-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康

以前我们科普过肠道菌群在门级别水平分类的肠道细菌四大常见菌门——拟杆菌门,厚壁菌门,变形菌门,放线菌门。
详见:肠道细菌四大“门派”——拟杆菌门,厚壁菌门,变形菌门,放线菌门
但是随着研究范围以及样本的扩大,发现我们人体肠道内除了这些常见的菌群之外,还有许多小众门派的菌群在肠道平衡中也扮演着重要的角色,它们同样可以帮助我们消化食物、维持肠道健康、增强免疫力等等。这些微生物组通常作为宿主基因组的功能扩展,在宿主生理和新陈代谢的调节中起着至关重要的作用。
在本文中,我们将深入探讨这些小众门派的菌群,揭示它们的神秘力量和重要性,帮助我们更好地了解肠道菌群的多样性和复杂性。
我们根据谷禾肠道数据库人群检出比例的丰度排名依次介绍这些菌及其与肠道和人体健康的相关信息。
根据人群检出丰度依次为:
疣微菌门 Verrucomicrobia
梭杆菌门 Fusobacteria
蓝藻门 Cyanobacteria
酸杆菌门 Acidobacteria
软壁菌门 Tenericutes
绿弯菌门 Chloroflexi
互养菌门 Synergistetes
芽单胞菌门 Gemmatimonadetes
黏胶球形菌门 Lentisphaerae
浮霉菌门 Planctomycetes
硝化螺旋菌门 Nitrospirae
脱铁杆菌门 Deferribacteres
螺旋体 Spirochaetes
装甲菌门 Armatimonadetes
绿菌门 Chlorobi
迷踪菌门 Elusimicrobia
衣原体 Chlamydiae
这里我们对门层级中的常见肠道菌群逐个介绍。
疣微菌门(Verrucomicrobia)是一类革兰氏阴性细菌,是细菌分类学中的一个门级分类单元。这类细菌的细胞形态多样,包括球形、杆状、螺旋形等,常见于土壤、水体和动物肠道等环境中。
疣微菌门是细菌域内的一个新分支,1997年被列为一个门。它们代表系统发育树中的一个独特谱系,包含许多环境物种以及少量培养物种。
疣微菌门是一类广泛存在于自然环境中的细菌,包括多种典型的菌种,例如常听说的Akkermansia muciniphila,其他还有Opitutus terrae、Prosthecobacter debontii等。这些菌种在不同的环境中具有不同的代谢和生理特性。
▼
疣微菌门存在于肠道粘膜内层,在健康个体中大量存在,它们可以分解多糖类物质,如黏多糖和纤维素等,从而提供能量和营养物质。
疣微菌门还可以产生短链脂肪酸,如丙酸和丁酸等,这些物质对肠道健康和免疫系统的调节具有重要作用。
虽然说疣微菌门仅占肠道微生物群落总数的一小部分,但结果表明,一些疣微菌种系型对多糖和木聚糖的降解做出了重大贡献。基因组编码多种糖苷水解酶、硫酸酯酶、肽酶、碳水化合物裂解酶和酯酶,具有水解多种多糖的机制。
▼
在肠道微生物组中,疣微菌门是一个重要的菌群之一。 谷禾肠道数据库中大约56.29%的人群有检出。
⇒
研究表明,疣微菌门的丰度与肠道健康密切相关。疣微菌门有助于人体肠道的葡萄糖稳态, 具有抗炎特性,可进一步帮助肠道健康。研究表明 该菌与foxp3 基因之间存在正相关关系,foxp3 基因是一种在人类中表达抗炎和免疫力的基因。
⇒
一些疾病如肥胖症、炎症性肠病、睡眠障碍和2型糖尿病等,与疣微菌门的丰度降低有关。其他如哮喘、自闭症等疾病人群中丰度也会变化。
⇒
其中,Akkermansia muciniphila(简称AKK菌)是疣微菌门中研究较多的菌种之一。研究表明,Akk菌可以降低肥胖、糖尿病、肠炎、肠癌等疾病的风险。这是因为Akk菌可以促进肠道黏液层的生长和维护,增强肠道屏障功能,减少有害菌的生长,降低肠道内毒素的水平,从而保护肠道健康。
详见:
肠道重要菌属——Akkermansia Muciniphila,它如何保护肠道健康
梭杆菌门 (Fusobacteria)是细菌门之一,它们是一类革兰氏阴性菌,通常是长杆状或螺旋状的。在谷禾肠道数据中,该菌的检出率是49.16%.
梭杆菌门包括多种典型的种属,如:Fusobacterium nucleatum(具核梭杆菌)、
Fusobacterium varium(变异梭杆菌)、
Fusobacterium necrophorum等。
▼
梭杆菌门广泛存在于自然环境中,包括土壤、水体和动物肠道等。已证明梭杆菌门下的许多物种可以自由生活,而无需与周围环境中的其他生物结合。其中大部分包括在海洋环境中发现的物种,例如 Llyobacter 属和 Psychrilyobacter 属的成员。
Photograph by Hipersynteza
自由生活的梭杆菌属的最好例子之一是 Psychrilyobacter atlanticus 物种,它可以在大西洋的海洋沉积物中找到。
它们是一类厌氧菌,通常生长在肠道内的低氧环境中。梭杆菌门在人体内的分布广泛,不仅存在于肠道中,还存在于口腔、阴道和皮肤等部位。
▼
⇒
一些研究表明,梭杆菌门可能与肠道疾病有关。例如,一些研究发现,梭杆菌门在结肠癌组织中的含量明显高于正常组织。此外,梭杆菌门也与炎症性肠病(IBD)和肠道感染有关。
⇒
在口腔中,梭杆菌门的存在则与龋齿和牙周炎等口腔疾病有关。
然而,梭杆菌门在肠道菌群中也扮演着有益的角色。梭杆菌门可以产生一些对人体有益的代谢产物,如丙酮酸和丁酸等。一些研究表明,梭杆菌门可能参与了肠道菌群的稳态维持和代谢功能。
⇒
吸烟者的梭杆菌丰度显著更高(P = 0.009,FDR = 0.027 )。
⇒
每日食用面包的人群中,梭杆菌丰度也显着更高(P = 0.005,FDR = 0.015)。
⇒
饮用更多咖啡的个体表现出略显着更高的梭杆菌属丰度( P= 0.02,FDR = 0.20)。
总的来说,梭杆菌门在人体健康中的作用还需要进一步的研究和探索。虽然它们可能与一些疾病有关,但在肠道菌群中的作用也不容忽视。
扩展阅读:梭杆菌属Fusobacterium——共生菌、机会致病菌、致癌菌
蓝细菌门(Cyanobacteria),是一类原核生物,也被称为蓝藻或蓝藻菌门。
蓝绿藻植物菌门下物种又称蓝细菌、蓝绿菌、蓝藻或蓝菌,包括蓝鼓藻、蓝球藻等生物。过去曾长期被归于藻类,但实际上蓝菌与真核生物非常不同,例如没有核膜,没有细胞器,其遗传物质DNA也不构成染色体,这些都是细菌的特征,因此现时已被归入细菌域。
▼
蓝细菌分布极广,普遍生长在淡水、海水和土壤中,主要分布在含有机质较多的淡水中;在极端环境(如温泉、盐湖、贫瘠的土壤、岩石表面或风化壳中、冰雪上、植物树干等)中也能生长;蓝菌有些还可穿入钙质岩石(如钙藻类)或土壤深层中(如土壤蓝藻),故有“先锋生物”的美称。
▼
蓝菌是一类能透过产氧光合作用获取能量的细菌,但有些也能透过异营来获取能量。
蓝绿菌在地球上已存在约21亿年,是目前以来发现到的最早的光合放氧生物,对地球表面从无氧的大气环境变为有氧环境起了巨大的作用。通过刺激生物多样性和导致厌氧生物接近灭绝,显著的改变了在地球上生命形式的组成。根据内共生学说,在植物和真核藻类发现的叶绿体是从蓝细菌祖先通过内共生进化而来的。
▼
以下是一些常见的典型菌种:
Anabaena(鱼腥藻):这是一种常见的蓝藻,可以在淡水和海水中生长。它们通常形成长链,其中一些细胞可以进行氮固定,这对于生态系统的氮循环非常重要。
Microcystis(微囊藻):这是一种广泛分布的蓝藻,可以在淡水和海水中生长。它们通常形成大量的胞囊,这些胞囊可以释放出毒素,对水生生物和人类健康造成威胁。
Spirulina(螺旋藻):这是一种广泛应用于食品和保健品的蓝藻。它们富含蛋白质、维生素和矿物质,被认为具有多种健康益处。
▼
谷禾肠道数据库中大约39.06%的人群有检出,但一些研究表明,它们可能与肠道健康和一些疾病有关。
一些研究发现,蓝细菌在肠道中的丰度与炎症性肠病(如克罗恩病和溃疡性结肠炎)的发生有关。
蓝细菌中的一些代表性菌种,如前面提到的Anabaena sp.和Microcystis sp.,可能产生毒素,对人体健康造成威胁。
⇒
在一些污染的地区,其中绝大多数淡水湖泊中发现了大量的蓝藻水华。蓝藻毒素早就被认为通过肝脏影响与之相关的健康问题,食用受蓝藻污染的饮用水相关的健康问题也包括胃肠炎和肠道不适,其中肠道微生物组发挥了关键作用。但蓝藻毒素的作用机制仍然是一个关键的、大部分未被探索的问题。
肠道蓝细菌丰度与人类健康和疾病的潜在联系

doi.org/10.3390/life12040476
⇒
在谷禾肠道样本人群中,发现渐冻症部分群体里发现蓝藻门(蓝细菌)比较高。结合研究发现蓝菌门生物皆含有神经毒素BMAA(β-N-methylamino-L-alanine),并可能透过食物链不断累积产生生物放大作用,对人类的损害可能会逐渐增加。BMAA已证实会对动物产生强烈的毒性,加速动物脑神经退化、四肢肌肉萎缩等等,小量BMAA积累对小鼠已能选择性杀死神经元。
因此需要注意,在水体中的过度生长会导致水体富营养化,产生毒素,对人体健康和生态环境造成危害。需要加强对蓝细菌的监测和管理。
酸杆菌门 (Acidobacteria) ,虽然这个名字听起来都是嗜酸性的,但一些物种可以在中性和弱碱性环境中找到。
虽然一些研究表明酸杆菌门有超过 18 个类别,但只有三个类别是众所周知的。它们是:
(Holophagae 纲在某些文献中被描述为一个目)
▼
下面了解一下酸杆菌门下的3个常见属:
◗ 酸杆菌属
酸杆菌门下第一类重要的菌属——酸杆菌属(Acidobacterium Genus),通常存在于酸性环境中。
荚膜酸杆菌(Acidobacterium capsulatum)是该组中最受欢迎的成员之一(由大约 8 个菌株组成)。与该类别的其他成员一样,荚膜酸杆菌是一种革兰氏阴性细菌。
酸杆菌属也是好氧菌,它们本质上是嗜温的;在适中的温度(20~45°C之间)生长良好。
与这些细菌相关的一些其他特征包括:
◗ Terracidiphilus属
酸杆菌门下第二类重要的菌属——Terracidiphilus 属,由已知可产生用于分解几丁质和寡糖的细胞外酶的生物体组成。
该属中最受欢迎的物种之一是 Terracidiphilus gabretensis。这种细菌常见于针叶林中,它在碳汇中起着重要作用。
◗ Terriglobus 属
Acidobacteria 门下第三类重要的菌属——Terriglobus属,是革兰氏阴性菌,与 Granulicella 和 Adaphobacter 属密切相关。
该组的成员是土壤中常见的好氧化学有机异养生物。虽然这些生物通常存在于土壤(根际土壤)中,但在淡水生境中也有。
目前,该属有 5 个知名物种,其中包括:
该属的一些特征为:
图源:Biology LibreTexts
与这些细菌相关的一些其他特征包括:
▼
据估计,酸杆菌是土壤中主要菌群,约占土壤中所有微生物的 20%。
在环境中,酸杆菌在养分循环中起着重要作用。
碳循环 ——通过降解各种碳源(糖和蛋白质等),这些细菌将碳返回到环境中。然后,这些碳被植物和其他生物用于各种功能,循环继续。
氮循环 ——目前,Geothrix fermantans是酸杆菌门内唯一已知在氮循环中发挥一定作用的物种。通过减少硝酸盐来实现的。
硫循环 ——嗜热氯酸杆菌是需要硫来生长和发育的酸杆菌的一个例子。硫代谢有助于硫循环。
▼
与人体肠道和健康的关系方面,目前对于酸杆菌的研究还比较有限。
一些研究表明,酸杆菌门可能在人体肠道中存在,谷禾肠道数据库中大约33.05%的人群有检出。
肠道微生物群落的失衡与多种疾病的发生有关,而酸杆菌门的数量在某些疾病中可能会发生变化。
⇒
一些研究表明,肠道炎症性疾病患者的肠道中酸杆菌门的数量较低。
⇒
妊娠期糖尿病患者中酸杆菌门显著增加,并与血糖水平呈正相关。
⇒
在特发性肾病综合征患者中,酸杆菌门显著减少。
⇒
糖尿病肾病具有与健康对照不同的肠道微生物群,酸杆菌门在糖尿病肾病患者中增加。
⇒
一项研究中,幽门螺杆菌阳性受试者的微生物群落表明变形杆菌、酸杆菌和螺旋体的数量增加。
扩展阅读:正确认识幽门螺杆菌
但是,目前还需要更多的研究来探究酸杆菌门与人体肠道和健康之间的具体关系。
软壁菌门(Tenericutes),在谷禾肠道数据中,该菌的检出率是29.61%.
这些细菌通常是无细胞壁的,因此它们的形态非常多样化,也被称为无壁菌门。由质膜包围的细胞组成的革兰氏阴性菌。
软壁菌门由从厚壁菌门进化而来的细菌组成。尽管一些研究人员强烈认为软壁菌门应该被整合到厚壁菌门中,但两个显着特征使软壁菌门有别于厚壁菌门:
目前,软壁菌门的分类地位尚不确定。随着未来鉴定出更多新的软壁菌门细菌谱系,软壁菌门的分类定位和单系性可能会受到进一步挑战。
▼
软壁菌门普遍存在于许多环境中。16S rRNA 测序已经在包括深海在内的不同环境中识别出大量未知的软壁菌门进化枝,这表明这些细菌可能代表独立生活的微生物,其生活方式与宿主无关。
事实上,在深海冷泉和盐水池中分别发现了Candidatus Izemoplasma和 Haloplasma自由生活。这些深海自由生活的软壁菌门细菌表现出新陈代谢的多样性和适应性的灵活性,表明有可能从海洋甚至其他极端环境中分离出更多的软壁菌门细菌。
▼
⇒
一项关于老年2型糖尿病认知障碍的研究发现参与者中,认知障碍患者的血红蛋白和高密度脂蛋白水平较低,相对糖尿病对照组而言,认知障碍糖尿病患者的软壁菌门Tenericutes丰度较低。
⇒
在长寿村社区老年人的肠道菌群中,厚壁菌门、软壁菌门Tenericutes和放线菌门的相对丰度显著低于城市化城镇社区。
扩展阅读:健康长寿的步伐永不停歇
⇒
一项研究将86名儿童(5-15岁)被分为三组:
与对照组相比,在代谢性不健康肥胖受试者中,软壁菌门(Tenericutes)以及α和β多样性显著降低。与对照组相比,互养菌门(Synergistetes)和拟杆菌属在代谢健康肥胖人群中更为普遍。
总的来说,软壁菌门在肠道中的作用和其与健康的关系还需要进一步的研究和探索。
注意,软壁菌门在临床上比较难分辨。它们往往不太可能生长,也不太可能被经典微生物学技术识别,一般通常需要进行分子鉴定。属内的敏感性特征通常变化很大,这使得针对他们的特异性鉴定以及合理选择抗菌药物非常重要。
绿弯菌门(Chloroflexi)是一类光合细菌,可以利用光合作用产生能量。又称作绿非硫细菌,还有一部分称作热微菌的细菌也属于绿非硫细菌。绿弯菌门的细菌生活在海洋,淡水等环境中。
该门包括六类:
▼
绿弯菌门由不同的生物群组成,包括无氧光合自养生物、好氧化学异养生物、嗜热生物以及通过有机氯化化合物的还原脱卤获得能量的厌氧生物。
典型的绿弯菌门细菌是线形的,通过滑行来移动。它们是兼性厌氧生物,在光合作用中不产生氧气,不能固氮。利用3-羟基丙酸途径,而不是常见的卡尔文途径来固定二氧化碳。
图源:de-academic
所有已知的成员都是丝状的,具有不寻常的滑动机制作为一种运动方式,虽然大多数革兰氏染色呈阴性,但没有一个具有革兰氏阴性菌特有的脂多糖外膜。
绿弯菌门包含生态和生理上多样化的细菌群,已在越来越广泛的厌氧生境中检测到这些细菌,包括沉积物、温泉、产甲烷厌氧污泥消化池,它们在这些地方非常丰富,并发挥着重要的发酵作用有助于污泥粒化。绿弯菌门是固体废物和废水处理系统中最主要的门之一。特别是,Anaerolineae 类已被确定为全面厌氧反应器中的核心微生物种群之一。
绿弯菌门可能参与了肠道中的一些代谢过程,例如氨基酸、葡萄糖和脂肪酸的代谢。
▼
虽然绿弯菌门在人群中不是常见菌,但目前人体肠道和口腔中也逐步检测到绿弯菌门细菌,谷禾肠道数据库中大约25.28%的人群有检出。
⇒
在门水平上,利福昔明治疗后腹泻型肠易激综合征患者的绿弯菌门(Chloroflexi)(P=0.008)、Deinococcus-Thermus菌(P=0.038)和酸杆菌群(P=0.028)增加。
⇒
在门水平上,与格雷夫斯病相比,格雷夫斯眼眶病患者中Deinococcus-Thermus菌和 绿弯菌门(Chloroflexi) 的比例显着降低。
⇒
小型研究发现,新冠肺炎刚痊愈的人与健康对照相比,绿弯菌门(Chloroflexi)显著降低。
扩展阅读:阳康后是否会二次感染,长新冠与肠道菌群的关联,多种潜在的相关干预措施
此外,一些研究还发现,绿弯菌门的存在与一些肠道疾病,如炎症性肠病和肠道肿瘤等有关联,但具体的机制还需要进一步研究。
互养菌门(一般翻译为 Synergistetes),也有翻译为增效菌门或协同菌门。在谷禾肠道数据中,该菌的检出率是24.61%.
Synergistetes细菌是最近认识到的一个门,其中已分离出 40 种生物,并且有超过三百个 16S rRNA 序列可用。
这个门的分类学历史很短,最近才被确定为细菌域内的一个独立门。第一个代表性物种,Synergistes jonesii,最初从夏威夷山羊的瘤胃中分离出来,以其命名,最初被分类在 Deferribacteres 门中。
▼
来自该门的物种共有的表型特征包括它们的革兰氏阴性细胞壁、厌氧、杆状/弧菌状细胞形状
图源:researchgate
虽然脂多糖存在于双层细胞膜中是一个重要特征,但在互养菌门物种中尚未被报道,但它们确实含有参与脂多糖生物合成的各种蛋白质的基因。虽然有些物种不能分解糖,但所有互养菌门都具有发酵氨基酸的能力。
它们可以利用多种有机物作为碳源和能源。
一些Synergistetes菌属可以利用蛋白质、脂肪酸和多糖等有机物进行代谢,同时还可以参与肠道中的硫循环和氮循环等过程。
一些Synergistetes菌属还可以产生一些对人体有益的代谢产物,如短链脂肪酸等。
▼
互养菌门主要栖息在厌氧环境中,包括动物胃肠道、土壤、油井和废水处理厂,它们也存在于人类疾病部位,如囊肿、脓肿和牙周病区域。
由于它们存在于疾病相关部位,互养菌门被认为是机会性病原体,但它们也可以在健康个体的脐部微生物组和正常阴道菌群中发现。
该门的其他物种已被确定为厌氧消化池中用于生产沼气的污泥降解的重要贡献者,并且是通过生产氢气用于可再生能源生产的潜在候选者。
常见菌属:Aminiphilus是一类革兰氏阴性菌,通常生长在富含有机物的水体中。它们可以利用氨基酸和蛋白质等有机物作为碳源和能源。
▼
⇒
大多数人类培养的菌株来自感染部位,表明互养菌门在感染过程中增殖。互养菌门的不同分支和物种表现出不同的感染倾向:
⇒
在牙周炎患者中发现了一些互养菌门OTU,并且在牙周炎患者的患病部位龈下菌斑中比健康部位更丰富。
⇒
2型糖尿病和牙周炎患者的微生物群比非糖尿病牙周炎患者的微生物群显示出更少的互养菌门。
⇒
在坏死性溃疡性牙龈炎的病例中,互养菌门聚类A OTUs的检测水平和比例高于牙龈炎期间。
⇒
研究显示,精神病患者患抗精神病药引起的便秘的风险很高,互养菌门在便秘组的肠道微生物群中显著增高。
⇒
互养菌门可能保护甲状腺,在格雷夫斯病患者的肠道菌群中,互养菌门与促甲状腺激素受体抗体(TRAb)水平呈负相关。
芽单胞菌门(Gemmatimonadetes)是一类革兰氏阴性菌,包括:
芽单胞菌纲(Gemmatimonadetes)
芽单胞菌目(Gemmatimonadales)
芽单胞菌科(Gemmatimonadaceae)
芽单胞菌门目前仅有一属得到正式命名,即芽单胞菌属(Gemmatimonas),是一类革兰氏阴性细菌,通过出芽方式繁殖。
图源:alchetron
芽单胞菌门可以在各种环境中生长,包括土壤、淡水、海水和沉积物等。在肠道中,芽单胞菌门也是一种常见的微生物群落成员。
▼
芽单胞菌门可以利用多种有机物和无机物作为碳源和能源,包括葡萄糖、氨、硝酸盐、硫酸盐等。此外,芽单胞菌门还可以在低氧或缺氧条件下生长,并且能够耐受一定的重金属和有机污染物。
一些研究表明,芽单胞菌门可能在土壤和水体中发挥重要的生态功能,如有机物分解和氮循环等。此外,芽单胞菌门可能与一些环境污染物的降解有关。因此,芽单胞菌门在环境保护和生态平衡方面具有重要的作用。
芽单胞菌门的代表性菌种包括:
Gemmatimonas aurantiaca、
Gemmatimonas phototrophica等。
这些菌种具有一些特殊的代谢特征,例如Gemmatimonas aurantiaca可以利用多种有机物作为碳源和能源,同时还具有一定的光合作用能力。
▼
芽单胞菌门与人体健康的关系尚未得到充分研究。谷禾肠道数据库中大约20.11%的人群检出。
一些研究发现,芽单胞菌门可能与肠道炎症的发生有关。
⇒
可能与骨质疏松相关:
芽单胞菌门和绿弯菌门,在原发性骨质疏松症患者和 正常对照组,以及骨质减少患者和正常对照组之间存在显着差异 ( p < 0.01 ) 。
⇒
补充唾液酸Neu5Ac对小鼠肠道形态、肝功能和肠道微生物影响的研究表明,肠道微生物群组成呈剂量依赖性变化,在门水平上,芽单胞菌门显着增加。
注:N-乙酰神经氨酸 (Neu5Ac) 是人类唾液酸的主要形式。
需要进一步的研究来探究芽单胞菌门与人体健康的关系。
黏胶球形菌门(Lentisphaerae)是一个相对较小的门,球形或椭圆形,通常是厌氧或微好氧的,可以生长在不同的环境中,如土壤、淡水、海水、动物肠道等。在谷禾肠道数据中,该菌的检出率是18.42%.
▼
黏胶球形菌门的代谢特征是多样的,包括无氧呼吸、发酵、光合作用等。
一些菌属可以利用多种碳源和氮源进行生长,如Lentisphaera和Victivallis可以利用多种碳水化合物和蛋白质作为碳源和氮源。
一些Lentisphaerae菌属还具有产生酸和气体的能力,如Fibrobacteres可以产生乳酸和乙酸。
Lentisphaerae还被发现可以参与到肠道中的多糖代谢和蛋白质降解等代谢过程中。
在黏胶球形菌门内的Victavallales属,可以酶解唾液酸、岩藻糖、半乳糖和 N-乙酰氨基葡萄糖,降解粘蛋白,其糖基水解酶图谱与AKK菌非常相似。
▼
⇒
一项研究发现,黏胶球形菌门(Lentisphaerae)在健康人的肠道中的丰度较高,而在患有炎症性肠病和非酒精性脂肪性肝病 (NAFLD) 的患者中的丰度较低。
⇒
研究还发现更好的睡眠质量与更好的认知灵活性和更高比例的肠道微生物门Verrucomicrobia和Lentisphaerae有关。
⇒
也有研究发现黏胶球形菌门( Lentisphaerae)与帕金森疾病相关。
扩展阅读:肠道微生物与帕金森以及相关影响因素
⇒
可能与他汀类药物的代谢有关,高脂血症患者接受了 10 mg/天的瑞舒伐他汀治疗 4-8 周,蓝细菌门和黏胶球形菌门(Lentisphaerae)与低密度脂蛋白胆固醇水平呈正相关。
⇒
横断面研究发现,黏胶球形菌门(Lentisphaerae)在多发性硬化患者中显著降低。
扩展阅读:肠道微生物群在多发性硬化中的作用
⇒
经常食用面包的受试者的肠道微生物群中,互养菌门和黏胶球形菌门(Lentisphaerae)的相对丰度显着更高(分别为P = 0.009,FDR = 0.028 和P = 0.004,FDR = 0.011)。
浮霉菌门(Planctomycetes)形成了一个独特的细菌门,具有独特的特征组合,例如,缺乏肽聚糖的蛋白质细胞壁,以及在细胞质内形成独立隔室的细胞内膜。
浮霉菌门的细胞结构
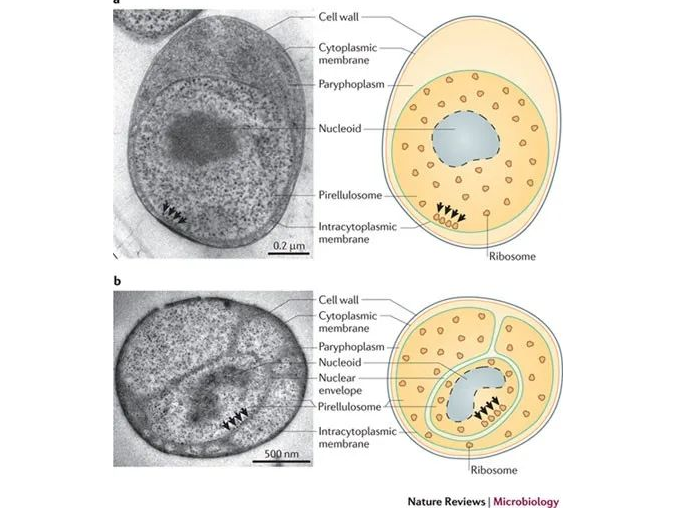
doi: 10.1038/nrmicro2578.
▼
浮霉菌门包括多个典型菌属,如Planctomyces、Gemmata、Pirellula、Rhodopirellula等。这些菌属通常是好氧或微好氧的,可以生长在不同的环境中,如海水、淡水、土壤、沉积物、动物肠道等。主要与大型藻类、海绵和地衣等颗粒或生物相关,具体取决于物种及其硫酸酯酶可代谢的多糖。大多数浮霉菌门生长在 pH 值从 3.4 到 11 不等的营养贫乏的贫营养环境中。
大多数浮霉菌门是嗜温的,但有一些 浮霉菌门是嗜热的(50°C -60°C)。通常添加的营养素有 N-乙酰氨基葡萄糖、酵母提取物、蛋白胨和一些微量元素和大量元素。
▼
一些浮霉菌属还具有产生酸和气体的能力,如Pirellula可以产生乳酸和乙酸。另外,浮霉菌门还具有一些特殊的代谢特征,如一些菌属可以利用甲烷和硫化氢作为能源和碳源,如Anammoxoglobus和Brocadia。
浮霉菌门包括一些具有非常不寻常的生理学的物种,比如一些浮霉菌可以合成甾醇,这是一种真核生物的典型能力,在细菌中并不常见。
浮霉菌门的另一个不寻常的代谢特征是它们拥有编码C1转移酶的基因。这些酶以前只在产生甲烷的古细菌和一组甲烷氧化的变形菌中被发现,它们在具有一个碳原子的化合物的代谢中发挥作用。比较基因组学和蛋白质组学表明,厌氧氨氧化和非厌氧氨氧化浮霉菌之间的区别超出了铵的代谢。
▼
浮霉菌门是人类消化道微生物群的一部分。谷禾肠道数据库中大约13.92%的人群有检出。它们的多样性因环境而异,包括个体的地理起源和抗生素治疗。
⇒
在两名患有白血病和再生障碍性中性粒细胞减少症的发热患者的血液中检测到与浮霉菌门密切相关的 DNA 序列。
⇒
G. massiliana的分离来自靠近这些患者的医院供水系统,可能支持消化道进入途径的假设,即摄入受污染的水然后在免疫功能低下的患者的血液中易位。Gemmata属的可能作为潜在的机会性病原体进入消化道。
硝化螺旋菌(Nitrospirae) 是革兰氏阴性菌,通常呈螺旋状。以其氧化亚硝酸盐和硝酸盐的能力而闻名。谷禾肠道数据库中大约11.04%的人群有检出。
硝化螺旋菌存在于各种环境中,包括土壤、淡水、海洋栖息地、污水处理厂等。在亚硝酸盐和硝酸盐等含氮化合物含量高的环境中,它们尤其丰富。
▼
硝化螺旋菌作为一种好氧化学自养亚硝酸盐氧化细菌,在硝化过程中发挥着关键作用。这些细菌通常与氨氧化细菌或古菌密切相关,这些细菌将氨转化为亚硝酸盐,亚硝酸盐被硝化螺旋菌进一步氧化为硝酸盐。
然而,在“相互喂食”的相互作用中,硝化螺菌也可以用尿素或氰酸盐释放的氨提供氨氧化剂,尿素或氰酸酯被进一步硝化。
图源:Mmolecular
硝化螺旋体成员甚至单独催化两个硝化步骤,因此被称为完全氨氧化剂或“comammox”生物体。这与传统的硝化细菌不同,传统的硝化细菌需要两种不同类型的细菌来完成这些步骤。
一些硝化螺旋菌菌株利用H2和甲酸盐等替代底物,使用氧气或硝酸盐作为末端电子受体,并可以在好氧亚硝酸盐氧化的同时利用这些能源。这种代谢的多样性使硝化螺旋菌能够在广泛的栖息地定居,并维持环境条件的变化,如氧气浓度的变化。
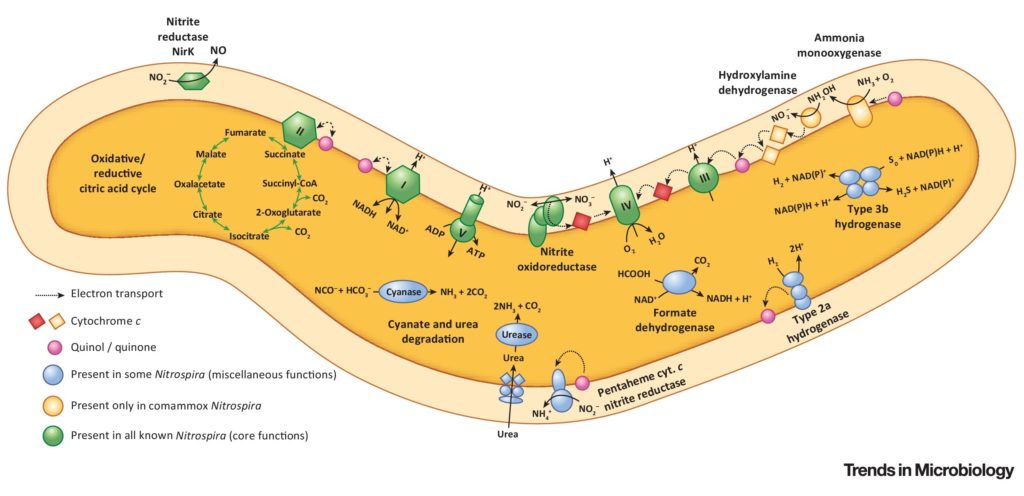
doi.org/10.1016/j.tim.2018.02.001
一些种类的硝化螺旋菌还能够利用硫化合物或铁等替代电子受体,进行厌氧呼吸。
已知一些种类的硝化螺旋菌参与有机物的降解,而另一些种类则参与甲烷的生产。
▼
⇒
比较墨西哥城儿童(西方化,高动物蛋白和精制糖饮食)和 Me’phaa 儿童(非西方化,高纤维饮食)的肠道菌群,这两个人群主要区别在于不同种类的饮食。研究发现,Me’phaa 儿童表现出更高的绿弯菌门(Chloroflexi)和硝化螺旋菌(Nitrospirae)。
注:来自格雷罗州“Montaña Alta”地区的 Me’phaa 是一个前西班牙土著群体,他们主要靠种植豆类和扁豆,玉米等为生。还收集野生食用植物,并种植一些水果和蔬菜。肉类几乎只是在特殊场合食用的,并不是日常饮食的一部分。其生活方式和墨西哥城形成鲜明对比。
⇒
但也有研究发现,耐药癫痫患者表现出硝化螺旋菌富集(Kruskal-Wallis检验:p<0.05)。
⇒
一项关于胃微生物群的研究发现,硝螺旋菌门存在于所有胃癌患者中,但在慢性胃炎患者中完全不存在。
⇒
乳铁蛋白可以降低硝化螺旋菌水平。
脱铁杆菌(Deferribacteres)是1999年首次被描述的一门细菌。这些细菌以其还原铁和其他金属的能力而闻名,它们存在于各种环境中,包括深海热液喷口、温泉和地下水。
脱铁杆菌是革兰氏阴性细菌,通常是杆状的。它们是厌氧菌,通常在低氧环境中被发现。脱铁杆菌也是嗜热的,一些种类的脱铁杆菌能够进行化能生长,这意味着它们可以通过氧化无机化合物(如铁或硫)来获得能量。
G. thiophilus 阴性染色细胞的电子显微照
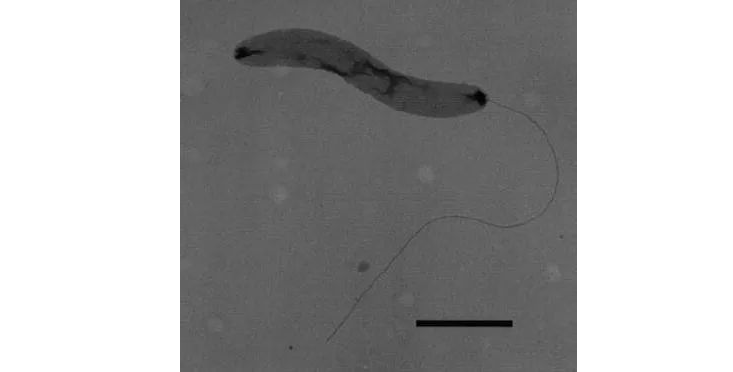
Janssen et al. 2002
脱铁杆菌中最著名的一种是脱铁杆菌脱硫菌,它能够还原铁和硫化合物,它被认为在深海环境中这些元素的循环中发挥着重要作用。
▼
脱铁杆菌属细菌最有趣的特征之一是它们能还原铁。这个过程包括将电子从铁转移到细菌,然后细菌可以利用这些电子产生能量。这一过程在许多环境中都很重要,因为它可以帮助维持生态系统中铁和其他金属的平衡。
除了还原铁,一些种类的脱铁杆菌还能还原其他金属,如锰和铀。脱铁杆菌属细菌在碳循环中也很重要,因为它们能够分解有机物并释放二氧化碳。一些种类的脱铁杆菌已知参与复杂有机化合物的降解,如木质素和纤维素。这一过程在许多环境中都很重要,因为它有助于循环营养物质,维持生态系统的平衡。
▼
谷禾肠道数据库中大约4.92%的人群有检出。
⇒
脱铁杆菌门可能与环境高温高湿导致的肠道菌群失调和轻微肠炎有关。
研究分为三组实验:
脱铁杆菌门是唯一在三组中具有差异丰度的门(P < 0.05),从正常对照组和高温高湿组的0.05%增加到广谱益生菌治疗组的1%。
⇒
一项研究发现,与正常对照和葡萄糖调节受损 (IGR) 患者相比,2型糖尿病患者中的脱铁杆菌门显着增加。
也发现脱铁杆菌门与镁摄入量呈负相关。
⇒
大鼠给予大麦或麦芽(7-8膳食纤维/100 g)4周,与对照大鼠相比,大麦组大鼠盲肠微生物群中的脱铁杆菌门的丰度低于对照组。
扩展阅读:谷物调节肠道菌群,促进代谢健康
在糖尿病中,关于脱铁杆菌门的研究不一致,有研究认为,糖尿病的改善与脱铁杆菌门相对丰度的降低有关,也有研究发现糖尿病的改善与脱铁杆菌门相对丰度的升高有关。
⇒
研究橄榄苦苷摄入对晚期2型糖尿病的缓解作用,发现橄榄苦苷可以增加Verrucomicrobia和脱铁杆菌门的相对丰度。
⇒
膳食菊粉治疗糖尿病组中脱铁杆菌门相对丰度下降。膳食菊粉通过抑制炎症和调节肠道微生物群来缓解2型糖尿病的不同阶段。
⇒
皮质酮治疗的小鼠中,拟杆菌门减少,脱铁杆菌门显著增加,水苏糖使拟杆菌门和脱铁杆菌恢复到正常水平。
⇒
较低的色氨酸补充量降低了脱铁杆菌门的丰度,而较高的色氨酸补充量不仅恢复了丰度,而且增加了丰度。
扩展阅读:色氨酸代谢与肠内外健康稳态
螺旋体(Spirochaetes),是革兰氏阴性菌,可运动的螺旋状细菌,断面呈圆形,以横向分裂繁殖,长度有 5 到 250 微米。螺旋体的独特之处在于,它们具有细胞内鞭毛。螺旋体属于双膜细菌门。
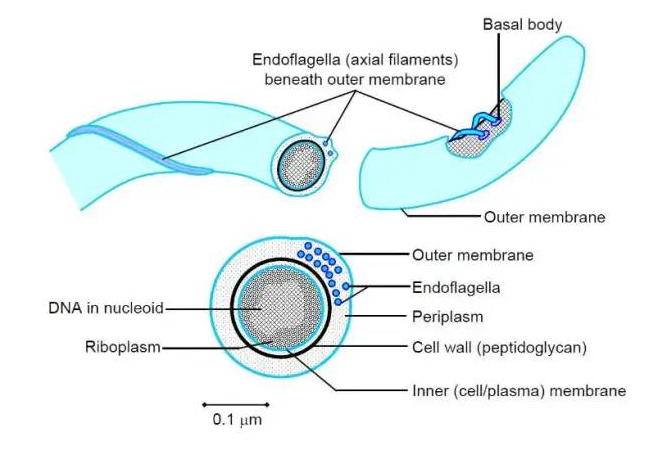
图片来源:crondon
螺旋体的细胞体被包裹在几层中。这些包括外膜和内膜、肽聚糖层以及细胞质膜。
其中一些是人类的严重病原体,会导致梅毒、雅司病、莱姆病和回归热等疾病。螺旋体属有螺旋体、密螺旋体、疏螺旋体、钩端螺旋体等。
▼
螺旋体可以在水(地表水/淡水)、湖泊、盐沼沉积物、泥浆、沉积物、深海喷口、血液和淋巴等各种栖息地中找到。
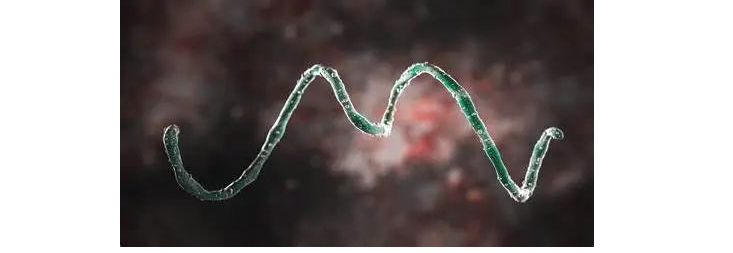
图源:Oxford Nanopore Technologies
▼
螺旋体本质上是化学异养的,能够在厌氧条件下繁衍生息。
螺旋体的一个很好的例子是 Spirochaeta isovalerica。它是专性厌氧菌,它们通过发酵碳水化合物产生乙酸盐、乙醇、二氧化碳和氢气来生存。
许多种类的螺旋体对环境有益,并在固氮和有机物分解等过程中发挥重要作用。密螺旋体的某些种类生活在牛胃的瘤胃中,在那里它们为宿主分解纤维素和其他难以消化的植物多糖。
一些物种也被用于生物技术和工业应用,如生物燃料和生物塑料的生产。
▼
谷禾肠道数据库中大约4.04%的人群有检出。
⇒
钩端螺旋菌病是由动物传染给人类的,一种常见的传播形式是让受污染的水接触到皮肤、眼睛和粘膜中未愈合的伤口。水由于与受感染动物的尿液接触而受到污染。——Leptospira
莱姆病 —— Borrelia burgdorferi,
Borrelia garinii, Borrelia afzelii
扩展阅读:夏季来临,警惕蜱虫叮咬感染疾病——莱姆病
回归热 ——复发性疏螺旋体(Borrelia recurrentis)
梅毒——梅毒螺旋体(Treponema pallidum)
雅司病(皮肤、骨骼和关节的热带感染)
—— T. pallidum subspeciespertenue
肠道螺旋体病——Brachyspira pirosicoli和Brachyspira aalborgi
文森特心绞痛—— Borrelia vincentii
⇒
在马来西亚北部的一个农村地区,与富裕人孩子相比,相对经济困难的土著儿童肠道微生物群表现出最多的微生物多样性。
Aeromonadales、拟杆菌门、瘤胃球菌科(Ruminococcaceae)、Deltaproteobacteria和螺旋体(Spirochaetes)富集,这与富含纤维的食物的分解有关。
装甲菌门(Armatimonadetes),以前被称为候选门OP10,其成员分布在各种环境中,包括土壤、岩石、淡水和海洋沉积物。谷禾肠道数据库中大约3.31%的人群有检出。
装甲菌门细菌的特征是其独特的细胞壁结构,其中包含一层肽聚糖和一层类似于革兰氏阴性菌的外膜层。它们还具有形成长而有分支的细丝的能力。绿弯菌门是与装甲菌门亲缘关系最密切的正式门。
已经发现一些种类的装甲菌门在碳和氮循环等环境过程中发挥着重要作用。
在装甲菌门中,属于Armatimonasis纲的菌株 YO-36 T和属于Chthoonomonadetes纲的菌株 T49 T是唯一有效命名的分离株。
T49 T中枢代谢和碳固定通过常规糖酵解和三羧酸循环进行。T49 T的一个有趣特征是它在 4.7 到 5.8 的窄 pH 范围内生长。
▼
很少有关于人体肠道菌群内装甲菌门的研究。
在一些关节炎患者的滑液中也检测到了Armatimonadetes门细菌,这些可能是游离污染物或机会性定植剂,而不是病原体。
在人体肠道中,装甲菌门的作用尚不清楚,待进一步研究和探索。
绿菌门(Chlorobi)是一类光合细菌,也被称为绿菌门,谷禾肠道数据库中大约1.9%的人群有检出。
绿菌门是一类光合细菌,能够利用光能进行光合作用,产生能量和有机物质。
▼
在人体肠道菌群中,Chlorobi包含以下几个菌属和菌种:
包括Chlorobium limicola、Chlorobium phaeobacteroides、Chlorobium tepidum等。
包括Prosthecochloris aestuarii、Prosthecochloris vibrioformis等。
包括Chloroherpeton thalassium等。
包括Chloronema giganteum等。
▼
⇒
有研究显示B 族链球菌定植的孕妇肠道菌群中检测到大量的绿菌门(Chlorobi),同时还有大量Lentisphaerae、Parcubacteria、Chloroflexi、Gemmatimonadetes、Acidobacteria、Fusobacteria 、 Fibrobacteres。
GBS感染孕妇的OTU水平与炎症指标存在显着相关性。表明包括绿菌门在内的多种菌改变与 GBS 阳性孕妇的炎症状态和新生儿血气指标有关。
⇒
一项研究发现,酒精性肝病患者肠道菌群中,携带的厚壁菌门(p=0.03)和绿菌门(Chlorobi)(p=0.009)的数量存在显著差异。
同样,携带绿菌门(Chlorobi)(p=0.01)和coprothermobacterota(p=0.03)的患者的肝脏失代偿严重程度也有显著差异。
扩展阅读:深度解析 | 肠道菌群与慢性肝病,肝癌
很少有关于人体肠道菌群内绿菌门的研究。可能其存在来源于饮食摄入或环境。在人体肠道中,绿菌门的作用尚不清楚,它们的作用和功能尚待进一步研究和探索。
迷踪菌门(Elusimicrobia),也称为”隐微菌门”,谷禾肠道数据库中大约1.44%的人群有检出。
它们是一类非常小的细菌,通常直径只有0.2-0.4微米。迷踪菌门的细胞壁非常薄,甚至可以说是缺乏细胞壁,这使得它们对抗生素的抵抗力较弱。
▼
迷踪菌门中的一些菌种是共生菌,与其他生物共同生活,例如Candidatus Endomicrobium trichonymphae与白蚁肠道中的Trichonympha寄生在一起,共同分解木质素。
迷踪菌门的代谢功能多样,包括产生氢气、甲烷、酒精等。迷踪菌门在生态系统中扮演着重要的角色,例如在土壤中参与有机物分解、在海洋中参与碳循环等。
▼
⇒
一项研究发现,在肥胖的2型糖尿病患者中,厚壁菌门丰度较高,而迷踪菌门(Elusimicrobia)丰度较低。
⇒
在两种大型急性辐射综合症动物模型的辐射暴露后的肠道菌群研究显示,哥廷根小型猪 (GMP)模型中的迷踪菌门在辐照后持续增加,表明它可用作肠道损伤的潜在生物标志物,以及对健康的潜在负面影响。
⇒
一项针对低质量睡眠和肠道菌群的研究显示Tenericutes 和 迷踪菌门(Elusimicrobia)在睡眠障碍患者中显著增加且与睡眠质量成正相关。
扩展阅读:肠道菌群与睡眠:双向调节
在人体肠道中,迷踪菌门的作用也还需要进一步研究。
衣原体门(Chlamydiae),革兰氏阴性细菌,是专性寄生菌,它们的生长完全在其它生物的细胞内进行。
衣原体门细菌比一般细菌小,有的比病毒小,直径约为0.2-1.5微米。通常呈球形或椭圆形,没有细胞壁,但具有外膜和内膜。
编辑
图源:Science Photo Library
最开始,科学界普遍认为衣原体门的细胞壁不含肽聚糖,然而最近已有研究显示其细胞壁上的确有肽聚糖存在,并成功辨认出几种蛋白质。
衣原体是一类专性真核细胞内寄生、具有独特发育周期、可以在多种真核生物宿主(包括人、动物、原虫等)中繁殖的细菌,不能自主生长和繁殖。
▼
在人体肠道菌群中,Chlamydiae包含以下几个菌属和菌种:
包括Chlamydia trachomatis、Chlamydia pneumoniae、Chlamydia psittaci 等。
包括Parachlamydia acanthamoebae、Parachlamydia boviseptica 等。
▼
衣原体门是一种常见的病原体,可以引起多种疾病,包括性传播疾病、肺炎、结膜炎等。
衣原体的类型和相关疾病已知的与人类疾病有关的衣原体有三种,分别是鹦鹉热衣原体、沙眼衣原体和肺炎衣原体。这三种衣原体均可引起肺部感染。
鹦鹉热衣原体可通过感染有该种衣原体的禽类,如鹦鹉、孔雀、鸡、鸭、鸽等的组织、血液和粪便,以接触和吸入的方式感染给人类。
沙眼衣原体和肺炎衣原体主要在人类之间以呼吸道飞沫、母婴接触和性接触等方式传播。
▼
在谷禾肠道菌群数据库中大约有0.14%的人群有检出。
如果肠道感染衣原体,因感染肠道黏膜细胞可能导致腹泻、腹痛、恶心和呕吐,还可能会引发发热;腹泻和呕吐可能导致脱水、营养不良。
总之,衣原体作为一类常见的病原体,引起多种疾病,需要引起足够的重视和预防。
更多关于衣原体的介绍详见:
肠道微生物群一直伴随着人类的进化,并在人类的健康生活和高质量长寿中扮演着不可忽视的重要角色。
随着人类活动范围的不断扩大,一些以前未曾接触过的微生物会通过各种途径进入人体肠道,从而有机会在人体内生存和繁殖,并在人际之间传播。
同时,随着工业化的发展,我们原有的肠道菌群也面临着挑战,逐渐接受新的菌群、新的食物和添加剂等。
肠道菌群中一些占比较少的菌属,对人体健康和疾病的影响虽然研究较少,但这并不意味着它们的作用微弱。这些小众门派的菌属在自然界经过数亿年的进化选择,能够适应更恶劣的环境和在更寡营养条件下生存。此外,一些菌属具有特定的代谢功能,这些功能对我们的身体产生何种影响,以及它们如何影响人类的大尺度进化,都值得关注和研究。
尽管这些菌门的研究相对较少,但已经发现了一些有意思的结果。例如:
Akkermansia可以帮助减轻肥胖、改善胰岛素抵抗和代谢综合征等问题;具核梭杆菌(Fusobacterium nucleatum)与口腔疾病和肠炎结直肠癌的发病关系。莱姆病的罪魁祸首为伯氏疏螺旋体(Borrelia burgdorferi)。
在大人群水平上研究和探索这些菌属的来源及其代谢与人体健康的关系,将有助于发现和完善肠道菌群对人体健康的作用。这将为我们提供更好的方法来预防和治疗与肠道菌群相关的疾病,从而提高人类的健康水平。
主要参考文献:
Heinz Schlesner, Cheryl Jenkins,James T. Staley.The Phylum Verrucomicrobia: A Phylogenetically Heterogeneous Bacterial Group. The Prokaryotes pp 881–896
Hu C, Rzymski P. Non-Photosynthetic Melainabacteria (Cyanobacteria) in Human Gut: Characteristics and Association with Health. Life (Basel). 2022 Mar 25;12(4):476. doi: 10.3390/life12040476. PMID: 35454968; PMCID: PMC9029806.
Verma D, Garg PK, Dubey AK. Insights into the human oral microbiome. Arch Microbiol. 2018 May;200(4):525-540. doi: 10.1007/s00203-018-1505-3. Epub 2018 Mar 23. PMID: 29572583.
Doocey CM, Finn K, Murphy C, Guinane CM. The impact of the human microbiome in tumorigenesis, cancer progression, and biotherapeutic development. BMC Microbiol. 2022 Feb 12;22(1):53. doi: 10.1186/s12866-022-02465-6. PMID: 35151278; PMCID: PMC8840051.
ohn D. Coates. (2010). Phylum XVII. Acidobacteria phyl. nov.
Miriam Gonçalves de Chaves. (2019). Acidobacteria Subgroups and Their Metabolic Potential for Carbon Degradation in Sugarcane Soil Amended With Vinasse and Nitrogen Fertilizers.
Sadaf Kalam et al. (2020). Recent Understanding of Soil Acidobacteria and Their Ecological Significance: A Critical Review.
Anna M. Kielak et al. (2016). The Ecology of Acidobacteria: Moving beyond Genes and Genomes.
Bong Suk Shim. (2011). Current Concepts in Bacterial Sexually Transmitted Diseases.
Jones and Bartlett Learning. Infectious Diseases Affecting the Respiratory System.
Yu-Jie Zhang, Sha Li, Ren-You Gan, and Tong Zhou. (2015). Impacts of Gut Bacteria on Human Health and Diseases.
Caitlin A. Brennan and Wendy S. Garrett. (2019). Fusobacterium nucleatum — symbiont, opportunist and oncobacterium.
K. W. Bennett and A. Eley. (1993). K. Fusobacteria: New taxonomy and related diseases Free.
Kevin Afra, Kevin Laupland, Jenine Leal, Tracie Lloyd, and Daniel Gregson. (2013). Incidence, risk factors, and outcomes of Fusobacterium species bacteremia.
Radhey Gupta and Mohit Sethi. (2014). Phylogeny and Molecular Signatures for the Phylum Fusobacteria and its Distinct Subclades.
John D. Coates. (2010). Phylum XVII. Acidobacteria phyl. nov.
Miriam Gonçalves de Chaves. (2019). Acidobacteria Subgroups and Their Metabolic Potential for Carbon Degradation in Sugarcane Soil Amended With Vinasse and Nitrogen Fertilizers.
Sadaf Kalam et al. (2020). Recent Understanding of Soil Acidobacteria and Their Ecological Significance: A Critical Review.
Anna M. Kielak et al. (2016). The Ecology of Acidobacteria: Moving beyond Genes and Genomes.
Zheng R, Liu R, Shan Y, Cai R, Liu G, Sun C. Characterization of the first cultured free-living representative of Candidatus Izemoplasma uncovers its unique biology. ISME J. 2021 Sep;15(9):2676-2691. doi: 10.1038/s41396-021-00961-7. Epub 2021 Mar 21. PMID: 33746205; PMCID: PMC8397711.
Bhandari V, Gupta RS. Molecular signatures for the phylum Synergistetes and some of its subclades. Antonie Van Leeuwenhoek. 2012 Nov;102(4):517-40. doi: 10.1007/s10482-012-9759-2. Epub 2012 Jun 19. PMID: 22711299.
Wang Z, Pu W, Liu Q, et al. Association of Gut Microbiota Composition in Pregnant Women Colonized with Group B Streptococcus with Maternal Blood Routine and Neonatal Blood-Gas Analysis. Pathogens. 2022;11(11):1297. Published 2022 Nov 4. doi:10.3390/pathogens11111297
He H, Lin M, You L, Chen T, Liang Z, Li D, Xie C, Xiao G, Ye P, Kong Y, Zhou Y. Gut Microbiota Profile in Adult Patients with Idiopathic Nephrotic Syndrome. Biomed Res Int. 2021 Feb 18;2021:8854969. doi: 10.1155/2021/8854969. PMID: 33681383; PMCID: PMC7910048.
Zhang J, Zhang X, Zhang K, et al. The Component and Functional Pathways of Gut Microbiota Are Altered in Populations with Poor Sleep Quality – A Preliminary Report. Pol J Microbiol. 2022;71(2):241-250. Published 2022 Jun 19. doi:10.33073/pjm-2022-021
Carbonero F, Mayta A, Bolea M, et al. Specific Members of the Gut Microbiota are Reliable Biomarkers of Irradiation Intensity and Lethality in Large Animal Models of Human Health. Radiat Res. 2019;191(1):107-121. doi:10.1667/RR14975.1
Lage OM, Bondoso J. Planctomycetes and macroalgae, a striking association. Front Microbiol. 2014 Jun 3;5:267. doi: 10.3389/fmicb.2014.00267. PMID: 24917860; PMCID: PMC4042473.
Kaboré OD, Godreuil S, Drancourt M. Planctomycetes as Host-Associated Bacteria: A Perspective That Holds Promise for Their Future Isolations, by Mimicking Their Native Environmental Niches in Clinical Microbiology Laboratories. Front Cell Infect Microbiol. 2020 Nov 30;10:519301. doi: 10.3389/fcimb.2020.519301. PMID: 33330115; PMCID: PMC7734314.
Bell A, Severi E, Owen CD, Latousakis D, Juge N. Biochemical and structural basis of sialic acid utilization by gut microbes. J Biol Chem. 2023 Feb 8;299(3):102989. doi: 10.1016/j.jbc.2023.102989. Epub ahead of print. PMID: 36758803; PMCID: PMC10017367.
Wang J, Wang Y, Gao W, Wang B, Zhao H, Zeng Y, Ji Y, Hao D. Diversity analysis of gut microbiota in osteoporosis and osteopenia patients. PeerJ. 2017 Jun 15;5:e3450. doi: 10.7717/peerj.3450. PMID: 28630804; PMCID: PMC5474093.
Bakhti SZ, Latifi-Navid S. Interplay and cooperation of Helicobacter pylori and gut microbiota in gastric carcinogenesis. BMC Microbiol. 2021 Sep 23;21(1):258. doi: 10.1186/s12866-021-02315-x. PMID: 34556055; PMCID: PMC8461988.
Glover JS, Ticer TD, Engevik MA. Characterizing the mucin-degrading capacity of the human gut microbiota. Sci Rep. 2022 May 19;12(1):8456. doi: 10.1038/s41598-022-11819-z. PMID: 35589783; PMCID: PMC9120202.
Wu N, Zhou J, Mo H, Mu Q, Su H, Li M, Yu Y, Liu A, Zhang Q, Xu J, Yu W, Liu P, Liu G. The Gut Microbial Signature of Gestational Diabetes Mellitus and the Association With Diet Intervention. Front Cell Infect Microbiol. 2022 Jan 14;11:800865. doi: 10.3389/fcimb.2021.800865. PMID: 35096649; PMCID: PMC8795975.
Castillo-Álvarez F, Pérez-Matute P, Oteo JA, Marzo-Sola ME. The influence of interferon β-1b on gut microbiota composition in patients with multiple sclerosis. Neurologia (Engl Ed). 2021 Sep;36(7):495-503. doi: 10.1016/j.nrleng.2020.05.006. Epub 2020 May 31. PMID: 34537163.
Zhang Y, Lu S, Yang Y, Wang Z, Wang B, Zhang B, Yu J, Lu W, Pan M, Zhao J, Guo S, Cheng J, Chen X, Hong K, Li G, Yu Z. The diversity of gut microbiota in type 2 diabetes with or without cognitive impairment. Aging Clin Exp Res. 2021 Mar;33(3):589-601. doi: 10.1007/s40520-020-01553-9. Epub 2020 Apr 16. PMID: 32301029.
Harakeh S, Angelakis E, Karamitros T, Bachar D, Bahijri S, Ajabnoor G, Alfadul SM, Farraj SA, Al Amri T, Al-Hejin A, Ahmed A, Mirza AA, Didier R, Azhar EI. Impact of smoking cessation, coffee and bread consumption on the intestinal microbial composition among Saudis: A cross-sectional study. PLoS One. 2020 Apr 29;15(4):e0230895. doi: 10.1371/journal.pone.0230895. PMID: 32348307; PMCID: PMC7190147.
Fuerst JA, Sagulenko E. Beyond the bacterium: planctomycetes challenge our concepts of microbial structure and function. Nat Rev Microbiol. 2011 Jun;9(6):403-13. doi: 10.1038/nrmicro2578. PMID: 21572457.
Gong X, Liu X, Chen C, Lin J, Li A, Guo K, An D, Zhou D, Hong Z. Alteration of Gut Microbiota in Patients With Epilepsy and the Potential Index as a Biomarker. Front Microbiol. 2020 Sep 18;11:517797. doi: 10.3389/fmicb.2020.517797. PMID: 33042045; PMCID: PMC7530173.
Yuan X, Chen R, McCormick KL, Zhang Y, Lin X, Yang X. The role of the gut microbiota on the metabolic status of obese children. Microb Cell Fact. 2021 Feb 27;20(1):53. doi: 10.1186/s12934-021-01548-9. PMID: 33639944; PMCID: PMC7916301..
Zhuang X, Tian Z, Li L, Zeng Z, Chen M, Xiong L. Fecal Microbiota Alterations Associated With Diarrhea-Predominant Irritable Bowel Syndrome. Front Microbiol. 2018 Jul 25;9:1600. doi: 10.3389/fmicb.2018.01600. PMID: 30090090; PMCID: PMC6068233.
Polo PG, Çolak-Al B, Sentürk H, Rafiqi AM. Gut bacteria after recovery from COVID-19: a pilot study. Eur Rev Med Pharmacol Sci. 2022 Nov;26(22):8599-8611. doi: 10.26355/eurrev_202211_30397. PMID: 36459041.
Dix C, Wright O. Bioavailability of a Novel Form of Microencapsulated Bovine Lactoferrin and Its Effect on Inflammatory Markers and the Gut Microbiome: A Pilot Study. Nutrients. 2018 Aug 17;10(8):1115. doi: 10.3390/nu10081115. PMID: 30126153; PMCID: PMC6115941.
Sánchez-Quinto A, Cerqueda-García D, Falcón LI, Gaona O, Martínez-Correa S, Nieto J, G-Santoyo I. Gut Microbiome in Children from Indigenous and Urban Communities in México: Different Subsistence Models, Different Microbiomes. Microorganisms. 2020 Oct 16;8(10):1592. doi: 10.3390/microorganisms8101592. PMID: 33081076; PMCID: PMC7602701.
Stewart OA, Wu F, Chen Y. The role of gastric microbiota in gastric cancer. Gut Microbes. 2020 Sep 2;11(5):1220-1230. doi: 10.1080/19490976.2020.1762520. Epub 2020 May 23. PMID: 32449430; PMCID: PMC7524314.
Chen S, Zheng Y, Zhou Y, Guo W, Tang Q, Rong G, Hu W, Tang J, Luo H. Gut Dysbiosis with Minimal Enteritis Induced by High Temperature and Humidity. Sci Rep. 2019 Dec 10;9(1):18686. doi: 10.1038/s41598-019-55337-x. PMID: 31822775; PMCID: PMC6904617.
Do MH, Lee HB, Oh MJ, Jhun H, Choi SY, Park HY. Polysaccharide fraction from greens of Raphanus sativus alleviates high fat diet-induced obesity. Food Chem. 2021 May 1;343:128395. doi: 10.1016/j.foodchem.2020.128395. Epub 2020 Oct 15. PMID: 33268179.
Phillippi DT, Daniel S, Pusadkar V, Youngblood VL, Nguyen KN, Azad RK, McFarlin BK, Lund AK. Inhaled diesel exhaust particles result in microbiome-related systemic inflammation and altered cardiovascular disease biomarkers in C57Bl/6 male mice. Part Fibre Toxicol. 2022 Feb 9;19(1):10. doi: 10.1186/s12989-022-00452-3. PMID: 35135577; PMCID: PMC8827295.
Nuli R, Cai J, Kadeer A, Zhang Y, Mohemaiti P. Integrative Analysis Toward Different Glucose Tolerance-Related Gut Microbiota and Diet. Front Endocrinol (Lausanne). 2019 May 27;10:295. doi: 10.3389/fendo.2019.00295. PMID: 31191448; PMCID: PMC6546033.
Zhong Y, Nyman M, Fåk F. Modulation of gut microbiota in rats fed high-fat diets by processing whole-grain barley to barley malt. Mol Nutr Food Res. 2015 Oct;59(10):2066-76. doi: 10.1002/mnfr.201500187. Epub 2015 Aug 26. PMID: 26184884.
Shafiee NH, Razalli NH, Muhammad Nawawi KN, Mohd Mokhtar N, Raja Ali RA. Implication of food insecurity on the gut microbiota and its potential relevance to a multi-ethnic population in Malaysia. JGH Open. 2022 Feb 1;6(2):112-119. doi: 10.1002/jgh3.12709. PMID: 35155820; PMCID: PMC8829104.
Lee KC, Herbold CW, Dunfield PF, Morgan XC, McDonald IR, Stott MB. Phylogenetic delineation of the novel phylum Armatimonadetes (former candidate division OP10) and definition of two novel candidate divisions. Appl Environ Microbiol. 2013 Apr;79(7):2484-7. doi: 10.1128/AEM.03333-12. Epub 2013 Feb 1. PMID: 23377935; PMCID: PMC3623213.

谷禾健康

人类肠道微生物组是一个多样化的生态系统,我们已经知道,它在多个器官系统健康中发挥着重要作用,肠道微生态失调可能导致各种常见疾病,如糖尿病、神经精神疾病、癌症等。
新的研究表明,肠道微生物组的改变与眼部疾病相关。
基于小鼠实验,已经开始对人类微生物组及其与眼部病理学的关系进行临床研究。
葡萄膜炎、年龄相关性黄斑变性、青光眼、干眼综合征和霰粒肿等病理学正在探索中,对微生物组的深入研究可能扩展这些疾病的治疗方案。

本文总结目前检查肠-眼轴的临床研究,尤其是改变微生物组来缓解眼部疾病的潜在治疗方法。
研究发现,10% 的炎症性肠病患者会出现眼部疾病(巩膜外层炎、葡萄膜炎、结膜炎等)。肠道和与之相对较远的眼睛之间有什么关联?我们从以下几个方面来看:
▼
视网膜是眼睛后部的一层,里面装满了神经细胞,可以捕获图像并将其发送到大脑。
在所有的眼组织中,从免疫的角度来看,视网膜被认为是一种特权组织。它有三层保护(内部的血液-视网膜屏障;外层血视网膜屏障;以及血水屏障),以及通过“抵抗”和“容忍”策略,来保护它免受来自内部和外部环境的伤害。
这些血液视网膜屏障的变化可能通过募集炎症细胞和随后的眼内炎症导致视网膜疾病的发展,例如葡萄膜炎。
此外,它还受到自身防御系统的保护,如小胶质细胞和补体系统,以维持视网膜稳态。
视网膜由于更新和修复能力差而非常脆弱,因此即使是轻微的损伤也会产生毁灭性的后果。
年龄相关性黄斑变性是一种全身免疫性疾病,局部表现为眼部免疫环境下调所致。免疫反应改变的迹象表现为先在视网膜色素上皮中逐渐积累,后在玻璃膜疣中逐渐积累的沉积物,构成有利于免疫系统显著激活的抗原刺激。
在衰老过程中,所有防御系统的效率都会降低,与年龄相关的形态功能和免疫变化伴随着慢性低水平炎症。“炎症”过程也会导致与年龄相关的视网膜疾病。
▼
眼睛的物理变化被认为是由于肠道内壁的炎症与肠道通透性增加有关。肠道通透性/肠漏综合征的变化允许细菌、毒素或免疫化合物穿过粘膜肠道屏障并传播到不同位置,包括眼睛表面。这些化合物直接影响眼睛或可能通过分子模拟引发眼睛的免疫反应。分子模拟是一种可能引发自身免疫性炎症的机制,因为保护屏障受损以及细菌或毒素的长期存在。
扩展阅读:
▼
在许多层面上,肠道微生物群和免疫化合物与眼睛的视网膜相互作用。不健康、失调的肠道微生物群和活化的免疫细胞会在眼睛中引发炎症,并影响视网膜、眼睛微生物组和眼睛润滑,从而导致眼部相关疾病。
肠道微生物组及其代谢物,尤其是短链脂肪酸,都可以通过直接或间接修改不同细胞类型的表观基因组来调节免疫细胞的关键功能。
损害眼睛的危及视力的免疫反应是眼内炎症性疾病的典型特征。葡萄膜炎、年龄相关性黄斑变性、与干眼症相关的干燥综合征、糖尿病视网膜病变、青光眼和感染性角膜炎与肠道微生物组异常有关。
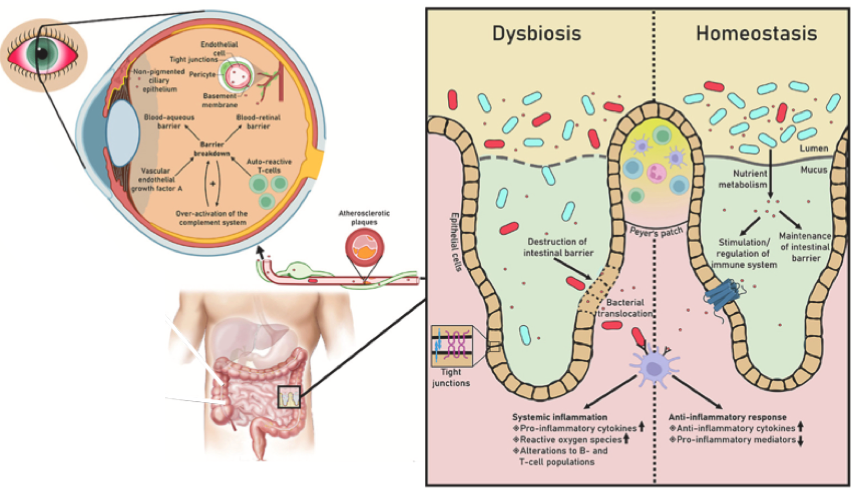
doi.org/10.1016/j.preteyeres.2022.101117
接下来,我们来看一下具体哪些肠道菌群与眼部相关疾病有关。
▼
在糖尿病视网膜病变中,与健康对照组相比,主要的分类门,包括拟杆菌门、放线菌门、粪杆菌门和梭菌门被耗尽。
在两项评估糖尿病视网膜病变队列中微生物组多样性的研究中,发现多样性下降,这与临床前小鼠模型一致。
▼
与对照小鼠相比,Bacteroides caecimuris在患病小鼠中显着增高。受影响的小鼠缺少健康肠道微生物组典型的菌群,如Rikenella,Muribaculaceae, Prevotellaceae UCG-001, Bacilli 等。
肠道微生物组变化与眼部疾病之间的联系可以通过多种机制来解释。肠道生态失调可以有利于增加肠道通透性,允许微生物及其代谢物诱导眼细胞炎症。微生物失衡也可能是血液视网膜屏障破裂和中枢神经系统氧化应激增加的原因。所有这些假设也可以解释视网膜色素变性小鼠模型中的神经炎症,氧化应激和细胞死亡。
▼
年龄相关性黄斑变性( ARMD )是一种多因素疾病,由遗传和环境因素的复杂组合引起。
与改变的肠道微生物群相关的肠道通透性增加,允许肠道代谢物和产物的更高易位,可能调节视网膜特异性免疫细胞。有趣的是,LPS 引起的慢性炎症会加速营养不良 P23H 大鼠的神经变性,导致营养不良视网膜的形态和生理紊乱恶化。
宏基因组测序评估了 ARMD 患者和对照组,研究人员发现 ARMD 患者中以下菌群含量较高 :
而以下菌群在对照组中含量较高:
研究人员推测可能与谷氨酸降解和精氨酸生物合成途径增加有关。谷氨酸是一种众所周知的视网膜兴奋性神经递质,因此其减少可能导致视网膜神经传递不足。
关于谷氨酸代谢详见:兴奋神经递质——谷氨酸与大脑健康
此外,患者也缺乏负责脂肪酸延伸途径的细菌。在这方面,长链多不饱和脂肪酸可能对视网膜生理学产生关键影响,并可能促进ARMD发展。
相比之下,对照组中Bacteroides eggerthii 的丰度可能对该疾病具有保护作用,因为它能够产生短链脂肪酸。这些代谢产物可能通过改变淋巴细胞从肠道向眼睛的迁移来调节眼内炎症。
▼
一项针对12名新生血管性年龄相关性黄斑变性(nAMD)患者的试验发现,与对照组相比,nAMD患者存在“微生态失调”。值得注意的是,研究人员发现了Anaerotruncus的增加,这也与小鼠模型中炎症信号的增加有关,这表明肠道微生物变化和nAMD进展相关的可能作用机制。
▼
在青光眼患者中,研究人员发现肠易激综合征是一种与微生物群失调相关的疾病,会显著增加患青光眼的几率(OR=5.84)。
除了发现青光眼患者与对照组的细菌谱存在差异外,还注意到视觉效果与巨单胞菌和Blautia属的丰度呈负相关。
扩展阅读:
肠道核心菌属——巨单胞菌属(Megamonas),不同人群差异大
肠道核心菌属——经黏液真杆菌属(Blautia),炎症肥胖相关的潜力菌
▼
一项视网膜研究了一组特发性颅内高压患者,再次发现与对照组相比微生物群存在差异。
有趣的是,使用乙酰唑胺治疗的患者发现乳酸杆菌增加,这被认为对肠道微生物健康有益。
总的来说,在眼部病理中,菌群可能发生改变或破坏,见下表:
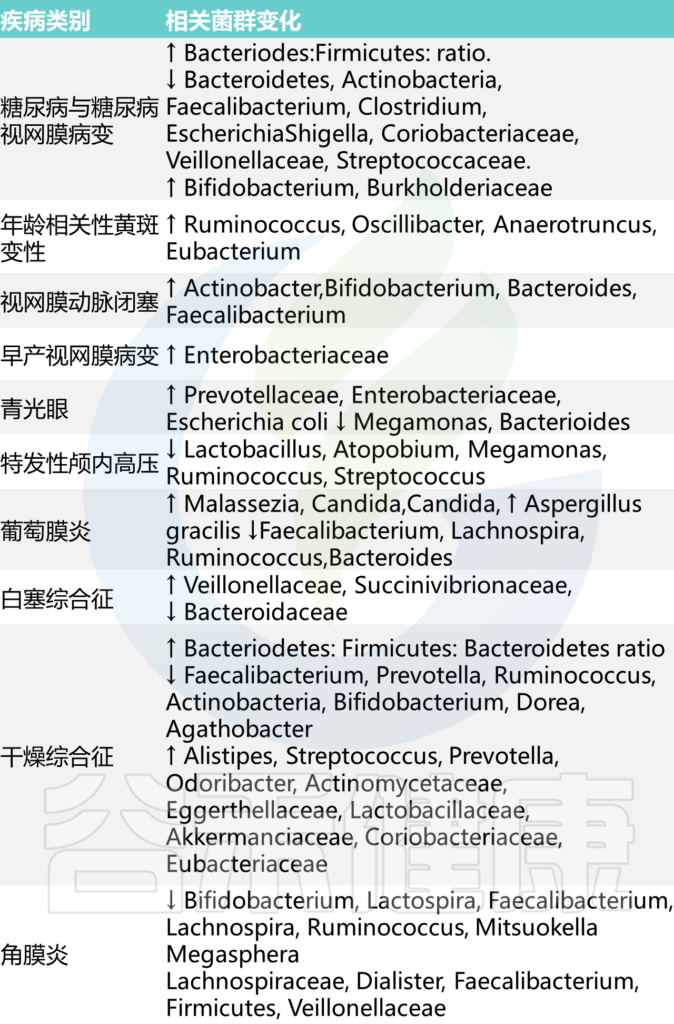
Russell MW, et al., Eye (Lond). 2023
肠道微生物组可以调节炎症信号的变化。因此,如果肠道组织受到影响,导致全身促炎状态,那么眼部后果可能是继发于或平行于肠道炎症轴的,或者可能在主要过程中有所不同。
▼
在葡萄膜炎的情况下检查了肠道失调,发现各种抗炎微生物群减少。但也有研究人员注意到葡萄膜炎的肠道失调,病例和对照组之间的没有显著差异,研究人员认为细菌可能与这种病理状态无关。
进一步探讨这种可能性,有研究人员发现,与对照患者相比,致病性念珠菌属和曲霉菌属增加了。另一项研究发现与对照患者相比,角膜炎患者的肠道失调,这两项研究都发现了标记的细菌群落变化。这些研究也检测了真菌的变化,注意到致病性曲霉、念珠菌和马拉色菌增加的趋势,这些真菌已被证明表现出抗真菌耐药性并参与其他疾病过程。
两项试验检测了白塞病葡萄膜炎患者的微生物群差异,发现病例和对照组之间存在显著差异。白塞病患者的微生物群多样性也显著降低。上述数据表明肠道健康和眼部病理之间可能存在联系。
然而,目前尚不清楚真菌和细菌是否直接介导眼部病理,是否与免疫系统有关,或者是否有其他未发现的途径在起作用。
扩展阅读:膳食真菌在癌症免疫治疗中的作用: 从肠道微生物群的角度
▼
研究人员发现,视网膜动脉阻塞(RAO)患者与健康对照相比,不同分类属的细菌有所增加。这项研究还发现RAO患者的三甲胺-N-氧化物(TMAO)显著增加,TMAO是一种微生物群衍生的代谢产物,已被发现是心血管不良事件、死亡率和血栓形成的独立风险因素。
在这项研究中,TMAO和阿克曼菌Akkermansia丰度呈正相关,表明微生物群和RAO之间存在潜在的机制联系。然而,必须注意的是,在其他研究中,Akkermansia被发现与TMAO浓度呈负相关,这表明TMAO可能不是病因,或者,TMAO本身可以在眼部病理学中发挥中介或主要作用。如上所述,这项研究并不是为了证明一种联系,也不是为了简单地假设一种联系的存在。
肠道微生态失调的眼部临床研究

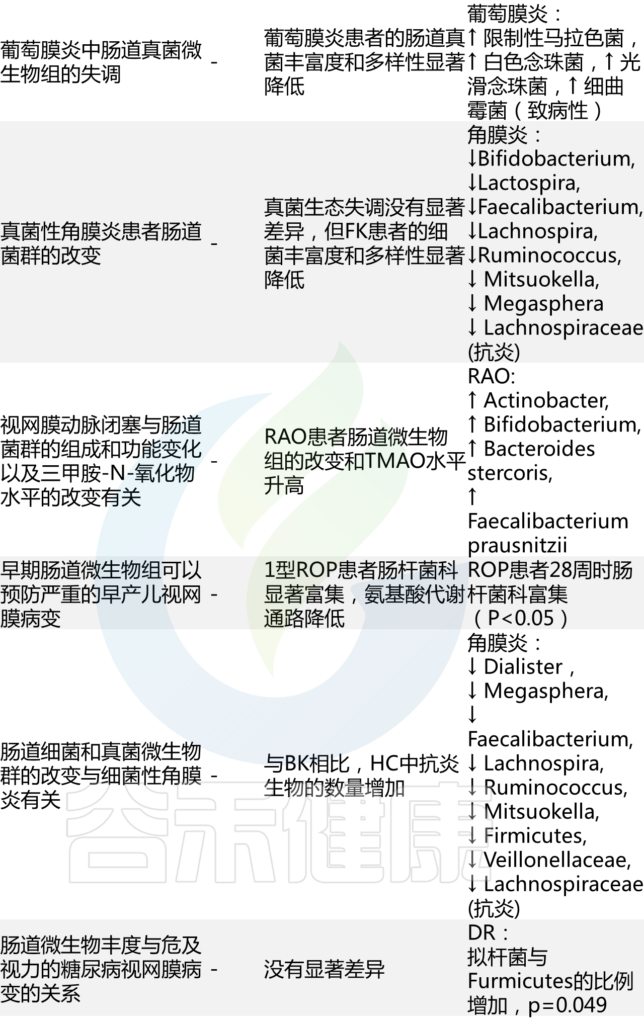
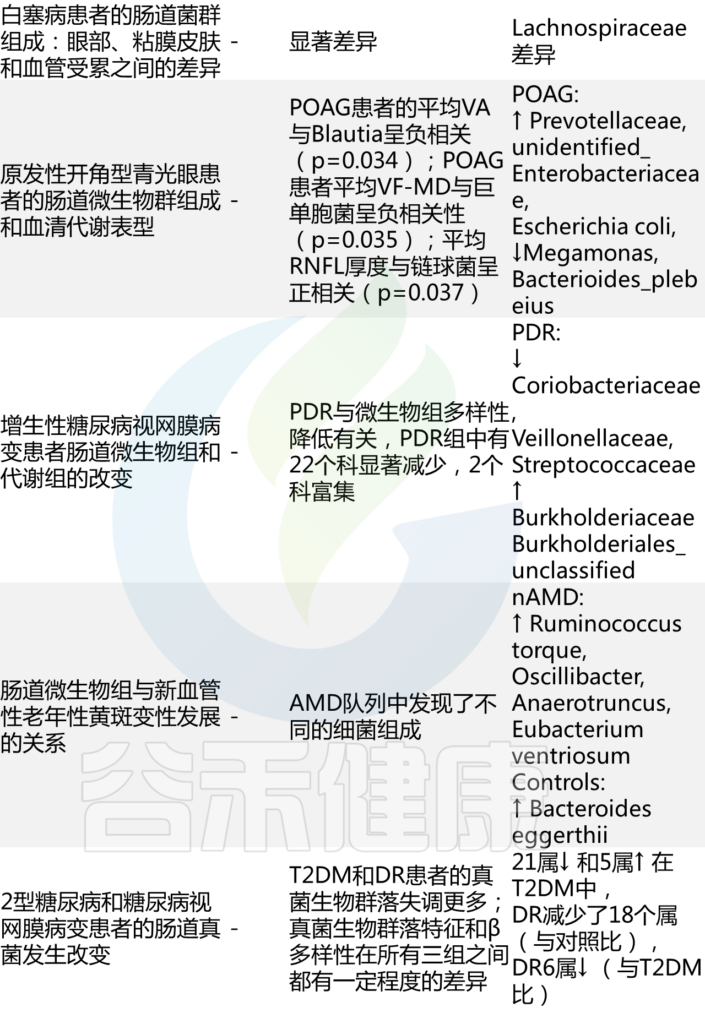
Russell MW, et al., Eye (Lond). 2023
有四项临床研究(≤23名患者)通过粪菌移植(FMT)或益生菌补充靶向肠道微生物群,来治疗眼部疾病。
▼
Watane等人于2021年对10例干燥综合征并发干眼症的患者进行了粪菌移植。粪菌移植后三个月,没有副作用报告,患者自我报告的干眼症症状在一半的队列中减轻了。
▼
Filippelli等人于2021和2022年对10名成人和13名儿童患者的益生菌补充剂及其治疗霰粒肿的疗效进行了研究。在这两项研究中,均使用了含有嗜热链球菌、乳酸乳球菌和德氏乳杆菌的益生菌。所有接受益生菌制剂治疗的成年患者霰粒肿消退时间显著缩短,而这种影响对于只有小于2.0mm的小霰粒肿的儿童来说也是如此。
Napolitano等人于2021报道了一例有三年前葡萄膜炎病史的患者的病例。患者服用了含有乳酸双歧杆菌、两歧双歧杆菌和短双歧杆菌的益生菌补充剂。两个月后,该患者的视觉功能增加,葡萄膜炎的临床症状减少。益生菌配方并不包括患者微生物组中不存在的物种。
扩展阅读:如果你要补充益生菌 ——益生菌补充、个体化、定植指南
除了以上的临床研究外,其他可能的干预措施:
▼
高纤维饮食会促进某些细菌在肠道中占据优势地位,这些细菌会产生短链脂肪酸,促进调节性 T 细胞分化,并降低发生眼部炎症的倾向。一些实验正在直接使用短链脂肪酸来测试它作为肠外自身免疫性疾病的治疗干预,已有研究人员发现,在小鼠身上,它对自身免疫性葡萄膜炎有保护作用。
▼
一项涉及衰老小鼠的研究中,高血糖饮食导致光感受器退化和视网膜色素上皮细胞萎缩,这在喂食正常饮食的小鼠中是看不到的。恢复到低血糖饮食可以逆转疾病的特征,并改变肠道中 AMD 保护因子(包括血清素)的水平。
▼
高脂饮食会导致肠道渗透性增加,从而使细菌产物如脂多糖和其他病原体相关分子模式的分子易位增加,它们通过先天免疫系统的模式识别受体 ( 特别是Toll样受体和Nod样受体)影响促炎信号转导,引起低度全身性炎症,加剧脉络膜新生血管形成,最终加重病理性血管生成。
在一项研究中,在4周龄的C57BL/6小鼠中研究了高脂肪饮食对泪腺功能的影响。结果显示,高脂饮食的小鼠表现出病理变化,包括眼泪分泌水平降低、炎症性CD4+T细胞增加 ,细胞浸润、TNF-α和IL-1β等促炎因子增加以及腺泡和肌上皮细胞凋亡增加。将标准饮食引入之前高脂肪饮食的小鼠后,泪腺的病理变化部分逆转,包括炎症细胞和促炎因子的减少以及抗炎胞质分裂素的上调。
▼
在啮齿动物模型中,在开始隔日禁食方案后的 1 周内,间歇性禁食已被证明可以降低血压和心率,这两者都是 糖尿病性视网膜病变等眼部血管疾病的已知危险因素。
另一项早期限时喂养(从早上 8 点到下午 2 点随意喂养,剩下的 18 小时禁食)被证明可以降低餐后胰岛素、血压、氧化应激和夜间食欲同时增加人类受试者的胰岛素敏感性和 β 细胞功能。这项研究和其他研究进一步支持间歇性禁食的有益作用,并表明它可能通过降低血压和胰岛素敏感性来治疗眼部血管疾病。
连续 7 个月的隔日禁食,增加了产生肠粘液的杯状细胞的数量,并降低了血浆 PGN 的浓度,表明肠血管屏障完整性得到改善。
▼
动物研究表明,锌通过减少氧化应激来改善视网膜的抗氧化过程。特别是,肠道菌群竞争锌的供应,锌对共生代谢途径和细菌毒力因子都很有用。然而,锌缺乏及其过量的存在都会改变微生物组的组成。
▼
膳食补充类胡萝卜素和锌可以预防或延缓眼部疾病的进展,可能是通过它们的抗氧化和抗炎特性。叶黄素和玉米黄质两种叶黄素,它们天然集中在人眼的黄斑中。它们充当蓝光的光学滤光片,并作为常驻抗氧化剂和自由基清除剂,以减少氧化应激引起的损伤。
由于人类无法合成类胡萝卜素,因此供应取决于含类胡萝卜素的食物,例如绿叶蔬菜、西兰花、豌豆、玉米和蛋黄。
▼
大量摄入 omega-3 长链多不饱和脂肪酸 (LCPUFA) 与年龄相关黄斑变性风险降低有关。而大量摄入 omega-6 LCPUFA 与风险增加有关。LCPUFAs高度集中在眼睛中,对视网膜的视觉功能至关重要。此外,它们是对氧化应激的促炎和抗炎免疫反应的重要调节剂。ω-3和ω-6 LCPUF之间的比例似乎对预防慢性低度炎症很重要。可以通过某些富含脂肪的鱼,亚麻籽和藻类等补充。
▼
膳食多酚可减少氧化应激,在视网膜色素上皮细胞中具有抗炎作用,并与各种白细胞介素和信号通路的调节有关。在丁香、浆果、红酒或绿茶中富含。
其他对眼部健康至关重要的营养物质包括:
维生素 A、番茄红素、硫辛酸、维生素 C、姜黄素、白藜芦醇、槲皮素、葡萄籽提取物、绿茶提取物等。
微生物组的复杂性对研究微生物变化有挑战,因为微生态失调可能归因于多种菌群的同时过度生长或损失。目前的文献并没有直接分析出因果关系。
除了目前的试验涉及的非特异性干预之外,其他干预措施也可能有效果。一项对36名患者进行的17周的随机前瞻性研究表明,通过逐步引入发酵食品等相对不那么激烈的措施,可以对免疫功能产生类似的影响。
关于发酵食品详见:肠道微生物群与健康:探究发酵食品、饮食方式、益生菌和后生元的影响
也有研究人员认为,应该谨慎采用通过改变微生物组来改善系统健康的干预措施。30名患者使用益生菌增加微生物多样性,然而却因这种补充而患上了小肠细菌过度生长和D-乳酸酸中毒。开始抗生素治疗后,患者症状减轻(P=0.005)。
由于许多原因,选择与疾病相关的正确益生菌并不容易。同一属和种的不同菌株可能对宿主产生完全不同的影响。应充分了解特定细菌菌株的特定特性和特征以及对宿主健康的影响。
因此,需要更大规模的前瞻性临床试验研究益生菌补充剂对各种眼部疾病的影响,对于进一步阐明这些干预措施的疗效至关重要。有必要研究在剂量和配方方面选择更合适的益生菌方案。
为了建立肠道菌群与眼部病变缓解之间的因果治疗关系,未来的研究可能考虑将微生物组-免疫-眼部效应与纯粹的微生物组-眼部效应分离开来。
临床试验检查了肠道微生物群和眼部病理之间的联系,显示了这两个系统之间的联系。
通过饮食、益生元和益生菌以及粪菌移植等方式调节肠道微生物群,可能都会成为预防和/或治疗眼部疾病的有效方案。
充分结合肠道菌群检测全面评估患者的菌群健康状况,可以考虑采用更有针对性的干预措施,而不仅仅是粪菌移植和益生菌补充剂。
大规模的随机对照临床试验可能会进一步证明这种联系,并阐明新的靶点治疗机制。
主要参考文献:
Zysset-Burri DC, Morandi S, Herzog EL, Berger LE, Zinkernagel MS. The role of the gut microbiome in eye diseases. Prog Retin Eye Res. 2023 Jan;92:101117. doi: 10.1016/j.preteyeres.2022.101117. Epub 2022 Sep 6. PMID: 36075807.
Napolitano P, Filippelli M, Davinelli S, Bartollino S, dell’Omo R, Costagliola C. Influence of gut microbiota on eye diseases: an overview. Ann Med. 2021 Dec;53(1):750-761. doi: 10.1080/07853890.2021.1925150. PMID: 34042554; PMCID: PMC8168766.
Russell MW, Muste JC, Kuo BL, Wu AK, Singh RP. Clinical trials targeting the gut-microbiome to effect ocular health: a systematic review. Eye (Lond). 2023 Mar 14. doi: 10.1038/s41433-023-02462-7. Epub ahead of print. PMID: 36918627.
Shivaji S. A systematic review of gut microbiome and ocular inflammatory diseases: Are they associated? Indian J Ophthalmol. 2021 Mar;69(3):535-542. doi: 10.4103/ijo.IJO_1362_20. PMID: 33595467; PMCID: PMC7942081.
Bai X, Xu Q, Zhang W, Wang C. The Gut-Eye Axis: Correlation Between the Gut Microbiota and Autoimmune Dry Eye in Individuals With Sjögren Syndrome. Eye Contact Lens. 2023 Jan 1;49(1):1-7. doi: 10.1097/ICL.0000000000000953. Epub 2022 Nov 11. PMID: 36544282.
Scuderi G, Troiani E, Minnella AM. Gut Microbiome in Retina Health: The Crucial Role of the Gut-Retina Axis. Front Microbiol. 2022 Jan 14;12:726792. doi: 10.3389/fmicb.2021.726792. PMID: 35095780; PMCID: PMC8795667.

谷禾健康

癌症是一种恶性肿瘤,它可以发生在人体的任何部位,包括肺、乳房、结肠、胃、肝、宫颈等。根据世界卫生组织的数据,全球每年有超过1800万人被诊断出患有癌症,其中约有1000万人死于癌症。癌症已成为全球范围内的主要健康问题之一。
癌症免疫疗法的概念在很早前就提出,但直到最近才被广泛接受为对抗癌症的新型选择。随着现代生物医学技术的发展,已经开发了各种类型的免疫治疗策略。
然而,癌症患者对免疫疗法的个体反应各不相同,并且经常观察到严重的副作用,这限制了这种新方法的进一步利用。
近年来的研究表明,肠道微生物群在免疫疗法的效果及副作用中的起到一定的介导作用,因为某些微生物物种或相关代谢物与癌症患者的反应密切相关。
已经对植物或动物天然产品的营养价值以及它们对肠道微生物群和肿瘤免疫疗法的调节进行了大量研究。膳食真菌因其丰富的营养价值和对人体的调节功能而成为近年来的研究热点。
本文将重点介绍天然产物中膳食真菌的摄入与肠道微生物群调节之间的关系,以及它们在癌症免疫治疗中的生物学作用和潜在机制。
本文出现的专业名词
CTLA-4(细胞毒性T淋巴细胞相关蛋白4),是一种蛋白受体,其作为免疫检查点起作用并下调免疫应答。
PD-1(程序性死亡受体1),是一种重要的免疫抑制分子。通过向下调节免疫系统对人体细胞的反应,以及通过抑制T细胞炎症活动来调节免疫系统并促进自身耐受。
PDL1(细胞程序性死亡-配体1),是一种跨膜蛋白,细胞程序性死亡受体-1(PD-1)与细胞程序性死亡-配体1(PD-L1)结合,可以传导抑制性的信号,减低淋巴结CD8+ T细胞的增生与免疫系统的抑制有关。
CAR-T细胞:技术人员通过基因工程技术,将T细胞激活,并装上定位导航装置CAR(肿瘤嵌合抗原受体),专门识别体内肿瘤细胞,并通过免疫作用释放大量的多种效应因子,它们能高效地杀灭肿瘤细胞,从而达到治疗恶性肿瘤的目的。
什么是癌症免疫疗法?
癌症免疫疗法是一种治疗癌症的新型方法,它利用人体自身的免疫系统来攻击癌细胞。传统的癌症治疗方法如化疗、放疗等是通过外源性方式杀死癌细胞来治疗癌症,但这些方法也会对正常细胞造成损伤。而免疫疗法则是通过激活或增强人体自身的免疫系统来攻击癌细胞,从而达到治疗癌症的目的。
癌症免疫疗法的优点是可以避免传统癌症治疗方法的副作用,同时可以提高患者的生存率和生活质量。然而,癌症免疫疗法仍然存在一些挑战和限制,如个体间治疗效果差异较大、治疗费用较高等问题,但其作为治疗癌症的新兴手段有着不错的前景。因此,目前仍需要进一步的研究和发展,以提高癌症免疫疗法的治疗效果和安全性。
免疫检查点阻断疗法(ICB)
免疫检查点阻断疗法(ICB)是一种新型的癌症免疫疗法,它通过阻断肿瘤细胞和免疫细胞之间的信号传导通路,从而激活免疫系统攻击癌细胞。
在正常情况下,免疫系统会通过检查点来控制和限制免疫细胞的活性,以避免对正常细胞的攻击。但是,癌细胞可以利用这种机制来逃避免疫系统的攻击,从而导致肿瘤的生长和扩散。 免疫检查点阻断疗法的主要作用是通过抑制免疫检查点分子的功能,从而激活免疫细胞攻击癌细胞。
免疫检查点阻断疗法(ICB)是基于“免疫监视”理论和在T细胞上发现免疫检查点分子(包括CTLA-4和PD-1等)的最具革命性的技术之一。
▸ 作用机理
CTLA-4与CD80/86细胞结合后转导的信号和PD-1与PD-L1结合后转导的信号抑制T细胞的“过度激活”,在预防慢性疾病的异常免疫反应中起重要作用。
阻断受体分子增强了肿瘤杀伤活性
然而,为了增强T细胞清除癌细胞的活性,需要取消这些信号。根据以往的研究,使用CTLA-4或PD-1/PD-L1单克隆抗体治疗的患者通过上调免疫活性产生了显著的抗肿瘤反应。
机理研究表明,CTLA-4或PD-1/PD-L1阻断显著增强了肿瘤特异性T细胞中的T细胞受体信号,从而导致更强的肿瘤杀伤活性,肿瘤微环境中T细胞的浸润和存活率也相应提高。
▸ 免疫检查点阻断适用于的癌症
目前,免疫检查点阻断已被批准用于各种类型的癌症,包括黑色素瘤、非小细胞肺癌、肾细胞癌、头颈部鳞状细胞癌、膀胱癌、肝细胞癌、霍奇金淋巴瘤,作为一线或二线治疗。
此外,许多其他靶向共刺激因子的激动性和拮抗性免疫检查点调节剂,如4-1BB、ICOS、GITR、OX-40、CD40等,目前正在研究中。
✦免疫检查点阻断疗法的局限性
随着临床实践的进展,免疫检查点阻断疗法仍有许多局限性。一个显著的问题是不同类型的癌症反应普遍较低。
•个体差异影响免疫检查点阻断疗法效果
虽然抗PD-1/PD-L1的有效性已经在黑色素瘤和非小细胞肺癌中得到明确证明,但来自其他类型癌症的结果不太明确,并且个体对免疫检查点阻断疗法的反应各不相同,这表明包括遗传、环境、行为甚至肠道微生物群在内的其他因素对免疫检查点阻断治疗效率的影响。
•会出现重度免疫相关毒副反应
免疫检查点阻断疗法的另一个限制是相关的副作用,称为重度免疫相关毒副反应(irAEs)。重度免疫相关毒副反应是免疫检查点阻断疗法诱导的过度炎症反应,可影响多个器官,在某些情况下甚至导致死亡。
据报道,免疫检查点阻断疗法总的重度免疫相关毒副反应发生率约为70-90%。irAEs最常见的症状涉及皮肤、胃肠道、肝脏、内分泌器官和肺,而它在不同类型的癌症和治疗中有所不同。
例如,结肠炎是胃肠道中最常见的irAEs类型,免疫检查点阻断治疗的10-20%的患者中发生。皮肤反应,包括皮疹、瘙痒和白癜风,也是免疫检查点抑制剂治疗中常见的副作用,其中大约50%的患者受到影响。
重度免疫相关毒副反应发生的确切机制仍在研究中,有人提出,过度激活的T细胞攻击正常组织,细胞因子不受控制的分泌,自身抗体的扩增,甚至免疫检查点抑制剂抗体与正常组织的结合(脱靶效应)是导致重度免疫相关毒副反应发生的原因。
肠道微生物影响ICB疗法
有趣的是,已经发现肠道微生物群可能影响免疫检查点阻断疗法的疗效以及相关重度免疫相关毒副反应的发生率。
•调节宿主免疫反应
粪便移植(FMT)已被证明可有效改善黑色素瘤或上皮肿瘤患者对PD-1治疗的总体反应,这表明肠道微生物群在PD-1治疗后调节宿主免疫反应中的重要作用。
•降低相关副作用发病率
另一方面,一项针对接受抗CTLA-4治疗的黑色素瘤患者的研究表明,拟杆菌(Bacteroidetes)的富集与结肠炎的发病率降低密切相关。
尽管有这些发现,但仍然迫切需要提高效率并消除免疫检查点阻断疗法的副作用,这依赖于对宿主与免疫检查点阻断反应的机制的深入理解,以及宿主免疫反应、免疫检查点阻断疗法和肠道微生物群之间相互作用。
嵌合抗原受体免疫疗法
嵌合抗原受体免疫疗法(CAR-T细胞疗法)是一种新型的癌症免疫疗法,它利用改造后的T细胞来攻击癌细胞。
CAR-T细胞疗法的基本原理是将患者的T细胞收集后,通过基因工程技术将其改造成能够识别并攻击癌细胞的CAR-T细胞,再将其注入患者体内。
▸ 作用机理
传统上,T细胞活化依赖于T细胞受体与细胞表面主要组织相容性复合体(MHC)呈递的特异性抗原(包括肿瘤细胞相关抗原)之间的相互作用,这种相互作用经常被肿瘤细胞下调。
为了克服这一问题,研究人员设计了一种嵌合抗原受体蛋白(由癌症抗原特异性B细胞的外结构域和T细胞的细胞内结构域组成),并在患者的正常T细胞中人工表达,以产生CAR-T细胞。
CAR-T细胞具有更强的杀伤性
与正常T细胞相比,CAR-T细胞在体外和体内对肿瘤细胞表现出更高的亲和力和更强的杀伤活性。CAR-T疗法最早用于治疗包括淋巴瘤和白血病在内的血癌,与传统疗法相比,CAR-T疗法表现出更好的效果。
✦嵌合抗原受体免疫疗法适用于的癌症
目前,CAR-T疗法已被批准用于治疗各种癌症,包括复发性或难治性多发性骨髓瘤、弥漫性大B细胞淋巴瘤、高级别B细胞淋巴瘤、原发性纵隔大B细胞淋巴瘤、急性淋巴细胞白血病等。
此外,CAR-T疗法治疗其他类型癌症的潜力也在临床和临床前研究中进行了评估。
✦嵌合抗原受体免疫疗法的局限性
•产生耐药性、抗原逃逸
CAR-T疗法也有明显的缺点,最具挑战性的问题之一是肿瘤对单一抗原靶向CAR构建体的耐药性。
尽管施用CAR-T细胞最初产生高反应率,但相当比例的患者经历了靶抗原表达的部分或完全丧失,这被称为抗原逃逸。
据报道,70-90%的急性淋巴细胞白血病患者在初始阶段对CD19 CAR-T治疗表现出持久的反应;但复发后CD19抗原表达下调或丢失的比例为30-70%
在CAR-T治疗的多发性骨髓瘤患者中,也观察到包括B细胞成熟抗原(BCMA)在内的其他靶点的下调。
•全身细胞因子释放综合征
CAR-T治疗在临床上的另一个挑战是全身细胞因子释放综合征(CRS),其特征是低血压、心功能障碍、循环衰竭、呼吸衰竭、肾衰竭、多器官系统衰竭等,如果控制不好可能会危及生命。
注意:促炎性白细胞介素1和白细胞介素6被认为是CAR-T治疗中CRS的关键介质;因此,IL-6/IL-6R阻断被认为是消除CRS的潜在途径。
然而,即使使用tocilizumab,一种食品药品监督管理局批准的用于治疗严重全身细胞因子释放综合征的IL-6R单抗,症状仍然持续并最终导致患者死亡。
迄今为止,仍然缺乏对抗CAR-T疗法诱导的全身细胞因子释放综合征的有效策略。
此外,CAR-T治疗实体瘤的疗效因组织浸润能力低而降低,导致治疗结果较差。利用局部注射代替全身给药来促进CAR-T细胞的肿瘤浸润,而这仅适用于单个肿瘤病变/低转移疾病。
✦肠道微生物可能在CAR-T疗法起作用
最近发现了肠道微生物群和CAR-T疗法的反应/毒性之间的相关性。
根据一项队列研究,肠道微生物谱与B细胞恶性肿瘤患者接受抗CD19 CAR-T细胞治疗后的反应和毒性密切相关,这表现在接受/未接受抗生素治疗的患者中不同的细菌分类和代谢途径,以及暴露于抗生素的患者中观察到的较差存活率和增加的神经毒性。
最新发表的一项研究表明,在开始CAR-T治疗前,患者的基线肠道菌群特征可以预测之后对治疗的应答情况,但前提是这些患者没有预先使用广谱抗生素,因为这会破坏菌群与治疗反应间的关联。
然而,关于肠道微生物群在CAR-T治疗结果中的作用,仍然有很大程度的未知,并且仍然缺乏对其机理的认识,需要更多相关研究阐述其机制。
其他免疫疗法
除了上述的主流癌症免疫疗法,还有其他几种已经开发或正在研究的免疫疗法。
细胞因子疗法
白细胞介素-2(IL-2)是细胞因子疗法的典型例子,并于1992年被批准用于治疗转移性肾细胞癌,但包括毛细血管渗漏综合征和多器官功能障碍在内的严重毒性限制了白细胞介素-2的使用。
T细胞受体工程疗法
T细胞受体工程(TCR-T)疗法是一种类似CAR-T的过继细胞转移(ACT)疗法,其特点是通过植入肿瘤抗原特异性TCR分子对T细胞进行基因修饰。
TCR-T的优势已在临床前和临床研究中得到充分证明
除此之外,癌症疫苗和溶瘤病毒疗法也被认为是治疗癌症的有效策略。然而,肠道微生物群在调节癌症免疫疗法的效率或毒性方面的作用仍然需要解决。
肠道微生物群是生活在消化道中的复杂微生物群落,与身体的任何其他部分相比,其数量和种类最多。
众所周知,人类肠道中的微生物对宿主的健康起着重要作用。微生物群成分之间的相互作用以及它们与宿主免疫系统的关系以多种方式影响疾病的发展。
例如,它通过定居粘膜表面和分泌各种抗微生物物质来保护宿主免受病原体的侵害,这有助于增强免疫反应。
此外,肠道菌群在消化代谢、控制上皮细胞增殖分化、调节胰岛素抵抗、脑肠联系等方面发挥着重要作用。
★ 肠道菌群对癌症免疫治疗的效果和副作用有显著影响
关于癌症免疫疗法,来源于肠道微生物群的组成、生物活性和代谢产物显示出对治疗的效率和副作用有显著影响。
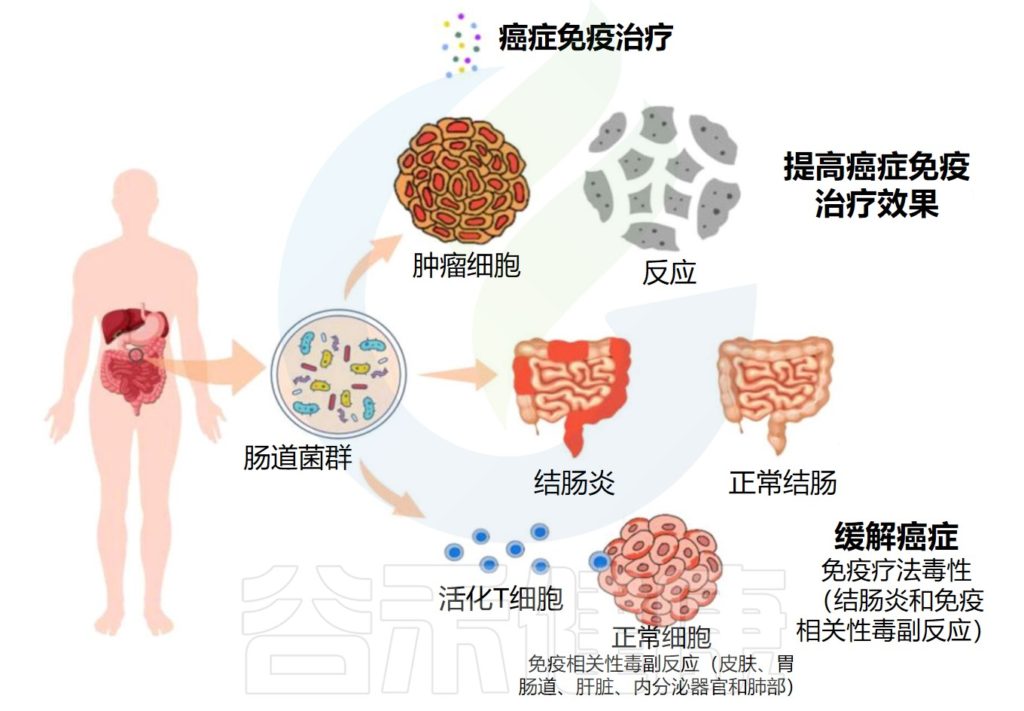
编辑
Wei Y,et al.Front. Oncol.2023
肠道菌群影响癌症免疫疗法效果
先前的一些研究已经揭示了黑色素瘤中肠道微生物群和抗PD-1功效之间的关系。
肠道菌群影响黑色素瘤细胞的生长
检查了在两个设施中饲养的遗传相似的小鼠中黑色素瘤的皮下生长。
他们发现,其中一组肿瘤生长更具侵略性,这与肿瘤内CD8+ T细胞积累显著降低有关,而这受肠道菌群组成的影响。
不同肠道微生物群接受癌症免疫治疗反应不同
一组研究人员检查了接受抗PD-1治疗的黑色素瘤患者的肠道微生物群,并观察到有反应者和无反应者之间肠道微生物群的多样性和组成有显著差异。
肠道微生物影响CAR-T治疗的患者生存率
此外,一项回顾性队列研究发现,在CAR-T治疗前四周暴露于抗生素可能会降低患者的生存率并增加神经毒性的发生率,这强调了肠道微生物群与CAR-T治疗效率之间的关联。
特定的肠道微生物在癌症免疫治疗中的参与也被发现。
✦双歧杆菌在免疫治疗中的抗肿瘤作用
在抗PD-1免疫治疗前从转移性黑色素瘤患者收集的粪便样本,发现长双歧杆菌(Bifidobacterium Longum)、产气柯林斯菌(Collinsella aerofaciens)以及屎肠球菌(Enterococcus faecium)在应答者中更丰富,这表明在PD-1免疫疗法的背景下双歧杆菌的抗肿瘤作用。
✦拟杆菌影响CTLA-4阻断剂的抗肿瘤作用
类似地,先前的研究表明CTLA-4阻断的抗肿瘤作用依赖于不同的拟杆菌物种,因为T细胞对多形拟杆菌(B.thetaiotaomicron)或脆弱拟杆菌(B.fragilis)
的特异性反应与CTLA-4阻断剂的疗效相关。
✦肠道菌群调节癌症免疫治疗效率
为了确定肠道菌群在调节患者对免疫检查点阻断治疗反应中的生物学作用,评估了粪菌移植(来自PD-1应答者)和抗PD-1给药联合治疗PD-1难治性黑色素瘤患者的疗效。
结果显示,应答者表现出先前被证明与抗PD-1反应相关的类群丰度增加,CD8+ T细胞激活增加,白细胞介素8表达髓系细胞频率降低。
应答者具有不同的蛋白质组和代谢组特征,跨域网络分析揭示了肠道微生物群在调节这些变化中的主导作用。这些结果证实了肠道微生物群在提高抗PD-1对黑色素瘤的效率方面的作用。
✦肠道微生物增强PD-1疗法对抗其他癌症
除此之外,还发现肠道微生物群在增强PD-1疗法对抗其他癌症类型。某些肠道微生物物种在调节癌症免疫疗法效率方面的生物学作用已经在以前的研究中得到了解决。
双歧杆菌(Bifidobacterium)在抗PD-1治疗过程中有利于促进抗肿瘤免疫应答。
脆弱拟杆菌(B.fragilis) 肠道定植与CTLA – 4阻断治疗之间的因果关系已通过粪便移植和脆弱拟杆菌植入得到很好的证明。
阿克曼菌提高了黑色素瘤患者PD-1的阻断效果
在一项类似的研究中,嗜黏蛋白阿克曼菌(Akkermansia muciniphila)在调节黑色素瘤患者对抗pd -1治疗的反应中的起作用。
在无应答者粪菌移植后口服嗜黏蛋白阿克曼菌(A.muciniphira),显著恢复了PD-1阻断的功效。
总之,这些发现强调了肠道菌群在提高癌症免疫治疗效率方面的重要性。
肠道微生物群改善癌症免疫疗法的毒性
关于癌症免疫治疗中肠道微生物群的另一个问题,是肠道微生物的组成或生物活性的改变如何影响免疫疗法相关毒性风险。
✦肠道菌群影响癌症免疫治疗后结肠炎的患病率
研究表明,肠道菌群的组成可以预测患者在接受阻断CTLA-4的单克隆抗体治疗后是否会发生结肠炎。
无结肠炎患者的拟杆菌门比例较高
16S rRNA基因测序/16S rDNA测序结果显示,在治疗前,结肠炎组和无结肠炎组的微生物组成相似,但治疗后仍无结肠炎的患者的拟杆菌门比例较高。
以下菌属在结肠炎耐药患者粪便中含量较高:
拟杆菌(Bacteroidaceae)、
巴恩斯氏菌科(Barnesiellaceae)
Rikenellaceae
注:上面三种细菌都是拟杆菌门下的生物
宏基因组测序分析进一步显示,与多胺转运和B族维生素生物合成相关的4个微生物模块在无结肠炎患者的微生物区系中更为丰富。
✦肠道微生物群缓解CAR-T疗法副作用
肠道微生物群也可用于预测抗PD-1/PD-L1疗法的副作用风险。
免疫相关毒副反应程度不同下的菌群丰度也不同
根据一项观察性研究,严重免疫相关毒副反应的患者以下菌属丰度较高:
链球菌(Streptococcus)
Paecalibacterium
Stenotrophomonas
轻度免疫相关毒副反应患者则富含Faecaliberium和毛螺菌(Lachnospiraceae)。
类似地,另一项临床研究显示,在经历临床显著性或不显著性irAEs的患者之间,肠道微生物群存在明显的组成差异。
拟杆菌丰度可能影响免疫治疗的相关副作用
在一项更全面的研究中,77名接受靶向PD-1和CTLA-4的联合免疫检查点阻断治疗的晚期黑色素瘤患者的血液、肿瘤和肠道微生物组被分析,在以结肠炎和黏膜白细胞介素1b上调为特征的毒性患者中发现肠拟杆菌丰度显著更高。
此外,如上所述,在接受CD19 CAR-T治疗的B细胞淋巴瘤和白血病患者中,抗生素暴露与神经毒性的高发相关。这间接证实了肠道菌群可以缓解CAR-T治疗的相关副作用。
肠道微生物治疗了免疫疗法引起的结肠炎
作为免疫检查点阻断疗法的另一个常见副作用,结肠炎通常用免疫抑制药物治疗,包括皮质类固醇或靶向肿瘤坏死因子-α (TNF-α)的药物,所有这些都具有明显的副作用。
显示结肠炎和炎症性肠病可以通过肠道微生物群操作成功治疗。
谷禾与浙一团队合作的研究,通过比较不同CAR-T治疗阶段的肠道菌群的多样性和组成,我们发现患者之间和治疗阶段之间的肠道菌群特征存在差异,并且可能反映复发/难治性多发性骨髓瘤(MM)、急性淋巴细胞白血病(ALL)和非霍奇金淋巴瘤(NHL)患者对治疗的反应。

这项研究对于理解肠道菌群在CAR-T治疗血液恶性肿瘤患者的治疗反应性中的生物学作用具有重要意义,并可能指导治疗干预以增加疗效。MM、ALL和NHL的治疗反应以及多发性骨髓瘤中严重细胞因子释放综合征的发生与特定的肠道菌群变化相关。
小结
总的来说,肠道微生物群在调节癌症免疫疗法的疗效和副作用方面的生物学作用已被充分证明,这预示着未来促进癌症免疫疗法的新策略的发展。
随着对肠道菌群的深入了解,最近也对优化患者和健康人群肠道微生物的潜在方法进行了评估。
粪菌移植和单菌移植(益生菌给药)在改善患者健康方面取得了良好的效果,但对暴露于粪菌移植的异体菌株的患者可能是有害的,并使人们容易患上自身免疫性疾病等慢性疾病。
相反,饮食干预或益生元补充可能更适合普通人群,因为它被认为危害较小,更容易被接受。同时,有必要阐明饮食干预对肠道微生物群调节机制。
不同饮食对肠道菌群的影响
许多膳食补充剂被确定具有“微生物调节”活性。
✦大量摄入动物性食品导致菌群结构不健康
动物性食品是动物来源的食物,包括畜禽肉、蛋类、水产品、奶及其制品等。
总的来说,动物性食品中氨基酸发酵产物含量较高,碳水化合物发酵产物含量较低,而氨基酸发酵产物含量与耐胆汁的拟杆菌(Bacteroides)和梭状芽孢杆菌(Clostridia)等微生物数量呈正相关。
相比之下,有益细菌,如双歧杆菌(Bifidobacteria)与动物食品的消费呈负相关。
高脂肪和动物性饮食可以促进沃氏嗜胆菌生长(Bilophila wadsworthia)——产生硫化氢的细菌,这可能是胃肠道炎症的原因。然而,大量摄入多不饱和脂肪会促进瘤胃球菌(Ruminococcus)在肠道内的生长。
✦富含纤维的饮食有助于维持肠道菌群稳态
几项研究揭示了纤维在植物性饮食中的作用。例如,富含碳水化合物和纤维的饮食增加了肠道微生物的多样性和丰富性,其特征是拟杆菌门(Bacteroidetes)数量增加,厚壁菌门/拟杆菌门比例降低。
高纤维摄入还促进了厚壁菌(Firmicutes)和变形菌(Proteobacteria)的生长,而这两种细菌在食用高脂肪饮食的受试者中通常很低。而大量摄入单糖而非纤维导致拟杆菌(Bacteroides)大量生长。
素食不含任何肉类或鱼类,但富含碳水化合物和纤维。食用素食导致短链脂肪酸的产生增加,这有利于预防胃肠道炎症和维持肠道微生物菌群的稳态。
✦优质的蛋白可以增加肠道菌群多样性
研究发现蛋白质的摄入增加了肠道菌群的多样性;然而,影响取决于蛋白质的来源。
值得注意的是,乳清和豌豆蛋白的摄入增加了双歧杆菌(Bifidobacterium)和乳酸杆菌(Lactobacillus)的水平,同时限制了脆弱拟杆菌(Bacteroides fragilis)和产气荚膜梭菌(Clostridium perfringens)的生长。
此外,豌豆蛋白增加了胃肠道中短链脂肪酸的水平。同时,动物蛋白饮食刺激耐胆汁厌氧菌。
植物化学物质对肠道菌群的调节
除了评估动物或植物性饮食对肠道微生物群的作用,还评估了植物化学物质以及益生元对肠道微生物群的调节活性。
包括多酚、类胡萝卜素、植物甾醇、木脂素、生物碱在内的植物化学物质已被证明对肠道微生物群的调节具有积极作用。
✦类胡萝卜素有助于维持肠道免疫稳态来提高抗肿瘤效率
补充类胡萝卜素如虾青素或视黄酸有助于通过诱导IgA产生来维持肠道免疫稳态。一项研究表明越橘花青素的消耗通过调节肠道微生物群的组成来促进免疫检查点阻断疗法的效率。
抗PD-L1抗体的抗肿瘤效率在口服越橘花青素后增强,同时在粪便中富集梭状芽孢杆菌(Clostridia)和约氏乳杆菌(Lactobacillus johnsonii)。
✦益生元通过调节共生微生物增强抗肿瘤效果
益生元,包括低聚果糖、低聚半乳糖、大豆低聚糖、菊粉等。通过增加共生菌如乳酸杆菌和双歧杆菌的数量,在调节肠道微生物群方面发挥作用。
一项研究报告称,口服菊粉凝胶可以通过调节共生微生物来增强α-PD-1治疗的抗肿瘤疗效,并通过激发记忆性CD8+T细胞反应来产生有效的长期抗肿瘤效果。
膳食真菌对肠道微生物群的影响
尽管在植物和动物的天然产物中发现了有趣的发现,但很少有研究研究来自膳食真菌的天然产物,这也可能对肠道微生物的组成和生物活性的调节有潜在影响。
1
香菇多糖调节了肠道微生物多样性
香菇是一种富含多种多糖的食用菌,研究表明,给予香菇多糖显著改变了小鼠小肠、盲肠、结肠和远端结肠(粪便)微生物群的多样性。具体而言,减少了拟杆菌(Bacteroidetes)与增加了变形菌(Proteobacteria)。
一项研究报道,香菇副产物(LESDF-3)可以促进拟杆菌(Bacteroides)的产生,表明香菇在调节肠道菌群方面的重要性。
与此同时,多项研究也证实了冬虫夏草多糖、黑木耳多糖、灵芝多糖、灰树花多糖、平菇多糖、猴头菌多糖、野生羊肚菌多糖在重塑肠道菌群、调节免疫等方面的生物学功能。
2
灵芝孢子油可以增强免疫活性,减少有害菌
灵芝长期以来被认为在药物治疗和饮食补充方面有价值。
根据最近的一项研究,灵芝孢子油具有很强的免疫增强活性,可导致几种有益细菌的丰度升高:
罗伊氏乳杆菌(Lactobacillus reuteri) ↑↑↑
肠乳酸杆菌(Lactobacillus intestinalis) ↑↑↑
Turicibacter ↑↑↑
Romboutsia ↑↑↑
并降低葡萄球菌(Staphylococcus)和幽门螺杆菌(Helicobacter)的丰度。
这些肠道菌群的改变进一步导致一系列关键代谢产物的分泌,如多巴胺、谷氨酰胺、蛋氨酸、L-苏氨酸、硬脂酰肉碱等,以增强巨噬细胞的吞噬能力和自然杀伤细胞的细胞毒性。
3
膳食真菌中的β-葡聚糖移植有害菌,上调
有益菌丰度
菊粉是一种存在于各种天然产品中的天然多糖,被认为是一种强有力的益生元,有趣的是,膳食真菌中的β-葡聚糖也表现出与菊粉相当的效果。
β-葡聚糖是由葡萄糖单位组成的多聚糖。它能够活化巨噬细胞、嗜中性白血球等,因此能提高白细胞素、细胞分裂素和特殊抗体的含量,全面刺激机体的免疫系统。
具体而言,β-葡聚糖可以通过抑制有害肠道微生物群的增殖,同时上调有益拟杆菌的丰度,从而调节肠道微生物群的结构和组成。
此外,β-葡聚糖和菊粉都能选择性地促进双歧杆菌(Bifidobacterium)的生长。来自蘑菇的D-葡聚糖对肠道菌群的调节也有类似的作用,因此可以认为是一种新型的益生元。
膳食真菌在调节肠道微生物群中的作用总结于下表:
膳食真菌中生物活性成分在调节肠道菌群中的作用

Wei Y,et al.Front. Oncol.2023
膳食真菌对癌症免疫治疗的影响
先前的研究已经调查了来自膳食真菌的关键成分在癌症免疫调节中的作用。
✦真菌中β-葡聚糖增强抗肿瘤免疫反应
真菌细胞壁的主要成分是β-葡聚糖,据报道,β-葡聚糖可作为有效的免疫调节剂,通过调节单核细胞骨髓源抑制细胞(MDSCs)的分化和功能来增强抗肿瘤免疫反应。
骨髓源抑制性细胞——它含义是骨髓来源的一群抑制性细胞,是树突状细胞、巨噬细胞或粒细胞的前体,具有显著抑制免疫细胞应答的能力。
姬松茸多糖刺激MDSC从M2向M1型分化,通过toll样受体2介导抑制肿瘤免疫逃避。
后来发现,自然杀伤细胞、巨噬细胞和树突状细胞负责介导真菌产物引起的抗肿瘤免疫反应。
✦双孢菇多糖激活自然杀伤细胞对结肠癌细胞的治疗作用
一项研究表明,双孢蘑菇多糖MH751906通过激活肠道自然杀伤细胞对结肠癌产生免疫治疗作用,这些激活的自然杀伤细胞对人结肠癌细胞具有稳定的杀伤作用。
✦灵芝中的成分在抗肿瘤过程中起到重要作用
灵芝多糖可部分或完全拮抗B16F10黑素瘤细胞对腹腔巨噬细胞活力的抑制,提示其在癌症免疫治疗中的潜在作用。
后续研究发现灵芝的抗肿瘤作用来自于在体内刺激树突状细胞成熟和启动对Th1辅助细胞极化的适应性免疫应答。
两项研究均表明灵芝介导的免疫调节机制在抗肿瘤过程中起作用。
✦其他一些膳食真菌成分的抗肿瘤作用
一项研究报告称牛肝菌RNA也可以刺激自然杀伤细胞对抗骨髓性白血病。
另一项研究揭示了孤苓多孔菌多糖-蛋白复合物的免疫调节活性,通过激活巨噬细胞介导的宿主免疫应答发挥抗肿瘤作用。
此外,云芝多糖肽(PSP)可增强巨噬细胞的吞噬作用,增加细胞因子和趋化因子的表达,促进树突状细胞和T细胞向肿瘤的浸润,具有免疫治疗肿瘤的作用。
膳食真菌在癌症免疫治疗中的确切作用除了少数报道了相关作用的研究外,在很大程度上仍然是未知的。
✦膳食真菌可以增强免疫阻断疗法效果
一项研究发现蛹虫草多糖通过抑制肿瘤相关巨噬细胞和T淋巴细胞之间的PD-1/PD-L1轴,将免疫抑制巨噬细胞转化为M1表型并激活T淋巴细胞,这可能提高抗PD-1/PD-L1免疫治疗的有效性。
另一项研究表明,人参多糖改变了肠道菌群和犬尿氨酸/色氨酸的比例,增强了PD-1/PD-L1免疫治疗的抗肿瘤作用。阐明了真菌多糖通过肠道菌群增强免疫检查点阻断疗法抗肿瘤作用。
✦提高癌症疫苗疗效
除了增强免疫检查点阻断疗法的治疗效果外,服用真菌产物也有提高癌症疫苗疗效的报道。口服香菇菌丝体提取物可增强肽疫苗的抗肿瘤活性,表明香菇提取物在癌症免疫治疗中具有重要作用。
总的来说,全面了解膳食真菌在癌症免疫治疗中的作用及其分子机制是非常有前景的,肠道菌群在这一过程中的参与也需要进一步研究。
膳食真菌在调节针对癌症的宿主免疫反应中的作用总结于下图:
膳食真菌在治疗各种类型癌症中的生物学作用和机制

Wei Y,et al.Front. Oncol.2023
膳食真菌对癌症免疫治疗的临床研究
迄今为止,已经进行了一些临床研究来评估真菌产物的抗肿瘤活性和潜在机制。
•灰树花抑制肺癌或乳腺癌转移
研究表明,灰树花能抑制肺癌或乳腺癌的转移,减小肿瘤的大小,这是通过增加自然杀伤细胞活性和促进Th1细胞反应实现的,而Th2细胞活性降低。
•灵芝多糖对晚期癌症的抗肿瘤作用
另一项研究表明,灵芝多糖对多种类型的晚期癌症具有抗肿瘤作用,这是通过刺激宿主免疫反应来实现的,包括白细胞介素2、白细胞介素6、人干扰素-g(IFN-g)的分泌增加,自然杀伤细胞活性增强,而白细胞介素-1b和肿瘤坏死因子的浓度较基线降低。
48名乳腺癌患者接受了灵芝孢子粉,并显示治疗后患者血清中肿瘤坏死因子的浓度显著降低,这伴随着肿瘤负荷的减轻。
•双孢蘑菇对前列腺癌具有一定抗肿瘤作用
此外,一项I期试验证明双孢蘑菇通过调节白细胞介素15水平和骨髓源抑制性细胞活性对前列腺癌具有抗肿瘤作用。
•平菇和羊角草也具有抗肿瘤活性
平菇,也被发现通过调节宿主免疫具有抗肿瘤作用。
通过临床试验研究了羊角草的抗肿瘤活性,发现血清中人干扰素-g和白细胞介素12水平随着自然杀伤细胞活性的升高而升高,提示Th1免疫应答参与了羊角草的靶向抗肿瘤活性。
临床上还评估了活性己糖相关化合物(AHCC,从担子菌蘑菇香菇中获得)对腺癌的抗肿瘤作用和免疫调节活性,治疗导致中性粒细胞增加,CD4+/CD8+、CD3+/CD16+/CD56+NK细胞的比例也相应增加,而淋巴细胞和单核细胞的数量减少。
总的来说,真菌产品的抗肿瘤作用以及分子机制已被很好地记录,然而,大多数结论仅基于小样本得出。
此外,尽管临床研究已经证实真菌产品可以增强化疗和放疗的抗肿瘤作用,但关于真菌产品在癌症免疫治疗中的有效性的临床证据仍然缺乏。因此,还需要额外的试验来研究真菌产品对癌症免疫治疗的临床效果。
膳食真菌在不同癌症中的作用总结下图:
膳食真菌治疗不同类型癌症的结果

Wei Y,et al.Front. Oncol.2023
近年来,随着高通量多组学技术(包括微生物扩增子、宏基因组、宏转录组、代谢组)的发展,肠道微生物群对人类健康的生物学相关性已得到充分认识。
肠道微生物在调节宿主免疫稳态方面具有多种功能,这有利于预防许多疾病,对改善癌症免疫治疗也有一定作用。
由于肠道微生物群与癌症患者在治疗时的临床特征密切相关,因此可通过评估其肠道微生物群来预测个体的情况。
食用真菌含有多种能被肠道微生物识别的营养物质,对肠道微生物群起到多种影响,并在癌症免疫治疗中起到作用。对膳食真菌和肠道微生物群的研究将为当前的免疫抗癌疗法提供非常有价值的帮助。
主要参考文献
Wei Y, Song D, Wang R, Li T, Wang H and Li X (2023) Dietary fungi in cancer immunotherapy: From the perspective of gut microbiota. Front. Oncol. 13:1038710. doi: 10.3389/fonc.2023.1038710.
Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat Rev Immunol (2020) 20(11):651–68. doi: 10.1038/s41577-020-0306-5.
June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. Car T cell immunotherapy for human cancer. Science (2018) 359(6382):1361–5. doi: 10.1126/science.aar6711.
Zhao Q, Jiang Y, Xiang S, Kaboli PJ, Shen J, Zhao Y, et al. Engineered tcr-T cell immunotherapy in anticancer precision medicine: Pros and cons. Front Immunol (2021) 12:658753. doi: 10.3389/fimmu.2021.658753.
Tan S, Li D, Zhu X. Cancer immunotherapy: Pros, cons and beyond. BioMed Pharmacother (2020) 124:109821. doi: 10.1016/j.biopha.2020.109821.
Yan Z, Zhang H, Cao J, Zhang C, Liu H, Huang H, et al. Characteristics and risk factors of cytokine release syndrome in chimeric antigen receptor T cell treatment. Front Immunol (2021) 12. doi: 10.3389/fimmu.2021.611366.
Fan Y, Pedersen O. Gut microbiota in human metabolic health and disease. Nat Rev Microbiol (2021) 19(1):55–71. doi: 10.1038/s41579-020-0433-9.

谷禾健康

人类微生物组有多种来源(比如通过母亲向婴儿的垂直传播,通过饮食等)。环境暴露允许人类与整个微生物生态系统相互作用,并且有新证据表明,生态系统中微生物的多样性可能影响人类微生物群的组成,从而对免疫功能和健康结果产生影响。
人类在富含微生物的环境中进化,病原体和共生菌共同调节着肠道微生物群和免疫系统。然而,城市化、土地利用变化、相关的生物多样性损失,以及室内时间的增加,都使我们减少了对更具多样性的环境微生物群的接触。
“卫生假说”的一个扩展假设是,如果减少与更多样的环境微生物群接触,可能导致人类微生物群的不利变化,从而导致各种疾病,比如自身免疫性疾病、过敏和哮喘。
近期的动物和人类研究,证明了环境微生物群暴露对宿主微生物组的影响;从而提供了环境微生物群对人体肠道微生物群和免疫功能的潜在调节作用的证据。
本文主要介绍研究人员提出的,环境微生物群暴露可能会改变人体肠道微生物群和免疫功能,且基于该理论的微生物群针对性改善措施。
目前有几项人类干预研究发现,与自然环境的相互作用(如接触土壤、植物物质)会影响皮肤、口腔、鼻腔和肠道微生物群,这里重点关注肠道微生物群,包括儿童和成人人群。4项研究总结如下:

doi.org/10.1016/j.mucimm.2023.03.001
通过这些研究,我们可以看到对影响肠道微生物组和/或免疫功能的干预措施研究包括:
直接接触堆肥和土壤混合物,在微生物富集的沙坑中玩耍(包括植物物质和商业园艺土壤);接触室外绿地和绿墙等。
沙坑
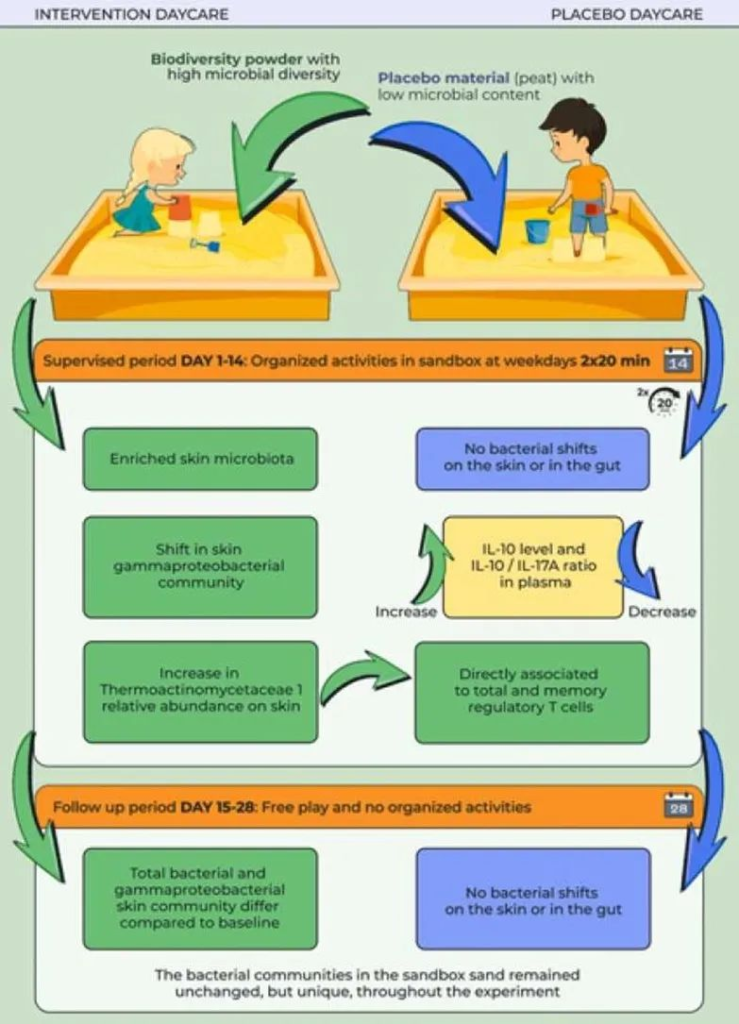
添加富含微生物的土壤粉末可使沙子微生物群多样化,影响儿童微生物群
一项双盲安慰剂对照研究,在干预组中,3-5岁的儿童暴露于富含微生物多样化土壤的操场沙子中,或者在安慰剂组中,视觉相似但微生物贫乏的泥炭沙子(n=13)。孩子们每天在沙盒中玩两次,每次20分钟,持续14天。
肠道变形杆菌多样性仅在干预组0至14天之间下降(线性混合效应模型简称LMM:P = 0.02, Q = 0.03),而安慰剂组则保持在基线水平(LMM:P > 0.6)。
在第14天,与安慰剂组相比,干预中的肠道变形杆菌辛普森多样性较低(t检验P<0.016,Q = 0.06),但在第28天不再降低(t检验P = 0.23)。
肠道菌群潜在变化与免疫标志物之间的关联
进一步探讨了肠道菌群测量值是否与线性混合效应模型的免疫参数相关。
——干预组中血浆中IL-17A的水平降低
干预组的肠道变形细菌多样性(辛普森和香农)降低,这种减少与血浆IL-17A水平降低有关。
注:IL-17是一种促炎细胞因子,与几种免疫介导的疾病有关,包括炎症性肠病,类风湿性关节炎和多发性硬化症。
——干预组中血浆中IL-10的水平增加
其他几种已知健康结果的肠道菌群与血浆IL-10水平相关。其中,厚壁菌门与拟杆菌门的比值(肥胖相关)与IL-10呈负相关,拟杆菌属的相对丰度与血浆中IL-10水平呈正相关。
注:IL-10是一种抗炎细胞因子,可通过限制促炎细胞因子的分泌以及调节巨噬细胞,T细胞和B细胞来预防自身免疫性疾病。
扩展阅读:肠道细菌四大“门派”——拟杆菌门,厚壁菌门,变形菌门,放线菌门
Faecalibacterium和Roseburia相对丰度的变化直接相关,而Romboutsia相对丰度的变化与IL-10水平的变化呈负相关。
扩展阅读:肠道核心菌属——普拉梭菌(Faecalibacterium Prausnitzii),预防炎症的下一代益生菌
编辑
doi.org/10.1016/j.ecoenv.2022.113900
总的来说,该研究支持免疫介导疾病的生物多样性假说,干预组儿童的免疫应答与安慰剂组儿童的免疫应答不同。环境微生物群可能有助于儿童健康。
绿地
邻近自然绿地对儿童特异性过敏具有保护作用
来自加拿大健康婴儿纵向队列研究的数据(699名婴儿)表明,婴儿早期生活在邻近自然绿地,在3岁时对多种吸入性特应性过敏的发展具有保护作用(优势比 = 0.28[95%CI 0.09,0.90])。4 个月大时肠道微生物群的放线菌门多样性介导了这种关系。
自然植被与更大的物种丰富度相关,但降低了小婴儿肠道中放线菌门的辛普森多样性。
这听起来有点矛盾。
我们知道,双歧杆菌作为益生菌可以预防特应性疾病,双歧杆菌属于放线菌门,为什么减少放线菌多样性值,反而可能降低过敏风险有利健康?
实际上,放线菌多样性辛普森测量值较低,表明物种分布不均匀,且少数优势双歧杆菌富集。
越来越多的证据表明,当大量的成人双歧杆菌相关物种,如B. catenulatum和青春双歧杆菌(B. adolescentis)在幼年时主导肠道菌群时,特异反应的风险更大。
也就是说,本来放线菌的辛普森多样性与吸入物致敏状态之间的正相关,但这种相关性因接近自然绿地而降低,据推测,这是婴儿主要双歧杆菌物种(如婴儿双歧杆菌)富集的结果。
鉴于早期双歧杆菌定植和低多样性的肠道微生物组的重要性,自然植被通过婴儿特异性双歧杆菌富集对吸入物致敏的保护作用值得进一步研究。
总之,该研究强调了促进自然城市绿地保护的重要性,通过降低特应性疾病的易感性来改善儿童健康。
扩展阅读:肠道核心菌属——双歧杆菌,你最好拥有它
绿墙
办公场所空气流通的绿墙会改变微生物组并调节上班族的免疫系统
最近的一项研究发现,暴露在办公场所的绿墙(带有植物)14天后,可能增加皮肤微生物组的多样性,绿墙研究中观察到的变化与对人类健康有益的变形杆菌和乳酸杆菌有关。
这种变化与血液样本中细胞因子转化生长因子βmRNA的表达有关,表明环境微生物群在免疫功能中的潜在作用。
由于没有直接接触绿墙,这些发现表明,皮肤微生物组和免疫功能可能与接触三种治疗的空气生物群落有关;然而该研究并未对空气微生物组进行评估。
空气微生物组的研究
不过呢,这一想法得到了一项小鼠研究的支持,在这项研究中,小鼠暴露于无土壤、低多样性土壤和高多样性土壤的空气微生物群中,在暴露7周后,各组之间的粪便和盲肠微生物群的群落组成存在显著差异。
关于空气微生物组对肠道微生物组和免疫功能的潜在影响的证据尚处于起步阶段,由于目前对低生物量样本的微生物组研究的局限性,在人类中建立这种关系将具有挑战性。
方法偏差
迄今为止,包含比较组的人类研究相对较少,也存在一系列潜在的方法偏差。例如,两项研究中的分配偏差,没有将微生物群暴露与干预措施的其他因素(如自然因素)隔离,干预措施的相对短期性质以及干预措施停止后的随访期。需要更多研究。
样本量
所有研究中的样本量相对较小。
精细化程度
目前微生物组分析侧重于相对较高的分类群,在DNA提取和测序技术以及生物信息学方面,通常缺乏明确性和精细度。还需要对免疫功能进行更全面的评估。
普遍性
在确定这些发现如何适用于所调查的场景之外时,还必须考虑这些发现的普遍性。
由于暴露环境组成的潜在差异以及暴露人群的行为差异(如室内时间、饮食、体力活动),此类干预措施调节肠道微生物群的普遍性尚不清楚。
对于健康状况较差的人,目前还没有关于环境微生物群如何影响其肠道生态和免疫功能的数据。关于暴露于环境微生物群作为调节肠道微生物群的工具的一个潜在问题是安全性。虽然我们总是与环境微生物群接触,但在建议改变这种接触时,我们必须注意潜在的感染风险增加(例如环境中的病原体),特别是对于免疫系统受损的人群。
虽然以上不足可能会影响我们对环境微生物群暴露如何影响肠道微生物群的理解,但研究提供了初步证据,表明这些暴露可能会改变人体肠道微生物群和免疫功能。
关于剂量、治疗持续时间和干预停止后变化的持续性,仍有许多问题,但随着这一领域研究的增长,预计将有更多明确的研究证据提供。
研究人员认为,可以采取几种方法来改善对更多样环境微生物群的暴露。具体如以下几个方向:
行为改变:
土壤摩擦、园艺、更多时间在绿地中户外活动;
优化人们常接触空间中的环境微生物群:
在室外环境中,更大的植被多样性与更多样的土壤微生物多样性和更多样的空气生物群落相关联;
而在室内环境中,环境微生物群可以通过建筑设计特征来改变,包括允许阳光和通风的窗户、控制湿度和温度以及减少室内污染物。
均质化、富含微生物的土壤可用于重建城市游乐场。
微生物接种的油漆和瓷砖:
这可能正在成为一种新的策略,作为一种更具针对性的方法,可以在不太可能有绿地的环境中,调节环境微生物群,从而调节人类微生物群。(例如边界生物安全、潜艇或太空旅行)
与其他基于微生物群干预措施并用:
环境微生物组暴露干预措施可与其他基于微生物组的干预措施一起使用,以实现协同增效,从而增强和/或延长任何益处。例如,如果暴露于更多样的环境微生物群延长其效力,粪菌移植给药的频率可能会降低。
暴露于环境微生物群可能会改变人体肠道微生物群和免疫功能,这一领域的研究尚处于起步阶段,需要进行更多的研究,以更好地了解暴露的类型和剂量、不同人群发现的普遍性,并确保此类干预措施的安全性。
这些研究为我们拓宽视野,应对城市居民免疫介导疾病的高发病率,新的可持续的基于自然的预防性干预措施正在涌现,增加户外活动与自然重新建立联系,生物多样性粉末用来制造沙子,重建城市栖息地,或者绿墙的建筑改造也可以成为新的选择。
主要参考文献:
Buchholz V, Bridgman SL, Nielsen CC, Gascon M, Tun HM, Simons E, Turvey SE, Subbarao P, Takaro TK, Brook JR, Scott JA, Mandhane PJ, Kozyrskyj AL. Natural Green Spaces, Sensitization to Allergens, and the Role of Gut Microbiota during Infancy. mSystems. 2023 Feb 15:e0119022. doi: 10.1128/msystems.01190-22. Epub ahead of print. PMID: 36790181.
Soininen L, Roslund MI, Nurminen N, Puhakka R, Laitinen OH, Hyöty H, Sinkkonen A; ADELE research group. Indoor green wall affects health-associated commensal skin microbiota and enhances immune regulation: a randomized trial among urban office workers. Sci Rep. 2022 Apr 20;12(1):6518. doi: 10.1038/s41598-022-10432-4. PMID: 35444249; PMCID: PMC9021224.
Stanhope J, Weinstein P. Exposure to environmental microbiota may modulate gut microbial ecology and the immune system. Mucosal Immunol. 2023 Mar 9:S1933-0219(23)00015-6. doi: 10.1016/j.mucimm.2023.03.001. Epub ahead of print. PMID: 36906178.
Roslund MI, Puhakka R, Grönroos M, Nurminen N, Oikarinen S, Gazali AM, Cinek O, Kramná L, Siter N, Vari HK, Soininen L, Parajuli A, Rajaniemi J, Kinnunen T, Laitinen OH, Hyöty H, Sinkkonen A; ADELE research group. Biodiversity intervention enhances immune regulation and health-associated commensal microbiota among daycare children. Sci Adv. 2020 Oct 14;6(42):eaba2578. doi: 10.1126/sciadv.aba2578. PMID: 33055153; PMCID: PMC7556828.
Sacks D, Baxter B, Campbell BCV, Carpenter JS, Cognard C, Dippel D, Eesa M, Fischer U, Hausegger K, Hirsch JA, Shazam Hussain M, Jansen O, Jayaraman MV, Khalessi AA, Kluck BW, Lavine S, Meyers PM, Ramee S, Rüfenacht DA, Schirmer CM, Vorwerk D. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int J Stroke. 2018 Aug;13(6):612-632. doi: 10.1177/1747493018778713. Epub 2018 May 22. PMID: 29786478.
谷禾健康
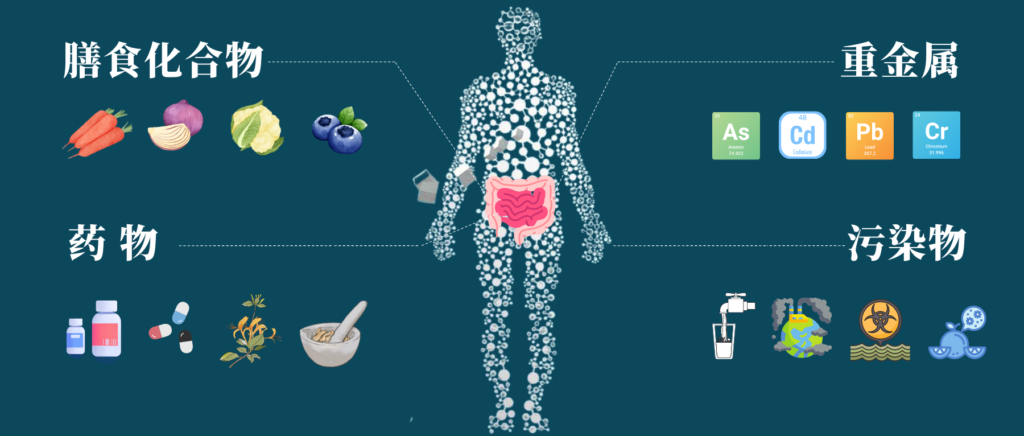
肠道菌群和人体健康息息相关,我们经常讲饮食、生活方式等都可以影响肠道菌群的组成,除了这些耳熟能详的因素之外,其他异源物如环境中的污染物,重金属,药物等都会影响肠道菌群,反过来,细菌也可以通过生物积累或化学修饰影响这些化合物。
通过皮肤接触、吸入或摄入等方式,我们每天都在和异生素接触,可能通过补充剂或药物自愿摄入异源物,或者通过受污染的食物和水被动摄入异源物。
肠道微生物群可以在吸收之前或之后与摄入的化合物相互作用。
了解肠道微生物群如何处理膳食成分的分子机制,是用“功能性食品”、益生菌和益生元治疗不同疾病的理论基础,有利于根据患者的代谢状况和肠道微生物群进行个性化营养定制。
与饮食类似,对肠道微生物群改变重金属、污染物、药物毒性进行研究,可以从肠道菌群代谢角度打开视野,进一步探讨从体内去除有害化合物并预防疾病的方法。
本文主要介绍了肠道微生物群对基本营养物质、重金属、污染物、药物、中草药等产生的代谢作用,并提供了基于肠道菌群的改善方式,以减轻这些有毒元素造成的损害。
什么是异源物?
异源物(xenobiotics),也就是说外源物质,来自希腊语,是指使人体从外界(包括肠道中的细菌作用产物)摄入体内的化学物质,例如药物、毒物、食品添加剂、环境污染物等。
人类肠道微生物群是一个多样而复杂的微生物群落,与宿主一起进化。肠道微生物群是宿主代谢过程的组成部分。
有时候会把肠道微生物群比作“器官”,可以干扰调节代谢的宿主基因。
人体肠道微生物群和异源物之间的大多数相互作用发生在胃肠道粘膜上。由于消化道的范围不同,不同的区域为微生物和代谢过程提供了不同的栖息地。
当我们说异源物的微生物代谢时,应该在宿主代谢过程的背景下进行。后者往往是同时发生的和相互竞争的。
▼ 异源物是怎么进入人体代谢的?
异源物经口服、吸收后在肠上皮细胞之间或通过肠上皮细胞传递,可被宿主酶处理或不被宿主酶处理。之后,它们可以通过门静脉输送到肝脏。
在肝脏中,异源物受到许多代谢酶的影响。因此,外源性代谢产物进入全身循环,从而使其分布到组织中并影响其他器官。
代谢产物可以与消化道的上皮细胞局部相互作用。当外源性药物通过静脉注射时,它们跳过了“第一道”代谢,立即进入全身循环。
而循环的外源性代谢产物进一步代谢并通过胆汁排泄排出,然后通过肠肝循环在小肠中重新吸收,或通过肾脏排出到尿液中。
▼ 异源物怎么和肠道菌群相遇?
可以通过多种途径与肠道微生物相遇。
小肠外源物中未被吸收或吸收不良的物质,继续进入大肠,被肠道微生物转化。
容易吸收或静脉注射的外源性物质最终可以通过胆汁排泄,到达肠道细菌。
▼ 肠道菌群影响异源物的生物转化
除了上面说的代谢过程外,肠道微生物群还可以影响异源物的生物转化,从而影响其功能和毒性。
同样,异源物可以诱导基因表达在人类肠道微生物群的直接参与异源物的代谢。结果表明,即使在短期暴露于异源物期间,这种表达也会上调。
异源物和微生物群之间的复杂相互作用
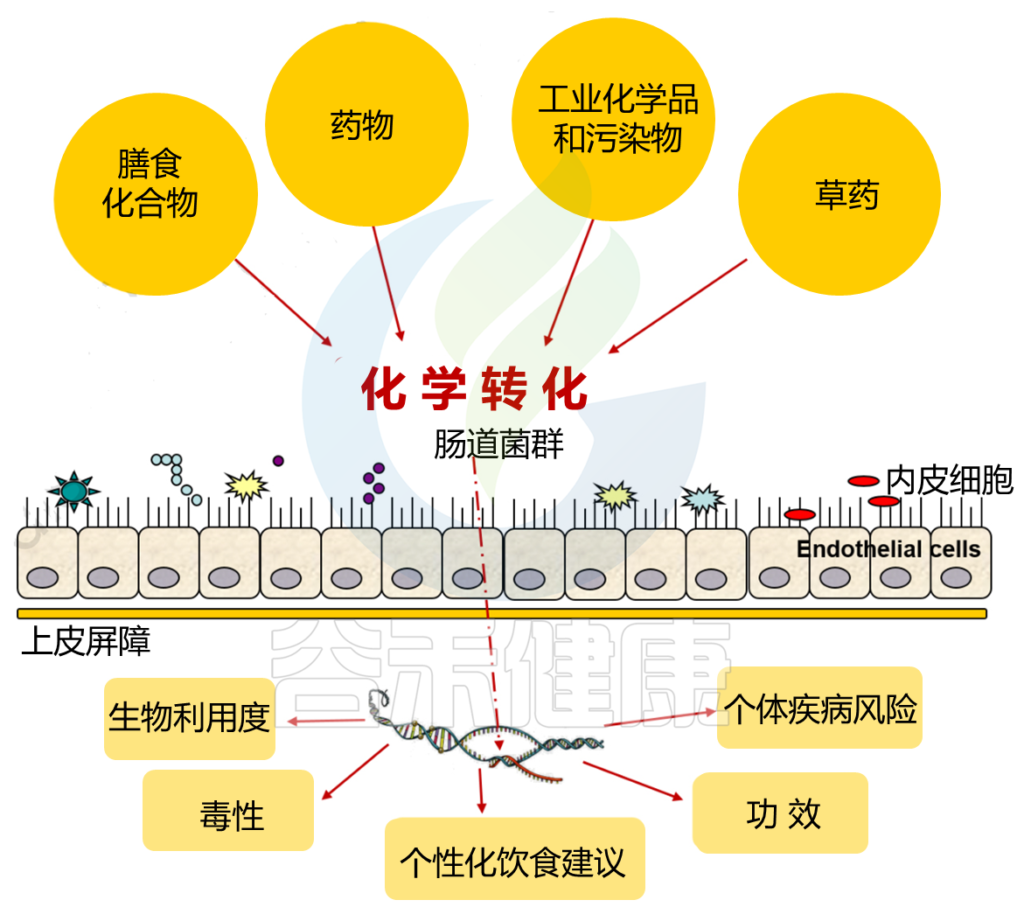
DOI: 10.2174/1389200221666200303113830
肠道微生物群对几种外源性物质的生物转化已通过体外和体内模型进行了广泛研究。然而,由于许多其他因素,例如在到达肠道细菌更普遍的肠道下部之前的有效吸收,或肝脏中的异源代谢,防止肠道微生物群遇到特定代谢产物,人类的异源物生物转化可能会发生很大变化。
同样,暴露于异源物质时,肠道菌群的结构和功能会发生特定变化。
摄入的外源物质的这些直接代谢导致了几种情况:
人类微生物群和异源物质相互作用是一个复杂的代谢网络,影响着双方。
饮食是人类健康的基石,目前已有许多研究将饮食模式与健康结果联系起来,这其中离不开肠道菌群的深度参与。在我们之前的多篇文章中也有相关阐述,这里我们简要回顾一下膳食化合物的代谢,重点关注重金属,污染物,药物代谢等异源物,逐一了解肠道菌群对异源物代谢过程,及其与健康之间的关联。
肠道菌群可以从多种膳食化合物中提取营养和能量,该过程的程度和类型在个体之间差异很大,主要是因为肠道微生物酶的存在和丰度不同,这里主要关注膳食蛋白质、脂类和多酚的转化。
膳食蛋白质可以用来做什么?
它可以体液平衡、细胞修复、血液凝固、激素、产生酶等。它也是肠道菌群的食物,主要作为氨基酸来源,可用于蛋白质合成和代谢能量的产生。
而肠道微生物群参与蛋白质代谢,它们在营养利用和宿主反应之间的相互作用中起着关键作用。
关于肠道微生物群参与蛋白质代谢详见:
小肠中的蛋白酶和许多肽酶对于将膳食蛋白质消化为氨基酸和寡肽是重要的。
肠道微生物群具有利用氨基酸的专门酶。
肠上皮细胞参与氨基酸分解代谢,并通过调节肠道屏障功能在先天免疫和适应性免疫中发挥重要作用。
肠道微生物生活在肠腔中,与粘蛋白密切相互作用,其任何变化都可能改变粘蛋白分泌。据推测,肠道微生物群利用必需和非必需的管腔氨基酸。
➤ 肠道菌群的组成及位置影响氨基酸代谢
肠道菌群的组成及其在肠道中的位置,对于确定膳食蛋白质的生产速率和氨基酸的代谢命运至关重要。
约氏乳杆菌(Lactobacillus johnsonii)
约氏乳杆菌是小肠中的常见居民,这种菌缺乏编码参与氨基酸生产的生物合成途径的基因。
约氏乳杆菌不利用氨,也不参与硫同化的代谢途径。但约氏乳杆菌产生一种胞外蛋白酶、3种寡肽转运蛋白、超过25种胞浆肽酶和20种氨基酸渗透酶型转运蛋白。这些有助于约氏乳杆菌吸收外源氨基酸或肽用于蛋白质合成。
➤不同菌群蛋白水解活性可能直接导致人类疾病
这里拿乳糜泻来举例,肠道微生物群与乳糜泻有关。乳糜泻是一种常见的自身免疫性疾病,其特征是对小麦基食物中的膳食面筋产生炎症反应。
这种富含脯氨酸的蛋白质避免了宿主蛋白酶的完全消化,从而产生了高分子量的免疫原性肽。肠道微生物群可能通过改变面筋蛋白水解来影响乳糜泻。
乳糜泻患者的肠道菌群是如何引发疾病的?
健康人和乳糜泻患者的粪便悬浮液,对面筋蛋白和免疫原性肽的处理方式不同。
一般健康人通过乳酸杆菌产生肽,而乳糜泻患者不一样,乳糜泻患者会多一些铜绿假单胞菌,这种铜绿假单胞菌产生的面筋衍生肽,更加易于穿过小鼠肠道易位,并引发增强的面筋特异性免疫应答。
所以说,鉴定负责肠道微生物面筋加工的特定蛋白酶,不仅可以更好地理解乳糜泻,还可以为该疾病的治疗干预提供信息,包括酶或益生菌治疗。
肠道微生物还可以将从膳食蛋白质中获得的氨基酸(包括l-苯丙氨酸、l-酪氨酸和l-色氨酸)代谢为一系列生物活性产物。例如,肠道细菌可以将l-色氨酸代谢为多种产物,包括抗氧化剂吲哚-3-丙酸、神经递质色胺和吲哚,后者可以通过肝脏酶进行羟基化和硫酸化,生成尿毒症毒素硫酸吲哚。
关于色氨酸代谢详见:
肠道微生物对脂质吸收很重要。
➤ 胆固醇的吸收
饮食胆固醇是西方饮食中与心血管疾病风险增加相关的主要成分。吃进去的胆固醇,被小肠吸收,随后经历胆汁排泄和肠肝循环。
➤ 肠道微生物群对胆固醇的影响
粪甾醇不能被重新吸收并被排出体外。通过肠道微生物的减少,胆固醇产生了不可吸收和排泄的粪甾醇。因此,这种转变有效地去除了循环中的胆固醇。
人类粪便中高达50%的类固醇是粪甾醇。高胆固醇降低患者和低胆固醇降低患者体内微生物群,分别定植的无菌小鼠,产生不同数量的粪甾醇。
还有研究表明,降低胆固醇的细菌可能会降低血清胆固醇。
➤ 降低胆固醇的细菌
对降低胆固醇的肠道细菌——产粪甾醇真细菌的研究表明,粪甾醇合成可能涉及氧化为5-胆甾-3-酮,然后烯烃异构化为4-胆甾-3-烯酮,共轭还原和酮还原。
确定负责这些修饰的酶并确定其在患者体内的丰度可能是关键,因为抑制胆固醇重吸收是降低胆固醇水平的有效方法。
关于脂质代谢,详见:
多酚是天然植物化合物,是人类饮食中最丰富的抗氧化剂,可以预防慢性退行性疾病。
多酚包括植物源性食物中存在的一类次级代谢产物,当它们作为纯化合物或富含多酚的提取物被纳入饮食中时,可以通过调节肠道微生物群的组成产生健康影响。当每天摄入高达2g时,多酚是重要的膳食生物活性成分。
它们的微生物调节是预期的,它们的化学结构,包括许多酚基团,表明了潜在的抗菌作用。
即使是不可吸收的聚合物化合物也会被肠道微生物群转化为可吸收的生物活性代谢物。
膳食多酚的生物利用度主要取决于肠道微生物群的组成:
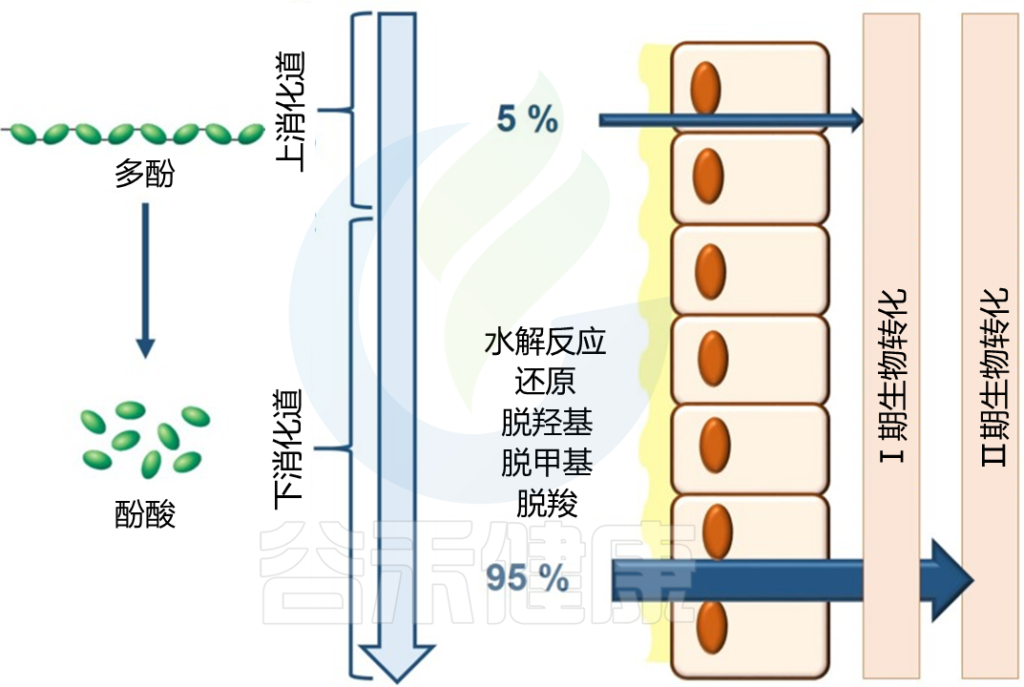
Westfall S, et al., Front Neurosci. 2019
肠道微生物群进行的三个主要分解代谢过程是水解、裂解和还原反应。在这些分解代谢反应之后,释放的苷元可能进行 II 期代谢并被肠道微生物群转化为简单的酚类衍生物,从而促进身体吸收。
关于肠道菌群代谢多酚详见:
近年来,人们越来越关注环境污染物对健康带来的影响。虽然说大多数污染物不直接针对肠道微生物群,但其中一些污染物可以通过不同途径进入人体,并与肠道微生物相互作用。
肠道菌群 ⇔ 污染物
肠道菌群活动可以改变这些化学品的毒性和生物利用度,并延长宿主接触有害化合物的时间。
暴露于环境污染物会改变肠道菌群的组成,导致能量代谢、免疫系统功能、营养吸收和/或产生其他毒性症状。
在评估这些物质的安全性时,必须考虑肠道微生物代谢的后果。这里讨论了几种与人类疾病风险有关的化学物质——重金属、持久性有机污染物和杀虫剂,有证据表明微生物代谢会影响其毒性。
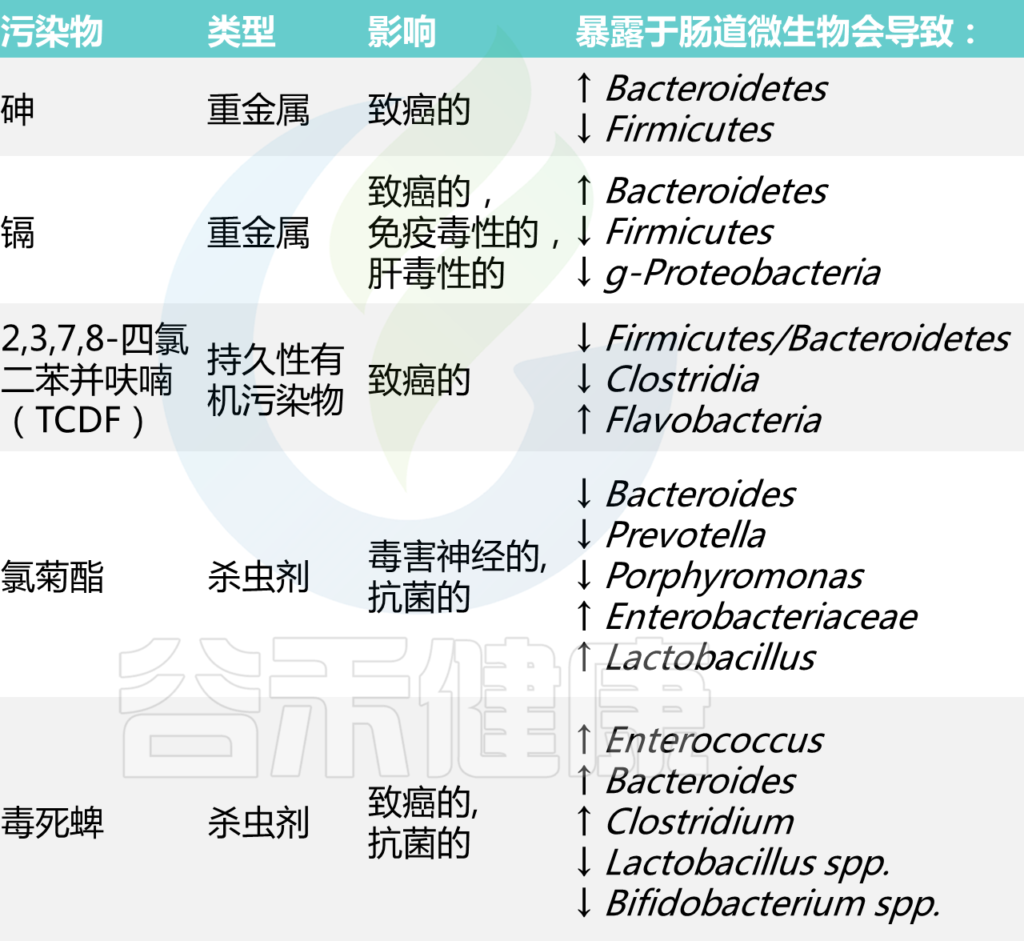
DOI: 10.2174/1389200221666200303113830
环境中的重金属与许多有害影响有关,包括致癌、氧化应激和对免疫系统的影响。几项研究表明,接触重金属也可能导致肠道微生物群失调。我们将以砷、镉、铅为例,展示重金属如何与肠道微生物相互作用,又会带来哪些健康后果。
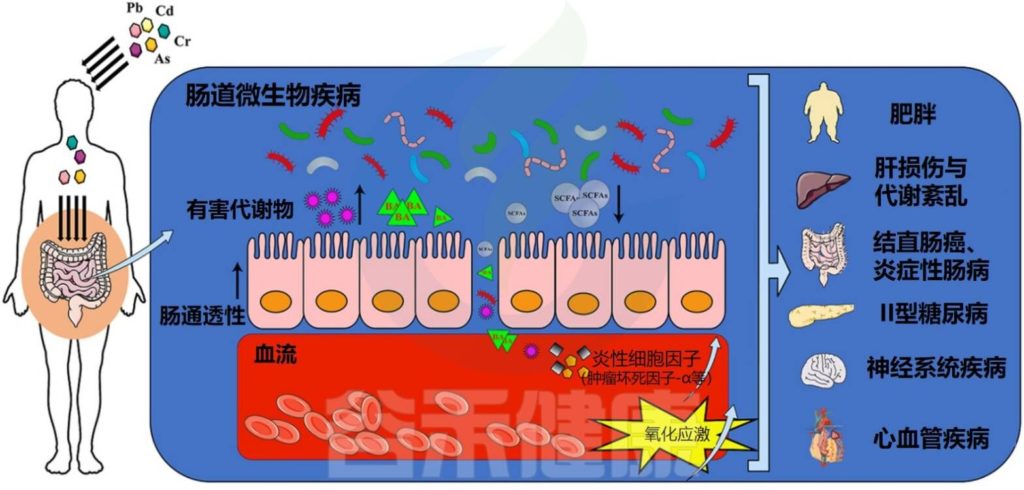
Liu X, et al.,Environ Pollut. 2023
砷(As)
砷(As)是最常见的有毒环境化合物之一。它具有已知的致癌作用。砷能够以砷酸盐[As(V)]的形式附着于固体表面。As(V)摄入并暴露于肠道微生物群后,会诱导硫化和甲基化。
研究发现,小鼠仅4周暴露于10mg/L的砷,就会显著干扰肠道微生物群的组成和代谢组学特征,拟杆菌门的丰度显著增加,而厚壁菌门的丰度则显著减少。这些变化与含吲哚的代谢产物、脂质代谢产物、异黄酮代谢产物和胆汁酸代谢产物有关。
➤砷暴露通过影响菌群及其代谢产物,损伤肝脏,从而影响健康
砷暴露会导致肠道微生物死亡,从而进一步损害肠道中的砷代谢。一旦砷不能代谢出体外,宿主的健康就会受到影响。砷暴露导致的肝脏损伤可能是由于肠道微生物代谢产物(如LPS)的变化所致。这种代谢产物是肝脏损伤的重要指标,并间接激活肠-肝轴。这表明肝脏是砷毒性的主要靶器官之一。
➤砷暴露影响脂质代谢,碳水化合物代谢
砷暴露极大地影响血清和肝脏的脂质稳态,主要影响脂质代谢,脂质代谢失衡与神经系统疾病有关,例如帕金森氏症、阿尔茨海默氏症和肌萎缩侧索硬化症;而肠道菌群在宿主脂质代谢中起着极其重要的作用,因为细菌产生的短链脂肪酸是宿主脂质合成、脂肪酸氧化和脂肪分解稳态的主要调节剂。
扩展阅读:
砷暴露还显着影响碳水化合物代谢途径。在砷暴露小鼠的肠道基因组中,参与淀粉利用系统的基因丰度显著增加。
➤食源性砷暴露不同于水源性砷暴露,会延长砷在胃肠道中的滞留时间
研究发现,小鼠粪便样本中接触30天砷,厚壁菌门和拟杆菌门的比例没有变化,该比率在暴露后 60 天显着降低,并且糖酵解、糖异生和肌醇磷酸代谢等碳水化合物代谢相关基因显著下调。
食源性砷暴露的影响是持久的。砷会导致参与丙酮酸代谢的几种酶水平下降,乙酸激酶和3-羟基丁基辅酶a脱氢酶水平下降,从而导致能量代谢异常。淀粉利用系统中susB、susC、susD和susR基因的增加,虽然缓解了这一情况,但碳水化合物代谢最终受到影响。
镉(Cd)
镉(Cd)用于电池、塑料、金属镀层和颜料的生产。镉以其致癌性、免疫毒性和肝毒性而闻名,它还诱导氧化应激。
研究人员发现雄性小鼠暴露于低剂量的镉(饮用水中为10mg/L)10周后,厚壁菌门和g-变形菌门的丰度降低,盲肠和粪便中拟杆菌门的丰度增加。肠道微生物组组成的这些变化与雄性小鼠血清中脂多糖(LPS)水平升高、肝脏炎症和能量代谢失调有关。
➤镉暴露影响脂肪代谢,引起“肠漏”,减少短链脂肪酸,引发炎症
镉暴露会引起代谢功能的显着变化,影响脂肪代谢并最终导致脂肪堆积。
镉暴露引起的肠道菌群失调会增加 FITC-葡聚糖,FITC-葡聚糖是肠道通透性的重要生物标志物,肠道通透性的增加会改变炎症和新陈代谢。
镉暴露会导致产短链脂肪酸菌数量急剧减少,并降低丁酸和丙酸的含量,短链脂肪酸减少会导致能量代谢受损,也会诱发炎症反应。
铅(Pb)
铅暴露没有明确的阈值。研究发现,血液中任何浓度的铅都会对成人和儿童产生毒性作用。铅中毒可直接影响中枢神经系统、肾脏和血压。
铅通过消化系统在体内蓄积,并对肠道微生物的组成产生显着影响,除了改变肠道多样性、菌群的组成之外,在一定程度上也改变代谢功能。铅对菌群影响主要是,厚壁菌门丰度减少,拟杆菌门丰度增加:
扩展阅读:
铅暴露还会导致某些代谢物和代谢途径发生变化。主要涉及胆汁酸、维生素、氮代谢、氧化应激、防御机制和能量代谢。而且,宿主释放有害代谢产物等变化可能会加剧铅造成的损害。
氮代谢的主要变化是UreE的激活;亚硝酸盐加速向一氧化氮生成,一氧化氮过量可能导致氧化应激、神经毒性、免疫抑制等问题。
注:UreE是一种激活尿素酶的共蛋白,导致尿素酶升高,最终导致肠道细菌中尿素的减少。
重金属暴露后肠道菌群紊乱引起的主要代谢变化
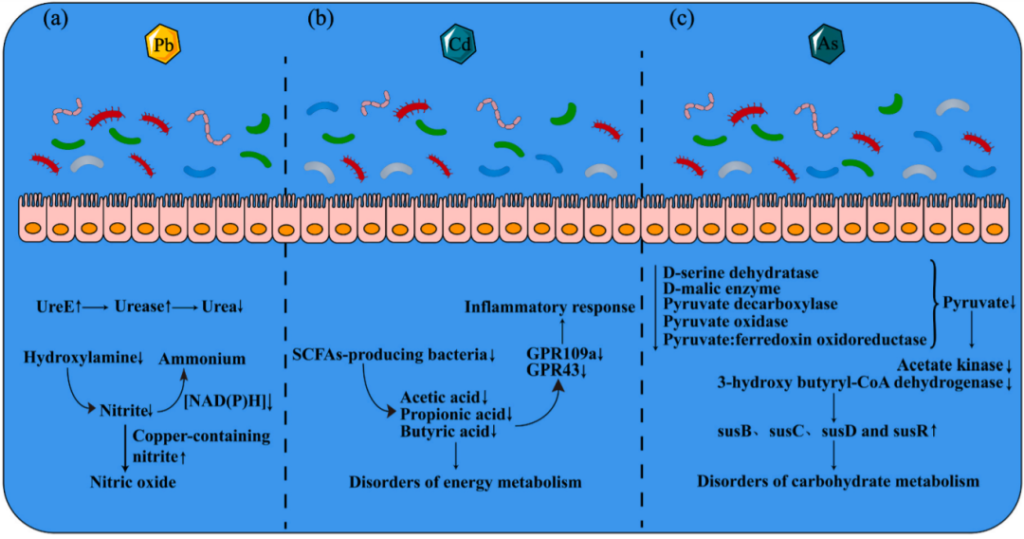
Liu X, et al.,Environ Pollut. 2023
肠道微生物群的研究为评估重金属毒性机制提供了一种新的途径。
持久性有机污染物(POP),包括有机氯农药、多氯联苯(PCB)、多溴二苯醚和多环芳烃(PAHs),是一种持久性和高毒性的合成化合物,可在生物组织中积累,可能与肥胖、糖尿病、自身免疫性疾病和某些发育障碍发病率上升相关。
扩展阅读:
通常,持久性有机污染物在摄入食物或水后会暴露于肠道微生物群,因此需要了解它们对肠道微生物的影响。
多氯联苯(PCB)
多氯联苯具有优异的介电财产和化学稳定性,因此用于制造冷却液、变压器、液压油和润滑剂。多氯联苯被认为是致癌物,可在小鼠暴露2天内显著改变肠道微生物群组成。肠中的产气荚膜梭菌Clostridium perfringens 和贝氏梭菌Clostridium beijerinckii可以降解多氯联苯,通过去除氯原子并打开苯环。
暴露于2,3,7,8-四氯二苯并呋喃(TCDF)(一种PCB)至少5天,可降低厚壁菌门/拟杆菌门的比例,降低梭状芽胞杆菌的水平,并增加盲肠内容物中的黄杆菌Flavobacteria水平。上述肠道微生物群的改变与胆汁酸代谢的变化有关。此外,TCDF还可以抑制法尼素X受体(FXR)信号通路,从而引发细菌发酵导致的宿主的严重炎症和代谢紊乱。
扩展阅读:环境污染物通过肠脑轴影响心理健康,精神益生菌或将发挥重要作用
杀虫剂
在食品材料、水和土壤中检测到多种农药残留,这使得农药成为一个严重的环境问题。一些杀虫剂具有抗菌活性,因此能够改变肠道微生物组的组成。
氯氰菊酯(PEM)
氯氰菊酯(PEM)低剂量用于大鼠,结果降低了拟杆菌、普雷沃氏菌和卟啉单胞菌的丰度,增加了肠杆菌科和乳杆菌的丰度。这些菌群变化可能促进氯氰菊酯的神经毒性。
毒死蜱(CPF)
毒死蜱(CPF)是一种有机磷杀虫剂,通常用于水果、蔬菜和葡萄园。毒死蜱可以通过肝脏或肠道中的细胞色素P450酶代谢。毒死蜱增加了肠球菌、拟杆菌和梭菌的水平,但降低了大鼠肠道中乳酸杆菌和双歧杆菌的水平。
毒死蜱诱导的肠道失调导致粘膜屏障受损,细菌易位增加,并激活先天免疫系统。
以上是重金属等异源物对肠道菌群带来的影响,肠道菌群的变化也会给宿主健康造成影响。基于肠道菌群的层面进行干预,在一定程度上,可以帮助人体免受有害异源物积累带来的健康困扰,这些干预措施包括益生菌,饮食,运动等。
益生菌对重金属的作用
两种重金属解毒策略如下:
✔ 益生菌对重金属诱导的肠道细菌失调的保护
➦ 传统益生菌:
研究表明,鼠李糖乳杆菌(Lactobacillus rhamnosus sp.shermanii JS)和丙酸杆菌(Propionibacterium freudenreichii)能够在体内和体外成功解毒重金属。
有研究发现,含有鼠李糖乳杆菌GR-1 的益生菌降低了孕妇血液中砷和汞的含量。
以下菌株通过肠道重金属隔离和刺激肠道蠕动促进重金属的粪便排泄,从而减少重金属在肠道中的吸收,并逆转重金属引起的肠道菌群变化:
植物乳杆菌TW1-1
植物乳杆菌CCFM8610
乳酸片球菌(Pediococcus acidilactici strain BT36)
蜡样芽孢杆菌
植物乳杆菌CCFM8661
植物乳杆菌CCFM8610
扩展阅读:客观认识植物乳杆菌 (L. plantarum) 及其健康益处
罗伊氏乳杆菌P16
扩展阅读:认识罗伊氏乳杆菌(Lactobacillus reuteri)
短乳杆菌23017
➦ 下一代益生菌:
普拉梭菌Faecalibacterium prausnitzii 是一种可以生产短链脂肪酸能力的人类肠道共生菌,参与微生物的解毒作用。
扩展阅读:肠道核心菌属——普拉梭菌(Faecalibacterium Prausnitzii),预防炎症的下一代益生菌
➦ 重组细菌:
由于一些天然益生菌的不足,重组菌成为重金属污染微生物解毒研究的热点。重组乳酸乳球菌pGSMT/MG1363,产生一种类似于谷胱甘肽 S-转移酶的融合蛋白,具有解毒功能。将重组乳酸乳球菌菌株喂给新生大鼠后,血铅水平显著降低。
Pb结合结构域(PbBD)是已证明具有吸附Pb2+能力的蛋白质之一。通过构建大肠杆菌PbBD并将其引入草鱼体内,结果表明,大肠杆菌PbBD可通过与Pb2+()结合,来减少组织中的Pb积累。
益生菌解毒重金属的机制
与细胞壁上的多糖的金属络合,以降低重金属浓度,通过转运蛋白的重金属离子,与细胞内金属螯合蛋白结合形成沉淀,以及通过酶反应转化为低毒形式等。
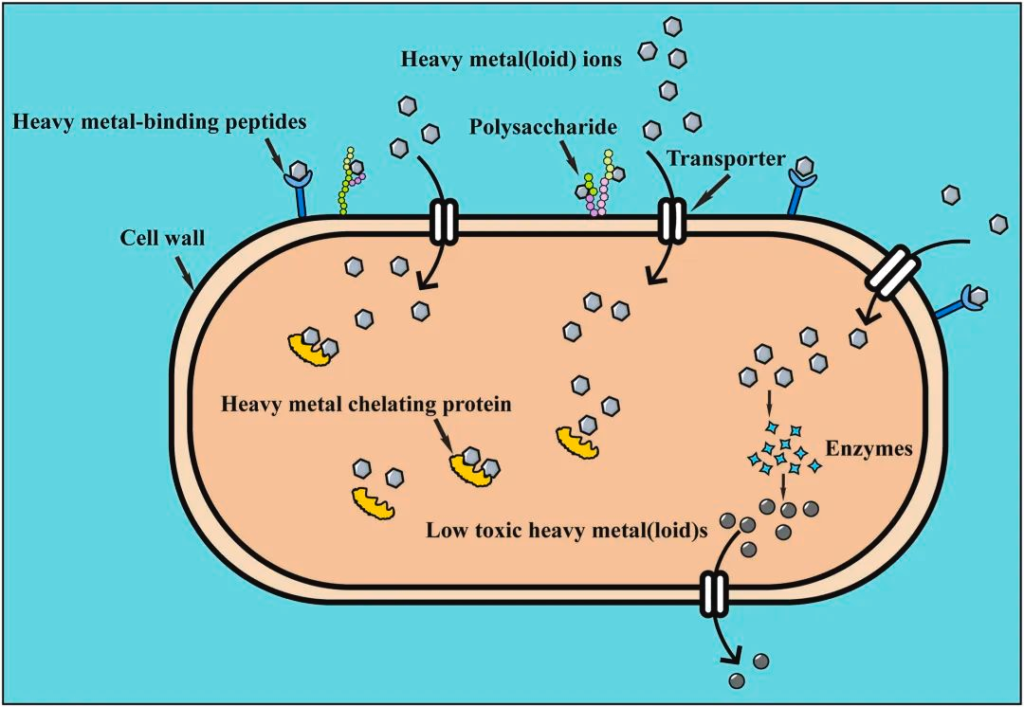
doi.org/10.1016/j.envpol.2022.120780
以上是第一种解毒策略,通过益生菌和重组细菌可以显著减少重金属对肠道微生物的影响,逐渐恢复健康的肠道菌群。接下来看第二张解毒策略:
✔ 肠道菌群防御反应的存在减少重金属的吸收
拮抗菌群的增加,可以减少宿主组织中镉的积累,从而降低发生一系列镉诱导疾病的风险。这种防御反应可能通过增加细胞外磷的浓度来增加铅的沉淀,从而降低肠道中的铅浓度。
——肠道微生物组可以直接影响砷代谢
在暴露于急性砷的小鼠肠道中,第一种反应被发现是一种防御机制,其特征是携带砷抗性基因或参与砷解毒机制的细菌数量增加。
动物研究也表明,肠道细菌通过将砷转化为粪便排泄,限制了砷在宿主体内的积累,降低了砷对宿主的影响。无机砷通过细胞膜上的磷转运蛋白或水通道蛋白进入生物体,并通过生物还原和生物甲基化进一步转化为各种其他形式的砷。
绝大多数无机砷的转化是由肠道菌群进行的,但肠道微生物对砷的处理在肠道的不同位置有所不同:
——肠道菌群抵抗铬吸收,有助于防止铬毒性
肠道微生物中的Cr(VI)耐受细菌,如副乳杆菌CL1107,在体外表现出Cr(VI)的减少,将Cr(IV)还原为Cr(III),其在中性或碱性条件下更难溶解。Cr(III)的低溶解度及其不能透过细胞,增加了粪便中铬的排泄量。
其他可能有用的措施
1、饮食
与排出重金属有关的食物包括:
螺旋藻、香菜、蓝莓等;
富含果胶的食物,如苹果、梨和柑橘类水果;
富含硫的食物,如西兰花、洋葱和大蒜等。
新鲜蔬菜榨汁:
绿叶蔬菜富含叶绿素,叶绿素在身体的解毒过程中非常重要,有助于清除血液中的重金属和毒素。同时,蔬果汁有利于增强肝功能,肝脏能够有效地处理和消除体内的毒素。
2、净化水源
例如:通过滤水器,帮助净化水中的重金属、有毒化学物质和污染物;或者饮用矿泉水等方式。
3、适量运动
出汗是一种自然的身体过程,可以帮助我们清除体内的毒素和废物。确保定期锻炼身体,让身体出汗排毒。
对于身体来说,药物是一种异源物质。身体可能会限制药物在胃肠道的吸收;加剧其在肠道和肝脏中的新陈代谢。
为什么不同人用药效果不一样?
药物明明是用于治病的,为什么可能带来所谓的副作用或者说毒性,甚至更严重后果?
肠道菌群是如何影响药物代谢,从而对身体带来不同影响?
用于神经系统疾病的药物,也与肠道菌群相关?
一个人服用多种药物,为什么会叠加健康风险?
本章节我们用常见的抗炎药,止泻药,癌症药物,神经系统药物,抗病毒药物来举例,从肠道菌群的角度来了解药物代谢的相关问题。
➤ 微生物对前药转化率不同,会影响患者疗效
抗炎药,尤其是针对胃肠道的抗炎药,由肠道细菌直接(化学修饰)或间接(通过微生物群和宿主细胞之间的相互作用)转化。此外,这些药物中的一些作为前药摄入,依赖微生物代谢将非活性前体转化为药物活性化合物。
肠道微生物将柳氮磺吡啶还原为磺胺吡啶后,柳氮磺嗪转化为活性抗炎剂5-ASA,然后进一步代谢为非活性N-乙酰基5-ASA。偶氮还原率和乙酰化率的差异,可以解释柳氮磺吡啶对患者的不同治疗效果。
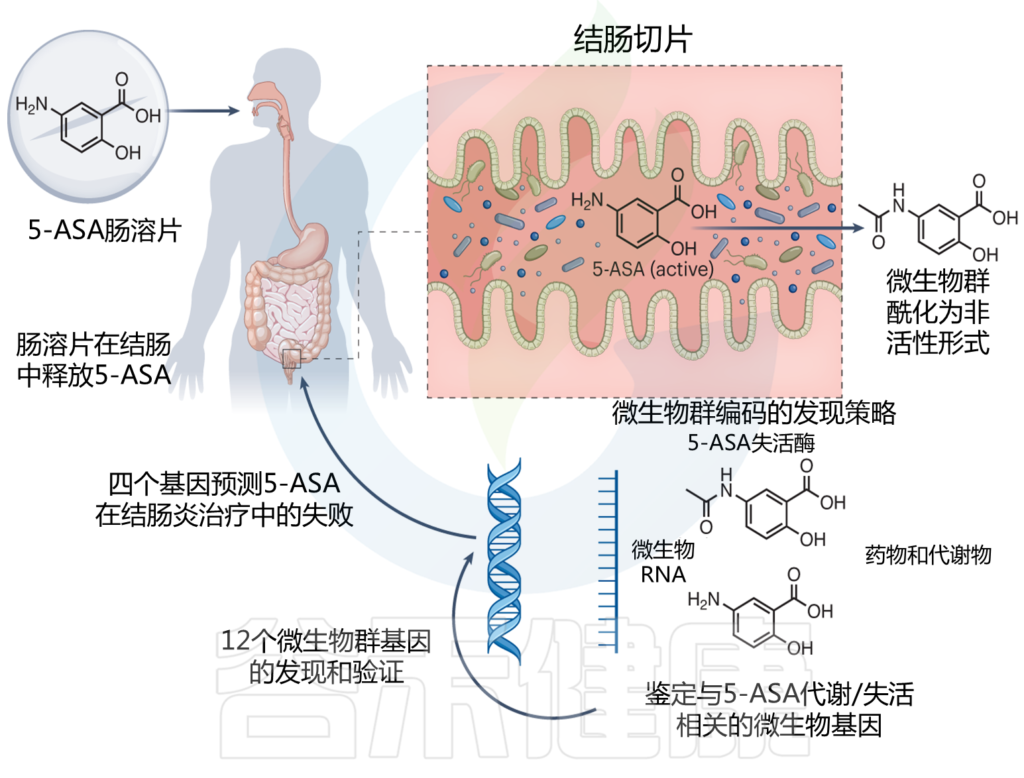
Macpherson AJ, et al., Nat Med. 2023
止泻药,洛哌丁胺的N-氧化物的减少也是肠道微生物活性激活前药的一个例子。
然而,对患者体内负责这些活动的特定生物和酶有更好的理解,有助于管理药物选择和给药等治疗。
个体之间对癌症化疗的治疗反应的一些差异,例如临床疗效和不良反应,也可能是由于肠道微生物群与药物相互作用的现象。肠道细菌可以直接改变化疗药物及其代谢产物的结构,从而改变与宿主细胞的相互作用。
➤ 癌症治疗:药物效果因人而异,可能与肠道菌群或肿瘤相关微生物对药物的结构改变有关
肠道菌群改变常用癌症药物的化学结构。在体外研究中,由于直接的化学修饰,大肠杆菌或魏氏李斯特菌(Listeria welshimeri)等细菌可以提高或降低30种抗癌药物的疗效。大肠杆菌也可以通过类似代谢活动来改变体内化疗的效果。
已知肠道微生物会降低化疗药物吉西他滨的活性浓度。大肠杆菌也可以阻止吉西他滨的疗效,因为它被微生物胞苷脱氨酶转化为其非活性代谢物。有意思的是,胞苷脱氨酶也作为嘧啶挽救途径的一部分存在于人类细胞中。因此,微生物酶在这种抗癌剂代谢中的真正影响仍然不确定。
➤ 神经药物作用于大脑,却也受控于肠道菌群
除了影响局部作用的药物外,肠道菌群代谢还会影响靶向远处器官系统的治疗效果。一些药物,如用于帕金森病治疗的口服左旋多巴,在穿过血脑屏障后,左旋多巴在大脑中被宿主酶局部代谢,以恢复多巴胺水平。然而,在到达大脑之前,药物在肠道中仍有广泛的代谢,导致浓度和患者反应发生显著变化。
一些短乳杆菌菌株,以及粪肠球菌、迟缓埃格特菌(Eggerthella lenta )能够在体外脱羧左旋多巴,但人类中大多数控制左旋多巴转化的肠道微生物和酶尚不清楚,需要更多的研究来揭示肠道菌群与各种神经药物之间的相互作用。
➤ 怎么看待同时服用不同种类药物,死亡风险升高?
拿抗病毒药物举例,如带状疱疹药物索罗夫定。据观察,服用索罗夫定的人,如果同时服用抗癌药物5-氟尿嘧啶,死亡风险会增加,这是为什么?
可能是因为索罗夫定溴戊酸脲的肠道微生物代谢产物减少,使二氢嘧啶脱氢酶失活,这反过来促进5-氟尿嘧啶的积累,导致更显著的毒性。
也可能与不同人群中酶的活性的显著差异有关。因此,同时服用这些药物带来药物不利影响的风险系数较高。
➤ 基于对肠道菌群药物代谢的了解,开发相应的方法提高疗效或减轻不良反应
地高辛主要用于治疗心力衰竭和心房颤动等心脏疾病,但是肠道菌群可以让它失活。地高辛具有狭窄的治疗窗口,用药过量会导致中毒反应,因此在临床上使用地高辛需要仔细监测以防止可能的心脏毒性。
当迟缓埃格特菌(Eggerthalla lenta)转化为没有治疗作用的代谢产物时,心力衰竭和心房颤动的治疗受到损害。
可能的机制与地高辛暴露时,迟缓埃格特菌中双基因细胞色素编码操纵子的上调有关。这种基因表达的蛋白质产物是Cgr1-Cgr2(心苷还原酶)复合物,它可以与地高辛结合并,将其还原为非活性代谢物。
既然我们了解到了这一点,那么有没有什么办法能阻止肠道菌群让地高辛失活?
研究人员认为,高蛋白饮食可以通过精氨酸防止肠道微生物群对地高辛的失活。也就是说饮食控制可以用来改变微生物的药物代谢,因为知道了细菌有独特的营养需求。不过随着对肠道细菌转化药物的机制了解的增加,也可以开发其他治疗方法来提高药物疗效或减轻不良反应。
所有这些研究都强调了揭示药物微生物转化的所有方面的重要性,可以从研究的药代动力学中明显地影响药物的作用。了解异源代谢过程是有必要的,这可能会影响药物的疗效、剂量和避免毒性。
草药在全世界范围内用于预防和治疗各种疾病已有数千年的历史。随着对天然药物的需求不断增长,中草药在世界范围内的重要性正日益增加。
肠道微生物群将中医药化学物质生物转化为代谢物,这些代谢物具有与其前体不同的生物利用度和生物活性/毒性。
接下来,我们从生物利用度和毒性这两方面,来详细了解,肠道菌群在中草药代谢中发挥什么样的作用,如何提高中草药的生物利用度,对毒性产生的正面或负面影响如何,用什么方法可以利用肠道菌群来降低中草药毒性等。
人在服用中草药后可分为两个阶段:
◗ 草药的生物利用度低,容易到达结肠与肠道菌群相遇
在草药中,许多成分的口服生物利用度非常低,因为它们是高极性化合物,亲脂性差。因此血流中的浓度水平有限。比如说,多酚的生物利用度通常低于 10%,人参皂苷的生物利用度可低至0.1%。
由于生物利用度低,这些化合物可以很容易地通过小肠并到达结肠。未被吸收的化合物就不可避免地与肠道菌群接触,并产生相互作用。
◗ 肠道菌群和中草药之间的相互作用是如何发生的?
首先,肠道微生物群与中草药之间的直接相互作用是双向的:
除了直接相互作用之外,中药与肠道菌群之间还会发生间接相互作用。在这类反应中,宿主的免疫和代谢系统起到了连接两端的桥梁作用。
免疫系统可以合成和释放一系列物质,如免疫球蛋白 A 和其他抗菌肽,可以调节肠道菌群组成。吸收的中草药可以调节胃肠道的免疫系统,最终导致肠道菌群发生变化。
比如说,一种中药化合物杜鹃花醇,可以改变牛乳腺上皮细胞中抗菌肽的表达,从而增强对金黄色葡萄球菌感染的防御能力。
◗ 在草药口服生物利用度低的问题中,肠道菌群能发挥作用吗?
出于患者方便和治疗依从性考虑,大多数药物是口服给药的。优化新药开发的主要特性之一是口服吸收度高。
糖苷,如三萜苷和黄酮苷,是许多草药含量中最常见的代表之一。由于氢键增加、糖部分的分子柔性和极性表面积,它们的肠道吸收有限。肠道微生物群可以将这些分子修饰成更亲脂性和极性更小的分子。
◗ 肠道菌群可以根据化合物的结构性质催化一系列代谢反应
草药中的糖苷首先在肠道中通过逐步水解(去糖基化和酯化)代谢。该过程产生的次生糖苷和/或苷元通常具有更好的肠道吸收和更好的生物利用度。它们通过骨架保留修饰(例如三萜苷)、骨架分裂(例如类黄酮苷)或骨架重排(例如环烯醚萜苷)进一步转化。
许多细菌门,其中拟杆菌门和厚壁菌门占优势,编码有丰富的糖苷水解酶基因,因此,它们用来进行糖苷水解。
◗ 不同肠道细菌可以合作处理单个化合物的代谢,单个细菌菌株也能够转化不同的化合物
梭杆菌K-60能使人肠道菌群中的槲皮苷去糖基化,而四种细菌菌株(Pedicoccus Q-5、链球菌S-3、拟杆菌JY-6、双歧杆菌B-9)参与了苷元(槲皮素)的进一步代谢。
丁酸梭菌通过各种反应机制,如脱糖基化、脱水、缩合、分子内环化和/或脱氢,能够转化属于不同结构类型的几种化合物,即紫草素、乌头碱和栀子苷。
然而说到中草药颇有争议,并不是所有人都认可,所谓“是药三分毒”,有人认为它会带来药物性肝损伤,这是一种常见的药物不良反应,可导致肝功能衰竭等问题。
近年来研究表明,中草药可以与肠道菌群相互作用,这种相互作用可以极大地影响其毒性和疗效。
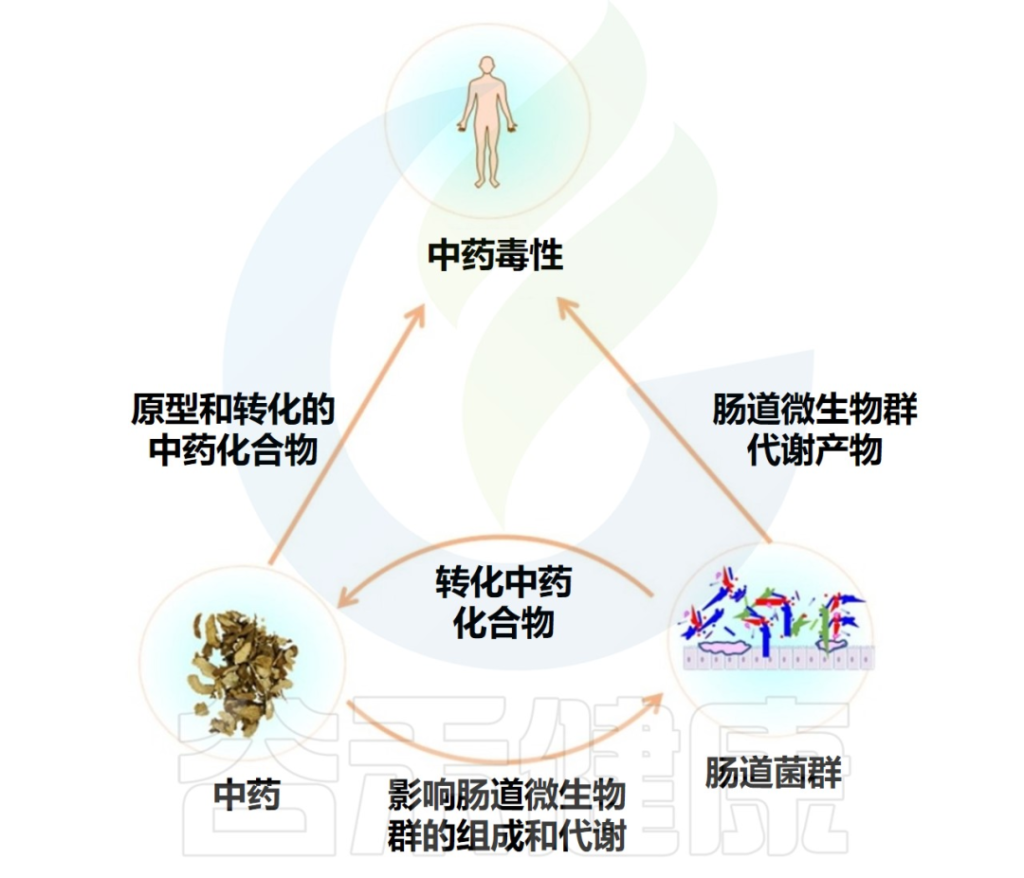
doi.org/10.1016/j.biopha.2020.111047
肠道菌群可以合成和释放一系列具有代谢异源物能力的酶,这些酶可以转化中药化合物,从而直接改变中药的毒性,这种改变包括增加毒性、减少毒性、解毒、激活毒性。
注:这些酶例如,β-葡萄糖醛酸酶、 β-葡萄糖苷酶、 β-半乳糖苷酶、α-鼠李糖苷酶等。
接下来从这四方面展开了解,肠道菌群对中药毒性的直接影响。
► 增加毒性
苦杏仁苷是一种在杏仁中发现的化合物,杏仁是一种用于治疗呼吸系统疾病如支气管炎、肺气肿的中药,但也可能有毒性,表现为恶心、呕吐、胸闷、头晕。
而肠道菌群可以水解苦杏仁苷的糖苷键,从而释放扁桃腈,扁桃腈是一种可以自发分解产生苯甲醛和有毒氢氰酸的化合物。因此,肠道菌群对苦杏仁苷毒性的修饰具有重要作用。
除了苦杏仁苷外,肠道菌群也可以代谢熊果苷、葛根素【与消化链球菌属 (Y-10)有关】、大豆苷【与双歧杆菌属 (K-111)有关】等其他中药成分,产生毒性更强的代谢物。
► 降低毒性
乌头(附子)的侧根是临床广泛用于治疗心力衰竭的中药,但常对神经和心血管系统产生毒性作用。
乌头生物碱是影响附子药理活性和毒性的主要化合物。在临床上有个办法来降低毒性,附子通常经过数小时的煮沸处理,然后再给患者开处方。
在胃肠道中,肠道菌群可以通过脱酰、酯化和去除甲基羟基以类似的方式转化乌头生物碱,参与的菌群与脆弱拟杆菌、肺炎克雷伯菌、丁酸梭菌有关。因此,肠道菌群降低了其毒性。注意肠道的这种减少的程度是有限的,口服大剂量依然会导致急性毒性。
除了乌头生物碱之外,肠道菌群还会对黄芩苷、对羟基苯甲酸丁酯转化为毒性较小的化合物。
► 解毒
这里的排毒,和前面章节的药物代谢机制类似,将活性中药化合物转化为非活性产物。
草酸盐是一种草药化合物,存在于许多中药中,能够诱发高草酸尿症、肾结石和心脏传导障碍。肠道菌群可以在甲酰辅酶 A转移酶和草酰辅酶 A脱羧酶的帮助下,解毒草酸盐。
► 激活毒性
这与前面解毒相反,激活是指肠道菌群将无毒化合物转化为有毒化合物。
苏铁甙是苏铁科植物中的一种偶氮糖苷,具有肝毒性和致癌能力。肠道菌群可以将苏铁甙转化为其苷元(甲氧基甲醇),苷元可以进一步转化为重氮甲烷,一种具有肝毒性和致癌作用的有毒化合物。
中药与肠道菌群的直接相互作用,不仅会导致中药化合物的转化,还会导致肠道菌群的组成和代谢发生变化,从而进一步影响中药的毒性。
以上我们可以看到,肠道菌群对毒性的影响并不是固定的,可能是增加毒性或者减小毒性。那么当发现某些肠道菌群可能会增加中草药毒性时,我们能做些什么尽可能阻止毒性增加呢?
由于肠道微生物群在调节中药毒性方面的重要作用,靶向肠道微生物群已成为控制中药毒性的新前沿。这里总结了靶向肠道菌群降低中药毒性的可能方法。
1、炮制减毒
许多中药在用于临床或制造中药之前要经过煮沸、漂、蒸等特殊处理,也就是“炮制”。
炮制的增效和降毒作用机制是中药现代化的重要问题。研究表明,煮沸等炮制可以将有毒化合物转化为无毒或毒性较小的化合物,从而改变中药的毒性,如前面说的乌头根。
近年来,研究人员证明炮制可以影响肠道微生物群,从而影响中药的药理作用。
甘遂是一种有毒的中药,用于治疗水肿、腹水和哮喘。由于其强烈的毒性,如肝脏和胃肠道毒性,通常用醋煎烤以降低毒性。最近的一项研究表明,醋制甘遂显著减少了甘遂中的10种有毒化合物,如甘遂素B、甘遂素C和甘遂素E。同时,醋制甘遂可以显著改变肠道微生物群的组成,显著提高了短链脂肪酸水平。
注:醋制是是中药炮制中重要的炮制方法。
考虑到肠道微生物群产生的短链脂肪酸具有抗炎和肠粘膜保护作用,可以推断醋制可以调节肠道微生物群的组成和代谢,从而降低甘遂的毒性。
2、中药组合
肠道微生物群释放的β-葡萄糖醛酸酶是一种重要的酶,可以将草药中的糖苷水解为苷元,这一过程可以增加草药化合物和药物的毒性。
非甾体抗炎药和β-葡萄糖醛酸酶抑制剂等化学药物的组合可以降低化学药物的毒性。
一些中药化合物(如黄芩苷)可以抑制细菌β-葡萄糖醛酸酶的活性,其与化学药物(如CPT-11)的组合可以降低CPT-11的毒性。
槲皮素的细菌代谢产物3,4-二羟基苯乙酸可以减少对乙酰氨基酚引起的肝损伤。因此,可以推断,具有抑制细菌酶的能力的中药可以与其他有毒中药或化学药物结合,以降低其毒性。
3、饮食
我们已经知道,饮食在调节肠道微生物群组成和功能中具有重要作用。同样,研究人员也想办法寻找饮食和药物之间的关联。上一章节我们已经了解到,高蛋白饮食可以抑制地高辛失活,并可用于降低某些患者(原本要更高剂量的患者)的地高辛毒性风险。
4、其他(FMT)
其他方法包括粪便微生物群移植(FMT)、益生元和益生菌的补充以及肠道微生物群的工程都有可能降低药物的毒性。
粪菌移植可以显著改善难治性免疫检查点抑制剂诱导的结肠炎,还可以显著改善FOLFOX(一种含有5-氟尿嘧啶、亚叶酸和草酸铂的方案)诱导的大肠癌癌症动物模型中的肠粘膜炎和腹泻。
预防性服用一种益生菌菌株干酪乳杆菌鼠李糖亚种,可显著降低FOLFOX诱导的结肠癌症动物模型中的肠道粘膜炎和腹泻,但不会改变FOLFOX的抗肿瘤作用。
TGF-β阻断剂和大肠杆菌Nisle 1917的组合增强了Galunisertib的肿瘤抑制作用。
肠道微生物群工程是直接治疗疾病和改善药物疗效的新兴前沿,如苯丙酮尿症和左旋多巴。尽管这些方法尚未用于控制中草药的毒性,但可以预见,这些方法将在不久的将来会引入,从而改善中草药毒性。
➭ 肠道微生物组、宿主因素和异源代谢的多方面相互作用是复杂的。微生物异源代谢可导致生物活化、解毒,通过加强肠粘膜屏障来防止吸收。肠道微生物的各种酶反应拓宽了我们对外来生物如何代谢的看法,代谢产物可以发挥新的活性。
➭ 破译异源物的肠道微生物转化,特别重要的是从患者处获取关于饮食或外源性暴露的广泛信息,有助于其与特定健康结果联系起来。
准确评估异源物质引起的肠道微生物群扰动,是将肠道微生物群失调与宿主健康状况联系起来和开发微生物群导向疗法的前提和基础。
➭ 重金属等环境污染物的暴露对肠道微生物群的扰动,会影响代谢和生理功能,部分导致代谢疾病的病因或进展。当然还需要更多深入、系统的研究,来确定重金属等污染物暴露对肠道微生物群的和时间效应。
➭ 了解肠道菌群对药物毒性和疗效的调节作用背后的分子机制,有助于改进药物开发和精准医学,更有效地评估风险,并开发个性化营养。
在不同的临床环境中为个体指导个性化营养膳食及肠道干预,或者制定相应益生菌补充方案,是提高中草药价值的重要研究方向。
将肠道微生物如何转化药物的知识整合到药物开发和实施的所有阶段,将有助于设计合适的治疗方案并改善患者对药物的临床反应。
主要参考文献
Liu X, Zhang J, Si J, Li P, Gao H, Li W, Chen Y. What happens to gut microorganisms and potential repair mechanisms when meet heavy metal(loid)s. Environ Pollut. 2023 Jan 15;317:120780. doi: 10.1016/j.envpol.2022.120780. Epub 2022 Nov 29. PMID: 36460187.
Mehta RS, Mayers JR, Zhang Y, Bhosle A, Glasser NR, Nguyen LH, Ma W, Bae S, Branck T, Song K, Sebastian L, Pacheco JA, Seo HS, Clish C, Dhe-Paganon S, Ananthakrishnan AN, Franzosa EA, Balskus EP, Chan AT, Huttenhower C. Gut microbial metabolism of 5-ASA diminishes its clinical efficacy in inflammatory bowel disease. Nat Med. 2023 Feb 23. doi: 10.1038/s41591-023-02217-7. Epub ahead of print. PMID: 36823301.
Koppel N, Maini Rekdal V, Balskus EP. Chemical transformation of xenobiotics by the human gut microbiota. Science. 2017 Jun 23;356(6344):eaag2770. doi: 10.1126/science.aag2770. PMID: 28642381; PMCID: PMC5534341.
Lindell AE, Zimmermann-Kogadeeva M, Patil KR. Multimodal interactions of drugs, natural compounds and pollutants with the gut microbiota. Nat Rev Microbiol. 2022 Jul;20(7):431-443. doi: 10.1038/s41579-022-00681-5. Epub 2022 Jan 31. PMID: 35102308.
Macpherson AJ, Sauer U. Secrets of microbiota drug metabolism. Nat Med. 2023 Feb 23. doi: 10.1038/s41591-023-02227-5. Epub ahead of print. PMID: 36823300.
Zhang Q, Bai Y, Wang W, Li J, Zhang L, Tang Y, Yue S. Role of herbal medicine and gut microbiota in the prevention and treatment of obesity. J Ethnopharmacol. 2023 Apr 6;305:116127. doi: 10.1016/j.jep.2022.116127. Epub 2023 Jan 2. PMID: 36603782.
Feng W, Liu J, Huang L, Tan Y, Peng C. Gut microbiota as a target to limit toxic effects of traditional Chinese medicine: Implications for therapy. Biomed Pharmacother. 2021 Jan;133:111047. doi: 10.1016/j.biopha.2020.111047. Epub 2020 Dec 4. PMID: 33378954.
Chen F, Wen Q, Jiang J, Li HL, Tan YF, Li YH, Zeng NK. Could the gut microbiota reconcile the oral bioavailability conundrum of traditional herbs? J Ethnopharmacol. 2016 Feb 17;179:253-64. doi: 10.1016/j.jep.2015.12.031. Epub 2015 Dec 23. PMID: 26723469.
Collins SL, Patterson AD. The gut microbiome: an orchestrator of xenobiotic metabolism. Acta Pharm Sin B. 2020 Jan;10(1):19-32. doi: 10.1016/j.apsb.2019.12.001. Epub 2019 Dec 10. PMID: 31998605; PMCID: PMC6984741.
Feng Y, Cao H, Hua J, Zhang F. Anti-Diabetic Intestinal Mechanisms: Foods, Herbs, and Western Medicines. Mol Nutr Food Res. 2022 Jul;66(13):e2200106. doi: 10.1002/mnfr.202200106. Epub 2022 May 11. PMID: 35481618.
Teschke R. Aluminum, Arsenic, Beryllium, Cadmium, Chromium, Cobalt, Copper, Iron, Lead, Mercury, Molybdenum, Nickel, Platinum, Thallium, Titanium, Vanadium, and Zinc: Molecular Aspects in Experimental Liver Injury. Int J Mol Sci. 2022 Oct 13;23(20):12213. doi: 10.3390/ijms232012213. PMID: 36293069; PMCID: PMC9602583.

谷禾健康

在20世纪初,Elie Metchnikoff(著名生物学家,酸奶之父)发现了有益的肠道微生物,该微生物可使肠道健康正常化并延长寿命,后来被称为“益生菌”。益生菌是指“以适当的剂量给予宿主健康有益的活生物体” 。
益生菌如今越来越受欢迎,主要是因为它们对我们整体健康的重要性。益生菌主要存在于人体肠道内,通过维持肠道微生物平衡,在宿主体内发挥有益作用。
不久前发表于《自然•化学》的一项最新研究成果表明,“吃土”也能调节肠道微生物组和治疗肠炎。
究其原因是许多益生菌来源于土壤微生物 (SBO)。细菌在土壤中有着惊人的多样性和丰富性,它们在土壤生态系统中的作用方式与它们在肠道生态系统中的作用相似。
注:该研究中的“吃土”并不是直接食用土壤,而是“人造土”,通过人工合成的方法构建的土壤仿生材料。事实上,自然界土壤中的微生物与肠道菌群中的微生物并不完全相同,且土壤中可能存在有害物质,如重金属、农药等,因此,直接食用土壤可能会引起不必要的健康问题。
考虑土壤微生物的多样性和复杂性,为了让大家更好地了解来源于土壤微生物的益生菌,本文将介绍其中最常见的土壤益生菌及其作用。希望对未来益生菌的选择和恢复肠道微生态提供一定价值。
▼
土壤微生物一词涵盖了土壤中高度多样化的细菌(和其他生命形式),这些细菌在土壤中自然存在。在引入工业化和现代农业之前,我们每天都会定期接触这些细菌。近年来,一些土壤微生物已被分离并用作益生菌。
许多土壤微生物的一个关键特征是它们可以形成孢子。当条件不太有利时,土壤微生物会形成小孢子,这是细菌的一种休眠形式,具有坚硬的保护性外涂层。在这种形式下,细菌对热、酸和大多数抗生素具有高度抵抗力。
土壤微生物和肠道微生物组息息相关
doi: 10.3390/microorganisms7090287
▼
• 通过触发抗体来帮助调节免疫系统;
• 对抗肠易激综合征,调节肠道功能;
• 减轻炎症;
• 治疗腹泻、腹痛和腹胀;
• 帮助消化和营养吸收;
• 防止感染;
• 支持情绪和心理健康;
• 支持平衡肠道微生物群;
• 促进新陈代谢,加速脂肪流失;
• 缓解过敏症状
▼
以下是益生菌中最常用的八种土壤微生物:
Bacillus coagulans(凝结芽孢杆菌)
Bacillus subtilis (枯草芽孢杆菌)
Bacillus clausii(克劳氏芽孢杆菌)
Bacillus indicus (印度芽孢杆菌)
Bacillus licheniformis(地衣芽孢杆菌)
Enterococcus faecium (屎肠球菌)
Enterococcus faecalis(粪肠球菌)
Clostridium butyricum(丁酸梭菌)
注:印度芽孢杆菌是Bacillus属的微生物,原产地为中国。
在下文中介绍了八种来源于土壤微生物的益生菌在人类临床试验中的作用,以确定哪些菌株有较好的治疗前景。
01
凝结芽孢杆菌 (Bacillus coagulans)
凝结芽孢杆菌(Bacillus coagulans)是一种产生L-乳酸的革兰氏阳性细菌。最适生长温度为45-50℃,最适pH为6.6-7.0。它表现出乳酸杆菌科的许多典型特征,但与大多数乳酸菌不同,凝固芽孢杆菌可以形成孢子。
图源:JBMbio
六种凝结芽孢杆菌菌株在人体临床试验中得到充分研究:
✦六种不同菌株显示能够调节肠道微生物群、改善肠易激综合征
GBI-30, 6086
Bacillus coagulans GBI-30, 6086 (“Ganeden BC30”):通过至少10项人类随机对照试验,这是研究最深入的土壤菌株之一。GBI-30、6086已被证明可以改善肠易激综合征 、免疫功能并且可能有益地调节肠道微生物群。
体外研究还表明,它可能有助于果糖和乳糖的消化。
LBSC (DSM 17654)
Bacillus coagulans LBSC (DSM 17654):两项随机对照试验表明该菌株对肠易激综合征患者和急性腹泻或腹部不适患者有益。
MTCC 5856
MTCC 5856(“LactoSpore”):两项随机对照试验表明,该菌株可显著减轻肠易激综合征患者的腹胀、腹痛和抑郁症状。
SANK 70258
Bacillus coagulans SANK 70258 (“Lacris-S”):
一项随机对照试验和一项开放研究发现,该菌株可改善健康成人的大便频率,减少大便次数,且无不良影响。
最近一项使用人体肠道模型系统的研究发现,该菌株抑制了肠杆菌科中的促炎细菌,并增加了健康个体中产丁酸盐的毛螺菌科的丰度。
注:自1966年以来,该菌株已在日本用作食品成分。
SNZ 1969
Bacillus coagulans SNZ 1969:一项安慰剂随机对照试验发现,这种菌株显著改善了结肠转运时间并减少了轻度间歇性便秘人群的肠道不适。它也被证明对细菌性阴道病有效。
另一项使用SNZ 1969以及克劳氏芽孢杆菌和枯草芽孢杆菌的随机对照试验发现,胃肠道不适的成年人的嗳气、腹胀、疼痛和总体症状有所减轻。
Unique IS-2
Unique IS-2(“ProDURA”):四项人类随机对照试验发现该菌株对儿童和成人的细菌性阴道病和肠易激综合征均有效。该菌株还被证明可以在体外代谢果糖。
所有这六种菌株都作为食品成分进行了安全评估,并获得了食品药品监督管理局的“公认安全”(GRAS) 状态。
▸ 小结
这些凝结芽孢杆菌菌株似乎是安全的、耐受性良好的,并且可能对患有细菌性阴道病或肠易激综合征的患者特别有益。
枯草芽孢杆菌(Bacillus subtilis)是一种形成孢子的革兰氏阳性细菌,被认为是人类肠道的正常居民。
枯草芽孢杆菌天然存在于纳豆中,纳豆是一种由发酵大豆制成的日本传统食品。
在引入抗生素之前,枯草芽孢杆菌培养物就已在世界范围内广泛用作免疫刺激剂,以帮助治疗泌尿道和胃肠道疾病。
B.subtilis可以产生一种降解草酸盐的酶,也可能产生少量维生素K2。

图源:eol.org
三种枯草芽孢杆菌菌株已证明对人类有治疗作用,并已通过作为食品成分的安全性测试:
✦三种不同菌株显示能够改善人体健康状况
DE111
Bacillus subtilis DE111:在训练期间对运动员进行的两项随机对照试验发现枯草芽孢杆菌DE111具有良好的耐受性。女运动员的体脂率下降幅度更大;男性运动员发现血液中的炎症标志物减少,对身体机能没有影响。
其他安慰剂随机对照试验发现枯草芽孢杆菌DE111改善了肠道不规律,增加了抗炎免疫细胞群的活性,降低了空腹血糖并降低了健康成人的总胆固醇。
该菌株被食品药品监督管理局公认为安全。
CU1
Bacillus subtilis CU1(“LifeinU”):一项随机对照试验发现,为期10天的B. subtilis CU1补充剂耐受性良好,并改善了老年人的免疫功能(粪便和唾液 中的免疫球蛋白)。
它没有表现出任何抗生素耐药性,并被证明在体外不存在产毒活性。
MB40
Bacillus subtilis MB40 (“OPTI-BIOME”) :
一项随机对照试验发现,该菌株在健康成人中具有良好的耐受性,可降低男性受试者的腹胀强度和胃肠道症状,并适度改善总体健康状况。
该菌株被食品药品监督管理局公认为安全 。
在亚洲对另外两种菌株进行了深入研究:
R0179
Bacillus subtilis R0179(“Medilac-S”):
该菌株存在于亚洲益生菌制剂Medilac-S中,该制剂还含有Enterococcus faecium R0026。几项安慰剂随机对照试验发现,两种菌株的制剂减少了肠易激综合征相关的腹痛,并改善了结肠镜检查的肠道。
最近一项针对临床试验的荟萃分析得出结论,Medilac-S也可有效诱导溃疡性结肠炎的缓解。
两项随机对照试验还发现,分离的枯草芽孢杆菌R0179具有良好的耐受性,并在通过人体胃肠道后存活下来。
TO-A
Bacillus subtilis TO-A (“BIO-THREE”) :
这种枯草芽孢杆菌菌株与E. faecalis T-110和C. butyricum TO-A相结合,作为益生菌在亚洲得到了相当深入的研究。在随机对照试验中,这三种菌株已被证明可以减少手术患者的术后感染并缩短急性腹泻患儿的住院时间。
▸ 小结
通常研究的枯草芽孢杆菌菌株似乎是安全的、耐受性良好的,并且没有不良反应的报道。一些菌株似乎对改善免疫功能和肠道规律性特别有益。
克劳氏芽孢杆菌(Bacillus clausii)是一种形成孢子的革兰氏阳性细菌,以其对金黄色葡萄球菌和艰难梭菌的抗菌活性而闻名。它被认为是一种共生微生物,已从健康个体的小肠和粪便中回收。
只有两种菌株组合已在人类身上进行过研究:
✦两种不同菌株组合显示对过敏、肠易激综合征有益
Bacillus clausii OC、NR、SIN和T
自1958年以来,这四种菌株已被广泛研究并用作药物制剂Enterogermina(美菌纳)。这些菌株已被证明具有酸和胆汁抗性,并且在人类单次口服给药后可在粪便中恢复4-12天。
这些菌株可能对那些有过敏症、减少鼻塞和抗组胺药需求的人特别有益。
它也被证明可以减少幽门螺杆菌抗生素治疗期间的副作用。
一项针对40名诊断为“小肠细菌过度生长”患者的研究发现,服用美菌纳一个月可使 47%的患者的葡萄糖呼气试验结果恢复正常。
值得注意的是,这个比率与许多用于小肠细菌过度生长治疗的抗生素所见的正常化率相当。只有一名患者报告了便秘的副作用。
尽管美菌纳有许多记录在案的益处和总体安全性,但仍有零星的克劳氏芽孢杆菌败血症病例报告。大多数报告都是在老年人、重病患者或免疫功能低下的个体中使用益生菌。
Bacillus clausii UBBC-07
在一项针对印度急性腹泻儿童的安慰剂对照随机试验中,该菌株被证明可以改善粪便稠度并缩短腹泻持续时间。该菌株经过了毒理学研究,被确定可供人类安全食用。
▸ 小结
克劳氏芽孢杆菌已在许多随机、安慰剂对照试验中证明了疗效,并且可能对过敏或肠易激综合症患者特别有益。
脓毒症的个别病例报告表明,老年人、重病或免疫功能低下的人以及最近服用广谱抗生素的人可能禁忌使用它。
印度芽孢杆菌(Bacillus indicus)是一种形成孢子的细菌,因其能够有效刺激免疫系统并产生高水平的类胡萝卜素、维生素和喹啉而受到吹捧。
✦暂未有毒性或致病性的发现
体外和动物研究未能发现B.indicus HU36的任何潜在毒性或致病性,该菌株是益生菌中最常用的菌株,并且没有关于人类感染的报道。
▸ 小结
关于人类印度芽孢杆菌的科学研究,目前还缺乏人类证据。
地衣芽孢杆菌(Bacillus licheniformis)是一种革兰氏阳性、产芽孢的细菌,通常用于工业酶生产。
在所有基于土壤使用的益生菌中,地衣芽孢杆菌可能是最具争议的。
✦可能具有一定毒性
根据一些报道,地衣芽孢杆菌可能是一种机会性病原体,可导致免疫功能低下的宿主感染。
然而,其他报告得出的结论是,地衣芽孢杆菌的毒力非常低。地衣芽孢杆菌存在于韩国传统消费的许多发酵食品中,并且已从健康人类志愿者的胃肠道中分离出来,因此至少某些菌株似乎是共生的。
虽然已经出现了与大量该物种的分离相关的急性自限性胃肠炎病例, 对肠上皮细胞的毒性或直接影响尚未得到证实。很难确定这些报告病例中的物种(数量相当有限)是否积极参与了感染,或与一种不明病原体一起分离。
只有四项关于分离的地衣芽孢杆菌的人体研究,没有一项提供菌株信息,并且只有一项是随机和安慰剂对照的。
▸ 小结
虽然含有地衣芽孢杆菌的传统发酵食品几乎是安全的,但目前没有足够的证据支持大剂量补充地衣芽孢杆菌。最好谨慎行事,避免使用含有该物种的益生菌。
✦发酵碳水化合物
Enterococcus faecium是革兰氏阳性菌。虽然它不形成孢子,但它可以耐受多种环境条件。E. faecium是人类肠道的正常居民,发酵碳水化合物并产生乳酸作为副产品。
粪肠球菌的一些菌株可以在传统发酵食品中找到。
by Dennis Kunkel Microscopy
✦可能致病
然而,粪肠球菌也可能致病,引起新生儿脑膜炎和心内膜炎等疾病。这种细菌的许多菌株已经产生了抗生素抗性和毒力因子,使其能够聚集并形成生物膜。
在美国,80-90%的医疗器械相关感染可归因于耐抗生素粪肠球菌。因此,人们担心将其用作益生菌。
已经研究了许多不同的菌株,但只有两种菌株值得强调,它们已经在多项人体试验中进行了研究:
M-74
Enterococcus faecium M-74:该菌株有许多与之相关的“随机”临床试验,但每一个菌株都服用益生菌和50微克硒。
许多益生菌指出这些研究表明粪肠球菌可以降低胆固醇,但缺乏硒对照组意味着硒单独提供治疗效果是完全合理的。
R0026
Enterococcus faecium R0026 (“Medilac-S”):
该菌株存在于亚洲益生菌制剂Medilac-S中,该制剂还含有枯草芽孢杆菌R0179,并在国内得到了很好的研究。
如前面所述,多项随机对照试验发现,双菌株制剂可减少肠易激综合征相关的腹痛,并改善结肠镜检查的肠道准备,还可以有效诱导溃疡性结肠炎的缓解。
▸ 小结
除了Medilac-S中的菌株外,很少有精心设计的随机对照试验证明了粪肠球菌的功效。
鉴于其可能获得多重抗生素耐药性并引起感染。谨慎使用高剂量的分离粪肠球菌。
Enterococcus faecalis是一种革兰氏阳性菌。与Enterococcus faecium类似,它不能形成孢子,但对恶劣的环境条件相当耐受。
✦在肠道免疫中起重要作用
粪肠球菌是人类肠道的正常居民,存在于大约90-95%的人中。这种细菌通常是生命早期定植于人类胃肠道的首批微生物之一,在肠道免疫系统的发育中发挥着重要作用。
T-110
Enterococcus faecalis T-110:这种粪肠球菌菌株与枯草芽孢杆菌TO-A和丁酸梭菌TO-A结合使用作为益生菌(BIO-THREE)。这三种菌株一起可以减少手术患者的术后感染并缩短急性腹泻患儿的住院时间。
YM0831
Enterococcus faecalis YM0831:一项交叉研究发现,对健康人类受试者单次施用该菌株可显着改善血糖对蔗糖耐量试验的反应。
▸ 小结
除了BIO-THREE中的菌株外,很少有精心设计的随机对照试验证明了Enterococcus faecalis作为益生菌的功效,还需要更多的研究。
✦丁酸梭菌是肠道重要的菌属
丁酸梭菌(Clostridium butyricum)是人类胃肠道的天然居民。它是肠道中发酵膳食纤维并产生有益的短链脂肪酸丁酸盐的众多细菌之一。丁酸盐作为结肠上皮细胞的能量来源,有助于维持肠道屏障,具有抗炎作用,并可预防结肠癌。
doi.org/10.1007/s00535-015-1084-x
三种菌株已在人体中得到充分研究:
✦三种不同菌株显示能够调节肠道稳态、减轻肠炎
CBM 588
Clostridium butyricum CBM 588:这种丁酸梭菌菌株早在1963年就用作益生菌,用于一种名为Miyairisan的药物制剂中。它已经过安全性评估,不携带任何编码任何已知毒素或毒力因子的基因。
两项随机对照试验发现,该菌株可减少接受幽门螺杆菌根除治疗的患者的腹部症状和腹泻发生率的副作用。
另一项针对溃疡性结肠炎患者的安慰剂对照随机对照试验发现,接受MIYAIRI 588治疗的受试者中,患上肠结肠炎的人数较少。
尽管其影响在统计学上并不显著,而安慰剂组则服用乳糖,这会加重许多人的结肠炎。
TO-A
Clostridium butyricum TO-A:这种丁酸梭菌菌株与粪肠球菌T-110和枯草芽孢杆菌TO-A结合使用,作为益生菌在亚洲得到了广泛使用和深入研究。
A Tai Ning
该菌株包含在一种名为阿泰宁的产品中,最近在一项大规模、多中心、的随机对照试验中显示,该菌株对以腹泻为主的肠易激综合征具有显著益处。
该研究包括200名患者。4周后,与安慰剂组相比,益生菌组在整体肠易激综合征症状、大便频率和生活质量方面有显著改善。
几项研究发现丁酸梭菌降低了早产儿败血症和腹泻的发生率,并改善了抗原特异性免疫治疗。在动物研究中,丁酸梭菌已被证明可以改善肠道稳态并减轻结肠炎。
▸ 小结
丁酸梭菌是肠道重要的菌属,在随机试验中似乎是安全且耐受性良好的,并且帮助增加丁酸盐水平的同时,极少引起毒性。
➣土壤基益生菌的安全性仍存在争议,值得进一步关注
支持者声称它们是益生菌,可以使肠道功能正常化、帮助消化、有益地刺激免疫系统并帮助重新播种肠道微生物群。他们还称赞土壤微生物具有抵抗胃酸的能力并且不需要冷藏。
反对者认为,由于它们形成孢子的性质,它们会迅速增殖,与我们常驻的肠道微生物竞争,在某些情况下,它们甚至可能致病。由于它们的孢子形成能力和对大多数抗生素的天然抗性,意外的过度生长将很难治疗。
在随机、安慰剂对照的人体临床试验中,许多单独的土壤微生物菌株已被证明是有益的,几乎没有报告不良反应。
然而,其他菌株的临床证据有限或没有临床证据,可能会导致免疫系统受损的人感染。我们需要进行更细致的讨论——考虑每个特定物种、菌株和配方,而不是广泛地给土壤微生物贴上好或坏的标签。
基于土壤的微生物得到了很好的研究,并且已经在随机临床试验中显示出对一些症状有效,但仍需更多研究来确定其安全性和有效性。建议选择使用高质量和经过临床测试的菌株,在服用益生菌制剂前最好咨询医生的建议。
每个人对益生菌的反应都不同。反应将取决于服用的益生菌、剂量、肠道中已有的微生物以及肠道和免疫系统的健康状况。
可以通过微生物测序技术来检测使用益生菌前后的肠道菌群组成及相关健康状况。再选择针对自身特定健康状况的益生菌菌株。
同时也期待更多关于土壤微生物的单个菌株和配方在不同情况下影响肠道微生物群和肠道环境的研究。
注:本账号内容仅作交流参考,不作为诊断及医疗依据。
主要参考文献:
Mu Y, Cong Y. Bacillus coagulans and its applications in medicine. Benef Microbes. 2019 Jul 10;10(6):679-688. doi: 10.3920/BM2019.0016. Epub 2019 Jun 17. PMID: 31203635.
Jäger R, Purpura M, Farmer S, Cash HA, Keller D. Probiotic Bacillus coagulans GBI-30, 6086 Improves Protein Absorption and Utilization. Probiotics Antimicrob Proteins. 2018 Dec;10(4):611-615. doi: 10.1007/s12602-017-9354-y. PMID: 29196920; PMCID: PMC6208742.
Acosta-Rodríguez-Bueno CP, Abreu Y Abreu AT, Guarner F, Guno MJV, Pehlivanoğlu E, Perez M 3rd. Bacillus clausii for Gastrointestinal Disorders: A Narrative Literature Review. Adv Ther. 2022 Nov;39(11):4854-4874. doi: 10.1007/s12325-022-02285-0. Epub 2022 Aug 26. PMID: 36018495; PMCID: PMC9525334.
Ianiro G, Rizzatti G, Plomer M, Lopetuso L, Scaldaferri F, Franceschi F, Cammarota G, Gasbarrini A. Bacillus clausii for the Treatment of Acute Diarrhea in Children: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Nutrients. 2018 Aug 12;10(8):1074. doi: 10.3390/nu10081074. PMID: 30103531; PMCID: PMC6116021.
Kovács ÁT. Bacillus subtilis. Trends Microbiol. 2019 Aug;27(8):724-725. doi: 10.1016/j.tim.2019.03.008. Epub 2019 Apr 15. PMID: 31000489.
Marzorati M, Van den Abbeele P, Bubeck S, Bayne T, Krishnan K, Young A. Treatment with a spore-based probiotic containing five strains of Bacillus induced changes in the metabolic activity and community composition of the gut microbiota in a SHIME® model of the human gastrointestinal system. Food Res Int. 2021 Nov;149:110676. doi: 10.1016/j.foodres.2021.110676. Epub 2021 Aug 30. PMID: 34600678.
Kawarizadeh A, Pourmontaseri M, Farzaneh M, Hosseinzadeh S, Ghaemi M, Tabatabaei M, Pourmontaseri Z, Pirnia MM. Interleukin-8 gene expression and apoptosis induced by Salmonella Typhimurium in the presence of Bacillus probiotics in the epithelial cell. J Appl Microbiol. 2021 Jul;131(1):449-459. doi: 10.1111/jam.14898. Epub 2020 Dec 7. PMID: 33058340.
Muras A, Romero M, Mayer C, Otero A. Biotechnological applications of Bacillus licheniformis. Crit Rev Biotechnol. 2021 Jun;41(4):609-627. doi: 10.1080/07388551.2021.1873239. Epub 2021 Feb 16. PMID: 33593221.
Gudiña EJ, Teixeira JA. Bacillus licheniformis: The unexplored alternative for the anaerobic production of lipopeptide biosurfactants? Biotechnol Adv. 2022 Nov;60:108013. doi: 10.1016/j.biotechadv.2022.108013. Epub 2022 Jun 22. PMID: 35752271.
Gök ŞM, Türk Dağı H, Kara F, Arslan U, Fındık D. Klinik Örneklerden İzole Edilen Enterococcus faecium ve Enterococcus faecalis İzolatlarının Antibiyotik Direnci ve Virülans Faktörlerinin Araştırılması [Investigation of Antibiotic Resistance and Virulence Factors of Enterococcus faecium and Enterococcus faecalis Strains Isolated from Clinical Samples]. Mikrobiyol Bul. 2020 Jan;54(1):26-39. Turkish. doi: 10.5578/mb.68810. PMID: 32050876.
Stoeva MK, Garcia-So J, Justice N, Myers J, Tyagi S, Nemchek M, McMurdie PJ, Kolterman O, Eid J. Butyrate-producing human gut symbiont, Clostridium butyricum, and its role in health and disease. Gut Microbes. 2021 Jan-Dec;13(1):1-28. doi: 10.1080/19490976.2021.1907272. PMID: 33874858; PMCID: PMC8078720.
Chen D, Jin D, Huang S, Wu J, Xu M, Liu T, Dong W, Liu X, Wang S, Zhong W, Liu Y, Jiang R, Piao M, Wang B, Cao H. Clostridium butyricum, a butyrate-producing probiotic, inhibits intestinal tumor development through modulating Wnt signaling and gut microbiota. Cancer Lett. 2020 Jan 28;469:456-467. doi: 10.1016/j.canlet.2019.11.019. Epub 2019 Nov 14. PMID: 31734354.

谷禾健康

肠道微生物组与脂质代谢:超越关联
脂质在细胞信号转导中起着至关重要的作用,有助于细胞膜的结构完整性,并调节能量代谢。
肠道微生物组通过从头生物合成和对宿主和膳食底物的修饰产生了大量的小分子。
最近的研究表明,由肠道微生物组从头生物转化和生物合成的脂质具有重要的结构和信号功能,可通过代谢和免疫途径影响宿主细胞。肠道微生物群既可以转化和合成脂质,也可以分解膳食脂质,生成具有宿主调节特性的次级代谢产物。
微生物来源的脂质可以被宿主直接感知,从而调节先天性和适应性免疫途径,并调节代谢途径,所有这些都可以影响慢性炎症、自身免疫性疾病、心血管疾病和代谢综合征的进展。
本文基于对宿主和微生物组如何作为一个完整社区相互作用的思考,从更精细的层面介绍了肠道细菌衍生的脂质在生理学中的功能,特别强调免疫和新陈代谢。
▼ 脂质调节生物学功能
脂质是细胞膜的主要结构成分。作为能量储存分子,它们储存的能量几乎是从蛋白质或碳水化合物分解代谢中释放的能量的两倍。
脂质调节许多基本的生物学功能,包括细胞内信号传导过程。
例如,鞘脂 (SP),尤其是神经酰胺,在调节细胞信号和细胞凋亡中发挥作用。
其他脂质,例如甘油二酯 (DG),充当能量代谢的中间体和信号分子。
总的来说,脂质代谢在多个层面表现出空间和动态的复杂性。因此,脂质紊乱具有影响人类健康的重要生理后果也就不足为奇了 。
人体肠道中存在代谢活跃的微生物群,它们对脂质的吸收、消化、代谢和排泄具有深远影响,甚至会改变宿主的代谢状态。
▼ 肠道菌群和循环脂质密切相关
在人类中,肠道微生物组的多样性与 BMI 和血清甘油三酯 (TG) 呈负相关。妊娠期糖尿病合并症与特定的肠道微生物群组成和循环脂质组密切相关。其中Faecalibacterium和Prevotella的相对丰度显示与循环脂质有关,特别是与溶血磷脂酰乙醇胺和磷脂酰甘油有关。
膳食脂质和肠道菌群之间的相互作用会影响宿主生理
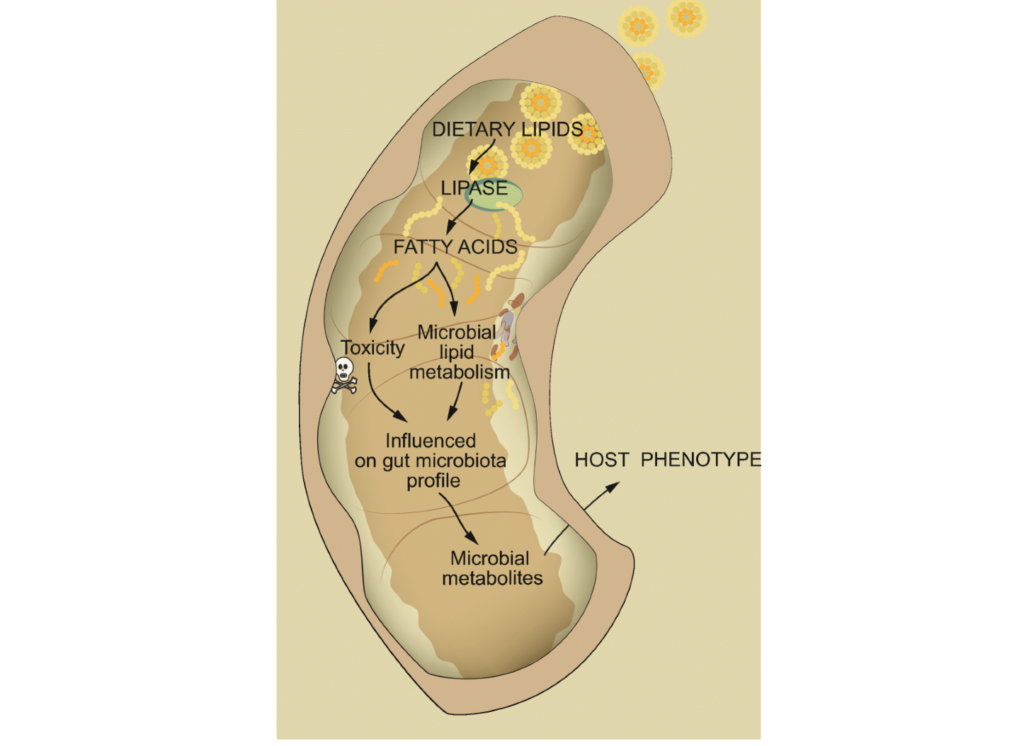
doi: 10.1007/s11154-019-09512-0.
▼ 饮食改变代谢状态,改变菌群
富含阿拉伯木聚糖的饮食改变了宿主的代谢状态,包括神经酰胺和胆碱水平,这随后影响了肠道中的普氏菌属和梭菌属。
▼ 肠道菌群可以调节宿主的脂质组
接受老年小鼠粪便细菌的小鼠表现出总单不饱和脂肪酸增加,以及大脑皮层中胆固醇和总多不饱和脂肪酸的相对量减少。
将微生物群从老年小鼠转移到年轻小鼠,会改变不同脂质类别的相对丰度和肝脏的脂肪酸含量。
在添加或不添加猪油或棕榈油的情况下,与棕榈油相比,猪油 + 富含含初级胆汁酸的饮食增加了定殖小鼠的脂肪量,但无菌小鼠的脂肪量没有增加。随后,这些影响与葡萄糖耐量受损和移居小鼠肝脏中甘油三酯、胆固醇酯和单不饱和脂肪酸升高有关。
长期以来,人们一直在研究细菌脂质在以下方面产生重要作用:
每种细菌的脂质特征都是独一无二的,反映了基因编码的生物合成机制和细菌的生活方式;然而,关于肠道细菌脂质生物合成的大部分知识都来自对大肠杆菌(Escherichia coli)的研究。
迄今为止,我们对细菌膜中发现的主要脂质类别的理解依赖于对少量模型细菌生物体的研究。
这些主要类别包括磷脂,例如:磷酸乙醇胺 (PE)、磷酸丝氨酸 (PS)、磷酸胆碱 (PC)、磷酸肌醇 (PI) 和磷酸甘油 (PG);甘油脂,例如甘油二酯(DAG)和甘油三酯(TAG);和心磷脂 (CL)。
还有糖脂,例如:脂多糖 (LPS),它们是连接有多种头基(例如糖)的大酰化脂质部分(例如脂质 A)。
其他脂质类别是特定细菌门或类群的特征。这些包括鞘脂,主要由共生拟杆菌菌株合成,如鞘氨醇、二氢神经酰胺 (DHCer)、神经酰胺磷酸乙醇胺 (CerPE)。
厌氧肠道微生物组的一个子集合成缩醛磷脂;然而,这些并不局限于特定的分类群,而是由拟杆菌门和厚壁菌门的不同成员产生的。
在一些肠道细菌中也发现了磺酸脂,包括拟杆菌属(Bacteroides)、Flavobacterium strains、Alistipes。每种脂质类别都有独特的结构,因此赋予细菌膜不同的结构特征和功能。
许多也是可以被宿主模式识别受体感知的信号分子,例如 Toll 样受体 (TLR)、NOD 样受体 (NLR)、C 型凝集素受体 (CLR) 和 G 蛋白偶联受体 (GPCR)。
细菌能够合成的脂质种类繁多。
最近人们才才刚刚开始意识到肠道微生物组的这种多样性,因为通过代谢组学技术在粪便中检测到的大部分脂质仍未注释且功能未知。
正在进行大量工作以生成培养的微生物组菌株的脂质组学概况,并将以前的结构数据与质谱相结合的脂质类别拼凑在一起。许多这些努力都围绕着宿主脂质注释,但最近已经开始解决细菌群落脂质组中的未知数。
在接下来的部分中,我们将讨论肠道微生物组中发现的主要脂质类别,并举例说明它们在宿主信号传导中的作用。
肠道微生物群生物合成的膜脂及其已知的宿主信号传导功能
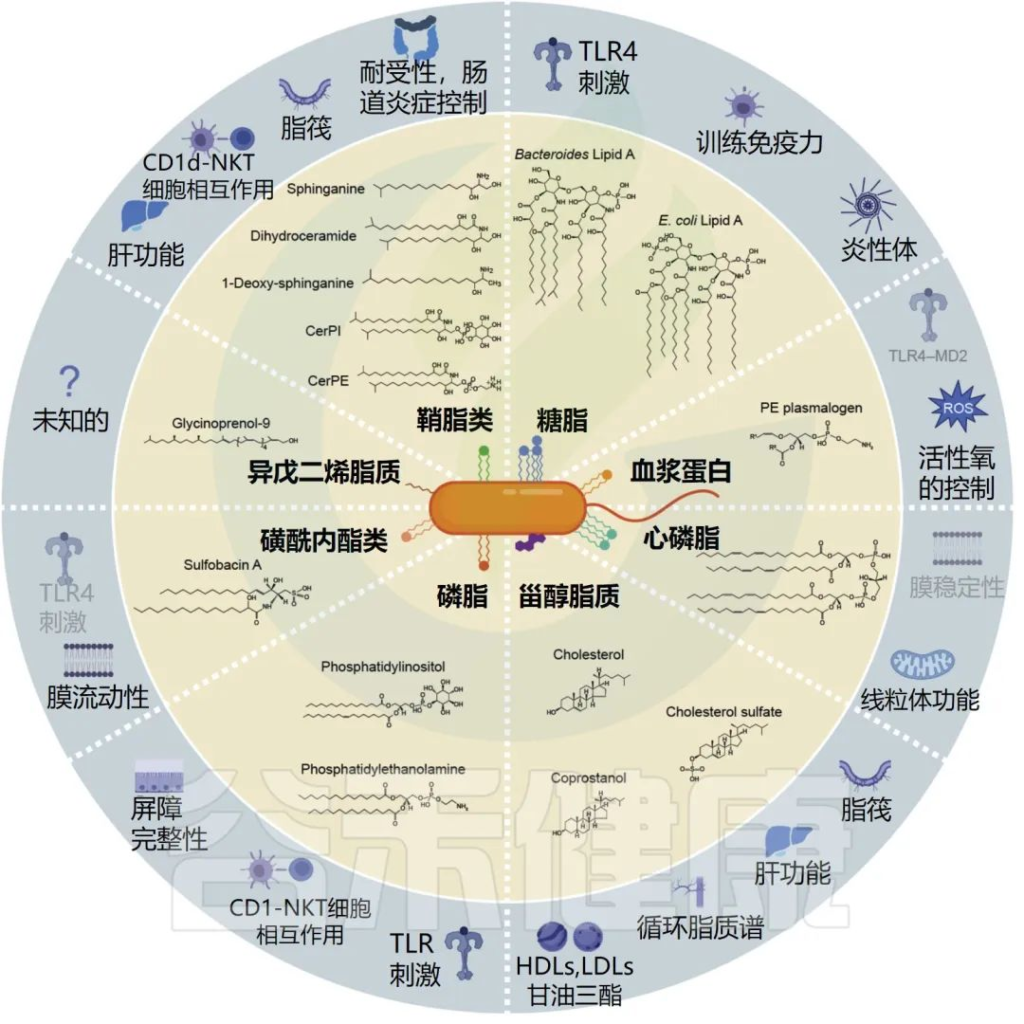
Brown et al.Cell Host Microbe, 2023.
细菌磷脂具有高度多样性,是从经过充分研究的细菌的细胞膜中回收的最丰富的脂质。它们的生物合成途径在模式生物大肠杆菌中已基本得到解决。
PE(磷酸乙醇胺) 是已知由肠道细菌广泛合成的主要膜磷脂。对宿主细胞微生物组代谢物的筛选尚未发现能够刺激细胞反应的多种磷脂。例如,大型 GPCR 筛选未能将微生物组磷脂识别为孤儿或已知受体的激动剂。
然而,最近的一项研究利用生物活性引导的分馏方法来筛选由常见肠道细菌Akkermansia muciniphila 产生的免疫调节分子,并鉴定出一种具有两条支链 (a15:0-i15:0 PE) 的二酰基磷脂酰乙醇胺,可激活模式识别由 TLR2-TLR1 组成受体 (PRR) 异二聚体。
与常见的 TLR2 激动剂相比,这种相互作用具有独特的信号特性,包括抑制人单核细胞中的促炎性 IL-12/IL-23 反应,这暗示了 Akkermansia 对人体生理学已知影响的分子机制。
▼ 细菌严格控制其膜磷脂的特性,使其广泛生长
细菌可以严格控制其膜磷脂的生化和生物物理特性,使它们能够在广泛的环境中茁壮成长。这包括改变酰基链长度和异构支化的能力,以及改变不饱和度和多样化头部基团的能力。这种响应肠道环境压力的修饰如何影响宿主免疫系统的信号传递能力尚不清楚。收集到的关于真核脂质的数据表明,异分支可以决定与蛋白质受体的结合强度。
对病原体的研究表明,微生物脂质的氧化、异构分支和饱和会影响宿主受体的识别。
▼ 肠道菌群影响细胞膜特性,膜磷脂变化导致肠道通透性增加
无菌小鼠宿主细胞中的磷脂水平与常规小鼠宿主细胞中的磷脂水平不同,这表明肠道微生物组也会影响哺乳动物细胞膜的特性。
膜磷脂化学的变化会导致肠道通透性增加,从而使细菌在宿主体内传播,从而产生许多病理后果。未来的研究需要解决微生物组合成的磷脂在肠道屏障功能中的潜在作用。
▼ 细菌磷脂可以调节免疫力,被自然杀伤 T 细胞识别
通过对病原体的研究中得出一些结论,即细菌磷脂会调节免疫力。结核分枝杆菌和李斯特菌磷脂,通常是 PG,被自然杀伤 T 细胞(NKT 细胞)识别,通过与呈递脂质抗原的非经典 MHC 分子 CD1d 结合。
共生磷脂是否在 CD1 信号转导或呈递给 T 细胞中具有相似的作用尚不清楚,微生物与宿主磷脂的 CD1 加载的生化决定因素也是如此。
▼ 肠道菌群可以合成磷酸肌醇
最近的研究已经确定肠道细菌可以合成 PI(磷酸肌醇),这是一种以前仅归因于真菌的能力。
鉴于 PI 在细菌和植物先天免疫之间的相互作用以及自噬中的肌醇信号传导中的作用,可以合理地假设这些脂质具有宿主特异性作用。作为真核细胞膜的一个小组成部分,肌醇信号对许多与细菌传感相关的重要生物信号通路有很大影响,包括 GPCR 信号。
糖脂是一种细菌糖脂,其中脂肪酸与糖骨架共价连接,以几何定义的方式定位酰基链。这些结构可以存在于膜双层中,并且在革兰氏阴性细菌中普遍存在,最典型的是 LPS(脂多糖)。
▼ 合成脂多糖 A 部分的生物合成机制可变
糖脂不是由真核生物产生的,因此被先天免疫系统识别为 MAMP(微生物相关分子模式)。在经典观点中,细菌 LPS 激活宿主细胞上的 PRR TLR4 以启动促炎反应。然而,激活取决于脂质 A 组分的酰基链结构,最近的研究表明,与深入研究的大肠杆菌LPS 相比,不同细菌的脂质 A 结构发生了变化。这种多样性源于用于合成脂多糖 A 部分的可变生物合成机制,并影响宿主感知代谢物的方式。
从拟杆菌属的微生物组菌株中分离出的 LPS可以通过 TLR2 和 TLR4 发出信号,并且由于其改变的脂质 A 酰基链而不会刺激有效的炎症反应。
普氏菌属和拟杆菌属 LPS 的免疫抑制也有报道。这些结构差异会影响生命早期的全身免疫启动、免疫发育和训练有素的免疫力。
▼ 糖脂(包括 LPS),通过CLR 发出信号
糖脂,包括 LPS,也可以通过哺乳动物细胞上的 C型凝集素受体发出信号。凝集素受体信号传导在调节肠道屏障功能和启动抗真菌或抗细菌免疫方面具有重要作用。通过许多 CLR 的信号也调节炎性体,介导 IL-1 家族细胞因子和警报素的释放。
注:CLR是免疫系统中重要的信号传导分子。
目前除 LPS 类似物外,没有证据表明微生物组中存在糖脂。有必要对来自微生物组菌株的不同 LPS 进行进一步的结构分析,并详细了解脂质糖对 C 型凝集素的参与。
想象鞘脂是细胞膜的“保护墙”,它可以由原核生物和真核生物从头合成。在真核生物中,膜鞘脂的存在无处不在,而只有少数细菌类群具有制造它们的酶促能力。
▼ 拟杆菌可从头合成鞘脂,缺乏鞘脂可能导致生长缺陷
拟杆菌在用spt酶催化的关键步骤从头合成鞘脂方面具有独特的能力,CerPI 是拟杆菌中最丰富的鞘脂之一,在赋予肠道细菌适应性方面具有重要作用。缺乏鞘脂的拟杆菌属虽然有活力,但表现出生长缺陷、膜结构改变和对氧化应激的易感性增加。
▼ 拟杆菌产生的α-GC,对免疫系统很重要
拟杆菌还能够产生一种类似于来自鞘氨醇单胞菌的 alpha-GC 的脂质,这对宿主的免疫系统可能具有重要作用,抗炎并减少结肠 NKT 细胞的数量。
鉴于 CD1d 限制性脂质特异性免疫细胞在各种炎症性疾病中的重要性,α-GC 是否是唯一能够与 CD1d 受体结合的鞘脂是一个需要解决的关键问题。
▼ 微生物群鞘脂影响宿主炎症和代谢途径
当无菌小鼠被鞘脂缺陷细菌定植时,导致肠道炎症和宿主神经酰胺库发生变化,提供了微生物群鞘脂影响宿主炎症和代谢途径的进一步证据。 拟杆菌外膜中鞘脂的存在有利于耐受性免疫反应。
OMV 中的鞘脂可作为巨噬细胞中 TLR2 信号的激动剂,并且在限制炎症信号方面很重要。
注:OMV 细菌细胞外膜的囊泡
在人类中,来自宿主的粪便鞘脂是炎症性肠病患者中最显着增加的代谢物类别,而微生物组鞘脂则显着减少。
微生物组鞘脂不仅发挥局部作用,而且通过运输到肠外器官和改变宿主鞘脂信号传导而发挥全身作用。
疾病关联、共同进化联系以及宿主和微生物鞘脂之间的直接生化串扰的结合,使其成为未来研究的一个值得探索的领域。
磺酸脂在结构上与磷酸化鞘脂(本质上是磷酸神经酰胺的硫类似物)相关,并且通常在拟杆菌门的成员中发现,例如Alistipes、Odoribacter、Flavobacterium、Chryseobacterium。
▼ 磺基脂类的生物合成
已知磺基脂类以类似于拟杆菌鞘脂类的方式进行生物合成,但最近的一项研究发现了它们合成的第一个酶促步骤:半胱氨酸酰基-酰基载体蛋白转移酶sulA。人们对它们的功能知之甚少。在Flavobacteria物种中,磺酸脂使细菌能够进行滑行运动。
已表征的主要磺脂是磺胺杆菌素,它与宿主唯一已知的相互作用是与 von Willebrand 因子受体结合。
▼ 磺脂对巨噬细胞和树突细胞有促炎作用
还有证据表明磺基杆菌素是一种与 MD-2 结合的 TLR4 激动剂,它与 TLR4 信号传导物理相关并增强 TLR4 信号传导,且来自金黄杆菌的磺脂对小鼠巨噬细胞具有强烈的促炎作用。具有聚糖头基的磺脂在人树突细胞中具有促炎作用。
▼ 产磺脂类的常见菌群——Alistipes
小鼠的膳食摄入对Alistipes的磺基脂类合成有很强的影响,高脂肪饮食喂养的小鼠显示出肠道硫杆菌素的丰度显着增加。
在人类中,Alistipes是已知可产生磺脂的最常见的人类肠道微生物组物种,并且在 IBD 患者的粪便中显着减少。
心磷脂和缩醛磷脂是脂质类别的其他例子,它们可以在相关途径中由原核生物和真核生物从头合成,再次表明趋同进化。
▼ 心磷脂的结构和功能
心磷脂是由甘油桥接的两个磷脂酰基组成的脂质二聚体。宿主心磷脂是仅在线粒体内膜中发现的标志性脂质,从进化的角度来看很有趣,因为它们被假设为细菌和真核生物的内共生融合体。
作为线粒体许多生理作用的关键脂质,例如高能 ATP 产生和线粒体蛋白质功能,心磷脂失调与许多疾病有关,包括衰老、代谢综合征、心力衰竭和癌症。
▼ 心磷脂调节免疫,稳定细胞膜
宿主心磷脂还可以调节免疫和细胞死亡途径,因为暴露于免疫系统会激活 NLRP3 炎性体和半胱天冬酶,并结合 CD1 分子呈递给 NKT 细胞,可能作为一种检测线粒体损伤的方法。
在细菌中,心磷脂似乎在稳定细胞膜方面发挥作用,因为心磷脂合酶的缺乏使细菌更容易受到渗透压的影响,并且它会积聚在细菌膜的两极。
▼ 细菌心磷脂信号
哪些微生物菌株含有心磷脂通常是未知的,有报道称它们在链球菌和大肠杆菌中富集。
人们对宿主免疫中的细菌心磷脂信号知之甚少。一项检查微生物组中 LPS 信号传导拮抗剂的研究发现,心磷脂是能够减少 LPS 与 TLR4 信号通路中 CD14 和 MD-2 结合的主要代谢物。
另一项研究发现了一种来自鲍曼不动杆菌(Acinetobacter baumannii)的脂质分子,该分子被认为是一种心磷脂,可通过 TLR2 介导的途径在哺乳动物细胞中诱导炎症和细胞死亡。
未来研究评估来自微生物组的心磷脂如何调节、改变或模拟宿主心磷脂通路将很重要。
▼ 缩醛磷脂:保护细胞免受氧化应激
缩醛磷脂是含有乙烯基醚键而不是酯键的甘油磷脂。缩醛磷脂具有许多重要功能,包括保护细胞免受氧化应激,并且在神经元和心血管细胞膜中的浓度最高。
▼ 缩醛磷脂:与神经系统疾病和炎症相关
虽然它们在人类中的全部功能尚不清楚,但缩醛磷脂合成和丰度的缺陷是许多神经系统疾病的基础,包括阿尔茨海默氏症。
缩醛磷脂也是活性氧的重要介质,因此被认为在引发或解决慢性炎症中发挥作用。
▼ 梭状芽孢杆菌:缩醛磷脂的生物合成酶
来自肠道微生物组的一部分细菌能够合成缩醛磷脂。最近,在梭状芽孢杆菌中发现了负责缩醛磷脂合成的酶,这些生物合成酶的同系物映射到许多不同的肠道微生物组物种。
膳食脂质的摄入对人类生命至关重要。从出生到幼年,人类的饮食由母乳组成,基于热量含量,母乳由约 55% 的脂质组成。
母乳甘油三酯中的脂质种类及其饱和度是影响婴儿健康发育的重要变量。
根据美国的最新数据,年龄较大的儿童和成人的饮食通常含有较少的脂肪热量,平均下降至 32%。
▼ 膳食脂质的来源
大多数脂质能够由真核细胞自身合成,因此提供了除饮食之外的第二来源。例外情况是 omega-3 和 omega-6 脂肪酸,哺乳动物不编码从头合成所需的酶,它们的前体必须来自饮食。
▼膳食脂质的健康益处
一般来说,膳食脂质为身体器官和细胞提供许多健康益处,例如,在细胞再生、蛋白质信号、能量平衡、膜稳定性和代谢途径的稳态维持方面。
▼ 肠道菌群类似肝脏,可以分解脂质
肠道菌群遇到摄入的所有膳食脂质,因此,肠道菌群中的酶就像第二个肝脏一样分解、转化和解毒膳食成分,这可能对宿主健康产生有益和有害的影响。由于肝酶具有分解膳食和外源性脂质的功能,因此微生物组酶在肠道中起着类似的作用。
微生物组酶对膳食脂质的生物转化
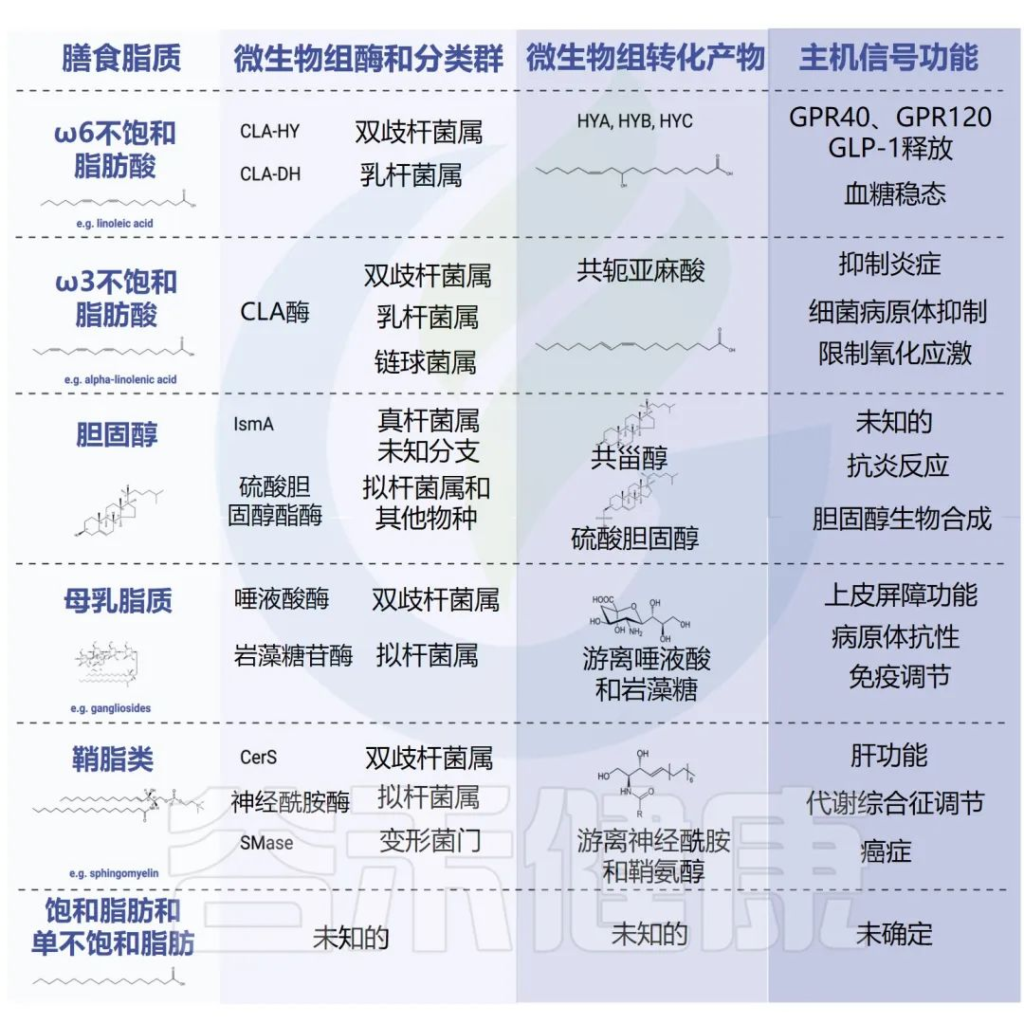
doi: 10.1016/j.chom.2023.01.009
注:由于我们的饮食中含有各种各样的脂质,因此上表并不代表所有生理上重要的膳食脂质清单。✔
从很小的时候起,人类就通过母乳接触饮食中的外源性鞘脂,随后食用乳制品、肉类、蛋和许多植物性食物。
摄入的常见类型的鞘脂包括神经酰胺、鞘磷脂、脑苷脂和神经节苷脂。
▼ 鞘脂的功能:尤其是大脑和神经生长很重要
鞘脂在整个胃肠道中被水解和吸收,它们在调节细胞生长、分化、免疫和新陈代谢方面起着重要作用。
鞘脂对大脑功能和神经生长很重要,神经节苷脂在脑细胞信号传导中起着重要作用,仅存在于动物脂肪中,因为植物缺乏合成这些脂质的酶。
▼ 拟杆菌:合成和转化鞘脂
在微生物组中,拟杆菌菌株不仅合成鞘脂,还通过从饮食中摄取简单的类鞘脂来生物转化膳食鞘脂。
它们还拥有许多聚糖降解酶,可以分解摄入的神经节苷脂。
这些包括唾液酸酶,它可以分解含唾液酸的代谢物(例如,神经节苷脂)并释放有助于免疫和预防感染的游离唾液酸。
神奇的是,不编码鞘脂合成所必需的酶的双歧杆菌菌株可以导入并利用鞘脂来产生 DHCer。
生命早期的宏基因组队列研究表明,双歧杆菌还编码预测分解复杂鞘脂的酶。目前的研究还很有限,但可以推测微生物组的酶活性与循环的鞘脂种类之间存在直接联系。
在肠道的局部水平上,增加复合鞘脂的摄入已被证明可以改善屏障功能并减少细菌毒素的伤害,这一过程可能受微生物组合成的酶功能影响的过程。
膳食鞘脂加工对哺乳动物生物学的影响才刚刚开始被发现。这些相互作用在富含鞘脂的母乳中尤为重要。鞘脂生物合成途径通常在早期微生物组中上调,这种上调是健康的预测指标。
拟杆菌和双歧杆菌是非常丰富的生命早期微生物组菌株,它们影响鞘脂代谢的能力可能会影响人类发育。
根据婴儿发育量表的评估,最近的一项研究发现婴儿肠道中肠道拟杆菌的存在与神经发育增强之间存在密切联系。随着肠道微生物群-脑轴正成为一个更被接受的范例,应进一步探索膳食和肠道来源的鞘脂在大脑发育中的作用。
胆固醇是真核细胞中类固醇激素和细胞膜的重要组成部分,哺乳动物无需膳食胆固醇即可合成胆固醇。这些甾醇脂质的信号传导和调节可以影响许多对人类健康和疾病很重要的免疫和代谢途径。
▼ 肠道微生物群胆固醇影响循环胆固醇
循环胆固醇被用作人类健康的重要生物标志物,可在肠道中吸收的游离胆固醇的量会影响循环中的胆固醇水平。
大约 100 年前提出了肠道微生物组胆固醇代谢影响血清胆固醇水平的观点,然而,直到最近的一份报告明确证实人类微生物组中的这种现象之前,研究这种联系的研究相对较少。
肠道微生物对胆固醇的代谢影响肠道和循环中的胆固醇浓度
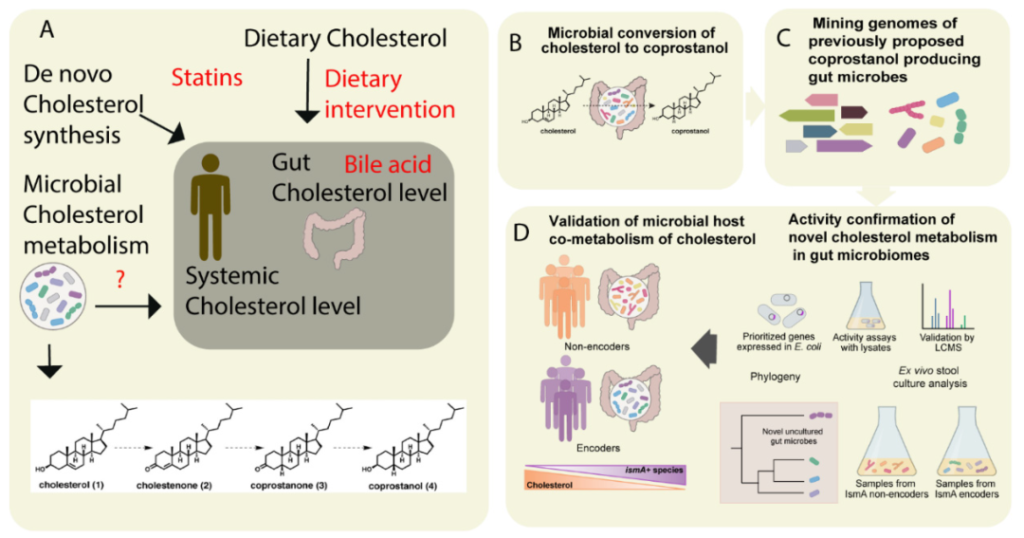
doi.org/10.3390/metabo11010055
血液中的胆固醇水平可能会受到肝脏中合成的新胆固醇的影响,也可能来自于饮食等外源性来源。内源性胆固醇水平也可能受到他汀类药物或胆汁酸代谢改变的影响。肠道微生物对胆固醇的代谢也可作为维持胆固醇稳态的检查点。
▼ 厚壁菌中的微生物酶影响血清胆固醇
这项研究在以前未培养的肠道微生物群的厚壁菌门物种中发现了微生物酶 IsmA,该酶催化胆固醇转化为胆甾烯酮,并最终转化为粪前列腺素。
此外,这种酶在人体中的活性和存在会影响他们的总血清胆固醇水平。
这种酶的影响具有临床相关性,因为它影响血清胆固醇浓度的比值比相当于依折麦布( FDA 批准的肠道胆固醇转运蛋白小分子抑制剂,是经临床验证的降低血液胆固醇的方法)。这些表明肠道微生物调节胆固醇具有很大的潜力。
注:依折麦布是第一个,也是唯一个胆固醇吸收抑制剂,通过选择性抑制小肠胆固醇转运蛋白,有效减少肠道内胆固醇吸收,降低血浆胆固醇水平以及肝脏胆固醇储量。
▼ 拟杆菌磺化胆固醇
最近表明,肠道微生物组菌株对膳食胆固醇的生物转化比之前认为的更为普遍。
许多共生微生物,包括拟杆菌属,都能够磺化胆固醇。含有磺基转移酶的拟杆菌属基因簇会影响小鼠的血清胆固醇水平,为膳食胆固醇被吸收到循环中的能力增加了另一个变量。
该基因簇还可以硫酸化类固醇激素,如维生素 D3 类似物、异异石胆酸、粪前列腺素和其他膳食甾醇,如 ß-谷甾醇。
▼ 不同人群肠道菌群不同,代谢胆固醇的微生物酶分布不均
在 IBD 和肠道炎症中,基因簇显着减少,考虑到最近归因于 T 细胞中固醇信号的免疫功能,这很有意思。
代谢胆固醇的微生物酶在人群中分布不均;因此,受试者的肠道基因组可以解释胆固醇、胆固醇衍生物和血脂组的一些变异性。
▼ 未来研究:可能发现更多胆固醇衍生物由微生物群合成
尽管次级胆汁酸等硫酸化甾醇衍生物与健康衰老有关,来自微生物组的粪便甾烷醇和硫酸化胆固醇代谢物的生物学功能尚未完全阐明,关于其途径的未来研究充满无限想象。
由于有数百种可能的修饰可以添加到甾醇骨架中,因此可以想象许多其他胆固醇衍生物能够由微生物组通过膳食胆固醇合成。
循环甘油三酯结合高密度脂蛋白和低密度脂蛋白水平通常用作代谢健康的指标。然而循环胆固醇在多大程度上直接影响心血管疾病的发展是有争议的,未来的研究有必要评估治疗干预中的这些途径,可能作为联合疗法。
人类能够合成除亚油酸和α-亚麻酸以外的所有必需脂肪酸,这必须来自饮食。它们分别是 omega-6 和 omega-3 多不饱和脂肪酸 (PUFA) 的前体。
▼ Omega-3 和 omega-6与炎症
omega-6 和 omega-3 多不饱和脂肪酸的下游代谢产物对膜成分至关重要,对调节炎症也很重要。
Omega-3脂肪酸具有抗炎作用,而 omega-6 脂肪酸具有促炎作用。然而,每一种都可以很容易地被氧化,并通过脂质过氧化作用导致一般的氧化应激。
▼omega-6:omega-3 比例高与代谢综合征相关
血液中的 omega-6:omega-3 比例是心血管健康的重要标志,高 omega-6 读数可能是代谢综合征的征兆。
摄入 Omega-6 脂肪酸会增加胰岛素抵抗,这是代谢综合征的常见机制。
有证据支持维持 omega-6:omega-3 脂肪酸摄入量的平衡比例对于预防代谢综合征、心血管疾病和癌症的重要性。西方饮食中 omega-3 和 omega-6 脂肪酸的摄入不平衡,omega-6 脂肪酸含量是古代不含加工食品的饮食的 10 倍多。
人类脂肪组织和母乳中的亚油酸含量在过去 100 年中一直在稳步增加,这会对儿童发育产生负面影响。
▼ 肠道微生物组的酶对脂质生物转化
来自肠道微生物组的酶可以在这些脂质进入宿主代谢途径之前对其进行生物转化,从而调节它们对宿主-脂质代谢的影响。
双歧杆菌和乳杆菌含有 CLA-HY,这是一种将亚油酸转化为共轭亚油酸的酶,然后转化为可结合 GPR40 和 GPR120 的分子,从而产生抗炎信号并限制亚油酸转化为下游产物的量。
CLA 酶在人类微生物组中很常见,可能导致对代谢综合征和肥胖的易感性发生变化。
膳食多不饱和脂肪酸也可以被常见的肠道微生物酶饱和,从而限制双键的数量和氧化潜力。
▼ 肠道 omega-6高,促进炎症细菌生长
功能不同的微生物群可改变饮食中的多不饱和脂肪酸的炎症潜力。
一项研究发现,肠道中高浓度的 omega-6 可以杀死通常与健康相关的细菌,并促进与炎症性疾病相关的细菌(例如变形杆菌)的生长。
▼ 肠道菌群影响多不饱和脂肪酸代谢物水平
此外,常见的多不饱和脂肪酸代谢物在 IBD 患者的肠道微生物组中存在显着差异,从超高加工食品中摄取它们会增加 IBD 风险。
另一项研究发现,大豆油中亚油酸摄入量的增加会改变肝脏鞘脂代谢物的平衡,肠道微生物组的存在会影响肝脏鞘脂的变化及其饱和度水平。
▼ 多摄入omega-3可以抗炎
大量证据表明,在饮食中摄入更多的 omega-3 脂肪酸可以起到消炎和抗炎作用。随机、双盲研究表明,摄入鱼油和富含 omega-3 的食物可以改善关节炎等炎症性疾病患者的全身炎症。
膳食中的 omega-3 脂肪酸可以改变人类和小鼠体内的微生物组组成,这已被证明对宿主具有积极的抗炎作用。
Omega-3 脂肪酸及其与肠道菌群的相互作用
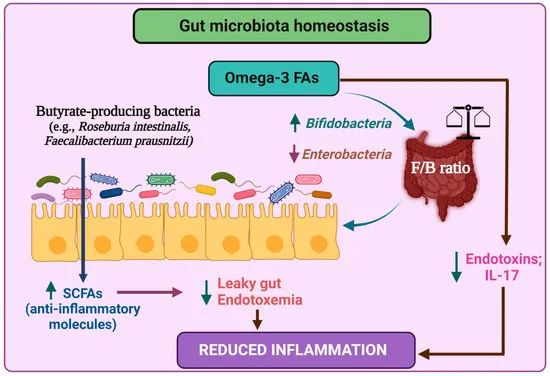
doi.org/10.3390/nu14091723
▼omega-3益处:通过肠道菌群产生共轭脂肪酸
omega-3多不饱和脂肪酸可能有益地影响宿主代谢的一种可能机制是,通过肠道微生物组产生共轭脂肪酸。据报道,α-亚麻酸的共轭异构体具有抗炎、抗癌和抗肥胖的特性。
体外研究表明,双歧杆菌、乳杆菌和丙酸杆菌菌株能够将 omega-3 脂肪酸、α-亚麻酸生物转化为共轭亚麻酸异构体。
此外,与无菌小鼠相比,常规小鼠结肠内容物中的共轭亚油酸异构体和非共轭代谢物有所增加,表明肠道微生物组有助于体内omega-3 脂肪酸代谢。
用源自肠道微生物组的 α-亚麻酸代谢物短期喂养小鼠会影响肠道免疫稳态。需要做更多的工作来确认肠道微生物组对这些代谢物的贡献及其对人类的有益作用。
最终,多不饱和脂肪酸的摄取和代谢不仅取决于饮食摄入,还取决于肠道中存在的微生物。肠道微生物组酶对膳食多不饱和脂肪酸的调节可能是易患炎症性疾病、代谢综合征和心血管疾病的重要因素。
▼酰基链饱和水平对脂肪的生化特性影响重要
哺乳动物消耗的植物和动物脂肪中的甘油三酯和磷脂通常根据酰基链的饱和水平进行分类,因为酰基链的饱和水平赋予脂肪重要的生化特性,比如氧化还原电位和进入炎症通路的能力。
▼ 饱和脂肪酸不一定有害
饱和脂肪酸,例如硬脂酸和棕榈酸(分别具有 18 个和 16 个碳骨架),在哺乳动物饮食中含量最丰富。尽管它们曾被认为是有害的并会导致心血管疾病,但有关饱和脂肪酸的最新数据并不支持这一观点。
摄入最多的单不饱和脂肪酸是 omega-9 脂肪酸,油酸,通常被认为是中性的或对人体健康有益。对能够生物转化或代谢饱和或单不饱和脂肪的肠道微生物菌株或酶的研究是有限的。一项研究表明,硬脂酸和油酸可以在体外选择性地改变某些细菌菌株的生长。
▼ 脂肪酸的饱和水平影响细胞功能和体内平衡
使用受控饮食的小鼠研究表明,膳食脂肪酸含量的饱和水平会对线粒体功能、肠道通透性、肠道运动和肠道微生物组组成产生重大影响,大豆油中的氧化脂多不饱和脂肪酸会导致肥胖增加。
细胞膜和线粒体中脂肪酸的饱和水平会影响细胞功能和体内平衡。
鉴于这些影响,了解肠道微生物菌株对膳食饱和和单不饱和脂肪酸以及与宿主途径相互作用的全面影响将很重要,因为它们在对饮食和炎症触发的个性化反应中发挥作用。
宿主微生物脂质代谢和炎症
慢性炎症是困扰全世界的众多疾病的基础,包括代谢综合征和自身免疫。与数十万年人类历史消耗的脂质相比,人类消耗的脂质的数量、平衡和类型在过去 50-100 年发生了巨大变化。
由于我们的基因适应这种快速变化的速度很慢,因此更加依赖共生微生物来进化以适应饮食变化。
▼微生物合成的脂质可以成为免疫的有效刺激物
人们普遍认为,膳食脂肪的数量和类型的改变会导致全身炎症增加。作为进一步的结果,由哺乳动物微生物组直接合成的脂质可以成为粘膜和全身免疫的有效刺激物,并且这种炎症的引发可以反馈改变脂质吸收和代谢。
绝大多数脂质在小肠中被吸收,小肠是肠道的一个区域,微生物衍生的脂质与免疫系统直接接触。
在肠上皮细胞中,消耗能量用于免疫与新陈代谢之间存在权衡,适应性 IgA 反应可防止微生物衍生的脂质过度刺激先天免疫系统,从而抑制脂质吸收。
▼脂质吸收促进微生物组介导的肠道炎症
脂质吸收还可以通过上皮细胞中促炎性膳食脂质的积累来促进微生物组介导的肠道炎症,这一过程由清道夫受体 CD36 促进并由 T 细胞信号传导调节。
在 T 细胞中,脂质谱和代谢维持调节细胞和表达 Th17 程序的倾向。来自微生物组的甾醇衍生物已被证明可以直接影响这些途径,影响在肠道炎症中被破坏的 T 细胞平衡。
最后,生命早期接触膳食和微生物衍生的脂质对于哺乳动物免疫系统的启动、训练和发育至关重要,并对以后的疾病易感性产生重要的影响。
总体而言:以下三方面之间的相互作用解释了脂质代谢变化引起的慢性炎症易感性的主要变化:
人类膳食脂质代谢的变化影响代谢综合征和自身免疫发展的风险
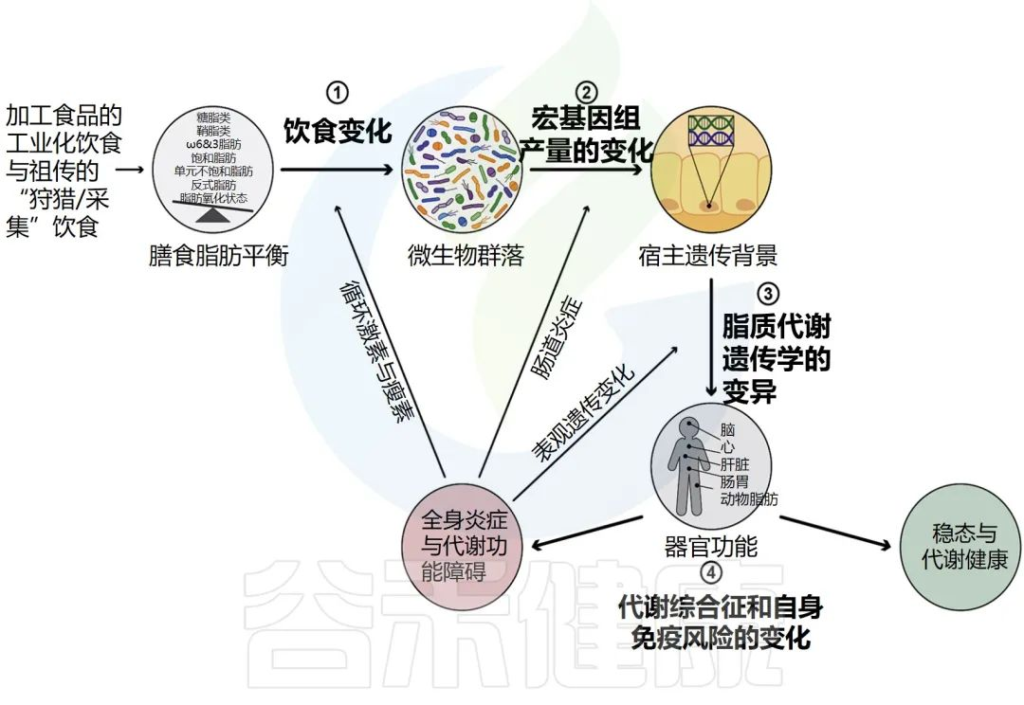
Brown et al., Cell Host Microbe.2023.
在上面三个变量中,微生物组酶产生的脂质产物及其对全身炎症和循环脂质信号传导的影响的特征最少。许多研究将基因和遗传多态性与人类的特定疾病和循环脂质谱联系起来;然而,人们对驱动临床结果的个体之间微生物组酶功能的变化知之甚少。
目前我们开始了解到,微生物来源的脂质及其对脂质代谢的影响在宿主许多器官的生物学功能中具有系统性作用。对健康人血清的深度代谢组学分析发现了数千种未知的细菌来源代谢物,其中许多被预测为脂质。
▼ 肠道菌群脂质代谢促炎或抗炎
随着时间的推移,这些脂质组谱中的每一个对于个体来说都是独一无二的,并且变化可以引发或解决炎症。例如,亚油酸和花生四烯酸代谢的副产物(类花生酸和消炎素)可能对哺乳动物炎症的发生或消退产生深远影响,而肠道中的微生物组酶可以控制它们的循环。
脂肪酸去饱和酶基因FADS1和FADS2的多态性,与许多炎性疾病的风险增加有关,也控制了循环类花生酸水平。
因此,必须考虑微生物组和宿主遗传学的结合,以应对由脂质代谢失调引起的炎症风险。在许多情况下,在 IBD 或风湿病等慢性炎症性疾病的发生过程中,环境因素的影响超过了宿主遗传易感性。
▼微生物组的脂质代谢响大脑中的慢性炎症
除了在肠道内,微生物组的脂质代谢可能会影响大脑中的慢性炎症,可能导致许多神经退行性疾病。大脑主要由脂质组成,微生物组脂质代谢是否影响脑脂质化学尚不清楚。将帕金森病等神经系统疾病视为始于肠道的疾病是该领域的一个想法。
大脑富含鞘脂和缩醛磷脂,已知这两种物质都是由微生物在肠道中产生的。NKT 细胞通过响应脂质抗原环境在维持大脑免疫耐受方面发挥重要作用。无菌小鼠表现出许多神经学特征,包括较低的焦虑和行为改变,其中许多可以在不同微生物的肠道定植后被诱导。未来的研究应该系统地评估哪些微生物来源的脂质影响肠脑轴。
肠道微生物组在脂质代谢(消耗和生成)中的作用正在成为人类健康和疾病的主要决定因素。未来的努力应该扩大我们在将各种脂质与生物功能联系起来方面取得的适度进展,并将我们的理解应用于人类疾病。
影响健康的微生物组酶可以被抑制或引入,具体取决于环境,并且可以使用代谢组学和遗传分析筛选受试者,以将代谢特征与个体微生物组分布联系起来。
我们可以使用这些数据对预防、诊断和治疗进行更明确的分析。一般地说,我们应该了解在我们体内循环的许多脂质都是微生物来源的知识,这些努力需要几个方面的努力。
➧ 首先是微生物组代谢物的大规模注释
➧ 其次是计算和机器学习提供脂质鉴定方法
计算和机器学习方法可以提供一种更具可扩展性的脂质鉴定方法,该方法可以从推定的理论结构中更好地预测脂质的功能和结构等。
➧ 最后将脂质与微生物和宿主-微生物相互作用联系起来
需要一个更完整的脂质组学目录,该目录来自与健康和疾病相关的组织相关微生物。此外,介导脂质代谢的微生物酶的遗传操作也很重要。
为了扩展对微生物组脂质与宿主相互作用的了解,优先对那些在对人类健康更重要的菌群进行基因敲除更适用,例如Akkermansia、双歧杆菌、毛螺菌科和梭菌目菌株。
总之,肠道微生物群及其衍生的脂质和宿主脂质代谢之间存在复杂的串扰。需考虑特定食物和营养素组合在塑造微生物特征方面的作用,饮食、肠道菌群结构和脂质代谢之间的关系需要在大量人群中进行研究,以制定治疗策略。鉴于肠道菌群组成的个体差异,这些策略很可能需要患者分层和个体化治疗。
主要参考文献:
Brown EM, Clardy J, Xavier RJ. Gut microbiome lipid metabolism and its impact on host physiology. Cell Host Microbe. 2023 Feb 8;31(2):173-186. doi: 10.1016/j.chom.2023.01.009. PMID: 36758518.
Yoon H, Shaw JL, Haigis MC, Greka A. Lipid metabolism in sickness and in health: Emerging regulators of lipotoxicity. Mol Cell. 2021 Sep 16;81(18):3708-3730. doi: 10.1016/j.molcel.2021.08.027. PMID: 34547235; PMCID: PMC8620413.
Schoeler M, Caesar R. Dietary lipids, gut microbiota and lipid metabolism. Rev Endocr Metab Disord. 2019 Dec;20(4):461-472. doi: 10.1007/s11154-019-09512-0. PMID: 31707624; PMCID: PMC6938793.
Lamichhane S, Sen P, Alves MA, Ribeiro HC, Raunioniemi P, Hyötyläinen T, Orešič M. Linking Gut Microbiome and Lipid Metabolism: Moving beyond Associations. Metabolites. 2021 Jan 15;11(1):55. doi: 10.3390/metabo11010055. PMID: 33467644; PMCID: PMC7830997.
Ma J, Zheng Y, Tang W, Yan W, Nie H, Fang J, Liu G. Dietary polyphenols in lipid metabolism: A role of gut microbiome. Anim Nutr. 2020 Dec;6(4):404-409. doi: 10.1016/j.aninu.2020.08.002. Epub 2020 Oct 9. PMID: 33364456; PMCID: PMC7750795.
谷禾健康

肠道微生物群之间编织了一个复杂的相互作用网络,影响人体的营养吸收和代谢,免疫功能等,对我们的健康状态有很大的影响。
我们知道,肠道微生物群具有多样性,平衡性,稳定性等特征,但同时也具有异质性,也就是说不同个体之间存在差异。不同人群对食物,膳食补充剂的健康需求不同。
特定的营养素、食物、整体饮食结构等诸多因素都会影响特定的肠道菌群,从而影响整体健康。
随着当前研究技术的不断进步,我们对肠道菌群和健康之间的关系认知不再停留在初级关联阶段,而是逐步走向精细化调节层面。
怎样通过这些看起来日常的饮食,益生菌补充剂等方式,去操纵肠道菌群以达到获得健康的目的,是一个值得深入探讨的话题。
本文,我们从肠道菌群的角度来了解一下,饮食模式、发酵食品、益生菌、益生元和其他相关化合物对我们健康的影响。
本文主要从五个方面讲述
●发酵食品对肠道菌群和健康的影响
●不同饮食模式与菌群及健康
●益生菌调节肠道微生物群和人体健康
●益生元化合物对肠道菌群的影响
●后生元和精神生物制剂与健康
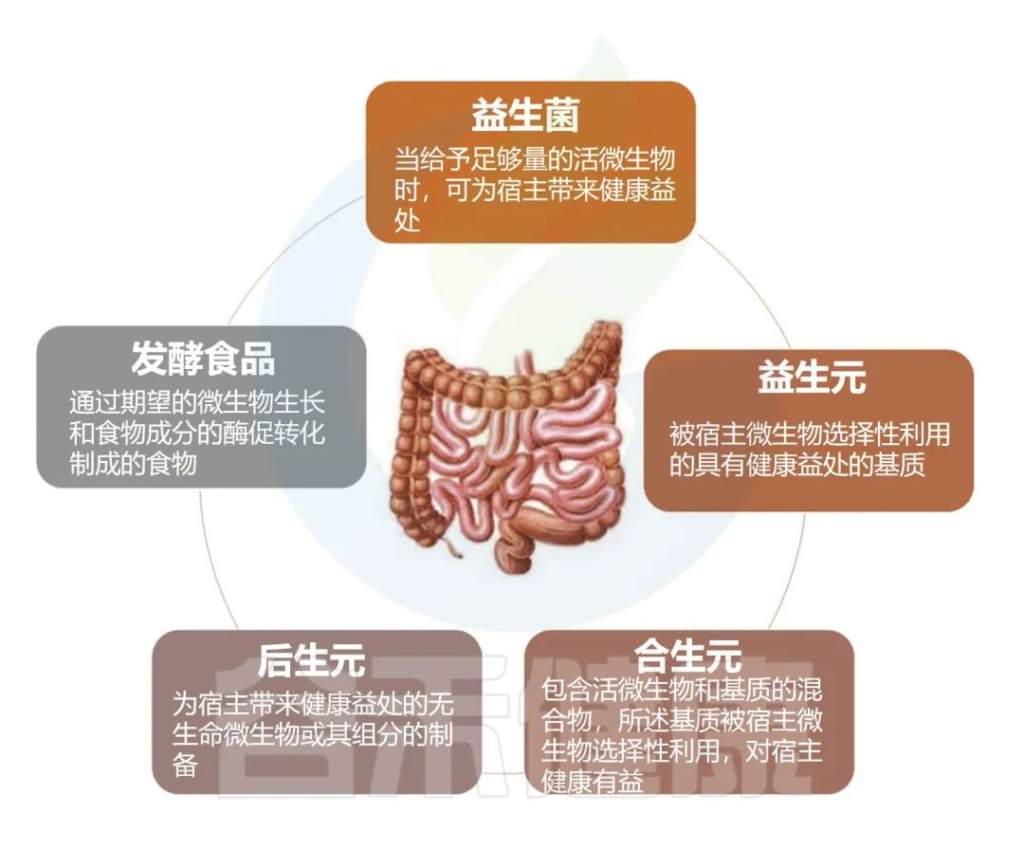
Nma B,et al.Elsevier Inc.2022
发酵食品被定义为“通过理想的微生物生长和食物成分的酶促转化制成的食品”。
许多发酵食品已被证明通过不同的方式对人体具有促进健康的作用,包括生物活性分子的合成、肠道微生物群的调节以及与免疫系统的相互作用。
✦发酵食品产生多种代谢物,促进有益作用
在食品发酵过程中,微生物进行酶促转化,提供多种具有不同生理活性的代谢产物(多肽、低聚糖、游离氨基酸、改性多酚、有机酸等)。
这些转化发生在食物摄入之前,发酵食品也是微生物进入我们肠道的绝佳载体,与我们的微生物群的其他成员一起,可以转化和代谢我们肠道中的食物成分,产生具有不同功能的微生物代谢物(短链脂肪酸、维生素、细菌素等)。
据报道,某些发酵食品或其成分能够以某种方式促进有益的效果,而这些效果有时与我们微生物群的变化有关。在本节中,我们将讨论发酵食品对微生物群的影响及其可能的有益作用。
发酵乳制品对健康的影响已经被深入研究了一个多世纪,但直到最近,随着新的组学技术和大规模测序方法的使用,我们才能够详细了解食用发酵乳制品对微生物群的影响。
1 酸奶

•酸奶有助于改善乳糖消化不良
酸奶是研究最多的发酵乳制品之一。它是通过两种乳酸菌(德氏乳杆菌保加利亚亚种和嗜热链球菌)在牛奶中的联合活性获得的。欧洲食品安全局已经认识到酸奶发酵剂的有益作用,认为活酸奶培养物可以改善乳糖消化不良患者的乳糖消化。
•饮用酸奶增加了嗜热链球菌,减少了拟杆菌丰度
一些动物模型的临床前研究表明,长期摄入酸奶改变了小鼠菌群中拟杆菌门(Bacteroidetes)和厚壁菌门(Firmicutes)之间的比例,并增加了链球菌科(Streptococcaceae)成员的数量,这可能是由于摄入了酸奶中存在的嗜热链球菌(S. thermophilus)。
此外,一些在人体中进行的干预试验表明,饮用酸奶会增加人体微生物群中的一些细菌数量:其中,乳酸菌的丰度较高,而拟杆菌(Bacteroides)的种类则相应减少。
2 奶酪

奶酪,也是一种发酵的牛奶制品,其性质与酸奶有相似之处,都是通过发酵过程来制作的,也都含有可以保健的乳酸菌,但是奶酪的浓度比酸奶更高,近似固体食物,营养价值也因此更加丰富。
•食用奶酪可以增加产丁酸盐细菌丰度,可能缓解特应性皮炎
研究了奶酪和乳制品引发肠道微生物群变化并促进相关健康影响的能力。最近进行了一项健康食品选择与肠道微生物群组成之间的广泛关联研究,表明α多样性与低脂奶酪之间存在强烈的正关联。
临床前研究报告称,给小鼠服用奶油奶酪后,产生丁酸盐的细菌水平增加,T细胞(Treg)介导的免疫反应和IgE水平降低,表明对缓解特应性皮炎有潜在的有益作用。
•奶酪中的发酵菌可以存在人体一段时间
在人类中,两项干预试验评估了食用卡门贝尔奶酪对不同肠道菌群成员的影响,结果表明,食用奶酪后粪肠球菌(Enterococcus faecalis)的丰度显著增加,并且卡门贝尔奶酪中存在的微生物,如乳酸乳球菌(Lactococcus lactis)、肠膜明串珠菌(Leuconostoc mesenteroides)和白地土菌(Geotricum candidum),在食用奶酪期间出现在个体的粪便中。在停止干预15天后,粪便样本中存在肠膜明串珠菌。
3 开菲尔
开菲尔是一种由微生物混合物发酵的产品,通常包括几种乳酸菌和酵母,其发酵和生产过程根据其生产的地理区域和传统的细化过程而显著变化。
因此,很难对开菲尔的效果得出一般性的结论,更谨慎的做法是根据所研究的开菲尔的类型来具体说明这些效果。
•改善机体代谢,抗肥胖
有证据表明,食用开菲尔可能有有益的代谢作用。开非尔能够调节啮齿动物体内的微生物群组成。向肥胖的人类微生物群相关大鼠喂食西藏开菲尔牛奶表明,微生物群变化与胆汁酸和氨基酸代谢有关。
此外,连续四周每天给小鼠服用克非尔显示了对厚壁菌门/拟杆菌门比率的影响,以及对性能和身体疲劳的改善。
另一项研究通过建立社区特征和几种生物标志物之间的相关性,证明了开菲尔在小鼠体内的抗肥胖作用,这表明食用开菲尔诱导的微生物群和真菌群调节可能与预防肥胖和脂肪酸代谢有关。
•改善炎症性肠病、代谢综合征
一项针对炎症性肠病患者的随机对照试验表明,定期食用开非尔可以改善生活质量,减少腹胀。
此外,一项平行组随机对照干预试验显示,代谢综合征患者接受开菲尔12周后,在开菲尔组中放线菌的相对丰度增加,对代谢综合征标志物有一些有利的影响。
•还可能影响宿主行为和免疫状态
对小鼠施用开非尔还被证明可以增加短乳杆菌(Lactobacillus brevis)的流行率和微生物群产生γ-氨基丁酸的能力,影响宿主行为和免疫状态。
这些临床前证据表明,开非尔对宿主的微生物群和代谢有影响,包括肥胖、糖尿病、肝脏和心血管疾病、免疫和神经系统变化。
除了发酵乳制品外,关于其他动物性发酵食品调节微生物群的信息较少。然而,有证据表明植物发酵食品对肠道微生物群的影响,以及其与健康影响的潜在关系。
在这方面,酸菜、泡菜、红茶菌和其他发酵的植物性食物已经被研究过,并显示出对肠道微生物群的不同影响,尽管需要更多的证据来建立它们对人类健康的明确联系。
1 酸面包
•低蛋白饮食相关细菌减少、影响升糖反应
酸面包是一种发酵食品,也显示出了显著的效果。采用元蛋白质基因组学方法分析了酸面包对大鼠微生物组的影响,结果显示低蛋白质饮食相关的细菌类群减少。
在另一项研究中,食用手工酸面包引起的升糖反应与食用工业白面包不同。这些反应是个体特异性的和微生物相关的。
每个人的血糖反应可以根据干预研究前确定的微生物组谱进行预测。
2 发酵豆制品
•促进脂肪酸分解代谢
关于发酵豆制品,一些临床研究显示了微生物群调节活性和各种生理效应。在啮齿动物中研究了大豆发酵产品对代谢过程的影响,表明发酵大豆能够促进小鼠脂肪酸分解代谢和主要细菌门的变化。
•影响其他代谢物水平
鼠李糖乳杆菌发酵的豆浆通过增加一些细菌类群,如拟杆菌(Bacteroides),对小鼠微生物群有影响,这是一种通过尿液排泄的异黄酮代谢物的来源。
此外,用解淀粉芽孢杆菌(Bacillus amyloliquefaciens)发酵的大豆降低了2型糖尿病大鼠模型中的高血糖,引起疣微菌(Verrucomicrobiales)种群的增加和肠杆菌(Enterobacteriales)的减少,以及其他微生物群的变化。
•改善认知功能
值得注意的是,在小鼠中摄入植物乳杆菌发酵的大豆后,观察到对认知功能的积极影响,以及乳酸菌和双歧杆菌种群的增加。
含有植物乳杆菌(L.plantarum C29)的发酵产品在一组轻度认知障碍患者中进行的为期12周的人类临床试验中显示出认知能力的改善。
•可能改善皮肤状况
最后,摄入含有干酪乳酸菌代田株(Lactobacillus casei Shirota)的发酵豆乳对健康绝经前日本女性的皮肤状况有显著影响,乳酸菌和双歧杆菌的数量有增加的趋势,而肠杆菌科和紫单胞菌科(Porphyromonadaceae) 的数量则有下降的趋势。
由于各种食物已经证明了调节特定细菌和肠道微生物群整体结构的能力,因此短期和长期的饮食模式可以影响肠道微生物组的构成和良好功能。
★ 饮食对肠道菌群的多样性至关重要
我们肠道菌群的多样性可能反映了我们饮食的多样性,因此,饮食可能是我们控制和平衡肠道菌群组成和代谢的最强大的武器。
以下部分旨在概述过去几年来产生的关于不同饮食和饮食习惯对调节肠道菌群的潜力的一些现有知识。
西式饮食、地中海饮食和素食这几种可能是世界上最常见的人类饮食模式,每一种都与一些特定的健康/疾病状况有关。
✦不同饮食模式的差异主要是碳水化合物和纤维的含量
宏量和微量营养素方面的显著差异决定了这些饮食模式,其中复合碳水化合物和纤维的含量可能是最显著的之一。
事实上,复杂的碳水化合物和纤维可能是产生最确凿证据的食物成分,它们是肠道菌群的有益调节器,在碳水化合物消耗仅24小时后就会发生快速转变。
西式饮食,是一种以高含量精加工糖和碳水化合物、高含量饱和脂肪酸、高含量动物蛋白以及低含量膳食纤维为特征的一种现代饮食方式,不能否认的是这种饮食好吃还容易上瘾。
✦西式饮食导致易患许多代谢疾病
这种饮食模式在人类历史上是最近才出现的,通常与城市生活方式有关,通常与炎症和代谢疾病有关,包括2型糖尿病和肥胖等。
✦西式饮食下的肠道微生物显著改变
此外,在将祖先生活方式和农业人口的饮食与西方化生活方式的饮食进行比较时,观察到肠道微生物群的组成发生了显著变化,总体特征是纤维降解细菌的减少和蛋白质代谢细菌的增加。
变形菌、拟杆菌等蛋白质代谢菌较丰富
最近的研究已经指出,与这些生活方式相关的一些疾病生物标志物可能是通过西方饮食对肠道微生物群的干扰作用介导的,其特点是多样性低。
蛋白质和胆汁代谢细菌占主导地位,包括产生三甲胺n-氧化物(TMAO)的物种,变形菌和拟杆菌通常更为丰富。
事实上,在动物模型中,西方化饮食的引入会迅速转化为微生物群的变化,并增加感染和代谢疾病的风险,在向新动物移植不良微生物群后,这些表型可以重现。
这表明,饮食模式影响人类健康的一些机制是通过调节肠道微生物群来实现的,并表明通过饮食调节肠道微生物群可能会降低一些疾病风险。
地中海饮食的特点是大量摄入新鲜水果、蔬菜、豆类和全谷物,同时摄入少量动物蛋白、加工食品和饱和脂肪。
✦地中海饮食改善代谢、降低慢性病患病率
长期以来,这种饮食习惯与改善代谢和心血管健康、健康老龄化、降低死亡率和慢性疾病患病率有关。
纤维降解细菌丰度增加、短链脂肪酸增加
其中一些影响与肠道菌群特征及其相关代谢组和免疫调节的特定变化有关。就地中海饮食对肠道菌群的具体影响而言,它通常与纤维降解物种的高度多样性和代表性有关,如普雷沃氏菌(Prevotella)和毛螺菌属(Lachnospira)。
其代谢导致短链脂肪酸的产生;以及一些瘤胃球菌的减少。事实上,严格坚持地中海饮食的人的短链脂肪酸水平似乎更高。
值得注意的是,与地中海饮食相关的有益肠道菌群调节并不完全与其长期食用有关,因为饮食习惯向这种模式转变,即使是相对较短的时间,通常也会改善微生物和健康生物标志物,这在各种炎症、代谢和认知障碍的动物模型以及人类临床试验中都得到了证明。
例如,对一组超重和肥胖的参与者进行了8周的地中海饮食干预,即使在保持能量摄入的情况下,也导致微生物群多样性显著增加,纤维降解细菌的表现包括普拉梭菌、一些拟杆菌属和一些罗氏菌属、颤螺菌属和毛螺菌,这些细菌负责短链脂肪酸的产生,包括丁酸盐生产者。其中一些还被认为具有抗炎特性。
✦有效改善炎症
此外,在饮食干预后,潜在的促炎细菌(如Ruminococcus gnavus)减少,胆固醇水平、炎症标志物和胰岛素抵抗全面改善。
地中海饮食对肠道菌群结构的好处也在其他健康受试者的队列中被揭示出来。在欧洲各国的老年人群中进行了一项大型饮食干预试验,表明在12个月的时间内坚持地中海饮食模式有利于调节微生物群中的关键物种,导致与较低虚弱和改善认知功能标志正相关的类群的丰富,并与炎症负相关,包括以下几种菌属:
普拉梭菌(Faecalibacterium);
罗氏菌属(Roseburia);
优杆菌属(Eubacterium);
多型拟杆菌(Bacteroides thetaiotaomicron);
普雷沃氏菌(Prevotella)。
素食
素食是一种不食肉等动物产品的饮食方式,有时也戒食奶制品和蜂蜜。在另一个极端,许多研究评估了长期素食和杂食饮食对微生物群、代谢组和疾病风险的影响。
✦素食对健康促进的具体作用暂不明确
长期以来,素食饮食一直被认为具有促进健康的作用,但与杂食性饮食相比,素食饮食也可以培养不同的肠道微生物群结构,总体上增加了多样性和丰富性;拟杆菌(Bacteroidetes)经常以较高的相对频率出现。
然而,报告了一些相互矛盾的结果,一些控制良好的喂养研究发现,生活在同一地理区域的杂食性和素食者之间的肠道微生物群只有适度的差异。不过两组在代谢组中都表现出很大的差异,这可能部分归因于微生物群产生的代谢物。
低FODMAP饮食
还有一种与潜在微生物群调节能力相关的饮食模式是低FODMAP饮食。
什么是FODMAP?
FODMAP是一组人体吸收较差的短链碳水化合物,包括果糖、果聚糖、乳糖、多元醇及半乳糖寡糖等。
其特点是可发酵低聚糖、双糖、单糖和多元醇的含量降低,虽然其中一些成分可能作为益生元对健康受试者的肠道菌群进行有益的调节,但它们也可能引发肠易激综合征(IBS)和其他肠道疾病患者的不良反应。
✦缓解肠易激综合征
低FODMAP饮食长期以来可以证明缓解了肠易激综合征患者的症状,但其有益效果似乎强烈依赖于患者的基础微生物群特征。
✦影响健康促进有关细菌的丰度
在微生物区系水平上,FODMAP消耗的减少通常会导致一些与健康促进有关细菌的肠道减少,如:
普拉梭菌(Faecalibacterium Prausnitzii)↓↓↓
双歧杆菌(Bifidobacterium)↓↓↓
嗜黏蛋白阿克曼菌(Akkermansia)↓↓↓
巨球型菌属(Megasphaera)↓↓↓
片球菌属(Pediococcus)↓↓↓
放线菌(Actinobacteria)↓↓↓
以及产生丁酸盐的细菌减少
同时伴随着其他气体消耗细菌的增加,如雌马酚产生者Adlercreutzia,甚至是Ruminococcus torques。细菌通常在肠易激综合征患者中检测到高丰度。
因此,低FODMAP饮食可以导致症状的改善,尽管所涉及的机制尚不清楚,但是同时,也可以诱导更明显的肠道菌群失调。
有趣的是,低FODMAP饲粮降低了结肠pH值和细菌总数,而短链脂肪酸浓度不受影响,这进一步支持了低FODMAP饮食下肠道菌群代谢中存在重要的重组。
注意
虽然人们普遍认为地中海和素食饮食可以改善肠道微生物群的多样性和纤维降解细菌的表现,但在不同的研究中,一些具体的类群有所不同,这阻碍了在饮食模式、微生物群变化和健康反映之间建立具体的因果关系。
这可以归因于不同食物组合的可变影响,以及对饮食干预的反应可能高度依赖于基础肠道微生物群组成,以及其他个体遗传和环境特征、宿主遗传、季节、地理和文化差异。
在考虑将饮食干预作为精确调节肠道微生物群的手段时,另一个需要考虑的关键方面不仅是宏观和微量营养素的具体组成,还包括摄入次数、饮食行为和饮食干预的持续时间。
一般来说,虽然相对短期的饮食干预足以对肠道菌群产生可测量的影响,但在恢复习惯性饮食后,这些变化会迅速恢复,甚至,根据基础菌群配置,可能需要更大的干预才能产生明显的效果;建议永久性的饮食适应,以确保提供持续的基质来源,为有益的肠道种群提供燃料及其相关的长期益处。
因此,虽然在该领域已经取得了重大进展,但为了建立旨在通过调节肠道微生物群来改善人类健康的个性化饮食策略,有必要进行进一步的研究。
益生菌被定义为“当施用足量时,对宿主的健康有益的活微生物”。
益生菌已被认为在不同水平上发挥其有益功能,包括:
•营养物质的代谢,以促进消化,产生维生素或具有全身作用的分子;
•神经系统信号的改变;
•免疫调节的诱导;
•对生理应激的保护;
•直接和间接的病原体拮抗;
•改善肠道上皮的屏障功能;
•调节微生物
益生菌可以在不同程度上对宿主产生有益作用
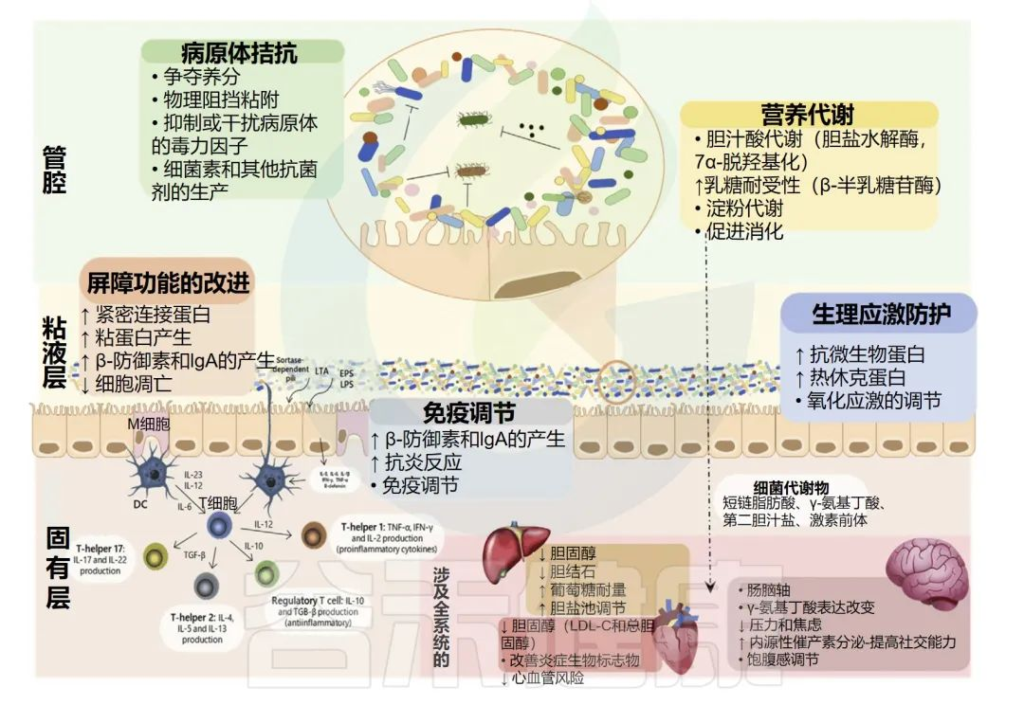
Nma B,et al.Elsevier Inc.2022
这些功能可以是接触依赖的或由表面分子介导的,如脂磷酸和梭酸依赖菌毛,或由分泌分子介导的,如短链脂肪酸和细菌素。
总的来说,突出了免疫调节和病原体拮抗活性,它们对于维持肠道微生物组中细菌群落与宿主之间的平衡至关重要。
一些研究,不仅在体外和动物模型中,而且在人类志愿者中,分析了益生菌对与人类疾病相关的特定细菌病原体或群体的抑制作用,如产气荚膜梭菌 (Clostridium perfringens)、肠炎沙门氏菌(Salmonella enteritidis)和大肠杆菌(Escherichia coli);以及其对生理或炎症标志物和免疫特征的影响。
益生菌也被认为能够调节整体肠道微生物群。事实上,有人提出所需的益生菌效果与肠道中的微生态变化有关。
✦益生菌治疗改变了肠道微生物组成
对动物模型的不同研究表明,用不同菌株的乳酸菌和双歧杆菌以及其他微生物(如布拉氏酵母菌)进行治疗,明显影响小鼠肠道的微生物群组成,促进微生物群落的结构和功能的变化。
注:这些研究大多使用细菌计数或qPCR分析来研究肠道微生物群分布的变化,只获得了生态系统的一小部分图像。在这方面,随着下一代测序技术和生物信息学工具的发展,我们可以对肠道微生物组进行更深入的研究,不仅可以从结构和组成层面,还可以从功能层面深入研究益生菌对微生物群落的影响。
缓解肥胖相关的肠道微生物失调
在动物模型上报道了鼠李糖乳杆菌、嗜酸乳杆菌、植物乳杆菌、两歧双歧杆菌、长双歧杆菌和粪肠球菌对饮食诱导的肥胖小鼠肠道微生物群的调节作用,缓解了饮食诱导的肥胖和相关的肠道微生物群失调。
此外一些益生菌,如茯砖茶中的冠突散囊菌(Eurotium cristatum),也显示出调节肠道真菌和细菌群落的能力,表现出与健康动物中观察到的情况更接近的特征,并减轻饮食引起的肥胖症状。
下面是益生菌调节肠道菌群的一些证据:
调节抑郁症的肠道生态失调
肠道生态失调
在抑郁症小鼠模型中,使用含有L.plantarum LP3、L.rhamnosus LR5、Bifidobacterium lactis BL3、Bifidobacterium breve BR3和Pediococcus pentosaceus PP1的益生菌配方进行干预,可对微生物组产生调节作用,使其与健康动物中观察到的情况相似,从而减少与疾病相关的肠道生态失调。
结肠炎生态失调得到改善
同样,在诱导结肠炎小鼠模型中,摄入布拉迪酵母菌(Saccharomyces boulardii)或两株发酵乳杆菌(Lactobacillus fermentum )已显示出能够调节肠道微生物组,增加生物多样性,并显著改善结肠炎动物的肠道生态失调特征。
结直肠癌的致病菌得到抑制,有益细菌增加
另外,在结直肠癌小鼠模型中,丁酸梭菌(Clostridium butyricum)的干预减弱了致病菌的增加,促进了有益菌的生长,并改变了次生胆盐和短链脂肪酸等微生物源性代谢产物。
在小鼠哮喘模型、抗生素诱导的生态失调小鼠和健康动物中也报道了这些调节作用,显示生物多样性和有益细菌丰度的增加。
益生菌对微生物群的调节在人类中并不明显,因为有的研究显示了相互矛盾的结果。可能还需一系列深入研究来了解益生菌对人体肠道微生物群的具体影响。
下面是存在争议的一些数据:
在最近的一项工作中,研究表明,以嗜酸乳杆菌(L. acidophilus)、乳酸乳杆菌(B. lactis)、长双歧杆菌(B. longum)、双歧双歧杆菌(B. bifidum)、和半乳糖低聚糖混合物组成的共生生物作为益生元进行干预,显示出在一组肥胖患者中调节肠道微生物群落的能力,观察到不同健康相关菌群(如双歧杆菌、毛螺菌和乳杆菌)的比例有所增加;以及与肥胖正相关的慢性炎症相关群体的减少,如瘤胃球菌科(Ruminococcaceae),普雷沃氏菌(Prevotella)和巨球型菌属(Megasphaera) 。
然而,先前对肥胖患者的研究显示了相互矛盾的结果,因此其中一些研究报告称,在益生菌干预后,肠道微生物群落没有变化,质疑这种对肠道微生物组的调节作用的存在。
与此同时,对影响全球数百万人的炎症性肠病患者的研究也显示出有争议的结果。尽管在动物模型中已经报道了对肠道微生物群落的调节作用,但在一组溃疡性结肠炎患者中,用含有短双歧杆菌(B. breve)的某品牌益生菌治疗,在研究过程中,两个治疗组之间没有显著差异。
另一方面,摄入含有L.acidophilus La-5和B. animalis亚种的益生菌酸奶。促进了一组炎症性肠病患者中乳酸菌、双歧杆菌和拟杆菌水平的增加,尽管这项工作没有从宏基因组的角度分析微生物组。
✦特定菌属水平增加,整体菌群组成变化不大
一些研究报道,益生菌不影响整体菌群组成,但可以改变特定属的比例。
例如,在高危特应性疾病婴儿中,在产前最后阶段和出生后第一年摄入益生菌混合物促进了双歧杆菌和乳酸杆菌水平的增加,但在肠道微生物组中没有检测到显著变化。
同样,在一组有功能性便秘症状的个体中,混合使用益生菌的干预促进了瘤胃球菌(Ruminococcus)水平的增加和丹毒丝菌科(Erysipelotrichaceae) 成员的减少,但α多样性指数和整体微生物组组成显示出与安慰剂组检测到的水平相似。
在一组HIV感染个体中,在使用鼠李糖酵母菌GG干预8周后,也描述了相同的趋势。
✦增加与健康相关共生细菌,减少病原体相关细菌
其他研究表明,益生菌可以引起肠道微生物种群的变化,增加与健康相关的共生细菌的水平,减少与病原体相关的群体,甚至恢复被改变的微生物群落的平衡。
在这方面,干酪乳杆菌(Lactobacillus casei Lcr35)在一组患有急性腹泻的儿童中,促进了拟杆菌、粪杆菌和瘤胃球菌属的增加,并降低了大肠杆菌和梭状芽孢杆菌的水平。
在一项针对乳糖不耐受患者的研究中,使用B.longum BB536和L.rhamnosus HN001进行30天的干预也导致肠道微生物组成发生积极变化。
在感染幽门螺旋杆菌的患者中也观察到了这种趋势。他们报告了多重耐抗生素的屎肠球菌LAB制剂与抗生素联合使用时,在正常肠道微生物群方面的有效效果,防止α多样性值的下降,并恢复受抗生素治疗影响的微生物群的水平。
同样,在一组患有早期败血症的成年人中,益生菌的干预导致α多样性的增加以及益生菌的增加,如不同种类的乳酸菌(Lactobacillus)和屎肠杆菌(E.faecium)。
此外,一些研究表明,益生菌不仅可以引起特定细菌种群的变化,还可以引起相关微生物群代谢物的变化。
✦肝硬化患者肠道失调得到改善
在肝硬化患者中,摄入8周鼠李糖乳杆菌GG不仅能调节肠道微生物组,降低肠杆菌科(Enterobacteriaceae)和紫单胞菌科(Porphyromonadaceae)的相对丰度,增加共生菌毛螺菌科(Lachnospiraceae)和瘤胃球菌科(Ruminococcaceae)的丰度,还能调节代谢物的分布,从而改善肠道生态失调和微生物-代谢组的联系。
在摄入增强结肠短链脂肪酸生成的发酵牛奶后,也有类似的趋势被报道;在乙型肝炎诱导的肝硬化患者中摄入含有丁酸梭菌(C.butyricum)和婴儿双歧杆菌(Bifidobacterium infantis)的益生菌混合物3个月后,促进了不同肠粘膜屏障完整性生物标志物的改善。
考虑到来自动物模型和人体试验的这一证据,最近有人提出将益生菌作为预防和治疗结直肠癌的新策略。
注:迄今为止,缺乏针对人类患者的研究,专注于分析益生菌对与CRC相关的肠道菌群紊乱的可能调节作用。
✦健康人群病原相关菌丰度下降
就健康人群而言,只有少数干预研究分析了益生菌摄入量对肠道微生物群的影响,而肠道微生物群可以根据参与者的年龄进行分类。
在一项对3至12个月大的婴儿的研究中,使用三种益生菌菌株进行干预(B.longum subsp. infantis R0033, Lactobacillus helveticus R0052和B. bifidum R0071)。8周后,一些病原相关类群如柯林斯菌属(Collinsella),肠球菌(Enterococcus)和克雷伯氏菌(Klebsiella),粪杆菌(Faecalibacterium)和瘤胃球菌(Ruminococcus)水平下降。
此外,在干预期间,一些细菌群保持稳定,而在安慰剂组中,它们增加了,如大肠杆菌志贺氏菌(Escherichia-shigella)和韦荣氏球菌属(Veillonella)的情况下
增加有益细菌,减少病原体相关菌的一些证据:
在另一项对健康婴儿的研究中,在生命的第一年摄入补充了4种双歧杆菌菌株的配方,促进了脆弱拟杆菌(Bacteroides fragilis)和经黏液真杆菌属(Blautia)水平的下降,以及代谢物谱的差异,尽管没有检测到整体微生物组的变化。
在成人人群中,有研究表明,益生菌摄入量对肠道微生物群落的影响与肠道菌群的基础组成密切相关。每天食用干酪乳杆菌(L.casei)14天,增加了某些有益细菌的相对丰度,如乳酸杆菌(Lactobacillus)、罗氏菌属(Roseburia)、粪球菌属(Coprococcus),而一些有害细菌的水平则降低了。
在用Lactobacillus kefiri LKF01治疗后,以及B.longum BB536和L.rhamnosus HN001进行干预后,也报告了类似的结果,显示出强大的调节肠道微生物群组成的能力,导致有益细菌的增加,并显著减少直接参与促炎反应和胃肠道疾病发作的潜在有害菌群。
✦产丁酸盐细菌增加
此外,每天食用两歧歧杆菌(B.bifidum strain Bb)4周会影响粪便微生物群中优势类群的相对丰度,并调节粪便丁酸盐水平,因此作者观察到普雷沃氏菌(Prevotellaceae)的数量减少,而瘤胃球菌(Ruminococcaceae)和Rikenellaceae的比例更高。
这些研究支持了之前的研究,即益生菌对肠道菌群的调节作用。有趣的是,最近有人提出了益生菌“ salami”的潜力,尽管微生物组的整体结构没有发现显著变化,但摄入益生菌萨拉米会促进产生丁酸盐的细菌的增加。
在益生菌摄入过程中检测到粘附相关基因的差异表达和大量参与细菌运动性的基因,提示益生菌干预可以通过调节其表达模式来调节肠道微生物组。
小结
总之,在过去的几十年里,大量的科学报告已经证实了益生菌对宿主健康的积极作用。然而,尽管存在不同的动物模型和人体研究,显示益生菌可能对肠道微生物群落及其功能具有调节作用,但在人体试验中报道的还存在一定的争议。
现有的研究使用了不同的益生菌菌株、广泛的年龄范围、疾病和疾病状态,以及摄入不同饮食和营养素的志愿者;因此,研究组表现出较高的个体内部和个体间的变异性,使得阅读结果变得困难。
另一方面,正如前面提到的,我们必须考虑到益生菌的作用不仅取决于菌株,还取决于基础微生物群,而最常用的益生菌(主要属于乳酸杆菌和双歧杆菌)不一定在肠道定植,并且可以在益生菌干预后释放,从而使确定益生菌调节宿主肠道微生物群的具体机制变得更加复杂。
为了阐明益生菌对肠道环境和微生物群影响的分子机制,我们应该进一步研究更大、更均匀的群体,也许还应该研究从人类肠道环境中分离出来的下一代益生菌,它们能够在肠道粘膜上定植,产生长期效应。
益生元概念首次定义为“不易消化的食物成分,通过选择性地刺激一种或少数已经存在于结肠中的细菌的生长或活性而有益地影响宿主”。
然而,这一概念多年来一直在发展。当前的国际益生菌和益生元协会共识小组提出了以下益生元的定义:“一种被宿主微生物选择性利用的对健康有益的底物”。
各种益生元
应该指出的是,目前确立的益生元是基于碳水化合物的,尽管其他物质如酚类化合物和多不饱和脂肪酸转化为各自的共轭脂肪酸可能符合这一定义。
几十年来,只有少数几类碳水化合物被认为是益生元,包括人乳寡糖(HMOs)、菊粉和低聚果糖(FOS)、低聚半乳糖(GOS)和乳果糖。
一些益生元及相关研究
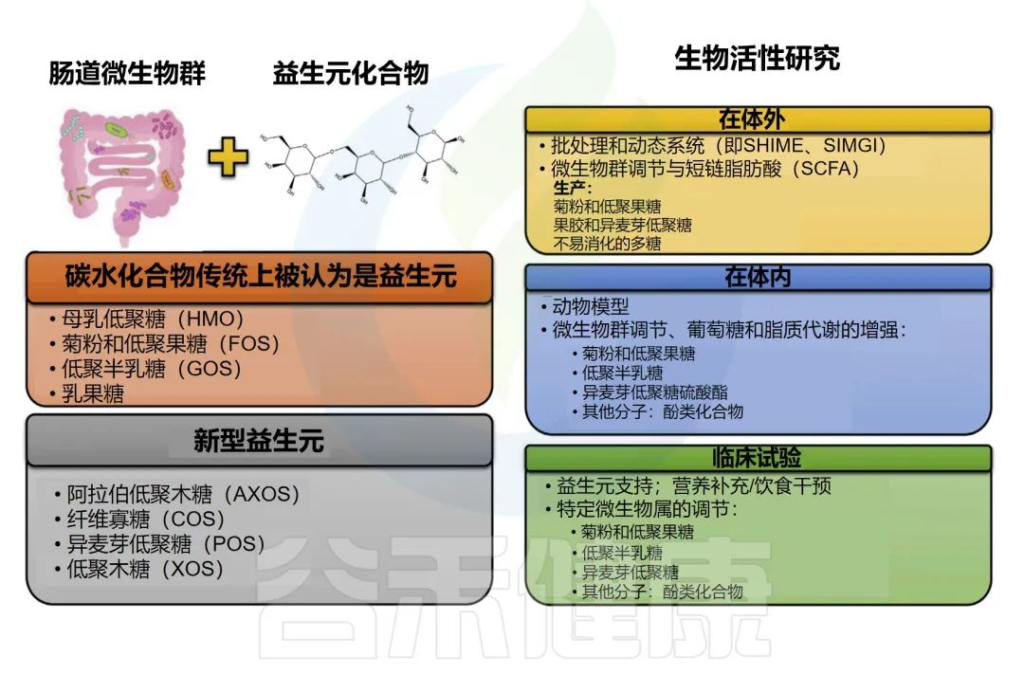
Nma B,et al.Elsevier Inc.2022
•人乳寡糖
人乳寡糖包括由5个单体(葡萄糖、半乳糖、n-乙酰葡萄糖胺、焦糖和唾液酸)通过多达12个不同的a-和b-糖苷键结合形成的广泛的低聚糖结构。
人乳寡糖对新生儿肠道菌群发育有很大影响,促进双歧杆菌(Bifidobacteriaceae)和拟杆菌(Bacteroidaceae)生长。
•低聚果糖
另一方面,低聚果糖是通过蔗糖的果糖转基化或菊粉的部分水解获得的,是唯一主要由果糖单位组成的碳水化合物。
•低聚半乳糖
相比之下,低聚半乳糖包括含有b键的b-GOS和来自乳糖延伸的b-GOS,以及存在于各种蔬菜来源中的a-GOS,如棉子糖和水苏糖。
•其他益生元
其他潜在的益生元低聚糖包括异麦芽糖-低聚糖(IMOS),由糖化植物淀粉中获得的由a键连接的葡萄糖单位形成的分支结构;
低聚木糖(XOS)和阿拉伯低聚糖(AXOS),由木聚糖和阿拉伯低聚木聚糖水解产生的含有阿拉伯糖酰和葡萄糖醛基分支的低聚木糖物组成;
纤维寡糖(COS),由木质纤维素生物质酶解获得,由b键连接的葡萄糖单位形成;
果胶寡糖(POS),由果胶部分水解产生,由半乳糖醛酸线性链形成,可能显示含有鼠李糖、阿拉伯糖、木糖和半乳糖等中性糖的分支结构域。
许多研究通过动物模型来评估体内益生元化合物的潜在生物活性。
✦增强代谢和生物利用度
通过16S rRNA基因测序和鸟枪法测序,大鼠给予低聚果糖和低聚半乳糖可导致普雷沃氏菌(Prevotella)水平升高,并由于普雷沃氏菌的代谢功能而增强了人参总苷的生物转化和生物利用度。
深度宏基因组测序实验已经报道了低聚果糖在高脂饮食喂养小鼠中促进了以下菌群的生长:
Allobaculum ↑↑↑
Oribacterium ↑↑↑
普雷沃氏菌(Prevotella) ↑↑↑
并降低了以下菌群:
嗜胆菌属(Bilophila) ↓↓↓
Butyrivibrio LE30 ↓↓↓
鸟枪测序分析还显示,菊粉处理的高脂饲料饲养的小鼠后代的葡萄糖和脂质代谢途径增强。
以下菌群水平增加:
B. breve ↑↑↑
B. acidifaciens ↑↑↑
Clostridium sp. CAG 343 ↑↑↑
Eubacterium sp. CAG 786 ↑↑↑
下列菌群水平下降:
B. massiliensis ↓↓↓
Oscillibacter sp. 1-3 ↓↓↓
Ruminococcus gnavus CAG 126 ↓↓↓
✦降低致病菌丰度
在肉鸡等其他动物模型中测试了低聚果糖补充剂作为抗生素的替代品,导致致病性脱硫弧菌属(Desulfovibrio)和幽门螺杆菌(Helicobacter)的丰度降低。
研究集中在其他寡糖家族,如异麦芽低聚糖,揭示了绿茶与异麦芽低聚糖联合使用,通过预防小鼠肠道生态失调来对抗高脂肪饮食诱导的代谢改变,显示了有益细菌(如Akkermansia muciniphilia,双歧杆菌,乳酸菌和罗氏菌属)丰度的增加。
✦非碳水化合物类益生元可以改善肥胖和肠道炎症
非碳水化合物的调节作用也在体内进行了研究,并通过元分类学评估了微生物种群的变化。已有研究证明,富含多酚的蔓越莓提取物可预防饮食诱导的肥胖、胰岛素抵抗和肠道炎症,并增加小鼠肠道微生物群中嗜黏蛋白阿克曼菌(Akkermansia muciniphila)丰度。
同样,槲皮素对高脂饮食诱导的非酒精性脂肪肝疾病的保护作用,以及类黄酮通过调节厚壁菌门/拟杆菌门比率来改善肥胖诱导的小鼠肠道生态失调的潜力已被描述。
临床试验
已经进行了几项临床试验,以测试不同益生元化合物的潜在作用。
✦增加新生儿双歧杆菌丰度
经荧光原位杂交测定,含有低聚半乳糖和低聚果糖混合物(比例为9:1)的配方可促进婴儿体内双歧杆菌的生长,而补充相同比例的短链低聚半乳糖和长链低聚果糖可导致丙型肝炎病毒感染母亲的新生儿粪便双歧杆菌和乳酸菌数量增加。
此外,肠内补充80%短链低聚半乳糖和长链低聚果糖(比例9:1)和20%酸性果胶寡糖的益生元混合物可导致早产儿双歧杆菌和出生后肠道定植增加。
✦抑制了艾滋病患者中一些有害菌的生长
在人类免疫缺陷病毒1型(HIV-1)感染的成人中,类似的益生元混合物促进了双歧杆菌的生长,并抑制了以下细菌的生长:
Clostridium coccoides ↓↓↓
直肠真杆菌(Eubacterium rectale) ↓↓↓
Clostridium lituseburense ↓↓↓
Clostridium histolyticum ↓↓↓
✦改善肥胖人群肠道菌群构成
同样,服用低聚半乳糖导致50岁以上男性和女性双歧杆菌丰度增加,并增加双歧杆菌数量,减少超重成人粪便样本中拟杆菌和Clostridium histolyticum的数量。
临床试验还报告了低聚果糖和果聚糖的益生元效应,结果表明菊粉型果聚糖导致双歧杆菌和普拉梭菌(F. prausnitzii)的增加;根据16S rRNA基因测序数据,这两种细菌都与血清脂多糖水平呈负相关,并且还降低了肥胖女性的肠道拟杆菌、Bacteroides vulgatus和丙酸杆菌(Propionibacterium)。
✦增加健康成年人体内双歧杆菌含量
此外,根据对健康成年人粪便微生物群的分析,菊粉给药增加了铁含量低的女性的粪便双歧杆菌,大豆寡糖、棉子糖和水苏糖可以被双歧杆菌选择性代谢。
✦其他益生元的作用
研究还评估其他益生元化合物的潜在效应。
食用聚葡萄糖可增加健康男性体内戴阿利斯特杆菌属 (Dialister)、普拉梭菌(Faecalibacterium)和考拉杆菌属(Phascolarctobacterium)的丰度。
这几类细菌的作用在谷禾之前的文章中有具体讲述
根据两项研究中获得的16S rRNA基因测序数据,B. animalis subsp. lactis 420促进了阿克曼菌、Christensenellaceae和Methanobrevibacter的生长,并降低了健康超重或肥胖个体中Paraprevotella的生长。
在健康个体中,部分水解瓜尔胶的饮食干预促进了拟杆菌、普拉梭菌、Fusicatenibacter和瘤胃球菌的生长,并减少了经黏液真杆菌属、毛螺菌科和罗氏菌属的生长。
值得注意的是,这些影响在男性参与者中更为明显。
同样,根据16S rRNA基因测序数据,女性不孕症患者口服部分水解瓜尔胶可导致双歧杆菌丰度增加,拟杆菌丰度降低,改善肠道生态失调和妊娠成功。
每日饮用红酒多酚可增加健康男性中的菌群数量,包括以下菌属:
拟杆菌属(Bacteroides) ↑↑↑
双歧杆菌属(Bifidobacterium) ↑↑↑
肠球菌属(Enterococcus) ↑↑↑
普雷沃氏菌属(Prevotella) ↑↑↑
Blautia coccoides ↑↑↑
Eggerthella lenta ↑↑↑
直肠真杆菌(E.rectale) ↑↑↑
益生菌定义中的关键词是“活的”,这意味着微生物必须是活的(在施用时)才能发挥有益的作用。
但是,在过去的几年里,越来越多的证据表明,不可活的细菌,它们的成分或它们分泌的代谢物,也可能在对健康的积极影响中发挥关键作用。
“后生元”一词于2011年首次提出,指的是摄入后对健康有益的灭活、不可存活的微生物细胞。
这一概念是基于观察到非活细菌或其提取物(破碎的细菌)具有调节宿主免疫反应的能力。
✦后生元相较益生菌的优势
将后生元与益生菌进行比较发现,后生元生产的复杂性较低,具有更好的长期稳定性。
对于某些高炎症状态的患者,使用后生元可能是更安全的替代方案,避免了使用活微生物时反应加剧。
在对术语进行分类的尝试中,提出了“true probiotics”(活的和活跃的细胞)、“pseudo-probiotic”(活的和不活跃的细胞,无论是营养细胞还是孢子细胞)和“ghost probiotics”(非活的或死亡的细胞,完整的或破裂的。
此外,活微生物对食物基质成分的活性也可能释放对健康有积极影响的副产物。例如,牛奶蛋白中加密的生物活性肽。
✦后生元的组成
后生元的组成可以是可变的。除了灭活的(无生命的)微生物生物量,细胞壁的成分(肽聚糖、蛋白质、糖蛋白、磷壁酸)、细胞质膜(磷脂、蛋白质)或细胞外成分,如胞外聚合物(EPS)。此外,分泌到培养基上清液或特定发酵食品的代谢物也可能存在。
后生元的成分示例及显微镜下图像
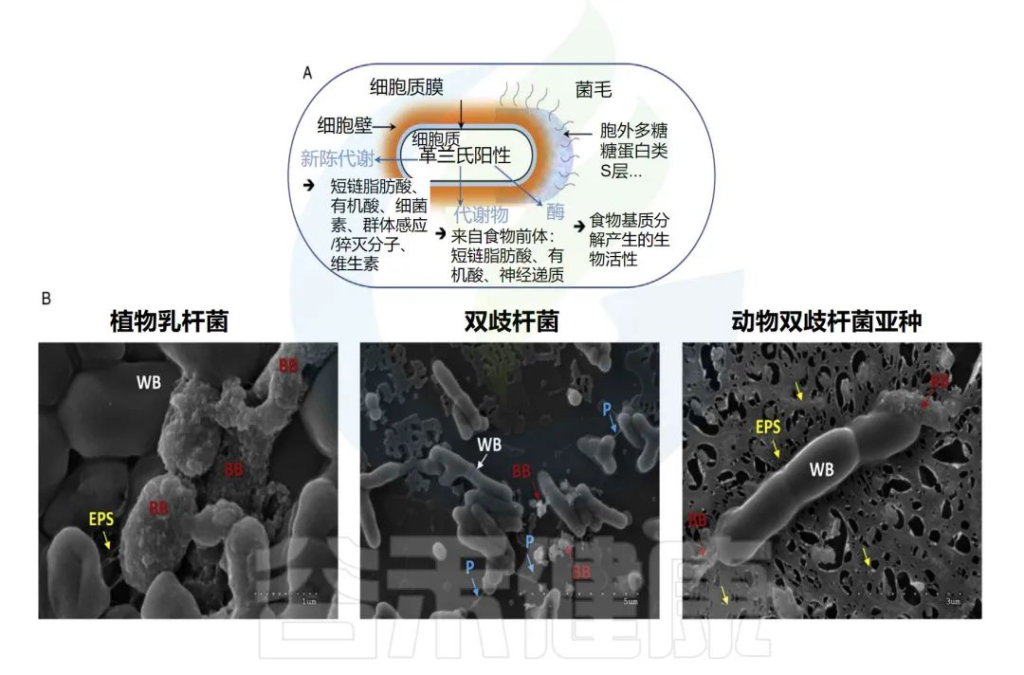
Nma B,et al.Elsevier Inc.2022
✦后生元的作用
后生元作用机制与益生菌的类似,即调节有益菌群的能力、对病原体的拮抗作用、增强肠上皮屏障功能的能力以及调节免疫反应或宿主的其他生理功能。
•刺激免疫反应
双歧杆菌产生的EPS,作为报道的对产生细菌有益作用的关键成分之一。无论是被聚合物包围的紫外线灭活双歧杆菌还是纯化的EPS本身,都能够以不同的方式刺激免疫反应,这取决于聚合物的物理化学特性。这些细菌EPS能够与肠上皮细胞的TLR4相互作用。
•促进脂质代谢和葡萄糖稳定
此外,双歧杆菌EPS是能够积极调节菌群组成的碳水化合物,因为它们被用作肠道某些细菌的选择性发酵底物。特异性EPS还促进饮食诱导的肥胖小鼠模型的脂质代谢和葡萄糖稳态的变化。
含有能够释放抗菌化合物的活益生菌,也可以被视为后生物制剂,可能有助于提高发酵产品的安全性。
将后生元应用于食品,虽然是改善我们健康的一种新方法,但仍然具有挑战性。
精神生物制剂
关于肠道菌群在维持我们生理系统(被称为微生物-肠道轴)方面的知识越来越多,这促使了与调节这种双向对话的饮食策略相关的新概念的发展。这些轴之一是研究的热点,即微生物-肠道-大脑轴。
这是基于对肠道微生物群活动可能参与神经疾病和随着年龄增长而维持认知功能。
▸ 精神生物制剂
针对这目标,提出了与益生菌相关的新定义:精神生物制剂是一种“活的有机体,当摄入足量时,会对患有精神疾病的患者产生健康益处”。
最近,这一定义扩大到包括能够调节肠-脑轴影响肠道微生物群的底物;因此,精神生物制剂是“有益的细菌(益生菌)或对影响细菌-大脑关系的细菌(益生元)的支持”。
研究强调,某些微生物代谢物可以作为某些代谢紊乱的调节因子。同样,在肠道-大脑相互作用的机制中,微生物代谢物也是关键角色。
✦改变行为
已经证明一些微生物来源的精神生物制剂能够有效地改变行为。在一种优雅的实验中,产生酪胺的Providencia能够以一种对共生和宿主都有益的方式操纵秀丽隐杆线虫(Caenorhabditis elegans)的“感官”决定;在食物选择测定中,被Providencia定植的线虫选择了含有这种细菌的食物。
然而,肠道微生物群代谢组的操纵非常复杂,因为肠道微生物的大量多样性之间存在着一种微生物代谢物生产者和消费者之间持续反馈的串扰。
例如,γ-氨基丁酸GABA——(中枢神经系统的主要抑制递质)可以由肠道微生物从饮食前体(如谷氨酸)合成,但它被用作支持该生态位中其他生物生长的营养素。
✦减轻疼痛
神经调节的精神生物学已经被提出,通过体外实验和在大鼠模型上的进一步验证,表明嗜酸乳杆菌菌株(Lactobacillusacidophilus NCFM)能够在肠道水平诱导鸦片样物质和大麻素受体的合成,从而作为一种镇痛药减轻疼痛。
✦调节焦虑
对无菌小鼠口服乳酸菌(Lactiplantibacillus plantarum PS128) 可增加小鼠大脑特定区域的多巴胺和血清素水平,并调节动物的焦虑样行为。
关于能够产生γ-氨基丁酸(中枢神经系统的主要抑制递质)的益生菌,已经在一些乳酸菌和双歧杆菌中描述了不同的代谢途径,其中Levilactobacillus是迄今为止量化产量最高的物种。
不同的策略,如优化培养基组成,可用于提高其他物种的γ-氨基丁酸产量。此外,其中一些乳酸菌也被用于发酵乳制品,以改善这种神经递质在食物中的合成。
//建议
通过γ-氨基丁酸前体的生物转化将食物与γ-氨基丁酸进行天然生物强化,以及使用食物或补充剂作为在肠道中输送能够合成γ-氨基丁酸的益生菌的载体,可能是调节大脑活动的一种策略。
上述的一些证据已经证明,发酵食品、健康的饮食模式、益生菌、益生元以及后生元等都是促进肠道健康的重要因素。通过食用这些食物和补充这些营养素,可以增加肠道内有益菌的数量和种类,并在一定程度上抑制病原相关菌群的丰度,维持肠道菌群的平衡,从而促进肠道健康。
饮食在塑造肠道微生物群的组成和活性方面起着重要作用,对营养饮食与健康肠道微生物组之间相互作用的深刻理解,将为我们理解其在疾病预防和治疗中的作用奠定基础。
任何饮食的好处都将在很大程度上取决于此人的微生物组,每个人根据自己独特的肠道微生物群摄入不同的饮食的方式,将会使我们迎来新时代的饮食模式。
除了肠道健康之外,针对多个健康维度例如提升免疫力,改善情绪,体重管理,皮肤管理等领域产品和食品的需求都在日益提升,这些与肠道微生物组之间又都存在密不可分的关联。因此基于肠道微生物组重要性的开创性研究,可能会从根本上改变消费者偏好及健康食品行业的走向。
主要参考文献
Nma B , Csa C , Ica C , et al. Mechanisms of Gut Microbiota Modulation by Food, Probiotics, Prebiotics and More – ScienceDirect.
Agus, A., Clément, K., Sokol, H., 2021. Gut microbiota-derived metabolites as central regulators in metabolic disorders.
Ali, A., Kamal, M., Rahman, H., Siddiqui, N., Haque, A., Saha, K.K., Rahman, A., 2021. Functional dairy products as a source of bioactive peptides and probiotics: current trends and future prospectives. J. Food Sci. Technol.
Barros, C.P., et al., 2020. Paraprobiotics and postbiotics: concepts and potential applications in dairy products. Curr. Opin. Food Sci. 32, 1–8.
Bellini, M., Tonarelli, S., Nagy, A.G., Pancetti, A., Costa, F., Ricchiuti, A., de Bortoli, N., Mosca, M., Marchi, S., Rossi, A., 2020. Low FODMAP diet: evidence, doubts, and hopes.Nutrients 12 (1), 148.
Cunningham, M., Acarate-Peril, M.A., Barnard, A., et al., 2021. Shaping the future of probiotics and prebiotics. Trends Microbiol.
Fiore, W., Arioli, S., Guglielmetti, S., 2020. The neglected microbial components of commercial probiotic formulations. Microorganisms 8, 1177.

谷禾健康

许多流行病的爆发都是病毒引起的,面对新的传染性基因组出现的最佳策略是及时识别,以便于在感染开始时立即实施相应措施。
目前可用的诊断测试仅限于检测新的病理因子。适用于同时检测存在的任何病原体的高通量方法可能比使用基于当前方法的大量单独测试更有优势。
宏基因组学测序、全基因组测序和靶向深度测序是目前用于病毒遗传鉴定和表征的最佳工具。通过使用这些技术,可以正确的对病毒进行分类,确定其变异性,识别与毒性相关的病毒遗传标记,并在现有知识的基础上考虑抗原性和对抗病毒药物的易感性。
尽管宏基因组学领域取得了巨大进步,但对于具体数据分析任务应使用各种方法中的哪一种,仍缺乏共识。
本文重点描述了宏基因组生物信息数据处理所需要的工具,以便于改善使用宏基因组学识别动物来源样本中新出现、再出现和未知的新病毒。
什么是宏基因组学?
宏基因组学是下一代测序的一个领域,可以识别微生物群落,以及基因检测、识别和表征致病因子。它已被证明是病毒遗传特征的关键因素,并导致了使用传统培养技术无法完成的发现。
目前的分子检测使用特定的引物或探针针对有限数量的病原体,而宏基因组学可以接近样本中存在的所有 DNA 和 RNA 分子,从而能够分析相应的宿主基因组及其微生物集合。
在宏基因组组装中鉴定病毒有五个主要步骤:
宏基因组分箱是在物种注释之前可选的附加步骤。分箱的目的是根据根据序列的起源对其进行聚类。
根据这些步骤,列举出以下目前使用较多的主流工具。
宏基因组学的第一步将是执行序列QC,因为从分析中消除技术错误是必不可少的。
此步骤的主要目的是识别不需要的接头序列、过短的序列、低质量的序列或核苷酸以及其他可能存在的数据。根据数据类型,在这一步中可以使用以下几种工具:

对于短读,可以使用FastQC执行质检 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) ,它可以检查序列的质量并生成总结报告。
其他QC程序也可以提供相同类型的报告,如MultiQC,它具有与FastQC相同的功能,但有一个主要区别,它可以同时合并多个fastq的QC报告,生成一个总的报告。
对于长读,可以使用longQC或MinionQC来检查序列质量,这两个工具已经应用于从纳米孔的MinION或其他长读取测序仪中获得的数据。
—— 低质量序列修剪工具
序列质检后,就需要修剪工具,删除低质量序列和接头序列。可供使用的工具如下图。

对于短读,常用的是Trimmomatic,其次是Cutadapt和Fastp。
对于长读,NanoPack可用于处理长读数据并可视化QC结果。与Nanopack功能相同的是SequelTools。
——删除测序数据中非靶向或污染序列的工具
删除不感兴趣的序列,这些序列可以从各种来源获得。在对病毒序列的分析中,必须删除宿主序列和被污染序列,它可以减少假阳性,并可以防止嵌合病毒-宿主序列的组装。
如下图,通常使用序列比对的工具:

对于短读,可以使用BWA、bowtie2和BBMap等。
其它工具如FastQ-Screen,可以以fastq格式比对自定义参考序列。
(https://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/)
对于长读,可以使用BWA和BBMap,也可以选择特定的minimap2。
也有专门用于识别和修剪特定微生物类群序列的工具,这类工具通常已包含参考基因组序列,一旦比对上,将通过内置的过滤程序丢弃掉。比如VirusHunter(https://bio.tools/virushunter),用于识别NGS数据中的病毒序列。
某些情况下,可能需要从宏基因组数据中删除非靶向分类群的其他RNA序列类型,如核糖体(rRNA),线粒体(mtRNA),或mRNA类型。这时可以用RiboDetector (https://github.com/hzi-bifo/RiboDetector),因为它专门识别rRNA,从而可以过滤掉rRNA以改进后续分析。
另一种方法是在组装前对序列进行物种注释。使用这种策略,可以过滤掉病毒以外的序列,保留病毒序列以供进一步分析。可以使用kraken2和kaiju。
为了更好的进行物种注释和识别存在的病毒,对序列进行组装,生成contigs,以提供更长的连续序列。宏基因组学中使用的组装类型主要为de novo,即从头基因组组装。
可使用的工具如下图:
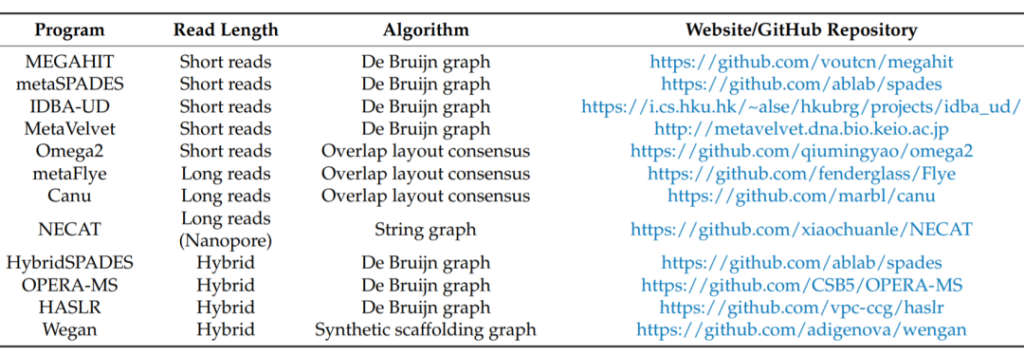
对于短读,推荐MEGAHIT,这是一个针对宏基因组优化的生物信息学组装工具,或者metaSPADES和IDBA-UD,它们也针对宏基因组进行了优化。
除了de novo,还有一种基于参考的组装,也可以用于宏基因组学。只是,并不是在所有情况下都可以获得合适的参考基因组,而且这种方法不能识别新的病毒或以前没有测序的病毒。
对于长读,推荐metaFlye、Canu和NECAT,这些工具可以用于各种技术下产生的数据格式,从纳米孔测序到PacBio,甚至在高保真序列。
对于混合组装,也就是将短读和长读的两个特性结合起来的组装,推荐OPERA-MS和HybridSPADES工具,它们都是用De Bruijn图算法实现的。
宏基因组组装完成,就应该确定组装的质量。用于此目的的工具可以分为两大类:
一类是需要参考基因组的工具,例如MetaQUAST,它使用参考来计算组装的统计信息。一般而言,在宏基因组学研究中,可能很难使用参考基因组,因为通常没有可用的参考基因组或参考基因组的质量很差。
不需要参考基因组的方法,例如DeepMAsED,它使用机器学习来识别错误装配,或者REAPR,是一种使用映射的配对端读长来评估基因组组装准确性的工具。常用的还有BUSCO和CheckM。
最后,VALET(https://github.com/marbl/VALET)可以用于检测宏基因组数据中的误组装,因为它可以根据覆盖范围对contigs进行分类,并避免由于覆盖深度不均匀而导致的假阳性和假阴性。

在宏基因组分析中识别病毒的一个重要步骤是进行物种注释。实现这一步骤的主要方法有两种:
两种方法各有优缺点:
在使用contig(即使用组装序列)进行的物种注释中,分类的对象是较长的序列,它存在一些contigs可能是嵌合的风险。
而直接对reads进行物种注释的统计学意义较小,虽然分析了大量的序列,但序列较短,这种方法可以提供更多样化的结果,只是计算成本会更高。
对于已知病毒的识别,一种是基于k-mer,直接使用参考数据库与reads/contigs进行比对,如kraken2、bracken、CLARK和Centrifuge,
另一种是先翻译序列,然后与参考蛋白质数据库进行比对,如kaiju、DIAMOND和MMseqs2。
还有基于算法的,如BLAST或DUDes,它们使用DUD(Deepest Uncommon Descent)算法。
使用基因标记的,如MetaPhlAn4、IGGsearch和GOTTCHA。
也有专门用来研究病毒组的工具,如VirusTaxo、Metavir2和DeepVirFinder,其主要算法是卷积神经网络(CNN)。
其中如MetaPhlAn4和MCP (Microbiota Community Profiler),包含未知的宏基因组组装基因组的序列,而MCP只能用于识别微生物区研究中的细菌、古菌、真核生物和病毒序列。
由于每个用于物种注释的工具的性能都不同,且都使用了各种算法和参考数据库,所以这种多样性也会导致不一样的结果、耗时和计算成本。
▪ 基于k-mer的物种注释工具似乎是计算效率最高的,虽然它们需要很大的内存。
▪ 基于标记的,对内存的要求较低,但它们只能对来自特定区域的reads/contigs进行注释分类。
▪ 基于比对的工具要比其他的计算成本更高。
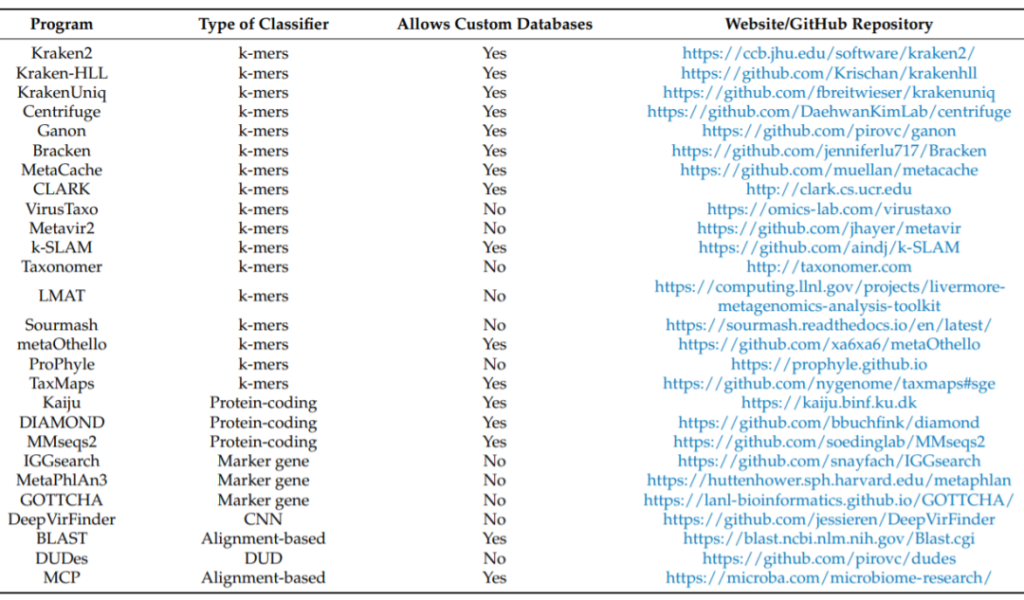
对于新型病毒的识别,现在也有不需要任何参考就能识别病毒序列的工具,即:
• VirSorter(https://github.com/simroux/VirSorter)
• VirFinder(https://github.com/jessieren/VirFinder)
VirFinder是一个基于k-mer的R包,可以以较好的预测识别病毒的contigs;
而VirSorter可以在不同的微生物数据集中识别新的病毒序列。
宏基因组分箱
在物种注释之前可以选择是否执行分箱(binning)。
分箱的主要目的是根据物种对contig进行聚类。根据数据类型,可使用的工具如下图:

CONCOT,它可以根据核苷酸组成和覆盖率数据对宏基因组contigs进行聚类。
GraphBin,它使用组装的连通性信息对contig进行集群化。
但宏基因组分箱并不局限于contigs,对于长读,可使用MEGAN-LR、BusyBee或LRBinner。
近年来,宏基因组学领域取得了许多进展,新技术可以帮助研究人员发现新的病毒,预测疫情,诊断某些疾病等。
长读测序平台也在快速发展,以得出更可靠的结果助力宏基因组分析。虽然已有许多工具和流程被开发出来以便更快更简单地进行数据分析,但还需要进一步发展,例如在数据处理分析中的通用指南的建立,因为虽然出于同一种目的而开发的工具,但由于计算过程不一样,它们在不同任务中的性能缺乏共识。此外,重要的是保持相关数据库的更新与维护。
宏基因组学检测人类样本中任何基因组(包括细菌、病毒、寄生虫和真菌)的能力,对于传染病的诊断具有重要意义。宏基因组学方法也已应用于其他几个研究领域:环境研究(如海洋样本、土壤、污水、农场灰尘) ;7000 年前青铜时代人类样本中的病毒感染;健康、疾病和法医调查中人体肠道微生物组的特征;临床研究 ; 以及新病毒病原体的发现,例如 SARS-CoV-2等。
Ibañez-Lligoña M, Colomer-Castell S, González-Sánchez A, Gregori J, Campos C, Garcia-Cehic D, Andrés C, Piñana M, Pumarola T, Rodríguez-Frias F, Antón A, Quer J. Bioinformatic Tools for NGS-Based Metagenomics to Improve the Clinical Diagnosis of Emerging, Re-Emerging and New Viruses. Viruses. 2023 Feb 20;15(2):587. doi: 10.3390/v15020587. PMID: 36851800; PMCID: PMC9965957.
谷禾健康
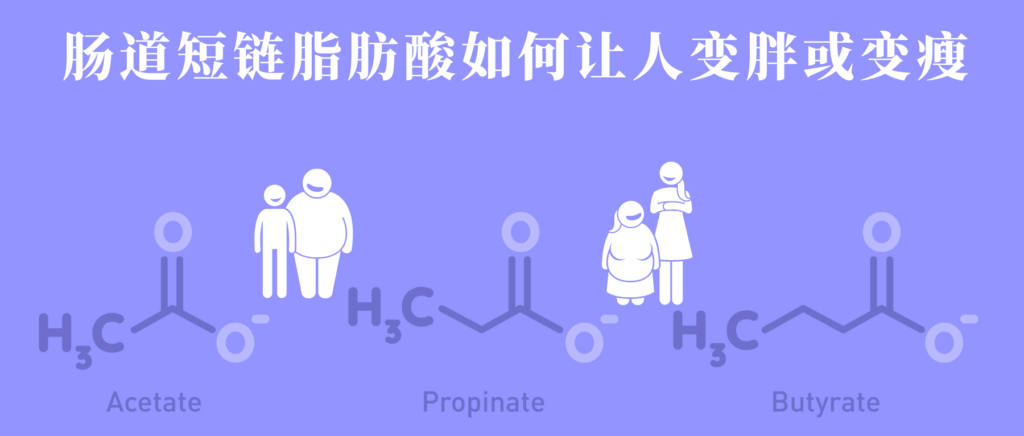
在目前的审美中,无论男性或女性的肥胖都是不太加分项。除此之外,肥胖还被认为是几种疾病的重要标志物,特别是高血压、2 型糖尿病 (T2DM) 和代谢综合征,肥胖在这些疾病中发挥着明确而重要的病理作用。
肥胖的发生有多种病因,其中遗传倾向、错误的饮食习惯(脂肪食物)和生活方式(缺乏运动)是重要的。除此之外,某些激素失衡和某些药物的副作用也有助于肥胖的形成和发展。但可惜这些致病因素无法解释所有肥胖的原因。
因此,世界范围内的研究正在继续寻找和探究,旨在找出上述因素以外的因素,以便对肥胖实行更好的管理和补救措施,从而防止这一世界性流行病的发展和由于其导致的严重病理负担。

治疗肥胖有多种方法,包括饮食控制、基于激励的锻炼计划和胃绕道手术等,但并不适用于所有人。
最新研究进展揭示了肠道微生物群的构成和代谢与肥胖发生之间存在的关系。肠道微生物会帮助消化大部分食物,将其转化为营养物质、神经递质、维生素、激素等。肠道微生物组通过这些代谢物,可以影响几乎所有代谢活动。
短链脂肪酸作为肠道菌群代谢产物之一,在肥胖中发挥重要作用,本文一起来了解一下。
短链脂肪酸(SCFA)是身体和饮食中的一种脂肪酸。链长是指化合物主链中的碳原子数 (C)。
短链脂肪酸(SCFA):5 个或更少的碳原子
中链脂肪酸(MCFA):6 至 12 个碳原子
长链脂肪酸(LCFA):13 至 21 个碳原子
极长链脂肪酸(VLCFA):22 个或更多碳原子
短链脂肪酸是少于 6 个碳 (C) 原子的脂肪酸。
其中乙酸盐 (C2)、丙酸盐(C3)和丁酸盐 (C4)是主要的短链脂肪酸,是在结肠中碳水化合物的细菌发酵过程中形成的。
短链脂肪酸的存在会使回肠(小肠末端)到盲肠(大肠起点)的肠道 pH 值降低,从而防止有害细菌(如肠杆菌科和梭状芽胞杆菌)过度生长。
短链脂肪酸有助于修复“肠漏”,通过增加粘蛋白 2 (MUC-2) 的分泌来加强肠壁,从而防止LPS穿过屏障。
目前肠道微生物组,已经被视为是免疫系统和部分能量调节器的 “器官”。肠道微生物群促进消化和食物吸收以产生宿主能量 ,并提供维生素和短链脂肪酸。
短链脂肪酸是由肠道中的友好细菌产生的,它们是结肠细胞的主要营养来源。
在结肠中存在的总短链脂肪酸中,健康人体内的90%-95%是乙酸盐、丙酸盐和丁酸盐。
大多数短链脂肪酸在结肠中被吸收,与碳酸氢盐交换。短链脂肪酸是酸性的,而碳酸氢盐是碱性的。
关于短链脂肪酸和体重的信息存在一些相互矛盾的信息:
总体而言,丁酸盐似乎对肥胖具有广泛的保护作用,丙酸盐具有混合关联,而乙酸盐与体重增加有关。
肥胖个体的粪便中短链脂肪酸浓度比瘦的个体高 20%。根据一些研究人员的说法,这可能反映了一种针对肥胖的补偿性保护机制,其中更多的量从粪便中排出。这将防止肠道中短链脂肪酸的积累增加,这可能导致体重增加。
丁酸盐和丙酸盐等短链脂肪酸会增加肠道激素胰高血糖素样肽 1 ( GLP-1 ) 和多肽 YY ( PYY ) 的形成。这些通过降低食欲来减少食物摄入量。丁酸盐和丙酸盐主要是抗肥胖的。
▼丁酸盐
丁酸盐主要被结肠细胞用作主要的能量来源。
丁酸盐的产生受产丁酸菌数量和大肠 pH 值的影响。丁酸盐主要由肠道中的厚壁菌门细菌产生,会影响大脑中的基因表达。产丁酸菌似乎在更酸性的环境(较低的 pH 值)中茁壮成长,而乙酸和丙酸盐细菌似乎在更碱性的环境(更高的 pH 值)中茁壮成长。
在结肠细胞的线粒体中,70%~90%的丁酸被氧化成乙酰辅酶A,随后通过三羧酸循环产生大量的ATP。丁酸盐已被发现可增加线粒体活性,预防代谢性内毒素血症,提高胰岛素敏感性,抗炎潜力,增加肠道屏障功能并防止饮食引起的肥胖。
除此之外,研究人员还在研究丁酸盐对抗自身免疫、癌症和心理障碍等方面的潜力。
▼丙酸盐
丙酸盐还通过门静脉循环到达肝脏,用于产生葡萄糖。丙酸盐是肝脏进行糖异生的主要能量来源,激活三羧酸循环,改变下丘脑食欲调节神经肽的表达谱,也能刺激脂肪组织增加瘦素的表达与释放。
丙酸盐已观察到可能促进或抑制肥胖的作用,但总体而言,它似乎对肥胖具有保护作用。
丙酸盐可抑制胆固醇合成,从而拮抗乙酸盐的胆固醇增加作用,并抑制脂肪细胞中抵抗素的表达。而且,已发现这两种短链脂肪酸通过其对厌食性肠道激素的刺激作用和增加瘦素的合成来引起体重调节。
▼乙酸盐
在所有三种短链脂肪酸中,乙酸盐似乎显示出更多的致肥胖潜力。它充当脂肪生成和胆固醇合成的底物。高脂肪饮食增加了 LPS 的吸收,而 LPS 又被发现与代谢性内毒素血症有关,并诱发炎症导致肥胖。
肠道中相当大比例的乙酸盐很容易被吸收并到达肝脏(通过门静脉循环),在那里它被用来制造胆固醇。
人体研究表明,在6名志愿者的饮食中给予乳果糖(经微生物群代谢产生大量乙酸盐)2周后,总胆固醇和LDL胆固醇、ApoB和血液中的乙酸盐水平均显著升高。
虽然乙酸盐主要是致胖的,但它也有一些可以防止肥胖的特性,其作用低于丁酸盐和丙酸盐。根据一些研究人员的说法,它可能通过增加GABA来对抗下丘脑(弓状核)的体重增加。
肥胖动物和人的微生物群落多样性较低,拟杆菌门、疣微菌的百分比较低,厚壁菌门和放线菌的百分比较高。但是其中一些结果没有并不能在所有研究中重现。
种属层面:

<来源:谷禾健康肠道菌群检测数据库>
以下微生物导致肥胖的证据较少:
与较瘦的受试者相比,肥胖小鼠和人类体内的产甲烷古菌含量更高。
M. smithii与B. thetaiotaomicron的共定植导致膳食果聚糖发酵成乙酸盐,从而显着增加脂肪生成。
M. smithii存在于 70% 的人类中,它会产生甲烷。已发现通过去除氢原子来增强多糖和其他碳水化合物的发酵,导致更多的短链脂肪酸产生,从而增加它们的吸收。这些短链脂肪酸作为额外的能量来源发挥作用,可能导致体重增加和随后的肥胖。
与瘦受试者和胃绕道手术后的受试者相比,肥胖个体的产氢普雷沃菌科(一种拟杆菌门)和甲烷杆菌目(古细菌,它们是一种氧化产甲烷菌)的数量处于较高水平。研究人员假设氢气减少了,这使得短链脂肪酸的产量更高。这导致更多的能量被肠道吸收。
已发现肥胖人类和小鼠的肠道菌群特征如下:
▼直接含有短链脂肪酸的食物
主要来源是乳制品,黄油等,其中含有丁酸盐。例如,黄油大约含有 3% 至 4% 的丁酸。这听起来可能不多,但它比大多数其他食物都多。推而广之,酥油还含有丁酸。
其他类型的乳制品也很重要。开菲尔和酸奶通常含有益生菌。这些益生菌可以帮助提高短链脂肪酸水平,只要没有乳糖吸收问题。
然而需要注意的是,以上食物如黄油类的并不能多吃,而通过食物改善肠道菌群组成,从而增加短链脂肪酸是相对有效和安全的方式。
因此,饮食方式对于调节体内短链脂肪酸较为重要,低碳水化合物、高脂肪(或高蛋白)饮食可能降低丁酸盐的产量,因为这样的饮食方式可能会剥夺肠道中产丁酸的细菌最喜欢的食物。那么具体该怎么吃?
▼通过菌群调节增加短链脂肪酸的食物
吃大量富含纤维和抗性淀粉类的食物,例如水果、蔬菜和豆类,与短链脂肪酸的增加有关。其中每一种都是由许多不同的底物(食物中的元素)产生的,并且影响不同的肠道微生物,进而影响不同的短链脂肪酸的产生,在调节体重方面发挥着不同的作用。
一项针对 153 人的研究发现,植物性食物摄入量增加与粪便中短链脂肪酸含量增加之间存在正相关关系。
▼纤维摄入量和类型间接影响短链脂肪酸生成
摄入的纤维量和类型会影响肠道细菌的组成,进而影响短链脂肪酸的产生。例如,研究表明,多吃纤维会增加丁酸盐的产量。
以下类型的纤维最适合在结肠中产生短链脂肪酸:
菊粉:进食菊粉刺激肠道菌群中芽孢杆菌属和拟杆菌属的细菌,产生短链脂肪酸显著提高。菊粉还能维持肠黏膜屏障的稳态,激活AMPK,缓解糖脂代谢紊乱,恢复免疫监控,防止肥胖等代谢性综合征的出现。
低聚果糖 (FOS):低聚果糖主要用作替代甜味剂。
抗性淀粉:抗性淀粉具有许多优点,与肠道健康特别相关,一些研究人员认为,吃富含抗性淀粉的饮食可以促进产丁酸菌的生长。抗性淀粉如绿色香蕉,还可以帮助降低血糖水平。
果胶:水果是果胶的最主要来源——通常含有 5% 至 10% 的果胶。
阿拉伯木聚糖:阿拉伯木聚糖存在于谷物中。
阿拉伯半乳聚糖,也称为甘露半乳聚糖,是一种有助于为肠道提供短链脂肪酸的糖类。
瓜尔豆胶跻身提供短链脂肪酸的食品之列。瓜尔豆胶是一种增稠剂,来自一种豆类。极少量的瓜尔胶对肠道有益,但大量会导致腹胀和胀气。
目前还有短链脂肪酸补充剂。最常见的类型是丁酸盐,如丁酸钠。这些补充剂是直接提供,而不是让身体去产生它。可能存在的问题是,使用这种类型的补充剂,丁酸盐通常在化合物到达结肠之前很久就被吸收了。这可能意味着带来的健康效果不佳,但是不能排除部分情况下补充剂可能仍然有用。
每种食物都有自己独特的营养成分,保持饮食多样性可以最大程度避免某些营养物质的缺乏。
一般人对饮食和补充剂的反应各不相同,因为每个人都有独特的“健康密码”。可能有些人吃了抗性淀粉或者高膳食纤维食物会出现腹胀,胀气等问题,可以进行肠道菌群健康检测,查看是哪些菌群超标可能带来的消化道问题。
需要深度挖掘自己的健康信息,并找到可能导致慢性健康问题的原因,然后根据个性化的建议调整,从而帮助达到最佳健康状态。
主要参考文献
Kallus SJ, Brandt LJ. The intestinal microbiota and obesity. J Clin Gastroenterol. 2012 Jan;46(1):16-24. doi: 10.1097/MCG.0b013e31823711fd. PMID: 22064556.
Chakraborti CK. New-found link between microbiota and obesity. World J Gastrointest Pathophysiol. 2015 Nov 15;6(4):110-9. doi: 10.4291/wjgp.v6.i4.110. PMID: 26600968; PMCID: PMC4644874.
Chakraborti CK. New-found link between microbiota and obesity. World J Gastrointest Pathophysiol. 2015 Nov 15;6(4):110-9. doi: 10.4291/wjgp.v6.i4.110. PMID: 26600968; PMCID: PMC4644874.
Blaak EE, Canfora EE, Theis S, Frost G, Groen AK, Mithieux G, Nauta A, Scott K, Stahl B, van Harsselaar J, van Tol R, Vaughan EE, Verbeke K. Short chain fatty acids in human gut and metabolic health. Benef Microbes. 2020 Sep 1;11(5):411-455. doi: 10.3920/BM2020.0057. Epub 2020 Aug 31. PMID: 32865024.
How Your Gut Microbiota Can Make You Fat (or Thin),Last updated: August 24, 2022,selfhacked
Ríos-Covián D, Ruas-Madiedo P, Margolles A, Gueimonde M, de Los Reyes-Gavilán CG, Salazar N. Intestinal Short Chain Fatty Acids and their Link with Diet and Human Health. Front Microbiol. 2016 Feb 17;7:185. doi: 10.3389/fmicb.2016.00185. PMID: 26925050; PMCID: PMC4756104.