-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康
帕金森病(PD)是全球第二常见的神经退行性疾病,预计到2050年,全球患者可能达到约1200万。传统诊断依赖运动症状评估,往往在疾病晚期才发现,因此急需可用于早期识别的生物标志物。
近年来,研究发现PD患者常有肠道症状(如便秘、肠道炎症),这些症状可能早于运动问题出现,甚至与肠道微生物组变化相关。
既往研究中,关于PD肠道微生物组的研究和小规模整合分析虽提示产短链脂肪酸菌减少等共性,但研究间差异大、缺乏在跨队列层面验证可复现的“微生物特征”并评估其诊断价值,尤其尚未系统评估机器学习模型跨研究、跨人群的可迁移性与疾病特异性。
针对上述研究空白,近日,一个由德国欧洲分子生物学实验室(EMBL)等国际顶尖机构组成的研究团队,通过对帕金森病(PD)微生物组数据进行大规模跨队列分析,建立了一套旨在提高模型稳健性、泛化能力和可复现性的最佳实践框架。其相关研究的成果发表于《Nature Communications》上,为未来的相关研究提供了清晰的路线图。
原文研究通过大规模荟萃分析和机器学习评估,回答两类关键问题:
作者的研究思路可以归纳为两个部分:
这项覆盖11国4大洲22项病例对照研究,共计4489份样本,结合机器学习的荟萃分析,得到的主要结论是:
接下来,我们深入了解该研究的方法学细节和主要结论验证过程。详细解析该研究在数据准备、特征工程、模型训练和多维度评估等关键环节上采取的具体策略和宝贵经验。
下图展示了研究中使用的22项病例对照研究概述:
16S数据集:
宏基因组数据集(SMG):
每项病例对照研究都必须≥30个样本,且元数据足够。除此之外,还纳入了多发性硬化(MS)与阿尔茨海默病(AD)的16S数据用于交叉疾病验证。
初步研究,聚焦于β多样性和批次效应评估。基于Bray-Curtis距离的冗余分析(db-RDA)与PERMANOVA(置换多元方差分析),分别在16S与SMG上评估研究来源(Study)、疾病状态(Disease)因素解释的变异性。
分析结果显示,去除批次效应后,样本未根据疾病状态明显聚类,PD与对照组仅呈现弱分离(下图c,d,灰色是对照)。研究来源解释了较大方差(下图a,b,颜色区分不同研究,形状区分疾病状态,16S:R2= 19.9%;SMG: R2=7.7%;p-value<0.001),这表明批次效应是主要变异来源。
物种分类和功能注释
统计检验应用
机器学习应用
在R包SIAMCAT的帮助下,研究人员应用了线性模型(Ridge、LASSO、Elastic Net)和非线性模型(Random Forest)。
训练前对特征数据做标准化处理,log.std或clr,筛选出至少在5%样本中检出的特征,GBM和GMM不做过滤。
训练时的特征筛选主要使用嵌套监督式特征选择,基于 Wilcoxon–Mann–Whitney 检验识别差异特征,在训练集内选择效应量最大的特征数量(如 KO 层面 ,保留范围500–4000个特征,步长500,训练多个模型,选择中位数 AUC 最优的那个模型,最终保留2500个特征用来构建最终模型)。
统一以 AUC 评估,并在 CV、CSV、LOSO 三层验证策略中对比模型的准确率与泛化性能。
PD相关的微生物组组成变化
在初步研究中发现不同研究间群落构成差异显著,疾病状态仅解释≤1%的变异,标识批次效应影响大于疾病。继续分析菌群发现,与对照组相比:
PD组中显著减少的菌群有:
PD组中显著富集的菌群有:
虽然大部分菌群在合并所有研究数据的分析基础上有显著变化(上图a),但是仍有不少菌群仅在个别单项研究中呈显著变化(上图b)。
控制协变量进行分析,发现在PD组中可能受性别、年龄或用药混杂的分类群为少数(<23%),且具有显著差异的分类群总体不受这些协变量影响,单变量方向与Ridge模型系数方向总体一致(上图c)。
PD相关的微生物功能变化(基于SMG)
与对照组相比,重点在于以下四大类的变化:
碳水化合物与复杂多糖相关功能
氨基酸与神经递质相关通路
与宿主互作和致病相关功能
外源性物质代谢相关功能
外源性物质(Xenobiotics)代谢相关功能的多条通路在PD组中富集,例如:
机器学习疾病分类性能与泛化性
比较模型AUC指标发现,单个研究内验证(CV)模型准确性高,但模型泛化性能差,跨研究(CSV)验证,普遍AUC下降。LOSO相较CSV显著提升。
研究可能影响 LOSO AUC 的因素,发现LOSO AUC在不同测试集之间存在显著差异,测试集本身解释了相当比例的方差(ICC=0.19)。LOSO AUC与训练集样本数呈正相关(p<0.01,解释约15%方差)。
寻找帕金森的通用微生物特征:分类学特征优于功能特征
在特征探索方面,研究人员发现存在一个相对“通用而精简”的肠道微生物特征子集,模型的判别能力(PD vs CTR)近似使用全部特征进行训练的模型。
在SMG数据集中使用LOSO策略,遍历训练集,筛选出绝对效应量最高且FDR校正p值<0.05的20个特征,构建模型,结果如下图a,其总体平均LOSO AUC几乎与全特征模型一致,分别为72.3%和72.4%。图b热图中的白色方块表示相应模型中未包含的物种。
同时,在SMG数据集基础上,比较了基于分类学特征(taxonomic)和功能特征(KO、KEGG module、KEGG pathway、GMM、GBM)训练的机器学习模型性能。分析结果显示基于功能特征的模型总体上表现略逊于基于分类特征构建的模型。
跨疾病验证:LOSO模型显著降低对其他神经退行性疾病的误判率
最后,研究人员又进行了外部验证,检验这些模型在多大程度上会错误地将患有其他神经退行性疾病的患者预测为PD,以阿尔茨海默病(AD)和 多发性硬化症(MS)为例。
由于其他神经退行性疾病的SMG数据稀缺,仅使用16S数据进行此项验证。使用假阳性率(FPR)评估,将AD和MS样本的FPR与PD内部对照组的10% FPR进行比较。
分析结果显示单个PD研究构建的ML模型(Single-study models)在其他神经退行性疾病上的交叉预测率差异极大,FPR从0%到近100%不等,平均FPR为35.1%,远高于预期的10% FPR水平。
使用LOSO模型时,跨疾病预测表现显著改善,平均FPR从35.1%大幅降至18.7%,18.7%的FPR仅略高于PD内部对照组的预期10% FPR,表现出较好的疾病特异性。
小 结
事实上,PD疾病与肠道菌群的相关研究不少,对比同类研究,该篇研究的优势在于:
为什么初步研究得出结论“批次效应差异大于疾病状态”,但是“多研究合并训练 + LOSO”却能提升模型泛化性能与疾病特异性?
关键在于数据层面的差异分析与机器学习分类任务的本质区别,原文中批次/研究效应在“整体群落差异”(方差、β多样性)上确实大于疾病效应,但这不等于模型“无法分类”。
微生物组分类依赖的是特定分类群的丰度模式,而非整体群落结构的相似性。
没有完美的模型,只有相对完美
原文中,单研究训练的跨研究验证(CSV)平均 AUC 约 61%,合并训练的 LOSO 提升到约 68%,但仍低于研究内 AUC(约 72%)。这说明,合并训练确实提高了泛化,但并未消除强烈的研究/批次差异,只是更好的对冲了其负面影响。
在机器学习中,泛化能力与拟合精度之间存在固有张力。合并训练实际上是在研究内高精度(可能包含过拟合)与跨研究稳定性之间找到最优平衡点。这不是缺陷,而是在真实世界复杂性面前的理性选择。
虽然也可以通过其它策略来补足缺陷,例如集成学习策略(如漏斗型、投票型模型),但是这也有损失,就是会累加错误。但在临床转化上,这种相对完美是具有实际价值的,因为临床诊断工具必须适用于不同人群,所以模型泛化性能也就很重要。
基于原文经验,利用肠道微生物组数据训练疾病预测模型的技术路线
一、 数据准备与质量控制
模型训练通常要准备数据集,建立包含训练集、验证集和独立测试集的完整数据架构。对于小样本数据集,可采用交叉验证替代独立验证集,但必须保留独立测试集用于最终性能评估。
基于原文经验,先做一些统计检验,对一些影响因子评估效应,例如批次、年龄、性别、用药、地理位置等,这种系统性的统计检验与效应量评估为后续的特征筛选和模型训练奠定了坚实的数据质量基础,确保识别的疾病关联特征具有真实的生物学意义而非技术或混淆因子的产物。
数据标准化推荐使用log变换(log.std)或中心化对数比值变换(clr),这两种方法在原文验证中均表现良好。
二、 关键特征筛选
数据集确定好后,要开始特征筛选,通常都会先基于存在与否做初步筛选,原文中的经验是保留在至少5%样本中检出的分类群,优先选择在多个研究中表现一致的特征,这是实现良好泛化性能的关键。
特征维度方面,对于高维特征空间(如KOs),建议通过嵌套监督特征选择将特征数控制在2500个左右,这在原文中获得了最佳的中位AUC(75.3%)。
三、 算法选择与训练策略
特征筛选完成后,要开始选择算法和制定训练策略,基于原文大规模比较,SMG数据优选Ridge回归或LASSO,16S数据可选择Random Forest,但为保证可比性,建议统一使用Ridge回归。
训练策略推荐单研究内训练+多研究合并训练,前者适用于初步模型构建,可获得较高的内部准确性(平均AUC 71.9%),后者显著提升泛化能力(LOSO平均AUC 68%)。可能有同学会疑惑为什么会用回归模型,原文使用的是SIAMCAT包的LibLinear,这是一个专门用于大规模线性分类的库,Ridge和LASSO在其中是作为线性分类器实现的,不是传统的回归模型,它们通过线性决策边界进行二分类预测。
四、 多层次性能评估
最后是选择评估模型性能的方法和指标。原文采用多层次性能评估:
五、 研究规模适配策略
应根据研究的具体规模调整策略重点:
主要参考文献
Romano, S., Wirbel, J., Ansorge, R. et al. Machine learning-based meta-analysis reveals gut microbiome alterations associated with Parkinson’s disease. Nat Commun 16, 4227 (2025).

谷禾健康

尽管地球上微生物类群的繁多,但只有一小部分得到了培养和有效命名。因为大多数菌无法在非常特定的条件下培养分离鉴定。
在过去十年中,宏基因组研究的重要性已经凸显,因为它能够评估细菌基因库并发现当前实验室培养技术无法掌握的新细菌基因组。这些数据对于扩大我们对地球上微生物多样性的理解至关重要。
由于宏基因组测序数据由来自多个物种和菌株的 DNA 序列片段组成,通常有数千个来自不同生命领域,因此此类分析的主要挑战是正确确定每个 DNA 序列片段的真实来源。不幸的是,这些步骤容易出错,因此必须对结果进行严格审查,以避免发布不完整和低质量的基因组。
最近,比利时研究人员新开发MAGISTA,这是一种评估宏基因组基因组组装质量的新方法,基于随机森林的方法估计MAGs的完整性和污染度,解决了当前基于参考基因的方法经常被忽视的一些缺陷。
MAGISTA是基于宏基因组bins内的contig片段之间的无对齐距离分布,而不是一组参考基因。该方法利用了来自整个 bin 的信息。为了正确评估此方法,并说明基于参考的工具的缺点,最近,比利时研究人员构建了一个高度复杂的 DNA 模拟群落,由 227 个细菌菌株组成,并且具有不同程度的相似性。

方 法
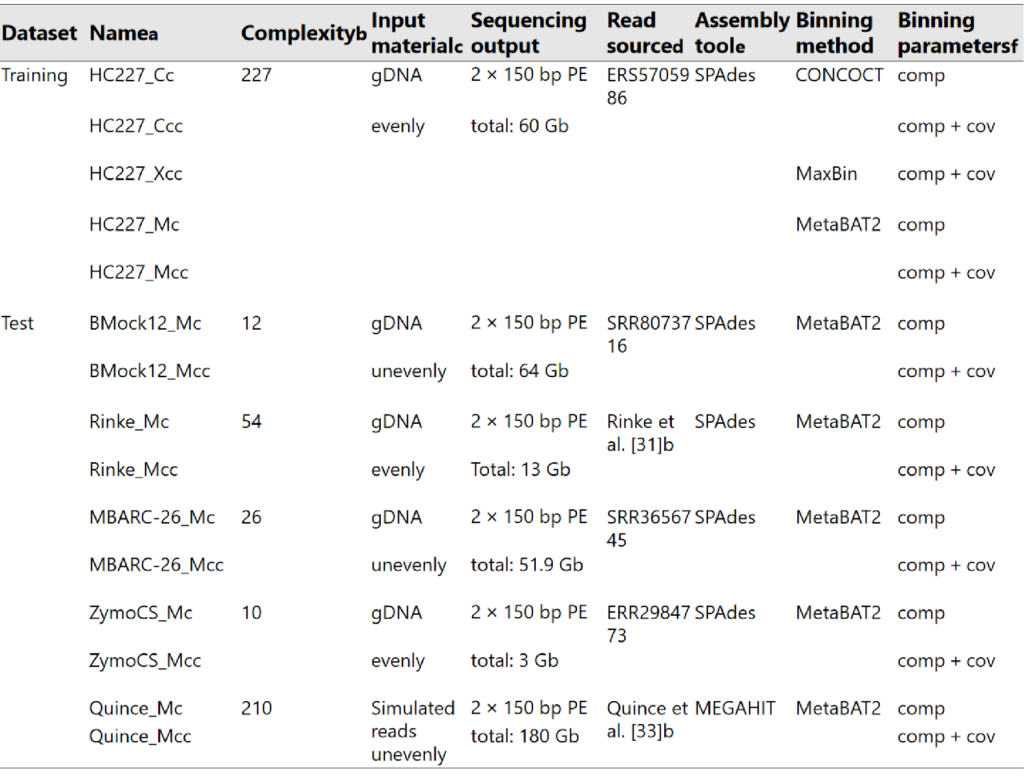
训练集来(HC227)自 227 个细菌菌株,测试数据集由五个公开可用的短读(short reads)子集构成,其中四个含有来自复杂度相对较低的基因组 DNA 模拟群落的reads。具体情况如下图所示。
Complexity列指示菌株数;Assembly tool列表示所使用的用于组装的软件;Binning method列表示所使用的用于分箱的工具;Binning parameters列表示所使用的用于评估分箱质量的指标,comp为完整度,cov为覆盖率。
MAGISTA计算步骤:
输入binning后的每个bins
-●-
第 1 步:选择适合的片段大小与距离计算方法
-●-
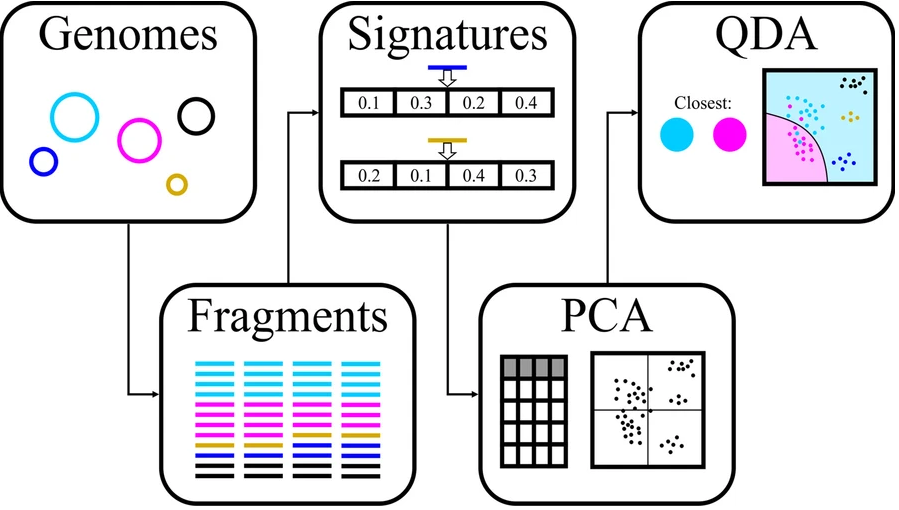
首先将每个 bin 中的每个 contig 拆分为固定长度的片段,然后使用四种不同的方法(即 PaSiT4、MMZ3、MMZ4 和 Freq4)计算一个 bin 中的片段之间的所有距离。对于每种方法,都选择了特定的片段长度,以便为不同的生物产生不同的特征分布。
每种方法的最终片段长度的选择是通过不同方法分析整合决定的,方法如下图所示。每组的设计中至少两个基因组来自同一个家族,两个基因组来自相同的顺序但来自不同的家族。这些基因组被人为地分成所需长度的片段,并为每个片段计算目标特征。
对于每组五个基因组,混合所有片段并根据它们的特征进行主成分分析(PCA),然后进行二次判别分析,用于生成分类器,旨在区分每组中重叠最多的两个基因组。对该分类器的准确度取平均值,结果用于选择方法和片段长度的最终组合。
-●-
第 2 步:模型中特征变量的选择
-●-
为每种方法选择片段长度后,使用平均值、标准差、偏度、峰度和中位数以及 2.5%、5%、10%、90%、95% 和 97.5% 百分位数计算距离分布。此外,还计算了 1 kb 片段的 GC含量分布。以及每个bin的大小,共计66个特征变量。
-●-
第3步:模型构建
-●-
使用 R (v 4.0.3) 包“RandomForest”中的“RandomForest”函数和默认参数训练随机森林模型。同时使用R包lm再建立一个线性模型执行线性回归,输入经对数转换后的特征变量值,用于交叉验证分析。
主 要 结 果
一个高度复杂的基因组DNA模拟群落
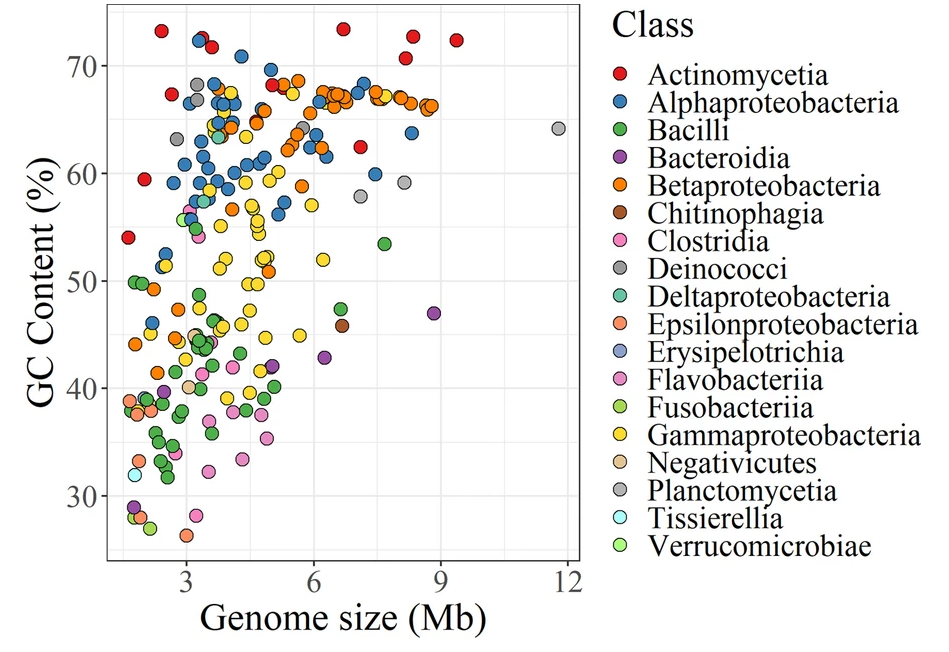
由来自 227 个细菌菌株的基因组 DNA 组成,这些菌株属于8 个门(Actinobacteria, Bacteroidetes,Deinococcus-Thermus, Firmicutes,Fusobacteria,Planctomycetes, Proteobacteria和Verrucomicrobia),18 类,47目,85科,175属,197种。
编辑
上图为模拟群落中的细菌菌株的基因组大小和GC含量(从26.3%到73.4%)散点图;
编辑
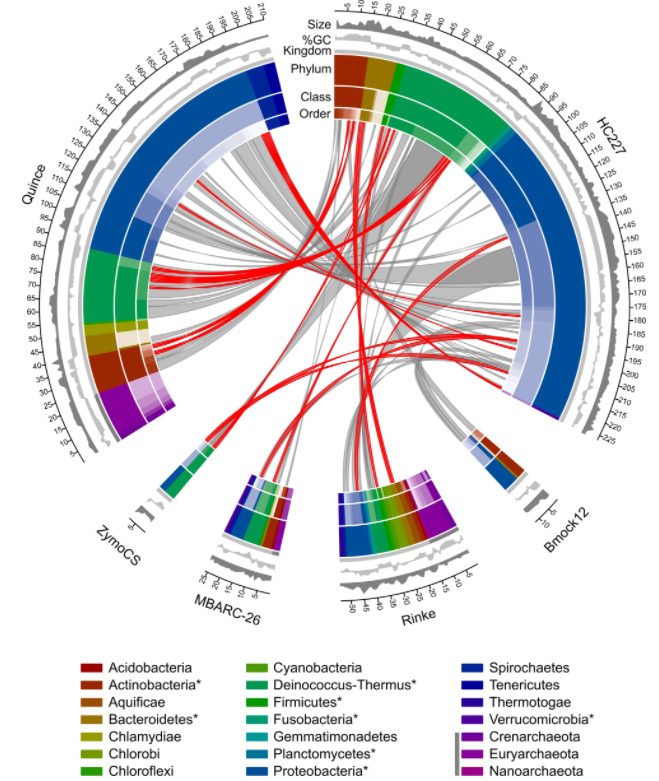
图为训练集与测试集中物种之间的关系图。红色线条表示在训练集中存在的菌种,灰色线条表示在训练集中存在的菌属。环状图中的不同颜色代表不同分类水平。图例中存在于训练集中的菌门用*标记,存在于古生菌的菌门用深灰色色带标记。
CheckM中基于单拷贝标记基因(SCMG)来评估 bin 质量的存在的缺陷
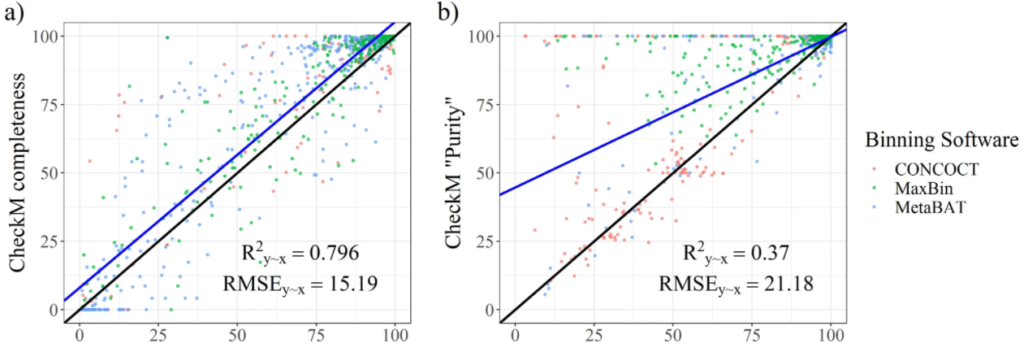
图a和b分别为从CheckM中输出的完整性指标和污染度。使用R^2y∼x(解释方差的百分比),RMSE(相对于实际值的均方根误差)两个参数评估结果。结果表示CheckM高估了bin的质量。许多受污染的bins被预测为接近未受污染。
使用MAGISTA分析模拟群落中的bins
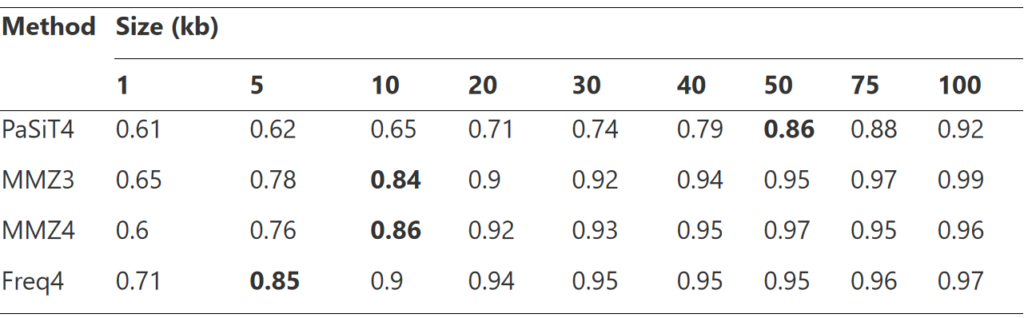
首先选择最佳片段大小用于计算距离分布,如上图所示,考虑了 1、5、10、20、30、40、50、75 和 100 kb 的片段,最终选择了粗体所示的片段大小。
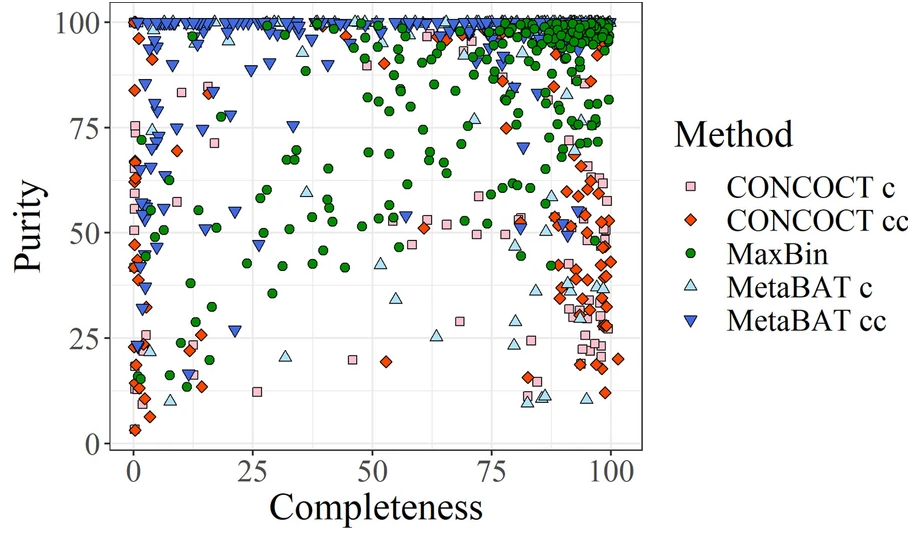
图为concont、MetaBAT和MaxBin产生的bins的完整性和污染度信息。
由于通过模拟生成这样的数据集并不能准确地表示真实的结果,所以使用了binning软件的结果,提供了一组不同质量的真实的bins。训练数据集的完整性和未污染度均在90%以上。
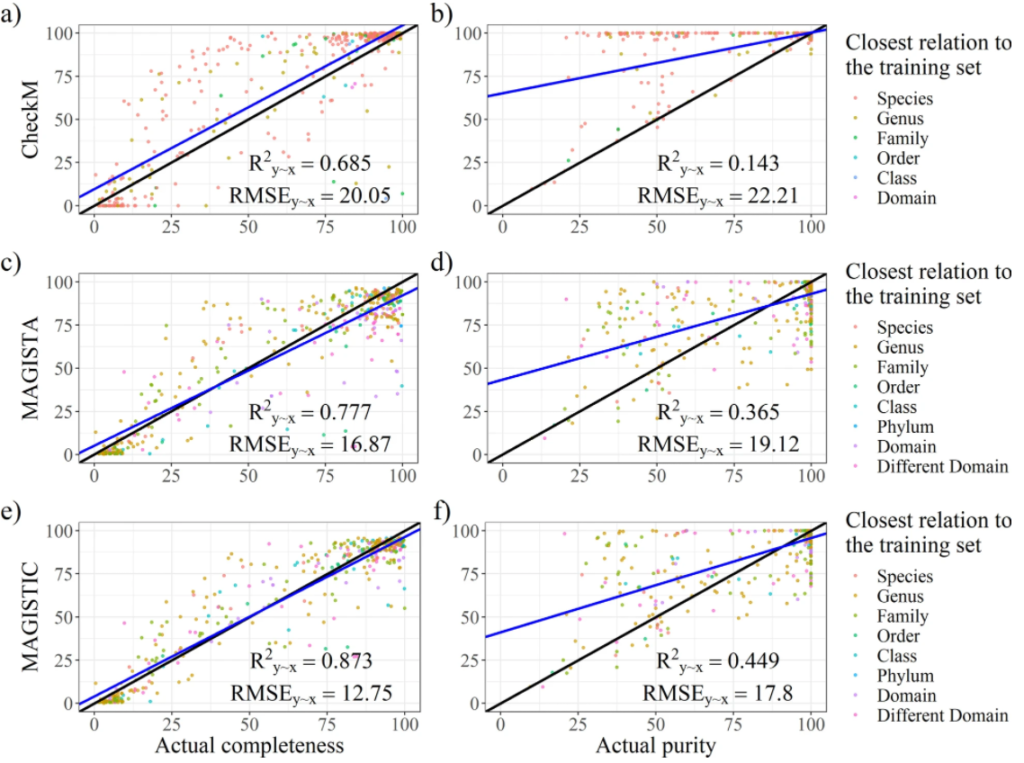
最后是模型构建,建立完整性和污染度的预测模型。并进行了模型评估,如图所示。分别对CheckM、MAGISTA 和 MAGISTIC测试了其性能。CheckM是现在主流的一款评估bin质量的工具。MAGISTIC是一款结合了CheckM和MAGISTA 的工具。使用解释方差的分数(R2y∼x)和均方根误差(RMSE)作为评估性能的指标。对于完整性的预测,MAGISTA 优于 CheckM。对于污染度的预测,MAGISTA 的表现优于 CheckM。
结 论
研究人员开发了一种新的用于预测高度复杂的宏基因组组装基因组bin的质量的方法,MAGISTA。是基于 SCMG 的低复杂性宏基因组方法的一个同样好的替代方法。除了MAGISTA之外,还通过结合CheckM的结果,使用MAGISTIC生成了一个更准确的预测。
研究人员在文章中指出MAGISTA 和 CheckM 都没有达到足够的准确度来被认为是可靠的。MAGISTIC 产生了比 MAGISTA 更好的结果。
在附加分析中,将测试集分为了两个子集,从真实和模拟reads中获得的bins,对此再进行分析,结果表示,CheckM 对于“真实”子集表现良好(但相比MAGISTA 和 MAGISTIC还是较差),对于“模拟”子集部分表现较差。而MAGISTIC相比MAGISTA会更准确些。但是文章中并没有详细说明MAGISTIC的工作流程。
查看作者在github上公开的软件说明,地址如下。但是没有说明和给出输出文件的内容。个人认为还不太成熟。
https://github.com/LM-UGent/MAGISTA
参考文献:
Goussarov G, Claesen J, Mysara M, Cleenwerck I, Leys N, Vandamme P, Van Houdt R. Accurate prediction of metagenome-assembled genome completeness by MAGISTA, a random forest model built on alignment-free intra-bin statistics. Environ Microbiome. 2022 Mar 5;17(1):9. doi: 10.1186/s40793-022-00403-7. PMID: 35248155; PMCID: PMC8898458.

谷禾健康


在现代测序技术的帮助下,微生物组研究的范围被扩大,通过16S rRNA测序或鸟枪法宏基因组测序可以生成大量的微生物组数据。而微生物群落研究中的一个重要问题是对这些微生物的归类,模拟和分析人类微生物群。
通常使用16S rRNA技术量化微生物群落的组成,但量化后的数据是偏斜的,带有过多的0。目前还缺乏对复杂的微生物群落测序数据的标准化的聚类分析方法。
近日,加拿大多伦多大学研究人员在《Microorganisms》上发表的一篇研究,针对上述问题构建了一个参数化的混合模型用于计算聚类分析的距离度量,模型根据观察到的OTU计数和估计的混合权重产生sample-specific的分布。这个方法可以准确的估计真实的0比例,从而构建一个精确的beta多样性度量。
大量的模拟研究表明,与一些被广泛使用的距离度量方法相比,当存在较大比例的0时,该方法取得了较好的聚类效果。
该研究人员提出了一种具有特定beta多样性度量的聚类算法,该算法可以解决稀疏计数数据遇到的有无偏差问题且能有效的度量样本距离,达到分层的目的。
微生物群落研究中的一个重要问题是对这些微生物的归类,它们是否能被划分为亚群。如果有,有多少组亚群,如何解释这个亚群。例如,这种分类是否区分了治疗方法、疾病或遗传类型。
为了回答这些问题,需要测量两个微生物群落之间的相似性。beta多样性是为了适应不同的目的而提出的,在评估群落之间的差异时提供不同的结果。对于微生物组成,beta多样性根据测量丰度来衡量群落之间的距离,丰度可以是观察到的计数,也可以是相对丰度,这些丰度是根据不同或距离度量计算出来的,以量化样本之间的相似性。
现如今,已经有许多非参数统计方法来量化距离度量。例如Euclidean和Manhattan距离是最常用的。其它beta多样性指标,例如Bray-Curtis距离、Jensen-Shannon距离、Jaccard指数、UniFrac距离(未加权的、加权的和广义的)也经常用于微生物组研究。
除了距离度量之外,还引入了用于生态关联推理的稀疏逆协方差估计(SPICE-EASI)的图形网络模型。然而这些方法都会有一定的局限性,例如SPIEC-EASI方法依赖于单一的方差-协方差矩阵,由于微生物群落结构复杂,可能无法完全恢复底层OTU网络。
于是,研究人员开发了一种创新的聚类方法,以混合模型而不是beta多样性度量作为距离度量,并将聚类算法应用于微生物群落数据来表征亚群。该算法还包括根据选择的内部指标选择最优聚类数,并将结果在几种距离度量和不同评估方法之间进行比较。通过全面的模拟研究和一个真实的帕金森病肠道微生物群数据集对该算法的性能进行了评估。
1. 构建混合模型
混合模型是一种概率模型,用于表示在无监督学习中经常使用的总体内的子群体。该模型关注单个OTU在种群中的分布,可以解决样本间的稀疏性问题。它参数化地模拟了计数的潜在分布,包括低计数OTU和极高计数。对于个体样本之间的成对距离,在L2范数距离中使用公式化的混合概率。
2. 距离度量
在确定混合模型分布后,使用概率分布通过样本之间的两两距离计算距离度量。为了进行比较,考虑了基于L2范数的三种距离度量(L2-PDF、L2CDF、L2-DCDF、L2-CCDF)。
除此之外还选择了一些其他的距离度量进行比较,即Manhattan距离和Euclidean距离,以及微生物组分析中特有的三个距离度量:Bray-Curtis距离、加权UniFrac距离和广义UniFrac距离。本研究不考虑未加权的UniFrac距离,因为它不包含类群丰度信息。
3. 聚类分析验证指数
这些指数用于衡量集群在集群内部和集群之间的可分离性表现很好。验证指标可以分为内部指标和外部评估,许多内部验证指标被用来选择最优聚类数。外部评估分数是在假设标签在建模阶段没有使用时,通过直接将划分结果与之前的标签进行比较来计算的。
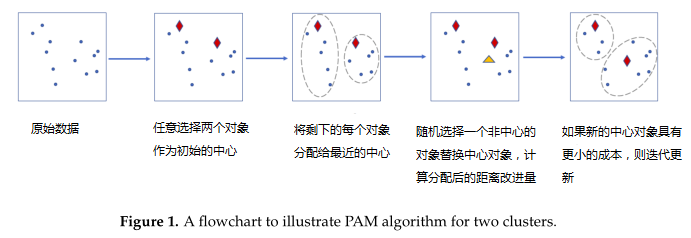
4. 用于聚类的分区算法(PAM)

使用混合分布的聚类过程的详细步骤:
为了测试该方法在聚类中的表现如何,研究人员推导了其准确性和Jaccard指数。
准确性是指聚类结果与真实的聚类指数的接近程度。它被定义为正确聚集的受试者所占的比例。
Jaccard指数衡量聚类结果与原始聚类标签之间的相似性,原始聚类标签定义为正确分类的主题数量(预测集与真实集的交集 )与两组总样本量(两集的并集)之比。
研究人员用类标签模拟数据来模拟OTU计数及其复杂的结构。研究人员考虑两个有两个子类和三个子类的场景,每个子类包含200个样本,总样本量分别为400和600。所有的结果被重复了100次。
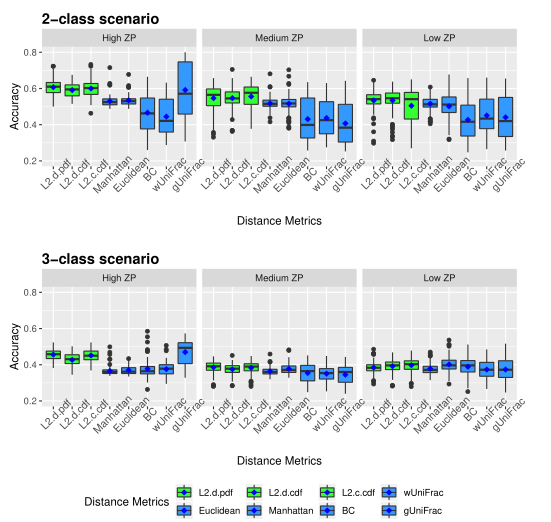
下图展示了模拟数据的准确性。评估了三种不同的0的比例(ZP)情况,左中右分别为高ZP、中等ZP、低ZP。
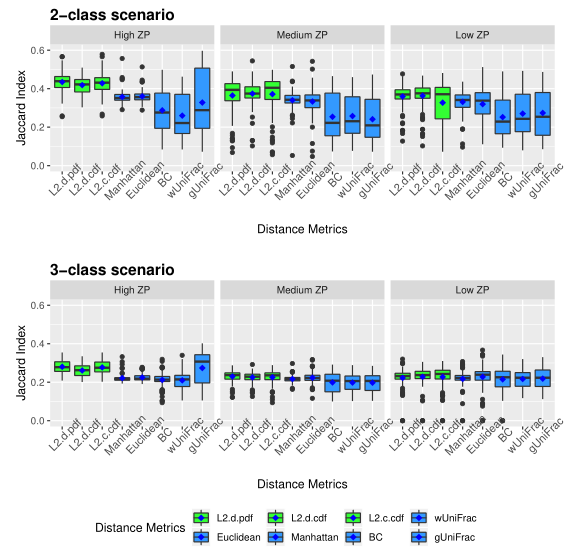
下图展示了模拟数据的Jaccard指数。同上图一样评估了三种不同的0的比例。
以上两图显示了具有不同ZP和子类数量的不同场景下模拟数据集的聚类结果。通过准确率和Jaccard指数对基于距离的算法性能进行了评估。填充颜色为绿色的箱形图为研究人员建议使用的距离度量。所有的距离都是根据相对丰度计算的。
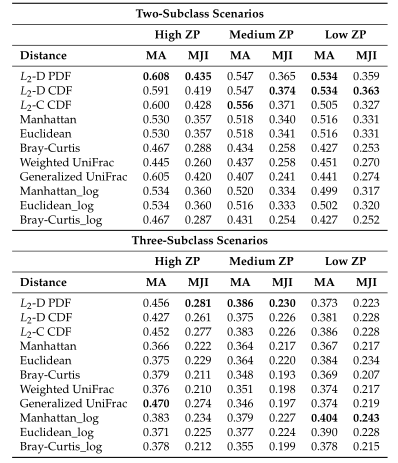
Table1平均准确率(MA)和平均Jaccard指数(MJI)估计。粗体表示每个方案下的最佳情况。Log表示对输入的模拟数据进行了对数变换。
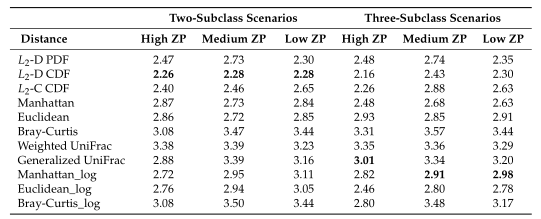
Table2所有模拟场景的平均集群数。根据Dunn内部指数计算出每次重复的最优聚类数。
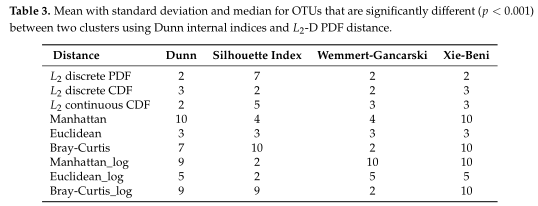
Table1 的结果是通过对每个场景中的100个重复结果求平均值计算得出的。观察得到在聚类算法中实现的距离度量(即绿色标识的箱形图)的准确率和Jaccard指数都优于其他距离度量,特别是在数据集中包含大量0时(高ZP)。在MA和MJI方面,L2范数也显示了其优势,在基于100次重复计算的L2范数在两个子类场景下的;平均准确率约为0.6秒,在三个子类场景下的平均准确率为0.45。而广义Unifrac距离(gUniFrac)在准确性估计中有很大的波动变化。
数据集为197名PD患者和130名对照的粪便样本的16S rRNA测序数据。首先过滤掉在80%的OTU中相对丰度都为0的OTU,因此,此次分析中共使用280个OTUs。将基于相对丰度计算的L2范数与其他三个距离度量进行了比较,包括对数变换和不变换的比较。
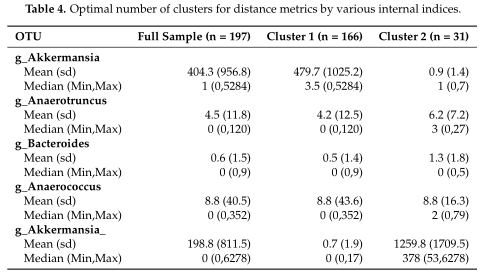
如Table3所示,距离度量采用各种内部验证指标(列名)进行灵敏度分析。对于Dunn和Xie-Beni指数,l2范数倾向于将数据聚类为两到三个亚群,而在有和没有对数变换的情况下,除了未变换的欧氏距离外,Manhattan、Euclidean和Bray-Curtis更倾向于聚类更多的亚群。(设置了最多聚类数目为10)
接着选择L2-D PDF范数作为进一步分析的例子。
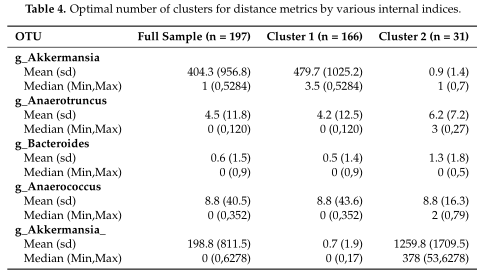
结果在Table4中展示,对数据集中的两个集群之间的OTUs进行了探索,得到了聚类之间差异最大的前5个OTU。
研究认为该聚类算法在高ZP和中等ZP的情况下表现最好,因此,当数据中出现大量的0时,建议使用。并且,在PAM框架下,文章中列出的那些距离度量都可以用作聚类的输入。
如模拟研究中显示的那样,在各种场景下,由混合模型计算的成对距离比其他距离度量表现的更好。但是与所有聚类方法一样,都有一个局限性,就是很难为每个新数据选择合适的内部指标,因此很难获得最优和最稳健的集群数。
此外,对于L2范数距离,在聚类中无法进行变量选择。但不可否认,该聚类算法结合了微生物测序数据的特殊距离,所介绍的聚类算法除了目前使用的方法之外,还可以被看作是分析微生物数据的一个很好的辅助工具。
研究人员指出,下一步会基于Dirichlet-Polyomial等模型进行分区,与文章中的方法进行比较,并努力将该方法扩展到其他微生物群落和疾病相关的数据上。
【参考文献】
Yang D, Xu W. Clustering on Human Microbiome Sequencing Data: A Distance-Based Unsupervised Learning Model. Microorganisms. 2020 Oct 20;8(10):E1612.