-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
越来越多小伙伴开始关注我们产品的同时也会对产品的准确性,检测使用的技术等产生强烈的好奇日常提问:
检测后能得到什么有用的信息?
这个结果的准确性如何?
通过检测能直接区分患者和健康人吗?
同个人过一段时间做菌群变化大吗?
取样时间不同影响大吗?
疾病之间会不会相互干扰?
……各种问题层出不穷今天就为大家详细解答
以下是分享原文。

非常高兴有机会在这里跟大家分享我们这几年在肠道菌群检测应用于临床和健康管理方面的尝试,以及我们的一些经验。

谷禾成立于 2012年,是最早从事肠道菌群健康事业的公司,技术骨干源自浙大。建立了完整的pcr分子实验室。
我们比较专注,一直在做肠道菌群检测,从整个样本量来说,我们更关注来自于临床的有病理的信息,以及临床辅助诊断的数据样本。所以我们在这方面积累了相当大一部分的数据集。
而且这些临床样本相对来说,对于我们后续将肠道菌群检测应用于临床辅助诊断当中有很大的帮助。

我们其实不只是检测一下肠道菌群的构成,以及哪些菌有异常,我们是希望将肠道菌群检测直接做成一个临床辅助诊断的产品。
不只是告诉你可能有哪些疾病风险,我们希望可以直接给出包括结直肠癌、胃癌、甚至肝癌、抑郁症、自闭症的临床辅助诊断提示。
从这个角度上来说,可能跟目前已有的检测,包括这几天嘉宾分享的一些研究,可能就会有很大的不同。
你可以通过检测找到很多肠道菌群构成的显著性差异,比如自闭症患者跟健康人之间有相当大一部分的菌是存在丰度上的显著差异,但是:
能不能准确告知,哪个是自闭症患者,哪个是健康人,或者是他甚至有中间型的状态?
那么这个过程当中就需要解决几个问题。
第一个问题,首先是准确度
不仅仅是告诉你一个概率的问题,而且需要临床辅助诊断,那么就需要提高样品的处理本身,以及疾病诊断模型的准确度。
第二个是稳定性
大家都知道肠道菌群其实受的影响因素非常多,你的饮食方式、生活、地域、健康状态、甚至情绪状态都可能对菌群有巨大的影响。
这种巨大的影响的来源有这么多的情况,那么如何保证无论你什么时候检测,都能够是可靠稳定的?
假设一个结直肠癌患者,他今天做了,和隔了一个礼拜之后再去做,从病理的状态上来说,他还是个结直肠癌患者,但是他的菌群状态可能会产生巨大的变化。
那么这些变化本身是否会对我们的检测和临床结论产生巨大的影响,这种波动如何去消除,所以这个是个稳定性的问题。我等会也会讲到。
再一个是可解释性
因为菌群相对来说,实际算是一个大的数据。我们现在如果采用高通量测序的方式来做,一次性可以拿到大量的数据集。
这些数据集本身会有各种各样的菌的构成差异,我们的经验是差不多每个人,从婴幼儿开始到成年人,大概两岁以上的婴幼儿,菌群构成会从200到2000种菌不等,也就是说每个人会有这么大的菌的种类。
但是总的数据集有多大呢?我们自己有几万例的人群的样本,构建了一个人的肠道菌群的参考数据集,这个数据集里目前包括7500多种菌。但是我们自己的评估,全人类的肠道当中可能出现的定植菌应该会超过10万例。
那么这么大的数据量当中的异常菌如何去进行解释,如何给临床上提供更有意义的,病理上也好,或者机制上的一种解释,以及可以量化的干预方案,这就是可解释性。
最后一个是经济性
因为如果希望肠道菌群检测能够作为一个临床辅助诊断的项目,或者是针对具体的临床疾病的辅助诊断来说,它不仅仅要做到准确,它要具有足够大的经济性。
也就是说成本必须要得到控制,几千块钱的项目可能能做,但是它无法做到普及,也无法在临床当中被大量的应用,所以如何控制成本也是个巨大的一个问题。

我们做了很多的工作,尝试在上面提到的方向去努力实现在临床应用当中的可能,以下几方面我后面会逐一讲一下我们所做的工作。
第一是取样和储存运输,然后是如何大规模、低成本、高效准确的去处理样品。
再一个是参考数据库,完整的数据库的建立,其实也是非常重要的。
然后是大规模人群队列和临床数据。我们的核心经验,由于肠道菌群的多样性,以及受各种因素的影响比较多,那么大规模的人群队列就变成一个非常重要的点。如何去构建大规模的临床队列,以及从这个大规模的临床队列当中,我们能不能拿到一些信息和有用的经验。
再有是全方位的解析,我们等会儿会讲到不只是在菌群层面上,也不只是在代谢层面上,我们甚至可以基于肠道菌群,把代谢营养,生理生化指标,免疫指标,我们都是来自于临床的,包括血常规,尿常规,我们都能够进行解析。
还有重要的一点是如何使用人工智能的高可用性的模型,从这么大的数据当中精细化的提高检出率的同时,又能够保证它的特异性和敏感度,这是个巨大的一个挑战,这个我后面也会讲到。

第一个方面,可能我们现在采用的这个取样方式,应该相对来说最简便和最小的一个取样方式,我们直接可以用棉签从厕纸上蘸取,直接洗脱在取样管当中。
你可以看到取样颜色达到左侧第二个管子的这种颜色的粪便量,我们就可以完成整个检测,从使用体验上来说,会比较简便,而且需要量少。
样本保存也可以在室温下至少可以保存一个月,运输过程当中就不需要涉及到冷链,可以直接快递,便捷性也会大大提高。
有了这个取样管之后,实际上从临床和门诊当中可以快速的拿到大量的方便的样品,因为不需要采用非常复杂的取样和储存的方案。
我们讲第二个方面,刚才提到菌群的构成特点是很多样性的,而且跟很多因素包括取样时间有关,比如说早上取、晚上取,取的粪便的部位以及取样的量的多少,可能出来的菌群构成都会有一些区别。
如何再将这些区别和波动有效地控制,并且从中提取稳定准确的信息
这就涉及到一个我们能够从菌群数据当中能拿到一些什么结果,我主要从几个维度来讲。
——首先是菌群丰度和菌群结构
你首先可以知道每一种菌大概有多少的量,相对比例是多少。你还会知道菌群构成,也就是说都有些什么菌。
——然后是菌群结构。
所谓菌群结构就是说,有一些菌它会共同出现。甚至你会发现你检测到了有几种菌,并不代表其他的菌可能就没有出现。我们的肠道菌群总共可能会有七万多种菌,每个人差不多200到2000种,但是在99%的人当中都出现的菌,可能不超过30几种。你的肠道当中有这种菌,但是很多人当中都没有这种菌,那么很多的信息是稀疏的。但是通过构建菌群结构之后,你会发现这两种菌,可能一个在你这里有,一个在另外一个人当中有,但是这两种菌它其实代表的意义和内涵是类似的。
——再一个方面,我们通过数据的模型的挖掘,可以拿到更高维的特征。
这些特征反应的是生理的,比如说你的尿酸量,你的尿蛋白,你的血液当中的高密度脂肪酸,包括一些代谢的指标,这些指标的生理的特征和病理的特征,我们也可以通过菌群结构来进行提取。
那么从信息的维度上提升了之后,你可以看到数据的稳定性在不断提高。
最底层数据菌种的丰度波动性是非常大的。前一天的饮食如果有改变,跟你日常的饮食习惯有一些稍微的改变,第二天的检测,菌群的构成丰度上,波动甚至会达到30%。这种菌群丰度的变化,就代表如果你单纯依据少量的几种菌的丰度变化去检测它的异常,或者是这个疾病的状态的话,稳定性是很差的。那么你就需要控制各种场景,各种条件,才能拿到一个稳定的结果。但是菌种丰度又代表了一个非常高的信息量。
那我们尝试的更多的是从第二个维度开始,就菌群的构成,菌群的结构以及高维的菌群特征这个角度,因为它的稳定性更好。我们通过不断的去加入各种各样的临床病例的数据的方式来提取这些菌群的附加信息。
这就涉及到第二个问题,我们要把这更多维度的信息量能够提出来的话,你就必须要有涉及到非常大规模的样本人群,包括疾病状态、年龄、社会生活区域、饮食方式等各种因素的情况。
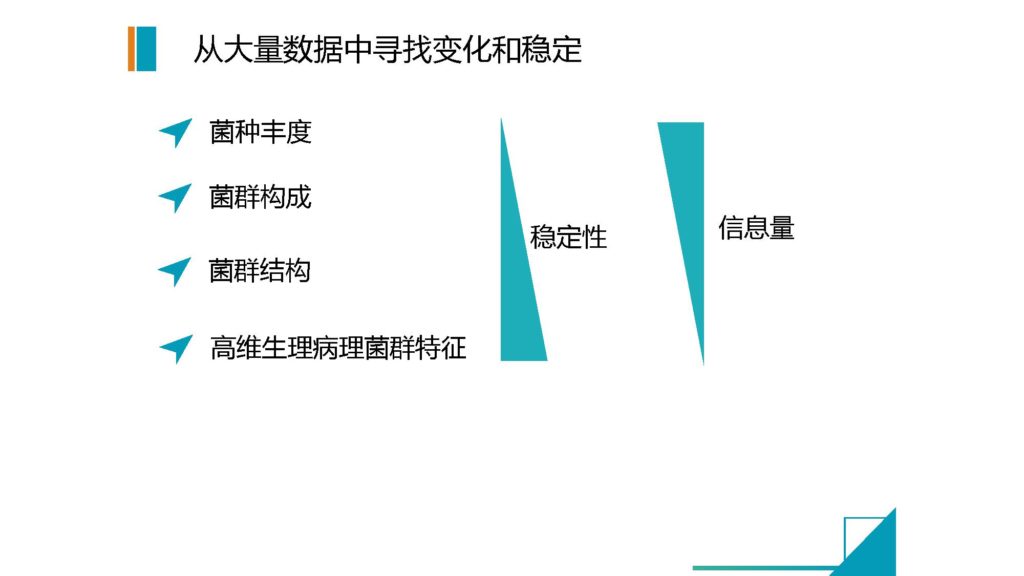
那么大量样本的话,我前面提到我们在取样盒上的改进,对应的我们还提供了一个快速的提取方式。就是通过磁珠法,来完全全自动化的来配合我们的取样盒,来做到大规模,自动化的,低成本的快速提取。
因为一般来说像MoBio这一类的试剂盒它对于样本的起始量有一个比较高的要求,并不适用于我们前面那种非常低当量的一个量。
我们自己改进之后的方法,稳定性和可靠性也是相当高的,这样是极大地降低了我们的整个实验处理过程当中的成本,同时又能够有效地保证这个检测结果的可靠性。我们的方法已经有文章发表。

那么当有了大量的样本之后,第二个问题就是
需要你更精准,更精细化的去提取这些数据
提取这些数据的过程当中我们自己也做过比较。用公共数据库包括Greengene、RDP或者HMP这些数据参考集,我们大概也就只能最多到95%的数据是能比对上去的,到种属的鉴定甚至会更低一些。
我们自己大概用了24000例的来自全球各地的样本,包括我们自己大概测了将近有17000多例的我们早期测的样本,还有各种来源的,特征的,包括疾病状态的,包括我们纳入了从各种基因组数据库拿到的肠道疾病和人体病原物的这些菌的数据,总共汇总之后,我们有24000多人的样本。
最后,我们构建了一个全新的人体肠道的一个参考集,这个参考集大概有75000多种OTU的菌,然后我们做了大量的注释,超过99.5%的菌是都能够完成比对的,这就大大的提高了对于菌属和样品当中菌构成的分辨率。
我们目前总的样本量已经接近快20万了,估计今年应该会超过20万例。
多种相关疾病互相存在干扰
这个是遇到的另外一个问题。
当我们解析了这些菌之后,我们尝试去做不同的疾病状态下菌的构成丰度和这些菌的特征信息,我们去尝试做疾病的分类。
前期做的时候效果相对还是非常不错的,因为它的特征菌比较明显。但是实际上面对临床的时候会遇到第二个问题,临床当中没有一个人的样品是非常干净,他可能会有结直肠癌,但是同时又会有高血压,或者是有消化道的疾病。
这些样本在你做检测之前你其实不知道他的状态,在试验或者研究型论文当中,你可能做的队列一个是健康人,再加一个某种疾病的患者,那么这两类的样本做出来,统计差异是非常明显的。但是如何在临床样本当中做到非常精准地将这两类人区分,而且不受任何中间因素的干扰,比如说阴性干扰样本的这个影响,这是需要面临的问题。
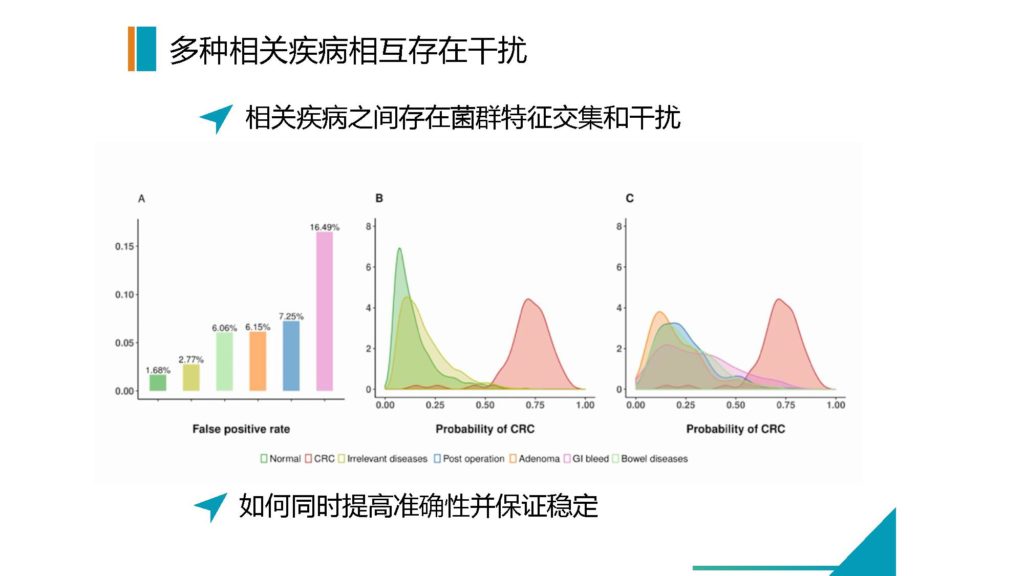
图解
上面图的左侧,我们自己做了一个结直肠癌的模型。结直肠癌我们现在检测的准确度可以达到非常高了。
但是,最开始做的时候,其实我们做了一下测试,单纯的模型去做预测分析的时候,会有其他中间疾病的大量干扰。尤其是消化道出血的情况下,会对整个菌群状态有非常大的影响。
包括腺瘤的阶段,刚才几位也都提到肠癌,肠癌其实是一个菌群变化要早于癌症发生的过程。但是菌群变化和癌症的阶段是有一些特征性的影响的,那么腺瘤的阶段跟肠癌是有大量的菌群特征是重叠的。
我们前期由于收集来自于各个来源的病例样本,所以可以大量的去寻找哪一些疾病是和我们要检测的目标疾病存在干扰因素的,我们可以挑选出这些疾病作为一个控制集,那么可以大大的减少假阳性和干扰的因素。
这也就是另外一个因素,就是我们在构建人群队列的时候,务必不能以一个相对干净的样本集去做。对于研究来说,它可能是很好的一个方式,你可以做前瞻性来寻找这是否可能以及效果。但是实际临床过程当中,你需要纳入大量的,可能影响你这个菌群,或者跟这个疾病有相互干扰和影响的相应的疾病来作为控制集,才能够提高它的可靠性和准确度。
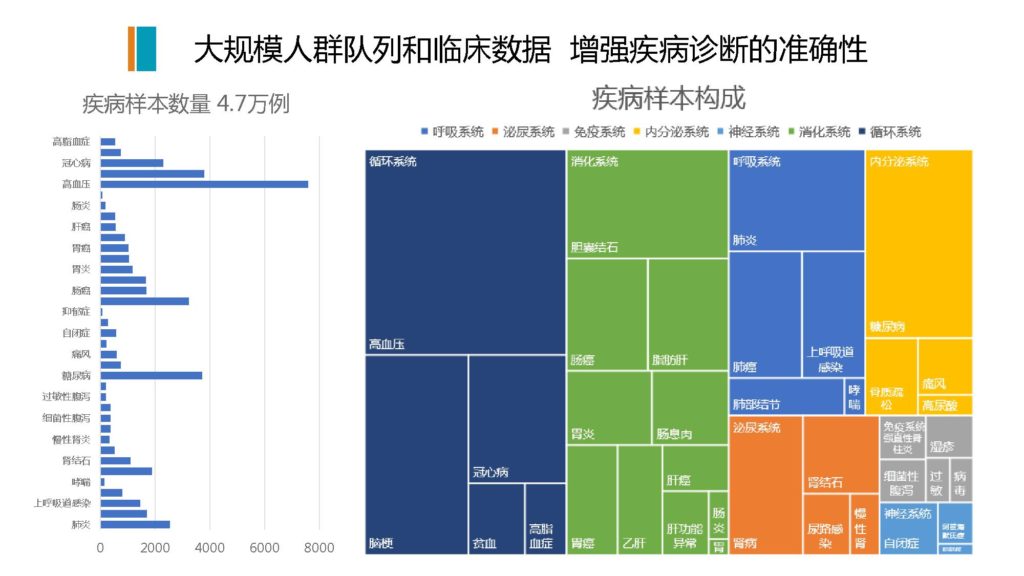
图解
这个图是我们自己有完整的临床病例,我们跟大量的医院和研究所在合作,我们自己构建了大量的人群队列规模,全部都是住院病人,有明确的临床的诊断和所有的病例信息,这个样本规模差不多有4.7万例。
图的左侧是各种疾病的类型,我们也通过各种疾病和菌群的模型构建,分析了七大类系统,包括呼吸系统,泌尿系统,免疫系统,内分泌系统,神经系统和消化系统,还有循环系统,跟肠道菌群能够有相对可靠的临床应用和检测,用于临床疾病的辅助诊断的可能性的。
右边这里是一个疾病的构成,其中有很多病跟菌群的关系目前甚至都没有发表过论文,就是说并不知道肠道菌群跟它有关。我们实际通过大规模的临床样本的实际筛查和模型构建之后,发现有很多病,通过肠道菌群可以做到非常精准。
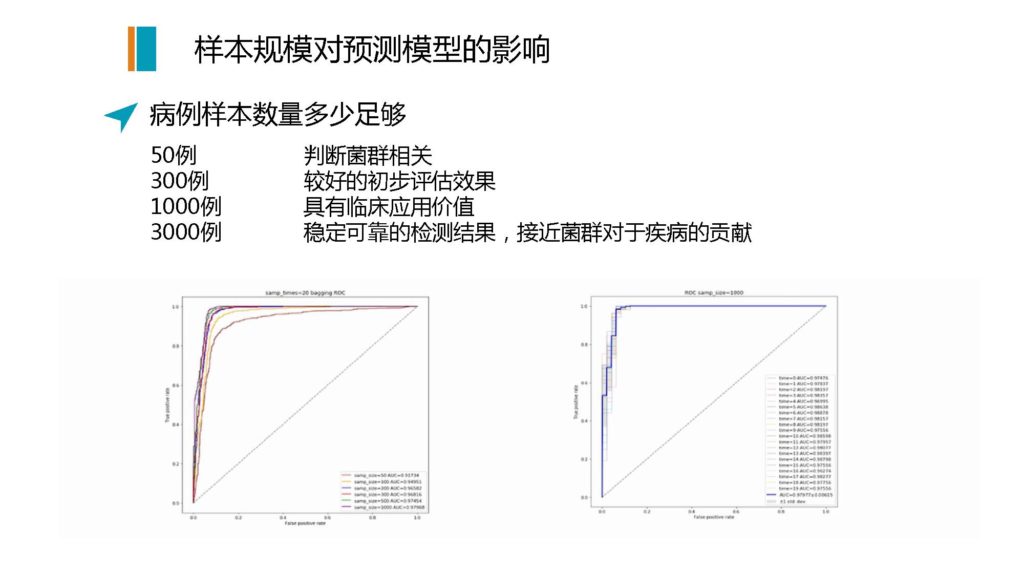
另外一个问题就是,我们对于一个病的预测也好,或者进行辅助诊断也好,基于肠道菌群
需要多大样本的量才能够做到足够的准确度
来看一个我们自己做的一个模型,是拿实际真实临床样本的数据来做的
图解
这个图实际上是拿二型糖尿病的患者的诊断来做的,可以看到不同的曲线丰度。
我们自己做了从50例、300例、1000例到3000例,这些都是病人的样本量,对照集的样本量一般会在两到三倍的量来进行构建模型。
从我们自己的这个模型数据来看,50例的样本,你可以有效地判断菌群到底能否对这个疾病进行一个相对较好的评估;那么如果是300例,你基本上可以拿到一个相对可用的模型,进行初步评估。
如果是要达到一个相对稳定的有临床应用价值的模型,我们认为差不多要1000例。因为你要纳入各种来自于不同临床疾病状态的样本,因为可能这个患者虽然有这个病,但是他同时还会有其他的疾病,包括不同的年龄和饮食习惯的这些背景因素要做控制,至少要1000例。
如果想要得到稳定可靠的检测结果,而且因为不是所有的病,菌群都是在其中起到绝对性的作用,有些是属于间接的,那么你希望菌群本身的检测,它需要有一个贡献度的上限,也就是说,菌群本身与这个疾病的参与度和关联性的上限。那么要达到这个上限,我们认为差不多要3000例的临床的阳性样本,就是病例患者的样本,可以达到上限的结果。
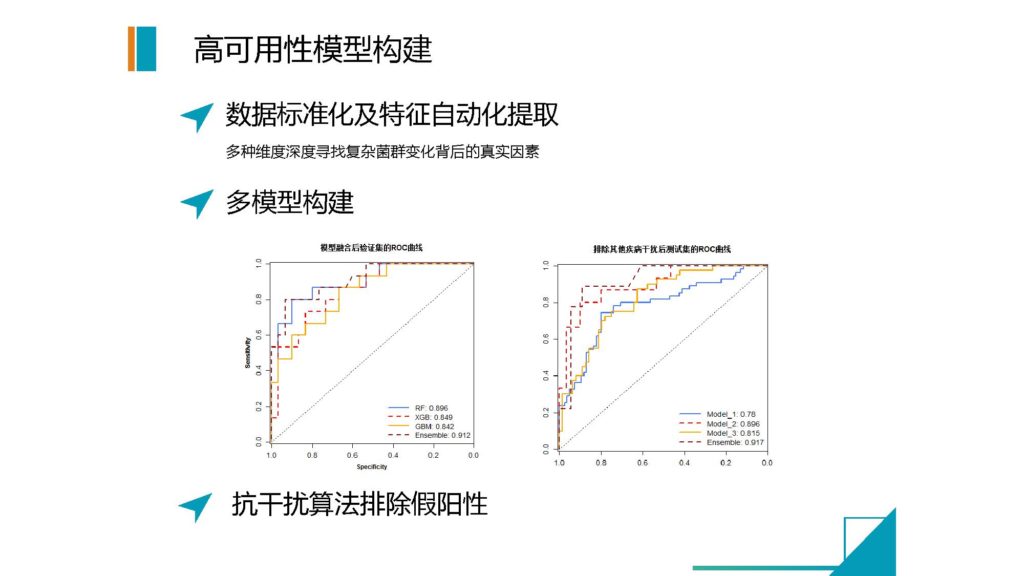
再下来,就是我们需要构建可靠的模型。
因为菌群是一个相对数据源,你的各种生活方式,疾病状态,营养健康的情况都会影响它。可能这个菌既在肠癌当中属于特征菌,同时也是一个炎症性疾病的特征菌,那么这些状态都会影响同一个菌的结果。
如何将这个菌的结果的变化反馈到去解释它到底是哪一个病的问题呢?
我们通过数据标准化和多维度的提升来构建一个判断的模型。
我们用人工智能和深度学习的方法,通过足够大的样本数据,来提取各种各样的菌群特征,并不直接用菌群自身的信息,而是用高维度自主学习的方式来提取这些菌群特征。
然后纳入各种各样数据,
比如有营养学的数据,有质谱的数据,
也有一些病理的数据,包括一些生理生化的数据,
都纳入之后,我们去解析它。
而且我们并不是用一个模型来做,我们现在是用三个模型来做。
我们第一轮是将所有的可能的干扰性疾病和有影响的疾病,全部作为一个病的类型,来进行模型的分析,筛出所有可能有问题的人。
然后第二轮我们需要精准化的去提取,到底哪些病是明确就是单一这种疾病的。
第三个模型,就是我们要对目标疾病与其他干扰的疾病进行区分。
通过多个模型之后,我们可以极大地提高菌群检测的稳定性,以及这个疾病当中的特异性程度。

图解
这个图是我们自己在做的一些疾病的检测结果。从目前来看,很多疾病的稳定性和效果都相当不错,这里每一个病至少都有将近500例病人的样本数据,来去做一个验证。每一个疾病的类型,我们差不多都有两到三个中心的检测结果数据去汇总。
通过多维度之后,我们就可以探寻不同的菌群变化背后,它可能真实驱动的因素。
我们还加入了营养的部分,这些营养其实我们是通过营养调查和一些质谱的数据,然后通过机器学习的方式来去把它解析出来。
我们也加入了像血常规,尿常规,生化,免疫组化,代谢组产物指标,肿瘤标志物,还有激素水平。我们将这些数据纳入之后,通过构建模型,可以将菌群的结果转换为这些生理生化相应的指标。也就是说,你如果只给我菌群的数据,我可以将这些生理生化的相应指标也能够给你体现出来。
甚至还有包括艾滋病的特征的,以及另外一些其它疾病,这里没有列出来,但是效果也是相当不错。
但是你可以看到,它的解释并不是指这个菌群直接的特征变化。我们通过菌群解析,像艾滋病,我们有免疫组化的数据解读了之后,我可以明确告诉你,CD4和CD8的比值会有特征性的差异。但它本身的菌群特征上并没有直接体现出这个东西,是通过生理生化指标的转换之后,我就可以告诉你,菌群特征的变化在具体哪些生理生化方面产生一些影响。
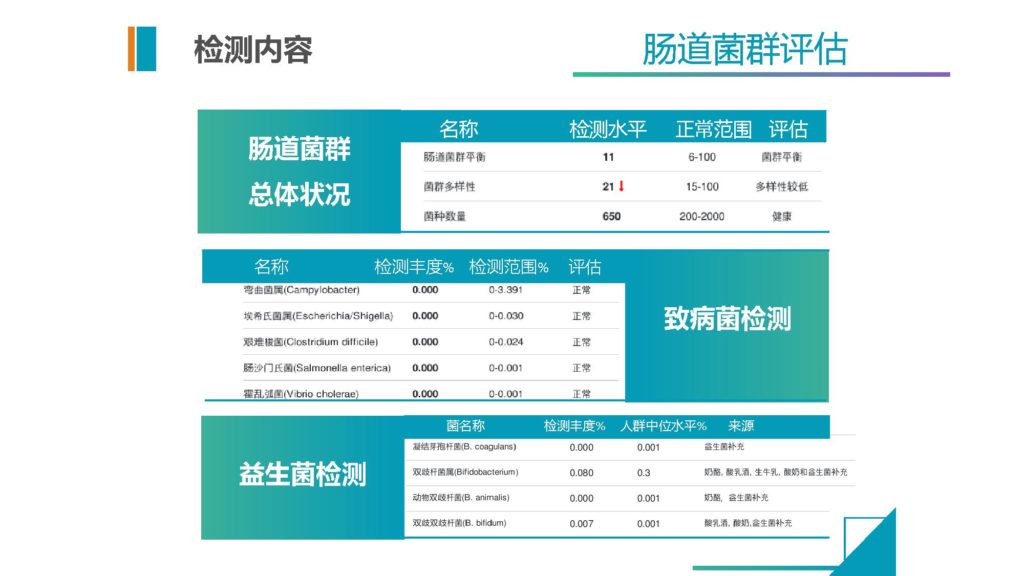
这个是我们现在提供给包括临床和健康检测的一些基本的内容。
可以提供菌群的总体状况,以及致病菌的情况,益生菌的情况。因为本身测的就是菌群,所以它直接就能提供这些最基本的一些信息。当然正常范围都是我们基于将近上万人的健康人群来做的正常范围的评估。
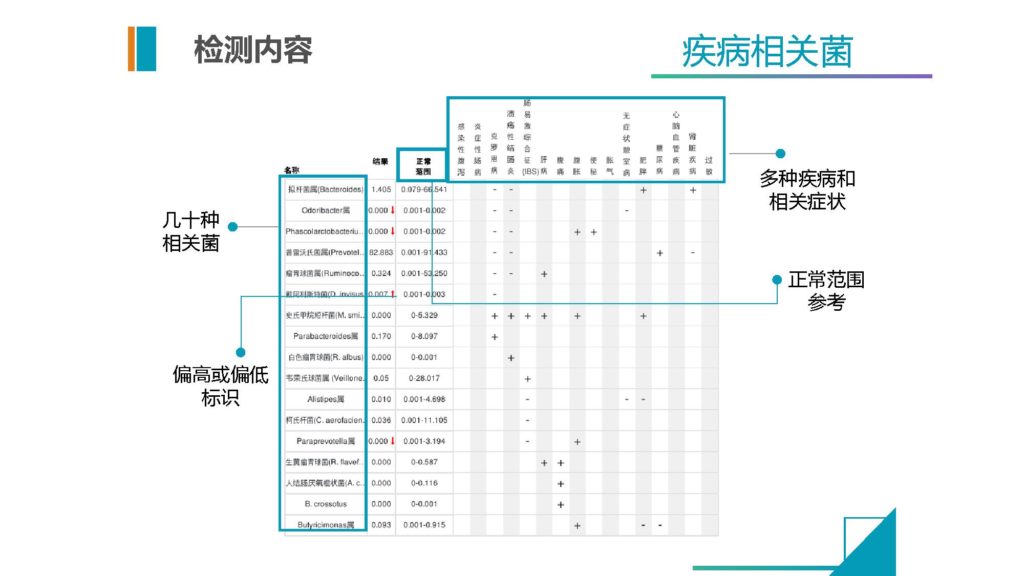
这个是除了来自文献之外,我们自己通过算法提取到的相关的菌,每一种菌在这个疾病内的相关性情况,以及它在正常人群的基本的正常范围是多少。然后我们通过检测这个是否异常,在临床当中给医生来做快速判断的一个内容和信息。
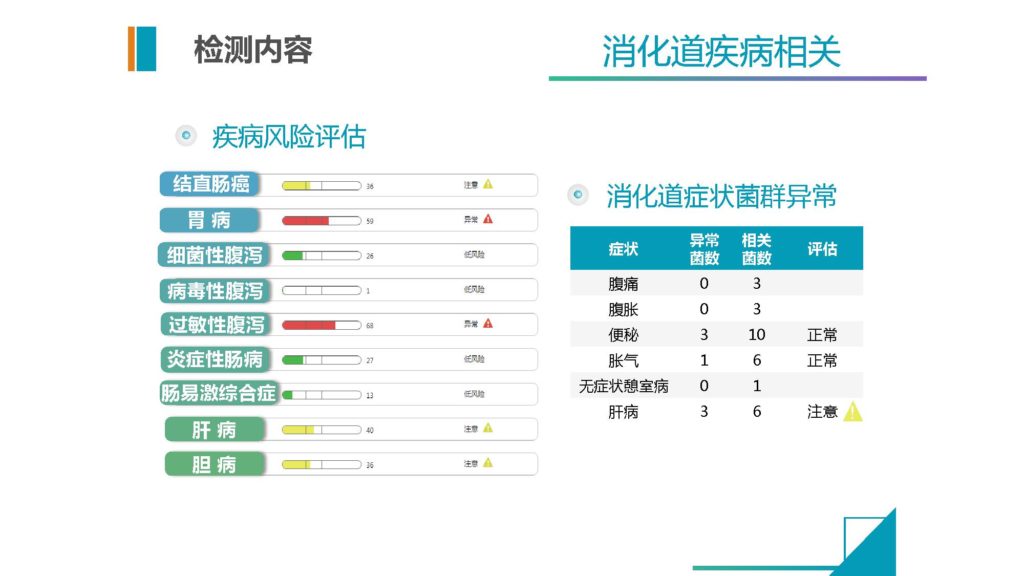
再有一个我们现在给一个疾病的辅助诊断,这些部分它可以相对有效的提供整个维度的不只是一个专科的信息,可以给到包括我们前面讲到七个系统的相关疾病的一个提示。
这些提示可以帮助我们来做一些专科性的疾病辅助诊断,能排除一些其他科室可能漏掉的一些疾病症状。另外包括消化道症状的解读,我们也会有一个菌群异常的评估提示。
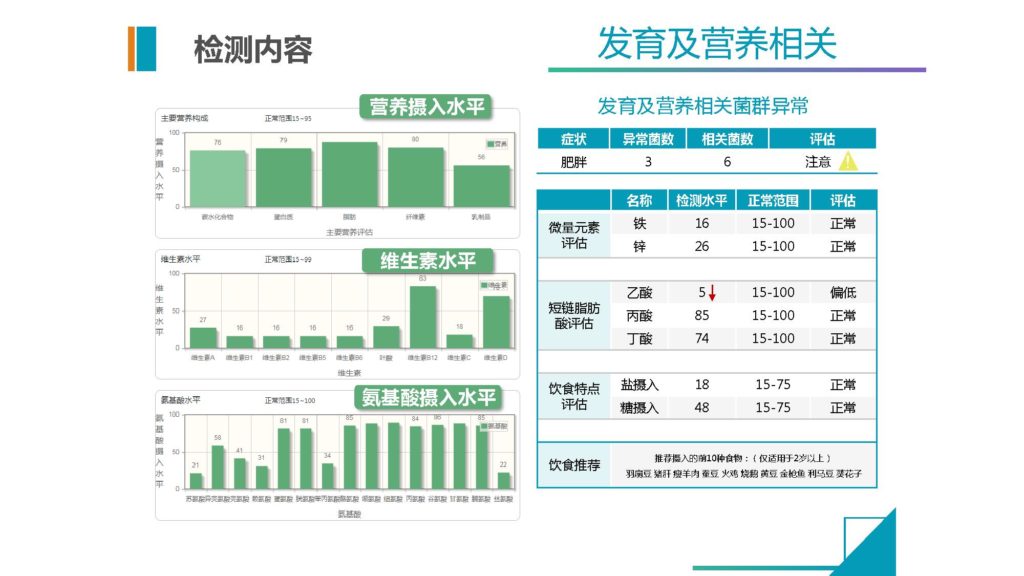
包括营养的部分,我们也单独加入营养摄入水平,维生素摄入水平,氨基酸摄入水平。
这个值目前来说我们是基于人群分布数据,就是说我们通过菌群来预测模型构建之后,评估出人群基础的水平,然后再基于人群的营养调查的水平,我们做拟合。现在来看,准确度还是相当的高。
那另外也包括微量元素,现在重金属的部分我们已经完成了,也很快会加入包括饮食特点、盐摄入、精制糖摄入对应的信息,还有短链脂肪酸的这些指标。

另外这里还有包括有心脑血管、神经系统的疾病的风险评估,包括过敏的一些问题。
过敏的话现在我们也在开展一个比较大的多中心的项目。关于过敏Broad Institute(博德研究所)去年还是今年有一篇文章,他们做的是一个大的欧洲的队列。
也就是通过菌群的数据,从刚出生开始持续采集,差不多到六岁,然后再去评估过敏以及过敏原。目前他那个数据我们做过测试,完全基于菌群的数据,对于过敏包括过敏体质的评估,我们差不多现在能到0.78左右。那如果你是做特异性的过敏原的检测的话,我们甚至也能够进行过敏源的评估,完全基于菌群数据。
所以我们自己的大量的数据检测完了之后,会发现,通过菌群数据本身,不止是能够告诉你菌群的结果,甚至能够反映非常非常多原来你意想不到的,需要用其他手段来进行检测的结果。
另外,我们现在的检测全部是基于16S,16S大家知道它本身的精细度可能还是有缺陷,就是它并不能到菌株;另外它只测细菌的部分,你的病毒和真菌是没有的。
但是我们这里可以看到有一项检测是病毒性腹泻,就是说我测的是肠道菌群,但是我们能够发现,这个病毒的感染,也会对整个肠道菌群产生一个巨大的影响。
所以我们通过整个菌群结构的特征变化,能够只检测细菌,也仍然能够发现病毒性的这种变化。

最后我说一下挑战。
第一个挑战,到目前为止我们做了这么多的数据和这么多样本量之后,竟然发现要真正去完整的解析整个菌群的特征,我们需要更大规模和全面多维度的数据集。不只是菌群检测本身的或者疾病的信息,我们需要纳入比如代谢组,以及其他的一些数据的情况,来构建更完整的数据集。
第二,我们发现不同的疾病,它的诊疗需求和特点是不一样的。虽然信息很多,但如何去跟临床辅助诊疗特定的疾病去做对接和结合是很重要的一点。
第三,我们现在的肠道菌群干预的手段其实也蛮多的,但是这些干预手段呢,现在缺乏量化,就是如何去评估每个人的干预手段,包括饮食的习惯,益生菌的菌株的水平的评估,益生元的效应,甚至粪菌移植的配体。这些量化的方面需要有大量的工作去做。
以上是我们这么多年做的实际临床大量样本的菌群检测的一些经验,跟大家分享一下,谢谢。