-

 CNAS L23010
CNAS L23010

 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室

谷禾健康
链读测序(Linked-read sequencing)通过将相同的barcode与长DNA片段(10-100kb)的序列连接在一起,能够消除其中的一些错读,从而改进宏基因组组装。但目前还不清楚在使用链读测序时参数的选择对组装的质量的影响如何。
近日,香港浸会大学研究人员发表文章 “通过链读测序对宏基因组组装全面研究”。


模拟数据和模拟菌群中的分析结果表明,模拟数据(simulated data)中读取深度(C)与组装序列的长度呈正相关,但对组装序列的质量影响不大,模拟菌群的研究中读取深度(C) 对组装序列的质量以及被注释为基因组草图的bin的比例有轻微影响。
另一方面,宏基因组组装质量受CR(每个短读长片段的平均深度)和CF(由长DNA片段计算的基因组的平均物理深度)的影响。对于相同的读取深度,较深的CR 会产生更多的基因组草图,而较深的CF 会提高基因组草图的质量。
还发现μFL(未加权的DNA片段的平均长度)对组装有边际效应,而NF/P(每个分区的片段数)对局部组装涉及到的偏离目标读数(off-target reads)有影响,即较低的NF/P值会通过减少off-target序列的错读而有更好的组装效果。
总体而言,与Illumina的短读长相比,使用链读改善了组装中重叠群的N50,但与PacBio CCS的长读长相比则没有改善。
人体微生物群是一个复杂的系统,在生理活动和疾病中起着重要的作用。对微生物群中的微生物基因组进行测序可以帮助我们研究其功能。
然而,微生物基因组序列很难获得,微生物群中的绝大多数微生物不能被分离出来进行单个测序。目前的宏基因组项目中使用短读长测序对混合的微生物基因组进行测序。
这些结果在基因组组装过程中是有错读的,导致微生物基因组的完整性和重叠群的连续性结果不理想。长读长测序已经被用来尝试减轻这些问题,如Nicholls等人和Sevim等人的研究。特别是Moss等人的研究,其成果优化了纳米孔测序的长读长文库制备方案,并获得了更完整的细菌基因组。
但实际应用中,长读长测序是昂贵的。虽然链读序列(linked-reads)的基因组组装的质量无法与PacBio CCS的长读长相提并论,但其低成本和高碱基质量的优点是值得去使用的。
01 三组链读序列数据集的来源及构成:
模拟数据(simulated data):
从MBARC-26数据集中下载了23个细菌和3个古细菌菌株,按丰度分类,L-sim,低丰度微生物,摩尔浓度<10-15;M-sim,中等丰度微生物,10-15 < 摩尔浓度 < 10-14;H-sim,高丰度微生物,摩尔浓度 > 10-14
模拟菌群(mock community):
(ATCC MSA-1003)是一个由20个菌株组成的池,同样按丰度分类,L-mock,低丰度微生物;M-mock,中等丰度微生物;H-mock,高丰度微生物;UH-mock,超高丰度微生物。
人类肠道菌群:
一份来自健康的中国人粪便样本
02 DNA提取、文库制备和测序:
对于模拟菌群,从ATCC 20菌株交错的混合基因组材料中提取DNA,不进行大小选择。
对于人类肠道菌群,用Qiagen QiAaMP粪便迷你试剂盒提取DNA,去掉5kb以下的DNA片段。
脉冲场凝胶电泳后,按照厂商的说明制备10x Chromium文库。使用Illumina XTen双端2x150bp测序。人类肠道微生物组的DNA也被用于标准的Illumina XTen短序列测序。
03 DNA长片段重建和链读序列二次抽样:
Long Ranger v2.2.1用于纠正barcode碱基错误,计算PCR重复率,并完成barcode感知的链读序列比对。
使用BWA-MEM v0.7.17比对短序列和没有barcode的链读序列。根据映射得到的具有共同的barcode的短序列的坐标重建DNA长片段。
链接序列首先按barcode排序,然后按它们的映射坐标排序。如果最近的barcode序列大于50kb,则终止延伸长DNA片段。每个片段必须包括至少两个具有共同barcode的成对序列,并且最小长度为2kb。
04 宏基因组组装:
对于链读序列的组装,没有 barcode 的链读序列首先由 metaSPAdes v3.11.1使用默认参数组装为“seed”重叠群,并通过BWA-MEM v0.7.17与重叠群比对。
最后使用 Athena-meta v1.3 通过汇集在 scaffold 中的两个“seed”重叠群里共享相同 barcode 的序列进行局部组装。
05 组装效果评估:
MaxBin v2.2.4将长于1kb的重叠群分组到bins中,并通过CheckM v1.0.12评估其完整性和污染率。
Quast v5.0.0统计了基础信息,如重叠群的N50、NG50、NGA50、总比对长度(total aligned length)和基因组覆盖率(genomic coverage)。
Kraken v0.10.6基于内置数据库MiniKrakenDB为bins做物种注释。每个bins都作为一个基因组草图,被分类为高质量的(完整性>90%,污染率<5%),中等质量的(完整性≥50%,污染率<10%),低质量的(完整性<50%,污染率<10%)
来自人类肠道菌群和Illumina短序列链读序列二次抽样的组装效果统计
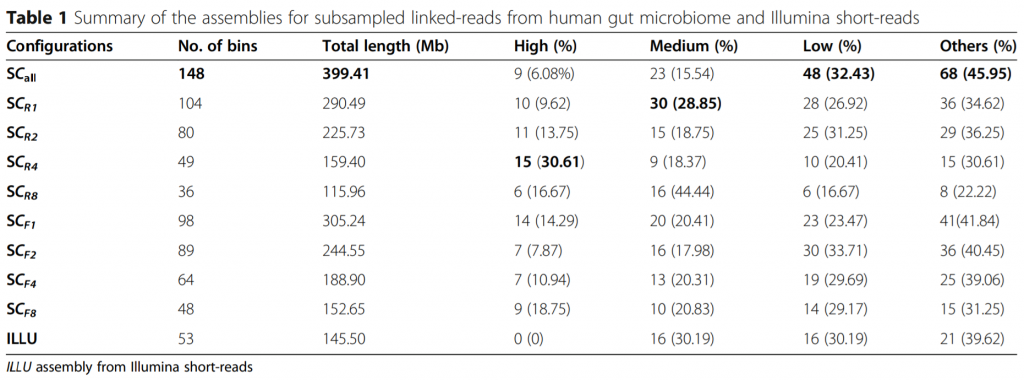

ILLU,Illumina短序列的组装
SC-all,模拟菌群和人类肠道菌群总共的两个测序lane链读序列
在链读测序中,有四个关键参数可能会影响宏基因组组装,如下图。
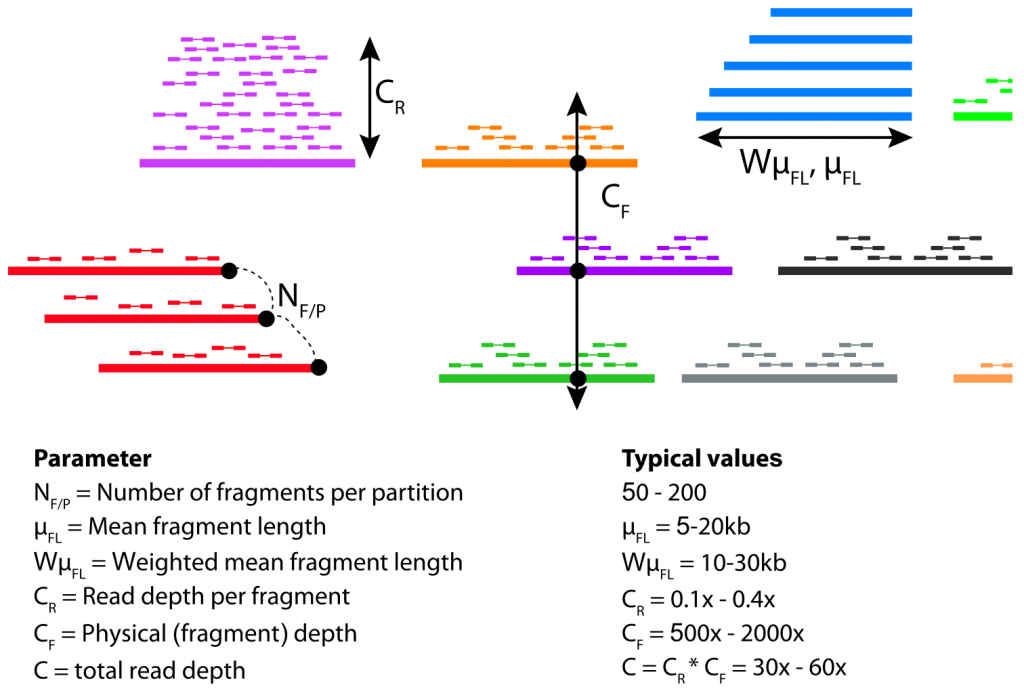

这些参数中有几个是相互依赖的。例如,输入DNA的量越大,CF和NF/P都会增加,CR就会降低;CF和CR的绝对值是由总读取深度(C)增加多少来设置的,因为CR×CF=C。
L-sim,模拟数据中的低丰度微生物,青色
M-sim,模拟数据中的中等丰度微生物,蓝色
H-sim,模拟数据中的高丰度微生物,红色
L-mock,模拟菌群中的低丰度微生物
M-mock,模拟菌群中的中等丰度微生物
H-mock,模拟菌群中的高丰度微生物
UH-mock,模拟菌群中的超高丰度微生物
“-”表示测序lane的倒数,例如MSCR4/MSCF4表示四分之一测序lane的序列被二次采样
MSCR-,模拟菌群中的短序列
MSCF-,模拟菌群中的长DNA片段
MSC-1,模拟菌群和人类肠道菌群总共的一个测序lane链读序列
SC-all,模拟菌群和人类肠道菌群总共的两个测序lane链读序列
相关阅读:
参考文献:
Zhang L, Fang X, Liao H, Zhang Z, Zhou X, Han L, Chen Y, Qiu Q, Li SC. A comprehensive investigation of metagenome assembly by linked-read sequencing. Microbiome. 2020 Nov 11;8(1):156. doi: 10.1186/s40168-020-00929-3. PMID: 33176883; PMCID: PMC7659138.
He S, Chandler M, Varani AM, Hickman AB, Dekker JP, Dyda F: Mechanisms of evolution in high-consequence drug resistance plasmids. MBio 2016;7(6): e01987–16.
Peng Y, Leung HC, Yiu SM, Chin FY. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth.Bioinformatics. 2012;28(11):1420–8.
Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast singlenode solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31(10):1674–6.
Nurk S, Meleshko D, Korobeynikov A. Pevzner PA: metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27(5):824–34.
Nicholls SM, Quick JC, Tang S, Loman NJ. Ultra-deep, long-read nanopore sequencing of mock microbial community standards. Gigascience. 2019;8(5): 1–9.
Sevim V, Lee J, Egan R, Clum A, Hundley H, Lee J, Everroad RC, Detweiler AM, Bebout BM, Pett-Ridge J, et al. Shotgun metagenome data of a defined mock community using Oxford Nanopore, PacBio and Illumina technologies. Sci Data. 2019;6(1):285.