-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
谷禾健康
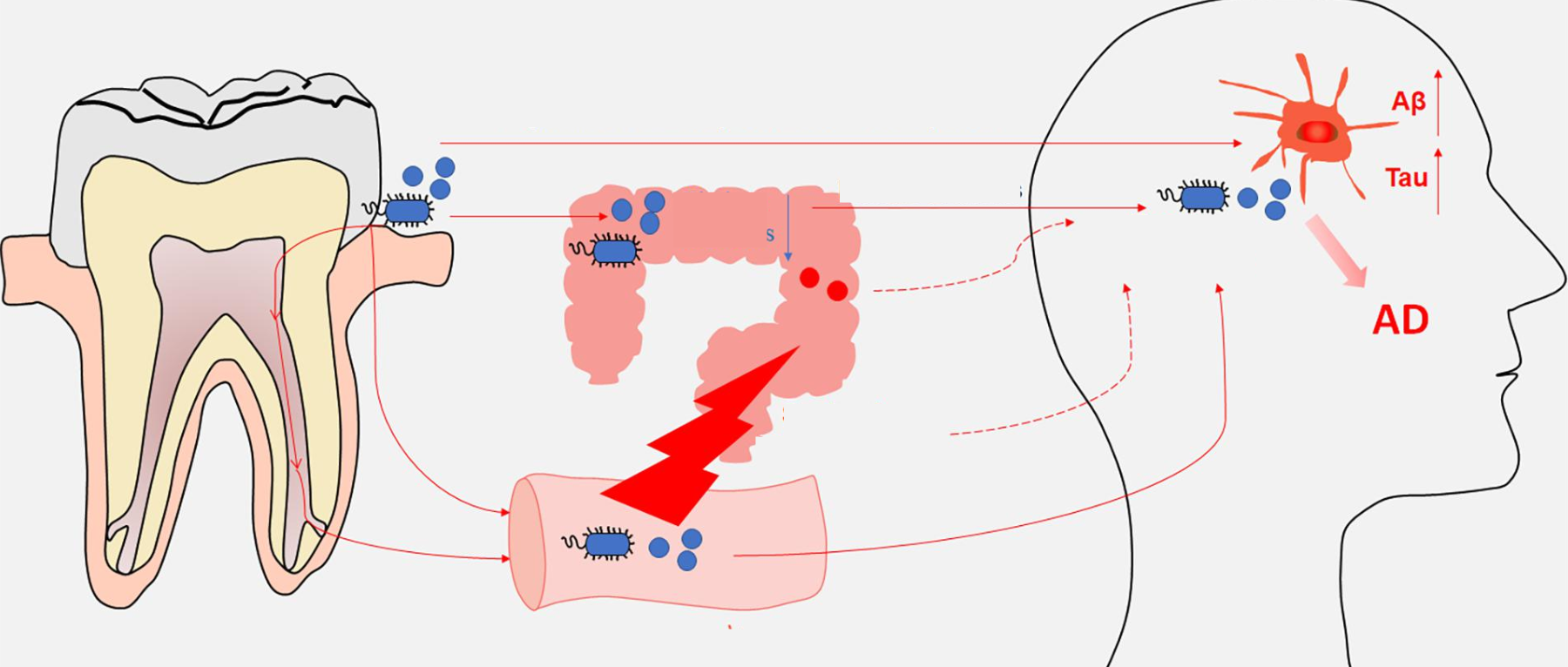
在个体中,每个微生物栖息地都表现出独特的微生物种群模式。迄今为止,关于微生物组相关疾病的研究主要集中在器官特异性微生物组上。然而,器官间的微生物网络正逐渐成为生理功能和病理过程中的重要调节因子和治疗机会。
在正常情况下,人体维持动态平衡,各个身体部位的微生物可以通过直接相互作用或间接作用于系统循环中的炎性物质、细胞因子和代谢物来相互影响。在这其中,口腔和肠道是人体中两个最为重要的微生物栖息地,它们在微生物组相关疾病中起着关键的作用。
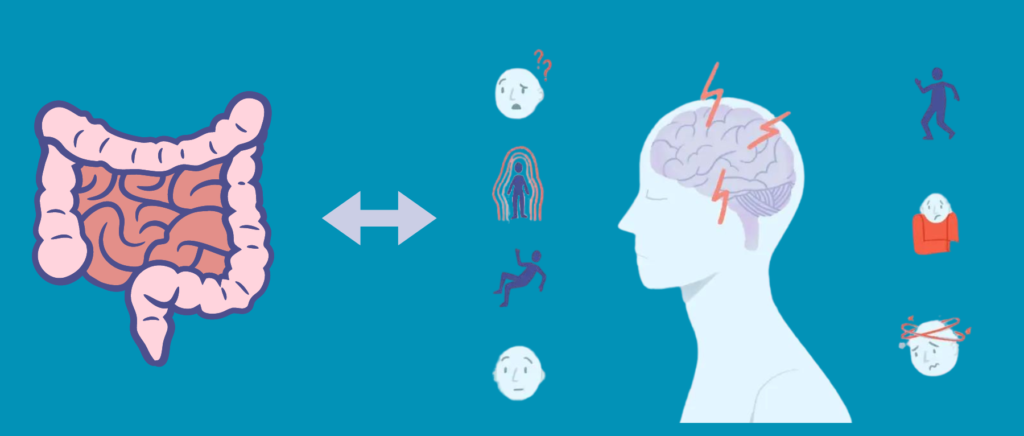
近年来,随着网络的发展和生活水平的提高,大众对心理健康产生了极大关注,同时精神类疾病也面临巨大挑战,患病率高、致残率高、治疗率低。许多患者无法获得及时有效的救治,导致病情日益严重。药物治疗只能暂时缓解症状,容易出现复发,甚至形成对药物的依赖,带来更加严重的后果。因此,对于人体微生物与精神健康之间的研究越来越多,尤其是以肠道菌群为研究靶点,探索和验证以肠道菌群为靶点治疗精神疾病的潜力。
此外,大量高质量的研究证据以及实践检测发现,精神疾病往往与功能性胃肠病共存,并可能相互影响和形成负向循环,其共同的病理生理基础是“菌-肠-脑轴”的异常互动。例如,精神疾病患者通常会出现胃肠道症状,特别是腹疼痛、饱胀、恶心、反酸、消化不良和腹泻等,以至于他们会在消化内科或外科咨询就诊。反过来,功能性胃肠疾病患者则伴随着睡眠障碍、社交障碍、焦虑和抑郁等症状,他们将前往心理科寻求帮助。同时,研究也发现患有精神类疾病的人除了胃肠道症状外,常常还伴有口腔问题,如牙周病或口臭,而患有牙周炎或牙周病的人更容易出现认知或精神障碍。
目前,在功能性胃肠病、口腔疾病与精神疾病的临床诊疗中存在一些问题,医患双方对于其临床表现的认知和重视程度不高,对病理生理机制的理解不够全面,对干预方式的整体观念和综合视角不足,导致目前该类疾病的疗效不理想。急需让精神疾病患者和医务工作者了解这些相关联系,并在治疗过程中予以关注。
在讨论和传播关于脑肠轴与精神疾病相关的知识和科普的基础上,本文主要拓展了对于口-肠轴或口-肠-脑轴在精神健康障碍中的重要性的理解,尤其是探讨了基于口肠微生物群的联合治疗神经精神疾病的解决方案和预防措施等。
口腔是继肠道之后微生物组的第二大定植区。此外,它还是呼吸道和消化道的门户。口腔微生物群的改变可能导致或预示各种口腔和全身疾病。
▼
食物的消化很少发生在口腔中。然而,通过咀嚼或咀嚼的过程,食物才能很好的通过上消化道运输到胃和小肠。咀嚼是食物经历的第一个机械过程。咀嚼时下颌的运动是由咀嚼肌(咬肌、颞肌、翼内肌、翼外肌和颊肌)。包围和支撑牙齿的牙周膜的敏感性,而不是咀嚼肌肉的力量,决定了咬合的力量。
理论上来说,咀嚼对于充分消化来说并不是必需的,但是咀嚼对于吞咽和运输又有很大的影响,从这个意义上说咀嚼确实有助于消化,因为它可以将食物切成小颗粒并将其与唾液腺分泌的唾液混合。唾液润滑和润湿干燥的食物,而咀嚼则使唾液分布在整个食物团中。舌头抵住硬腭和脸颊的运动有助于形成圆形的食物团。
嘴唇和脸颊
嘴唇是围绕嘴的两个肉质褶皱,外部由皮肤组成,内部有粘膜。粘膜含粘液分泌腺,它们与唾液一起确保言语和咀嚼的充分润滑。
脸颊,即嘴的两侧,与嘴唇连续,具有相似的结构。脸颊的皮下组织(皮肤下的组织)有一个明显的脂肪垫;这种垫对于婴儿来说特别大,被称为吸吮垫。在每个脸颊的内表面上,与第二上臼齿相对的地方有一个轻微的隆起,标志着腮腺管的开口,从位于耳朵前面的腮腺唾液腺引出。在这个腺体的后面有四到五个分泌粘液的腺体,其导管在最后一颗臼齿的对面打开。
口腔顶部
口腔顶部呈凹形,由硬质和软质形成上颚。硬腭由两块腭骨的水平部分和上颌骨或上颌的腭部分形成。硬腭覆盖着厚厚的、略显苍白的粘膜,该粘膜与牙龈连续,并通过坚固的纤维组织与上颌骨和腭骨结合。软腭与前面的硬腭是连续的。其后面与覆盖鼻腔底部的粘膜连续。软腭由坚固、薄的纤维片、腭腱膜、舌腭肌和咽腭肌组成。
口腔底
只有当舌头抬起时才能看到口底。中线是一个突出的、升高的粘膜皱襞(舌系带)将每个嘴唇与牙龈结合在一起,其两侧都有一个轻微的褶皱,称为“舌系带”舌下乳头,下颌下唾液腺导管从中开口。从每个舌下乳头向外和向后延伸的是一个脊(舌下皱襞),标志着舌下(舌下)唾液腺的上边缘,该腺体的大部分导管都通向此处。
牙龈
牙龈由粘膜组成,粘膜通过厚纤维组织与颌骨周围的膜连接。牙龈膜上升,在每颗牙齿的牙冠基部(暴露部分)周围形成一个颈圈。牙龈组织富含血管,接收来自牙龈的分支肺泡动脉;这些血管由于与牙槽或牙槽的关系而被称为牙槽,也供应牙齿和上颌和下颌的松质骨,牙齿位于其中。
牙齿
牙齿是口腔中坚硬的白色结构。不同脊椎动物的牙齿通常用于咀嚼,有时是专门化的牙齿。咀嚼对于食肉动物来说并不像对于草食动物那么重要。人类是杂食动物(吃植物和动物组织),其牙齿在功能和结构上属于食肉动物和食草动物的牙齿所达到的极端专业化之间。
每颗牙齿有牙冠和压根。它们具有不同的功能。口腔的不同部位以及不同动物的牙冠和牙根的形状各不相同。下颌一侧的牙齿本质上是另一侧牙齿的镜像。上牙与下牙不同,但又互补。
舌头
舌头是位于口底的肌肉器官,是一个极其灵活的结构,是言语、咀嚼和吞咽等运动功能的重要辅助器官,也是口腔微生物聚集的地方。它与脸颊一起,能够引导和保持食物在上牙和下牙之间,直到咀嚼完成。舌头的运动有助于在口腔内产生负压,从而使婴儿能够吸吮。作为一种外周感觉器官,舌头尤其重要,它含有一组特殊的上皮细胞,称为味蕾,将刺激从口腔传送到中枢神经系统。此外,舌头的腺体会产生一些吞咽所需的唾液。
舌头由大量交织的横纹肌组成,其中散布着脂肪。覆盖舌头的粘膜在不同区域有所不同。舌头通过其外在肌肉附着在下颌、舌骨(下颌和喉部之间的U 形骨)、颅骨、软腭和咽部。它与口腔底部和口腔相连会厌(作为喉盖的一块软骨板)由粘膜褶皱形成。
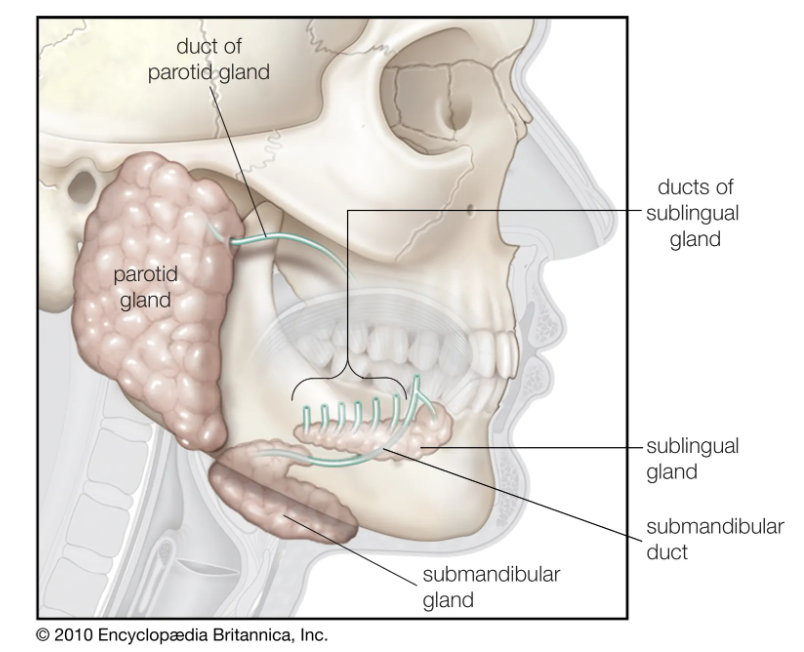
唾液腺
食物被品尝并与几组腺体分泌的唾液混合。除了许多分泌唾液的微小腺体外,还有三对主要的唾液腺:腮腺、颌下腺和舌下腺。唾液腺是最大的一对,位于面部侧面、每只耳朵的下方和前方。腮腺被包裹在鞘中,当发炎时,如腮腺炎,腮腺可以限制肿胀的程度。下颌下腺呈圆形,位于下颌骨内侧附近,位于胸骨乳突肌(下颌突出的肌肉)前面。这舌下腺直接位于覆盖舌头下方口腔底部的粘膜下方。
唾液腺中含有很多分泌细胞,分泌细胞在圆形囊中呈簇状排列,称为腺泡,附着于导管的自由分支系统。分泌细胞可以是浆液型或粘液型。后一种类型分泌粘蛋白,粘蛋白的主要成分粘液; 前者是含有淀粉酶的 水状液体。
腮腺的分泌细胞为浆液型;颌下腺,有浆液性和粘液性两种类型,浆液性细胞与粘液性细胞的数量为四比一。舌下腺的腺泡主要由粘液细胞组成。
唾液腺由自主神经系统的两个部分(交感神经和副交感神经)控制。这副交感神经 供应调节腺泡细胞的分泌并导致血管扩张。受规管的职能交感神经包括腺泡细胞的分泌、血管的收缩以及可能的肌上皮细胞的收缩。
正常情况下,无论口腔中是否有食物,唾液的分泌都是恒定的。24小时内分泌的唾液量通常为1-1.5升。当有东西接触牙龈、舌头或口腔内壁的某些区域时,或者咀嚼时发生时,唾液分泌量增加。刺激物质不一定是食物——嘴里的干沙,甚至在空嘴时移动下巴和舌头都会增加唾液流量。这种对口腔粘膜的直接刺激与唾液分泌增加的结合被称为无条件唾液反射。当一个人了解到特定的视觉、声音、气味或其他刺激通常与食物相关时,仅该刺激就足以刺激唾液流量增加。这种反应被称为条件性唾液反射。
唾液
唾液溶解一些咀嚼的食物并充当润滑剂,促进后续食物顺利通过消化道。唾液还含有一种称为淀粉酶(ptyalin)的淀粉消化酶,它可以启动酶水解过程;它将淀粉(一种含有许多连续链结合的糖分子的多糖)分解成双糖麦芽糖分子。
许多食肉动物,例如狗和猫,其唾液中没有淀粉酶;因此,他们的天然饮食中含有很少的淀粉。物质必须处于溶液中才能刺激味蕾;唾液的成分各不相同,但其主要成分是水、与血浆中常见的无机离子,以及许多有机成分,包括唾液蛋白质、游离氨基酸以及溶菌酶和淀粉酶。尽管唾液呈弱酸性,但其中所含的碳酸氢盐和磷酸盐可充当缓冲剂,并在正常条件下保持唾液的pH 值或氢离子浓度相对恒定。
唾液中碳酸氢盐、氯化物、钾和钠的浓度与其流速直接相关。碳酸氢盐浓度与血液中二氧化碳分压之间也存在直接关系。血液中氯化物的浓度从低流速时的 5 毫摩尔/升到高流速时的 70 毫摩尔/升不等。类似情况下的钠浓度从每升 5 毫摩尔到每升 100 毫摩尔不等。血液中钾的浓度通常高于血浆中的浓度,每升高达 20 毫摩尔,这就是唾液在快速流动时产生尖锐金属味的原因。
唾液的持续流动使口腔和牙齿保持湿润,并且相对清除食物残渣、脱落的上皮细胞和异物颗粒。通过去除可用作细菌培养基的物质,唾液抑制细菌的生长。唾液具有保护功能,因为溶菌酶具有溶解或溶解某些细菌的能力。因此唾液的分泌与口腔菌群构成和变化紧密。
唾液的分泌还提供了一种机制,使某些有机和无机物质可以从体内排出,包括汞、铅、碘化钾、溴化物、吗啡、乙醇以及某些抗生素,如青霉素、链霉素和金霉素。虽然唾液不是生命所必需的,但唾液的缺乏会导致许多不便,包括口腔粘膜干燥、细菌过度生长导致口腔卫生不良、味觉大大减弱以及言语困难。
▼
与牙周炎有关:
如普氏菌属、卟啉单胞菌属、密螺旋体属、聚集杆菌属、梭杆菌属等。
与龋齿有关:
如变形链球菌 、乳酸菌、奈瑟菌属等。
与精神障碍潜在相关的主要口腔细菌种类如下:
doi.org/10.3390/microorganisms9071450
口腔和肠道之间存在密切的解剖和生理联系。口腔和肠道都是消化系统的一部分。由于胃酸的pH值较低,恶劣的化学环境使得许多口腔细菌难以在肠道内定殖。然而,疾病、药物、衰老等各种情况都会促进口腔细菌在肠道中的定植。
▼
研究人员已经提出了细菌从口腔转移到肠道的两种可能途径:
胃肠道途径
人类每天要吞下大约1.5升唾液,这些唾液富含口腔细菌。来自牙周炎患者和健康对照者的唾液微生物可以在小鼠肠道中存活至少 24 小时,暗示口腔菌群影响肠道菌群的重要途径。然而,胃酸和碱性胆汁对口腔微生物群在肠道中的定位构成了很大的瓶颈,关于口腔微生物群是否可以通过肠内途径定植肠道存在激烈争论。最近的一项研究表明,没有证据表明口腔细菌在健康成人的远端肠道中定植。相反,另外的研究得出的结论是,至少三分之一的口腔微生物群可以解决健康成年人的肠道问题,肠癌和类风湿性关节炎患者比健康人有更多的口腔到肠道微生物传播。胃炎、炎症性肠病、结直肠癌等肠道疾病,使易位的口腔细菌在肠道内定植和扩张。
由上所述,唾液含有粘液(由水、脂质和蛋白质如粘蛋白组成),可以保护微生物群免受胃酸影响,从而在胃肠道中存活。口腔管饲与牙周炎相关的唾液可加重小鼠糖尿病模型、结肠炎、精神症状、老年痴呆症和骨质疏松症。据估计,患有严重牙周炎的患者每天会吞咽大约大量的牙龈卟啉单胞菌 (P. gingivalis),如果进入肠道定制会改变肠道菌群 。
然而,由于胃肠道的屏障功能和胃的酸性,摄入的口腔细菌很少到达并定植在健康的肠道中。但是这两道屏障也可能受损,以下三种情况下,口腔菌群可以趁机进入肠道。
方式一:肠道微生物群破坏
我们知道,肠道微生物群作为一个重要的屏障,可以阻止摄入的口腔细菌在肠道中定植,一旦健康肠道微生物群被破坏,则会导致口腔细菌在肠道的定植增加。例如,抗生素万古霉素用于治疗细菌感染,扰乱肠道微生物组成,从而为口腔细菌在肠道中定植和扩张提供便利。
方式二:胃功能不全
由于长期使用质子泵抑制剂而导致胃功能不全的患者,其口腔细菌如嗜血杆菌属、链球菌属、韦荣氏球菌属的肠道定植显著增加。
胃炎和胃手术也可能导致摄入的口腔细菌暴露于胃液的减少,研究表明,患有胃炎或接受过胃手术的人的肠道微生物组成发生了改变,特定口腔微生物组分类群的相对丰度在肠道中显著增加,如链球菌属、韦荣氏球菌属、肠杆菌科等。
方式三:某些菌耐酸
某些口腔细菌,如牙龈卟啉单胞菌,具有耐酸性,因此可以通过胃屏障进入肠道。
血液途径
当一个人患上牙周病时,牙周袋表皮的撕裂、日常的口腔卫生习惯(激烈刷牙/用牙线)、侵入性的牙齿手术等情况,都可能导致口腔细菌传播至全身循环(菌血症)。
此外,日常牙科活动(如用力咀嚼、刷牙)和牙科手术(如洗牙和牙根平整、牙齿矫正、拔牙)造成的口腔机械损伤可能会使口腔细菌扩散到体循环中。此外,牙周炎导致牙周袋血管化和牙龈溃疡,使牙周病原体很容易进入血液。血液途径可能是口腔梭杆菌到达结肠肿瘤的首选途径,而不是肠道途径。
研究表明,牙周炎引发口腔细菌传播至肝脏和脾脏。此外,口腔细菌具有入侵免疫细胞(如巨噬细胞和树突状细胞)并在其内部存活的能力,这就好像口腔细菌能够利用宿主免疫细胞作为特洛伊木马,从口腔传播到肠道粘膜。
以上是口腔菌群进入肠道的几种方式。研究发现,绝大多数口腔细菌可转移到肠道。
免疫细胞迁移路线
一些口腔细菌可以在树突状细胞和巨噬细胞等免疫细胞内存活,表明口腔细菌可能劫持宿主免疫细胞作为“木马”从口腔粘膜传播到肠道粘膜。此外,来自口腔淋巴结引流的免疫细胞可以迁移到其他淋巴组织,包括但不限于肠道。口腔致病菌反应性 T 辅助细胞 17(Th17) 可以迁移到发炎的肠道。在肠道中,口腔来源的 Th17 细胞可被易位的口腔致病菌激活并导致结肠炎的发展。
以上我们了解到口腔-肠道传播是影响肠道微生物组成的重要过程,因此可能通过改变肠道微生物群间接影响中枢神经系统功能,我们在下面展开阐述。
▼
口腔-肠道轴在正反馈回路中具有引发全身炎症的倾向。失调的口腔微生物群不仅直接引发炎症,还可以改变肠道微生物群的组成、功能和微生物代谢产物,从而导致促炎级联反应,进一步加剧口腔炎症。
口腔微生物群也有可能通过代谢产物和细菌素的释放影响肠道微生物群。在自然环境中,细菌产生具有抗菌活性的细菌素和肽,以便与其他细菌争夺营养物质。
唾液微生物组影响肠道微生物群
将重度牙周炎患者的唾液灌胃移植到小鼠体内可改变肠道菌群,肠道菌群的β多样性与对照组有显着差异,卟啉单胞菌和梭杆菌增多,阿克曼菌减少,表明唾液微生物群可以通过肠道途径改变肠道微生物群。
通过灌胃将牙周炎患者的唾液移植到患有结肠炎的小鼠体内可以加速结肠炎,并改变与炎症性肠病相关的微生物群,如Blautia、幽门螺杆菌和瘤胃球菌。
牙龈卟啉单胞菌影响肠道菌群
牙龈卟啉单胞菌是牙周炎最重要的致病菌之一。它也是研究最多的影响肠道菌群的口腔致病菌。研究牙龈卟啉单胞菌对肠道菌群的影响,首先要明确以下问题:
下面将从这三个方面进行讨论。
牙龈卟啉单胞菌定植于小鼠口腔并影响口腔微生物群
一般认为牙龈卟啉单胞菌可以在小鼠口腔内定植。接种P. gingivalis 7 天后,小鼠口腔内可检测到P. gingivalis 。在小鼠牙龈停止外用牙龈卟啉菌4周和8周后口腔内仍可检测到牙龈卟啉单胞菌DNA,提示牙龈卟啉单胞菌可在小鼠牙龈内定植增殖。
在小鼠口腔中接种牙龈卟啉单胞菌会导致可培养的共生细菌总量增加,并改变口腔微生物群的质量组成,增加口腔微生物多样性并允许潜在的机会性物种的定植。
牙龈卟啉单胞菌可以在肠道定植吗?
由于胃液和胆汁的恶劣环境,牙龈卟啉单胞菌能否在肠道定植尚不确定。为了模拟体外胃环境,将牙龈卟啉单胞菌暴露于人工胃液 (AGJ) 中。在 pH 5 下暴露于 AGJ 2 小时后,只有 1% 的牙龈卟啉单胞菌浮游细胞存活,这相当于饭后立即的 pH 值。牙龈卟啉单胞菌生物膜的形成显着提高了存活率。几乎 100% 的细胞在 pH 5 下存活。因为牙龈卟啉单胞菌在口腔内与多种细菌形成复杂的生物膜,所以这个体外实验的结果不能推导到体内。
有的研究人员试图通过使用无菌小鼠来解决这个问题。他们通过在无菌小鼠口中擦拭人类唾液,开发了人类口腔微生物群相关小鼠模型(HOMA),还通过灌胃无菌人类粪便悬浮液开发了人类微生物群相关小鼠模型(HMA)老鼠。然后,他们cohoused HOMA 和HMA 模型。与 HMA 模型相比,同居模型显示小肠中的卟啉单胞菌增加,Turicibacter减少这个结果表明卟啉单胞菌在与肠道微生物群竞争小肠定植方面发挥了关键作用。
牙龈卟啉单胞菌影响肠道菌群
大量研究表明,牙龈卟啉单胞菌可以改变肠道菌群组成, 持续时间范围为P. gingivalis口服灌胃后 2 天一次到 10 周的重复应用P. gingivalis。
大多数研究使用口服管饲,而有些人将P. gingivalis应用于口腔,一项研究使用静脉注射。三项研究报告α多样性没有显著变化,并且有两个报告称 α多样性降低。五项研究表明β多样性存在显著差异,一项研究表明β多样性没有差异。
P. gingivalis 属于拟杆菌门,在门水平上,一些研究显示拟杆菌的比例增加,而有些人则相反。厚壁菌门是肠道微生物群中的另一个主要门,一项研究显示厚壁菌门的丰度增加,以及其他显示厚壁菌门减少的研究。
有趣的是,牙龈卟啉单胞菌在野生型小鼠 (WT) 和链脲佐菌素诱导的小鼠 (STZ) 中诱导了一些肠道微生物群的相反变化。乳酸杆菌的丰度在 WT 小鼠中减少,但在 STZ 小鼠中增加。Turicibacter的丰度在 WT 小鼠中增加,但在 STZ 小鼠中减少。这表明高血糖可能会影响细菌生长并改变小鼠肠道菌群的组成。
具核梭杆菌影响肠道菌群
据报道,具核梭杆菌( F. nucleatum ) 参与牙周病和根尖病变的发展,会影响肠道微生物群。具核梭杆菌感染牙髓诱发大鼠磨牙根尖周炎后, 2周时可在肠道中检测到具核梭菌,并改变肠道菌群,证实大肠感染。
伴放线放线杆菌影响肠道菌群
伴放线放线杆菌Aggregatibacter actinomycetemcomitans (A. actinomycetemcomitans )经常在严重牙周炎中检测到,并与局部侵袭性牙周炎有关。在管理A之后。给小鼠灌胃 6 周伴随放线菌,肠道中的Turicibacter属显着减少。该属与丁酸的生产有关,丁酸盐的减少与胰岛素抵抗有关。
肠道微生物群影响口腔微生物群
由于口腔与肠道在物理上相连,一些研究人员报告说,肠道微生物群的变化也会影响口腔微生物群。发现不同类型的肠道微生物群与不同类型的口腔微生物群相关。经过长期富含脂肪的饮食,小鼠的肠道微生物群概况可分为三种类型:糖尿病抵抗型、中间型和糖尿病敏感型。只有糖尿病敏感小鼠的牙周微生物群显示出丰富的普氏菌属和坦纳氏菌属,它们是主要的牙周病原体,表明肠道微生物群和口腔微生物群存在相互作用。同样,糖尿病导致口腔微生物群变得更具致病性。高血糖发作后,口腔微生物群中肠杆菌科、气球菌、肠球菌和葡萄球菌的水平升高,这些菌群通常与牙周炎有关。
在了解口腔微生物群是如何影响精神障碍类疾病之前,我们先来看一下,口-肠轴是如何影响大脑的。
这部分内容我们分成几个部分来详细阐述:
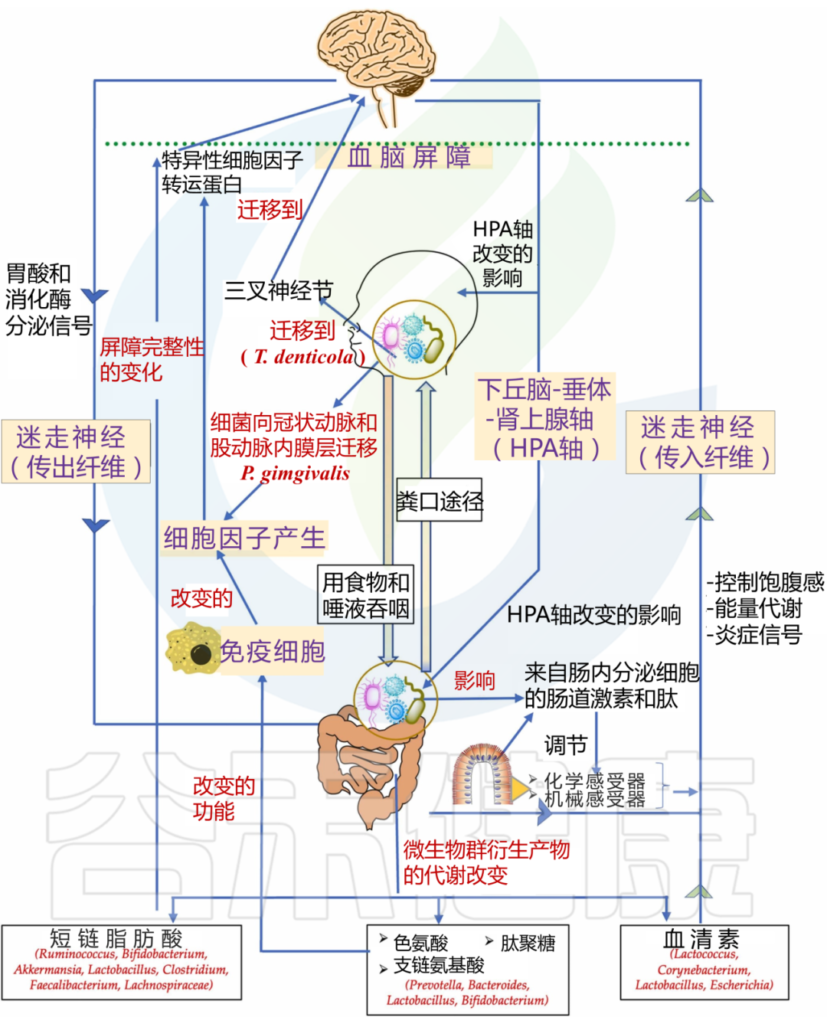
doi.org/10.1016/j.jdsr.2022.11.003
口腔微生物群如何影响大脑?
1
血液途径(细菌或其产物破坏血脑屏障)
口腔微生物群的改变可能导致炎症反应的增加,破坏血脑屏障,使得炎症介质和细菌代谢产物进入大脑,从而影响神经元的功能和存活。
如牙龈卟啉单胞菌通过血流迁移到冠状动脉和股动脉的内膜层。这种迁移可能会在体内产生急性炎症状态,导致炎性细胞因子的产生,这些细胞因子可以通过特定的细胞因子转运蛋白,穿过血脑屏障进入大脑。
2
神经途径
迷走神经和三叉神经复合体是口腔与大脑之间的重要神经连接。口腔微生物群的变化可能通过这些神经连接与中枢神经系统进行交流,影响大脑的功能和健康。
在阿尔茨海默病患者的三叉神经节和海马中发现了一种口腔细菌,即齿状密螺旋体(Treponema denticola)。这一发现在另一项针对小鼠的临床前研究中得到了支持,其中口腔T. denticola感染诱导了海马中淀粉样蛋白-β的产生。目前尚不清楚这些细菌是如何迁移到海马体的,人们认为口腔细菌可能是通过三叉神经途径到达大脑的。
牙周炎会诱发全身炎症,促炎细胞因子可以激活表达 TNF-α 和 IL-1 受体的内皮细胞,进而向紧邻脑内皮细胞的血管周围巨噬细胞发出信号。这些血管周围巨噬细胞随后与小胶质细胞通讯,从而导致小胶质细胞激活和随后的神经炎症。
牙周细菌细胞外囊泡,如外泌体,是免疫系统的有力刺激物,增加炎症负担。外泌体存在于大多数体液中,包括唾液,一项研究发现唾液中 CD9/CD81 外泌体水平降低与牙周病的发病机制有关。
因此,牙周细菌具有多种机制,可以将包括牙周炎在内的周围炎症转化为神经炎症,从而影响中枢神经系统的功能和行为。
牙周血管系统的渗透性增加导致脂多糖“泄漏”。脂多糖可激活下丘脑-垂体-肾上腺轴,从而增加应激激素和/或神经递质。这会影响肠道生理、栖息地、微生物组组成和细菌基因表达。
肠道微生物群的改变可能导致全身炎症,这不仅影响中枢神经系统,而且加剧其他炎症病理。
口腔微生物群影响和促进神经精神疾病结果的机制
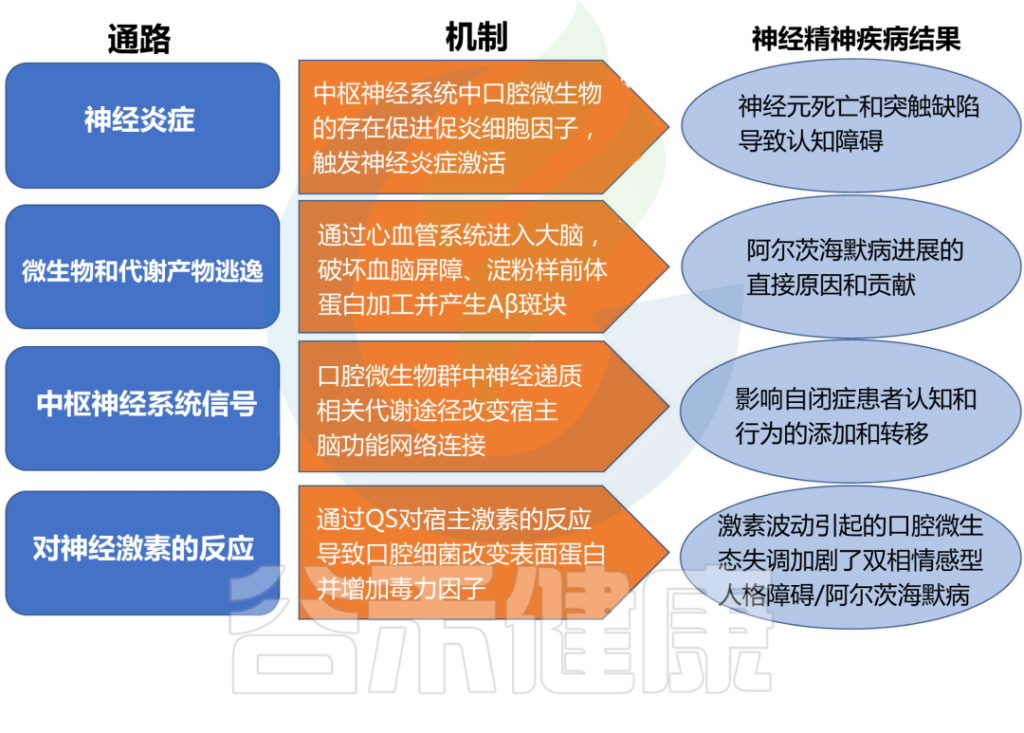
doi.org/10.3389/fpsyt.2022.810008
以上是口腔微生物群影响神经精神疾病的一些机制,口腔微生物群还可以通过肠道间接和大脑产生交流。
肠道微生物群如何影响大脑?
肠道作为“第二大脑”影响情绪和行为,大脑和肠道直接通过迷走神经和脊髓中的自主神经系统进行沟通。细菌通过迷走神经和肠神经系统传入神经元的刺激,在大脑和胃肠道微生物群之间建立直接的神经联系。
肠-脑轴的相互作用对我们的情绪和行为产生影响。肠道微生物的失衡和肠道疾病与焦虑、抑郁等精神疾病的发生和发展密切相关。
微生物群与大脑之间通过肠脑轴的分子通讯途径
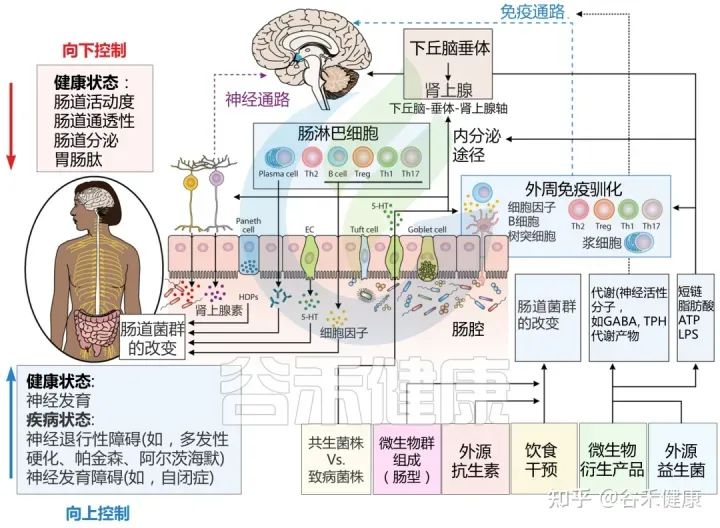
Sorboni SG, et al.,Clin Microbiol Rev. 2022
肠-脑轴的相互作用是一个复杂的系统,涉及到肠道微生物群、肠道黏膜屏障、免疫系统、神经递质和代谢产物等多种机制的相互作用和影响。这些在我们之前的很多文章已经详细阐述,此处就不展开,详见:
口腔和肠道微生物群之间的相互交流如何影响大脑?
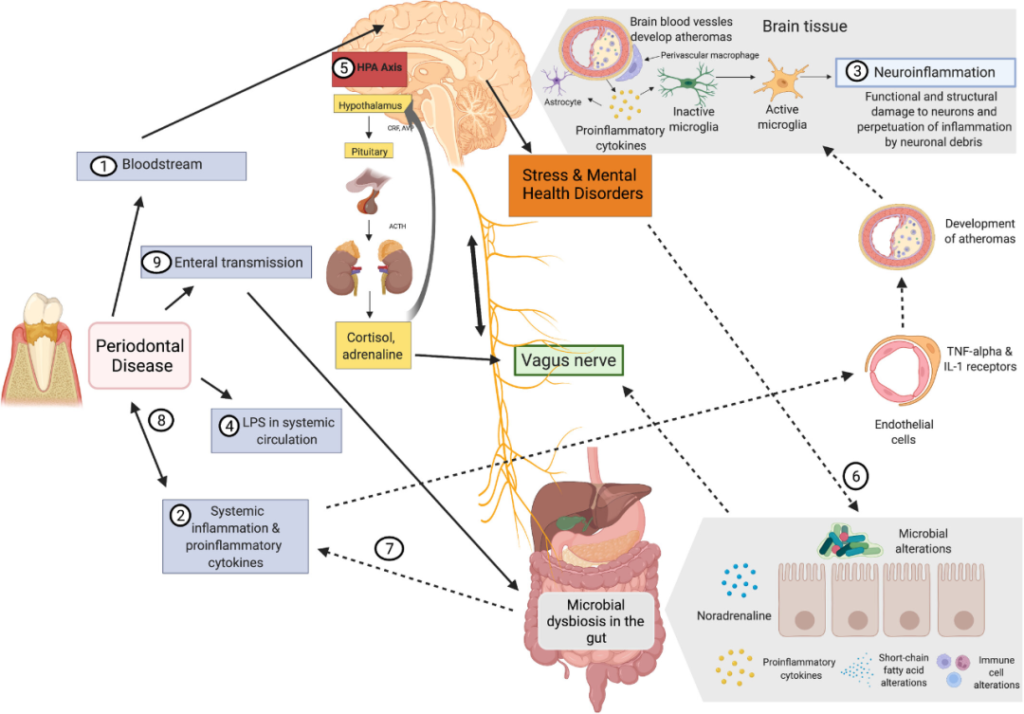
doi.org/10.1111/prd.12452
1) 牙周病原体可以通过血液和受损的血脑屏障直接到达大脑。
2) 促炎细胞因子激活内皮细胞可以间接影响中枢神经系统。
3) TNF-α和IL-1内皮受体的表达激活小胶质细胞,导致炎症。
4) 牙周血管系统通透性的增加导致脂多糖的“渗漏”。
5) 脂多糖可以激活下丘脑-垂体-肾上腺轴,从而增加应激激素和/或神经递质。
6) 这会影响肠道生理、栖息地、微生物组组成和细菌基因表达。
7) 肠道微生物组的变化可能导致进一步的全身炎症,从而增强对中枢神经系统的影响。
8) 此外,它可能通过增加炎症负担来影响牙周病。
9) 口腔细菌通过唾液传播到肠道也可能影响肠道微生物组的组成和功能。
10)激素途径,例如,神经肽Y (NPY)、糖皮质激素、胰高血糖素样肽-1 (GLP-1)、食欲素-A、瘦素,口肠菌群的变化导致这些激素稳态或受体响应,进而影响精神系统。
以上,我们可以看到,口-肠轴对于大脑,也就是心理健康来说非常重要。这也就说明了口腔健康和心理健康之间存在着很大关联。
值得注意的是,口腔-肠道微生物组轴改善了胃肠道系统的发病机理和预后的预测。荟萃分析表明,口腔微生物组的变化与胃肠道癌的风险有关,包括CRC,PDAC和HCC,这可能是早期发现的潜在指标。已经验证了PDAC特定的口腔微生物模式作为PDAC生物标志物。两种口腔细菌物种长奈瑟菌(Neisseria elongata)和 轻型链球菌(Streptococcus mitis),同时富集可以将PDAC患者与健康受试者区分开来。
龋齿、严重牙周病、牙齿脱落是主要口腔疾病。口腔健康是一个全球被低估的健康问题。
口腔健康问题带来的经济负担
2017年,全球每年所有口腔疾病的负担达到了1830万,较1990年增加了19.9%;而全球经济负担为5440亿美元,其中1870亿美元是由生产力损失造成的。
间接成本,如生产力损失,可能归因于口腔健康对社交退缩和孤立、疼痛和咀嚼功能减弱、自尊心、缺乏口腔健康意识,对口腔医生的不信任所产生的影响。
口腔健康带来的健康问题与心理障碍并存
口腔健康对一般健康有影响,有证据表明口腔健康与冠心病、呼吸系统疾病、中风和糖尿病密切相关。这些疾病在患有心理障碍的人群中常见并存。
口腔健康问题和心理问题之间关联
据报道,与普通人群相比,严重的精神障碍导致缺牙的风险高2.8倍。口腔健康不佳的预测因素和决定因素涉及心理障碍和生活方式因素,说明了心理和口腔健康中影响因素的复杂相互作用。
口腔健康状况不佳的预测因素
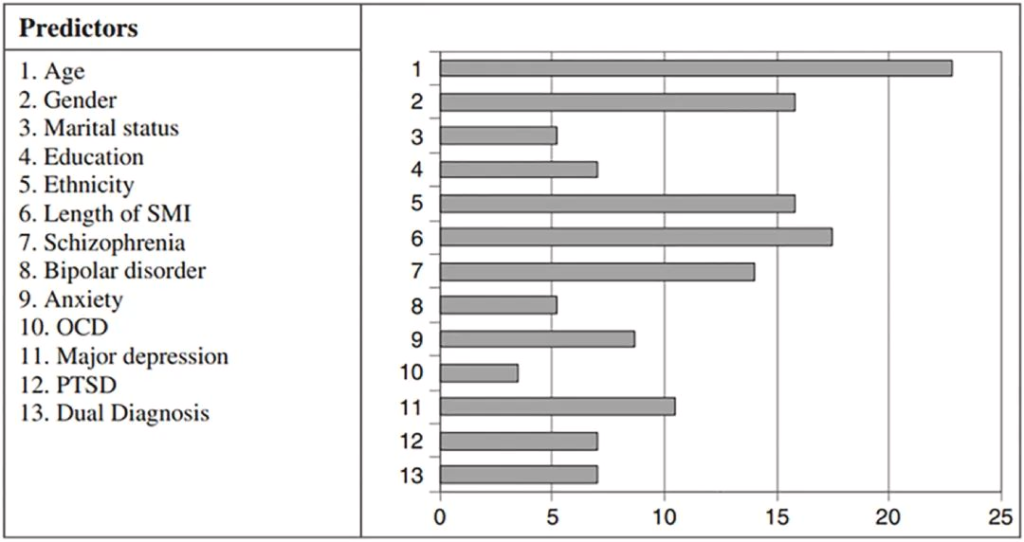
doi.org/10.1016/j.jobcr.2023.06.003
口腔健康状况不佳的决定因素
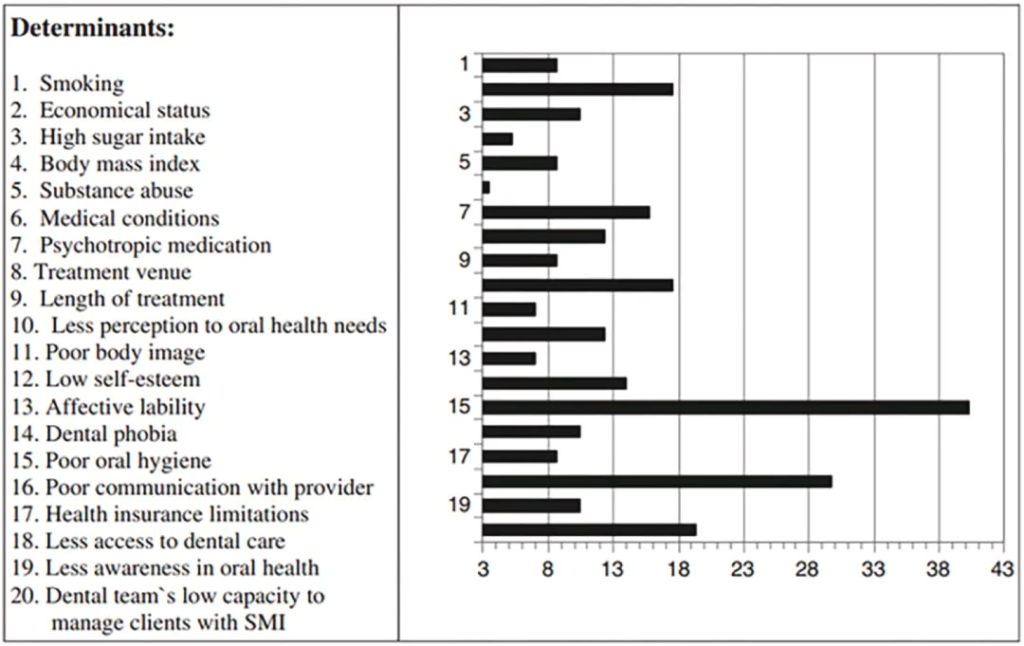
doi.org/10.1016/j.jobcr.2023.06.003
诊断口腔疾病时发现异常,应考虑是否存在精神障碍
常见的情况,如牙齿表面物质的损失,可能归因于许多基于其表现的精神障碍。
以上我们了解了口腔健康与心理健康之间的关联,接下来进一步深入探讨精神障碍与牙科疾病之间的关系。研究表明,精神障碍患者更容易出现牙科问题,而牙科疾病也可能与精神健康问题相关联。
▼
精神障碍涉及一个人的行为或心理模式;基于 DSM-5 中的标准。
最常见的疾病是抑郁症和焦虑症,影响世界人口的约 3.8%。最近,全球青少年抑郁和焦虑的患病率估计为 25-31%。心理健康状况不佳是全球日益严重的负担。这不仅仅涉及药物和住院等直接成本,还包括因失业或迅速退休而导致生产损失的收入损失。
常见的精神障碍包括抑郁症、焦虑症、躁郁症、精神分裂症、痴呆症、酒精和药物滥用障碍等。
常见的心理健康障碍及其常见症状

doi.org/10.1016/j.jobcr.2023.06.003
▼
牙周炎常见且危害较多
牙周炎是一种慢性疾病,影响牙齿结构周围的组织,伴有炎症障碍、退化,最终导致牙齿脱落。
除了牙齿脱落和随后的咀嚼功能障碍,牙周炎还影响整体健康。在全球范围内,牙周炎是最常见的疾病之一,有20-50%的人口受到影响。随着人口老龄化和越来越多的老年人选择保留天然牙齿,牙周炎的患病率预计将增加。
牙周炎是一种慢性炎症性疾病
牙周炎会使宿主全身长期暴露于促炎细胞因子和急性期蛋白中,中度至重度牙周炎患者的全身C反应蛋白水平升高。评估牙周炎全身炎症影响的研究表明,前列腺素E2、IL-1β、IL-6和TNF-α水平升高,牙周炎产生的炎症介质可以通过系统和神经途径延伸到大脑。牙周袋为大量牙周细菌进入系统循环和神经组织提供了独特的机会。
精神障碍及其与口腔疾病,特别是牙周炎的关系,在研究界越来越受到关注。其中一些关系被认为是双向的,这为未来的治疗、诊断和预防措施开辟了道路。
阿尔茨海默病
牙周炎和阿尔茨海默病之间存在显著关联
许多研究支持这种联系,并提出了几种解释,细菌移位导致全身炎症的机制似乎是合理的。
牙龈卟啉单胞菌(牙周炎的主要病原体之一)的DNA以及针对几种牙周炎相关细菌的抗体支持了这一点。
口腔微生物组通过血管、炎症/免疫、神经毒性和葡萄糖代谢途径,在已建立的生活方式因素和阿尔茨海默病风险之间是一个合理的因果中介:
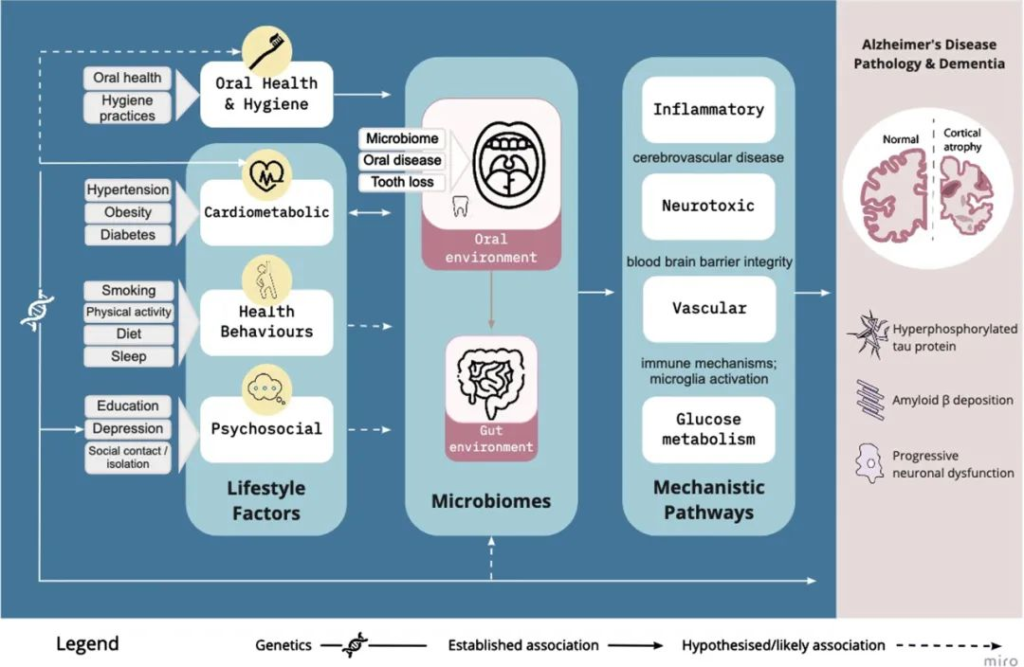
Loughman A, et al.,J Alzheimers Dis. 2023
牙周炎的严重程度与阿尔茨海默病之间存在关联
对这一证据的进一步支持归功于动物研究,其中小鼠受试者被给予活的牙龈卟啉单胞菌或其内毒素脂多糖。给药导致动物海马中学习和记忆功能明显下降,淀粉样蛋白-β斑块(一种与阿尔茨海默病相关的典型组织学发现)减少。
这些发现激发了抑制牙龈卟啉单胞菌蛋白酶的银杏蛋白酶抑制剂的开发。抑制剂导致斑块形成、细菌体积减少,并对海马细胞产生保护作用。
注:一个例子是用于治疗阿尔茨海默病的银杏蛋白酶抑制剂COR388,目前正处于2/3期临床试验中。
总之,文献表明,牙周炎是痴呆症的一个可改变的风险因素(特别是阿尔茨海默病),因此可以作为治疗和预防措施的目标。
抑郁症
细菌可能在牙周炎和抑郁症之间的关系中发挥作用。
抑郁症患者的口腔微生物组存在显著差异
一项16s高通量测序研究比较了符合 DSM-IV 抑郁症标准的年轻人 (n = 40) 和匹配对照 (n = 43) 的唾液微生物组的结构和组成,健康受试者和抑郁受试者之间的分类群在丰度上不同,奈瑟菌属(Neisseria)和变黑普雷沃氏菌(Prevotella nigrescens)的水平升高。
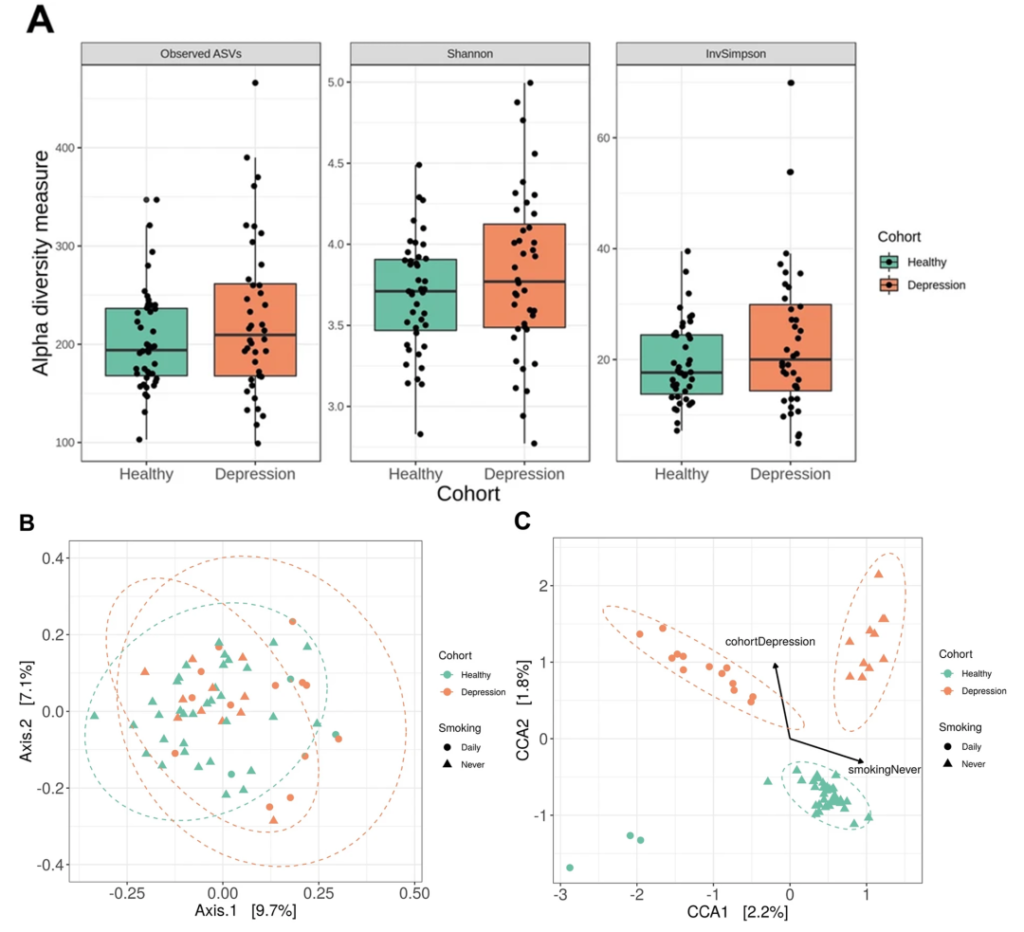
Wingfield B, et al.,Sci Rep. 2021
动物研究支持这样一种观点,即:
口服脂多糖或活的牙周炎相关病原体会导致包括大脑在内的系统炎症标志物升高和抑郁样行为。
有人提出了一种遗传关系。一项研究调查了串扰基因和神经肽在这两种疾病中的作用。
神经肽肾上腺髓质素、胰岛素样生长因子2、强啡肽原和抵抗素在牙周炎和抑郁症中相互表达,也在识别抑郁症中发挥作用。
牙周炎和抑郁症共病
抑郁症受到心理和社会因素的影响。牙周炎症状,如口臭、口腔卫生不良、缺牙、牙龈退缩,可能会出现社会孤立、羞耻、自尊下降等,产生负面影响,从而导致抑郁。
种植体周围炎也可能是抑郁症的危险因素
当牙齿丢失时,可能会被种植牙替代,然而,种植牙也可能发展为种植体周围炎,类似于牙周炎。最终,植入物可能会丢失。从牙周炎到种植体周围炎,可以推断出几种影响和疾病关系。牙周炎和种植体周围炎可能是抑郁症的可改变的危险因素,如果是这样,简单的牙周干预和口腔卫生指导可以预防或帮助治疗抑郁症。
双相情感障碍(躁郁症)
慢性炎症是双相情感障碍的一个因素。
2001年-2012年对双相情感障碍与牙周炎之间的关系进行了研究:
这些研究支持了双相情感障碍和牙周炎之间的可能关系,值得进一步研究。
帕金森病
一些研究报告了帕金森氏症患者牙周炎患病率的升高。
帕金森病会导致运动障碍和认知障碍,这是由于大脑黑质中产生多巴胺的神经元的神经元细胞死亡所致。
手抖和僵硬是常见的症状,这就很难保持足够的日常口腔卫生。这种疾病本身可以说是牙周炎的一个危险因素,然而,流行病学证据支持牙周炎会增加患帕金森病的风险。
与完全没有接受治疗或连续5年没有接受治疗的患者相比,在连续5年接受牙周治疗的患者中观察到了对帕金森病的保护作用。作者推测:
牙周炎相关病原体进入大脑,引发和维持的炎症最终会导致帕金森症。
进一步研究其机制关系,以及牙周治疗对已确定的帕金森氏症的影响,可能是未来的方向。
精神分裂症
关于牙周炎和精神分裂症之间关系的支持性文献很少。
一些研究报告称,精神分裂症患者患牙周炎的风险较高,服用抗精神病药物的患者患牙周炎风险更高。
血管紧张素转换酶基因的D等位基因是对抗精神分裂症和牙周炎的保护因子,并可能被证明是一种生物学联系。
口咽微生物组、唾液微生物组和牙周炎的作用被认为可能与精神分裂症有关,牙周炎强化了炎症在精神分裂症病理生理学中的作用。
口腔微生物群与精神分裂症之间的联系
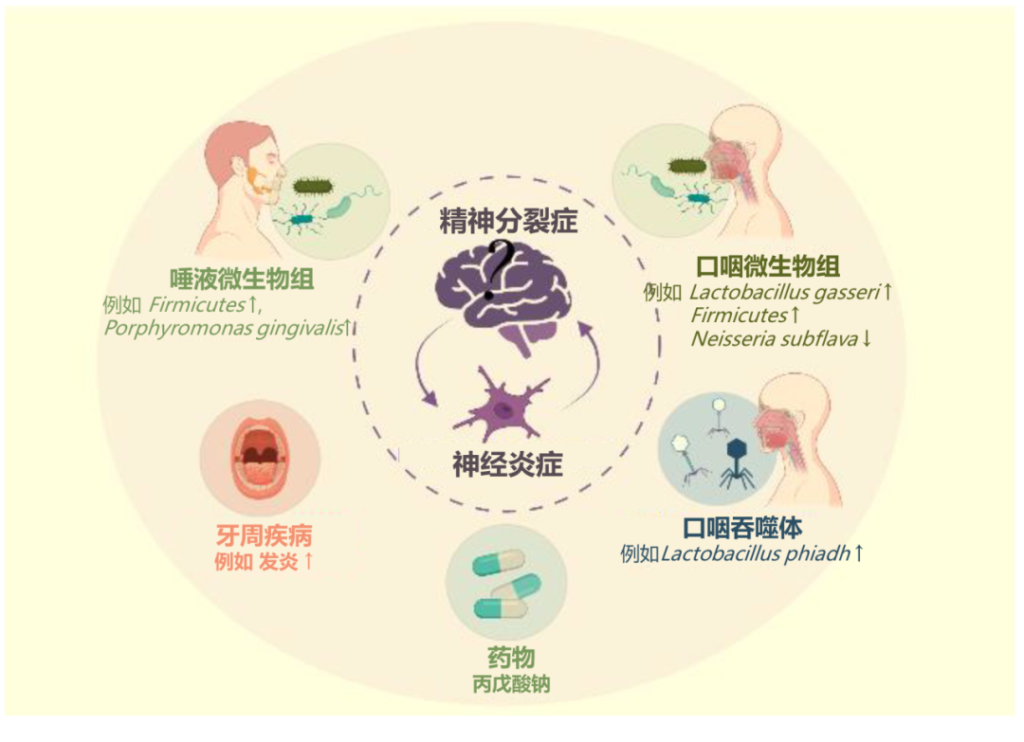
doi.org/10.3390/ijms23020846
唾液是一种对多种疾病具有诊断意义的体液,也可能用于精神分裂症的诊断,目前的证据仍然有限,还需更多研究。
精神分裂症患者唾液牙龈卟啉单胞菌较多
一项横截面研究发现,精神分裂症患者唾液中牙龈卟啉单胞菌的数量显著更高。此外,牙龈卟啉单胞菌细胞的数量与精神分裂症的精神病理学严重程度呈正相关。牙龈卟啉单胞菌可能导致神经炎症状态。
患有精神病发作的精神分裂症患者血清中炎症细胞因子的浓度升高,包括IL-12、干扰素 γ、肿瘤坏死因子α 和 C反应蛋白。低度慢性炎症状态可能会导致免疫系统异常,从而使精神分裂症患者易患全身性疾病。
精神分裂症患者口腔菌群在肠道定植
口腔常驻细菌口腔乳杆菌(Lactobacillus oris)、唾液链球菌(Streptococcus salivarius)可能在精神分裂症患者的肠道中定殖,导致口腔常驻细菌显著富集。
也有宏基因组研究发现,与精神分裂症相关的肠道细菌之间的共现相关性,大多数物种起源于口腔,口腔常驻细菌可能以协同的方式在精神分裂症患者的肠道定植。
与口腔疾病相关的精神障碍的管理
精神健康障碍患者应接受全面的口腔和牙周健康信息、卫生指导、教育和定期随访,以提高患者的意识、习惯。
多学科干预可以进一步改善依从性、牙齿恐惧、口腔健康和习惯,并有助于更积极的预后。
牙科医生应接受有关精神障碍的教育,以便更好地管理、沟通和识别这些患者,并与其他卫生专业人员合作。牙科应与现有的精神康复和预防计划相结合,实现一个全面、方便的多学科计划,将人体内部的整体关联考虑在内。
“胃肠精神病学”涉及两大系统,涉及的病种、症状均较多,而且各个病种间可能交互出现,因此症状也可能隐匿、多变,这给诊断和治疗带来巨大的困难,因此需要包括消化系统内外科、精神心理、营养及影像科的多学科讨论。
医生应该了解当前的研究状况,在治疗过程中应该详细了解患者的饮食习惯、生活习惯和肠道健康状况对患者的发病原因进行综合评价,并就基于肠道菌群的干预措施向患者提准确而明智的建议,将改善饮食、生活方式等改善肠道健康的方式纳入患者的治疗方案之中。以下给大家一些在选用常见的菌群改善或营养饮食方面的简单建议。
肠道菌群作为治疗精神疾病的潜在靶点,一直是近年来精神病学研究的热点。肠道细菌通过影响神经、免疫和内分泌,在肠道和大脑的交流中发挥关键作用。
微生物-肠-脑轴为精神病学的研究和治疗提供了一种新的范式。一项大规模的人群研究证实,许多精神疾病患者,特别是精神分裂症、双相情感障碍和重度抑郁症等重度精神疾病患者,比一般人群摄入的致肥胖营养物质和炎症性饮食更高。自闭症患儿也大多存在严重的偏食和挑食等不良饮食习惯。
虽然关于肠道菌群在精神类疾病发生中的具体作用还有待发现,但是营养和肠道健康领域已经成为精神疾病整体治疗中的一个重要组成部分。如在自闭症儿童中,存在严重的食物过敏现象,限制麸质饮食和酪蛋白饮食有助于自闭症症状的改善;诸多精神类疾病也存在偏食症状,如偏食促发自闭症的发生发展。IBS患者可能有与饮食成分消化不良相关的腹胀,如可发酵的低聚糖、双糖或单糖和多元醇饮食有关。因此,建议精神障碍人群的精准个性化的营养饮食。
益生菌无论是在精神性疾病或者胃肠功能性疾病都表现良好的治疗效果,而且存在双向调节作用。近年来,关于肠-脑轴的研究则提供了更加充分的理论支持。因此,肠-脑-菌群轴被认为是能为精神病患者提供创新疗法的基础,其重点是在临床层面上系统性鉴别出精神益生菌(psychobiotics)。在临床前研究中发现,精神益生菌对行为、肠道通透性、神经活性有益处和减少促炎性及应激反应。
在啮齿动物中的实验表明,精神病益生菌可通过迷走神经、脊髓、神经内分泌系统起到抗抑郁及抗焦虑的作用。精神病益生菌在IBS患者中被广泛研究,他的作用包括缓解抑郁症状及慢性疲劳综合征等,可能与益生菌的抗炎及减少下丘脑-垂体-肾上腺轴活性相关。
虽然领域还处于起步阶段,存在着许多挑战,但是鉴于目前已有的初步证据,在精神类疾病的治疗中,补充益生菌或加入一些营养配方是合理且具有极大潜力的。
临床报道益生元对于慢性便秘、腹泻、IBS及焦虑、抑郁、自闭症等具有良好的疗效。益生元是被宿主微生物选择性利用以促进健康的物质,发挥抵抗病原体、调节免疫、增加矿物质吸收、改善肠道功能、影响代谢和饱腹感等作用。
益生元与益生菌还存在协同和互养的作用,如果聚糖、低聚半乳糖、抗性淀粉、维生素、植物多酚、海藻等益生元可通过不同机制维持益生菌在肠道中的活性,包括增强益生菌对氧气及活性氧的抗性、增强其对胃酸和胆汁酸的抗性。益生元可通过多种机制增强益生菌在肠道菌群中的益生功能,包括发酵产生的短链脂肪酸促进益生菌的定殖、与致病菌竞争性结合宿主上皮细胞上的受体。
无论是基础研究还是临床研究均表明,肠道菌群移植在治疗胃肠功能的同时,对肠道外疾病如精神神经系统包括焦虑和抑郁症、自闭症、帕金森病、阿尔兹海默症等均有良好的疗效。
上海市第十人民医院在《Lancet Gastroenterol Hepatol 》杂志上发表成果显示,8547例菌群移植治疗肠道功能合并自闭症、焦虑和抑郁等疾病的成果,均表现出良好的临床疗效和安全性。此外,在对IBS的5年长期随访中发现,菌群移植在改善胃肠功能障碍的同时,对精神心理也有很好的疗效。
菌群移植通过纠正肠道菌群的失衡,增加菌群的多样性,促进有益菌的定殖等,在改善胃肠道功能的同时,肠道内环境的改善,肠道菌群及其代谢产物可以通过自主神经系统、免疫系统调节中枢的活性,从而改善精神行为的异常症状。近年来,人源化的无菌动物模型的构建,更进一步证明肠道菌群移植对精神神经系统的干预作用,即将自闭症、焦虑或者抑郁症患者的粪便移植至无菌小鼠内,无菌小鼠产生临床类似的精神症状,而通过健康小鼠共喂养或者将健康人群的粪便移植至模型小鼠,其症状可以得到明显的改善。
无锡第二人民医院柳老师团队前期的研究结果显示,对菌群移植可以显著改善睡眠障碍合并抑郁症的患者,菌群移植治疗后,睡眠和抑郁症同时得以明显的改善。因此,菌群移植在功能性胃肠病合并精神症状的机体中,具有双向调节作用,为该类患者提供了整体治疗思路。
对于复杂胃肠疾病,心理医师的介入是非常关键的,需要心理医师,参与筛查、评估、诊断、治疗和随访。
同时患者家庭,亲属和朋友对“胃肠精神共病”需要有充分的认知和参与,认知越高参与越积极,治疗效果越好。如父母因素在儿童慢性便秘的病理生理和预后中有重要作用,神经质和存在抑郁症状的父母及父母的养育态度和培养行为与便秘严重程度相关;在这种情况下,基于家庭的认知干预也许有好处。
▼
抗菌药物是我们对抗病原体感染的第一道防线,致力于消除特定物种(即噬菌体治疗),或整个微生物群(即抗生素治疗)。
去除与神经精神疾病相关的微生物,可能是未来解决神经精神疾病症状或严重程度的新方法。口服抗生素已被证明对口腔微生物群影响很小,在口腔中局部或直接施用抗生素可能更有效。
▼
一项关于饮食与牙周炎之间关系的10000名NHANES参与者的横断面研究发现,富含水果、蔬菜、沙拉、水、茶的饮食模式,一定程度限制摄入可发酵碳水化合物、脂肪酸、蛋白质和高糖饮料的摄入,患牙周病的程度较低。
▼
益生菌作用的主要益生菌机制包括:
体外和体内证实了乳酸杆菌和双歧杆菌对调节与精神障碍相关的口腔微生物群(动物双歧杆菌、副干酪乳杆菌、嗜酸乳杆菌、鼠李糖乳杆菌、德氏乳杆菌)的作用。细菌竞争排除了一些病原体而不破坏生物膜结构(具核梭杆菌、牙龈卟啉单胞菌等)。
乳酸杆菌和双歧杆菌属可以帮助控制口腔中致龋链球菌的生长。
▼
使用各种益生元化合物(硝酸盐、β-甲基-d-半乳糖苷、N-乙酰基-d-甘露糖胺等)对口腔微生物组进行营养刺激,可能会诱导牙齿生物膜的组成和有益口腔细菌的生长,减少致病菌(P.gingivalis、A. actinomycetemcomitans、F.nucleanum)。
虽然益生菌的使用可能作为精神障碍患者的补充治疗手段,但有必要注意口腔微生物组稳态的多因素特征。
有研究发现,海藻提取物、n-3 PUFA、海参提取物和海洋细菌代谢物等海洋生物活性成分具有抑制口腔致病菌、消除炎症和抗肿瘤的作用。这一发现为通过使用这种生物活性成分(例如以口香糖或无糖片剂的形式包装)来预防和稳定精神障碍开辟了有趣的研究前景。
▼
来自健康捐赠者的微生物群被移植或播种到接受者体内。口腔微生物移植(OMT)可能能够作为预防龋齿的第一道防线或用于治疗牙周病,但这尚未在人体中进行过测试。未来应探索 OMT 在缓解神经精神疾病等全身性疾病症状方面的应用。
▼
牙膏和漱口水等口腔卫生产品通过限制某些物种的生长来管理口腔微生物群落,并且可能是促进所需口腔微生物定殖的一种方法。
例如,当前的牙膏采用具有抗菌特性的化学物质(例如氟化物)配制,除了促进牙釉质健康之外,氟化物已被证明可以降低总体微生物负荷和多样性。许多漱口水含有酒精来杀死微生物。
虽然这些是日常卫生习惯中用于预防和减少口腔疾病的工具,但它们也可能是帮助调节与神经精神疾病相关的微生物的工具。新的研究正在检查其他化合物,它们可以在不破坏微生物群共生平衡的情况下保持口腔卫生。
▼
在研究口腔疾病的预防措施和干预措施时,需要考虑的一个重要组成部分是微生物群产生的生物膜。
生物膜是口腔微生物在牙齿坚硬表面形成的细胞外基质。这些生物膜附着在牙齿表面,将微生物群包裹在分泌聚合物的保护层中,使微生物能够抵抗环境变化。
微生物群还能够通过基因表达模式改变生物膜表型以响应变化。正因为如此,口腔微生物生物膜能够抵抗去除和抗生素或用于抗菌治疗。因此,生物膜可能在移植成功或抗菌产品功效中发挥重要作用,并可能在神经精神疾病的发展和治疗反应中发挥潜在作用。
▼
口腔微生物群可以通过改变生活方式或饮食来调节,或者可能通过改变环境暴露来调节。
通过饮食和水源将环境微生物引入口腔是优先的,尽管这方面的研究有限。环境暴露可能在我们如何考虑调节口腔微生物群以解决系统健康问题方面发挥作用。
例如,城市规划通过引入自然“绿地”来增加土壤微生物的多样性,可能有助于增加对有益微生物的接触,因为接触这些环境微生物可能在神经精神疾病的治疗中发挥关键作用。
具体而言,暴露于土壤细菌分枝杆菌已被证明对宿主具有抗焦虑作用,因为宿主的免疫反应释放抗炎细胞因子,对减少身体和大脑中的炎症具有积极效果,这在焦虑和抑郁中是一个重要因素。
然而,通过口腔微生物群促进的机制尚未确定。尽管如此,实施提供健康和环境暴露的社会政策(例如,要求儿童在上学期间每天安排一点时间接触这些空间),可以进一步确保人们能够受益于环境微生物多样的地方。
口腔健康与精神障碍之间的相互作用可以从微生物群-口肠-脑轴的几个角度进行解释,包括微生物群失调、细菌迁移和神经炎症等。目前对于微生物组在口腔-肠道-脑轴中的作用已经有了一定的认识,这是一个重要的研究方向,从病理生理学到调节肠道微生物组对精神疾病的临床影响。进一步了解微生物组与精神疾病的发展和预后之间的紧密联系,还需要探究地理、种族、饮食模式、过往医疗状况、口腔护理以及胃肠道手术等因素如何改变微生物组的情况。
口肠微生物组还可被视为精神疾病潜在治疗的目标之一。例如,通过改变饮食习惯、使用益生菌或抗生素等手段来调节口肠微生物组,可能对精神疾病的治疗产生积极的影响。谷禾正在整合口腔和肠道微生物组数据,在提高检出率等方面获得了更全面的视角。
总的来说,人体微生物群检测有望为精神疾病的早期辅助诊断、辅助治疗和预防提供新的方法和策略。在一些细分疾病领域中仍需要进一步的研究和临床实践来推动其在临床上的应用。
相关阅读:
环境污染物通过肠脑轴影响心理健康,精神益生菌或将发挥重要作用
主要参考文献:
Skallevold HE, Rokaya N, Wongsirichat N, Rokaya D. Importance of oral health in mental health disorders: An updated review. J Oral Biol Craniofac Res. 2023 Sep-Oct;13(5):544-552. doi: 10.1016/j.jobcr.2023.06.003. Epub 2023 Jun 19. PMID: 37396968; PMCID: PMC10314291.
Wingfield B, Lapsley C, McDowell A, Miliotis G, McLafferty M, O’Neill SM, Coleman S, McGinnity TM, Bjourson AJ, Murray EK. Variations in the oral microbiome are associated with depression in young adults. Sci Rep. 2021 Jul 22;11(1):15009. doi: 10.1038/s41598-021-94498-6. PMID: 34294835; PMCID: PMC8298414.
Loughman A, Adler CJ, Macpherson H. Unlocking Modifiable Risk Factors for Alzheimer’s Disease: Does the Oral Microbiome Hold Some of the Keys? J Alzheimers Dis. 2023;92(4):1111-1129. doi: 10.3233/JAD-220760. PMID: 36872775; PMCID: PMC10200234.
Ball J, Darby I. Mental health and periodontal and peri-implant diseases. Periodontol 2000. 2022 Oct;90(1):106-124. doi: 10.1111/prd.12452. Epub 2022 Aug 1. PMID: 35913583; PMCID: PMC9804456.
Maitre Y, Mahalli R, Micheneau P, Delpierre A, Guerin M, Amador G, Denis F. Pre and Probiotics Involved in the Modulation of Oral Bacterial Species: New Therapeutic Leads in Mental Disorders? Microorganisms. 2021 Jul 6;9(7):1450. doi: 10.3390/microorganisms9071450. PMID: 34361886; PMCID: PMC8306040.
Maitre Y, Micheneau P, Delpierre A, Mahalli R, Guerin M, Amador G, Denis F. Did the Brain and Oral Microbiota Talk to Each Other? A Review of the Literature. J Clin Med. 2020 Nov 28;9(12):3876. doi: 10.3390/jcm9123876. PMID: 33260581; PMCID: PMC7760025.
Scassellati C, Marizzoni M, Cattane N, Lopizzo N, Mombelli E, Riva MA, Cattaneo A. The Complex Molecular Picture of Gut and Oral Microbiota-Brain-Depression System: What We Know and What We Need to Know. Front Psychiatry. 2021 Nov 2;12:722335. doi: 10.3389/fpsyt.2021.722335. PMID: 34819883; PMCID: PMC8607517.
Bowland GB, Weyrich LS. The Oral-Microbiome-Brain Axis and Neuropsychiatric Disorders: An Anthropological Perspective. Front Psychiatry. 2022 Mar 30;13:810008. doi: 10.3389/fpsyt.2022.810008. PMID: 35432038; PMCID: PMC9005879.
Martínez M, Postolache TT, García-Bueno B, Leza JC, Figuero E, Lowry CA, Malan-Müller S. The Role of the Oral Microbiota Related to Periodontal Diseases in Anxiety, Mood and Trauma- and Stress-Related Disorders. Front Psychiatry. 2022 Jan 27;12:814177. doi: 10.3389/fpsyt.2021.814177. PMID: 35153869; PMCID: PMC8833739.
Paudel D, Uehara O, Giri S, Yoshida K, Morikawa T, Kitagawa T, Matsuoka H, Miura H, Toyofuku A, Kuramitsu Y, Ohta T, Kobayashi M, Abiko Y. Effect of psychological stress on the oral-gut microbiota and the potential oral-gut-brain axis. Jpn Dent Sci Rev. 2022 Nov;58:365-375. doi: 10.1016/j.jdsr.2022.11.003. Epub 2022 Nov 17. PMID: 36425317; PMCID: PMC9678961.
Tan HE. The microbiota-gut-brain axis in stress and depression. Front Neurosci. 2023 Apr 14;17:1151478. doi: 10.3389/fnins.2023.1151478. PMID: 37123352; PMCID: PMC10140437.
Martin S, Foulon A, El Hage W, Dufour-Rainfray D, Denis F. Is There a Link between Oropharyngeal Microbiome and Schizophrenia? A Narrative Review. Int J Mol Sci. 2022 Jan 13;23(2):846. doi: 10.3390/ijms23020846. PMID: 35055031; PMCID: PMC8775665.
谷禾健康
运动可以说是最有效和可行的生活方式因素,个人可以利用它来保护自己免受各种疾病的侵害,包括代谢性、心血管、神经退行性和肿瘤性疾病。
世界卫生组织建议,每周进行150-300分钟的中等强度运动。
运动的好处具体不用多说了,我们大家都知道,但你有没有想过,为什么只有一小部分人能坚持呢?

让我们挪不开腿的除了以上理由之外,有没有可能存在什么生理瓶颈,阻止了我们进行定期的体育活动呢?
由Lenka Dohnalová领导的宾夕法尼亚大学的研究团队,开始使用一大群基因多样性的野生小鼠来回答这个问题。
结果发现,肠道微生物组占据了很大的因素,也就是说,“不想运动”这个想法很可能不是因为懒,而是与肠道微生物群相关。
肠道微生物群是如何影响到我们的神经系统,促进运动的进行呢?
本文我们来详细了解一下。
研究人员观察到,个体间运动能力的显著差异,表现最好的动物比最弱的动物快10倍以上。
为了探索这些差异的原因,研究人员对这些小鼠进行了深入的分子和生理学分析。神奇的是,研究人员发现:
我们来看一下研究人员的探索过程。
– 研究人员首先关注基因组,发现遗传对运动能力个体间变异的贡献很小。
– 转向非遗传参数并评估它们对运动表现的影响。动物队列中的血清代谢组、肠道微生物组组成和代谢参数差异很大。
– 使用机器学习方法来识别对运动表现有很强预测作用的变量。
– 发现 仅基于肠道微生物群 16S rDNA 测序结果的预测所达到的准确度与血清代谢组或所有测量参数组合所实现的准确度几乎一样高。
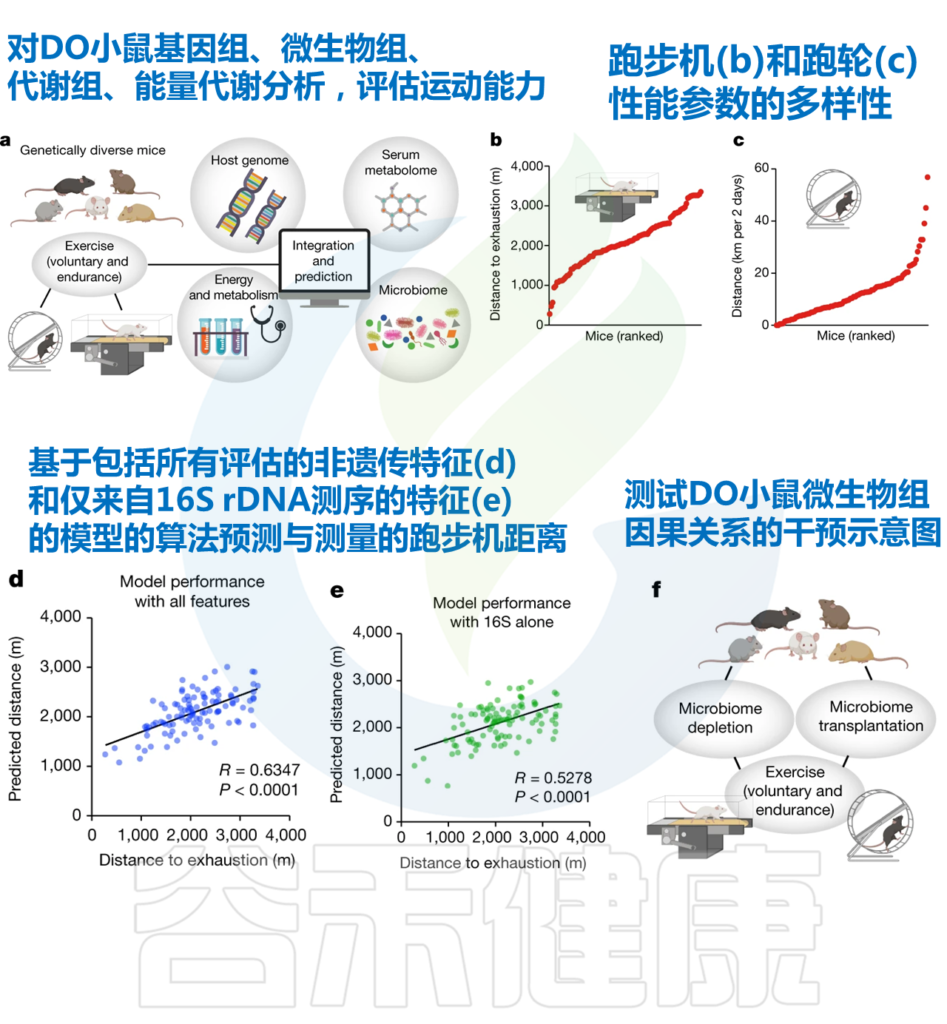
Dohnalová L, et al., Nature. 2022
研究人员探究了肠道菌群在运动表现中的功能重要性,并在无菌或接受抗生素治疗的小鼠中进行了运动测量实验(即在确定的微生物群落环境下)。
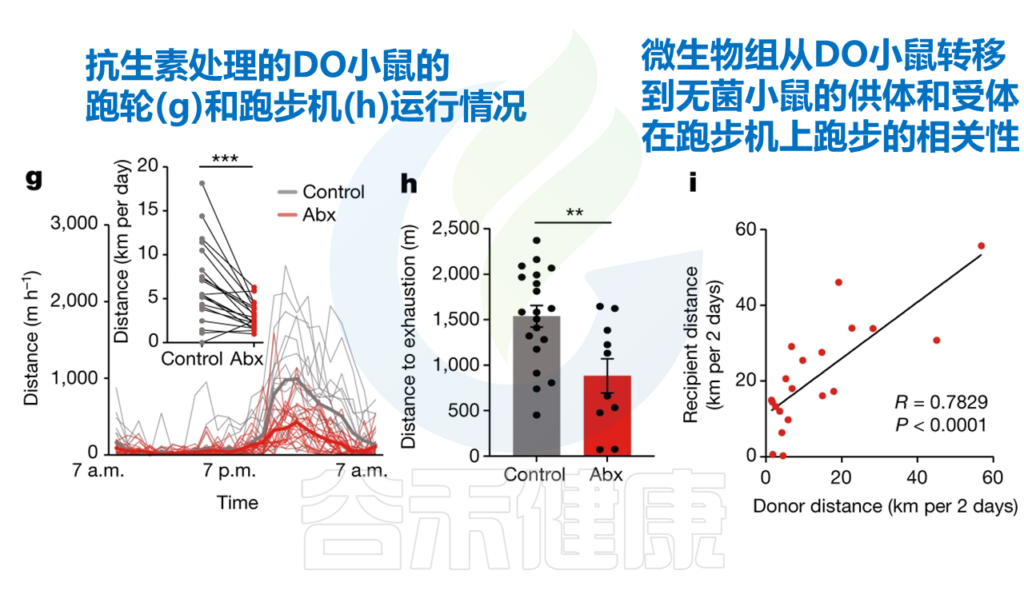
Dohnalová L, et al., Nature. 2022
发现在无菌或接受抗生素治疗的小鼠中(也就是缺乏微生物群的小鼠),自愿运动和耐力运动能力降低了50%左右。
与特定菌群相关——丹毒丝菌科和毛螺菌科
研究人员使用 SHapley Additive exPlanation (SHAP)评估了预测模型中各个微生物组特征的贡献。
所有微生物群特征的SHAP值排序有助于预测DO小鼠的运动表现
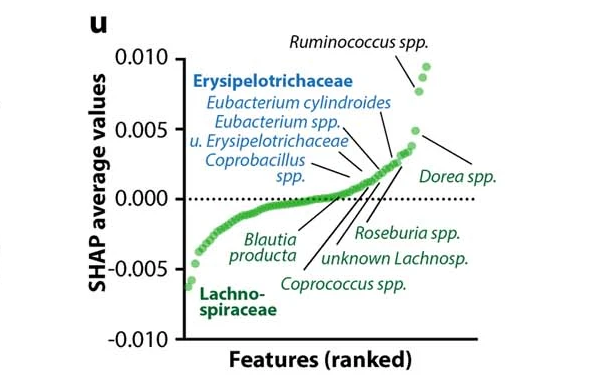
Dohnalová L, et al., Nature. 2022
研究人员确定,这一影响是由特定菌群引起的,包括毛螺菌科(Lachnospiraceae)和丹毒丝菌科(Erysipelotrichaceae)的成员。
这些实验证明,肠道菌群可能是个体运动能力差异的一个重要影响因素。
与研究人员的预期不同,肠道菌群对身体活动的影响并不是通过与运动经典相关的组织,如骨骼肌来介导的。相反,肠道菌群似乎根本上影响了运动对大脑的影响。
剧烈的体育活动会在大脑中引起多种神经化学反应,包括纹状体中多巴胺释放的激增。有趣的是,在没有肠道菌群的情况下,这种激增严重减弱了。
我们知道,多巴胺的释放会导致愉悦、动力和奖励感,这也是为什么有些人在剧烈运动后所经历的“跑步快感”现象。
基于这些发现,推测:
微生物群通过调节运动诱导的多巴胺反应来增强运动能力。
通过一系列药理学和化学遗传学的介入实验,发现多巴胺缺乏确实是导致微生物群消失动物运动能力降低的原因。与此一致的是,恢复多巴胺信号完全恢复了它们的运动能力。
微生物群与大脑之间的通信可以通过微生物分子进入全身循环,或者通过肠道和大脑之间的直接神经连接进行。
研究人员没有找到证据表明微生物群通过全身介质影响运动能力。相反,抑制与肠道神经传导相关的感觉神经元,可以重现微生物群消失对运动的影响,而刺激感觉神经元则可以在没有微生物群的情况下恢复运动能力。
为了确定微生物群在运动过程中如何影响感觉神经元的活动,研究人员结合了微生物遗传学、代谢组学和神经元记录技术。
结果发现,微生物通过合成脂肪酸酰胺,增强感觉神经元的活动,通过内源性大麻素受体CB1介导。这种神经元活动的增加进而增强了运动过程中的多巴胺信号(见下图)。
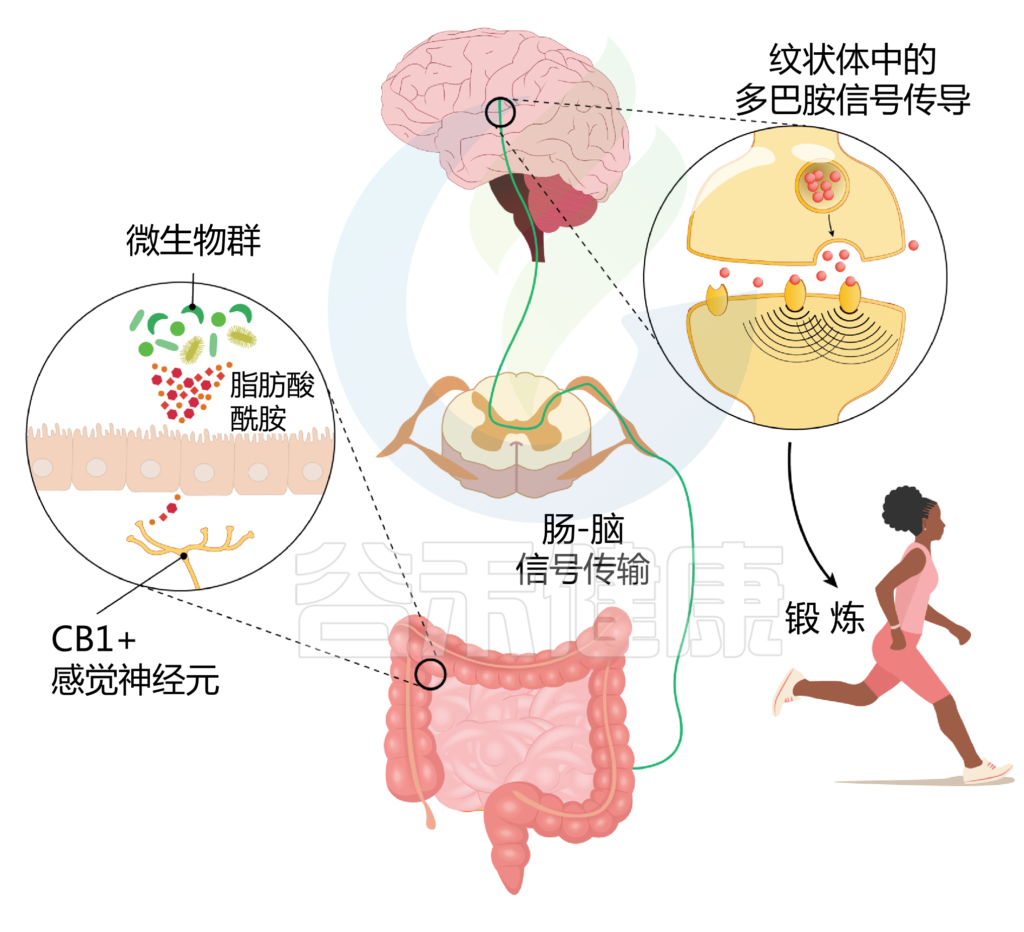
Thaiss CA. Science. 2023
这种途径可以通过胃肠道干预来调节运动表现
例如,用基因工程产生脂肪酸酰胺的细菌定植无菌小鼠,或用含有脂肪酸酰胺的饮食喂养抗生素治疗的小鼠,可以恢复它们的运动能力。
然而,使用外周CB1抑制剂或中枢多巴胺受体阻断剂时,这些干预的积极效果就会被取消掉。
该研究的重要发现:
微生物群代谢产生脂肪酸酰胺,由肠道神经支配神经元上的内源性大麻素受体CB1感知,这增强了它们在运动中的活性。感觉神经元的激活,反过来增强了纹状体中的多巴胺信号,从而推动了运动表现。
这项发现有几个重要的启示:
– 胃肠道信号作为大脑中运动反应神经元活动的调节因子,有可能促进“跑步快感”的产生
这种肠-脑途径的进化目的尚待确定,推测它可能将动物决定长期进行体育活动与胃肠道的营养状况联系起来。无论是精英运动员还是业余跑步者,心理状态在运动参与和表现中都起着至关重要的作用。
– 微生物组依赖性途径,可能有助于更好地理解促使某些个体达到巅峰表现的因素
微生物组及其代谢产物的可塑性,为在全球范围内增加运动参与及其健康益处提供了一条潜在途径。
– 纹状体多巴胺是运动以外的动机行为的关键介质
这项发现开启了利用这一途径影响其他多巴胺依赖性大脑功能的可能性,如学习、情绪、成瘾等领域都可能应用。
总的来说,这项研究为我们打开了一扇了解肠道微生物与运动之间关系的窗户,为相关领域的深入研究提供了新的启示。对影响神经元活动的微生物组衍生分子的广阔宇宙的进一步探索,将为胃肠道塑造思维的潜力提供有价值的见解。
当然,这也让我们看到了肠道菌群检测在个体运动能力评估中具有巨大潜力,通过微生物群干预,为提升运动积极性,发展个性化的运动方案和训练策略提供了新的思路和可能性。
参考文献:
Dohnalová L, Lundgren P, Carty JRE, et al., A microbiome-dependent gut-brain pathway regulates motivation for exercise. Nature. 2022 Dec;612(7941):739-747. doi: 10.1038/s41586-022-05525-z

谷禾健康

在漫长的历史中,一种神秘而令人不安的疾病一直困扰着人类,那就是癫痫。
癫痫是最常见的神经系统疾病之一,影响着全世界近7000万人。它会导致突发性的、不可控制的、反复发作的痉挛和意识丧失。
突如其来的发病行为,不仅让患者和他们的家人感到恐惧和困惑,也给他们的生活带来了巨大的不便和影响。
虽然二十世纪以来出现了大量抗癫痫药物,但许多的癫痫患者对药物治疗没有反应,并且病情仍然不受控制。
近年来,动物研究和人类病例的证据表明,肠道微生物群与癫痫有关。
本文将带大家初步认识癫痫这一疾病,并阐述了肠道微生物群如何在癫痫患者和动物模型触发、加重或调节这种疾病的病程。
此外,肠道微生物群可以用作癫痫诊断和预后的潜在生物标志物以及难治性癫痫患者的新治疗靶点。
目录

癫痫(epilepsy)是大脑神经元突发性异常放电,导致短暂大脑功能障碍的一种慢性疾病。
▼
▸ 根据发病特点分类
•全面性癫痫
涉及到整个大脑的神经元异常放电。这种类型的癫痫通常会导致全身抽搐和意识丧失。
常见的全面性癫痫包括肌阵挛性癫痫和失神癫痫等。
•局灶性癫痫
起源于大脑的一部分区域,只涉及到局部神经元的异常放电。这种类型的癫痫可能会导致局部肌肉抽搐、感觉异常或意识丧失。
常见的部分性癫痫包括颞叶癫痫和顶叶癫痫等。
▸ 根据病因分类
•继发性癫痫
继发性癫痫是由已知的病因或病变引起的。这些病因可以是脑部的结构异常、脑损伤、感染、代谢紊乱、中毒、药物副作用等。
注:继发性癫痫可以发生在任何年龄。
•特发性癫痫
特发性癫痫是指没有明确病因或病变可以解释的癫痫。在这种情况下,癫痫发作的原因可能是由于遗传因素、发育异常或其他未知因素引起的。
注:特发性癫痫通常在儿童或青少年时期开始,且没有明显的脑部结构异常。
▼
由于异常放电的神经元在大脑中的部位不同,而有多种临床表现。
✦全面性发作
这种类型的癫痫患者,往往在发作初期就会失去意识,完全意识不到自己发生了什么。
如下细分不同类型还会有各自对应的特点:
•强直-阵挛性发作
患者在发作早期,不仅失去意识,而且还会跌倒。此时患者大多会尖叫一声,全身抽搐,持续10~20秒后,发生阵挛。每一次的阵挛都会有一个间歇期,发作频率逐渐变慢、间歇期也越来越长。
在一次剧烈阵挛后,发作停止。这时候会观察到患者的瞳孔散大,唾液分泌物等增多,以及呼吸停止。之后患者会慢慢恢复,上述体征逐渐恢复正常,整个过程大概5~15分钟,有些患者在发作期还会发生牙关紧闭和大小便失禁。
患者醒来后,一般会觉得头痛,全身酸痛,很想睡—觉。
•强直性发作
此类型的患者多见于弥漫性脑损伤的患者。发作时可能是局部或全身的骨骼肌强烈而持续的收缩,能将患者固定于某一个特殊的姿势。
•阵挛性发作
此类型主要多见于新生儿和婴儿,发作时患儿会意识丧失。
•失神发作
意识丧失突然发生并迅速终止,是本类型癫痫发作的主要特征。患者可能会突然间活动停止,发呆、手上拿着的东西滑落到地板上,对旁人的呼叫无应答。也有些患者可能机械重复原有的简单动作。
•肌阵挛性发作
这是—种突然发生的类似于触电—样的不自主运动,发作时间一般比较短暂。
•失张力发作
这一类的患者往往会突然跌倒,也有些不太严重的患者会突然间低头,以及胳膊突然间下垂。
•伴有或不伴失神的眼肌阵挛性发作
此类患者的发作主要与眼部相关,大多在持续的光线下眼睑闭合后发生,间歇性的闪光刺激也可能诱发癫痫发作。发作时,患者的眼睛看起来半开半闭,有时候还会伴有手部的抽动。
✦局灶性发作
这—类的患者在癫痫发作时神志清楚,发作后能描述刚刚自己发生了什么。
一般分为以下几种类型:
•局灶性运动性发作
患者癫痫发作时,主要是某一个身体部位的不自主抽动,大多是一侧眼睑、口角、手或者足趾,也可能是一侧面部或肢体。严重的话,患者在发作过后可能发生短暂性的肢体瘫痪。
有些患者还会出现与人体的运动系统相关的异常动作,诸如不自主地重复发作前的单词或者单个音节,伴有身体或眼睛的旋转等。
•局灶性感觉性发作
这—类患者发作时,往往存在感觉异常。诸如味觉、嗅觉、听觉的异常,出现幻觉等。
•自动症
这一类的主要特征是患者出现存在意识障碍,会做一些看起来有目的,但实际上没有目的的动作,比如反复咀嚼、反复搓手或无目的地开门、关门等,发作后无法回忆起发作细节。
提醒
癫痫发作期千万不要强行约束患者,以免自己被误伤,也避免造成患者骨折、脱臼。
▼
癫痫是一种常见的神经系统疾病,全球范围内都有发病。根据世界卫生组织统计,全世界有超过6500万癫痫患者。癫痫的发病率在不同地区和人群之间有所差异。
★ 中低收入国家癫痫发病人数较高
在高收入国家,癫痫患病率约为每1000人6.4例,年发病率为每100000人中出现67.8例。在低收入和中等收入国家 ,这些数字几乎翻了两倍。其中约80%的患者生活在中低收入国家。
中国癫痫的发病率在5‰~7‰之间,全国有650万~910万患者。每年,我国会有40万~60万人被新确诊为癫痫患者。
★ 青少年和老人易发癫痫
癫痫可发生在各个年龄人群,但儿童患者和老年患者比较常见。儿童和青少年是癫痫的高发人群,其中大约有一半的癫痫病例在20岁以下发病。
此外,在孕期女性中,癫痫发作的比例约为0.3%~0.7%。
▼
✦结构性病因
结构性病因是指神经影像学上的异常发现,合理推断导致患者癫痫发作,并与电子临床评估或临床发现一致。
结构性病因的原因包括缺氧缺血性脑病、中风、外伤和感染。在结构性病因中,值得注意的是在内侧颞叶癫痫发作中相对频繁地发现海马体硬化。
✦遗传性病因
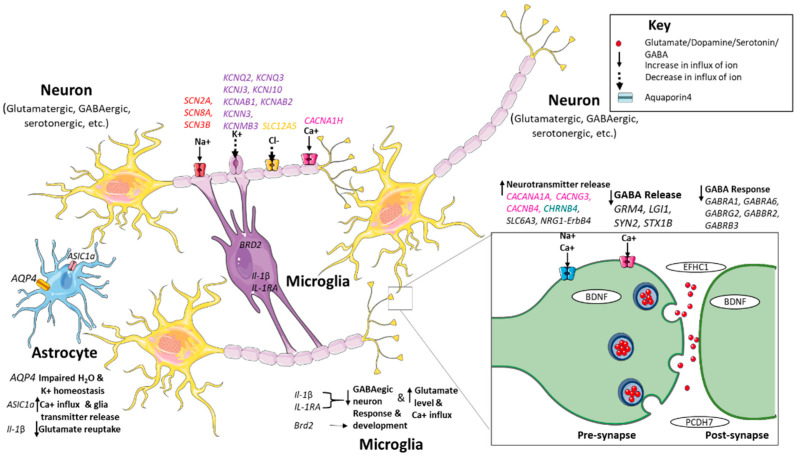
癫痫被认为受到遗传的影响。一项研究发现977个基因与癫痫有关。
这些基因包括癫痫基因(引起癫痫或以癫痫为核心症状的综合征的基因)、神经发育相关的癫痫基因(与大脑发育畸形和癫痫相关的基因)、癫痫相关基因(与身体或其他方面相关的基因)。
与此相关的基因有SCN2A、SCN8A、SCN3B、KCNJ3、KCNJ10、KCNN3、KCNMB3、CACNA1H、AQP4。
基因如CACANA1A、CACNG3、CACNB4、CHRNA4、GRM4、LGI1、ASIC1a、STX1B、SYN2、SLC12A5 ME2、ALDH5A、Il-1β和IL-1RA以及GABA-A和GABA-B受体基因直接或间接影响神经递质的合成或释放,导致兴奋性和抑制性神经递质的不平衡,导致神经元过度兴奋。
影响癫痫的一些基因
Thakran S,et al.Int J Mol Sci.2020
✦感染性病因
中枢神经系统感染是癫痫的主要危险因素,也是世界某些地区最常见的癫痫病因。据报道,发达国家中枢神经系统感染幸存者人群中无端癫痫发作的风险为6.8%至8.3%,中低收入国家的风险更高。
例如囊尾蚴、人类免疫缺陷病毒、巨细胞病毒、弓形虫、结核分枝杆菌和恶性疟原虫等都会感染中枢神经系统。
✦代谢性病因
一些代谢性疾病可表现为细胞变性和髓鞘形成障碍以及神经元迁移障碍,通过对细胞或器官功能产生负面影响而间接促进癫痫发生。
尽管大多数代谢性癫痫都有遗传基础,但有些可能是后天获得的,例如吡哆醇依赖性癫痫发作和脑叶酸缺乏症。
✦免疫性病因
当不明原因的癫痫患者神经特异性抗体血清呈阳性并且有自身免疫介导的中枢神经系统炎症的证据时,可以怀疑其免疫病因。
根据基于人群的研究,自身免疫性癫痫的发病率约为所有癫痫的5-7%。这种病因的识别具有治疗意义,因为自身免疫性脑炎引起的癫痫发作应该通过免疫疗法而不是传统的抗癫痫药物疗法来治疗。
免疫反应也与癫痫的诱发和癫痫的发展有关。癫痫脑中的先天性和适应性免疫反应均由常驻免疫细胞及其分泌的介质以及从外周渗透的白细胞激活。致病性神经炎症过程可以是外周起源的,也可以是中枢起源的。外周炎症通过离子和谷氨酸稳态的变化以及促炎分子从外周炎症灶迁移到血脑屏障来增强癫痫放电。
回顾了越来越多的临床前和临床证据,表明肠道微生物群会影响癫痫。
Amlerova J,et al.Int J Mol Sci.2021
动物实验
// 肠道菌群失衡增加了癫痫的易感性
研究人员发现,将肠道微生物群从长期应激的老鼠身上移植到幼年老鼠身上会促进癫痫的发作。这表明肠道菌群失衡,尤其是在慢性压力的影响下,增加了对癫痫的易感性。
实验结果表明,接受来自癫痫动物的微生物群的小鼠比对照组更容易出现癫痫持续状态,这表明微生物群介导了癫痫发作的易感性。
还有研究人员预测,移植癫痫小鼠的微生物群可能会通过增加健康小鼠的大脑兴奋性来诱发癫痫。
// 肠道炎症会增加癫痫发作
对肠道炎症与癫痫之间关系的进一步研究表明,肠道炎症会增加癫痫小鼠的癫痫发作活动。由此推断:肠道炎症可能是癫痫控制的有效目标,也可能是癫痫易感患者癫痫发作的一个因素。
// 益生菌降低了癫痫的严重程度
研究了益生菌混合物对大鼠戊四氮触发的大脑攻击活动、认知能力以及总脑组织抗氧化能力的影响。
结果表明,益生菌大大降低了癫痫发作的严重程度。同时,口服益生菌也部分改善了大鼠的空间学习和记忆。
虽然神经递质的抑制/兴奋以及抗氧化剂和氧化剂之间的失衡是癫痫发作的主要原因,但益生菌治疗增加了γ-氨基丁酸活性并改善了大鼠抗氧化剂和氧化剂之间的平衡。
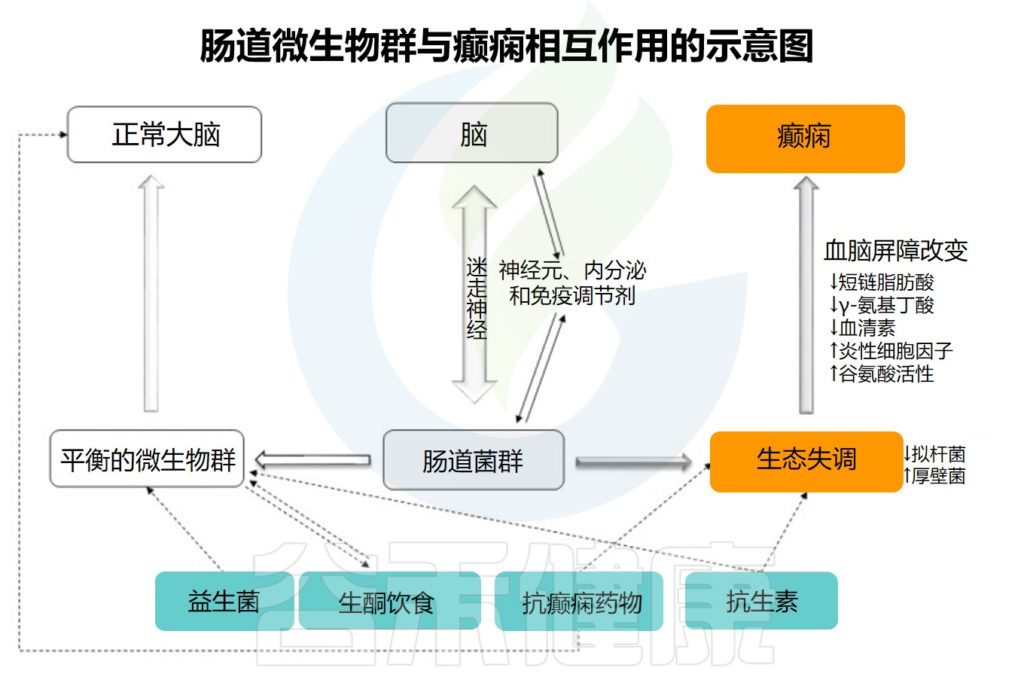
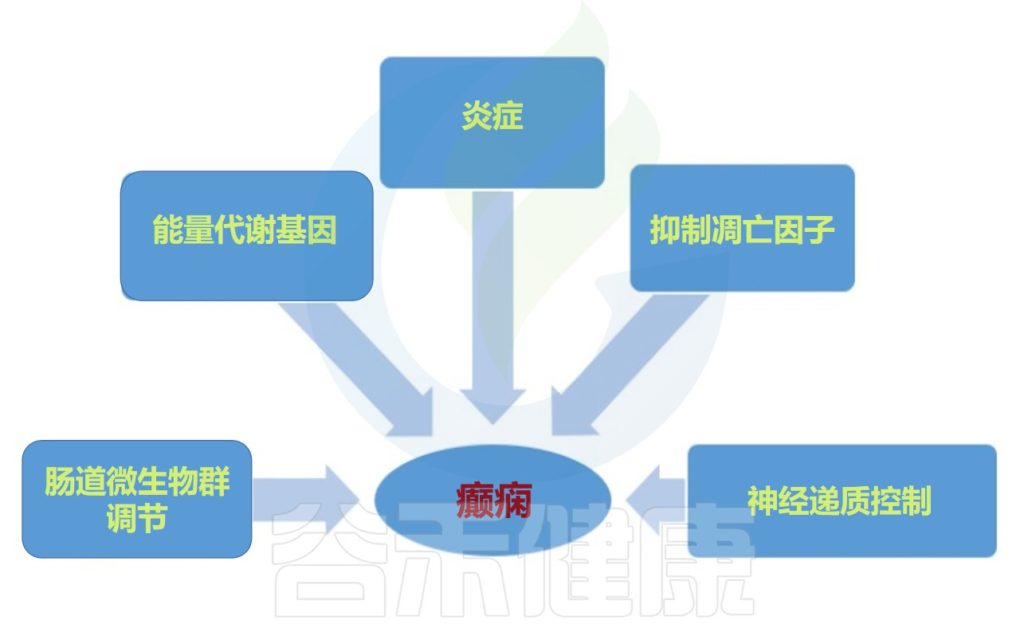
微生物群-肠-脑轴与癫痫
微生物群-肠-脑轴由中枢神经系统 、肠神经系统和 肠道微生物群构成,通过向上传导和向下传导进行通信。
// 健康的肠道菌群会产生良好的代谢产物
不良的肠道菌群会上调促癫痫代谢物的产生、炎症因子的分泌等,导致γ-氨基丁酸/谷氨酸比例异常,进而诱发癫痫。
慢性压力可能是这一过程的触发因素。
健康的肠道微生物群可以产生良好的代谢产物,例如短链脂肪酸和血清素,可以抑制癫痫的发生。下丘脑-垂体-肾上腺轴、肠神经系统和迷走神经系统也参与肠道菌群与癫痫之间的相互作用。
癫痫中的微生物群-肠-脑轴
Ding M,et al.Front Immunol.2021
重点来了
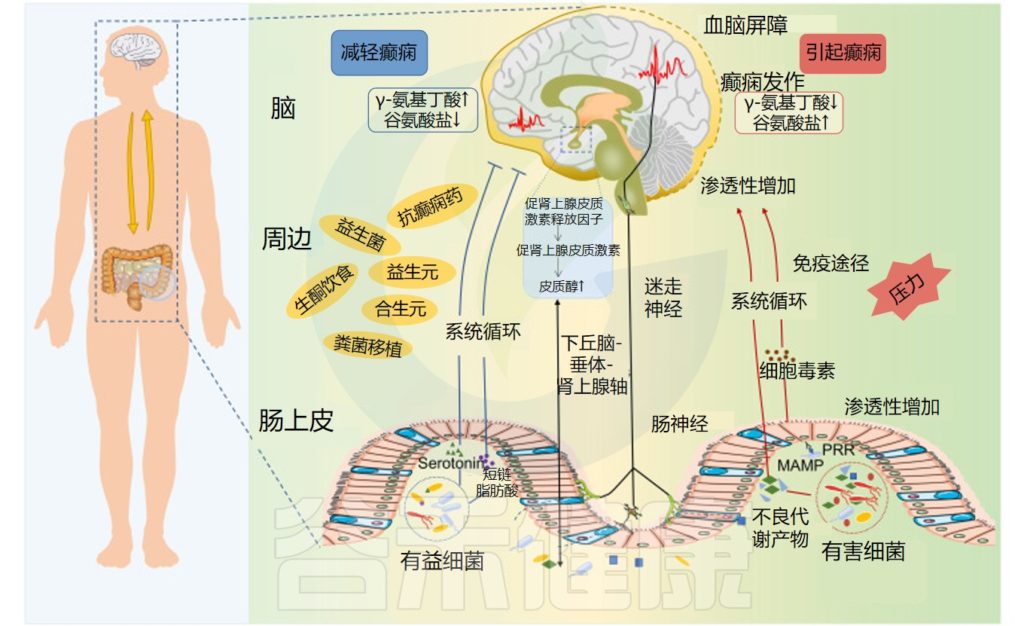
具体地说,对于癫痫,癫痫发作和癫痫发生可能会受到肠道微生物群通过以下方式的影响:
1) 肠道产生神经递质,如γ-氨基丁酸、谷氨酸和血清素;
2) 通过免疫系统介导的促炎作用,释放细胞因子和趋化因子,以及脂多糖水平的增加,导致肠和血脑屏障通透性增加和神经炎症增加;
3) 通过改变肠道源性代谢物的量,例如主要以中枢神经系统保护作用而闻名的短链脂肪酸。
此外,神经和神经内分泌下丘脑-垂体-肾上腺轴,以及内源性大麻素系统和脑源性神经营养因子的水平可以受肠道微生物群干扰癫痫发作机制的影响。
癫痫患者的肠道微生物群
已经有多项关于癫痫患者和健康对照之间肠道微生物群差异的临床研究。
但目前人体临床研究主要关注两个方面:一是癫痫患者肠道菌群与健康人的差异,二是癫痫患者服用益生菌或粪菌移植后症状的改善。
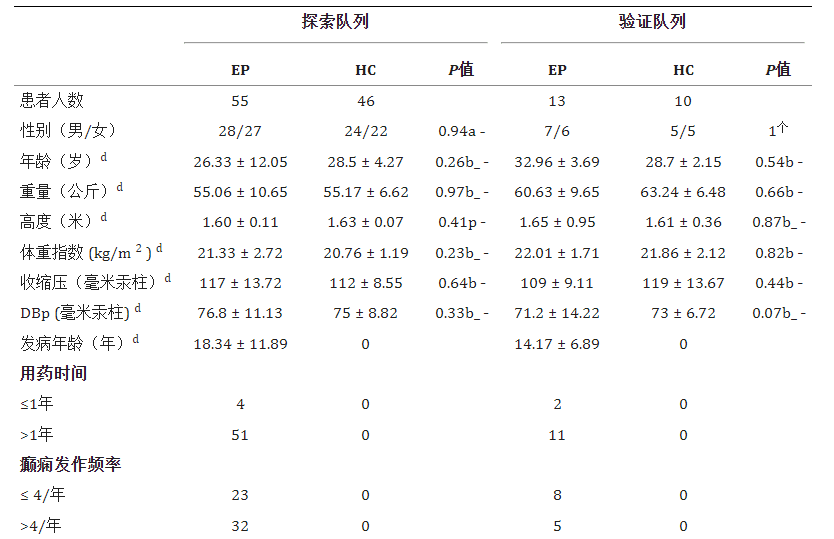
本文主要采用了华西医院神经内科的研究,该研究的样本量较大,纳入55名被诊断确认超过三年的癫痫患者和46名来自同一家庭的健康对照。
与不生活在同一家庭的人相比,夫妻之间的肠道细菌群落往往更加相似。因此,我们在研究的探索和验证队列中纳入了健康配偶作为对照。仅包括一起生活并饮食相似至少10年的夫妇。

Gong X,et al.Front Microbiol.2020
此外,该研究中还排除了一些可能影响癫痫的风险因素,使结果的可信度更高。
癫痫患者和健康对照的排除标准如下:(1)最近3个月内接受过抗生素、益生菌、益生元或合生元治疗;(2)近3个月内有胃肠炎病史;(3)有其他自身免疫性疾病史(多发性硬化症、视神经脊髓炎、系统性红斑狼疮、类风湿性关节炎、1型糖尿病等);(4)肠道手术史;(5) 怀孕或哺乳期;(6)有神经或精神疾病史(帕金森病、阿尔茨海默病、焦虑症、抑郁症、自闭症谱系障碍、精神分裂症等);(7)6个月内除抗癫痫药物外的其他方案摄入史(维生素、蛋白质、不饱和脂肪酸等);(8) 严重营养不良或感染或吸毒或酗酒。
研究结果
// 癫痫患者的肠道微生物α多样性下降
通过使用 16S rDNA 测序,发现癫痫患者组的α多样性指数远低于健康对照组。最近,相对较低的微生物多样性与儿童药物难治性癫痫以及与中枢神经系统改变相关的其他疾病有关,包括阿尔茨海默病、多发性硬化症和帕金森病。
// 梭杆菌等具有致病性作用的菌群在癫痫患者中过度生长
结果表明,一些细菌门,包括梭杆菌(Fusobacteria)、疣微菌(Verrucomicrobia)和硝化螺旋菌(Nitrospirae),在疾病组中生长过度。而厚壁菌门和Saccharibacteria在疾病组中数量较少。
梭杆菌
现有研究表明,梭杆菌对脊椎动物具有致病性,在人类结直肠癌和发炎的肠道粘膜中普遍存在。一些研究人员将梭杆菌属物种描述为病原体,因为它们具有侵入性,并且能够转移到血液中并导致全身疾病状态。
疣微菌
疣微菌门以大量产生短链脂肪酸和粘蛋白降解的微生物而闻名。疣微菌可以降解粘蛋白,这可能会扰乱肠道屏障的完整性以及随后的细菌易位。
硝化螺旋菌
硝化螺旋菌可以增加亚硝酸盐的毒性,最终可能导致血脑屏障功能障碍和通透性增加,并有助于癫痫的作用机制。
拓展:A. muciniphila过高的危害
在我们的检测中发现一名56岁女士肠道内该菌的丰度占比超50%,菌群构成如下:

过量的Akkermansia将过度消耗粘液蛋白而存活下来,这是大多数其他细菌所缺乏的生存优势。
在这种情况下,非粘液消耗物种的数量显著减少,导致物种多样性减少, Akkermansia增殖异常,从而可能导致肠道屏障损伤,诱发肠道炎症、脂多糖进入血液的增加、自身免疫性疾病、神经退行性疾病等有关。
// Blautia、双歧杆菌等菌属丰度增加
预后不良的患者中,经黏液真杆菌属(Blautia)、双歧杆菌属、Subdoligranulum、普雷沃氏菌(Prevotella)、戴阿利斯特杆菌属 (Dialister)和Anaerostipes增加。
较高丰度的普雷沃氏菌会引发炎症
普雷沃氏菌为肠道核心菌,但是较高丰度的普雷沃氏菌会促进炎症。较高丰度的普氏菌可能导致肠道中持续产生IL-6,从而引发炎症反应。此外,据报道,普雷沃菌属会改变肠道通透性。
扩展阅读:肠道重要基石菌属——普雷沃氏菌属 Prevotella
癫痫患者碳水化合物代谢增加可能导致双歧杆菌丰度较高
此外,我们研究中的功能分析还显示,疾病组的碳水化合物代谢显著增加。已经确定碳水化合物代谢物的紊乱可能在癫痫发生机制中发挥潜在作用。双歧杆菌消化复杂的碳水化合物并表现出最大的预测糖生物组之一。
// 耐药性癫痫与药物敏感性癫痫患者的肠道微生物也不同
此外,耐药性癫痫患者的肠道微生物组可能与药物敏感性癫痫患者不同。
与药物敏感性癫痫患者相比,耐药性癫痫患者的α多样性和主要属于厚壁菌门的细菌的相对丰度有所增加。
注:在难治性癫痫组中,粪肠球菌(Enterococcus faecalis)、长双歧杆菌(Bifidobacterium longum)和迟缓埃格特菌(Eggerthella lenta)是潜在的生物标志物。
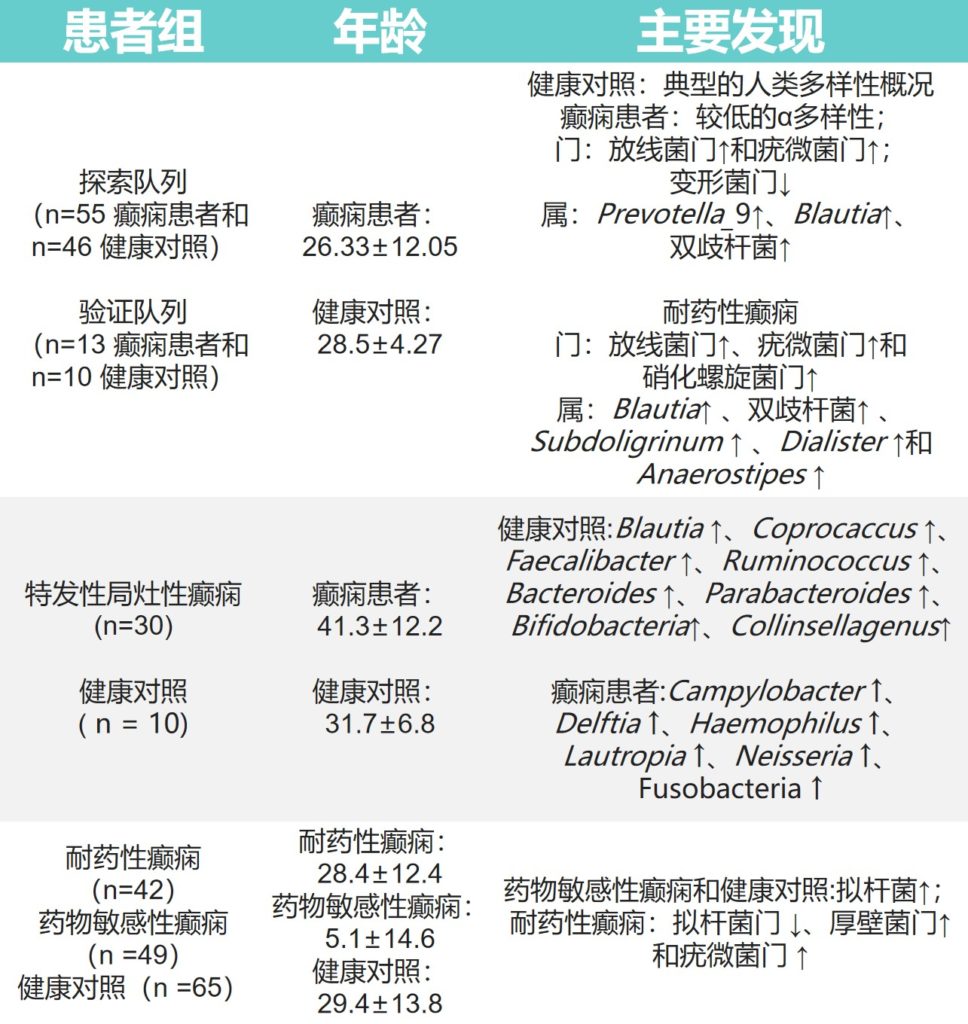
还有一些关于癫痫患者肠道微生物群的其他研究,主要研究成果展示在下表:

Ding M,et al.Front Immunol.2021
小结
这些研究表明肠道微生物群与癫痫之间有着千丝万缕的联系。肠道微生物群的组成可能会影响癫痫的易感性。
需要注意的是,癫痫患者之间的差异以及所使用的研究方法存在局限性。考虑到影响肠道微生物组的变量较多,如年龄、饮食和生活环境等的差异,需要在合理控制变量的基础上进行更大样本的分析。
▼
癫痫的发病机制与神经免疫和神经炎症有关。越来越多的证据表明,脑肠轴的免疫和炎症通路可能参与癫痫的发病机制。
小胶质细胞和星形胶质细胞是中枢神经系统中主要的炎症细胞,其炎症状态会促进癫痫的发生。
√肠道微生物群通过调节免疫影响癫痫
肠粘膜的淋巴组织含有体内所有免疫细胞的70%–80%。
肠道微生物群会影响免疫细胞:例如,无菌小鼠表现出免疫异常,T细胞和B细胞数量减少,细胞因子产生减少。
此外,肠道微生物群似乎是小胶质细胞成熟以及星形胶质细胞激活的最重要因素之一。肠道微生物群调节先天免疫、适应性免疫和炎症机制,以调节癫痫的发展。
√肠屏障和血脑屏障损伤影响大脑
肠粘膜屏障和血脑屏障共同作用,防止肠道微生物群及其分泌物进入大脑。
“肠漏”综合征的特点是肠道通透性增加,导致细菌、有毒代谢物和小分子转移到血液中。在肠道炎症下,细菌可以直接将因子释放到体循环中,从而激活外周免疫细胞,改变血脑屏障完整性,从而改变转运速率,甚至可以诱发“漏脑”。
压力会增加肠粘膜通透性,管腔内的脂多糖和其他细胞因子进入血液循环刺激Toll样受体,产生炎症细胞因子,增加血脑屏障通透性并损害大脑。
√神经免疫与癫痫的发生
小胶质细胞和星形胶质细胞通过释放过量的细胞因子参与癫痫的发病机制,并相互作用。
星形胶质细胞是大脑中最丰富的神经胶质细胞,具有多种功能,包括调节血脑屏障的完整性、神经递质的循环利用以及参与免疫反应。
小胶质细胞是中枢神经系统的常驻巨噬细胞,介导先天免疫反应。
小胶质细胞和星型胶质细胞相互作用的机制
小胶质细胞可以调节星形胶质细胞的表型和功能,例如小鼠小胶质细胞可以通过VEGF-B(促进星形胶质细胞的致病反应和炎症反应)和TGF-α(促进相反的反应)来调节星形胶质细胞的行为。
肠道微生物将膳食色氨酸代谢为芳基碳氢化合物受体激动剂,并与其受体相互作用,控制小胶质细胞活化以及TGF-α和VEGF-B表达,从而调节星形胶质细胞的致病活性。
星形胶质细胞释放的炎症细胞因子和趋化因子增强小胶质细胞的活性,包括迁移、凋亡细胞的吞噬作用和突触修剪。
血脑屏障通透性增加易引起神经炎症
星形胶质细胞和小胶质细胞之间的相互作用导致促炎细胞因子的产生和血脑屏障通透性增加,从而导致外周血免疫细胞和细胞因子渗入中枢神经系统,以及随后的慢性神经炎症。
无菌和抗生素处理的动物也改变了小胶质细胞的形态以及成熟、激活和分化方面的缺陷,导致对多种病原体的免疫反应不足,而这种免疫反应可以在肠道微生物群重新定植后修复,这表明肠道微生物多样性对于小胶质细胞和中枢神经系统功能至关重要。
肠道微生物群通过先天免疫诱发癫痫
肠道微生物群可以通过先天免疫途径诱发癫痫。在无菌小鼠的整个生命周期中,血脑屏障通透性不断增加,这与内皮细胞中occludin和claudin-5蛋白表达的降低有关。
肠道微生物群失调会减少紧密蛋白的产生并增加肠壁的通透性,导致微生物、代谢物和毒素从肠腔中逸出。肠道微生物群失调还会减少短链脂肪酸,从而增加血脑屏障通透性并促进神经炎症。
如果这两个屏障被打破,微生物群释放的免疫细胞和炎症因子就会进入大脑并诱发癫痫发作。
肽聚糖是细菌细胞壁的成分,主要存在于人体肠道中。肽聚糖作为慢性脑炎的驱动因素,也在大脑小胶质细胞中检测到。因此,我们得出结论,肽聚糖可能通过促进肠漏和脑漏从肠道转移到中枢神经系统,导致慢性炎症并诱导癫痫的发生。
肠道微生物群通过适应性免疫促进癫痫的发生
肠道微生物群还通过诱导适应性免疫来促进癫痫的发生。肠道微生物群可以诱导免疫细胞产生细胞因子,通过肠粘膜和血脑屏障进入大脑,激活大脑免疫细胞参与免疫反应。
辅助T细胞17(Th17)是是适应性免疫的关键组成部分,IL-17是由Th17细胞产生的细胞因子,可以通过特定的肠道微生物群(例如拟杆菌门)进行调节。
最近发现,癫痫患者的脑脊液和外周血中IL-17水平均高于对照组,并且与癫痫发作的频率和严重程度高度相关。因此,肠道微生物群可以通过介导IL-17影响癫痫的发生。
此外,共生微生物群的缺失会下调IgA和IgG1,并上调 IgE,从而导致疾病易感性增加。
因此,肠道微生物群可以通过肠-脑轴诱导免疫反应,从而导致癫痫发生。
然而,只有少数研究直接关注肠道、免疫反应和癫痫之间的关系,许多问题仍有待探索。
▼
在大脑和肠道之间传递信息的重要途径之一是通过自主神经纤维。
√肠道刺激通过自主神经系统调节大脑活动
给小鼠口服空肠弯曲杆菌会导致脑干迷走神经感觉神经节和初级感觉中c-fos表达增加,表明肠道刺激可以通过自主神经系统调节大脑活动。
√神经足细胞与迷走神经元接触影响神经系统
迷走神经刺激已成为癫痫的常规治疗方法。据报道,迷走神经传入纤维的电刺激可以改变大脑中血清素、γ-氨基丁酸和谷氨酸的浓度,从而解释了其在癫痫中的用途。
以前,肠道内分泌细胞和脑神经被认为只能通过激素进行交流;然而,最近发现称为神经足细胞的肠内分泌细胞可以与迷走神经元突触,以转导肠腔信号,使用谷氨酸作为神经递质将肠腔连接到脑干。
神经足细胞的发现为通过调节肠道微生物群来治疗神经系统疾病提供了强有力的理论支持。
▼
√肠内分泌信号和神经递质与癫痫密切相关
神经递质失衡与癫痫密切相关。癫痫病灶存在神经递质失衡,如γ-氨基丁酸活性低下、谷氨酸活性亢进、多巴胺和去甲肾上腺素活性亢进、血清素活性低下。
肠道微生物影响神经递质的产生
在胃肠道中,神经递质可由肠道微生物群直接分泌或由胃肠细胞在肠道微生物群代谢物的刺激下产生。
不同的肠道微生物群可以产生不同的神经递质(肠球菌属、链球菌属和埃希氏菌属产生血清素;乳杆菌属和双歧杆菌属产生γ-氨基丁酸;埃希氏菌属和芽孢杆菌属可以产生多巴胺)。
肠道微生物群产生的各种神经递质可以通过肠粘膜,但很少通过血脑屏障,γ-氨基丁酸除外。在海马损伤或癫痫状态下,肠道微生物群产生的γ-氨基丁酸会导致γ-氨基丁酸和谷氨酸系统之间的不平衡,从而引起癫痫发作。
一些肠道菌群调节氨基酸水平从而影响癫痫
嗜黏蛋白阿克曼菌(Akkermansia muciniphila)和Parabacteroides定植可以改变血清和肠腔中的氨基酸水平,从而调节海马中与癫痫发作相关的神经递质(例如 GABA 和谷氨酸)的水平,从而为小鼠提供保护性抗癫痫作用。
血清素可以改善癫痫患者的发作
肠嗜铬细胞产生大约90%的血清素。在小鼠中,某些肠道微生物群,例如形成孢子的梭菌类群,可以通过上调结肠色氨酸羟化酶1(一种血清素生产的限速酶)来促进肠道中血清素的生物合成。
先前的研究表明,颞叶癫痫患者存在血清素缺乏症。增加血清素的药物组合,例如选择性血清素再摄取抑制剂,可以改善癫痫患者的癫痫发作控制。
去甲肾上腺素对癫痫发作具有双重作用
去甲肾上腺素对癫痫发作具有双重作用,具体取决于其含量,低剂量的去甲肾上腺素具有促癫痫作用,而高剂量的去甲肾上腺素可以抑制癫痫。
√短链脂肪酸癫痫密切相关
短链脂肪酸包括乙酸盐、丙酸盐和丁酸盐,可以由一些肠道细菌(主要是拟杆菌属和厚壁菌门)通过不溶性膳食纤维的发酵产生。
短链脂肪酸在小胶质细胞成熟、肠脑神经系统、血脑屏障通透性以及通过直接或间接途径的应激反应中发挥着重要作用,所有这些都与癫痫密切相关。
不同短链脂肪酸对癫痫的保护作用
在癫痫小鼠模型中进一步研究了不同短链脂肪酸对癫痫的保护作用和机制。
丁酸盐可能通过减轻肠道炎症和氧化应激,表现出抗癫痫作用。丁酸盐还可以通过Keap/Nrf2/HO-1途径改善线粒体功能障碍并保护脑组织免受氧化应激和神经元凋亡的影响,从而提高癫痫阈值并降低癫痫强度。
丙酸盐治疗可以通过减少线粒体损伤、海马细胞凋亡和神经缺陷来减轻癫痫发作强度并延长癫痫发作潜伏期。
这些研究表明,短链脂肪酸在癫痫模型中减少,并且通过不同的机制对癫痫具有保护作用。
▼
压力可促进癫痫的诱发,癫痫患者糖皮质激素水平较高。
下丘脑-垂体-肾上腺轴(HPA轴)是应激反应的核心,包括促肾上腺皮质激素释放因子、促肾上腺皮质激素的分泌以及随后糖皮质激素和儿茶酚胺下游途径的释放。
不同的激素可能有不同的作用:例如,大多数脱氧皮质酮是抗惊厥药,而促肾上腺皮质激素释放激素和皮质酮可诱发癫痫发作。
√肠道微生物调节HPA轴影响癫痫
尽管HPA轴和肠道微生物群之间存在相关性,但具体机制尚未阐明。慢性压力可能会上调糖皮质激素,从而增强谷氨酸信号传导并诱发癫痫发作。肠道微生物群可以通过改变循环细胞因子水平或其他途径影响下丘脑的功能,从而调节HPA轴。
小鼠的应激反应表明,肠道微生物群调节应激依赖性垂体和肾上腺激活,并改变结肠中调节促肾上腺皮质激素释放激素途径的基因表达。慢性压力可能通过肠道微生物群影响HPA轴并促进癫痫。
注:HPA轴、肠道微生物群和癫痫之间的具体关系仍需进一步研究。
健康人和不同类型癫痫患者之间的肠道微生物及其代谢物差异使其成为癫痫鉴别诊断、预后和治疗监测的潜在代谢标志物。
这里将讨论一些涉及癫痫或是神经活性的物质。
1
使用啮齿动物模型的实验研究检验了短链脂肪酸对宿主神经系统的调节作用。短链脂肪酸调节多种受体,包括普遍存在的多效性G蛋白偶联受体,不仅存在于肠上皮细胞上,而且还存在于脑组织中。
▷调节G蛋白偶联受体影响神经系统
G蛋白偶联受体参与激活抑制性调节性T细胞、辅助性T细胞1和辅助性T细胞17,增强FOXP3转录因子的基因表达并下调促炎细胞因子,例如IL-12,肿瘤坏死因子和核因子-κB(NF-κB)。
短链脂肪酸还能够通过将乙酸盐转化为细胞营养和能量代谢所需的乙酰辅酶A来激活整合mTOR途径。
▷调节组蛋白去乙酰酶影响癫痫
此外,乙酸盐、丙酸盐和丁酸盐可能通过抑制组蛋白去乙酰酶 (HDAC) 或触发乙酰化来引起表观遗传修饰。
组蛋白去乙酰酶已被证明参与神经发生、突触传递、可塑性、脑源性神经营养因子水平的调节、神经胶质细胞发育、与学习和记忆相关的高级脑功能以及抑郁症和精神分裂症等神经系统疾病。
最近的研究已经证实了组蛋白乙酰化在失神性癫痫啮齿动物模型中癫痫发生和相关精神疾病中的作用。在这项研究中,在早期长期给予丁酸盐和丙戊酸(单独或联合)后,大鼠表现出致癫痫活动的数量和持续时间显著减少。
此外,组蛋白乙酰化的增强可以改善抑郁样行为和记忆表现等症状。这些发现强调了组蛋白去乙酰酶抑制剂作为癫痫治疗新策略的能力。
▷促进血清素和儿茶酚胺的合成
短链脂肪酸还可以促进色氨酸羟化酶1和酪氨酸羟化酶基因的转录,从而分别促进肠道血清素和血清儿茶酚胺(即多巴胺、去甲肾上腺素和肾上腺素)的生物合成,这些血清素具有通过自主受体调节中枢神经系统的关键作用。
小鼠下丘脑的谷氨酸和γ-氨基丁酸水平在暴露于醋酸盐或鼠李糖乳杆菌和长双歧杆菌的特定菌株后会发生变化,表现出抑郁和焦虑样表型的减弱。
综上所述,短链脂肪酸作为癫痫的生物标志物,通过影响神经信号传导、血脑屏障和免疫系统等多个途径参与癫痫的发病机制。
2
胆汁酸是胆汁的主要有机成分,可作为胆固醇、胆红素和异生物质的生物洗涤剂。
胆酸和鹅去氧胆酸是肝脏产生的初级胆汁酸,一旦进入肠道,肠道微生物群就会通过脱羟基作用将胆汁酸转化为次级胆汁酸,包括脱氧胆酸和石胆酸。
在小鼠中,已证明次级胆汁酸是法尼醇X受体的内源性配体,法尼醇X受体是调节肝脂肪生成、胰岛素敏感性和葡萄糖稳态的重要传感器。法尼醇X受体的激活还可以通过增加抗菌基因的表达和阻止细菌易位来抑制肠道微生物的过度生长。
法尼醇X受体在肝脏、肠和皮质神经元中表达。在中枢神经系统中,法尼醇X受体影响γ-氨基丁酸、去甲肾上腺素和血清素的神经传递。
▷胆汁酸与癫痫的炎症程度和发作频率有关
一些研究发现,癫痫患者的胆汁酸代谢存在异常。具体来说,癫痫患者的胆汁酸合成和转运过程可能受到影响,导致胆汁酸浓度的改变。
此外,胆汁酸与肠道微生物群之间存在相互作用,微生物群可以通过代谢胆汁酸来影响其浓度和代谢途径。
一项发表于2020年的研究发现,癫痫患者的血液和脑脊液中的胆汁酸水平明显降低。此外,胆汁酸的降低与癫痫的严重程度和发作频率呈负相关。
这些发现表明,胆汁酸可能在癫痫的发生和发展中发挥一定的作用。
需要指出的是,胆汁酸作为癫痫标志物的研究仍处于初步阶段,尚需进一步的研究来验证这些发现,并深入探索其在癫痫发病机制中的具体作用。
了解胆汁酸与癫痫之间的关系有助于我们更好地理解癫痫的病理生理过程,并为癫痫的预防和治疗提供新的思路。
3
色氨酸是一种重要的氨基酸,它在人体内起着多种生物学功能。它是各种激素、维生素、某些脑神经递质以及抗菌活性所需蛋白质的前体。最近的研究表明,色氨酸代谢异常可能与癫痫有关。
▷色氨酸及其代谢物参与抗癫痫作用
一项对16名难治性癫痫儿童进行的研究表明,色氨酸代谢物可能参与抗癫痫作用。
马吲哚胺2,3-双加氧酶1的激活(分解谷氨酸代谢物)通过改变犬尿氨酸/谷氨酸比例与慢性颞叶癫痫大鼠模型中的抑郁样行为直接相关。
另一项在两种癫痫小鼠模型中进行的重要研究指出,α-乳清蛋白通过提高血浆和大脑中色氨酸的浓度来发挥抗惊厥活性,从而通过微生物群和血清素能受体改善神经传递。
在不同的啮齿动物模型中长期使用α-乳清蛋白治疗可以保护或抑制癫痫发作。
此外,据推测,色氨酸代谢物可作为N-甲基-D-天冬氨酸受体的拮抗剂,从而抑制兴奋性途径。
肠道微生物群的调节可能是癫痫的潜在治疗方法。一方面,调节肠道微生物群可以通过调整与癫痫相关的机制来减少癫痫的发作。
另一方面,药物可以通过直接或间接的方式被肠道微生物群转化为代谢物。对于癫痫患者来说,调整肠道微生物组的组成可能会促进药物代谢和吸收,提高抗癫痫药物的反应性。
肠道微生物群在癫痫中的假设机制和作用
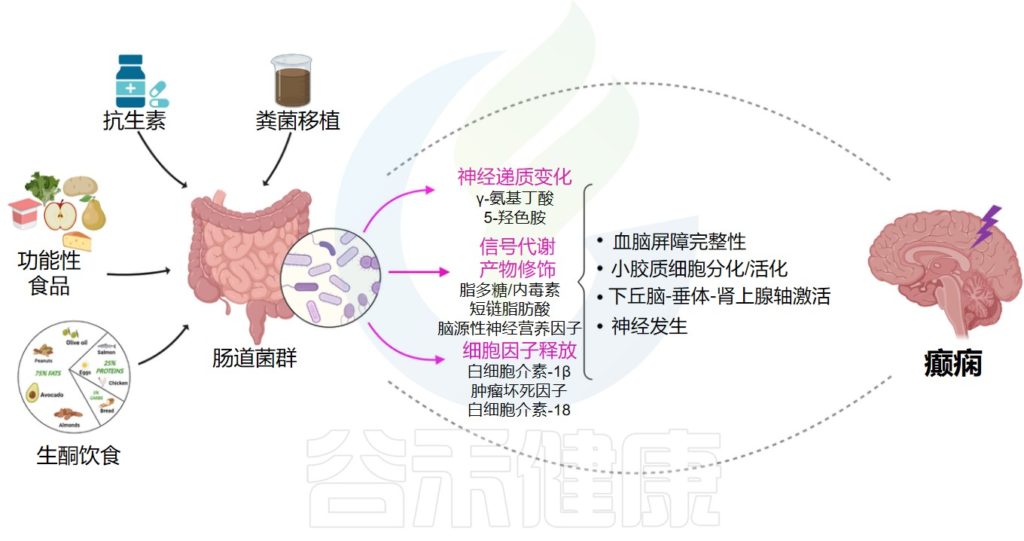
Iannone LF,et al.Neurobiol Dis.2022
在本节中,谷禾将阐述一些基于肠道微生物群的癫痫潜在疗法。如生酮饮食,施用抗生素、益生菌、益生元以及抗癫痫药物和粪便移植对癫痫的影响。
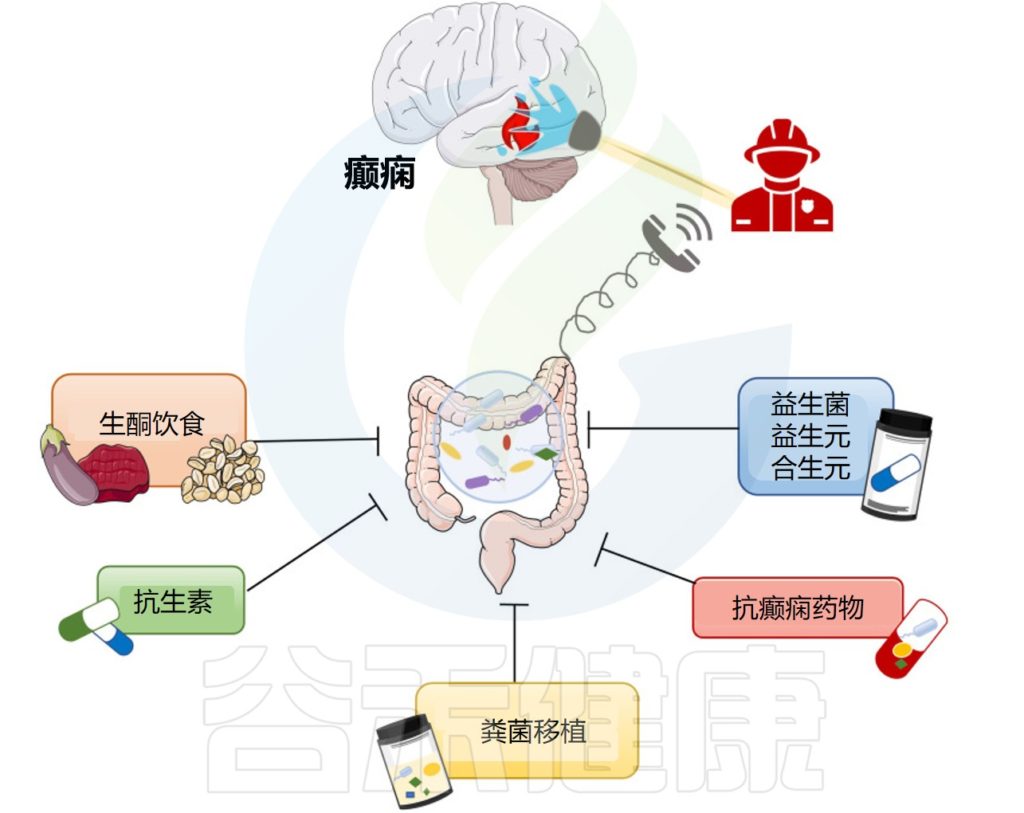
Ding M,et al.Front Immunol.2021
▼
饮食,尤其是生酮饮食,可以通过塑造肠道微生物群来调节癫痫的发生,生酮饮食是一种高脂肪、低碳水化合物和充足蛋白质的饮食,自1921年以来一直用于治疗难治性癫痫患者。
注:生酮饮食对其他神经系统疾病也有积极作用,如多发性硬化症、帕金森病、阿尔茨海默病和偏头痛。
关于癫痫发作频率,在生酮饮食治疗期间,10%的患者没有癫痫发作,40%的患者癫痫发作频率降低至50%。
生酮饮食对癫痫发作活动的可能影响
Ułamek-Kozioł M,et al.Nutrients.2019
生酮饮食中脂肪与蛋白质和碳水化合物的经典比例为4:1,这会引发代谢模式从葡萄糖代谢转向脂肪酸代谢。
经典的生酮饮食可以通过多种途径缓解癫痫,包括调节神经递质、脑能量代谢、氧化应激、离子通道和肠道菌群。
•不饱和脂肪酸具有抗癫痫作用
在生酮饮食中,饱和脂肪一直是主要使用的脂肪;然而,动物和人类研究已经证明多不饱和脂肪酸,尤其是Omega-3脂肪酸具有抗癫痫作用。
膳食Omega-3存在于亚麻籽、坚果、深海鱼和海洋哺乳动物中。 Omega-6主要来源于动物产品和植物油,构成现代西方饮食中多不饱和脂肪酸的大部分。
二十二碳六烯酸是大脑中主要的多不饱和脂肪酸,通过多种途径参与神经功能的调节,例如与离子通道的相互作用和神经递质的释放。一项病例对照研究表明,癫痫儿童血清Omega 3/Omega 6 比率低于健康儿童。
体外和体内研究都表明,富含Omega 3脂肪酸的饮食有利于癫痫控制,2021年七项临床试验研究的荟萃分析表明,补充Omega-3可显著降低癫痫发作频率,并且对成人比儿童更有效。
•肠道菌群对生酮饮食抗癫痫治疗具有促进效果
一些临床和实验研究探讨了肠道微生物群对生酮饮食抗癫痫疗法的作用。
通过使用 16S rRNA 测序方法,分析了接受生酮饮食治疗一周的中国儿童耐药性癫痫患者的分类肠道微生物群变化。
生酮饮食调节了不同肠道微生物群的相对丰度
结果表明,生酮饮食增加了特定门的相对表达,包括拟杆菌门和普雷沃氏菌门,同时降低了克罗诺杆菌属、丹毒杆菌属、链球菌属、另枝菌属(Alistipes)、瘤胃梭菌属(Ruminiclostridium)、Barnesiella和肠球菌属的相对表达。
研究人员将这些改变与癫痫发作减少联系起来,因为21%的患者没有癫痫发作,43%的患者癫痫发作减少了 50%。
这些结果得到了后续临床研究的证实。检查了接受生酮饮食治疗的中国儿童耐药性癫痫患者的粪便样本。发现经过六个月的生酮饮食治疗后,β多样性与基线水平有所不同。生酮饮食后,拟杆菌门的相对丰度增加,厚壁菌门和放线菌的比例显著降低。
有趣的是,在对生酮饮食反应较少的剩余50%患者中,梭状芽胞杆菌、瘤胃球菌科、毛螺菌科、另枝菌属和文肯菌科(Rikenellaceae)的相对丰度较高。
这些研究虽然规模较小,但却强化了这样一个假设:通过生酮饮食调节肠道微生物群可以对癫痫患者发挥治疗作用。
生酮饮食对癫痫的影响

Mejía-Granados DM,et al.Seizure.2021
最近的一项研究表明,高脂肪:碳水化合物+蛋白质比例对于癫痫的治疗并不是必不可少的。低脂肪:蛋白质+碳水化合物比例的新组合饮食,包括中链甘油三酯、多不饱和脂肪酸、低血糖指数碳水化合物和高支链氨基酸/芳香族氨基酸比例,也可减少兴奋性驱动并防止啮齿动物模型中的癫痫发作。
饮食干预是控制癫痫有效且有前景的方法,特别是生酮饮食。对微生物群-肠-脑轴的进一步研究将有助于开发更有效的饮食疗法。
▼
•肠道微生物群影响抗癫痫药物的功效和毒性
肠道微生物含有丰富的药物代谢酶,可能会影响其药理学,导致药物功效和毒性存在人群差异。
例如,氯硝西泮是一种抗惊厥和抗焦虑药物,被肠道微生物群还原和代谢,导致药物毒性。
•抗癫痫药物会改变微生物群组成
非抗生素药物会在一定程度上改变肠道微生物群。在一项涉及1197种非抗生素药物对肠道微生物群影响的大型研究中,24%的人类靶点药物在体外抑制了菌株的生长。
抗癫痫药物,如卡马西平、丙戊酸和拉莫三嗪影响肠道微生物群组成。小鼠怀孕期间丙戊酸治疗导致粪便微生物群改变,厚壁菌门增加,拟杆菌门减少,这可能与后代的自闭症谱系障碍行为有关。而拉莫三嗪可能通过抑制细菌核糖体生物合成来减少大肠杆菌的生长。
抗癫痫药物或生酮饮食治疗的患者肠道微生物群变化

Amlerova J,et al.Int J Mol Sci.2021
进一步研究抗癫痫药物与肠道微生物的关系,将有助于开发基于肠道微生物调控原理的新型抗癫痫药物。调整肠道微生物成分可以改变抗癫痫药物的代谢过程,从而提高其疗效并减少副作用。
▼
由于肠道微生物组似乎在包括癫痫在内的几种神经病理性疾病中发生了改变,使用广谱抗生素已成为恢复肠道微生物群生态的可行替代方案,在病理生理学方面显示出有益的结果。
•抗生素治疗可以阻止部分患者的癫痫发作
一些报告表明,抗生素治疗,可能有助于阻止癫痫患者和动物模型的癫痫发作。
用地塞米松(4毫克/天)和米诺环素(50毫克/天)治疗一名患有星形细胞瘤的53岁耐药性癫痫患者,使患者在长达2个月的时间里没有癫痫发作。当地塞米松或米诺环素治疗停止时,癫痫发作频率突然增加至每周7次癫痫发作。
对六名接受抗生素治疗的难治性癫痫患者的回顾性研究表明,某些抗生素可以在短期内减少癫痫发作的频率。
抗生素治疗对癫痫的影响

Mejía-Granados DM,et al.Seizure.2021
•部分抗生素可能也会诱发癫痫
抗生素可能通过干扰肠道菌群和肠-脑轴来诱导癫痫发作或降低癫痫发作频率。然而,某些抗生素也会诱发癫痫:例如,内酰胺类抗生素,包括青霉素、头孢菌素和碳青霉烯类,最有可能引起癫痫发作。
一些青霉素如第四代头孢菌素、亚胺培南和环丙沙星,可能会导致症状性癫痫发作的风险增加。
因此,在这些患者中使用抗生素时应密切监测血清水平和脑电图。未来还需要进一步研究明确各种抗生素对癫痫的具体作用和机制。
▼
最近研究了补充益生菌在癫痫发作和癫痫中的效果。一项临床试验中,由43名西班牙耐药性癫痫患者组成的队列接受了益生菌补充剂的治疗。
益生菌补充剂包括每日两剂乳杆菌(嗜酸乳杆菌、植物乳杆菌、干酪乳杆菌、瑞士乳杆菌、短乳杆菌和乳杆菌)、乳双歧杆菌和唾液链球菌,持续4个月。
•补充益生菌降低了部分癫痫患者的发作频率
研究报告称,补充益生菌可使28.9%的患者癫痫发作频率减少高达50%。其中76.9%的改善患者在停药4个月后仍保持较低的癫痫发作频率。然而,48.9%的患者对补充益生菌没有反应。
此外,益生菌反应组的生活质量得分显著改善。有趣的是,补充益生菌似乎可以使γ-氨基丁酸血清水平升高和白细胞介素-6水平降低。
•益生菌还可以减轻癫痫发作的严重程度
在戊四氮诱导的化学点燃小鼠模型中,益生菌补充组没有表现出完全点燃,并且小鼠脑组织中的γ-氨基丁酸增加,这表明补充益生菌可以显著降低癫痫发作的严重程度。
化学性点燃是用亚惊厥剂量的兴奋剂连续间隔投药逐渐诱发癫痫发作。
在治疗戊四氮诱导的小鼠癫痫发作时与生酮饮食、合生元或发酵乳杆菌(Lactobacillus fermentum MSK 408) 合用可以减少生酮饮食的副作用,而不影响其抗癫痫作用。
生酮饮食和Lactobacillus fermentum MSK 408均通过调节肠道微生物群来增加γ-氨基丁酸代谢。
补充益生菌对癫痫的影响

Mejía-Granados DM,et al.Seizure.2021
这些研究是补充益生菌治疗难治性癫痫的初步观察,还需要在更大规模的安慰剂对照试验和更严格的动物实验中进行进一步的理论验证和机制探索。
益生菌有潜力成为难治性癫痫的补充治疗,并可与生酮饮食疗法联合使用以减少副作用。
扩展阅读:如何调节肠道菌群?常见天然物质、益生菌、益生元的介绍
▼
粪便微生物群移植包括将健康捐赠者的粪便微生物群溶液注入接受者的肠道中,以恢复正常的肠道微生物群落。
粪菌移植已广泛应用于多种神经系统疾病,包括阿尔茨海默病、帕金森病、自闭症、多发性硬化症和癫痫,对这些疾病均显示出有益的作用。
•粪菌移植后癫痫发作频率和症状缓解
一些研究评估了粪菌移植对癫痫患者和动物模型的影响。对一名患有慢性病和癫痫的17岁女孩进行3次粪菌移植治疗后,癫痫发作频率得到了有益的影响。粪菌移植20个月后,患者癫痫发作完全缓解,不再需要抗癫痫药物。
此外,衡量这种疾病严重程度的疾病活动指数也从361点显著下降至131点。粪菌移植治疗前(约70天)失调的月经周期在粪菌移植治疗后(约30天)也得到恢复。
粪菌移植对癫痫的影响

编辑
Mejía-Granados DM,et al.Seizure.2021
扩展阅读:粪菌移植——一种治疗人体疾病的新型疗法
注意
尽管有证据表明健康的粪菌移植对癫痫有益,但仍有一些问题需要在进一步的研究中解决。例如,粪菌移植发挥其抗癫痫作用的机制、其有益作用的持久性以及参与此类结果的细菌属或门。
此外,粪菌移植可能会破坏基线微生物群多样性,导致对多种有害微生物的定植抵抗力崩溃。因此,在大规模临床应用之前,还需要更长期的随访研究来确定粪菌移植对癫痫患者的有效性和安全性。
随着研究的深入,肠道微生物群与癫痫之间千丝万缕的联系逐渐被揭示,人们对它们的功能有了更深入的了解。
目前,一些临床研究已证实难治性癫痫患者、药物敏感患者和健康对照者的肠道菌群存在差异。同时,肠道菌群还在癫痫的治疗中影响生酮饮食、抗癫痫药物等的治疗效果。
总体而言,未来微生物组特异性治疗可能是治疗癫痫的有效选择。发现肠道微生物群和癫痫之间的关系将有助于我们更好地了解癫痫的发病机制,从而提高癫痫患者的生活质量。
相关阅读:
环境污染物通过肠脑轴影响心理健康,精神益生菌或将发挥重要作用
主要参考文献
Wang Y, Zhuo Z, Wang H. Epilepsy, gut microbiota, and circadian rhythm. Front Neurol. 2023 May 18;14:1157358.
Iannone LF, Gómez-Eguílaz M, De Caro C. Gut microbiota manipulation as an epilepsy treatment. Neurobiol Dis. 2022 Nov;174:105897.
Liu T, Jia F, Guo Y, Wang Q, Zhang X, Chang F, Xie Y. Altered intestinal microbiota composition with epilepsy and concomitant diarrhea and potential indicator biomarkers in infants. Front Microbiol. 2023 Jan 11;13:1081591.
Zeng Y, Cao S, Yang H. Roles of gut microbiome in epilepsy risk: A Mendelian randomization study. Front Microbiol. 2023 Feb 27;14:1115014.
Ułamek-Kozioł M, Czuczwar SJ, Januszewski S, Pluta R. Ketogenic Diet and Epilepsy. Nutrients. 2019 Oct 18;11(10):2510.
Dahlin M, Prast-Nielsen S. The gut microbiome and epilepsy. EBioMedicine. 2019 Jun;44:741-746.
Mejía-Granados DM, Villasana-Salazar B, Lozano-García L, Cavalheiro EA, Striano P. Gut-microbiota-directed strategies to treat epilepsy: clinical and experimental evidence. Seizure. 2021 Aug;90:80-92.
Ding M, Lang Y, Shu H, Shao J, Cui L. Microbiota-Gut-Brain Axis and Epilepsy: A Review on Mechanisms and Potential Therapeutics. Front Immunol. 2021 Oct 11;12:742449.

谷禾健康

已有研究证明宿主微生物在癌症预防和治疗反应中的关键作用,了解宿主微生物和癌症之间的相互作用,可以推动癌症诊断和微生物治疗(即用微生物作为药物)。
然而肿瘤内微生物组数据通常是复杂的,想要厘清相互关系也是极为困难的,有许多可能导致虚假关联的混杂因素,还需要足够多样本的大型数据集进行分析。
近期,研究人员开发了一个生物信息学工具——MEGA,这是一个基于深度学习的Python包,用于识别癌症相关的肿瘤内微生物。
该模型使用ORIEN(Oncology Research Information Exchange Network)的RNA-seq测序数据进行训练,以识别与12种人类癌症最相关的微生物。本文我们来详细了解一下。
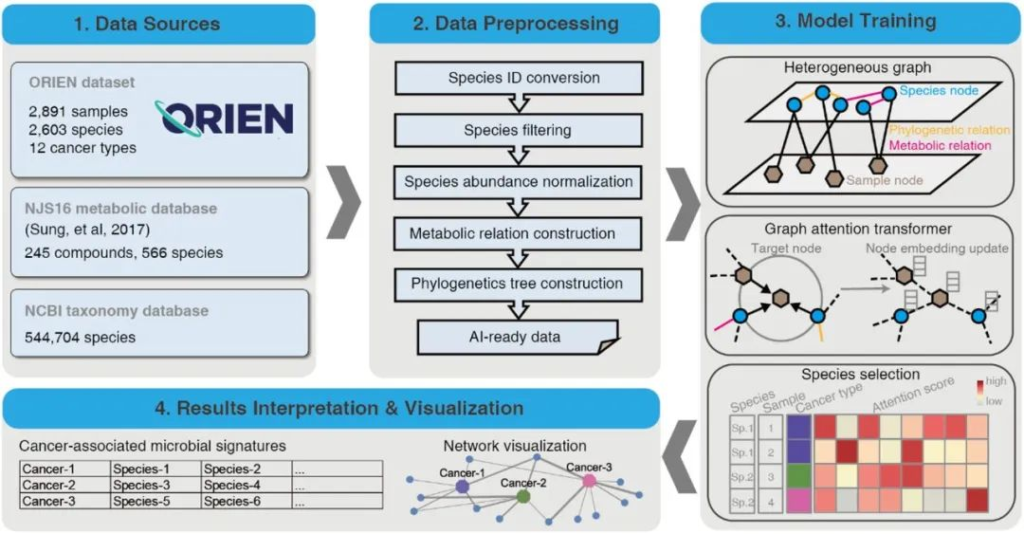
1. 使用ORIEN数据集和两个数据库依赖项作为数据源
ORIEN数据集包括2891份样本中的2603个种水平物种以及对应的癌症类型元数据;
NJS16代谢数据库是通过对大量文献的整理和分析构建的,旨在提供关于微生物种间相互作用和代谢活动的信息,内含约570种微生物物种和3种人类细胞类型;
NCBI数据库,从中提取ORIEN数据集中物种的系统发育关系。
2. 生成用于图神经网络训练的人工智能准备数据
准备数据包括筛选后的数据归一化的相对丰度矩阵(相对丰度大于0.1%的物种)、代谢关系网络和系统发育关系网络。
3. 深度学习模型训练后,根据样本水平上每个物种的attention scores,选择与癌症相关的微生物特征
利用之前开发的基于PyTorch(v1.4.0)实现的heterogeneous graph转换模型进行训练。
heterogeneous graph转换模型是一种用于处理不同类型节点和它们之间关系的模型,在这个场景中,节点代表了微生物物种和样本,而边表示它们之间的关系。
为了训练这个模型,使用了两个自编码器来生成每个节点的密集向量,每个向量都是256维,这些向量作为深度学习模型的输入值,用于学习样本和物种之间的关系。
训练中,使用Adam优化器,并设置学习率为0.003,其他超参数的默认设置为:
Focal Loss函数用于量化预测的癌症类型标签与真实标签之间的差异。当评估指标连续5个epoch没有改善时,学习率会降低0.5倍。最终生成attention score值作为重要的训练结果。
这个分数表示源节点对目标节点的重要性。
较高的分数表示该物种在样本中具有较高的代表性,然后通过计算具有较高分数的每个物种在癌症类型中的样本数量,确定与癌症类型显著相关的物种,p值小于0.05的物种被认为与癌症类型显著相关。
4. 最终识别出的与癌症相关的微生物群落结果将输出为以tab分隔的文件,可用于后续的可视化操作
结果可以以UpSet图进行展示,也可以通过Cytoscape软件生成网络图。
MEGA的Github地址:
MEGA在ORIEN数据集中鉴定出了来自12种癌症类型的73种独特的微生物群落。
分析结果显示,在12种癌症类型的微生物群落中有15个物种是共有的。而在结肠腺癌、直肠腺癌和其它结直肠癌中,有8种物种是独属于它们的。
下图展示了已确定的物种和癌症类型的分布。
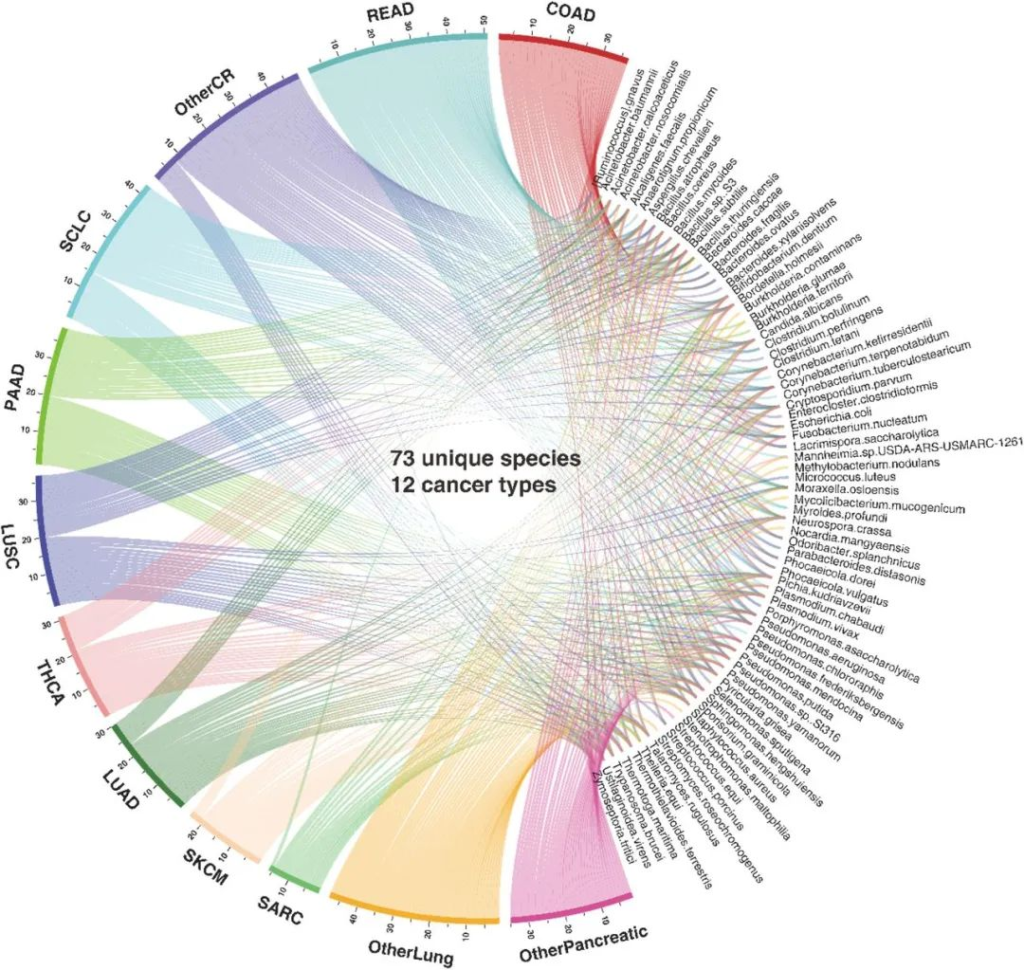
条带的宽度指示该癌症中检测到的物种总数,并且与各自存在的物种相连。
COAD(结肠腺癌); 肺腺癌(LUAD); LUSC(肺鳞状细胞癌);
OtherCR(未指明的其他结直肠癌类型);
OtherLung(未指明的其他肺癌类型);
OtherPancreatic(未指明的其他胰腺癌类型);
胰腺腺癌(PAAD); READ(直肠腺癌); SARC(肉瘤);
小细胞肺癌(SCLC); 皮肤黑色素瘤(SKCM);
THCA(甲状腺癌)
为了展示MEGA的数据分析和解释能力,研究人员重点研究了结肠腺癌和甲状腺癌的案例。
分析发现,有8种物种是只在结直肠相关癌症类型中共享的,分别为:
其中的Bacteroides fragilis, Ruminococcus gnavus,Bacteroides ovatus 这3个物种与之前的验证实验结果一致,这表明MEGA仅通过整合代谢和系统发育关系就成功鉴别出了这些物种。
通过整合物种与代谢之间的关系,发现在结肠腺癌中,Fusobacterium nucleatum具有较强的代表性,而在小鼠模型的研究中,它通过改变黏膜微生物群和结肠转录组促进了结直肠癌的进展。
Ruminococcus gnavus与结肠腺癌的相关性较弱,其丰度与结直肠癌肿瘤数量和疾病评分呈显著负相关。
然而,这两种菌Fusobacterium nucleatum和Ruminococcus gnavus 共享了同一种代谢物——N- Acetylneuraminate acid,它参与的细胞间的黏附事件在结直肠癌的血管生成、转移和生长控制中可能起着重要作用。
Ruminococcus gnavus还与Bacteroides fragilis 共享了同一种代谢物L-Fucose,而最近的研究发现,Bacteroides fragilis毒素可能有助于结直肠癌的形成。
在甲状腺癌中,发现 Pseudomonas aeruginosa和Staphylococcus aureus与代谢物甘油三酯相关。而最近的研究表明,甘油三酯水平可能与甲状腺癌的发生风险相关。
通过整合物种的系统发育关系,能够发现与癌症相关性较弱的物种之间的关联。
例如,Bacteroides ovatus(卵形拟杆菌)在以往的研究被证明是结肠癌中的代表物种之一,但在MEGA的分析中,它与结肠腺癌的关联较弱,很有可能被遗漏,但通过分析Bacteroides fragilis的系统发育,依旧被识别出了。
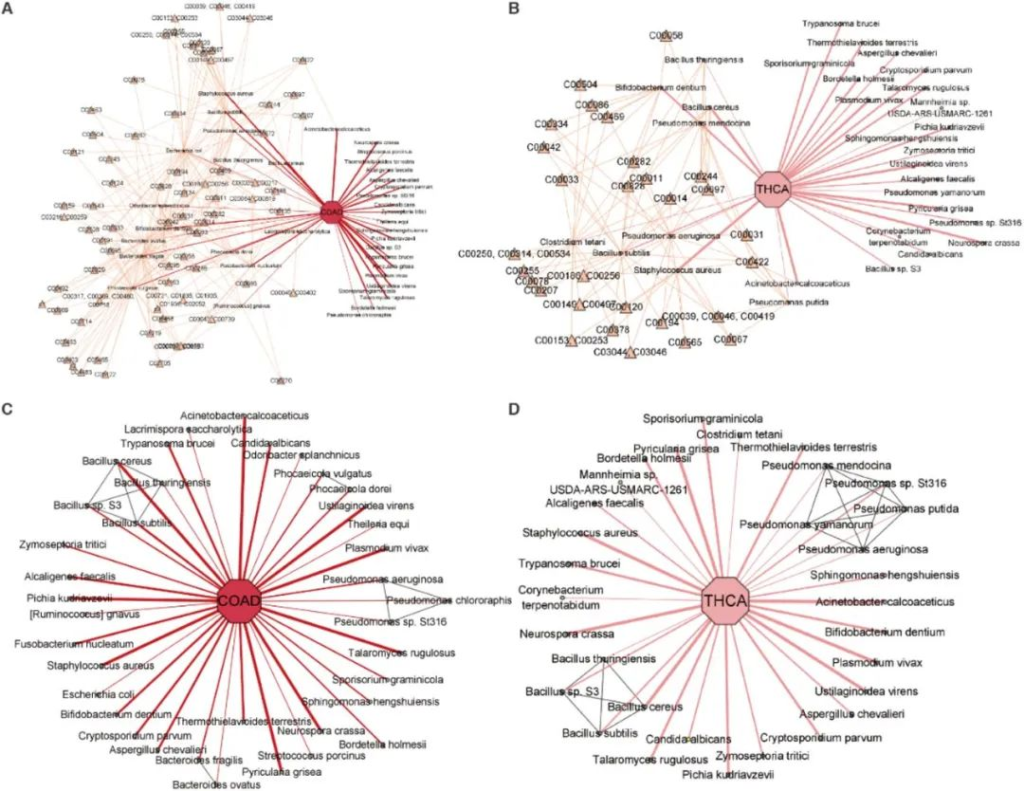
图为结肠腺癌和甲状腺癌中已鉴定微生物群落的网络可视化。圆形节点指代微生物物种,黄色三角形节点指代代谢物,线条厚度表示物种与癌症之间关系的强度,灰色线条表示系统发育关系。
A) 结肠腺癌相关微生物与代谢物间的关联。
B) 甲状腺癌相关微生物与代谢物间的关联。
C) 结肠腺癌相关微生物与系统发育关系的关联。
D) 甲状腺癌相关微生物与系统发育关系的关联。
MEGA的开发代表着在识别和解读与癌症相关的肿瘤内微生物方面,迈出了重要一步。
研究中提出的深度学习模型可以识别与12种不同癌症类型相关的微生物特征,并将相关性的强弱通过attention scores进行了量化,通过网络图直观展示,从而可以更全面、更细致地理解相互关系。
此外,研究人员认为将MEGA应用于单细胞RNA-seq数据,可以更详细地了解微生物群落与肿瘤细胞在细胞水平上的相互作用,从而为基于肿瘤内微生物多样性的肿瘤异质性表征提供新的视角,也可能为癌症的治疗干预提供新的靶点。
参考文献:
Wang C, Ma A, McNutt ME, Hoyd R, Wheeler CE, Robinson LA, Chan CHF, Zakharia Y, Dodd RD, Ulrich CM, Hardikar S, Churchman ML, Tarhini AA, Singer EA, Ikeguchi AP, McCarter MD, Denko N, Tinoco G, Husain M, Jin N, Osman AEG, Eljilany I, Tan AC, Coleman SS, Denko L, Riedlinger G, Schneider BP, Spakowicz D, Ma Q. A bioinformatics tool for identifying intratumoral microbes from the ORIEN dataset. bioRxiv [Preprint]. 2023 May 24:2023.05.24.541982.

谷禾健康

在我们日常的护肤和美容过程中,我们经常听到关于皮肤的各种话题,从保湿到抗衰老,从痘痘到过敏…
随着科学的不断进步和技术的发展,人们开始逐渐发现,皮肤上隐藏着一个神秘的世界——皮肤微生物群。它在维护我们的皮肤健康方面扮演着举足轻重的角色。
皮肤微生物群由各种细菌、真菌等微生物组成,它们聚集在毛囊、汗腺、皮脂腺等地方,形成一个庞大的生态系统。它们在皮肤表面形成了一道坚固的屏障,阻止了有害菌的入侵。除了提供保护作用外,皮肤微生物群还参与调节角质层的代谢,协助皮肤的水分平衡,并对免疫系统起到了重要的调节作用。
皮肤微生物群的平衡易受到许多因素的干扰。个人的生活方式(过度清洁)、饮食习惯(高糖高脂的饮食)等可能直接影响皮肤微生物的结构和组成,进而引发皮肤问题。外界环境中的污染物、紫外线辐射、气候变化等也会对皮肤微生物群产生影响,从而引发皮肤干燥、过敏、炎症等问题。肠道微生物群的失衡可能导致身体免疫系统的异常反应,进而影响皮肤的健康。
了解皮肤微生物群的特征及其与其他因素的相互关系,对于制定精确的治疗和护肤策略具有重要意义。

图源:Getty Images
本文我们来了解一下整个生命中皮肤微生物组,探讨皮肤微生物群的功能,包括保护屏障、免疫调节等,阐述了皮肤微生物与宿主的相互关系,微生物群在皮肤病中的影响,同时也介绍一些基于微生物群的保持皮肤健康的方法,以及皮肤微生物群在不同领域的应用前景和潜力。
-本文主要内容如下-
编辑
-正文-
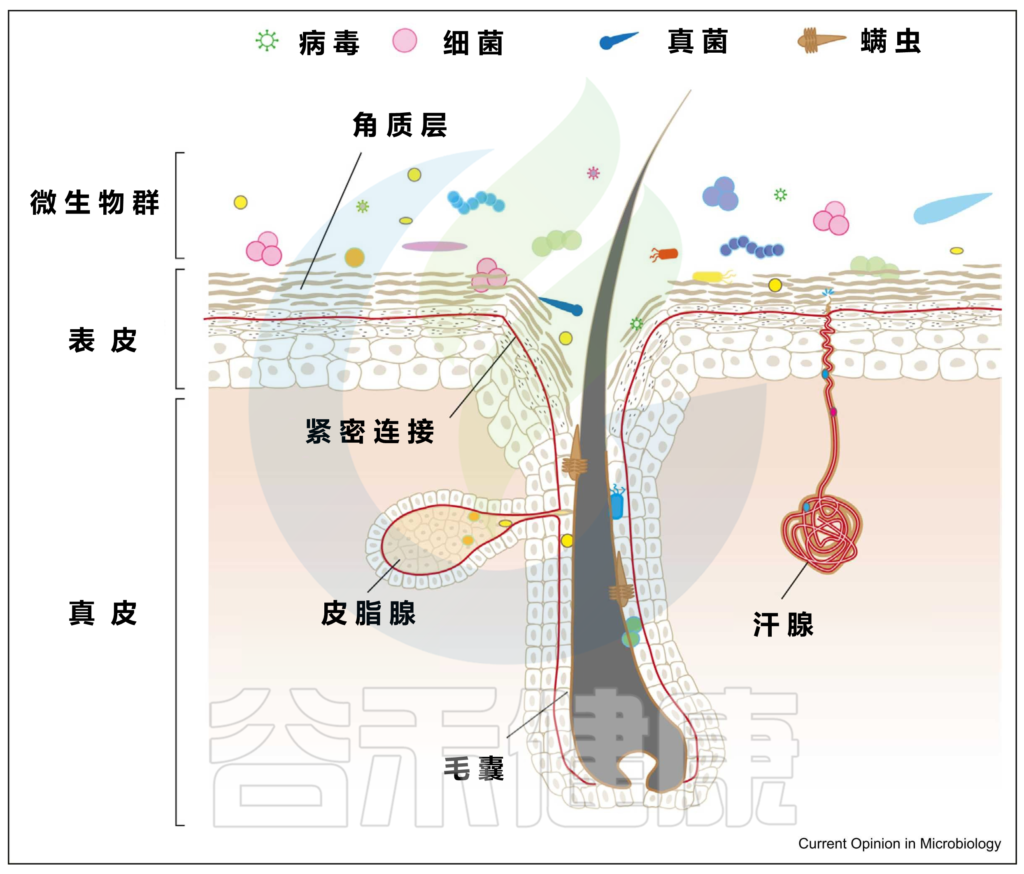
皮肤是暴露于外界环境的重要器官,它可以调节体温、防止感染、保护内脏器官等。
皮肤表面是一个酸性、富含盐分、干燥、有氧的环境,而形成毛囊皮脂腺单位的内陷则相对厌氧,甚至富含脂质。
▼
皮肤是身体最大的器官。一个成年人的皮肤平均面积约为1.5-2.0平方米。皮肤除了作为外界与生物体之间的物理和化学屏障的功能外,还作为许多微生物的栖息地。通常,一个人的皮肤上有大约 1000 种细菌。
皮肤微生物组由多种微生物组成,包括细菌、真菌、病毒、螨虫等。
皮肤微生物群通过参与皮肤中发生的基本生理过程,对于维持皮肤屏障、抵御病原体入侵、增强免疫系统、分解天然产物等方面发挥着重要作用。
皮肤和微生物群的结构

编辑
上图可以看到,皮肤由两层组成,即真皮和表皮,具有不同的、专门的生态位或微环境。
▼
皮肤微生物成员和功能可能因皮肤的各种特殊生态位或微环境而异:
具有高密度的毛囊和皮脂腺,例如面部(额头、鼻翼、耳后)、胸部和背部。通常呈高酸性,其特点是细菌可以消耗脂质,需要或可以在厌氧条件下生存,例如:
Corynebacterium minutissimum(微小棒状杆菌)
Cutibacterium
肘部,膝盖,生殖器,肚脐,腹股沟等部位。温和的酸性环境,温度和湿度较高,导致体味的细菌喜欢在这样的环境生活,例如:
Corynebacterium (棒状杆菌)
Staphylococcus (葡萄球菌)
例如手掌等部位。生物量最低,但细菌多样性却最高。
最不稳定的是足部微生物群。足部皮肤上细菌的平均数量从足背表面的103CFU/cm2到第四趾裂处的107CFU/cm2不等。
脚跟底部的真菌居多, 例如:
Malassezia(马拉色菌属)
Aspergillus (曲霉属)
Cryptococcus (隐球菌属)
Rhodotorula (红酵母属)
Epicoccum (附球菌属)
▼
在一生中,随着个人皮肤免疫系统的成熟和激素驱动汗液和皮脂腺的发育,皮肤的生理机能会发生变化。这些变化与突出的皮肤微生物群的相对丰度的变化和整体微生物群落多样性的变化有关。
作为与环境的直接接触面,皮肤也不断地与我们周围的地方和人分享微生物。下图总结了人类一生中皮肤微生物组的变化,并强调了在与年龄相关的关键阶段皮肤微生物组的破坏会影响疾病发展的风险。
皮肤及其微生物组在整个生命周期中的动态平衡
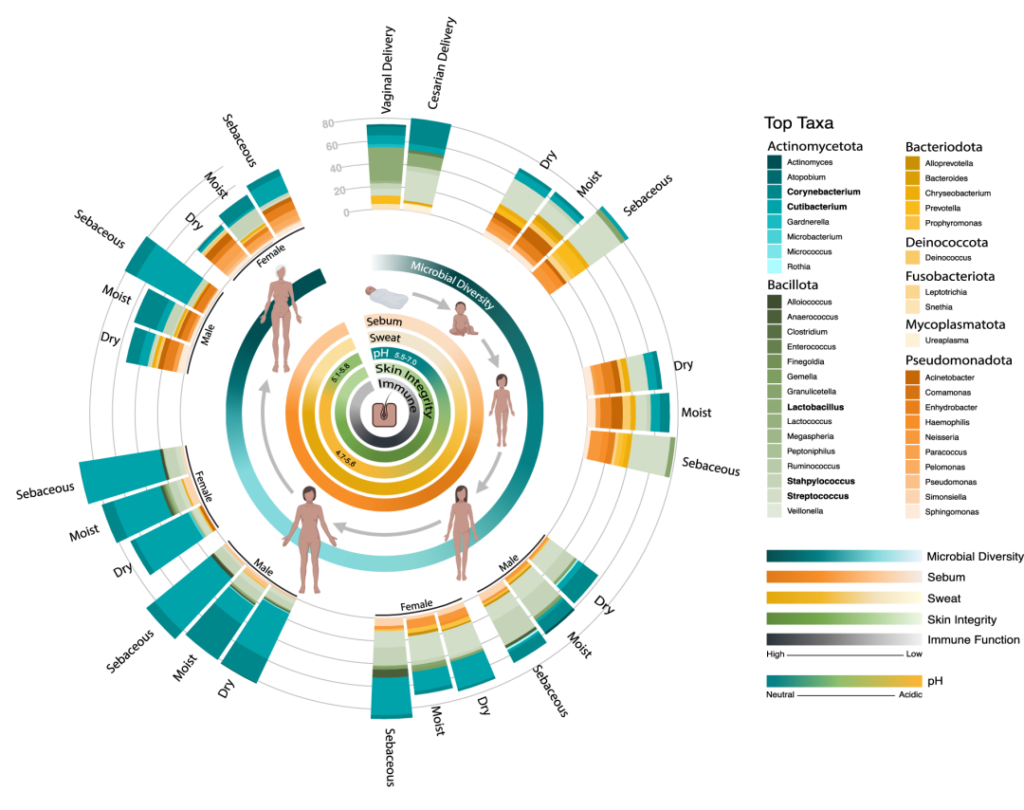
doi.org/10.1042/BST20220216
内圈代表相对微生物多样性、皮脂生成、汗液生成、表面pH值、皮肤完整性和终身免疫功能。微生物组16s测序数据显示了每组前 10 个微生物类群的平均相对丰度。
出生
皮肤微生物组在出生时就已开始定植,并受到多种因素的影响,如:分娩方式、母亲微生物群、抗生素治疗、卫生条件、营养缺乏、住房、动物/宠物接触和环境暴露等。
阴道分娩新生儿的皮肤微生物组以阴道相关菌群为主,主要是乳杆菌,普雷沃氏菌,白色念珠菌。
剖宫产新生儿的微生物群中含有母体皮肤相关微生物,包括葡萄球菌、链球菌、棒状杆菌,Cutibacterium等。
这些初始群落是短暂的,不过物种定殖的顺序和时间会影响菌株后面的相互作用。这些优先效应可以塑造未来的菌群结构,并对皮肤、微生物组和整体健康产生长期影响。
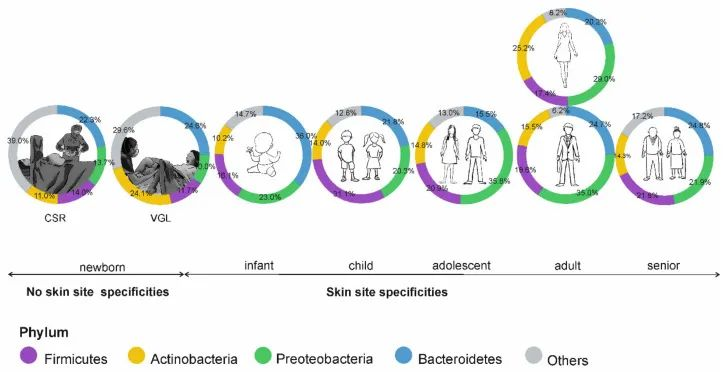
doi: 10.3390/microorganisms9030543
皮肤微生物组的年龄依赖性特异性;CSR剖宫产,VGL阴道分娩。
婴儿期和儿童期
婴儿期,最初接触微生物会促进免疫发育,并通过促进角质形成细胞的适当分化和表皮修复来加强皮肤屏障。
新生儿和婴儿皮肤含水量更高,pH值更高,皮脂生成受到抑制,表皮更新更快,抗菌性能更强。在3-6个月内,微生物分类群与皮肤代谢功能(如脂质生成和pH)之间的联系建立起来。
早期皮脂生成减少与棒状杆菌、Cutibacterium、马拉色菌丰度降低,葡萄球菌、链球菌增加以及以念珠菌为主的真菌生物群落有关。
随着儿童年龄的增长,皮肤进一步酸化并产生更多的皮脂脂质,这促使了酸敏感链球菌(acid-sensitive streptococci)的逐渐减少和整体群落多样性的增加。
在整个儿童时期,皮肤会继续携带来自照顾者的不同微生物群。然而随着年龄的增长,年龄较大的孩子具有更高的皮肤微生物多样性,以及更多来自农村或城市环境的微生物,母婴微生物组之间的相似性逐渐下降。
一旦这种平衡破坏,则可能与更大的炎症有关,并可能增加儿童患特应性皮炎和过敏的风险。
青春发育期
青春期标志着皮肤微生物群的下一个重大转变。驱动身体和性发育的激素也直接促进皮肤的结构和功能变化,如皮脂和顶泌汗液的产生,导致了随后微生物组成的变化。
横断面和纵向研究都表明,Tanner阶段的皮肤微生物组组成发生了明显变化。与V期的年轻人相比,I期的儿童链球菌、拟杆菌和假单胞菌的相对丰度更高,细菌和真菌的多样性也更高。
在年轻成人的皮肤微生物组中主要存在亲脂菌群,如棒状杆菌、痤疮角质杆菌和马拉色菌。与皮脂生成和痤疮相关。
青春期早期和晚期皮肤、微生物组和体味产生的差异
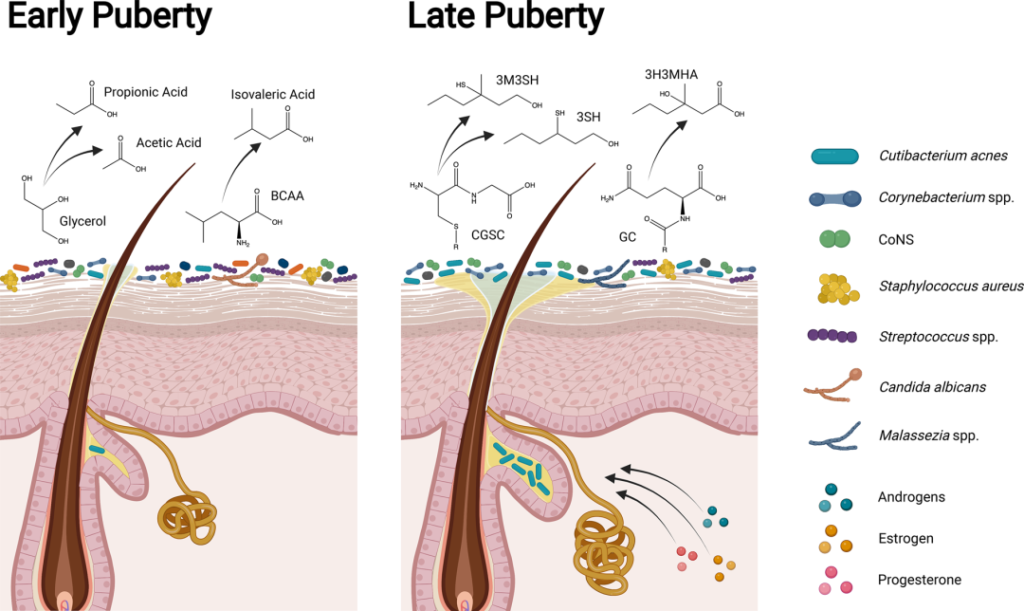
doi.org/10.1042/BST20220216
在儿童期和青春期早期(Tanner阶段I至II),皮肤微生物组高度多样化,体味与凝固酶阴性葡萄球菌属(如表皮葡萄球菌和人型葡萄球菌)产生挥发性脂肪酸(如丙酸、乙酸和异戊酸;酸味)和硫(臭鸡蛋味)有关。随着青春期的发展,类固醇激素促进皮脂腺和顶泌汗腺的发育,改变皮脂中的脂质类型,增强皮肤屏障。
在青春期后期(Tanner IV至V期),脂质生成增加和脂质含量改变与亲脂性类群主导的皮肤微生物组有关。虽然汗液和皮脂成分仍会分解为挥发性脂肪酸,但年轻人的体味与棒状杆菌属更为相关。皮脂和汗液成分代谢为硫烷基烷醇(如3-硫烷基己醇和3-甲基-3-磺基己醇;洋葱味)和挥发性有机化合物(如3-羟基-3-甲基己酸;类孜然味)。
成年期
成年皮肤微生物组在几年内是稳定的。微生物-微生物相互作用网络、持久的成人皮肤生理学和有弹性的皮肤免疫力维持了平衡的成人皮肤微生物群。
成年皮肤微生物群以角质杆菌、棒状杆菌、葡萄球菌、马拉色菌为主。
一旦成年后,成熟和持久的皮肤生理机能,会促进皮脂的产生、汗液成分和表面pH值的一致性,这些共同提供了稳定的身体部位微环境和营养库。免疫系统那时候也成熟了,这些内在特征使皮肤上的大部分微生物群能够在日常环境变化的情况下持续存在。
年龄增长
随着年龄的增长,皮肤会发生明显的变化,包括胶原蛋白合成下降、细胞外基质断裂和皮肤细胞再生减少,皮肤皱纹也就出现了。
随着皮肤屏障的变化,它可能会失去保持水分的能力,导致天然保湿因子(NFM)产生的补偿性增加。NMFs既能吸收水分,又能促进细菌增殖和粘附在皮肤上。随后,NMFs的增加与许多分类群的更丰富有关,如棒状杆菌、微球菌、链球菌、厌氧球菌,同时角质杆菌的减少。皮肤微生物多样性也广泛增加。
女性更年期后皮脂细胞面积和皮脂生成的减少,与角质杆菌的减少以及棒状杆菌、链球菌、不动杆菌和棒状杆菌丰度的增加有关。
在男性中,皮脂分泌下降的速度明显较慢,因此随着年龄的增长,它们保持着更丰富的角质杆菌。
随着年龄增长,免疫系统功能也会慢慢下降。老年人维持低度炎症状态,免疫防御受损和潜在致病菌(如β-溶血性链球菌)增加,皮肤感染的风险大幅增加,难以清除感染。
衰老会改变皮肤结构、功能和微生物定植
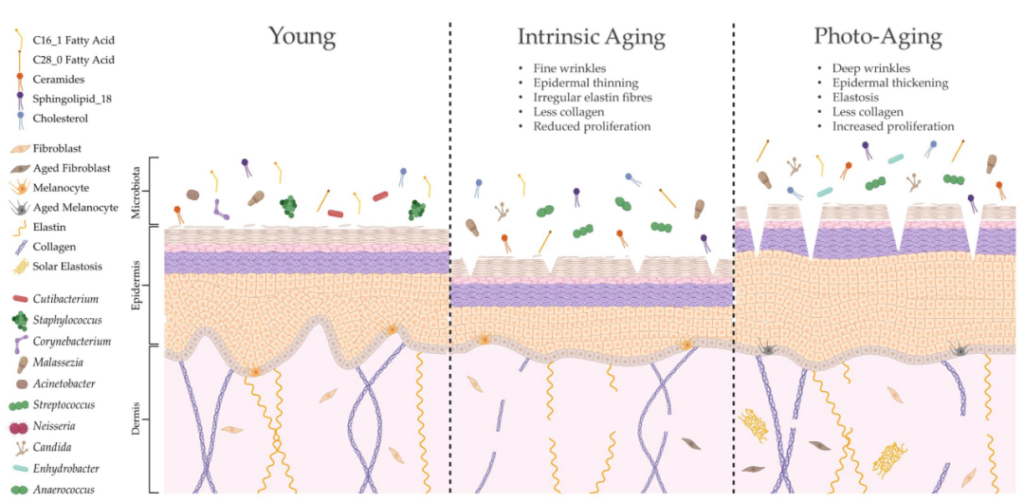
doi.org/10.3390/ijms24043950
内在衰老和光老化会导致皮肤结构和生理的不同变化,导致微生物组成的显著变化。这种改变的皮肤微生物组可能是由脂质成分的特定修饰形成的,这可能进一步导致与年龄相关的皮肤异常。
以上是皮肤微生物组在整个生命周期中的变化情况,那么皮肤微生物组是稳定的吗?它有可能受到哪些因素的影响?我们来看下一章节。
持续暴露于各种外在和内在因素会影响这个皮肤生态系统的平衡。
皮肤结构决定了皮肤微生物组的组成,个体特征取决于宿主的年龄、性别和健康状况等。个人生活方式和所处环境也会影响皮肤上微生物的数量和组成。微生物组的组成可能会随着宿主健康状况的恶化、衰老、甚至居住或职业的改变而改变。皮肤的物理和化学特性影响特定微生物群的优势、它们的比例以及它们之间的相互关系。
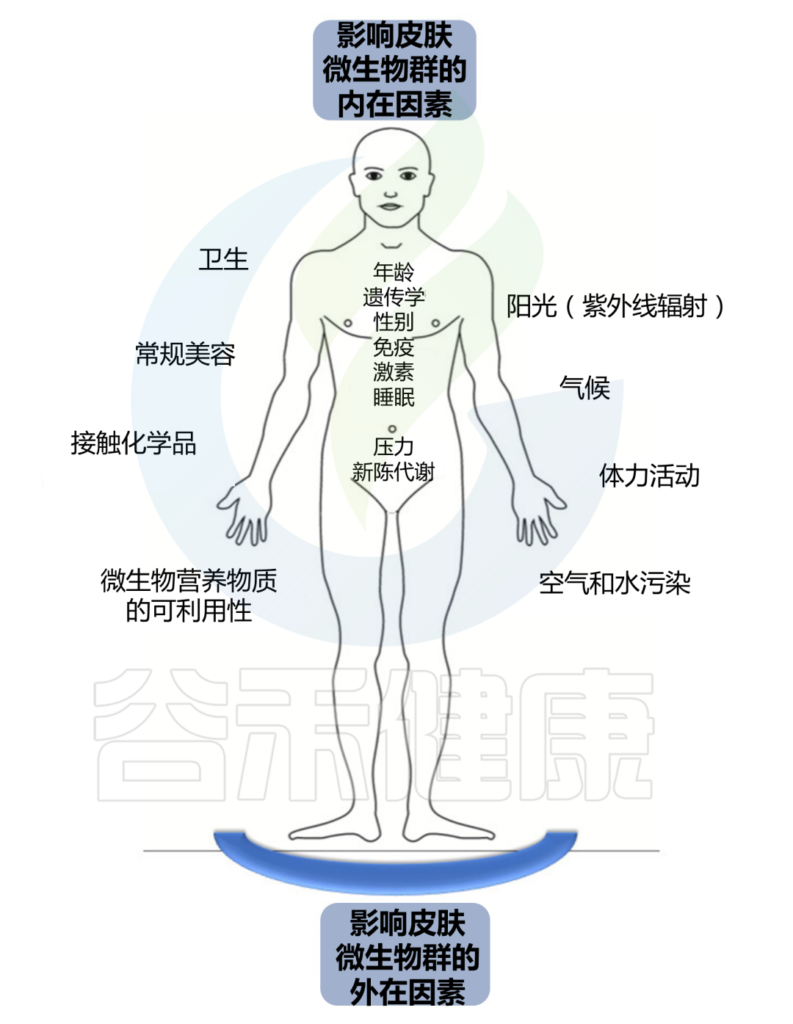
Skowron K, et al., Microorganisms. 2021
我们分为外在因素和内在因素两大块。
外 在 因 素
▼
紫外线辐射,对皮肤细胞有破坏和抗菌作用。大多数与年龄相关的皮肤病是由光老化引起的。皮肤光老化表现为:皱纹、局部色素沉着、毛细血管扩张、干燥和粗糙。这些与表皮和真皮中各种细胞和组织的病理生理变化有关。
皱纹作为光老化最明显的临床特征,主要是由于真皮成纤维细胞减少,以及胶原蛋白和弹性蛋白合成速度减慢但分解速度加快所致。皮肤光老化不仅影响美观,还会损害正常的皮肤屏障功能,增加皮肤炎症性疾病甚至恶性肿瘤的风险。
紫外线:破坏作用
皮肤强烈暴露于紫外线辐射可能会增加其感染的易感性,并加剧相关症状,例如单纯疱疹病毒。
紫外线辐射也可能影响皮肤微生物群的遗传变异,扰乱健康的微生物组结构。
皮肤暴露在紫外线下导致蓝藻菌数量总体增加,乳酸杆菌科和假单胞菌科数量减少。
紫外线:抗菌作用
阳光和紫外线也有效抑制了金黄色葡萄球菌和痤疮丙酸杆菌的生长。痤疮丙酸杆菌数量的减少与卟啉的产生减少有关。
微生物也可以抗紫外线辐射
皮肤微生物组对太阳辐射和紫外线辐射的抵抗力各不相同。一些细菌可以保护皮肤免受紫外线辐射的破坏。皮肤表面的蓝细菌和乳酸杆菌降低了色素沉着的强度和光老化相关损伤的发生。
共生马拉色菌对紫外线辐射表现出高度敏感性,尽管它们有能力合成类似紫外线过滤器的物质——pityriacitrin。
关于紫外线辐射对皮肤微生物群的影响详见谷禾之前的文章:
▼
化学空气污染物,包括 O3、颗粒物(PM 2.5:≤2.5 μm;PM 10:≤10 μm)、挥发性有机化合物和二氧化氮(NO2)等温室气体,是已知的外部暴露组的组成部分,增加过敏性疾病发生和恶化的风险。
空气污染物N2O干扰共生微生物,在对头葡萄球菌和结核棒状杆菌的负面影响大于对金黄色葡萄球菌的负面影响的情况下,有可能发生微生态失调。
烧烤烟雾中较多的成分——多环芳烃,在推动皮肤微生物群分化成不同类型中的作用
多环芳烃来源可以分为自然源和人为源:自然源指火山爆发、森林火灾等自然现象释放到环境介质中的;人为源则是由于人类生产生活活动中化石燃料(煤、油等)不充分燃烧造成的。
我们生活中例如室内外烧烤烟雾中存在较多,在烧烤的过程中,燃料的不完全燃烧或肉类食品脂肪的高温热解均可以产生大量多环芳烃类化合物。
一项研究揭示了多环芳烃暴露与皮肤微生物组分化成不同皮肤类型之间的关联。
皮肤微生物组分化为两种细胞类型(cutotype 1 和 cutotype2)。Cutotype 2与45岁以下受试者的皮肤干燥和色素沉着过度有关。多环芳烃暴露量高与皮肤干燥和cutotype 2有关,cutotype 2富含具有潜在生物降解功能的物种,相关网络结构完整性降低。
cutotype 1中精氨酸生物合成途径中的优势类群、关键功能基因和代谢产物之间的正相关性表明,来自细菌的精氨酸有助于合成聚丝蛋白衍生的天然保湿因子(NMFs),为皮肤提供水合作用,并可解释正常皮肤表型。
这项研究揭示了多环芳烃在推动皮肤微生物群分化成不同类型中的作用,这些类型在分类学和代谢功能上存在广泛的差异,并可能随后导致皮肤与微生物之间的相互作用变化,从而影响人体皮肤的健康。
也就是说:暴露于空气污染后皮肤微生物组组成的变化,可能导致皮肤干燥和炎症的恶化。
▼
全球变暖和极端天气事件等气候变化相关因素,会影响皮肤维持体内平衡的能力,在许多皮肤疾病的发病机制中发挥作用。
全球变暖可能破坏皮肤微生物组
温度和湿度的升高与皮肤上细菌的总体生长有关。
较高的气温和金黄色葡萄球菌的生长之间可能存在关联:
在一项以人群为基础的每月皮肤和软组织感染(SSTI)发病率研究中,SSTI 的时间变化与平均温度和比湿度显着相关。在美国 SSTI 的回顾性分析中(n = 616,375),在气温较高的南部地区,社区获得性耐甲氧西林金黄色葡萄球菌的感染率较高。
温度每升高1˚C,皮脂的产生就会增加10%,这反过来可能会增加微生物的生长,包括角质杆菌和马拉色菌。
极端天气可能引发皮肤病
气候变化导致极端天气事件发生的频率不断增加,包括热浪、干旱、野火、暴雨、洪水和飓风。
例如,洪水的最初影响阶段,经常有创伤与继发性伤口感染的相关风险,包括:嗜水气单胞菌( Aeromonas hydrophila)、创伤弧菌、副溶血性弧菌、Burkholderia pseudomallei等感染。
除了对皮肤病的直接影响外,极端天气事件的额外影响还包括冲突加剧、被迫迁移、心理健康恶化以及传染病的更大传播,所有这些都进一步增加了皮肤病的风险。
▼
农村和城市居民皮肤微生物组的差异,可能与不同程度地接触农业或畜牧业中的土壤、水和生物质中的微生物有关。即使皮肤与土壤和植物材料的短期接触,也会导致手部微生物组的变化以及酸杆菌Acidobacteria和拟杆菌的丰度增加。
在芬兰进行的一项研究结果表明,城市和乡村环境对 1-4 岁儿童的皮肤微生物群有显著影响。这种效应在青少年(14岁)中消失,这直接归因于该年龄段的户外活动时间有限。然而,在其他国家获得的研究结果并未证实这种趋势,表明其他因素(文化差异)也影响皮肤微生物组。
角质杆菌属在农村成年人的背部皮肤上更常见,而Trabulsiella属细菌在城市居民的手和前臂上更丰富。
农村环境的特点是微生物多样性很高
棒状杆菌和角质杆菌属数量的减少,以及假单胞菌和不动杆菌数量的增加,主要发生在与各种农场动物接触的农场工人身上。
封闭空间环境中的微生物有城市和工业区的特点
随着室内城市化的发展,与人类皮肤相关的真菌和细菌的相对丰度也在增加。此外,潜在致病真菌的数量也在增加,包括曲霉菌、马拉色菌、念珠菌等。
由于卫生习惯和西方生活方式,皮肤的细菌多样性降低。许多皮肤共生菌(如表皮葡萄球菌、乳酸杆菌、伯克霍尔德菌Burkholderis、痤疮梭菌)消失,取而代之的是葡萄球菌、棒状杆菌、角质杆菌(Cutibacterium)和微球菌Micrococcus。
▼
不同的动物物种含有独特的微生物群,与动物的持续接触会影响健康人皮肤细菌群落的组成和多样性。例如家养狗和家庭主人共享微生物群。菌群结构受季节的影响,但不受狗的性别、年龄、品种或皮毛类型的影响。
宠物肠道菌群与主人的肠道菌群也会产生关联,详见:
▼
皮肤与衣服的长时间接触也很重要,这会导致微生物的传播,并形成所谓的纺织品和挥发性微生物组。反过来,织物微生物组的组成会受到洗涤和干燥的影响。附着在纤维上的微生物可以利用污垢或皮脂化合物作为基质,并产生挥发性物质作为副产品,从而产生难闻的气味。
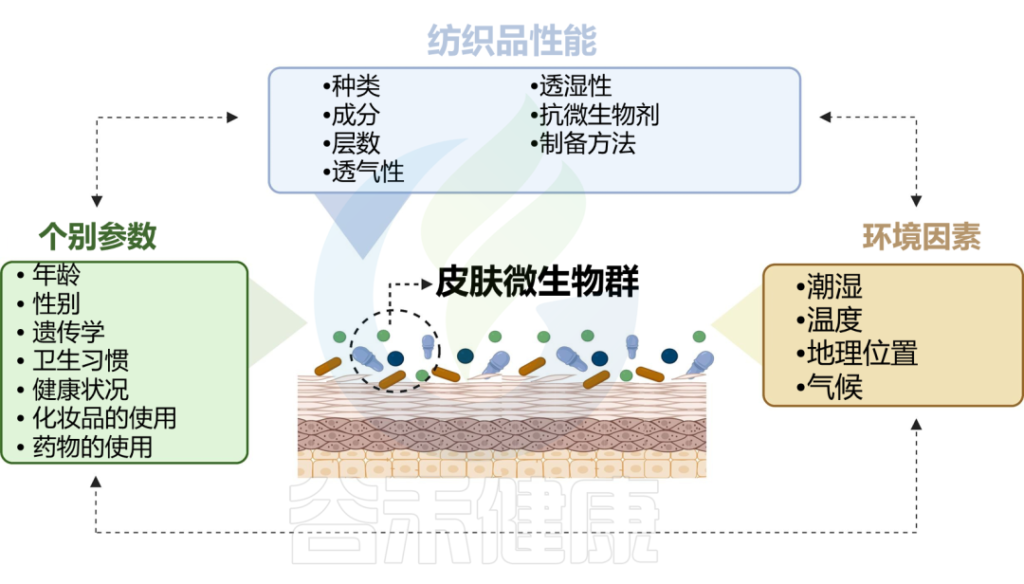
doi.org/10.1016/j.ejpb.2023.05.004
纺织纤维的性质可以直接影响微生物的附着、生长和定植
葡萄球菌属在几乎所有纺织纤维中显示出显着的固定性。Staphylococcus hominis对棉花的亲和力较高,在粘胶纤维和羊毛中不生长。
羊毛促进了许多菌群生长,包括表皮葡萄球菌、Enhydrobacter、角质杆菌、微球菌属。
聚酯为角质杆菌、Enhydrbecter、微球菌属提供了最大的生长环境。
棒状杆菌属无法在棉花、丙烯酸、羊毛、粘胶、尼龙、羊毛和聚酯上进行竞争,这解释了只有少量棒状杆菌属才能从破旧的衣服中分离出来。
合成纤维由于其疏水性和较差的吸附能力,通常抵抗微生物定植。
天然纤维更容易受到微生物定植的影响,因为它们具有高保湿性能,并且它们的聚合物键更容易被微生物酶获取。天然纤维可以以碳水化合物或蛋白质的形式为微生物提供营养和能量来源,支持微生物生长和定植。
与棉花相比,亚麻纺织品对金黄色葡萄球菌和表皮葡萄球菌表现出强烈的抑制作用,同时对角质形成细胞产生细胞毒性。
▼
化妆品旨在改善皮肤,减缓衰老过程。这些产品可能有助于皮肤微生物组的多样化,尤其是当定期或长期使用时。
化妆品中含有的活性成分可能有利于或抑制某些微生物的生长
N-乙酰氨基葡萄糖是刺激皮肤微生物群的化合物之一,它是透明质酸的前体,常见于护肤品中。
保湿产品可以降低皮肤水分流失的强度,并可以增加皮肤微生物群多样性(α多样性高是健康皮肤微生物群的标志),同时减少皮肤细胞剥落。它们的脂质化合物促进亲脂性细菌的生长,如葡萄球菌和角质杆菌。另一方面,皮肤水合水平的提高会降低皮脂含量,并可能减少角质杆菌数量。
化妆品成分的作用持续数周,个体的反应可能差异很大。不合适的化妆品或不合适的应用会减少皮肤微生物组的多样性,从而对其产生负面影响,导致生态失调。洗发水或面霜等化妆品也可能会导致感染,有时会导致严重的健康后果,尤其是在儿童或免疫力下降的人群中使用。
内 在 因 素
▼
皮肤表面呈微酸性(pH值5.6左右)且干燥,但温度比体内低。
表皮细胞自身脱落机制影响菌群组成
表皮外层不断释放角质化皮肤细胞,导致皮肤每四个星期自我更新一次。每小时有 500-3000 个细胞从1cm2的皮肤脱落,这意味着一个成年人每小时释放 600,000-100 万个或更多细胞。由于约 10% 的脱落细胞含有细菌,这种机制可能会显着影响微生物组的组成。
皮肤的厚度、表面褶皱的深度和位置,毛囊和腺体的密度都是影响宿主微生物群的关键因素。腺体释放的分泌物以不同的方式影响微生物,创造刺激或抑制微生物发育的条件。
皮脂腺:确保专性和兼性厌氧菌的最佳环境。这些腺体分泌的皮脂在皮肤上形成保湿、疏水的保护层,并且是微生物使用的脂质的来源。这些脂质水解产生的游离脂肪酸有利于细菌粘附到腺体表面并降低皮肤pH值,抑制金黄色葡萄球菌和化脓性链球菌等病原体的生长。
水分含量:潮湿的区域为许多微生物创造了有利的条件,如棒状杆菌属、葡萄球菌属等。相对干燥且温度波动较大的皮肤部位主要含有变形菌、拟杆菌、放线菌等。微生物的数量随着深层皮肤层中营养物质和水分含量的增加而增加。
▼
男性和女性微生物群之间物种组成的差异是由皮肤的性别特异性特性造成的,即皮肤厚度、毛发、汗液和皮脂腺的数量。女性多样性高于男性。更薄的皮肤、更低的 pH 值和更少的出汗量会导致更多的多样性。
对手部表面的微生物进行的一项研究表明,女性的物种多样性高于男性。在女性手上,肠杆菌和乳杆菌科的数量显著较高(300-400%),而在男性中,观察到更高浓度的角质杆菌和棒状杆菌。
关于不同年龄皮肤菌群构成不同,在前面第一章节已经详细阐述。
▼
在形成皮肤微生物组的遗传因素中,种族是次要的,但也有一定影响。最主要的是不同生活方式的差异。非洲和拉丁美洲男性头皮和腋下的Cutibacterium数量低于其他种族(高加索、非洲裔、东亚和南亚)。中国人皮肤微生物组与其他人群存在差异,比如Enhydrobacter在中国人的皮肤上较为常见。
▼
抑制细菌和减少炎症病变
口服米诺环素(用于治疗痤疮)降低了Cutibacterium、棒状杆菌、普雷沃氏菌、乳酸杆菌和卟啉单胞菌的丰度。
多西环素显著减少痤疮梭菌的数量(治疗6周后为1.96倍)。Snodgrassella alvi的数量也减少了(3.85倍)。另一方面,观察到Cutibacterium granulosum的数量显著增加(4.46倍)。
大环内酯类、四环素类和克林霉素用于治疗痤疮。用利美环素进行的脸颊皮肤治疗减少了角质杆菌的存在,并增加了链球菌、葡萄球菌、微球菌和棒状杆菌的数量。反过来,二甲胺四环素导致微生物组紊乱。
虽然氟喹诺酮类药物(培氟沙星)和大环内酯类药物(红霉素)显著减少了痤疮梭菌的数量,但只有纳氟沙星对凝固酶阴性葡萄球菌表现出抑制活性。
导致出现抗生素耐药性物种
例如痤疮梭菌和表皮葡萄球菌。大环内酯类药物的长期治疗痤疮,增加了痤疮梭菌分离株的数量,但对大环内酯的影响的敏感性降低。
据估计,红霉素和阿奇霉素耐药菌株的比例可能分别达到50%,甚至100%。从感染皮肤分离的G+细菌中,77.5%对青霉素耐药,28%对甲氧西林耐药。在所有测试的菌株中,31.9%对三种以上的抗生素不敏感。
儿童皮肤分析结果显示,36.4%的从皮肤表面分离的金黄色葡萄球菌菌株对甲氧西林有耐药性。此外,耐甲氧西林葡萄球菌(MRSA)是医院感染最常见的原因之一。
▼
肠道内表面和皮肤表面有一些有趣的相似之处:两者都被上皮细胞覆盖,上皮细胞维持着体内与外部环境之间的重要联系,充当第一道防线,在抵御外部病原体、调节免疫反应和抑制分解代谢物方面发挥着重要作用。
肠道和皮肤组织是宿主原核和真核共生微生物的两个主要生态位,因为它们的高细胞周转率决定了定植微生物组的低粘附和感染。
皮肤健康与肠道屏障的完整性有关。一些饮食代谢物可以直接吸收到皮肤中,其他通过肠道微生物代谢来做到这一点,这两者都可能有助于皮肤健康。
由于肠道通透性增加,肠道菌群或其代谢产物可能从肠道迁移到循环系统中并在皮肤中积聚,这可能会损害皮肤屏障并使其容易发炎。
肠道微生物群的变化还可能引发系统性炎症和异常免疫反应,从而破坏皮肤健康。
皮肤或肠道微生物群失调与免疫应答改变密切相关,与多种皮肤病相关,包括特应性皮炎、牛皮癣、寻常痤疮、甚至皮肤癌等,这在下一章节会详细讲述。
饮食强烈影响肠道微生物组的组成,影响代谢和免疫功能,间接影响皮肤健康。关于如何通过饮食调整在最后章节会讲到。
以上是影响皮肤微生物群的外在和内在因素,那么皮肤微生物群会如何影响人体健康呢?我们来看下一章节。
这里我们分为两个部分来阐述:
皮肤微生物群直接影响皮肤健康
我们知道,皮肤是由角质形成细胞的分层角质化上皮组成,这些上皮经历终末分化。这些物理结构通过增强屏障的化学和免疫学特征得到进一步强化。
皮肤微生物群影响皮肤屏障的各个方面,同时也直接与表面遇到的共生微生物和病原微生物相互作用。
皮肤微生物群介导多种屏障功能
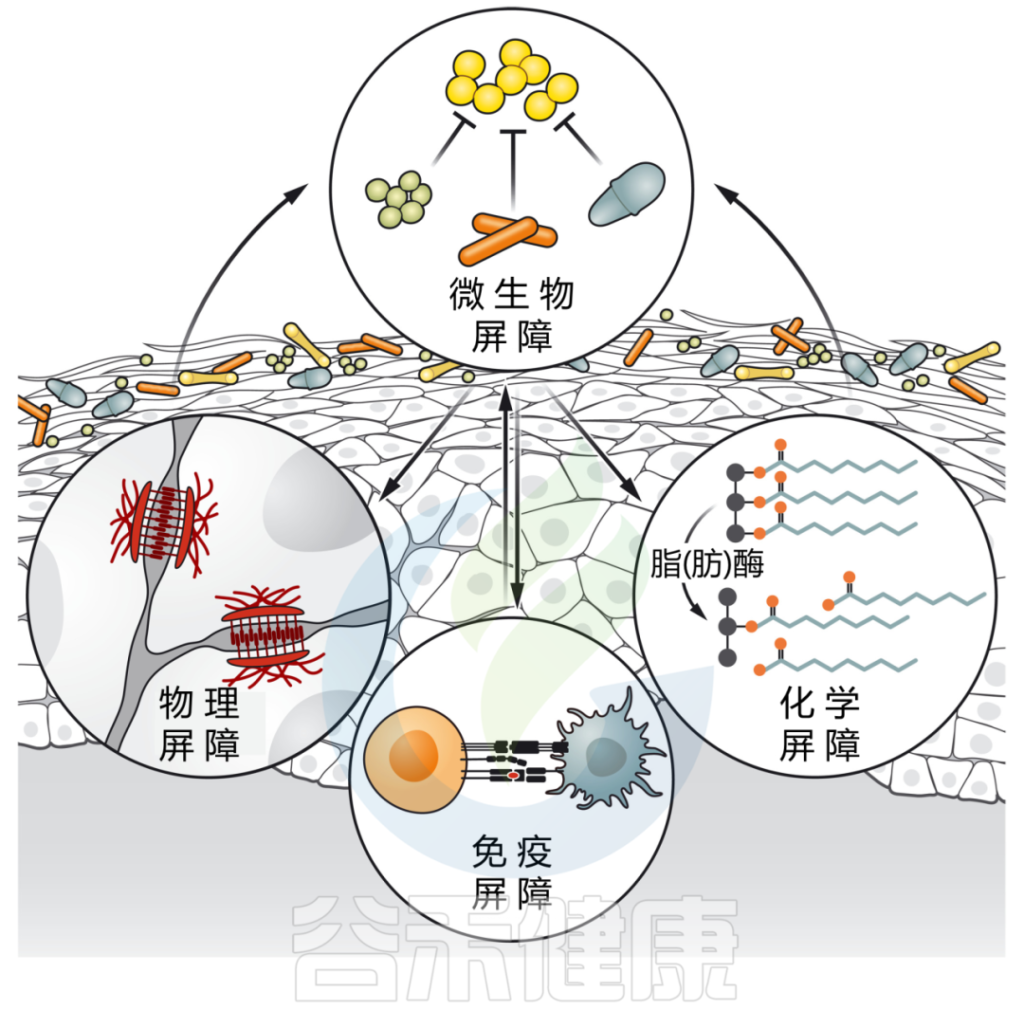
DOI: 10.1126/science.abo0693
微生物群强化皮肤屏障的多个方面:
皮肤微生物通过各种定殖抗性机制,包括资源排斥、直接抑制和/或干扰,形成对抗环境的第一道屏障。
皮肤微生物群也有助于物理皮肤屏障的分化和上皮化。微生物通过产生脂肪酶来增强皮肤的化学屏障,脂肪酶将皮脂甘油三酯消化为游离脂肪酸,从而增强皮肤的酸性,并限制瞬时和致病物种的定植。
最后,微生物刺激先天和适应性免疫防御,如抗菌肽的释放、新生儿耐受性的诱导和保护性免疫的发展。
接下来我们讨论微生物群到底如何与皮肤屏障的微生物、化学以及先天和适应性免疫成分相互作用。
▼
皮肤微生物群本身是抵御外来微生物和病原微生物入侵、定植和感染的屏障。
——直接竞争关系
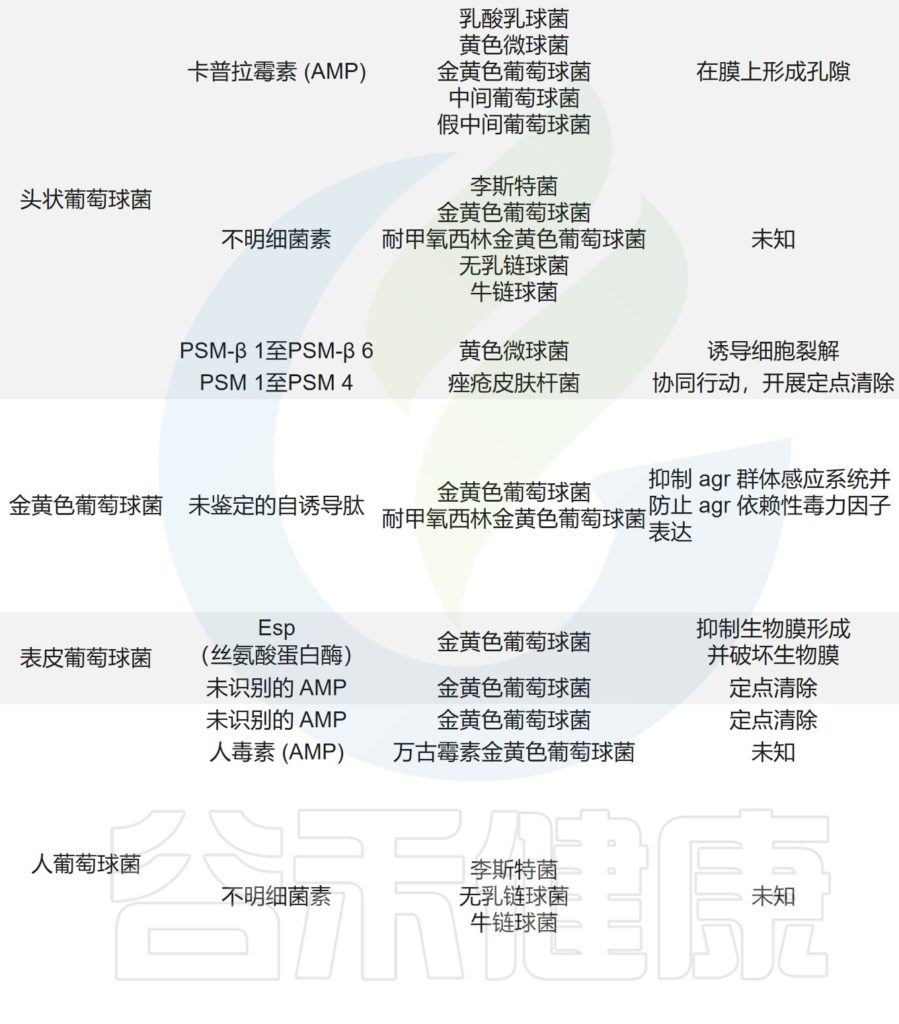
皮肤微生物争夺资源,并进化出直接对抗对手的机制。
多种CoNS物种(凝固酶阴性葡萄球菌),如人葡萄球菌,产生具有独特化学性质的抗生素,并抑制皮肤病原体金黄色葡萄球菌。
其他物种,如头葡萄球菌,通过干扰金黄色葡萄球菌毒力所需的辅助基因调节因子(agr)群体感应途径来拮抗金黄色葡萄菌。
——拮抗机制与宿主抗菌反应协同作用
人型葡萄球菌和表皮葡萄球菌,可以产生共生衍生的AMPs,其发挥选择性抗菌活性,并与宿主衍生的AMPs协同作用,以抑制皮肤病原体的存活。
痤疮角质杆菌与产生硫肽抗生素角质霉素的特定菌株竞争,以维持其在人类毛皮脂腺单元中的生态位,从而限制金黄色葡萄球菌的定植。
皮肤微生物组内微生物之间的相互作用,可以驱动整体微生物群结构。
主要皮肤菌群产生的抑制其他微生物群,和/或潜在病原体的突出和最近鉴定的抗菌分子汇总在下表,分子作用机制也包括在内。
皮肤上关键的微生物与微生物相互作用

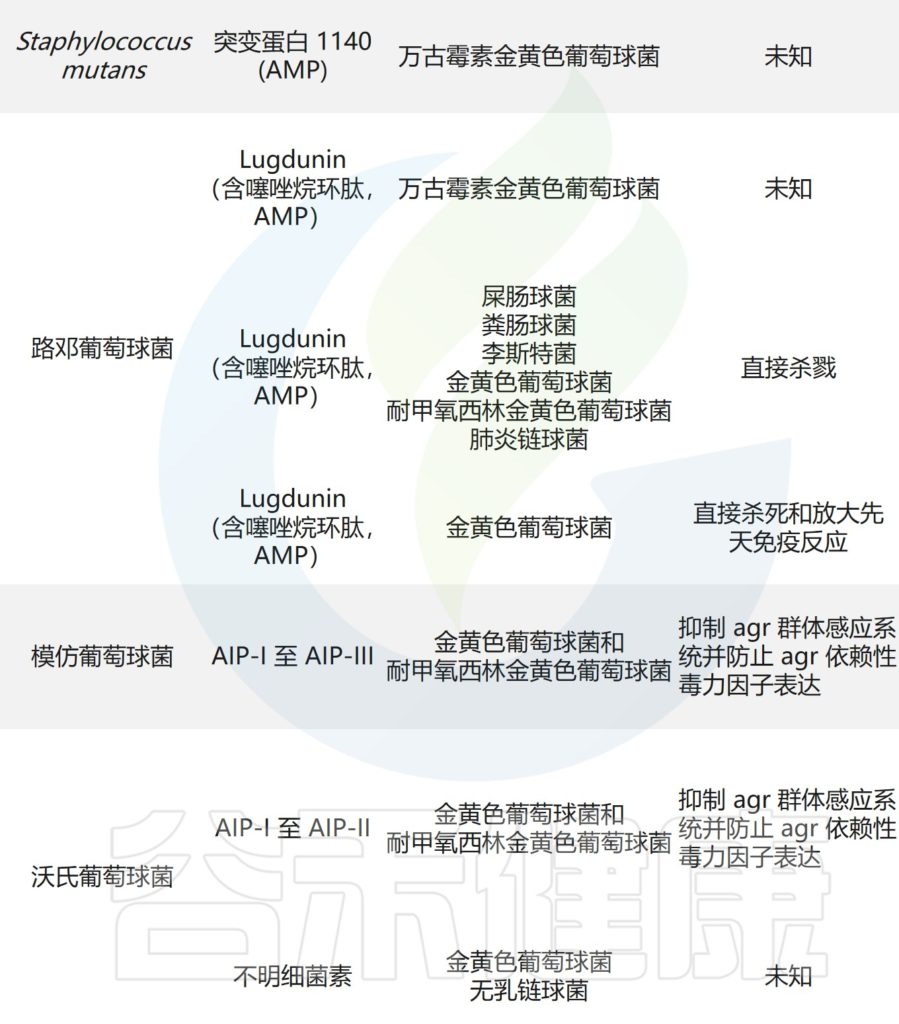
doi.org/10.1042/BST20220216
▼
角质细胞经历严格调控的终末分化程序,形成角质层,该过程由微生物群介导。微生物群通过角质形成细胞芳香烃受体(AHR)的信号传导促进分化和上皮完整性;还分泌鞘磷脂酶,将层状脂质加工成神经酰胺,神经酰胺是角质层的关键成分。
▼
酸性皮肤表面还产生了限制细菌定植的化学环境。痤疮角质杆菌和棒状杆菌都分泌脂肪酶,水解皮脂中甘油三酯中的游离脂肪酸。游离脂肪酸通过直接抑制细菌和刺激人β-防御素2(hBD-2)的表达,进一步增强皮肤免疫力。痤疮角质杆菌也直接与游离脂肪酸结合,这表明游离脂肪酸的存在促进了痤疮角质杆菌的定植。
▼
微生物可以刺激多种与先天免疫反应有关的反应,通常取决于代谢和炎症环境。例如,念珠菌的菌丝和酵母形式在皮肤中刺激不同的免疫反应。S. epidermidis在皮肤中引起的T细胞反应,需要菌体表面特定糖蛋白与宿主先天免疫细胞上的C型凝集素相互作用。
氧气的可用性也会影响皮肤表面宿主与微生物的相互作用。微氧耐性细菌痤疮角质杆菌生成短链脂肪酸,抑制组蛋白去乙酰化酶,后者可作为免疫系统的表观遗传调节因子,从而刺激炎症。
注:在皮肤中,短链脂肪酸具有促炎作用,这点和肠道中不同。SCFAs通过抑制HDAC8和HDAC9以及通过TLR信号通路刺激炎症。
皮肤微生物还通过刺激宿主产生抗菌肽和蛋白来增强皮肤免疫力,这些抗菌肽和蛋白起到天然抗生素的作用。
皮肤微生物群落还在创伤修复过程中协调先天免疫反应。在皮肤中的共生微生物群落会引发I型干扰素(IFN)反应。作为对微生物信号的反应,中性粒细胞会表达CXCL10,吸引活化浆细胞样树突状细胞(pDC)到损伤部位。pDC会产生I型干扰素,通过刺激成纤维细胞和巨噬细胞增长因子反应来加速创伤修复。
实际上,抗原呈递细胞向皮肤的募集是微生物群依赖性的。微生物通过需要IL-1R-MYD88信号传导的过程,在伤口修复和毛囊新生中增强皮肤再生。
▼
皮肤是各种适应性免疫细胞的家园,其中包括大量的常驻记忆T细胞,随时准备对各种环境刺激做出反应,包括致病微生物和共生微生物。
在婴儿早期,暴露于皮肤共生表皮葡萄球菌介导调节性T细胞(Tregs)流入皮肤。这种Treg迁移波与毛囊发育同时发生,需要毛囊角质形成细胞产生趋化因子。Tregs,以及皮肤中的许多其他免疫细胞亚群,最终位于毛囊附近,对在这个发育窗口期间检测到的微生物抗原具有特异性。
在一个平行的过程中,粘膜相关不变T细胞(MAIT)是在婴儿期在类似的时间限制的发育窗口中获得的。MAIT细胞在无菌小鼠中是不存在的,它们的发育需要维生素B2代谢产物,而这些代谢产物仅由细菌和真菌产生,而不是哺乳动物细胞。
在胸腺中,暴露于5-(2-oxopropylideneamino)-6-d-ribitylaminouracil(一种维生素B2的细菌代谢产物,从粘膜部位运输到胸腺),介导MAIT细胞扩增并靶向皮肤和粘膜部位。
微生物细胞表面分子也可以作为宿主的信号。大多数棒状杆菌的细胞膜中含有霉菌酸。棒状杆菌属霉菌酸在稳定状态下可以以IL-23依赖的方式促进γδT细胞的积累。然而,这种相互作用取决于环境,因为高脂肪饮食反而会促进皮肤炎症。因此,微生物暴露时存在的炎症环境影响皮肤内的免疫反应。
这些发现突出了微生物在皮肤免疫细胞的募集和刺激中发挥的关键作用。
以上是皮肤微生物群从物理、化学、免疫等多角度与皮肤之间的关联,如果说上述对局部组织微环境的相互作用,那么接下来我们从更系统的角度来看,皮肤微生物群通过与其他器官的交流,对全身健康产生的影响。
皮肤微生物群通过影响其他器官间接影响
越来越多的证据表明,皮肤损伤和致敏会影响其他屏障部位,如肠道、肺部等。
▼
皮肤和肠道之间存在双向沟通。
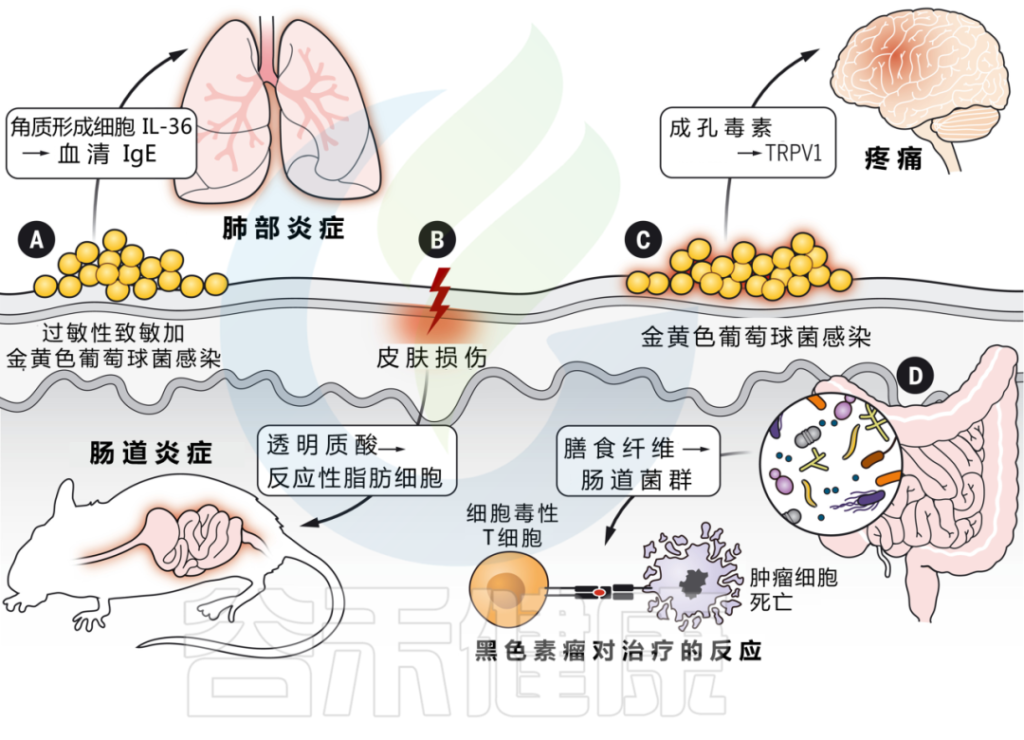
为什么浅表皮肤损伤会引起肠道炎症?
浅表皮肤损伤会导致角质形成细胞全身释放 IL-33。IL-33 与 IL-25 协同作用,触发肠道内 ILC2 的激活,产生 IL-4。这反过来又刺激肠道中肥大细胞的扩张,在那里它们准备对食物过敏原做出反应并介导过敏反应。
在模拟炎症性肠病的小鼠模型中,皮肤受伤还会加剧肠道炎症。
皮肤和肠道之间的相互作用取决于损伤期间真皮中产生的透明质酸片段的产生,这些片段刺激肠道成纤维细胞,通过反应性脂肪生成的过程分化为促炎脂肪细胞。这些反应性脂肪细胞通过产生 AMP 和其他炎症介质来传播肠道炎症。
肠道微生物群变化也会影响皮肤炎症
在这两种情况下,肠道免疫网络的激活都会影响皮肤中炎症信号的振幅。
因此,肠道微生物组的改变可能会影响皮肤免疫力。
研究表明,饮食对肠道微生物组的影响,尤其是膳食纤维,对系统免疫有重要影响。皮肤先天免疫反应也与肠道有关,肠道中保护细菌性皮肤感染的AMPs的充分表达,取决于饮食中的维生素A。这些发现加强了我们对饮食在宿主免疫发展中重要性的分子理解。
▼
流行病学证据表明,许多患者经历了“特应性进军”,首先出现特异性皮炎,随后发展为过敏性鼻炎、食物过敏、哮喘。它们的先后出现意味着存在什么样的关联?
皮肤微生物群失调和金黄色葡萄球菌定植增加,与特应性皮炎的发作有关。
表皮暴露于金黄色葡萄球菌刺激角质形成细胞产生IL-36,从而提高血清IgE水平。
而缺乏IL-36受体的小鼠对金黄色葡萄球菌的反应不会产生升高的IgE,并且也可以免受过敏原特异性肺部炎症的影响。这些发现支持了皮肤暴露于微生物病原体作为全身炎症的起始。
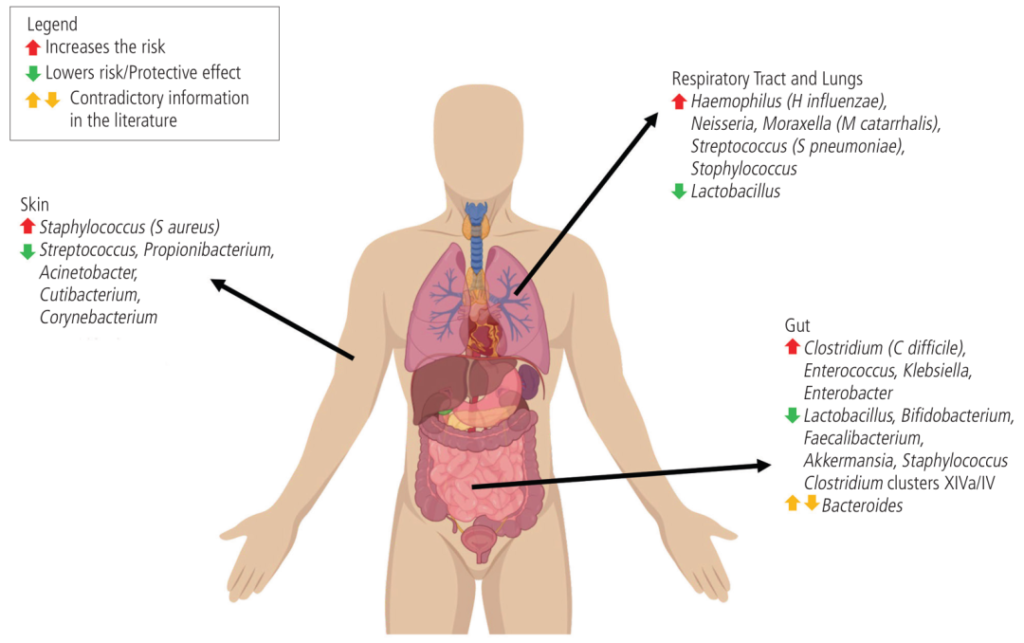
微生物组的变化与哮喘、过敏性鼻炎、特应性皮炎和食物过敏的风险有关
doi: 10.18176/jiaci.0852
在气道中,卡他莫拉克菌(Moraxella catarrhalis)、流感嗜血杆菌(Haemophilus influenzae)和肺炎链球菌水平较高与婴儿哮喘有关。
肠道艰难梭菌的比例高于双歧杆菌,这与更高的食物过敏率有关。
▼
神经免疫相互作用中的皮肤病原体
细菌可以直接激活皮肤中的感觉神经元,并通过产生造孔毒素引起疼痛。菌株水平的变化驱动着可变的反应,这取决于特定毒素和群体感应系统的存在。
关于群体感应,详见:
真菌(白色念珠菌)也可以直接激活皮肤中的感觉神经元。γδT细胞免疫需要刺激才能通过释放神经肽CGRP来控制皮肤念珠菌感染。
相反,引起坏死性筋膜炎的病原体化脓性链球菌,通过分泌链球菌溶血素S直接激活伤害感受器神经元,进而促进神经肽CGRP的释放并抑制化脓性链球菌的杀伤。在这种情况下,CGRP拮抗作用可防止坏死性感染。
皮肤与其他器官系统的交互作用是由微生物群介导的
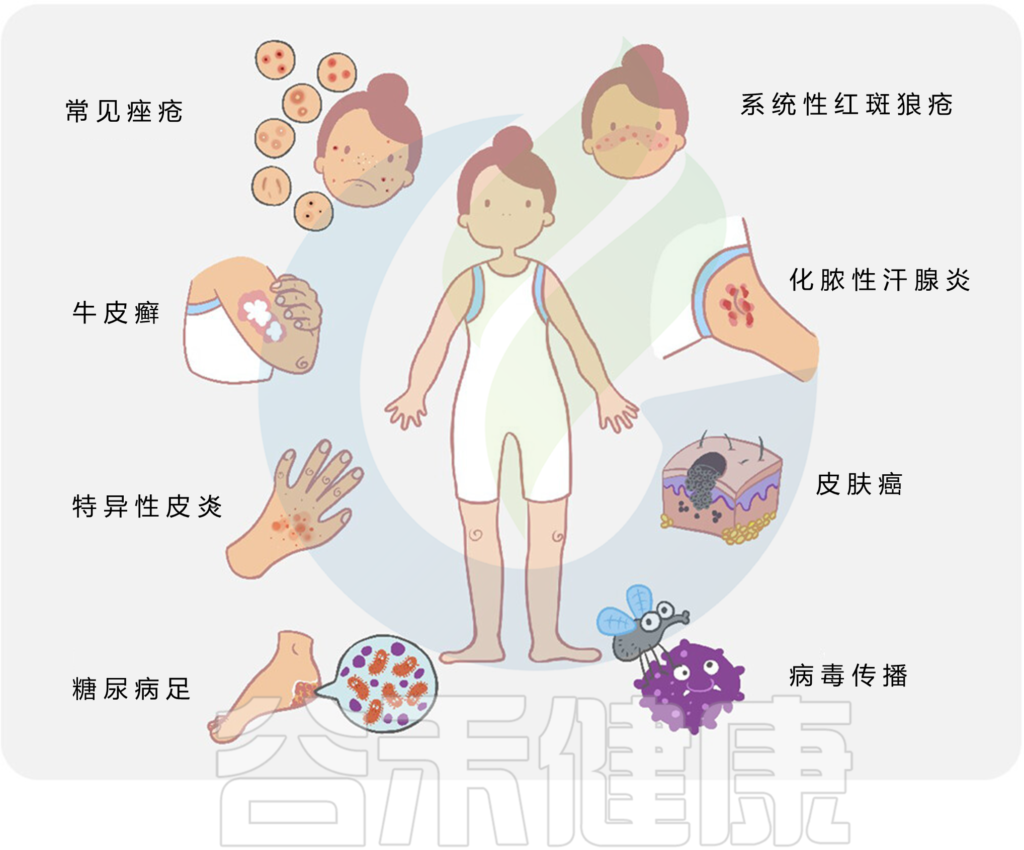
DOI: 10.1126/science.abo0693
微生物与宿主相互作用和皮肤疾病
doi.org/10.1002/mlf2.12064
▼
痤疮患者,特别是那些症状严重的患者,表现出α多样性增加,四种革兰氏阴性细菌(即粪杆菌属、克雷伯氏菌属、臭杆菌属和拟杆菌属)的比例更高。
痤疮角质杆菌(C. acnes)的过度生长与痤疮发病机制有着长期的关联。宏基因组分析表明,痤疮患者中痤疮丙酸杆菌的菌株结构与健康个体不同,IV 型和 V 型菌株在受痤疮影响的皮肤中特别普遍。
痤疮丙酸杆菌通过多种不同方式参与痤疮发病机制的调节,它参与:
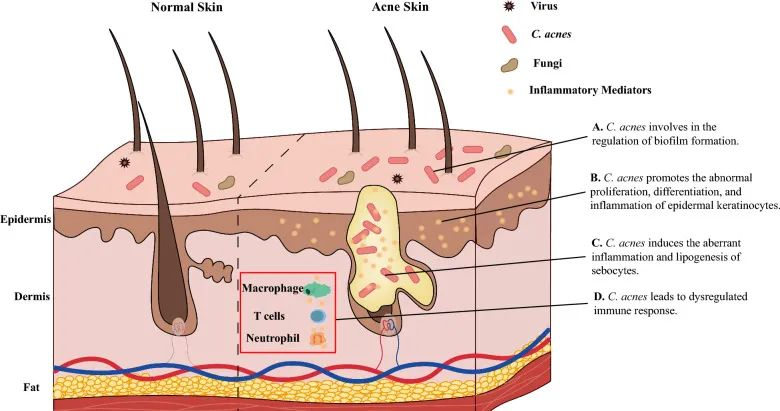
doi: 10.1186/s13578-023-01072-w
痤疮丙酸杆菌和表皮葡萄球菌在痤疮以及炎症后色素沉着过度中具有病理生理作用。
肠道微生物群在皮肤炎症和情绪之间起着中介作用
痤疮和胃肠道功能障碍之间的联系可能起源于大脑。支持这一假设的是压力引起的痤疮加重。实验动物和人类研究表明,压力会损害正常的肠道菌群,尤其是乳酸杆菌和双歧杆菌。心理应激源导致肠道微生物群产生神经递质(即乙酰胆碱、血清素、去甲肾上腺素),这些神经递质穿过肠粘膜进入血流,导致全身炎症。
痤疮中肠-脑-皮肤轴的拟议模型
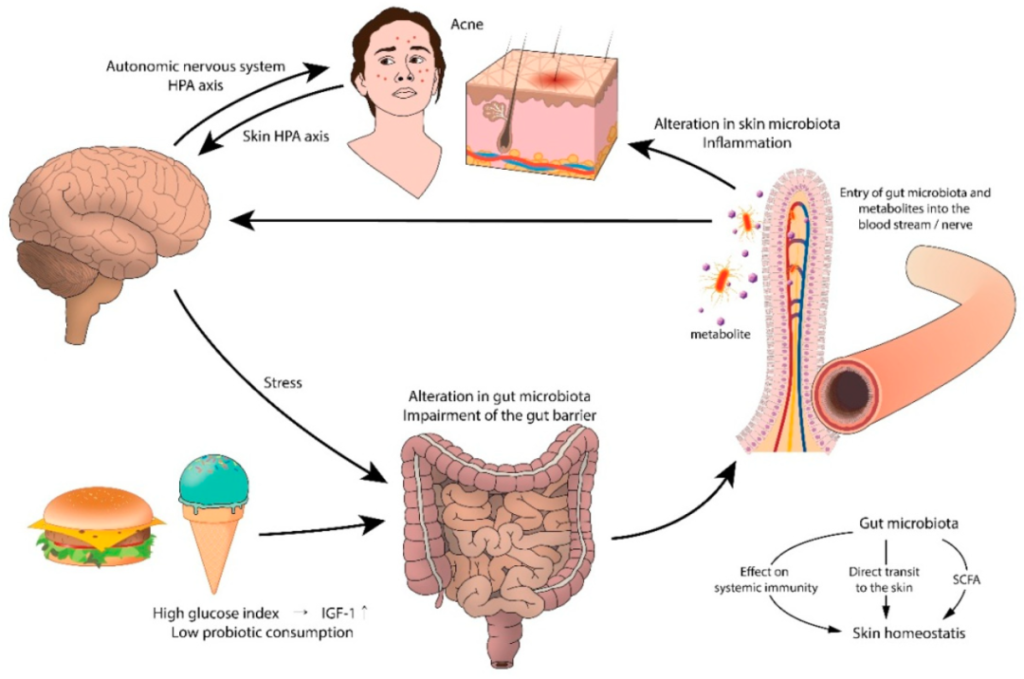
doi.org/10.3390/jcm8070987
西方饮食包括乳制品、精制碳水化合物、巧克力、饱和脂肪等,这些物质可能通过激活营养来源的代谢信号来加重痤疮。高脂肪饮食会降低肠道菌群水平,增加脂多糖的浓度,通过损害结肠上皮完整性和屏障功能、降低粘液层厚度和增加促炎细胞因子的分泌来引起全身炎症。
扩展阅读:
▼
皮肤干燥、斑块发痒和反复出现的湿疹是特异性皮炎的标志。
特异性皮炎引起的皮肤耀斑通常与更多的金黄色葡萄球菌丰度有关,金黄色葡萄球菌定殖的增加与CoNS数量的减少相关,CoNS本来会产生抗菌蛋白,它在特异性皮炎患者中数量少。
金黄色葡萄球菌在病变的真皮中更为普遍,这表明在剥皮过程中更容易接触到更深的皮肤层。
是什么引起金黄色葡萄球菌定植增加呢?
表皮葡萄球菌、痤疮杆菌和棒状杆菌属的丰度降低,它们通常对金黄色葡萄菌的入侵起作用。
特异性皮炎的菌群多样性低。共生细菌数量减少而导致的共生产生的AMPs的缺失,抵御病原体如金黄色葡萄球菌的能力下降,金黄色葡萄球菌定植增加。
与特异性皮炎相关的皮肤屏障缺陷会损害层状膜的完整性,改变皮肤的微生物群,并可能使金黄色葡萄球菌等有害细菌滋生。厌氧微生物的缺乏可能会降低关键的皮肤屏障活性,并促进潜在的感染。
金黄色葡萄球菌分泌毒力因子
金黄色葡萄球菌分泌几种毒力因子,包括纤连蛋白结合蛋白1(FBP1),α-和δ-溶血素,酚溶性调节素(psm)的蛋白家族等,所有这些毒素都会导致更高的炎症反应和更严重的症状。
皮肤稳态取决于复杂的宿主-微生物相互作用,包括金黄色葡萄球菌和特异性皮炎宿主细胞之间的相互作用,微生态失调会导致疾病的发展。
特异性皮炎的其他微生物群变化包括痤疮角质杆菌、棒状杆菌、Dermacoccus、微球菌、CoNS减少,链球菌和一些马拉色菌属增加。这些微生物变化似乎是暂时的,在特异性皮炎发作之前和期间,群落多样性丧失,金黄色葡萄球菌优势更大,在炎症消退后逐渐恢复到基线。
潜在益生菌治疗和预防AD的临床试验

doi.org/10.1016/j.phymed.2023.154824
▼
一般来说,皮肤破裂会导致炎症级联活动;然而,这种免疫反应在糖尿病皮肤中被破坏,也就是无法有效引起免疫反应。微生物组的改变可能会加剧疾病的严重程度。
糖尿病皮肤的菌群特征
糖尿病早期患者的皮肤细菌微生物群与健康人的非常相似。随着疾病的恶化,物种多样性和丰度发生动态变化。总的来说,糖尿病足的皮肤细菌微生物群的多样性低于健康足。因此,不太常见的微生物种类的变化,其中大多数只在健康的足部皮肤中发现,可以用来预测是否患有糖尿病。
糖尿病足皮肤中葡萄球菌的含量通常较低,金黄色葡萄球菌的比例较高。金黄色葡萄球菌的大量存在破坏皮肤微生物群平衡,可能会导致炎症变化,并增加皮肤感染的风险。
慢性溃疡相关菌群
铜绿假单胞菌和厌氧菌通常与深部慢性溃疡有关,但金黄色葡萄球菌通常与急性浅部溃疡有关。比较有慢性感染和没有慢性感染的糖尿病患者的微生物组的研究可以提供有关诊断标志物的信息,这些标志物可以用作发展为慢性损伤的可能性的指标。
▼
牛皮癣患者由于慢性炎症性皮肤病而出现中度至持续性皮肤斑块。许多遗传和环境变量之间的复杂组合导致皮肤过度活跃的炎症反应是病因。
牛皮癣皮肤菌群特征
牛皮癣患者皮肤样本在α、β多样性明显低于正常皮肤。下列菌群相对丰度和分类性能显著下降:
棒状杆菌在牛皮癣的发病机制中发挥重要作用
大量的研究表明,棒状杆菌属丰度上升,棒状杆菌有可能干扰干扰素信号系统,这可能导致皮肤微生物组的微生态失调。
乳制品和糖类的摄入是牛皮癣最常见的诱因之一,而肉类和鸡蛋则被列为次要的常见诱因之一。
关于牛皮癣与肠道菌群之间也存在很多相关性,此处不展开阐述,详见:
▼
皮肤黑色素瘤
黑色素瘤和正常皮肤样本之间的微生物组成和多样性存在显著差异。黑色素瘤样本中的梭杆菌和Trueperella水平较高。
棒状杆菌属与疾病严重程度相关,棒状杆菌水平与IL-17之间存在关系,IL-17可以通过增加IL-6和信号转导器和STAT-3来促进黑色素瘤细胞增殖。
相反,痤疮角质杆菌的细菌上清液增加了黑色素细胞的凋亡。
角质细胞皮肤癌
以皮肤微生物群为代表的生物屏障通过分泌抗微生物肽(AMP)(如组织蛋白酶LL-37和人β-防御素)来抑制病理生物和病原体入侵,从而与角质形成细胞和免疫细胞产生串扰。
研究人员认为金黄色葡萄球菌和鳞状细胞癌之间的联系不是偶然的,皮肤溃疡是有利于外源性搪塞/感染的致病过程的结果。金黄色葡萄球菌也可能参与鳞状细胞癌的发病机制,引起慢性局部炎症,涉及不同的致瘤阶段,包括促进生存、增殖、细胞转化、侵袭、血管生成、转移。
葡萄球菌毒素-α决定了参与炎症过程的局部细胞的分泌,进而导致活化B细胞的NF-Kβ的激活,从而增加不同细胞因子和趋化因子的表达,包括IL-1β、IL-6和IL-12。
其他因素如紫外线辐射(尤其是UVB)也是皮肤癌发生的主要危险因素之一。紫外线照射会改变皮肤微生物群,导致大量形成活性氧、细胞凋亡和炎症,与皮肤癌相关。
总的来说,许多常见的皮肤病,如痤疮、特异性皮炎、牛皮癣、皮肤癌等,都与皮肤微生物群的变化有关。
皮肤病中的关键微生物发现如下:

编辑
doi.org/10.1002/mlf2.12064
●
饮食对肠道微生物群的影响较大,皮肤和肠道微生物群是内在相关的,由宿主免疫系统介导。因此,肠道和皮肤可以通过饮食、微生物代谢产物、神经内分泌途径和中枢神经系统等途径相互作用,也就是说,饮食对皮肤也会产生较大影响。
饮食结构
西方饮食已被证明会破坏微生物组并导致皮肤病,从而对皮肤健康产生负面影响。相反,植物性饮食与更健康的皮肤有关。
以植物为基础的饮食是一种由多种蔬菜、水果、豆类、扁豆、豆类、坚果、种子、真菌和全谷物组成的饮食模式,并且限制或不摄入动物产品、加工食品或糖果。
这种饮食的饱和脂肪、反式脂肪和花生四烯酸含量较低,而抗氧化剂和 omega-3 脂肪酸含量较高,再加上其直接治疗作用,可减少炎症和皮肤症状。
植物性饮食与皮肤健康/疾病之间的关联
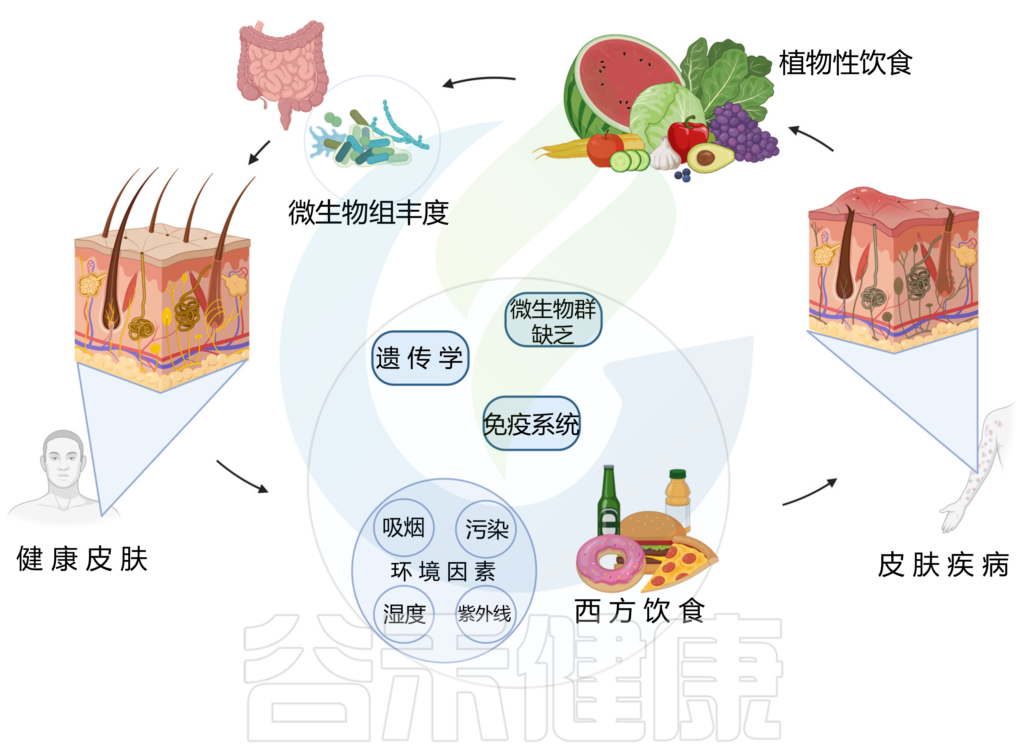
doi.org/10.3390/nu15132842
多项研究发现,植物性饮食对缓解牛皮癣、特异性皮炎、痤疮等皮肤问题有益。


doi.org/10.3390/nu15132842
食 物
植物性功能性食品增强皮肤健康,减少皮肤老化迹象,并改善整体外观。下图是芒果、杏仁、牛油果及其对皮肤健康的积极影响。
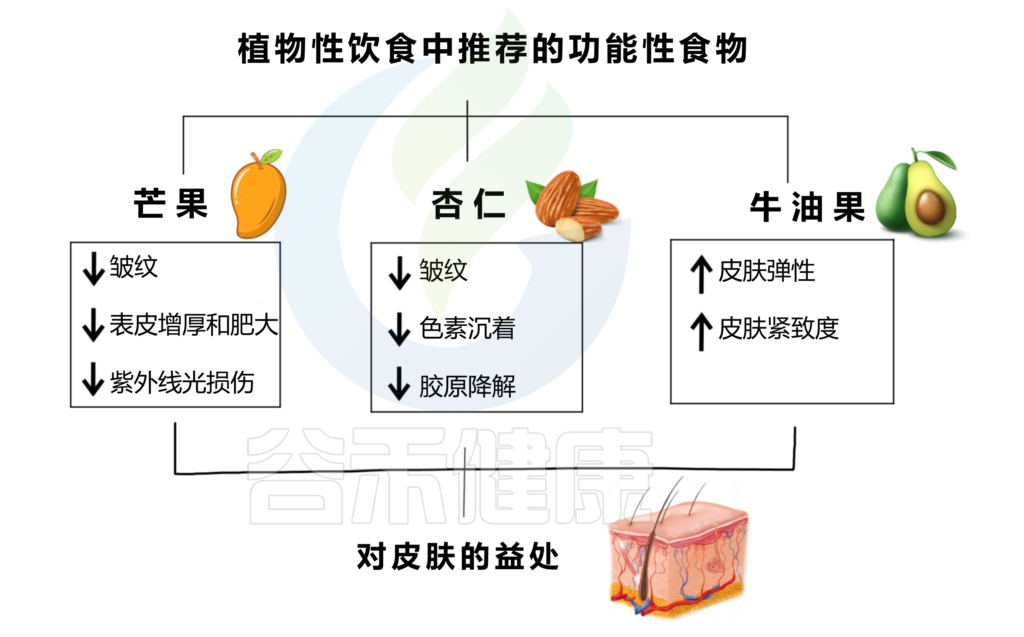
编辑
doi.org/10.3390/nu15132842
芒果能够减少皱纹、表皮变薄和肥厚,防止 UVB 损伤。无论是果肉还是果汁,芒果酚酸的抗氧化特性和生物利用度都会得到保留,而果汁的呈现可能会增强其特性。从芒果干中提取的芒果提取物也可以减少UVB辐射引起的皱纹的形成。
杏仁富含α-生育酚(或维生素E)、脂肪酸、多酚,因此是一种具有抗氧化特性的食物,可以减少皱纹、色素沉着和胶原蛋白降解。
牛油果含有类胡萝卜素、单不饱和脂肪酸、酚类化合物,某些基因的表达,如胶原蛋白和弹性蛋白基因,在进行饮食调整后被诱导,因此可以促进皮肤弹性和紧致度的增加。
限制饮酒和甜食
酒精会使你的身体和皮肤脱水,这可能会使皮肤看起来更加干燥或有皱纹,许多含有酒精的混合饮料也富含糖,这都不利于皮肤健康。糖可能使胶原蛋白变硬,从而使皮肤老化,也可能带来炎症。
喝 水
对于每日饮水量较低的人(即那些本来就脱水的人)来说,增加饮水量对皮肤外观有积极影响,有助于维持皮肤水合水平。同时,尝试在食物中多加入黄瓜、芹菜、西葫芦、西瓜、草莓和花椰菜等,也可以适当补水。
●
适当的清洁和保湿可用于维持皮肤的生理pH值。据报道,早在1995年,与使用 pH8 的普通肥皂相比,使用酸性合成皂(pH 5.5-5.6)可显著减少非炎症和炎症病变。
从那时起,pH 值的变化通过皮肤屏障的完整性与痤疮的发病机制联系起来,建议使用 pH 值约为 5.5 的皮肤清洁剂。
为什么洗脸很重要?
脸每天面对风吹、紫外线、化妆/护肤品、屏幕等刺激,会积聚污垢、油脂和其他碎屑,如果不及时清除,可能会导致刺激和其他皮肤问题。
合适的洗脸方式
注意:
应该多久洗一次脸?
没有既定的指导方针,一般来说,最好每天洗两次脸。
如果皮肤干燥或敏感,可以在晚上用清洁去除污垢,然后在早上用温水冲洗脸。
即使当天不化妆或者不出门,污垢、油脂和其他不需要的碎屑仍然会在一天中积聚在皮肤上,因此最好在睡觉之前洗脸。
如果刚在健身房、参加高温瑜伽课或在户外徒步旅行,并且出汗较多,最好马上洗脸。
如果存在敏感问题或其他特殊的皮肤状况,请与医生沟通。
●
痤疮是一种慢性炎症性皮肤病,对于痤疮,护肤品有多种作用机制,包括:
1) 保护和改善皮肤屏障
2) 保护皮肤微生物组
3) 维持健康的皮肤 pH 值
4) 抵御紫外线伤害
保护皮肤屏障是皮肤化妆品改善痤疮管理的重要机制,临床上,屏障功能障碍表现为皮肤干燥、刺痛/烧灼/刺痛、紧绷、疼痛或刺激性皮炎等形式。这些被认为与经表皮失水 (TEWL) 有关,并且可以通过使用保湿剂至少部分缓解。
特别适用于痤疮的成分包括烟酰胺、视黄醇衍生物、水杨酸、神经酰胺、甘油、温泉水、泛醇等。
护肤品中可能存在的活性成分及其针对性作用
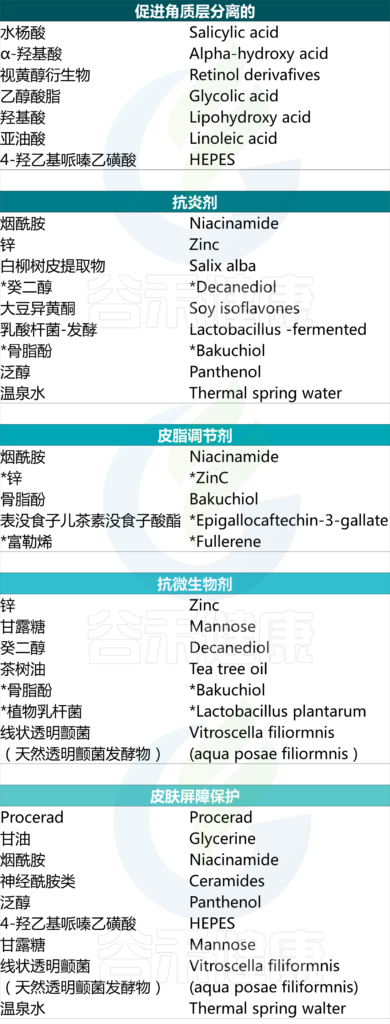
Kurokawa I, et al., Dermatol Ther (Heidelb). 2023
而皮肤微生物组的核心作用与表皮屏障功能一起,为优化护肤提供了强有力的支持。可以通过尝试恢复微生物组的多样性并通过下调先天免疫来抑制炎症。
总的来说,现有文献结果的总体趋势表明,护肤品可以改善整体皮肤健康,减少痤疮皮损,在处方治疗后维持痤疮清除,并且可能对减少表面皮肤油腻具有有益作用。
然而需要进一步研究才能更好地理解这一作用。在此情况下可能有益的成分包括但不限于:乙醇酸、LHA、亚油酸、烟酰胺、锌、吡罗克酮乙醇胺、procerad、Vitroscella filiformis.
注意:
痤疮的治疗管理需谨慎,一些基于类维生素A的治疗方案,可能会加剧皮肤干燥和刺激,这不仅可能导致屏障功能改变,而且还会增加深色皮肤患者继发性妊娠高血压综合征的可能性。
●
温泉水已被证明对膜流动性、皮肤屏障修复、抗自由基、抗氧化、抗炎和免疫调节特性以及增殖活性和衰老和保湿过程的调节有影响。
温泉水的水微生物群中的生物活性化合物可以改善特应性皮炎或红斑痤疮等皮肤病,并改善瘙痒和干燥症;还能增加对紫外线的防护,强化屏障功能,维持皮肤防御良好的稳态,修复受损皮肤,促进伤口愈合,改善皮肤状况,减少皮肤色素不均匀,防止皮肤老化。
未来,护肤品研发人员、水文学家、温泉中心之间的合作,将推动该行业更好地了解温泉水的水生生物群落对皮肤病的作用,并考虑将这种水生生物群落的衍生物纳入皮肤病配方(以发酵罐、裂解物、提取物等形式)。
●
某些益生菌菌株及其代谢物可能带来许多益处,如:改善皮肤屏障功能、减少炎症、改善易长粉刺或易湿疹的皮肤、抗皮肤光老化等。因此,近年来益生菌已成为护肤品中的流行成分。
益生菌分为口服和外涂。
口服益生菌
肠道微生物群的变化可能会引发全身炎症和异常免疫反应,从而破坏皮肤健康。口服益生菌直接作用于肠道微生物群,帮助恢复肠道微生物群的稳态,这在皮肤稳态中发挥着至关重要的作用。
益生菌在皮肤光老化中的作用
含有约氏乳杆菌和营养类胡萝卜素的膳食补充剂的摄入对紫外线暴露的长期和反复影响有益处,并且对光老化更有针对性。
含有长双歧杆菌和低聚半乳糖的膳食补充剂由于其抗炎和抗氧化特性,保护皮肤免受UVB诱导的光老化。它们也提高了血清中短链脂肪酸和乙酸盐的水平,可以增加和激活依赖于组蛋白乙酰化的皮肤固有Treg。
口服植物乳杆菌HY7714通过抑制JNK/AP-1信号通路的激活,降低了UVB损伤细胞中过量的MMP-13转录水平和MMP-2和MMP-9的活性。
口服清酒乳杆菌可以通过阻断MAPK信号通路来抑制AP-1的表达,以增加真皮成纤维细胞中的胶原蛋白并延缓皮肤光老化。
益生菌通过多种途径对抗皮肤光老化的作用
编辑
doi: 10.2147/CCID.S388954
局部益生菌(外涂)
局部益生菌于1912年首次被提出作为皮肤疾病的治疗方法。
在特定条件下,益生菌可以持续存在并成功定殖皮肤:
虽然益生菌对皮肤健康有一定益处,但益生菌使用的安全性可能存在一些限制,主要针对免疫系统较弱的人群,如婴儿、孕妇、老年人等。需要进一步的研究来证明益生菌作为皮肤病治疗和护肤品的功效,作用机制以及主要是局部使用益生菌的安全性。
●
过氧化苯甲酰 (BPO) 治疗可以调节痤疮患者的皮肤微生物群,治疗后细菌种类的数量和多样性均减少,接近健康组。也有研究人员认为,BPO治疗虽然降低了GAGS评分并降低了微生物多样性,但它也损害了痤疮的表皮屏障,这可以被认为是一种副作用。
抗生素
针对痤疮丙酸杆菌的抗生素一直是痤疮治疗的支柱。其中,大环内酯类、克林霉素和四环素类药物的处方最为广泛。
红霉素、罗红霉素、克拉霉素和阿奇霉素是大环内酯类药物。常用于痤疮的四环素类药物是多西环素、四环素和米诺环素。
异维甲酸是一种全反式维甲酸原药,是严重顽固性痤疮患者的最终选择,它抑制皮脂生成,它可以使痤疮患者的C.acnes/TLR-2介导的先天免疫反应正常化,也就是说,异维甲酸会间接影响皮肤微生物。
其他,比如抗生素治疗会降低皮肤伤口中的细菌密度并改变细菌组成,其次是RegIIIγ表达的降低,这可能有助于延迟伤口修复。
●
几项研究表明,互惠共生对维持微生物物种之间的新陈代谢很重要。我们不仅需要关注微生物组的转移,还需要关注潜在的交叉喂养和共同居住。
有研究表明,整个原始皮肤微生物组从一个皮肤部位移植到另一个部位。研究人员将能够在腋下产生气味的细菌转移到受试者的前臂,前臂上培养的双菌群样本产生强烈的气味,这表明引起气味的细菌可以从腋下传播到前臂。
这项研究表明,通过皮肤微生物组移植重塑人类气味,降低对传染病媒介的吸引力,从而阻断病毒传播,为传染病预防和控制提供了一条新的途径。
●
尽可能选择天然纤维材料,如棉、亚麻或丝绸等,这些材料通常具有良好的透气性和吸湿性,可以帮助皮肤保持干爽。同时,天然纤维材料也更加温和,减少与皮肤的摩擦和刺激。
避免合成纤维材料:尽量减少使用合成纤维材料,如尼龙或涤纶等。
保持衣物的清洁和卫生非常重要。定期清洗和消毒衣物,避免细菌、真菌和其他有害微生物的滋生。
●
吸烟会使皮肤最外层的微小血管变窄,从而减少血液流动并使皮肤更苍白。这也耗尽了皮肤对皮肤健康很重要的氧气和营养物质。吸烟还会损害胶原蛋白,也可能会增加患鳞状细胞皮肤癌的风险。
压力会通过肠-脑-皮肤轴影响皮肤健康,皮肤细胞的代谢和分泌作用可能发生变化,导致皮肤微生物群失去原本的平衡状态,皮肤更加敏感,并引发痤疮爆发和其他皮肤问题。
长期的压力会导致免疫系统的紊乱,使皮肤更容易受到各种外界因素的侵害,从而引发炎症、过敏等症状。
睡眠可以促进皮肤细胞的修复和再生,有助于提升肤色的均匀度。同时,在深度睡眠状态下,身体会产生更多的生长激素,促进皮肤中胶原蛋白和弹力纤维的生成,有助于减少皱纹的产生。因此,保证充足的睡眠对皮肤健康也相当重要。
适度的身体运动可以促进血液循环和新陈代谢,有助于清除毒素和废物,使皮肤更加健康。选择适合自己的运动方式,如散步、跑步、瑜伽等,坚持每周进行几次。
随着年龄的增长,我们的皮肤微生物群组成发生了很大变化且可预测。未来的研究将继续阐明动态皮肤微生物组在整个生命周期中的积极作用。
皮肤微生物群扮演着重要角色,它不仅是阻止致病菌进入皮肤的屏障,还可以通过调节免疫系统减少炎症等方式对皮肤疾病起到积极作用。肠道微生物组的研究为我们探索皮肤微生物群在其他生理系统中的作用提供了途径。确定哪些微生物及其代谢产物对维持人类健康和疾病至关重要。
了解皮肤微生物群和肠道微生物群之间的相互关系可以帮助我们更好地理解肠-皮肤轴的作用机制。
未来的研究需要更深入地了解皮肤微生物组的分子基础,包括微生物-微生物相互作用、微生物-宿主-微生物相互作用、环境因素-微生物相互作用以及不同细菌菌株之间的互动对宿主健康的影响,为调控皮肤微生物群的紊乱提供更专业的治疗方法。
微生物群研究的相关应用领域
护肤品可以改变皮肤上的分子和菌群多样性及微生物在皮肤上的动力学和结构。微生物群在护肤中的应用是一种先进、前沿的方法。
服装和皮肤之间的关系已成为探索纺织品如何通过调节皮肤微生物群,来治疗或缓解皮肤疾病的新领域。
考虑到生物活性纺织品的广泛应用,需要寻找创新技术和产品。对于抗菌纺织品,根据其对纺织材料和人体皮肤微生物群的主要影响,可以在下述生物医学研究方向中找到有价值的用途:
这些应用使得生物活性纺织品更加绿色环保、安全、高性能,能够提供更多的功能性和舒适性,满足人们对健康和舒适的需求。
通过利用皮肤微生物穿透皮肤屏障的能力,或许可以开发微生物活化免疫细胞来传递细胞因子、小分子化学物质或疫苗。
微生物在皮肤上引发免疫反应,并与皮肤免疫细胞进行相互作用,调节免疫应答和免疫耐受。这为开发新的免疫调节治疗策略提供了新的思路。
通过调节肠道微生物组来靶向皮肤健康是一种很有前景的替代疗法。对于一些慢性皮肤病患者来说,肠道微生物群的紊乱可能是病情加重或难以治愈的原因之一,而通过调整饮食、生活习惯、环境等因素,同时加入益生菌或其他补充剂等,调节肠道微生物群的平衡,增强皮肤对各种病原体的抵抗力;同时通过肠道菌群调节影响自身免疫反应,改善皮肤疾病的症状。
通过皮肤、肠道微生物群联合检测可以探索和评估微生物之间的相互关系,并在皮肤问题的中提供更全面有效、个性化的治疗方案。
主要参考文献:
Townsend EC, Kalan LR. The dynamic balance of the skin microbiome across the lifespan. Biochem Soc Trans. 2023 Feb 27;51(1):71-86.
Kurokawa I, Kobayashi M, Nomura Y, Abe M, Kerob D, Dreno B. The Role and Benefits of Dermocosmetics in Acne Management in Japan. Dermatol Ther (Heidelb). 2023 Jul;13(7):1423-1433.
Harris-Tryon TA, Grice EA. Microbiota and maintenance of skin barrier function. Science. 2022 May 27;376(6596):940-945.
Belzer A, Parker ER. Climate Change, Skin Health, and Dermatologic Disease: A Guide for the Dermatologist. Am J Clin Dermatol. 2023 Jul;24(4):577-593.
Patra V, Bordag N, Clement Y, Köfeler H, Nicolas JF, Vocanson M, Ayciriex S, Wolf P. Ultraviolet exposure regulates skin metabolome based on the microbiome. Sci Rep. 2023 May 3;13(1):7207.
Skowron K, Bauza-Kaszewska J, Kraszewska Z, Wiktorczyk-Kapischke N, Grudlewska-Buda K, Kwiecińska-Piróg J, Wałecka-Zacharska E, Radtke L, Gospodarek-Komkowska E. Human Skin Microbiome: Impact of Intrinsic and Extrinsic Factors on Skin Microbiota. Microorganisms. 2021 Mar 5;9(3):543.
Trompette A, Ubags ND. Skin barrier immunology from early life to adulthood. Mucosal Immunol. 2023 Apr;16(2):194-207.
Alashkar Alhamwe B, López JF, Zhernov Y, von Strandmann EP, Karaulov A, Kolahian S, Geßner R, Renz H. Impact of local human microbiota on the allergic diseases: Organ-organ interaction. Pediatr Allergy Immunol. 2023 Jun;34(6):e13976.
Xu H, Li H. Acne, the Skin Microbiome, and Antibiotic Treatment. Am J Clin Dermatol. 2019 Jun;20(3):335-344.
Zubeldia-Varela E, Barker-Tejeda TC, Obeso D, Villaseñor A, Barber D, Pérez-Gordo M. Microbiome and Allergy: New Insights and Perspectives. J Investig Allergol Clin Immunol. 2022 Oct;32(5):327-344.
De Almeida, C.V.; Antiga, E.; Lulli, M. Oral and Topical Probiotics and Postbiotics in Skincare and Dermatological Therapy: A Concise Review. Microorganisms 2023, 11, 1420.
Flores-Balderas, X.; Peña-Peña, M.; Rada, K.M.; Alvarez-Alvarez, Y.Q.; Guzmán-Martín, C.A.; Sánchez-Gloria, J.L.; Huang, F.; Ruiz-Ojeda, D.; Morán-Ramos, S.; Springall, R.; et al. Beneficial Effects of Plant-Based Diets on Skin Health and Inflammatory Skin Diseases. Nutrients 2023, 15, 2842.
Suellen Ferro de Oliveira C, Kekhasharú Tavaria F. The impact of bioactive textiles on human skin microbiota. Eur J Pharm Biopharm. 2023 Jul;188:66-77.
Santiago-Rodriguez, T.M.; Le François, B.; Macklaim, J.M.; Doukhanine, E.; Hollister, E.B. The Skin Microbiome: Current Techniques, Challenges, and Future Directions. Microorganisms 2023, 11, 1222.
Fernandes A, Rodrigues PM, Pintado M, Tavaria FK. A systematic review of natural products for skin applications: Targeting inflammation, wound healing, and photo-aging. Phytomedicine. 2023 Jul;115:154824.
Lee, H.-J.; Kim, M. Skin Barrier Function and the Microbiome. Int. J. Mol. Sci. 2022, 23, 13071.
Ito Y, Amagai M. Dissecting skin microbiota and microenvironment for the development of therapeutic strategies. Curr Opin Microbiol. 2023 Apr 3;74:102311.
Mourelle, M.L.; Gómez, C.P.; Legido, J.L. Hydrobiome of Thermal Waters: Potential Use in Dermocosmetics. Cosmetics 2023, 10, 94
Azzimonti, B.; Ballacchino, C.; Zanetta, P.; Cucci, M.A.; Monge, C.; Grattarola, M.; Dianzani, C.; Barrera, G.; Pizzimenti, S. Microbiota, Oxidative Stress, and Skin Cancer: An Unexpected Triangle. Antioxidants 2023, 12, 546
Leung MHY, Tong X, Shen Z, Du S, Bastien P, Appenzeller BMR, Betts RJ, Mezzache S, Bourokba N, Cavusoglu N, Aguilar L, Misra N, Clavaud C, Lee PKH. Skin microbiome differentiates into distinct cutotypes with unique metabolic functions upon exposure to polycyclic aromatic hydrocarbons. Microbiome. 2023 Jun 1;11(1):124.
Kengmo Tchoupa A, Kretschmer D, Schittek B, Peschel A. The epidermal lipid barrier in microbiome-skin interaction. Trends Microbiol. 2023 Jul;31(7):723-734.

谷禾健康

人类微生物组包含约 1000多 个常见物种,微生物细胞数量达 10-100 万亿个。微生物大多数遗传库分布在人类胃肠道内,与许多生理过程的发育和功能有内在联系,例如,肠道屏障完整性和稳态、营养、免疫和神经心理行为等。
人类肠道的菌群失调(即共生肠道微生物群的变化对宿主健康产生影响)通常与一系列疾病相关,其中许多疾病无法使用完善的医疗方案进行治疗。
在这种情况下,将健康捐赠者的粪便物质直接移植到受体的胃肠道中(称为粪便微生物群移植,简称粪菌移植,FMT)代表了一种操纵受体微生物群并赋予健康益处的治疗方法。许多国家食品和药物管理局最近的批准表明,粪菌移植和治疗性益生菌鸡尾酒疗法目前正在扩大研究和应用领域。
这种方法成功的关键是能够在患病患者的胃肠道内重建并将肠道微生物群恢复到理想的健康状态。据报道,粪菌移植在临床中治疗艰难梭菌感染成功率接近 90%。
最近发表了几项通过粪便微生物移植(FMT)来治疗炎症性肠病(IBD),特别是溃疡性结肠炎的随机临床试验,但是研究设计存在重大差异。这些包括给药剂量、给药途径和频率、安慰剂类型和评估终点的差异。总体结果看起来很有前景,但它们高度依赖于捐赠者和接受者因素。
近日,来自意大利、法国、英国、美国等 25 名IBD、免疫学和微生物学领域专家多次召开会议,通过对当前可用和/或已发布的数据进行深入评估来制定基于IBD 中与 FMT 相关证据的指南。所有成员对声明进行评估和投票,最终形成全体共识会议并生成拟议指南。
声明包括:
这份共识目的是为使用 FMT 评估、管理和潜在治疗 IBD 制定基于共识的声明和建议,以实现标准化实践。
今天谷禾与大家分享该共识或指南内容,并结合以往研究结果和相关经验阐述说明。我们也相信,在临床研究中明确考虑供体和受体的原有肠道微生物生态原则可以更好地指导FMT试验的设计并有助于提高 FMT 的疗效。
★
生态学是一门核心学科,研究集中构建生物群落的过程和机制,并调节其在空间和时间上的变化。
虽然说,生态学和微生物学在研究复杂群落的方向经常会有交叉部分,但两者在历史上都是作为独立的科学学科发展起来的,在概念、术语、综合和理论的使用上往往有所不同。
临床研究中的 FMT,个体之间粪便物质的移植符合社区合并的概念。这个概念指的是:
整个生物群落及其环境的大规模混合
虽然在宏观生物生态学(即植物和动物)中构成罕见事件,但群落合并在微生物系统中很常见,实际上,群落经常作为一个单元迁移。
这一原则与 FMT 治疗相一致,尽管该术语似乎尚未在临床研究中得到很好的确立。然而,合并后群落重新排列的结果(即植入)更常用于跟踪 FMT 功效。
基于来自供体、FMT前、FMT后接受者的粪便微生物组的微生物菌群监测,才能更好的确定与较高供体菌株植入相关的最重要的临床变量,通过不同的上消化道和下消化道引入的混合途径(即通过胶囊、肠镜检查、鼻胃管、鼻十二指肠管或上内窥镜检查和结肠镜检查),单独或结合进行 FMT 给药,会导致 FMT 后供体菌株植入率更高。
这一发现支持了繁殖压力(即引入的个体数量与独立引入的数量的组合)对于在新环境中成功引入非本地物种的重要性。
发生这种情况主要是因为:
i)初始微生物物种种群的大小可以直接影响其在非本地环境中持续存在的能力
ii)即使是短暂的入侵者也会对微生物组产生影响,有时会促进其在后续环境中的引入入侵尝试(例如,重复入侵尝试的重要性)
其次,在 FMT 给药前接受抗生素的患者中,可能供体菌株植入率更高。已知抗生素的使用会导致肠道微生物群失衡(即非生物紊乱)。这种干扰降低了受体胃肠道微生物群的生物抗性,有利于随后的供体群落植入。重要的是,这个个别研究结果,在推广这一原则时需要谨慎,因为 FMT 之前的抗生素剂量、类型和时间都会导致与 FMT 最终结果的差异。
第三,患有传染病的患者在接受单途径 FMT 给药后,微生物组植入量更大。这与以下观点一致:驻留微生物组的先前不稳定(即,在这种情况下,生物干扰)也会对肠道生物抵抗力产生负面影响。特别是,胃肠道中非本地物种(例如感染期间的致病菌)的存在可以促进其他非本地物种的后续入侵。
★
粪便微生物群移植(FMT)被定义为将健康捐赠者的粪便输注到受体的胃肠道中,以治疗与疾病相关的肠道菌群失调。
根据几项随机对照试验和荟萃分析的报告,FMT是治疗复发性艰难梭菌感染的一种既定且高效的治疗选择,最终制定了国际指南,以标准化其使用艰难梭菌感染的可行治疗方式。
继成功治疗艰难梭菌感染后,FMT 也在炎症性肠病 (IBD) 患者中进行了研究,首先是在非随机研究中,随后在随机对照试验中,尽管 FMT 方案和程序存在显着差异,但两者均显示出有希望的结果。
然而,采用 FMT 治疗 IBD 受到一些限制,包括招募捐赠者、准备粪便材料、确定最佳给药途径以及缺乏明确和既定的监管框架。
解决这些问题的潜在策略包括识别和使用可持续、可重复和标准化的方案,最终目标是改变肠道微生物组的组成。因此,建立最佳的FMT整体框架对于IBD的未来管理具有重要意义。
▼
共识过程是按照以下步骤制定的:
确定成员
确定了 25 名共识成员,他们在微生物学、免疫学、FMT 和 IBD 领域拥有公认的专业知识,并全部参加了专家小组。
注:德尔菲法是在20世纪40年代由O.赫尔姆和N.达尔克首创,经过T.J.戈尔登和兰德公司进一步发展而成的。
德尔菲法作为一种主观、定性的方法,不仅可以用于预测领域,而且可以广泛应用于各种评价指标体系的建立和具体指标的确定过程。
分工作小组
根据个人专业知识,每位成员被分配到四个工作组之一:
每个工作组提出了一份关键问题清单,并制定了与指定主题相关的声明。对于每个关键问题,最佳现有证据是通过对相关文献进行系统审查而获得的。声明以专家意见 (EO) 的形式发布。
详细陈述已上传至在线投票系统(http://scott.armstrong.delphi.stlouisintegration.com/delphi2/),并分发给专家组。
专家评分
每轮审查后,都会收集、处理并与专家小组分享专家的答复。
对于每项陈述,专家们被要求对他们的同意程度进行评分:
如果至少 80% 的受访者对每项陈述表示强烈同意或同意保留意见,则达成共识。
未通过此门槛的声明将在后续轮次投票中进行修改和再次评级,直至达成共识。小组专家于 2022 年 6 月 25 日齐聚罗马,对总体声明进行完善和最终批准。
最终达成共识
经过三轮投票,最终的累积声明达成共识。第一轮和第二轮后,分别有67%和79%的陈述通过了80%的同意门槛,而第三轮后100%的陈述达到了目标水平。
★
声明 A1
IBD 的确切病因目前尚不清楚;然而,其发病机制是多因素的,受遗传易感性、宿主粘膜免疫反应和环境(包括饮食和肠道微生物群)的影响。
评论:
IBD,如克罗恩病 (CD) 和溃疡性结肠炎 (UC),是一种慢性、复发性消化道炎症性疾病,是由于遗传易感个体肠道免疫系统和肠道微生物群之间失去稳态而导致的。由于对肠道微生物群的耐受性失调或将微生物与底层组织分开的上皮屏障破坏,导致不适当的粘膜免疫反应,可能会导致 IBD 的发展或持续。
声明 A2
肠道微生物群的组成、相对丰度、多样性和功能的改变(即菌群失调)促进IBD的发生和进展。
评论:
越来越多的证据表明,肠道微生物组组成的不平衡或“生态失调”是促进 IBD 发展影响最大的环境因素之一,因为改变的微生物群与宿主的相互作用可以触发和促进免疫与 IBD 相关的改变。大量证据表明,IBD 患者肠道微生物组存在特定的共同改变,这些改变与许多功能受损相关,包括短链脂肪酸代谢、氨基酸生物合成、氧化应激调节和毒素,可以单独或共同促进 IBD 的发展。
声明 A3
溃疡性结肠炎和克罗恩病患者的肠道微生物组特别缺乏普拉梭菌,这种细菌因其潜在的抗炎特性而被认可。
关于普拉梭菌详见:
肠道核心菌属——普拉梭菌(F. Prausnitzii),预防炎症的下一代益生菌
评论:
越来越多的临床和实验数据表明,共生微生物群在维持人类和实验性 IBD 的炎症过程中发挥着关键作用。
一些报告显示,IBD 期间微生物多样性下降,尤其是厚壁菌门和拟杆菌门。有趣的是,厚壁菌门的成员普拉梭菌显着减少,并且具有公认的抗炎活性。
相反,活动性 IBD 中的变形菌和放线菌通常升高,以及特定的大肠杆菌菌株也如此。在这种情况下,肠道微生物组的成分在炎症性肠病中发挥着至关重要的作用,这可能代表了一种与细菌加工相关的疾病。因此,肠道微生物组和宿主免疫反应之间存在功能失调的关系,从而引发和维持 IBD 的慢性炎症。
声明 A4
IBD 患者感染艰难梭菌的风险高于一般人群。
评论:
IBD 特异性危险因素,例如免疫抑制、炎症的严重程度和扩展以及 IBD 中观察到的肠道菌群失调,被认为是IBD 患者艰难梭菌感染高风险的主要原因。
IBD 患者并发艰难梭菌感染会导致住院时间延长以及结肠切除率和死亡率增加。鉴于这种情况可能会危及生命的并发症,建议在 IBD 发作期间和早期治疗干预期间筛查艰难梭菌。
艰难梭菌的选择较高毒力、抗生素耐药性和反复感染率增加的菌株使得IBD 中艰难梭菌感染的治疗更具挑战性。因此,建议采用个体化治疗方法来控制IBD 发作期间的艰难梭菌感染。
★
声明 B1
适合 IBD 实验性 FMT 的供体应接受血液和粪便检测,测试符合目前通过 FMT 治疗艰难梭菌感染的国家和国际指南,并且一般用于临床实践。
评论:
以 FMT 为目的的供体筛查的主要原则是避免潜在的传染病传播。用于筛查艰难梭菌感染 FMT 的指定血液和粪便参数已在溃疡性结肠炎患者的多项随机对照试验中被证明是安全的。应提供包含待测试的强制性国际参数的列表,而其他参数则应取决于地理区域(例如,热带地区)、患者的医疗状况或捐赠者的病史(例如,粪便钙卫蛋白增加史)。
除了血液和粪便检测外,还应通过多项调查问卷监测总体健康状况、饮食和心理状态,以避免任何潜在的非传染性不良事件。
声明 B2
粪便捐赠应该是自愿的,并且应告知捐赠者捐赠的潜在风险和/或好处。此外,每位患者必须提供书面知情同意书。
评论:
由于粪便采集程序是非侵入性的,并且可以在不受控制的环境中进行,因此不应允许捐赠者直接从粪便捐赠中受益,以避免样本欺诈。然而,根据国家规定,捐赠者可以获得时间和旅行费用的补偿。此外,捐赠者应了解捐赠的风险和益处,因为筛查过程可能会发现以前未知的疾病(例如艾滋病毒、结直肠癌)的诊断结果或其他疾病的易感性(例如与微生物群改变相关的疾病) 。此外,捐赠者应该知道他们可以随时撤回同意。
声明 B3
可以通过粪便库对供体进行管理,以供实验使用,符合适用于艰难梭菌感染的国家和国际指南和法规,并且一般用于临床实践。
评论:
粪便库能够以标准化方式处理粪便捐赠,这适合 IBD 的进一步临床和实验程序。在粪便库中,捐献者在给患者施用粪便之前经过严格筛选,FMT 制备是标准化的,与临床环境相比,制备 FMT 样本所需的成本和时间都可能减少。此外,粪便库拥有专业知识,因此总体上可能有助于优化 FMT 样本的质量。最后,粪便库可以为无法提供此项服务的 IBD 中心提供 FMT 样本。
声明 B4
捐赠者的粪便最好在粪便库现场或进行实验程序的地点收集,遵循国家和国际指南和法规。
评论:
捐赠者应收到有关如何收集粪便的明确说明,最好是在现场或粪便库收集。如果无法做到这一点,收集的粪便应储存在 4°C 下,并在收集后 6 小时内运送到临床地点或粪便库,由经过培训的人员进行处理。
声明 B5
根据实验方案,每个捐赠者都可以参与捐赠不同的 FMT 制剂。
评论:
供体粪便可用于制备不同的 FMT 制剂(例如,新鲜样品与冷冻样品,以及每个制剂的单个供体与多个供体)。到目前为止,建议使用冷冻 FMT 样品而不是新鲜制剂,主要是因为安全性。冷冻 FMT 样本可以被隔离,直到捐赠期结束时完成全面的捐赠者筛查。对于新鲜的 FMT,在完成完整的筛选过程之前对材料进行管理。因此,对于新鲜的 FMT,建议进行更定期的供体常规筛查和菌群评估。
声明 B6
应根据国家和国际准则和法规维护和存储捐助者信息登记册。
评论:
与 FMT 流程相关的捐赠者信息登记应由国家和国际医疗保健机构监管。来自捐赠者和接受者的信息应保存至少 10 年,或符合国家和国际法规。这些数据应提供给粪便库以获得长期安全数据。
声明 B7
患者不应直接使用粪便库来治疗 IBD。FMT 样本的提供应始终在治疗医疗保健提供者的指导下进行,并符合国家和国际指南和法规。
评论:
粪便库的使用应仅限于医疗保健提供者,因为 FMT 管理需要记录和适当的后续行动,以应对任何潜在的不良事件,而这些不良事件只能在医生的监督下安全地发生。因此,患者直接使用粪便库是不合适的。
声明 B8
FMT 的整个过程(从捐赠者筛选到粪便收集和 FMT 样本管理)应具有清晰的可追溯性。因此,应保留每个 FMT 样品的等分试样以进行测试,以防发生意外的不良事件。
评论:
FMT 过程的所有步骤都应进行登记,捐赠者样本的等分试样应保留并储存在 -80°C 下,以便在发生任何意外不良事件(例如病原体感染)时进行回溯。
据报道,FMT 后出现耐药大肠杆菌传播,这表明保留供体粪便以便在可能的不良事件发生后进行进一步分析的重要性,并保证在此类情况下及时干预。
声明 B9
关于供体 FMT 制备技术方面的共同协议,将有助于在全球范围内提供程序标准化和优化,从而促进结果的解释。
评论:
尽管研究设计不同,FMT 已显示出有希望的结果,尤其是在溃疡性结肠炎中。方案的变化包括不同的 FMT 输注途径(例如,鼻十二指肠、直肠、口服)、给药频率、对照安慰剂(例如,水、自体粪便材料)和终点测量。关于冷冻粪便材料制备、储存和给药量的剂量标准化将有助于未来 FMT 研究的解释和比较,包括结果。
声明 B10
需要进行研究,来确定与粪菌移植作为治疗炎症性肠病选择的临床疗效和整体治疗效果更好相关联的供体特征。
评论:
必须进行研究来确定供体标志物,以实现 FMT 在 IBD 中的最佳治疗效果和总体成功。因此,应该对微生物群、饮食模式(问卷)和其他方面(如药物使用、家族病史、心理状态和遗传背景)进行表征,以确定改善临床结果的潜在趋势。除了特定的供体标记外,还应该研究供体-受体的植入情况。
★
声明 C1
以前进行的随机对照试验一般规模较小,且方法学上存在异质性;因此,目前还不能得出明确的结论。
评论:
在成功用于治疗艰难梭菌感染后,FMT 也在溃疡性结肠炎患者中进行了研究,首先是在非随机研究中,然后在随机对照试验中,均取得了有希望的结果,尽管存在FMT 程序和测量结果存在显着差异。事实上,尽管这些研究和随后的荟萃分析强调供体 FMT 施用后令人满意的缓解率,已发表和/或可用的随机临床试验通常规模较小且方法学上异质,因为它们在粪便输注的时间、数量和途径、FMT 供体粪便与对照(假冒)的特征方面存在差异,使得结果难以从整体上解释结果,无法得出明确的结论。
声明 C2
对于患有IBD的轻度和重度复发或难治性难免性艰难梭菌感染,建议采用FMT作为治疗选择。
评论:
FMT 对于治疗无 IBD 患者的复发性艰难梭菌感染有效, 以及患有溃疡性结肠炎和克罗恩病的患者。然而,针对溃疡性结肠炎患者的研究报告称,单剂量 FMT 无法预防溃疡性结肠炎发作。没有足够的证据推荐 FMT 作为IBD首次艰难梭菌感染的治疗方法。
声明C3
FMT 可有效诱导轻度至中度溃疡性结肠炎的缓解;然而,没有足够的证据推荐 FMT 作为常规临床实践中溃疡性结肠炎的治疗方法,其使用通常应仅限于研究环境。
评论:
迄今为止,FMT 在诱导轻度至中度溃疡性结肠炎患者缓解方面显示出有希望的结果。然而,这些研究是在溃疡性结肠炎患者队列中进行的,样本量相对较小,而且研究设计之间存在差异,使得研究之间的比较难以协调。因此,没有足够的数据支持临床常规使用 FMT 来诱导溃疡性结肠炎患者缓解。但专家们一致认为,FMT可在特定情况下使用,应具体情况具体考虑,并与有关各方详细讨论。
声明C4
随机对照试验表明,在 FMT 后获得缓解的溃疡性结肠炎患者通常在 FMT 治疗后不会持续缓解超过 1 年。
评论:
溃疡性结肠炎中的 FMT 研究缺乏有关治疗效果和持久性的长期随访数据。因此,需要对治疗后的时间进行更持续的疾病监测随访。现有数据表明,复发是可能的,为了实现长期疗效,维持治疗可能是强制性的。然而,诱导治疗和维持治疗的输注次数和剂量均应进一步研究。
声明C5
反复输注和供体-受体移植可能对于 FMT 治疗溃疡性结肠炎的成功非常重要。
评论:
来自对溃疡性结肠炎进行 FMT 的现有随机对照试验的数据表明,重复输注很重要;然而,目前对于 FMT 成功所需的最少管理次数尚未达成共识。此外,FMT 在溃疡性结肠炎中的疗效似乎也依赖于受体,表明供体-受体移植至关重要。除了鉴定对 FMT 成功可能重要的供体标记外,还应研究受体标记及其与供体粪便物质/粪便抗原相互作用的重要性。
声明C6
FMT 后肠道微生物组组成多样性的增加,可能是 溃疡性结肠炎反应的标志。
评论:
除了临床和内镜缓解等临床结果外,还应评估微生物标志物,包括微生物组多样性的改变(即增加),并将其与未来研究中 FMT 的成功或失败相关联。这些标记物可用于预测患者的反应,这有可能实现精准医疗方法的个性化治疗。
声明C7
现有数据表明,FMT 对于轻度至中度溃疡性结肠炎诱导缓解的风险较低;然而,在使用 FMT 治疗 IBD 时已有严重不良事件的报道,包括疾病恶化。
评论:
已进行的随机对照试验中对患者安全性的评估显示出良好的结果,不良事件非常有限,其中大部分与给药方式有关。一般来说,鼻十二指肠给药比直肠给药灌肠产生的不良事件相对较多;因此,后者已被普遍认为是更安全的递送途径。然而,迄今为止,尚缺乏溃疡性结肠炎患者的长期 FMT 安全性数据。
事实上,在随机对照试验中,在有限数量的患者(<10%)中观察到了严重的不良事件。这些包括当通过上消化道给药进行 FMT 时误吸和疑似小肠穿孔。与 FMT 手术相关的最常见的严重不良事件是疾病恶化,需要住院治疗,在少数情况下需要结肠切除术。
在 Costello等人的试验中,观察到 43 名接受自体 FMT 的患者中有 2 名病情恶化,而 38 名接受捐赠者 FMT 的患者中有 1 名病情恶化。
在 Moayyedi 的试验中,38 名患者中有 2 名出现斑片状炎症和脓肿。其他显着的严重不良事件包括肺炎、艰难梭菌感染或其他形式的小肠结肠炎。
声明C8
随机对照试验尚未证明 FMT 和对照组在疾病恶化或轻微或严重不良事件引起的症状方面存在显着差异。
评论:
溃疡性结肠炎中的 FMT 试验对不良事件进行了评论;然而,由于对照组和治疗组之间没有发现显着差异,因此这些不良事件被认为是给药程序的结果,而不是加工后的供体粪便材料本身。
声明 C9
溃疡性结肠炎中 FMT 后,常见的不良事件是短暂的轻微胃肠道症状,如腹胀、腹泻和肠胃气胀。
评论:
FMT 试验中高达 83% 的患者观察到轻微不良事件,包括胃肠道症状,如短暂性腹泻、肠鸣、腹痛、腹胀和肠胃气胀。也有短暂发烧的报道。大多数不良事件在手术后几天内自然消失。
声明 C10
在临床实践中,没有足够的证据推荐 FMT 作为克罗恩病的治疗方法。迄今为止,其使用应仅限于研究环境。
评论:
克罗恩病中 FMT 的可用数据非常有限,主要包括病例报告和试点研究,而不是大型随机对照试验。这些研究显示不良事件,主要包括胃肠道症状,疾病发作报告为与 FMT 相关的严重不良事件。
Vermeire等人的一项初步研究显示,六名难治性 克罗恩病患者在 FMT 后第 8 周没有差异。
在评估 FMT 在维持克罗恩病缓解方面的效果时,Sokol等人报告称,与假手术组相比,FMT 组的急性发作发生率没有显着降低。
进一步的研究应包括优化该患者群体的缓解诱导和维持。大型随机对照试验必须推荐 FMT 作为克罗恩病患者的可行治疗方法。
声明 C11
在临床实践中,没有足够的证据推荐 FMT 作为贮袋炎(Pouchitis)的治疗方法。迄今为止,其使用应仅限于研究环境。
注:贮袋炎(Pouchitis)是保留肛门的大肠全切除术(或次全切除术)术后发生在患者回肠贮袋的非特异性炎症,是溃疡性结肠炎行回肠贮袋-肛管吻合术后最为常见的并发症。
本病病因不明。大约有1/3的患者在术后5年内可能出现这种难以解释的炎症,其中2/3的患者有复发的可能性。
评论:
据信,患有贮袋炎的患者也会出现菌群失调。目前,只有有限的研究使用 FMT 来治疗贮袋炎。然而,在已发表的文献和病例报告中,该手术大多被报道是安全的,但并不有效。
与溃疡性结肠炎中 FMT 的随机对照试验类似,所进行的研究也具有异质性,并评估了不同的结果测量。因此,需要进一步研究和优化方案来确定 FMT 在贮袋炎中的潜在用途。
声明 C12
关于 FMT 在克罗恩病和贮袋炎中的安全性数据不足。
评论:
基于缺乏大型随机对照试验和长期随访数据,目前无法得出FMT治疗克罗恩病和储袋炎的安全性结论。
声明 C13
需要进一步的研究来确定 FMT 在克罗恩病和贮袋炎中的有效性和安全性。
评论:
需要对克罗恩病和储袋炎的 FMT 进行进一步研究和优化,以评估疗效并生成(长期)安全性数据,以便将 FMT 引入临床环境。
★
展望D1
未来的研究需要确定精准的 FMT 供体和受体用于治疗 IBD 的最佳特征。
评论:
FMT 已被证明是一种有前景的溃疡性结肠炎治疗策略。然而,有效率似乎受到供体特异性、受体特异性和手术特异性特征的影响。此外,供体-患者移植获得了更多支持,有利于确定通用的“超级供体”。因此,需要进一步的研究来定义理想的患者和捐赠者特征及其最佳移植。
展望D2
有必要进行对照FMT试验,以优化IBD特定表型的疗效。
评论:
未来的研究在考虑 IBD 中 FMT 的结果时应考虑严格定义的患者表型。这些研究有可能确定 FMT 给药后与阳性反应或缺乏反应相关的特定表型。
展望D3
需要确定预测 IBD 中 FMT 反应的生物标志物。
评论:
除了确定供体和受体以及患者表型可能的最佳特征外,还需要确定免疫学和微生物生物标志物来预测对治疗的反应,以避免因 FMT 治疗无反应而造成时间、成本和不良事件的损失。
此类生物标志物可以通过 16S rRNA 扩增子测序、鸟枪法宏基因组测序、蛋白质组学和/或转录组分析来检测。通过识别此类生物标志物,有可能为 IBD 的 FMT 治疗设计更有针对性的策略,从而朝着更加精确、个性化的医疗方法发展。
展望D4
未来的研究需要确定基于 FMT 的 IBD 治疗的最佳配方和给药途径。
评论:
提高 FMT 功效的最佳剂量尚未确定。迄今为止,鼻十二指肠管输送以及通过结肠镜检查或直肠灌肠进行 FMT 输注已成为 IBD 研究最多的给药途径。
然而,最近的一项研究利用含有冻干粪便微生物群的口服胶囊作为其递送系统。这种给药途径侵入性较小,因此可能更受患者青睐。需要更多的随机对照试验来优化剂量和给药途径。
展望D5
需要进行研究来积累有关使用补充策略来提高 FMT 疗效的循证信息。
评论:
通过 FMT 调节肠道微生物组成的成功率可以通过补充策略来提高,例如捐赠者和接受者的支持性抗炎饮食, 通过优化FMT前的肠道准备,也可以通过抗生素预处理。如益生菌、益生元、合生元和后生元补充。
扩展阅读:
展望 D6
需要进行研究来评估 FMT 作为 IBD 的独立治疗方法或与当前可用的治疗方式相结合的作用。
评论:
除了评估补充策略(D5)之外,还需要进一步研究 FMT 与目前使用的 IBD 联合疗法(从皮质类固醇到生物制剂和 JAK 抑制剂)的组合。
这种针对免疫反应和肠道微生物组成的组合方法可能比单独的每种策略带来更高的缓解率。此外,应研究预防发病和/或术后复发,以及 FMT 对癌症治疗的影响,反之亦然。
★
本手稿的作者代表 IBD 各个方面的国际专家组,他们一致认为,在将 FMT 推广为公认的 IBD 治疗策略之前,有必要进行进一步的研究。人们普遍认为 FMT对于 IBD 患者是安全的,特别是对于溃疡性结肠炎患者。
文献中报道的大多数并发症主要与粪便输注的给药途径有关,而不是与感染传播有关。然而,为了避免不良事件的负担,应遵循针对艰难梭菌感染的 FMT 治疗现有的国际指南,来严格筛选捐赠者。
IBD 中的粪便微生物群移植 (FMT) 途径:从健康供体到恢复疾病患者的肠道微生物群
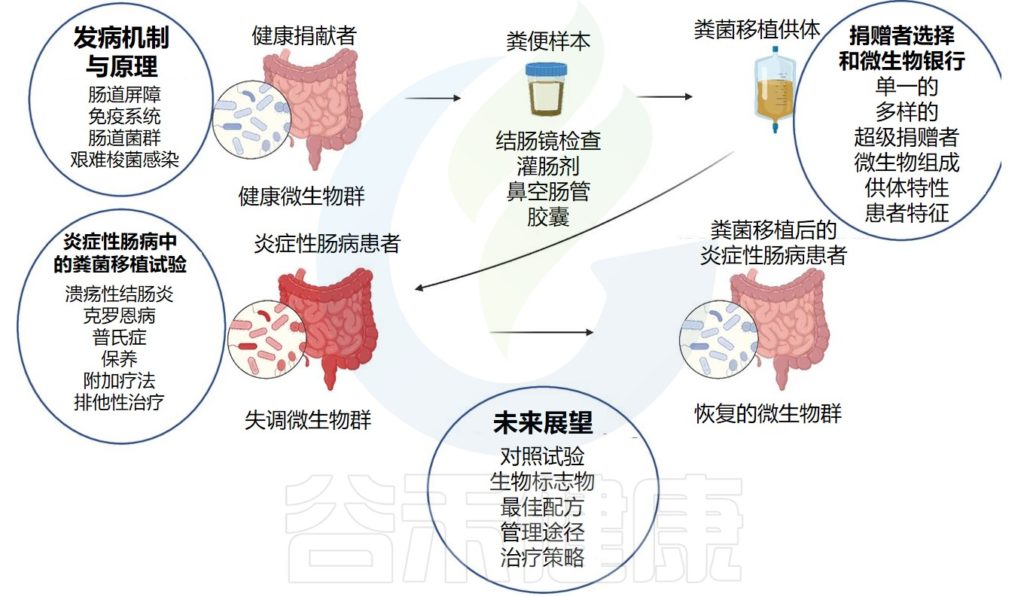
doi: 10.1136/gutjnl-2023-329948.
应投入粪便库以促进FMT研究,并应考虑通过实施永久捐赠者登记、通过微生物群特征和捐赠者健康状况筛选捐赠者粪便材料以及优化捐赠者粪便材料、粪便样本的储存,来治疗IBD的可能性。
粪便库应登记所有捐献者和患者数据,以便有效追踪并监测 FMT 施用后健康状况的变化(例如缓解/发作、心理状态)。
已经进行了几项 FMT 试点研究和随机对照试验来治疗 IBD,但使用的是异质研究设计。现有的结果,特别是在溃疡性结肠炎方面,是有希望的,但似乎依赖于供体和患者。然而,要将 FMT 纳入日常胃肠道实践中,还需要进一步研究来优化短期和长期成功率,并进一步评估安全性。
该方法应确定:最佳给药途径、剂量、频率、供体-受体植入、患者表型,以及 FMT 反应的免疫学和微生物组生物标志物的识别。
总而言之,这样将有助于 FMT 的标准化及其治疗溃疡性结肠炎的临床应用。对于克罗恩病和储袋炎,必须进行进一步的研究来评估其使用的(长期)安全性和有效性。尽管如此,这一系列研究可能会从优化其在溃疡性结肠炎中的使用所采取的措施中受益匪浅。
未来的工作包括对供体微生物群进行严格表征,以及调查 FMT 前后对 IBD 接受者的影响,这可用于最大限度地提高 FMT 功效并阐明作用机制。
通过研究供者和受者的支持性饮食、肠道准备、抗生素预处理、益生菌、益生元、合生元和生元后支持,以及与 IBD 治疗同时进行的联合治疗,进一步提高 FMT 的疗效至关重要。此外,识别与 FMT 成功预测相关的特定微生物菌株,可能会开发出明确的单菌株或多菌株益生菌。
参考文献:
Dini-Andreote F, Custer GF. Ecological principles of fecal microbiota transplantation. Trends Microbiol. 2023 Jun 8:S0966-842X(23)00162-2.
Lopetuso LR, Deleu S, Godny L, Petito V, Puca P, Facciotti F, Sokol H, Ianiro G, Masucci L, Abreu M, Dotan I, Costello SP, Hart A, Iqbal TH, Paramsothy S, Sanguinetti M, Danese S, Tilg H, Cominelli F, Pizarro TT, Armuzzi A, Cammarota G, Gasbarrini A, Vermeire S, Scaldaferri F. The first international Rome consensus conference on gut microbiota and faecal microbiota transplantation in inflammatory bowel disease. Gut. 2023 Jun 20:gutjnl-2023-329948.

谷禾健康

我们知道,人体的皮肤、口腔、肺部、肠道、阴道等都是微生物的栖息地,每个部位都有独特的微生物群组成。微生物群受到基因、饮食、环境和生活方式等多种因素的影响。
当然,人体微生物群的组成也会随着年龄的增长而发生变化。从婴儿期到老年阶段,微生物群的种类和数量都会发生变化,这些微生物与人体形成了错综复杂的共生关系,这对我们的健康和免疫系统功能产生重要影响。
人体微生物群与发育、免疫、营养、神经、代谢稳态等方面有密切关联。反过来,宿主也会提供营养并促进健康和有弹性的微生物群的发展。
了解以上这些可以帮助我们更好地理解微生物与人类的共生关系,拓展对微生物多样性和生态系统的认知,为预防和治疗相关疾病提供新的思路。
本文讲述了不同年龄和不同部位的人体微生物群、影响微生物组成的各种因素、微生物与宿主的相互作用(包括对生理、疾病的影响及相关治疗中的作用)、及其对于健康管理和疾病治疗的价值。
目录
•人体不同部位的微生物群
•不同年龄下的微生物群
•影响微生物组成的因素
•微生物群对宿主生理的作用
•微生物群与疾病的关联
•微生物群与医学治疗
•结语
不同环境提供了不同的营养资源、温度、湿度、pH值等生态因素,这些因素会影响各种微生物的生存和繁殖。
多样化的全球微生物数据集
Hogeweg P,et al.Nat Ecol Evol.2023
a:样本来自截然不同的带注释的生物群落和研究设计;
b:样本的地理分布;
c:每个样本的分类注释读数总数(n = 22,518 个样本);
d:来自相似注释生物群落的样本根据 t-SNE 可视化中的分类概况(困惑度 = 500)聚集在一起;
e:类群丰富度因注释的生物群落和分类等级而异
人体微生物群是指在人体内外生活的微生物群落,包括细菌、真菌、病毒等。这些微生物群落分布在人体的不同部位,如口腔、皮肤、肠道、生殖道、大脑等。人体的不同部位提供了不同的环境条件,适合不同类型的微生物生长和繁殖。
每个部位的微生物群落都有其独特的组成和功能,它们与人体之间存在着相互作用和影响。
注:肠道微生物是人体内最丰富、最多样化、功能最大的微生物群落
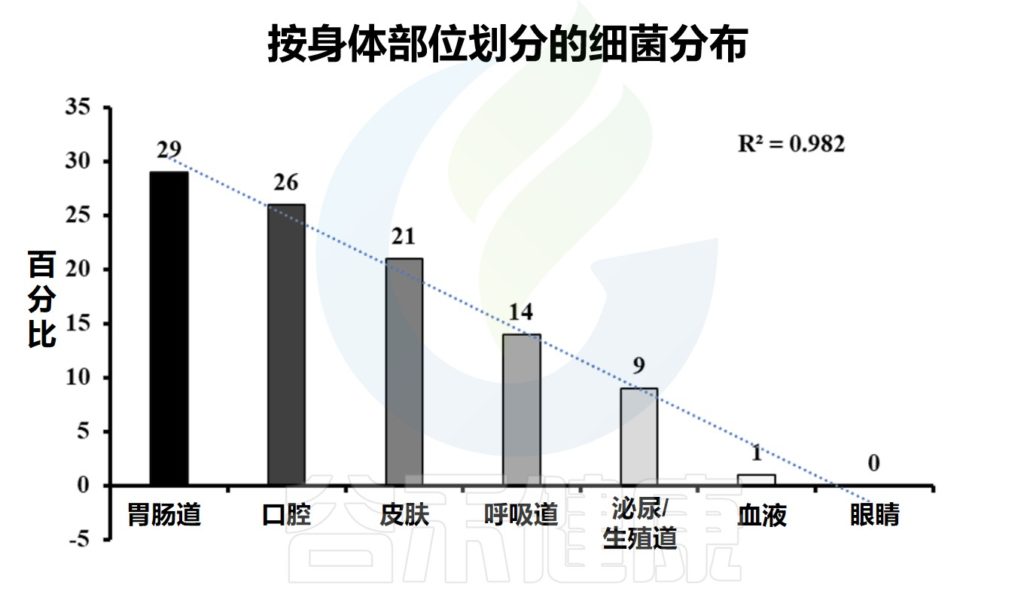
Ayariga JA,et al.Arch Microbiol.2022
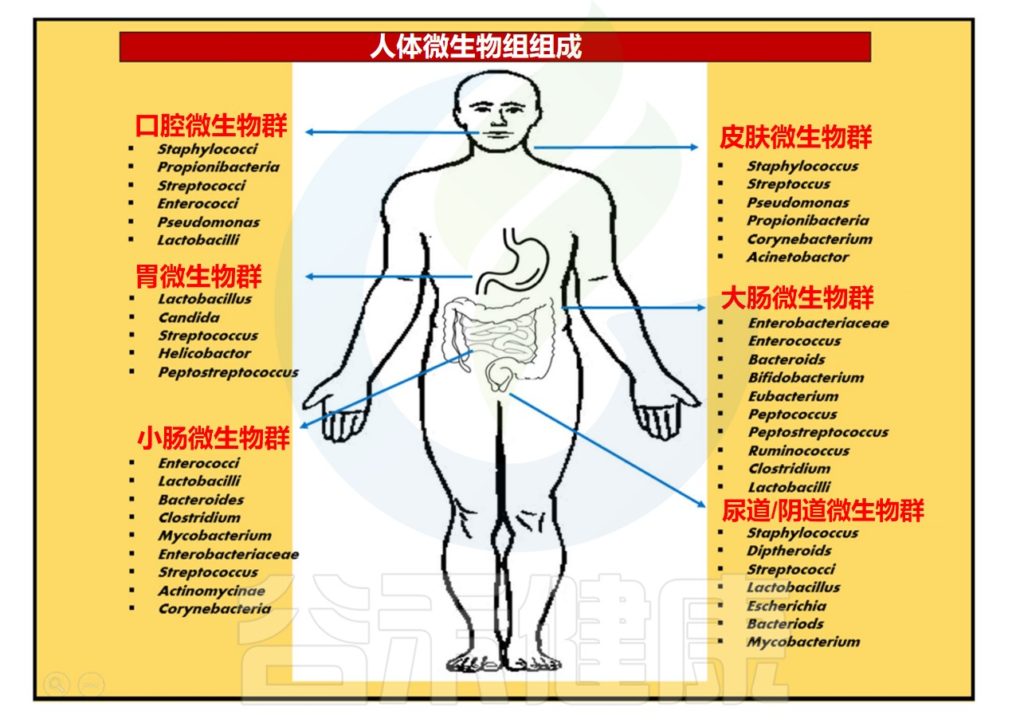
Zaidi S,et al.Arch Microbiol.2023
在人体内,微生物群主要包括以下几个方面:
▼
肠道提供了温暖、潮湿、酸性和富含营养物质的环境,适合多种菌群的繁殖。此外,肠道还有大量的食物残渣和纤维素,为益生菌提供了生长的基质。
肠道中居住着高度多样化的微生物群落,其肠道内容物密度达到10^12个微生物/毫升,包含超过1000万个基因。
一般来说,肠道首先由兼性厌氧菌如肠球菌(Enterococci)和肠杆菌(Enterobacteria)定植,然后由专性厌氧菌定植。
肠道微生物群所拥有的基因编码了数千种微生物酶和代谢物。它们在消化、降解、消除有毒化合物、将难消化的复合糖聚合物转化为短链脂肪酸和维生素等多种功能中发挥着关键作用。
肠道菌群的分布
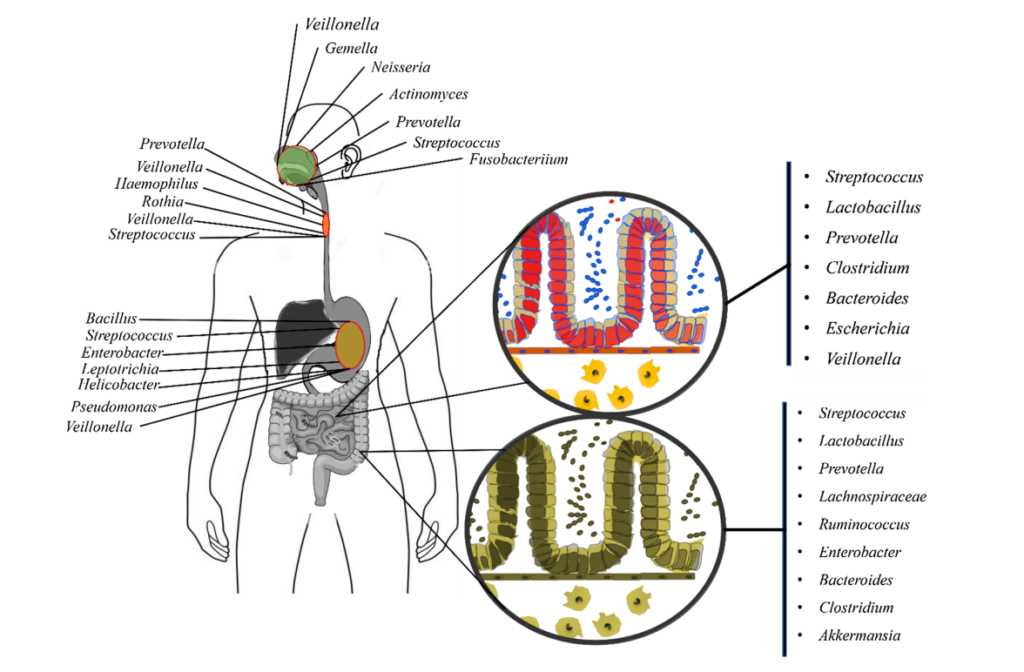
Ayariga JA,et al.Arch Microbiol.2022
▸ 空肠和回肠中主要的微生物群
通过分析空肠和回肠内容物,发现最丰富的群落是链球菌(Streptococci)、乳杆菌(Lactobacilli)、γ-变形杆菌、肠球菌(Enterococcus)和拟杆菌(Bacteroides)。
随着小肠远端向回肠推进,微生物群落变得更加复杂。回肠末端以梭菌科、毛螺菌科、消化链球菌科、瘤胃球菌科、肠杆菌科和拟杆菌科较丰富。
此外,十二指肠还含有与胃相似的菌属,包括肠杆菌科、链球菌科、韦荣氏球菌科和假单胞菌科。
▸ 结肠中主要的微生物群
结肠中栖息的微生物群丰富多样,主要包括放线菌门、拟杆菌门、厚壁菌门、变形菌门和疣微菌门。
与小肠相比,结肠黏液有更明确的层状组成。结肠黏液层具有物理清除细菌的内黏液层,并含有针对微生物群的免疫效应细胞。外层是松散的,为许多微生物提供了一个定植点。
嗜黏蛋白阿克曼菌(Akkermansia)、瘤胃球菌(Ruminococcus)和一些拟杆菌属是肠道黏液外层的居民。
★ 肠道核心微生物群
此外,从肠腔到粘膜存在氧梯度,并通过结肠向下移动,对结肠微生物组成产生影响。大部分细菌种类总是存在并形成稳定的核心微生物群。
这些核心微生物包括另枝菌属(Alistipes)、拟杆菌(Bacteroides)、经黏液真杆菌属(Blautia)、粪杆菌(Faecalibacterium)、瘤胃球菌属(Ruminococcus)、罗氏菌属(Roseburia) 、 普拉梭菌(Faecalibacterium prausnitzii)和颤螺菌属 (Oscillospira)。
▼
口腔是微生物群落多样性排名第二的地方,大约有700种不同亚群的细菌。
人体口腔包含牙龈、面颊、扁桃体、舌头、牙齿、软硬腭等多种微生物环境。口腔提供了温暖、潮湿和富含碳水化合物的环境,适合细菌的繁殖。此外,口腔还含有唾液,其中的酶可以帮助控制微生物的生长。
由于口腔内含有众多菌群,因此有自己的数据库——人类口腔微生物组数据库。健康人的唾液中含有Gemella、韦荣氏球菌属(Veillonella)、奈瑟菌属(Neisseria)、梭杆菌属(Fusobacterium)、链球菌属(Streptococcus)、普氏菌属(Prevotella)、Pseudomonas、放线菌属(Actinomyces) 等多个属,占总分类群的96%。
▼
早期的理论认为胃是一个无菌器官,不适合细菌生存,然而胃腔内幽门螺杆菌的发现打破了这一观点。
采用16S rRNA测序技术的研究进一步表明,胃内存在着链球菌(Streptococcus)、假单胞菌(Pseudomonas)、肠球菌、葡萄球菌(Staphylococcus)、以及变形菌门、放线菌门、厚壁菌门、拟杆菌门和梭杆菌门。
▼
呼吸道包括鼻腔、咽喉和肺部等部位。这些部位通常比较干燥,但仍然存在微生物的定居。
▸ 鼻腔中的微生物群
鼻腔是人体呼吸道的入口,也是微生物的第一个定居地。鼻腔内存在多种细菌,如葡萄球菌、链球菌等。这些细菌可以与宿主共生,帮助抵御潜在的病原体侵袭。
鼻腔内还有纤毛和黏液,可以帮助清除微生物
▸ 咽喉处的微生物群
咽喉是连接鼻腔和气管的部位,也是呼吸道的一部分。咽喉内存在多种细菌,包括厌氧菌和革兰氏阴性菌等。这些细菌参与了呼吸道的免疫调节和防御功能。
▸ 肺部的微生物群
正常情况下,肺部是相对无菌的环境。然而,在某些情况下,如免疫系统受损或存在呼吸道感染时,肺部可能会受到微生物的感染。常见的肺部微生物包括肺炎链球菌、流感病毒等。
▼
皮肤被认为是人体最大的器官。皮肤是一个动态的、复杂的生态系统,其中含有许多共生细菌。皮肤是人体最外层的保护屏障,同时也是微生物的栖息地。皮肤表面有油脂和汗液分泌物,提供了微生物生长所需的水分和营养物质。
注:研究表明,皮肤的生理特征,如温度、湿度、pH值、皮脂含量等,会影响和塑造微生物群。皮肤微生物群落的变异性和多样性还受到人口统计学、遗传学、区域环境波动等因素的影响,从而导致微生物群落结构的改变。因此,皮肤微生物群应该是独一无二的,因此可以作为“微生物指纹”。
最近的研究,在毛囊深处发现了大量细菌。棒状杆菌(Corynebacterium)和葡萄球菌(Staphylococcus)在特定的身体部位如脚底和腘窝繁殖良好。
✦湿润和干燥皮肤下的微生物群不同
大多数不同种类,以及不同相对丰度的厚壁菌门、变形菌门、拟杆菌门和放线菌门都被发现存在于干燥的皮肤中。
注:变形杆菌定植于深层的皮肤区域,可能参与控制宿主和环境之间的皮肤稳态。
棒状杆菌(Corynebacterium)是一种普遍存在于湿润和干燥皮肤的菌属,在表皮区比真皮区数量更丰富。
此外,Pelomonas spp是皮肤群落的核心共生生物之一。对金黄色葡萄球菌、表皮葡萄球菌、痤疮丙酸杆菌、马拉色菌等皮肤相关微生物进行分析,有助于阐明其复杂的分子机制及与皮肤的关联。
注:后三种被发现在湿润的皮肤占主导地位。
▼
阴道酸性较高,含有乳酸菌等益生菌。阴道微生物群被认为是预防许多泌尿生殖系统疾病的关键,可以防止病原体在阴道内定植。如艾滋病毒、细菌性阴道病和酵母菌感染。
然而,与月经周期相关的激素变化可以显著改变微生物群的组成,并在阴道微生物群的动态中发挥主导作用。
✦女性生殖道微生物群
测序表明,主要的乳杆菌属,如卷曲乳杆菌(L.crispatus)和惰性乳酸杆菌(L.iners)构成了一个“健康”的阴道微生物群。
这些物种产生乳酸、抑菌和杀菌分子,创造一个低pH的生态位,并通过竞争排斥提供保护。阴道微生物群的一部分在分娩时传递给婴儿,这反过来又推动了新生儿消化道中微生物群的初始定植。
✦男性生殖道微生物群
与女性生殖道和其他身体部位相比,男性生殖道微生物群的鉴定和研究一直较少。
男性下生殖道(即尿道和冠状沟)的微生物群主要由放线菌门、梭菌门、厚壁菌门、拟杆菌门和变形菌门组成,尽管受试者之间存在很大的差异。
注:与未行包皮环切术的个体相比,人工干预包皮环切术导致革兰氏阴性菌和厌氧菌的丰度减少。
▼
一系列突破性研究表明神经和神经胶质细胞内存在微生物。然而,生活在大脑中的共生细菌比肠道中的要少的多。
大脑常驻微生物的RNA测序显示,这些微生物属于肠道中常见的门,即厚壁菌门、变形菌门和拟杆菌门,它们可能影响情绪、行为或使个体易患神经系统疾病。
注:先前的研究表明,弓形虫可以侵入大脑,但不会引起明显的疾病。
大脑微生物群主要在黑质、海马体和前额叶皮层的星形胶质细胞内,这些发现有助于未来研究与神经精神疾病的关联。
▼
人类的血液通常被认为是无菌的,然而最近的研究表明,健康的人有一个血液微生物群。
✦血液中的微生物主要是其他部位转移而来
来自不同队列的测序数据描述了9770名健康人血液中的微生物。过滤污染物后,血液中有117种微生物,它们主要是来自胃(n=40)、口腔(n=32)和泌尿生殖系统(n=18)的共生菌,而不是医院血液培养中发现的病原体。
这些发现不支持人类血液固有稳定核心微生物群的观点。相反,它支持共生微生物从其他身体部位暂时和偶尔转移到循环中。
越来越多的证据表明,年龄与人类微生物群之间的关联很大,随着年龄的增长,人体内的微生物群落会发生变化。
人类相关微生物群从出生到死亡的变化
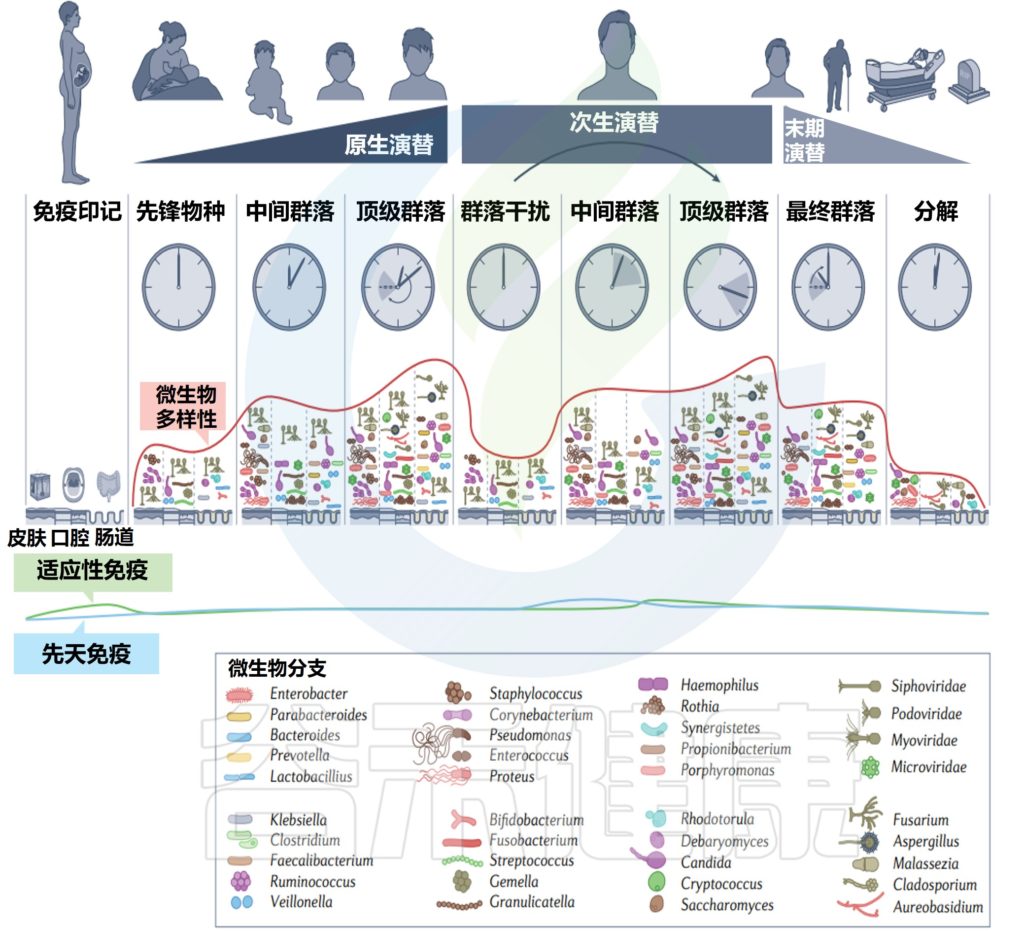
Martino C,et al.Nat Rev Microbiol.2022
美国一项研究集中测量了从儿童到老年人的粪便(a部分)、口腔(b部分)和皮肤(c部分)微生物群的细菌多样性,该项目包含21919个粪便、1920个口腔和998个皮肤微生物群样本。
α多样性,一种对样本中不同类型微生物数量的定量测量,通过Faith的系统发育多样性(PD)α多样性度量跨年龄测量。
UniFrac β多样性主坐标分析,一种用于比较微生物群落相似性的方法,其中空间上接近的点表示相似的样本,空间上远离的点表示不同的样本,按年龄着色。
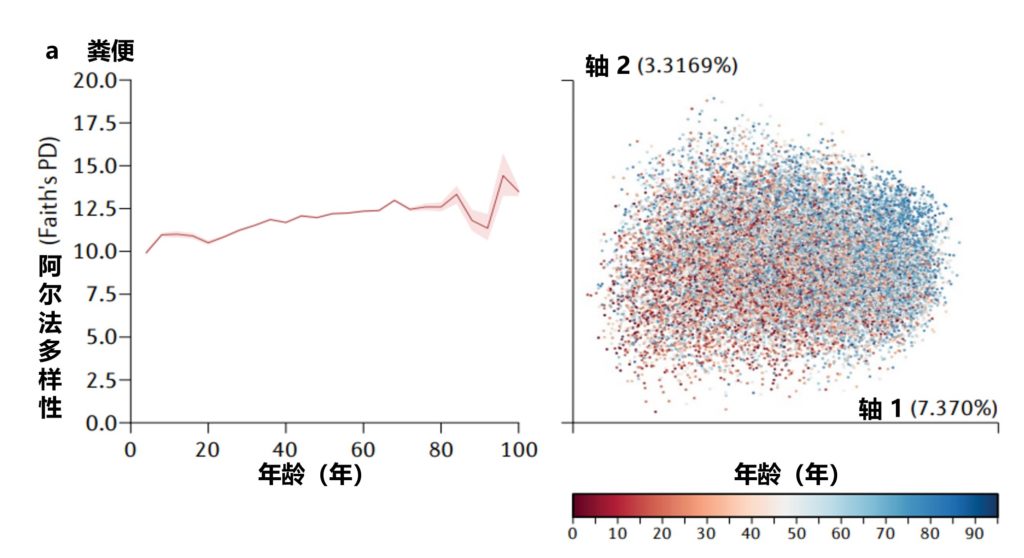
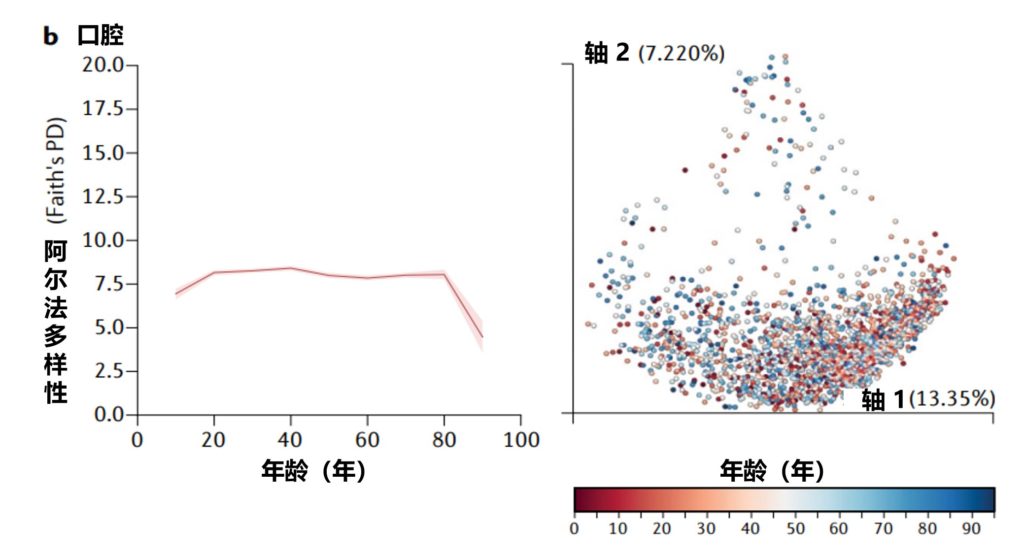
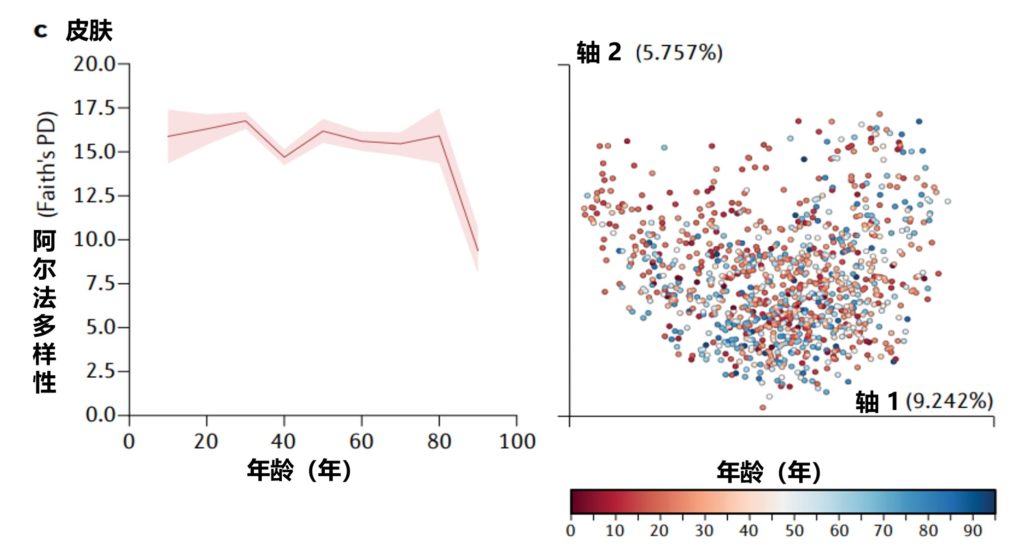
Martino C,et al.Nat Rev Microbiol.2022
▼
关于出生时获得的微生物群是否通过混合来源于阴道和粪便,或者阴道微生物群本身在出生时是否具有多能性,是否是微生物先驱的主要来源,存在一些争议。
无论确切的母体来源如何,这一阶段的特征是先锋细菌种类。包括下列菌群:
•Lactobacillus
•Enterobacter
•Escherichia
•Bacteroides
•Parabacteroides
•Prevotella
这些细菌定居在常规身体部位:肠道、口腔和皮肤。
许多先锋细菌是兼性厌氧菌,它们会消耗氧气,从而使专性厌氧菌能够在以后的环境中定居。先锋细菌进驻后,生命早期的微生物群逐渐开始形成。
双歧杆菌属(Bifidobacterium)在婴儿刚出生时占主导地位,直到在生命的第一年结束时,它们被双歧杆菌、梭状芽孢杆菌和拟杆菌的组合所取代。拟杆菌属的丰度增加,而双歧杆菌属等物种的丰度相对减少。
▼
健康成年人中的微生物群也会随着时间的推移而进化,不过功能和组成的进化以较稳定状态发生。饮食或是疾病会在一定时间内改变成年人的微生物群构成。
•不同季节饮食对微生物的影响
一个经过充分研究发生在几周到几年范围内变化的例子是饮食驱动的肠道微生物群的改变。
例如,坦桑尼亚哈扎部落在旱季食用富含肉类和块茎的饮食,但在雨季食用富含蜂蜜和浆果的饮食,拟杆菌等属中表现出较大的季节波动。
饮食对微生物群的影响也可能在人类健康中发挥作用,许多工作致力于了解特定的饮食成分和总体饮食模式如何影响微生物群及其对健康的影响。
•疾病在短期内改变微生物群
肠道中的许多疾病,如炎症性肠病,破坏了微生物群落,但没有达到新的稳定群落组成,而是在没有干预的情况下继续长期不稳定。
在皮肤上,特应性皮炎的特征是免疫介导的炎症引起的金黄色葡萄球菌大量繁殖和细菌多样性减少。在金黄色葡萄球菌大量繁殖期间观察到马拉色菌属的数量减少,反之亦然,真菌数量增加导致金黄色葡萄菌数量减少。
除了饮食和疾病,还有许多其他因素会在一定时间内影响成年人的微生物群,包括地理因素、压力、代谢情况等。
注:谷禾将在下一章节中具体讲述影响人体微生物群的因素
▼
衰老会影响细胞功能的各个方面,微生物群也不例外。随着年龄的增长,微生物群α多样性减少,β多样性增加。
•肠道
一般而言,老年人肠道中观察到的微生物群变化是年轻成年人中占优势和普遍的细菌属丰度减少,如双歧杆菌(Bifidobacteria), 拟杆菌(Bacteroides),乳杆菌(Lactobacillus), 抵御机会细菌爆发的能力降低。
•皮肤
在65岁及以上的人群中,genera Cutibacterium和Staphylococcus的皮肤细菌数量减少,同时观察到更多的Corynebacterium。
•口腔
在口腔部位,Rothia和Streptococcus spp.是核心口腔细菌群落,Porphyromonas,Treponema和Faecalibacterium spp.的数量持续减少。
由于微生物组的动态特性,它在空间和时间上会不断变化,还与个体的健康状况有关。
这些变化的程度和后果取决于性质、持续时间、扰动强度和微生物群的结构和稳定性。
已经发现许多因素影响微生物群的组成,如分娩方式,年龄、性别、地理位置、饮食、怀孕、昼夜节律、宿主遗传和社会经济地位,药物以及其他一些因素(益生元和益生菌补充剂,手术和非手术治疗)。
Zaidi S,et al.Arch Microbiol.2023
下面谷禾列举了其中一些对微生物群组成有重要影响的因素。
1
上一章节讲述了微生物群会随着年龄而变化。微生物群经历了一系列的发育阶段,它们的复杂性和丰富性提高,从新生儿期到断奶后的明显稳定。
这样的初级共生群落不断进化,变得更加多样化和稳定。一旦儿童达到3岁,其微生物群变得与成人的微生物群相似。
2
出生方式会决定了最初的微生物。在剖腹产和顺产婴儿之间有惊人的微生物差异。
√阴道分娩的婴儿微生物群和母亲更相似
在阴道分娩时,孩子接触到女性产道的微生物,导致母亲和孩子的微生物组成相似。这些婴儿的肠道中含有大量的乳杆菌(Lactobacillus)和普雷沃氏菌(Prevotella)。
√剖腹产分娩的婴儿微生物群主要来自母亲皮肤
相反,在剖腹产分娩的母亲和婴儿之间没有观察到大量微生物群重叠。通过剖腹产出生的新生儿从母亲的皮肤中获得细菌群,从而导致链球菌(Streptococcus)、棒状杆菌(Corynebacterium)和丙酸杆菌(Propionibacterium)较多。
剖腹产出生的婴儿在头六个月中乳杆菌的丰度较低,不像顺产的婴儿,在前六个月的时间里乳杆菌的百分比都在增加。
不过一旦儿童满3岁,乳杆菌检出率的这种差异就会消失。产后,尤其是拟杆菌和双歧杆菌在剖腹产出生的婴儿肠道内的定植也较晚,相反,他们的艰难梭菌水平增加。
注:剖腹产似乎是婴儿早期微生物群落破坏的原因之一。这种在剖腹产时定植的躁动扰乱了微生物与宿主的相互作用,这可能进一步表现为代谢紊乱的形式。在剖腹产分娩后的最初两年里,婴儿患特应性疾病的比例更高。
3
一旦婴儿出生,影响微生物群组成的最重要因素是婴儿饮食,要么是配方奶,要么是母乳。
√母乳喂养有助于诱导肠道微生物群成熟
饮食塑造了早期的微生物群,尤其是肠道中的微生物群。母乳中含有低聚糖,容易被乳酸菌和双歧杆菌(普遍存在于母乳喂养的婴儿肠道中)代谢,从而导致短链脂肪酸浓度上升。
这些短链脂肪酸进一步控制免疫系统过度表达免疫球蛋白G,并诱导新生儿肠道微生物群的成熟。
而在用配方奶粉喂养的婴儿中,常见的种类是肠球菌、肠杆菌、拟杆菌、梭菌和链球菌。
√母乳喂养下得婴儿免疫系统更完善
婴儿时期的微生物群定植似乎在整个儿童生长阶段的早期免疫发展中起着关键作用。因此,初始微生物群的组成是重要的,因为它可以防御可能由于免疫力低下而引起的多种疾病。
许多研究比较了母乳喂养和配方奶喂养的新生儿的肠道微生物群和粘膜免疫反应。观察到母乳喂养导致更稳定和更好的粘膜免疫反应。
相反,依赖配方奶粉的婴儿在以后的生活中发现免疫系统发育受损以及代谢不正常。在哺乳期间,影响母乳成分的生理和激素波动也可能影响微生物群的组成。
4
抗生素会扰乱微生物群结构。它们不仅对消化道上下段微生物的系统发育组成有不同的影响,而且对去除抗生素后微生物群落的恢复也有不同的影响。
√抗生素会减少微生物多样性
抗生素的使用导致肠道微生物多样性的减少,耐药物种的增加,宿主的应激反应和噬菌体基因的表达。
使用抗生素是一把双刃剑:它消除了病理微生物和有用微生物,最终导致生态失调。研究表明,一些抗生素如克林霉素、克拉霉素、甲硝唑和环丙沙星对微生物群结构的影响是长期的。
下面列举了使用一些抗生素后的微生物变化:
克林霉素可以持续2年而不恢复拟杆菌的多样性;同样,使用克拉霉素对抗幽门螺杆菌导致放线菌数量减少,然而环丙沙星已被提出导致鲁米诺球菌数量减少。
万古霉素是治疗艰难梭菌感染的最佳药物,但它也会引起肠道微生物群的改变,导致艰难梭菌感染的复发性感染,并诱导致病性大肠杆菌菌株的生长。
此外,万古霉素还会导致拟杆菌(Bacteroidetes)、Fuminococcus、普拉梭菌(Faecalibacterium)等肠道微生物群的减少,以及变形菌门(Proteobacteria)种类的增加。
特定抗生素对肠道菌群的影响和恢复时间取决于个体的生理状况。此外,围产期给孕妇服用抗生素也会影响新生儿的微生物群,因为其中一些抗生素可以穿过胎盘。
√抗生素的作用取决于身体部位
此外,抗生素的作用取决于身体部位。例如,与肠道相比,在抗生素治疗后,喉咙和唾液在更短的时间内恢复了最初的共生多样性。
抗生素还会干扰微生物组和免疫系统的相互作用,导致免疫紊乱,并增强宿主对病原体的易感性。抗生素的广泛使用推动了病原微生物耐药性的进化,导致耐药基因的流行增加。
5
膳食成分除了影响微生物组的功能外,还能调节其组成。
√不同饮食成分下的微生物群组成不同
饮食对于确定微生物群的形态、结构和多样性至关重要。素食饮食与健康、多样的微生物群有关,其特征是能够代谢不溶性碳水化合物的物种占优势,即瘤胃球菌(Ruminococcus)、罗氏菌属(Roseburia)和真杆菌(Eubacterium)。
而非素食饮食与厚壁菌门比例下降和拟杆菌门比例增加有关。随着肉类的摄入,微生物群代谢氨基酸,以短链脂肪酸的形式产生能量源,但也会形成产生不利影响的化合物。
在一项研究中,测定了150名健康的杂食性、素食性和纯素食性志愿者粪便中存在的微生物群的组成结构以及代谢组。研究表明,富含蔬菜的食物增加了纤维降解细菌的丰度,并导致粪便短链脂肪酸的产生。
对地中海饮食依从性降低的志愿者拥有较高百分比的有害微生物代谢产物,如酚类和吲哚衍生物,以及三甲胺N-氧化物。
这些例子表明,饮食调节微生物群的组成和功能,从而影响个体的代谢状态。
6
不同身体部位的微生物组成是不同的,那么当微生物群的位置从身体的一个部位交换到另一个部位时,优势微生物物种的生态或流行如何受到影响,这是一个有趣的研究。可见,器官相关微生物群既具有动态性,又具有可塑性。
器官特异性微生物群可以跨界到与身体其他部位相关的其他生态位,在此过程中,微生物承受与身体各器官相关的pH、温度、毒素、免疫细胞等变化。
然而,在给定的生态位上,微生物群的结构组成基本上没有受到干扰。
微生物群在不同器官内混合的机制在很大程度上是未知的。
√器官间的微生物联系可能有利于宿主平衡
从空间和生长的角度来看,跨生态位的微生物对特定器官的优势微生物群体构成了挑战,但不同微生物物种之间的竞争是有利于宿主的微妙平衡。宿主细胞的器官特异性微环境有利于与该器官相关的微生物群的优势种群,并防止微生物群生态中的无意干扰。
宿主和微生物群之间存在着复杂的相互作用。宿主提供了微生物群生存和繁殖的环境,而微生物群则对宿主的生理状态和代谢产生着重要的影响。
由于微生物群与其宿主之间存在高水平的串扰,因此对微生物与宿主之间相互作用的研究仍然具有挑战性,尽管如此,以代谢物为中心的研究已经认识到对宿主健康重要的各种微生物靶点。
▼
微生物群对人体食欲的影响是非常复杂的,因为不同类型的微生物群会产生不同的代谢产物,一些研究表明,肠道微生物群可以通过产生短链脂肪酸等代谢产物来影响人体的食欲和能量代谢。
短链脂肪酸是肠道微生物群代谢产物的一种,主要包括丙酸、丁酸和乙酸等。
▷短链脂肪酸影响神经系统进而影响食欲
关于短链脂肪酸在调节能量摄入和食欲中的作用已经有了详细的研究。研究表明,这些化合物也可以影响外周和中枢神经系统的活动。
不过目前尚不清楚是单一的短链脂肪酸驱动,还是这些化合物的组合被利用。
目前关于可发酵纤维在食欲调节中的作用的研究有限,但增加每日纤维摄入量在16-35克/天范围内可以帮助改善这种调节。
虽然短链脂肪酸在食欲调节中的确切作用机制尚不清楚,但已有研究表明,人类体内短链脂肪酸的存在可以触发短期食欲调节。
例如,人类结肠丙酸盐通过PYY和GLP-1介导的机制诱导短期食欲调节。
▼
短链脂肪酸对脂肪代谢的影响也是显著的。
▷调控脂质积累和瘦素分泌
研究表明丙酸盐可以防止脂肪和胆固醇生成,它可以通过抑制FARE 2信号的活性来阻止脂肪细胞中的脂质积累。
还有研究表明,乙酸盐可以刺激脂肪细胞中瘦素激素的分泌。这一关键信号调节食欲和能量平衡。其他研究表明,抑制脂肪分解可以减少游离脂肪酸从脂肪细胞向肝脏的转运。
在脂肪肝疾病中,已知来自脂肪细胞的脂肪积累贡献了肝脏中总脂肪酸的60%。直肠输注丙酸和乙酸显示血清脂肪酸水平降低40%。
因此,重要的是保持丙酸与乙酸的比例,以确保结肠乙酸对脂质储存的最佳贡献。
▼
几项临床研究指出,肠道中细菌过度增殖与骨矿物质密度(BMD)降低之间存在关联。小肠细菌过度生长综合征患者的骨矿物质密度值低,骨软化,其中一些患者具有高水平的促炎细胞因子TNF-α和IL-1,以及破骨细胞活化增加。
肠道微生物组和骨骼之间的联系
doi.org/10.1016/j.jbspin.2018.02.008
近年来,一些横断面的临床研究以及系统评价和荟萃分析均发现肠道菌群改变与调节骨量、骨髓生成,骨骼发育、骨代谢、骨质疏松、骨骼炎症、骨折风险以及骨癌有关。
▼
微生物群对宿主免疫系统的影响非常重要,可以通过多种机制来影响宿主的免疫系统。
▷直接影响免疫细胞功能
微生物群中的某些成分可以直接影响宿主免疫细胞的功能,如调节巨噬细胞和树突状细胞的活性,从而影响宿主的免疫反应。
▷调节免疫细胞分化和增殖
微生物群中的一些成分可以影响免疫细胞的分化和增殖,如调节T细胞的分化和功能,从而影响宿主的免疫反应。
▷影响肠道黏膜屏障
微生物群可以通过影响肠道黏膜屏障的完整性和功能来影响免疫系统。肠道黏膜屏障是宿主体内与外部环境之间的主要屏障,它可以防止有害物质和微生物进入宿主体内。微生物群通过增强肠道黏膜屏障的功能来促进免疫系统的正常功能。
总之,微生物群对宿主免疫系统的影响是非常重要的,它们可以影响宿主的免疫反应、调节免疫细胞的分化和增殖、以及影响肠道黏膜屏障的完整性和功能。
肠道菌群及其代谢产物对人体还有其他影响,谷禾罗列在下表中:


Ayariga JA,et al.Arch Microbiol.2022
微生物群不仅影响宿主的生理功能,研究发现病理状况也与微生物群的组成、功能和生长动态密切相关。
如肥胖、高血压、2型糖尿病、非酒精性脂肪肝以及胃肠道疾病、过敏、自闭症、神经退行性疾病甚至癌症等都被发现与微生物群生态失调相关。
Zaidi S,et al.Arch Microbiol.2023
▼
研究表明肠道微生物群的存在可以影响血压调节。
在高血压大鼠中,观察到肠道中的微生物多样性和丰富度显著下降。这种情况已知是由血管收缩和血管阻力引起的。
在无菌小鼠中观察到血管紧张素II对血压的影响,表明肠道微生物群在调节血压方面发挥作用。尽管肠道微生物群调节血压的机制尚不完全清楚,但人们认为这种情况可能是导致高血压发展的一个因素。
动物中的特定肠道微生物代谢物,如短链脂肪酸,可能是导致高血压的一个因素。
来自HELIUS队列研究表明,克雷伯氏菌属和链球菌属与血压呈正相关。已显示,Lactobacillus coryniformis可以改善血管功能和胰岛素敏感性。
注:乳杆菌(Lactobacillus)治疗不仅可以改善心血管疾病,还可以改善实验性自身免疫性疾病。
▼
一些研究表明,微生物群的失调可能与某些癌症的发生有关。
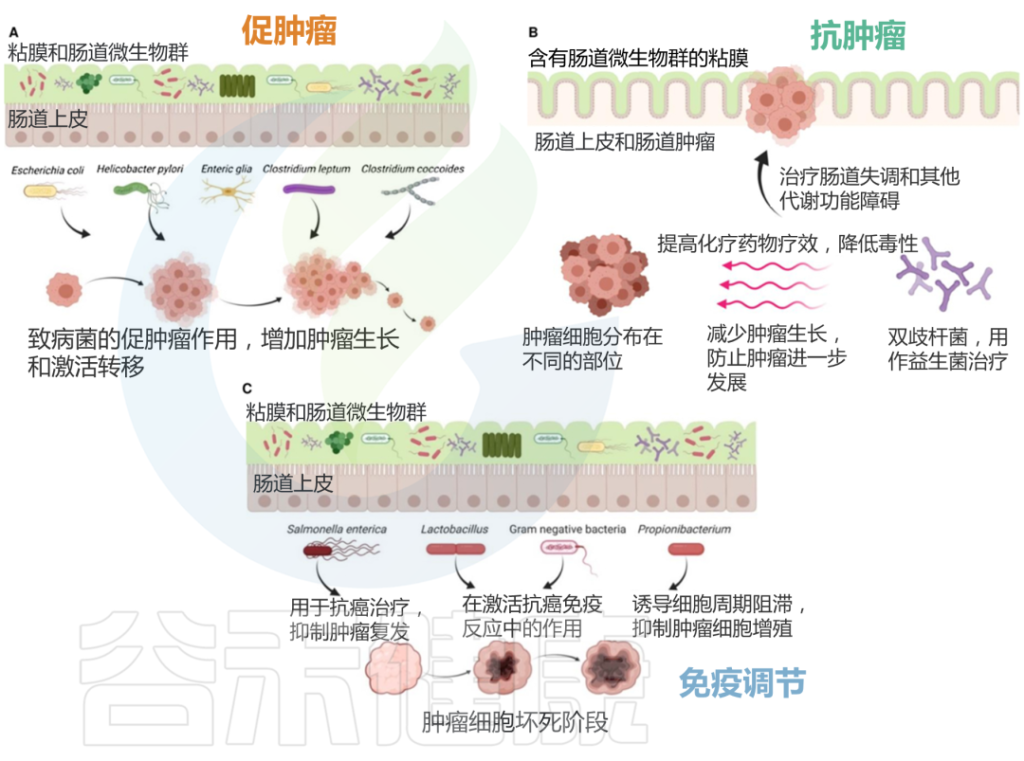
•结直肠癌
在与肠道微生物群相关的各种癌症中,对结直肠癌的研究最为广泛。
已经确定了肠道微生物群中的几种菌,这些细菌除了它们的致病性之外,还被假设对结肠直肠癌具有致癌作用,包括幽门螺杆菌、肝螺杆菌(Helicobacter hepaticus)、牛链球菌(Streptococcus bovis)、大肠杆菌、脆弱拟杆菌、败血梭菌(Clostridium septicum)、粪肠球菌、具核梭杆菌、厌氧消化球菌(Peptostreptococcus anaerobius)和牙龈卟啉单胞菌(Porphyromonas gingivalis),所有这些细菌都显示出潜在的致癌作用。
•肺癌
肺癌是常见的恶性肿瘤之一,迫切需要制定有效的肺癌治疗策略。研究表明,肠道和肺部微生物群之间通过淋巴和血液循环系统在双向轴上存在复杂的联系。
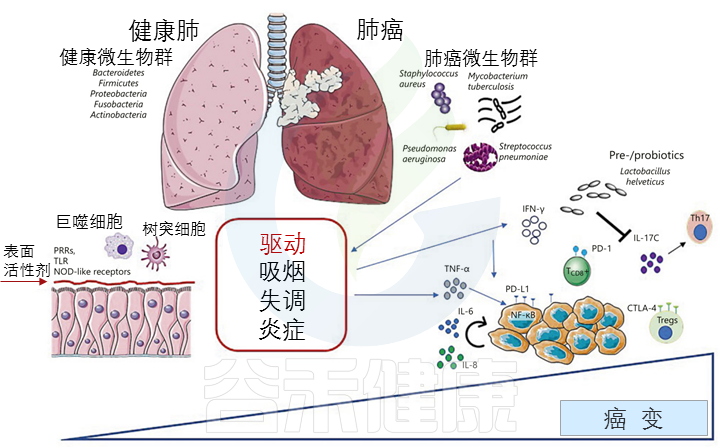
Martins D,et al.Pathobiology.2020
•乳腺癌
一项关于绝经后妇女的研究调查了乳腺癌与肠道代谢组学改变的相互关系。发现健康对照受试者和绝经后乳腺癌患者的肠道微生物组的组成和生物活性存在差异,其中绝经后乳腺癌患者的肠道宏基因组具有编码β-氧化、铁复合物转运系统和脂多糖生物合成的基因。
体外研究提供了支持肠道微生物群与乳腺癌转移进展之间联系的功能证据,其中微生物代谢物可以通过血液传播,影响乳腺癌细胞和免疫细胞的功能。
除此之外,分析唾液微生物组组成的变化有助于早期发现胰腺癌。另一种被称为产肠毒素脆弱拟杆菌的菌株与肠上皮细胞的致癌性有关。
▼
肠道微生物群的扰动可能导致炎症性肠病。同样,厚壁菌门相关细菌的不足和某些变形菌门数量的升高,可能导致粘膜免疫功能受损,这是引发慢性肠道炎症的主要原因,从而导致炎症性肠病的发生。
•肠易激综合征
肠易激综合征中厚壁菌门(Ruminococcus和Clostridium)数量增加,普拉梭菌和双歧杆菌种类数量减少。
•克罗恩病
此外,与克罗恩病患者或健康患者相比,回肠克罗恩病患者的普拉梭菌(Faecalibacterium Prausnitzii)数量明显减少,而大肠杆菌(Escherichia coli)数量过多。
▼
已经证明某些微生物可以通过感染特定的组织而引起疾病。
•细菌性相关组织感染
最早被人们了解的传染病是那些由制造毒素的细菌引起的传染病。白喉、肉毒杆菌和破伤风毒素分别与白喉棒状杆菌(Corynebacterium diphtheria)、肉毒梭菌(Clostridium botulinum)和破伤风梭菌(Clostridium tetani)引起的局部感染相关。
•细菌性腹泻和败血症
大肠杆菌、沙门氏菌、志贺氏菌、葡萄球菌和霍乱弧菌产生的肠毒素可导致由这些微生物引起的腹泻病。
革兰氏阴性菌脂多糖的脂质A部分具有强大的生物活性,可引起革兰氏阴性细菌性败血症的许多临床表现,包括发热、肌肉蛋白水解、血管内凝血失控和休克。
大多数致病的病原体都经过类似的途径,如呼吸道、胃肠道和生殖器官,这些途径被认为是通过与身体直接接触传播的;然而,其中一些微生物也可以通过与环境的间接接触获得,例如通过血液或水。
注:外科手术中的一些植入物也有可能引发细菌感染。
外科植入物引起的相关细菌感染
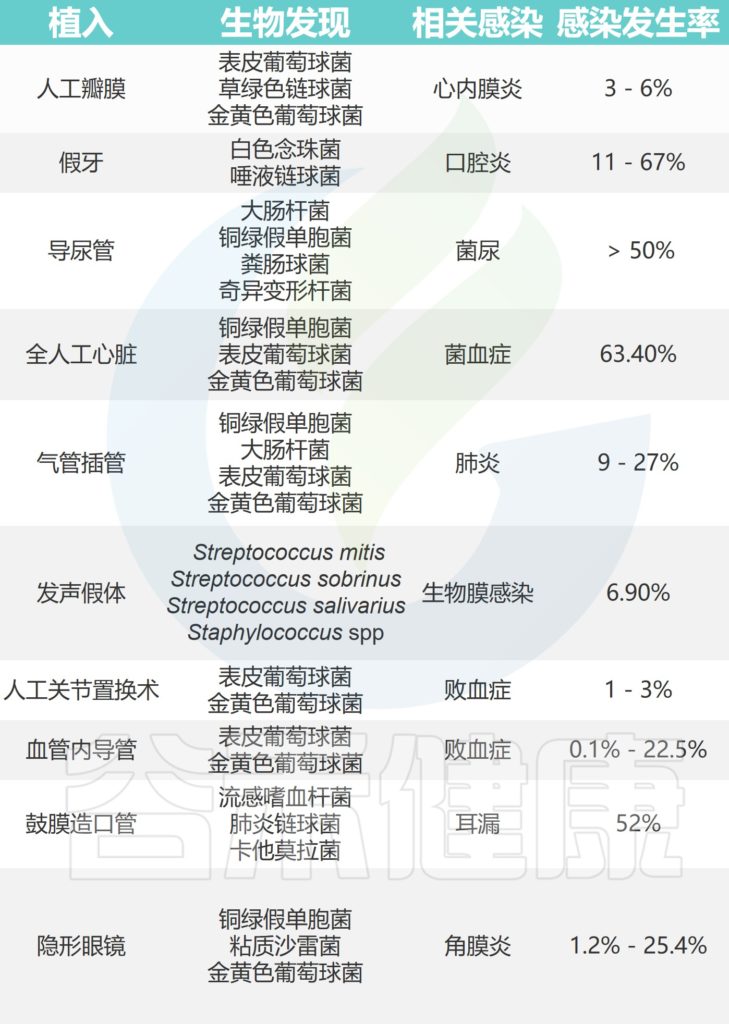
Zaidi S,et al.Arch Microbiol.2023
微生物群与其他一些疾病:
过敏
最近的研究表明Faecalibacterium、Bifidobacterium或Akkermansia的减少,加上Rhodotorula或念珠菌数量的增加,可能通过影响T细胞分化而使新生儿易过敏。
哮喘
同样,已发现罗氏菌属(Rothia)、毛螺菌属(Lachnospira)、韦荣氏球菌属(Veillonella)或普拉梭菌(Faecalibacterium )数量的减少会增加患哮喘的风险。
帕金森病
帕金森病最一致的发现是阿克曼菌(Akkermansia)的丰度增加。
其他组成特征包括双歧杆菌(Bifidobacterium)和乳杆菌(Lactobacillus)的丰度增加,丁酸生产菌(Roseburia)、(Faecalibacterium)和(Blautia)的丰度减少。
注意
微生物群评估有望在早期发现不同的疾病,如癌症、神经退行性疾病、代谢性疾病和自身免疫性疾病等。它采用非侵入性采样技术,同时降低了分析成本,从而使诊断过程变得可行。
对人类生理学、基因组学更好理解使我们的重点转向了针对患者的个性化/精准诊断和治疗。
个性化医疗的标志之一是对易感亚人群的特定疾病风险进行评估,从而可以对人群进行分层,提供更准确和更具成本效益的治疗。
微生物群在人类疾病和健康中的作用致使新的生物疗法发展,可以解决特定的疾病机制。微生物和免疫系统之间的相互作用是复杂的,它们的治疗可以提高患者的生活质量。
▼
每个人对医疗药物的反应也有很大的差异,可能与体内微生物群不同有关。不同微生物组在药物吸收、代谢、功效和毒性方面起着关键作用,并具有重大的健康影响。
地高辛
例如,地高辛是一种心脏糖苷类药物,专门用于治疗充血性心力衰竭。在微生物组研究进展之前,人们注意到一些患者能够化学还原地高辛,从而导致其失活,抗生素的使用导致血浆地高辛浓度增加两倍。
研究人员推断肠道细菌能够调节地高辛的代谢。最近的报告表明地高辛可被Eggerthella lenta灭活,而抗生素的摄入可使这种作用最小化,从而导致其在血浆中的浓度大幅增加。
对乙酰氨基酚
同样,对乙酰氨基酚,一种存在于许多镇痛药物中的化合物,在个体的临床作用中表现出明显的差异。这种个性化的反应最近被解释为与微生物组功能的差异有关。
他汀类药物
另一个微生物组驱动的个体化药物案例是他汀类药物和化疗药物,它们也被发现由于共生菌的作用而受到显著影响。
另一方面,药物可以通过微生物群转化为衍生物,这些衍生物可能具有非靶标效应。用于生产这些营养物质的底物的变化会影响肠道的代谢组学特征。这可能会对宿主产生不同的影响。
▼
根据上述数据,针对患者量身定制的微生物组操作似乎是多因素疾病更精确的微生物组特异性治疗的最佳替代方案。
如饮食干预、抗生素的应用、益生菌和益生元的使用以及粪便微生物群移植可用于调节微生物群以获得有利的反应。
微生物疗法的原理是通过调节宿主的微生物群,来促进身体健康和预防疾病。它可以包括多种形式,例如口服益生菌、粪菌移植、皮肤微生物移植等。
★ 肠道微生物群检测
要对人体的微生物群进行干预,首先要做的就是对体内的微生物群进行检测。肠道微生物群检测是一种通过分析肠道微生物群的组成和数量来评估患者的健康状况的方法。这种方法已经被证明对诊断肠道炎症、炎症性肠病等多项疾病有效。
口服益生菌改善健康
口服益生菌可以帮助恢复肠道菌群的平衡,从而改善肠道健康和免疫系统功能。
口服补充乳杆菌,可以提高耐受性,减轻儿童对牛奶的过敏,并通过过敏患者粪便中产生丁酸盐的细菌的活性恢复丁酸盐的最佳水平。
此外,酵母菌和乳酸菌可以将抗生素相关疾病的风险降低50%。益生菌持续治疗多种胃肠道和肠外疾病,如阴道感染、肠易激综合征、炎症性肠病和免疫增强。
食用合生元改善健康
此外,合生元是微生物群靶向治疗的另一种有效方法。包括在原始微生物群中引入新的微生物,补充足够的底物以促进新的所需微生物的生长。
例如,植物乳杆菌(益生菌)与低聚果糖(益生元)一起生长被发现可以减轻新生儿因败血症而死亡的数量。
麦角硫因是一种在蘑菇、豆类和谷物等食物中发现的著名抗氧化剂。研究证明麦角硫因被幽门螺杆菌利用来保护它免受宿主胃组织的侵害。
麦角硫因对人体有抗炎作用。更重要的是,麦角硫因的减少与心血管疾病、自身免疫性疾病和神经系统疾病的风险增加有关,这意味着肠道中的细菌会影响人类健康。
微生物移植改善健康
粪菌移植疗法的工作原理是通过引入健康的微生物群来替代患者肠道中的有害菌群,从而恢复肠道菌群的平衡。
这可以改善肠道健康和免疫系统功能,从而减少肠道炎症和其他与肠道微生物群失调相关的疾病的发生。
除了治疗肠道疾病外,粪菌移植疗法还可以用于治疗其他疾病,例如自身免疫性疾病、代谢性疾病和神经系统疾病等。皮肤微生物移植可以用于治疗某些皮肤疾病,例如顽固性湿疹和痤疮。
微生物群在我们身体的许多部位中存在着,如肠道、皮肤、口腔和生殖道等。这些微生物群与我们的身体密切相连,对我们的健康和疾病起着重要作用。
了解人体微生物群的组成、功能和影响因素,对于我们更好地了解自身健康以及疾病的预防和治疗具有重要意义。
主要参考文献
Zaidi S, Ali K, Khan AU. It’s all relative: analyzing microbiome compositions, its significance, pathogenesis and microbiota derived biofilms: Challenges and opportunities for disease intervention. Arch Microbiol. 2023 Jun 6;205(7):257.
Ayariga JA, Ibrahim I, Gildea L, Abugri J, Villafane R. Microbiota in a long survival discourse with the human host. Arch Microbiol. 2022 Nov 28;205(1):5.
Abenavoli L, Scarpellini E, Colica C, Boccuto L, Salehi B, SharifiRad J, Aiello V, Romano B, De Lorenzo A, Izzo AA, Capasso R (2019) Gut microbiota and obesity: a role for probiotics. Nutrients 11(11):2690.
Apparao Y, Phan CW, Kuppusamy UR, Sabaratnam V (2022) Ergothioneine and its prospects as an anti-ageing compound. Exp Gerontol 170:111982.
Aarnoutse R, Ziemons J, Penders J, Rensen SS, de Vos-Geelen J, Smidt ML (2019) The clinical link between human intestinal microbiota and systemic cancer therapy. Int J Mol Sci 20:4145.
Aggarwal N, Kitano S, Puah GR, Kittelmann S, Hwang IY, Chang MW (2022) Microbiome and human health: Current understanding, engineering, and enabling technologies. Chem Rev 123:31.
De Angelis M et al (2020) Diet influences the functions of the human intestinal microbiome. Sci Rep 10:1–15.

谷禾健康

维生素C是一种广泛存在于自然界中的水溶性维生素。维生素C在人体新陈代谢中具有多种重要功能,包括抗氧化、参与胶原蛋白合成、增强铁的吸收等。由于其众多生理益处,维生素C被广泛地应用于修复伤口、治疗感冒、癌症等多种疾病。
人体无法自行合成维生素C,需要从膳食来源(如水果、蔬菜)中获取。维生素C的代谢过程涉及多个酶和转运蛋白。肠道微生物群可能通过影响这些酶和蛋白的活性或表达,来影响维生素C的代谢吸收过程。
维生素C也可以直接调节肠道微生物群,或通过修复肠道屏障、改变氧化还原电位等方式间接对肠道微生物群的平衡起到调节作用。
个体之间的差异、饮食习惯和生活方式等因素各不相同,这些都可能对维生素C与肠道菌群的相互作用产生影响。
本文将从维生素C的结构、功能、吸收和代谢、与肠道菌群的关联等多角度,全面探讨维生素C的作用及其对人体健康的影响,同时介绍了一些维生素C的食物来源、人体需要的剂量、如何补充、注意事项等。
▼
本文主要内容:

编辑
维生素C,也称为抗坏血酸,是一种水溶性维生素,对人体健康非常重要。它在许多身体功能中起着关键作用,包括增强免疫力、抗氧化、胶原蛋白的合成等。
▼
维生素C的化学名称是L-抗坏血酸,它是一种有机化合物。无臭,味酸,易溶于水,微溶于乙醇,不溶于乙醚。
维生素C结构简单,化学式为C6H8O6:
这种结构使维生素C具有抗氧化性质,能够捕捉自由基,并保护细胞免受氧化损伤。
注:体内的分子暴露于环境污染物、吸烟和慢性炎症等情况时,它们会变成自由基。自由基是不稳定的分子,会破坏细胞并导致疾病,维生素C可以通过中和自由基来减缓或预防某些健康问题。
由于其与葡萄糖的结构相似,维生素C可以在许多化学反应中取代葡萄糖,并且可以防止蛋白质的非酶糖基化。
▼
维生素C参与胶原蛋白、激素、肉碱的合成,促进铁离子的吸收,此外,它在免疫系统的功能和调节中发挥着重要作用,对维持内部环境的平衡和中枢神经系统的正常功能极为重要。
维生素C的大部分功能是由于其作为抗氧化剂和辅助因子的能力。由于人类缺乏L-gulono-1, 4-lactone氧化酶,无法自行合成维生素C,因此完全依赖于维生素C的饮食摄入。
▼
从食物中获取营养总是最好的。大约90%的日常需求来自蔬菜和水果,它们是这种维生素的极好来源,例如奇异果、橙子、芒果、草莓、红椒、青椒等。
一些维生素C含量较高的食物
注:单位“杯”是一个常见的非正式计量单位,美规和英规略有区别,大约是237毫升-250毫升左右,涉及到果蔬的份量时,一杯通常是指将果蔬切碎后填满一杯容器的量。
▼
维生素 C 对大脑健康非常重要。大脑在长期缺乏维生素 C 的情况下以牺牲其他组织为代价来保留维生素 C,并且可以维持比其他器官(例如肝脏和肾脏)高很多倍的浓度,如下图。
维生素C的分布在身体各器官之间差异很大
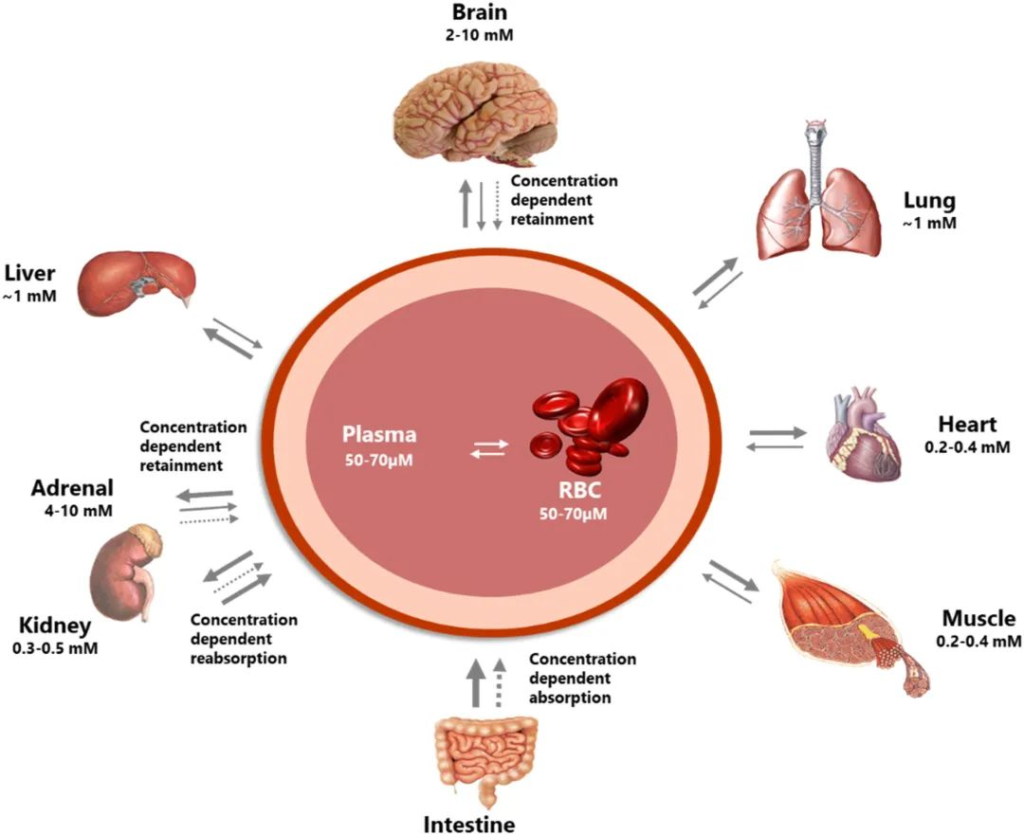
doi.org/10.1016/j.redox.2020.101532
维生素C供应不足时,首先保证大脑里的浓度,那么维生素C对于大脑而言有什么作用?
▼
维生素 C 对胶原蛋白的稳定作用对于形成整个身体的结缔组织框架至关重要;包括皮肤、骨骼、软骨、肌腱、韧带、血管等。
胶原蛋白生产的最后步骤取决于维生素 C,维生素 C 在前胶原脯氨酰和赖氨酰残基的羟基化中充当电子供体。
▼
维生素 C 可增强其他营养素的生物利用度,例如维生素 E 和非血红素铁,这可能会增强含维生素 C 的食物的健康效果。
维生素C经常添加到含铁的口服制剂中,以增加铁的吸收。
▼
维生素 C 是否可以预防或减轻包括普通感冒在内的感染的严重程度是一个有争议的话题。大多数证据都支持其好处。
▼
维生素C对正常骨骼发育至关重要。维生素C水平与骨骼健康之间存在正相关关系,如骨密度、骨折概率、骨转换标志物等。
▼
维生素 C 有助于维持健康的皮肤。
▼
肺部的维生素C水平是血液中的30倍。
维生素C在抵御氧化剂的同时也会被消耗,这表明即使是单剂量的维生素C,也能有效抵御肺部氧化应激的急性增加。
▼
根据现有证据,摄入足够的维生素 C 可能有助于保持健康的情绪。
焦虑
抑郁
▼
人类慢性低维生素 C 状态与神经退行性疾病有关。但是,尚未确定因果关系。
▼
维生素 C 可以通过抑制炎性细胞因子来减轻炎症。
▼
除上述主要的功能之外,在部分小型研究中提到的关于维生素C的功能如下:
助孕育:
助减肥:
降血压、防中风:
降血糖:
助排毒:
助抗癌:
牙周健康:
水溶性维生素在人体内储存较少,从肠道吸收后进入人体的多余的水溶性维生素大多从尿中排出。因此,摄入较多的水溶性维生素一般不会引起中毒现象,但是若摄入量过少,则会很快出现缺乏症状。
▼
刚缺乏的时候症状不明显,饮食中缺乏VC需要大约一个月的时间才会出现症状。
体内维生素C总含量低于300-400mg会出现明显症状。
维生素C的缺乏会出现什么症状?
由于维生素C功能的复杂性及其被不同还原剂的部分替代性,维生素C与坏血病症状的直接联系不容易确定。
如果发展为坏血病,典型症状是:
肌肉无力、牙龈肿胀和出血、牙齿脱落、瘀点出血、自发性瘀斑、贫血、愈合障碍、角化过度、虚弱、肌痛、关节痛和体重减轻(也可能因肿胀而出现矛盾的体重增加),而早期表现包括嗜睡、倦怠、易激惹,甚至呼吸困难等。
在生化上,维生素C血浆水平低于11μM被认为与坏血病的临床症状一致。
在专业医疗人员的指导下,补充维生素C可以轻松有效地逆转坏血病。许多症状可以在几周内轻松解决。富含维生素C的饮食将防止坏血病的发展。
什么人群更容易发生维生素C缺乏?
▼
NIH 认为,成人可耐受的上限是每天 2000 毫克,仅仅靠含有维生素 C 的食物几乎不可能达到这一上限,所以食物可以放心吃。服用补充剂则需注意剂量,可能存在过量的风险。
更高的剂量更有可能导致副作用。
维生素C过量可能会出现什么症状?
单次口服5-10克维生素C会产生短暂的渗透性腹泻和/或腹胀伴疼痛,不建议这样做。
随食物一起摄入可减少这些不良反应。
每天超过 2000 毫克的剂量可能会增加腹泻和肾结石的风险。如果有肾结石病史,每天摄入超过 1000 毫克可能会增加患结石的几率。
那么到底应该怎么补充?每日摄取多少维生素C 合适呢?
▼
科学界对维生素C的最佳剂量方案(摄入量和频率)最健康存在持续争论。
对于大多数健康人来说,通过食物可以获得足量的维生素 C。
维生素C摄入量标准在不同地区有所不同:
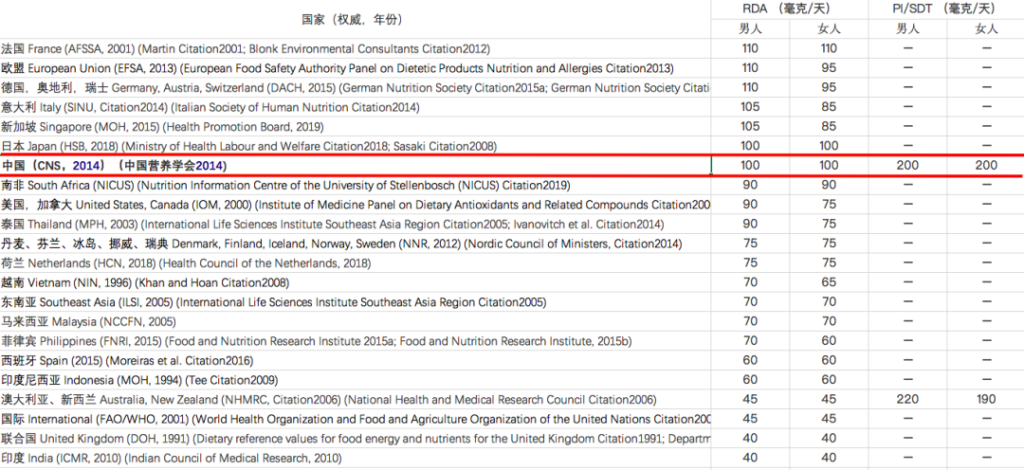
注:RDA – 推荐膳食摄入量,
PI – 建议摄入量,SDT – 建议膳食目标
在中国营养学会编著的《中国居民膳食指南》2022版中对维生素C的推荐摄入量:
中国居民膳食指南2022版
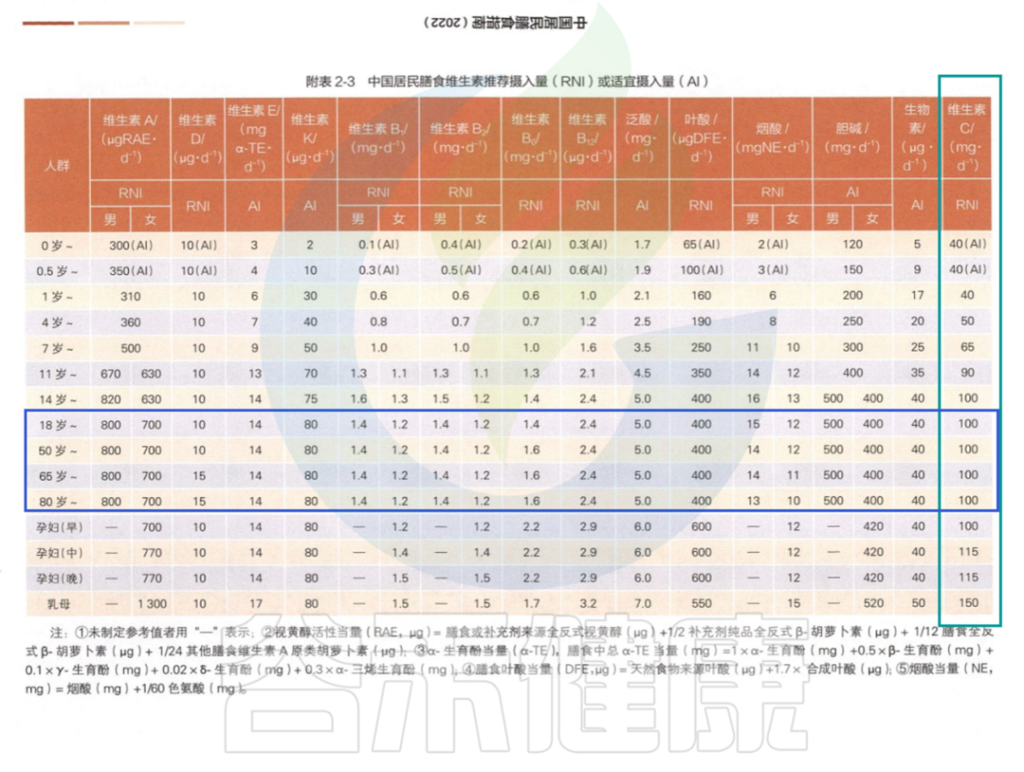
为什么不同地区的标准不一样呢?
这主要是由于RDA标准的基本前提从预防坏血病(~45 mg/d)至健康优化(~200 mg/d)。许多权威机构建议使用最低量的维生素C,但这可能无法满足不同亚群的健康需求。
例如,吸烟者和肥胖者比普通人群有更高的要求。一些国家的吸烟率继续上升,有证据表明,吸烟者每天至少摄入200 mg的维生素C。
随着全球肥胖率的增加,以及与肥胖相关的合并症,如代谢综合征、糖尿病和心血管疾病,需要更多地考虑适当的建议,以优化快速增加的亚健康人群中的维生素C状态。
以上是维生素C的摄入量标准,具体该如何补充,应该注意什么?详见下一章节。
▼
食物:如何才能最大程度地保留其维生素C?
——烹饪方式
长时间烹饪,特别是用大量水煮沸会导致维生素C浸出到水中,显著降低食物中的维生素C含量。
在少量水中蒸或煮,且持续时间较短,是保存维生素C的更温和的方法。
即使将所有外部因素消除到最低限度,也会发生损失,因为由于抗坏血酸氧化酶的存在,材料内部会发生氧化反应。因此,理想的加工方法是用最少量的水快速热灭活酶,然后快速冷却。
——长期保存条件
温度对储存稳定性也有很大影响。随着温度的升高,会出现更显著的损失。然而,在长期储存期间,即使维持短期储存期间仅发生少量损失的条件,维生素C的量也会显著减少。
损失主要是由于酶催化的氧化反应,其程度尤其取决于pH、材料完整性和温度。
总之,为了使水果和蔬菜的维生素C含量保持更长时间,最温和的方法是深度冷冻。
补充剂:一天中分几次服用
食物和许多补充剂中的维生素 C 是一种抗坏血酸的形式。
当肠道面临较低水平的抗坏血酸(即低于约 400 毫克)时,主动运输系统会吸收维生素 C(即,将营养物质通过肠道并进入血液,到身体需要的地方)。
一旦这些主动运输变得不堪重负,被动扩散就会接管吸收其余的维生素 C(这是一个相当低效的过程)。吸收并不像听起来那么容易,事实上抗坏血酸有吸收上限。
身体一次可以处理大约 300 – 400 毫克的纯抗坏血酸形式的维生素 C,更多量一下子难吸收。
所以如果能记得的话,一天中分几次服用比较合适。
▼
维生素C补充剂并非适合所有人。如果遇到以下任何情况,请首先与医生联系:
也不要认为维生素C服用越多越好,每天服用 1000 毫克或更多,实际上会使吸收率降低约 50%。
▼
维生素C可以增加某些药物的吸收,例如:
服用维生素C可以增加含铝药物(如磷酸盐粘合剂)对铝的吸收。这可能对有肾脏问题的人有害。抗酸剂中含铝:不要同时服用维生素C和抗酸剂。服用维生素C后至少等待两个小时,然后再服用抗酸剂。服用抗酸剂后等待四个小时服用维生素C。
维生素C可能会增加左旋甲状腺素的吸收。
补充维生素C会降低一些药物的疗效:
口服维生素C可能会降低这些抗病毒药物的作用。
当与维生素C一起服用时,烟酸和他汀类药物的影响可能会降低,这可能有益于高胆固醇的人。
高剂量的维生素C可能会降低人体对这种抗凝剂的反应。
其他还包括:
维生素C增加或减少药物副作用的风险
如果服用雌激素或基于雌激素的避孕药,维生素C可能会增加激素副作用的风险。这是因为维生素C可能会减缓雌激素离开身体的速度。
一些早期的研究认为, 维生素C可能有助于预防阿司匹林和非甾体抗炎药引起的胃部不适。
▼
维生素 C 和益生菌对肠道健康和免疫力有不同的好处,它们可以很好地互补,这意味着它们可以安全地一起服用。
一项针对学龄前儿童的双盲、随机、安慰剂对照初步研究中,发现益生菌与维生素 C 联合预防呼吸道感染的功效(URTI;33%,P =0.002)。
Lab4 益生菌和维生素 C 组合补充 6 个月的儿童显示,上呼吸道感染症状的发生率和持续时间有所减少,降低感染的严重程度。
注:Lab4 益生菌包含:嗜酸乳杆菌CUL21(NCIMB 30156)和CUL60(NCIMM 30157),双歧杆菌CUL20(NCIMB 30153)和动物双歧杆菌乳亚种CUL34(NCIMM 30172)。
注意:根据说明书剂量服用或遵医嘱。
以上并不是维生素C可能发生的相互作用的完整列表。在开始补充维生素C或调整摄入量之前,请与医生或药剂师沟通,让医生知道你正在服用的所有药物,包括处方药和非处方药、其他维生素或微量营养素、草药补充剂等。
▼
静脉注射和口服给药,这两者可能具有不同的药代动力学特征。
药理学模型显示,口服维生素C,即使是在非常大和频繁的剂量下,也只能适度地增加血浆浓度,从0.07 mM增加到最大0.22 mM。
而静脉注射剂量预计会导致血浆维生素C峰值水平比口服剂量高60倍以上,尿液浓度比口服剂量低140倍。
分子的实际生物利用度由许多因素控制,包括肠道和其他组织的吸收、肾脏的吸收和排泄以及其他患者特异性因素。
除了通过静脉给药和口服给药的浓度差异外,口服给药将维生素C直接输送到肠道微生物组,而不是通过血液;因此,它对肠道微生物的影响可能与动力学和浓度有关,这与影响血浆水平的动力学和浓度完全不同。
静脉注射维生素C常用于临床医疗环境中,用于治疗某些疾病或特殊情况下的高剂量补充,如感染、外伤、手术恢复等。专业医生会给予相应的建议。
口服给药适用于一般的日常维生素C补充,维持正常的维生素C水平。
有几种方法可以评估人体中的维生素C状态。这些包括测量血浆、尿液、组织、粪便中维生素 C 的浓度。
▼
检测血浆维生素C,血浆样品中维生素C的定量测定常见的有两种方法:酶法和色谱法。
酶促维生素C测定
有几种基于维生素C的酶促转化的商业试剂盒,产生可以用光光谱法检测的信号。通常,抗坏血酸氧化酶用于这种类型的测定。这些测定的常见方法是酶联免疫吸附测定(ELISA),它非常适合分批处理样品,但不太方便立即测定少数样品中的值。
根据欧洲外部质量评估计划(Instand EQAS)中报告的方法,基于酶的分析方法在医院中并不常规使用。如果临床对立即测定维生素C的需求增加,由于其直接的技术性质,这些基于酶的测定可用于护理点或集中平台。
色谱法测定维生素C
抗坏血酸和DHA的定量测量目前是通过高效液相色谱(HPLC)方法进行的。如果必须分析具有相似性质的多种化合物,或者如果存在许多可能干扰感兴趣化合物定量的物质,则HPLC方法是优越的。
将酸化样品注射到HPLC仪器中后,通过通过基于化合物的物理性质不同地保留化合物的柱来分离化合物。结果,在分离柱的末端,可以选择性地检测抗坏血酸和DHA,而不受其他化合物的干扰。
目前有两种方法可以检测分离后的抗坏血酸和DHA,一种是电化学检测,另一种是紫外线检测。这两种检测方法给出的结果相同,但由于相对技术简单,紫外线检测更广泛地用于日常检测。
其他检测技术,如荧光检测,需要在柱前对抗坏血酸和DHA进行化学改性,但使用较少。比色/荧光法可能会产生更高的DHA浓度,因为该方法缺乏特异性。
▼
可以查看近期体内维生素状况。
肠道菌群可以影响食物中的营养物质的吸收和利用。肠道菌群的失调可能会影响维生素C吸收,从而出现维生素C缺乏,引发一系列健康问题。因此,检测肠道菌群的状况,可以帮助我们更好地了解维生素C的吸收和利用情况。
与抽血检测不同,肠道菌群的评估更加反映一段时间 ( 一般2周左右 ) 的长期状态,如部分维生素无法在体内留存,需要每日补充,血液检测波动较大。
肠道菌群与维生素C的水平之间存在怎样的关联?
为什么肠道菌群检测报告可以了解维生素情况?
我们来看下一章节。
我们在日常生活会看到,同样吃食物,有些人的维生素吸收状况比较好,有些人就容易缺乏,这是为什么呢?
为什么大剂量补充对一些人的身体有益,而少数人因为过量出现了肾结石呢?
这其实都与肠道微生物群相关。
这里我们分为两个方面来讨论:
一个是肠道微生物群对维生素C的影响,
一个是维生素C的补充对肠道微生物群的影响。
维生素C在人体代谢过程中的吸收和利用,与肠道微生物群相关。了解肠道微生物群对维生素C的吸收和利用的影响,可以帮助我们更好地理解其与人体健康之间的关系。
▼
微量营养素使用各种特定的吸收途径和机制,既可以是被动的,也可以是主动的。
膳食维生素 C 很容易通过钠依赖性维生素 C 转运蛋白(SVCT1 和 SVCT2)在肠道中吸收,其他比如维生素A 、维生素D 的吸收通过小肠中的被动扩散发生。
肠道微生物群是人体肠道中的有效生物反应器,可将各种化合物转化为有益或有害的代谢物,因此对其生物利用度起着至关重要的作用。
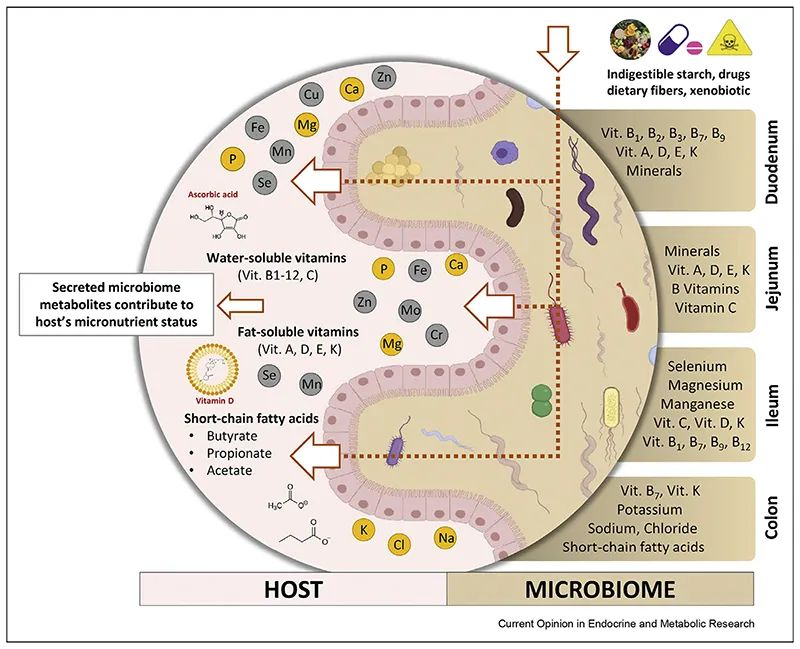
doi: 10.1016/j.coemr.2021.100285
胃肠道各部分理化特性的差异,以及位点特异性受体的存在,使得不同的维生素和矿物质能够沿胃肠道吸收。不同微生物在每个不同部分的定殖会影响当地环境,从而对微量营养素的生物利用度产生积极或消极的影响。
微生物可以干扰维生素C的吸收
微生物可以通过干预生物合成过程和调节吸收,来调节微量营养素的水平,包括维生素C。
来自革兰氏阴性菌的脂多糖降低SVCT-1的表达,进而降低SVCT-1-介导的维生素C的摄取。
Chmiel JA, et al.,Nat Rev Urol. 2023
大肠杆菌可以通过释放脂多糖来减少宿主对抗坏血酸的摄取,从而增加NF-κB依赖性TNF的产生,其进而通过抑制SLC23A1和SLC23A2启动子(分别编码SVCT1和SVCT2),来降低钠依赖性维生素C转运蛋白SVCT1与SVCT2的表达。从而对维生素C的吸收率产生负面影响。
细菌可以与宿主竞争维生素C
大肠杆菌抗坏血酸转运蛋白对抗坏血酸的亲和力高于哺乳动物SVCT1,这表明细菌可以与宿主竞争维生素C。
因此,肠道菌群中大肠杆菌等革兰氏阴性菌占比较多的情况,可能不利于维生素C的吸收,这在谷禾肠道菌群检测报告中也是可以反映的。
▼
肠道细菌如大肠杆菌和乳酸杆菌代谢维生素C。
利用ula基因簇,大肠杆菌等细菌可以将抗坏血酸代谢为D-木酮糖,宿主细胞可以进一步加工木酮糖以产生草酸盐。
在肺炎链球菌、沙门氏菌、福氏志贺菌、粪肠球菌、肺炎克雷伯菌等病原菌中,也发现了相同的ula基因簇。
带ula的致病菌与草酸盐:携手制造结石
结石形成者(比如肾结石患者)微生物群更常见地富含携带ula基因簇的致病菌,并且这些患者在接受口服维生素C时,草酸盐增加的水平比非结石者增加得更多。高草酸盐水平增加了结石的风险。
因此,如果在肠道菌群检测报告中发现以上提到的致病菌占比较多,则有可能在代谢维生素C的时候产生的草酸盐过多,增加了结石的风险。
Chmiel JA, et al.,Nat Rev Urol. 2023
“
以上通过微生物群代谢维生素的机制,有助于我们更好地理解结石形成的机制。
既然有促进结石的细菌,自然也有降解的细菌:
一些乳酸杆菌可以将维生素C转化为乙酸盐和乳酸盐,这是一种无毒的代谢产物,通过生物能量途径增加微生物组的功能,并可能促进这些草酸降解细菌的定植。
Oxalobacter formigenes是一种革兰氏阴性厌氧细菌,可降解肠道草酸盐并促进原发性高草酸条件下肠道草酸盐的分泌。该菌在肠道定植可降低尿液或血浆中的草酸盐浓度。
Barone M, et al., Biofactors. 2022
我们看到生活中有人认为不能吃生菠菜,会得肾结石,但有些人每天吃生菠菜也没事…其实可能是因为人家的肠道菌群中致病菌较少,而降解草酸的菌群又在拼命干活…
因此,健康的肠道菌群结构非常重要,菌群在该干活的时候各司其职,井井有条,那么你的身体抵抗疾病的能力也随之提升。
以上是肠道菌群对维生素代谢吸收的影响,反过来,维生素C的补充也可以影响肠道菌群的组成。
二、
补充维生素C可以直接调节肠道微生物群,也可以通过改变氧化还原电位、修复肠道屏障,改善肠道条件,支持部分有益菌生长,防止有害菌泄漏到身体其他部位。
▼
维生素C直接调节肠道菌群
与安慰剂组相比,补充维生素C已被证明可以显着增加微生物生态系统的多样性,以及Collinsella的相对丰度和粪便水平的短链脂肪酸,特别是丁酸盐和丙酸盐。
在健康受试者中,每日高剂量维生素C补充(1000 mg/天):
下列菌群的相对丰度升高
下列菌群的相对丰度下降
一项观察性研究探讨了微量营养素维生素C对肠道微生物组组成和多样性的影响。结果表明,维生素C增加了肠道中双歧杆菌属的丰度,在科水平上,毛螺菌科和双歧杆菌科显著增加。
双歧杆菌属的成员是有益菌,例如增加ATP生成、调节免疫系统、黏膜屏障完整性、短链脂肪酸的产生,对健康有益,维生素C通过增加肠道有益菌促进健康。
研究发现,囊性纤维化患者维生素C摄入量的增加与厚壁菌门的丰度呈正相关,与拟杆菌门的丰度呈负相关。
▼
改变氧化还原电位
调节微生物组的机制类似于维生素B2:通过改变氧化还原电位,改善消化道中的厌氧/耐氧平衡,从而选择性地支持微生物生长,改善肠道条件。
与安慰剂相比,维生素C组粪便样本中的粪便pH值和氧化还原平衡降低。
▼
修复肠道屏障
我们知道,肠道屏障受损,细菌和有害物质有可能会穿过屏障进入血液循环,对人体产生各种负面影响,导致诸如过敏、炎症、自身免疫疾病等多种反应。
关于肠漏可以详见我们之前的文章:
而维生素 C 具有维持肠粘膜屏障完整性和修复粘膜屏障的作用。
这里介绍两种修复肠屏障机制。
——通过激活Notch 信号
Notch 信号影响细胞正常形态发生的多个过程,与许多人类疾病有关,包括IBD,因此被认为是癌症治疗的潜在靶点。Notch 信号通路的激活会改变紧密连接蛋白的表达并影响其分布的连续性,从而降低细胞屏障通透性。
豚鼠结肠组织相关基因检测表明,低剂量的维生素C可强烈激活Notch/Hes-1信号通路,对DSS诱导的结肠炎豚鼠的肠粘膜具有一定的保护作用。肠上皮受损时,Notch-1表达增加可促进上皮细胞增殖,有利于损伤部位的修复和重建。
——通过增加肠道胶原蛋白合成
增加维生素 C 摄入量的另一个潜在好处是肠道胶原蛋白合成增加,从而改善屏障功能。这一提出的机制与抗坏血酸的辅酶功能一致,即羟基化脯氨酸和赖氨酸以交联胶原蛋白。
例如,对具有吲哚美辛诱导的屏障功能障碍的 T84 人隐窝样上皮细胞系的研究表明,细菌通过跨细胞途径穿过上皮细胞,维生素 C 治疗可消除该途径。因此,肠道中抗坏血酸状态不佳可能会加剧屏障功能障碍,从而增加 LPS 衍生的革兰氏阴性菌的易位,从而加剧炎症。
维生素C修复肠道屏障后,肠道屏障可以正常发挥吸收、分解和转换营养物质等功能,同时可以帮助促进健康微生物群栖息和生长,从而促进整体健康。
以上我们了解到,维生素C可以通过多种方式影响肠道菌群,从而促进整体健康。那么当我们看到肠道菌群报告中菌群多样性较低,部分菌群失调,尤其是上面提到的毛螺菌科、肠球菌属、Collinsella、Gemmiger formicilis等,可以考虑通过补充维生素C来调节。
我们首先可以考虑通过食物补充,如卡姆果、针叶樱桃等维生素C含量很高的食物,或者一些常见的蔬菜水果例如:猕猴桃、番石榴(芭乐)、青椒、羽衣甘蓝等。也可以考虑通过维生素C补充剂调节。
人体维生素研究面临着许多挑战,维生素通常是通过食物摄入的,而食物中同时存在多种营养素,这使得难以研究单一维生素的作用;某些高剂量维生素的使用可能存在潜在的风险,进行研究时涉及到人体试验和干预,需要遵循伦理准则,并确保研究的安全性;不同人对维生素的需求和代谢能力存在个体差异,基因、环境和生活方式等因素都可能影响维生素的吸收、利用和代谢。
肠道菌群研究可以揭示不同个体之间菌群组成的差异,这有助于理解个体差异对维生素代谢和利用的影响。可以通过检测肠道菌群的组成和丰度,了解维生素的代谢情况,从而辅助评估维生素水平。
综合运用多种研究方法和技术,结合肠道菌群检测,可以考虑多个因素对维生素代谢和利用的综合影响,进一步理解维生素的作用机制,对人体健康具有重要意义。
注:本账号内容仅作交流参考,不作为诊断及医疗依据。
主要参考文献
Barone M, D’Amico F, Brigidi P, Turroni S. Gut microbiome-micronutrient interaction: The key to controlling the bioavailability of minerals and vitamins? Biofactors. 2022 Mar;48(2):307-314.
Chmiel JA, Stuivenberg GA, Al KF, Akouris PP, Razvi H, Burton JP, Bjazevic J. Vitamins as regulators of calcium-containing kidney stones – new perspectives on the role of the gut microbiome. Nat Rev Urol. 2023 May 9:1–23.
Pham VT, Dold S, Rehman A, Bird JK, Steinert RE. Vitamins, the gut microbiome and gastrointestinal health in humans. Nutr Res. 2021 Nov;95:35-53.
Li XY, Meng L, Shen L, Ji HF. Regulation of gut microbiota by vitamin C, vitamin E and β-carotene. Food Res Int. 2023 Jul;169:112749.
Yang Q, Liang Q, Balakrishnan B, Belobrajdic DP, Feng QJ, Zhang W. Role of Dietary Nutrients in the Modulation of Gut Microbiota: A Narrative Review. Nutrients. 2020 Jan 31;12(2):381.
Steinert RE, Lee YK, Sybesma W. Vitamins for the Gut Microbiome. Trends Mol Med. 2020 Feb;26(2):137-140.
Gomes-Neto JC, Round JL. Gut microbiota: a new way to take your vitamins. Nat Rev Gastroenterol Hepatol. 2018 Sep;15(9):521-522.
Rowland I, Gibson G, Heinken A, Scott K, Swann J, Thiele I, Tuohy K. Gut microbiota functions: metabolism of nutrients and other food components. Eur J Nutr. 2018 Feb;57(1):1-24. doi: 10.1007/s00394-017-1445-8. Epub 2017 Apr 9. PMID: 28393285; PMCID: PMC5847071.
Traber MG, Buettner GR, Bruno RS. The relationship between vitamin C status, the gut-liver axis, and metabolic syndrome. Redox Biol. 2019 Feb;21:101091.
Rozemeijer S, Spoelstra-de Man AME, Coenen S, Smit B, Elbers PWG, de Grooth HJ, Girbes ARJ, Oudemans-van Straaten HM. Estimating Vitamin C Status in Critically Ill Patients with a Novel Point-of-Care Oxidation-Reduction Potential Measurement. Nutrients. 2019 May 8;11(5):1031.
Otten AT, Bourgonje AR, Peters V, Alizadeh BZ, Dijkstra G, Harmsen HJM. Vitamin C Supplementation in Healthy Individuals Leads to Shifts of Bacterial Populations in the Gut-A Pilot Study. Antioxidants (Basel). 2021 Aug 12;10(8):1278.

谷禾健康

“铲屎官”们都希望自己的宠物有一个健康的身体。但是猫狗都不会说话,平时我们只能从它们的精神状态来判断它们是否健康,但这并不准确。去宠物医院又不太方便,很多猫咪和狗狗还会对抽血等检查有所抗拒。
肠道微生物检测在人类中的应用已经相对成熟,而猫咪和狗狗与人同为哺乳动物,身体结构具有一定的相似性,近年来关于猫狗等宠物肠道微生物的研究也越来越多。
宠物体内具有大量的微生物,包括细菌、真菌以及病毒,稳定的微生物生态平衡对健康成长意义重大。肠道微生物群有助于宿主新陈代谢、抵御病原体、影响免疫系统,并通过这些方式直接或间接影响宿主的行为、情绪等。
许多因素会影响微生物群的生态平衡,包括饮食、年龄、种族、是否绝育等。当体内微生物平衡被打破或者病原微生物入侵时,宠物的机体便遭受破坏,进而影响健康。

本文主要从以下四个方面讲述
Part 1: 猫狗体内的微生物群
Part 2: 影响猫狗微生物群的因素
Part 3: 宠物肠道微生物群影响健康
Part 4: 恢复宠物肠道菌群的措施
微生物群是微生物的复杂集合,包括细菌、病毒、真菌、古细菌和原生动物。猫狗体内的微生物群包括肠道微生物群和皮肤微生物群和口腔微生物群等。
宠物胃肠道微生物群对其健康的影响最为重要,也是数量最庞大的微生物群落。它们可以帮助宠物消化食物、合成维生素、维持肠道黏膜屏障等。
胃中的细菌计数在10^4和10^5 CFU/ml 之间;在十二指肠和空肠中,细菌计数通常较低 (10^5 CFU/ml),但在某些狗和猫中可达到10^9 CFU/mL。回肠含有越来越多的不同微生物群,大多数为10^7 CFU/mL;结肠中的细菌计数介于10^9和10^11 CFU/g。
狗狗身上的微生物群落
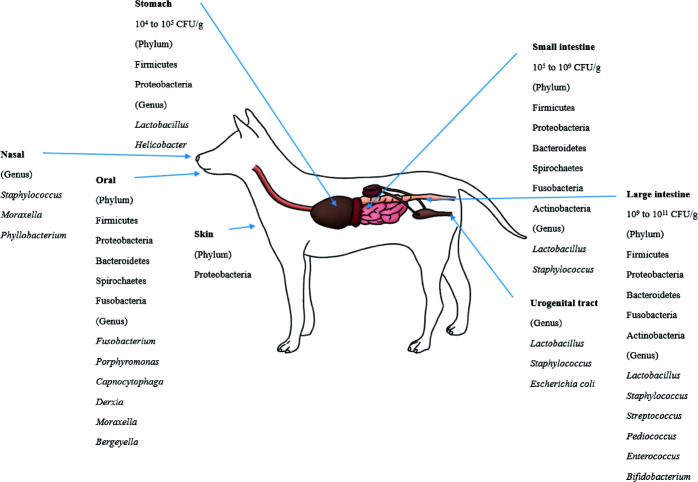
Lee D,et al.J Anim Sci Technol.2022
猫咪身上的微生物群落

Lee D,et al.J Anim Sci Technol.2022
狗肠道内的细菌
狗的核心细菌主要五个主要门组成:厚壁菌门(Firmicutes)、梭杆菌门(Fusobacteria)、拟杆菌门(Bacteroidetes)、变形杆菌门(Proteobacteria)和放线菌门(Actinobacteria)。
狗肠道菌群的一个特征是梭杆菌丰度高
狗与人类和小鼠具有相似的变形杆菌和放线菌的相对丰度,但它是三个物种中唯一具有高丰度梭杆菌的。使其成为狗肠道微生物群一个独有的特征。


Garrigues Q,et al.Front Vet Sci.2022
•厚壁菌
厚壁菌门是肠道微生物群中最丰富的三大细菌门之一,具有高度的物种多样性。它们的主要功能之一是在肠道中产生丁酸盐,丁酸盐被结肠细胞用作能量来源。
厚壁菌门的另一重要类别是杆菌,主要由乳杆菌属和链球菌属组成。乳酸杆菌产生乳酸和乙酸,能够刺激免疫功能并在抗原耐受性中发挥重要作用。
•拟杆菌
狗中第二个最主要的细菌是拟杆菌。
拟杆菌能够使用各种类型的底物进行发酵(其中包括蛋白质和各种碳水化合物),拟杆菌还可以使用聚糖与肠道组织相互作用,提供保护免受病原体侵害。
注:在患有炎症性肠病的狗中观察到该菌的丰度减少。
•梭杆菌
与人类不同,梭杆菌是成年犬肠道微生物群的三个主要细菌门之一,更具体地说是梭杆菌属,约占总相对丰度的20%。
虽然梭杆菌与人类的胃肠道疾病有关,但这种门在健康的狗中很常见。此外,由于梭杆菌在狗和猫体内的丰度高于人类,并且由于它们能够将蛋白质降解为氨基酸和肽,因此推测梭杆菌(Fusobacterium)是食肉动物肠道代谢的关键细菌。
•变形菌
变形菌门多种多样,包括一些机会性病原体,如大肠杆菌(Escherichia coli)、沙门氏菌(Salmonella)和弯曲杆菌(Campylobacter),它们对宿主的健康有潜在影响。
虽然变形菌的丰度增加确实与生态失调和炎症性疾病有关,但这些细菌也被证明在健康的狗中大量存在。
变形菌具有多种功能,包括蛋白质、碳水化合物和维生素的代谢,但与拟杆菌一样,它们的主要功能似乎是维持肠道厌氧环境以实现正常微生物组功能。
•放线菌
放线菌门是狗中数量最少的门,约占成年狗微生物群的4%。该门的相对丰度在幼犬中甚至更少,研究发现56日龄以下幼犬的粪便中放线菌的含量不到1%。
该门的一个重要属是双歧杆菌。在人类中,双歧杆菌是婴儿肠道的首批定植者之一,在宿主的全身和粘膜免疫以及母乳低聚糖降解中发挥关键作用。
注:虽然在1至7周大的幼犬中观察到了双歧杆菌,但在任何年龄较大的犬中均未检测到,这表明它是幼犬肠道的特定细菌。
狗其他部位的微生物群
•口腔微生物群
在口腔微生物中,拟杆菌门(60%)是最主要的,其次是变形菌门(20.8%)、厚壁菌门(11.4%)、梭杆菌门 (4.7%)和螺旋体门 (1.7%)。
在属水平上,口腔微生物群包括卟啉单胞菌(Porphyromonas)(39.2%)、 梭杆菌(4.5%)、二氧化碳嗜纤维菌(Capnocytophaga)(3.8%)、德克斯氏菌(Derxia)(3.7%)、莫拉菌(Moraxella)(3.3%) 和伯格菌(Bergeyella)(2.7%)。
•鼻腔微生物群
在健康狗的鼻腔微生物群中, 莫拉氏菌(Moraxella)是最丰富的物种,其次是叶杆菌属(Phyllobacterium)、葡萄球菌属(Staphylococcus)和心杆菌(Cardiobacteriaceae)。
•皮肤微生物群
在皮肤微生物群中,最主要的细菌是变形杆菌和草酸杆菌科(Oxalobacteraceae)。
•阴道微生物群
从狗的阴道中分离出最常见的细菌是乳杆菌、大肠杆菌和假中间葡萄球菌(Staphylococcus pseudointermedius)。
猫肠道内的细菌
关于健康猫肠道细菌的研究较少。家猫是专性食肉动物,依赖大量摄入动物组织来满足其营养需求。这导致其对低葡萄糖和高蛋白质代谢的适应。
与人类或其他哺乳动物相比,猫不太依赖肠道微生物群通过微生物发酵获取能量。尽管如此,稳定和平衡的微生物群对于维持肠道健康仍然至关重要。
猫胃肠道中厚壁菌门(68%)占主导地位,其次是变形菌门(14%)、拟杆菌门(10%),梭杆菌(5%)和放线菌(4%)。
猫胃肠道中的主要细菌群

Lyu Y,et al.Front Microbiol.2020
然而,这些细菌群的百分比通常因物种和个体具有差异。这些差异可能是由动物的生活环境或使用的不同实验方法引起的。
★ 猫和狗的肠道菌群差异
一项研究比较了喂养适合的饮食下狗和猫的粪便微生物群,发现猫的物种数量比狗多,暗示其多样性更高;然而,需要更多的研究来证实这一发现。
具体来说,变形菌在狗体内更丰富,而放线菌在猫中的相对丰度更高。
猫其他部位的微生物群
一项研究调查猫口腔微生物的水平。主要是变形菌门(75.2%),其次是拟杆菌门(9.3%)、厚壁菌门(6.7%)、螺旋体门(1.8%)、梭杆菌门(1.3%)和放线菌门(0.6%)。
猫狗身上的真菌
在真菌水平,在狗粪便中检测到子囊菌(Ascomycota)、担子菌(Basidiomycota)、球囊菌(Glomeromycota)和接合菌(Zygomycota) 。
而子囊菌门是唯一在猫肠道内检测到的真菌。
•一些真菌感染对猫狗有害
皮肤癣菌病是指皮肤和毛发的浅表真菌感染,常见病原体有:
•犬小孢子菌(影响猫、狗和人类皮肤上层的真菌)
•须毛癣菌、疣状毛癣菌和毛癣菌(影响30%的犬,尤其是梗类犬)
•石膏样小孢子菌(是一种与土壤有关的皮肤癣菌)
•马拉色菌是猫狗动物皮肤菌群中最普遍的真菌。M.pachydermatis被称为存在于皮肤微生物组中的酵母,但它也可以充当可引起皮炎的病原体。
•隐球菌是一种常见的猫狗真菌感染,主要通过呼吸道感染,可以引起呼吸道症状、眼部症状等。
•曲霉菌是一种常见的环境真菌,可以在猫狗身上生长,引起皮肤感染、呼吸道感染等。
需要注意的是,不同的真菌感染症状和治疗方法可能不同,如果发现宠物出现异常症状,应及时就医。
猫狗身上的病毒
家养的猫狗身上一般不存在病毒,或是之前通过疫苗产生免疫,已经存在抗体。猫狗身上的病毒对其基本都是有害的。
•狗狗容易感染的病毒
犬瘟热病毒、犬细小病毒、犬副流感病毒、犬腺病毒1型(传染性肝炎)、犬腺病毒2型(传染性喉气管炎)、犬冠状病毒、狂犬病毒。
一些病毒感染的临床表现
(1)犬瘟热病毒(狗瘟)
潜伏期3—9天,传播方式:直接接触,间接接触
双相热:体温先升高,再降低,然后再升高。 体温升高到39.5~41度,持续1~3天 ,然后下降到正常之后再升高,持续时间不定。
呼吸道症状:眼鼻流脓性粘液、喷嚏、咳嗽、严重时出现肺炎、以腹式呼吸为主。
消化道症状:食欲降低、呕吐、排水样便或粘便,严重时血便,病犬极度脱水,消瘦。
神经症状:抽搐、痉挛 。可能先从嘴角开始,表现为嘴角皮肤抽搐。也可能先从四肢开始抽搐,开始是一肢轻微抽搐,然后加重,频率增加,最后四肢或全身抽搐。
皮肤角化症:鼻部角化干裂、脚垫角化形成硬脚垫病。
眼睛损伤:临床上以角膜炎、结膜炎为特征 ,角膜变白,重者可以出现穿孔、角膜溃疡、失明。
(2)犬细小病毒
出血性肠炎型:接触感染潜伏期3~14天,平均5~7天。呕吐,粪便稀薄,呈喷射状或出现血便,味腥臭。
心肌炎型:40日龄左右的幼犬 ,无明显临床症状,突然衰竭死亡是幼犬患本病的唯一体症。
呼吸道传染病,主要感染幼犬, 发病急,传播快。出现咳嗽、流涕、发热
(4)犬腺病毒1型(传染性肝炎)
潜伏期2~5天。体温升高,随后降低。呕吐,腹痛腹泻。多数两周内死亡,致死率10~25%。
肝炎病变:蓝眼睛(角膜水肿和前葡萄膜的炎症)。肝脏增大,色淡;胆囊壁增厚,出血,呈黑红色。
潜伏期在7天内
呼吸道症状:持续性高热、先是阵发性干咳,后是湿咳并有痰液,浆液性至粘液性鼻漏、扁桃体炎、喉气管炎和肺炎。
潜伏期1~8天,感染途径是消化道。
胃肠道症状:反复呕吐 ,粪便由糊状、半糊状至水样,橙色或绿色,含黏液或血液 (血便),随着日龄的增长,死亡率降低。
•猫咪容易感染的病毒
猫瘟(猫泛白细胞减少症)、猫病毒性鼻气管炎、猫杯状病毒和猫冠状病毒。
一些病毒感染的临床表现
猫瘟(猫泛白细胞减少症 ):
潜伏期为2-9天 ,传染途径是接触带病毒的尿粪或经吸血昆虫及蚤类
发热为40℃左右,顽固性呕吐 ,腹泻后期带有血液,呈咖啡色 ,脱水、眼球下陷 ,循环血液中的白细胞减少 。
猫病毒性鼻气管炎:
潜伏期约2~6天
阵发性喷嚏和咳嗽 ,羞明、流泪、结膜炎,鼻腔分泌物增多 ,鼻液和泪液初期透明,后变为粘脓性。
急性病历通常持续10~14天。成年猫死亡率较低。
猫杯状病毒:
潜伏期约2—3天 ,传播方式:直接接触,间接接触。
浆液性和粘液性鼻漏。口腔溃疡是该病特有的症状,并且有时是唯一的症状。
注意
猫狗的真菌病和病毒通常只在宠物之间传播,但也有可能会对人类健康造成影响。
这些真菌和病毒可以通过接触患病宠物或宠物的环境而传播给人类,引起皮肤感染、呼吸道感染或是消化系统感染等疾病。
特别是对于免疫力较弱的人群,如老年人、儿童、孕妇等,感染的风险更高。
例如近日陕西西安,一名9岁女孩突然发高烧到40度,伴随寒颤四肢发凉,头痛精神差。
经检查确诊猫抓病,猫抓病是由汉氏巴尔通体感染引起的感染性疾病,主要宿主就是猫狗,部分孩子被抓咬也很容易感染,被抓伤后一定要及时处理。

因此,为了自己和家人的健康,我们需要注意以下几点:
1.定期带宠物进行检查,确保宠物健康;
2.及时给宠物接种疫苗,预防宠物感染病毒;
3.定期给宠物清洁并驱虫,避免宠物身上携带寄生虫;
4.在和宠物玩耍后洗手,避免细菌病毒通过手传播。
饮食、年龄、种族、生长环境、绝育、疾病状况和相关疗法在内的多种因素已被证明会影响猫狗的微生物组成。
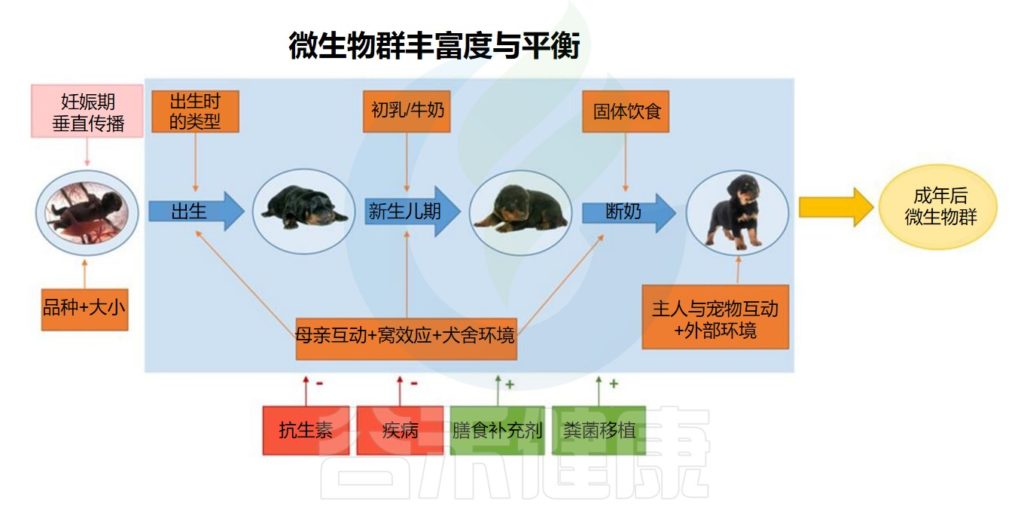
Garrigues Q,et al.Front Vet Sci.2022
1
饮食
饮食已被认为是影响哺乳动物肠道微生物群生物多样性和功能特征的主要驱动因素之一。
家犬目前被认为是杂食动物,商业宠物食品的配方旨在提供均衡的营养摄入,同时补充高浓度的纤维和碳水化合物。
而结肠较短的家猫仍然被认为是专性食肉动物。尽管如此,家猫的商业饲料富含植物来源的成分。
✦常量营养素含量对狗微生物群组成影响显著
一项研究发现,与具有相似常量营养素含量的传统(动植物混合)膨化饮食相比,仅用植物来源的蛋白质制备的膨化饮食不会显著改变狗的微生物组。
另一项针对健康狗的研究测试了宏量营养素成分的重大变化,其中包括4种为减肥、肾脏疾病、低脂肪或抗过敏而配制的处方饮食。减肥饮食在常量营养素(更高的蛋白质和纤维)方面发生了最剧烈的变化,并导致微生物群组成发生了最大的变化。
✦高蛋白低碳水饮食更适合猫
研究评估了断奶后使用高蛋白/低碳水化合物饮食 (HPLC)的小猫与断奶后使用中等蛋白质/中等碳水化合物 (MP/MC) 饮食的小猫的微生物组。
发现高蛋白/低碳水饮食增加了物种多样性,在高蛋白/低碳水喂养的小猫中增加5个属是已知的丁酸盐生产者。
罐头饮食是一种额外的高蛋白替代品,通常单独喂养或与膨化饮食结合使用。研究表明,猫食用罐头会增加水的摄入量,从而减少自主能量摄入量并降低尿液比重,这可能对某些健康状况有益。
高蛋白饮食对猫肠道微生物组成的影响


Pilla R,et al.Vet Clin North Am Small Anim Pract.2021
✦以生肉为主的饮食
近年来,用生肉饮食(RMBD)代替更传统的商业干粮来喂养猫狗已经变得越来越流行。生肉饮食提供重要的健康益处,包括减少牙齿疾病和清新口气、缓解关节炎、增强免疫反应、更健康的皮肤和闪亮的皮毛。
然而生肉饮食也会改变宠物体内的微生物组成,增加接触人畜共患病原体的风险。
以生肉为主的饮食对猫狗肠道微生物的影响
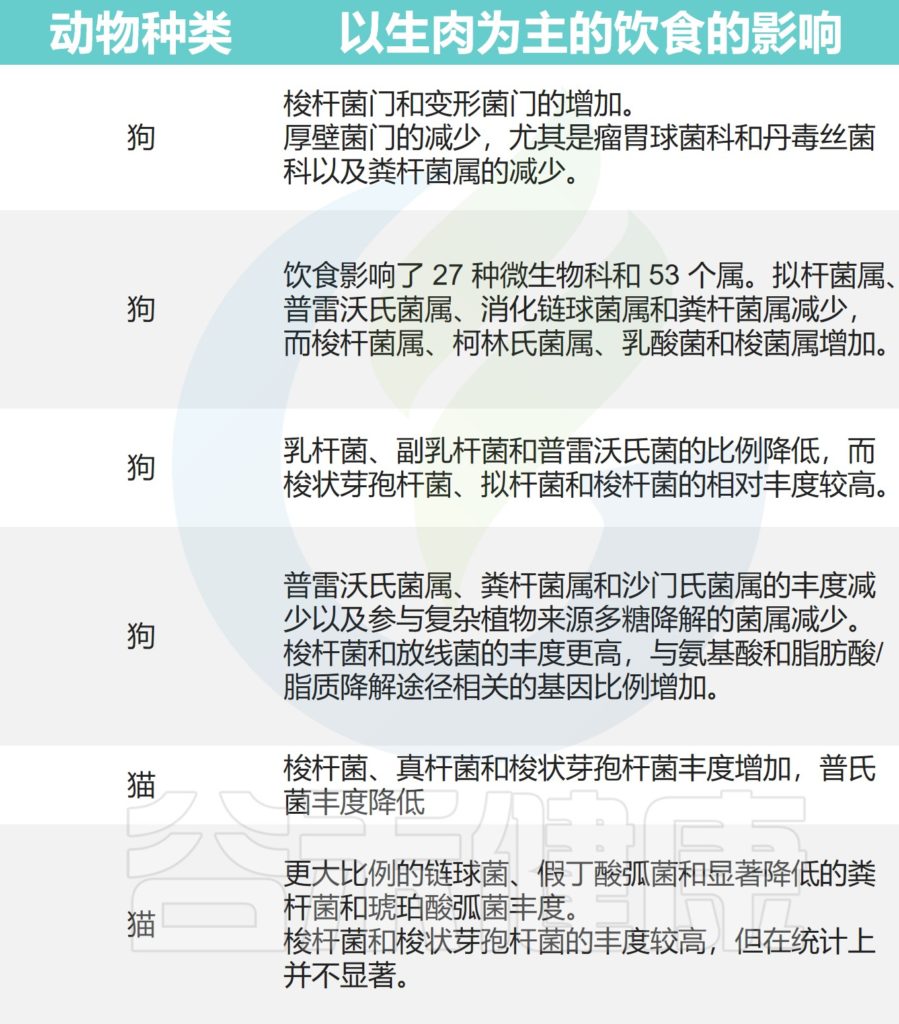
Alessandri G,et al.Microb Biotechnol.2020
总而言之,这些研究证据强调了不同饮食成分对猫狗肠道菌群的塑造作用。需要根据宠物具体的状况选择合适的饮食。
2
年龄
在影响肠道微生物群的众多因素中,年龄是对微生物组成影响最大的因素之一。肠道微生物群的发育在出生时就开始了,并且其组成会随着宿主生命的不同阶段而不断演变。
关于与年龄相关的差异,常见的发现是随着年龄的增加,肠道微生物组多样性减少,乳酸杆菌增多。在一些研究中,观察到梭杆菌随着年龄的增长而显著下降,这与在人类中的结果相反。
梭杆菌在人类百岁老人中的流行率(阳性样本的百分比)并不低于老年人和年轻人。
接下来以狗为例,具体表述一下不同生命阶段微生物群的差异。
幼犬从出生到成年菌群的变化
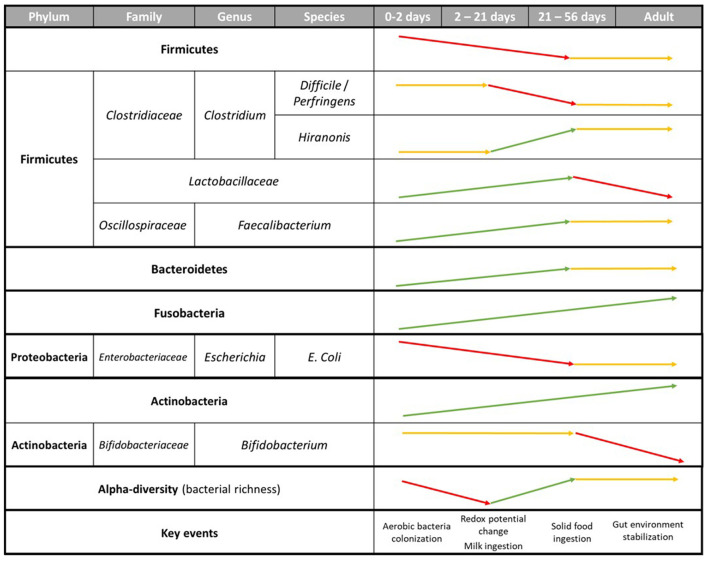
Garrigues Q,et al.Front Vet Sci.2022
✦产前暴露和出生时肠道定植
最初人们认为,哺乳动物的子宫内胎儿期是无菌的,在分娩后的最初几个小时内,通过接触母亲的阴道、皮肤和摄入母乳来接种微生物。
细菌可能在子宫内就从母体转播给胎儿
这种想法最近受到了挑战,因为分子技术的出现允许检测不同哺乳动物胎盘、子宫或羊水中的细菌,细菌可能在子宫内从母亲传播给胎儿。
通过分析胎粪和胎盘样本的微生物群组成,探索了狗胎儿宫内细菌定植的可能性:在出生后立即收集的86.5%的胎粪样本和57%的胎盘样本中检测到细菌。
在幼犬和人类中,分别来自厚壁菌门和变形菌门的葡萄球菌属、链球菌属和Neisseria zoodegmatis是从胎粪和胎盘中分离出的最常见的细菌。
葡萄球菌似乎是母犬子宫内膜微生物群中最常见的属之一,而链球菌更多地存在于母犬的阴道中,这证实了通过阴道分娩出生的幼犬胎粪微生物群部分类似于母犬阴道,并支持微生物的潜在经胎盘转移。
✦幼崽哺乳期乳杆菌丰度增加
出生后,新生小狗的胃肠道很快被微生物定植并且非常不稳定。在生命的前2天,微生物群大约60%的细菌群落的厚壁菌门为主。
注:这个时期微生物群的低微生物丰度和多样性促进了外部细菌的潜在定植。
虽然厚壁菌门在最初2日时主导肠道,但在出生后的头几周其相对丰度显著下降,梭状芽孢杆菌属从2日龄幼犬中鉴定出的总序列的10%开始下降, 在3周时下降到1%。
尽管厚壁菌门的相对丰度总体下降,但幼犬肠道中乳杆菌科的丰度却增加了100倍。结合这些细菌消化牛奶低聚糖和产生乳酸的能力,这表明幼犬肠道丰度的增加不仅与氧稳态有关,还与幼犬在整个新生儿期摄入的母乳有关。
从2天到21天,细菌丰富度显著增加。这些信息表明,幼犬胃肠道细菌的重要变化发生在生命的最初几周,甚至在幼犬开始吃固体食物之前。
✦断奶引起的微生物群变化
狗的断奶被描述为幼犬饮食从母乳逐渐过渡到固体饮食,通常发生在3周大左右,结束于8周左右,此时小狗与母亲分开并且没有无法再喝奶了。
断奶标志着幼犬肠道菌群建立和发展的重要一步,因为新型食物的到来促进了某些菌群的丰度和活动。
拟杆菌丰度增加
如前所述,拟杆菌从第2天的 <1% 丰度增加到第56天的39%, 并一直增加到成年。
拟杆菌是狗肠道中一种多糖降解菌,多糖对于断奶后的幼犬来说是必不可少的,因为它们的饮食开始主要由富含复合碳水化合物的宠物食品组成。
梭杆菌丰度增加
虽然不像拟杆菌那么重要,梭杆菌在断奶后也看到其相对丰度增加,与狗的年龄呈正相关。
梭杆菌可以发酵蛋白质和氨基酸以产生短链脂肪酸和支链挥发性脂肪酸。可以假设断奶后梭杆菌丰度的增加与肉类产品的摄入有关。
✦成年后肠道微生物群丰度趋于稳定
犬科动物和猫科动物的肠道微生物群在整个成年期保持稳定,到老年时会发生进一步变化。衰老通常与营养、生活方式和生理学的持续改变有关,伴随着免疫系统的衰退(免疫衰老),导致慢性低度炎症,并对肠道微生物群落产生影响。
3
品种
品种是微生物群个体间变异的主要来源。专门探索狗粪便微生物群差异的研究发现不同品种之间的α或β多样性没有差异,但微生物组成存在差异。
✦相同饮食和居住条件下不同犬菌群丰度不同
在马尔济斯犬中,梭杆菌(Fusobacteria)数量丰富,而在贵宾犬中,厚壁菌(Firmicutes)和放线菌(Actinobacteria)数量丰富。即使在相同条件下饲养并接受相同饮食时也是如此。
然而,由于在这些研究中观察到的大多数狗都超过1岁,该品种对成长中狗肠道细菌群的影响仍需要更多探索。
✦同一窝的幼崽菌群更相似
此外,与来自不同窝的幼崽相比,来自同一窝的小狗显示出更相似的双歧杆菌种群。这些发现支持这样一种观点,从母亲到她的后代的垂直传播在新生儿肠道微生物群的生物多样性和组成方面发挥着关键作用。
4
绝育
绝育对猫狗健康的影响是一个备受关注的话题,目前有一些研究表明,绝育可能会对宠物的肠道微生物组成产生影响。
一项研究发现,绝育后的雄性猫狗肠道中的某些菌群数量会发生变化,如乳杆菌(Lactobacillus)、双歧杆菌(Bifidobacterium)等菌群的数量会减少,而肠球菌(Enterococcus)、大肠杆菌(Escherichia)等菌群的数量会增加。这些变化可能与代谢、免疫等方面有关。
需要注意的是,目前绝育对宠物肠道微生物的影响还需要更多的研究来证实,而且不同的绝育方式(如手术绝育、化学绝育等)可能会对肠道微生物产生不同的影响。
此外,宠物的饮食、生活环境等因素也会对肠道微生物产生影响,因此在绝育后需要注意宠物的饮食和生活环境,以维护宠物的肠道健康。
5
健康状况及病原体感染
消化系统疾病如腹泻、便秘、胃肠炎等会对猫狗微生物群产生影响,破坏肠道微生物群的平衡,导致有益菌的减少,有害菌的增加。
免疫系统疾病如自身免疫性疾病、过敏等也会对猫狗微生物群产生影响,破坏肠道微生物群的平衡,导致免疫系统功能异常。
炎症性肠病期间肠道微生物群的紊乱
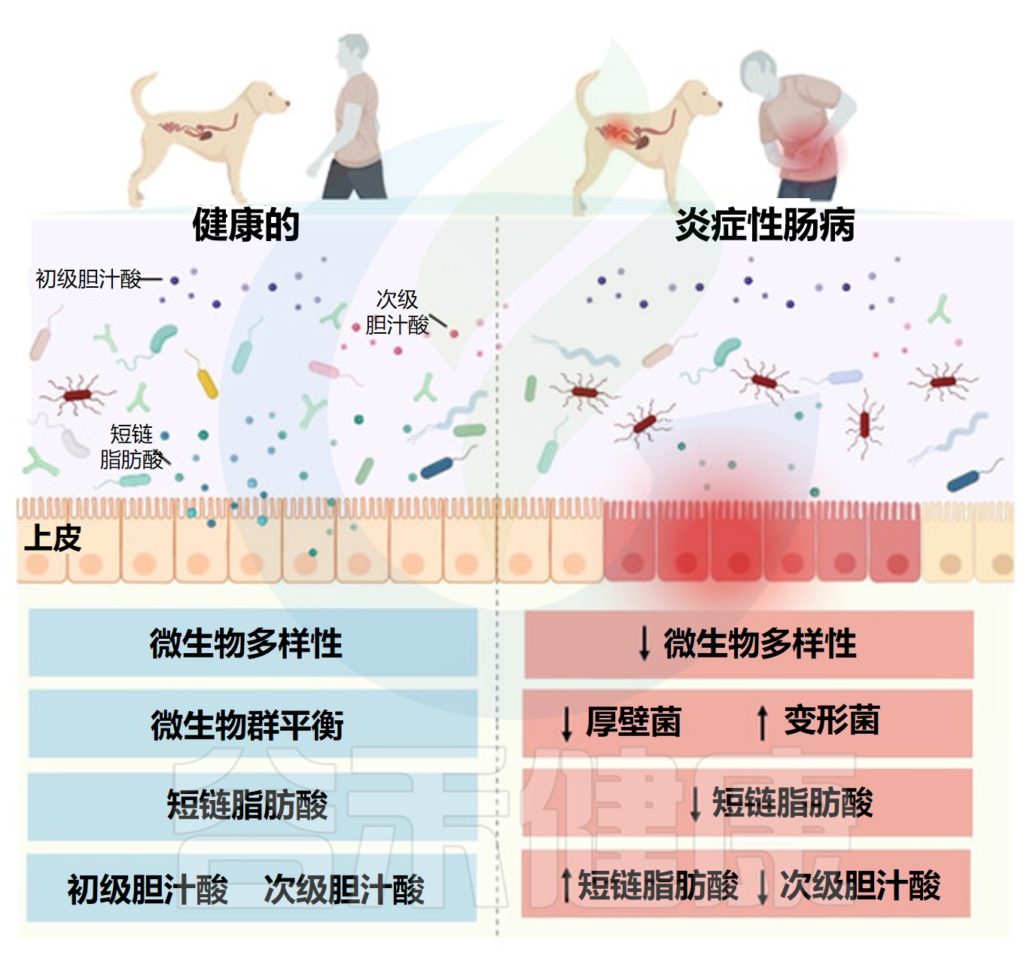
Hernandez J,et al.Microorganisms.2022
✦感染寄生虫改变肠道微生物群
许多肠道寄生虫被证明会引起宠物肠道微生物群的显著改变,其中贾第鞭毛虫是一种普遍存在的肠道寄生虫,可导致腹泻,其带来的改变最为明显。
贾第鞭毛虫与许多细菌群落的丰度呈正相关,例如普雷沃氏菌(Prevotella)和厌氧螺菌(Anaerobiospirillum succiniproducens)。这些细菌会导致肠道屏障粘液的脆弱化。这种脆弱化使肠贾第鞭毛虫更容易切割屏障并允许更多肠道病原体在肠道定植。
此外,在22周大的幼犬中,贾第鞭毛虫的高负荷也与约氏乳杆菌(Lactobacillus johnsonii)的减少相关。
这种细菌是幼犬特有的,并且由于免疫调节、病原体抑制和上皮细胞附着特性,可能在幼犬肠道健康的早期发育中发挥重要作用。
✦病毒感染影响肠道微生物群
犬细小病毒是影响狗的最常见病原体之一,会导致腹泻、出血性肠炎和幼犬死亡。在一项研究中,四只幼犬在6周大时自然感染了犬细小病毒,肠道微生物群发生严重改变,变形杆菌丰度增加,主要是肠杆菌科(Enterobacteriaceae),拟杆菌(Bacteroidetes)和梭杆菌(Fusobacteria)丰度减少。
有趣的是,犬细小病毒阳性幼犬的这些细菌变化并不是永久性的。在感染后2周(一旦从临床细小病毒病中恢复),微生物组成又恢复到与非犬细小病毒感染组相似的组成。
6
其他外源化学物质
宠物受益于先进的兽医护理,并接受与人类相同类别的药物治疗(如抗生素、抗炎药、疫苗等)。与此同时,其中的化学成分也会对宠物的肠道微生物群产生影响。
✦药物改变了宠物肠道菌群丰度
近年来,一些研究评估了某些异生素对人类和宠物狗肠道微生物群的影响。给健康狗服用奥美拉唑会导致厚壁菌门和梭杆菌门的比例升高,胃幽门螺杆菌数量减少,十二指肠总细菌数量增加。
双酚A的影响可以推断到人类,因此,宠物被认为是人类健康的生物哨兵。
✦食品内的化学物质对宠物肠道菌群也有影响
除了出于医学原因服用外源性药物外,人类和宠物狗每天都会接触到影响宿主和肠道微生物群的环境化学物质。研究证明了双酚A(一种广泛存在于食品罐头中的内分泌干扰化学物质)对宠物肠道细菌组成的影响。
在许多细菌属和物种中发现了干扰,包括拟杆菌属、均匀拟杆菌、瘤胃球菌属、罗氏菌属、巨单胞菌属、梭杆菌属、链型杆菌属(Catenibacterium)和普氏粪杆菌。
这里的健康我们分为两部分来讲,一个是影响宠物自身的健康,一个是与主人之间的健康关联。

1-影响宠物自身健康
微生物群可以影响宿主的许多方面,包括生理、行为、繁殖和健康。
胃肠道微生物群促进食物分解以及代谢物的产生,如短链脂肪酸、次级胆汁酸等。微生物群还将营养物质和代谢物释放到体内,影响免疫细胞和炎症反应等。
这里我们从宠物的肠道菌群参与消化吸收、影响肠道屏障、影响代谢、免疫、神经系统等多方面来了解肠道菌群具体如何影响宠物健康。
参与消化吸收
肠道微生物可以分解和代谢宠物食物中的一些难以消化的成分,如纤维素、淀粉等,从而帮助宠物消化和吸收营养物质。
例如,碳水化合物发酵导致短链脂肪酸的产生,短链脂肪酸可以为肠道上皮细胞的生长发育提供营养支持,促进肠道上皮的生长与分化。
蛋白质发酵产生短链脂肪酸、氨基酸和支链脂肪酸。
√ 影响微量元素的合成吸收
同时,肠道菌群还参与合成动物生长发育必需的氨基酸、维生素等,如B族维生素、维生素K、烟酸、泛酸等。
此外,肠道中微生物还能够能促进机体对钙、铁、镁、锌等多种离子的吸收,这些离子对于促进身体某些结构的生长与发育,如:骨骼、牙齿等,对体内氧的输送等有重要作用。
影响肠道屏障
胃肠道微生物群促进定植抵抗,通过竞争排斥提供针对潜在病原体的微生物屏障。这层“菌膜屏障”就像是一层保护伞,对于不慎食入的病原菌进行抑制并排斥,维持体内微生态的平衡状态。
宠物肠道中一些有益菌可以与肠道上皮细胞相互作用,促进上皮细胞的修复和再生,增强肠道屏障的完整性,减少有害物质的渗透。
影响免疫功能
√ 影响先天免疫和适应性免疫
例如,在健康成年猫中补充嗜酸乳杆菌(Lactobacillus acidophilus)可增加外周血粒细胞的吞噬能力并降低血浆中内毒素的浓度。
此外,双歧杆菌(Bifidobacterium)能通过刺激免疫细胞产生重要的细胞因子白介素来促进动物机体内重要的免疫细胞——淋巴细胞的增殖、分化、成熟,增强免疫细胞的对病原体的杀伤力。
√ 刺激宿主产生各种抗菌化合物
例如抗菌肽。微生物代谢物也可以诱导抗菌肽表达:例如,短链脂肪酸和石胆酸可通过不同途径诱导抗菌肽LL-37的表达,这在哺乳动物针对侵袭性细菌感染的先天免疫防御中起着关键作用。
肠道微生物可以与宠物的免疫系统相互作用,调节免疫系统的功能,从而帮助宠物应对病原体的侵袭。
影响代谢
肠道微生物可以产生一些有益的代谢产物,如短链脂肪酸等,这些代谢产物可以维护肠道黏膜屏障的完整性,抑制有害菌的生长,从而维护肠道健康。
肠道微生物组进行的代谢对宿主的影响


Ziese AL,et al.Vet Clin North Am Small Anim Pract.2021
√ 影响代谢相关疾病
•肥胖
肠道微生物群通过其对肠道的直接影响及其对远端器官的间接影响,与肥胖的发展有关。
肠道微生物群会影响胆汁酸的代谢;细菌代谢产生的游离胆汁酸可以抑制细菌种群的生长,例如乳酸杆菌和双歧杆菌,它们被认为可以预防肥胖。
•糖尿病
各种微生物研究表明,胃肠道微生物群在糖尿病等肠外疾病中发挥作用。改变的肠道微生物群组成与猫和狗的糖尿病的发展有关。
例如,最近的一项研究表明,患有1型糖尿病的狗肠道菌群失调,粪便中浓度发生变化。
此外,与同龄的健康猫相比,患有糖尿病的猫的肠道微生物多样性显著降低,产生丁酸盐的细菌也减少了。
•肾脏疾病
慢性肾脏病是猫和狗最常见的疾病之一,有研究发现肾脏病与肠道微生物群之间也存在关联。
与健康猫相比,患有慢性肾脏病的猫的粪便微生物组的丰富性和多样性降低,这与先前对患有慢性肾脏病的人类肠道微生物的研究一致。
影响神经系统
越来越多的证据表明肠道微生物群与身体的主要神经内分泌系统,即下丘脑-垂体-肾上腺(HPA)轴之间的密切相互作用,并将其称为“脑-肠微生物轴”。
√ 影响情绪和行为
该系统控制应对压力的各种身体过程,从而影响宠物的行为和情绪,如焦虑、抑郁等。有研究发现肠道微生物群的失衡会导致猫狗出现抑郁情绪。
√ 记忆力较弱的狗放线菌相对较多
在一项研究中,测量了宠物狗的肠道微生物群组成,并检查了它与年龄和记忆力之间的联系。
表现较差(即记忆力较弱)的狗的粪便样本中放线菌(Actinomycetes)相对较多。这一结果与人类阿尔茨海默病患者中某些放线菌的高丰度一致。
注:该研究的局限性包括犬数量相对较少、用于测试行为关联的样本量较小以及品种和喂养方式差异较大。
因此,宠物的肠道微生物群作为一项重要的健康指标,可以比较全面地了解宠物的肠道微生物组种类、丰度及整体健康状况,包括菌群失调、疾病风险的评估等;还可以在一定条件下判断宠物的情绪和行为。
这些数据可以帮助兽医或宠物主人更好地了解宠物的状况,并根据检测结果制定相关的治疗或饮食计划。
注:相比于普通的检查,肠道微生物检测只需提取少量粪便,不用带宠物去医院,更加方便与安全。
2-宠物菌群与主人健康的关联
研究发现宠物的肠道微生物群与其主人的肠道微生物群存在一定的相似性,因此,通过观察宠物的肠道微生物群情况,可以一定程度上反映出主人的肠道微生物状况及健康。
具体来说,以下是一些可能的方式:
√通过宠物的肠道微生物群反映主人的菌群
研究表明,宠物和主人的肠道微生物群中存在一些共生菌群,如双歧杆菌、乳酸菌等,因此可以通过宠物的肠道微生物群反映主人的肠道微生物群状况。
√通过宠物的微生物群反映主人的疾病风险
宠物与其主人共享家庭环境,因此暴露于相同的环境因素。一些研究表明,与没有疾病的狗相比,患有非传染性疾病的狗的主人更容易患上述疾病。据报道,与非糖尿病犬相比,患有糖尿病的犬的主人患2型糖尿病的风险增加。
宠物和主人的肠道微生物群健康状况与一些疾病的风险存在一定的关联,如肥胖、糖尿病、心血管疾病等,因此可以通过宠物的肠道微生物群反映主人的疾病风险。
√反映主人的生活方式
宠物和主人的生活方式也会对肠道微生物群产生影响,如饮食、运动、应激等,因此可以通过宠物的肠道微生物群反映主人的生活方式。
在谷禾肠道菌群检测报告中,就有这样的特殊案例:
报告显示,该宠物样本菌群构成非常单一,实际测序深度很高,达到10万,但仅检出167种菌,绝大部分是大肠杆菌。
下面的表是根据人的菌群结果评估的,可以看到构成多样性非常低。
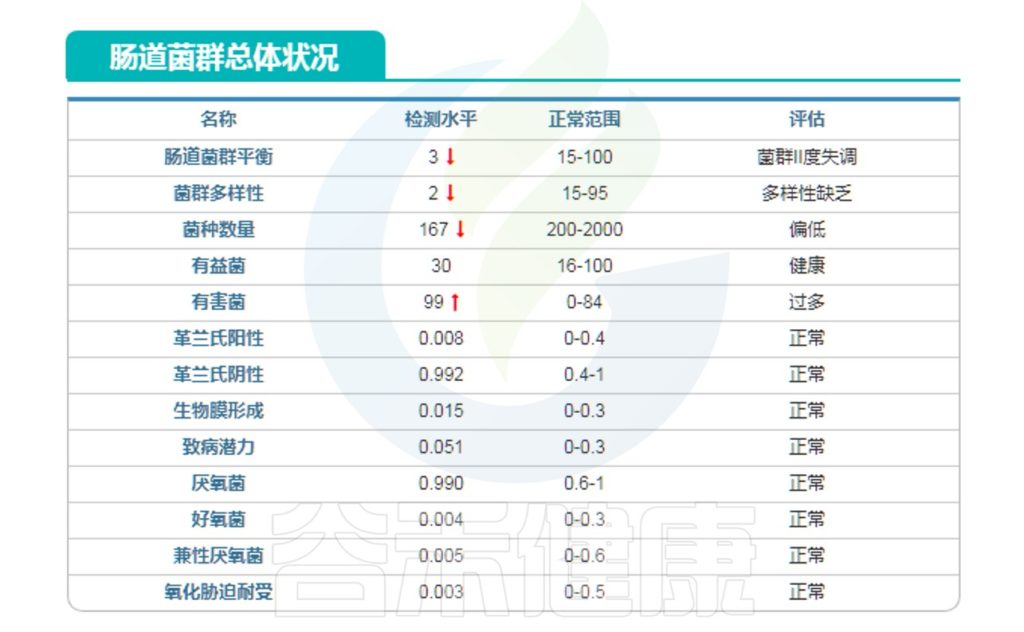
下面是主要菌门和属还有种的构成,种部分列出了注释有菌名称的丰度大于0.1%以上的菌。
下图是菌属构成表:
可以看到,菌属构成中弯曲杆菌占比较高。
除此之外,其他的病原和机会致病菌主要检出了大肠杆菌,占比77%,属于严重超标,空肠弯曲杆菌,产气夹膜菌和痢疾志贺氏菌都有超标:
注:空肠弯曲菌(上图中红色箭头指向菌Campylobacter jejuni),有内毒素能侵袭小肠和大肠黏膜引起急性肠炎。
经过沟通后,我们了解的情况如下:
主人反映该宠物猫有腹泻,其主人也有长期腹泻情况,因此送检了其本人和宠物猫的粪便样本做肠道菌群检测,了解菌群构成,找出腹泻原因,以便后续治疗。
因此,宠物的微生物群组成在一定程度上可以算是主人的健康哨兵。
随着年龄、生活方式或药物的影响,健康宠物的肠道微生物组平衡可能受到影响甚至会被破坏,从而引发各种疾病。我们应该采取何种方法来恢复其健康呢?
肠道微生物群可以通过饮食调整、抗菌剂、益生元和益生菌以及粪菌移植进行调节。这些治疗方法每一种都具有不同的优点和副作用。因此必须考虑潜在的疾病,因为没有针对所有类型生态失调的治疗方法。
饮食是影响宠物肠道微生物群的重要因素之一,不同的饮食组成会对宠物肠道微生物群的生态平衡产生不同的影响。
根据不同宠物制定相对应的饮食
蛋白质、碳水化合物和脂肪是大部分宠物必需的营养物质,是促进机体生长的重要营养素。合理控制这些营养素的摄入是维持肠道健康的关键。
不同年龄段的宠物对营养素的吸收能力不同。老年犬猫由于肠道消化功能逐渐退化,不能较好地吸收,需要根据不同的宠物状态制定相对应的饮食。
适量的功能性脂肪酸,如Omega-3脂肪酸可以增强宠物机体免疫力,预防炎症,减轻其胃肠道疾病的发生。
注意宠物粮成分
宠物食品制造商在其产品中添加的一些成分会对猫狗的肠道菌群和整体健康产生负面影响。
人工色素和抗氧化剂等食品添加剂减少肠道细菌的数量。在购买前最好能阅读食物的配料表,选择适合自家宠物当前阶段的粮食。
谷禾在此提出了两种饮食建议供大家参考
•“无谷物狗粮”
存在关于无谷物狗粮的争论。有些兽医认为不应该用任何谷物喂养宠物,有些人则说它可以适量喂养。
不建议吃的原因包括体重增加、胃肠道炎症增加、过敏等。如果狗不耐受谷物,可能会出现皮肤发痒、呕吐、胃部不适、脱毛、腹泻、感染和胀气。
换句话说,如果出现上述症状,可以尝试一下无谷物食物并监测症状是否有所改善。
•在饭菜中加入骨汤
骨汤含有多种有益营养素,但与肠道健康最相关的是胶原蛋白。胶原蛋白是一种结缔组织,有助于维持包括肠道和皮肤在内的身体器官的结构。
狗狗的肠道内部衬有一层叫肠粘膜的组织。粘膜的作用是防止食物分子和细菌等危险物质从肠道进入血液。
然而,粘膜可能会受损,导致肠道变得可渗透。换句话说,像细菌这样的物质可以通过。这就是所谓的“肠漏”。
通过给狗狗吃富含胶原蛋白的食物,比如骨头汤,可以加强粘膜中的结缔组织,从而减少肠漏程度。
研究发现压力会引起体内激素的变化,从而引起肠道微生物组的变化。特别是,压力会触发皮质醇激素的释放。皮质醇对身体有多种负面影响,包括血压升高和肠道健康受损。
保持良好的心情有利于宠物健康成长,以下是一些有助于宠物减轻压力的方法:
1.提供足够的运动和活动:适当的运动和活动可以帮助宠物消耗能量,缓解压力。主人可以带宠物出门散步、玩耍等。
2.提供安全感:宠物可能会因为分离焦虑或其他原因感到不安。主人可以给宠物提供一个安全的环境,例如提供一个温暖舒适的床铺、一个安静的角落等。
3.提供适当的社交环境:宠物需要与其他宠物和人类进行社交互动。主人可以带宠物参加社交活动,例如去公园或参加宠物聚会等。
4.适当的训练:训练可以帮助宠物建立自信和独立性,从而减少压力。
益生菌、益生元的施用也会影响和改变微生物群的组成。益生菌和益生元已经在人类中广泛运用,在宠物中也同样适用,现在许多宠物粮中已经添加了益生菌。
许多在宠物健康方面研究的益生菌属于乳杆菌属、双歧杆菌属和肠球菌属。
益生菌可以通过多种机制改变常驻微生物组,包括通过代谢相互作用刺激常驻细菌的生长,改变病原菌的丰度,或通过与宿主上皮细胞和上皮免疫系统的相互作用间接影响健康。
益生菌对狗的治疗作用

Mondo E,et al.Open Vet J.2019
一些研究报告了在宠物饮食中添加益生元的好处。事实上,它们能调节肠道微生物群并保护动物免受肠道感染。
猫和狗接受益生元的治疗效果

Mondo E,et al.Open Vet J.2019
注意
一些人建议益生菌中使用的细菌最好来自宿主同一物种的肠道;然而,对于目前市场上用于猫和狗的大多数益生菌而言,情况并非如此,并且没有研究将狗或猫来源的益生菌的功效与来自其他物种的市售菌株进行比较。
对市售兽医益生菌的审查揭示了质量问题,包括标签不准确和生存能力差; 其他人对安全性提出了担忧,还需要更多研究来了解在猫和狗中使用益生菌的效果。
当饮食调整不起作用时,患有慢性肠病的狗和猫会接受抗生素治疗。
常用的抗生素:
甲硝唑
甲硝唑对粪便微生物组和代谢组有显著影响,无论是单独使用还是与水解蛋白饮食结合使用。
施用甲硝唑后,微生物组丰富度降低。微生物组的变化伴随着粪便乳酸杆菌增加、粪便和血清中氧化应激标志物增加以及胆汁酸转化受损。
泰乐菌素
泰乐菌素用于治疗对泰乐菌素有反应的慢性腹泻,这通常会影响成年犬。许多研究强调了它的效率,但其作用机制仍然未知。
恩诺沙星
恩诺沙星是另一种用于治疗肠病的抗生素。它是一种氟喹诺酮,可用于治疗肉芽肿性结肠炎。
使用抗生素同时还会减少有益菌
需要注意的是,使用抗生素治疗某些疾病存在禁忌症。事实上,大量使用抗生素可能会减少有益菌的数量,促进潜在病原体数量的增加,并促进抗菌素耐药现象的发生。
在停用抗生素后,微生物的变化可能会持续数年,而一些细菌类群的数量可能永远不会恢复到初始状态。
一些研究人员表明,抗生素实际上会改变微生物组成,从而降低细菌多样性,因此它们会导致生态失调的发生。在过去的几年中,抗菌素耐药性的现象不断增加,并具有重要意义。人类与宠物的密切关系甚至会导致抗菌素耐药性细菌转移人体。
调节肠道微生物群的一种新方法是“粪便微生物群移植”。粪菌移植是指将来自供体的粪便物质溶液施用到受体中,主要是为了改变受体的微生物组成。
该手术可通过十二指肠镜检查、鼻胃管/鼻空肠管、结肠镜检查、灌肠或口服胶囊进行。
患有肠病的狗经粪菌移植后得到改善
报告证实一只患有难治性炎症性肠病的贵宾犬在粪菌移植后也产生了积极作用。这只狗通过灌肠接受了九次粪菌移植。6个月后,狗的临床炎症性肠病活动指数和粪便稠度得到改善。
另一项研究描述了一只患有慢性结肠炎和艰难梭菌感染的8个月大的法国斗牛犬。这只狗接受了单次口服粪菌移植。排便频率和粪便稠度在2至3天后显著改善,至少6个月未观察到复发。
对患有急性腹泻的犬只进行一次粪菌移植效果非常成功。使用粪菌移植代替抗生素可以预防负面后果,例如微生物多样性降低、特定细菌类群发生变化、丰度和代谢转变。
粪菌移植对宠物其他慢性疾病也具有治疗效果
粪菌移植在治疗患有慢性肠病或胰腺外分泌功能不全等慢性疾病的狗时也显示有效的结果。需要注意的是,在粪便移植几天后病情有所好转,但随后往往会复发。因此,在大多数情况下可能需要多次粪菌移植。
如今,在动物实践中,粪菌移植有可能改善与生态失调相关的急性和慢性疾病的健康状况。由于对其使用的研究较少,其标准化需要进一步研究。
噬菌体是一种能够侵染细菌的病毒,是生物圈中广泛分布,但是关于这方面的研究和应用还较少。
噬菌体可以裂解消灭有害菌
噬菌体通过尾丝蛋白与宿主菌表面受体特异性结合侵染细菌使其裂解,精准消灭有害菌,不影响有益菌。
30分钟内可杀灭99.9%的宿主菌,且不受细菌耐药性限制。
面对细菌耐药、抗生素残留等问题,噬菌体作为天然的细菌杀手,是防治细菌性疾病的最佳选择。
有研究表明,噬菌体可以裂解肠道中的有害菌例如沙门氏菌和大肠杆菌,降低了肠道有害菌数量,有益于肠道有益菌(如乳酸菌类)的繁殖和生长,起到了调节肠道菌群的作用。
噬菌体还有利于免疫器官的发育和体液免疫功能的提高。
研究宠物的微生物群不仅有益于其自身的健康,也有益于它们主人的健康,因为伴侣动物与人类具有相同的生活环境、相似的饮食模式和微生物群落。
对于猫狗的肠道菌群仍然有很多未知和亟待发掘的意义,有望成为一种非常有价值的检测和干预渠道。谷禾欢迎更多相关领域的合作伙伴共同推进和完善猫狗相关肠道菌群的研究和应用。
主要参考文献
Tian T, Zhou Y, Xu Y, Xu Y. Intestinal microbial 16S sequencing and LC-MS metabonomic analysis revealed differences between young and old cats. Heliyon. 2023 May 19;9(6):e16417.
Sinkko H, Lehtimäki J, Lohi H, Ruokolainen L, Hielm-Björkman A. Distinct healthy and atopic canine gut microbiota is influenced by diet and antibiotics. R Soc Open Sci. 2023 Apr 26;10(4):221104.
Garrigues Q, Apper E, Chastant S, Mila H. Gut microbiota development in the growing dog: A dynamic process influenced by maternal, environmental and host factors. Front Vet Sci. 2022 Sep 2;9:964649.
Deschamps C, Humbert D, Zentek J, Denis S, Priymenko N, Apper E, Blanquet-Diot S. From Chihuahua to Saint-Bernard: how did digestion and microbiota evolve with dog sizes. Int J Biol Sci. 2022 Aug 1;18(13):5086-5102.
Li Y, Ali I, Lei Z, Li Y, Yang M, Yang C, Li L. Effect of a Multistrain Probiotic on Feline Gut Health through the Fecal Microbiota and Its Metabolite SCFAs. Metabolites. 2023 Feb 3;13(2):228.
Pilla R, Suchodolski JS. The Gut Microbiome of Dogs and Cats, and the Influence of Diet. Vet Clin North Am Small Anim Pract. 2021 May;51(3):605-621.
Balouei F, Stefanon B, Sgorlon S, Sandri M. Factors Affecting Gut Microbiota of Puppies from Birth to Weaning. Animals (Basel). 2023 Feb 6;13(4):578.
Kubinyi E, Bel Rhali S, Sándor S, Szabó A, Felföldi T. Gut Microbiome Composition is Associated with Age and Memory Performance in Pet Dogs. Animals (Basel). 2020 Aug 24;10(9):1488.
You I, Kim MJ. Comparison of Gut Microbiota of 96 Healthy Dogs by Individual Traits: Breed, Age, and Body Condition Score. Animals (Basel). 2021 Aug 18;11(8):2432.

谷禾健康

人类微生物组研究正在从描述关联发展到了解整个微生态对人类的影响。虽然存在挑战,但在应用数据驱动的微生物组诊断和干预方面正在取得进展,这可能会在未来十年内带来精准医学的突破。
本文我们来探讨关于微生物组的研究进展及其对人类健康的影响及应用。
随着对微生物组的理解深入,发现巨大的潜力
过去 20 年,我们对微生物组在人类宿主生理学中作用的理解发生了重大转变。
开创性研究表明,与人类细胞和基因相比,原核生物为人类“全生物”贡献了相同数量的细胞和多倍的基因,同时影响哺乳动物从消化和新陈代谢过程到免疫甚至大脑功能。
微生物组的变化与越来越多的人类疾病有关,包括癌症。这些发现使人们普遍希望,积累的关于宿主-微生物组相互作用的知识,将迅速转化为人类疾病的治疗方法。
对微生物组的认识开始客观,同时意识到复杂性
随着该领域的稳步成熟,科学家们开始更好地了解整个微生物组。他们意识到这并不像他们最初想的那么简单。随着他们开始了解微生物之间的相互作用是多么复杂,以及将微生物组用于人类治疗有多么大的挑战,最初围绕它的兴奋和炒作开始平息。
面临挑战,需要综合多方面的努力
要克服这些挑战,许多研究者需要进行长期、数据驱动的研究。他们要综合各种看起来不相干的科学和医学学科,包括进化论、人类学和营养学等等,还要结合新技术的开发和优化,以及注意事项和混杂因素的识别和最小化,共同推动了这一探索。他们的目标是要弄清楚宿主和微生物组之间的互动关系和分子机制,以期找到因果关系。虽然充满了挑战,但只要大家协作一致,一定可以获得成果。这些成果将在未来十年内实现对可利用的治疗性微生物组目标进行更广泛和可重复的识别和调节。
微生物组研究中的挑战和新兴方法
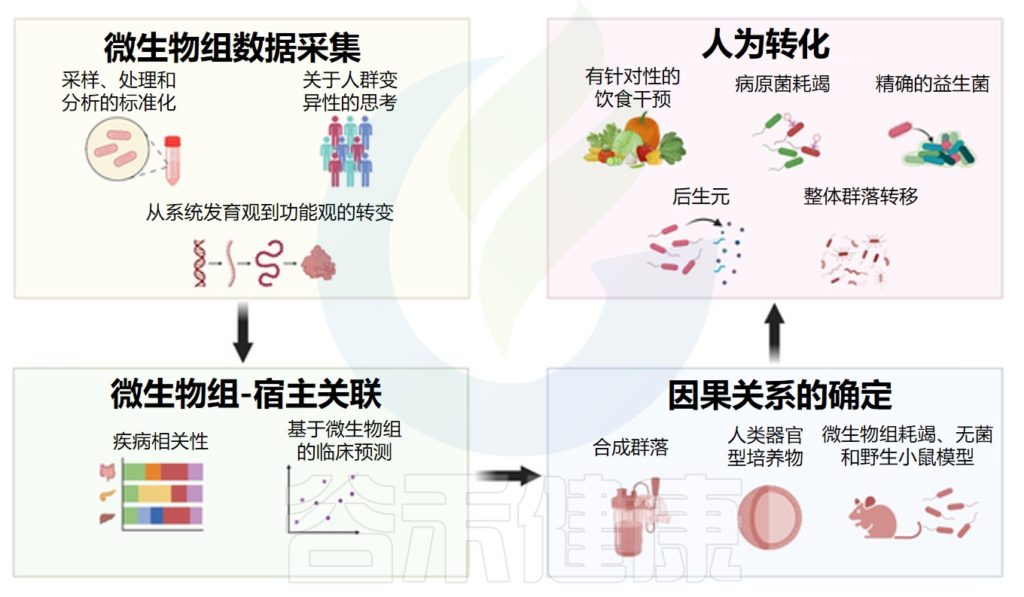
doi: 10.1371/journal.pbio.3002053
本世纪第一个十年,研究微生物组结构表征、对生理功能的重要性,由此衍生到各个领域
事实上,本世纪微生物组研究的第一个十年,集中于证明肠道微生物组对生理功能的重要性,以及在各种生理和疾病背景下基于下一代测序的微生物组群落结构表征。
这带来了微生物组与各种健康结果之间的关联、相关性和预测的识别,报告规模和细节的不断提升。与此同时,该领域广泛描述了微生物特征响应多种因素的动态调节,例如饮食、宿主免疫 、基因组学和昼夜振荡。
▼
随着这些重大进展,人们也认识到了主要的技术和概念障碍,这些障碍对微生物组发现的解释、概括和转化到临床具有挑战性。
使微生物组相关观察结果的解释复杂化的技术挑战之一是:不同研究之间样本收集、处理和分析方法的广泛差异。
此外,在微生物组处理和分析级联的每个步骤中发生的污染和注释错误可能会引入伪影和偏差,这些伪影和偏差难以解决并使真实和虚假生物信号之间的区别复杂化。
解决方案:将真实信号与混杂因素分开
了解与这些步骤中的每一个相关的范围、限制和混杂因素,对于将真实信号与混杂因素分开至关重要,特别是在微生物丰度低或无微生物丰度的样本。更好地协调这些方法,加上多种技术和生物控制,可以更准确地解释可以在人群、地域、性别和种族之间推广和复制。
然而,如果不冒技术可变性和多样性减少的风险,微生物组处理和分析标准化就不能过度执行,而技术可变性和多样性构成了推动研究创新的引擎。
▼
大多数人类微生物组研究面临的一个概念挑战是,微生物群落结构中内在的和生理上重要的个体间变异性。
如何在大样本人群数据上去识别和消除这些差异,应该是目前微生物组特征分析面临的巨大分析挑战,因为它们代表了一种固有的、难以解决的“噪音”。
解决方案:基于深度人工智能的方法等创新方法
可以识别和利用不同的微生物组配置来探索微生物组对个体人类表型的贡献,范围从血糖反应到补充剂和食物携带相同疾病易感位点的个体之间的不同疾病表现。
总而言之,使用基于深度人工智能的方法等创新方法应对技术和生物变异性的挑战,将加深微生物组知识与精准医学的合理整合。
▼
一个同样艰巨的挑战涉及从微生物组-人类特征关联到因果关系证明的进展。事实上,在建议与临床结果相关、相互关联和预测的许多微生物群中,只有少数基因产物和代谢物可以证明对生理和疾病状态有因果影响,而我们预计一部分微生物特征是先发于疾病病理改变的特征,而有一部分特征是继发于以疾病状态为特征的环境变化。
越来越多的工具正在开发中,通过揭示因果关系和机制洞察力,将前者“驱动”微生物组改变与后者“乘客”改变区分开来。
解决方案:无菌或使用抗生素的动物模型或其他改进的技术,测试单个细菌菌株或群落的影响
一种经常使用的方法依赖于微生物群耗尽(通过抗生素治疗)或微生物群缺乏(无菌)小鼠模型,这些模型允许引入单个细菌菌株或群落以测试它们对宿主生理学的影响。
抗生素杀菌模型局限性:多因素混杂,难以预测
抗生素是现代医学的基石,因为它们能够在感染期间抑制病原体的生长。抗生素的广泛特征是它们的抑制机制是抑菌(生长停止)或杀菌(致死),并且它们的活性范围是宽的或窄的。大多数抗生素的作用主要在物种单一培养中进行了研究,尽管临床环境中的治疗不可避免地会对人类肠道上的多物种群落产生意想不到的后果。抗生素会对肠道细菌造成附带损害,并降低肠道微生物群的多样性,由此可能出现艰难梭菌感染倾向增加和癌症治疗后死亡率增加。
抗生素扰动期间的群落动态很难预测,因为环境可以通过种间相互作用、pH 调节和代谢转化改变抗生素的作用。使用多种治疗方案,例如可变剂量、顺序安排多种化合物和同时使用鸡尾酒治疗使预测进一步复杂化。
同时多种药物可产生协同或拮抗作用, 由此杀死的程度分别大于或小于单独药物的总和,但是他们对肠道菌群的扰动和影响研究还甚少,检测中面临这些用药信息准确跟踪记录也较困难。而且抗生素导致的多重耐药细菌的定植也会导致抗生素耐药基因转移到微生物群中,最终会导致对各种有效抗生素的耐药性增加,并增加致命感染的风险。
其他新技术:改进的体内模型、类器官和片上器官平台
随着更多具有人类代表性的实验平台的出现,野鼠体内的微生物群与圈养外的家鼠相似,正迅速受到关注。这些改进的体内模型得到了合成人类共生群落的发展、以前“无法培养”微生物在菌株水平分辨率下的改进培养和表征,以及通过非靶向代谢组学和蛋白质组学对生物活性微生物效应因子或代谢物的无偏见阐明的补充。
类器官和片上器官平台能够研究人体组织环境中的单个细菌、细菌群落及其生物活性产物。
注:
类器官(organoid)是指由人体组织或干细胞等体外培养出的三维结构,能够模拟某些人体器官的结构和功能。类器官的制备通常是通过将干细胞或组织细胞在特定的培养条件下进行分化和自组装,形成类似于人体器官的结构。类器官的制备可以为疾病研究和药物筛选提供更真实、更可靠的模型。例如,肝脏类器官可以用于测试药物代谢和毒性,肠道类器官可以用于研究肠道微生物和肠道疾病,而大脑类器官可以用于研究神经退行性疾病等。
片上器官平台(organ-on-a-chip platform)是一种仿生学技术,它是通过微流控技术、微加工技术和3D打印技术等手段,将人体组织和器官的细胞培养在微型芯片上,模拟人体器官的生理、病理过程,以实现高通量、高精度的药物筛选、毒性测试和疾病研究。这种技术可以模拟人体器官的微环境,如细胞间相互作用、细胞信号传递、细胞分化、细胞迁移、细胞凋亡等生理和病理过程,从而更好地研究疾病的发生和发展,以及评价药物的安全性和有效性。
凭借这些新技术获得的能力,该领域正在揭示基因组测序之外的功能读数,解码难以培养和低生物量微生物组的贡献,并评估重要但研究不足的非细菌共生王国及其错综复杂的相互作用组网络。
事实上,对共生和机会病毒(包括噬菌体)、真菌和寄生虫的潜在功能及其对细菌共生生态系统和人类宿主的影响的不断探索,构成了正在推进的研究领域,当然肠道细菌还是最主要的。对这些特征不明确的共生微生物的研究需要进一步开发各种工具,包括改进的计算参考数据集、分子开发工具和体内定植模型。
通过对微生物分子和功能见解的不断扩展,可能会在微生物组研究的下一个时代开发广泛的诊断、预后和治疗人类应用。
治疗性微生物组使用的一个有前景的例子:
粪便微生物组移植 (FMT)
除了治疗常见的艰难梭菌感染之外,显示出对肝病、炎症和免疫相关疾病的积极改善效果
促进微生物组研究领域的蓬勃发展
失败的试验和风险强调了对机制的了解的重要性
为了避免人体微生物研究领域中早期技术失误:
为了辅助人类疾病的诊断和治疗需要:
作为向前迈出的积极一步,微生物组研究能够做到以下:
早期检测
识别和利用微生物组粘膜,免疫和全身信号,以便通过离散的疾病特征进行早期检测和患者分层。
治疗层面
“一刀切”的传统观念正在发生改变,人们不再简单地认为所有人的微生物组都可以通过相同的方式进行调节,而是转向将微生物组视为“信号中心”的观点,也就是说更加注重研究和借鉴微生物群落的信号传递机制,以更好地治疗相关疾病。
微生物组被视为特定环境的中继或缓冲器,整合环境和内源性信号,影响携带健康或疾病易感遗传特征的个体的生理和疾病表现。
一个备受关注的话题是“益生菌干预法”。证据表明益生菌可能有助于治疗和预防传染病,改善过敏,降低血糖,联合免疫治疗提高治疗效果。益生菌治疗会改变腔内微生物群,这种改变进一步会显着影响全身代谢,包括胰岛素抵抗,炎症,代谢等。
将微生物组作为一个高度模块化的“指纹”,有助于开发合理的、针对具体情况的和数据驱动的干预措施,例如个性化营养、精准益生菌、代谢物补充(“后生元”疗法)和定向病原体抑制模式。
这些方法的验证和整合,加上对大量微生物群及其相关生物活性产品对人类宿主的影响的持续探索,将有助于实现该领域改变人类健康的巨大前景。
参考文献
Puschhof J, Elinav E. Human microbiome research: Growing pains and future promises. PLoS Biol. 2023 Mar 17;21(3):e3002053.