-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康

编辑
微生物组和组学数据集,由于其生物学性质,通常是高维的,特征常以各种成分,如基因、OTU、RNA转录本等的计数为特征。这些数据统称为成分数据。
这类数据分析的中心概念是对数转换,而其中最简单的策略是ALR(Additive log ratio)方法。对于高维数据,ALR方法有一下几个特点:
(a) 次要成分都是相干的
(b)可以解释100%的总对数方差
(c)测量结果非常接近于等距。
最近,来自西班牙科学团队的一篇题为“Compositional Data Analysis of Microbiome and Any-Omics Datasets: A Validation of the Additive Logratio Transformation” 的文章指出:
ALR对数转换可以有效提供一组简单的变量来表示整个成分数据集,其关键节点在于选择哪个成分为参考,并使用三个高维组学数据集进行验证。
通过ALR方法的理论和推导公式(这里不详述,推荐看原文),分别计算总对数方差(The total logratio variance 总结了采样点在多维空间中的分散程度),Logratio Geometry和Procrustes分析,以此找到有效的参照特征。再与其它对数转换方法对比,如CLR对数转换。
1. 兔子数据集
数据集为非零数据集,89个样本,3937个特征。
总对数方差为0.1601,Procrstes相关系数最高为0.9991,对应的基因数为856。该基因在3937个基因中的相对丰度排名第201位。
图一为所有3937个特征的Procrstes相关性直方图。为了直观地显示ALR变量接近等距的程度。
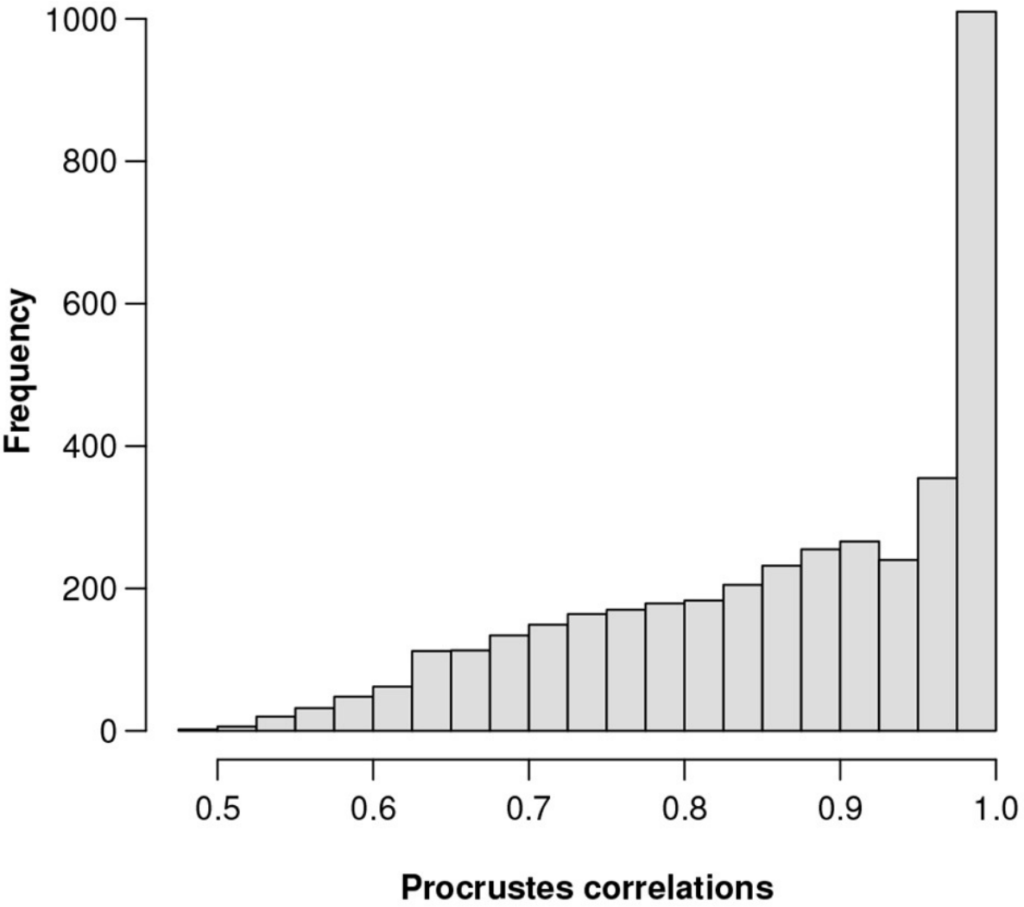
图一
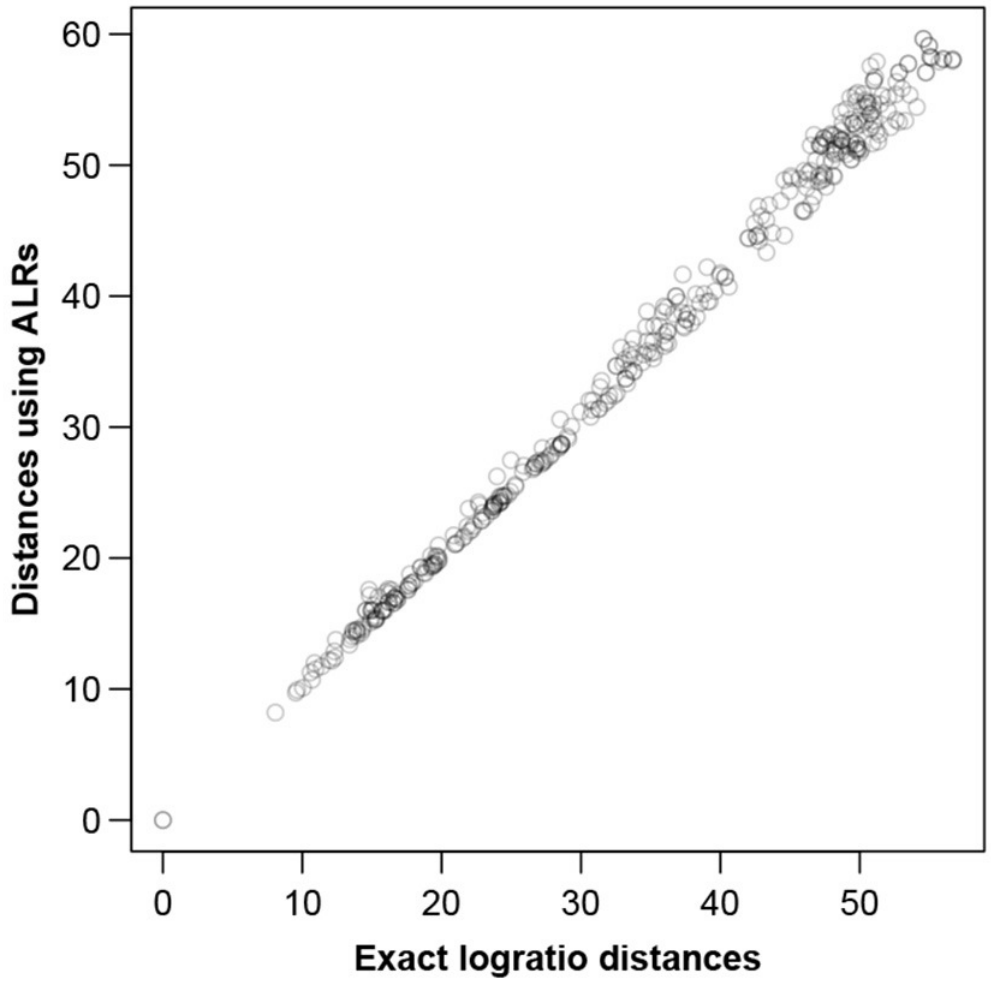
图二显示了在ALR上计算的所有样本间距离,基于所有成对对数的对数距离或同等情况下的所有CLR绘制相应的精确对数距离。
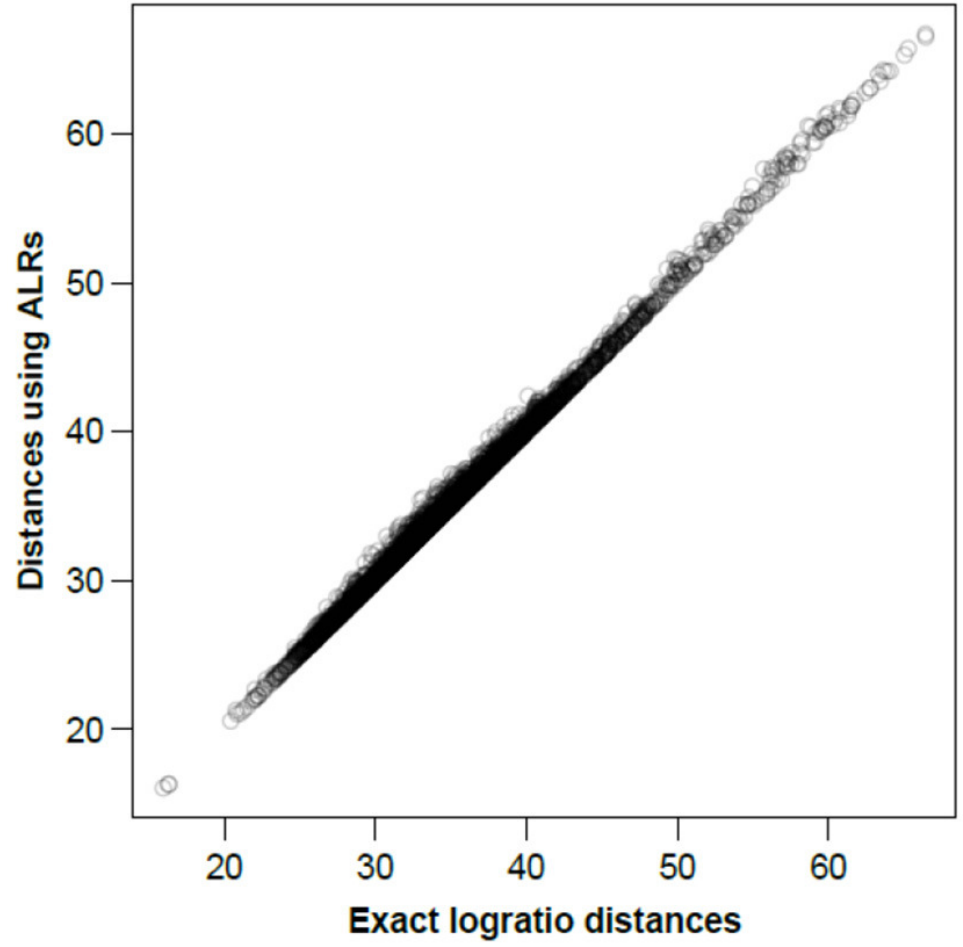
图二
图三为对于数据集的89个样本,参考基因编号856的计数与计数总和之间成正比。
图三
下图四展示了整个数据集的LRA(是所有成对对数的主成分分析(PCA),相当于所有CLR的主成分分析,以加权或非加权的形式)。
而图五中展示了具有参考基因856的ALR的对应PCA。主成分分析与参考成分微生物基因编号为856时,其几何形状实际上与确切的直线几何形状相同(Procrstes相关=0.9991)。字母S和F代表进行测序的两个实验室,显示出明显的分离。
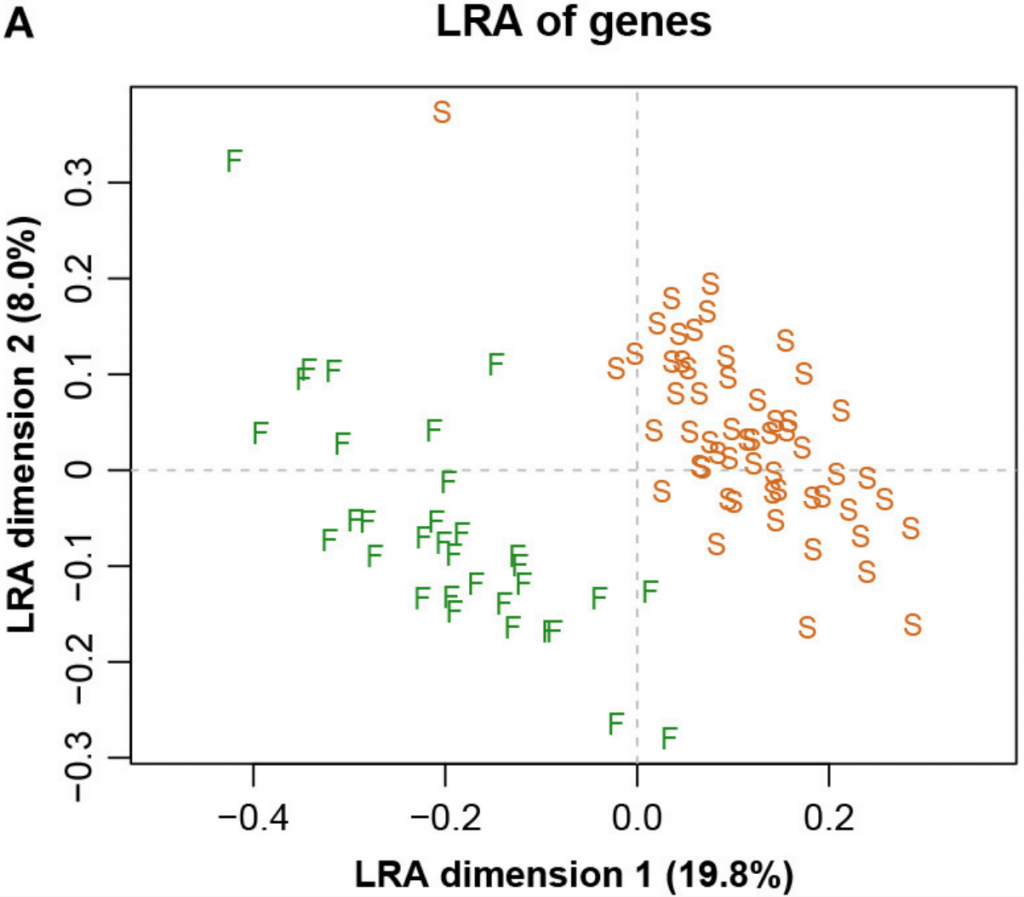

图四
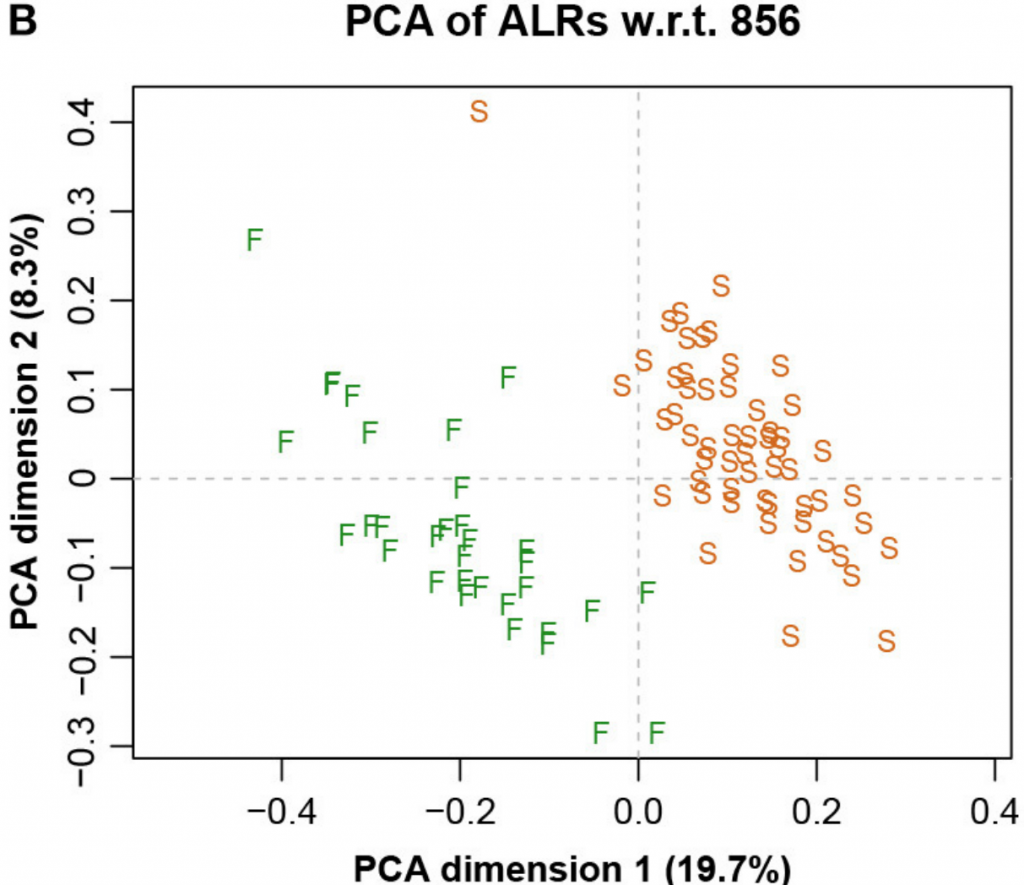

图五
2. 小鼠数据集
数据集大小,28个样本,3147个特征。此数据集中有34个零,使用R包zComposition中的函数cmultReplin替换。
总对数方差 0.2099,Procrustes相关系数最高为0.9977,对应转录本编号1318,其中转录本编号1179的Procrustes相关系数也与其相似。
图六
图七
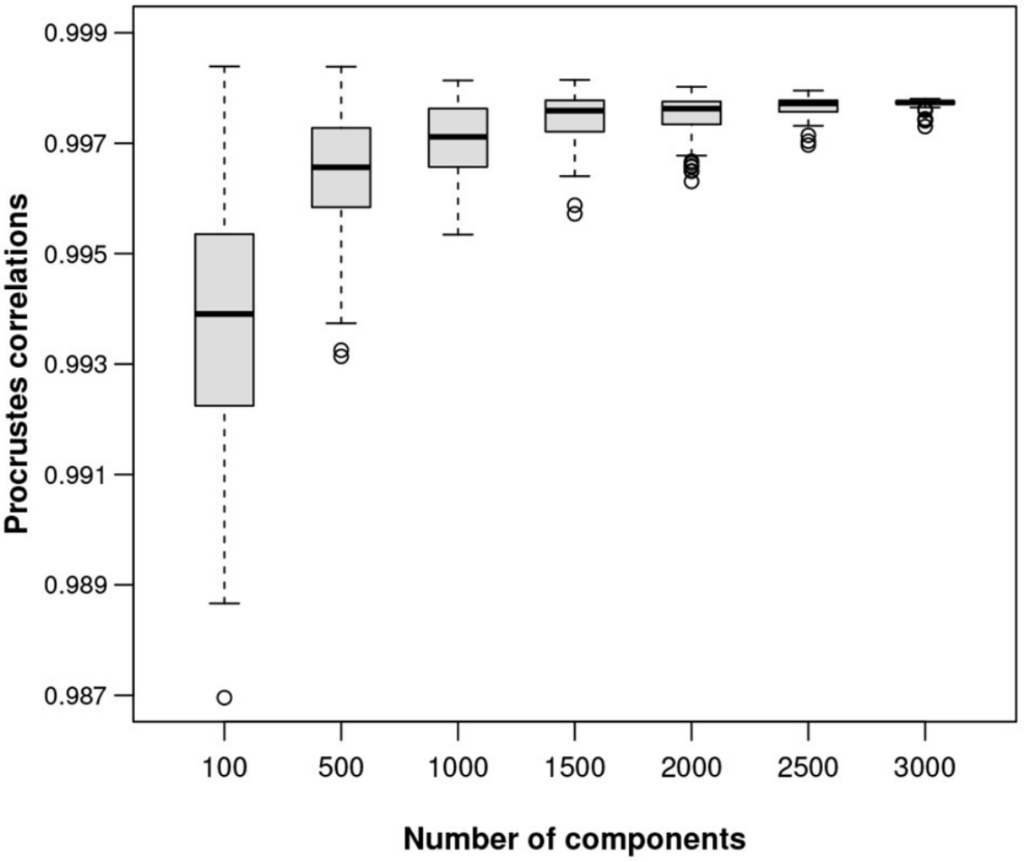
图六显示了在ALR上计算的样本间距离。为了显示任意大小数据集的ALR变换的质量,对MICE数据进行了模拟研究,从数据中随机抽取不同大小的样本,将每个样本作为独好的立的样本,并为该特定数据集的ALR变换找到最佳参考。
对于100、500、1,000、1,500、2,000、2,500、3,000和3,500个转录本的子集,以及每个子集的100个随机样本,绘制最佳的Procrstes相关性,如图七展示。ALR变换的等距质量随着可能的参考成分特征数量的增加而提高。
图八展示完整数据集的LRA,图九展示了参考转录本编号1179的ALR的PCA。它们实际上是相同的,只是有很小的差异,而在这之前的Procrstes相关系数结果就已经指示出了。标签代表两种不同的处理(L和M)和7种不同的时间(0、1、2、4、6、9和12h)。
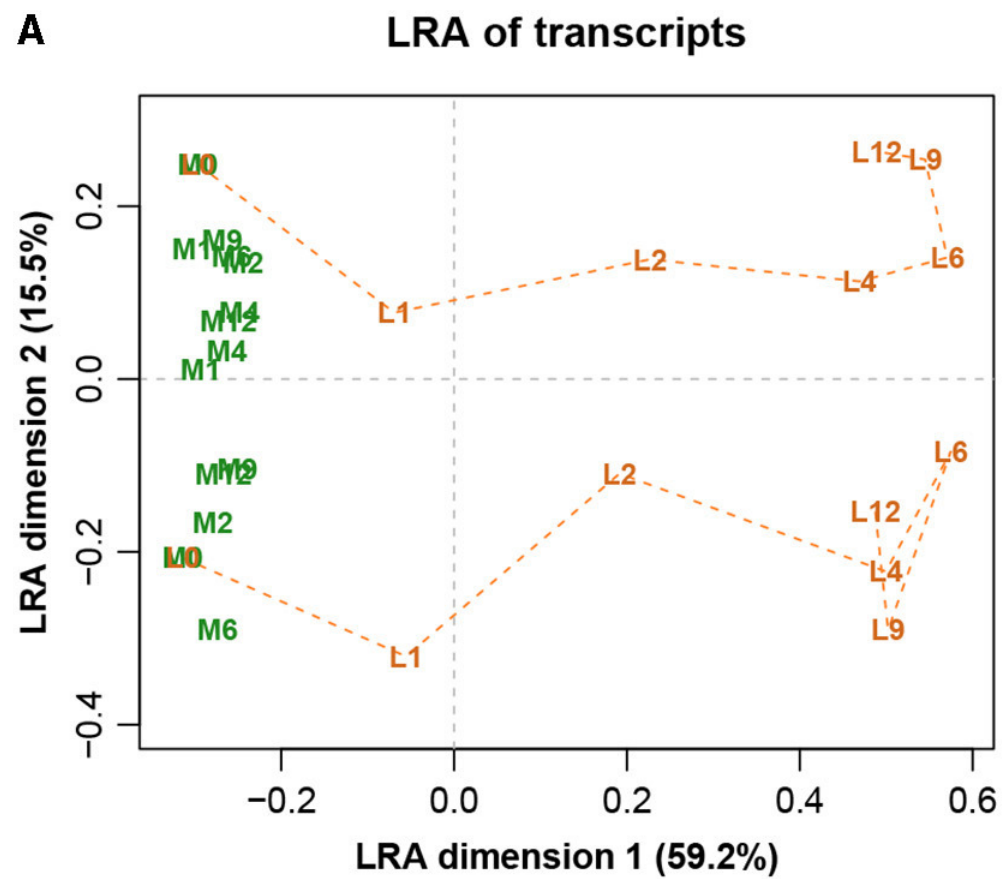
图八
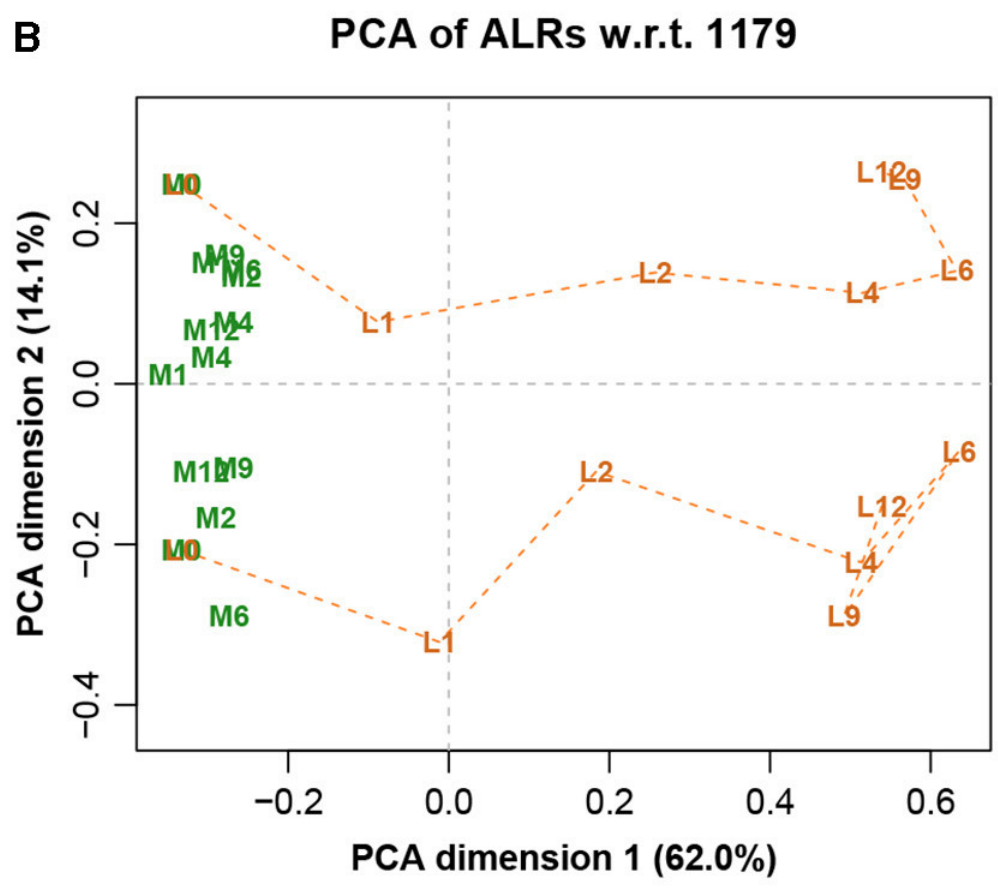
图九
3. 奶牛数据集
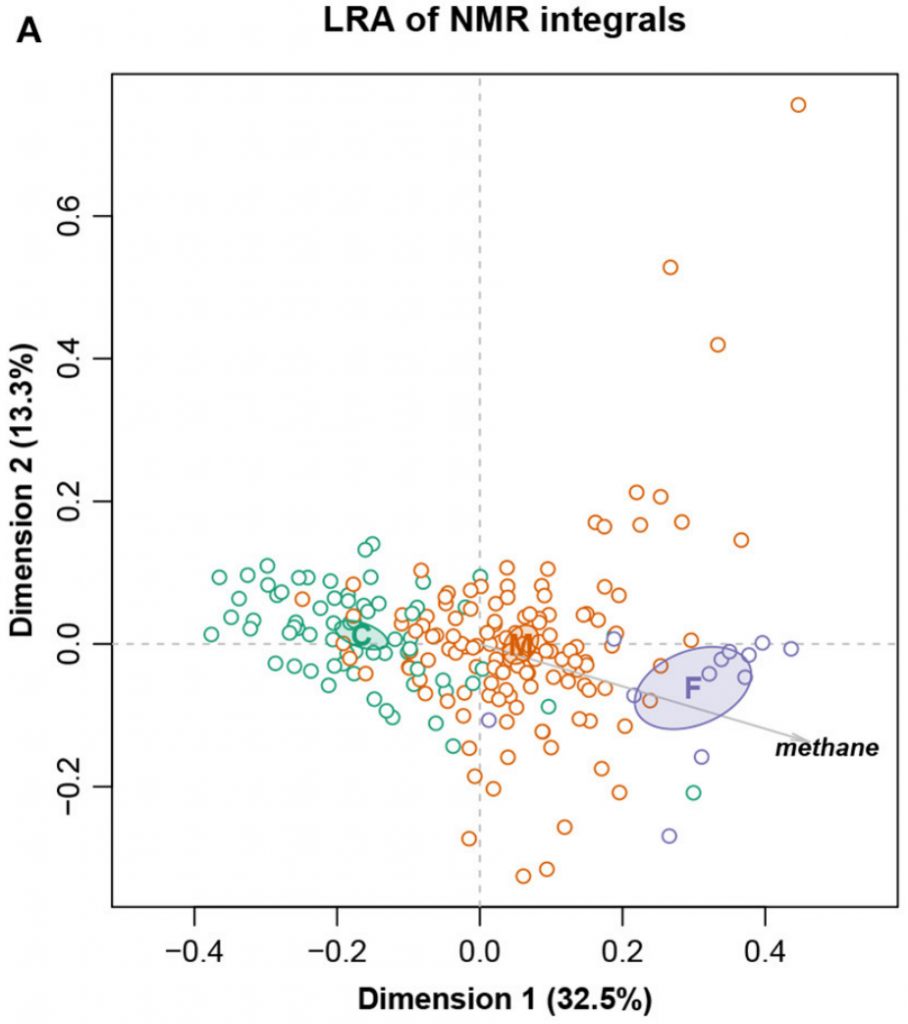
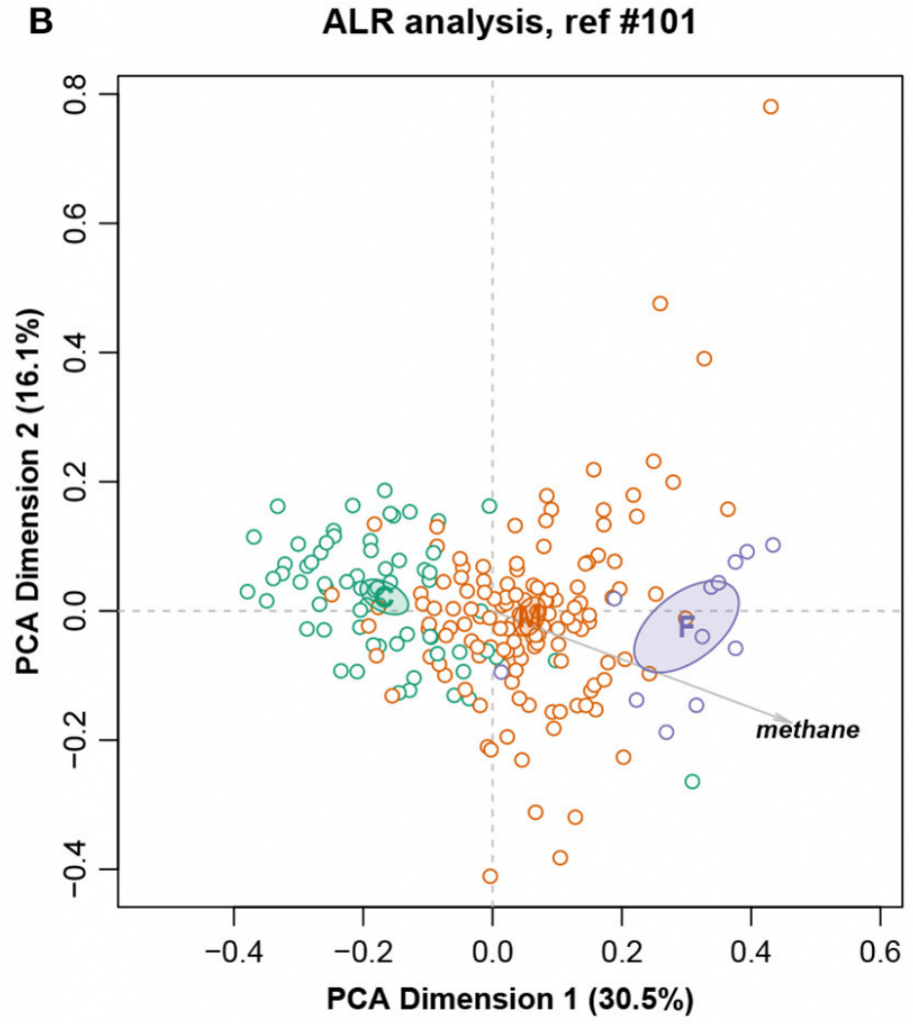
这是一个大小为211个样,127个特征的核磁共振强度数据集。样本被分成三个饮食组:精料组、混合组和饲草组,还测量了甲烷产量。

图十

图十一
总对数方差0.09128,Procrustes相关系数最高为0.9902,对应于编号101。图十展示完整数据集的LRA,图十一展示了编号101的ALR的PCA。标签C(精料)、M(混合)和F(饲料)。
从以上三个数据集的验证分析不难看出,对于高维数据,使用ALR对数转换也能得到对全部特征使用CLR对数转换方法的结果,关键在于找到有效的参考特征(成分)。
文章中作者建议将其作为此类高维数据成分数据分析的第一步。作者公开了部分数据集的存放地址,以及用于数据处理的部分代码。可以自己尝试看看是否适用。
扩展:数据集位置及实用脚本
兔子数据集: https://www.ebi.ac.uk/ena/browser/view/PRJEB46755
小鼠数据集:http://doi.org/10.5281/zenodo.3270954
其它数据集及脚本:https://github.com/michaelgreenacre/CODAinPractice
在这个github中有详细列出文中所使用的用于数据处理的各个R源码,以及目前这些数据处理的相关函数。
而这些脚本现已被整合为R包,easyCODA,可以从CRAN中直接下载。在Rstudio中调用“install.packages(“easyCODA“)”。
Tips
在对成分数据(composition data)进行分析时,通常会对原始数据进行矫正,也可以理解为一种标准化方法。比较常用的对数转换方法是CLR(Centered Log-Ratio),其次是ALR(Additive Log-Ratio,也就是文章主要推荐的方法)和ILR(Isometric Log-Ratio)。
每种方法都有优缺点,对于后续统计分析的适用程度,CLR>ALR>ILR。个人建议先使用CLR和ALR对数据进行转换,然后使用PCA或其他降维分析方法查看其类群分布,搭配adonis查看其统计显著性水平。只要能达到预期结果就都能使用。如果CLR和ALR数据转换后结果差异不大,那推荐使用CLR。
参考文献:
Greenacre M, Martínez-Álvaro M, Blasco A. Compositional Data Analysis of Microbiome and Any-Omics Datasets: A Validation of the Additive Logratio Transformation. Front Microbiol. 2021 Oct 11;12:727398. doi: 10.3389/fmicb.2021.727398.

谷禾健康
ResistoXplorer基于Web的耐药基因组数据可视化,统计和探索性新分析工具。
对宏基因组测序后的数据进行抗生素耐药性基因组的注释与分析,逐渐成为一条必经之路。过去,人们需要自己下载相关数据库再用比对工具进行比对,然后去冗余,再进行下游分析。这通常需要学习编程并熟练应用,对于一些临床医生或科研人员是一个很大的挑战。
最近有一款新的工具,用于对耐药基因组数据的成分分析,功能分析和比较分析。
ResistoXplorer,一款Web程序,地址:http://www.resistoxplorer.no
ResistoXplorer的主要功能包括:
1.支持多种常用和先进的方法,用于成分分析、可视化和探索性数据分析
2.全面支持各种数据归一化方法,包括标准的和最新的统计和机器学习算法
3.支持对配对数据集进行垂直数据综合分析的多种方法
4. ARG功能注释及其微生物和表型关联,基于10多个参考数据库的对比结果
5.功能强大且齐全的网络可视化,直观展现ARG于微生物的关联
打开网址后的界面:

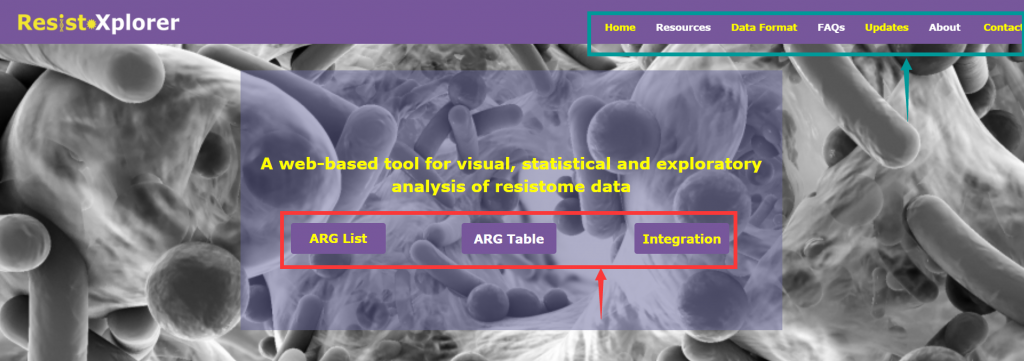
由三个主要分析模块组成(上图红色箭头所指框内):
“ARG List”:探索给定的ARG信息的功能和微生物宿主的关联,可视化网络。
“ARG Table”:对从宏基因组组学研究中获得的耐药基因组丰度文件进行功能分析,α多样性分析,排序分析,差异丰度分析等。
“Intergration”:综合分析,进一步探索潜在的联系,并结合新的生物学见解和假说,相似性分析,成对微生物-ARG相关分析等
上图绿色箭头所指框内:
“DataFormat”和“About”: 提供了关于注释表的格式、结构和数据库统计信息的详细描述
“FAQs”:提供了一些问题的答疑
“Resources”:分为“Manuals”和“Downloads”两个模块
Manuals是使用手册,对用户进行操作指导,建议仔细阅读。

Downloads,提供了示例上传文件和单个数据库的下载
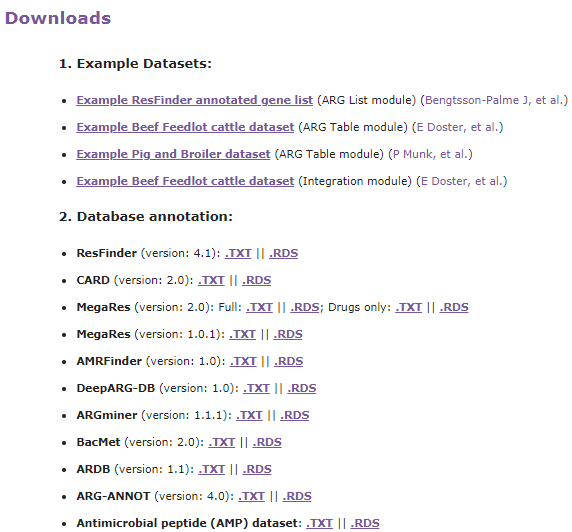
分析流程
ResistoXplorer接受抗性基因列表和ARG/taxa丰度表作为输入数据。然后是数据处理、数据分析和结果输出三个步骤。数据处理包括数据过滤和标准化,数据分析包括成分分析,比较分析和综合分析。结果输出以可视化图形,表格或html格式输出。
ResistoXplorer的功能注释使用的参考数据库来自9个通用的AMR数据库,CARD、ResFinder、MEGARes、AMRFinder、SARG、DeepARG-DB、ARGminer、ARDB和ARG-ANNOT。
此外,研究人员还从BacMet数据库和抗菌肽(AMP)耐药基因数据集中手动构建了功能注释信息,使用户能够对抗菌药物/金属和AMP抗性基因进行功能分析和下游分析。
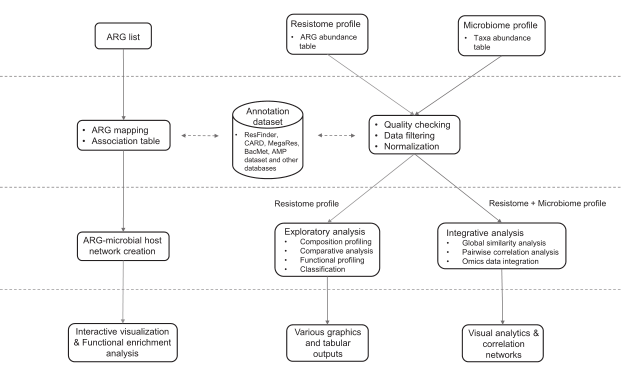
数据处理、分析及结果
数据过滤和标准化
默认情况下,低质量的特征会根据样本流行度及其丰度水平进行过滤。默认值是其他工具所使用的值,大多数在文献中可以找到。用户可以根据分位数间范围、标准差或变异系数排除这些低变异特征。
除alpha多样性和稀疏性分析外,过滤后的数据大多数用于下游分析。在综合分析的情况下,用户还可以对分类注释和耐药基因组丰度数据选择不同的数据筛选标准。
过滤后的数据还需要normalization(归一化)。ResistoXplorer提供了三种数据归一化方法,rarefying, scaling和transformation(稀疏、缩放和转换)。此外还支持其他归一化方法,如中心对数(CLR)和加性对数比(ALR)变换,以便于成分数据分析。方法的选择取决于要执行的分析类型。归一化后的数据用于探索性数据分析,包括排序、聚类和综合分析。用户可以自行探索适合的参数。
成分分析
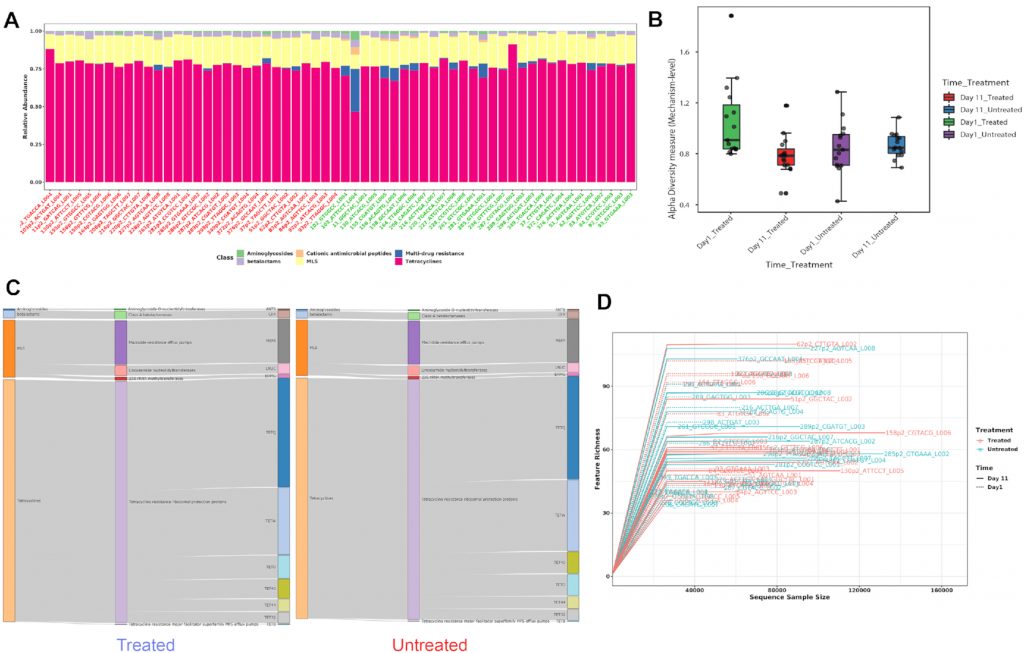


A) 显示各样本在不同分类水平下的ARG丰度。
B) Shannon多样性指数
C) 桑基图。显示了各组内的包括类别,机制和分组的ARG丰度分布。
D) 稀疏曲线。评估样本中估计的多样性的可靠性,在稀疏曲线中,识别的唯一特征(ARG)的数量与序列样本大小相对应。
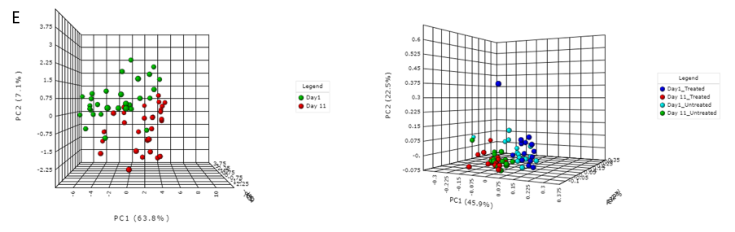
E) 排序分析。左边是基于时间点的带有样本颜色的3D PCA图。右边是根据不同的治疗组和时间点绘制3D PCoA图。目前,支持三种通用的排序方法, PCoA、NMDS和 PCA。结果表示为2D和3D样本图。
比较分析
差异丰度分析
使用DESeq2、Edger、metagenomeSeq、Lefse,以及单变量分析方法,比如ALDEx2和ANCOM。DESeq2和Edger说明计数数据的特征,相比之下metagenomeSeq使用推荐的CSS规范化,在更大的分组规模下具有更高的性能。
Lefse使用标准的非参数检验统计显著性,结合线性判别分析来评估差异丰富特征的效应大小。
ALDEx2对来自数据的模型化概率分布的对数比值执行参数或非参数统计测试,并返回统计测试的期望值以及效应大小估计。
ANCOM使用非参数统计检验来检验所有特征对的对数比丰度,以找出均值差异。结果以表格样式展现。
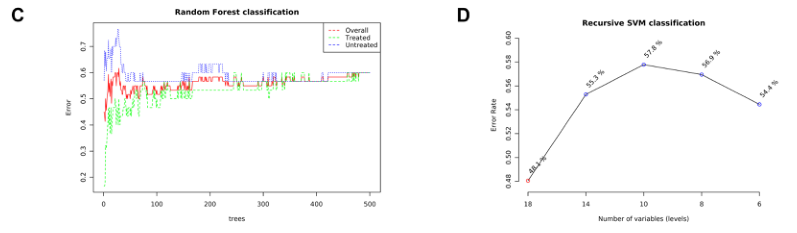
基于机器学习的分类
提供了两种功能强大的监督分类方法–随机森林和支持向量机(SVM),以识别潜在的生物标志物。
C)随机森林
D)展示了SVM在特征(变量)数量减少的情况下的分类性能
其他的一些可视化分析

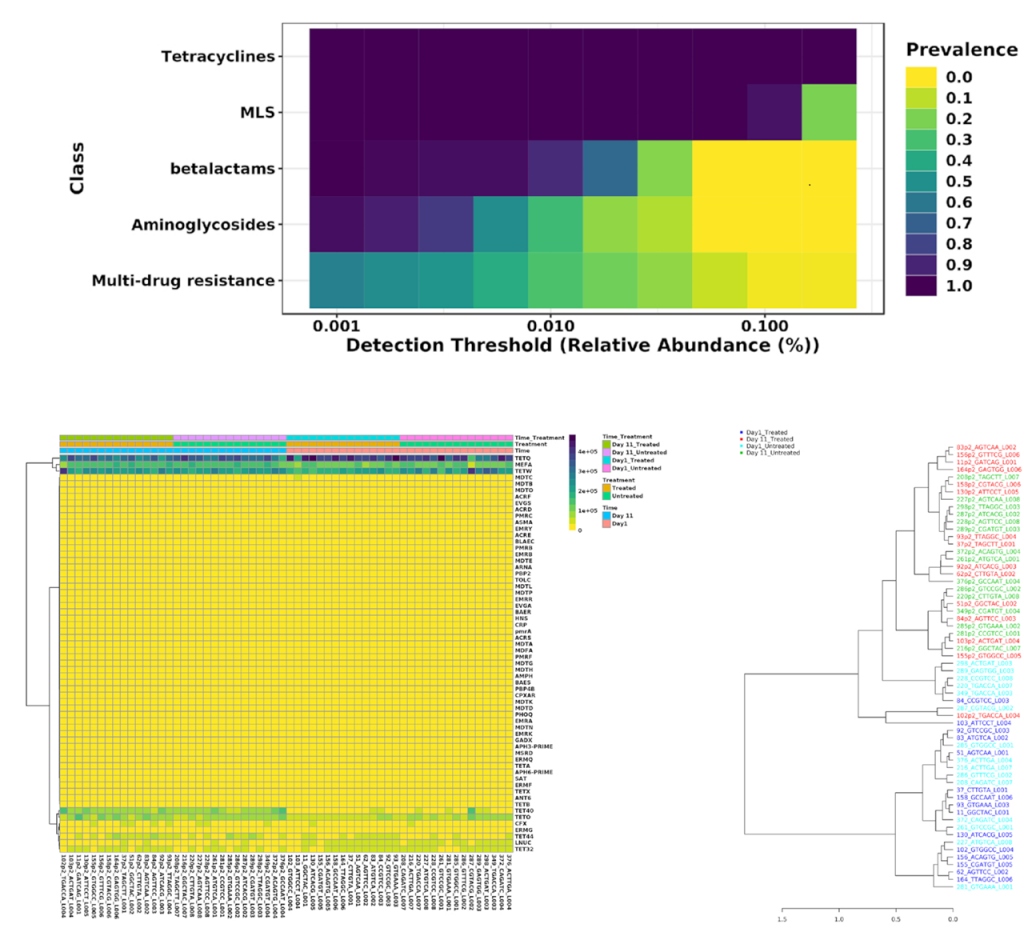
用户可以根据样本的丰度和流行程度,执行核心抗性分析来检测样本或样本组中存在的核心特征集,以热图的形式展现;以及关联分析和层次聚类,使用热图或者树状图可视化。
综合分析
使用各种综合数据分析方法来探索和揭示微生物群和抗性群之间潜在的潜在关联,这种分析大多用于探索不同环境中细菌和ARGs之间的联系。目前,为数据集成和相关分析提供了几种领先的、常用的单变量和多变量统计方法。所有这些分析都是在过滤和归一化数据集上执行的。
全局相似性分析
用两种基于多变量相关性的方法来确定微生物组和AMR数据集之间的总体相似性,分别为普鲁克分析(PA)和协惯量分析(CIA),在各种功能和分类级别上执行分析。相似系数和P值用于评估两个数据集之间的关联的强度和显著性,相似性系数在0到1之间,0表示两个数据集之间的完全相似,而1表示两个数据集之间的完全不相似。可视化结果用2D和3D排序图表示,如下图

A) 来自普鲁克分析的3D NMDS图,包含与数据集相关的样本、形状和颜色。
B) 来自协惯量分析的3D PCoA图,其中连接两点的线的长度表示两个数据集之间的样本的相似性。
组学数据集成方法
基于多变量投影的探索性方法,如正则化典型相关分析(RCCA)和稀疏偏最小二乘法(SPLS),用于微生物组和AMR数据的集成。这些方法旨在突出高维“组学”数据集之间的相关性。
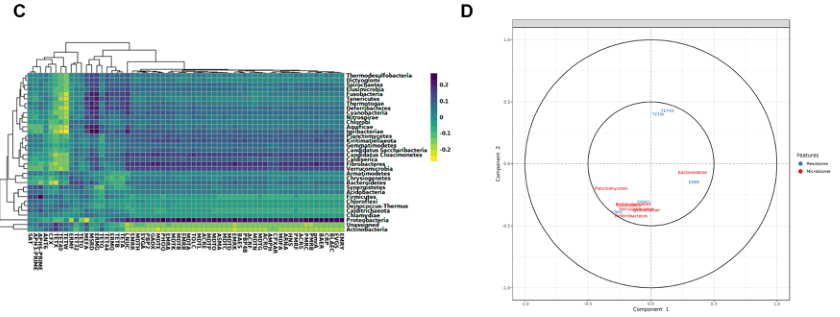
A 门水平微生物群落与ARGs(组水平)之间的聚类图像热图
B 显示存在于两个数据集中的特征(分类群/参数)的相关结构的相关圆图
成对微生物-ARG相关分析
使用单变量相关分析来确定单个菌群和ARGs(耐药基因组)之间是否存在强相关。使用Spearman、Pearson、CCLasso和最大信息系数(Maximal Information Coefficient)四种方法。用户可以使用绝对相关系数和调整p值的组合来选择强且显著的成对相关性。结果如下图,每个节点表示一个菌或ARG。用户可以双击一个节点,以突出显示网络中相应的相关节点。边缘的宽度和颜色表示两个节点之间相关性的强度和方向。
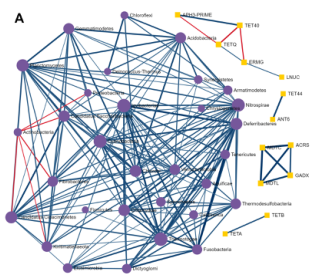
探索ARGs-微生物宿主网络
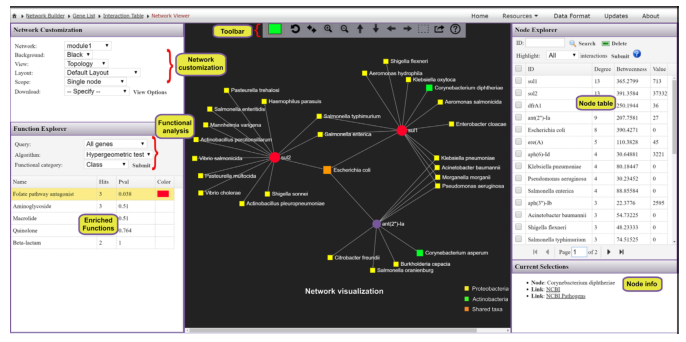
基于网络的可视化分析系统,提供了解ARGs和微生物宿主之间复杂的“多对多”关系的可能性。例如,通过查找在多个微生物中发现的ARGs或通过识别同时包含多个感兴趣的ARGs的微生物,可以直接从网络的角度找到承上启下的关键点。
从ResistoXplorer程序中涵盖的数据库中搜集ARGs-微生物宿主信息,构建的关联表用于网络可视化和功能分析。如下图,它由三个主要组件组成:中央网络可视化区、左侧的网络定制和功能分析面板,包含节点表的右侧面板。
用户可以使用带滚轮的鼠标直观地查看和操作中心区域的网络。例如,可以滚动滚轮来放大和缩小网络,将鼠标悬停在任何节点上以查看其名称,单击节点以在右下角显示其详细信息,或双击节点以将其选中。
顶部的水平工具栏显示了操纵网络的基本功能。第一个是颜色选择器,能够为下一次选择选择高亮颜色。还可以使用工具栏中的虚线方形图标选择并拖动多个节点。对当前网络中存在的ARGs进行功能富集分析,使用超几何测试方法,这种方法与网络可视化系统相结合,在解释AMR耐药机制和提供ARGs的可能传播路径信息可能会有更好的效果。
文章中为了展示该工具的可用性,在已发表的一些研究中,选择了1个研究进行抗性分析,“利用商业饲养牛检验图拉霉素(抗菌药物)对肠道微生物组和耐药性的影响”,分析的内容就如同上面展示的那样,这里就不多加赘述。
与其他工具的比较,文章中也列举了一个表格,分别与AMR++Shiny、resistomeAnalusis、WHAM!在分析模块上进行了比较。实际上大同小异,主要的分析模块以及使用的数据库都是相似的,只是谁的数据库更强大,搭载的分析模块更多的区别。
哪款软件的算法和统计分析匹配你的实验数据,或者它能为你提供更多的数据信息,就是适合你的。
这款在线分析抗生素耐药性基因组的程序值得探索一下,统计分析方法和数据库内容都挺强大的,交互式的使用也免去了对编程语言的探索,并且开发人员也表示会持续更新和精选数据库以达到更准确的下游分析。
参考文献
Dhariwal A, Junges R, Chen T, Petersen FC. ResistoXplorer: a web-based tool for visual, statistical and exploratory data analysis of resistome data. NAR Genom Bioinform. 2021 Mar 24;3(1): lqab018.
Interagency Coordination Group on Antimicrobial Resistance No time to wait–securing the future from drug-resistant infections. Rep. Secret. Gen. Nations. 2019.
Simonsen G.S., Tapsall J.W., Allegranzi B., Talbot E.A., Lazzari S. The antimicrobial resistance containment and surveillance approach-a public health tool. Bull. World Health Organ. 2004; 82:928–934.
Cecchini M., Langer J., Slawomirski L. Antimicrobial Resistance in G7 Countries and Beyond: Economic Issues, Policies and Options for Action. Paris: Organization for Economic Co-operation and Development. 2015; 1–75.
Xia Y., Zhu Y., Li Q., Lu J. Human gut resistome can be country-specific. PeerJ. 2019; 7:e6389.
Forslund K., Sunagawa S., Kultima J.R., Mende D.R., Arumugam M., Typas A., Bork P. Country-specific antibiotic use practices impact the human gut resistome. Genome Res. 2013; 23:1163–1169.

谷禾健康


微生物多样性测序(扩增子测序)是基于二代高通量测序对16S/18S/ITS等序列进行测序。可以同时检测样本中的优势物种、稀有物种及一些未知物种的检测,获得样本的微生物群落组成以及相对丰度。
相信关注我们的小伙伴对此并不陌生。
这次我们整合了大家平时会遇到的一些问题,在原有的基础上对报告进一步完善。



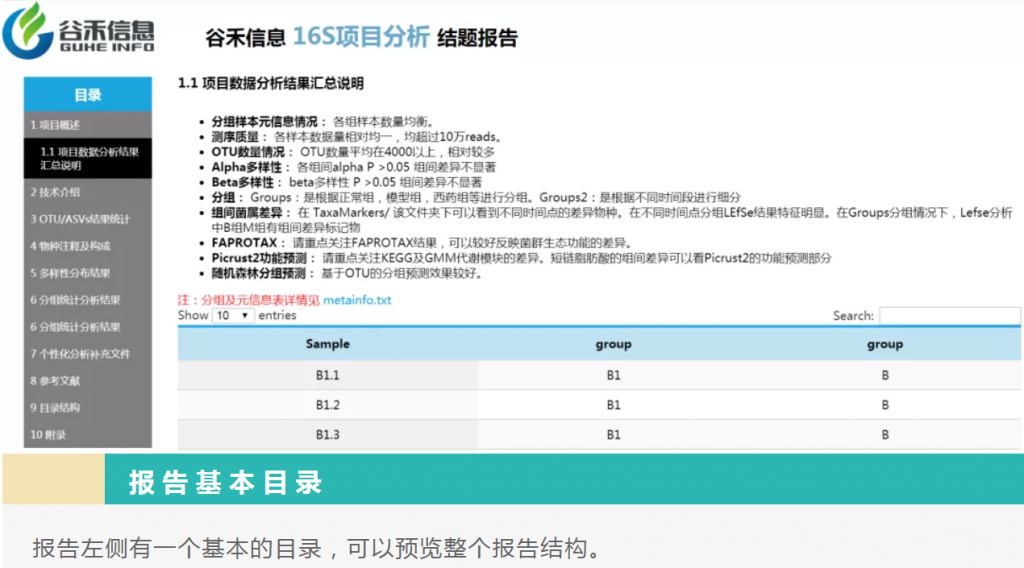
重要指数 :★★★★★
这部分内容必看。
主要是汇总信息,包括样本数据量,测序质量,重复性效果评估,分组信息,组间差异评估,代谢途径上差异,功能预测等。
这里会给出本项目中的一些重要提示,帮你从众多的报告信息中获取关键的部分。


重要指数 :★★★
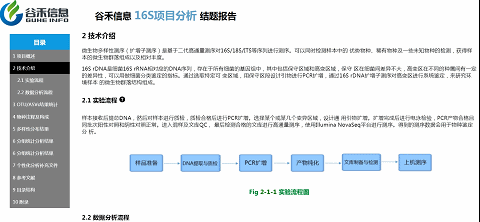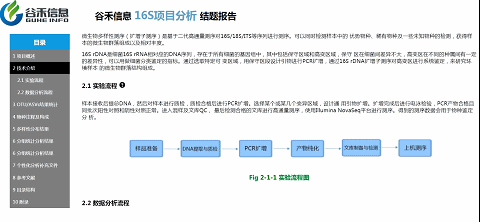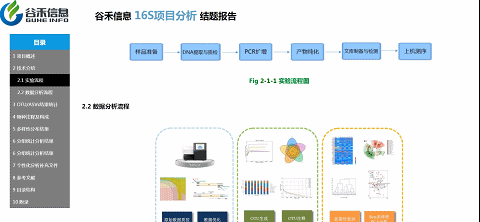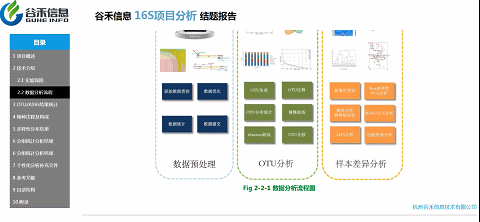
技术介绍这部分内容,就是说我们基于是怎么样一个测序平台、什么方法来获得的最后的数据。

如果你担心
这么直观的报告,
会不会不够详细?
小问号里有宝藏!


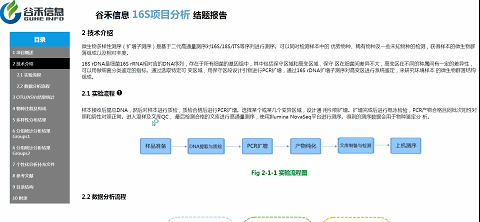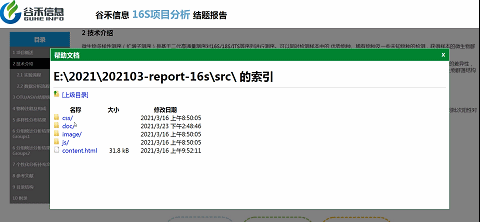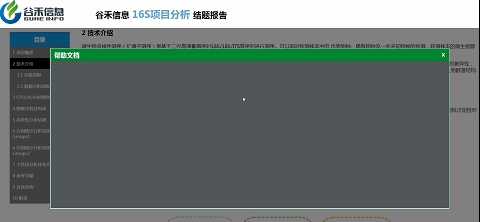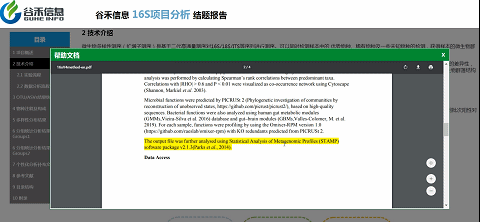

如上图,点击实验流程旁边的小问号,弹出的文件夹里就有详细的英文版方法介绍。
重要指数 :★★★★
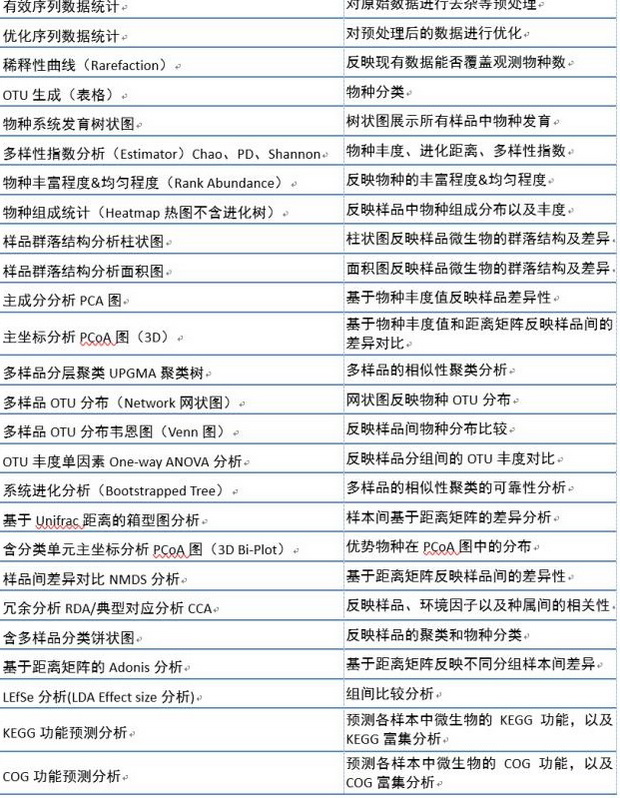
这部分内容主要是数据统计的图表:
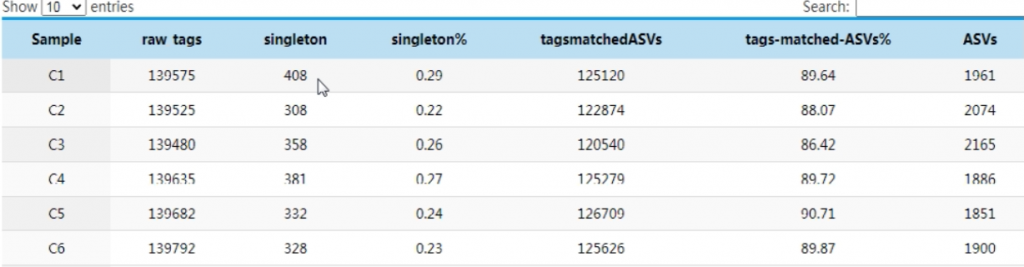

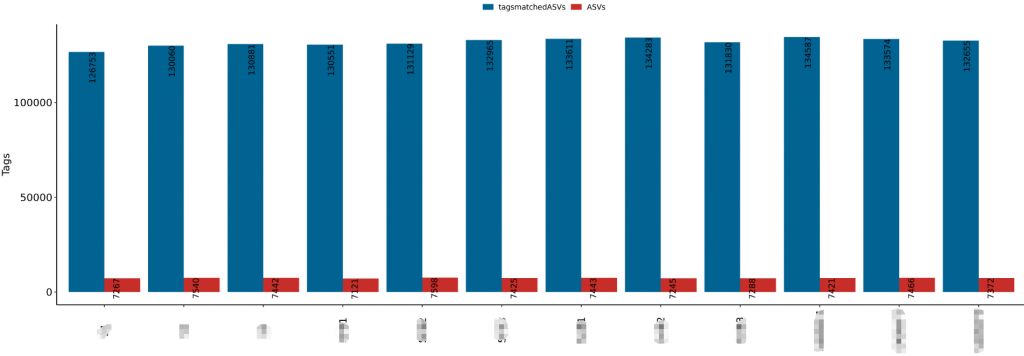
Raw-tags: 样本的原始序列数据
Singleton: 无完全匹配的单条序列数量
tagsmatchedASVs: 比对到最终ASVs的序列数据
ASVs:以及ASVs的种类个数



重要指数 :★★★★
经过SILVA138数据库的注释,得到ASVs的物种注释结果。
这一部分可以看到每个样本的物种构成比例,Taxonomic Level 可以选择Level1 ~ Level7 界门纲目科属种,不同分类水平下的物种构成。
这里选择level2就是“界”层级(可根据需求自选),另外比如选一个groups分组,如下:
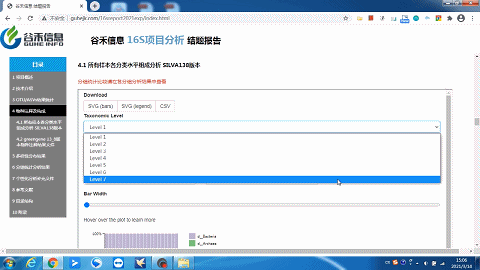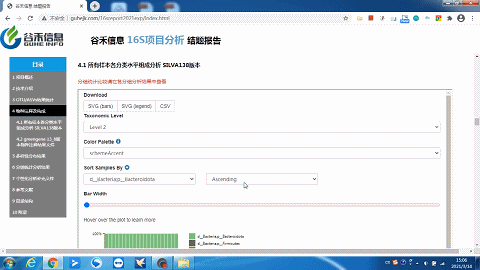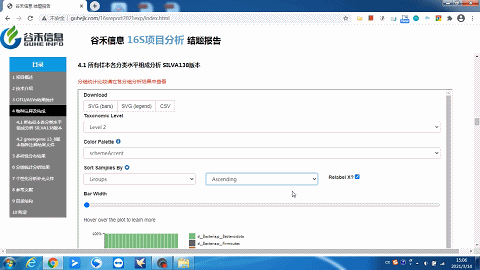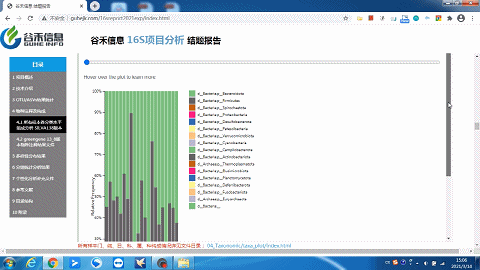

柱状图太宽?太窄?
一拉即可调整!
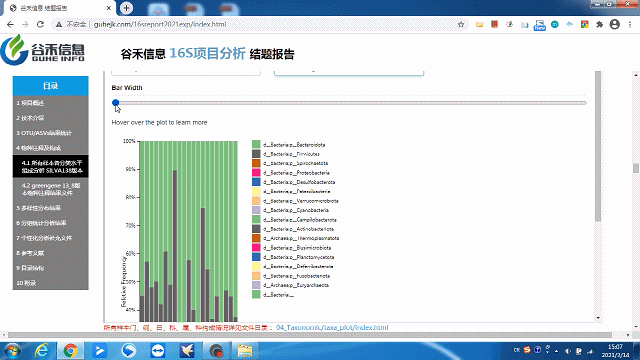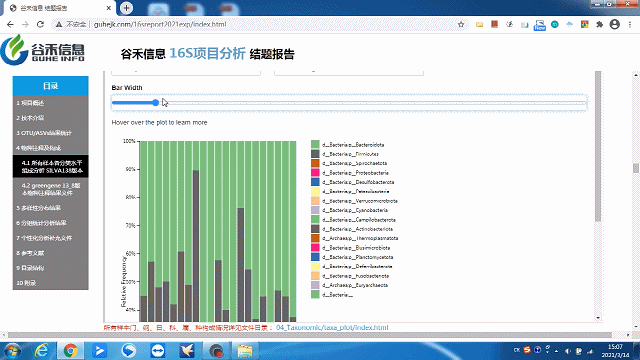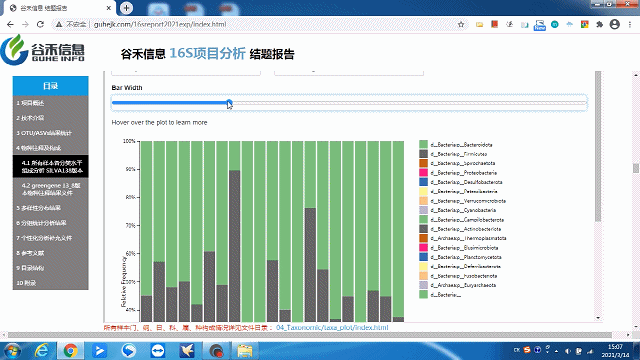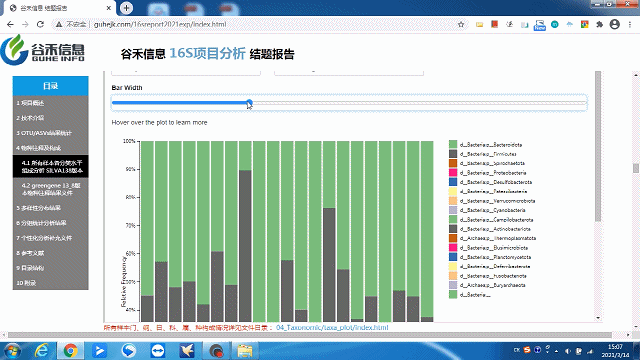

同时给出了各分类水平的相关原始数据,可以到对应路径进行查看。


重要指数 :★★★★
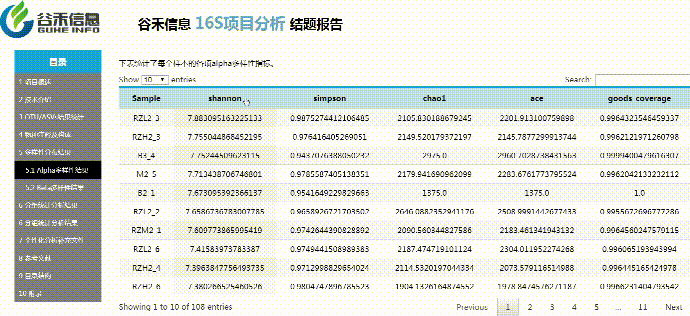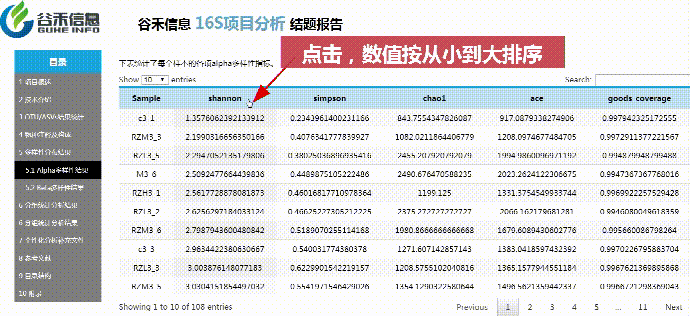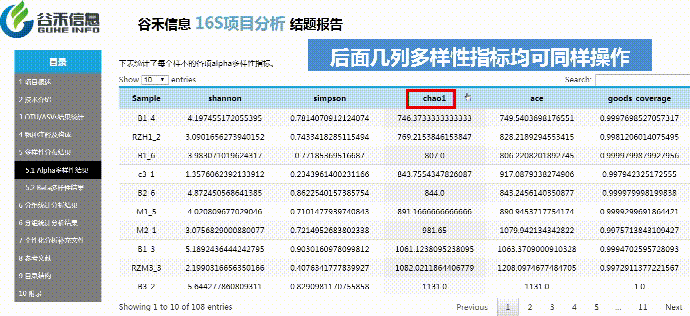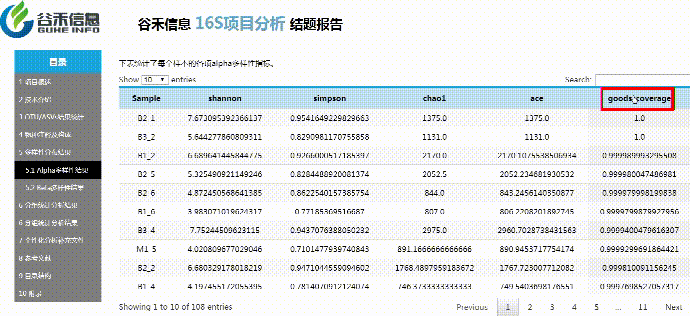
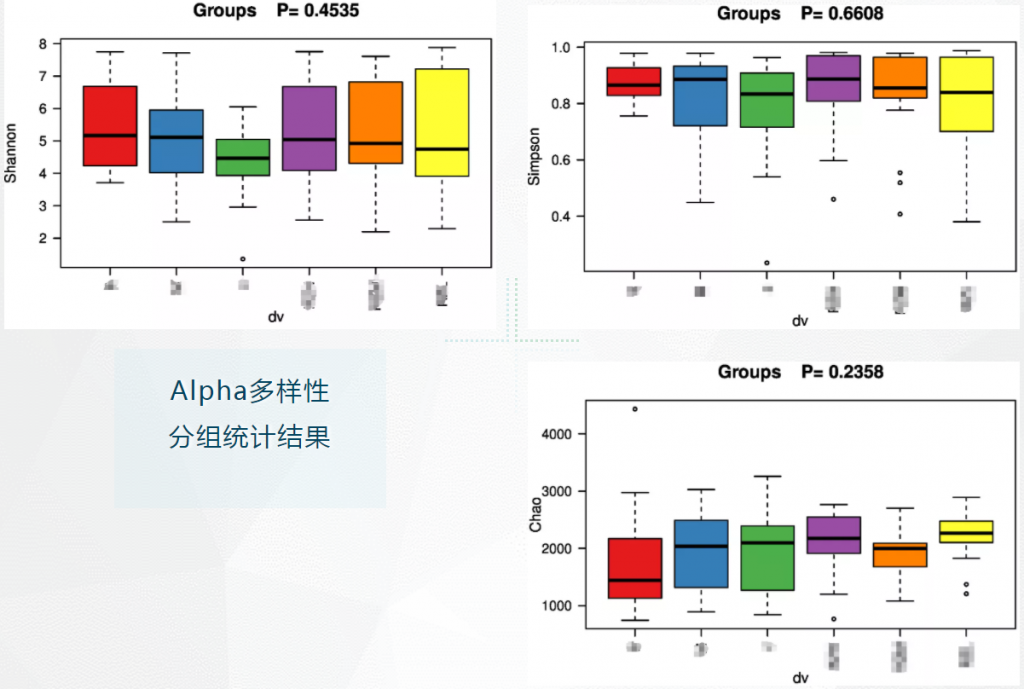
α多样性
评估单个样本内的物种构成的丰度情况
使用Qiime2进行α多样性分析,分别计算获得simpson,ace,shannon,chao1以及goods_coverage数据统计结果。

β多样性
通过降维的方法来考察样本与样本之间的相似度和关系,种属构成特征。
三种聚类方式:
Beta多样性PCA、非加权距离的PcoA、加权距离的PcoA的3D图。
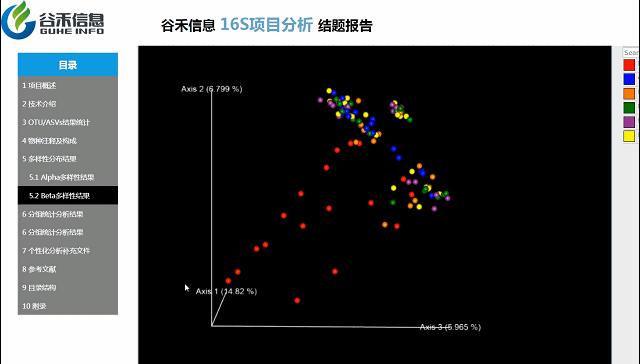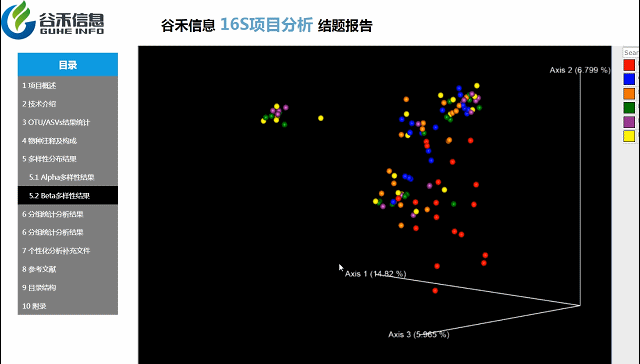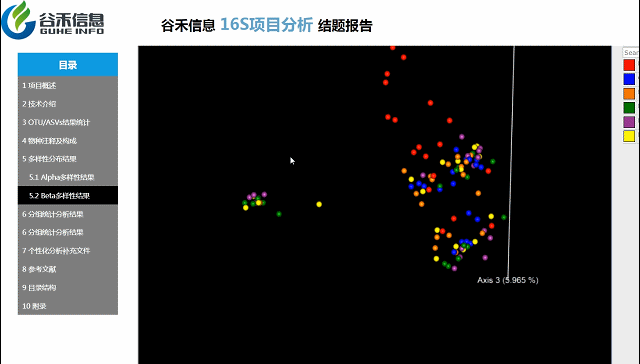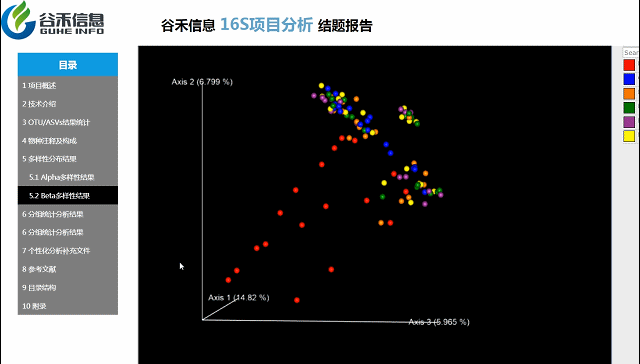

按住鼠标随意拖动,可以看到任意角度的三维坐标自由变换。

大小可自行调整
多色系任你挑选
总有你想要的图
重要指数 :★★★★★
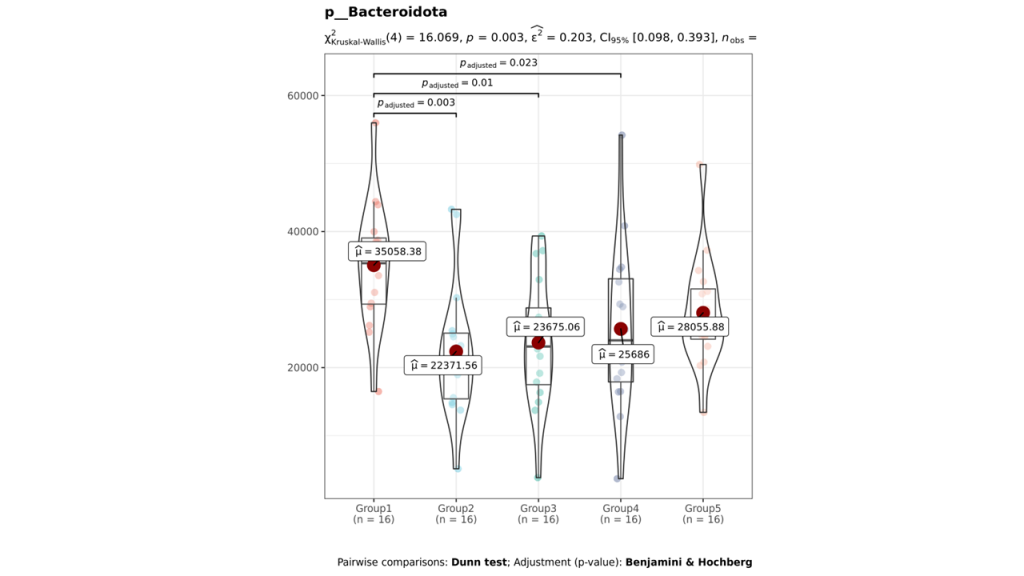
按照你填写的样本信息单,对各分组情况,进行统计学差异分析。
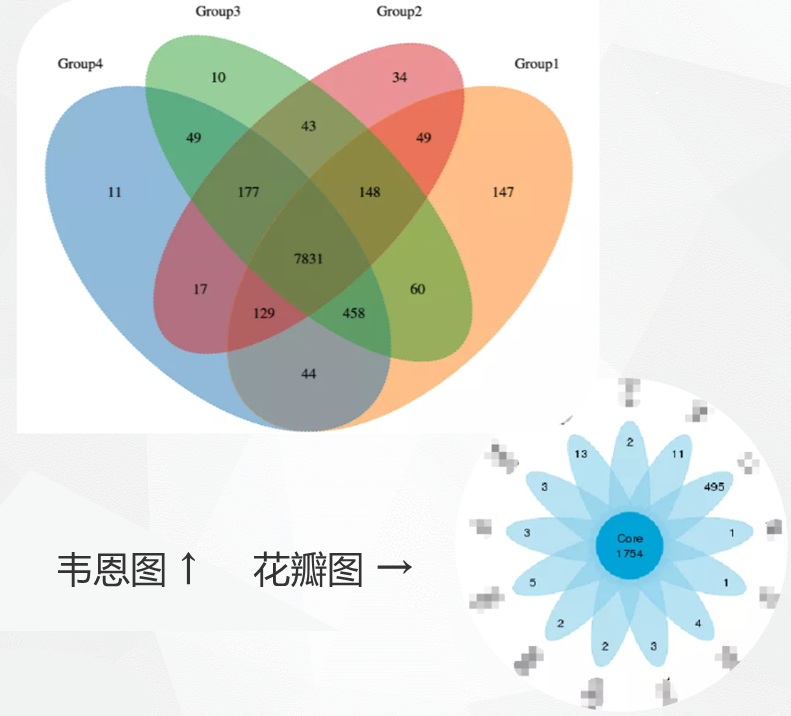
分组Venn图
OTU/ASVs比较韦恩图(样本数/分组数<=5个样本,若分组数大于5出花瓣图)

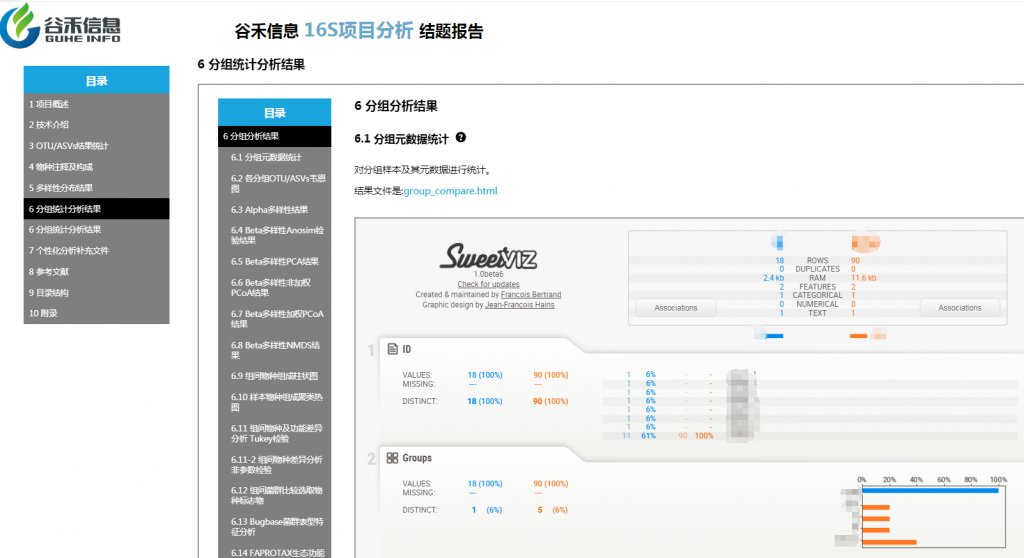
分组元信息统计
对分组样本及其元数据进行统计

α多样性
分组之间alpha多样性指数使用非参数统计检验

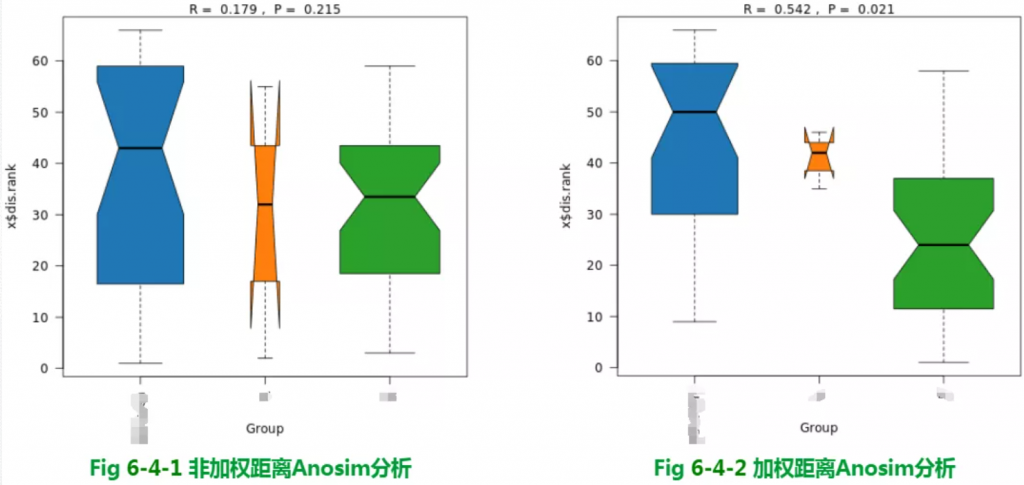
分组是否有意义?——β多样性
Beta多样性分组Anosim检验结果
Anosim分析是一种非参数检验,用来检验组间的差异是否显著大于组内差异,从而判断分组是否有意义。

要PCA结果图?
要PCoA结果图?
要NMDS结果图?
要加权?非加权?
… …
全部都有
Beta多样性PCA结果
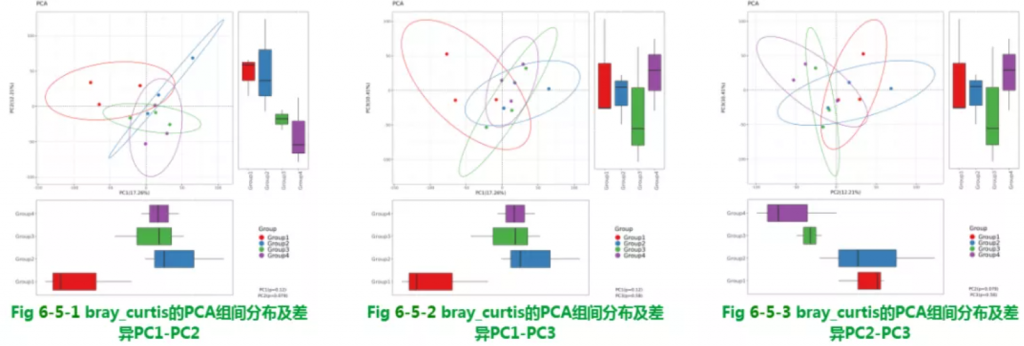

使用bray_curtis的PCA组间分布及差异
Beta多样性非加权PCoA结果
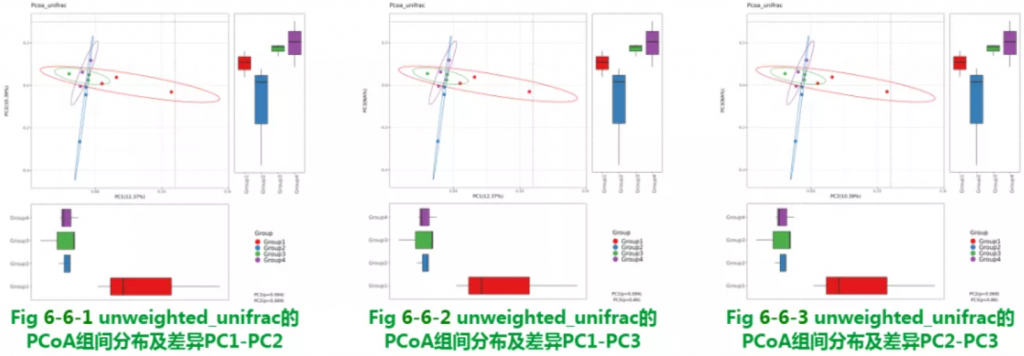

使用unweighted_unifrac的PCoA组间分布及差异
Beta多样性加权PCoA结果
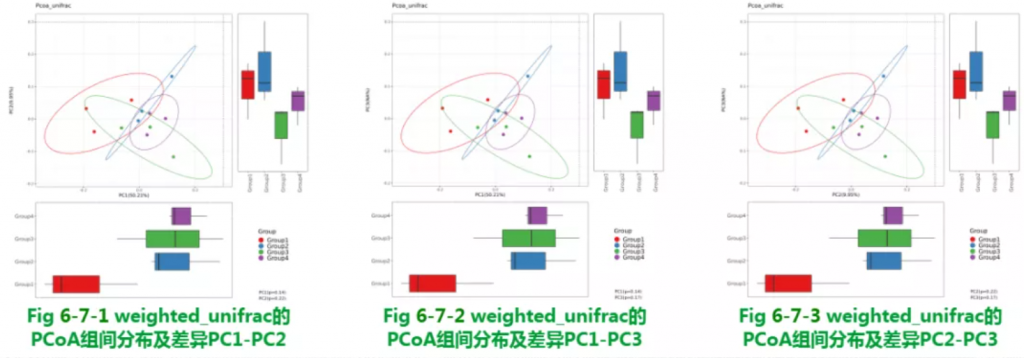

使用weighted_unifrac的PCoA组间分布及差异
Beta多样性NMDS结果
非度量多维尺度分析 NMDS 分析与 PCoA 类似,也是一种基于样本距离矩阵的分析方法,通过降维处理展现样本特定的距离分布。
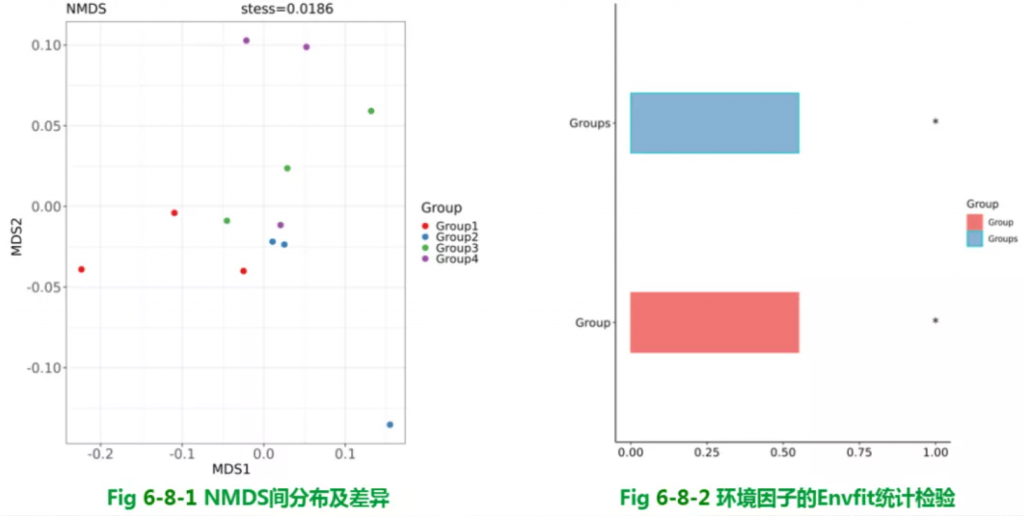

通过对样本距离进行等级排序,使样本在低维空间中的排序尽可能符合彼此之间的距离远近关系(而非确切距离数值)。因此,NMDS 分析不受样本距离的数值影响,对于结构复杂的数据排序结果可能更稳定。


你想要的层级或分组都有——组间物种构成柱状图


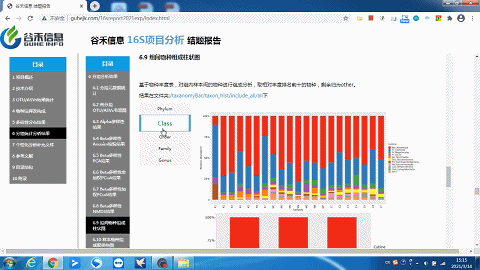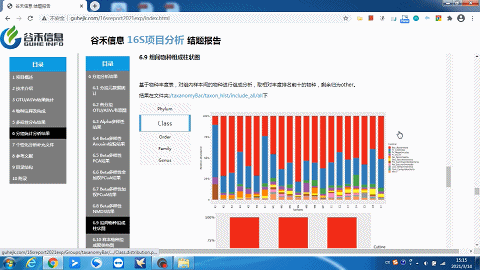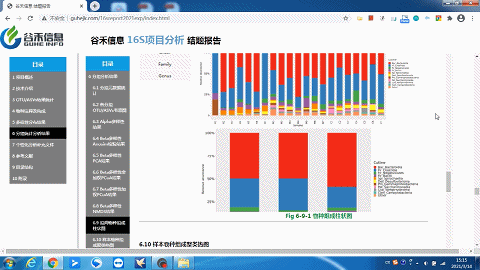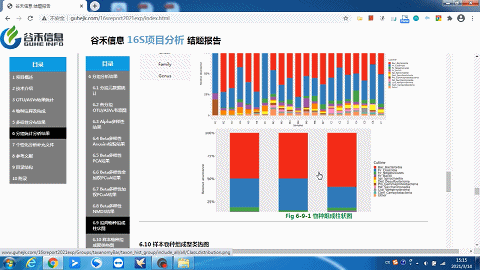

样本及分组之间聚类热图
了解样品之间的相似性以及属水平上的群落构成相似性。
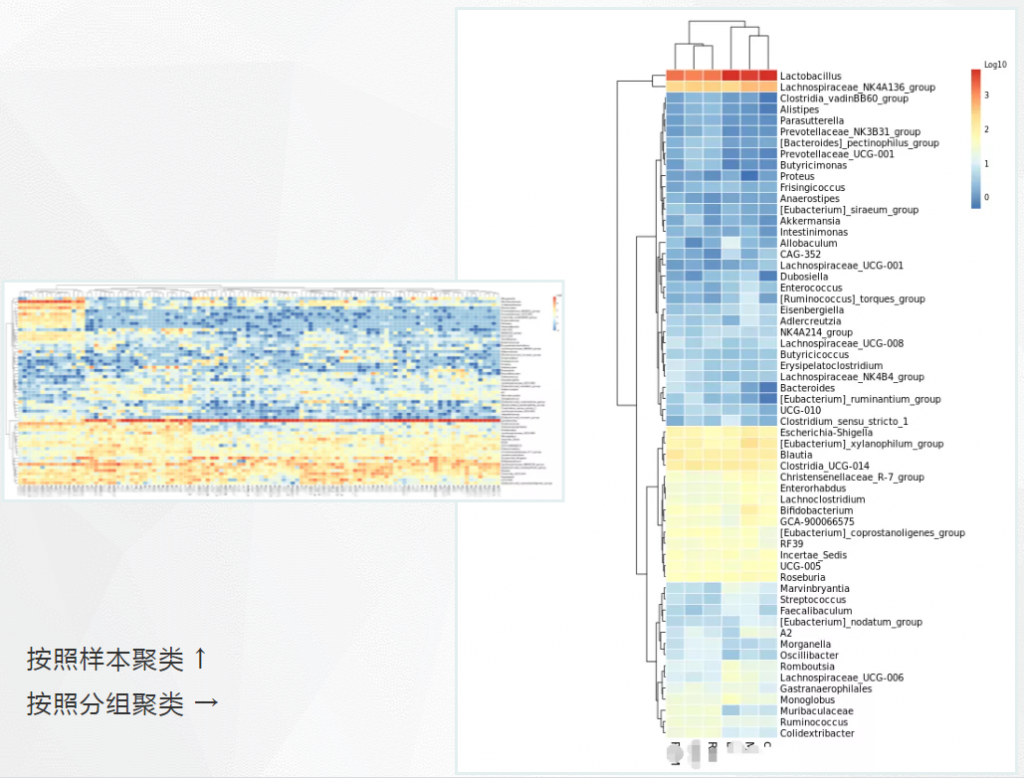

组间各物种分类水平及功能差异
Tukey检验
如果样本每个分组是完全均等的情况(比如说每个组各有10个样),适合用Tukey检验。

优势:
可以快速在图中表现出多个分组之间,哪两个之间存在显著差异。
组间各物种分类水平
非参数检验
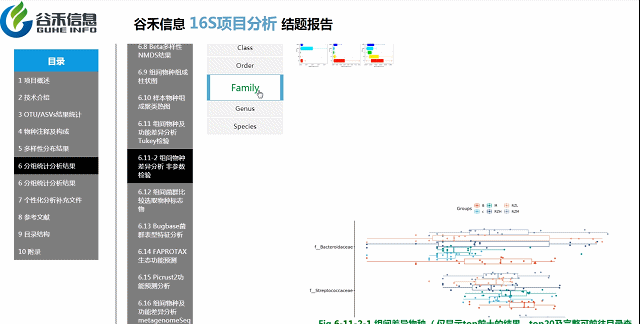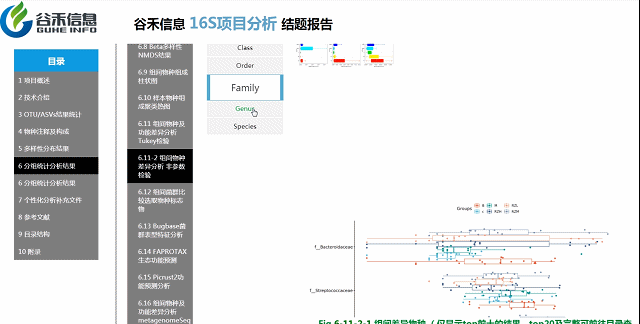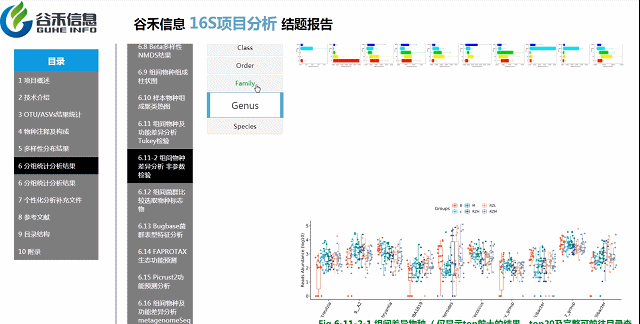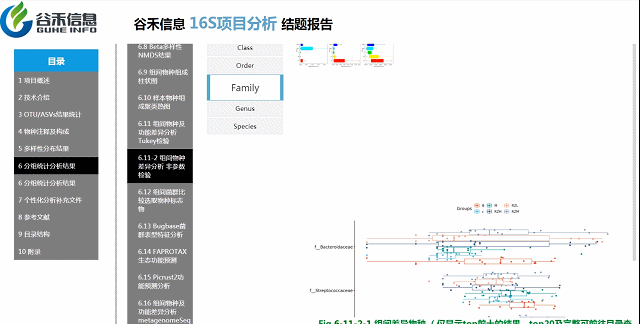

各个层级均有相对应的图展示。
组间菌群比较选取物种标志物
Lefse分析
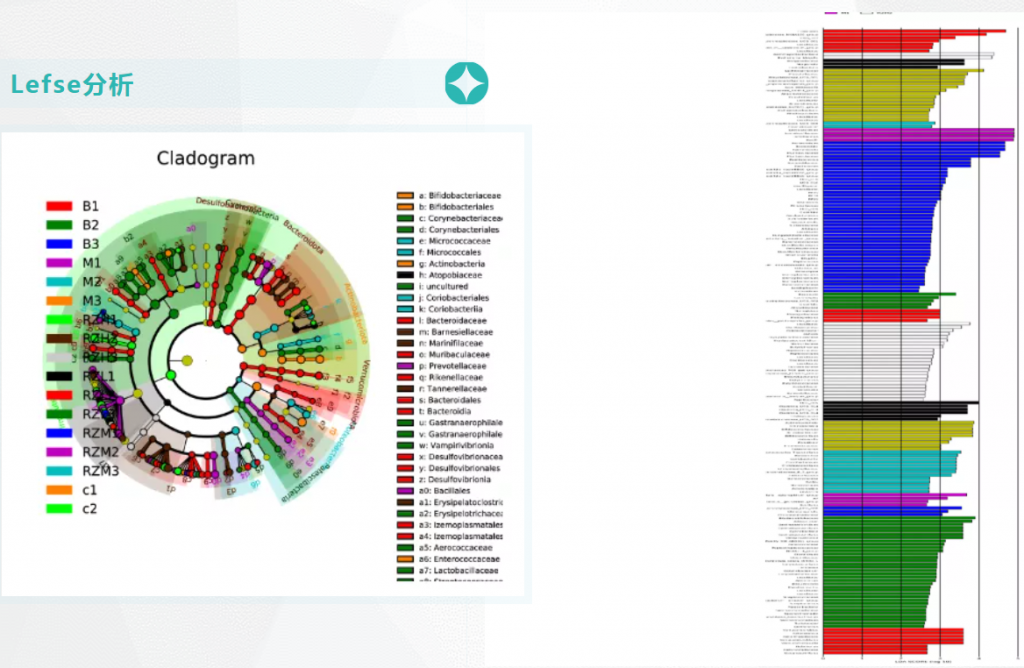

基于线性判定的方式,筛选组与组间的生物标记物——也就是说找到组间存在特别显著的高丰度的菌属。
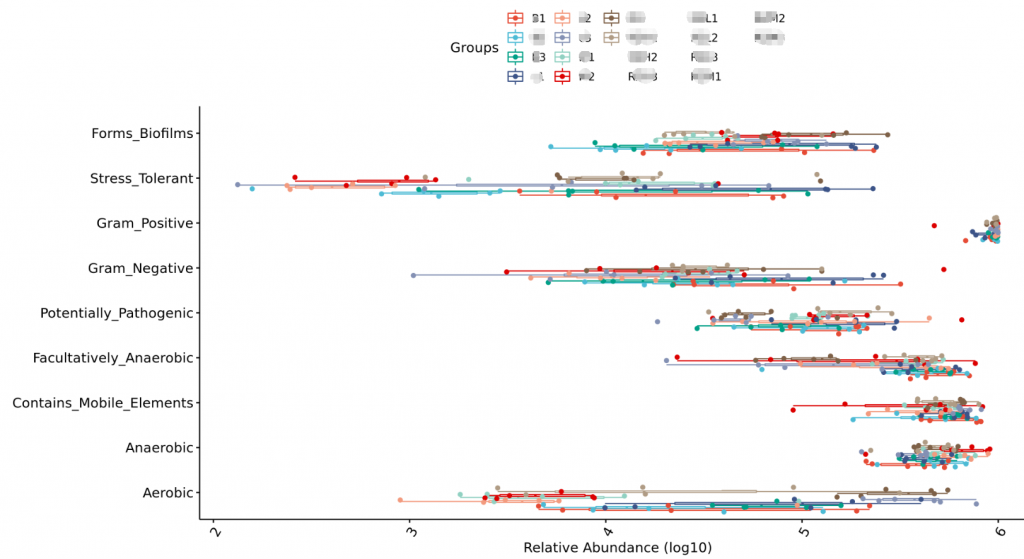
Bugbase菌群表型特征功能预测分析
基于文献的一些分类,对菌属进行菌群表征,包括对厌氧/好氧,革兰氏阴性/阳性,生物膜形成等分类。

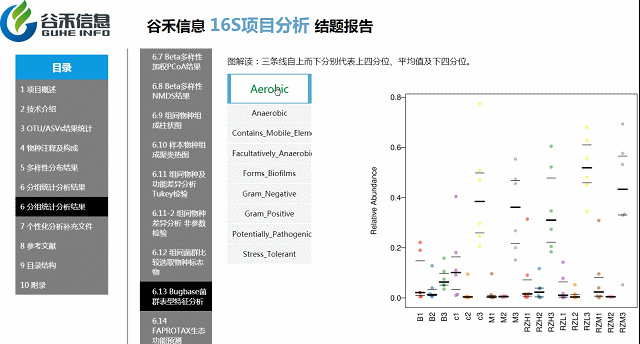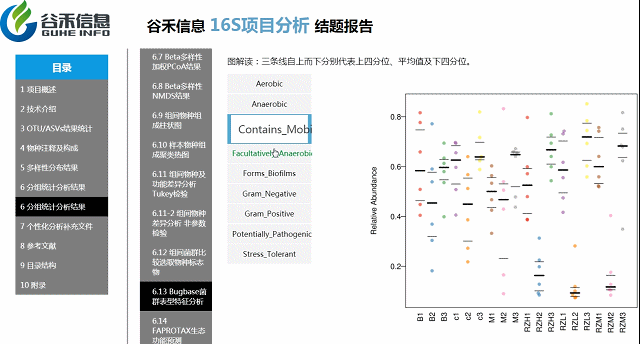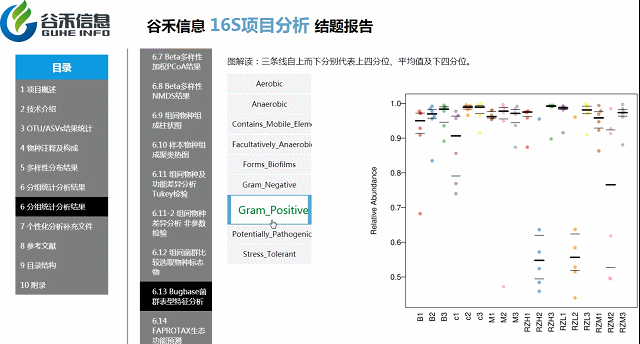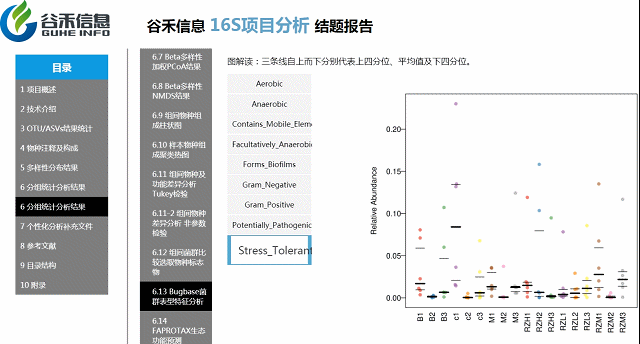

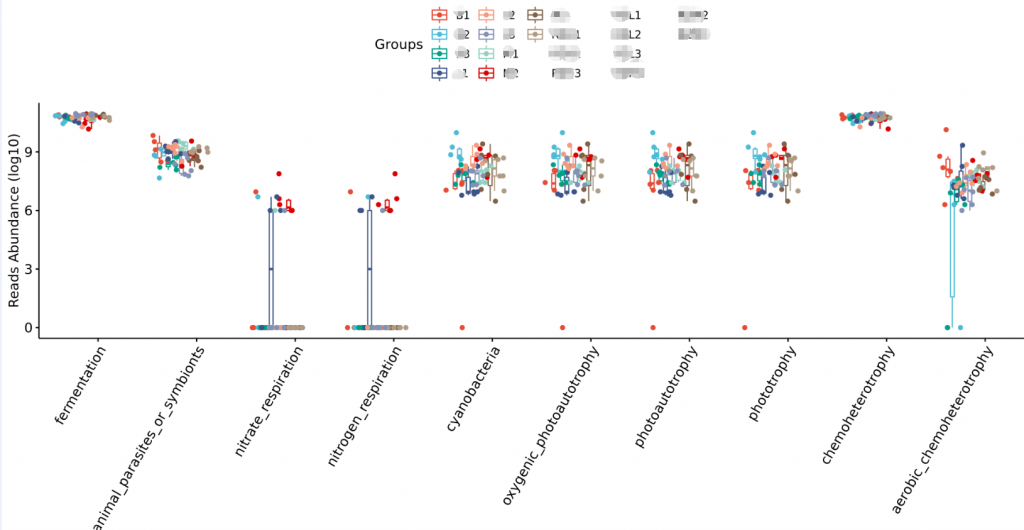
环境样本工具?——FAPROTAX生态功能预测
整合文献原核功能数据库,偏向于代谢和生物学功能的注释。比较适合环境样本,比如说碳、氢、氧、氮、硫等元素的代谢循环的能力。



基因功能预测?——Picrust2功能预测分析
随着研究的不断深入,很多菌的基因组数据有了,基于基因组数据一旦能确定其物种来源,可以推测它具有的基因的拷贝数、代谢通路的构成特征。

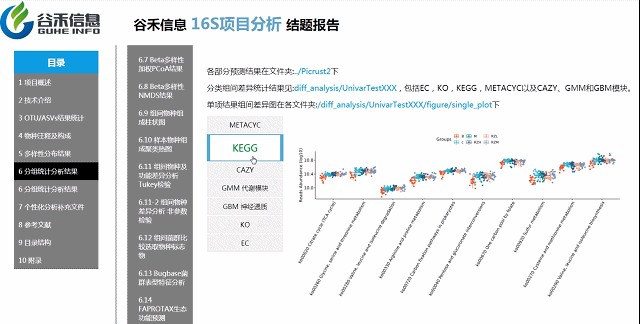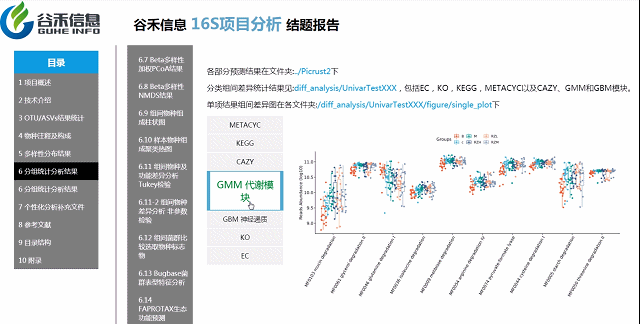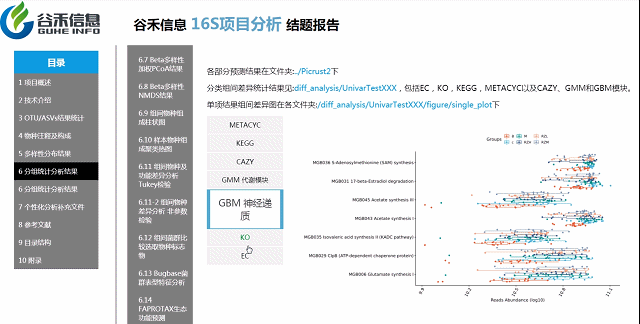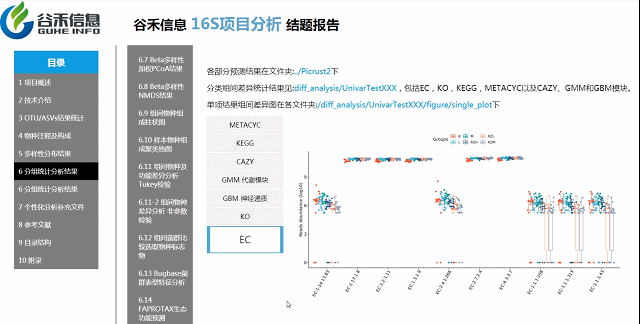
2万多的物种,基因覆盖更完整
还包括了CAZY,GMM,GBM等模块
具体差异的意义要结合你的实际研究目标解释
组间各物种分类水平及功能差异
MetagenomeSeq分析
更保守,结果可靠性更高


组间物种及功能差异热图
基于上面MetagenomeSeq的结果中,找到差异的物种种属和代谢通路做的热图。

差异菌属与代谢通路之间有什么关系?
差异菌属和功能代谢关联分析

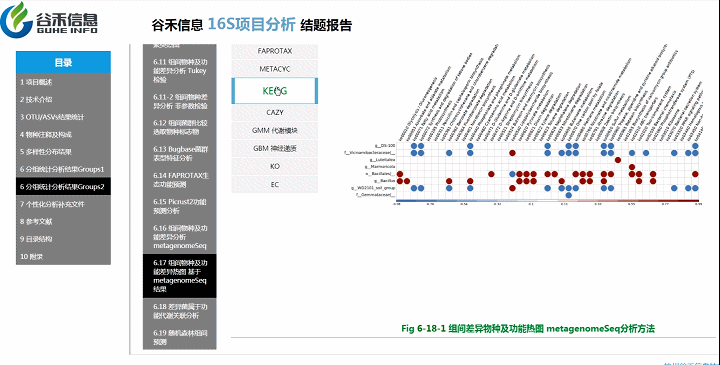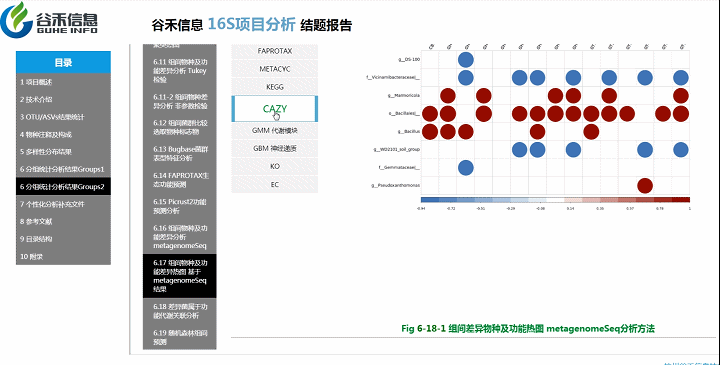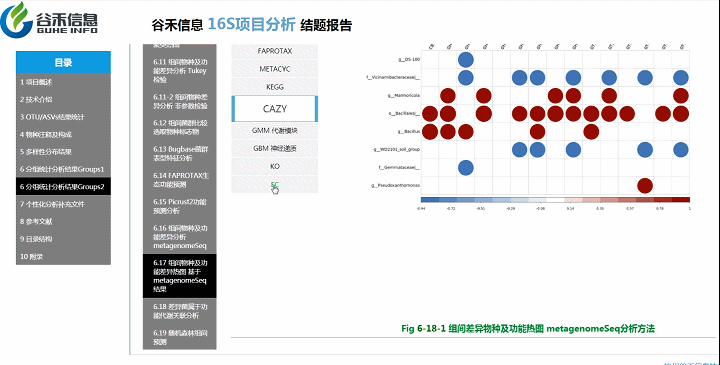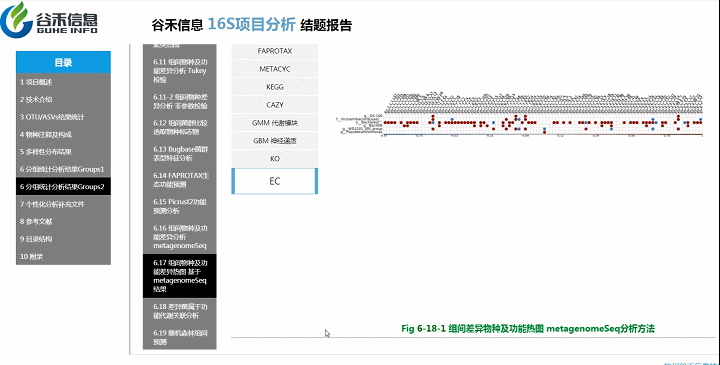
从菌属上的差异,代谢通路的差异等来看,到底是如何关联,是什么类的菌或代谢通路作出贡献。
不同分组之间相对明确区别的模型?
随机森林预测
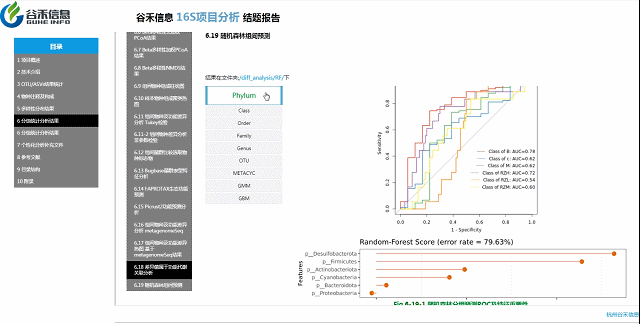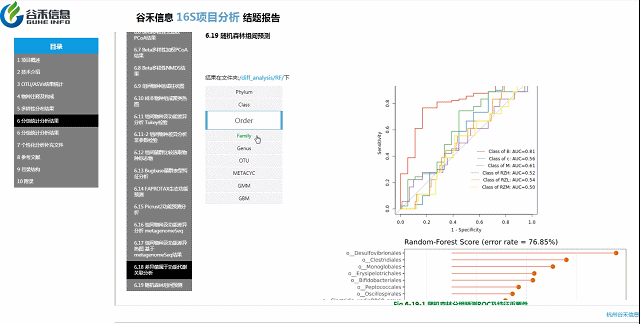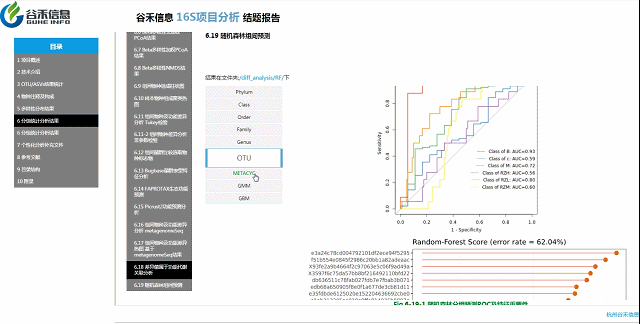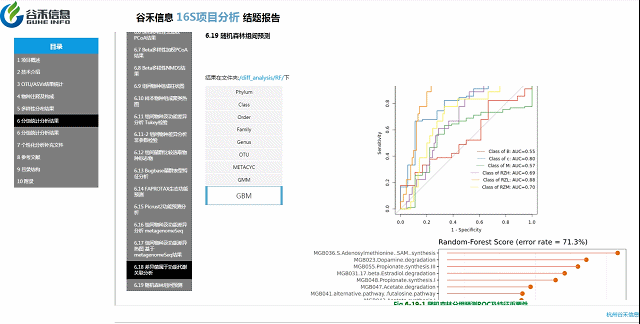

判断是哪个层面上的数据能最大程度作为分组样本的区分,以及区分效果。
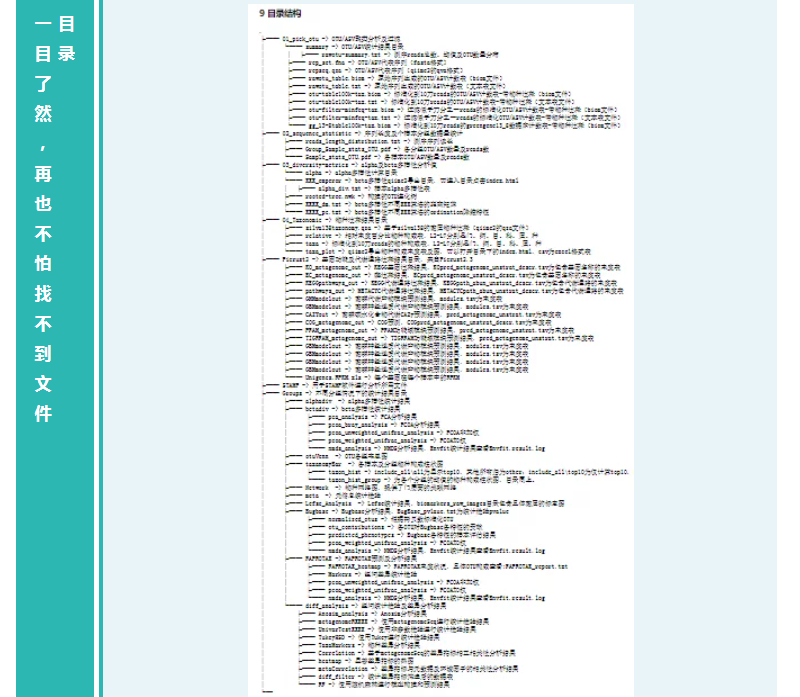



我们提供的基础分析包括以下所有内容:


相关阅读:

谷禾健康
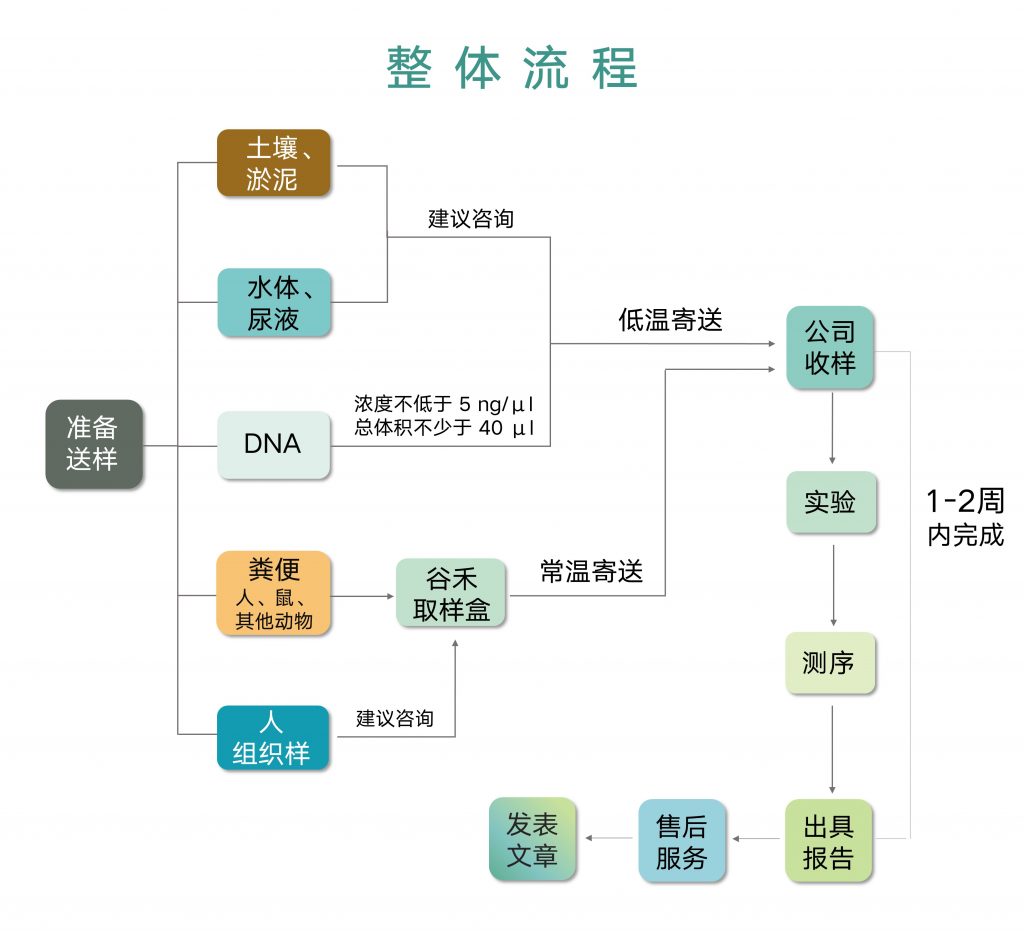
肠道菌群检测的第一步是取样,取样的重要程度不言而喻,实验人员只有在拿到合格样本后才能开展后续实验。
如果储存和运输不当菌群结构就会发生变化,进而导致菌群测序不准确,因此,便捷可靠是关键。
谷禾经过多年肠道菌群检测实践和研发,开发出适用于肠道菌群取样和常温储存的取样管,可以采集并稳定DNA,用于定量肠道菌群组成分析。

整个取样盒包括:
取样管(内含裂解液和稳定液);
无菌棉签;
回寄袋;
每个取样管上均有唯一条码。
主要特点:
在家中轻松自行采样高质量样品
起始样品需要量低至0.01g,快速且稳定
常温保存运输
标准样品适合手动或高通量自动处理
获得适用于16S ,qPCR,宏基因组的高质量DNA
条形码化全样本可追溯性
谷禾取样管的独特特点使得取样变的异常简便,下面是取样演示:
仅需使用棉签从厕纸上沾取粪便,然后洗脱到取样管的保存液中即可,使保存液可见粪便颜色即表示取样量足够。

适用于-20°C至65°C下保持DNA完整性
室温下有效存储长达60天
与新鲜样本一致的菌群构成特征
低成本
下面来看一下取样管在不同条件下的保存效果,我们使用凝胶电泳来检测不同保存处理条件下提取菌群DNA的状态:
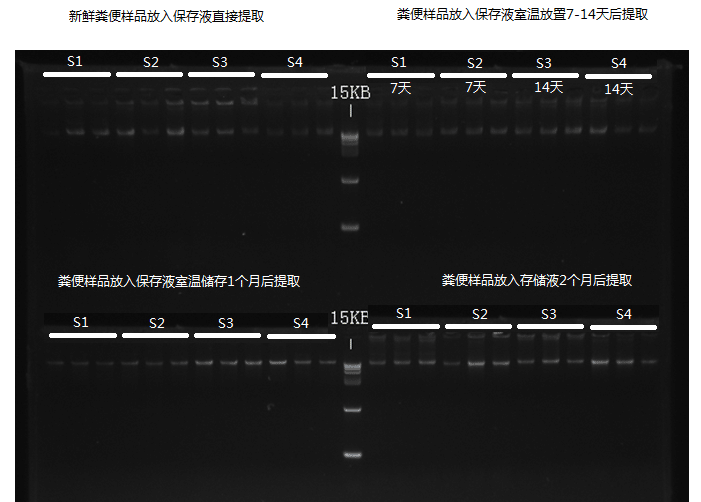
可以看到,使用谷禾保存管的DNA样品即便在存储至60天仍然没有出现明显的DNA降解情况。
专利号:ZL201511009389.7
配合谷禾肠道菌群取样保存管
适用于提取极低当量菌群DNA
具备以下特点:
磁珠法-适用于自动化高通量提取
起始量限制低
与MoBio试剂盒一致性高
现有样本处理量450例/天

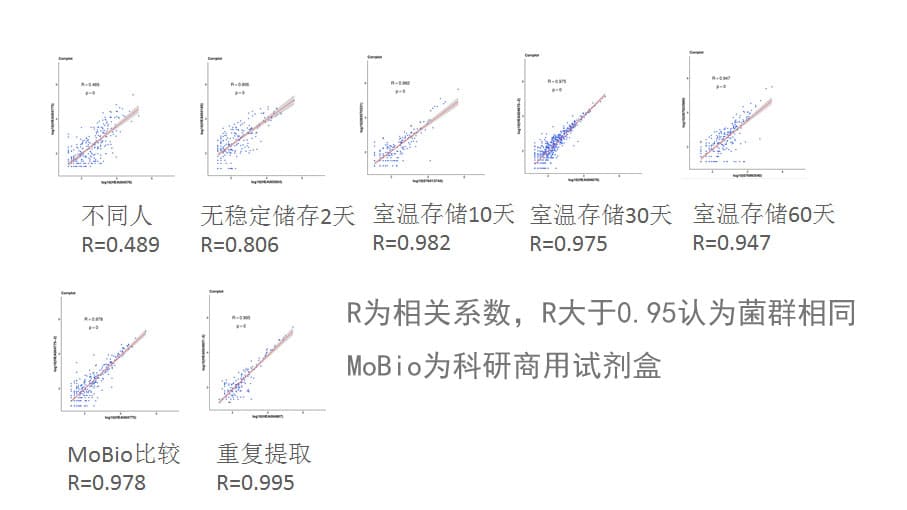
下图可以看到我们使用谷禾提取方法与MoBio试剂盒比较以及重复提取的菌群相关性。另外同时比较了使用谷禾取样管保存不同天数后的提取菌群结果。
专业的实验环境
让整个实验操作
得以高效可靠运行
二级生物安全实验室

注:生物安全实验室的分级

生物安全实验室一般实施两级隔离。一级隔离通过生物安全柜、负压隔离器、正压防护服、手套、眼罩等实现;二级隔离通过实验室的建筑、空调净化和电气控制系统来实现。

谷禾健康
基因组规模代谢网络模型(Genome-scale metabolic model,GEM),是一种包含了某种特定生物或者是细胞基因组范围代谢反应,及其酶及基因关联的数学模型。
这里,我们基于文章的描述,介绍一款新软件——MetaGEM。
研究者认为,目前代谢建模的工作流程仍然是倾向于依赖参考基因组作为重建和模拟GEMs的起点,这忽略了微生物群落中存在的物种内和物种之间的多样性。也限制了对已知参考基因组空间中的代谢网络的分析和解释。
可能导致假阳性(即在参考基因组中存在但在群落中的变量中缺失的通路)或假阴性(即在参考基因组中缺失但在群落变量中存在的通路)结果,最终导致对个别物种代谢通路以及交互营养共生(cross-feeding)相互作用的不准确预测。
也就是说当前的代谢建模方法很可能无法捕捉特定物种在不同环境中的特定代谢特征,例如具有不同疾病状况的个体的微生物群。为了克服这一局限,研究者们开发了MetaGEM。
MetaGEM可以不依赖参考基因组,直接从短读的宏基因组数据中重建样本特定的代谢模型。
下图是该软件的流程图,图中蓝底白字的部分是该流程中所使用到的软件,都是已经由他人开发完成的。
研究者们自己开发的部分有两个:
一是end-to-end的框架,能够进行群落水平的代谢交互模拟;
二是一个来自宏基因组生物群落的14,000多个MAGs,包括3750份高质量的MAGs,以及来自人类肠道微生物组研究和全球微生物组项目的相应的随时可用的GEMs。
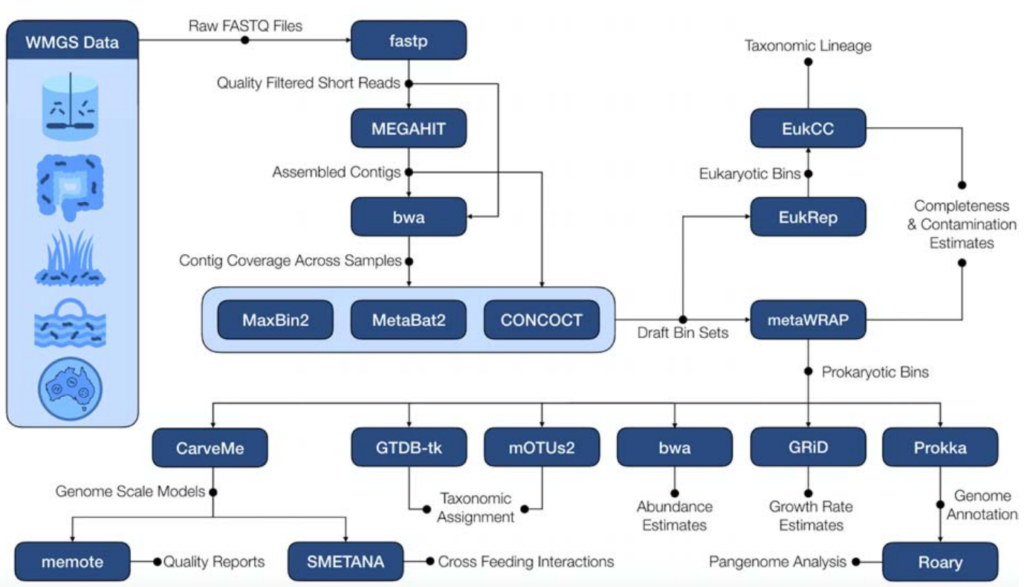
整个流程使用Snakemake实现,从原始的宏基因组的fastq文件开始,质控、组装、估计contig覆盖率、binning、Bin的改进和重组、MAG丰度定量和物种分类、CarveMe进行基因组规模代谢模型重建及质量报告,Smetana模拟重建的基因组规模代谢模型的肠道微生物群落。
(这里只简单介绍了处理步骤,文章中的“Methods”部分有给出使用的参数)
除了以上的必备选项,该流程还有一些附加功能可供用户选择。可以使用GRID估计中和高覆盖率的MAGs的增长率。
Prokka可以对MAGs做功能注释,并且其结果可以提供给Roary,获得一组MAGs的核心MAG和泛基因组的可视化结果。
EukRep可以用于寻找真核生物的MAGs。
EukCC可以对真核生物的bins做后续的分析。
MetaGEM流程具有两个特点:
一是直接从宏基因组获得高质量的代谢重建;
二是可以为个性化的人类肠道群落建模,研究者通过两个实验进行了描述:
MetaGEM模型与EMBL、AGORA、KBase和Bigg模型相比较
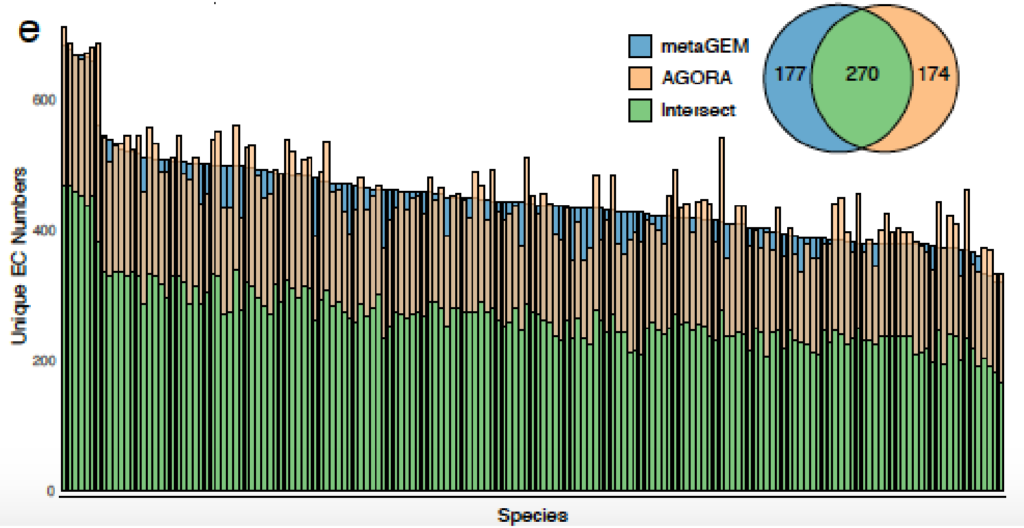
用MetaGEM基于宏基因组短读序列构建MAGs,分为HQ(高质量的),MQ(中等质量的),并以此进行代谢重建,总共获得14087个GEMs,然后将它们与高度精选的基于参考基因组的BIGG模型、AGORA、EMBL和KBase模型进行了比较。
利用基于定位的方法(方法)生成的丰度估计值与基于标记基因的丰度估计值完全相关;
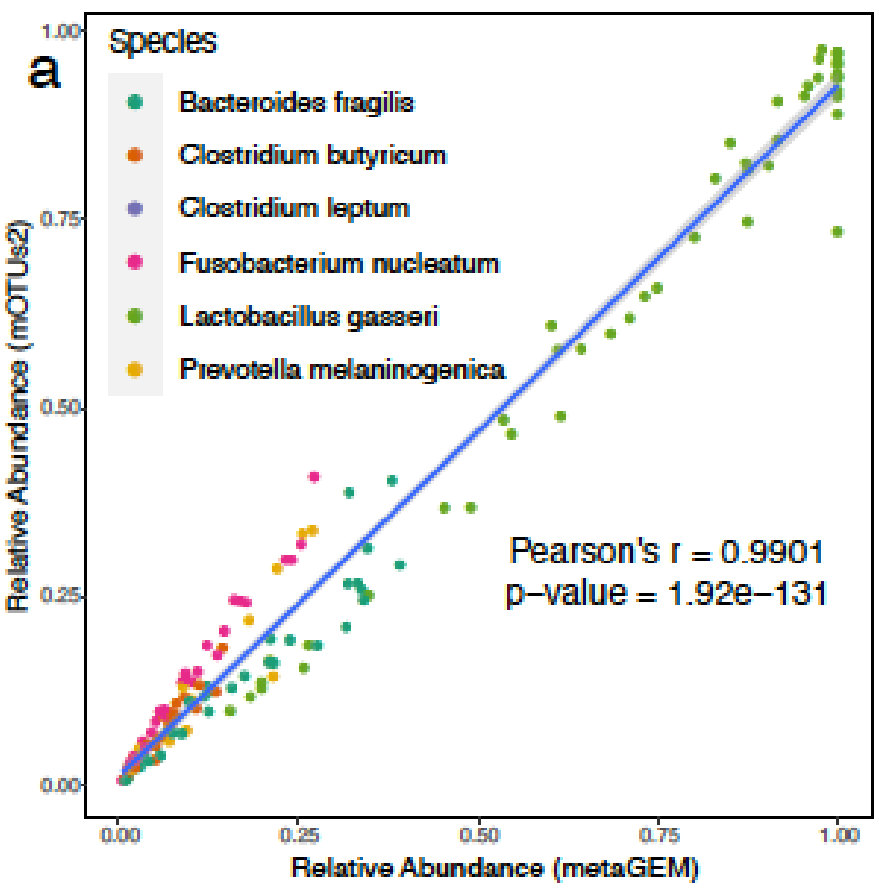
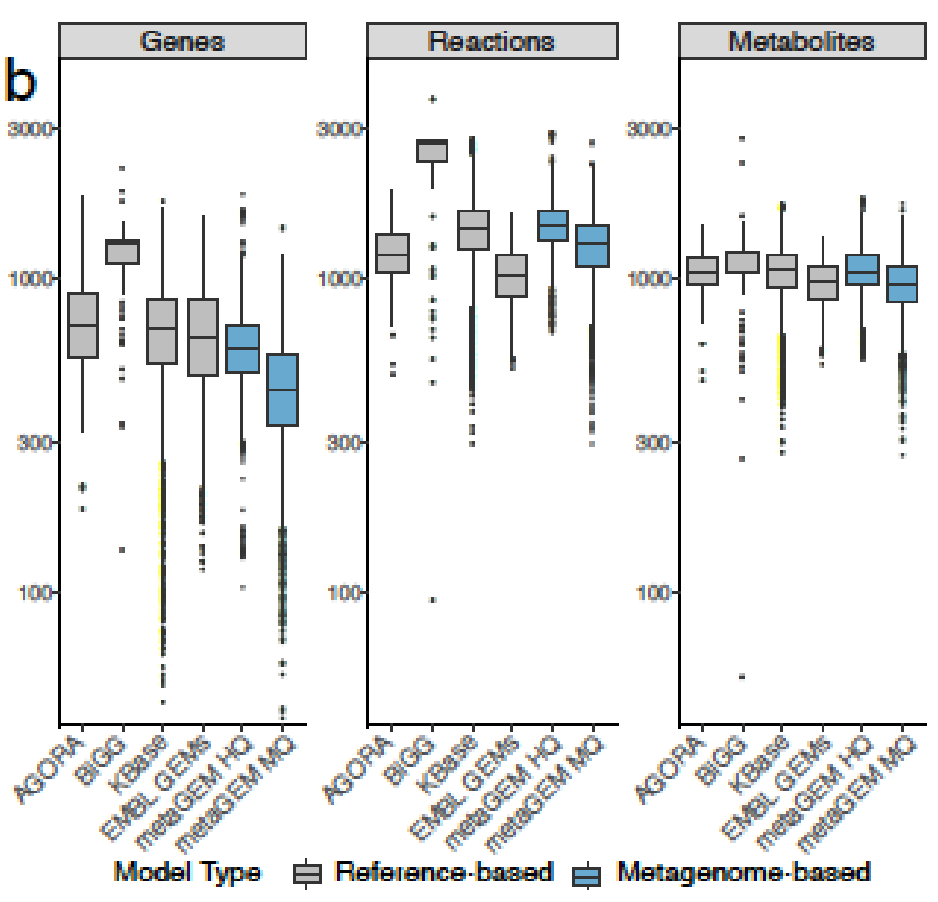
MetaGEM和其他模型都具有类似数量的反应和代谢物,但基因数量相比较少;
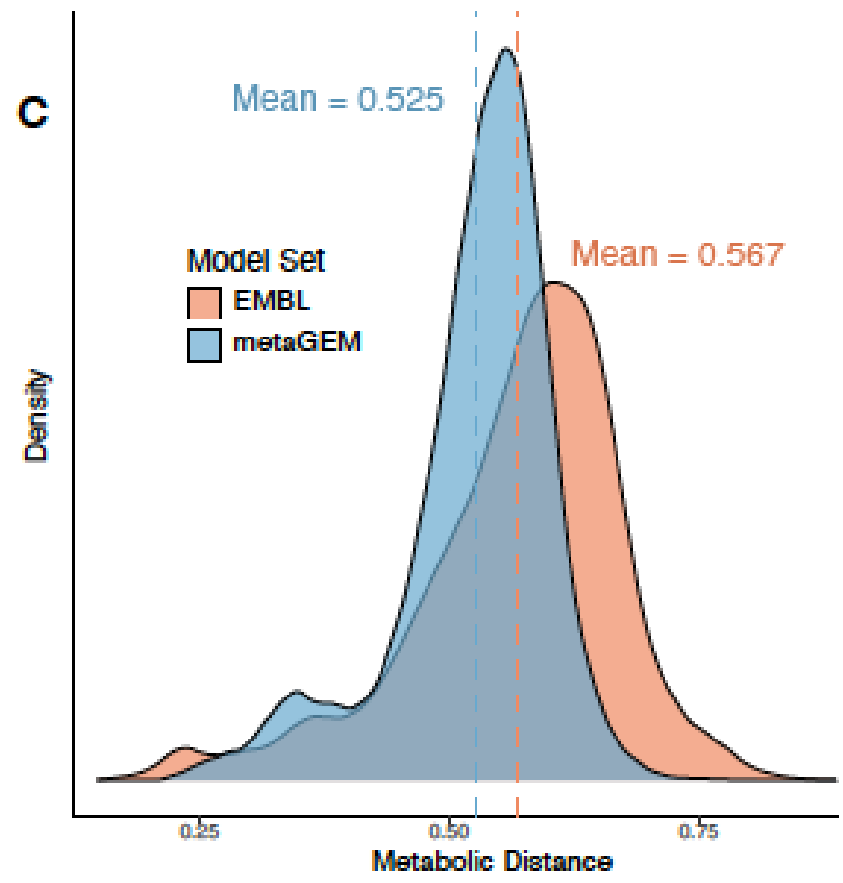
通过计算模型之间成对的代谢之间的距离,发现MetaGEM具有相似的酶多样性分布;
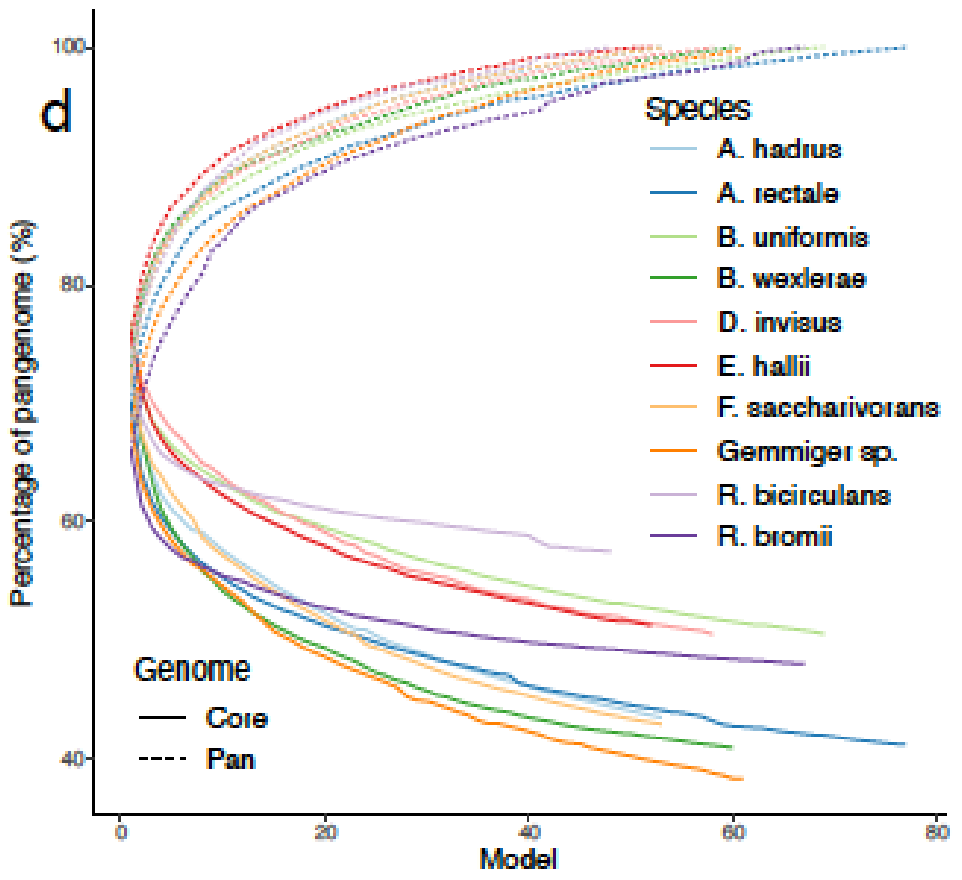
可以捕捉到种水平物种间的显著的代谢差异。高达60%的代谢多样性存在于物种泛基因组中,metaGEM模型捕获的物种内代谢变异程度显著
与基于参考基因组的肠道物种代谢模型AGORA比较,发现基于参考的模型引入的代谢反应不一定存在于每个宏基因组环境中,而MetaGEM模型是完全基于实际的宏基因组在特定环境下重建的代谢模型。
AGORA和MetaGEM模型的EC数的交集在48.9%到69%之间,其中53.9%的情况下MetaGEM模型比相应的AGORA模型包含更多的EC数。
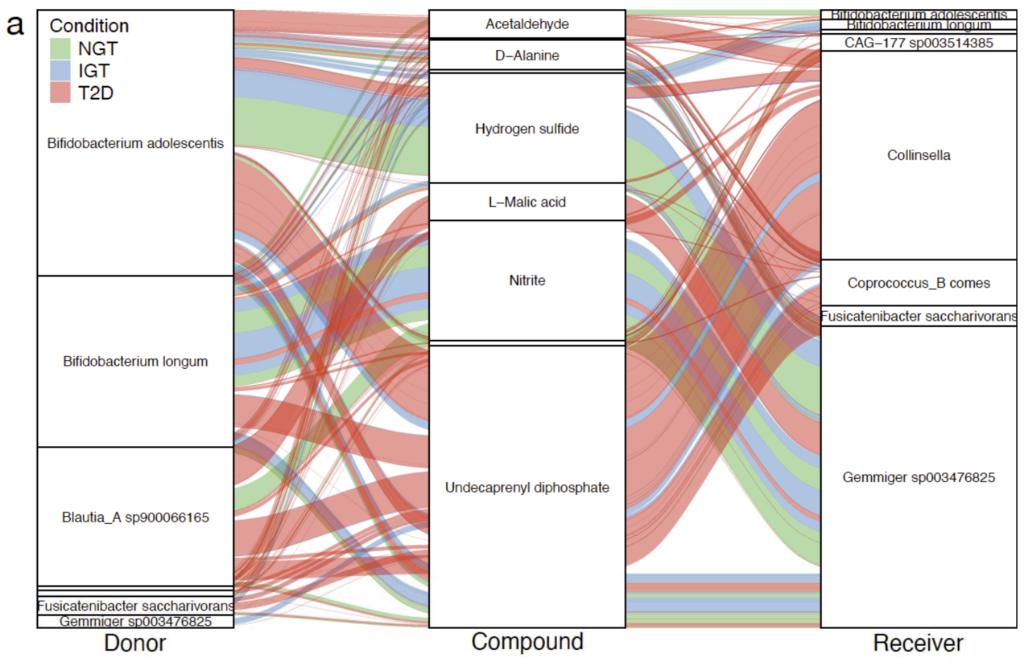
研究健康和代谢受损的2型糖尿病患者肠道微生物群落中潜在的微生物代谢相互作用。
使用metaGEMs通过137个宏基因组数据重建了4127个个性化的GEMs。
根据疾病状况分类,即正常糖耐量(NGT,n=42)、糖耐量受损(IGT,n=42)、 2型糖尿病(T2D,n=53),然后应用Smetana软件模拟微生物群落中的物种间依赖关系,Smetana为每个群落输出一个分数表,对应于在给定条件下为支持群落成员的成长而应发生的交叉喂养相互作用强度的度量,即物种A生长的可能性取决于物种B的代谢物X。
不同的2型糖尿病疾病组(NGT、IGT、T2D)相对应的肠道代谢基因组产生具有不同代谢结构的群落。
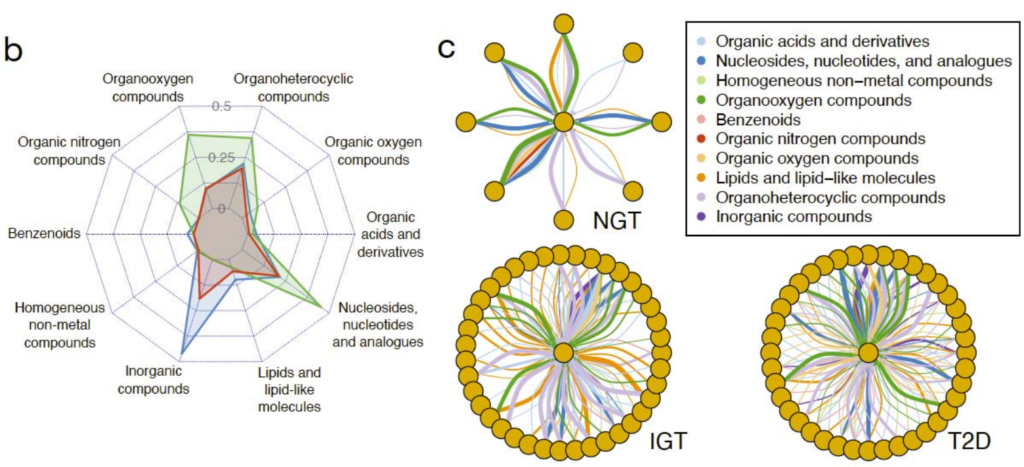
MetaGEM具有完善的流程,搭载的工具也是生物信息分析中常用的处理工具,下载很方便,用conda就能完成。无需参考基因组,这也意味着不需要下载动辄几十Gb的文件。使用Snakemake做流程的自动化管理,运行命令简单,也可以分步骤运行。
总体而言,MetaGEM可以直接从宏基因组数据中研究复杂微生物群落中特定样本(sample-specific)的新陈代谢。
【附录】
关于文中MetaGEM流程搭建所应用到的宏基因组分析软件,这其中也有我们常用的软件,比如fastp、MEGAHIT、bwa、SAMtools、metaWRAP,它们在处理数据时非常的方便也易于上手。
参考文献:
Zorrilla F, Patil K R, Zelezniak A. metaGEM: reconstruction of genome scale metabolic models directly from metagenomes[J]. bioRxiv, 2021: 2020.12. 31.424982.
相关阅读:

谷禾健康
迄今为止,已经有了许多对呼吸道微生物组通过16S rRNA高通量测序的研究。这其中所有基于扩增子的研究的共同之处就是PCR的应用:
一是扩增待测序的目标标记基因,
二是为多样本的混合测序添加必要的索引序列。
这些步骤可以通过一步PCR或两步PCR完成,但没有研究说明两步PCR方案相关的实验室处理步骤是否会使样品比一步PCR方案更容易受到来自实验室的细菌DNA污染的影响。
本文
试图确定对16S rRNA V3V4与V4基因区域的一步或两步PCR的建库方案对上呼吸道和下呼吸道微生物组的影响
对收集的样本进行了三个设置下的lllumina MiSeq测序
设置1(两步PCR,V3V4区域)
设置2(两步PCR,V4区域)
设置3(一步PCR,V4区域)
分别对这三个设置产生的测序数据进行分析
结论
PCR步骤数量的差异会影响对呼吸道微生物群落的物种组成分析,且对上呼吸道(高细菌载量)的影响小于下呼吸道(低细菌载量),这表明PCR设置的偏差与样本生物量有关。
通过三个实验,即对模拟群落样品HM-783D、NCS样品、呼吸道样品采用三种PCR方案进行建库后的测序结果分析,研究这三种建库方案对其菌群描述的影响。
模拟群落样品HM-783D,来自20种不同细菌物种(17个属)的基因组DNA。
阴性对照样本NCS
呼吸道样品,从Bergen COPD微生物组研究中选择了23名研究对象,其中9名健康,4名患哮喘,10名患COPD(慢阻肺)。上呼吸道样本以漱口水(OW)为代表,下呼吸道样本以标本刷(PSB)和支气管肺泡灌洗液(PBAL)为代表。
三种PCR方案:
细菌DNA提取后通过三种不同的建库设置进行MiSeq测序,分别为
Setup1(两步PCR,V3V4区域);
Setup2(两步PCR,V4区域);
Setup3(一步PCR,V4区域)
下图完整的展示了三个PCR设置下的使用呼吸道样本的生物信息学过滤步骤:
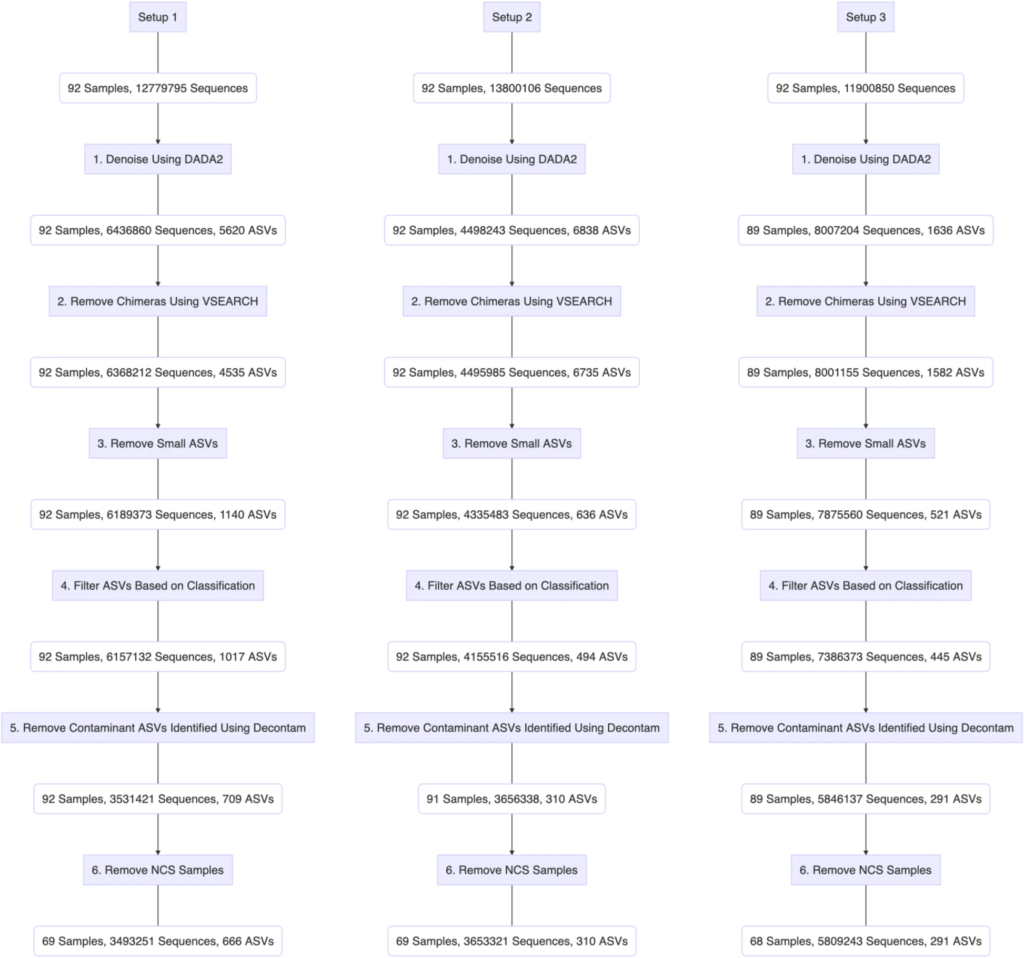
最终:
设置1:得到了666个ASVs
设置2:得到了310个ASVs
设置3:得到了291个ASVs
1. 对模拟群落样品HM-783D的分析
在设置1中进行了四次测序,设置2和设置3分别进行了一次。
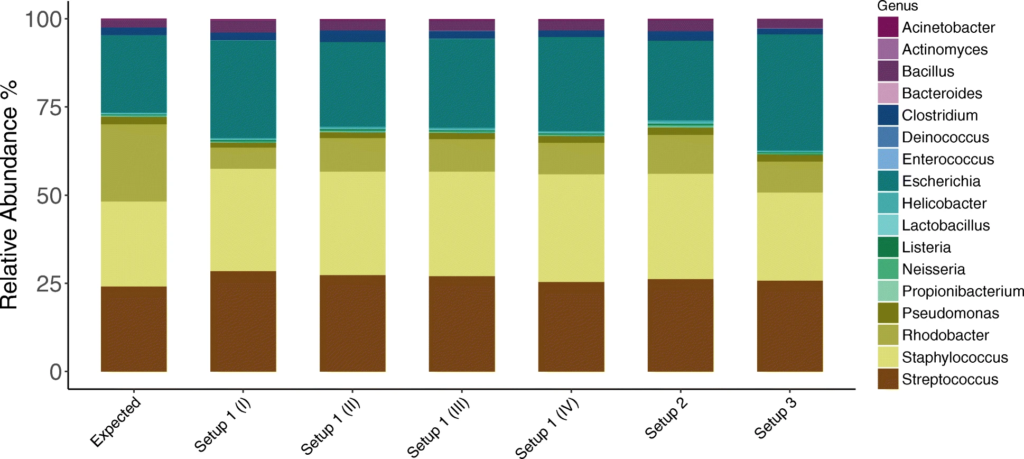
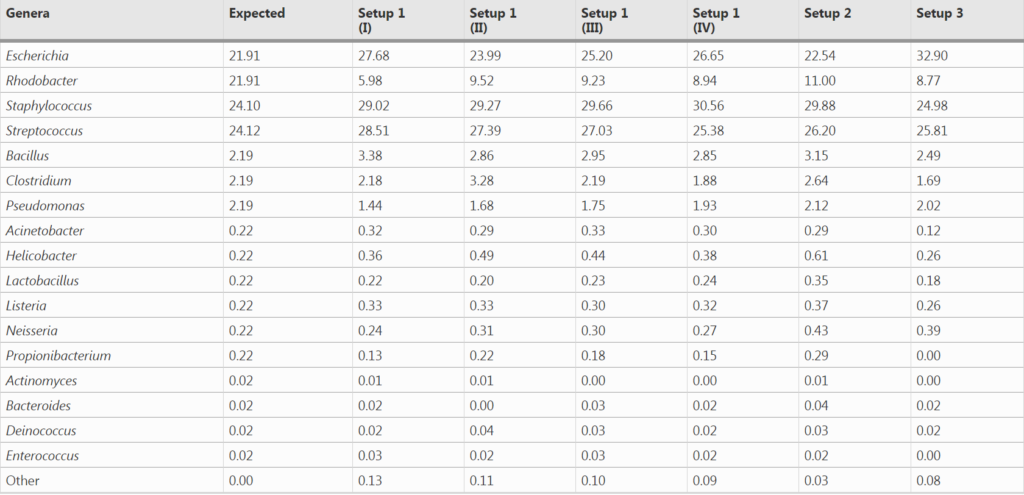
与预期丰度(Expected)相比,柱状图中观察到三种PCR设置下的各菌属的相对丰度与预期丰度相差不大,表中数据显示,三种PCR设置都在恢复高丰度物种方面具有最高的效率,但设置3在回收低丰度物种时的效率最低。
2. 对阴性对照样品的分析
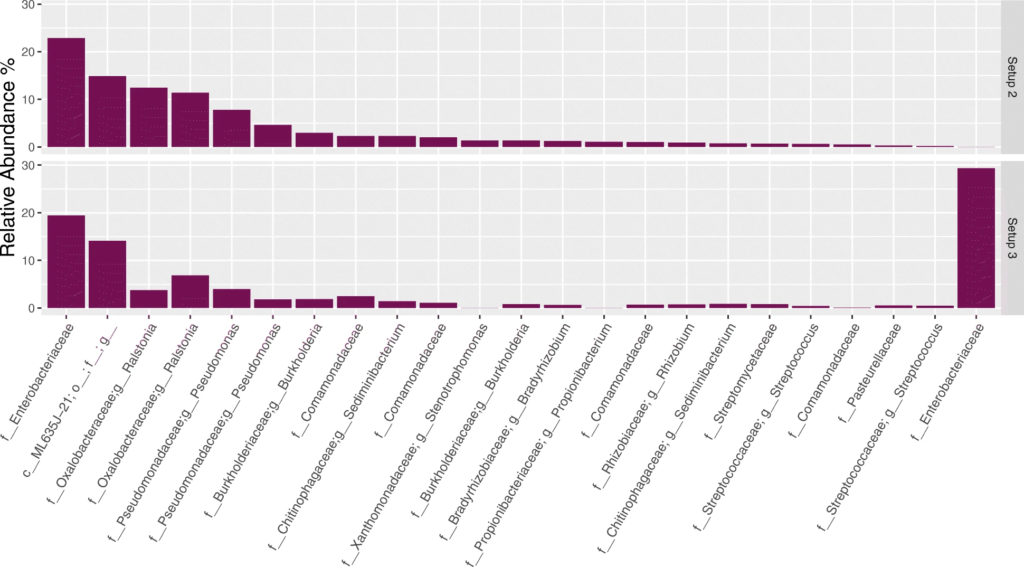
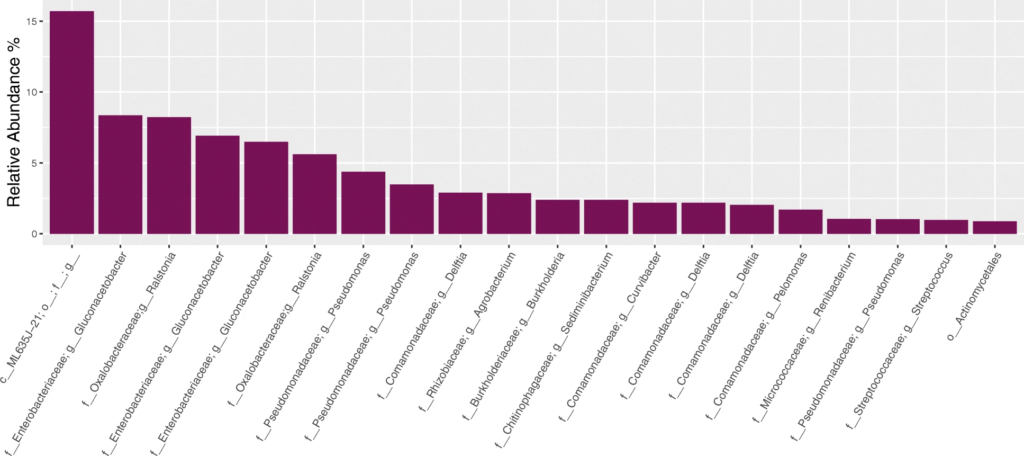
从上至下分别为设置123测序后,在NCS样品中观察到的20种最丰富的ASV。通过R包Decontam去除污染物,在设置23之间差异最大的是属于肠杆菌科的ASV,与后续的对水样品进行设置23下的测序分析结果相比较,发现大肠杆菌ASV就是在建库步骤中使用设置3的试剂时引入的污染物。
3. 对采集的呼吸道样品的分析
在去除污染物前后,代表为呼吸道菌群的链球菌,普雷伏氏菌,Veillonella和Rothia属的相对丰度变化不大,而去除污染物后,预测作为污染物代表的数量较少的物种被滤出。基于主坐标分析,发现高细菌载量的OW样品聚集在一起,低细菌载量PBAL,PSB一句设置23分离开。
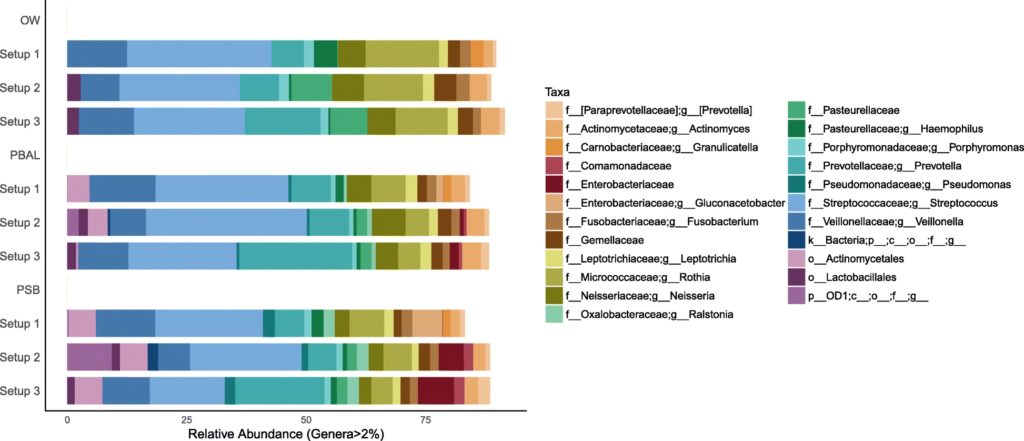
去除污染物之前的三种类型样品的三种PCR设置下的物种分类
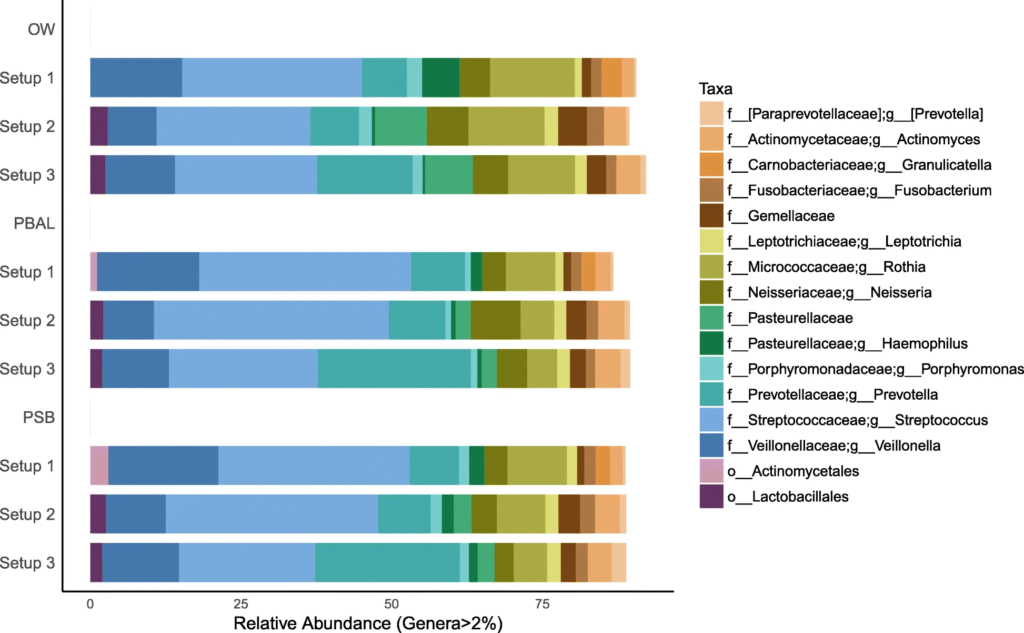
去除污染物后的
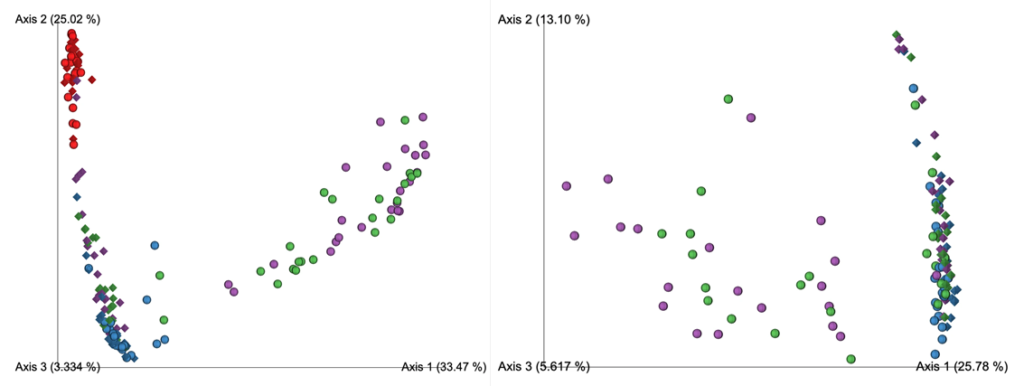
从左至右分别为去除污染物前后的未加权UniFrac距离的主坐标分析
OW:蓝色; PBAL:绿色;PSB:紫色;NCS:红色。
设置2(球形),3(菱形)
文章作者给出的结论是文库制备和测序方法的选择会对呼吸道微生物组的分析产生影响,且对上呼吸道的影响小于下呼吸道。靶向扩增子区域的差异(16S rRNA基因V3 V4与V4)并未表现出对细菌群落描述的重大影响。对于整篇研究存在的主要的局限性在于仅研究了DNA提取后的PCR步骤,污染或影响也可能来自于更前期的处理。
编者按
在使用测序技术进行的微生物研究中,测序偏差和污染物是一直存在的问题,也因此诞生了许多工具和计算方法用于尽可能的消除或降低这方面的影响。这篇研究也提醒了我们,在呼吸道微生物组的研究中,要注意上呼吸道与下呼吸道的菌群差异或相似可能不仅仅来源于样本自身,还可能掺杂着PCR方法选择上的影响。
参考文献:
Drengenes C, Eagan TML, Haaland I, Wiker HG, Nielsen R. Exploring protocol bias in airway microbiome studies: one versus two PCR steps and 16S rRNA gene region V3 V4 versus V4. BMC Genomics. 2021 Jan 4;22(1):3.
相关阅读:
谷禾健康
宏基因组中短序列的注释是理解测序微生物群落潜在功能的重要步骤之一。单纯利用局部匹配的注释容易混淆那些蛋白同源性且局部序列非常相似的序列,进而不能真实准确反映复杂蛋白质家族中多变的结构和功能域。
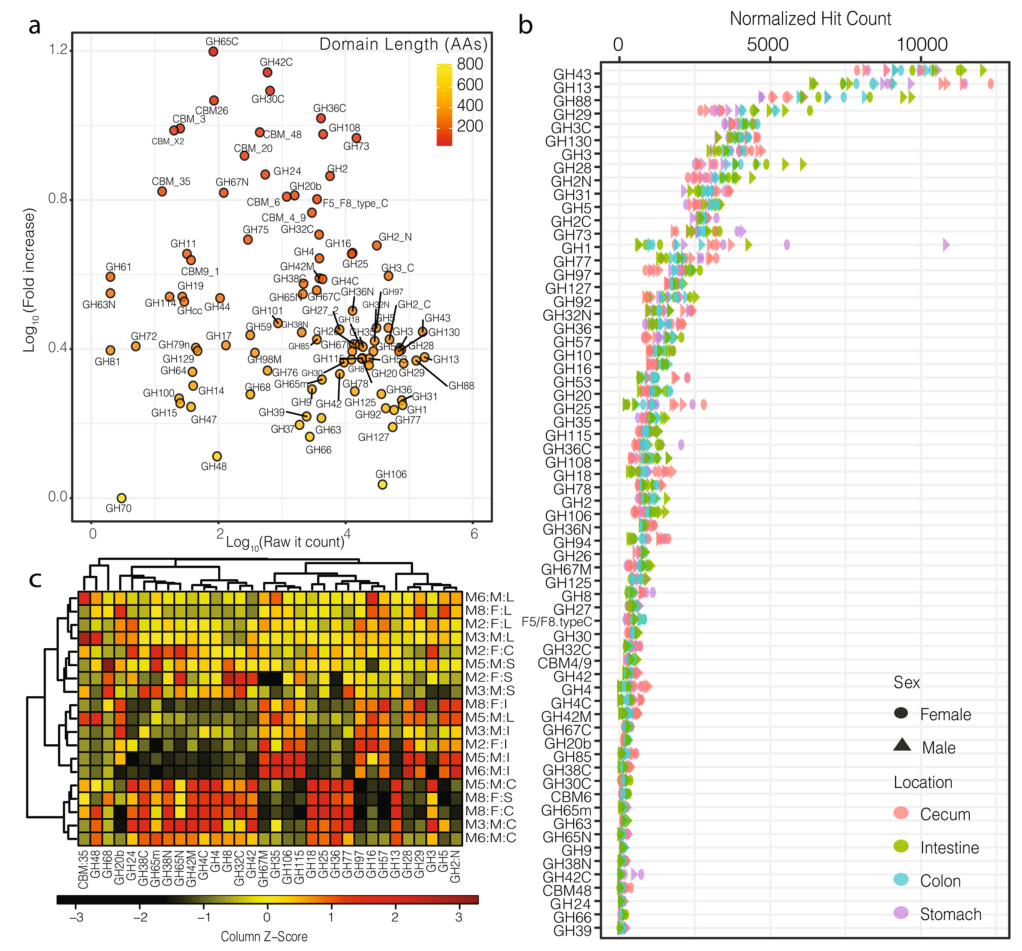

今天我们介绍一种新方法MetaGeneHunt,该方法可以识别特定的蛋白质结构域,并根据结构域的长度对hit-counts进行标准化。使用MetaGeneHunt对MG-RAST对公开获取的宏基因组进行分析,包括哺乳动物微生物群和Twin Gut肠道菌群研究,以评估短序列中含GH蛋白的频率和位于GH区域的匹配频率。
在对糖苷水解酶(GHs)的研究,发现在所有样本中4726,023条含有GH区域蛋白匹配的短读序列中,有58.3%的序列位于目标区域之外。接下来,在比较样本之前,将匹配到目标区域的hit-counts标准化,以说明对应的域长度。肠道和盲肠中的菌群显示出与不同微生物组合相匹配的GH谱特征。
相反,胃和结肠的菌群在结构和功能上显示出更多样性和多变性。在样本中,尽管有波动,但碳水化合物处理的潜在功能变化与群落组成的变化相关。这表示,在利用MG-RAST平台处理宏基因组测序序列时,MetaGeneHunt是一种能快速准确地识别短序列宏基因组中离散蛋白结构域的新方法。
在过去的几十年里,宏基因组DNA的高通量测序已经产生了大量的序列,这些序列的特征为我们了解微生物群落的结构和功能提供了许多认知。例如,截至2019年12月,MG-RAST托管了约40万个可公开访问的带注释的数据集。在数据处理过程中,不考虑目标区域(或蛋白质)的长度会导致两个主要的系统偏差。
首先,目标区域越长,他们的频率就越容易被高估。其次,如果数据处理涉及稀疏性,较短的、不太丰富的域,尽管重要,也可能被丢弃。为了解决这些问题,研究人员设计了MetaGeneHunt来精确注释从MG-RAST检索到的短序列宏基因组中的蛋白质结构域。MetaGeneHunt将MG-RAST提供的短序列局部比对与M5nr数据库中精确的基于PFam的蛋白质结构域识别相结合,以在公共可访问数据集中识别蛋白质结构域。
MetaGeneHunt简要说明:
MetaGeneHunt的设计基于MG-RAST平台注释的数据集的。在使用GeneHunt创建的M5nr数据库中,MetaGeneHunt使用了糖苷水解酶和辅助结构域(如CBMs)的精确的特定结构域注释(PFam)作为参考注释表(RAT)。
首先,MetaGeneHunt使用MG-RAST应用程序接口从MG-RAST(“330”和“650”文件)检索M5nr注释的宏基因组。接下来,使用来自RAT的注释命中的MD5id,在文件“650”中识别与潜在的GHs匹配的序列。
接下来,对于这些局部匹配,将精确对齐位置与RAT中特定于域的注释进行比较。如果查询中的>20AAs与特定的蛋白质结构域(考虑到RAT中的HMM-envelope位置)对齐,则该结构域注释被转移到查询中。
相反,如果查询的>20AAs匹配在目标区域之外(例如,在连接域、辅助域、信号肽中),则该注释被认为是否定的。用户可以随意修改重叠(overlapping)的阈值。接下来,从序列聚集文件( “330”文件)中检索每个识别出的命中的实际序列计数。最后,在后续的数据处理和标准化过程中,根据Pfam数据库中蛋白质结构域的大小,对每个蛋白质结构域的命中计数进行标准化。
方法验证:
文中使用的原始数据和预处理数据可在MG-RAST服务器上公开访问。在mgp20861项目中可获得对应于〜555百万个100 bp序列的小鼠微生物组数据。使用MG-RAST API 检索了哺乳动物微生物组数据(mgp116)和双肠肠道菌群研究(mgp10)其他数据集。哺乳动物微生物组研究糖苷水解酶(GHs)和相关酶的附加注释表是从Brian Muegge(直接对应)获得的。使用MG-RAST API检索了预处理的数据,包括从门到属水平的读物分类注释。数据分析和统计使用R统计语言。
1. 糖苷水解酶的识别,识别蛋白质结构域并考虑其长度产生了一个健壮的功能注释系统,对hit-count的标准化反应了目标区域的实际分布。

a).横轴为目标区域的原始hit-count,纵轴为标准化后的hit-count,图中的颜色阶梯表示目标区域的长度。这种标准化主要影响长度短的域(例如,GH78、GH25)、小的亚域(例如,GH31N、GH36C)和目标区域的附属域(例如,CMB5_12)。
b).小鼠胃肠道中目标区域的标准化后的hit-count(仅显示大于100的hit-count的区域),可见,标准化后的hit-count与结构域长度无关(附加文件中有对两者做相关分析,结果分别为P.pearson=0.38,P.spearman=0.33)
c).热图显示了小鼠胃肠道中最受样本来源影响的被稀疏标准化的GH区域的分布(two-way方差分析)。纵轴的注释列Mx:F/M:S/I/C/L分别表示小鼠(样本号):雌性/雄性:胃/肠/盲肠/结肠
2. 小鼠肠道菌群的结构,与盲肠中的微生物群落相比,结肠与肠道中的微生物群落结构更相似,结肠和胃中的微生物群落有较高的相似性。

a).对受样本来源影响较大的样本根据属水平进行样本聚类(Bray-Curtis距离指数,complete linkage)。
b).样本间的微生物群落组成,只展示了相对丰度至少占群落中1%的属水平物种(V:疣微菌门,B:拟杆菌门,A:放线菌门,F:厚壁菌门)。
c).NMDS分析(2D stress=0.020),展示了在样本聚类中都存在的这些菌属,在b)中的主要类群用标签指示,不同门水平按颜色区分,点的大小反映该属在样本中的最大频率。
微生物组中的结构-功能关系,多样性仍然与潜在功能高度相关。胃和盲肠的群落在结构和功能上是最多样化的。其次,肠道中的群落组成和功能大多是保守的,而与保守的微生物群落相关的大肠则显示出可变功能潜力。
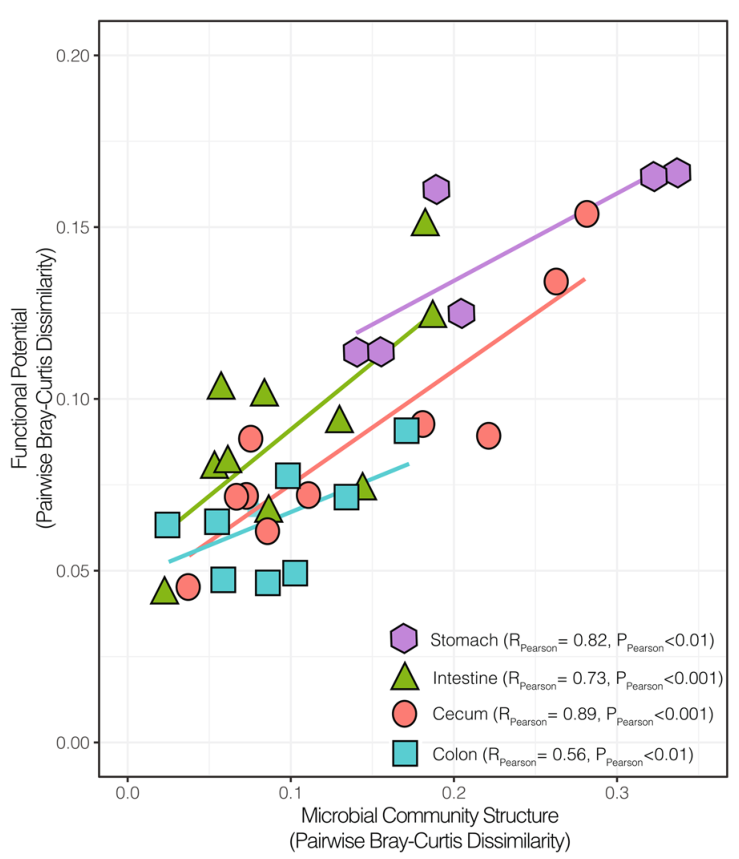

对同一位置的样本的微生物群落结构和功能差异进行成对比较(Bray-Curtis),线条为线性回归的结果。在胃,肠,盲肠和结肠中,属水平群落结构的变化与多糖解构功能的相关性分析结果表示除大肠外,其余的P.pearson的值都在0.001以下。胃和盲肠的群落在结构和功能上是最多样化的,尽管多样性仍然与功能潜力高度相关。其次,肠道中的群落组成和功能大多是保守的,而与保守的微生物群落相关的大肠则显示出可变的功能潜力。
MetaGeneHune提供了一种新的方法来识别短序列宏基因组中的GHs及其相关结构域。识别结构域而不是蛋白质是至关重要的,因为GH结构域与许多可变结构域相关。这种新方法基于GeneHunt注释方法,并对其进行补充,旨在分析MG-RAST中的短序列宏基因组。因此,它不需要大型计算机基础设施。
通过这种新方法对小鼠胃肠道菌群的GHs研究发现,在胃中,虽然富含碳水化合物处理的酶,但相对于胃肠道的其他部分,胃中没有特定酶可供选择;在肠道中,出现了更保守的菌群,最为富集的是拟杆菌门,它们的潜在功能主要在多糖处理上;来自结肠和胃的菌群虽然是距离最远的,但在结构和功能上却表现出高度的相似性。
在未来,利用GeneHunt和MetaGeneHunt相结合创建新的专用参考注释表将为研究宏基因组的潜在功能提供新的更有效的途径。
MetaGeneHunt和GH的RAT可在GitHub上公开访问。(https://github.com/renober/MetaGeneHunt)
参 考 文 献
Berlemont R, Winans N, Talamantes D, Dang H, Tsai HW.MetaGeneHunt for protein domain annotation in short-read metagenomes. Sci Rep.2020 May 7;10(1):7712. doi: 10.1038/s41598-020-63775-1. PMID: 32382098; PMCID:PMC7205989.
Muegge BD, et al. Diet drives convergence in gut microbiomefunctions across mammalian phylogeny and within humans. Science.2011;332:970–4. doi: 10.1126/science.1198719
Turnbaugh PJ, et al. A core gut microbiome in obese and leantwins. Nature. 2009;457:480–484. doi: 10.1038/nature07540.
Berlemont R, Martiny AC. Glycoside Hydrolases acrossEnvironmental Microbial Communities. PLOS Comput. Biol. 2016;12:e1005300. doi:10.1371/journal.pcbi.1005300.
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity,stability and resilience of the human gut microbiota. Nature. 2012;489:220–30.doi: 10.1038/nature11550.
Sharpton TJ. An introduction to the analysis of shotgunmetagenomic data. Front. Plant Sci. 2014;5:209. doi: 10.3389/fpls.2014.00209.

谷禾健康
链读测序(Linked-read sequencing)通过将相同的barcode与长DNA片段(10-100kb)的序列连接在一起,能够消除其中的一些错读,从而改进宏基因组组装。但目前还不清楚在使用链读测序时参数的选择对组装的质量的影响如何。
近日,香港浸会大学研究人员发表文章 “通过链读测序对宏基因组组装全面研究”。


模拟数据和模拟菌群中的分析结果表明,模拟数据(simulated data)中读取深度(C)与组装序列的长度呈正相关,但对组装序列的质量影响不大,模拟菌群的研究中读取深度(C) 对组装序列的质量以及被注释为基因组草图的bin的比例有轻微影响。
另一方面,宏基因组组装质量受CR(每个短读长片段的平均深度)和CF(由长DNA片段计算的基因组的平均物理深度)的影响。对于相同的读取深度,较深的CR 会产生更多的基因组草图,而较深的CF 会提高基因组草图的质量。
还发现μFL(未加权的DNA片段的平均长度)对组装有边际效应,而NF/P(每个分区的片段数)对局部组装涉及到的偏离目标读数(off-target reads)有影响,即较低的NF/P值会通过减少off-target序列的错读而有更好的组装效果。
总体而言,与Illumina的短读长相比,使用链读改善了组装中重叠群的N50,但与PacBio CCS的长读长相比则没有改善。
人体微生物群是一个复杂的系统,在生理活动和疾病中起着重要的作用。对微生物群中的微生物基因组进行测序可以帮助我们研究其功能。
然而,微生物基因组序列很难获得,微生物群中的绝大多数微生物不能被分离出来进行单个测序。目前的宏基因组项目中使用短读长测序对混合的微生物基因组进行测序。
这些结果在基因组组装过程中是有错读的,导致微生物基因组的完整性和重叠群的连续性结果不理想。长读长测序已经被用来尝试减轻这些问题,如Nicholls等人和Sevim等人的研究。特别是Moss等人的研究,其成果优化了纳米孔测序的长读长文库制备方案,并获得了更完整的细菌基因组。
但实际应用中,长读长测序是昂贵的。虽然链读序列(linked-reads)的基因组组装的质量无法与PacBio CCS的长读长相提并论,但其低成本和高碱基质量的优点是值得去使用的。
01 三组链读序列数据集的来源及构成:
模拟数据(simulated data):
从MBARC-26数据集中下载了23个细菌和3个古细菌菌株,按丰度分类,L-sim,低丰度微生物,摩尔浓度<10-15;M-sim,中等丰度微生物,10-15 < 摩尔浓度 < 10-14;H-sim,高丰度微生物,摩尔浓度 > 10-14
模拟菌群(mock community):
(ATCC MSA-1003)是一个由20个菌株组成的池,同样按丰度分类,L-mock,低丰度微生物;M-mock,中等丰度微生物;H-mock,高丰度微生物;UH-mock,超高丰度微生物。
人类肠道菌群:
一份来自健康的中国人粪便样本
02 DNA提取、文库制备和测序:
对于模拟菌群,从ATCC 20菌株交错的混合基因组材料中提取DNA,不进行大小选择。
对于人类肠道菌群,用Qiagen QiAaMP粪便迷你试剂盒提取DNA,去掉5kb以下的DNA片段。
脉冲场凝胶电泳后,按照厂商的说明制备10x Chromium文库。使用Illumina XTen双端2x150bp测序。人类肠道微生物组的DNA也被用于标准的Illumina XTen短序列测序。
03 DNA长片段重建和链读序列二次抽样:
Long Ranger v2.2.1用于纠正barcode碱基错误,计算PCR重复率,并完成barcode感知的链读序列比对。
使用BWA-MEM v0.7.17比对短序列和没有barcode的链读序列。根据映射得到的具有共同的barcode的短序列的坐标重建DNA长片段。
链接序列首先按barcode排序,然后按它们的映射坐标排序。如果最近的barcode序列大于50kb,则终止延伸长DNA片段。每个片段必须包括至少两个具有共同barcode的成对序列,并且最小长度为2kb。
04 宏基因组组装:
对于链读序列的组装,没有 barcode 的链读序列首先由 metaSPAdes v3.11.1使用默认参数组装为“seed”重叠群,并通过BWA-MEM v0.7.17与重叠群比对。
最后使用 Athena-meta v1.3 通过汇集在 scaffold 中的两个“seed”重叠群里共享相同 barcode 的序列进行局部组装。
05 组装效果评估:
MaxBin v2.2.4将长于1kb的重叠群分组到bins中,并通过CheckM v1.0.12评估其完整性和污染率。
Quast v5.0.0统计了基础信息,如重叠群的N50、NG50、NGA50、总比对长度(total aligned length)和基因组覆盖率(genomic coverage)。
Kraken v0.10.6基于内置数据库MiniKrakenDB为bins做物种注释。每个bins都作为一个基因组草图,被分类为高质量的(完整性>90%,污染率<5%),中等质量的(完整性≥50%,污染率<10%),低质量的(完整性<50%,污染率<10%)
来自人类肠道菌群和Illumina短序列链读序列二次抽样的组装效果统计
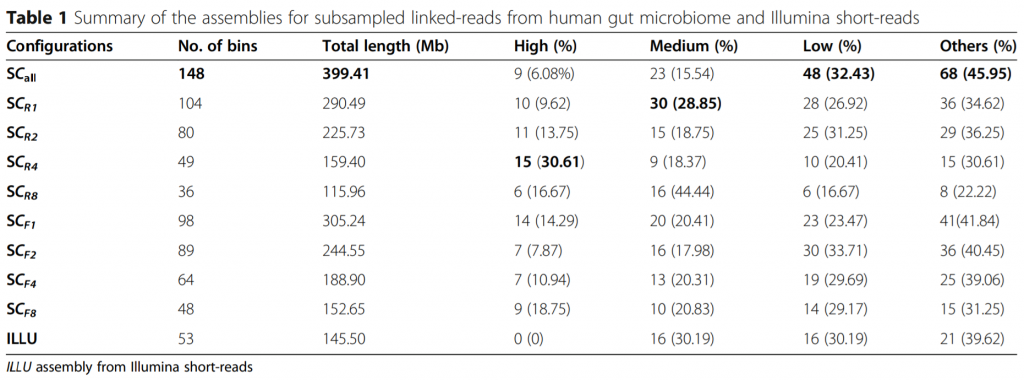

ILLU,Illumina短序列的组装
SC-all,模拟菌群和人类肠道菌群总共的两个测序lane链读序列
在链读测序中,有四个关键参数可能会影响宏基因组组装,如下图。
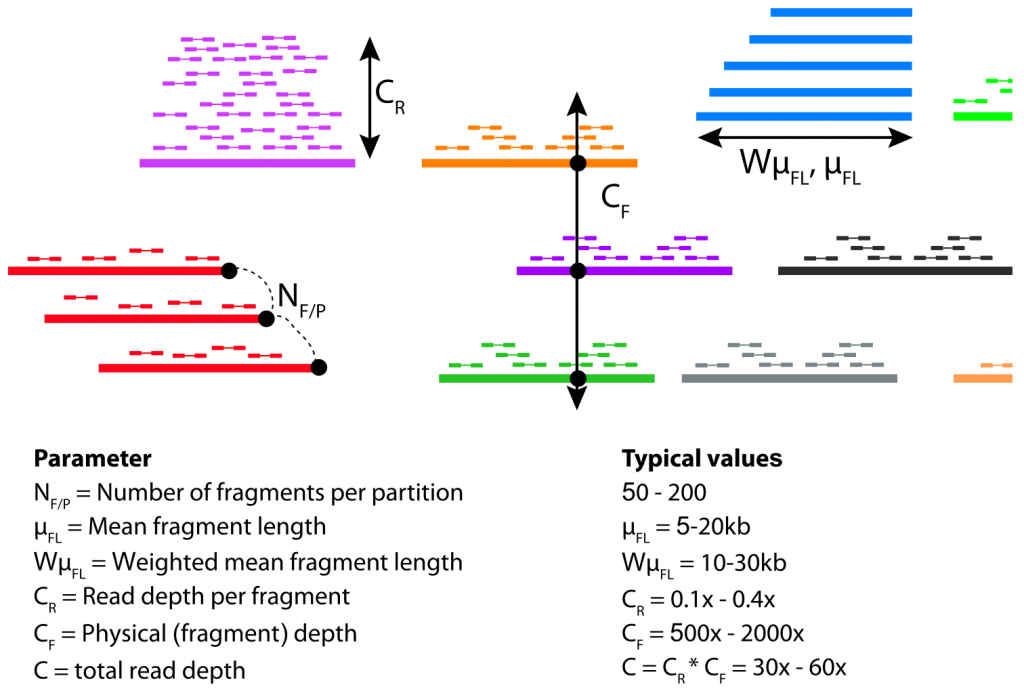

这些参数中有几个是相互依赖的。例如,输入DNA的量越大,CF和NF/P都会增加,CR就会降低;CF和CR的绝对值是由总读取深度(C)增加多少来设置的,因为CR×CF=C。
L-sim,模拟数据中的低丰度微生物,青色
M-sim,模拟数据中的中等丰度微生物,蓝色
H-sim,模拟数据中的高丰度微生物,红色
L-mock,模拟菌群中的低丰度微生物
M-mock,模拟菌群中的中等丰度微生物
H-mock,模拟菌群中的高丰度微生物
UH-mock,模拟菌群中的超高丰度微生物
“-”表示测序lane的倒数,例如MSCR4/MSCF4表示四分之一测序lane的序列被二次采样
MSCR-,模拟菌群中的短序列
MSCF-,模拟菌群中的长DNA片段
MSC-1,模拟菌群和人类肠道菌群总共的一个测序lane链读序列
SC-all,模拟菌群和人类肠道菌群总共的两个测序lane链读序列
相关阅读:
参考文献:
Zhang L, Fang X, Liao H, Zhang Z, Zhou X, Han L, Chen Y, Qiu Q, Li SC. A comprehensive investigation of metagenome assembly by linked-read sequencing. Microbiome. 2020 Nov 11;8(1):156. doi: 10.1186/s40168-020-00929-3. PMID: 33176883; PMCID: PMC7659138.
He S, Chandler M, Varani AM, Hickman AB, Dekker JP, Dyda F: Mechanisms of evolution in high-consequence drug resistance plasmids. MBio 2016;7(6): e01987–16.
Peng Y, Leung HC, Yiu SM, Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth.Bioinformatics. 2012;28(11):1420–8.
Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast singlenode solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31(10):1674–6.
Nurk S, Meleshko D, Korobeynikov A. Pevzner PA: metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27(5):824–34.
Nicholls SM, Quick JC, Tang S, Loman NJ. Ultra-deep, long-read nanopore sequencing of mock microbial community standards. Gigascience. 2019;8(5): 1–9.
Sevim V, Lee J, Egan R, Clum A, Hundley H, Lee J, Everroad RC, Detweiler AM, Bebout BM, Pett-Ridge J, et al. Shotgun metagenome data of a defined mock community using Oxford Nanopore, PacBio and Illumina technologies. Sci Data. 2019;6(1):285.