-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

16S rRNA 基因是编码原核生物核糖体小亚基的基因,长度约为1542bp,其分子大小适中,突变率小,是细菌系统分类学研究中最常用和最有用的标志。
16S rRNA基因序列包括9个可变区和10个保守区,保守区序列反映了物种间的亲缘关系, 而可变区序列则能体现物种间的差异。
16S rDNA是细菌染色体上编码16SrRNA相对应的DNA序列, 16S rRNA是由16s rDNA转录来的 , 一般扩增检测和分析的对象都是16s rDNA 。
16S rRNA基因测序以细菌16S rRNA基因测序为主,核心是研究样品中的物种分类、物种丰度系统进化、功能预测以及微生物与环境互作关系等。
粪便、动物肠道内容物、皮肤、组织、痰液、血液、唾液、牙菌斑、尿液,阴道分泌物、发酵物,瘤胃,废水,火山灰,冻土层、病害组织、淤泥、土壤、堆肥、污染河流,养殖水体、空气等有微生物存在的样本都可以用于16s测序分析
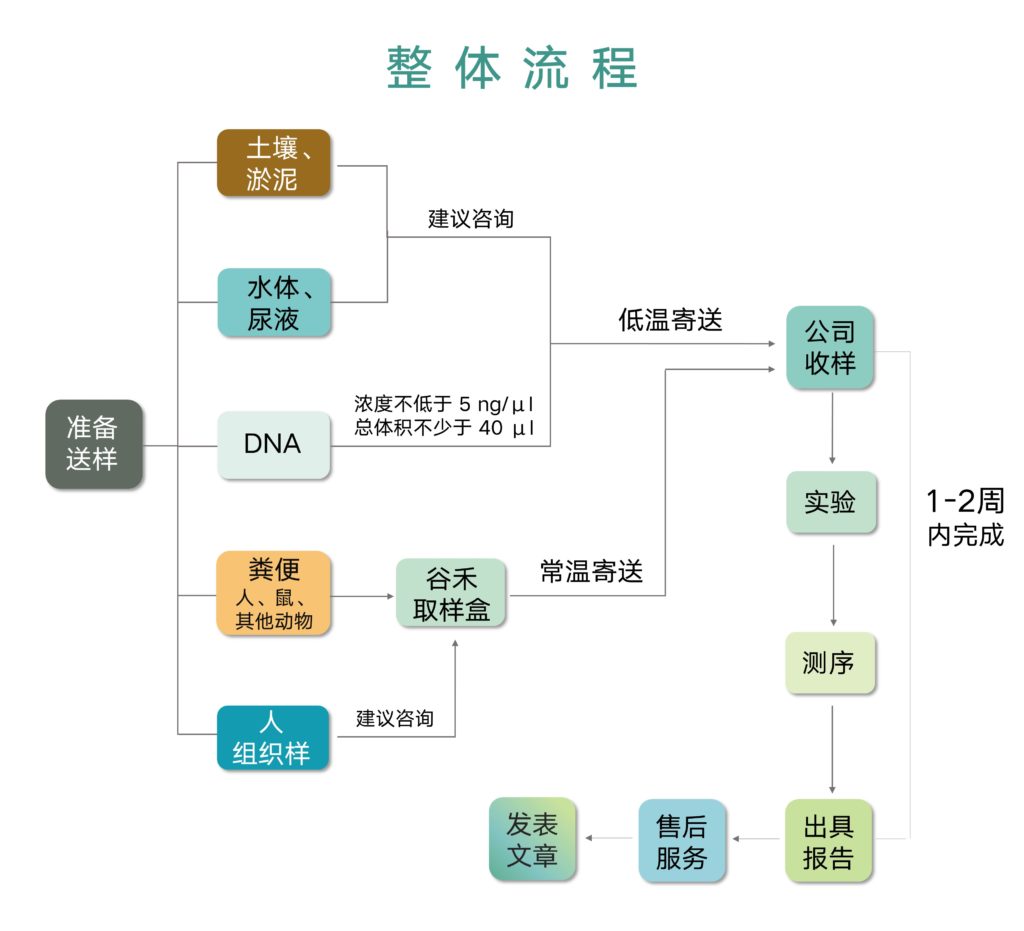
取样:人/动物粪便,口腔,唾液,阴道,尿液,皮肤等可提供免费常用保存运输取样盒,其他样本可直接送样
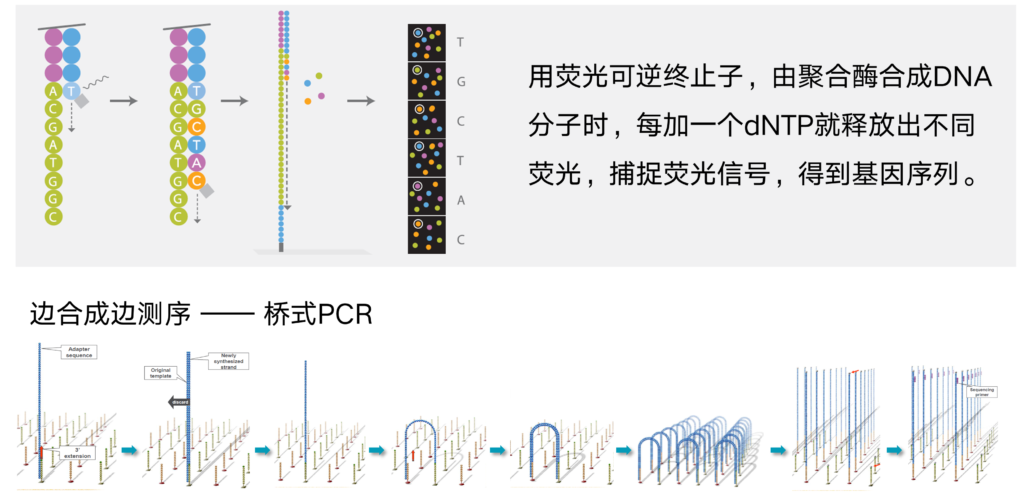
测序平台:Illumina Novaseq
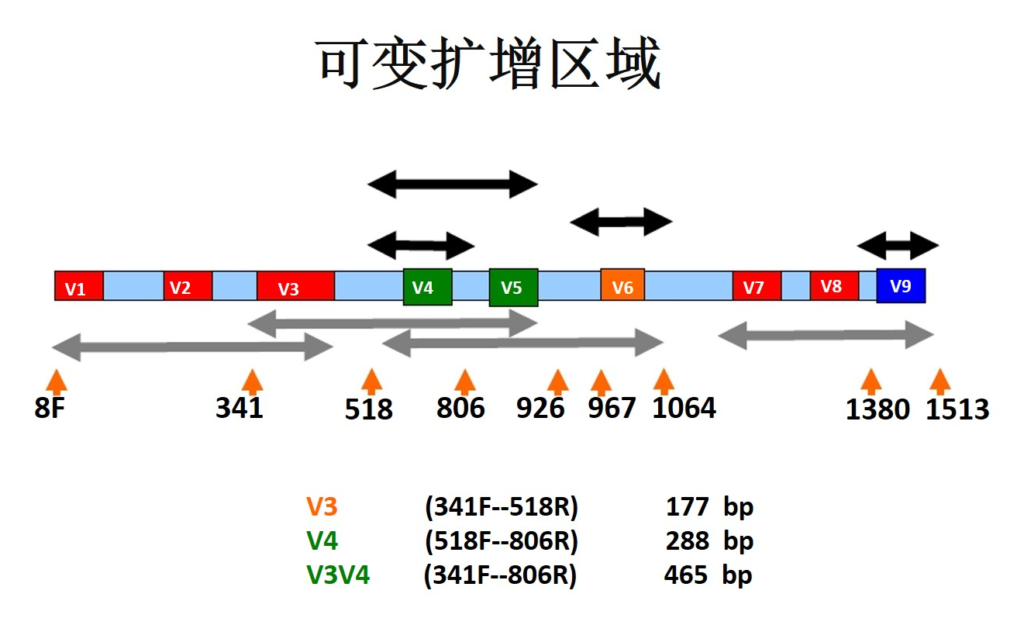
测序区域:V4,V3V4
测序数据量:10万 reads(V4); 5万 reads(V3V4)
周期:1-2周(V4);2-4周(V3V4)
①粪便样本包括肠道内容物:我们提供专门取样盒(免费)。人、大鼠、猪等,直接用取样盒里的棉签沾取约绿豆至黄豆大小的粪便至粪便保存液即可。颗粒状粪便,如小鼠,可根据粪便大小取几颗至粪便保存液即可。
备注:取样盒里有详细的粪便取样操作说明。
②人或者动物其他部位:例如口腔,鼻腔,阴道等:我们提供专门取样盒(免费)。取样方式也是用棉签沾取相应部位菌至保存液。但是根据研究项目,取样部位以及方式略有不同,这个不能一概而论,特殊项目最好单独咨询便于提供最佳方案。
③土壤,底泥水,污泥:需要5-10g的鲜样,土壤,底泥样若有沙石等需要先过筛后再送样。
④水体样,包括河流,湖畔,自来水等:需要先过滤膜,根据水体中含菌量选择一定体积的水体过滤膜,如自来水,一般需要5-15升水过滤膜,然后将滤膜送过来即可。
⑤DNA:浓度不低于5ng/ul, 总体积不少于40ul。


解决了从前期准备到怎么看报告、如何利用数据等问题,包括个性化图表的制作,离发表文章也就不远了,就像长跑已经能看到终点。
但仍然会有零星小问题,如何 “跑赢最后一公里”?
我们能做的就是为大家创建一个良好的交流环境,提供的交流平台致力于用最少的时间,最高效地解决问题。

售后服务:


售后服务:



样本需求量低:常规宏基因组建库建议样本量在500ng以上,公司研发实现了低当量微生物样本提取和建库,保证提取丰度以及片段完整性同时,样本量需求低于同行其他公司要求;对于样本获取困难的样本,也可以选择微量建库,样本量可低至10ng。
免费取样盒和针对性取样建议:粪便及环境样本提供取样盒助力临床/科研取样,人体口腔、痰液、腹水、脑脊液、尿液、皮肤等高寄主细胞含量样本可根据我们的处理方案简单处理后大幅降低宿主DNA比例。
严格标准的实验流程:自动化样品处理平台辅助,每轮设置阳性对照,上轮检测样本对照,阴性对照。评估污染,轮次比对,最大化减少误差,保证样本重复性和稳定性
Illumina测序平台:宏基因组测序(PE150)采用先进的Illumina Novaseq测序平台,快速、高效地读取高质量的测序数据、结合样品特点和数据的产出,充分挖掘环境样品中的微生物菌群和功能基因
大数据分析流程质量流程控制严格:优化的数据质量控制,包括过滤比对质量低、非特异性扩增、覆盖度低、低复杂度的序列,从而快速准确获得样本中微生物信息及其丰度信息,最大化提高质量数据
分析内容丰富全面:物种分析,基因预测与分析,多样性和相似性分析,功能分析,网路互作分析,代谢网络,关联分析等
完整详细的报告:提供质检实验报告,分析统计报告,分析报告解读,原始数据
高效个性化服务:在线项目系统方便您及时查看项目动态和下载报告以及与分析人员高效交流,支持个性化图表修改以及重新分组出报告。
价格低,周期快:包括提取,测序到分析,最快一周出报告。
大数据分析团队和多中心大项目分析经验(团队主要源自浙江大学,包括生物信息学,计算机,微生物以及统计分析等专业,积累了多年的大健康项目多中心项目分析经验,有助于宏基因组大数据,多样本,多表型,多组学联合分析
兼容性强的合作模式:有专门团队负责,提供切实可行的项目方案,兼顾临床和科研双需求模式。

研究背景:全球塑料产量飞速增长,而且呈持续上升的趋势,因此导致大量塑料废物排放到环境中,从沿海河口到大洋环流,从东大西洋到南太平洋海域。塑料废弃物具有化学稳定性和生物利用率低的特点,可长期存在于海洋中,从而影响海洋环境包括海洋生物的生存。
作为一个独特的底物,塑料碎片可以吸附海洋中的微生物并形成个“塑性球”。以生物膜形式存在于塑料碎片上的微生物群落。许多研究表明,无论是在海洋还是淡水生态系统中,附着在塑料碎片上微生物群落的组成明显不同于周围环境(水和沉积物),而且易受位置、时间和塑料类型的影响。
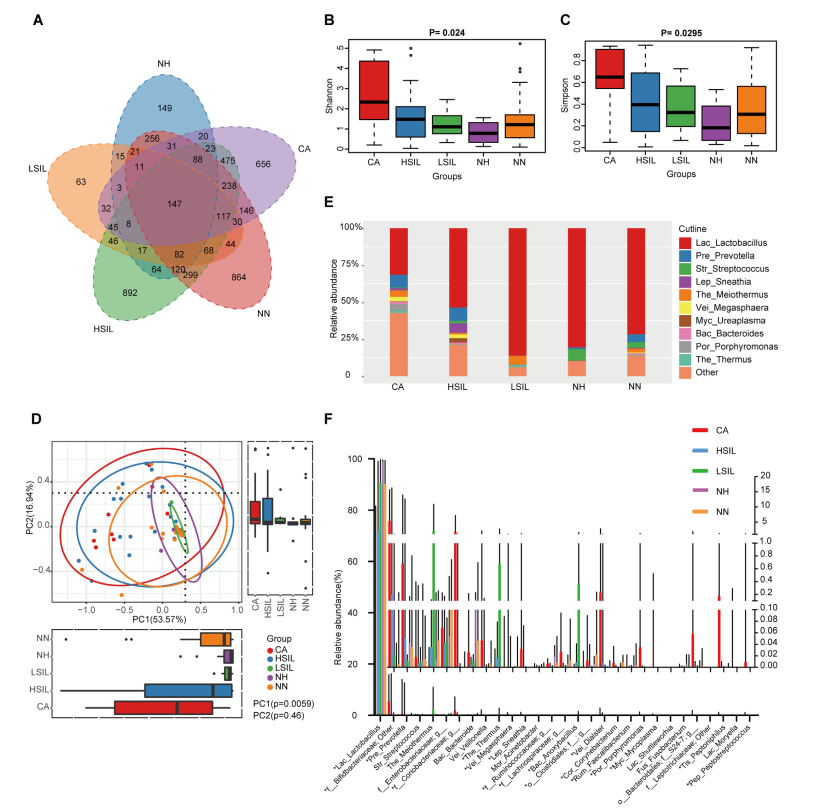
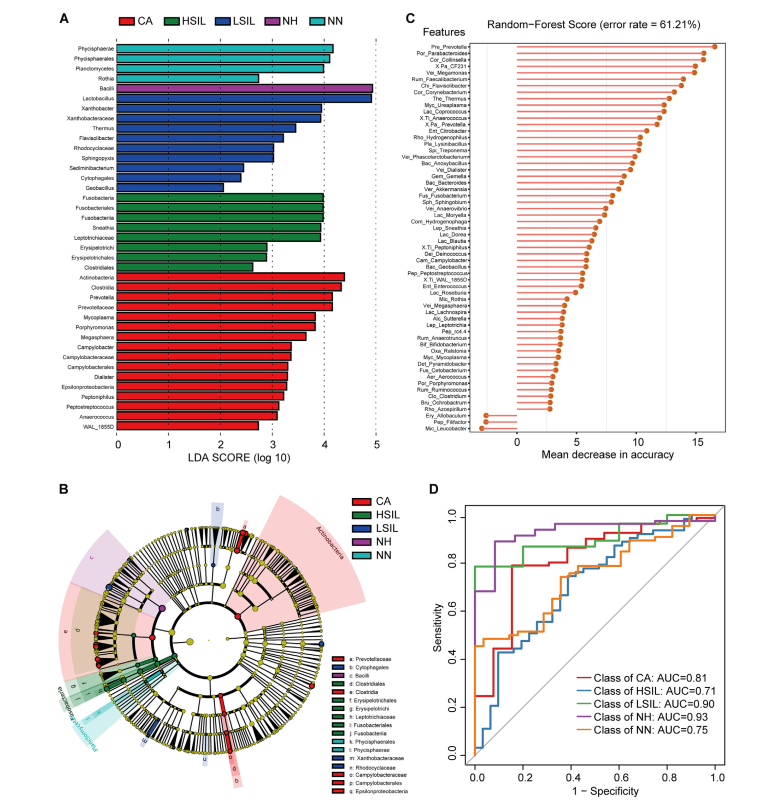
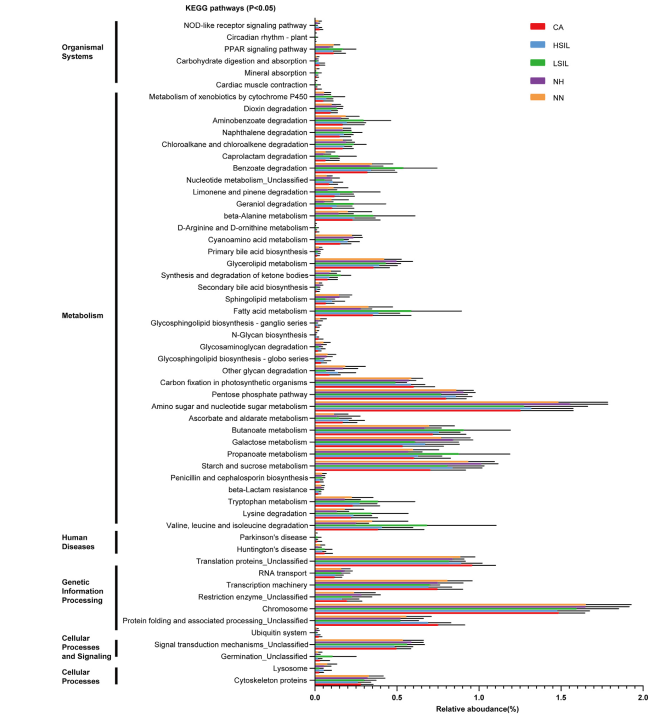
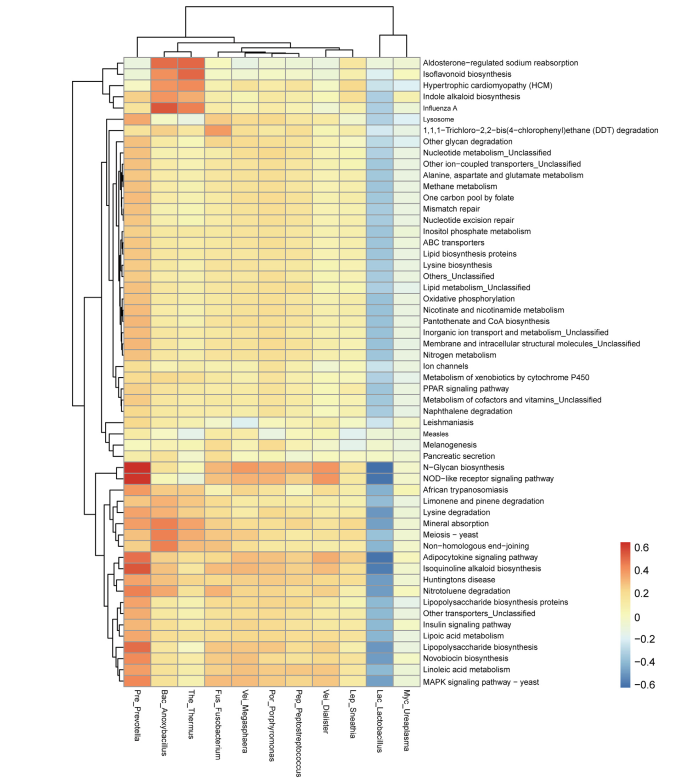
主要图表
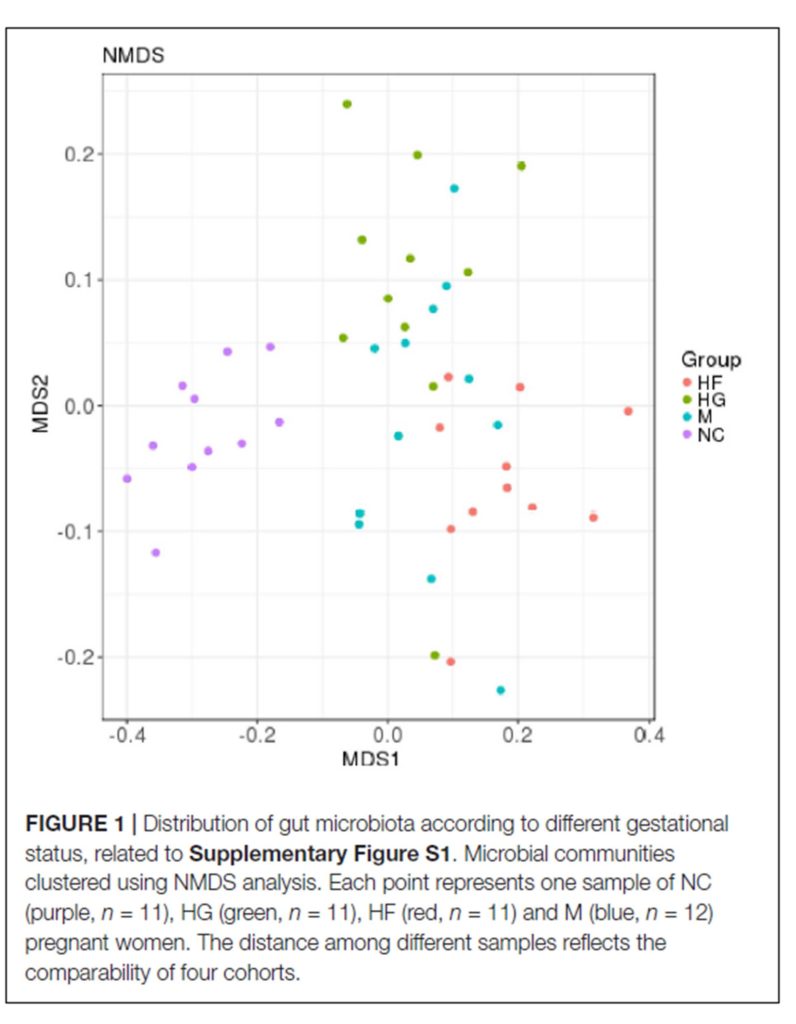
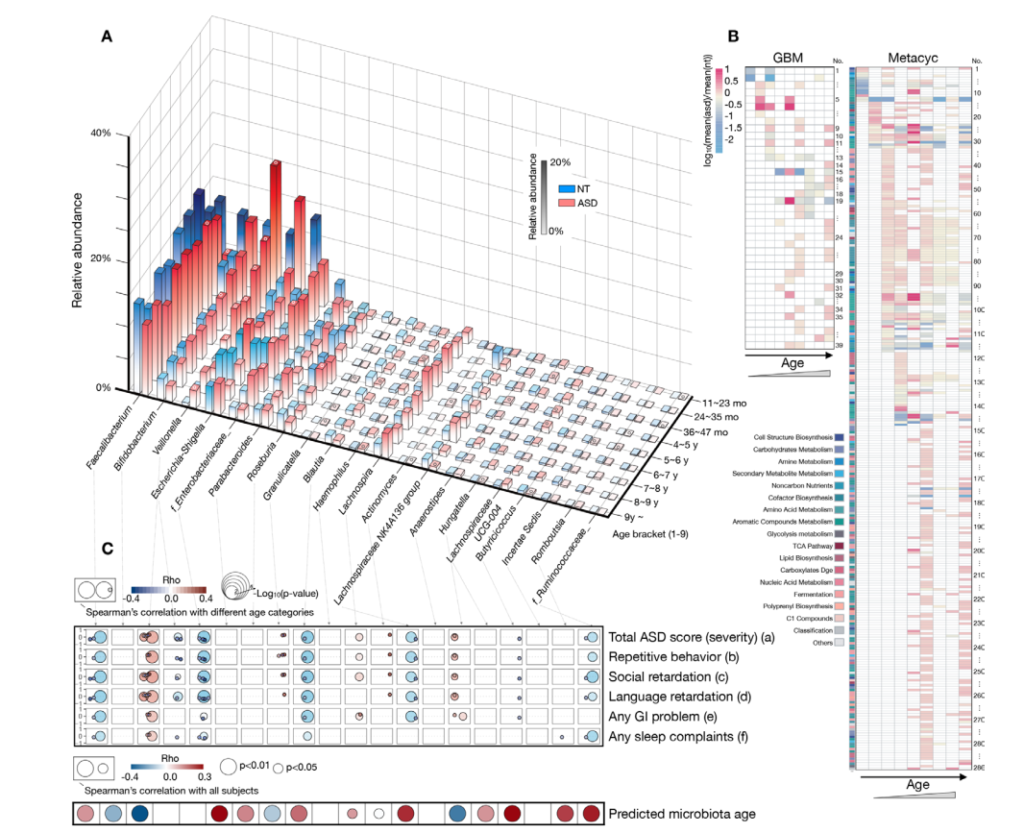
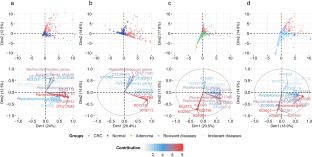
两两群落差异指数的PCoA图
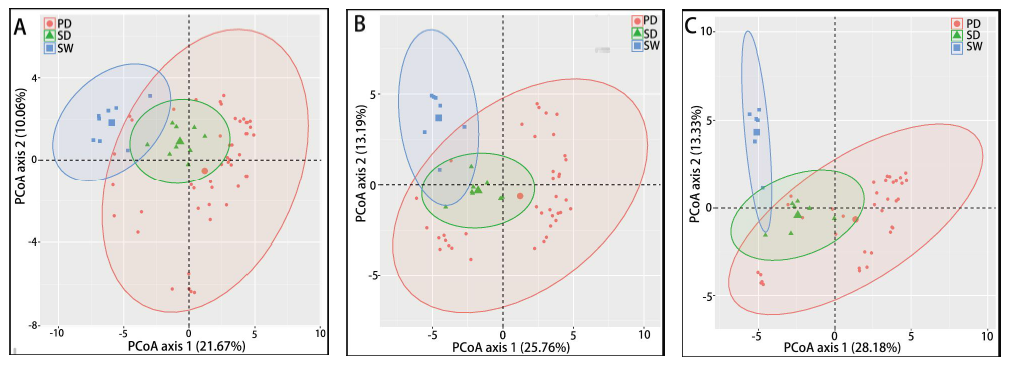
PCoA 图可以清楚地看到,SW区细菌群落的置信椭圆与pd和sd的置信椭圆有显著的偏差(p<0.05),而sd上细菌群落的置信椭圆几乎覆盖了pd的置信椭圆(p>0.05),这表明pd和sd上的细菌群落有相似之处。
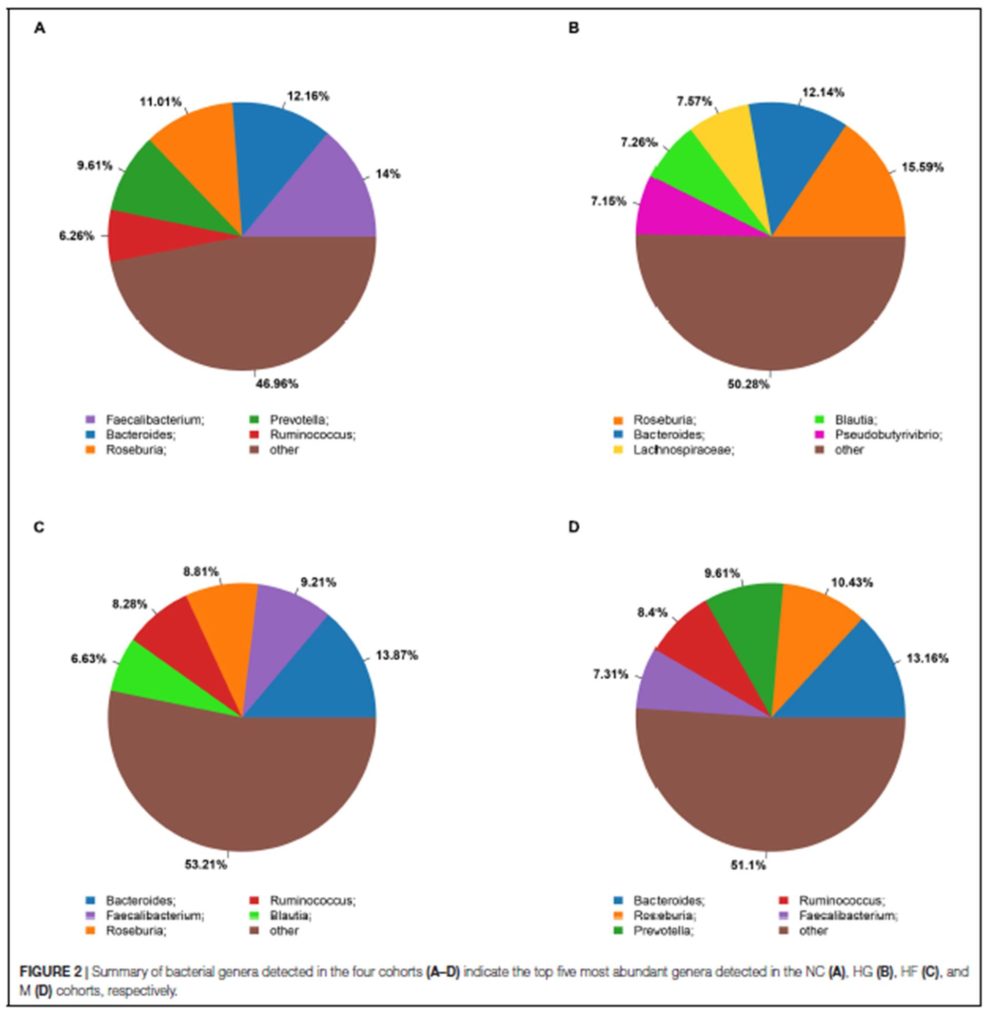
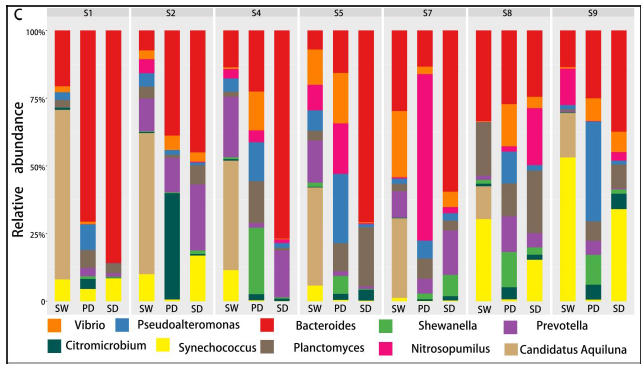
不同样本和处理下的细菌群落( 前 10 位)丰度分布
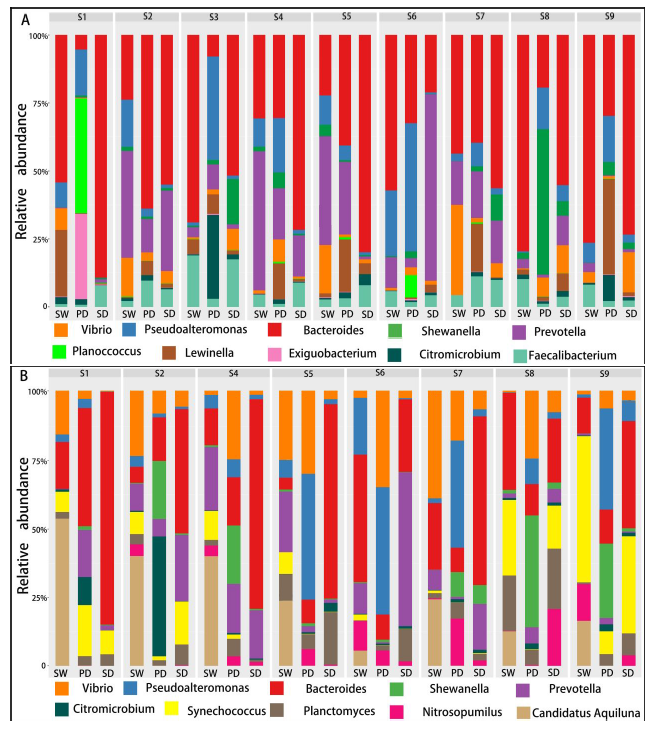
底物(SW、SD和Pd)上的主要属为细菌和假互斥单胞菌,暴露两周后,这些菌可能是分布广泛和适应性强的三种底物(SW、SD和PD)。暴露4周后,弧菌相对丰度增加.此外,暴露6周后,自养细菌(如扁平菌和硝酸菌)的数量增加。这三种底物上个细菌群落的生长模式也与3.2的结果一致。图5还显示,在6个星期内,在429个原位点中,假单胞菌在pd上的相对丰度高于sw和sd(anova,p<0.05)。
研究结论:首先,营养物质 (TN 和 TP) 与生物膜的平均生长速率呈正相关,而盐度与生物膜的平均生长速率呈负相关。盐度是影响PD的个细菌多样性的主要因素,而温度、溶解氧和养分(TN和TP)在类似的盐度条件下可能具有二次效应。尽管种聚合物类型对PD上的细菌群落的多样性具有较少的影响,但是在细菌群落中的一些属显示对PD的聚合物类型的选择性,并且倾向于将其优选的基质定殖。大的相对丰度SW、PD、SD间属显著差异。盐度是改变河口地区Pd条件致病菌富集的主要因素。另外,在种病原物种丰富的基础上,PD具有较高的致病性。

研究背景:研究表明遗传和环境影响都在I型糖尿病的发展中起作用,增加的遗传风险不足以引起疾病,环境因素也是需要的,而且起着至关重要的作用。肠道菌群也许就是这个重要的环境因素,肠道菌群在免疫系统的成熟中起重要作用,此外还影响自身免疫疾病发展。

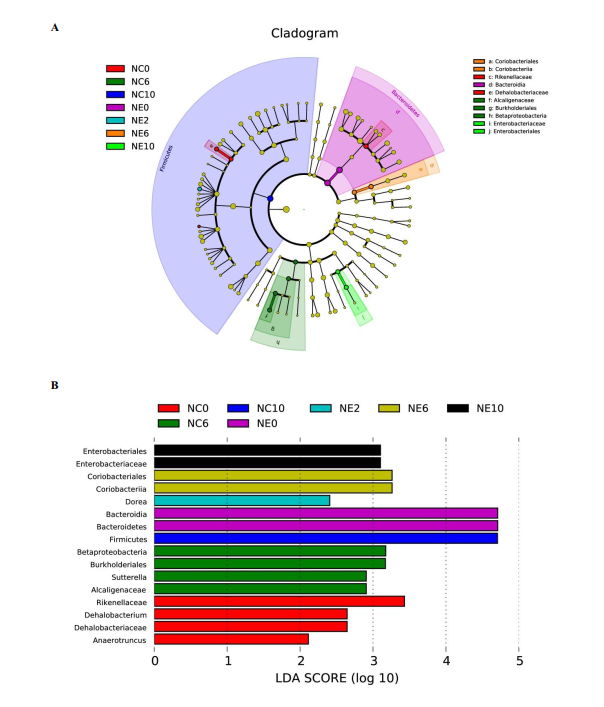
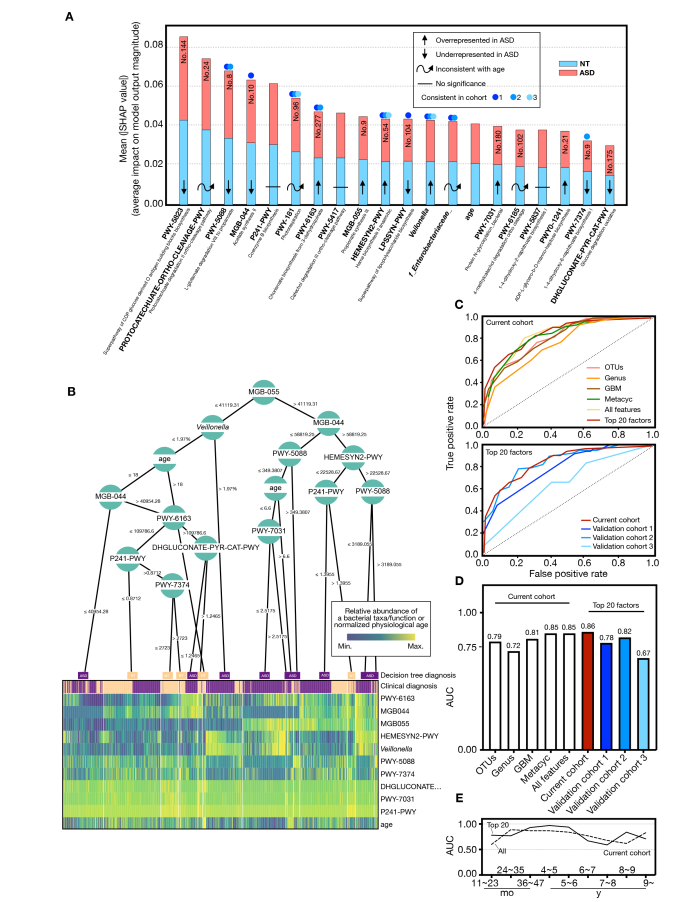
不同遗传风险儿童的LDA差异菌群
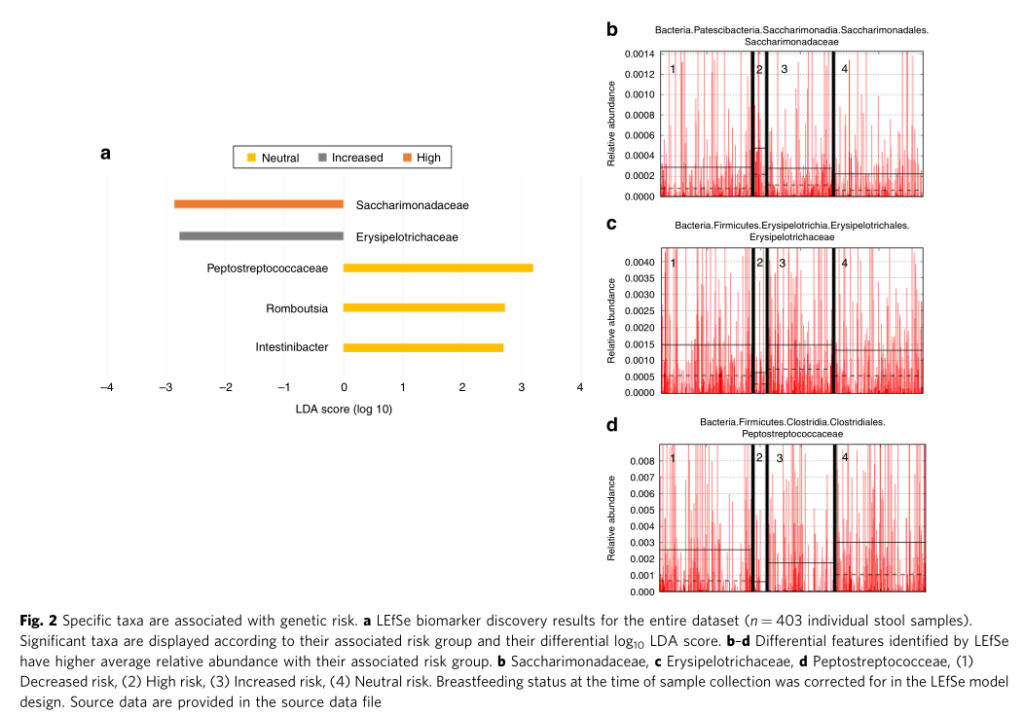
不同遗传风险分组中包含的常见菌属,部分存在特定分组中
PCoA分析揭示不同遗传风险儿童肠道菌群的在不同地域样本中均存在显著差异
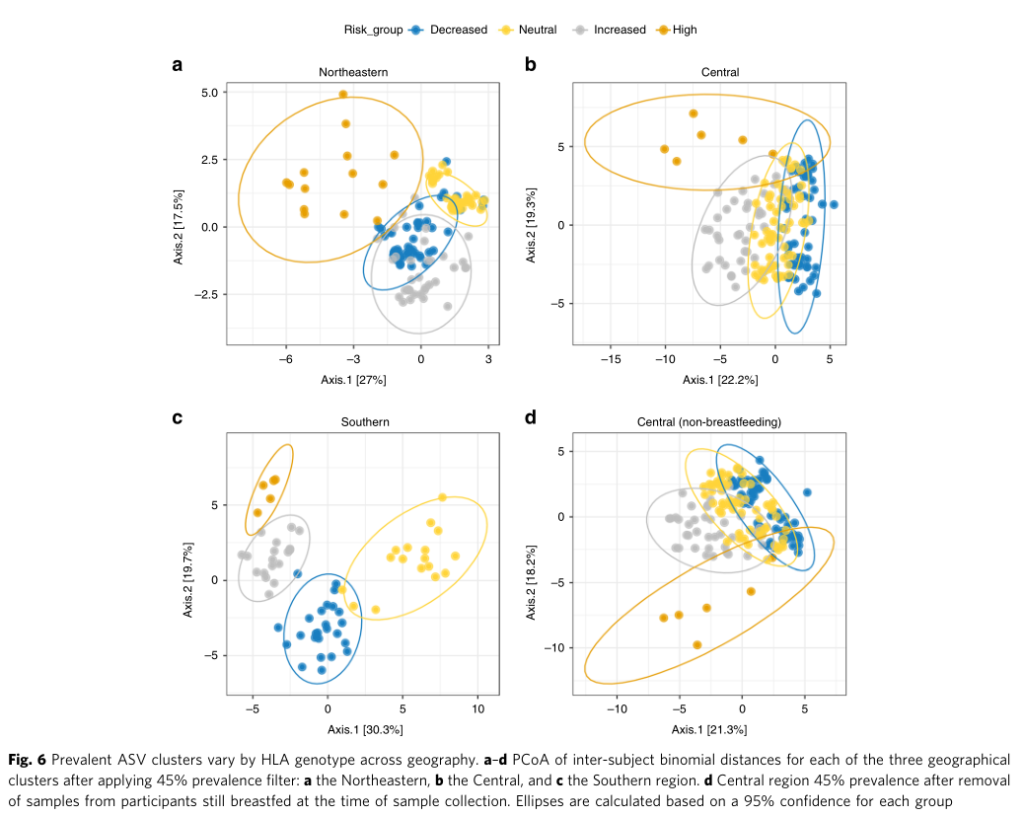

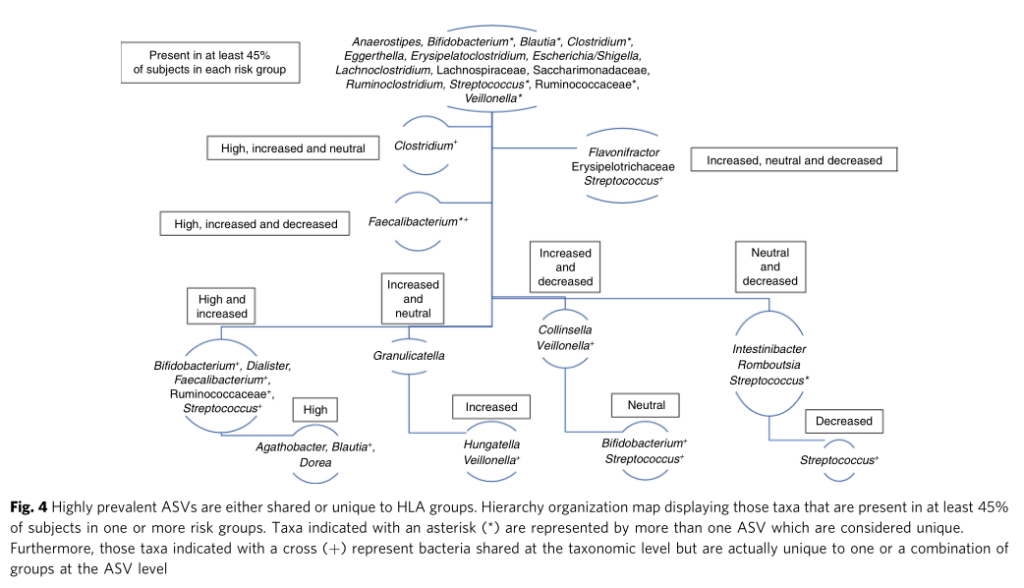
点评:针对I型糖尿病疾病发生过程中遗传HLA分型风险和对应肠道菌群菌的关联分析,揭示了特定肠道菌群与宿主特定遗传风险共同作用推进疾病发生。某些特定菌属可能无法在遗传高风险儿童肠道内定植,可能对疾病发生存在特定作用。此外对于其他遗传风险的自身免疫疾病也具有重要提示意义,例如乳糜泻和类风湿性关节炎。
Xu S, Xiong J, Qin X, et al. Association between gut microbiota and perinatal depression and anxiety among a pregnancy cohort in Hunan [J]. China Brain Behav Immun. 2025 , 125:168-177.
Cai Z, Che C, Li D, et al. Common Gut Microbial Signatures in Autism Spectrum Disorder and Attention Deficit Hyperactivity Disorder [J]. Autism Res. 2025, 18(4):741-751.
Jiang J, Deng J, Zhao Y, et al. Heterophyllin B alleviates cognitive disorders in APP/PS1 model mice via the spleen-gut microbiota-brain axis [J]. Int Immunopharmacol. 2025, 8;154:114591.
Wen J, Wu Y, Zhang F, et al. Neonatal hypoxia leads to impaired intestinal function and changes in the composition and metabolism of its microbiota [J]. Sci Rep. 2025, 1;15(1):15285.
Cheng Q, Xie M, Ying H, et al. A spatiotemporal probiotic spore-loaded oxygen generator Resumes gut microbiome balance and Improves hypoxia for treating viral pneumonia [J]. Chemical Engineering Journal. 2025, 159706
Zhang X, Wang L, Khan AI, et al. Lentinan’s effect on gut microbiota and inflammatory cytokines in 5-FU-induced mucositis mice [J]. AMB Express. 2025, 22;15(1):11.
Hou Q, Zhang J, Su Z, et al. Clinical Trial of Ozonated Water Enema for the Treatment of Fibromyalgia: A Randomized, Double-Blind Trial [J]. Pain Physician. 2025, 28(1):E13-E22.
Zhang D, He X, Wang Y, et al. Hesperetin-Enhanced Metformin to Alleviate Cognitive Impairment via Gut-Brain Axis in Type 2 Diabetes Rats [J]. Int J Mol Sci. 2025, 23;26(5):1923.
Fu X, Ren X, Zhao M, et al. Disruption of intestinal barrier and dysbiosis of gut microbiota in an experimental rhesus macaque model with 6-year diabetes mellitus [J]. Exp Anim. 2025, 17.
Rana MS, Alshehri D, Wang RL, et al. Effect of molybdenum supply on crop performance through rhizosphere soil microbial diversity and metabolite variation [J]. Front Plant Sci. 2025, 28;15:1519540.
Chen X, Feng Y, Zhang D, et al. Orally administered hydrogel containing polyphenol@halloysite clay for probiotic delivery and treatment of inflammatory bowel disease [J]. 2025, 62.
Li X, Wu Q, Wang Y, et al. UHPM dominance in driving the formation of petroleum-contaminated soil aggregate, the bacterial communities succession, and phytoremediation. J Hazard Mater [J].2024,5;471:134322.
Lan J, Zhang Y, Jin C, et al. Gut Dysbiosis Drives Inflammatory Bowel Disease Through the CCL4L2-VSIR Axis in Glycogen Storage Disease. Adv Sci [J]. 2024 Aug;11(30):e2309471.
Li X, Wu Q, Wang Y, et al. UHPM dominance in driving the formation of petroleum-contaminated soil aggregate, the bacterial communities succession, and phytoremediation. J Hazard Mater [J].2024,5;471:134322.
Lan J, Zhang Y, Jin C, et al. Gut Dysbiosis Drives Inflammatory Bowel Disease Through the CCL4L2-VSIR Axis in Glycogen Storage Disease. Adv Sci. 2024 Aug;11(30):e2309471.
Li X, Wu Q, Wang Y, et al. UHPM dominance in driving the formation of petroleum-contaminated soil aggregate, the bacterial communities succession, and phytoremediation. J Hazard Mater [J].2024,5;471:134322.
Lan J, Zhang Y, Jin C, et al. Gut Dysbiosis Drives Inflammatory Bowel Disease Through the CCL4L2-VSIR Axis in Glycogen Storage Disease. Adv Sci. 2024 Aug;11(30):e2309471.
Chen Y, Le D, Xu J, J et al. Gut Microbiota Dysbiosis and Inflammation Dysfunction in Late-Life Depression: An Observational Cross-Sectional Analysis. Neuropsychiatr Dis Treat [J]. 2024 27;20:399-414.
Yang T, Dai M, Tang G, et al. Effect of an electro-assisted biochemical cycle reactor on bio-oxidation of gold ore. Minerals Engineering [J]. 2024,209, 108630
He C, Jiang J, Liu J, et al. Pseudostellaria heterophylla polysaccharide mitigates Alzheimer’s-like pathology via regulating the microbiota-gut-brain axis in 5 × FAD mice. Int J Biol Macromol [J]. 2024, 270(Pt 2):132372.
Zhao J, Yuan J, Zhang Y, et al. Bifidobacterium pseudonumeratum W112 alleviated depressive and liver injury symptoms induced by chronic unpredictable mild stress via gut-liver-brain axis. Front Nutr [J]. 202419;11:1421007.
Hu L, Ye W, Deng Q, et al. Microbiome and metabolite mnalysis insight into the potential of shrimp head hydrolysate to alleviate depression-like behaviour in growth-period mice exposed to chronic stress. Nutrients [J]. 2024, 19;16(12):1953.
Han YL, Li XY, Li J, et al. Vaginal microbiome dysbiosis as a novel noninvasive biomarker for detection of chronic endometritis in infertile women. Int J Gynaecol Obstet [J]. 2024, 167(3):1034-1042.
Lai J, Gong L, Liu Y, et al. Associations between gut microbiota and osteoporosis or osteopenia in a cohort of Chinese Han youth. Sci Rep [J]. 2024, 9;14(1):20948.
Li H, Hu Y, Huang Y, et al. The mutual interactions among Helicobacter pylori, chronic gastritis, and the gut microbiota: a population-based study in Jinjiang, Fujian. Front [J]. 2024,14;15:1365043.
Fan Z, Han D, Fan X, et al. Analysis of the correlation between cervical HPV infection, cervical lesions and vaginal microecology. Front Cell Infect Microbiol. 2024 16;14:1405789.
Lai J, Gong L, Liu Y, et al. Associations between gut microbiota and osteoporosis or osteopenia in a cohort of Chinese Han youth. Sci Rep [J]. 2024, 9;14(1):20948.
Zhao H, Shang L, Zhang Y, et al. IL-17A inhibitors alleviate Psoriasis with concomitant restoration of intestinal/skin microbiota homeostasis and altered microbiota function. Front Immunol [J]. 2024, 28;15:1344963.
Sun Y, Liu X, Wang R, et al. Lacticaseibacillus rhamnosus HF01 fermented yogurt alleviated high-fat diet-induced obesity and hepatic steatosis via the gut microbiota-butyric acid-hepatic lipid metabolism axis. Food Funct [J]. 2024, 22;15(8):4475-4489.
Qiao S, Bu S, Wang H. Effects of stroke on the intestinal biota in diabetic mice and type 2 diabetic patient biota. J Appl Microbiol [J]. 2024, 19:lxae015.
Zhao R, Han B, Yang F, et al. Analysis of extracellular and intracellular antibiotic resistance genes in commercial organic fertilizers reveals a non-negligible risk posed by extracellular genes. J Environ Manage [J]. 2024,354:120359.
Ali M, Ullah H, Farooqui NA, et al. NF-κB pathway activation by Octopus peptide hydrolysate ameliorates gut dysbiosis and enhances immune response in cyclophosphamide-induced mice. Heliyon [J]. 2024, 24;10(19):e38370.
Geng X, Lin R, Hasegawa Y, et al. Effects of Scallop Mantle Toxin on Intestinal Microflora and Intestinal Barrier Function in Mice. Toxins (Basel) [J]. 2024, 27;16(6):247.
Chen Y, Li J, Le D, et al. A mediation analysis of the role of total free fatty acids on pertinence of gut microbiota composition and cognitive function in late life depression. Lipids Health Dis [J]. 2024, 29;23(1):64.
Wei Q, Zhou Y, Hu Z, et al. Function-oriented mechanism discovery of coumarins from Psoralea corylifolia L. in the treatment of ovariectomy-induced osteoporosis based on multi-omics analysis. J Ethnopharmacol [J]. 2024,15;329:118130.
Li Y, Wang N, Guo J, et al. Integrative Transcriptome Analysis of mRNA and miRNA in Pepper’s Response to Phytophthora capsici Infection. Biology (Basel) [J]. 2024,14;13(3):186.
Hussain A, Rui B, Ullah H, et al. Limosilactobacillus reuteri HCS02-001 Attenuates Hyperuricemia through Gut Microbiota-Dependent Regulation of Uric Acid Biosynthesis and Excretion. Microorganisms [J]. 2024, 22;12(4):637.
Wen Y, Ullah H, Ma R, et al. Anemarrhena asphodeloides Bunge polysaccharides alleviate lipoteichoic acid-induced lung inflammation and modulate gut microbiota in mice. Heliyon. 2024 Oct
Khan A, Rehman A, Ayub Q, et al. The composition of the blood microbiota and its relationship to osteoporosis-related clinical parameters [J]. Medicine in Microecology, 2023, 100097.
Xu S, Liu W, Gong L, et al. Association of ADRB2 gene polymorphisms and intestinal microbiota in Chinese Han adolescents [J]. Open Life Sciences, 2023,18(1), 20220646.
Wang X, Weng Y, Geng S, et al. Maternal procymidone exposure has lasting effects on murine gut-liver axis and glucolipid metabolism in offspring
[J]. Food and Chemical Toxicology, 2023,174, 113657.
Liu H, Xing Y, Wang Y, et al. Dendrobium officinale Polysaccharide Prevents Diabetes via the Regulation of Gut Microbiota in Prediabetic Mice [J]. Foods. 2023 , 8;12(12):2310.
Yan X, Yan J, Xiang Q, et al. Early-life gut microbiota in food allergic children and its impact on the development of allergic disease [J]. Ital J Pediatr. 2023, 9;49(1):148.
Xiang Q, Yan X, Lin X, et al. Intestinal Microflora Altered by Vancomycin Exposure in Early Life Up-regulates Type 2 Innate Lymphocyte and Aggravates Airway Inflammation in Asthmatic Mice [J]. Inflammation. 2023, 46(2):509-521.
Qiu J, Zhao L, Cheng Y, Chen Q, et al. Exploring the gut mycobiome: differential composition and clinical associations in hypertension, chronic kidney disease, and their comorbidity [J]. Front Immunol. 2023, 14;14:1317809.
Jiang S, Zhang B, Fan X, et al. Gut microbiome predicts selenium supplementation efficiency across different Chinese adult cohorts using hybrid modeling and feature refining [J]. Front Microbiol. 2023, Oct 17;14:1291010.
Liu T, Liu J, Wang P, et al. Effect of slurry ice on quality characteristics and microbiota composition of Pacific white shrimp during refrigerated storage
[J]. Journal of Agriculture and Food Research, 2023,14, 100792.
Chen B, Xu J, Lu H, et al. Remediation of benzo[a]pyrene contaminated soils by moderate chemical oxidation coupled with microbial degradation [J]. Sci Total Environ. 2023 , 1;871:161801.
Shouhua, Z., & Meilan, L. Microbial Diversity and Abundance in Pulmonary Tissue of Patients with Early-Stage Lung Cancer
[J]. Jundishapur Journal of Microbiology, 2023, 16(5).
Wei T, Liao Y, Wang Y, et al. Comparably Characterizing the Gut Microbial Communities of Amphipods from Littoral to Hadal Zones [J]. Journal of Marine Science and Engineering, 2023, 11(11), 2197.
Ji C, Luo Y, Yang J, et al. Polyhalogenated carbazoles induce hepatic metabolic disorders in mice via alteration in gut microbiota [J]. J Environ Sci (China). 2023, 127:603-614.
Han B, Yang F, Shen S, et al. Effects of soil habitat changes on antibiotic resistance genes and related microbiomes in paddy fields [J]. Sci Total Environ. 2023, 15;895:165109.
Li S, Niu Z, Wang M, et al. The occurrence and variations of extracellular antibiotic resistance genes in drinking water supply system: A potential risk to our health [J]. Journal of Cleaner Production, 2023, 402, 136714.
Gao X, Zhao J, Chen W, et al. Food and drug design for gut microbiota-directed regulation: Current experimental landscape and future innovation
[J]. Pharmacol Res. 2023,194:106867.
Han B, Shen S, Yang F, et al. Exploring antibiotic resistance load in paddy-upland rotation fields amended with commercial organic and chemical/slow release fertilizer [J]. Frontiers in Microbiology, 2023,14, 1184238.
Alsholi DM, Yacoub GS, Rehman AU, et al. Lactobacillus rhamnosus Attenuates Cisplatin-Induced Intestinal Mucositis in Mice via Modulating the Gut Microbiota and Improving Intestinal Inflammation [J]. Pathogens, 2023, 11;12(11):1340.
Xie X, Xu H, Shu R, et al. Clock gene Per3 deficiency disrupts circadian alterations of gut microbiota in mice [J]. Acta Biochim Biophys Sin (Shanghai). 2023,15; (12):2004-2007.
Xiao W, Zhang Q, Zhao S, et al. Citric acid secretion from rice roots contributes to reduction and immobilization of Cr(VI) by driving microbial sulfur and iron cycle in paddy soil [J]. Sci Total Environ. 2023, 1;854:158832.
Li R, Liu R, Chen L, et al. Microbiota from Exercise Mice Counteracts High-Fat High-Cholesterol Diet-Induced Cognitive Impairment in C57BL/6 Mice [J]. Oxid Med Cell Longev. 2023, 20;2023:2766250.
Liao J, Dou Y, Yang X, et al. Soil microbial community and their functional genes during grassland restoration [J]. Journal of Environmental Management, 2023, 325, 116488.
Liu F, Du J, Lin H, et al. The bladder microbiome of chronic kidney disease with associations to demographics, renal function, and serum cytokines
[J]. medRxiv, 2023-05.
Ullah H, Deng T, Ali M, et al. Sea Conch Peptides Hydrolysate Alleviates DSS-Induced Colitis in Mice through Immune Modulation and Gut Microbiota Restoration [J]. Molecules. 2023, 28;28(19):6849.
Zou S, Liu R, Luo Y, et al. Effects of fungal agents and biochar on odor emissions and microbial community dynamics during in-situ treatment of food waste [J]. Bioresour Technol. 2023; 380:129095.
Wu J, Zhang D, Zhao M, et al. Gut Microbiota Dysbiosis and Increased NLRP3 Levels in Patients with Pregnancy-Induced Hypertension
[J] . Curr Microbiol. 2023, 6;80(5):168.
Li R, Liu R, Chen L, et al. Microbiota from Exercise Mice Counteracts High-Fat High-Cholesterol Diet-Induced Cognitive Impairment in C57BL/6 Mice
[J]. Oxid Med Cell Longev. 2023, 20:2766250.
Xiao W, Zhang Q, Zhao S, et al. Citric acid secretion from rice roots contributes to reduction and immobilization of Cr(VI) by driving microbial sulfur and iron cycle in paddy soil [J]. Sci Total Environ. 2023, 16:158832.
Wang X, Weng Y, Geng S, et al. Maternal procymidone exposure has lasting effects on murine gut-liver axis and glucolipid metabolism in offspring [J]. Food Chem Toxicol. 2023, 174:113657.
Liao J, Dou Y, Yang X, et al. Soil microbial community and their functional genes during grassland restoration [J]. J Environ Manage. 2023, Jan 1;325(Pt A):116488.
Li S, Niu Z, Wang M, et al. The occurrence and variations of extracellular antibiotic resistance genes in drinking water supply system: A potential risk to our health [J]. Journal of Cleaner Production. 2023, 20, 136714.
Hu Y, Li J, Ni F, et al. CAR-T cell therapy-related cytokine release syndrome and therapeutic response is modulated by the gut microbiome in hematologic malignancies[J]. Nature communications, 2022, 13(1): 1-14.
Lou M, Cao A, Jin C, et al. Deviated and early unsustainable stunted development of gut microbiota in children with autism spectrum disorder [J]. Gut, 2021 Dec 20:gutjnl-2021-325115.
Chen C, Du Y, Liu Y, et al. Characteristics of gastric cancer gut microbiome according to tumor stage and age segmentation[J]. Applied Microbiology and Biotechnology, 2022, 106(19): 6671-6687.
Chen C, Shen J, Du Y, et al. Characteristics of gut microbiota in patients with gastric cancer by surgery, chemotherapy and lymph node metastasis[J]. Clinical and Translational Oncology, 2022, 24(11): 2181-2190.
Shen J, Jin CL, Zhang YY, et al. A multiple-dimension model for microbiota of patients with colorectal cancer from normal participants and other intestinal disorders[J]. Applied Microbial and Cell Physiology, 2022, February 26
Wang YB, Shang LC, Zhong CH, et al. Transplantation of feces from mice with Alzheimer’s disease promoted lung cancer growth [J]. Biochemical and Biophysical Research Communications. 2022, 600: 67-74
Zhang G, Pang Y, Zhou Y, et al. Effect of dissolved oxygen on N2O release in the sewer system during controlling hydrogen sulfide by nitrate dosing [J]. Science of The Total Environment. 2022, Apr 10;816:151581.
Zhao Y, Huang J, Li T, et al. Berberine ameliorates aGVHD by gut microbiota remodelling, TLR4 signalling suppression and colonic barrier repairment for NLRP3 inflammasome inhibition[J]. Journal of Cellular and Molecular Medicine, 2022, 26(4): 1060-1070.
Hang, ZC, Cai SL, Lei T, et al. Transfer of Tumor-Bearing Mice Intestinal Flora Can Ameliorate Cognition in Alzheimers Disease Mice[J]. J Alzheimers Dis. 2022, 86(3):1287-1300.
Zhu W, Yan M, Cao H, et al. Effects of Clostridium butyricum Capsules Combined with Rosuvastatin on Intestinal Flora, Lipid Metabolism, Liver Function and Inflammation in NAFLD Patients[J]. Cellular and Molecular Biology. 2022, 68(2): 64-69.
Yang F, Wang Z, Zhao D, et al. Food-derived Crassostrea gigas peptides self-assembled supramolecules for scarless healing[J]. Composites Part B: Engineering, 2022, 246: 110265.
Yu T, Ji L, Lou L, et al. Fusobacterium nucleatum Affects Cell Apoptosis by Regulating Intestinal Flora and Metabolites to Promote the Development of Colorectal Cancer[J]. Frontiers in microbiology. 2022, 13.
Qin Z, Yuan X, Liu J, et al. Albuca Bracteata Polysaccharides Attenuate AOM/DSS Induced Colon Tumorigenesis via Regulating Oxidative Stress, Inflammation and Gut Microbiota in Mice [J]. Frontiers in pharmacology. 2022, 13: 833077.
Wang R, Deng Y, Zhang Y, et al. Modulation of Intestinal Barrier, Inflammatory Response, and Gut Microbiota by Pediococcus pentosaceus zy-B Alleviates Vibrio parahaemolyticus Infection in C57BL/6J Mice[J]. Journal of Agricultural and Food Chemistry. 2022, 70(6): 1865-1877.
Hu L, Sun L, Zhou J, et al. Impact of a hexafluoropropylene oxide trimer acid (HFPO-TA) exposure on impairing the gut microbiota in mice. Chemosphere, 2022: 134951.
Yang T, Tang G, Li L, et al. Interactions between bacteria and eukaryotic microorganisms and their response to soil properties and heavy metal exchangeability nearby a coal-fired power plant. Chemosphere. 2022, 302: 134829.
Chen G, Zeng R, Wang X, et al. Antithrombotic Activity of Heparinoid G2 and Its Derivatives from the Clam Coelomactra antiquata. Marine Drugs, 2022, 20(1): 50.
Khan A I, Rehman A U, Farooqui N A, et al. Effects of shrimp peptide hydrolysate on intestinal microbiota restoration and immune modulation in cyclophosphamide-treated mice. Molecules. 2022, 27(5): 1720.
He L, Xing Y, Ren X, et al. Mulberry Leaf Extract Improves Metabolic Syndrome by Alleviating Lipid Accumulation In Vitro and In Vivo[J]. Molecules, 2022, 27(16): 5111.
Rehman A U, Siddiqui N Z, Farooqui N A, et al. Morchella esculenta mushroom polysaccharide attenuates diabetes and modulates intestinal permeability and gut microbiota in a type 2 diabetic mice model[J]. Frontiers in Nutrition, 2022, 9.
Wang X, Hu L, Wang C, et al. Cross-generational effects of maternal exposure to imazalil on anaerobic components and carnitine absorption associated with OCTN2 expression in mice[J]. Chemosphere, 2022, 308: 136542.
Zhang G, Wang G, Zhou Y, et al. Simultaneous use of nitrate and calcium peroxide to control sulfide and greenhouse gas emission in sewers[J]. Science of The Total Environment, 2022, 855: 158913.
Ma C J, He Y, Jin X, et al. Light-regulated nitric oxide release from hydrogel-forming microneedles integrated with graphene oxide for biofilm-infected-wound healing[J]. Biomaterials Advances, 2022, 134: 112555.
Dong Y, Sui L, Yang F, et al. Reducing the intestinal side effects of acarbose by baicalein through the regulation of gut microbiota: An in vitro study[J]. Food Chemistry, 2022, 394: 133561.
Ji C, Luo Y, Yang J, et al. Polyhalogenated carbazoles induce hepatic metabolic disorders in mice via alteration in gut microbiota[J]. Journal of Environmental Sciences, 2022, 127: 603-614.
Hu J, Wei S, Gu Y, et al. Gut Mycobiome in Patients With Chronic Kidney Disease Was Altered and Associated With Immunological Profiles[J]. Frontiers in immunology, 2022, 13.
Xiao W, Zhang Q, Zhao S, et al. Citric acid secretion from rice roots contributes to reduction and immobilization of Cr (VI) by driving microbial sulfur and iron cycle in paddy soil[J]. Science of The Total Environment, 2022, 854: 158832.
Gu Y, Chen H, Li X, et al. Lactobacillus paracasei IMC 502 ameliorate type 2 diabetes by mediating gut microbiota‐SCFAs‐hormone/inflammation pathway in mice[J]. Journal of the Science of Food and Agriculture, 2022.
Sun D, Wang C, Sun L, et al. Preliminary Report on Intestinal Flora Disorder, Faecal Short-Chain Fatty Acid Level Decline and Intestinal Mucosal Tissue Weakening Caused by Litchi Extract to Induce Systemic Inflammation in HFA Mice. Nutrients. 2022, 14(4): 776.
Zhou S P, Zhou H Y, Sun J C, et al. Bacterial dynamics and functions driven by bulking agents to enhance organic degradation in food waste in-situ rapid biological reduction (IRBR)[J]. Bioprocess and Biosystems Engineering, 2022, 45(4): 689-700.
Wen Y, Feng S, Dai H, et al. Intestinal dysbacteriosis-propelled T helper 17 cells activation mediate the perioperative neurocognitive disorder induced by anesthesia/surgery in aged rats[J]. Neuroscience Letters, 2022, 783: 136741.
Gu Y, Li X, Chen H,et al. Antidiabetic effects of multi-species probiotic and its fermented milk in mice via restoring gut microbiota and intestinal barrier. Food Bioscience. 2022, Volume 47
Wang R, Deng Y, Zhang Y, et al. Modulation of Intestinal Barrier, Inflammatory Response, and Gut Microbiota by Pediococcus pentosaceus zy-B Alleviates Vibrio parahaemolyticus Infection in C57BL/6J Mice. J Agric Food Chem. 2022, 16;70(6):1865-1877.
Zhang M, Sun Q, Chen P, et al. How microorganisms tell the truth of potentially toxic elements pollution in environment[J]. Journal of Hazardous Materials, 2022, 431: 128456.
Wang Q, Guan C, Han J, et al. Microplastics in China Sea: analysis, status, source, and fate[J]. Science of The Total Environment, 2022, 803: 149887.
Sun D, Wang C, Sun L, et al. Gooneratne R. Preliminary Report on Intestinal Flora Disorder, Faecal Short-Chain Fatty Acid Level Decline and Intestinal Mucosal Tissue Weakening Caused by Litchi Extract to Induce Systemic Inflammation in HFA Mice. Nutrients. 2022, Feb 12;14(4):776.
Sun Y, Ling C, Liu L, et al. Effects of Whey Protein or Its Hydrolysate Supplements Combined with an Energy-Restricted Diet on Weight Loss: A Randomized Controlled Trial in Older Women. Nutrients. 2022, 28;14(21):4540.
Ye S, Wang L, Li S, et al. The correlation between dysfunctional intestinal flora and pathology feature of patients with pulmonary tuberculosis. Front. Cell. Infect. Microbiol. 2022,
Ma CJ, He Y, Jin X, et al. Light-regulated nitric oxide release from hydrogel-forming microneedles integrated with graphene oxide for biofilm-infected-wound healing. Biomater Adv. 2022, 134:112555.
Wang X, Hu L, Wang C, et al. Cross-generational effects of maternal exposure to imazalil on anaerobic components and carnitine absorption associated with OCTN2 expression in mice. Chemosphere. 2022, 308(Pt 3):136542.
Xiao W, Zhang Q, Zhao S, et al. Citric acid secretion from rice roots contributes to reduction and immobilization of Cr(VI) by driving microbial sulfur and iron cycle in paddy soil. Science of The Total Environment, 2022, 16:158832.
Gu Y, Chen H, Li X, et al Lactobacillus paracasei IMC 502 ameliorates type 2 diabetes by mediating gut microbiota-SCFA-hormone/inflammation pathway in mice. J Sci Food Agric. 2022, 11.
Liao J, Dou Y, Yang X, et al. Soil microbial community and their functional genes during grassland restoration. J Environ Manage. 2023, 1;325(Pt A):116488.
Ou-Yang YN, Yuan MD, Yang ZM, et al. Revealing the Pathogenesis of Salt-Sensitive Hypertension in Dahl Salt-Sensitive Rats through Integrated Multi-Omics Analysis. Metabolites. 2022, 7;12(11):1076.
Dong Y, Sui L, Yang F, et al. Reducing the intestinal side effects of acarbose by baicalein through the regulation of gut microbiota: An in vitro study. Food Chem. 2022, 15;394:133561.
Zhang G, Wang G, Zhou Y, et al. Simultaneous use of nitrate and calcium peroxide to control sulfide and greenhouse gas emission in sewers. Sci Total Environ. 2023, 10;855:158913.
Fang Z, Chen Y, Li Y, et al. Oleic Acid Facilitates Cd Excretion by Increasing the Abundance of Burkholderia in Cd-Exposed Mice. Int J Mol Sci. 2022, 25;23(23):14718.
Siddiqui NZ, Rehman AU, Yousuf W, et al. Effect of crude polysaccharide from seaweed, Dictyopteris divaricata (CDDP) on gut microbiota restoration and anti-diabetic activity in streptozotocin (STZ)-induced T1DM mice. Gut Pathog. 2022, 17;14(1):39.
Hu J, Wei S, Gu Y, et al. Gut Mycobiome in Patients With Chronic Kidney Disease Was Altered and Associated With Immunological Profiles. Front Immunol. 2022, 16;13:843695.
Li X, Wu Q, Wang Y, et al. UHPM dominance in driving the formation of petroleum-contaminated soil aggregate, the bacterial communities succession, and phytoremediation. J Hazard Mater [J].2024,5;471:134322.
12 comments so far
许佳扬Posted on4:25 下午 - 12月 3, 2020
咨询16S和18S高通量测序一个DNA样本的价格
谷禾健康Posted on10:21 上午 - 3月 31, 2021
您致电询问一下 13336028502/400-161-1580
zhaoPosted on12:32 下午 - 1月 14, 2021
16S土壤样品价格
qingPosted on8:37 上午 - 2月 2, 2021
有没有联系方式
吴Posted on9:54 下午 - 3月 27, 2021
你好,想问一下送检24份动物肠道内容物大概要花多少钱
谷禾健康Posted on10:19 上午 - 3月 31, 2021
您可以联系谷禾牛博 13336028502
时Posted on10:39 下午 - 5月 6, 2021
粪菌16s测序价格和周期,30个样本,直接送样基因组DNA
谷禾健康Posted on1:24 下午 - 5月 7, 2021
周期是1-2周,价格您咨询牛博13336028502/400-161-1580
丁文銮Posted on8:57 下午 - 9月 30, 2021
想咨询一下小鼠肠道菌群的价格和相关信息
谷禾健康Posted on1:34 下午 - 10月 7, 2021
嗯,电话联系400-161-1580或联系牛博 13336028502
房禧霖Posted on4:28 下午 - 3月 21, 2023
您好,我想进行一个药理实验后续的肠道微生物16s 测序实验,区域可能是V3-V4,分为空白、疾病和给药组,每个组6个平行。想问您下肠道内容物、肠道组织或者粪便样品分别的测序的送样要求和价格,以及有没有最低送样数量。
谷禾健康Posted on10:52 上午 - 3月 24, 2023
您好,每份样本肠道内容物在200mg左右,价格您可以电话400-161-1580询价,谢谢关注