-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康
麦角硫因 (EGT) 是一种含硫的抗氧化剂,由某些微生物合成,大量存在于蘑菇、发酵食品和其他膳食产品中。人体无法自主合成,需要通过饮食摄取和积累。
麦角硫因(EGT)由哺乳动物细胞输入,可以在哺乳动物组织中积累到低毫摩尔浓度,在那里它与保护健康作用有关。 同样,许多宿主相关微生物输入EGT,EGT通过其抗氧化特性增强细胞内氧化还原稳态。某些细菌物种也编码降解 EGT 的酶。事实上,最近的研究发现 EGT 可以被人类粪便细菌的复杂群落代谢。 粪便群落的 EGT 代谢因人而异,表明微生物组组成的个体间差异可能影响 EGT 代谢。
许多与宿主相关的微生物都会吸收EGT,通过其抗氧化特性增强细胞内氧化还原稳态。某些细菌物种也编码降解 EGT 的酶。因此研究人员提出假设并进行验证:EGT是否会先被部分菌种代谢生成某种可用物质,再由后续菌种接力还原,从而获能(促进ATP合成与生长)?
为此,研究人员结合群落模型、分离菌株和共培养实验、代谢物时间序列实验、多队列宏基因组数据再分析等技术进行了研究和验证。完整重现了Clostridium symbiosum和Bacteroides xylanisolvens这两种细菌在厌氧环境下”EGT→TUA(thiourocanic acid)→还原产物(3-(2‑thione‑imidazol‑4‑yl)‑propionic acid)”的代谢接力过程。在对24份健康人粪便群落进行48小时培养的功能表型测试中,有18/24样本显示EGT代谢活性并产生TMA,且其中11/18检测到还原TUA产物;相应的宏基因组分析显示,具EGT代谢活性的样本中ergothionase基因显著富集。在四个独立结直肠癌队列的粪便宏基因组数据分析中,发现ergothionase基因在其中两队列中显著富集,在另外两队列中呈增加趋势。
这些发现揭示了饮食抗氧化分子在肠道微生物能量代谢与潜在疾病风险差异中的功能纽带,为未来通过调控微生物EGT代谢改善肠道健康提供了方向。
1
确定麦角硫因代谢菌和酶
已知麦角硫因酶(EGT trimethylammonia lyase)可将EGT裂解为TMA与TUA。Treponema denticola SP33 ergothionase (TdETL) 是已被表征的“参照”,用其氨基酸序列做同源搜索,在人肠道可培养菌株库里寻找麦角硫因(ergothionase)同源物,从而锁定了C. symbiosum CLOSYM_01531和C. symbiosum CLOSYM_03165。
通过LC-MS/LC-MS-MS对C. symbiosum+EGT-d9的培养上清与细胞组分进行非靶与靶向代谢组学分析,验证其将EGT裂解为TMA与TUA,并显示TUA主要分泌到胞外。异源表达验证实验(将CLOSYM_01531这个基因装进E. coli里表达),观察到EGT-d9在约6小时内被完全转化为TMA-d9与TUA。
2
小鼠粪便菌群
来自不同来源的雌性6周龄的C57BL/6小鼠(JAX、TAC、CR)做粪便群落48小时厌氧培养+EGT-d9,比较EGT代谢能力差异。对CR群落做非靶代谢组学,鉴定得到新的代谢物(m/z 173.0379),经标准品比对,确认为还原的TUA产物3-(2‑thione‑imidazol‑4‑yl)‑propionic acid
3
细菌共培养
通过对CR 和 TAC 粪便微生物组的16S rRNA 测序分析,发现CR群落中Bacteroides acidifaciens富集。研究人员选取与 B. acidifaciens 亲缘关系较近、且在人肠道常见并已知具备多糖与宿主营养代谢能力的代表物种 B. ovatus 与 B. xylanisolvens 作为候选,检验其是否具有将 TUA 还原为 3-(2thioneimidazol4yl)propionic acid 的活性。并测试还原过程对能量代谢与生长的影响。
单培养与共培养添加EGT-d9的C.symbiosum和B.xylanisolvens时,进行长时间跟踪。同时用表达CLOSYM_01531的工程E.coli替代C.symbiosum重复共培养过程。进一步证明接力关键在于EGT产生的胞外TUA与下游还原步骤的耦合。
4
健康人队列粪便菌群
对24份健康粪便样本做48小时厌氧培养+EGT-d9,查看EGT代谢能力和产物,发现有18/24个样本把EGTd9代谢掉了。在这18个样本中,对其中3个TUA还原能力强的样本做了时间跟踪,还原代谢轨迹。同时将其中23份宏基因组数据分为两组(有代谢活性 vs 无代谢活性),做配对分析,查看ergothionase基因富集情况。
5
结直肠癌队列宏基因组功能分析
基于EGT稳态与结直肠癌(CRC)的文献线索,汇总四个已发表CRC粪便宏基因组数据集,采用统一流程定量ergothionase基因丰度,然后进行组间差异分析。
▸ C.symbiosum可将EGT代谢为TMA与TUA,且TUA主要分泌到胞外
如上图所示,经EGT-d9处理的培养物的细胞沉淀物(C. symbiosum ATCC 14940)中检测到EGT-d9和TMA-d9,表明C. symbiosum ATCC 14940导入并代谢EGT。
在C. symbiosum ATCC 14940培养上清液中检测到一种单一的代谢物(m/z 171.0223,图C),对比未经EGT-d9处理的样品,m/z 171.0223显著上调(Log2(fold change) ≥ 2, p ≤ 0.05) ,经标准品对比(图D、E),确认为TUA。用或不用EGT-d9处理C. symbiosum培养物48小时后的EGT-d9、TMA-d9和TUA含靶向定量结果显示(图F),EGT‑d9下降与TMA‑d9、TUA上升在计量上匹配,符合EGT代谢为“两段”——TMA与TUA的表现。
将CLOSYM_01531异源表达于E. coli(图C),发现其可在约6小时内将EGT‑d9完全转为TMA‑d9与TUA(图D),空载对照(Ec_EV)无此活性(图G)。此外,C. symbiosum中还鉴定到第二个同源物CLOSYM_03165,异源表达同样具活性。
▸ 小鼠粪便菌群的代谢组学分析鉴定出“还原产物”
不同来源小鼠展现出群落差异,如图A,发现CR群落48小时内可将EGT‑d9完全代谢为TMA‑d9,TAC群落仅中等程度代谢并积累TUA,JAX群落则基本无代谢。
编辑
但在CR培养物上清液中并未检测到TUA,而是发现了一个新的代谢物(m/z 173.0379),且呈显著上调(log2(fold change) ≥ 3, p ≤ 0.05),经标准品比对,确认是3‑(2‑thione‑imidazol‑4‑yl)‑propionic acid,由TUA进行2e−/2H+加氢还原所得(图E)。CR群落可将外源TUA完全转为该还原产物,而JAX、TAC不能(图G)。
▸ TUA还原增强了B.xylanisolvens在厌氧条件下的ATP合成和生长
16S rRNA 分析CR和TAC样本的菌群,发现它们具有相似的菌群多样性(图A、B),且Bacteroides acidifaciens在CR群落中显著富集(图C)。研究人员又提出假设, B. ovatus和B. xylanisolvens或许可以将TUA还原为3‑(2‑thione‑imidazol‑4‑yl)‑propionic acid。
事实也的确如此,在添加了TUA的培养基中于厌氧条件下培养这两种菌48h,定量结果显示两种菌株都消耗了培养基中的TUA,并产生了等量的3‑(2‑thione‑imidazol‑4‑yl)‑propionic acid(下图A)。B. xylanisolvens约6小时内可将TUA完全转化,且产物主要在胞外(上图E、F)。在缓冲体系中,B. xylanisolvens因TUA或TUA+甲酸钠而ATP合成约提升4倍(下图B),并伴随还原产物累积(下图C);在厌氧环境中且缺乏其它电子受体(也就是能增强ATP合成的化合物)的最小培养基中,发现TUA显著提升其生长(下图D)。
▸ C.symbiosum和B.xylanisolvens互相利用EGT的代谢产物
鉴于C. symbiosum和B. xylanisolvens可以分别代谢EGT和TUA,研究人员先假设这两个菌种可以共同将EGT转化为3‑(2‑thione‑imidazol‑4‑yl)‑propionic acid,然后通过共培养实验进行验证。验证结果显示,C.symbiosum在单独培养时会将EGT‑d9转为TMA‑d9和TUA,不产生3‑(2‑thione‑imidazol‑4‑yl)‑propionic acid,这表明C. symbiosum不能还原TUA。B.xylanisolvens则对EGT‑d9无作为。两者共培养时,EGT‑d9消耗程度与C.symbiosum单独培养时相当,但TUA几乎不积累,反而是还原产物显著积累。(图E培养4天、图F培养7天)
代谢组火山图显示细胞沉淀和培养上清液中分别仅富集TMA‑d9与还原产物,符合跨物种接力还原产物的代谢模式“EGT→TUA→3‑(2‑thione‑imidazol‑4‑yl)‑propionic acid”(图I、L)。
用表达CLOSYM_01531的E.coli与B.xylanisolvens共培养,也再现了“EGT→TUA→3‑(2‑thione‑imidazol‑4‑yl)‑propionic acid”反应(图G)。
▸ 人群普遍性和个体差异
研究人员筛选了24名健康成人,获得其粪便样本。在添加EGT-d9培养基中共同培养48小时,观察EGT代谢活性差异。发现24个样本中,有18个样本在48小时内显著代谢EGT‑d9,且均产生TMA‑d9(图A-D),这表明麦角硫因介导的EGT代谢的一个特征是消除TMA。虽然在24个样本中都没有检测到TUA,但能够代谢EGT‑d9的18个样本里,有11个样本积累了还原产物(图D)。
对3个TUA还原能力强的样本做时间序列分析,发现6–12小时内TUA短暂出现并随EGT‑d9下降而上升,随后被转化为还原产物,这直接展示了跨物种接力还原产物的代谢模式。
▸ 肠道EGT稳态的改变可能与结直肠癌有关
编辑
研究人员量化了来自四个独立结直肠癌队列和健康对照队列的粪便宏基因组数据集中ergothionase基因的相对丰度,发现有两个CRC组存在ergothionase基因显著富集的现象,另两队列呈上升趋势,这提示EGT代谢能力可能是癌症相关肠道菌群的功能特征之一。
过去更多讲的是“单个细菌把EGT彻底分解”,现在发现EGT还能被“分工协作”重塑成能增强ATP合成的化合物。这项“分工协作“的主角分别是C. symbiosum和B. xylanisolvens,重点工作内容是,C.symbiosum将EGT代谢为TUA和TMA-d9,B.xylanisolvens将TUA还原为3-(2-thione-imidazol-4-yl)-propionic acid,同时提高了自己的ATP产量,促进了自己的生长。B. xylanisolvens作为一种益生菌,目前被认为与宿主健康密切相关。根据现有发现,通过添加TUA或在有“产TUA菌”背景下添加EGT,理论上有助于B. xylanisolvens的定植。
研究中虽然提示EGT→TUA轴与疾病生态有关,但当前缺乏动物与人体定植/功能结果的直接验证。
总而言之,如果在合适的群落背景下提升B.xylanisolvens的定植或TUA还原能力,理论上可以带来:更稳的肠道生态位占据、更高效的厌氧能量代谢、潜在更好的底物利用与有益代谢物输出,并可能通过改变供给-利用格局而影响与疾病相关的微生态失衡。
主要参考文献
Zhou Z, Jiang A, Jiang X, Hatzios SK. Metabolic cross-feeding of a dietary antioxidant enhances anaerobic energy metabolism by human gut bacteria. Cell Host Microbe. 2025 Aug 13;33(8):1321-1332.e9.
谷禾健康

最近的Nature 和 Nature Medicine 连发表了好几篇关于肠道菌群的文章,包括肠道菌群与神经互作,和基于这个原理的针对自闭症的临床治疗方案。心血管疾病的微生物组和代谢特征等。
今天我们主要介绍心血管疾病中冠状动脉疾病的相关重要研究发现和意义。
复杂的疾病,如冠状动脉疾病(CAD),往往是多因素的,由多种潜在的病理机制引起。尽管冠状动脉疾病在预防、诊断和治疗方面取得了巨大进展,但仍然是世界范围内发病率和死亡率的主要原因。目前对冠状动脉疾病的治疗基于传统的和可控制的冠状动脉疾病风险因素,只能取得部分成功。
冠状动脉疾病的发展包括血管壁上动脉粥样硬化斑块的逐渐生长,这通常与代谢状态受损有关。人体接触环境分子的主要部位是胃肠道,其中膳食成分被微生物群转化,利用产生代谢物传播到全身器官。
血液充当体内分子的液体输送器, 特别是数以千计的循环代谢小分子,它们可以帮助我们了解体内生物过程状况,并且是研究冠状动脉疾病多因素性质疾病的宝贵来源。肠道微生物组积极参与血液代谢物的代谢。
几种肠道微生物群衍生的循环代谢物与心血管疾病相关:
三甲胺 N-氧化物
三甲胺 N-氧化物被确定为人类心血管疾病的标志物,进一步的证据表明在小鼠模型中具有促动脉粥样硬化性和促血栓形成。
硫酸吲哚酚
硫酸吲哚酚在细菌色氨酸酶降解色氨酸后在肝脏中产生,并被证明与动脉僵硬和外周血管疾病有关。
对甲酚
对甲酚是苯丙氨酸和酪氨酸的结肠细菌发酵产物,显示与心血管事件增加相关。
近期,以色列科学家招募了下列人群,采集其粪便和血清样本进行了全面的多组学分析,同时调查详细的医疗、生活方式和营养问卷等。
通过对粪便样本宏基因组测序(每个样本1000万 reads,约3G/样本)和对血清样本的进行非靶向质谱LC-MS测量了 961 种代谢物的水平,包括脂质、氨基酸、异生物质、碳水化合物、肽、核苷酸和大约 30% 的未命名化合物。
通过 Nightingale Health 的质子核磁共振 ( 1 H-NMR) 平台测量了另外 228 种血浆代谢物和比率,并使用了一个独立宏基因组数据集MetaCardis进行验证(该数据集样本来自于北欧血统队列,在地里区域上与该研究样本来源不同,这样可以分析遗传,饮食差异变量)。
MetaCardis数据集主要由四个主要群体组成:缺血性心脏病、健康对照组、代谢匹配的对照组和未经治疗的代谢受损对照组(详细数据集描述可以参看原文)
一、ACS的肠道微生物组特征
1. ACS 患者的变形杆菌丰度更高
这与之前的大多数研究结果一致,变形菌增多会导致处于炎症状态,是生态失调的标志。
20个在 ACS 或对照个体中显着富集的细菌,包括产丁酸盐的细菌如:梭菌属(Clostridium)、Anaerostipes hadrus嗜热链球菌(Streptococcus thermophilus)和Blautia菌属,以及Odoribacter splanchnicus 和大肠杆菌。
2. ACS患者队列中一种梭菌科的细菌物种 SGB 4712缺乏
在20 个显着富集的基因组中,鉴定到了一种以前未知的梭菌科细菌物种,索引为 SGB 4712。为了进一步验证该结果稳定和实用性,使用另外一个来自北欧血统地理上分布不同的队列,MetaCardis宏基因组数据集进行验证,与该研究结果一致,该物种的相对丰度随着具有 CAD 传统风险因素的种群逐渐减少。
3. SGB 4712关联15种显著差异的代谢物,其中包括降低心血管疾病风险的独立标志物——麦角硫因(ergothioneine,天然氨基酸)
对照组相比, 鉴定到SGB 4712 菌种与15 种循环代谢物的水平显着相关,在 MetaCardis 研究中,所有 15 种代谢物与 SGB 4712 的相关系数均可以重复,其中 10 种相关性仍然显著。
值得注意的是,SGB 4712与麦角硫因呈正相关,麦角硫因是一种天然存在的氨基酸,在体外显示对细胞应激源具有抗氧化和细胞保护能力,最近被证明是降低心血管疾病和人类死亡率风险的独立标志物。
此外,SGB 4712 与七种化学结构未知的化合物有关。其中包括 X-11315 和 X-24473,预测它们来自饮食,并与 SGB 4712 呈正相关。
图一 ACS 的微生物组和血清代谢组学特征
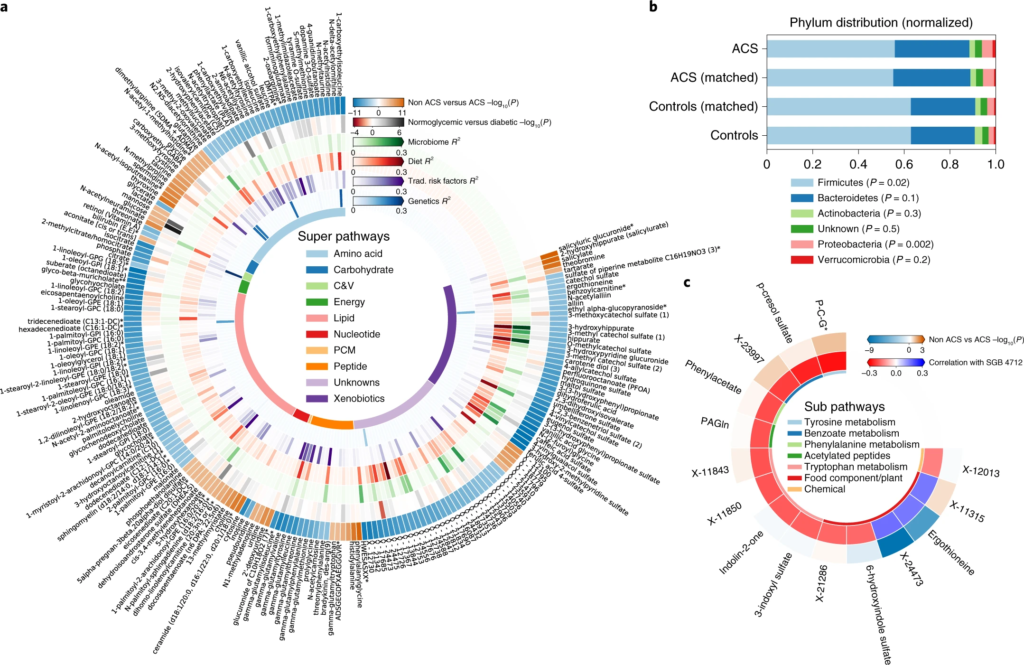
圆形热图显示 ACS 和非 ACS 对照组之间显着差异的前 200 种代谢物,与年龄、性别、BMI、吸烟状况和 DM 相匹配(方法)。每个切片代表一个代谢物,其名称显示在图表的外层周围。
这些结果突出了SGB 4712菌种在 CAD 发展中具有潜在的保护作用,由一系列循环血液代谢物介导,其中一些以前被证明在元生物途径中发挥核心作用,而另一些则未知。
因此,在实验研究中进一步验证后,这些代谢物可能会形成降低 CAD 风险的新目标。
二、ACS 的代谢特征因人而异
1. ACS 患者的血清代谢物水平个体化差异较大
虽然 CAD 患者具有共同的内表型,但他们通常表现出生物学上不同的疾病特征。为了更好地了解 ACS 的个体水平变异性,作者试图检查与非 ACS 对照的代谢偏差,并询问它们是否是个体特异性的。
计算了他们的个体偏差,并根据之前根据饮食、微生物组、传统风险因素和遗传学估计的 EV 对每个个体的前 100 个偏差代谢物进行加权。最后发现ACS 患者与其匹配对照的代谢偏差是因人而异的。
急性冠脉综合征患者的血清谱在血清代谢物水平上表现出广泛的扰动,包括533种显著改变的代谢物。
ACS的血清代谢组遵循一种主要的消耗模式,因为在对照组参与者中,358种代谢物(67%)的平均测量值较高。然而,这一趋势在主要的生物途径中并不一致。但是,与富含 ACS 的代谢物相比,饮食和微生物组在与 ACS 耗尽代谢物的偏差相关联方面更为显着(双尾 Mann–Whitney U-检验,P-value小于10 -20),这表明微生物组对 CAD 起保护作用。
值得注意的是,超过 90% 的显着扰动的代谢物无法用血糖状态来解释,这表明这种变化背后还有其他机制。所以进一步分析了其他系列综合因素(包括宿主遗传学、微生物组和饮食),得到一个重要发现就是:饮食和微生物组可以更好地解释 ACS 缺乏或含量低的代谢物,而传统的风险因素可以更好地解释 ACS 富集的代谢物。
图2 代谢偏差由潜在决定因素解释,并与临床参数相关
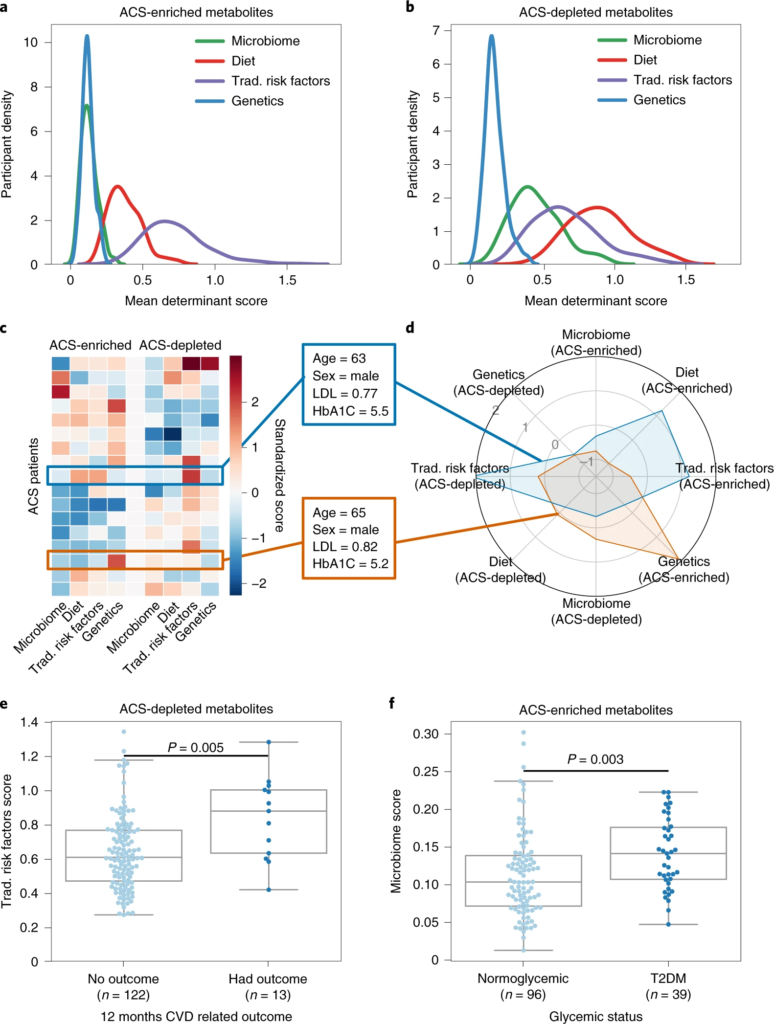
a、b、密度图显示 ACS 参与者的分布(y轴)与代谢物的潜在决定因素(微生物组、饮食、传统风险因素或遗传学)的平均加权R 2 ( x轴);富含 ACS 的代谢物。
2. 相似的临床特征,但其动脉粥样硬化负担的代谢机制却不同
虽然一些患者可能具有相似的临床特征,但他们的潜在生理状态和疾病轨迹可能不同。为了强调这种 CAD 患者的变异性,作者选择了 ACS 患者的常规危险因素的同质亚组。其中包括 17 名 60 至 70 岁的男性患者,低密度脂蛋白 (LDL) 在 0.70–1.30 mg ml -1范围内,糖化血红蛋白 (HbA1C) 低于 6%。尽管具有相似的临床特征,但该 ACS 患者亚组在代谢偏差方面表现出异质性。
三、微生物组在CAD早期阶段发挥作用
动脉粥样硬化是一种经过多年发展的进行性疾病,其中动脉粥样硬化斑块形成的每个阶段的特点是不同的病理过程。在早期阶段,血管壁上的动脉粥样硬化斑块的生长通常与代谢状态的损害有关。
为了解释每个代谢成分在 CAD 发展的时间轴上的参与,作者将个体代谢偏差的分析应用于代谢受损的对照(定义为 T2DM、高血压或血脂异常的诊断,或 BMI > 35),以及到非 ACS 个体的随机子集。
在比较这三组的分数时,我们发现分数分布存在一致的差异。与微生物组和饮食相关的代谢异常呈现出渐进的趋势,与对照组的随机子集相比,代谢受损的对照参与者的代谢物存在显着偏差。
这表明,微生物组和饮食对ACS的贡献可能是通过受损的代谢状态介导的,而不是代谢受损个体中尚未表现出的与传统风险因素和遗传学相关的代谢物异常。
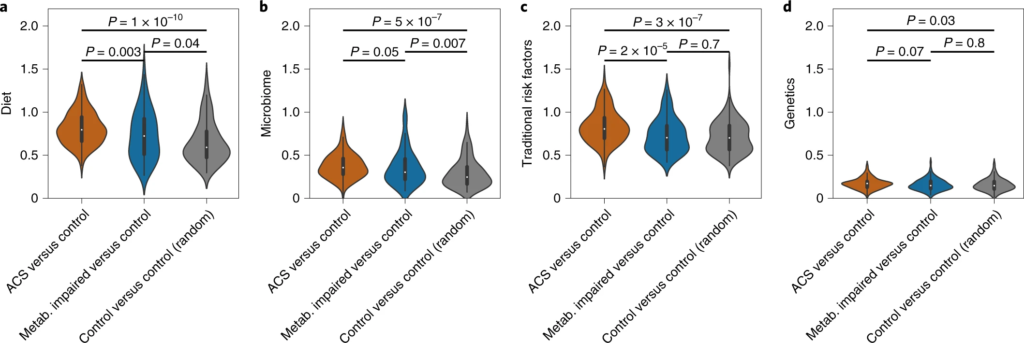
a – d,归因于饮食 ( a )、微生物组 ( b )、传统风险因素 ( c ) 和遗传学 ( d ) 的代谢偏差分数计算三个亚组:(1) ACS 个体 ( n = 135) 与非 ACS 对照与年龄、性别和 BMI 相匹配(橙色);(2) 患有代谢障碍的非 ACS 对照(定义为:诊断为 T2DM、高血压或血脂异常,或 BMI > 35;n = 102)与其他年龄、性别和 BMI 匹配的非 ACS 对照(蓝色);(3) 一组随机的非 ACS 个体 ( n = 132) 与其他匹配年龄、性别和 BMI(灰色)的非 ACS 对照。
四、血清代谢组学预测ACS患者 BMI 更高
肥胖是 CAD 的主要独立危险因素,影响已知的危险因素,如血脂异常、高血压、葡萄糖耐受不良和炎症状态,以及可能尚未认识到的机制。BMI 测量被用作肥胖的标志和代谢健康的指标。
为了研究肥胖作为 CAD 的独立危险因素,该研究设计并彻底验证了基于血清代谢组学的 BMI 模型,并表明较高的预测 ΔBMI 对应于更广泛的动脉粥样硬化疾病。
作者分析了CAD 患者的 BMI-代谢组平衡是否以及如何被破坏。使用了梯度提升决策树 (GBDT) 算法预测 BMI,结果表明在非ACS受试者中发现的代谢组-BMI模式在ACS患者中受到干扰。
为了研究这些扰动,作者测试了对照组和 ACS 测试集中预测和测量 BMI 之间的差异,这里称为 ΔBMI。结果发现,与非 ACS 受试者相比,该研究的模型预测 ACS 的 ΔBMI 更高。
为了验证这些结果的稳健性,作者试图根据其他类型的代谢组学数据和独立队列来复制这些发现。将相同的预测程序应用于基于 NMR 的代谢组学数据,并观察到ACS 和对照之间 ΔBMI 的更大差异,应用于为发表的MetaCardis 队列数据中得出在所有 BMI 范围内,与血糖正常的缺血性心脏病患者相比,患有糖尿病的缺血性心脏病患者的 ΔBMI 显着更高。
进一步分析推断哪些特定代谢物是 ACS 患者高 ΔBMI 的主要驱动因素,发现两种脂质在对照组中与 BMI 呈负相关,后者在患有更广泛疾病的患者中也显着减少,这两种脂质分别是:
1-(1-enyl-palmitoyl)-2-oleoyl-GPC (P-16:0/18:1)
1-(1-enyl-palmitoyl)-2-linoleoyl-GPC (P-16:0/18:2)
最近的研究表明,脂质1-linoleoyl-GPC (18:2) 与肥胖和 T2DM呈负相关,并且脂质水平的增加显着降低了T2DM的风险。该研究发现 1-linoleoyl-GPC (18:2) 和 1-(1-enyl-palmitoyl)-2-linoleoyl-GPC (P-16:0/18:2) 在对照组中与 BMI 呈负相关,并且在患有更广泛 CAD 的患者中显着耗尽,这表明这些代谢物可能作为降低 CAD 风险的潜在靶点。
此外,两种代谢物都含有一条亚油酸链,一种必需脂肪酸,与 T2DM 风险呈负相关。然而,这些假设应在干预性研究中进一步检验。
迄今为止,大多数研究都集中在寻找在 CAD 患者中增加的新代谢物,而该研究对 199 名 ACS 患者进行了全面的多组学分析结果强调, ACS 的代谢组学特征是缺乏多种血清代谢物,其中许多与饮食和微生物组有关。
其中一个重要的发现是以前未知的细菌物种 SGB 4712,它在 ACS 患者和独立验证队列中都显着缺乏或偏低。通过进一步将这种细菌与心脏毒性和心脏保护代谢物的水平联系起来,证明了特定细菌基因组的缺失可能与 CAD 风险增加相对应,并提出在后续干预研究中评估的具体目标。总体而言,这些发现因此为 CAD 患者的预测甚至治疗提供了一种新方法。
迄今为止,大多数研究都对 CAD 患者进行了批量分析,寻找人群水平的风险因素,而不是关注个体水平的生物变异性。在这项研究中,作者使用全面的代谢组学和微生物组分析,呈现了 CAD 内部变异性的深度映射。总之,结果揭示了新的范式和治疗方向。
参考文献:Talmor-Barkan Y, Bar N, Shaul AA, Shahaf N, Godneva A, Bussi Y, Lotan-Pompan M, Weinberger A, Shechter A, Chezar-Azerrad C, Arow Z, Hammer Y, Chechi K, Forslund SK, Fromentin S, Dumas ME, Ehrlich SD, Pedersen O, Kornowski R, Segal E. Metabolomic and microbiome profiling reveals personalized risk factors for coronary artery disease. Nat Med. 2022 Feb;28(2):295-302. doi: 10.1038/s41591-022-01686-6. Epub 2022 Feb 17. PMID: 35177859.