-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
谷禾健康
近年来,免疫治疗在癌症治疗领域展现出了巨大的潜力,特别是针对PD1/PDL1通路的免疫检查点抑制剂(ICIs)已在多种实体瘤患者中取得显著疗效。然而,治疗反应的异质性和耐药性的出现依然是当前面临的主要挑战之一。越来越多的研究表明,肠道菌群作为人体内一个庞大的微生态系统,不仅参与机体代谢和免疫调控,而且在调节肿瘤免疫反应方面发挥着关键作用。
让我们快速了解一下肠道菌群如何通过细胞水平、分子信号通路和代谢产物来影响PD1/PDL1肿瘤免疫疗法的疗效。
1 核心菌群与免疫调控机制
多项临床与实验室研究均显示,特定菌群对PD1/PDL1免疫检查点抑制剂治疗具有显著的正向作用。
例如,Bifidobacterium属菌群被发现能显著促进抗PDL1疗法的抗肿瘤作用;而Akkermansia muciniphila在PD1抑制治疗中也表现出与治疗反应正相关的趋势。此外,研究还发现,黑色素瘤患者中较高的微生物多样性和特定菌群如Ruminococcaceae和Faecalibacterium的丰富度均与更长的无进展生存期相关。
肠道菌群对免疫调控的机制主要表现在以下几个方面:
促进树突状细胞成熟:某些菌群(如Bifidobacterium)可通过激活树突状细胞,从而增强抗原呈递能力,提高CD8+ T细胞的活性。
调节T细胞免疫状态:特定菌群通过影响细胞因子分泌和T细胞亚群分布,调节包括CD8+效应T细胞和调节性T细胞(Treg)在内的免疫平衡,从而实现抗肿瘤效应。
通过代谢产物发挥作用:菌群代谢产物,如短链脂肪酸(SCFAs)和次级胆汁酸,对免疫细胞储存、功能激活具有直接调控作用。
这些作用机制不仅在单一免疫治疗模式中发挥效应,同时也对联合治疗(例如PD1/PDL1抑制剂与CTLA-4抑制剂联合使用)产生协同增效作用。
2 关键代谢物及其信号调控路径
肠道菌群通过发酵膳食纤维等底物,产生大量的短链脂肪酸(SCFAs),如丁酸盐和丙酸盐,这些代谢物在调节宿主免疫功能中起到关键作用。研究发现,短链脂肪酸不仅参与维持肠道上皮屏障的稳定,还能调节T细胞分化和促炎/抗炎反应的平衡。此外,菌群代谢的次级胆汁酸也被证明在免疫抑制和调控细胞因子水平中起到重要作用。
值得注意的是,不同菌群通过生成不同的代谢产物,对免疫系统的影响可能存在正负两方面的效应。例如,有研究显示高浓度的丁酸盐和丙酸盐可能会在某些条件下限制CTLA-4抑制剂的疗效,从而提示适度平衡菌群代谢产物对于免疫治疗的成功至关重要。同时,某些菌群如Prevotellaceae和Rikenellaceae则可能通过降低丁酸盐水平来促进促炎性巨噬细胞M1型的极化,从而间接影响免疫治疗的疗效。
3 不同肿瘤类型中的菌群特征与免疫应答
肠道菌群对免疫治疗的影响在不同肿瘤类型中可能存在显著差异。下面介绍一下在黑色素瘤、非小细胞肺癌(NSCLC)、肝细胞癌(HCC)以及结直肠癌(CRC)中的相关发现。
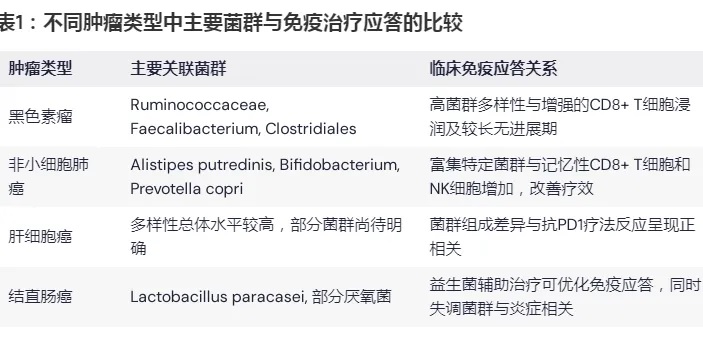
表1说明:各肿瘤类型中显示出菌群多样性和特定菌群丰度与PD1/PDL1免疫治疗效果之间存在明显相关性,该表对比了不同肿瘤中的主要菌群与免疫应答情况。
4 肠道菌群调控机制的分子与细胞通路
肠道菌群通过多条分子信号通路及细胞间相互作用调控宿主的免疫反应,进而影响PD1/PDL1免疫治疗的疗效。
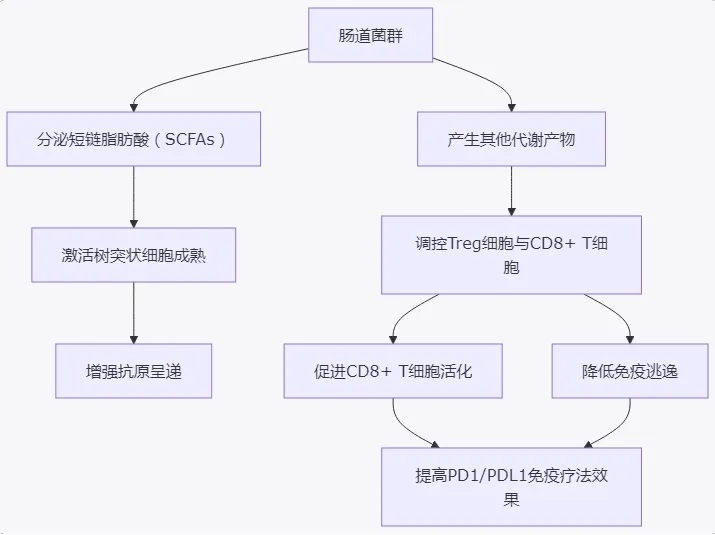
图1:肠道菌群通过分泌代谢产物调控树突状细胞成熟、T细胞活性与调节性T细胞平衡,从而增强PD1/PDL1免疫治疗效果。
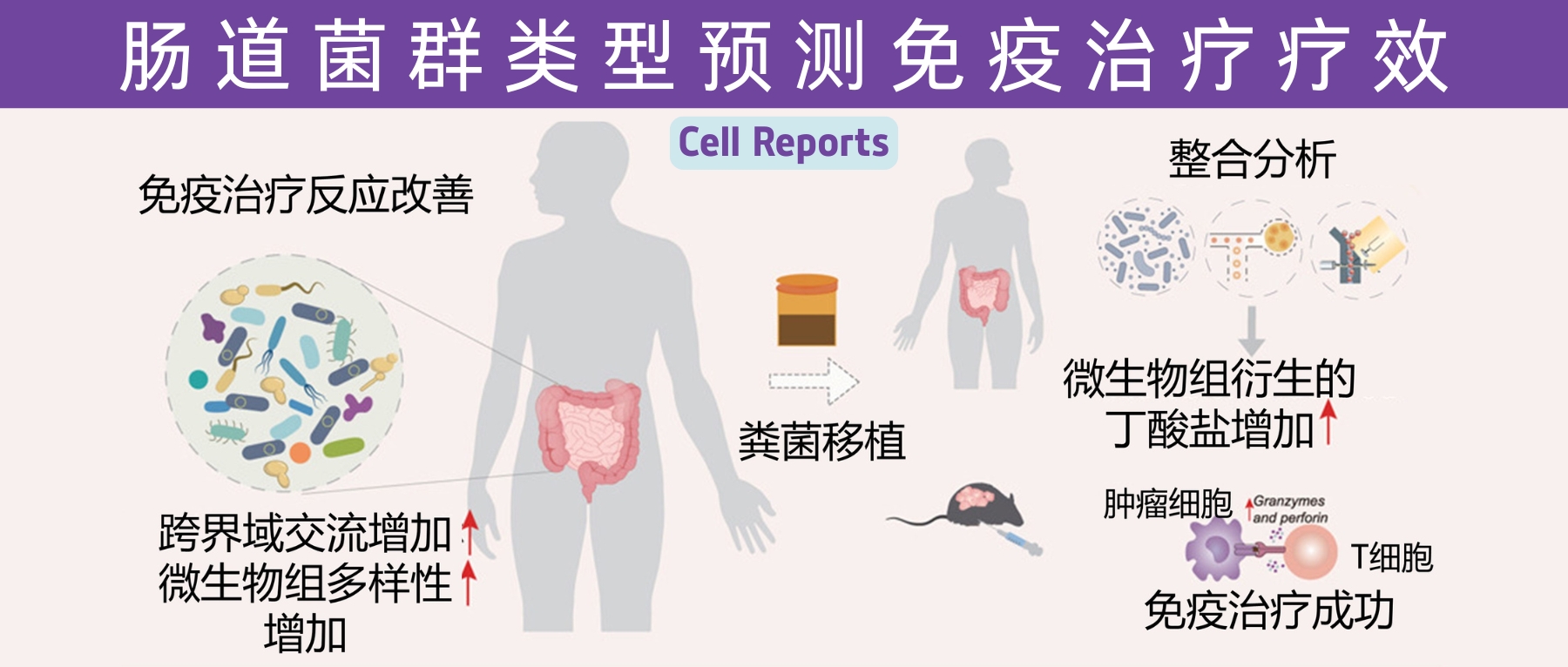
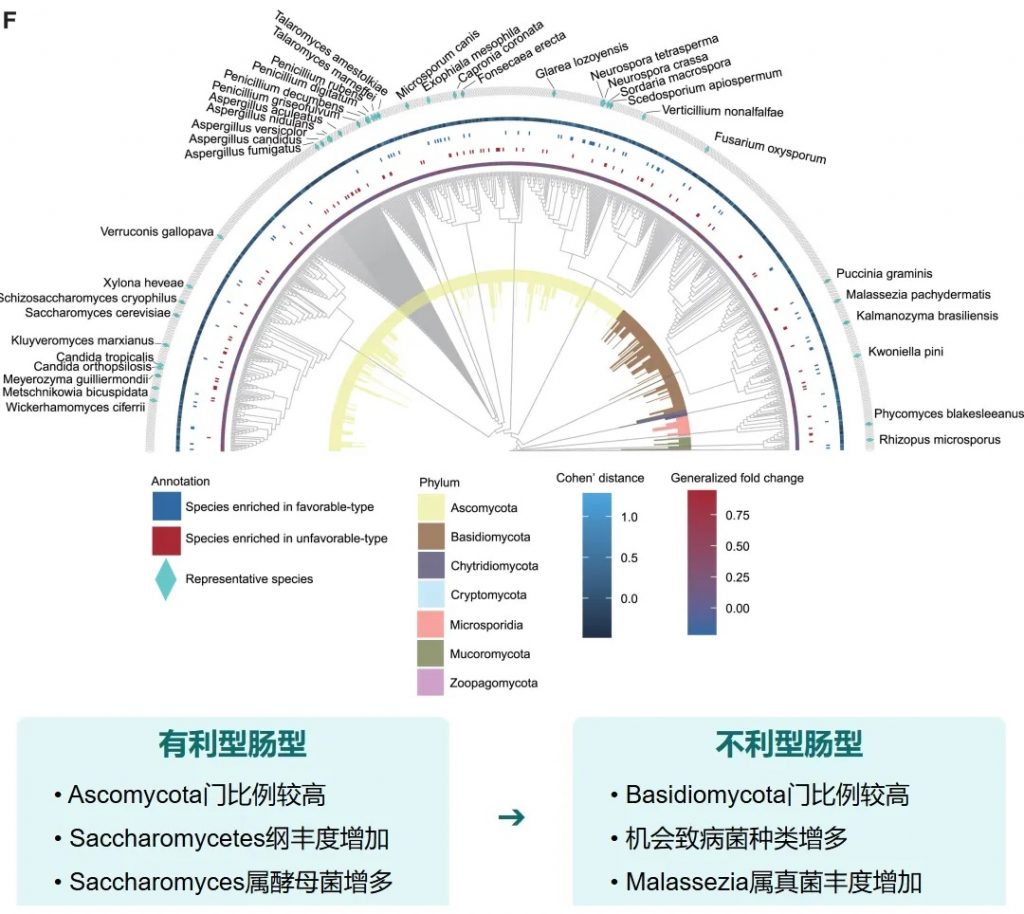
近日,一项来自上海交通大学医学院研究团队的成果发表在《Cell Reports》,通过整合多组学数据与临床队列,揭示了基于肠道菌群分类(有利型与不利型)与抗PD-1/PD-L1免疫治疗疗效的显著关联。

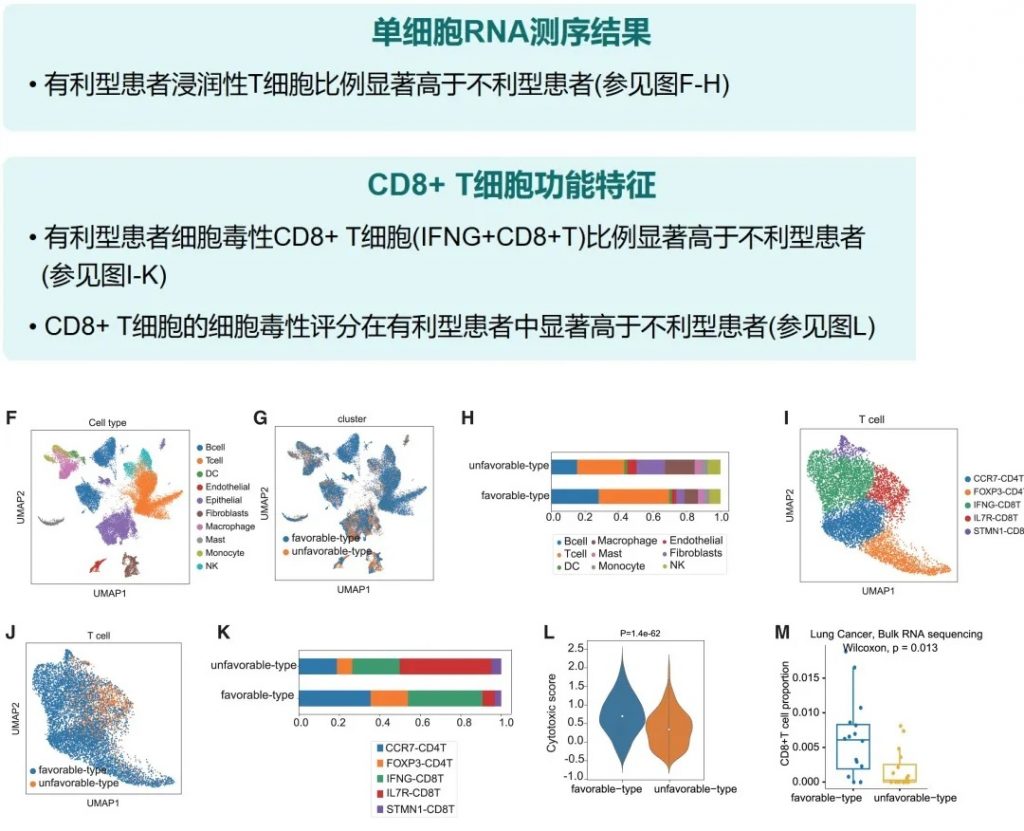
其中,有利型患者表现出更高的真菌与细菌多样性、富集丁酸代谢通路及促免疫菌群(如Faecalibacterium prausnitzii),以及肿瘤微环境中细胞毒性CD8+ T细胞浸润增强的特征,且在多癌种队列中显著关联于抗PD-1/PD-L1治疗的临床缓解与生存获益。
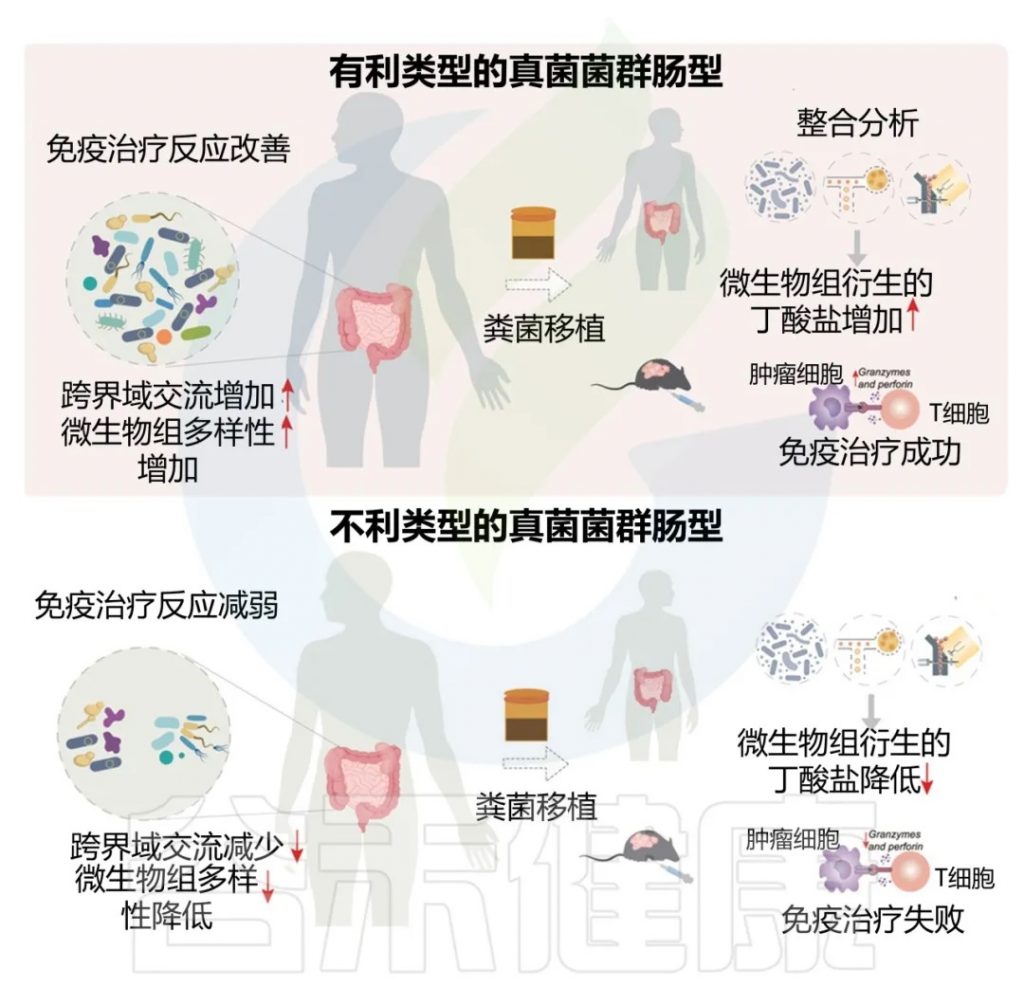
进一步通过FMT实验证实,移植来自有利型供体的粪便可显著提升受体对免疫治疗的敏感性,并重塑肠道菌群代谢功能。
这些发现不仅深化了对肠道真菌组-细菌互作网络的理解,为优化个体化免疫治疗提供了新型生物标志物,更为优化免疫治疗分层策略及FMT供体筛选提供了科学依据,具有重要的临床转化潜力。

多组学关联分析与肠道菌群类型特征
详细对比了两种肠道菌群类型的微生物群落特征差异,包括多样性、产丁酸菌含量、代谢通路等,并展示了它们与临床疗效的关联。
粪便微生物移植实验验证
呈现了临床FMT研究和动物模型验证结果,证明来自有利型供体的FMT可显著提升受体对免疫治疗的敏感性。
总结了研究的创新点及其在预测分层和干预优化方面的临床应用价值。
肠道菌群类型:免疫治疗疗效的新生物标志物
免疫检查点抑制剂(如抗PD-1/PD-L1)显著改变了癌症治疗格局,但患者反应差异大。
该研究团队采用无监督聚类方法分析肠道菌群组成,在多个独立队列中成功识别并验证了两种截然不同的类型——“有利型”与“不利型”,这一发现揭示了肠道菌群对免疫治疗应答的预测价值。

肠道菌群的类型不仅与临床反应相关,还与患者总生存期密切相关,为个体化治疗策略提供了新思路。
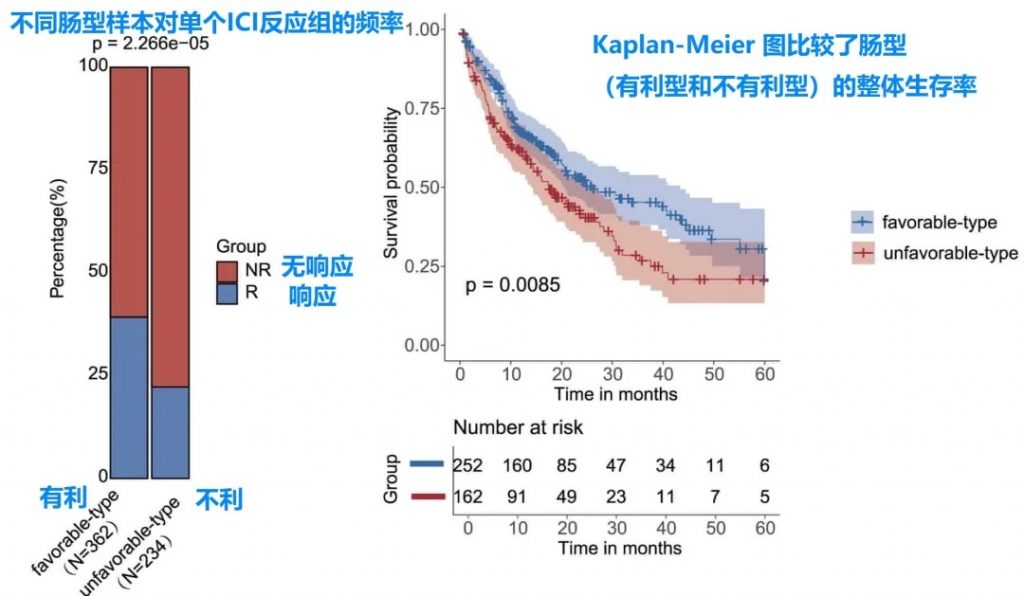
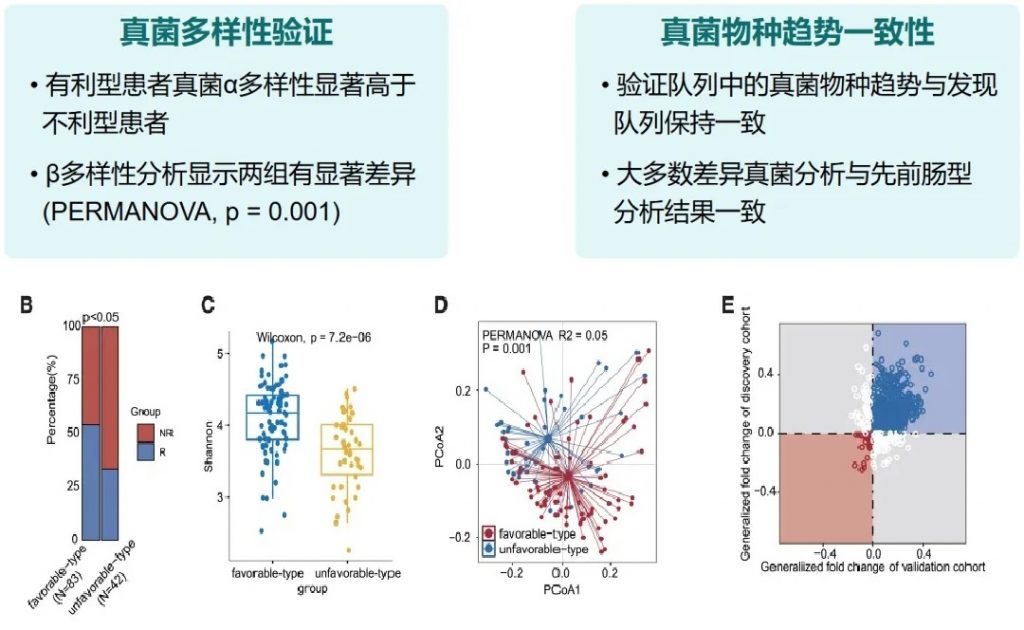
有利型患者对抗PD-1/PD-L1免疫治疗的响应率显著高于不利型 (p=2.266e−5),总生存期也更长 (p=0.0085)。
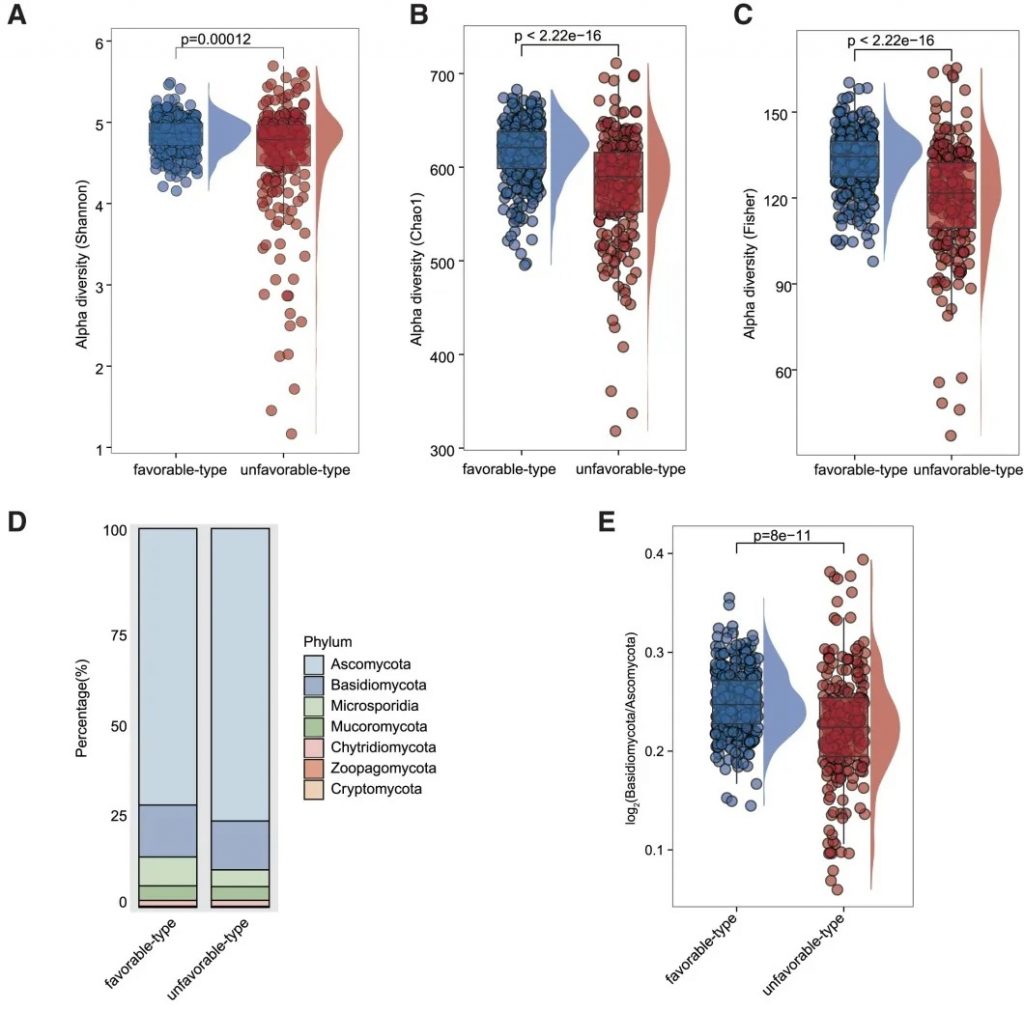
“有利型”和“不利型”肠道菌群差异 细菌、真菌
细菌 多样性
——α多样性差异
有利型患者表现出显著更高的细菌α多样性指数(Shannon、Chao1、Simpson),这与既往研究发现的免疫治疗响应者多样性更高的结论一致。
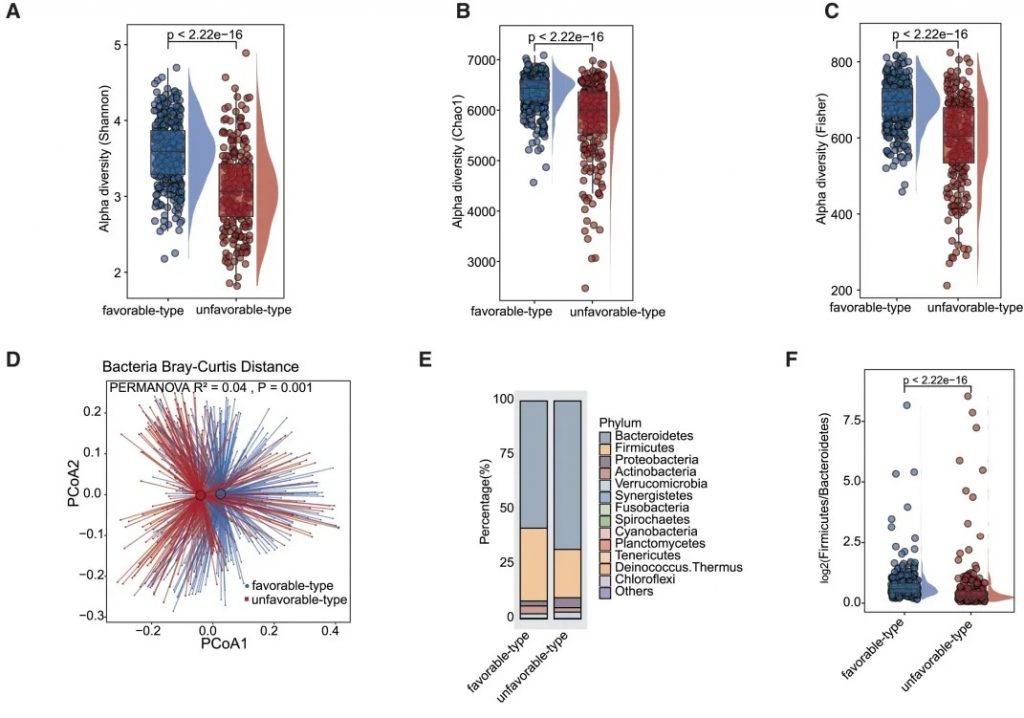
——β多样性差异
细菌 主要构成
有利型患者表现出更高的Firmicutes/Bacteroides比值,这被认为是肠道菌群平衡的重要指标,与免疫治疗的良好反应相关。
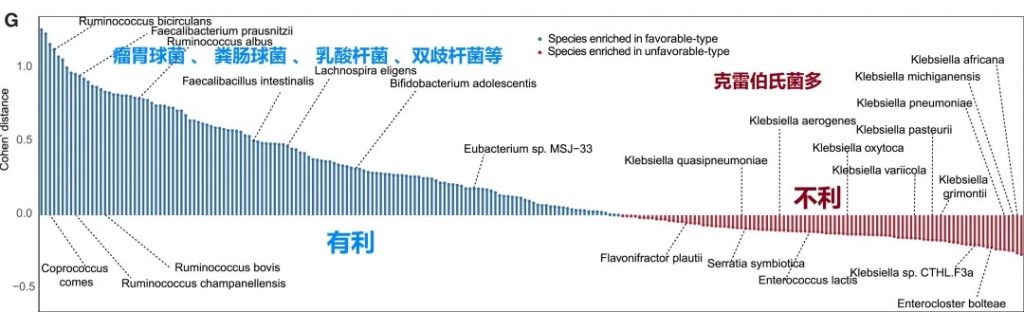
有利型的患者肠道中富含产丁酸盐的细菌,特别是Faecalibacterium prausnitzii、Lachnospira eligens,这些细菌通过产生丁酸盐调节肠道微环境,增强抗肿瘤免疫反应。
真菌 多样性
有利型表现出更高的Shannon指数和OTU丰富度,真菌物种多样性更丰富,这与对免疫治疗的更好反应相关联。
真菌 主要构成
关键真菌与细菌的共现分析
有利型样本在真菌-细菌界中表现出增加的正相互作用,而不利型样本在菌-细菌界表现出更多的负相互作用。
关键真菌-细菌互作对有利型中的关键互作真菌-产丁酸盐细菌正向互作:
有利型的真菌群落结构似乎与某些细菌表现出更积极的共生关系。真菌-细菌间相互作用可能在抗PD-1/PD-L1 ICI的反应中发挥关键作用。
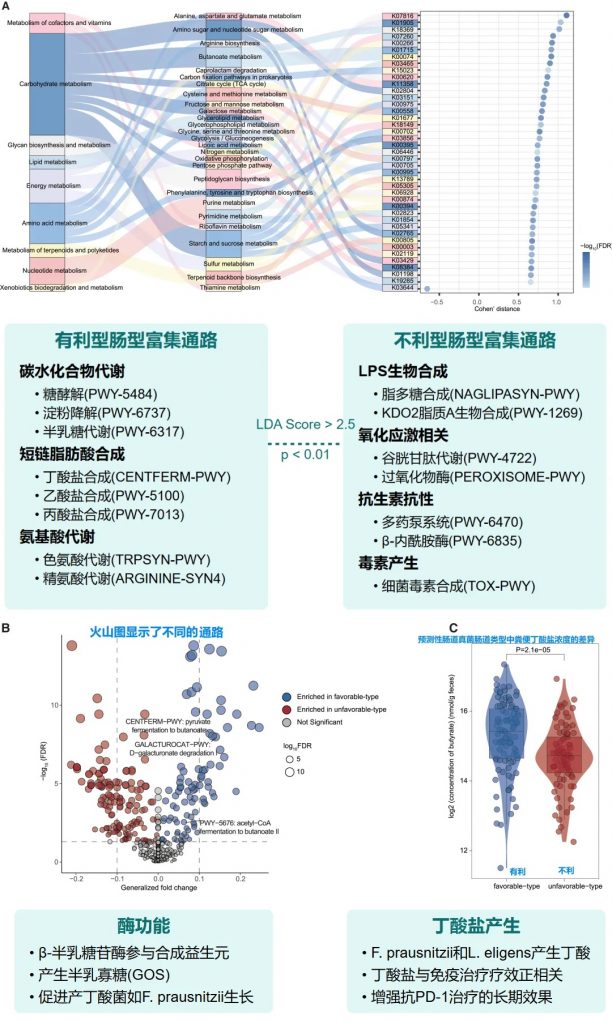
基于特定菌群类型的微生物功能分析
肠道菌群类型的外部验证与多组学分析
验证队列特征
• 125名接受PD-1抗体治疗的泛癌症患者
• 基于随机森林分类器(RandomForestClassifier)进行肠道菌群类型的预测
• 预测结果: 83名有利型患者,42名不利型患者
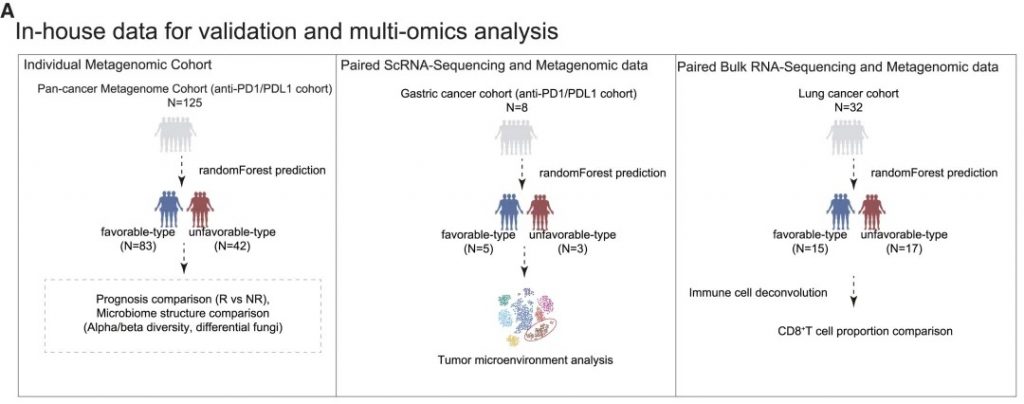
验证分析方法
• 基于机器学习的肠道菌群类型预测模型
• 随访评估对免疫治疗的临床反应
丁酸可促进CD8+ T细胞浸润肿瘤微环境,直接增强抗肿瘤免疫。
FMT供体选择:优化免疫治疗疗效
整合数据构成
• 3个近期发表的临床试验数据
• 12名FMT供体 (5名有利型, 7名不利型)
• 43名黑色素瘤患者接受者
• 包含宏基因组数据和临床结果
FMT临床疗效分析
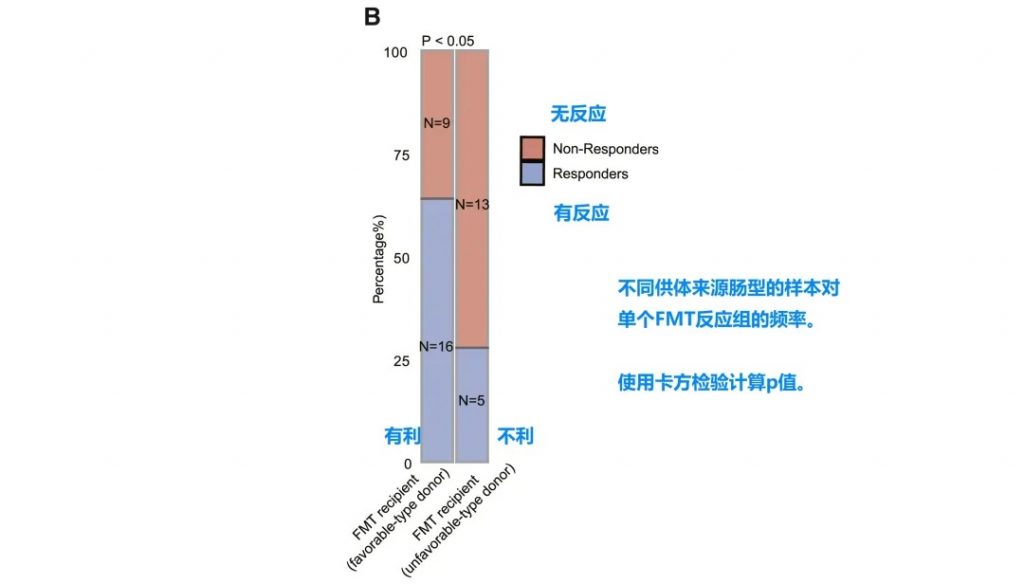
• 接受有利型供体FMT的患者对抗PD-1/PD-L1抗体免疫治疗的反应明显改善
• 卡方检验证实统计学显著差异 (p < 0.05)
接受“有利型”供体FMT的患者显示出对抗PD-1/PD-L1抗体免疫治疗的显著改善反应(p < 0.05)。这一发现具有临床指导意义,为FMT供体筛选提供了新的标准。
FMT前后微生物多样性变化
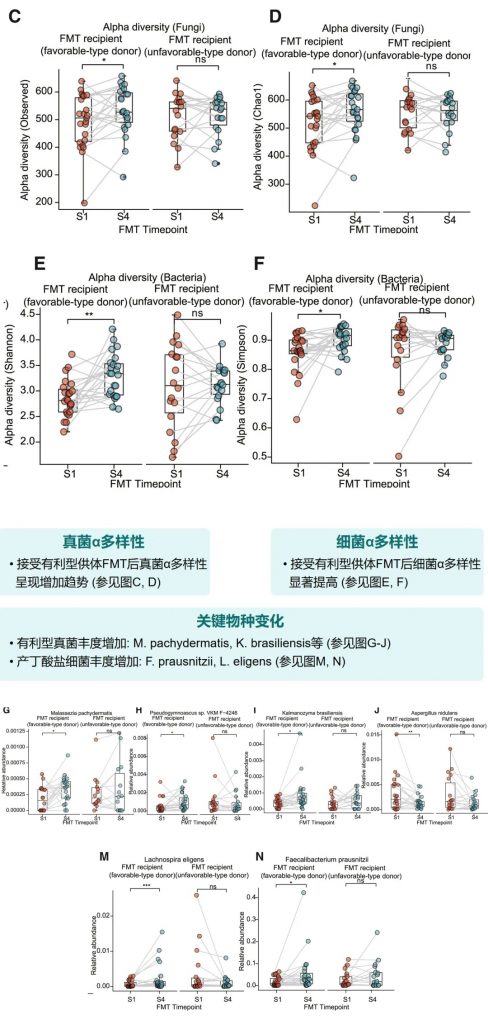
接受有利型供体FMT后,受者的真菌和细菌α多样性均有明显提高。特别是,有利型特征真菌和产丁酸菌的丰度显著增加,这与增强的免疫治疗效果相关。
体内验证实验结果
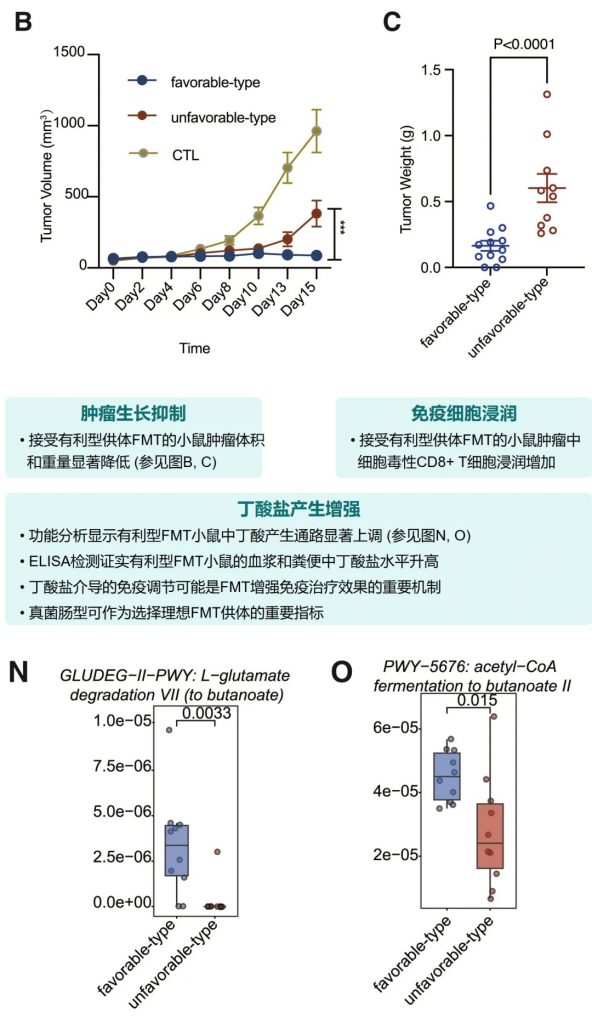
小鼠实验进一步确认了有利型供体FMT可以增强抗PD-1治疗效果,表现为肿瘤生长抑制、CD8+ T细胞浸润增加,以及丁酸盐产生增强。这为FMT增强免疫治疗提供了机制解释。
该研究揭示了肠道微生物群类型与免疫检查点抑制剂(ICI)疗效之间的密切关系,为优化癌症免疫治疗策略提供了新的视角。
肠道菌群与免疫治疗响应之间存在显著关联,且这种关联可通过粪菌移植等干预手段加以调控,为克服免疫治疗耐药提供新策略。
以下是几个具有前景的研究和临床应用方向:
辅助诊断的开发与实施
随着肠道菌群类型与免疫治疗疗效之间关联的确立,开发肠道微生物群分析作为免疫治疗的伴随诊断工具已成为一个明确的临床需求。
粪菌移植(FMT)作为辅助治疗手段
对于被确定为不利型的患者,粪菌移植提供了一种有前景的干预手段,可能显著提高免疫治疗的响应率。
更大规模的临床验证研究
为了将这些发现转化为临床实践,需要进行更大规模的验证研究:
未来,肠道微生物群检测有望成为免疫治疗患者的常规伴随诊断工具,帮助临床医生识别潜在的治疗无应答人群。
也就是说做免疫治疗的人群可以做肠道菌群评估作为辅助诊断,如果免疫治疗没有效果的病人,考虑通过FMT调整菌群后,提高治疗效果。来自有利型供体的粪菌移植可能成为提高治疗效果的有效辅助手段。
这一整合策略不仅能够优化现有免疫治疗的应用,也为开发新型微生物组靶向干预手段提供了方向,最终实现免疫治疗疗效的最大化和个体化。
在癌症精准医疗的框架下,肠道微生物组评估和干预将与基因组学、免疫学等多组学手段一起,共同构成未来肿瘤免疫治疗的个体化决策基础,为患者带来更好的临床获益。
主要参考文献
Hu M, Zhu X, Huang X, Hua L, Lin X, Zhang H, Hu Y, Tong T, Li L, Xuan B, Zhao Y, Zhou Y, Ding J, Ma Y, Jiang Y, Ning L, Zhang Y, Wang Z, Fang JY, Zhang Y, Xiao X, Hong J, Chen H, Li J, Chen H. Optimizing anti-PD-1/PD-L1 therapy efficacy and fecal microbiota transplantation donor selection through gut mycobiome-based enterotype. Cell Rep. 2025 Apr 20;44(5):115589.

谷禾健康

随着技术的发展,高通量测序技术已成为研究微生物群落的重要工具。这种技术使得科学家们能够解析巨量微生物DNA序列,从而获得丰富的微生物组数据,包括16S rRNA基因、ITS序列和宏基因组。然而,这些数据只是迈向揭示微生物群落复杂性的第一步。
通过对环境样本的可变区域如16S、18S、ITS序列进行高通量测序获得的原始序列数据,再对其进行聚类,数据分析,统计学差异比较等得到微生物多样性分析报告。那么,什么是微生物群落多样性?
微生物群落多样性(Microbial Community Diversity)是指在特定环境中存在的微生物种类的数量和分布情况,它不仅包含不同种类微生物的丰度,还包括它们之间的相互关系。多样性可以从不同角度进行评价,主要分为以下几种:
α多样性(Alpha Diversity): 这是衡量某一特定样本内部多样性的一种指标。常用的α多样性指标包括物种丰富度(Species Richness)、香农指数(Shannon Index)和辛普森指数(Simpson Index)。这些指标可以帮助我们了解样本内部的复杂性和均一性。
β多样性(Beta Diversity):不同样本之间的多样性比较被称为β多样性。常用的β多样性指标包括Bray-Curtis距离和Jaccard指数,通过这些指标可以探索样本之间的相似性和差异性,揭示不同环境或条件下微生物群落的变化模式。
γ多样性(Gamma Diversity): 这是指在一个更大尺度、多个样本的总体多样性,通常用以评估一个较大区域的整体多样性水平。
为理解这些多样性指标,我们可以借助一些简单的比喻来形象解释。例如,α多样性就像是在观察一个花园的花卉种类和数量;β多样性则是比较不同的花园之间的相似性或不同之处;而γ多样性则是对一个城市中所有花园的总览评价。
在接下来的部分中,我们将深入探讨这些多样性指标的详细内涵,以及从多个角度展示如何通过高通量测序技术解析微生物群落中的这些多样性规律。
下图是实验上机测序流程,提取的样本总DNA经过质检、PCR扩增、建库等步骤进行高通量测序得到测序原始数据。

原始数据经过Reads拼接、tags过滤、去嵌合体等步骤得到有效数据clean data。在特定的相似度下进行聚类得到OTU/ASV,报告中通过降噪方法得到ASV表,一切后续分析都围绕ASV表来进行。根据ASV表可以继续做物种分类注释、丰度计算、多样性分析、差异分析、功能预测等。所以ASV特征表是微生物多样性分析中关键数据结果。
OTU和ASV的区别
OTU和ASV是微生物组学中用来表示微生物多样性的两个不同概念。两者都是从环境样本中获得的DNA序列数据,通过一定的分析方法分类得到的用于表示微生物种类或种群的单位。它们之间的主要区别在于定义的精确度和建立的方法。
– OTU(Operational Taxonomic Units):
OTU是一种将序列通过相似度聚类的传统方式,来表示相似序列组成的种群。通常,这种聚类方法会将序列之间相似度达到97%(或其他设定的阈值)的序列分到同一个OTU。OTU聚类通常不考虑序列中的单个变异位点,而是基于整体相似度。
由于使用阈值聚类,OTU不能准确反映序列之间的实际差异,可能会将生态学意义上不同的微生物序列归为一个OTU。OTU分析可能过于简化,有时无法捕获低水平的微生物多样性。
– ASV(Amplicon Sequence Variants):
ASV采用较新的降噪方法,可以精确地解析序列中的每一个核苷酸差异,简单来说就是以100%相似度进行聚类,对低质量序列进行去除和校正,这种方法可以生成“零半径OTU”,即互不相同的基于序列的变体。
ASV通常使用误差校正算法来排除测序错误,从而提供更精确的序列变体识别。ASV方法对单一核苷酸变异敏感而能提供更细粒度的微生物多样性解析。ASV为每一种变异提供更一致、可复制的标识符,这在比较不同研究之间的微生物群落组成时非常有用。
简而言之,ASV方法提供了比OTU更高分辨率、更精准的序列变体检测。换句话说,ASV提供了一种微生物组多样性分析的“高清”视角,它更可能捕捉到微生物群落内变化的微妙差异,尤其是在不同环境或时间点间的比较中。
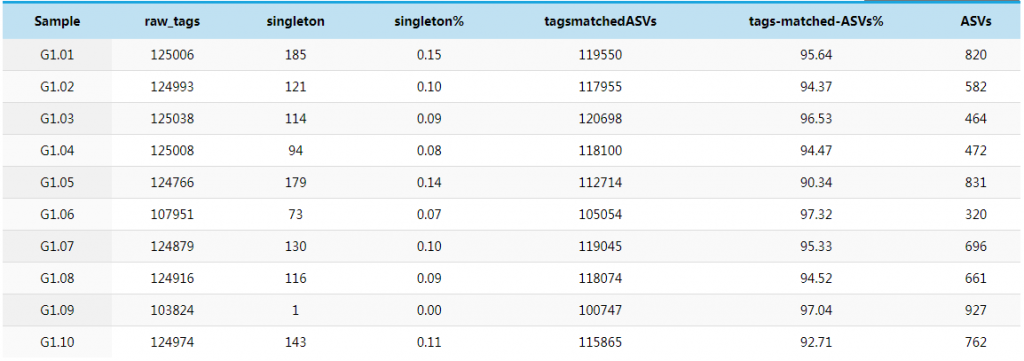
原始序列数据(raw tags)经过质控、过滤、去嵌合体,最终得到有效数据(effective tags)。再对有效数据进行UNOISE降噪处理,得到ASV特征表。数据处理过程中各步骤得到的序列进行途径统计,可以直观的反映每个样本的数据量和物种丰度。
文件目录:
01_pick_otu/summary/sumOTUPerSample.txt
raw-tags:每个样本的原始序列数据;
singleton :每个样本中无完全匹配的单条序列的数量。singleton ASV 是指只有单条代表序列的 ASV,可能由于测序错误,或者是来自于PCR过程中产生的嵌合体;
tagsmatchedASVs: 每个样本中比对到ASVs的最终有效序列数据 及其比例,聚类的同时vsearch会根据UCHIME算法将singleton ASV及嵌合体去除,得到最终的有效序列数据 Effective Tags;
ASVs:每个样本的ASVs数量。
一般文献中的测序原始数据量raw-tags 要求达到3万条以上,可以满足数据分析的基本要求。绝大多数文献数据量平均在5万条左右。世面上不同公司承诺的数据指标有所不同,谷禾测序得到的原始数据一般可以达到10万 reads左右,足够满足当前文章发表要求的参考数据量。
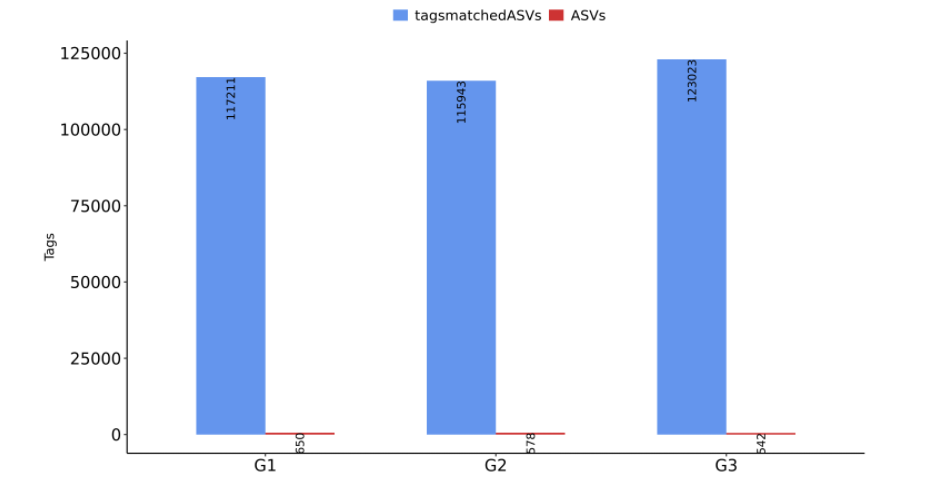
若原始数据量低于1万条,尤其是少于3000条reads以下,则很有可能受环境污染的杂带较多,建议重新上机补测数据。ASVS列可以反映每个样本的物种多样性,一般一个ASVs就代表一个物种。因此可以用ASV数量来代表物种数量。将每个样本的有效原始数量和ASVs数据可视化做成柱状图,可以更直观的观察每个样本/分组数据量的变化。
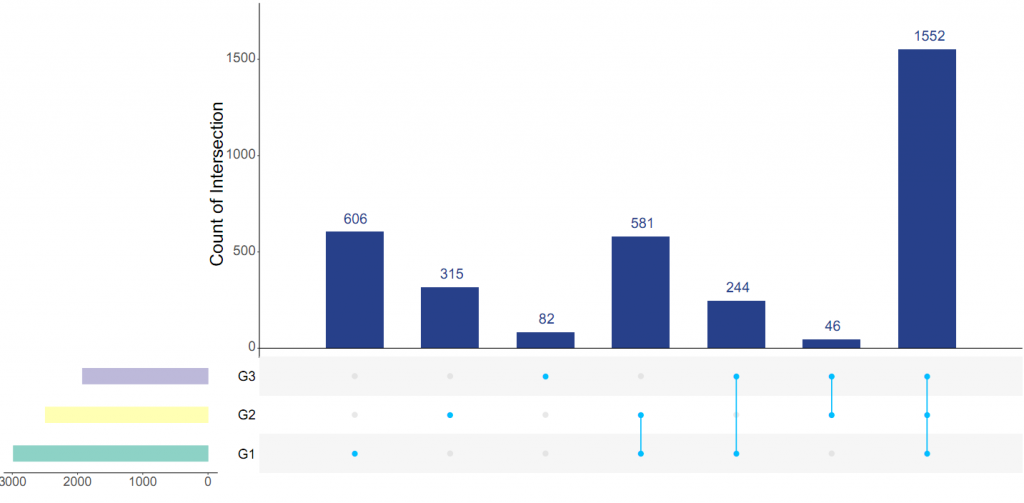
每个样本/分组可能会有一些共有的和独有的ASV,通常用韦恩图或花瓣图表示(样本数/分组数<=5个样本用维恩图,数量大于5出花瓣图)。除了用Venn图将几个数据集之间的交集进行可视化,还可以使用upset图表示。
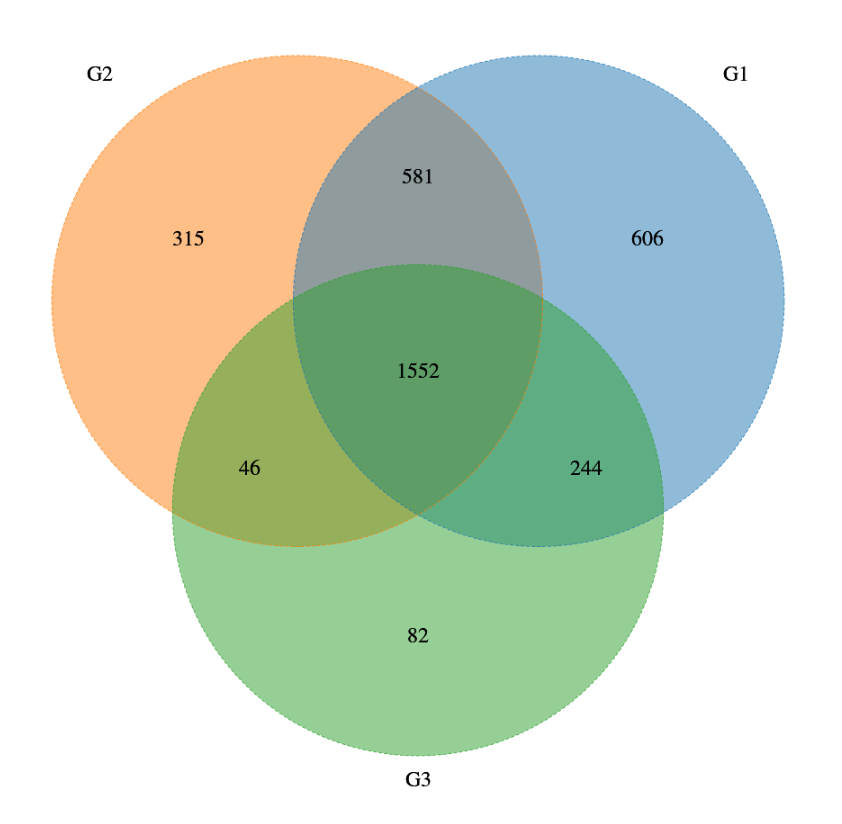
韦恩图中不同颜色的圆圈代表一个样本/分组,圈之间的重迭区域表示样本/分组间共有的ASVs,每个区域的数字大小表示该区域对应的ASVs数目。
UpSet图主要包含三个部分:上部分为各个分组独有和共有的ASV数量,下部分为各个分组独有和共有的分类情况,左部分每一个行代表一个分组。
alpha多样性主要用来衡量单个样本内的菌群多样性,不涉及样本之间的比较。alpha多样性与两个因素相关,分别是:一、丰富度(richness),二、多样性(diversity)。
丰富度指的是单个样本物种的种类数目;而多样性是指菌群在个体中分配的均匀度。样本的丰度高不一定就代表菌群的多样性丰富,丰度高如果是因为里边含有较多低丰度的杂带,这些可能是来源于环境的污染物导致的,这些低丰度的物种并不会使菌群的多样性增加。
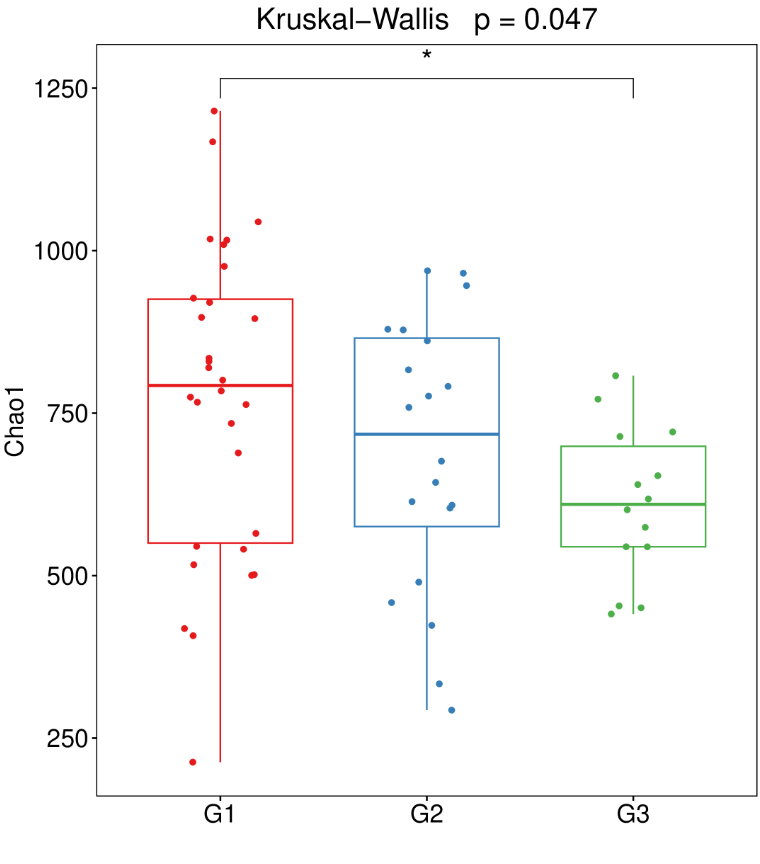
alpha多样性有三类相关指数,其中包括菌群丰度指数(Chao1和ACE)、菌群多样性指数(shannon和simpson)和测序深度指数(Goods coverage和Observed spieces)。
Chao1:Chao1算法用于评估样本中所含ASV数目的指数,Chao1在生态学中常用来估计物种总数,由Chao(1984)最早提出。通过计算群落中只检测到1次和2次的ASV数估计群落中实际存在的物种数。chao1指数可以评估一个样本中的ASV数量,chao指数越大,ASV数目越多,说明该样本物种数越多。
计算公式如下:
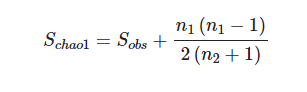
编辑
其中:
Schao1=估计的OTU数;
Sobs=观测到的OTU数;
n1=只有一条序列的OTU数目(如“singletons”);
n2=只有两条序列的OTU数目(如“doubletons”)。
ACE:用来估计群落中含有ASV数目的指数,由Chao提出,是生态学中估计物种总数的常用指数之一,与Chao1的算法不同。预设将序列量10以下的ASV都计算在内,从而估计群落中实际存在的物种数。
计算公式如下:
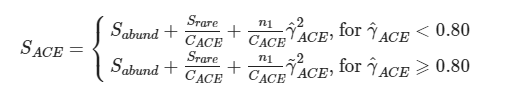
其中
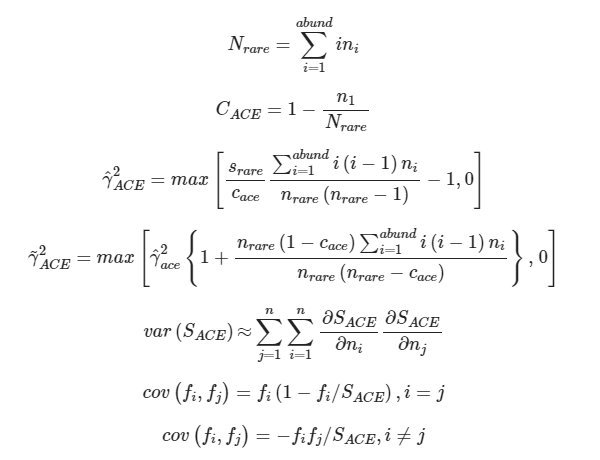
ni=含有i条序列的ASV数目;
Srare=含有“abund”条序列或者少于“abund”的OTU数目;
Sabund=多于“abund”条序列的OTU数目;
abund=被视为“优势”的ASV的阈值,默认为10。
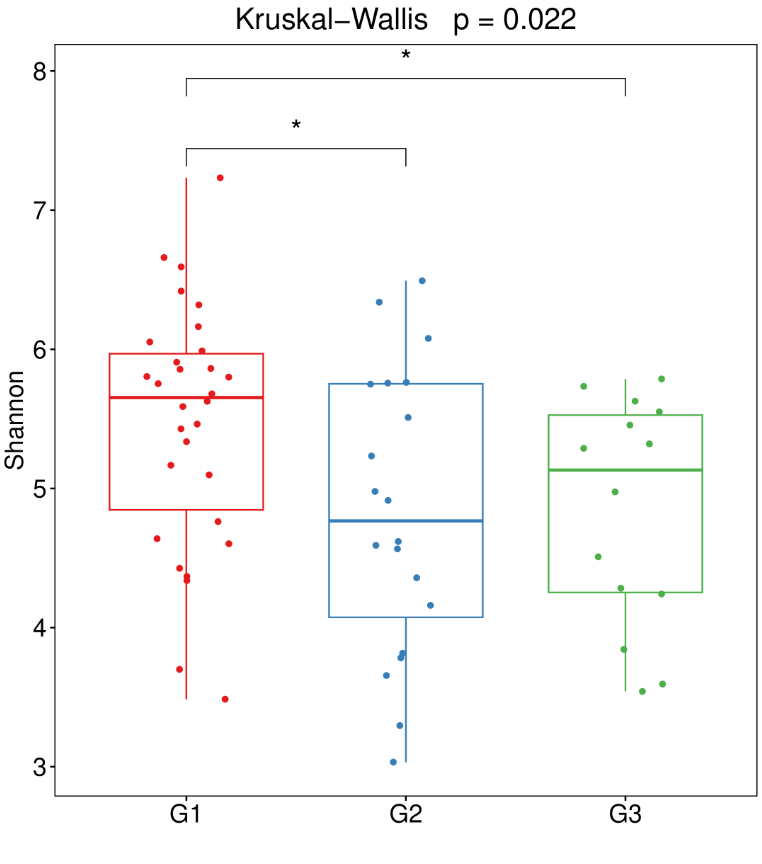
Shannon:香农-威纳指数综合考虑了群落的丰富度和均匀度,是用来评估样本中微生物多样性指数之一。Shannon指数值越高,表明群落的多样性越高。
计算公式如下:
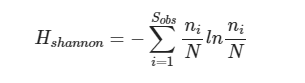
其中:
Sobs=观测到的ASV数目;
ni=含有i条序列的ASV数目;
N=所有的序列数。
Simpson:辛普森多样性指数对菌群多样性评估,Simpson指数值越高,表明群落多样性越高。由EdwardHugh Simpson(1949)提出,在生态学中常用来定量描述一个区域的生物多样性。一般而言,Shannon指数侧重对群落的丰富度以及稀有ASV,而Simpson指数侧重均匀度和群落中的优势ASV。
计算公式一如下:
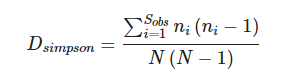
计算公式二如下:
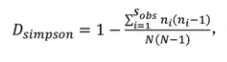
此时,Simpson指数越大,说明群落多样性越大。报告中用到的是计算公式二。
其中:
Sobs=观测到的ASV数目;
ni=含有i条序列的ASV数目;
N=所有的序列数。
Coverage:是指各样品克隆文库的覆盖率,其数值越高,则样品中序列被测出的概率越高,而没有被测出的概率越低。该指数反映本次测序结果是否代表了样品中微生物的真实情况。
计算公式如下:
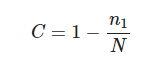
其中:
n1=只含有1条序列的ASV数目;
N=所有的序列数。
下表统计了每个样本的各项alpha多样性指标:
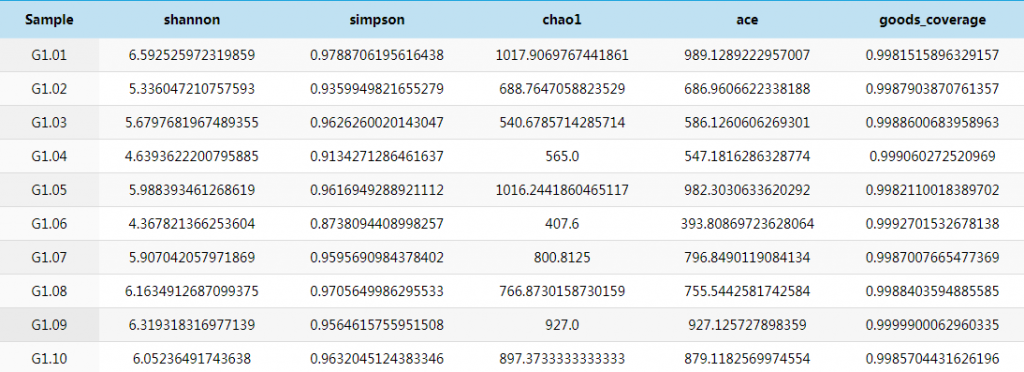
结果目录:
03_diversity-metrics/alpha/alpha_div.txt
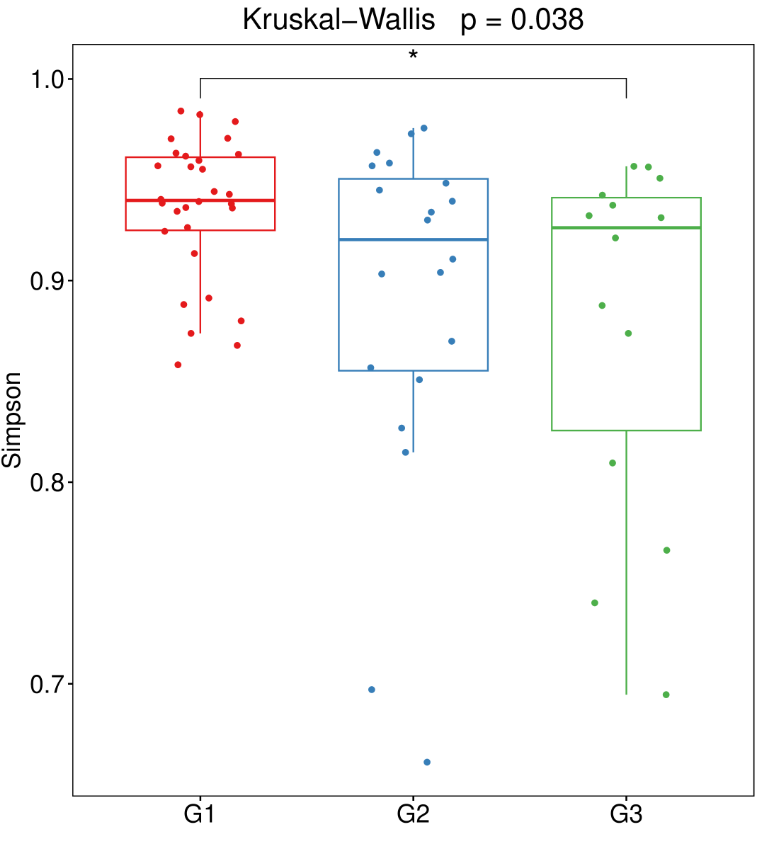
可以选择不同的alpha多样性指数进行显著性差异比较,一般常用丰富度指数Chao1,多样性指数Shannon、simpson,比较不同组间指数是否有显著差异。Alpha多样性分析将样本的菌群群整体研究并转换为具体的指数与p值,来说明群落的变化与差异。
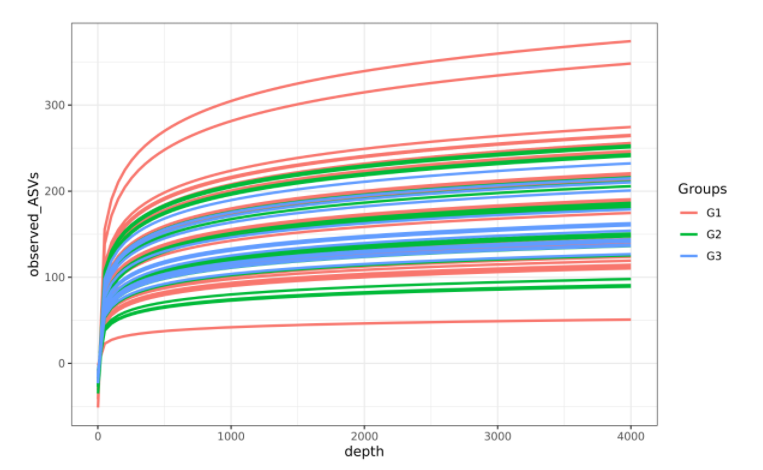
•稀释性曲线(Rarefaction curve)
稀释曲线是从每个样本中随机抽取一定数量的序列,统计这些序列所代表的ASV数目,以随机抽取的序列数与ASV数量来构建曲线。可以用来比较不同样本中的物种多样性,也可以用来说明样本出测序数据量是否足以反映环境中的物种多样性。
•
菌群多样性指数(shannon和simpson)
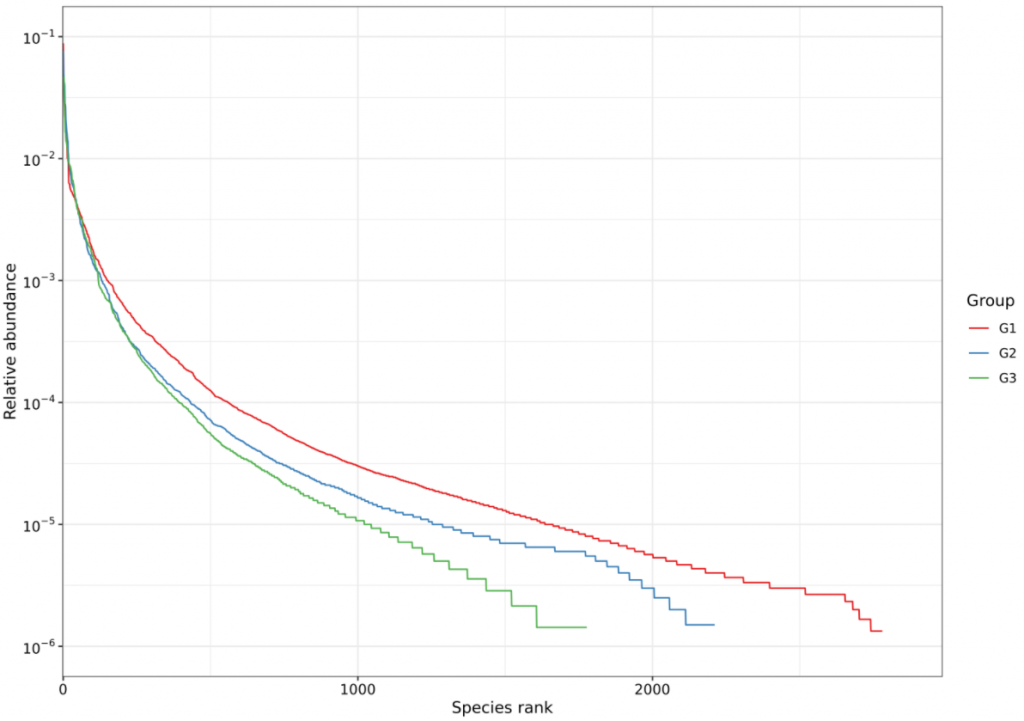
丰度等级曲线(Rank abundance curve)是分析多样性的一种方式。构建方法是统计单一样品中,每一个OTU所含的序列数,将OTU按丰度(所含有的序列条数)由大到小等级排序,再以OTU等级为横坐标,以每个OTU中所含的序列数(也可用OTU中序列数的相对百分含量)为纵坐标做图。
Rank-abundance曲线可用来解释多样性的两个方面,即物种丰度和物种均匀度。在水平方向,物种的丰度由曲线的宽度来反映,物种的丰度越高,曲线在横轴上的范围越大;曲线的形状(平滑程度)反映了样品中物种的均度,曲线越平缓,物种分布越均匀。
Beta多样性指的是样本间多样性,Beta多样性是衡量个体间菌落构成相似度的一个指标。通过计算样本间距离可以获得beta多样性距离矩阵,Beta多样性计算主要基于OTU的群落比较方法,有欧式距离、bray curtis距离等,这些方法优势在于算法简单,考虑物种丰度(有无)和均度(相对丰度),但其没有考虑OTUs之间的进化关系,认为OTU之间不存在进化上的联系,每个OTU间的关系平等。
另一种算法Unifrac距离法,是根据系统发生树进行比较,并根据16s的序列信息对OTU进行进化树分类,因此不同OTU之间的距离实际上有“远近”之分。而其他距离算法认为OTU之间的关系是平等的。Unifrac距离分为加权距离和非加权距离。
1
欧式距离(Euclidean distance):
欧几里得距离是空间中两点间“普通”(即直线)距离。
2
Bray-Curtis距离:
Bray-Curtis距离是生态学中用来衡量不同样地物种组成差异的测度。由J. Roger Bray and John T. Curtis 提出。其计算基于样本中不同物种组成的数量特征(多度,盖度,重要值等)。
计算公式为:
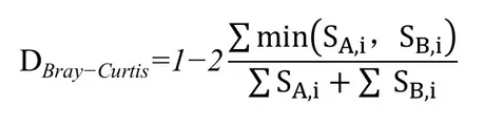
SA,i=表示A样本中第i个OTU所含的序列数;
SB,i=表示B样本中第i个OTU所含的序列数。
3
Unweighted UniFrac距离:
非加权距离包含特征之间的系统发育关系的群落差异定性度量。
4
Weighted UniFrac距离:
加权距离包含特征之间的系统发育关系的群落差异定量度量。
两者的区别在于:Weighted Unifrac 距离是一种同时考虑各样品中微生物的进化关系和物种的相对丰度,计算样品的距离,而Unweighted Unifrac则只考虑物种的有无,忽略物种间的相对丰度差异。
一般采用PCA、PCoA、NMDS等进行图像化展示,区分样本间的菌群组成差异。其原理是利用降维思想把样本平铺到二维平面上,使得相似的样品距离相近,相异的样品距离较远。
PCA图是基于ASV table的欧式距离,PCoA是基于两两样品之间的距离矩阵(有Bray-Curtis距离、加权距离、非加权距离),基于距离矩阵的统计检验方法有ANOSIM相似性分析和Adonis多元方差分析。
Anosim分析是一种非参数检验,用来检验组间差异是否显著大于组内差异,从而判断分组是否有意义。对 Anosim 的分析结果,基于两两样本之间的距离值排序获得的秩(组间的为 between,组内的为 within),这样任一两两组的比较可以获得三个分类的数据,并进行箱线图的展示(若两个箱的凹槽互不重迭,则表明它们的中位数有显著差异)。
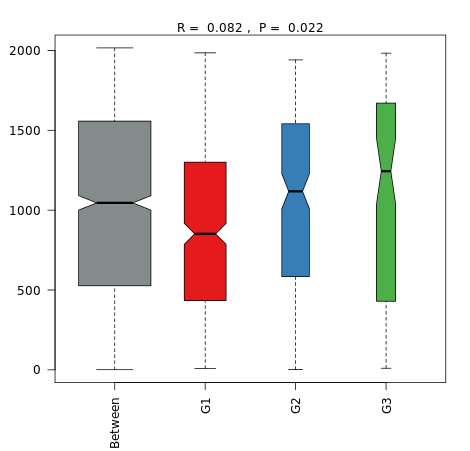
该方法主要有两个数值结果:R值,用于比较不同组间是否存在差异;P值,用于说明是否有显著差异。
R-value 介于(-1,1)之间,R-value > 0,说明组间差异大于组内差异。R-value < 0,说明组间差异小于组内差异, R只是组间是否有差异的数值表示,并不提供显著性说明。统计分析的可信度用 P-value 表示,P< 0.05 表示统计具有显著性。
Adonis检验,多元方差分析,其实就是PERMANOVA,亦可称为非参数多元方差分析。其原理是利用距离矩阵(比如基于Bray-Curtis距离、Unifrac距离)对总方差进行分解,分析不同分组因素对样品差异的解释度,并使用置换检验对其统计学意义进行显著性分析。它与Anosim的用途相似,也能够给出不同分组因素对样品差异的解释度(R值)与分组显著性(P值)。
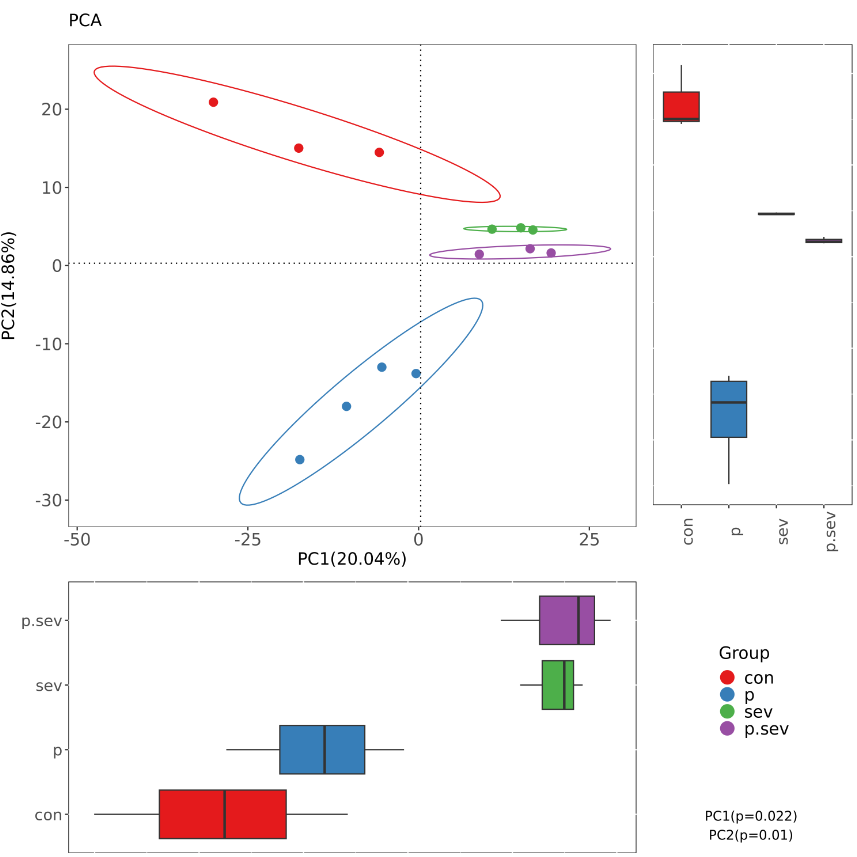
PCA(Principal Components Analysis)即主成分分析,首先利用线性变换,将数据变换到一个新的坐标系统中;然后再利用降维的思想,使得任何数据投影的第一大方差在第一个坐标(称为第一主成分)上,第二大方差在第二个坐标(第二主成分)上。
这种降维的思想首先减少数据集的维数,同时还保持数据集的对方差贡献最大的特征,最终使数据直观呈现在二维坐标系。经过一系列的特征值和特征向量进行排序后,选取PCA分析得到的前三个主成分(PC1、PC2和PC3)中的任意两个数据作图。通过PCA 可以观察个体或群体间的差异。
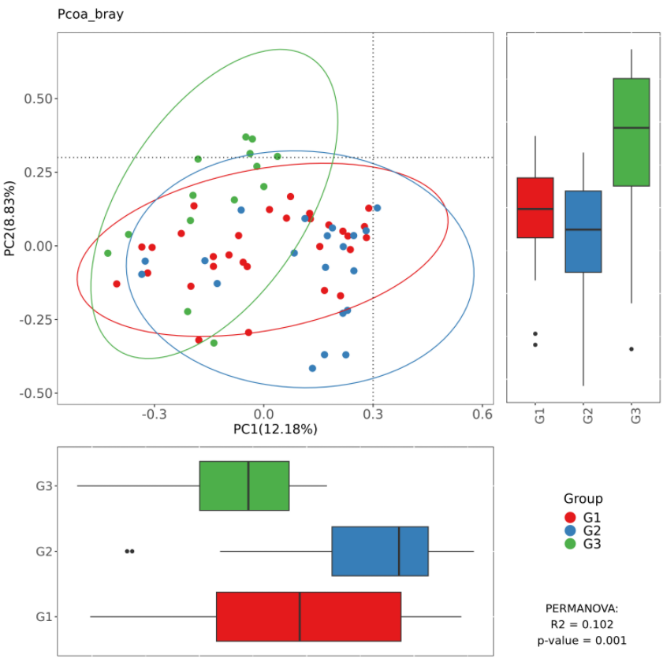
主坐标分析 PCoA (Principal component analysis)是一种非约束性的数据降维分析方法,可用来研究样本群落组成的相似性或相异性。通过 PCoA 可以观察个体>或群体间的差异。
它与PCA类似,两者的区别为PCA是基于样本的相似系数矩阵(如欧式距离)来寻找主成分,而PCoA是基于距离矩阵(欧式距离以外的其他距离)来寻找主坐标。我们基于Bray-Curtis 距离、 Weighted Unifrac 距离和Unweighted Unifrac 距离来进行 PCoA 分析。
该图是基于Bray-Curtis距离做的PCoA图,图中右下角的P值就是基于Adonis检验得到的结果:
编辑
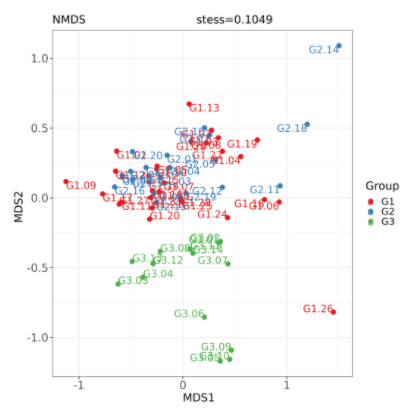
非度量多维尺度分析 NMDS 分析(Nonmetric Multidimensional Scaling)与上述 PcoA 分析类似,也是一种基于样本距离矩阵的分析方法,通过降维处理展现样本特定的距离分布。
与 PcoA 的区别是 NMDS 分析不依赖于特征根和特征向量的计算,而是通过对样本距离进行等级排序,使样本在低维空间中的排序尽可能符合彼此之间的距离远近关系(而非确切距离数值)。因此,NMDS 分析不受样本距离的数值影响,对于结构复杂的数据排序结果可能更稳定。

谷禾健康

在持续的肠道菌群检测实践过程中,我们收到很多新的问题反馈和对肠道菌群检测在具体问题中的疑问。在此谷禾基于长期和大规模样本群的经验以及实验分析,对部分常见问题进行汇总和整理。

一次肠道菌群检测好比一场健康考试,你拿到报告的那一刻,等同于拿到了你考的那张卷子,那么你首先会关心自己考了多少分。
在肠道菌群检测报告中,同样也有基于肠道菌群的健康评估分数,即健康总分。
基于大数据和整体性评估,报告中会给出健康总分这项指标。这个健康总分是如何计算得出的?
还是拿我们最熟悉不过的考试举例,一场语文考试可能包括了拼音词语、阅读理解、写作等模块,所以最后你的总分是综合各个模块的测试之后得到的(比如说拼音写错了扣1分,阅读理解错了一题扣5分……),通过各模块测评后得到的总分反映的是你的综合能力。
健康总分也是一样,综合计算了三个部分:肠道菌群健康状况、疾病风险情况和营养饮食均衡情况综合评估计算。总分100分,采取扣分制,疾病风险和营养不均衡以及菌群失衡都会相应的减分。
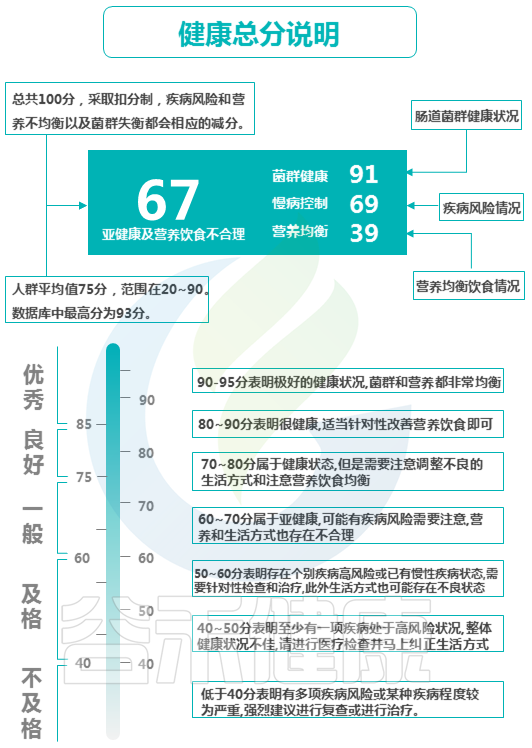
以上是具体的评分标准。
健康总分可以说是非常直观的一个指标,除此之外,整体性评估指标还有一个:肠道预测年龄。
生理年龄是指人达到某一时序年龄时生理和其功能所反映出来的水平,是从医学、生物学角度来衡量的。
谷禾肠道预测年龄是基于超过6万人群队列的深度学习模型构建的,对健康人群的肠道年龄预测与真实生理年龄吻合度很好。
肠道预测年龄和生理年龄就像齿轮运作,井井有条匹配状态,身体这个系统运作起来相对健康轻松。
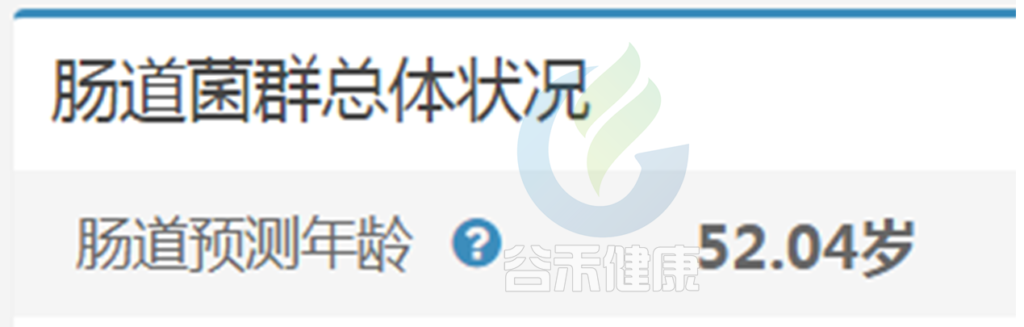
疾病人群或菌群紊乱人群,肠道年龄会较大偏离真实年龄,也就是这个齿轮系统出现一些偏差问题。
如果肠道菌群多样性下降,且以大肠杆菌为主,可能会被预测为10岁以下儿童,也就是预测年龄远小于真实年龄。
如果存在较多病原菌,则预测年龄会偏向远大于真实年龄。
如果菌群预测年龄和实际生理学年龄相差很大,如何解读?
还是用考试来说,每个年龄段都应具备该年龄段的能力。如果你是一个初中学生,那么就应该答出初中阶段学生该会的题,这时候给你做个测评,发现还停留在幼儿园水平或者已经到了大学生水平,要么太幼稚要么太早熟,都不符合健康的身心发展规律。
肠道预测年龄同样,如果肠道预测年龄偏离实际年龄很大,两种情况,一种是偏大,另一种是偏小。
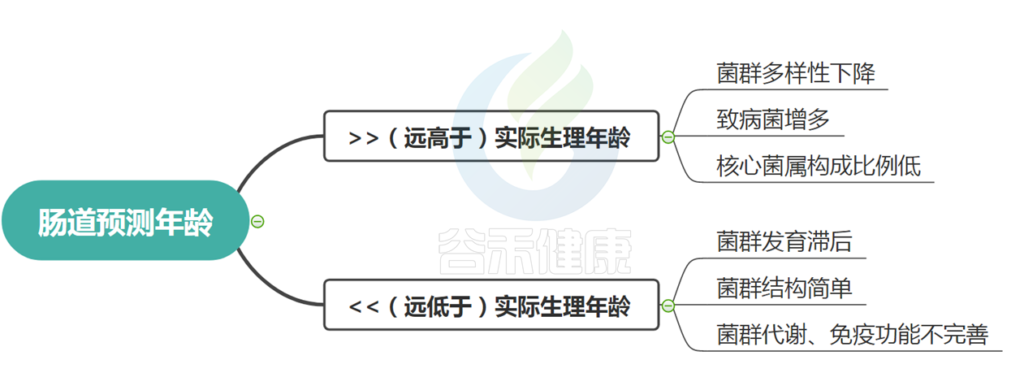
这两种情况均表明菌群发育成熟偏离了实际生长发育,我们均认为其代表菌群状况不太好,存在菌群异常或不健康状况。
如果偏小,即肠道年龄远小于生理学年龄,一般菌群发育滞后或者偏幼龄,菌群构成简单,代谢以及免疫功能不完善。
如果偏大,即肠道年龄远大于生理学年龄,一般菌群多样性下降,变形菌、肠杆菌等致病菌增多,核心菌属构成比例低等。
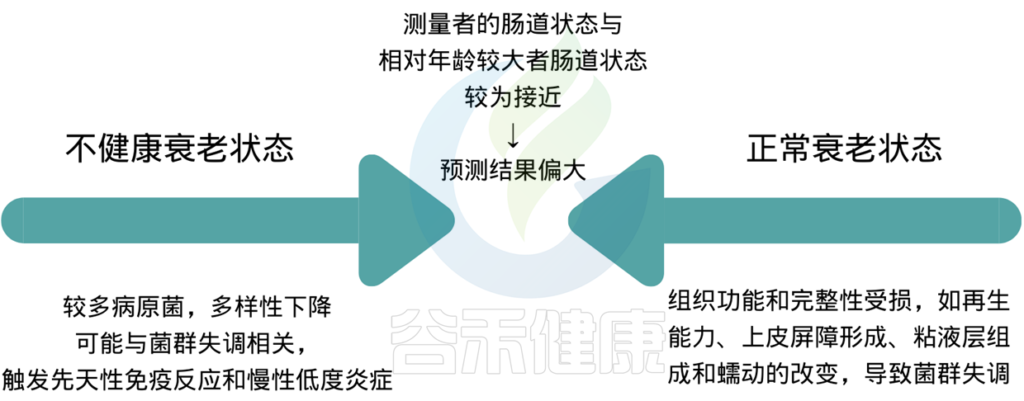
而在正常范围内,肠道预测年龄小于生理学年龄,那么表示菌群发育正常,菌群构成和代谢偏向于更年轻,比较好。那么什么是正常范围呢?
谷禾肠道年龄预测如下范围内表示正常:
0~2岁:偏差小于3个月
3~5岁:偏差在6个月以内
6~15岁:偏差在1岁左右
16~50岁:偏差在3岁以内
50岁以上:偏差在5岁以内
真实年龄与肠道预测年龄在范围内的差异可以反映其肠道菌群的发育和衰老状况。以下情况可能会导致肠道预测年龄完全偏离真实年龄,包括:
▪ 肠道菌群紊乱
▪ 菌群结构过于单一
▪ 近期服用可能严重干扰菌群的药物(如抗生素)
▪ 病原菌感染或者处于疾病状态
▪ 长期补充益生菌
由于肠道年龄考虑了整体的肠道菌群结构,如果肠道年龄严重偏离真实年龄,通过干预调整或去除上述干扰因素肠道年龄是能够恢复正常范围,但该干预周期一般需要1个月以上。
有益菌
有益菌包括益生菌,益生菌主要来自两个菌属:
分别是双歧杆菌属和乳杆菌属,目前已获得批准的有效益生菌菌株均来自这两个细菌属。
其中双歧杆菌可有效改善肠道状况,而特定的乳杆菌菌株可以改善精神健康,包括焦虑和情绪,也能改善肠道健康。双歧杆菌和乳杆菌也是人体肠道菌群中常见的菌。
虽然说是常见菌,却不见得它们数量多。在成年人肠道菌群中,双歧杆菌的比例较低,在1%左右,乳杆菌更是低于1%,甚至很多人(20~40%)的肠道菌群中比例低至万分之一。
下表是谷禾检测的益生菌列表,列出了主要的常见益生菌。
除了上述益生菌,有益菌还包括下列种属,这些菌属是构建肠道菌群的核心菌属,在评估有益菌水平时根据菌属对肠道菌群结构的重要性会给予不同的权重。
Faecalibacterium、Ruminococcus、Roseburia
Phascolarctobacterium、Prevotella、Parabacteroides
Oscillospira、Megamonas、Lachnospira
Lachnoclostridium、Gemmiger、Eubacterium
Coprococcus、Dorea、Dialister
Clostridium、Blautia、Bacteroides
Akkermansia、Alistipes、Agathobacter
通常益生菌的检出率比较低,一般在益生菌补充一周左右在报告中可以体现。从大数据来看,益生菌检出的同时,菌群的相关指标也会有所提升,比如说有害菌降低,改善菌群平衡状况。
有害菌
有害菌和肠道内的其他共生菌共同构成菌群微生态,也是大部分人群肠道内常见的菌群。
有害菌是相对而言的,正常肠道菌群也包含许多这些菌属的菌,但有害菌比例或个别菌属丰度超标可能预示着肠道菌群的健康状况受到破坏。这些菌过多会影响肠道内环境,如pH值,含氧量以及肠道内毒素等,可能会导致出现一些机会感染和机会致病菌入侵,进而诱发炎症和疾病。
我们报告中的有害菌包含了致病菌和条件致病菌,以及属内主要菌种为致病菌的属。为便于统计,我们在计算的时候统一按照属层级进行计算比例。
报告中的有害菌包括了以下的菌属:韦荣氏球菌属、葡萄球菌科、变形菌属、弓形菌属、弯曲菌属、螺杆菌属、厌氧螺菌属以及弧菌属等。
在肠道菌群检测报告中会有对有益菌,有害菌的整体评估。
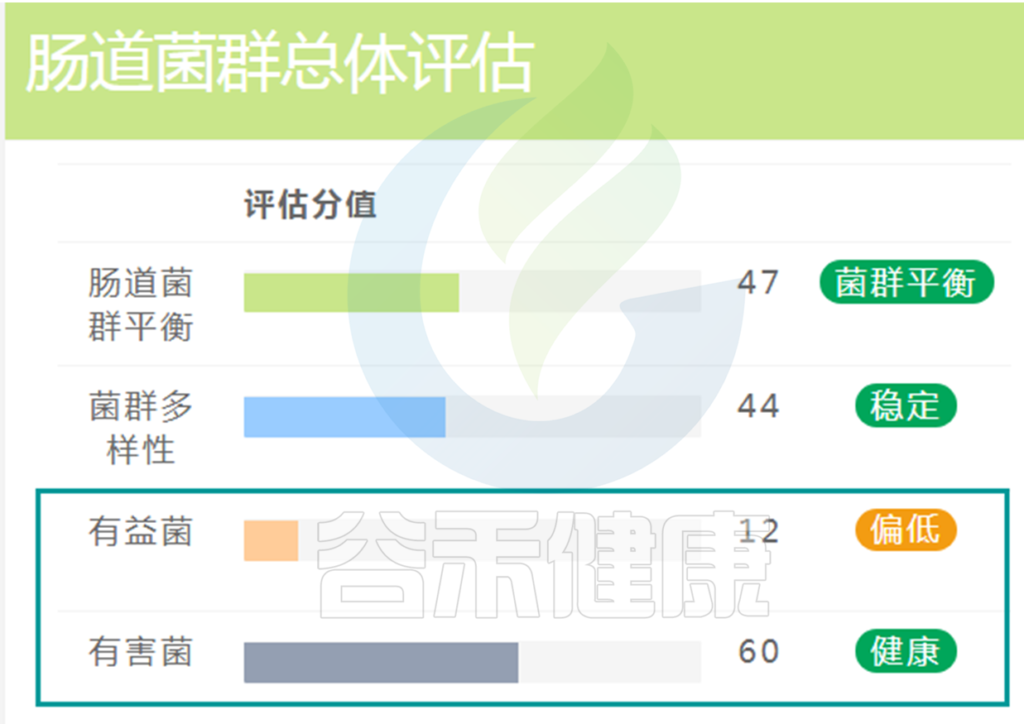
如果有害菌过多,通常建议服用益生菌或益生元的方式首先增加有益菌的比例,相应的有害菌比例就会降低。想要持久的改善菌群结构降低有害菌水平就需要改善生活方式,适当增加抗性淀粉等膳食纤维并规律饮食和睡眠,增加运动等。
整个生态系统平衡对于地球而言十分重要,同理,肠道菌群平衡对于我们人体健康也很重要。健康的肠道菌群丰富且多样性高。
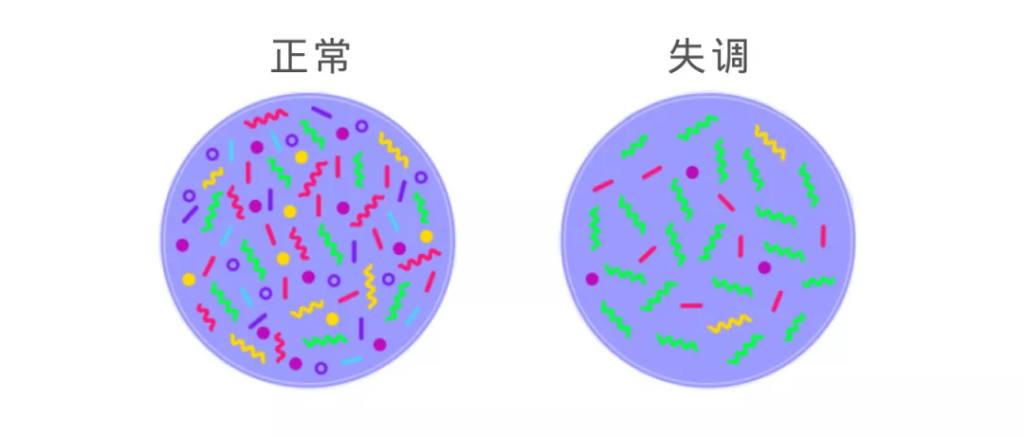
菌群失调是指体内微生物群不平衡,这可以表现为某些细菌的出现率较高,细菌的出现率较低,细菌的多样性不足,有害菌,有益菌比例失调等。
通常临床上采用大便常规检查,通过显微镜下观察统计染色细菌中杆菌和球菌以及革兰氏阴性和阳性菌的比值是否超标来判别的。
其中致病菌多为球菌和革兰氏阴性菌,而肠道有益菌多为杆菌和阳性菌,因而在传统临床上简单比较两者的比值评估是否菌群紊乱,是相对比较粗放的。
谷禾菌群检测报告中的菌群失调:
基于高通量测序可以精准的检测低至万分之一水平的菌,甚至可以分类到种水平,因此可以更加精细化评估菌群是否出现紊乱和异常。
基于谷禾超过30万人群的菌群数据库分析结果,我们将在90%的人群都有检出,且人群平均丰度1%以上的菌属做为核心菌属。这些核心菌属通过长期与人类共生,在帮助消化复杂碳水化合物和产生短链脂肪酸外还影响整个肠道环境,抑制病原微生物的定植生长。因此当这些核心菌属占总肠道菌群比例低于60%时,肠道菌群很可能处于紊乱状态。
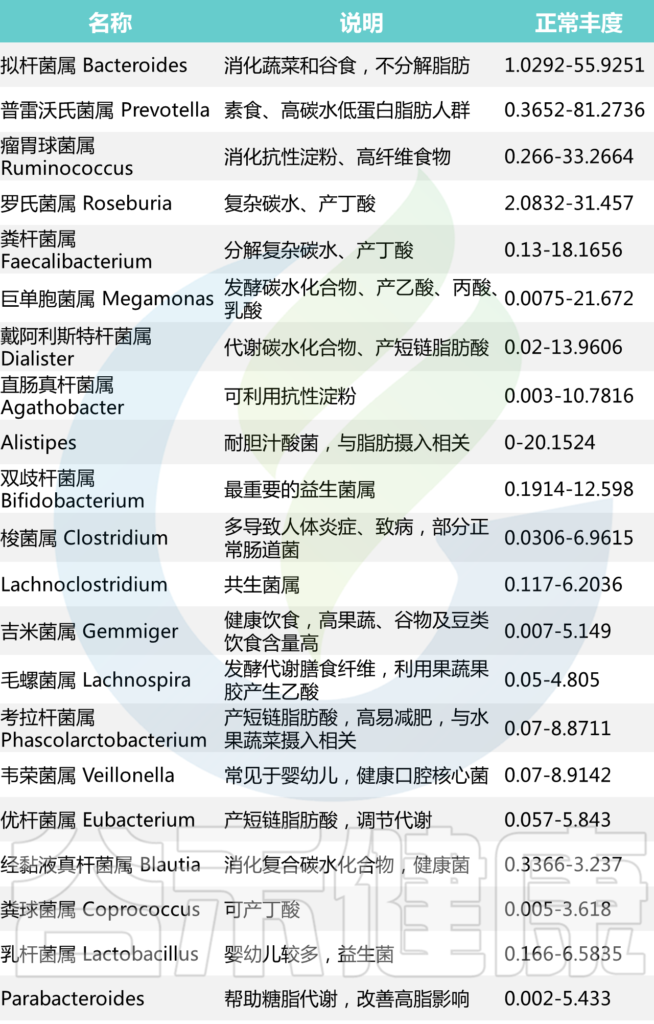
【谷禾健康菌群数据库】
如果出现菌群严重失衡,例如致病菌占了相当大比例,那么首先应考虑针对致病菌使用相应的抗生素治疗,然后再通过益生菌补充及饮食、生活方式的改变进行调理,直到菌群恢复平衡。
多样性包含两个维度。
一个是肠道菌群种类,人群中肠道菌群的种类参考范围在100~2000种,种类数量越多多样性越高。
另一个维度是均匀性,即各个菌种的含量丰度较为均一没有出现单一菌种占据绝大部分的情况。
多样性的评估一般通过一个叫做香农-维纳多样性指数的指标来进行评估,计算公式为:
H=-∑(Pi)(log2Pi)
其中Pi为每个菌的占比例,值越大代表物种种类越多,均匀性也更好相应的多样性也越高。正常人群中香浓指数在2~9之间,一般大于3以上表明具有一定多样性。
换句话说,肠道菌群多样性表现在:微生态系统的稳定性,以及面对外界致病菌等入侵的抵御能力。
在一定范围内,更高的多样性通常代表饮食更加丰富多样,同时也意味着更健康的身体状况。
菌群多样性高可能与下列情况有关:
环境,农村儿童比城市儿童菌群多样性高;
饮食,低脂饮食与菌群多样性较高有关;
年龄,长寿老人的菌群多样性较高;
……
多样性低不代表一定有疾病,但是更容易受到饮食,环境或疾病的影响,包括更易发生水土不服或更容易因饮食不洁导致腹泻等。
多样性低可能与下列情况有关:
分娩方式,剖腹产宝宝菌群多样性较低;
饮食营养,营养不良的孩子菌群多样性会下降;
药物,抗生素的使用会大幅降低菌群多样性,并且需要一段时间才能恢复。其他药物也会降低菌群多样性,如治疗胃溃疡和反酸的质子泵类药物也会导致菌群多样性降低;
环境,医院的ICU病房、更衣室等消毒严格,可能导致环境菌群多样性下降。
此外,神经系统、代谢、免疫等慢性疾病也与多样性下降有关。
你可以通过在饮食中增加纤维素,从高脂饮食逐渐转为低脂饮食来提高菌群多样性,另外规律运动也可增加多样性。
另外,我们在实际检测中会发现有这样一种情况:
多样性指标虽然很高,但是整体看起来健康总分并不理想。甚至还有很多慢性疾病风险,这是为什么呢?
这种情况可能是核心菌群丰度不够,核心菌群在代谢、免疫等方面都发挥重要作用,一旦核心菌群丰度下降,则可能造成外源物质侵入。感染、旅行等可能会出现这种情况。
看过我们检测报告的可能会发现,报告里有包括肠道致病菌和病原菌,分别代表什么?

<篇幅关系,此处仅展示部分>
肠道致病菌列出了最主要和常见的感染类肠道致病菌。(注意这里重点是肠道)

病原菌中给出的包括几十种人体的致病菌,不仅仅是肠道的。<如果没有检出就没有列出>
病原菌和条件致病菌的区别是什么?
病原菌一般极少存在于健康人的肠道菌群,正常范围很小,条件致病菌一般会在正常人群的肠道内存在,丰度较高或菌群结构单一到一定程度会引发疾病。如大肠杆菌和肺炎克雷伯氏菌正常人群中都会有检出,但当丰度较高是就会导致肠道菌群紊乱或疾病。
报告中如果出现病原菌超标的情况,不一定直接认为有病,需要结合症状。
如果出现相应的腹泻等症状,需要考虑是不是因为这些病原菌导致的。单纯超标如果没有症状只是表面有病原菌摄入,注意一下饮食和生活卫生,无须过于担心。
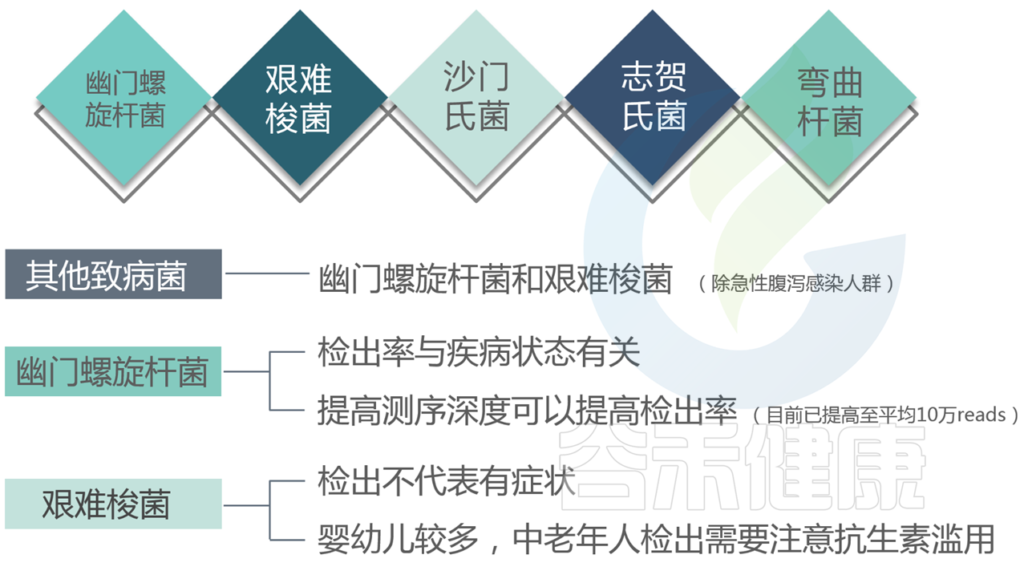
★ 幽门螺杆菌
为什么在医院检查出幽门螺杆菌感染,而报告中并未显示?
注意:本检测未检出并不代表完全不存在该致病菌感染,可能由于比例或其他因素未能达到检测丰度或未检出。
如果肠道菌群检测报告中检出幽门螺杆菌,是否需要去医院进行幽门螺杆菌呼气检测?
如果肠道菌群检测报告显示该项为超标,且同时存在胃部不适或其他胃酸、胃胀等症状,建议前往医院进行幽门螺旋杆菌检测,及早发现治疗。
★ 沙门氏菌
在食物中毒案例中,通常伴随着沙门氏菌,沙门氏菌粘附到肠上皮表面是发病机制中重要的第一步,并且是其在肠道定植的核心。
关于沙门氏菌的治疗及预防详见:食物中毒一文
扩展阅读:细菌大盘点(二) | 葡萄球菌、沙门氏菌、弯曲杆菌
通过以上部分,我们大概了解了菌群的构成及其扮演的角色,那么我们能利用检测到的这些菌的信息,给我们的健康带来什么帮助呢?
很重要的几个点:
第一,也就是前面所述的,菌群的构成本身就可以反映你的肠道内的环境是不是健康菌群,如果紊乱,它会带来很多的问题,比如说儿童菌群紊乱,可能会营养不良,因为菌群紊乱本身会影响营养吸收。
第二,对病原物的抵抗,也就是说身体是不是比较容易出一些状况,比如说腹泻,感染等问题。
第三,它还会诱发一些长期的慢性疾病,比如说糖尿病,实际上当然饮食是一个问题,但是有一些炎症相关的菌群,会诱发慢性的持续的炎症,从而导致慢性疾病的发展。
这就是我们接来下要讲的,疾病风险这块内容。
目前我们疾病风险检测部分包括16类主要疾病,根据疾病检测准确度和稳定性,我们将检测疾病的水平分为三个等级:低风险、中风险和高风险。
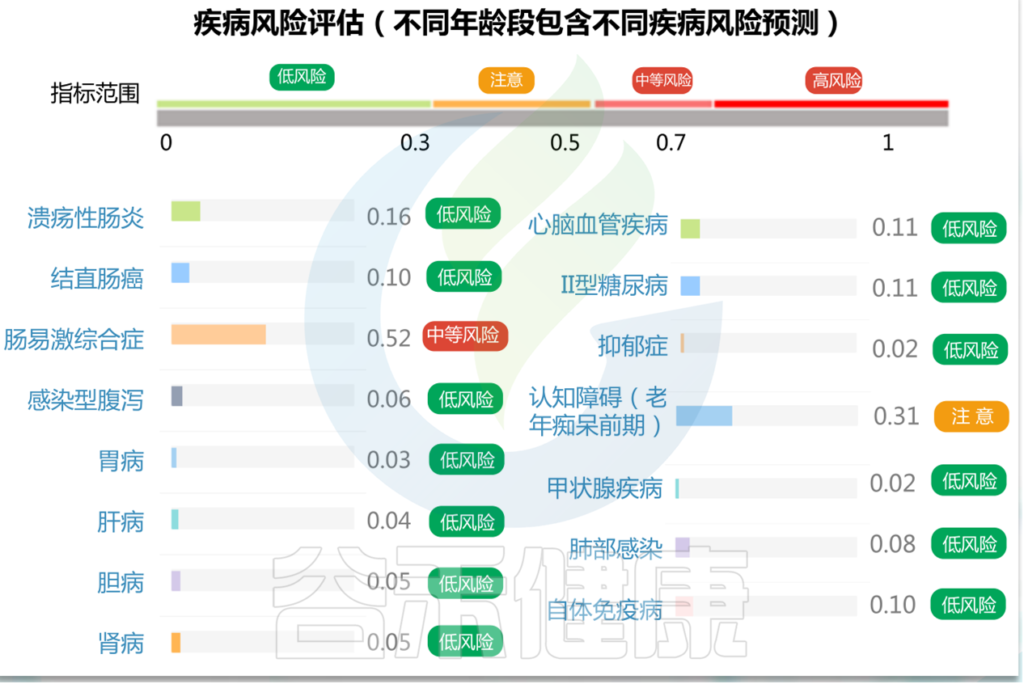
根据每种病的分值,0~0.3归为低风险,0.3~0.5评估为注意,0.5~0.7为中等风险,超过0.7为高风险。
目前报告中提供的疾病均经过大量病例样本检验并且准确率超过90%,虽然不作为疾病的诊断依据,但是其分值的高低仍然具有很强的指示作用。
0-0.3
如果某种疾病的风险值低于0.3以下表明菌群状态提示疾病风险较低,不同身体条件和生活方式下会有0.05的波动。
0.3-0.5
如果某种疾病的风险值位于0.3~0.5之间我们认为属于疾病前期阶段,通过饮食调理和相应的注意就可以降低风险。
0.5-0.7
如果某种疾病的风险值位于0.5~0.7之间表明可能患有该疾病或处于疾病风险阶段,这时候我们建议最好前往医院相关科室进行检查,如果不便前往医院也可根据建议先进行饮食调理和相应的注意,一般一个月后再进行一次检测查看疾病风险是否下降到正常范围,如果仍然较高甚至升高建议最好前往医院复查。
0.7- 1
如果某种疾病的风险值超过0.7表明有很大可能已患有该疾病,且分值越高表明风险越高。因此我们强烈建议去医院进行相应检查并听从医生建议。
注意:本检测目前尚不属于医疗诊断,疾病分值作为提示,低分值不代表完全没有疾病,只表示风险较低,也可能存在一定的未检出。高分值只表示存在很大疾病风险,疾病的确诊和精确诊断需要通过进一步的医疗检查确认。
说到这里,可能有人对以上这个0.3,0.5…这些风险值有所不解,风险值是你们自己确定的吗?如何计算得出这个值的呢?有参考依据吗?
这里我们来了解一下风险值的计算。
通过模型的构建和大规模人群队列的测试和学习,现在大概已经有几十种病,我们可以比较好的通过菌的构成,来预测到底有没有这个疾病。虽然现在它还做不到直接确诊,但它可以起到一个很好的提示作用,以及对病程进展的评估。
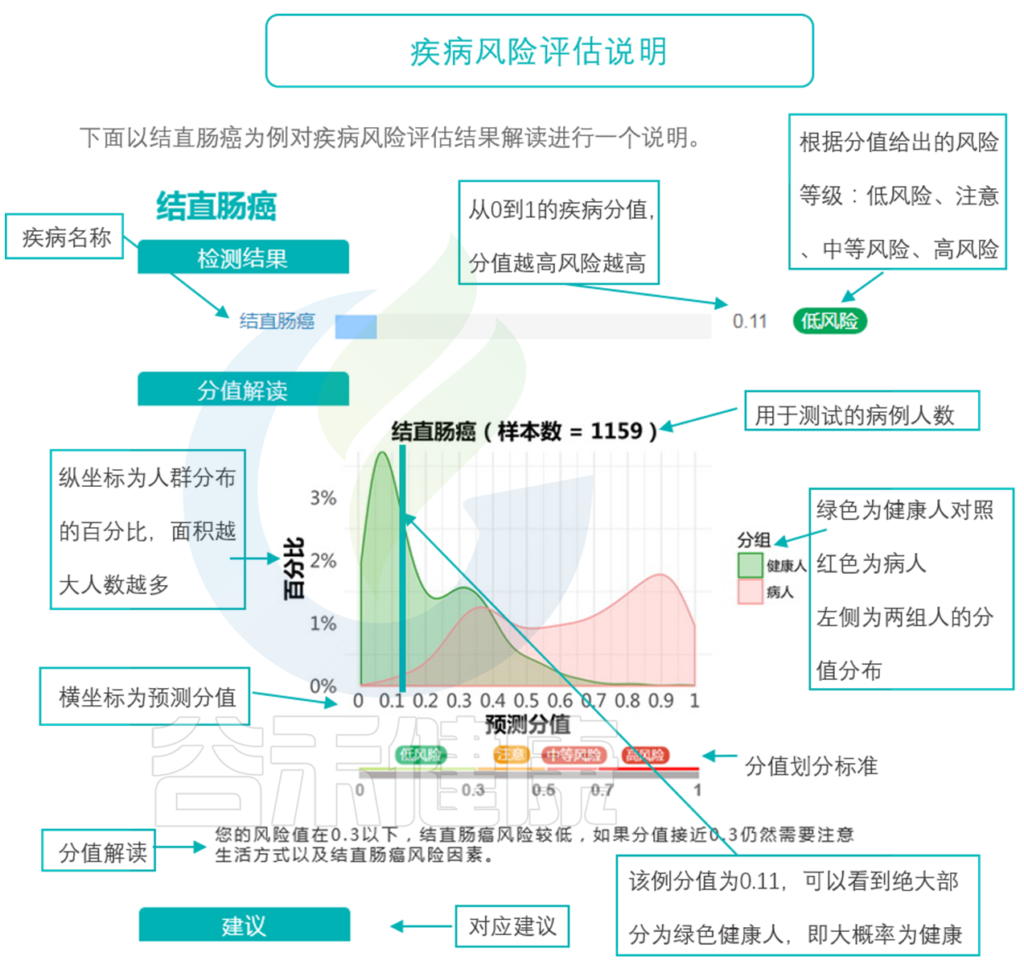
那么,具体哪些方面的疾病跟菌群有重要的关系,并且能够用菌群来预测和评估呢?
消化系统疾病
首先当然是消化道疾病,这很好理解,因为菌群本身就在消化道环境内。像肠炎,就包括克罗恩病,溃疡性结肠炎之类的,还有消化性的腹痛、腹胀这些问题,可能是由于菌群的特征变化造成。
炎症性肠病中的菌群失调
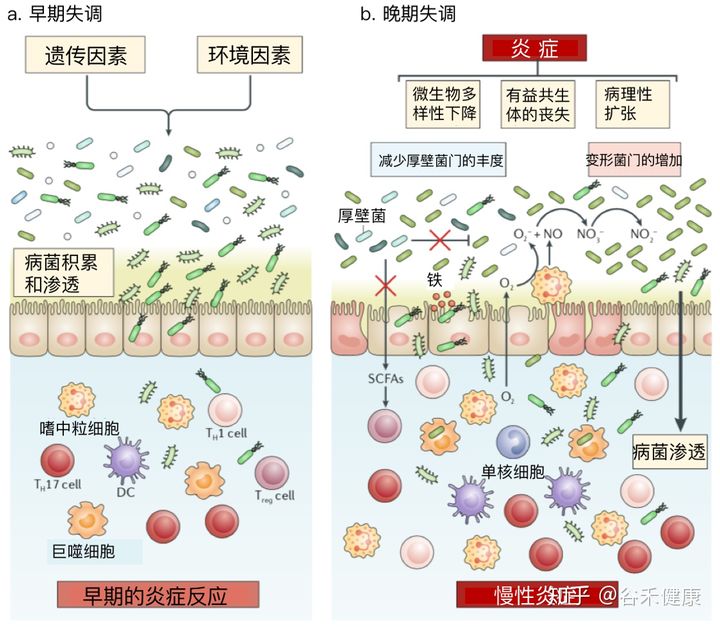
详见:炎症性肠病一文
还有过敏性腹泻,有人可能对一些食物过敏,吃完之后会导致一些腹泻,菌群特征变化很明显,包括甚至一些肠道病毒的感染,比如说诺如病毒、轮状病毒的感染。它也会体现出非常特定的菌群变化特征。
在肠道菌群检测报告中,这类疾病风险呈现如下:
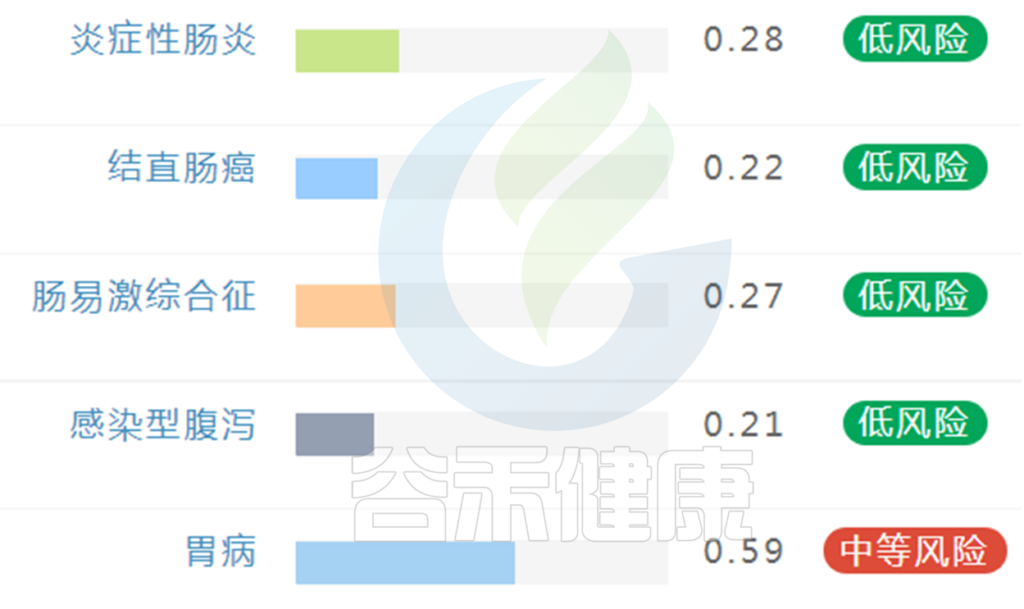
上图样本可以看到胃病有中等发现,其备注信息里有填:胃痛,可能要开始注意这方面的疾病隐患,通过饮食等调理一段时间,或前往医院就诊。
★ 胃癌
胃部更严重一点的疾病就是胃癌,胃癌与肠道菌群之间也有很大关系,最近,在“谷禾开放基金项目”中,也有相关论文也已发表。
肠道菌群在区分胃癌患者和健康人方面具有高度的敏感性和特异性,表明肠道微生物群是胃癌诊断的潜在无创工具。
胃炎与胃癌具有某些微生物群特征,化疗降低了胃癌患者的微生物丰度和多样性。乳酸杆菌Lactobacillus和巨球菌Megasphaera,是胃癌的预测标志物。
★ 结直肠癌
现在已经有多项研究表明,通过菌群可以做一个很好的标志物。虽然做不到所有的结直肠癌患者都能够被检出,但是最终的准确率相对来说还是挺高的,甚至比一些,包括肿瘤标注可能还要更高一些。
我们现在大概能做到70%多的肿瘤患者是能被筛查出来。并且准确度其实能够到90%,作为普筛或者健康评估来说,已经是一个比较有效的标志物了。
化疗与手术会大幅降低风险分值,但仍比健康人高。
此外,结直肠癌会经历从息肉到腺瘤到癌症多个阶段,应结合年龄和家族史判断息肉和结直肠癌。
肝胆类疾病
肝脏类疾病,比如说非酒精性脂肪肝跟肠道菌群有相当大的关系。
不同肝病有不同的菌群特征,尤其是脂肪肝的严重程度,肝功能异常的严重程度。
扩展阅读:深度解析 | 肠道菌群与慢性肝病,肝癌
因为菌群会产生大量的刺激代谢物,这些代谢物本身可能会加重肝脏的负担,并且诱发一些肝脏的疾病,但反过来肝脏的代谢能力的减弱和一些慢性肝脏疾病进展又会反映在菌群的构成上,所以它们是相互的。当然也可以用菌群的构成来反映具体肝病的特征。

由于不同阶段肝功能异常,脂肪肝等情况都统一归类在肝病这个大类,因此目前还无法判断确切的疾病分类,后续如果有更多细分疾病的样本用于建模,报告也会随之迭代更新。
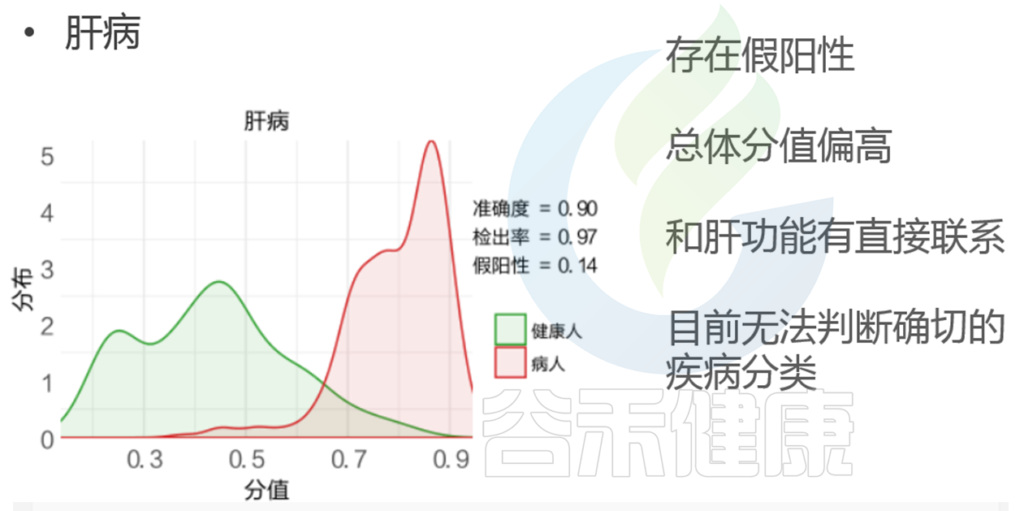
代谢类疾病
代谢类疾病,比如糖尿病,肥胖等,都与肠道菌群有密切关联。
★ 2型糖尿病
2型糖尿病的发病率越来越高,也有更多人开始关注菌群与2型糖尿病的关系。很多文献都有报道它们之间的关联性。
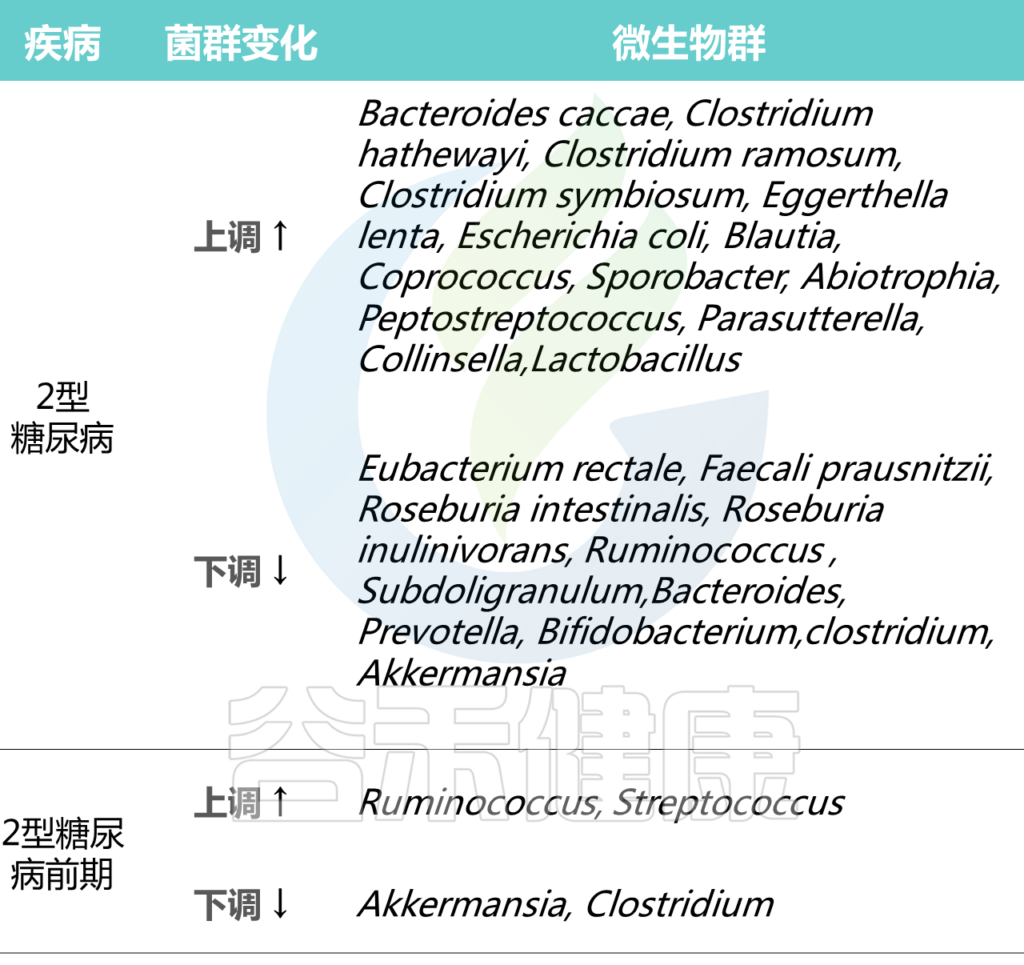
2型糖尿病人群中个体微生物群的差异
Cunningham A L et al., Gut Pathog, 2021
在2型糖尿病患者普遍具有相对高丰度的特定属:Blautia、Coprococcus、Sporobacter、Abiotrophia、Peptostreptococcus、Parasutterella、Collinsella。
2型糖尿病患者中,产生丁酸菌特别缺乏,特别是梭菌目,包括:
Ruminococcus、Subdoligranulum,Eubacterium rectale、Faecali prausnitzii、Roseburia intestinalis 、
Roseburia inulinivorans
通过肠道菌群检测,一方面健康人群可以查看是否有患病风险,另一方面如果已经患病人群,也可以查看菌群是否异常,推测是否是因菌群显著变化导致的,从而能进行更有针对性的干预。
肠道菌群检测报告中疾病风险预测如下:
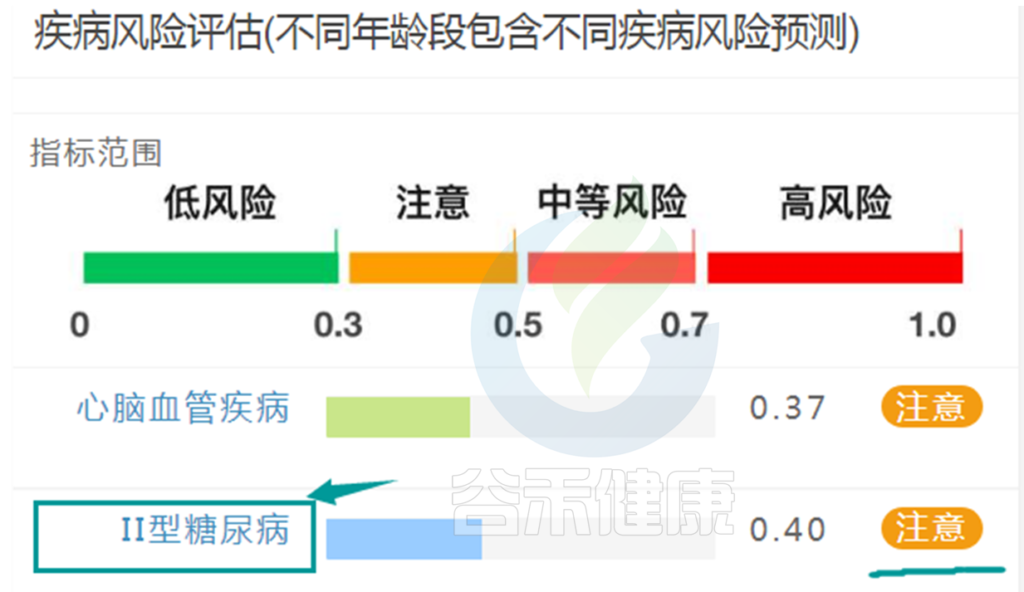
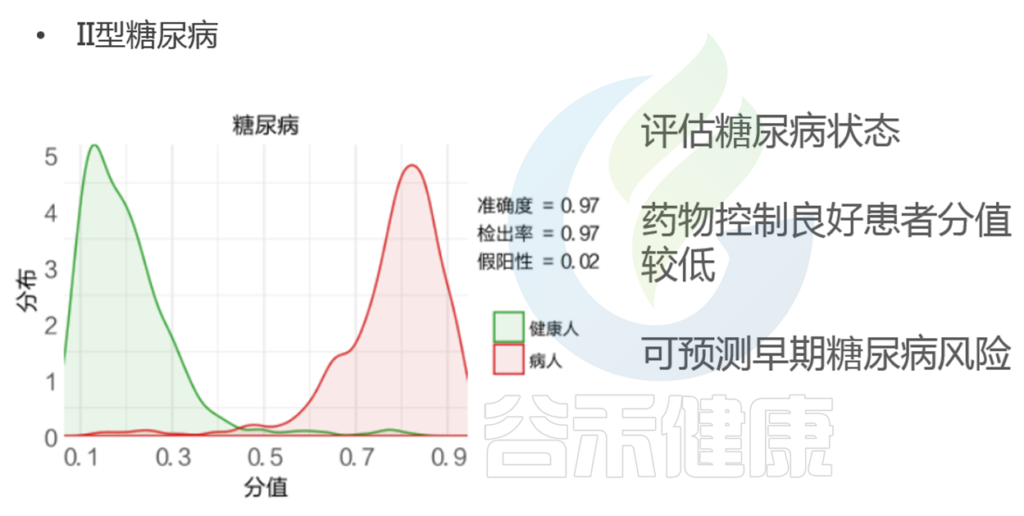
2型糖尿病的检出率相对较高,可以达到95%以上,准确的也较高,可以预测早期糖尿病风险。
★ 肥胖
目前已有很多关于肠道菌群和肥胖之间关系的研究。
人体摄入大量营养素、肠道菌群与肥胖的关系
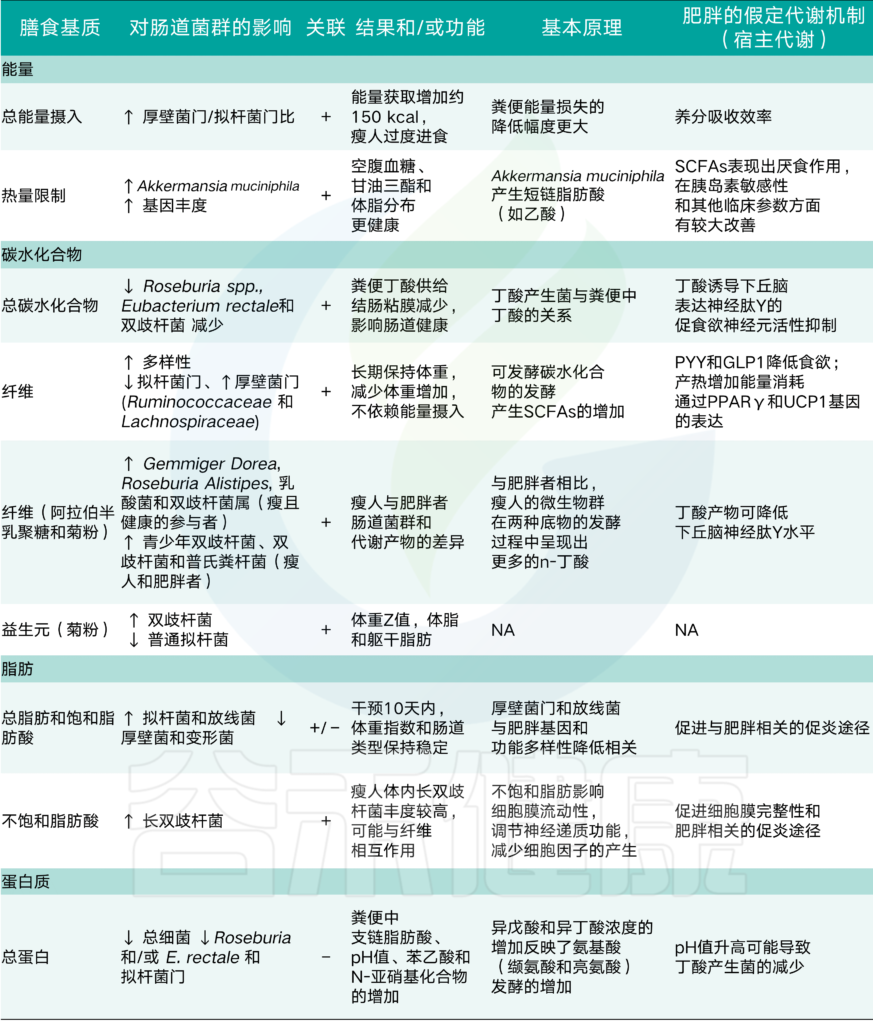
↑, 增加;↓,减少;NA,不可用;第三列:营养物质和/或饮食基质与肠道微生物群之间的关系
有人说,为什么我们的肠道菌群报告没有判别测试者是否肥胖?
首先,肥胖不肥胖这个症状是肉眼可见的,也就是说测试者自身已经了解,这种情况下用模型来判别没有意义。
而我们更希望通过肠道菌群检测来可以告诉你,可能是什么因素造成的肥胖,饮食结构的,还是某些菌属代谢问题。
通过菌群知道营养构成,以及是否存在一些特定代谢菌的异常,比如说Akk菌,它是一种在一定程度上帮助减肥的菌群。
如果在你的肠道内该菌特别少,那么可能同样减肥,控制饮食,别人一个月假设瘦十斤,你就不一定能达到这个效果。这些都是菌群可以提供的一些信息。
在肠道菌群检测报告中,会列出肥胖正负相关菌群,及其是否超标。
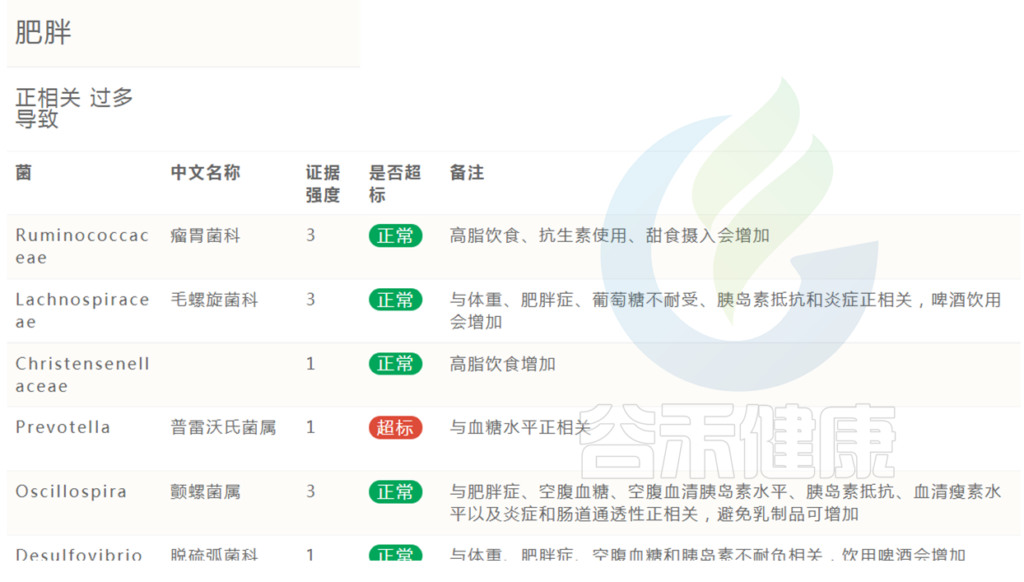
同理,其他各类肉眼可见的症状(包括腹泻、便秘、腹胀、过敏、皮肤状况等)正负相关菌群都会在报告中呈现,此处就不一一列举。
神经系统疾病
听起来神经系统好像没什么关系,但实际上很多肠道菌群能代谢产生大量神经递质及其他代谢产物。
肠道菌群会影响HPA轴的发育,该轴调节压力反应并参与皮质醇的释放。在抑郁和长期处于压力下的人中,HPA轴可能失调,导致过量的皮质醇(一种压力激素)被循环。
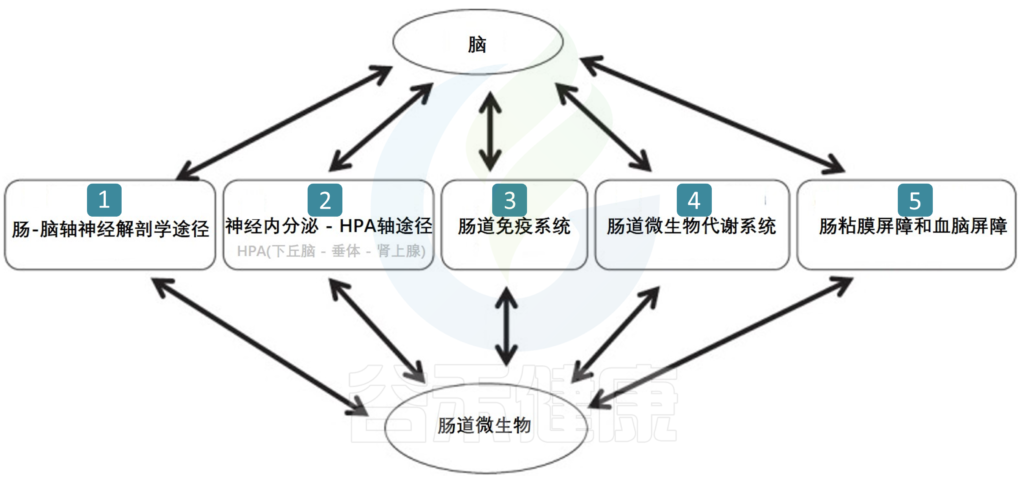
肠道菌群的部分代谢物质也会通过免疫系统影响神经系统。促炎性细胞因子的失衡可导致慢性炎症和自身免疫性疾病,通常与抑郁症同时发生。
通过肠道菌群检测,可以了解体内血清素水平及激素水平,同时也可以了解神经系统相关疾病风险,包括自闭症,抑郁症,阿尔兹海默症等。

肺部疾病
宿主,微生物组和环境之间的三重相互作用在健康功能中维持了肺稳态。
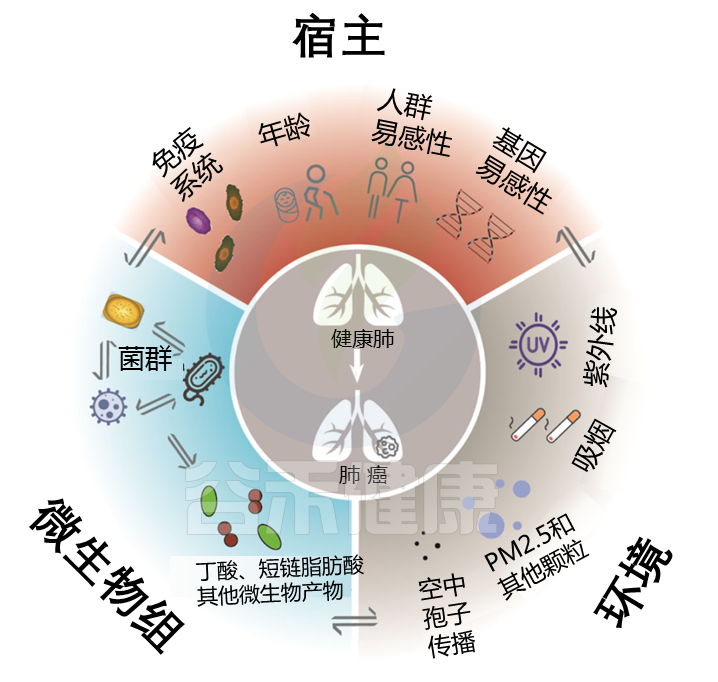
Liu NN, et al., NPJ Precis Oncol. 2020
在大量的临床样本数据当中可以发现,肺部感染,包括社区性肺炎,慢性阻塞性肺疾病,通过血氧浓度和全身的免疫反应,一定程度上是可以反映在肠道菌群上。
另外像肺部的感染,比如说在肺炎链球菌之类的感染中,肺部的病原菌可以通过痰或者是呼吸进入到肠道,所以我们在肠道当中是能检测到这些肺部的感染菌,并且随着其严重程度和感染进程,菌群的丰度会越来越高。
肠道菌群检测报告中也有对肺部相关疾病风险提示。

免疫疾病
肺部感染会出现咳嗽等症状,但咳嗽不一定仅是肺部感染,也可能是哮喘。
★ 哮喘
在哮喘中,微生物群是导致肺和肠道之间相互作用的重要因素。肠道微生物可以影响肺部的免疫反应,而肺部刺激可以导致肠道反应。
在一项研究中,来自加拿大的三个月大婴儿哮喘高风险的粪便样本中观察到 Lachnospira, Veillonella, Faecalibacterium, Rothia显著下降。这种菌群特征在1岁时不再明显,同时伴随着粪便乙酸的减少和肝肠代谢物失调。
肠道微生物对哮喘的影响部分是由细菌代谢物介导的,1岁时粪便中含有大量丁酸和丙酸的儿童,其特应性敏感性明显降低,3至6岁之间哮喘的可能性较小。此外,哮喘患者的粪便中Akkermansia muciniphila 菌水平均有所降低。
★ 过敏
已知的婴儿期与过敏性疾病相关的微生物群改变如下:
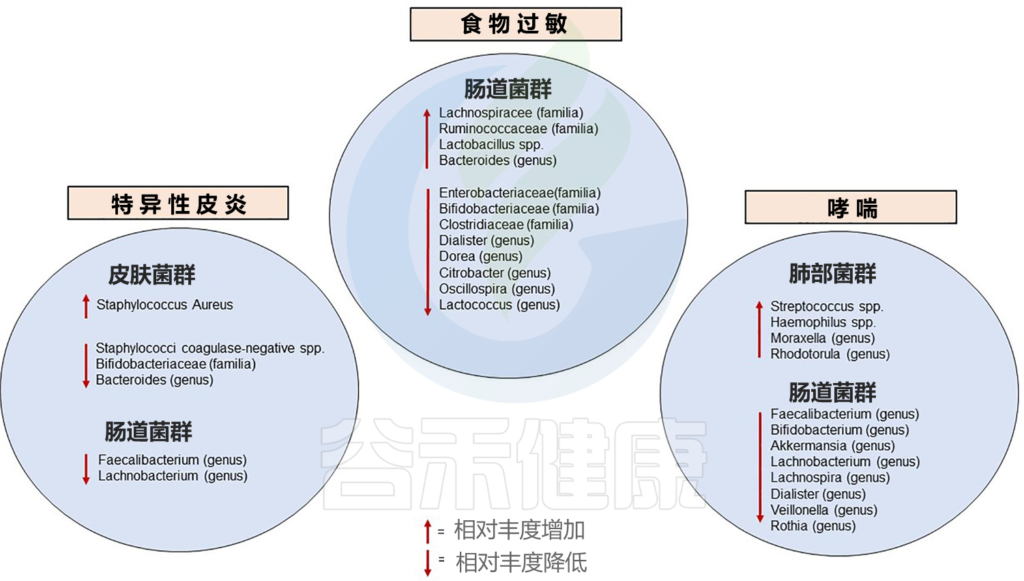
Diego G. Peroni et al, Front.Immunol. 2020
肠道菌群检测报告中有列出与过敏正负相关菌群,及是否超标。
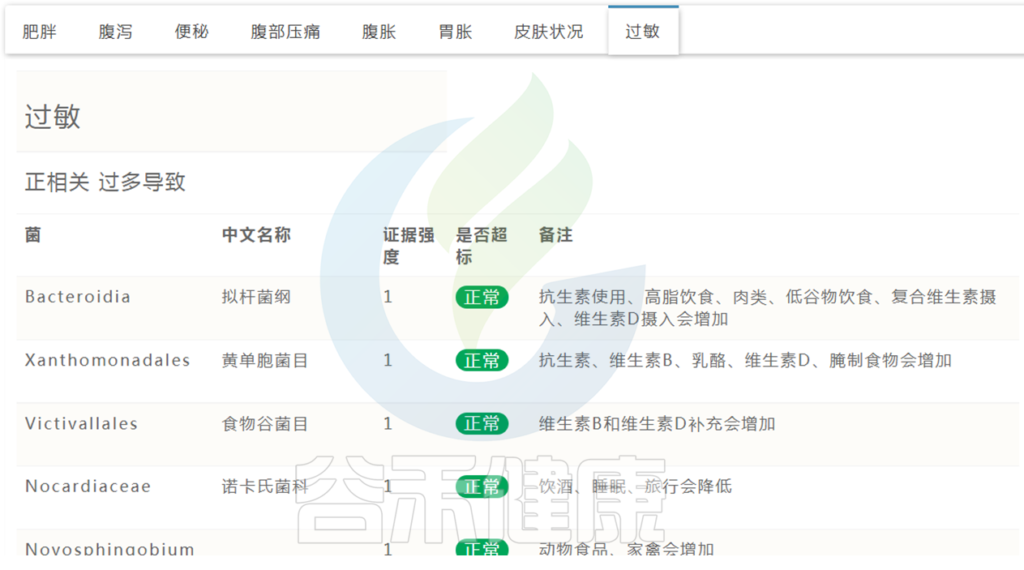
< 篇幅关系,此处仅展示部分 >
菌群生长需要养分,它的食物来源取决于你的肠道,有句话叫:you are what you eat (在我们这篇文章中有详细解释它们之间的关系 深度解读 | 饮食、肠道菌群与健康)。
也就是说,你吃的食物会帮助构建你的专属菌群。有的菌擅长代谢碳水化合物,有些菌擅长代谢脂肪,所以饮食结构不同,也就是食物来源比例不同,最后会塑造不同的菌。
那反过来,如果知道你的菌群的构成,就可以相对数量化的去了解你的饮食构成,包括营养摄入具体是什么样子,所以菌群很大的另外一个作用就可以反映你的营养饮食摄入状况。
这部分内容在我们报告中的呈现如下:
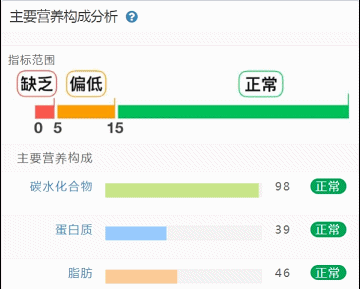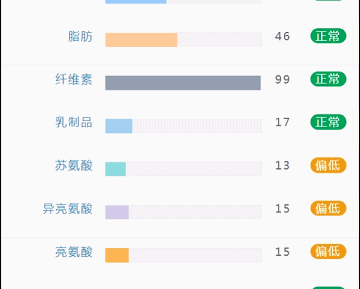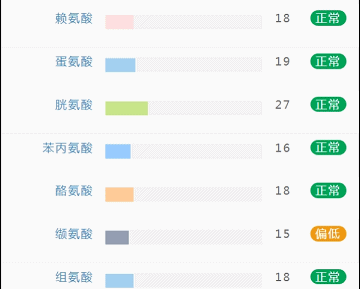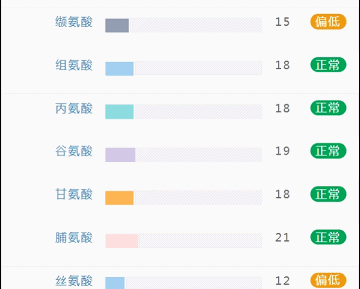
那么这里可能又会有疑惑,以上这些数值是什么意思,如何计算的呢?
不同的细菌有不同的代谢能力,需要不同的营养物质进行繁殖。通过评估特定营养供给下的偏好菌群的比例,即可反映不同营养物质的摄入比例。所以报告中的主要营养代谢分值评估的是主要营养物质摄入的比例在人群中的分布水平。
因此不会出现所有主要营养物质均高或均低的情况,也因此主要营养指标的最佳分值在70,且更关注不同营养物质的均衡性。
单项营养物质的分值低于5,表明摄入比例在人群中属于最低的5%,评估为缺乏,低于15评估为偏低。
而如果某项指标达到或超过95,则表明该项可能摄入比例偏高,通常对应会有其他营养成分较低。只需要针对性的增加缺乏或偏低的营养成分摄入,维持不同营养成分相对一致即达到营养均衡的目标。
为什么会出现所有的营养指标都很低?
这可能是菌群失调引起的。营养指标的评估是基于菌群构成特征和菌群代谢生成特定营养素的途径来评估的,如果菌群结构异常,将导致后续的预测失常,例如大量氨基酸都评估缺乏的情况。
这时候需要先调节菌群,等菌群指标恢复到一定水平后再次检测,评估营养指标。
我们日常摄入的除了上一小节提到的宏量营养素之外,还包括微量元素和维生素等。有些维生素比如说B族维生素中有相当一部分,甚至百分之六七十需要通过肠道菌群对初始原料进行代谢之后才会产生,也就是说有些细菌会代谢我们食物中的一些成分,转换成B族维生素。
而你的菌群构成和代谢B族维生素的能力,会直接决定是否缺乏该类维生素。当然也有部分受基因影响,因此肠道菌群相应的基因和代谢途径的丰度水平也会直接反映这些维生素的摄入水平。
总的来说,菌群在这其中起重要作用。在我们报告中呈现如下:

微量营养元素和维生素的评估分值与主要营养物质不同,是通过调查人群的单项营养成分水平,然后寻找与该项成分异常相关的菌群,并基于这些菌群和代谢途径计算丰度并转换为人群分布后的值。
简单来说,报告中的微量营养元素的分值即代表该营养元素的摄入水平。
菌群检测营养状况与血液检测有什么区别吗?
通过肠道菌群评估的维生素一般反映一段周期内的维生素状况,因为肠道菌群在没有突发疾病的情况下相对稳定,受一段周期的饮食影响为主,一般是2周。B族维生素是水溶性维生素,每日摄入后会通过尿液代谢排出,通过血液检测,不同时间检测波动较大。
菌群评估营养和血液检测营养趋势是一样的,在极端缺乏和极端过量是吻合的,中间档可能在数值上不是完全吻合,血液反映的营养水平比较及时。
★
当了解了体内的营养素和维生素是否缺乏,以及哪方面的缺乏,就可以进行有针对性地补充。菌群也是需要营养物质的,这就离不开我们的日常饮食,那么该如何补充呢?
我们的肠道菌群检测报告中有个体化饮食推荐表。

<篇幅关系,此处仅展示部分>
以上食物推荐表是怎么来的?
这是经过综合考虑疾病风险和营养缺乏状况计算得到的。主要是计算每种食物的营养构成与目前营养状况的匹配度,以及特定疾病需要避免的食物。
该表推荐的食物分数从-100~100,排序为不推荐到强烈推荐,日常饮食可以参考这个推荐表。推荐分值,表示基于目前的菌群和营养状况对食物的推荐指数,正数分值越大,建议优先选择,同时也是对改善最有帮助;负数分值越大,并不表示不能吃,而是目前状况下不优先推荐或尽量少吃。
p.s. 如果有特殊疾病需要忌口的,优先遵医嘱。
该表包括几百种日常食物,如下图。

<个体化饮食推荐,建议用电脑查看,目前手机端展示不太美观>
对于长期调理菌群而言,饮食无疑是最主要的驱动因素之一。
下一步我们将利用更全和详细的菌群结构,食物营养,人群膳食构成以及营养数据库推出个性化膳食营养升级方案,特别会针对个别菌属的异常和失衡状况以及营养元素异常和缺乏问题。
前面章节我们知道,通过菌群可以反映你的饮食状况,那么反过来,如果你吃了一个东西,会对菌群检测造成影响吗?是不是菌群就变了,那检测就不准了?
这也是比较重要的一部分,也就是肠道菌群检测的准确性,它能允许多大范围内的变化?什么因素会影响?
其实,菌群变化算快,也不算快。饮食对菌群是有一定影响没错,但这种影响呢,一般来说是前一天的饮食会影响第二天的菌群结构的百分之十几,也就是说,假设你昨天吃大餐,大量吃肉,蛋白摄入非常高,而你之前是以碳水化合物为主的,那么第二天饮食当中,你的蛋白质相关的这部分菌的比例可能会有15%,最高到20%可能会有,但一般来说是在15%以内,会有一个波动。
然而,总体的核心菌群构成,不会因为你今天一顿大餐,就直接从素食的变成肉食的菌群结构,核心菌是相对稳定的,那么多久会发生变化呢?
一般来说坚持两周,饮食结构的变化,核心菌群就会发生一个迁移改变。但两周只是一个短暂的周期,如果你两周后又换回先前的那种饮食方式,菌群也会随之改变到之前的状态。那要怎么样才能持久改变菌群呢?
这个时间线可能要拉长到两个月。
这是在我们的菌群干预中,很多人会遇到的一个周期性的问题。也就是如果你想有效改善菌群,至少需要两周会见到相对明显的菌群结构变化,那如果把干预延伸到持续两个月的周期,甚至是持续干预周期更长,那效果会更好。
取样前饮食会不会造成影响?
前面我们知道,菌群会受检测前一天饮食的影响,造成15~30%的菌群改变,同样也会反映在营养状况的评估上,因此建议检测前一天尽量保持近期正常饮食,这样能更好的反映真实的营养饮食状态。
此外,如果你是在调理一段时间后再次检测,想要和上次比较的话,最好在检测前保持饮食大体相似(意思是不要突然吃和平时不一样的食物或者吃完大餐后取样)。
取样过少会怎么样?
取样不能太少,如果太少的话,可能会影响DNA提取,另外会导致一些低丰度的菌检测不到。
取样过多会怎么样?
如果说取样太少导致样本不合格可以理解,那么取样过多为什么也会有问题呢?
我们的采样管中有保存液,可以将菌群固定在采样的瞬间,但是如果取样过多的话,可能导致部分粪便无法完全溶解于粪便,这部分样不能正常保存可能会使其中的大肠杆菌等兼性厌氧菌开始在管内繁殖。
正确合格取样量(黄豆大小,约200mg,如果是稀便,反复沾取)

只需棉签沾取少量,混匀于保存液,固体粪便取样不能超过管子1/5体积(右图刻度线)。且保存液带有粪便颜色即可。(右图所示)

详见:肠道菌群取样方法
注 意 事 项
如3天内使用过抗生素类、质子泵类胃药、阿片类精神药物请停药3天后进行检测(如果长期服用某种药物,如降压、降血糖药等,不建议停药,检测反映的是用药控制的菌群和身体状况)。
感冒、腹泻或其他症状期间不影响取样,拉稀或稀便可以用棉签反复沾取粪便至取样管。
★
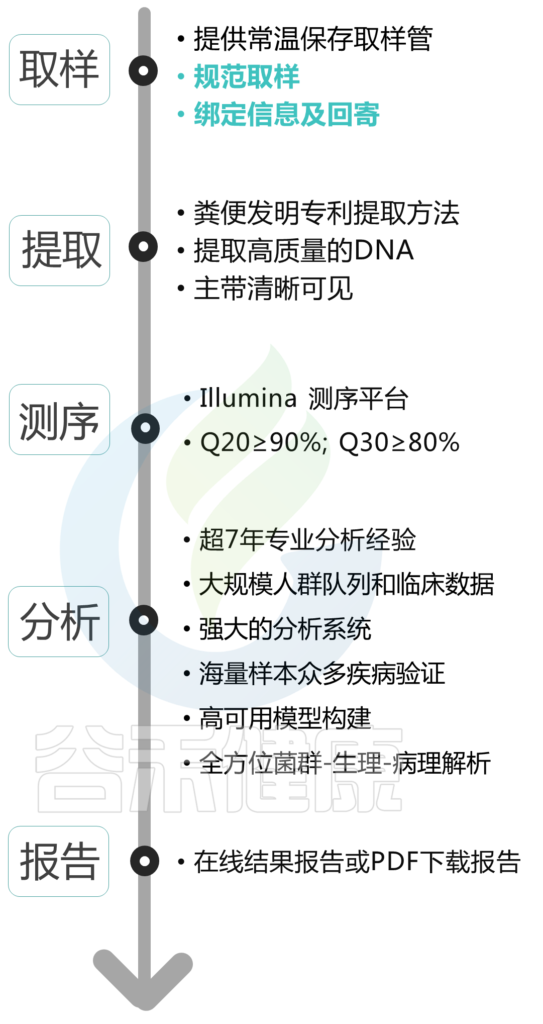
总的来说,取样虽然很重要,但也只是其中一个环节。每一个样本的结果呈现都凝聚了我们与你共同的努力。那么,从取样到结果报告呈现的那一刻,中间经历了什么?
在你取完样之后,把样本用快递寄到我们这里之后,它会经历提取->测序->分析->报告到你手上。下图绿色标注部分是你需要完成的。

近年来,我国将全面健康和预防作为国家重点领域。我们致力于将信息技术(IT)与生物技术(BT)相融合,发展推动肠道菌群基因检测进入成为精准和预防医学时代下的“生命健康新基建”,尽管目前的菌群检测,包括疾病关系,算法,数据库,后端干预均在成长积累阶段,但是菌群检测正在进入大数据时代,菌群基因中蕴藏海量对人体生命和健康的重要数据,我们致力于将这些数据和实际应用相结合,最终转化为疾病预防、改善健康的有效方案。
前沿技术正在不断创新发展,报告也在迭代更新中,谷禾肠道菌群健康检测在辅助判别慢病风险、精准营养、亚健康管理、临床治疗干预中显示出其广泛的社会需求和指导价值。
你问我答
不同部位间的样本(如前段/中段/后段),检测结果差异性有多大?
答:会有不同的,不过主要反映在具体的菌种丰度上,有无这种菌的差异不大。另外慢病的评估也影响不大慢病模型中使用了高维特征,丰度的变化波动对结果的影响没那么大。营养和代谢部分受菌群丰度影响相对大一些,同一个人的前后两天的取样最大可能有15%左右的差异。
肠道菌群在肠道内不同部位以及粪便的不同部分其实都存在差异,含水量、连续几餐的饮食构成和排便周期的长度都会对菌群各个菌种的丰度造成影响。单纯从绝对丰度上来看是一个动态变化的过程,各个菌属在继承之前的构成比例的情况下因各种因素的变化增长或降低。因此并不存在一个绝对的菌群构成以及完全准确的单一指标。肠道菌群检测获取的丰度含量本身信息量很大,但是稳定性和一致性并不很高。
更高层级的菌群相对比例顺序则相对稳定一些,之后具体包含的菌种也相对稳定。目前我们使用的疾病预测模型主要通过高维的菌群结构特征,并不单纯依靠每个菌的绝对丰度来评估,稳定性很高。针对一些特定的病原菌或问题菌,需要通过与人群范围比较,在正常范围内并无问题。
日常多添加有益菌或益生菌的酸奶,可以改善肠道菌群状况吗?
答: 大范围人群调查显示添加益生菌的酸奶可以改善肠道健康,但效应因人和状态而定。总体而言我们支持服用益生菌酸奶有益,但需要注意酸奶饮料可能包含果糖,游离糖等,其作用仍然非常有限。
同一份样本,不同批次的实验环节如上机测序,差异有多大?这种差异率是否有一个范围呢?
答:不同批次上机影响很小,菌群数据相关性不低于98%。我们会在每轮设置一个阳性对照,一个上轮检测样本对照,一个阴性对照。评估污染,轮次比对。理论上不同的实验室,扩增引物,方法都会带来对不同菌丰度的系统误差,我们尽力保证本实验体系下各个轮次之间最小化的实验误差。另外使用的引物是经过大量验证的标准化引物。
实际患者建不建议送检,我们这个产品主要针对健康体检,还是也可以辅助诊断和预后治疗呢?
答:产品主要针对健康体检,如果临床诊断判断可能菌群异常或疾病症状与菌群相关,产品可以通过菌群检测提供临床参考,用于辅助诊断和治疗方案的评估。产品关于疾病和菌群相关指标的评估仅限于菌群相关方面,以临床诊断为准,不适用于单独使用产品进行疾病诊断。
抗生素是如何影响菌群的,菌群的敏感性和抗性基因是什么?
答:广谱抗生素会杀死细菌,但是部分细菌在抗生素选择或滥用的情况下会在抗生素靶点基因产生突变或携带耐药基因,从而对特定抗生素产生耐药。不同菌目前的耐药菌比例以及携带的耐药基因水平不同,对应的抗生素耐药水平和种类也有不同。
有在吃富含某种事物或者相关营养素,为什么报告显示缺乏?
答:营养指标的评估是基于菌群构成特征和菌群代谢生成特定营养素的途径来评估的,直接的营养素补充会反映在相关菌群构成上,但部分营养素因为吸收部位不同以及菌群代谢途径上下游的影响,预测可能有一定差异。另外菌群构成异常的情况也会导致营养指标预测失常,如大量氨基酸都评估缺乏的情况。
有人说长期服用益生菌,会让肠道自己产生的益生菌的能力减弱或者可以说是肠道自主平衡的能力减弱,不能长期服用。这种说法是否有依据?长期服用一种益生菌,也容易产生耐药性,那么是否建议定期更换或者调整益生菌的菌种和数量呢?
答:持续服用单一或特定组合的益生菌确实会存在效力减退的情况,主要是菌群具有适应性,如果不配合生活方式和饮食结构的改变,会较快失效。可以根据菌群检测结果来调整益生菌的方案。
样品的稳定性对于那些数据的影响是比较大的哪些是影响比较小的?
答:越是直接和具体菌相关的指标变化越快越大,和菌群结构相关的指标,比如一些慢病风险还有总体饮食结构一类的变化较稳定。
从波动性排序来看,具体菌丰度>多样性>微量营养(锌 铁 氨基酸 维生素)>消化道疾病风险评估 (受当前状态影响较大)>肠龄>宏量营养素(碳水 蛋白 脂肪 纤维素 乳制品)>抗生素水平 >菌属是否出现>其他慢病风险
中大龄儿童小孩检测到自闭症风险高,如何解读?
答:肠道菌群在1-3岁期间主要是菌群发育滞后会影响神经发育和营养,3~6岁左右菌群参与的神经递质代谢异常会加剧自闭症的程度,但这个年龄段已有的神经发育滞后不光靠菌群改善就能解决了。
所以如果是0~2岁的如果这个风险值较高,不管有没有症状都建议改善菌群。如果是3~6岁甚至6岁以上,如果就风险值高没有相应的神经或行为异常,就问题不大,可能是菌群代谢构成不太好,不会导致自闭症的。如果有症状那改善菌群有助于改善症状。
肠道菌群平衡,为何多样性指数是低的?
答:菌群平衡和多样性指数是2个不同指标;
多样性仅仅评估肠道菌群的种类数量和丰度分布,与具体是有益和有害无关。多样性主要与饮食摄入,药物如抗生素类以及疾病状态有关。
菌群平衡对应的异常称为肠道菌群失调,临床上有I度失调和更严重的II度失调。大便常规检查是通过显微镜下观察统计染色细菌中杆菌和球菌以及革兰氏阴性和阳性菌的比值是否超标来判别的。本报告同时提供了另一评估算法,通过有益菌/有害菌的总体情况来评估菌群平衡状态,低于2为重度失衡,低于5为失衡,同时分值也提示菌群平衡水平,越高越正常。
菌群失衡如何调整?
从菌群失衡的评估角度来看,首先就是快速增加有益菌特别是双歧杆菌的丰度可有效改善该项指标。因此临床上通常提供多联的益生菌制剂来快速补充益生菌,可以短期有效改善菌群平衡比例。
菌群平衡和多样性分值都高的,但是肠道年龄预测比实际大,年龄预测模型是不参考多样性和平衡性参数的?
答:肠道年龄是靠机器学习和人群大队列做的,不是只根据菌群平衡和多样性,每个年龄段都有核心和标致的菌群特征,比如婴儿的双歧杆菌,老年人瘤胃球菌等,这几个指标都是表征菌群的状态和健康的。
END
声明
谷禾专注于提供肠道菌群基因检测和基于此的健康评估咨询,肠道菌群对人体健康的影响和关联性已被广泛研究和认可,但基于对健康的慎重和法规,谷禾重申其提供的肠道菌群基因检测目前不用于临床疾病诊断,仅作为菌群状况构成检测和健康评估以及基于菌群的科研。分析报告中疾病风险和健康相关评估来自于公开研究数据和谷禾构建的大人群队列数据分析的预测评估结果,涉及临床诊断和医疗建议请遵照临床诊断和医生的医嘱。由于技术进步和样本数据不断积累,报告中可能存在尚未完全涵盖的因素或状况,不可避免的存在一定概率部分风险未被完全检出的情况。

谷禾健康

本文原创:谷禾健康
自闭症谱系障碍是一种神经发育疾病,其特征是社交和沟通困难、限制性和重复性行为以及异常的感觉反应。
自闭症的具体发病机制尚不能明确,但目前为止许多研究表明,自闭症与肠道微生物组之间存在很大关联性。
最新,Yap等人发表于Cell的一篇题为“Autism-related dietary preferences mediate autism-gut microbiome associations”的论文就自闭症与肠道菌群的关联给出了他们的研究成果。
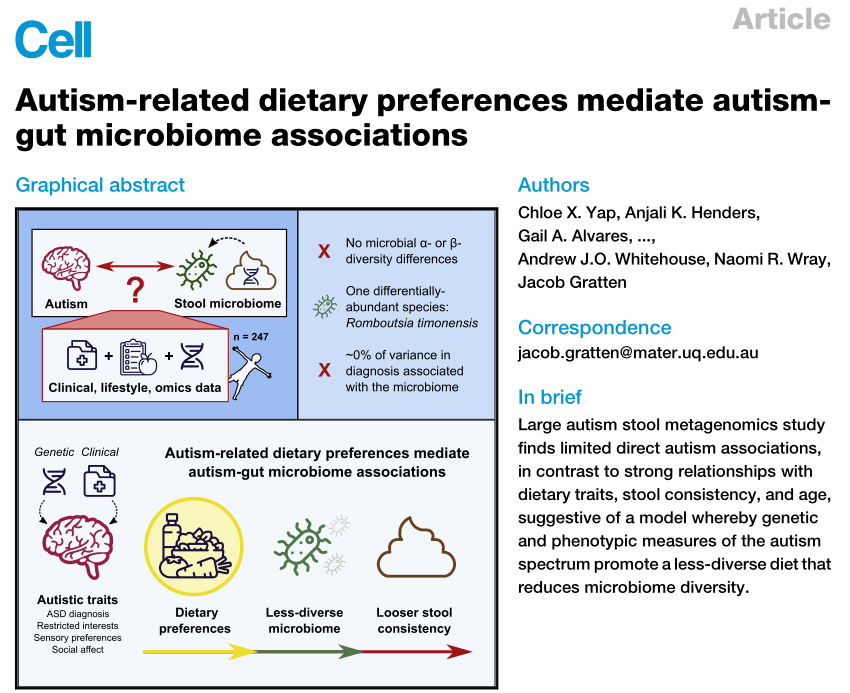
其核心结论是:
肠道菌群与自闭症之间没有直接联系。自闭症儿童与正常儿童的肠道菌群差异是由于自闭症症状导致患儿的饮食多样性下降,饮食类型狭窄,从而导致肠道菌群多样性减少,进而引发便秘和消化道症状。
我们来看看其研究设计情况。
关于肠道菌群这方面的研究,很关键的一个点是研究的样本数量。
首先,这项研究涵盖了共247名儿童(2-17岁),其中自闭症患者99名,51名患者的兄弟姐妹,97名非自闭症儿童,样本来自澳大利亚自闭症生物银行Australian Autism Biobank (AAB)。
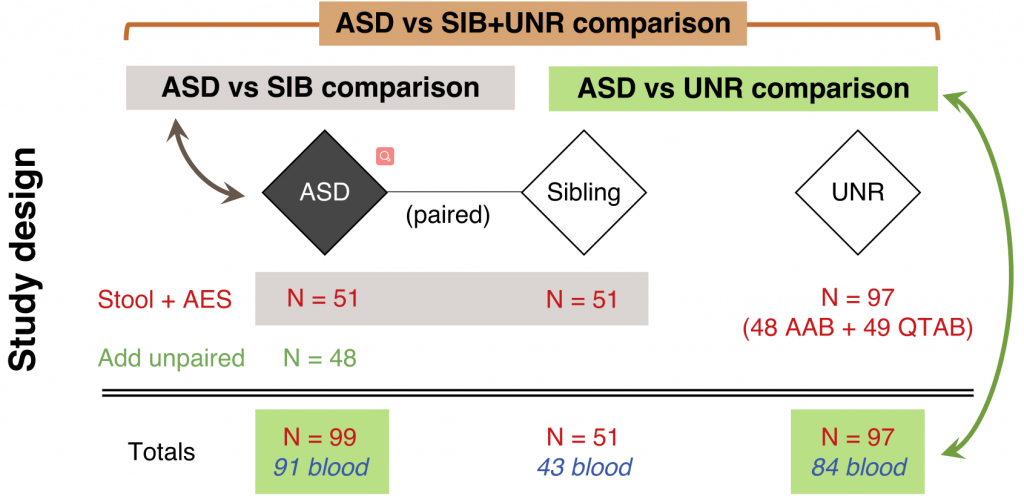

我们观察到样本人群相对于宏基因组来说样本数量还可以,但是99例自闭症患者样本还是让整个研究的统计效力及研究的适用范围有很大限制。
自闭症属于神经发育疾病,虽然其病因复杂,但是疾病的发生阶段绝大部分在出生到3岁左右,主要影响了儿童早期的神经系统发育,导致出现神经发育滞后、刻板行为和社交障碍。
类似的疾病还有注意力缺陷ADHD以及多动症等。越早期的干预其愈后和改善就越明显,因为早期神经系统发育是阶段性的,错过了发育阶段,很难在后期通过行为学等方面获得明显改善。
进一步查看研究样本的年龄分布我们发现,该研究的样本年龄均值在8.7岁。
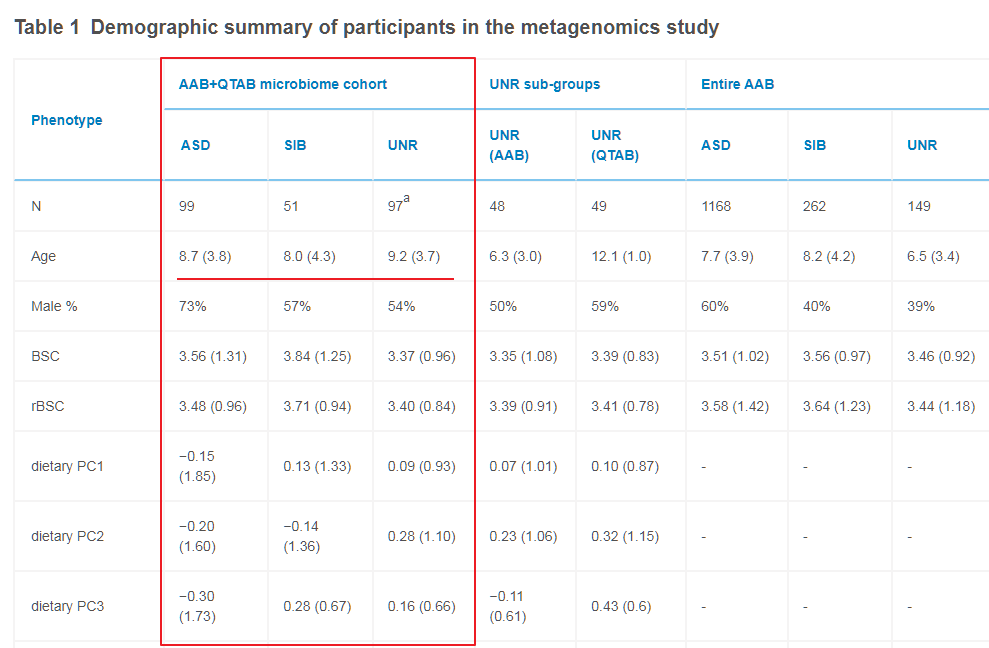
虽然范围在2-17岁,但是和自闭症发病阶段3岁以下的各组样本分别是7例、7例和8例,2岁以下的样本仅有1例。
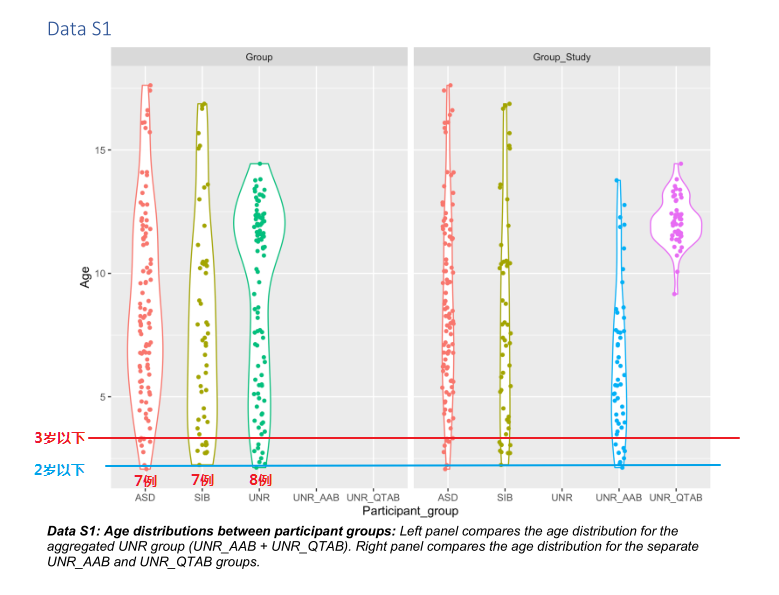
也就是说,研究涉及的自闭症患者虽然仍然有着自闭症的诊断和行为表现,但是绝大部分样本均不是处于神经发育的最核心阶段,而且大部分样本应该是经历过多年的包括行为干预或其他治疗。
因为自闭症与早期行为发育相关,大部分确诊儿童可能其行为表现和社交能力直到成年可能仍然没有完全恢复或达到正常水平,可能在多年后即便其引发自闭症的病因(主要是环境或生理因素)已经消失,但症状或诊断仍然没有变化,这就意味着这些样本可能不能反映真实的自闭症发生时的神经发育和菌群状况,因而也不能说明菌群在自闭症的发病和发展过程中并无联系。
更重要的是肠道菌群的组成变化尤其是生命早期与年龄和发育阶段密切相关,3岁之前的肠道菌群基本上每个月龄都存在变化,3岁之后的肠道菌群会趋向于接近成年人的菌群构成,并逐渐成熟。
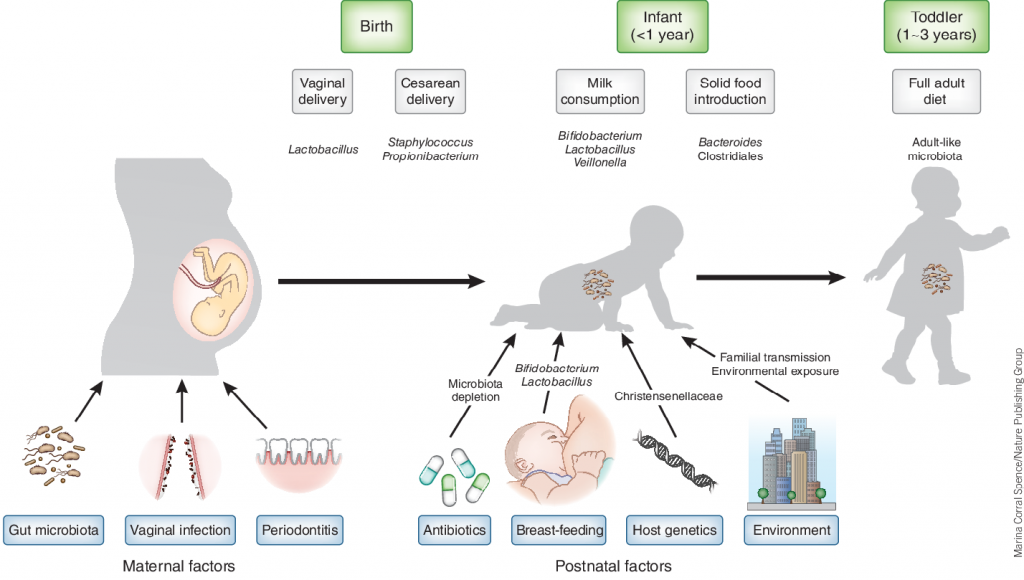
还需要注意的是,在6个月左右,由于固体辅食的引入,婴幼儿的饮食结构会发生重要变化,相对应的肠道菌群也会发生重要的转变,从乳制品代谢为主的韦荣氏菌、双歧杆菌、大肠杆菌为主逐渐进入以碳水化合物和蛋白质代谢为主的拟杆菌或普雷沃氏菌属等成年人常见核心菌群为主的菌群构成。这一变化阶段恰恰是自闭症对应早期神经发育的最重要阶段,而该研究基本没有这个阶段的样本。
研究中也明确提及肠道菌群构成和年龄存在较强的相关性,在分析中是将年龄和性别作为协变量进行控制,但我们认为这种统计方式不足以解决儿童肠道菌群在不同年龄阶段的变化差异,需要进一步对不同年龄阶段或年龄的儿童进行单独分组分析,但是这样该研究的样本数量就严重不足以获得足够的统计效力。
研究中包含有来自同一家庭的非自闭症兄弟姐妹,作为对照能较好的控制包括饮食、生活方式及居住环境等变量,因此很自然我们希望看到针对成对家庭兄弟姐妹的比较分析。
在论文的补充材料方法部分有描述了使用成对样本进行比较的内容,一个102个样本,形成51对样本。对于这样的成对样本分析,比较简单的方式是直接进行成对T检验。
然而,论文中并没有这么做,比较奇怪的将family ID作为随机变量从而控制成对样本的差异检验。但是家庭ID本身除了家庭之外并没有类似年龄或分层等信息量,作为随机变量加入后并不能有效实现成对分析的效果。

另外根据论文的结论,饮食结构单一引起了菌群的变化,进而诱发肠道问题,那么在成对家庭成员样本之间,自闭症儿童相较于同家庭的兄弟姐妹在相同饮食习惯和环境下是否饮食结构明显单一呢?
我们期待看到自闭症儿童的饮食多样性要显著低于其兄弟姐妹,且基本集中于低多样性的区间。
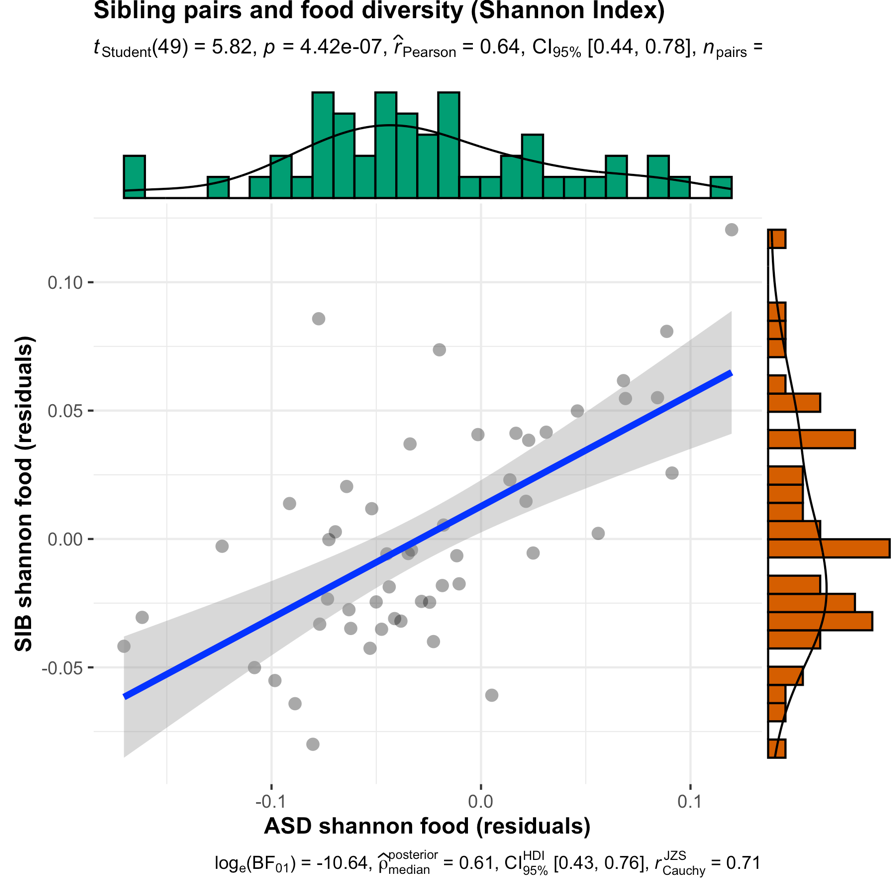
论文补充材料部分的下面这张图显示,同家庭兄弟姐妹之间的饮食多样性是显著相关的。
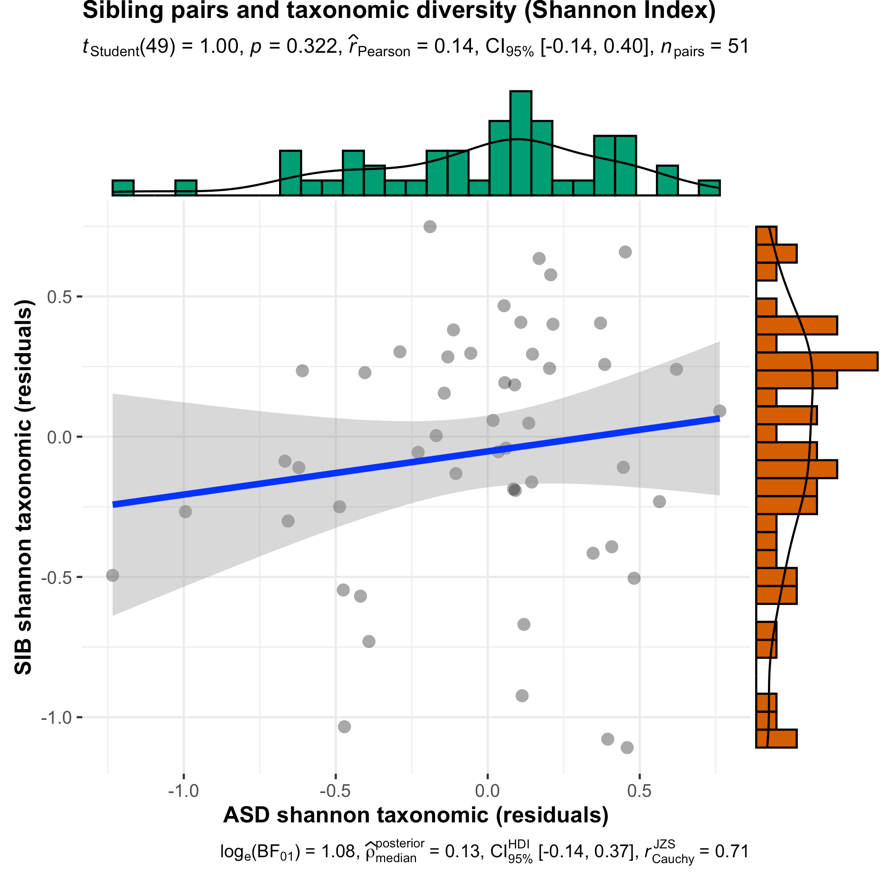
那么对应的菌群多样性呢?下面的图显示,基本没有相关性。
由于论文没有进行成对样本的检验,因此我们尝试下载数据进行单独分析,很遗憾,论文中提供的数据仅包括100例样本的数据,表型和分组等信息只有50例样本的,无法进行单独分析。
针对论文结论的自闭症儿童的饮食类型狭窄的问题,我们认为在早期婴幼儿期饮食构成本身就是相对单一的,而且非自闭症儿童中也存在相当一部分饮食结构单一的,单以饮食结构问题来解释自闭症儿童的菌群差异还不具有足够的说服力。
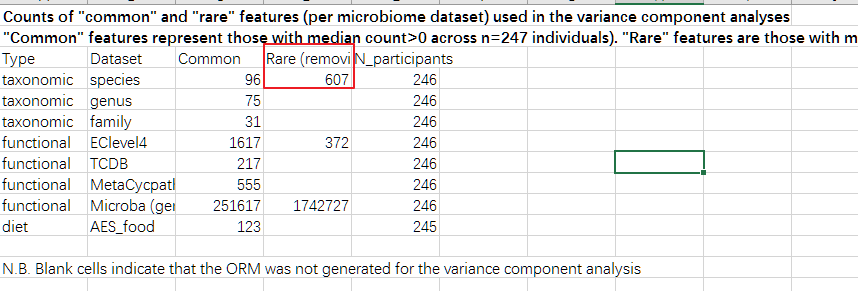
另外我们注意到,该研究将菌种和后续的基因及代谢途径分为常见和罕见两组,其中种部分中位数大于0的作为常见的,一共96个,其他的有607个种作为罕见。

另外在后续对功能基因的分析时也是将分析集中于前面发现的Romboutsia timonensis菌种相关的基因。
当然这是受限于样本数量的因素,聚焦于普遍的高丰度的菌属和基因,但是也有很大可能丢失了可能的联系。
综上,文章否定的是菌群与自闭症之间的直接关联,与之相关文章识别到了自闭症与健康儿童间的差异菌(Romboutsia timonensis,经过年龄、性别、饮食偏好调整之后),以及菌群与重复刻板行为存在显著相关(Fig. 4H)。
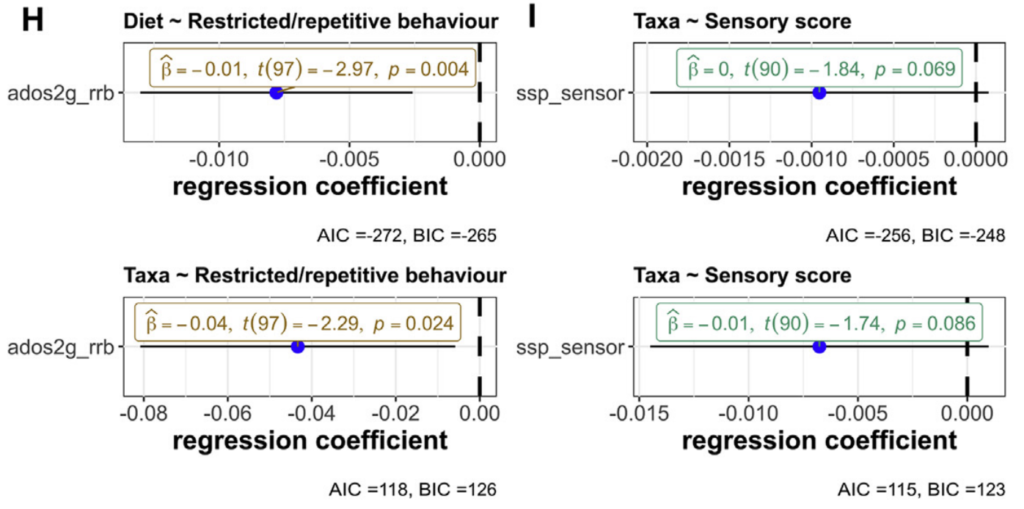
在这样的事实前面,文章依然要强行否认自闭症与菌群的关系,作者的行为很让人费解。
对此,网友们也各抒己见,就该文发表了一些见解:
他们的研究甚至没有试图确定:微生物群是否在自闭症谱系障碍中起驱动作用

他们自己的研究需要收费,这让事情更糟糕。人们必须付费去看他们的研究…
我们认为,以下系列问题仍有待回答
1. 他们的自闭症儿童都属于主要集中在轻度或者边缘程度,这个样本选择是否能代表自闭症的全部群体还存疑;
2. 如果将饮食归因于挑食等问题,那么在临床实践中我们也经常看到正常孩子也有挑食。研究者如果要说明菌群和挑食等行为有关而不是自闭有关,那么应该要设置一组挑食的健康对照儿童,才能彻底屏蔽这个因素的可能影响;因为作者明确表示饮食和自闭症有关,而不认为菌群和自闭症有关;
3. 这些样本的分布是否有跨地区特点?如果有,那么区域也会带来极大的差异,如菌群、饮食习惯等等,如何规避这个的影响?
《cell》原文:doi.org/10.1016/j.cell.2021.10.015