-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康
当”健康中国2030″战略深入人心,当消费者从”治病”思维转向”防病”理念,整个医疗健康行业正在迎来前所未有的发展契机。
随着精准医疗时代的到来,用户需求正呈现出前所未有的细分化和专业化特征:
– 临床端:医生迫切需要具备更高灵敏度和特异性的诊断早筛等工具,以实现疾病的早期识别和精准分层;
– 机构端:健康管理机构正在寻求具有独特技术壁垒和差异化优势的检测产品,以构建竞争差异化;
– 消费端:用户不再满足于标准化的方案,而是渴望针对个体症状或后端干预的精准指导检测。
在这样的市场需求下,谷禾作为菌群检测行业先行者和深耕者,经过不断的技术测试和研发,完成了产品的全面升级和多形态布局。
从最初的16SrRNA测序技术科研应用起步,
到如今构建起涵盖
肠道菌群检测专业版
16S+tNGS靶向检测
宏基因组精准健康检测
针对特定人群的检测报告的完整产品矩阵,
再到技术平台向 女性阴道微生态
宠物肠道健康 等新兴领域的拓展,
谷禾以”场景导向、需求先行“的产品策略,
持续拓宽微生态检测技术的应用疆域。
本文将带您深入了解这一产品生态背后的技术逻辑与应用思考,每一个产品的诞生,都见证着微生态检测技术从”标准化”走向”个性化”、从”通用型”迈向”专业化”的发展轨迹。
01
肠道菌群检测专业版(16S rRNA测序),作为谷禾健康最早推向市场、历经十余年打磨的经典产品,凭借其成本效益、高效性与成熟度,为大规模人群的健康筛查、慢病风险评估、营养干预以及诸多科研项目基线建立,提供了坚实、可靠且极具价值的数据基础。
16S检测专注于细菌和古菌的16S rRNA基因,能够快速勾勒出肠道菌群的整体结构,包括多样性、核心菌属构成、有益菌与有害菌比例等关键菌群相关指标,评估肠道菌群失衡风险,评估健康风险、营养代谢及免疫情绪等多维度健康指标,提供个性化健康管理建议。
基于这一庞大数据资源,我们能通过菌群信息判别菌群的平衡或失调状态,进而判别个体的菌群与健康风险及与饮食、生活方式等关联。
数据的力量
在样本量达到一定量级时,
就会发生质的飞跃
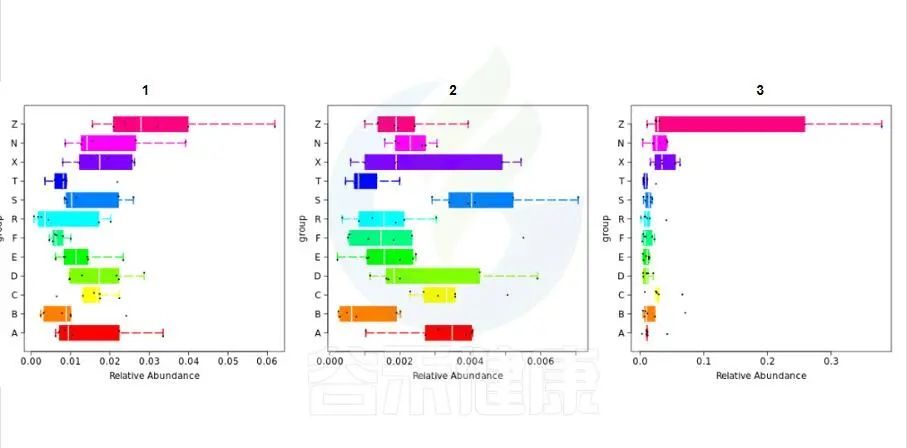
📊 以结直肠癌模型为例:
<来源:谷禾健康肠道菌群检测数据库>
这也就是大样本量赋予16S检测
从 “观察” 到 “预见” 的核心能力
主要包括:健康总分评估、慢病风险预警、
肠道屏障及代谢物、神经递质分析、
个性化营养评估等内容。
健康总分评估
报告提供综合性的健康评分,直观反映客户肠道微生态的整体状态。
菌群整体分析
整体评估肠道菌群平衡、菌群多样性、有益菌、有害菌等指标,还包括核心菌群的丰度,这些核心菌群的减少往往与免疫力下降、肠漏、炎症等问题直接相关。
慢病风险预警
依托海量数据库和先进算法模型,16S检测能够评估与消化系统疾病、代谢类疾病(如肥胖、2型糖尿病)、心血管疾病、肝病甚至部分精神心理问题(自闭症、抑郁症等)等多种疾病相关风险。这为健康管理提供了强有力的早期干预工具,将健康管理从治疗推向预防。
肠道屏障及代谢物、神经递质分析
个性化营养评估
报告能分析菌群对不同营养物质(如膳食纤维、蛋白质、脂肪、维生素、微量元素)的代谢能力,为用户提供饮食建议。
无论是院内还是院外的健康管理,“先检后干预”的科学思路是健康管理的基础理念。通过全面系统的健康检测,准确评估个体健康状况,制定针对性的干预措施。谷禾的肠道菌群检测报告,针对菌群异常、营养代谢失衡以及疾病风险等问题,都会提供相应的干预建议。
市场应用场景
适用人群
专业应用
健康管理升级
多维度健康评估+个性化干预方案,提升服务深度
临床辅助
基于大样本数据库疾病预测模型,助力辅助诊断,辅助用药
渠道共赢
周期短(3-5天)+全流程可控+资质保障,实现快速业务整合
科研赋能
海量200万数据库+研究成果,支持学术前沿探索
谷禾肠道菌群检测专业版
凭借其10多年的发展和应用
已成为理解肠道微生态的成熟且普及的工具
这为许多常规健康管理和慢病预防场景
是谷禾所有产品中不可或缺的基石
通过多种技术分析方法的持续迭代
特别是依托我们的国家发明专利技术
谷禾实现了重要的技术突破
我们能够将16S的物种识别精度大幅提升
在成本可控的前提下
实现了更高的检测精度
⚠️ 技术局限性
但是我们同样坦诚其技术局限性和边界,由于16S针对细菌或古菌的保守基因的扩增,它没有覆盖病毒、真菌、寄生虫等,16S技术还是很难更进一步精确到“菌株”水平,也难以精确到毒力/耐药基因的判别。
🔄 技术进化:从局限到突破
正是基于对16S技术边界的清醒认知,特别是对于个别需要判断毒力基因或者幽门螺杆菌、艰难梭菌、致病性大肠杆菌等病原体以及其特定毒株分型的临床需求,考虑到宏基因组检测的高成本现状,谷禾历经两年研发,成功开发了粪便样本的靶向消化道测序技术(16S+tNGS),为精准病原体检测提供了更加经济高效的解决方案。
02
谷禾16S+tNGS技术结合了超多重PCR和高通量测序的优势,旨在提供比传统16S rRNA测序和宏基因组测序更优、更全面的病原体及耐药基因检测方案。
传统16S + 病原体精准分型 = 全新升级
它在保留16S报告的基础上
以接近16S的成本和周期
用靶向测序技术在原16S的基础上
增加了125 种消化道病原体的检测
还包括耐药基因和毒力基因等
如幽门螺杆菌、大肠杆菌、艰难梭菌分型
弥补了传统16S无法检测
非细菌/古菌病原体的不足
这是一款突破传统16S检测瓶颈而生的产品
编辑
值得一提的是,谷禾在tNGS技术的研发道路上并非一帆风顺。这项看似成熟的技术,在不同应用场景下却面临着截然不同的挑战难度。
tNGS对血液和上呼吸道样本检测较简单,因其主要含病原体。但在消化道特别是粪便样本检测时,技术难度大幅增加。
“两年磨一剑,突破粪菌检测技术壁垒”
这两年谷禾团队需要解决一系列前所未有的技术难题:
这正是消化道微生态检测的技术壁垒所在。与呼吸道样本不同,肠道环境的复杂性要求我们必须在技术层面实现更精准的信号识别与干扰排除。
经过无数次的实验优化和迭代
我们最终突破了这一技术瓶颈
为大家带来真正可靠的
消化道病原体检测解决方案
谷禾16S+tNGS产品特点
弥补16S检测技术边界
以接近16S的成本
实现靶向病原体精准检测
谷禾16S+tNGS报告内容涵盖所有16S报告的内容,即包括健康总分评估、慢病风险预警、肠道屏障及代谢物、神经递质分析、个性化营养评估等。此外还包括常见消化道病原体,例如:
细菌病原体
…
病毒
真菌、寄生虫、其他病原体
…
毒力基因
…
耐药基因
…
也包括相关病原微生物的解释
…
例如,通过检测幽门螺杆菌毒力基因组合,可判断是否需立即治疗,避免对弱毒株患者的不必要抗生素使用。强毒株感染会损伤胃黏膜,增加胃炎和溃疡风险,早期预警能在胃黏膜不可逆损伤前提供治疗窗口期。
注: 由于该技术是检测粪便中的幽门螺杆菌,当浓度低于检测下限(50 copies/mL)时,可能出现假阴性结果。因此,对于临床症状明显但检测结果为阴性的患者,建议结合其他检测方法。
✎ 谷禾16S+tNGS精准检测
一次检测即可实现对消化道病原体的全面筛查和耐药基因的精准识别,提供科学依据,最大化治疗效果,最小化治疗风险。
03
宏基因组检测项目以环境中所有微生物基因组为研究对象,通过对样本中的全基因组DNA进行高通量测序,能够多维度全面解析肠道微生物组。
基于持续积累的20万+肠道宏基因组数据库,我们可以系统性地挖掘微生物群的功能基因谱,并预测其代谢潜力。
结合自主研发的多模态模型和机器学习算法,该技术可以建立微生物特征与宿主表型的关联,实现肠龄预测、菌群恢复力评估、定植能力分析等功能。这些分析结果有助于为个性化益生菌干预、精准营养调控及FMT供体筛选等应用场景提供分子水平的参考依据。
宏基因组流程——从随机打断到精准重构
优 点
缺 点
不适合大规模筛查
宏基因组更适合宿主含量不高的样本
谷禾以往在科研领域深耕十余年,积累了丰富的科研项目经验和数据分析能力,几年前,谷禾成功实现了从科研到大健康应用的技术转化,推出了谷禾宏基因组精准检测。
对于一些特定需要深度检测的应用场景,如健康管理机构的差异化服务需求,或者临床应用需求,宏基因组精准健康检测提供了另一种专业的技术选择。
宏基因组数据库的物种涵盖范围和菌株构成,直接影响着宏基因组物种鉴定分类的准确性和分类精度。
针对宏基因组数据库不完善的问题,谷禾整合了最新的NCBI refseq数据库,涵盖细菌、病毒、真菌和寄生虫,结合自研多元统计模型和机器学习算法,极大提升了物种鉴定和功能注释的准确性。
物种精准鉴定
分辨率更加精细,可达“种”和“菌株”水平,并对复杂的多菌种感染进行精细化解构。
肠道功能评估
肠道基础功能:包括蛋白质发酵能⼒、消化吸收效率、肠道产气情况、肠道屏障完整性、肠道炎症状态等,在菌群整体评估指标中也增加了包括菌群恢复力、革兰氏阴性菌、好氧菌等指标。
功能基因分析
– 全面评估菌群的基因功能潜力
如次生代谢产物合成通路、维生素合成能力、碳水化合物利用能力等,为个性化营养和精准干预提供科学证据。
耐药基因
– 分析耐药基因
不局限于已知靶点,可鉴定出各种已知和新型耐药基因,全面评估耐药基因的种类和数量,例如,在人体肠道宏基因组中发现了大量β-内酰胺酶等耐药基因。
– 追踪耐药基因的传播途径
通过比较不同环境(如土壤、水体、动物和人体)中耐药基因的分布情况,可推测耐药基因的来源和传播途径。
– 辅助指导耐药风险评估和防控策略
宏基因组学评估环境和宿主中耐药基因的分布特点,识别高风险区域和人群,为制定针对性监测和干预措施提供依据。
毒力基因
宏基因组检测技术在病原微生物毒力基因研究中具有独特优势,可在基因组水平系统分析其毒力基因组成及调控网络,加深对致病机理的理解。
免疫炎症分析
肠道菌群通过调控免疫平衡维持健康,⽽炎症标志物则作为评估机体炎症程度的关键指标。
宏基因组测序对测序深度要求较高,当数据量不足时,一些低丰度的真菌、寄生虫等病原体可能覆盖不到;然而,若要获得足够的测序深度来确保全面覆盖,则会显著增加测序成本,同时对分析能力和计算资源提出更高要求。
因此,宏基因组检测看似”简单粗暴”,只要更多数据量,实则真正的挑战在于,如何在成本与深度之间找到最优平衡。
谷禾持续迭代升级自有数据库
整合最新的NCBI refseq数据库
并结合十余年积累的临床样本数据
让相对较小的测序量
也能获得高精度的物种鉴定结果
同时,谷禾致力于深入挖掘
数据背后的生物学意义
通过专业团队的生物信息学分析
从宏基因组数据中构建
炎症状态、消化功能异常等关键健康指标
通过机器学习算法将海量基因信息
转化为实用的健康评估结果
通过这种”数据挖掘+算法迭代“
尽可能为大家控制成本的同时提供
媲美高深度测序的检测精度
极力追求技术创新与商业价值的完美结合
宏基因组报告中的解读更详细,还整理了一些评估指标,检测指标的总结等。
其他谷禾肠道菌群检测专业版的内容,宏基因组报告里面也都涵盖了,包括慢病风险、菌群代谢物及神经递质代谢、个性化营养等板块。
…
…
个性化饮食板块也在谷禾16S版本的基础上进行了迭代升级。
…
宏基因组检测并不常用于常规检测,其高昂的成本和复杂的数据分析决定了它更适用于关键时刻。
特定菌群感染的判别
对于一些复杂的多菌种感染,宏基因组能够更精细化鉴定感染菌群的构成,为临床辅助诊疗提供依据。
真菌与病毒感染的深度判别
相比传统培养,宏基因组学诊断真菌感染的敏感性和特异性更高,适用于一些真菌感染疾病。也可能鉴定出可疑的新病原体,为后续的病原学研究、药物和疫苗开发奠定基础。
一图看懂以上谷禾三大产品线
04
在肠道菌群检测系列产品成熟之后,谷禾健康将深耕多年的微生物组学技术平台,延伸至关乎女性全生命周期健康的另一核心领域——阴道微生态。
还包括子宫颈沙眼衣原体、HPV、HSV、EB病毒、巨细胞病毒等。
谷禾阴道菌群检测报告引入科学前沿的菌群状态分型(CST)概念,将复杂的菌群构成归纳为几种易于理解的健康状态类型。
例如,以卷曲乳杆菌为主的CST-I型代表健康的稳定状态,而以加德纳菌等多种厌氧菌为主的CST-IV型则与细菌性阴道病高度相关。这为临床判断和干预效果评估提供了科学支持。
谷禾阴道菌群检测报告中包括阴道菌群总体评估、CST分型、致病菌表(细菌性阴道病,需氧菌性阴道炎,外阴念珠菌病等)、列出异常菌群及相关说明,菌群详细构成等。
…
…
…
…
阴道菌群检测让我们能够更全面地了解阴道微生物组的组成及其变化,以及它是如何随着时间的推移或对各种因素(如环境、激素变化、性活动和抗生素使用等)的反应而变化的。
注:本产品可辅助评估和筛查,不用作临床诊断。
05
随着“它经济”的蓬勃发展和“科学养宠”理念的深入人心,宠物已成为家庭的重要成员。然而,面对“毛孩子”们无法言说的病痛,如反复腹泻、顽固皮肤病、食欲不振、呕吐等,传统兽医诊断往往面临挑战。
从宠物医院的实际经营来看,慢性疾病正成为他们面临的核心挑战。慢性肾病、老年痴呆、精神类疾病等病症不仅治疗费用昂贵,而且现有手段往往无法覆盖,特别是小型诊所更是心有余而力不足。
在与许多B端合作伙伴的深度交流中我们发现
宠物腹泻,肾病以及其他疾病等正在增加
后期医疗费用高昂让宠物主人无能为力
异常行为严重影响生活质量和主人养宠体验
情感难舍却不得不放弃…
因此,迫切需要一种更加
科学、经济、精准的健康管理方式
既能降低医疗成本
又能提供个性化的健康方案
还能避免过度医疗
这就需要我们从根本上
重新思考宠物健康管理的方法论
从”治疗导向“转向”预防导向“
✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲
谷禾凭借在人类健康领域积累的深厚微生物组学经验,战略性延伸至动物医学领域,推出宠物菌群精准检测服务。
我们致力于解码宠物肠道微生态的奥秘,为兽医临床、宠物营养和家庭养护提供科学依据,开启宠物健康管理的精准化新时代。
从人类微生态到宠物微生态,不是简单的复制,而是技术能力的升维应用。人类肠道微生态的复杂性研究为我们提供了强大的算法基础和数据分析能力,以及多年来在宠物菌群科研中的持续投入,这些经验在宠物领域的应用中展现出了独特的技术优势。
研究表明,宠物肠道菌群不仅影响消化吸收功能,更与免疫调节、神经系统、皮肤健康等多个生理系统密切相关,成为宠物整体健康状况的重要晴雨表。
谷禾正有序推进构建涵盖不同品种、年龄、健康状况犬猫的肠道菌群数据库,结合最新的机器学习算法,实现对宠物肠道微生态健康状况和营养进行精准评估。
宠物菌群报告展示采用更温馨活泼的配色,通过可视化图表和情感化设计,让复杂数据更直观,帮助主人轻松了解爱宠健康。
谷禾宠物菌群检测报告中包括菌群评估(整体指标)、肠道基础功能评估(屏障功能、炎症水平、代谢状态)、菌群代谢物评估(短链脂肪酸等)、炎症免疫评估(促炎、抗炎等指标)、营养饮食评估(维生素、微量元素)等。
…
…
症状相关菌群分析,包括腹泻、呕吐、过敏等。
…
菌群代谢物评估(短链脂肪酸等)。
…
…
从多维度全面评估宠物肠道健康状况,为宠物主人提供科学的健康管理依据和个性化调理建议。
自2012年成立以来,谷禾健康始终扎根于肠道微生态领域。我们不仅是国家高新技术企业和专精特新企业,更是通过中国合格评定国家认可委员会(CNAS)认可评审,成为CNAS认可的微生物检测实验室。同时,谷禾也拥有几十项国家发明专利以及在国际顶级期刊发表的研究成果,已经服务和合作150多家顶级医院与机构,积累了超过200万的样本数据库,这既是我们的底气,也是您成功的保障。
作为菌群检测行业的先行者和深耕者,谷禾始终坚守科学严谨的初心,在技术研发、质量管控、数据安全等各个环节持续深化建设。
从样本储存运输的标准化流程,
到阳性对照、阴性对照的严格设置;
从仪器校准溯源的精准把控,
到人员素质培训的持续提升,
从数据安全保障的多重防护,
到人机料法环的全方位管控,
每一处细节都是我们
以科研匠心在守护谷禾检测命脉。
正是出于对科学研究价值的深度认同,谷禾设立了『人体肠道菌群开放基金』,从心梗脑梗风险监测到儿童自闭症干预,从肿瘤免疫治疗到妊娠期健康管理,通过阶段性的递进式合作模式,我们已成功孵化近百个前沿研究项目,并在国际权威期刊
《Gut》、《Advanced Science》、《Clin Transl Oncol》等发表突破性成果。
已开展申请项目
未来,谷禾将持续投入研发力量,在青少年抑郁症、代谢综合征、心脑血管、老年阿尔茨海默病、过敏相关免疫疾病以及特定肿瘤(如胰腺癌、肝癌)等前沿领域继续深耕,进行模型的深度开发与优化。
同时,谷禾正积极推进临床营养检测评估中心建设,通过与首科等权威机构合作,深入开展儿童精神发育、老年营养监测等特定人群研究,未来加入社区筛查项目,长期追踪社区人群的菌群状况,通过对稳定人群的菌群纵向研究更加深度挖掘菌群与健康之间的关联性,为精准医疗和个体化营养干预提供更加坚实的科学依据。
从科研到应用的全链条能力建设,让我们能够更好地赋能下游产业。通过携手更多科研院所、医疗机构和行业伙伴,谷禾将持续探索菌群检测技术在医疗大健康生态系统中的创新应用,与业界伙伴携手共进,推动行业健康发展。
欢迎有相关方向的人群或者
科研、临床、干预机构咨询合作

谷禾健康
肠道菌群检测临床版(16S rRNA测序),作为谷禾健康最早推向市场、历经十余年打磨的经典产品,凭借其成本效益、高效性与成熟度,为临床端肠道微生态评估以及诸多科研项目基线建立,提供了坚实、可靠且极具价值的数据基础(注:仅用于菌群科学研究和辅助参考,不直接用于临床诊断 )。

临床版对报告版式进行简化,以更符合临床检验的形式呈现分析结果,便于临床医生快速查看和判断异常。
临床版是主要面向临床和医疗机构的版本,主要用于临床科室已有明确症状或诊断,需要对肠道菌群进行进一步分析以为临床提供辅助判断。
该版本减少了基本介绍和文字说明,并对部分指标的异常判定范围和计算方式进行调整,更加适应临床需求。
报告内容截图:


谷禾健康

随着“它经济”的蓬勃发展和“科学养宠”理念的深入人心,宠物已成为家庭的重要成员。然而,面对“毛孩子”们无法言说的病痛,如反复腹泻、顽固皮肤病、食欲不振、呕吐等,传统兽医诊断往往面临挑战。
从宠物医院的实际经营来看,慢性疾病正成为他们面临的核心挑战。慢性肾病、老年痴呆、精神类疾病等病症不仅治疗费用昂贵,而且现有手段往往无法覆盖,特别是小型诊所更是心有余而力不足。
在与许多B端合作伙伴的深度交流中我们发现
宠物腹泻,肾病以及其他疾病等正在增加
后期医疗费用高昂让宠物主人无能为力
异常行为严重影响生活质量和主人养宠体验
情感难舍却不得不放弃…
因此,迫切需要一种更加
科学、经济、精准的健康管理方式
既能降低医疗成本
又能提供个性化的健康方案
还能避免过度医疗
这就需要我们从根本上
重新思考宠物健康管理的方法论
从”治疗导向“转向”预防导向“
✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲✲
谷禾凭借在人类健康领域积累的深厚微生物组学经验,战略性延伸至动物医学领域,推出宠物菌群精准检测服务。
我们致力于解码宠物肠道微生态的奥秘,为兽医临床、宠物营养和家庭养护提供科学依据,开启宠物健康管理的精准化新时代。

从人类微生态到宠物微生态,不是简单的复制,而是技术能力的升维应用。人类肠道微生态的复杂性研究为我们提供了强大的算法基础和数据分析能力,以及多年来在宠物菌群科研中的持续投入,这些经验在宠物领域的应用中展现出了独特的技术优势。
研究表明,宠物肠道菌群不仅影响消化吸收功能,更与免疫调节、神经系统、皮肤健康等多个生理系统密切相关,成为宠物整体健康状况的重要晴雨表。
谷禾正有序推进构建涵盖不同品种、年龄、健康状况犬猫的肠道菌群数据库,结合最新的机器学习算法,实现对宠物肠道微生态健康状况和营养进行精准评估(注:仅用于菌群科学研究和辅助参考,不直接用于临床诊断 )。
宠物菌群报告展示采用更温馨活泼的配色,通过可视化图表和情感化设计,让复杂数据更直观,帮助主人轻松了解爱宠健康。
谷禾宠物菌群检测报告中包括菌群评估(整体指标)、肠道基础功能评估(屏障功能、炎症水平、代谢状态)、菌群代谢物评估(短链脂肪酸等)、炎症免疫评估(促炎、抗炎等指标)、营养饮食评估(维生素、微量元素)等。
…
…
症状相关菌群分析,包括腹泻、呕吐、过敏等。
…
菌群代谢物评估(短链脂肪酸等)。
…
…
从多维度全面评估宠物肠道健康状况,为宠物主人提供科学的健康管理依据和个性化调理建议。


谷禾健康
在肠道菌群检测系列产品成熟之后,谷禾健康将深耕多年的微生物组学技术平台,延伸至关乎女性全生命周期健康的另一核心领域——阴道微生态。
还包括子宫颈沙眼衣原体、HPV、HSV、EB病毒、巨细胞病毒等。
谷禾阴道菌群检测报告引入科学前沿的菌群状态分型(CST)概念,将复杂的菌群构成归纳为几种易于理解的健康状态类型。
例如,以卷曲乳杆菌为主的CST-I型代表健康的稳定状态,而以加德纳菌等多种厌氧菌为主的CST-IV型则与细菌性阴道病高度相关。这为临床判断和干预效果评估提供了科学支持。
谷禾阴道菌群检测报告中包括阴道菌群总体评估、CST分型、致病菌表(细菌性阴道病,需氧菌性阴道炎,外阴念珠菌病等)、列出异常菌群及相关说明,菌群详细构成等(注:仅用于菌群科学研究和辅助参考,不直接用于临床诊断)。
…
…
…
…
阴道菌群检测让我们能够更全面地了解阴道微生物组的组成及其变化,以及它是如何随着时间的推移或对各种因素(如环境、激素变化、性活动和抗生素使用等)的反应而变化的。
注:本产品可辅助评估和筛查,不用作临床诊断。
05

谷禾健康
谷禾16S+tNGS技术结合了超多重PCR和高通量测序的优势,旨在提供比传统16S rRNA测序和宏基因组测序更优、更全面的病原体及耐药基因检测方案。

传统16S + 病原体精准分型 = 全新升级
它在保留16S报告的基础上
以接近16S的成本和周期
用靶向测序技术在原16S的基础上
增加了125 种消化道病原体的检测
还包括耐药基因和毒力基因等
如幽门螺杆菌、大肠杆菌、艰难梭菌分型
弥补了传统16S无法检测
非细菌/古菌病原体的不足
这是一款突破传统16S检测瓶颈而生的产品
值得一提的是,谷禾在tNGS技术的研发道路上并非一帆风顺。这项看似成熟的技术,在不同应用场景下却面临着截然不同的挑战难度。
tNGS对血液和上呼吸道样本检测较简单,因其主要含病原体。但在消化道特别是粪便样本检测时,技术难度大幅增加。
经过无数次的实验优化和迭代
我们最终突破了这一技术瓶颈
为大家带来真正可靠的
消化道病原体检测解决方案
谷禾16S+tNGS产品特点
弥补16S检测技术边界
实现靶向病原体精准检测
谷禾16S+tNGS报告内容涵盖所有16S报告的内容,即包括健康总分评估、慢病风险预警、肠道屏障及代谢物、神经递质分析、个性化营养评估等。此外还包括常见消化道病原体(注:仅用于菌群科学研究和辅助参考,不直接用于临床诊断)。
例如:
细菌病原体
…
病毒
真菌、寄生虫、其他病原体
…
毒力基因
…
耐药基因
…

也包括相关病原微生物的解释
…
例如,通过检测幽门螺杆菌毒力基因组合,可判断是否需立即治疗,避免对弱毒株患者的不必要抗生素使用。强毒株感染会损伤胃黏膜,增加胃炎和溃疡风险,早期预警能在胃黏膜不可逆损伤前提供治疗窗口期。
注: 由于该技术是检测粪便中的幽门螺杆菌,当浓度低于检测下限(50 copies/mL)时,可能出现假阴性结果。因此,对于临床症状明显但检测结果为阴性的患者,建议结合其他检测方法。

开始写作或按/来选择区块

谷禾健康
谷禾以往在科研领域深耕十余年,积累了丰富的科研项目经验和数据分析能力,几年前,谷禾成功实现了从科研到大健康应用的技术转化,推出了谷禾宏基因组精准检测。
对于一些特定需要深度检测的应用场景,如健康管理机构的差异化服务需求,或者临床应用需求,宏基因组精准健康检测提供了另一种专业的技术选择。

宏基因组数据库的物种涵盖范围和菌株构成,直接影响着宏基因组物种鉴定分类的准确性和分类精度。
针对宏基因组数据库不完善的问题,谷禾整合了最新的NCBI refseq数据库,涵盖细菌、病毒、真菌和寄生虫,结合自研多元统计模型和机器学习算法,极大提升了物种鉴定和功能注释的准确性。
物种精准鉴定
分辨率更加精细,可达“种”和“菌株”水平,并对复杂的多菌种感染进行精细化解构。
肠道功能评估
肠道基础功能:包括蛋白质发酵能⼒、消化吸收效率、肠道产气情况、肠道屏障完整性、肠道炎症状态等,在菌群整体评估指标中也增加了包括菌群恢复力、革兰氏阴性菌、好氧菌等指标。
功能基因分析
– 全面评估菌群的基因功能潜力
如次生代谢产物合成通路、维生素合成能力、碳水化合物利用能力等,为个性化营养和精准干预提供科学证据。
耐药基因
– 分析耐药基因
不局限于已知靶点,可鉴定出各种已知和新型耐药基因,全面评估耐药基因的种类和数量,例如,在人体肠道宏基因组中发现了大量β-内酰胺酶等耐药基因。
– 追踪耐药基因的传播途径
通过比较不同环境(如土壤、水体、动物和人体)中耐药基因的分布情况,可推测耐药基因的来源和传播途径。
– 辅助指导耐药风险评估和防控策略
宏基因组学评估环境和宿主中耐药基因的分布特点,识别高风险区域和人群,为制定针对性监测和干预措施提供依据。
毒力基因
宏基因组检测技术在病原微生物毒力基因研究中具有独特优势,可在基因组水平系统分析其毒力基因组成及调控网络,加深对致病机理的理解。
免疫炎症分析
肠道菌群通过调控免疫平衡维持健康,⽽炎症标志物则作为评估机体炎症程度的关键指标。
宏基因组测序对测序深度要求较高,当数据量不足时,一些低丰度的真菌、寄生虫等病原体可能覆盖不到;然而,若要获得足够的测序深度来确保全面覆盖,则会显著增加测序成本,同时对分析能力和计算资源提出更高要求。
因此,宏基因组检测看似”简单粗暴”,只要更多数据量,实则真正的挑战在于,如何在成本与深度之间找到最优平衡。
谷禾持续迭代升级自有数据库
整合最新的NCBI refseq数据库
并结合十余年积累的临床样本数据
让相对较小的测序量
也能获得高精度的物种鉴定结果
同时,谷禾致力于深入挖掘
数据背后的生物学意义
通过专业团队的生物信息学分析
从宏基因组数据中构建
炎症状态、消化功能异常等关键健康指标
通过机器学习算法将海量基因信息
转化为实用的健康评估结果
通过这种”数据挖掘+算法迭代“
尽可能为大家控制成本的同时提供
媲美高深度测序的检测精度
极力追求技术创新与商业价值的完美结合

宏基因组报告中的解读更详细,还整理了一些评估指标,检测指标的总结等。
其他谷禾肠道菌群检测专业版的内容,宏基因组报告里面也都涵盖了,包括慢病风险、菌群代谢物及神经递质代谢、个性化营养等板块。
…
…
个性化饮食板块也在谷禾16S版本的基础上进行了迭代升级。
…
宏基因组检测并不常用于常规检测,其高昂的成本和复杂的数据分析决定了它更适用于关键时刻。
特定菌群感染的判别
对于一些复杂的多菌种感染,宏基因组能够更精细化鉴定感染菌群的构成,为临床辅助诊疗提供依据。
真菌与病毒感染的深度判别
相比传统培养,宏基因组学诊断真菌感染的敏感性和特异性更高,适用于一些真菌感染疾病。也可能鉴定出可疑的新病原体,为后续的病原学研究、药物和疫苗开发奠定基础。

<来源:谷禾宏基因组精准检测报告>
缺点:
总的来说,宏基因组测序仍存在技术瓶颈和生物学解释的局限性。然而,针对某些特殊情况研究需要,宏基因组测序也是一种有用的微生物组学研究工具。
特殊应用场景
对于一些复杂的多菌种感染,宏基因组能够更精细化鉴定感染菌群的构成,为临床辅助诊疗提供依据。
与传统方法相比,宏基因组学诊断真菌感染的敏感性和特异性更高,适用于一些真菌感染疾病。也可能鉴定出可疑的新病原体,为后续的病原学研究、药物和疫苗开发奠定基础。
以上是谷禾宏基因组精准检测报告的一些节选,其全面、精准、个性化分析肠道菌群的组成和功能,可帮助评估菌群失衡的风险和预后,为个性化诊疗和健康管理提供科学依据。
注:报告仅用于菌群科学研究和辅助参考,不直接用于临床诊断 。

谷禾健康

16S科研项目是一个完整的闭环,前期的课题项目设计方案、取样和重复实验设置决定了后期分析报告的数据完整性和项目类型。
想要拿到一手有利用价值的科研报告和项目数据,前期的实验方案设计和后续的分析都起着关键性的作用。
然而有时候拿到报告不知道如何去解读,这里为大家梳理一下16s科研项目的全过程,帮助大家更好的了解报告内容,快速获取关键信息。


实验方案设计就像一个总工程的设计图纸,决定了未来科研分析报告的类型走向,并且前期的分组设计的越详细,各种理化指标、生化指标、代谢物等信息准备越充分,后续报告的完整度越高。


明确项目课题类型
第一步要做的就是明确项目课题类型:
最常见的就是多分组之间差异分析比较:例如,要比较对照组、模型组、实验组,之间的差异结果。
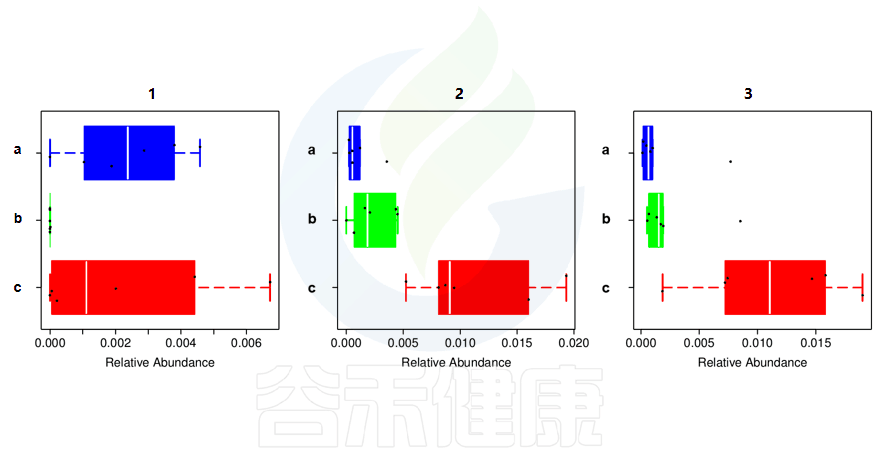

还有多分组中,任意两组之间比较:例如某实验设计了正常组、疾病组、用药组服用奥氮平、阿立哌唑、氨磺必利、利培酮,像比较不同的用药组和疾病组之间的菌群的差异结果,就用到了分组之间两两差异比较。
✦举个例子
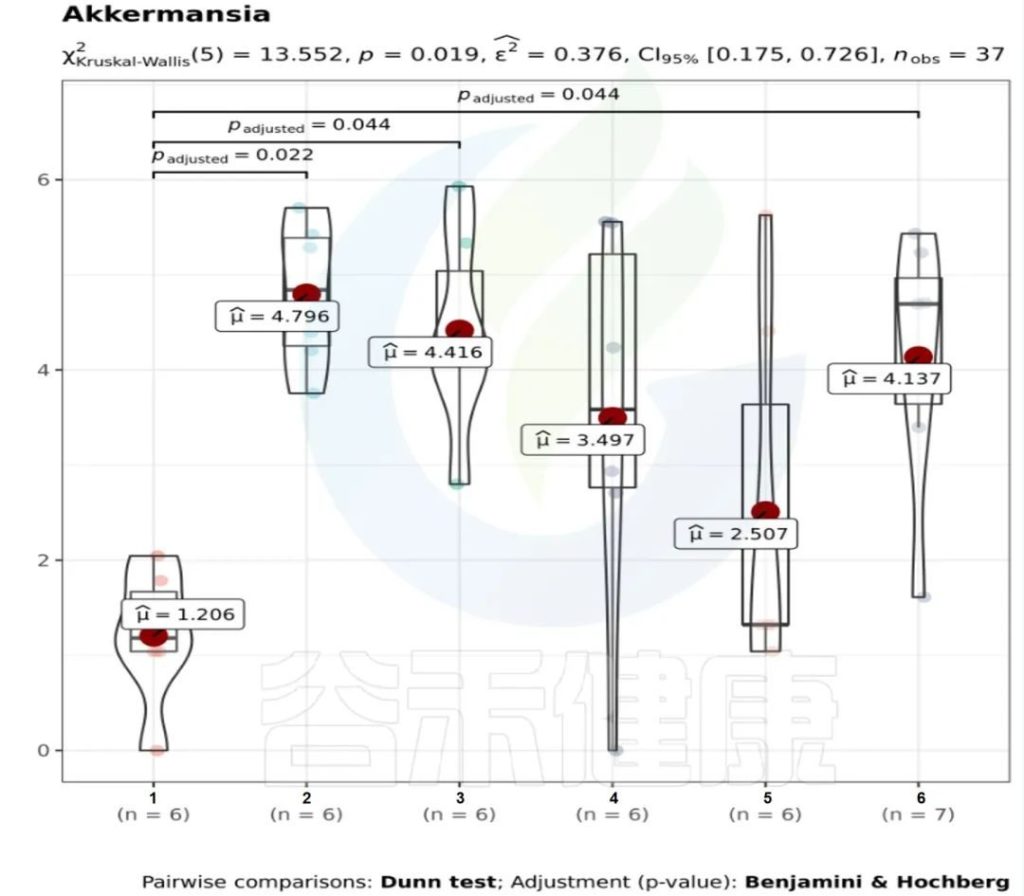
图中1组与3组、4组、6组 组间差异显著
还有随时间的变化比较菌群之间的变化规律:例如在用药不同时间段包括3天,5天,2周,1个月,2个月,观察菌群的变化情况。如下图所示:
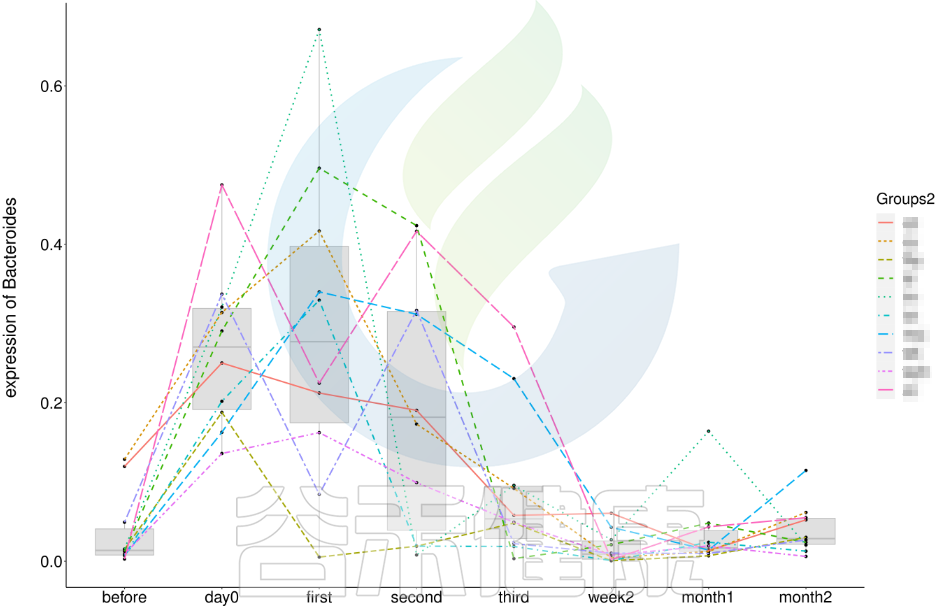
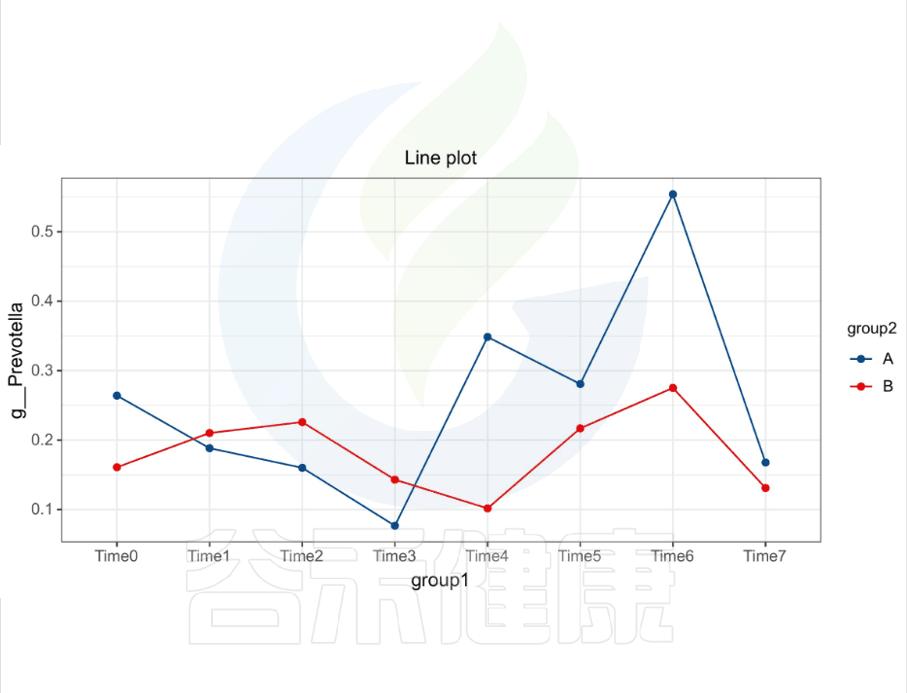
收集理化指标非常重要
如果前期搜集好每个样本的相关理化指标,还可以计算这些指标与菌群之间是否具有相关性。
✦举个例子
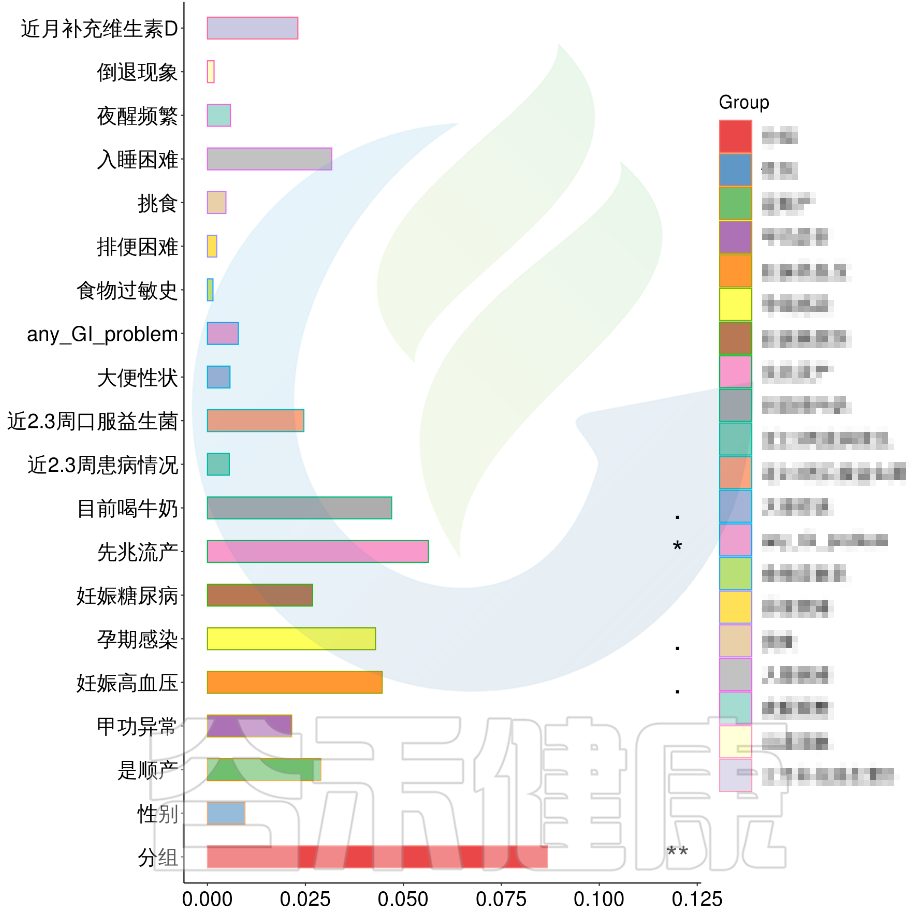
例如该项目比较自闭症儿童与正常儿童的菌群差异。客户在样本信息单里还详细搜集了母孕期的各种详细指标,例如孕期天数、出生体重、白细胞介素6、肿瘤坏死因子a、五羟色氨等数值型理化指标。
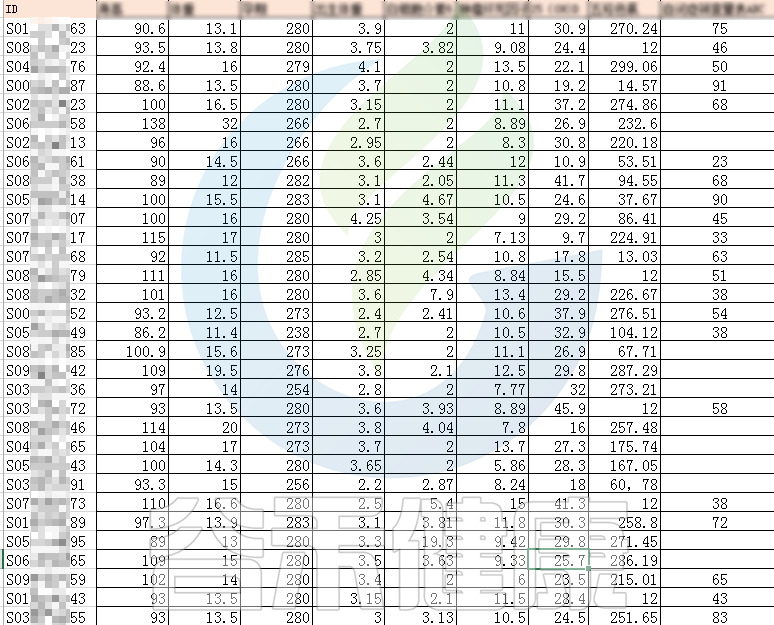
还搜集了是否顺产、是否妊娠高血压、是否孕期感染、是否妊娠糖尿病、是否先兆流产等因子型理化指标。其中0代表否,1代表是:
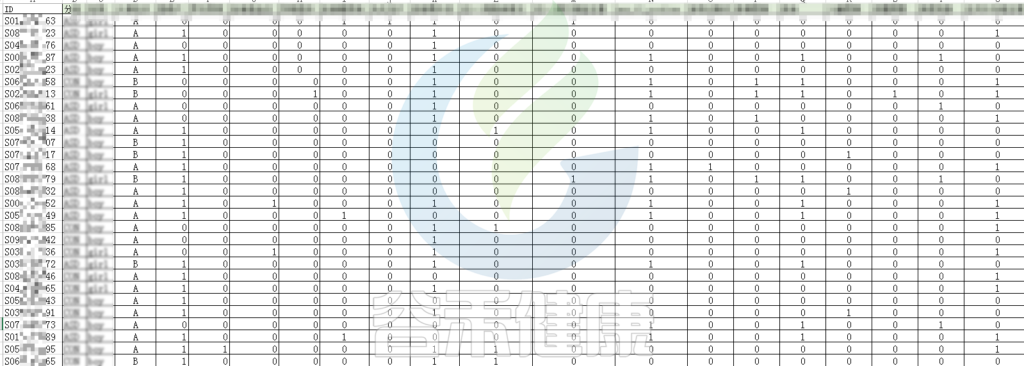
根据这些理化指标与菌群数据做相关性分析,从因子型的结果可以看出,自闭症(ASD)与正常儿童之间的分组与菌群之间相关性极显著**,其次是否有先兆流产的分组与菌群之间有显著相关性*,其他的包括是否喝牛奶、孕期是否感染、妊娠高血压都与菌群有相关性。
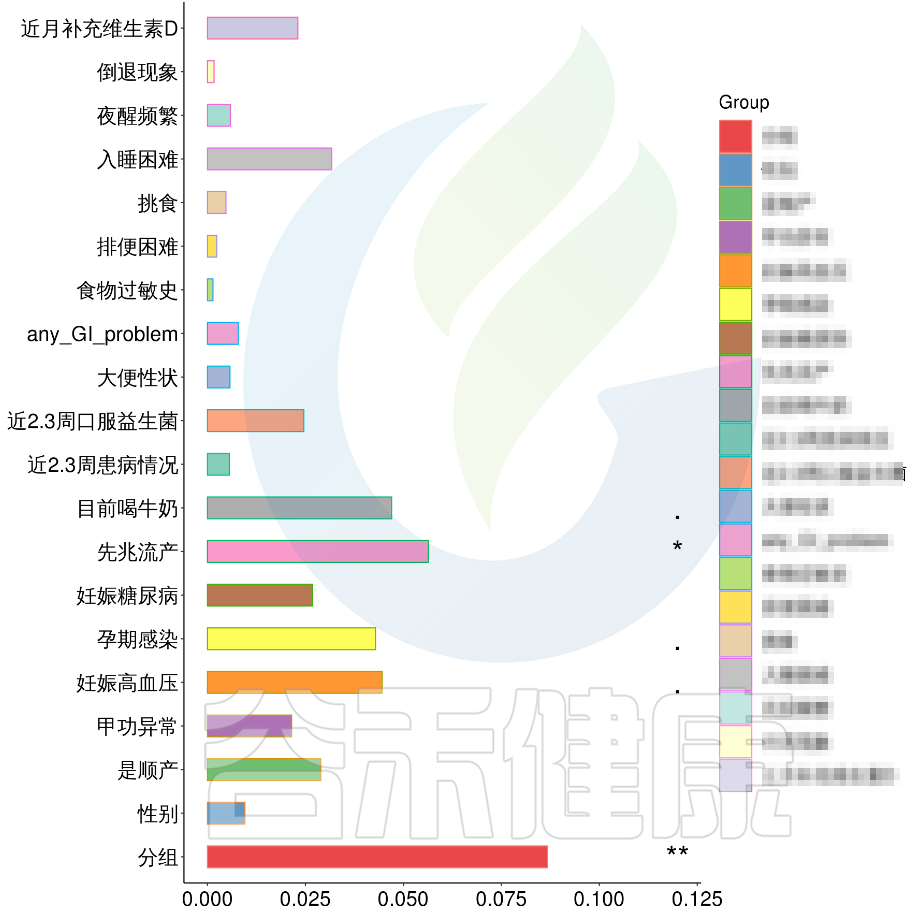
在数值型理化指标中,孕期的天数与菌群之间相关性显著*,其次是白细胞介素6与菌群之间有相关性。
小结
因此,前期搜集相关资料越详细充分,对分析报告的完整性也会有帮助,分析人员也会根据您的样本信息单提供的相关内容,做出个性化的分析和售后指导建议。
首先基于样本类型,最常见的环境样本来源是人体、动物、土壤、水体等。而人体中的肠道菌群样本是目前研究最广泛,可鉴定的物种也最为丰富,谷禾在肠道菌群与人体健康方面有深入研究,目前已完成超20万例临床肠道菌群样本检测,并构建了超过60万各类人群粪便样本数据库。
其他样本类型还包括人体/动物唾液样本、组织样本、尿液样本等。
▸ 粪便样本
目前粪便样本从采样到提取数据分析技术较为成熟、应用较为广泛,谷禾最早在15年就开发了针对粪便样本的取样管,也是最早致力于研发粪菌取样盒的公司,方便实验室、个人日常取样需求,实现了粪菌样本的常温运输。
谷禾取样管常温保存,取样也较为方便卫生,在家就可以轻松完成,相较于传统取样方法都有所升级。并且该取样管也有专利证书。该取样方法被大量客户采用并接纳,大大降低了采集粪便样本的难度,缩短了搜集样本的时间周期。
取样示意图

▸ 其他样本
土壤样本也相对较为容易提取出DNA,但需要注意的是土壤样本的菌群特征容易受植物腐殖质基因的影响和干扰,所以提取时要进行纯化。
而口腔、组织、尿液等样本,由于DNA含量较少,在实验阶段提取相对较为困难,所以提前准备样本时,尽量多取一些,并且可以多取几个重复,尽量避免扩增不出来的情况。
并且这些样还很容易受到环境样的污染,所以在实验阶段,可以取空白样本,和阳性样本ST做对照,数据分析时可以用来纯化样本,排除来自环境的干扰序列。
✦组间差异分析需重复取样
要做组间差异分析时,每组要重复取样,才能做组与组之间的统计检验。理论上,每个组至少3个样就满足基本的统计差异分析需求。所以在重复取样时,每个分组至少取3个样。取样时要保证每个分组内部的样本一致性,如果组内样本之间的个体差异性较大,则会影响后期组间差异结果分析。
✦举个例子
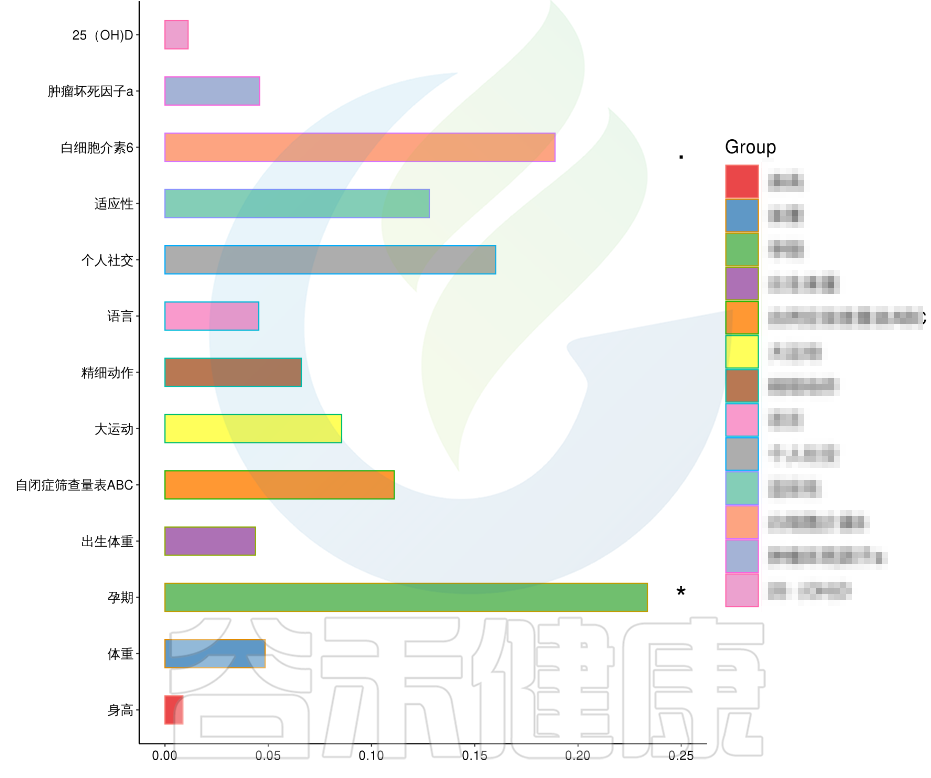
例如从该图可以看出,分组之间组间差异较大,并且组内的样本之间较为接近和相似。
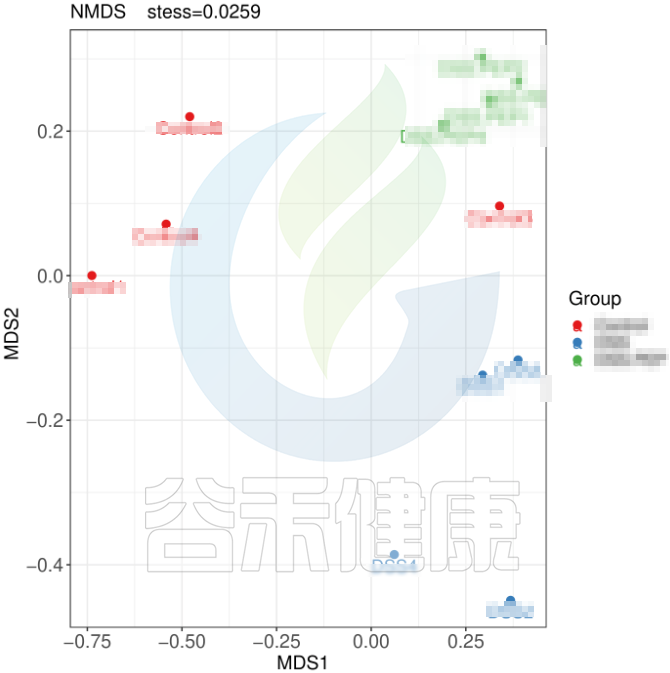
但从该图可以看出,Control组中Control3样本明显与组内的其他样本差异较大,与DSS组内的样本较为相近,这样就对后期组间差异分析的时候会产生影响,需要将该样本去除。
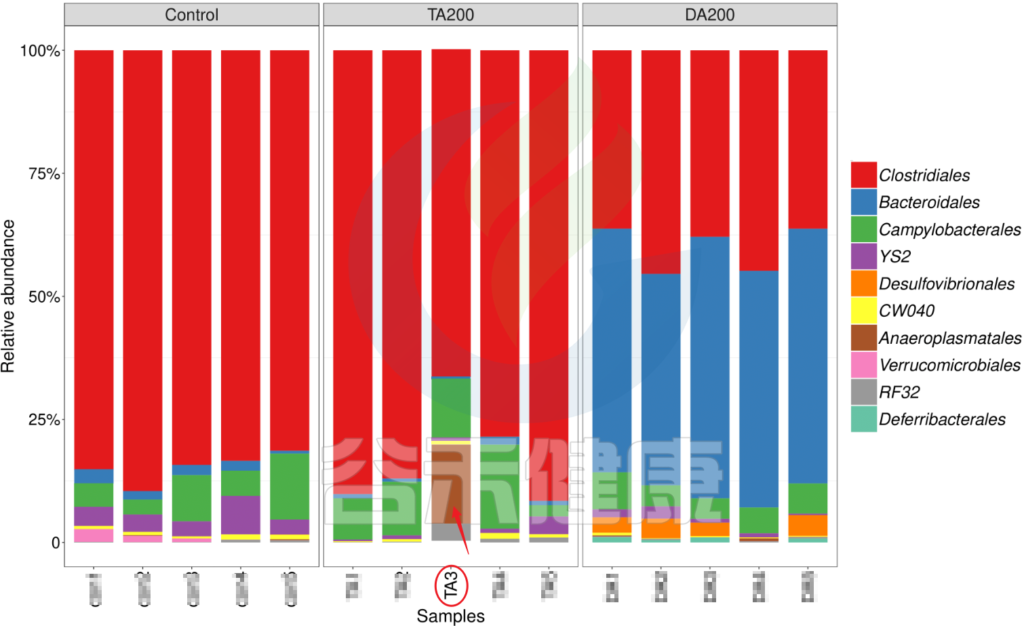
又例如在该图中,TA200组中的TA3样本的Anaeroplasmatales物种丰度含量非常高,该样本与组内的其他样本明显差异较大,该样本可能受到环境污染等其他因素干扰,这样就没有办法保证组内样本的均一性,也会影响分组之间的差异分析统计结果,再后期分析的时候建议把该样本去掉重新分析。
建议
为了便于后期数据整理修改,每个分组需要保留一定量的重复样本,假如每个分组只取了3次重复,假如其中有一个样本质量不好需要去除,该分组只剩2个样本,则不满足每组至少3个样的分组条件,整体就没有办法做组间差异分析统计。
所以这里建议每个分组至少取5个样做重复,一般6到10个样就能分析出比较完善的结果。具体分组和组内的重复取样数量视具体的实验设计方案而定。
在经费允许的情况下,建议多取一些重复。假设每组取50到100个重复或者以上,得到的分析结果就基本可以涵盖该分组情况所有的菌群构成情况,可以较为全面的研究分组之间的菌群构成差异情况。
当拿到16S科研分析报告以后,面对纷繁复杂,各式各样的图表分析结果犯了难,不知道如何从这么多的图表中入手,快速找到报告中需要的图表结果。
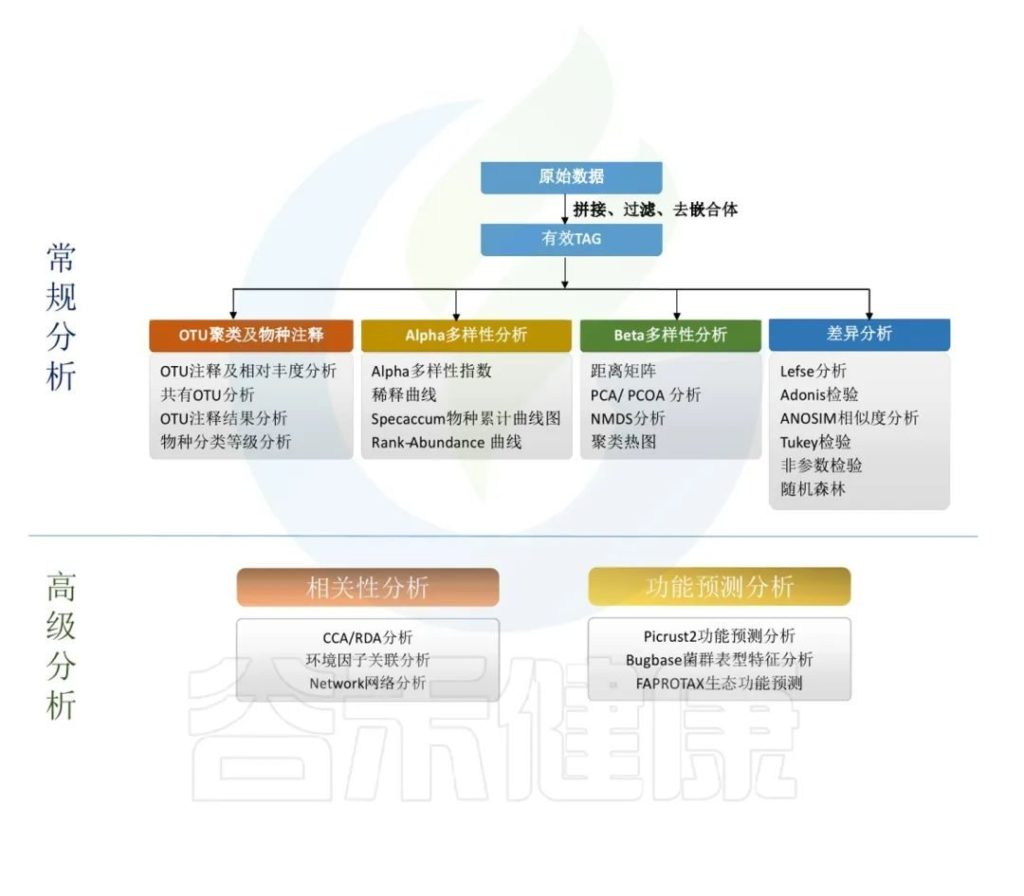
这里对16S科研分析结果抽丝剥茧,概括出报告中的主要几大内容板块。
•16S科研分析究竟是在做什么?
16S rDNA 是一种对特定环境样品中所有的细菌进行高通量测序,以研究环境样品中微生物群体的组成,解读微生物群体的多样性、丰富度及群体结构,探究微生物与环境或宿主之间的关系的技术。
主要是对原始数据进行拼接过滤得到的优化序列,降噪方法得到ASV,再对ASV进行物种注释,注释到门、纲、目、科、属、种各层次上的分类结果。
通过ASV表计算Alpha多样性,样本内的多样性指数,Beta多样性,样本间相似性的指标。
对ASV表进行功能预测,例如Picrust2功能预测分析、Bugbase菌群表型特征分析,FAPROTAX生态功能预测等。
得到的每个样的数据结果,根据客户提供的分组情况和理化指标,进一步做组间差异分析,以及和环境理化指标之间做关联分析,相关性分析,比较分组之间是否有差异,差异是否显著,来验证分组是否合理,和环境宿主之间是否有关联性。
原始数据处理
Illumina NovaSeq测序平台测序得到的双端数据Raw PE,经过拼接和质控,根据一定的标准过滤掉低质量数据、接头或PCR错误,得到Raw Tags。再经过去重复序列,去singleton序列,过滤嵌合体,得到可用于后续分析的有效数据 Effective Tags。
OTU(ASV) 表生成
微生物多样性分析中最重要的就是OTU特征表,一切后续分析都围绕OTU表来进行。生成OTU除了传统的聚类的方法(一般按照97%的相似度进行聚类),现在最新用到的技术的是降噪的方法得到ASV。
简单来讲ASV就是在去除了错误序列之后,将Identity的标准设为100%进行聚类,常见的有DADA2、Deblur、Unoise三种降噪方法。项目里用到的是UNOISE2降噪方法获得ASV数据。
物种的分类与注释
采用QIIME2训练分类器方法对ASVs代表序列进行分类学注释,默认选用SILVA138数据库进行物种注释。并在各个分类水平上:domain(域),phylum(门),class(纲),order (目),family(科),genus(属),species(种)对每个样本的群落组成统计。
alpha多样性
Alpha多样性主要反映样本内多样性。对ASV表进行计算可以获得每个样本的simpson,ace,shannon,chao1以及goods_coverage等指数,alpha多样性指数用来来评估样本菌群物种的丰富度(richness)和多样性(diversity)
beta多样性
Beta多样性反映的是样本间多样性,Beta多样性是衡量个体间微生物组成相似性的一个指标。通过计算样本间距离可以获得β多样性矩阵,基于OTU的群落比较方法报告中给出了,欧式距离、bray curtis距离、Unweighted UniFrac距离和Weighted UniFrac距离等。
功能预测
得到群落的微生物组成之后,也可以对群落功能组成进行预测,常用的16S功能预测的相关软件有PICRUSt2、FAPROTAX、BugBase。
PICRUSt2用来预测功能,通常指的是基因家族,PICRUSt2支持基于多个基因家族数据库的预测,报告中包括了KEGG同源基因,KO直系同源物,EC酶分类编号,MetaCyc途径的丰度,CAZy碳水化合物活性酶数据库,GMM是肠道代谢模块和GBM是肠脑模块。
FAPROTAX是原核的微生物注释代谢或其他生态相关的功能(例如硝化,反硝化,发酵)的一个数据库和软件。FAPROTAX预测的功能主要集中在海洋、湖泊等环境样本微生物的功能,特别是硫、碳、氢、氮的循环功能。
BugBase能进行表型预测,其中表型类型包括革兰氏阳性(Gram Positive)、革兰氏阴性(Gram Negative)、生物膜形成(Biofilm Forming)、致病性(Pathogenic)、移动元件(Mobile Element Containing)、氧需求(Oxygen Utilizing,包括Aerobic、Anaerobic、facultatively anaerobic)及氧化胁迫耐受(Oxidative Stress Tolerant)等7类。
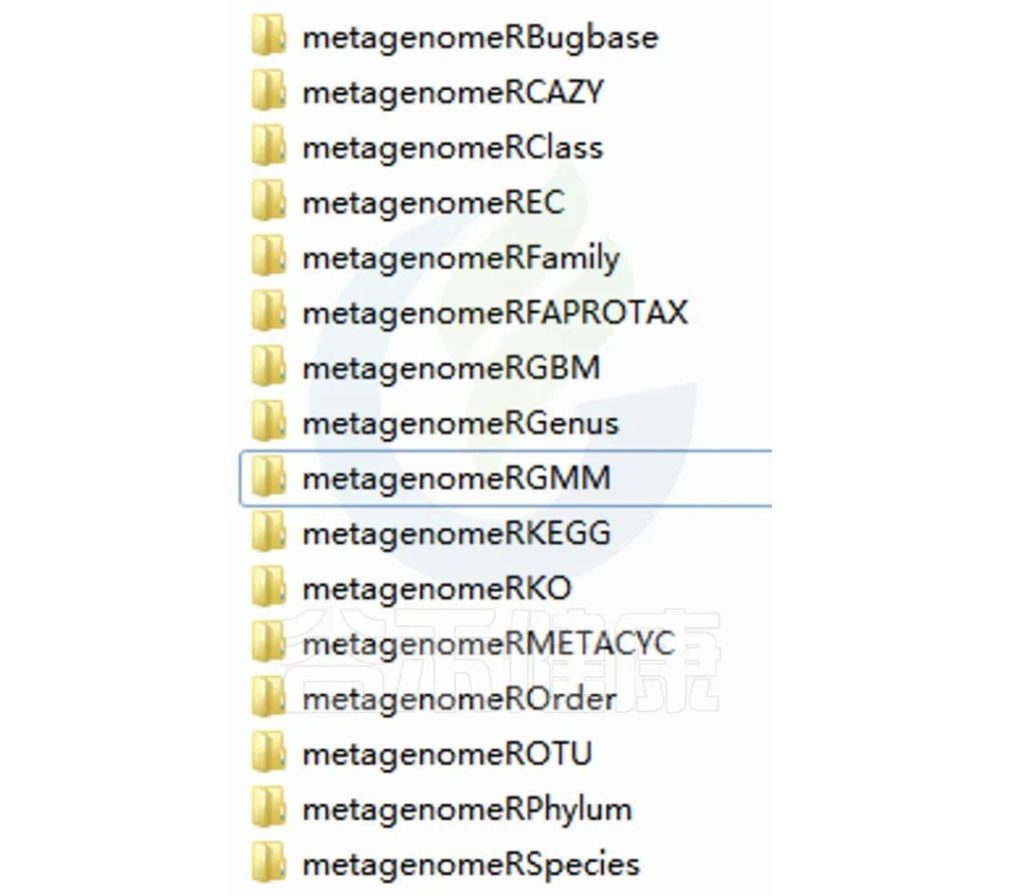
以上这些部分,我们通过数据处理分析,得到了每个样本相关的大量数据结果,包括每个样本的序列统计、ASVs表格、物种分类注释统计、alpha多样性指数、beta多样性指数、功能预测等。这些数据主要集中在报告里的这些内容:
▸ 科研分析报告结果文件夹
01_pick_otu/ 文件夹主要是对样本ASV表格统计
02_sequence_statistic/ 文件夹是对样本序列数据的统计
03_diversity-metrics / 文件夹是对样本的alpha多样性指数、beta多样性指数的统计
04_Taxonomic/ 文件夹是对物种分类注释的统计(门到种水平)
Picurst2/ 文件夹是Picrust2功能预测得到的每个样本的相关功能预测数据
Groups/ 文件夹下是对组间差异分析结果

红框是样本个体的相关数据统计,Group是分组比较
根据以上常规分析得到的相关数据进行作图,其路径也在对应文件夹下,可以打开 分析报告.html 有相关分析的图表和对应文件的详细介绍和路径说明。
★拿到样本后需要进行统计分析
当我们拿到这些样本大量的数据结果,之后关键的一步就是做对这些数据进行处理,做统计分析,比较分组之间的差异结果,找出菌群和环境之间的关联性等,对数据进一步做研究,找出课题方案对应的结果。
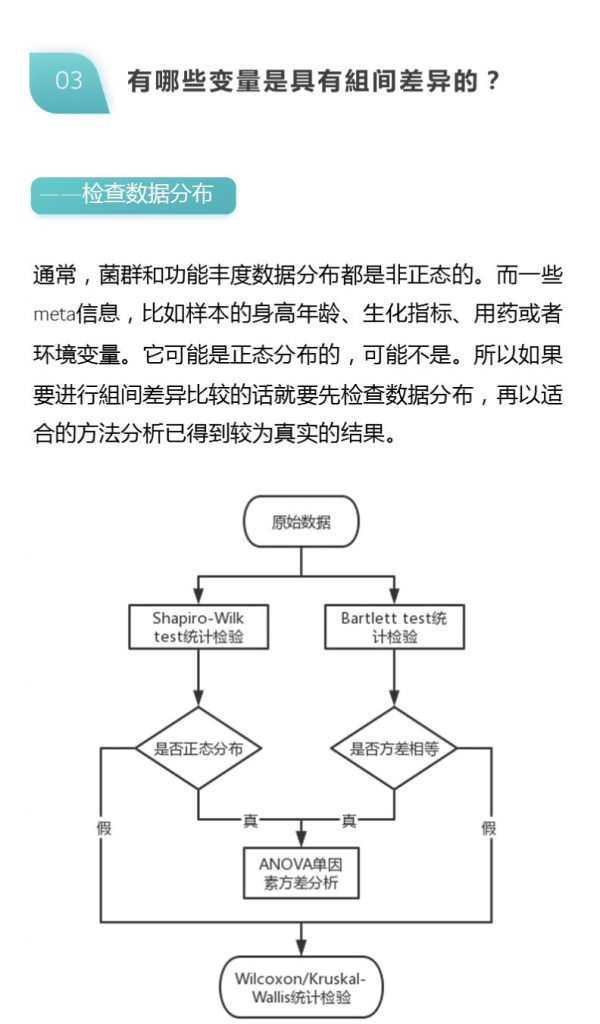
不同的数据用到的统计检验方法也不太一样,接下来我们对报告中的不同的分析结果对应的统计差异分析方法进行介绍说明。
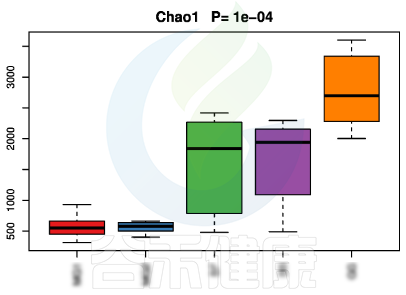
▸ alpha多样性
alpha多样性指数组间差异统计分析用到的检验方法是:单因素方差分析(如果只有两个分组,用Wilcoxon秩和检验,3个及以上的分组用Kruskal-Wallis 检验),图上方显示P值
▸ beta多样性
beta多样性指数的统计检验方法有ANOSIM相似性分析和Adonis多元方差分析,这两种都是基于距离矩阵的检验方法。
✦Anosim相似性分析
Anosim分析是一种非参数检验,用来检验组间的差异是否显著大于组内差异,从而判断分组是否有意义。
报告中给出了加权距离和非加权距离的Anosim结果图,图中给出了R值和P值。
R值用于比较不同组间是否存在差异,R-value 介于(-1,1)之间,R-value > 0,说明组间差异大于组内差异。R-value < 0,说明组间差异小于组内差异。R只是组间是否有差异的数值表示,并不提供显著性说明。
统计分析的可信度用 P-value 表示,P< 0.05 表示统计具有显著性。
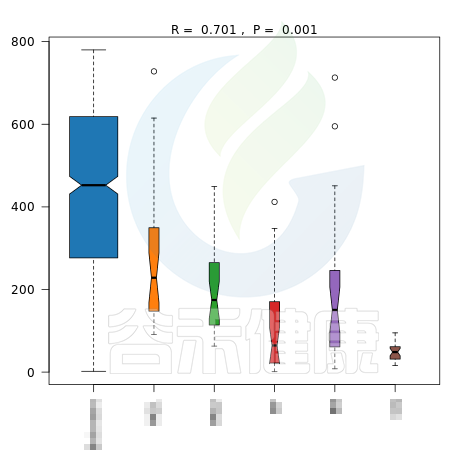
图中能看出R>0,说明组间差异大于组内差异,P<0.05 ,说明差异显著,证明该分组情况效果较好。
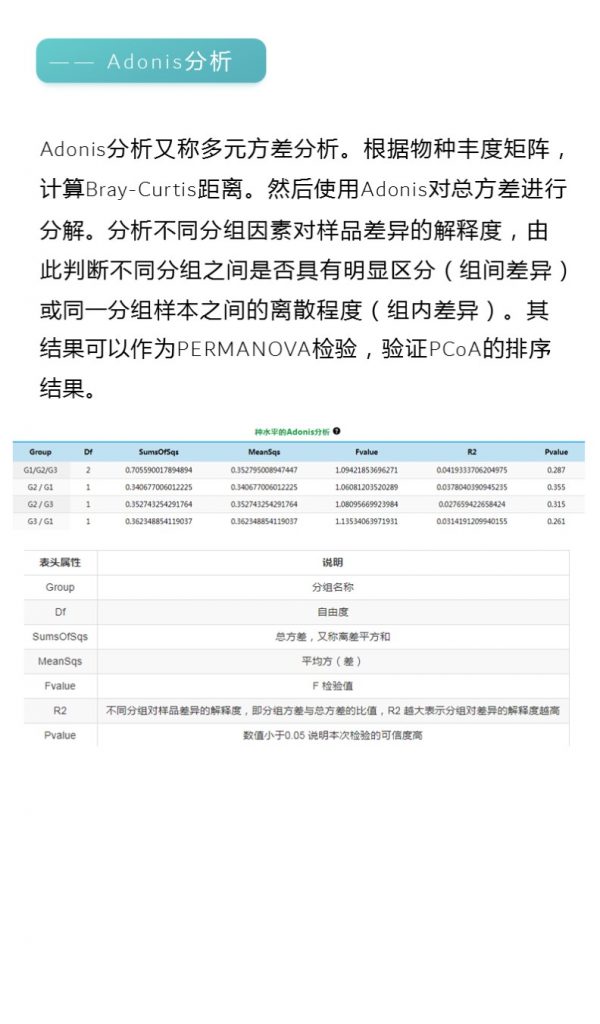
✦Adonis多元方差分析
Adonis多元方差分析,其实就是PERMANOVA,亦可称为非参数多元方差分析。
其原理是利用距离矩阵(比如基于Bray-Curtis距离、Euclidean距离)对总方差进行分解,分析不同分组因素对样品差异的解释度,并使用置换检验对其统计学意义进行显著性分析。
它与Anosim的用途相似,也能够给出不同分组因素对样品差异的解释度(R值)与分组显著性(P值)。
报告中PCoA bray距离、PCoA weighted_unifrac距离、PCoA unweighted_unifrac距离的图片右下角有给出PERMANOVA检验的P值和R值。
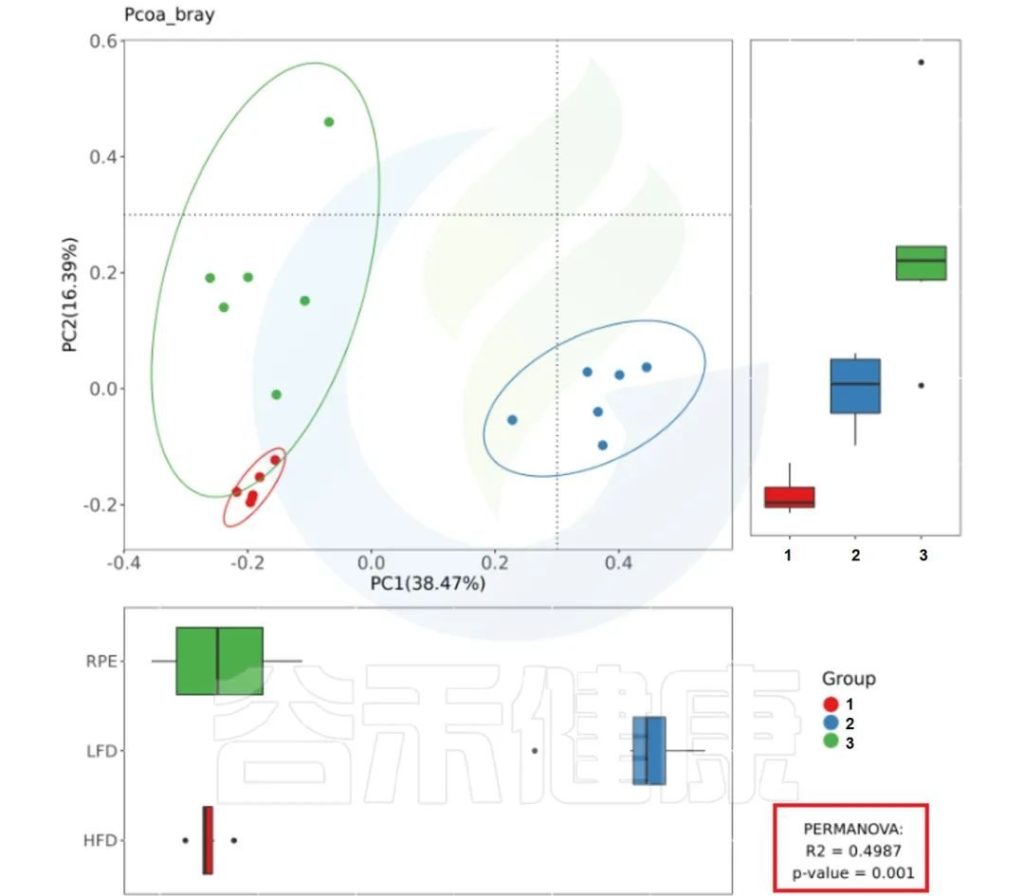
图中看出PCoa bray距离得到的检验P<0.05 组间差异显著,并且分组之间区分较为明显。
PCoa bray距离的PERMANOVA检验结果路径:
多组间检验结果:
Groups/betadiv/pcoa_bray_analysis/PERMANOVA.result_all.csv
两组间检验结果:
Groups/betadiv/pcoa_bray_analysis/ PERMANOVA_paired_result.csv

不同分类水平下的检验方法
在很多分析报告当中,例如在不同疾病的肠道菌群分组中,本身样本个体之间肠道菌群的物种多样性,丰富度差异并不大,alpha多样性组间差异并不显著,beta多样性分组间区分不是很明显,这样就需要进一步找出分组之间的差异物种或者差异功能来进行分析。
对于不同分类水平的物种和功能预测结果用到以下几种检验方法:
Tukey检验
Tukey主要应用于3组或以上的多重比较,适合于各组例数相等的每两两分组之间比较。
Tukey检验的一个重要的优点是非常简单,而且所需实验样本相对较少。
其检验结果的可信度达到95%的置信水平时,最少的情况下只需6个样本进行验证(改善前3个样本、改善后3个样本)。
•举个例子
图中的字母代表显著性差异的字母表示法,只要含有相同的字母,就表明两组之间没有显著性差异。
例如a和ab含有相同字母“a”,表示两组之间没有显著性差异。ab中的“b”表示这一组和其他含有字母b的组(比如bc)没有显著性差异,但是a和bc就有显著性差异了。
图中只展示Tukey检验差异显著的物种或功能,如果数量较多,则只展示前10个。
路径:Groups/diff_analysis/TukeyHSD/
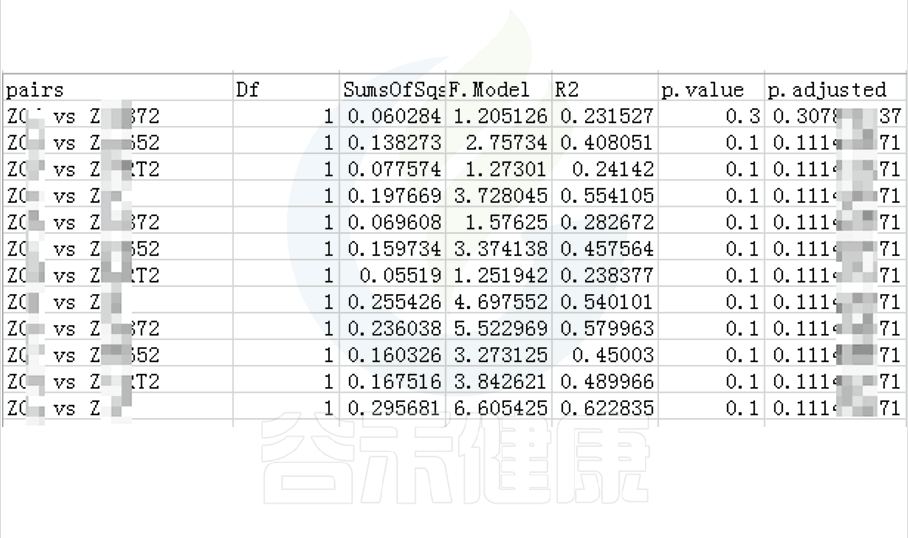
图中显示的都是Tukey检验组间差异显著的物种,依次按照丰度从高到底排列,如果差异结果较大,则显示前10个物种。例如在该图中,Tukey检验结果,门水平物种Actinobacteriota在BB与MG1组、BB与MG2、BF与GG组、BF与MG1组、BF与MG2组,这些分组之间组间差异显著。
组间差异箱型图
组间差异箱型图用到的检验方法是通过单因素方差检验(只有两个分组,用的是Wilcoxon秩和检验,3个及以上的分组用的是Kruskal-Wallis 检验),Var检验和one-way相结合,筛选出组间差异性物种。
路径:Groups/diff_analysis/TaxaMarkers
图中每一个箱型图代表一个组间差异显著的物种
图中显示的都是统计方法得到的差异显著的物种,图中能看出这3个物种分组之间差异显著。
命名格式是,例如:Cen_Nitrosopumilus 指的是,当前分类水平(属水平)的名字 g__Nitrosopu 加上一级分类水平(科水平)的名字 f__Cenarchaeaceae 的前 3 个字母简写Cen,如果当前水平没有注释到名字则以全称的名字表示。
统计结果表:Groups/diff_analysis/TaxaMarkers/ xxx.Groups.sig.meanTests.csv
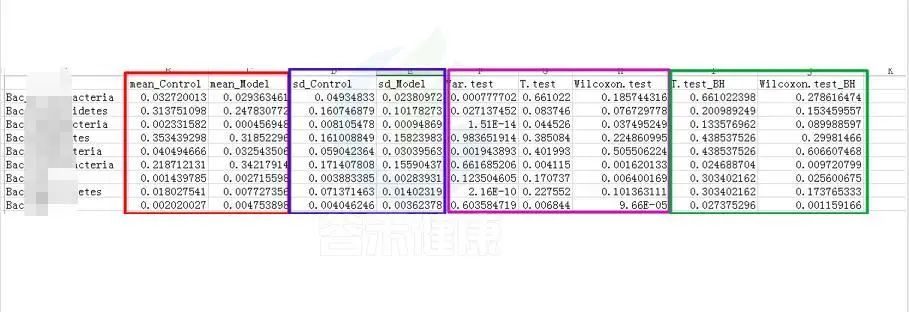
例如这是一个表格的截图
红框 mean_ 是分组组间的平均值
蓝框 sd_ 代表组间的标准差
粉色 .test 代表不同统计检验结果的P-value P值,这里有var检验 T 检验 Wilcoxon检验(或Kruskal-Wallis 检验)
绿色 _BH 例如Wilcoxon.test_BH代表Wilcoxon.test检验BH矫正的Q-value,Q值
UnivarTest检验(单因素方差分析)
单因素方差分析是指如果只有两个分组,用Wilcoxon秩和检验,3个及以上的分组用Kruskal-Wallis 检验。
路径:Groups/diff_analysis/UnivarTestXXX
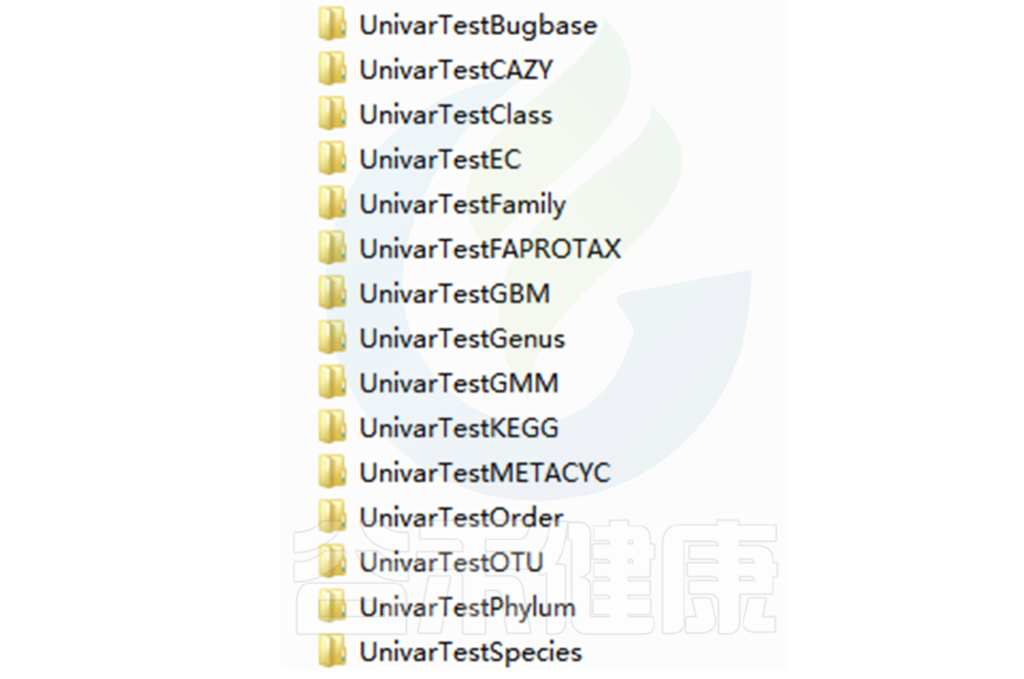
Groups\diff_analysis\UnivarTestKEGG\figure 文件夹下有做成柱状图、箱型图和单个物种之间的图,其中有横着排列和竖着排列的,有用原始值计算的,还有对原始值取log后进行统计的。图中只展示Univar 检验组间差异显著的物种/功能。
统计结果表:Groups/diff_analysis/UnivarTestXXX/ UnivarTest_sign.txt
•举个例子
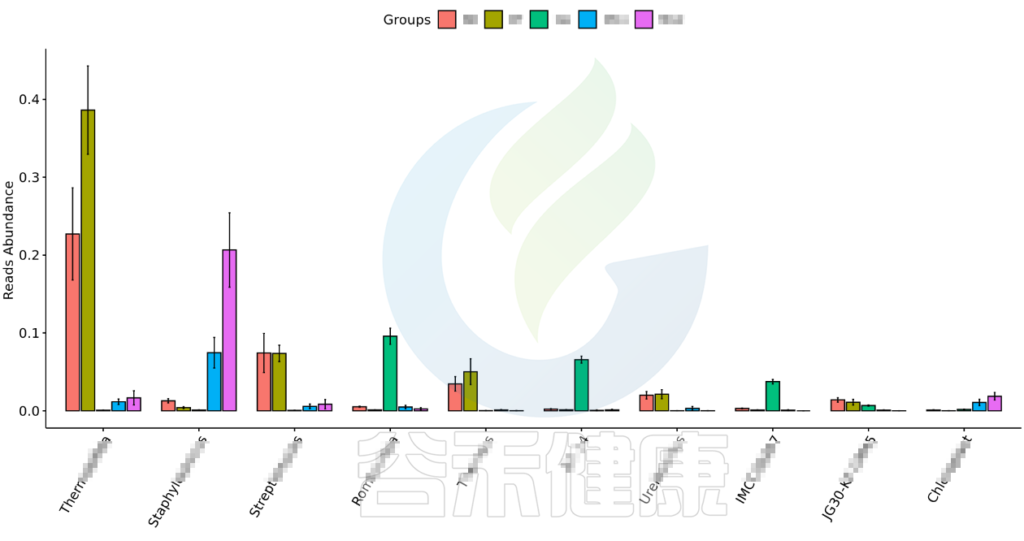
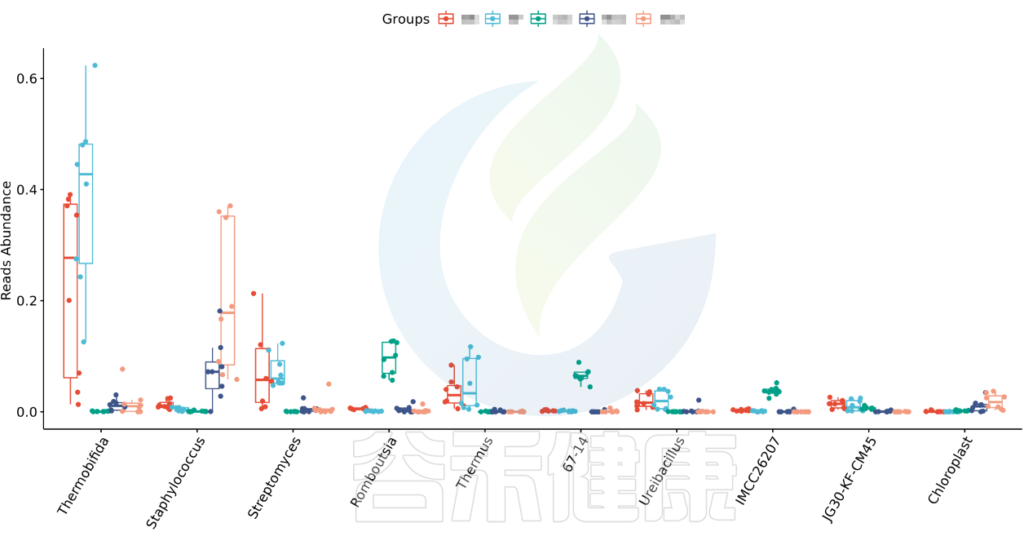
图中显示的是该统计检验差异显著的物种的柱状图或箱型图,按照丰度从高到低排列,如果差异物种/功能较大,则只显示前10个。例如该图中Therobifida、Staphylococcus、Streptomyces等物种用Kruskal-Wallis 检验得到的组间显著差异物种。
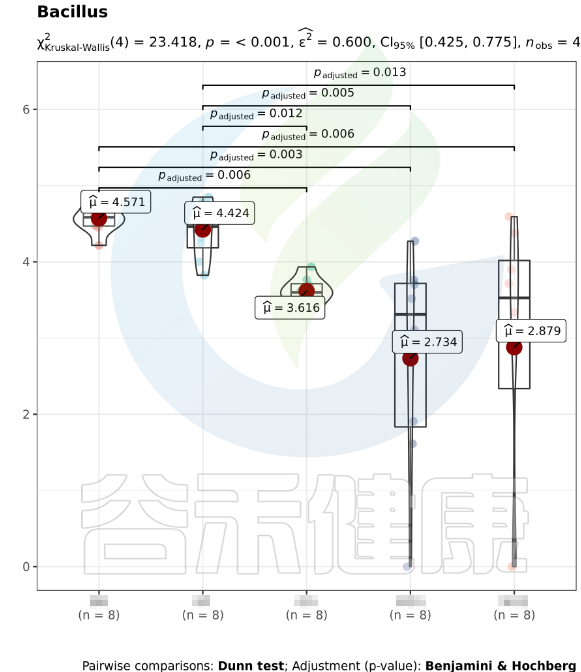
该图展示了Bacillus物种Kruskal-Wallis 检验差异结果,所有分组中P<0,001 多组间差异显著,两组间BB与GG、BB与MG1、BB与MG2、BF与GG、BF与MG1、BF与MG2,组间差异显著。
LEfse分析
LEfse分析即LDA Effect Size分析,是一种用于发现和解释高维度数据生物标识(基因、通路和分类单元等)的分析工具,可以进行两个或多个分组的比较,它强调统计意义和生物相关性,能够在组与组之间寻找具有统计学差异的生物标识(Biomarker)。
LEfSe用到的统计分析方法是将线性判别分析与非参数的Kruskal-Wallis以及Wilcoxon秩和检验相结合。
LEfse分析结果中一般会出现两个图,一张表( LDA值分布柱状图、进化分支图以及特征表)。
LDA值分布柱状图
这个条形图主要为我们展示了LDA score大于预设值的显著差异物种,即具有统计学差异的Biomaker,默认值为2.0(看横坐标,只有LDA值的绝对值大于2才会显示在图中);柱状图的颜色代表各自的分组,长短代表的是LDA score,即不同组间显著差异物种的影响程度。
路径:
Group/Lefse_Analysis/out_formant.cladogram.png
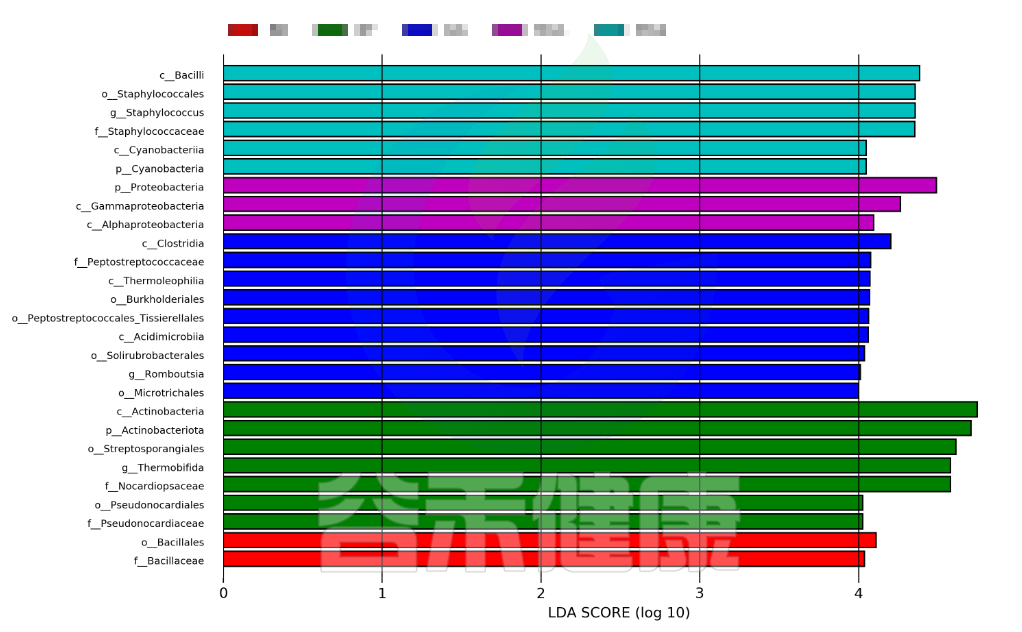
图中展示了不同分组特有的Lefse组间差异标记物,例如BB组的标记物是目水平的Bacillales和科水平的Bacillaceae,不同的分组标记物也不同,图中如果只展示了部分分组,则代表只有部分分组通过Lefse分析筛选出组间差异标记物。
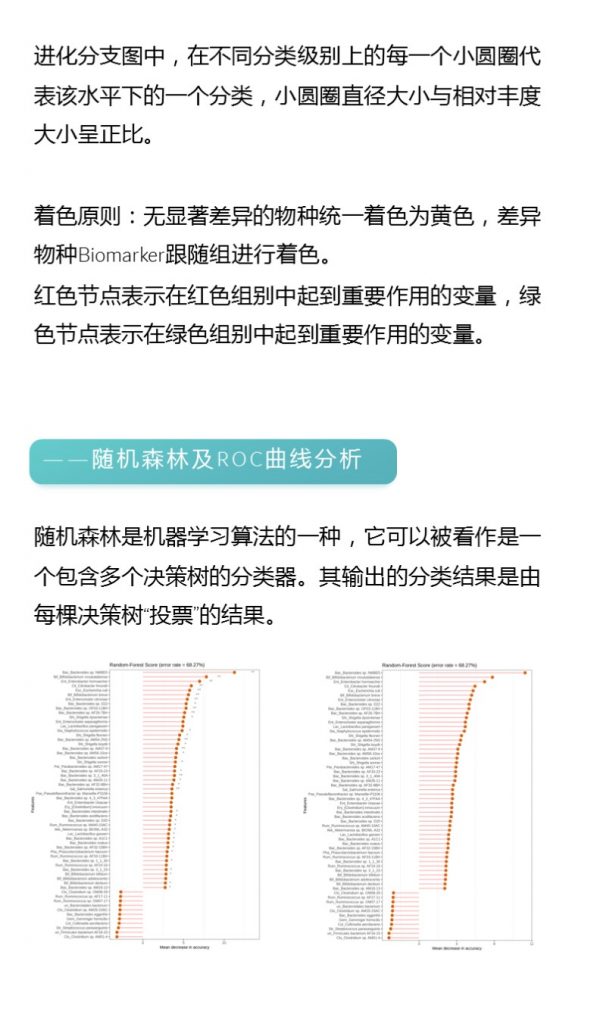
进化分支图
小圆圈: 图中由内至外辐射的圆圈代表了由门至属的分类级别(最里面的那个黄圈圈是界)。不同分类级别上的每一个小圆圈代表该水平下的一个分类,小圆圈的直径大小代表了相对丰度的大小。
颜色: 无显著差异的物种统一着色为黄色,差异显著的物种Biomarker跟随组别进行着色,红色节点表示在红色组别中起到重要作用的微生物类群,蓝色节点表示在蓝色组别中起到重要作用的微生物类群。
未能在图中显示的Biomarker对应的物种名会展示在右侧,字母编号与图中对应(为了美观,右侧默认只显示门到科的差异物种)。
路径:Group/Lefse_Analysis/out_formant.png
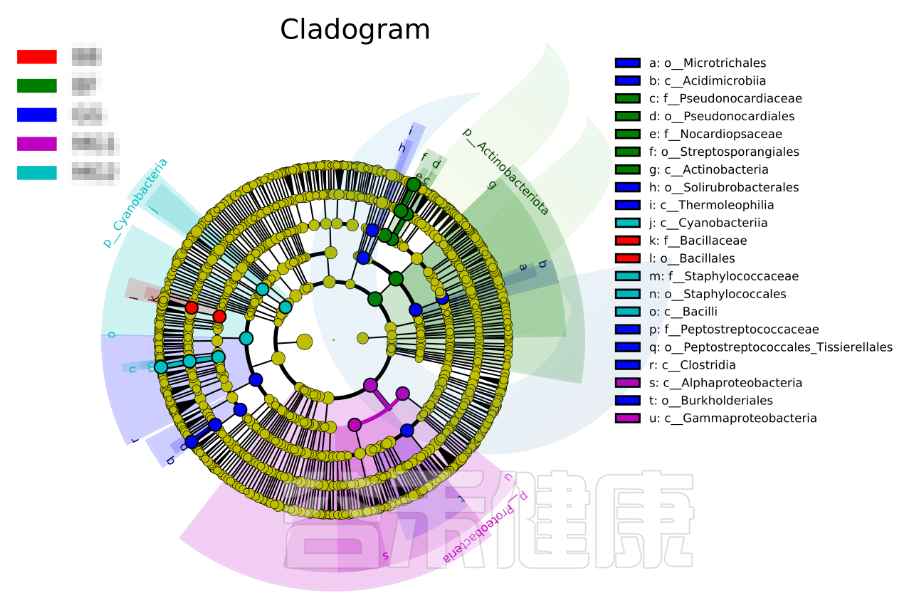
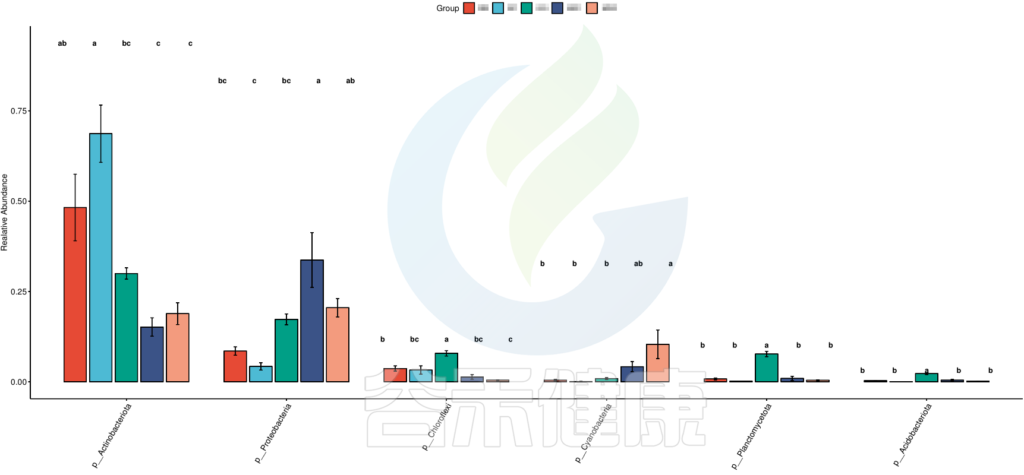
图中右侧展示了分支图中的字母对应的物种信息,例如a 代表GG组的标记物目水平的Microtrichales ,b代表GG组的标记物刚水平的Acidimicrobiia。在分支图的最外层显示的是各分组门水平物种的标记物,例如BF组的是Actinobacteriota、MG1组的是Proteobacteria、
MG2组的是Cyanobacteria
特征表
路径:Group/Lefse_Analysis/out_formant.res.csv
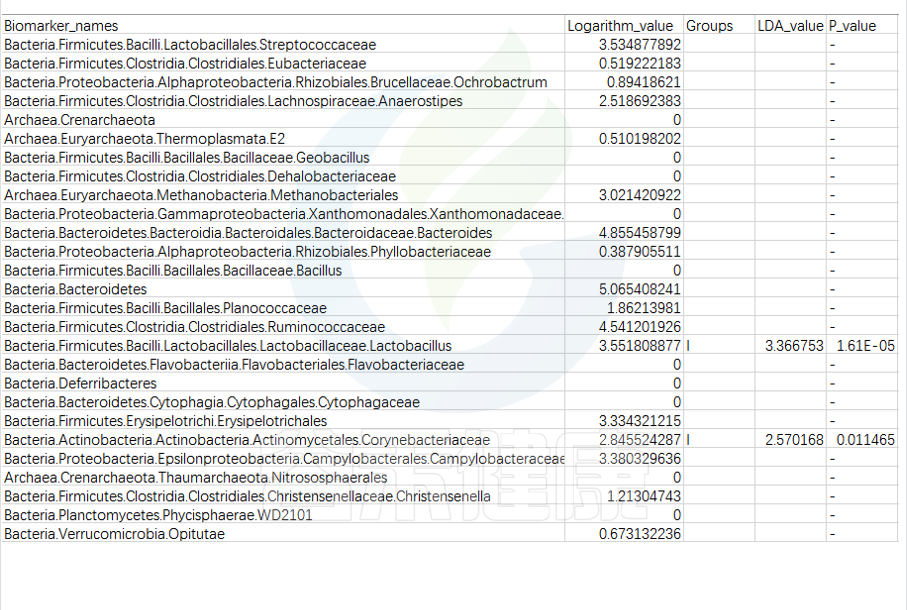
第一列是样本中从门到属水平所有分类单位的列表
Lefse会逐一判断这些分类单位的在分组之间是否具有统计学显著性差异。
第二列:各组分丰度平均值中最大值的log10,如果平均丰度小于10的按照10来计算;如果该分类单位未体现出显著组间差异,则后三列为空。
对于具有统计学差异的分类单位:
第三列:差异基因或物种富集平均丰度最高的分组组名;
第四列:LDA差异分析的对数得分值;
第五列:Kruskal-Wallis秩和检验的p值,若不是Biomarker用“-”表示。
默认LDA>2,P<0.05
通常根据第4列的LDA差异分析对数得分值和第五列的P值,可以描述组间具有显著差异的分类单位统计学效力强弱。
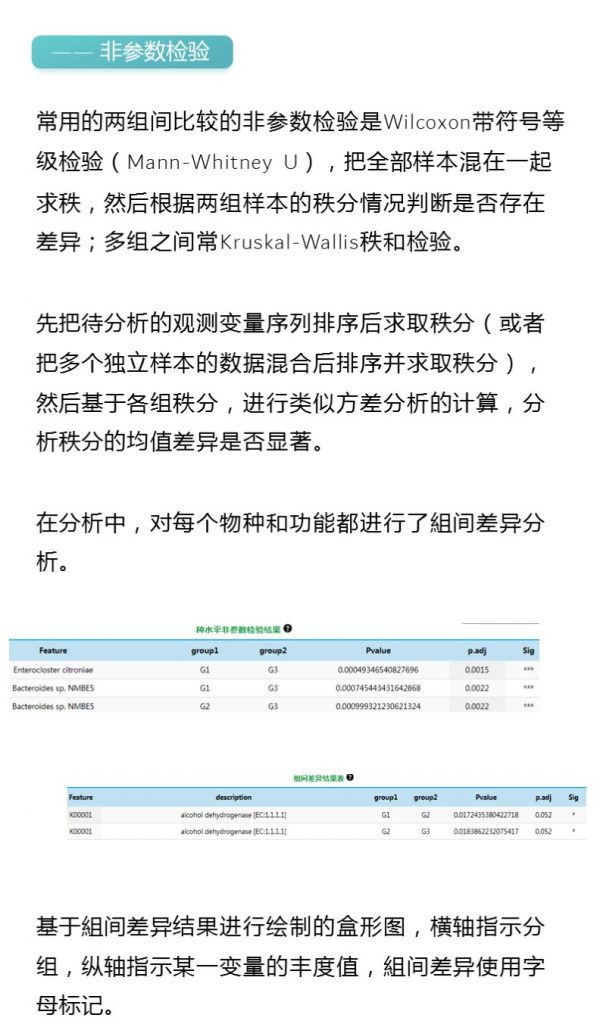
metagenomeSeq
metagenomeSeq是用R开发的一个包,metagenomeSeq的基本思想,用normalization实现分类注释时的biases处理,同时用零膨胀高斯分布(zero-flated Gaussian distribution)处理了测序深度所带来的影响,在此基础上,利用线性模型找到存在的差异所在。
路径:Groups/diff_analysis/ metagenomeRXXX
metagenomeSeq 差异显著物种/功能 热图
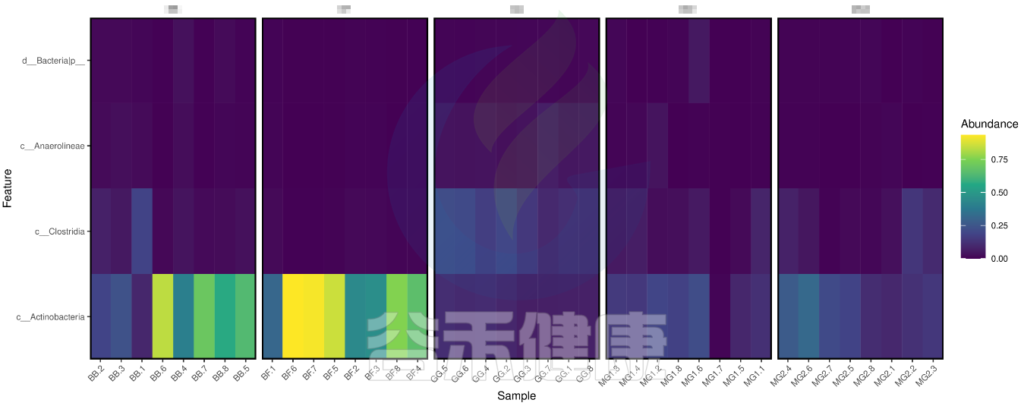
图中颜色越深相关性越小,颜色越接近黄色相关性越大,从图中能看出Actinobacteria物种与BB组和BF组相关性较大。
metagenomeSeq差异菌属于功能代谢关联分析
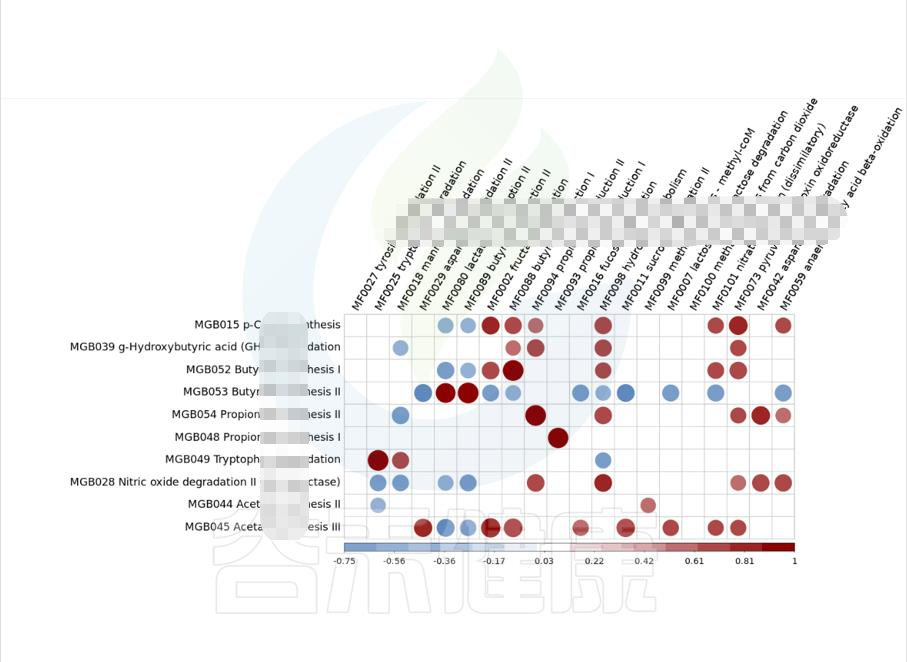
图中红色代表正相关,蓝色代表负相关,颜色越深,圆圈越大,相关性也越大,例如图中能看出MGB049余MF0025 之间成正相关,且相关性较大。
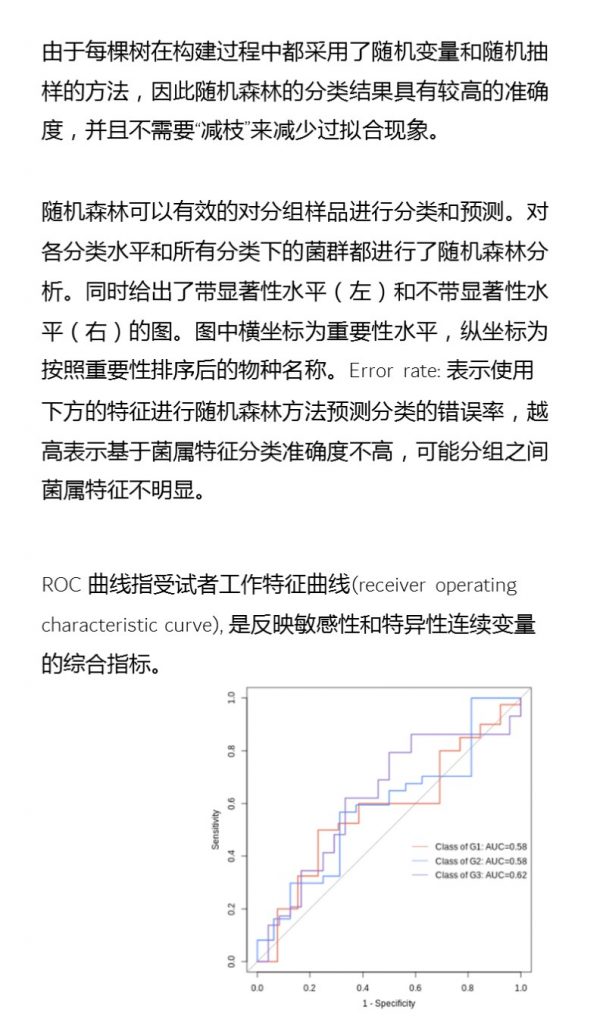
随机森林模型
一种非线性分类器,随机森林属于集成类型的机器学习算法,挖掘变量之间复杂的非线性的相互依赖关系。通过随机森林重要性点图,可以找出分组间差异的关键物种/功能。
反映了分类器中对分类效果起主要作用的特征,按重要性从大到小排列。
Error rate:表示使用下方的特征进行随机森林方法预测分类的错误率,数值越高表示基于特征分类准确度不高,可能分组之间特征不明显。分值越低证明分组效果比较好。
•举个例子
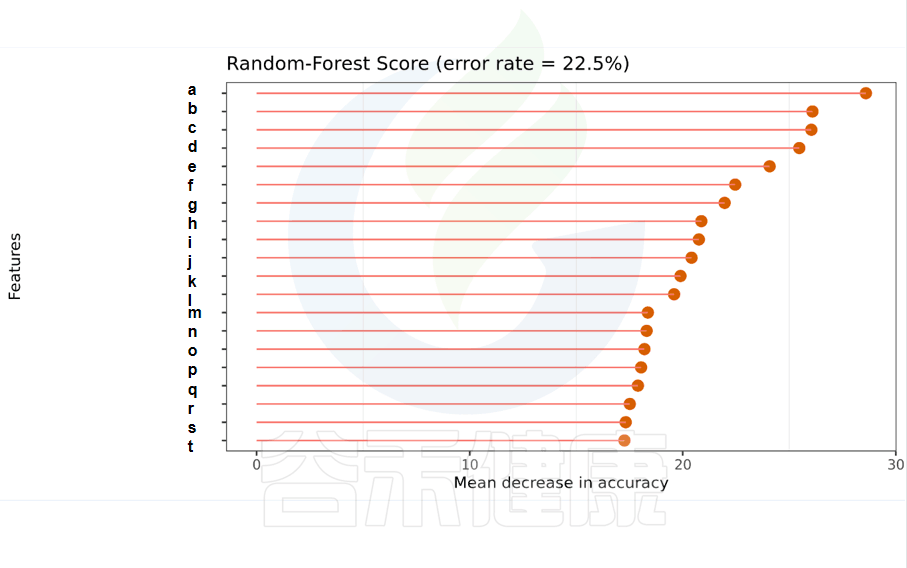
图中按照随机森林模型效果筛选出的对分组效果有重要性作用的物种,按照重要性从高到低进行排列,例如图中最终要的是a,依次往下是b、c等。错误率较小,表明该分组效果较好。
ROC曲线
ROC曲线分析是一种常用的统计学分析方法,在医学研究中主要用于评价诊断试验的效能。在16S测序报告中,我们通过绘制ROC曲线,并计算ROC曲线下面积(AUC),来确定分组对于菌群是否有诊断价值。
ROC曲线图是反映敏感性与特异性之间关系的曲线。ROC曲线下的面积值在1.0和0.5之间。在 AUC>0.5的情况下,AUC越接近于1,说明诊断效果越好。
AUC在0.5~0.7时有较低准确性,AUC在0.7~0.9时有一定准确性,AUC在0.9以上时有较高准确性。AUC=0.5时,说明诊断方法完全不起作用,无诊断价值。AUC<0.5不符合真实情况,在实际中极少出现。
•举个例子
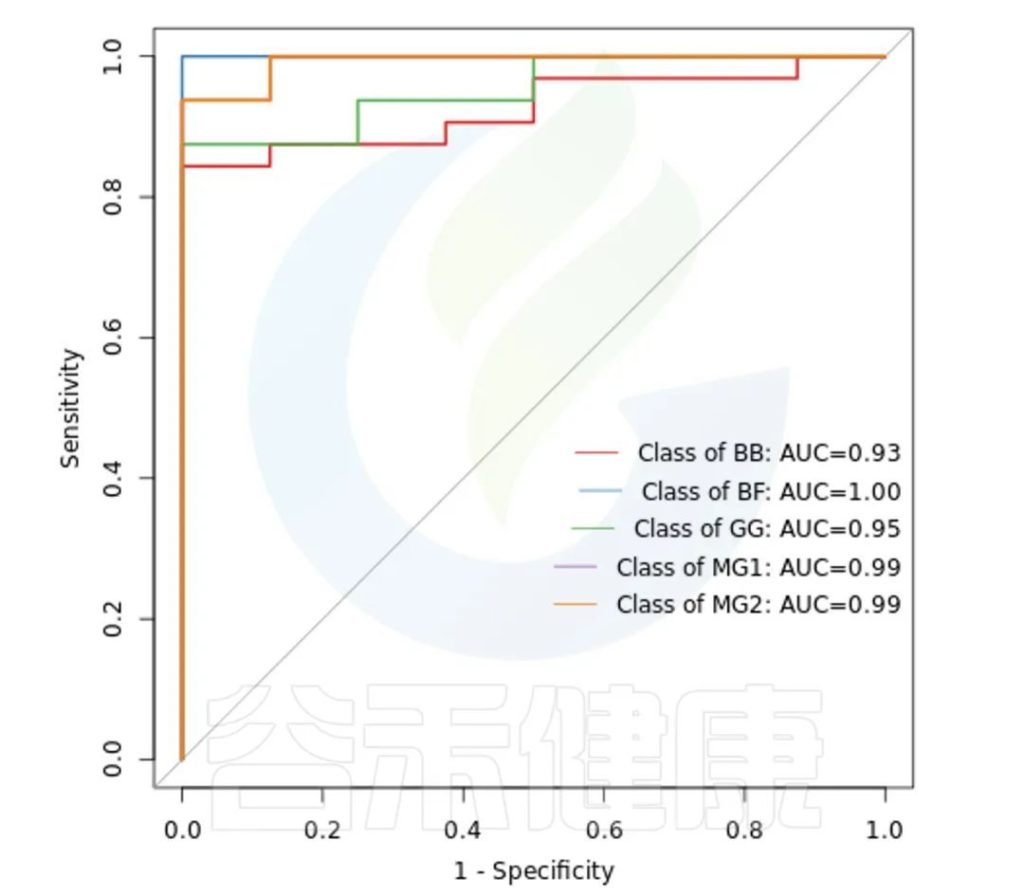
从图中能看出各分组的AUC都大于大于0.9,各分组的分组效果较好,BF组AUC等于1,该分组效果最好,可能样本之间较为相近,并且跟其他分组组间差异也比较大。
以上是组间统计差异的方法介绍,其他的还包括关联分析。
例如客户提供了每个样的相关理化指标数据,想计算这些指标与均属之间有什么相关性,就可以做一下分析。
关联性分析
✦相关性热图
图中X轴代表属水平物种,Y轴代表代谢指标,红色代表正相关,蓝色代表负相关,**代表相关极显著P<0.01,* 代表相关性显著P<0.05相关性具有统计学意义。
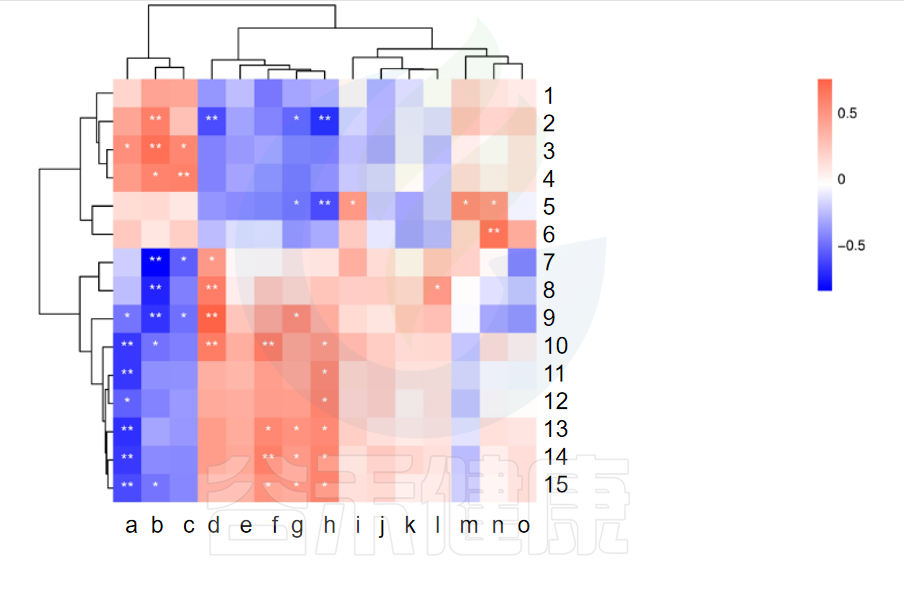
例如从该图中能看出6与n物种成正相关,并且相关性极显著**,7与b物种成负相关,并且相关性极显著**。
可以得到表格:任意菌属和代谢的相关性的值和P值
✦CCA图
可以分析样本、菌群、理化指标之间的关联关系。图中使用点代表不同的样本,从原点发出的箭头代表不同的环境因子。
箭头的长度越长,表示环境因子的影响越大;夹角越小,代表相关性越高。样本点与箭头距离越近,该环境因子对样本的作用越强。
图像中坐标轴标签中的数值,代表了坐标轴所代表的环境因子组合对物种群落变化的解释比例。
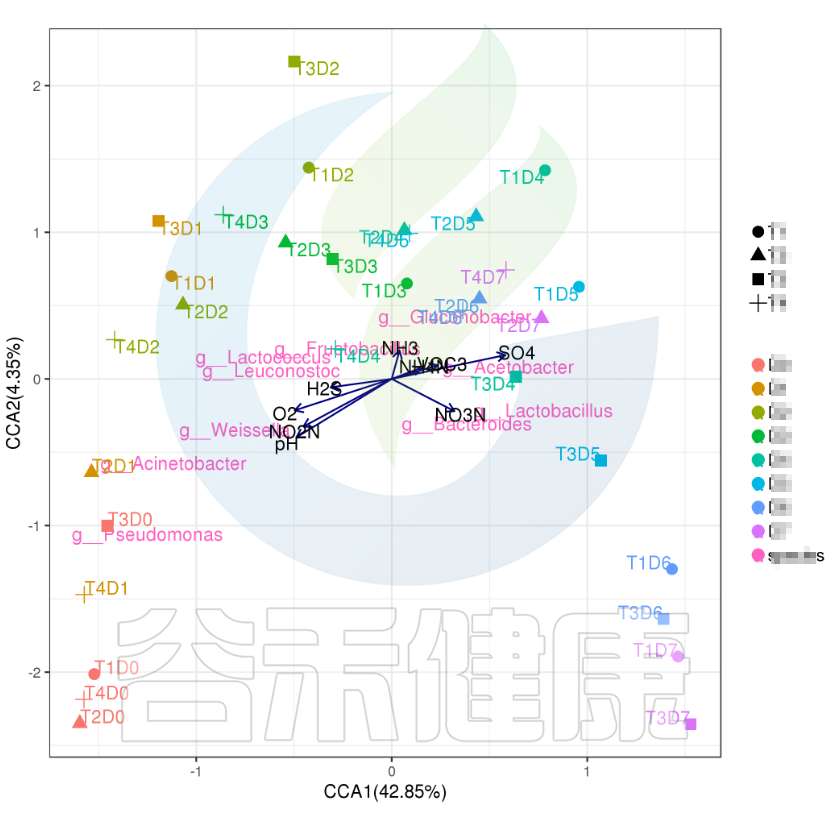
例如从图中能看出pH 、NO2N、02与 Acinetobacter、Weissella等物种成正相关,与T3D0、T1D0、T4D0等D0组的样本成正相关。
✦RDA 冗余分析

例如从图中能看出pH与Helicobacer物种成正相关,相关性较大,pH与NC组有一定的相关性。
✦Envfit分析
回归拟合分析结果:
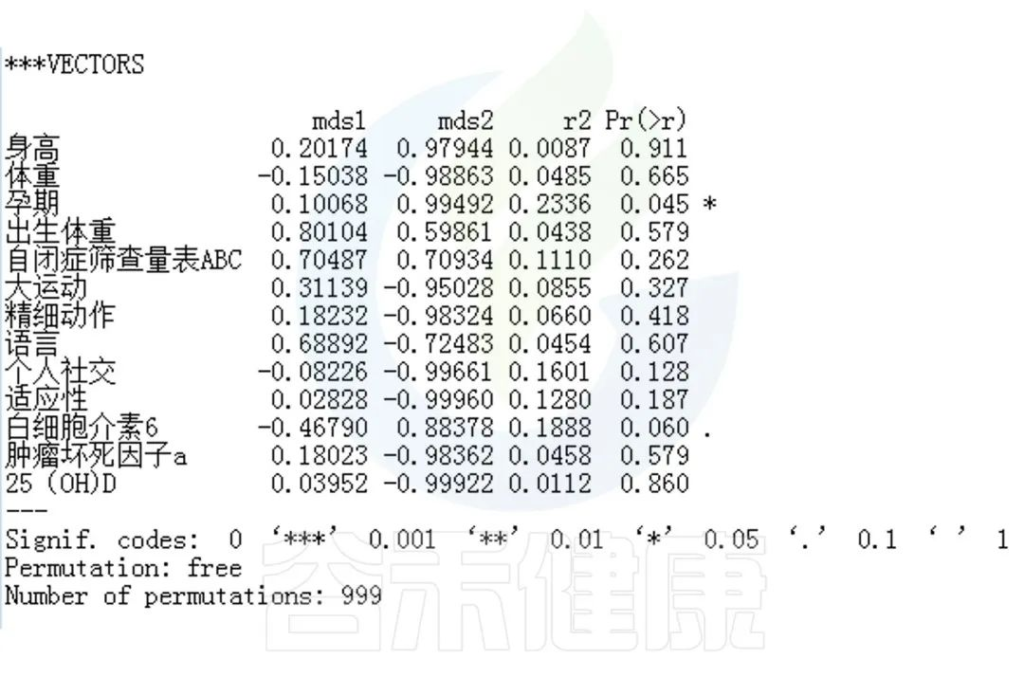
图中能看出ASD与正常儿童之间的分组与菌群之间相关性极显著**,其次是否有先兆流产的分组与菌群之间有显著相关性*,其他的包括是否喝牛奶、孕期是否感染、妊娠高血压都与菌群有相关性。
环境因子与功能/物种的相关性线形图P<0.05显著,图中红色点代表正相关,绿色点代表负相关,灰色相关性不显著。
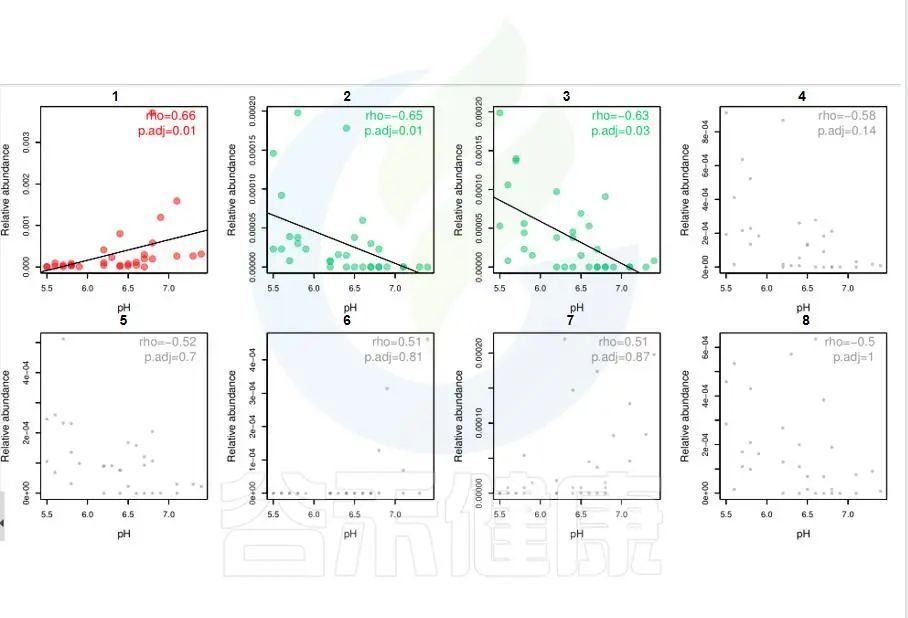
图中能看出pH 与Candidatus Rhabdochlamydia 之间成正相关,且相关性显著,pH 与Sinorhizobium、Euzebya 之间成负相关,切相关性显著。
✦Network网络分析
还可以做菌属之间的网络分析关联图,共发生网络图为研究复杂微生物环境的群落结构和功能提供了新的视角。
由于不同环境下微生物的共发生关系截然不同,通过物种共发生网络图,可以直观看出不同环境因素对微生物适应性的影响,以及某个环境下占互作主导地位的优势物种、互作紧密的物种群,这些优势物种以及物种群往往对维持该环境的微生物群落结构和功能稳定发挥着独特以及重要的作用。
•举个例子
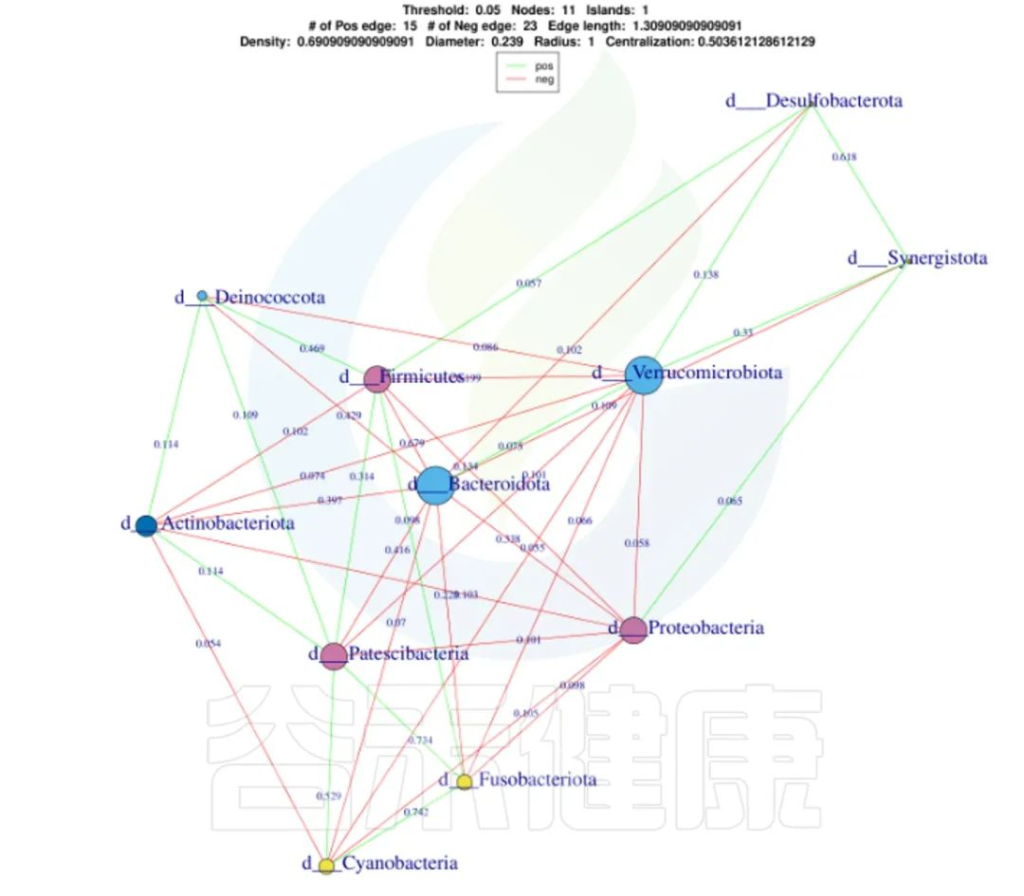
图中展示了相关性的物种,例如Bacteroidota、Actinobacteriota、Proteobacteria 这些物种与其他物种相关较大,图中这些物种与其他物种连线较多,字体比较大也代表相关性较强,例如Actinobacteriota与Deinococcota连线是绿色的代表这两个物种是负相关。
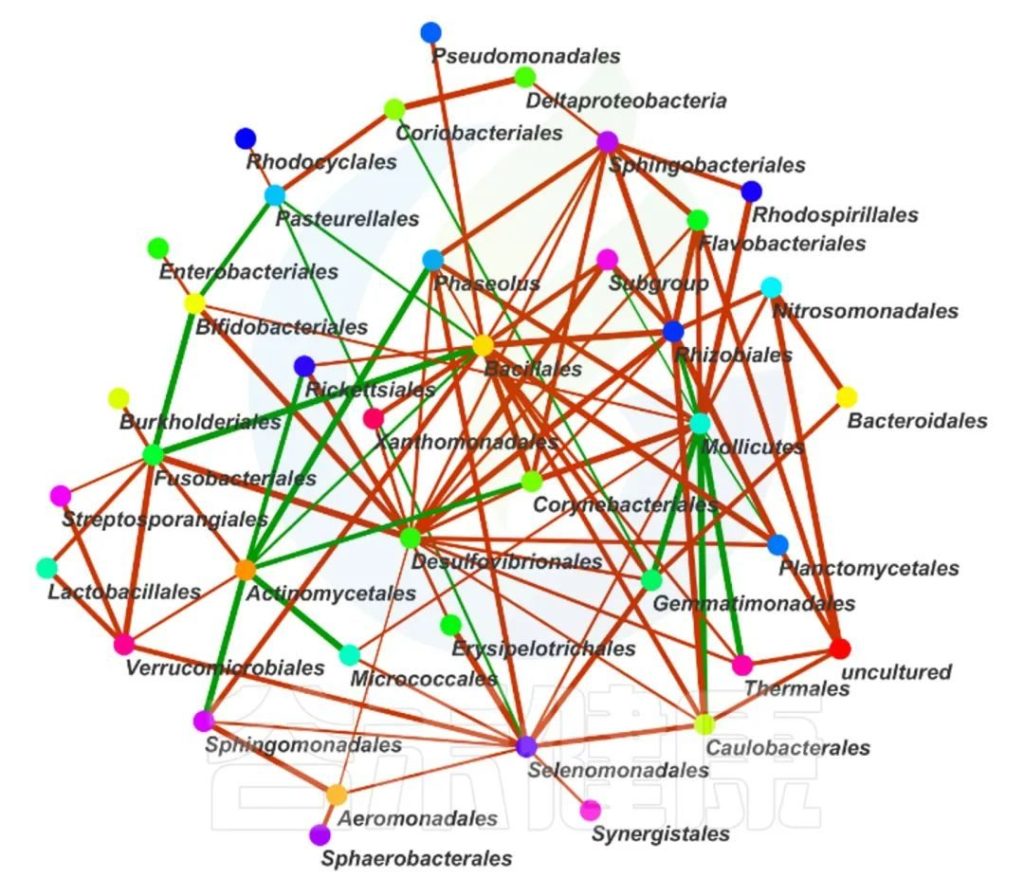
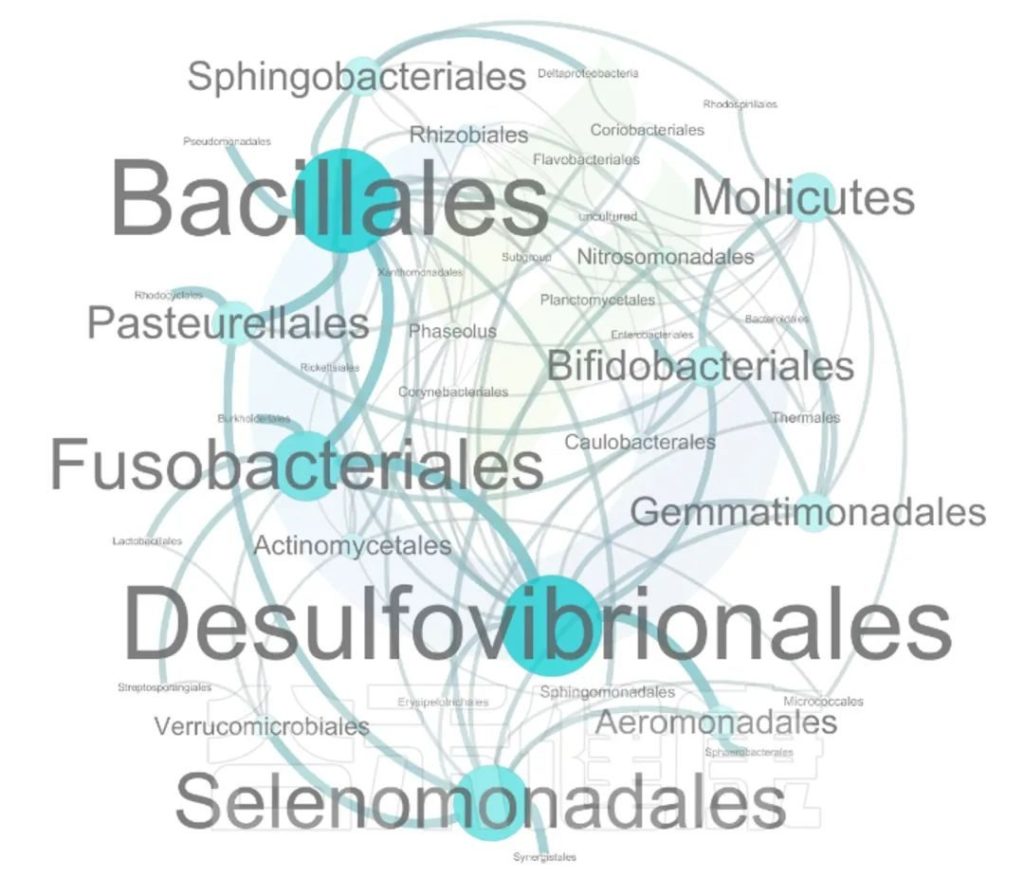
这两个图类似的物种相关性的图,用同一个数据做出来的,图中能看出Bacillales、Desulfovibrionales、Selenomonadales与其他物种相关性较强。
报告中已经基本都涵盖了16S科研数据分析所需要的图表、差异统计,以及相关性分析结果。如果在几种不同类型的统计方法对比之下有略微的差异结果,选取其中一组差异结果即可。
报告里涵盖了大部分16S所需要的图片,不过也有个别个性化的图需要单独用到软件去做,可以单独完成个性化图表生成。
随着16s分析报告的不断升级,报告中的图表以及相应的解读也会越来越精细完善,谷禾也将尽可能为大家的科研之路带来更多便利。


谷禾健康
宏基因组测序可以使我们深度全面地了解微生物群的构成,对于缺乏深度研究和高质量参考基因组的样本,宏基因组获得的较为完整的基因组不仅可以丰富参考基因组数据库,同时可以提供更加准确的物种分类。
关于宏基因组的介绍可见我们之前的文章:

在宏基因组分析过程中,可能遇到的问题,及问题相关解决思路如下:






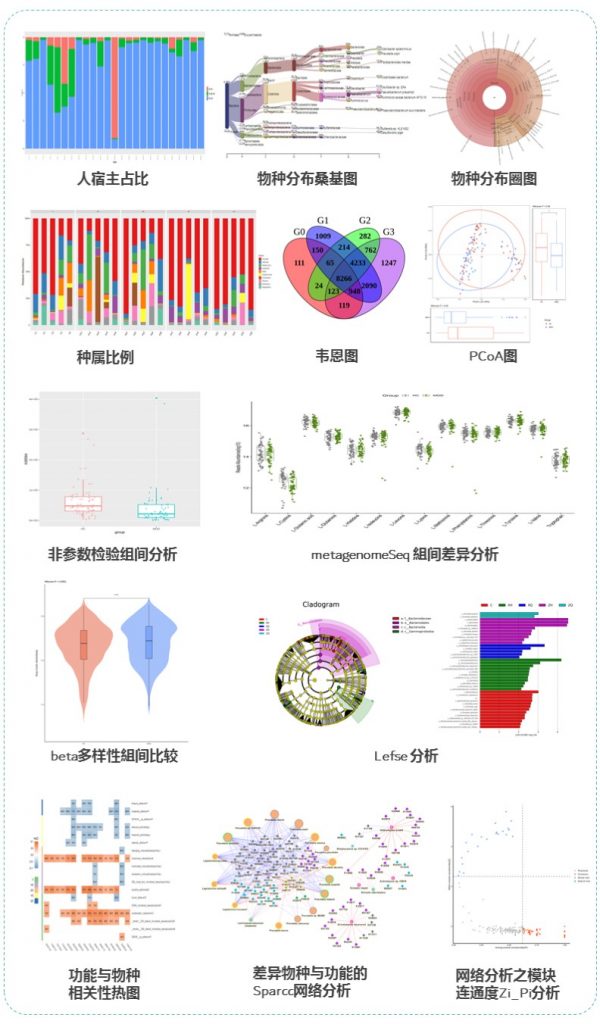








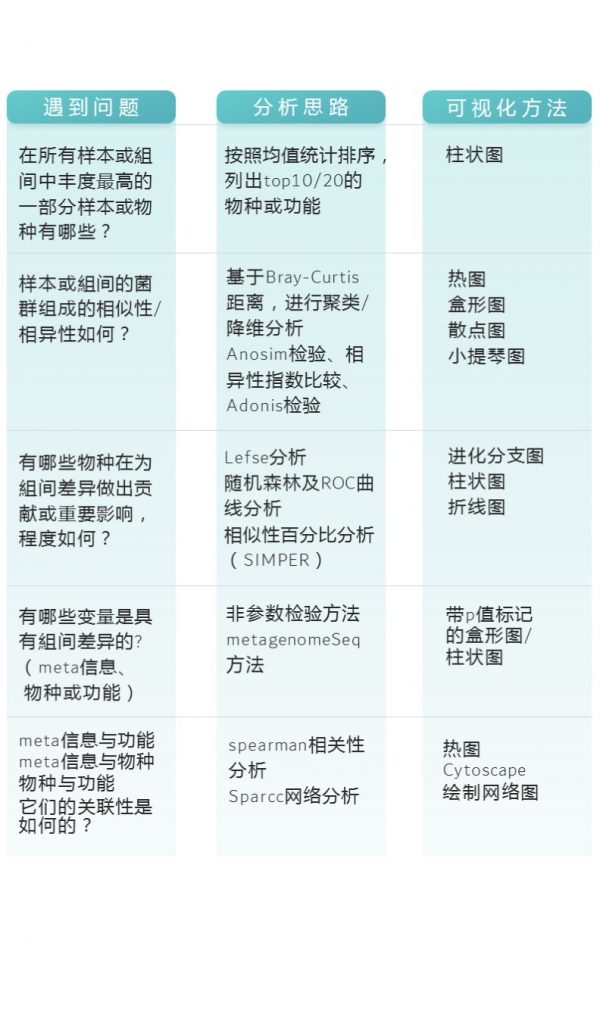
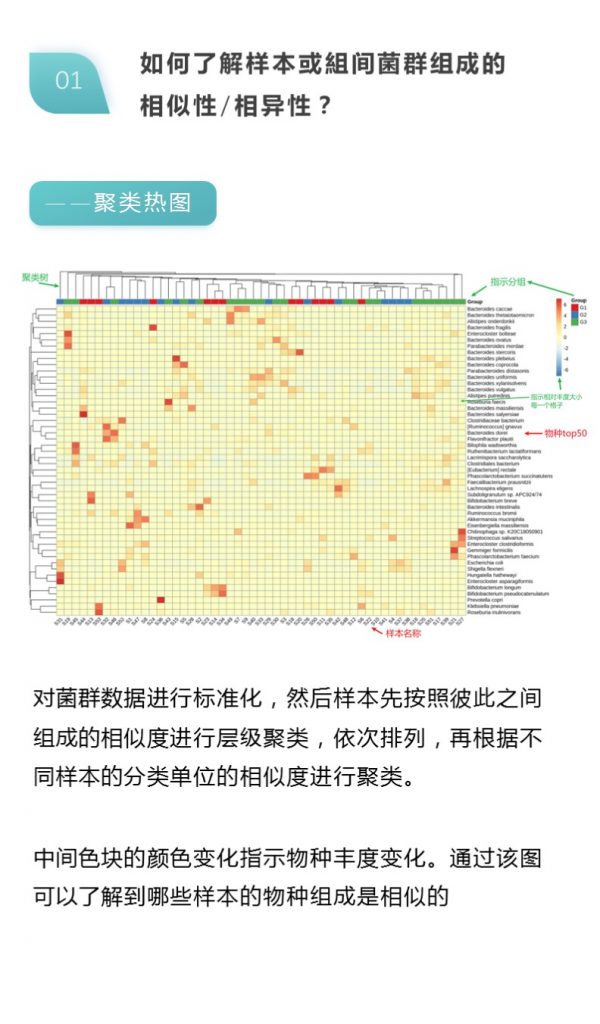
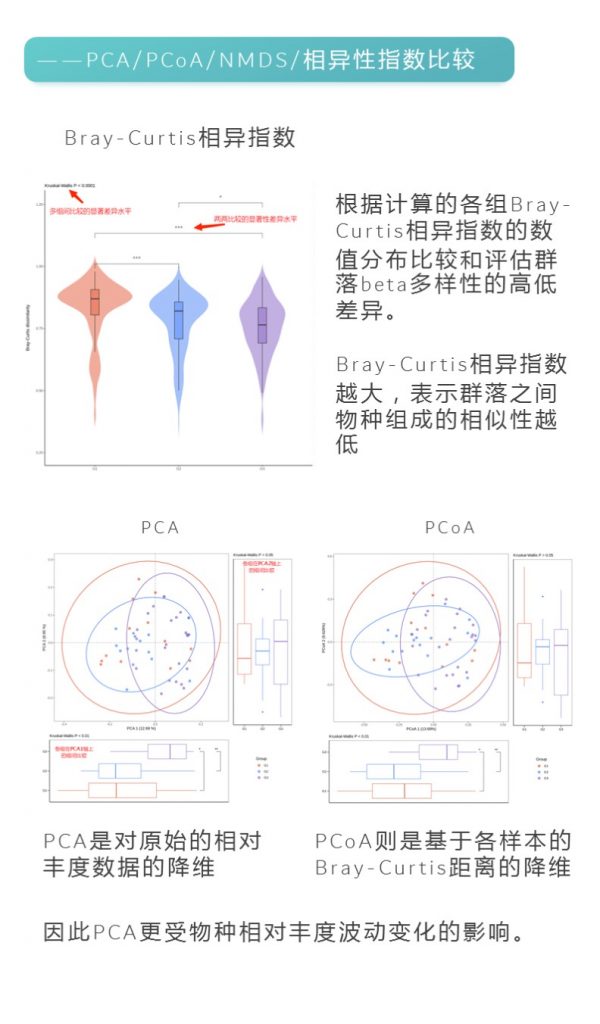
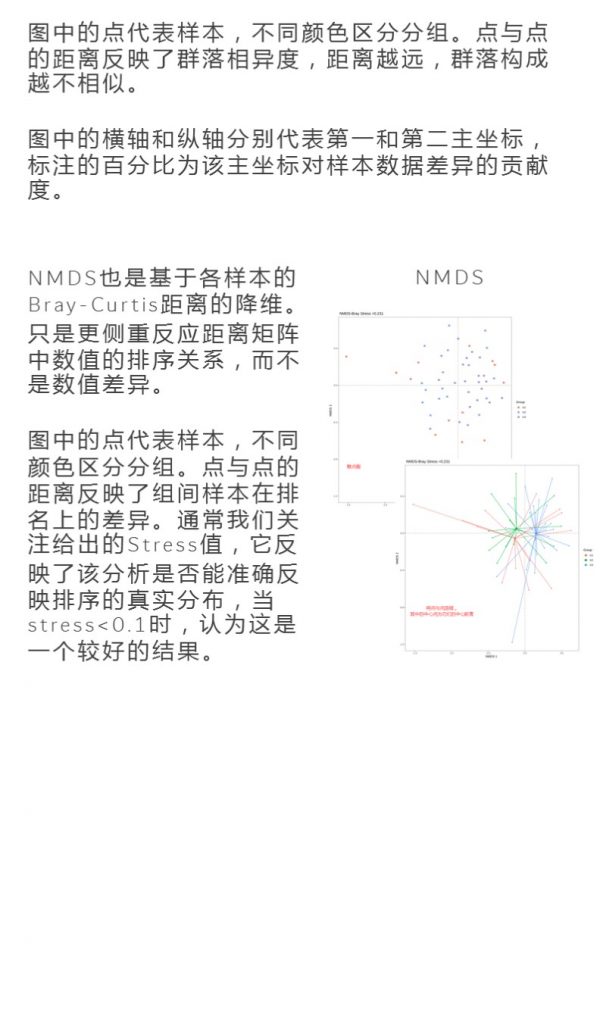






























更多关于宏基因组科研服务详询:
商务经理:13336028502(微信同号)
近年的研究热点集中于环境和生物体相互作用的微生物群体,而大量复杂的微生物群体存在培养困难,构成复杂(包括细菌、古菌、真菌、原生生物、病毒甚至小型真核生物)。 为了克服传统纯培养技术的不足,充分挖掘此类未培养微生物所蕴涵的巨大潜能,发展了 宏基因组学的研究方法,该方法绕过纯培养技术来研究微生物的多样性及功能。
宏基因组测序也就是shotgun测序, 以环境中所有微生物基因组为研究对象,通过对环境样品中的全基因组DNA进行高通量测序,获得单个样品的饱和数据量,基于denovo组装进行微生物群落结构多样性, 深度全面的了解微生物群体的构成,甚至获得单个菌株的完整基因组,在这些高质量基因组序列的基础上可以更加精细化开展其基因构成、分布,次生代谢合成,抗生素耐药基因及其演化, 微生物群体基因组成及功能,特定环境相关的代谢通路等分析 甚至菌株的生态演变和水平转移事件等研究。 进一步发掘和研究具有应用价值的基因及环境中微生物群落内部、微生物与环境间的相互关系。
此外对于缺乏深度研究和高质量参考基因组的样本,如土壤和特殊环境下的样本,宏基因组获得的较为完整的基因组不仅可以丰富参考基因组数据库,同时可以提供更加准确的物种分类。
建库流程
1) 将检测合格的基因组DNA样品用酶切随机打断成400bp-500bp的片段并末端修复加A;
2) 将测序引物连接到DNA片段上;
3) 进行PCR扩增引入测序index
4) 对构建的文库进行质量检测;
5) 将质量检测合格的文库上机测序。
粪便、动物肠道内容物、皮肤、组织、痰液、血液、唾液、牙菌斑、尿液,阴道分泌物、发酵物,瘤胃,废水,火山灰,冻土层、病害组织、淤泥、土壤、堆肥、污染河流,养殖水体、空气等有微生物存在的样本都可以用于宏基因组测序。
测序平台:Illumina Novaseq,PE150,默认:5-6G/样,每增加一个G加100元。
周期:2-3周
粪便、动物肠道内容物、皮肤、组织、痰液、血液、唾液、牙菌斑、尿液,阴道分泌物、发酵物,瘤胃,废水,火山灰,冻土层、病害组织、淤泥、土壤、堆肥、污染河流,养殖水体、空气等有微生物存在的样本都可以用于宏基因组测序。




第一项研究是关于肥胖患者减肥手术后的宏基因组和代谢数据的分析研究。
研究纳入了61名严重肥胖的受试者,他们是可调节胃束带术(AGB,n = 20)或Roux-en-Y胃旁路术(RYGB,n = 41)的候选人。减肥手术后1、3和12个月随访24名受试者。使用宏基因组学测序和LC-MS分析肠道菌群和血清代谢组。另外纳入了10人和147人分别作为宏基因组和代谢检测的验证集。
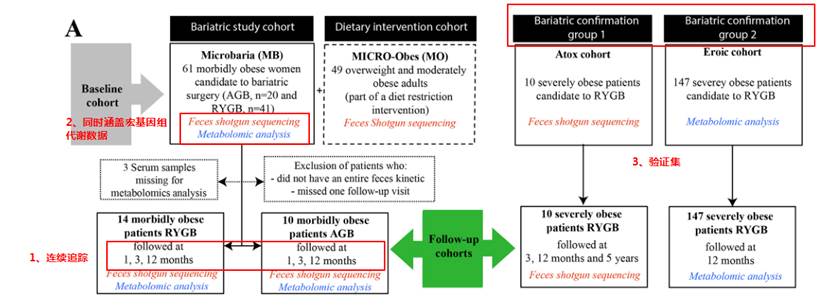
研究思路
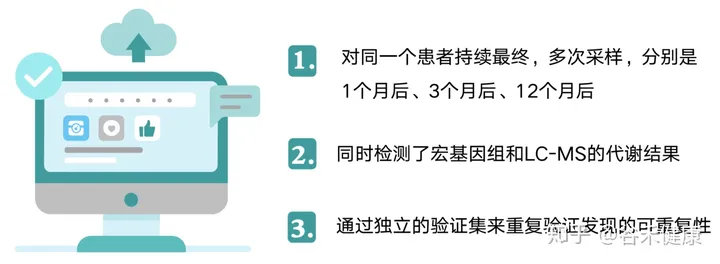

这样的设计分别有什么作用?
第一点持续的动态采样可以获得持续变化情况,尤其是在一个特定变化后(减肥手术),持续的最终采样有助于确认菌群的变化出现和特定事件或生理病理变化的前后,尤其是在确定因果中有重要帮助。
第二点获得多维的数据有助于帮助我们全方位的了解菌群变化背后的带来的生理和代谢变化以及之间的关联。
第三点独立验证集的存在将大大增强研究的可信度,尤其是该研究纳入的样本量并不多,无法全面有效的控制无关因素,使得很多统计检验的效力无法显现。这也导致该研究仅在基因总量和多样性上获得较好的重复效果,而更多的菌群精细特征以及具体基因和代谢通路没有得到深入分析。但是独立验证集保证了核心结论的可靠性和重复性,这点在宏基因组研究中非常重要。
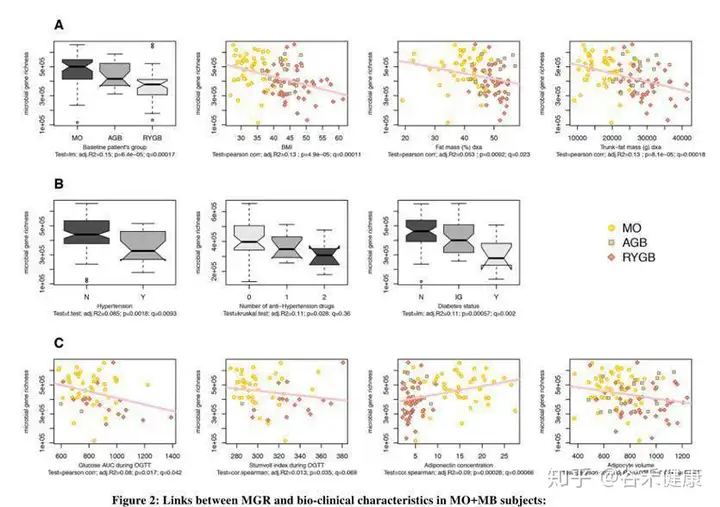
从下图可以看到研究针对样本的总基因多样性水平与生理指标和疾病状态进行相关性分析和组间差异分析,图中给出了显著相关和差异的指标。
使用的统计检验方法是pearson和sperman相关和t-test以及Kruskal-Wallis检验。
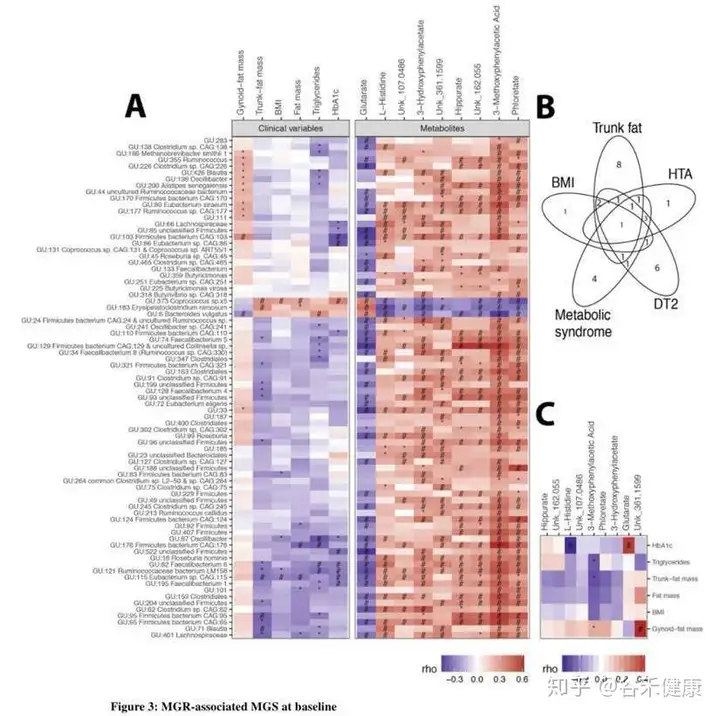
下图是研究将MAGs与各项生理和代谢值进行相关性分析后的热力图。该研究由于测序较早,并未独立拼接,而是直接使用了之前一项人类肠道菌群研究获得组装基因组参考序列。
进一步研究分析了术后特定变化模式的MAGs以及它们与代谢生理指标的相关性,见下图:
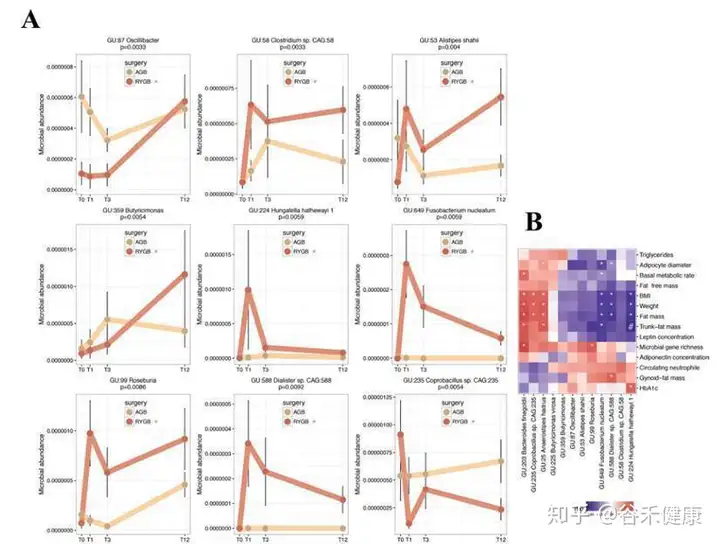
上图的研究可以通过pattern search的方法寻找特定变化模式的菌种。
研究的主要结论发现是低基因丰富度(LGC)存在于75%的患者中,并且与躯干脂肪质量和合并症(2型糖尿病,高血压和严重程度)增加相关。LGC改变了78种宏基因组种(MGS),其中50%与不良的身体成分和代谢表型有关。九种血清代谢产物(包括谷氨酸盐,3-甲氧基苯基乙酸和L-组氨酸)和含有参与其代谢的蛋白质家族的功能模块与低MGR密切相关。术后一年,BS会增加MGR,但尽管RYGB患者的代谢改善比AGB患者大,但术后一年的MGR仍然很低。
总体而言该项研究可以使用浅宏基因组来完成所有测序和分析,进一步扩大样本数量,如果能同时获得人的转录组数据甚至能更加明确的找到菌群变化与特定代谢通路的关联关系。
< 案例二 > 食物与人类肠道微生物组
第二项研究是Dan Knights实验室发表在Cell Host & Microbe,2019的一篇针对34个人17天每日饮食和菌群变化的相关研究,试图揭示日常食物选择与人类肠道微生物组组成之间的精细关系。
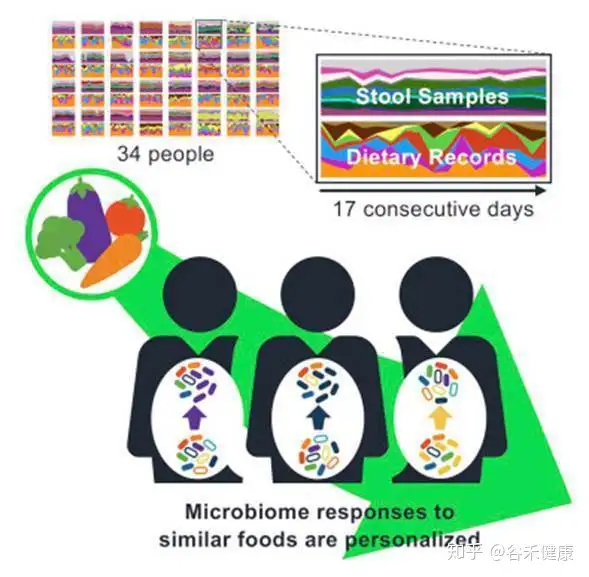
可以看到,研究同时记录了粪便样本的菌群宏基因组和每日的饮食记录。研究的核心在于将每日饮食的食物通过营养构成进行量化,并构建类似物种进化树的食物物候树。
此外由于有每日的数据,可以通过前一日的食物与第二日的菌群数据进行时间序列分析,构建食物与菌之间的关联以及时间相关性。
最后基于菌群数据和前一日饮食来构建模型预测判断后一日的菌群状态,帮助我们了解食物对于个体菌群的影响因素并实现定量和预测。
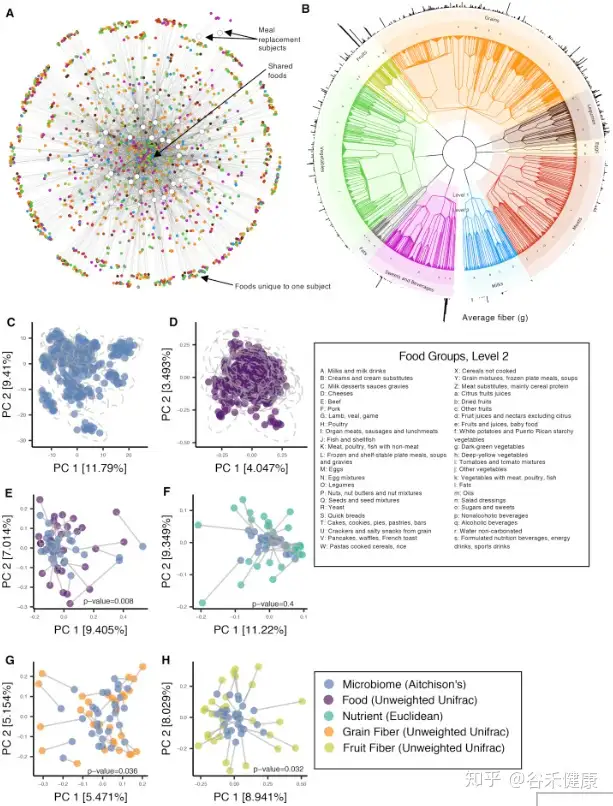
研究中对数据的处理过滤标准如下:删除所有具有低读取计数(每个样品<23,500个读取)的样品。物种级别的分类表仅限于研究对象中至少存在25%的研究日,且在10%的研究样本对象中发现的那些物种。
最后,相对丰度<0.01%的稀有物种被丢弃,将物种数量限制为290个注释。将得到的分类表汇总到较高的分类级别(即属,科,门等),以进行下游分析。
菌群和饮食以及营养构成的堆叠图很好展现了变化和对应。
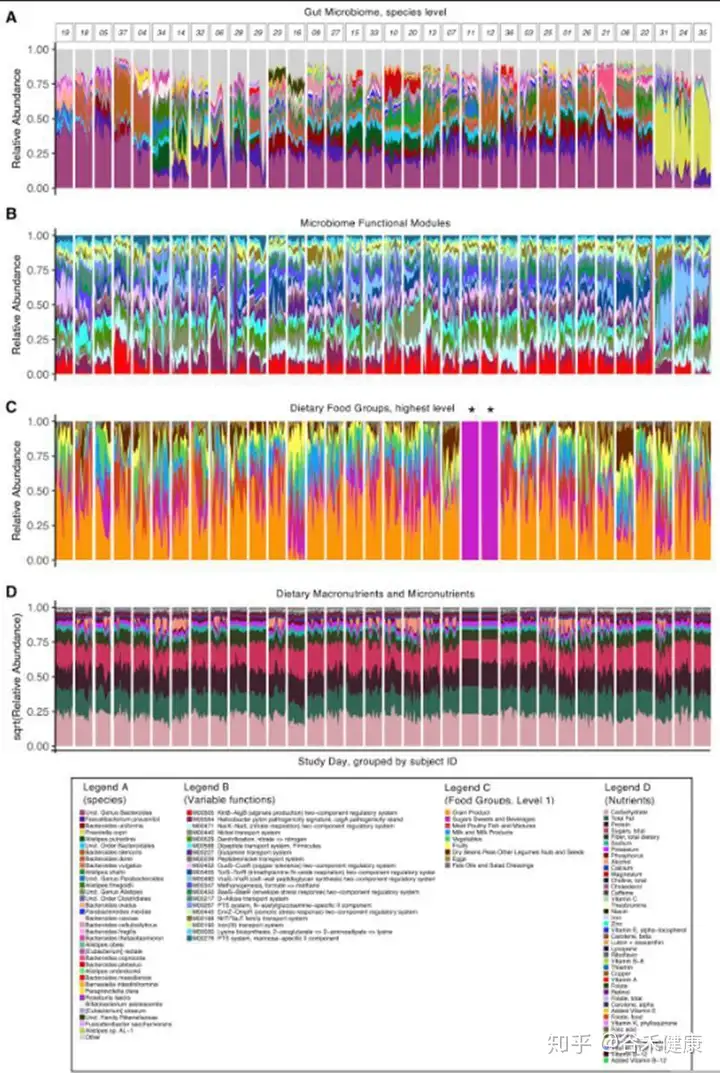
下面这张图很好的显示了饮食食物的变化与菌群变化之间的时间变化关系:
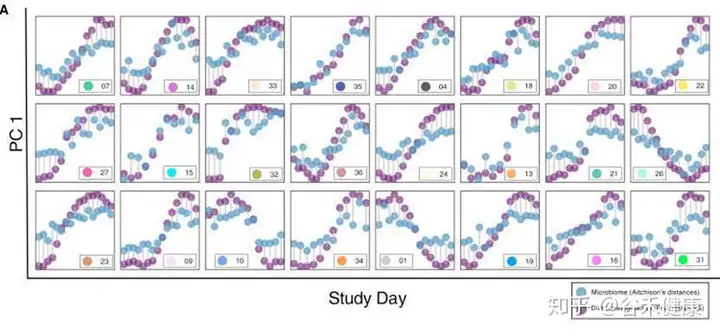
下面这张图通过对每个人单独进行菌属与食物的Spearman相关,展现了菌与食物之间的关联的个体化差异,在特定菌属对应相同食物不同人会出现完全不同方向的变化,这也正是这项研究所揭示的,这种关联关系的复杂性。
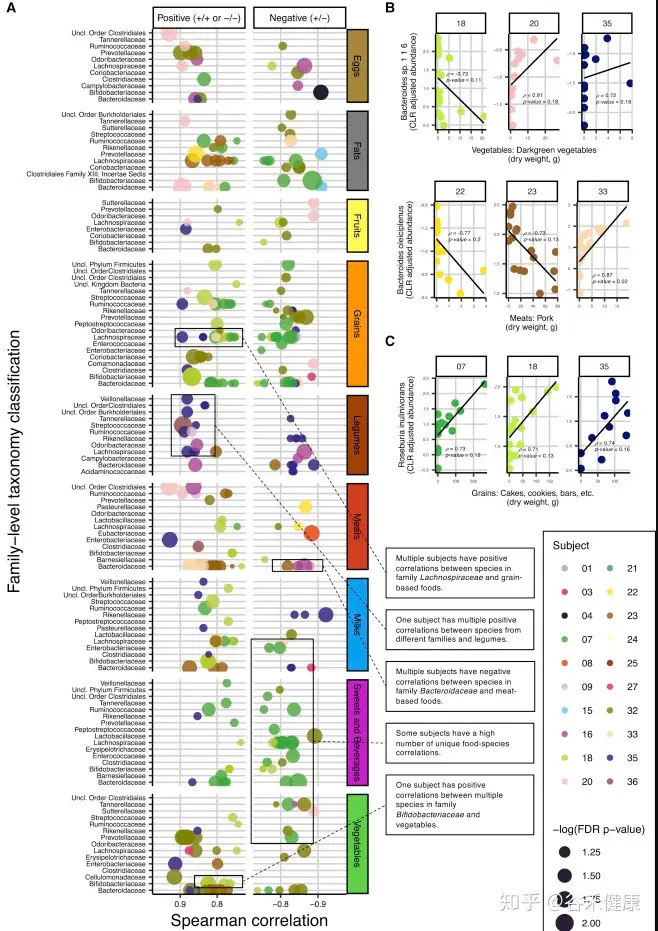
本研究虽然有大量样本,但并未进行组装,而是直接使用了Refseq的细菌完成基因组序列作为参考。研究由于样本数量众多,测序深度也很有限,类似研究也可以使用浅宏基因组方式完成。
接下来的一个研究是比较典型的宏基因组组装并与疾病进行关联分析的案例,研究的是类风湿关节炎人群的肠道微生物组的全基因组关联研究。
研究使用较高深度的宏基因组shotgun测序(每个样本平均13 Gb)对日本人群(病例 = 82,对照 = 42)进行了RA肠道微生物组的MWAS分析。MWAS由三个主要的生物信息学分析渠道(系统发育分析、功能基因分析、途径分析)组成。
使用了之前研究中6139个完成拼接日本人肠道宏基因组作为参考序列以及其他几项研究的参考基因组,在过滤部分种过多的基因组之后,最后一共使用了7881个参考基因组。
将QC后的序列直接比对到参考基因组,并根据基因组长度计算对应物种的相对丰度。
基因方面选择denovo组装,使用MegaHIT,然后再contig上完成ORF预测和CD-HIT聚类去冗余,最后与UniRef和KEGG数据库比对。
最后使用bowtie2将测序序列比对到注释后的unigene序列上获得基因丰度,经过KEGG注释得到代谢途径的丰度。研究的数据处理流程图如下:
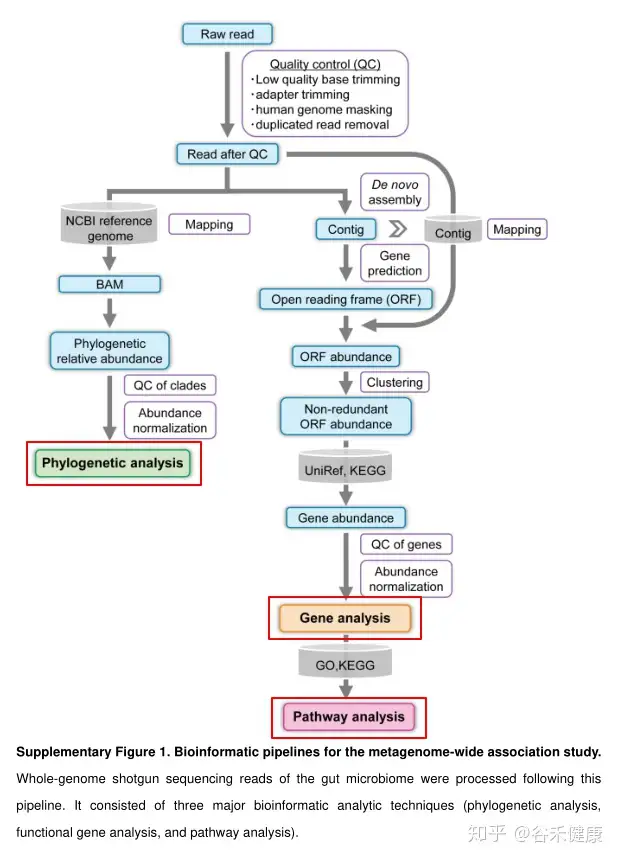
在数据分析流程和方案选择上人体肠道菌群由于研究众多,以及有多个深度测序拼接完成的Binning参考基因组数据集,确实可以直接使用参考基因组直接比对。
对于其他一些环境或来源的样本这个深度的数据量可以考虑独立拼接和分箱。该研究中使用已有参考基因组,大概88%的序列能比对到参考基因组,如果直接组装这个比例应该可以更高一些。另外在获得基因丰度是可以考虑使用Salmon,比对获得基因丰度更为方便。
获得相应数据后对相对丰度,该研究使用Box-Cox transformation对数据进行标准化,并过滤了一些低丰度的菌属。Case-control的相关性分析使用的R的glm2模块,将年龄、性别和测序上机分组作为协变量。
对于菌属的关联分析,最终将显著性结果以火山图和GraPhlAn图的形式展现如下:
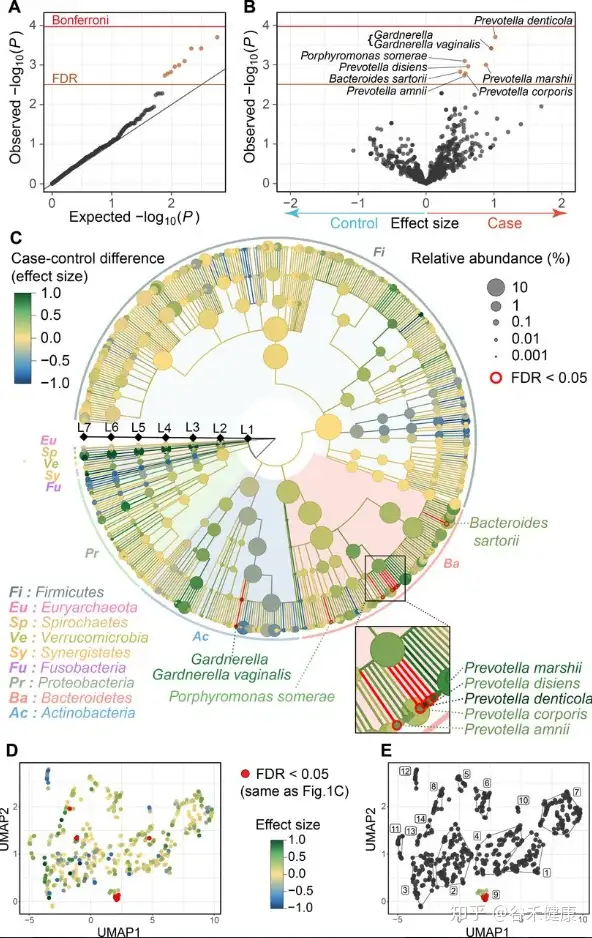
上面其中D图使用每个菌的丰度进行UMAP分析,并映射关联效应的展示比较有意思。
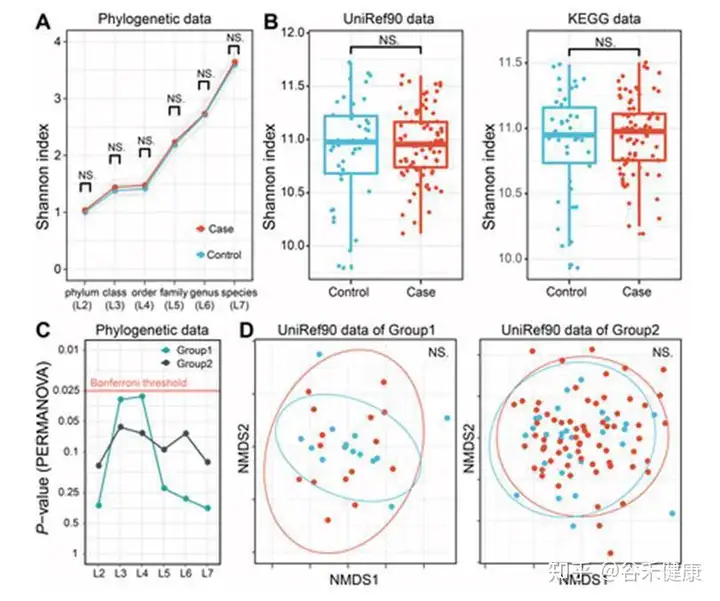
不过在基因层面上并未找到相应的关联,可以看到下图UniRef90的NMDS分布图两组之间无法有效区分,多样性也没有显著差异。
这项研究在菌层面发现了多个普雷沃氏菌属的菌在日本人群中与类风湿性关节炎存在关联,不过除此之外其他方面的发现并不多,仅找到一个基因存在显著关联,涉及的临床调查也相对有限,且人群队列数量不算多,并无独立验证集,因此亮点并不多。如果能纳入免疫相应指标可能能研究的更细致一些。
最后这项研究是对来自永冻土融化梯度的214个样品的宏基因组测序组装了1,529个基因组,揭示了参与有机物降解的关键种群,包括其基因组编码先前未描述的木糖降解真菌途径的细菌。
通过宏基因组denovo组装和分箱Binning,最终获得了1529个永冻土菌群基因组。基于这些数据描绘了永冻土融化梯度下的菌群构成特征,如下图。
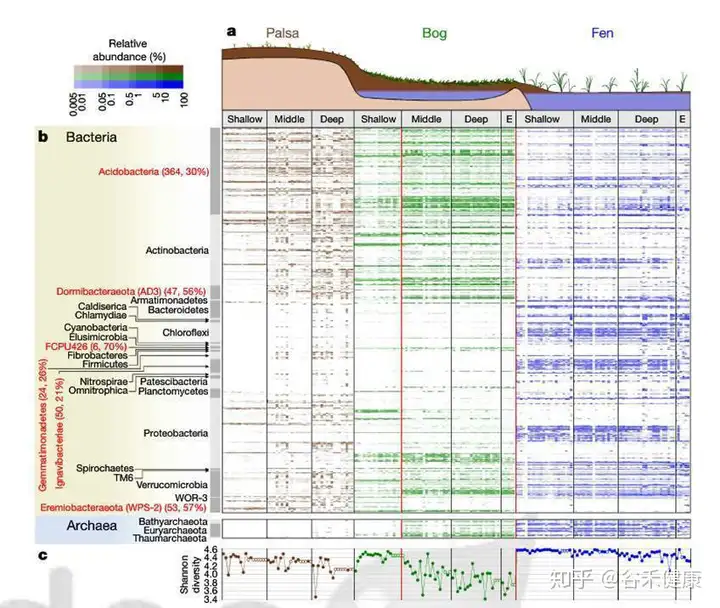
论文是2018年发表的,测序是在2011和12年测的,使用的是CLC Genomics Workbench 较早的4.4版分单样本组装,然后使用MetaBAT进行分箱,最后的标准是70%完成度和低于10%的污染。
其中糖苷水解酶基因使用dbCAN数据库的HMM进行预测,碳代谢使用KEGG数据。
研究还同时选择了部分样本进行了宏转录组和宏蛋白组的测序,对碳代谢同时结合转录组和蛋白组的数据,展现了特定通路下不同永冻土的菌群构成和表达丰度差异。
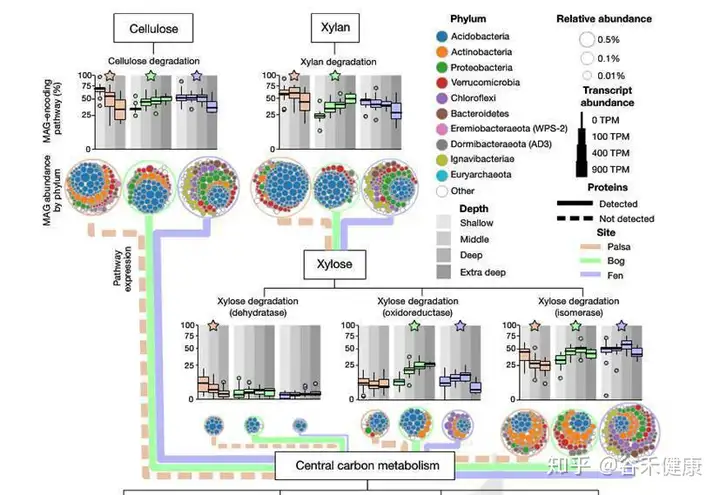
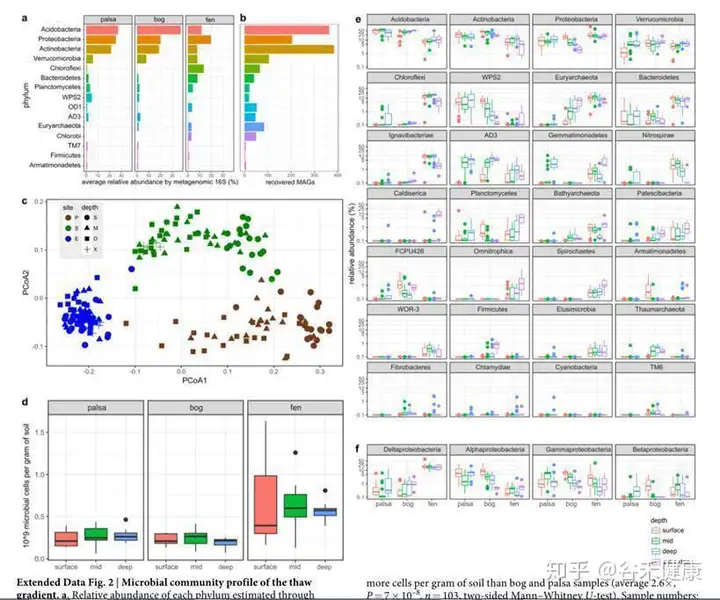
基因组拼接的分布情况,以及不同地域样本分布和菌属丰度情况如下:
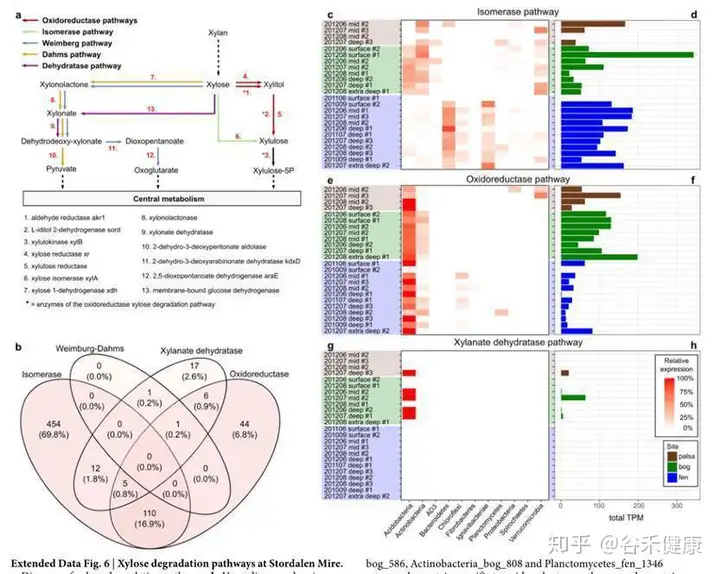
木糖降解途径在每个样本中的分布和维恩图,另外详细的展现了主要门对每个代谢途径的贡献和基因表达丰度,如下图:
这张图分析了特定菌与地理位置和CO2以及甲烷的浓度的关联性,如下图:
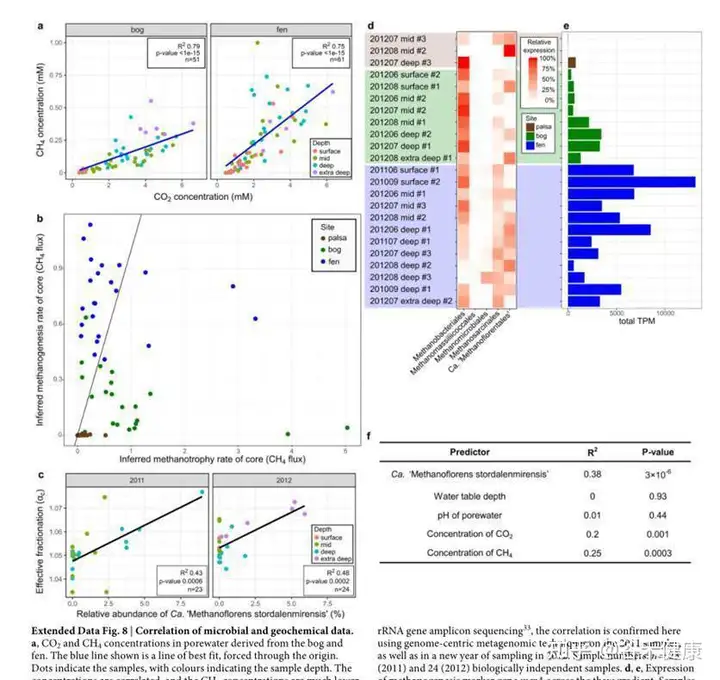
对关键物种的CH 4 :CO2浓度比相关代谢途径的重建,以及相应基因的基因家族分析。
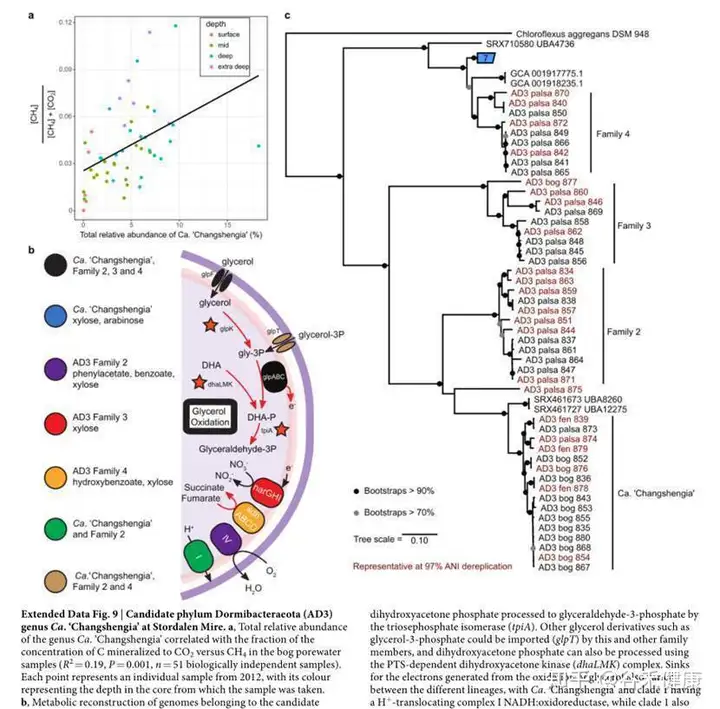
总结一下这项研究,永冻土的菌群参考基因组数据缺乏,该研究从大量地点采集样本重建了1500多个参考基因组。
首先从物种构成特征上与永冻土融化阶段特征进行分析,并与重要环境因子进行分析,锁定重要的特征菌。
然后针对重要的代谢途径和关键基因结合宏转录组和宏蛋白组全面解析代谢途径的分布和差异变化。对关键的物种重建了相关代谢途径并对其相关基因家族进行分析。
研究基本上从头构建了一个生态环境下的菌群结构数据,并利用获得的基因组深度解析特定代谢途径和基因的构成和表达变化,应该说既宽又深。很多样本采集和测序是2011年和 2012年就开展的,虽然测序技术远不如现在成本低和成熟,但是其独特的研究对象和全面深入的分析仍然使整项研究和目前的一些研究相比完成的更加出色。



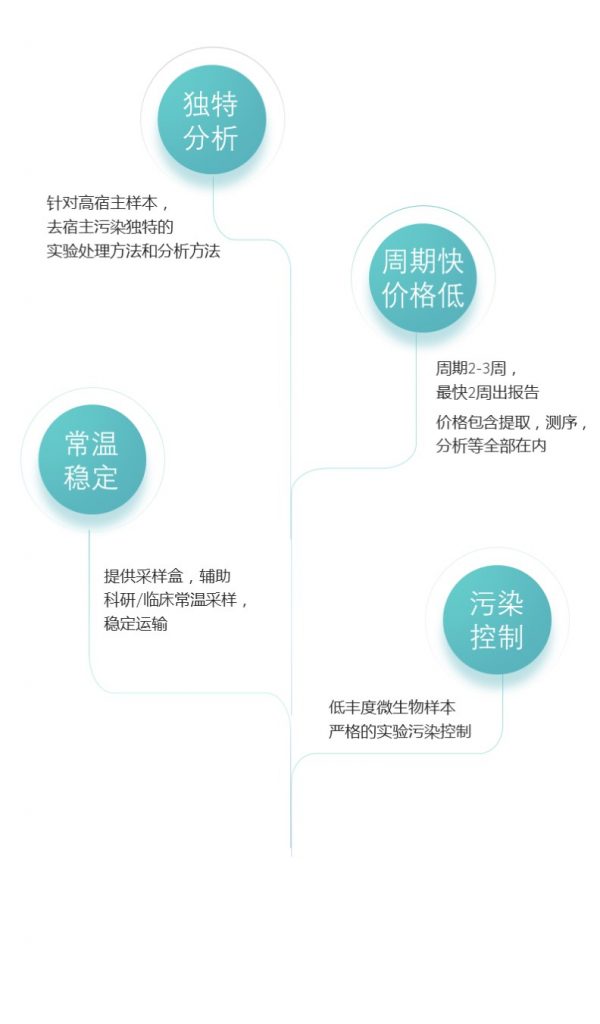
样本需求量低:常规宏基因组建库建议样本量在500ng以上,公司研发实现了低当量微生物样本提取和建库,保证提取丰度以及片段完整性同时,样本量需求低于同行其他公司要求;对于样本获取困难的样本,也可以选择微量建库,样本量可低至10ng。
免费取样盒和针对性取样建议:粪便及环境样本提供取样盒助力临床/科研取样,人体口腔、痰液、腹水、脑脊液、尿液、皮肤等高寄主细胞含量样本可根据我们的处理方案简单处理后大幅降低宿主DNA比例。
严格标准的实验流程:自动化样品处理平台辅助,每轮设置阳性对照,上轮检测样本对照,阴性对照。评估污染,轮次比对,最大化减少误差,保证样本重复性和稳定性
Illumina测序平台:宏基因组测序(PE150)采用先进的Illumina Novaseq测序平台,快速、高效地读取高质量的测序数据、结合样品特点和数据的产出,充分挖掘环境样品中的微生物菌群和功能基因
大数据分析流程质量流程控制严格:优化的数据质量控制,包括过滤比对质量低、非特异性扩增、覆盖度低、低复杂度的序列,从而快速准确获得样本中微生物信息及其丰度信息,最大化提高质量数据
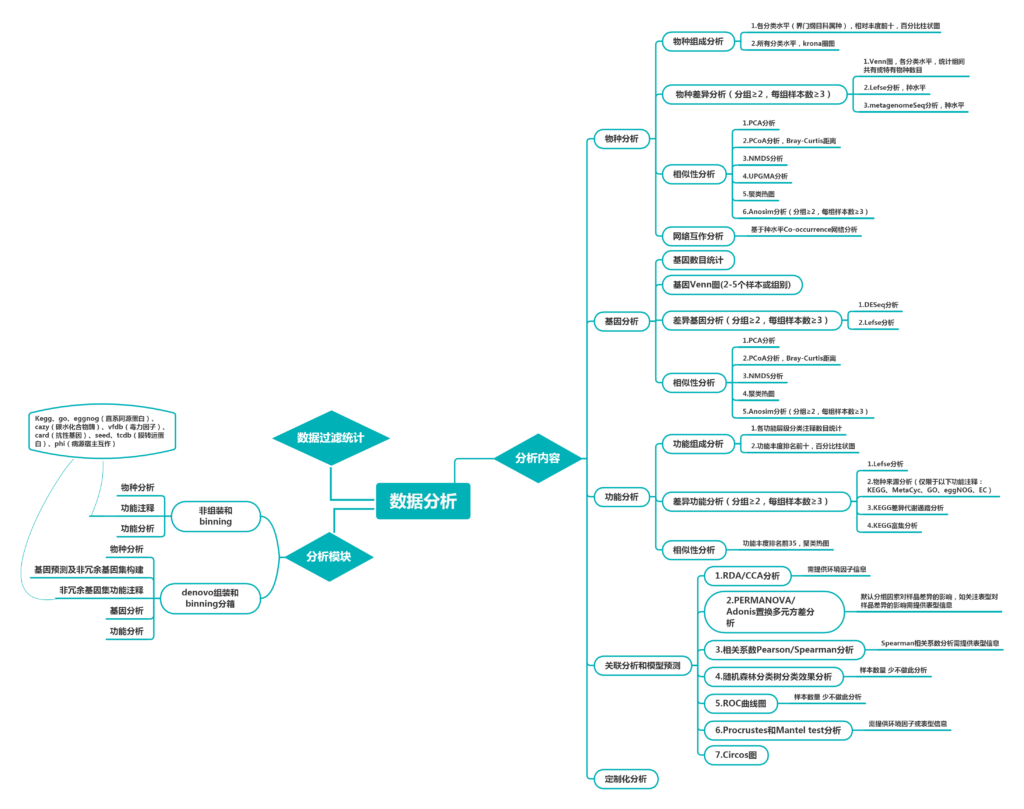
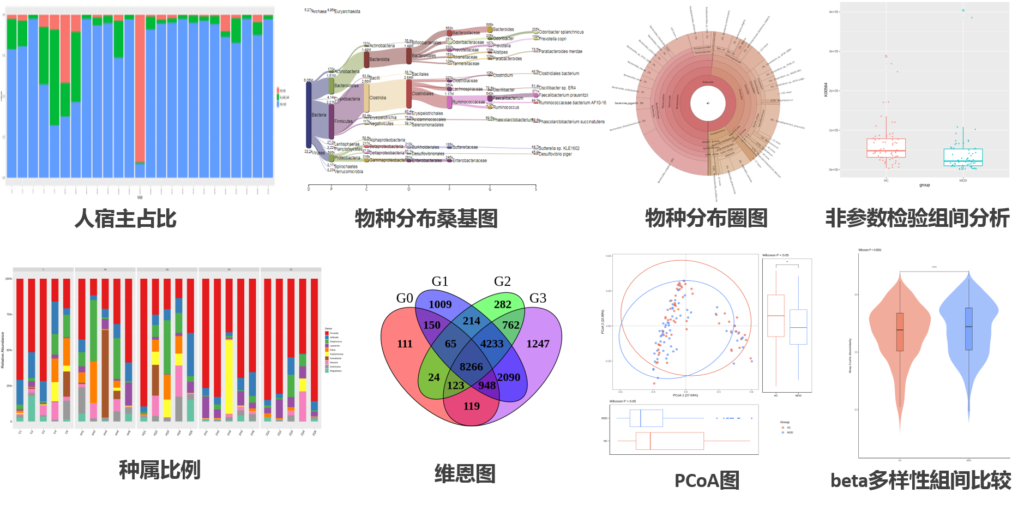
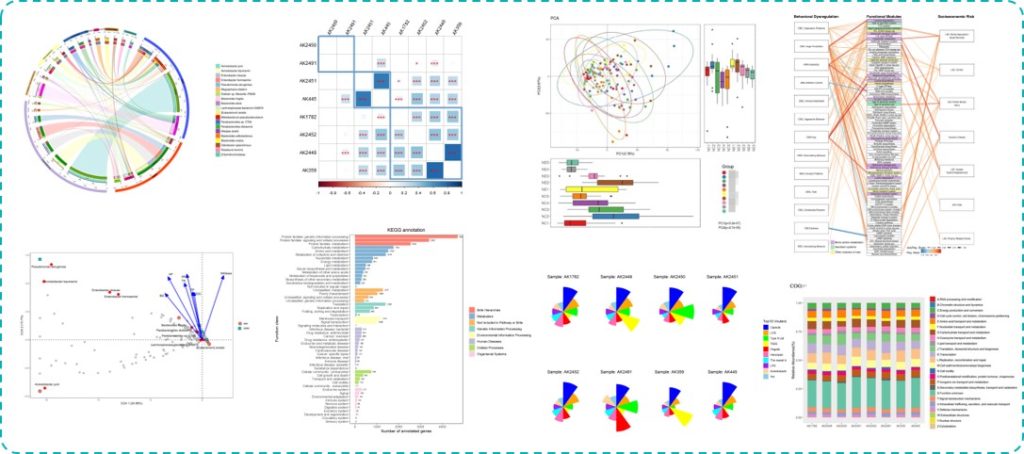
分析内容丰富全面:物种分析,基因预测与分析,多样性和相似性分析,功能分析,网路互作分析,代谢网络,关联分析等
完整详细的报告:提供质检实验报告,分析统计报告,分析报告解读,原始数据
高效个性化服务:在线项目系统方便您及时查看项目动态和下载报告以及与分析人员高效交流,免费支持个性化图表修改以及重新分组出报告。
价格低,周期快:包括提取,测序到分析,最快2周出报告。
大数据分析团队和多中心大项目分析经验(团队主要源自浙江大学,包括生物信息学,计算机,微生物以及统计分析等专业,积累了多年的大健康项目多中心项目分析经验,有助于宏基因组大数据,多样本,多表型,多组学联合分析
兼容性强的合作模式:有专门团队负责,提供切实可行的项目方案,兼顾临床和科研双需求模式。