-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康

16S科研项目是一个完整的闭环,前期的课题项目设计方案、取样和重复实验设置决定了后期分析报告的数据完整性和项目类型。
想要拿到一手有利用价值的科研报告和项目数据,前期的实验方案设计和后续的分析都起着关键性的作用。
然而有时候拿到报告不知道如何去解读,这里为大家梳理一下16s科研项目的全过程,帮助大家更好的了解报告内容,快速获取关键信息。


实验方案设计就像一个总工程的设计图纸,决定了未来科研分析报告的类型走向,并且前期的分组设计的越详细,各种理化指标、生化指标、代谢物等信息准备越充分,后续报告的完整度越高。


明确项目课题类型
第一步要做的就是明确项目课题类型:
最常见的就是多分组之间差异分析比较:例如,要比较对照组、模型组、实验组,之间的差异结果。
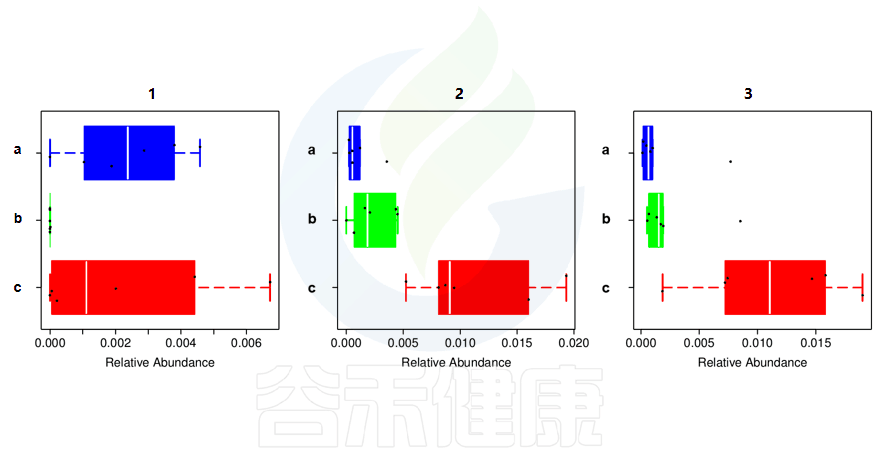

还有多分组中,任意两组之间比较:例如某实验设计了正常组、疾病组、用药组服用奥氮平、阿立哌唑、氨磺必利、利培酮,像比较不同的用药组和疾病组之间的菌群的差异结果,就用到了分组之间两两差异比较。
✦举个例子
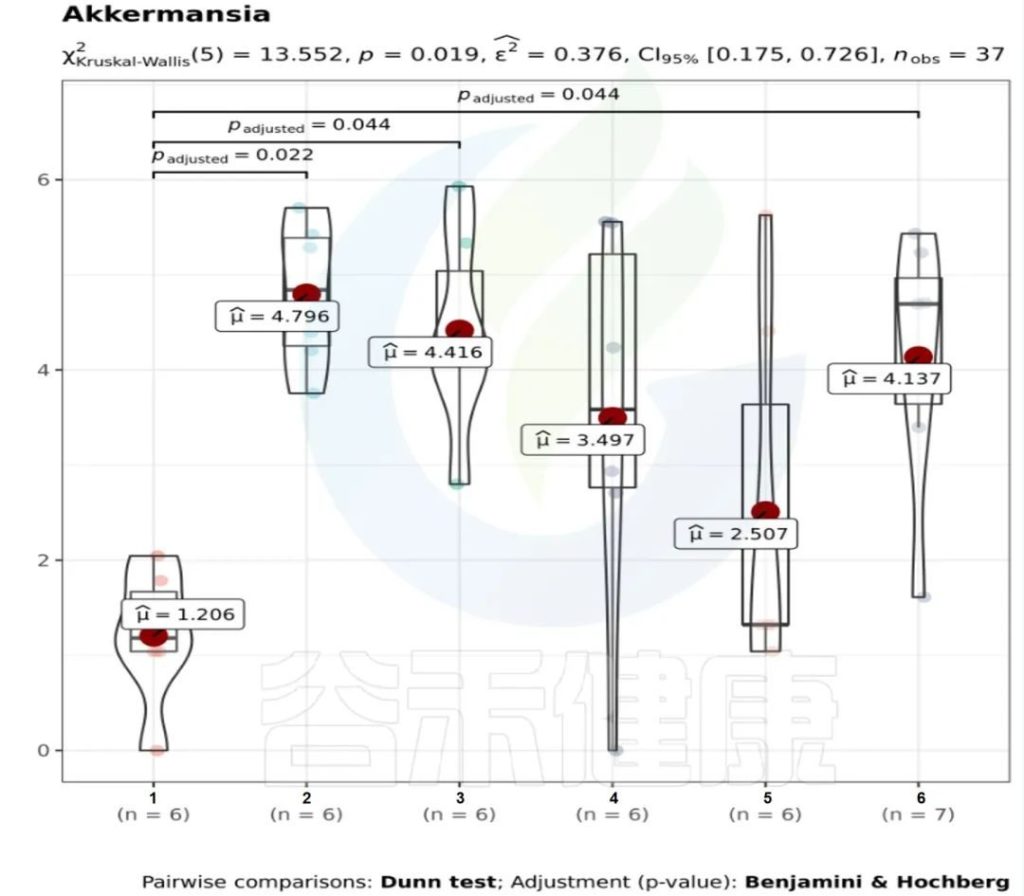
图中1组与3组、4组、6组 组间差异显著
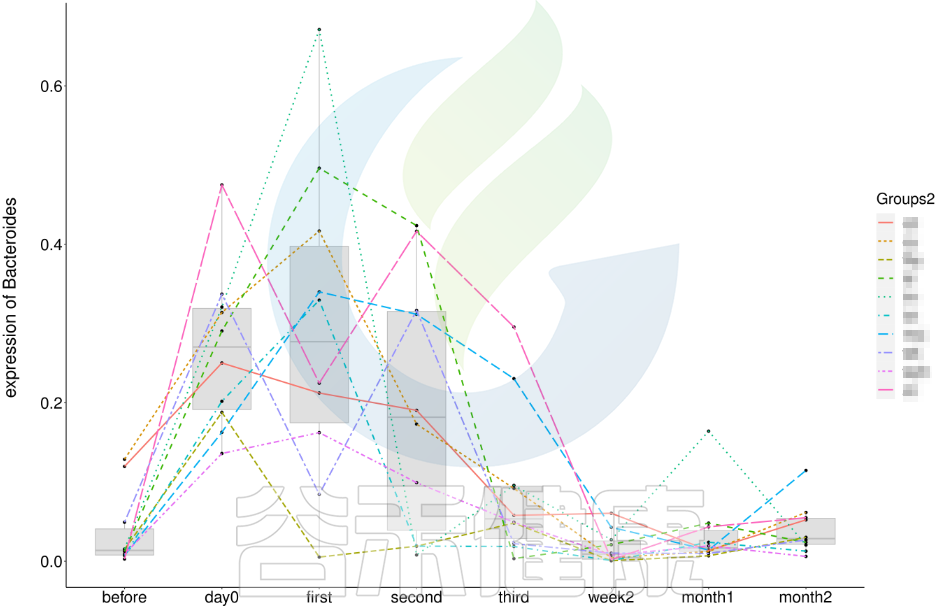
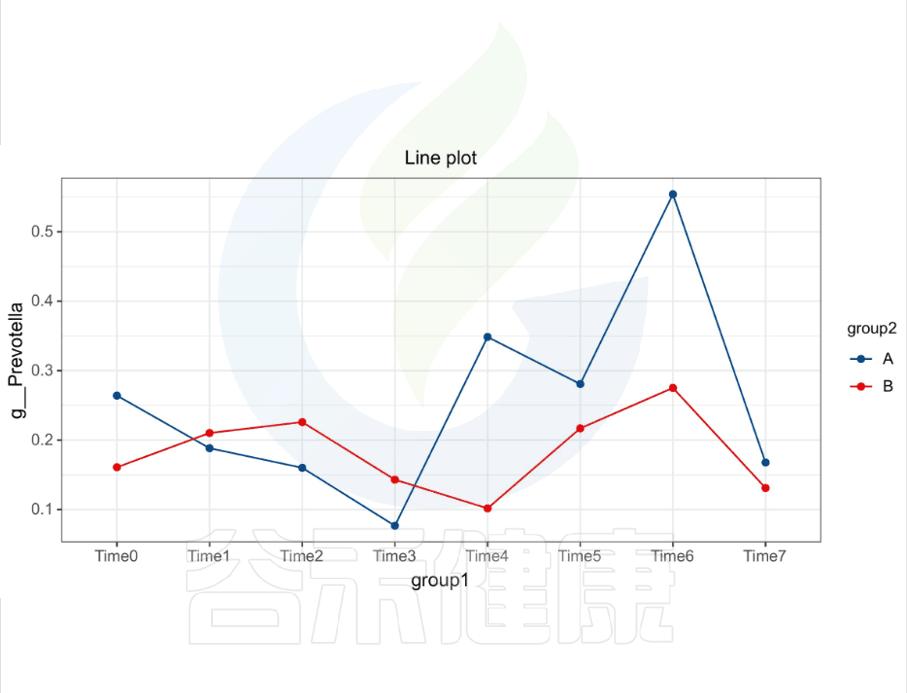
还有随时间的变化比较菌群之间的变化规律:例如在用药不同时间段包括3天,5天,2周,1个月,2个月,观察菌群的变化情况。如下图所示:
收集理化指标非常重要
如果前期搜集好每个样本的相关理化指标,还可以计算这些指标与菌群之间是否具有相关性。
✦举个例子
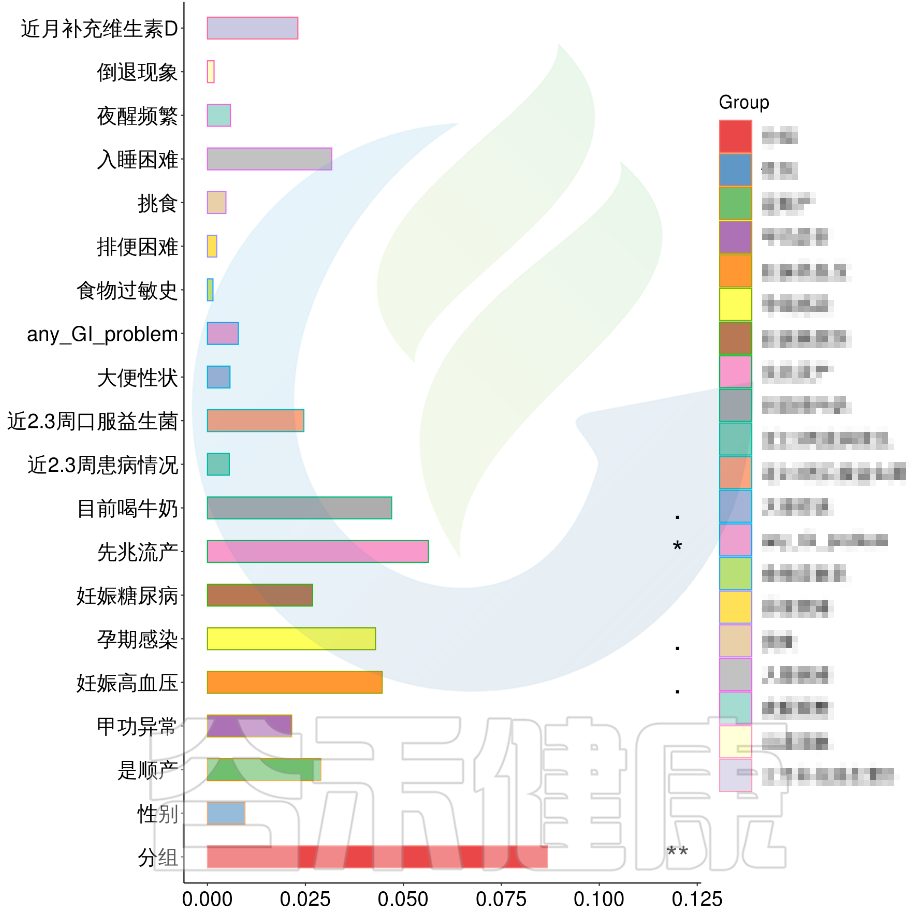
例如该项目比较自闭症儿童与正常儿童的菌群差异。客户在样本信息单里还详细搜集了母孕期的各种详细指标,例如孕期天数、出生体重、白细胞介素6、肿瘤坏死因子a、五羟色氨等数值型理化指标。
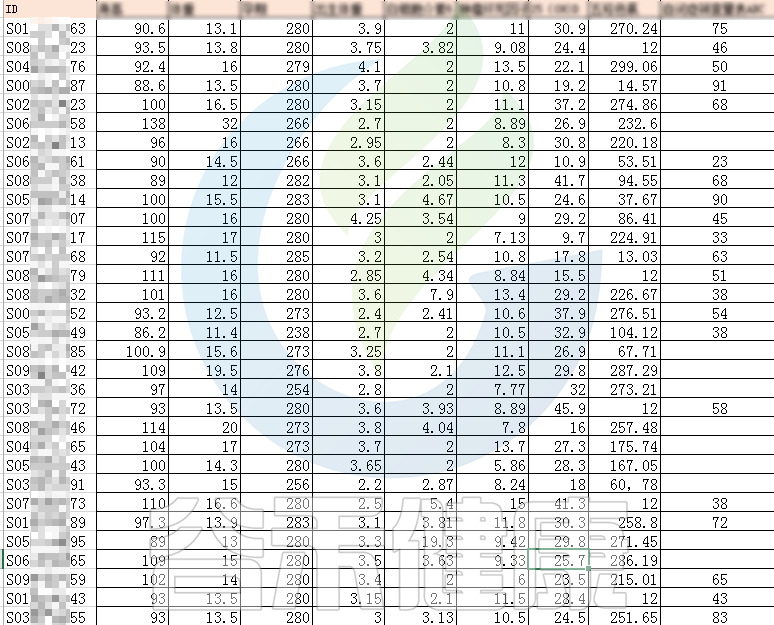
还搜集了是否顺产、是否妊娠高血压、是否孕期感染、是否妊娠糖尿病、是否先兆流产等因子型理化指标。其中0代表否,1代表是:
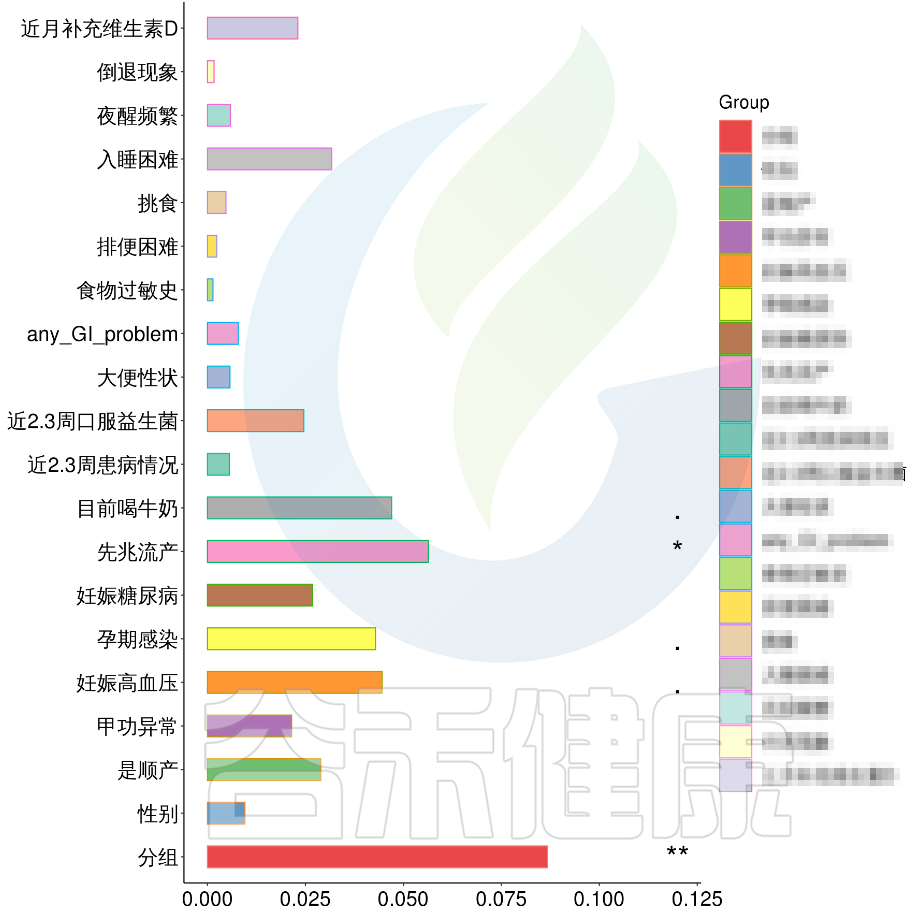
根据这些理化指标与菌群数据做相关性分析,从因子型的结果可以看出,自闭症(ASD)与正常儿童之间的分组与菌群之间相关性极显著**,其次是否有先兆流产的分组与菌群之间有显著相关性*,其他的包括是否喝牛奶、孕期是否感染、妊娠高血压都与菌群有相关性。
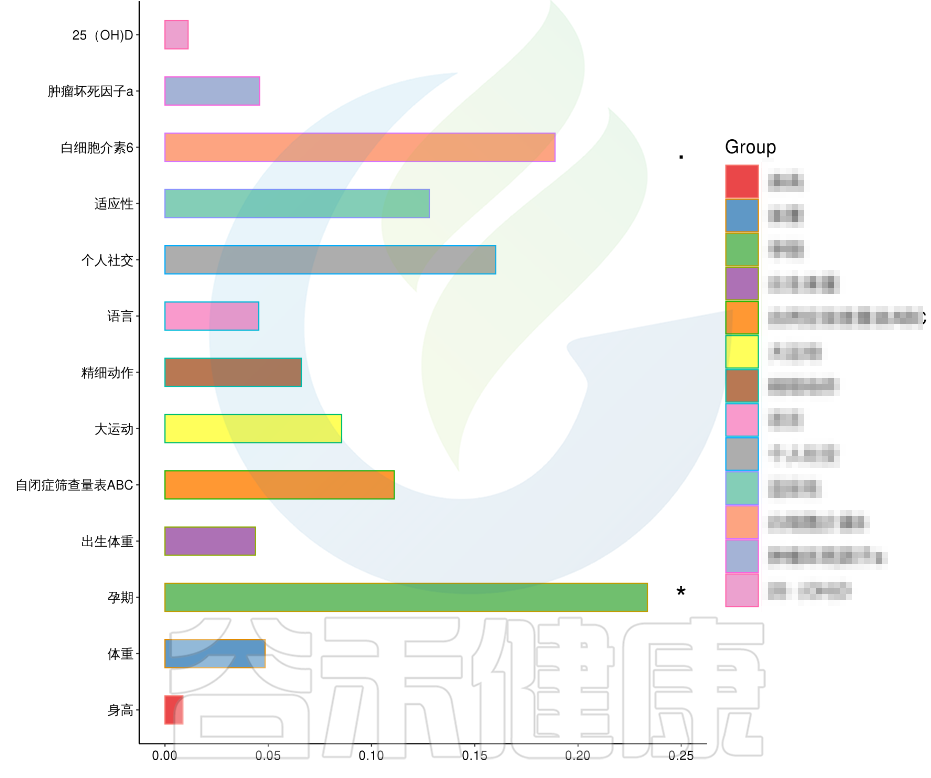
在数值型理化指标中,孕期的天数与菌群之间相关性显著*,其次是白细胞介素6与菌群之间有相关性。
小结
因此,前期搜集相关资料越详细充分,对分析报告的完整性也会有帮助,分析人员也会根据您的样本信息单提供的相关内容,做出个性化的分析和售后指导建议。
首先基于样本类型,最常见的环境样本来源是人体、动物、土壤、水体等。而人体中的肠道菌群样本是目前研究最广泛,可鉴定的物种也最为丰富,谷禾在肠道菌群与人体健康方面有深入研究,目前已完成超20万例临床肠道菌群样本检测,并构建了超过60万各类人群粪便样本数据库。
其他样本类型还包括人体/动物唾液样本、组织样本、尿液样本等。
▸ 粪便样本
目前粪便样本从采样到提取数据分析技术较为成熟、应用较为广泛,谷禾最早在15年就开发了针对粪便样本的取样管,也是最早致力于研发粪菌取样盒的公司,方便实验室、个人日常取样需求,实现了粪菌样本的常温运输。
谷禾取样管常温保存,取样也较为方便卫生,在家就可以轻松完成,相较于传统取样方法都有所升级。并且该取样管也有专利证书。该取样方法被大量客户采用并接纳,大大降低了采集粪便样本的难度,缩短了搜集样本的时间周期。
取样示意图

▸ 其他样本
土壤样本也相对较为容易提取出DNA,但需要注意的是土壤样本的菌群特征容易受植物腐殖质基因的影响和干扰,所以提取时要进行纯化。
而口腔、组织、尿液等样本,由于DNA含量较少,在实验阶段提取相对较为困难,所以提前准备样本时,尽量多取一些,并且可以多取几个重复,尽量避免扩增不出来的情况。
并且这些样还很容易受到环境样的污染,所以在实验阶段,可以取空白样本,和阳性样本ST做对照,数据分析时可以用来纯化样本,排除来自环境的干扰序列。
✦组间差异分析需重复取样
要做组间差异分析时,每组要重复取样,才能做组与组之间的统计检验。理论上,每个组至少3个样就满足基本的统计差异分析需求。所以在重复取样时,每个分组至少取3个样。取样时要保证每个分组内部的样本一致性,如果组内样本之间的个体差异性较大,则会影响后期组间差异结果分析。
✦举个例子
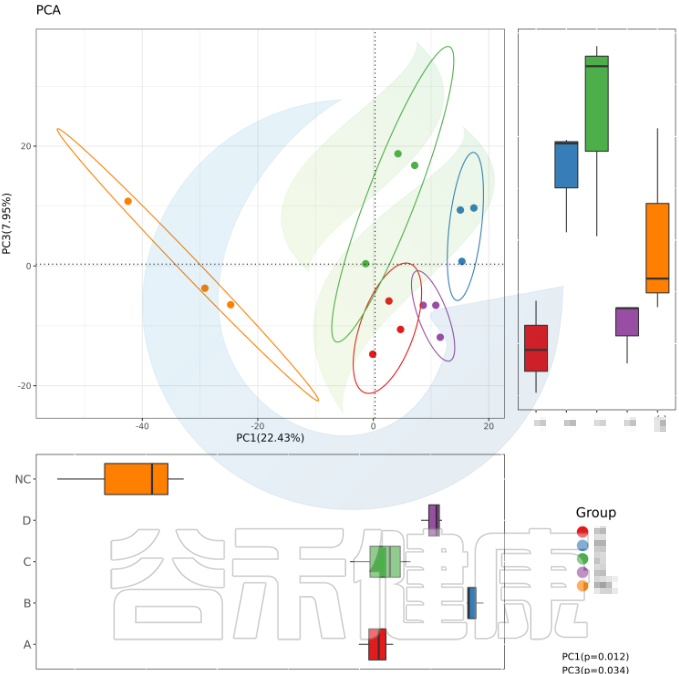
例如从该图可以看出,分组之间组间差异较大,并且组内的样本之间较为接近和相似。
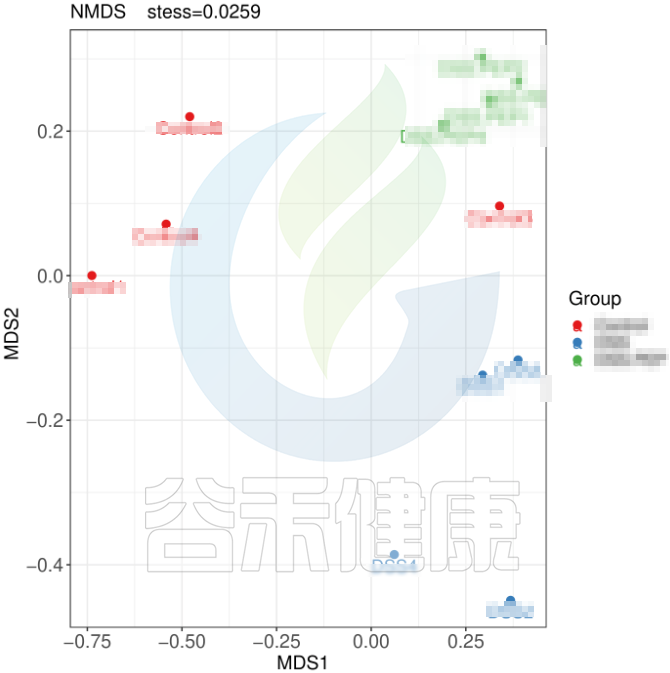
但从该图可以看出,Control组中Control3样本明显与组内的其他样本差异较大,与DSS组内的样本较为相近,这样就对后期组间差异分析的时候会产生影响,需要将该样本去除。
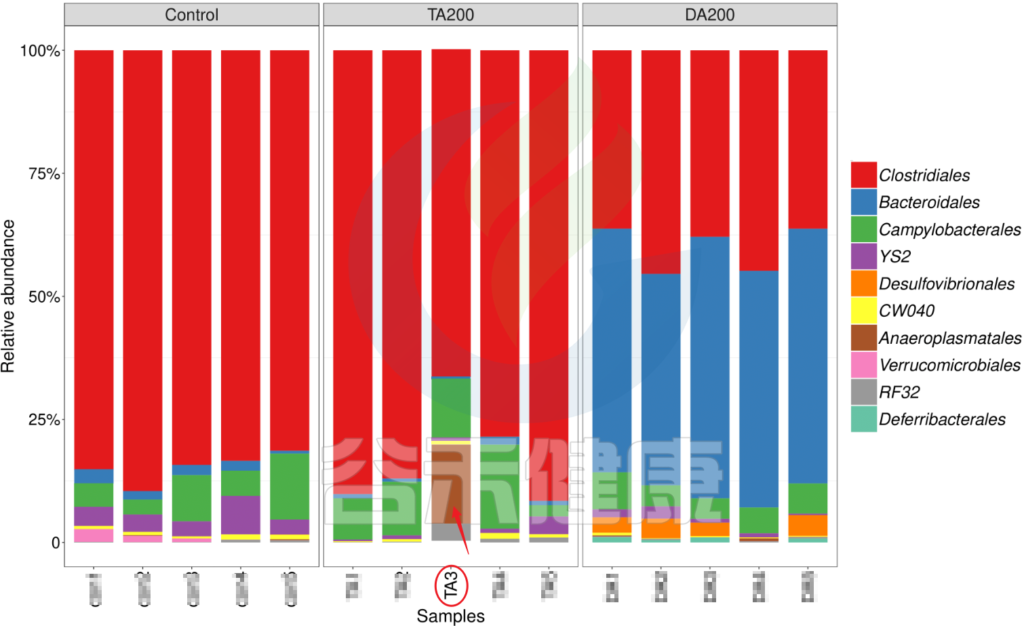
又例如在该图中,TA200组中的TA3样本的Anaeroplasmatales物种丰度含量非常高,该样本与组内的其他样本明显差异较大,该样本可能受到环境污染等其他因素干扰,这样就没有办法保证组内样本的均一性,也会影响分组之间的差异分析统计结果,再后期分析的时候建议把该样本去掉重新分析。
建议
为了便于后期数据整理修改,每个分组需要保留一定量的重复样本,假如每个分组只取了3次重复,假如其中有一个样本质量不好需要去除,该分组只剩2个样本,则不满足每组至少3个样的分组条件,整体就没有办法做组间差异分析统计。
所以这里建议每个分组至少取5个样做重复,一般6到10个样就能分析出比较完善的结果。具体分组和组内的重复取样数量视具体的实验设计方案而定。
在经费允许的情况下,建议多取一些重复。假设每组取50到100个重复或者以上,得到的分析结果就基本可以涵盖该分组情况所有的菌群构成情况,可以较为全面的研究分组之间的菌群构成差异情况。
当拿到16S科研分析报告以后,面对纷繁复杂,各式各样的图表分析结果犯了难,不知道如何从这么多的图表中入手,快速找到报告中需要的图表结果。
这里对16S科研分析结果抽丝剥茧,概括出报告中的主要几大内容板块。
•16S科研分析究竟是在做什么?
16S rDNA 是一种对特定环境样品中所有的细菌进行高通量测序,以研究环境样品中微生物群体的组成,解读微生物群体的多样性、丰富度及群体结构,探究微生物与环境或宿主之间的关系的技术。
主要是对原始数据进行拼接过滤得到的优化序列,降噪方法得到ASV,再对ASV进行物种注释,注释到门、纲、目、科、属、种各层次上的分类结果。
通过ASV表计算Alpha多样性,样本内的多样性指数,Beta多样性,样本间相似性的指标。
对ASV表进行功能预测,例如Picrust2功能预测分析、Bugbase菌群表型特征分析,FAPROTAX生态功能预测等。
得到的每个样的数据结果,根据客户提供的分组情况和理化指标,进一步做组间差异分析,以及和环境理化指标之间做关联分析,相关性分析,比较分组之间是否有差异,差异是否显著,来验证分组是否合理,和环境宿主之间是否有关联性。
原始数据处理
Illumina NovaSeq测序平台测序得到的双端数据Raw PE,经过拼接和质控,根据一定的标准过滤掉低质量数据、接头或PCR错误,得到Raw Tags。再经过去重复序列,去singleton序列,过滤嵌合体,得到可用于后续分析的有效数据 Effective Tags。
OTU(ASV) 表生成
微生物多样性分析中最重要的就是OTU特征表,一切后续分析都围绕OTU表来进行。生成OTU除了传统的聚类的方法(一般按照97%的相似度进行聚类),现在最新用到的技术的是降噪的方法得到ASV。
简单来讲ASV就是在去除了错误序列之后,将Identity的标准设为100%进行聚类,常见的有DADA2、Deblur、Unoise三种降噪方法。项目里用到的是UNOISE2降噪方法获得ASV数据。
物种的分类与注释
采用QIIME2训练分类器方法对ASVs代表序列进行分类学注释,默认选用SILVA138数据库进行物种注释。并在各个分类水平上:domain(域),phylum(门),class(纲),order (目),family(科),genus(属),species(种)对每个样本的群落组成统计。
alpha多样性
Alpha多样性主要反映样本内多样性。对ASV表进行计算可以获得每个样本的simpson,ace,shannon,chao1以及goods_coverage等指数,alpha多样性指数用来来评估样本菌群物种的丰富度(richness)和多样性(diversity)
beta多样性
Beta多样性反映的是样本间多样性,Beta多样性是衡量个体间微生物组成相似性的一个指标。通过计算样本间距离可以获得β多样性矩阵,基于OTU的群落比较方法报告中给出了,欧式距离、bray curtis距离、Unweighted UniFrac距离和Weighted UniFrac距离等。
功能预测
得到群落的微生物组成之后,也可以对群落功能组成进行预测,常用的16S功能预测的相关软件有PICRUSt2、FAPROTAX、BugBase。
PICRUSt2用来预测功能,通常指的是基因家族,PICRUSt2支持基于多个基因家族数据库的预测,报告中包括了KEGG同源基因,KO直系同源物,EC酶分类编号,MetaCyc途径的丰度,CAZy碳水化合物活性酶数据库,GMM是肠道代谢模块和GBM是肠脑模块。
FAPROTAX是原核的微生物注释代谢或其他生态相关的功能(例如硝化,反硝化,发酵)的一个数据库和软件。FAPROTAX预测的功能主要集中在海洋、湖泊等环境样本微生物的功能,特别是硫、碳、氢、氮的循环功能。
BugBase能进行表型预测,其中表型类型包括革兰氏阳性(Gram Positive)、革兰氏阴性(Gram Negative)、生物膜形成(Biofilm Forming)、致病性(Pathogenic)、移动元件(Mobile Element Containing)、氧需求(Oxygen Utilizing,包括Aerobic、Anaerobic、facultatively anaerobic)及氧化胁迫耐受(Oxidative Stress Tolerant)等7类。
以上这些部分,我们通过数据处理分析,得到了每个样本相关的大量数据结果,包括每个样本的序列统计、ASVs表格、物种分类注释统计、alpha多样性指数、beta多样性指数、功能预测等。这些数据主要集中在报告里的这些内容:
▸ 科研分析报告结果文件夹
01_pick_otu/ 文件夹主要是对样本ASV表格统计
02_sequence_statistic/ 文件夹是对样本序列数据的统计
03_diversity-metrics / 文件夹是对样本的alpha多样性指数、beta多样性指数的统计
04_Taxonomic/ 文件夹是对物种分类注释的统计(门到种水平)
Picurst2/ 文件夹是Picrust2功能预测得到的每个样本的相关功能预测数据
Groups/ 文件夹下是对组间差异分析结果

红框是样本个体的相关数据统计,Group是分组比较
根据以上常规分析得到的相关数据进行作图,其路径也在对应文件夹下,可以打开 分析报告.html 有相关分析的图表和对应文件的详细介绍和路径说明。
★拿到样本后需要进行统计分析
当我们拿到这些样本大量的数据结果,之后关键的一步就是做对这些数据进行处理,做统计分析,比较分组之间的差异结果,找出菌群和环境之间的关联性等,对数据进一步做研究,找出课题方案对应的结果。
不同的数据用到的统计检验方法也不太一样,接下来我们对报告中的不同的分析结果对应的统计差异分析方法进行介绍说明。
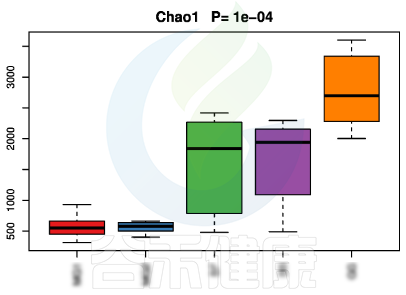
▸ alpha多样性
alpha多样性指数组间差异统计分析用到的检验方法是:单因素方差分析(如果只有两个分组,用Wilcoxon秩和检验,3个及以上的分组用Kruskal-Wallis 检验),图上方显示P值
▸ beta多样性
beta多样性指数的统计检验方法有ANOSIM相似性分析和Adonis多元方差分析,这两种都是基于距离矩阵的检验方法。
✦Anosim相似性分析
Anosim分析是一种非参数检验,用来检验组间的差异是否显著大于组内差异,从而判断分组是否有意义。
报告中给出了加权距离和非加权距离的Anosim结果图,图中给出了R值和P值。
R值用于比较不同组间是否存在差异,R-value 介于(-1,1)之间,R-value > 0,说明组间差异大于组内差异。R-value < 0,说明组间差异小于组内差异。R只是组间是否有差异的数值表示,并不提供显著性说明。
统计分析的可信度用 P-value 表示,P< 0.05 表示统计具有显著性。
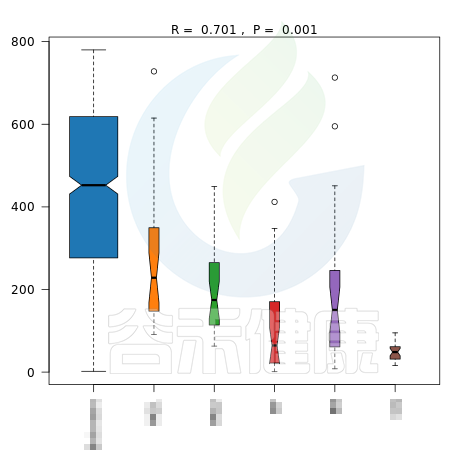
图中能看出R>0,说明组间差异大于组内差异,P<0.05 ,说明差异显著,证明该分组情况效果较好。
✦Adonis多元方差分析
Adonis多元方差分析,其实就是PERMANOVA,亦可称为非参数多元方差分析。
其原理是利用距离矩阵(比如基于Bray-Curtis距离、Euclidean距离)对总方差进行分解,分析不同分组因素对样品差异的解释度,并使用置换检验对其统计学意义进行显著性分析。
它与Anosim的用途相似,也能够给出不同分组因素对样品差异的解释度(R值)与分组显著性(P值)。
报告中PCoA bray距离、PCoA weighted_unifrac距离、PCoA unweighted_unifrac距离的图片右下角有给出PERMANOVA检验的P值和R值。
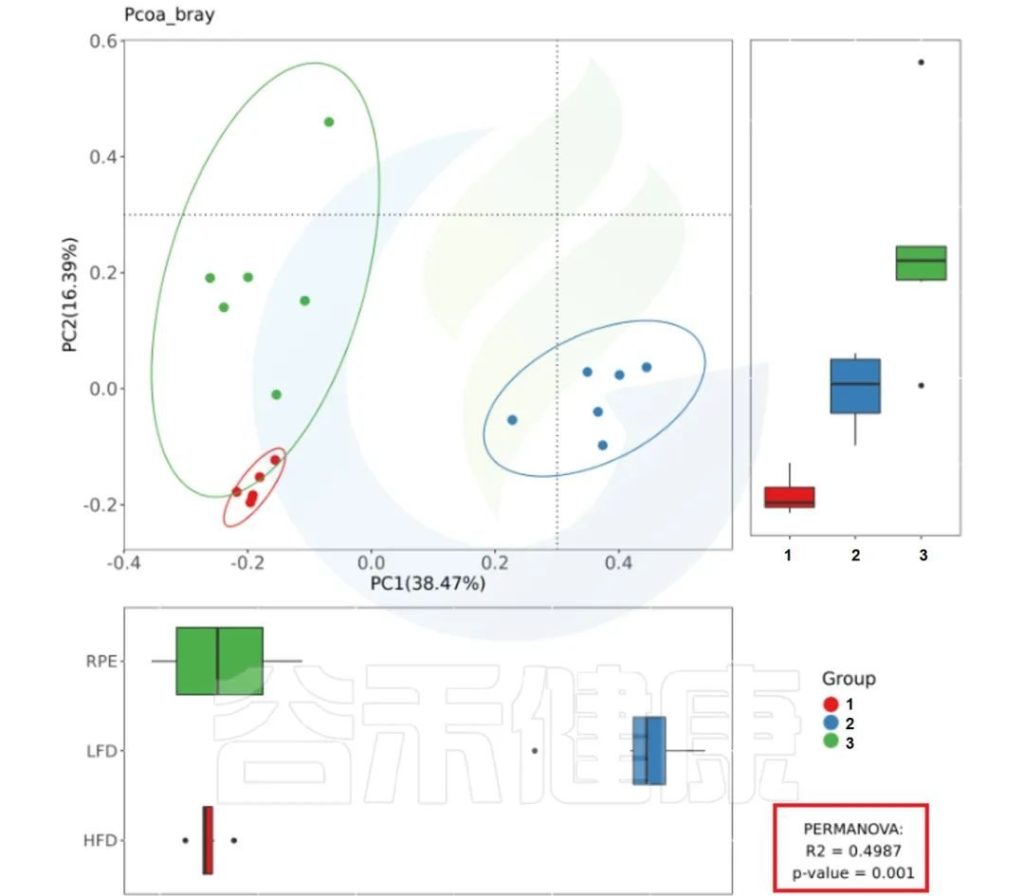
图中看出PCoa bray距离得到的检验P<0.05 组间差异显著,并且分组之间区分较为明显。
PCoa bray距离的PERMANOVA检验结果路径:
多组间检验结果:
Groups/betadiv/pcoa_bray_analysis/PERMANOVA.result_all.csv
两组间检验结果:
Groups/betadiv/pcoa_bray_analysis/ PERMANOVA_paired_result.csv

不同分类水平下的检验方法
在很多分析报告当中,例如在不同疾病的肠道菌群分组中,本身样本个体之间肠道菌群的物种多样性,丰富度差异并不大,alpha多样性组间差异并不显著,beta多样性分组间区分不是很明显,这样就需要进一步找出分组之间的差异物种或者差异功能来进行分析。
对于不同分类水平的物种和功能预测结果用到以下几种检验方法:
Tukey检验
Tukey主要应用于3组或以上的多重比较,适合于各组例数相等的每两两分组之间比较。
Tukey检验的一个重要的优点是非常简单,而且所需实验样本相对较少。
其检验结果的可信度达到95%的置信水平时,最少的情况下只需6个样本进行验证(改善前3个样本、改善后3个样本)。
•举个例子
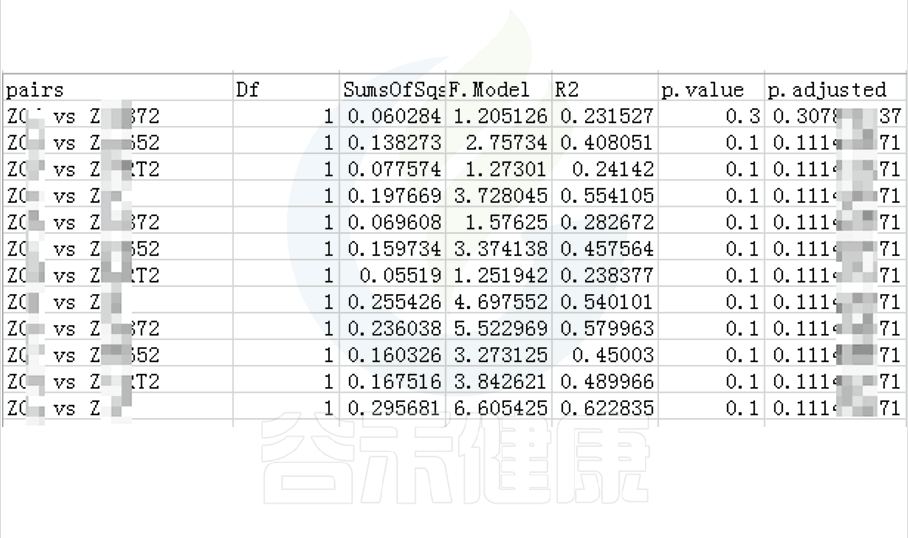
图中的字母代表显著性差异的字母表示法,只要含有相同的字母,就表明两组之间没有显著性差异。
例如a和ab含有相同字母“a”,表示两组之间没有显著性差异。ab中的“b”表示这一组和其他含有字母b的组(比如bc)没有显著性差异,但是a和bc就有显著性差异了。
图中只展示Tukey检验差异显著的物种或功能,如果数量较多,则只展示前10个。
路径:Groups/diff_analysis/TukeyHSD/
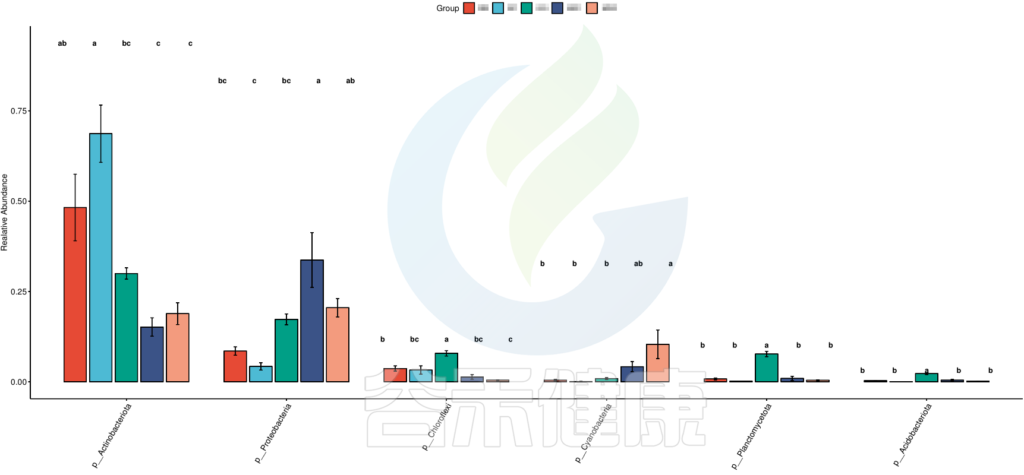
图中显示的都是Tukey检验组间差异显著的物种,依次按照丰度从高到底排列,如果差异结果较大,则显示前10个物种。例如在该图中,Tukey检验结果,门水平物种Actinobacteriota在BB与MG1组、BB与MG2、BF与GG组、BF与MG1组、BF与MG2组,这些分组之间组间差异显著。
组间差异箱型图
组间差异箱型图用到的检验方法是通过单因素方差检验(只有两个分组,用的是Wilcoxon秩和检验,3个及以上的分组用的是Kruskal-Wallis 检验),Var检验和one-way相结合,筛选出组间差异性物种。
路径:Groups/diff_analysis/TaxaMarkers
图中每一个箱型图代表一个组间差异显著的物种
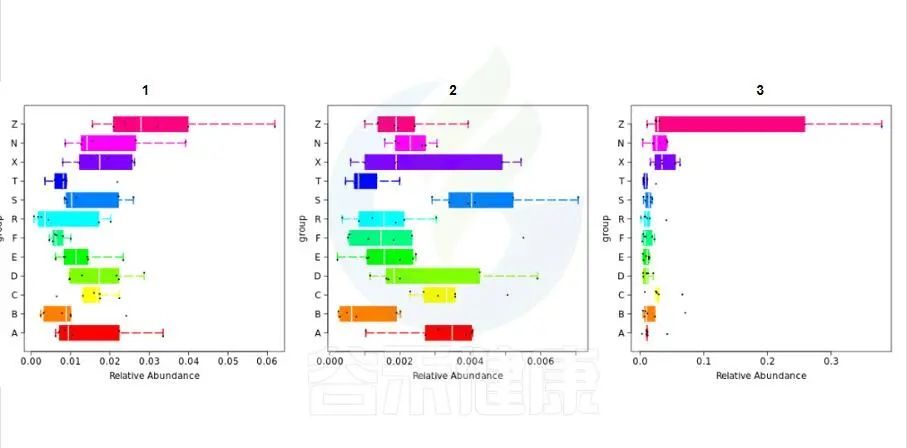
图中显示的都是统计方法得到的差异显著的物种,图中能看出这3个物种分组之间差异显著。
命名格式是,例如:Cen_Nitrosopumilus 指的是,当前分类水平(属水平)的名字 g__Nitrosopu 加上一级分类水平(科水平)的名字 f__Cenarchaeaceae 的前 3 个字母简写Cen,如果当前水平没有注释到名字则以全称的名字表示。
统计结果表:Groups/diff_analysis/TaxaMarkers/ xxx.Groups.sig.meanTests.csv
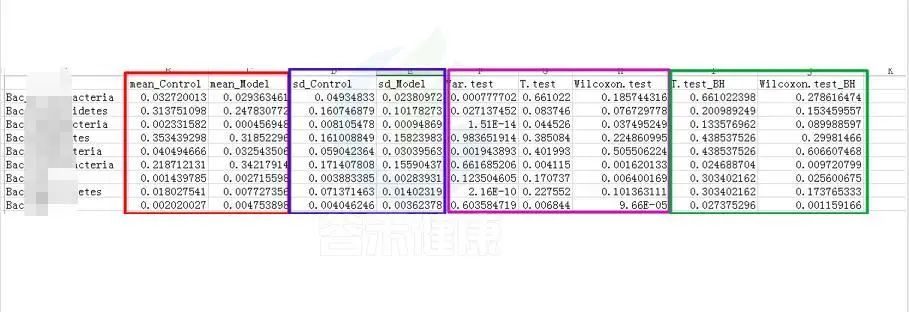
例如这是一个表格的截图
红框 mean_ 是分组组间的平均值
蓝框 sd_ 代表组间的标准差
粉色 .test 代表不同统计检验结果的P-value P值,这里有var检验 T 检验 Wilcoxon检验(或Kruskal-Wallis 检验)
绿色 _BH 例如Wilcoxon.test_BH代表Wilcoxon.test检验BH矫正的Q-value,Q值
UnivarTest检验(单因素方差分析)
单因素方差分析是指如果只有两个分组,用Wilcoxon秩和检验,3个及以上的分组用Kruskal-Wallis 检验。
路径:Groups/diff_analysis/UnivarTestXXX
Groups\diff_analysis\UnivarTestKEGG\figure 文件夹下有做成柱状图、箱型图和单个物种之间的图,其中有横着排列和竖着排列的,有用原始值计算的,还有对原始值取log后进行统计的。图中只展示Univar 检验组间差异显著的物种/功能。
统计结果表:Groups/diff_analysis/UnivarTestXXX/ UnivarTest_sign.txt
•举个例子
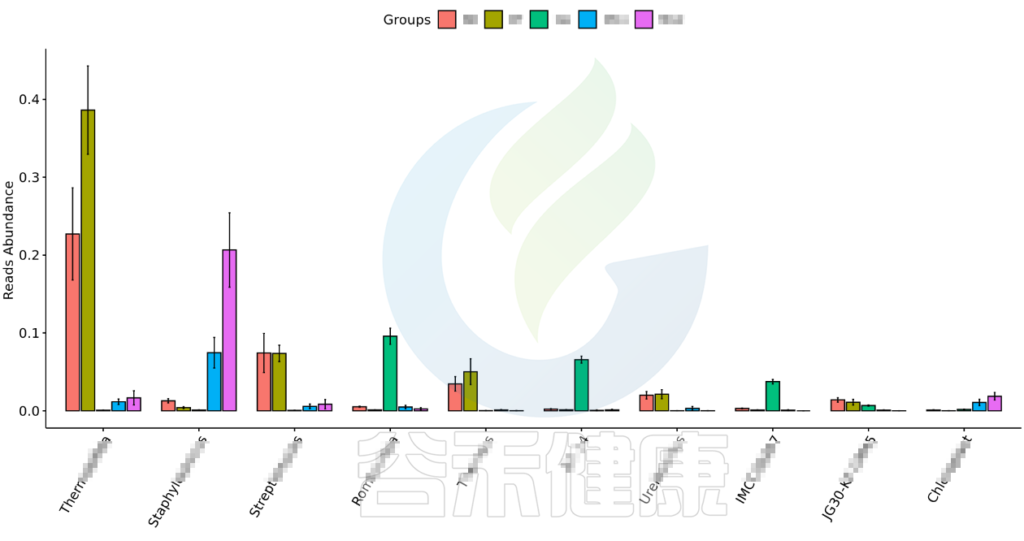
图中显示的是该统计检验差异显著的物种的柱状图或箱型图,按照丰度从高到低排列,如果差异物种/功能较大,则只显示前10个。例如该图中Therobifida、Staphylococcus、Streptomyces等物种用Kruskal-Wallis 检验得到的组间显著差异物种。
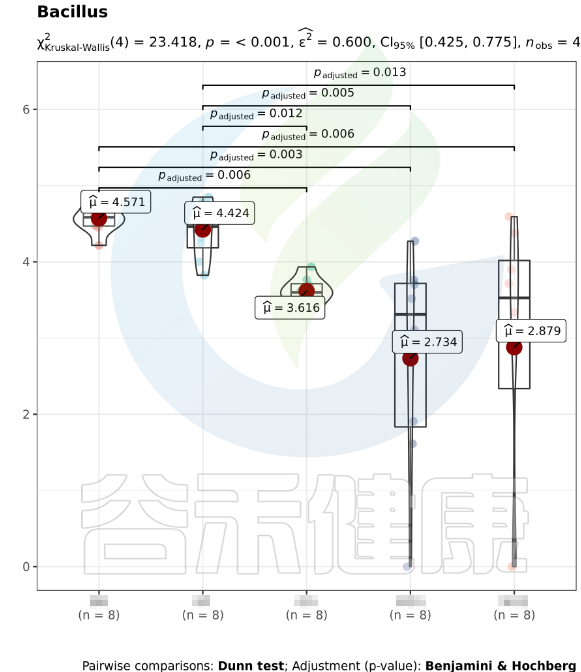
该图展示了Bacillus物种Kruskal-Wallis 检验差异结果,所有分组中P<0,001 多组间差异显著,两组间BB与GG、BB与MG1、BB与MG2、BF与GG、BF与MG1、BF与MG2,组间差异显著。
LEfse分析
LEfse分析即LDA Effect Size分析,是一种用于发现和解释高维度数据生物标识(基因、通路和分类单元等)的分析工具,可以进行两个或多个分组的比较,它强调统计意义和生物相关性,能够在组与组之间寻找具有统计学差异的生物标识(Biomarker)。
LEfSe用到的统计分析方法是将线性判别分析与非参数的Kruskal-Wallis以及Wilcoxon秩和检验相结合。
LEfse分析结果中一般会出现两个图,一张表( LDA值分布柱状图、进化分支图以及特征表)。
LDA值分布柱状图
这个条形图主要为我们展示了LDA score大于预设值的显著差异物种,即具有统计学差异的Biomaker,默认值为2.0(看横坐标,只有LDA值的绝对值大于2才会显示在图中);柱状图的颜色代表各自的分组,长短代表的是LDA score,即不同组间显著差异物种的影响程度。
路径:
Group/Lefse_Analysis/out_formant.cladogram.png
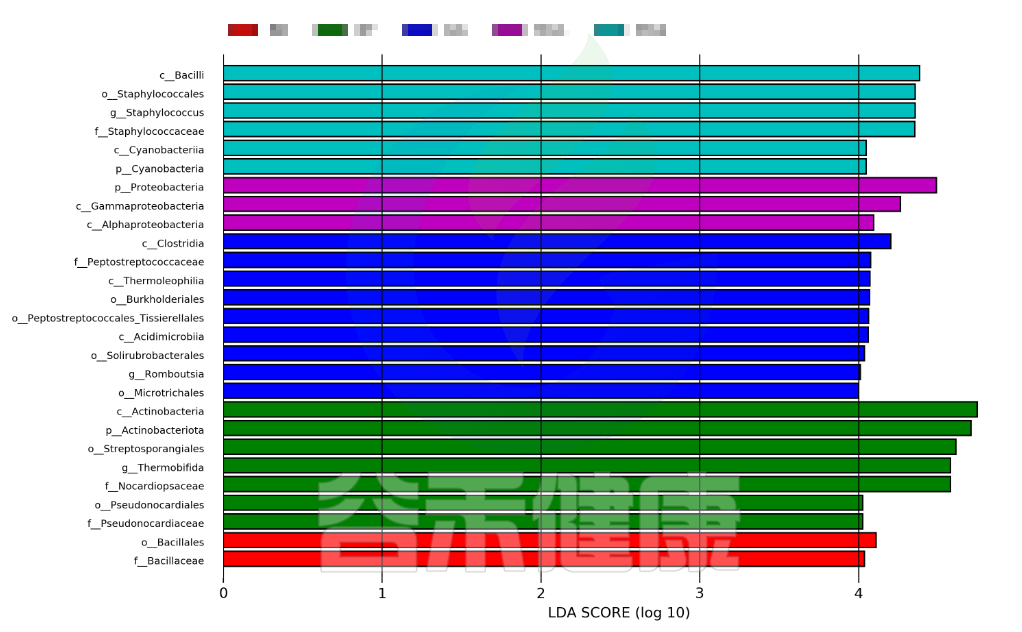
图中展示了不同分组特有的Lefse组间差异标记物,例如BB组的标记物是目水平的Bacillales和科水平的Bacillaceae,不同的分组标记物也不同,图中如果只展示了部分分组,则代表只有部分分组通过Lefse分析筛选出组间差异标记物。
进化分支图
小圆圈: 图中由内至外辐射的圆圈代表了由门至属的分类级别(最里面的那个黄圈圈是界)。不同分类级别上的每一个小圆圈代表该水平下的一个分类,小圆圈的直径大小代表了相对丰度的大小。
颜色: 无显著差异的物种统一着色为黄色,差异显著的物种Biomarker跟随组别进行着色,红色节点表示在红色组别中起到重要作用的微生物类群,蓝色节点表示在蓝色组别中起到重要作用的微生物类群。
未能在图中显示的Biomarker对应的物种名会展示在右侧,字母编号与图中对应(为了美观,右侧默认只显示门到科的差异物种)。
路径:Group/Lefse_Analysis/out_formant.png
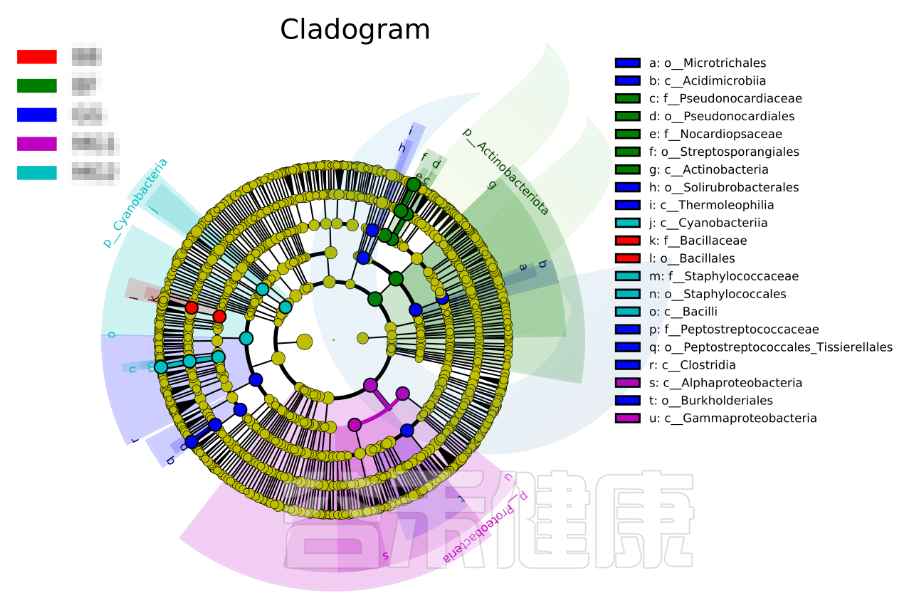
图中右侧展示了分支图中的字母对应的物种信息,例如a 代表GG组的标记物目水平的Microtrichales ,b代表GG组的标记物刚水平的Acidimicrobiia。在分支图的最外层显示的是各分组门水平物种的标记物,例如BF组的是Actinobacteriota、MG1组的是Proteobacteria、
MG2组的是Cyanobacteria
特征表
路径:Group/Lefse_Analysis/out_formant.res.csv
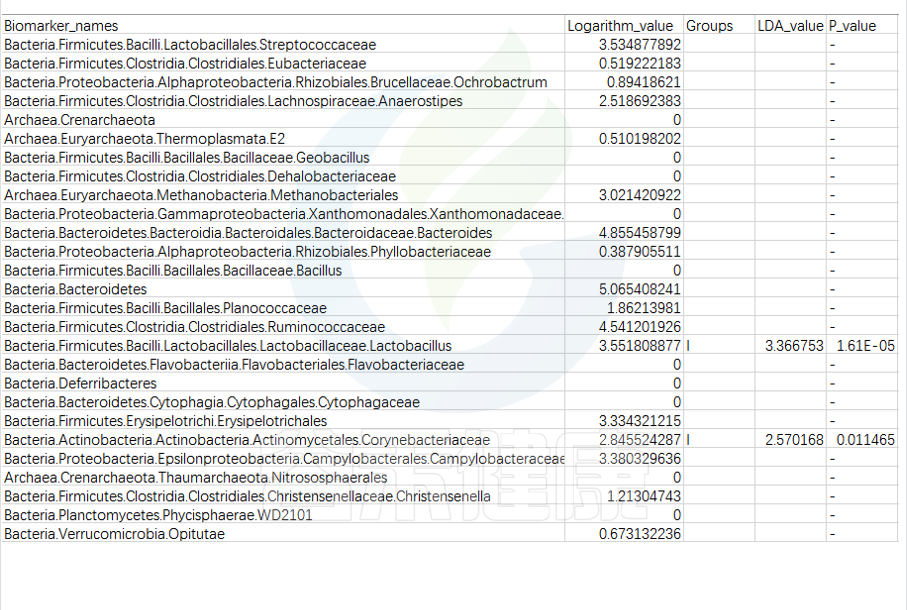
第一列是样本中从门到属水平所有分类单位的列表
Lefse会逐一判断这些分类单位的在分组之间是否具有统计学显著性差异。
第二列:各组分丰度平均值中最大值的log10,如果平均丰度小于10的按照10来计算;如果该分类单位未体现出显著组间差异,则后三列为空。
对于具有统计学差异的分类单位:
第三列:差异基因或物种富集平均丰度最高的分组组名;
第四列:LDA差异分析的对数得分值;
第五列:Kruskal-Wallis秩和检验的p值,若不是Biomarker用“-”表示。
默认LDA>2,P<0.05
通常根据第4列的LDA差异分析对数得分值和第五列的P值,可以描述组间具有显著差异的分类单位统计学效力强弱。
metagenomeSeq
metagenomeSeq是用R开发的一个包,metagenomeSeq的基本思想,用normalization实现分类注释时的biases处理,同时用零膨胀高斯分布(zero-flated Gaussian distribution)处理了测序深度所带来的影响,在此基础上,利用线性模型找到存在的差异所在。
路径:Groups/diff_analysis/ metagenomeRXXX
metagenomeSeq 差异显著物种/功能 热图
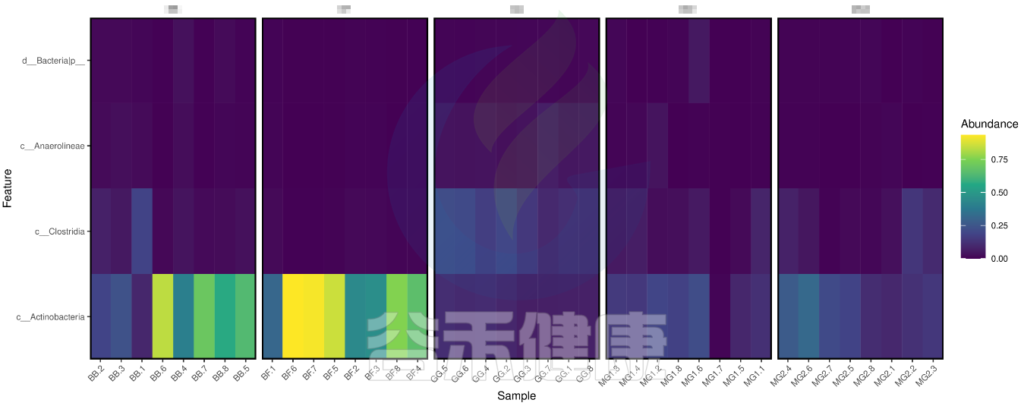
图中颜色越深相关性越小,颜色越接近黄色相关性越大,从图中能看出Actinobacteria物种与BB组和BF组相关性较大。
metagenomeSeq差异菌属于功能代谢关联分析
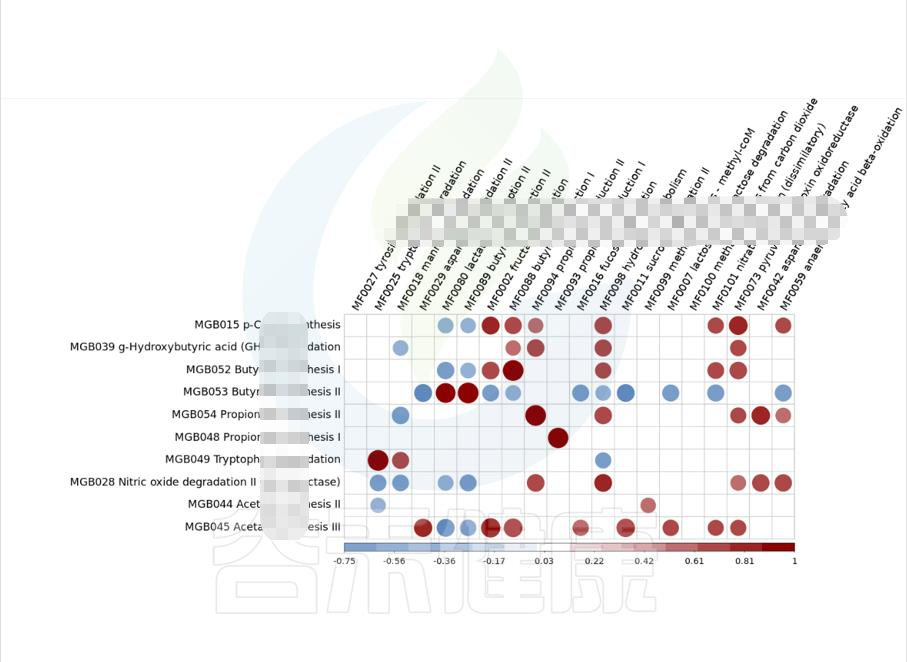
图中红色代表正相关,蓝色代表负相关,颜色越深,圆圈越大,相关性也越大,例如图中能看出MGB049余MF0025 之间成正相关,且相关性较大。
随机森林模型
一种非线性分类器,随机森林属于集成类型的机器学习算法,挖掘变量之间复杂的非线性的相互依赖关系。通过随机森林重要性点图,可以找出分组间差异的关键物种/功能。
反映了分类器中对分类效果起主要作用的特征,按重要性从大到小排列。
Error rate:表示使用下方的特征进行随机森林方法预测分类的错误率,数值越高表示基于特征分类准确度不高,可能分组之间特征不明显。分值越低证明分组效果比较好。
•举个例子
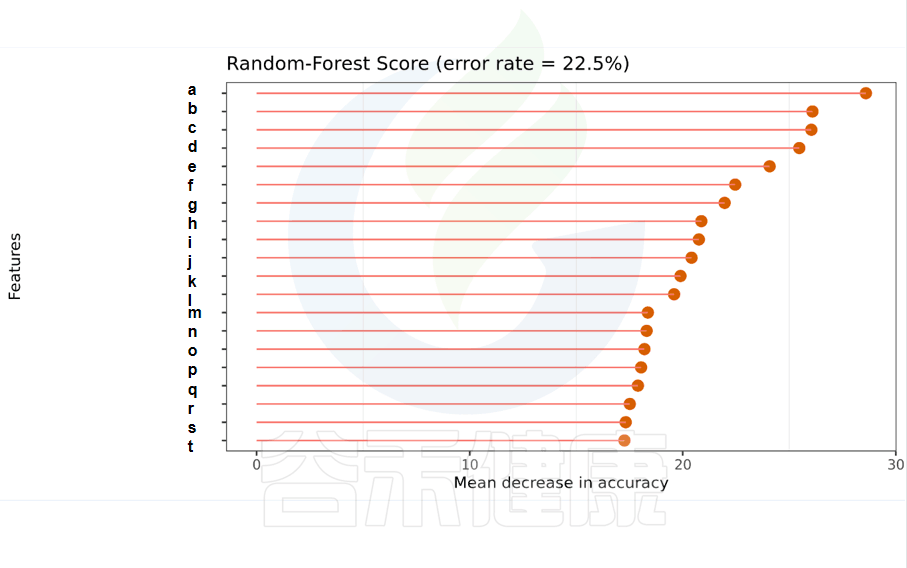
图中按照随机森林模型效果筛选出的对分组效果有重要性作用的物种,按照重要性从高到低进行排列,例如图中最终要的是a,依次往下是b、c等。错误率较小,表明该分组效果较好。
ROC曲线
ROC曲线分析是一种常用的统计学分析方法,在医学研究中主要用于评价诊断试验的效能。在16S测序报告中,我们通过绘制ROC曲线,并计算ROC曲线下面积(AUC),来确定分组对于菌群是否有诊断价值。
ROC曲线图是反映敏感性与特异性之间关系的曲线。ROC曲线下的面积值在1.0和0.5之间。在 AUC>0.5的情况下,AUC越接近于1,说明诊断效果越好。
AUC在0.5~0.7时有较低准确性,AUC在0.7~0.9时有一定准确性,AUC在0.9以上时有较高准确性。AUC=0.5时,说明诊断方法完全不起作用,无诊断价值。AUC<0.5不符合真实情况,在实际中极少出现。
•举个例子
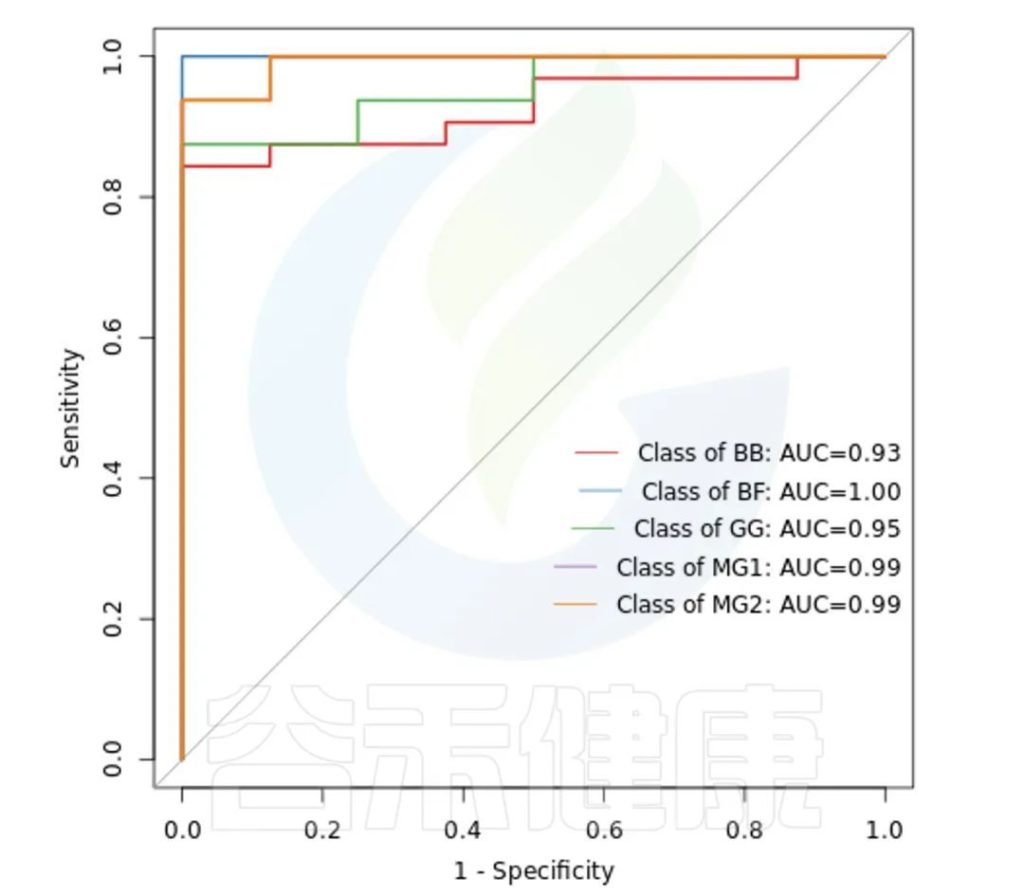
从图中能看出各分组的AUC都大于大于0.9,各分组的分组效果较好,BF组AUC等于1,该分组效果最好,可能样本之间较为相近,并且跟其他分组组间差异也比较大。
以上是组间统计差异的方法介绍,其他的还包括关联分析。
例如客户提供了每个样的相关理化指标数据,想计算这些指标与均属之间有什么相关性,就可以做一下分析。
关联性分析
✦相关性热图
图中X轴代表属水平物种,Y轴代表代谢指标,红色代表正相关,蓝色代表负相关,**代表相关极显著P<0.01,* 代表相关性显著P<0.05相关性具有统计学意义。
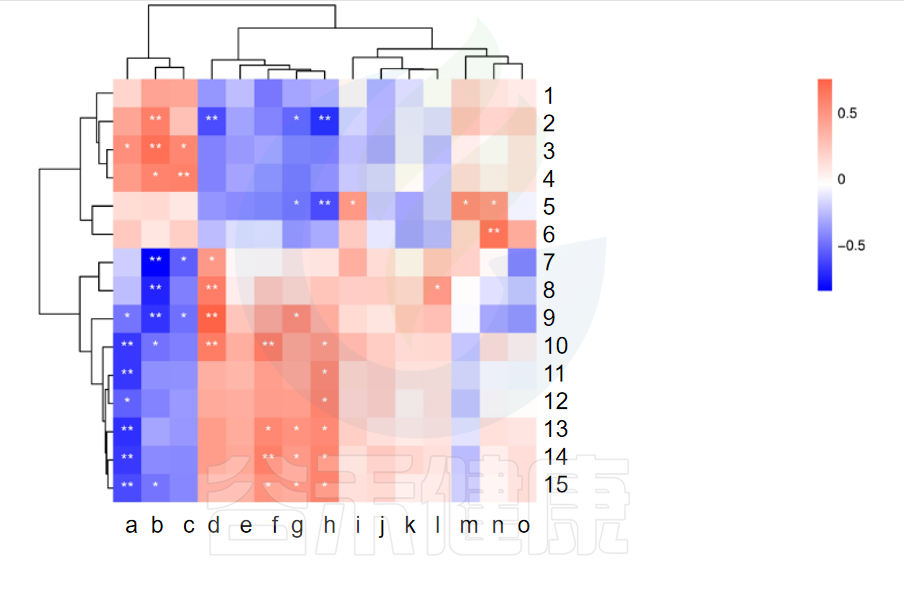
例如从该图中能看出6与n物种成正相关,并且相关性极显著**,7与b物种成负相关,并且相关性极显著**。
可以得到表格:任意菌属和代谢的相关性的值和P值
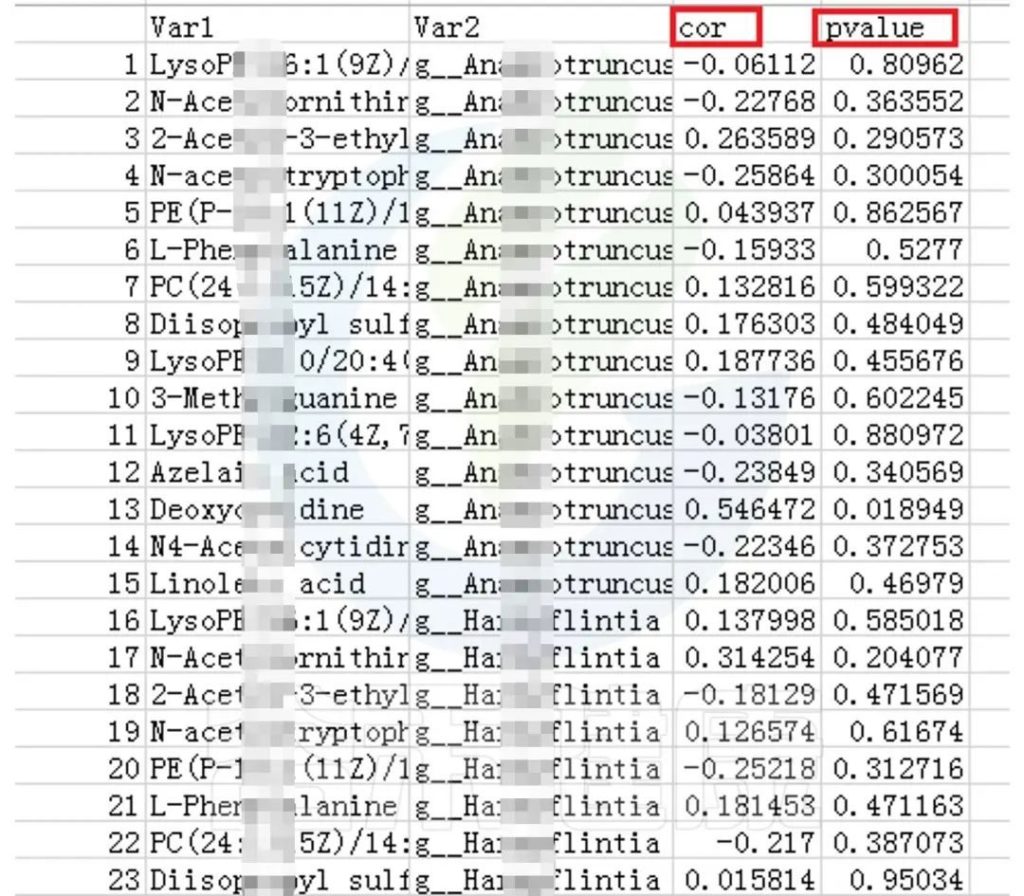
✦CCA图
可以分析样本、菌群、理化指标之间的关联关系。图中使用点代表不同的样本,从原点发出的箭头代表不同的环境因子。
箭头的长度越长,表示环境因子的影响越大;夹角越小,代表相关性越高。样本点与箭头距离越近,该环境因子对样本的作用越强。
图像中坐标轴标签中的数值,代表了坐标轴所代表的环境因子组合对物种群落变化的解释比例。
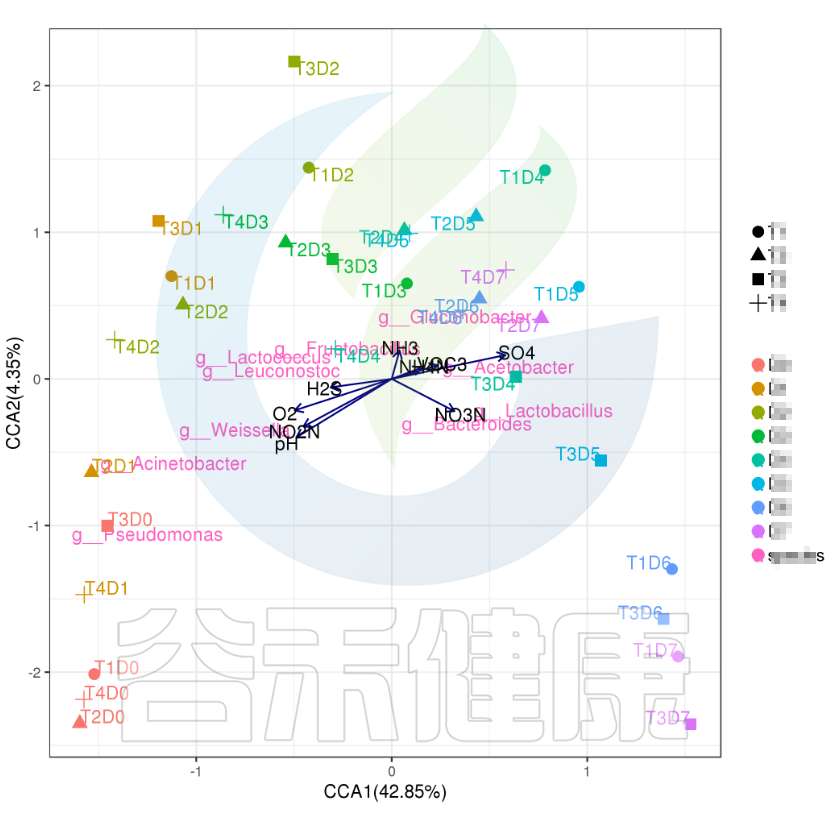
例如从图中能看出pH 、NO2N、02与 Acinetobacter、Weissella等物种成正相关,与T3D0、T1D0、T4D0等D0组的样本成正相关。
✦RDA 冗余分析

例如从图中能看出pH与Helicobacer物种成正相关,相关性较大,pH与NC组有一定的相关性。
✦Envfit分析
回归拟合分析结果:
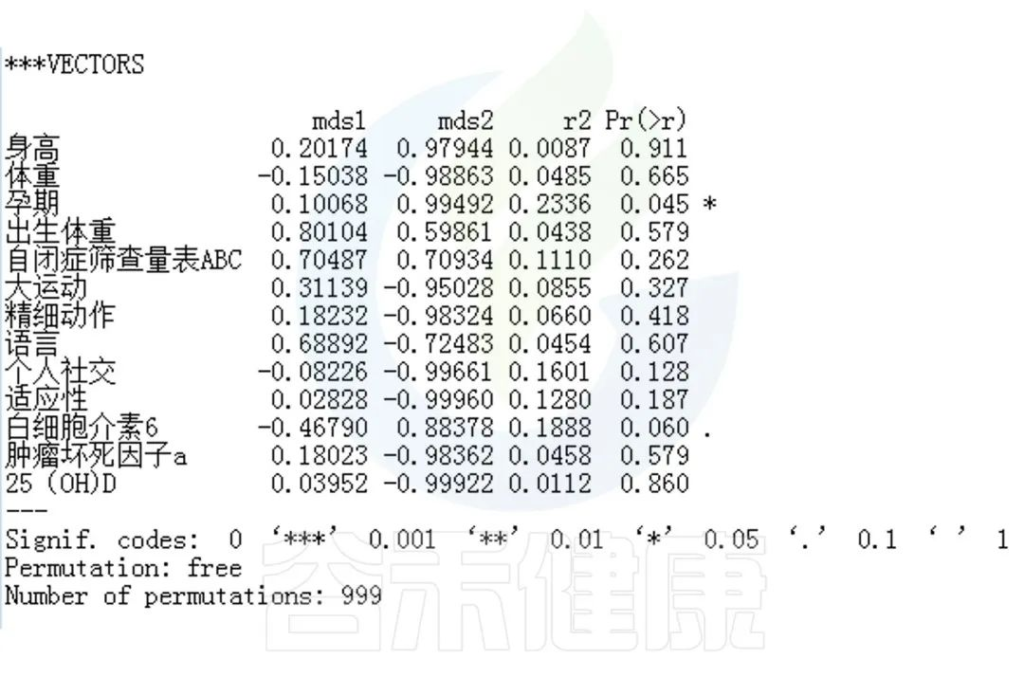
图中能看出ASD与正常儿童之间的分组与菌群之间相关性极显著**,其次是否有先兆流产的分组与菌群之间有显著相关性*,其他的包括是否喝牛奶、孕期是否感染、妊娠高血压都与菌群有相关性。
环境因子与功能/物种的相关性线形图P<0.05显著,图中红色点代表正相关,绿色点代表负相关,灰色相关性不显著。
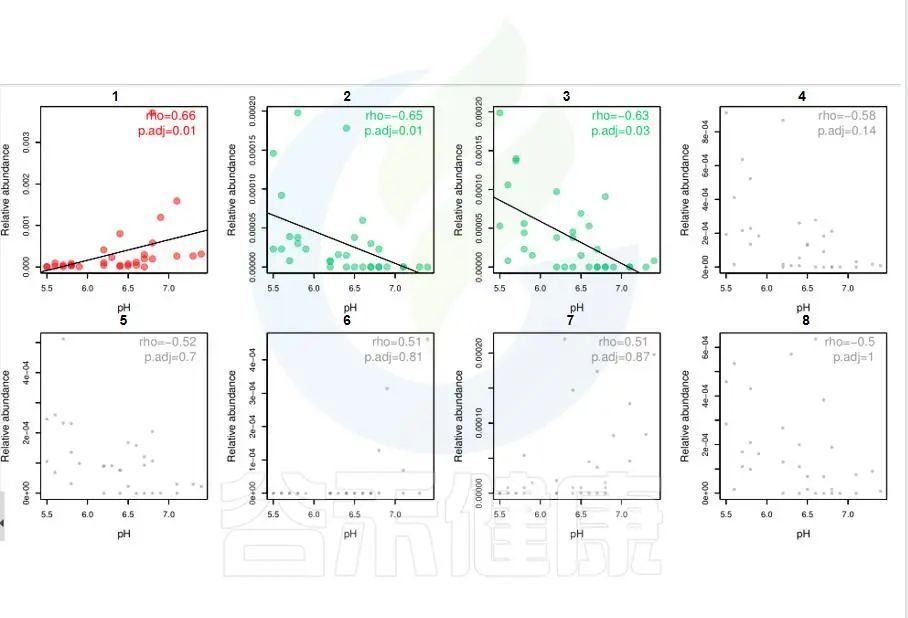
图中能看出pH 与Candidatus Rhabdochlamydia 之间成正相关,且相关性显著,pH 与Sinorhizobium、Euzebya 之间成负相关,切相关性显著。
✦Network网络分析
还可以做菌属之间的网络分析关联图,共发生网络图为研究复杂微生物环境的群落结构和功能提供了新的视角。
由于不同环境下微生物的共发生关系截然不同,通过物种共发生网络图,可以直观看出不同环境因素对微生物适应性的影响,以及某个环境下占互作主导地位的优势物种、互作紧密的物种群,这些优势物种以及物种群往往对维持该环境的微生物群落结构和功能稳定发挥着独特以及重要的作用。
•举个例子
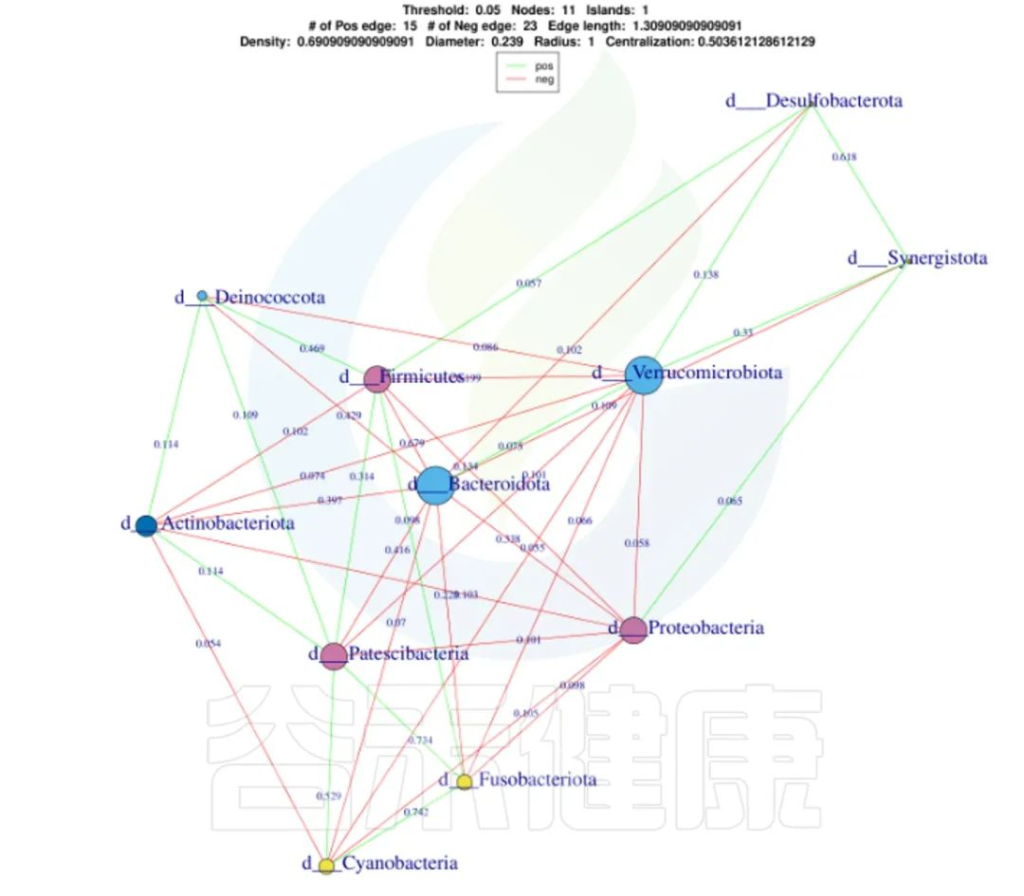
图中展示了相关性的物种,例如Bacteroidota、Actinobacteriota、Proteobacteria 这些物种与其他物种相关较大,图中这些物种与其他物种连线较多,字体比较大也代表相关性较强,例如Actinobacteriota与Deinococcota连线是绿色的代表这两个物种是负相关。
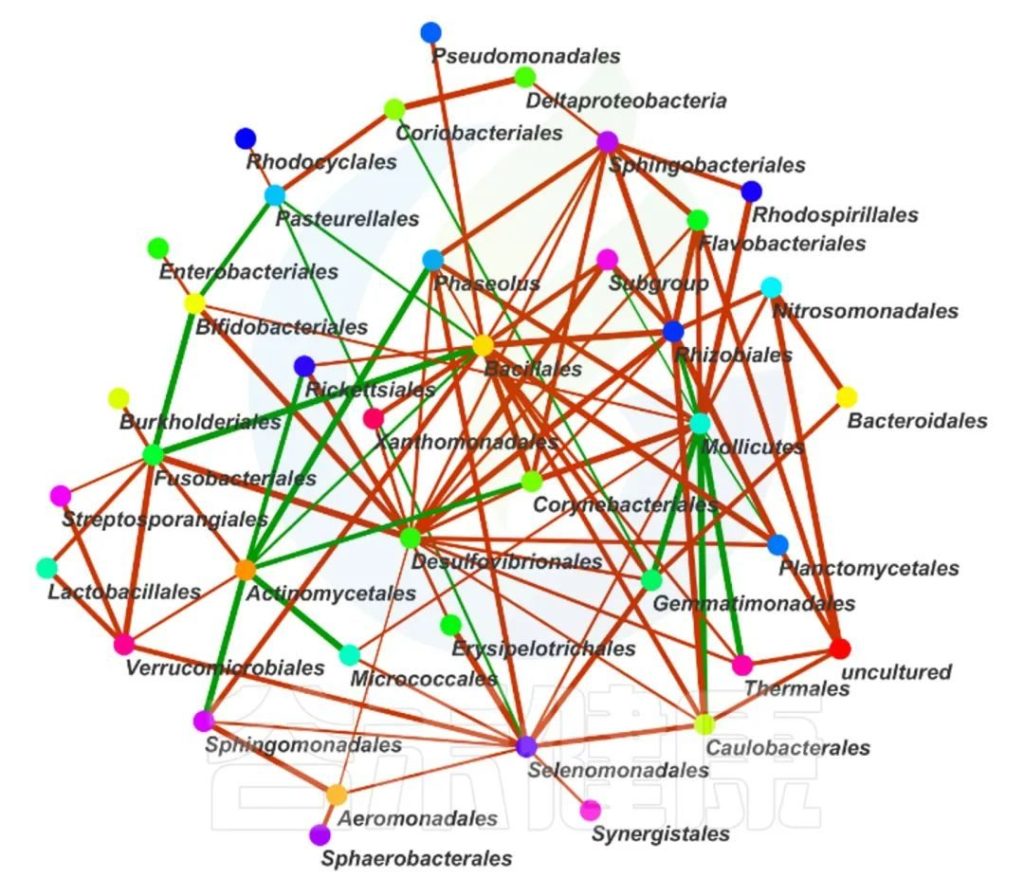
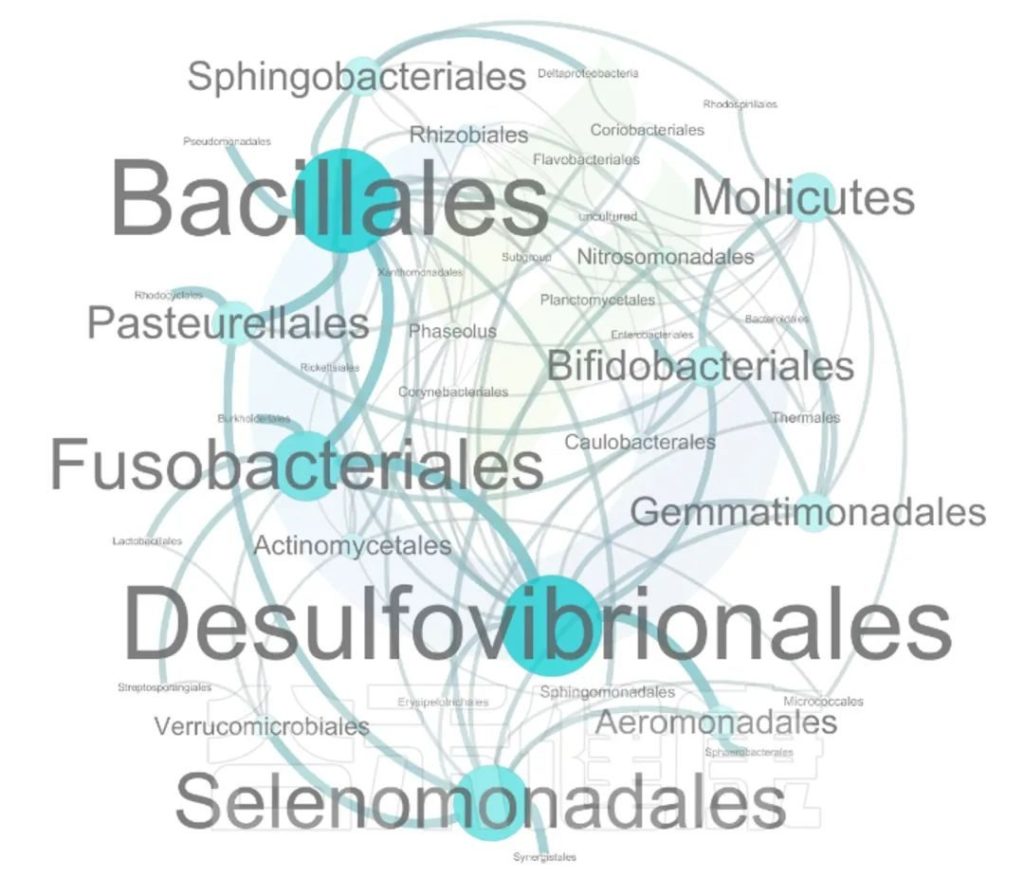
这两个图类似的物种相关性的图,用同一个数据做出来的,图中能看出Bacillales、Desulfovibrionales、Selenomonadales与其他物种相关性较强。
报告中已经基本都涵盖了16S科研数据分析所需要的图表、差异统计,以及相关性分析结果。如果在几种不同类型的统计方法对比之下有略微的差异结果,选取其中一组差异结果即可。
报告里涵盖了大部分16S所需要的图片,不过也有个别个性化的图需要单独用到软件去做,可以单独完成个性化图表生成。
随着16s分析报告的不断升级,报告中的图表以及相应的解读也会越来越精细完善,谷禾也将尽可能为大家的科研之路带来更多便利。


谷禾健康

统计和科学编程已迅速成为科学中的一项必要技能,而这一般都需要用到——R语言。
R语言以其简单易学、免费开源的特性,正在各个领域发挥着越来越重要的作用。


由于其强大的统计计算和数据可视化两大功能,可以说在生信领域, R语言是干活的法宝。


本文分享的是来自一些自学R语言的研究人员认为有用的技巧,希望能为已经上路的R语言的自学者,看不到清晰道路的R语言小白、以及广大想要学习R语言的生物科研人员提供一些帮助。
注意:
这10条技巧并不是提供使用 R 的技术指导,而是提供了构建或磨练 R 编程技能的实用策略。
学习 R 就是学习一门新语言,包括词汇、语法、句法,甚至可能是一种新的思维方式,打开一个新的世界。
可以这样说,学习 R 语言很困难,因为它涉及经常报错,所以这个学习的过程要做好准备,识别这些错误,并最终学习如何修复。


▸不怕犯错,永远没有完美
即使是最有经验的人,仍然会犯错误、忘记函数参数,或转向互联网搜索以刷新他们可能已完成多次的任务。
R 语言的流畅度不是永远不会收到错误消息,而是当你收到错误消息时感觉有能力修复它们(图 1)。当然,有些 bug 的发生并不是因为输入错误的代码,而是因为对函数参数或输出的误解。
虽然避免错误消息是很好的第一步,但确保脚本完成预期任务的最佳方法是:
仔细阅读文档,一次运行一行代码。
学习正确的编程实践的一个好方法是看书。书的一个优点是它们通常代表专家的声音、社区的技能,或两者兼而有之。大多数学习 R 编程的好书都包含代码示例,你可以使用这些示例来提高技能。
▸不管哪本书,都要运行代码示例并检查输出,积极分享
当书本后面没有答案时,开始或加入阅读小组会很有帮助,而且更有趣(后面第八条会讲到加入 R 社区)。与同事一起尝试练习,并利用彼此作为网络来提出问题、比较方法并通过复杂的任务进行集体思考。
教科书解决方案也定期在个人博客或网站上在线共享。在线发布自己的解决方案可以让你能够获得反馈,使整个 R 社区受益(参见第9条)。
▸专业书籍可以成为提升特定领域技能的绝佳资源
R 用户其实很幸运,你们已经拥有大量高质量的已出版书籍作为学习资源,其中包括:
RStudio 网站
( https://www.rstudio.com/resources/books/ ) 上精选的十多种书籍,以及无数其他作者由知识渊博的 R 贡献者提供。
比较推荐的 R 一些书籍包括“R for Data Science”和“Advanced R”,它们通过tidyverse介绍了 R 的现代方法(参见第5条关于风格)。
▸确立目标,按需去学
根据你学习 R 的目的,去看涵盖与你兴趣相符的,更具体主题的书可能会有所帮助。
例如,如果你打算将 R 用于高级统计,可以去看这类书,如《使用 R 的生态学中的混合效应模型和扩展》或《广义相加模型:R 简介》,每个都鼓励对核心编程和统计技能的深刻理解。
如果你想用 R 来画出高质量的 Web 图形,从而传达结果,可能会需要看《Interactive web-based data visualization with R, plotly, and shiny》,其中涵盖了先进的基于 Web 的数据可视化技术的介绍。
那么到底应该买多少本书?
应该选修哪些编程的课程?
是不是需要很大的金钱成本?
……
别担心,下一章节我们提供一些免费资源供参考。
幸运的是,许多高质量的R资源在线免费提供,涵盖了你可能所需的一切。
▸电子书,网站
例如,许多电子书在线免费提供,包括RStudio网站上的电子书。为了更快地参考,RStudio还提供了几个单页备忘单,每个备忘单都涵盖了一个特定包或编程任务的基本知识,可以作为很好的提醒。
如果你觉得看书枯燥,不容易看懂,那么也可以参考一些别的形式:
▸互动教程、视频课程、博客等
在Coursera、edX和freeCodeCamp等网站上也有许多关于R、统计和数据科学的免费课程,The Carpentries的免费培训材料,甚至YouTube和Twitch等网站上的免费视频教程。
一些地区和全球R团体,如R-Ladies和ROpenSci(更多信息请参见第8条),提供多种语言的博客文章、研讨会和资源。此处维护了R-Ladies博客列表(https://github.com/rladies/awesome-rladies-blogs); 包括Julia Silge和Danielle Navarro等。
其他精选的免费资源列表可以在inSileco博客和r-directory网站上找到。
无论是通过博客、互动教程还是视频课程,如果有一种在线方式你觉得最适合的,那么很可能会有这种方式的R资源,只需要搜索一点就可以找到。
▸对于R技术领域的更多具体培训,许多机构提供免费教程、研讨会和在线课程。
例如,魁北克生物多样性科学中心(QCBS)R研讨会系列以英语和法语提供了关于数据可视化、线性模型、多元分析等的介绍性和高级研讨会,并在其网站上免费提供PPT、代码和配套书籍。
爱丁堡大学的编程俱乐部为生态学家和环境科学家提供广泛的课程,从数据处理和统计到地理空间分析和机器学习。
EcoDataScience是一个以加州大学圣巴巴拉分校为中心的组织,提供一系列生态相关研究技术的技能分享和培训。
以上这些只是高质量学习小组的几个例子;可以与你所在的机构、公司或同事联系讨论,获得最相关或最方便的建议。
▸用“只是为了好玩”的项目来练习
如果你有时间和灵活性,一个很好的培养技能的方法就是通过“只是为了好玩”的项目来练习R。
– 想学习如何使用ShapeFile进行映射吗?
尝试制作一张你最喜欢的城市的海报,作为墙壁艺术印刷。
– 通过API抓取数据?
尝试在Spotify上找出最受欢迎的艺术家的共同品质。
– 文本分析?
尝试使用R的文本挖掘包来比较你喜爱的书籍或电视节目的情感关联。
– 定制数据可视化?
尝试使用ggplot形状和美学来复制你最喜爱的艺术作品(参见Twitter上的#RecreationThurday)。

▸ 低风险环境更解压
仅仅为了好玩,项目可以成为培养关键技能的非常有价值的环境。
低风险的环境会减轻你成功的压力,但当你成功时,会给你新的产品和工作代码,供你下次尝试类似的任务时参考。
▸ 用各种活动,趣味竞赛的方式边玩边学
参与有趣的社区活动、Twitter挑战或R编码竞赛(更多信息,请第8和9条)将帮你建立坚实的基础,为你的编程道路打开大门,同时选择你喜欢的项目。
如果你更愿意让你的R学习更多的“任务”工作,你可以对现有的项目进行低压力的增加,从而获得新的技能。
尝试通过向现有图添加自定义文本注释来练习HTML呈现,或者通过创建用于交互式数据探索的闪亮应用程序,来升级日常工作流程。
在改进现有工作的同时,寻找扩大技能的机会将有助于你成为更全面的R用户。
培养和维护项目的编程方法和组织系统有助于你和他人的代码清晰一致。
▸ 在开始编程项目时,需要考虑你的心态
如果在开始新任务时对所有未来步骤都进行了精心规划,那么你就可能会从使用虚拟程序代码开始项目中获益。
*虚拟程序代码是计划完成的操作的简单语言描述列表,在文档中写出,然后逐行翻译为代码。
这种方法的一个好处是,它允许你从头到尾对项目进行概念化,并为你将要编写的每一行代码提供一个离散的目标。
如果说,你更喜欢边做边学,可能更喜欢直接编写代码,观察每一行的输出,并在文本中注释最后一行完成的内容,以较小的增量达到最终目标。
在这两种情况下,对代码进行注释是很重要的,确保将来能够理解当前的想法。


▸ 接下来,需要决定在编码时使用什么样式
在本文中,编程风格是编写代码时使用的特定函数、包和语法策略的集合。如果你认为R是一种语言,可能会认为你的风格是一种方言;两种风格看起来可能不同,但它们的含义是相同的。
在R中,主要的样式划分通常围绕着:你是使用基本R的典型样式,还是采用R的管道方法以及其他tidyverse原则。
3 种代码样式的示例


虽然这些样式并不相互排斥,但它们包含不同的函数集,通常适合不同的语法策略来构造代码。根据你的背景,你可能更喜欢其中一种语法。
例如,如果有在C++或Java等程序中编写代码的经验,则顺序或嵌套语法可能会让你熟悉,而管道函数可能会使代码读起来更类似于用英语编写的句子。
(请参阅tidyverse样式指南:https://style.tidyverse.org/)
在大多数情况下,只要适合你的,什么样式都可以。

当然,匹配协作者中最常见的代码风格或规程中的标准,对阅读、共享和排除代码故障很有帮助。
有时,可能需要切换样式,以便于最好地完成某项任务,但选择尽可能保持样式一致将有助于确保你和其他人可以解释代码。
▸ 最后,将文件放入严格的目录结构中非常重要
R生态系统提供了许多工具来促进项目组织。大多数集成开发环境(IDE)都有“项目目录”的概念;这对于领先的R IDE、RStudio和大多数其他(例如,VScode和Atom)都是如此。
还可以使用包来简化项目目录中的文件路径命名方案。根据你所在的领域,文件夹组织结构可能会有所不同,但随着项目列表的增长,对系统进行批判性思考将简化生活。
此外,保持适当的项目组织将有助于你学习“版本控制”的实践,这是一种跟踪更改和备份代码的系统,正在成为跨科学领域的专业标准。
R的大多数功能都捆绑在特定于任务的包中,但很难知道哪些包存在,哪些包最适用于某些任务。
在R中,用户开发包的主要存储库是CRAN,即the Comprehensive R Archive Network,综合R存档网络。
虽然软件包也可以来自其他来源,但CRAN软件包具有通过R’s install.packages功能轻松访问,确保软件包已在多个平台上测试的优势。
当搜索完成特定任务所需的新包时,最好从CRAN的任务视图开始(https://cran.r-project.org/web/views/).
任务视图允许用户按主题浏览已发布的包,包括多元统计、时空分析、元分析等。任务视图浏览器列出每个主题的相关包,并提供指向每个特色包的扩展文档的链接。
该资源对于R初学者以及正在寻找应对新挑战的方法的更有经验的R用户非常有用。
自学R时,你可能会遇到自己还不知道如何解决的问题。

在这些情况下,寻求帮助可能会为你节省大量时间,缓解头痛。
知道如何以有针对性的方式寻求帮助,这样你就能够查明问题的根源,并且在理想情况下,帮你避免将来出现类似问题。
▸ 如何以有针对性的方式寻求帮助?
当你第一次遇到问题时,复制和粘贴遇到的错误到搜索引擎中,可能会让你访问Stack Overflow、GitHub或R-bloggers等网站。
-通常情况下,有人会遇到与你相同的问题,并且可能会找到你可以使用的解决方案。
-如果没有的话,你可能需要发布自己的求助。
在这种情况下,有一些指导原则可以使帖子更高效:
在寻求帮助时,可复制的示例至关重要。你的目标应该是尽可能少地展示你的问题,并且在发布你的问题之前需要花一些时间隔离有问题的代码。
最有用的方法是在示例中生成一个玩具数据集,或者使用内置于R中的数据集(请参阅R中的data),并逐步删除代码中与要解决的问题无关的部分。
还应该在最小可复制示例中附带有关R会话和相关包版本的信息。所有这些步骤都可以通过reprex软件包实现。
▸ 水平提高后,帮助他人
随着技能的提高,你可能会发现自己处于一个可以帮助他人的位置。
在Twitter或Stack Overflow这样的地方回答问题是回馈R社区的好方法。
你甚至可以回过头来回答你自己过去问过的问题,这样你的帖子标记为“已解决”,并为下一个遇到同样问题的人留下一个有效的解决方案,帮助自己的同时也能给他人带来便利。
建立R语言流利性的最佳方法之一是与他人一起学习。
R社区充满活力,为学习R技能举办了会议、聚会和定期在线活动。
一个很好的起点是寻找你所在的区域的R用户或建立兴趣小组,经常举办技能分享、讲座、编程实践会议。
当然,你也可以虚拟加入R社区。The R for Data Science(R4DS)社区为学习者和导师提供了分享研究技能和协作的空间。R4DS Slack频道拥有超过10000名成员,成员可以通过讨论频道寻求帮助、分享胜利、网络等。
如果你不属于任何特定群体,R社区在许多社交网络上也很活跃。
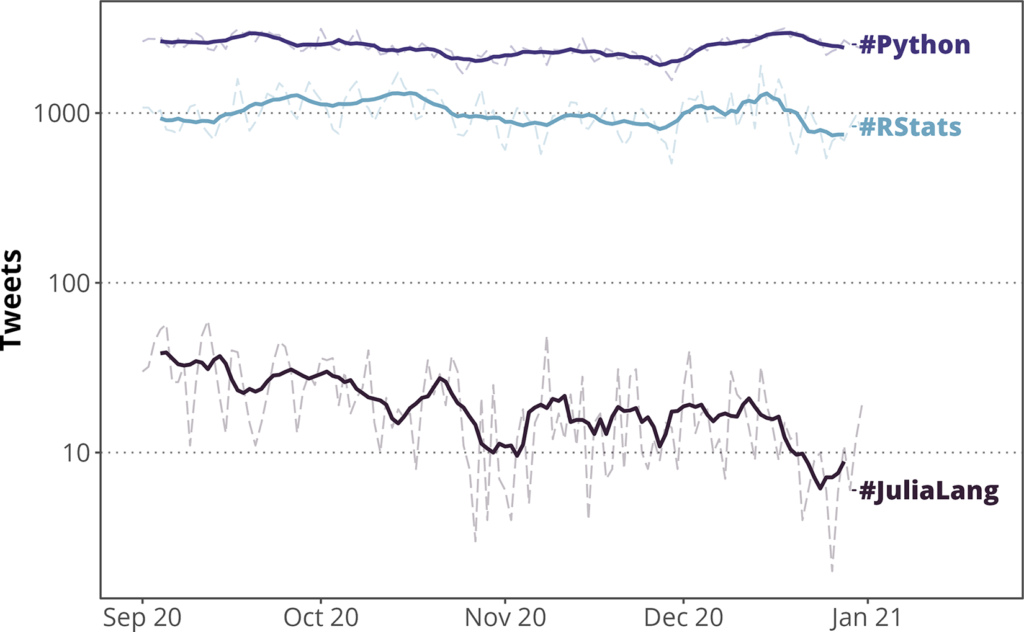
R通常在Twitter上与#RStats标签讨论如下图。
一些常见科学编程语言的Twitter讨论流行度
社区的一个很好的切入点可能是以下帐户:
@rstudio、@Rbloggers、@icymi_r、@RLadiesGlobal、@R4DS、@rOpenSci
在Facebook和Reddit等网站上也存在一般和特定领域的R组。你甚至可以参加在线活动,如Twitch上的数据科学节目“切片”,观看专家们在R编程挑战(以及其他语言)中的竞争。
关注多产的R博客也是了解R的好方法;一些推荐的R博客作者包括Maëlle Salmon、Jacqueline Nolis、Miles McBain。
在线找到社区都是了解新功能、了解最新软件包、遇到其他地方可能看不到的提示和技巧的好方法。
最重要的是,保持积极性,继续编写代码。

R的开源文化为代码共享提供了丰富的资源。
▸ 阅读和运行来自公开来源的代码,对于发现新函数、优化处理速度和向专家学习非常有价值
一些R项目,如#TidyTuesday R社交数据可视化项目,鼓励在线代码共享,以帮助用户获得和磨练技能。
其他活动,如每年一度的RStudio Shiny竞赛,为用户制造的R产品提供了一个友好竞争的渠道,最终免费提供代码,允许用户阅读、下载和复制获奖应用程序。
如果你发现一个特别好的R产品提供了代码,请自己逐行运行代码,以了解每一行或函数的确切意思,具体起到什么作用。你可能会学到一个新的功能,可以应用于自己的项目,或者有许多方法可以完成相同的任务。
▸ 已发表的论文提供了另一个R代码源代码,以实现更多以研究为中心的目的
现在,许多学术期刊要求出版物中包含公开可用的数据和代码,其中许多(尤其是自然科学领域)都是用R编写的。通过从已发表的文献中下载材料,你可以了解所在领域的专家如何进行分析,使用哪些软件包,以及他们如何组织代码。
▸ 学会分享:在线共享你的R代码
当然,代码共享是双向的;除了从其他人那里获取公开可用的材料,您还应分享您自己的材料。
代码共享的通用平台包括GitHub、GitLab和开放科学框架(OSF)。
在线共享你的R代码将进一步增强R社区的开放性,并帮助你成为一名更加专注的代码编写者。
发布代码可能会让人感到害怕,但请记住,对科学界来说,可用代码总是比不可用代码更有价值。
如果你的代码没有完全优化或完全干净,不要过分在意。如果它能运行起来,可能会教给别人一些新的东西,是值得分享的。
R是一个非常棒的工具,可以用于统计、数据操作、可视化等,但它不一定是编程旅程的终点。
▸ 流利的R语言帮你获得未来适用于其他程序、语言或领域的技能
例如,为Shiny Applications设计用户界面(ui)将帮助你建立前端web开发的基础,R的各种文本插件(例如ggtext包)将帮助你练习HTML语法,R的向量操作(apply和purrr::map函数族)将构建概念框架以转移到其他编程语言,如Julia或Python。
▸ 这是双向的:如果你已经掌握了其他编程语言的技能,它们也会帮助你学习R语言
当你继续将编程技能应用于更广泛的任务时,你可能会发现对于某些任务,不同的工具会更有效或更合适。

在这些情况下,你在学习R时建立的信心和技能可能是下一次编程努力的有用跳板。
通过学习R获得的知识,以及你通过自学获得的经验,将使你受益远远超过你手头的任务。
学习R语言,可能是一个充满挫折、自我怀疑和缺乏继续动力的过程。在这里列出的10条是帮助你克服挑战、掌握新技术的最佳策略,甚至可能在这一过程中获得一些乐趣。

也希望可以帮助初学者,无论是研究生、业余爱好者还是渴望学习新工具的研究人员。
当然也不需要一次尝试所有这些规则,也不局限于这十条,你只需要找到最适合你的方法即可。
主要参考文献:
Lawlor J, Banville F, Forero-Muñoz N-R, Hébert K, Martínez-Lanfranco JA, Rogy P, et al. (2022) Ten simple rules for teaching yourself R. PLoS Comput Biol 18(9): e1010372. https://doi.org/10.1371/journal.pcbi.1010372

谷禾健康

在过去的几十年里,生物技术和计算技术的巨大进步彻底改变了我们对微生物群落的理解。特别是,基于16S rRNA和宏基因组测序的研究进一步验证了一个事实,即微生物群不是静态的实体,而是动态的生态系统,身在其中的组成成员之间,以及成员与环境之间,都在以各种方式进行相互作用。
在这些研究过程中产生了大量的关于微生物相互作用的机制和环境依赖性的数据,也出现了许多专门储存单一类型或进行了部分分类整合的数据库。但由于是以不同的标准去整理的数据,所以很难共享。
//
本文研究人员提出了一套体系,用于微生物组数据管理,简称FAIR (findable, accessible, interoperable, and reusable),即可查询、可访问、可交互和可重复使用。这些原则于2016年首次正式提出。
首先基于可查询和可访问的原则,需要数据库,它能存储现有数据也能继续转化新的数据。这个平台可以是自己从零开始设计,可以是使用现成的数据库构建工具作为辅助(比如mako软件),也可以是直接使用已有的数据库,比如GloBI。
关于数据获取,文中列出了一些资源,都是目前广泛使用或者比较独特的,如下表:

其次基于交互性和可重复使用的原则,需要将大数据拆分为一个个子集,每一个子集都应该是能满足大量用户的第一需求或者说是第一想了解的信息点。基于此,研究人员提出了四类元数据作为起点:
1. 微生物实体
应提供参与相互作用的每种微生物的物种(和菌株,如果相关)名称。
示例:
2. 相互作用的推理方法
使用不同的方法来指示高度相关的元数据。
示例:

3. 相互作用的上下文语境
一些环境背景,如生物群落(例如,宿主相关的、合成的)。

示例:

4. 属性,用于定义互作关系的类型
如合作、对抗、关联、成对或高阶等。
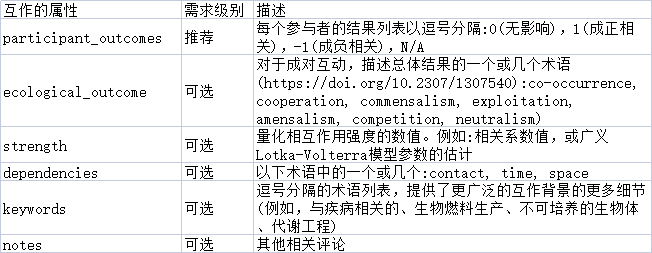
示例:

最后,文章中给出了依照FAIR体系研究微生物互作关系的可拓展性。
如下图,通过研究微生物相互作用和相关性(A部分)而产生的多种类型的元数据(B部分),这些元数据(DT),例如真菌细菌或噬菌体细菌的培养实验数据(DT1),扩增子序列或OTU计数的相关性(DT2),两个或两个以上物种基因组尺度代谢网络的通量平衡模型(DT3),直接比对到特定数据库(DT4)。
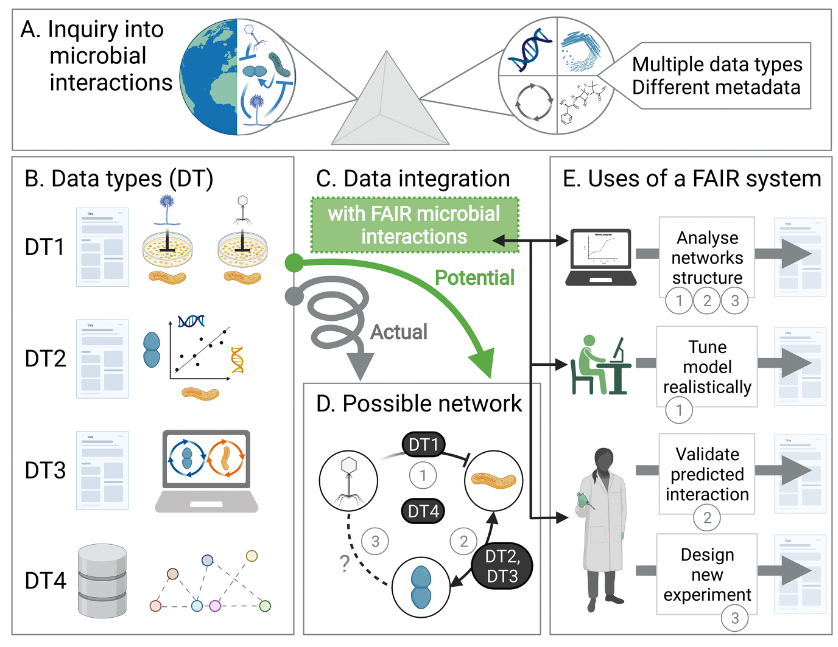
遵循FAIR里的四个原则来解析微生物互作内容(C部分),解析结果如D部分,得到一个可能的相互作用网络关系图谱。
同时,C部分的内容可以横跨不同领域,为不同领域的专家提供信息,例如E部分,建模者可以更容易确定具体的互作以便真实的模拟生态动力学,而实验人员可以评估是否有新的互作在其他宿主或上下文中被报告。
以上内容,是研究人员基于自身经验归纳总结的,研究人员也指出在此概述的实践、标准和示例并不是详尽无遗的,文章旨在促进进一步讨论如何改进微生物相互作用及其属性数据的访问和可用性。希望能进一步的激励各位科学家,共同创建一个共享开放的,拥有统一标准的微生物相互作用资源站。
参考文献:
Pacheco AR, Pauvert C, Kishore D, Segrè D. Toward FAIR Representations of Microbial Interactions. mSystems. 2022 Aug 25:e0065922. doi: 10.1128/msystems.00659-22. Epub ahead of print. PMID: 36005399.
谷禾健康
为了使研究更系统更全面,越来越多的研究人员追求在多组学背景下解释分子数据。
OmicsNet (www.omicsnet.ca) 顺应而生,这是一个在线平台,允许用户轻松地构建、可视化和分析多组学网络。
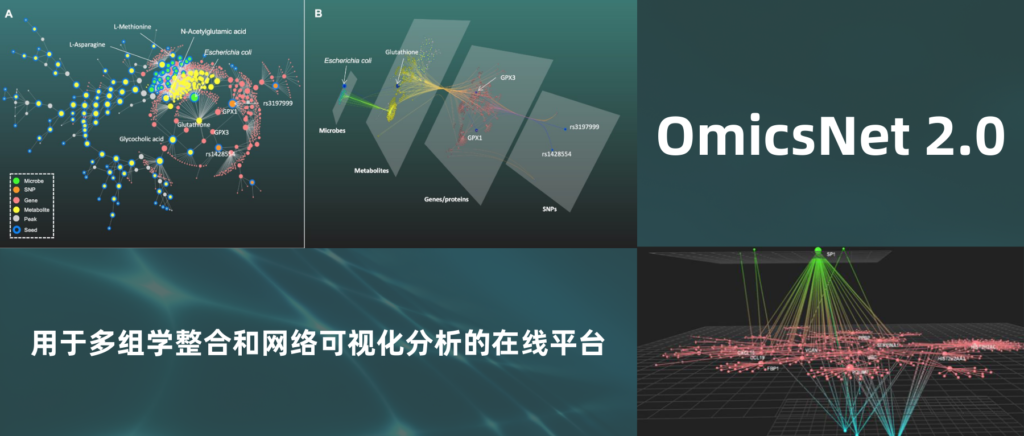
OmicsNet 2.0在原有基础上进行了升级,主要改进了三个方面:
(1) 可视化分析中2D图形布局选项增加了11个,以及一个新颖的3D模块。
(2) 支持三种新的组学类型:
(3) 分析时将同时输出使用的R包OmicsNetR和历史命令,并生成共享链接可在网页查看和交互操作,从而提高研究的可重复性。
研究中使用OmicsNet 2.0对炎症性肠病(IBD)的数据进行多组学分析。
OmicsNet 2.0工作流程

如图,主要为4个步骤:
OmicsNet 2.0 的改进
1、更新了分子相互作用数据库
包括PPI数据库(STRING,InnateDB和IntAct),TF-target数据库(TRRUST和JASPAR), miRNA-target数据库(TarBase和miRTarBase),代谢数据库(KEGG, Recon3和AGORA)。
2、支持三种新的组学类型
可支持来自遗传变异研究的SNP矩阵、来自非靶向代谢组学的 LC-MS 峰和来自微生物组分析的物种丰度矩阵数据的输入及互作分析。
对于那些对影响基因调控的变异感兴趣的用户,可以分别基于ADmiRE和SNP2TFBS将SNPs映射到miRNAs或TF结合位点。由此产生的网络可以通过proteins、miRNAs或TFs进一步扩展,以了解潜在的影响。
对于代谢组学分析,使用最近发布的NetID算法对LC-MS峰(m/z, retention time, intensity和p-value)进行数据处理,可选三种数据库(KEGG, PubChemLite_BioPathway和HMDB)进行注释。
使用Rcpp/C++ 引擎重新编写了核心算法,使其速度提高了10 倍以上。使用 lpsymphony 包进一步优化了整数线性规划。
直接上传物种丰度矩阵,可以使用贝叶斯逻辑模型预测潜在代谢物,该模型使用超过 6000 个高质量基因组规模的代谢模型进行训练。
3、代码开源
开放R包OmicsNetR(https://github.com/xia-lab/OmicsNetR),显示分析期间所执行的R命令。结果可输出为网页链接,有效期一个月。
4、减少假阳性
对于来自人类和小鼠的数据,提供了一个基于ENCODE、基因型-组织表达(GTEx)或人类蛋白图谱(HPA)的基因表达数据的过滤器,这些过滤器帮助研究人员专注于与生物相关的相互作用,并减少假阳性。
5、支持 2D 和 3D 网络可视化
2D模块可支持11种图形布局,3D模块可支持交互。图形化主要基于 igraph 包和 graphlayouts 包。
使用OmicsNet 2.0进行的IBD多组学研究
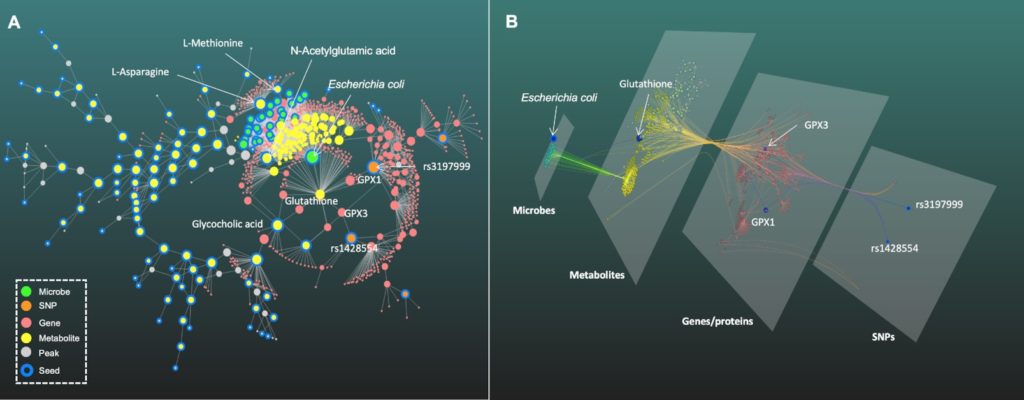
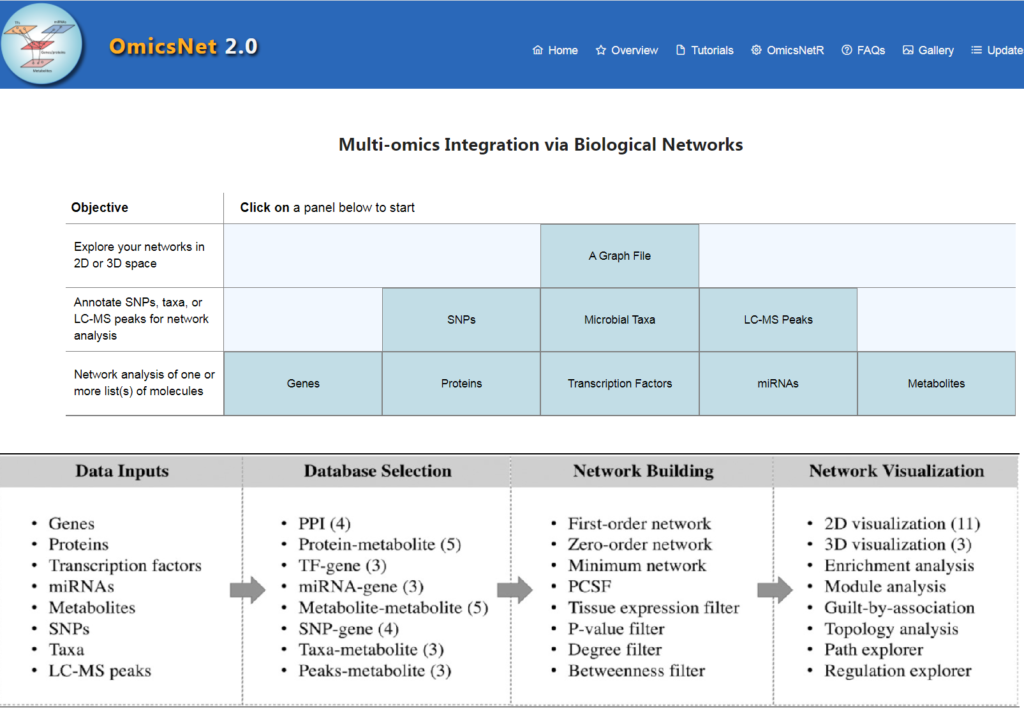
图A为2D视图,图B为3D视图
将获得的物种丰度矩阵,SNP矩阵和LC-MS峰数据上传至OmicsNet 2.0平台。
利用AGORA数据库预测潜在的微生物代谢物(potential score:0.9)。
利用PhenoScanner对基于eQTLs的基因进行SNP定位。
利用KEGG数据库对LC-MS峰进行注释。
通过添加代谢物-蛋白质相互作用,从SNPs和LC-MS峰生成的个体网络进一步扩大,这样三个网络可以在代谢组学层合并。
p-value过滤:(cut-off: 0.2)来排除p值较大的LC-MS峰值所贡献的节点。其网络连接基于其相关代谢物、基因或蛋白质。
如图A,生成的子网络1包含六种类型的节点(物种、SNPs、代谢物、SNPs或代谢物相关的基因/蛋白质),结果显示出glutathione是宿主-微生物相互作用中的重要交互作用点。
由此推测包括大肠杆菌在内的几种微生物会产生glutathione,两个SNPs(rs3197999和rs1428554)通过编码谷胱甘肽过氧化物酶(GPx)的基因GPX1和GPX3与代谢产物相关。
与此呼应的是,在以往的研究中证明大肠杆菌和相关物种在IBD中的过度表达可能是由于它们能更好地产生glutathione以抵抗氧化应激。
图B的3D分层网络提供了一个直观的多组学整合视角,突出显示了连接微生物组和宿主遗传的glutathione的流动路径。
OmicsNet 2.0与其它工具的比较
如下表,比较OmicsNet 2.0与其他基于web的多组学集成工具的关键特性。
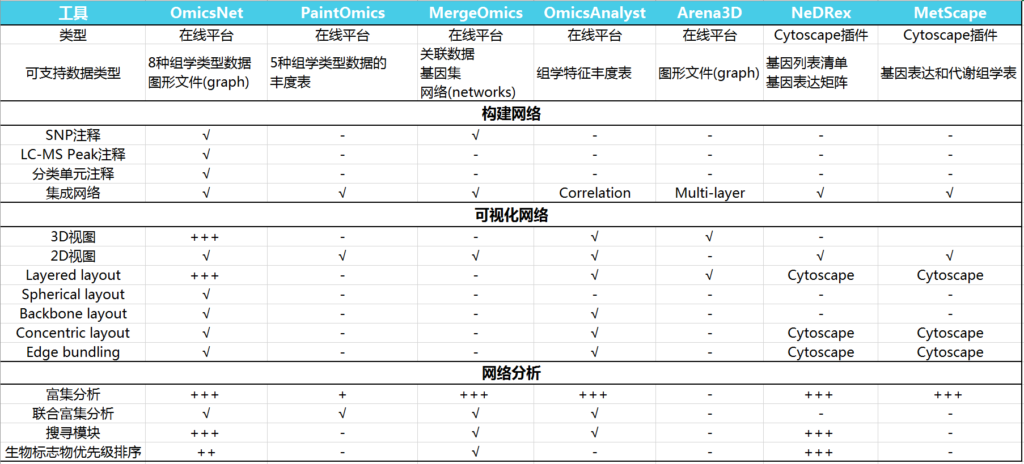
‘√’表示支持,‘-’ 表示缺失,“+”表示定量评估(“+”越多表示更好的支持)
附带各工具的官网及主要特点描述:
PaintOmics
(http://www.paintomics.org)
专注于在视觉上呈现多组学数据的探索分析,包括转录组学,代谢组学,表观基因组学,miRNA和转录因子,将其映射到KEGG通路联合分析。
MergeOmics
(http://mergeomics.research.idre.ucla.edu)
整合了来自单个组学层面的关联研究的汇总统计数据,以及不同功能基因组学数据,以获得机理上的见解,最近还加入了多组学信息的药物重新定位(drug repositioning)。
OmicsAnalyst
(https://www.omicsanalyst.ca)
基于输入的数据,利用多元统计、相关性分析和聚类方法,结合网络、热图和散点图进行多组学分析。
Arena3D
(https://www.arena3d.org)
擅长使用基于3D的分层布局对多层网络进行交互式可视化,适用于多组学网络数据。
NedRex
(https://nedrex.net)
是一个Cytoscape插件,专注于疾病模块识别和药物再利用,使用各种模块识别和优先排序算法。
MetScape
(http://metscape.ncibi.org)
是一个Cytoscape插件,通过构建和分析不同类型的含有酶、代谢物和/或反应的网络,专注于基因表达和代谢组学数据的集成和可视化。
OmicsNet
(https://www.omicsnet.ca)
通过将多个分子互作数据库与强大的2D/3D可视化网络分析相结合,对于多组学集成分析和结果解释有更好的理论基础,对以上工具的不足做了补充。
结 语
OmicsNet 2.0是一个基于网络的多组学分析平台,支持2D和3D网络可视化探索。
在1.0版本中强调基于web的3D网络可视化。
在2.0版本中,进一步改进了其可视化分析系统,添加了一个功能完整的2D网络可视化系统,并支持三种新的组学数据输入(SNPs、微生物分类单元和LC-MS峰),目前的生物信息学工具无法很好地支持这些数据。
除了富集分析、搜寻模块和最短路径分析,用户还可以使用重启随机游走算法(Random Walk with Restart)搜索候选疾病标记物。最后,2.0版本通过发布底层的R代码和共享链接,改进了工具的可再现性和透明度。
使用IBD多组学数据的案例研究表明,OmicsNet 2.0可以揭示与原始出版物和后续出版物以及IBD文献一致的有意义的模式、关系和功能。总之,OmicsNet 2.0解决了对多组学数据进行分析的一些需求。
参考文献:
Zhou G, Pang Z, Lu Y, Ewald J, Xia J. OmicsNet 2.0: a web-based platform for multi-omics integration and network visual analytics. Nucleic Acids Res. 2022 May 26:gkac376. doi: 10.1093/nar/gkac376. Epub ahead of print. PMID: 35639733.

谷禾健康

尽管地球上微生物类群的繁多,但只有一小部分得到了培养和有效命名。因为大多数菌无法在非常特定的条件下培养分离鉴定。
在过去十年中,宏基因组研究的重要性已经凸显,因为它能够评估细菌基因库并发现当前实验室培养技术无法掌握的新细菌基因组。这些数据对于扩大我们对地球上微生物多样性的理解至关重要。
由于宏基因组测序数据由来自多个物种和菌株的 DNA 序列片段组成,通常有数千个来自不同生命领域,因此此类分析的主要挑战是正确确定每个 DNA 序列片段的真实来源。不幸的是,这些步骤容易出错,因此必须对结果进行严格审查,以避免发布不完整和低质量的基因组。
最近,比利时研究人员新开发MAGISTA,这是一种评估宏基因组基因组组装质量的新方法,基于随机森林的方法估计MAGs的完整性和污染度,解决了当前基于参考基因的方法经常被忽视的一些缺陷。
MAGISTA是基于宏基因组bins内的contig片段之间的无对齐距离分布,而不是一组参考基因。该方法利用了来自整个 bin 的信息。为了正确评估此方法,并说明基于参考的工具的缺点,最近,比利时研究人员构建了一个高度复杂的 DNA 模拟群落,由 227 个细菌菌株组成,并且具有不同程度的相似性。

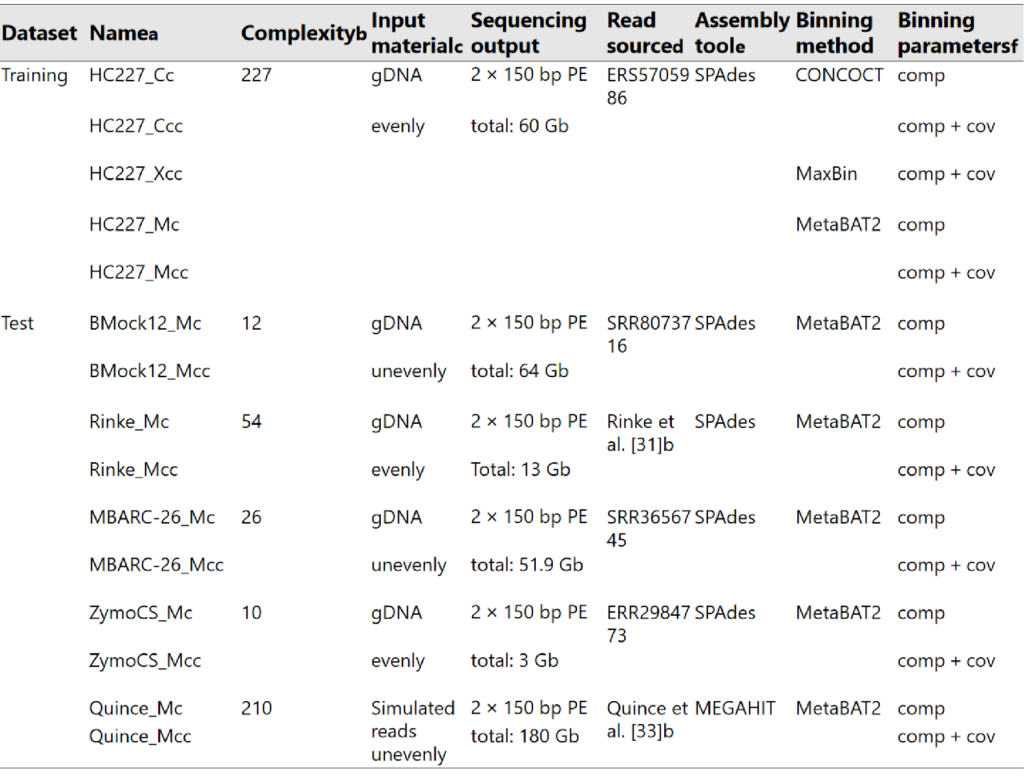
方 法
训练集来(HC227)自 227 个细菌菌株,测试数据集由五个公开可用的短读(short reads)子集构成,其中四个含有来自复杂度相对较低的基因组 DNA 模拟群落的reads。具体情况如下图所示。
Complexity列指示菌株数;Assembly tool列表示所使用的用于组装的软件;Binning method列表示所使用的用于分箱的工具;Binning parameters列表示所使用的用于评估分箱质量的指标,comp为完整度,cov为覆盖率。
MAGISTA计算步骤:
输入binning后的每个bins
-●-
第 1 步:选择适合的片段大小与距离计算方法
-●-
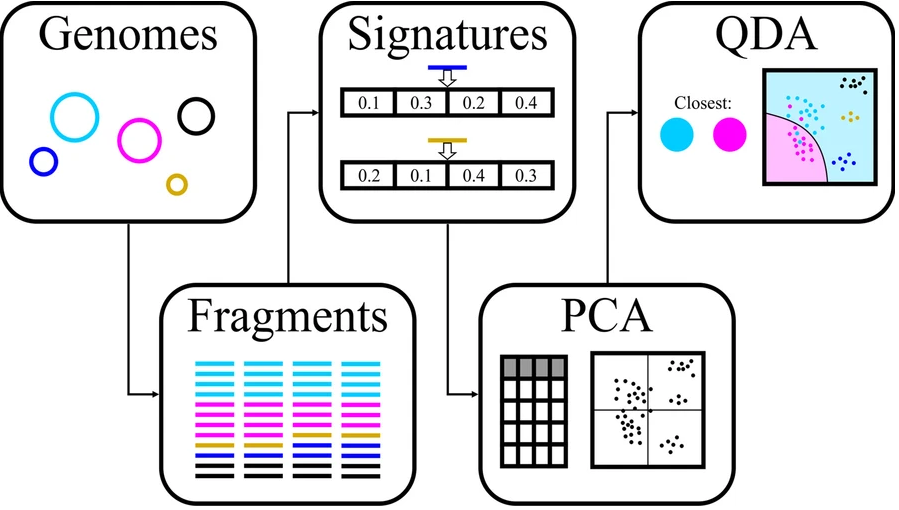
首先将每个 bin 中的每个 contig 拆分为固定长度的片段,然后使用四种不同的方法(即 PaSiT4、MMZ3、MMZ4 和 Freq4)计算一个 bin 中的片段之间的所有距离。对于每种方法,都选择了特定的片段长度,以便为不同的生物产生不同的特征分布。
每种方法的最终片段长度的选择是通过不同方法分析整合决定的,方法如下图所示。每组的设计中至少两个基因组来自同一个家族,两个基因组来自相同的顺序但来自不同的家族。这些基因组被人为地分成所需长度的片段,并为每个片段计算目标特征。
对于每组五个基因组,混合所有片段并根据它们的特征进行主成分分析(PCA),然后进行二次判别分析,用于生成分类器,旨在区分每组中重叠最多的两个基因组。对该分类器的准确度取平均值,结果用于选择方法和片段长度的最终组合。
-●-
第 2 步:模型中特征变量的选择
-●-
为每种方法选择片段长度后,使用平均值、标准差、偏度、峰度和中位数以及 2.5%、5%、10%、90%、95% 和 97.5% 百分位数计算距离分布。此外,还计算了 1 kb 片段的 GC含量分布。以及每个bin的大小,共计66个特征变量。
-●-
第3步:模型构建
-●-
使用 R (v 4.0.3) 包“RandomForest”中的“RandomForest”函数和默认参数训练随机森林模型。同时使用R包lm再建立一个线性模型执行线性回归,输入经对数转换后的特征变量值,用于交叉验证分析。
主 要 结 果
一个高度复杂的基因组DNA模拟群落
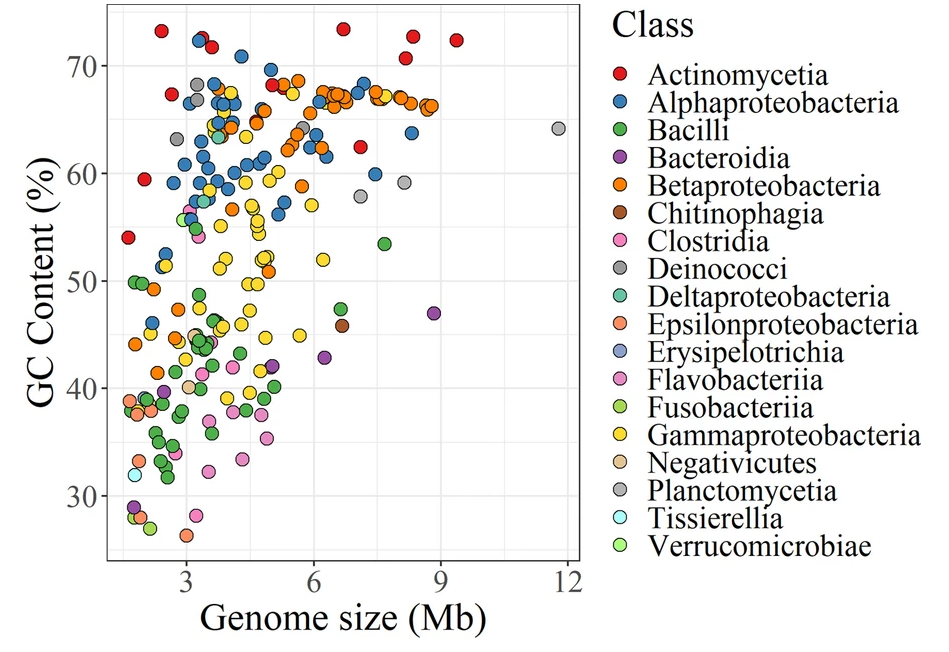
由来自 227 个细菌菌株的基因组 DNA 组成,这些菌株属于8 个门(Actinobacteria, Bacteroidetes,Deinococcus-Thermus, Firmicutes,Fusobacteria,Planctomycetes, Proteobacteria和Verrucomicrobia),18 类,47目,85科,175属,197种。
编辑
上图为模拟群落中的细菌菌株的基因组大小和GC含量(从26.3%到73.4%)散点图;
编辑
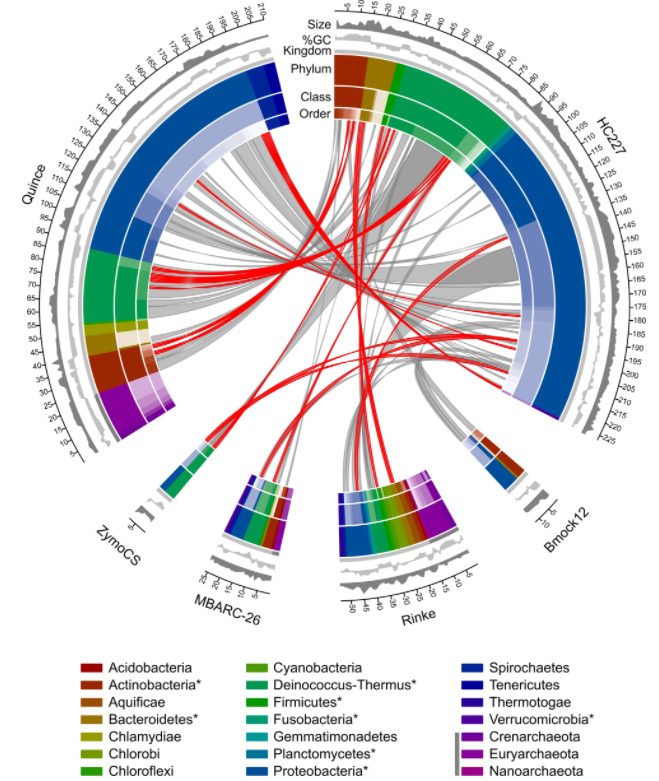
图为训练集与测试集中物种之间的关系图。红色线条表示在训练集中存在的菌种,灰色线条表示在训练集中存在的菌属。环状图中的不同颜色代表不同分类水平。图例中存在于训练集中的菌门用*标记,存在于古生菌的菌门用深灰色色带标记。
CheckM中基于单拷贝标记基因(SCMG)来评估 bin 质量的存在的缺陷
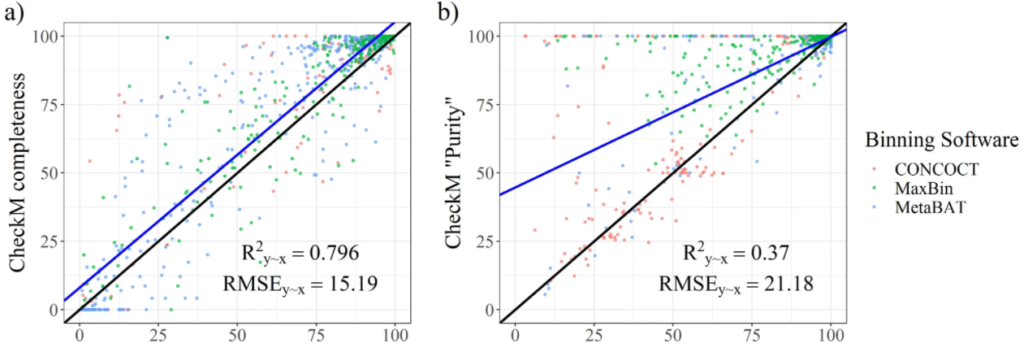
图a和b分别为从CheckM中输出的完整性指标和污染度。使用R^2y∼x(解释方差的百分比),RMSE(相对于实际值的均方根误差)两个参数评估结果。结果表示CheckM高估了bin的质量。许多受污染的bins被预测为接近未受污染。
使用MAGISTA分析模拟群落中的bins
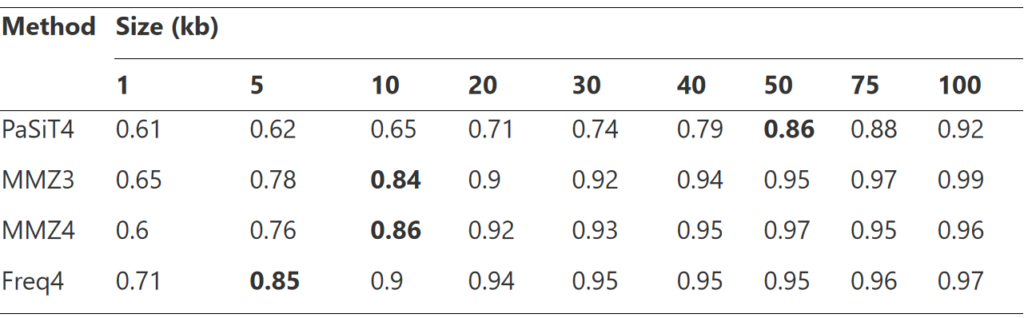
首先选择最佳片段大小用于计算距离分布,如上图所示,考虑了 1、5、10、20、30、40、50、75 和 100 kb 的片段,最终选择了粗体所示的片段大小。
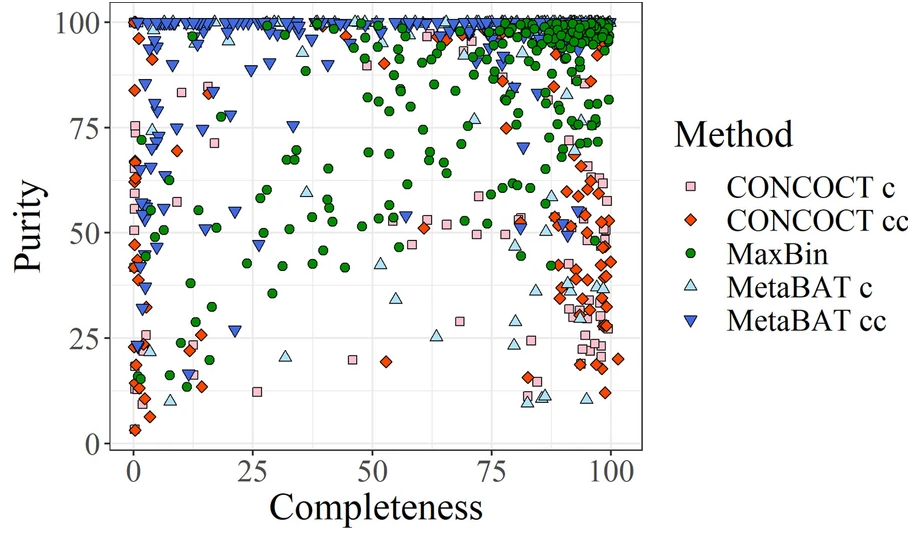
图为concont、MetaBAT和MaxBin产生的bins的完整性和污染度信息。
由于通过模拟生成这样的数据集并不能准确地表示真实的结果,所以使用了binning软件的结果,提供了一组不同质量的真实的bins。训练数据集的完整性和未污染度均在90%以上。
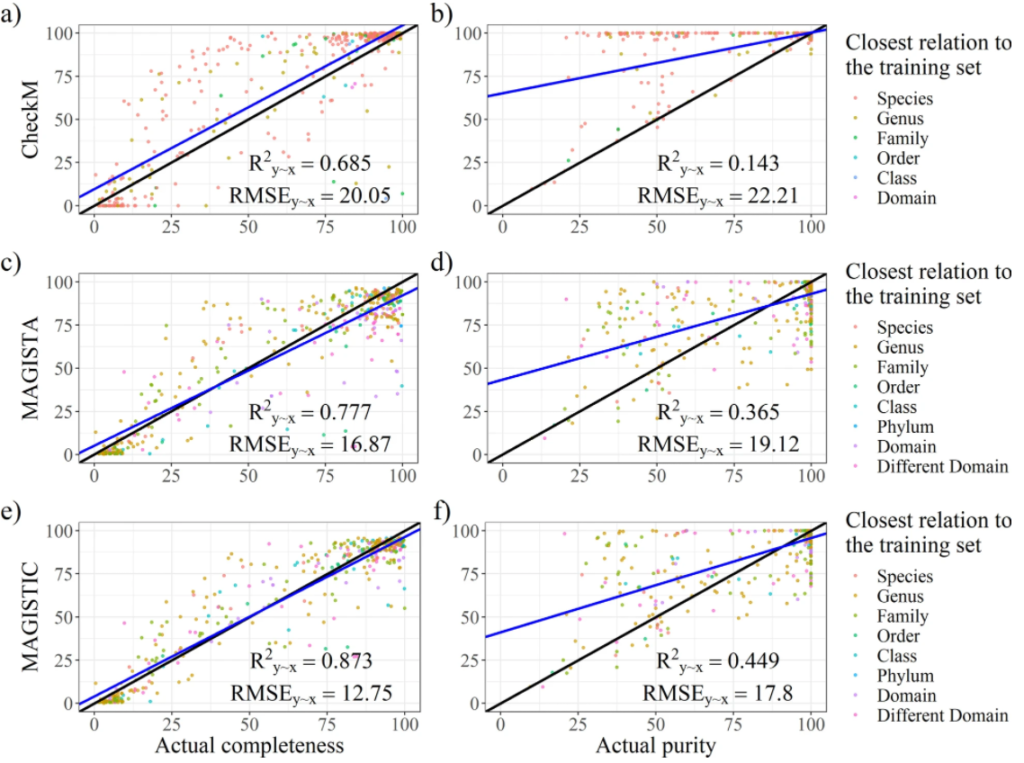
最后是模型构建,建立完整性和污染度的预测模型。并进行了模型评估,如图所示。分别对CheckM、MAGISTA 和 MAGISTIC测试了其性能。CheckM是现在主流的一款评估bin质量的工具。MAGISTIC是一款结合了CheckM和MAGISTA 的工具。使用解释方差的分数(R2y∼x)和均方根误差(RMSE)作为评估性能的指标。对于完整性的预测,MAGISTA 优于 CheckM。对于污染度的预测,MAGISTA 的表现优于 CheckM。
结 论
研究人员开发了一种新的用于预测高度复杂的宏基因组组装基因组bin的质量的方法,MAGISTA。是基于 SCMG 的低复杂性宏基因组方法的一个同样好的替代方法。除了MAGISTA之外,还通过结合CheckM的结果,使用MAGISTIC生成了一个更准确的预测。
研究人员在文章中指出MAGISTA 和 CheckM 都没有达到足够的准确度来被认为是可靠的。MAGISTIC 产生了比 MAGISTA 更好的结果。
在附加分析中,将测试集分为了两个子集,从真实和模拟reads中获得的bins,对此再进行分析,结果表示,CheckM 对于“真实”子集表现良好(但相比MAGISTA 和 MAGISTIC还是较差),对于“模拟”子集部分表现较差。而MAGISTIC相比MAGISTA会更准确些。但是文章中并没有详细说明MAGISTIC的工作流程。
查看作者在github上公开的软件说明,地址如下。但是没有说明和给出输出文件的内容。个人认为还不太成熟。
https://github.com/LM-UGent/MAGISTA
参考文献:
Goussarov G, Claesen J, Mysara M, Cleenwerck I, Leys N, Vandamme P, Van Houdt R. Accurate prediction of metagenome-assembled genome completeness by MAGISTA, a random forest model built on alignment-free intra-bin statistics. Environ Microbiome. 2022 Mar 5;17(1):9. doi: 10.1186/s40793-022-00403-7. PMID: 35248155; PMCID: PMC8898458.

谷禾健康

微生物物种的遗传变异研究通常包括单核苷酸多态性(SNPs)、结构变异(structural variants ,SV)和可移动遗传元件(mobile genetic elements,MGEs)。
在宏基因组中,SNP被用来量化种群结构、追踪菌株和鉴定微生物表型的遗传决定因素。然而,现有的基于比对的宏基因组SNP检测方法需要高性能的计算和足够的覆盖深度来区分SNP和测序错误。
为了解决这些问题,美国加利福尼亚大学研究人员使用高质量基因组,构建了 909 个人类肠道物种中 1.04 亿个 SNPs 的目录,并使用针对该目录的独特 k-mers 表征来自 7,459 个样本的肠道菌群的全球种群结构,开发了GenoTyper for Prokaryotes(GT-Pro),可以对宏基因组的这些 SNPs进行快速基因分型的方法。该研究成果近日公开在《Nature Biotechnology》发表。
该方法与使用读长对齐的方法相比,GT-Pro 更准确,速度快两个数量级,作者构建了一个GT-Pro数据库,基于大约25,000个宏基因组样本,并展示了GT-Pro如何用于数千种菌群的菌株水平探索,可以实现在个人电脑上快速高效地对数百万个SNP进行宏基因组分型。

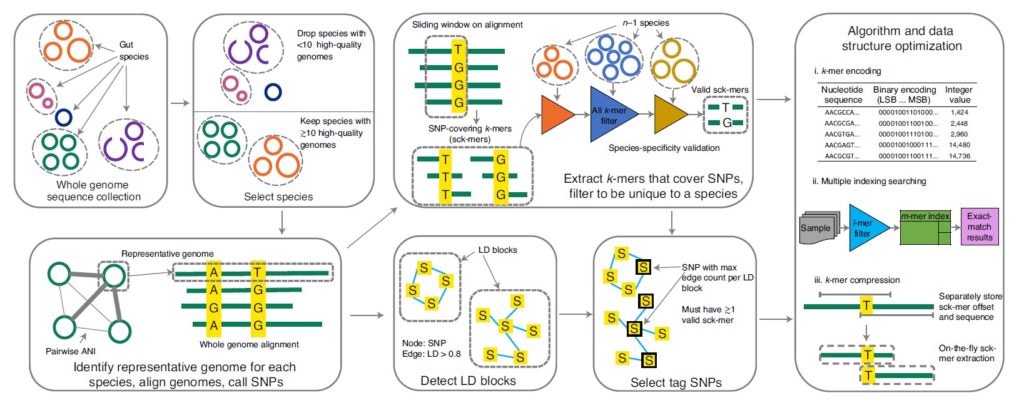
如图,按箭头方向所示。
首先从全基因组序列中识别高质量基因组的物种(去除<10 个高质量基因组的物种,高质量基因组:≥90% 的完整性和≤5% 的污染),对于每个物种,一个有代表性的基因组是根据平均核苷酸一致性(Average Nucleotide Identity,ANI)和组装质量指标选择的,确定代表性基因组后,对每个物种,通过MUMmer软件将每个同种基因组(conspecific genome )与代表性基因组比对,确定SNP,在这些SNP中选择常见的双等位基因SNP用于分型(site prevalence ≥90% and minor allele frequency >1%)。
接下来提取覆盖SNPs的k-mers(sck-mers),过滤出独有的物种,同时检测LD块,并选择具有物种特异性的sck-mers的SNPs和该块中其它SNP的最高LD。LD块为基于跨基因组的共现模式将 SNP 聚类成linkage disequilibrium block。检测LD块使用R2 阈值 (0.81) 。具有物种特异性的sck-mers即删除了两个或多个物种共有的任何sck-mer。
最右边的方框里简要是GT-Pro的算法和数据结构的优化方法。也是该研究的主要目标之一,正是利用了该方法构建的SNP索引才能实现快速地分型。
首先是k-mers编码,选择了k=31,以便使用64位整数编码,通过这一步骤,GT-Pro 数据库缩小了四倍。
其次是多索引检索和进一步压缩SNP数据结构。
优化后的GT-Pro数据库由两个表组成:
(1)10.6 GB的sck-mers表,包含每个k-mer的4字节条目;
(2)2.4 GB的sc-span表,包含每个等位基因的24字节条目。
所需的总存储空间为13 GB,是原始sck-mer表的bzip2压缩的两倍。也使得GT-Pro可以在个人计算机中高效运行。
1.从模拟宏基因组中准确识别SNP
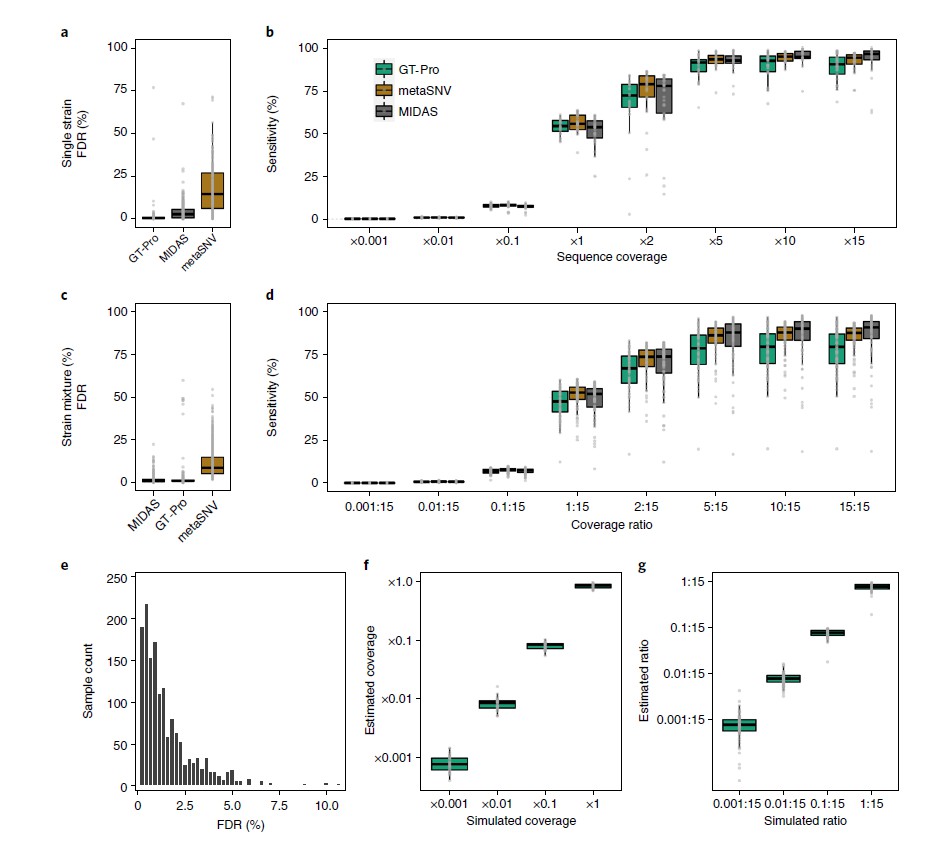
比较GT-Pro、MIDAS和metaSNV宏基因分型的准确性,使用232个未用于开发这些方法的人类肠道分离株的模拟宏基因组(大约2600万次reads)。
图a为FDR比较,假阳性指不正确的基因型,是由测序错误和读数映射到错误位点导致的。假阴性指缺失的基因型,在没有读数映射时产生。在宏基因组中,FDR最低的是GT-Pro(中位数,0.4%),而 metaSNV 最高(中位数,14.5%)。
图b为对图a的灵敏度调查,用于直接比较不同方法。敏感性是指在GT-Pro数据库中检测到分离株基因组(参考和非参考等位基因)中存在基因型的概率。结果表示,随着覆盖度的加深,GT-Pro的灵敏度损失较小。
图c为比较三个工具在一对同种分离株但不同覆盖率下的FDR,目的是检查宏基因组分型方法对菌株混合物的表现。其中一个菌株始终为15倍的覆盖率,另一个菌株的覆盖率从 0.001 到 15 倍不等。FDR包括纯合位点和杂合位点。
总体而言,GT-Pro的 FDR与 MIDAS 相似但低于 metaSNV。
图d为对图c的灵敏度调查,敏感性是指正确判断reads所模拟的基因组的基因型(纯合位点和杂合位点)的概率。GT-Pro 的灵敏度低于基于比对的方法,基于比对的宏基因分型通常使用覆盖率和等位基因频率过滤来减少错误的杂合性调用。
图e为基于图a中模拟的等位基因,从tag SNPs推算的基因型的FDR。结果表示大多数物种的 FDR 较高但仍低于 5%。
图f和图g,为了探索 GT-Pro 是否能用于定量估计物种丰度,使用从单个分离株和对同种分离株中模拟的宏基因组,比较了sck-mer匹配reads的平均数量和已知的基因组覆盖率。结果表示GT-Pro等位基因的调用和计数可以用一个小的校正因子来估计物种和菌株的相对丰度。
所有的结果表示,在模拟宏基因组的测试中,metaSNV 和 MIDAS 对于丰富的物种(>5×覆盖度)和保守位点表现良好,但 GT-Pro 对典型覆盖率值、非参考和杂合位点更准确和敏感,同时对错配和测序错误更为稳健。只是,与 metaSNV 和 MIDAS 相比,GT-Pro 无法检测其数据库中缺少的新 SNP。
结论是,在保守的基因组区域仔细选择sck-mers能使 GT-Pro 能对来自鸟枪法宏基因组数据的已知 SNPs 进行敏感和特异性的基因分型。
2.从模拟宏基因组中准确识别SNP
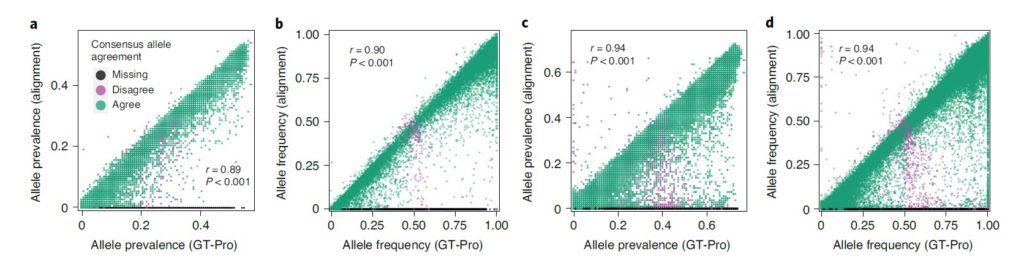
使用GT-Pro对肠道微生物组样本进行宏基因组分型,结果与基于比对的MIDAS宏基因组分型比较。
图a和b分别为流行率(prevalence)、平均等位基因频率(Average allele frequency)
图c和d类似图a和b,只是物种不同。
每个点代表一个 SNP,颜色表示两种方法的共有等位基因(即样品中最常见的)是否相同(绿色),两种方法都返回某些样品的基因型,但共有等位基因不同(紫色)或仅GT-Pro 返回基因型(黑色)。
结果表示对于高覆盖率物种,基于比对的方法能检测到GT-Pro数据库中没有的SNP,而GT-Pro 在中低覆盖率物种中检测到更多SNP位点。这部分结果也与模拟宏基因组测试时的结论一致。
1.使用GT-Pro的SNP估算结构变异
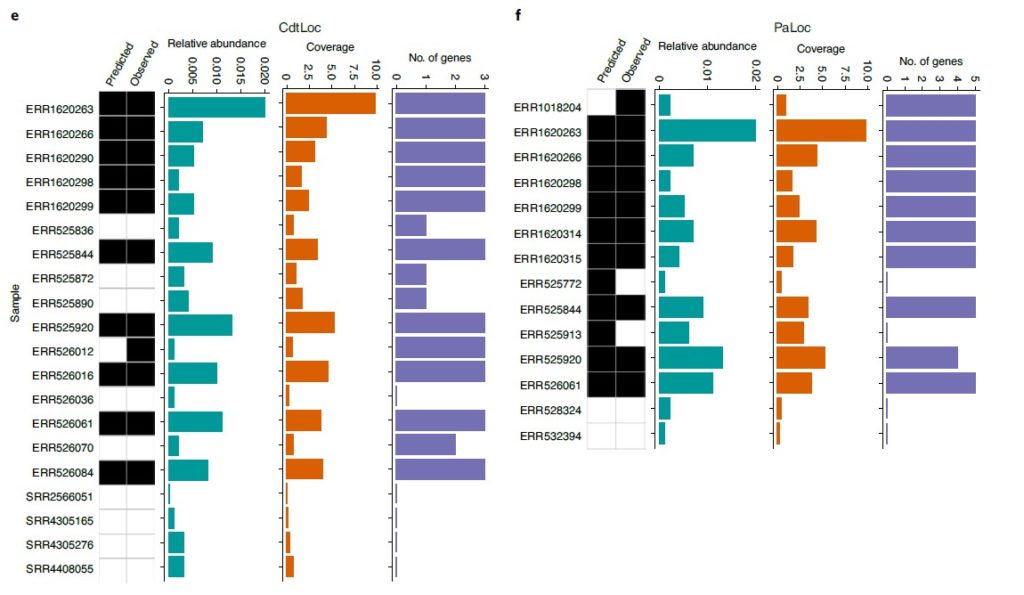
研究人员试图使用GT-Pro的SNP推断附近基因或操纵子的存在,从而作为结构变异的生物标志物。
首先对艰难梭菌的毒性控制位点CdtLoc和PaLoc的侧翼区域使用GT-Pro检索SNP。
接着用艰难梭菌的参考基因组训练了一个随机森林分类器,用于预测来自混合群组(n = 7,459)的人类肠道宏基因组中存在/不存在艰难梭菌毒素基因位点。
图e和f分别代表CdtLoc基因和PaLoc基因,对每个样本,最左边的热图,第一列为预测的,第二列为基于比对方法得到的,黑色表示存在,白色表示不存在。
从左到右的条形图分别指艰难梭菌的相对丰度、全基因组序列覆盖率,从毒素位点检测到的基因数目,所有这些都是通过比对到艰难梭菌的代表性基因组来估计的。结果表示预测到艰难梭菌毒素位点的概率>0.6。
对CdtLoc的几个预测与宿主的表型相关(P < 0.001),包括5名艰难梭菌阳性和CdtLoc(+)的克罗恩病患者,这与该人群对艰难梭菌病理的高易感性相一致。与此相反,CdtLoc基因座在大多数可检测到艰难梭菌的健康婴儿中没有被预测,这与婴儿期艰难梭菌常见的无症状定殖一致。这些结果表明,GT-Pro可以预测具有临床相关性的linked structural variants。
2. 使用 GT-Pro 捕获新的种内遗传结构
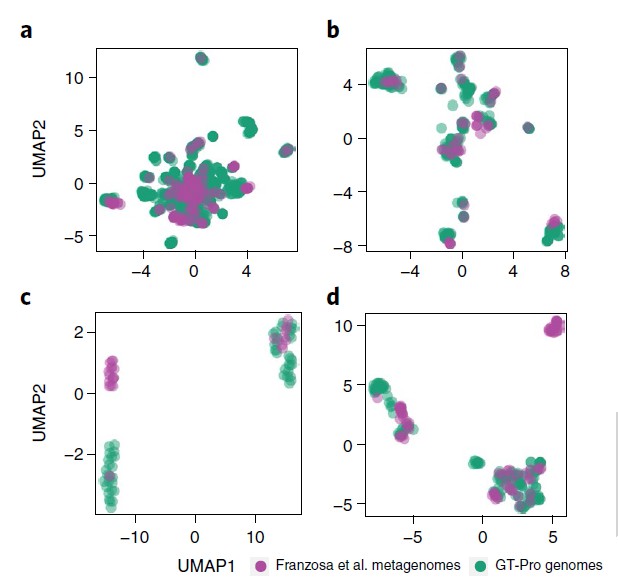
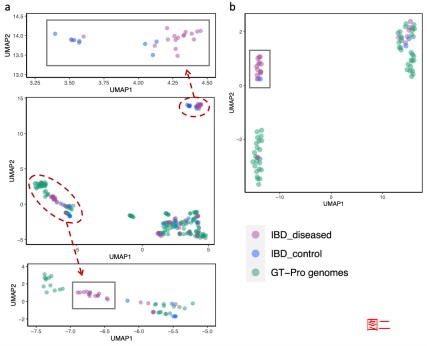
GT-Pro 可以对从参考基因组中鉴定的已知 SNP 进行宏基因组分型分析。但研究人员认为GT-Pro还有更广阔的发展,假设GT-Pro可以基于 SNP 等位基因的不同组合检测新的菌株变异。
为了验证该假设,研究人员使用GT-Pro 对最近发表的北美炎症性肠病 (IBD) 队列 的 220 个粪便宏基因组中发现的物种进行基因分型。使用UMAP降维分析,每个图都是将UMAP应用于一个物种GT-Pro SNPs基因型矩阵的结果。每个点代表该物种的一个菌株(杂合宏基因组的主等位基因)。紫色为队列样本,绿色为GT-Pro基因组。
结果表明 GT-Pro 的数据库代表了这些个体的常见菌株多样性,对于大多数物种,如图一的a和b,粪便样本组与参考基因组聚集在一起,相比之下,对于少数物种。
如图二的c和d,分别是新的亚种,观察到基因型与数据库中任何参考基因组不同的粪便样本群,包括一些富含IBD患者的样本。这说明可以使用 GT-Pro 常见 SNP 发现新的亚种遗传结构。
3. GT-Pro 探索全球人类肠道微生物组遗传变异
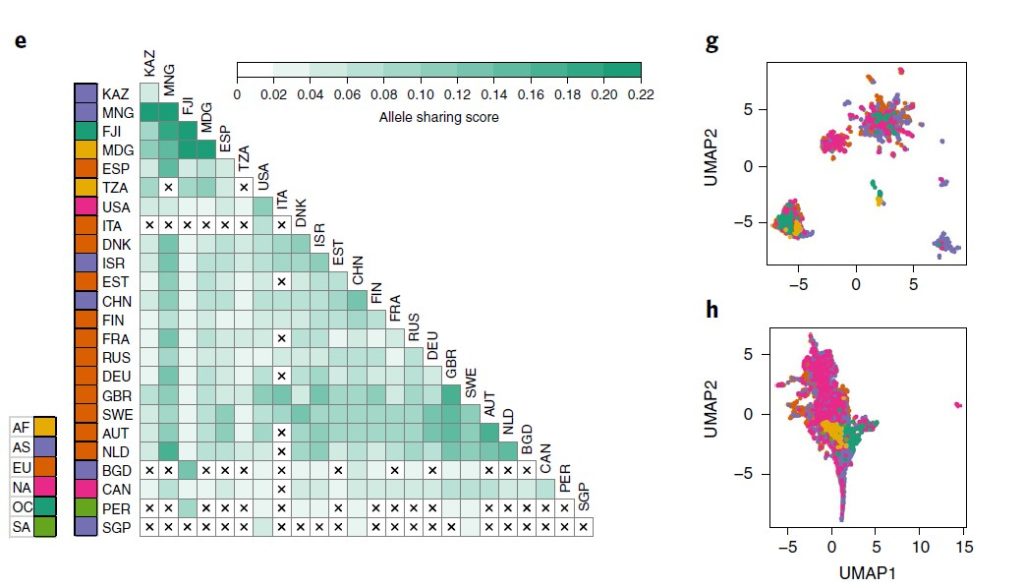
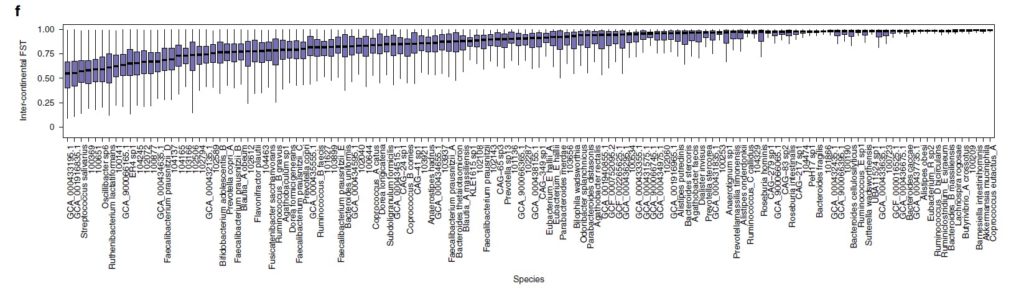
来自六大洲 31 个地点的 7,459 个肠道样本中发现的 881 个物种的 5180 万个 SNP的多个物种的种内遗传变异荟萃分析。
图e来自不同国家的宏基因组间的等位基因平均共享分数的热图。打叉单元格表示由于样本对不足(<5,000)而导致分数缺失。
图f为78 个常见物种的洲际种群分化分析(大陆内部与大陆之间的遗传相似性,用 F 统计检验测量亚种群 (FST) 中捕获的总遗传变异的比例)。
每个箱线图代表一个物种的洲际 FST 分布,按中位数排序。图g为通过直肠Agathobacter rectalis(物种ID 102492)的GT-Pro宏基因组基因型中的种内遗传变异捕获的地理模式的示例。
图h为为基于图g中相同样本的物种相对丰度的UMAP 分析。每个点都是一个宏基因组样本。颜色与图e示意一致。
结果表示,等位基因共享与工业化程度以及宿主关系明显关联;洲际种群间的分化程度有巨大差异,具有高FST的物种显示出明显的宿主集群,但不是所有宿主集群都与地理相关。
这与菌株在宿主中殖民的生活方式和环境的作用相一致。相比之下,在基于物种相对丰度的UMAP分析中,宿主间并没有明显集群,这表明宏基因组基因型可能揭示了在丰度分析中缺失的微生物生态学和微生物群落-宿主关系。
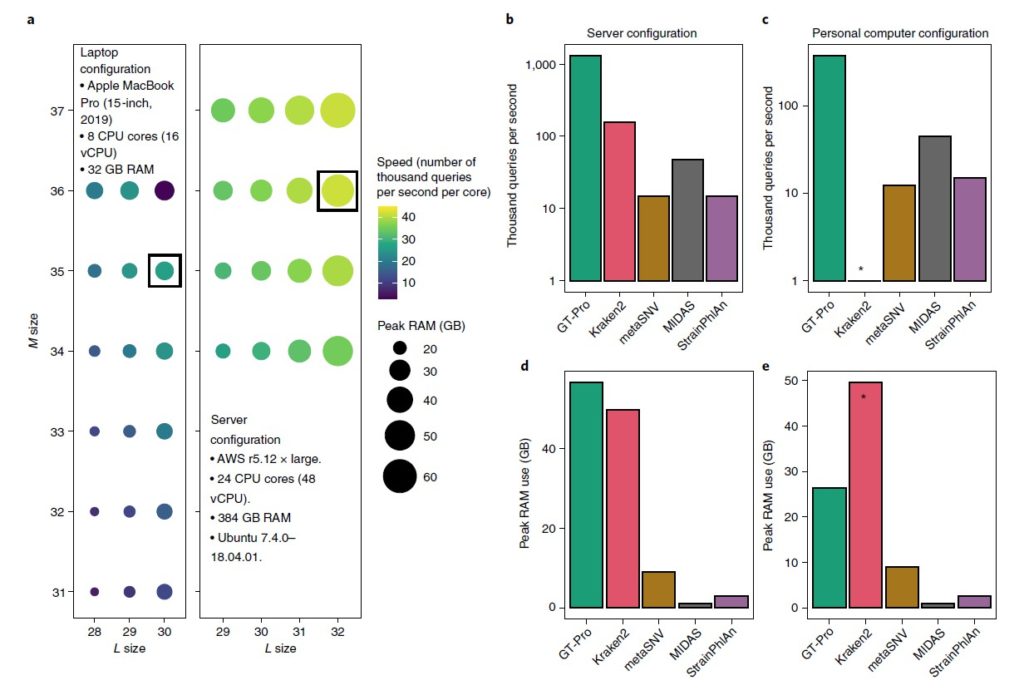
图a评估GT-Pro在笔记本电脑(左)和服务器环境(右)中的计算性能,以bits为单位。颜色表示处理速度,圆圈大小为RAM使用峰值。黑色方框表示最优状况。
图b-c为GT-Pro与metaSNV、MIDAS、StrainPhlAn、 Kraken2之间的速度比较,分别在服务器环境和笔记本电脑下比较。
图d-e为RAM使用峰值的比较,分别在服务器环境和笔记本电脑下比较。* 由于超出可用 RAM,Kraken2 无法在笔记本电脑中运行。
这些分析表明,与其他方法相比,GT-Pro 在服务器上大约快 8.5-570 倍,在笔记本电脑上快 8.3-163.6 倍。平均而言,处理每个宏基因组只需要在服务器上不到 4 秒,在笔记本电脑上大约需要 13 秒(平均为 497 万次读取)。虽然 GT-Pro 比其他方法更快,但它在服务器上需要 1.1-53.7 倍的 RAM和笔记本电脑上的 2.9-29.2 倍的 RAM(不包括内存不足的 Kraken2)。因此,只要计算机具有足够的 RAM,GT-Pro 数据结构和算法就可以极大地加速宏基因组分型。
研究人员在该文章中使用GT-Pro大约分析了2.5万个宏基因组,展示了GT-Pro是如何快速准确的识别SNP以及探索结构变异、种内遗传变异等。GT-Pro不使用基于比对的方法,而是类似于Kraken2,通过编码k-mers来快速检索,并适用于个人计算机或服务器环境。
但是它也不是完美的,目前GT-Pro存在的不足和如何应对:
第一,GT-Pro 数据库并未捕获所有人类肠道微生物多样性:但是通过基因组测序,会持续扩大SNPs的数量和涵盖的物种。
第二,GT-Pro 类似于基因分型阵列,因此不能识别新的 SNP,这需要其他方法,例如基于比对的宏基因组分型或单细胞基因组测序。
第三,由于基因组集合中存在高度相关的物种,少数物种缺乏物种特异性的 sck-mers。替代策略,例如使用更长的 k-mer 或不太常见的 SNP,可以对这些物种使用 GT-Pro 。
第四,尽管非常严格的挑选了用于构建GT-Pro的基因组和SNPs,但不可能完全排除错误(例如,不完整、污染和物种错误分类)。
最后,GT-Pro 不直接对结构变异进行基因分型。
“
考虑几个GT-Pro的未来发展方向,比如:
将GT-Pro与下游算法结合起来,以识别代表新微生物菌株的SNPs簇,或准确标记参考数据库中已知菌株的SNPs;
将GT-Pro的计算框架扩展到其他微生物环境中;为短插入缺失和结构变异开发无比对宏基因组分型;
将微生物组应用于精准医学,综合识别与疾病或其他特征(如致病性、抗菌耐药性、药物降解)相关的SNPs;
将GT-Pro用于检测污染、重组和跟踪变化,比如变异或菌株随时间、宿主生活方式和地理位置的变化。
主要参考文献
Shi ZJ, Dimitrov B, Zhao C, Nayfach S, Pollard KS. Fast and accurate metagenotyping of the human gut microbiome with GT-Pro. Nat Biotechnol. 2021 Dec 23. doi: 10.1038/s41587-021-01102-3. Epub ahead of print. PMID: 34949778.
谷禾健康
分类分析的研究,依赖于高质量的序列分类参考数据库,然而,目前已有记录公共序列数据库中出现错误,这些错误可能导致下游结果出错。不同的参考数据库对生物数据的分类结果差别很大,但缺乏客观评价单个数据库质量的标准。
有人选择自行构建特定于环境的数据库,但生成这样的数据库在技术上具有挑战性,导致了研究人员难以获取适当参考材料,或者对专有资源和服务有很大的依赖性。
为了满足可重复的生物信息学工作流程,以简化数据库生成和管理,来自阿肯色大学的Michael等人开发了一款新的工具——RESCRIPt. 该文章最近发表在《PLOS COMPUTATIONAL BIOLOGY》上。

RESCRIPt是一个独立的python3软件包,也是QIIME 2插件。用于参考序列分类数据库的可重复构建和管理,主要功能是格式化主流的公共数据库内序列用以自建分类数据库,由于处理步骤是透明化的,所以用户可以为不同的研究应用创建参考材料。
次要功能有评估、比较和交互探索参考数据库的定性和定量特征的功能。RESCRIPt使用QIIME 2文件格式,对每个处理步骤都生成专一的文件存储,使用户可以随时追溯任一计算步骤。
文章中,作者使用RESCRIPt对几个常用的16S rRNA基因、ITS和COI序列的参考数据库利用RESCRIPt进行了评估,并探讨了RESCRIPt目前存在的问题和未来的目标。
RESCRIPt处理和管理参考数据库的工作流程
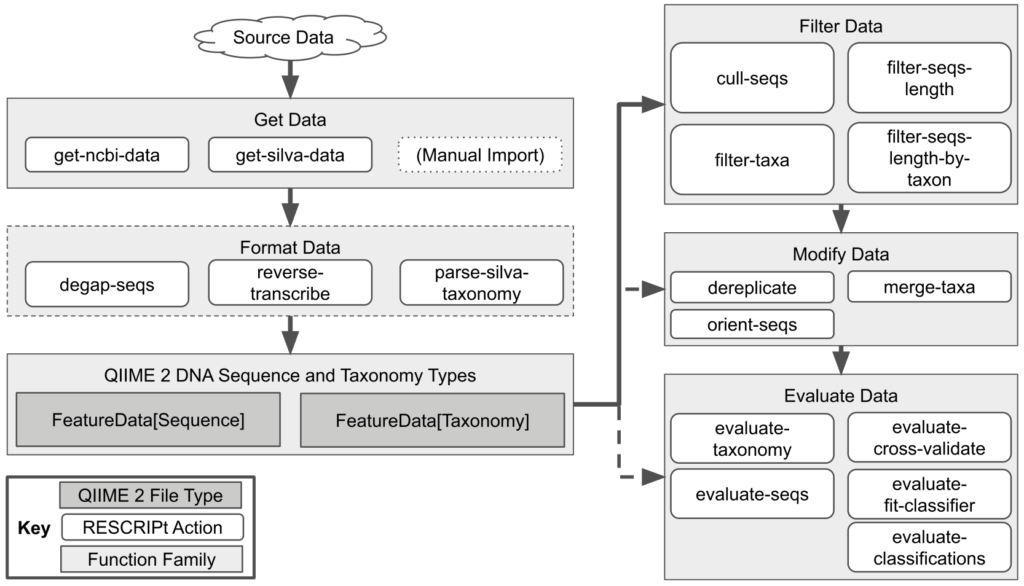
实线箭头表示建议的流程。虚线的箭头和边框表示自定义工作流程时的可选步骤。
RESCRIPt可以有效和透明的构建任何存在源数据的扩增子的参考数据库,以及来自NCBI的全基因组。
“Get Data”:获取源数据,可以直接从SILVA和NCBI GenBank数据库中自动下载序列和分类。
“Format Data”:格式化数据,包括基本的序列操作、逆转录和解析分类。
“Filter Data”:过滤数据,根据序列的质量或长度过滤以及根据分类和分类单元所在的序列长度过滤。
“Modify Data”:修改数据,去重复、合并分类或聚类。
“Evaluate Data”:评估, 对序列的一般质检,以及对分类准确率的评估。
详细的操作命令,见:
bokulich-lab/RESCRIPt: REference Sequence annotation and CuRatIon Pipeline (github.com)
RESCRIPt比较评估目前常用的四种16S rRNA基因数据库,分别为SILVA、Greengenes、GTDB和NCBI-RefSeq
从结果上看,在这些数据库中,SILVA数据库展示了最多的唯一序列和物种数,但是SILVA缺乏种水平的分类管理,其在种水平的分类准确率为0.73,远远低于其他16S rRNA基因数据库。相比之下,SILVA在属水平上的分类准确率要高得多。
NCBI-RefSeq的参考序列质量最高,分类准确率为0.94。
GTDB表现出略低的分类准确率0.92。
Greengenes13_8含有大量独特的序列和与SILVA相似的序列信息熵,但在属(54%)和种(90%)水平上有许多没被注释的序列。这表明该数据库中的大量序列在遗传上相似(≥98%),但在分类上是不同的,产生了不明确的标签。
各数据库的序列信息
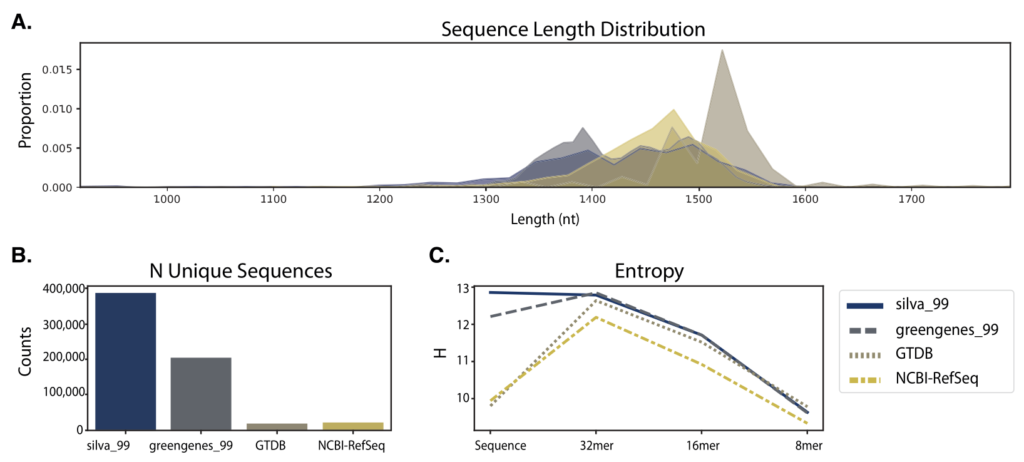
图A. 序列长度分布(去除异常值后);
图B. 每个数据库中唯一序列的数量;
图C. 每个数据库中全长序列和不同kmer长度的熵。
各数据库的分类信息和模拟分类的准确率比较
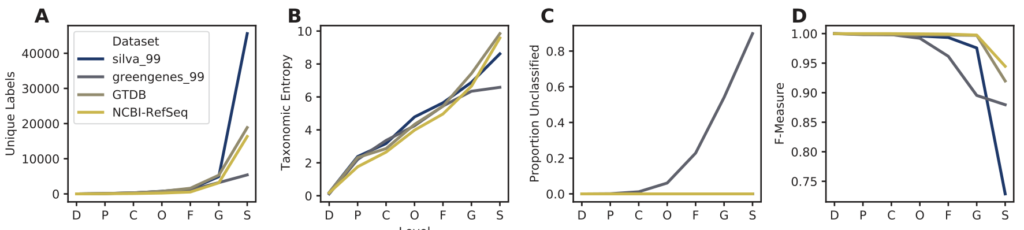
图A.唯一分类标签的数量。 图B.分类熵。
图C.在每一层级上未分类物种的比例。 图D.分类准确率。
横轴表示分类水平域门纲目科属种。
各数据库的分类覆盖率比较
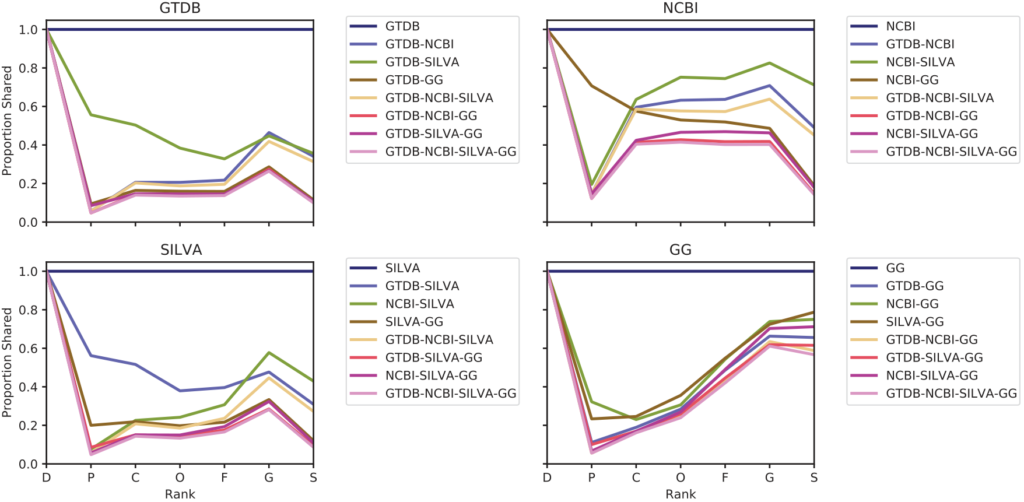
每张子图表示该数据库与其他数据库在每个分类水平上共享的分类群比例。图例指出了要相互比较的数据库。
RESCRIPt比较评估不同过滤步骤对16S rRNA基因SILVA数据库的影响
RESCRIPt使用get-silva-data命令获取SILVA序列和分类文件。“get-silva-data”命令允许选择下载哪个版本的数据库,是否下载LSU、SSU序列或SSU NR99序列,以及使用哪个分类水平和分类解析的选项等其它选项。
对16S rRNA基因SILVA数据库中每个连续序列使用不同RESCRIPt的质量过滤步骤后的序列信息比较
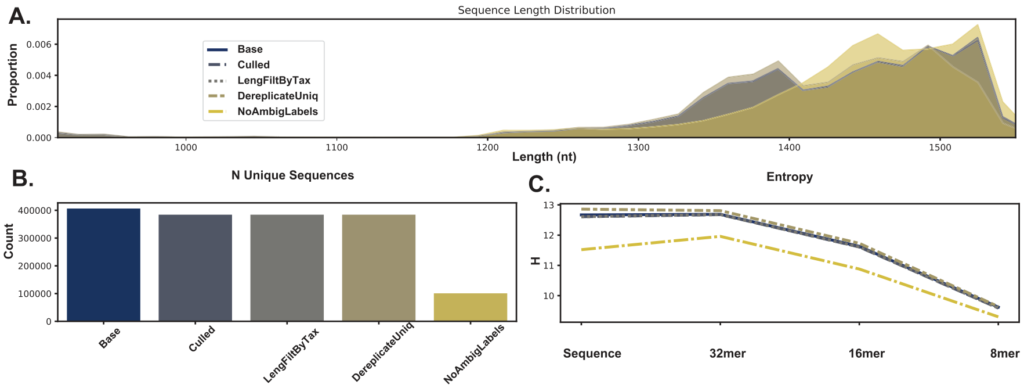
图A.序列长度分布。图B.唯一序列的数量。
图C.全长序列和不同kmer长度的熵。
图例中Base指完整的NR99 SILVA数据库;Culled指在序列中去掉8个或更多的均聚物(homopolymers)和/或5个具有歧义的碱基(ambiguous bases);
LengFiltByTax指基于分类学对数据进行序列长度过滤,即去除长度小于900 bp和小于1200 bp的古菌和细菌序列;
DereplicateUniq指使用“uniq”模式对分类和序列去重,即任何具有不同分类的相同序列将不会被合并;
NoAmbigLabels指任何与具有歧义的标签(通常在较低的分类级别) 相关的序列都从数据集中删除。
结果表示Culled和LengFiltByTax步骤对序列的影响是有益的,而NoAmbigLabels方法会过多丢失序列信息。
各过滤步骤下序列分类信息和模拟分类准确率的比较
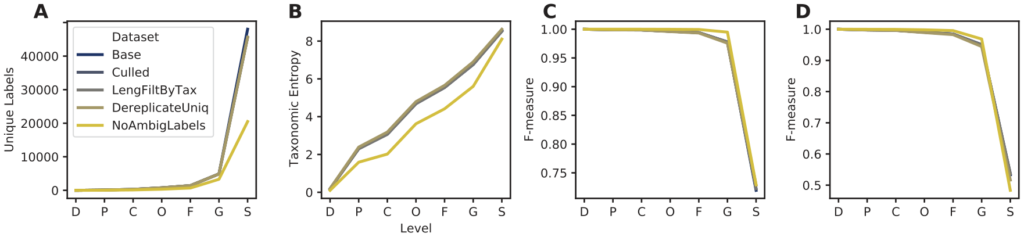
图A. 唯一分类标签的数量。图B.分类熵。
图C. 无需交叉验证的最佳分类准确率(当真实标签已知,但分类准确率可能被数据库中其他类似的命中混淆时,模拟可能的最佳分类准确率)。
图D. 使用交叉验证的分类准确率(在不知道正确标签的情况下模拟真实的分类任务)。
横轴表示分类水平域门纲目科属种。除了NoAmbigLabels的分类注释外,质量过滤对分类准确率的影响微乎其微。
RESCRIPt评估在多个OTU%相似性阈值下聚类的Greengenes数据库(13_8版本)的多个数据库质量特征
结果表示相似性阈值的降低导致了信息丢失,在属和种水平上,唯一分类标签的数量迅速减少。相反,相似性阈值的增加使得分类准确率上升。
这表明,即使选择了认为合适的相似度阈值也可能对数据库的信息内容和分类准确率产生负面影响。但作者还是建议不要在任何标记基因序列数据库中使用相似度<99%的OTU聚类。
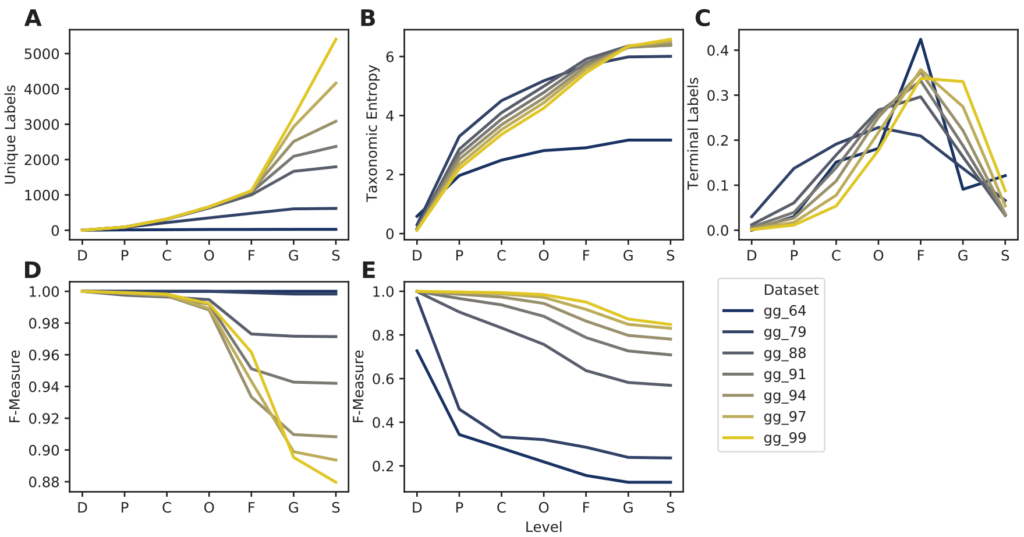
图A. 唯一分类标签的数量。 图B.分类熵。
图C. 在每一层级里分类单元的数目。
图D. 无需交叉验证的最佳分类准确率(当真实标签已知,但分类准确率可能被数据库中其他类似的命中混淆时,模拟可能的最佳分类准确率)。
图E. 使用交叉验证的分类准确率(在不知道正确标签的情况下模拟真实的分类任务)。
横轴表示分类水平域门纲目科属种。图例指示不同的OTU%相似性阈值。
RESCRIPt评估不同处理步骤下的UNIT ITS真菌序列数据库
结果表示OTU聚类方法里,97%比99%比动态聚类,对结果的影响最小。含所有真核生物的数据库所包含的序列是仅含真菌序列数据库的两倍多,但其分类准确率是最低的。
而只含目水平或更低级别分类水平的真菌序列数据库在分类准确率上提升最大。
对UNIT ITS数据库的三种类型UNIT_97,UNIT_99,UNIT_dynamic数据库分别进行划分
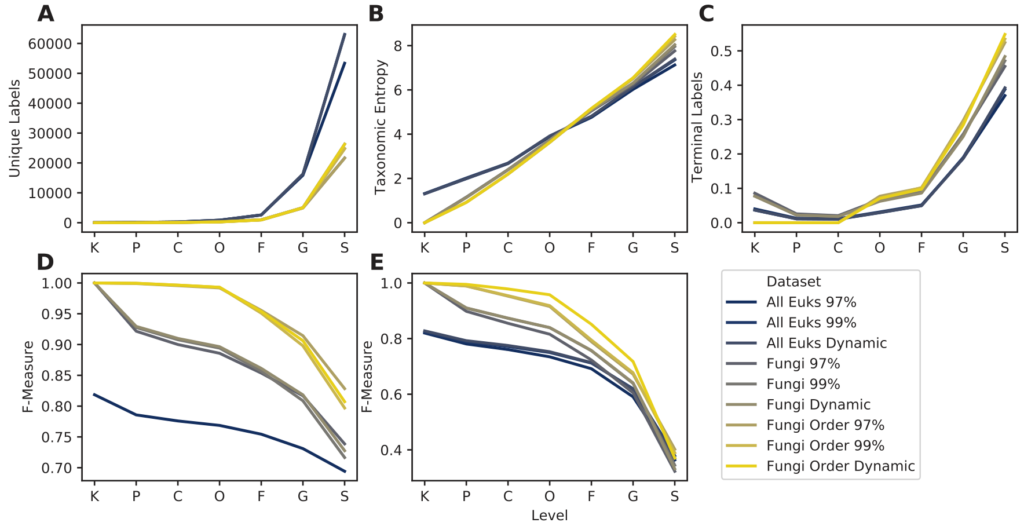
Euks表示含所有真核生物序列,Fungi表示只含真菌序列,Fungi Order表示只含目水平或更低级别分类水平的真菌序列。
图A. 唯一分类标签的数量。 图B. 分类熵。
图C. 在每一层级里分类单元的数目。
图D. 无需交叉验证的最佳分类准确率(当真实标签已知,但分类准确率可能被数据库中其他类似的命中混淆时,模拟可能的最佳分类准确率)。
图E. 使用交叉验证的分类准确率(在不知道正确标签的情况下模拟真实的分类任务)。横轴表示分类水平域门纲目科属种。
RESCRIPt评估用于后生动物分类鉴定的COL基因数据库
首先比较评估了不同序列处理步骤下的BOLD COL基因数据库(BOLD全称Barcode of Life Data Systems)。
结果表示聚类序列大大减少了未修剪和引物修剪的BOLD COI数据集中唯一序列的数量,经引物修剪也会降低唯一序列的数量。且在种水平上表现最明显。聚类和引物修剪也降低了分类准确性。数据表明OTU聚类不利于COI基因分类。
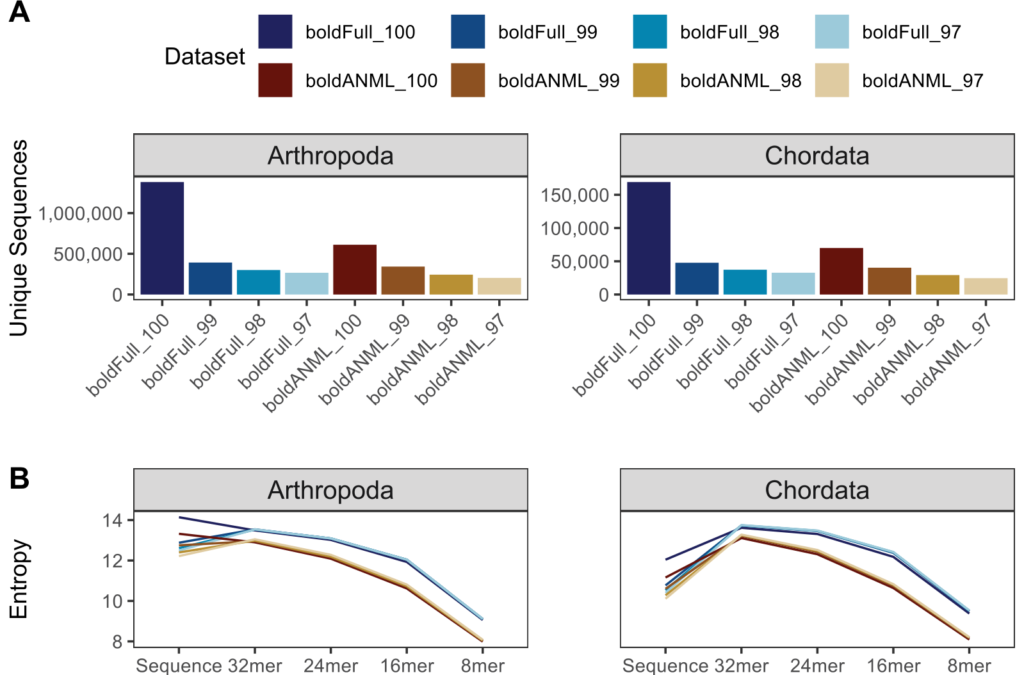
图例中Full表示未修剪的全长序列,ANML表示经引物修剪后的序列,后边接的数字表示相似性聚类阈值。Arthropod指节肢动物,chordate指脊索动物。图A.唯一分类标签的数量。图B.不同kmer长度的分类熵。横轴表示不同数据库。
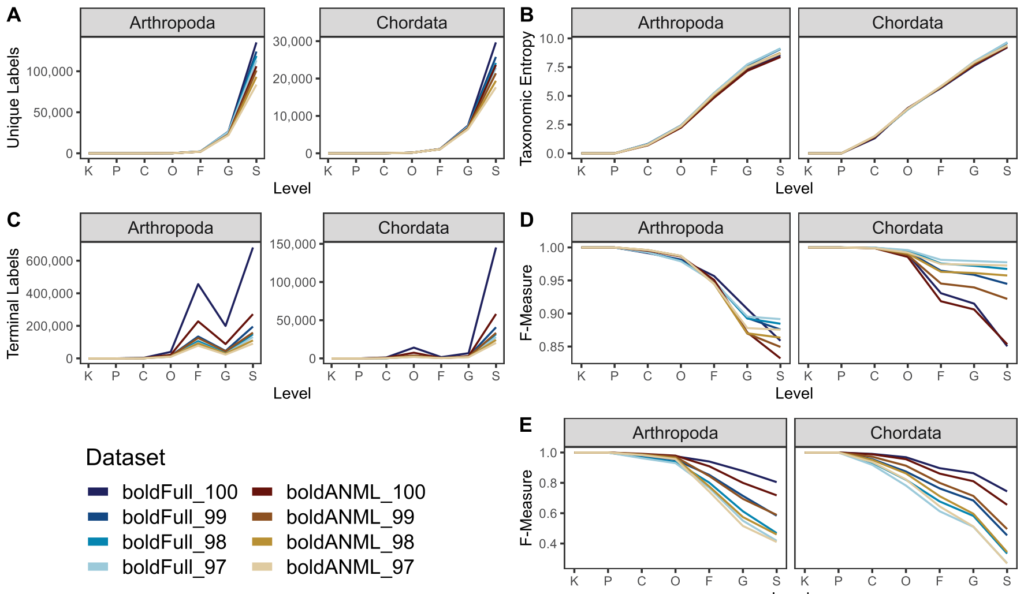
图A.唯一分类标签的数量。图B.分类熵。
图C.在每一层级里分类单元的数目。
图D. 无需交叉验证的最佳分类准确率(当真实标签已知,但分类准确率可能被数据库中其他类似的命中混淆时,模拟可能的最佳分类准确率)。
图E. 使用交叉验证的分类准确率(在不知道正确标签的情况下模拟真实的分类任务)。横轴表示分类水平域门纲目科属种。
其次评估比较了从BOLD或NCBI GenBank获得的去重复和引物修剪的COL基因数据库。
数据表明,整体看NCBI的唯一序列较少,但局部看,NCBI在属水平和种水平上有更多唯一序列。从分类准确率看,NCBI相对于BOLD,从科到种水平都有提高。
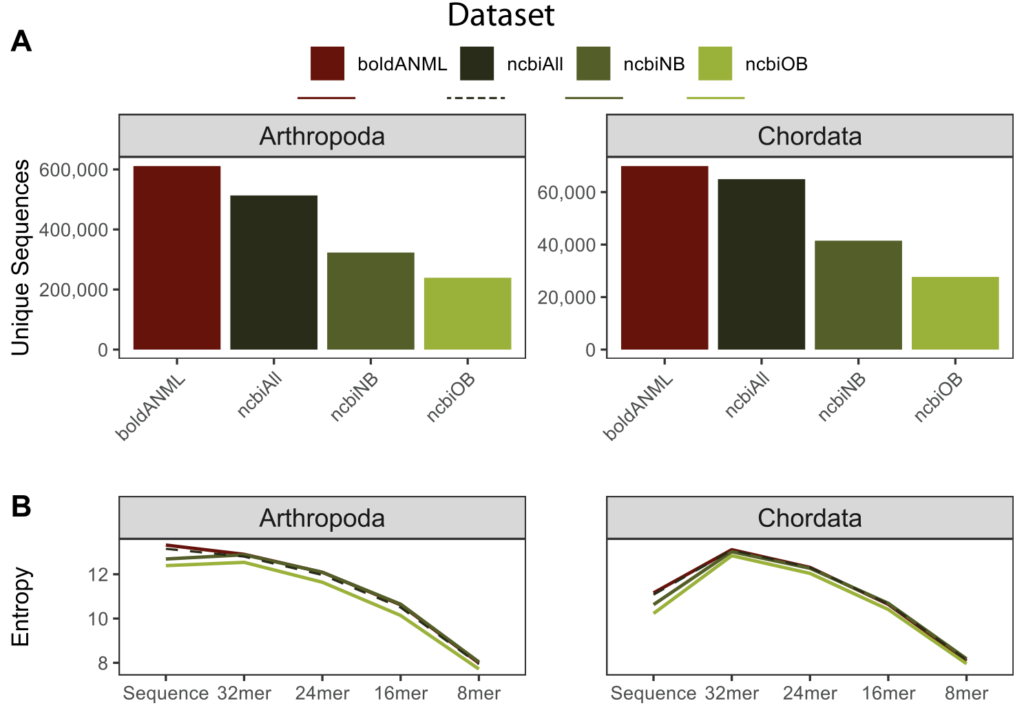
数据集分别为boldANML(BOLD COL基因数据库)、ncbiAll(ncbiNB与ncbiOB的集合)、ncbiNB(不含BOLD COL基因序列的NCBI GenBank COL基因数据库)、ncbiOB(含BOLD COL基因序列的NCBI GenBank COL基因数据库)。图A.唯一分类标签的数量。图B.不同kmer长度的分类熵。横轴表示不同数据库。
图A. 唯一分类标签的数量。 图B. 分类熵。
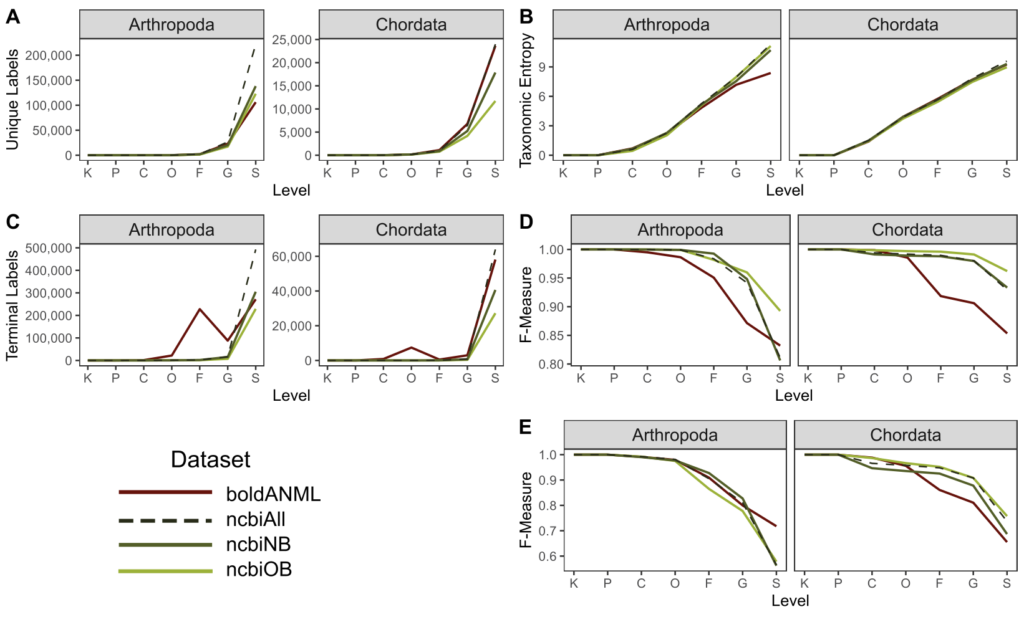
图C. 在每一层级里分类单元的数目。
图D. 无需交叉验证的最佳分类准确率(当真实标签已知,但分类准确率可能被数据库中其他类似的命中混淆时,模拟可能的最佳分类准确率)。
图E. 使用交叉验证的分类准确率(在不知道正确标签的情况下模拟真实的分类任务)。横轴表示分类水平域门纲目科属种。
RESCRIPt旨在为研究人员提供可重现的核苷酸序列和分类学数据库生成、整理和评估的工具。它不是一个数据源,也不是分类学、系统学或数据质量方面的权威,并且RESCRIPt生成的评估结果也不是质量或准确性的可靠指标。
与任何生物信息学方法一样,RESCRIPt输出的质量取决于其输入的质量和用户作出的处理决策。一般来说,用户应该使用多个指标来指导他们对RESCRIPt结果的解释,但在对数据库质量作出结论之前,还需要了解输入数据的组成。
RESCRIPt目前的版本已经兼容宏基因组数据库。未来将计划提供更多的基因组和宏基因组功能。例如用于(元)基因组距离估算的ANI和MASH方法,以及用于(元)基因组数据库分类精度估算的方法。会增加从学界里常用的公共在线数据库中获取序列和分类的方法。
RESCRIPt作为一个Python3软件包和QIIME 2插件,可以用conda安装也可以docker运行,或者在已有的qimme2环境中安装。
通过RESCRIPt工具可以独立完成序列的获取、修剪、过滤、去重、聚类,整合为数据库,并且可以对多个数据库进行评估比较。每个处理步骤会有独立的日志文件生成和中间文件生成,便于溯源和重现该流程。只是庞大的数据库和庞大的功能在计算资源消耗这方面肯定不容小觑,虽然文章中没有提及这方面的内容,但作为使用者不能忽视。
关于安装和测试使用还是要仔细阅读官方手册,地址:
参考文献:
Robeson MS 2nd, O’Rourke DR, Kaehler BD, Ziemski M, Dillon MR, Foster JT, Bokulich NA. RESCRIPt: Reproducible sequence taxonomy reference database management. PLoS Comput Biol. 2021 Nov 8;17(11):e1009581. doi: 10.1371/journal.pcbi.1009581. PMID: 34748542; PMCID: PMC8601625.

谷禾健康

在细菌全基因组测序数据分析中,区域(regional)和功能注释已经成为常规操作。一个完整的基因组注释是下游分析的基础,而注释的准确性和全面性往往影响研究结果。
最近来自德国一个生物信息学团队们开发了Bakta,一款新的命令行工具,用于自动化和标准化的细菌基因组注释。该文章最近在《MICROBIAL GENOMICS》公开发表。

作者认为现有的各种注释软件工具都在以下问题留下了改进的空间:
1. 尽管早在20年前就发现了以前被忽视的保守的短ORF(sORF),但它们既不能预测也不能检测到短于29个氨基酸的小蛋白的编码序列(CDS),因为在基因预测工具中实施了基因长度截断,以减少错误的从头预测的数量。
2. 它们不识别存储在公共数据库(如RefSeq和UniRef100)中的已知蛋白质序列,因此不能分配数据库交叉引用(dbxref),即稳定的公共数据库标识符,方便与更详细的数据库互连。
3. 对于跨越人工序列边的CDS结构注释,没有考虑附加的序列信息,即完整性和拓扑结构。
为了解决这些问题,团队们开发了Bakta。它为编码和非编码基因提供了全面的注释工作流程,并加之CRISPR阵列、gaps、oriC和oriT特征的预测。与其他轻量级注释管道不同,Bakta能够通过自定义的sORF提取和过滤步骤来检测和注释小分子蛋白。而CDS注释流程通过一种基于哈希的、无需比对的蛋白质序列识别方法。
注:CRISPR,clustered regularly interspaced short palindromic repeats
此外,这种新的方法便于通过稳定的标识符交叉引用公共数据库来标注CDS。
软件工作流程如下图。
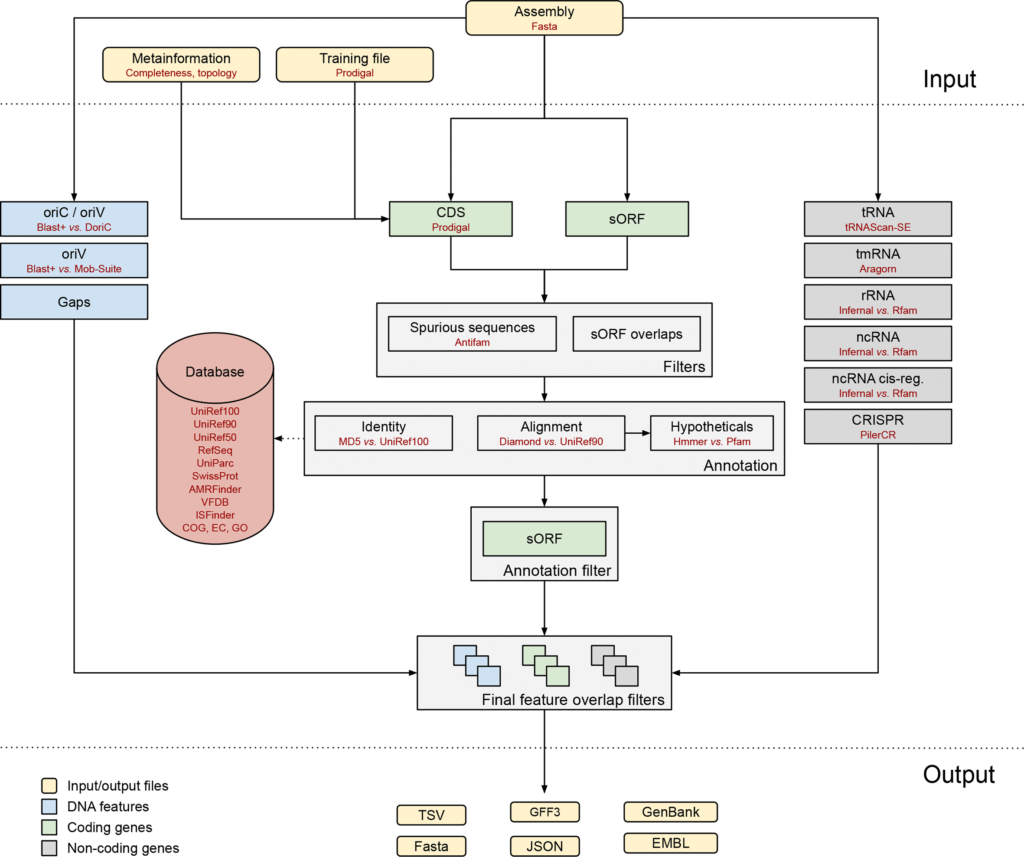
输入文件为fasta格式的基因组组装序列。
可选择输入序列元数据文件或Prodigal软件提供的training文件。非编码基因,比如tRNA使用tRNAscan-SE预测和注释。
gaps、oriC和oriT等特征的预测用BLAST+工具。利用Prodigal预测CDS,使用BioPython提取短于30个氨基酸的小蛋白的sORF。
HMMER和AntiFam分别过滤假阳性序列和重复的sORF。为了加快对CDS和sORF的注释,使用无比对序列鉴定(AFSI),即通过全长蛋白序列MD5哈希算法和相关蛋白质序列长度检查进行鉴定的组合过程。使用Diamond和UniRef90比对识别剩余的未识别蛋白质序列。
Bakta有自建的SQLite数据库,用于识别查找UniRef100, UniRef90、UniRef50、RefSeq、COG、EC 、GO、耐药基因、VFDB等。对于还是没有明确注释的CDS标记为假设蛋白(Hypotheticals),通过HMMER使用Pfam HMM图谱筛选蛋白质结构域。
通过与其它软件工具进行基准测试,评估Bakta的性能。
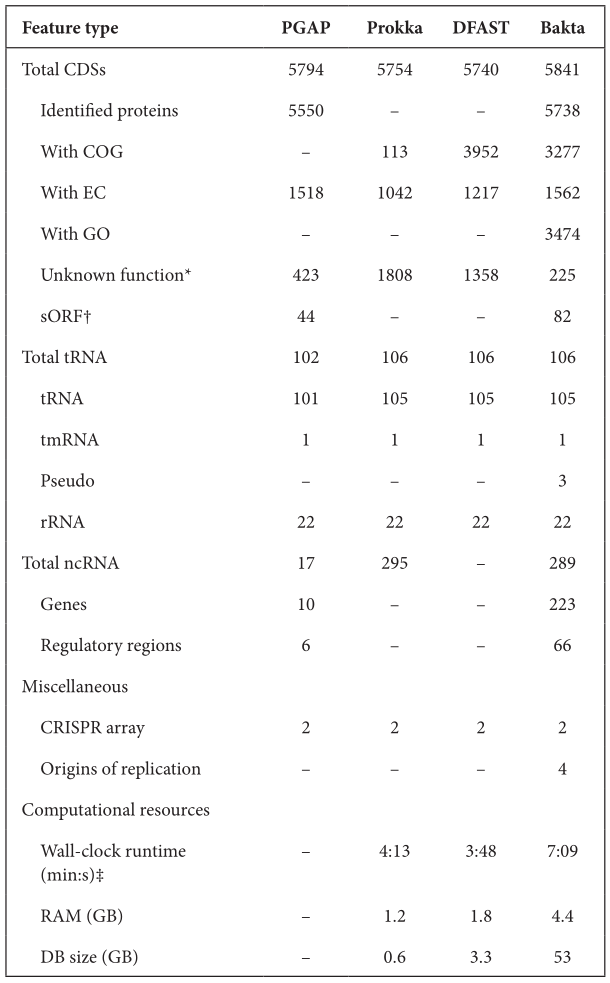
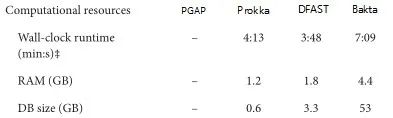
首先是注释得到的特征结果之间的比较。作者选择E. coli O26 : H11 strain 11368菌株的基因组,分别使用Prokka、DFAST、PGAP与Bakta比较,如下表。对于CDS,PGAP和Bakta预测到更多的基因。在CDS序列的功能注释方面,PGAP和Bakta表现最好,且Bakta是唯一一个分配到GO术语的工具。

其次是功能注释的性能基准测试。选择来自RefSeq的35个不同分类的细菌基因组进行注释。统计其假设蛋白占总CDS的比例,如小提琴图。
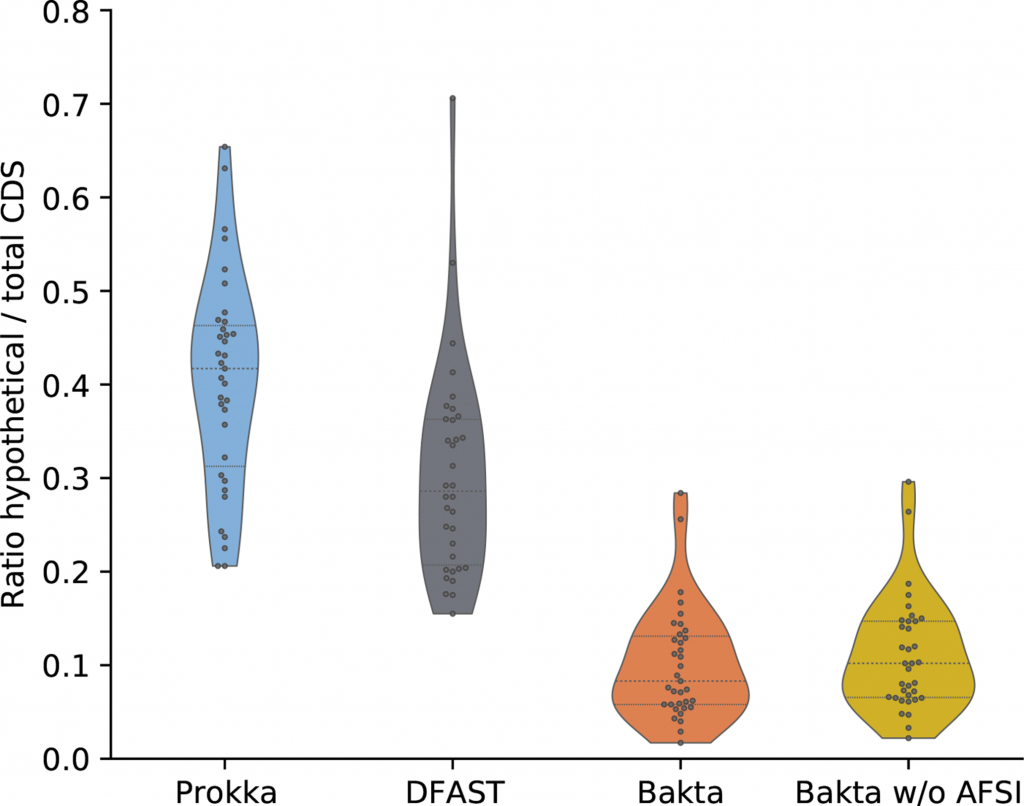

同时统计了在没有用AFSI(Bakta w/o AFSI,只是用Diamond比对序列)和使用AFSI的假设蛋白占总CDS的比例,两者之间的差异只有0.9%。
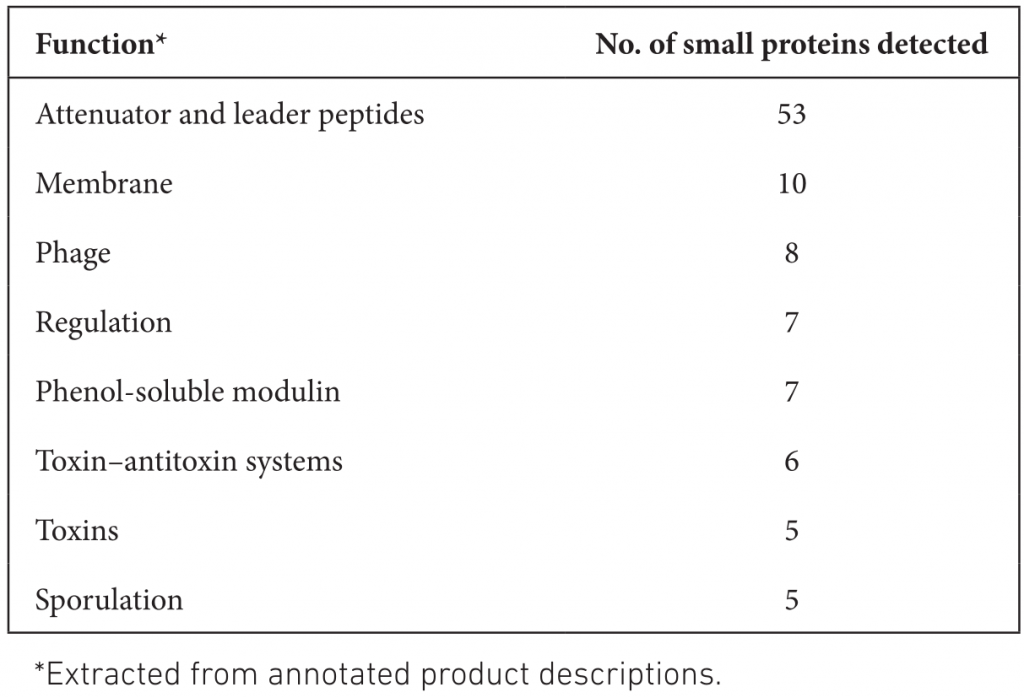
由此得出,AFSI对RefSeq中检测到的小蛋白的功能注释贡献很小。表格中展示了Bakta检测到的小蛋白参与的一系列与致病性高度相关的重要过程,以及更一般的细胞内务管理过程。

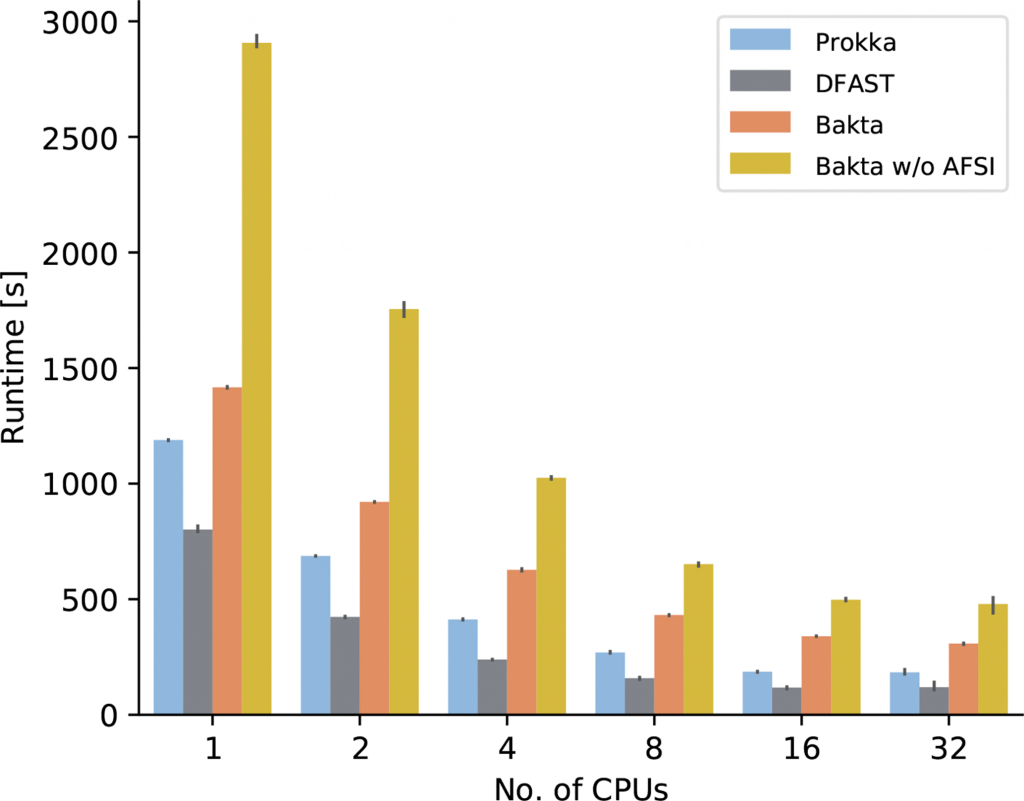
最后比较了Bakta的运行时间、内存消耗和存储需求。在具有4个Intel Xeon E5-4627CPU和总共40个核的服务器机器上,使用不同数量的CPU连续三次测量注释E.coli O26:H11 strain11368的PROKKA、DFAST、Bakta和不使用AFSI的Bakta的运行时间。
结果如下图和表格。Bakta虽然运行时间时最慢的,但它所包含的数据库内容是最多的,其分析深度也有很大提高。对比没有使用AFSI的Bakta,在同等条件下,使用AFSI大大提高了序列注释的速度。

Bakta可以对宏基因组的MAG进行注释,在与DFAST和Prokka的比较中,Bakta依旧是假设蛋白占总CDS比例最低的;注释结果格式符合INSDC标准;在线版Bakta应用程序,提供交互式GUI向导,可输入数据与命令行工具一样,适合不太熟悉命令行操作的研究员,地址:bakta-web-ui (computational.bio)
通过以上的工作流程介绍和性能评估,该软件有如下优势:
1 Bakta在已知和未知物种的分类范围内对CDS序列的功能注释方面优于现有工具
2 Bakta能够检测和注释当代预测工具无法预测的小蛋白,比如在使用Prodigal和MetaGeneAnnotator工具预测时
3 Bakta能够精确识别已知的蛋白质序列,并分配RefSeq和UniProt数据库标识符
4 新的AFSI方法加速了Bakta的功能注释工作流程
5 Bakta利用序列元数据改进了CDS的结构预测
6 Bakta以功能类别(COG、EC和GO)为CDS提供了同等或更全面的注释。
目前看来,较为明显的缺点就是运行时间长,虽然提供了Web版本,但如果样本数量较大,还是需要在linux上运行。
参考文献:
Schwengers O, Jelonek L, Dieckmann MA, Beyvers S, Blom J, Goesmann A. Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb Genom. 2021 Nov;7(11). doi: 10.1099/mgen.0.000685. PMID: 34739369.
谷禾健康
人类微生物组的变化与许多疾病和健康状况有关。然而,看懂人类微生物组研究结果的报告具有挑战性,因为它通常涉及微生物学、基因组学、生物医学、生物信息学、统计学、流行病学等领域的方法。
人类微生物组的研究与其他类型的分子流行病学研究具有许多共同特征,但它们也需要独特的考虑因素,具有自己的方法学最佳实践和报告标准。除了流行病学研究设计的标准要素外,独立于培养的微生物组研究还涉及生物标本的收集、处理和保存;不断发展的实验处理方法,具有更高的批次效应潜力;生物信息学处理;稀疏、异常分布、高维数据的统计分析;报告可能的数千种微生物特征的结果等。
由于微生物组研究没有公认的金标准方法,而且该领域还没有就这些方面形成共识,但是各个领域的人员共同努力逐渐形成适用于广泛的人类微生物组研究报告的规范流程是微生物领域快速发展的基础。
参 与 者
对研究参与者的描述中,应描述入组的人群,以及如何从人群中抽取参与者。
参与者的特征,例如环境、生活方式行为、饮食、生物医学干预、人口统计和地理都可能引起微生物组的显着差异,因此应该包含这些基本描述。
时间背景也很重要,因此应说明招聘、跟进和数据收集的开始和结束日期。
此外,还应包括用于评估潜在参与者是否符合研究资格的具体标准,包括纳入标准和排除标准的详细信息。纳入和排除标准是用于选择研究参与者的预先确定的特征,描述这些标准对于了解研究的目标人群至关重要。
应描述收集的有关可能影响微生物组的抗生素或其他治疗的任何信息,以及是否有任何排除标准包括最近使用抗生素或其他药物。
应说明最终的分析样本量,以及在招募、随访或实验室过程的任何步骤中排除参与者的原因。
建议使用流程图来显示参与者被排除在研究之外的时间和原因。例如如下流程说明。

Mirzayi C, et al., Nat Med. 2021
如果参与者在纵向研究中失访或未完成所有评估,则应说明如何进行随访的详细信息,并应报告特定时间点的样本量。
此外,将病例与对照进行匹配的研究应描述在匹配中使用了哪些变量。
实 验 方 法
应描述实验室样品的处理,包括样品采集、运输和储存的程序。
由于 DNA 提取可能是跨研究技术差异的主要来源,因此应描述 DNA 提取方法。如果进行了人类 DNA 去除和微生物 DNA 富集的描述,也应包括在内。同样,如果使用阳性对照、阴性对照或污染物减轻方法,则应对其进行识别和描述。
应描述报告与测序相关的方法。这包括引物选择和 DNA 扩增(包括16S rRNA 基因可变区,如果适用)。测序完成的主要单位(公司或者检测机构),例如鸟枪法或扩增子测序。最后,应解释用于确定相对丰度的方法。
批次效应应作为潜在的混杂来源进行讨论,包括为确保批次效应不与暴露或感兴趣的结果重叠而采取的步骤。如果进行宏转录组学、宏蛋白质组学或代谢组学,应提供这些方法的详细信息。
数 据 源 / 测 量
对于非微生物组数据(例如,健康结果、参与者的社会经济、行为、饮食和生物医学特征,包括疾病位置和活动以及环境变量),应描述每个变量的测量和定义。例如,参与者的性别和年龄可以从电子病历或分发给参与者的问卷中获得,那么应该清楚描述这个数据源的获得方式。还可以讨论测量的局限性,包括由于错误分类或丢失数据导致的潜在偏差,以及为解决这些测量问题所做的任何尝试。
因果推断的研究设计注意事项
在没有直接观察到假设的因果关系的情况下,观察数据通常用于测试旨在进行因果推断的关联。
方法包括,例如,使用多变量分析或匹配来调整假设暴露(例如微生物分类群的丰度)与研究中的疾病或病症之间的混杂变量。混杂因素可以被认为是暴露和研究结果的常见原因,可以导致暴露和结果之间的虚假关联。例如,年龄可能是一个常见的混杂因素,因为它会影响微生物组和大多数健康结果的风险。
如果不采取措施避免批次间条件的不平衡,实验室批次效应也可能混淆微生物组与感兴趣条件之间的关系。试图控制测量混杂的常用方法是调整或分层混杂。应为因果推断的回归模型中包含或排除的变量提供理由,因为对非混杂变量进行调整或分层会引入偏差。作为这一理论论证的一部分,作者应考虑包括一个有向无环图,显示假设的感兴趣的因果关系。
除了考虑本研究的理论动机外,还应讨论可能会扭曲微生物组与感兴趣变量之间观察到的关系的选择或生存偏差的可能性。例如,这种偏倚可能是由于失访(在纵向研究中)或由于疾病本身而没有将参与者纳入研究(例如,死于侵袭性结直肠癌和还没有幸存下来,无法参与结肠直肠癌微生物组的假设研究)。检查表中其他地方的其他项目可能与因果推断问题直接相关,包括假设、研究设计、匹配、偏倚和普遍性。鼓励调查因果问题的作者在因果推断的背景下考虑他们对这些项目的报告。
生物信息学和统计方法
对生物信息学和统计方法的充分描述对于生成严谨且可重复的研究报告至关重要。
应描述数据转换(例如标准化、稀疏和百分比)。应充分披露质量控制方法,包括过滤或删除读数或样本的标准。应说明用于分析数据的所有统计方法,包括如何选择感兴趣的结果(例如,使用P值、q值或其他阈值)。
应详细描述分类、功能分析或其他序列分析方法。为了重现性,所有用于数据预处理和分析的软件、软件包、数据库和库都应该被描述和引用,包括版本号。
可重复的研究
可重复的研究实践作为出版过程中的质量检查以及进一步的透明度和知识共享,如 Schloss 提出的标题中所详述。期刊越来越多地实施可重复的研究标准,包括数据和代码的发布,并且在可能的情况下应遵循这些指南。
如果可能,原始数据和处理过的数据,应存放在独立维护的公共存储库中,这些存储库可提供长期可用性,例如由 NCBI 或 EMBL-EBI 维护的公共存储库。Zenodo ( https://zenodo.org/ ) 或 Publisso (https://www.publisso.de/en/ ) 可用于为处理后的数据集提供 DOI。
如果数据或代码不公开或不能公开,即使在提供限制访问选项的存储库中,也应提供感兴趣的读者如何访问数据的描述。至少应描述任何受保护的信息,以及如何访问此类数据。
描 述 性 数 据
应报告关于研究人群的描述性统计数据。至少,应描述研究人群的年龄和性别,共享数据文件中应包括每位参与者的年龄和性别,但应尽可能报告其他重要的参与者特征,包括药物使用或生活方式因素,例如饮食。
作者应考虑在描述性统计表中如何报告这些数据。例如R 软件中的 table1 包等包,使创建这样的表不那么复杂。
结 果 数 据
研究的主要结果应该是详细的,包括描述性信息、感兴趣的发现和任何额外分析的结果。
应为每个组和每个时间点报告描述性微生物组分析(例如,降维如主坐标分析、多样性测量和总分类组成)。
这应为读者提供了差异丰度分析的结果。当报告差异丰度测试结果时,应明确说明每个可识别的标准化分类单元的差异丰度的大小和方向。其他类型分析的结果,如代谢功能、功能潜力、MAG 组装和 RNA-seq,也应在结果中描述。
附加结果(例如,非显著结果或完全差异丰度结果)可以包含在补充中,不应完全排除。
虽然这个问题已经存在了几十年,许多领域的期刊都认识到发表偏倚的问题,但在出版物中包含此类结果将有助于降低这种偏倚的严重程度,并改进未来的系统评价和荟萃分析。
讨论应包括对本研究和相关方法的局限性的讨论。应讨论偏差的可能性以及它们将如何影响研究结果。
许多形式的偏倚,例如残差/未测量混杂、与成分分析相关的偏倚、测量偏倚或选择偏倚,都可能影响对研究结果的解释,在讨论中承认潜在的偏倚来源很重要。
还应考虑研究发现的普遍性,以及这些发现是否适用于目标人群或其他人群。如果不同形式的偏见没有被评估或假设,可以忽略不计,但应说明这一点。
主要参考文献
Mirzayi C, Renson A; Genomic Standards Consortium et al. Reporting guidelines for human microbiome research: the STORMS checklist. Nat Med. 2021 Nov;27(11):1885-1892. doi: 10.1038/s41591-021-01552-x.
Wirbel, J. et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 25, 679–689 (2019).
Simoneau, J., Dumontier, S., Gosselin, R. & Scott, M. S. Current RNA-seq methodology reporting limits reproducibility. Brief. Bioinform. 22, 140–145 (2021).
Ten Hoopen, P. et al. The metagenomic data life-cycle: standards and best practices. Gigascience 6, 1–11 (2017). – PubMed – PMC
Yilmaz, P. et al. Minimum information about a marker gene sequence (MIMARKS) and minimum information about any (x) sequence (MIxS) specifications. Nat. Biotechnol. 29, 415–420 (2011). – PubMed – PMC