-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康

16S科研项目是一个完整的闭环,前期的课题项目设计方案、取样和重复实验设置决定了后期分析报告的数据完整性和项目类型。
想要拿到一手有利用价值的科研报告和项目数据,前期的实验方案设计和后续的分析都起着关键性的作用。
然而有时候拿到报告不知道如何去解读,这里为大家梳理一下16s科研项目的全过程,帮助大家更好的了解报告内容,快速获取关键信息。


实验方案设计就像一个总工程的设计图纸,决定了未来科研分析报告的类型走向,并且前期的分组设计的越详细,各种理化指标、生化指标、代谢物等信息准备越充分,后续报告的完整度越高。


明确项目课题类型
第一步要做的就是明确项目课题类型:
最常见的就是多分组之间差异分析比较:例如,要比较对照组、模型组、实验组,之间的差异结果。
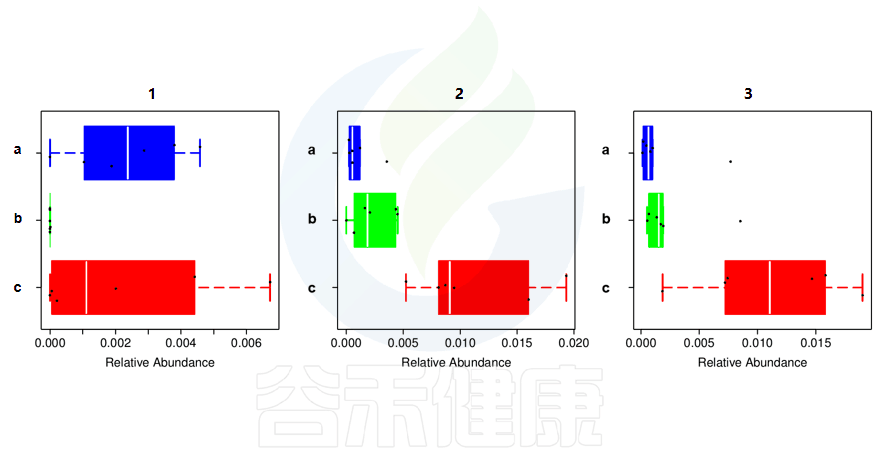

还有多分组中,任意两组之间比较:例如某实验设计了正常组、疾病组、用药组服用奥氮平、阿立哌唑、氨磺必利、利培酮,像比较不同的用药组和疾病组之间的菌群的差异结果,就用到了分组之间两两差异比较。
✦举个例子
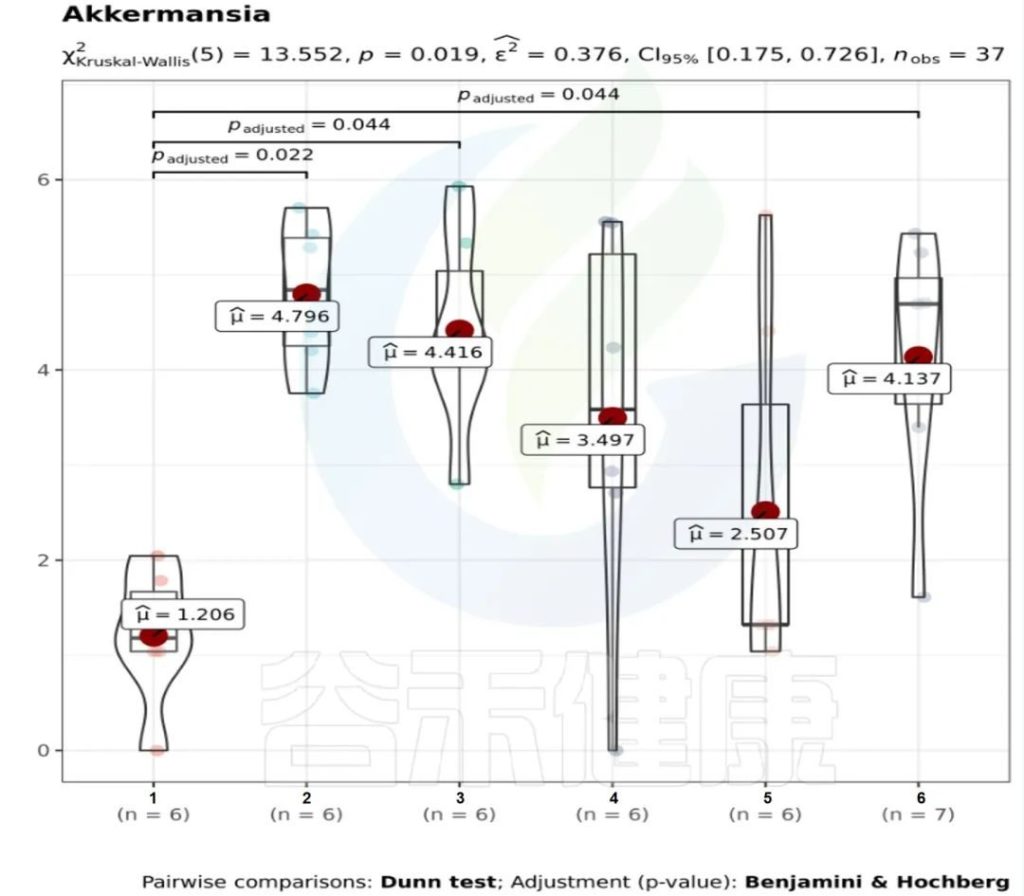
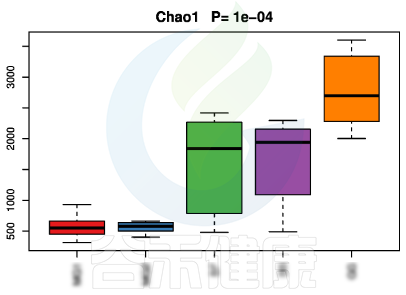
图中1组与3组、4组、6组 组间差异显著
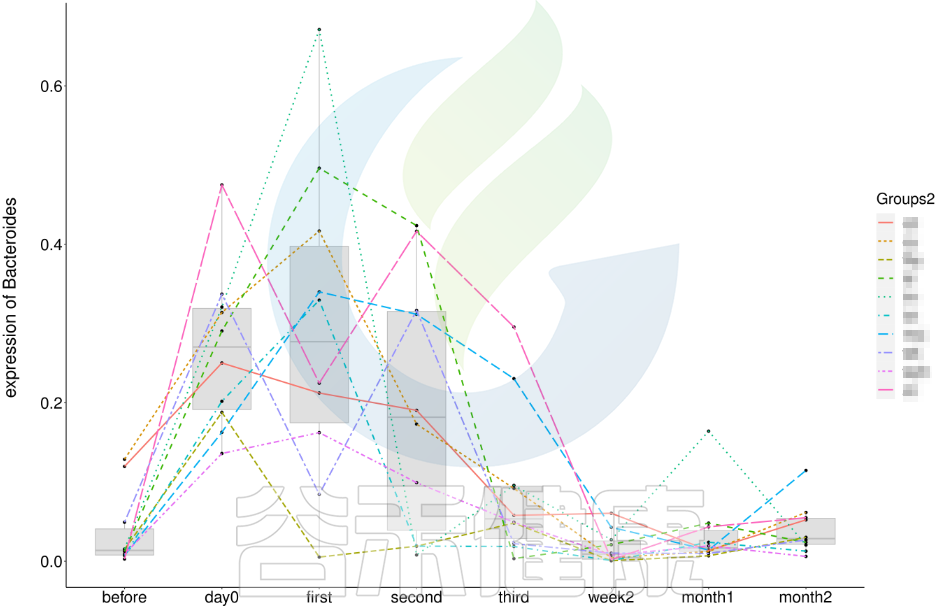
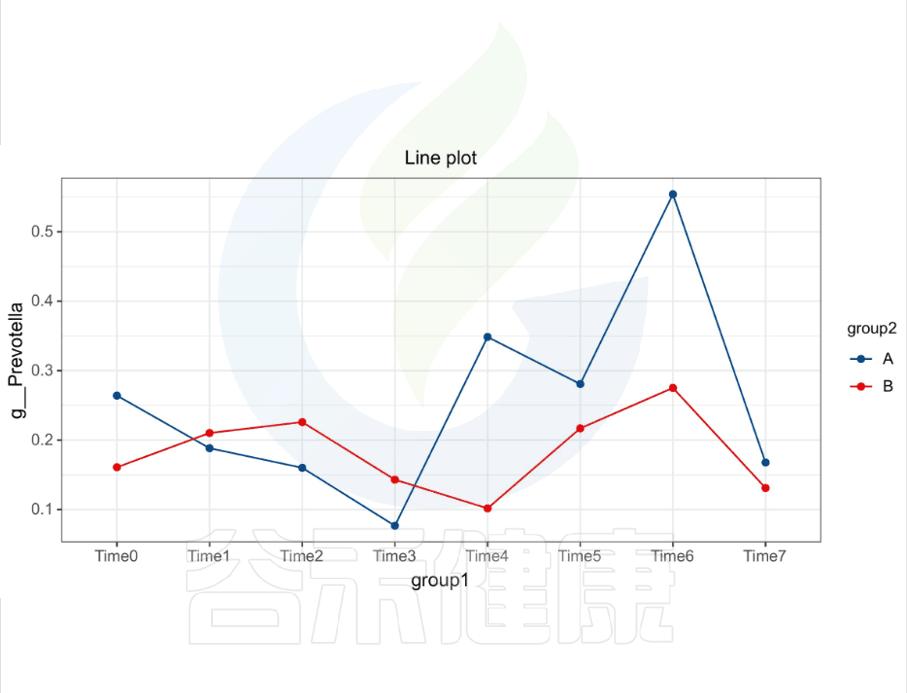
还有随时间的变化比较菌群之间的变化规律:例如在用药不同时间段包括3天,5天,2周,1个月,2个月,观察菌群的变化情况。如下图所示:
收集理化指标非常重要
如果前期搜集好每个样本的相关理化指标,还可以计算这些指标与菌群之间是否具有相关性。
✦举个例子
例如该项目比较自闭症儿童与正常儿童的菌群差异。客户在样本信息单里还详细搜集了母孕期的各种详细指标,例如孕期天数、出生体重、白细胞介素6、肿瘤坏死因子a、五羟色氨等数值型理化指标。
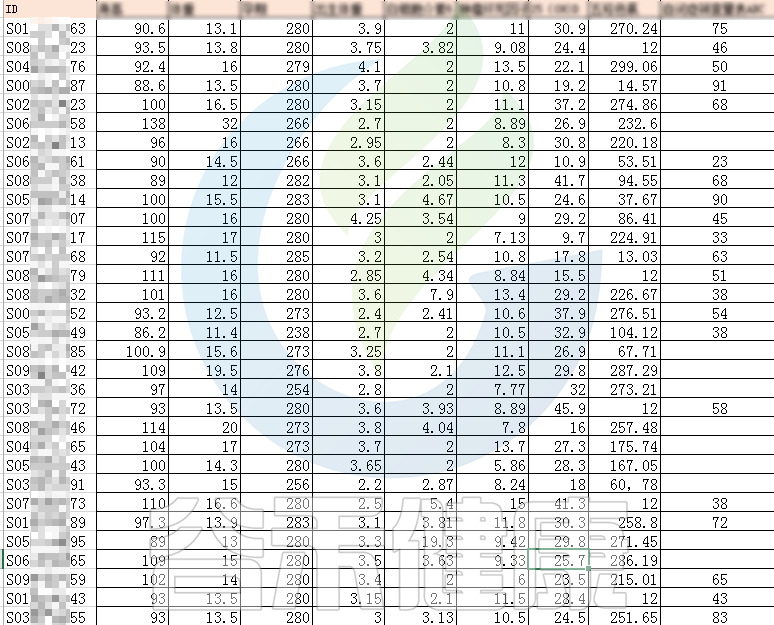
还搜集了是否顺产、是否妊娠高血压、是否孕期感染、是否妊娠糖尿病、是否先兆流产等因子型理化指标。其中0代表否,1代表是:
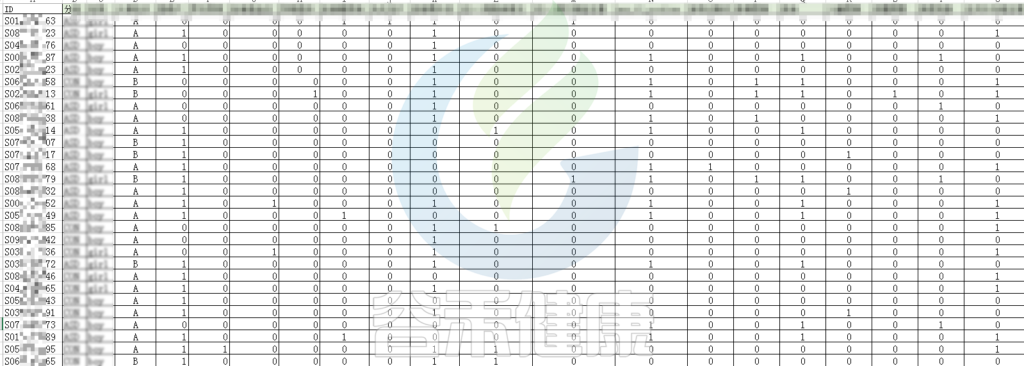
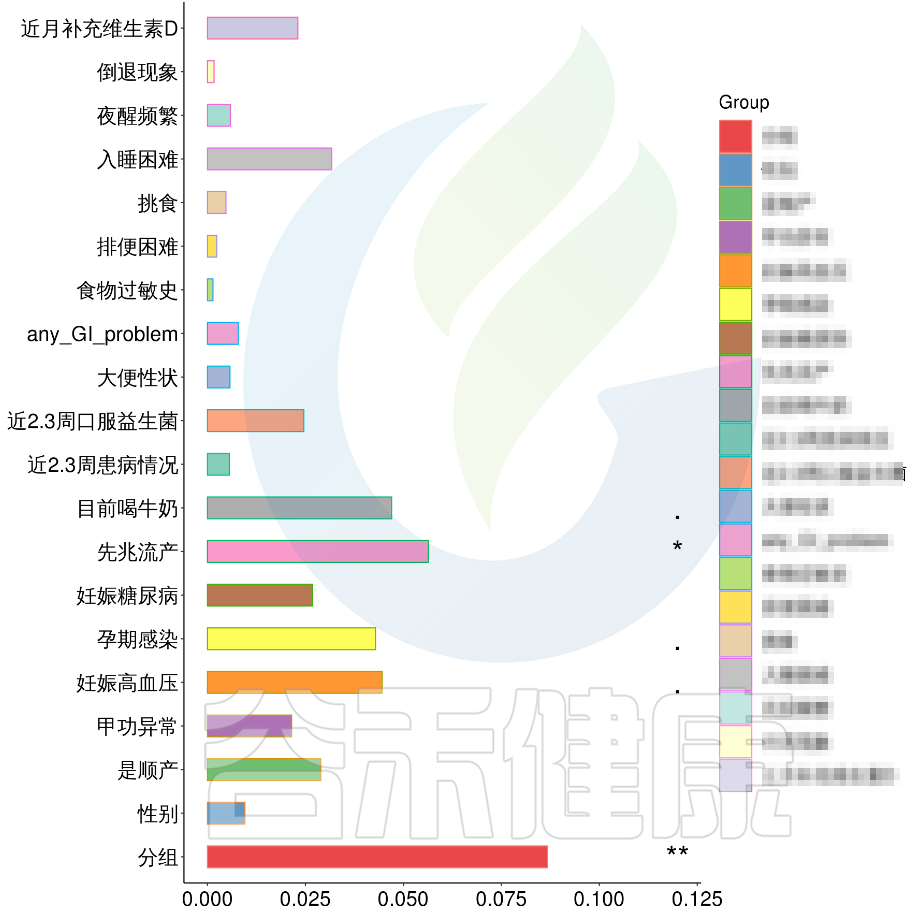
根据这些理化指标与菌群数据做相关性分析,从因子型的结果可以看出,自闭症(ASD)与正常儿童之间的分组与菌群之间相关性极显著**,其次是否有先兆流产的分组与菌群之间有显著相关性*,其他的包括是否喝牛奶、孕期是否感染、妊娠高血压都与菌群有相关性。
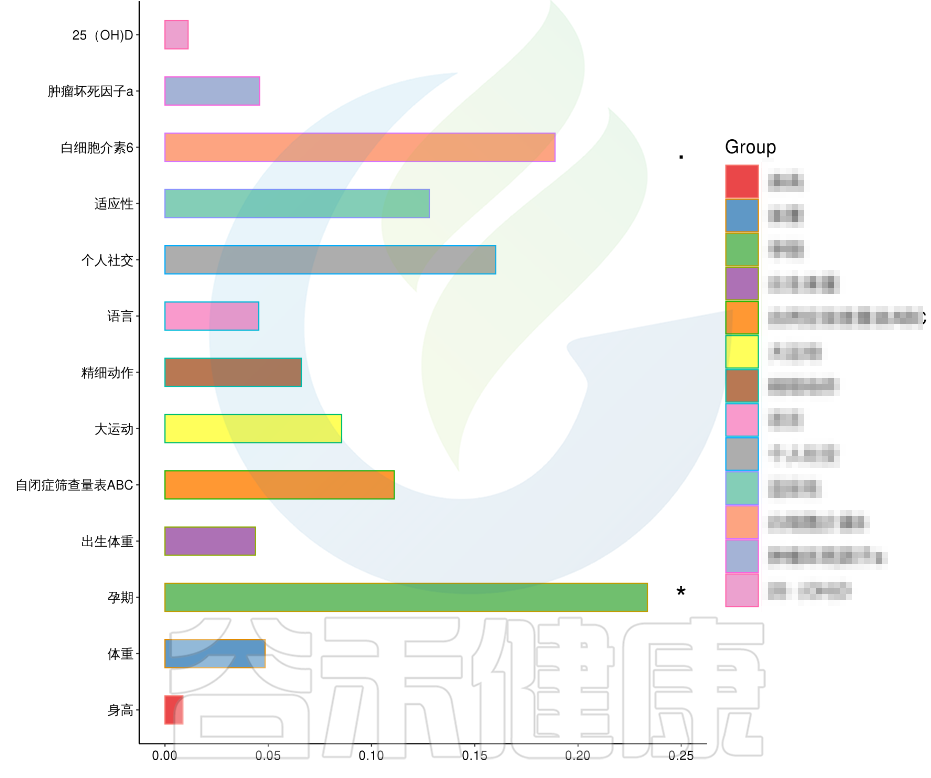
在数值型理化指标中,孕期的天数与菌群之间相关性显著*,其次是白细胞介素6与菌群之间有相关性。
小结
因此,前期搜集相关资料越详细充分,对分析报告的完整性也会有帮助,分析人员也会根据您的样本信息单提供的相关内容,做出个性化的分析和售后指导建议。
首先基于样本类型,最常见的环境样本来源是人体、动物、土壤、水体等。而人体中的肠道菌群样本是目前研究最广泛,可鉴定的物种也最为丰富,谷禾在肠道菌群与人体健康方面有深入研究,目前已完成超20万例临床肠道菌群样本检测,并构建了超过60万各类人群粪便样本数据库。
其他样本类型还包括人体/动物唾液样本、组织样本、尿液样本等。
▸ 粪便样本
目前粪便样本从采样到提取数据分析技术较为成熟、应用较为广泛,谷禾最早在15年就开发了针对粪便样本的取样管,也是最早致力于研发粪菌取样盒的公司,方便实验室、个人日常取样需求,实现了粪菌样本的常温运输。
谷禾取样管常温保存,取样也较为方便卫生,在家就可以轻松完成,相较于传统取样方法都有所升级。并且该取样管也有专利证书。该取样方法被大量客户采用并接纳,大大降低了采集粪便样本的难度,缩短了搜集样本的时间周期。
取样示意图

▸ 其他样本
土壤样本也相对较为容易提取出DNA,但需要注意的是土壤样本的菌群特征容易受植物腐殖质基因的影响和干扰,所以提取时要进行纯化。
而口腔、组织、尿液等样本,由于DNA含量较少,在实验阶段提取相对较为困难,所以提前准备样本时,尽量多取一些,并且可以多取几个重复,尽量避免扩增不出来的情况。
并且这些样还很容易受到环境样的污染,所以在实验阶段,可以取空白样本,和阳性样本ST做对照,数据分析时可以用来纯化样本,排除来自环境的干扰序列。
✦组间差异分析需重复取样
要做组间差异分析时,每组要重复取样,才能做组与组之间的统计检验。理论上,每个组至少3个样就满足基本的统计差异分析需求。所以在重复取样时,每个分组至少取3个样。取样时要保证每个分组内部的样本一致性,如果组内样本之间的个体差异性较大,则会影响后期组间差异结果分析。
✦举个例子
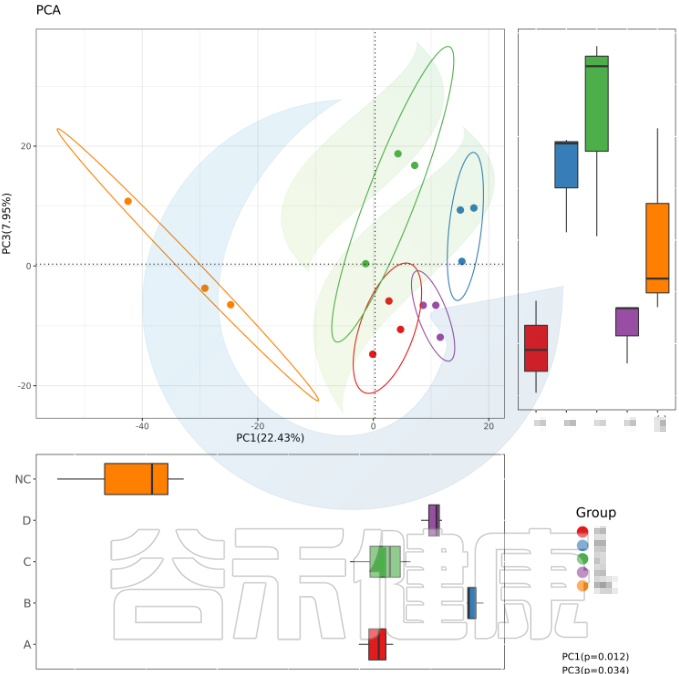
例如从该图可以看出,分组之间组间差异较大,并且组内的样本之间较为接近和相似。
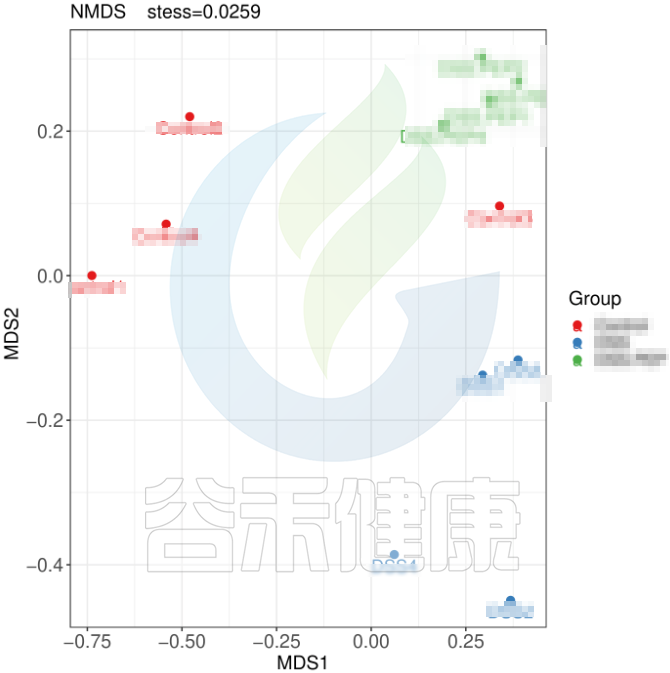
但从该图可以看出,Control组中Control3样本明显与组内的其他样本差异较大,与DSS组内的样本较为相近,这样就对后期组间差异分析的时候会产生影响,需要将该样本去除。
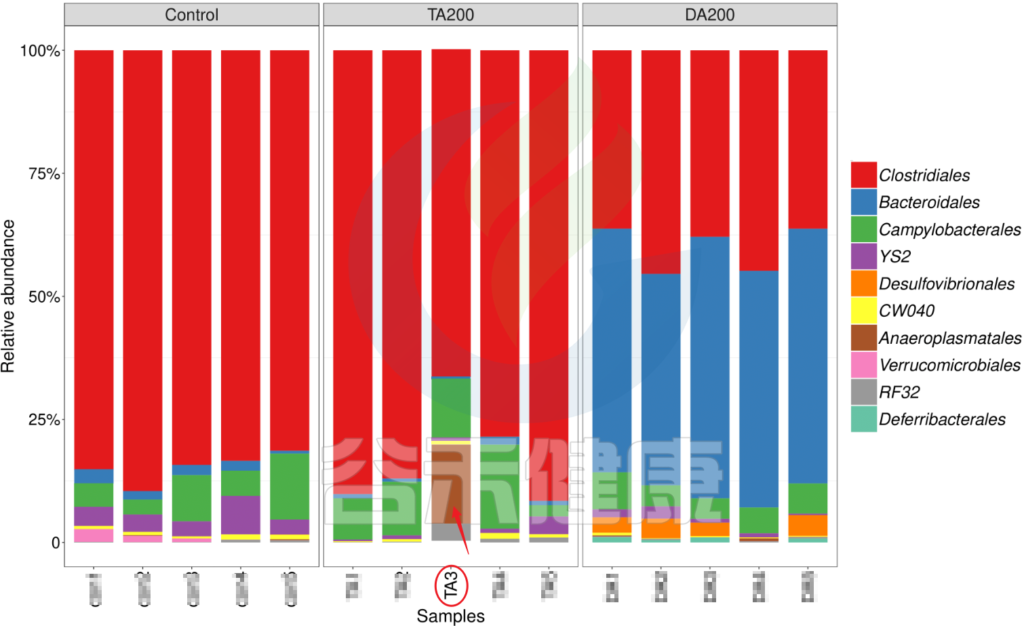
又例如在该图中,TA200组中的TA3样本的Anaeroplasmatales物种丰度含量非常高,该样本与组内的其他样本明显差异较大,该样本可能受到环境污染等其他因素干扰,这样就没有办法保证组内样本的均一性,也会影响分组之间的差异分析统计结果,再后期分析的时候建议把该样本去掉重新分析。
建议
为了便于后期数据整理修改,每个分组需要保留一定量的重复样本,假如每个分组只取了3次重复,假如其中有一个样本质量不好需要去除,该分组只剩2个样本,则不满足每组至少3个样的分组条件,整体就没有办法做组间差异分析统计。
所以这里建议每个分组至少取5个样做重复,一般6到10个样就能分析出比较完善的结果。具体分组和组内的重复取样数量视具体的实验设计方案而定。
在经费允许的情况下,建议多取一些重复。假设每组取50到100个重复或者以上,得到的分析结果就基本可以涵盖该分组情况所有的菌群构成情况,可以较为全面的研究分组之间的菌群构成差异情况。
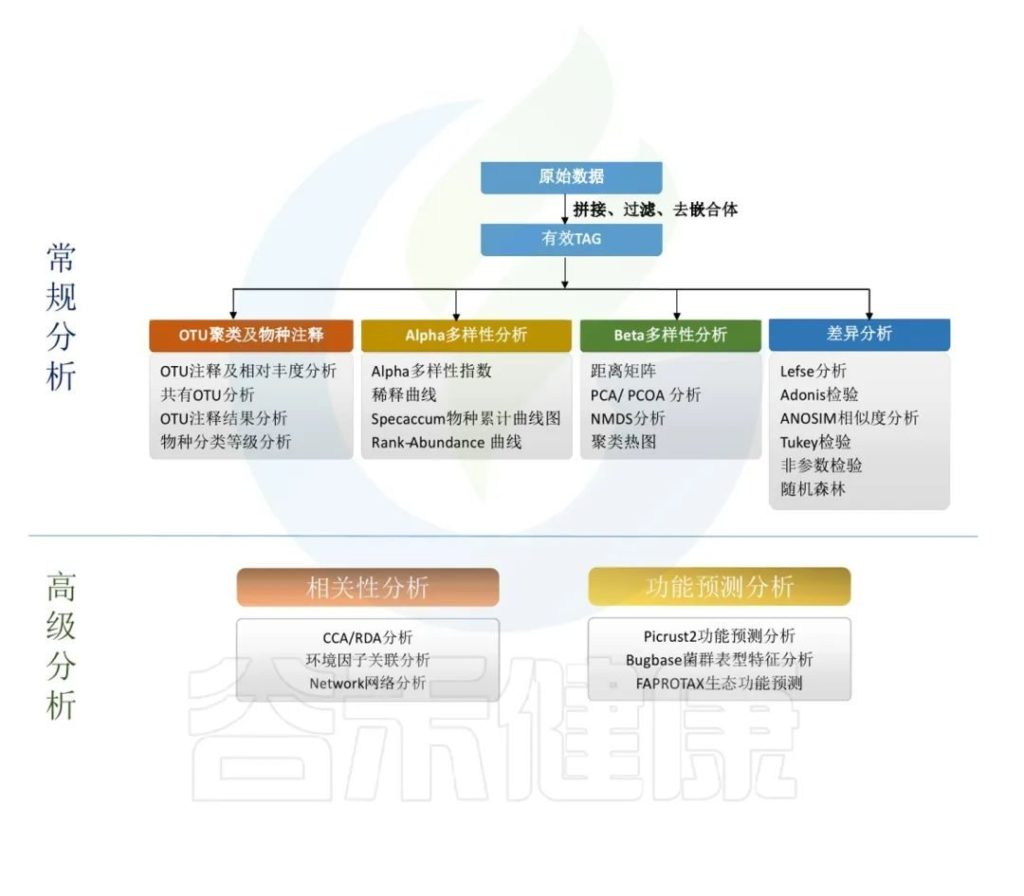
当拿到16S科研分析报告以后,面对纷繁复杂,各式各样的图表分析结果犯了难,不知道如何从这么多的图表中入手,快速找到报告中需要的图表结果。
这里对16S科研分析结果抽丝剥茧,概括出报告中的主要几大内容板块。
•16S科研分析究竟是在做什么?
16S rDNA 是一种对特定环境样品中所有的细菌进行高通量测序,以研究环境样品中微生物群体的组成,解读微生物群体的多样性、丰富度及群体结构,探究微生物与环境或宿主之间的关系的技术。
主要是对原始数据进行拼接过滤得到的优化序列,降噪方法得到ASV,再对ASV进行物种注释,注释到门、纲、目、科、属、种各层次上的分类结果。
通过ASV表计算Alpha多样性,样本内的多样性指数,Beta多样性,样本间相似性的指标。
对ASV表进行功能预测,例如Picrust2功能预测分析、Bugbase菌群表型特征分析,FAPROTAX生态功能预测等。
得到的每个样的数据结果,根据客户提供的分组情况和理化指标,进一步做组间差异分析,以及和环境理化指标之间做关联分析,相关性分析,比较分组之间是否有差异,差异是否显著,来验证分组是否合理,和环境宿主之间是否有关联性。
原始数据处理
Illumina NovaSeq测序平台测序得到的双端数据Raw PE,经过拼接和质控,根据一定的标准过滤掉低质量数据、接头或PCR错误,得到Raw Tags。再经过去重复序列,去singleton序列,过滤嵌合体,得到可用于后续分析的有效数据 Effective Tags。
OTU(ASV) 表生成
微生物多样性分析中最重要的就是OTU特征表,一切后续分析都围绕OTU表来进行。生成OTU除了传统的聚类的方法(一般按照97%的相似度进行聚类),现在最新用到的技术的是降噪的方法得到ASV。
简单来讲ASV就是在去除了错误序列之后,将Identity的标准设为100%进行聚类,常见的有DADA2、Deblur、Unoise三种降噪方法。项目里用到的是UNOISE2降噪方法获得ASV数据。
物种的分类与注释
采用QIIME2训练分类器方法对ASVs代表序列进行分类学注释,默认选用SILVA138数据库进行物种注释。并在各个分类水平上:domain(域),phylum(门),class(纲),order (目),family(科),genus(属),species(种)对每个样本的群落组成统计。
alpha多样性
Alpha多样性主要反映样本内多样性。对ASV表进行计算可以获得每个样本的simpson,ace,shannon,chao1以及goods_coverage等指数,alpha多样性指数用来来评估样本菌群物种的丰富度(richness)和多样性(diversity)
beta多样性
Beta多样性反映的是样本间多样性,Beta多样性是衡量个体间微生物组成相似性的一个指标。通过计算样本间距离可以获得β多样性矩阵,基于OTU的群落比较方法报告中给出了,欧式距离、bray curtis距离、Unweighted UniFrac距离和Weighted UniFrac距离等。
功能预测
得到群落的微生物组成之后,也可以对群落功能组成进行预测,常用的16S功能预测的相关软件有PICRUSt2、FAPROTAX、BugBase。
PICRUSt2用来预测功能,通常指的是基因家族,PICRUSt2支持基于多个基因家族数据库的预测,报告中包括了KEGG同源基因,KO直系同源物,EC酶分类编号,MetaCyc途径的丰度,CAZy碳水化合物活性酶数据库,GMM是肠道代谢模块和GBM是肠脑模块。
FAPROTAX是原核的微生物注释代谢或其他生态相关的功能(例如硝化,反硝化,发酵)的一个数据库和软件。FAPROTAX预测的功能主要集中在海洋、湖泊等环境样本微生物的功能,特别是硫、碳、氢、氮的循环功能。
BugBase能进行表型预测,其中表型类型包括革兰氏阳性(Gram Positive)、革兰氏阴性(Gram Negative)、生物膜形成(Biofilm Forming)、致病性(Pathogenic)、移动元件(Mobile Element Containing)、氧需求(Oxygen Utilizing,包括Aerobic、Anaerobic、facultatively anaerobic)及氧化胁迫耐受(Oxidative Stress Tolerant)等7类。
以上这些部分,我们通过数据处理分析,得到了每个样本相关的大量数据结果,包括每个样本的序列统计、ASVs表格、物种分类注释统计、alpha多样性指数、beta多样性指数、功能预测等。这些数据主要集中在报告里的这些内容:
▸ 科研分析报告结果文件夹
01_pick_otu/ 文件夹主要是对样本ASV表格统计
02_sequence_statistic/ 文件夹是对样本序列数据的统计
03_diversity-metrics / 文件夹是对样本的alpha多样性指数、beta多样性指数的统计
04_Taxonomic/ 文件夹是对物种分类注释的统计(门到种水平)
Picurst2/ 文件夹是Picrust2功能预测得到的每个样本的相关功能预测数据
Groups/ 文件夹下是对组间差异分析结果

红框是样本个体的相关数据统计,Group是分组比较
根据以上常规分析得到的相关数据进行作图,其路径也在对应文件夹下,可以打开 分析报告.html 有相关分析的图表和对应文件的详细介绍和路径说明。
★拿到样本后需要进行统计分析
当我们拿到这些样本大量的数据结果,之后关键的一步就是做对这些数据进行处理,做统计分析,比较分组之间的差异结果,找出菌群和环境之间的关联性等,对数据进一步做研究,找出课题方案对应的结果。
不同的数据用到的统计检验方法也不太一样,接下来我们对报告中的不同的分析结果对应的统计差异分析方法进行介绍说明。
▸ alpha多样性
alpha多样性指数组间差异统计分析用到的检验方法是:单因素方差分析(如果只有两个分组,用Wilcoxon秩和检验,3个及以上的分组用Kruskal-Wallis 检验),图上方显示P值
▸ beta多样性
beta多样性指数的统计检验方法有ANOSIM相似性分析和Adonis多元方差分析,这两种都是基于距离矩阵的检验方法。
✦Anosim相似性分析
Anosim分析是一种非参数检验,用来检验组间的差异是否显著大于组内差异,从而判断分组是否有意义。
报告中给出了加权距离和非加权距离的Anosim结果图,图中给出了R值和P值。
R值用于比较不同组间是否存在差异,R-value 介于(-1,1)之间,R-value > 0,说明组间差异大于组内差异。R-value < 0,说明组间差异小于组内差异。R只是组间是否有差异的数值表示,并不提供显著性说明。
统计分析的可信度用 P-value 表示,P< 0.05 表示统计具有显著性。
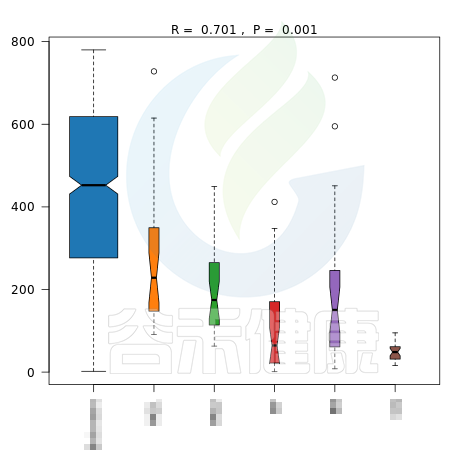
图中能看出R>0,说明组间差异大于组内差异,P<0.05 ,说明差异显著,证明该分组情况效果较好。
✦Adonis多元方差分析
Adonis多元方差分析,其实就是PERMANOVA,亦可称为非参数多元方差分析。
其原理是利用距离矩阵(比如基于Bray-Curtis距离、Euclidean距离)对总方差进行分解,分析不同分组因素对样品差异的解释度,并使用置换检验对其统计学意义进行显著性分析。
它与Anosim的用途相似,也能够给出不同分组因素对样品差异的解释度(R值)与分组显著性(P值)。
报告中PCoA bray距离、PCoA weighted_unifrac距离、PCoA unweighted_unifrac距离的图片右下角有给出PERMANOVA检验的P值和R值。
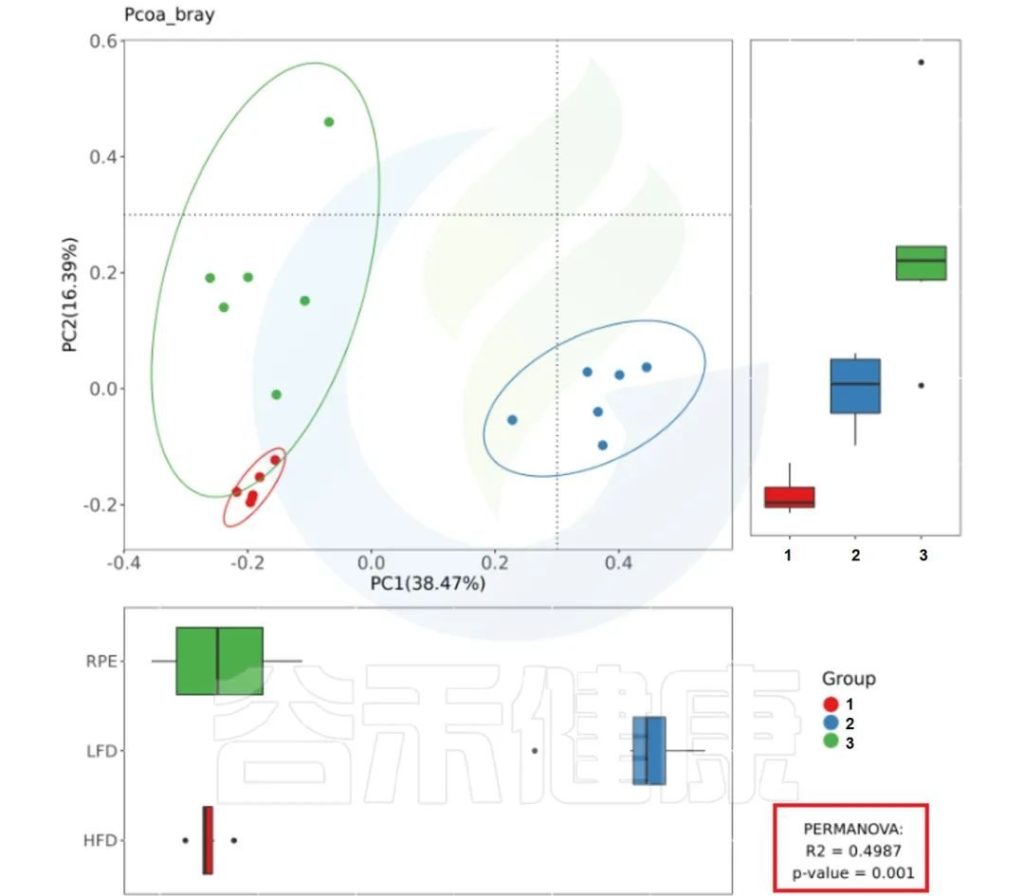
图中看出PCoa bray距离得到的检验P<0.05 组间差异显著,并且分组之间区分较为明显。
PCoa bray距离的PERMANOVA检验结果路径:
多组间检验结果:
Groups/betadiv/pcoa_bray_analysis/PERMANOVA.result_all.csv
两组间检验结果:
Groups/betadiv/pcoa_bray_analysis/ PERMANOVA_paired_result.csv

不同分类水平下的检验方法
在很多分析报告当中,例如在不同疾病的肠道菌群分组中,本身样本个体之间肠道菌群的物种多样性,丰富度差异并不大,alpha多样性组间差异并不显著,beta多样性分组间区分不是很明显,这样就需要进一步找出分组之间的差异物种或者差异功能来进行分析。
对于不同分类水平的物种和功能预测结果用到以下几种检验方法:
Tukey检验
Tukey主要应用于3组或以上的多重比较,适合于各组例数相等的每两两分组之间比较。
Tukey检验的一个重要的优点是非常简单,而且所需实验样本相对较少。
其检验结果的可信度达到95%的置信水平时,最少的情况下只需6个样本进行验证(改善前3个样本、改善后3个样本)。
•举个例子
图中的字母代表显著性差异的字母表示法,只要含有相同的字母,就表明两组之间没有显著性差异。
例如a和ab含有相同字母“a”,表示两组之间没有显著性差异。ab中的“b”表示这一组和其他含有字母b的组(比如bc)没有显著性差异,但是a和bc就有显著性差异了。
图中只展示Tukey检验差异显著的物种或功能,如果数量较多,则只展示前10个。
路径:Groups/diff_analysis/TukeyHSD/
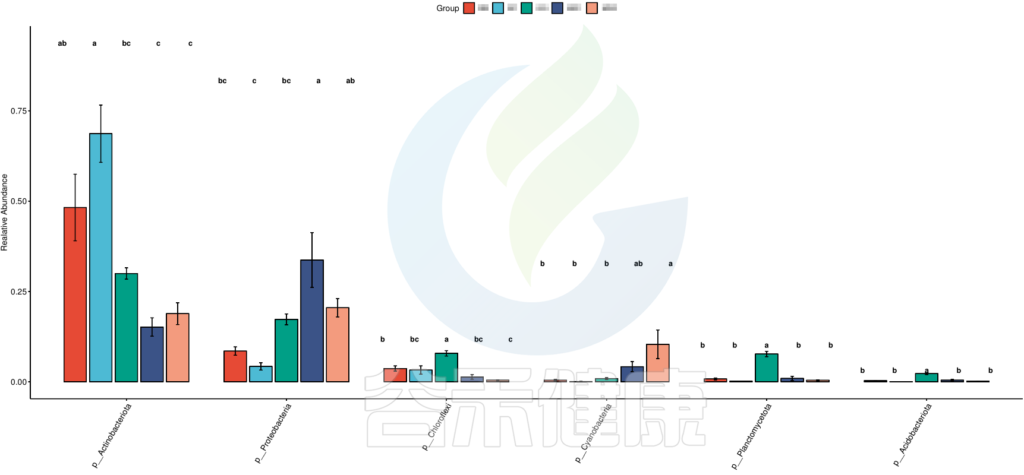
图中显示的都是Tukey检验组间差异显著的物种,依次按照丰度从高到底排列,如果差异结果较大,则显示前10个物种。例如在该图中,Tukey检验结果,门水平物种Actinobacteriota在BB与MG1组、BB与MG2、BF与GG组、BF与MG1组、BF与MG2组,这些分组之间组间差异显著。
组间差异箱型图
组间差异箱型图用到的检验方法是通过单因素方差检验(只有两个分组,用的是Wilcoxon秩和检验,3个及以上的分组用的是Kruskal-Wallis 检验),Var检验和one-way相结合,筛选出组间差异性物种。
路径:Groups/diff_analysis/TaxaMarkers
图中每一个箱型图代表一个组间差异显著的物种
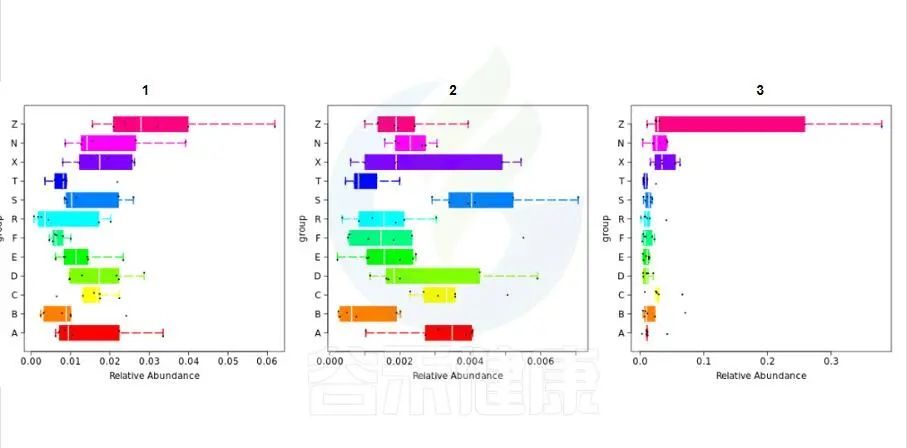
图中显示的都是统计方法得到的差异显著的物种,图中能看出这3个物种分组之间差异显著。
命名格式是,例如:Cen_Nitrosopumilus 指的是,当前分类水平(属水平)的名字 g__Nitrosopu 加上一级分类水平(科水平)的名字 f__Cenarchaeaceae 的前 3 个字母简写Cen,如果当前水平没有注释到名字则以全称的名字表示。
统计结果表:Groups/diff_analysis/TaxaMarkers/ xxx.Groups.sig.meanTests.csv
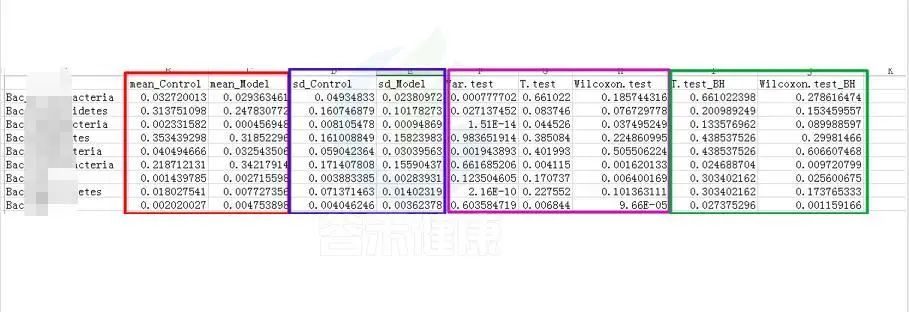
例如这是一个表格的截图
红框 mean_ 是分组组间的平均值
蓝框 sd_ 代表组间的标准差
粉色 .test 代表不同统计检验结果的P-value P值,这里有var检验 T 检验 Wilcoxon检验(或Kruskal-Wallis 检验)
绿色 _BH 例如Wilcoxon.test_BH代表Wilcoxon.test检验BH矫正的Q-value,Q值
UnivarTest检验(单因素方差分析)
单因素方差分析是指如果只有两个分组,用Wilcoxon秩和检验,3个及以上的分组用Kruskal-Wallis 检验。
路径:Groups/diff_analysis/UnivarTestXXX
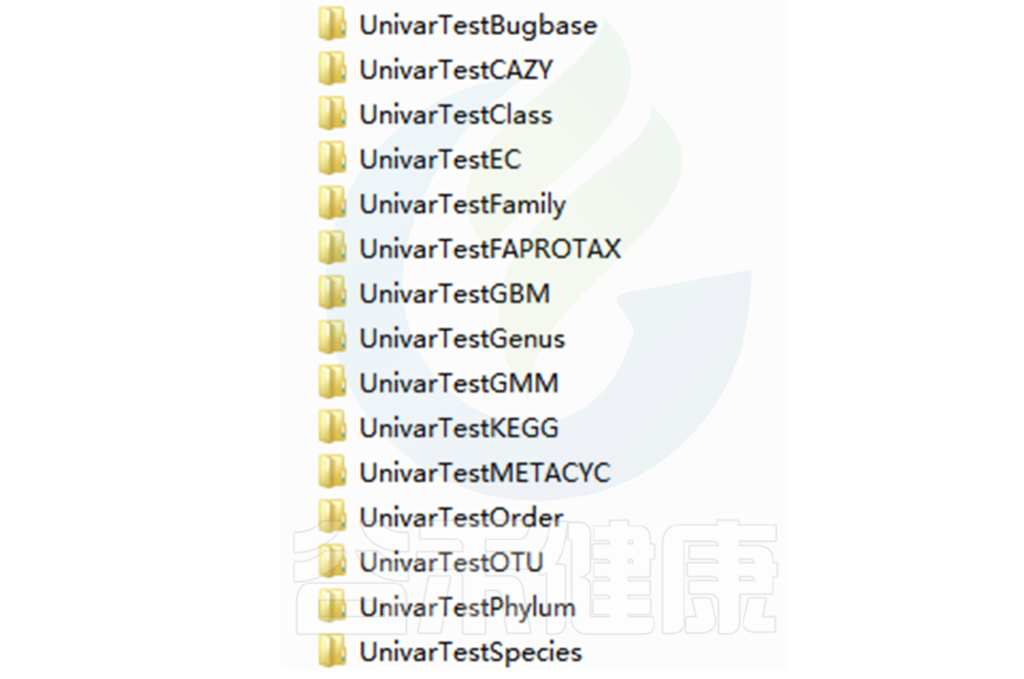
Groups\diff_analysis\UnivarTestKEGG\figure 文件夹下有做成柱状图、箱型图和单个物种之间的图,其中有横着排列和竖着排列的,有用原始值计算的,还有对原始值取log后进行统计的。图中只展示Univar 检验组间差异显著的物种/功能。
统计结果表:Groups/diff_analysis/UnivarTestXXX/ UnivarTest_sign.txt
•举个例子
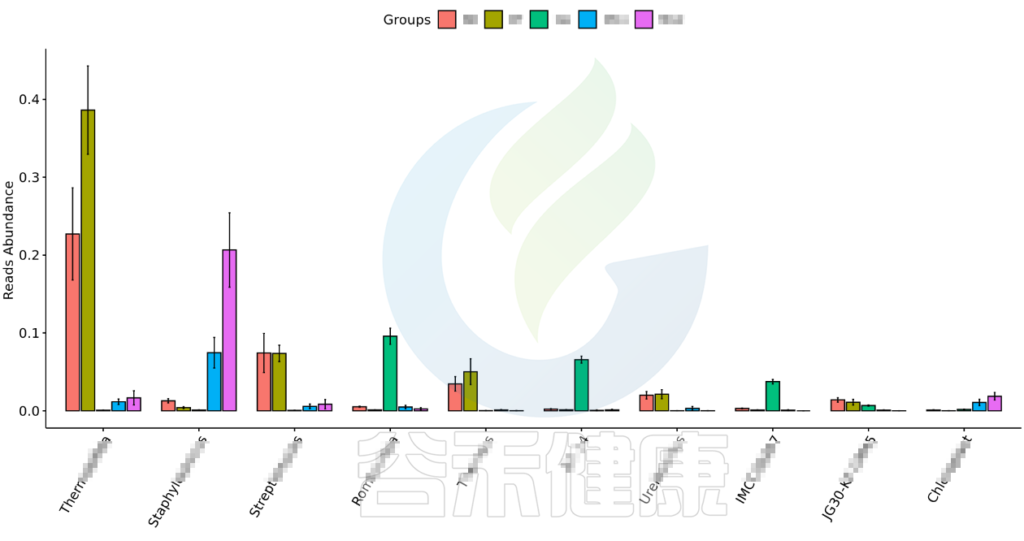
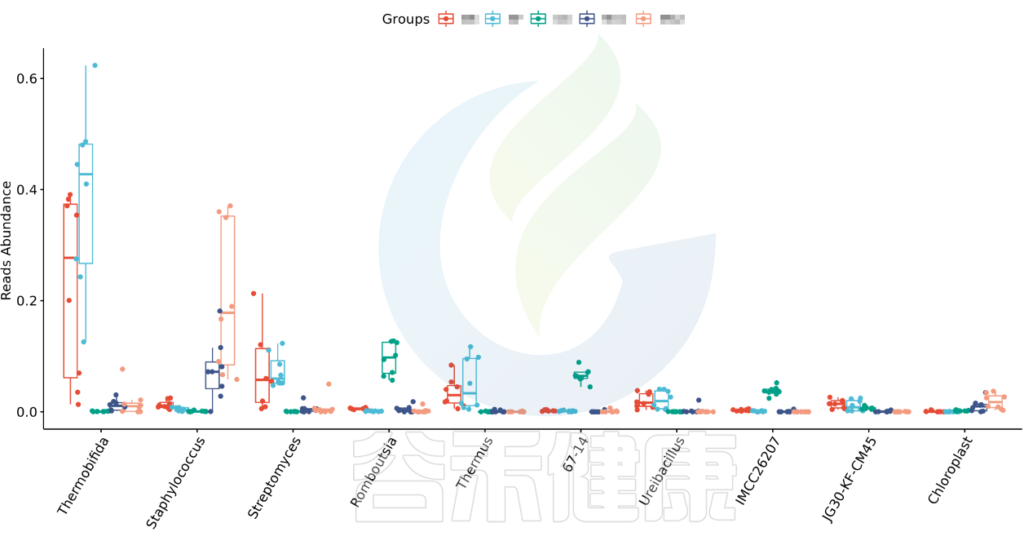
图中显示的是该统计检验差异显著的物种的柱状图或箱型图,按照丰度从高到低排列,如果差异物种/功能较大,则只显示前10个。例如该图中Therobifida、Staphylococcus、Streptomyces等物种用Kruskal-Wallis 检验得到的组间显著差异物种。
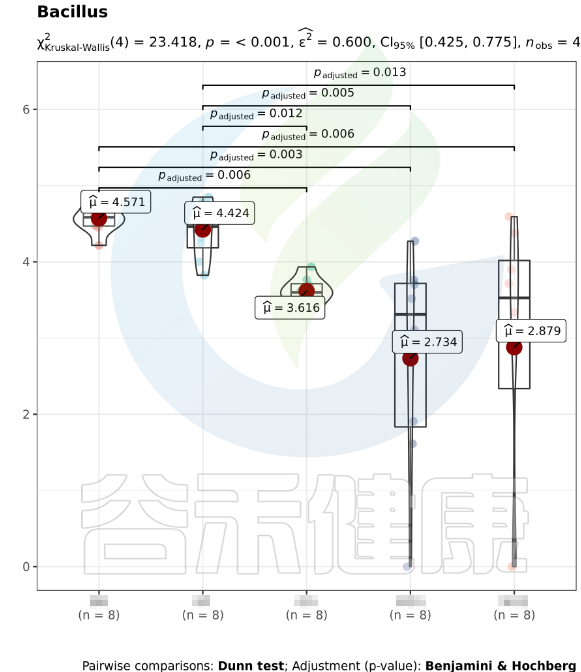
该图展示了Bacillus物种Kruskal-Wallis 检验差异结果,所有分组中P<0,001 多组间差异显著,两组间BB与GG、BB与MG1、BB与MG2、BF与GG、BF与MG1、BF与MG2,组间差异显著。
LEfse分析
LEfse分析即LDA Effect Size分析,是一种用于发现和解释高维度数据生物标识(基因、通路和分类单元等)的分析工具,可以进行两个或多个分组的比较,它强调统计意义和生物相关性,能够在组与组之间寻找具有统计学差异的生物标识(Biomarker)。
LEfSe用到的统计分析方法是将线性判别分析与非参数的Kruskal-Wallis以及Wilcoxon秩和检验相结合。
LEfse分析结果中一般会出现两个图,一张表( LDA值分布柱状图、进化分支图以及特征表)。
LDA值分布柱状图
这个条形图主要为我们展示了LDA score大于预设值的显著差异物种,即具有统计学差异的Biomaker,默认值为2.0(看横坐标,只有LDA值的绝对值大于2才会显示在图中);柱状图的颜色代表各自的分组,长短代表的是LDA score,即不同组间显著差异物种的影响程度。
路径:
Group/Lefse_Analysis/out_formant.cladogram.png
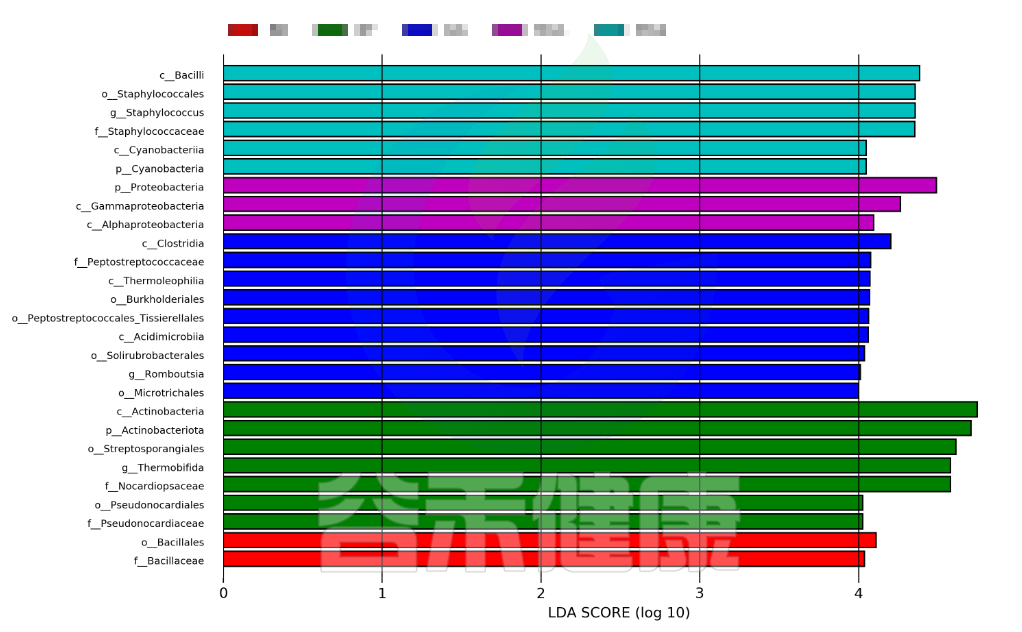
图中展示了不同分组特有的Lefse组间差异标记物,例如BB组的标记物是目水平的Bacillales和科水平的Bacillaceae,不同的分组标记物也不同,图中如果只展示了部分分组,则代表只有部分分组通过Lefse分析筛选出组间差异标记物。
进化分支图
小圆圈: 图中由内至外辐射的圆圈代表了由门至属的分类级别(最里面的那个黄圈圈是界)。不同分类级别上的每一个小圆圈代表该水平下的一个分类,小圆圈的直径大小代表了相对丰度的大小。
颜色: 无显著差异的物种统一着色为黄色,差异显著的物种Biomarker跟随组别进行着色,红色节点表示在红色组别中起到重要作用的微生物类群,蓝色节点表示在蓝色组别中起到重要作用的微生物类群。
未能在图中显示的Biomarker对应的物种名会展示在右侧,字母编号与图中对应(为了美观,右侧默认只显示门到科的差异物种)。
路径:Group/Lefse_Analysis/out_formant.png
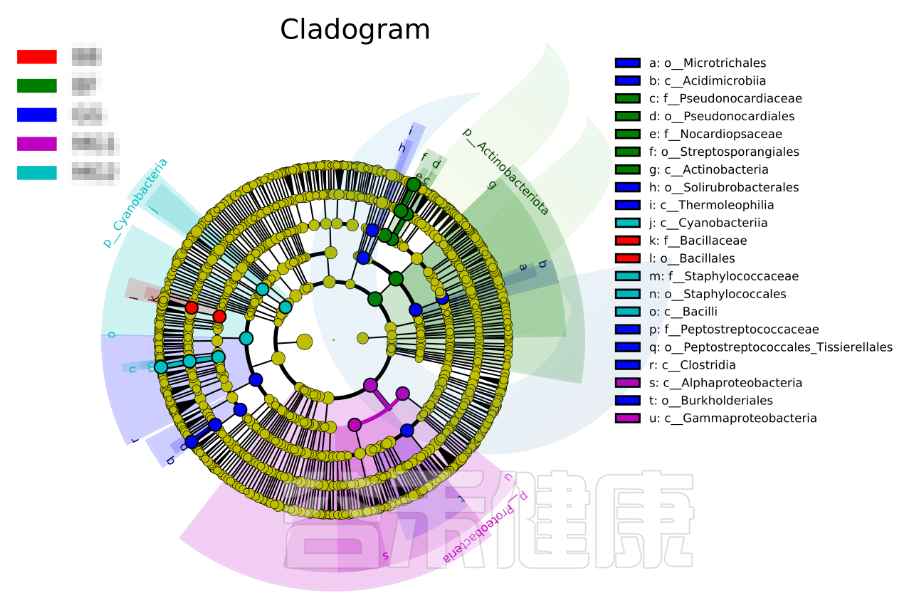
图中右侧展示了分支图中的字母对应的物种信息,例如a 代表GG组的标记物目水平的Microtrichales ,b代表GG组的标记物刚水平的Acidimicrobiia。在分支图的最外层显示的是各分组门水平物种的标记物,例如BF组的是Actinobacteriota、MG1组的是Proteobacteria、
MG2组的是Cyanobacteria
特征表
路径:Group/Lefse_Analysis/out_formant.res.csv
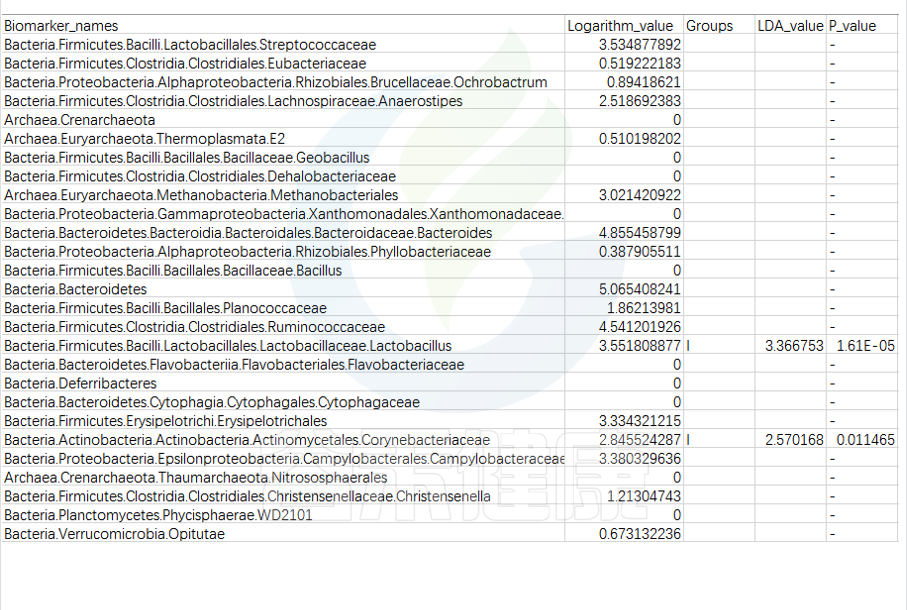
第一列是样本中从门到属水平所有分类单位的列表
Lefse会逐一判断这些分类单位的在分组之间是否具有统计学显著性差异。
第二列:各组分丰度平均值中最大值的log10,如果平均丰度小于10的按照10来计算;如果该分类单位未体现出显著组间差异,则后三列为空。
对于具有统计学差异的分类单位:
第三列:差异基因或物种富集平均丰度最高的分组组名;
第四列:LDA差异分析的对数得分值;
第五列:Kruskal-Wallis秩和检验的p值,若不是Biomarker用“-”表示。
默认LDA>2,P<0.05
通常根据第4列的LDA差异分析对数得分值和第五列的P值,可以描述组间具有显著差异的分类单位统计学效力强弱。
metagenomeSeq
metagenomeSeq是用R开发的一个包,metagenomeSeq的基本思想,用normalization实现分类注释时的biases处理,同时用零膨胀高斯分布(zero-flated Gaussian distribution)处理了测序深度所带来的影响,在此基础上,利用线性模型找到存在的差异所在。
路径:Groups/diff_analysis/ metagenomeRXXX
metagenomeSeq 差异显著物种/功能 热图
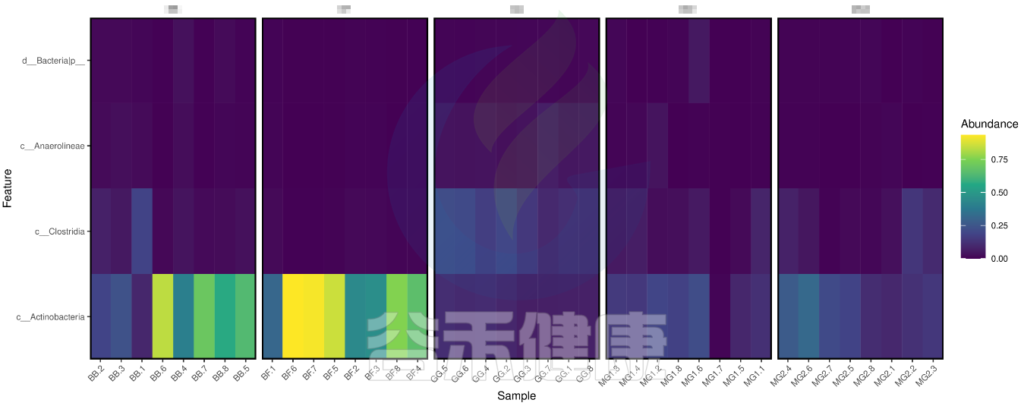
图中颜色越深相关性越小,颜色越接近黄色相关性越大,从图中能看出Actinobacteria物种与BB组和BF组相关性较大。
metagenomeSeq差异菌属于功能代谢关联分析
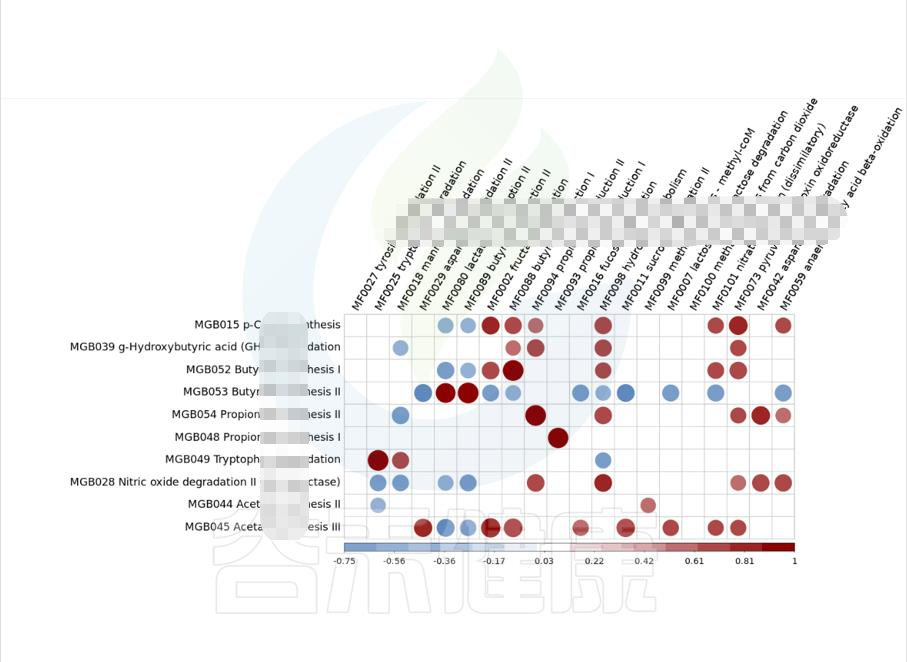
图中红色代表正相关,蓝色代表负相关,颜色越深,圆圈越大,相关性也越大,例如图中能看出MGB049余MF0025 之间成正相关,且相关性较大。
随机森林模型
一种非线性分类器,随机森林属于集成类型的机器学习算法,挖掘变量之间复杂的非线性的相互依赖关系。通过随机森林重要性点图,可以找出分组间差异的关键物种/功能。
反映了分类器中对分类效果起主要作用的特征,按重要性从大到小排列。
Error rate:表示使用下方的特征进行随机森林方法预测分类的错误率,数值越高表示基于特征分类准确度不高,可能分组之间特征不明显。分值越低证明分组效果比较好。
•举个例子
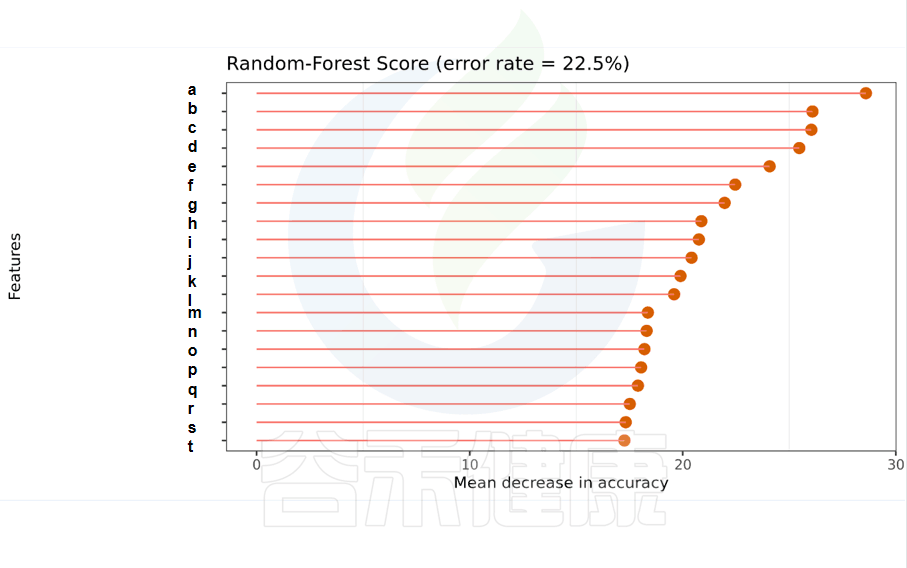
图中按照随机森林模型效果筛选出的对分组效果有重要性作用的物种,按照重要性从高到低进行排列,例如图中最终要的是a,依次往下是b、c等。错误率较小,表明该分组效果较好。
ROC曲线
ROC曲线分析是一种常用的统计学分析方法,在医学研究中主要用于评价诊断试验的效能。在16S测序报告中,我们通过绘制ROC曲线,并计算ROC曲线下面积(AUC),来确定分组对于菌群是否有诊断价值。
ROC曲线图是反映敏感性与特异性之间关系的曲线。ROC曲线下的面积值在1.0和0.5之间。在 AUC>0.5的情况下,AUC越接近于1,说明诊断效果越好。
AUC在0.5~0.7时有较低准确性,AUC在0.7~0.9时有一定准确性,AUC在0.9以上时有较高准确性。AUC=0.5时,说明诊断方法完全不起作用,无诊断价值。AUC<0.5不符合真实情况,在实际中极少出现。
•举个例子
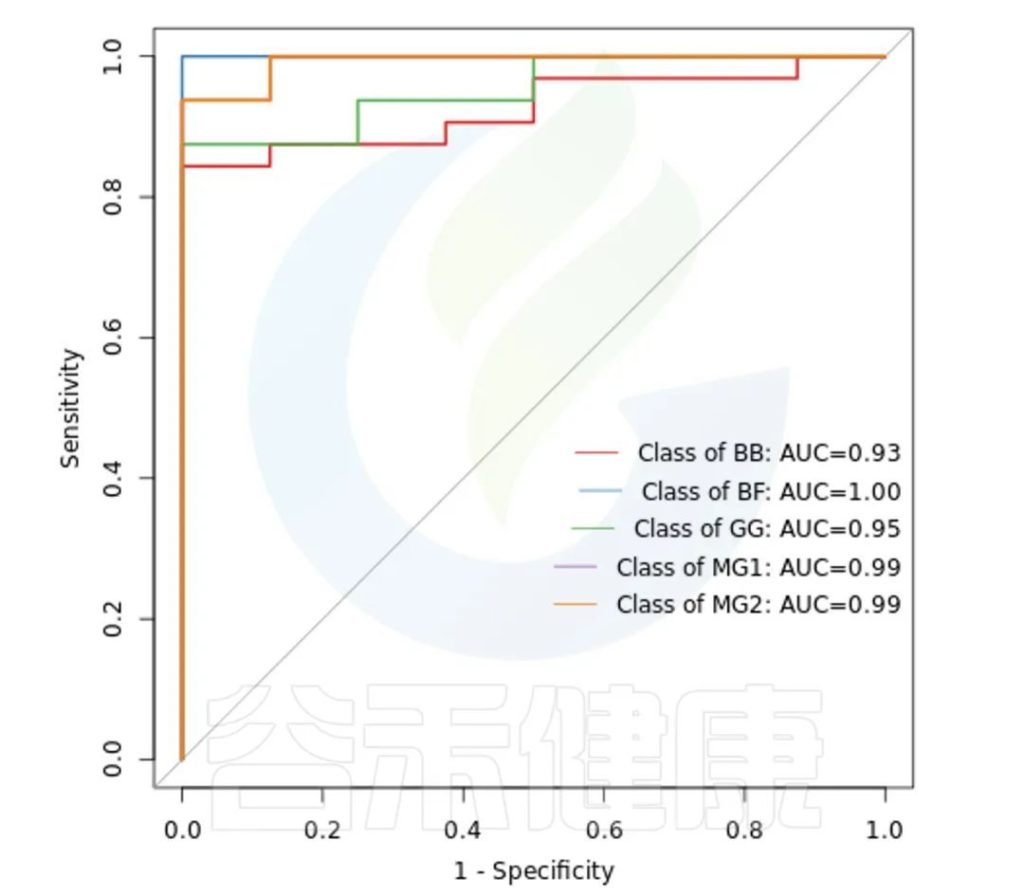
从图中能看出各分组的AUC都大于大于0.9,各分组的分组效果较好,BF组AUC等于1,该分组效果最好,可能样本之间较为相近,并且跟其他分组组间差异也比较大。
以上是组间统计差异的方法介绍,其他的还包括关联分析。
例如客户提供了每个样的相关理化指标数据,想计算这些指标与均属之间有什么相关性,就可以做一下分析。
关联性分析
✦相关性热图
图中X轴代表属水平物种,Y轴代表代谢指标,红色代表正相关,蓝色代表负相关,**代表相关极显著P<0.01,* 代表相关性显著P<0.05相关性具有统计学意义。
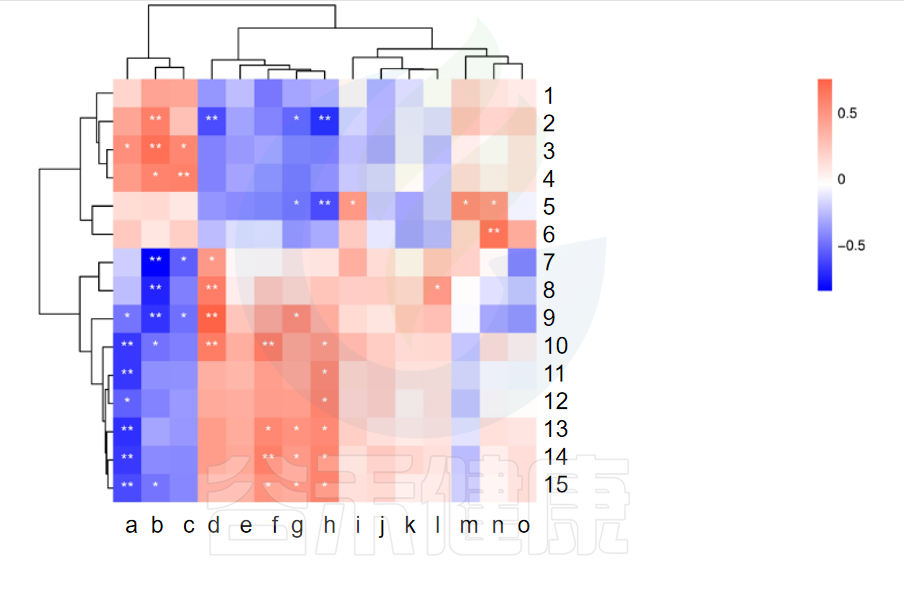
例如从该图中能看出6与n物种成正相关,并且相关性极显著**,7与b物种成负相关,并且相关性极显著**。
可以得到表格:任意菌属和代谢的相关性的值和P值
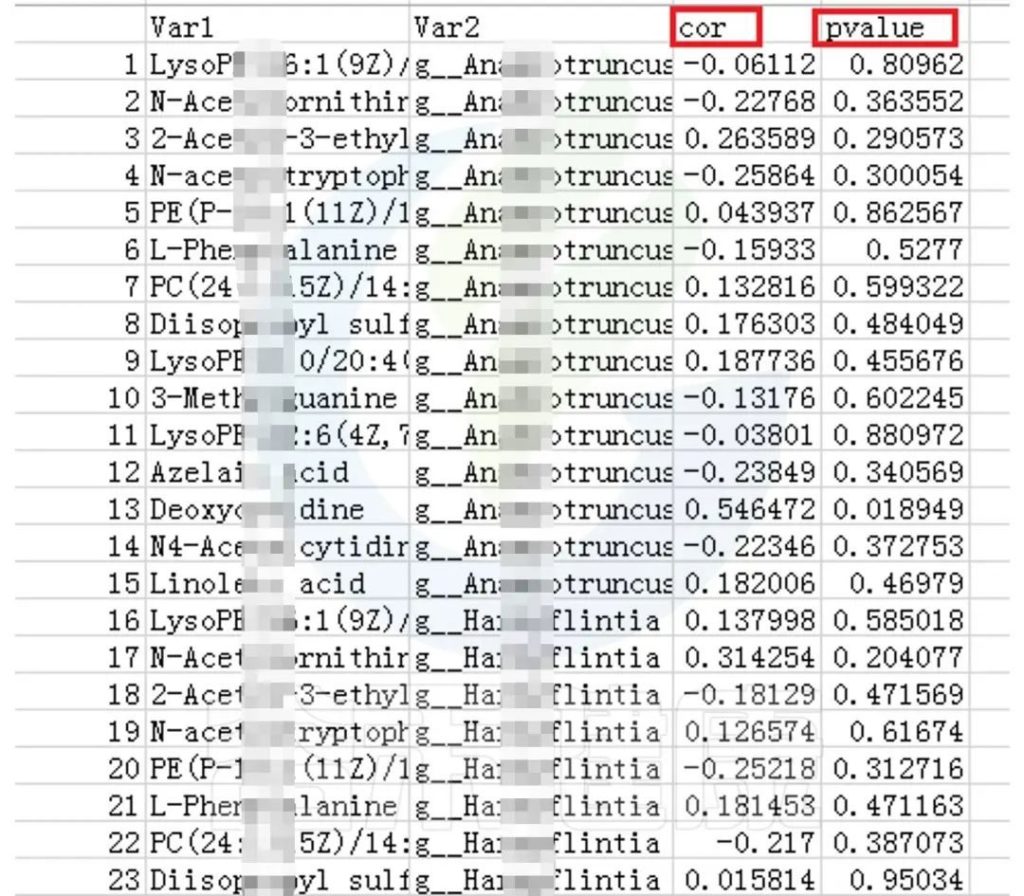
✦CCA图
可以分析样本、菌群、理化指标之间的关联关系。图中使用点代表不同的样本,从原点发出的箭头代表不同的环境因子。
箭头的长度越长,表示环境因子的影响越大;夹角越小,代表相关性越高。样本点与箭头距离越近,该环境因子对样本的作用越强。
图像中坐标轴标签中的数值,代表了坐标轴所代表的环境因子组合对物种群落变化的解释比例。
例如从图中能看出pH 、NO2N、02与 Acinetobacter、Weissella等物种成正相关,与T3D0、T1D0、T4D0等D0组的样本成正相关。
✦RDA 冗余分析

例如从图中能看出pH与Helicobacer物种成正相关,相关性较大,pH与NC组有一定的相关性。
✦Envfit分析
回归拟合分析结果:
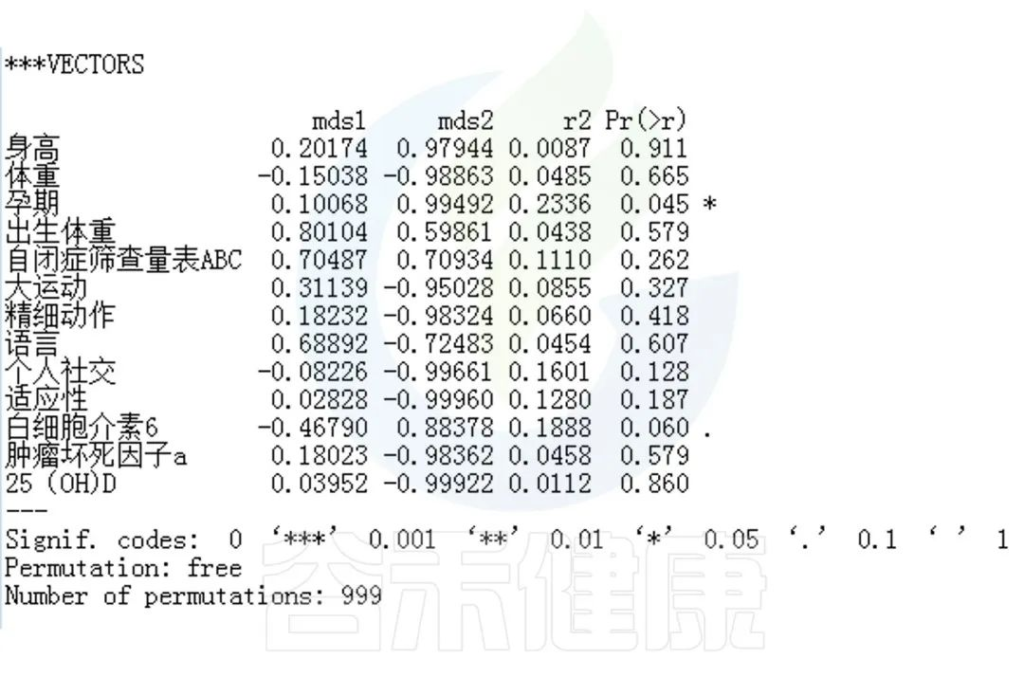
图中能看出ASD与正常儿童之间的分组与菌群之间相关性极显著**,其次是否有先兆流产的分组与菌群之间有显著相关性*,其他的包括是否喝牛奶、孕期是否感染、妊娠高血压都与菌群有相关性。
环境因子与功能/物种的相关性线形图P<0.05显著,图中红色点代表正相关,绿色点代表负相关,灰色相关性不显著。
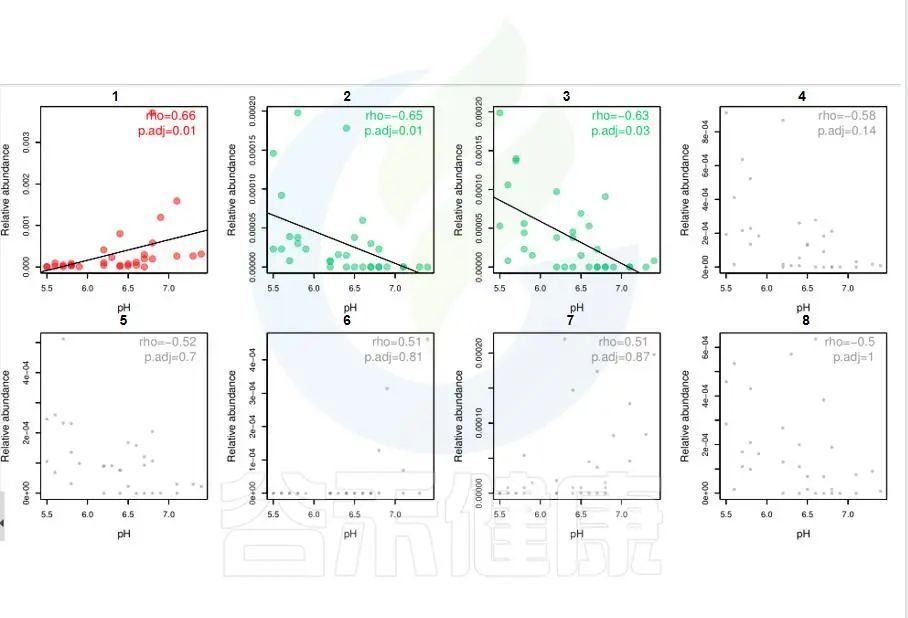
图中能看出pH 与Candidatus Rhabdochlamydia 之间成正相关,且相关性显著,pH 与Sinorhizobium、Euzebya 之间成负相关,切相关性显著。
✦Network网络分析
还可以做菌属之间的网络分析关联图,共发生网络图为研究复杂微生物环境的群落结构和功能提供了新的视角。
由于不同环境下微生物的共发生关系截然不同,通过物种共发生网络图,可以直观看出不同环境因素对微生物适应性的影响,以及某个环境下占互作主导地位的优势物种、互作紧密的物种群,这些优势物种以及物种群往往对维持该环境的微生物群落结构和功能稳定发挥着独特以及重要的作用。
•举个例子
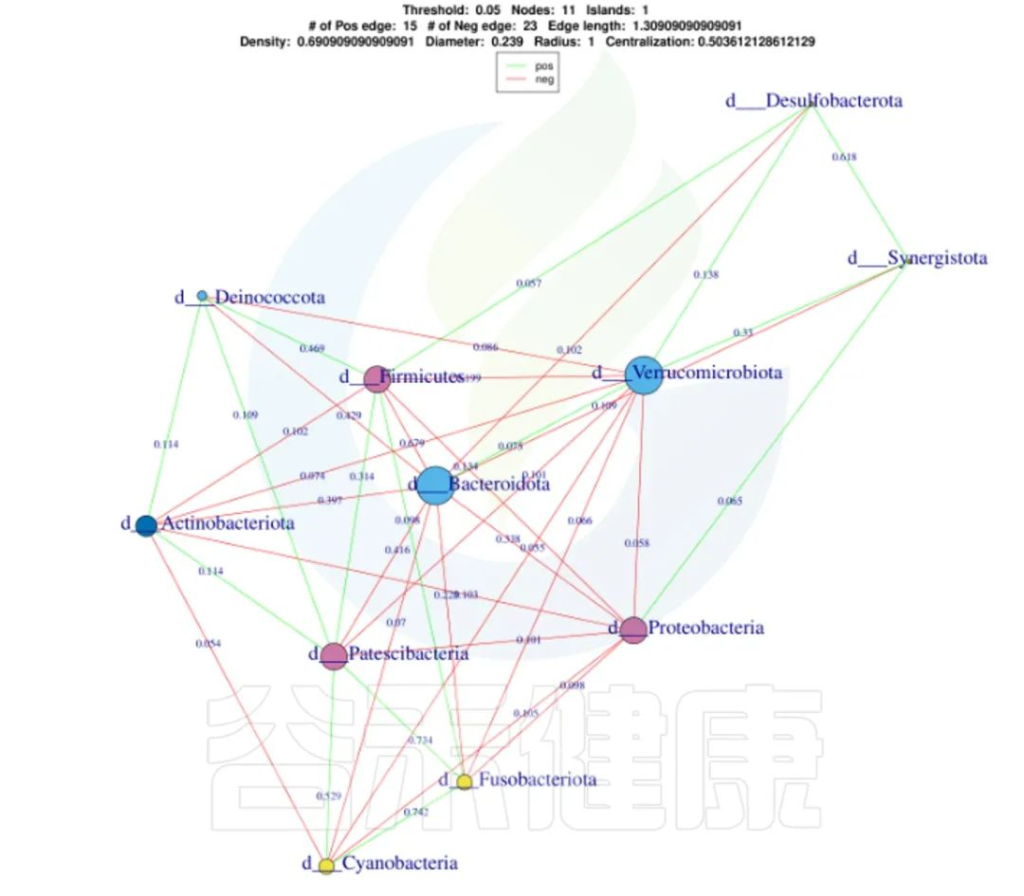
图中展示了相关性的物种,例如Bacteroidota、Actinobacteriota、Proteobacteria 这些物种与其他物种相关较大,图中这些物种与其他物种连线较多,字体比较大也代表相关性较强,例如Actinobacteriota与Deinococcota连线是绿色的代表这两个物种是负相关。
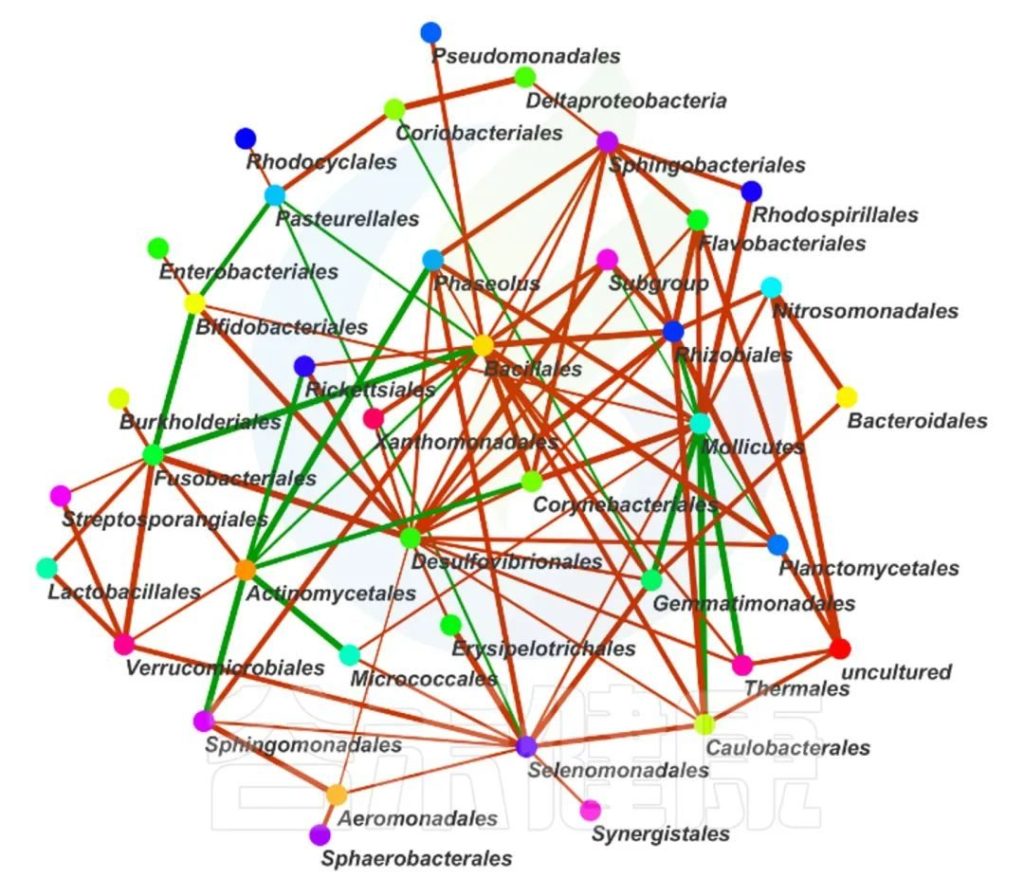
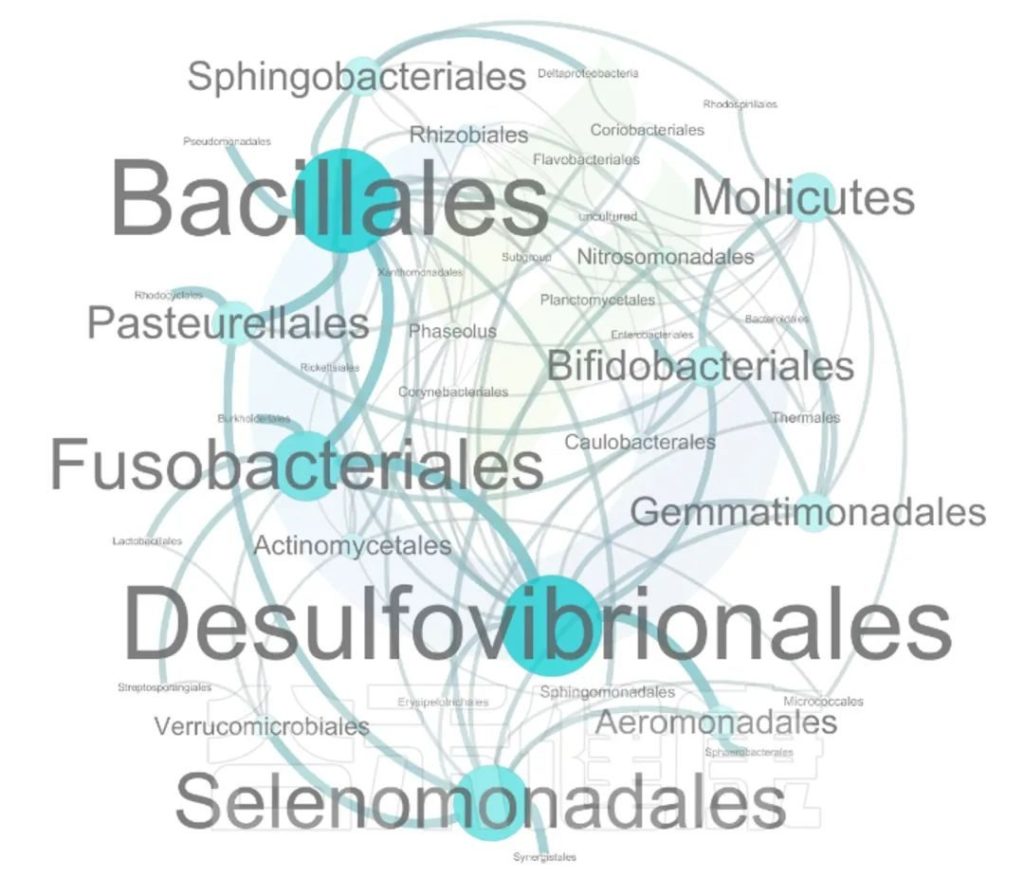
这两个图类似的物种相关性的图,用同一个数据做出来的,图中能看出Bacillales、Desulfovibrionales、Selenomonadales与其他物种相关性较强。
报告中已经基本都涵盖了16S科研数据分析所需要的图表、差异统计,以及相关性分析结果。如果在几种不同类型的统计方法对比之下有略微的差异结果,选取其中一组差异结果即可。
报告里涵盖了大部分16S所需要的图片,不过也有个别个性化的图需要单独用到软件去做,可以单独完成个性化图表生成。
随着16s分析报告的不断升级,报告中的图表以及相应的解读也会越来越精细完善,谷禾也将尽可能为大家的科研之路带来更多便利。

谷禾健康
大千世界多样的微生物,在各自的世界,在自己的方寸空间中生长,跨物种间便有了庞大和渺小。
我们总是善于去观察自己能看到的那些,而忽略肉眼看不到的。微生物存在于任何物理条件允许的地方。湖水在肉眼看来是透明的,但一升水可以容纳十亿个细菌,一克土壤也可以包含超过十亿个细菌。微观世界是一个无穷无尽的迷人之地,里面物种繁多,形态各异。
在地球存在的 46 亿年中,微生物已经在这里生活了至少 37 亿年。在植物和动物出现之前,微生物已经存在超过 30 亿年的时间,可以说它们帮助创造了生物圈,并支持地球上的生命过程。因此,微生物的多样性(从遗传、代谢和生理方面),远大于在植物和动物中发现的多样性也就不足为奇了。
那么,它们是如何生存的?
它们如何创造了这个世界?
它们给我们的生活带来怎样的便利?
……
近年来,微生物生态学出现了爆炸性发展,特别是在分子水平上。本文带大家更好地去认识和了解这个迷人的微生物世界,也能使我们更好地理解微生物在极端环境下的生存状态,帮助我们更好地与它们共存,将它们应用到生活中,并为我们的健康带来益处。
极端环境是指比人类或其他生物体发育的最佳范围环境条件更恶劣的栖息地。
极端环境的特点是各种不利条件,包括高温或低温、高压或低压以及酸性或碱性pH值。
对于一个被认为是极端的区域,必须认为环境的某些条件或方面非常难以让不同的生命形式生存。
一些极端环境的例子包括极地、沙漠、火山区、深海海沟、外太空,以及太阳系中除地球以外的所有其他行星。
一些常见的极端环境包括碱性、酸性、极热或极冷、高盐浓度、没有水或氧气的区域。
不同的极端环境
▸ 极端温度
极端温度可以描述两种极端环境:极冷和极热。
极冷环境是环境温度低于5°C的环境。这些可以在深海,高山峰顶或极地地区找到。
极热环境的特点是环境温度高于45°C。这些环境受到地热活动的影响,如大陆火山区或深海喷口的间歇泉和喷气孔。
▸ 极端pH值
极端环境也可根据其pH值分为酸性或碱性。
极端酸性环境是pH 值低于5的自然栖息地;而极端碱性环境是指pH值高于9的环境。
▸ 高盐环境
高盐环境是离子浓度高于海水(大于3.5%)的环境。
▸ 极端压力
极端压力环境是指处于极端水压力或岩石压力下的环境,例如2000米或以上深度的水生栖息地或地下深处的生态系统。
什么是极端微生物?
Microbe Notes
极端微生物:是由于不同的生理和分子适应能力,能够在极端环境中生存和繁衍的生命体。
注:这些生物在极端生态位、冰和盐溶液以及酸性和碱性条件下茁壮成长。有些可能生长在有毒废物、有机溶剂、重金属或其他几个被认为不适合生命生存的栖息地。
极端微生物可分为两类:需要一种或多种极端条件才能生存的极端微生物;以及即使它们在中性条件下最佳生长,也能耐受一种或多种物理参数的极端条件的极端耐受生物。
极端微生物包括所有三个生命领域的成员;细菌、古细菌和真核生物。大多数极端微生物是原核生物,属于古生菌的比例很高,但一些生物可能是真核生物,例如原生生物(例如藻类、真菌和原生动物)和多细胞生物。
根据它们生长的条件进行分类:嗜热菌和超嗜热菌(分别在高温或非常高温下生长的生物)、嗜冷菌(在低温下生长的生物)、嗜酸菌和嗜碱菌(在酸性或碱性环境中生长的生物)、嗜压菌(在压力下生长最好的生物)和嗜盐菌(在高盐环境中生长良好的生物)。
极端温度下的微生物
▸ 嗜冷菌
嗜冷菌,字面意思是喜冷,是适应低温生长的生物。嗜冷菌成功地克服了低温生长过程中出现的两个主要挑战:首先,低温,因为温度的任何降低都会以指数方式影响生化反应的速率;其次,水环境的粘度。
超过80%的地球生物圈永久低于5°C,适合嗜冷菌的环境非常普遍,其中包括海水、永久冻土、冰川、南极岩石、雪原和极地冰盖。嗜冷微生物已成功地在从深海到山区和极地地区的所有永久寒冷环境中定殖。
原核嗜冷菌对氧气的耐受性各不相同,包括(严格)好氧、(严格)厌氧和兼性厌氧。
除了极端寒冷,许多嗜冷菌还能忍受或在某些情况下需要其他极端环境条件才能生长和生存。例如,深海嗜冷菌可能需要高压才能生长,从而使它们成为嗜压嗜冷菌。
✦利于嗜冷菌生存的结构
蛋白质和酶适宜低温生存
嗜冷菌产生在低温下发挥最佳功能的蛋白质和酶。
已经观察到,平均而言,冷活性酶具有更多量的α-螺旋和较少量的β-折叠二级结构。因为这些β-折叠倾向于形成更刚性的结构,冷活性酶的α-螺旋含量使蛋白质在低温下具有更大的灵活性。
用于增加蛋白质灵活性的另一种机制是通过减少形成多个氢键和盐桥, 并降低构象灵活性的精氨酸和脯氨酸含量的产生。
嗜冷菌还产生抗冻蛋白,这些蛋白能够通过一个大的互补表面与冰晶结合,从而产生热滞后并降低生物体可以生长的温度。
膜的流动性好
嗜冷菌的细胞质膜含有较高量的不饱和脂肪酸,有助于维持细胞膜的半流体状态。
已经提出的增加膜流动性的进一步适应包括增加大脂质头基、蛋白质和非极性类胡萝卜素色素的含量。
一些嗜冷菌的脂质还含有多不饱和脂肪酸,以及具有多个双键的长链碳氢化合物,增加了脂质膜的流动性。
✦嗜冷菌的应用:食品、洗衣去污
在嗜冷菌中,脂酶和蛋白酶具有相当大的应用潜力。酯酶可应用于许多方面,如作为食品的风味改变酶、去污添剂添加物等;蛋白酶也可被大量应用于食品工业、洗衣业以及对X光胶片上银的回收等。
★常见的嗜冷菌
嗜冷菌种最常见的品种有耶氏菌、李斯特菌和假单胞菌(pseudomonas)。
▸ 嗜热菌
嗜热菌是在高于维持大多数生命形式的温度下生长的生物。通常,嗜热菌在高于45°C的温度下表现出最大的生长速率。
高温环境包括陆地和海底环境。世界各地不同的高温栖息地都适合嗜热菌的生长。嗜热菌通常存在于热喷口、温泉和沸腾的蒸汽喷口等区域。
嗜热菌进一步分为不同的组:兼性嗜热菌和专性嗜热菌。兼性嗜热菌可以在高温和中等温度下茁壮成长,而专性嗜热菌需要高温才能生长。
✦利于嗜热菌生存的结构
与嗜冷菌一样,嗜热菌也具有不同的生理和分子适应能力,使生物体能够在较高的温度下生存。
酶和蛋白质更稳定
这些生物体中的酶和其他蛋白质比中温微生物中的酶和其他蛋白质对热更稳定。酶中一个或几个位置的关键氨基酸取代允许它们以符合热稳定性的方式折叠。
此外,某些溶质如磷酸二肌醇和甘油单酯会大量产生,有助于稳定蛋白质免受热降解。
膜稳定性更强
除了酶和细胞的其他成分外,嗜热菌的细胞质膜还需要稳定。嗜热菌通常具有富含饱和脂肪酸的脂质,因此允许膜在高温下保持稳定和功能。
饱和脂肪酸比不饱和脂肪酸形成更强的疏水环境,这也增加了膜的稳定性。
✦嗜热菌的作用:降解有机物、生产酶制剂、用于基因研究
嗜热菌通常存在于堆肥、干草堆和碎木堆等高温环境中,有助于一些有机物的降解。利用嗜热菌对废水废料进行厌氧处理,可提高反应速度,消灭污水污物中的病原微生物。
嗜热菌可用于生产多种酶制剂,例如纤维素酶、蛋白酶、淀粉酶、脂肪酶、菊糖酶等,由这些微生物中产生的酶制剂热稳定性好、催化反应速率高,易于在室温下保存。
此外,嗜热菌研究中最引人注目的成果之一就是将水生栖热菌中耐热的Taq DNA聚合酶用于基因的研究和遗传工程的研究以及基因技术的广泛应用中。
★常见的嗜热菌
嗜热菌的一些常见例子包括Methanosarcina thermophila、Methanobacterium wolfei、Methanobacterium thermoautotrophicum、Archaeglobus profundus、Alicyclobacillus acidoterrestris、A. acidocaidarius等。
▸ 超嗜热菌
超嗜热菌是可以在极高温度(80°C以上)下生存和生长的生物。一般超嗜热菌的最适生长温度为80°C,但它们可以在高于100°C的温度下生存。
大多数超嗜热菌能够承受其他极端条件,如更高的pH值和更高的压力。
注:大多数超嗜热生物存在于温泉和沸腾的蒸汽喷口中,即使是中等嗜热生物也无法生存或繁衍。
超嗜热菌大多属于古细菌组,极少数物种是细菌物种。
✦利于超嗜热菌生存的结构
蛋白质和酶的热稳定性
超嗜热古菌在其嗜热适应过程中使用基于结构的物理机制来增加其蛋白质的热稳定性。
由于结构中盐桥(在氨基酸残基之间桥接电荷的阳离子)数量的增加,提高了超嗜热菌中蛋白质和酶的稳定性。
此外,各种氨基酸的正电荷和负电荷之间的离子键数量的增加也使蛋白质稳定。增加的离子键在蛋白质内部形成了密集的高度疏水性内部,从而阻止了蛋白质在高温下的展开。
醚键使膜更加稳定
大多数嗜热古细菌的膜中没有任何脂肪酸。相反,它们具有带有分支烃链的脂质。这些链由五碳化合物异戊二烯的重复单元组成,通过醚键相互结合。
醚键是更稳定的键,这反过来又稳定了膜以防止热断裂,而支化降低了膜的流动性。膜的整体结构形成了比中温生物的脂质双层更耐热的脂质单层。
★常见的超嗜热菌
超嗜热生物的一些常见例子是Thermoproteus uzoniensis、Staphylothermus marinus、Pyrodictium abyssi、Pyrococcus furiosus、Hypothermus butylicus、Pryococcus woesei、Pyrodictium brockii、Pyrodictium occultum等。
极端pH环境的微生物
▸ 嗜酸菌
嗜酸菌是可以在高酸性条件下(通常在pH2.0)下生存和繁衍的生物。
嗜酸微生物在极低pH值的自然和人造环境中茁壮成长,例如酸性湖泊、酸性硫酸盐土壤、硫化风化层和矿石,以及受金属和煤矿影响的环境。
天然酸性环境:包括火山区、热液源、深海通风口、金属矿区和动物的胃。
研究最广泛的嗜酸菌是氧化还原铁和硫的原核生物。它们可以催化黄铁矿等金属硫化物矿物的氧化溶解,从而严重酸化它们赖以生存的环境(通常pH值低于3)。
✦嗜酸菌的种类
嗜酸生物属于三个领域;古生菌、细菌和真细菌,但古生菌代表了最大的嗜酸生物群。
在生理上,嗜酸菌非常多样化:有需氧菌和兼性厌氧菌、化能自养菌以及不同类型的异养原核生物、光合自养真核生物、捕食性原生动物等。
✦利于嗜酸菌生存的结构
细胞膜的不渗透性
有助于维持嗜酸菌细胞内pH值的适应性之一是细胞膜的不渗透性,它限制了质子流入细胞质。
在嗜酸古细菌中,细胞膜中四醚脂质的存在与对酸性pH值的耐受性之间存在很强的关联。
已经提出减小膜通道的孔径作为维持pH稳态以防止质子进入细胞的另一种机制。另一个是加入了超长链二羧酸脂肪酸,占膜脂肪酸的50%以上。这些专门的机制防止质子进入细胞和膜的酸水解。
细胞质可以缓冲并维持pH稳态
嗜酸菌通过不同机制从细胞质中去除多余的质子来维持pH稳态。
对于酸热芽孢杆菌(Bacillus acidocaldarius)和嗜酸芽孢杆菌(T. acidophilum)等细菌而言,它们能够主动将质子泵出细胞质以维持pH稳态,这一过程与呼吸链有关。
众多质子驱动的二级转运蛋白也可以适应在低pH环境下生存。嗜酸菌细胞质的缓冲能力也是嗜酸菌生理适应的重要手段。这些细胞含有缓冲分子,这些分子含有赖氨酸、组氨酸和精氨酸等碱性氨基酸,这些分子有助于去除质子。
有机酸的降解
通过质子化形式扩散到细胞中,有机酸(如乙酸或乳酸)在低pH值下充当呼吸链的解偶联剂。这些有机酸的降解可能会被异养嗜酸菌用来防止它们的有害影响。
✦嗜酸菌的作用:保持肠道菌群平衡、促进发酵、促吸收、生物湿法冶金
在医学方面,嗜酸菌,是有益的肠内菌之一。可以保持肠道菌群平衡,有效抑制肠道内不良微生物的增生,减少腹泻等问题的出现。
多种嗜酸菌组合在一起还能促进发酵,使身体产生乳酸、醋酸等多种物质。提升钙元素、磷元素利用率和吸收率,维持身体健康。
工业方面,可以利用嗜酸菌将贫矿和尾矿中金属溶出并回收,这种方法称为生物湿法冶金。
★常见的嗜酸菌
嗜酸生物包括乳酸杆菌、硫叶硫杆菌、酸热芽孢杆菌、嗜酸嗜热原体、嗜酸铁原体、嗜酸铁原体、嗜酸硫杆菌、钩端螺旋体、酸杆菌属、硫杆菌等。
▸ 嗜碱菌
嗜碱菌是一组能在pH值极高(9-13)的环境中生存和繁衍的极端微生物,一般最适pH值为10。
嗜碱菌有两种类型;专性嗜碱菌仅在pH高于9的环境中生长,兼性嗜碱菌可在中性pH和碱性条件下生存。
迄今为止,大多数被描述为在碱性条件下生长的生物都是原核生物,包括真细菌的异质集合和一些古细菌的例子。
✦利于嗜碱菌生存的结构
产生酸来促进pH稳态
嗜碱菌通过糖发酵和氨基酸脱氨酶产生代谢酸。酸的产生主要通过增加细胞质氢离子浓度来促进pH稳态。此外,酸的产生,除了防止细胞质碱化外,还可以增加细胞附近氢离子的可用性。
细胞膜减少氢离子的损失
嗜碱菌的细胞膜由次生细胞壁聚合物组成,这些聚合物富含带负电荷的残基,例如天冬氨酸、半乳糖醛酸、谷氨酸和磷酸。
嗜碱菌的细胞膜由约90%的支链脂肪酸组成,这些脂肪酸通过减少氢离子泄漏来帮助实现pH稳态。
高度带负电荷的细胞壁结构与阳离子如氢离子相互作用,可以延缓细胞表面氢离子的快速损失,增强嗜碱菌的生物能。
修复损伤提高耐受
嗜碱菌还采用不同的损伤修复系统来维持功能性生物分子的必要水平,修复细胞质碱化造成的损伤。
伴侣蛋白水平的增加和蛋白质损伤修复酶的释放是一些经过充分研究的机制。已知伴侣参与重折叠由压力展开的蛋白质,从而提高耐受水平。
✦嗜碱菌的应用:生产酶制剂、处理人造纤维废物、洗涤剂添加剂、保健品添加剂
嗜碱菌在发酵工业中,可作为许多种酶制剂的生产菌。例如嗜碱芽孢杆菌产生的弹性蛋白酶适宜作用弹性蛋白,而且在高pH条件下裂解该种蛋白质的活性可以大大提高。
由嗜碱细菌产生的蛋白酶具有碱性条件下催化活力高、热稳定性强之优点,常作为洗涤剂的添加剂。
由嗜碱芽孢杆菌产生的木聚糖酶能够水解木聚糖产生木糖和寡聚糖,因此可用来处理人造纤维废物,而碱性β甘露聚糖酶降解甘露聚糖产生的寡糖可作为保健品的添加剂。
★常见的嗜碱菌
嗜碱菌的一些例子包括嗜碱芽孢杆菌(Bacillus alkaliphilus), Bacillus pasteurri, Bacillus halodurans, Halobacterium, Clostridium paradoxum, Halomonas pantelleriensis, Alkaliphilus hydrothermalis
极端盐浓度下的微生物
▸ 嗜盐菌
嗜盐菌是一组嗜盐菌,它们的生存和生长需要高盐浓度。嗜盐微生物构成高盐生态系统的天然微生物群落,广泛分布于世界各地。范围从高盐土壤、泉水、盐湖到海洋沉积物。
嗜盐微生物的一般特征是营养需求低,耐高浓度盐,具有平衡环境渗透压的能力。嗜盐菌有两种类型;要求盐浓度为3%或更高的专性嗜盐菌,以及在平均盐浓度和更高浓度下都能存活的耐盐菌。
它们在生理上是多样的;主要是需氧的,以及厌氧的、异养的、光养的和化学自养的。
这些生物存在于三个领域,即古生菌、细菌和真核生物。嗜盐细菌在特定的系统发育亚群中更为丰富,其中大部分属于变形杆菌科的盐单胞菌。
✦利于嗜盐菌生存的策略
为了避免在高盐条件下过度失水,嗜盐菌采用两种不同的策略来增加其细胞质与外部环境的渗透活性,要么产生相容的有机溶质,要么在其细胞质中积累大量盐浓度以达到平衡状态。细胞内的总盐浓度与环境的盐浓度相关。
高盐策略平衡盐浓度
高盐策略是另一种适应技术,可保护嗜盐菌免受盐环境的影响,在盐环境中,嗜盐菌在细胞内积累无机离子,以平衡其环境中的盐浓度。
这一过程涉及氯离子泵,仅在嗜盐菌中发现,将氯离子从环境输送到细胞质中。精氨酸和赖氨酸位于通道的两端,以促进氯离子的吸收和释放。
极端嗜盐菌通过在细胞内浓缩钾离子来维持其渗透平衡。这是通过膜结合质子泵细菌视紫红质、ATP合酶和钠离子逆向转运体的联合作用实现的,其产生了驱动细胞吸收钾离子的电位。
有机盐策略增加耐受性
高盐策略可能不适合在盐度波动的栖息地繁衍生息的中度嗜盐菌的生存。
有机盐策略包括在嗜盐生物中演化出惰性、相容的有机溶质(渗透物)。这些渗透物保护微生物蛋白质在低盐浓度的水中不变性,同时增强它们对外部盐水环境剧烈波动的耐受性。
酶在高盐环境下更能保持活性
高盐环境显著影响蛋白质的溶解度和稳定性,从而影响其功能。
嗜盐菌的蛋白质和酶在其表面上具有较大比例的谷氨酸和天冬氨酸,这导致大量的蛋白质电荷和增加疏水性。
这两种机制对嗜盐酶的适应起作用。由于它们的多极端特性,嗜盐酶比它们的非嗜盐酶更稳定。这些酶在高盐环境、耐热和嗜碱环境中保持活性。
✦嗜盐菌的应用:提高原盐产量,处理废水
嗜盐菌能使提高盐田中的原盐产量;还能处理含盐的有机工业废水;嗜盐菌上的菌紫质蛋白是未来光生物材料。
同时嗜盐生物正被开发为胡萝卜素、相容溶质、甘油和药用表面活性剂的潜在来源。
★嗜盐菌的例子
嗜盐菌对盐的需求分为三组;低 (1-3%盐浓度)、中等 (3-15%盐浓度) 和极端 (15-30%盐浓度)。
轻度嗜盐:Erwinia, Bacillus hunanensis, Halomonas zhaodongensis, Alkalibacterium thalassium
中度嗜盐:Erwinia, Bacillus hunanensis, Halomonas zhaodongensis, Alkalibacterium thalassium
重度嗜盐:Halococcus salifodinae, Halobacterium salinarum, Limimonas halophilia, Lentibacillus kimchii, Sporohalobacter salinus
极端压力下的微生物
▸ 嗜压菌
嗜压菌被定义为在高于大气压的压力下以最佳方式生长和繁殖的生物。
大多数嗜压生物倾向于嗜冷,因此不能在高于 20°C 的温度下培养。嗜压细菌已从世界各地的各种深海环境中分离出来,并在低温和高压下迅速生长。
高压会影响微生物的生存,从而影响细胞的膜结构和功能。深海环境中的高压和低温会降低脂质的流动性,甚至会抑制生物膜的功能。
✦利于嗜压菌生存的结构
膜流动性较低
高压可能会导致形成凝胶状膜,从而降低养分的吸收和加工。
嗜压菌在脂质中产生更高水平的不饱和脂肪酸,并且脂肪酸不饱和程度的增加可以在高压、低温或两者兼有的条件下将膜保持在功能性液晶状态。
降低的流动性使膜具有确定的结构,从而支持细胞的正常功能。
蛋白质灵活性较高
适用于嗜压菌的高压条件会导致抑制其功能的蛋白质的构象变化。为了防止这种变化,嗜压蛋白通常具有较低浓度的脯氨酸残基和较高浓度的甘氨酸残基。
脯氨酸残基具有破坏α-螺旋的环状侧链,而甘氨酸残基具有具有高构象灵活性的小侧链。增加的灵活性可防止α螺旋的破坏并保护此类蛋白质的功能。
✦嗜压菌的作用:生产高压生物反应器
嗜压菌及其嗜压酶可用于生产高压生物反应器,以及食品加工中的高压灭菌。
嗜压菌在揭示海洋环境变迁和元素的地球化学循环中也起重要作用。
★常见的嗜压菌
嗜压微生物的一些常见例子是Shewanellabenthica、Moritella yayanosii、Shewanella violacea、Photobacterium profundum、Moritella japonica、Sporosarcina spp
极端辐射中的微生物
▸ 嗜放射菌
嗜放射性菌是一组能够在极端形式的辐射(如电离辐射、伽马射线和紫外线辐射)中幸存下来的极端微生物。
对放射性物质的研究非常有限,因为它们要与其他行星的外太空等极端环境隔离开来。
这些生物的多样性很低,所有生物都属于古细菌和细菌家族。放射性物质可以是耐辐射的或抗辐射的。耐辐射微生物可以耐受有害辐射一段时间,而抗辐射微生物可以耐受更长的时间。
辐射对中性粒细胞有害,因为它们会因电离而破坏各种重要的生物分子,如DNA、蛋白质和酶。
反过来,非电离辐射会导致形成像超氧化物这样的活性氧物质,然后影响这些细胞的新陈代谢。
✦嗜放射菌的适应方式
对于电离辐射和非电离辐射,嗜放射微生物使用的自适应机制可能不同。
// 电离辐射
电离辐射主要负责生物体基因组中的双链断裂。然而,它也被证明会损害蛋白质和脂质并诱导持续的氧化应激。
因此,电离辐射抗性生物已经开发出所有或不同策略的组合,如新的和适应性DNA修复机制、抗氧化和酶防御系统以及浓缩的类核。
基因组的快速和准确修复对于幸存的电离辐射剂量是必不可少的,这是通过使用核苷酸切除修复途径在放射性物质中完成的。
其他形式的氧化应激预防和耐受机制包括通过消除氧化大分子来清洁细胞,选择性保护蛋白质免受氧化损伤,以及抑制活性氧的产生。
浓缩的类核也被证明可以提高DNA修复的效率和准确性,并限制辐射产生的DNA片段的扩散。
// 非电离辐射
与伽马辐射不同,紫外线辐射通过形成环丁烯嘧啶二聚体以更微妙的方式损害DNA。
为了修复这些DNA损伤,生物体使用光活化基因、核苷酸切除修复、碱基切除修复和同源重组的组合。
这些生物还开发了一套光保护装置,以保护自己免受持续暴露于紫外线辐射。类胡萝卜素、超氧化物歧化酶和氢过氧化物等产品以及通过多倍体进行的基因复制过程可作为光保护装置。
双嘧啶序列数量减少的生物体的基因组组成也提供了对暴露的保护。
✦嗜放射微生物的作用:环境修复
抗辐射微生物是十分重要的生物资源,可以直接作为环境修复特别是核废料的处理工具。
抗辐射微生物中含有丰富的基因资源,具有应用于作物抗干旱研究的潜在价值。
为治疗肿瘤提供了新药物和新方法。
★ 常见的嗜放射菌
嗜放射菌主要有Deinococcus radiodurans, Brevundimonas, Rhodococcus, Halomonas, Herbaspirillum, Hymenobacter, Rhodobacter。
藻类是光合生物,主要存在于淡水或海洋资源中。大多数藻类都含有色素,可帮助生物体生产食物或氧气。
Onview.net Ltd
藻类的结构与植物和动物等其他生物有很大不同。一些藻类是微观的,而一些大型的却有200英尺长。
绿藻门
绿藻,含叶绿素a、叶绿素b,具有与高等植物相同的色素和贮藏物质,因此通常把它们认为是陆地植物的祖先。
绿藻门不同于其他真核藻类,它的储存物质在叶绿体而非细胞质中合成,通常在蛋白核的参与下合成淀粉。叶绿体周围没有叶绿体内质网。
✦显微镜下的结构
在显微镜下,绿藻被视为封闭在以链状排列的隔室中的绿色结构。在每个这样的隔间内,观察到一个大液泡,还可以看到两层细胞壁。
藻类的形状和大小因属而异。一些藻类是能动的,而一些是不能动的。
✦可用作饲料或生化材料,还具有净化作用
小球藻和珊藻富含蛋白质可供人食用和作动物饲料。
绿藻是藻类生理生化研究的材料及宇宙航行的供氧体,有的可制藻胶。绿藻在水体自净中起净化和指示生物的作用。
黄藻门
✦结构
这组藻类几乎没有链状结构的物种,而是呈鼓形、变形虫或梨形结构。
有些物种有毛发状的附属物或鞭毛,有时比生物本体还要长。
一些藻类的形状和大小可能会在其一生中发生变化,具体取决于生命阶段和栖息地。
✦影响水中氧气,造成水体污染
黄藻是一种水生浮游植物,发生的适宜气象条件为气温高、降水少、日照长,它的主要组成物是双星藻、转板藻和水棉三个属的藻类。
这种黄色藻类生长旺盛,大量消耗水内氧气,对鱼类和其他水生植物生长造成影响。黄藻使得鸟赖以生存的食物被覆盖和污染,对在此栖息生存的水禽构成严重危害,也造成水体严重污染,渔业资源遭到破坏。
隐藻门
✦结构
隐藻门是一大类的藻类,大都具有色素体,淡水中常见。细胞大小约为10-50μm,形状扁平,有两个稍微不等长的鞭毛。
该组中的藻类呈逗号形,带有红色或类似的色素。有些种类的细胞膜上可能有凹槽,而另一些则没有。色素通常位于侧面,而细胞核位于液泡附近的中心。
✦能将藻胆素带给宿主
一个特征是能寄生于红藻中,形成一种内共生关系,并把藻胆素带给宿主。
同时隐藻在海洋浮游生物群落中占有一定地位。隐藻喜生于有机物和氮丰富的水体,是我国传统高产肥水鱼池中极为常见的鞭毛藻类。有隐藻的鱼池,白鲢生长好,快,产量高,隐藻是水肥、水活、好水的标志。
红藻门
红藻门是藻类植物的一门。该科多数是多细胞的,少数是单细胞的,该门只有红藻纲一纲,约有760属,4410余种。
红藻纲又分两个亚纲:紫菜亚纲和真红藻纲。该门绝大多数海产,少数生于淡水;分布于世界各地,包括极地。
✦结构
红藻是丝状的,其中身体的特征是具有钙质沉积物的菌体,从而形成固体结构。有机体的颜色范围从粉红色到紫色、红色、黄色、绿色,甚至是白色。
有些物种是光合作用的,因此绿色色素沉积在细胞壁内部。
✦重要经济价值
红藻门的经济价值很高。在红藻类中,紫菜是一种食用藻类,它含有丰富的蛋白质,不仅营养丰富,而且味道鲜美。此外石花菜、海萝等均可食用。
鹧鸪菜和海人草是常用的小儿驱虫药。从石花菜属、江篱属、麒麟菜属植物中提取的琼胶,被应用在医药工业和纺织工业上,并广泛作为培养基。
甲藻
✦结构
由于存在金棕色质体,这些单细胞生物呈现金棕色。它们有凹陷的细胞膜,并展现出游动的能力。
甲藻的细胞核相当大,有可见的染色体。并且有两个从细胞膜突出的不同鞭毛。
注:有些甲藻是肉眼可见的,即使没有任何显微镜也能看到。
✦富集会污染水体
甲藻是具有双鞭毛的单细胞集合群植物,形状不定,常分布于淡水和海水中。
有些甲藻的活动是有害的,它们的生存会带有一些特殊的气味。有的则会形成“赤潮”和“藻花”,使局部海水呈现红色、黄色或棕色。
赤潮:水体中某些微小的浮游植物、原生动物或细菌,在一定的环境条件下突发性地增殖和聚集,引起一定范围内一段时间中水体变色现象。
藻花:又称“水花”,是淡水水体中某些蓝藻类过度生长所产生的现象。
眼虫(裸藻)
眼虫是眼虫属生物的统称,在植物学中称裸藻,也称绿虫藻,是一类介于动物和植物之间的单细胞真核生物。
✦结构
在显微镜下,它们有一个大而细长的绿色结构。形状可能会从一种物种变为另一种。在它们的细胞质中有两到四个带有叶绿体沉积物的鞭毛。
在眼虫中,可以看到外围有一个橙色的斑点,称为有机体的眼斑。
✦环境污染的生物指标
研究眼虫不仅对遗传变异理论的探讨有意义,而且对了解有色、无色鞭毛虫类动物间的亲缘关系,对了解动、植物的亲缘关系都有重要意义。
眼虫也有被作为有机物污染环境的生物指标,用以确定有机污染的程度,另外眼虫对净化水的放射性物质也有作用。
病毒
NIAID (Flickr)
病毒可以被认为是强制性寄生的一种粒子,因为它们不会在活生物体外生长或存活。
✦病毒体型十分微小
病毒的大小范围从直径20nm到200-450nm。与细菌相比,病毒很小。
因此病毒无法用复合显微镜观察,需要使用荧光显微镜或透射电子显微镜等高倍显微镜。
荧光显微镜下的病毒
在荧光显微镜下,病毒呈现出所用荧光颗粒的颜色。
但是依然很难区分病毒的结构,但这种技术对于病毒的定量估计很有用。
荧光染料对某些蛋白质具有特异性,从而使它们能够检测所需的颗粒。
透射电子显微镜下的病毒
透射电子显微镜更适合观察病毒,它们可提供高达1000倍的粒子放大倍率。
通过这种显微镜,可以观察生物细胞内的病毒。与荧光显微镜一样,该技术还利用病毒中蛋白质的特异性染料,从而使病毒可视化。
在观察病毒的结构时,它可能是二十面体或螺旋形。每种病毒的形状和结构各不相同,但成分相似。
所有病毒都有遗传物质,可以是包裹在蛋白质外壳内的DNA或RNA。
在噬菌体病毒的情况下,尾部和尾部纤维也是可见的,并且被发现附着在细菌细胞表面。
通过本文的介绍,相信大家对微观世界有了一定的认识和了解。但是微生物世界远比我们现在所了解的庞大的多。
科学家还在不断地对微生物世界进行探索,识别微生物的种类并了解其结构和作用,有助于构建更好的生存环境,创造更健康的身体。对其研究探索的过程,为农业、医学、工业、生物修复等提供了新的机遇,将对社会产生深远的积极影响。

谷禾健康

随着一日三餐米面肉蛋菜等一些列食物的食用,数百种化学成分会进入我们的消化道。在那里,它们被肠道微生物组进一步代谢,这是数千种微生物物种的独特集合。
因此,肠道微生物组在决定营养如何影响健康方面发挥着重要作用。然而到目前为止,微生物组中的许多微生物的代谢能力仍然是未知的。这意味着我们不知道它们以什么物质为食,以及它们是如何处理这些物质的。
近期,来自普林斯顿大学的研究人员在《CELL》期刊上发表了最新的文章:
“Gut bacterial nutrient preferences quantified in vivo”,研究人员使用同位素追踪定量研究了小鼠肠道微生物群的输入和输出。
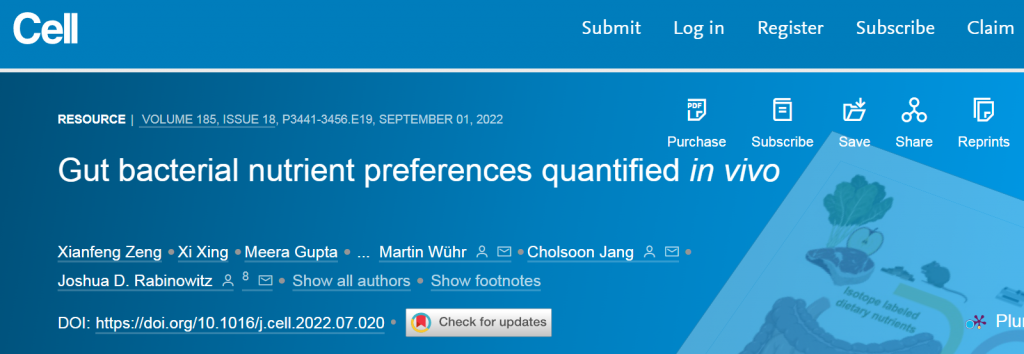

微生物碳水化合物发酵的主要输入是膳食纤维,支链脂肪酸和芳香代谢物的主要输入为膳食蛋白质。此外,循环宿主乳酸、3-羟基丁酸和尿素(但不是葡萄糖或氨基酸)为肠道微生物群提供食物。
肠道菌群拥有巨大的酶多样性,超过哺乳动物基因组的数量100多倍。这些酶的能力能使摄入的膳食营养物质加工成一些列微生物代谢物。
为了复制自身和释放代谢产物,肠道细菌需要营养输入。这些形式包括摄入的食物、宿主合成的肠道粘液和宿主循环代谢物。
//
在本文中,研究人员通过对肠道菌群及其进入宿主循环系统的代谢物进行了大规模的定量评估。
研究了膳食淀粉、纤维和蛋白质的贡献以及宿主粘液的贡献,也研究了大多数主要的循环宿主营养素,发现乳酸、3-羟基丁酸和尿素在从宿主传递到肠道微生物群中表现突出。基于对细菌特异性肽序列的测量,评估了不同细菌属的营养偏好,并表明这些偏好与响应改变饮食的微生物组分变化一致。
同位素追踪能够定量测量代谢物和生物量的输入。与质谱检测相结合的稳定同位素示踪剂,使得能够测量特定下游产物的标记。通过注入氮标记的苏氨酸来标记宿主粘液,研究人员能够比较饮食和粘液蛋白对肠道微生物群的贡献,并观察到喂食低蛋白饮食的小鼠中粘液贡献的变化。
从小鼠尾部静脉抽取血样;
使用注射器从小鼠膀胱采集尿液;
所有血清样品在没有抗凝剂的情况下置于冰上 15 分钟,并在 4°C 下以 16,000 x g 离心 15 分钟。
用预冷的Wollenberger钳在液氮中快速分离并快速冷冻(< 5秒)获得组织;夹紧前取出肠内容物;盲肠内容物取样时,先将小鼠盲肠取出并在表面切开,然后用镊子将盲肠内容物挤出。
取新鲜粪便,轻揉小鼠腹部诱导排便。将血清、组织和粪便样本保存在 -80 ºC 直至进一步分析。
为了测定血清和组织样本中的代谢物浓度,进行了同位素标配(isotope spike-in)或标准标配(standard spike-in )。
对于前者将已知浓度的同位素标记标准品加入血清或组织提取液中,通过标记与未标记代谢物的比值计算浓度。
当没有同位素标准品时,加入连续稀释的非标记标准品,测量的总离子计数与加入的标准品浓度之间产生线性拟合。然后通过拟合线的x截距确定内源代谢物的浓度;蛋白质氨基酸组成采用酸水解法测定。
首先,使用13C同位素标记的不同营养物质,通过口服管饲法对小鼠进行灌胃采集小鼠的血清、组织和粪便样本。对粪便和肠内容物进行16S rRNA测序获得细菌分类。
首先使用代谢组学方法测定盲肠内容物中游离氨基酸13C-或15N标记。
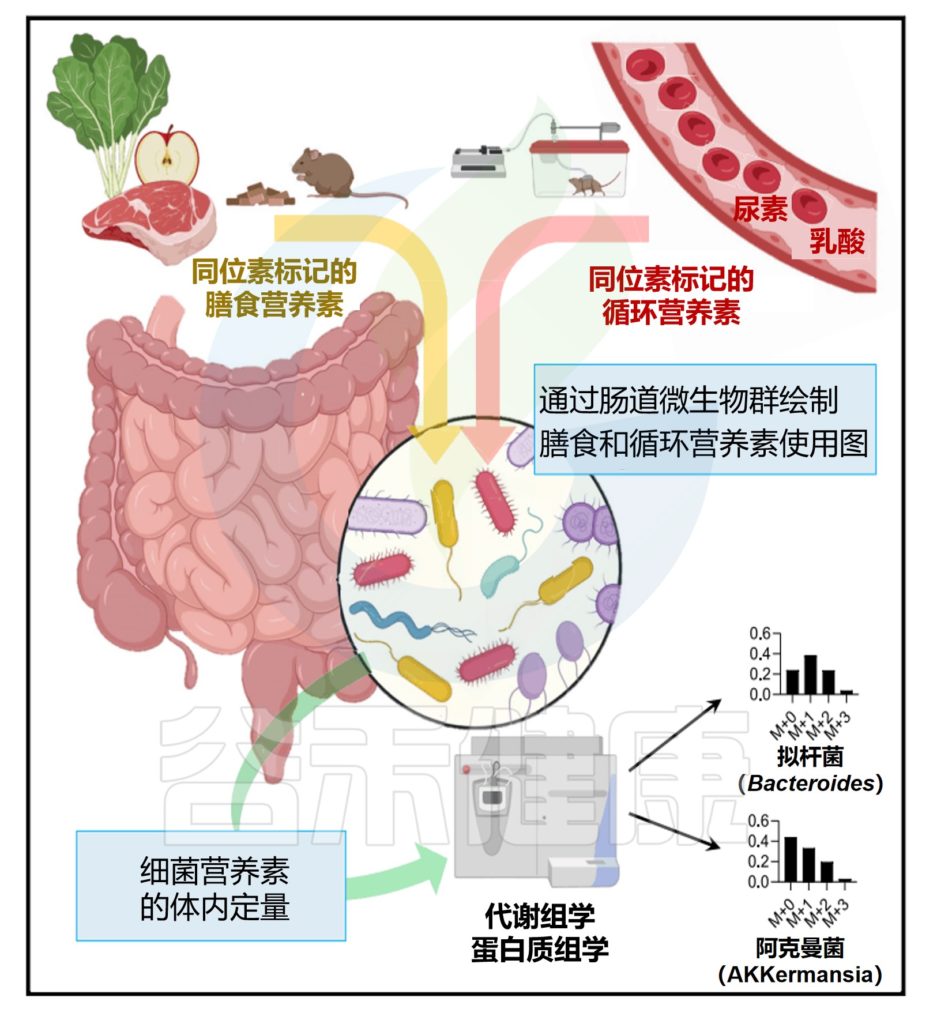
然后,对于每个肽,模拟了未标记(Iunlabeled)和由游离盲肠氨基酸(Ifree)合成的肽的同位素包膜模式。标量γ可以通过将测量的肽同位素分布(Imeasured)与Iunlabeled和Ifree的线性组合拟合来确定。
注意,当一个菌属使用的特定营养素超过该营养素对盲肠游离氨基酸的贡献时,γ将大于1。
具体来说,测量的每个肽的γ如下:
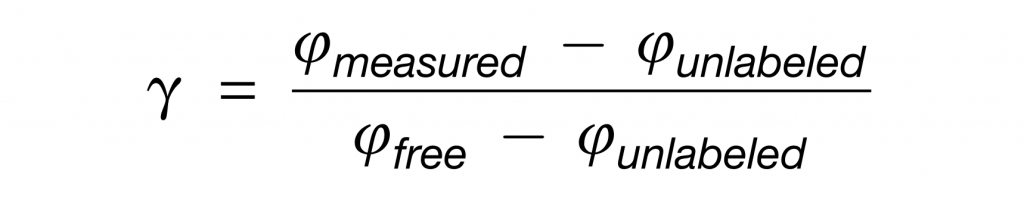

对于细菌属水平的原料贡献程度的测量,分析中只保留测量超过3个肽的属,多肽的中位数为γ-genus。
对于细菌科水平,仅分析在蛋白质组学中始终检测到的属,以及在 16S rRNA 基因扩增子测序中检测到 (> 0.5%) 的属的上一级科。
每种营养物质对菌属的贡献程度的定量公式如下:
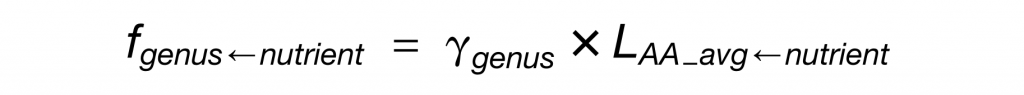

LAA_avg-nutrient为各营养物质对细菌蛋白质的贡献程度,其计算公式如下:
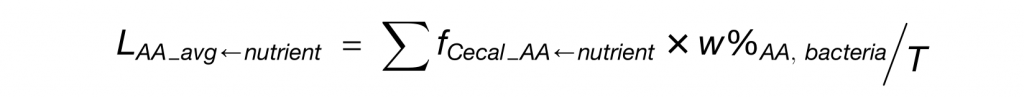

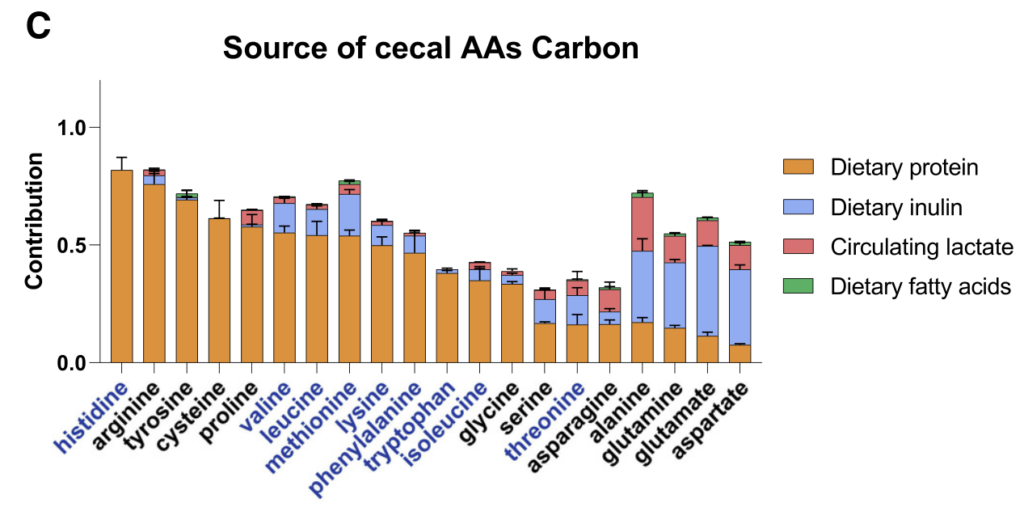
1 微生物组消耗较少的可消化膳食成分
微生物群影响宿主生理学的主要机制是通过分泌代谢产物。研究人员在门静脉和体循环以及盲肠内容物中测量了微生物衍生的50多种代谢产物的绝对浓度。
微生物群相关代谢物的绝对浓度和来源
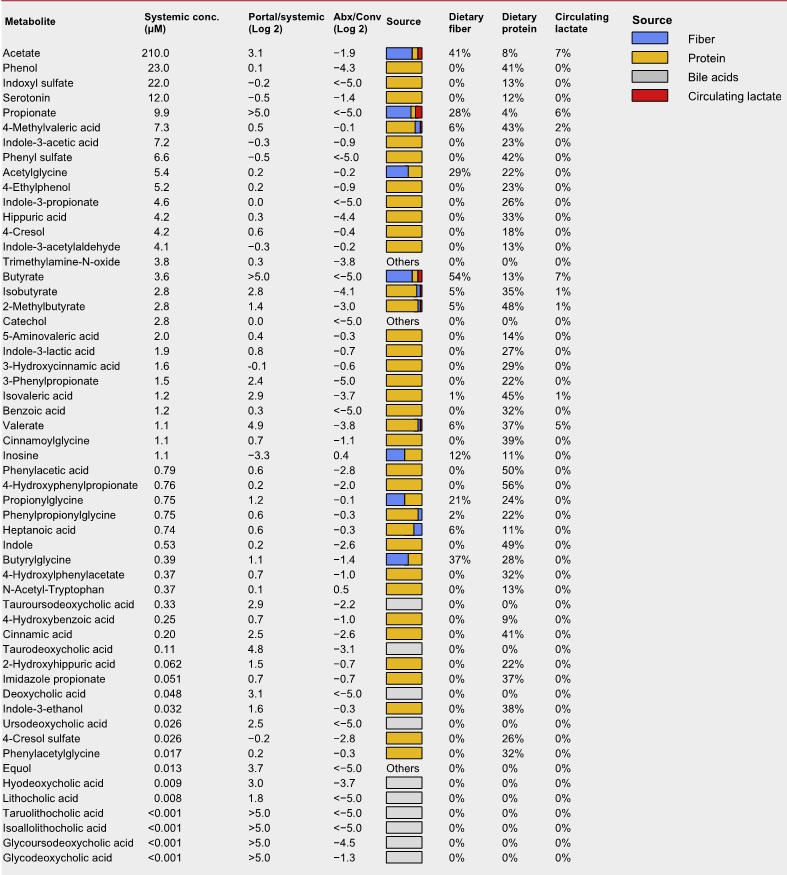
上表可以看到,与全身血液相比,大多数在门静脉循环中升高,除两种(肌苷和N-乙酰色氨酸主要来源于宿主)外,其余均被抗生素治疗耗尽。
门静脉血中主要排泄产物是短链脂肪酸。
其他相对丰富的微生物群产物是芳香族氨基酸发酵产物(苯酚、吲哚硫酸盐和3-苯丙酸盐)和支链脂肪酸(戊酸盐、异戊酸盐,4-甲基戊酸、异丁酸盐和2-甲基丁酸盐)。
探索肠道微生物产物的膳食输入:淀粉、菊粉
研究人员通过口服管饲法、淀粉(易消化葡萄糖聚合物)和菊粉(易消化果糖聚合物,即可溶性纤维)喂养小鼠:
13C淀粉灌胃后,标记的葡萄糖、乳酸和丙氨酸迅速出现在门脉循环中,并占大多数淀粉碳(约75%)。
13C菊粉和13C淀粉有什么不同?
13C菊粉灌胃后,没有观察到大量标记的果糖、葡萄糖、乳酸和丙氨酸,取而代之的是标记的门静脉代谢产物以短链脂肪酸的形式缓慢出现,约40%的菊粉碳成为短链脂肪酸,其余未消化并随粪便排出。
膳食菊粉,而不是淀粉,在盲肠内容物中广泛标记糖酵解和TCA中间体和氨基酸。
藻类蛋白大量标记了微生物群衍生的门静脉代谢物:短链脂肪酸、支链脂肪酸和芳烃(吲哚、吲哚-3-丙酸盐和3-苯丙酸盐)。
“难以消化的碳水化合物和蛋白质直接为微生物组提供营养,并通过微生物产物间接为宿主提供营养。”
研究中发现宿主循环系统中的乳酸,3-羟基丁酸以及尿素能为肠道细菌提供营养。
如图A,将同位素标记的营养物质通过静脉输注到小鼠的全身血液循环中。 2.5 小时后收集血清和粪便以量化每种营养物质对相应菌群代谢物的碳贡献。
图BCD表示了13C标记的各种营养物质在小鼠的血液和粪便中的含量,可见乳酸和 3-羟基丁酸有进入肠道菌群中,而其余大部分营养物质如柠檬酸盐、葡萄糖、氨基酸等都没有进入到肠道菌群中。
图F为15N标记的营养物质,可见尿素也同样被菌群大量利用。
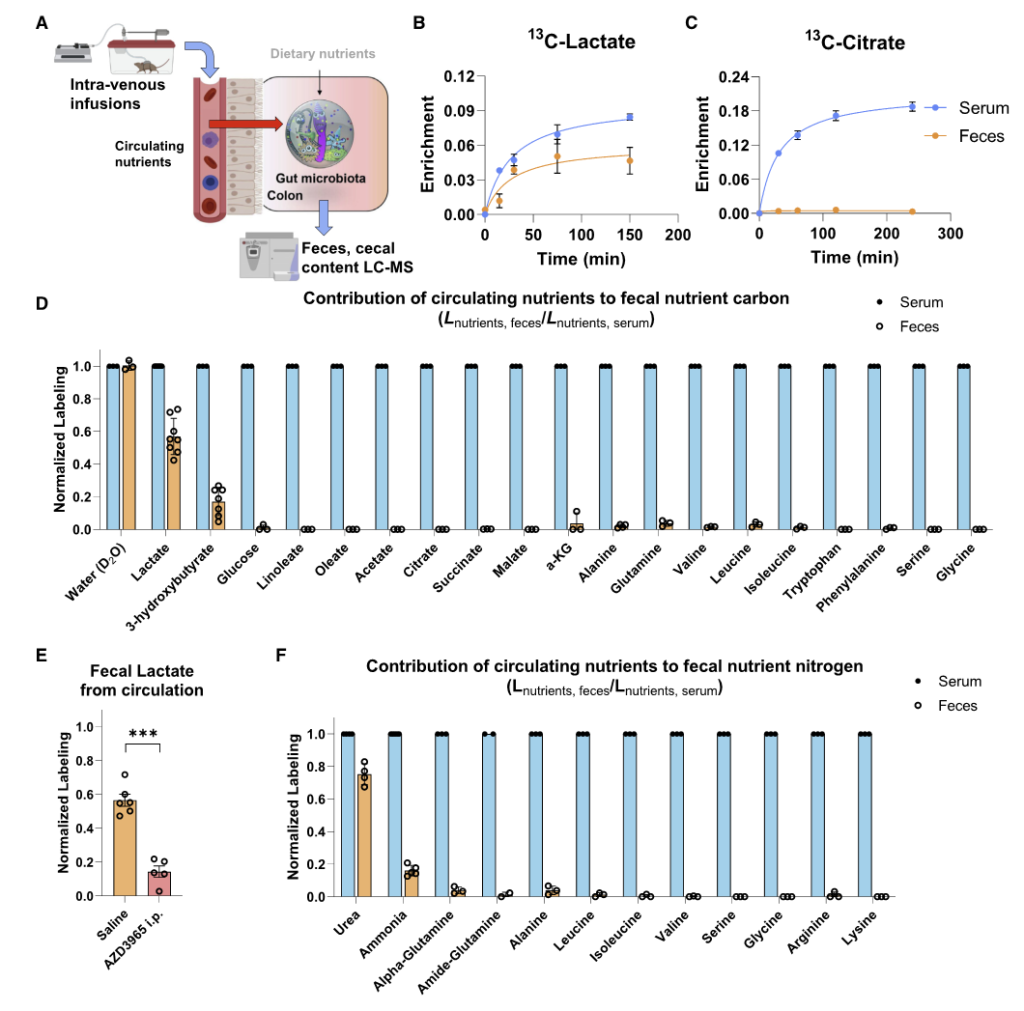
为了定量确定微生物代谢物的来源,研究人员给小鼠喂食部分纤维、脂肪或蛋白质13C标记的标准食物,盲肠标记在12小时内达到稳定状态。
为了说明循环营养输入,研究人员还注入了13C乳酸或3-羟基丁酸。
这些研究确定了大多数微生物群中心代谢物中的碳供给:
接下来,研究人员检查了微生物组游离氨基酸的输入,并用15N标记的膳食蛋白和注入的尿素进行追踪。
与哺乳动物不同,大多数肠道细菌具有合成所有20种蛋白质氨基酸的生物合成能力。
然而,研究人员观察到“必需氨基酸”主要来源于膳食蛋白质,哺乳动物无法制造,需要在细菌中表达广泛的生物合成途径。
“非必需氨基酸”主要在肠道微生物群中合成,使用膳食菊粉和循环乳酸作为碳源。
抗生素或无菌小鼠中的微生物群消耗有利于盲肠中氨基酸的积累(基于同位素追踪研究),这些氨基酸主要来自膳食蛋白质和微生物合成的氨基酸的消耗。
膳食蛋白质是必需氨基酸和非必需氨基酸的主要氮源,宿主尿素对非必需氨基酸也有很大贡献。
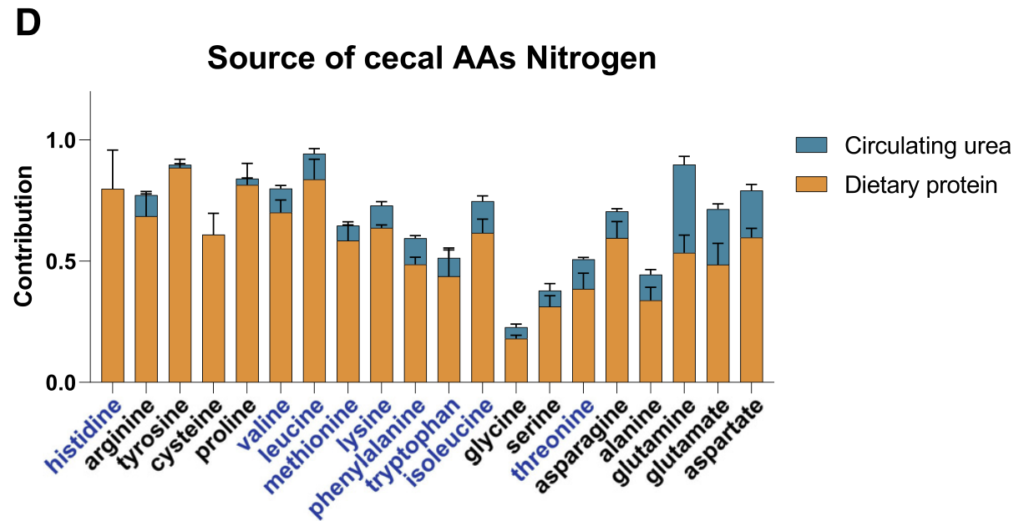
研究人员的发现如下:
【1】必需氨基酸,尽管能够由微生物群合成,但主要来自饮食,不经历任何碳重排;
【2】与TCA连接最紧密的非必需氨基酸基本上由微生物群合成,使用来自纤维的碳,通过中心代谢反应与其他碳争夺;
【3】转氨反应部分地将来自饮食衍生氨基酸的氮与来自宿主尿素的氮混合。
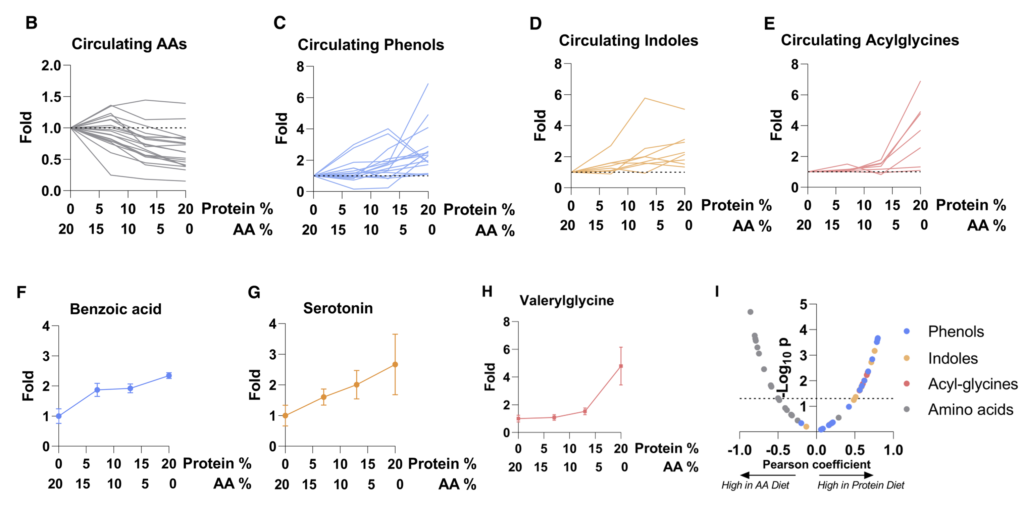
研究人员发现,许多微生物来源的代谢物来源于到达结肠的未吸收膳食蛋白。假设这些代谢物的循环水平将取决于膳食蛋白质到达结肠微生物群的程度。
为了控制这一点,研究人员给小鼠喂食的食物中,一部分蛋白质(酪蛋白,部分到达结肠微生物群)被游离氨基酸(基本上在小肠中完全吸收)取代。
2周后对全身血液进行代谢组学研究。含有较少完整蛋白质和更多游离氨基酸的饮食往往会增加循环氨基酸水平。
重要的是,蛋白质衍生的循环微生物代谢物(酚类、吲哚类和酰基甘氨酸)串联下降。
“微生物代谢物营养来源的知识可用于操纵其系统水平。”
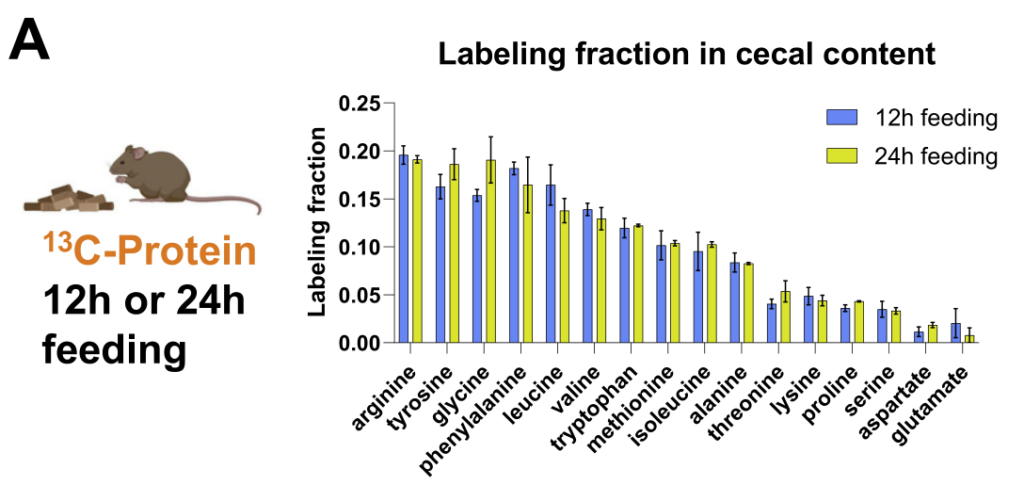
研究人员通过结合13C营养标记和蛋白质组学来定量不同微生物的碳原料。
每种13C标记的营养素(膳食菊粉、膳食藻蛋白或循环乳酸)提供24小时,这足以在肠道细菌中实现稳态标记。
如同B-D,分别计算了在膳食中使用的菊粉和蛋白质以及乳酸在各细菌内的喜好程度,这个喜好程度也就是将在细菌特异性肽上被同位素标记的程度进行了量化。
结果可见:
拟杆菌属和梭状芽胞杆菌利用菊粉的程度是 Akkermansia、Muribaculum 或 Alistipes 的 4 倍多。
总体而言,厚壁菌门下的菌属比拟杆菌门的使用膳食中的蛋白质(厚壁菌0.237±0.052;拟杆菌0.175±0.031,p=0.02)。
Akkermansia通常被认为是一种促进健康的肠道微生物,使用的菊粉和蛋白质最少。相比之下,它使用了来自宿主的循环乳酸最多。
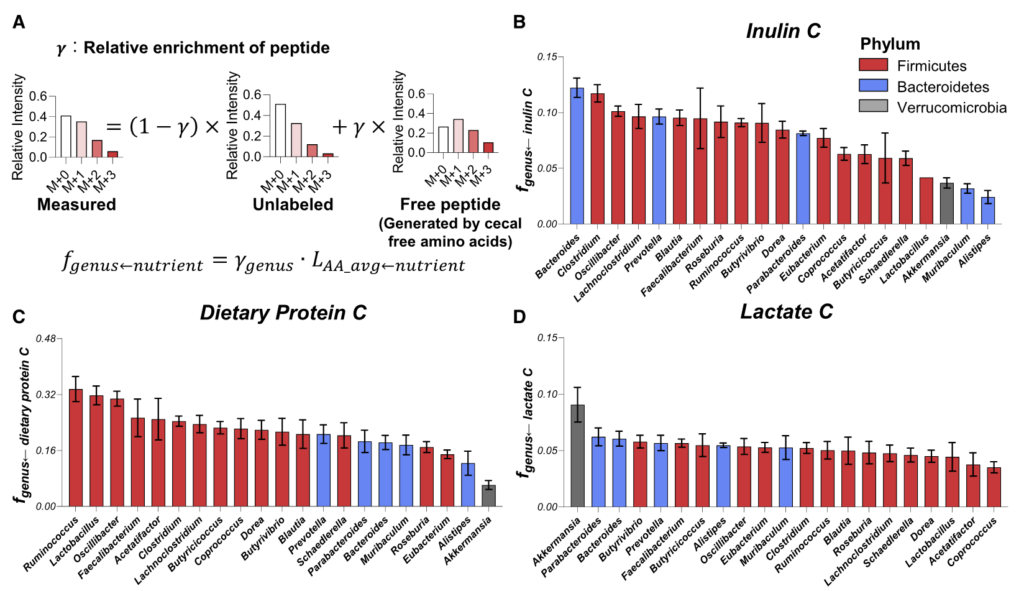
为了知晓这些细菌的营养偏好是否能预测饮食变化后的肠道菌群的组成变化。研究人员给小鼠喂食富含菊粉或藻类蛋白的饮食 2 天,并通过 16S rRNA 测序测量微生物组的组成。
结果如图F和I:
利用最多菊粉的拟杆菌属在高菊粉饮食后增加了4倍;
另一种利用较多菊粉的梭状芽胞杆菌也增加了2倍;
利用较少菊粉的菌属要么没有变化,要么略有下降;
富含藻类蛋白饮食的实验结果同理。
图G和J计算了这两种营养物与对其利用程度最高的前两名菌属相对丰度的相关性,p<0.05呈显著相关。
“不同肠道细菌的营养偏好有助于解释饮食操作后微生物组分的变化。”
最后,研究人员转向不同肠道细菌的氮源偏好,比较15N标记的膳食蛋白喂养和15N尿素输注。
高度利用膳食蛋白质中碳的细菌属也高度利用膳食蛋白中的氮,这与细菌蛋白质组中完整吸收的膳食蛋白质中的氨基酸一致。
厚壁菌喜欢从膳食蛋白质获取氮
在厚壁菌门成员中,偏好尿素氮的属往往是菊粉的疯狂使用者,即使用菊粉和尿素合成自己的氨基酸。这包括一些脲酶阴性菌属,它们可能通过交叉喂养获得尿素氮。
此外,在厚壁菌中也看到了一些属更喜欢从膳食蛋白质中获得氮,而其他属更喜欢循环尿素。
静脉注射尿素以提高循环尿素浓度后,偏好尿素的厚壁菌以及阿克曼菌的丰度大幅增加。
拟杆菌喜欢从宿主分泌的蛋白质中获取氮
与厚壁菌相比,拟杆菌对膳食蛋白质和循环尿素氮的利用率较低,这提出了一个关键问题:
拟杆菌如何获得氮?
肠道微生物群的一些成员(如拟杆菌和阿克曼菌)能够消化宿主分泌的蛋白质,如粘蛋白。
假设宿主分泌的蛋白质是拟杆菌氮的关键来源。为了探索这种可能性,研究人员进行了长期15N标记的赖氨酸和精氨酸输注(12、18和36小时),以标记结肠中的宿主蛋白。
尽管没有直接给微生物组喂食,但在36小时输注后,赖氨酸和精氨酸确实起作用,这与通过宿主蛋白进行的标记一致。这种标记优先发生在拟杆菌和阿克曼菌中。
膳食和分泌宿主蛋白的氮贡献呈负相关,与某些肠道细菌优先消耗膳食蛋白和其他宿主蛋白一致。
“膳食蛋白质和循环尿素是厚壁菌的主要氮原料,而分泌的宿主蛋白质为拟杆菌提供氮。”
研究人员开发了定量同位素追踪方法来测量肠道细菌的营养偏好。除了膳食纤维和分泌的宿主蛋白外,还将膳食蛋白和循环宿主乳酸、3-羟基丁酸和尿素确定为喂养肠道细菌的重要营养素。排除了其他循环宿主营养素(如葡萄糖和氨基酸)对结肠微生物群的直接贡献。
一项关键技术成就是能够从不同碳源和氮源追踪到细菌特异性肽,从而揭示复杂和竞争性肠腔环境中不同细菌的营养偏好。
厚壁菌门倾向于从膳食蛋白质获得氨基酸,而拟杆菌门更多地依赖宿主分泌蛋白。同样,一些厚壁菌门(如梭菌属)大量利用纤维(菊粉),而其他厚壁菌门则不利用纤维。
动物饮食干预实验发现,拟杆菌属和梭菌属是转化纤维最活跃的菌属。宿主循环代谢物水平也可能影响微生物组的营养获取和最终组成。
本文提供了关于哪些营养素喂养肠道微生物群以及哪些细菌更喜欢哪些营养素的基础知识。
文中所开发的方法具有广泛的应用前景,最终将有助于全面和定量地了解饮食-微生物-健康的关系。
参考文献:Zeng X, Xing X, Gupta M, Keber FC, Lopez JG, Lee YJ, Roichman A, Wang L, Neinast MD, Donia MS, Wühr M, Jang C, Rabinowitz JD. Gut bacterial nutrient preferences quantified in vivo. Cell. 2022 Sep 1;185(18):3441-3456.e19. doi: 10.1016/j.cell.2022.07.020. PMID: 36055202; PMCID: PMC9450212.

谷禾健康

统计和科学编程已迅速成为科学中的一项必要技能,而这一般都需要用到——R语言。
R语言以其简单易学、免费开源的特性,正在各个领域发挥着越来越重要的作用。


由于其强大的统计计算和数据可视化两大功能,可以说在生信领域, R语言是干活的法宝。


本文分享的是来自一些自学R语言的研究人员认为有用的技巧,希望能为已经上路的R语言的自学者,看不到清晰道路的R语言小白、以及广大想要学习R语言的生物科研人员提供一些帮助。
注意:
这10条技巧并不是提供使用 R 的技术指导,而是提供了构建或磨练 R 编程技能的实用策略。
学习 R 就是学习一门新语言,包括词汇、语法、句法,甚至可能是一种新的思维方式,打开一个新的世界。
可以这样说,学习 R 语言很困难,因为它涉及经常报错,所以这个学习的过程要做好准备,识别这些错误,并最终学习如何修复。


▸不怕犯错,永远没有完美
即使是最有经验的人,仍然会犯错误、忘记函数参数,或转向互联网搜索以刷新他们可能已完成多次的任务。
R 语言的流畅度不是永远不会收到错误消息,而是当你收到错误消息时感觉有能力修复它们(图 1)。当然,有些 bug 的发生并不是因为输入错误的代码,而是因为对函数参数或输出的误解。
虽然避免错误消息是很好的第一步,但确保脚本完成预期任务的最佳方法是:
仔细阅读文档,一次运行一行代码。
学习正确的编程实践的一个好方法是看书。书的一个优点是它们通常代表专家的声音、社区的技能,或两者兼而有之。大多数学习 R 编程的好书都包含代码示例,你可以使用这些示例来提高技能。
▸不管哪本书,都要运行代码示例并检查输出,积极分享
当书本后面没有答案时,开始或加入阅读小组会很有帮助,而且更有趣(后面第八条会讲到加入 R 社区)。与同事一起尝试练习,并利用彼此作为网络来提出问题、比较方法并通过复杂的任务进行集体思考。
教科书解决方案也定期在个人博客或网站上在线共享。在线发布自己的解决方案可以让你能够获得反馈,使整个 R 社区受益(参见第9条)。
▸专业书籍可以成为提升特定领域技能的绝佳资源
R 用户其实很幸运,你们已经拥有大量高质量的已出版书籍作为学习资源,其中包括:
RStudio 网站
( https://www.rstudio.com/resources/books/ ) 上精选的十多种书籍,以及无数其他作者由知识渊博的 R 贡献者提供。
比较推荐的 R 一些书籍包括“R for Data Science”和“Advanced R”,它们通过tidyverse介绍了 R 的现代方法(参见第5条关于风格)。
▸确立目标,按需去学
根据你学习 R 的目的,去看涵盖与你兴趣相符的,更具体主题的书可能会有所帮助。
例如,如果你打算将 R 用于高级统计,可以去看这类书,如《使用 R 的生态学中的混合效应模型和扩展》或《广义相加模型:R 简介》,每个都鼓励对核心编程和统计技能的深刻理解。
如果你想用 R 来画出高质量的 Web 图形,从而传达结果,可能会需要看《Interactive web-based data visualization with R, plotly, and shiny》,其中涵盖了先进的基于 Web 的数据可视化技术的介绍。
那么到底应该买多少本书?
应该选修哪些编程的课程?
是不是需要很大的金钱成本?
……
别担心,下一章节我们提供一些免费资源供参考。
幸运的是,许多高质量的R资源在线免费提供,涵盖了你可能所需的一切。
▸电子书,网站
例如,许多电子书在线免费提供,包括RStudio网站上的电子书。为了更快地参考,RStudio还提供了几个单页备忘单,每个备忘单都涵盖了一个特定包或编程任务的基本知识,可以作为很好的提醒。
如果你觉得看书枯燥,不容易看懂,那么也可以参考一些别的形式:
▸互动教程、视频课程、博客等
在Coursera、edX和freeCodeCamp等网站上也有许多关于R、统计和数据科学的免费课程,The Carpentries的免费培训材料,甚至YouTube和Twitch等网站上的免费视频教程。
一些地区和全球R团体,如R-Ladies和ROpenSci(更多信息请参见第8条),提供多种语言的博客文章、研讨会和资源。此处维护了R-Ladies博客列表(https://github.com/rladies/awesome-rladies-blogs); 包括Julia Silge和Danielle Navarro等。
其他精选的免费资源列表可以在inSileco博客和r-directory网站上找到。
无论是通过博客、互动教程还是视频课程,如果有一种在线方式你觉得最适合的,那么很可能会有这种方式的R资源,只需要搜索一点就可以找到。
▸对于R技术领域的更多具体培训,许多机构提供免费教程、研讨会和在线课程。
例如,魁北克生物多样性科学中心(QCBS)R研讨会系列以英语和法语提供了关于数据可视化、线性模型、多元分析等的介绍性和高级研讨会,并在其网站上免费提供PPT、代码和配套书籍。
爱丁堡大学的编程俱乐部为生态学家和环境科学家提供广泛的课程,从数据处理和统计到地理空间分析和机器学习。
EcoDataScience是一个以加州大学圣巴巴拉分校为中心的组织,提供一系列生态相关研究技术的技能分享和培训。
以上这些只是高质量学习小组的几个例子;可以与你所在的机构、公司或同事联系讨论,获得最相关或最方便的建议。
▸用“只是为了好玩”的项目来练习
如果你有时间和灵活性,一个很好的培养技能的方法就是通过“只是为了好玩”的项目来练习R。
– 想学习如何使用ShapeFile进行映射吗?
尝试制作一张你最喜欢的城市的海报,作为墙壁艺术印刷。
– 通过API抓取数据?
尝试在Spotify上找出最受欢迎的艺术家的共同品质。
– 文本分析?
尝试使用R的文本挖掘包来比较你喜爱的书籍或电视节目的情感关联。
– 定制数据可视化?
尝试使用ggplot形状和美学来复制你最喜爱的艺术作品(参见Twitter上的#RecreationThurday)。

▸ 低风险环境更解压
仅仅为了好玩,项目可以成为培养关键技能的非常有价值的环境。
低风险的环境会减轻你成功的压力,但当你成功时,会给你新的产品和工作代码,供你下次尝试类似的任务时参考。
▸ 用各种活动,趣味竞赛的方式边玩边学
参与有趣的社区活动、Twitter挑战或R编码竞赛(更多信息,请第8和9条)将帮你建立坚实的基础,为你的编程道路打开大门,同时选择你喜欢的项目。
如果你更愿意让你的R学习更多的“任务”工作,你可以对现有的项目进行低压力的增加,从而获得新的技能。
尝试通过向现有图添加自定义文本注释来练习HTML呈现,或者通过创建用于交互式数据探索的闪亮应用程序,来升级日常工作流程。
在改进现有工作的同时,寻找扩大技能的机会将有助于你成为更全面的R用户。
培养和维护项目的编程方法和组织系统有助于你和他人的代码清晰一致。
▸ 在开始编程项目时,需要考虑你的心态
如果在开始新任务时对所有未来步骤都进行了精心规划,那么你就可能会从使用虚拟程序代码开始项目中获益。
*虚拟程序代码是计划完成的操作的简单语言描述列表,在文档中写出,然后逐行翻译为代码。
这种方法的一个好处是,它允许你从头到尾对项目进行概念化,并为你将要编写的每一行代码提供一个离散的目标。
如果说,你更喜欢边做边学,可能更喜欢直接编写代码,观察每一行的输出,并在文本中注释最后一行完成的内容,以较小的增量达到最终目标。
在这两种情况下,对代码进行注释是很重要的,确保将来能够理解当前的想法。


▸ 接下来,需要决定在编码时使用什么样式
在本文中,编程风格是编写代码时使用的特定函数、包和语法策略的集合。如果你认为R是一种语言,可能会认为你的风格是一种方言;两种风格看起来可能不同,但它们的含义是相同的。
在R中,主要的样式划分通常围绕着:你是使用基本R的典型样式,还是采用R的管道方法以及其他tidyverse原则。
3 种代码样式的示例


虽然这些样式并不相互排斥,但它们包含不同的函数集,通常适合不同的语法策略来构造代码。根据你的背景,你可能更喜欢其中一种语法。
例如,如果有在C++或Java等程序中编写代码的经验,则顺序或嵌套语法可能会让你熟悉,而管道函数可能会使代码读起来更类似于用英语编写的句子。
(请参阅tidyverse样式指南:https://style.tidyverse.org/)
在大多数情况下,只要适合你的,什么样式都可以。

当然,匹配协作者中最常见的代码风格或规程中的标准,对阅读、共享和排除代码故障很有帮助。
有时,可能需要切换样式,以便于最好地完成某项任务,但选择尽可能保持样式一致将有助于确保你和其他人可以解释代码。
▸ 最后,将文件放入严格的目录结构中非常重要
R生态系统提供了许多工具来促进项目组织。大多数集成开发环境(IDE)都有“项目目录”的概念;这对于领先的R IDE、RStudio和大多数其他(例如,VScode和Atom)都是如此。
还可以使用包来简化项目目录中的文件路径命名方案。根据你所在的领域,文件夹组织结构可能会有所不同,但随着项目列表的增长,对系统进行批判性思考将简化生活。
此外,保持适当的项目组织将有助于你学习“版本控制”的实践,这是一种跟踪更改和备份代码的系统,正在成为跨科学领域的专业标准。
R的大多数功能都捆绑在特定于任务的包中,但很难知道哪些包存在,哪些包最适用于某些任务。
在R中,用户开发包的主要存储库是CRAN,即the Comprehensive R Archive Network,综合R存档网络。
虽然软件包也可以来自其他来源,但CRAN软件包具有通过R’s install.packages功能轻松访问,确保软件包已在多个平台上测试的优势。
当搜索完成特定任务所需的新包时,最好从CRAN的任务视图开始(https://cran.r-project.org/web/views/).
任务视图允许用户按主题浏览已发布的包,包括多元统计、时空分析、元分析等。任务视图浏览器列出每个主题的相关包,并提供指向每个特色包的扩展文档的链接。
该资源对于R初学者以及正在寻找应对新挑战的方法的更有经验的R用户非常有用。
自学R时,你可能会遇到自己还不知道如何解决的问题。

在这些情况下,寻求帮助可能会为你节省大量时间,缓解头痛。
知道如何以有针对性的方式寻求帮助,这样你就能够查明问题的根源,并且在理想情况下,帮你避免将来出现类似问题。
▸ 如何以有针对性的方式寻求帮助?
当你第一次遇到问题时,复制和粘贴遇到的错误到搜索引擎中,可能会让你访问Stack Overflow、GitHub或R-bloggers等网站。
-通常情况下,有人会遇到与你相同的问题,并且可能会找到你可以使用的解决方案。
-如果没有的话,你可能需要发布自己的求助。
在这种情况下,有一些指导原则可以使帖子更高效:
在寻求帮助时,可复制的示例至关重要。你的目标应该是尽可能少地展示你的问题,并且在发布你的问题之前需要花一些时间隔离有问题的代码。
最有用的方法是在示例中生成一个玩具数据集,或者使用内置于R中的数据集(请参阅R中的data),并逐步删除代码中与要解决的问题无关的部分。
还应该在最小可复制示例中附带有关R会话和相关包版本的信息。所有这些步骤都可以通过reprex软件包实现。
▸ 水平提高后,帮助他人
随着技能的提高,你可能会发现自己处于一个可以帮助他人的位置。
在Twitter或Stack Overflow这样的地方回答问题是回馈R社区的好方法。
你甚至可以回过头来回答你自己过去问过的问题,这样你的帖子标记为“已解决”,并为下一个遇到同样问题的人留下一个有效的解决方案,帮助自己的同时也能给他人带来便利。
建立R语言流利性的最佳方法之一是与他人一起学习。
R社区充满活力,为学习R技能举办了会议、聚会和定期在线活动。
一个很好的起点是寻找你所在的区域的R用户或建立兴趣小组,经常举办技能分享、讲座、编程实践会议。
当然,你也可以虚拟加入R社区。The R for Data Science(R4DS)社区为学习者和导师提供了分享研究技能和协作的空间。R4DS Slack频道拥有超过10000名成员,成员可以通过讨论频道寻求帮助、分享胜利、网络等。
如果你不属于任何特定群体,R社区在许多社交网络上也很活跃。
R通常在Twitter上与#RStats标签讨论如下图。
一些常见科学编程语言的Twitter讨论流行度
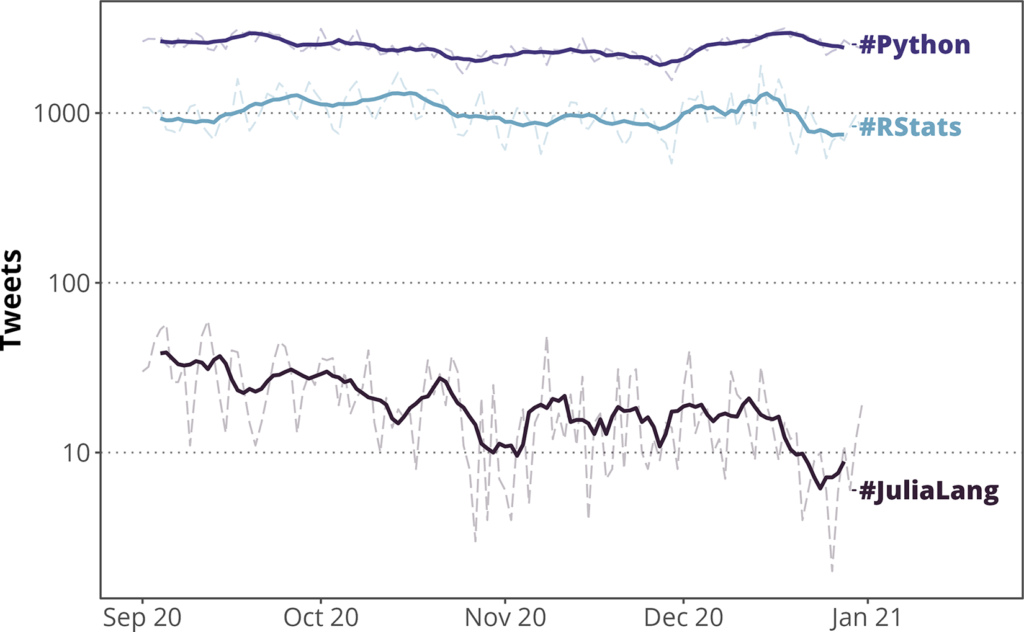
社区的一个很好的切入点可能是以下帐户:
@rstudio、@Rbloggers、@icymi_r、@RLadiesGlobal、@R4DS、@rOpenSci
在Facebook和Reddit等网站上也存在一般和特定领域的R组。你甚至可以参加在线活动,如Twitch上的数据科学节目“切片”,观看专家们在R编程挑战(以及其他语言)中的竞争。
关注多产的R博客也是了解R的好方法;一些推荐的R博客作者包括Maëlle Salmon、Jacqueline Nolis、Miles McBain。
在线找到社区都是了解新功能、了解最新软件包、遇到其他地方可能看不到的提示和技巧的好方法。
最重要的是,保持积极性,继续编写代码。

R的开源文化为代码共享提供了丰富的资源。
▸ 阅读和运行来自公开来源的代码,对于发现新函数、优化处理速度和向专家学习非常有价值
一些R项目,如#TidyTuesday R社交数据可视化项目,鼓励在线代码共享,以帮助用户获得和磨练技能。
其他活动,如每年一度的RStudio Shiny竞赛,为用户制造的R产品提供了一个友好竞争的渠道,最终免费提供代码,允许用户阅读、下载和复制获奖应用程序。
如果你发现一个特别好的R产品提供了代码,请自己逐行运行代码,以了解每一行或函数的确切意思,具体起到什么作用。你可能会学到一个新的功能,可以应用于自己的项目,或者有许多方法可以完成相同的任务。
▸ 已发表的论文提供了另一个R代码源代码,以实现更多以研究为中心的目的
现在,许多学术期刊要求出版物中包含公开可用的数据和代码,其中许多(尤其是自然科学领域)都是用R编写的。通过从已发表的文献中下载材料,你可以了解所在领域的专家如何进行分析,使用哪些软件包,以及他们如何组织代码。
▸ 学会分享:在线共享你的R代码
当然,代码共享是双向的;除了从其他人那里获取公开可用的材料,您还应分享您自己的材料。
代码共享的通用平台包括GitHub、GitLab和开放科学框架(OSF)。
在线共享你的R代码将进一步增强R社区的开放性,并帮助你成为一名更加专注的代码编写者。
发布代码可能会让人感到害怕,但请记住,对科学界来说,可用代码总是比不可用代码更有价值。
如果你的代码没有完全优化或完全干净,不要过分在意。如果它能运行起来,可能会教给别人一些新的东西,是值得分享的。
R是一个非常棒的工具,可以用于统计、数据操作、可视化等,但它不一定是编程旅程的终点。
▸ 流利的R语言帮你获得未来适用于其他程序、语言或领域的技能
例如,为Shiny Applications设计用户界面(ui)将帮助你建立前端web开发的基础,R的各种文本插件(例如ggtext包)将帮助你练习HTML语法,R的向量操作(apply和purrr::map函数族)将构建概念框架以转移到其他编程语言,如Julia或Python。
▸ 这是双向的:如果你已经掌握了其他编程语言的技能,它们也会帮助你学习R语言
当你继续将编程技能应用于更广泛的任务时,你可能会发现对于某些任务,不同的工具会更有效或更合适。

在这些情况下,你在学习R时建立的信心和技能可能是下一次编程努力的有用跳板。
通过学习R获得的知识,以及你通过自学获得的经验,将使你受益远远超过你手头的任务。
学习R语言,可能是一个充满挫折、自我怀疑和缺乏继续动力的过程。在这里列出的10条是帮助你克服挑战、掌握新技术的最佳策略,甚至可能在这一过程中获得一些乐趣。

也希望可以帮助初学者,无论是研究生、业余爱好者还是渴望学习新工具的研究人员。
当然也不需要一次尝试所有这些规则,也不局限于这十条,你只需要找到最适合你的方法即可。
主要参考文献:
Lawlor J, Banville F, Forero-Muñoz N-R, Hébert K, Martínez-Lanfranco JA, Rogy P, et al. (2022) Ten simple rules for teaching yourself R. PLoS Comput Biol 18(9): e1010372. https://doi.org/10.1371/journal.pcbi.1010372

谷禾健康
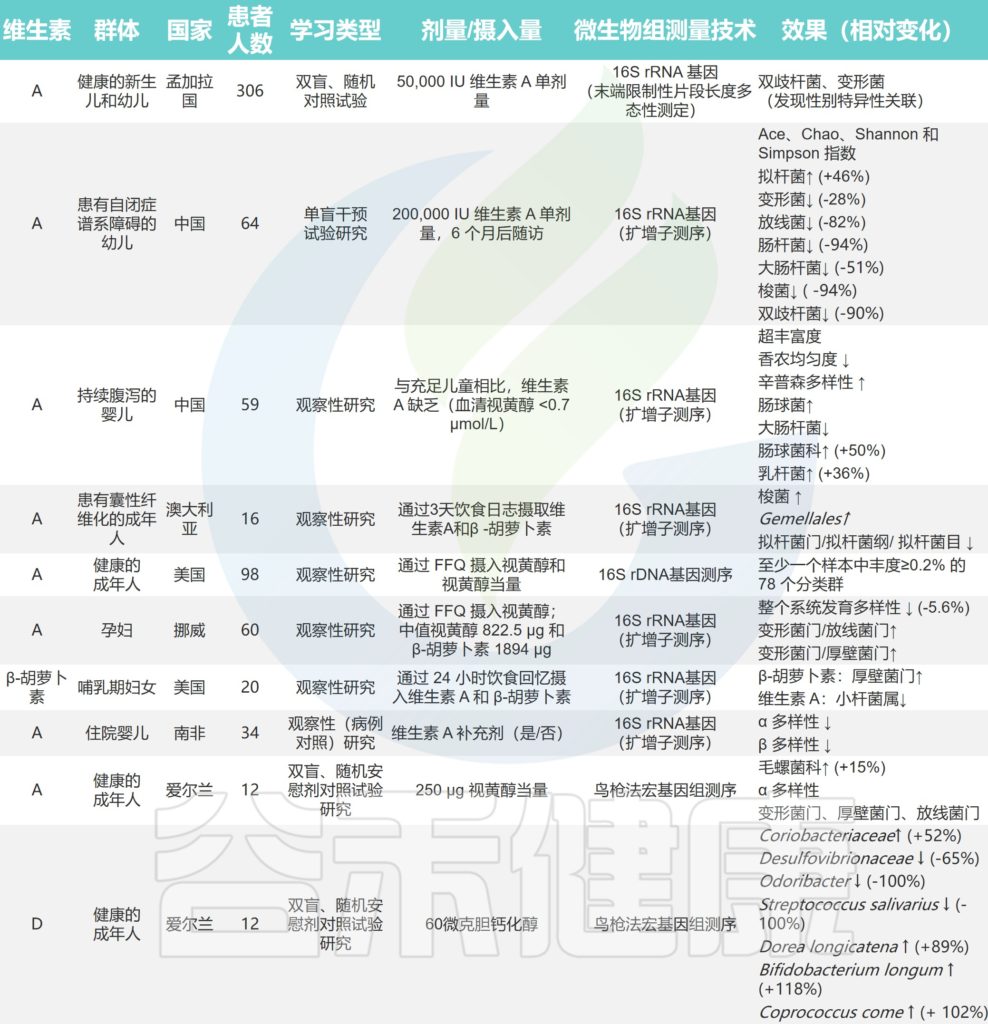
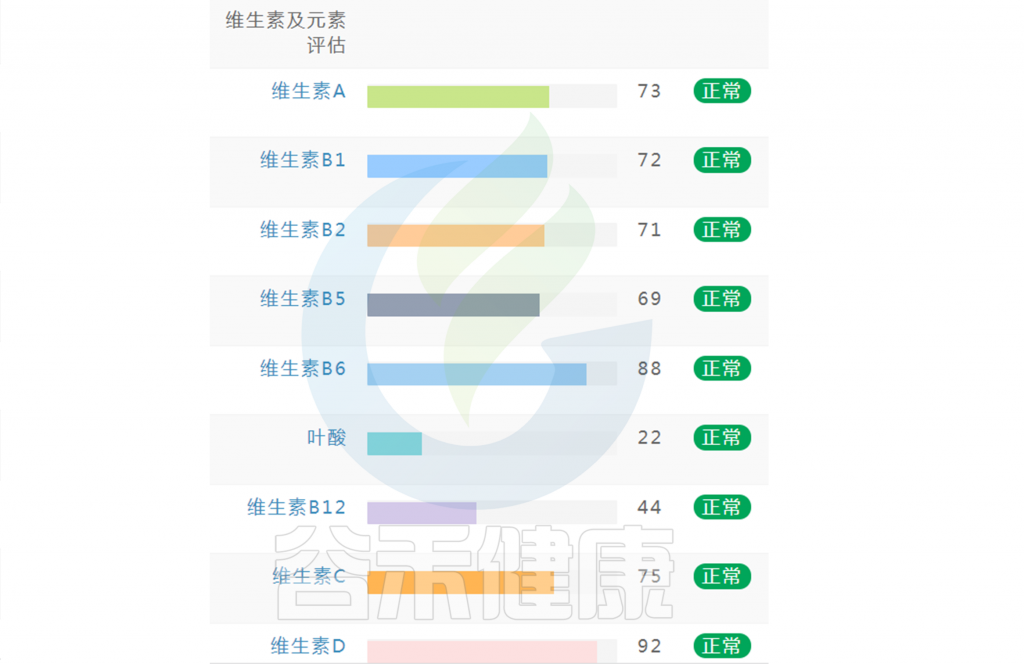
在谷禾肠道菌群健康检测中,我们会看到结果报告中关于维生素的评估如下:

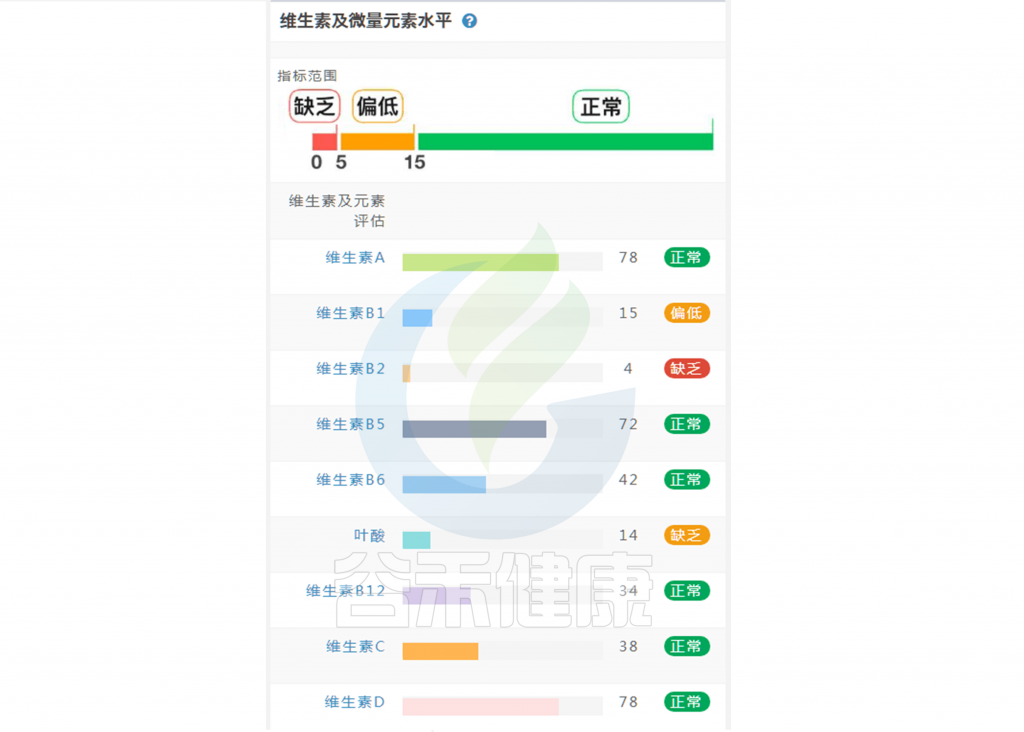
摄入水平建议保持在70-80分之间最佳,如果单项指标低于5表明摄入比例在人群中属于最低的5%,评估为缺乏,如上图中维生素B2;低于15评估为偏低,如上图中叶酸;达到或超过95则表明该项指标可能摄入比例偏高,可适当减少摄入;其余则为正常范围。
上图可以看到,像维生素C这项指标分值在38,虽然正常但相对于最佳来说是偏低的。
一些小伙伴可能会存在这样的疑惑:
为什么肠道菌群检测可以评估维生素?
这些维生素指标的分值代表着什么含义?
肠道菌群和维生素之间有什么样的关联?
它们如何影响人体健康/疾病?
如何判断维生素是否缺乏?
该如何补充?
…
本文就以上问题进行详细解答,同时也包括维生素-微生物群之间的相互作用,维生素维持肠道菌群稳态和减少肠道炎症以预防癌症的机制,产生维生素的益生菌,补充调节维生素的方式包括饮食、益生菌等。
在阅读本文之前,可以先了解一下各类常见的维生素功能,缺乏导致的症状。


每个维生素的详细介绍可以点开以下查看(请在谷禾健康微信公众号找到这篇文章查看)。
维生素B1(硫胺素)
维生素B2(核黄素)
维生素B3(烟酸)
维生素B5(泛酸)
维生素B6(吡哆醇)
维生素B7(生物素)
维生素B9(叶酸)
维生素B12(钴胺素)
以上每个都有关于该维生素的详细介绍,包括:
—正文—
维生素是一种微量营养素,在人体的生长、新陈代谢和发育中起着至关重要的作用。
在谷禾肠道菌群健康检测报告中,维生素分值即代表该维生素的膳食摄入水平和菌群代谢能力(报告中显示的分值是经过一系列计算得到的一个相对值)。
其中B族维生素很多需要通过肠道菌群对初始原料进行代谢之后才会产生,因此肠道菌群相应的基因和代谢途径的丰度水平也会直接反映这些维生素的摄入水平。
我们知道维生素的缺乏可能引起一些不良后果,导致维生素缺乏的原因有很多,摄入不足,吸收不良等都会导致维生素缺乏。
我们日常主要从饮食中获取维生素,肠道是主要吸收部位。例如,维生素 A 主要在近端空肠吸收,维生素 D 在远端空肠吸收最佳,维生素 E 和 K 主要在回肠吸收。因此,肠道功能受损可能会影响维生素的吸收。当然,影响维生素吸收的其他原因还包括年龄,某些疾病,药物等因素。
那么肠道菌群和维生素之间有什么关联?
肠道菌群是人体生理和健康的重要决定因素。肠道菌群帮助吸收营养,并参与维生素代谢。
肠道有益菌:乳酸菌和双歧杆菌,可以重新合成B族和K族维生素,为宿主提供约30%的每日摄入量。与从食物中获得的维生素不同,微生物产生的维生素主要在结肠中吸收。接下来了解一下具体哪些菌群,如何产生维生素。
前一章节我们知道,除了通过饮食提供维生素外,人体肠道中的细菌也可以产生一些维生素,如果吸收得当,可以部分满足人体的需要。
可以把这些细菌微生物想象成小小的维生素工厂。细菌确保为自己和与他们共生的微生物朋友提供维生素,同时也会为人体提供维生素。
合成的B族维生素的菌群较多
研究人员估计了人体肠道细菌可以提供维生素每日参考摄入量的百分比,得出的结论是可以提供:
40-65% 的人体肠道菌群具有合成 B 族维生素的能力。两种最常见的合成维生素是维生素B2和B3,预测分别有 166 和 162 个生产者。
可以合成 B 族维生素的细菌以及B 族维生素缺乏对肠道健康的影响
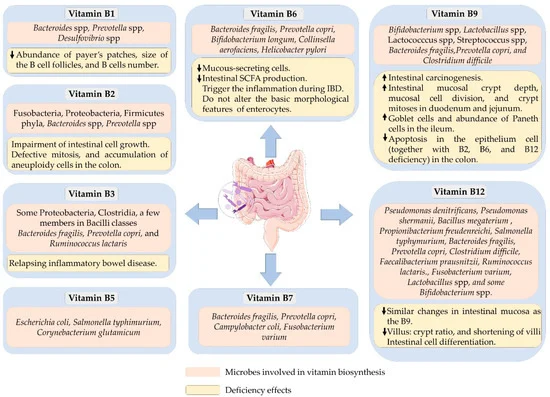
doi.org/10.3390/microorganisms10061168
大部分肠道菌群都参与维生素的合成
随着基因组注释方法的不断完善,研究人员可以预测维生素代谢途径并评估维生素生物合成潜力。通过检索 UniProt 数据库,研究人员发现:
厚壁菌门是维生素的主要代谢相关菌,其次是变形菌门,再然后是拟杆菌和放线菌。这四种菌群是人体肠道菌群的主要组成部分,占总菌群的60%-90%.
下表列出了参与合成B族维生素的肠道菌群,以及相应的代谢机制。


以上是肠道菌群对维生素产生的影响,而维生素和肠道菌群之间的作用是双向的,维生素也会影响肠道菌群,下一章节我们详细了解维生素对肠道菌群的影响。
维生素通过调节免疫力、细菌生长和新陈代谢来改变肠道微生物群的组成。
例如,膳食补充剂中的维生素 B、C、D 和 E 通过有利于双歧杆菌、乳酸杆菌和罗斯氏菌等有益菌属的肠道黏膜扩张和定植,在很大程度上有助于微生物组的组成。
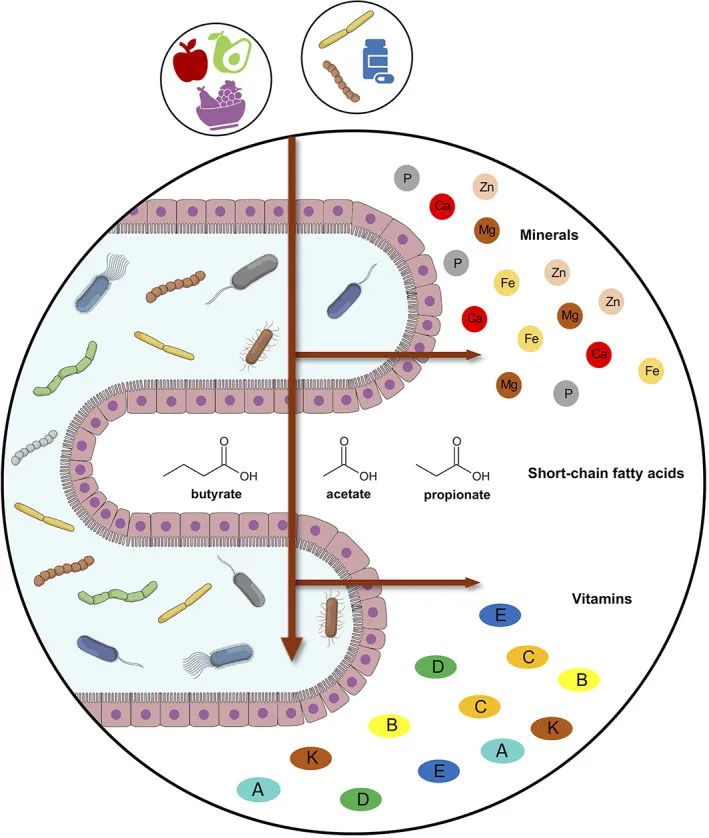
肠道微生物组和宿主之间的微量营养素交换
doi: 10.1002/biof.1835
一项研究调查了 96 名健康志愿者,结果表明:
补充维生素 B2 后肠道微生物的种类数量显着增加;联合补充维生素 B2 和 C 导致Sutterella显着减少,但Coprococcus数量增加;
维生素C显着提高肠道微生物的α多样性;
服用维生素D后,促进放线菌的生长和抑制拟杆菌的生长。
补充维生素对人体肠道菌群的影响
doi.org/10.3390/nu14163383
/ 维生素A /
维生素 A 的充足状态可能与微生物多样性增加有关。在小鼠实验中,普通拟杆菌(Bacteroides vulgatus )在维生素 A 缺乏期显着增加。维生素 A 缺乏导致的粘膜反应受损,粘蛋白和防御素 6 表达减少,可能使病原菌更容易穿透肠道屏障。
维生素A缺乏使厚壁菌门中毛螺菌_NK4A136_群、厌氧菌、颤杆菌的数量减少,毛螺菌的含量也降低;然而,Parasutterella呈上升趋势。TLR4 可能参与了维生素 A 调节微生物群的过程。
/ B族维生素 /
研究人员在一个小的成年志愿者群体中进行了一项试点研究,该群体补充了过量核黄素(100mg),持续14天。他们发现,在补充期间,每克粪便中的Faecalibacterium prausnitzii数量增加。作者还注意到厌氧菌Roseburia 增加,大肠杆菌减少。
其他关于B族维生素对肠道菌群的影响详见:
/ 维生素C /
补充维生素 C 可减少肠杆菌科细菌的数量,增加乳酸杆菌的丰度,抑制有害菌的生长,促进有益菌的增加。
也有研究表明,维生素 C 服用4周导致 α 多样性增加,短链脂肪酸浓度增加。

/ 维生素D /
维生素 D 和肠道微生物群的相互作用对免疫稳态至关重要。补充高水平的维生素 D 增加了普氏菌,减少了韦荣氏菌和嗜血杆菌。
婴儿饮食中补充维生素 D 对早期微生物组成的变化有重要影响,而儿童缺乏维生素 D 会导致细菌多样性降低。
最近的一项研究表明,维生素D的活性代谢物1,25-二羟基胆钙化醇,维生素D受体的配体(VDR),影响美国不同地区老年人肠道菌群的α -和β -多样性。
研究人员报告了通过食物频率问卷评估的微量营养素摄入量与孕妇微生物群组成之间的相关性。他们观察到,高脂溶性维生素,特别是维生素D的膳食摄入量与微生物α多样性降低有关(P值<0.001),维生素D和视黄醇与变形菌相对增加有关,变形菌门是一个已知包含多种病原体并具有促炎特性的门。
/ 维生素E /
维生素E对变形菌有抑制作用,而维生素E(和纤维)的摄入量较低与Sutterella水平较高相关,据报道,自闭症和某些胃肠道疾病婴儿的Sutterella水平大量增加。
体外维生素E 可以防止几种人类病原体的生物膜形成,特别是金黄色葡萄球菌和表皮葡萄球菌。
/ 维生素K /
一项动物实验表明,缺乏维生素 K 的小鼠的肠道中,瘤胃球菌、毛螺菌科、Muribaculaceae的含量较多。
关于维生素对人体肠道微生物组直接影响的研究
doi.org/10.1016/j.nutres.2021.09.001
饮食是维生素的主要来源,通过饮食补充维生素也会影响菌群。
注:由于测试饮食干预效果所需的随机试验的样本量和持续时间,相关发病率的研究具有挑战性。此外,由于特定的营养素不是孤立地消耗的,而是作为饮食模式的一部分,并且饮食成分之间相互作用,因此饮食带来的实际影响可能只有作为一个整体考虑时才会变得明显。
因此这里我们主要考虑饮食模式,例如地中海饮食等饮食方式。
地中海饮食是营养均衡饮食的典型代表,其特点是大量且频繁地摄入重要的纤维来源(谷物、蔬菜、豆类、水果和坚果)和具有抗氧化特性的化学成分(维生素、类黄酮、植物甾醇、矿物质、萜烯和酚类)。

同时地中海饮食还富含复杂和不溶性纤维含量。我们知道,大量摄入膳食纤维可促进肠道中有益菌群的生长,例如增加拟杆菌、普雷沃氏菌属、罗斯氏菌属、瘤胃球菌属、普拉梭菌等菌属的丰度,从而在肠道中产生高水平的短链脂肪酸,包括丁酸盐。
响应地中海饮食而增殖的细菌可以充当“基石”物种,也就是说它们对于稳定的“肠道生态系统”至关重要。这些变化主要是由于膳食纤维和相关维生素和矿物质的增加,特别是维生素C、B6、B9、铜、钾、铁、锰和镁。
总之,维生素似乎是微生物-宿主间代谢相互作用的重要媒介。
越来越多证据表明,维生素缺乏会导致肠道菌群紊乱,进而引发肠道疾病,甚至促进炎症和肿瘤的发展。下一章节详细讨论,维生素-微生物群相互作用对健康/疾病的影响。
最近的几项观察表明,微生物群失调和维生素缺乏是相互关联的。
维生素对宿主健康的影响

doi.org/10.1016/j.nutres.2021.09.001
这种关系可能直接影响宿主健康:例如,克罗恩病恶化与参与抗炎介质核黄素、硫胺和叶酸生物合成的微生物基因减少有关。
此外,2型糖尿病受试者在与微生物介导的维生素代谢相关的基因丰度谱中显示出显著变化。
营养不良儿童的微生物群显示,参与B族维生素代谢的多种途径(包括烟酸/NADP生物合成)显著减少。
在经历饮食振荡以诱导急性短期维生素A缺乏的灵长类小鼠模型中,Hibberd等人观察到细菌群落结构和宏转录组的调节,其中Bacteroides vulgatus是显著的应答者,在缺乏维生素A的情况下其丰度增加。有趣的是,B.vulgatus是在人类肠道微生物群的灵长类小鼠模型中鉴定的一种生长差异物种。
所有这些观察结果表明,维生素缺乏可能会改变肠道微生物群,从而影响人体健康。
下面我们以肠道疾病和精神类疾病两大类疾病为例,来具体了解维生素-微生物相互作用及其在疾病中的影响。
维生素 A 和 D 分别在近端和远端空肠吸收。维生素E和K主要在回肠吸收;微生物产生的维生素主要在结肠中吸收。维生素缺乏会加重肠道炎症,甚至通过多种机制促进癌症。
肠道菌群->维生素->肠道疾病中的作用
慢性 IBD 发生和发病机制中的关键作用是微生物(尤其是共生菌群)对宿主黏膜免疫功能的影响。同时,肠道微生物群和慢性炎症已被证明与肿瘤发生密切相关。
维生素具有调节肠道菌群和保护肠道的功能。因此,维生素和微生物群的相互作用可能在 IBD 和结直肠癌的治疗中具有巨大的潜力。
维生素A通过促进黏膜愈合、促进产生ASCFA的相关菌增加、降低UC相关菌的水平来达到治疗UC的效果。
费氏丙酸杆菌ET-3 产生维生素 K2 的前体,即 1,4-二羟基-2-萘甲酸 (DHNA),可激活芳烃受体 (AhR) 以改善结肠炎并调节肠道微生物群。
维生素 D 的缺乏会增加拟杆菌门、变形杆菌门和螺杆菌科的丰度,降低厚壁菌门和去铁细菌的丰度门,并且还影响 E-钙粘蛋白表达并减少耐受树突状细胞的数量。
然而,在治疗 IBD 时,维生素 D 与利福昔明的共同给药会影响肠道菌群和利福昔明的疗效。维生素 D 促进A. muciniphila的生长以保护肠粘膜屏障,这些作用对于对抗结直肠癌的发展尤为重要。
研究表明,维生素 E 及其代谢物在调节肠道菌群、减少炎症和抑制癌变方面具有巨大潜力。此外,维生素 Eδ-生育三烯酚 (δTE) 及其代谢物δTE-13′-羧基色原酚 (δTE-13′) 增加了肠道中的乳球菌和拟杆菌,并抑制炎症因子的产生。
维生素->肠道菌群->肠道疾病中的作用
▸维生素在IBD和结直肠癌中的作用不容忽视
大量临床研究表明,缺乏维生素 B 和维生素 D 的人群中结直肠癌的患病率较高。同时,IBD 的长期不愈合使患者面临更高的结直肠癌风险。维生素 D 水平低的 IBD 患者疾病严重程度和预后较差。
▸为什么肠道炎症容易导致癌症高风险?
在炎症背景下,敲除 IKKbetaβ(炎症与癌症之间的联系)可减少由于上皮细胞凋亡增加而导致的癌症发生。在一项关于结肠炎相关癌前癌 (CApC) 的研究中,IL-6 反式信号转导的存在增加了炎症性致癌的风险。如果不及时治疗,由肠道菌群紊乱和维生素缺乏引起的肠道炎症最终可能发展为癌症。
维生素 A 在肠道炎症和癌症中的作用
维生素 A 及其活性代谢物视黄酸 (RA) 在人体免疫系统中发挥着关键作用,并可能对辅助 T 细胞的分化产生影响。
炎症下:视黄酸从保护转变为破坏作用
在非炎症条件下,视黄酸能够抑制 IL-6 受体的表达和 Th1/Th17 的产生。
在炎症条件下,视黄酸从对粘膜的保护作用转变为破坏作用;这反映在活动期 IBD 患者黏膜中视黄酸水平显着升高,伴随着 CD4 和 CD8 分泌的 IL-17 和 IFN-γ 的上调。
维生素A及其代谢物:发挥抗炎作用
维生素 A 及其代谢物通过阻断 Th1 和 Th17 的激活,抑制 IL-17、INF-γ 和 TNF-α 的产生而显示出抗炎作用。同时,它们可以通过与TGF-β协同作用,提高Foxp3的水平,发挥免疫功能,从而促进抗炎因子的发挥。
一项数据显示,低水平的维生素 A 会激活核 NF-kB 并促进胶原蛋白的形成,从而加剧结肠炎的炎症。补充维生素后,肠道炎症明显缓解。
全反式维甲酸 (AtRA) 可降低 UC 和结直肠癌患者结肠黏膜分泌的 TNF-α 和一氧化氮合酶 2 (NOS2) 蛋白的表达。

维生素A保护肠黏膜屏障,其潜在机制是拮抗LPS的肠道破坏作用
在一项关于维生素 A 缺乏对结肠炎和结直肠癌发展的影响的检查中,研究人员使用葡聚糖硫酸钠 (DSS) 诱导小鼠结肠炎;此外,偶氮甲烷 (AOM) 预注射和 DSS 结肠炎的组合诱导了结直肠癌。缺乏维生素的小鼠肠道炎症水平较高,黏膜愈合较慢,免疫反应增强,更容易发生结直肠癌。
AtRA具有抗癌作用,结直肠癌中AtRA 水平降低
在结直肠癌小鼠模型中,肠道细菌引起的炎症影响 AtRA 代谢;这导致其水平下降。在 UC 及其相关结直肠癌的临床样本中发现 AtRA 代谢酶活性降低和 AtRA 水平降低。同时,AtRA通过激活CD8 + T细胞发挥抗癌作用;这为 CAC 的治疗提供了新的见解。
视黄醇和视黄醇结合蛋白(RBP)的结合激活致癌基因STRA6,促进结直肠癌的发生;Holo-RBP/STRA6 通路可通过促进成纤维细胞的致癌作用进一步发挥致癌作用。
在一项关于维生素 A 缺乏对结肠炎和结直肠癌发展影响的动物实验中,当维生素 A 处于低水平时,小鼠体内的维生素 A 脂滴会被降解,免疫反应会增强,结肠炎症会加重,癌变进程将加快。
维生素 B12 和叶酸在肠道炎症和癌症中的作用
IBD 患者缺乏维生素 B12 和叶酸的原因有很多,包括回肠和空肠微生物过度生长、维生素 B12 摄入不足或身体需求增加、维生素肠道破坏增加或吸收能力降低、某些药物(如甲氨蝶呤或柳氮磺胺吡啶)的不良反应、一些病理原因例如蛋白丢失性肠病、肝功能异常、回肠相关病变或手术切除、肠瘘等。
维生素 B12 缺乏不会影响健康的肠道微生物群组成;然而,它会导致实验性结肠炎中肠道菌群失调,并促进条件致病菌的生长。出乎意料的是,维生素 B12 缺乏减少了结肠组织的损伤;这可能与抗炎细胞因子 IL-10 的增加有关。

对甲基缺乏饮食 (MDD) 的潜在作用进行了一项研究,该饮食可降低维生素 B12 和叶酸的血浆浓度,并提高同型半胱氨酸水平,对 DSS 诱导的小鼠结肠炎的影响。喂食 MDD 的 DSS 治疗小鼠比其他治疗组患有更严重的结肠炎。
尽管超氧化物歧化酶和谷胱甘肽过氧化物酶活性保持稳定,但 caspase-3 和 Bax 的水平受到影响。除Bcl-2表达增加外,炎症相关标志物如胞质磷脂酶A2和环氧合酶2的表达也有明显增加趋势;这伴随着金属蛋白酶组织抑制剂(TIMP)3蛋白的表达降低。因此,维生素 B12 缺乏可能会加重实验性 IBD 的炎症程度。
高维生素 B12 水平可通过减少 DNA 甲基化来降低结直肠癌的风险
在结直肠癌患者中,与低血清维生素 B12 组相比,高维生素 B12 组的肿瘤区域和外周血单个核细胞 (PBMC) 中长散布的核元素 1 (LINE1) 甲基化被证明是降低的;肿瘤区域的LINE1甲基化水平也低于周围的非肿瘤区域。
氧化应激是结直肠癌发病机制之一;此外,叶酸和维生素 B12 的水平与体内抗氧化剂谷胱甘肽的水平呈正相关。提高 AOM 诱导的结直肠癌中的叶酸和维生素 B12 水平显示出显着的抗凋亡、抗氧化应激和抗 AOM 细胞毒性。
在对 4517 名 IBD 患者的系统评价和荟萃分析中,补充叶酸被证明可以降低 IBD 患者的结直肠癌风险并防止结直肠癌发展。
有趣的是,有证据表明缺乏甲基供体营养素叶酸、胆碱、蛋氨酸和维生素 B12 会抑制 Apc 突变小鼠的肿瘤发展。总而言之,维生素B12和叶酸在肠道疾病中的作用需要更深入的研究。
维生素 D 在肠道炎症和癌症中的作用
流行病学和动物实验表明,维生素 D 缺乏是 IBD 和 结直肠癌的高危因素。维生素 D 补充剂有助于降低疾病严重程度,可能通过多种机制,包括调节免疫细胞运输和分化,以及抗菌肽合成。
维生素D可以维持肠黏膜屏障的正常功能,提高机体的先天性和适应性免疫
1α,25-二羟基维生素 D3(骨化三醇)是维生素 D 的活性形式,可与 TGF-β 结合,提高 IL-2 水平,调节 T 细胞抑制炎性细胞因子的产生,增强 Foxp3 + Treg 细胞的存活和功能。

维生素D受体(VDR)是维生素D调节免疫和发挥抗炎作用的重要途径
相关资料显示,VDR对肠道有保护作用;它可以通过调节 JAK/STAT 通路来维持肠道稳态并预防癌症。
在 IBD 患者 中,结肠上皮中VDR的含量明显低于正常人。在实验性结肠炎模型中,与缺乏 VDR 的小鼠相比,表达 hVDR 的转基因小鼠的结肠炎症较少。用 hVDR 转基因恢复上皮 VDR 表达可减轻严重结肠炎并降低死亡率。内在机制是 VDR 通过抑制 NF-κB 活化发挥抗凋亡作用,以保护肠道屏障缓解结肠炎。
肠道菌群通过犬尿氨酸通路(合成维生素),在精神健康方面发挥作用
关于肠道细菌在心理健康方面的作用的关键方面,是它们通过犬尿氨酸通路参与调节色氨酸代谢。微生物群能够合成犬尿氨酸途径 (KP) 的酶促辅助因子,如维生素 B2 和 B6。

犬尿氨酸是主要的色氨酸代谢途径,其中 95% 的这种氨基酸被代谢为各种免疫和神经调节犬尿氨酸/色氨酸分解代谢物 (TRYCAT),在大脑中,犬尿氨酸途径主要在神经胶质细胞中分隔。
犬尿氨酸通路在精神、神经退行性和神经系统疾病中的作用是至关重要的,包括重度抑郁症,双相情感障碍,精神分裂症,阿尔茨海默病,亨廷顿病和帕金森病,与 HIV 感染相关的痴呆,手术后认知能力下降,肌萎缩侧索硬化(ALS) 等。
精神病理学和炎症中维生素缺乏与高同型半胱氨酸血症有关
精神病理学和炎症中维生素缺乏的另一个关键机制与高同型半胱氨酸血症(hHcy)有关,这可能是由叶酸、维生素 B6 和 B12 缺乏引起的。
高同型半胱氨酸血症和维生素 B 缺乏在重度抑郁症、精神分裂症、双相情感障碍、自闭症、焦虑症和痴呆症(包括阿尔茨海默病和帕金森病)中起关键作用。
同型半胱氨酸(Hcy)是在蛋白质消化过程中获得的另一种氨基酸蛋氨酸代谢过程中形成的氨基酸和中间体。该反应需要维生素 B12 作为酶促辅因子和叶酸衍生物(5-甲基四氢叶酸)作为甲基供体。
注:Hcy-同型半胱氨酸,是人体内含硫氨基酸的一个重要的代谢中间产物,可能是动脉粥样硬化等心血管疾病发病的一个独立危险因子。
此外,Hcy 可以在需要维生素 B6 作为酶辅因子参与的途径中转化为半胱氨酸。
因此,Hcy 被认为是叶酸和维生素 B12 缺乏的敏感标志物。
高同型半胱氨酸血症导致神经和精神病理学的机制包括:
促进免疫炎症反应、增加肠道和血脑屏障通透性、NMDA受体激动和神经毒性、诱导神经元凋亡、氧化应激、线粒体功能障碍和由于甲基化受损导致的单胺能神经递质合成失调。
目前对体内维生素水平的检测例如:
抽取血液检测其中维生素的含量水平,可以判断是否存在维生素的缺乏情况。
其他,例如通过肠道菌群健康检测,也可以查看近期体内维生素状况。
与通过血液进行维生素检测不同,肠道菌群的评估更加反映一段时间 ( 一般2周左右 ) 的长期状态,如部分B族维生素无法在体内留存,需要每日补充,血液检测波动较大。
注:菌群会受检测前一天饮食的影响,造成15~30%的菌群改变,同样也会反映在营养状况的评估上,因此建议检测前一天尽量保持近期正常饮食 ,这样能更好的反映真实的营养饮食状态。
在了解补充维生素的干预措施之前,我们先从肠道菌群的角度,来了解一下影响维生素合成吸收的因素。
人类基因的变异与肠道结构和微生物组组成有关。人类肠道微生物群中存在不同的维生素 B 生物合成途径支持人类遗传变异影响维生素 B 合成的观点。
维生素的合成吸收不仅需要靠饮食补充,还与吸收相关。而维生素的吸收涉及到相关基因,例如:
MTHFR 基因的突变会影响我们产生加工维生素 B9的酶——亚甲基四氢叶酸还原酶。
亚甲基四氢叶酸还原酶是叶酸代谢通路中的一种重要的辅酶,亚甲基四氢叶酸还原酶基因缺陷,容易造成叶酸在体内的代谢障碍,MTHFR基因最主要的两种突变为C677T、A1298C基因多态性。该两种位点同时突变可显著降低MTHFR活性进而降低叶酸水平。
VDR基因(维生素 D 受体):维生素 D(来自阳光、食物或补充剂)经过转化步骤后,活性形式骨化三醇 (1,25(OH)2D3 ) 可以通过VDR在细胞内发挥作用,是打开或关闭基因的转录因子。该基因突变可能导致维生素D缺乏引起的佝偻病。
维生素缺乏是一个严重的问题,尤其是在老年人中。随着年龄的增长,营养需求会随之变化。
由于食物中的维生素B12 需要胃酸及胃蛋白酶的作用才能释放出来被吸收,而老年人胃酸及胃蛋白酶分泌减少,就会影响维生素B12 的吸收。
患有维生素B12缺乏症的老年人可能出现神经精神或代谢缺陷。
一些药物会改变营养物质的吸收或代谢方式。例如,抗惊厥药也会减少叶酸的吸收。
肠道菌群通过各种代谢途径影响维生素的合成,例如拟杆菌属、肠球菌属和双歧杆菌属等人类肠道共生菌可以从头合成维生素 K 和大多数水溶性 B 族维生素,这在前面第二章节的表已经详细阐述。
在 B 族维生素合成中暴露于抗生素的反应因使用的抗生素类型而异。例如,在饮食中添加青霉素和金霉素会增加雄性大鼠的肝脏维生素 B2 浓度,以及 B2 和 B3 在尿液中的排泄。然而,链霉素和放线菌酮的施用降低了肝脏中维生素 B9 和 B12 的浓度。维生素合成对抗生素暴露的混合反应尚不清楚,但它们可能是由肠道微生物群的选择性改变引起的。
自由基是含有不成对电子的化学物质,可以诱导氧化应激。一个这样的例子是一氧化氮,它与金属离子形成复合物,包括钴,维生素 B12 的一种结构成分,因此使其无法用于细菌维生素 B12 的生物合成。此外,维生素生产者(如脆弱拟杆菌)暴露于过氧化氢等自由基会抑制其生长 ,从而降低维生素的生物合成能力。
维生素主要在小肠中吸收,其生物利用度取决于食物成分,相关相互作用等。
饮食和膳食的组成会通过影响肠道转运时间和/或混合胶束的肠道形成来影响某些维生素的吸收。
饮食中足量的水和膳食脂肪对于分别吸收水溶性和脂溶性维生素至关重要。
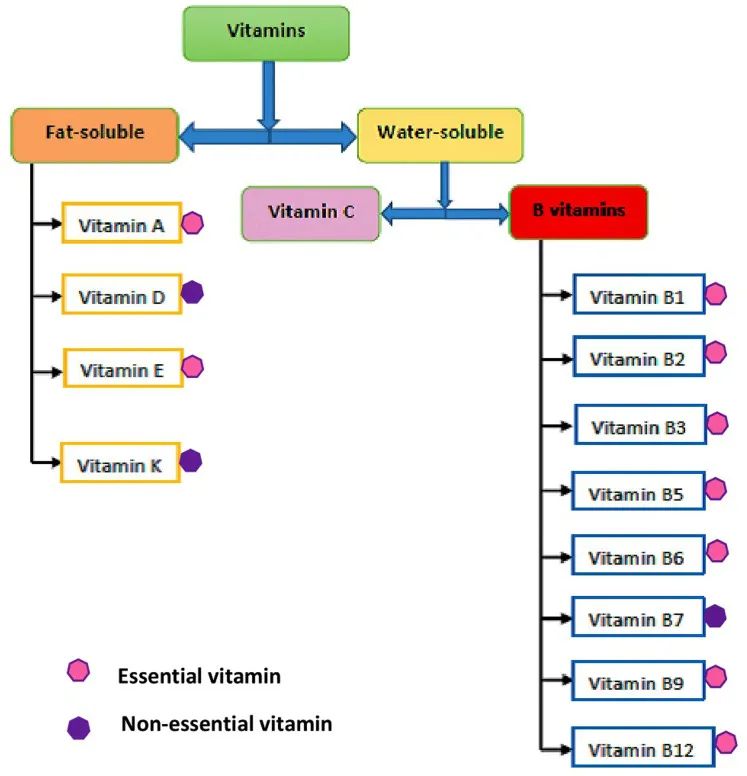
*水溶性维生素包括:B族维生素,维生素C;
脂溶性维生素包括:维生素 A、D、E 、K.
doi: 10.7717/peerj.11940
由于脂溶性维生素可以溶解在脂肪中,因此与膳食脂肪一起食用时最容易被吸收。例如,一种富含维生素 A 的小胡萝卜,如果单独食用,将在食物中获取维生素 A,但如果它是在含有一些膳食脂肪的食物成分中(比如说,橄榄油),将增加体内维生素 A 的吸收。
食物的性质(物理状态)也会影响维生素的吸收效率。例如,存在于可消化性较差的纤维植物材料中的类胡萝卜素已被证明相对于维生素A表现出较低的生物利用度。
当我们看到维生素缺乏的时候,可能希望通过饮食来补充相应缺乏的维生素,下表列出了常见的维生素的食物来源,可供参考。
此外,宿主饮食作为肠道中细菌的底物,其对肠道微生物分布的影响已被广泛研究。含有益生元和其他膳食营养素(如微量营养素和多酚)的饮食可以显着影响有益细菌的生长,包括维生素生产菌。
一些维生素,如核黄素,可作为氧化还原介质并刺激营养缺陷菌(如Faecaibacterium prauznitsii)的生长。
在即将形成共生关系的环境中,限制这些基质会增加微生物和微生物与宿主之间的竞争。
除了通过饮食直接补充之外,我们还可以通过补充益生菌来调节维生素水平,从而改善疾病。
双歧杆菌
在健康成人中补充益生菌菌株青春双歧杆菌DSM 18350、青春双歧杆菌DSM 18352 和假链双歧杆菌DSM 18353,导致粪便中叶酸浓度显着增加。
乳酸菌
乳酸菌通过不同的机制抑制炎症过程,包括调节IBD患者肠道菌群紊乱、保护肠道屏障和黏膜的正常功能、调节人体免疫反应等。乳酸菌通过产生核黄素(维生素 B2)和叶酸发挥抗炎和抗氧化作用。
产维生素的乳酸菌不仅对急性肠炎有抗炎作用,还能有效缓解复发性结肠炎。此外,在与美沙拉秦合用过程中,可有效降低不良反应,提高疗效。
研究人员发现注射产生叶酸的乳酸菌会缓解 5-FU 引起的肠炎小鼠的腹泻,改善结肠组织的结构和功能。这一发现降低了癌症化疗期间发生的肠黏膜炎症的严重程度,并提高了药物有效性;因此,这提高了患者的生活质量。
此外,乳酸菌 和 5-FU 的联合使用可减少 5-FU 引起的血细胞计数减少,并使患者获得完整的治疗周期。
产维生素的益生菌在肠道疾病中的作用

doi.org/10.3390/nu14163383
研究人员从 150 个收集的人类粪便样本中分离出三种产生核黄素和叶酸的益生菌;他们用它们来治疗乙酸引起的大鼠结肠炎。他们发现这些益生菌可以保护结肠黏膜,促进溃疡性病变的愈合;此外,它们具有抗炎和抗氧化应激作用。
一种新分离的具有产生叶酸能力的细菌——清酒乳杆菌LZ217,具有促进丁酸产生和改善肠道菌群组成的作用。
Akkermansia muciniphila 是肠道中的一种常见细菌,可调节 CLT 以保护肠道免受炎症和肿瘤侵袭;它还产生维生素 B12 以缓解 IBD 患者的维生素缺乏症。
研究发现,丙酸杆菌菌株 P. UF1 合成维生素 B12;这对肠道免疫和肠道健康有积极的调节作用。
大肠杆菌通过产生维生素来缓解 IBD. 使用大肠杆菌生产两种产生β-胡萝卜素的菌株来治疗维生素A缺乏症。这些结果显示出巨大的临床潜力。
维生素 A 及其代谢物与短乳杆菌KB290 的组合提高了 CD11c + MP/CD103-DC 比率;因此,这在结肠炎的治疗中起着积极的作用。
此外,肠道中的分段丝状细菌 (SFB) 可以产生 AtRA,以抵消感染对肠道的损害。
益生菌对维生素D及其受体活性有积极作用,如鼠李糖乳杆菌GG(LGG)和植物乳杆菌(LP);同样在沙门氏菌结肠炎模型中,使用 VDR (-/-) 小鼠验证 LGG 对 IBD 的缓解作用是通过 VDR 信号通路。
此外,胆汁盐水解酶 (BSH)活性罗伊氏乳杆菌NCIMB 30,242 可调节血浆中的活性维生素 D 水平。磷虾油 (KO)、益生菌罗伊氏乳杆菌和维生素 D 的混合物显着降低病理评分和炎症因子的释放,促进黏膜愈合并减少机会性感染的发生。
经益生菌 VSL#3 预处理后,VDR 水平显着提高,共同保护肠黏膜,防止损伤;这对预防CRC的发展起到一定的作用。
用从韩国泡菜中分离的乳酸菌条件培养基处理 HCT116 细胞或肠类器官后,其分泌的蛋白质 P40 和 P75 与 VDR 的表达增加有关;它们还增强自噬反应,共同具有抗炎作用。肠道微生物合成的石胆酸 (LCA) 充当连接 VDR 与微生物的桥梁,从而提高维生素 D 水平。
益生菌配方有助于抑郁症患者维生素水平的增加
一项随机对照试验中,重度抑郁症患者接受了多种益生菌配方,其中含有双歧杆菌W23、乳双歧杆菌W51、乳双歧杆菌W52、嗜酸乳杆菌W22、干酪乳杆菌W56、副干酪乳杆菌W20、植物乳杆菌W62、唾液乳杆菌W24、乳酸乳杆菌W19。
此外,益生菌组和安慰剂组的患者接受了相同剂量的维生素 B7。在两组中,抑郁症的临床参数都有所改善,然而,益生菌干预组与安慰剂组相比,仅在微生物 β 多样性方面存在差异,临床结果指标没有差异。有趣的是,尽管两组都接受了相同剂量的生物素,但接受益生菌的那组维生素 B6 和 B7 的合成上调。
多种益生菌相结合通过增加叶酸和维生素 B12血浆水平,改善精神疾病
八周的个性化饮食与含有多种益生菌的菌株相结合:婴儿双歧杆菌DSM 24737、长双歧杆菌DSM 24736、短双歧杆菌DSM 24732、嗜酸乳杆菌DSM 24735 、德氏乳杆菌、保加利亚乳杆菌DSM 24734、副干酪乳杆菌DSM 24733、植物乳杆菌DSM 24730 、嗜热链球菌DSM 24731 (VSL#3),在健康老年人中增加了叶酸和维生素 B12 血浆水平并降低了 Hcy 血浆水平。
此外,益生菌的添加导致粪便双歧杆菌浓度增加,这种变化与叶酸和维生素 B12 水平呈正相关。
在精神病患者中引入高同型半胱氨酸的评估和治疗可能非常有价值,益生菌可能成为治疗工具之一。
维生素是相互关联的、具有协同作用的微量营养素,当它们处于适当的平衡状态时,它们的全部潜力就会得到充分发挥。
因此,在食用益生菌和发酵食品时,应考虑维生素生产者与代谢者之间复杂的相互作用。
除了以上方式干预菌群之外,也可以通过良好的生活方式调理菌群,从而使维生素达到一个相对健康稳定的水平,减少各类疾病风险。
▸在服用维生素的同时可以服用益生菌吗?
可以。在大多数情况下,服用益生菌不会影响其他补充剂的效果。
一项 2021 年对临床试验的系统评价发现,益生菌可以改善健康人群的微量营养素水平,特别是维生素 B12、叶酸(维生素 B9)、钙、铁和锌。
2017 年的一项非随机临床试验发现,服用益生菌和铁补充剂的参与者比不服用益生菌的铁吸收明显更多。
有研究表明,维生素 D 和益生菌之间存在协同关系。
随机对照试验发现,维生素 D 补充剂与益生菌一起可以改善多囊卵巢综合征患者和同时患有冠心病的糖尿病患者的各种心理健康参数、一般健康状况、代谢和炎症标志物。
2019 年对随机对照试验的系统评价和荟萃分析发现,维生素 D 强化酸奶(富含益生菌嗜酸乳杆菌)有助于改善维生素 D 和胆固醇水平、代谢功能和身体测量值。
然而以上研究都没有单独研究维生素 D 和益生菌的作用,因此尚不清楚结果是否与两者的综合影响有关。
研究人员认为,无论有没有维生素,服用益生菌的时间很重要。作为一般规则,服用益生菌的最佳时间是空腹,大约在进食前 30 分钟。
研究人员担心胃酸的存在会影响益生菌的生存能力。在餐前或餐后几个小时服用时,当胃酸自然降低时,益生菌可以进入肠道,从而提高其生存几率。
何时服用维生素取决于维生素的种类。复合维生素通常最好在早上第一时间服用,非常适合搭配早餐前的益生菌。脂溶性维生素,如 A、D、E 和 K 以及一些矿物质,包括铁和镁,最好与食物一起服用。否则可能会导致胃部不适。
▸应该从食物中补充维生素还是通过维生素补充剂?
2020 年的一篇文献综述发现,与浓缩补充剂相比,许多微量营养素在其全食物形式中的生物利用度更高。因此提倡补充方式以食物为先。
一般认为,对于健康人来说,营养均衡的饮食可以提供身体需要的维生素,不需要额外补充,但对于可能存在免疫功能、肠道健康、吸收不良等问题的人群,可以考虑维生素补充剂进行补充,具体补充剂量请遵医嘱。
下表是维生素易缺乏的高风险人群:

▸可以长期服用维生素补充剂吗?补充过量会带来副作用吗?
一般健康人不需要长期服用维生素补充剂。
对于服用复合维生素片,多余的维生素会被排出体外,因此不用过于担心会带来危害。但是如果长期十倍以上的用量,对身体是有危害的。

doi: 10.7717/peerj.11940
在发现维生素缺乏的症状的时候,我们可能希望通过补充相应的维生素补充剂来改善健康。然而服用任何补充剂之前,我们应该寻找其根本原因而不是直接根据症状盲目补充。
通过肠道菌群健康检测可以了解维生素缺乏状况,且可以根据各类菌群丰度来推断维生素的菌群代谢状况。如果是由于菌群的代谢异常,可能直接补充并没有太大效果,这时候优先调节菌群或许是更好的选择。
如果维生素指标都显示正常没有缺乏(如下图),保持常规饮食不需要刻意补充。还想要更健康,指标更接近70的话,可以在数值略小的指标上,针对性地通过饮食进行补充调理。

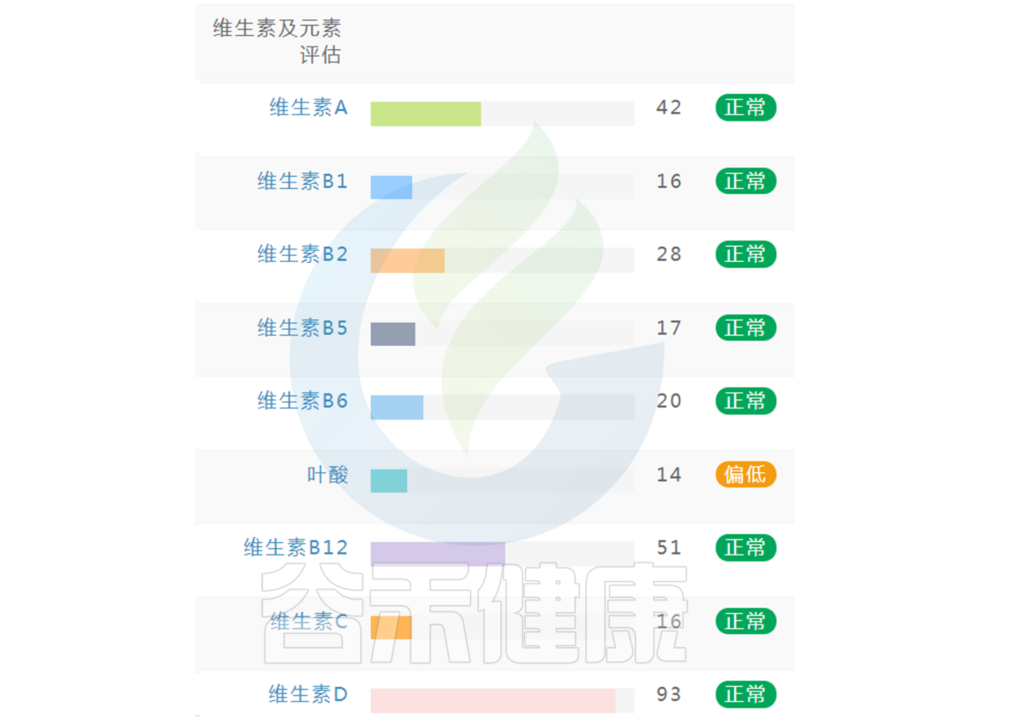
如果维生素指标中出现个别指标缺乏或偏低(如下图),可以通过饮食针对性地进行改善调整,如果已经出现对应症状,例如缺乏维生素A,同时出现干眼症或者夜盲症等相应的症状,可以使用相应的维生素补充剂进行干预,或者根据菌群代谢通路判别,通过菌群调理进行相应干预。
如果维生素指标中出现缺乏或偏低的指标较多,则需要选用复合维生素,各类维生素之间可能存在协作关系,同时配合饮食、菌群进行干预。
选择补充剂,应优先考虑生产规范良好的产品,比如说可以查看是否有“OTC”标志。
注:本账号内容仅作交流参考,不作为诊断及医疗依据。
主要参考文献:
Zhai Z, Dong W, Sun Y, Gu Y, Ma J, Wang B, Cao H. Vitamin-Microbiota Crosstalk in Intestinal Inflammation and Carcinogenesis. Nutrients. 2022 Aug 17;14(16):3383. doi: 10.3390/nu14163383. PMID: 36014889; PMCID: PMC9414212.Zhai Z, Dong W, Sun Y, Gu Y, Ma J, Wang B, Cao H. Vitamin-Microbiota Crosstalk in Intestinal Inflammation and Carcinogenesis. Nutrients. 2022 Aug 17;14(16):3383. doi: 10.3390/nu14163383. PMID: 36014889; PMCID: PMC9414212.
Bellerba F, Muzio V, Gnagnarella P, Facciotti F, Chiocca S, Bossi P, Cortinovis D, Chiaradonna F, Serrano D, Raimondi S, Zerbato B, Palorini R, Canova S, Gaeta A, Gandini S. The Association between Vitamin D and Gut Microbiota: A Systematic Review of Human Studies. Nutrients. 2021 Sep 26;13(10):3378. doi: 10.3390/nu13103378. PMID: 34684379; PMCID: PMC8540279.
Ofoedu CE, Iwouno JO, Ofoedu EO, Ogueke CC, Igwe VS, Agunwah IM, Ofoedum AF, Chacha JS, Muobike OP, Agunbiade AO, Njoku NE, Nwakaudu AA, Odimegwu NE, Ndukauba OE, Ogbonna CU, Naibaho J, Korus M, Okpala COR. Revisiting food-sourced vitamins for consumer diet and health needs: a perspective review, from vitamin classification, metabolic functions, absorption, utilization, to balancing nutritional requirements. PeerJ. 2021 Sep 1;9:e11940. doi: 10.7717/peerj.11940. PMID: 34557342; PMCID: PMC8418216.
Steinert RE, Lee YK, Sybesma W. Vitamins for the Gut Microbiome. Trends Mol Med. 2020 Feb;26(2):137-140. doi: 10.1016/j.molmed.2019.11.005. Epub 2019 Dec 17. PMID: 31862244.
Pham VT, Dold S, Rehman A, Bird JK, Steinert RE. Vitamins, the gut microbiome and gastrointestinal health in humans. Nutr Res. 2021 Nov;95:35-53. doi: 10.1016/j.nutres.2021.09.001. Epub 2021 Oct 21. PMID: 34798467.
Hossain KS, Amarasena S, Mayengbam S. B Vitamins and Their Roles in Gut Health. Microorganisms. 2022 Jun 7;10(6):1168. doi: 10.3390/microorganisms10061168. PMID: 35744686; PMCID: PMC9227236.
Rudzki L, Stone TW, Maes M, Misiak B, Samochowiec J, Szulc A. Gut microbiota-derived vitamins – underrated powers of a multipotent ally in psychiatric health and disease. Prog Neuropsychopharmacol Biol Psychiatry. 2021 Apr 20;107:110240. doi: 10.1016/j.pnpbp.2020.110240. Epub 2021 Jan 9. PMID: 33428888.
Barone M, D’Amico F, Brigidi P, Turroni S. Gut microbiome-micronutrient interaction: The key to controlling the bioavailability of minerals and vitamins? Biofactors. 2022 Mar;48(2):307-314. doi: 10.1002/biof.1835. Epub 2022 Mar 16. PMID: 35294077; PMCID: PMC9311823.
谷禾健康
儿童呼吸系统疾病,包括呼吸道感染、反复喘息和哮喘,是儿童及其以后年龄发病和死亡的重要原因。
而哮喘是其中比较典型的一种,哮喘是全球最常见的慢性疾病之一,是一种复杂的、异质性的免疫介导的紊乱集合,以气道重塑和慢性气道炎症为特征。
▸ 哮喘的危险因素
哮喘的发病机制仍不清楚,但该疾病与多种遗传、环境、感染和营养因素有关。
哮喘的许多危险因素,包括生命早期的抗菌素暴露、配方奶喂养、以及母体接触抗生素怀孕期间,集中在产前和产后早期,儿童过敏性哮喘的发生可能与微生物和免疫发育关键时期的早期肠道微生物群落有关。
动物模型提供的证据表明,生命早期肠道微生物群的组成可能会影响呼吸道免疫以及对哮喘和呼吸道感染的易感性。
在这里我们总结了婴儿(0-12 个月大)肠道微生物群组成与儿童(0-18 岁)呼吸道疾病(即呼吸道感染、喘息或哮喘)之间的关联。
谷禾健康希望通过研究数据,找到更利于儿童健康的菌群数量与种类,有助于为未来的干预研究提供信息和构建更好的健康。
本文主要内容
●生命早期的肠道微生物
●生命早期肠道微生物群对儿童呼吸道疾病的影响
●早期肠道微生物的调理方法
●青少年哮喘的预防与治疗
每个人都可以被视为一个岛屿,由各种栖息地组成,这些栖息地被微生物群落定殖,并遵循创造和塑造当地组合多样性的规则。
不同婴儿身体部位的微生物群组成
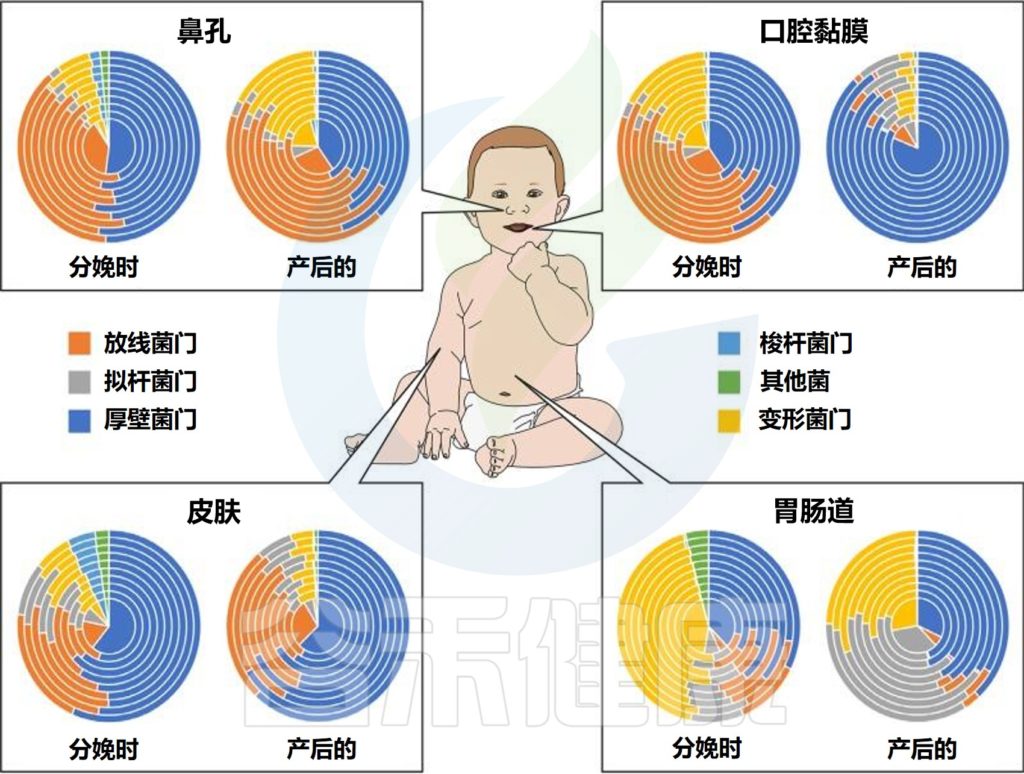
Milani C,et al.Microbiol Mol Biol Rev.2017
该图显示了婴儿微生物群组成的关键门在不同身体部位和生命早期不同阶段的相对丰度的全局概览。同心饼图示意性地表示个体间的可变性。
肠道微生物群是体内最大和最多样化的微生物群,包含数十亿细菌(主要生物)、古细菌、真核生物和病毒。
肠道菌群定植从出生时就开始了,在生命的最初几年是高度动态的,在1-3年后趋于稳定。
婴儿肠道核心微生物群
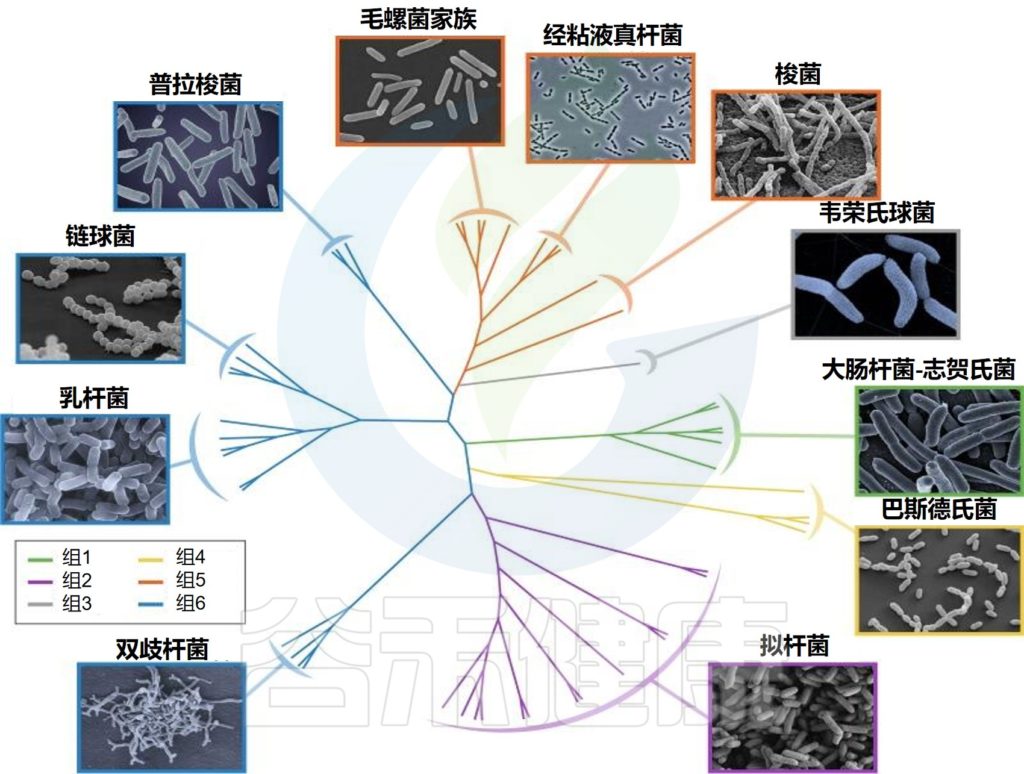
Milani C,et al.Microbiol Mol Biol Rev.2017
该图显示了涉及婴儿细菌核心微生物群的基于 16S rRNA 基因的树。树枝的颜色表示婴儿肠道微生物群的六个主要系统发育组。显示树的每个分支的关键婴儿肠道细菌分类群的电子显微镜图像。
相对于成人或年龄较大的儿童(>1岁)的肠道菌群,婴儿肠道菌群的多样性较低,菌群结构通常不稳定且高度动态。
双歧杆菌通常大量存在于婴儿,特别是母乳喂养的婴儿中,因此被认为是婴儿肠道微生物群的关键成员 。
尽管从婴儿肠道微生物群的初始组合到成人肠道微生物群的建立期间,个体水平的差异很大,但婴儿肠道微生物群可分为六种主要类型。
这种婴儿肠道微生物群的类型是根据肠道微生物群的组成和优势菌群的出现来确定的。详细地说,这些主要群体包括以下:
第1类,由肠杆菌目组成;
第2类,由拟杆菌目和疣微菌目组成;
第3 类,包括Selenomonadales以及梭菌目Pseudoflavonifractor、Subdoligranum和Desulfovibrio的成员;
第4类,包括所有巴斯德氏菌目;
第5类,包括大多数梭菌目;
第6类,包括梭状芽孢杆菌属、厌氧菌属和粪杆菌属、乳酸杆菌属和双歧杆菌属。
Bifidobacterium, Veillonella,
Streptococcus, Citrobacter,
Escherichia, Bacteroides, Clostridium
以上这些菌群在不同个体中主导婴儿肠道微生物群,它们在成人肠道微生物群中也很丰富。
▸ 梭状芽孢杆菌
梭状芽胞杆菌属的成员最近被重新分类为几个属,它们都属于梭状芽胞杆菌纲。这些物种通常存在于婴儿肠道微生物群中的微生物类群中。
▸ 拟杆菌
拟杆菌属。拟杆菌属的成员是成人肠道微生物群的主要成分,尽管它们也可能存在于婴儿肠道微生物群中,它们的存在似乎受到母乳低聚糖(HMO)的调节,其方式类似于双歧杆菌。
母乳低聚糖(HMO)——是母乳中第三丰富的固体成分(仅次于脂肪和乳糖),含量为5~15g/L,具有调节免疫,帮助大脑发育及调节肠道菌群等功能,有助于婴幼儿成长发育。
在小鼠实验中,已显示拟杆菌属的肠道定植。是宿主免疫系统识别和选择的结果,通过Toll样受体 (TLR) 和其他特定微生物-宿主相互作用。该属的成员被归类为能够代谢宿主产生的聚糖(例如HMO和粘蛋白)以及复杂的植物多糖(例如淀粉、纤维素、木聚糖和果胶)的糖破碎细菌。
●拟杆菌的作用
由于细胞外蛋白酶的作用,拟杆菌属物种通常具有蛋白水解活性。拟杆菌属成员利用的其他关键代谢功能包括胆汁酸的去结合。
在拟杆菌属中,脆弱拟杆菌被描述为可以产生多种荚膜多糖的成员,称为多糖A(PSA),是肠道菌群定植、宿主-微生物串扰或免疫调节的重要介质。
在各种拟杆菌属物种中,预计荚膜多糖会改变细胞表面的物理特性,并在宿主细菌共生中发挥关键作用。
▸ 韦荣氏球菌和链球菌
韦荣氏球菌和链球菌是婴儿肠道微生物群的一个次要成分。
这些细菌具有糖分解作用,利用其他婴儿肠道细菌(如链球菌和双歧杆菌)的碳水化合物发酵的最终产物(如乳酸)产生丙酸,形成重要的营养链。
这种短链脂肪酸被认为是肠道菌群的有益产物,因为它表现出抗炎特征,影响葡萄糖和能量稳态,增加胰岛素敏感性。
链球菌属的特定成员也构成婴儿肠道核心微生物群的一部分,并且是婴儿肠道中最早建立的细菌之一,可以在出生后的最初24小时内被识别出来。
▸ 乳酸杆菌
已知乳酸杆菌存在于婴儿肠道微生物群中,尽管它们在大肠中的数量低于上述细菌属,但在分娩后不久就存在。
乳酸杆菌的后续研究表明,与阴道分娩婴儿相比,剖腹产婴儿在生命的前6个月内的不同时间点的乳酸菌属检出率显著降低。
▸ 阿克曼氏菌
阿克曼氏菌自生命早期就存在于人类肠道中,但是水平非常低,阿克曼氏菌的存在与肠道完整性相关,已知其相对丰度和绝对数量会随着年龄的增长而迅速增加,特别是在断奶后。
小鼠实验证实了阿克曼氏菌对肠道屏障功能的影响,并证明其给药可防止饮食引起的肥胖。最近,还提供了涉及通过特定菌毛相关蛋白的TLR信号传导的机制解释。
临床、母体、喂养方式和环境因素共同塑造了生命早期的肠道微生物群。 对身体更方面都产生了一定影响。
考虑了生命早期肠道微生物群与儿童呼吸道疾病(包括呼吸道感染)之间的关联,在这里汇总了一些之前对肠道菌群与呼吸道疾病直接关联的研究。
肠道微生物的测量:通常通过收集粪便样本来测量,并且可以从多样性和丰度方面进行广泛描述。多样性描述了社区内不同分类群的数量。
探索哮喘或特应性喘息的研究
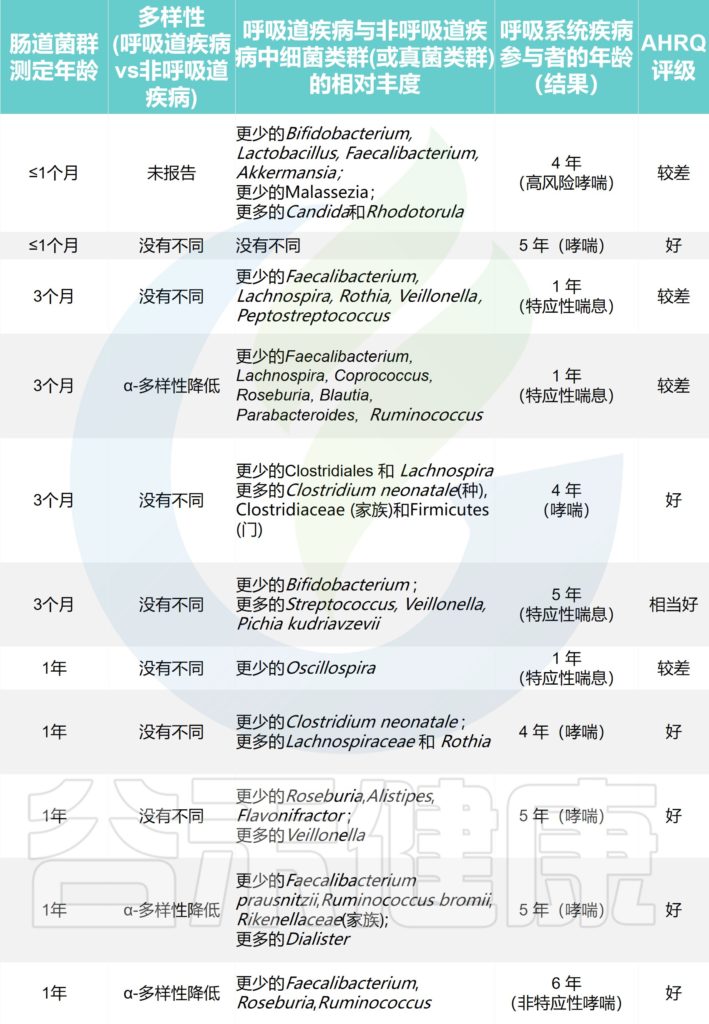
Alcazar CG,et al.Lancet Microbe.2022
探索呼吸道感染的研究

Alcazar CG,et al.Lancet Microbe.2022
喘息是与感染、过敏或后来的哮喘诊断相关的呼吸道症状。我们发现较高的α多样性与的喘息之间存在关联,这主要发生在阴道分娩的婴儿中。
α-多样性是指每个样本检测到的分类群数量,而β-多样性表示样本之间的组成差异。更细微的比较确定了不同分类水平下细菌或真菌的特定相对丰度。
大型研究(>700 名参与者)报告说,高 α 多样性对哮喘和喘息有保护作用。
探索了α-多样性与哮喘或特应性喘息之间的直接关联:与较低的α-多样性相比,生命第一年较高的肠道微生物群α-多样性与1岁时没有特应性喘息显著相关,并且在5岁和6岁时没有哮喘。
一项研究报告称,5周龄时肠道微生物群成熟度增加,肠道微生物群成熟度下降与6-11岁的哮喘高风险相关;还有两项研究报告了12个月时未成熟的肠道微生物群与5-6岁时哮喘风险增加之间存在一定关联。
基于细菌类群组成随时间的变化探索了健康参与者肠道微生物成熟度,并将该微生物群成熟度与儿童呼吸道疾病参与者的微生物群成熟度进行了比较。
总体而言,有证据表明双歧杆菌在3个月前婴儿的粪便中的相对丰度较低,与1岁时的呼吸道感染和4-5岁时的哮喘有关。
在3-12个月时的粪便样本中粪杆菌属、罗氏菌属和瘤胃球菌的丰度较低,与1-6岁时的哮喘和特应性喘息有关。
注意
然而,存在重要的研究限制,包括异质的结果定义和随访时间、残余混杂、小样本量以及异质的生物信息学和统计方法,大多数研究没有报告效果估计。
还有一些研究报告说,在1周龄时,非共生肠道细菌(如克雷伯氏菌和肠球菌)的相对丰度较高与1岁时的呼吸道感染有关; 3 个月时链球菌的相对丰度与 5 岁时的特应性喘息有关;1岁时Rothia或Dialister的高相对丰度与4-5岁时哮喘相关。但是具体作用机制目前还不清楚。
▸ 双歧杆菌增强呼吸道的免疫
双歧杆菌属(Bifidobacterium)是出生后4个月内儿童肠道中最丰富的细菌之一。并且已被证明通过体外和体内的表面相关分子和微生物群衍生代谢物调节个体的全身免疫反应。
在哮喘和呼吸道感染小鼠模型中,特定的双歧杆菌已被证明会影响呼吸道疾病的易感性。
一项研究表明,婴儿双歧杆菌的肠道定植可调节Th1和Th2反应之间的平衡,从而减少诱导小鼠模型中特应性哮喘的症状。
Th1(辅助型T淋巴细胞1)主要是增强吞噬细胞介导的抗感染免疫,特别是抗胞内病原体的感染。
Th2(辅助型T淋巴细胞2)Th2细胞的主要效应是辅助B细胞活化,其分泌的细胞因子可以促进B细胞增殖、分化和抗体的生成。
另一项研究报告称,当受到流感病毒的攻击时,与肠道丰度较低的小鼠相比,肠道丰度较高的双歧杆菌和拟杆菌的小鼠通过增强的CD8 T细胞和调节良好的巨噬细胞反应来提高流感存活率,从而防止过多的气道中性粒细胞流入。
▸ 梭状芽胞杆菌降低呼吸道炎症
Faecalibacterium、Ruminococcus、Lachnospira、Roseburia和Veillonella属于梭状芽孢杆菌类,在4-6个月大的儿童肠道中丰度较高。
已经描述了Roseburia和Faecalibacterium的潜在免疫调节机制,它们产生丁酸盐。
丁酸盐——一种在动物和体外模型中具有抗炎特性的细菌代谢物。
研究发现上呼吸道感染与婴儿粪便样本中丁酸梭菌的丰度降低有关。梭状芽胞杆菌可促进调节性T细胞产生并抑制炎症细胞因子,其中一些与人类全身感染有关。因此,丁酸梭菌对婴儿对感染的免疫反应的潜在抑制作用需要进一步研究。
另一项研究表明,给无菌小鼠接种毛螺菌属、韦荣氏菌属、粪杆菌属和罗氏菌可改善这些小鼠成年后代的气道炎症,但是这些细菌在呼吸系统疾病中的机制作用了解还是较少。
▸ 韦荣氏球菌刺激免疫分化
在我们的研究中,韦荣氏球菌,特别是小韦荣氏球菌(Veillonella parvula),与上呼吸道感染呈正相关,尤其是剖宫产婴儿。Veillonella parvula常见于口腔菌群中,它在口腔和肠道生态系统中都可以观察到。
一项针对120名荷兰婴儿的前瞻性研究发现,在1周大的婴儿中使用 16S V4 rRNA 测序发现了大量的韦荣氏菌操作分类单位,这与出生后第一年的呼吸道感染数量增加有关。
在研究中,Veillonella parvula在人体肠道中产生丙酸盐,这可能会刺激产生IL10的调节性T细胞分化;在小肠中,它会诱导产生IL-8、IL-1β、IL-10和 TNF- α37来影响呼吸道以至于全身的免疫。
▸ 棒状杆菌为呼吸道的致病菌
在剖宫产婴儿中,较高的棒状杆菌属(Corynebacterium)物种相对丰度与较高的上呼吸道感染风险相关。
棒状杆菌属物种通常被列为呼吸道中的致病菌。病例系列表明,痰中的假白喉棒状杆菌是肺部感染的驱动因素,一项来自法国的鼻咽微生物组病例对照研究发现,与健康对照组相比,病毒性呼吸道感染患者的假白喉棒状杆菌富集。
在3个月和1岁收集的粪便中,Faecalibacterium、 Roseburia和Ruminococcus相对丰度较低,与1-6岁的哮喘和特应性喘息相关。
3个月时Lachnospira的相对丰度较低,但1岁时的相对丰度增加也与1-6岁时哮喘和特应性喘息相关。
一项研究显示3个月时Veillonella的相对丰度较低与1岁时的特应性喘息相关,而两项研究报告称3个月和1岁时Veillonella的相对丰度较高与5岁时的哮喘和特应性喘息相关。
✦真菌与哮喘的关系
三项研究探索了真菌和哮喘之间的关系。
在一项研究中,在 1 个月大时测量的念珠菌和红酵母菌的相对丰度较高,而马拉色菌类群的丰度较低。
在另一项研究中,3月龄时Pichia kudriavzevii的相对丰度增加,与4-5岁时的哮喘和特应性喘息相关。
第三项研究却发现真菌成熟度与6岁儿童哮喘之间没有关联。
肠道菌群对哮喘的影响至少部分是由细菌代谢物介导的,这些代谢物可能会影响身体远端的免疫反应。
✦短链脂肪酸降低哮喘致敏性
在人类气道炎症中具有保护作用的最知名代谢物是 短链脂肪酸。1岁时粪便中含有大量丁酸盐和丙酸盐的儿童的特应性致敏性显著降低,并且在3至6岁之间不太可能患哮喘。
✦组胺和氧化脂质影响肺部炎症
与非哮喘志愿者相比,哮喘患者粪便样本中分泌组胺的细菌数量显著高于非哮喘志愿者。
此外,分泌组胺的细菌数量与疾病严重程度相关。然而,在过敏性气道炎症模型中,细菌来源的组胺降低了支气管肺泡液中的总细胞数和肺匀浆中IL-4、IL-5和IL-13的量。
相反,在蟑螂抗原小鼠气道炎症模型中,用12,13-diHOME(一种氧化脂质)对小鼠进行腹腔内治疗会减少肺部调节性T细胞的数量并增加肺部炎症。
建议
越来越多的证据表明细菌在哮喘中的作用,但需要进一步的研究来更清楚地定义所涉及的最重要的物种,并了解哮喘背景下的细菌生态失调是否是疾病的原因或影响。
有必要进行更详细的机制研究,以充分了解生命不同阶段肺和肠道微生物群组成和代谢与特定类型的哮喘炎症之间的复杂关联。
最后,未来的工作应该集中在继续详细描述在哮喘中介导细菌与宿主之间交流的细胞和分子机制。
通常用于预防或治疗不一定由特定病原体引起的感染的抗生素可以有效地消耗肠道微生物群。患有 NEC的新生儿感染肠道微生物的风险很高,抗生素通常用于预防或治疗这些感染。
NEC——新生儿坏死性小肠结肠炎(NEC)为一种获得性疾病,是多种原因引起的肠黏膜损害,使之缺血、缺氧,导致小肠、结肠发生弥漫性或局部坏死的一种疾病。
对于儿童炎症性肠病的治疗,使用单一抗生素对有并发症的患者有益,例如瘘管和脓肿,而广泛的抗生素组合可能会改善临床结果。
注意
在幼儿中使用抗生素存在很大风险。大量证据表明,抗生素会影响我们抵抗感染的能力、免疫系统的功能以及我们加工食物的能力。
肠道微生物群的破坏可能导致长期的健康后果,包括维生素产量减少、营养吸收减少以及糖尿病、哮喘、肥胖和感染风险增加。
口服益生元和益生菌是影响生命早期肠道微生物群发育的最常见方法。
益生元被定义为“选择性刺激肠道微生物群中一种或多种微生物属或物种的生长和活性,从而为宿主带来健康益处”的化合物,而益生菌被定义为“赋予宿主健康益处的活微生物”。摄入足量时会对宿主产生健康影响。
目前可用的益生元包括人乳低聚糖、菊粉、低聚果糖和低聚半乳糖;可用的益生菌包括双歧杆菌和乳酸杆菌属。
✦益生元和益生菌的作用
通过调节肠道微生物群,益生元对宿主产生健康影响。益生菌通过对粘膜和上皮的竞争性粘附、粘膜 IgA反应、抗菌物质的分泌、促炎途径的下调、抗炎细胞因子的产生和免疫系统的调节来增强肠道上皮屏障。
最近的研究表明,益生菌可以预防儿科疾病和障碍的进展,包括过敏、胃肠道感染、肥胖,甚至上呼吸道感染。
干预研究进一步表明,益生菌可以减轻某些疾病的严重程度,但对每种疾病的最佳干预仍然知之甚少。
虽然益生菌在某些情况下可以缓解过敏症状,但它们通常不能有效调节肠道微生物的组成。有证据表明,嗜热链球菌和双歧杆菌的组合可有效预防儿童抗生素相关性腹泻。
此外,合成生物学使益生菌和共生微生物的工程具有新的治疗功能。例如,融合蛋白HSP65-6P277的表达减少了非肥胖糖尿病小鼠中1型糖尿病的发病,而在非肥胖糖尿病鼠中口服重组乳酸乳球菌可改善糖耐量并显著减少胰岛素炎。
建议
尽管益生元和益生菌的应用前景不错,但在未来的研究中,应讨论益生菌和益生素的施用时间、不同菌株和菌株组合的效果、工程、安全性以及这些益生菌与益生素组合是否更有效的确定。
营养素可以通过塑造微生物菌群的组成,对婴儿肠道微生物的定植模式产生短期和长期影响。
越来越多的证据表明,摄入的膳食成分与炎症性肠病、2型糖尿病和动脉粥样硬化的发展有关。肠道菌群的最大变化发生在固体食物的引入,这表明饮食应被视为肠道菌群的核心决定因素。
✦饮食疗法
有趣的是,在中医中,早就有“药食同源”的概念。该概念的一个方面是食物是一种药物,适当的饮食或某些食物可以维持身体的平衡和健康,并预防或减轻某些疾病的发展。
在现代医学中,饮食改变越来越被认为是一种通过改变肠道微生物群来改变全身炎症的相对简单的方法。
早期肠内营养 (EEN) 是一种饮食疗法,已被用作儿童克罗恩病的一线疗法,通过用仅由液体营养素组成的配方代替正常饮食成分,旨在使炎症标志物正常化并诱导临床缓解。
✦高膳食纤维可以减轻呼吸道疾病
此外,另一项研究报告称,喂食高纤维饮食的小鼠可以产生独特的肠道微生物群,从而导致短链脂肪酸乙酸酯水平升高。
高纤维或醋酸盐喂养通过减少与人类哮喘和小鼠哮喘模型相关的某些基因在小鼠胎肺中的表达,显著抑制过敏性气道疾病。
此外,发现肠道微生物不仅与哮喘有关,而且还降低了与哮喘严重程度和炎症表型相关的气道微生物群的多样性和群落组成。
抗炎特性
此外,最近的一些研究证明,膳食纤维具有抗炎特性,这可以部分解释纤维对肠道微生物菌群的影响。 使用临床前模型的几项研究表明,可发酵纤维补充剂通过微生物群诱导的特定抗炎代谢物产生的变化来改变疾病结果。
然而,还需要更多的研究来增加我们对不同饮食如何塑造微生物群和改变健康结果的理解。
粪便微生物群移植(FMT)被定义为将健康供体的粪便悬浮液输注到受体患者的胃肠道中,以恢复肠道微生物群的正常多样性和功能。
粪菌移植的方式:粪便微生物群可通过结肠镜检查、鼻胃管或鼻十二指肠管、灌肠剂或口服胶囊置于患者体内。
由于肠道微生物群宏基因组测序的技术进步以及对其组成和功能的日益了解,粪菌移植近年来引起了越来越多的兴趣和关注。尽管粪菌移植仍然知之甚少,但它不再被认为是一种“替代”和最后的医疗实践,现在作为一种具有生物学合理性的有价值的疗法正在获得主流接受。
此外,这种疗法已被证明能够重建正常运作的微生物群落。通过为患者提供来自合适供体的平衡微生物群,纠正了在艰难梭菌感染 (CDI)发病机制中起重要作用的不平衡肠道微生物菌群。
在一系列关于复发性CDI的研究中,85%接受粪菌移植的患者出现症状缓解。此外,考虑到肠脑轴和肠道微生物之间的相互作用,粪菌移植被认为是治疗某些精神疾病的可能方法,例如自闭症谱系障碍。
建议
然而,粪菌移植的微生物组成尚未完全确定。因此,需要澄清与改善临床结果相关的微生物结构或功能特征,以确定优选的组合。
未来的研究应侧重于确定“健康”微生物菌群的范围以及制定评估最佳组成的标准。
5岁至14岁的哮喘患病率约为10%,使其成为全球儿童时期最普遍的慢性病。尽管下呼吸道感染带来了巨大的健康负担,但目前还没有专门针对它们或儿童哮喘被广泛许可的预防策略,所以暂时只能用一般呼吸道疾病的方法来预防。
✦注意空气卫生
注意室内的清洁和空气流通,因为空气中的灰尘和细菌是哮喘病发的主要致敏原,所以应该勤加打扫,减少空气中的尘埃。尽量减少暴露于空气污染的室内和室外。
✦良好饮食习惯
坚持每天喝水,喝水是排出身体毒素的最佳的方法。在日常生活中注意饮食习惯,一日三餐要按时就餐,少吃油腻。
✦良好生活,避免螨虫
哮喘病人要在日常生活中每天要保持良好的生活态度,放松心情。不要在家里养猫、狗、花、鸟等。经常晾晒被褥、换洗床单,避免螨虫孳生。
✦加强自我管理
对于5岁及以上被诊断为哮喘的儿童或青少年,提供哮喘自我管理计划,包括书面的个性化行动计划和教育。
说明污染会引发或加剧哮喘,并在个性化的行动计划中包含尽量减少暴露于室内和室外空气污染的方法。
注:哮喘好发于青少年和儿童,一旦患病,如防治不当,很容易反复发病,随着发病频度的增加,病情会逐渐加重,必将严重影响生活质量和学习工作能力,给个人家庭和社会造成沉重负担。
我们结合当前的研究与认知,提出了一些适用于儿童和青少年新诊断哮喘或当前治疗无法控制哮喘的治疗建议。
✦药物治疗
•SABA
β2受体激动剂(SABA)是一类能够分布在气道平滑肌上的β2受体产生支气管扩张作用的哮喘治疗药物。这类药物属于支气管扩张药,是哮喘急性发作(气道痉挛)的首选药物,能够迅速改善哮喘急性发作时的呼吸困难、咳嗽等的症状。
对新诊断为哮喘的儿童和年轻人(5至16岁),可以提供SABA作为缓解疗法。
对于患有哮喘的儿童和青少年(5至16岁),他们很少出现短暂的喘息和正常的肺功能,也可以考虑单独使用SABA缓解疗法进行治疗。
•ICS
吸入性糖皮质激素(ICS)是目前控制哮喘病的气道炎症最有效的药物,以定量气雾剂、干粉剂或溶液吸入。
在哮喘炎症表型中,通常接受高剂量吸入性皮质类固醇 (ICS) 的中性粒细胞性哮喘患者表现出较少的细菌负荷,其中嗜血杆菌和莫拉菌属、变形杆菌门的成员相对富集,而链球菌的相对丰度降低。
ICS已经成为目前哮喘治疗的第一线治疗,对病人是最为重要的治疗,任何哮喘患者,只要诊断正确,都应该接受ICS的治疗,这是一个长期维持治疗,可以起到气管局部抗炎的效果,改善病情,预防哮喘急性发作。
为儿童和青少年(5至16岁)提供儿科低剂量ICS作为一线维持治疗。
就诊时出现明显表明需要维持治疗的症状(如导致夜间醒来)或单独使用SABA无法控制的哮喘也应使用ICS治疗。
✦风险分层
同时使用风险分层来识别预后不良风险增加的哮喘患者,并使用此信息优化他们的护理。
根据诸如不依从哮喘药物、心理社会问题和反复发作的哮喘计划外护理等因素进行风险分层。
总体而言,有观察证据表明,在生命的第一年,肠道共生细菌属的低α多样性和相对丰度与随后的呼吸系统疾病,尤其是哮喘有关。因此在婴儿早期关注和了解肠道菌群状况对于后面哮喘发生和预防非常重要,进一步研究哮喘患者的肠道和下呼吸道微生物群可能有助于开发更有效的方法来预防和治疗哮喘。
主要参考文献
Alcazar CG, Paes VM, Shao Y, Oesser C, Miltz A, Lawley TD, Brocklehurst P, Rodger A, Field N. The association between early-life gut microbiota and childhood respiratory diseases: a systematic review. Lancet Microbe. 2022 Aug 18:S2666-5247(22)00184-7. doi: 10.1016/S2666-5247(22)00184-7. Epub ahead of print. PMID: 35988549.
Milani C, Duranti S, Bottacini F, Casey E, Turroni F, Mahony J, Belzer C, Delgado Palacio S, Arboleya Montes S, Mancabelli L, Lugli GA, Rodriguez JM, Bode L, de Vos W, Gueimonde M, Margolles A, van Sinderen D, Ventura M. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol Mol Biol Rev. 2017 Nov 8;81(4):e00036-17. doi: 10.1128/MMBR.00036-17. PMID: 29118049; PMCID: PMC5706746.
Zhuang L, Chen H, Zhang S, Zhuang J, Li Q, Feng Z. Intestinal Microbiota in Early Life and Its Implications on Childhood Health. Genomics Proteomics Bioinformatics. 2019 Feb;17(1):13-25. doi: 10.1016/j.gpb.2018.10.002. Epub 2019 Apr 12. PMID: 30986482; PMCID: PMC6522475.
Barcik W, Boutin RCT, Sokolowska M, Finlay BB. The Role of Lung and Gut Microbiota in the Pathology of Asthma. Immunity. 2020 Feb 18;52(2):241-255. doi: 10.1016/j.immuni.2020.01.007. PMID: 32075727; PMCID: PMC7128389.
Ver Heul A, Planer J, Kau AL. The Human Microbiota and Asthma. Clin Rev Allergy Immunol. 2019 Dec;57(3):350-363. doi: 10.1007/s12016-018-8719-7. PMID: 30426401; PMCID: PMC7449604.
Moroishi Y, Gui J, Hoen AG, Morrison HG, Baker ER, Nadeau KC, Li H, Li Z, Madan JC, Karagas MR. The relationship between the gut microbiome and the risk of respiratory infections among newborns. Commun Med (Lond). 2022 Jul 14;2:87. doi: 10.1038/s43856-022-00152-1. PMID: 35847562; PMCID: PMC9283516.

谷禾健康

在过去的几十年里,生物技术和计算技术的巨大进步彻底改变了我们对微生物群落的理解。特别是,基于16S rRNA和宏基因组测序的研究进一步验证了一个事实,即微生物群不是静态的实体,而是动态的生态系统,身在其中的组成成员之间,以及成员与环境之间,都在以各种方式进行相互作用。
在这些研究过程中产生了大量的关于微生物相互作用的机制和环境依赖性的数据,也出现了许多专门储存单一类型或进行了部分分类整合的数据库。但由于是以不同的标准去整理的数据,所以很难共享。
//
本文研究人员提出了一套体系,用于微生物组数据管理,简称FAIR (findable, accessible, interoperable, and reusable),即可查询、可访问、可交互和可重复使用。这些原则于2016年首次正式提出。
首先基于可查询和可访问的原则,需要数据库,它能存储现有数据也能继续转化新的数据。这个平台可以是自己从零开始设计,可以是使用现成的数据库构建工具作为辅助(比如mako软件),也可以是直接使用已有的数据库,比如GloBI。
关于数据获取,文中列出了一些资源,都是目前广泛使用或者比较独特的,如下表:

其次基于交互性和可重复使用的原则,需要将大数据拆分为一个个子集,每一个子集都应该是能满足大量用户的第一需求或者说是第一想了解的信息点。基于此,研究人员提出了四类元数据作为起点:
1. 微生物实体
应提供参与相互作用的每种微生物的物种(和菌株,如果相关)名称。
示例:
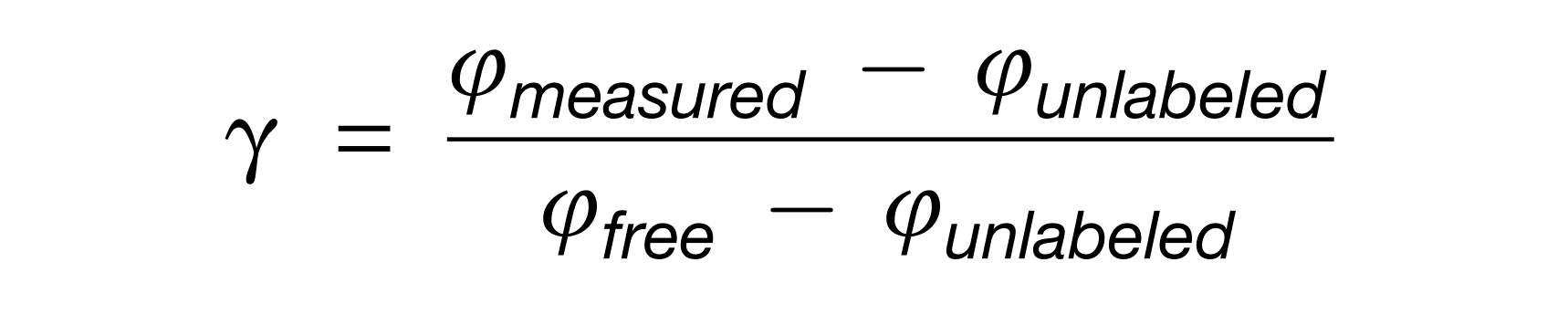
2. 相互作用的推理方法
使用不同的方法来指示高度相关的元数据。
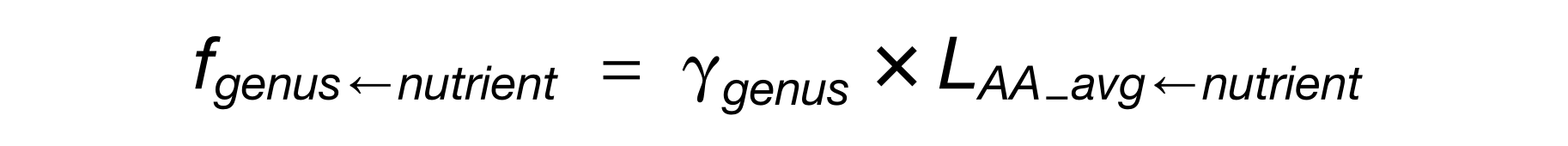
示例:

3. 相互作用的上下文语境
一些环境背景,如生物群落(例如,宿主相关的、合成的)。

示例:

4. 属性,用于定义互作关系的类型
如合作、对抗、关联、成对或高阶等。
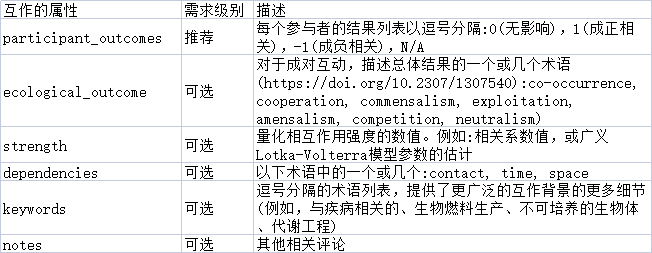
示例:

最后,文章中给出了依照FAIR体系研究微生物互作关系的可拓展性。
如下图,通过研究微生物相互作用和相关性(A部分)而产生的多种类型的元数据(B部分),这些元数据(DT),例如真菌细菌或噬菌体细菌的培养实验数据(DT1),扩增子序列或OTU计数的相关性(DT2),两个或两个以上物种基因组尺度代谢网络的通量平衡模型(DT3),直接比对到特定数据库(DT4)。
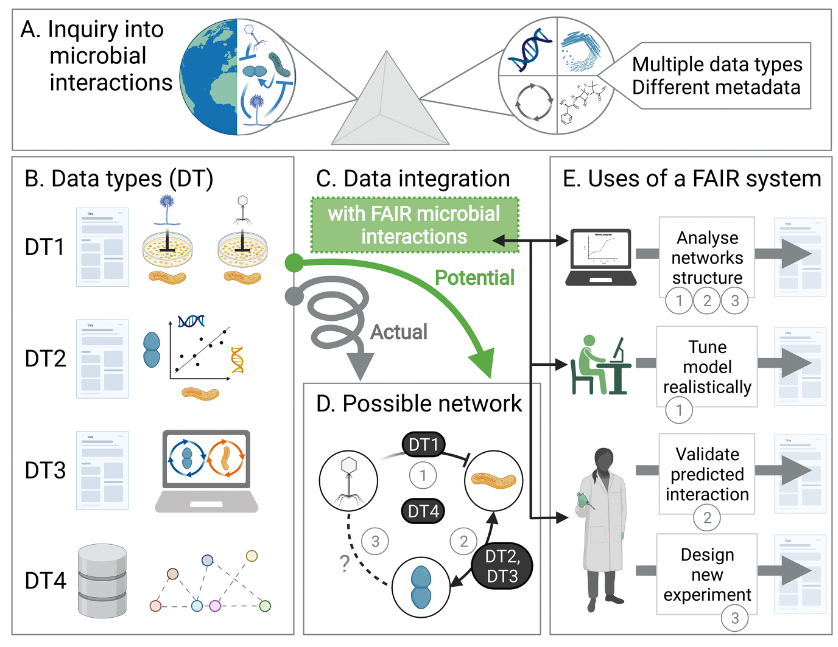
遵循FAIR里的四个原则来解析微生物互作内容(C部分),解析结果如D部分,得到一个可能的相互作用网络关系图谱。
同时,C部分的内容可以横跨不同领域,为不同领域的专家提供信息,例如E部分,建模者可以更容易确定具体的互作以便真实的模拟生态动力学,而实验人员可以评估是否有新的互作在其他宿主或上下文中被报告。
以上内容,是研究人员基于自身经验归纳总结的,研究人员也指出在此概述的实践、标准和示例并不是详尽无遗的,文章旨在促进进一步讨论如何改进微生物相互作用及其属性数据的访问和可用性。希望能进一步的激励各位科学家,共同创建一个共享开放的,拥有统一标准的微生物相互作用资源站。
参考文献:
Pacheco AR, Pauvert C, Kishore D, Segrè D. Toward FAIR Representations of Microbial Interactions. mSystems. 2022 Aug 25:e0065922. doi: 10.1128/msystems.00659-22. Epub ahead of print. PMID: 36005399.

谷禾健康

在过去的几十年里,肥胖患病率的持续快速增长。成为了许多国家的主要医疗保健问题,尤其是在2019年新冠状病毒时代以来。
肥胖是包括心血管疾病在内的一系列疾病不断扩大的风险因素。2型糖尿病、慢性肾病、非酒精性脂肪肝病, 负重过大导致的关节炎,甚至许多癌症都与肥胖有关。
▸ 肥胖的定义
肥胖定义为身体脂肪过度积累到可能对健康产生不利影响的程度。
一般使用体重指数(BMI;体重(千克)除以身高(米)的平方)进行评估。
我国规定的BMI正常范围在18.5-23.9之间,24-27.9为超重,超过28则为肥胖。
肥胖不是单纯的体重增加,而是体内脂肪组织积蓄过剩的状态。肥胖是指一定程度的明显超重与脂肪层过厚,是体内脂肪,尤其是甘油三酯积聚过多而导致的一种状态。
主要原因是由于能量摄入过多或机体代谢的改变而导致体内脂肪积聚过多造成体重过度增长并引起人体病理、生理改变或潜伏。
▸ 引起肥胖的因素
肥胖是一个多因素问题,不仅限于饮食或缺乏运动的原因,还包括遗传、环境和心理社会因素,这些因素通过能量摄入和消耗的生理介质起作用。
肠道微生物组是这些环境因素之一;大约 20年前,在小鼠研究中已经确定了脂肪储存和肠道微生物组之间的联系。粪便微生物群移植研究提供了更切实的证据。
本文结合了最新的学术研究和谷禾健康数据库,涵盖了不同的角度,既关注单个细菌的作用,也特别强调整个微生物组的组成,以试图解开肠道微生物组与肥胖的关系。
让人们更好地了解肥胖以及其发病机制,在此基础上提出一些预防和治疗肥胖的建议,使人们拥有更健康的生活。
本文主要从以下几个方面讲述
●肠道微生物对肥胖发病机制的影响
●菌群代谢物对肥胖的影响
●健康与肥胖人群中的细菌比例
●肥胖与肠道微生物的研究分类
●微生物多样性与人体健康有关
●肥胖与肠道微生物的未来研究方向
●预防和治疗肥胖的一些建议
学术专业用词缩写
PRR—模式识别受体
NOD2—核苷酸结合寡聚化结构域2
FXR—法尼醇X受体
TLR5—TOLL样受体5重组蛋白CDI—复发性艰难梭菌感染
BSH—胆盐水解酶
GLP1—胰高血糖素样肽-1
GPR—G蛋白偶联受体
01
肠道微生物对肥胖发病机制的影响
研究肥胖的发病机制,有助于我们更好地了解肥胖,并以此制定相应的治疗方案。实验研究发现肠道微生物对肥胖的发病机制存在一定的影响。
许多研究已经确定了肠道微生物群与宿主免疫系统之间的关联。其中一个发现是肥胖与肠道微生物引起的慢性低度炎症有关。
肠道微生物群和肠道细胞之间的密切接触是由微生物相关分子模式介导的,这些分子模式可以与上皮细胞和免疫细胞中的模式识别受体 (PRR) 结合。
这些识别受体属于先天免疫系统,控制炎症和免疫反应。PRR还可以检测宿主细胞释放的损伤相关分子模式。
✦革兰氏阴性菌中的脂多糖易引起炎症
脂多糖 (LPS)是革兰氏阴性菌外膜的一种特有成分,由脂质和多糖构成,似乎会引起小鼠的低度炎症。
在这里列举了一些常见的革兰氏阴性菌:
大肠杆菌、变形杆菌、痢疾杆菌
肺炎杆菌、布氏杆菌
需要注意的是,大部分革兰氏阴性菌对人体都有害
在一项人体研究中进行了类似的观察,其中能量摄入与内毒素血症和伴随的炎症有关。
事实上,与健康对照组相比,在患有2型糖尿病的受试者中,革兰氏阴性菌的数量明显更多。
脂多糖通过脂多糖分化受体14(CD14)和辅助受体 toll样受体 (TLR4)引起炎症,这反过来又导致脂肪细胞产生的促炎细胞因子增加。
●饮食在脂多糖中起重要作用
果胶可抑制脂多糖诱导的单核细胞或树突状细胞中的TLR4活化,而果糖或高脂肪饮食导致含有脂多糖的变形菌增加,瘦素信号与饱腹感和能量平衡紊乱有关,因此失调。
在此列举了一些高果糖高脂食物:
1.蜂蜜和市面上一些甜的饮料果糖含量较高;
2.淀粉类:经油炸加工的馅饼、油条、葱油饼、油糕等食物中,含有大量脂肪与糖分;
2、肉类:用糖汁、糖煎、糖烧的方法进行烹调的红烧肉、炸鸡等,也为高糖高脂食物;
3、奶油制品食物:如奶油蛋糕、奶茶、泡芙等甜品,主要原材料为淀粉与黄油等物质,所以也有较高的糖分与脂肪。
同时还表明,分泌型脂蛋白脂肪酶(LPL)抑制剂血管生成素样蛋白4(一种禁食诱导的脂肪因子)可被微生物群抑制,进而导致分泌型脂蛋白脂肪酶活性增加和白色脂肪组织中的脂肪储存。
✦肽聚糖影响体内平衡
另一个例子是肽聚糖,它是细菌细胞壁的一种成分,对人体内平衡很重要。
核苷酸结合寡聚化结构域2 (NOD2) 是肽聚糖的产物,是一种位于上皮细胞和免疫细胞内的胞质 ,能够感知胞壁酰二肽。
这种胞质对于病原体入侵和几种炎症性疾病期间的免疫反应至关重要,从而调节粘膜细菌定植。
// 一些关于NOD2的研究案例
NOD2缺乏的小鼠在高脂饮食期间显示出脂肪组织、肝脏炎症和胰岛素抵抗增加。因此经常用于糖尿病研究。
在具有功能性NOD2受体的肥胖小鼠中,胞壁酰二肽识别显示可减少脂肪炎症和胰岛素抵抗,而不会减轻体重或改变肠道微生物群组成。
上述案例在一定程度上可以说明NOD2对于减轻肥胖和肠道微生物群稳定具有一定作用。
✦Toll样蛋白受体影响免疫
——Toll样受体5(TLR5)重组蛋白是免疫系统的关键成分,还是单体鞭毛蛋白的传感器,可以检测细菌感染并启动宿主抗菌的防卫反应。
肠道微生物群也通过位于上皮细胞上的TLR5与免疫系统相关联。
免疫系统通过TLR5感知肠道微生物群的组成和肠道微生物群的定位,以避免共生肠道微生物群传播到肠外器官、产毒成员的过度生长以及机会性病原体的过度生长和入侵。TLR5检测鞭毛蛋白会导致白细胞介素-22的产生,从而预防与肠道炎症相关的疾病。
// 关于TLR5影响免疫在小鼠中的研究
与野生型无菌小鼠相比,TLR5缺陷小鼠的胰岛素抵抗和肥胖水平增加。肠道微生物群从这些TLR5缺陷小鼠转移到野生型无菌小鼠也导致这些野生型小鼠代谢综合征的相似特征转移。
一项调查缺乏TLR5受体的小鼠的研究,观察到鞭毛蛋白特异性免疫球蛋白的丢失导致鞭毛细菌增加,包括许多变形杆菌,以及粘膜屏障破坏和炎症增加。
肠道微生物影响宿主免疫的推定机制
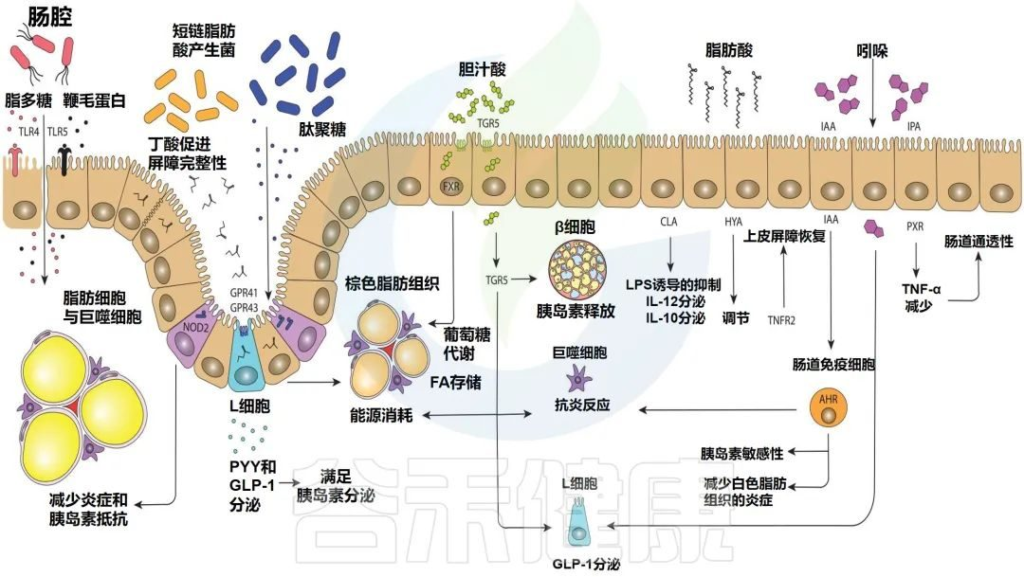
Levin E,et al.Therap Adv Gastroenterol.2022
部分肠道微生物群的鞭毛蛋白和脂多糖可以与toll样受体重组蛋白结合,而细胞内NOD2感知肽聚糖。几种短链脂肪酸的产生可以与GPR41和GPR43(2种特异性短链脂肪酸受体)结合,导致PYY(肽YY(一种新的胃肠道激素,具有抑制胃肠运动和胃酸分泌等作用))和GLP-1(胰高血糖素样肽-1)的表达增加。
胆汁酸激活TGR5和FXR(一种胆汁酸的受体)),影响脂质和葡萄糖代谢。脂肪酸,如HYA,调节TNFR2,参与上皮屏障恢复。吲哚通过GLP-1调节和AHR的激活以及与PXR 的结合影响宿主。
注意
事实上,与瘦的人相比,肥胖的人往往有的粪便鞭毛蛋白、更少的粪便抗鞭毛蛋白IgA和更高水平的慢性肠道炎症。
02
菌群代谢物对肥胖的影响
短链脂肪酸
短链脂肪酸(SCFA)主要是微生物厌氧发酵的衍生终产物,对宿主具有多种影响。它是一组少于六个碳的羧酸,包括乙酸盐、丙酸盐和丁酸盐。这些短链脂肪酸及其比例在几种不同的组织中具有多种有益的作用。
✦短链脂肪酸有利于肠道环境的稳态
短链脂肪酸被认为是人类宿主的能量来源和能量调节剂,但它们也有助于维持肠道环境的稳态。短链脂肪酸的细胞外活性主要由G蛋白偶联受体(GPRs)介导。
这些受体在多种细胞上表达,包括肠上皮细胞、脂肪细胞、肠内分泌L细胞、先天免疫细胞和体细胞感觉神经节的神经元。
✦短链脂肪酸会影响饱腹感
短链脂肪酸参与L细胞产生的肽YY和胰高血糖素样肽1(GLP1) 激素的调节。这两种激素都调节神经系统的饱腹感,GLP1在葡萄糖刺激的胰岛素敏感性和分泌中也起作用。
饱腹感也由丙酸盐通过激活脂肪细胞中的游离脂肪酸受体3(FFAR3)来控制,因为这些脂肪细胞会产生瘦素。微生物衍生的丁酸盐和丙酸盐都会诱导肠道糖异生,进而诱导对葡萄糖和能量稳态的有益影响。
✦短链脂肪酸促进能量消耗
研究显示丁酸盐通过游离脂肪酸受体2(FFAR2)的活化刺激棕色脂肪组织的活化,从而显著促进能量消耗。并且脂肪积累被丁酸盐诱导的白色脂肪组织中的游离脂肪酸受体2活化抑制。最后,丁酸盐通过降低肠屏障的通透性来减少上皮细胞中的细菌易位。
在肠道内,短链脂肪酸的产生通过各种中间体发生。不同的物种,在产生这些中间体和最终产物的每个步骤中使用不同的酶,都参与了这个过程。
●2型糖尿病中产丁酸盐菌丰度较低
在2型糖尿病中,许多研究看到的一个共同趋势是,糖尿病患者的丁酸盐生产者(如Roseburia和Faecalibacterium)的丰度低于对照组,这可能取决于饮食。
在肥胖症中也可能如此,短链脂肪酸的过量生产可能会导致更高的能量可用性和摄入量。
事实上,一项比较肥胖与瘦的受试者的研究表明,肥胖者的总短链脂肪酸水平较高,但必须指出,肥胖与丙酸盐水平特别相关。
胆汁酸
胆汁酸是胆汁的重要成分,在脂肪代谢中起着重要作用。 胆汁酸主要存在于肠肝循环系统并通过再循环起一定的保护作用。
许多研究报告了肠道微生物组、胆汁酸和肥胖或肥胖相关疾病之间存在联系。
初级胆汁酸通过两种途径在肝细胞中产生:
产生大部分胆汁酸的经典途径是由细胞色素P450中的胆固醇7α-羟化酶启动的。
替代途径由细胞色素P450中的27α-羟化酶启动。
注:细胞色素P450——一个很大的可自身氧化的亚铁血红素蛋白家族,属于单氧酶的一类,因其在450纳米有特异吸收峰而得名。它参与内源性物质和包括药物、环境化合物在内的外源性物质的代谢。
7α-羟化酶和27α-羟化酶都属于细胞色素P450中的成员。
经典途径中的一种中间体胆固醇7α-羟化酶与总血浆甘油三酯浓度相关,表明肝胆汁酸合成对于调节肥胖者的血浆甘油三酯水平很重要。
胆汁酸的作用途径
产生的初级胆汁酸是胆酸、鹅去氧胆酸和猪胆酸。这些初级胆汁酸与甘氨酸或牛磺酸结合。餐后,这些结合物被分泌到胆汁中并释放以促进膳食脂肪的溶解和吸收。
此后,肠道微生物群使用胆盐水解酶(BSHs)去结合初级胆汁酸。
Bifidobacterium spp., Lactobacillus spp., Enterococcus spp.和Methanobrevibacter spp.,这些细菌中都含有这些胆盐水解酶。
接下来,这些去结合的初级胆汁酸随后被转化为次级胆汁酸。
注:这是通过肠道微生物群的脱氨基作用和7α-羟化酶的脱羟基化来完成的。
在最后阶段,胆汁酸被回肠远端吸收,完成肠肝循环。产生的次级胆汁酸是脱氧胆酸和石胆酸。这些胆汁酸参与调节能量消耗,以及炎症和葡萄糖代谢和脂质代谢。
这表明这些胆汁酸在肥胖的病理生理学中非常重要,因为与肥胖相关的肠道微生物群的改变包括胆汁酸池大小和组成的变化。
✦不同胆汁酸具有不同的作用
不同的胆汁酸对各种肠道受体具有不同的亲和力,例如与膜结合的蛋白偶联受体(TGR)以及法尼醇X受体(FXR) 。
注:TGR5—是一种G蛋白偶联受体,不仅是胆汁酸的受体,也是多种选择性合成激动剂的受体。
法尼醇X受体(FXR):一种胆汁酸受体,被特定胆汁酸代谢物激活后发挥转录因子作用,参与调控胆汁酸的合成和肠肝循环,影响机体的糖脂代谢。
在小鼠中,已经表明肠道菌群通过FXR受体促进饮食诱导的肥胖。
在脂肪组织中,脂肪细胞分化受FXR通过促进过氧化物酶体增殖物激活受体γ活性,进而调节脂肪酸储存和葡萄糖代谢。
在棕色脂肪组织中,能量消耗因胆汁酸与TGR5结合而增加,随后产生的环磷酸腺苷会增加参与能量稳态的甲状腺激素活化。
在巨噬细胞中,胆汁酸激活TGR5会导致抗炎反应,因为抑制了NF-κb通路和NLRP3依赖性炎症小体活性。FXR和TGR5受体都存在于相似的细胞中,例如胰岛β细胞和肠内分泌L细胞。
在胰岛β细胞中,正向调节合成和葡萄糖诱导的胰岛素分泌。在肠内分泌L细胞中,观察到相反的效果。FXR的激活导致GLP-1分泌的抑制,而TGR5的激活诱导GLP-1的分泌。
✦饮食会影响胆汁酸的含量
几项研究已经将特定的肠道微生物群改变以及胆汁酸成分的改变与肥胖联系起来,同时考虑到饮食的类型。
与富含精制谷物的饮食相比,富含全谷物的饮食导致血浆胆汁酸含量显著增加,包括牛磺鹅去氧胆酸、甘胆酸和牛磺石胆酸。
这被假设为激活FXR和TGR5受体并影响葡萄糖稳态。事实上,高膳食纤维的纯素饮食与Prevotella丰度较高相关被证明可以增强法尼醇X受体的信号通路
与杂食动物相比,纯素食者的粪便胆汁酸含量也显著降低。当杂食动物的饮食中膳食纤维增加时,观察到粪便胆汁酸显著减少。
//研究证明高脂饮食胆汁酸水平升高
在小鼠中,高脂饮食引起的肥胖导致粪便中脱氧胆酸水平升高。此外,高脂肪饮食略微增加总胆汁酸池,特别是增加肝脏和血浆中的脱氧胆酸和牛磺脱氧胆酸水平。
这些变化与以下菌群的丰度增加相关:
Blautia ↑↑↑
Coprococcus ↑↑↑
Intestinimonas ↑↑↑
Lactococcus ↑↑↑
Roseburia ↑↑↑
Ruminococcus ↑↑↑
另一项小鼠研究调查了胆盐水解酶对法尼醇X受体胆汁酸拮抗剂牛磺-β-鼠胆酸的影响,因为法尼醇X受体抑制会导致对肥胖的抵抗。他们发现,乳酸杆菌水平降低与BSH水平降低相关,因此与牛磺酸-β-鼠胆酸水平升高相关。
事实上,从小鼠盲肠中分离出的L.johnsonii被发现表达产生胆盐水解酶的基因,这些基因专门针对牛磺-β-鼠胆酸,提供了肠道微生物群变化与调节法尼醇X受体和胆盐水解酶基因表达之间的机制联系。
然而,与其他产生类似胆盐水解酶的肠道微生物相比,乳酸杆菌对法尼醇X受体拮抗剂浓度的贡献仍不清楚。
一项调查肥胖受试者的人体研究发现了毛螺菌科的瘤胃球菌家族与甘氨脱氧胆酸的比例和血浆中次级胆汁酸与初级胆汁酸的比例呈正相关。
除此之外,Faecalibacterium prausnitzii与粪便中的异石胆酸水平呈负相关。
一项调查肥胖受试者的研究发现,该组的非12-OH胆汁酸比例降低。在同一项研究中,高脂饮食抗肥胖小鼠的这些非12-OH胆汁酸水平升高。
在高脂饮食易肥胖的小鼠中,这些胆汁酸减少并与肠道微生物群的改变有关。在这里,梭状芽孢杆菌减少的很明显,肥胖与肠道微生物群通过胆汁酸池的大小和组成有关,但在单个细菌、特定胆汁酸剖面和肥胖表型之间还没有明确的联系。
因此,还需要进行更多的研究,以将肥胖与胆汁酸谱和胆汁酸池大小与特定细菌组成谱联系起来。
脂肪酸
除了产生胆汁酸外,一些细菌,包括Lactobacilli和Bifidobacteria,还通过多不饱和脂肪酸的饱和代谢产生代谢物。这会产生中间脂肪酸,如羟基、氧代、共轭和部分饱和反式脂肪酸。
结果表明,与无菌小鼠相比,无特定病原体小鼠的羟基脂肪酸水平要高得多,这表明肠道微生物组的脂质代谢会影响宿主体内的脂肪酸组成,因此会影响宿主的健康。
✦增强抗炎能力,促进屏障恢复
此外,共轭脂肪酸组中的一些脂肪酸对健康有益。体外对树突状细胞的实验表明,共轭亚油酸的异构体抑制脂多糖诱导的白细胞介素12产生并增强抗炎细胞因子白细胞介素10的产生。
一个例子是10-hydroxy-cis-12-octadecenoic acid(HYA),因为它部分调节肿瘤坏死因子受体2 (TNFR2),从而促进上皮屏障恢复作用。
注:HYA是不饱和脂肪酸的代谢过程中,肠道微生物产生的中间体游离脂肪酸。HYA能够改善与一些细胞中成熟标志物表达相关的抗氧化/解毒防御能力。
✦保护宿主,减少肥胖
另一项研究展示了HYA如何通过G蛋白偶联受体40(GRP40)和G蛋白偶联受体120(GRP120)分泌胰高血糖素样肽-1来减轻高脂饮食诱导的小鼠肥胖。
此外,他们还证实了几种乳酸杆菌属,如
Lactobacillus salivarius和
Lactobacillus gasseri,能够产生相似水平的 HYA,保护宿主免受高脂饮食引起的肥胖。
吲哚
吲哚是吡咯与苯并联的化合物,细菌产生吲哚对人体健康具有重要意义。
✦饮食类型影响吲哚的产生
吲哚是通过降解肠中芳香族氨基酸如酪氨酸、苯丙氨酸和色氨酸的分解代谢产生的。因此,肠道吲哚水平取决于饮食类型。
富含蛋白质的饮食会促进吲哚的产生。然而,富含糖的饮食可能会降低吲哚合成,因为过度消耗糖可能会导致小肠饱和,从而导致更多剩余的糖进入大肠。
由于碳水化合物发酵优于蛋白水解活性,因此抑制色氨酸酶活性导致吲哚合成速率降低。吲哚通过以下途径影响宿主代谢L细胞对GLP-1分泌的调节,表明在2型糖尿病等代谢疾病中发挥作用。
吲哚丙酸(3-Indolepropionic acid)由Clostridium sporogenes产生,它与膳食纤维摄入量呈正相关。
•2型糖尿病会影响吲哚水平
事实上,一项研究发现较高的血浆吲哚丙酸水平与降低患2型糖尿病的风险之间存在关联。
另一项研究发现,与瘦对照相比,患有2型糖尿病的肥胖受试者的吲哚丙酸水平降低。吲哚丙酸显示通过与孕烷X受体结合并随后下调肿瘤坏死因子α来调节炎症。
✦吲哚具有抗肥胖等特性
研究显示吲哚丙酸可降低饮食诱导的肥胖小鼠的肠道通透性。吲哚丙酸也被证明在小鼠中具有抗肥胖活性。
在肠道中,色氨酸可以被肠道菌群用作底物来产生吲哚,但也可以被宿主代谢。在低度肠道炎症(肥胖的一种慢性症状)期间,巨噬细胞中的吲哚胺2,3-双加氧酶活性增加,导致犬尿氨酸的产生水平升高,从而将生产从微生物衍生的吲哚转移。
注:吲哚胺2,3-双加氧酶是人体内色氨酸代谢中的关键酶,可通过介导色氨酸耗竭及其代谢产物调节机体抗肿瘤免疫。
与正常饮食的小鼠相比,高脂肪饮食的小鼠显示出吲哚胺2,3-双加氧酶活性增加。然而,与高脂饮食的野生型小鼠相比,在这种酶被敲低的小鼠中观察到胰岛素耐受性有所改善。
微生物衍生的吲哚,如吲哚乙酸激活芳烃受体,但犬尿氨酸抑制其激活。微生物衍生的吲哚乙酸进一步限制了巨噬细胞中脂肪酸的积累和炎症标志物的产生。
谷氨酸
除了吲哚,谷氨酸也可以影响人体。
——谷氨酸是一种多功能氨基酸,谷氨酸在生物体内的蛋白质代谢过程中占重要地位。除此之外,谷氨酸也是人体兴奋神经递质,不仅参与消化系统和免疫系统,还是大脑健康密切相关。现在强有力的证据表明肠道微生物产生神经活性分子,如神经递质(即去甲肾上腺素、多巴胺、血清素、GABA 和谷氨酸)和代谢物(即,色氨酸代谢物,短链脂肪酸等)维持宿主和细菌之间跨界跨区域交流。谷氨酸代表了在这种跨界交流中活跃的众多神经活性分子之一。
根据对肥胖和瘦受试者的队列进行的全基因组关联分析显示,谷氨酸盐具有潜在危害。
通过进行途径分析,谷氨酰胺/谷氨酸转运系统在肥胖个体中高度富集。这与拟杆菌属(包括B.thetaiotaomicron)的物种呈负相关。事实上,与瘦受试者相比,肥胖者体内这种细菌的数量减少。因此谷氨酸与人体之间也存在一定联系。
•拟杆菌的在高脂饮食中的研究
对多形拟杆菌(B.thetaiotaomicron)在高脂饮食小鼠中的作用的研究表明,编码参与脂肪生成的蛋白质的基因表达较低,而编码参与脂肪酸氧化和脂肪分解的蛋白的基因表达较高。此外,炎症相关标志物的表达也降低。
关于发现与肥胖相关的B.thetaiotaomicron,其效应可能是由于与某些其他物种的相互作用,例如B. uniformis,已知其部分恢复了高脂肪饮食诱导的肥胖效应。
03
健康与肥胖人群中的细菌比例
有研究发现,健康人群和肥胖人群中的拟杆菌门和厚壁菌门比例存在不同。但是将健康受试者与肥胖受试者用拟杆菌与厚壁菌的比例区分开来的一个有争议的话题。
•支持的证据
一项研究调查了遗传易感肥胖小鼠及其接受相同多糖饮食的正常野生型同胞的盲肠微生物群之间的差异。
在肥胖小鼠中,拟杆菌数量减少,而厚壁菌的相对丰度较高。一年后,在比较肥胖和正常时发现了类似的结果。
•反对的证据
然而,同一组在比较正常人和肥胖人双胞胎时观察到了有争议的结果。然而,此处观察到拟杆菌显著减少,与厚壁菌没有关联。
除此之外,使用16s rRNA基因的类似管道和区域重新分析前面提到的文章的数据集和其他公开可用的数据也导致了与拟杆菌与厚壁菌比率相关的矛盾结果。
鉴于人类肠道中这两个门所代表的目、科、属的物种众多,这些门水平上相互矛盾的肠道微生物群结果并不令人惊讶。
另一方面,厚壁菌门是如此广泛,以至于说某个菌属于厚壁菌门,但是不同菌的功能差别很大。
此外,这些门中分类上不同的细菌具有截然不同的属性。拟杆菌门中最重要的例子是普氏杆菌属和拟杆菌属,它们往往相互排斥。当比较多个研究时,将每个门的细菌汇集在一起时,预计会出现相互矛盾的结果。
因此,目前还不鼓励使用拟杆菌与厚壁菌的比例来区分健康人群与肥胖人群。我们在检测实践中也发现部分肥胖人群拟杆菌比例较高。
Prevotella与Bacteroides的比例
在引入肠型后,在拟杆菌门内做出了更合适的区分,即Prevotella和Bacteroides的比率。
与Bacteroides相比,Prevotella个体在食用左旋肉碱时血浆氧化三甲胺浓度较高。
以Prevotella为主的肠道微生物群往往与素食主义或非工业化的富含膳食纤维的饮食有关。这些例子可以在非洲、南美洲或者东南亚狩猎采集者或农村人口进行的几项研究中找到。
✦Prevotella与Bacteroides更利于减肥
研究很好地说明了饮食和环境导致的从普氏杆菌向更为拟杆菌主导的肠道微生物群的转变,来自泰国农村的人移民到了美国。不出所料,这种转变也伴随着体重的增加。
关于减肥方案,这一比例很重要,因为普氏杆菌与拟杆菌比例较高的受试者在膳食纤维含量较高的情况下更容易减肥。
研究发现,给予辣椒素时,拟杆菌量较多的受试者体重减轻更多,在此强调了个性化营养的必要性。
04
肥胖与肠道微生物的研究分类
为了更好更有条理地研究肥胖与肠道微生物之间的关系,需要将微生物进行研究分类。
大多数关于肥胖与肠道微生物群之间关系的研究通常将个体分类群与病理生理途径联系起来,以建立与肥胖的联系。
影响微生物的因素
细菌并不存在于真空中,所以它们的生长速度以及它们能够进行的代谢活动取决于外部环境因素。
这些外部因素包括pH、胆汁酸和底物可用性。所有这些反过来又取决于微生物组分本身;这意味着一种细菌的功能受其周围所有其他细菌的影响。
更直接地说,各种细菌种类依赖于其他细菌种类为它们提供中间底物(其他细菌的废物),并且反过来,依赖于将消耗其自身废物(发酵产物)的其他细菌,以使其从中获得能量的生化转化在能量上有利。
同一物种的不同菌株可能存在很大差异
通常使用不同的分类水平(门/科/属/种)来归因特定的特征和关联,而物种的功能甚至在同一属内,甚至是目前被认为属于不同菌株的细菌。相同的物种,可以有很大的不同。
因此,旨在通过查看更高的分类级别来限制分类组数量的降维策略通常应该优选地限制在类属级别。
同一物种的不同菌株可能具有也可能不具有归因于它们的特定功能,正如在碳水化合物活性酶中观察到的那样。如果高度相似的基因存在于多种细菌中,则可能还会出现冲突模式。
越来越多的研究人员在过去几十年中得出结论,与肥胖相关的有益影响应归因于肠道微生物群中的多个参与者协同工作。而这种关联的紊乱可以被视为生态失调的一种形式。
微生物成员分组
——由于上述个体分类群分析的缺陷使得难以找到特定于健康结果的具有生物学意义的模式,因此创造了两个不同的术语来将个体微生物组成员分组。
▸ 微生物“聚类”
应用了“guild”这个术语,这在宏观生态学中已经众所周知。它包括“以类似方式利用同一类环境资源的一组物种”,后来成为“功能组”的同义词。
通过构建基于微生物丰度协变的共丰度组,给出了一个框架,以更生态有意义的方式解开肠道微生物组与人类健康之间的关系。这将克服目前对基于分类单元的分析和以基因为中心的分析存在问题的各种缺点。
▸ 营养网络
另一个术语称为“营养网络”,营养网络被定义为微生物种群形成代谢相互依赖的生物体的食物网,随着时间的推移以相关的方式稳定地建立。
小结
通过观察微生物聚类或特定的营养网络,可以实现对与健康和肥胖相关的肠道生态学的更有意义的解释。
此外,将数百个分类群聚集到有限数量的微生物聚类或营养网络中将有助于降低维度,从而有可能应用经典统计数据来限制与校正多重测试相关的问题。
尽管基于微生物聚类的方法似乎是一种有前途的方法,在了解肥胖儿童的体重调节方面观察到了附加价值,但与肥胖本身的相关性仍有待阐明。
05
微生物多样性与人体健康有关
α多样性与疾病状态有关
——在区分肥胖受试者和健康受试者时,一个常见的观察结果是他们平均较低的α-多样性。
在许多其他疾病中也观察到相同的情况,例如克罗恩病、肠易激综合征和结肠直肠癌。因此,微生物多样性的丧失通常与各种疾病状态有关。可以说,断奶后肠道α多样性降低是与各种人类状况相关的普遍特征。
在成年人中,较高丰度的细菌(如Akkermansia muciniphila和F. prausnitzii)通常与较高的α多样性相关。
丰富的A. muciniphila与BMI、炎症标志物、脂质合成和总脂肪组织重量呈负相关。
▸ α多样性是什么?
α多样性主要关注局域均匀生境下的物种数目,因此也被称为生境内的多样性。α-多样性是由扩散、局部多样化、环境选择和生态漂移共同形成的。
多样性本身不仅仅是健康的指标,因为多种高丰度的病原体持续存在一般不会让肠道感觉 “幸福”。
相反,更高的α多样性应该被视为存在发育良好和扩展的微生物营养网络,它们共同导致发酵能力的提高。
✦低α多样性下的肠道微生物
富含拟杆菌的微生物群倾向于具有较低的α-多样性值、较简单的营养网络,并且更容易下降。
这种低α-多样性组合物通常富含诸如肠杆菌科、梭杆菌属、链球菌属、瘤胃球菌属和各种拟杆菌属物种的物种。
这种益生菌组合物在肠型方面与拟杆菌2肠型最为相似,最终会是肥胖和2型糖尿病的危险因素。
营养网络被破坏导致α多样性减低
研究表明营养网络的彻底破坏以及由此导致的α-多样性、基因丰富度和肠道发酵能力的极大降低。
调查了(抗生素治疗)危重儿童的肠道微生物群、粪便短链脂肪酸和胆汁酸谱。由于缺乏代谢和发酵能力,这些儿童的初级胆汁酸与次级胆汁酸的比例较高,但短链脂肪酸的产量极低,而碳水化合物发酵的中间产物,如乳酸盐和琥珀酸盐与健康对照儿童相比含量增加。
后一项发现,加上剩余的未发酵糖组分、较高水平的未接触蛋白质和更松散的粪便,突出了肠道中剩余的发酵仍然处于糖分解阶段。
Christensenellaceae营养网络
——一个与高α-多样性和健康相关的特定营养网络
与肥胖受试者相比,体重指数正常的健康受试者的Christensenellaceae水平更高。
Christensenellaceae和寄主BMI之间的关联被认为是最稳健的关联之一。在无菌小鼠体内移植来自人类供体的富含菊苣科植物的粪便可减少肥胖。在富含瘤胃球菌科或厚壁菌的肠型的人中,Christensenellaceae通常很丰富。
如上所述,不应将Christensenellaceae视为一个独特的独立实体,因为它始终与其他细菌和古细菌形成营养网络。
✦Christensenellaceae与古细菌的关联
Christensenellaceae与一种古细菌——Methanobrevibacter smithii 的关联可能是这一营养网络最典型的部分。
M.smithii 从微小梭菌产生的氢气中产生甲烷。如果这种营养网络与低BMI之间存在因果关系,则仍然相当不确定。
除了M. smithii是这一营养网络的一部分外,一项比较意大利瘦弱和肥胖老年人的研究发现,Christensenellaceae、Rikenellaceae和Porphyromonadaceae之间存在相关性。
在日本的一个队列中,调查了不同地区健康成年人的粪便样本,Christensenellaceae与各种其他细菌也与BMI呈负相关。
注意
鉴于α-多样性、瘦弱性和Christensenellaceae细菌营养网络之间的紧密联系,未来将继续从机制上研究这种联系。还应注意的是,该营养网络对于短链脂肪酸生产的重要性尚未确定。
虽然Christensenellaceae和Methanobrevibacter可能仅占总微生物群的一小部分,但它们所代表的核心指示物种的营养网络在不同种族中绝不是一个小角色。这种营养网络,其中各种物种彼此之间非常密切相关,具有肠型定义潜力。
Prevotella stercorea营养网络
另一个营养网络,通常在工业化国家的人们中代表性不足,是Prevotella stercorea营养网络,它可以被视为Prevotella肠型组成中的一个重要因素。
这个营养网络的建立首先是通过观察冈比亚儿童正在发育的肠道微生物群来广泛描述的。P. stercorea与Succinivibrio dextinosolvens和Paraprevotella xylaniphila等形成一个大型营养网络,并且类似地与高α-多样性相关。
✦肠道Prevotella的特征
肠道普雷沃氏菌是一个完美的例子来展示微生物“聚类”和营养网络之间的区别。
在人群范围内的研究中,例如使用多民族队列研究的数据,被定义为肠型普氏杆菌的人通常具有非常高的P. stercorea水平和与P.stercorea营养网络相关的高水平物种。
当在分层聚集的热图中可视化时,P.copri和P.stercorea营养网络中的物种聚集在一起。然而,这种共同发生主要是由于粪便中的Prevotella(包括P.copri、P.stercorea和其他许多普氏杆菌属)和Bacteroides/Phocaeicola.之间的强烈拮抗作用。
P.copri和P.stercorea营养网络在同一环境中表现良好(Bacteroides贫乏),但P.copri的高丰度完全独立于P.stercorea营养网络发展,这可以通过跟踪儿童在前3个年的肠道微生物群成熟情况看出多年生活在一个每个人都会发展出富含Prevotella的肠道微生物群的环境中。
12个月后,P.copri成为优势种并保持优势,而与P.stercorea营养网络相关的物种丰度在生命的前30个月以相互依赖的方式缓慢增加,直到达到稳定水平。推测在P. stercorea的营养网络中存在着各种代谢产物的交换,值得进一步研究,特别是与Prevotella肠型生产短链脂肪酸的能力增加有关。
✦Prevotella与健康相关
与肥胖率上升最快的工业化国家相比,肠型拟杆菌相关的拟杆菌和种类在冈比亚并不多见。
肠道中的Prevotella本身也与较低的BMI相关,并且已观察到低密度脂蛋白胆固醇与肠道Prevotella呈负相关,这表明在非工业化国家,肠道Prevotella与健康有关。
06
肥胖与肠道微生物的未来研究方向
尽管使用大型队列的关联研究对于试图解开与肥胖相关的肠道微生物组的极端复杂性至关重要,但其他几种研究途径也具有潜力,其中一种是粪菌移植。
粪菌移植
▸ 定义
粪菌移植,是将粪便从瘦供体转移到受体。也称为“人类肠道微生物群转移”、“粪便移植”和“粪便细菌疗法”。
✦粪菌移植的作用
粪菌移植已被证明是比抗生素更有效的复发性艰难梭菌感染 (CDI) 治疗方法。然而,与肥胖不同,从病理学的角度来看,CDI是一种相对简单的疾病,其中肠道微生物群的因果关系是明确的。
在一项对患有胰岛素抵抗的肥胖受试者进行的粪菌移植试验中。受试者接受自己的粪便(自体)或瘦供者粪便(同种异体)。短期内在接受瘦供体粪菌移植的受试者中观察到对胰岛素敏感性的有益影响。
进一步研究表明基线肠道菌群有利于粪菌移植的成功。在这里,当接受同种异体粪菌移植时,在α-多样性降低的受试者中,粪菌移植成功率更高。
总的来说,与那些肠道微生物组组成尚未严重恶化的受试者相比,那些α-多样性较低的受试者有更大的改进空间。
✦其他影响粪菌移植的因素
一项研究,其中包括几个调查不同疾病的粪菌移植队列,显示生态变量(如低α-多样性)与临床变量(如抗生素治疗和灌洗)一起在植入成功中发挥作用。
他们进一步表明,通过合并供体样本来增加α-多样性预计不会增加供体菌株的植入,这表明合并供体样本在功能上并不等同于单个高α-多样性供体样本。
对队列进行的分析表明,P.copri对接受同种异体粪菌移植的受试者具有有益的影响。P.copri与BMI、C反应蛋白和空腹胰岛素水平进一步呈负相关。
此外,肠道微生物群的变化可能与特定血浆代谢物水平和血浆单核细胞中DNA甲基化的变化有关,为肠道微生物群影响肥胖相关疾病的机制提供了额外线索。
验证细菌植入的生物学工具
最近开发了几种工具来帮助解开粪菌移植中肠道微生物组与肥胖之间的关系。
为了验证来自瘦供体的菌株是否已移植到受体中,需要进行菌株跟踪分析。比较了七种不同的生物信息学工具,用于在数据集上进行应变跟踪。
减轻肥胖和相关疾病负担有前景的方法
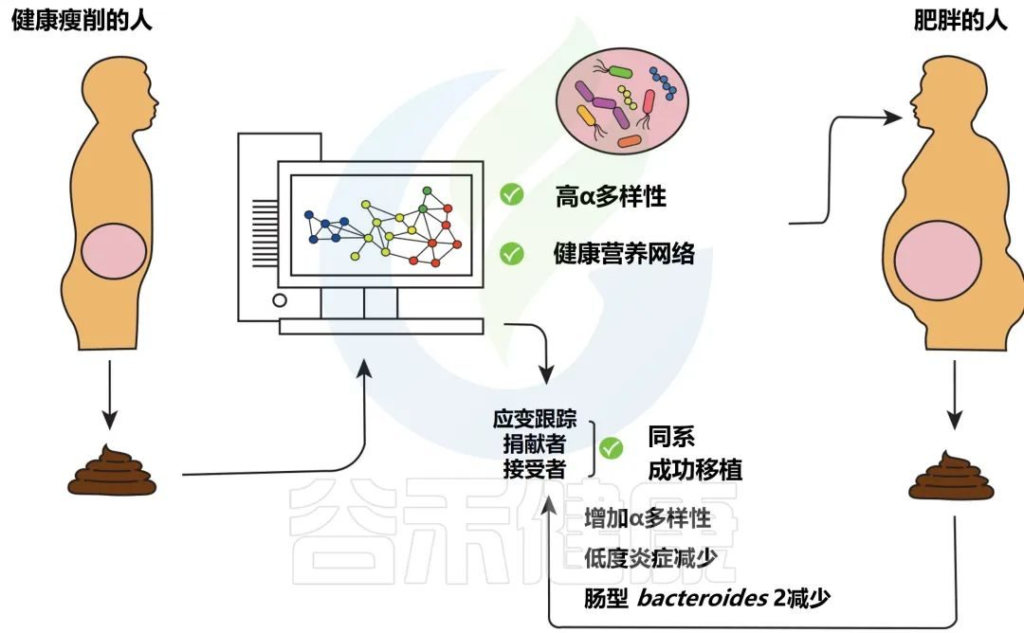
Levin E,et al.Therap Adv Gastroenterol.2022
分析健康瘦供体粪便的微生物组成,以选择具有高 α 多样性(以及其他)的供体,这可以被视为存在复杂的健康相关营养网络的标志。
如果合适,然后将高α多样性供体的粪便转移到肥胖的接受者身上,这可能会减轻低度炎症。在粪菌移植之后,使用菌株追踪在接受者的粪便中追踪肠道微生物群基因组中特定位置的特定SNP的供体菌株验证。
在这里,观察到概率工具在宏基因组测序数据上表现最好。然而,随着最近开发的两种新的应变跟踪工具,这一技术领域仍在快速发展。
其中一个工具是基于物种特异性标记基因中的单核苷酸变体跟踪菌株,另一个是先前发布和改进的进一步构建工具,应用应变跟踪方法。
在接受粪菌移植后调查了受体中的菌株植入,观察到供体和受体特异性菌株可以共存。与此同时,发现肥胖受试者的粪菌移植胶囊会导致微生物群落组成发生变化,从而导致受试者从一种肠型转变为另一种肠型。这随后改变了菌群的代谢潜力。微生物组向供体的转变与α多样性呈正相关。
此外,肠道微生物群组成的变化在治疗后持续26周。本研究结合了多个供体的粪便,并表明一些供体具有用于移植的高效微生物群,这意味着供体粪便的组成和整个营养网络的转移,而不是添加单个分类群的重要作用。
07
预防和治疗肥胖的建议
预防肥胖
——鉴于肥胖症如此普遍,并且考虑到治疗的难度,预防尤为重要。
为预防超重和肥胖,人们应该根据自己的营养需求进食和饮水,定期锻炼,定期检查体重。
•少吃高热量食物
就营养而言,他们应该少吃高能量密度的食物,多吃低能量密度的食物。由于水分或纤维含量高而能量密度低的食物,如全麦制品、水果和蔬菜,相对来说更能饱腹,能量含量也较低。地中海饮食有助于预防超重和肥胖。
还应减少酒精、快餐和含糖饮料的消费。快餐通常含有高比例的脂肪和糖,因此能量很高。不仅是加糖的饮料,还有果汁和果汁饮料,含糖量也很高。
•避免久坐或不活动
经常坐着看电视或上网和类似活动的不活跃生活方式会促进体重增加。在日常活动和休闲活动中进行锻炼具有预防作用。这个目标最好通过每周2小时以上的以耐力为重点的体育锻炼(使用大肌肉群)来实现。
肥胖的治疗方法
✦饮食疗法
为了减轻体重,目标应该是遵循减量饮食,这将产生约500kcal/天的热量缺口,或在个别情况下更多。
每天500至600kcal的能量缺口将使体重减轻,以约0.5kg/周的速度发生,持续12周最多24周。
低碳水化合物饮食在开始时会比其他饮食导致更剧烈的体重减轻,但一年后就看不到差异了。过去几年的几项大型研究表明,常量营养素组成(脂肪、碳水化合物和蛋白质的比例)与减肥无关。各种减脂饮食可在1至2年内减掉约4公斤。个人经验、知识和资源比营养关系更重要。
✦益生菌帮助减肥
已经证明几种益生菌,单独使用或以共生混合物的形式使用,能够通过物种和菌株特异性机制(例如,肠道微生物群调节、降低胰岛素抵抗、更强的饱腹感)来治疗肥胖。
更具体地说,乳酸杆菌和双歧杆菌物种由于其低致病性和低水平的抗生素耐药性而已成功用于成熟的肥胖动物模型。
益生菌对减肥作用的一些实验
Abenavoli L, et al. Nutrients.2019
与安慰剂组相比,这些治疗导致不同程度的体重增加减少和脂肪累积减少。
所以在一些时候,我们可以利用例如乳酸杆菌等益生菌来帮助我们减肥。
✦增加运动
有效的减肥需要>150分钟/周的运动,能量消耗率为1200至1800kcal/周。单独的力量训练对于减轻体重作用不大。
运动中消耗的能量常常被高估。当使用大肌肉群,强度适中到高,运动时间长时,体重减轻是可以预期的。对照良好的研究和荟萃分析显示,在6至12个月内体重减轻了约2公斤,腹部脂肪减少了约6%.
应该向超重和肥胖的人解释运动的健康益处(代谢、心血管和社会心理),无论体重减轻如何,这些益处都会产生。即使在肥胖个体中,增加运动的健康价值也不仅仅体现在体重减轻上。
✦行为矫正干预
在团体或个人中,基于行为方法的干预应成为减重计划的一部分。
干预的主要目的是改变营养和运动方面的生活方式,并且可以由合格的非心理治疗师进行。如果伴随超重或肥胖的症状更严重,精神科医生或心理治疗师应参与患者管理,并应支持患者进行饮食治疗和锻炼。
08
结语
肥胖和肠道微生物群以多种方式交织在一起。饮食的类型及其数量会影响能量的可用性并因此影响肥胖,但也会强烈影响肠道微生物组,这反过来又可以放大饮食的致肥胖特性,或另一方面提供各种保护性益处。
许多微生物衍生的代谢物,包括短链脂肪酸、胆汁酸、吲哚和其他氨基酸,对健康同样至关重要。过量或缺乏这些,或者更具体地说,在任何这些方式中改变的整体组成,都可能是致肥胖的。
通过本文更好地了解肥胖以及其发病机制与微生物组之间的关系,有助于在日后的生活中更好地应对肥胖,使人人都有一个健康的身体。
主要参考文献
van der Vossen EWJ, de Goffau MC, Levin E, Nieuwdorp M. Recent insights into the role of microbiome in the pathogenesis of obesity. Therap Adv Gastroenterol. 2022 Aug 9;15:17562848221115320. doi: 10.1177/17562848221115320. PMID: 35967920; PMCID: PMC9373125.
Canfora, EE, Meex, RCR, Venema, K, et al. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat Rev Endocrinol 2019; 15: 261–273.
Abenavoli L, Scarpellini E, Colica C, Boccuto L, Salehi B, Sharifi-Rad J, Aiello V, Romano B, De Lorenzo A, Izzo AA, Capasso R. Gut Microbiota and Obesity: A Role for Probiotics. Nutrients. 2019 Nov 7;11(11):2690. doi: 10.3390/nu11112690. PMID: 31703257; PMCID: PMC6893459.
GBD 2015 Obesity Collaborators . Health effects of overweight and obesity in 195 countries over 25 Years. N Engl J Med 2017; 377: 13–27.
Chauhan, S, Jena, KK, Mehto, S, et al. Innate immunity and inflammophagy: balancing the defence and immune homeostasis. FEBS J. Epub ahead of print 26 November 2021.
Beukema, M, Faas, MM, de Vos, P. The effects of different dietary fiber pectin structures on the gastrointestinal immune barrier: impact via gut microbiota and direct effects on immune cells. Exp Mol Med 2020; 52(9): 1364–1376.

谷禾健康

每个人的一生都会经历很多,从出生到长大,健康到衰老疾病。你的出生、遗传、家庭环境、很大程度上决定的人生起点,日常的饮食、行为习惯决定你的身体成长,一些不同的选择或意外的事件又会让人生有很多起伏和不同。
每个人的菌群和我们的人生一样也是独一无二的,我们菌群的特点反映着不同人各自生活的烙印。从母亲的腹中开始影响和决定了我们最初的菌群,出生方式、喂养的食物、用药等都决定了我们的菌群基数。当我们开始从喝奶到开始摄入辅食,我们的菌群也同样迎来巨大的演变。当我们生病、感染、运动、饮食、社交、虚弱、衰老这些同样反映在我们菌群的变化和演替上。
相对的,当我们更多的了解我们的菌群,善待和改善它们,同样的变化也会出现在我们的身体和生活中。
越来越多的证据表明,年龄与人类微生物群之间的关联很大,肠道微生物群是许多年龄相关变化的核心,包括免疫系统失调和疾病易感性。几个身体部位的微生物组成可以相对准确地预测人类的年龄。
谷禾健康肠道菌群检测数据库中,也有关于肠道年龄预测:
谷禾健康-肠道年龄预测模型图
<来源:谷禾健康肠道菌群数据库>
可以看到,肠道年龄和生理年龄基本是符合的。健康人的肠道菌群年龄恰恰是最符合真实年龄的,与真实年龄差异大意味着肠道菌群出现偏离。
健康的人存在更多样化且平衡的肠道菌群。微生物群中与年龄相关的变化归因于生理,生活方式和健康状况。这些因素中的每一个都与某些菌群的相对丰度变化有关。
例如,饮食、卫生、兄弟姐妹、宠物、过敏、儿童疾病和抗生素是影响儿童微生物组的一些突出因素。到了成年期微生物群相对稳定,而到了老年期,一些有益菌开始逐渐下降,菌群又向另一个阶段过渡。
在从出生到死亡和分解的每个生命阶段,微生物群落都是身体的动态组成部分。研究微生物群的自然和诱导变化有可能彻底改变我们对人类生物学的理解。
本文介绍了健康人的微生物群在一生中的变化,讨论了从出生时菌群构成,到疾病或抗生素使用时的变化,再到死亡时的微生物扩展的各个阶段,以及这些阶段在身体部位和组成(细菌、真菌或病毒)上的差异。了解微生物群与年龄关系的未来研究方向,以此对人体微生物群及基于此的干预有更好的了解。
微生物群落存在于人体的每个粘膜表面,人的每个身体部位都有一个独特的生态学。每个人的微生物群像指纹一样,都是独特的。
在个体内,特定的身体部位、地理位置和个体的年龄与健康微生物群具有极强的关系。年龄驱动人类微生物群的α多样性和β多样性。
在了解各个阶段的微生物群变化之前,我们先了解一个概念:微生物演替。
微生物演替是指微生物群落中一种或多种生物的存在、相对丰度或绝对丰度的变化。
在正常或健康衰老期间,微生物演替的三个主要阶段自然发生在人类生活中。
✦初级演替(出生时先锋菌群定植,快速变化直到童年晚期)
第一阶段,初级演替,从先锋物种首次建立群落时开始,随后微生物群落发生快速变化。从出生到童年,变化率降低,许多中间物种存在于出生到童年晚期之间。
初级演替结束于顶级群落的形成,在青春期实现,并在很大程度上持续到成年;该群落的特征是其相对稳定。
虽然成年期的微生物群比儿童期更稳定,但仍然存在变异,这引发了关于人类微生物群中是否存在顶级群落的争论。成年微生物群的自然变异存在于小时(昼夜节律)到年(老化)的时间尺度上,但微生物群相对稳定,除非存在干扰,如饮食或药物的改变。
✦次生演替(菌群的改变,重建)
下一个阶段,即次生演替,发生在一个先前存在的稳定群落一部分被改变或移除之后,然后群落再生到相同的状态或不同的状态。这可以通过抗生素等医疗手段人为实现,也可以通过霍乱弧菌感染等疾病自发实现。
人类的次生演替的特征是至少有一段时间的随机过程占主导地位。在诱导条件下,如单疗程抗生素,群落遵循类似于初级演替的过程,其中现有微生物群落的一部分充当“微生物记忆”,帮助重建一个类似于以前存在的群落。
这一过程被认为是由核心微生物群驱动的,而不是驱动初级演替的先锋微生物。
✦末期演替(自然衰老和死亡阶段)
最终的末期演替是宿主自然衰老和死亡的一部分。在老年期间,微生物群落再次以更高的变化率,成功产生了一个由更少成员组成的群落,通常变形菌门(也称为假单胞菌)的相对丰度增加,有时占总优势。
研究演替的每个阶段使研究人员能够解决与人类相关的微生物群落是如何形成和维持的。通过了解这些过程,我们可以更好地了解微生物群随着年龄的增长的变化及其与人类健康的关系,了解如何管理微生物群。
人类相关微生物群从受孕到死亡的变化
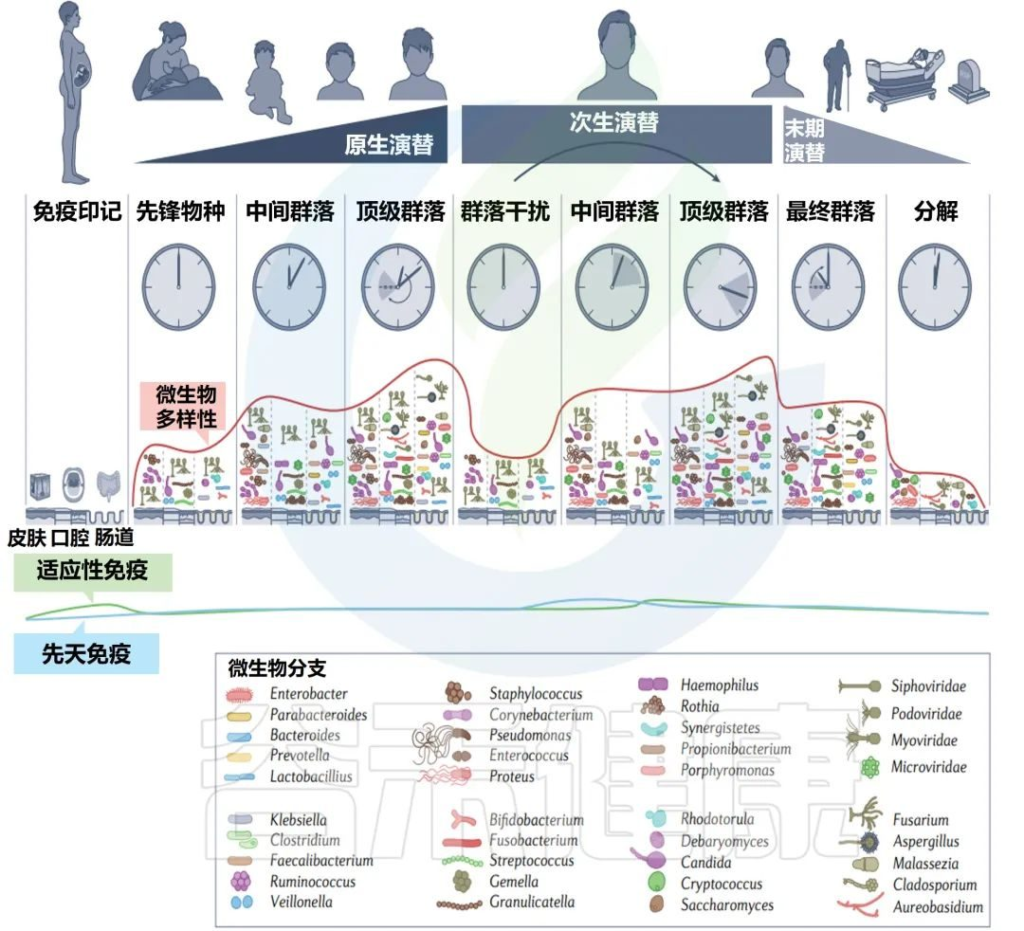
Martino C,et al.Nat Rev Microbiol.2022
常驻细菌、真菌和病毒的多样性在人类生命的各个阶段都会发生变化。模拟时钟代表每个微生物群落阶段发育的宿主年龄的相对时间。
免疫印记在出生前通过母亲的微生物群及其代谢物开始(第一栏)。先锋物种的初始定殖始于出生,身体部位特定的微生物群落出现(第二栏)。这些群落的复杂性不断增加,直到它们达到相对稳定的群落结构(第三列和第四列)。
这些微生物群落的次生演替可能来自内部和外部扰动(第五栏)。中间微生物重新建立初始群落,并再次达到稳定状态(第六列和第七列)。
在晚年,随着寄主接近自然死亡,群落经历了最后的演替和变化(第八栏)。微生物演替的最后阶段发生在腐败和分解阶段。在此阶段,多样性进一步下降,在最初的24-48小时内,许多人类微生物群结构保持不变,但随后很快开始侵蚀分解(第九栏)。
绿线和蓝线分别显示了微生物演替不同阶段的适应性免疫和先天免疫的相对强度。
不同年龄段的细菌多样性测量
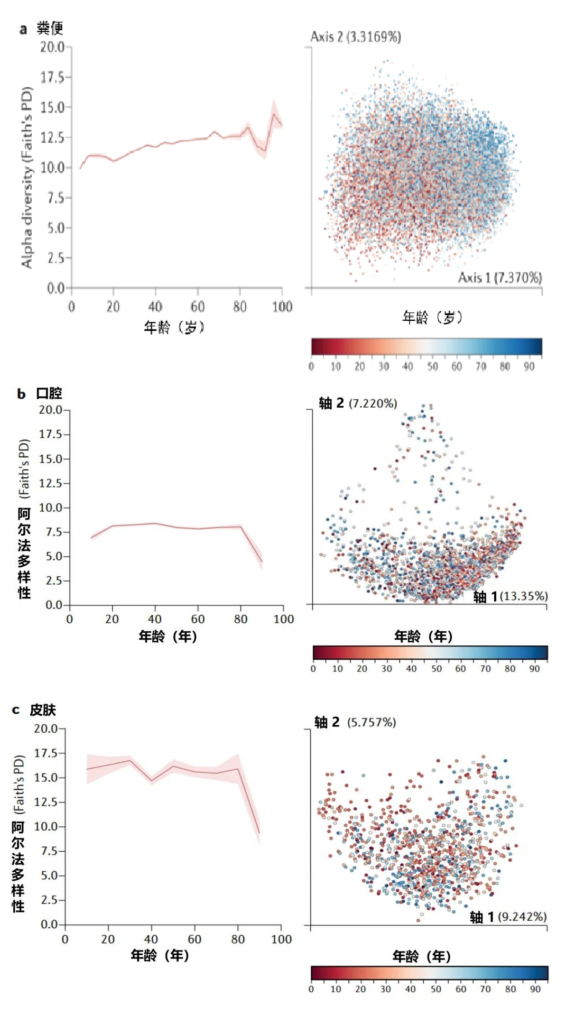
Martino C,et al.Nat Rev Microbiol.2022
美国一个肠道项目集中测量了从儿童到老年的人类粪便(a部分)、口腔(b部分)和皮肤(c部分)微生物群的细菌多样性和系统发育史,该项目包含21919个粪便、1920个口腔和998个皮肤微生物群样本,带有16S核糖体RNA基因扩增子序列。
α多样性,一种对样本中不同类型微生物数量的定量测量,通过Faith的系统发育多样性(PD)α多样性度量跨年龄测量。
UniFrac β多样性主坐标分析,一种用于比较微生物群落相似性的方法,其中空间上接近的点表示相似的样本,空间上远离的点表示不同的样本,按年龄着色。
✦胎儿时期——菌群及代谢物影响免疫发育
塑造人类微生物群的第一个因素来自胎儿发育过程中的母亲。
胎儿通过胎盘接触到母亲微生物群落产生的代谢物,这些代谢物会影响其免疫系统,并会影响正常微生物群和后期病理学的各个方面。代谢物,如短链脂肪酸(乙酸盐)和其他微生物化合物,可以通过胎盘转移到胎儿体内,并影响免疫发育。母亲的饮食和健康也会影响这些代谢物。
胎儿组织中的乙酸盐影响与成人调节性T细胞生成相关的表观遗传印记,其与防止生命后期哮喘的发展相关。
✦出生后——菌群受出生模式,饮食,环境等影响
出生后,微生物群落根据身体部位迅速分化。
在最初的时候,先锋物种和未来4年的群落发展可能会受到出生模式和妊娠时间的影响。中间群落由饮食影响,如母乳或配方奶粉的消费,以及环境。
最后,饮食和环境再次塑造了稳定的顶级群落。主要由真菌、细菌和病毒组成。
子宫内和生命早期的主要演替
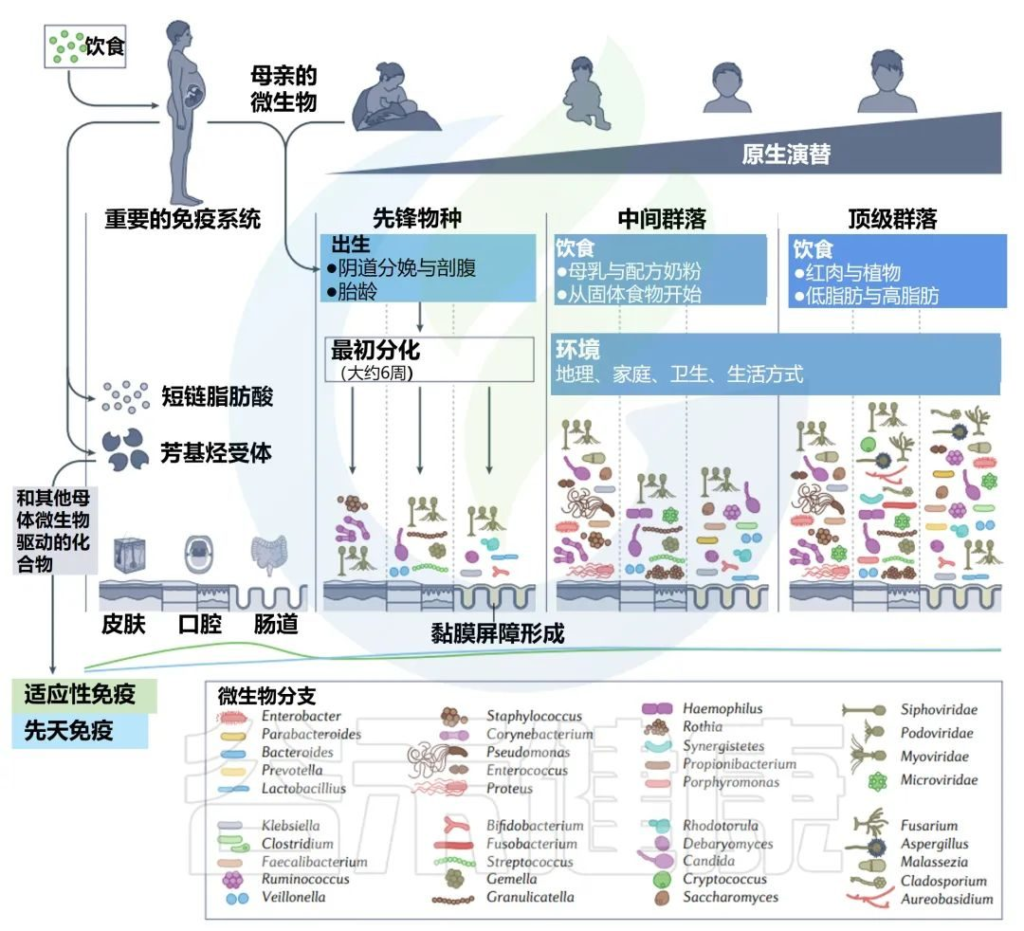
微生物代谢物和配体调节宿主芳基烃受体,这有助于塑造新生儿微生物和免疫发育。母亲使用抗生素和胃肠道相关疾病,如炎症性肠病,也被认为会通过胎儿免疫系统的印记增加后代的病理风险。
然而,这些联系仅在非人类实验中研究过。在一个案例中,由患有炎症性肠病的孕妇或其新生儿的微生物群所定殖的无菌小鼠继续发展出异常微生物群和指示炎症性肠病的免疫发育。
✦怀孕期间母体的微生物群与免疫系统的变化
在怀孕期间,母亲的微生物群和免疫系统也发生了改变。母亲的阴道微生物群变得更加多样化,通常由在其他身体部位发现的许多微生物群组成。
孕期母体免疫系统与胎儿形成协同作用,包括通过胎盘转移IgG抗体。
关于出生时获得的微生物群是否通过混合来源于阴道和粪便,或者阴道微生物群本身在出生时是否具有多能性,是否是微生物先驱的主要来源,存在一些争议。
无论确切的母体来源如何,这一阶段的特征是先锋细菌种类。包括下列菌群:
然后这些细菌定居在常规身体部位:肠道、口腔和皮肤。
许多先锋细菌是兼性厌氧菌,它们会消耗氧气,从而使专性厌氧菌能够在以后的每个环境中定居。起初,新生儿的每个身体部位都相对未分化,但先锋微生物很快开始启动身体部位依赖性微生物多样性的级联,至少在生命的第4到第6周,每个部位的细菌都可以很容易地区分。
先锋细菌进驻后,生命早期的微生物群逐渐开始形成。接下来的章节我们来了解生命早期的肠道,口腔,皮肤等各部位的微生物群(包括细菌、真菌、病毒等)。
✦肠道细菌群——双歧杆菌主导
人类肠道细菌群落的发展已经得到了很好的研究。
双歧杆菌属一直占主导地位,直到在生命的第一年结束时,它们被双歧杆菌、梭状芽孢杆菌和拟杆菌属的组合所取代。拟杆菌属的丰度增加,而双歧杆菌属等物种的丰度相对减少。
双歧杆菌分解母乳低聚糖,开始终生影响免疫系统
最近,一项研究发现,双歧杆菌等细菌含有母乳低聚糖分解代谢所需的基因,与婴儿免疫发育之间存在功能联系。特别是,接受Bifidobacterium infantis EVC001极化初始T细胞的婴儿的粪便水与来自对照组的粪便水平不同,其方式与减少肠道炎症有关。
其他菌属也可降解母乳低聚糖(如拟杆菌、阿克曼菌)
到3-6岁时,肠道细菌群落汇聚到整个成年期持续的顶级群落。这一微生物群是已知的密度最大、多样性最强的生态群落之一。通常,在这段时间内,普通健康人中只有两个细菌门占优势:厚壁菌门和拟杆菌门。
✦肠道其他微生物群——真菌、古细菌、病毒
在人类肠道发育过程中,对病毒组、真菌组和古菌组的研究远远少于细菌组。在整个生命周期中,真菌群落所占的总数远远少于细菌组或病毒组。
//真菌群落
真菌群落在生命的最初几天含有大量的Rhodotorula和Debaryomyces,接下来的一个月则是CandidaCryptococcus和Saccharomyces spp.。
到成年时,主要的真菌属是Aspergillus, Candida和Saccharomyces。
//古细菌群落
发育期间肠道的古细菌群落尚不清楚,但古细菌是一些最早的移生菌落,但丰度较低。
早期定植的古细菌包括Methanosphaera和Methanobrevibacter。
// 病毒群落:噬菌体家族在出生后就开始流行
主要由噬菌体组成的病毒群落在出生后的第一周数量众多。噬菌体家族Siphoviridae、Podoviridae和Myoviridae在出生后立即流行,主要以溶原形式整合到细菌基因组中。
到生命的第四个月,有尾噬菌体目大量生长,成员更常为裂解型(传染性噬菌体颗粒或主动复制的噬菌体)。
在成人中,Caudovirales和Microviridae在肠道噬菌体群落中占主导地位,但噬菌体肠道病毒组对个体具有高度特异性,其演替仍有许多未知之处。
与噬菌体不同,感染真核病毒的肠道病毒组主要与儿童和成人的病理相关。最近,在健康儿童和健康成人中也观察到一些感染真核细胞的病毒丰度较低,但其发生时间和流行率尚不清楚。
✦口腔细菌群:出生后几个月逐渐趋于稳定,牙齿形成后再次转变
在出生时,口腔细菌群在以下菌属中的流行率很高:
在接下来的几个月里,Lactobacillus和Fusobacterium也开始流行。Staphylococcus的丰度在出生后3个月左右达到峰值,然后稳步下降,让位与更高丰度的GemellaGranulicatella, Haemophilus和Rothia spp.
牙齿形成后,口腔微生物群再次转变,在成年期具有更高丰度的梭杆菌门, Synergistetes, Tenericutes, Saccharibacteria (TM7), SR1 。
✦口腔其他微生物:成年口腔含产甲烷菌,最常见的噬菌体群是尾状病毒
口腔真菌群落被认为比皮肤和内脏的真菌多样性少。Candida spp.是口腔的第一批真菌定植菌。对中级口腔真菌群落知之甚少,但成年人CandidaCladosporiumAureobasidium
AspergillusFusarium和Cryptococcus spp.的丰度较高。
发育过程中的口腔古菌体尚不清楚,但成年口腔中含有许多古菌产甲烷菌,包括甲烷杆菌属。
目前对人类婴儿口腔中病毒的知之甚少。在成年人中,与肠道类似,最常见的噬菌体群是尾状病毒。
口腔病毒群在本质上通常被视为病理性的(例如柯萨奇A病毒、麻疹病毒、红疹病毒和人乳头瘤病毒),并且没有对病毒群落组成进行纵向研究。然而,在无症状和健康成人中也观察到许多真核病毒分类群。
✦皮肤细菌群落:出生时母亲阴道乳杆菌属占据较多,4-5周与成人相似
皮肤细菌群落在出生时含有大量的母亲阴道乳杆菌属。到第4-5周,婴儿皮肤微生物群与成人皮肤微生物群相似,但在青春期继续变得更具位点特异性。
Staphylococcus和Corynebacterium在不同位点PseudomonasEnterobacterEnterococcus,
Proteus和Klebsiella在特定位点(如腋窝与前臂)。
✦皮肤其他微生物:马拉色菌占比较高,古细菌占4%左右
在皮肤真菌群落中,MalasseziaCandida和 Saccharomyces在生命的前30天最为普遍。对于中间群落的确切组成知之甚少,但成年真菌群落中Malassezia的丰度通常很高,估计约占真菌群落总组成的75%至90%。
关于皮肤古细菌群落的发育情况了解较少,但古细菌约占成年人菌群的4%。大体上,成年人皮肤古细菌群由Thaumarchaeota门和Euryarchaeota门代表。在成人皮肤上也发现了Halobacteriaceae和 Methanobrevibacter。
与肠道和口腔不同,健康的皮肤微生物群拥有相对较少的已知病毒多样性,很少有对其进行研究,可能是由于与低生物量样本相关的技术限制。不过,皮肤上有一些自然存在的病毒群。
以上了解关于生命早期肠道、口腔、皮肤的微生物群,那么哪些因素会给生命早期的微生物群发展带来影响?
在生命的最初几年中,有几个因素塑造并区分了微生物群落的发展。
✦出生方式和母体抗生素的使用
出生方式和母体抗生素的使用是影响人类微生物群落的研究最好、最清楚的因素之一。然而,微生物的发育可能会导致独特的结果,即使是在同居的同卵双胞胎中,这可能是由于许多未知或随机的过程。
通过剖腹产和围产期和新生儿抗生素暴露,自然微生物群落的建立过程可能会在所有身体部位受到干扰。这一发现突出了阴道微生物群落的重要性,阴道微生物群落自然含有大量Lactobacillus spp.,但在青春期发生改变,对女性健康至关重要。
一些最佳样本的婴儿发育研究,通常缩写为DIABIMMUNE ECAM和TEDDY,在婴儿出生后的前2年和3年进行了随访,重点关注抗生素使用或出生方式的影响。
在上述所有研究中,阴道分娩的婴儿的拟杆菌属相对丰度高于剖腹产婴儿。
由于缺乏建立微生物群落的天然先锋微生物群,导致可变的群落组成被认为是由随机过程而不是确定性过程驱动的,出生模式对微生物群落组成的影响直到生命的第四年仍然可见。
出生模式影响的一个例外是早产,可能是由于在出生后的头几天大量使用抗生素,其特点是无论出生模式如何,微生物发育都不稳定。婴儿微生物群自然发育的这种改变与感染、免疫疾病、肥胖和神经内分泌异常的风险增加相关。
✦母乳喂养:母乳低聚糖给菌群带来稳定性
其次,与其他因素相比,母乳喂养对微生物群的发育有很大影响。与母乳喂养相比,配方奶粉的使用导致了更高的多样性和更不确定的微生物群落。
例如,考虑到出生时肠道中双歧杆菌科的自然优势,缺乏某些母乳低聚糖作为主要营养源可能会导致初始定植的不稳定性。然而,微生物群、牛奶代谢组和免疫系统发育的多组学整合是一个活跃且快速发展的研究领域。
除了母乳低聚糖,母乳还含有其他免疫调节化合物,例如革兰氏阴性细菌的脂多糖、分泌性IgA、先天免疫因子、抗菌肽和益生元因子。
最后,所有这些因素都会影响人类免疫发育。微生物相关分子模式识别受体与微生物群衍生分子相互作用,代谢物如短链脂肪酸(与GPR43、GPR41和GPR109相互作用)和次级胆汁酸(与FXR相互作用)直接影响免疫发育。
//
这些因素加在一起,有助于形成一个独特的、相对稳定的细菌、真菌和病毒微生物群落,这种微生物群落在人类生命的大部分时间都持续存在。
前面章节了解了婴儿期初级演替期间发生的巨大变化,与之相比,成年期微生物群基本上是稳定的(15-65岁),但该群落可能会受到干扰,因此本章节从以下三方面展开讨论:
健康成年人中某些细菌的基因组随着时间的推移而进化,表明在次生演替中,功能和组成进化以稳定状态发生。
• 昼夜节律影响菌群变化
成人微生物群也会发生自然的短期变化,时间尺度为一天到数月或数年。
短期变化的一个典型例子是微生物群落组成的昼夜节律。与昼夜节律相关的人类基因表达和免疫激活,以及肠道微生物群中细菌的丰度和组成也遵循这种模式。
在小鼠中表现出昼夜循环的细菌家族包括瘤胃球菌科、毛螺菌科、Muribaculaceae和疣微菌科,但对人体的等效周期知之甚少。
青春期和成年生活中的二次演替
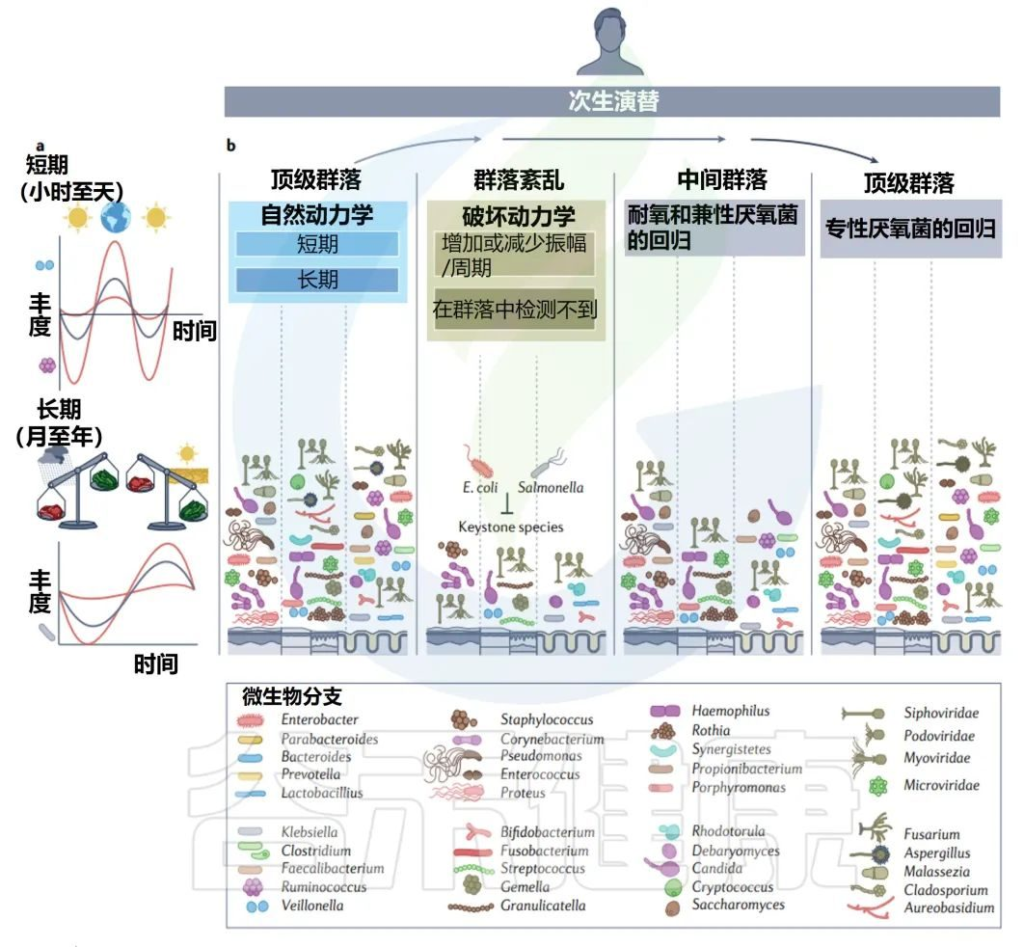
Martino C,et al.Nat Rev Microbiol.2022
• 口腔和皮肤的微生物群随清洗而变化
在口腔中,整组真菌和细菌的每日振幅与刷牙频率一致。在皮肤上,真菌和细菌每天的变化也与洗涤频率一致,并依赖于个人护理产品。
• 饮食会影响肠道微生物群
一个经过充分研究的发生在几周到几年范围内的变化的例子是饮食驱动的肠道微生物群的改变。饮食对微生物群落有很大影响,可以包括群落中的自然和可逆变化。
例如,坦桑尼亚哈扎部落在旱季食用富含肉类和块茎的饮食,但在雨季食用富含蜂蜜和浆果的饮食,在拟杆菌等属中表现出较大的季节波动。
饮食对微生物群形成的巨大影响也可能在人类健康中发挥作用,许多工作致力于了解特定的饮食成分和总体饮食模式如何影响微生物群及其对健康的影响。
肠道细菌喜欢大量的水果、蔬菜、全谷物、橄榄油等健康食物。研究表明,饮食主要由富含纤维的食物(如地中海饮食)组成的人具有更大的微生物组多样性,并且通常更健康。
此外例如,西方饮食中红肉含量高,这与全因死亡率有关。肠道微生物群可能以有害的方式将红肉中富含的左旋肉碱转化为三甲胺,而肝脏则将三甲胺转化为三甲胺氮氧化物,据推测这会促进动脉粥样硬化。
肠道微生物群也可以起到保护作用,例如,在红肉被肠道吸收之前将其分解,以防止炎症。除了饮食,还有许多其他因素有助于形成成年微生物群,包括遗传学、地理、宿主因素,如代谢病和药物。
扩展阅读:深度解析 | 炎症,肠道菌群以及抗炎饮食
• 抗生素对微生物群的影响巨大
由于微生物群的破坏而发生的次生演替已被广泛研究和审查。在破坏微生物群的众多因素中,抗生素是最强的,治疗后的恢复率往往各不相同。
抗生素治疗后肠道微生物群反弹的能力被认为取决于特定的群落成员,如拟杆菌和青春双歧杆菌。
扩展阅读:抗生素对微生物组及对人体健康的影响
细菌的天敌抗生素,如何用好这把救命的双刃剑?
疾病本身也会破坏微生物群,无论这种变化是由微生物群落内部、宿主还是多种因素共同引起的。
• 疾病破坏菌群
——肠道:炎症破坏菌群
肠道中的许多其他疾病,如炎症性肠病,破坏了微生物群落,但没有达到新的稳定群落组成,而是在没有干预的情况下继续长期不稳定。
——皮肤:炎症引起金黄色葡萄球菌大量增殖
在皮肤上,特应性皮炎的特征是免疫介导的炎症引起的金黄色葡萄球菌大量繁殖和细菌多样性减少。在金黄色葡萄球菌大量繁殖期间观察到马拉色菌属的数量减少,反之亦然,真菌数量增加导致金黄色葡萄菌数量减少,这部分可能是由于真菌产生蛋白酶的能力,蛋白酶消化金黄色葡萄球菌生物膜并降低细菌逃避免疫系统的能力。
——口腔:细菌和真菌间的竞争和协同
口腔中也存在类似的跨界相互作用;例如,真菌白色念珠菌的定殖依赖于细菌生物膜,但同时,Pseudomonas和Staphylococcus等细菌属分别形成竞争和协同关系。
这些例子强调了微生物群落的相互作用和演替是如何跨域和与宿主作用的,但由于其高阶相互作用的复杂性质,仍然没有完全理解。
干扰后微生物群落恢复的障碍导致许多研究人员探索有针对性地恢复微生物群落的干预措施的可能性。微生物群落恢复包括定向重新播种或某些物种的富集或耗竭,旨在促使微生物群落恢复到接近扰动前的水平。
这可以通过益生菌、益生元、抗生素或其他药物、从健康个体移植完整的微生物联合体或这些的组合来尝试。
尽管这些疗法在某些特征明确的环境中可以非常有效地恢复健康的微生物群落,但它们往往因缺乏与现有群落相互作用的机理知识,或因其仅短暂移植的能力而受到限制。
为了解决这些,研究集中在两个领域:
第一个领域涉及更好地了解群落是如何组合的。例如,对人类发育的研究有助于确定微生物群落在发育过程中如何聚集,以及这种聚集在生命后期的影响。
其次,正在开发新方法,通过探索微生物群落相互作用来确定机制,包括计算和实验,包括高通量共培养和微生物群落的基因组编辑。
为了解决瞬时性问题,采用了两种主要方法:
首先,微生物群疗法的短暂和个性化影响是由每个人的微生物群的个体性质决定的。因此,精准医学将群落改变的目标定位于每个人独特的微生物群,前景广阔。例如,基于微生物群落组成的个性化营养在盲法随机对照干预中有效地改善了餐后血糖。
另外,超越细菌组,探索病毒组和真菌群落及其之间的相互作用,具有巨大的前景。例如,噬菌体疗法已经用于严重的耐药细菌感染,并且对目标细菌菌株具有高度特异性。但大多数此类干预措施仍处于初步研究阶段,且规模成本高昂。
前面章节我们了解了成年微生物群的变化,以及变化后的恢复情况等,成年稳定微生物群在老年时转变为最终群落,本章节来详细了解老年微生物群。
“老年”的确切时间尺度取决于其他几个与宿主相关的因素,如疾病,但迄今为止大多数文献将“老年人”定义为65岁及以上的人。
接近寿命终点的晚期演替
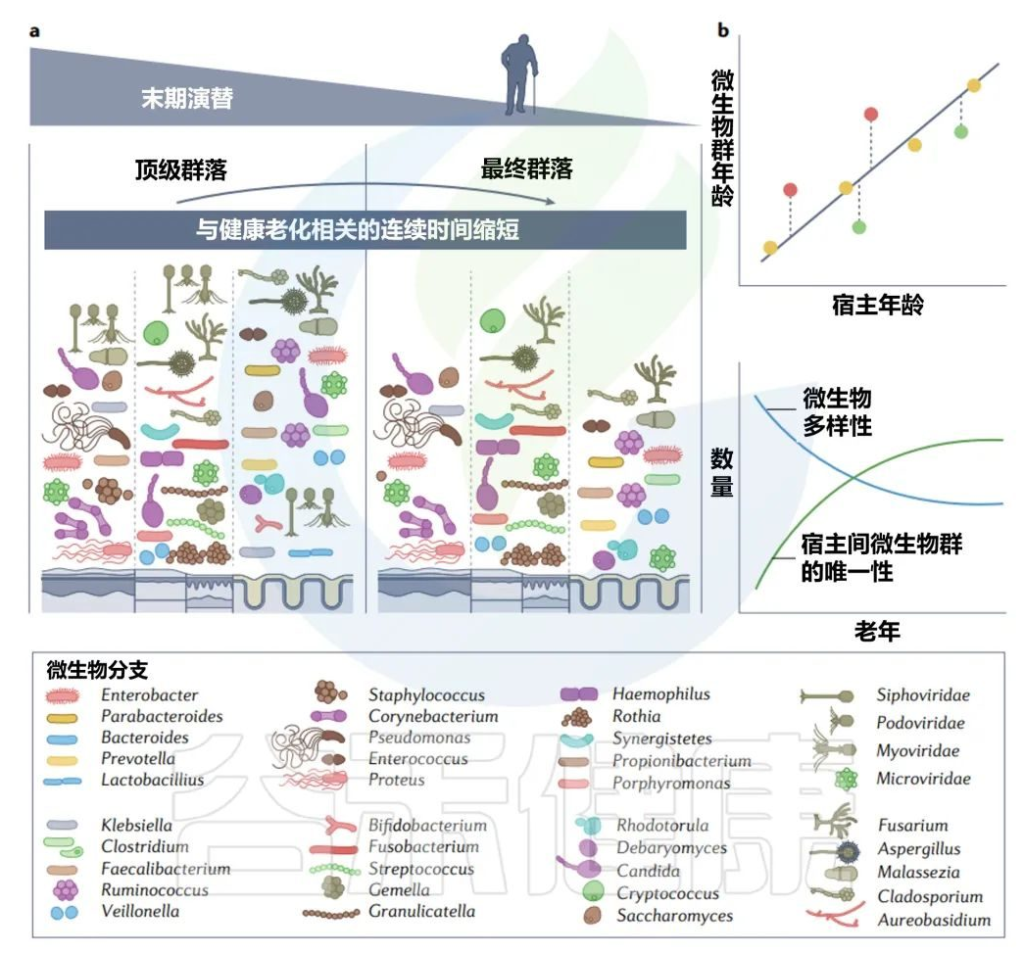
由于生物编程和生命中损伤的累积而导致的衰老影响细胞功能的各个方面,微生物群也不例外。随着年龄的增长,肠道微生物群α多样性减少,β多样性增加。
关于老年微生物群,仍有许多未知之处,而文献也有些矛盾(一项报告称65岁及以上成年人拟杆菌数量增加,与其他研究相矛盾),大多数研究都集中在肠道细菌上。
老年微生物群:年轻优势菌丰度减少
一般而言,肠道中观察到的群落演替是年轻成年人中占优势和普遍的细菌属丰度减少,如Bifidobacteria, Bacteroides, Lactobacillus, 抵御机会细菌爆发的能力降低。
• 皮肤
在65岁及以上的人群中,genera Cutibacterium和Staphylococcus的皮肤细菌数量减少,同时观察到的Corynebacterium。
• 口腔
在口腔部位,Rothia和Streptococcus spp.是核心口腔细菌群落,PorphyromonasTreponema和Faecalibacterium spp.的数量持续减少。
• 肠道
老年期肠道真菌群落的特征是Penicillium, CandidaAspergillus和Saccharomyces spp.的优势度增加。
在皮肤和口腔部位的研究很少,但老年期皮肤上的Malasseziaspp.和口腔内的Candidaspp.丰度减少。
在肠道噬菌体中,成年期的Siphoviridae占主导地位,而老年期的Microviridae和Podoviridae则占主导地位。与肠道细菌、真菌和噬菌体群体相比,真核病毒的多样性在童年后和整个余生中保持不变。
研究重点
由于个体之间的高度变异性,老年微生物演替的研究重点主要是比较健康和不健康的衰老。
目前尚不清楚微生物群是否在健康衰老中起着机械作用,还是仅仅是其他变量的一个有力指标,如饮食、运动和药物。然而,在那些长寿健康的人中,可以观察到在健康成年人中高度流行的菌群的持续保留方面的共同点。
然而,百岁老人表现出更独特的微生物群,α多样性增加,群落组成的个体间差异更大,使“健康”和“不健康”年龄之间的比较复杂化。次生胆汁酸在百岁老人中含量丰富,也可能在健康老龄化中发挥作用。尽管前景看好,但这一研究领域仍处于起步阶段。
扩展阅读:肠道菌群与健康长寿
• 微生物的演替不会随着个体的死亡而结束
宿主的死亡可以视为微生物群的生态干扰。心脏停止后,组织立即因缺氧而开始分解。细胞功能持续,直到所有剩余的氧气耗尽,二氧化碳不再能够从组织中运输为止。细胞内二氧化碳的积累创造了一个缺氧的酸性环境,导致细胞破裂。
细胞成分,例如酶会泄漏到周围环境中,在被称为“自溶”的过程中进一步促进组织分解。自溶通过消除免疫系统、松开细胞连接并为微生物群提供营养,触发了一系列负责组织分解的微生物过程。
死亡后的微生物群
Martino C,et al.Nat Rev Microbiol.2022
• 死亡后微生物群分解
人类微生物群在死亡后的前24-48小时内相对稳定,具有不同的身体部位微生物生态、年龄的α多样性模式和可识别的个性化皮肤微生物群特征。
在分解的最初几天到几周内,腐败主要由细菌进行,但随着分解的进行,真菌的作用增加。然而,在这个过程中,对病毒组的演替和功能作用了解甚少。
随后,环境变化促进了微生物的演替,改变了人体和微生物群,不再像活着的个体(除非身体被冷冻)。
由于缺乏宿主生活中先前遇到的环境限制,使得微生物的相对丰度发生了快速变化以及在身体各部位的移动。迁移的细菌群成为从肠道转移到肠外部位的先锋物种,根据身体部位参与初级演替或次级演替。
• 死亡微生物群——生物指示器
死亡微生物群因其对法医调查的影响而引起了越来越多的关注。与多个个体和身体部位相关的一致的时间序列模式证明,死后微生物群可以作为死后间隔的生物指示器。
每个尸体的死后微生物群都是独一无二的,并且根据死亡时间、死因、环境、死亡地点和年龄以及开始时身体部位之间的差异,尸体之间的微生物群是不同的。
当微生物演替包括群落成员的快速更替时,在分解的早期阶段(即死亡后的前2-3周),死后时间间隔估计更为准确,但在分解的后期阶段(例如骨骼)仍然有用,因为几乎没有证据可以估计死后时间间期。
• 死亡原因与微生物群存在联系
还证明了与死亡原因和微生物群存在的联系。例如,在死于心脏病的个人的口腔微生物群中发现了的Rothia spp.。
此外,皮肤微生物群脱落可能通过将个人与他们接触过的物品联系起来,从而有助于追踪证据;然而,这一独特特征能够准确匹配到个体的时间取决于对象的材料和用途。
人类微生物群是动态的。考虑到这一点,设计一种能够捕捉微生物群的时间和空间变异性的采样策略非常重要,特别是当这些波动与所提出的科学问题相关时。
✦测量时间不同:多个时间点的样本采集
横断面研究从每个个体收集一个样本,而重复测量研究在多个时间点或身体部位收集样本。随着时间的推移,采样频率应该调整到研究人员试图观察的现象。
例如,小鼠昼夜节律研究通常每2-4小时收集一次粪便样本;而在炎症性肠病中,在一周内对患者进行三到五次采样可以改善疾病分类。
在其他应用中,例如研究特定治疗对个体微生物群的影响,这可能与进行“一对一”研究有关,在该研究中,同一参与者被反复检测其微生物群的结果变化;治疗前采集的样本被视为个体水平的对照。
✦测量空间不同:城市化/农村环境不同
同样重要的是要考虑到人口的微生物群高度依赖于地理和种族。
例如,在一个大型中国群体中,一种与年龄高度相关的微生物在一个美国大型群体中根本没有检测到。
另一个具体的例子涉及城市化社会的“建筑环境”;城市化人群通常较少接触环境微生物,更多地使用家用抗菌剂,与来自农村社会的人类微生物群相比,这导致了重大变化。
这些考虑因素与微生物群领域尤其相关,因为大多数公共微生物群数据来自城市化的北美和欧洲人。因此,现有数据集的结论可能无法很好地推广到全球人口。
从人类微生物群和微生物群研究中生成的测序数据的主要类别是扩增子测序数据和鸟枪测序数据。
✦扩增子测序
在扩增子测序中,对已建立的高变区的PCR产物(扩增子)进行深度测序,从而能够通过与个体“条形码”匹配来识别和测量群体成员。
这里有两种选择:要扩增的基因和该基因的哪一部分要扩增。微生物基因组的常见扩增区域包括:细菌的16S核糖体RNA基因、真核微生物的18S核糖体DNA基因和真菌的内部转录间隔区。
每个特定基因中高变区的选择取决于要捕获的特定微生物,但广泛使用的高变区包括来自地球微生物组项目的V4区。
✦肠道微生物群参与人体的调节
在鸟枪测序中,所有微生物DNA都被测序,而不仅仅是PCR产物,从而能够对微生物进行更具体的分类。由于鸟枪测序不依赖于任何标记基因,因此与扩增子测序相比,它对某些微生物的偏向性较小。
然而,鸟枪测序的成本要高得多,并且需要更大的计算能力,这使得在不需要提高鸟枪序列分辨率的情况下,扩增子测序具有吸引力。
结合其他技术进行扩增子或宏基因组测序可以丰富对微生物群和宿主的理解。定量PCR和荧光激活细胞分选等技术通过将相对丰度锚定到可靠的绝对丰度测量值,为相对丰度提供了更多的背景。
酶联免疫吸附试验和单细胞测序可以通过提供宿主细胞类型或宿主免疫信息与宏基因组测序很好地配对。
培养组学使研究人员能够通过实验验证功能或活性的基因组预测,并将微生物转化为益生菌。微生物产生的代谢物或蛋白质,即微生物群的下游效应物,可以分别通过代谢组学和蛋白质组学进行探测。
最后,宿主基因组学和转录组学越来越多地与扩增子或宏基因组学数据配对,以深入了解宿主基因表达和微生物群之间的联系。
最后,从被调查的参与者那里收集数据至关重要。一般微生物群研究的一些重要元数据类别包括人口统计、临床信息和饮食信息;然而,使用的确切元数据因研究而异。应采用产生标准化元数据的实践,以便结果可重复使用和再现。
本文描述了目前对不同年龄和不同身体部位的人类常驻微生物群落组成的研究现状。
人类健康与微生物群组成之间存在许多联系,对肠道菌群的干预可能改善健康。侧重于整个微生物群而不是单一物种的富集或消除的干预措施,需要了解这些群落是如何形成和维持的。
不同人群年龄,不同部位的微生物群需要依托于大样本数据库的构建,这为微生物群研究的准确性提供了保障。
通过研究人类整个生命周期中的微生物群,我们可以更好地了解这些微生物群复杂的相互作用,以及如何有效地将微生物群推向宿主所需的组成。此外也正应用于除人类健康外的其他领域,如法医学。随着微生物群的相关研究不断突破,将给人类生命健康和生产生活带来巨大的影响。
主要参考文献:
Martino C, Dilmore AH, Burcham ZM, Metcalf JL, Jeste D, Knight R. Microbiota succession throughout life from the cradle to the grave. Nat Rev Microbiol. 2022 Jul 29. doi: 10.1038/s41579-022-00768-z. Epub ahead of print. PMID: 35906422.
Lim, A. I. et al. Prenatal maternal infection promotes tissue-specific immunity and inflammation in offspring. Science 373, eabf3002.
Al Nabhani, Z. & Eberl, G. Imprinting of the immune system by the microbiota early in life. Mucosal Immunol. 13, 183–189.
Helve, O. et al. 2843. Maternal fecal transplantation to infants born by cesarean section: safety and feasibility. Open. Forum Infect. Dis. 6, S68.
Seppo, A. E. et al. Infant gut microbiome is enriched with Bifidobacterium longum ssp. infantis in old order mennonites with traditional farming lifestyle. Allergy 76, 3489–3503.
报告新增以下内容:
菌群构成层级
给出丰度千分之一以上菌门、属及种的桑基层级图。便于快速直观了解菌群构成。

核心菌属构成表
我们将在90%人群检出,人群平均丰度1%以上的菌属为核心菌属,属于人体肠道菌群中最常见和主要的菌属以及主要的益生菌菌属,包含21个属。核心菌属及有益菌累加占总肠道菌群比例低于60%的可能出现肠道菌群紊乱。
表中包含了检测丰度,正常范围,处于人群水平以及正常人群检出比例,以及菌属的说明。如果出现菌属超标,会在表下给出改善方式。
有害菌属构成表
有害菌涵盖了多个包含机会致病菌或与肠道菌群紊乱相关的菌属。
表构成与核心菌属一致
其他重要菌属构成表
其他重要菌属包括人群平均丰度不高,但对肠道菌群健康重要的菌属。