-

 CNAS L23010
CNAS L23010

 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室

谷禾健康

癌症——这个困扰人类几个世纪的疾病,至今仍是全球死亡率最高的疾病之一。
国际癌症研究机构调查显示,2022年,全球有超过 1900 万新诊断的癌症病例和近1000 万癌症死亡病例,无数家庭因此经历痛苦。
癌症是一种复杂的多步骤慢性疾病,由活跃分裂细胞在 DNA 复制过程中获得的自发突变积累而成,环境也对癌症风险产生了相当大的影响。
传统的癌症治疗方法——手术切除、放射治疗和化学疗法,虽然在过去几十年中挽救了无数患者的生命,但它们的局限性也日益明显:手术无法完全清除微小转移灶;放疗和化疗的副作用严重,往往让患者雪上加霜;而且,即便是相同类型、相同分期的癌症患者,对同一治疗方案的反应也可能截然不同。
传统的癌症生物学理论难以完全解释这些差异。
这些问题引发了研究人员的深思:也许我们需要超越单纯关注癌细胞本身,将目光投向更广阔的“宿主环境”——患者的整体生理状态、免疫系统功能,以及体内的微生态系统。
从结直肠癌到肝癌,从乳腺癌到胰腺癌,微生物的”指纹”无处不在,它们通过复杂的代谢网络和免疫调节机制,参与肿瘤的发生、发展、转移和治疗反应全过程。
例如,肠道微生物群与宿主黏膜表面之间存在相互作用,后者在肿瘤发生中起着主要作用。微生物可能通过在胃黏膜中诱导氧化应激来损伤 DNA,增强上皮炎症并破坏黏膜屏障,从而增加肿瘤发生风险。
本文我们主要来了解微生物与癌症之间错综复杂的关系,探讨微生物如何成为癌症研究的新焦点,以及这一领域如何有可能彻底改变我们对癌症的认知和治疗方法。
随着年龄增长,我们的身体就像一台逐渐磨损的精密机器,各个部件的功能开始下滑。这种时间依赖性的细胞和生理功能退化过程,被称为”衰老“,它是多种重大疾病的主要风险因素之一,也包括癌症。
衰老的基本特征
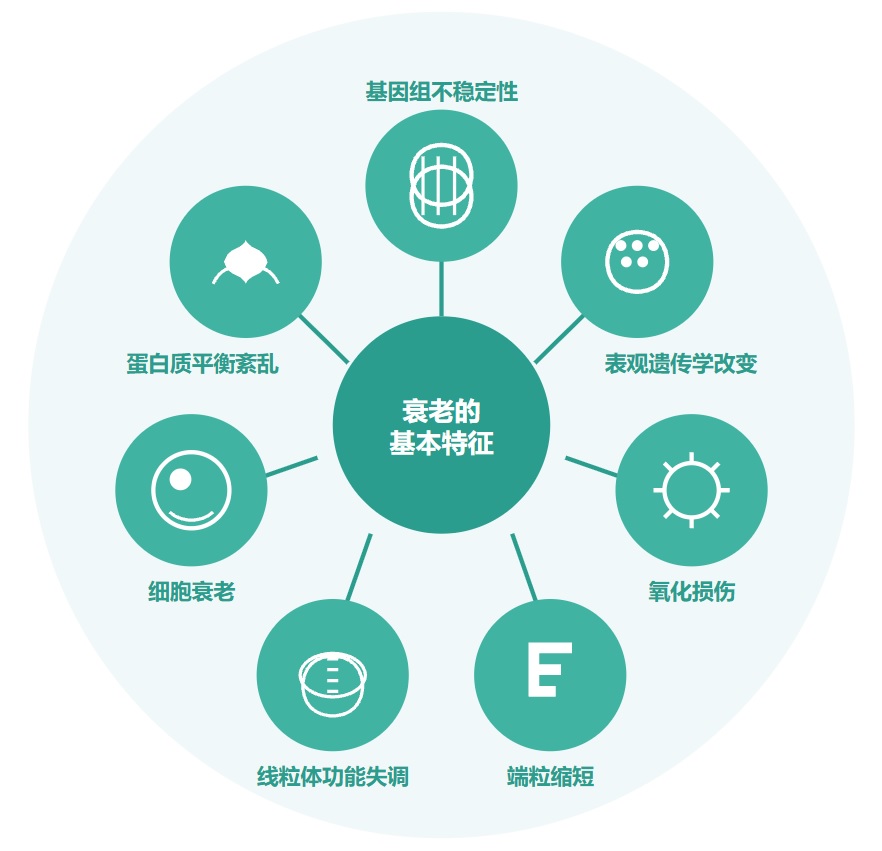
有人提出,尽管存在显著的个体间异质性和生活方式、饮食和治疗等外部因素的影响,但人类的肠道微生物组成在衰老过程中会逐渐变化。
多项报告表明,与有益菌相比,微生态失调在衰老过程中会增加,并导致促炎共生菌的大量存在,这些菌的富集,可以滋养生理性炎症。
对人类老龄化队列的研究表明,包括 Akkermansia、Anaerotruncus、Eggerthella、Bilophila 在内的一组属的丰度随着年龄的增长而显著增加,而 Faecalibacterium、Prevotella、Bacteroides 的丰度在老年人中相对较低。
衰老可能导致癌症,癌症治疗可能导致过早衰老

doi.org/10.1186/s12935-025-03787-x
微生物代谢物如何影响细胞衰老和健康?
短链脂肪酸,包括丁酸,可以帮助减缓细胞衰老。它们通过抑制组蛋白脱乙酰化酶来实现这一点,这调节代谢过程,增加胰岛素分泌,并调节免疫反应。
相比之下,致病菌的代谢产物,如脂多糖(LPS),通过加速炎症和增加氧化应激来增强细胞衰老,这是衰老的一个标志。
此外,铜绿假单胞菌和幽门螺杆菌产生的毒素会导致宿主 DNA 损伤并增加氧化应激。
因此,这加剧了 DNA 损伤反应、基因组不稳定性和细胞衰老。
菌群失调和细菌毒素促进衰老人群中衰老细胞的积累、DNA 损伤和促炎微环境的形成。这导致代谢紊乱,并创造一个促进肿瘤细胞生存和增殖的 肿瘤微环境,最终导致癌症。
癌症如何开始?
癌症起源于普通细胞中的遗传变化。当细胞DNA受到致癌物损伤时,会触发一系列变化。这些变化可能不会立即表现出来,但会逐渐积累。关键的基因突变会赋予细胞生长优势和抵抗死亡的能力,导致异常增殖,最终形成肿瘤。

癌细胞有什么特别之处?
癌细胞最显著的特征之一是它们的代谢方式发生了改变,也就是“代谢重编程”。正常细胞和癌细胞使用能量的方式完全不同。癌细胞即使在氧气充足的环境中也会进行糖酵解,这种现象称为”有氧糖酵解“。
此外,它们还进行大分子合成、维持氧化还原平衡,谷氨酰胺分解代谢,所有这些都有助于细胞快速增殖和生长。所有这些都有助于癌细胞快速分裂、扩散并侵入身体不同部位。
一个具体例子是D-2-羟基戊二酸的累积,这个代谢物抑制细胞分化,加速癌症发展。这是由于异柠檬酸脱氢酶-1/2 的连续突变。这表明通过抑制分化直接促进癌症的发生和进一步发展。
一个肿瘤内部并非完全一致,不同区域的细胞可能有不同的代谢特性,这种现象称为肿瘤异质性,这使得癌症治疗更加复杂。

肿瘤微环境:癌症的”生态系统”
肿瘤微环境(TME)是另一个在癌症发生和进展中发挥关键作用的组成部分。
肿瘤微环境是围绕癌细胞形成的高度组织化区域,就像一个小型社会,其中癌细胞只是众居民之一。其他成员如下图:

这个微环境如何影响癌症发展?
癌细胞能够改造邻居,使整个环境有利于肿瘤生长。
比如,癌细胞怎样获得养分和氧气?它们促使血管内皮细胞形成新血管。肿瘤相关巨噬细胞(TAMs)被招募后,会分泌生长因子和细胞因子,刺激血管生成和肿瘤侵袭。
癌细胞甚至能重新编程癌细胞相关成纤维细胞(CAFs),使它们分泌ECM蛋白和血管生成因子(如VEGF-A)。ECM则作为信号分子的储存库,增强细胞间通讯、粘附和迁移。
因此,TME 的不同组成部分通过影响各种细胞过程,有助于癌症的发生、进展和迁移。更深入地了解调节癌症进展和可塑性的分子机制,有助于开发精确和针对性的癌症治疗疗法,并预防复发。
微生物如何影响癌症发展?
最直接的联系是某些微生物直接致癌的能力:
动物研究显示,微生物可促进多种癌症的发展,包括:

doi.org/10.1186/s12935-025-03787-x
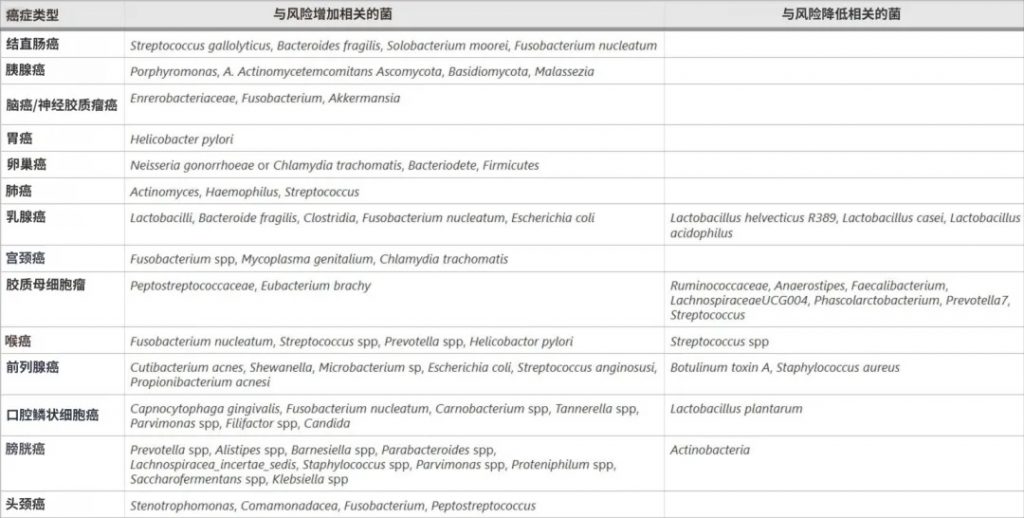
微生物群如何变化?
高通量DNA测序揭示了癌症相关的微生物群落变化:
doi.org/10.1186/s12935-025-03787-x
微生物如何促进癌症?
微生物可能通过多种机制促进肿瘤发生:
了解微生物与癌症的关系,可能帮助我们开发新的癌症预防策略、提高治疗效果,甚至预测哪些患者可能对特定治疗产生耐药性。
实际上,微生物群可以同时扮演促进肿瘤发展和抑制肿瘤生长的双重角色,这取决于肿瘤进展的阶段以及微生物群的功能和分布位置,那么,哪些微生物会致癌?又是如何作用的?
微生物的促癌作用
结直肠癌
脆弱拟杆菌、大肠杆菌、Streptococcus gallolyticus、Enterococcus faecalis、Fusobacterium nucleatum、Parvimonas micra等在结直肠癌患者肠道中大量存在,通过分泌短链脂肪酸等代谢物、DNA甲基化、组蛋白修饰等表观遗传机制影响肿瘤形成。
胰腺癌
牙龈卟啉单胞菌、奈瑟菌、放线菌、链球菌、双歧杆菌、拟杆菌、梭杆菌等与胰腺癌的发生有关,它们通过引起炎症和免疫抑制影响肿瘤生长。
口腔微生物群和幽门螺杆菌通过诱导炎症而成为胰腺癌症的危险因素。
乳腺癌
对人类乳腺癌症组织16S rRNA基因序列的分析显示,与早期乳腺肿瘤相比,晚期乳腺肿瘤中卟啉单胞菌、Lacibacter、Ezakiella、Fusobacterium的丰度更高。
具核梭杆菌(Fusobacterium nucleanum)和脆弱拟杆菌通过定植于乳腺肿瘤和促进癌症细胞的自我更新来加重乳腺癌症的生长和转移。
脑癌
Enterobacteriaceae、Fusobacterium、 Akkermansia是脑癌患者肠道中的主要菌群,它们可能通过免疫抑制、激活炎症、限制细胞死亡以及促进血管生成和侵袭性来影响胶质瘤。
微生物虽然不能穿过血脑屏障,但能释放具有穿透能力的细胞外囊泡,这些囊泡有能力穿过屏障进入大脑。
其他
其他癌症相关的微生物还包括:
这些菌在相应的癌症中大量存在,并通过改变代谢和免疫反应、增强炎症和毒性以及改变信号通路来提高肿瘤易感性。
微生物的抗癌作用
一些微生物如乳杆菌、双歧杆菌、Faecalibaculum rodentium、Streptococcus thermophiles等具有抗肿瘤特性,它们主要通过以下机制抑制肿瘤生长:
调节免疫系统
一组共生肠道细菌,如Paraprevotella xylaniphila、Bacteroides dorei、 Parabacteroides distasonis,可以诱导 CD8 T 细胞产生 IFNγ,抵抗单核细胞增生李斯特菌感染,并增强免疫检查点抑制剂在小鼠体内的疗效。
产生抑制性毒素
其他细菌,如铜绿假单胞菌、鼠伤寒沙门氏菌、艰难梭菌,分别在黑色素瘤、胰腺癌和乳腺癌癌症中表现出抗肿瘤特性。它们通过产生抑制增殖、使细胞停滞在G1-S期并诱导细胞凋亡的毒素来实现这一点,从而促进抗癌活性。
嗜热链球菌还抑制细胞增殖,引发细胞周期阻滞,增强体外结直肠细胞的凋亡,并减少结直肠癌异种移植物的生长。此外,肠道微生物群通过抑制结肠lncRNA Snhg9和上调p53表达来限制小鼠结直肠癌癌症的进展。
产生有益代谢物
– 微生物本身及其毒素能抗癌
科利毒素:赋予宿主抗肿瘤免疫力。
产气荚膜梭菌毒素(CPE)能识别乳腺癌、前列腺癌等细胞表面高表达的claudin-3/4蛋白,与这些蛋白结合后直接引发癌细胞死亡。
Akkermansia muciniphila在肝癌小鼠模型中减少免疫抑制细胞(单核MDSC、M2巨噬细胞),同时增强PD-1免疫治疗的疗效。
– 微生物代谢产物:短链脂肪酸抗癌
丁酸钠,在非小细胞肺癌中消除肿瘤细胞生长,诱导细胞周期阻滞,促进细胞凋亡,并改变免疫反应。
异丁酸,通过减少肿瘤体积来提高癌症结肠癌小鼠模型中抗PD-1免疫疗法的效率。
色氨酸,是人体必需氨基酸,其代谢途径包括:
拟杆菌、Clostridium sporogenes、 Eubacterium、Ruminococcus gnavus等菌群通过吲哚途径产生吲哚-3-乳酸、吲哚-3-丙酸、吲哚-3-乙酸等代谢物,这些物质既能抑制肿瘤免疫逃逸,也可能促进癌细胞生长,具体作用取决于代谢物种类和浓度。
吲哚代谢物可改善化疗和免疫治疗效果,但菌群失衡可能导致色氨酸代谢紊乱,反而加速癌症进展。
微生物与肿瘤微环境的相互作用
具核梭杆菌的Fap2蛋白可与抑制性受体TIGIT结合,抑制NK细胞和T细胞清除肿瘤的能力。
幽门螺杆菌与微环境中的巨噬细胞互动,诱导其极化为M2样巨噬细胞,促进胃癌进展。
肿瘤内微生物也参与肿瘤生成和进展,例如:
也有报道称,一些瘤内微生物可以增强抗肿瘤免疫反应,从而抑制肿瘤进展。
了解微生物在癌症中的双重作用,可能为开发新型抗癌疗法提供思路,包括调节微生物群落和利用微生物产物来增强现有治疗方法的效果。
转移是癌症细胞的一个关键标志,大多数癌症类型的晚期通常以转移开始为特征。
转移,是指癌症细胞通过血液循环从原发肿瘤转移到其他器官的继发部位。它涉及上皮间质转化(EMT)、迁移、侵袭、外渗和继发部位的定植。
转移过程包括哪些步骤,癌细胞面临哪些挑战?
这个过程对癌细胞来说充满了物理和化学挑战,包括需要穿越坚硬的细胞外基质、承受液体剪切应力、逃避免疫监视,并最终在可能与原发部位环境截然不同的部位建立殖民地。
微生物如何协助癌细胞转移?
– 乳腺癌:细菌随转移扩散
– 具核梭杆菌的多重促转移机制
有核梭杆菌可诱导乳腺癌、结直肠癌、喉癌的转移。它是口腔中常见的革兰氏阴性菌,通常与牙周病、口臭有关。
结直肠癌

doi.org/10.1186/s12935-025-03787-x
喉癌
– 抗生素治疗的启示
当然,微生物并不总是促进转移,已知许多微生物群也能抑制转移,从而为防止癌症进展提供了一种自然的方法。
微生物如何阻止转移?
– 抑制转移的菌群代谢物
– 调节肿瘤微环境
膀胱癌中的菌群关联:
– 抗癌微生物的鉴定
前列腺癌数据分析:
肿瘤内微生物种类在转移中的重要性在其他癌症中也得到了证实(下表)。

doi.org/10.1186/s12935-025-03787-x
这些研究表明,癌症中微生物的特异性靶向可以抑制肿瘤的转移,从而成为抗癌联合治疗的重要组成部分。未来需揭示菌群与宿主互作的具体机制,探索“以菌治癌”的精准策略,为癌症治疗提供新突破口。
微生物不仅参与癌症的发生和发展,还可能影响癌症治疗的效果。为什么有些患者对治疗产生耐药性?微生物可能是其中一个关键因素。
微生物如何影响抗癌疗效?
微生物群主要通过两种方式来影响抗癌疗效:
这样一来,药物的疗效和对癌细胞的毒性都会发生变化。
以环磷酰胺(CTX)为例,它是一种常用的抗癌药,用于治疗各种血液系统恶性肿瘤和实体瘤。
环磷酰胺不仅能杀伤癌细胞,还能调节肠道微生物的组成。它会促使一些特定的革兰氏阳性细菌从肠道转移到次级淋巴器官,而这些细菌到达那里后,会激活致病性 T 辅助细胞,从而增强环磷酰胺的抗癌活性。
但如果通过某种方式消除肠道微生物,比如用无菌小鼠或给小鼠喂食抗生素,特别是针对Barnesiella intestinihominis、Enterococcus hirae等特定菌群时,就会发现一个很棘手的问题:环磷酰胺的抗癌效果会变差,耐药性出现了。这说明肠道微生物群在化疗耐药性中扮演着重要的角色。
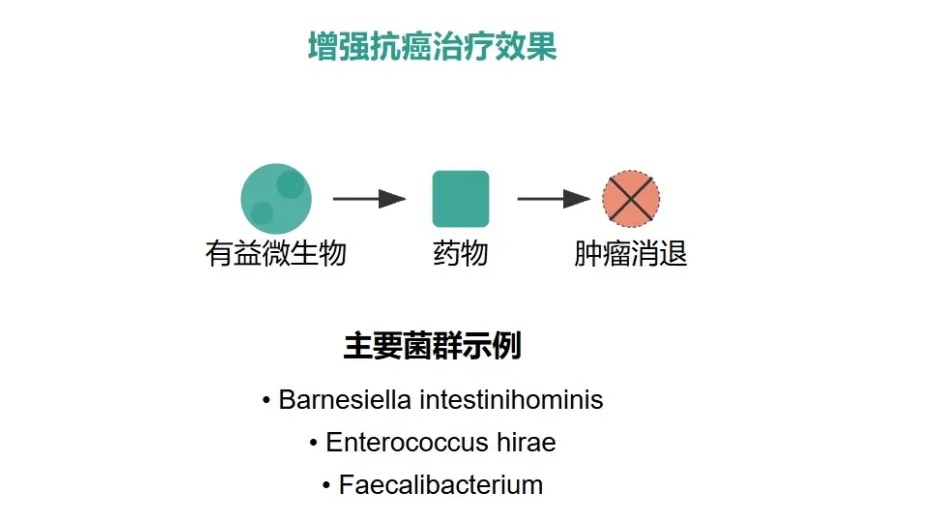
不过,这里又出现了一个违反我们直觉的情况。抗生素虽然可以杀菌,但用多了反而会导致细菌耐药性增加。
研究还在不断深入,科学家们发现:
微生物群调节有助于提高癌症治疗的效率,并通过改变代谢和免疫反应促进更好的预后。
比如,García-González 等人做了一项很有意思的研究,大肠杆菌可改变线虫体内代谢通路,增强5-氟尿嘧啶(FUDR)的疗效。
同样,在黑色素瘤患者的治疗中,研究发现粪杆菌Faecalibacterium能帮助增加免疫细胞和抗原呈递,从而让细胞毒性 CD8 + T 细胞在肿瘤床的浸润程度更高,引发大规模的免疫反应,这对抗 PD-1治疗很有帮助。
基于这些发现,有人提出一个大胆的想法:
肠道微生物群也可以用作生物标志物,来预测患者化疗和免疫治疗的治疗反应和疗效。
肝癌患者的肠道菌群失衡指数(Ddys) 反映了HCC患者粪便样本中的微生物干扰,可用于预测治疗效果。
失衡指数是根据HCC患者粪便样本中有益菌与有害菌的相对丰度计算的。
工程菌成为抗癌新武器
直接杀伤癌细胞:
靶向递送药物:
激活免疫:
现有研究揭示了微生物群在癌症治疗中的多重价值:增强药效、预测疗效、直接作为治疗载体。需深入解析不同癌症特异的菌群特征,明确特定菌种的作用机制,同时解决工程菌的生物安全性问题。
化疗不仅直接作用于肿瘤,同时也会对人体内的微生物群落产生显著影响。肠道微生物在应对化疗时,会通过代谢和免疫调节来影响化疗的疗效和毒性。
化疗如何破坏肠道菌群平衡?
化疗不仅攻击癌细胞,还会显著扰乱肠道微生物的组成。例如,在肠道中,化疗可能会破坏黏液层,使部分肠道微生物能够穿透黏膜层,引发免疫反应。
在结直肠癌患者中,化疗会降低肠道菌群的多样性,这种变化可能反向影响治疗效果——菌群结构越失衡,化疗耐药风险可能越高。
菌群网络的重构:竞争还是合作?
梭杆菌、拟杆菌、粪杆菌分别与肿瘤标志物CEA、CA724、CA242相关,但它们的丰度不受不同化疗阶段的影响。这提示特定菌群可能成为化疗反应的独立预测指标。
注:CEA是一种糖蛋白,最初在胚胎组织和结肠癌组织中发现。作为最早被应用于临床的肿瘤标志物之一,CEA主要用于消化系统肿瘤的辅助诊断和监测。
CA724是一种高分子量糖蛋白,是胃癌较为特异的肿瘤标志物。
CA242是一种新型的消化系统肿瘤标志物,主要用于胰腺癌和胆道系统肿瘤的辅助诊断。
菌群如何“对抗”化疗药物?
某些细菌通过代谢转化直接削弱药物活性:
菌群失衡是否会加剧化疗副作用?
5-氟尿嘧啶(5-FU)为例
5-氟尿嘧啶用于治疗乳腺癌、结直肠癌、胃癌、胰腺癌、胃癌等。其作用方式涉及DNA损伤,导致细胞凋亡或RNA合成抑制。
菌群也能增强化疗效果吗?
奥沙利铂和丁酸盐为例
奥沙利铂是晚期结直肠癌患者的一线化疗药物。
研究表明,肠道微生物产生的代谢物丁酸盐能增强奥沙利铂的抗癌效果。
丁酸盐通过依赖ID2的方式激活CD8 + T细胞。在结直肠癌患者中,对奥沙利铂有反应的患者血清丁酸盐水平高于无反应患者。这表明,肠道微生物产生的丁酸盐可能是决定患者对奥沙利铂反应的关键因素。
临床转化方向
预测性生物标志物
精准调控策略
化疗与肠道菌群存在双向影响:药物破坏菌群平衡,菌群则通过代谢和免疫调控反作用于药物疗效。未来研究需结合菌群检测,在分子机制与临床干预之间架起桥梁,最终实现精准治疗策略。
多项研究表明,放射治疗和微生物群之间存在相互交织的关系。虽然放射治疗可以杀死有益的微生物群,但某些微生物群也可以通过影响患者的免疫系统来提高放射治疗的敏感性。
例如,口腔中的具核梭杆菌会被转移到结直肠肿瘤部位,从而降低放疗的治疗效果。
而甲硝唑是一种针对具核梭杆菌的抗生素,研究发现,在结直肠癌小鼠模型中,甲硝唑可以作为一种放疗增敏剂,提高放疗的效果。
微生物群导致放疗抵抗
丁酸盐在肠道健康中通常被认为有益,为何在放疗中反而有害?
由Lachnospiraceae合成的丁酸盐与放疗抵抗有关
细菌和真菌对放疗的影响相反
小鼠模型(黑色素瘤、乳腺癌)研究发现:
关键分子:免疫受体Dectin-1(感知真菌感染)高表达的乳腺癌患者生存率较差。抑制Dectin-1可提升放疗疗效。
临床转化方向
精准菌群干预:
免疫微环境重塑:
传统癌症治疗方法存在很多局限性,例如对正常细胞的附带损伤、产生治疗耐药性的可能性以及无法完全穿透肿瘤等。因此,迫切需要开发新的、更好的癌症治疗方法。有文献表明,对癌症患者的肠道微生物群进行干预可以增强当前抗癌药物的疗效,如化疗和免疫疗法。
微生物群提高各种肿瘤免疫疗法的效率
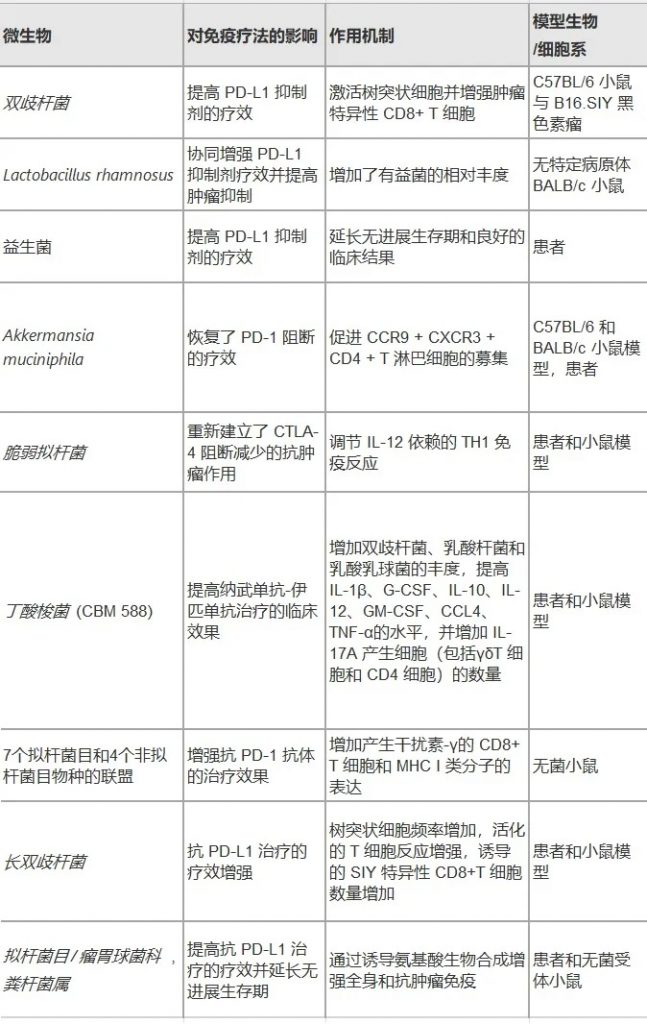
doi.org/10.1186/s12935-025-03787-x
细菌疗法
早在1868年,William Coley发现细菌感染可导致肿瘤消退,开创了细菌疗法的先河。现代研究揭示了细菌靶向肿瘤的独特机制:
研究人员改造了专性厌氧鼠伤寒沙门氏菌菌株YB1,并通过在神经母细胞瘤小鼠模型的肿瘤核心注射这种修饰的细菌观察到对肿瘤生长的抑制。
重组减毒沙门氏菌菌株SL7207被用作在黑色素瘤小鼠模型中递送工程肿瘤疫苗的载体。活细菌也可以与纳米粒子结合,形成有效的药物递送系统。
挑战:
细菌介导的抗癌疗法带来了一些挑战,包括半衰期短、DNA不稳定性和微生物的内在致病潜力。
基因工程有助于删除致病菌株的一些毒力基因,从而可以控制其抗肿瘤活性、特异性和定植。
目前,一些临床试验正在确定功能化鼠伤寒沙门氏菌菌株的效果。这些菌株要么通过各种遗传技术进行工程改造,要么通过纳米粒子或其他试剂进行表面修饰,以显示出所需的肿瘤靶向和定植。
粪菌移植 (FMT)
最近的研究表明,使用FMT重塑微生物群失调可以潜在地抑制癌症进展,特别是结直肠癌。
结直肠癌(CRC)
移植健康小鼠粪便至CRC模型,逆转菌群失衡,增加CD8+ T细胞浸润,减少促炎因子(IL-6、IL-17),抗炎细胞因子IL-10增加,抑制肿瘤进展。
临床试验进展

注:部分试验中FMT导致腹泻等副作用,提示需优化供体筛选。
宏基因组分析显示,在反应者中,FMT后Prevotella copri、Ruminoccocaceae、Eubacterium丰富。此外,与非反应者相比,反应者在FMT后一个月的粪便样本在小鼠模型中抑制肿瘤生长的能力更强。
涉及粪便微生物移植的临床试验

doi.org/10.1186/s12935-025-03787-x
关于 FMT的研究我们之前也写过,详见:
益生菌
益生菌如何帮助对抗癌症?
最近的一项队列研究中,发现益生菌的低等和中等摄入量与癌症死亡率的降低显著相关。
益生菌通过以下机制发挥作用:
鼠李糖乳杆菌(LGG)通过释放脂磷壁酸(LTA)激活TLR2信号,保护肠道干细胞免受放疗损伤。
另一种益生菌Prohep由鼠李糖乳杆菌GG(LGG)、具有活性的大肠杆菌Nissle 1917(EcN)和热灭活VSL3组成,用于在HCC小鼠模型中通过减少Th17细胞和IL-17细胞因子来减少肿瘤生长。
工程化益生菌有何突破?
益生菌要适量
益处:适量补充益生菌可降低癌症死亡率。
风险:过量可能干扰免疫治疗,如黑色素瘤模型中PD-1抑制剂疗效下降。
益生元
益生元——激活菌群抗癌潜能的“燃料”
益生元选择性地促进有益菌(如产丁酸菌)增殖,协同抗癌治疗。
益生元如何优化药物递送?
临床启示与风险管控
个体化菌群干预:
动态监测必要性:
风险提示:
免疫缺陷患者慎用活菌制剂,优先选择灭活益生菌或纯化代谢产物。
总的来说,益生菌双刃剑效应显著,需严格把控种类和剂量;而益生元通过调节菌群代谢,相对安全增强化疗/免疫疗效。
益生菌+益生元+传统疗法可能成为癌症治疗新方向,但需更多临床验证。
抗癌药物
为什么同种药物在不同患者中效果差异显著?
肠道微生物群还参与抗癌药物的生物转化和代谢,导致这些药物的差异吸收和生物利用度。在药物的生物转化过程中,微生物群采用各种机制,如脱氨基、水解、去甲基化、葡萄糖醛酸化和其他反应。
伊立替康(CPT11):
5-氟尿嘧啶(5-FU):
如何减轻菌群介导的副作用或耐药?
抑制特定酶活性:
调控菌群组成:
壳寡糖通过降低肠球菌、大肠杆菌-志贺氏菌、Turicibacter的密度以及促进丁酸产生菌的生长,对结直肠癌具有保护潜力。

临床潜在方向
未来,建立“菌群-药物代谢”数据库,结合人工智能预测个体化用药方案。例如,对高β-葡萄糖醛酸酶活性的患者,优先选择不受该酶影响的化疗药物(如奥沙利铂)。
关于常用药物和肠道菌群之间复杂的双向相互作用,我们之前的文章也写过,详见:
为什么药物对人效果不一?探索药物-微生物群相互作用对效果的影响
全球癌症治愈率依然不高,肠道微生物群在癌症发生、发展、转移和药物反应中的关键作用正逐渐成为研究热点,为精准抗癌策略提供了全新思路。
菌群在癌症治疗中的关键作用
微生物通过多种机制影响癌症治疗效果:
doi.org/10.1186/s12935-025-03787-x
当前挑战:从实验室到临床的鸿沟
– 研究方法的“碎片化”
样本收集(粪便 vs 肿瘤组织)、测序技术(16S rRNA vs 宏基因组)的差异导致结果难以比较;
解决方案:建立全球统一的微生物组分析标准。
– 个体化差异
年龄、饮食、地理因素使菌群组成差异巨大——同一疗法在不同人群中的响应率波动;
突破口:开发基于AI的个体化菌群图谱,预测治疗敏感性与毒性风险。
– 治疗复杂性的叠加
在现有化疗/免疫治疗基础上引入菌群调控,可能引发不可预见的药物-微生物相互作用;
未来方向:迈向精准菌群医学
– 精准菌群分层
通过多组学技术(宏基因组+代谢组)构建个体化菌群图谱,识别“促癌菌”与“抑癌菌”。
针对不同化疗方案调整菌群结构,丁酸水平低者优先使用奥沙利铂联合益生元。
– 联合干预策略
饮食-菌群协同:高纤维饮食联合特定益生元(如菊粉),可使MEK抑制剂疗效提升。
个性化膳食(如高纤维饮食)联合益生菌,改善肠道健康并增强治疗响应。
结合基因、饮食等多维度数据定制治疗方案。
工程菌开发:设计靶向降解耐药相关酶(如β-葡萄糖醛酸酶)的工程菌。
– 临床转化路径
推动大规模临床试验,验证菌群移植(FMT)、益生元/菌在特定癌种中的疗效。
探索菌群标志物(如丁酸盐水平)作为治疗响应预测指标。
开发微生物群-人工智能联合预测模型,优化治疗决策。
将菌群检测纳入癌症辅助诊疗,实现早筛与疗效监控。
总的来说,微生物组研究不仅扩展了我们对癌症发生发展的认知,更为癌症治疗提供了新的可能性。通过系统化的菌群检测与精准干预,跨学科协作与技术创新,未来有望实现真正的个体化治疗方案,提高抗癌疗效并改善患者生活质量。这一领域的进步,代表着癌症治疗从单一靶向向生态系统整体调控的深刻范式转变。
主要参考文献
Adlakha YK, Chhabra R. The human microbiome: redefining cancer pathogenesis and therapy. Cancer Cell Int. 2025 Apr 28;25(1):165.
Shi Z, Li Z, Zhang M. Emerging roles of intratumor microbiota in cancer: tumorigenesis and management strategies. J Transl Med. 2024 Sep 11;22(1):837.
Murayama M, Hosonuma M, Kuramasu A,et al., Isobutyric acid enhances the anti-tumour effect of anti-PD-1 antibody. Sci Rep. 2024 May 17;14(1):11325.
Jia D, Kuang Z, Wang L. The role of microbial indole metabolites in tumor. Gut Microbes. 2024 Jan-Dec;16(1):2409209.
Sun X, Shan X, Zhu B, Cai Y, et al., 5-Fluorouracil Loaded Prebiotic-Probiotic Liposomes Modulating Gut Microbiota for Improving Colorectal Cancer Chemotherapy. Adv Healthc Mater. 2025 Feb;14(4):e2403587.
Zhang H, Xu Z. Gut-lung axis: role of the gut microbiota in non-small cell lung cancer immunotherapy. Front Oncol. 2023 Nov 22;13:1257515.
Wang S, Yin F, Guo Z, Li R, Sun W, Wang Y, Geng Y, Sun C, Sun D. Association between gut microbiota and glioblastoma: a Mendelian randomization study. Front Genet. 2024 Jan 4;14:1308263.

谷禾健康
肠道微生物群通过代谢发酵膳食纤维等不被人体直接吸收的物质,产生了一类重要的代谢产物——短链脂肪酸。
短链脂肪酸主要包括乙酸盐、丙酸盐、丁酸盐,它们不仅仅是肠道上皮细胞的重要能量来源,更是联结肠道-免疫-代谢-大脑轴的信号分子。通过与特定受体(如GPR41/GPR43)结合,它们能调节肠道蠕动、维护肠屏障完整性、调节免疫反应、影响食欲和能量代谢,甚至可能影响神经系统功能。
随着微生物组研究的深入,我们逐渐认识到肠道微生物与人体健康之间存在着错综复杂的关系网络,而短链脂肪酸恰是这一网络中的核心。短链脂肪酸并非独立存在的简单分子,而是复杂生态系统平衡的产物和指标。它们反映了微生物群落的结构与功能,同时也塑造着宿主的生理状态。正是这种动态平衡的特性,使得短链脂肪酸在不同条件下表现出迥异的生物学效应。
因此,短链脂肪酸的健康效应高度依赖于机体状态、剂量水平以及具体应用场景,这远比简单的”好”与”坏”复杂得多。以丁酸盐为例,在健康状态下,它是肠道健康的守护者;但在肠道屏障受损或炎症状态下,同样的丁酸盐可能延迟伤口修复,甚至加重炎症反应。丙酸盐也展现类似特性:低浓度时可能有益肠道健康,但高浓度时可能产生毒性效应,甚至与自闭症等神经发育障碍存在争议性关联。
本文将全面梳理短链脂肪酸的生物学特性及健康影响,探讨短链脂肪酸异常的相关干预措施。更全面地理解短链脂肪酸,不仅有助于我们认识肠道微生物组与人体健康的深层联系,也为未来的个性化营养干预、精准医疗和健康管理提供了理论基础。
* 本研究参考并整合了微生物学领域知名专家Lucy博士和MacFabe博士的部分研究总结与学术观点。作为肠道微生物与人体健康研究方向的权威学者,他们长期致力于探索肠道菌群、营养代谢与疾病发生机制之间的科学关联。
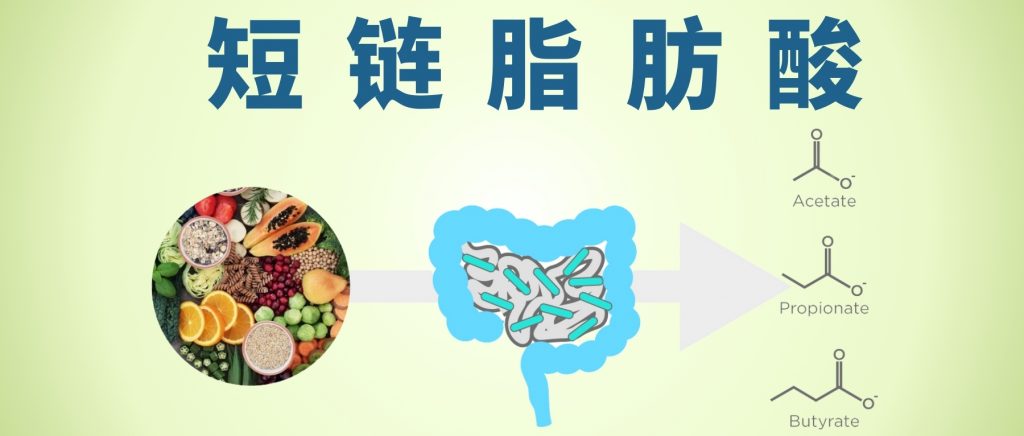
短链脂肪酸是包含 6 个或更少碳分子的脂肪酸的一个子集。它们包括乙酸盐(C2)、丙酸盐 (C3)、丁酸盐(C4)、戊酸(C5)、己酸(C6)。最近的科学进展主要集中在乙酸盐、丙酸盐、丁酸盐上,因此这三个将是本系列文章的主要关注点。
可以在下面看到它们的化学结构:
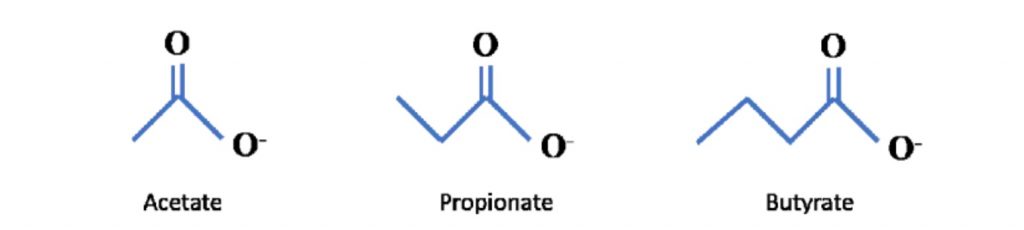
哺乳动物消化道不擅长代谢某些纤维。相反,菊粉、低聚果糖和抗性淀粉等膳食纤维很容易被肠道中的微生物发酵,短链脂肪酸是这种发酵的主要产物。
短链脂肪酸去哪里?
短链脂肪酸要么被肠道上皮细胞局部使用,要么通过肠道上皮运输到门静脉。
丁酸盐主要被结肠上皮细胞用作能量来源,而丙酸盐和乙酸盐主要通过门静脉运输到肝脏。
丙酸盐被肝细胞迅速代谢。乙酸盐可以留在肝脏中或释放到外周循环中。
信号分子和 HDAC 抑制剂
短链脂肪酸是有效的信号分子,与专门的 G 蛋白偶联受体(GPCR)结合,并最终改变细胞和组织的生物化学性质。这些受体可以在免疫细胞、神经细胞、甲状腺、肾脏、胰腺、脾脏、肝脏、其他组织上找到。
注:G蛋白偶联受体(G Protein-Coupled Receptors, GPCR)是一类重要的膜蛋白,在细胞信号转导中扮演关键角色。作为人体内最大的膜蛋白受体家族,GPCR参与调控众多生理过程。
它们也是基因表达的有效修饰因子,影响各种细胞类型的表观遗传学。特别是丁酸盐,它是组蛋白脱乙酰酶的有效抑制剂,组蛋白脱乙酰酶是负责确定 DNA 卷曲的紧密程度以及因此转录成 RNA 的程度的酶。
注:组蛋白脱乙酰是由组蛋白去乙酰化酶(HDACs)催化的过程,该过程通过调节染色质结构和基因表达参与几乎所有关键生物过程,包括基因表达,细胞分化,DNA修复和生长发育方面,是表观遗传调控的核心机制,也是重要的疾病治疗靶点。
SCFA 具有广泛的影响
通过这些机制,SCFA 能够对宿主生理产生广泛的影响。SCFA 决定结肠运动、血流和胃肠道 pH 值,这会极大地影响电解质和其他营养物质的摄取和吸收。它们也是结肠健康和肠道屏障完整性的重要促进剂,并在维持正常的肠道和免疫功能方面发挥着重要作用。
SCFA 也被证明会影响神经系统和大脑。丁酸盐已被证明可以调节小胶质细胞(大脑的免疫细胞) 的活性,而丙酸盐则被认为与自闭症谱系障碍的发展和进展有关,这个将在本系列的下面 部分中详细介绍这个主题。
SCFA 的产生也可能在塑造肠道微生物生态学中发挥重要作用。SCFA 在低浓度下表现出广谱抗菌活性。有趣的是,SCFA 对产生它们的细菌种类是相对惰性的抗菌剂,但对其他微生物具有相当强的抗菌活性。
注意,SCFA 总是好的吗?不一定。
SFCA 对宿主健康的总体影响存在广泛争议。例如,尽管刺激饱腹感信号,但 SCFA 也与增加从饮食中收集的能量有关。短链脂肪酸在高浓度或特定疾病条件下也可能具有毒性,将在下面详细讨论。
改变的SCFA水平会是疾病的原因吗?
SCFA 可能影响多种疾病的发病机制,包括过敏、哮喘、癌症、肥胖、代谢性疾病、自身免疫性疾病和神经系统疾病。但是请考虑以下事项:
一些研究仍在试图确定这些关联背后的机制。粪便 SCFA 测量有一些主要局限性,并且 SCFA 水平改变是这些疾病的原因还是结果仍然未知。更好地了解 SCFA 对于描述肠道和肠道微生物群在慢性疾病中的作用至关重要。
丁酸盐是肠道中产生的一种代谢物,对大脑、皮肤、免疫系统等都有好处,其分子结构如下:
编辑
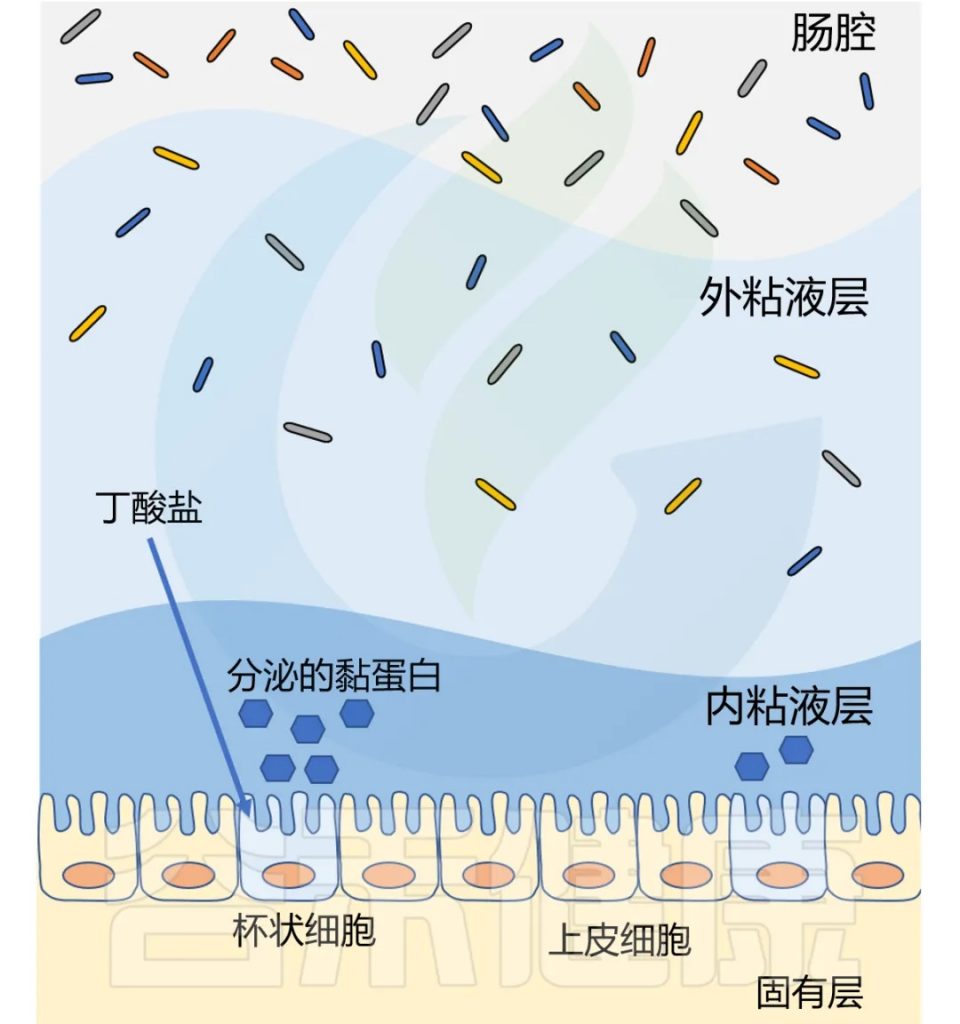
丁酸盐保护肠道
丁酸盐是结肠细胞的首选底物,可提供结肠上皮细胞 60-70% 的能量需求。
丁酸盐抑制炎症、调节免疫、促进紧密连接蛋白组装,增强肠道屏障功能。
丁酸盐还影响粘液层,健康的结肠上皮覆盖着双层粘液。厚厚的内层致密无菌,保护上皮细胞免受微生物侵袭;外层松散,容纳以粘液层糖蛋白为食的细菌。
两个粘液层均由杯状细胞分泌的MUC2粘蛋白构成。研究表明,补充生理浓度的丁酸盐可增加人杯状细胞系中MUC2基因的表达和MUC2的分泌。
丁酸盐改善代谢功能
丁酸盐还影响新陈代谢。在肥胖小鼠模型中,补充丁酸盐被证明可以提高胰岛素敏感性、增加能量消耗和减少肥胖。它还增加了线粒体的数量及其在骨骼肌和棕色脂肪组织中的活性。一系列研究证实丁酸盐反应受体在脂肪组织中高度表达。
但是这种表达随着年龄的增长而下降,这可能解释了与年龄相关的胰岛素敏感性下降。丁酸盐还诱导饱腹感激素的产生,从而减少食物摄入量。
丁酸盐也可能对其他代谢疾病有益。研究表明,生命早期口服丁酸盐可延缓大鼠糖尿病的发展。丁酸盐补充剂还可以通过改善肠道屏障功能和纠正微生物失调来减轻饮食诱导的小鼠脂肪肝疾病。
丁酸盐改变基因表达
虽然我们的基因构成基本上是不可改变的,但基因的表达方式却具有高度可塑性。
在我们身体每个细胞的细胞核中,DNA 缠绕在组蛋白周围。
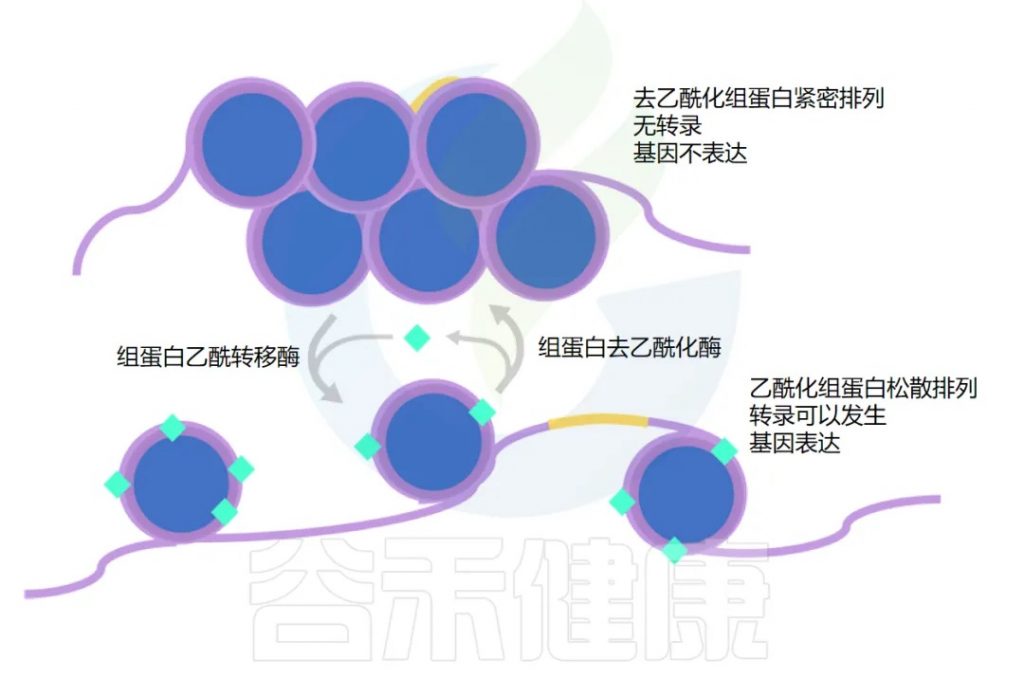
那么什么决定了基因是乙酰化还是去乙酰化呢?
组蛋白乙酰转移酶(HAT)负责添加乙酰基,而组蛋白去乙酰化酶(HDAC)负责去除。细胞内的许多信号分子可以影响这些酶的活性,丁酸盐就是其中之一。
丁酸盐通过两种不同的方式增加乙酰化,用于不同的基因集。
这两种机制都有助于增加乙酰化并保持基因“开启”。下一节将探讨其如何在癌细胞中发挥作用。
丁酸盐有抗癌作用
长期以来,人们一直知道丁酸盐可以刺激上皮细胞增殖,但同时对结肠癌具有保护作用。换句话说,丁酸盐可以在健康和癌变的结肠细胞中发挥不同的作用。
研究揭示了丁酸盐悖论背后的机制,实验表明 Warburg 效应是其解释的关键。Warburg效应描述了快速分裂的癌细胞的代谢特点。
注:Warburg效应是由德国生物化学家Otto Warburg于20世纪20年代发现的一种代谢现象,描述了肿瘤细胞的特殊能量代谢方式:即使在氧气充足的环境中,癌细胞也主要通过糖酵解而非有氧呼吸产生能量。
◆ 丁酸盐促进增殖
正常上皮细胞主要通过线粒体三羧酸循环(TCA循环)代谢丁酸盐等脂肪酸,产生能量。TCA循环通量增加,导致胞质溶胶中柠檬酸盐积累,柠檬酸盐可在细胞核中转化为乙酰辅酶A。该乙酰辅酶A为组蛋白乙酰转移酶(HAT)提供乙酰基,乙酰化调控上皮细胞增殖的基因。

◆ 丁酸盐的抑制作用
另一方面,癌细胞往往具有功能失调的线粒体,这意味着它们无法代谢脂肪酸(如丁酸盐)以获取能量。正因为如此,它们在很大程度上依靠葡萄糖代谢来产生能量,而丁酸盐等脂肪酸往往会在细胞中积累。这种细胞丁酸盐浓度的增加导致 HDAC 抑制增加,以及阻止癌细胞复制的基因表达。
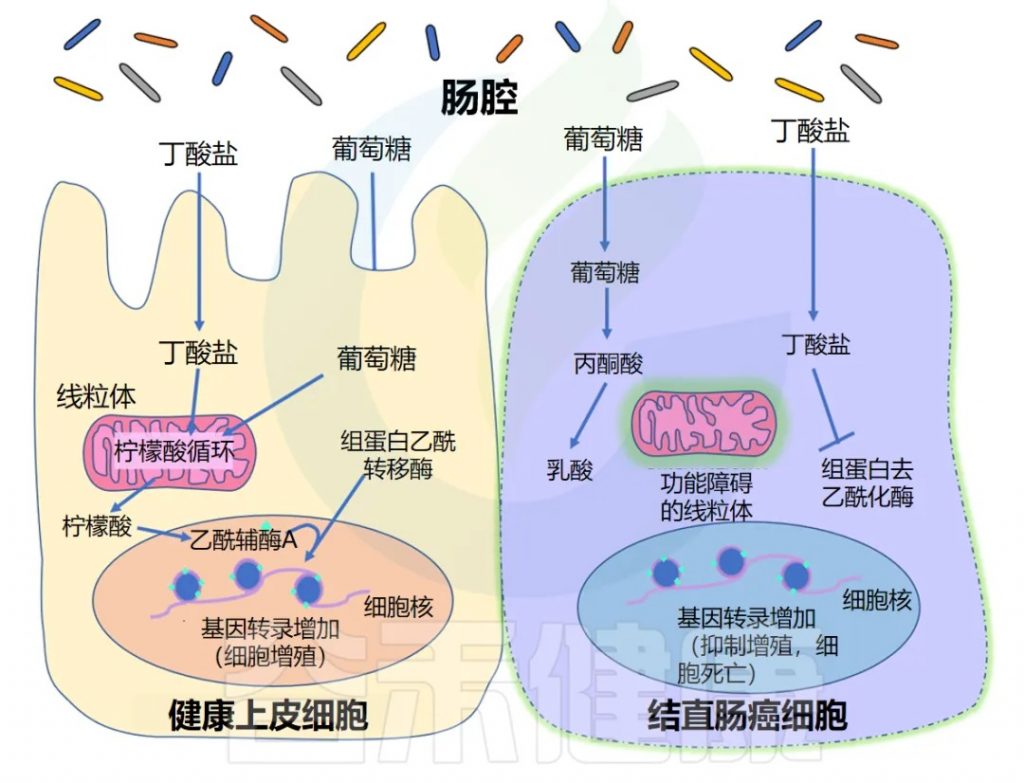
这种现象并非结肠癌所独有。丁酸盐已被确定在多种人类癌细胞系中具有抗癌活性,包括舌癌、前列腺癌、 肝癌、乳腺癌、肺癌、神经母细胞瘤等。
丁酸盐可能有益于大脑
丁酸盐对大脑健康的影响已在动物模型中得到广泛证实。
◆ 脑损伤保护
研究表明,给予丁酸钠或产丁酸梭菌的小鼠,在缺血性脑损伤后表现出神经发生增加、氧化应激减少和恢复改善。丁酸钠还能恢复血脑屏障的完整性,并减轻创伤性脑损伤后的神经功能缺损,同时可预防脊髓性肌萎缩症中的神经变性。
◆ 抗抑郁效应
在抗抑郁方面,丁酸盐可改变小鼠海马体和额叶皮层中脑源性神经营养因子(BDNF)等基因的表达,并增加血清素水平,同时减少药物诱导的双相情感障碍模型中的躁狂样行为,增加神经营养因子表达,并在慢性轻度应激和母体分离模型中减轻了认知障碍。
◆ 记忆增强作用
丁酸盐对记忆形成和保留也具有潜在作用。研究表明,丁酸盐通过抑制 HDAC 可以减轻海马胃泌素释放肽受体(GRPR)信号阻断导致的认知障碍,并使大脑处于“可塑性准备状态”,类似于运动的效果。
注:海马胃泌素释放肽受体 (GRPR) 的紊乱可能会抑制适当的记忆形成和消退,并导致与神经发育障碍相关的认知障碍。
最后,丁酸盐可能对自闭症谱系障碍有益,相关讨论将在SCFA不平衡和丙酸盐-自闭症连接部分展开。
丁酸盐调节免疫系统
丁酸盐在调节免疫系统方面也起着重要作用,几乎在所有类型的免疫细胞上都发现了丁酸盐反应受体。
丁酸盐信号传导产生抗炎效应,抑制促炎细胞因子的产生并上调抗炎细胞因子的产生,即使在脂多糖(LPS)等炎症刺激下也能发挥作用。这可能是通过 HDAC 抑制 NF-kB 通路来实现的,
注:NF-kB 通路是参与炎性细胞因子释放的主要通路。已在许多不同的免疫细胞类型中观察到丁酸盐介导的 HDAC 抑制。
丁酸盐还促进结肠和外周调节性T 细胞 (Treg) 的产生,这有助于抑制免疫反应,影响 Treg 和效应 T 细胞功能,并诱导抗菌肽素(Cathelicidin)的活性,从而增强对细菌感染的先天防御能力。
注:cathelicidin 是一种有效的抗菌剂,已知在结肠和肺上皮细胞中对细菌感染的先天防御中起重要作用。
丁酸盐和皮肤
由于很多朋友的小孩湿疹病史和对肠道皮肤轴的实验研究,小编感兴趣的是丁酸盐对皮肤健康的影响。
丁酸盐充当皮肤免疫系统的调节剂。皮下或局部应用丁酸盐被证明可以减少接触超敏反应,这可能是通过观察到调节性 T 细胞的增加和炎症效应 T 细胞的减少来解释的。丁酸盐还诱导胶原蛋白的合成,胶原蛋白是皮肤的重要结构成分。
丁酸盐改善骨骼健康
骨骼在成骨细胞(骨骼构建)细胞活性和破骨细胞(骨分解)细胞活性之间的微妙平衡中不断重塑。存在于骨髓中的间充质干细胞(MSC)可以分化成骨细胞(造骨细胞)或脂肪细胞(脂肪细胞)。
﹝仅供参考﹞
增加丁酸盐的方法
我们已经了解到丁酸盐是基因表达的有效修饰剂,对宿主健康有广泛的益处。想要增加丁酸盐,有几种方法可以做到这一点:
增加产生丁酸盐的细菌的丰度。具有特别高丁酸盐生产能力的细菌种类属于厚壁菌门和拟杆菌门,包括梭菌门、真杆菌属(Eubacterium)、罗氏菌属(Roseburia)、粪球菌属(Coprococcus)和普拉梭菌(Faecalibacterium prausnitzii)等。
双歧杆菌(Bifidobacteria)和乳酸杆菌(Lactobaccilus)等属也可能通过产生乙酸盐和乳酸盐来促进丁酸盐的产生,然后其他细菌可以将其转化为丁酸盐。
增加产生丁酸盐的细菌可用的底物。这意味着吃含有可发酵纤维的益生元食品,这些纤维会刺激丁酸盐的产生。一些最有效的产丁纤维包括抗性淀粉和低聚果糖。
补充剂。这应该非常谨慎,使用正确的补充剂形式。下面将对此进行说明。
过量的丁酸盐有毒吗?
目前各种形式的口服丁酸盐补充剂。但我们真的应该服用丁酸盐吗?丁酸盐过多是一件坏事吗?
查阅很多文献,发现一些研究表明,低浓度的丁酸盐是有益的,但高浓度可能是有害的,尤其是对于那些肠道受损的人。
在本文中,我们将分析几项阐明丁酸盐悖论的研究。首先,我们先来了解肠道解剖结构,这对于了解丁酸盐对肠道的影响至关重要。
肠道结构与丁酸盐的关联
◆ 肠道解剖学:隐窝和绒毛
肠道上皮在人的一生中不断更新。肠壁内陷形成隐窝,而向外突出形成绒毛(villi)。隐窝有点像一个 “山谷”,把绒毛想象成 “山”。
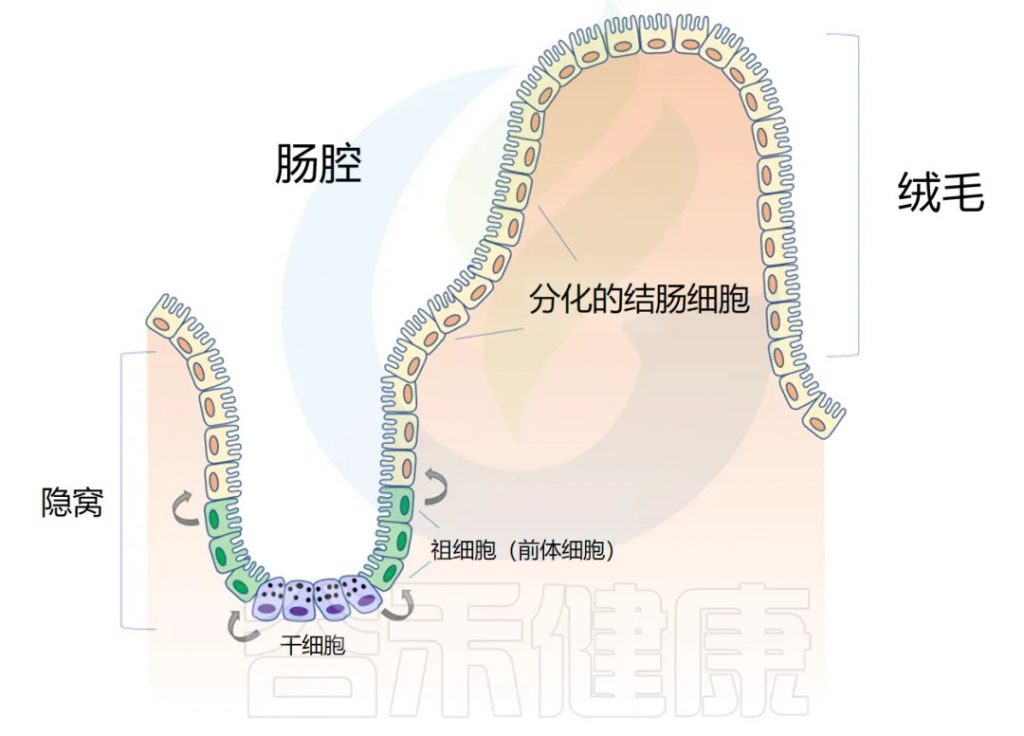
干细胞位于隐窝底部,周期性产生祖细胞,这些祖细胞在绒毛中向上移动时反过来分化为成熟的结肠细胞(colonocytes)。这补充了在顶部脱落的旧细胞。通过这个过程,整个肠道上皮大约每 4-5 天翻新一次。
注:肠道内壁覆盖着一层称为上皮的细胞组织,这些细胞负责吸收营养物质并形成保护屏障。这些上皮细胞不会长期存在,而是会定期死亡并被新细胞替代。这个更新过程非常快速 – 大约每4-5天,肠道内所有的上皮细胞都会被完全更换一遍。这种快速更新有几个重要作用:
这是人体中细胞更新最快的系统之一,反映了肠道作为消化和吸收器官以及防御屏障的重要性。
◆ 隐窝结构对干细胞的保护功能
人们已知隐窝已有数个世纪,但其结构功能直到近些年才被阐明。1974年,Cheng和Leblond提出隐窝可能保护快速分裂的干细胞和祖细胞,使其免受肠腔内的病原体和毒素侵害。

2016年,华盛顿大学的研究小组于《CELL》上发表的研究进一步佐证了该假设。他们利用高通量筛选方法研究微生物代谢物对干细胞增殖的抑制作用,结果他们发现在所有代谢物中,发现丁酸盐是抑制干细胞和祖细胞增殖最有效的抑制剂。
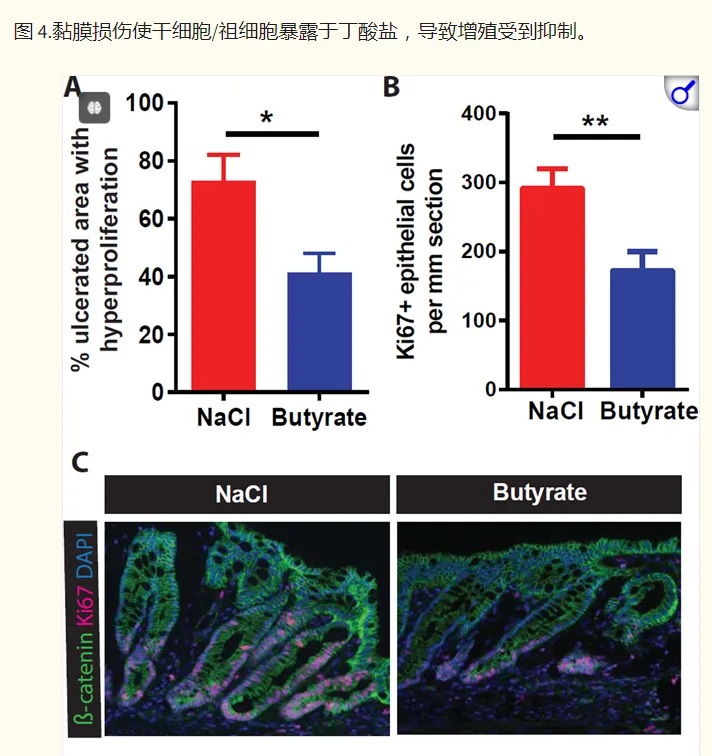
doi:10.1016/j.cell.2016.05.018
隐窝底部干细胞缺乏丁酸盐代谢酶 → 避免暴露于高浓度丁酸盐
然而,在活体生物中,丁酸盐并未导致这种抑制。他们想知道是否隐窝底部的干细胞没有代谢丁酸盐。为了找出答案,他们使用含有放射性标记13C的丁酸盐来评估不同位置丁酸盐代谢的速率。
他们发现,与隐窝底部的干细胞和祖细胞相比,隐窝顶部细胞中13C标记的乙酰辅酶A(丁酸盐氧化的最终产物)水平高出3倍。

作者总结了这些发现:
位于隐窝顶部的分化结肠细胞可以将丁酸盐作为能量来源代谢,从而可能防止干细胞生态位暴露于高水平的管腔丁酸盐。
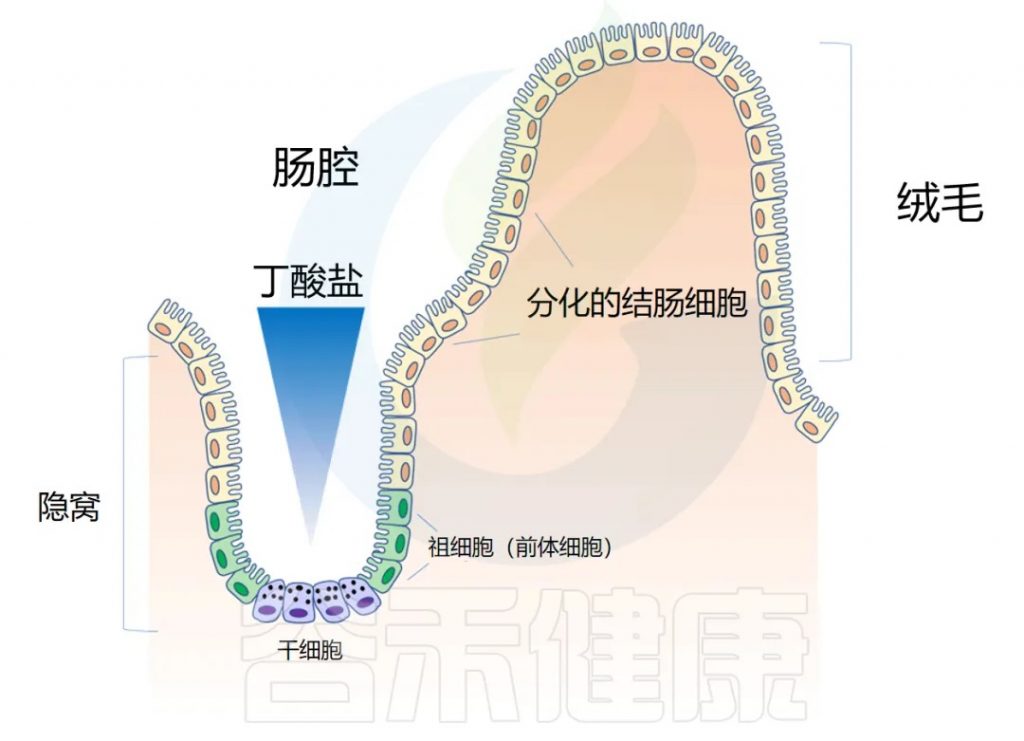
炎症性肠病中的丁酸盐
◆ 丁酸盐延迟炎症模型中的伤口修复
然后,研究人员转向结肠炎的小鼠模型,以了解在丁酸盐存在下隐窝结构的破坏如何影响干细胞增殖。抑制干细胞增殖将阻止愈合粘膜损伤所需的上皮细胞更新。
他们发现,给予丁酸盐可减少与结肠溃疡直接相邻的隐窝中的上皮增殖,并延迟伤口修复。
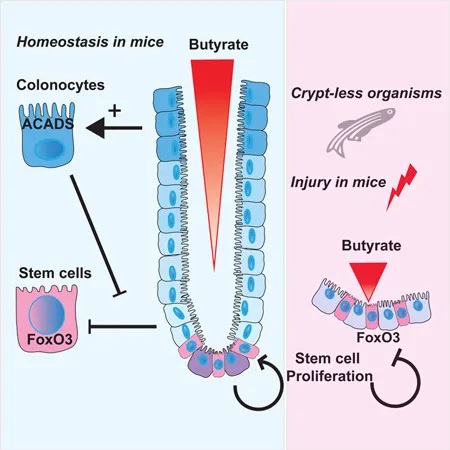
◆ 产丁酸菌与可发酵纤维在IBD中的双重效应
在另一项实验中,他们发现使用抗生素敲除产生丁酸盐的微生物可以减少结肠炎损伤后的溃疡大小。重新引入丁酸盐或产生丁酸盐的微生物导致大溃疡复发。
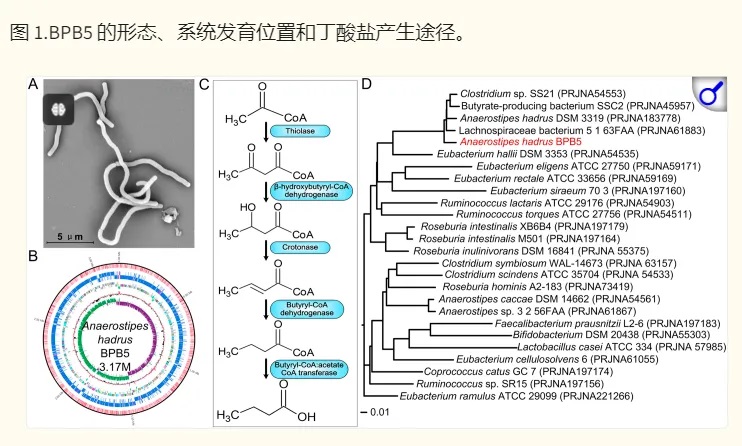
上海交大一项类似的研究(如下),将一种源自人类的产丁酸盐细菌株Anaerostipes hadrus BPB5 引入患有和不患有结肠炎的小鼠中。

另一项研究发现,补充已知可刺激丁酸盐产生的可溶性纤维菊粉导致 IBD 易感(IL-10 敲除)小鼠结肠中的促炎基因表达增加。这在对照野生型小鼠中没有发生。
总之,这些研究表明,丁酸盐、丁酸盐生产菌,甚至潜在的可发酵纤维的好处,都取决于肠道的状况。这些数据也可能部分解释为什么一些炎症性肠病患者在减少 FODMAP 摄入量后似乎有所改善。
同理,上海交大研究团队还指出:对健康个体可能有益的细菌也可能会成为入侵物种,从而对肠道微生物群被疾病破坏的宿主造成致命后果。这样的案例在免疫功能低下个体中引起感染甚至败血症、溃疡性结肠炎、短肠综合征、癌症等的报道均时有发生。
◆ IBD中,产丁酸菌丰度低:原因还是后果?
IBD 患者产丁酸菌水平降低,长期以来被认为是疾病的诱因。然而,一项研究引发了疑问:
缺乏丁酸盐生产者是否仅仅是疾病的结果?
炎症肠道有没有可能为了保护干细胞,抵抗丁酸盐生产菌,从而促进干细胞增殖和粘膜修复?
作者对自身研究的解读:
短期内,丁酸盐可能不利于伤口修复,但长期来看,它可能使宿主受益。通过抑制上皮损伤后干细胞的快速扩增,防止干细胞在接触遗传毒性管腔内容物时分裂。
尽管这个解释有其道理,但更严谨的科学需要不断验证。长期以来,丁酸盐都被认为是有益的,因此理解丁酸盐在肠道完整性受损时可能有害这一新范式颇具挑战。当然如果更多研究就更好了。
◆ 溃疡性结肠炎患者的丁酸盐氧化受损
体外和体内研究的证据表明,溃疡性结肠炎 (UC)患者丁酸盐的氧化受损。De Preter 等人发现,UC 活检中的平均丁酸盐氧化率降低到几乎是对照活检的一半。

根据疾病严重程度分层,非活动性和轻度疾病的丁酸盐氧化率与对照组无异,但中度和重度疾病的丁酸盐氧化率显著降低。值得注意的是,对照组之间的丁酸盐氧化也存在显著差异。
进一步研究丁酸盐动力学表明,丁酸盐浓度并非限制因素。对照组和UC活检的管腔丁酸盐浓度增加至1 mM以上并未提高丁酸盐氧化,表明超过1 mM的饱和点后,丁酸盐浓度不再是限制因素。
作者写道:
“…局部应用较高浓度的丁酸盐并不能克服UC中的粘膜能量不足,并且通过不可消化的碳水化合物或丁酸盐灌肠剂刺激丁酸盐的产生可能对治疗这种疾病无效。”
一项后续的研究表明,丁酸盐氧化减少是由于发炎粘膜对丁酸盐的吸收减少,这与促炎细胞因子下调 MCT1 的表达有关。MCT1 是一种转运蛋白,负责包括丁酸盐在内的多种分子的转运。此外,丁酸盐对 IBD 患者的抗炎作用也降低。
临床试验、动物研究和细胞培养实验显示结果喜忧参半
事实上,当在溃疡性结肠炎中测试丁酸盐灌肠剂或丁酸盐治疗时,动物研究和人体临床试验得出了相互矛盾的结果。
人类研究:丁酸盐灌肠剂研究的矛盾结果
在两项小型非安慰剂对照研究以及两项对照交叉研究中发现了丁酸盐灌肠剂的有益作用。
一项针对轻度至中度结肠炎患者的小型随机安慰剂对照研究发现,含有乙酸盐、丙酸盐和丁酸盐的 SCFA 灌肠剂具有有益作用

目前为止,大型的随机安慰剂对照试验(91名患者)表明,SCFA灌肠对结肠炎没有治疗价值,即使是病情较轻、发作短暂的患者也未能显著获益。
另一项大型随机安慰剂对照试验(38名患者)也未发现生理盐水安慰剂与丁酸盐灌肠之间存在显著差异,接受盐水灌肠的患者甚至比接受丁酸盐灌肠的患者临床改善比例更高(9/19 vs 7/19)。这些研究都没有显示丁酸盐的明确有害影响,但也没有评估其潜在粘膜毒性。

动物研究中的剂量依赖性效应
一项研究发现,口服丁酸钠可减轻小鼠的结肠炎。
但高浓度丁酸盐灌肠可诱导小鼠细胞凋亡和 UC 样病变。
在结直肠癌的小鼠模型中,菊粉和直肠内丁酸盐都已被证明可以改变结肠上皮并增加肿瘤的形成。
在小鼠中,慢性口服 SCFA 诱导 T 细胞介导的输尿管组织炎症,导致肾积水,即肾脏中的积液。
摘要如下:
“当 SCFA 以高于生理水平的水平全身给药时,会导致肾脏系统中的 T 细胞反应失调和组织炎症。”

细胞培养:浓度与环境决定效果
值得注意的是,现在有几项研究表明,SCFA 可以促进免疫调节或免疫激活,具体取决于炎症环境。换句话说,在正常状态下,SCFA 促进免疫调节;在炎症状态下,它们会促进免疫反应的放大。
此外,以低浓度(2mM)施用丁酸盐到上皮培养模型通过加速紧密连接蛋白的组装来改善肠道屏障完整性。然而,在高浓度(8mM)下,丁酸盐会增加屏障通透性。
丁酸盐还促进致病性大肠杆菌中毒力因子的表达。因此,这些大肠杆菌倾向于在丁酸盐水平最高的结肠中定植。
我们应该考虑补充丁酸盐吗?
有很多人发现了丁酸盐补充剂的好处,就如前面提到的。大部分试验中的一些患者确实看到了丁酸盐灌肠的临床改善,丁酸盐当然有很多好处。然而,目前还没有在人类中进行口服丁酸盐补充剂的双盲、安慰剂对照试验,所以需要谨慎的更细致入微的方法。
严重粘膜损伤时应避免直接补充
如果您有严重的粘膜损伤,例如高度活动性溃疡性结肠炎,倾向于建议不要使用丁酸盐灌肠剂或补充剂。
那些较轻的病例、缓解期或难治性结肠炎患者,或没有胃肠道疾病的人可以考虑丁酸盐。尽管如此,避免超生理水平还是很重要的。
直到最近,最广泛可用的丁酸盐形式是丁酸盐,例如丁酸钠或丁酸钙/镁。然而,丁酸盐可能会部分溶解在口中。高浓度口服丁酸盐与牙周炎有关,并可能抑制牙龈上皮细胞的增殖。
益生元:更自然的丁酸盐来源
由于肠道发酵的性质,益生元纤维也可能在结肠中提供更缓慢、更分散的丁酸盐释放。从肠道解剖结构和肠道生态学是在富含可发酵纤维的饮食中进化而来的,这是我们肠道最习惯的丁酸盐前体。
除了产生丁酸盐之外,益生元还有许多好处,包括对肠道上皮的抗炎作用和有益微生物的增殖。要注意的是,在严重的肠道炎症和粘膜损伤的情况下,即使是大量的益生元也可能不利于愈合。
健康状态下,丁酸盐通过多种机制促进肠道健康,包括增强肠屏障功能、促进黏蛋白MUC2的产生、调节免疫反应、抑制炎症,有抗癌潜力。它也是肠道上皮细胞提供主要能量来源。
然而,在肠道损伤或炎症状态下,丁酸盐的作用可能完全相反。研究表明,当肠隐窝结构受损,高浓度丁酸盐直接接触干细胞时,会抑制细胞增殖,延迟伤口修复。在炎症性肠病患者中,丁酸盐的代谢和转运机制受损,使得补充更多丁酸盐可能无法发挥预期效果,甚至可能加重病情。
这种复杂性表明,丁酸盐补充不应一刀切,而需根据个体健康状况、肠道完整性以及具体需求进行个性化评估和应用。肠道菌群检测成为个体化营养干预的重要工具。
对于产丁酸菌缺乏但肠道功能正常的个体,可适度补充丁酸盐;对于肠道屏障功能受损者,则应谨慎使用丁酸盐,可能需先修复屏障再考虑补充。
未来研究应关注丁酸盐作用的剂量-反应关系,明确不同肠道状态下的最佳干预策略,并将肠道菌群分析结果整合到个体化方案中,以实现丁酸盐益处的最大化和风险的最小化,从而更精准地应对不同人群的健康需求。
丙酸盐(Propionate)含有三个碳原子,羟基(-OH)位于第二个碳原子上。
而前面讲的丁酸盐(Butyrate)含有四个碳原子,羟基(-OH)位于第四个碳原子上。
丙酸盐通常由肠道细菌通过发酵L-鼠李糖、聚葡萄糖、阿拉伯木聚糖、D-塔格糖、甘露寡糖、昆布多糖等糖类物质产生。
丁酸盐可以通过肠道微生物群发酵富含抗性淀粉和果聚糖的食物来增加,如菊粉、马铃薯、洋葱等。
丙酸盐的生理作用
肠道健康与代谢调节
丙酸盐刺激肠道平滑肌收缩,增加黏液分泌,促进抗菌肽表达,扩张结肠动脉,并通过释放血清素调节肠道内分泌功能。它还能通过调节胆囊簇细胞促进胆汁释放,防御肠道细菌入侵。
糖代谢:丙酸盐是肝脏糖异生的主要能量来源,可激活三羧酸循环,影响下丘脑食欲调节神经肽,增加瘦素释放,抑制食欲。
脂代谢:抑制胆固醇合成,拮抗乙酸盐的促胆固醇作用,并降低脂肪细胞抵抗素表达,可能对肥胖有保护作用。但在某些研究中,丙酸盐可能通过升高胰高血糖素、去甲肾上腺素等激素增加糖尿病和肥胖风险,提示作用存在个体差异。
免疫调节与抗炎作用
丙酸盐通过激活GPR43/GPR41受体,促进调节性T细胞分化,抑制辅助T细胞活性,减少全身炎症反应。
心血管保护
丙酸盐减少促炎细胞因子(如TNF-α、IL-6),改善心脏纤维化和心功能。
血脑屏障保护
丙酸盐通过Nrf2信号通路减少氧化应激,增强紧密连接蛋白表达,保护血脑屏障完整性。
在前文中,我们看到了四碳丁酸盐,一种有效的抗炎和维持肠道屏障功能的关键分子,在高浓度下可能是有害的,特别是在粘膜炎症的情况下。有趣的是,类似的悖论可能适用于三碳丙酸盐,低浓度有益,但高浓度有毒。
生理双重性
在小鼠中,丙酸盐已被证明可以诱导肠道中产生饱腹感激素,以减少食物摄入并防止饮食诱导的肥胖。 最近的一项研究发现,短链脂肪酸丙酸的管理显著减弱脊柱关节炎。
然而,过量的丙酸盐可能是有问题的。在丙酸血症(代谢的遗传错误)中,丙酸盐的积累与酸中毒、发育迟缓、癫痫发作、氧化应激增加、线粒体功能障碍和胃肠道症状发作有关。肠易激综合征中也有过量丙酸的报道,当然还有自闭症。
丙酸盐与自闭症
近几十年来,自闭症和其他神经精神疾病的患病率急剧上升。环境因素肯定会发挥作用,包括肠道微生物群及其非常重要的代谢产物短链脂肪酸(SCFA)。
自闭症是一种复杂的神经生物学状况,影响多个身体系统,包括免疫和神经系统,表现为沟通和感官理解能力受损,通常在孩子很小的时候被诊断出来,且对男孩的影响更大。
近年来,自闭症的发病率呈上升趋势,在谷禾数据库,自闭症样本量也积累了超过5000例的样本。其特征包括社交和沟通障碍、感觉异常和重复行为等。越来越多的证据表明,自闭症不仅是一种大脑疾病,更是一种“全身”疾病,涉及代谢和免疫功能异常、胃肠道症状等。
多因素病因学挑战传统认知
遗传因素仅解释10%-20%病例,参与线粒体功能,免疫调节和神经回路形成的基因与自闭症有关。
环境因素在自闭症的发病中起着重要作用,肠道菌群失调、抗生素使用、饮食西化等环境因素显著增加ASD风险其中肠道微生物群及其代谢产物短链脂肪酸受到关注。
注:饮食西化这一点在从不发达国家向发达国家移徙的人口中尤其明显。每一个移民到西方国家的索马里人都注意到他们社区的ASD患病率增加。他们称之为“西方病”,因为他们在东非的家乡没有获得这种疾病。
关于自闭症我们以前的文章也有写过相关:
儿童神经发育异常的脑肠轴视角 – 自闭症早期风险判别和干预新路径

这意味着环境因素起着主要作用,自闭症是由基因与环境的复杂相互作用引起的。最近的证据表明,肠道微生物代谢物,特别是SCFA丙酸盐,可能发挥作用。
在动物模型中丙酸盐诱导自闭症样行为
在一项研究中,他们将丙酸盐直接注射到老鼠大脑的脑脊液中。与接受安慰剂输注的大鼠相比,在短短2-30分钟内,丙酸盐治疗的大鼠表现出重复行为,多动增加,社会行为受损和癫痫发作活动的证据。虽然丁酸盐和乙酸盐处理的大鼠确实显示出一些异常,但丙酸盐引起了最强烈的ASD样行为。

在另一项研究中,他们通过外周注射怀孕大鼠及其后代来观察系统性丙酸盐的影响。丙酸盐增加焦虑样行为和重复行为,特别是在出生前和出生后暴露的大鼠中。
丙酸盐与人类自闭症相关的证据
动物模型结果的外推需谨慎,需通过纵向人类研究逐步验证这些生物标志物与自闭症消退或临床改善的关联。
肠道微生物群改变是ASD相关症状的原因还是结果,尚不明确。
然而,普遍认为剖腹产、早期感染和抗生素暴露可能改变肠道微生物群,并增加ASD风险。
研究表明,ASD患儿粪便中丙酸盐水平升高,且ASD相关细菌包括多种丙酸盐产生菌,如梭菌、拟杆菌、脱硫弧菌等。
近期临床研究发现,ASD患者胃肠道活检中梭菌代谢物尿液标志物升高,碳水化合物代谢也发生改变。另有研究报告,使用抗生素治疗后暂时的行为改善,这些抗生素可降低产丙酸菌的水平。
机制:丙酸盐如何导致自闭症
✦【 间隙连接功能 】
MacFabe博士推测,丙酸盐的许多效应可能源于其关闭间隙连接的能力。间隙连接连接着相邻细胞的细胞质,允许小分子和离子在它们之间通过。这对于同步神经电活动至关重要,并在早期大脑发育中扮演着关键角色。间隙连接耦合的减少也可能抑制皮质修剪,这与 ASD 患者中发现的神经元密度增加的现象一致。
关于 Dr. MacFabe 与自闭症研究
Dr. MacFabe是Kilee Patchell-Evans自闭症研究小组的主任。他是温哥华不列颠哥伦比亚大学 iTARGET 自闭症倡议的核心成员。
该小组的核心重点是肠道微生物组的代谢产物如何控制自闭症患者的大脑功能和行为,以及相关的神经精神疾病,如强迫症、焦虑症、运动、饮食和学习障碍。尤其对肠道细菌短脂肪酸代谢物的作用及其在自闭症中的作用以及开发新的临床生物标志物和疗法以预防、识别、筛选和治疗该疾病特别感兴趣 。
此外,间隙连接“基因敲除”小鼠(即经过基因工程改造,不表达任何间隙连接蛋白的小鼠)表现出异常的大脑发育和行为、癫痫性疾病,以及对神经毒性损伤的过度反应。
✦【 线粒体功能障碍 】
ASD 通常伴有与线粒体功能障碍一致的遗传和生化变化。有证据表明,丙酸盐可能通过干扰线粒体 TCA 周期来引起这些变化。

线粒体三羧酸(TCA)循环是细胞能量产生的关键步骤。TCA循环的产物是 NADH 和 FADH2,它们将高能电子带到电子传递链(ETC),在那里它们的还原能力用于产生 ATP(细胞能量)。
从下图中可以看出,一轮TCA循环产生3个NADH分子和1个FADH2分子。丙酸盐通过转化为琥珀酰辅酶A进入TCA循环。少量有助于维持TCA循环中间体,并有利于细胞能量的产生。
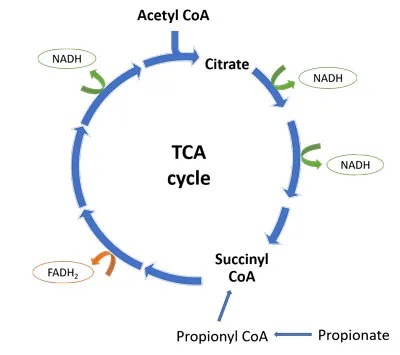
然而,进入TCA循环的大量丙酸绕过前四种TCA酶,并可能导致循环的转变。在循环的后半部分通量增加,产生柠檬酸盐积聚,而由于琥珀酰辅酶 A 的反馈抑制,前半部分则因“拥堵”而减缓。
这有几个后果:
首先,这改变了NADH : FADH2的比例,导致产量为1:1而不是3:1。当这些能量载体到达电子传递链时,NADH进入ETC复合物I,FADH2进入复合物II。
每个NADH分子导致产生3个ATP分子,而每个FADH2分子导致产生2个ATP分子。因此,丙酸盐诱导的TCA循环通量的转变将导致较少的NADH产生,以及复合物I处能量载体的缺乏,导致较少的总体ATP形成。事实上,ASD儿童已被证明缺乏ETC复合物。
其次,线粒体柠檬酸盐的积累将导致柠檬酸盐被转运到细胞胞质溶胶中。柠檬酸盐抑制磷酸果糖激酶,糖酵解的关键调节步骤。柠檬酸盐还增加了丙二酰辅酶A的形成,丙二酰辅酶A抑制CPT-1,CPT-1是将脂肪酸运送到线粒体中的转运蛋白。这有效地阻止脂肪酸氧化。
丙酸盐与线粒体功能:浓度与环境依赖性效应
为了更多地了解丙酸盐在线粒体功能障碍中的作用,McFabe博士实验室的学生培养了ASD和对照患者的免疫细胞,这些患者具有不同浓度的丙酸盐,有和没有活性氧(ROS)。
奇怪的是,他们发现如果ROS不存在,丙酸盐会改善线粒体功能;然而,在ROS存在的情况下,它会对线粒体功能产生负面影响。
换句话说:丙酸盐可以对线粒体功能产生有益和毒性作用,这取决于浓度,暴露时间和微环境。
作为一种弱酸,丙酸盐的摄取在细胞内酸化的条件下增加,并且可以在细胞内变得更加浓缩。像脱硫弧菌这样的菌群,它们产生丙酸和硫化氢。硫化氢的存在可能会增加丙酸盐损害线粒体功能的能力,可能是通过酸化细胞。
✦【 肉碱代谢障碍 】
肉碱代谢障碍也可能在神经发育障碍如ASD中发挥作用。肉碱是最有名的参与脂肪酸β-氧化。脂肪酸不能自由地穿过线粒体膜被代谢,而是必须与肉毒碱分子结合以穿过线粒体膜运输。
然而,肉碱也在脂质合成、胆碱能神经传递、膜稳定性和抗氧化活性中发挥作用。因此,肉碱对细胞能量,大脑发育和大脑功能很重要。

许多ASD患者有相对的肉碱缺乏症,这可能是由于遗传和后天因素。例如,已知某些抗生素可降低肉碱水平。口服肉碱及其衍生物乙酰-L-肉毒碱已被证明具有神经保护作用,并有望作为ASD和其他神经发育障碍的治疗剂。
如果肉碱缺乏,柠檬酸盐的积累阻止脂肪酸被运送到细胞中,不仅脑细胞会受到影响,肠道上皮细胞也会受到影响,后者依赖脂肪酸氧化(特别是丁酸代谢)来获得70%的能量,以维持肠道完整性。缺乏脂肪酸氧化会导致肠道通透性增加。研究表明,36%的ASD患者肠道通透性明显增加,近一半患者存在某种胃肠道症状。
进一步的研究可以探索肉碱补充在改善ASD患者的神经行为和胃肠道症状方面的具体益处,并确定最佳的剂量和给药方案。此外,还需要考虑个体化治疗方法。
关于丙酸盐调节的干预措施
有几种方法可以潜在地减少胃肠道丙酸盐的产生和丙酸盐的细胞水平。
抗生素
甲硝唑和万古霉素已用于ASD患者,取得了一定的成功。这些抗生素对革兰氏阳性菌具有广谱活性,包括产丙酸盐的梭菌属成员。在这两种药物中,万古霉素是更安全的,因为在正常情况下,口服万古霉素不会被显著吸收到循环中,而甲硝唑被全身吸收,可能具有不良的全身副作用。
然而,任何一种抗生素对丙酸盐产生的影响可能是短暂的。
一项为期8周的小型部分盲法临床试验发现,万古霉素对11名儿童中的8名ASD暂时有效,但这种益处并不持久。停止抗生素治疗后,梭菌很快又重新出现,可能是由于它们的孢子形成特性。因此,从长远来看,单独使用抗生素不足以降低丙酸盐的产量。
恢复胃肠道pH值和SCFA比值
改变肠道的pH值对肠道微生物群组成有重大影响。对人类粪便微生物群落的研究发现,在pH 5.5时,有益的丁酸盐产生菌占总菌群的20%。当pH值上升到6.5时,这些细菌几乎完全消失,产生乙酸盐和丙酸盐的细菌占主导地位。当然,恢复ASD患者的结肠pH值并非易事,目前也不是一种可行的治疗选择。

另一方面,丁酸盐已显示出治疗ASD和其他神经系统疾病的一些前景。当然,丁酸盐可以改善屏障功能,调节免疫系统,并可能有助于降低肠道的pH值,选择丙酸盐生产者。丁酸盐对转运蛋白的竞争也可能减少从肠道吸收到循环中的丙酸盐的量。需要更多的研究来确定SCFA比率如何影响ASD。
纠正营养缺乏症
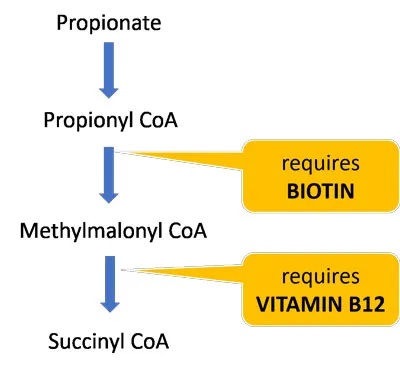
生物素和维生素B12是分解丙酸盐并使其进入TCA循环的酶的重要辅助因子。这些维生素的饮食缺乏可能进一步损害丙酸盐和肉毒碱代谢,并导致线粒体功能障碍(参考自lucy 博士博客内容)。
注:Dr lucy 以优异的成绩获得Kalamazoo College生物学学士学位和伊利诺伊大学厄巴纳-香槟分校营养科学博士学位,她花大量的时间研究饮食和运动对健康和疾病肠道微生物群的影响。
编辑
大约一半的生物素是由肠道微生物群产生的,因此那些肠道生态失调的人特别容易缺乏。
一项研究发现,与对照组相比,ASD儿童的生物素水平显着降低。一项后续研究发现,补充生物素水平与ASD症状改善高度相关,表明许多ASD患者可能受益于生物素补充剂。

无麸质、无酪蛋白饮食
在过去的十年中,无麸质,无酪蛋白(GFCF)饮食已成为ASD患者越来越受欢迎的治疗方法。虽然从饮食中去除这些免疫原性蛋白质可能具有独立的益处,但GFCF饮食也可能对丙酸盐产生影响。
值得注意的是,丙酸盐是小麦和乳制品中主要的动物青贮饲料和食品防腐剂,如丙酸钠或丙酸钙盐,并天然存在于一些乳制品中,如奶酪。

丙酸盐也由许多ASD相关细菌直接或间接产生,如梭菌属和脱硫弧菌属,来自精制糖和小麦糖的发酵。换句话说,当你喂这些细菌某些谷物和精制碳水化合物,他们开始制造更多的丙酸盐。
去除这些食物可能对丙酸代谢和肠道屏障功能有好处。上面提到的同一项研究发现,自闭症儿童的肠道通透性增加,发现那些GFCF饮食的肠道通透性评分低于对照组。
丙酸盐的其他饮食来源
除了小麦和乳制品,丙酸盐也被添加到许多精制食品中作为防腐剂。食品工业和农业越来越多地使用丙酸盐和相关的化学衍生物。硝基丙酸盐是许多植物和真菌产生的丙酸盐的衍生物。它是甘蔗、酱料和加工大米的潜在污染物,也是一种强效的线粒体和神经毒素。
其他食物刺激丙酸盐的产生。例如,人工甜味剂如糖精和糖精已被证明会显著增加啮齿动物肠道丙酸盐水平。
我们之前有写过关于食品添加剂相关的文章:
你的焦虑可能与食品添加剂有关,警惕食品添加剂引起的微生物群变化
2005年的一项研究发现,配方奶粉喂养的婴儿的丙酸盐水平比母乳喂养的婴儿高2.5倍。在配方中添加低聚糖仅部分改善了过量丙酸盐的产生。

GAPS饮食和碳水化合物
Natasha Campbell-McBride博士因其著作《肠道与心理综合征》而在自闭症社区中广为人知。麦克布赖德博士认为,包括自闭症在内的神经精神疾病是在肠道中产生的。她帮助许多儿童和成人扭转自闭症行为,并使用特定碳水化合物饮食(SCD)的修改版本(称为GAPS饮食)恢复正常的日常功能。
GAPS饮食在一段时间内从饮食中去除所有可发酵的碳水化合物,使炎症消退,肠道屏障愈合和密封。有趣的是,GAPS饮食排除了几乎所有的饮食丙酸盐来源和产丙酸菌的底物,这至少可以部分解释饮食对治疗神经系统疾病的成功。
在最初的消除阶段之后,重新引入富含菊粉的蔬菜,如西兰花和花椰菜,可能会优先喂养丁酸盐生产者。
GAPS饮食也是生酮的,特别是在早期阶段。生酮饮食对丙酸代谢的影响尚不清楚,尽管由于碳水化合物限制,丙酸产量可能要低得多。生酮饮食似乎还增加了整体NADH的产生和通过TCA循环右侧的通量,可能改善了由过量丙酸引起的左侧性。最后,酮与丙酸盐和其他SCFA使用相同的转运蛋白,因此可能通过竞争转运减少细胞中丙酸盐的积累。

回顾一下本章节主要的要点:
自闭症和其他神经系统疾病越来越普遍,这不能用遗传影响来解释。
丙酸盐在自闭症中的研究最多,但它也可能对其他神经系统和神经系统相关疾病有影响。即使是患有脑雾或疲劳的人也可能会有过量丙酸盐的影响。
丙酸盐在动物模型中可诱导ASD样行为。人类ASD患者的产丙酸盐菌丰度增加,粪便丙酸盐水平升高。
丙酸盐不是自闭症的唯一原因,但它可能在ASD患者的一个子集中起着重要作用。丙酸盐诱导间隙连接功能障碍、线粒体功能障碍、肉毒碱功能障碍,特别是在炎症和酸中毒的情况下。
抗生素可以暂时击倒丙酸盐生产者,但不是长期解决方案。
补充乙酰-L-肉毒碱、生物素、甲基维生素B12和/或丁酸盐可能有帮助。
丙酸盐在食品中的广泛使用需要注意。去除小麦、乳制品和其他丙酸盐的饮食来源可能会改善症状。
婴儿配方奶粉、精制碳水化合物和人造甜味剂会刺激产丙酸菌的生长。
GAPS饮食可以通过在一段时间内去除所有可发酵的碳水化合物来帮助调节丙酸盐的产生。之后,战略性地重新引入合适的纤维可能能够刺激有益的产丁酸菌的增长,并使丙酸盐生产者处于困境。
乙酸盐是含两个碳原子的短链脂肪酸,由肠道菌群发酵膳食纤维产生,也可通过外源性摄入。
乙酸盐的生理功能
能量代谢:作为细胞燃料参与三羧酸循环(TCA)。
抑制炎症反应:乙酸盐通过激活G蛋白偶联受体(GPR43),抑制促炎因子(如IL-6、TNF-α)表达。
调节肠道免疫:通过GPR43激活,促进肠黏膜屏障修复,减少细菌易位,并通过调节IgA与菌群结合维持肠道稳态。
神经调节:作为星形胶质细胞的主要能量源,参与神经递质合成(如GABA)。
维持肠道屏障:乙酸盐促进肠道上皮细胞增殖,改善血供,增强黏液分泌,保护黏膜完整性。
乙酸盐会让你发胖?
研究已经知道,微生物群的改变与代谢综合征和体重增加有关。
2006年,研究人员发现,遗传肥胖小鼠的微生物组具有更强的从饮食中收集能量的能力。此外,将肥胖小鼠的粪菌移植到无菌小鼠体内,会使无菌小鼠体重和脂肪增加。
在人类中,粪便乙酸盐水平升高与肥胖相关,但相关性不等于因果关系。
耶鲁大学研究人员开展了一项题为 “乙酸盐介导微生物组 – 脑 – β – 细胞轴以促进代谢综合征” 的研究,发表在《自然》杂志上,主要发现如下:
◆ 肥胖大鼠代谢更多乙酸盐,分泌更多胰岛素
在导致肥胖的饮食 3 天或 4 周后,大鼠的全身乙酸盐周转、血液乙酸盐和粪便乙酸盐浓度显著增加,肠道微生物群是乙酸盐升高的来源。喂食正常饮食并将乙酸盐输注到胃中的大鼠复制了肥胖大鼠中 GSIS 的增加。
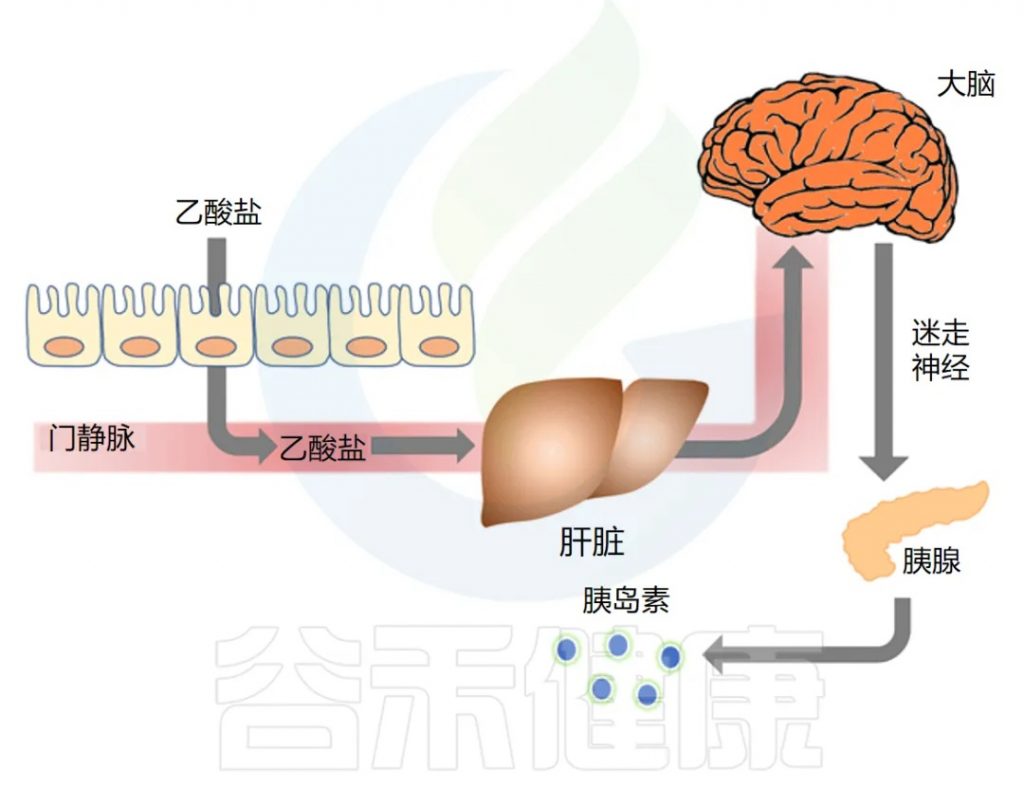
◆ 乙酸盐激活迷走神经以驱动胰岛素分泌
作者接下来研究了乙酸盐是否可以通过直接影响 β 细胞(负责胰岛素分泌的胰腺细胞)来刺激 GSIS。他们发现乙酸盐不会刺激离体胰岛中的 GSIS,但通过影响副交感神经系统,特别是激活迷走神经,增加了 GSIS。
注:副交感神经系统调节β细胞胰岛素分泌。这通常被称为“休息和消化”神经系统,因为它会减慢心率并增加肠道活动。
研究人员发现,输注乙酸盐 60 分钟后,副交感神经活动增加了三倍。脑乙酸盐浓度也增加,证实了乙酸盐穿过血脑屏障的能力。
刺激迷走神经也被证明可以驱动胰岛素分泌。迷走神经从脑干开始,几乎支配每个腹部器官,包括肠道和胰腺。
乙酸盐是否激活了迷走神经?为了找出答案,他们比较了乙酸盐对迷走神经完整的大鼠和手术切除迷走神经的大鼠的影响。
研究发现,输注乙酸盐后,副交感神经活动和脑乙酸盐浓度增加,将乙酸盐直接输注到大脑也导致 GSIS 大幅增加。切断迷走神经的大鼠对乙酸盐的反应中血浆胰岛素浓度显著降低。
下图总结了他们的发现,显示了肠-脑-β细胞轴:
◆ 长期乙酸盐输注会导致肥胖和代谢综合征
最后,研究人员想确定乙酸盐本身会导致肥胖。他们给瘦大鼠连续输注乙酸盐到胃中10天,结果发现,接受乙酸盐输注的大鼠胰岛素分泌和胰岛素抵抗增加,副交感神经系统活动增加,血浆 grehlin (一种调节食物摄入的激素)激素增加了3倍,每日热量摄入量增加,体重显著增加,血浆、肝脏和骨骼肌甘油三酯含量增加。而迷走神经切断术的大鼠未出现这些作用。再次证明乙酸盐的作用是通过迷走神经介导的。
所以肠道来源的乙酸盐会导致肥胖,对吧?
还没那么快下定论。
这项研究的结果与体外研究和多项动物研究形成鲜明对比,那些研究认为乙酸盐对新陈代谢有益。
体外研究
乙酸盐与受体 GPR43 结合,在培养的肠道上皮细胞中,导致分泌一种调节能量代谢的肠道来源的激素;在脂肪组织中,激活 GPR43 抑制胰岛素信号传导并抑制脂肪堆积,从而提高胰岛素敏感性。
动物研究
缺乏乙酸受体 GPR43 的小鼠在喂食正常饮食时变得肥胖,而过表达 GPR43 的小鼠即使在喂食致肥胖饮食时也保持苗条。
给饮食诱导的肥胖小鼠纳米颗粒递送的乙酸盐。在肝脏中,乙酸盐减少了脂质积累,改善了肝功能,并提高了线粒体效率。在白色脂肪组织中,乙酸盐抑制脂肪分解,但会诱导“褐变”,增加代谢能力并导致体内脂肪减少。
菊粉、低聚果糖等益生元增加了乙酸盐的产生,并导致 Grehlin 的产生减少,从而导致食物摄入量、体重和脂肪量减少。
在饮食诱导的肥胖小鼠中,每隔一天禁食 (EODF)升高血清乙酸盐和乳酸水平,这与白色脂肪组织的褐变和代谢改善有关。
乙酸盐已被证明还有其他好处:
耶鲁大学的研究为理解乙酸盐在肥胖中的作用提供了一个新视角,但同时也引发了更多的疑问,为什么在不同研究条件下,乙酸盐对肥胖和代谢综合征的影响会出现如此大的差异呢?这促使我们进一步探讨可能影响乙酸盐作用的各种因素。
不同模型和条件下乙酸盐的差异
模型差异
Perry 等人使用大鼠进行研究,而其他关于乙酸盐和肥胖或代谢综合征的研究大多使用小鼠品系,不过大鼠和小鼠的生理机能相似,这一因素不太可能解释所有差异。
方式差异
胃内(进入胃)和口服或结肠乙酸盐之间可能存在差异。正如 Canfora 等人指出的那样,“连续胃内供应的乙酸盐可能会影响胃的 pH 值,从而可能影响胃排空和食物释放、消化和吸收以及激素调节。”
此外,乙酸盐受体的表达在整个胃肠道中并不均匀,不同位置的乙酸盐可能产生不同影响。稍后将讨论一些人体研究,这些研究发现根据乙酸盐的位置存在不同的影响。
剂量差异
正如我们在丁酸盐中看到的那样,SCFA 的剂量非常重要。Perry 等人使用的大剂量乙酸盐可能与肠道微生物群正常产生或在饮食中消耗的乙酸盐水平不符,剂量不同可能导致不同结果。
SCFA 比率差异
在大多数健康人中,SCFA 以 3:1:1 的比例存在,仅通过提供乙酸盐可能扭曲这个比率,导致不同的代谢效应。
那么,超生理剂量的胃内乙酸盐会引起代谢综合征吗?是的。Perry 等人证明了这一点。
但是肠道菌群产生的乙酸是否会导致代谢综合征?可能不是。
我们可以从 Perry 等人的研究中学到什么?
首先,乙酸盐周转受肠道影响,在肥胖时升高。其次,乙酸盐可能确实会激活迷走神经以引起胰岛素分泌,因为将乙酸盐注射到大脑中证实了胃内输注的结果。至少在动物中是这样的。
人类和动物受体对乙酸盐的反应不同?
无论如何,至少有三项研究表明,GPR43 是乙酸盐的主要受体,对小鼠和人类的刺激反应不同:
这只是动物模型无法准确模拟人类生理的常见案例之一,所幸已有相当数量的人体研究关注了乙酸盐。
需要进一步的研究来明确阐明乙酸盐在人类中的作用。
关于乙酸盐和代谢综合征的动物研究结果喜忧参半,这可能是由于给药方式、剂量和 SCFA 比率的差异。
乙酸对大脑和免疫系统有益。
人类和动物对乙酸的响应不同 ,因此我们不能依赖动物研究来了解乙酸在人类中的作用。
有限的临床试验似乎表明乙酸对新陈代谢有有益作用。
乙酸盐的位置似乎很重要。在结肠更远端产生的乙酸盐似乎具有最有益的效果。
短链脂肪酸这些看似简单的分子,无疑是解读“微生物-宿主”对话的重要密码。短链脂肪酸研究引领我们重新思考健康的本质——健康并非某种单一指标的最优化,而是复杂生态系统的动态平衡。
正如肠道微生物群的多样性对健康至关重要,短链脂肪酸的种类、比例和浓度的平衡同样重要。未来的健康管理将从单一指标的”正常化”,转向生态系统的”平衡化“。
肠道菌群检测与短链脂肪酸代谢评估将成为个体健康”生态地图”的重要组成部分,谷禾肠道菌群检测报告中也有关于菌群代谢的短链脂肪酸相关指标。
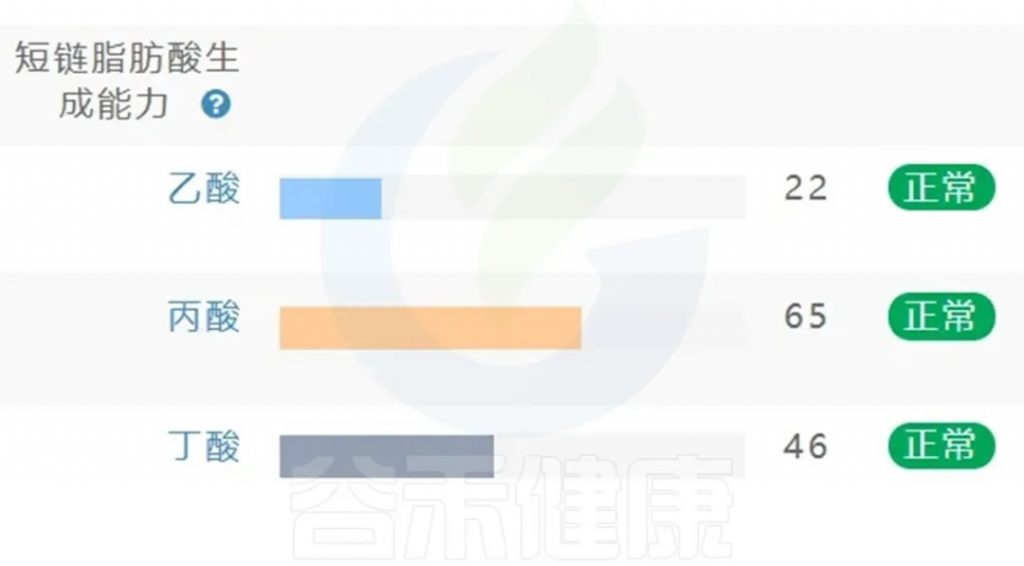
<来源:谷禾肠道菌群检测数据库>
随着对短链脂肪酸双面性理解的加深,未来的干预策略将更加精准和个性化。
针对短链脂肪酸不足的个体,可能会有靶向益生菌组合或特定结构的益生元配方,从小剂量开始逐步增加,避免肠道不适,有利短链脂肪酸的产生。但同时也需要结合其他指标,比如说肠道屏障功能是否正常,这在谷禾肠道菌群检测报告中也有相关指标。在肠道屏障薄弱的情况下,可以考虑优先修复肠道屏障,再补充短链脂肪酸。
而对于那些特定短链脂肪酸过高或对其敏感性增加的人群,可能需要定制化的饮食方案或特异性的益生菌干预,鉴别过度生长菌群比如说梭菌,可能需要优先考虑维持菌群平衡的相关干预策略,例如考虑低FODMAP饮食、减少添加剂摄入等方式,同时补充关键辅助因子如B族维生素、辅酶Q10等支持线粒体能量产生。
通过精准检测和个性化管理,我们可以逐步优化肠道微生态平衡,使短链脂肪酸保持在有利于健康的水平。
在药物开发方面,短链脂肪酸受体调节剂、缓释型短链脂肪酸制剂以及能够在特定肠道环境中释放活性成分的”智能”递送系统,都可能成为下一代肠道健康干预工具。
在日益关注肠道健康的今天,短链脂肪酸研究无疑将成为连接微生物学、营养学、临床医学的重要桥梁。
注:本账号内容仅供学习和交流,不构成任何形式的医疗建议。
主要参考文献
Singh, N. et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 40, 128–139 (2014).
Chang, P. V., Hao, L., Offermanns, S. & Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. U. S. A. 111, 2247–2252 (2014).
Zhou, D. et al. Sodium butyrate attenuates high-fat diet-induced steatohepatitis in mice by improving gut microbiota and gastrointestinal barrier. World J. Gastroenterol. 23, 60–75 (2017).
Donohoe, D. R. et al. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol. Cell 48, 612–626 (2012).
Lucy Mailing, PhD., A brief introduction to short-chain fatty acids
Decrypting the butyrate paradox: can excess butyrate be toxic?
Does acetate make you fat? The skinny on acetate and metabolic syndrome
The propionate-autism connection
Sun, J. et al. Clostridium butyricum pretreatment attenuates cerebral ischemia/reperfusion injury in mice via anti-oxidation and anti-apoptosis. Neurosci. Lett. 613, 30–35 (2016).
Li, H. et al. Sodium butyrate exerts neuroprotective effects by restoring the blood-brain barrier in traumatic brain injury mice. Brain Res. 1642, 70–78 (2016).
Sun, J. et al. Antidepressant-like effects of sodium butyrate and its possible mechanisms of action in mice exposed to chronic unpredictable mild stress. Neurosci. Lett. 618, 159–166 (2016).
Kaiko, G. E. et al. The Colonic Crypt Protects Stem Cells from Microbiota-Derived Metabolites. Cell 165, 1708–1720 (2016).
Zhang, Q. et al. Accelerated dysbiosis of gut microbiota during aggravation of DSS-induced colitis by a butyrate-producing bacterium. Sci. Rep. 6, (2016).
Kuo, S.-M., Chan, W.-C. & Hu, Z. Wild-type and IL10-null mice have differential colonic epithelial gene expression responses to dietary supplementation with synbiotic Bifidobacterium animalis subspecies lactis and inulin. J. Nutr. 144, 245–251 (2014).
Machiels, K. et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 63, 1275–1283 (2014).
Belcheva, A. et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell 158, 288–299 (2014).
Park, J., Goergen, C. J., HogenEsch, H. & Kim, C. H. Chronically Elevated Levels of Short-Chain Fatty Acids Induce T Cell-Mediated Ureteritis and Hydronephrosis. J. Immunol. Baltim. Md 1950 196, 2388–2400 (2016).
Park, J. et al. Short-chain fatty acids induce both effector and regulatory T cells by suppression of histone deacetylases and regulation of the mTOR-S6K pathway. Mucosal Immunol. 8, 80–93 (2015).
Chambers, E. S. et al. Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut 64, 1744–1754 (2015).
Asquith, M. et al. Intestinal Metabolites Are Profoundly Altered in the Context of HLA-B27 Expression and Functionally Modulate Disease in a Rat Model of Spondyloarthritis. Arthritis Rheumatol. Hoboken NJ 69, 1984–1995 (2017).
MacFabe, D. F. Enteric short-chain fatty acids: microbial messengers of metabolism, mitochondria, and mind: implications in autism spectrum disorders. Microb. Ecol. Health Dis. 26, 28177 (2015).
Foley, K. A., Ossenkopp, K.-P., Kavaliers, M. & MacFabe, D. F. Pre- and Neonatal Exposure to Lipopolysaccharide or the Enteric Metabolite, Propionic Acid, Alters Development and Behavior in Adolescent Rats in a Sexually Dimorphic Manner. PLOS ONE 9, e87072 (2014).
Finegold, S. M. State of the art; microbiology in health and disease. Intestinal bacterial flora in autism. Anaerobe 17, 367–368 (2011).
Rossignol, D. A. & Frye, R. E. Mitochondrial dysfunction in autism spectrum disorders: a systematic review and meta-analysis. Mol. Psychiatry 17, 290–314 (2012).
Frye, R. E. et al. Modulation of mitochondrial function by the microbiome metabolite propionic acid in autism and control cell lines. Transl. Psychiatry 6, e927 (2016).
Reichardt, N. et al. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 8, ismej201414 (2014).
Suez, J. et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 514, 181–186 (2014).
Napoli, E., Dueñas, N. & Giulivi, C. Potential Therapeutic Use of the Ketogenic Diet in Autism Spectrum Disorders. Front. Pediatr. 2, (2014).
Fernandes, J., Su, W., Rahat-Rozenbloom, S., Wolever, T. M. S. & Comelli, E. M. Adiposity, gut microbiota and faecal short chain fatty acids are linked in adult humans. Nutr. Diabetes 4, e121 (2014).
Kimura, I. et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 4, 1829 (2013).
Sahuri-Arisoylu, M. et al. Reprogramming of hepatic fat accumulation and ‘browning’ of adipose tissue by the short-chain fatty acid acetate. Int. J. Obes. 2005 40, 955–963 (2016).
Frost, G. et al. The short-chain fatty acid acetate reduces appetite via a central homeostatic mechanism. Nat. Commun. 5, (2014).
Everard, A. et al. Microbiome of prebiotic-treated mice reveals novel targets involved in host response during obesity. ISME J. 8, 2116–2130 (2014).
Li, G. et al. Intermittent Fasting Promotes White Adipose Browning and Decreases Obesity by Shaping the Gut Microbiota. Cell Metab. 26, 672–685.e4 (2017).
Wu, W. et al. Microbiota metabolite short chain fatty acid acetate promotes intestinal IgA response to microbiota which is mediated by GPR43. Mucosal Immunol. 10, 946–956 (2017).
Long, P. M. et al. Acetate supplementation as a means of inducing glioblastoma stem-like cell growth arrest. J. Cell. Physiol. 230, 1929–1943 (2015).
Trent, C. M. & Blaser, M. J. Microbially Produced Acetate: A ‘Missing Link’ in Understanding Obesity? Cell Metab. 24, 9–10 (2016).
Canfora, E. E. & Blaak, E. E. Acetate: a diet-derived key metabolite in energy metabolism: good or bad in context of obesity and glucose homeostasis? Curr. Opin. Clin. Nutr. Metab. Care 20, 477–483 (2017).
Ang, Z. et al. Human and mouse monocytes display distinct signalling and cytokine profiles upon stimulation with FFAR2/FFAR3 short-chain fatty acid receptor agonists. Sci. Rep. 6, srep34145 (2016).
Canfora, E. E. et al. Colonic infusions of short-chain fatty acid mixtures promote energy metabolism in overweight/obese men: a randomized crossover trial. Sci. Rep. 7, 2360 (2017).

谷禾健康

姜黄素是姜黄根茎中所含的一种亲脂性多酚,其很早就被亚洲地区的人们用作食品中的香料和染料。除此之外,姜黄素被发现还具有抗氧化、抗炎、抗菌、抗肿瘤、调控血糖、神经保护等多种药理作用,目前市场上许多功能性食品中都包含这一成分。
然而,尽管姜黄素具有多种药理活性,但其水溶性较差,在口服后的肠道吸收率较低,并且在肝脏中迅速代谢,加之化学不稳定性,导致其生物利用度非常低,这些都限制了它的治疗效果和临床应用。
值得注意的是,许多研究证实口服姜黄素会在肠道中与微生物群发生双向相互作用。姜黄素的代谢转化不仅发生在肠上皮细胞和肝细胞中,一些肠道菌群如大肠杆菌、长双歧杆菌也具有能够代谢姜黄素的酶。将其转化为多种活性代谢物,它们具有特异性且往往更有效。有助于提高姜黄素的生物利用度。
同时,姜黄素也可以作为一种益生元对肠道菌群有改善作用。其增加了产丁酸盐菌等有益菌的丰度,并改善了糖尿病和代谢综合征等患者的肠道菌群。姜黄素不仅可以影响肠道微生物群的成分,还能够增强肠道屏障,抑制促炎介质的激活和表达,减轻肠道炎症和氧化应激。
姜黄素与肠道微生物群之间相互作用,在临床治疗中具有多种健康益处。包括降低炎症水平、缓解炎症性肠病、减少结肠炎和结肠癌等胃肠道疾病的风险,改善代谢功能障碍如肥胖、调节血糖水平、减轻糖尿病症状,辅助治疗阿尔兹海默病神经系统疾病等。
在本文中,我们介绍了姜黄素的生物学特性及药理作用,重点关注它与肠道微生物群的相互作用。由于个体差异,人们对姜黄素的反应各不相同。肠道微生物群检测可了解个体菌群结构、优势菌群和多样性,从而评估姜黄素吸收代谢的潜在差异,更有效地利用姜黄素促进健康。
什么是姜黄素?
姜黄素(Curcumin),也称为二阿魏酰甲烷,是一种源自姜黄植物的天然多酚类成分,也是姜黄主要的生物活性成分。
注:还有另外两种被称为姜黄素的化合物,即“姜黄素II”(去甲氧基姜黄素)和“姜黄素III”(双去甲氧基姜黄素),它们在芳香环上的甲氧基数量不同。它们分别占总姜黄素类化合物的10-20%和3%,具有不同的药理活性。
✔ 富含姜黄素的产品已遍布生活中
姜黄素呈亮橙黄色,具有独特的色泽与风味,是咖喱粉中的主要香料之一。它在全球获得广泛认可,应用领域多样:在印度,含有姜黄素的姜黄已被用于制作咖喱;在日本,它被装在茶里;在泰国,它用于化妆品;在中国,它被用作着色剂和功能性食品等;在韩国,它被装在饮料里;在马来西亚,它被用作防腐剂;在巴基斯坦,它被用作抗炎剂;在美国,除了胶囊和粉末形式外,它还用于芥末酱、奶酪、黄油和薯片中,用作防腐剂和着色剂。姜黄素产品形式多样,包括胶囊、片剂、软膏、能量饮料、肥皂和化妆品等。
生姜和姜黄有什么区别?
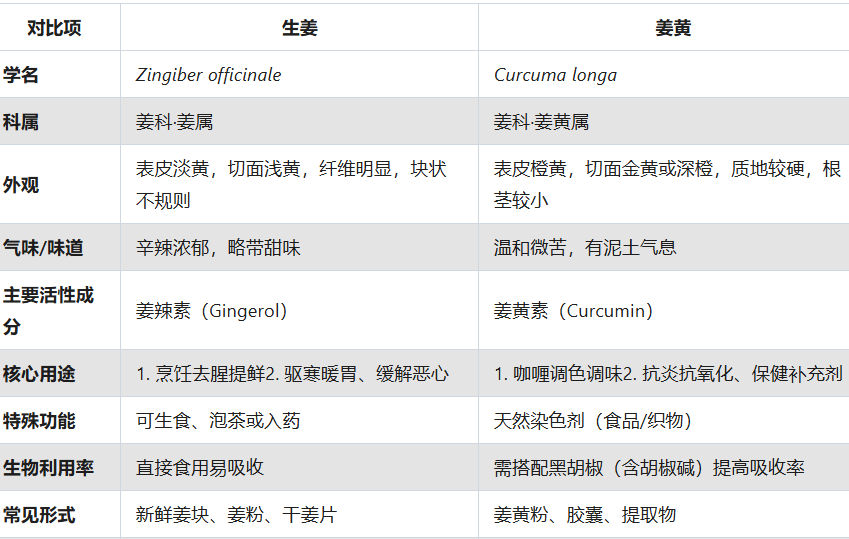
姜黄素的生物益处
姜黄素不仅具有独特的色泽与风味,还具有许多生物学益处。其抗菌特性于1949年首次得到证明,随后的研究表明,它还具有抗炎特性、抗氧化特性和其他一些显著的好处。
✔ 抗氧化
氧化应激(OS)是活性氧产生与机体抗氧化保护系统间的失衡。这种不平衡可能导致细胞功能障碍和损伤。
研究表明,姜黄素可以作为一种抗氧化剂。首先,它与活性物质直接反应,中和它们并防止进一步的损害。它可以清除不同形式的自由基,例如活性氧和氮物质(分别为ROS和RNS)。
其次,姜黄素诱导各种细胞保护和抗氧化蛋白的上调,增强身体对氧化应激的防御能力。同时,通过激活细胞保护蛋白受体核因子红细胞系相关因子2(Nrf2)信号通路来调节抗氧化酶的表达,从而稳定活性氧水平。
这种转录因子通过控制抗氧化酶和解毒蛋白的基因表达,从而保护细胞免受氧化损伤,在细胞对氧化应激的反应中发挥关键作用。它可以调节在中和自由基中活跃的谷胱甘肽(GSH)、过氧化氢酶和超氧化物歧化酶(SOD)的活性;此外,它还可以抑制产生活性氧的酶,如脂氧合酶/环氧合酶和黄嘌呤氢化酶/氧化酶。这些细胞保护蛋白发挥抗氧化活性,保护细胞免受氧化损伤。
注:姜黄素是一种亲脂性化合物,这使其成为过氧自由基的有效清除剂,与维生素E一样,姜黄素被认为是一种链破坏型抗氧剂。链破坏型抗氧剂即链终止型抗氧化剂。可以终止氧化过程中自由基链的传递与增长。
此外,姜黄素能够激活AMP活化蛋白激酶(AMPK),这是细胞能量稳态的重要调节剂。姜黄素的这种激活有助于减轻氧化应激引起的肠道屏障和线粒体损伤。
✔ 抗炎
炎症过程与氧化应激密切相关,因为活性氧的产生与机体抗氧化防御失衡导致细胞功能障碍和炎症反应。
炎症反应被发现参与多种慢性疾病发展,包括阿尔茨海默病、帕金森病、多发性硬化症、癫痫、脑损伤、心血管疾病、代谢综合征、癌症、过敏、哮喘、支气管炎、结肠炎、关节炎、肾缺血、银屑病、糖尿病、肥胖、抑郁、疲劳等。
炎症反应表现为显著的病理改变,其特征是炎症指标水平升高,如肿瘤坏死因子-α(TNF-α)、白细胞介素-6(IL-6)、白细胞介素-10(IL-10)、C反应蛋白(CRP)、单核细胞趋化蛋白-1(MCP-1)或血管细胞粘附分子-1(VCAM-1)。
姜黄素抑制氧化应激并改善炎症途径
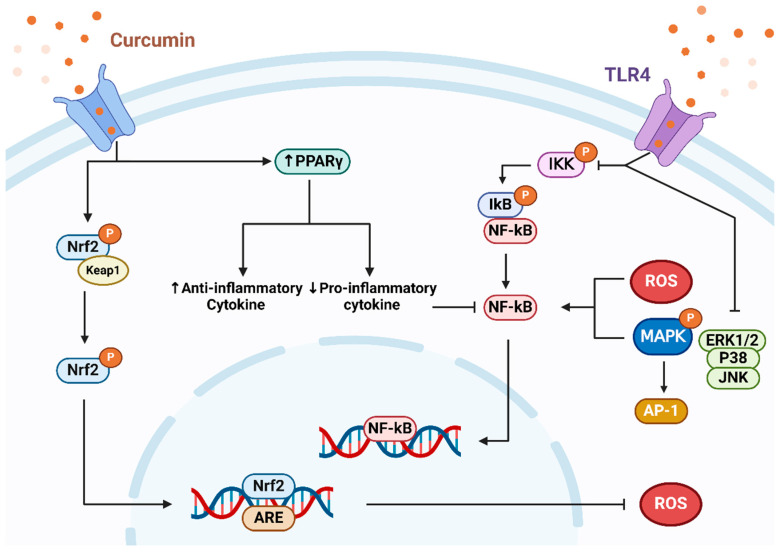
Servida S,et al.Int J Mol Sci.2024
研究发现姜黄素通过降低促炎介质水平减轻炎症反应。这可能因其附着于toll样受体(TLR)并控制NF-κB、MAPK和AP-1等信号通路。其中,NF-κB作为关键转录因子在诱导炎症中至关重要。姜黄素抑制NF-κB,减少IL-1β和IL-6等炎症因子释放。
在葡萄糖硫酸钠(DSS)诱导的结肠炎小鼠模型中,给予适量姜黄素有效抑制了NF-κB抑制蛋白(IκB)的磷酸化,从而抑制了肠道中的NF-κB,这最终减轻了炎症反应。
✔ 抗癌
通过多项研究,姜黄素被证明是一种有效的抗癌候选物质,特别是在以下方面:
NF-κB通路抑制:姜黄素能抑制核因子κB的活化,从而减少炎症因子如IL-1β和IL-6的释放,这些炎症因子与肿瘤发生、发展密切相关。
调控肿瘤抑制基因:研究表明姜黄素可以上调p53表达,人类结直肠癌患者服用姜黄素后p53表达增加,增强了机体对癌细胞的抑制能力。
COX-2抑制:姜黄素能特异性抑制环氧合酶-2(COX-2)的表达,这在HT-29人类结肠癌细胞中已得到证实。
除此之外,姜黄素还具有以下药理作用:
姜黄素的药理作用
Balaji S,et al.World J Exp Med.2025
姜黄素在各种疾病中的治疗作用
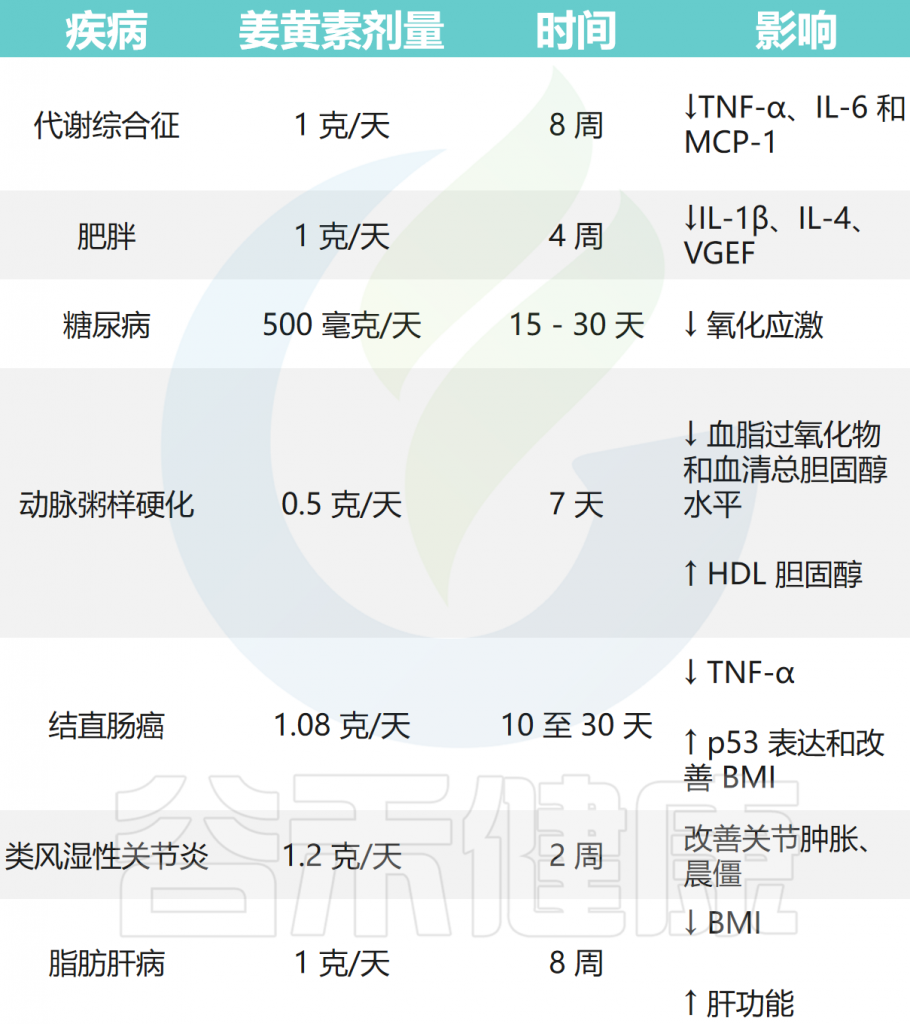
doi: 10.3390/microorganisms12040642.
✔ 姜黄素本身的生物利用度较低
尽管姜黄素的药理活性多样,但其治疗应用受到严重限制,姜黄素在口服后的肠道吸收率较低,并且在肝脏中迅速代谢并通过胆囊排泄,加之其水溶性低和化学不稳定性,导致生物利用度非常低。
姜黄素的吸收及代谢过程
作为第一步,摄入的姜黄素先通过胃,在那里几乎没有被吸收。由于其对低pH值的抵抗力,姜黄素在没有任何化学修饰的情况下到达大肠并经历广泛的I期和II期代谢。
口服后姜黄素的药代动力学
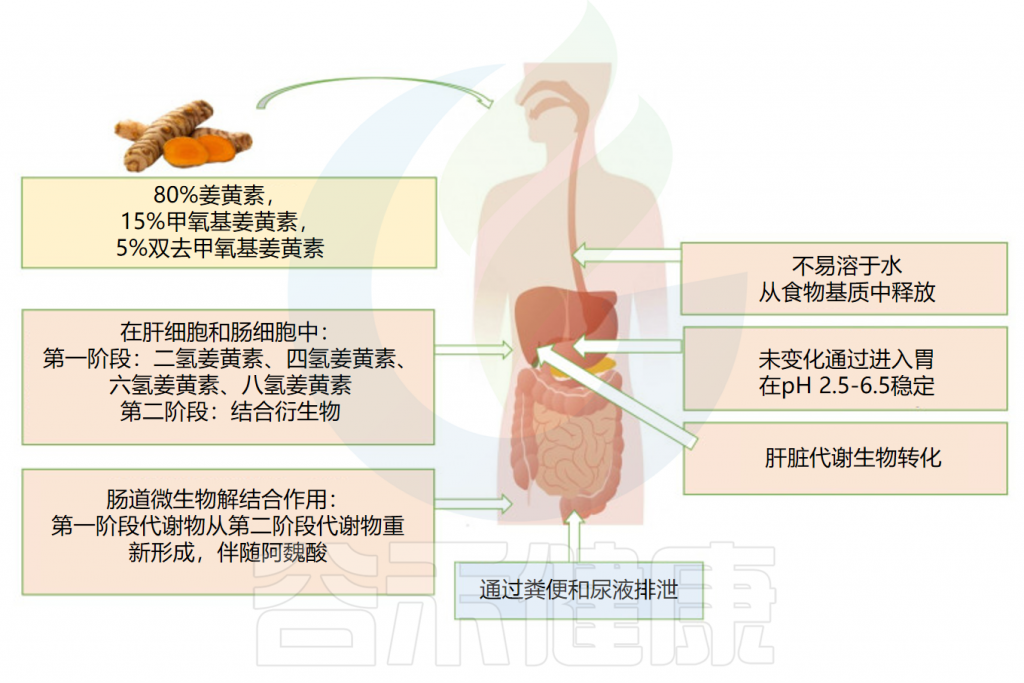
Servida S,et al.Int J Mol Sci.2024
✔ I期代谢发生在肠细胞和肝细胞中
首先,它被I期酶代谢:不同的还原酶在其底物中引入反应基团和极性基团,产生活性代谢物,即二氢姜黄素、四氢姜黄素(DHC)和六氢姜黄素(THC)。姜黄素的这种还原性代谢反应广泛发生在肠细胞和肝细胞中。
✔ 代谢物的活性低于其底物
在I期形成的代谢物被转运到肠道和肝脏胞质溶胶,在那里它们被转化(II 期)成偶联衍生物(即偶联姜黄素、偶联DHC、偶联THC和偶联八氢姜黄素)。
葡萄糖醛酸化是偶联的主要途径,姜黄素葡糖苷酸是器官和细胞体液中存在的主要代谢物(约占血浆姜黄素的99%),其分子的活性低于其底物且分子量更高。
口服后姜黄素的代谢
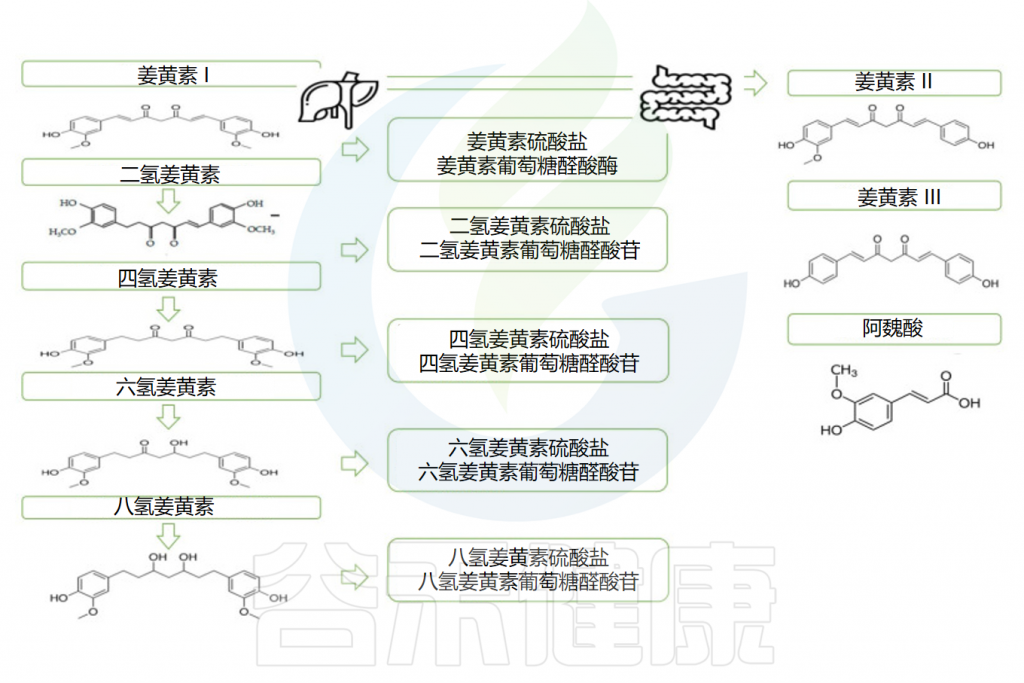
Servida S,et al.Int J Mol Sci.2024
通常口服给药后,在1至2小时内观察到姜黄素峰值血液浓度,并在大约12小时后变得检测不到。
肠道菌群提高姜黄素的生物利用度
值得注意的是,姜黄素主要作用于肠道,其代谢转化不仅发生在肠上皮细胞和肝细胞中,还由肠道微生物群产生的酶进行,通过这些酶产生多种活性代谢物。
肠道衍生代谢物的生物活性与天然姜黄素不同,它们具有特异性且往往更有效。因此,肠道微生物组成会影响姜黄素代谢物的生物利用度。
✔ 大肠杆菌、长双歧杆菌等能够代谢姜黄素
已鉴定出多种能代谢姜黄素的肠道细菌:人类粪便微生物分析显示,大肠杆菌通过NADPH依赖性姜黄素/二氢姜黄素还原酶表现出最高的姜黄素代谢活性。这种酶能够将姜黄素转化为二氢姜黄素,再转化为四氢姜黄素。
其他微生物,如长双歧杆菌(Bifidobacteria longum)、假小链双歧杆菌(Bifidobacteria pseudocatenulaum)、粪肠球菌(Enterococcus faecalis)、嗜酸乳杆菌(Lactobacillus acidophilus)和干酪乳杆菌(Lactobacillus casei)是能够代谢姜黄素的相关细菌菌株。
✔ 姜黄素的有益作用可能取决于肠道菌群组成
与花青素等其他膳食多酚类似,姜黄素的生物活性不仅与吸收率相关,还与肠道菌群消化产生的活性代谢物有关。姜黄素的生物学特性取决于这些微生物代谢物的活性。
肠道微生物群的姜黄素代谢途径包括还原、甲基化、脱甲氧基化、羟基化和乙酰化,主要产物为四氢姜黄素、二氢阿魏酸和1-(4-羟基-3-甲氧基苯基)-2-丙醇。
此外,姜黄素还可以被毕赤酵母代谢成四种主要代谢产物,包括1,7-双(4-羟基-3-甲氧基苯基)庚烷-3,5-二醇、5-羟基-1,7-双(4-羟基-3-甲氧基苯基)庚烷-3-酮、5-羟基-1,7-双(4-羟基苯基)庚烷-3-酮和5-羟基-7-(4-羟基-3-甲氧基苯基)-1-(4-羟基苯基)庚烷-3-酮。
因此,姜黄素的有益作用不仅取决于姜黄素的饮食摄入量,还取决于个人代谢姜黄素的能力,即最终取决于每个人肠道微生物群的组成。
提高姜黄素效果的其他方式
✔ 给药形式会影响姜黄素的疗效
研究显示,给药形式会影响姜黄素的生物利用度。使用脂质体、聚合物纳米颗粒、环糊精包封、脂质复合物或合成聚合物-姜黄素复合物等给药形式可提高姜黄素的活性和生物利用度,增强其对癌症和肝病等疾病的治疗效果。
姜黄素与胶体纳米颗粒分散的新制剂能通过刺激GLP-1(胰高血糖素样肽1)和胰岛素分泌改善高血糖,表明其可用于糖尿病治疗,且可能对炎症和骨关节炎有效。
此外,纳米气泡姜黄素提取物补充剂对小鼠健康和运动表现有益,帮助克服身体疲劳。
✔ 新鲜或粉状姜黄生物利用度更高
最近的一些论文还显示了食物基质在姜黄素吸收中的重要性,强调与补充剂相比,当它作为新鲜或粉状姜黄食用时,生物利用度更高,这可能是由于与其他姜黄化合物的协同活性或姜黄基质效应。
共给药是提高姜黄素生物利用度的重要方法。研究显示,与黑胡椒碱(piperine)联合使用可显著抑制姜黄素首过代谢,增加血液浓度。2克姜黄素与5毫克黑胡椒碱联用可使生物利用度提高三倍以上,主要通过抑制葡萄糖醛酸转移酶活性,减少肝脏和肠道代谢。
此外,与其他具协同作用的抗氧化剂、抗炎剂联合应用能放大姜黄素药理效应,改善临床疗效。
值得注意的是,姜黄素和肠道微生物群之间的相互作用是双向的。如上所述,姜黄素在口服给药后优先在胃肠道中积累,一方面,肠道微生物群通过多种酶促途径(如还原、去甲基化、羟基化等)将姜黄素代谢转化为具有独特生物活性的代谢物。
与此同时,姜黄素能够调节肠道菌群的丰富度、多样性和组成,而这些受影响的菌群又反过来影响姜黄素的吸收、代谢和治疗效果。
利于肠道中有益菌株的生长
越来越多的研究证明肠道菌群失调与各种疾病的发生之间存在密切关系,姜黄素已被证明可以调整失衡菌群中有益细菌的比例,促进有益菌株的生长。
✔ 增加了产丁酸盐菌等有益菌的丰度

连续15天给小鼠施用100mg/kg姜黄素后发现其对肠道菌群有调节作用,姜黄素组显示普雷沃氏菌属的丰度显著降低,拟杆菌科和理研菌科(Rikenellaceae)的丰度显著增加。其他动物模型研究也表明,口服姜黄素增加了有益细菌(如双歧杆菌、乳酸菌和产生丁酸盐的细菌)的丰度,同时减少了普雷沃氏菌属、拟杆菌科等细菌的数量。
✔ 姜黄素调节脂肪肝病中的菌群失衡
高脂肪饮食的人易出现肝脏代谢改变,伴随着肠道微生物群组成改变和肠道通透性增加。在饮食中添加姜黄素可增强肝脏代谢,增加有益菌,并减少与高脂肪饮食引起的菌群失调有关的有害细菌菌株。
使用姜黄素治疗成功减少了36种与肝脂肪变性呈正相关的潜在有害细菌菌株。姜黄素对柯林斯氏菌属 、链球菌属、萨特氏菌属、Thalassospira、Gordonibacter和放线菌属具有富集作用,这些是人体肠道的核心菌属或益生菌。同时对密螺旋体、Alloprevotella、瘤胃球菌属、另枝菌属、Elusimicrobium、Anaerofilum和Papillibacter具有抑制作用。
✔ 姜黄素调节阿尔茨海默病等疾病的重要菌群
在阿尔茨海默病小鼠中,姜黄素改善空间学习记忆能力,减少海马体淀粉样斑块,并显著改变拟杆菌科、普雷沃氏菌科和乳杆菌科等与阿尔茨海默病相关的关键菌株丰度。
另一研究中,姜黄素(100mg/kg/天,12周)能部分逆转卵巢切除导致的肠道菌群多样性变化。给结肠癌小鼠高剂量姜黄素(162mg/kg/天)可减少肿瘤负荷,增加乳杆菌并减少Coriobacterales。姜黄素还能减少瘤胃球菌,其增加与结直肠癌发生相关。
✔ 姜黄素增加了细菌的多样性
在一项人体随机安慰剂对照试验中,调查了姜黄和姜黄素膳食补充剂与安慰剂相比对30名健康受试者(每组10名)的影响。
姜黄片剂含有1000毫克姜黄和1.25毫克胡椒碱提取物;姜黄素片剂含有1000毫克姜黄素和1.25毫克胡椒碱提取物;受试者被指示随餐口服3片,每天两次(每天总共6000毫克)。在基线和治疗8周后进行微生物群分析。
所有受试者都表现出微生物群组成的随时间的显著变化和对治疗的个体化反应。肠道菌群因人而异,个体对治疗的反应并不均匀。然而,比较治疗前后每组存在的细菌种类数量,安慰剂组显示物种总体减少15%,而姜黄和姜黄素处理组分别增加7%和 69%。
这些研究表明姜黄素的保护作用可能源于其促进肠道菌群从失衡转变为平衡的能力,减少了致病菌并增加有益菌的丰度。
姜黄素对肠道微生物群的影响总结
Servida S,et al.Int J Mol Sci.2024
姜黄素增强肠道屏障功能
姜黄素不仅可以影响肠道微生物群的成分,还能够增强肠道屏障。
✔ 姜黄素能够增强中和脂多糖内毒素的能力
肠道屏障由四种不同类型的核纤层蛋白组成。其完整性的任何缺陷都会引起细菌侵入正常结肠组织,导致肠上皮细胞失调和随后的局部炎症。
第一层包含碱性磷酸酶(IAP),IAP具有中和细菌内毒素脂多糖的能力。研究表明,口服姜黄素可以将IAP活性提高三倍,并降低循环内毒素脂多糖(LPS)水平,从而直接证明姜黄素对肠道屏障初始层的调节作用。
✔ 姜黄素减少了粘蛋白的分解
构成第二层的肠粘膜层对于将管腔内容物与上皮细胞分离并防止病原菌进入至关重要。随着第二层的消失,肠上皮细胞将直接与管腔细菌相互作用,导致肠道炎症加剧。
在姜黄素的驱动下,肠道酸性粘蛋白的增加促进了合成,并最大限度地减少了肠粘膜层的分解,从而保留了其结构。
✔ 姜黄素可增强抗菌肽的产生
第三层由肠上皮细胞之间的紧密连接组成,它们阻止外来抗原、微生物和毒素等有害物质从肠腔转移,同时允许重要的营养物质、电解质和水从肠腔流入血液。通过跨上皮以及跨细胞和旁细胞运输,建立了针对细菌内毒素的防御机制,有助于保持肠道屏障的完整性。
在最后一层发现的抗菌肽可防止细菌突破肠道屏障。α-防御素和β-防御素具有杀菌特性,其中α-防御素在体内具有显著影响,该因素影响着肠道微生物群的组成。研究表明,姜黄素可增强抗菌肽的产生。
✔ 体外和动物研究也证实姜黄素可以恢复肠道屏障
体外研究也显示姜黄素可恢复受损肠道通透性。在CaCo2细胞中,姜黄素减轻肠上皮屏障损伤,抑制脂多糖诱导的IL-1β分泌,保护紧密连接蛋白,并通过抑制p38 MAPK激活减少紧密连接蛋白异常磷酸化。
这些结果也在动物模型中得到证实:高脂饮食大鼠经姜黄素处理(200mg/kg/日)后,肠道紧密连接结构改善,血清TNF-α和LPS水平降低,肠粘膜occludin表达上调。同样,西式饮食小鼠补充姜黄素(100mg/kg/日)显著改善肠道屏障功能,恢复肠碱性磷酸酶活性及ZO-1和claudin-1表达。
鉴于紧密连接蛋白表达下降在非酒精性脂肪肝(NAFLD)发病中的关键作用,姜黄素(200mg/kg/日,4周)被证明能恢复NAFLD大鼠远端回肠中ZO-1和occludin的表达,表明姜黄素通过改善肠道屏障完整性可能成为NAFLD新疗法。
这些研究提供有力证据表明姜黄素有助于维持肠道屏障完整性,可作为肠道疾病预防/治疗的新工具。
姜黄素减轻肠道炎症
✔ 减轻了炎症和氧化应激
一项随机对照人体试验中,58名非酒精性脂肪性肝病(NAFLD)患者接受含50mg/天纯姜黄素的或安慰剂。代谢组学显示姜黄素对氧化应激和炎症标志物有益,减轻了患者中的炎症反应,并抑制了NAFLD进展过程中某些细菌的增长。
一项动物研究报道,一种新开发的纳米颗粒姜黄素通过抑制促炎介质的表达和诱导Treg扩张来积极改善小鼠的炎症,这还伴随着粪便丁酸盐水平的增加。
✔ 可抑制促炎介质的激活和表达
含0.2%(w/w)纳米颗粒姜黄素的啮齿动物饮食可抑制小鼠结肠上皮细胞中NF-κB激活和促炎介质表达。或者,姜黄素可以通过抑制TLR4/MyD88/NF-κB信号通路的激活来减轻脂多糖诱导的炎症。此外,姜黄素已被证明可以抑制NF-κB核易位,并减轻癌症中过度激活的其他促炎基因的表达。
研究证明,断奶仔猪饲喂300mg/kg姜黄素28天可通过抑制大肠杆菌增殖和下调TLR4表达缓解炎症。
缓解炎症性肠病
大量研究表明,姜黄素可以通过调节肠道微生物群的组成和多样性,对胃肠道系统健康产生有益影响。
✔ 姜黄素调节乳酸菌并改善肠道屏障
炎症性肠病(IBD)与肠道菌群稳定性密切相关。研究发现,补充姜黄素可增加乳酸菌相对丰度,通过提高sIgA水平增强粘膜免疫并改善肠道屏障功能。
注:IgA是一种在改善肠道微生物疾病中起重要作用的免疫球蛋白。
✔ 姜黄素调节信号通路并减少炎症因子
炎症性肠病的发生与TLR4/NF-κB/AP-1信号上调有关。在结肠炎的动物模型中,姜黄素被发现可以通过减少TLR4信号传导来改善炎症。姜黄素通过与细胞外TLR4结构域结合蛋白髓样分化蛋白2(MD-2)结合来抑制脂多糖引起的免疫反应并减少炎症因子的释放。
作为IBD发病机制主要贡献者,NF-κB可被姜黄素通过调节NF-κB/IκB通路抑制。姜黄素干扰IκB激酶信号,阻止IκB降解,抑制NF-κB激活,降低TNF-α、IL-1、IL-6等细胞因子释放,减轻炎症反应。研究表明肠道炎症严重程度与NF-κB p65含量相关,IBD患者肠道中NF-κB p65含量较高。
✔ 抑制氧化应激
此外,姜黄素能够降低肿瘤坏死因子(TNF-α)表达水平,同时显著减少一氧化氮(NO)的产生,从而抑制氧化应激并对炎症性肠病产生有益影响。
其次,研究表明姜黄素能够通过选择性阻断环氧合酶-2(COX-2)受体来抑制炎症。给予有效剂量的姜黄素可以抑制iNOS/COX-2的表达并减弱p38 MAPK的激活,p38 MAPK在调节炎症因子的转录和释放中具有重要作用。
✔ 姜黄素对一些其他胃肠道疾病也有改善作用
姜黄素通过调节Th17/Treg细胞的平衡和恢复肠道微生物群组成来改善糖尿病患者的结肠炎。补充姜黄素可以将肠道微生物群组成转变为富含短链脂肪酸产生细菌的成分,从而促进肠道粘膜保护并减轻与肠道疾病相关的炎症。
姜黄素对肠道微生物的调节作用还可能影响结直肠癌,姜黄素和富含生育三烯酚的部分的组合改变了结直肠癌细胞中的微生物多样性,在抑制结肠癌细胞生长方面具有潜在的治疗协同作用。
姜黄素对胃肠道疾病的影响

Balaji S,et al.World J Exp Med.2025
综上所述,姜黄素能够通过调节肠道菌群、修复肠道屏障、抑制炎症信号通路等多种机制来缓解炎症性肠病,其与肠道菌群的互作有望成为促进胃肠道系统健康和改善一系列胃肠道疾病的天然治疗剂,也为基于肠道微生物组的姜黄素靶向治疗策略提供了理论基础。
改善肥胖
✔ 调节了肥胖的重要指标(厚壁菌/拟杆菌)比值
肠道菌群组成与肥胖发病密切相关,肥胖患者肠道中厚壁菌门与拟杆菌门比例(F/B比值)升高。然而,在施用有效剂量的姜黄素后,观察到F/B比值显著降低。这种减少还伴随着毛螺菌科(Lachnospiraceae)和瘤胃球菌科(Ruminococcaceae)数量的减少,以及拟杆菌科、Riskenellaceae 和普雷沃氏菌科(Prevotellaceae)丰度的增加。
姜黄素还增加了双歧杆菌、乳酸杆菌和嗜粘蛋白阿克曼菌等在人类抗肥胖过程中发挥关键作用的细菌丰度。
✔ 姜黄素还能够抑制成脂基因,减少脂肪积累
此外,一些研究证实姜黄素在多种器官包括脂肪组织中发挥多种生物学功能。姜黄素通过抑制丝裂原活化蛋白激酶(ERK、JNK和p38)活性抑制3T3-L1脂肪细胞分化,并通过抑制PPARγ和C/EBPα表达抑制成脂基因。
适量姜黄素可减少室管膜脂肪组织、增加能量消耗、减少体内脂质积累,同时阻止吞噬细胞浸润脂肪组织并增加脂质运载蛋白产生,从而减轻脂肪组织炎症。在高脂肪饮食诱导的肥胖小鼠中,0.2g/d姜黄素显著减少了白色脂肪组织。
✔ 临床证实姜黄素具有减重和降低甘油三酯水平的功效
临床效果表明,姜黄素可使超重人群BMI恢复正常并显著降低血清甘油三酯水平。姜黄素还增强高脂饮食诱导的胰岛素敏感性,阻断脂肪生成。此外,姜黄素可通过调节脂质转运蛋白的表达和活性,维持胆固醇稳态。
调节血糖水平,减轻糖尿病
姜黄素可以通过改善肠道屏障功能、影响肠道激素分泌、调节抗炎细胞因子及减少与胰岛素抵抗相关的炎症分子来调控宿主葡萄糖稳态。
姜黄素对血糖稳态的影响
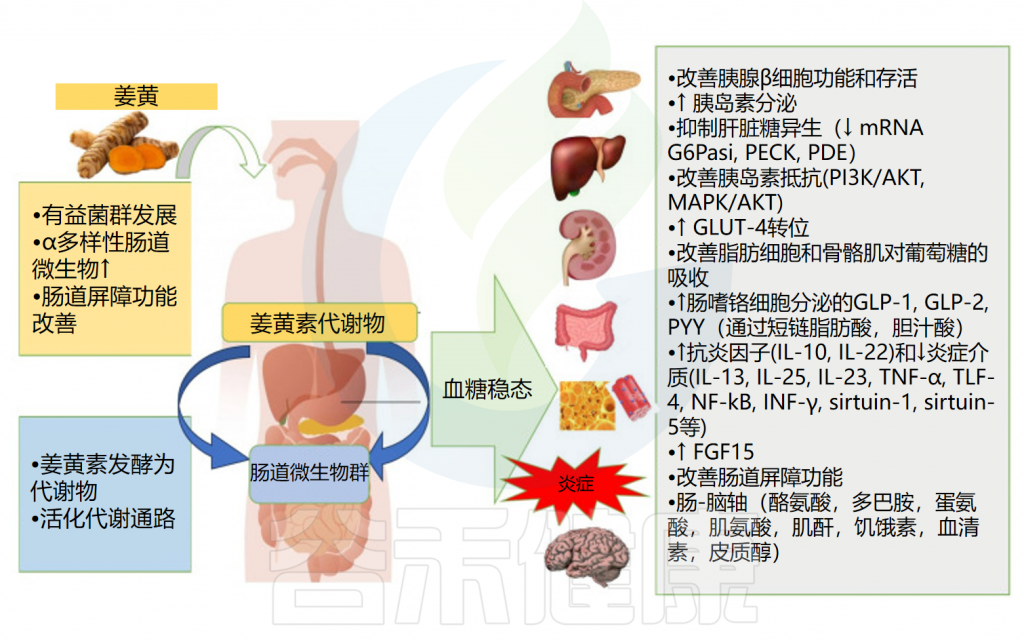
Servida S,et al.Int J Mol Sci.2024
✔ 姜黄素调节与糖尿病发作相关的菌群丰度
服用姜黄素可增加有益细菌(乳酸杆菌、双歧杆菌和产生丁酸盐的细菌)的数量,同时减少条件性致病菌(肠杆菌、Prevotellaceae和Rikenellaceae)。特别是,姜黄素增加了Muribaculaceae科细菌丰度,这类产生琥珀酸、乙酸和丙酸的细菌减少与炎症性肠病和1型糖尿病发病相关。
✔ 姜黄素及肠道菌群通过多种信号通路调节血糖
姜黄素及其衍生物通过多种信号通路调节血糖,包括PI3K/Akt通路(对氧化应激敏感的主要信号转导系统),调控细胞生长和死亡。姜黄素还激活AMPK通路调节能量代谢与细胞稳态,并通过Akt/Nrf2通路上调抗氧化机制。
肠道微生物群以相似机制影响血糖调节。姜黄素增加的干酪乳杆菌通过PI3K、AMPK2、Akt2和肝糖原合成途径改善胰岛素抵抗,并通过胆道途径降低高血糖,同时减少Caco-2细胞中的胰岛素降解酶和脂肪组织中的IGFBP-3。
IGFBP3(胰岛素样生长因子结合蛋白3)是胰岛素样生长因子(IGF)系统中的关键调节蛋白,主要功能是结合并调控IGF-1和IGF-2的活性,影响细胞生长、代谢及分化。
✔ 姜黄素与肠道菌群影响葡萄糖吸收和糖原合成
肠道微生物群通过调节GLUT-4表达和易位直接影响葡萄糖代谢。姜黄素增加的乳双歧杆菌促进糖原合成,抑制肝糖异生基因,改善胰岛素刺激的葡萄糖吸收和GLUT-4易位。加氏乳杆菌BNR-17增加肌肉GLUT-4表达,显著降低血糖。
体外研究表明姜黄素改善Akt磷酸化,促进GLUT-4易位,减少炎症因子。姜黄素与GLUT-1结合可即时、可逆地抑制葡萄糖重吸收,并调节缺氧脂肪细胞中葡萄糖转运蛋白表达,其效果取决于剂量和暴露时间。长期用药可代偿性上调GLUT蛋白。2型糖尿病肥胖大鼠接受姜黄素治疗(80mg/kg/天,8周)后改善血糖参数、胰岛素敏感性和血脂,降低肝胰丙二醛水平,降血糖作用与GLUT-4基因增加相关。
✔ 姜黄素改善血糖水平的机制与微生物活动相关
姜黄素通过增加GLP-1分泌影响血糖水平。其机制可能与抑制降解GLP-1的二肽基肽酶-4活性有关,或通过激活Ca²⁺/钙调蛋白依赖性激酶II通路直接刺激GLP-1分泌。这两种机制均与微生物活动相关,且需要足够给药时间以便调节菌群。
姜黄素通过影响含胆汁盐水解酶的拟杆菌调节胆汁酸代谢。它恢复脂多糖引起的菌群紊乱,增加产丁酸菌,减少致病菌,提高抗炎细胞因子水平。产丁酸盐的细菌促进GLP-1、PYY和GLP-2从L细胞释放,通过GPCR41/43和胆汁酸/TGR5通路发挥作用。
姜黄素还增加FXR基因表达,促进次级胆汁酸通过FXR和GPRC5调节脂质和碳水化合物代谢,并增加回肠GPRC5A/B及去乙酰化酶表达,维持碳水化合物稳态。
成纤维细胞生长因子15(FGF15)是连接菌群、宿主与姜黄素降血糖作用的关键分子。FGF15改善胰岛素敏感性,抑制肝糖异生关键酶,其表达受FXR调节。
姜黄素通过多种机制调控葡萄糖稳态,其中肠道菌群扮演着重要角色。多项随机双盲对照研究都证实姜黄素能降低血清葡萄糖、甘油三酯、低密度脂蛋白(LDL)、糖化血红蛋白(HbA1c)、瘦素,增加脂联素水平,来预防2型糖尿病。
辅助治疗神经系统疾病
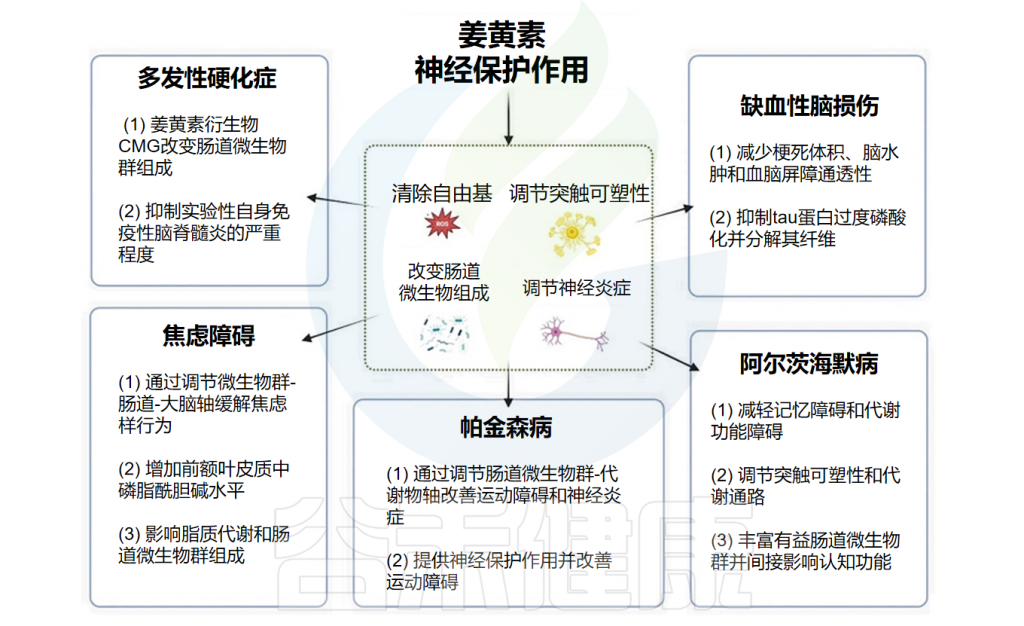
姜黄素及其肠道细菌代谢物展现出神经保护作用,在阿尔茨海默病、帕金森病、多发性硬化症、缺血性脑损伤和焦虑症等神经系统疾病中具有治疗潜力。
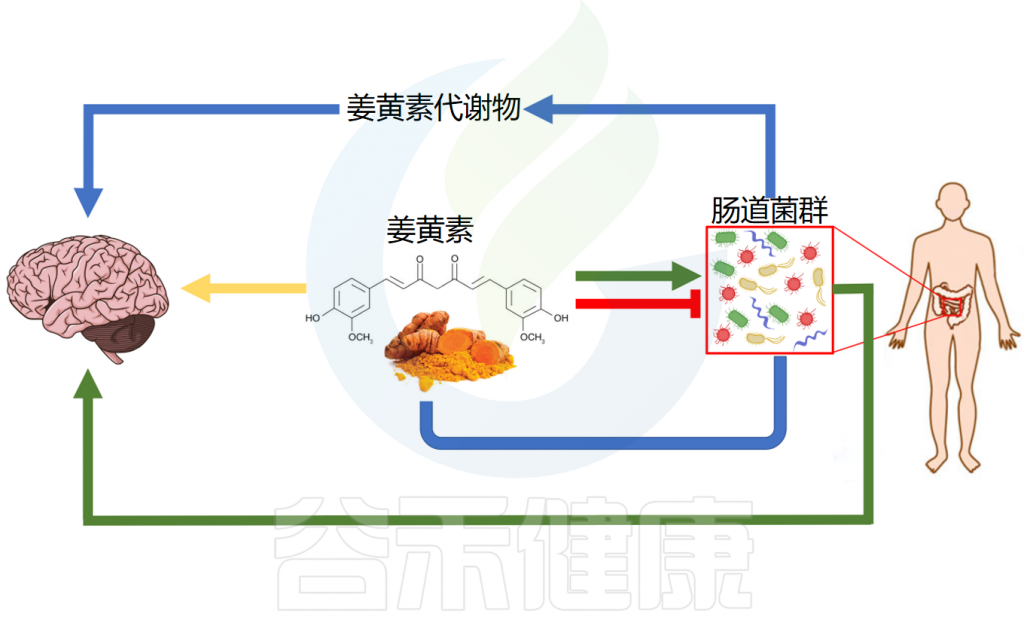
✔ 姜黄素清除自由基发挥神经保护作用
神经退行性疾病表现为特定神经元群功能的进行性丧失,导致神经缺陷和认知障碍。虽然其确切机制尚未完全阐明,但氧化应激和炎症被认为是主要致病因素。高水平活性氧(ROS)会损害所有细胞,神经元对较低ROS水平也特别敏感。ROS是大脑衰老的主要因素,与神经退行性疾病的发生发展密切相关。
姜黄素通过直接和间接清除自由基提供神经保护。它增强超氧化物歧化酶活性,将超氧化物转化为过氧化氢和氧气,并提高过氧化氢酶活性促进过氧化氢分解,展现抗氧化作用。
四氢姜黄素是研究最广泛的细菌修饰姜黄素衍生物,也能减少氧化应激和神经元凋亡,激活自噬,抑制脑损伤后线粒体凋亡。它对Aβ-寡聚体毒性有保护作用,调节神经炎症,降低β-淀粉样蛋白触发的活性氧水平和线粒体膜电位,抑制caspase激活。在脑损伤中,四氢姜黄素通过上调Nrf2通路防止神经元凋亡并改善神经行为功能。
✔ 姜黄素与肠道菌群互作改善多种神经系统疾病
在阿尔茨海默病(AD)模型中,姜黄素通过减轻记忆障碍和代谢功能障碍来发挥神经保护作用。此外,它调节突触可塑性和代谢途径,有可能改善AD相关症状。此外,姜黄素丰富了有益的肠道微生物群,从而间接影响认知功能。
在帕金森病(PD)中,姜黄素通过调节肠道微生物群-代谢物轴来改善运动缺陷和神经炎症。在多发性硬化症(MS)中,姜黄素衍生物CMG会改变肠道微生物群组成,从而抑制自身免疫性脑脊髓炎的严重程度。这种抑制与粪便和回肠内容物中特定细菌种类丰度的变化相关。
在缺血性脑损伤中,姜黄素可减少梗死体积、脑水肿和血脑屏障通透性。此外,它还可以改善缺血后的认知缺陷和神经系统结局。姜黄素治疗表明,小鼠的大脑连接和社会行为得到显著改善,同时肠道微生物群组成的改变。
在焦虑症中,姜黄素通过调节微生物群-肠-脑轴和增加前额叶皮层中的磷脂酰胆碱水平来缓解焦虑样行为。此外,它还影响脂质代谢和肠道微生物群组成以缓解焦虑症状。
姜黄素的神经保护作用
Balaji S,et al.World J Exp Med.2025
姜黄素通过清除自由基、调节突触可塑性和神经炎症以及改变肠道菌群组成等多种机制发挥神经保护作用,使其成为治疗神经系统疾病极具前景的候选药物。
姜黄素可能存在的不足
姜黄素的主要不足是单独服用时吸收率较低,且可能会引起轻微不良反应。有小部分研究中姜黄素可致肠胃胀气、胃部刺激、促进胆汁分泌和胆管炎,尤其高剂量时可能出现恶心、腹泻和头痛。
研究中,7名服用500-12000mg姜黄素的受试者72小时内出现了腹泻、头痛、皮疹和黄便症状。另一项研究显示,部分服用0.45-3.6g/天姜黄素持续1-4个月的受试者报告恶心、腹泻,并且血清碱性磷酸酶和乳酸脱氢酶水平升高。
✔ 一些肝病患者和酗酒者应谨慎使用
姜黄素可能与非甾体抗炎药、利血平和抗凝剂相互作用,肝病患者(如肝硬化、胆道梗阻、胆结石)和酗酒者应避免使用或在医师指导下使用。
应该如何服用姜黄素?
如果你正在服用补充剂,医生可能会建议每天两次,每次500毫克姜黄,与食物同服。(但并不是越多越好)
每天摄入量最高可达 8 克,但一般人群每天 500 至 1000 毫克。
可以尝试将姜黄与优质脂肪如油、鳄梨、坚果、种子等一起食用。
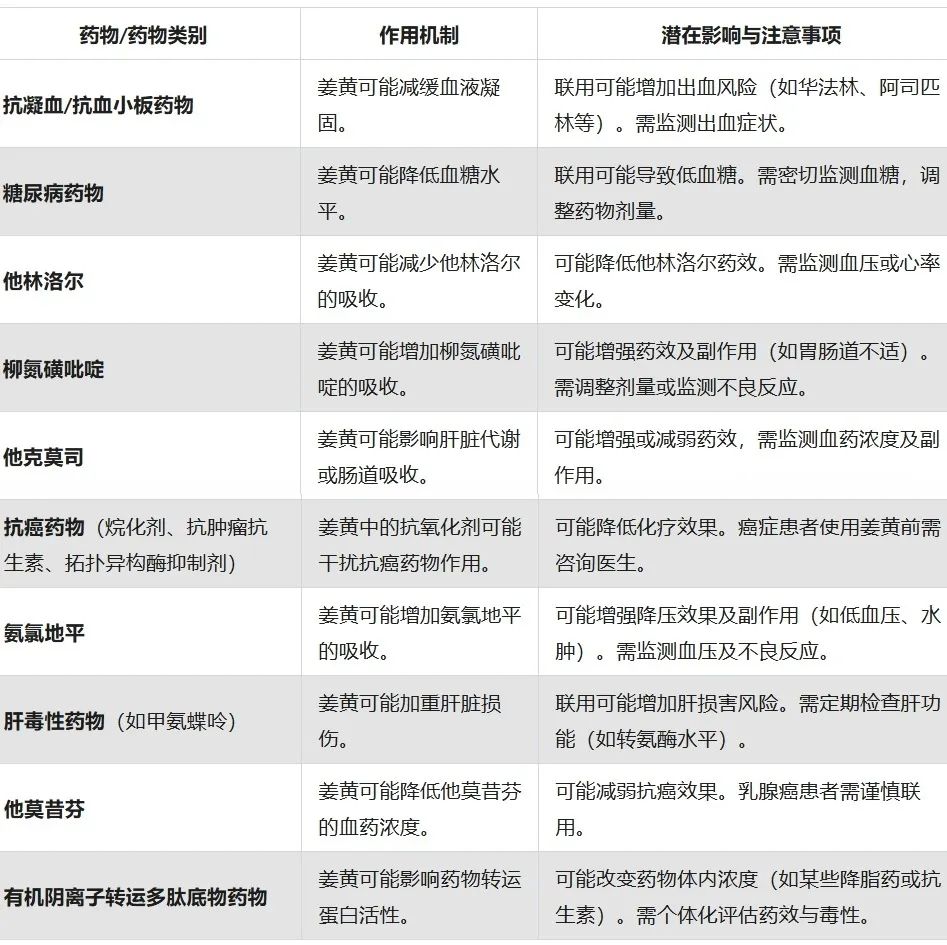
姜黄与药物之间的相互作用
中度相互作用
轻度相互作用
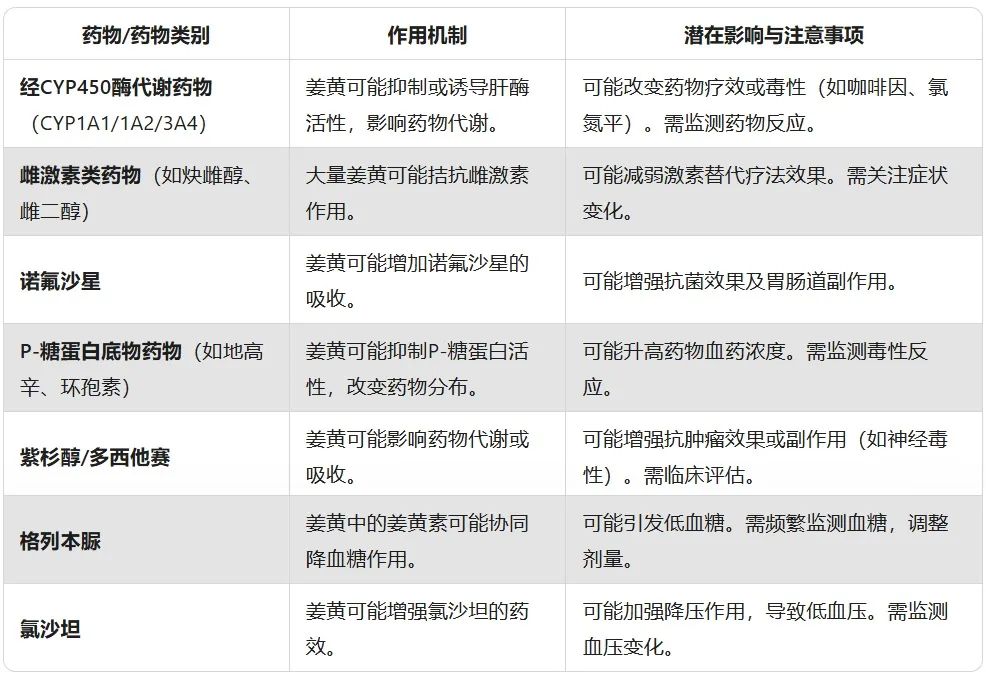
此列表可能不完整,许多其他药物如草药产品等也可能影响姜黄。
避免与其他可能影响凝血的草药/健康补充剂一起使用姜黄,包括当归、辣椒、丁香、蒲公英、丹参、 月见草 、大蒜、姜、银杏、 马栗 、人参、白杨、红车轴草等。
避免与其他可能降低血糖的草药/健康补充剂一起使用姜黄,如 α-硫辛酸 、铬、达米安娜、 熊掌草 、鹰嘴豆、大蒜、瓜尔胶、马栗、人参、车前子等。
如何更好地利用姜黄素?
姜黄素虽有多种药理活性,但因口服后肠道吸收率低,其治疗应用受到严重限制。以下是几个可能的策略和思路,旨在使人体更好地利用姜黄素,实现其抗炎、抗氧化、抗肿瘤以及其他保护作用。
✔ 个体化肠道微生物群检测与评估
通过粪便菌群测序等手段,了解个体肠道菌群的整体结构、优势菌群和多样性情况。这有助于判断个体在姜黄素吸收和代谢过程中的潜在差异,因为肠道菌群在姜黄素的化学转化中可能起到双向调控作用(既可能通过代谢生成更有活性的代谢物,也可能助推姜黄素的降解)。
依据检测结果,对肠道菌群中与药物代谢、炎症调节和屏障功能相关的菌群比例进行评估,从而判断是否需要额外进行菌群调节干预。
✔ 利用益生菌和益生元改善姜黄素代谢环境
有研究提示,共给药策略(例如与黑胡椒碱联合使用)能够显著提高姜黄素的生物利用度。同理,合理补充某些益生菌(如乳杆菌、双歧杆菌)和益生元可改善肠道微生态平衡,优化肠屏障功能;这不仅有助于减少姜黄素在肝脏首过效应中的代谢转化,还可能促进姜黄素在肠道内的活性释放。
根据个体菌群失衡的具体情况,可以设计联合微生态干预方案,例如在姜黄素给药前后,先行或同步补充针对性益生元,从而改善消化道环境,增强姜黄素的吸收和转化效果。
✔ 给药策略的个性化优化
由于姜黄素本身具有低水溶性和较高亲脂性,目前已有纳米技术、脂质体、固体分散体等多种新型剂型用于提高其生物利用度。结合个体的肠道菌群特点,可以选择或定制适合个体微生态环境的姜黄素制剂。例如,对于部分菌群功能较弱的个体,使用纳米载体不仅可以增加姜黄素的稳定性,也可以延缓其在肠道内的降解过程,从而为肠道菌群与姜黄素之间的互作提供足够的时间。
此外,若检测发现个体肠内特定菌群(例如参与代谢姜黄素转化的菌群)数量较低,可能需要重点采用辅佐用药策略,抑制姜黄素过快的首过代谢(比如结合黑胡椒碱)与微生态调节进行联合应用,以获得更高的药效浓度。
✔ 饮食和生活方式的干预
饮食习惯对肠道菌群有显著影响,个体化的饮食调整(例如增加富含膳食纤维、益生元的食物)可以促进有益菌群的发展,改善肠道环境,从而间接提高姜黄素的吸收和生物转化。
此外,合理的饮食还能减少慢性炎症状态,增强机体对姜黄素抗炎、抗氧化作用的反应。因此,制定一套综合性的生活方式干预方案,将姜黄素的服用与膳食、运动等措施相结合,有望发挥协同增效作用。
✔ 未来的个性化药物方案探索
随着精准医学的发展,可以通过多组学(如基因组、代谢组、微生物组)的综合分析进一步解析姜黄素与个体肠道菌群之间的交互机制,从而设计出针对不同疾病状态(如炎症性疾病、肿瘤或代谢性疾病)的个性化姜黄素使用方案。
临床上可设计小规模试验,通过定期监控个体肠道菌群变化、姜黄素血药浓度和临床指标,进一步验证联合微生态调控和个性化姜黄素给药的效果,逐步形成标准化的治疗模式。
姜黄素与肠道微生物群的双向互作开辟了天然药物对健康影响的新视角。一方面,肠道菌群通过多种酶促途径(还原、去甲基化、羟基化等)将姜黄素转化为具有独特生物活性的代谢物,显著提高其生物利用度;另一方面,姜黄素作为天然益生元调节菌群丰度与多样性,增加有益菌如乳酸菌的比例,抑制有害菌繁殖,改善肠道屏障功能并减轻炎症水平。
这种协同互利的关系使姜黄素能够在临床治疗中发挥多种健康功效,从炎症性肠病、结直肠癌等胃肠道疾病,到肥胖、糖尿病等代谢性疾病,甚至阿尔兹海默病等神经系统疾病。
随着精准医学发展,未来结合肠道微生物组测序与个性化给药策略,有望开发出更精准、高效的药物应用方案,不仅提高其生物利用度,更能充分发挥其治疗潜力。姜黄素与肠道微生物群的协同作用只是连接传统草药与现代精准医学的一个例子,为健康产品市场提供了极具价值的科学依据和创新方向。
主要参考文献:
Balaji S, Jeyaraman N, Jeyaraman M, Ramasubramanian S, Muthu S, Santos GS, da Fonseca LF, Lana JF. Impact of curcumin on gut microbiome. World J Exp Med. 2025 Mar 20;15(1):100275.
Zhu J, He L. The Modulatory Effects of Curcumin on the Gut Microbiota: A Potential Strategy for Disease Treatment and Health Promotion. Microorganisms. 2024 Mar 23;12(4):642.
Obrzut O, Gostyńska-Stawna A, Kustrzyńska K, Stawny M, Krajka-Kuźniak V. Curcumin: A Natural Warrior Against Inflammatory Liver Diseases. Nutrients. 2025 Apr 18;17(8):1373.
Scazzocchio B, Minghetti L, D’Archivio M. Interaction between Gut Microbiota and Curcumin: A New Key of Understanding for the Health Effects of Curcumin. Nutrients. 2020 Aug 19;12(9):2499.
Zam W. Gut Microbiota as a Prospective Therapeutic Target for Curcumin: A Review of Mutual Influence. J Nutr Metab. 2018 Dec 16;2018:1367984.
Servida S, Piontini A, Gori F, Tomaino L, Moroncini G, De Gennaro Colonna V, La Vecchia C, Vigna L. Curcumin and Gut Microbiota: A Narrative Overview with Focus on Glycemic Control. Int J Mol Sci. 2024 Jul 14;25(14):7710.
Hewlings SJ, Kalman DS. Curcumin: A Review of Its Effects on Human Health. Foods. 2017 Oct 22;6(10):92.
Pluta R, Januszewski S, Ułamek-Kozioł M. Mutual Two-Way Interactions of Curcumin and Gut Microbiota. Int J Mol Sci. 2020 Feb 5;21(3):1055.
Di Meo F, Margarucci S, Galderisi U, Crispi S, Peluso G. Curcumin, Gut Microbiota, and Neuroprotection. Nutrients. 2019 Oct 11;11(10):2426.
谷禾健康
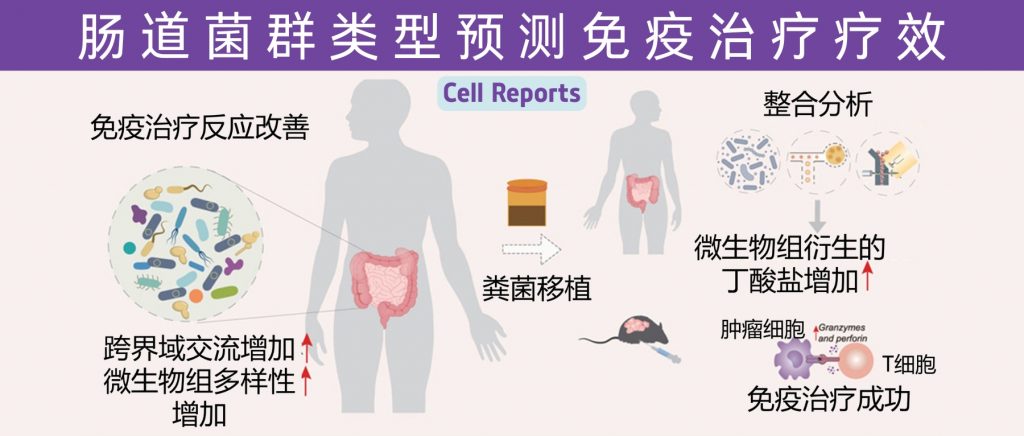
近年来,免疫治疗在癌症治疗领域展现出了巨大的潜力,特别是针对PD1/PDL1通路的免疫检查点抑制剂(ICIs)已在多种实体瘤患者中取得显著疗效。然而,治疗反应的异质性和耐药性的出现依然是当前面临的主要挑战之一。越来越多的研究表明,肠道菌群作为人体内一个庞大的微生态系统,不仅参与机体代谢和免疫调控,而且在调节肿瘤免疫反应方面发挥着关键作用。
让我们快速了解一下肠道菌群如何通过细胞水平、分子信号通路和代谢产物来影响PD1/PDL1肿瘤免疫疗法的疗效。
1 核心菌群与免疫调控机制
多项临床与实验室研究均显示,特定菌群对PD1/PDL1免疫检查点抑制剂治疗具有显著的正向作用。
例如,Bifidobacterium属菌群被发现能显著促进抗PDL1疗法的抗肿瘤作用;而Akkermansia muciniphila在PD1抑制治疗中也表现出与治疗反应正相关的趋势。此外,研究还发现,黑色素瘤患者中较高的微生物多样性和特定菌群如Ruminococcaceae和Faecalibacterium的丰富度均与更长的无进展生存期相关。
肠道菌群对免疫调控的机制主要表现在以下几个方面:
促进树突状细胞成熟:某些菌群(如Bifidobacterium)可通过激活树突状细胞,从而增强抗原呈递能力,提高CD8+ T细胞的活性。
调节T细胞免疫状态:特定菌群通过影响细胞因子分泌和T细胞亚群分布,调节包括CD8+效应T细胞和调节性T细胞(Treg)在内的免疫平衡,从而实现抗肿瘤效应。
通过代谢产物发挥作用:菌群代谢产物,如短链脂肪酸(SCFAs)和次级胆汁酸,对免疫细胞储存、功能激活具有直接调控作用。
这些作用机制不仅在单一免疫治疗模式中发挥效应,同时也对联合治疗(例如PD1/PDL1抑制剂与CTLA-4抑制剂联合使用)产生协同增效作用。
2 关键代谢物及其信号调控路径
肠道菌群通过发酵膳食纤维等底物,产生大量的短链脂肪酸(SCFAs),如丁酸盐和丙酸盐,这些代谢物在调节宿主免疫功能中起到关键作用。研究发现,短链脂肪酸不仅参与维持肠道上皮屏障的稳定,还能调节T细胞分化和促炎/抗炎反应的平衡。此外,菌群代谢的次级胆汁酸也被证明在免疫抑制和调控细胞因子水平中起到重要作用。
值得注意的是,不同菌群通过生成不同的代谢产物,对免疫系统的影响可能存在正负两方面的效应。例如,有研究显示高浓度的丁酸盐和丙酸盐可能会在某些条件下限制CTLA-4抑制剂的疗效,从而提示适度平衡菌群代谢产物对于免疫治疗的成功至关重要。同时,某些菌群如Prevotellaceae和Rikenellaceae则可能通过降低丁酸盐水平来促进促炎性巨噬细胞M1型的极化,从而间接影响免疫治疗的疗效。
3 不同肿瘤类型中的菌群特征与免疫应答
肠道菌群对免疫治疗的影响在不同肿瘤类型中可能存在显著差异。下面介绍一下在黑色素瘤、非小细胞肺癌(NSCLC)、肝细胞癌(HCC)以及结直肠癌(CRC)中的相关发现。
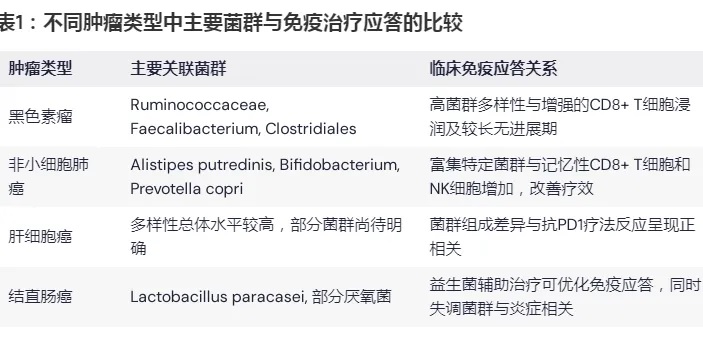
表1说明:各肿瘤类型中显示出菌群多样性和特定菌群丰度与PD1/PDL1免疫治疗效果之间存在明显相关性,该表对比了不同肿瘤中的主要菌群与免疫应答情况。
4 肠道菌群调控机制的分子与细胞通路
肠道菌群通过多条分子信号通路及细胞间相互作用调控宿主的免疫反应,进而影响PD1/PDL1免疫治疗的疗效。
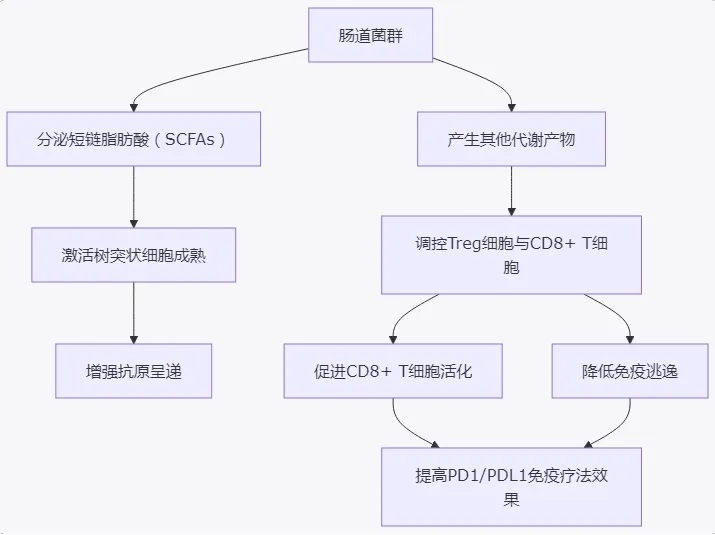
图1:肠道菌群通过分泌代谢产物调控树突状细胞成熟、T细胞活性与调节性T细胞平衡,从而增强PD1/PDL1免疫治疗效果。
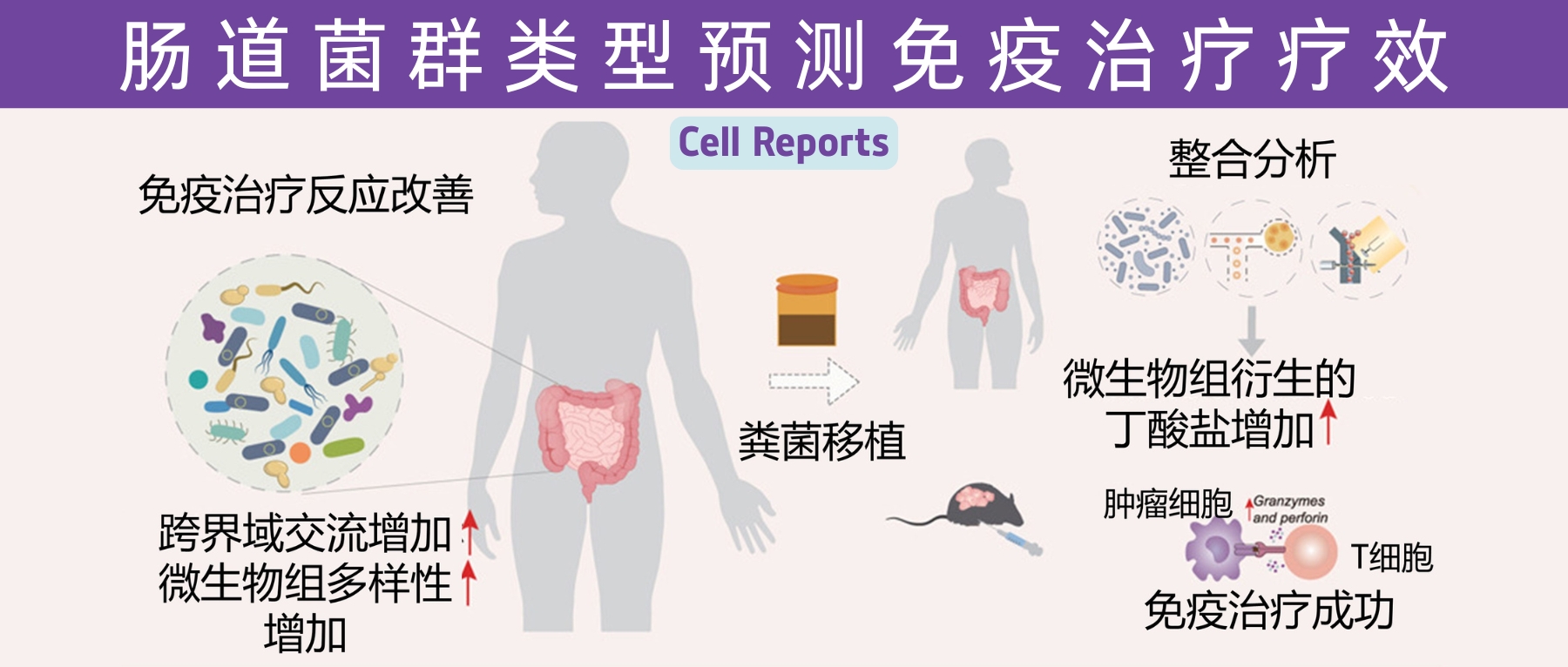
近日,一项来自上海交通大学医学院研究团队的成果发表在《Cell Reports》,通过整合多组学数据与临床队列,揭示了基于肠道菌群分类(有利型与不利型)与抗PD-1/PD-L1免疫治疗疗效的显著关联。

其中,有利型患者表现出更高的真菌与细菌多样性、富集丁酸代谢通路及促免疫菌群(如Faecalibacterium prausnitzii),以及肿瘤微环境中细胞毒性CD8+ T细胞浸润增强的特征,且在多癌种队列中显著关联于抗PD-1/PD-L1治疗的临床缓解与生存获益。
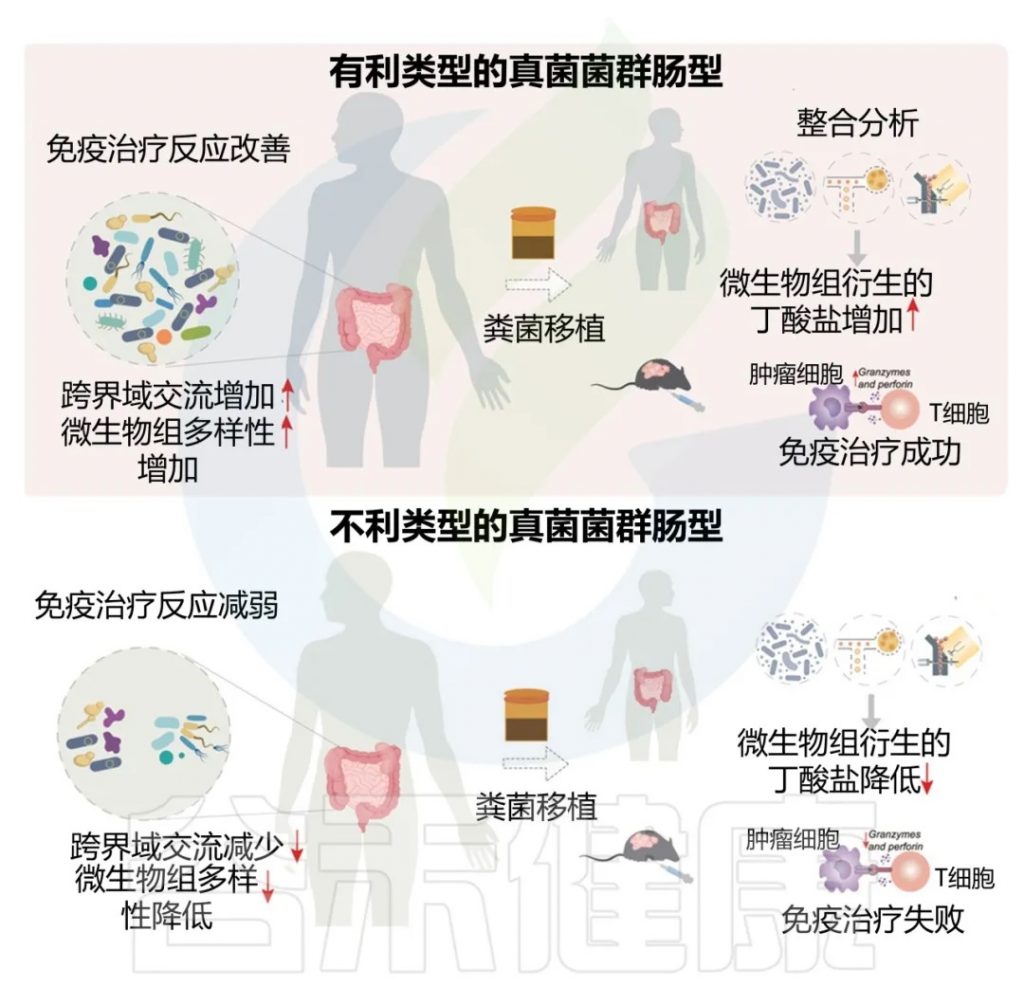
进一步通过FMT实验证实,移植来自有利型供体的粪便可显著提升受体对免疫治疗的敏感性,并重塑肠道菌群代谢功能。
这些发现不仅深化了对肠道真菌组-细菌互作网络的理解,为优化个体化免疫治疗提供了新型生物标志物,更为优化免疫治疗分层策略及FMT供体筛选提供了科学依据,具有重要的临床转化潜力。

多组学关联分析与肠道菌群类型特征
详细对比了两种肠道菌群类型的微生物群落特征差异,包括多样性、产丁酸菌含量、代谢通路等,并展示了它们与临床疗效的关联。
粪便微生物移植实验验证
呈现了临床FMT研究和动物模型验证结果,证明来自有利型供体的FMT可显著提升受体对免疫治疗的敏感性。
总结了研究的创新点及其在预测分层和干预优化方面的临床应用价值。
肠道菌群类型:免疫治疗疗效的新生物标志物
免疫检查点抑制剂(如抗PD-1/PD-L1)显著改变了癌症治疗格局,但患者反应差异大。
该研究团队采用无监督聚类方法分析肠道菌群组成,在多个独立队列中成功识别并验证了两种截然不同的类型——“有利型”与“不利型”,这一发现揭示了肠道菌群对免疫治疗应答的预测价值。

肠道菌群的类型不仅与临床反应相关,还与患者总生存期密切相关,为个体化治疗策略提供了新思路。
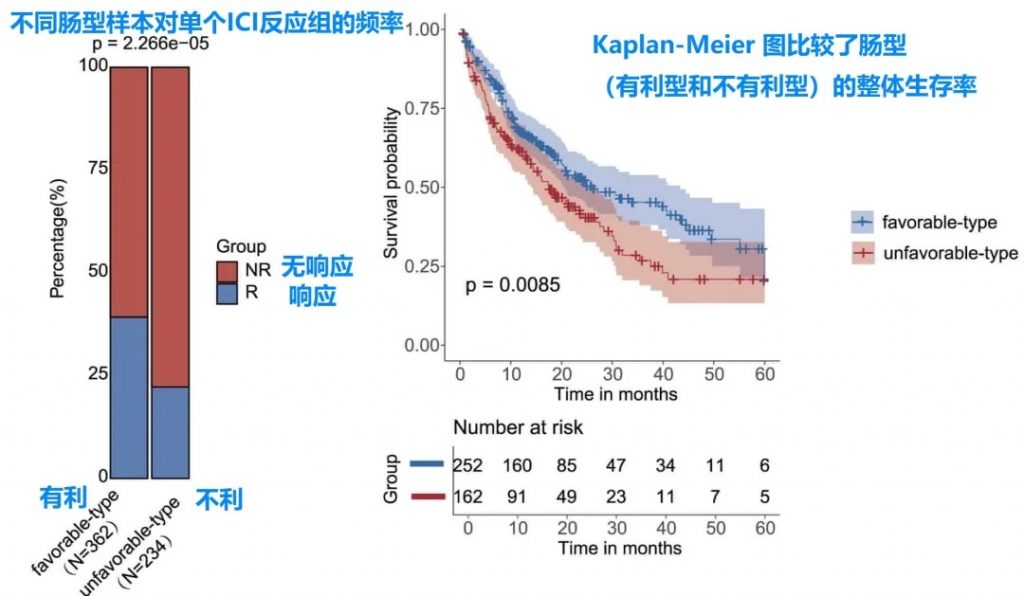
有利型患者对抗PD-1/PD-L1免疫治疗的响应率显著高于不利型 (p=2.266e−5),总生存期也更长 (p=0.0085)。
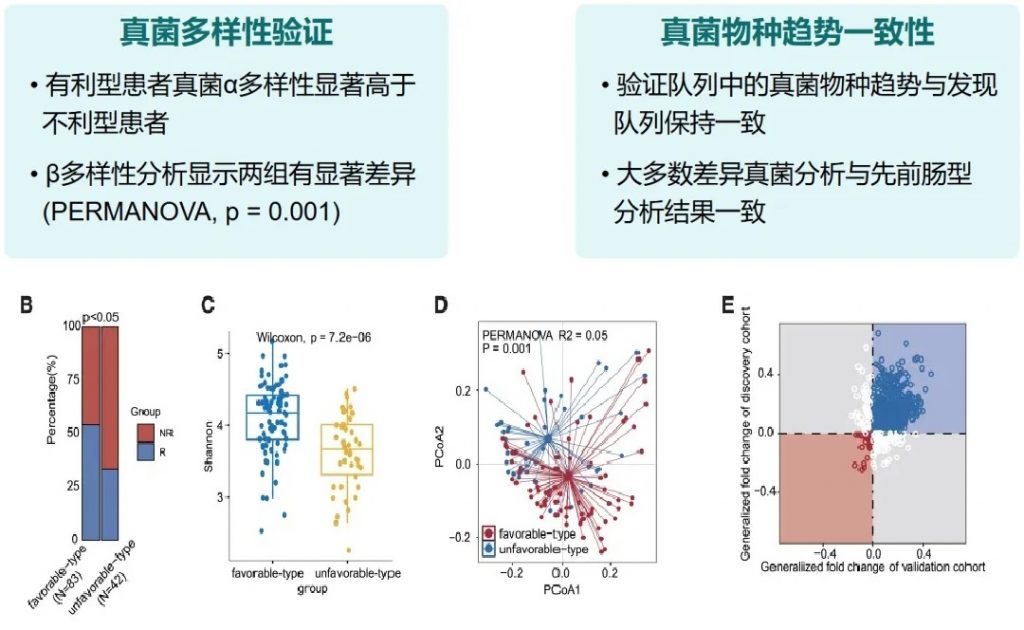
“有利型”和“不利型”肠道菌群差异 细菌、真菌
细菌 多样性
——α多样性差异
有利型患者表现出显著更高的细菌α多样性指数(Shannon、Chao1、Simpson),这与既往研究发现的免疫治疗响应者多样性更高的结论一致。
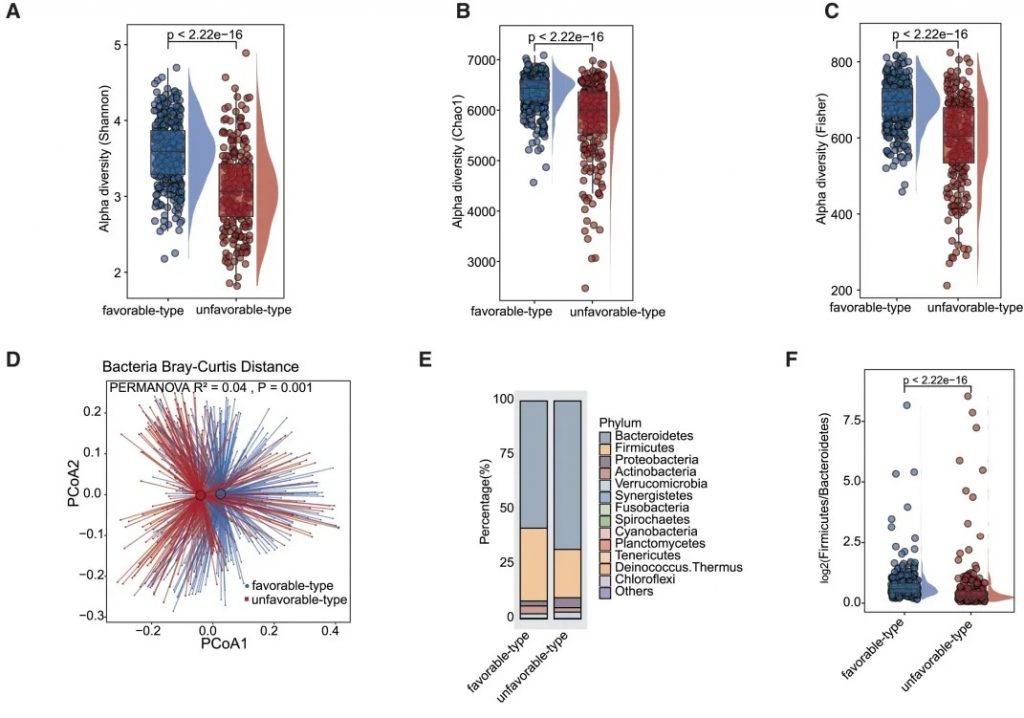
——β多样性差异
细菌 主要构成
有利型患者表现出更高的Firmicutes/Bacteroides比值,这被认为是肠道菌群平衡的重要指标,与免疫治疗的良好反应相关。
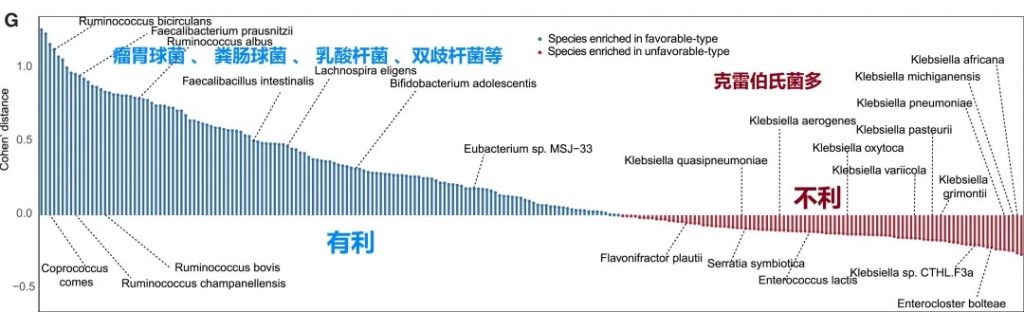
有利型的患者肠道中富含产丁酸盐的细菌,特别是Faecalibacterium prausnitzii、Lachnospira eligens,这些细菌通过产生丁酸盐调节肠道微环境,增强抗肿瘤免疫反应。
真菌 多样性
有利型表现出更高的Shannon指数和OTU丰富度,真菌物种多样性更丰富,这与对免疫治疗的更好反应相关联。
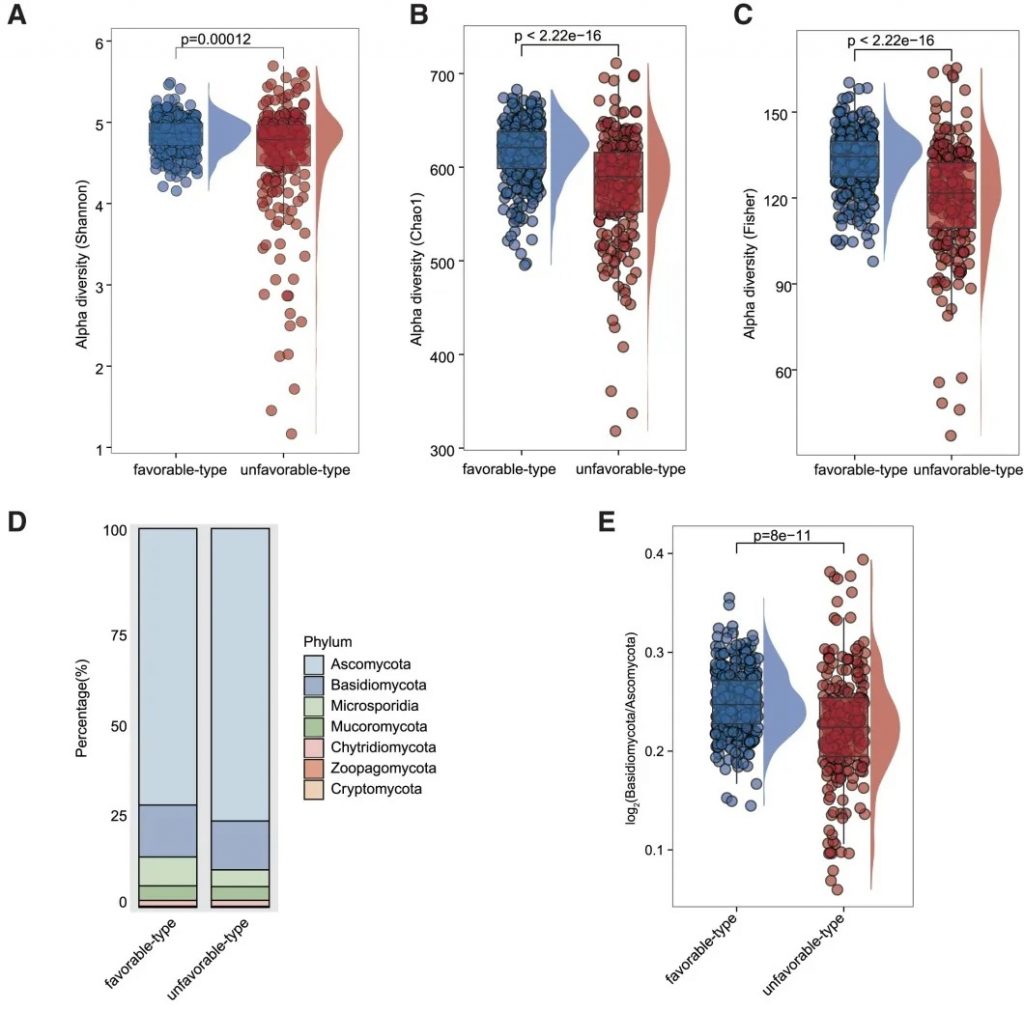
真菌 主要构成
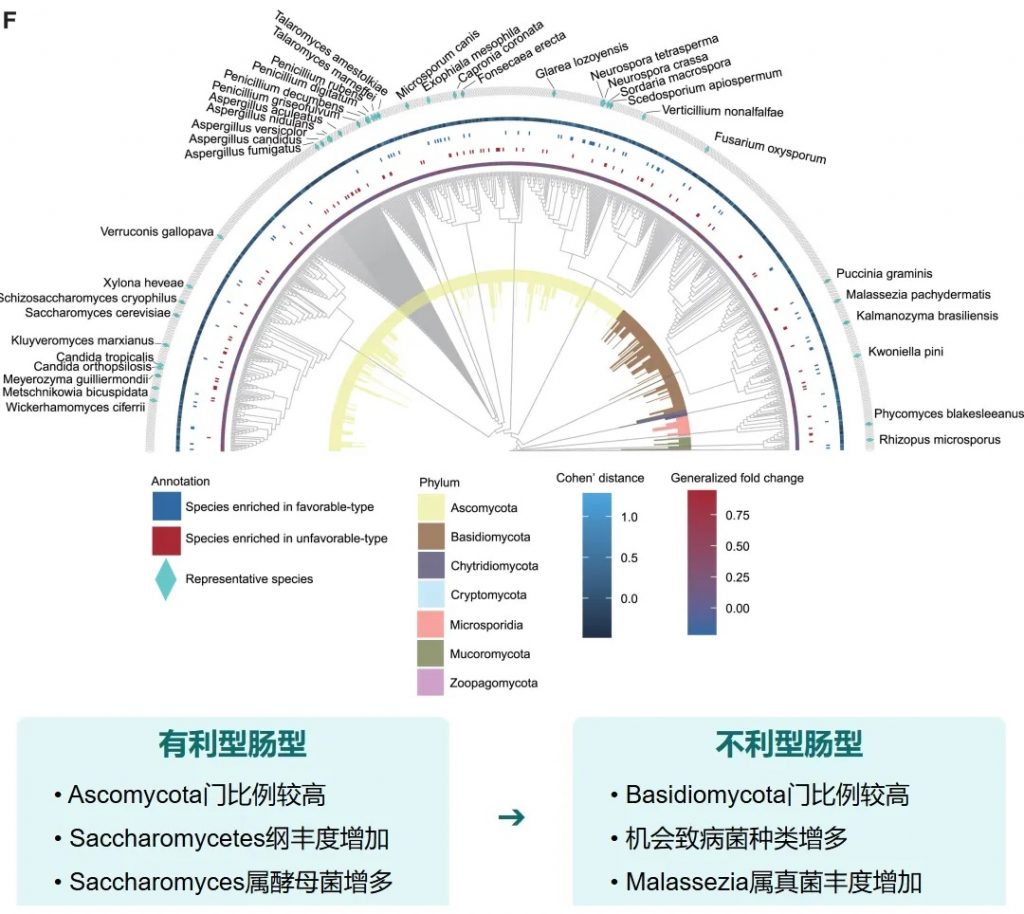
关键真菌与细菌的共现分析
有利型样本在真菌-细菌界中表现出增加的正相互作用,而不利型样本在菌-细菌界表现出更多的负相互作用。
关键真菌-细菌互作对有利型中的关键互作真菌-产丁酸盐细菌正向互作:
有利型的真菌群落结构似乎与某些细菌表现出更积极的共生关系。真菌-细菌间相互作用可能在抗PD-1/PD-L1 ICI的反应中发挥关键作用。
基于特定菌群类型的微生物功能分析
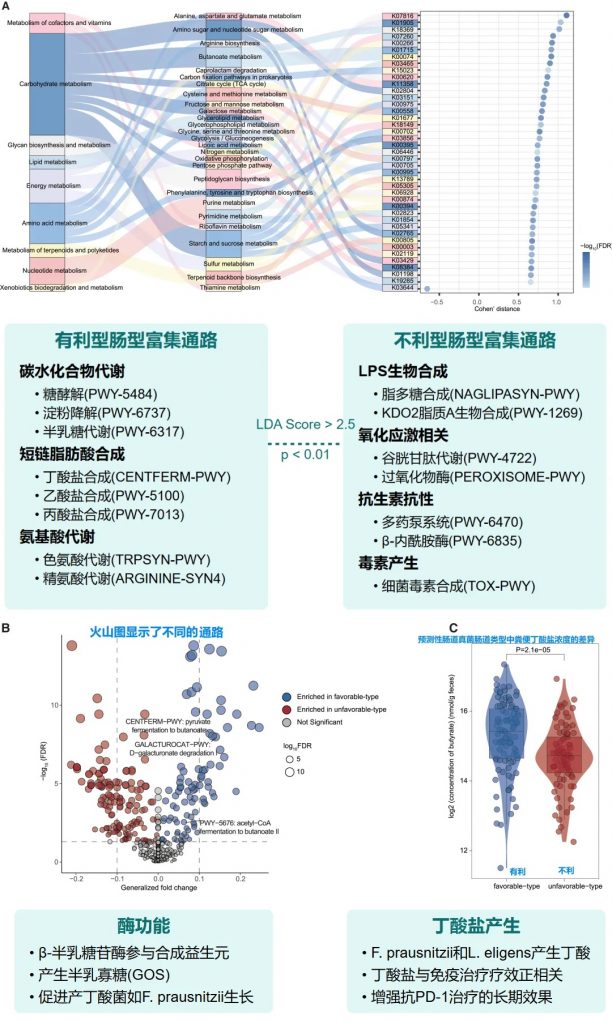
肠道菌群类型的外部验证与多组学分析
验证队列特征
• 125名接受PD-1抗体治疗的泛癌症患者
• 基于随机森林分类器(RandomForestClassifier)进行肠道菌群类型的预测
• 预测结果: 83名有利型患者,42名不利型患者
验证分析方法
• 基于机器学习的肠道菌群类型预测模型
• 随访评估对免疫治疗的临床反应
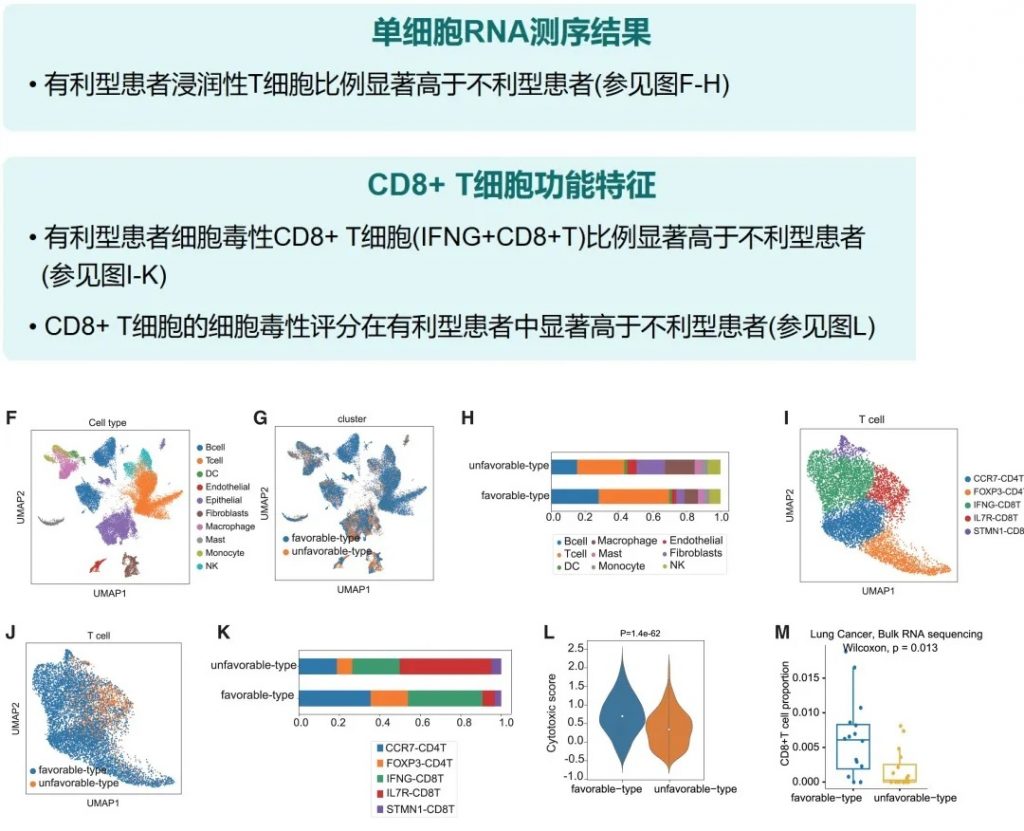
丁酸可促进CD8+ T细胞浸润肿瘤微环境,直接增强抗肿瘤免疫。
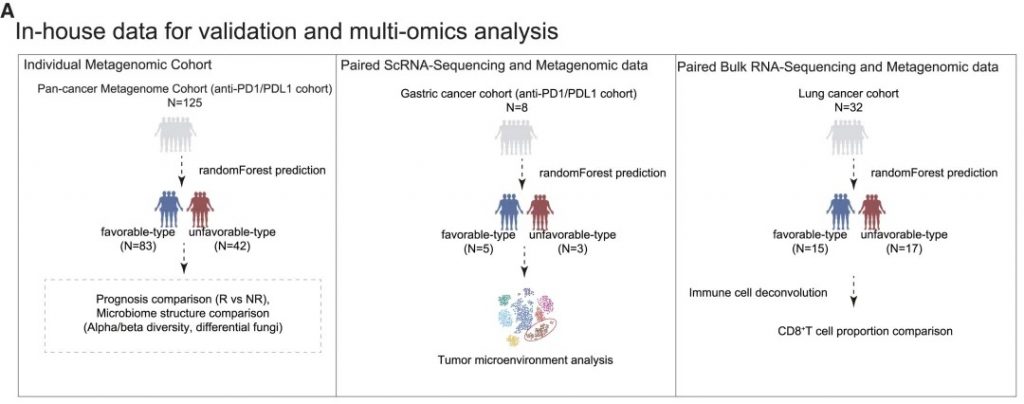
FMT供体选择:优化免疫治疗疗效
整合数据构成
• 3个近期发表的临床试验数据
• 12名FMT供体 (5名有利型, 7名不利型)
• 43名黑色素瘤患者接受者
• 包含宏基因组数据和临床结果
FMT临床疗效分析
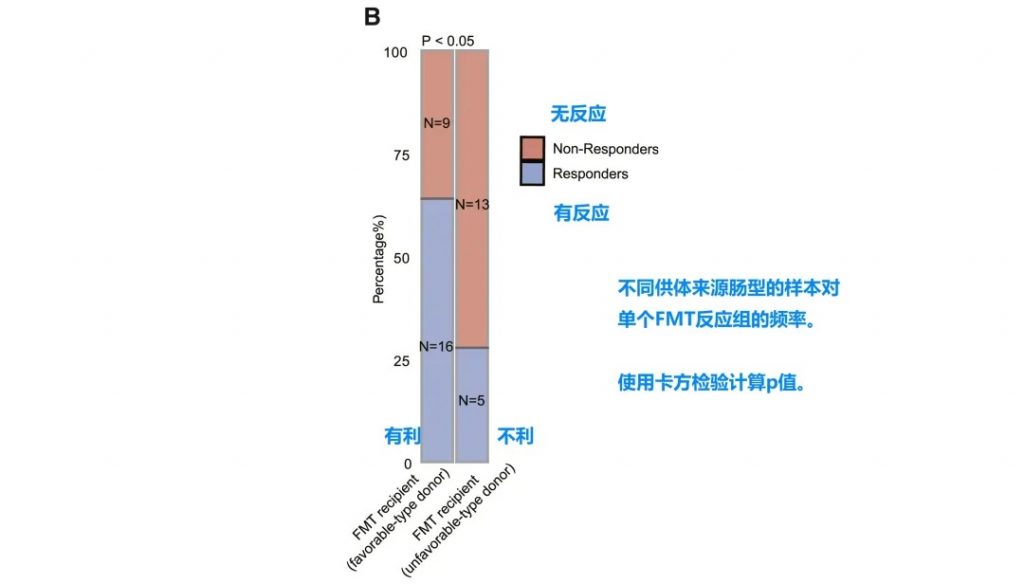
• 接受有利型供体FMT的患者对抗PD-1/PD-L1抗体免疫治疗的反应明显改善
• 卡方检验证实统计学显著差异 (p < 0.05)
接受“有利型”供体FMT的患者显示出对抗PD-1/PD-L1抗体免疫治疗的显著改善反应(p < 0.05)。这一发现具有临床指导意义,为FMT供体筛选提供了新的标准。
FMT前后微生物多样性变化
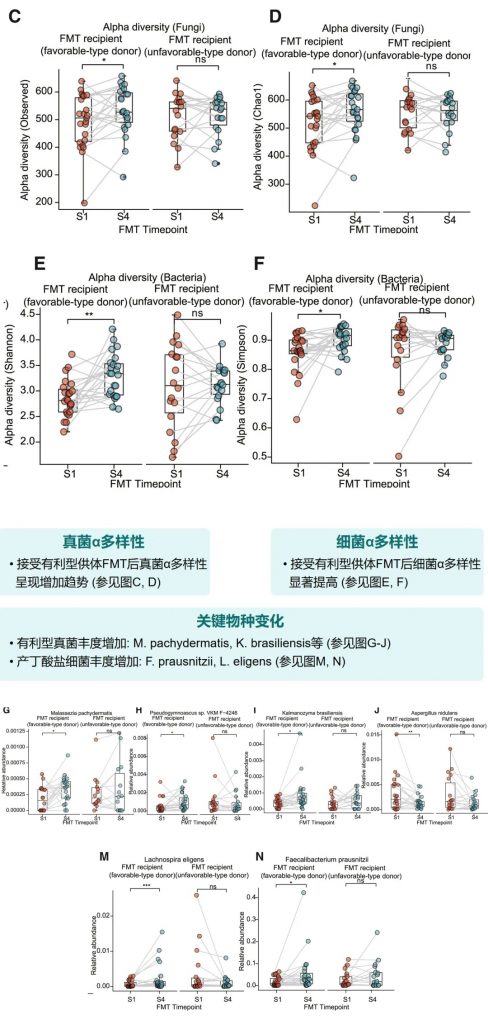
接受有利型供体FMT后,受者的真菌和细菌α多样性均有明显提高。特别是,有利型特征真菌和产丁酸菌的丰度显著增加,这与增强的免疫治疗效果相关。
体内验证实验结果
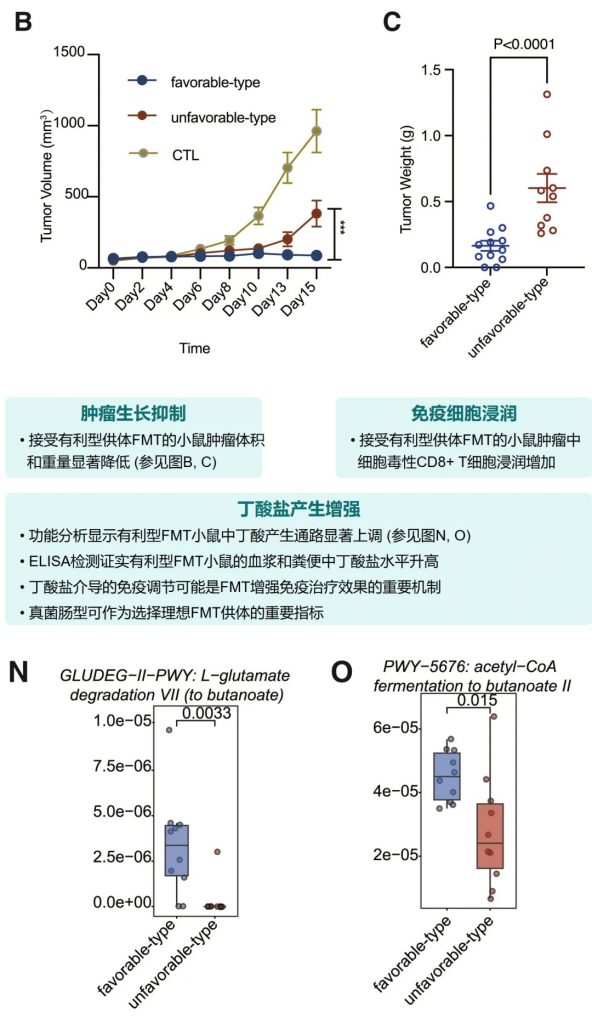
小鼠实验进一步确认了有利型供体FMT可以增强抗PD-1治疗效果,表现为肿瘤生长抑制、CD8+ T细胞浸润增加,以及丁酸盐产生增强。这为FMT增强免疫治疗提供了机制解释。
该研究揭示了肠道微生物群类型与免疫检查点抑制剂(ICI)疗效之间的密切关系,为优化癌症免疫治疗策略提供了新的视角。
肠道菌群与免疫治疗响应之间存在显著关联,且这种关联可通过粪菌移植等干预手段加以调控,为克服免疫治疗耐药提供新策略。
以下是几个具有前景的研究和临床应用方向:
辅助诊断的开发与实施
随着肠道菌群类型与免疫治疗疗效之间关联的确立,开发肠道微生物群分析作为免疫治疗的伴随诊断工具已成为一个明确的临床需求。
粪菌移植(FMT)作为辅助治疗手段
对于被确定为不利型的患者,粪菌移植提供了一种有前景的干预手段,可能显著提高免疫治疗的响应率。
更大规模的临床验证研究
为了将这些发现转化为临床实践,需要进行更大规模的验证研究:
未来,肠道微生物群检测有望成为免疫治疗患者的常规伴随诊断工具,帮助临床医生识别潜在的治疗无应答人群。
也就是说做免疫治疗的人群可以做肠道菌群评估作为辅助诊断,如果免疫治疗没有效果的病人,考虑通过FMT调整菌群后,提高治疗效果。来自有利型供体的粪菌移植可能成为提高治疗效果的有效辅助手段。
这一整合策略不仅能够优化现有免疫治疗的应用,也为开发新型微生物组靶向干预手段提供了方向,最终实现免疫治疗疗效的最大化和个体化。
在癌症精准医疗的框架下,肠道微生物组评估和干预将与基因组学、免疫学等多组学手段一起,共同构成未来肿瘤免疫治疗的个体化决策基础,为患者带来更好的临床获益。
主要参考文献
Hu M, Zhu X, Huang X, Hua L, Lin X, Zhang H, Hu Y, Tong T, Li L, Xuan B, Zhao Y, Zhou Y, Ding J, Ma Y, Jiang Y, Ning L, Zhang Y, Wang Z, Fang JY, Zhang Y, Xiao X, Hong J, Chen H, Li J, Chen H. Optimizing anti-PD-1/PD-L1 therapy efficacy and fecal microbiota transplantation donor selection through gut mycobiome-based enterotype. Cell Rep. 2025 Apr 20;44(5):115589.

谷禾健康
不夸张的说,微生物是我们星球的统治者,维持着每一个宏观生物的运作。它们调控地球元素循环和大气碳平衡,并且许多关键步骤仅由微生物进行(例如,涉及无机氮转化、甲烷产生和氧化、金属硫化物氧化、铁氧化和还原等的许多过程)。在过去的20年里,大量的研究证实人类健康行为到心理方面也依赖于每个人的微生物组。
由于自然界和人体都是一个高度竞争的环境,微生物的生长通常受到严格限制和管制,因此微生物丰度和生物量在生态系统和地球的承载能力如何达到平横值得我们关注和研究。想要搞清楚这个问题,首先要弄清楚微生物是如何生长的,它们的生长需要什么营养素,如何吸收,影响因素是什么,生长速率和趋势是什么?
我们知道微生物生长速率是微生物细胞在变化的环境中所面临的充足或不利条件下营养物质可利用性的结果。自然界以及人体肠道中存在的高微生物丰度和多样性有助于产生高度复杂的系统。
例如,据报道:
土壤中含有约细菌1010/g,约有30000种不同的微生物分类群;
清洁水可能含有约细菌106/mL,可能有数千种不同的分类群;
人每克结肠内容物中含有约1011-1012个微生物。
不同的微生物由群落内特定比例的细胞代表,它们的丰度和活性会随时间变化,取决于环境的变化。这些变化是动态物理(例如,温度、湿度等)、化学(有机和无机营养物的可用性、pH值、污染物的抑制作用等)和生物(竞争、捕食、多样性等)自然环境中的因素。目前,微生物学面临的主要挑战是评估微生物群落的生长状态和活性及其各组分的贡献。
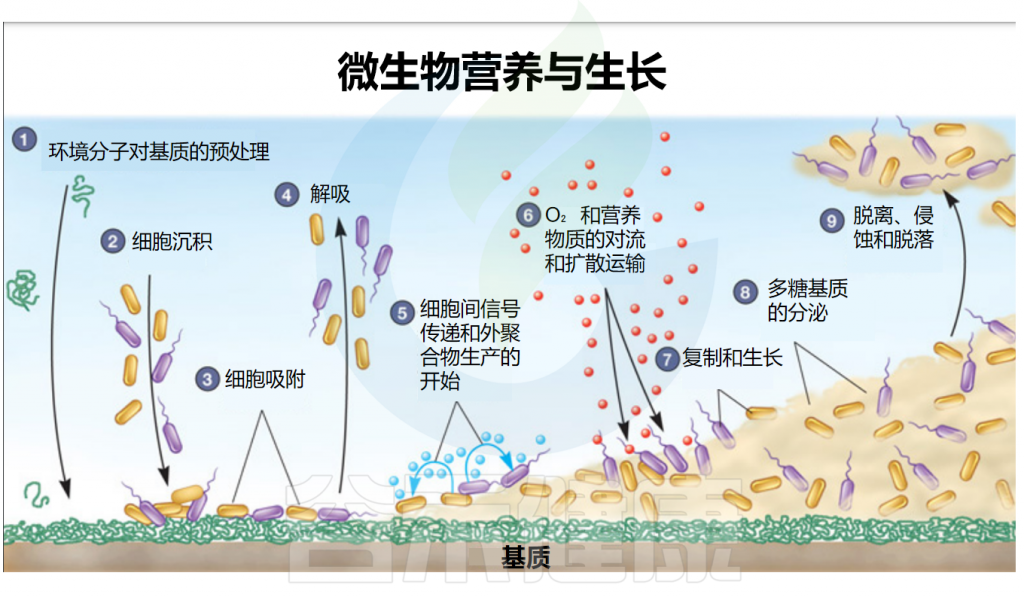
本文从微生物生长所需的营养和影响因素入手,了解评估微生物群落的生长状态和活性等。
▸ 宏量和微量营养素
宏量营养素是大量且必需的
微生物细胞由碳、氧、氢、氮、硫、磷、钾、钙、镁和铁等多种元素组成。这些也被称为宏量元素或宏量营养素,因为微生物需要大量这些元素。其中,C、H、O、N、S和P是碳水化合物、脂质、蛋白质和核酸所需的主要元素。
除此之外,还发现其他常量营养素具有多种生物学功能。例如,钾离子(K+)参与多种酶的活性,钙(Ca2+)是细菌内生孢子的重要元素,镁(Mg2+)作为不同酶的辅助因子参与等。
微量营养素需要少量但也有重要作用
另一方面,微生物在小范围内也需要其他几种元素,这些元素被称为微量元素或微量营养素。包括锰、锌、钴、钼、镍和铜。
这些不是微生物生长所必需的元素,但它们以多种方式参与生物功能。例如,锌(Zn2+)存在于几种酶的活性位点,锰(Mn2+)参与磷酸基团转移的催化,钼(Mo2+)对于固氮至关重要等。
▸ 对碳、氢、氧和电子的需求
每个生物体生长和发育都需要碳、氢、氧和电子
有机分子对微生物至关重要,这些有机分子的主要成分是碳、氢和氧。
让我们首先关注碳:碳是所有生物的基础元素,构成了蛋白质、碳水化合物、脂质和核酸等生命大分子的核心骨架。
C、H和O的需求通常可同时满足,因为大多数碳源分子中含有氢和氧。异养生物不仅从有机分子获取碳、氢和氧,还获取电子,用于能量产生和生物合成反应。
电子有两个主要功能,即通过电子传递链的运动,在其他氧化还原反应中可以提供能量用于细胞工作,并且在生物合成过程中也需要电子来还原分子。
由于这些有机碳源提供的电子可用于电子传输以及其他氧化还原反应,因此许多异养生物也使用它们的碳源作为能源。
生物获取能量的主要途径是光能和化学能
生物获取能量有两种途径:光能或化学能。光能来自太阳,而化学能可以来自有机或无机化学物质。那些使用光能的生物体被称为光养生物(“光食者”),而那些使用化学能的生物体被称为化学营养生物(“化学食者”)。
化学能可以来自无机源或有机源。使用无机来源的生物体被称为“无机营养生物”(lithotroph),而使用有机来源的生物体被称为“有机营养生物”(organotroph)。
▸ 氮、磷、硫的需求和作用
氮是一种必需元素,在氨基酸、嘌呤、嘧啶、一些碳水化合物和脂质的合成中起重要作用。许多微生物可以使用氨基酸中的氮。也可以通过谷氨酸脱氢酶或谷氨酰胺合成酶和谷氨酸合酶等酶的作用直接利用氨。
磷存在于核酸、磷脂、ATP等核苷酸、几种辅助因子、一些蛋白质和其他细胞成分中。几乎所有微生物都使用无机磷酸盐作为其磷源并直接掺入。
水生环境中的低磷酸盐水平常限制微生物生长。大肠杆菌等微生物能同时利用有机和无机磷酸盐。己糖6-磷酸盐等部分有机磷酸盐可被细胞直接吸收,而其他有机磷酸盐则需在周质中被碱性磷酸酶水解为无机磷酸盐后,才能穿过质膜进入细胞。
硫是合成氨基酸如半胱氨酸和蛋氨酸、一些碳水化合物、生物素和硫胺素等物质所必需的。大多数微生物使用硫酸盐作为硫的来源,并通过同化硫酸盐还原来减少硫;少数微生物需要还原形式的硫,例如半胱氨酸。
▸ 生长因子
一些微生物仅需碳源和无机盐即可从头合成所需有机分子,而其他微生物则依赖环境中的特定有机化合物。这些不能被生物体合成但又必需的有机化合物称为生长因子,可分为三类:
1)氨基酸(蛋白质的结构单元);
2)嘌呤和嘧啶(核酸的结构单元);
3)维生素(酶辅因子)。
一些微生物需要许多维生素;例如,粪肠球菌需要八种不同的维生素才能生长。此外,流感嗜血杆菌需要血红素(源自血红蛋白或细胞色素),而部分支原体则需要胆固醇等其他生长因子。
了解微生物的生长因子需求具有重要实用价值,特别是在利用具有特定需求的微生物或能大量产生维生素等物质的微生物方面。
为了维持其活动,微生物细胞必须通过细胞膜从环境中获取营养物质。细菌和古细菌拥有多种不同的物质运输机制。
▸ 被动扩散(Passive Diffusion)
被动或简单扩散允许简单分子和气体(如CO2、O2和H2O)穿过细胞膜。在这种情况下,必须存在浓度梯度,其中细胞外的物质浓度高于细胞内的物质浓度。随着更多的物质被转运到细胞中,浓度梯度降低,扩散速率减慢。
▸ 易化扩散(Facilitated Diffusion)
易化扩散是膜蛋白介导的被动扩散。物质通过膜上特殊蛋白质(载体、通道)的介导,沿电-化学梯度进行跨膜转运。主要有两种方式:载体介导的易化扩散和通道介导的易化扩散。这些易化扩散属于被动转运,其特点是转运过程本身不需消耗能量,仅在膜蛋白”帮助”下沿浓度梯度或电位梯度进行跨膜转运,是一个”被动”过程。
如果浓度梯度消失,分子进入细胞的通道就会停止。并且每种载体蛋白通常表现出特异性,仅转运特定类型的分子或密切相关的分子。
▸ 主动转运(Active Transport)
很多营养摄取需要细胞逆浓度梯度(细胞内浓度高于细胞外)运输物质。这时,细胞必须利用代谢能通过膜内载体蛋白进行物质运输,这种过程称为主动运输。所有主动运输都依赖载体蛋白完成。
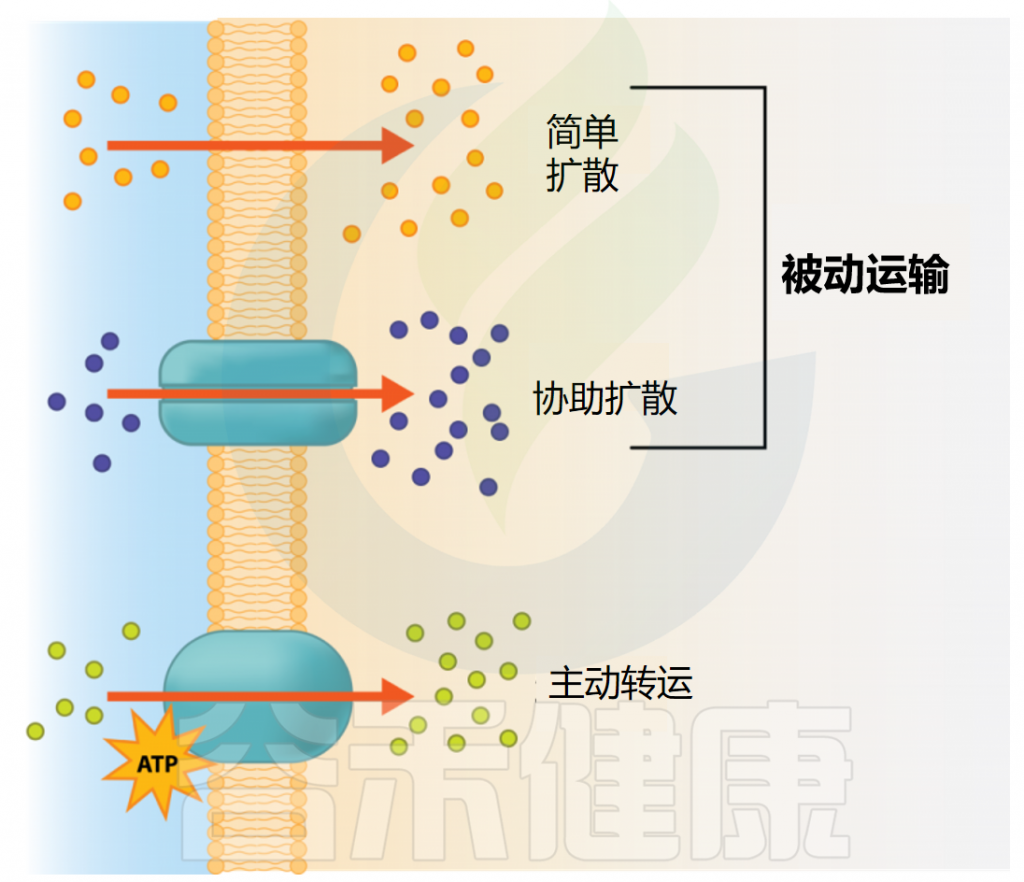
图源:open.oregonstate
主动转运有三个主要的例子:初级主动转运、次级主动转运和基团移位。
•初级主动转运(Primary active transport)
初级主动转运利用ATP等化学能驱动物质运输,如ABC系统就使用ATP结合盒转运蛋白。
每个ABC转运蛋白包含三个组分:
1)形成跨膜孔的跨膜蛋白(载体蛋白)
2)水解ATP提供能量的ATP结合区
3)结合并运送待转运物质到跨膜蛋白的底物结合蛋白
在革兰氏阴性细菌中,底物结合蛋白位于周质中;而在革兰氏阳性细菌中,则附着于细胞膜外部。
ABC转运器结构
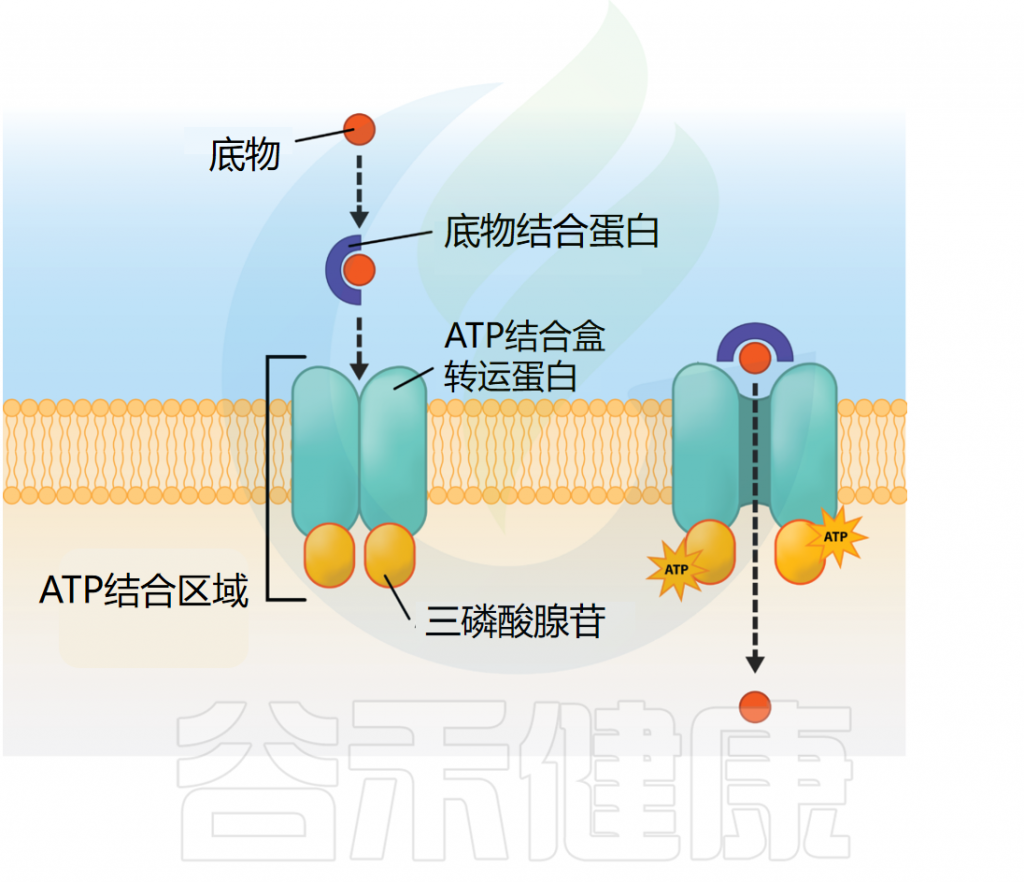
图源:open.oregonstate
•次级主动转运(Secondary active transport)
次级主动转运利用质子动力(PMF)提供能量。PMF是电子传输链节能过程中产生的离子梯度,正电荷质子在带负电荷的电池外部积累,形成质子梯度。
包括三种类型:单向转运、同向转运和反向转运,各使用不同蛋白质转运体:单向转运蛋白将单一物质穿过膜转入或转出。
协同转运蛋白同时将两种物质(通常是一个质子与另一分子配对)穿过细胞膜转运。
反向转运蛋白也能转运两种物质,但方向相反—一种物质进入细胞时,另一种物质被运出。
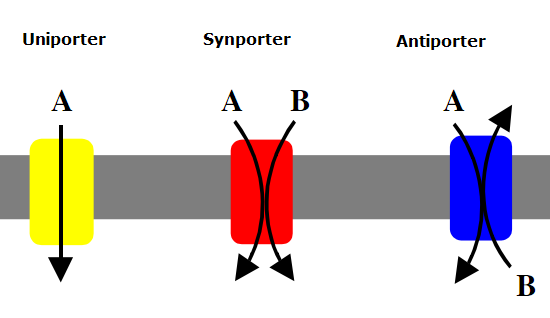
Uniport Synport Antiport.By Lupask,via Wikimedia Commons
•基团移位(Group Translocation)
基团移位是一种特殊的主动运输方式,不使用ATP而是利用富能有机化合物的能量。与简单转运和ABC转运蛋白不同,基团移位过程中被转运物质会被化学修饰。
最典型的例子是磷酸烯醇式丙酮酸:糖磷酸转移酶系统(PTS),它利用高能分子磷酸烯醇式丙酮酸(PEP)的能量将糖转入细胞。在此过程中,磷酸基团从PEP转移到进入的糖分子上。
通过PTS进行基团移位
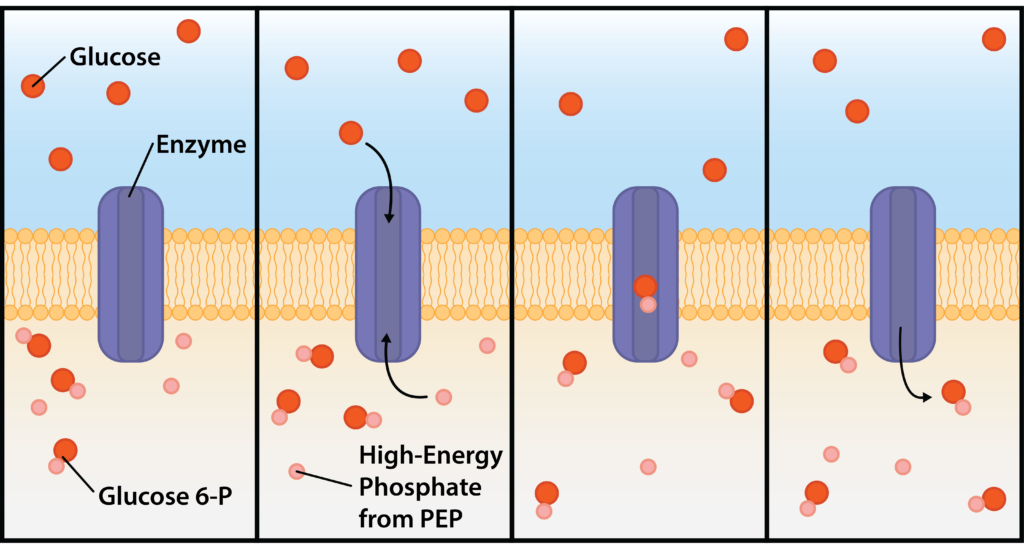
▸ 铁摄取
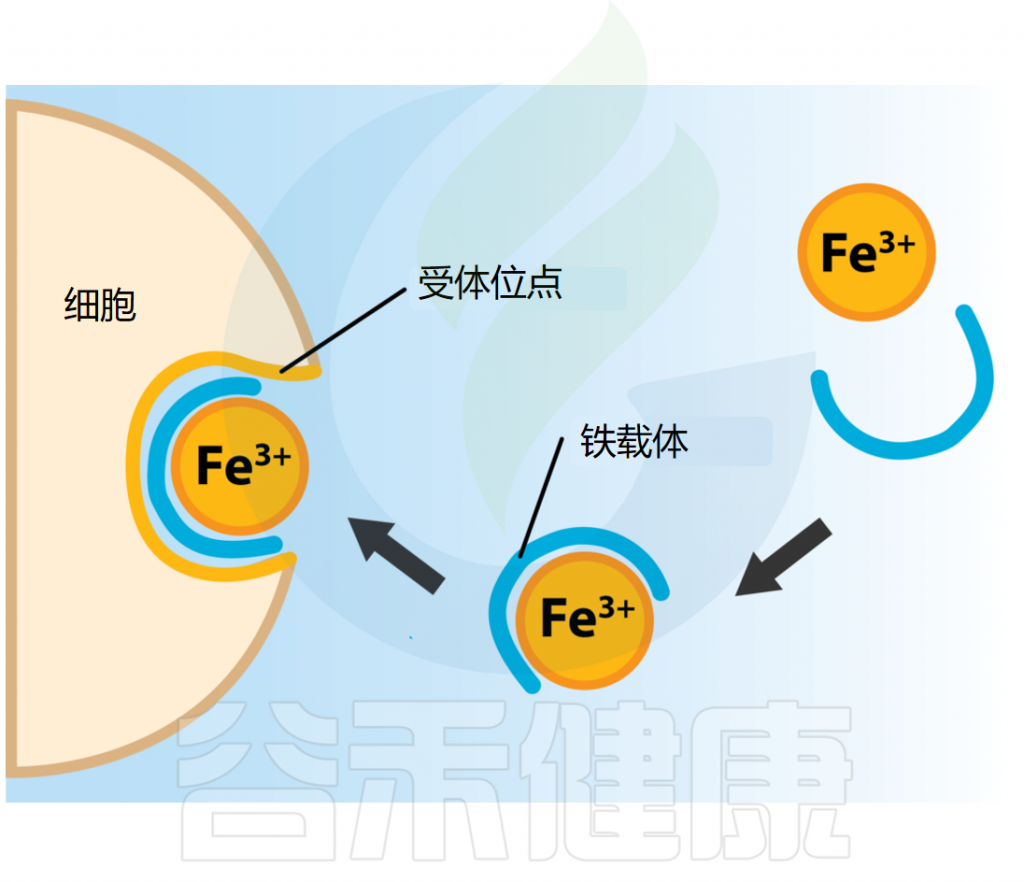
图源:open.oregonstate
铁是微生物细胞色素和酶功能必需的限制性微量营养素,但由于其不溶性,环境中几乎不存在游离态铁。
许多细菌进化出铁载体——一种能高亲和力螯合或结合三价铁的有机分子。这些铁载体被释放到环境中,结合可用的三价铁。随后,铁-铁载体复合物被细胞表面的特定受体识别结合,使铁得以被运输入细胞。
微生物的生长指的是细胞数量的增加,而非细胞体积的增加。细菌通过二分裂法生长和分裂,这是一种快速且相对简单的过程。
影响细菌生长的主要条件包括以下几种:
▸ 物理条件和分类
•温度
微生物根据其偏好的温度范围大致分为几个类别。
1) 嗜冷菌:“喜冷的”,能在0°C下生长。可分为两组,真嗜冷菌对超过20°C的温度敏感。最适生长温度为15°C或以下。存在于极冷环境(北极、海洋深处)。很少引起疾病或食物腐败。
2) 耐冷菌:最适生长温度为20至30°C。是大多数低温食物腐败的责任菌。
3) 嗜温菌:“喜欢中等温度的”。大多数细菌都属于此类。
包括大多数病原体和常见的腐败微生物。
最佳生长温度在25至40°C之间。
最适生长温度通常为37°C。
许多已适应在动物体内生存。
4) 嗜热菌:“喜热的”。最适生长温度在50至60°C之间,许多不能在45°C以下生长。
适应在阳光照射的土壤、堆肥和温泉中生存。
一些嗜热菌形成极其耐热的内生孢子。
5) 极端嗜热菌(超嗜热菌):最适生长温度在80°C或更高。属于古菌。大多生活在火山和海底热泉口。
•pH值
根据pH值,微生物可分为以下几类:
1) 嗜酸微生物:“喜爱酸性”
酸性会抑制大多数微生物生长,常用于食品保存(例如:腌制)。但这类细菌可以在极低pH值(0.1至5.4)下生长,例如乳酸菌产生乳酸,能耐受轻度酸性环境。
2) 中性微生物
在pH值5.4至8.5之间生长,大多数细菌偏好中性pH值(6.5-7.5),包括很多人类病原体。例如霉菌和酵母菌能够在更广泛的pH范围内生长,但偏好pH值在5和6之间。
3) 嗜碱微生物:“喜爱碱性”
可以在碱性或高pH值(7至12或更高)环境中生长。例如霍乱弧菌(Vibrio cholerae)和粪碱杆菌(Alkaligenes faecalis)的最适pH值为9,而土壤细菌根癌农杆菌(Agrobacterium)能在pH值12的环境中生长。
•渗透压
细胞由80-90%的水组成,高渗溶液具有高渗透压,会从细胞中抽取水分,导致细胞膜收缩(质壁分离)。常用于控制腐败和微生物生长,如果酱中的糖和肉类上的盐。
低渗溶液具有低渗透压,导致水分进入细胞。通常细胞壁能防止水分过度进入,但若细胞壁较弱,微生物可能溶解或破裂。
按渗透压分类,细菌可分为:
1) 嗜盐菌:需要中等到大量的盐浓度。大多数海洋中的细菌。
2)极端嗜盐菌或专性嗜盐菌:需要非常高的盐浓度(20至30%)。如死海、盐水池中的细菌。
3) 兼性嗜盐菌:生长不需要高盐浓度,但能耐受2%或更高的盐度。
▸ 化学需求和分类
•碳
碳构成细胞干重的50%,是所有有机化合物的结构骨架。化能异养生物从能量来源(脂类、蛋白质和碳水化合物)获取碳;化能自养生物和光能自养生物则从二氧化碳中获取碳。
•氮
构成细胞干重的14%,用于形成氨基酸、DNA和RNA。
氮的来源:
蛋白质: 大多数细菌;
铵: 存在于有机物质中;
氮气(N₂): 直接从大气中获取氮;
硝酸盐: 解离产生NO₃⁻的盐类。
•硫
硫用于形成蛋白质和某些维生素(硫胺素和生物素)。
硫的来源:
蛋白质: 大多数细菌;
硫化氢;
硫酸盐: 解离产生SO₄²⁻的盐类。
•磷
磷用于形成DNA、RNA、ATP和磷脂。
来源:主要是无机磷酸盐和缓冲剂。
•其他元素
钾、镁和钙通常作为酶辅助因子被需要,钙还是革兰氏阳性菌细胞壁合成所必需的。
微量元素:
许多微量元素用作酶的辅因子,通常存在于自来水中,包括:
•氧气
使用分子氧(O₂)的生物体比厌氧生物能从营养物质中获取更多能量。微生物可按氧气需求分类为:
1). 专性需氧菌:需要氧气才能生存缺点:氧气并非存在于所有环境中,且在水中溶解度较差。
例子:铜绿假单胞菌,常见的医院内感染病原体。
2). 兼性厌氧菌:倾向于使用氧气,但在缺氧情况下也能生长。拥有复杂的酶系统。
例子:大肠杆菌、葡萄球菌、酵母菌和许多肠道细菌。
3). 专性厌氧菌:不能使用氧气,并且会被有毒形式的氧气伤害。
例子:引起破伤风和肉毒中毒的梭状芽胞杆菌(Clostridium bacteria )。
4). 耐氧厌氧菌:不能利用氧气,但能耐受其存在。能分解有毒形式的氧。
例子:乳杆菌,无论是否存在氧气都能进行发酵。
5). 微需氧菌:需要氧气,但只能在低浓度下。对有毒形式的氧气敏感。
例子:弯曲菌(Campylobacter)。
氧气的有毒形式:
1.单线态氧:极具反应活性的氧形式,存在于吞噬细胞中。
2.超氧自由基(O₂⁻·):具有高毒性和反应活性的氧形式。所有在大气氧中生长的生物必须产生超氧化物歧化酶(SOD)清除它们。需氧菌、兼性厌氧菌和耐氧厌氧菌产生SOD,而厌氧菌或微需氧菌则不产生。
反应:O₂⁻+ O₂⁻+ 2H⁺+SOD—> H₂O₂+O₂
3.过氧化氢(H₂O₂):过氧化物离子具有毒性,是几种抗微生物药物的活性成分(如过氧化苯甲酰)。有两种不同的酶可以分解过氧化氢:
A.过氧化氢酶:将过氧化氢分解为水和氧气。常见,可由人类以及许多细菌产生。
2H₂O₂+过氧化氢酶—>2H₂O+O₂
B.过氧化物酶:将过氧化氢转化为水。
H₂O₂+2H⁺+过氧化物酶—>2H₂O
二元裂变是微生物细胞分裂的过程。通过增长曲线可了解微生物群体生长情况。微生物在液体培养基中通常采用分批培养或封闭系统,即在密闭容器中与单批培养基一起培养。
由于孵育过程中不添加新鲜培养基,营养物质浓度降低,废物浓度增加。二元裂变繁殖的微生物生长可绘制为活细胞数量与孵育时间的对数关系图,形成的曲线包含四个不同阶段。
▸ 滞后阶段
初始阶段,微生物被引入新鲜培养基后不会立即繁殖,细胞分裂不会立即开始。原因可能是老细胞缺乏足够ATP分子、核糖体、必需辅助因子,或培养基特性变化、微生物受损需恢复等。
滞后阶段的长度随微生物和培养基而变化
滞后阶段长度因微生物状况和培养基性质而异。接种物来自老旧或冷藏培养物时,此阶段较长;将培养物转移至不同化学性质的培养基也会延长滞后期。而年轻、生长旺盛的指数期培养物转移到相同成分的新鲜培养基中时,滞后期会很短或不存在。
▸ 指数或对数相位
这是微生物生长曲线的第二阶段。此阶段微生物以最高速率快速分裂,分裂速率取决于培养基特性、生物体的遗传组织和环境因素。指数阶段的增长率保持恒定,微生物数量以固定间隔分裂和加倍。由于各个体在略有不同时刻分裂,生长曲线呈平滑上升而非离散跳跃。
指数期微生物增长率通常保持恒定
指数期的均匀生长有助于研究者进行物理或化学活性研究。指数增长是平衡增长,所有细胞成分以相对恒定速率合成。若营养水平或环境条件变化,会导致生长不平衡。此阶段以产生初级代谢物著称,如氨基酸、核酸、维生素等,这些物质对生物体生长必需,对执行生理功能至关重要。
▸ 固定相
固定相阶段活细胞数量保持稳定
对数阶段后,由于营养物质耗尽和有毒产物积累,细菌生长几乎停止。新形成的细胞数量仅能补充死亡细胞,活细胞数量保持稳定,垂死与新生细胞几乎平衡。
微生物进入固定相有多种原因:
首要因素是营养限制;必需营养素严重耗尽导致细菌增长放缓。需氧微生物常受O₂可用性限制。氧气溶解度低且消耗快,可能仅培养物表面的O₂浓度足够支持生长。若不摇动或通气,表面以下细胞将无法生长。
有毒废物积累也可能导致增长停止,这限制了许多厌氧培养物的生长。例如,链球菌通过糖发酵产生大量乳酸和有机酸,使培养基变酸并抑制生长。链球菌培养物也可能因糖耗尽而进入固定相。此外,有证据表明当达到临界种群水平时,增长可能停止。
在此阶段,生物体产生次生代谢物,这些化合物与生物体生长无直接关系,如抗生素、萘、生物碱等。
▸ 死亡阶段
固定期后,因细胞死亡导致细菌种群减少,进入死亡阶段。该阶段由营养物质耗尽、毒性产物和自溶酶积累引起。活菌计数下降,而总数保持不变;对于自溶细菌,总数也会呈现下降趋势。
微生物在维持生态系统和人体健康方面扮演着关键角色。本文系统介绍了微生物生长与营养的基本原理及影响因素。微生物的生长速率取决于环境条件和营养物质的可利用性,通过宏量营养素(如碳、氧、氢、氮、硫、磷等)和微量营养素(如锰、锌、钴等)的摄取来满足生长需求。不同微生物对营养的需求各异,有些需要特定的生长因子(如氨基酸、嘌呤和嘧啶、维生素)才能生存。
微生物通过多种物质运输机制(包括被动扩散、易化扩散等)从环境中获取营养物质。其生长还受到多种物理和化学条件的影响,如温度(嗜冷菌、耐冷菌、嗜温菌、嗜热菌、极端嗜热菌)、pH值(嗜酸、中性、嗜碱微生物)、渗透压(嗜盐菌、极端嗜盐菌、兼性嗜盐菌)以及氧气需求(专性需氧菌、兼性厌氧菌等)。
微生物的生长曲线可划分为四个阶段:滞后阶段、指数或对数阶段、固定相和死亡阶段,每个阶段具有不同的特征和代谢活动。
了解这些生长规律和营养需求对于控制微生物在自然环境和人体中的生长至关重要,同时也为生物技术应用和疾病防控提供了科学基础。通过研究微生物的生长状态和活性,能够更好地理解微生物群落如何在复杂环境中维持生态平衡,并为人类健康和环境保护提供新的见解和解决方案。
主要参考文献:
Gonzalez JM, Aranda B. Microbial Growth under Limiting Conditions-Future Perspectives. Microorganisms. 2023 Jun 23;11(7):1641.
open.oregonstate.education/generalmicrobiology/chapter/microbial-nutrition/
Somak Banerjee and Sagar Aryal. 2023. Microbial Growth and Nutrition. Microbenotes.
gcwk.ac.in/econtent_portal/ec/admin/contents/154_P18ZC311_2020120205400254.pdf
tbiokem.lth.se/fileadmin/_migrated/content_uploads/3__Microbial_Nutrition_and_Growth_07.pdf

谷禾健康

抗生素的诞生彻底改写了人类对抗微生物感染的历史,从远古先民偶然利用霉变物质疗伤,到现代医学精准调控分子机制,抗生素通过抑制细菌细胞壁合成、干扰蛋白质生成等机制,成为治疗感染的核心武器。
它们按来源可分为天然(如青霉素)、半合成(如阿莫西林)和全合成(如喹诺酮类);按作用谱分为广谱(覆盖革兰氏阳性与阴性菌)和窄谱(针对特定菌种)。
然而,抗生素的过度使用(如农业促生长、医疗误用)导致耐药性急速发展。细菌通过β-内酰胺酶分解青霉素、修饰核糖体靶点逃避大环内酯类药物、外排泵排出喹诺酮类等策略,使药物失效。
耐药基因通过质粒、噬菌体在环境中扩散,甚至存在于未接触抗生素的偏远人群肠道中。通过评估个体肠道菌群中耐药基因的分布特点(如喹诺酮耐药基因的丰度),临床可优先选择对特定菌株敏感的抗生素,减少盲目使用广谱药物带来的生态风险。
本文谷禾通过查阅文献以及指南等,带大家一起深入理解抗生素的分类体系和作用机制,以及如何根据来源、作用谱和效果来认识不同的抗生素,及其针对不同微生物的使用策略探讨,在理解和允许的情况下结合感染或致病菌合理选择抗生素,并知晓其可能的副作用和耐药防控。
“抗生素”一词源自希腊语“anti”(反对)和“bios”(生命),指通过特异性作用于细菌靶点来抑制或杀灭微生物的有机分子。
天然抗生素最初由微生物(如真菌、细菌)产生,例如青霉素由青霉菌合成,链霉素来自链霉菌。现代抗生素的定义已扩展至包括全合成或半合成化合物,如磺胺类、喹诺酮类等。
抗生素的发现是医学史上的里程碑。

图源:tansuo
抗生素的现代角色抗生素在治疗细菌感染(如肺炎、尿路感染、皮肤感染)、预防术后感染及控制流行病中不可或缺。例如,β-内酰胺类抗生素(如青霉素)至今仍是许多感染的一线药物。
回顾抗生素的发展历程,这些微观世界的”分子武器”究竟是如何发挥作用的?它们为何能在细菌的生命过程中制造如此精准的”障碍”?
按来源分类
■ 天然抗生素
抗生素的天然来源涉及从生物成分中生产抗生素,其中包括微生物作为主要来源。
天然来源可能包括从细菌、真菌或放线菌中提取抗生素。
■ 半合成抗生素
这包括使用微生物生产从发酵中获得的物质,然后通过添加官能团进行化学修饰,从而产生变异。
例如:青霉素-氨苄西林、阿莫西林、甲氧西林。
■ 合成抗生素
属于这一类的抗生素是通过化学合成获得的。
示例:磺胺类药物、喹诺酮类药物。
按活性谱分类
■ 广谱抗生素
这些对革兰氏阳性菌和革兰氏阴性菌均具有活性。
例子包括:四环素类、氟喹诺酮类、“第三代”和“第四代”头孢菌素类。
■ 窄谱抗生素
它们的活性有限,主要仅对特定种类的微生物(革兰氏阳性或革兰氏阴性)有用。
例如,糖肽和杆菌肽仅对革兰氏阳性菌有效,而多粘菌素通常仅对革兰氏阴性菌有效。
氨基糖苷类和磺胺类药物仅对需氧微生物有效,而硝基咪唑通常仅对厌氧菌有效。
按抗菌活性分类
■ 抑菌剂
指那些阻止或抑制细菌生长的抗生素,也就是说细菌不会繁殖或产生,但它们不会杀死细菌。
例如:四环素、氯霉素
■ 杀菌剂
指根据所使用的抗生素,通过任何机制杀死细菌的抗生素。
示例:氨基糖苷类
基于行为模式的分类
细胞壁抑制剂:β-内酰胺、杆菌肽
细胞膜抑制剂:聚霉素、唑类抗真菌药物
蛋白质合成抑制剂
核酸合成抑制剂:喹诺酮类、利福平、呋喃妥因
叶酸合成抑制剂:甲氧苄啶、磺胺类

Uddin TM, et al., J Infect Public Health. 2021
按化学成分分类
■ β-内酰胺类抗生素
在引入后的20年里,青霉素是唯一一类β-内酰胺类抗生素。20世纪60年代中期,青霉素类抗生素与头孢菌素类抗生素并列,20世纪末,碳青霉烯类抗生素和单杆菌类抗生素并列。
这些抗生素具有相同的抗菌作用机制,并且都具有β-内酰胺环作为其结构的组成部分,但它们在其他特征上存在很大差异。
这些抗生素的作用机制是抑制细胞壁。
如:青霉素、头孢菌素、碳青霉烯类、单巴坦
■ 四环素类抗生素
这些是一类具有四个环核的抗生素,都是从土壤放线菌中获得的。
四环素最早是在20世纪40年代和50年代开发的,有几种至今仍在使用,如四环素本身、土霉素和金霉素。
多西环素和米诺环素是分别于1966年和1972年发现的更有效的半合成类似物,此后直到2005年引入替加环素(一种甘氨酰甘氨酸衍生物)才有了重大进展,替加环蛋白通常比其他四环素更有效,并对一些对该组早期成员产生耐药性的生物体保持活性。
四环素在实验室浓度下可能具有杀菌性,但体内安全浓度下仅为抑菌剂。它们通过结合敏感生物的30S核糖体来抑制蛋白质合成。
■ 氨基糖苷类
这些是一组天然和半合成的抗生素,具有与两个或多个氨基糖残基糖苷连接的多元氨基。
氨基糖苷类抗生素具有杀菌作用,在碱性条件下活性更强。它们通过干扰细菌蛋白质合成发挥作用,主要针对需氧革兰阴性杆菌,但抗菌谱各异。
氨基糖苷类的所有成员都具有杀菌作用,在碱性pH值下更具活性。
它们通过干扰细菌蛋白质合成来发挥作用。
它们主要对需氧革兰阴性杆菌有活性,但光谱不同。
如:链霉素、新霉素、卡那霉素、阿米卡星、庆大霉素

■ 多肽
低分子量阳离子多肽抗生素,均为强效杀菌剂,但因毒性较高,极少全身使用。
■ 糖肽
目前仅有的两种重要糖肽抗生素是万古霉素和替考拉宁。
它具有复杂的三环糖肽结构,其大分子量意味着它不能穿透大多数革兰氏阴性菌的外膜,因此它的使用实际上仅限于治疗需氧或厌氧革兰氏阳性菌的感染。
万古霉素在接近其最低抑菌浓度(MIC)的浓度下对大多数易感细菌具有杀菌作用,并且是细菌细胞壁肽聚糖合成的抑制剂,其位置与β-内酰胺类抗生素不同。
■ 大环内酯类
如: 红霉素、克拉霉素、阿奇霉素
大环内酯类抗生素是由12-16碳内酯环组成的大分子,通过糖苷键与氨基糖连接。
红霉素于1952年首次发现,至今仍是重要抗生素。它常用于治疗呼吸道、皮肤及软组织感染。大环内酯类药物通过抑制细菌蛋白质合成发挥抗菌作用,通常为抑菌剂,但在高浓度下可能具有杀菌活性。红霉素的抗菌活性具有pH依赖性,随pH值升高而增强,最佳pH值约为8.5,该特性在其他大环内酯类药物中也不同程度地存在。
■ 利福霉素
利福平口服吸收良好,蛋白结合率高(80-90%),能穿透细胞壁作用于细胞内病原体(如结核杆菌)。利福昔明因肠道吸收率低,主要用于局部感染。
如:利福平、利福巴林
■ 链霉素
普利司他霉素、维吉尼亚霉素
奎奴普汀/达福普汀是以30:70比例混合的两种链阳霉素类抗生素。它们源自链霉菌,经化学修饰后,通过抑制蛋白质合成发挥作用。
■ 抗真菌药物
灰黄霉素、多烯类
■ 合成药物
喹诺酮类、咪唑类、硝基呋喃衍生物
■ 氯霉素
氯霉素最初于1947年从委内瑞拉链霉菌中分离得到,后经化学合成。它是一种黄白色结晶固体,可溶于水。其抗菌活性可能源于其硝基苯结构。氯霉素的作用机制是抑制蛋白质合成。
■ 夫西地酸
夫西地酸是一种类似类固醇的杀菌抗生素,主要用于对抗葡萄球菌。
它对青霉素耐药的金黄色葡萄球菌菌株有活性,包括MRSA,可以与红霉素或克林霉素联合使用,用于严重的葡萄球菌感染。
■ 克林霉素
克林霉素是另一种对革兰氏阳性球菌具有显著抑菌活性的抗生素,尽管对革兰氏阴性球菌的活性较低,对肠杆菌的活性完全没有。
克林霉素在结构上与大环内酯类无关,但具有相似的作用机制,因此它们之间可能会发生交叉耐药性。
■ 林可霉素
它是克林霉素的前身,具有类似的抗菌和毒性特性,但效力较弱。
以上,我们已基本了解了抗生素的分类体系及其作用机制,这些分子武器本应是人类对抗微生物感染的坚固屏障。然而,当这些强大的武器被不当使用时,细菌迅速发展出层出不穷的耐药策略,将医学奇迹逐渐转变为临床危机。
全球健康的紧迫威胁
许多与人类疾病流行相关的细菌病原体在抗生素使用后已演变成多重耐药(MDR)形式。例如,多重耐药结核分枝杆菌(MDR M. tuberculosis)是发展中国家和工业化国家都发现的主要病原体,成为20世纪版本的古老病原体。
其他严重感染包括医院相关感染,如:
“超级细菌”指那些因多重突变而具有增强致病性和死亡率的微生物。

超级细菌的崛起:抗生素滥用的致命代价
近十年来,结核病的治疗遭遇了重大危机,广泛耐药结核菌(XDR-TB)和完全耐药结核菌(TDR-TB) 接连出现,这些“超级结核菌”对至少四种一线抗生素完全失效,且在全球快速传播。更令人担忧的是,像大肠杆菌、肺炎克雷伯菌等常见致病菌,也因抗生素滥用纷纷进化出耐药性。其中,青霉素类抗生素(如阿莫西林)和头孢类抗生素的过度使用,直接促使细菌发展出“β-内酰胺酶”这种分解抗生素的酶,让药物彻底失效。
医院里的危险分子也在增加:
某些超级细菌的爆发竟是抗生素滥用的直接后果。例如,艰难梭菌引发的严重腹泻,高毒力产毒素菌株已被确认,作为一种革兰氏阳性孢子形成菌,很容易通过医院人员、设备和气溶胶传播。它的重新突出被认为是医院广泛使用抗生素的结果,如广谱头孢菌素,新的青霉素和氟喹诺酮类药物,导致革兰氏阴性肠道菌群的显著消耗,从而提高了艰难梭菌。
在感染数量和后果方面,霍乱弧菌仍是头号杀手。亚洲和南美洲的疫情仍因缺乏有效监测系统难以控制。这些案例无不警示:抗生素的每一次误用,都在加速催生更危险的敌人。
耐药机制:细菌的“生存绝招”
细菌对抗生素的耐药性,本质上是它们演化出的保命技能。科学家发现,这些技能涉及基因突变、酶分解药物、改变自身结构等多种手段,甚至能通过“共享基因”在细菌间传播耐药能力。
以下是几种典型的耐药策略:
1
分解抗生素的“盾牌”——β-内酰胺酶
β-内酰胺酶是一种酶,能像剪刀一样切断青霉素、头孢类抗生素的结构(β-内酰胺环),让药物失效。β-内酰胺酶基因可能是分布最广泛的耐药基因。
这种酶的基因广泛存在于细菌的质粒(可移动的基因片段)上,甚至能在不同细菌间“快递传播”。
例如,新的超广谱β-内酰胺酶(CTX-M酶)最初在土壤中的Kluyvera菌里被发现,并于20世纪90年代出现在临床上,后来传播到致病的大肠杆菌中,导致头孢类抗生素失效。
2
改变“药物靶点”——大环内酯类耐药
红霉素等药物,广泛用于革兰氏阳性感染治疗。
通过结合细菌的核糖体(蛋白质工厂)来杀菌。而耐药菌通过修改核糖体的RNA或蛋白质结构,让药物无法锁定目标。
结果:即使更换新型大环内酯类药物,细菌仍能通过类似机制抵抗,形成“交叉耐药”。
3
升级“防御系统”——喹诺酮类耐药
当这类高效抗生素在1987年首次推出时,一些专家过于乐观地认为:”这种新型抗生素几乎不可能产生耐药性,因为细菌需要同时发生至少两种基因突变才能对它产生明显的抵抗力。”
然而,现实很快就打破了这些预测:
科学家感叹,永远别低估细菌的适应能力。
4
天生的“耐药基因”——内在耐药性
内在耐药性指细菌基因组中存在可能产生耐药表型的基因,即原始或准耐药性。不同属、种、菌株等表现出不同范围的抗生素反应表型。例如,某些肠道菌天生对磺胺类药物不敏感。
潜在威胁:这些基因平时潜伏,一旦被其他致病菌获取,可能引发新的耐药危机。
科学家们开发了一种方法来预测潜在的耐药基因。通过系统性地敲除细菌基因组中的每一个基因,观察哪些基因缺失会使细菌对抗生素变得更敏感。
逻辑很简单:如果缺少某个基因会使细菌更容易被抗生素杀死,那么这个基因过度表达可能会使细菌更具抗药性。
这种前瞻性研究已在多种细菌中进行:
5
环境中的“耐药基因库”
长期以来,人们知道可以通过在实验室中将环境细菌培养在含抗生素的培养基上来分离耐抗生素的细菌菌株。
产抗生素细菌的自我保护机制
对于那些能够产生抗生素的细菌(如放线菌),它们拥有耐药性并不令人惊讶。这是一种自我保护机制,细菌必须有能力抵抗自己生产的”武器”。在许多情况下,科学家们已经确定这种耐药机制是通过特定的酶来修饰抗生素,使其失去活性。
链霉菌:β-内酰胺酶的重要来源
链霉菌(Streptomycetes)能产生多种β-内酰胺酶(一种能破坏青霉素类抗生素的酶)。这很可能是如今临床上出现的一些β-内酰胺类抗生素耐药性的源头。同样,环境中的Kluyvera菌已被证实是CTX-M基因(一种重要的耐药基因)的起源。
多重耐药机制的进化蓝图
在产抗生素的细菌中,多重耐药机制十分常见,如四环素生产者Streptomyces rimosus就具有这种特性。根据生化和遗传相似性分析,这些天然存在的耐药机制预示了后来在致病细菌中发现的耐药机制。换句话说,致病菌的耐药策略很可能是从环境细菌那里”借鉴“而来的。
启示:耐药性为何难以控制?
对其他产抗生素细菌的进一步研究,如芽孢杆菌科、假单胞菌、蓝藻和多种放线菌科(这是一个已知能产生许多生物活性小分子的细菌群体),将有助于我们更全面地了解自然界中存在的耐药基因的本质和多样性。这些知识对于预测和应对未来可能出现的临床耐药问题至关重要。
当我们越来越理解这场微生物与药物之间的分子博弈,便不得不直面一个问题:如何在保持抗生素治疗效力的同时,为抗生素的未来开辟一条可持续发展的道路?下一章节我们来继续了解。
常见感染与抗生素选择
1. 尿路感染 (UTI)
无并发症尿路感染
– 首选药物:甲氧苄啶/磺胺甲噁唑(Bactrim Ds)、呋喃妥因(硝基呋喃类)
– 替代方案:头孢氨卡(Keflex,第一代头孢菌素)、阿莫西林/克拉维酸(Augmentin)
复杂性或上尿路感染(如肾盂肾炎)
– 首选药物:氟喹诺酮类(如环丙沙星Cipro、左氧氟沙星Levaquin)
– 替代方案:第三代头孢菌素(如头孢曲松)、氨基糖苷类(庆大霉素)
用药注意事项
– 磺胺类药物可能引起结晶尿,需充分水化
– 氟喹诺酮类因肌腱损伤和神经毒性风险,禁用于儿童及孕妇
– 氨基糖苷类雲监测肾功能及耳毒性
2. 肺 炎
社区获得性肺炎(CAP)
– 非重症患者:阿奇霉素(Zithromax,大环内酯类)或多西环素(Vibramycin,四环素类)
– 合并基础疾病或耐药风险:阿莫西林/克拉维酸(Augmentin)联合大环内酯类
医院获得性肺炎(HAP)
– 首选药物:第三代头孢菌素(如头孢他啶)或碳青烯类(如美罗培南)联合氟喹诺酮类
用药注意事项
– 大环内酯类(如阿奇霉素)可能延长QT间期,禁用于心律失常患者
– 碳青霉烯类需螫惕超广谱β-内酰胺酶(ESBL)耐药菌感染
3. 皮肤及软组织感染
轻度感染(如蜂窝织炎)
– 首选药物:头孢氨苄(Keflex)或双氯西林(耐酶青霉素)
疑似耐甲氧西林金黄色葡萄球菌(MRSA)感染
– 首选药物:复方磺胺甲噁唑(Bactrim DS)、克林霉素(Cleocin)
– 替代方案:万古霉素(糖肽类)或达托霉素(环脂肽类)
用药注意事项
– 需警惕艰难梭菌感染克林霉素可能引起伪膜性肠炎
– 万古霉素需根据肾功能调整剂量,监测血药浓度
4. 链球菌性咽炎 (Strep Throat)
首选药物
– 青霉素V钾(Penicillin VK)或阿莫西林(Amoxil)
青霉素过敏患者
– 阿奇霉素(Zithromax)或克拉霉素(Biaxin)
用药禁忌
– 四环素类(如多西环素)对链球菌活性低,不推荐作为一线治疗
– 氟喹诺酮类对链球菌无效
禁忌症与用药原则
病毒性感染的绝对禁忌
流感、普通感冒、急性病毒性鼻窦炎等无需抗生素治疗
滥用抗生素将增加耐药风险:60%的急性支气管炎为病毒性,仅需对症处理
特殊人群剂量调整
肝肾功能不全者:氨基糖苷类需减少剂量;磺胺类慎用于肾功能减退患者
孕妇与哺乳期妇女:禁用四环素类(影响胎儿骨骼发育)、氟喹诺酮类(致畸风险)
药物相互作用风险
大环内酯类(如红霉素)抑制CYP3A4酶,可能升高华法林、他汀类药物浓度
复方磺胺甲噁唑(Bactrim)增强磺脲类降糖药作用,需监测血糖
不合理用药的后果
耐药性加速:如氟喹诺酮类滥用导致铜绿假单胞菌外排泵基因突变频发
生态失衡:广谱抗生素(如第三代头孢菌素)破坏肠道菌群,增加艰难梭菌感染风险
经济负担:非必要使用增加医疗成本
抗生素的选择需基于病原体敏感性、感染严重程度及患者个体化因素。
临床实践中应严格区分细菌性与病毒性感染,优先使用窄谱抗生素。通过规范用药,可最大限度延缓耐药性蔓延。
通过肠道菌群检测识别抗生素耐药基因
宏基因组测序可获得细菌群体的全部基因组信息,通过序列比对和功能注释,可鉴定出各种已知和新型耐药基因,全面评估耐药基因的种类和数量。
例如,在人体肠道宏基因组中发现了大量β-内酰胺酶等耐药基因。
2021年一项研究显示,印度某社区爆发的碳青霉烯耐药肺炎克雷伯菌感染,其耐药基因与当地养殖场废水中分离的菌株高度同源,证实耐药基因通过水源传播至人体。
数据驱动的防控策略:从预警到干预
基于宏基因组分析的耐药风险评估,可制定精准防控措施:
对畜牧从业人员、医护人员等暴露于耐药基因环境的人群定期检测肠道菌群,早期发现耐药定植。
临床用药指导:建立区域耐药基因数据库,实时更新病原体耐药谱,帮助医生相对个性化地选择的窄谱抗生素(如用头孢他啶替代广谱碳青霉烯类药物)。
谷禾宏基因组肠道菌群检测报告中,就有抗生素耐药基因检测相关内容。
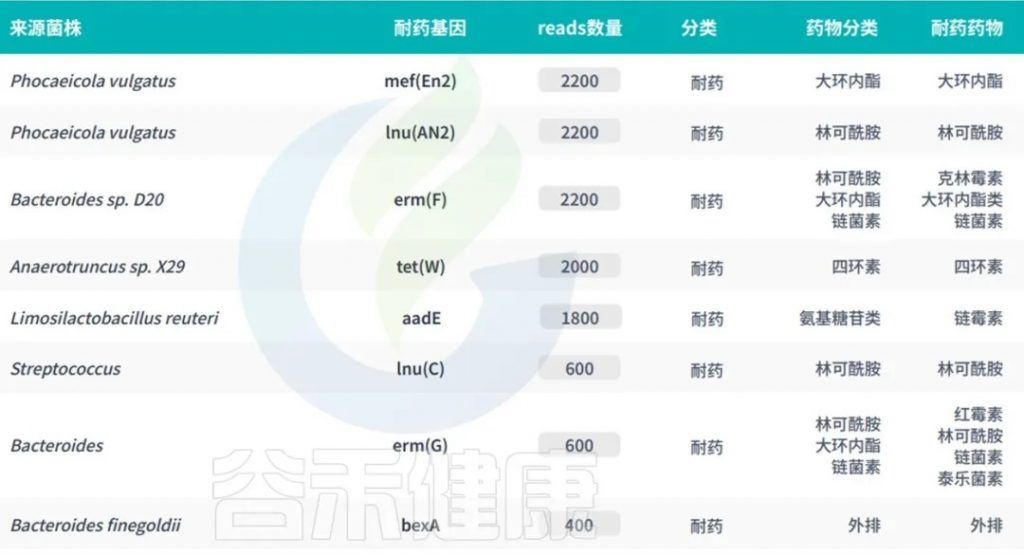
<来源:谷禾宏基因组肠道菌群健康检测数据库>
筛查应用:如对术前患者进行肠道耐药基因检测,若检出 vanA(万古霉素耐药基因),可优先选择替考拉宁或达托霉素替代万古霉素,降低术后感染风险。
用药方案优化:在复杂性尿路感染治疗中,若患者肠道中qnrS(氟喹诺酮耐药基因)丰度较高,应避免使用左氧氟沙星,转而选择磷霉素或呋喃妥因。
存在争议
支持证据
Cochrane综述表明,在抗生素后服用益生菌的儿童腹泻较少。
注:Cochrane综述是由国际非营利组织Cochrane协作网制作的系统性综述,被公认为循证医学领域的最高标准,旨在通过系统性方法评估医疗干预措施(如药物、手术、诊断测试)的有效性、安全性及适用性,为临床决策和政策制定提供可靠证据。
Cochrane对23项研究(3938名参与者)的综述调查了给予含有以下一种或多种的益生菌:
22/23项报告抗生素相关性腹泻发病率的试验结果显示,与活性、安慰剂或无治疗对照组相比,益生菌具有显著益处(益生菌组为8%,对照组为19%)。
报告副作用的16项试验(n=2455)均未记录任何可归因于益生菌的严重副作用,最常见的副作用是皮疹、恶心、胀气、肠胃气胀、腹胀、腹痛、呕吐、痰增、胸痛、便秘、味觉障碍和食欲低下。作者的结论是,益生菌对预防抗生素相关性腹泻具有保护作用。相对风险为0.46(95%CI 0.35至0.61),NNT为10。
作者认为,鼠李糖乳杆菌或布拉氏酵母菌的菌落形成单位为50亿~400亿个/天是最合适的选择。他们还评论,尽管在这些试验中,在其他健康儿童中没有观察到严重的不良事件,但在具有潜在风险因素(如中心静脉导管使用)的严重衰弱或免疫功能低下的儿童中观察到了严重的不良事件,并建议在进行进一步研究之前,应避免在有不良事件风险的儿科人群中使用益生菌。
反对观点
以色列魏茨曼科学研究所的科学家发现,当患者服用抗生素后,如果连续四周补充含11种菌株的复合益生菌,肠道菌群需要整整六个月才能恢复正常。尽管这些益生菌成功在肠道“安家落户”,但新引入的细菌和酵母菌株反而干扰了原有菌群的恢复。
对比发现:未服用益生菌的患者,停用抗生素后仅需三周,肠道菌群就自行恢复正常。
关键提示:
替代方案?
科学家正在探索其他恢复肠道的方法:
注:目前异体粪便移植目前是主流,但自体粪便移植可避免供受体不匹配的问题,部分研究仍处于早期阶段,需进一步验证长期安全性和有效性。
实用建议
如果你决定服用含有抗生素的益生菌,请在开始服用抗生素的同一天开始服用,但不要与抗生素完全同时服用。
选择高活性菌株,如鼠李糖乳杆菌GG、布拉氏酵母菌(每日5-400亿菌落单位)。
间隔服用时间,抗生素服用后2小时再服益生菌,避免活性被抑制。
抗生素的未来发展需在科学突破、临床实践与健康管理之间找到平衡点。面对耐药性危机,人类正通过多维度创新重塑感染治疗的格局。
1. 新型抗生素研发:从基因靶点到智能设计
宏基因组学驱动,通过分析环境样本(如深海沉积物、极端环境微生物)的宏基因组数据,挖掘新型抗生素合成基因簇。
合成生物学技术:利用CRISPR-Cas9基因编辑改造放线菌的代谢通路,定向生产改良抗生素。如改造链霉菌以高产泰斯巴汀(一种可靶向细胞壁脂质的广谱抗生素)。
噬菌体-抗生素联用,穿透生物膜杀灭耐药菌。例如,噬菌体搭载 β-内酰胺酶抑制剂,可恢复青霉素对产酶菌的活性。
缩短新药研发周期(从传统10-15年压缩至5-8年),降低开发成本,并为“无药可治”的超级细菌感染提供解决方案。
2. 精准医疗:从“一刀切”到个体化治疗
通过宏基因组测序分析患者肠道耐药基因谱(如 CTX-M、NDM-1),指导抗生素选择。例如,若检测到高丰度氟喹诺酮耐药基因(qnr),则优先选择磷霉素或呋喃妥因替代左氧氟沙星。
针对抗生素导致的菌群失衡,开发“益生菌-益生元-后生元”组合产品。例如,含 Akkermansia muciniphila 的微生态制剂可加速肠道屏障修复,减少艰难梭菌定植风险。
催生“感染精准医疗”细分市场,涵盖检测试剂、数据分析平台和微生态调节产品,预计2030年全球市场规模将突破520亿美元。
3. 耐药性防控:环境-医院-社区的联防联控
建立全球耐药基因数据库(如 ResistoMap),追踪耐药基因热点区域。例如,印度恒河流域因高浓度 blaNDM-1 基因被列为“耐药红色预警区”。
医院综合当地耐药数据、患者过敏史、肝肾功能、菌群检测报告等各类数据,个性化推荐窄谱抗生素。
设立“微生物学-药学-数据科学”交叉学科,开展耐药知识科普,提高公众对不当使用抗生素危害的认知,利用菌群检测手段辅助判别耐药情况,指导居民合理用药。
通过环境、医院、社区的协同联动,构建一个全方位的耐药性防控体系,减缓耐药菌的传播速度,保障公众健康。
抗生素的未来不仅是科学问题,更是社会命题。从基因编辑实验室到养殖场,从医院处方系统到家庭药箱,每个环节的革新都在重塑人类与微生物的关系。将技术创新、精准医疗和全球治理相结合,让抗生素这一“医学奇迹”持续守护生命,而非成为耐药危机的推手。
主要参考文献
Denyer S.P, Hodges N, Gorman S.P and Gilmore B.F. 2011. Hugo and Russell’s Pharmaceutical Microbiology. Eighth edition. A John Wiley & Sons, Ltd. Publication. Page no.169-193.
Antibiotics: Comprehensive Guide
Last updated: August 10, 2022 by Sagar Aryal
Davies J, Davies D. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev. 2010 Sep;74(3):417-33.
Uddin TM, Chakraborty AJ, Khusro A, Zidan BRM, Mitra S, et al., Antibiotic resistance in microbes: History, mechanisms, therapeutic strategies and future prospects. J Infect Public Health. 2021 Dec;14(12):1750-1766.
谷禾健康
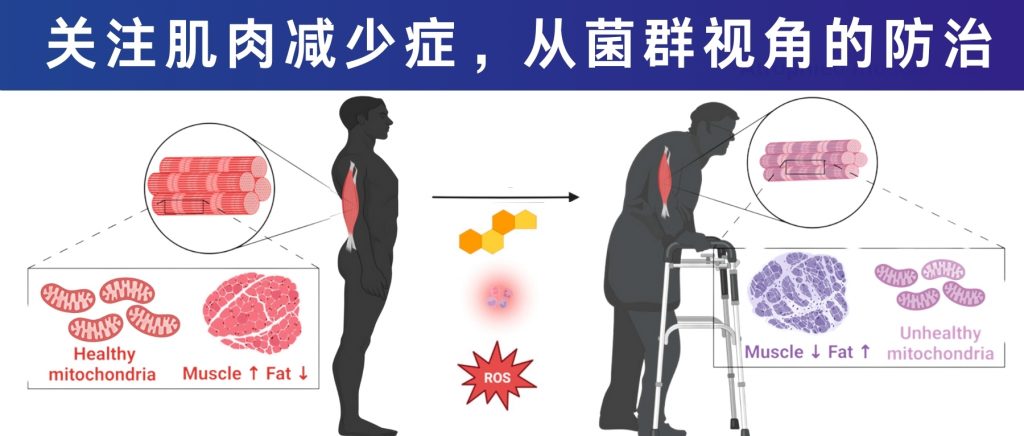
我国的老龄化问题日益突显,老年群体的生活质量是一个高度关注的焦点。许多老年人都会出现行走缓慢、起身或是爬楼梯困难、易失去平衡而摔跤、肌肉明显萎缩等症状。这很多时候都源于一种疾病——肌肉减少症。
肌肉减少症是一种与年龄相关的骨骼肌疾病,其特征是肌肉质量进行性丧失和肌肉力量下降。大约从30岁以后,肌肉质量会因为老化开始逐渐流失;到了40岁后,肌肉会开始以每10年约8%左右的速度流失,如果没有营养和运动干预,70岁时,人体肌肉质量下降约40%,到了80岁,肌肉大约会流失掉50%。据估计,60-70岁人群肌肉减少症的发病率为5-13%,80岁及以上人群更是高达11-50%。
肌肉作为生命的”隐形盔甲”,——它保护关节、储存营养、影响代谢等,许多人把肌肉流失当作“自然衰老”,直至跌倒骨折才认识其严重性。肌肉减少症对健康老龄化构成威胁,因为它不仅影响日常活动能力,还会损害代谢与免疫功能,增加肥胖、心血管等疾病病风险,甚至导致死亡率的升高。并且肌肉减少症不仅影响老年人,也威胁中、青年健康。肌肉减少会降低代谢率,易导致肥胖和三高,并与糖尿病、心梗和脑中风密切相关。
如今肌肉减少症已不仅限于老年人,部分年轻人也受其影响。长期”葛优躺”、”咸鱼瘫”、”办公坐”。无论是白天上班,还是晚上回家,很多人动都不愿多动一下,只想瘫着。长此以往后发现,不仅爬个楼梯就膝盖痛,甚至有时连拿个东西都觉得无力。肌肉减少症的主要原因是自然衰老,但缺乏运动、不良的饮食、慢性炎症、肥胖等一些疾病也都会促进肌肉减少症的发生,而这些正是当代年轻人普遍的生活特征。
如何提前预防、减缓或治疗改善肌肉减少症?除了常规的抗阻运动和充足且均衡的营养摄入外。不少研究发现肠道微生物群在肌肉减少症中发挥了不可忽视的作用,并提出了“肠道-肌肉轴”。紊乱的微生物组成可能导致肌肉老化和肌肉减少症的发展。肌肉减少症患者肠道菌群多样性和丰富度都较低,表现为放线菌门和梭杆菌门比例下降,普雷沃氏菌(Prevotella)与拟杆菌(Bacteroidetes)比率(P/B)降低,以及粪球菌属(Coprococcus)和毛螺菌科(Lachnospiraceae)的等产生短链脂肪酸的细菌丰度降低。
肠道微生物群似乎通过多种途径影响肌肉系统:包括调节饮食效果与营养传导,改变肠道通透性和炎症状态,影响肌肉合成代谢/分解代谢平衡,调节线粒体功能与激素分泌,以及影响骨骼肌纤维组成和神经肌肉传递等。
肠道微生物群分析有望帮助我们提前评估营养状况和预测肌肉减少症风险。谷禾肠道微生物检测报告不仅分析菌群状况,还提供营养评估和相关健康指标,有助于早期风险预警。同时有研究证实补充益生菌可以改善肌肉减少症,因此通过调节肠道微生物群可能成为治疗该症的新靶点。
肌肉减少症的定义
肌肉减少症(Sarcopenia),临床上也将其称为“肌少症”。医学定义是肌肉质量、力量和功能逐渐丧失而引起的综合症。
下面是肌肉减少症的一些最常见症状:
•肌无力
•活动时失去耐力或持久力
•行走缓慢
•难以进行日常活动
•爬楼梯困难
•易失去平衡
•肌肉明显萎缩
患病情况
▸ 主要发生在老年人,随年龄增长发病率增高
肌肉减少症通常发生在60岁及以上的老年人群,发病率随年龄的增长明显上升。60-70岁人群发病率为5-13%,80岁及以上人群高达11-50%。
依据2019年亚洲肌少症工作组制订的诊断标准,一项基于我国社区人群的流行病学调查研究结果显示,60岁以上人群的肌少症患病率为14.7%,其中男性肌少症患病率为17.3%,女性为12.5%。
▸ 不良生活习惯导致年轻人群患病增加
然而,近年来有统计发现,我国30岁以下的年轻人,竟有超过两成患“肌少症”。随着生活节奏加快,很多年轻人久坐少动、饮食不规律。
此外,再加上”白幼瘦”等审美观念影响,种种因素相加,增加了年轻人的肌肉流失,使这种原本的老年病,也变得越来越年轻化。肌少症已成一种全球性健康威胁。
肌少症的危害
肌肉和身体健康之间紧密相关,肌肉作为人体最重要的器官之一,意味着力量和生命力。有研究显示,肌肉减少对人体的影响和危害巨大:
肌肉减少10%左右,就可能出现机体免疫功能下降,感染风险增加;
肌肉减少20%左右,就可能出现肌肉无力,日常生活能力下降,跌倒风险增加,伤口愈合延迟;
肌肉减少30%左右,就可能出现日常生活能力下降甚至失能,还易发生压疮;
肌肉减少40%以上,则死亡风险明显增加。
肌肉减少症是一个严重的问题。一旦你失去了大量的肌肉和力量,你可能很难做一些事情,比如从椅子上站起来、打开罐子或搬运物品。也可能变得虚弱,跌倒、骨折、残疾和死亡的风险更高。我们总结肌肉减少症的危害主要有以下几方面:
▸ 身体功能下降与日常生活能力丧失
•跌倒风险增加;
•骨折风险增加;
•行动受限,步行变慢,难以上下楼梯;
•逐渐丧失独立生活能力,无法自理基本日常活动。
▸ 代谢与免疫功能受损
•影响糖代谢,胰岛素敏感性降低;
•炎症水平升高,导致”炎症老化”状态;
•免疫功能下降,增加感染风险;
•肌肉脂肪浸润增加,导致肌肉质量和功能持续下降。
▸ 共病风险增加
•肥胖和三高:中年后很容易出现肥胖等代谢问题。以前大家都把它归咎于吃太多,运动不足,但肌肉减少,基础代谢率降低也是关键所在;
•心血管疾病风险增加:肌少型肥胖者的高血压风险,会高出正常者两倍之多;
•呼吸功能下降:影响呼吸肌功能和肺活量;
•认知功能下降:肌肉减少症与认知障碍有关联;
•术后并发症增加:手术恢复更慢,术后风险更高;
•全因死亡率增加:研究显示肌肉减少症与死亡率有一定的关联。
导致肌肉减少症的原因
▸ 自然衰老
肌肉减少症最常见的原因是自然衰老。尽管并非所有长寿者都患此症,但大约从35岁以后,肌肉质量就会因老化开始逐渐流失。到了40岁后,肌肉会开始以每10年约8%左右的速度流失,如果没有营养和运动干预,70岁时,人体肌肉质量下降约40%,到了80岁,肌肉大约会流失掉50%。
随着年龄的增长,我们身体中会发生一些可能导致肌肉减少症的事情:
•负责从大脑向肌肉发送信号以开始运动的神经细胞减少;
•某些激素的浓度较低,包括生长激素、睾酮和胰岛素样生长因子;
•将蛋白质消化利用的能力下降;
•炎症增加,部分原因是疾病。
除了生理性衰老外,还有其他风险因素,例如缺乏运动、不良饮食和慢性疾病,这些因素都会导致肌肉和力量的丧失。
▸ 缺乏运动
研究表明,保持活跃可显著降低肌肉减少症风险。久坐卧床时间越长,肌肉质量和力量流失越多。即使在固定时间锻炼,长时间不活动仍会导致肌肉和力量损失。
▸ 不良饮食
劣质饮食可能导致肌肉减少症,蛋白质摄入不足被认为是主要原因,因为老年人体内蛋白质转化能力下降。部分研究显示蛋白质摄入不足与肌肉减少症相关。水果蔬菜摄入不足也可能是影响因素。富含超加工食品(高糖、高盐、多添加剂和不健康脂肪)的饮食同样与低肌肉质量有关。
一般来说,营养不良的老年人,无论是摄入量不足还是营养质量不佳,都面临更高的肌肉减少症风险,且病情恶化速度更快。
▸ 肥胖
肥胖似乎会加重肌肉减少症。高水平的体脂会增加炎症并改变胰岛素反应,这两者都可以加速肌肉流失。肥胖还会降低活动能力,形成肌肉流失和脂肪堆积的恶性循环。
▸ 慢性疾病
患有慢性阻塞性肺病(COPD)、肾病、糖尿病、癌症或艾滋病等慢性疾病会增加患肌肉减少症的风险。
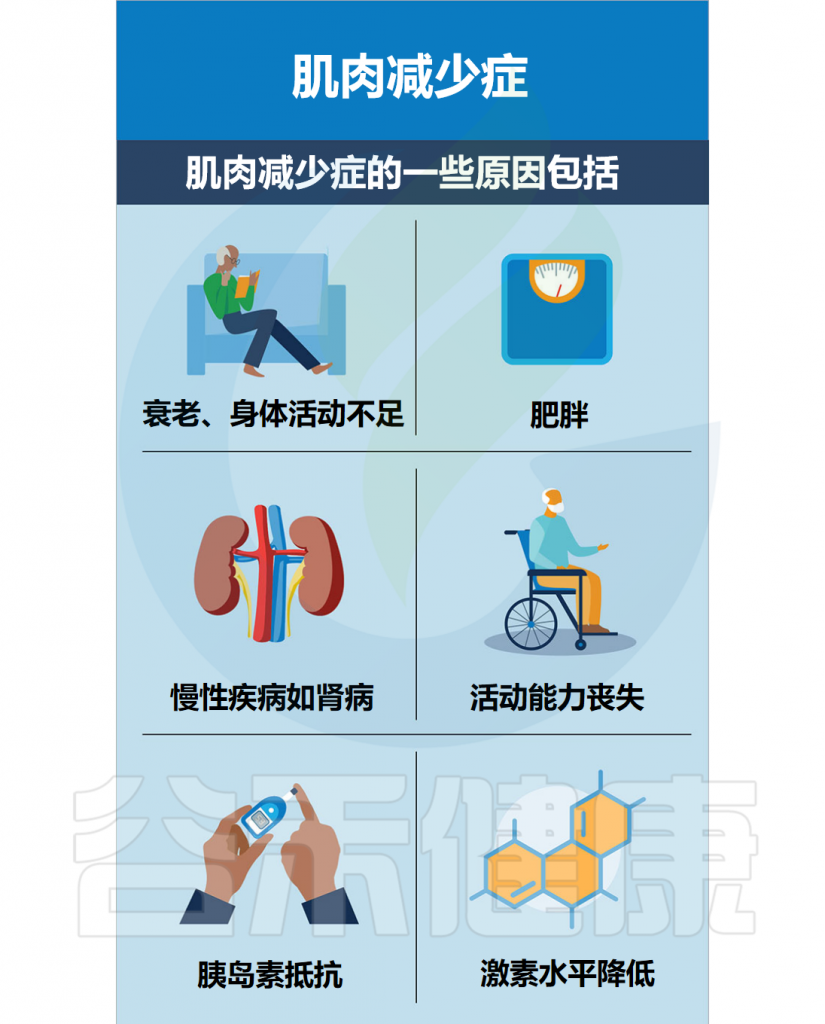
人们有时会将肌肉减少症与其他疾病混淆,以下是两种症状相似且易混淆的疾病。
▸ 肌肉减少症与恶病质的区别
病因不同:恶病质主要由癌症、感染、肾病等慢性消耗性疾病引起;肌肉减少症主要由自然老化、激素水平和蛋白质合成减少相关。
表现差异:恶病质除肌肉流失外,还伴有明显的非自愿性体重减轻和脂肪组织丢失,是全身性消耗,并且进展迅速,常在数周或数月内显著恶化;而肌肉减少症进展缓慢,通常在数年间发展,且主要影响肌肉组织。
危害:肌肉减少症主要是增加跌倒和骨折风险、降低日常生活自理能力;而恶病质严重影响治疗效果,是晚期疾病的不良预后标志,显著增加死亡风险。
▸ 肌肉减少症与身体虚弱的区别
病因不同:身体虚弱源于多系统生理储备减退,涉及肌肉、神经、内分泌和免疫系统等,且心理社会因素在发病过程中具有重要影响。
表现差异:表现为虚弱表型五项特征(非自愿体重减轻、自我报告的疲劳感、肌力下降、步行速度减慢、体力活动水平降低),虽与肌肉减少症高度重叠但不等同,范围更广,涵盖全身各系统功能衰退。
危害:身体虚弱会导致全方位健康状况恶化的风险更高,面对应激时缺乏稳态解决能力,对各种医疗干预的耐受性下降。
我们知道,肠道微生物群在免疫、内分泌、能量代谢稳态和整体健康等方面都起着至关重要的作用。肠道菌群失调和肌肉减少症又都常见于老年人,那么两者之间是否存在一定的联系呢?
近年来许多研究聚焦于肌肉减少症与肠道菌群组成之间关系,发现菌群失调与骨骼肌量和功能降低相关。并有研究发现,肠道微生物群及其代谢物可以作用于肌肉,还提出了“肠-肌肉轴”理论 。
参与“肠肌肉轴”发病机制的关键驱动因素
Nardone OM,et al.Front Immunol.2021
2024年一项涉及4307名43至87岁参与者的17项随机对照研究荟萃分析,基于16S rRNA基因测序揭示了肌肉减少症患者肠道菌群特征变化。

肌肉减少症患者的肠道菌群变化
▸ 肠道菌群的多样性和丰富度低于正常人
研究发现,肌肉减少症患者肠道菌群的多样性和丰富度低于健康对照组。在门水平上,放线菌门和梭杆菌门的比例下降(梭杆菌门减少20%至28%,放线菌门减少7%至10%),而其他门没有显著变化。
在属水平上,Akkermansia、Dialister、Dorea、Lachnospiraceae、Ruminococcaceae、Coprococcus、Faecalibacterium、Megamonas、Phascolarctobacterium、Prevotella、Lachnoclostridium、Paraprevotella、Ruminococcus_2、Streptococcus和Subdoligranulum的比例下降,瘤胃球菌科、粪杆菌属、普雷沃氏菌属、 Lachnoclostridium 显著减少(下调3%),而其他细菌略有减少(下降1%)。
Alistipes、Bacteroidota、Escherichia、Eubacterium_rectale_group、Flavonifractor、g-Bacteroides、 g-Lactobacillus、o-Clostridiales、g-Parabacteroides和Shigella的比例增加,其中拟杆菌属的增加幅度最大(上调12%),副拟杆菌属和志贺氏菌的比例增加2%,其他细菌增加1%。
▸ 菌群的变化可能是肌少症的早期迹象或标志物
具体来说,普雷沃氏菌(Prevotella)与拟杆菌(Bacteroidetes)比率(P/B)的降低,以及粪球菌属(Coprococcus)和毛螺菌科(Lachnospiraceae)的丰度降低,是重要的指标。
普雷沃氏菌(Prevotella)和拟杆菌(Bacteroidetes)参与膳食纤维的发酵和短链脂肪酸(SCFA)的产生,这对维持肌肉质量和功能至关重要。较低的P/B比率表明SCFA的产生能力降低,这可能会对肌肉健康产生负面影响。同样,粪球菌属(Coprococcus)和毛螺菌科(Lachnospiraceae)也是产生SCFA的细菌,它们的减少与SCFA水平降低有关,导致肌肉萎缩和无力。因此,监测这些特定的细菌标志物可以提供肌肉减少症的早期迹象,从而允许及时干预。
肠道菌群和肌肉力量
肠道菌群和肌肉功能参数也显示出了一定的关联,肌肉力量下降是肌肉减少症的一个重要特征。通过使用手持测力计测量优势侧握力评估肌肉力量。
注:该参数在生理上会随着年龄的增长而下降,但在满足肌肉减少标准的患者中,其降低通常更为明显。
▸ 肌肉力量下降的人群体内促炎微生物更丰富
在最近的一项观察性研究中发现,过度饮酒的人们的握力较低,变形菌门、萨特氏菌属、梭状芽胞杆菌属和霍尔德曼氏菌属的相对丰度较高,而粪杆菌属的相对丰度较低。
粪便微生物群的这些变化伴随短链脂肪酸水平降低,表明存在促炎微环境。这些结果表明微生物群可能对肌肉力量产生一些影响。
▸ 调整肠道微生物群有助于改善握力等肌肉功能
一项对60名疗养院老人的随机对照试验显示,菊粉和低聚果糖益生元干预13周后,握力显著提升。虽未直接评估肠道菌群,但菊粉和低聚果糖对肠道微生物群组成有已知有益的影响。补充菊粉可以提高人体中双歧杆菌、嗜胆菌的相对丰度,而低聚果糖具有选择性增加双歧杆菌丰度的能力。益生元还刺激微生物合成SCFAs,从而改善钙吸收和骨矿化。
正如最近的一项系统评价所表明的那样,骨骼健康和钙稳态与肌肉减少症发作密切相关。因此,握力改善可能由肠道菌群变化调节炎症和代谢平衡所致。
综上,人类肠道菌群与肌肉力量可能相关,双歧杆菌和粪杆菌被认为是主要参与者。这两个属之间存在相互作用,粪杆菌合成丁酸盐的能力是由双歧杆菌的丰度介导的,在可能的肌肉力量-微生物群关联中也应考虑。
肠道菌群和步行速度
步行速度是衡量老年肌肉减少症的一项参数,能预测行动不便和死亡率,且与多种慢性疾病进展相关。慢步态(≤0.8m/s)与事故残疾显著相关,尽管临界值因环境和人群而异。
注:步行速度通常在4米直线路径上以患者平常行走速度测量,推荐使用电子设备以提高精度和可重复性。
▸ 肠道菌群可以影响运动功能
肠道菌群与步行速度关系的研究较少。一项动物实验显示,无菌状态或抗生素使用导致运动过度活跃,比肠道菌群正常的移动更快。向无菌果蝇添加特定乳酸菌后恢复正常运动行为,表明肠道菌群可通过调节糖代谢和章鱼胺能系统(相当于人类去甲肾上腺素系统)影响运动功能。
一项对32名65岁以上久坐女性的非随机比较试验显示,躯干肌肉训练与有氧运动后肠道菌群组成发生特异性变化,拟杆菌属丰度与6分钟步行测试中步态速度增加呈正相关。另一项绝经前健康女性研究表明,拟杆菌属丰度与心肺健康相关,但未直接评估步态速度。
最后,在一项随机对照试验中,在36名肝硬化患者中测试了多菌株益生菌与安慰剂的效果发现,益生菌给药诱导的肠道微生物群有益修饰与步态速度的改善有关。
这些研究表明肠道菌群组成可能与步行速度相关,但均未针对肌肉减少症老年人开展,研究未能确立菌群与步态速度变化间的因果关系。
此外,步行速度作为功能参数不仅依赖肌肉力量和功能,还取决于中枢神经系统功能。衰老过程中,微生物群通过多种机制影响大脑生理学,这些机制可能影响老年人步行速度和功能依赖性。肠道细菌产生神经递质(γ-氨基丁酸、去甲肾上腺素和多巴胺)的能力及调节宿主产生血清素的能力,似乎与菌群组成和步行速度的关联有关。因此,这些关联可能不仅取决于肠道-肌肉轴,还与微生物对大脑的影响有关。
改善微生物群对肌肉的影响
▸ 肠道菌群失调可能导致炎症引发神经肌肉功能受损
评估肠道菌群改善对肌肉质量和功能影响的研究较少,多集中于动物模型。一项研究证明,向小鼠癌症模型给予含罗伊氏乳杆菌(一种FoxN1调节剂)的益生菌可抑制恶病质发展并维持肌肉质量。
部分益生菌具有明显抗炎作用,可能通过促进合成代谢有益肌肉健康。例如,含普拉梭菌(主要SCFA生产者之一)的益生菌制剂能改善小鼠肝脏合成代谢并减少全身炎症。同样,螺旋藻(一种食品添加剂或补充剂)治疗可改善老年小鼠炎症和氧化应激生物标志物。
此外,动物研究还评估了抗生素引起的肠道菌群失调的全身影响。肠道菌群失调与骨骼强度和机械性能受损相关,可能因成骨减少导致合成代谢刺激不足。抗生素诱导的菌群失调会干扰小鼠神经肌肉传递,可能促进肌肉蛋白质分解代谢。
越来越多证据显示肠道菌群可能参与肌肉减少症的病理生理学。但肠道微生物群及其代谢物究竟是如何影响肌肉减少症的?
我们通过查阅文献并汇总,认为肠道菌群可能通过以下方式影响肌肉减少症:
微生物群作为营养信号的传感器
▸ 肠道微生物群会影响饮食的效果、营养的传导
肠道微生物群会影响宿主代谢平衡。例如无菌小鼠即使在喂食高脂肪饮食时也表现出持续的瘦表型。即使在均衡饮食的情况下,将营养不良人类的粪便微生物群移植到无菌小鼠也会导致生长缺陷。
将猪肠道菌群移植到瘦无菌小鼠中,导致肌肉纤维结构发生显著变化,类似于猪的典型变化。因此,微生物群可以作为向宿主发送营养信号的基本转导,而饮食则塑造微生物群组成和功能。
肠道菌群与营养和肌肉功能
doi: 10.3390/nu9121303.
饮食会影响微生物群的组成;反过来,微生物群将一些营养物质(包括纤维和蛋白质)代谢成介质,例如短链脂肪酸,进入体循环。
这些介质通过调节炎症和促进胰岛素敏感性产生的多种信号通路,对肌细胞,即对它们的线粒体产生已知影响。图下半部分显示体育锻炼能直接调节肠道菌群组成,并标明了相关参与因子。
▸ 肠道微生物群对氨基酸吸收和肌肉合成很重要
蛋白质摄入对骨骼肌具有公认的促合成代谢作用,有利于肌肉质量的沉积与体育锻炼协同作用。这种影响可能是由肠道微生物群介导的。例如在肉鸡中,在类似的饮食方案下,肌肉质量的增长速度深受特定肠道微生物群代谢型的影响,这表明肠道微生物群在氨基酸吸收和促进肌肉合成代谢方面发挥着重要作用。
一项对38名超重人群的随机对照试验显示,与麦芽糖糊精对照组相比,接受酪蛋白和大豆蛋白3周补充后,细菌代谢明显转向氨基酸降解和发酵。这表明肠道菌群可通过增加氨基酸生物利用度并刺激骨骼肌中胰岛素分泌和反应来促进蛋白质合成代谢。动物研究表明,微生物产生的支链氨基酸(BCAA)增加与胰岛素敏感性和蛋白质合成改善相关。
▸ 但过度高蛋白饮食并不一定对肌肉有利
需要注意的是,高蛋白饮食对微生物群的影响可能并不总是对肌肉有利。高蛋白饮食小鼠的肠道菌群表现为厚壁菌门/拟杆菌门比率降低、病原体(如肠杆菌科)增加,同时产生代谢调节剂(如SCFA)的菌群减少,导致体重减轻并可能对肌肉代谢产生负面影响,包括炎症调节减少和胰岛素抵抗增加。一项人体随机对照试验也发现,耐力运动员长期服用牛肉蛋白补充剂导致健康相关菌群(包括双歧杆菌、Roseburia和Blautia)减少。
因此,蛋白质摄入与微生物群组成的关系及其对宿主代谢的影响复杂且尚未完全明确。蛋白质摄入对肠道菌群组成及肌肉质量沉积的影响可能取决于蛋白质质量和微生物群代谢型,微生物群介导的蛋白质摄入促合成代谢反应可能因个体差异而不同。
肠道菌群影响肌肉质量
肠道微生物群参与氨基酸的代谢、吸收和活力。膳食蛋白质被宿主和细菌的蛋白酶/肽酶水解成肽和氨基酸,这些产物能够调节全身能量状态,支持肠道菌群生长和生存。
▸ 肠道菌群可以影响肌肉合成对蛋白质的需求
有证据表明,肠道微生物群组成的改变可以决定肌肉合成对蛋白质的更高需求,那些老年肌细胞的特征现象(即所谓的“合成代谢抵抗”)。合成代谢抵抗是导致肌肉蛋白质合成减少和随之而来的肌肉生理改变的原因,促进肌肉减少症发生。
合成代谢抵抗现象与参与肌肉合成的蛋白质基因表达改变有关,受损的氨基酸转运到肌肉中,使流向骨骼肌的营养血流失调,减弱蛋白质消化和吸收,肌肉蛋白分解增加和骨骼肌干细胞的损失。
▸ 短链脂肪酸等菌群代谢物会调节肌肉合成代谢
证据支持肠道-肌肉轴的存在,其中肠道微生物群组成可以通过产生介质来影响肌肉质量合成代谢和宿主的功能。一般来说,随年龄增长,肠道菌群中短链脂肪酸(SCFA)生产者减少。SCFA能够调节蛋白质调节途径,增加ATP的产生,通过调节全身合成代谢/分解代谢平衡来影响骨骼肌蛋白质沉积刺激骨骼肌葡萄糖摄取,并影响胰岛素敏感性和炎症等功能。
肠道菌群影响氨基酸进入门静脉循环供全身使用。菌群失调会降低膳食蛋白质和促进肌肉蛋白合成的特定氨基酸(如色氨酸)的生物利用度。
肠道菌群代谢物可能参与骨骼肌功能

doi: 10.3390/nu9121303.
▸ 肠道微生物群本身也可以合成一些氨基酸
肠道微生物群本身可以合成氨基酸,这是肌肉蛋白质合成代谢的关键底物,衰老会损害微生物群合成赖氨酸、异亮氨酸、亮氨酸和缬氨酸的能力,从而增加其蛋白水解功能。肠道微生物群还参与维生素的合成,包括叶酸、维生素B12和核黄素,介导这些营养素对骨骼肌细胞的促合成代谢作用。
▸ 肠道菌群变化会改变骨骼肌纤维组成并影响肌肉力量
此外,肠道菌群的年龄相关变化能诱导肌内脂肪浸润。动物模型显示,特定菌群会改变骨骼肌纤维组成。值得注意的是,随年龄增长,肌肉力量下降幅度大于肌肉质量,主要由于肌肉纤维分布改变(I型纤维比例增高、II型纤维萎缩)以及肌纤维间脂肪积累。
肠道与肌肉的交流可能是双向的。研究表明,运动训练与有益的菌群谱(生物多样性和代谢活性物质产生)相关。
炎症性衰老导致肌肉减少
老年人肠道菌群特征是由促炎与抗炎途径失衡引起的慢性低度炎症(”炎症性衰老”)所促成。炎症性衰老部分源于免疫衰老现象,表现为免疫功能失调、CD-28共刺激分子丧失和端粒缩短。
▸ 肠道菌群及其代谢物影响炎症性衰老
肠道菌群及其代谢物在炎症性衰老中扮演重要角色。衰老的肠道菌群抵抗有害微生物、清除代谢物的能力下降,循环内毒素水平升高。因此,调整肠道菌群的饮食改变可能影响促炎/抗炎介质释放,维持表型变化。
免疫系统受菌群与肠道细胞间互动调节。健康肠道菌群调控免疫系统发育和稳态,维持炎症平衡并抑制慢性炎症。短链脂肪酸通过减少促炎细胞因子和趋化因子分泌及巨噬细胞浸润发挥抗炎作用。特别是,丁酸盐诱导IL-10、视黄酸和TGF-β分泌,并刺激抗炎调节性T细胞产生。
▸ 炎症反应会通过影响合成代谢导致肌肉减少
肠道菌群通过维持肠屏障功能调控脂多糖和促炎细菌内毒素的吸收,参与炎症调节。动物研究表明,年龄相关的肠道菌群变化与肠道通透性增加相关,这主要由上皮紧密连接损伤引起。屏障功能的这种改变促进宿主全身炎症。肠道菌群失调会刺激肠上皮细胞分泌粘蛋白,增加病原体进入肠粘膜的几率。丁酸盐则通过增强紧密连接组装减轻炎症,阻止内毒素易位。
这些机制导致炎症反应改变,可能通过在骨骼肌促进分解代谢并抑制合成代谢来加速肌肉流失。
菌群失调导致线粒体功能障碍
肠道菌群能影响肠道屏障稳态,实验表明,在肠道菌群存在时,肠道上皮能产生生理水平的氧化应激。氧化应激与肠道菌群组成、功能及肠壁通透性相互作用,从而调节外源分子进入体循环的可能性。
▸ 不健康的菌群会使线粒体损伤进而改变肌肉稳态
不健康的肠道菌群抗氧化能力下降,可能导致年龄相关的肌细胞线粒体质量降低。线粒体损伤释放的分子激活线粒体DNA诱导的炎症途径,促使炎症细胞释放细胞因子、趋化因子、活性氧和一氧化氮,造成慢性炎症持续并形成恶性循环,从而改变肌肉稳态。
线粒体功能障碍影响肌肉减少症
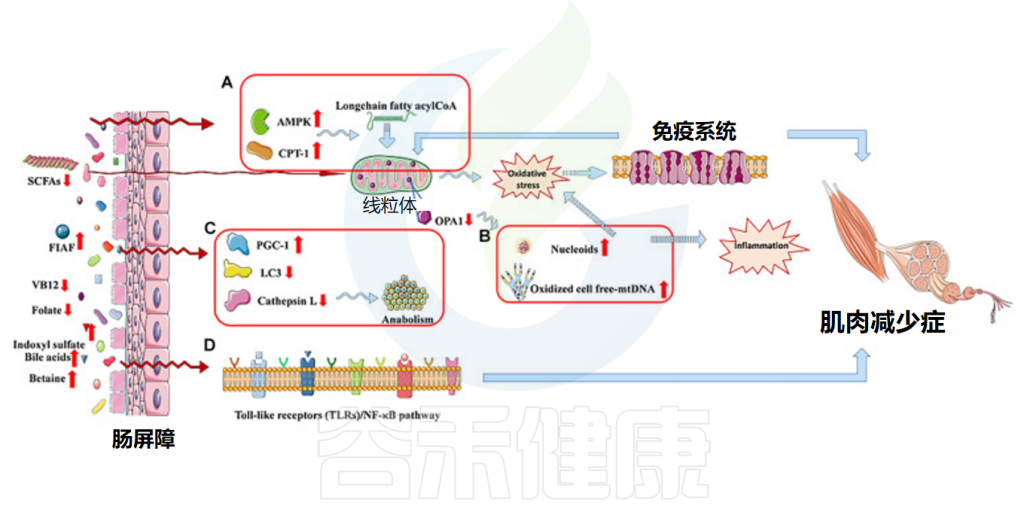
doi: 10.3389/fbioe.2020.590869.
研究表明,肌细胞质量控制过程的变化可能参与肌肉减少症的发生。老年肌细胞中线粒体功能障碍和自噬信号分子表达降低是衰老现象的组成部分。这些变化可能导致受损线粒体清除效率低下和功能失调细胞器积累,引起肌肉萎缩。肠道菌群产生的短链脂肪酸等代谢物可能积极参与骨骼肌中的线粒体生物发生。
肠道微生物群和骨骼肌的分子信号通路
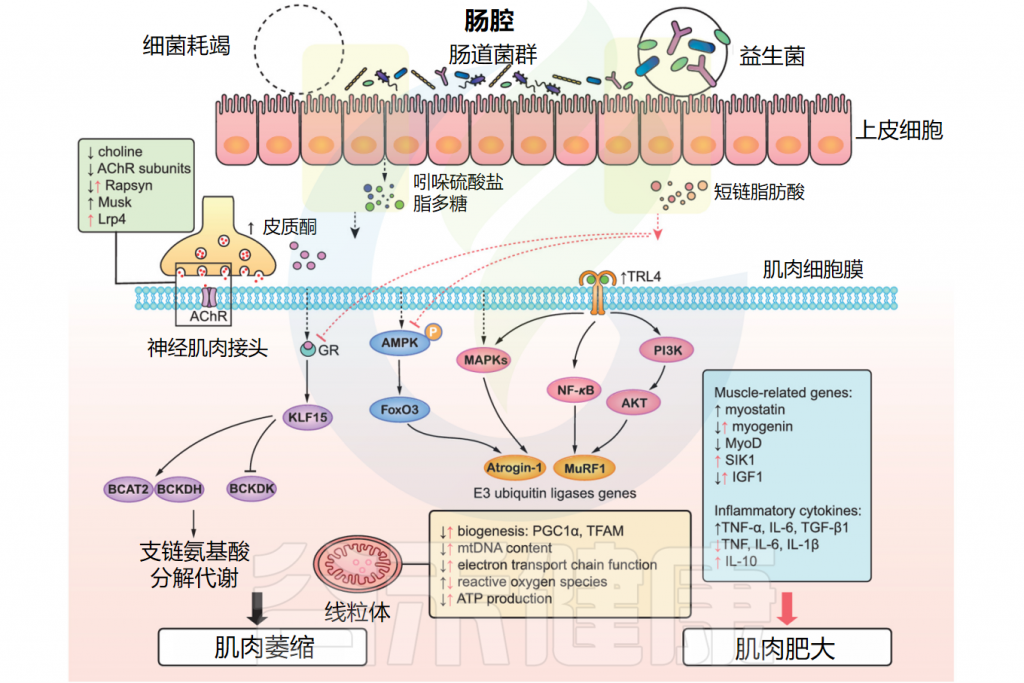
doi: 10.1002/jcsm.12784.
有害细菌代谢物(硫酸吲哚酯和脂多糖)和肠道菌群缺失诱导肌肉萎缩。这些物质通过激活PI3K/AKT、NF-κB和MAPKs信号通路,上调E3泛素连接酶基因(Atrogin-1/MAFbx和MuRF1)及炎症因子,导致肌肉萎缩和炎症。
细菌耗竭条件下,AMPK-FoxO3-Atrogin-1/MuRF1级联反应和BCAAs分解代谢被激活,同时IGF1、肌生成素和MyoD表达降低,肌肉生长抑制素升高,神经肌肉接头和线粒体功能受损。
胰岛素抵抗加速肌肉损失
健康肠道菌群可降低胰岛素抵抗。肠道菌群失调和肠屏障改变导致短链脂肪酸和次级胆汁酸减少,同时脂多糖和支链氨基酸的吸收与循环水平增加,这些变化共同导致胰岛素抵抗。
短链脂肪酸增强胰岛素敏感性,在调节葡萄糖摄取和代谢中发挥关键作用。它们提高能量消耗,改善葡萄糖耐量。次级胆汁酸则通过激活胰高血糖素样肽-1分泌防止胰岛素抵抗,影响葡萄糖稳态。脂多糖结合并激活toll样受体4信号通路,诱导胰岛素抵抗、亚临床炎症和肥胖。循环支链氨基酸水平升高与2型糖尿病发病率增加5倍相关。
▸ 胰岛素抵抗会加速肌肉质量和力量的流失
胰岛素抵抗和糖尿病与肌肉质量和力量的加速损失有关。胰岛素在刺激肌细胞中线粒体蛋白合成和抑制蛋白水解中起关键作用。由于信号系统的改变,响应胰岛素的蛋白质合成正常增加受损,促进了肌细胞中与年龄相关的合成代谢抵抗。
众所周知,肌肉减少症(即肌肉生长和再生的损害)的特征是生长激素(GH)和胰岛素样生长因子1(IGF-1)浓度下降。有证据表明,短链脂肪酸促进全身性 IGF-1 释放,这表明由于细菌失调而改变这些微生物群介质可能会影响肌肉健康。
肠道微生物群与肌肉健康密切相关,构成”肠道-肌肉轴“。这一关系使肠道菌群可能成为肌肉减少症早期诊断、预防和辅助治疗的重要指标和潜在干预靶点。
谷禾的肠道微生物群检测可为肌肉减少症的早期诊断和预防提供哪些关键信息?让我们接着往下看。
作为肌肉减少症的生物标志物
许多研究表明,肌肉减少症患者肠道菌群的多样性和丰富度低于健康对照组,这种变化先于临床症状出现。
▸ 关键菌属丰度变化
•放线菌门和梭杆菌门的比例下降;
•普雷沃氏菌(Prevotella)和粪球菌(Coprococcus)等有益菌群减少;
•普雷沃氏菌(Prevotella)与拟杆菌(Bacteroidetes)比率(P/B)的降低;
•变形菌门尤其大肠杆菌等机会病原菌增加;
•产丁酸盐菌群(如Lachnospiraceae和Ruminococcaceae科细菌)减少。
▸ 菌群代谢物作为生物标志物
短链脂肪酸:尤其是丁酸盐,在肌肉健康中起关键作用。检测报告中显示的短链脂肪酸水平可作为评估风险的生物标志物。
次级胆汁酸:次级胆汁酸水平改变与胰岛素敏感性和肌肉质量相关。
色氨酸代谢物:肠道菌群参与色氨酸代谢,其代谢产物(如血清素等)与系统性炎症和肌肉功能相关。
评估营养状况
营养不足是肌肉减少症的主要原因之一,谷禾肠道微生物检测报告中有专门的营养评估内容,以及一些与营养相关的健康指标,这些有助于提前预测肌肉减少症风险。
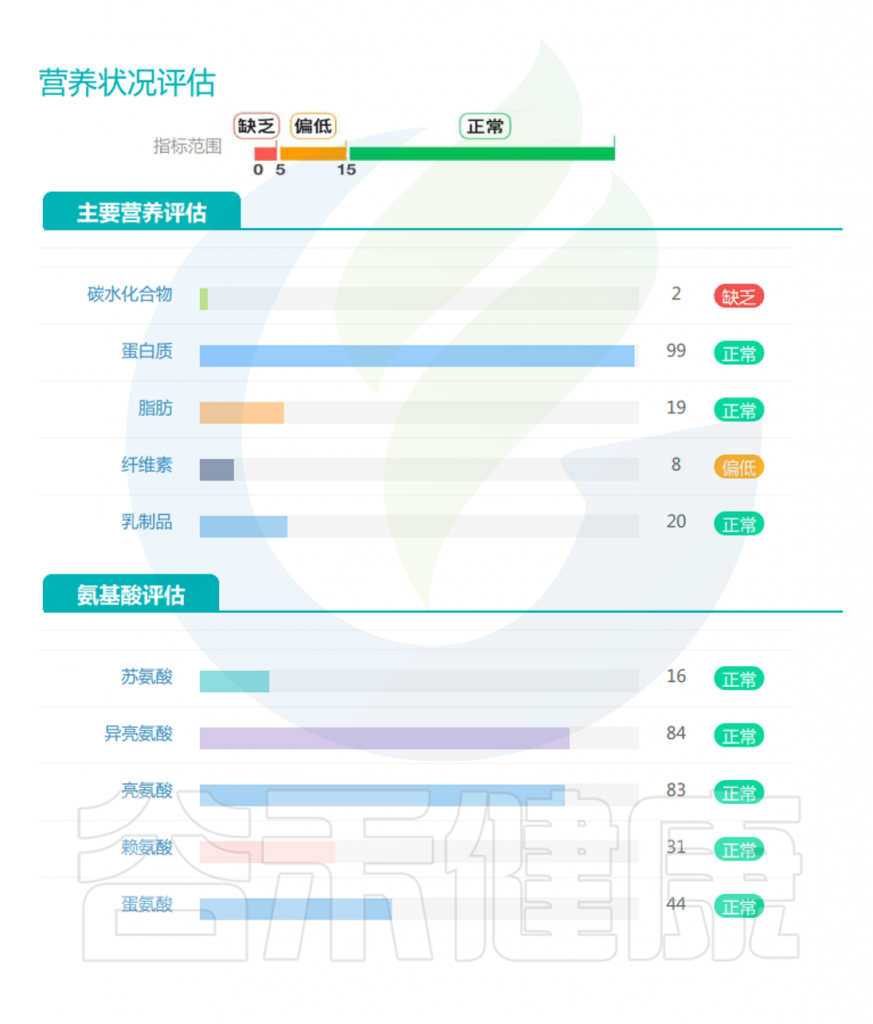
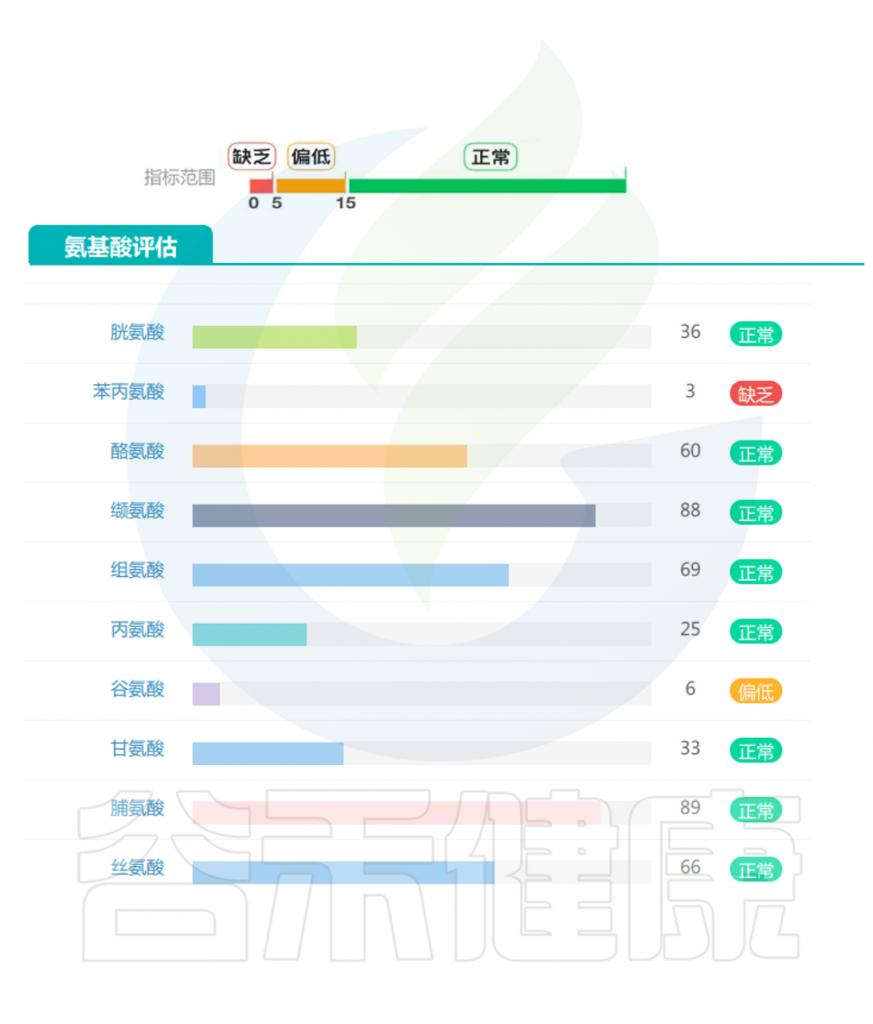
<来源:谷禾健康肠道菌群检测数据库>
▸ 根据营养指标评估是否营养不良
蛋白质和氨基酸:这是与肌肉减少症最直接相关的重要指标。作为构成肌肉组织的基本单位,蛋白质及其组成成分氨基酸的代谢状况直接影响肌肉的合成与分解平衡,因此在预测和诊断肌肉减少症风险中具有核心意义。
短链脂肪酸(SCFAs):作为膳食纤维发酵的主要产物,肠道SCFAs水平直接反映了膳食纤维摄入状况。丁酸盐、丙酸盐和乙酸盐的比例和浓度在肌肉健康中起关键作用。
氨基酸代谢物:支链氨基酸(BCAAs)代谢异常与肌肉减少症相关。
次级胆汁酸:胆汁酸代谢反映肠道对脂质消化吸收的能力,次级胆汁酸水平的变化与胰岛素敏感性和肌肉质量维持相关。
注:谷禾健康与临床机构合作成立的临床营养检测评估技术中心,致力于使营养检测更具体、更直观,扩展肠道菌群检测应用,克服传统营养评估方法局限,为临床提供更有价值的数据支持。
▸ 宏基因组反映消化吸收能力
肠道微生物的功能基因组学分析也可提供营养代谢能力的证据:
KEGG通路富集分析:与蛋白质消化、氨基酸代谢和能量转换相关的功能通路在肌肉减少症患者中表现异常。
碳水化合物活性酶谱:反映消化复杂碳水化合物的能力。
蛋白酶和肽酶活性:与蛋白质消化吸收效率相关。
除了在预防和提前诊断中发挥作用外,肠道菌群还能够在改善或治疗肌肉减少症患者中起到一定辅助作用。
补充益生菌改善肌肉减少症
已经有不少研究证实使用益生菌可以改善肌肉质量和力量。2021年一项随机双盲临床试验证实,口服植物乳杆菌(Lactobacillus plantarum TWK10)六周可有效提升虚弱老年人的肌肉质量和功能,预防肌肉减少症。
同年另一项研究表明,植物乳杆菌TWK10通过提高肌肉组织糖原浓度和调节肠道菌群,预防小鼠因衰老导致的肌肉无力、骨质流失和认知障碍。
此外研究发现,补充干酪乳杆菌(L.casei Shirota)(LcS)可通过肠道肌肉轴减轻衰老小鼠的肌肉减少症。LcS通过改变肠道菌群组成、改善线粒体功能、降低炎症和活性氧水平及维持短链脂肪酸水平,减缓与衰老相关的肌肉质量损失。
从一位举重金牌运动员肠道分离出的长双歧杆菌(Bifidobacterium longum OLP-01),连续补充4周可剂量依赖性地提高小鼠握力和耐力。
肌肉减少症的发展可以受肠道微生物群调节
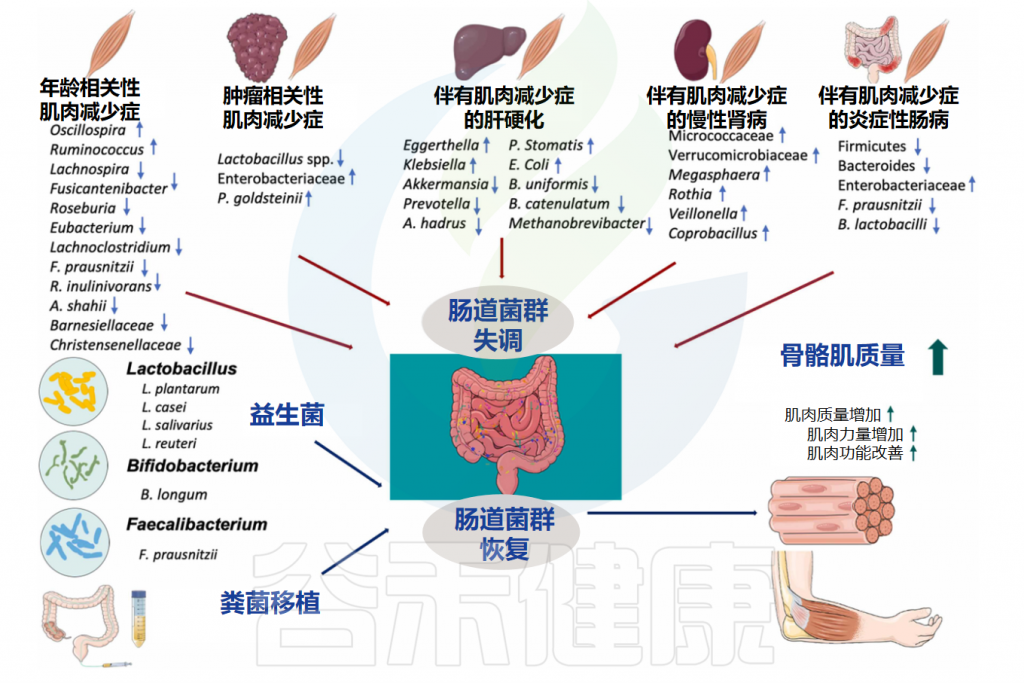
Zhang T,et al.Ageing Res Rev. 2022
益生菌改善肌肉减少症的作用机制可能通过以下几点:
1.提高蛋白质分解吸收效率
肌肉减少症的一个关键问题是营养吸收不足,特别是蛋白质摄入和利用效率下降。研究表明,益生菌能够显著提高蛋白质的消化吸收率,尤其是对植物性蛋白质。
2020一项安慰剂对照、随机、双盲、多中心交叉研究中证实,益生菌补充能显著增加植物蛋白中氨基酸的吸收率。这对素食者或依赖植物蛋白为主要蛋白质来源的老年人尤为重要。
2.增强肠道屏障功能
肠漏被认为与肌肉减少症有关,由于肠道屏障的破坏,其通透性增加,调节有害物质转移的能力降低,从而触发免疫系统和炎症反应。低度慢性炎症是被认为导致合成代谢抵抗和肌肉减少症发展的因素之一 。
而特定益生菌能够增强肠道上皮细胞间的紧密连接,减少肠道通透性。
3.作为肌肉营养传感器
肠道核心菌属普拉梭菌(Faecalibacterium prausnitzii)干预显著增加高脂肪饮食小鼠的肌肉质量,可能与增强线粒体呼吸、ATP合成酶水平提高、肠道菌群改变及肠道完整性改善有关。
肠道微生物群可能还是肌肉营养的调节传感器,肠道菌群的产物,尤其是短链脂肪酸(SCFA)被发现可以改善肌肉质量。喂养SCFA的年轻无菌小鼠的新证据表明,与未治疗的对照相比,骨骼肌质量和力量增加。
粪菌移植
与补充益生菌相比,粪菌移植(FMT)是一种更激进的治疗选择。
▸ 来自健康供体的粪菌移植后改善了原本的肌肉质量
许多研究表明,粪菌移植可能是一种改善骨骼肌质量和功能的方法。与肠道菌群正常的无病原体小鼠相比,缺乏肠道菌群的无菌小鼠表现出骨骼肌萎缩增加,以及与骨骼肌生长和线粒体功能相关的基因表达降低。
在将无病原体小鼠的肠道微生物群移植到无菌小鼠体内后,观察到肌肉质量增加,肌肉萎缩改善,肌肉氧化代谢能力增强,神经肌肉连接相关基因Rapsyn 和 Lrp4 表达增加。
注:Rapsyn 和 Lrp4是肌肉发育和功能中极其重要的两个基因,尤其是在突触后膜乙酰胆碱受体(AChR)聚集和信号转导方面。
上述结果表明肠道菌群在调节骨骼肌质量和功能中的重要作用。当肠道稳态受损时,通过益生菌或粪菌移植(FMT)干预可增强肌肉功能并缓解疾病症状,从而为宿主带来益处。
常规的防治措施
除了通过肠道微生物群干预外,肌肉减少症的常规防治方法我们也应掌握。
▸ 营养充足且均衡的饮食
营养不良和肌肉减少症在老年患者中经常重叠,老年人的营养和能量摄入通常会随着年龄的增长而下降,因此预防和治疗肌肉减少症的主要方法之一是促进营养充足。
蛋白质、维生素D、抗氧化营养素和长链多不饱和脂肪酸的充足摄入量受到特别重视,因为这些营养素能够抵消合成代谢抵抗,促进蛋白质合成并调节炎症,从而防止其对肌肉细胞的有害后果。
营养不良或肌肉减少症老年人的推荐蛋白质摄入量(1.2-1.5 g/kg/天)需高于健康活跃老年人(1.0-1.2 g/kg/天),以满足更高能量需求并防止肌肉损失。
▸ 运动锻炼
目前没有专门用于治疗肌肉减少症的药物,其治疗方法还有肌肉强化和步态训练的物理疗法。
长期研究证实,运动训练(尤其是阻力训练)是改善老年人肌肉质量和力量的最有效方法,几乎所有临床试验均证实了运动对预防肌肉减少症的积极作用。
此外,运动对肠道微生物组有显著影响,研究表明运动能增加肠道菌群多样性并促进有益代谢功能菌群的增长。
随着人口老龄化加剧和不健康生活方式,肌肉减少症已从传统的老年疾病逐渐年轻化,成为威胁全民健康的重要问题。本文深入探讨了肌肉减少症的定义、症状、危害及其与肠道菌群的密切关系,揭示了这一隐形健康杀手的多重面貌。
本文主要从“肠道-肌肉轴”的新视角重新审视肌肉减少症的发生机制及防治。研究表明,肠道菌群通过多种途径影响肌肉系统:调节饮食效果与营养传导,影响肌肉合成代谢/分解代谢平衡,改变肠道通透性和炎症状态,调节线粒体功能与激素分泌,以及影响骨骼肌纤维组成和神经肌肉传递。
并且肌肉减少症患者普遍存在肠道菌群多样性和丰富度降低,表现为特定菌群比例失调,尤其是产生短链脂肪酸细菌的减少。
在日益重视健康老龄化的时代背景下,深入理解肠道菌群与肌肉健康的关联至关重要。肠道微生物群分析有望成为肌肉减少症的早期预警手段,益生菌等菌群调节手段可能成为创新治疗靶点。未来研究需聚焦个体化肠道菌群干预策略,为各类人群提供精准肌肉和营养健康管理方案,使”肌”不可失的健康意识深入人心。
主要参考文献:
Song Q, Zhu Y, Liu X, Liu H, Zhao X, Xue L, Yang S, Wang Y, Liu X. Changes in the gut microbiota of patients with sarcopenia based on 16S rRNA gene sequencing: a systematic review and meta-analysis. Front Nutr. 2024 Jun 28;11:1429242.
Zhang T, Cheng JK, Hu YM. Gut microbiota as a promising therapeutic target for age-related sarcopenia. Ageing Res Rev. 2022 Nov;81:101739.
Liao X, Wu M, Hao Y, Deng H. Exploring the Preventive Effect and Mechanism of Senile Sarcopenia Based on “Gut-Muscle Axis”. Front Bioeng Biotechnol. 2020 Nov 5;8:590869.
de Marco Castro E, Murphy CH, Roche HM. Targeting the Gut Microbiota to Improve Dietary Protein Efficacy to Mitigate Sarcopenia. Front Nutr. 2021 Jun 21;8:656730.
Prokopidis K, Cervo MM, Gandham A, Scott D. Impact of Protein Intake in Older Adults with Sarcopenia and Obesity: A Gut Microbiota Perspective. Nutrients. 2020 Jul 30;12(8):2285.
Zhao J, Huang Y, Yu X. A Narrative Review of Gut-Muscle Axis and Sarcopenia: The Potential Role of Gut Microbiota. Int J Gen Med. 2021 Apr 13;14:1263-1273.
Ticinesi A, Nouvenne A, Cerundolo N, Catania P, Prati B, Tana C, Meschi T. Gut Microbiota, Muscle Mass and Function in Aging: A Focus on Physical Frailty and Sarcopenia. Nutrients. 2019 Jul 17;11(7):1633.
Sarcopenia Cruz-Jentoft, Alfonso J et al.The Lancet, Volume 393, Issue 10191, 2636 – 2646.
Casati M, Ferri E, Azzolino D, Cesari M, Arosio B. Gut microbiota and physical frailty through the mediation of sarcopenia. Exp Gerontol. 2019 Sep;124:110639.
Ticinesi A, Lauretani F, Milani C, Nouvenne A, Tana C, Del Rio D, Maggio M, Ventura M, Meschi T. Aging Gut Microbiota at the Cross-Road between Nutrition, Physical Frailty, and Sarcopenia: Is There a Gut-Muscle Axis? Nutrients. 2017 Nov 30;9(12):1303.

谷禾健康

青春双歧杆菌(Bifidobacterium adolescentis)是人类肠道微生物组的关键成员,在健康成人中的存在率约为70-90%。于1963年首次被发现。是一种革兰氏阳性、厌氧、不产芽孢的细菌。
青春双歧杆菌主要存在于16至45岁左右人体肠道内,这一阶段恰是人体最青春的时期,而在儿童和老年人中丰度较低。有趣的是,研究发现百岁老人体内该菌丰度显著增高,表明它可能是健康长寿相关菌群的重要成员。
该菌丰度与碳水化合物摄入量呈正相关,能代谢抗性淀粉、纤维素、半乳糖、木聚糖和果胶等复杂碳水化合物,产生乙酸和乳酸,以及叶酸等B族维生素。此外,它是人类肠道微生物群中GABA(γ-氨基丁酸)的主要生产者。
青春双歧杆菌具有多种健康益处:增强肠道屏障功能、调节肠道功能和健康、抗炎免疫调节、改善代谢紊乱、缓解焦虑和抑郁。研究表明其与炎症性肠病、肠易激综合征、乳糜泻、囊性纤维化、幽门螺杆菌感染、糖尿病、代谢综合征、非酒精性脂肪肝和某些过敏疾病密切相关。
本文将全面介绍并深入讲述青春双歧杆菌的多维度健康益处:从改善肠道屏障功能到调节免疫反应,从改善精神心理健康到预防特定疾病如肌肉减少症和脂肪肝。这种微生物通过产生乙酸、乳酸、GABA和叶酸等关键代谢物,以及与其他微生物和宿主的互动,共同构建人体健康微生物基础。
随着研究的深入,青春双歧杆菌已成为功能性食品、保健品和微生物疗法的明星菌种,广泛应用于酸奶、乳酸饮料和膳食补充剂,为人类健康开创新可能。
doi: 10.1163/18762891-20230030.
1
基本信息
青春双歧杆菌(Bifidobacterium adolescentis)于1963年被首次发现,是人类肠道中重要的双歧杆菌属成员之一。该属还包括短双歧杆菌、动物双歧杆菌、婴儿双歧杆菌和两歧双歧杆菌等其他重要肠道共生菌。
青春双歧杆菌是一种革兰氏阳性、厌氧、不产芽孢的细菌,最适生长温度35-37℃左右,超过46.5℃和低于20℃不生长。它通过发酵多种碳水化合物产生乙酸和乳酸,还是人类肠道微生物群产生GABA的关键成员。
注:青春双歧杆菌对氧气极为敏感,在有氧条件下,青春双歧杆菌的生长完全受抑制,而婴儿双歧杆菌、短双歧杆菌和长双歧杆菌则不受影响。
青春双歧杆菌是肠道益生菌,通常出生后即在肠道定植。因主要存在于16至45岁人体肠道内(人体最富青春活力的时期)而得名。年长后,其数量会因饮食、压力和抗生素等因素而减少。
2
代谢能力
青春双歧杆菌能够发酵复杂碳水化合物(如抗性淀粉、纤维素、半纤维素半乳糖、木聚糖和果胶等),对18个菌株的泛基因组分析发现36个糖苷水解酶家族、12个糖苷转移酶家族和4个碳水化合物酯酶家族,表明其代谢主要面向植物碳水化合物。
• 能够发酵多种复杂碳水化合物
所有菌株都能发酵半乳糖、葡萄糖、低聚半乳糖、乳糖、乳果糖、麦芽糖、麦芽三糖、蜜二糖和棉子糖,而对低聚果糖、果糖、甘露醇、山梨醇、海藻糖和木糖的利用则因菌株而异。
• 能够产生乙酸和乳酸,产生ATP比乳酸菌更多
青春双歧杆菌与其他双歧杆菌一样,通过”双歧途径“代谢己糖,利用果糖-6-磷酸酮醇酶生成ATP,主要产物为乙酸和乳酸。该途径从葡萄糖中产生的ATP比乳酸菌的发酵途径要多。
尽管善于发酵饮食中的植物碳水化合物,但它无法利用人类粘蛋白和乳汁中的糖苷。
3
抗生素耐药性
许多抗生素会减少肠道双歧杆菌数量。青春双歧杆菌对多粘菌素、链霉素、庆大霉素、新霉素、四环素、氯霉素、羧苄西林、苯唑西林和利福平具有抵抗力,但对卡那霉素、米诺环素、阿莫西林、氨苄西林、克拉维酸、环丙沙星和硝唑酮敏感。
• 能够促进抗生素治疗后的微生物群恢复
值得注意的是,青春双歧杆菌被确认为促进抗生素治疗后微生物群恢复的关键成员。这些个体微生物组表现为增强的碳水化合物降解能力和更高的细菌群落生长率,但与抗生素耐药基因无关。
在小鼠实验中,同时补充青春双歧杆菌与多形拟杆菌或单形拟杆菌可加速微生物恢复并减轻抗生素治疗后的结肠组织病变。
4
与人体的相互作用
• 主要产物为乙酸盐和乳酸
与其他双歧杆菌一样,青春双歧杆菌通过果糖-6-磷酸酮醇酶途径理论上以3:2比例产生乙酸盐和乳酸盐,实际比例可能取决于能源物质和消耗速率。
乙酸盐和乳酸盐具有多种健康益处,包括改善黏膜屏障、免疫功能、代谢和矿物质吸收。
• 青春双歧杆菌是人类肠道微生物群中产生GABA的关键成员

一项研究发现,在82株青春双歧杆菌体外筛选中,79%的菌株能将谷氨酸转化为GABA,其中23%为高产GABA菌株,转化效率超过65%。
与植物乳杆菌(Lactiplantibacillus plantarum)、短双歧杆菌(Lactobacillus brevis)、角双歧杆菌(Bifidobacterium angulatum)和齿双歧杆菌(Bifidobacterium dentium)菌株相比,青春双歧杆菌菌株通常是GABA产量最高的菌株之一,一些菌株在48小时后的GABA浓度高达6000mg/l培养基。
γ-氨基丁酸(GABA)是哺乳动物主要抑制性神经递质,通过作用于中枢神经系统受体抑制神经元兴奋性,与兴奋性递质谷氨酸形成平衡,两者失衡可导致神经精神疾病。
研究表明补充青春双歧杆菌可缓解小鼠长期固定应激导致的焦虑抑郁症状,但GABA在这一过程中的具体作用尚待确定。
• 一些青春双歧杆菌能够较稳定地产生叶酸
叶酸(维生素B9)是哺乳动物必须从饮食或肠道菌群获取的重要维生素。青春双歧杆菌等几种双歧杆菌能在无叶酸环境中产生并积累叶酸,其中青春双歧杆菌产量尤为突出。虽然某些菌株的叶酸产生会受到培养基中叶酸或前体物质的抑制,但青春双歧杆菌MB239菌株不受此影响,其叶酸产量也不受pH值或碳水化合物源类型影响。
健康志愿者服用两株产叶酸的青春双歧杆菌30天后,粪便中游离叶酸浓度显著增加(分别增加50和80微克/天),同时总双歧杆菌水平也相应提高。
注:一些研究发现青春双歧杆菌可以产生维生素B1、B2、B6、B12及丙氨酸、缬氨酸、天冬氨酸和苏氨酸等人体必需的营养物质,对于人体具有重要的营养作用。
5
青春双歧杆菌在人群中的分布
青春双歧杆菌是健康成年人中重要的双歧杆菌物种。早期研究显示该菌在90%的受试者粪便中存在,浓度高达每克108-1010个细胞。进一步研究证实其在70-90%成年人肠道中定植,密度约为每克粪便109-1010个细胞。谷禾数据库表明超过95%的人群能检出该菌,但其分布明显受年龄和饮食因素影响。

编辑
<来源:谷禾肠道菌群健康检测数据库>
• 青年、中年群体的青春双歧杆菌丰度更高
虽然青春双歧杆菌不能利用人类粘蛋白或人乳低聚糖,但其丰度与血型抗原分泌状态相关。分泌型个体比非分泌型个体更易被青春双歧杆菌定植。随年龄增长,青春双歧杆菌丰度从60-70岁开始下降,可能与粘液粘附能力降低有关。
• 长寿老人体内青春双歧杆菌也较高
然而,意大利百岁老人研究发现,与老年(68-88岁)和年轻(21-48岁)个体相比,极老年人粪便中青春双歧杆菌丰度显著增高,可能是健康长寿相关菌群的重要成员。
尽管主要被视为”成人型”双歧杆菌,但青春双歧杆菌也存在于婴幼儿中,但数量较少(每克粪便106-107个细胞)且流行率低。研究证实该菌可在阴道分娩和母乳喂养过程中从母亲传给婴儿,约80%的母乳中检测到活的青春双歧杆菌。非分泌型母乳缺乏岩藻糖化寡糖,反而促进婴儿肠道中青春双歧杆菌的存在。
注:人体肠道中的青春双歧杆菌菌株具有高度稳定性,可持续定植长达43个月。一个人体内可同时存在多个遗传相似但表型不同的青春双歧杆菌菌株,它们在自聚集能力和碳水化合物亲和力等特性上存在显著差异。
6
丰度受饮食碳水化合物影响明显
研究发现,青春双歧杆菌是饮食影响最显著的微生物种类之一。其丰度与碳水化合物摄入量呈正相关,显示对淀粉的偏好;而与高蛋白或高脂摄入呈负相关。因此,高蛋白低碳水饮食的运动员体内青春双歧杆菌水平明显低于饮食均衡的人群。
多种益生元如抗性淀粉、低聚半乳糖、菊粉、果寡糖、木寡糖和阿魏酸低聚糖能有效刺激青春双歧杆菌生长。这些非消化性碳水化合物到达结肠后被微生物发酵,产生乙酸、丙酸和丁酸等短链脂肪酸,对肠道和整体健康具有重要意义。青春双歧杆菌能利用多种益生元,反映其丰富的糖转运体和降解酶系统。
• 马铃薯抗性淀粉能促进青春双歧杆菌丰度增加
临床研究表明,摄入马铃薯抗性淀粉(RS2)后,青少年双歧杆菌增长最显著。携带者每日摄入48克淀粉12天后,粪便中该菌丰度大幅上升并大量定植于淀粉颗粒表面。尽管双歧杆菌不直接产丁酸,但其作为淀粉主要分解者的代谢产物可促进产丁酸菌生长,从而提高丁酸水平。
• 低聚半乳糖促进青春双歧杆菌增长
人体研究证明摄入低聚半乳糖(GOS)能剂量依赖性促进双歧杆菌生长。乳糖不耐受者连服36天高纯度GOS(1.5-15克/天)后,青少年双歧杆菌增长45倍之多,β-半乳糖苷酶活性增强,并显著缓解乳糖不耐受症状。
• 菊粉等低聚果糖、低聚木糖也促进其丰度增加
菊粉是植物中储备性多糖,低聚果糖(FOS)是较短的果糖聚合物,通常由菊粉水解获得。β-呋喃果糖苷酶能切割末端果糖残基,是降解菊粉类化合物的关键酶。青少年双歧杆菌具有此酶活性,部分菌株还产生胞外酶降解长链果聚糖。
临床研究证实,健康人群、肥胖女性、糖尿病患者和超重儿童摄入菊粉型果聚糖后,青少年双歧杆菌丰度均显著增加。
低聚木糖(XOS)是常带分支的β-1,4木糖聚合物。青春双歧杆菌产生多种木糖苷酶,研究显示XOS使ATCC 15703T菌株生长率较木糖高1.5倍,同时促进碳水化合物代谢基因表达。人体结肠模型研究证实,添加甘蔗源XOS能剂量依赖性增加青少年双歧杆菌丰度。
青春双歧杆菌(Bifidobacterium adolescentis)是人类肠道微生物组的关键成员,对维持宿主健康至关重要。该菌通过调节肠道生态平衡、参与营养代谢、增强免疫功能及抵抗病原体等机制,对人体健康产生全面深远的影响。
下面我们来讲讲青春双歧杆菌在影响肠道功能及健康、免疫及过敏反应的调节、焦虑、抑郁等精神心理健康及肝脏或肌肉等特定疾病预防与治疗中的应用价值与潜力,以科学阐述这一重要微生物对人体健康的多维度影响。
1
改善肠道屏障
肠道上皮在保障营养吸收的同时,负责排除、中和或解毒肠道内有害物质,包括微生物。上皮层及紧密连接等物理结构对维持肠道屏障至关重要,肠道屏障功能障碍与多种疾病密切相关,可能存在因果关系。
• 青春双歧杆菌能保护肠道屏障

多项动物研究证实,青春双歧杆菌能保护肠道屏障。硫酸葡聚糖钠(DSS)是一种对肠道上皮细胞有毒的硫酸化多糖,可诱导啮齿类动物产生类似人类溃疡性结肠炎的病变。在DSS结肠炎模型中,补充青春双歧杆菌ATCC 15703T增加了结肠粘液层厚度和Muc2、Muc3基因表达,同时提高了紧密连接蛋白occludin的表达水平。
在闭合股骨骨折模型中,与对照组相比,补充青春双歧杆菌ATCC 15703T的小鼠Muc2、Ocln、粘附分子C(Jam3)、闭锁小带蛋白1(ZO-1)(Tjp1)、紧密连接蛋白3(Cldn3)和紧密连接蛋白15(Cldn15)的表达上调,而形成孔洞的紧密连接蛋白2(Cldn2)的表达下调。股骨骨折显著增加了肠道通透性,直至骨折后10天,但青春双歧杆菌在骨折前2周开始补充可以减少骨折引起的通透性。改善的肠道屏障降低了血清内毒素水平和对骨折的系统炎症反应。
• 恢复上皮细胞紧密连接,改善肠道通透性
在关节炎模型中,青春双歧杆菌减轻了临床症状并改善了小肠ZO-1和occludin表达下降。在非酒精性脂肪肝病小鼠模型中,补充青春双歧杆菌DSM 20086通过恢复上皮细胞中ZO-1和occludin,显著抵消了高脂饮食引起的肠道通透性增加,并降低血清脂多糖水平。
另一种菌株,青春双歧杆菌 CGMCC 15058,从在D-氨基半乳糖(D-GalN)诱导的肝损伤模型中改善了肠道屏障。用B.adolescentis CGMCC 15058预处理减轻了D-氨基半乳糖诱导的回肠末端组织学异常,包括上皮下Gruenhagen间隙的发生率降低、刷状边界破坏和组织学评分显著降低。还增加了肠道Muc4和Tjp1表达,促进了ZO-1在肠道上皮细胞近端区域的均匀分布。
2
与肠道功能和健康紧密相关
• 炎症性肠病中青春双歧杆菌减少
炎症性肠病(IBD)特征为肠道组织慢性炎症,伴有复发和缓解周期。微生物群失调与这些疾病相关,许多研究显示青春双歧杆菌减少。

多项队列研究发现,克罗恩病(CD)、溃疡性结肠炎(UC)和肠易激综合征(IBS)患者粪便中青春双歧杆菌数量减少。新诊断CD儿童的粪便中,该菌数量比健康对照减少10倍,表明微生物失调在疾病早期已存在。
前瞻性研究显示UC复发患者粪便中青春双歧杆菌少于缓解期患者。然而,静止期IBD患者采用低发酵性碳水化合物饮食虽缓解症状,但显著降低了青春双歧杆菌丰度和粪便短链脂肪酸水平。
• 功能性腹胀和便秘患者中丰度降低
患有功能性腹胀和便秘(FABD)但不符合肠易激综合征(IBS)标准的个体的粪便微生物群与健康对照组相比有显著变化。一个主要差异在于青春双歧杆菌(B.adolescentis),其在健康个体中的平均相对丰度为5.4%,而在FABD患者中降至0.2%。
• 乳糜泻患者中检测不到青春双歧杆菌
乳糜泻(CeD)是一种影响小肠黏膜的慢性炎症性疾病。研究发现,CeD患者口咽、十二指肠和结肠的微生物群发生了改变。在患有CeD的儿童中,粪便细菌多样性的减少,且在任何样本中都未检测到B.adolescentis,而健康儿童平均检出率为40%。
然而,在另一项研究中发现了相反的情况:活动期CeD儿童小肠活检中该菌检出率达50%,而治疗后儿童和正常对照组均未检出。
• 改善减轻结肠炎症状
研究证实,在DSS诱导的结肠炎模型中,补充青春双歧杆菌显著改善了多项炎症指标,包括体重变化、腹泻评分、脾脏重量、结肠长度和组织学评分。
其保护机制主要通过增强肠道屏障功能,抵抗严重肠道炎症。健康人粪便微生物群移植降低了无菌小鼠对DSS诱导结肠炎的敏感性,效果优于溃疡性结肠炎患者粪便移植。接受健康供体粪便的小鼠表现出较高的青春双歧杆菌丰度,而UC患者粪便移植既未显著减少炎症标志物,也未增加抗炎因子IL-10的分泌。青春双歧杆菌丰度与IL-10和FOXP3(T调节细胞标志物)呈正相关,表明其在免疫调节中的潜在作用。这引发了一个疑问:该菌是否属于抗炎微生物群的一部分?
• 幽门螺杆菌携带者青春双歧杆菌极低
幽门螺杆菌感染可引起慢性胃炎、溃疡及胃癌,虽然大多数感染(80-90%)为良性。但研究发现携带者与非携带者肠道微生物群存在差异,这可能影响疾病严重程度。幽门螺杆菌携带者双歧杆菌数量减少,严重胃病患者青春双歧杆菌和长双歧杆菌极低。幽门螺杆菌根除治疗使用的抗生素组合显著改变肠道微生物群,尤其降低青春双歧杆菌丰度。
• 降低了使用非甾体抗炎药后溃疡的发生率
非甾体抗炎药(NSAIDs)已知可促进人类胃十二指肠溃疡形成。小鼠研究显示NSAIDs引起小肠溃疡,而抗生素治疗减少溃疡发生,表明微生物在溃疡形成中发挥作用。NSAID处理的无菌鼠未出现溃疡,但当这些无菌鼠饮水中添加单一菌株大肠杆菌或黏液真杆菌(Eubacterium limosum)时则出现回肠溃疡。
值得注意的是,抗生素处理后接种青春双歧杆菌ATCC 15703T的小鼠未发生溃疡,给特定病原体自由小鼠补充该菌同样未观察到溃疡。与普通饮用水相比,含有该菌的饮用水降低了回肠溃疡发生率。后续研究发现,在饮用水中添加无菌大鼠细胞培养基也能产生类似效果,证明青春双歧杆菌ATCC 15703T产生的代谢物在预防溃疡中起关键作用。
3
与免疫系统的相互作用

多项体外和小鼠研究表明,青春双歧杆菌具有免疫调节作用,通过影响ZO-1、occludin蛋白表达,并发现外周血单核细胞细胞因子与肠道微生物群结构功能相关。该菌与TNF-α刺激响应呈负相关,显示抗炎特性。
• 具有活性的青春双歧杆菌减少炎症因子的产生
在体外实验中,青春双歧杆菌能诱导人单核细胞衍生树突状细胞成熟,促使CD4+T细胞向产生IFN-γ和IL-10的Th1细胞极化。菌株CGMCC 15058能缓解单核细胞衍生树突状细胞异常。在脂多糖刺激的大鼠巨噬细胞中,该菌减少了炎症因子产生,但热灭活的菌株反而增加未刺激巨噬细胞的促炎细胞因子分泌。
• 增加Th17细胞数量,改善炎症反应
单一菌落培养的无菌小鼠研究发现,青春双歧杆菌增加小肠固有层Th17细胞数量和频率,但不引发肠道或全身炎症。Th17细胞对黏膜保护至关重要,而调节性T细胞(Tregs)抑制免疫反应和炎症,两者平衡对健康至关重要。
注:辅助性T细胞17(Th17)是一种能够分泌白介素17(IL-17)的T细胞亚群,在自身免疫性疾病和机体防御反应中具有重要的意义。
研究表明,部分青春双歧杆菌菌株可恢复饮食或DSS诱导性肠炎小鼠的Tregs/Th17平衡。这种能力因菌株而异,可能与胞外多糖(EPS)等表面分子有关。产高分子量EPS的菌株能促进CD4+ T细胞向Tregs细胞分化,减轻DSS结肠炎模型炎症标记物,而低EPS产生菌株效果较差。多项啮齿类动物研究证实,青春双歧杆菌主要通过促进调节性T细胞扩增和抑制炎症来影响免疫系统。
• 抑制核因子κB激活,具有免疫保护作用
核因子κB(NF-κB)是一种主要的转录因子,通过上调免疫反应相关基因而调控炎症过程。研究表明,青春双歧杆菌能抑制多种因素引起的NF-κB激活:包括由脂多糖、肠出血性大肠杆菌和小肠结肠炎耶尔森氏菌诱导的炎症反应,以及动物模型中由高脂饮食或慢性应激引起的NF-κB激活。
这表明青春双歧杆菌能降低NF-κB激活及随后的炎症反应,对过度和持续的免疫激活具有保护作用。
• 青春双歧杆菌还能调节体液免疫
青春双歧杆菌对体液免疫的影响研究较少,但显示其具调节作用。研究发现,青春双歧杆菌单一菌落定植的啮齿类动物血清免疫球蛋白或脾细胞因子变化很小或无变化。
在青春双歧杆菌(B.adolescentis)定植后,发现啮齿类动物粪便中有增加的青春双歧杆菌特异性分泌型IgA。已显示B.adolescentis菌株减弱了针对B.thetaiotaomicron定植的IgA、IgG和IgE反应,以及胶原诱导性关节炎和2,4-二硝基氟苯(DNFB)诱导的特应性皮炎的反应。
▸ 抑制病原菌和病毒

多项研究已观察到青春双歧杆菌(B.adolescentis)菌株在体外对多种人类致病菌具有杀菌和抑制效果,包括大肠杆菌、沙门氏菌、金黄色葡萄球菌、铜绿假单胞菌、普通变形杆菌、单核细胞增生李斯特菌、痤疮丙酸杆菌、牙龈卟啉单胞菌、变异链球菌以及白色念珠菌。分泌抗菌物质如短链脂肪酸(SCFAs)和竞争特定营养物质被认为与这些效应有关。
在体外实验中,青春双歧杆菌菌株的活细菌、无细胞上清液和细胞提取物显示出抗病毒效果,针对柯萨奇病毒、乙型肝炎病毒、诺如病毒、单纯疱疹病毒、人乳头瘤病毒和轮状病毒。抗病毒效果被认为与阻断病毒复制或细胞附着以及糖介导的病毒与细菌结合有关。
4
与过敏反应存在一定联系
关于青春双歧杆菌(B.adolescentis)与过敏之间关联的研究结果存在矛盾,可能源于人群差异、样本量小、微生物群采样时间及过敏类型不同。
• 在过敏反应中的作用存在一些矛盾
一项涵盖21项研究的系统综述显示,过敏儿童肠道微生物相比非过敏儿童通常具有较低的微生物多样性,以及较低的B.adolescentis、B.bifidum和乳杆菌检出率,但研究B.adolescentis检出率的研究较少。
一项为期5年、涉及47名婴儿的研究显示,出生后一周内被B.adolescentis和特定乳杆菌定植的婴儿患过敏性疾病风险较低——研究中16名婴儿发展为过敏性皮炎、哮喘或过敏性鼻炎/结膜炎。2.5岁患特应性皮炎的儿童中,同时患食物过敏的子群(n=20)可通过粪便微生物组特征识别,特点是B.adolescentis、B.breve、F.prausnitzii和A.muciniphila较少。
相反,也有一些其他研究发现B.adolescentis是过敏儿童(患特应性皮炎、过敏性哮喘以及2-7月龄特应性湿疹婴儿)中主要的双歧杆菌物种。
5
有助于改善代谢紊乱相关疾病
代谢紊乱相关疾病(如2型糖尿病、代谢综合征和肥胖)与肠道菌群失调相关。肠道微生物群可影响多种代谢过程,包括饮食能量利用、能量消耗、脂质代谢、食欲调节及慢性炎症。
• 2型糖尿病患者青春双歧杆菌显著减少
与健康对照组相比,2型糖尿病(T2D)患者的粪便微生物群中发现双歧杆菌数量显著减少,包括青春双歧杆菌在内。在高脂肪饮食和链脲佐菌素诱导的糖尿病小鼠中,几种青春双歧杆菌菌株降低了空腹血糖浓度,改善了胰岛素抵抗,但效果不同。更有效的菌株倾向于将肠道微生物群结构向对照组小鼠的微生物群转移,增加粪便中短链脂肪酸的丰度,并缓解与T2D相关的炎症。
• 影响胰高血糖素样肽-1浓度,调节葡萄糖稳态
在人类中,粪便中B.adolescentis相对丰度与胰高血糖素样肽-1(GLP-1)的血浆浓度呈正相关。GLP-1是一种由肠内分泌L细胞产生的激素,调节葡萄糖稳态。二肽基肽酶-4(DPP-IV)是一种丝氨酸外肽酶,可降解GLP-1,并负责该激素在血浆中的2-5分钟半衰期。因此,抑制DPP-IV活性可以增加血浆中的GLP-1水平。体外实验显示,B.adolescentis的无细胞提取物具有DPP-IV抑制活性,而在人类中发现的B. adolescentis-GLP-1正相关性可能归因于DPP-IV的抑制作用。
• 患有高血压的糖尿病儿童青春双歧杆菌减少
高血压糖尿病儿童相比正常血压糖尿病儿童和健康对照组,肠道微生物群独特且多样性降低。B.adolescentis、B.bifidum和B.longum丰度减少是其菌群紊乱特征之一。虽然发现多个微生物途径发生改变,但需更多研究阐明其在高血压中的潜在作用。
• 青春双歧杆菌丰度与腰围、甘油三酯负相关
代谢综合征(MetS)患者(一种多组分、常与肥胖相关的疾病)与健康对照组相比,青春双歧杆菌(B.adolescentis)相对丰度降低是其不良微生物组的特征之一。MetS患者遵循地中海饮食2年后,B.adolescentis显著增加。该菌及其他菌种丰度与腰围、血糖和甘油三酯呈负相关,与高密度脂蛋白胆固醇呈正相关。
在高脂饮食动物模型中,补充B.adolescentis可改善肠道菌群失调、减少肝脂肪变性和肠系膜脂肪积累、增强肝脏抗氧化活性并改善胰岛素敏感性,但这些效果可能因菌株而异。
6
抗焦虑和抑郁作用
抑郁症是一种常见且反复发作的情绪障碍,并伴有行为缺陷。近年来,新出现的证据表明肠道微生物群参与炎症、大脑发育和行为。压力会扰乱肠道微生物群的平衡并刺激炎症到大脑的机制,进而引发焦虑和抑郁障碍。青春双歧杆菌(B.adolescentis)显示出明显的抗炎特性,并表现出抗焦虑和抗抑郁效果。

• 抗焦虑和抑郁的行为学证据
研究表明,B.adolescentis可有效预防慢性约束应激(CRS)诱导的焦虑和抑郁样行为。研究使用不同剂量(0.25×10⁹、0.5×10⁹和1×10⁹ CFU/kg)的B.adolescentis均显示出抗抑郁效果,表现为强迫游泳测试中不动时间的显著减少。
B.adolescentis(0.25×10⁹ CFU/kg)处理使小鼠在开放场测试中心区域停留时间增加,以及在高架十字迷宫开放臂的进入次数百分比增加,表明其具有抗焦虑作用。
• 青春双歧杆菌逆转束缚应激诱导的菌群失衡
为验证青春双歧杆菌(B.adolescentis)的抗焦虑和抗抑郁作用是否与肠道微生物群平衡重建相关,研究分析了盲肠微生物群落多样性。
增加乳杆菌丰度:B.adolescentis处理显著增加了慢性约束应激(CRS)小鼠粪便中乳杆菌(Lactobacillus)的丰度,而乳杆菌减少与焦虑/抑郁相关。
降低拟杆菌丰度:研究发现,拟杆菌(Bacteroides)比例的增加与抑郁相关,B.adolescentis可逆转CRS导致的拟杆菌增加。
改善菌群结构:B.adolescentis增加了因慢性应激减少的厚壁菌门(Firmicutes)比例,同时降低了增加的拟杆菌门(Bacteroidetes)比例,恢复菌群平衡。
• 降低促炎细胞因子水平
青春双歧杆菌(B.adolescentis)显著抑制了CRS导致的海马区炎症因子水平升高,包括:白细胞介素-1β (IL-1β)、肿瘤坏死因子-α (TNF-α)、核因子-κB (NF-κB)、小胶质细胞标记物Iba1,表明其可抑制微胶质细胞活化,从而减轻神经炎症。
此外,B.adolescentis处理增加了CRS小鼠海马区脑源性神经营养因子(BDNF)的表达,而BDNF水平降低与抑郁相关。
• 可能的肠-脑轴调节机制
青春双歧杆菌(B.adolescentis)通过肠-脑轴发挥作用,其可能的机制包括:
1.调节肠道免疫系统,减少促炎细胞因子释放进入循环系统;
2.改变肠道微生物组结构,增加有益菌如乳杆菌的丰度,这些菌株可产生对精神状态有积极影响的代谢产物;
3.通过抑制肠道中的DPP-IV活性,增加血浆中GLP-1的水平,从而改善代谢和精神状态。
4.青春双歧杆菌作为人类肠道微生物群产生GABA的关键成员,GABA是哺乳动物关键抑制性神经递质,通过抑制神经元活性与谷氨酸形成平衡,维持神经系统稳态。青春双歧杆菌产生GABA有效调节这一神经递质平衡。
综上所述,青春双歧杆菌通过调节肠道菌群(增加乳杆菌比例、降低拟杆菌比例)和抑制炎症反应(降低海马体IL-1β、TNF-α、p-NF-κB p65和Iba1表达,同时提高BDNF表达)发挥抗焦虑抗抑郁作用。
7
调节肝脏炎症,减轻肝损伤
青春双歧杆菌对肝脏健康也具有重要影响,研究显示,非酒精性脂肪性肝炎(NASH)儿童与健康对照组相比,青春双歧杆菌丰度降低。研究发现该菌与血浆甘油三酯、胆固醇和低密度脂蛋白胆固醇水平呈显著负相关。
在啮齿类动物和兔子的非酒精性脂肪肝病模型中,补充青春双歧杆菌改善了肝脏组织病理和脂质代谢,减轻了肝脏炎症。其作用机制包括调节短链脂肪酸、增加肠道粘液层厚度、降低血浆内毒素水平、预防肝脏脂质过氧化、增加成纤维细胞生长因子21表达及抑制NFκB激活。
• B.adolescentis CGMCC 15058缓解肝损伤

还有研究发现了青春双歧杆菌在缓解肝损伤中的作用,青春双歧杆菌能显著缓解D-半乳糖胺诱导的肝脏退化、坏死和炎症浸润。肝功能检测显示,接受青春双歧杆菌处理的大鼠血清丙氨酸转氨酶(ALT)和总胆汁酸(TBA)水平较对照组明显降低,表明其对肝损伤具有保护作用。
在肝损伤中,B.adolescentis通过多种机制发挥保护作用。以下是其主要机制:
减少炎症细胞因子:B.adolescentis能够降低肝脏和血浆中的促炎细胞因子水平,如肿瘤坏死因子-α(TNF-α)和白细胞介素-6(IL-6)。同时,它增加了抗炎细胞因子白细胞介素-10(IL-10)的表达。
增强肠道屏障完整性:B.adolescentis通过增加紧密连接蛋白(如ZO-1)和粘蛋白(如MUC4)的表达,增强了肠道屏障的完整性,减少了细菌易位。
降低内毒素水平:通过减少肠道通透性,B.adolescentis降低了血液中的内毒素水平,从而减轻了肝脏的炎症反应。
调节肠道微生物群组成:急性肝损伤大鼠的肠道微生物结构受青春双歧杆菌影响显著。口服该菌可改善D-半乳糖胺诱导的丹毒丝菌科(Erysipelotrichaceae)减少情况。与对照组相比,青春双歧杆菌组中Desulfovibrionaceae、 Erysipelotrichaceae、Peptostreptococcaceae和 Bacteroides科明显富集。
在属水平上,急性肝损伤广泛影响肠道微生物组成。对照组中常见病原体如Proteus和Runella丰度增加。补充青春双歧杆菌则显著促进了与脂质和芳香氨基酸代谢相关的Coriobacteriaceae、Bacteroidales和Allobaculum的丰度。
8
改善肌肉减少症

临床研究发现,青春双歧杆菌在肌肉减少症个体中显著耗尽,与肌肉质量和功能降低相关。
多组学证据:宏基因组学和代谢组学分析表明,青春双歧杆菌丰度与肌肉减少症呈显著负相关。
• 补充青春双歧杆菌可改善老年群体肌肉质量和功能
青春双歧杆菌与骨骼肌质量和功能呈正相关。该菌与多项肌肉健康指标显著相关,包括附肢骨骼肌质量指数(ASMI)、握力、小腿围及5次椅子站立测试(5TCS)。在区分肌肉减少症与健康个体的随机森林模型中,青春双歧杆菌是贡献最大的菌种。
• 青春双歧杆菌对骨骼肌的保护机制
青春双歧杆菌可以通过抑制FoxO3/Atrogin-1/Murf-1信号通路及下调Atrogin-1和Murf-1表达,有效抑制肌肉萎缩。同时促进肌肉再生,增加肌肉干细胞(MUSCs)数量并提高PAX7阳性细胞表达。该菌还能增强线粒体功能,通过NAD+激活SIRT1/PGC-1α信号轴,促进线粒体生物发生,增强骨骼肌氧化代谢(尤其在慢肌纤维中)并提高ATP含量。
烟酸(NA)代谢通路是关键联系机制。青春双歧杆菌产生的烟酸是影响宿主骨骼肌健康的关键代谢物,作为NAD+前体,可恢复肌肉中NAD+水平,对能量代谢至关重要。代谢组学研究显示,肌肉减少症患者血清中烟酸等多种有益代谢物水平显著降低。
Zhang Z,et al.Cell Rep.2025
9
其他健康益处
还有一些研究发现了青春双歧杆菌的其他益处:
• 减轻骨折诱导的全身性后遗症
一项研究发现了青春双歧杆菌(Bifidobacterium adolescentis)补充对骨折诱导系统性影响的调节作用。骨折会导致肠道通透性增加和肠道屏障功能受损,持续约7天后恢复。补充B.adolescentis可增强肠道紧密连接蛋白表达,减少系统性炎症,改变肠道微生物群落结构,加速软骨愈合,并防止创伤后骨质流失。这些发现表明益生菌补充可能是一种有效策略,可减轻骨折引起的系统性病理变化
doi: 10.1016/j.biopha.2020.110831.
• 可能加强新冠疫苗的效果
2021年的时候,香港中文大学医学院和香港大学医学院进行的全球首个人体研究发现,一种名为”青春双歧杆菌”(Bifidobacterium adolescentis)的微生物与新冠疫苗的有效性密切相关。
研究显示,肠道内缺乏青春双歧杆菌的人士接种新冠疫苗后,抗体水平普遍不达预期标准。这项突破性发现意味着通过补充青春双歧杆菌可提高疫苗效力,为未来疫苗接种策略提供了新方向。
青春双歧杆菌(Bifidobacterium adolescentis)作为人体肠道微生物组的重要成员,与其他益生菌共同使用时展现出显著的协同健康效应。研究表明,这些协同作用不仅增强了单一菌株的益处,还能产生更为全面的健康保护作用,具有重要临床和市场应用价值。
1
与罗伊氏乳杆菌共同减轻焦虑、抑郁等

该研究探讨了来自人类肠道菌群的益生菌对压力诱导的焦虑/抑郁和结肠炎的预防和治疗效果。研究人员从健康人粪便中分离出罗伊氏乳杆菌(Lactobacillus reuteri NK33)和青春双歧杆菌(Bifidobacterium adolescentis NK98),并检验了它们对固定压力(IS)诱导的小鼠焦虑/抑郁和结肠炎的影响。
结果表明,NK33和NK98显著抑制脂多糖诱导的BV-2细胞NF-κB激活,口服后能防治小鼠焦虑/抑郁症状,减少海马小胶质细胞浸润,降低血液炎症标志物,提高海马BDNF表达。同时,这些益生菌抑制压力引起的结肠炎症反应,通过调节肠道菌群和减少脂多糖产生,协同改善神经精神和肠道症状。
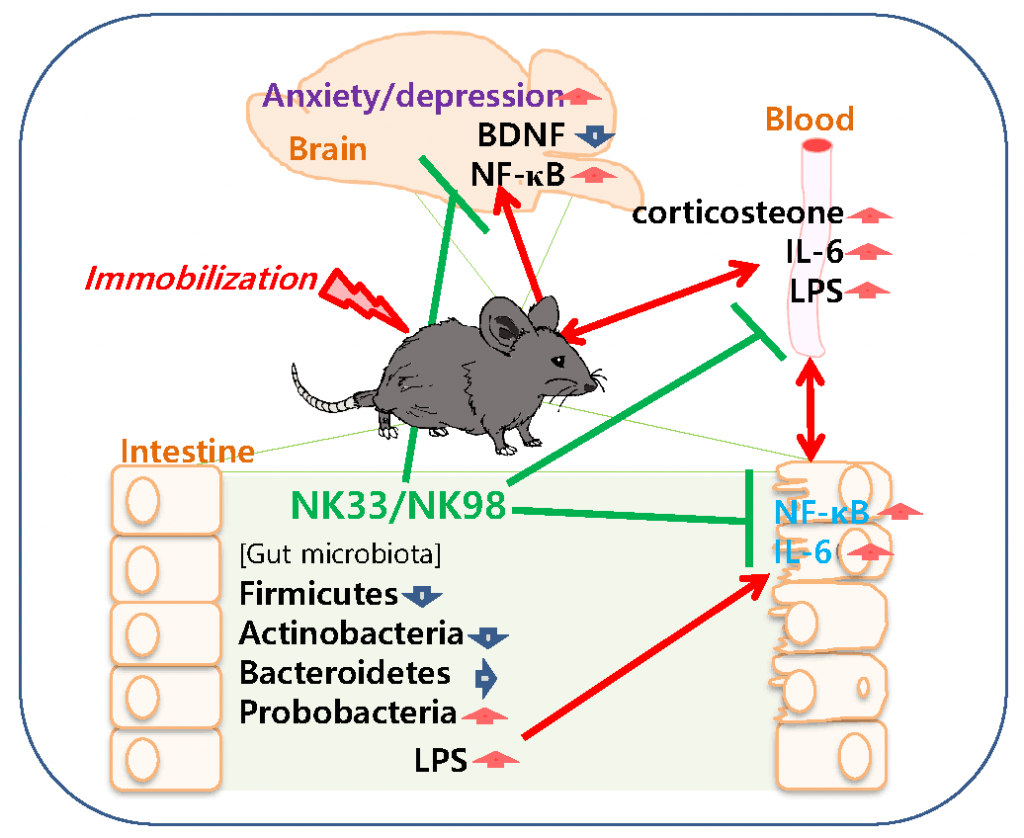
doi: 10.3390/nu11040819.
2
与鼠李糖乳杆菌缓解非酒精性脂肪肝病
肠道菌群失衡与非酒精性脂肪肝病(NAFLD)的发展密切相关。调节肠道菌群可以通过减少有害菌群的比例、调节代谢物水平和降解胆固醇来缓解NAFLD。双歧杆菌和鼠李糖乳杆菌被认为是缓解NAFLD的潜在益生菌。
研究表明,青春双歧杆菌(Bifidobacterium adolescentis)和鼠李糖乳杆菌(Lactobacillus rhamnosus)通过调节不同的肠道微生物群依赖途径来缓解高脂高胆固醇饮食诱导的非酒精性脂肪肝病(NAFLD)。以下是具体的机制:
1.短链脂肪酸(SCFAs)的调节:
青春双歧杆菌的不同菌株通过增加肠道中短链脂肪酸(如乙酸、丙酸、丁酸和异丁酸)的浓度来缓解NAFLD。这些SCFAs的增加有助于改善能量代谢和脂质代谢,减少肝脏脂肪积累,并降低血清中的内毒素水平。
2.肠道微生物群的调节:
青春双歧杆菌和鼠李糖乳杆菌都能通过调节肠道微生物群来缓解NAFLD。研究发现,这些益生菌能够阻止高脂高胆固醇饮食引起的厚壁菌门(Firmicutes)与拟杆菌门(Bacteroidetes)比例的升高,并改变肠道中的优势菌种。
3.能量代谢和脂质代谢的调节:
鼠李糖乳杆菌的不同菌株表现出不同的缓解机制。例如,鼠李糖乳杆菌GG(LGG)通过调节能量摄入来缓解NAFLD,而鼠李糖乳杆菌L7-1则通过增加SCFAs的浓度来缓解NAFLD。鼠李糖乳杆菌L10-1可能通过减少肝脏炎症来缓解NAFLD。
4.抗炎作用:
青春双歧杆菌和鼠李糖乳杆菌都能减轻肝脏炎症。通过降低血清中促炎细胞因子(如TNF-α、IL-1β和IL-6)的浓度,这些益生菌有助于减轻肝脏炎症反应。
5.肠道屏障的保护:
这些益生菌通过增加紧密连接蛋白(如ZO-1和occludin)的转录水平来保护肠道屏障的完整性,减少肠道通透性,从而减少内毒素进入血液的机会。
综上所述,青春双歧杆菌和鼠李糖乳杆菌通过多种机制缓解非酒精性脂肪肝病(NAFLD),包括调节SCFAs的产生、改善肠道微生物群平衡、调节能量代谢和脂质代谢、减轻肝脏炎症以及保护肠道屏障。这些机制共同作用,显示出青春双歧杆菌等益生菌在预防和治疗NAFLD中的潜力。
3
与其他厚壁菌门细菌的协同作用
• 可以增加产短链脂肪酸细菌的丰度
青春双歧杆菌与产丁酸菌等其他厚壁菌门细菌的联合使用也表现出重要的协同效应。青春双歧杆菌产生的醋酸、乳酸和可溶性分子可交叉喂养产丁酸菌。研究表明,青春双歧杆菌补充可以增加肠道中Clostridia(梭菌)的相对丰度,这类细菌是主要的短链脂肪酸(SCFA)产生者。这些产生的SCFAs(主要是丁酸盐)在维持和恢复肠道屏障完整性方面发挥关键作用,通过控制紧密连接蛋白的表达增强肠道屏障功能。
青春双歧杆菌补充还会增加肠道中毛螺菌科(Lachnospiraceae)、粪球菌属(Coprococcus)和梭菌目(Clostridiales)细菌的相对丰度,这些细菌是重要的短链脂肪酸产生者,共同促进肠道健康和免疫调节。
4
在抑制肠道炎症中的协同作用
青春双歧杆菌与其他益生菌在抑制肠道炎症方面表现出显著的协同作用。研究表明,青春双歧杆菌与乳杆菌(如乳酸乳杆菌)的培养上清液共同使用,可通过抑制需氧细菌的不平衡生长和脂质过氧化,有效抑制非甾体抗炎药诱导的大鼠回肠溃疡形成。
此外,青春双歧杆菌与其他双歧杆菌(如Bifidobacterium bifidum)联合使用,可协同增强肠道上皮紧密连接,改善肠道屏障功能。黄酮类化合物(如槲皮素、黄芩素和非瑟酮)可增强青春双歧杆菌产生抗炎物质的能力,表明饮食因素可以进一步增强青春双歧杆菌与其他益生菌的协同抗炎作用。
注:双歧杆菌与乳酸杆菌一起是益生菌补充剂中使用最广泛的细菌之一,它们也被用于酸奶的添加剂。
非瑟酮(Fisetin)是一种天然的黄酮类化合物,属于多酚类物质。它是一种存在于多种水果和蔬菜中的植物色素。
小结
青春双歧杆菌与其他益生菌或益生元协同使用时展现出明显的健康增益效应,这种组合应用方式不仅能够优化肠道菌群结构,还能增强各自的生理功能。随着消费者对肠道健康认知的不断提升,这类配方产品在保健品、功能性食品及医药领域具有广阔的商业价值和市场发展潜力。
青春双歧杆菌与人类健康的关系日益凸显。研究证实,其丰度减少与多种疾病相关,包括炎症性肠病、肠易激综合症、代谢综合征及肌肉减少症等。
作为主要的碳水分解者,青春双歧杆菌能通过代谢并促进产丁酸菌等有益菌群生长,增强肠道屏障功能,调节免疫系统,并产生GABA和叶酸等重要代谢物。值得注意的是,青春双歧杆菌与其他益生菌或益生元协同使用时展现出更显著的健康增益效应,这种组合应用能优化肠道菌群结构,增强各自的生理功能。
随着对其研究的深入,以及更多的青春双歧杆菌功能菌株被发现,未来包含青春双歧杆菌的配方产品在保健品、功能性食品及医药领域具有广阔的应用前景和市场潜力。
主要参考文献:
T. Leser and A. Baker. “Bifidobacterium adolescentis – a beneficial microbe..” Beneficial microbes, 14 6 (2023): 525-551 .
Zhang Z, Guo Q, Yang Z, Sun Y, Jiang S, He Y, Li J, Zhang J. Bifidobacterium adolescentis-derived nicotinic acid improves host skeletal muscle mitochondrial function to ameliorate sarcopenia. Cell Rep. 2025 Feb 25;44(2):115265.
Li B, Wang H, Wang M, Liang H, Hu T, Yang J, Li S, You X, Xia B, Yuan Y, Zou Y, Miao Y, Sun Y. Genome analysis of Bifidobacterium adolescentis and investigation of its effects on inflammation and intestinal barrier function. Front Microbiol. 2025 Jan 22;15:1496280.
Duranti S, Ruiz L, Lugli GA, Tames H, Milani C, Mancabelli L, Mancino W, Longhi G, Carnevali L, Sgoifo A, Margolles A, Ventura M, Ruas-Madiedo P, Turroni F. Bifidobacterium adolescentis as a key member of the human gut microbiota in the production of GABA. Sci Rep. 2020 Aug 24;10(1):14112.
Qian X, Si Q, Lin G, Zhu M, Lu J, Zhang H, Wang G, Chen W. Bifidobacterium adolescentis Is Effective in Relieving Type 2 Diabetes and May Be Related to Its Dominant Core Genome and Gut Microbiota Modulation Capacity. Nutrients. 2022 Jun 15;14(12):2479.
Guo Y, Xie JP, Deng K, Li X, Yuan Y, Xuan Q, Xie J, He XM, Wang Q, Li JJ, Luo HR. Prophylactic Effects of Bifidobacterium adolescentis on Anxiety and Depression-Like Phenotypes After Chronic Stress: A Role of the Gut Microbiota-Inflammation Axis. Front Behav Neurosci. 2019 Jun 18;13:126.
Jang HM, Lee KE, Kim DH. The Preventive and Curative Effects of Lactobacillus reuteri NK33 and Bifidobacterium adolescentis NK98 on Immobilization Stress-Induced Anxiety/Depression and Colitis in Mice. Nutrients. 2019 Apr 11;11(4):819.
Roberts JL, Liu G, Darby TM, Fernandes LM, Diaz-Hernandez ME, Jones RM, Drissi H. Bifidobacterium adolescentis supplementation attenuates fracture-induced systemic sequelae. Biomed Pharmacother. 2020 Dec;132:110831.
Long X, Liu D, Gao Q, Ni J, Qian L, Ni Y, Fang Q, Jia W, Li H. Bifidobacterium adolescentis Alleviates Liver Steatosis and Steatohepatitis by Increasing Fibroblast Growth Factor 21 Sensitivity. Front Endocrinol (Lausanne). 2021 Dec 30;12:773340.
Tan TG, Sefik E, Geva-Zatorsky N, Kua L, Naskar D, Teng F, Pasman L, Ortiz-Lopez A, Jupp R, Wu HJ, Kasper DL, Benoist C, Mathis D. Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. Proc Natl Acad Sci U S A. 2016 Dec 13;113(50):E8141-E8150.

谷禾健康

人体肠道微生物组对消化、免疫调节、代谢平衡、抵抗病原体及整体健康至关重要。在儿童发育中,生命最初两到三年的肠道微生物组发展是影响终生健康的关键期。
出生时几乎无菌的婴儿肠道迅速发展为复杂的微生物生态系统,经历连续的群落更替和成熟。这一过程不仅是对环境的被动适应,更是与宿主生理发育协同的积极进程,对免疫系统和代谢功能的正常发展具有决定性作用。
近年来,随着高通量测序技术的进步,我们对婴儿肠道微生物组的理解已经从描述性分析转向更深入的功能和临床相关性研究。然而,目前关于婴儿微生物组发育轨迹的大规模纵向研究依然有限,尤其缺乏将微生物群落动态变化与远期健康结果相关联的研究。
婴儿微生物组发展能否预测未来健康状况?是否存在”健康”或”最优”的微生物组发展模式?哪些环境因素和早期暴露会影响微生物组的发展方向?

近期发表的一项大规模纵向队列研究或许能解答此问题。研究人员对芬兰赫尔辛基地区967名3周至24月龄婴儿进行肠道微生物组纵向跟踪(谷禾肠道菌群数据库也纳入了该队列数据),旨在揭示其婴幼儿生长发育的规律,并通过建立和评估微生物组健康指数(MWI)探明与其远期健康的关联性。
研究发现肠道微生物组的发展遵循可预测的轨迹,这些轨迹受到出生方式、喂养类型和早期抗生素暴露等因素的调节,并与儿童期的健康状况具有重要关联。通过这项研究,我们期望为理解肠道微生物组在早期生命中的作用提供关键见解,并为指导干预措施优化婴儿肠道微生物组提供科学依据。
★ 研究意义
1.证明肠道微生物群的重要性
2.对早期生活的影响
3.健康预测
★ 研究特点
1.大规模队列:研究使用了一个包含近1000名婴儿的大型纵向队列,监测了从出生到5岁的健康数据。
2.多时间点采样:收集了婴儿前两年内的6203份粪便样本,进行肠道微生物群的分析。
3.微生物群发展轨迹:通过聚类和轨迹建模,识别出五种不同的肠道微生物群发展轨迹。
4.健康指数:创建了一个基于健康发展轨迹的肠道微生物群健康指数,用于评估婴儿的整体健康状况。
5.关键微生物:研究发现双歧杆菌和拟杆菌是早期的关键微生物,指导微生物群的发展,并持续预测积极的健康结果。
6.影响因素:研究分析了多种因素(如分娩方式、饮食、抗生素使用)对肠道微生物群发展的影响。
★ 研究目标
1.描述婴儿前两年内肠道微生物群的发展模式。
2.识别与健康相关的微生物群发展轨迹。
3.创造一个肠道微生物群健康指数,用于预测婴儿的健康风险。
本研究旨在提供对婴儿肠道微生物群发展的深入理解,说明肠道微生物群在预测健康方面起着重要的作用,并为改善婴儿健康提供新的工具和方法。
研究使用了来自芬兰赫尔辛基的一个纵向出生队列,共收集了984名婴儿的粪便样本和健康数据。样本在婴儿3周、6周、3个月、9个月、12个月和24个月时采集。
儿童健康、发育和健康相关数据是从父母填写的问卷中收集的,由两种类型的变量组成:
(a)2岁时医生诊断的过敏性疾病,或5岁时确诊的特应性皮炎、过敏性鼻炎或哮喘;表型过敏,即基于ISAAC问卷的上述疾病在2或5岁时的症状;2或5岁时按世卫组织标准的异常身高或体重(>2或<-2SD);下呼吸道和上呼吸道感染、胃肠感染、耳部感染、痘病毒感染或发热的发生率。罕见感染(N<5)被排除。
(b)正常变异和主观评估:2或5岁时轻度异常身高或体重(>1或<-1SD);父母用VAS量表(0-100mm)对儿童健康状况较低评分;2岁前胃肠功能(排便、胃痛、胀气)表现;3个月龄时基于哭闹时间的婴儿分类。
•纵向出生队列
-采用纵向队列研究设计,跟踪婴儿从出生到2岁,甚至到4-5岁,以观察肠道微生物群随时间的变化。
•样本收集
在婴儿的3周、6周以及3、9、12、18和24个月时收集粪便样本。
•微生物组测序
对粪便样本进行16S rRNA基因测序,以分析肠道微生物群的组成。
•数据分析
-使用统计软件(如R语言)进行数据分析。
-应用线性混合模型(LMM)来分析微生物群随时间的变化。
-使用机器学习算法(如随机森林)来识别与过敏性疾病相关的微生物特征。
•问卷调查
通过在线问卷收集关于母亲和婴儿的信息,包括生活方式、环境暴露、儿童饮食、健康等。问卷在多个时间点进行,以跟踪变化。
•过敏性疾病诊断
根据国际疾病分类(ICD-10)代码或医生诊断记录来确定过敏性疾病的发生。包括食物过敏、哮喘、湿疹和过敏性鼻炎。
•统计分析
-使用多变量回归分析来评估肠道微生物群与过敏性疾病之间的关系。
-考虑潜在的混杂因素,如母亲的过敏史、抗生素使用、分娩方式等。
通过这些综合方法,能够全面评估婴儿肠道微生物群的发展轨迹,并探索其与过敏性疾病风险及健康结果之间的关联。
1
影响婴儿肠道微生物组的因素
在肠道微生物群组成中观察到一个明显的年龄梯度,婴儿肠道微生物群在生命的前两年逐渐接近,但没有达到成人样组成。
在最初的6个月(26周),婴儿肠道微生物群的组成在个体之间差异很大,但此后趋于一致。
984名婴儿的肠道微生物群
doi: 10.1038/s41467-024-52561-6.
注:主坐标分析展示了对数转换微生物属相对丰度的Pearson相关距离。图中不同颜色代表3至104周的婴儿年龄,父母微生物群以黑色表示。图b和图c分别显示PC组件2和PC组件1与年龄和分娩方式的关系,组中值用大圆圈标识。
肠道菌群组成的最重要决定因素是前26周的出生方式、抗生素暴露、第一年的排便率以及1至2岁(52-104周)的饮食和家庭组成。母亲特征在所有时间点都有适度且一致的影响,并且母亲微生物群组成在26周时变得有影响力,随着时间的推移而增加。
婴儿肠道微生物组的影响因素
doi: 10.1038/s41467-024-52561-6.
2
婴儿微生物群落类型
研究者使用K-means聚类和log-pearson距离方法,在属水平上对婴儿样本进行分析,确定了四种主要的微生物群落类型(C1-C4),这些类型随着婴儿年龄发展而呈现规律性变化。
•C1型(绿色)
主导菌群:双歧杆菌属(39.2%)和拟杆菌属(12.8%);
特点:放线菌门和拟杆菌属成员覆盖超过50%的相对丰度;
时间分布:主要在婴儿生命的前26周出现;
常见于:未接触抗生素的阴道分娩婴儿;
粪便特征:多为黄色粪便。
•C2型(蓝色)
主导菌群:几乎没有双歧杆菌(4.8%),梭菌科(13.4%)和肠杆菌科(25.7%)相对丰度高;
特点:微生物丰富度最低,潜在致病菌相对丰度最高;
时间分布:主要在婴儿生命的前26周出现;
常见于:剖腹产婴儿和接触抗生素的阴道分娩婴儿;
粪便特征:相比C1更容易出现绿色粪便。
•C3型(橙色)
主导菌群:双歧杆菌科(27.3%)、乳杆菌科(18.5%)和韦荣球菌科(20.1%);
时间分布:39-52周(9-12个月)时常见;
常见于:大多数婴儿在9个月左右;
粪便特征:棕色。
•C4型(红色)
主导菌群:毛螺菌科(30.0%)和瘤胃球菌科(30.0%)为主;
特点:微生物丰富度最高;
时间分布:52周(12个月)后常见;
粪便特征:多为棕色粪便,反映固体食物增加。
微生物群落类型
doi: 10.1038/s41467-024-52561-6.
微生物丰富度存在差异
微生物丰富度在群落类型之间差异很大,C4最高,C2最低,并且通常显示与婴儿年龄的相关性越来越大。潜在致病菌的相对丰度在C2中最丰富。
群落决定因素和健康关联
doi: 10.1038/s41467-024-52561-6.
(a)微生物群落类型决定因素的分区树;
(b)使用负广义线性模型分析了不同年龄段微生物群落类型之间的时间演变及其与5岁时健康结果的关联。
不同微生物类型与健康结果相关
分析各时间点微生物群落类型与2岁和5岁健康结果的关联发现,C2型与不良健康结果尤其是过敏性疾病风险增加相关。
6个月前属于C3型的儿童过敏性疾病风险增加,5岁时身高Z评分低于-1标准差。12个月前过渡到C4型与2岁时身高Z评分低于-1标准差相关,但C4型与2岁时哮喘诊断呈负相关。12个月时,C1型与胃肠道感染有关。
3
微生物组发展轨迹
婴儿肠道微生物组发展高度可预测,遵循五种轨迹之一。
T1轨迹(最常见,47%)
特征:前6个月C1成员稳定,9个月过渡到C3,12-18个月过渡到C4;
菌群:这些婴儿的双歧杆菌的初始相对丰度很高,其相对丰度逐渐下降,最初被韦荣氏球菌属取代,然后被粪杆菌和毛螺菌科的成员所取代;
相关因素:未接触抗生素的阴道分娩、母乳喂养、有兄弟姐妹;
健康关联:与较好的健康结果相关;
微生物稳定性:最稳定的微生物群组成。
T2轨迹(11%)
特征:最初位于C1,但在转移到C3之前转移到C2;
菌群:双歧杆菌迅速减少,梭菌和克雷伯菌短暂增加;
微生物稳定性:9个月大时表现出最大的波动性
T3轨迹(9%)
特征:开始于C1,但在前6个月在C1和C2之间反复振荡;
微生物稳定性:3-12周时表现出最高的波动性
T4轨迹(18%)
特征:T3的反向模式,从C2开始的婴儿在前6个月在C1和C2之间波动,在6-9个月时双歧杆菌达到峰值。
T5轨迹(14%)
特征:前6个月持续处于C2状态;
菌群:梭菌和克雷伯氏菌相对丰度较高。
微生物群发展轨迹、决定因素和与健康结果的关联
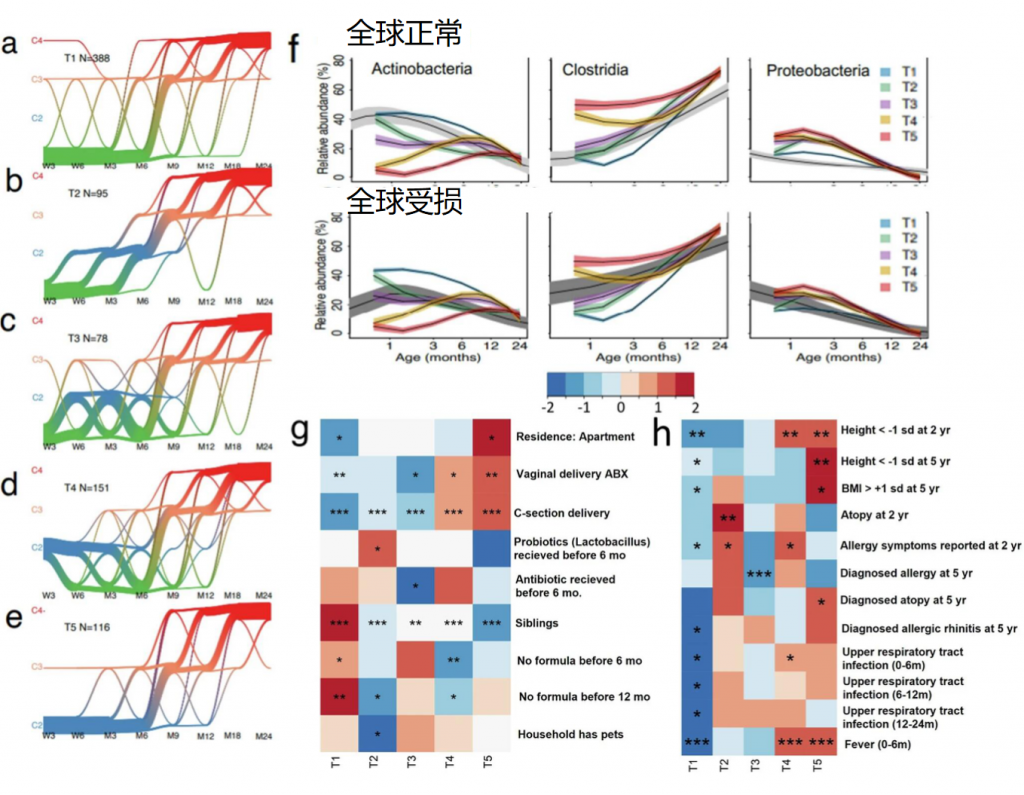
doi: 10.1038/s41467-024-52561-6.
影响微生物发展轨迹的因素:
轨迹T1-T3与阴道分娩相关,T4和T5则与剖腹产及阴道分娩预防性抗生素使用相关。T1独特地与有兄弟姐妹、住单户住宅和前12个月纯母乳喂养相关。
T2婴儿向C2型过渡可能由配方奶喂养或无兄弟姐妹促成,这些婴儿更常接受益生菌。T3中C1和C2间波动主要与缺乏兄弟姐妹及较低社会经济地位相关。T4的微生物群自发校正可能由母乳喂养或其他高社会经济地位相关因素驱动。
不同微生物轨迹的健康结果:
T1与多项健康指标呈负相关:前2年过敏症状和上呼吸道感染风险降低,0-6个月发烧报告减少,5岁时过敏性鼻炎诊断减少,5岁时ISO-BMI Z评分不超过1标准差,2岁和5岁时身高Z评分不低于-1标准差。
T2与2岁时特应性风险和前2年父母报告的过敏症状增加相关。
T3与2岁时父母报告的过敏症状减少有关。
T4和T5均与2岁时身高Z评分低于-1标准差,以及0-6个月间上呼吸道感染和发热相关。由于T4的微生物群校正,这些婴儿5岁时未表现出T5所见的生长改变或过敏性鼻炎风险增加。T5还与5岁时特应性风险增加相关。
总体而言,微生物组轨迹与健康结果的相关性强于单个时间点的微生物组类型,表明纵向发育分析提供更多信息。
与年龄相关的细菌属:
数据分析中,我们识别出一组随年龄变化的细菌属。这些分类群在各发育轨迹中展现相似模式,主要分为早期(放线菌门、拟杆菌属、肠杆菌门、Negativicutes、Bacilli)和晚期(主要是梭状芽胞杆菌)两组。
然而,某些关键类群,如双歧杆菌属和拟杆菌属,在不同的轨迹中表现出不同的时间模式。
4
微生物组健康指数
微生物组健康指数(MWI)是研究者基于大型婴儿队列(近1000名婴儿)的健康发展轨迹开发的一种评估指标,用于预测婴儿整体健康状况。该指数代表了基于微生物群落组成估计婴儿属于健康参考组的概率。
★ 关键有益指示菌:
最强的总体积极关联见于双歧杆菌(Bifidobacterium)和拟杆菌(Bacteroides),这两类菌群一致地指示健康参考微生物组。
随年龄增加积极关联增强的菌群:Eisenbergiella, Oscillibacter, Parabacteroides, Anaerostipes, Streptococcus。
★ 负面指示菌:
随年龄增加负面关联增强的菌群:Lachnospira, Faecalicatena, Lacrimispora, Klebsiella, Sutterella。
★ 年龄依赖性:
大多数指示菌显示年龄依赖性关联;
某些菌群展示暂时性负面关联(Roseburia, Faecalibacterium);
某些菌群展示暂时性正面关联(Citrobacter, Blautia, Gemmiger, Hungatella)。
微生物群健康指数
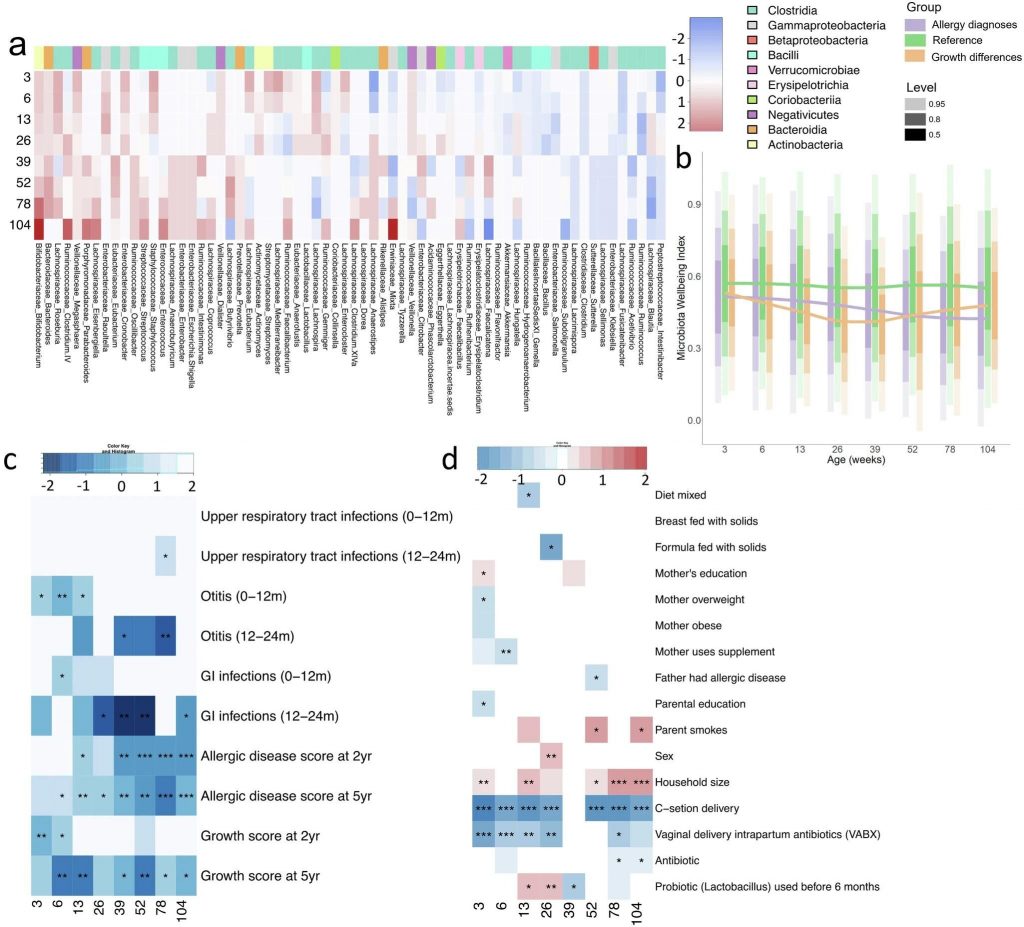
doi: 10.1038/s41467-024-52561-6.
★ 健康预测能力:
MWI在患有过敏性疾病或生长差异的婴儿中显著降低;
在详细分析中,MWI与多种健康结果相关,包括从过敏性疾病到2岁和5岁时的生长指标;
可预测感染发生率。
★ MWI的优势:
整体评估:超越单个菌群评估,考虑整个微生物群落结构对宿主健康的影响;
发展视角:将肠道菌群演替视为婴儿生理发育的一部分,与免疫系统成熟有关;
预测能力:可预测前5年的总体健康状况,有助于早期识别健康风险;
实用性:相比成熟度指数,能更好地区分不同的发育轨迹和捕捉各种健康关联。
通过对近1000名婴儿的前瞻性纵向队列研究,首次系统阐明了人类婴儿肠道微生物组发展的规律性和可预测性。确定了四种主要的微生物群落类型(C1-C4),这些类型随着婴儿年龄发展而呈现规律性变化。并且不同微生物类型与健康结果相关。
婴儿肠道微生物组发展遵循五种主要轨迹的预设路径,非随机发生,并受出生方式、喂养类型及抗生素使用等因素显著影响。研究确立了双歧杆菌和拟杆菌作为关键早期定植微生物,它们在引导整个微生物组健康发展中起着决定性作用。
基于这些发现,建立的微生物组健康指数(MWI)不仅能够评估婴儿当前肠道微生物组的健康状态,还能预测未来5年的整体健康结果,包括过敏性疾病风险、生长发育状况和感染易感性。这一指数的建立将微生物组分析从描述性研究提升至具有临床预测价值的工具,为精准医疗提供了新的维度。
需要注意的是,该研究仅使用了来自芬兰地区的婴幼儿样本,其开发的微生物健康指数(MWI)目前仅适用于北欧地区的婴幼儿样本,更大范围的应用还需要纳入更多地区和年龄的样本。
主要参考文献:
Hickman B, Salonen A, Ponsero AJ, Jokela R, Kolho KL, de Vos WM, Korpela K. Gut microbiota wellbeing index predicts overall health in a cohort of 1000 infants. Nat Commun. 2024 Sep 27;15(1):8323.

谷禾健康

在当代生命科学研究领域,微生物学以其广泛的应用前景和深远的理论意义持续引领着科学前沿。随着技术的迅猛发展,微生物学研究已从传统的形态学描述拓展至分子机制解析、微生物组学、系统生物学等多维度探索。
在这一背景下,高质量学术期刊作为知识传播与学术交流的重要平台,对推动学科发展具有不可替代的作用。
本文汇总了微生物领域常见且有影响力的26个期刊,以下是期刊的名称和一些描述及其网站链接和基于经验和GPT给出的投稿建议,希望可以微生物领域的大家投稿决策提供一些参考。
影响因子: 69.2
期刊特色:Nature旗下顶级综述期刊,发表微生物学各领域的权威综述。
网址:www.nature.com/nrmicro/
投稿建议:
– 主要接受邀请稿,自主投稿接受率极低
– 综述需覆盖领域前沿进展,8000-10000字
– 图表要求高度专业和艺术性
– 建议先与编辑邮件沟通投稿意向

影响因子: 20.6
期刊特色:Cell出版集团期刊,专注微生物与宿主相互作用研究。
网址:
www.cell.com/cell-host-microbe/home
投稿建议:
– 偏好机制创新性研究,尤其是病原菌-宿主互作
– 多组学整合研究受青睐
– 临床相关性研究更具竞争力
– 文章结构需严格遵循Cell系列风格

影响因子: 20.5
期刊特色:发表微生物学领域具有重大突破的研究论文。
网址:www.nature.com/nmicrobiol/
投稿建议:
– 研究必须具有原创性和重大科学意义
– 数据质量和完整性要求极高
– 通常需要3-4轮修改
– 附信(Cover Letter)非常重要,需清晰说明创新点和影响力

影响因子: 19.1
期刊特色:发表高质量、全面的微生物学领域综述。
网址:cmr.asm.org/
投稿建议:
– 多为邀请稿,自荐需提前联系编辑
– 综述需系统性强,聚焦于当前知识状态,并提供对未来研究方向的见解
– 选择与临床微生物学家和感染病专家相关的主题,关注具有临床应用价值的研究领域
– 参考文献需全面且新近
影响因子: 14.0
期刊特色:是 Cell Press 旗下的一本高影响力综述期刊,为微生物学的各方面讨论提供了一个多学科论坛:从细胞生物学和免疫学到遗传学和进化,范围涵盖病毒学、细菌学、原生动物学和真菌学。
网址:
www.cell.com/trends/microbiology/home
投稿建议:
– 采用预投稿咨询制度
– 全面分析特定微生物学主题的最新研究进展,提供对现有知识的整合性视角
– 对微生物学领域特定问题提出新颖视角或整合分析
– 加入批判性分析,而非仅罗列已知事实
– 稿件将经过严格的同行评审,评审重点包括科学准确性、时效性和写作质量

影响因子: 13.8
期刊特色:专注微生物组研究,包括人体、动物和环境微生物组。
网址:
microbiomejournal.biomedcentral.com/
投稿建议:
– 大型队列研究或多组学整合分析优先
– 必须有规范的生物信息学分析流程
– 方法学创新或领域新概念更易接受
– 开放获取期刊,版面费约2800美元
影响因子: 12.2
期刊特色:专注于肠道微生物研究的国际权威期刊、高影响力、国人友好
网址:www.tandfonline.com/toc/kgmi20/current
投稿建议:
– 明确研究如何填补领域空白,聚焦肠道微生物与宿主互作机制,突出临床转化价值
– 实验设计严谨,若涉及临床样本,需说明伦理审批和队列代表性
– 优化语言与图表呈现,规避格式雷区
– 采用单盲同行评审,审稿平均3-5个月,避开6-8月投稿高峰
影响因子: 10.8
期刊特色:国际微生物生态学会官方期刊,微生物生态学领域权威。
网址:www.nature.com/ismej/
投稿建议:
– 环境微生物学和生态学研究是核心
– 新型生态模型或概念框架受欢迎
– 技术文章需展示实际生态学应用
– 数据共享和透明度要求高

影响因子: 10.1
期刊特色:欧洲微生物学联合会综述期刊。
网址:academic.oup.com/femsre/
投稿建议:
– 接受约稿和自荐稿件
– 综述需有明确主题和系统性
– 欧洲地区研究者接受率略高
– 审稿严格,修改要求详尽

影响因子: 8.5
期刊特色:是微生物学领域的排名靠前的期刊之一,专门发表综述性文章的同行评审学术期刊。
网址:
www.annualreviews.org/journal/micro
投稿建议:
– 采用”邀请制”,编辑会直接联系被选中的专家学者发出邀请
– 综述应全面涵盖所讨论主题的关键方面、作用方式、最新进展和临床应用
– 不仅总结现有研究,还应提供对文献的分析和见解
– 指出研究领域的未来方向和挑战
影响因子: 8.2
期刊特色:发表有关从业者和研究人员感兴趣的各种主题文章。主题范围从感染的临床描述、公共卫生、微生物学和免疫学到感染的预防、当前和新疗法的评估以及诊断和治疗最佳实践的推广。
网址:academic.oup.com/cid
投稿建议:
– 关注具有临床相关性的传染病研究
– 强调研究结果对临床实践的潜在影响,选择新颖且有实用价值的研究方向
– 简洁明了,避免冗长叙述
– 直接引用与主题高度相关的参考文献
– 图表说明应清晰且独立于正文仍能理解

影响因子: 8
期刊特色:权威综述期刊,侧重分子微生物学。
网址:journals.asm.org/journal/mmbr
投稿建议:
– 几乎只接受邀请稿
– 综述极为全面,通常10000字以上
– 需包含历史背景、现状和未来展望
– 图表需原创且高质量

影响因子: 6.7
期刊特色:发表关于原核和真核微生物的研究,如酵母、真菌、细菌、古菌和生动物。也涵盖了病原微生物与其环境或宿主之间相互作用的研究。
网址:
www.sciencedirect.com/journal/microbiological-research
投稿建议:
– 详细展示具有创新性的微生物学研究成果,特别欢迎具有创新性和多学科性质的研究
– 注重研究的应用前景和跨学科特性
– 清晰呈现实验结果,避免重复描述图表内容
– 投稿成功率很高,文章审稿大多在1-3个月左右,最快的审稿周期仅需7天,审稿效率较高。

影响因子: 6.1
期刊特色:是临床微生物学领域的权威期刊。重视具有临床相关性的微生物学研究,包括诊断方法的开发与评估、抗微生物药物敏感性测试、分子诊断等多个方向。
网址:journals.asm.org/journal/jcm
投稿建议:
– 强调研究的临床相关性和实用价值,关注能直接影响临床微生物实验室工作的研究
– 确保研究设计合理,方法学严谨
– 清晰阐述研究的临床意义,讨论研究结果如何影响临床实践,避免纯理论研究
– 高质量的实验设计、充分的数据支持以及清晰的写作

影响因子: 6.0
期刊特色:该期刊专注于发表微生物学领域的系统性综述与批判性分析,涵盖病原微生物、环境微生物及工业微生物等方向。
网址:
www.tandfonline.com/journals/imby20
投稿建议:
– 选择具有时效性和重要性的微生物学主题
– 对微生物学特定领域的全面且批判性综述,应提供深入分析而非仅是文献汇编
– 包含对研究领域的发展、现状和未来方向的评述
– 准备封面信,简要介绍文章的意义和创新点
– 高质量的图表和流畅的语言表达,保持逻辑连贯性和论证深度
影响因子: 5.9
期刊特色:权威观点综述期刊,主题鲜明。
网址:
www.sciencedirect.com/journal/current-opinion-in-microbiology
投稿建议:
– 主要为邀请稿,按特定主题组织
– 综述篇幅适中,4000-6000字
– 强调个人观点和前沿展望
– 图表需原创且概念清晰

影响因子: 5.2
期刊特色:ISME Journal的姊妹期刊,开放获取。
网址:
academic.oup.com/ismecommun?login=true
投稿建议:
– 接受范围与ISME Journal类似但门槛略低
– 审稿速度较快,一般4-6周
– 适合具有一定创新性的微生物生态学研究
– 数据要求与ISME Journal一致

影响因子: 5.1
期刊特色:美国微生物学会旗舰期刊,覆盖面广。
网址:journals.asm.org/journal/mbio
投稿建议:
– 接受范围广,适合创新性但未达Nature级别的研究
– 审稿速度相对较快,一般6-8周
– 开放获取,ASM会员有折扣
– 编辑决策通常基于2位审稿人意见

影响因子: 5
期刊特色:系统微生物学研究的专业期刊。
网址:journals.asm.org/journal/msystems
投稿建议:
– 系统生物学和多组学研究是核心
– 计算方法和模型构建研究优先
– 大数据分析和可视化要求高
– 开放获取,审稿相对快速

影响因子: 4.3
期刊特色:环境微生物学领域的权威期刊。
网址:
enviromicro-journals.onlinelibrary.wiley.com/journal/14622920
投稿建议:
– 环境样本微生物研究,功能比描述重要
– 生态功能和环境适应机制研究优先
– 创新性分析方法更受欢迎
– 可接受Special Issue投稿,提高接受率

影响因子: 4.2
期刊特色:专注真菌学研究的开放获取期刊。
网址:www.mdpi.com/journal/jof
投稿建议:
– 真菌病原性和抗性研究优先
– 真菌分类学和生态学研究也受欢迎
– 方法学创新是加分项
– 版面费较高
影响因子: 4
期刊特色:微生物基因组学研究专业期刊。
网址:
www.microbiologyresearch.org/content/journal/mgen
投稿建议:
– 基因组学和比较基因组学研究为主
– 基因组功能注释和演化研究受欢迎
– 大数据分析方法需创新
– 开放获取,版面费约2000英镑
影响因子: 4
期刊特色:开放获取期刊,分区明确。
网址:
www.frontiersin.org/journals/microbiology
投稿建议:
– 选择合适的分区(section)投稿
– 接受率相对较高,但质量要求仍严格
– 审稿速度快,一般1-2月出结果
– 版面费约2950美元
影响因子: 3.9
期刊特色:微生物学应用研究的重要平台。
网址:journals.asm.org/journal/aem
投稿建议:
– 应用导向研究,解决实际问题
– 方法学必须详尽可重复
– 工业和环境应用研究优先
– 数据分析需规范,统计方法恰当

影响因子: 2.7
期刊特色:美国微生物学会期刊,专注细菌学研究。
网址:journals.asm.org/journal/jb
投稿建议:
– 细菌生理、代谢和遗传学研究是主体
– 基础研究为主,机制解析清晰
– 方法学部分要详尽,便于重复
– 审稿周期适中,通常2个月内

影响因子: 2.6
期刊特色:关注病原微生物与宿主互作。
网址:
www.sciencedirect.com/journal/microbes-and-infection
投稿建议:
– 感染性疾病机制研究优先
– 免疫应答研究是重点
– 临床相关性强的研究更具竞争力
– 审稿周期一般在2-3个月

影响因子: 2.6
期刊特色:专注微生物分子机制研究。
网址:
onlinelibrary.wiley.com/journal/13652958
投稿建议:
– 分子水平机制研究是核心
– 实验设计需严谨,多角度验证结论
– 遗传学和分子生物学方法需现代化
– 与模型微生物相关研究更易接受
