucT 改善了几种心脏代谢风险因素,包括胰岛素抵抗,而不增加 GLP1 的循环水平,也不影响 DPP4 活性,DPP4 是一种参与 GLP1 降解的酶。
有趣的是,无论使用何种形式的A. muciniphila MucT——活的或巴氏杀菌的,甚至是蛋白Amuc\U 1100,文献中的所有数据都通过作用于不同的关键标记物来强化肠道屏障。
事实上,所有的治疗都指向更高的粘液产量,紧密连接蛋白的恢复,抗菌因子的恢复,以及最终加强肠道屏障。
特异性调节
此外,一部分人通过对用活的或巴氏杀菌的 Akkermansia治疗的人体进行脂质组学和代谢组学分析。已经发现A. muciniphila MucT治疗诱导了不同生物活性脂质的特异性调节,这些被鉴定为 PPARα 激动剂(2-PG和1-PG)。
重要的是,在人类中使用非靶向代谢组学分析,他们已经能够重建一条指向通过β-氧化激活脂肪酸氧化的代谢途径,并且所有鉴定的代谢物都在PPARα的控制下趋向于增加线粒体活性,这些数据也在啮齿动物身上得到了证实。
除了对肠道屏障的影响,这可能涉及对几种疾病(即糖尿病、肥胖症、NAFLD和/或非酒精性脂肪性肝炎、炎症性肠道疾病、多发性硬化症)的保护作用,A. muciniphila对癌症的作用也是与依赖免疫系统的其他机制有关。
例如,使用检查点抑制剂(抗-PD1)的免疫疗法与动物数据中涉及 IL-12 依赖性效应的特定机制方面有关。此外,数据表明A. muciniphila菌株对抗-PD1反应的辅助作用增加了特定T细胞向肿瘤床的募集。
在炎症性肠病、T1DM或肠癌的小鼠模型中,A. muciniphila MucT或特定蛋白增加Treg细胞群的分化或降低结肠中浸润性巨噬细胞和CD8+细胞毒性T淋巴细胞的水平。
A. muciniphila MucT的代谢和抗炎作用是强大的,因为来自不同研究团队的许多结果已经证实了A. muciniphila菌株的各种健康特性。
A. muciniphila的生理和有益作用是多效性的(例如,能量、脂质、葡萄糖代谢、炎症、免疫、脑功能),重要的是要强调有许多汇聚的作用模式,可能是由于其特异性特性与其粘蛋白代谢的特化有关。
事实上,已经确定了几种常见的途径,都指向调节肠道屏障功能(即粘液产生和免疫系统)。肠道屏障的恢复,也有助于恢复几种改变的途径的正常功能,包括例如线粒体活性、肝脏代谢、脂肪组织和大脑活动。
最后,A. muciniphila MucT的有益作用已从临床前观察转化为代谢综合征背景下的人类干预。这种情况是独一无二的,是其他下一代微生物无法比拟的。
需要注意的是,虽然A. muciniphila具备各种有益特性,但是该菌丰度并不是越多越好,要保持在合适范围。谷禾肠道菌群检测结果中曾经有案例,发现该菌丰度过高,占比超50%。
该菌过量将过度消耗粘液蛋白而存活下来,这是大多数其他细菌所缺乏的生存优势,该菌增殖异常,从而可能导致肠道屏障损伤,诱发肠道炎症、LPS 进入血液的增加、自身免疫性疾病有关。
该样本检测报告也显示菌群多样性低,肠炎和几项慢病注意风险:
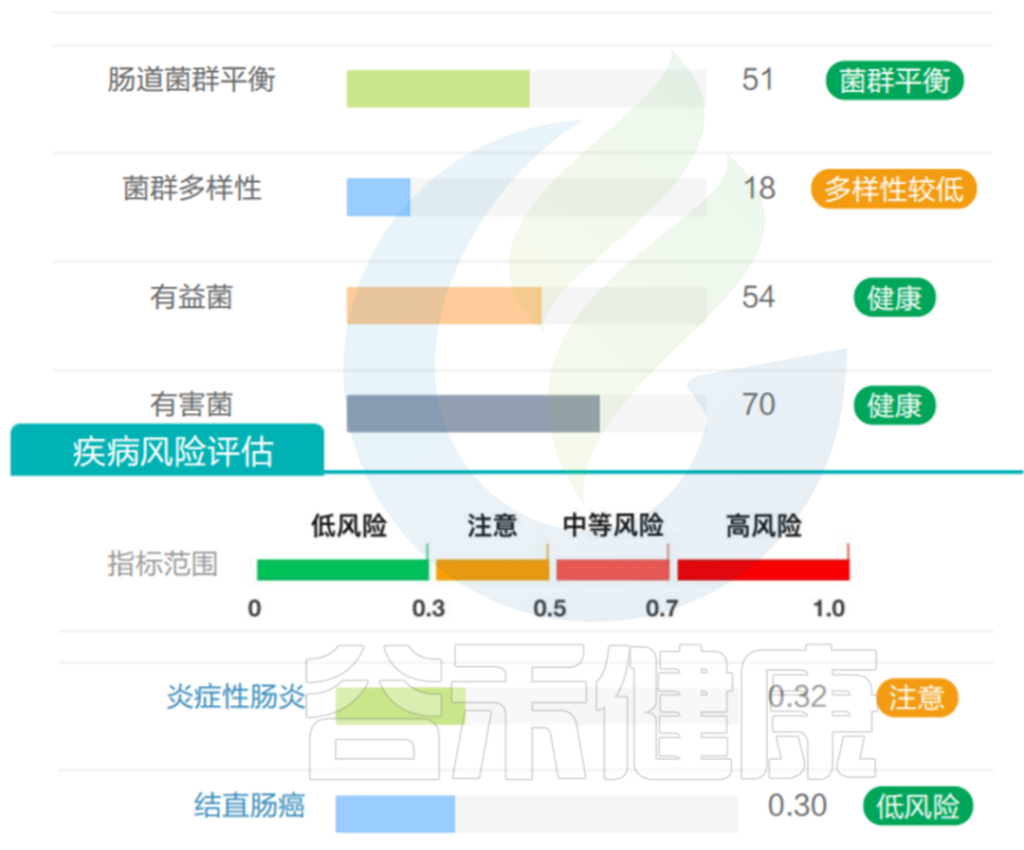
<来源:谷禾健康数据库>
总的来说,在合理范围内,A. muciniphila 带来有益影响。
当然也需要更多的研究来支持A. muciniphila的有益特性及临床治疗应用:
主要参考文献
Cani PD, Depommier C, Derrien M, Everard A, de Vos WM. Akkermansia muciniphila: paradigm for next-generation beneficial microorganisms. Nat Rev Gastroenterol Hepatol. 2022 May 31. doi: 10.1038/s41575-022-00631-9. Epub ahead of print. PMID: 35641786.
Kostopoulos I, Elzinga J, Ottman N, Klievink JT, Blijenberg B, Aalvink S, Boeren S, Mank M, Knol J, de Vos WM, Belzer C. Akkermansia muciniphila uses human milk oligosaccharides to thrive in the early life conditions in vitro. Sci Rep. 2020 Aug 31;10(1):14330. doi: 10.1038/s41598-020-71113-8. PMID: 32868839; PMCID: PMC7459334.
Yin J, Song Y, Hu Y, Wang Y, Zhang B, Wang J, Ji X, Wang S. Dose-Dependent Beneficial Effects of Tryptophan and Its Derived Metabolites on Akkermansia In Vitro: A Preliminary Prospective Study. Microorganisms. 2021 Jul 14;9(7):1511. doi: 10.3390/microorganisms9071511. PMID: 34361945; PMCID: PMC8305782.
Paone P, Cani PD. Mucus barrier, mucins and gut microbiota: the expected slimy partners? Gut. 2020 Dec;69(12):2232-2243. doi: 10.1136/gutjnl-2020-322260. Epub 2020 Sep 11. PMID: 32917747; PMCID: PMC7677487.
谷禾健康
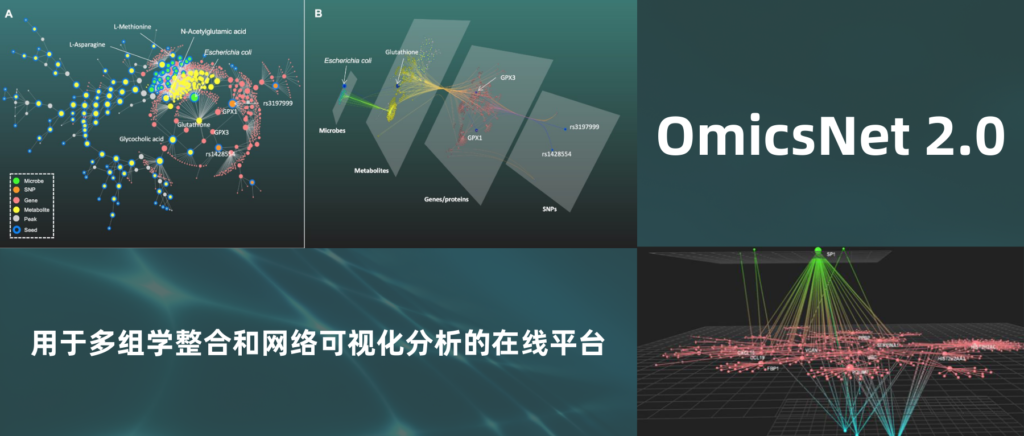
为了使研究更系统更全面,越来越多的研究人员追求在多组学背景下解释分子数据。
OmicsNet (www.omicsnet.ca) 顺应而生,这是一个在线平台,允许用户轻松地构建、可视化和分析多组学网络。
OmicsNet 2.0在原有基础上进行了升级,主要改进了三个方面:
(1) 可视化分析中2D图形布局选项增加了11个,以及一个新颖的3D模块。
(2) 支持三种新的组学类型:
(3) 分析时将同时输出使用的R包OmicsNetR和历史命令,并生成共享链接可在网页查看和交互操作,从而提高研究的可重复性。
研究中使用OmicsNet 2.0对炎症性肠病(IBD)的数据进行多组学分析。
OmicsNet 2.0工作流程
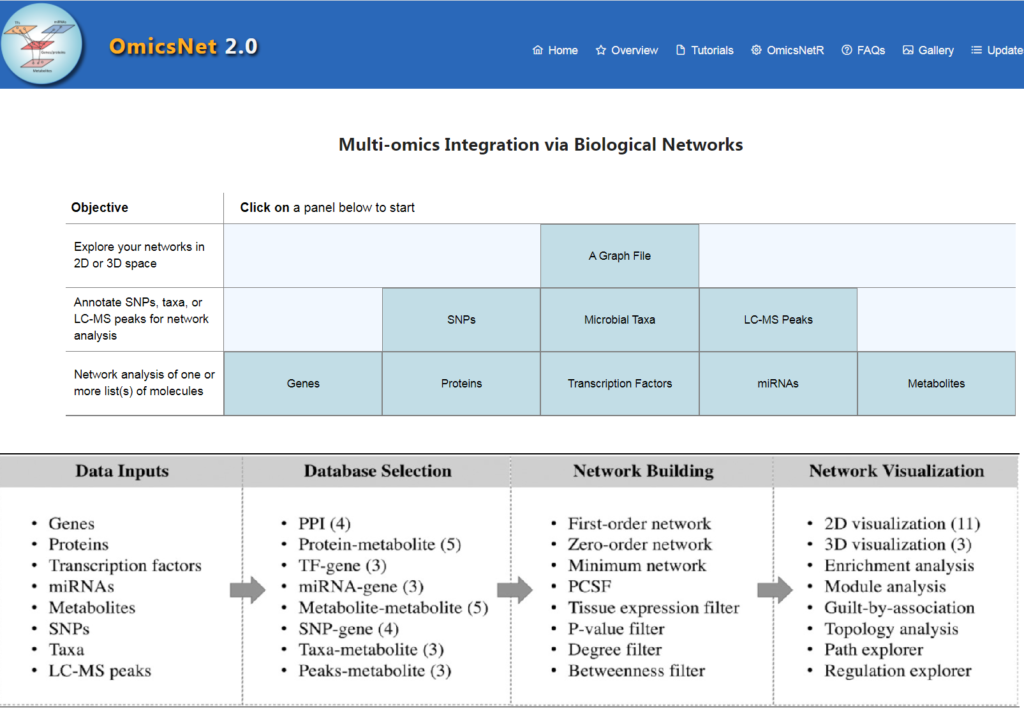

如图,主要为4个步骤:
OmicsNet 2.0 的改进
1、更新了分子相互作用数据库
包括PPI数据库(STRING,InnateDB和IntAct),TF-target数据库(TRRUST和JASPAR), miRNA-target数据库(TarBase和miRTarBase),代谢数据库(KEGG, Recon3和AGORA)。
2、支持三种新的组学类型
可支持来自遗传变异研究的SNP矩阵、来自非靶向代谢组学的 LC-MS 峰和来自微生物组分析的物种丰度矩阵数据的输入及互作分析。
对于那些对影响基因调控的变异感兴趣的用户,可以分别基于ADmiRE和SNP2TFBS将SNPs映射到miRNAs或TF结合位点。由此产生的网络可以通过proteins、miRNAs或TFs进一步扩展,以了解潜在的影响。
对于代谢组学分析,使用最近发布的NetID算法对LC-MS峰(m/z, retention time, intensity和p-value)进行数据处理,可选三种数据库(KEGG, PubChemLite_BioPathway和HMDB)进行注释。
使用Rcpp/C++ 引擎重新编写了核心算法,使其速度提高了10 倍以上。使用 lpsymphony 包进一步优化了整数线性规划。
直接上传物种丰度矩阵,可以使用贝叶斯逻辑模型预测潜在代谢物,该模型使用超过 6000 个高质量基因组规模的代谢模型进行训练。
3、代码开源
开放R包OmicsNetR(https://github.com/xia-lab/OmicsNetR),显示分析期间所执行的R命令。结果可输出为网页链接,有效期一个月。
4、减少假阳性
对于来自人类和小鼠的数据,提供了一个基于ENCODE、基因型-组织表达(GTEx)或人类蛋白图谱(HPA)的基因表达数据的过滤器,这些过滤器帮助研究人员专注于与生物相关的相互作用,并减少假阳性。
5、支持 2D 和 3D 网络可视化
2D模块可支持11种图形布局,3D模块可支持交互。图形化主要基于 igraph 包和 graphlayouts 包。
使用OmicsNet 2.0进行的IBD多组学研究
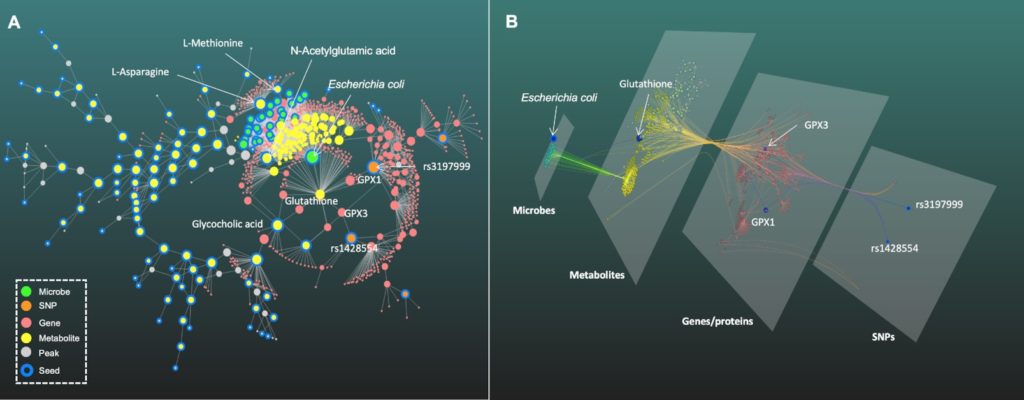
图A为2D视图,图B为3D视图
将获得的物种丰度矩阵,SNP矩阵和LC-MS峰数据上传至OmicsNet 2.0平台。
利用AGORA数据库预测潜在的微生物代谢物(potential score:0.9)。
利用PhenoScanner对基于eQTLs的基因进行SNP定位。
利用KEGG数据库对LC-MS峰进行注释。
通过添加代谢物-蛋白质相互作用,从SNPs和LC-MS峰生成的个体网络进一步扩大,这样三个网络可以在代谢组学层合并。
p-value过滤:(cut-off: 0.2)来排除p值较大的LC-MS峰值所贡献的节点。其网络连接基于其相关代谢物、基因或蛋白质。
如图A,生成的子网络1包含六种类型的节点(物种、SNPs、代谢物、SNPs或代谢物相关的基因/蛋白质),结果显示出glutathione是宿主-微生物相互作用中的重要交互作用点。
由此推测包括大肠杆菌在内的几种微生物会产生glutathione,两个SNPs(rs3197999和rs1428554)通过编码谷胱甘肽过氧化物酶(GPx)的基因GPX1和GPX3与代谢产物相关。
与此呼应的是,在以往的研究中证明大肠杆菌和相关物种在IBD中的过度表达可能是由于它们能更好地产生glutathione以抵抗氧化应激。
图B的3D分层网络提供了一个直观的多组学整合视角,突出显示了连接微生物组和宿主遗传的glutathione的流动路径。
OmicsNet 2.0与其它工具的比较
如下表,比较OmicsNet 2.0与其他基于web的多组学集成工具的关键特性。
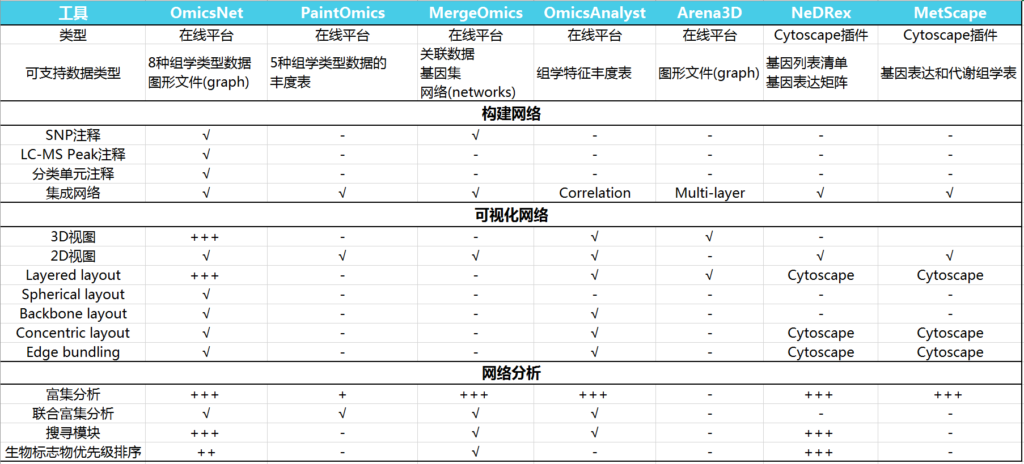
‘√’表示支持,‘-’ 表示缺失,“+”表示定量评估(“+”越多表示更好的支持)
附带各工具的官网及主要特点描述:
PaintOmics
(http://www.paintomics.org)
专注于在视觉上呈现多组学数据的探索分析,包括转录组学,代谢组学,表观基因组学,miRNA和转录因子,将其映射到KEGG通路联合分析。
MergeOmics
(http://mergeomics.research.idre.ucla.edu)
整合了来自单个组学层面的关联研究的汇总统计数据,以及不同功能基因组学数据,以获得机理上的见解,最近还加入了多组学信息的药物重新定位(drug repositioning)。
OmicsAnalyst
(https://www.omicsanalyst.ca)
基于输入的数据,利用多元统计、相关性分析和聚类方法,结合网络、热图和散点图进行多组学分析。
Arena3D
(https://www.arena3d.org)
擅长使用基于3D的分层布局对多层网络进行交互式可视化,适用于多组学网络数据。
NedRex
(https://nedrex.net)
是一个Cytoscape插件,专注于疾病模块识别和药物再利用,使用各种模块识别和优先排序算法。
MetScape
(http://metscape.ncibi.org)
是一个Cytoscape插件,通过构建和分析不同类型的含有酶、代谢物和/或反应的网络,专注于基因表达和代谢组学数据的集成和可视化。
OmicsNet
(https://www.omicsnet.ca)
通过将多个分子互作数据库与强大的2D/3D可视化网络分析相结合,对于多组学集成分析和结果解释有更好的理论基础,对以上工具的不足做了补充。
结 语
OmicsNet 2.0是一个基于网络的多组学分析平台,支持2D和3D网络可视化探索。
在1.0版本中强调基于web的3D网络可视化。
在2.0版本中,进一步改进了其可视化分析系统,添加了一个功能完整的2D网络可视化系统,并支持三种新的组学数据输入(SNPs、微生物分类单元和LC-MS峰),目前的生物信息学工具无法很好地支持这些数据。
除了富集分析、搜寻模块和最短路径分析,用户还可以使用重启随机游走算法(Random Walk with Restart)搜索候选疾病标记物。最后,2.0版本通过发布底层的R代码和共享链接,改进了工具的可再现性和透明度。
使用IBD多组学数据的案例研究表明,OmicsNet 2.0可以揭示与原始出版物和后续出版物以及IBD文献一致的有意义的模式、关系和功能。总之,OmicsNet 2.0解决了对多组学数据进行分析的一些需求。
参考文献:
Zhou G, Pang Z, Lu Y, Ewald J, Xia J. OmicsNet 2.0: a web-based platform for multi-omics integration and network visual analytics. Nucleic Acids Res. 2022 May 26:gkac376. doi: 10.1093/nar/gkac376. Epub ahead of print. PMID: 35639733.

谷禾健康

现代化疗,放射疗法在摧毁癌细胞的同时,对健康细胞也造成了伤害,引发相关毒性,反应例如便秘,腹泻,疲劳,恶心,呕吐等。
癌症患者的营养状况可能是癌症治疗相关毒性的核心决定因素,也是癌症症状的指标。
一些临床前研究和初步临床试验表明,饮食干预可能会减轻某些癌症治疗相关的症状和毒性。可能机制包括影响炎症、氧化应激、肌肉质量、心脏健康和调节肠道菌群。
本篇涵盖了三大块内容,包括:
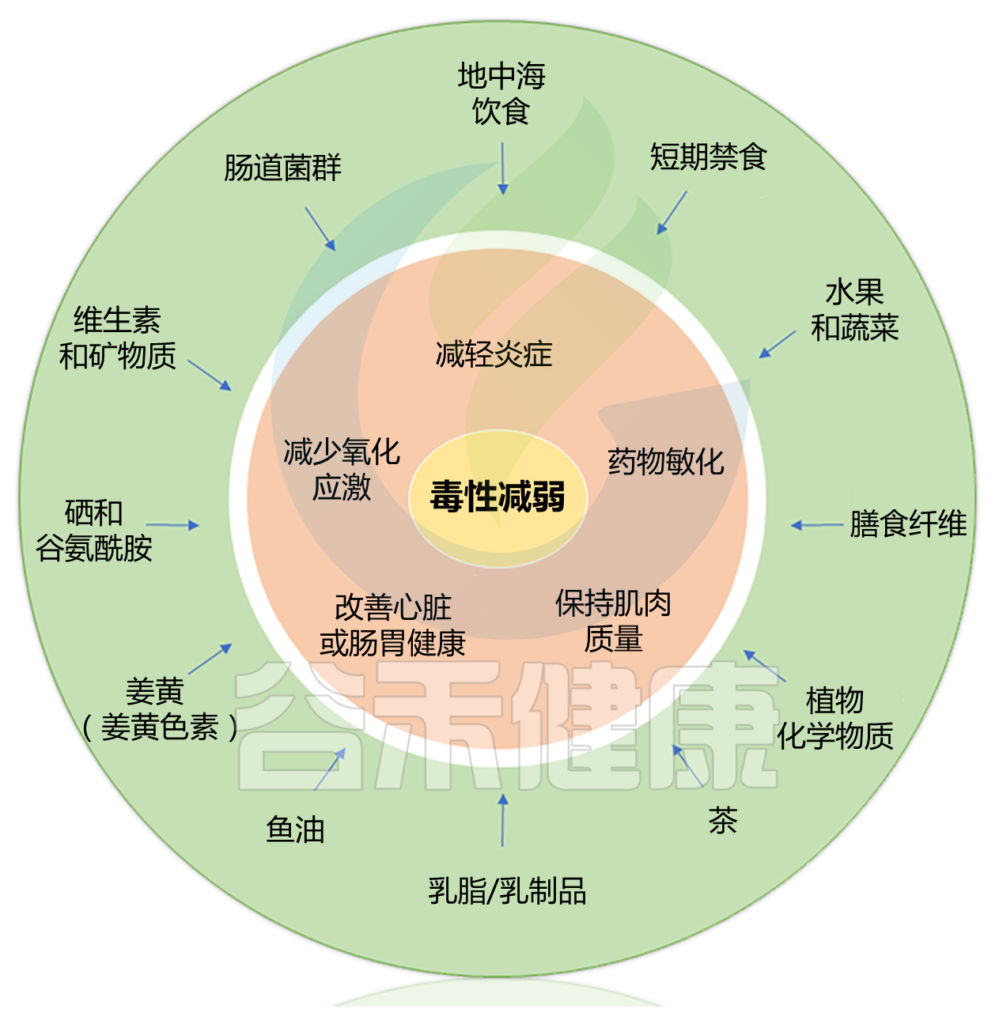
Alan J. Kim et al.,Cancer Treatment Reviews, 2022
地中海饮食模式,其特点是大量摄入水果、蔬菜、豆类、橄榄、全谷物、不饱和脂肪、坚果和鱼类;适度饮酒;减少红肉和加工肉类以及高脂肪乳制品的摄入,对减少炎症,改善心血管健康,抗肥胖,改善血脂以及对肠道菌群和免疫调节有积极作用。
↓↓ 降低心源性猝死
在一项大型前瞻性研究中,调查了激素替代疗法、钙和维生素D或饮食调整对健康绝经后妇女的影响,发现地中海饮食模式与降低心源性猝死的风险有关。
↓↓ 缓解癌症相关的疲劳
一项随机对照试验中,23名前列腺癌男性患者接受了至少3个月的雄激素剥夺治疗,被随机分成两组,一组接受12周常规治疗,另一组接受地中海饮食并同时接受六次个性化营养咨询,与常规治疗相比,坚持地中海饮食与缓解癌症相关的疲劳以及提高总体生活质量有关。
↓↓ 减轻妇科肿瘤患者症状
一项对22名接受铂类化疗的妇科肿瘤患者的观察研究表明,坚持地中海饮食的患者胃肠道毒性较小,恶心、胃痛、腹胀和干扰日常活动的频率和严重程度差异有统计学意义。
周期性禁食即在给定的时间内部分或完全不吃除水以外的食物和饮料。在动物模型和人类患者中,周期性禁食与降低癌症治疗中的毒性密切相关。
↓↓ 延缓肿瘤进展
例如,在一项临床前研究中证明,禁食条件增加了对化疗药物的敏感性,并延缓了肿瘤的进展。还有人发现,在荷尔蒙受体阳性乳腺癌的小鼠模型中,周期性禁食或类似禁食的饮食(FMD)可增强激素治疗的活性(如他莫昔芬和氟维司群),并通过降低循环中胰岛素、瘦素和IGF1的水平以及抑制AKT-mTOR信号传导来促进长期的肿瘤消退。
↓↓ 提升生活质量
在一项针对131名HER2阴性的II/III期乳腺癌患者的II期临床研究证实了4天FMD的潜在益处。在新辅助化疗前3天和新辅助化疗当天,接受了植物性、低氨基酸替代饮食(包括肉汤、汤、液体、维生素片和茶)的患者报告称,与对照组(常规饮食)相比,总体幸福感有所提高,情绪、身体、认知和社会功能都有所改善。
↓↓ 减少化疗相关毒性
在一项对照交叉初步研究中,调查了30名接受化疗的妇科癌症患者,结果表明,短期禁食的改良形式减少了化疗相关的毒性,包括口腔炎、头痛、虚弱和总体毒性。
据报道,FMD是安全可行的,可以减少脂肪量,降低循环中胰岛素生长因子1、胰岛素和瘦素的水平。禁食强烈影响新陈代谢和细胞途径,导致循环中类胰岛素一号生长因子(IGF-1)和葡萄糖水平下降。这些变化反过来影响几个癌基因,包括RAS和AKT信号通路,导致细胞生长和增殖的下调。
禁食和FMD(低卡路里、低蛋白质和低糖的饮食)可能与减少化疗副作用有关,即当营养素缺乏时,正常细胞,而不是肿瘤细胞,可以切换到抑制生长和增殖途径的保护模式,这一过程被称为差异应激抵抗。
↓↓ 增强免疫
此外,禁食和/或FMD已被证明可以增强免疫系统,减少炎症,减缓小鼠的骨密度损失,并减少HER2阴性的II期乳腺癌患者化疗引起的淋巴细胞DNA损伤。
生酮饮食的特点通常是碳水化合物消耗量低,占每日总热量摄入量的5%-10%,但其他产生能量的大量营养素,即脂肪和蛋白质的含量却不同。生酮比用来定义饮食的生酮能力,定义为脂肪克数与碳水化合物和蛋白质克数之和的比值。
在癌症中研究最多的生酮饮食包括经典生酮饮食(CKD),其特征是生酮比为4:1或3:1,每日87%-90%的热量来自脂肪)和中链甘油三酯生酮饮食[MCTKD,其中30%-60%的总热量来自中链脂肪酸,如己酸(C6)、辛酸(C8)、葵酸(C10)和月桂酸(C12)]。
↓↓ 血糖,体重更可控,生活质量高
在接受放化疗和辅助化疗的胶质母细胞瘤患者中,那些生酮饮食患者的血糖水平低于那些标准饮食的患者。有人研究发现,在接受放疗的非转移性乳腺癌患者中,以天然食物为基础的生酮饮食与未指定标准饮食相比,体重和脂肪减少得更多,生活质量水平更高。
↓↓ 降低癌症治疗相关毒性
生酮饮食降低癌症治疗相关毒性的机制可以用瓦氏效应(Warburg effect)来解释,在瓦氏效应中,癌细胞利用糖酵解而不是氧化磷酸化,能预防由活性氧引起的氧化损伤。
生酮饮食(通常是高脂肪低葡萄糖),可以利用这种代谢差异,要么让癌细胞挨饿,要么迫使它们转而利用氧化磷酸化。与正常细胞相比,癌细胞中氧化应激的增加可以使它们对化疗和放射更加敏感,从而减少治疗所需的剂量,降低与治疗相关的毒性。
↓↓ 注意高脂肪生酮饮食的副作用
有限的依据支持在临床实践中使用生酮饮食,而且,根据总含量和相对常量营养素组成,不同类型的生酮饮食可能对治疗相关的毒副作用产生不同的影响。例如,长期食用高脂肪生酮饮食可能会增加心血管或脑血管疾病的风险,特别是与特定的抗癌药物联合使用时。高脂生酮饮食还可能增加严重脂肪性肝炎的风险,促进肝纤维化的进展。此外,研究表明,高脂肪生酮饮食增加了酮体乙酰乙酯的循环水平,并促进了异种移植小鼠中表达BRAF-V600E致癌基因的人类黑色素瘤细胞的肿瘤生长潜力。
因此,根据目前对营养的理解,接受癌症治疗的患者的饮食应该包括所有的常量营养素,以降低营养不良的风险。
膳食纤维与多种肿瘤类型的胃肠道毒性和症状的预防有关。在一项随机对照试验中,在放疗期间服用高剂量膳食纤维(18克/天)的盆腔癌患者报告称辐射引起的胃肠道毒性发生率较低,证明了膳食纤维在预防放射治疗相关毒性方面存在潜在影响。
膳食纤维还可以促进健康的肠道菌群,从而降低毒性并增强治疗效果,例如纤维与促进免疫的普拉梭菌和短链脂肪酸丁酸盐的有关。有人发现在黑色素瘤患者中,膳食纤维会影响肠道菌群,并与免疫检查点阻断反应的增强有关。

Spencer CN, et al.,Science. 2021
tips
对于腹膜癌、肠癌进展或原发性胃肠癌的患者来说,可能需要低膳食纤维饮食,以降低肠梗阻的风险。
增加水果和蔬菜的摄入量与改善胃癌幸存者的身体和认知功能、减少疲劳和食欲不振有关。
水果和蔬菜含有过多的植物化学物质,并且已经检查了几种植物化学物质的影响,并在下表中进行了总结。

Alan J. Kim et al.,Cancer Treatment Reviews, 2022
例如,葡萄产品的抗氧化和抗炎作用归因于它们的植物化学物质,即芪类、花青素和原花青素,包括白藜芦醇。
其他几种水果,包括黑醋栗、李子、石榴和苹果,已经被证明具有抗癌和细胞毒性作用,这归因于植物化学物质,特别是多酚和黄酮类物质的抗氧化和抗炎作用。
tips
对于肠梗阻风险较高的患者,应避免过量摄入水果和蔬菜相关的膳食纤维。
据报道,在70多种植物中发现的一种植物化学物质白藜芦醇可以增加结直肠癌细胞系对化疗药物的敏感性,包括阿霉素、索拉非尼、5-氟尿嘧啶、依托泊苷、丝裂霉素、奥沙利铂和姜黄素。
关于白藜芦醇在该文有详细介绍:
注意:高剂量(每天>2.5克)会引起轻微的毒性(包括腹泻、胃肠道症状和前额头痛),即使在健康的人身上也是如此。
其他多酚也可能具有抗炎和抗氧化作用。
▸槲皮素是浆果中一种常见黄酮醇,它通过作用于炎症介质,包括白细胞介素6、白细胞介素8、干扰素γ、诱导型一氧化氮合酶、环氧合酶2和肿瘤坏死因子α来诱导抗炎作用,并在对促凋亡刺激敏感的癌细胞系中提供促凋亡作用。
▸非瑟素是一种存在于草莓、苹果和柿子中的类黄酮类化合物,在结构上与槲皮素相似,具有类似的抗炎和抗癌活性。
▸番茄红素是番茄和红色水果(包括木瓜和西瓜)中的一种植物化学物质,具有高抗氧化活性,并通过减少氧化应激以及染色体和膜的异常来减轻致癌损害。此外,番茄红素具有神经保护作用,并被认为可以通过减少氧化应激和神经炎症来预防神经毒性。
喝茶,尤其是绿茶,与抗癌作用、减轻药物引起的毒性以及对化疗药物的敏感性有关。
↓↓ 抑制继发性肿瘤
茶中存在的儿茶素在促进健康的作用中起着重要作用。表没食子儿茶素没食子酸酯在体外和体内乳腺癌模型中显示了抗氧化和抗炎活性(尤其是在他莫昔芬诱导的氧化应激情况下),能够抑制头颈部或盆腔癌症患者的放射性皮炎,并抑制继发性肿瘤的继发发展。
↓↓ 防止辐射带来的不良反应
据报道,表没食子儿茶素没食子酸酯还可用于预防暴露于γ辐射或以顺铂为基础的治疗后的唾液腺细胞功能障碍,防止辐射引起的不良血液学变化(如贫血、血小板减少),以及预防博莱霉素引起的肺纤维化。
↓↓ 减少药物引起的毒性
此外,表没食子儿茶素没食子酸酯可减少伊立替康治疗期间的胃肠紊乱、顺铂引起的肾毒性和耳毒性,以及阿霉素和柔红霉素治疗引起的心脏毒性。
在药物致敏方面,绿茶显示了许多与化疗的协同作用,其中一些包括与4-羟基他莫昔芬联合使用可以提高细胞毒性水平,通过降低大B细胞淋巴瘤(Bcl-xL)基因(编码抗凋亡蛋白)的表达使得MCF7细胞对5-氟尿嘧啶增敏,以及4只接种T1的Balb/c小鼠对紫杉醇的敏感性。这种致敏作用通过降低所需化学治疗药物的剂量有效地降低了药物引起的毒性的严重程度。
乳制品/奶制品与癌症治疗之间的关系一直存在争议。尽管据报道在Balb/c小鼠中,乳制品/乳脂可以增强紫杉醇治疗的有效性,并减少与该制剂相关的毒性,例如,器官损伤、腓肠肌丧失、附睾脂肪组织减少、红细胞和白细胞损失以及空肠形态、绒毛长度和肠 γ-谷氨酰转肽酶活性的破坏。
tips
有研究表明,在被诊断患有早期浸润性乳腺癌的女性中,高脂牛奶与乳腺癌、全因和非乳腺癌死亡率的增加有关,以及与绝经前妇女乳腺癌进展的风险增加有关。
鱼油富含omega-3脂肪酸,如二十碳五烯酸(EPA)和二十二碳六烯酸(DHA),在减少癌症和癌症治疗相关症状和毒性方面的作用已被广泛研究。
↓↓ 减少化疗引起的毒性
一项随机临床试验表明,在宫颈癌患者(n=40)中,补充鱼油可以减少化疗引起的毒性,如厌食、恶心、口干和味觉障碍。一项对88例癌症患者补充精氨酸、谷氨酸和鱼油的研究表明,补充精氨酸、谷氨酸和鱼油可显著减少3-4级血液毒性,并提高两年总生存率。
↓↓ 增强抗肿瘤作用
一些体外和体内研究表明,与单纯化疗相比,化疗期间给予EPA和DHA可以增强抗肿瘤作用,减少化疗对正常组织的毒性,抑制全身炎症,改善癌症患者的营养状况。同样,在紫杉醇和顺铂/卡铂治疗期间,在等卡路里饮食中添加EPA与非小细胞肺癌患者的疲劳减轻、食欲改善和化疗引起的神经病变减少有关。
↓↓ omega-3脂肪酸减少化疗毒性
omega-3脂肪酸还可以减少癌症相关的恶病质,增加免疫调节作用,从而增强结直肠癌和食道癌患者的化疗与放疗,减少食道癌患者化疗引起的毒性(如口腔炎,3/4级腹泻,以及天冬氨酸转氨酶和丙氨酸转氨酶水平的升高)。
已经提出了多种机制来解释omega-3脂肪酸在降低癌症治疗相关毒性方面的作用。其中一种假设是细胞膜的组成,这表明不饱和脂肪酸掺入癌细胞膜中,导致细胞膜组成模式与正常细胞不同。考虑到细胞膜是细胞内信号传导和基因表达调控的中心,癌细胞和正常细胞之间细胞膜上脂肪酸分布的不同可能导致信号通路的不同激活(例如,PKC激活和NF-KB 通路)。
尽管omega-3脂肪酸在临床试验中显示出了益处,但在临床前模型中,omega-3脂肪酸十六碳-4,7,10,13-四烯酸已证明可以抑制铂化合物的肿瘤导向细胞毒性,这可能会对患者造成潜在的伤害。
因此,在建议临床食用鱼油之前,有必要进一步研究普通鱼油成分与纯化的EPA/DHA对特定化疗药物的细胞毒性的影响。
姜黄是一种开花植物,可从中提取姜黄素。它们与预防和治疗化疗和放疗相关的不良事件有关。姜黄/姜黄素对癌症的有益作用归因于它们的抗氧化和抗炎特性,以及它们在抑制细胞增殖和肿瘤干细胞发展方面的作用,以及它们对肠道菌群和免疫系统的积极作用。
↓↓ 姜黄油降低化疗4级毒性发生率
姜黄油具有保肝作用,并缓解刀豆蛋白A诱导的氧化应激和炎症,从而减少人类患者的多种症状和毒性。同样,与其他草本物质:发酵大豆提取物、绿茶提取物、樟芝菌丝体、螺旋藻和葡萄籽提取物相结合,可显著降低接受亚叶酸钙/5-氟尿嘧啶化疗患者的4级毒性发生率。
↓↓ 姜黄/姜黄素减轻粘膜炎严重程度
一项评估的结论是,局部应用姜黄和姜黄素可以控制接受化疗和/或放疗的癌症患者的口腔粘膜炎,而接受姜黄/姜黄素治疗的患者报告称疼痛较轻,红斑强度较低,溃疡区域较少。一项评估32名接受放射治疗的头颈部癌症患者的临床试验也得出结论,口服纳米胶束姜黄素显著减轻了放疗引起的粘膜炎的严重程度。
↓↓ 姜黄/姜黄素有助于控制癌症相关的疼痛
一项针对绝经后乳腺癌患者的多中心临床试验(n=45)显示,联合应用羟基酪醇(一种在橄榄油中发现的具有强大抗氧化作用的酚类植物化学物质)、omega-3脂肪酸和姜黄素可以减轻患者报告的疼痛,并降低炎症生物标志物的水平。
↓↓ 姜黄与常见化疗药物的协同 / 拮抗作用
姜黄素在I期临床试验中协同增强化疗药物FOLFOX(5-氟尿嘧啶、亚叶酸钙、奥沙利铂)和达沙替尼对经FOLFOX处理的HCT116和HT-29细胞有抗增殖作用。
尽管这些结果提示了有希望的进一步研究领域,但对人乳腺癌细胞株(即MCF-7、MDA-MB-231和BT-474)的体外实验表明,姜黄素与以伊立替康或环磷酰胺为基础的化疗之间存在拮抗作用,饮食补充姜黄素可能会抑制基于化疗的肿瘤消退。这些发现表明,需要更多的研究来确定乳腺癌患者是否应该在化疗期间避免补充姜黄素。
谷氨酰胺是巨噬细胞、淋巴细胞和肠细胞的主要燃料来源,具有多种有益作用,包括改善免疫系统、减少炎症和分解应激状态。
↓↓ 化疗配合谷氨酰胺补充,降低血液学毒性发生率
一项针对接受同步放化疗的癌症患者的随机研究表明,除了常规饮食外,还接受精氨酸、谷氨酰胺和鱼油营养补充剂的患者与未接受补充剂的患者相比,发生3级或4级血液学毒性的发生率较低。
↓↓ 补硒改善免疫系统
据报道,补硒在脂质过氧化方面具有抗氧化作用,刺激自然杀伤细胞的细胞毒活性,减少肿瘤内血管生成,并在体外改善免疫系统。硒与重金属的解毒作用有关。这些影响的产生可能是因为硒是硒蛋白和酶的重要组成部分,这些硒蛋白和酶有助于抗氧化防御、减少炎症、甲状腺激素产生、DNA合成。
一项对关于硒和放射治疗的文章进行了评估,得出的结论是,每天服用300-500微克的硒,持续10天到6个月,可以减少放疗的副作用,包括腹泻、唾液腺损伤和辐射伤口,不会产生不良影响。
tips
高剂量的硒(>400微克/天)会产生严重的副作用,像指甲变脆、脱发、胃肠功能障碍、皮疹、神经紊乱等。
维生素补充剂在减少癌症和癌症治疗相关症状和毒性方面的功效研究显示出相互矛盾的结果。
例如,尽管许多研究表明维生素C补充剂具有潜在的抗癌作用和降低毒性作用,但对接受化疗的癌症患者的维生素C进行的系统审查发现,没有明确的证据表明服用维生素C补充剂可以减少毒性或改善治疗的抗癌效果。
同样,尽管维生素D补充剂已被证明可以预防癌症治疗引起的骨质流失,并恢复许多早期乳腺癌患者经历的维生素D不足,但在各种研究中报告的维生素D的抗肿瘤效果仍然很差。
tips
由于维生素补充剂在癌症治疗期间的效果尚不清楚,强烈建议患者遵守饮食建议,通过水果和蔬菜等天然食物摄入维生素,而不是依赖膳食补充剂。
一个健康人体胃肠道中的各种复杂的微生物群已显示出显著的生理益处,如增强肠道功能和消化能力、抵御病原体和调节免疫力。
虽然“健康”肠道菌群的定义并不明确,但数据表明,具有高度功能冗余度的多样化和稳定的微生物群是健康状态的关键标志。
肠道菌群对癌症患者的治疗反应有显著影响。例如,白血病或淋巴瘤患者在造血干细胞移植后具有高水平的粘液真杆菌,其复发或肿瘤进展的可能性较低。
↓↓ 肠道菌群失调影响抗PD-L1治疗效果
人类、动物和体外研究表明,肠道菌群的免疫调节影响靶向免疫治疗的疗效,如细胞毒性T淋巴细胞相关4(CTLA-4)阻断和抗PD-L1治疗。与这些发现一致的是,一部分接受抗PD-L1治疗和广谱抗生素治疗的上皮性肿瘤患者经历了失败治疗,其原因可能是导致了微生物群的失调。
进一步支持这些发现的是,从应答者和无应答者患者向有免疫活性小鼠的粪便转移导致小鼠对抗PD-L1抗体产生了与相应粪便移植供体相同的反应,从而证明了肠道菌群对抗PD-L1治疗的效果。
↓↓ 肠道菌群调节化疗的疗效和毒性
化疗药物环磷酰胺和阿霉素能诱导革兰氏阳性菌(约氏乳杆菌、鼠乳杆菌和海氏肠球菌)转移到小鼠的次级淋巴器官中。一旦转移到淋巴器官,微生物就会刺激17型和1型T辅助细胞反应的积累,从而增强免疫反应。
临床前和临床证据都表明,抗生素会降低化疗的疗效。例如,长期使用抗生素已被证明会降低环磷酰胺治疗荷瘤小鼠P815肥大细胞瘤的疗效,支持了肠道细菌易位的重要性。
一项对C57BL/6(B6)和129SvEv(129)小鼠化疗所致周围神经病变的临床前研究表明,肠道细菌在确定紫杉醇诱导的疼痛敏感性中起主导作用;在对紫杉醇诱导的疼痛敏感和抵抗的小鼠之间,观察到肠道微生物群组成的显著差异。
↓↓ 肠道菌群与胃肠道不良反应的发生率有关
一项对接受放化疗的宫颈癌患者进行的临床研究(n=35)得出结论,肠道微生物多样性与胃肠道毒性的发生率呈负相关。
同样,一项针对儿童急性淋巴细胞性白血病患者(n=51)的临床研究确定,化疗期间肠道微生物群的变化与胃肠道不良反应的发生率有关,如全身炎症和肠道粘膜炎。Toll样受体(TLRs)和肠道微生物之间的免疫调节相互作用可能调节结肠的炎症和愈合,防止甲氨蝶呤化疗的毒性。
↓↓ 肠道菌群从根本上与粘膜炎的发病机制相关
粘膜炎是一种常见的胃肠道毒性,会导致腹泻、疼痛、体重减轻和剂量限制。例如,伊立替康是一种已知会引起严重腹泻的化疗剂。这种毒性的一种机制可能是某些细菌β-葡萄糖醛酸酶的作用,这些酶已被证明通过将伊立替康的活性代谢物SN-38释放到肠腔中来诱导腹泻。
与这一假设一致,通过喹诺酮类抗生素环丙沙星抑制此类酶可抑制伊立替康治疗小鼠的腹泻,从而证明肠道微生物群的调节可以降低伊立替康的毒性。
↓↓ 肠道菌群影响癌症治疗相关的心理神经症状
一项系统评价评估了肠道微生物群与化疗之间关系的研究,得出的结论是,肠道微生物群可能会影响癌症治疗相关的心理神经症状,例如疲劳、焦虑、抑郁和睡眠障碍。
因此,相对健康的肠道微生物群可以改善癌症患者的健康,通过增强治疗效果和减少免疫疗法和化学疗法的副作用以及通过免疫调节等方式。
饮食影响肠道微生物种类的组成和多样性。膳食纤维的高摄入量有利于膳食纤维消化细菌的增加。作用机制是消化膳食纤维的细菌产生丁酸等短链脂肪酸,滋养肠道上皮细胞,从而加强肠道黏膜屏障,增强黏膜和全身免疫。
↓↓ 食品补充剂调节微生物群驱动的化学治疗毒性
由于肠道屏障和微生物群稳态的潜在损害,化疗通常与肠道菌群的改变有关。因此,预防和治疗与化疗相关的肠道菌群改变可能有助于预防与化疗相关的胃肠道毒性。
▸从鱿鱼墨汁中提取的多糖在小鼠给药环磷酰胺后可富集双歧杆菌,并减少拟杆菌,从而改善肠道微生物群功能障碍。
▸人参化合物可以增强化疗药物5-氟尿嘧啶对结直肠癌细胞系的作用,特别是当这些化合物被肠道微生物群菌群代谢时。
▸鞣花酸,一种常见于草莓、葡萄和黑莓等蔬菜和水果中的多酚,被肠道菌群代谢释放尿石素,这对人类结肠癌具有抗增殖作用。
↓↓ 益生元和益生菌在癌症治疗中发挥作用
益生元(促进有益肠道微生物生长的难消化的食物成分,例如香蕉、芦笋和朝鲜蓟)和益生菌(引入体内以发挥有益作用的微生物,例如酸奶、克非尔、酸菜、豆豉,和泡菜)与肠道菌群的组成密切相关,这些物质在癌症治疗过程中的作用很关键。
许多动物和人类研究表明,益生元、益生菌在预防化疗期间的粘膜炎方面具有强大的作用。
对于接受同步放化疗的鼻咽癌患者,益生菌与放射治疗相结合,可以通过改变肠道菌群,显着增强宿主免疫力,缓解放化疗相关的口腔黏膜炎。
VSL #3(包括Streptococcus thermophiles, Bifidobacterium breve, B. longum, B. infantis, Lactobacillus paracasei, L. delbrueckii subsp. bulgaricus, L. acidophilus, L. plantarum),在伊立替康治疗期间,减少大鼠腹泻和体重减轻。
L. casei, L. rhamnosus, B. bifidum 通过抑制肿瘤坏死因子α、白细胞介素-1b和白细胞介素-6 mRNA的表达减少化疗诱导的小鼠腹泻。
鼠李糖乳杆菌GG补充可减少人类大肠癌患者在5-氟尿嘧啶化疗期间的严重腹泻和腹部不适。
在接受化疗的儿童中使用养乐多的短双歧杆菌菌株可预防发烧并减少静脉注射抗生素的频率。
膳食补充益生元低聚果糖和菊粉可放大药物对小鼠的作用(5-氟尿嘧啶、多柔比星、长春新碱、环磷酰胺、甲氨蝶呤、阿糖胞苷),从而证明了益生元与化疗之间的协同作用。
编辑
大量临床前数据以及有限的临床证据表明,饮食因素可能在预防和/或治疗癌症以及癌症治疗相关的症状和毒性方面发挥作用,寻求特定饮食干预措施的数据仍在不断涌现。
人们对肠道菌群的性质和对癌症治疗的影响也越来越感兴趣。期待更大样本量的随机对照试验,进一步研究饮食干预措施。
最好的饮食是患者愿意并且能够坚持的饮食,因此在将这些策略引入临床时可能需要一定程度的个性化。
如果能开发出,通过调节肠道菌群来改善治疗效果的药物,其潜力是巨大的。
主要参考文献
Spencer CN, McQuade JL, Gopalakrishnan V, McCulloch JA, Vetizou M, Cogdill AP, Khan MAW, Zhang X, White MG, et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science. 2021 Dec 24;374(6575):1632-1640. doi: 10.1126/science.aaz7015. Epub 2021 Dec 23. PMID: 34941392; PMCID: PMC8970537.
Yu ZK, Xie RL, You R, et al. The role of the bacterial microbiome in the treatment of cancer. BMC Cancer. 2021;21(1):934. Published 2021 Aug 19. doi:10.1186/s12885-021-08664-0
Kim AJ, Hong DS, George GC. Dietary influences on symptomatic and non-symptomatic toxicities during cancer treatment: A narrative review. Cancer Treat Rev. 2022 May 13;108:102408. doi: 10.1016/j.ctrv.2022.102408. Epub ahead of print. PMID: 35623220.
Barrea L, Caprio M, Tuccinardi D, Moriconi E, Di Renzo L, Muscogiuri G, Colao A, Savastano S; Obesity Programs of nutrition, Education, Research and Assessment (OPERA) group. Could ketogenic diet “starve” cancer? Emerging evidence. Crit Rev Food Sci Nutr. 2022;62(7):1800-1821. doi: 10.1080/10408398.2020.1847030. Epub 2020 Dec 4. PMID: 33274644.
Baguley BJ, Skinner TL, Jenkins DG, Wright ORL. Mediterranean-style dietary pattern improves cancer-related fatigue and quality of life in men with prostate cancer treated with androgen deprivation therapy: A pilot randomised control trial. Clin Nutr. 2021 Jan;40(1):245-254. doi: 10.1016/j.clnu.2020.05.016. Epub 2020 May 25. PMID: 32534948.
革兰氏阳性和阴性菌
在日常生活中,我们常常会看到药物或抗菌产品适应症会这样写到,对革兰氏阳性菌有效,对革兰氏阴性菌敏感,或者说对革兰氏阴性菌有效,对革兰氏阳性菌无效。可能很多人不是很清楚或搞不懂二者的区别。
本文主要介绍革兰氏阳性和阴性菌,它们的区别,代表性菌种以及针对用药等。
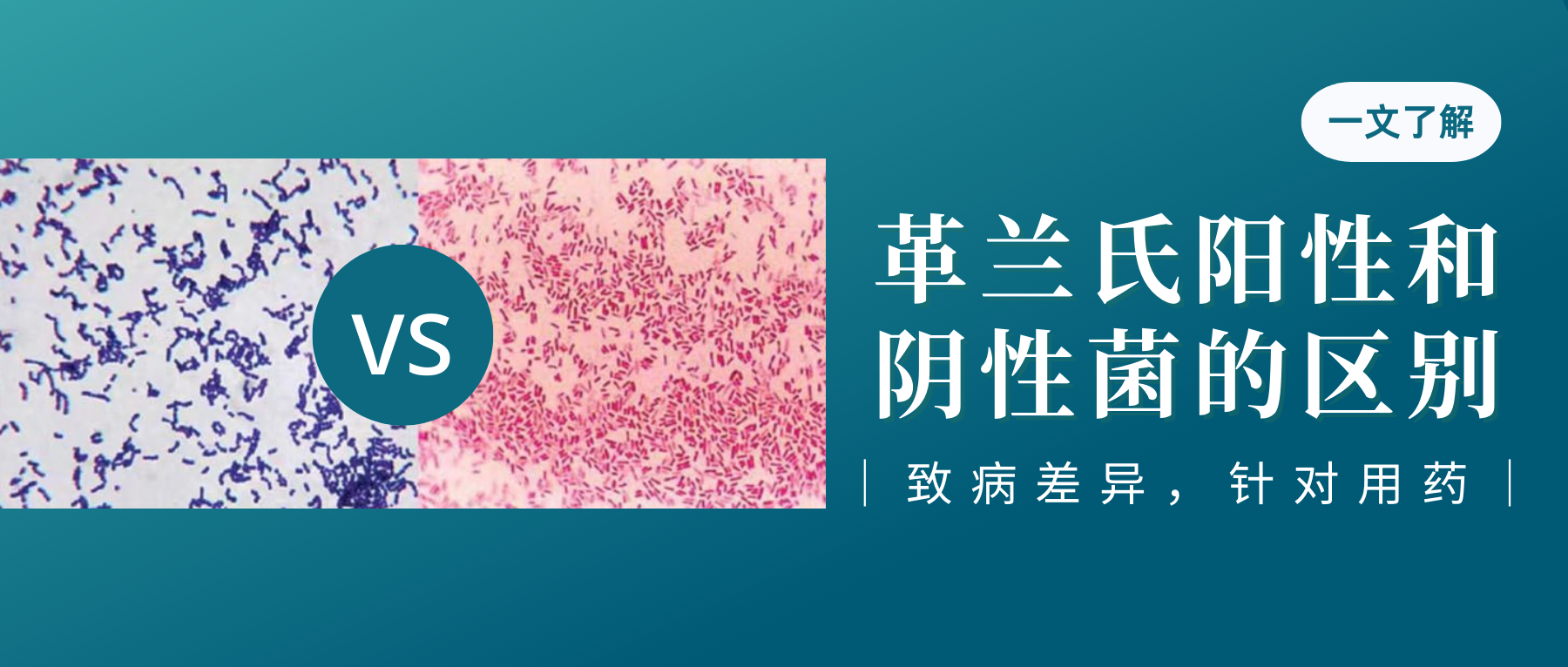
1884年,细菌学家Hans Christian Gram发明了革兰氏染色法来鉴别区分细菌。这种技术将细菌分成两大类,即革兰氏阳性菌(G+)和革兰氏阴性菌(G-)。区分主要是这两类细菌细胞壁成分不同,因而着色也不同所致。
这两类细菌的生理结构,疾病原因以及抗菌作用不一,因此,区分病原菌是革兰氏阳性菌,还是阴性菌,在临床确定感染和选择用药方面意义重大。
革兰氏阴性菌致病多由于患者有基础疾病或者体质比较差,肠道细菌感染引起的腹泻多是由肠道菌群中的革兰氏阴性菌所致,如大肠杆菌、沙门氏菌、志贺氏菌,布氏杆菌等,治疗这类细菌感染,一般使用三代头孢菌素以及喹诺酮类抗生素。注意大多数革兰氏阴性菌对青霉素耐药或不敏感。
大多数化脓性球菌都属于革兰氏阳性菌,它们能产生外毒素使人致病,常见的菌种有葡萄球菌、链球菌、肺炎双球菌、李斯特菌、炭疽杆菌、白喉杆菌、破伤风杆菌等。尤其在人体肠道内,革兰氏阳性致病菌致病几率更大。大多数由革兰氏阳性菌引起的感染可以用相当少量的抗生素治疗。青霉素、氯唑西林和红霉素足以覆盖 90% 的革兰氏阳性感染。
此外,某些广谱抗生素对革兰氏阳性菌和革兰氏阴性菌都有抗菌作用,如氨苄青霉素、庆大霉素、土霉素、磷霉素及环丙沙星等,但是作用效果可能不是最优。此外,磺胺类药物也属于广谱抑菌药物。
临床应用时,如果对细菌感染比较明确,尽量使用窄谱抗菌药物,如不太明确,可选用广谱抗菌药物。因此,临床治疗疾病时,首先要对药物的作用与用途要详细了解,然后再根据感染类型或诊断结果合理选择药物,这样才能取得最佳治疗效果。否则,药物选择不当,将会出现无效或越治越重的结果。
革兰氏阴性菌和革兰氏阳性菌之间的主要区别在于肽聚糖层的厚度和外部脂质膜的存在与否。
不同细菌的革兰氏染色
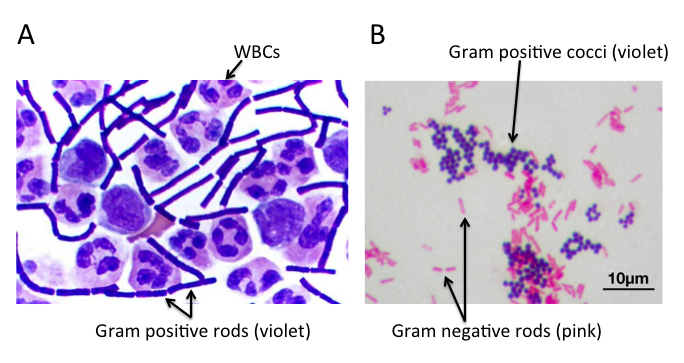
A) 脑脊液样本中的杆状革兰氏阳性炭疽杆菌(导致炭疽),也含有白细胞。B) 革兰氏阳性金黄色葡萄球菌(紫色或紫色)和革兰氏阴性杆菌大肠杆菌(粉红色)的革兰氏染色,它们是最常用的革兰氏染色参考细菌。参考来源:Wikipedia commons 和 tmedweb)
// 缺乏外膜,更容易受抗生素影响
革兰氏阳性菌的细胞壁含有肽聚糖、脂质、磷糖醛酸和磷壁酸。这种结构成分不同于由肽聚糖和外膜(由脂质、蛋白质和脂多糖组成)组成的革兰氏阴性细菌细胞壁。尽管革兰氏阳性菌具有较厚的肽聚糖层,但它们比革兰氏阴性菌更容易受到某些靶向细胞壁的抗生素的影响,因为它们缺乏外膜。
// 常见的致病菌
最常见的革兰氏阳性细菌包括葡萄球菌、链球菌、芽孢杆菌、梭状芽孢杆菌、李斯特菌、棒状杆菌等。这些革兰氏阳性菌的代表性物种是致病的,并可能引起多种疾病。
// 可用于治疗的抗生素
青霉素是影响革兰氏阳性菌的主要抗生素之一。
红霉素是另一种用于治疗革兰氏阳性细菌感染的强效抗生素。红霉素属于一类称为大环内酯类的抗生素,与阿奇霉素和克拉霉素同属一类。它通常用于对青霉素过敏的人。
甲氧苄啶/磺胺甲恶唑,克林霉素,克林霉素,强力霉素,万古霉素也可以用于特定的革兰氏阳性菌的感染。
// 层层抵抗更难杀死:细胞壁更硬,可改变外膜
革兰氏阴性菌有坚硬的保护外壳。它们的肽聚糖层比革兰氏阳性杆菌薄得多。
当它们的细胞壁受到干扰时,革兰氏阴性细菌会释放内毒素,症状更糟。同时,大多数抗生素为了接近它们的目标,必须通过外膜。例如,亲水性抗生素通过孔蛋白。革兰氏阴性菌可以通过改变它们的疏水特性或通过孔蛋白的突变来改变它们的外膜。这对这些细菌细胞产生了抵抗力。
// 暗藏玄机:比革兰氏阳性菌更危险
与革兰氏阳性菌相比,革兰氏阴性菌作为疾病生物体更危险,因为存在覆盖外膜的荚膜或粘液层。通过这种方式,微生物可以隐藏表面抗原,这个抗原可以触发人体免疫反应。
革兰氏阴性菌是一组臭名昭著的细菌,可导致多种疾病,包括肺炎、脑膜炎、淋病、细菌性痢疾、霍乱、胃炎等。在重症监护病房 (ICU)的患者,处于发病和死亡的高风险中,更容易遇见这类细菌,因此它们在医院具有重要的临床意义。
// 可用于治疗的抗生素
已经开发了许多不同种类的抗生素来杀死革兰氏阴性菌,例如头孢菌素、叶酸拮抗剂、哌拉西林-他唑巴坦、脲青霉素、内酰胺-β-内酰胺酶抑制剂、碳青霉烯类和喹诺酮类。它们是专门针对革兰氏阴性细菌而开发的,不过有时也对某些革兰氏阳性细菌有效。
革兰氏阳性菌与革兰氏阴性菌区别总结

参考来源:microbenotes
以上是革兰氏阳性菌和阴性菌的主要区别,接下来我们针对革兰氏阳/阴性菌,从细菌特征,形状表征,细胞结构等方面,进行更详细的介绍。
革兰氏阳性菌的定义是基于它们在革兰氏染色中用酒精短暂洗涤后保留结晶紫染料的能力。革兰氏阳性菌呈紫色。
这些细菌具有非常独特的特征,可以将其与其他类型的细菌区分开来。这些包括:
在革兰氏染色过程中,革兰氏阴性细菌在用酒精清洗后将失去结晶紫染料的颜色,并吸收反染物藏红花的粉红色/红色。
几乎在地球上的每个生活区域都可能发现革兰氏阴性细菌。
尽管大多数细菌是通过革兰氏染色染料进行区分的,但显微镜下的观察揭示了更多可用于定义和表征这些细菌的特征。
根据形状的定义,革兰氏阳性菌可分为两类:
革兰氏阳性细菌形成的其他特殊形状包括:
革兰氏阴性菌细胞的显微镜观察范围从杆状到芽孢杆菌,从球菌到螺旋状,螺旋状是最常见的形状。然而,有些表现出特殊的形状,如球杆菌、四分体、栅栏、毛状体等。例如:
革兰氏阳性菌的细胞壁含有肽聚糖、脂质、磷糖醛酸和磷壁酸。这种结构成分不同于由肽聚糖和外膜(由脂质、蛋白质和脂多糖组成)组成的革兰氏阴性细菌细胞壁。因此,革兰氏阳性菌的细胞壁很厚,并吸收了革兰氏染色的结晶紫染料。因此,显微镜下的革兰氏阳性菌呈紫色。
革兰氏阳性细菌细胞壁的结构特征
革兰氏阳性细菌具有由肽聚糖组成的厚的多层细胞壁(因为它含有肽和糖的混合物)。由于肽聚糖存在于大多数细菌中,但不存在于哺乳动物细胞中,因此它是抗菌药物的良好靶标(例如细胞壁合成抑制剂,包括青霉素、头孢菌素和万古霉素)。
这些抗生素会干扰转肽酶(也称为青霉素结合蛋白或PBP 的活性) 在细胞壁中催化相邻聚糖链之间的交联。
细胞壁还含有磷壁酸纤维,有助于细菌附着在宿主细胞膜(例如粘膜细胞)上,释放后会引起感染性休克,类似于革兰氏阴性菌释放的内毒素 (LPS) 产生的感染。
β-内酰胺酶(青霉素酶)是由细菌产生的酶家族,可水解 β-内酰胺抗生素(其中许多也是细菌来源)的四原子 β-内酯环,从而使其抗菌性能失活。
细菌细胞膜还可以包含ABC 外排泵这可能导致抗生素耐药性和对那些具有细胞内作用机制的药物(例如 DNA 促旋酶抑制剂或蛋白质合成抑制剂)的多药耐药性 (MDR)。
结晶紫染料附着在革兰氏阳性菌细胞壁的厚肽聚糖层上,在光学显微镜下观察时将它们染成紫色或紫色。
肽聚糖
它也被称为胞壁质(murein),占细菌细胞壁含量的 90%。
——维持形状,并保持细胞壁强度和弹性
它是一种优质聚合物,由两种相同的糖衍生物(N-乙酰氨基葡糖和 N-乙酰胞壁酸)以及 L-氨基酸链和蛋白质中很少发现的三种不同 D-氨基酸(即 D-谷氨酸、D-丙氨酸和内消旋二氨基庚二酸)组成,可保护细胞壁免受肽酶的攻击。
D-氨基酸和L-氨基酸连接到N-乙酰壁酸,L-氨基酸特别是赖氨酸可以取代中二氨基丙烯酸。
肽聚糖亚基的这种相互连接使肽聚糖具有很强的维持细菌形状和完整性的能力,并具有弹性和延展性。
肽聚糖也具有渗透性,允许分子进出细菌细胞。
——肽聚糖的生物合成
抑制细菌细胞壁肽聚糖层的合成是许多抗菌药物的分子靶点,包括 β-内酰胺类抗生素(青霉素、头孢菌素、碳青霉烯类和单环内酰胺类)和糖肽类抗生素(万古霉素和其他较新的类似物)。
这些药物的两个主要分子靶标是转肽酶,也称为青霉素结合蛋白 (PBP),因为它们与青霉素结合,而糖基转移酶 (GT) 可被万古霉素等糖肽抑制。
PBPs 有许多亚型,给定的细菌菌株可以表达多种 PBPs。这些 PBP 在其生理特性和与抗生素相互作用的敏感性方面可能有所不同。取决于 PBP 亚型,糖基转移酶可以作为单独的酶或作为与转肽酶相关的二聚体存在。
革兰氏阳性细胞壁生物合成
图片来源自(Wikipedia Commons 和 tmedweb)
细菌细胞壁由重复的 N-乙酰氨基葡萄糖 (NAG) 和 N-乙酰胞壁酸 (NAM) 亚基链组成。NAM 亚基附有短肽链。
肽链的组成因细菌而异,但近端的丙氨酸通常是 L-Ala,而远端的两个通常是 D-Ala。也与青霉素结合的细胞壁转肽酶(青霉素结合蛋白:PBPs)在肽侧链之间形成键,并从肽侧链之一排出末端 D-丙氨酸。
一旦形成交联,PBP 就会从壁上解离。用糖基转移酶 (GT)分离酶结构域NAM 和 NAG 残基之间的活性形成联系。一些高分子量 PBP(例如 PBP2)是含有转肽酶和糖基转移酶结构域的酶复合物。
磷壁酸纤维存在于革兰氏阳性菌的细胞壁中,由磷酸甘油或磷酸核糖醇的聚合物组成。它们参与细菌与黏膜细胞的附着,可诱发感染性休克,类似于革兰氏阴性菌释放的 LPS(内毒素)。
磷壁酸
这是由甘油共聚物组成的加固墙。
磷壁酸是水溶性的,占细菌细胞壁总干重的 50%。
它要么直接与肽聚糖共价连接,要么与细胞膜(脂磷壁酸)连接。通过 6-羟基 N-乙酰胞壁酸与肽聚糖直接相连。
带负电荷,延伸到肽聚糖表面,使细菌细胞壁带负电荷。
它还有助于维持细胞壁的结构。
它在革兰氏阴性菌中完全不存在。
脂质
它们在肽聚糖下方有一层薄薄的脂质,大约 2-5%,其作用是锚定细菌细胞壁。
细胞壁
——细胞壁非常复杂
结合细胞外膜的主要作用,加上一层肽聚糖,其功能特性复杂,这里是对细胞壁及其功能部分的描述。
革兰氏阴性细菌的细胞壁具有一层2-7nm的肽聚糖薄层和7-8nm厚的外膜。
——周质空间较大
显微镜下,细胞膜和细胞壁之间有一个空间,称为由周质组成的周质空间。在革兰氏阴性菌和革兰氏阳性菌中均能发现,但在革兰氏阴性菌中,周质空间较大。
革兰氏阴性细菌细胞壁
在结构上,革兰氏阴性细胞壁由细胞膜外部的两层组成:一层薄薄的肽聚糖(太薄而无法吸收大量甲基紫染色)和一层外膜(革兰氏阴性细菌独有),通常含有促进小(<700 Da)亲水分子(例如糖、氨基酸和维生素)扩散的孔蛋白。
许多抗生素(例如许多青霉素和头孢菌素)也可以通过孔蛋白扩散到达它们的作用部位。
然而,万古霉素 (1449 Da) 的质量太大,无法透过孔蛋白到达其作用部位,这使其对革兰氏阴性细菌无效。因此,外膜为革兰氏阴性菌提供了对某些抗生素的固有“内在抗性”,可以通过改变孔蛋白的表达水平或改变孔蛋白的孔特性以降低抗生素的渗透性来进一步修饰。
革兰氏阴性菌的外膜还含有脂多糖 (LPS)或内毒素,可被细菌排出,引起宿主强大的免疫反应。
周质空间
革兰氏阴性菌的周质空间由几种蛋白质组成,这些蛋白质有助于获取营养,例如攻击核酸和磷酸化分子的水解酶,以及积极协助将物质运输到细菌细胞中的结合蛋白。周质空间还具有合成肽聚糖和修饰可能对细胞造成伤害的有毒元素的酶。
肽聚糖
革兰氏阴性细菌细胞壁有一层薄的肽聚糖层,位于质膜上方,约占细胞干重的5%。厚度不超过4纳米,一些细菌如大肠杆菌只有2纳米厚的肽聚糖。
外膜和脂多糖
革兰氏阴性菌还有第二层脂质双层,位于肽聚糖层的外部。这种外膜通过布劳恩脂蛋白与肽聚糖相连。外膜和肽聚糖之间的紧密连接是维持外膜作为许多有毒分子和抗生素的不渗透屏障所必需的。
外膜上的粘附位点也加强了革兰氏阴性细胞壁,这些粘附位点在允许细胞接触和膜融合方面发挥作用。物质通过这些粘附位点进入细胞。
革兰氏阴性菌结构

图源:Jeff Dahl,wikipedia
外膜主要由脂多糖 (LPS)组成,脂多糖是由脂质和碳水化合物组成的大型复杂分子。脂多糖由3个单元组成:脂质A、核心多糖和O侧链。
脂质A由两种氨基葡萄糖糖衍生物组成,每个衍生物含有三种脂肪酸和焦磷酸盐,脂多糖的任何剩余部分都会伸出膜表面。
O侧链也称为O抗原,是从核心向外延伸的链。它由导致细菌菌株之间变异的糖组成。这些 O 抗原也负责细菌逃避抗体反应。
// 外膜及其脂多糖的作用
——脂多糖负责保护细胞壁免受外部攻击
LPS 带有负电荷,使电池表面带负电荷。因此,这稳定了膜结构。
脂质A是脂多糖的有毒成分,因此它起到内毒素的作用。
——防毒素进入,防成分丢失
外膜及其脂多糖有助于防止抗生素、胆汁盐和其他有毒元素进入并破坏细胞。
外膜由孔蛋白组成,使其具有渗透性,允许小分子(如葡萄糖)进入。维生素 B12 等较大的分子通过特定的载体运输穿过外膜。
外膜还有助于防止成分丢失,特别是来自周质空间。
下表描述了主要的革兰氏阳性致病菌,它们的基本形态特征以及它们在人类中引起的疾病。
已知革兰氏阴性菌是正常菌群,部分会导致严重的人类感染,从社区获得性感染到医院感染。
革兰氏阴性菌外膜的结构是其众多显着特征之一。脂多糖 (LPS) 存在于膜的外叶上,其脂质 A 部分用作内毒素。
// 革兰氏阴性菌感染:严重时可危及生命
如果由于某种原因,革兰氏阴性细菌能够到达动物的循环系统,脂多糖将激活免疫系统,并触发先天免疫反应,产生细胞因子和激素调节剂。这会引起炎症,并可能导致毒性反应,从而导致发烧、呼吸急促和低血压。这就是已知革兰氏阴性细菌会导致危及生命的休克的原因。
内毒素休克的一些症状:
发烧和发冷或体温下降、发炎、皮疹、呼吸急促、心率加快、低血压、多器官衰竭等。
下表给出了一些革兰氏阴性致病细菌的例子以及它们在人类宿主中引起疾病和感染时表现出的临床特征。

如上表所述,已知革兰氏阳性细菌会引起多种感染,如果不及时和适当地治疗和管理,可能对人类造成灾难性的影响。
革兰氏阳性杆菌感染用抗生素治疗。青霉素、氯唑西林和红霉素可治疗 90% 以上的革兰氏阳性菌。
常见的革兰氏阳性抗菌药物及作用机制

然而,抗生素耐药性正在成为革兰氏阳性感染的一个严重问题。研究人员正在开发新的药物来帮助解决这个问题。只有在绝对需要时才应使用抗生素。需要严格遵循感染控制标准,以防止抗生素耐药性感染的发展和传播。
由于它们的外膜,这些细菌对溶菌酶和青霉素具有抗性。这是因为存在保护内膜和细胞壁的外壁。
在周质空间(两个细胞膜之间的区域)中也发现了分解或改变抗生素的酶。用于治疗革兰氏阴性菌感染的治疗方法包括羧基、氨基和脲基青霉素。为了对抗可以消化这些药物的酶,有时将它们与β-内酰胺酶抑制剂结合使用。β-内酰胺酶是一种存在于周质中的酶。
针对细菌病原体的抗菌剂被称为抗生素。这些抗生素启动针对细菌细胞的阻断或抑制机制,以诱导细菌细胞增殖和复制。
用于对抗革兰氏阴性菌的抗生素示例

抗生素耐药性是现在世界上的一个主要临床问题。
耐多药细菌在人群中变得越来越普遍,如果不进行有效治疗,这种感染可能会导致肾功能衰竭、败血症,甚至死亡。
微生物以多种方式抑制临床治疗中使用的许多抗菌剂。这些包括改变药物结合位点的方法,改变药物构象的方法,改变膜通透性的方法,可以导致耐药机制失活。
例如革兰氏阴性菌中有两层膜,外膜和内膜。脂多糖被认为是一种非常强的免疫反应诱导剂,它具有三个重要成分:脂质 A、亲水性多糖、抗原 O 的疏水域。
疏水域在细胞膜的外部表达。它是疏水成分脂质a,它负责内毒素作用。LPS 在细菌中是可变的,并且由于遗传变异,一些细菌只产生一种不被 Toll 样受体识别的弱抗原。然而,有大量的革兰氏阴性菌团体可能会引起这样的反应。免疫系统也被一些 toll 样受体 4 (TLR4) 激活,这些受体存在于与免疫系统有关的众多细胞中,如巨噬细胞、单核细胞、中性粒细胞和树突状细胞。
由 LPS 和 TLR4 受体介导的先天免疫反应的激活导致反应增强,产生细胞因子、趋化因子和干扰素等。
免疫系统的反应取决于感染过程的严重程度以及侵袭性细菌中 LPS 的结构,这与菌的毒力有关。因此,虽然一些细菌(如大肠杆菌)可以诱导免疫系统,但其他细菌(如幽门螺杆菌)仅具有弱抗原性。
// 对付耐药性细菌新思路:根据电荷相互作用原理设计新药
2017 年,伊利诺伊大学化学教授和当前研究的合著者Paul Hergenrother(ACPP 负责人/MMG)在《自然》杂志上报告说 ,发现的一个关键是,如果向它们添加带正电荷的基团,例如胺,一些抗生素可以使用特定的膜孔穿透革兰氏阴性细菌的细胞膜。
这项工作表明,抗生素上带正电荷的胺基与细菌孔内的负电荷有良好的相互作用。这些吸引力使带有胺基的抗生素以一种更有利于能量的方式排列,因为它穿过收缩区的孔的最狭窄部分。不含胺的抗生素面临更高的能量屏障去通过孔隙。
这或许意味着未来可以设计新药(或修改旧药),以攻击和杀死对抗生素治疗具有耐药性的微生物。
问
革兰氏阳性细胞和革兰氏阴性细胞之间的三个区别是什么?
答
革兰氏阳性菌有一层厚的肽聚糖作为它们的细胞壁,而革兰氏阴性菌有一层薄薄的肽聚糖和外膜。
革兰氏阴性菌有脂多糖(LPS),而革兰氏阳性菌没有。
一些革兰氏阳性细菌含有霉菌酸,它会在细胞壁上形成一层蜡质层。
问
什么是革兰氏阳性感染?
答
由革兰氏阳性菌引起的感染,如耐甲氧西林金黄色葡萄球菌 (MRSA)、耐万古霉素肠球菌 (VRE) 和艰难梭菌是常见的多重耐药菌感染。
问
革兰氏阳性菌更容易治疗吗?
答
革兰氏阳性细菌,即那些具有肽聚糖外层的物种,更容易被杀死——它们的厚肽聚糖层很容易吸收抗生素和清洁产物。因此,某些容易杀死革兰氏阳性菌的洗涤剂不会破坏革兰氏阴性菌。
问
肠道内很多革兰氏阳性菌致病吗?
答
是的,人体肠胃道的大多数革兰氏阳性菌都是条件致病菌。包括微球菌、肠球菌、金黄色葡萄球菌、表皮葡萄球菌、腐生葡萄球菌、肺炎链球菌、草绿色链球菌、酿脓链球菌、无乳链球菌、破伤风梭菌、肉毒杆菌、产气荚膜梭菌、产气荚膜梭菌、艰难梭菌, 单核细胞增生李斯特菌等。
问
革兰氏阳性菌在哪里发现?
答
根据革兰氏阳性菌种,它们可以在人类的土壤、水生沉积物、灰尘、皮肤、口腔、肠道或生殖道中找到。
问
革兰氏阳性球菌危险吗?
答
革兰氏阳性菌可能是球菌或杆菌。这些称为常驻菌群的细菌通常不会引起疾病。革兰氏阳性杆菌引起某些感染,包括:炭疽。
问
革兰氏阳性菌对抗生素的抵抗力更强吗?
答
不是。与革兰氏阳性菌相比,革兰氏阴性菌对多种抗生素的耐药性更强。由于它们的外膜,它们对抗生素的抵抗力更强。
革兰氏阳性细菌更容易被杀死,因为它们的厚肽聚糖层很容易吸收抗生素和清洁剂。另一方面,革兰氏阴性细菌具有薄的肽聚糖层,不会吸收周围的任何异物。
问
革兰氏阳性菌有内毒素吗?
答
不会。内毒素与革兰氏阳性菌无关。这些细菌没有内毒素,因为它们没有外膜。另一方面,革兰氏阴性细菌会产生内毒素。
这些内毒素是革兰氏阴性细菌细胞外膜的一部分,只有当细胞裂解或细菌死亡时才会释放出来。内毒素是形成革兰氏阴性菌细胞壁结构的热稳定性脂多糖-蛋白质复合物。
问
为什么革兰氏阳性菌对抗生素更敏感?
答
尽管革兰氏阳性菌具有较厚的肽聚糖层,但它们比革兰氏阴性菌更容易受到某些靶向抗生素的细胞壁的影响,因为它们缺乏外膜。
大多数抗生素为了接近它们的目标,必须通过外膜。例如,亲水性抗生素通过孔蛋白。因此,革兰氏阴性菌可以通过改变它们的疏水特性或通过孔蛋白的突变来改变它们的外膜。这对这些细菌细胞产生了抵抗力。革兰氏阳性菌缺乏这一因素,因此革兰氏阴性菌对抗生素的抵抗力比它们强。
问
革兰氏阳性菌是否致病?
答
是的,大多数革兰氏阳性菌都是致病菌。致病性革兰氏阳性菌的实例包括微球菌、肠球菌、金黄色葡萄球菌、表皮葡萄球菌、腐生葡萄球菌、肺炎链球菌、草绿色链球菌、酿脓链球菌、无乳链球菌、破伤风梭菌、肉毒杆菌、产气荚膜梭菌、产气荚膜梭菌、艰难梭菌, 单核细胞增生李斯特菌等。
问
革兰氏阳性菌引起的常见感染有哪些?
答
炭疽、白喉、腹泻、脑膜炎、恶心、皮肤感染、尿路感染。
问
哪种抗生素对革兰氏阳性菌有效?
答
对革兰氏阳性菌有效的抗生素是青霉素、氯唑西林和红霉素,几乎覆盖了 90% 的革兰氏阳性菌感染。其他还有万古霉素、甲氧苄啶/磺胺甲恶唑,克林霉素,克林霉素等。
问
革兰氏阴性菌引起的人类常见疾病有哪些?
答
革兰氏阴性菌会在医疗机构中引起感染,包括肺炎、血流感染、伤口或手术部位感染以及脑膜炎。此外还有霍乱、鼠疫、伤寒、脑膜炎和尿路感染是人类常见的细菌性疾病。
问
为什么革兰氏阴性菌比革兰氏阳性菌更有害?
答
革兰氏阴性菌细胞壁坚硬,不易对抗生素敏感,在抗生素作用下会释放内毒素。
问
什么会杀死革兰氏阴性菌?
答
这些抗生素包括头孢菌素类(头孢曲松-头孢噻肟、头孢他啶等)、氟喹诺酮类药物(环丙沙星、左氧氟沙星)、氨基糖苷类(庆大霉素、阿米卡星)等
问
革兰氏阴性菌的分泌系统是什么?
答
革兰氏阴性菌具有广泛封闭的分泌系统,可以转移微小分子、DNA、氨基酸、蛋白质。
问
革兰氏阴性菌从哪来?
答
革兰氏阴性细菌随处可见,几乎遍布地球上所有支持生命的环境。革兰氏阴性菌包括模式生物大肠杆菌,以及许多致病菌,如铜绿假单胞菌、淋病奈瑟菌、沙眼衣原体和鼠疫耶尔森菌。
问
如何自然去除革兰氏阴性菌?
答
天然抗生素。一些天然抗生素是大蒜、蜂蜜、卷心菜、葡萄柚籽提取物、生苹果醋、特级初榨椰子油、发酵食品等。
免责声明:本公众号内容仅作交流参考,不作为诊断及医疗依据。主要参考文献:
Oliveira J, Reygaert WC. Gram Negative Bacteria. 2022 Mar 26. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan–. PMID: 30855801.
Carroll K.C., & Hobden J.A., & Miller S, & Morse S.A., & Mietzner T.A., & Detrick B, & Mitchell T.G., & McKerrow J.H., & Sakanari J.A.(Eds.), (2019). Jawetz, Melnick, & Adelberg’s Medical Microbiology, 27e. McGraw Hill.
Acheson DWK (2015): Patient information: Food poisoning (foodborne illness) (Beyond the Basics). In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/1/16
Apicella M (2015): Treatment and prevention of meningococcal infection. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/1/16
Baum SG (2016): Mycoplasma pneumoniae infection in adults. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/2/16
Crowe SE (2016): Bacteriology and epidemiology of Helicobacter pylori infection. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/1/16.
Crowe SE (2016b): Treatment regimens for Helicobacter pylori. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/1/16.
File TM (2016): Treatment of community-acquired pneumonia in adults in the outpatient setting. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/1/16
Ghanem KG (2016): Clinical manifestations and diagnosis of Neisseria gonorrhoeae infection in adults and adolescents. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/1/16
Hicks CB, Clement M (2016): Syphilis: Treatment and monitoring. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/2/16
Kanafani ZA, Kanj SS (2014): Acinetobacter infection: Epidemiology, microbiology, pathogenesis, clinical features, and diagnosis. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/10/16
Kanafani ZA, Kanj SS (2016): Acinetobacter infection: Treatment and prevention. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/10/16
Kelly CP, Lamont JT (2015): Clostridium difficile in adults: Treatment. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/2/16
Koulenti D et al (2009): Spectrum of practice in the diagnosis of nosocomial pneumonia in patients requiring mechanical ventilation in European intensive care units. Critical Care Med 37(9):2360-2369. doi: 10.1097/CCM.0b013e3181a037ac
Lamont JT (2016): Clostridium difficile in adults: Epidemiology, microbiology, and pathophysiology. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/2/16
Li X-Z, Nikaido H (2004): Efflux-mediated drug resistance in bacteria. Drugs. 64(2):159–204.
Lowy FD (2016): Methicillin-resistant Staphylococcus aureus (MRSA) in adults: Treatment of skin and soft tissue infections. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/1/16
Pegram PS, Stone SM (2016): Botulism. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/1/16
Riley LW (2015): Natural history, microbiology, and pathogenesis of tuberculosis. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/2/16
Sauvage E et al (2008): The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 32:234–258. DOI:10.1111/j.1574-6976.2008.00105.x
Southwick F (2008): Infectious Diseases. A Clinical Short Course. McGraw Hill/Lange.
Sterling TR (2016): Treatment of pulmonary tuberculosis in HIV-uninfected adults. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/2/16
Stevens DL, Bryant A (2015): Group A streptococcus: Virulence factors and pathogenic mechanisms. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/1/16
Stevens DL (2016): Group A streptococcal (Streptococcus pyogenes) bacteremia in adults. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/1/16
Swygard H et al (2016): Treatment of uncomplicated gonococcal infections. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/1/16
Wanke CA (2015): Pathogenic Escherichia coli. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 7/28/16
Wanke CA (2015b): Travelers’ diarrhea: Clinical manifestations, diagnosis, and treatment. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/1/16
Yeh S (2015): Microbiology, epidemiology and treatment of Haemophilus influenzae. In: UpToDate, Basow, DS (Ed), Waltham, MA. Cited 8/1/16
Sagar Aryal,Gram-Positive Vs Gram-Negative Bacteria- 31 Differences With Examples. Microbenotes.2022,January 9
Jawertz M., Alderbergs., Medical Microbiology 28th Edition.
Prescott M. L., Microbiology. 5th Edition
Lippincott Microbiology in review: 3rd edition
Faith Mokobi,Gram-Positive Bacteria- Cell Wall, Examples, Diseases, Antibiotics,2021, April 15
Faith Mokobi,Gram-Negative Bacteria- Cell Wall, Examples, Diseases, Antibiotics ,2021 ,April 15

谷禾健康

对微生物组的深入研究,有望为困扰我们的健康问题提供新的解决之道。
然而目前有大多数微生物基因组尚未被培养,就算在已发现的基因组序列中,也有很多无法进行功能注释。因此还不能充分捕捉微生物系统发育树的功能多样性,这限制了我们对生物序列高级特征进行建模的能力。而模型的构建又是微生物组研究中重要的一块。
这里我们介绍一个新发布的深度学习模型,为大家的微生物研究提供一些新的思路。

研究人员搭建了LookingGlass程序,应用RNN循环神经网络和LSTM长短期记忆神经网络学习方法,学习序列中的每个核苷酸字符,以达到能预测分类不同功能、同源性和环境起源的reads的目的。
LookingGlass模型还具有迁移学习的能力,经过微调后可以执行一系列不同的任务:例如识别新的氧化还原酶,预测酶的最适温度,以及识别氨基酸序列。
LookingGlass模型能够对其他未知和未注释的序列进行功能相关表征,从而挖掘微生物暗物质。
代表性细菌和古细菌基因组序列的分类由GTDB51(89.0 版)确定。
完整的基因组序列通过 NCBI Genbank ftp下载。这产生了 24,706 个基因组,包括 23,458 个细菌基因组和 1248 个古细菌基因组。
为了确定它们实际的序列长度,使用MetaSeek API下载了它们的测序元数据。去除长度<60bp或>300bp的样本,最终获得了平均序列长度为136bp的共计7909个样本。
LookingGlass模型的训练集、验证集和测试集都是在纲水平上划分的,在该分类水平下三者之间没有重叠的部分。
主要应用RNN循环神经网络和LSTM长短期记忆神经网络。
LookingGlass使用三层LSTM编码器模型,每个隐藏层有1152个单元,根据超参数调整的结果,embedding大小为104。
LookingGlass以自我监督的方式进行训练,根据序列中前面的核苷酸的上下文,预测一个被掩盖的核苷酸。
对于训练集序列中的每个reads,考虑多个训练输入,将被掩盖的核苷酸沿序列长度从第二位置移动到最后位置。因为它是一个字符级模型,线性解码器从可能的词汇“A”、“C”、“G”和“T”中预测序列中的下一个核苷酸,并带有“开始阅读”的特殊标记、“未知核苷酸”(对于不明确序列的情况)、“读取结束”(在LookingGlass训练期间仅对“读取开始”进行标记)和“填充”标记(仅用于分类)。
LSTM 的正则化和优化利用 dropout 和梯度下降方法以获得最佳性能,使用fastai 库进行训练。
硬件方面,LookingGlass在 Microsoft Azure 上,内存为 16GB的 Pascal P100 GPU 进行训练。总共训练了 12 天,共 75 个 epoch,根据超参数优化的结果逐渐降低学习率:15 个 epoch,学习率为 1e-2,15 个 epoch,学习率为2e-3,并以 1e-3 的学习速率进行 45 个 epoch。
通过随机搜索调整超参数,主要调整:
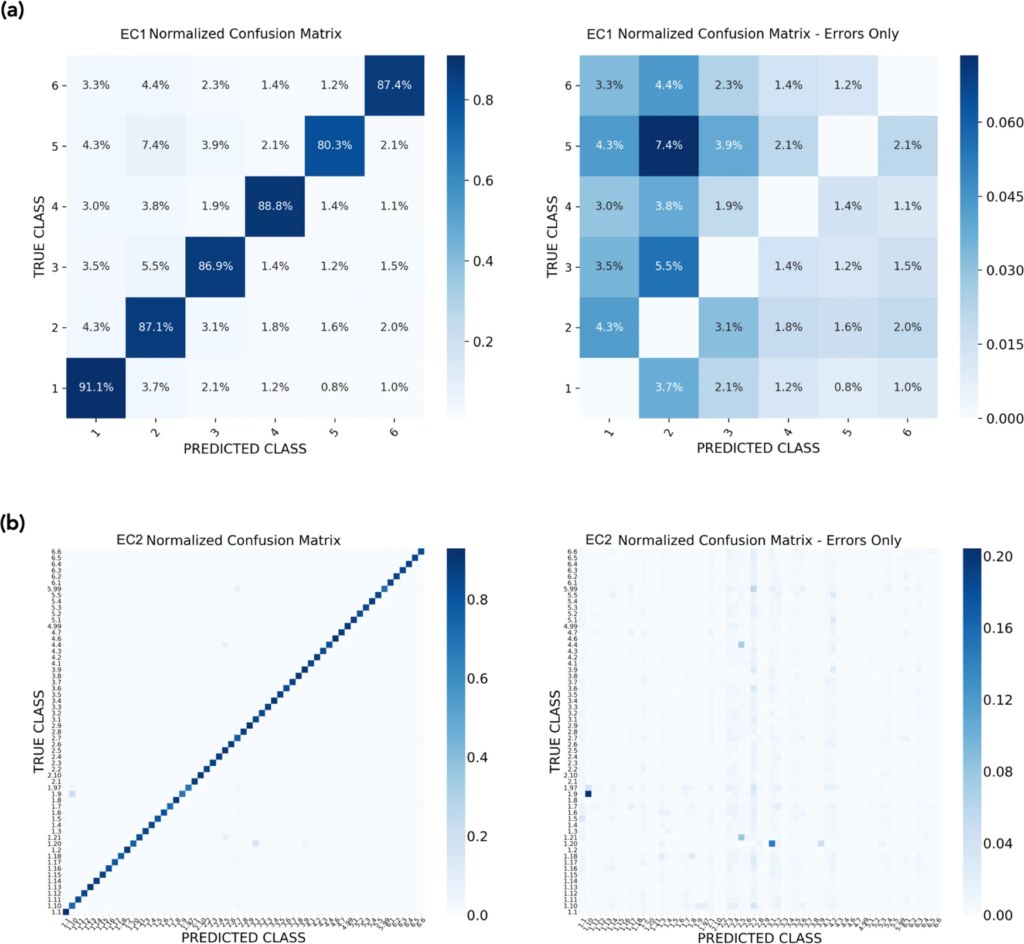
功能注释预测的多分类混淆矩阵。横轴表示真实值,纵轴表示预测值。方块内的数值为归一化后的预测百分比,左边为预测正确的,右边为预测错误的。
图a表示对验证集中EC功能编号的第一个位置的预测,图b表示对第二个位置的预测,显示准确率都在80%以上。

LookingGlass在门水平上识别同源序列对。蓝色为同源(Homologous),红色为非同源(Nonhomologous)。
图a为embedding相似性度的组间比较,同源组显著高于非同源组,embedding相似度计算为embedding向量之间的余弦相似度。
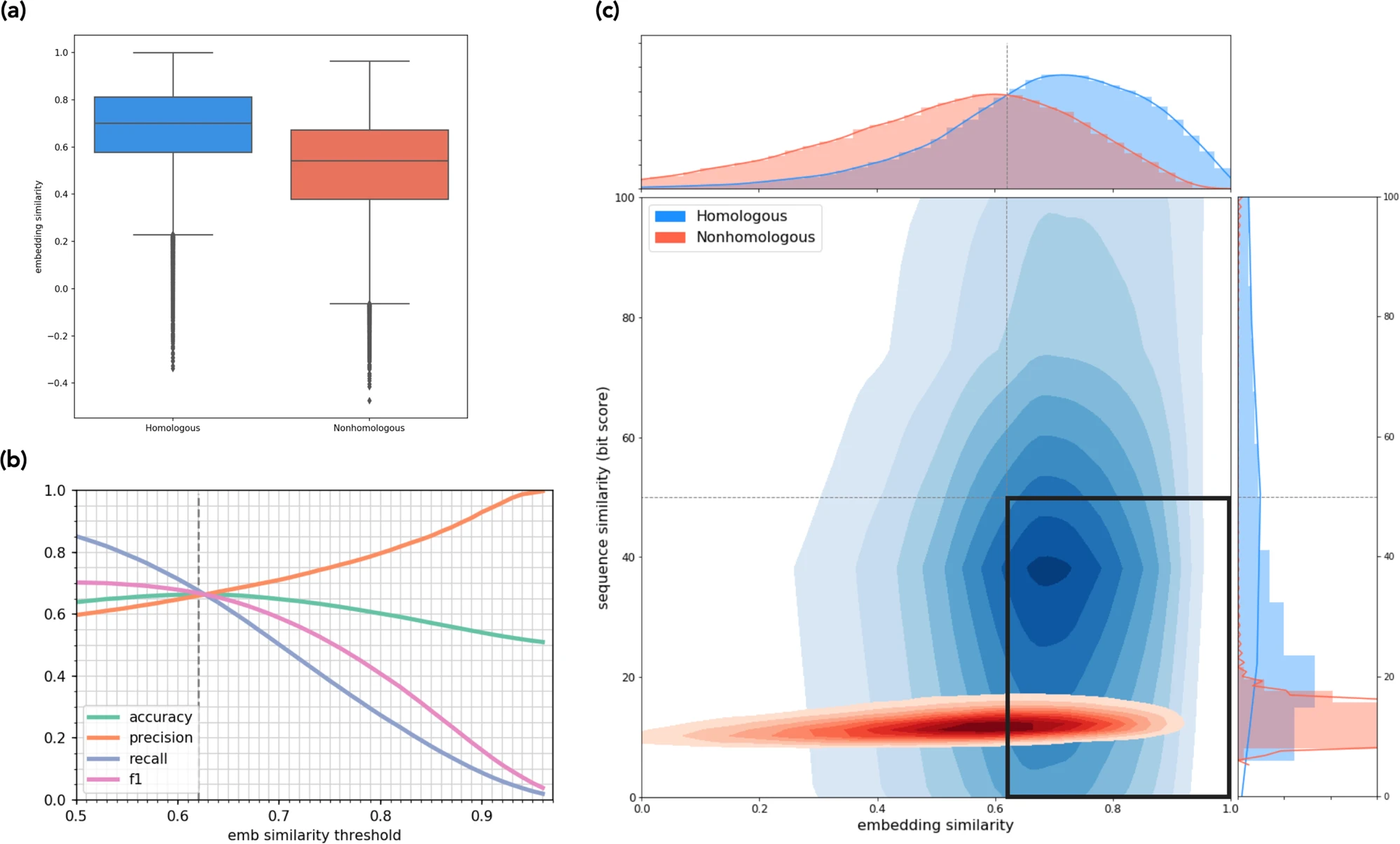
图b为准确度、精确度、召回率和 F1分值的变化,可见在embedding相似性阈值为0.62时其准确度(accuracy)最高,达到了66.4%,这是指门水平的。文中表示在纲水平上达到了68.3%,在目水平上达到了73.2%,在科水平上达到了76.6%,在属水平上达到了78.9%。LookingGlass使用embedding方法区分同源和非同源序列,而不依赖它们的序列相似性(Smith-Waterman比对)。
图c比较了这两种方法在搜索同源物时的结果,图中的黑框表示被LookingGlass 正确识别的同源序列,但使用比对时遗漏了。可见许多同源物具有非常低的序列相似性(bit score<50),不能被基于比对的方法捕获到,但LookingGlass可以。LookingGlass识别同源基因的高精度,与它们的序列相似性无关,表明它捕捉到了高水平的特征,可能反映了序列之间的在系统发育上的关系。

来自100个不同环境样本的宏基因组功能注释集作为验证集。从中对每个环境组别中随机抽取20000个序列计算embedding相似度。发现组间的embedding相似性通常低于组内的,即来自相同环境背景的序列通常聚集在一起。
以LookingGlass的底层架构作为起点,微调预训练模型,以执行不同任务。
1. 预测分类氧化还原酶

对LookingGlass功能注释分类模型进行微调后,执行氧化还原酶的预测分类任务。
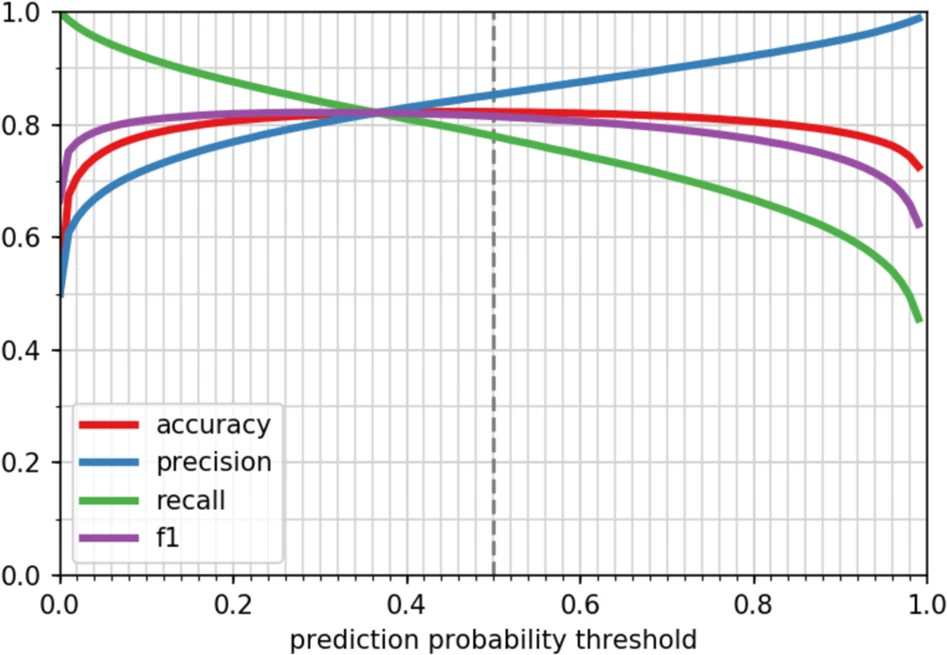
上图为对序列相似性(bit score<50)的序列,预测分类是否为氧化还原酶的编码基因的准确度、精确度、召回率和 F1分值的变化,结果表示默认阈值为0.5时,其准确度(accuracy)最高,为82.3%。
基于LookingGlass模型能够区分不同环境背景下的序列这一优势,研究人员使用来自16个海洋宏基因组作为测试集,样品覆盖范围从纬度(从-62 度到 76 度),海洋深度(从表层~5米到中层~ 200-1000米)以及氧浓度(包括来自氧最低区的4个中上层样品),并从中对每个宏基因组随机抽取 2000 万reads。
挖掘其中的氧化还原酶序列,并证明LookingGlass对氧化还原酶的分类优于传统的基于同源性的方法。

图a为LookingGlass在海洋表层区(surface)、中层区(mesopelagic)以及氧浓度最低区(OMZ)预测分类为氧化还原酶序列的比例。
图b为在海洋表层区组中,纬度与氧化还原酶的相关性,结果表示存在显著相关(R2 = 0.79,P = 0.04)。
图c为分别使用LookingGlass、MG-RAST和 mi-faser工具搜索氧化还原酶序列,并统计其预测为氧化还原酶(oxidoreductases)、非氧化还原酶(not oxidoreductases)和未注释(unannotated)的序列比例。结果为MG-RAST 注释了 26.7-50.3% 的reads,其中 0.01-4.0% 被鉴定为氧化还原酶。Mi-faser 注释了 0.17-2.9% 的reads,其中 0.04-0.59% 被鉴定为氧化还原酶。可见,LookingGlass更具优势。
2. 使用LookingGlass识别氨基酸序列
LookingGlass直接从CDS预测翻译帧起始位置(1、2、3、-1、-2 或 -3),准确率达到了97.8%,但目前仅用于非编码DNA比例较低的原核序列。
3. 从 DNA 序列片段预测酶的最佳温度
酶的最佳温度部分取决于 DNA 序列特征,但难以预测,尤其是短读长。将温度划分为嗜冷(<15°C)、嗜温(20-40°C)或嗜热(>50°C),微调LookingGlass程序后,输入序列预测最佳温度类别,准确率达70.1%。
LookingGlass程序能够不通过比对参考数据库来预测表征DNA序列,从而获得功能注释和系统发育相关信息。同时,LookingGlass的迁移学习框架能够快速学习、训练和收敛以适用不同分类任务,这对于未来复杂生物系统建模提供了一些贡献。
预测分类氧化还原酶可以挖掘位置序列的潜在功能,未来还会扩大可预测的酶类。预测酶的最佳温度可以用于指导蛋白质设计所需的功能和最佳温度。
总而言之,这是一个不错的探索。作者已经将本文所用到的模型功能封装为python库—fastBio,能够直接使用本文使用到的数据集进行模型训练,可在以下地址获得:github.com/ahoarfrost/fastBio/
主要参考文献:Hoarfrost A, Aptekmann A, Farfañuk G, Bromberg Y. Deep learning of a bacterial and archaeal universal language of life enables transfer learning and illuminates microbial dark matter. Nat Commun. 2022 May 11;13(1):2606. doi: 10.1038/s41467-022-30070-8. PMID: 35545619; PMCID: PMC9095714.

谷禾健康

有人在的地方就有江湖,这也同样适用于细菌。
单个细菌的行动往往只是徒劳,然而当它们在一起的时候,集体行动的能力令人刮目相看。
细菌使用化学物质作为它们的“语言”,使用化学通讯来区分自己的物种和其他物种。
实际上它们看不到,并不知道彼此在那里,但它们可以测量化学物质的浓度。
当这些化学物质的水平达到临界水平时,会向细胞内部传递一个信号,该信号会提醒每个细菌细胞及其他在附近的细菌同胞,告诉它们已达到“法定人数”。

然后,整个细菌群作为一个大的、协调的群体,去执行单个细菌无法完成的任务。
以上就是所谓的“ 群体感应 ”。
微生物细胞群体密度的增加使微生物之间的细胞间通讯成为可能,从而产生群体感应信号。
生物发光、毒性因子产生、次级代谢产物产生、DNA摄取能力、生物膜形成等,这些都离不开群体感应。
如果有一两个细菌进入我们体内,它们释放出一些有毒物质或毒素,对于我们几乎没啥影响。
但是如果它们“略施计谋”,等待并计算自己的数量,等到大量细菌一起分泌毒素,攻击人体,那么就可能会压倒人的免疫系统。
它们可以利用群体感应启动致病性。
干扰群体感应有可能阻止有害细菌的致病。当然,促进群体感应,特别是有益细菌,也可以使我们更健康,并可能产生有价值的药品和工业产品。
本文我们详细了解一下细菌世界里的各种故事,群体感应期间会发生什么,群体感应如何影响宿主,群体感应在生物膜的形成中发挥什么作用,与哪些疾病相关,该如何利用群体感应控制疾病从而利于人体健康。
群体感应(简称QS)是细菌细胞与细胞之间的交流过程,涉及到细胞外信号分子自诱导剂(AIs)的产生、检测和响应。AIs使细菌能够感知和响应时间和连续环境,并通过改变基因表达来协调菌落的行为。
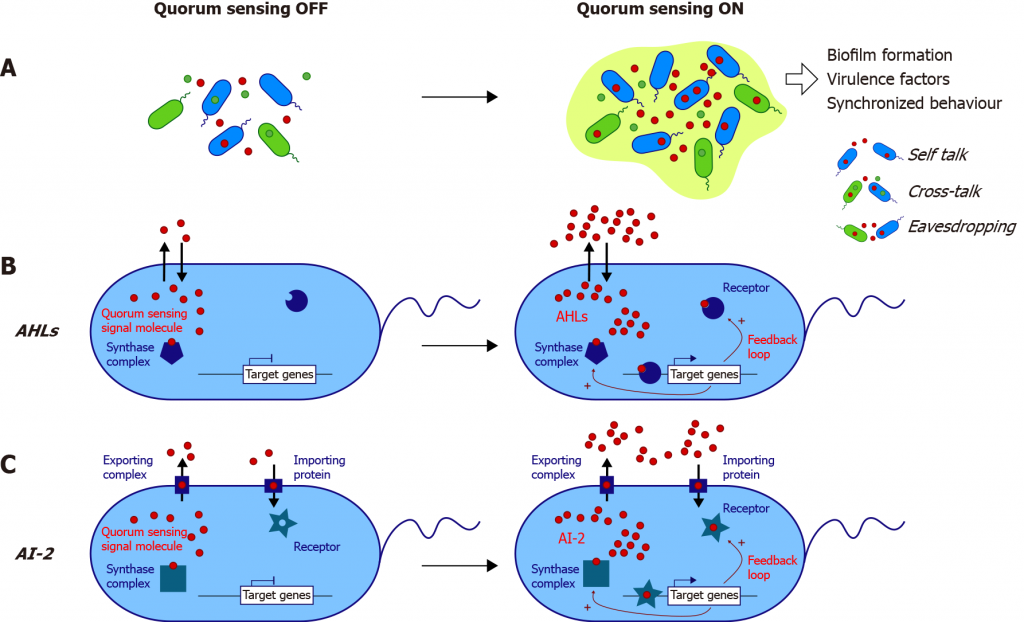

doi: 10.3748/wjg.v27.i42.7247
说起群体感应,就要了解以下三个主要的信号分子。
1. AHL(酰基高丝氨酸内酯Acyl-Homoserine Lactones)
在革兰阴性菌中由AHL介导
AHL是由合成酶复合物产生的,AHL可以通过膜自由扩散。AHL被其细胞内受体识别,复合物与靶基因调控元件结合。
许多革兰氏阴性细菌利用LuxI/LuxR型群体感应系统,产生一系列AHL信号,当与同源LuxR同源物结合时,这些信号可调节控制多种性状的基因的表达。LuxR同系物可与一系列AHL结合。
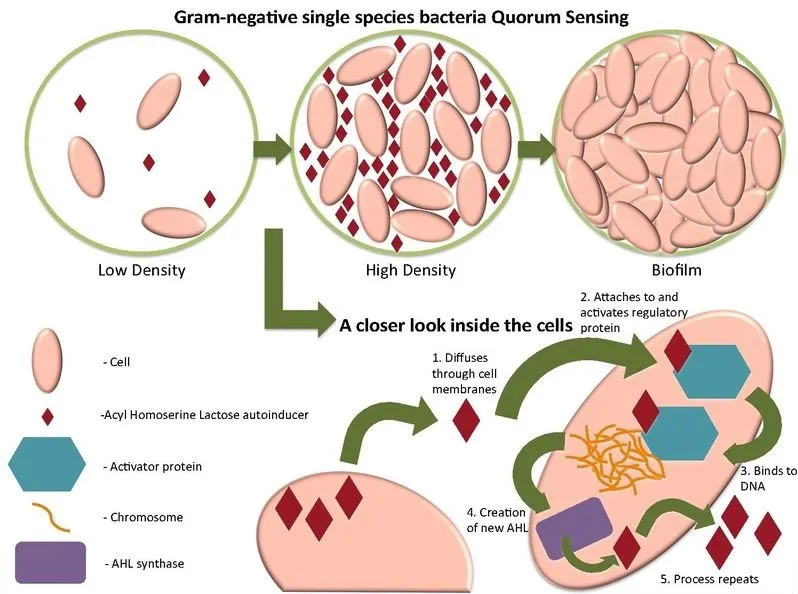
图片来源:Wikimedia Commons
2.自诱导肽 AIP(Autoinducing Peptides)
在革兰阳性菌中的由AIP介导
在革兰氏阳性细菌中,群体感应通常由称为自动诱导肽(AIPs)的小分泌肽控制。AIP由核糖体作为前体肽合成,然后加工并主动运出细菌细胞。
AIP传感涉及与细菌膜中的传感器激酶信号受体结合,然后使控制靶基因转录的细胞质反应调节器磷酸化。
AIP控制革兰氏阳性细菌的一系列细菌功能,包括枯草芽孢杆菌的产孢和能力,以及金黄色葡萄球菌的毒力。
3. AI-2(Autoinducer 2)
在革兰阴性菌和革兰阳性菌中均能由AI-2介导,细菌世界里的“通用语言”
Al-2存在于一些革兰氏阴性和革兰氏阳性细菌中。AI-2需要一种转运蛋白才能进出细胞。现在人们普遍认为,AI-2在革兰氏阳性和革兰氏阴性细菌中都是一个重要的信号,但其信号活性仅限于具有特定AI-2受体的细菌。
AI-2控制多种细菌功能,包括哈维氏弧菌的发光和许多细菌的生物膜形成。
除了以上三种之外,当然也有其他一些信号分子,例如:
研究人员从luxS/AI-2细菌性肠出血性大肠杆菌中纯化了一种假定的自诱导信号AI-3。进一步的研究证实,AI-3的合成独立于LuxS(AI-2合酶)。
AI-3是属于吡嗪酮家族的几种产物。苏氨酸脱氢酶(Tdh)介导的AI-3信号产生和氨基酰基tRNA合成酶相关的自发环化是两个基本反应。
不同细菌种类采用的主要群体感应系统


doi.org/10.3390/microbiolres12040068
肠道是一个特别动态的环境,许多观点表明群体感应是肠道生态系统中的一个新角色。
微生物群具有极其稳定的结构。肠道微生物群的稳定性取决于群体感应。
▸在正常生理条件下,群体感应可能有助于肠道细菌的空间分布
肠道细菌分布和群体感应信号
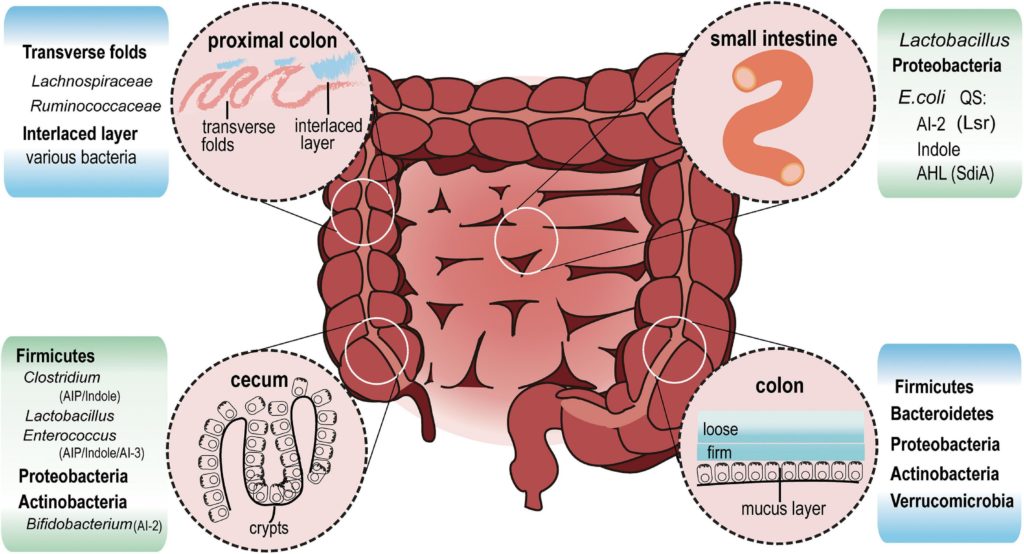
doi.org/10.3389/fmicb.2021.611413
在回肠(小肠)中,乳酸杆菌和变形杆菌是最丰富的细菌。大肠杆菌属于变形杆菌,通过AI-2、吲哚和AHL信号与其他细菌进行通信。
在盲肠隐窝中,可以发现厚壁菌,包括梭状芽孢杆菌、乳酸杆菌、肠球菌、变形杆菌和放线杆菌。该区域可能存在AIP、AHL、吲哚、AI-2和AI-3。
在近端结肠的远端,一条薄而致密的带(交错层)将细菌与上皮分离。近端结肠的交错层主要为毛螺菌科和瘤胃菌科。
从横结肠到直肠,粘液增加并分为两层。厚壁菌、拟杆菌、放线菌、变形菌和疣微菌门生活在松散的黏液层中。
▸肠道菌群如何利用群体感应来生存?
根据已发表的研究,可以推断,盲肠和近端结肠的隐窝和横向皱襞中的聚集物的大小大于肠腔中的聚集物,在肠腔中,粪便不断被肠道运动挤压。生活在这里的厚壁菌可能会产生足够的群体感应信号,通过AI-2影响自己的群落以及邻近物种。
厚壁菌
约83.33%的厚壁菌含有LuxS蛋白直系同源物,这是一种产生AI-2的必要合成酶,而拟杆菌中仅在16.83%发现。这种AI-2生产能力的显著差异可能加强厚壁菌的竞争优势,使其能够支配盲肠和近端结肠。
拟杆菌
在拟杆菌中,B. thetaiotaomicron从膳食中植物多糖或无多糖的黏液聚糖中获取碳源。它们有很高的代谢能力,在粘液和管腔中同样复制自己。然而,由于缺乏LuxS直系同源物(KEGG碱基),它们发现很难与其他物种竞争,如梭状芽孢杆菌和乳酸杆菌等,它们利用AI-2形成生物膜或自我生长。
▸同样是大肠杆菌,生活在粘液比管腔获取更多铁,这离不开群体感应AI-2
粘液驻留细菌和管腔共生细菌种类繁多,通常分布在不同的生态位中,营养素的利用方式不同。
同样是大肠杆菌,它在外粘液中能够快速复制;而在肠腔中,由于有限的糖苷水解酶,大肠杆菌保持在固定相。
粘液是分离细菌和肠上皮的物理保护屏障,并不断更新。内粘液层周转迅速,最终转化为松散的外粘液层。外层通过肠道运动与结肠内容物一起排出。下一轮的食物伴随着潜在的粘液,使细菌重新定居。
AI-2分子调节生物膜的形成,并允许一些细菌(如双歧杆菌和乳酸杆菌)粘附和富集。在粘液周转期间,生活在粘液中的大肠杆菌细胞选择性增殖,而不是从管腔内容物中重新定居。
此外,AI-2信号与细菌的铁代谢调节有关,包括放线杆菌、弧菌和双歧杆菌。
研究表明,相对管腔而言,定植于粘液中的大肠杆菌可以利用更多的铁,这可能是由于在粘液中暴露更多的AI-2信号所致。
群体感应信号可能有助于共栖肠道细菌相互协作,增强抵抗入侵者定殖的能力,并通过调节某些物种的相对丰度来保持动态平衡。当然这是一个需要进一步探索的研究领域。
在肠道这样一个网络化、复杂的生态系统中,宿主不断与数以万亿计菌群相互作用。 细菌的交流必须从一个大的角度来看待。群体感应如何影响宿主?
可以通过两种方式:直接或间接。
细菌和宿主之间通过群体感应分子的对话
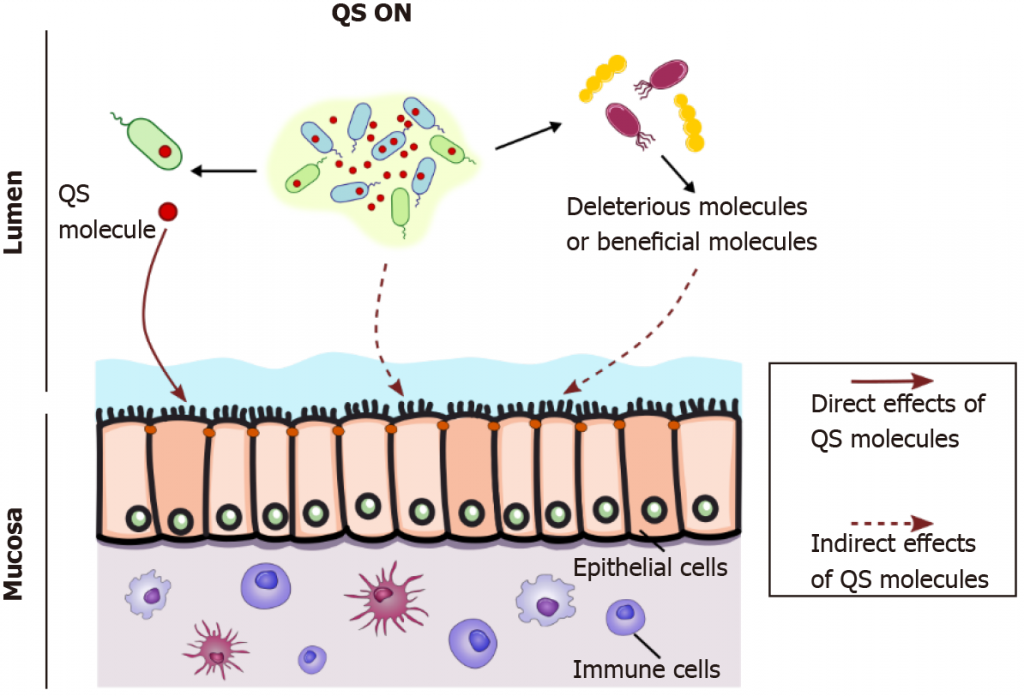

doi: 10.3748/wjg.v27.i42.7247
▸直接的方式:
群体感应分子可以通过与宿主细胞(如上皮细胞或免疫细胞)的直接接触(全箭头,左)影响宿主,如铜绿假单胞菌群体感应分子3-oxo-C12-HSL那样,它可以自由进入哺乳动物细胞。
▸间接的方式:
当达到细菌群落内的阈值浓度时,群体感应自动诱导剂会同步群体行为,如肠出血性大肠杆菌中的毒力和附着消除策略,从而间接影响宿主(上图虚线箭头,中间)。
此外,群体感应分子可以通过对具有不同代谢特性的其他细菌种群的影响,间接改变宿主(右虚线箭头)
细菌代谢改变有益的代谢产物,如短链脂肪酸和胆汁酸。通过调节肠道微生物群组成,群体感应可以通过促进有害或有益细菌间接影响肠道生理学。
例如,AI-2通过促进厚壁菌生长来调节失调菌群。体外和体内的一些研究描述了肠道病原体如何通过群体感应向共生体发出信号,并触发毒素、毒力因子和生物膜的表达。
此外,AI-3控制使肠出血性大肠杆菌通过附着和清除过程引起损伤的基因。
以上是群体感应影响宿主的途径。接下来我们来看,肠道细菌的群体感应给宿主带来的影响具体表现在哪些方面?
目前研究其对宿主的影响,主要包括屏障功能、炎症过程、致癌作用这三方面。
群体感应分子对肠上皮屏障功能不同参数的影响


doi: 10.3748/wjg.v27.i42.7247
铜绿假单胞菌群体感应分子3-oxo-C12-HSL诱导多种细胞类型的凋亡,包括上皮细胞,促进肠道屏障的破坏。此外,3-oxo-C12-HSL破坏紧密连接,从而导致细胞旁通透性增加,并影响粘蛋白的产生。而3-oxo-C12:2-HSL和色氨酸代谢物吲哚保护紧密连接。
与OCTN2结合的枯草芽孢杆菌CSF也通过激活HSP27信号来减少细胞死亡,从而促进肠道屏障的完整性。
群体感应分子对肠道屏障功能和免疫反应的影响
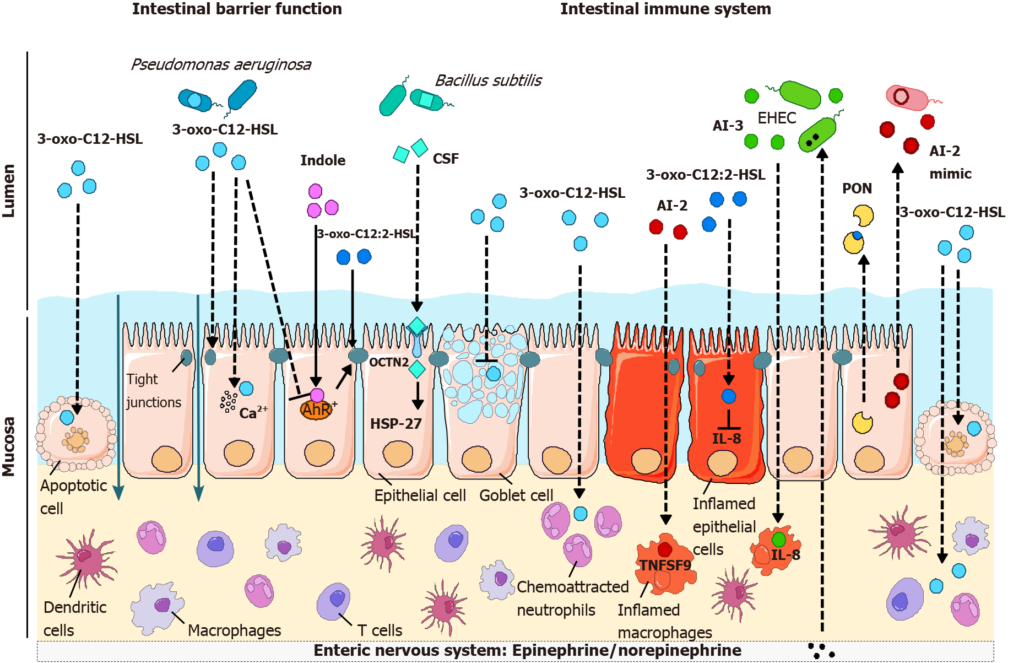
doi: 10.3748/wjg.v27.i42.7247
3-oxo-C12-HSL刺激中性粒细胞的化学吸引和吞噬并诱导细胞死亡,其对免疫细胞的促炎或抗炎作用更为复杂。
除了对肠道屏障的作用外,群体感应分子在肠道免疫室的不同因子也产生作用,该免疫室参与与上皮室的复杂串扰,以维持对肠腔内容物的适当免疫反应。
群体感应分子对不同细胞类型炎症的影响
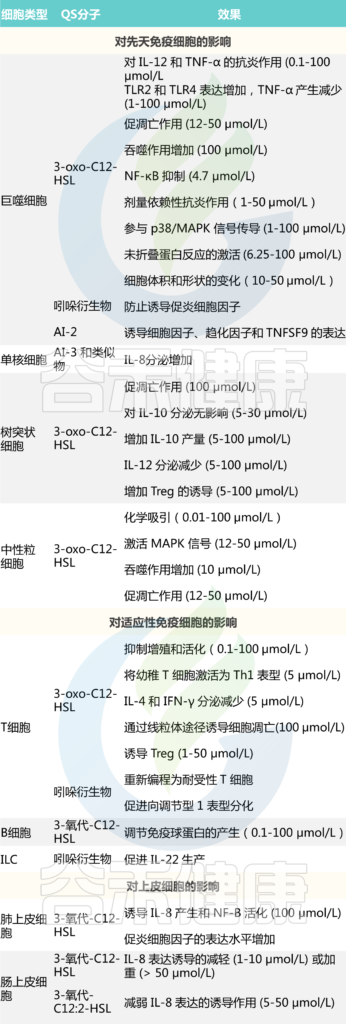
doi: 10.3748/wjg.v27.i42.7247
自体诱导剂AI-2和AI-3通过分别诱导免疫介质TNSF9和白细胞介素(IL-8)的表达,对巨噬细胞产生促炎作用,而3-oxo-C12:2-HSL可减少上皮细胞产生IL-8。
所有这些群体感应分子如何在生理环境中穿过肠道屏障和/或到达体内免疫细胞仍有待澄清。
越来越多的证据表明,肠道微生物群失调在大肠癌的发展中起着重要作用。条件致病菌较多促进肠道炎症,这是大肠癌发病的驱动因素之一。
一些驱动病原体,如脆弱拟杆菌,可以促进强烈的Th17炎症反应。这种促炎微环境可能有利于条件致病菌(如梭杆菌属)的定植。因此,梭杆菌优势生物膜与人类结直肠癌相关。
总之,这些发现支持多微生物相互作用和细胞间通讯可能在大肠癌的发展中发挥重要作用。然而,在大肠癌期间,细菌如何与自身和宿主进行沟通仍不清楚。
最近的研究,为群体感应分子AI-2在大肠癌细胞间通讯中的作用提供了新的见解。
首先,与人类大肠癌周围正常组织相比,肿瘤中的AI-2浓度增加。根据CRC TNM(肿瘤结节和转移)评分,这些水平也与疾病的进展相关。
CD4-TnAi的表达与肿瘤细胞的免疫反应呈负相关。
在分子水平上,已证明AI-2通过TNFSF9信号通路诱导U987衍生巨噬细胞的体外M1极化。
这些发现表明,AI-2可能是与免疫肿瘤微环境相关的一个重要因素,并阐明了群体感应系统在大肠癌发展和进展中的作用。
有趣的是,哺乳动物上皮细胞能够产生模拟AI-2效应的AI-2类似分子,这说明了细菌-宿主串扰的复杂性。因此,更好地了解参与肿瘤发生的群体感应分子,可能是提高我们对大肠癌发生机制的认识的一个机会。
以上是群体感应对宿主的影响,那么对于群体感应的影响,宿主有没有什么回应?下面章节我们继续来看。
宿主细胞除了受到细菌群体感应分子的调控外,还通过反击对群体感应信号作出反应。
肠上皮细胞分泌模拟AI-2的信号类似物
当受细菌衍生的可溶性分子影响时,肠上皮细胞可以分泌模拟AI-2的信号类似物,从而影响肠道细菌。当缺乏AI-2信号产生的LuxS突变株与从结肠组织分离的上皮细胞共培养时,发现通常由AI-2诱导的lsr基因转录增加。这归因于肠上皮细胞生产的AI-2模拟物。
此外,这些信号与上皮紧密连接损伤有关。上述结果表明,宿主衍生的AI-2模拟物可能与肠道细菌粘附和上皮屏障破坏有关。
宿主分泌的分子可以被细菌利用,作为肠道微环境中的群体感应信号
儿茶酚胺等宿主激素可促进细菌生长。Epi/NE可由群体感应系统的QseC受体感知。最近的研究表明,其他肾上腺素能受体包括QseE和CpxA在内,它们与 QseC 在系统发育图谱上存在显着差异,它们也充当受体发挥作用。
1-辛酰基-rac-甘油(OCL),一种在哺乳动物胃肠道中含量丰富的单酰甘油,形成三酰甘油,并作为化学伴侣稳定大肠杆菌中的SdiA,使其具有基础活性。
脂肪酸抑制生物膜,影响群体感应
脂肪酸(FAs)广泛存在于各种生物体中,其化学结构与扩散信号因子(DSF)家族相似。一些革兰氏细菌将DSF用作生物膜形成和毒力的群体感应信号。FAs与DSFs相似,可抑制细菌生物膜或其他群体感应依赖性基因的表达,并影响AHL和AI-2信号转导。
常见的人体病原体,包括伯克霍尔德菌、铜绿假单胞菌、弧菌、幽门螺杆菌和沙门氏菌,都利用DSF。其中一些药物专门针对胃肠系统。在小肠缺乏物理屏障的情况下,化学屏障在分离小肠中的细菌和上皮细胞,从而保护宿主免受病原体感染方面起着关键作用。胆汁中的FAs还可以模拟群体感应信号来调节细菌生物膜的形成。
以上是细菌通过群体感应和宿主之间的互相交流,具体到疾病中,这些交流是怎么运作的?
铜绿假单胞菌是一种作用于人体组织的条件致病菌。它通过三个主要的群体感应系统起作用,包括两个AHL依赖的LuxI/LuxR型系统和一个假单胞菌喹诺酮类信号(PQS)系统。
铜绿假单胞菌利用AHL信号家族中的N-3-氧代-十二烷基-高丝氨酸内酯(3O-C12-HSL)和N-丁基-L-高丝氨酸内酯(C4-HSL)控制300多个基因,其中许多基因参与毒力调节。PQS系统与生物膜的形成有关。
当铜绿假单胞菌感染人体时,上述群体感应信号与人体细胞相互作用,导致包括中性粒细胞、巨噬细胞以及上皮细胞在内的免疫细胞发生生理和功能变化。
与缺乏AHL产生的突变铜绿假单胞菌菌株相比,含有3O-C12-HSL和C4-HSL的野生型菌株促进巨噬细胞吞噬。3O-C12-HSL导致细胞体积增加,这与水通道蛋白9(AQP9)的上调有关,AQP9是炎症性肠病(IBD)的一种慢性炎症标记物。
研究分析了健康受试者和出现炎症发作或病情缓解不足的IBD患者的粪便。使用液相色谱法和质谱法检测这些样品中的AHL。
在AHL中,3-oxo-C12:2在健康组中显著富集,与伴有FLARE的IBD患者相比,IBD缓解组中含量更高。与3-oxo-C12(3O-C12-HSL)不同,3-oxo-C12:2可以减少IL-1β刺激的肠上皮细胞中IL-8的分泌,但对上皮细胞旁通透性没有影响(图2)。
此外,3-oxo-C12:2水平与粪杆菌、类球梭状芽孢杆菌和细梭状芽孢杆菌的较高计数呈正相关。这些细菌种类很罕见,属于厚壁菌,尤其是普拉梭菌F.prausnitzii。事实上,一项早期研究报告称,口服活的 F.prausnitzii 或其上清液可降低2,4,6-三硝基苯磺酸(TNBS)结肠炎的严重程度。F.prausnitzii的抗炎作用部分是由于其分泌的代谢物阻止IL-8的产生。
与CDI阴性腹泻患者相比,艰难梭菌感染(CDI)患者的粪便中含有高水平的吲哚和艰难梭菌毒素诱导的群体感应信号肽。这表明艰难梭菌利用群体感应信号调节其在胃肠道的感染,并且该群体感应信号与吲哚的产生有关。
然而,艰难梭菌缺乏色氨酸酶基因,这有助于吲哚的生成。随后的研究结果表明,艰难梭菌可能利用毒素诱导的群体感应信号来调节吲哚产生菌的相对丰度,并为其生存创造有利的环境。
同时,共生肠道细菌的吲哚耐受性低于艰难梭菌,共生肠道细菌的恢复将受到抑制,从而为艰难梭菌的定植和扩张提供更有利的环境。
理论上,AHL仍然是使用群体感应中的天然分子调节微生物群组成和肠道炎症的良好候选方法。AHL信号可能涉及有助于控制肠道炎症的不同途径,例如抑制NF-κB、调节、抑制MAPK激活、增加调节性T细胞诱导、减少促炎细胞因子和调节上皮屏障中的连接复合物。
事实上,使用群体感应分子可以在代谢和炎症疾病中的肠道生态系统紊乱的两个组成部分(肠道微生物群和宿主反应)中发挥作用。基于AHL的群体感应已经作为治疗应用存在,用于动态控制革兰氏阴性菌群,尤其是在传染病中。其他群体感应分子可以扩展为潜在的临床治疗方法,用于治疗与肠道微生物群相关的疾病,尤其是涉及生物膜形成和抗生素耐药性相关。
当然以上只是从群体感应的角度来了解其与疾病之间的关联,我们知道,疾病的发生发展不止这些因素,还包括其他的,比如说致病菌生物膜的存在,会使某些致病菌难以清除,与疾病的发生有关。
为什么生物膜的存在会使某些致病菌难以清除?我们前面阐述的群体感应与生物膜是不是类似?它们之间又有什么样的联系?
下一章节我们继续了解。
在了解群体感应与生物膜的关联之前,我们先了解一下生物膜是什么?
生物膜是粘附在表面上的细胞外基质,由核酸、蛋白质、多糖和脂质的复合物组成。
约 80% 导致慢性感染的细菌都能产生生物膜,它是一种重要的毒力机制,可诱导对抗菌剂的耐药性和逃避宿主免疫系统。
许多细菌物种,包括病原菌都能产生生物膜,如金黄色葡萄球菌、铜绿假单胞菌等,它们通过产生生物膜,变得对细胞外应激条件更具抵抗力,更能生存下去,所以生物膜是微生物的一种有用的适应。
它们更好的生存能力对人类来说可不是好事,一般来说,生物膜内的微生物比以单细胞存在时更难根除。这主要是由于生物膜相关的细胞外网络、代谢休眠和其他潜在机制介导的耐受性。
参与生物膜形成的细菌种类及其生物效应的例子

doi: 10.15190/d.2019.13
形成生物膜的细菌嵌入在由细胞外聚合物(EPS)构成的自生黏液基质中。这种生长模式可以改变细菌的生物学和生理特性,如繁殖、生长、基因转录率和对抗生素的耐药性。
生物膜的形成需要五个成熟阶段:
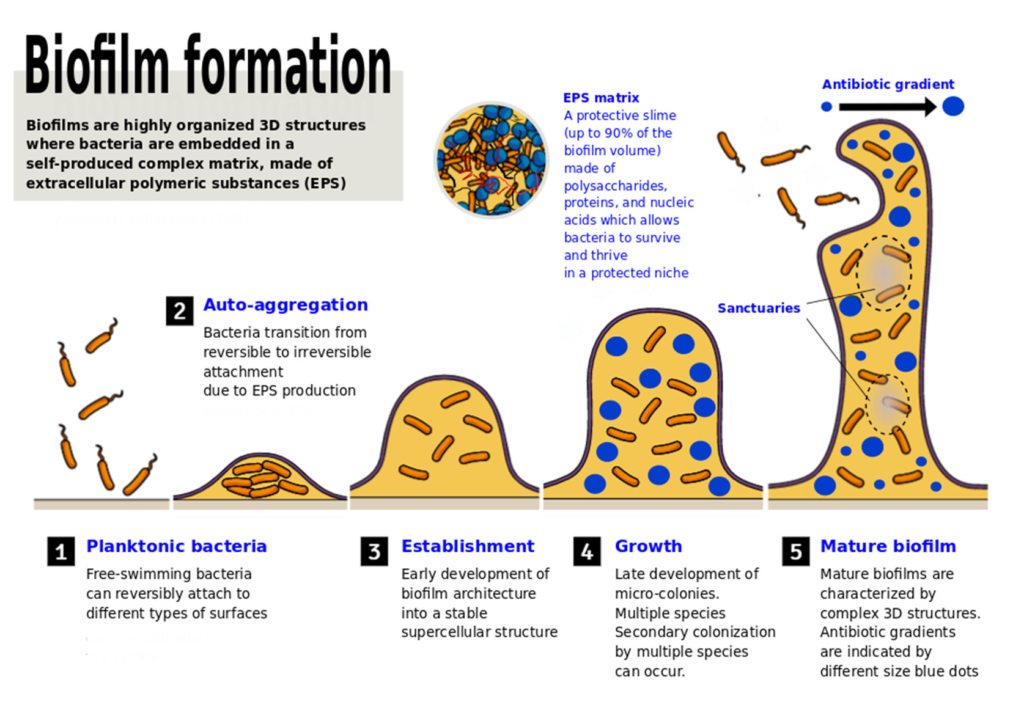
doi.org/10.3390/microbiolres12040068
小蓝点和大蓝点代表抗生素浓度不同的区域(表示存在梯度),灰色区域是细菌可以在低浓度抗生素下存活的“避难所”,有利于产生耐药性。
(i) 浮游细菌在表面的初始附着(可逆)
(ii)产生和分泌EPS和/或其他对接方式,以及驱动从可逆到不可逆的过渡附着的特定粘附素(例如,鞭毛、自转运蛋白、菌毛、卷曲纤维和F型结合菌毛)
(iii)作为超级细胞结构的生物膜结构的早熟
(iv)微菌落的晚熟和进化为成熟生物膜
(v) 细胞从生物膜上分离并分散到周围环境中
所有这些过程都受到不同细胞间信号分子的严格调控,这些信号分子负责种群密度依赖的基因表达,这些基因表达会深刻影响生物膜的形成过程。
生物膜的成熟与细胞外聚合物物质的积累并行。最后一步涉及到细菌菌株从微菌落分离,可能导致在不同位置形成一个新的生物膜菌落。
以上我们可以看到,群体感应和生物膜形成是细菌的两种群体行为。
由于生物膜聚集体中的细胞非常相似,并通过自生的细胞外基质相互连接,因此生物膜代表了与群体感应相关的生态环境。
一些研究表明,群体感应信号分子在革兰氏阳性和革兰氏阴性细菌的生物膜形成中起着重要作用。
S. oralis 34 产生的AI-2信号分子已被证明是口腔链球菌34 和Antinomyces naeslundiiT14V 形成生物膜所必需的。AI-2介导的群体感应也参与大肠杆菌生物膜的形成、运动基因的调节、鞭毛的合成和趋化性。
乳糖可诱导蜡样芽孢杆菌生物膜,观察到AI-2产生的乳糖以剂量依赖性方式增加。而其他研究表明,添加外源AI-2可以削弱金黄色葡萄球菌生物膜的形成。
低浓度的AI-2可以促进猪链球菌生物膜的形成,但高浓度的AI-2显示出抑制作用。
细菌生物膜的形成是一个动态的分层过程。粘附在宿主表面是细菌生物膜形成的第一个也是关键的阶段。细菌通常有两种粘附表面的方式:
(i)通过外膜粘附蛋白与宿主表面结合;
(ii)胞外多糖(EPS)对宿主表面的粘附,如多糖细胞间粘附(PIA)。
以目前研究较多的铜绿假单胞菌为例,我们具体来看群体感应对生物膜形成的影响。
铜绿假单胞菌的群体感应非常复杂,可以由转录调节器(MvaT和RsaL)、转录后调节器(RsmA)、σ因子因子(RpoN和RPO)甚至其他群体感应系统(PQS系统)进行调节。
在LasI/LasR-RhlI/RhlR系统调节的功能中,有几种与生物膜形成相关的毒力因子,如鼠李糖脂、凝集素和铁载体。鼠李糖脂被证明是维持生物膜聚集所必需的,它影响一种称为“聚集”的表面运动,这种运动与生物膜的形成有关。
LecA和LecB是两种依赖群体感应的碳水化合物结合凝集素,已被证明会影响生物膜的形成,突变菌株无法形成成熟的生物膜。
群体感应控制的铁载体,如吡啶,不能产生这种影响生物膜聚集体形成的铁螯合剂。
与野生型铜绿假单胞菌形成的生物膜聚集性大的特点不同,lasI突变株产生的生物膜结构均匀,结构平坦,多层细胞密集。
在生物膜形成过程中,PQS群体感应系统负责增加细胞外DNA(eDNA)的产生,eDNA可与基质中带正电的EPS相互作用,产生生物膜。在PQS缺陷突变体中,生物膜聚集体不能充分发育。
因此,铜绿假单胞菌的三个群体感应调节系统可以影响生物膜的形成,并具有一定的促进作用。
微生物感染与群体感应介导的生物膜密切相关,群体感应信号分子参与各种细菌生物膜的形成、成熟和功能调节。那么,治疗微生物生物膜相关的感染可以从群体感应入手,群体感应抑制可作为对抗生物膜感染的有效工具。
通常,群体感应可通过以下三种方式受到抑制:
(1) 延迟或阻断群体感应信号分子的产生(细菌利用QSI阻断AIs);
(2) 使用QSMs同系物阻断受体。例如,从革兰氏阴性菌Rheinhemiera aquimaris QSI02中获得的活性二酮哌嗪环(Trp-Ser),不仅可以降低由QS调节的紫罗兰素的生物合成能力(67%),还可以降低弹性蛋白酶活性、铜绿假单胞菌生物合成和PAO1的生物膜形成能力。可能是环(Trp-Ser)比通常的革兰氏阴性信号分子(AHLs)更容易与lasR受体结合;
(3) 在氧化压力下,群体感应信号分子的酶降解,例如通过酰化酶和乳糖酶降解类似于AHL的群体感应信号更有效。这些策略已被证明是降低细菌致病性和生物膜的有效方法,有可能提高细菌对抗生素和噬菌体等抗菌剂的敏感性。
群体感应抑制剂影响生物膜的形成

doi.org/10.1016/j.foodres.2020.109742
以上是群体感应通过影响生物膜生成来阻止细菌感染,其他只要能干扰群体感应的物质,也可以作为微生物感染的治疗方式。
通过靶向细菌群体感应系统,可以开发新的治疗策略。
与健康人相比,大肠腺瘤和结直肠癌(CRC)患者的结肠粘膜和粪便中观察到更高的AI-2水平。重要的是,发现AI-2浓度随着大肠癌的进展而增加,这表明其有可能成为大肠癌临床筛查的新标志物。
另一项关于烧伤部位感染的最新研究集中于铜绿假单胞菌群体感应系统。烧伤常伴有肠道菌群失调、肠道完整性受损、免疫失调和细菌肠外移位。铜绿假单胞菌是烧伤后感染的主要病原体。其毒性产物可延长肠道功能障碍,加重全身感染。
研究人员观察到,在使用MvfR拮抗剂抑制MvfR转录因子后(一种控制该菌株毒力的重要群体感应相关转录调节因子),肠道功能障碍得到改善,铜绿假单胞菌传播减少。
事实上,抗菌治疗策略已经开始从使用抗生素扩展到开发基于群体感应系统的抑制剂,如抗毒性或抗生物膜。这些抑制剂或类似物内源性存在于细菌或真核细胞中。
一个例子是鼠尾草酸,它是金黄色葡萄球菌群体感应系统的一种特殊抑制剂。它存在于迷迭香叶中,可以在低浓度下抑制Agr表达和金黄色葡萄球菌毒力。
综上,这些研究指出了难治的传染病药物开发的新潜力。下面列举一些小分子物质,通过干扰细菌群体感应系统,从而达到抑制致病菌的效果。
具有酚类结构的黄酮类化合物有槲皮素、山柰酚、黄芩素等。黄酮类单体对细菌群体感应和生物膜均显示出抑制活性。
研究表明,这是由于黄酮类骨架中存在的两个羟基基团,阻止LasR/RhlR受体与信号分子的结合,从而抑制群体感应系统。
▸槲皮素
槲皮素是一种天然存在的黄酮醇,普遍存在于蔬菜和水果中。64μg/mL槲皮素显著抑制铜绿假单胞菌生物膜的形成,并且当槲皮素的浓度为16μg/mL时与阿奇霉素浓度为32 μg/mL具有相同效应的生物被膜抑制作用。
PCR显示lasI、 lasR、rhlI 和 rhlR等群体感应相关基因与相关毒力因子基因的mRNA表达水平均显著降低。
利用计算机分子对接技术,观察到槲皮素与LasR受体蛋白通过竞争性结合方式与LasR受体蛋白进行结合,且槲皮素比信号化合物结合更为牢固。
▸山柰酚
研究显示,64 μg/mL山奈酚对生物膜形成的抑制率为 80%,粘附降低约75%,说明山柰酚可影响生物被膜形成的粘附阶段。
柚皮素、山柰酚和槲皮素均显著抑制信号分子诱导的生物发光,槲皮素和柚皮素对哈维氏弧菌生物膜的形成有抑制作用。
▸黄芩素
研究了40种中药及天然来源的黄酮化合物,对耐药鲍曼不动杆菌菌株的体外抑菌作用和生物膜形成能力的影响,结果显示黄芩素和汉黄芩苷具有良好的抑菌效果。
其最低抑菌浓度(MIC)分别为0.0625和0.125 mg/mL, 黄芩素对鲍曼不动杆菌生物膜的形成具有明显抑制作用。
32、64 μg/mL黄芩素下调群体感应系统调节因子agrA、RNAIII和sarA等基因的mRNA表达水平,明显抑制生物膜的形成。
有研究报道,一些苯甲酸衍生物如没食子酸、香草酸、肉桂酸等可通过群体感应系统调节细菌的致病性和毒力。
▸肉桂酸
亚MIC的肉桂酸(CA)显著影响细菌的群集运动和生物被膜完整性, 减少EPS的产生。CA对LasR和RhlR受体均有拮抗作用,从而影响细菌群体感应活性。
▸没食子酸
Luis等研究结果显示,4 μg/mL的没食子酸可使金黄色葡萄球菌菌株的生物膜形成减少约40%。Srivastava等研究结果表明,没食子酸、香草酸等 中药单体的合成衍生物可抑制群体感应系统和生物膜的形成。
萜类化合物是甲戊二羟酸衍生的一类化合物。从植物中提取的萜类化合物有香芹酚、丁香酚、芳樟醇,甘草次酸、熊果酸和白桦脂酸等。
▸香芹酚
百里香精油中含有较高的单萜类化合物香芹酚和麝香草酚,可抑制紫色杆菌的紫色菌素产生。亚MIC的香芹酚和麝香草酚可以显著减少紫色杆菌CV026中信号分子的产生。
L-香芹酮是一种单萜,是传统香料植物的主要成分。亚MIC的L-香芹酮可显著减少蜂房嗜血杆菌在聚丙烯和锌表面形成生物膜。
▸丁香酚
丁香酚可以显著降低紫色杆菌CV026中的紫色菌素生成;亚MIC的丁香酚可抑制铜绿假单胞菌弹性蛋白酶、绿脓菌素的产生和生物膜的形成。
将丁香酚转化为纳米乳状态后对群体感应的抑制显著增强,亚MIC的丁香酚纳米乳剂对紫色杆菌中的紫色菌素生成可抑制约50%。
Joshi等通过分子对接技术将丁香酚与高丝氨酸内酯合成酶(ExpR)和调节蛋白(ExpI)进行对接,观察到丁香酚与受体蛋白的对接效果优于呋喃酮C-30等已知的抑制剂,因此推断丁香酚的作用机制可能是通过与ExpI/ExpR蛋白的结合,抑制信号分子的产生。
▸芳樟醇
芳樟醇具有显著的抗菌活性,MIC值在2 μg/mL~8 μg/mL之间;亚MIC的芳樟醇对紫色杆菌的群体感应系统有抑制作用。此外,芳樟醇可以减少鲍曼不动杆菌生物膜的形成和降低其粘附性。
100 μg/mL香豆素及其衍生物能够显著抑制紫色杆菌CV026的紫色菌素产生和铜绿假单胞菌生物膜的形成;在7种香豆素衍生化合物中,含6,7二羟基和7-羟基的化合物对生物膜形成的抑制活性更强。
▸小檗碱
小檗碱亦称黄连素,是中药黄连抗菌作用的主要有效成分。1/2MIC和1/4MIC小檗碱显著下调大肠杆菌群体感应系统相关基因luxS、pfS、hflX、ftsQ和ftsE,对生物膜的形成表现出明显的抑制作用。扫描电镜结果显示,菌株经处理后,粘附性下降,菌体形态也发生改变。
▸苦参碱
苦参碱明显抑制大肠杆菌群体感应系统;2.56 mg/mL苦参碱能显著降低大肠杆菌的LuxS 、 sdiA等群体感应相关基因和生物被膜相关基因的mRNA表达水平;在对大肠杆菌外膜蛋白ompA基因和群体感应基因表达的双重抑制作用下,使细菌不易产生聚集和粘附。
除上述提及的中药单体外,硫类化合物、苯丙素类化合物和单宁类等中药单体均具有群体感应抑制作用。
作用于细菌群体感应系统的中药单体的抗细菌感染,与传 统的抗菌中药相比具有剂量明确、副作用少等特点。
但是,中药单体抗细菌感染的研究主要局限于体外实验,研究方式过于单一,具体作用机制的研究还不够深入,研究多集中在观察与验证上;但细菌感染机制是复杂的,中药单体的作用机制也是多样性的,因此需要更进一步研究。
此外,大部分中药单体通常在较高的浓度发挥抗菌或抗毒力作用,这也增加了产生毒性的风险并且在体内也难以达到有效的浓度,对其进行适当的结构修饰合成中药单体衍生物则表现出对群体感应活性抑制更高,同时降低了药物浓度,这为后续抗菌药物研发提供新的思路。
由于目前临床常规使用的抗菌药物不足以对抗耐药性细菌感染,因此抗菌药物与中药单体联合用药是提高耐药细菌感染疗效的新策略。
小檗碱与亚胺培南和美罗培南联用时89%表现为协同抗菌作用;黄芩苷联合替加环素则100%协同抗菌作。
姜黄素降低头孢他啶和环丙沙星的MIC;姜黄素和头孢他啶组合具有协同抗菌效应,姜黄素和环丙沙星组合具有相加效应。
头孢他啶单独和与姜黄素联合使用时,群体感应系统的lasR基因的mRNA表达水平显著降低。
文献报道,阿奇霉素具有群体感应拮抗活性,2 μg/mL阿奇霉素可能通过影响自身诱导分子的合成,抑制铜绿假单胞菌PAO1的群体感应并能减少毒力因子的产生。1/4和1/16MIC的阿奇霉素、庆大霉素和姜黄素单独和联合用药均可显著降低生物膜形成能力。
阿奇霉素单独和与姜黄素联合作用时,lasR基因的表达水平显著下降。阿奇霉素单独给药和与姜黄素联合给药使群体感应系统的rhlI基因的mRNA表达分别降低60%和67%.
头孢吡肟、头孢他啶和亚胺培南等抗菌药物具有群体感应抑制活性。
亚MIC的阿奇霉素、美罗培南、头孢吡肟和哌拉西林/他唑巴坦对群体感应依赖性毒力因子的影响,结果所有菌株的群体感应依赖性毒力因子(如生物被膜、绿脓
菌素、蛋白酶、溶血素和DNase生成)均明显降低。
亚MIC多西环素显著降低紫色杆菌的紫色菌素产量(70%),4 μg/mL多西环素显著抑制生物被膜的生成
(72.8%),并显著降低铜绿假单胞菌的弹性蛋白酶(67.2%)、绿脓菌素(69.1%)产量以及群集运动(74%) 。
亚MIC红霉素对紫色杆菌群体感应系统的抑制率为84%,红霉素抑制群体感应的机制可能是与AbaI自身诱导剂合成酶结合而阻止信号分子合成。
许多中药单体通过作用于细菌群体感应系统而发挥抗感染作用;当与抗菌药物联合应用于细菌感染,不仅可产生协同抗菌和协同抗群体感应作用,还能提高抗感染的疗效、降低单一药物的剂量、减少不良反应,逆转细菌的耐药性。
将群体感应抑制剂中药单体与抗菌药物联合应用,成为恢复抗菌药物对细菌敏感性的一种新策略。中药单体种类繁多,抗感染作用机制复杂多样,还需要对其更深入研究,为临床多重耐药菌引起的感染提供新的治疗药物。
除中药外,益生菌也能作为群体感应的干扰剂,通过群体感应来预防病原体定植和黏膜感染。
另一方面,对于益生菌,我们更多的是希望延长它们在肠道中的停留时间并最大限度地发挥益生菌的作用。
生物膜的形成依赖于粘附、自聚集和共聚集作为细菌的重要特征。益生菌最理想的特性是其良好的粘附性,可延长它们在肠道中的停留时间,从而有效增强屏障功能,维持肠上皮稳态,增加黏液分泌、改善肠蠕动来保护或治疗肠道疾病。
益生菌对肠道菌群和宿主生理具有恢复或保护作用,例如,在存在生态失调或微生物群受到干扰的情况下,缓解胃肠道症状。
IBD患者菌落生物膜的平均密度比健康人高100倍。核梭杆菌以侵袭性生物膜的形式引起肠道疾病。同时,在感染CRC和IBD的邻近健康组织中也出现了成熟的生物膜。
生物膜可能是肠道疾病的早期预警信号。在某种程度上,生物膜提供了一个保护性环境,促进宿主防御机制的逃避,并进一步加剧疾病。虽然抗生素可以去除大多数有害细菌的生物膜,但生物膜在慢性伤口愈合过程中可以快速再生,表明生物膜中存在持久性细胞。
厚的多微生物致病性粘膜生物膜的生长标志着健康微生物群和疾病微生物群之间的过渡。
大肠杆菌Nissle 1917具有良好的生物膜形成能力,其生物膜形成能力强于肠致病性大肠杆菌(EPEC)和肠毒性大肠杆菌(ETEC),并在生物膜形成过程中与这些菌株竞争。因此,大肠杆菌Nissle 1917可作为一种益生菌用于治疗各种肠道疾病。
双歧杆菌
双歧杆菌是人类健康中最重要的益生菌之一,具有LuxS/AI-2 群体感应系统,产生包括AI-2在内的群体感应信号分子,并促进生物膜的形成。
添加碳水化合物后,双歧杆菌中AI-2的生成量正增加至89.45%。对感染产志贺毒素大肠杆菌(STEC)O157:H7的小鼠施用短双歧杆菌,通过产生高浓度的乙酸(56 mM)抑制STEC的Stx毒素表达,显示出强大的抗感染活性。
乳酸杆菌
植物乳杆菌中也存在LuxS/AI-2 群体感应系统和细菌素的产生,植物乳杆菌是一种控制一些重要区域(如肠道和阴道)微生态平衡的益生菌,在保持食品质量方面也有实际应用。
一些病原体对植物乳杆菌群体淬灭系统敏感(例如,铜绿假单胞菌PAO1/ATCC 27853,耐甲氧西林金黄色葡萄球菌ATCC 43300),该系统对金黄色葡萄球菌的生物膜形成和铜绿假单胞菌的绿脓菌素生成表现出最大的活性。
用铜绿假单胞菌感染小鼠烧伤皮肤模型,并用植物乳杆菌上清液处理。结果显示,感染后5、10和15天,铜绿假单胞菌在皮肤、肝脏和脾脏中的定植受到抑制,这表明局部益生菌给药已经阻止了病原体的血液传播。
体内研究表明,不同益生菌(例如干酪乳杆菌酪蛋白亚种ATCC 393、乳乳杆菌罗伊氏亚种ATCC 23272、植物乳杆菌植物亚种ATCC 14917和唾液乳杆菌ATCC 11741)对口腔病原体变形链球菌具有抗链球菌活性。
研究人员开发了一种牛奶模型,以研究LAB的抗李斯特菌活性(抗单核细胞增生李斯特菌),使用具有AI-2分子的沙克乳杆菌Lactobacillussakei和植物乳杆菌。
群体感应系统可能在生物膜的组织、形成和成熟阶段发挥关键作用;因此,它可以被视为开发新型抗菌剂的一个有吸引力的目标。
乳酸菌菌株——QS拮抗剂

doi.org/10.3390/microorganisms10020350
细菌群体感应和生物膜领域的研究迅速扩大,群体感应在细菌行为中发挥着关键作用,通过细菌-宿主串扰影响感染状态和疾病发展。目前很多临床难以治疗的感染或疾病与细菌的群体感应和生物膜形成有关。大多数病原菌病理反应受群体感应系统的调控。生物膜细菌通过屏障作用、群体感应系统、抗免疫清除机制、特殊的生长特性及独特的微环境、生物膜耐药基因开启等机制形成耐药,造成临床耐药菌株增多,给临床治疗带来严重困难。
上述我们可以看到,利用各种群体感应抑制剂或干扰病原菌群体感应的药物进行治疗,是一种合理且有前景的策略。例如通过群体感应靶向剂调节细菌 群体感应信号传导是控制细菌毒力因子产生和生物膜形成的有效策略。这种新型的非抗生素疗法可以抑制致病基因的表达,预防感染,降低细菌细胞耐药性的风险,近年来得到了广泛的应用。
还可以考虑群体感应分子作为菌群失调相关慢性疾病(如IBD或CRC)的可靠生物标志物。已证明,肠道生态系统中某些AI-1 群体感应分子的存在与细菌群大小直接相关。AHL可以代表细菌水平群体的生物标记物,作为菌群失调的放大镜。
此外,在腺瘤向结肠直肠转移和大肠癌进展期间,AI-2浓度增加。这为使用群体感应系统作为慢性病预防和随访的生物标志物开辟了前景。
主要参考文献:
Coquant G, Aguanno D, Pham S, Grellier N, Thenet S, Carrière V, Grill JP, Seksik P. Gossip in the gut: Quorum sensing, a new player in the host-microbiota interactions. World J Gastroenterol. 2021 Nov 14;27(42):7247-7270. doi: 10.3748/wjg.v27.i42.7247. PMID: 34876787; PMCID: PMC8611211.
Deng Z, Luo XM, Liu J, Wang H. Quorum Sensing, Biofilm, and Intestinal Mucosal Barrier: Involvement the Role of Probiotic. Front Cell Infect Microbiol. 2020 Sep 25;10:538077. doi: 10.3389/fcimb.2020.538077. PMID: 33102249; PMCID: PMC7546212.
Meroni, G.; Panelli, S.; Zuccotti, G.; Bandi, C.; Drago, L.; Pistone, D. Probiotics as Therapeutic Tools against Pathogenic Biofilms: Have We Found the Perfect Weapon? Microbiol. Res. 2021, 12, 916-937. doi.org/10.3390/microbiolres12040068
Li J, Zhao X. Effects of quorum sensing on the biofilm formation and viable but non-culturable state. Food Res Int. 2020 Nov;137:109742. doi: 10.1016/j.foodres.2020.109742. Epub 2020 Sep 22. PMID: 33233307.
Prazdnova, E.V.; Gorovtsov, A.V.; Vasilchenko, N.G.; Kulikov, M.P.; Statsenko, V.N.; Bogdanova, A.A.; Refeld, A.G.; Brislavskiy, Y.A.; Chistyakov, V.A.; Chikindas, M.L. Quorum-Sensing Inhibition by Gram-Positive Bacteria. Microorganisms 2022, 10, 350. doi.org/10.3390/microorganisms10020350
Wu L, Luo Y. Bacterial Quorum-Sensing Systems and Their Role in Intestinal Bacteria-Host Crosstalk. Front Microbiol. 2021 Jan 28;12:611413. doi: 10.3389/fmicb.2021.611413. PMID: 33584614; PMCID: PMC7876071.
曾利,凌保东.作用于细菌群体感应系统的抗菌中药单体[J].中国药理学与毒理学杂志,2021,35(10):802-803
Preda VG, Săndulescu O. Communication is the key: biofilms, quorum sensing, formation and prevention. Discoveries (Craiova). 2019 Sep 30;7(3):e100. doi: 10.15190/d.2019.13. PMID: 32309618; PMCID: PMC7086079.
Jiang Q, Chen J, Yang C, Yin Y, Yao K. Quorum Sensing: A Prospective Therapeutic Target for Bacterial Diseases. Biomed Res Int. 2019 Apr 4;2019:2015978. doi: 10.1155/2019/2015978. PMID: 31080810; PMCID: PMC6475571.

谷禾健康

写在前面
本文是谷禾健康微生物团队参考自英国大不列颠国家图书馆,美国微生物学会于 2003 年 在南卡罗来纳州查尔斯顿举办的一次微生物学座谈会,以及哈佛医学院微生物学系教授罗伯托·科尔特(Roberto Kolter)给Annual Review 期刊成立75 周撰写的微生物特刊等综合整理的,我们不是历史学家,更称不上微生物学专家,整理本文仅仅是向大家分享微生物学诞生和发展历程,以及全世界微生物学如何蓬勃发展、微生物相关技术给人类生命健康和生产生活等带来哪些影响和改变,我们相信未来微生物学的历史进程将会发生巨大的变化,因为从未见过像现在这样普遍的对微生物感兴趣。
我们应该思考随着技术的进步和人类对世界资源的压力越来越大,微生物科学正在发生怎样的变化?哪些主题值得探索,探索这些领域的障碍在哪里。
当我们站在基因组学、公众对生物恐怖主义、全球传染病爆发、前所未有的计算能力以及大规模生态灾难的可能性的交汇处,微生物学的最大机遇在哪里,什么是真正需要的,必须克服哪些障碍实现的机会?21世纪微生物学应该有哪些新方向?
总之,我们应该将此视为一个特殊的机会,致力于共同改善人们的生活和我们共同星球的健康。
前言
从当今科学飞速发展的角度来看,微生物学经历了漫长的发展。从安东尼·范·列文虎克(Antonie van Leeuwenhoek)对小动物的描述,到法国化学键路易斯·巴斯德(Louis Pasteur)创造的“微生物”一词已经过去了200多年。但是关于微生物学的累积知识和技术发展,大部分是在19世纪下半叶获得的,得益于多学科方法来研究地球上的各种微生物活动。
此时此刻,对于微生物学来说,是一个承上启下空前发展的阶段,尤其是2019 年冠状病毒病 (COVID-19) 大流后,我们越来越意识到微生物是生物圈的基础。它们是所有生物的祖先,也是所有其他生命形式的支持系统。
矛盾的是,某些微生物对人类健康以及植物和动物的健康构成威胁。作为生物圈的基础和人类健康的主要决定因素,微生物在地球上的生命中扮演着主要的、根本的角色。因此,研究微生物对于研究所有生物至关重要,而微生物学对于研究和理解这个星球上的所有生命至关重要。
微生物学研究正在迅速变化。该领域受到影响公众对微生物认知的事件的影响,例如全球大流行的新冠病毒出现和持续变异、生物恐怖主义的威胁、以前有效的抗生素和治疗微生物疾病的疗法越来越失败,以及大规模污染食物的事件。微生物研究正在利用开辟新研究领域的技术进步,特别是在基因组学方面。
微生物有着迷人的生活方式,许多生活在自然环境中的微生物会根据它们所处的环境发挥有益或有害的功能。微生物学从未像今天这样令人很多人感兴趣。强大的新技术,包括新颖的成像技术、基因组学、蛋白质组学、纳米技术、快速 DNA 测序和海量计算能力,已经融合在一起,使科学家能够深入研究以往认为无法或难以实现的研究。因此,几乎每一天都会有一项关于微生物的研究发表表明微生物具有核心重要性。
1
什么是微生物和微生物学?
微生物是在自然栖息地中共同生活的微生物群落,这些生物只能用显微镜才能看到,这些生命形式被称为微生物或微生物组。微生物包括细菌、古生菌、病毒、真菌、原生动物、朊病毒和酵母菌以及微观藻类。
微生物学是对肉眼无法看到的所有生物体的研究。微生物学研究涵盖了这些微生物的所有方面,例如它们的行为、进化、生态学、生物化学和生理学,以及它们引起的疾病的病理学。正如接下来的章节中所讨论的,微生物学在不同的流中扮演着非常重要的角色。
为什么微生物很重要?
在人类的发展史上,每个人都着延长寿命,1850年出生的婴儿只能指望活35年,然而目前的人类普遍的平均寿命都可以超过75年了。
那么人类是如何推翻掌握我们生命计算尺的预言,很大程度依赖过去,现在,未来在实验室战斗的无畏科学家们,尤其是微生物学家,比如,安东尼·飞利浦·范·列文虎克(发明显微镜,并观察到 “动物”(小动物)的微生物(细菌和原生动物)的人)、路易斯·巴斯德(他创造了“发酵”一词,是“现代微生物学之父/细菌学之父”)、罗伯特·科赫(提出科赫法则,验证了疾病的细菌理论,现代微生物的奠基人)、亚历山大·弗莱明安东(青霉素的发现者)、中国科学家伍连德(中国科学防疫第一人,世界鼠疫专家)、汤飞凡(著名微生物学家、沙眼衣原体发现人)等,他们抢救孕妇,拯救婴儿,对抗瘟疫,研制疫苗,发现抗生素等,大大扩展了人类对生命和微生物的认知,对延长人类寿命做出重要的贡献。
2
微生物和微生物学的核心作用
微生物影响着所有生命以及我们星球的物理和化学组成。从生命的起源开始,没有所有其他生物的生命是可能的,但没有微生物的生命是不可能的。
因此,科学家认为如果不考虑微生物的活动,就无法对生物学或地质学的任何分支进行深入研究。微生物是生物圈的主人,而我们存在的星球确实是微生物的星球。
微生物也是人类健康的决定因素,也是医疗和工业用途的关键材料的来源。因此,微生物学与生物化学、遗传学、进化或分子生物学一样,是生命研究的核心。
// 生命之树的根源
天体生物学中最大的问题之一是生命如何起源于地球。生命的基石和对生命至关重要的有机分子从原始地球开始就存在于地球上,在那里形成了生命的基本单位——细胞。这个过程是如何完成的,是天体生物学的主要研究领域之一。
微生物是现在地球上所有复杂多样的生物形式的祖先。植物和动物出现在微生物世界中,并与微生物保持着密切的联系和依赖。
作为生命之树的根,微生物是所有生命形成的原始模板,所有生命都与之密切相关。为了了解我们今天在生命之树的树枝顶端看到的生物的进化过程,有必要研究它们与祖先的关系以及这些祖先是什么样的。
通过研究当今生活的与第一批生命形式的特性相呼应的微生物,微生物学家试图了解创造我们全球生态系统的力量和过程。
此外,微生物是用于实验进化的卓越系统,因为它们为研究人员提供了快速的世代时间、遗传灵活性、无与伦比的实验规模和易于管理的研究系统。使用微生物的研究对所有物种的进化产生了开创性的见解。例如,对微生物相互关联性的研究首先揭示了地球上所有生命进化关联性的当前模型,即生命之树。
1970 年代发现了“生命的第三领域”。古生菌因其独特的生物学特性和大分子而被认为是非凡的研究对象。这一发现引发了许多关于生命起源和了解生物系统如何运作的潜力的问题,因为古细菌为研究提供了独特的资源,以揭示免疫系统的多样性和复杂性。
微生物学家开始拼凑生物系统如何从其组成部分工作,从而使科学家能够设计出执行定制功能的生物体。随着合成生物学领域的发展,现实世界的应用正在从思想领域和实验室范围内的研究转向产业化实践和产业。
最初,许多科学家的印象是深海通风口是生命起源的关键。在这里发现的化合物,可以在我们的海洋底部合成,尽管它是黑暗的,但有许多有机分子和微生物在那里发现了生命。这导致科学家们将他们的研究超越了我们的世界,对包括火星在内的行星进行了任务。鉴于我们知道微生物有能力在水中生存,他们一直在寻找确定“红色星球”过去是否有海洋。
// 维持地球上的生命
生命不仅始于微生物,地球上生命的持续存在完全依赖于不起眼的微生物。据估计,大约有5×10 31( 重达 50 万亿公吨)微生物细胞存在于这个星球上,它们对生物圈的重要性再怎么强调都不为过。
微生物负责循环生命中的关键元素,包括碳、氮、硫、氢和氧。通过在土壤中循环这些元素,微生物可以调节植物养分的可用性,从而控制土壤肥力并实现维持人类和动物生命的有效植物生长。
微生物在循环大气气体中也发挥着重要作用,包括造成“温室效应”的化合物,自相矛盾的是,它维持着我们星球上的生命,但通过全球变暖,对所有生物构成威胁。与绿色植物相比,微生物进行更多的光合作用。
事实证明,由于微生物可以吸收大型生物通常无法利用的营养物质和其他元素,因此微生物位于许多食物链的底部,在那里它们将以前惰性的无机材料转化到生物圈中。
微生物也是回收专家,它们降解生物废物并释放关键元素供其他生物使用。
目前,科学家才刚刚开始了解微生物如何适应环境,它们如何应对变化,以及它们如何与微生物群落的其他成员交流以执行维持生物圈的功能。了解这些现象将有助于我们更全面地了解我们的全球生态系统,并可能使科学家能够纠正人类对大大小小的生态系统的破坏。
// 对人类健康至关重要但危险
人类与微生物有着密切的关系。尽管它们对环境产生了很大的有益影响,但一小部分臭名昭著的细菌、真菌、寄生虫和病毒可能会导致疾病。
我们体内90%以上的细胞都是微生物;细菌和真菌遍布我们的皮肤、口腔、肠道和其他区域。微生物能够在我们的肠道中有效消化,合成必需的营养物质,并与身体器官保持良性甚至有益的关系。这些生物的存在会影响我们的身心健康。
微生物是如何引起疾病的?病原微生物和病毒具有各自的生态策略,该策略决定了它们攻击的位置以及它们对宿主的影响。
问题的根源之一是病原体在我们的免疫系统认为是“特权”的人体区域定殖。在进入这些地点或维持其菌落的过程中,微生物和病毒可能会对人体组织造成损害,产生疾病的迹象和症状。当免疫系统检测到微生物细胞或病毒时,疾病也可能开始。身体的免疫系统会对可能对身体本身造成伤害的外来生物进行攻击。在某些情况下,病原体在人体组织中造成的损害或对它们的免疫反应可以促进病原体向新宿主的传播。
因此,疾病的出现是一种新的致病微生物或病毒被识别或一种旧的微生物或病毒导致一种新的疾病——是当今的热门话题。从大肠杆菌O157 菌株到 SARS到COVID-19,每年都会发现新的疾病和新的病原体,这让公众感到恐惧。许多情况可能在疾病出现率的增加中起作用,包括各种宿主、环境和社会因素。
// 对工业和医学有用
工业和医学越来越依赖微生物来生产化学品、抗生素和酶,从而改善我们的世界并拯救生命。微生物正在被分子生物学工具驯化,以生产可生物降解的塑料和各种类型的新材料。
生物技术很快将成为各国工业基础的支柱,它以多种方式利用微生物和病毒,包括作物基因工程和基因治疗等不同应用。微生物学研究使这些成功的技术成为可能,而在工业和医学中使用微生物的未来进步依赖于今天进行有效的研究。
3
为什么微生物学研究很重要?
如上所述,微生物在我们的生活中起着至关重要的作用。事实上,我们不能没有他们,但他们可以没有我们。它们涉及许多过程,并且存在于多种环境中,其中一些是极端的。
由于它们的多功能性,微生物可以在许多方面发挥作用:疾病的原因和控制、制造救命药物、制造生物燃料、清理污染以及生产/加工食品和饮料,影响气候和生态环境。
// 医学微生物学的重要性
医学微生物学主要研究对人类和动物都有益和有害的微生物。医学微生物学的分支包括病毒学、细菌学、寄生虫学和真菌学等。
医学微生物学具有的一些重要特征,这些重要性在于它有助于病原微生物的鉴定、分离、诊断和治疗,还可以产生有益的生物体,如酵母菌和一些抗生素。
微生物学家研究微生物,而支撑现代社会的一些最重要的发现来自著名微生物学家的研究,例如詹纳和他的天花疫苗、弗莱明和青霉素的发现、马歇尔以及确定幽门螺杆菌之间的联系幽门螺杆菌感染和胃溃疡,以及 zur Hausen,他确定了乳头状瘤病毒与宫颈癌之间的联系。
生物医学研究源自生命和物理科学的许多领域,包括生物学。生物学家使用微生物学来开发预防疾病的新方法。微生物学提供的信息可进一步用于制造针对不同疾病的疫苗和治疗方法。
随着新细菌和感染的识别和分类,医学微生物学家的工作也在不断发展。生物学家在研究免疫系统时会使用从微生物学中获得的知识。
科学家在确定维生素补充剂对人类的影响时研究维生素效率。如果没有微生物学,科学家将无法看到细胞的内部结构并了解细菌、病毒和原生生物如何发育、生长和感染其他细胞。
微生物学在医疗设备中发挥着重要作用,例如荧光融合,用于快速准确地检测组织样本中的病原体。它是一种进行免疫荧光研究的技术,可用于在复杂的生物系统中寻找特定细胞。
医学微生物学的最大例子是给糖尿病患者注射从动物身上获得的胰岛素。但是由于大量需求以及由于它不是人类衍生的兼容性问题,因此需要一些其他来源的人胰岛素。然后采用涉及大肠杆菌的rDNA(Recombinant DNA)技术生产大量甚至安全的人胰岛素。
外科、医疗技术和药理学领域的发展促进了以前无法治愈的各种恶性和非恶性疾病的治疗。这些进步导致预期寿命和生活质量的提高。然而,也有不利的一面,包括因手术和其他侵入性手术、免疫抑制药物治疗或免疫系统老化而特别容易感染的患者数量激增。这些患者可能会感染多种微生物,包括那些通常不具致病性的微生物。
人们也越来越认识到感染的异常临床表现和病原体在它们通常不相关的身体区域中的存在。分子技术能够检测不可培养或难以培养的微生物,有望快速检测新的和不寻常的微生物。并非所有分子系统都是针对所有病原体设计的,但未来可检测微生物的范围可能会增加。
实验室检查是临床医生处理侵袭性感染患者的重要工具。脓毒症的发病率在世界某些地区有所增加,迫切需要快速识别致病微生物。与传统的培养和鉴定技术相比,多项研究已经确定现代分子方法的总体敏感性和特异性更高。检测时间也令人印象深刻,快速分子方法为 0.2-6 小时,而传统方法为 24-48 小时。
// 人体微生物学的重要性
人类胃肠道是极其密集的微生物群落的家园。这些微生物采用独特的策略在这种主要厌氧的环境中捕获能量。在分解源自饮食和宿主的底物的过程中,肠道微生物群会产生范围广泛的代谢产物,这些产物会在肠道中积累到高水平。越来越多的研究表明,这些化学物质通过作用于胃肠道内的细胞或进入循环并在体内远端部位发挥作用,从而影响宿主生物学。
鉴于肠道微生物群的高度功能多样性和多样化饮食,人体内微生物群衍生分子的组成部分因个体而异。因此,肠道中的微生物和它们产生的代谢终产物代表了一个表型杠杆,我们可以控制它来开发个性化医疗的新疗法。
越来越多的微生物组功能研究揭示了代谢物对人类健康和疾病的具体影响。随着对这些分子知识的扩展和生物学机制的阐明,将会出现一组新的治疗靶点。我们设想精确的健康策略将需要测量一个人的微生物组并将这些数据与代谢组以及蛋白组等分析相结合。然后,这些数据将指导利用微生物组的相对可塑性来调节微生物代谢物和促进健康的策略。
研究人员正在研发和推进控制微生物的个性化的策略,包括:
(a)改变微生物代谢的底物可用性
(b)通过饮食调节物种组成
(c)靶向微生物或代谢物的药物
(d)开发设计益生菌
(e)组装微生物群落
(f)进行粪便微生物群移植
….
这些策略存在于分子特异性的连续统一体上,从针对单个微生物中的单个酶的药物治疗方法到替代微生物组大部分代谢活动的粪便移植。
找到有效的策略将取决于对微生物和宿主新陈代谢的精确了解,同时需要足够大的人群数据库。这些与宿主修饰的化合物特别相关,在动物模型中控制代谢物通量的化合物可能不会在人体中这样做,因为 I 期和 II 期代谢不同。
// 环境微生物学的重要性
环境微生物学是对环境中微生物群落的组成和生理学的研究。在这种情况下,环境是指覆盖地球的土壤、水、空气和沉积物,还可以包括居住在这些区域上的动植物。
环境微生物学还包括对存在于人工环境(如生物反应器)中的微生物的研究。在这一领域工作的科学家通常强调微生物与植物生命和动物生命的相互作用,以及污染和季节变化等所有环境因素。
以下是微生物学的一些环境用途。它用于油的降解。
众所周知,石油是有毒的,石油造成的污染是一个重大的生态问题。沿海地区和公海的石油泄漏难以控制且难以缓解,但大部分石油可以通过微生物群落的碳氢化合物降解活动消除,特别是碳氢化合物细菌 (HCB)。这些生物可以帮助修复海洋栖息地石油污染造成的生态破坏。HCB 在生物塑料和生物催化领域也具有潜在的生物技术应用。
其次,它用于芳香族化合物的降解。从环境中分离出来的不动杆菌菌株能够降解多种芳香族化合物,也用于废物生物处理的分析。
分子生物学和基因组学在环境微生物学中的应用导致在自然微生物群落中发现了巨大的复杂性。在一系列分子和生物信息学技术的支持下,对自然种群的多样性调查、社区指纹识别和功能询问已经变得很普遍。
// 微生物学在食品工业中的重要性
微生物学也有助于保证我们的食品安全。微生物学帮助我们识别食物中存在的微生物。更好地了解这些微生物,帮助生物学家找出防止食物腐败变质和确保食物安全的方法。科学家使用好细菌对抗病原菌来防止食物污染。以下是微生物学在食品和农业工业中的优点。
专门研究食品细菌的微生物学家可以与食品和药物管理局合作,帮助识别可能对人类健康构成风险的食品。他们还调查食物中毒的爆发,旨在找出其原因,以防止再次发生。
微生物学也为我们提供了干净的水。这些活细菌有助于通过污水处理保持水的清洁。细菌分解污水中的有机物,在水释放回环境之前帮助净化水。
微生物学家研究微生物在土壤中的重要作用。一些专注于植物病虫害,开发控制它们的方法,甚至使用微生物来控制害虫和杂草。其他人则研究导致农场动物疾病的微生物。此外,微生物学帮助农民优化硝酸盐水平并最大限度地提高产量。
它有助于找出天然农药。很少有像细菌和病毒这样的微生物被用来对抗攻击农作物的害虫。因此,它们被称为天然农药。它们对害虫或昆虫如此特殊,不会对植物或动物和人类造成任何伤害。
帮助使用天然肥料。很少有像藻类和细菌这样的微生物通过固氮和保持土壤容量的水分来提高土壤肥力。因此,它们还保持适合植物生长的土壤微生物学。轮作是农民采用的一种技术,通过利用豆科植物根部的微生物来提高土壤肥力。
农业微生物学有助于分解废物。微生物分解农业土壤中的合成农药残留和其他有毒物质,从而保护农场免受毒素积累。
食品工业中微生物学应用的一个例子是乳酸链球菌素。它是一种抗菌剂,用于奶酪、肉类和饮料,通过抑制有害细菌的生长来延长保质期。
// 微生物学在日常生活中的重要性
微生物学在我们的日常生活中使用并在其中发挥着重要作用。这里讨论了我们日常生活中使用的微生物学的一些主要特征。日常生活中应用的微生物学,在食品生产、生物降解、商业产品生产、生物技术和基因工程方面。
有各种各样的菜肴需要微生物。例如,为了制作凝乳和奶酪,就需要微生物。一种叫做乳酸杆菌的细菌将牛奶中的乳糖转化为乳酸,从而将牛奶转化为凝乳。此外,酵母可以用来制作面包,在制作酸奶的过程中,细菌很重要。
此外,维生素K只能由人体内的微生物合成。除此之外,细菌用于合成具有商业价值的产品,例如用于制造一次性尿布和塑料的羟基丁酸。也在乙醇中,这是一种生物燃料,它们还合成氨基酸,这是非常常见的膳食补充剂。
因此,微生物学旨在通过研究微生物的形态、新陈代谢、生理学、繁殖和遗传学来获得和扩展我们对微生物的基本理解。这就是微生物学在不同领域发挥重要作用的方式。
在接下来的几年里,我们将看到微生物学的各种其他用途,这对我们在各个方面都非常有益。
1
诞生时代
▸微生物学一词是由法国化学家路易斯巴斯德 (Louis Pasteur, 1822-95) 提出的。
▸据说微生物学起源于 1850 年后生物科学的巨大扩展和发展。
▸Sedillot (1878) 首次使用微生物一词。
2
发展时代
▸17 世纪的英国科学家罗伯特·胡克是第一个使用镜头观察他称之为“细胞”的最小组织单位的人。不久之后,荷兰业余生物学家安东·范·列文虎克用他自制的显微镜观察了他所谓的“动物”。
▸荷兰代尔夫特的安东尼·范·列文虎克 (1632-1723)是第一个在 1676 年观察并准确描述称为“动物”(小动物)的微生物(细菌和原生动物)的人。他建造了 250 多台小型强大的显微镜,可以放大大约 50-300 倍。
▸列文虎克是第一个使用他自己制造的显微镜对细菌和原生动物进行精确和正确描述的人。由于对微生物学的这一非凡贡献,安东尼·范·列文虎克被认为是“微生物学之父”。
3
过渡时代
▸当已知微生物存在时,大多数科学家认为这种简单的生命形式肯定可以通过自发产生。也就是说,生命被认为是从泥泞、湖泊或任何营养充足的地方自发产生的。这个概念是如此引人注目,以至于它一直持续到 19 世纪后期。
▸Francesco Redi (1626-1697):自发产生的古老信念首先受到意大利医生 Redi 的挑战,他对腐肉及其自发产生蛆的能力进行了一系列实验。
▸John Needham (1713-1781):他可能是自发产生理论的最大支持者。他提出这种微生物是在他的羊肉汁中自发产生的。他像 Redi 那样用软木塞盖住烧瓶,甚至加热了一些烧瓶。微生物仍然出现在羊肉汤上。
▸拉扎罗·斯帕兰扎尼(Lazzaro Spallanzani,1729-1799):他是一位试图反驳李约瑟实验的意大利博物学家。他将牛肉汤煮了更长的时间,从烧瓶中抽出空气,然后密封容器。在温育之后,他在这些烧瓶中没有观察到生长。他表明,只要在脖子上开一个小裂缝,加热的营养物质在暴露在空气中时仍然可以生长出微生物。因此,斯帕兰扎尼反驳了自发产生的学说。
▸Nicolas Appert遵循了 Spallanzani 的工作理念。他是一位法国酿酒师,他证明了汤和液体可以通过在厚厚的香槟瓶中广泛加热来保存。
▸Ignaz Semmelweis和John Snow是对疾病传播方式的认识日益增强的两个人。
▸两位德国学者Schulze (1815-1873)和Theodor Schwan (1810-1882)认为空气是微生物的来源,并试图通过将空气通过热玻璃管或强化学物质进入烧瓶中煮沸的输液来证明这一点。两种情况下的输液都没有微生物。
▸George Schroeder和Theodor Von Dusch (1854)率先提出使用棉塞塞住微生物培养管的想法。
▸达尔文(1859 年)在他的著作《物种起源》中表明,人体可以被视为易受自然法则影响的生物。他认为疾病可能是一种生物学现象,而不是任何魔法。
4
黄金时代
▸微生物学的黄金时代始于路易斯巴斯德和罗伯特科赫的工作,他们拥有自己的研究所。更重要的是,全世界的科学界都接受了他们的工作,并愿意继续和扩展这项工作。在此期间,我们看到了微生物学作为生物学学科的真正开端。
▸法国化学家路易斯·巴斯德( Louis Pasteur)在一组涉及鹅颈烧瓶的启发性实验中最终搁置了自发产生的概念。当他在直颈烧瓶中煮肉汤并将其暴露在空气中时,有机体就会生长。当他用他的鹅颈烧瓶做这个时,什么都没有。第二个烧瓶的 S 形截留空气中的灰尘颗粒,防止它们到达肉汤。通过证明他可以让空气进入烧瓶而不是空气中的颗粒,巴斯德证明了在肉汤中生长的是灰尘中的有机体。
▸因此,巴斯德在 1858 年终于解决了自发产生与生物发生的争论,并证明微生物不是从无生命的物质自发产生的,而是从其他微生物产生的。
▸他还发现水果和谷物发酵产生酒精是由微生物引起的,并确定细菌是发酵过程中葡萄酒变质的原因。巴斯德在 1862 年提出,在 62.8°C (145°F) 温度下温和加热 30 分钟而不是煮沸足以在不破坏产品味道的情况下破坏有害生物,该过程称为巴氏杀菌。巴氏杀菌法于 1892 年在商业基础上引入美国。他的工作导致了疾病细菌理论的发展。
▸路易斯巴斯德被誉为“现代微生物学之父/细菌学之父”。
▸John Tyndall (1820 – 1893):一位英国物理学家,在 1877 年对自发产生进行了最后一击。他在一个无菌设计的盒子中进行了实验,以证明灰尘确实携带了细菌。他证明,如果不存在灰尘,无菌肉汤即使直接暴露在空气中,也可以无限期地保持没有微生物生长。他在干草的输液中发现了高度抗性的细菌结构,后来被称为内生孢子。需要长时间煮沸或间歇加热来杀死这些孢子,使输液完全灭菌,这一过程称为丁达尔化。
▸大约在巴斯德进行实验的同时,一位名叫罗伯特·科赫的医生正在努力寻找一些非常讨厌的动物疾病(首先是炭疽,然后是肺结核)的原因。他首次直接证明了细菌在引起疾病中的作用。他是一位德国医生,1876年首先分离出炭疽杆菌(Bacillus anthracis,引起炭疽病)。他完善了纯培养分离细菌的技术。他还在 1881 年通过使用明胶作为固化剂介绍了固体培养基的使用。1882年发现结核分枝杆菌. 他提出了科赫假设,该假设发表于 1884 年,是疾病细菌理论的基石,至今仍在用于证明传染病的病因(具体原因)。
科赫的四个假设是:
许多科学家的共同努力,最重要的是路易斯巴斯德和罗伯特科赫建立了疾病的细菌理论。看不见的微生物是疾病的原因的想法被称为细菌理论。
5
医学和外科的发展
▸一旦科学家们知道微生物会导致疾病,医疗实践的显着改善只是时间问题。手术曾经和不做任何事情一样危险,但一旦引入无菌(无菌)技术,康复率就会显着提高。洗手和对感染患者进行隔离减少了疾病的传播,并使医院成为接受治疗的地方,而不是死亡的地方。
▸约瑟夫·李斯特勋爵 (1827-1912):一位著名的英国外科医生,因其对预防和治疗伤口感染的抗菌治疗的显着贡献而闻名。Lister 得出结论,伤口感染也是由微生物引起的。
1867 年,他开发了一种消毒手术系统,旨在通过在手术敷料上使用苯酚来防止微生物进入伤口,有时还会喷洒在手术区域。
他还设计了一种方法,通过向空气中喷洒细小的石炭酸雾来破坏手术室中的微生物,从而产生消毒环境。因此,约瑟夫李斯特是第一个引入无菌技术,通过使用今天仍在使用的物理和化学试剂来控制微生物。由于这一显着的贡献,Joseph Lister 被称为防腐手术之父。
▸微生物的致病机理在很大程度上是环境微生物学的一部分。罗伯特·科赫的工作是世界观即将发生变化的主要驱动力。科赫证明细菌是重要传染病的病原体,这一观点的强大吸引力使微生物学的医学方面处于中心地位。知道细菌是传染病的病原体,长期以来一直是人类死亡的主要原因,意味着如果你能杀死负责的细菌,你就能治愈疾病。
▸到了20世纪初,微生物学主要集中在努力识别人类传染病背后的微生物。1918年流感大流行造成的可怕死亡人数只是加速了这些努力。由于直到20世纪30年代才发现流感病毒,在此之前,人们认为罪魁祸首是一种细菌,因此在大流行之后的几年里,许多致病细菌都得到了鉴定和深入研究。在这种情况下,关于微生物有益方面的研究基本上被搁置一旁。
▸在鉴定致病微生物的同时,许多微生物学家将注意力转向发现能够专门杀死这些微生物的化合物,这导致了20世纪人类医学史上最戏剧性的革命。
萨哈希罗·哈塔(Sahachiro Hata)和保罗·埃利希(Paul Ehrlich)的早期努力产生了第一种有机抗菌剂,即毒性较大的砷胂胺(Salvarsan),早在1910年就用于治疗梅毒。将染料用作抗菌灵丹妙药的长期努力导致了磺胺类药物的偶然发现,首先是磺胺嘧啶核苷(Prontosil),它在20世纪30年代进入临床使用。但毫无疑问,最有影响力的早期抗菌药物是青霉素。
从1928年亚历山大·弗莱明意外发现青霉素,到1942年霍华德·弗洛里、恩斯特·查恩、玛格丽特·詹宁斯等人在牛津大学的开创性工作,青霉素的纯化和有效注射使用,这种抗生素的开发仍然是人类最伟大的成就之一。
▸一种临床上有用的杀死其他微生物的微生物产品的发现对微生物学、医学和整个制药业的历史产生了持久的影响。为了发现更多的抗生素,出现了一场巨大而非常成功的热潮。
抗生素的临床成功导致其产量增加。这最初得益于20世纪40年代初的世界大战思维。由于青霉素被视为战争中的神奇药物,它的产量在很短的时间内急剧增加。1941年,全球的库存量只有几毫克,到1945年,每月生产近4000公斤。
由于与环境微生物学的分歧,医学微生物学很少关注生产和使用如此大量抗菌剂的生态后果。那些对根除病原菌的方法感兴趣的人和那些对微生物生态学感兴趣的人之间几乎没有什么合作和深入的交流对话。
6
疫苗开发
▸疫苗接种是在细菌理论之前发现的,但直到巴斯德时代才被完全理解。在 18 世纪后期,挤奶女工从他们挤奶的奶牛身上感染了非致命的牛痘病,而这些女工在周期性地肆虐英格兰的致命天花爆发中幸免于难。医生爱德华·詹纳(Edward Jenner)使用牛痘痂脓液为人们接种天花疫苗。
▸爱德华·詹纳(Edward Jenner,1749-1823)是第一个预防天花的英国医生。观察到农村挤奶女工在挤奶时接触了牛痘(牛痘是一种与天花密切相关的病毒引起的较轻的疾病),随后对天花产生了免疫力,这让他印象深刻。1796年 5 月 14日,他证明给人们接种牛痘病灶的脓液可以预防天花。
▸Jenner 于 1798 年发表了他对 23 名成功接种者的研究结果。最终这个过程被称为疫苗接种,基于拉丁词“Vacca”,意思是奶牛。
▸Jenner 的实验意义是由巴斯德实现的,他接下来将这一原理应用于预防炭疽病,并且奏效了。他将减毒培养疫苗(Vacca = 牛)和该过程称为疫苗接种。
受到通过疫苗接种成功预防炭疽热的鼓舞,巴斯德通过研制一种针对恐水症或狂犬病(一种通过狗和其他动物叮咬传播给人类的疾病)的疫苗,朝着为人类服务的方向前进。
与詹纳的天花疫苗接种一样,狂犬病的预防治疗原则也充分发挥了作用,为现代免疫计划奠定了针对白喉、破伤风、百日咳、脊髓灰质炎和麻疹等许多可怕疾病的免疫计划的基础。
▸Elie Metchnikoff (1845-1916)于 1883 年提出了免疫吞噬理论。他发现一些血液中的白细胞、白细胞 (WBC) 通过吞噬引起疾病的细菌来预防疾病。这些细胞被称为吞噬细胞和吞噬过程。因此,人体血细胞也具有免疫力,称为细胞免疫。
7
化疗药物、抗毒素和抗生素的开发
▸两位著名的法国细菌学家Emile Roux (1853-1933)和Alexandre Yersin证明了在白喉生物肉汤培养物的滤液中产生了毒素。
Emil von Behring (1854 -1917) 和 Shibasaburo Kitasato (1852-1931) 都是 Robert Koch 的同事,在 1890 年发现了破伤风(锁颚)抗毒素。在宣布发现破伤风抗毒素仅大约一周后,1890 年,冯·贝林 (Von Behring) 就用白喉抗毒素对白喉进行免疫治疗提出了反驳。毒素-抗毒素关系的发现对免疫学的发展具有重要意义。
▸Paul Ehrlich (1854-1915)在 1904 年发现染料台盼红对导致非洲昏睡病的锥虫具有活性,可用于治疗。这种具有抗菌活性的染料被称为“魔术子弹”。随后在 1910 年,埃利希与日本医生畑坂弘合作,推出了药物 Salvarsan(砷苯)作为治疗由梅毒螺旋体引起的梅毒。Ehrlich 的工作为未来的许多发展奠定了重要基础,Salvarsen 的使用标志着化学疗法的开端,以及选择性抑制或杀死病原体而不对患者造成伤害的化学物质的使用。
▸1935 年,德国的Gerhard Domagk用多种合成染料进行了实验,并报告说 Prontosil 是一种用于给皮革染色的红色染料,它对小鼠体内的病原体、链球菌和葡萄球菌具有活性,尽管它在试管中对相同的传染原没有作用。同年,两位法国科学家 Jacques 和 Therese Trefonel 表明,化合物 Prontosil 在动物体内被分解为真正的活性因子磺胺(磺胺药物)。Domagk 因发现第一种磺胺类药物而于 1939 年获得诺贝尔奖。
▸抗生素是在 1920 年代完全偶然发现的,当时在培养皿(称为盘子)中放置的固体培养物比平时更长。就像任何食物来源一样,它会发霉,长出一块毛茸茸的真菌。与平板其余部分的细菌相比,真菌菌落周围区域的菌落尺寸较小,并且似乎生长不良。被发现负责这种抗菌作用的化合物被命名为青霉素。第一种抗生素青霉素后来被用于治疗患有各种细菌感染的人,并预防烧伤患者的细菌感染,以及许多其他应用。就这样,亚历山大·弗莱明爵士在 1929 年发现了第一个抗生素青霉素。
▸美国罗格斯大学的瓦克斯曼在 1944年发现了另一种抗生素链霉素,它是由两种放线菌(灰色链霉菌)产生的。瓦克斯曼于 1952 年因发现用于治疗结核病的链霉素获得了诺贝尔奖,这是一种由结核分枝杆菌引起的细菌性疾病由 Robert Koch 于 1882 年发现。
▸到 1950 年,又鉴定出其他三种产生抗生素的微生物,例如Paul R. Burkholder 博士于 1947 年从委内瑞拉链霉菌中提取的氯霉素(氯霉素),以及BM 博士从金黄色葡萄球菌中提取的金霉素1948 年的挖掘机;Finlay、Hobby 和合作者于 1950 年从S. rimosus中提取土霉素。
▸1910 年罗伯特·科赫 (Robert Koch) 去世和第一次世界大战的到来标志着微生物学研究发生了巨大转变。巴斯德研究所关闭,德国实验室转而生产用于治疗战争感染的血液成分。许多人称之为微生物学的黄金时代就此结束。
8
20世纪:分子生物学时代
▸到 1900 年底,微生物学发展到青春期,并已成为更具包容性的生物学领域的一个分支。
晚年,微生物被作为研究各种生命过程的理想工具而被广泛采用,由此诞生了一个独立的微生物学学科——分子生物学。
微生物的相对简单性、它们的短寿命和遗传同质性为了解活生物体的生理、生化和遗传复杂性提供了一个真实的模拟模型。
分子生物学领域在理解遗传密码、如何调节 DNA 以及如何将 RNA 翻译成蛋白质方面取得了长足的进步。在此之前,研究主要集中在植物和动物细胞上,它们比细菌细胞复杂得多。当研究人员转而研究细菌中的这些过程时,基因和酶的许多秘密开始显露出来。
▸快速浏览一下从 1947 年到 1976 年分子生物学取得的进展,绝对会让人惊叹不已。科学从对基因性质的不确定状态转变为对复制、转录、翻译的分子基础以及如何调节这些过程的基础知识的相当完整的理解。大多数进展是细菌遗传学(Delbrück 和噬菌体小组的遗产)和生物化学(以 Avery 的纯化和表征方法为例)结合在一起的结果,生物物理学也做出了重要贡献。这一切的中心是大肠杆菌及其噬菌体。
很容易想象单看 DNA 结构就能揭示 DNA 复制和蛋白质合成的机制。但这种结构无疑提供了使用所有可用工具解决这些问题所需的所有动力。在不到十年的时间里,复制和蛋白质合成的一般机制被研究出来。
▸在提出 DNA 结构后不久,了解 DNA 中包含的信息如何被处理以产生蛋白质成为一个关键问题。1954 年,宇宙学家 George Gamow 提出了一种过程,即蛋白质合成通过钥匙锁机制在 DNA 表面发生,其中每个氨基酸都将作为一把钥匙,专门插入可能的 20 个孔或锁中的一个。
▸大部分关于 DNA 如何通过 RNA 聚合酶转录成 mRNA 以及 mRNA 如何在核糖体中翻译成蛋白质以及破译遗传密码的基础知识都发表于 1961 年,这是分子生物学非凡的一年。
▸核糖体于 1955 年被发现并被描述为真核细胞中蛋白质合成的场所。1958年报道了tRNA的存在。但到 1960 年底,几乎没有更多细节被公布。然后,在短暂的推进中,所有的拼图都慢慢凑齐了。基于对噬菌体突变体的遗传分析,Crick 与 Leslie Barnett、Sydney Brenner 和 Richard Watts-Tobin (23) 表明遗传密码是三联密码,三联密码不重叠,密码不包含逗号,每个基因序列都是从特定的起点读取的。
▸ 在基因活动调控这一主题上取得的进展使 1961 年发表的所有其他成就加冕。虽然研究了许多系统,但毫无疑问,在该主题上最有影响力的工作来自弗朗索瓦·雅各布和雅克·莫诺。通过应用遗传分析来研究大肠杆菌中β-半乳糖苷酶活性的诱导,他们开辟了一个关于基因如何关闭和开启的全新世界。
特别是,到 1961 年,他们发表了具有里程碑意义的论文,展示了他们积累的关于乳糖利用基因如何在大肠杆菌中受到调节的遗传证据。其中,他们提出了他们的操纵子模型,其中多个编码酶的基因由作用于操纵基因的阻遏基因调节。事实证明,他们的想法对分子生物学家如何进行基因调控研究产生了持久而极强的影响。
9
归为统一,21世纪微生物学
在 20 世纪的最后 20 年里,毫无疑问,研究宿主-病原体与模型系统和纯培养物相互作用的非常成功的方法使他们中的许多人专注于他们正在进行的研究。1999 年发表了使用非培养方法描述人类肠道微生物群高度多样性的最早报告之一。也许是因为它的资深作者 Joel Doré 曾接受过环境微生物学家的培训,这项开创性的工作几年来相对不为人所知。
在 Torsvik 描述土壤中的高细菌多样性 15 年后,Paul Eckburg、David Relman 及其同事在 2005 年发表了一篇广泛阅读的论文,描述了人类肠道中的高微生物多样性。这项工作让许多人大开眼界,它引发了人类微生物群研究的爆炸式增长。很快,几个实验室要么在纯培养中研究模型微生物,要么在动物模型中使用它们,转而使用独立于培养的方法研究人类微生物群的不同方面。这种转变恰逢测序方法的显着改进和计算能力的提高。
这个话题引起了科学家和公众的极大兴趣,因此美国国立卫生研究院于 2007 年启动了耗资 1.7 亿美元、为期 10 年的人类微生物组计划。尽管对因果关系有大量的夸张和广泛的不恰当解释,但将特定的微生物群落组成与功能联系起来有很多精彩的工作,这项新工作还包含生态学。
人类希望发展能够减轻与人类疾病相关的痛苦的医学实践,推动了继续进行基因研究的努力。世界各地的研究人员越来越多地以联盟和其他协作小组的形式聚集在一起,以完成生物学的重大成就。这些努力的第一个重大成功是人类基因组的测序,这是通过人类基因组计划(HGP) 完成的。
这种对理解人类与其相关微生物群落之间相互作用的兴趣的增加对整个微生物学产生了深远的,当然也是非常积极的影响。生态学家、进化生物学家、化学家、物理学家、计算生物学家和其他人已经进入该领域,因为他们的专业知识变得至关重要。现在,人们对微生物及其代谢途径的广泛多样性的研究重新引起了人们的关注,因为人们认识到所有生态系统都在其最基本的水平上存在微生物。生态和进化原理现在指导着微生物的研究。无论是研究人类肠道还是南极洲的冰冻湖泊,都是环境微生物学。
10
过去十年,微生物学发生了怎样的变化
在过去的 15 年里,这一统一的新微生物学出现了许多令人兴奋的新进展,因此不可能在此一一介绍。最近的历史仍在形成中,而且是最难评估的。
公众现在比历史上任何时候都更加了解微生物和病毒。不幸的是,这种公众意识通常伴随着焦虑和恐惧。过去十年的事件和媒体报道这些事件的基调助长了公众的恐惧,并产生了微生物世界风险增加的看法。这种“微生物恐惧症”导致消毒剂、抗菌肥皂和其他声称可以阻止疾病的产品的普及。
与传染病相关的话题对微生物学家来说当然不是什么新鲜事,但过去十年的发现和进步揭示了该领域的新视野,为提高人类生活质量提供了令人兴奋的机会。预测疾病出现的能力是一个特别关键的话题。对引发病原体出现的环境因素、驱动疾病迁移的因素以及疾病频率的季节性模式的研究可能会揭示影响新旧疾病如何在人群中出现和持续存在的因素。这些观察结果将使我们能够为新的和现有的病原体设计更好的治疗策略。
最近发现将人类疾病越来越多的与细菌或病毒原因联系起来,突出了其他具有神秘病因的慢性疾病也可能由微生物介导的可能性。候选药物包括炎症性肠病、糖尿病、类风湿性关节炎、结节病、系统性红斑狼疮和冠状动脉疾病。对这些疾病和其他疾病原因的研究将更加了解这些疾病及其诊断、预防和治疗。
生物恐怖主义以及炭疽或其他传染性微生物可能被用作对付无辜平民、农作物或牲畜的武器的明显可能性已经吓坏了全球人民。COVID-19、SARS、西尼罗河病等新型传染病的出现明显增加,也将微生物带入了公众视野。
最近,用于对抗某些传染病(包括艾滋病和肺结核)的常规健康疗法由于这些病原体的不断进化而失败,这加剧了公众对科学家和医生保护公众免受熟悉疾病侵害的能力的怀疑。一些曾经被认为是由遗传易感性或偶然性等因素引起的慢性疾病已被证明是由细菌或病毒引起的。而在很多国家,大规模食品污染事件呈上升趋势。
由于公众对生物战、传染病和食源性疾病的担忧,过去十年微生物学的进步经常被忽视。然而,过去十年的进步是不可否认的。药物研究现在严重依赖微生物和微生物学来进行药物发现和生产。2019年,美国科学家团队捕捉到细菌“间谍”:研究人员发现了一种有助于耐药细菌感知抗生素的蛋白质。肠道微生物作为隐藏的骑士:肠道微生物群被证明可以保护宿主免受轮状病毒感染。
使用微生物进行工业过程的绿色化学是解决化学相关行业安全性和可持续性问题的一种日益有效的策略。生物技术也依赖微生物技术和微生物基因来进行改良,以改善作物、牲畜品种和合成原料。
在农业中,微生物和微生物产品现在用于益生菌疗法、抗生素和害虫防治措施。
食品微生物学的进步提高了我们在超市和餐馆购买的食品的安全性,无疑每天都在挽救生命。
在危险废物处理场,微生物已经开始消化有毒化学物质——将它们代谢成无害物质,从而防止进一步污染土壤和水。
生物恐怖主义和疾病令人恐惧,但应牢记微生物学的进步和应用微生物正在解决看似棘手的人类问题。
在过去十年中,不仅公众对微生物学的看法发生了变化,微生物学研究的实践也发生了变化。过去十年的微生物学是充满活力和令人兴奋的,新的发现建立在过去非凡的工作之上。
1
更多的综合,更少的简化
微生物学曾经几乎只关注在人工条件下孤立生长的单个微生物,试图从实验室记录的微小观察中推断出对疾病或环境的理解。然而,今天,许多科学正在从还原论方法转向综合领域——将微生物、其环境以及其他生物体在许多尺度上的影响的测量和观察编织在一起,以创建一个综合图景的微生物活动。曾经不熟悉的细胞由相互作用的蛋白质网络组成的概念现在已经渗透到科学界。现在更加重视系统级研究,其中微生物在其栖息地被视为一系列相互关联的隔间、过程、和反馈。
将合成或系统镜头放在微生物学上可能具有很高的指导意义,并且与严格的还原论相比具有几个优点。希望在未来,综合方法将使微生物学家能够预测微生物结果,使他们能够查明人类健康或给定生态系统扰动的后果。
2
更加重视进化和生态
在过去的十年里,微生物学家越来越认识到生态和进化的重要性。实验生态学和进化论的研究提供了不仅适用于微生物的原理的证据,也可能适用于更大的生物体。生态思维已占主导地位,微生物学不再是过去的试管科学。一个例子是认识到微生物引起疾病的方式实际上是一个生态问题,需要了解微生物及其环境——疾病的宿主。
微生物学研究的调查方式也发生了变化。以前,由于技术限制或缺乏与微生物学相关的领域(如土壤科学、地质学或医学)的专业知识,许多调查潜力领域都被关闭了。在这些情况下,研究中的问题经常被重新定义以适应手头的技术和主要研究者的学术经验。今天,这些探究路线通常通过应用过去十年的技术进步进行正面探索,更重要的是,通常通过合作从其他学科招募专业知识和资源。
3
技术进步
技术进步改变了微生物学研究的格局,使长期存在的关于微生物的问题终于可以研究。在过去十年的重大进步中,最主要的是使基因组学成为可能的技术的发展,包括增强的计算能力、更快速的 DNA 测序和其他实验室技术。
基因组学利用基因组的全部或部分,即细胞的完整遗传体,来回答有关生物体的问题。尽管基因组学已经影响了大部分生命科学,并让人们对所有生命形式的功能和过程有了新的认识,但其最重要的影响还是对微生物学的影响,这一发展为微生物的生态学和进化开辟了新的认识。其他大规模研究,例如蛋白质组学或转录组学(基因表达的模式),也对微生物学研究的实践产生了很大影响。
信息技术的进步增加了所有领域研究人员之间的互动,使微生物学与其他学科之间的共同点得以持续对话。纳米技术和相关方法应该允许研究人员用单细胞进行实验,回答关于微生物生理学的长期问题。
最后,核磁共振成像 (NMR)、ESR 等高端成像技术允许对微生物细胞结构和微生物群落结构进行详细分析。信息技术的进步增加了所有领域研究人员之间的互动,使微生物学与其他学科之间的共同点得以持续对话。
纳米技术和相关方法应该允许研究人员用单细胞进行实验,回答关于微生物生理学的长期问题。最后,核磁共振成像 (NMR)、ESR 等高端成像技术允许对微生物细胞结构和微生物群落结构进行详细分析。
随着分子微生物学的出现,用于定义疾病微生物病因的传统方法(如科赫假设)已被发现不足,因为它们经常导致“假阴性”结论。研究人员一直在努力制定强有力的标准来识别超出科赫假设的微生物因果关系,并利用技术进步来识别因果关系,即使是无法在实验室培养的微生物也是如此。
本科阶段的培训
在本科阶段,微生物学教育有两个不同的方面:培养未来的微生物学家和培养其他领域的生物学家。关于培养未来的微生物学家,需要努力修改教科书,以反映关于微生物全球重要性的新知识,并克服可能仍然困扰一些微生物学课程的对记忆的强调。
很明显,所有生命科学家都应该接受微生物学培训,作为其核心课程的一部分。微生物生理学、进化、生物化学和遗传学等主题都应纳入生命科学本科生的课程。
幸运的是,有很多机会将适当的微生物学课程引入其他学科的课程。例如,几乎所有生物学学生都需要的有机化学课程将从微生物学和绿色化学的例子中受益,以证明从简单的前体合成复杂的化合物。即使是生命科学以外领域的学生也会从微生物学课程中受益,可能会在非专业的生物学探索课程中作为“微生物和你”部分进行介绍。
在许多大学中,微生物学被严格视为专业领域,而不是核心学科。鉴于微生物科学的根本意义,应该认识到拥有大量微生物科学教师的重要性。这样的教师不一定要设在微生物学系。地质学、化学、临床医学、工程学甚至历史学系都非常需要微生物学家的任命。这些教师可以跨越传统的部门障碍,在许多领域进行互动,有效地教育和培训下一代科学家。
提高微生物素养
回顾事实:微生物是最早的生命形式,它们是人类健康的重要决定因素,它们执行确保清洁饮用水和肥沃土壤的过程。它们是基因和生化最多样化的生命形式,也是地球上进化最快的生物。
微生物控制着世界营养物质和生命必需物质的环境循环。在每一个裂缝和每一个表面,从地壳深处,再到地球上每一种昆虫的肠道,微生物都在那里。
它们是所有生物系统的关键组成部分。鉴于这些事实,很明显微生物学家必须努力教育公众和政策制定者。然而,公共素养从事微生物学工作的人所经历的兴奋与公众的意识水平之间存在着严重的鸿沟。
然而,有证据表明,在艾滋病毒-艾滋病、西尼罗河病和 SARS 等重大公共卫生问题上对公共扫盲的支持水平提高会影响大学生对专业和研究项目的选择。换句话说,提高公共素养可能有助于引导学生进入最需要他们精力的领域。
此外,公众舆论可以通过提高对微生物学中未解决问题的认识来引导,从而影响领导者将资源投入到需要的领域。因此,旨在解决微生物学重大问题的培训计划应包括培养公众和政府意识的外展计划。
对公众进行微生物学教育的最佳方式是什么?迫切需要向公众宣传微生物学成功和紧迫的公共卫生问题的机制。
沟通问题
在向公众传达有关微生物学的信息时,首先要确定目标受众和适合传达给每个受众的信息类型是至关重要的。微生物外展的潜在目标受众包括一长串,包括商界领袖、各级学生和教师、公职人员、卫生专业人员(可能对微生物学不够熟悉)、农民、餐馆工作人员、联邦决策者机构等。
为了实现最有效的外展计划,教育公众的过程应该有明确的目标。具体的外展计划应该考虑到可以在指定时间段内实施的具体教育目标。这种方法将允许进行有针对性的评估,以确定外展计划在向公众传播微生物学方面是否有效。例如,一个项目的目标可能是在五到十年内对公众进行特定微生物学主题的教育,并且可以使用调查或其他指标来衡量目标受众的知识水平。
沟通的途径
向非科学家传达科学信息的最佳方式是什么?有许多途径可供外展。
例如,可以发起一项运动,向使用公共交通工具的人展示科学信息:公共汽车、火车、出租车或机场。
基于微生物主题的流行书籍的出版也将吸引大量读者。科学博物馆是教育年轻人的有力渠道,互动微生物展品可以以一种引人入胜的方式激发许多未来科学家的思想。也可以使用大众媒体。
以系统的方式研究目前用于向公众传播的与微生物学相关的材料的质量可能是有益的。目前缺乏公共知识可能是由于可用信息质量差,而不是可用性低。
改善向公众提供的知识需要让传播专业人士参与并通知他们。微生物组织应优先接触传播专业人士,并应帮助培训科学作家。
与决策者沟通
关于推进本报告中概述的目标,“决策者”一词包括联邦机构,例如国家科学基金会、卫生研究院、环境保护署、能源部、疾病控制和预防中心、农业部。其他包括地方教育委员会、国土安全部、食品和药物管理局、私人基金会等。
荧光显微镜、热循环仪、qPCR 机器、杂交炉、自动化专家系统、专用试剂——这些是现代微生物实验室更昂贵的要求。
在一些地区,新技术的吸收速度一直很慢。对于资源匮乏的地区,这些障碍似乎无法克服,因为必须为升级实验室基础设施和员工培训以及购买主要设备分配大量资金。同时,采购所需的设备、试剂供应和售后服务也很困难。
用于确认临床诊断、进行传染病监测和指导公共卫生保健政策的实验室拨款相对较少。获得良好实验室检测的机会有限导致对临床算法的依赖,但如果没有实验室确认,误诊可能很常见,导致治疗不充分、死亡率增加和缺乏对传染病真实流行率的了解。
受微生物科学进步影响但本身不是微生物学家的知情人士可能是微生物学的最佳倡导者之一。例子包括生物技术公司及其客户的代表、依赖训练有素的微生物学家技能的商业创始人、使用微生物修复财产的社区成员以及微生物疗法的受益者,包括细菌衍生的抗生素和其他药物。
建议
由于微生物对生命至关重要,并且在生物学研究中必须考虑到它们的活动,所有生物学家都必须具有扎实的微生物科学背景。微生物学课程应纳入生命和地球科学所有学生的核心课程。
在年轻学生中建立对微生物的了解,最终将提高公众对微生物对个人和地球日常健康重要性的认识。修改小学、初中和高中的学校科学课程,以包括微生物学课程和实验室练习。
通过与疾病相关的问题、生物技术、生物恐怖主义和食品安全,微生物和微生物学对公众产生了深远的影响。为了提高个人管理健康和对微生物科学做出明智判断的能力,与微生物学相关的专业协会应支持培养公共微生物素养的项目。
主要参考文献:
Microbiology in 20th Century. 2011. Link (accessed July 21, 2016).
Introduction to Microbiology. 2016. Link (accessed July 21, 2016).
Microbiology, American Society For. Significant Events in Microbiology 1861-1999. 2006. Link (accessed july 21, 2016).
Rhoads, Dan. History of Cell Biology. 2007. Link (accessed July 22, 2016).genbacteriology.
Bacterial Growth. 2015. Link (accessed July 22, 2016).
Utah.edu. PCR. 2016. Link (accessed July 22, 2016).
Innes, Centre John. Microscopy. 2016.
Mikell, Meredith. Understanding Cell Biology: History & Theories. 2003.
Pasteur, L. “”Animalcules infusoires vivant sans gaz oxygene libre et determinant des fermentations.”.” Compt. Rend. Acad. Sci., 1861: 344-347.
Pray, L. “Discovery of DNA Structure and Function: Watson and Crick.” Nature Education, 2009: 100-101
Microbiology in the 21st Century: Where Are We and Where Are We Going? This report is based on a colloquium sponsored by the American Academy of Microbiology held September 5–7, 2003, in Charleston, South Carolina. Washington (DC): American Society for Microbiology; 2004. PMID: 32809308.
Microbiology in the 21st Century: Where Are We and Where Are We Going?
American Academy of Microbiology held September 5–7, 2003, in Charleston, South Carolina.
Washington (DC): American Society for Microbiology; 2004.
Engelkirk, P. G., Duben-Engelkirk, J. L., & Burton, G. R. W. (2011). Burton’s microbiology for the health sciences. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins.
Levinson, W. (2014). Review of medical microbiology and immunology (Thirteenth edition.). New York: McGraw-Hill. Chicago
Cowan, M. Kelly.Herzog, Jennifer. (2013) Microbiology fundamentals :a clinical approach New York, NY : McGraw-Hill
Trivedi P.C., Pandey S, and Bhadauria S. (2010). Textbook of Microbiology. Pointer Publishers; First edition
Tortora, Gerard J., Funke, Berdell R.Case, Christine L.. (2013) Microbiology :an introduction Boston : Pearson.
Apurba Sankar Sastry and Sandhya Bhat K. 2018. Review of Microbiology and Immunology. 6th Edition. Jaypee Brothers Medical Publishers (P) Ltd.

谷禾健康

巨单胞菌属(Megamonas)
巨单胞菌属(Megamonas),厚壁菌门,梭状芽孢杆菌目的革兰氏阴性菌,发酵各种碳水化合物,终产物是乙酸、丙酸和乳酸。分离于人、动物和家禽的肠道。
巨单胞菌作为肠道核心种,可能是亚洲人种的特征。与炎症性肠病、结直肠癌、强制性脊柱炎(AS)、自闭症谱系障碍(ASD)、肥胖等疾病密切相关。
巨单胞菌属(Megamonas)革兰氏阴性,专性厌氧,嗜中温,亲糖,不产芽孢,杆状,不产芽孢,不运动。分离于人、动物和家禽的肠道。

doi.org/10.1099/ijs.0.65456-0
基于比较16S rRNA基因测序的系统发育分析表明,该菌种与“氨基酸球菌科(Acidaminococcaceae)” 的菌种聚为一类,故有研究者认为巨单胞菌属应归入厚壁菌门( Firmicutes )、巨单胞菌属的谱系中。
化能有机营养,发酵各种碳水化合物,终产物是乙酸、丙酸和乳酸。
属内成员有三个种:
Megamonas hypermegas(趋巨巨单胞菌)
Megamonas funiformis(单形巨单胞菌)
Megamonas rupellensis
此属的模式种为趋巨巨单胞菌 ( Megamonas hypermegale )。
目前该类型菌株M. funiformis JCM 14723、M. funiformis菌株1CBH44的完整基因组已被报道,从健康日本男性的人类粪便中分离出来。
Megamonas rupellensis sp. nov,一种从鸭子的盲肠中分离出来的厌氧菌。
结直肠癌
宏基因组和代谢组学揭示早期结直肠癌患者的肠道微生物群特征,对616名参与者进行了粪便宏基因组和代谢组学研究。在代谢组学研究中,他们发现:
在19.2%的患者(616名中的118名)中,巨单胞菌属(Megamonas)数量非常丰富。但在以往欧美受试者的肠道微生物群研究中,巨单胞菌没有被报道为优势属,只在中国个体的研究中发现,这表明该属可能是亚洲人群的特征;但在另外一篇研究中表明,炎症性肠病患者巨单胞菌属度显著降低。
Yachida S, et al., Nat Med. 2019
Megamonas funiformis 可以作为区分胆囊切除术后病人与普通人的生物指标。其丰度与胆囊切除术后结直肠癌的发展有关。
没有癌前期病变或结直肠癌的胆囊切除术后病人比起有癌前期病变或结直肠癌的胆囊切除术后病人其Megamonas. funiformis丰度更高。
此外,研究发现巨单胞菌属(Megamonas)相对丰度与结直肠息肉发病风险增大呈负相关。
Ren, X et al., Frontiers in oncology. 2020
强制性脊柱炎(AS)
研究共纳入207名研究对象(包括103名AS患者和104名健康对照),结果显示巨单胞菌属(Megamonas)和链球菌属(Streptococcus)是强直性脊柱炎组中相对丰度增幅最大的2个属,通过聚类分析发现,巨单胞菌属的相对丰度在 AS、溃疡性结肠炎(ulcerative colitis,UC)、RA 及银屑病等病种组间的欧氏距离较近,而与Ⅱ型糖尿病及腺瘤较远。
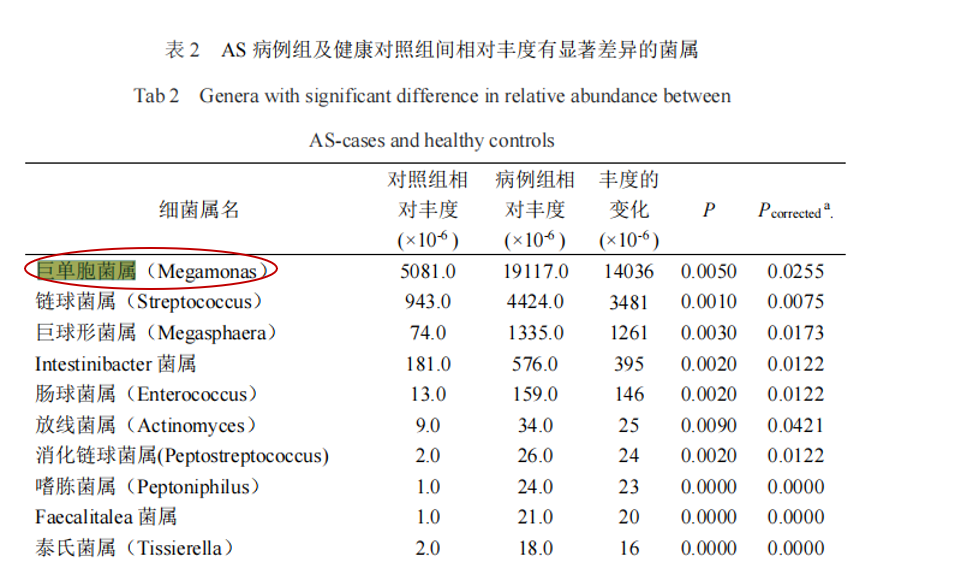
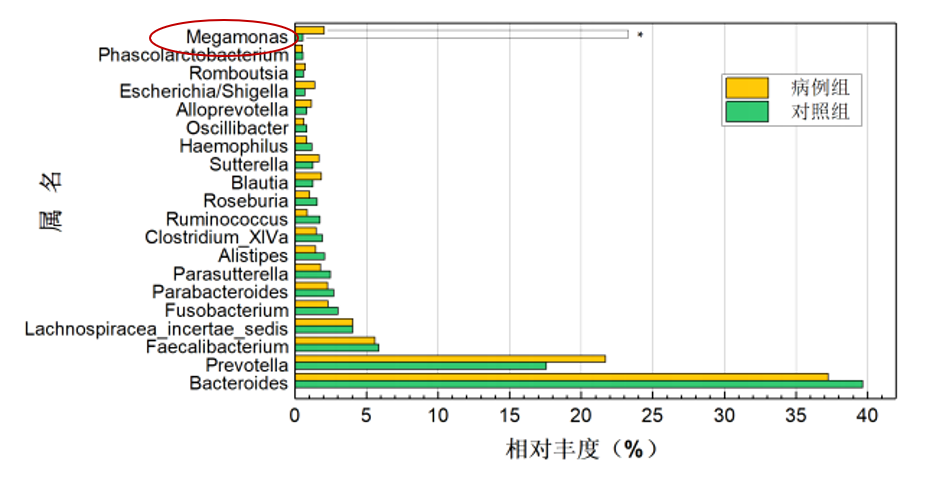
蒋光明,安徽医科大学,2021
自闭症谱系障碍(ASD)
肠道微生物群的改变可能会影响自闭症谱系障碍(ASD),患者中可能出现胃肠道(GI)生态失调。研究发现在自闭症儿童粪便中Megamonas丰度显著高于健康儿童。同时在矮身材儿童组的Megamonas丰度显著高于健康组,表明维持一定丰度在巨单胞菌属在儿童神经和体格生长发育方面的重要作用。

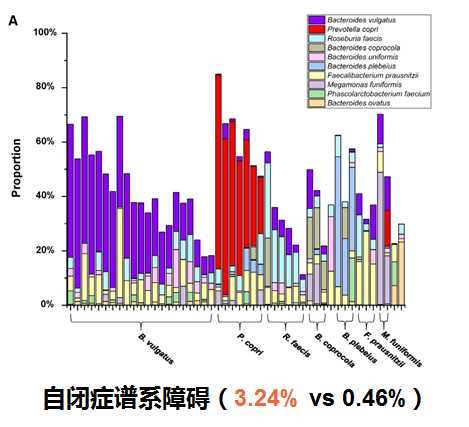
Zou R, et al., Autism Res. 2020
注意缺陷/多动障碍 (ADHD)
注意缺陷/多动障碍(ADHD)是一种神经发育障碍,其特征在于持续存在注意力不集中、多动和冲动的症状,导致个体生活两个或更多区域的功能(ADHD)组在属水平上显示出较高水平的Dialister和Megamonas以及较低的Anaerotaenia 和 Gracilibacter 丰度。
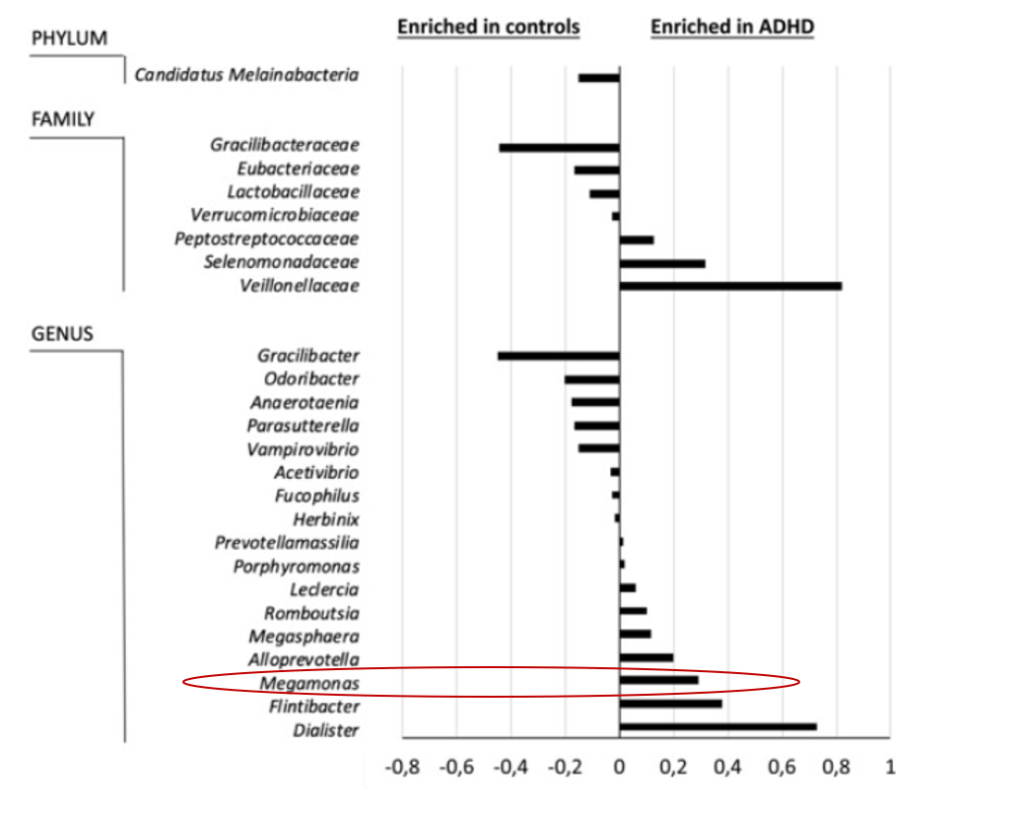
Richarte V, et al., Transl Psychiatry. 2021
注意缺陷多动障碍患者巨单胞菌丰度提高。同时作者还指出巨单胞菌可作为区分多动症患者与正常人的指标之一。
肥胖
研究发现在肥胖和对照受试者之间微生物群的显着差异。肥胖组的Prevotella、巨型单胞菌(Megamonas)、梭杆菌属和Blautia显著增加。
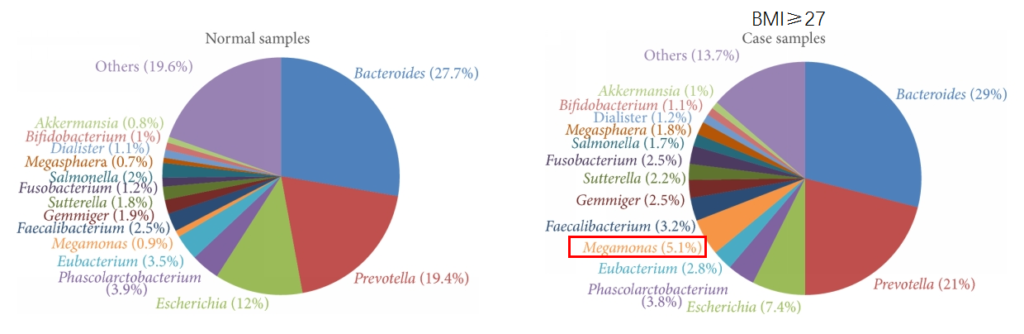
Chiu, C.M., et al., BioMed research international, 2014
另外一项研究表明体重降低或营养不良与巨单胞菌丰度减少有关联。巨单胞菌的丰度与体重减轻率呈负相关,Megamonas的丰度与肥胖犬减重的速度呈负相关。
急性缺血性脑卒中(AIS)
急性脑卒中(AIS)是一类脑部血液供应障碍引起局部组织缺血缺氧性坏死、相应神经功能出现缺损的不可逆性损害的临床综合征。同健康组比较,AIS组患者肠道中巨单胞菌属相对丰度上调,大肠杆菌属相对丰度下调。
通过将健康组和AIS患者肠道中巨单胞菌属和大肠杆菌属相对丰度进行分析,发现二者相对丰度及比值对潜在AIS具有一定诊断效能,可作为潜在的AIS诊断指标。
抑郁
部分研究报道巨单胞菌属的丰度在抑郁症患者中增加。脑卒中后抑郁患者巨单胞菌属水平上均显著高于对照组。巨单胞菌属与精神分裂症阳性和阴性症状量表(PANSS)总分呈正相关。
但也有个别文献检测到其丰度在抑郁症患者中下降。实际变化需要进一步研究。
其他
在最近的一项研究中,根据16S rRNA测序结果,发现Megamonas、放线杆菌属、Dorea和Ruminococcus与男性血清睾酮浓度呈正相关。
在另一项关于肠道微生物群性别差异的研究中,发现Megamonas、Prevotella、梭杆菌属和Megasphaera在男性中比在女性中更丰富。前列腺特异性抗原(PSA)水平高(G3)组的Megamonas丰度低于中等PSA水平(G2)组;此外,观察到PSA水平与先前报道的其他属之间没有关联。暗示Megamonas在雄激素代谢中具有潜在积极作用。
Kim HN, et al., J Pers Med. 2021
虚弱
虚弱是一种常见的老年综合征,主要根据症状进行诊断和分期。以确定这种综合征的微生物生物标志物收集了94名社区居住的老年人的血清和粪便样本,采用16SrRNA扩增子测序法测定粪便微生物群的粪便组成。
与对照组相比,来自虚弱组的粪便样本下列菌群具有较高的水平:
Akkermansia, Parabacteroides, Klebsiella
而共生属较低水平菌群如下:
Megamonas, Faecalibacterium, Prevotella, Roseburia, Blautia
推测其中Megamonas减少与老年人虚弱症状有关。
Xu Y, et al., Front Cell Infect Microbiol. 2021
炎症性肠病,白塞病,肝病等
IBD患者肠道中巨单胞菌属相对丰度显著降低。
与 正常个体相比,白塞病患者肠道中巨单胞菌物种的相对丰度显着降低。这可能与白塞病患者代谢物改变导致的 T 细胞畸变有关。
代偿期肝硬化患者巨单胞菌丰度下降。
老年血液透析(HD)患者巨单胞菌属减少。
血肌酐升高和血液透析可能影响肠道菌群的生存环境心力衰竭组与对照组相比,巨单胞菌属丰度降低。
▾ 该菌丰度较少相关:
动物脂肪摄入过多,肠道中产生短链脂肪酸(SCFAs) 的细菌(如Blautia、Megamonas)的丰度显着降低。
一项针对3500名加拿大儿童进行了从出生前直至青春期的持续追踪,其主要目标是为了发现过敏、哮喘、肥胖症及其它慢性疾病的根本原因。他们发现无论婴儿采用何种喂养方式(母乳喂养或配方奶喂养),直接补充维生素D滴剂的婴儿体内巨单胞菌属丰度都较低。
▴ 该菌丰度增加相关:
在体外发酵条件下,含牛肉蛋白和鸡肉蛋白组巨单胞菌属(Megamonas) 相对丰度显著增加,有益菌相对丰度增加。
饮食中豆类消费的高频率与Megamonas属呈正相关,但是目前该证据样本量太小,还需要进一步研究。
抗性淀粉(RS)在小肠中不能被酶解,大部分在结肠被肠道微生物发酵。研究显示玉米,马铃薯可以增加巨单胞菌属丰度。
岩藻糖基硫酸软骨素(fCS)是从海参中提取的一种独特的天然硫酸软骨素类似物,fCS-Sc显著增加了Megamonas(1.26倍)。
燕麦阿拉伯木聚糖 (AX) 刺激了鸭肠巨单胞菌和双歧杆菌的生长物种,其中巨单胞菌表现出最大的刺激。
在日粮中添加桑叶粉后,鸡肠道中的拟杆菌属、普氏菌和巨单胞菌属的相对丰度增加。
作为亚洲人肠道重要的菌属——巨单胞菌属,关于其与疾病的研究还处于开始阶段。部分数据能说明其与炎症性肠病、结直肠癌、强制性脊柱炎(AS)、肥胖、神经系统的相关性。但具体的因果关系与分子机制仍待研究,可能的研究方向有巨单胞菌的代谢产物短链脂肪酸在机体中的作用以及其与免疫反应互作反应。p.s. 本文感谢提供部分资料的各位同学。
主要参考文献:
Richarte V, Sánchez-Mora C, Corrales M, Fadeuilhe C, Vilar-Ribó L, Arribas L, Garcia E, Rosales-Ortiz SK, Arias-Vasquez A, Soler-Artigas M, Ribasés M, Ramos-Quiroga JA. Gut microbiota signature in treatment-naïve attention-deficit/hyperactivity disorder. Transl Psychiatry. 2021 Jul 8;11(1):382. doi: 10.1038/s41398-021-01504-6. PMID: 34238926; PMCID: PMC8266901.
Yachida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T, Watanabe H, Masuda K, Nishimoto Y, Kubo M, Hosoda F, Rokutan H, Matsumoto M, Takamaru H, Yamada M, Matsuda T, Iwasaki M, Yamaji T, Yachida T, Soga T, Kurokawa K, Toyoda A, Ogura Y, Hayashi T, Hatakeyama M, Nakagama H, Saito Y, Fukuda S, Shibata T, Yamada T. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat Med. 2019 Jun;25(6):968-976. doi: 10.1038/s41591-019-0458-7. Epub 2019 Jun 6. PMID: 31171880.
Ren, X., Xu, J., Zhang, Y., Chen, G., Zhang, Y., Huang, Q., & Liu, Y. (2020). Bacterial Alterations in Post-Cholecystectomy Patients Are Associated With Colorectal Cancer. Frontiers in oncology, 10, 1418. https://doi.org/10.3389/fonc.2020.01418
Duan, M., Wang, Y., Zhang, Q., Zou, R., Guo, M., & Zheng, H. (2021). Characteristics of gut microbiota in people with obesity. PloS one, 16(8), e0255446.
Chiu, C. M., Huang, W. C., Weng, S. L., Tseng, H. C., Liang, C., Wang, W. C., Yang, T., Yang, T. L., Weng, C. T., Chang, T. H., & Huang, H. D. (2014). Systematic analysis of the association between gut flora and obesity through high-throughput sequencing and bioinformatics approaches. BioMed research international, 2014, 906168. https://doi.org/10.1155/2014/906168
Elizabeth P. Cato, Ella M. Barnes. Designation of the Neotype Strain of Bacteroides hypermegas Harrison and Hansen. https://doi.org/10.1099/00207713-26-4-494
Zou R, Xu F, Wang Y, Duan M, Guo M, Zhang Q, Zhao H, Zheng H. Changes in the Gut Microbiota of Children with Autism Spectrum Disorder. Autism Res. 2020 Sep;13(9):1614-1625. doi: 10.1002/aur.2358. Epub 2020 Aug 24. PMID: 32830918.
Morotomi M, Nagai F, Sakon H. Genus Megamonas should be placed in the lineage of Firmicutes; Clostridia; Clostridiales; ‘Acidaminococcaceae’; Megamonas. Int J Syst Evol Microbiol. 2007 Jul;57(Pt 7):1673-1674. doi: 10.1099/ijs.0.65150-0. PMID: 17625216.
Shimizu, J., Kubota, T., Takada, E., Takai, K., Fujiwara, N., Arimitsu, N., Ueda, Y., Wakisaka, S., Suzuki, T., & Suzuki, N. (2019). Relative abundance of Megamonas hypermegale and Butyrivibrio species decreased in the intestine and its possible association with the T cell aberration by metabolite alteration in patients with Behcet’s disease (210 characters). Clinical rheumatology, 38(5), 1437–1445. https://doi.org/10.1007/s10067-018-04419-8
Romain Chevrot, Arnaud Carlotti, Valérie Sopena, Pierre Marchand, Eric Rosenfeld. Megamonas rupellensis sp. nov., an anaerobe isolated from the caecum of a duck. https://doi.org/10.1099/ijs.0.2008/001297-0
Cheung, S. G., Goldenthal, A. R., Uhlemann, A. C., Mann, J. J., Miller, J. M., & Sublette, M. E. (2019). Systematic Review of Gut Microbiota and Major Depression. Frontiers in psychiatry, 10, 34. https://doi.org/10.3389/fpsyt.2019.00034
Kelly, J. R., Minuto, C., Cryan, J. F., Clarke, G., & Dinan, T. G. (2017). Cross Talk: The Microbiota and Neurodevelopmental Disorders. Frontiers in neuroscience, 11, 490.
Kim HN, Kim JH, Chang Y, Yang D, Kim HL, Ryu S. Gut Microbiota Composition across Normal Range Prostate-Specific Antigen Levels. J Pers Med. 2021 Dec 17;11(12):1381. doi: 10.3390/jpm11121381. PMID: 34945854; PMCID: PMC8703440.

谷禾健康
微生物群代表宿主肠道中存在的整个微生物群。肠道内细菌界的“贫富差距”非常大,和人类社会创造的大部分的财富都流向少部分人口的现实类似,只有少数几十种的细菌分布在近乎90%的人群中。换句话说,大部分细菌都只能在特定的环境中生存,只有少数细菌适应能力超强,这可能也是我们需要重点关注的对象。
如果把不同细菌品种看作互相竞争的国家,那么细菌界的“超级大国”就属拟杆菌门和厚壁菌门了。当然它们都不是单独某一种细菌,而是一大类细菌的统称。
然而近年来随着患有肠内外疾病的人群越来庞大,变形菌门也逐渐被关注和研究,变形菌门是含有最丰富细菌的门,麾下包括多种“著名的”病原菌,如大肠杆菌、幽门螺杆菌、克雷伯氏菌、沙门氏菌、志贺氏菌、绿脓杆菌、霍乱弧菌、空肠弯曲菌、鼠疫杆菌、脑膜炎双球菌、淋球菌等,让其备受关注。
事实上,越来越多的数据将变形菌确定为疾病的可能微生物特征。目前主要证据涉及代谢紊乱和炎症甚至癌症。然而,最近的研究表明,在哮喘和慢性阻塞性肺病等肺部疾病中也有作用,有些疾病中变形菌不受控制扩张导致疾病易感和发生。
变形菌(proteobacteria)是细菌中最大、种类最多的一个门,它们在系统发育、生态和致病方面具有广泛的重要性。所有变形菌都是革兰氏阴性菌,外膜主要由脂多糖组成。
图源:esacademic
变形菌门主要是由核糖体RNA序列定义的,名称取自希腊神话中能够变形的神普罗透斯(这同时也是变形菌门中变形杆菌属的名字),因为该门细菌具有极为多样的形状,代谢特征等。
△ 形状:杆状和球菌、弯曲的、螺旋状的、环状的、丝状的和带鞘的细菌都有。
△ 新陈代谢:新陈代谢类型也多种多样,一系列代谢特征包括化学自养(从无机化合物的氧化中获取能量)、化学有机营养(从有机化合物的氧化中获取能量)和光养(从光中获取能量)。
△ 氧气利用:从严格厌氧菌和严格需氧菌到兼性厌氧菌和微需氧菌株的都有,但是大多数变形菌门的成员是兼性厌氧菌。
△ 运动:许多使用鞭毛移动,但有些不能移动或依赖细菌滑动,而一些细菌是不运动的。
△ 生态分布:变形菌门的成员具有极大的可变形态和多才多艺的生理学,这使它们在各种生态位中生存具有竞争优势。已观察到变形菌在不同生境中无处不在。
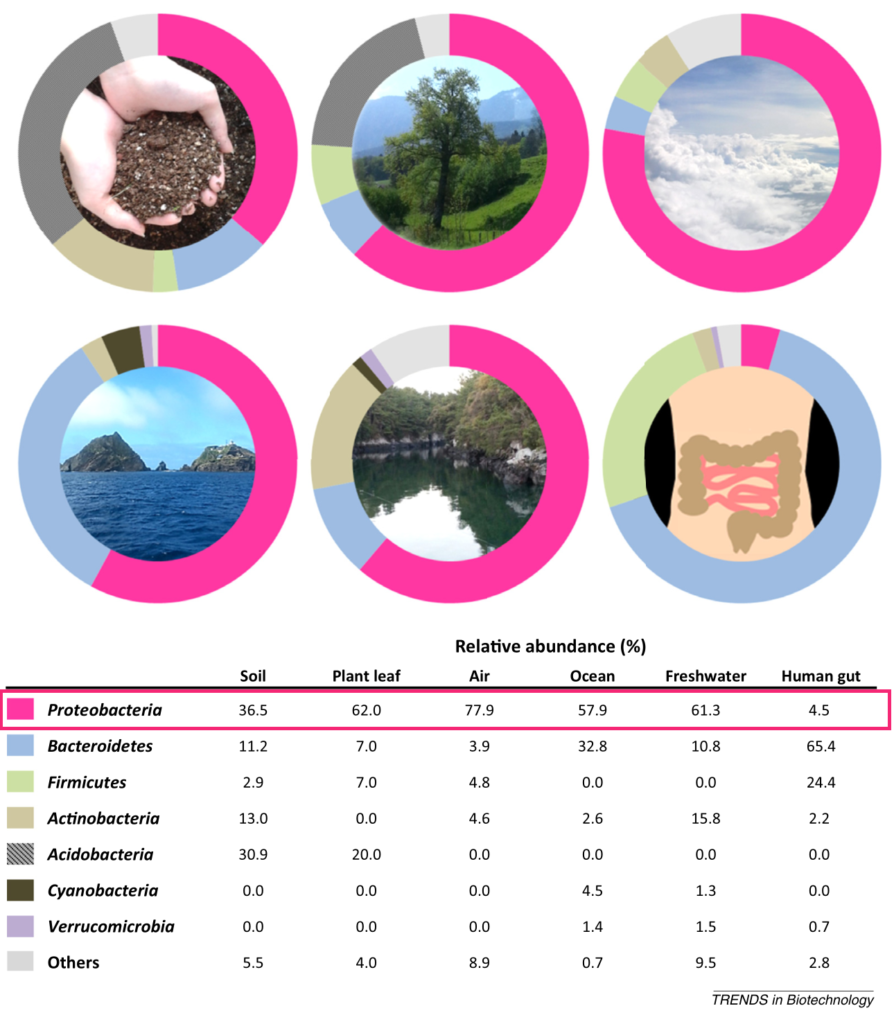
Shin NR, et al., Trends Biotechnol. 2015
植物 、海水、淡水 ,空气,以及人和动物的身体部位,包括肠道、口腔、皮肤、阴道。尽管存在研究间差异,但健康人口腔微生物群的变形菌相对丰度最高(17.2-36.8%),其次是皮肤(6.8-30.0%)、胃肠道(2.5-4.6%)和阴道(2.3%)。
在系统发育学上,变形菌是根据小核糖体亚单位RNA基因(16S rRNA)的测序定义的。这是一个巨大的革兰氏阴性原核生物门,原线粒体起源于此。
图片来源:Maria Lane,eportfolio
该门主要分为以下几大类:
最初,变形菌包括 α、β、γ 和 δ 四个亚类。ε变形菌 和 δ变形菌 通常被认为是最古老的变形菌群,因为它们包括利用硫化合物进行能量代谢的专性厌氧菌。
α变形菌(Alpha-proteobacteria)
第一类变形菌是α-变形菌。这一类的统一特征是它们是寡营养生物,能够生活在低营养环境中,如深海沉积物、冰川或深层地下土壤。同时α-变形菌是多样化的细菌分支之一,在生活方式、地理分布和基因组大小方面表现出极大的差异。
在 α-变形菌 中有两个重要分类群,衣原体和立克次体,它们是专性细胞内病原体,这意味着它们的部分生命周期必须发生宿主细胞内。由于它们无法合成自己的三磷酸腺苷 (ATP),因此,量需求依赖宿主于细胞。
立克次体属是人类很多严重疾病的病原体。例如,布鲁氏菌属、埃立克体属和立克次氏体。立克次氏杆菌会导致落基山斑疹热,这是一种威胁生命的脑膜炎(包裹大脑的膜发炎)。R. rickettsii 感染蜱,并可以通过被感染的蜱叮咬传播给人类。此外,布鲁氏菌科(Brucellaceae)和巴尔通氏菌科(Bartonellaceae)的细菌是人类病原体。
α-变形菌 还包括固氮细菌,例如固氮螺菌属和根瘤菌属。这两种细菌都使用一种称为固氮酶途径的复杂酶途径将大气中的氮 (N2) 转化为氨 (NH3)。此外,α变形菌还包括硝化细菌。这种类型的细菌将氨和铵 (NH4+) 还原为硝酸盐 (NO3–)。乙酸杆菌属和葡糖杆菌属的变形菌可用于生产乙酸。
β变形菌(Beta-proteobacteria)
与依靠最少量营养物质生存的 Alpha-proteobacteria 不同,Beta-proteobacteria 类是富营养生物,这意味着它们需要大量的有机营养物质。
Beta-proteobacteria 通常在需氧和厌氧区域之间生长(例如,在哺乳动物的肠道中)。一些属包括作为人类病原体的物种,能够引起严重的,甚至可能危及生命的疾病。例如,奈瑟球菌属包括淋病奈瑟菌( STI淋病的病原体)和脑膜炎奈瑟菌(细菌性脑膜炎的病原体)
β变形菌中的亚硝化单胞菌可以将亚硝酸盐还原为亚硝酸盐 (NO2–)。同时,硫杆菌属物种是将硫化氢 (H2S) 和元素硫氧化成硫酸盐 (SO42-) 的细菌,以及用于污水处理的菌胶团(Zoogloea)和Sphaerotilis 。
γ变形菌(Gamma-proteobacteria)
最多样化的革兰氏阴性细菌是γ-变形菌,它包括许多人类病原体。包括几个医学和科学上重要的细菌群,例如肠杆菌科、弧菌科和假单胞菌科。
此外,许多重要的病原体属于这一类,例如:
Richard B. Frankel
△ 铜绿假单胞菌
一个庞大而多样的科,假单胞菌科,包括假单胞菌属。铜绿假单胞菌在该属内,它是一种病原体,可以造成身体不同部位的各种感染。铜绿假单胞菌是一种严格需氧、不发酵、高度运动的细菌。
它通常可能造成伤口和烧伤感染,也可能是慢性尿路感染的原因,并且可能是囊性纤维化患者或机械呼吸机患者呼吸道感染的重要原因。
铜绿假单胞菌感染通常难以治疗,因为该细菌对许多抗生素具有抗性,并且具有形成生物膜的非凡能力。
△ 肠杆菌科
肠杆菌科是属于γ-变形菌 的一大类肠道细菌。它们是兼性厌氧菌,能够发酵碳水化合物。在这个家族中,微生物学家认识到两个不同的类别。
第一类,大肠杆菌,以其原型细菌种类大肠杆菌命名。大肠菌能够完全发酵乳糖(即产生酸和气体)。
第二类,非大肠杆菌,要么不能发酵乳糖,要么不能完全发酵(产生酸或气体,但两者不能同时产生)。
非大肠杆菌包括一些值得注意的人类病原体,例如沙门氏菌属,志贺氏菌,鼠疫耶尔森氏菌。
δ 变形菌(Delta-proteobacteria)
δ-变形菌(Delta-proteobacteria )包括基本好氧的形成子实体的粘细菌和严格厌氧的一些种类,如脱硫球菌属(Desulfococcus)、脱硫线菌属(Desulfonema)、硫酸盐还原菌(脱硫弧菌属(Desulfovibrio)、脱硫菌属(Desulfobacter)、和硫还原菌(如除硫单胞菌属Desulfuromonas),以及具有其它生理特征的厌氧细菌,如还原三价铁的Geobacter和互营菌属(Syntrophus)。
△ 蛭弧菌属:
δ-变形菌还包括蛭弧菌属,Bdellovibrio侵入宿主细菌的细胞,将自身定位在周质中,即质膜和细胞壁之间的空间,以宿主的蛋白质和多糖为食。这种感染对宿主细胞是致命的。
△粘细菌:
粘细菌(“粘液细菌”)是一组主要生活在土壤中并以不溶性有机物质为食的细菌。与其他细菌相比,粘细菌具有非常大的基因组,例如 9-1000 万个核苷酸。
Sorangium cellulosum 拥有最大的已知(截至 2008 年)细菌基因组,有 1300 万个核苷酸。
粘细菌产生许多在生物医学和工业上有用的化学品,例如抗生素。他们将这些化学物质输出到细胞外。
ε变形菌(Epsilon-proteobacteria )
ε-变形菌(Epsilon-proteobacteria) 是革兰氏阴性微需氧细菌(意味着它们在其环境中只需要少量氧气)。多数是弯曲或螺旋形的细菌,如沃林氏菌属(Wolinella)、螺杆菌属(Helicobacter)和弯曲菌属(Campylobacter)。它们都生活在动物或人的消化道中,为共生菌(沃林氏菌在牛中)或致病菌(螺杆菌在胃中或弯曲菌在十二指肠中)。
△ 弯曲杆菌:
变形菌门Epsilon-proteobacteria 中的两个临床相关属是弯曲杆菌属和螺杆菌属,它们都包括人类病原体。
弯曲杆菌可引起食物中毒,表现为严重的肠炎(小肠发炎)。这种由空肠弯曲杆菌引起的疾病在发达国家相当普遍,通常是因为食用了受污染的家禽产品。鸡通常携带空肠弯曲杆菌在胃肠道和粪便中,它们的肉在加工过程中可能会受到污染。
△螺杆菌:
螺杆菌是ε-变形菌的一个属,具有特征性的螺旋形状。它们最初被认为是弯曲杆菌属的成员,但自 1989 年以来,它们独立为自己的属。
螺杆菌属属于ε-变形菌,弯曲杆菌目,螺杆菌科,已经有超过 35 种。已经发现一些菌生活在上胃肠道的内壁,以及哺乳动物和一些鸟类的肝脏中。
该属中最广为人知的物种是幽门螺杆菌,它感染多达 50% 的人口。这种细菌的某些菌株对人类具有致病性,因为它与消化性溃疡、慢性胃炎、十二指肠炎和胃癌密切相关。它也作为该属的模式种。
幽门螺杆菌在胃的高酸性环境中存活的能力有些不同寻常。它产生脲酶和其他酶来改变其环境以降低其酸性。
幽门螺杆菌也有它存在的意义,可能抑制引起结核的细菌(结核分枝杆菌),预防哮喘,克罗恩病,食管反流,腹泻病以及食道癌。
❥ 识别微生物编码的基因,与特征相关联
栖息在哺乳动物肠道中的微生物编码了大量的蛋白质,这些蛋白质有助于广泛的生物功能,从调节免疫系统到参与新陈代谢。
我们从这些微生物中识别蛋白质编码基因并将基因水平与疾病、药物功效或副作用以及其他宿主特征相关联。
例如,与传统的高纤维农业饮食相关的人类肠道微生物群编码了参与纤维素和木聚糖水解的基因家族,而这些基因家族在吃典型西方饮食的人群(年龄匹配)中不存在。
一般编码适应肠道环境所必需的功能的微生物有很强的选择性,在不同宿主中具有大量冗余的基因库。然而,目前的研究和临床很容易忽略健康人类微生物组之间基因丰度的生理意义差异。
❥ 较少丰度的变形菌门,才是是跨宿主丰度变异性最大的基因的主要来源
人体肠道通常由拟杆菌门和厚壁菌门主宰,这些门内的进化枝(尤其是拟杆菌属、普氏菌属和瘤胃球菌科)是最常用于将个体聚集成“肠型”,因为它们解释了最多的分类变异。Bacteroidetes 与 Firmicutes 的比率也被推定为疾病或健康的潜在生物标志物。
有人提出,人类肠道微生物组中可能存在少量“肠型”,每一种都具有不同的分类组成。因此,虽然拟杆菌门和厚壁菌门可能对宿主之间的分类变异贡献最大,但变形菌门的丰度可能会捕获更多的功能变异。
与先前确定的肠型标记分类群相比,变形菌门的水平和可能的 Euryarchaeota 更好地解释了肠道微生物基因功能的人与人之间的差异。
在肠型研究中遗漏了这些不太丰富的门,可能是因为肠型是通过倾向于对高丰度分类群进行更多加权的方法鉴定的,并且肠型是从分类学而非功能数据中鉴定的。这对解释人类肠道微生物群的分类数据具有重要意义。
例如,变形菌门的过度生长与代谢综合征和炎症性肠病有关。通过 TLR5 敲除小鼠测试的肠道炎症关联到变形菌门(超过拟杆菌门和厚壁菌门),并且一些变形杆菌可以在这种背景下诱发结肠炎,可能导致反馈循环。因此,可变基因家族对解释人类肠道微生物群的分类数据具有重要意义。
备注:肠道受体蛋白TLR5参与积极地塑造新生小鼠肠道微生物群落的长期组成,敲除的Toll样受体(TLR5),是免疫系统识别鞭毛细菌(比如变形菌和梭状芽孢杆菌)的关键受体,缺乏它则机体可能不会在感知到细菌鞭毛时对细菌产生免疫应答。
肠道相关微生物群落组成的变化与许多人类疾病有关,但驱动这种不平衡(生态失调)的机制尚不完全清楚。
在肠道菌群失调期间观察到的最一致和最强大的生态模式是属于变形菌门的兼性厌氧细菌的扩张。
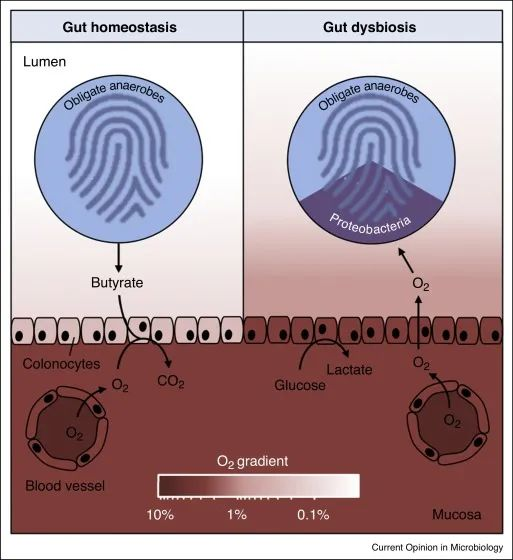
变形菌的菌群失调是上皮功能障碍的微生物特征
在肠道稳态期间(左),微生物群衍生的丁酸盐的 β 氧化导致上皮缺氧,从而维持大肠腔内的厌氧状态。反过来,腔内厌氧症导致肠道微生物群内专性厌氧菌占主导地位。
备注:丁酸(Butyrate acid,BA),俗称酪酸,是构成脂肪的一种脂肪酸,含有4个碳原子又称短链脂肪酸。人体的丁酸部分来自于食物中丁酸的吸收,主要的来自结肠厌氧菌的发酵产生。人体结肠产生的短链脂肪酸丁酸占比大部分)。
在肠道菌群失调期间(右),表面结肠细胞通过无氧糖酵解获得能量,从而导致上皮氧合增加,这种上皮功能障碍破坏了管腔中的厌氧菌,从而通过有氧呼吸推动兼性厌氧变形菌的扩张。
健康结肠的厌氧菌导致肠道微生物群的组成以专性厌氧菌为主,而菌群失调通常与兼性厌氧变形菌的丰度持续增加有关,这表明厌氧菌的破坏。
结肠上皮是缺氧的,但肠道炎症或抗生素治疗会增加结肠中的上皮氧合,从而破坏厌氧作用,通过有氧呼吸驱动兼性厌氧变形菌的菌群失调。
肠沙门氏菌(S. enterica)是一种食源性病原体,属于肠杆菌科,变形菌门,可引起小鼠结肠炎。在肠道沙门菌S. enterica诱导的结肠炎期间,肠腔内的氧气可用性增加,这表明结肠中病原体的氧气呼吸依赖性大量繁殖以及随之而来的专性厌氧梭状芽胞杆菌的丰度下降。
同样,结肠隐窝增生由鼠肠道病原体柠檬酸杆菌(肠杆菌科,变形菌门)引发,可提高肠腔内的氧气利用率,从而通过有氧呼吸推动变形菌病原体扩张。
这些观察结果表明,变形菌的菌群失调是上皮功能障碍的潜在诊断微生物特征,建议将变形菌负荷作为生态失调和疾病的潜在诊断标准,所以在谷禾即将更新的肠道菌群检测报告中,我们会加入变形菌门丰度和参考范围这一指标。
大肠中专性厌氧菌的优势可能是宿主环境的氧气限制严重的结果,这反过来又对用于营养物质的分解代谢途径产生重要影响。
避免被上消化道中的宿主酶降解的复合碳水化合物,可以被大肠中的专性厌氧细菌水解并发酵成更小的化合物。专性厌氧菌最终将许多发酵产物转化为短链脂肪酸,其中乙酸盐、丙酸盐和丁酸盐是最丰富的产物。宿主吸收了大约 95-99% 的微生物产生的短链脂肪酸,它到达血流以影响免疫发育。因此,大肠中专性厌氧菌的优势确保了维持肠道稳态的代谢物的产生。
变形菌是平衡的肠道相关微生物群落中的一个次要成分。然而,由遗传易感性、化学物质或肠道病原体感染引起的肠道炎症会导致小鼠模型中变形杆菌的管腔扩张不受控制。
同样,在患有严重肠道炎症的人类中,包括炎症性肠病、结直肠癌或坏死性小肠结肠炎的患者中观察到变形杆菌的丰度增加。此外,在包括肠易激综合征和代谢综合征在内的低水平肠道炎症条件下观察到大量变形菌。
肠道炎症增加了替代电子受体的可用性,这些电子受体通过厌氧呼吸支持兼性厌氧细菌的生长。肠道炎症过程中产生的活性氧可以将内源性硫化合物氧化为连四硫酸盐,这是一种电子受体,通过连四硫酸盐呼吸作用在鼠结肠中驱动类似肠沙门氏菌和Yersinia enterocolitica(一种属于肠杆菌科,变形菌门的病原体)的管腔扩张 。
一氧化氮由宿主酶产生化学诱导的结肠炎或由遗传易感性引发的结肠炎期间的诱导型一氧化氮合酶(iNOS) 。一氧化氮在肠腔内分解成硝酸盐,从而通过硝酸盐呼吸支持生长,从而增加小鼠结肠中共生大肠杆菌的丰度。类似,宿主衍生的硝酸盐的呼吸有助于在 S. enterica 诱导的小鼠结肠炎期间腔内病原体扩张。
有趣的是,即使在没有明显肠道炎症的情况下,例如在抗生素治疗期间,呼吸电子受体也有助于细菌群落从专性厌氧菌转变为兼性厌氧菌。为了支持这一观点,用链霉素治疗小鼠可将盲肠中的氧化还原电位提高到接近需氧培养液的水平。链霉素治疗通过硝酸盐呼吸和氧气呼吸的结合增加结肠中共生大肠杆菌或致病性肠杆菌的生长。
其他类似研究的结论也表明,氧气,单独或与其他呼吸电子受体结合,是广泛的胃肠道失衡中肠道菌群失调的常见驱动因素。因此,为了开发新的预防或治疗策略,必须了解在肠道菌群失调期间呼吸电子受体的可用性如何升高。
基于这些观察,有人提出变形菌的扩增是肠道菌群失调的微生物特征,而氧气、用药,遗传易感,肠炎驱动了变形菌的扩张,反过来加剧疾病的进展。
宿主遗传因素和外在环境因素,如饮食和生活环境,不断影响肠道微生物群的分类和功能组成。鉴于具有高度稳定性的平衡肠道微生物群与宿主的免疫系统具有共生相互作用,能够抑制变形杆菌失控的扩张,肠道中变形杆菌的大量繁殖可以反映肠道微生物群落结构的不稳定;这种不稳定的结构可以在非疾病状态下观察到(例如,新生儿期 和胃绕道手术后和疾病状态例如,代谢紊乱和肠道炎症)。
Shin NR, et al., Trends Biotechnol. 2015
在新生儿胃肠道的初始定植期间,兼性厌氧变形菌使肠道生态位有利于专性厌氧菌的定植;后者很快被专性厌氧的厚壁菌门和拟杆菌门所取代,它们在健康成年人的肠道微生物群中占主导地位。胃绕道手术导致的胃肠道重排可以改变 pH、胆汁流量和肠道激素,所有这些因素都会影响变形杆菌的丰度。
新生儿肠道中的变形菌
新生儿肠道中的微生物群备受关注,因为它不仅反映了细菌群落的脆弱结构,而且反映了哺乳动物肠道微生物群的真正起源。新生儿肠道中的细菌群落由于其快速的时间变化而不稳定。然而,这种脆弱性与更重要的肠道菌群定植有关,例如严格的厌氧菌。
具体来说,由于新生儿肠道中的氧气丰富,生命第一周的微生物群经常以兼性厌氧菌为主,主要是变形菌属(例如,埃希氏菌属、克雷伯氏菌属和肠杆菌属)。这些兼性厌氧菌通过消耗氧气、改变 pH 值、降低氧化还原电位并产生二氧化碳和营养物质,使栖息地适合严格的厌氧菌定殖。
因此,可以推测变形杆菌在为新生儿肠道准备好接受严格厌氧菌的连续定植方面发挥了作用,这些厌氧菌在健康成人的肠道中含量丰富。
最近对母体胎盘微生物组的一项研究描述了共生细菌群落的存在,其中大肠杆菌的丰度最高。尽管关于胎盘微生物群的活力和起源存在争议,但在母体胎盘中发现的这些有趣的细菌群落与来自母体羊水和新生儿胎粪的细菌群落重叠。
因此,新生儿肠道中的变形菌可能通过胎儿在子宫内吞咽羊水从母体胎盘传播。有趣的是,妊娠后期孕妇肠道中变形菌的比例增加。这意味着母亲微生物群中的这种特定细菌群转移到了新生儿身上。
在新生儿肠胃道中观察到的变形杆菌定植生长的持续时间很可能在母体控制之下。事实上,新生儿微生物群会受到各种母体因素的影响,例如分娩方式、饮食和怀孕期间接触抗生素。
最重要的是,新生儿肠道中变形菌的丰度受喂养类型的影响,这些细菌在配方奶喂养的婴儿中的频率更高,但在母乳喂养的婴儿中很少见。
人乳寡糖 和分泌型 IgA 的产生参与在最初的肠道定植过程中选择性抑制变形菌。因此,越来越多的人认为,及时减少变形菌的丰度是初始微生物定植的正常部分,而这种定植模式的紊乱与新生儿疾病的风险增加有关。
肠道中微生物群和宿主细胞之间的相互作用对于免疫系统的形成和调节至关重要,由于肠腔内有大量外源性抗原,免疫系统必须严格调节其反应以维持与共生菌的共生关系。共生体传递一种信号,诱导宿主免疫的耐受性反应。因此,宿主可以区分有益的本土微生物和有害病原体,并建立健康的微生物群。
变形杆菌的主要分类及其与IBD的关系
Mukhopadhya I, et al., Nat Rev Gastroenterol Hepatol. 2012
为了防止对共生细菌的炎症反应,肠道内的免疫细胞,如单核吞噬细胞(巨噬细胞和树突状细胞)和 CD4 + T 细胞,对微生物刺激反应迟钝或表现出共生反应。
同时,黏膜免疫系统负责清除病原体,这一过程需要积极的促炎信号级联反应。因此,不适当的免疫反应会破坏肠道稳态,引发生态失调,并导致局部和全身炎症和代谢功能障碍。
这种慢性进行性肠道炎症的状态在临床上被诊断为炎症性肠病 (IBD),其中包括溃疡性结肠炎 (UC) 和克罗恩病 (CD)。IBD 的确切病因仍然无法获得,但新出现的证据表明,肠道微生物群成为了这种疾病的主要嫌疑。
许多研究报告了动物和人类各种炎症持续条件下微生物群组成的改变。在这种情况下,通常发现变形菌在疾病中增加,变形菌在肠道炎症中的作用已在各种结肠炎小鼠模型中得到解决,与疾病呈正相关。
例如,使用易发炎症的小鼠模型,即鞭毛蛋白受体 TLR5 缺陷小鼠 (T5KO),发现,进展为结肠炎的小鼠表现出明确的微生物群特征,其特征是变形菌的水平增加,尤其是大肠杆菌属。并且一些作者已将其确定为微生物群不稳定性的潜在标志物,因此易诱发疾病发作。
与变形杆菌属大量繁殖的同时,结肠炎Tlr5-/- 小鼠表现出杂乱无章的结肠粘液层,与非结肠炎Tlr5-/- 同胞相比,感染性病原体的清除延迟。
这些结果表明,短暂不稳定的肠道微生物群,尤其是以变形菌为主的群落,会使遗传易感的小鼠易患慢性结肠炎。
先天免疫反应失调推动变形杆菌生长的假设这反过来又会促进肠道炎症,这一点得到了其他小鼠模型研究的支持,这些小鼠模型具有影响适应性免疫的突变,白细胞介素 (IL)-10 是对本地微生物群产生免疫耐受所需的主要免疫调节细胞因子。
IL-10 缺陷小鼠由于对肠道菌群不耐受而表现出自发性结肠炎。随着结肠炎症的发生和发展,在定植常规微生物群或缺乏特定病原体的微生物群的 IL-10-/- 小鼠中,变形杆菌和大肠杆菌比野生型小鼠多。
在另一项对 IL-10 缺陷小鼠的研究中,富含饱和乳脂的饮食扰乱了肠道微生物群,导致亚硫酸盐还原Delta-proteobacteriumBilophila wadsworthia 大量繁殖。这种病原菌在 IL-10 -/-小鼠中诱导促炎性黏膜免疫反应并促进自发性结肠炎的发生率和严重程度;它还在喂食高乳脂饮食的野生型小鼠中促进葡聚糖硫酸钠 (DSS) 诱导的结肠炎。
除了对结肠炎的易感性与肠道变形菌的相对丰度之间存在正相关性之外,对先天性和适应性免疫系统均缺陷的小鼠的研究提供了支持变形菌在肠道炎症中的致病作用的证据。
来源:谷禾健康肠道菌群数据库
谷禾健康肠道菌群检测大数据也显示,在炎症性肠病,结直肠癌等患者的肠道菌群检测报告中,85%以上的患者显示变形菌门超标或多项变形菌门病原菌超标或占比丰度偏高。
在最近的一项研究中重现了,结肠炎中变形杆菌的显着扩增,该研究比较了患有活动性结肠炎的 TRUC 小鼠的肠道微生物组与因庆大霉素、甲硝唑或抗肿瘤坏死因子 (TNF)-α 治疗而缓解的小鼠的肠道微生物组。
值得注意的是,从 TRUC 小鼠的粪便中分离出的两种肠杆菌科细菌(肺炎克雷伯菌和奇异变形杆菌)即使在没有任何遗传免疫缺陷的受体小鼠中也足以引发结肠炎。
然而,这两种微生物的致结肠潜力并未在无菌 TRUC 小鼠中复制,这表明结肠炎的发病机制需要其他共生成员。口服伤寒杆菌,另一种富含 TRUC 小鼠的变形菌,也会在非结肠炎 TRUC 小鼠中引发结肠炎,这些小鼠具有大量的促炎细胞因子(例如,TNF-α)。
遗传易患结肠炎的小鼠的生态失调与人类 IBD 特别相关,因为与 IBD 相关的风险等位基因或多态性与先天性和适应性免疫成分有关。与小鼠研究相似,两项人类研究表明,与健康受试者相比,IBD 患者肠道微生物群落的特点是微生物多样性低、变形菌门(尤其是肠杆菌科)的产物以及厚壁菌门的减少。
一项人类队列研究发现,核苷酸结合寡聚化结构域 (NOD)-2 风险等位基因剂量与 IBD 患者肠道标本中肠杆菌科的相对丰度呈正相关。
在 UC 患者中,与炎症的中度和轻度阶段相比,在严重阶段观察到的变形杆菌水平显着升高。
在新发 CD 的初治儿科患者和非 IBD 对照受试者之间,回肠和直肠活检(但不在粪便样本中)的粘膜相关微生物组存在明显差异。变形菌的相对丰度增加,包括肠杆菌科、巴氏杆菌科和奈瑟菌科,将 CD 相关细菌群落与健康对照组区分开来。与慢性炎症一致,伴随变形杆菌属优势的肠道微生物群落改变不仅见于传染性病原菌或原生动物寄生虫引起的急性炎症,而且见于实验性和人类结肠炎相关的结肠直肠癌。
最有趣的生物体,通过一个孤立的病例报告与 IBD 有关,该病例报告一名感染这种细菌的小男孩在放射成像上出现回肠增厚,这是克罗恩病的典型表现。
血清学研究表明,与健康对照相比,克罗恩病患者的大肠杆菌抗体数量增加。具体地说,已发现37-55 % 的克罗恩病患者、2-11% 的溃疡性结肠炎患者和 <5% 对照组患者的百分比。
此外,克罗恩病患者中这些抗体的存在与更严重的表型相关,其特征是小肠受累、疾病进展频繁、病程更长和对手术的需求更大,这表明它们可以用作克罗恩病的预后标志物。
饮食被认为是塑造肠道微生物结构的最关键的环境因素之一。
△ 肥胖:丰富的变形菌为特征
累积证据表明,人类和啮齿动物的健康和肥胖个体的肠道微生物群的分类和功能组成存在差异。
此外,肥胖表型通过粪便移植的传播能力表明肠道微生物群落的改变,作为主要触发因素,是因果关系而不是结果。
肠道微生物群的分类组成失衡,称为生态失调,在代谢紊乱中得到充分证明,并被视为厚壁菌门相对于拟杆菌门的相对丰度增加(F:B 比率)。尽管一致的研究结果普遍支持这一概念,但代谢紊乱期间的生态失调通常包括变形菌的患病率增加。
例如,一项对儿童肠道微生物群的研究发现,与低脂肪、高纤维饮食儿童相比,食用高热量、高脂肪、低纤维饮食的欧洲儿童中的变形杆菌数量更多。
这种差异揭示了肠道微生物群落对非洲儿童饮食的适应性,这可以提高他们从难消化的多糖中获取能量的能力。此外,一些导致有害代谢影响的因素,例如食用无热量的人造甜味剂和乳化剂(通常用作加工食品中的添加剂),也会损害血糖控制并诱发变形杆菌繁殖。
特别是,人造甜味剂介导的肠杆菌科和Delta-proteobacteria类相对丰度的升高与 2 型糖尿病 (T2DM) 患者的结果一致,表明葡萄糖稳态和肠道变形菌之间存在联系。相比之下,证明变形菌的丰度与糖尿病表型呈负相关,挑战代谢疾病患者中高丰度变形菌的概念。
为支持代谢紊乱与变形菌属的扩张之间的关系,变形杆菌属的致肥胖潜力已在无菌小鼠的单关联研究中被确定。
在对一名病态肥胖志愿者进行的减肥试验中,肠杆菌科的相对丰度逐渐减少,假设肠杆菌在代谢恶化中具有致病作用。用从肥胖的人类肠道中分离出来的阴沟肠杆菌B29对无菌小鼠进行单菌定植足以诱导肥胖和胰岛素抵抗。
这一发现支持了这样一个假设,即以丰富的变形菌为特征的不稳定的肠道微生物群落可能代表代谢紊乱的主动特征,而不是被动后果。
△ 营养不良儿童:变形菌成为优势菌
营养不良会导致其他健康问题,例如消瘦和夸希奥科病。在发展中国家,营养不良是威胁 5 岁以下儿童生命的疾病。
营养不良的主要病因是在孕期或产后头 3 年由于大量营养素缺乏和微量营养素缺乏导致的慢性能量负平衡。
然而,最近的研究表明,孟加拉国和马拉维营养不良儿童的肠道微生物群落结构和基因含量与营养良好的儿童不同。在这些研究中,在营养不良的儿童中普遍观察到变形菌的优势和肠道微生物群的低多样性,并被认为是肠道微生物群成熟的障碍。
此外,最近的一项研究揭示了肠杆菌科细菌与营养不良下的肠道黏膜免疫球蛋白 A (IgA) 反应之间存在机制上的相互关系,这会引发肠病并中断黏膜免疫的发展和健康微生物群的组装。
鉴于生态失调驱动的选择压力似乎干扰了微生物群的稳定性,变形菌随后借此机会增加了它们的适应性。微生物群落在异常代谢条件下的不稳定性已被解释为对定植的抵抗力受损。
当接种来自肥胖人类供体的培养细菌(“肥胖受体小鼠”)的无菌小鼠与携带来自瘦肉供体的细菌物种(低脂肪、高纤维饮食)的小鼠共同饲养时,它们被瘦肉有效定殖供体来源的细菌菌株及其肥胖表型得到改善。相比之下,瘦小鼠没有被来自肥胖小鼠的外源或外源细菌菌株定殖。
这一发现表明,生态失调的特点是传播能力减弱和对定植的抵抗力。鉴于 kwashiorkor 儿童的肠道微生物不成熟且富含肠道病原体营养不良被认为与对殖民化的抵抗力有缺陷有关。
总的来说,这一间接证据导致了这样一种观点,即肠道变形菌的扩张反映了宿主的能量不平衡和不稳定的微生物群。有趣的是,在非疾病状态下,如新生儿期和胃绕道手术后也观察到肠道微生物群落的不稳定结构和高丰度的变形菌。
与大多数细菌一样,在细胞外环境中对变形菌的初步识别是通过病原体识别受体 (PRRs) 发生的,PRRs 识别微生物相关分子模式 (MAMPs)——一个包括病原体相关分子模式 (PAMPs) 和危险相关分子模式的统称分子模式(DAMP)。
这些信号受体可分为三个家族:
尽管至关重要的是,只有 TLR 家族参与识别肠细胞表面的细菌配体。
存在于变形菌细胞表面的主要 MAMP 是脂多糖 (LPS) 和鞭毛蛋白,它们分别被 TLR4 和 TLR5 识别。其他参与细菌识别的TLR包括检测细菌脂蛋白的TLR2和检测未甲基化 CpG DNA 的细胞内受体 TLR9。
LPS 的产生和鞭毛组装是在原核生物中观察到的两个最动态的过程,这些结构组成的巨大差异反映在不同变形菌家族成员中观察到的先天免疫反应的强度和方向上。例如,弯曲杆菌和螺杆菌属LPS 与大肠杆菌LPS 的不同之处在于具有更长的酰基链和增加的链连接和脂质 A 磷酸基团的修饰。
在许多病原生物体(例如百日咳杆菌和幽门螺杆菌)中观察到脂质 A 锚中的一个或两个磷酸基团丢失,并且已被证明可提供对抗菌肽的抗性。
参与细菌识别的 TLR 的遗传变异与 IBD 相关。2010 年发表的一项荟萃分析表明,TLR4 Asp299Gly 和 Thr399Ile 变体都赋予白人患克罗恩病和溃疡性结肠炎的统计学显着风险。有趣的是,这两种变体都位于 LPS 结合域内 TLR4 的胞外域,并且被认为会影响蛋白质的二级结构。
这些功能变体的存在已被证明会影响 LPS 反应性,并使个体更容易受到革兰氏阴性菌的感染。证据还表明,这些遗传变异的存在可能会影响基础免疫状态。
因此,有理由推测,在 TLR4 基因变异的携带者中,在营养不良事件之前或期间发生的免疫反应改变,可能足以驱动 IBD 发生不可挽回的免疫反应改变。TLR9 中的遗传变异也与 IBD 易感性增加有关。证据不如TLR4那样令人信服,尽管这一警告可能反映了 TLR9 处理来自所有细菌的配体而 TLR4 反映革兰氏阴性菌易感性的事实。
变形菌门是肠道菌群中四个主要门(厚壁菌门、拟杆菌门、变形菌门和放线菌门)中最不稳定变化最快的门。变形菌门作为一线反应者,对环境因素(如饮食)反应敏感。
总的来说,迄今为止的许多研究都支持这样一个概念,即肠道中大量变形菌反映了生态失调或不稳定的肠道微生物群落结构。除了外源性肠致病性变形杆菌外,健康的哺乳动物肠道还含有数种属于该门的共生细菌,作为其天然肠道菌群。
这些细菌在比例较小时似乎是良性的,而在某些肠道环境下,它们会变成可引发炎症反应甚至代谢障碍。
然而,肠道中变形菌的长期富集可能代表不平衡的不稳定微生物群落结构或宿主的疾病状态。因此,时间顺序监测,而不是横断面研究,可能是根据肠道中变形菌的比例确定疾病风险的更好方法。
在健康肠道中,免疫系统严格调节其反应以维持与共生菌的共生关系。这种可能性表明存在正反馈循环。环境或宿主因素(例如低纤维饮食和急性或慢性炎症)破坏体内平衡,具有选择性并导致肠道内大量变形菌的生态失调。由于宿主无法保持共生的变形菌而导致变形菌的不受控制的扩张,在一小部分和微生物群落对定植的抵抗力降低的情况下,可以进一步促进炎症或外源性病原体的入侵。
因此,切断反馈回路的策略可能包括优化肠道微生物群和宿主之间的伙伴关系。鉴于大多数研究已经在与宿主生理学相关的背景下描述了微生物群落状态,因此对于未来的炎症和代谢干预治疗,首先需要判别变形菌的丰度以及是其是否不受控制扩张,另外需要确定变形杆菌大量繁殖的原因以开发有效的治疗方法。
主要参考文献:
Rizzatti G, Lopetuso LR, Gibiino G, Binda C, Gasbarrini A. Proteobacteria: A Common Factor in Human Diseases. Biomed Res Int. 2017;2017:9351507. doi: 10.1155/2017/9351507. Epub 2017 Nov 2. PMID: 29230419; PMCID: PMC5688358.
Mukhopadhya I, Hansen R, El-Omar EM, Hold GL. IBD-what role do Proteobacteria play? Nat Rev Gastroenterol Hepatol. 2012 Feb 21;9(4):219-30. doi: 10.1038/nrgastro.2012.14. PMID: 22349170.
Litvak Y, Byndloss MX, Tsolis RM, Bäumler AJ. Dysbiotic Proteobacteria expansion: a microbial signature of epithelial dysfunction. Curr Opin Microbiol. 2017 Oct;39:1-6. doi: 10.1016/j.mib.2017.07.003. Epub 2017 Aug 4. PMID: 28783509.
Shin NR, Whon TW, Bae JW. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015 Sep;33(9):496-503. doi: 10.1016/j.tibtech.2015.06.011. Epub 2015 Jul 22. PMID: 26210164.
Shin NR, Whon TW, Bae JW. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015 Sep;33(9):496-503. doi: 10.1016/j.tibtech.2015.06.011. Epub 2015 Jul 22. PMID: 26210164.
Rigottier-Gois L. Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. ISME J. 2013 Jul;7(7):1256-61. doi: 10.1038/ismej.2013.80. Epub 2013 May 16. PMID: 23677008; PMCID: PMC3695303.