-

 CNAS L23010
CNAS L23010

 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室

谷禾健康 原创
最近, 国际顶级方法学期刊《Nature Methods》发表了由加利福尼亚大学圣地亚哥分校儿科、加州大学计算机科学与工程系 、加州大学合作质谱创新中心以及加利福尼亚大学圣地亚哥分校微生物群创新中心 多学科合作研究的最新成果:“Learning representations of microbe–metabolite
Interactions(可用于分析微生物与代谢产物之间相互作用的人工神经网络)”,此项研究恢复微生物与代谢物之间关系的能力,并证明了该方法如何发现微生物产生的代谢产物与炎症性肠病之间的关系。


摘要
研究人员表示,整合多组学数据集对于微生物组研究至关重要。但是,推断整个组学数据集之间的交互具有多种统计学上的挑战。文章中通过使用神经网络(https://github.com/biocore/mmvec)来解决了此问题,其能够在存在特定微生物的情况下估算每个分子存在的条件概率。研究人员以已知的环境(沙漠土壤湿润生物结壳)和临床(囊性纤维化肺)实例为例,展示了这一方法恢复微生物与代谢物之间关系的能力,并证明了该方法如何发现微生物产生的代谢产物与炎症性肠病之间的关系。
背景
虽然已经有广泛的努力来开发整合多组学数据的方法,但一些概念上的挑战限制了整合不同组学数据的技术,例如,将微生物测序和非靶向质谱联系起来。因此,需要新的方法来处理不同的数据类型。为此,研究人员提出了“mmvec”(微生物-代谢物载体),一种神经网络,可以从单个微生物序列预测整个代谢物丰度曲线。通过迭代训练,mmvec可以学习微生物和代谢物之间的共现概率。微生物-代谢物相互作用可以通过标准的降维界面进行排序和可视化,从而产生可解释的结果。
主要结果
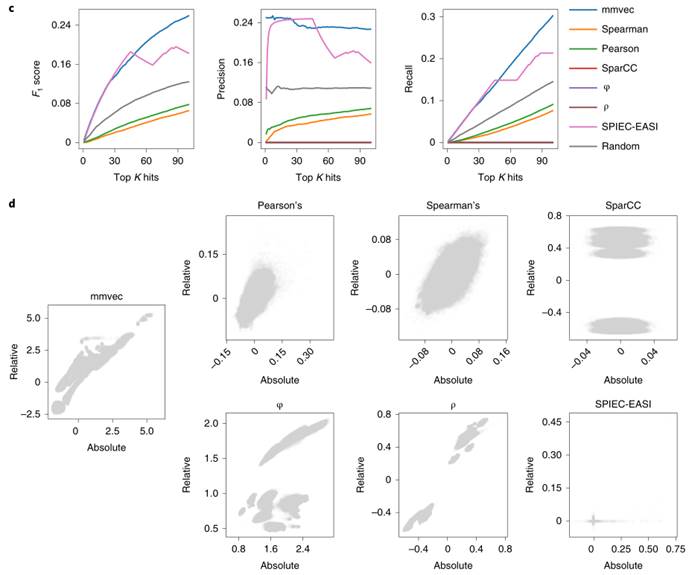
1.使用模拟囊性纤维化生物膜的数据集,将mmvec与Pearson’s、Spearman’s、SPIEC-EASI、SparCC和proportionality方法进行基准比较。证明了mmvec优于所有旨在推断成对微生物-代谢物丰度数据集之间相互作用的现有工具。
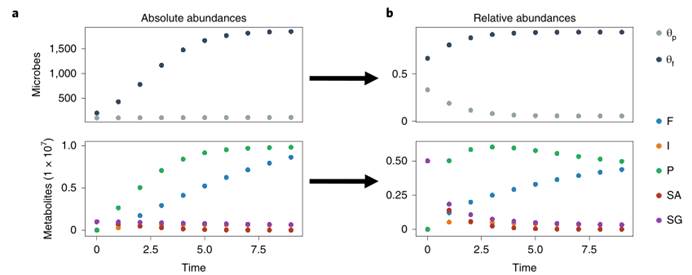
图a.两个微生物和多个分子之间的相互作用被模拟成单分子动力学和扩散过程,(发酵剂由θf表示,铜绿假单胞菌由θp表示)从推导的微分方程模拟的微生物和代谢物的绝对丰度,图b. 为图a.中绝对丰度的比例。这里模拟了五种代谢物,即糖(SG)、抑制剂(I)、酸(F)、铵(P)和氨基酸(SA)
图c. 在每种微生物的前100个代谢产物中,使用 F1 score、precision(精确率)和recall curves(召回率)比较了mmvec与Pearson’s、Spearman’s、SparCC、SPIEC-EASI(生态关联的稀疏逆协方差估计与统计推断)、比例度量(φ和ρ)。图中表示mmvec和SPIEC-EASI的随机表现(Random)优于其它所有工具,其中mmvec表现最好。
图d.从绝对丰度和从所有基准测试方法获得的相对丰度的系数比较。图中显示mmvec是唯一对比例偏差具有鲁棒性的方法。这对于保持绝对丰度和相对丰度之间的一致性至关重要,否则可能导致虚报假阳性和假阴性。
2.沙漠土壤生物润湿事件,测试mmvec是否可以解决微生物-代谢物相互作用中无法解释的差异。结果是mmvec解决了阴道分枝杆菌释放的体外验证代谢物与环境样品的测序和质谱分析之间的冲突发现。
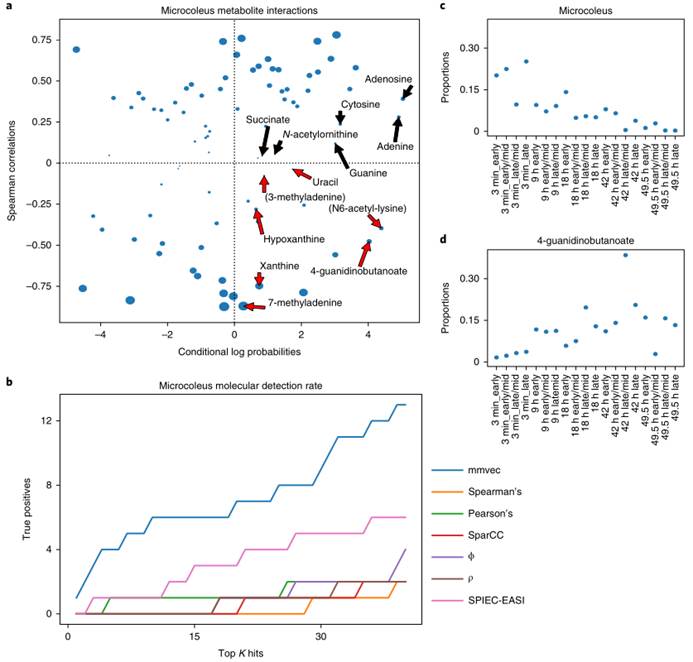
图a. 阴道分枝杆菌-代谢物相互作用的比较,根据Spearman‘s和mmvec估计(n = 19个样品)。由阴道分枝杆菌释放的所有经实验验证的代谢物都被标记。所有与生物润湿实验结果和体外实验结果相矛盾的代谢物都用红色突出显示。Spearman‘s标记的13个标签中有7个具有负相关性,表明这些分子被阴道分枝杆菌消耗而不是释放。
图b.经实验验证的分子在不同统计方法中的检测率的基准比较。mmvec具有相当高的真阳性率。
图cd. 阴道分枝杆菌(c)和4-胍丁酸(d)在生物润湿事件后的比例
MMVEC和Spearman‘s之间的冲突结果可以用生物润湿后微生物生物量的增长(c)和可用资源(d)的转移来解释。
3. 囊性纤维化患者的肺粘液微生物组研究,进一步验证mmvec是否可以检测已知的微生物-代谢物相互作用。结果表明mmvec可以可靠地识别由铜绿假单胞菌产生的所有经实验验证的感兴趣的分子
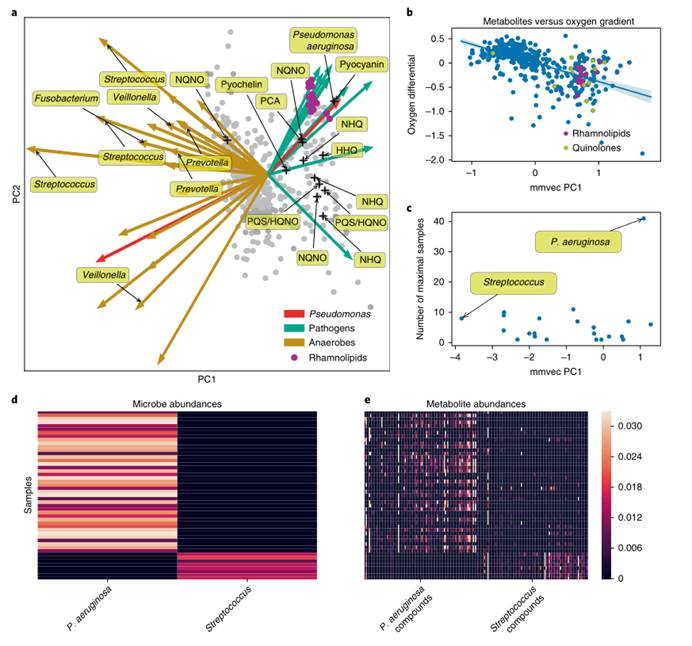
图a.依据mmvec在囊性纤维化数据集中估计的条件概率做的双标图。箭头代表微生物,圆点代表代谢物。x轴和y轴表示由mmvec (n = 138个样本)估计的微生物代谢产物的条件概率的奇异值分解(SVD)的主成分(PCs)。点之间的距离量化了代谢物之间的共现强度,较小的距离表明代谢物有很高的共现概率。箭头尖端之间的距离可以量化微生物之间的共现强度。箭头的方向性可以用来确定哪些微生物可以解释代谢产物的共现模式。绿色箭头表示推测的囊性纤维化病原体,黄色箭头表示已知的厌氧菌。只有铜绿假单胞菌产生的已知分子被标记。mmvec清楚地分离了厌氧菌和病原体,左侧是已知的厌氧微生物,右侧是显著的病原体。
图b.从mmvec学习到的第一主成分与代谢物在氧梯度上的对数倍数变化之间存在负相关 ( Pearson‘s r=−0.59,P=1.8×10−44,n=442个分子)。Pearson‘s法未发现氧梯度与第一微生物主成分之间的这种相关性(r=0.11,P=0.16,n=138个分子)。
图c. 第一主成分与样本数量的关系,其中分类群是该样本中最丰富的分类群。
图d. 铜绿假单胞菌和链球菌最丰富的样品的热图(log ratio t test = 6.51, P = 4.4 × 10−8, n = 49 个样本)。这提供了证据表明,在本研究的背景下,代谢组学特征在很大程度上受到最丰富的微生物的影响。
图e. 与铜绿假单胞菌和链球菌共生的前100个代谢物分子的热图。图中表示仅是预测铜绿假单胞菌代谢物谱就可以解释这些样品中10%的代谢物变异(r = 0.319, P = 1.18×10−11,n = 442个分子)。
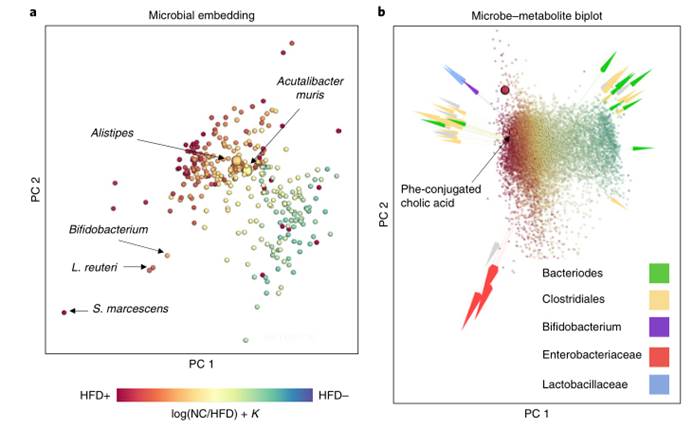
4. 胆汁酸研究。证明mmvec能够在复杂的生物系统中进行探索性分析,并简化特定代谢物的微生物来源的发现
图a. 微生物共生模式的可视化,其中点之间的距离近似于微生物之间的Aitchison距离,它量化了微生物发生的情况。较小的距离表明微生物具有很高的共生概率。微生物根据它们与HFD(高脂肪饮食)的关联被着色,这是通过多项式回归用差异丰度分析估计的。mmvec的使用显示了与HFD相关的不同微生物群
图b. 微生物-代谢物相互作用的双图,代谢物根据它们与HFD的关联而着色。HFD关联性通过多项式回归的差异丰度分析进行估计。点之间的距离近似代谢物之间的Aitchison距离,箭头间距近似微生物之间的Aitchison距离。表明mmvec根据饮食对质谱数据进行了清晰的分层。
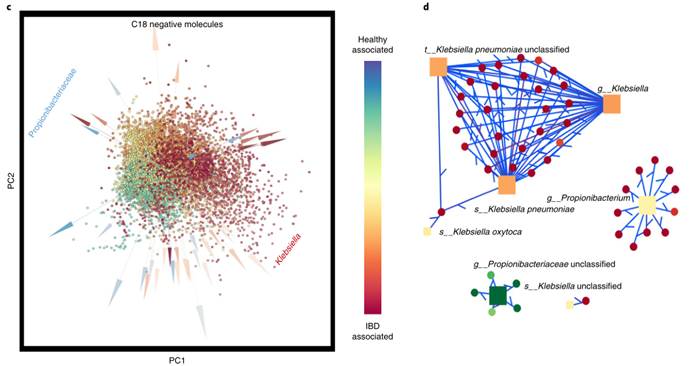
5.炎症性肠病中微生物-代谢产物的相互作用。结果表示mmvec能够确定IBD研究中对代谢物丰度最强的微生物贡献,并发现了在最初的研究中被遗漏的一种微生物(Propionibacterium)
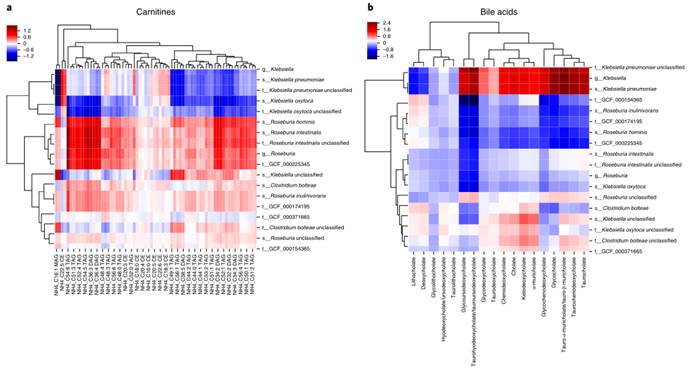


图a.和图b.分别为在三种菌属Klebsiella,Roseburia和Clostridium bolteae存在的情况下,推断各种胆汁酸(a)和肉碱(b)的条件概率的热图
图c.从宏基因组的概要文件和C18负离子模式LC-MS中学习到的微生物-代谢物相互作用的多组学双图。微生物(箭头)和代谢物(球体)根据多项式回归估计的差异着色。Klebsiella似乎与IBD密切相关,而Propionibacterium有强烈的负相关。
图d. 前300条边缘的网络,只有包含Klebsiella和Propionibacterium的边缘可见。
结论
作者表示,鉴于这些发现,目前的方法仍有局限性。目前还不清楚如何使用共现概率来获得相互作用的统计意义。同样,还不能计算每个微生物-代谢物相互作用强度的置信区间,还需要理论工作来处理连续值的输入。
Tips:文章末尾有关于mmvec算法的推导公式。