-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康

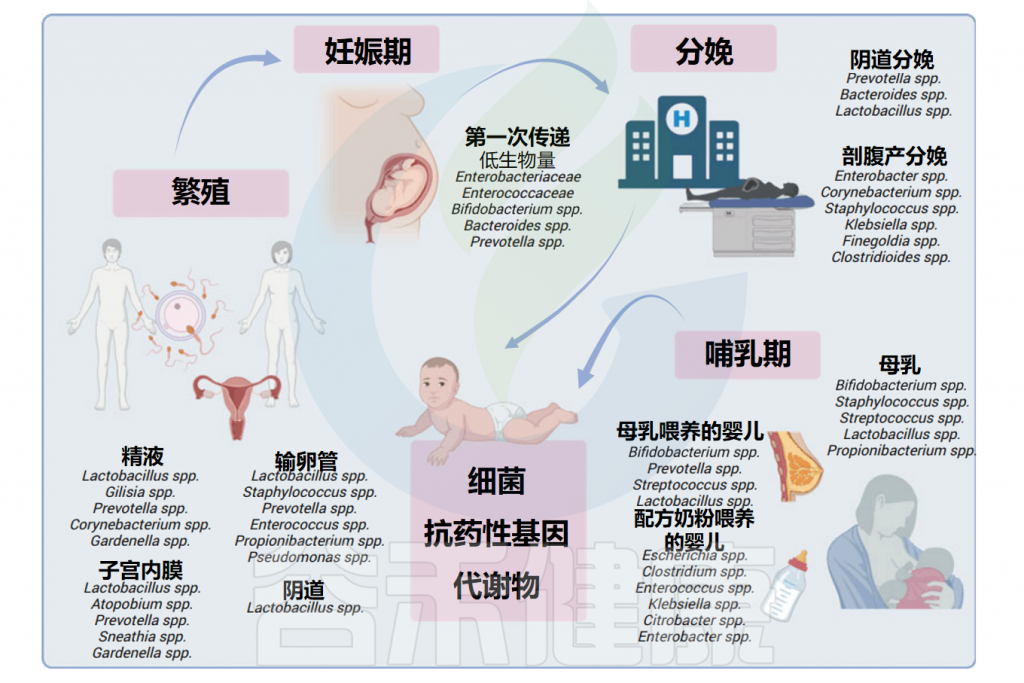
众多的文献和谷禾之前写过的文章都阐述了微生物群与人类健康密切相关。这种联系甚至可能在还未出生的时候就已经开始形成。微生物群的存在和活动从母体怀孕前到幼年的过程中,持续地参与和调节着人体的各种生理功能,从而在每一个生命阶段都扮演着至关重要的角色。
在受孕之前,男性和女性生殖道中的微生物就具有关键作用,它们可以在受孕期间产生影响。例如女性阴道、子宫颈内膜、子宫、输卵管和卵巢中的微生物群,其特征是生物量低,并且与生殖和子宫健康相关。精液微生物群可能会干扰受孕和早产。据报道,怀孕期间典型的精液细菌(如嗜酸乳杆菌)的存在率较高与早产有关,而另一种常见的精液细菌——卷曲乳杆菌(Lactobacillus crispatus)被确定为具有保护作用。
在怀孕期间,母体微生物的变化与所经历的生理和激素变化同步。阴道微生物多样性降低,乳杆菌成员增加,潜在地加强了它们的免疫保护功能,而较低水平的乳杆菌与早产风险增加相关。妊娠三个月后肠道中促炎的微生物更丰富,产短链脂肪酸的微生物减少。同时妊娠期口腔微生物群的特征是活菌数量增加,同时致病菌水平较高。需要注意的是,口腔生态失调与妊娠期糖尿病、先兆子痫和胎儿紊乱的风险增加相关。
此外,母婴间的垂直传播与婴儿健康相关。一些有益菌种(双歧杆菌、乳杆菌)可以从母亲传播到婴儿体内并定植。但与此同时,一些抗生素抗性基因也会随着微生物的转移,从而使新生儿对一些药物具有耐药性,这将不利于一些新生儿疾病的治疗。
因此了解从受孕到生命最初几年期间关键的微生物群非常重要。本文总结了有关人类生殖微生物群的现有证据,以及生命早期的微生物集群,重点是对婴儿发育和健康结果的潜在影响。
研究还发现:包括特定益生菌和其他益生菌混合物在内的一些饮食策略可能会成为潜在的工具,调节母婴微生物群,并在整个生命周期中实现健康结果。
★ 生命早期的微生物群对代谢和免疫非常重要
生命的前1000天(即从怀孕到出生后的前2年)对于先锋微生物在人体内的定植以及免疫系统的发育和成熟都是至关重要的。因此,这一时期被认为是一个“关键时期”,在此期间,任何事件都将对代谢、免疫和微生物行为产生关键影响,从而影响人类健康。
而对逐步形成的新生儿微生物群的任何改变和破坏都有可能在短期和长期增加个体患病的风险和倾向。
▸ 阴道微生物群
母体微生物群是新生儿最相关的产前和产后微生物来源。
阴道微生物群是一个复杂的生态系统,大约由200多种细菌组成,最主要的阴道种群是乳杆菌(Lactobacillus),在阴道中已检测出超过20种乳杆菌,占细菌群落的近70%。以及其他不太丰富的细菌,如普雷沃氏菌属、链球菌属、拟杆菌属和韦荣氏球菌属。
✦ 阴道微生物群主要分为五种类型
阴道微生物群主要分为五种类型,它们在组成和丰度方面都不同,每个类型都会以一种细菌为主导:
卷曲乳杆菌(CSTI),加氏乳杆菌(CSTII),惰性乳杆菌(CSTIII),多元组(CSTIV表现出较低的乳杆菌属丰度)和詹氏乳杆菌(CSTV)。
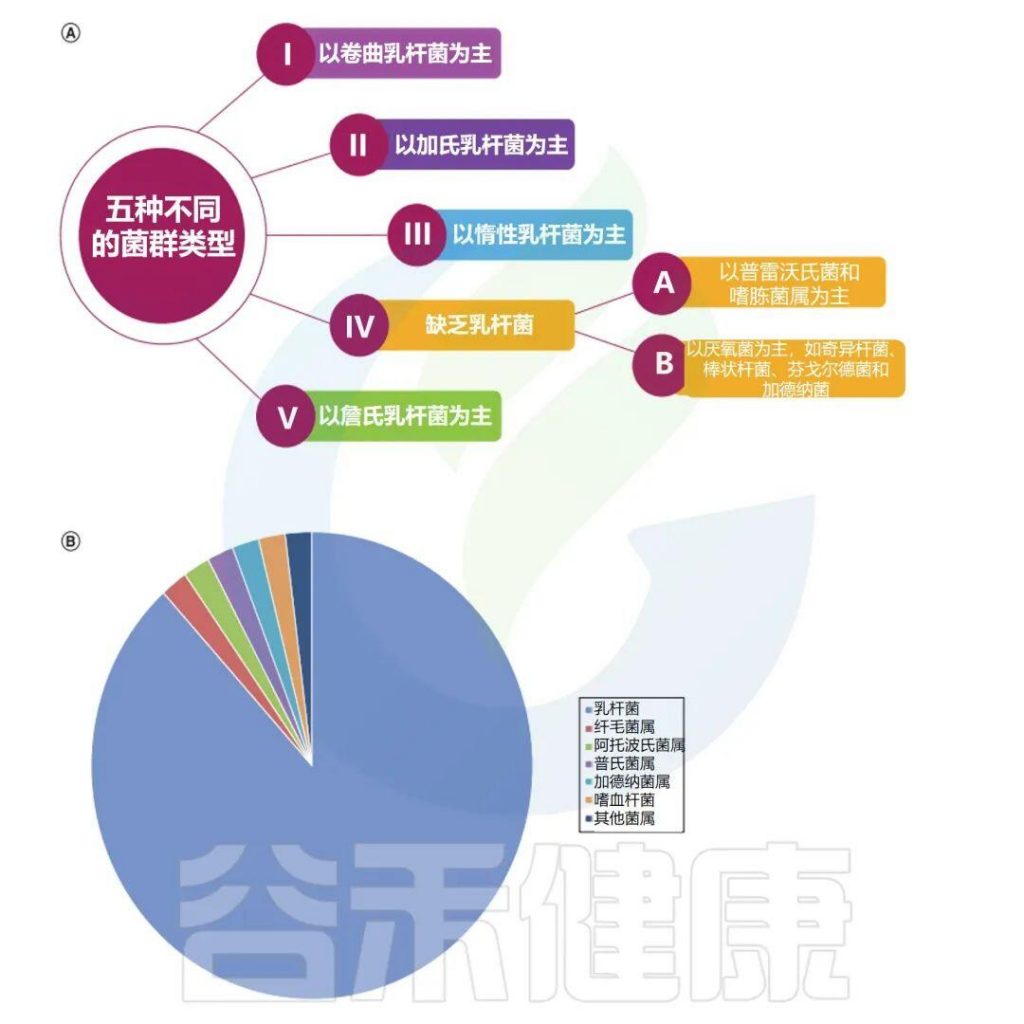
阴道微生物群会因地理位置、种族和卫生条件而异。例如,在美国的非裔美国人和西班牙裔妇女中CSTIV的丰度较高,这些成员在维持低pH值和代谢物分泌中起重要作用,以防止病原体在阴道中定植。
✦ 阴道微生物群会随激素水平、分娩、母乳喂养而变化
越来越多的研究证据表明阴道微生物群是阴道健康的重要生物标志物。然而,阴道微生物群并非一成不变。事实上,阴道微生物群显示出时间动态变化,有些群落发生变化,有些则相对稳定,这取决于核心菌群。

最近一项涉及比利时3345名妇女(18-98岁)的研究显示,阴道微生物群与激素变化(雌激素水平和避孕药的使用)正相关。
相反,阴道微生物群与分娩和母乳喂养等因素呈负相关。此外,同一项研究表明,富含阴道加德纳菌(Gardnerella)、普雷沃氏菌属(Prevotella)和拟杆菌(Bacteroides)的阴道微生物群与更年期、月经卫生和避孕药具使用有关。
▸ 子宫、输卵管的微生物群对生殖健康也有影响
女性生殖系统含有对女性健康有影响的细菌,这些细菌也存在于阴道之外。先前的几项研究表明,女性上生殖道(UFRT)中存在微生物群,包括子宫颈内膜、子宫、输卵管和卵巢,其特征是生物量低,并且与生殖和子宫健康相关。
✦ 子宫内膜也以乳杆菌为主,但与阴道菌群有所不同
子宫内膜微生物群的特征是大量乳杆菌(Lactobacillus),其次是加德纳菌(Gardnerella), 普雷沃氏菌属、Atopobium和Sneathia,这也在阴道中发现。然而,似乎子宫内膜细菌群与阴道中的细菌群有些不同,这表明这两种微生物群是相关的,但并不相同。
✦ 输卵管中的菌群
此外,输卵管中也有一些细菌定居,如乳杆菌属、葡萄球菌属、肠球菌属、普雷沃氏菌属和丙酸杆菌,与假单胞菌属。
▸ 男性生殖道的微生物群
之前很少有人研究男性生殖道中的微生物群。最近的研究显示,精液中存在微生物群,其中芽孢杆菌门(Bacillota)是最主要的门,其次包括拟杆菌门、假单胞菌门和放线菌门,其中最常见的属有乳杆菌属、普雷沃氏菌属、Gillisia、棒状杆菌属和加德纳菌。
✦ 男性的生殖道微生物群对生育健康有一定影响
阴道加德纳菌在阴道微生物群中的优势可能与男性炎症有关,这或与阴道定植/感染有关。有人认为,男性和女性生殖微生物群之间存在联系,影响生育、生殖健康和妊娠。
事实上,之前的大多数关于精液微生物组研究都集中在不孕不育问题上,突出了微生物特征和多样性的差异。据报道,男性微生物群受到包括环境在内的不同因素的影响。此外,精液微生物群可能会干扰受孕和早产。据报道,怀孕期间典型的精液细菌(如嗜酸乳杆菌)的存在率较高与早产有关,而另一种常见的精液细菌——卷曲乳杆菌(Lactobacillus crispatus)的优势已被确定为具有保护作用。
总之,女性和男性的生殖微生物组与生殖健康密切相关。研究显示,微生物可作为生物标志物,有助于开发诊断和治疗不孕症的新工具。
生殖过程发生在“微生物环境”下,这些微生物及其代谢物需得到更多关注,因为它们与孕前阶段密切相关。
在怀孕期间,妇女的生理和代谢有所变化,以便为胎儿提供最佳子宫内环境,促进其健康发育。妊娠期间的内分泌、免疫和代谢变化,会营造一种促炎状态,反映在阴道、肠道和口腔的微生物群变化中。
研究微生物标志物与妊娠结局之间的关系,有助于预防和调整与妊娠并发症相关的不利微生物变化。
▸ 阴道微生物群
据报道,怀孕后阴道微生物多样性降低,而乳杆菌成员增加,潜在地加强了它们的保护功能。
✦ 较低水平的乳杆菌与早产风险增加相关
就个别物种而言,卷曲乳杆菌与来自足月妊娠的样本相关。进一步证明,病史包括反复尿路感染的妇女表现出早产风险增加。在足月分娩和早产分娩之间,已经观察到阴道微生物群在组成、稳定性和多样性方面的差异。
加特纳菌(Gardnerella)、Atopobium和Uraplasma spp.的存在,以及较低水平的乳杆菌成员或较高水平的白色念珠菌的存在,已被发现与早产风险有关。
因此,在妊娠期间检测阴道微生物组内的异常可以用作预测早产可能性的微生物生物标志物,特别是在妊娠前三个月检查阴道微生物组。

最近的一项研究报告称,无抗生素的阴道微生物群移植(VMT)和供体移植解决了复发性流产后的阴道生态失调问题。这些研究表明,VMT是一种潜在的治疗具有不良生育和妊娠结局的严重阴道生态失调的方法,然而,还需要进一步的研究。
▸ 肠道微生物群
虽然在妊娠的前三个月期间,肠道微生物群的组成保持稳定,并且类似于孕前微生物群,但是从妊娠的前三个月结束起,其组成发生了根本的变化。
✦ 妊娠三个月后促炎的微生物更丰富,产短链脂肪酸的微生物减少
这些变化反映了更具促炎性的特征,包括妊娠晚期假单胞菌门(肠杆菌科成员)和放线菌门(主要是双歧杆菌成员)的增加,以及短链脂肪酸(SCFA)生产者的减少。
肠道微生物群与多种妊娠表型有关,包括体重增加、轻度炎症和胰岛素抵抗。
✦ 怀孕期间的饮食、营养、抗生素的使用会影响后代的微生物
已经有研究广泛证明,怀孕期间暴露于抗生素会改变母亲的微生物生态系统,从而改变后代的微生物生态系统。怀孕期间使用抗生素会增加阴道内葡萄球菌物种,此外,肠道以外的其他区域的潜在微生物转移可能与过敏和肥胖的风险增加有关。
此外,微生物群在怀孕期间受到母亲的饮食和营养状况的影响,也对其后代的微生物群组成产生影响。人体研究表明,妊娠期女性肠道微生物群的变化易受孕前母体最大体重指数(身体质量指数)的影响,也易受妊娠期体重增加的影响。较低的双歧杆菌属物种在超重和肥胖的母亲以及在怀孕期间体重增加过多的母亲中观察到。
另一项研究报告了怀孕期间体重状况的类似变化和较低水平的拟杆菌物种,以及更丰富的葡萄球菌和大肠杆菌物种在超重孕妇中被发现。
母体肠道微生物群的这种变化可能与通过阴道分娩出生的新生儿的肠道定植过程的差异有关。
▸ 口腔微生物群
怀孕还会引起口腔微生物群的变化。比较孕妇和非孕妇口腔中几种物种的丰度时,已经报道了显著的差异。
✦ 怀孕时口腔中的微生物数量增加,致病菌水平也较高
妊娠期口腔微生物群的特征是活菌数量增加,同时致病菌水平较高,例如牙龈卟啉单胞菌(Porphyromonas gingivalis),Aggregatibacter actinomycetemcomitans和假丝酵母(Candida genus)。
✦ 口腔生态失调与妊娠期并发症存在一定关联
越来越多的证据表明口腔感染和与妊娠并发症(包括早产)之间的联系。还有证据表明,妊娠期口腔生态失调与妊娠期糖尿病、先兆子痫和胎儿紊乱的风险增加相关。
这些数据表明,口腔微生物群在孕产妇和新生儿结局中也起着关键作用,包括妊娠并发症、早产风险和早期胎儿/新生儿微生物定植。
越来越多的证据表明了细菌从母亲到孩子的垂直传播。垂直传播是一个显著的过程,其中细菌菌株在母亲和新生儿之间交换。
研究人员探索了各种母体菌群来源,包括粪便、母乳、阴道微生物群和皮肤,以揭示这些关键细菌菌株的来源。影响婴儿肠道定植的关键因素包括出生时的胎龄、分娩方式、产前暴露、遗传、抗生素的使用和是否纯母乳喂养。
▸ 特定的垂直传播——例如双歧杆菌
双歧杆菌属(Bifidobacterium)是从母亲传播到婴儿肠道的最常见的微生物,主要传播源是母乳和粪便,出生时的胎龄、出生地、分娩方式和抗生素使用等变量可能会影响双歧杆菌的状态。
✦ 双歧杆菌等菌属从出生时自母亲传播到婴儿体内并定植
已知胃肠道微生物的早期正常定殖有助于免疫系统的发育,并有助于通过调节环境来维持体内平衡。

一项纵向研究表明长双歧杆菌(B.longum)和B. Brief的菌株似乎从出生到4个月一直存在于婴儿的微生物生态系统中。
通过宏基因组测序,我们深入了解了母体肠道到婴儿肠道的传播,外部因素的影响——地点、出生模式和抗生素的使用,每个因素都在形成婴儿微生物群中发挥作用。
双歧杆菌属物种表现出持久性和适应性。与母乳中发现的生物活性化合物(如母乳低聚糖)一起,在支持婴儿肠道微生物群和免疫系统的成熟方面发挥着至关重要的作用。
▸ 抗生素抗性基因转移
双歧杆菌等有益菌可以由母亲传递给婴儿并定植,与此同时,一些带有抗生素抗性基因的微生物也在母体到婴儿之间转移。
抗生素抗性微生物(ARMs)分布广泛,在人、动物之间传播,并通过动物性食物传播。它们携带抗生素抗性基因(ARG),整个细菌群体中的ARG组被称为抗性组。
✦ 耐药病原体可能从母体转移至新生儿体内
生命早期,尤其是围产期和产后,是婴儿肠道微生物群和免疫力发育的关键时期。此外,婴儿的免疫系统尚未成熟,因此他们特别容易获得和携带耐药病原体。这导致每年估计有214000名新生儿因抗生素抗性病原体引起的脓毒性感染而死亡。
婴儿肠道中抗生素耐药微生物菌株的主要来源尚不确定;虽然母体微生物群是主要来源,但其他个体和环境也是相关的,并可能导致婴儿耐药性。微生物在出生、哺乳和密切接触期间从母亲传播给后代。
✦ 母乳喂养、剖腹产等因素影响抗性基因的转移
各种围产期因素,如剖腹产、抗生素暴露、早产和母乳喂养会影响微生物从母亲到婴儿的垂直传播。一些研究阐明了抗生素抗性基因(ARG)在新生儿出生后不久就存在,表明ARMs的最初定殖发生在出生后不久,可能来自与母亲或医院环境的接触。一些研究表明,母亲向婴儿传播耐药细菌,包括各种抗生素耐药菌株,如大肠杆菌, 链球菌等。
众所周知,母乳喂养对婴儿健康有益,为婴儿提供营养和细菌。然而,对于发育中的婴儿来说,母乳似乎是耐药细菌和精氨酸的来源之一。在人乳中发现了耐药菌株。宏基因组分析揭示了母乳中高水平的精氨酸和遗传元素,类似于婴儿对母乳的抵抗力。这表明母亲通过抗性细菌的转移在发展婴儿的肠道抗性中起作用。
需要更多的研究来了解耐药细菌在母体与婴儿间的垂直传播,以理解其对婴儿最初肠道定植的影响。这种更深入的理解将有助于开发控制抗生素耐药基因在母婴之间传播的策略。
在出生时,除了来自环境的微生物之外,新生儿还会遇到来自阴道和母体肠道的数量巨大、种类繁多的微生物。
新生儿中的微生物群定植是一个与健康密切相关的敏感、动态过程。早期多样的微生物建立和特定短链脂肪酸的产生对免疫系统的成熟至关重要。
▸ 最早的新生儿微生物群
✦ 母体与婴儿之间的微生物接触最早可能始于子宫收缩时
不同的研究报告了通过培养依赖性和独立性方法确定的第一次粪便中存在细菌,即胎粪。一些研究人员认为,微生物接触可能最早始于子宫收缩和胎膜破裂期间,从而解释了胎粪中细菌的存在。
首个微生物群由变形菌门(Pseudomonadota)和芽孢杆菌门(Bacillota)的物种组成,如肠杆菌科(Enterobacteriaceae)和肠球菌科(Enterococcaceae)。
此外,还发现了其他菌属,包括双歧杆菌属、拟杆菌属和普雷沃氏菌属。与7天新生儿微生物群相比,首次通过的胎粪微生物群显示出更高的微生物多样性和丰富性,这表明新生儿出生时大量接触微生物,但只有少数能够在肠道中定植并持续存在,以便在出生后的第一年逐步进化。
▸ 影响新生儿微生物群的因素
肠道微生物群会随时间进化,变得更加多样。因此,新生儿定植是一个脆弱的、动态的和逐步的过程,它会受到许多母婴因素以及环境因素的影响。
其中,最相关的因素包括:孕龄、分娩方式(阴道分娩与剖腹产)、地点(医院与家庭)、喂养方式(母乳、混合膳食或婴儿配方奶粉)以及产妇健康状况、遗传、药物、接触异生物质或物理化学制剂的环境、兄弟姐妹、宠物以及人与人之间的水平微生物传播等。
✦ 抗生素的使用

分娩期抗生素预防可能会改变出生后第一周的肠道微生物群,包括放线菌的减少以及假单胞菌属和杆菌属比例的稳步增加。
另一项研究报道,分娩期间施用的抗生素也可以改变新生儿的口腔微生物群,从而导致在抗生素治疗后假单胞菌门的丰度更高。这可能会对儿童发育产生重要影响。
此外,据观察,在妊娠中期和晚期接受抗生素治疗的儿童患肥胖症的风险增加了84%,这可能是由于肠道微生物的变化引起的。
✦ 早产影响婴儿的肠道微生物群平衡
此外,有几项研究报道早产婴儿的微生物群是不平衡的。例如,已有研究表明,与足月婴儿相比,早产胎粪微生物组导致无菌小鼠粪便移植后免疫功能改变、生长受限和激素水平变化。
然而,早产儿有其他合并症,因为他们大多数是通过剖腹产出生的,在出生时和住院期间暴露于抗生素;他们也用母乳喂养,并在新生儿重症监护室呆了更长时间,这些因素可能都会影响早产婴儿的肠道微生物群。
✦ 母亲的健康状况也会影响婴儿的微生物群
也有越来越多的证据表明,子宫内炎症导致围产期并发症增加。婴儿健康和微生物群的母婴传播之间的关联可能受到诸如母亲糖尿病等因素的驱动、特应性疾病、饮食,身体质量指数和围产期抗生素使用。
研究人员观察到患有妊娠期糖尿病(GDM)前期的母亲所生婴儿的胎粪中含有丰富的细菌,这些细菌通常在成年糖尿病患者中观察到。母亲的高脂肪饮食影响婴儿微生物群,表现为拟杆菌属显著减少。
此外,婴儿微生物群影响免疫系统发育的分子机制尚未完全了解,尽管有越来越多的证据表明肠道微生物群在儿童期和成年期的疾病和健康中具有免疫调节活性。
✦ 顺产的婴儿微生物群与母亲肠道微生物更相似
先前已经证明,阴道出生的新生儿具有类似于阴道和肠道母体微生物群的微生物群,富含乳杆菌和普雷沃氏菌,尽管也存在其他细菌,如肠杆菌科,包括埃希氏菌属或克雷伯菌属。
而通过剖腹产出生的新生儿呈现出与口腔、皮肤和环境微生物群相似的独特微生物群。母体肠道似乎也是早期定植细菌的重要来源,因为在经阴道分娩的新生儿中,已发现72%的肠道细菌来自母体肠道,相比之下,经剖腹产出生的受试者中只有41%来自母体肠道,已显示双歧杆菌属在新生儿肠道定植过程中作为先锋细菌的成员,剖腹产出生的婴儿表现出拟杆菌属的延迟定植和多样性降低。
✦ 母乳喂养有助于母亲的微生物群在婴儿中定植
出生后,母乳喂养既支持了微生物的肠道定殖,又促进了免疫系统的成熟。据报道,母乳喂养和配方奶粉喂养的新生儿微生物群之间存在差异。
此外,母乳喂养的停止,而不是补充和固体食物的引入,对婴儿微生物群产生了关键影响。传统上,母乳被认为是无菌的。尽管如此,证据表明,母乳样本中存在活微生物,包括葡萄球菌和链球菌,是最丰富的微生物群,其次是乳酸菌、双歧杆菌和某些假单胞菌(Pseudomonas和Acinetobacter)。

最近的一项研究表明,婴儿肠道中27.7%的细菌来自母乳,10.3%来自乳晕皮肤,从而突显了母乳喂养对婴儿肠道微生物群的潜在影响。
此外,母乳和婴儿肠道之间共享的微生物和病毒种类也有被发现。
饮食是塑造我们肠道微生物群的一个关键因素。因此,建议进行饮食和营养咨询以及营养补充,以便在孕前和妊娠阶段达到适当的营养状态。最近有报道称,怀孕期间以及随后的母乳喂养期间,母亲的饮食对母亲和婴儿的微生物群都有重要影响。
★ 地中海饮食有助于改善母婴的健康水平
母亲的蛋白和纤维摄入量会影响母乳成分,如微生物群、脂质、母乳低聚糖(HMO)和microRNA表达谱。地中海饮食(MD)被证明在怀孕期间能够改善母婴健康结果,包括生长受限、胎儿和新生儿大脑发育、儿童神经发育。
妊娠期地中海饮食,主要是水果、蔬菜和豆类的摄入量增加,红肉的摄入量减少,与Acidaminoacaeae家族水平以及短链脂肪酸生产者的丰度较高有关。
植物蛋白、纤维和多酚的摄入量增加与瘤胃球菌属(Ruminococcus)、Christenselacae科、Dehalobacterium和真/优杆菌属(Eubacterium)以及短链脂肪酸生产菌的存在增加有关。
✦ 饮食不仅改善肠道微生物,对阴道、口腔微生物也有影响
大多数关于饮食-怀孕-微生物群的可用数据都集中在肠道微生物群上,但据报道,增加水果、维生素D、纤维和酸奶的摄入量与女性阴道卷曲乳杆菌(L.Crispatus)有关;纤维摄入与较高的异质性相关。
有限的证据表明饮食对阴道微生物群和妊娠结局的影响。在这种情况下,仍需确定哪些食物、食物成分、营养素和其他膳食化合物会影响围产期妇女的微生物群(肠道、口腔和阴道),以及对母婴结局的潜在影响。
对围产期饮食干预的研究兴趣日益增加,以及在怀孕期间使用益生菌、益生元和共生体来调节微生物群和促进“有利的微生物垂直传播”。有一些证据表明,在怀孕和哺乳期间服用益生菌可以起到保护作用,而益生元和其他益生菌的证据较少。
值得注意的是,脆弱和敏感人群需要特殊的安全性考虑,包括安全性和与妊娠、哺乳和婴儿期相关的不良反应的潜在报告。
▸ 益生菌对生育能力的影响
补充乳杆菌有利于重建阴道菌群平衡
阴道感染会影响乳杆菌的平衡,导致可能影响生育能力的细菌的生长。因此,补充乳杆菌应有助于重建平衡。
一项涉及健康女性的体外受精研究显示,在阴道定植卷曲乳杆菌(L.Crispatus)后,受孕结果很有希望。
补充益生菌有助于提高精子质量
在不孕症和辅助生殖医学的病例中,已报道了精液微生物群组成的差异,使用益生菌可能在精液特征和质量参数方面具有有益的效果。研究已经显示出Lacticaseibacillus rhamnosus CECT8361和B.longum CECT7347在弱精子症男性中,补充为期六周可以改善精子质量参数和活力,并降低DNA片段和活性氧(ROS)水平。
另一方面,有报道称,共生菌(Flortec, Lacticaseibacillus paracasei B21060 和阿拉伯半乳聚糖、低聚果糖和l-谷氨酰胺)对少弱畸形精子症患者精液质量和数量具有一定的改善作用。
此外,系统综述和荟萃分析表明,益生菌有利于精子活力。然而,关于益生菌对接受辅助生殖的女性的功效,存在相互矛盾的证据。这表明需要更多临床研究来证明益生菌对生育能力的影响。
▸妊娠期使用益生菌降低疾病风险
孕期服用益生菌为减少母婴健康风险提供了方案。现有研究主要集中在早产风险、减少生殖器感染、妊娠期糖尿病(GDM)和先兆子痫前期等方面,这些往往被视为次要健康结果。
益生菌有助于改善孕妇生殖器感染
孕妇经常患有复发性尿路感染(UTIs)、外阴阴道念珠菌病(VVC)和细菌性阴道病(BV)。这些问题与不利的母婴妊娠结果有关,包括早产风险和胎儿生长受限。益生菌已被提出作为治疗BV和UTI的预防性替代品,通过维持或恢复阴道微生物群或防止病原体在尿道定植。
先前的研究表明,益生菌组合,即嗜酸乳杆菌、乳双歧杆菌(B.lactis)和长双歧杆菌(B.longum),对孕妇B组链球菌(GBS)定植具有潜在的治疗作用。

此外,最近的研究表明益生菌菌株在治疗妊娠期GBS感染方面的有效性,其中一项临床研究报告称,补充鼠李糖乳杆菌GR-1和罗伊氏乳杆菌RC-14被证明对治疗GBS感染有效。
最近一项涉及36名GBS阳性女性的研究报告称阴道共生菌显著增加,对阴道GBS有影响。而一项对18项研究的系统综述和荟萃分析表明,抗生素和益生菌显著降低了细菌性阴道病(BV)的复发率,提高了治愈/缓解率。其他报告益生菌对阴道炎影响的研究也显示了类似的结果。
✦ 降低先兆子痫的风险

最近一项涉及70149名孕妇的研究发现,益生菌(嗜酸乳杆菌[LA-5],乳双歧杆菌[Bb12]还有鼠李糖乳酸杆菌GG [LGG])显著降低先兆子痫的风险。
这项研究还强调了“时机”(即何时进行益生菌干预)的相关性,因为在怀孕前或怀孕早期食用益生菌没有观察到任何影响。孕晚期(而非孕前或孕早期)摄入益生菌牛奶与降低先兆子痫风险显著相关(调整后的 OR:0.80(95% CI 0.68 至 0.94)p 值:0.007)。孕早期(而非孕前或孕晚期)摄入益生菌与降低早产风险显著相关(调整后的 OR:0.79(0.64 至 0.97)p 值:0.03)。
然而另一项系统综述报告称,与安慰剂相比,益生菌会导致肥胖受试者的先兆子痫风险增加。肥胖与先兆子痫的风险较高有关。因此,需要更多的研究来确定弱势群体中与益生菌相关的风险。
预防妊娠期糖尿病,改善胰岛素敏感性
鼠李糖乳杆菌(L.rhamnosus)GG和乳双歧杆菌(B.lactis)BB12与饮食相结合已被证明可以预防妊娠期糖尿病(GDM),降低血糖浓度和改善胰岛素敏感性。
另一种益生菌菌株,鼠李糖乳杆菌(L.rhamnosus) HN001菌株能够降低GDM的患病率,特别是在中老年妇女和那些以前患有妊娠期糖尿病的妇女中。
最近的一项系统综述包括30多项在怀孕期间进行的研究,表明益生菌干预对改善血糖控制、降低低密度脂蛋白胆固醇(VDL)和甘油三酯以及减少炎症标记物的潜在作用。
益生菌补充剂可能会降低妊娠期糖尿病(GDM)的发病率,并有助于控制健康孕妇和超重和肥胖妇女的血糖参数。
最近的荟萃分析显示,益生菌将GDM发病率降低了33%,当使用多种菌株益生菌组合时,效果更好。一项系统综述和荟萃分析还报告称,摄入酸奶(活微生物)可以降低GDM的风险,并降低空腹血糖。
减少产后压力和抑郁
越来越多的数据显示益生菌对抑郁、焦虑和情绪的潜在用途。一项研究(系统综述和荟萃分析)显示,益生菌在减少孕妇和哺乳期妇女的焦虑和抑郁方面具有积极作用。
其他研究报告了益生菌和对照组在抑郁评分方面没有差异,然而,在总体精神健康评分方面,报告了焦虑的降低。
▸ 出生后使用益生菌的有益作用
文献中有数据和研究表明特定益生菌的有益作用,例如布拉氏酵母菌,鼠李糖乳酸杆菌GG(LGG),罗伊氏乳杆菌和动物乳双歧杆菌(Bb12)。
婴儿早期使用益生菌的主要目标是:
1)预防腹泻和感染;
2)预防过敏;
3)预防坏死性小肠结肠炎和早产儿迟发性败血症、预防婴儿绞痛。
此外,一项系统综述强调了灭活益生菌的潜在益处,灭活益生菌在生命的早期和敏感期以及敏感人群是一种安全的替代品,如早产儿和儿科人群。
产后使用益生菌的另一个例子与患有乳腺炎的女性有关,包括感染性乳腺炎和葡萄球菌乳腺炎。
在探索微生物群与人类健康之间的复杂关系时,我们发现,从受孕到生命最初几年,微生物群在每一个生命阶段都扮演着不可或缺的角色。母体的微生物环境不仅影响着怀孕过程中的生理变化,还直接关系到婴儿的健康和发育。通过了解这些微生物的作用,我们可以更好地认识到早期干预的重要性,尤其是通过饮食和生活方式的调整来优化母婴微生物群。
随着科学研究的不断深入,我们有望开发出更有效的策略,以促进母婴健康,预防潜在的健康问题。未来的研究将继续揭示微生物群的奥秘,帮助我们在整个生命周期中实现更好的健康结果。
主要参考文献
Samarra A, Flores E, Bernabeu M, Cabrera-Rubio R, Bäuerl C, Selma-Royo M, Collado MC. Shaping Microbiota During the First 1000 Days of Life. Adv Exp Med Biol. 2024;1449:1-28.
Arzamasov AA, Nakajima A, Sakanaka M, Ojima MN, Katayama T, Rodionov DA, Osterman AL (2022) Human milk oligosaccharide utilization in intestinal Bifidobacteria is governed by global transcriptional regulator NagR. mSystems 7(5):e00343–e00322.
Calvo-Lerma J, Selma-Royo M, Hervas D, Yang B, Intonen L, González S, Martínez-Costa C, Linderborg KM, Collado MC (2022) Breast milk lipidome is associated with maternal diet and infants’ growth. Front Nutr 9:854786.
Canha-Gouveia A, Pérez-Prieto I, Rodríguez CM, Escamez T, Leonés-Baños I, Salas-Espejo E, Prieto-Sánchez MT, Sánchez-Ferrer ML, Coy P, Altmäe S (2023) The female upper reproductive tract harbors endogenous microbial profiles. Front Endocrinol 14:1096050.
Mikami K, Kimura M, Takahashi H (2012) Influence of maternal Bifidobacteria on the development of gut Bifidobacteria in infants. Pharmaceuticals 5(6):629–642.
Garcia-Mantrana I, Selma-Royo M, Gonzalez S, Parra-Llorca A, Martinez-Costa C, Collado MC (2020) Distinct maternal microbiota clusters are associated with diet during pregnancy: impact on neonatal microbiota and infant growth during the first 18 months of life. Gut Microbes 11(4):962–978.
García-Mantrana I, Bertua B, Martínez-Costa C, Collado MC (2016) Perinatal nutrition: how to take care of the gut microbiota? Clin Nutr Exp 6:3–16.
Li W, Tapiainen T, Brinkac L, Lorenzi HA, Moncera K, Tejesvi MV, Salo J, Nelson KE (2021) Vertical transmission of gut microbiome and antimicrobial resistance genes in infants exposed to antibiotics at birth. J Infect Dis 224(7):1236–1246.

谷禾健康
认识免疫疗法
免疫疗法是一种利用人体自身的免疫系统来对抗疾病的方法,尤其是在癌症治疗中,它通过激活或增强免疫系统来识别和攻击癌细胞。这一疗法的核心在于激发免疫系统对肿瘤细胞的特异性反应,从而达到治疗目的。
免疫疗法具有高度的选择性和靶向性,能够减少对正常细胞的伤害,降低传统化疗和放疗的副作用。免疫疗法的种类多样,包括单克隆抗体、免疫检查点抑制剂、肿瘤疫苗、过继细胞疗法等。
肠道微生物群与癌症治疗之间的关联已经成为癌症研究领域的热点。多项研究表明,肠道微生物群的组成和功能与癌症患者的免疫疗法反应性密切相关。肠道微生物群不仅在维持肠道健康中起着关键作用,而且在调节全身免疫反应、影响药物代谢和毒性反应中也扮演着重要角色。例如,某些特定的肠道细菌在调节免疫检查点抑制剂的疗效中起到关键作用,而抗生素的使用可能会干扰这一过程,影响治疗效果。
本文我们来了解肠道微生物组对 ICI (免疫检查点抑制剂) 疗效的影响,讨论了微生物组与先天和适应性免疫细胞相互作用以改善 ICI 反应的机制,微生物组衍生的代谢物和分子介导的抗肿瘤免疫反应对 ICI 的机制,同时也包括操纵肠道微生物组以提高 ICI 疗效的治疗策略和正在进行的临床试验。
•
首先了解下关于免疫疗法相关的一些关键知识和问题:
免疫疗法利用人体自身免疫系统对抗疾病主要通过以下几种方式:
免疫检查点抑制剂(ICI):
免疫检查点抑制剂通过阻断 PD-1 或 CTLA-4 等蛋白质,解除对免疫细胞的抑制,使免疫系统能够更有效地识别和攻击肿瘤细胞。
过继性细胞免疫治疗:
从患者体内提取免疫细胞(如 T 细胞),在体外进行基因修饰或培养扩增,使其具有更强的识别和攻击肿瘤细胞的能力,然后再回输到患者体内,发挥抗癌作用。
例如,CAR-T 细胞疗法就是通过对T细胞进行基因改造,使其表面表达能够特异性识别肿瘤抗原的嵌合抗原受体(CAR),从而增强对肿瘤细胞的杀伤能力。
肿瘤疫苗:
包括预防性疫苗(如预防某些病毒感染引起的癌症,如 HPV 疫苗预防宫颈癌)和治疗性疫苗。治疗性疫苗通过向患者体内引入肿瘤相关抗原,刺激免疫系统产生针对肿瘤细胞的特异性免疫反应。
单克隆抗体治疗:
利用人工制备的单克隆抗体特异性地结合肿瘤细胞表面的靶点,直接杀伤肿瘤细胞或通过标记引导免疫系统攻击肿瘤细胞。
总之,免疫疗法旨在激发、增强或调节人体自身的免疫系统,使其能够更有效地识别和消除异常细胞,从而达到治疗疾病的目的。
✦✧✦
免疫功能是一把双刃剑,过强或过弱都会危害人体健康,因此,需要对患者进行免疫功能的监测和评估。
人体免疫可以从三个层面进行探讨:
在免疫细胞中,淋巴细胞谱系与髓样细胞谱系,各占半壁江山,共同抵抗外界对人体的侵袭。
淋巴细胞是构成免疫系统的主要细胞类别,占外周血白细胞总数的 20%-45%,成年人体内约有 1012 个淋巴细胞。
淋巴细胞可分为许多表型与功能均不同的群体,如 T细胞、B细胞、NK细胞等,T 细胞和 B 细胞还可进一步分为若干亚群。淋巴细胞不同亚群的比例、数量以及功能会直接影响机体的免疫状态。
一,T淋巴细胞(简称T细胞)
起源于骨髓造血干细胞,在胸腺素及胸腺微环境影响下分化成熟为T细胞,故T细胞又称胸腺依赖性淋巴细胞。
T 细胞主要参与细胞免疫,表达CD3抗原,其中,T 细胞又包括辅助T细胞(Th)和细胞毒性T细胞(Tc),它们分别表达 CD4 和 CD8。
T淋巴细胞亚群的临床意义
它能反映机体当前的免疫功能、状态和平衡水平,并可以辅助诊断某些疾病,对分析发病机制,观察疗效及检测预后都有重要意义。
例如:CD4+/CD8+ 比值是指在血液中,CD4+ T细胞的数量与CD8+ T细胞数量的比率。这个比值对于评估免疫系统的健康状况非常重要。

CD4+/CD8+的其他临床应用场景
——器官移植
跟移植前相比,器官移植后CD4+/CD8+明显增加,则可能发生排斥反应。
——肿瘤病人
在肿瘤病人外周血中T淋巴细胞亚群数值都有异常,其特征是患者体内 CD3+细胞、CD4+细胞明显减少,而 CD8+细胞明显增加;CD4+/CD8+比值显著降低,说明肿瘤患者的细胞免疫功能处于免疫抑制状态,患者对识别和杀伤突变细胞的能力下降,形成了肿瘤的生长转移。
——再生障碍性贫血与粒细胞减少症
患者的外周血 CD4+细胞数减少,CD8+细胞数增多,CD4+/CD8+比值明显下降。
二、B/NK 淋巴细胞的临床意义
B 细胞主要参与体液免疫,表达 CD19 抗原;NK 细胞表达 CD16 和/或 CD56,在机体中不依赖抗原刺激自发地发挥细胞毒效应。淋巴细胞在免疫应答中起核心作用。
NK 细胞(CD3-CD16+和/或CD56+)能够介导对某些肿瘤细胞和病毒感染细胞的细胞毒性作用。
注:以上内容参考自 赛欧细胞
2007~2013 年
小鼠研究表明肠道菌群可以刺激抗肿瘤免疫反应。
2015年
两项临床前小鼠研究首次将肠道菌群与 ICI 反应联系起来。
2018 年
小鼠和人类研究表明,肠道菌群的组成和多样性可以预测对 ICI 免疫疗法的反应。将对 ICI 有反应的患者的粪菌移植到无菌或抗生素治疗的小鼠中,可以改善肿瘤控制并改善对 ICI 的反应。
2019~2020 年
前瞻性研究证实,非小细胞肺癌(NSCLC)、肝细胞癌 (HCC)、黑色素瘤、肾细胞癌 (RCC) 患者的肠道菌群与 ICI 结果之间存在显著相关性。
在非小细胞肺癌 (NSCLC) 和肾细胞癌 (RCC) 中,细菌多样性较高的患者对抗 PD-1 疗法更敏感。对ICI 无反应者进行 FMT 后口服补充Akkermansia muciniphila 可恢复抗 PD-1 治疗反应 。
在黑色素瘤患者中,肠道菌群的多样性和组成与抗 PD-1 治疗反应呈正相关。大多数情况下,对 ICI 有反应的患者肠道中粪杆菌和瘤胃球菌科的丰度较高,其外周CD4 + T 细胞和 CD8 + T 细胞数量增加。另一项针对转移性黑色素瘤患者研究表明,长双歧杆菌、产气柯林斯菌(Collinsella aerofaciens)、屎肠球菌(Enterococcus faecium)在有反应患者的基线粪便中更为丰富。
回顾性研究表明,抗生素与生存率下降和对 ICI 的反应减弱有关,支持抗生素引起的菌群失调与 ICI 疗效不佳之间存在因果关系。
2021 年
两项临床试验发现,ICI反应者的FMT联合抗PD-1疗法克服了黑色素瘤患者对 PD-1 阻断的耐药性。
肠道菌群和 ICI 疗效的时间表
doi:10.1186/s13045-022-01273-9
以上是肠道菌群对调节免疫检查点抑制剂(ICI)免疫治疗外,肠道菌群还可以影响细胞免疫转移(ACT)免疫治疗、CpG寡核苷酸(CpG-ODN)免疫疗法和基于细胞的免疫疗法。
ACT疗法
抗生素暴露降低了小鼠的ACT疗法疗效,而通过给予细菌脂多糖(LPS)来影响治疗效果,通过toll样受体4信号(TLR)传导恢复了治疗效果。另一项研究表明,肠道菌群通过增加CD8α+ DC的丰度和上调IL-1来维持ACT疗法的治疗效果。
注:过继性细胞转移(Adoptive Cell Transfer, ACT)疗法,是一种利用患者自身的免疫细胞进行癌症治疗的方法。它涉及从患者体内分离出T细胞,然后在体外进行改造,使其能够识别并攻击肿瘤细胞,最后再输回患者体内。包括TIL、TCR-T以及CAR-T等几种治疗方法。
CpG-ODN免疫疗法
在CpG-ODN免疫疗法中,肠道菌群激活TLR4,直接或间接启动肿瘤相关骨髓细胞对CpG-ODNs的TLR9依赖反应。CpG-ODN的有效性在无菌和暴露抗生素的小鼠中被削弱,影响了肿瘤坏死因子(TNF)和IL-12的产生。
注:CpG-ODN免疫疗法是一种利用CpG寡脱氧核苷酸(CpG ODN)来激活和调节免疫系统的治疗方法。它通过激活多种免疫细胞如自然杀伤细胞(NK细胞)、单核/巨噬细胞、树突状细胞(DC)、B细胞和T细胞等,来增强机体的免疫反应。
基于细胞的免疫疗法
肠道菌群介导的胆汁酸代谢增加了肝脏中CXCR6+自然杀伤T(NKT)细胞的丰度,并在肝细胞癌中发挥抗肿瘤作用。
总之,肠道菌群在调节免疫治疗方面发挥着重要作用,影响细胞免疫转移和各种免疫疗法效果,并通过不同机制影响治疗效果。这些研究揭示了肠道菌群对免疫疗法的关键影响,包括调节免疫细胞活性和相关信号通路,进而影响肿瘤治疗的效果。
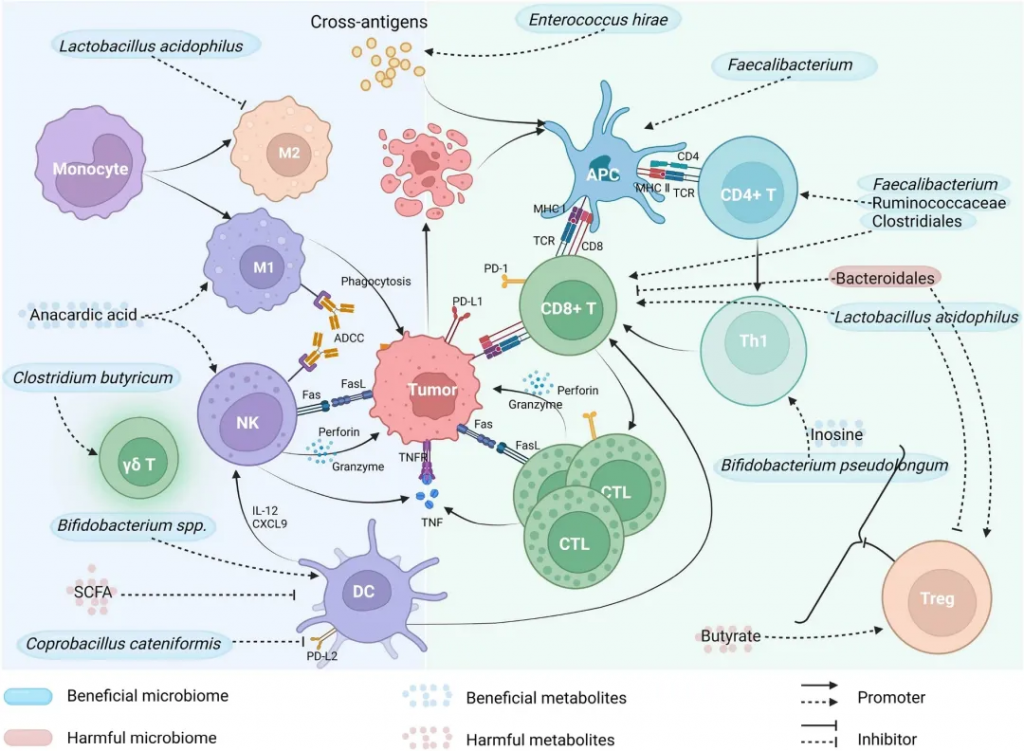
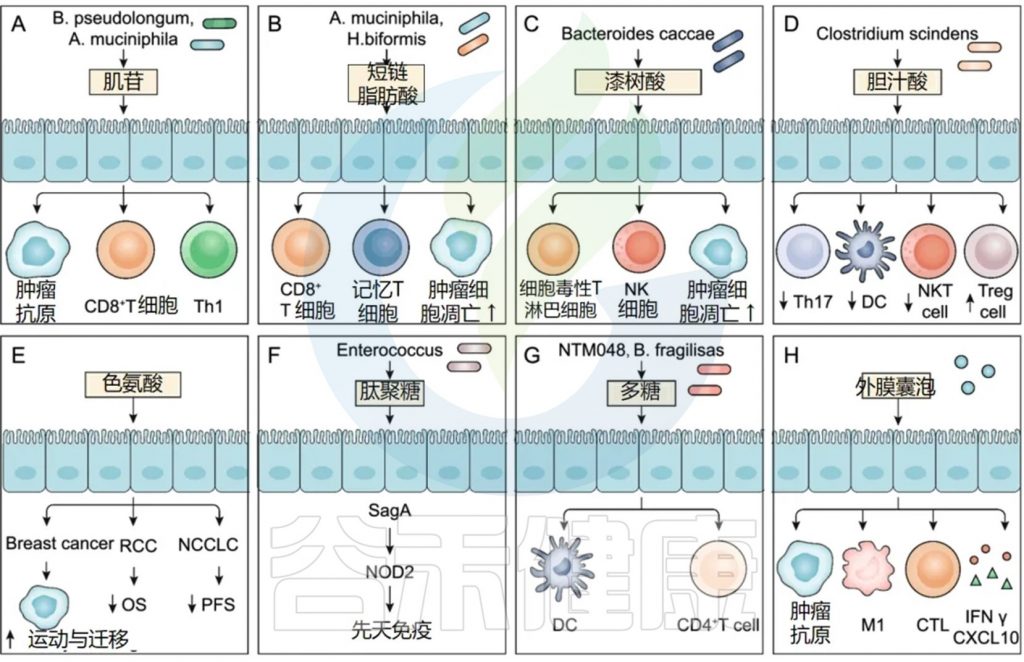
★ 毛螺菌科(Lachnospiraceae)
不可切除肝细胞癌(HCC)患者在接受抗PD-1药物治疗后,有客观临床反应的患者的粪便样本中Lachnoclostridium富集,且与胆汁酸等特定细菌代谢物的高浓度相关。类似的现象也在对抗PD-1治疗有反应的黑色素瘤患者中观察到。Lachnospiraceae的某些菌株与改善无进展生存期(PFS)相关。
★ 瘤胃球菌科(Ruminococcaceae)
Ruminococcaceae家族的共生菌通过降低肠道通透性促进宿主健康,并在产生短链脂肪酸如乙酸和丙酸中起重要作用。
临床研究表明,瘤胃球菌属在多种癌症类型中与ICI治疗反应性正相关,但其对ICI毒性的潜在影响及通过SCFA直接影响ICI效果的证据有限。不同瘤胃球菌属对ICI治疗反应的影响各异,表明需要进一步研究以开发基于肠道菌群组成的治疗反应预测生物标志物。
★ 颤螺旋菌科(Oscillospiraceae)
Faecalibacterium与ICI反应性:观察研究表明,Oscillospiraceae特别是F. prausnitzii的丰度与黑色素瘤患者对ICI的反应正相关,并减少不良事件。
Faecalibacterium刺激Tregs增殖并释放抗炎细胞因子如IL-10和IL-33,有助于维持肠道内抗炎因子平衡。
Faecalibacterium可能通过诱导CTLA-4+ Tregs的扩增增强抗CTLA-4治疗的效果,并作为增强疗效的辅助剂。
Faecalibacterium衍生的效果与免疫细胞频率增加相关,可能增强抗PD-1治疗的疗效,但研究结果存在差异。
★ 乳酸杆菌科(Lactobacillaceae)
乳酸菌作为共生菌具有免疫调节特性,常用作益生菌,临床前研究表明其能增强小鼠对anti-CTLA-4的反应,可能作为ICI治疗成功与否的生物标志物。
乳酸菌增强免疫应答:乳酸菌与小鼠树突细胞共培养可促进细胞成熟,增强免疫反应,提高IFN-γ和Granzyme B的产生,增加肿瘤中的CD8+ T细胞浸润,减缓肿瘤生长。
乳酸菌与PD-1抑制剂的协同效应:乳酸菌ZW18和L. reuteri的补充可优化肠道菌群组成,增强抗PD-1治疗的效果,通过不同机制提高CD8+ T细胞的活性和肿瘤组织中的IFN-γ表达。
★ 拟杆菌门
系统性抗生素治疗可能破坏其与厚壁菌门的比例,拟杆菌门增多,引起肠道菌群失衡和抗肿瘤免疫的负面影响。
负面影响:研究发现,拟杆菌门的丰度与黑色素瘤患者对免疫治疗的反应率呈负相关,高水平拟杆菌属与较差的ICI治疗反应有关。
拟杆菌门的增多可能抑制外周细胞因子反应,并促进免疫抑制细胞如Tregs和骨髓源抑制细胞的频率,从而影响ICI治疗的效果。
正面影响:尽管拟杆菌门的整体丰度与不良的ICI治疗反应相关,但某些物种如B. caccae、B. fragilis、B. thetaiotaomicron在转移性黑色素瘤患者中可能具有免疫刺激效应。
尽管拟杆菌门的某些物种对宿主免疫系统有益,但整体丰度与ICI治疗效果呈负相关。特定拟杆菌属的加入可能通过促进效应免疫反应来增强ICI治疗的效果,但需要更多研究来验证这一点。
★ 疣微菌科(Akkermansiaceae)
积极作用:Akkermansia muciniphila是一种在人体和动物消化系统中丰富的菌种,能分解黏蛋白,其丰度在对anti-PD-1/PD-L1治疗有反应的多种癌症患者中较高,有助于增强免疫反应和提高治疗效果。
增强免疫应答:A. muciniphila通过刺激树突细胞释放IL-12,减少免疫抑制Tregs的招募,增强Th-1相关免疫反应,与无进展生存期(PFS)相关。
注:该菌能通过其细胞膜中的脂质二酰基磷脂酰乙醇胺调节TLR1-TLR2分子途径,并在宿主代谢调节中发挥关键作用。
尽管A. muciniphila通常与正面的临床结果相关,但一些研究显示其在肠道微生物群中的主导地位可能预示着对anti-PD-1阻断的反应较差。
作为预测生物标志物和治疗策略:A. muciniphila的积极效果使其成为预测ICI治疗反应的潜在生物标志物,其口服给药在临床前模型中能将无反应者转变为有反应者,但剂量是关键因素。
★ 红蝽菌科(Coriobacteriaceae)
研究发现,对anti-PD-1治疗有反应的黑色素瘤患者中C. aerofaciens的丰度更高,且带有响应者菌群(包括C. aerofaciens)的无菌小鼠显示出更强的T细胞激活,导致增强的anti-PD-1效果和减缓的肿瘤生长。
C. aerofaciens的增加不仅促进T细胞激活,还通过提高IL-17A和CXCL1及CXCL5趋化因子的产生来促进炎症环境,同时减少紧密连接蛋白的表达,增加肠道通透性,这些免疫调节效应提示C. aerofaciens对ICI活性的重要性需要进一步探索。
★ 双歧杆菌科(Bifidobacteriaceae)
多项研究表明,双歧杆菌科的存在与增强的免疫介导的肿瘤抑制和ICI治疗的疗效相关。双歧杆菌与多种癌症(包括肾细胞癌、结直肠癌、黑色素瘤、非小细胞肺癌和三阴性乳腺癌)中ICI治疗的有效性增强有关。
双歧杆菌通过改变树突细胞的功能、促进CD8+ T细胞浸润、增加干扰素-γ的产生等机制,提高抗肿瘤反应。
双歧杆菌产生的肌苷通过T细胞特异性腺苷2A受体A2AR信号传导增强抗肿瘤能力,双歧杆菌通过抗原交叉反应,使表达特定新抗原的肿瘤对T细胞介导的破坏更加敏感。
双歧杆菌的这些特性使其成为潜在的预测ICI治疗反应的生物标志物,并且其作为单一治疗或与ICI治疗结合的策略在临床前模型中显示出前景。
临床前和临床研究中不同细菌对 ICI 治疗效果的积极和消极影响
doi.org/10.1016/j.xcrm.2024.101487
细菌种群与ICI治疗结果之间的关联
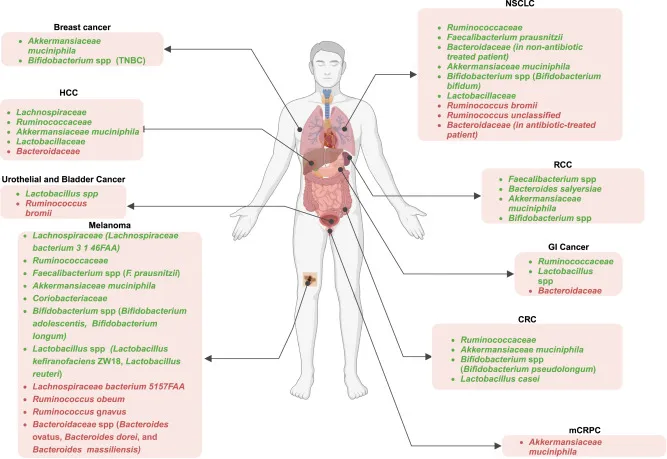
doi.org/10.1016/j.xcrm.2024.101487
注:癌症患者中绿色高亮细菌的存在或高丰度与不同癌症类型对ICI治疗的积极反应相关,而非应答患者中红色高亮细菌的流行率更高。
TNBC,三阴性乳腺癌;HCC,肝细胞癌;NSCLC,非小细胞肺癌;RCC,肾细胞癌;GI,胃肠道;CRC,结直肠癌;mCRPC,转移性去势抵抗性前列腺癌。
研究表明,肠道菌群调节 ICI 反应,有必要详细探索特定细菌种类和微生物代谢物对 ICI 的作用机制。
肠道菌群可调节先天性和适应性免疫,并影响 TME 中的抗肿瘤免疫反应。如下表,特定细菌种类在免疫背景下影响TME 以提高 ICI 疗效的复杂机制。

肠道菌群调节先天免疫、适应性免疫和肿瘤抗原,以改善 ICI 反应。
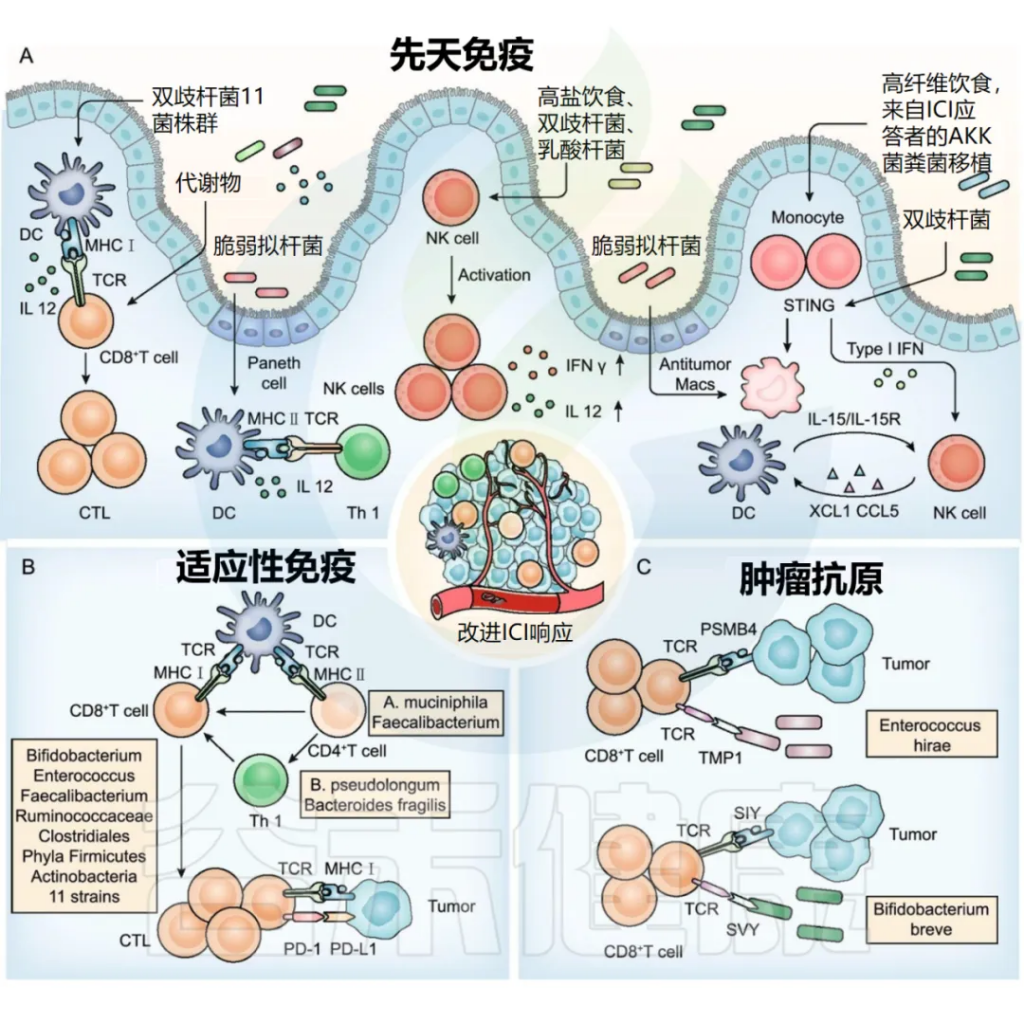
doi:10.1186/s13045-022-01273-9
■ 树突状细胞
双歧杆菌、十一种菌株及其代谢物和脆弱拟杆菌促进树突状细胞成熟或活化,随后活化 CD8 + T 细胞和 Th1 细胞。
树突状细胞 (DC) 是一组特殊的抗原呈递细胞,在 T 细胞活化和抗肿瘤免疫中起着至关重要的作用。肠道菌群抗原或含有免疫调节剂的代谢物可用于动员和激活树突状细胞 ,逆转未成熟树突状细胞诱导的免疫耐受。
例如,口服双歧杆菌可增加树突状细胞 (DC) 活化,进而支持改善肿瘤特异性CD8 + T细胞应答,并恢复抗 PD-L1 疗法在肠道菌群“不利”的小鼠中的治疗效果。
11 种菌株与 ICI 相结合,可强力诱导干扰素 (IFN) γ + CD8 + T 细胞,以依赖于固有层 cDC1 和主要组织相容性复合体 (MHC) I 类的方式抑制肿瘤生长。
脆弱拟杆菌通过触发树突状细胞成熟和刺激 IL-12 依赖性 Th1 细胞免疫反应,增强了 CTLA-4 阻断的抗肿瘤作用。
此外,万古霉素介导的肠道微生物组成调节通过增加 cDC1 和上调 IL-12,提高了抗肿瘤特异性效应 T 细胞的活性。
■ NK细胞
植物乳杆菌增加 NK 细胞活化;
高盐饮食增加肠道通透性和肿瘤内双歧杆菌的定位,并增强 NK 细胞活化以诱导抗肿瘤免疫。
NK 细胞可以调节TME 中的DC 和 CD8 + T 细胞丰度并影响对 ICI 的反应。越来越多的研究发现 NK 细胞和肠道菌群之间的相互作用。
注:NK 细胞也被称为自然杀伤细胞,是一类重要的淋巴细胞,主要起着免疫监视和细胞毒灭活的作用。这些细胞在体内寻找并摧毁受感染或变异的细胞,帮助维持身体的免疫功能。NK 细胞是免疫系统中的重要组成部分,对抗病毒感染和肿瘤细胞具有重要作用。
具有高微生物多样性的 NSCLC 患者在 PD – 1 阻断反应中具有更高丰度的独特记忆 CD8 + T 细胞和外周 NK 细胞亚群。
植物乳杆菌有效增加天然细胞毒受体(NCR)蛋白的表达并促进 NK 细胞活化以触发先天免疫。
次优剂量的 PD-1 阻断联合高盐饮食显著抑制了小鼠的肿瘤生长。
高盐饮食的双重作用
研究发现,高盐饮食会导致肠道通透性增加,以及双歧杆菌在肿瘤内部的定殖(即定居和繁殖)。这些变化会促进自然杀伤细胞(NK细胞)的活化,进而增强身体对肿瘤的免疫反应,从而发挥抗肿瘤作用。
由于高盐饮食与慢性炎症、心血管疾病和自身免疫性疾病有关,因此食用高盐饮食被认为是一种不健康的生活方式。免疫系统会转向促炎作用,然而,这种不良副作用被发现对肿瘤免疫有益。
高盐饮食增加了肠道的通透性,有利于双歧杆菌进入肿瘤部位,刺激自然杀伤细胞(NK细胞)并激活T细胞,导致肿瘤细胞的破坏并增加苯甲酸盐水平。肿瘤细胞通过程序性细胞死亡受体蛋白L1逃避破坏,L1与PD受体结合并使T细胞失活。
尽管抗PD-1疗法可以恢复T细胞功能,但由于某些肿瘤的PD-1分泌功能,它并非对所有肿瘤都有效。通过测量苯甲酸盐水平可以指导抗PD-1疗法的效果。高盐环境提供了一种避免肿瘤细胞检查点抑制的途径,通过干扰免疫介导的炎症,这可以通过调节盐水平被用作治疗方式。
总之,高盐饮食是一把双刃剑,但通过影响肠道微生物组和其他细菌产物,在肿瘤免疫中有希望的作用,需要进一步研究以揭示其潜在的好处。
■ 单核细胞/巨噬细胞
喂食高纤维饮食、用产生 cdAMP 的A. muciniphila单定植或从 ICI 反应者转移粪便菌群可触发单核细胞-IFN-I-NK 细胞-DC 级联;
双歧杆菌以 STING 信号和 IFN-I 依赖的方式促进基于 CD47 的免疫治疗;
脆弱拟杆菌诱导巨噬细胞表型极化为M1。
IFN-I (I型干扰素)和单核吞噬细胞水平的偏差导致免疫失调和免疫抑制性 TME。微生物诱导的 IFN-I 信号传导介导从先天免疫到适应性免疫的转变(上图)。
注:I型干扰素是一类干扰素,包括多种干扰素如IFN-α和IFN-β等,在免疫系统中起着重要的调节和抗病毒作用。
微生物衍生的干扰素基因刺激物 (STING) 激动剂(如 c-di-AMP)诱导肿瘤内单核细胞发出 IFN-I 信号传导,从而使单核吞噬细胞转向抗肿瘤巨噬细胞 (Macs) 并引发单核细胞-IFN-I-自然杀伤 (NK) 细胞-DC 串扰。
喂食高纤维饮食、用产生 cdAMP 的A. muciniphila进行单定植或从 ICI 应答者转移粪便微生物群可改善抗肿瘤反应和 ICI 疗效。
类似地,研究显示双歧杆菌优先定植于肿瘤部位,以 STING 信号和 IFN-I 依赖的方式促进基于 CD47 的免疫治疗。
脆弱拟杆菌诱导巨噬细胞表型极化为M1,并上调细胞中 CD80 和 CD86 的表达,促进先天免疫。
■ CD8 + T 细胞
双歧杆菌、肠球菌、粪杆菌、瘤胃球菌和梭菌目促进 CD8 + T 细胞浸润至肿瘤组织;
厚壁菌门和放线菌门可提高ICI 应答者外周血CD56 + CD8 + T 细胞的活化;
11 种菌株可增加体循环中效应 IFNγ + CD8 + T 细胞的比例。
多项研究已证实,特定肠道菌群可在体循环或 TME 中 诱导 CD8 + T 细胞(上图)。例如,具有相对丰富的有益菌群(包括梭菌目、瘤胃球菌科或粪杆菌)的黑色素瘤患者,其抗原呈递增加,外周血和 TME 中的效应 CD4 + T 细胞和 CD8 + T 细胞功能改善,从而改善了 ICI 的抗肿瘤功效。
临床试验的证据表明,厚壁菌门和放线菌门在 FMT 与 PD-1 阻断反应者联合治疗中富集。FMT 和 PD-1 阻断联合治疗可刺激外周血单核细胞 (PBMCs) 中的粘膜相关不变 T (MAIT) 细胞和 CD56 + CD8 + T 细胞,并上调肿瘤部位 CD8 + T 细胞上人类白细胞抗原 (HLA) II 类基因 CD74 和 GZMK 的表达。
同时,FMT 联合 PD-1 阻断治疗后,难治性转移性黑色素瘤中的肠球菌相对丰度增加,导致肿瘤内 CD8 + T 细胞浸润增加和肿瘤细胞坏死。
此外,双歧杆菌和 11 种菌株也可以增加依赖树突状细胞 (DC) 的 CD8 + T 细胞的丰度,以提高 ICI 治疗效果。
■ CD4 + T 细胞
B. pseudolongum和脆弱拟杆菌刺激 Th1 免疫反应;
A . muciniphila触发CCR9+CXCR3+CD4+T 淋巴细胞募集到瘤床;
粪杆菌可增加 CD4 + T 细胞比例。
在小鼠模型中,B. pseudolongum主要通过肠道微生物代谢物肌苷促进 Th1 转录分化和抗肿瘤免疫反应,从而提高 ICI 疗效。
脆弱拟杆菌通过促进固有层树突状细胞的动员来刺激 IL-12 依赖的 Th1 免疫反应,从而恢复对 ICI 的免疫反应。
FMT无反应小鼠口服补充A. muciniphila可通过触发 CCR9 + CXCR3 + CD4 + T 淋巴细胞募集到瘤床来恢复抗 PD-1 反应。
注:瘤床(Tumor Bed)是指在肿瘤手术切除后,原有肿瘤所在的部位,包括切除肿瘤组织后的创面以及周围组织。瘤床的特征和变化对于肿瘤的预后和治疗效果起着重要作用。
在人类患者中,粪杆菌增加了外周血中的 CD4 + T 细胞比例和血清 CD25 的产生,并降低了外周血中的 Treg 细胞比例,从而诱导了伊匹单抗的长期临床益处。
注:伊匹单抗(yervoy)是首个免疫检查点抑制剂,于2013月由FDA批准上市,用于治疗转移性黑色素瘤或无法手术切除的黑色素瘤。伊匹单抗是一种抗CTLA-4抗体。
CTLA-4表达于T细胞表面,是第一个用于临床研究的Chemicalbook抑制性受体靶点。CTLA-4在T细胞活化阶段发挥抑制功能:T细胞活化后,CTLA-4在T细胞表面上调表达,一方面与CD28竞争性结合共刺激分子B7,另一方面传导抑制性信号来终止细胞活化,CTLA-4在维持免疫系统内稳态中起至关重要的作用。
肠道菌群通过提供肿瘤交叉抗原来增强肿瘤细胞的免疫原性,从而改善 ICI 的疗效,包括抗原表位 TMP1 和抗原表位 SVY。
肿瘤免疫原性降低是肿瘤细胞抵抗T细胞杀伤的重要机制。
一方面,肠道菌群可以通过作用于肿瘤细胞表面的UBA6,直接增强肿瘤细胞的先天免疫原性,从而增强ICI反应。
另一方面,肠道菌群可以通过提供肿瘤交叉抗原来间接增加肿瘤细胞的免疫原性,从而促进ICI的疗效。已鉴定出肠道微生物表达的抗原与肿瘤细胞之间存在一些交叉反应性(上图C ) 。
Enterococcus hirae 基因组中的抗原表位尾长卷尺蛋白1(TMP1)与蛋白酶体亚基β4型(PSMB4)肿瘤抗原具有高度相似性。它们同时激活CD8 + T细胞,提高PD-1阻断疗法的疗效。
已证明共生菌短双歧杆菌表达的抗原表位SVYRYYGL(SVY)与肿瘤表达的抗原表位SIYRYYGL(SIY)相似,导致SVY特异性T细胞识别SIY并抑制肿瘤生长。
肠道微生物群及其代谢物对免疫疗法影响的机制
doi.org/10.1186/s40164-023-00442-x
肠道微生物群调节抗肿瘤免疫的主要方式之一是通过代谢物。肠道微生物群合成或转化大量代谢物,这些代谢物是可以从肠道中的原始位置扩散的小分子,影响局部和全身抗肿瘤免疫反应,从而促进 ICI 疗效。
这里进一步讨论不同肠道菌群代谢物的机制以及肠道菌群特征的其他特征介导抗肿瘤免疫反应。
肌苷是人体正常代谢产物,生理状态下参与核酸代谢、能量代谢和蛋白质合成,能激活免疫细胞、刺激新陈代谢。以往研究表明肌苷具有免疫抑制作用。而近年来更多研究发现肌苷可以重编程肿瘤微环境 (TME),改善ICI治疗的反应性。目前的研究表明,肌苷主要通过以下机制影响ICI疗效。
肠道微生物代谢物肌苷促进ICI疗效的潜在机制
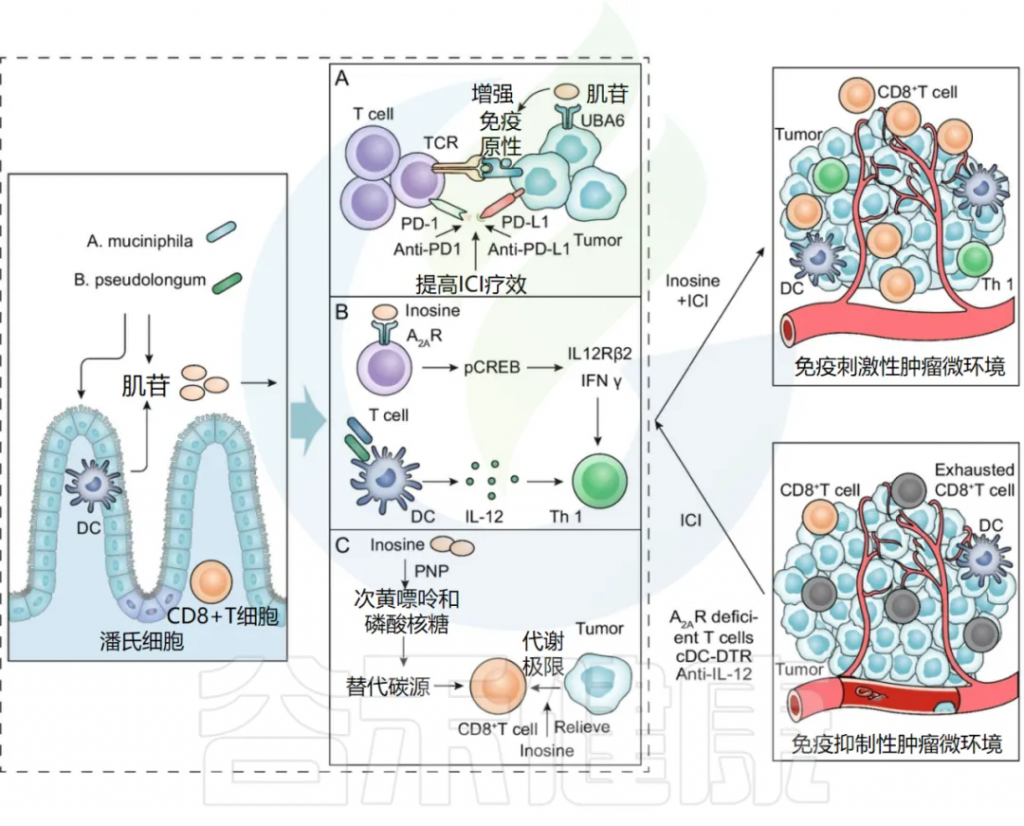
doi:10.1186/s13045-022-01273-9
肠道菌群A. muciniphila和B. pseudolongum的嘌呤代谢物肌苷,与ICI联合治疗可发挥协同抗肿瘤作用。
A) 肌苷增加肿瘤细胞的免疫原性
肌苷通过增强肿瘤抗原呈递和激活IFNγ及TNFα信号通路,提升肿瘤细胞的免疫原性,使免疫细胞更易识别并杀死肿瘤细胞。此外,肌苷还能通过抑制UBA6酶,增强T细胞介导的细胞毒作用,从而提高免疫检查点抑制剂(ICI)的疗效。
B) 肌苷促进免疫细胞活化
肌苷可能通过作用于肠癌、膀胱癌和黑色素瘤小鼠模型中T淋巴细胞上的A2AR,增强ICI疗效。它通过肌苷-A2AR-cAMP-PKA信号通路刺激cAMP反应元件结合蛋白(pCREB)的磷酸化,从而上调IL12Rβ2和IFNγ转录,促进Th1细胞分化和在TME中的聚集。
ICI 与肌苷联合使用的体内抗肿瘤作用需要共刺激物,例如 CpG 和 IL-12。肌苷还可以增强植物血凝素 (PHA) 介导的免疫反应,提高肿瘤抗原水平,并增强 T 淋巴细胞分化和增殖。此外,肌苷通过激活巨噬细胞刺激B淋巴细胞分化和抗体产生,发挥抗病毒和抗肿瘤作用。
C) 肌苷为CD8 + T细胞提供了替代碳源
肌苷可以在葡萄糖受限时作为CD8 + T细胞的替代碳源,缓解肿瘤细胞对CD8 + T细胞能量代谢的限制。
注:T 细胞通过嘌呤核苷磷酸化酶(PNP)将肌苷代谢为次黄嘌呤和磷酸化核糖。肌苷的核糖体亚基进入中央代谢途径,为糖酵解途径和戊糖磷酸途径(PPP)提供 ATP 和生物合成前体。
结肠厌氧菌从未消化和吸收的碳水化合物或肠道上皮细胞分泌的糖蛋白中产生短链脂肪酸 (SCFA)。
肠道微生物衍生的短链脂肪酸与实体瘤的纳武单抗或派姆单抗治疗之间的关联已得到证实。
注:纳武单抗OPDIVO(nivolumab)具有免疫检查点抑制活性和抗肿瘤活性,用于先前有过治疗的晚期鳞状NSCLC患者。
派姆单抗(Pembrolizumab),是一种人源化单克隆抗体,主要用于治疗多种实体瘤和淋巴瘤。
研究发现,短链脂肪酸是重要的物理和化学屏障,可刺激潘氏细胞和杯状细胞产生 AMP 和粘液,从而维持肠粘膜屏障的完整性。
短链脂肪酸在复杂的肠道微生物免疫和代谢网络中发挥关键作用,影响免疫细胞和肿瘤细胞的活性(下图)。
肠道菌群代谢物SCFAs增强ICI疗效的潜在机制
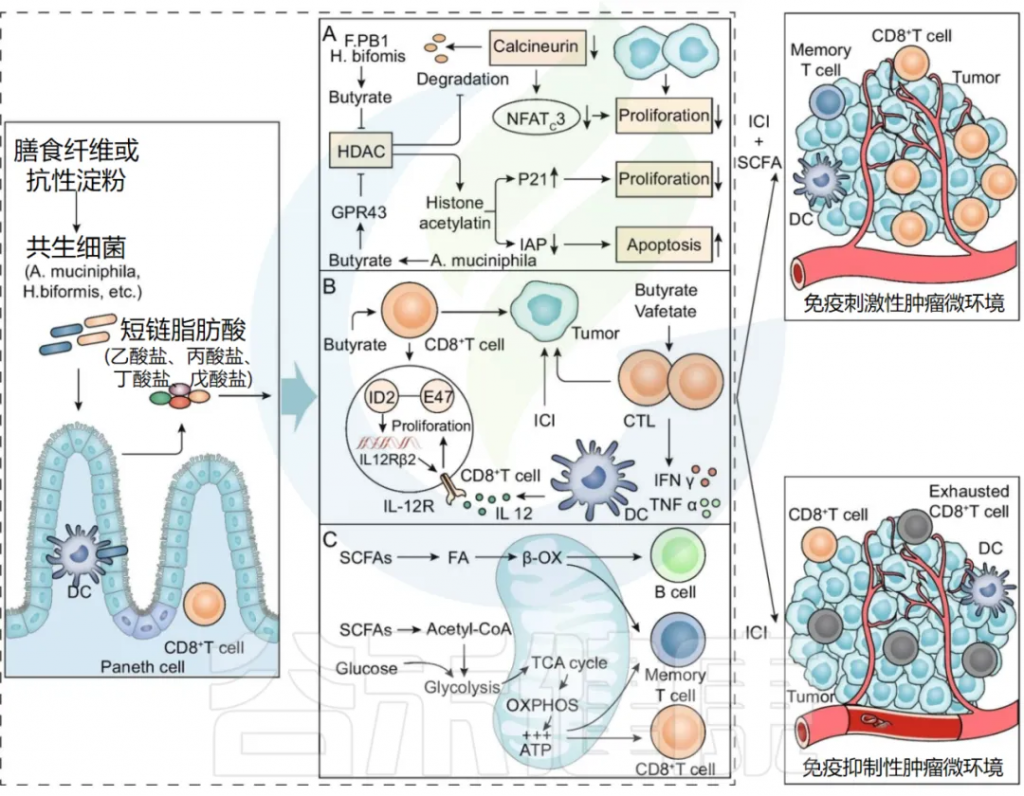
doi:10.1186/s13045-022-01273-9
A) SCFAs抑制癌细胞增殖并诱导癌细胞凋亡
Faecalibaculum rodentiumPB1和H. biformis的代谢产物SCFAs中的丁酸,可抑制HDAC的活性和钙调神经磷酸酶介导的NFATc3转录因子的激活,从而阻断肿瘤细胞的增殖。
A . muciniphila产生的丙酸通过GPR43激活细胞周期抑制剂p21并下调IAP抑制剂,从而抑制癌细胞增殖,诱导细胞凋亡,提高ICI的抗肿瘤作用。
B) SCFAs改善抗肿瘤免疫反应
丁酸可以通过IL-12信号诱导CD8+T细胞中ID2的表达,直接增强CD8 + T细胞的抗肿瘤细胞毒作用。
SCFA 中的戊酸和丁酸促进 IFNγ 和 TNFα 等效应分子的表达,增强 CTL 的抗肿瘤作用。
C) SCFA为免疫细胞提供能量
SCFA 通过调节糖酵解、TCA 循环和 β-氧化等代谢途径,为 B 细胞、记忆 T 细胞和效应 T 细胞提供能量,从而增强 ICI 疗效。
漆树酸能调节抗肿瘤免疫反应
在前瞻性研究中,对接受 ICI 治疗的黑色素瘤患者粪便细菌的代谢组学分析显示,Bacteroides caccae显著富集,ICI 反应组的漆树酸水平大幅升高(62 倍,P < 0.01)。
注:漆树酸是一类具有生物活性的有机化合物,主要存在于腰果壳液中(Cashew Nut Shell Liquid, CNSL),也可以在其他漆树科植物中找到。这类化合物以酚类结构为特征,并具有多种生物学和药理学活性,包括抗菌、抗病毒、抗氧化和抗炎作用。
漆树酸激活先天免疫
漆树酸通过磷酸化丝裂原活化蛋白激酶 (MAPK) 触发巨噬细胞中的经典活化途径,从而激活先天免疫。
漆树酸促进适应性免疫反应
漆树酸还能诱导中性粒细胞胞外陷阱( NET) 的产生,从而促进巨噬细胞、NK 细胞和 T 淋巴细胞产生肿瘤浸润免疫细胞,从而调节适应性免疫和抗肿瘤免疫。
临床前模型中,漆树酸的抗肿瘤作用
在乳腺癌模型中,漆树酸增加了肿瘤浸润的NK细胞和CTL的水平,并诱导了肿瘤细胞的凋亡(下图)。
doi:10.1186/s13045-022-01273-9
一些研究显示,肠道菌群代谢物,如次级胆汁酸或色氨酸,具有免疫抑制作用(上图5D、E)。
例如,胆汁酸代谢物3-氧化石胆酸和丰富的肠道代谢物异石胆酸可以抑制Th17细胞的分化。
此外,次级胆汁酸3β-羟基脱氧胆酸在DCs中表现出较弱的免疫刺激特性,从而诱导Foxp3的表达,增加Tregs的数量,促进免疫逃逸。
喂食次级胆汁酸或胆汁酸代谢细菌Clostridium scindens能减弱NKT细胞介导的肝脏选择性肿瘤抑制。
IL-2通过激活STAT5-5-羟色氨酸(5-HTP)-AhR轴,诱导CD8+ T细胞的耗竭,并强烈表达色氨酸羟化酶1。
色氨酸代谢物有效促进乳腺癌肿瘤细胞的运动和迁移。
在接受纳武单抗治疗的黑色素瘤和肾细胞癌患者中,血清中犬尿氨酸/色氨酸比值的增加与总体生存率的降低有关。
非小细胞肺癌患者中,低血浆色氨酸代谢物-3-羟苯甲酸水平与延长的无进展生存期(PFS)显著相关。
肽聚糖(PG)和多糖(PSA)
肠球菌(Enterococcus)表达和分泌同源NlpC/p60 PG水解酶SagA,可以促进先天免疫传感蛋白NOD2的表达,并增强ICI抗肿瘤效力。此外,以NOD1依赖的方式识别源自微生物的PG可以促进系统性先天免疫。
Leuconostoc mesenteroides NTM048或脆弱拟杆菌产生的PSA作为免疫刺激剂,能够增强粘膜屏障并影响系统性免疫反应。PSA可被小肠中的DCs识别,并激活CD4+ T细胞分泌细胞因子,促进T细胞增殖,改善Th1/Th2细胞失衡,并促进淋巴组织形成功能。
TLRs(如TLR9及其激动剂CpG-ODN)在病原体识别和免疫反应启动中起着重要作用。艰难梭菌毒素A结合的DNA激活了TLR9信号和先天免疫反应。
外膜囊泡(OMV)
由细菌自然分泌的微生物来源外膜囊泡(OMVs)可以重新编程肿瘤微环境,并已被开发用于肿瘤免疫治疗剂、细菌疫苗、佐剂和药物递送载体。
OMVs表达肿瘤抗原,诱导先天免疫反应和抗原特异性T细胞介导的抗肿瘤免疫;生物工程改造的OMVs可表达多种肿瘤抗原,触发协同抗肿瘤免疫反应。
带有磷酸钙(CaP)外壳的OMVs能促进细胞毒性T细胞渗透和M2型巨噬细胞向M1型极化,有效改善免疫抑制性TME。此外,全身给药的细菌OMVs能特异性靶向并积聚在肿瘤床中,随后诱导产生抗肿瘤细胞因子CXCL10和IFN-γ,有效增强抗肿瘤免疫反应。
免疫检查点抑制剂(ICI)治疗在杀死肿瘤细胞的同时可能扰乱宿主的免疫平衡,导致免疫相关性结肠炎、肺炎,甚至危及生命的心肌炎。
多项研究表明,肠道微生物群在免疫相关不良事件(irAEs)中的作用是一把双刃剑。
一些肠道细菌的保护作用
小鼠模型验证了双歧杆菌、脆弱拟杆菌和伯克氏菌可在抗CTLA-4治疗的背景下改善肠道免疫病理学。
一项前瞻性研究显示,拟杆菌门的增加和参与多胺运输及维生素B生物合成的微生物基因途径与治疗转移性黑色素瘤患者ipilimumab造成的结肠炎的抵抗性有关。
注:Ipilimumab是一种单克隆抗体,主要用于治疗某些类型的癌症,如黑色素瘤、肾癌等。其作用机制是通过阻断细胞毒性T淋巴细胞抗原-4(CTLA-4),从而增强免疫系统对癌细胞的攻击能力。
另一些肠道菌群与ICI引起的高风险毒性有关
在接受抗PD-1治疗的黑色素瘤患者中,Lachnospiraceae 和Streptococcus 的富集与irAEs的增加有关。
肠道菌群的双重作用
有趣的是,不同的肠道微生物基线可能同时与良好的抗癌反应和ICI引起的毒性有关。在26名接受ipilimumab治疗的转移性黑色素瘤患者中,基线时富含Faecalibacterium和Firmicutes的患者易同时发生免疫治疗引起的结肠炎和增强的ICI敏感性,称为ICI背景下的疗效-毒性耦合效应。
肠道菌群作为预测生物标志物
肠道微生物如拟杆菌,也可用作预测接受联合CTLA-4和PD-1阻断治疗的晚期黑色素瘤患者ICI治疗毒性的生物标志物。
肠道菌群的重构与治疗毒性
通过重构肠道微生物组和增加结肠黏膜中Tregs比例,FMT这种治疗方式已在两例患者中显示出对免疫治疗引起毒性的有益效果。
这些研究表明,肠道微生物组对ICI引起的毒性具有复杂的正负作用。在充分筛选和操控肠道微生物组以增强ICI反应和减少irAEs之前,还需要更多证据。
了解肠道微生物群及其代谢物对抗肿瘤免疫和免疫治疗反应的生物机制,对于合理调控微生物活动以提高免疫检查点抑制剂(ICI)的疗效至关重要。下面介绍了结合肠道微生物群与ICI的治疗策略。
doi:10.1186/s13045-022-01273-9
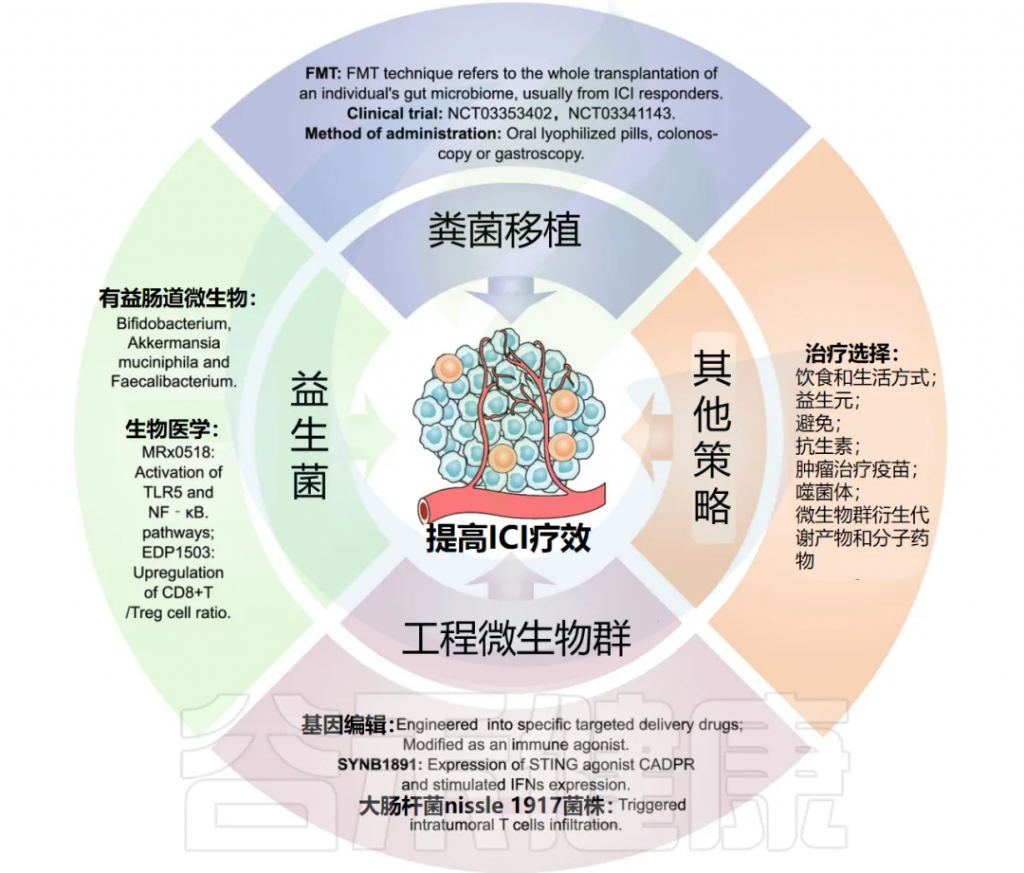
粪菌移植(FMT)治疗指的是将个体的全部肠道微生物群进行移植,通常从ICI响应者中获取。
FMT制剂可以通过口服冻干胶囊或通过结肠镜或胃镜直接给药。FMT最初用于治疗对传统疗法耐药的艰难梭菌感染。
FMT增强抗肿瘤效果
近年来,多项研究表明,FMT可以增强ICI的抗肿瘤效果并克服对免疫治疗的耐药性。
FMT的应用是逆转ICI免疫治疗耐药性和减少irAEs的一种新方法。
FMT与ICI治疗结合的临床试验
目前,一些评估FMT与ICI治疗结合的安全性和有效性的临床试验正在进行中。
武汉大学人民医院肿瘤中心陈永顺教授团队与善泰健康合作的题为”Fecal microbiota transplantation plus tislelizumab and fruquintinib in refractory microsatellite stable metastatic colorectal cancer: an open-label, single-arm, phase II trial (RENMIN-215) “前瞻性临床研究结果在线发表于《eClinicalMedicine》上。
注:《eClinicalMedicine》是LANCET(柳叶刀)的高端金色子刊,医学类一区TOP期刊,影响因子15.1。

该研究对至少二线系统治疗失败的微卫星稳定型(MSS)晚期结直肠癌患者进行了创新性治疗。
从2021年5月10日至2022年1月17日,共纳入20名患者。中位随访时间为13.7个月。该方案耐受性良好,无因不良反应导致死亡。通过“三联疗法”(肠道菌群移植 + PD-1单抗 + 靶向药)有效延长难治型结直肠癌患者的生存期,粪便中双歧杆菌丰度较低、毛螺菌科丰度高和外周血中扩增的 TCR 可能是预后预测和风险分层的宝贵分类因素。
这一突破性的临床研究成果具有重要的科研价值和临床应用前景,同时也充分证明了善泰健康坚持采集山区儿童供体制备优质肠道菌群的价值。
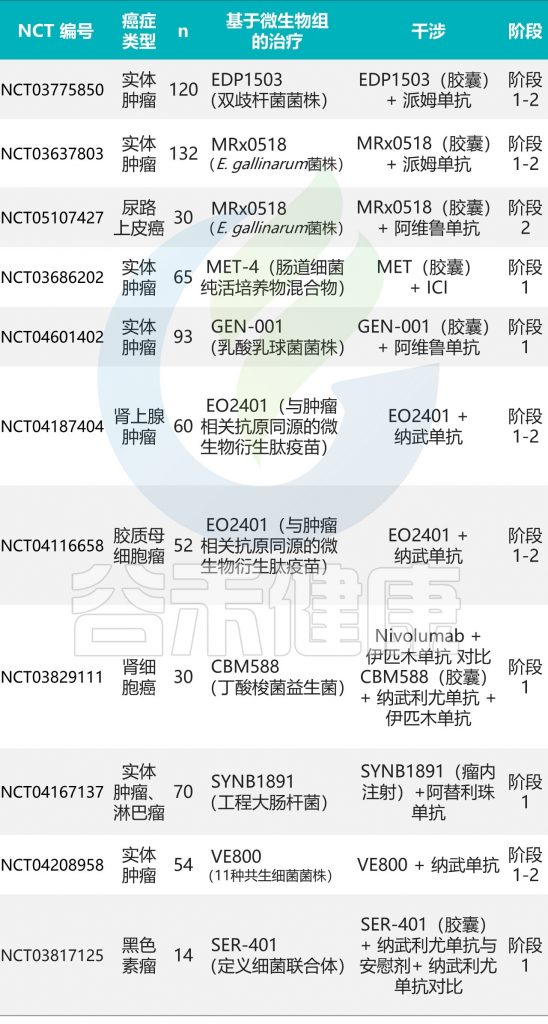
一些评估FMT联合ICI治疗安全性和有效性的临床试验正在进行中(下表)。

doi:10.1186/s13045-022-01273-9
FMT治疗的安全性考量
尽管FMT治疗在接受ICI治疗的患者中显示出有前景的结果,但其长期安全性仍存在担忧。
2019年报道的两项独立临床试验中,两名患者在接受来自同一供体的FMT后,感染了产超广谱β-内酰胺酶(ESBL)的大肠杆菌,其中一名患者死亡。这一研究促使美国食品药品监督管理局发布安全公告,警告FMT治疗的感染风险。
一项对供体粪便中多药耐药生物进行检测的回顾性队列研究显示,在66名受试者中,有6人(9%)在任何时间点检测出多药耐药生物。
因此,应定期筛查供体粪便,以严格限制可能导致不良感染事件的生物的传播,这对于免疫缺陷患者尤为重要。进一步的临床研究,对于更好地理解供体FMT的来源、移植程序和受体表型,对于成功的ICI-FMT联合治疗至关重要。
益生菌是指“当以适当剂量给予时,对宿主健康有益的活微生物群”。早期针对癌症患者的临床试验主要评估益生菌如何改变微生物群组成或调节抗肿瘤免疫。
一项针对乳腺癌患者的临床试验评估了益生菌(13种有益菌株)对TME中CD8+ T细胞浸润的影响(NCT03358511)。
接受益生菌(乳双歧杆菌和嗜酸乳杆菌)治疗的CRC患者在肿瘤、黏膜和粪便中有更多的丁酸盐生成细菌(NCT03072641)。
另一项试验表明,接受益生菌治疗的患者结肠黏膜中的IL-1b、IL-10、IL-23A mRNA水平较低(NCT01895530)。
益生菌作为免疫佐剂的作用
特定肠道微生物,如双歧杆菌、Akkermansia、肠球菌、粪杆菌、Ruminococcaceae,在ICI免疫治疗中起着免疫佐剂的作用。
多项临床试验正在评估益生菌与ICI联合使用的安全性和有效性。例如,MRx0518(粪肠球菌胶囊)主要依靠自由鞭毛蛋白激活TLR5和NF-κB信号通路,发挥抗肿瘤作用。
EDP1503是一种基于双歧杆菌的癌症免疫治疗新佐剂。临床试验(NCT03775850)显示,EDP1503与帕姆单抗联合使用是安全且耐受良好的,生物标志物研究发现,EDP1503通过上调CD8+ T细胞/Treg细胞的比例发挥作用。
doi:10.1186/s13045-022-01273-9
随着细菌基因工程技术的发展,通过改造肠道微生物群或其代谢物来改善免疫检查点抑制剂(ICI)的抗肿瘤反应变得可行。与FMT相比,基因工程药物具有特异性。
基因工程细菌的临床前模型应用
目前,经过基因改造的营养缺陷型和可诱导性大肠杆菌、双歧杆菌、李斯特菌和沙门氏菌已被用于静脉内、肿瘤内和口服给药途径的临床前模型中,显示出抗肿瘤效果。
利用工程菌作为药物载体
利用多种菌株(包括沙门氏菌和梭菌)在固体肿瘤中的靶向特性,使它们成为递送和诱导免疫刺激剂、延缓肿瘤生长和转移的理想载体。
工程化细菌的临床试验进展
SYNB1891是一种基于大肠杆菌生物学设计的双重固有免疫激动剂,被改造后表达STING激动剂CADPR,可刺激IFNs表达并实现抗肿瘤效果。正在进行临床试验以探索SYNB1891联合阿替利珠单抗在晚期实体瘤中的安全性和有效性。
工程化益生菌增强T细胞活性
L-精氨酸通过调节T细胞代谢来增强T细胞存活和抗肿瘤活性。
工程化益生菌大肠杆菌Nissle 1917菌株在肿瘤部位定植,并持续将代谢产物氨转化为L-精氨酸。在小鼠中,该菌株的肿瘤内注射增加了细胞内L-精氨酸浓度,触发肿瘤内CD4+ T细胞和CD8+ T细胞浸润,并在与抗PD-L1联合使用时产生协同抗肿瘤效果。
这些结果表明,工程化微生物治疗能够通过代谢调控肿瘤微环境,以增强免疫治疗的疗效。
通过调控肠道微生物群来增强免疫检查点抑制剂(ICI)疗效的治疗策略还包括调整饮食和生活方式、摄取益生元和避免使用抗生素。
肿瘤治疗疫苗的开发
利用肿瘤交叉抗原和肠道微生物来源的免疫激活剂,开发新的肿瘤治疗疫苗,如Ty21a、JNJ-64041809、VXM01。这些疫苗的开发为增强ICI疗效提供了新的途径。
微生物衍生药物的应用
EO2401是一种结构上与肿瘤相关抗原相同的微生物衍生多肽药物,EO2401联合纳武利尤单抗的临床试验正在胶质母细胞瘤和肾上腺皮质癌患者中进行。
口服微生物群衍生化合物(如噬菌体和微生物代谢物)可能比完整或特定活菌移植更加实际和精确。噬菌体作为一种治疗策略,可以调节肠道微生物群、免疫系统和肿瘤微环境。
短链脂肪酸戊酸(VPA)是一种微生物来源的代谢物。评估VPA与ICI联合使用在实体瘤患者中安全性和有效性的临床试验已经在进行中。
菌群影响ICI疗效的准确性
肠道菌群对肿瘤ICI治疗的影响是多方面的,甚至是双向的。未来研究需要准确分类针对免疫细胞或通路的有利和不利的菌群特征,并充分了解它们在不同肿瘤微环境中的作用。
提高治疗策略的精准度
了解菌群-代谢物-免疫轴有助于直接操纵来自肠道菌群的特定免疫刺激代谢物或化合物,而不是整个或独特的活菌移植,以改善ICI反应。其他促进抗肿瘤免疫反应的菌群衍生分子的作用可能成为未来ICI临床前或临床研究的方向。
肠道菌群作为预测有效性的生物标志物
患者的菌群数据可以与其他已知的相关预测标志物(如PD-L1表达和肿瘤突变负荷)相结合,以预测免疫治疗的潜在疗效或不良事件。
提高临床试验的有效性
在人体临床试验中,需要全面监测影响肠道菌群的因素,如饮食、益生菌、抗生素、药物、心理健康、宿主免疫系统、宿主遗传学、地理因素、肿瘤类型和分期等。
整合微生物学、遗传学、免疫学、代谢组学、分子病理学和流行病学的多层次多维度研究设计将成为未来个性化癌症治疗的一部分。
通过这些策略和研究方向,我们可以更深入地理解肠道微生物群在抗肿瘤免疫治疗中的作用,并开发出更有效的治疗方案。
肠道微生物组与免疫调节
肠道微生物组与先天性和适应性免疫细胞发生相互作用,增强先天性免疫细胞的中间效应,增强适应性免疫细胞的抗肿瘤作用,增加肿瘤细胞的免疫原性,从而重新编程TME的免疫力并改善ICI反应。
微生物代谢物与ICI效率
微生物衍生的循环代谢物调节多种人体生理,并从其在肠道中的原始位置扩散,介导局部和全身抗肿瘤免疫反应,从而促进ICI效率。
肠道微生物组与ICI相结合的治疗策略
例如适当的抗生素选择、益生菌摄入、FMT和细菌基因工程,可能为肠道微生物组及其代谢物成为ICI的优良佐剂提供新的可能性。进一步了解ICI与肠道微生物组协同作用的机制并准确识别免疫刺激和免疫抑制菌株或途径,有望开发更有效的ICI联合治疗策略并推进精准医疗策略。
肠道微生物群在癌症治疗中的应用前景广阔
首先,肠道微生物群的特性有望用于早期检测和预后评估。通过分析患者的粪便样本,可以评估其肠道微生物群的组成,预测对特定免疫疗法的反应性,从而为临床医生提供个性化的治疗方案。
其次,肠道微生物群的调节可能成为提高治疗效果的策略。例如,通过益生菌/益生元的补充、饮食调整或粪便微生物群移植,可以优化肠道微生物群的组成,增强免疫系统的抗癌能力。
肠道微生物群在癌症治疗中的作用日益受到重视,其在预测治疗反应、优化治疗策略和开发新型抗癌药物方面具有巨大潜力。
未来的研究需要进一步深入探索肠道微生物群与癌症治疗之间的具体作用机制,以及如何在临床实践中有效利用这些发现,为癌症患者带来更好的治疗效果和生活质量。
附录:
免疫疗法的治疗效果如何评估?
免疫疗法的治疗效果可以通过以下多种方式进行评估:
肿瘤大小和数量的变化
通过影像学检查,如 CT、MRI 等,观察肿瘤的大小、数量以及是否有新的病灶出现。这是评估治疗效果的常见指标之一。如果肿瘤缩小或消失,通常表明治疗有效。
肿瘤标志物水平
某些肿瘤会产生特定的蛋白质或生物标志物,检测血液中这些标志物的水平变化可以辅助评估治疗效果。
症状改善
患者的症状,如疼痛、呼吸困难、乏力等的减轻或消失,也可以反映治疗的效果。
生存时间
包括无进展生存期(PFS),即从治疗开始到肿瘤出现进展或患者死亡的时间;总生存期(OS),即从治疗开始到患者死亡的时间。较长的 PFS 和 OS 通常表示治疗效果较好。
免疫细胞的活性和数量
通过检测血液中免疫细胞(如T细胞、NK细胞等)数量、活性和表型,了解免疫系统的状态,评估免疫疗法对免疫系统的激活和调节作用。
免疫相关指标
如细胞因子水平、免疫检查点分子的表达等,也可以作为评估免疫治疗效果的参考。
患者的生活质量
通过问卷调查等方式评估患者在身体功能、心理状态、社会功能等方面的生活质量改善情况。
需要注意的是,免疫疗法的效果评估可能较为复杂,有时可能会出现假性进展或延迟反应等特殊情况,需要综合多种评估指标和患者的具体情况进行判断。
免疫疗法可以用于哪些癌症的治疗?
免疫疗法目前已被广泛应用于多种癌症的治疗,以下是一些常见的例子:
随着研究的不断深入,免疫疗法的应用范围还在不断扩大,有望为更多癌症患者带来希望。
免疫疗法的治疗周期一般是多久?
免疫疗法的治疗周期并没有一个固定的标准,会因癌症类型、患者个体情况、治疗方案以及治疗反应等多种因素而有所不同。
一般来说,免疫治疗可能需要持续几个月到几年不等。
对于某些癌症,如黑色素瘤,可能需要进行几个月的初始治疗,然后根据治疗效果和患者的耐受性,决定是否继续维持治疗。维持治疗的周期可能会持续数月甚至数年。
在肺癌的治疗中,免疫治疗的周期也会因病情和治疗方案而异。有的患者可能需要持续治疗 2 年左右,而有些情况下可能会更长。
对于一些治疗效果显著且耐受性良好的患者,医生可能会建议长期使用免疫治疗药物以防止癌症复发。
需要注意的是,医生还会定期评估患者的病情、治疗效果和不良反应,从而调整治疗方案和治疗周期。
免疫疗法治疗效果和患者耐受性有哪些关系?
免疫疗法的治疗效果和患者的耐受性密切相关。
首先,患者对免疫治疗的耐受性好,能够按计划完成治疗疗程,这有助于药物充分发挥作用,从而提高治疗效果。如果患者因耐受性差而频繁中断治疗或减少药物剂量,可能无法达到理想的治疗浓度和作用时间,影响对肿瘤细胞的抑制和杀灭效果。
其次,耐受性不佳可能导致严重的不良反应,如严重的免疫相关炎症等。这些不良反应不仅会影响患者的生活质量,还可能需要使用大量的药物来控制,这些药物之间的相互作用有时会干扰免疫治疗药物的疗效。
另外,耐受性差可能使患者心理负担加重,产生焦虑、恐惧等不良情绪,影响患者的治疗依从性和信心,进而间接影响治疗效果。
相反,良好的耐受性能够让患者保持积极的心态,更好地配合治疗,有助于增强免疫系统的功能,提高免疫治疗的效果。
总之,患者对免疫疗法的耐受性在很大程度上影响着治疗效果,而治疗效果的好坏也会反过来影响患者的耐受性和治疗依从性。
免疫疗法的优势和劣势
免疫疗法与其他传统疗法(如手术、放疗、化疗)相比,具有以下优势和劣势:
优 势
长期效果:免疫疗法有可能产生持久的免疫记忆,一旦有效,其疗效可能持续较长时间,甚至实现长期的肿瘤控制或治愈。
全身性治疗:能够激发全身性的免疫反应,不仅可以作用于局部肿瘤病灶,还可以对转移灶发挥作用,减少肿瘤复发和转移的风险。
低毒性:相比化疗等方法,免疫治疗的全身性副作用通常较轻,对正常细胞的损伤相对较小,患者的生活质量相对较高。
适用范围广:对于一些晚期或转移性肿瘤,以及传统治疗方法无效的患者,免疫疗法仍可能有一定的效果。
当然,免疫疗法也可以与其他治疗方法(如化疗、放疗或靶向疗法)结合使用,从而增强治疗效果。
劣 势
起效较慢:免疫治疗发挥作用通常需要一定的时间,不像手术能迅速切除肿瘤,也不像化疗有时能较快地缩小肿瘤体积。
个体差异大:不同患者对免疫治疗的反应差异较大,部分患者可能疗效不佳。
价格昂贵:免疫治疗药物的研发和生产成本较高,导致治疗费用昂贵,给患者带来较大的经济负担。
潜在的免疫相关不良反应:虽然相对较轻,但仍可能出现免疫相关的炎症反应,如免疫性肺炎、免疫性肝炎、免疫性肠炎等,需要密切监测和及时处理。
预测疗效困难:目前还缺乏准确可靠的方法来预测哪些患者能够从免疫治疗中获益,导致治疗的选择存在一定的盲目性。
随着研究的不断深入和技术的进步,免疫疗法的优势有望进一步扩大,劣势也有望逐渐得到改善。在实际治疗中,医生会根据患者的具体情况,综合考虑各种疗法的特点,制定最适合的个体化治疗方案。
免疫疗法的适用人群有哪些?
免疫疗法的适用人群主要包括以下几类:
晚期癌症患者
对于一些晚期癌症,如晚期黑色素瘤、非小细胞肺癌、肾癌、膀胱癌等,在传统治疗方法效果不佳时,免疫疗法可能成为一种有效的治疗选择。
具有特定基因突变的患者
例如携带微卫星高度不稳定(MSI-H)或错配修复缺陷(dMMR)的实体瘤患者,往往对免疫治疗有较好的反应。
肿瘤突变负荷(TMB)高的患者
TMB 高通常意味着肿瘤产生了更多的新抗原,更容易被免疫系统识别,免疫治疗效果可能更好。
部分血液系统恶性肿瘤患者
如某些类型的淋巴瘤等。
一线治疗后复发或进展的患者
在其他治疗方案失败后,免疫疗法可能为他们提供新的治疗机会。
然而,并非所有癌症患者都适合免疫疗法。医生会综合考虑患者的身体状况、肿瘤类型、基因突变情况等多种因素,来判断患者是否适合接受免疫治疗。同时,在治疗过程中也会密切监测患者的反应和副作用,以便及时调整治疗方案。
哪些人群不适合免疫疗法?
以下人群可能不适合免疫疗法:
自身免疫性疾病控制不佳的患者
如患有严重的类风湿关节炎、系统性红斑狼疮等,免疫治疗可能会加重自身免疫反应,导致病情恶化。
器官功能严重障碍的患者
例如存在严重的心肺功能不全、肝肾功能衰竭等,可能无法耐受免疫治疗带来的潜在副作用。
处于急性感染期的患者
身体正在对抗严重的感染,此时进行免疫治疗可能会进一步影响免疫系统的平衡,加重感染或影响治疗效果。
怀孕或哺乳期妇女
免疫治疗可能对胎儿或婴儿产生潜在的不良影响。
对免疫治疗药物成分过敏的患者
这会引发严重的过敏反应,危及生命。
患有精神疾病或无法配合治疗和随访的患者
免疫治疗需要患者密切配合治疗和定期随访,以监测疗效和不良反应。
对于每个患者是否适合免疫疗法,需要医生根据患者的具体情况进行综合评估和判断。
免疫疗法有哪些不良反应?
免疫疗法的副作用是一个重要的问题,因为它们可能会影响患者的生活质量和治疗效果。
免疫疗法的副作用包括疲劳、发热、恶心、呕吐、腹泻、皮疹、瘙痒、呼吸困难、头痛、肌肉疼痛、关节疼痛等。需要对这些副作用进行评估和管理。
免疫疗法可能会引起多种不良反应,免疫疗法的副作用可以分为两类:
与免疫疗法直接相关的副作用包括免疫相关不良事件(irAE)和免疫检查点抑制剂相关不良事件(ICI-AE)。
皮肤反应:如皮疹、瘙痒、白癜风等。
胃肠道反应:如腹泻、结肠炎、恶心、呕吐等。
内分泌系统紊乱:可能导致甲状腺功能异常(甲状腺功能减退或亢进)、垂体炎、肾上腺功能不全等。
肝脏毒性:表现为转氨酶升高、肝炎等。
肺部问题:如肺炎、咳嗽、呼吸困难等。
心血管系统不良反应:如心肌炎、心包炎、心律失常等,严重情况下可能危及生命。
关节和肌肉疼痛:可能出现关节炎、肌痛等症状。
神经系统症状:头痛、头晕、外周神经病变等。
这些不良反应的发生机制主要与免疫系统被过度激活,攻击自身正常组织和器官有关。不过,并非每个接受免疫治疗的患者都会出现不良反应,且不良反应的严重程度因人而异。在治疗过程中,医生会密切监测患者的身体状况,以便及时发现和处理这些问题。
免疫疗法在治疗周期内需要注意什么?
在免疫疗法的治疗周期内,需要注意以下几个方面:
饮食方面:保持均衡的饮食,摄入富含蛋白质、维生素和矿物质的食物,以增强身体的抵抗力。避免食用过多油腻、辛辣和刺激性食物。
作息规律:保证充足的睡眠,规律作息,避免过度劳累,有助于维持免疫系统的正常功能。
按时复查:按照医生的要求定期进行血常规、肝肾功能、肿瘤标志物等检查,以及影像学检查(如 CT、MRI 等),以便及时监测治疗效果和发现可能出现的不良反应。
关注身体变化:留意自身的身体状况,如是否出现发热、咳嗽、皮疹、腹泻、关节疼痛等异常症状。若有不适,及时告知医生。
用药管理:严格按照医嘱用药,不可自行增减药量或停药。
心理调节:保持积极乐观的心态,避免过度焦虑和紧张,可以通过适当的运动、与家人朋友交流等方式缓解心理压力。
避免感染:注意个人卫生,勤洗手,避免去人员密集、容易感染的场所。
运动适度:根据自身身体状况,进行适量的运动,如散步、瑜伽等,但避免剧烈运动和过度劳累。
告知医生其他用药:如果在治疗期间需要使用其他药物,包括保健品、中草药等,应提前告知医生,以防药物相互作用影响免疫治疗效果或增加不良反应风险。
主要参考文献
Zhao W, Lei J, Ke S, Chen Y, Xiao J, Tang Z, Wang L, Ren Y, Alnaggar M, Qiu H, Shi W, Yin L, Chen Y. Fecal microbiota transplantation plus tislelizumab and fruquintinib in refractory microsatellite stable metastatic colorectal cancer: an open-label, single-arm, phase II trial (RENMIN-215). EClinicalMedicine. 2023 Nov 14;66:102315.
Yousefi Y, Baines KJ, Maleki Vareki S. Microbiome bacterial influencers of host immunity and response to immunotherapy. Cell Rep Med. 2024 Apr 16;5(4):101487.
Lu Y, Yuan X, Wang M, He Z, Li H, Wang J, Li Q. Gut microbiota influence immunotherapy responses: mechanisms and therapeutic strategies. J Hematol Oncol. 2022 Apr 29;15(1):47.
Balan Y, Sundaramurthy R, Gaur A, Varatharajan S, Raj GM. Impact of high-salt diet in health and diseases and its role in pursuit of cancer immunotherapy by modulating gut microbiome. J Family Med Prim Care. 2024 May;13(5):1628-1635.
Kang X, Lau HC, Yu J. Modulating gut microbiome in cancer immunotherapy: Harnessing microbes to enhance treatment efficacy. Cell Rep Med. 2024 Apr 16;5(4):101478.
Zhang, M., Liu, J. & Xia, Q. Role of gut microbiome in cancer immunotherapy: from predictive biomarker to therapeutic target. Exp Hematol Oncol 12, 84 (2023).

谷禾健康

饮食在塑造肠道微生物群的组成、功能和多样性方面起着关键作用,各种饮食对肠道内微生物群落的稳定性、功能性和多样性有着深远的影响。了解不同饮食对微生物群的深远影响至关重要,改善代谢和肠道健康,预防和减缓由饮食不当引起的特定饮食相关疾病的发生。
在生命早期,分娩方式、喂养、饮食和环境等因素会塑造肠道微生物群。在成年期,虽然微生物群趋于相对稳定,但外界因素,尤其是饮食,会大大影响其组成和功能。营养素、微生物群和免疫系统之间的这种复杂相互作用是维持体内平衡和防御外部病原体的重要调节机制。
精准营养承认每个人对饮食的代谢反应会有所不同,因此针对人群健康的广泛饮食指南在个人层面上并不理想。一些大规模研究已开始将微生物组概念纳入精准营养,发现纳入肠道微生物组组成的预测模型远远优于仅基于宿主、饮食和身体活动因素的预测模型。
比如从控制体重来说,我们常常关注卡路里的摄入与消耗,却可能忽略了肠道菌群层面的理解。不同人群可以选择不同的方式,高纤维饮食可以促进产生短链脂肪酸的肠道细菌的生长,这些短链脂肪酸不仅有助于维持肠道健康,还可能通过调节食欲和能量代谢等方式来帮助控制体重。
鉴于测序和机器学习等方面技术的最新进展,极大地提高了人们对饮食及其对微生物群影响的理解。在此基础上,本文讨论了常见整个饮食方式(如地中海饮食、高纤维饮食、植物性饮食、高蛋白饮食、生酮饮食、西方饮食、间歇性禁食、热量限制饮食等)影响肠道微生物群的机制,还包括生命早期和成年期肠道微生物群相关的饮食相关慢性疾病,临床实践中用于缓解或预防疾病进展的特定饮食等。
微生物组研究成果的迅速扩展使多种长期营养原则变得复杂,同时也为干预提供了新的机会。更深入地了解饮食、宿主和微生物之间的因果关系,可以为开发精准营养和基于微生物组的疗法提供新的视角。
饮食对肠道微生物群的组成和功能有相当大的有益或负面影响。
下图是常见饮食方式对肠道菌群的影响,这在后面我们会详细展开阐述。
全膳食的常量营养素组成及其对肠道菌群的影响

doi.org/10.1038/s41579-024-01068-4
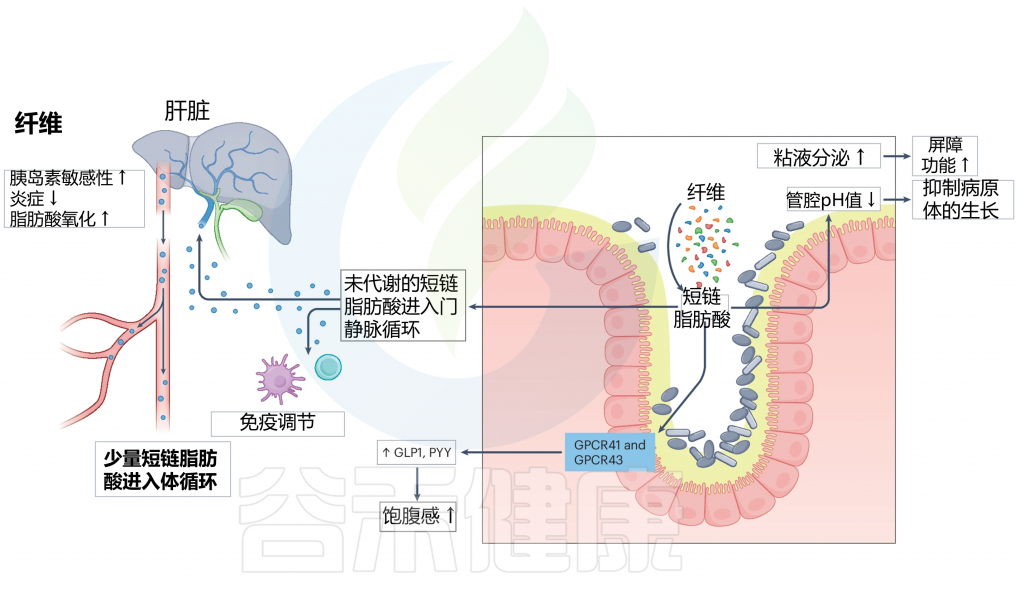
当膳食纤维到达肠道时,会经过肠道微生物群的发酵,产生如乙酸盐、丙酸盐和丁酸盐等短链脂肪酸(SCFA)。这些短链脂肪酸随后进入门脉循环,对宿主健康产生一系列积极影响。
激活GPCRs
短链脂肪酸激活G蛋白偶联受体GPCRs 41和43,这是它们发挥作用的初步机制。
触发肠道激素分泌
激活的受体进一步触发胰高血糖素样肽(GLP)和肽YY(PYY)等肠道激素的分泌。
注:GLP1和PYY在调节食欲、减缓胃排空和促进饱腹感方面起着关键作用。
增强肠道屏障功能
SCFAs通过增加粘液分泌和降低肠腔pH值来增强肠道屏障功能,保护肠道内壁,防止有害病原体进入血液。
抗炎与免疫调节作用
SCFAs具有抗炎和免疫调节作用,有助于维持整体肠道健康,并降低胃肠道疾病的风险。
肠道微生物群对纤维的分解及其对屏障功能和免疫力的影响
doi.org/10.1038/s41579-024-01068-4
在肠道中,膳食蛋白质经过肠道微生物群的代谢,这与拟杆菌属的增加有关。这导致产生各种代谢产物,包括短链脂肪酸、支链脂肪酸(BCFAs)和吲哚。
支链脂肪酸可以激活 GPCR41 和 GPCR43,从而触发 GLP1 和 PYY 等肠道激素的分泌。此外,BCFAs 可以增加粘液分泌并降低腔内 pH 值,从而增强肠道屏障功能并保护肠道内壁。
肠道微生物群对蛋白质的代谢以及SCFA和吲哚对人类健康的后续影响
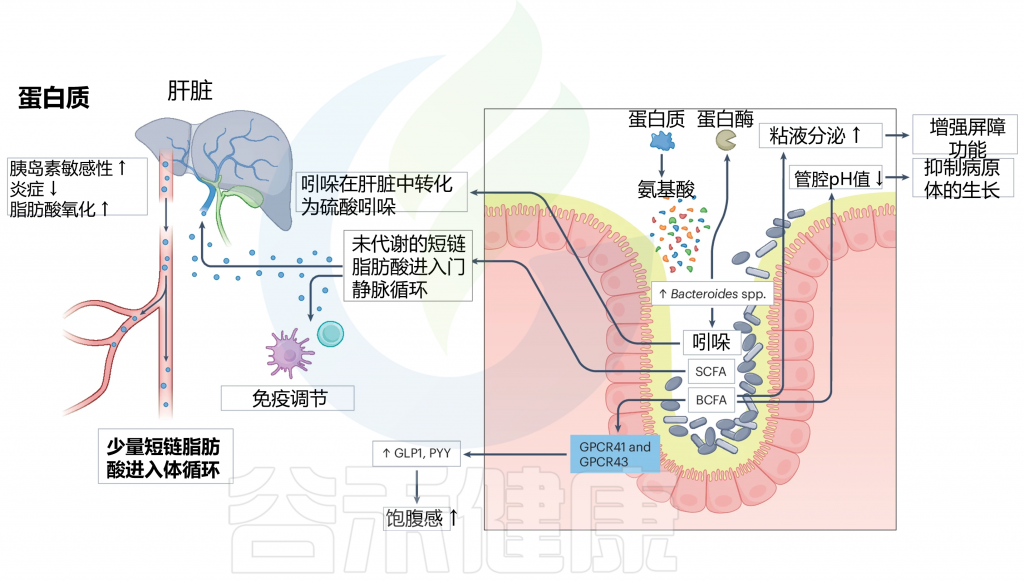
doi.org/10.1038/s41579-024-01068-4
SCFAs、BCFAs、GLP1 和 PYY 等肠道激素、粘液分泌和腔内 pH 对人体健康的影响,包括改善胃肠功能、调节食欲、减少炎症、改善胰岛素敏感性和脂肪酸氧化,从而促进整体肠道健康。
当膳食PUFAs到达肠道时,它们会被肠道微生物群代谢。这一过程增加了特定细菌的丰度,如双歧杆菌属和产丁酸菌。因此,产生了各种代谢产物,如短链脂肪酸,例如丁酸盐。
PUFAs可以减少促炎的肠杆菌属(Enterobacterium)的丰度,从而减少炎症并改善肠道屏障功能。这可能导致内毒素和IL-17的产生减少,进而减少炎症并改善对人类健康的影响。由PUFA代谢产生的未代谢SCFAs进入系统循环,在其中发挥免疫调节作用。它们可以通过改善胰岛素敏感性、减少炎症和改善肠道渗漏症内毒素血症来增强抵抗肥胖的能力。
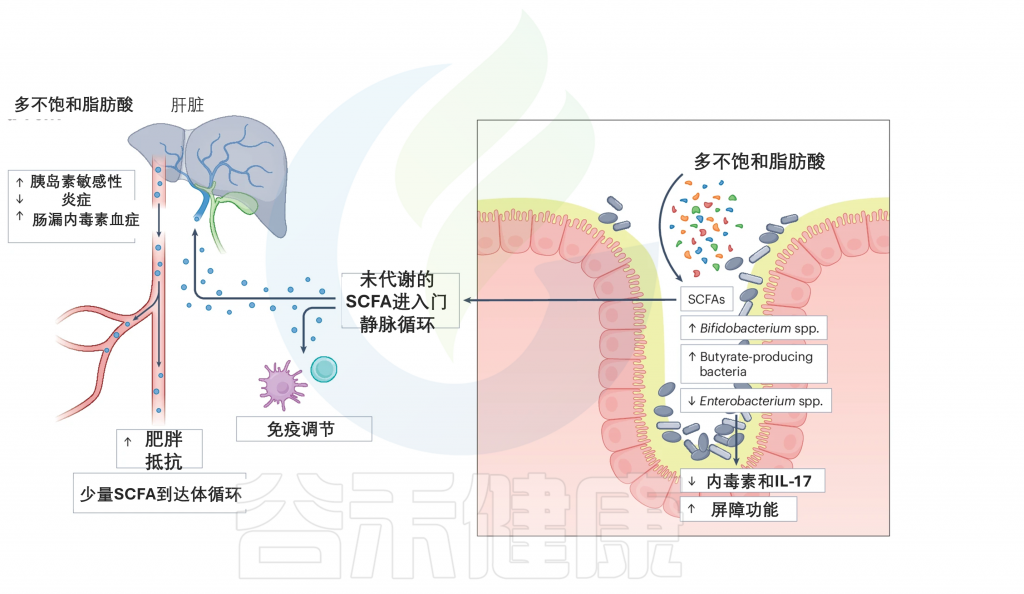
doi.org/10.1038/s41579-024-01068-4
多酚类物质被肠道细菌代谢,因此被分解成生物活性微生物代谢产物。多酚已被证明可以增加肠道腔中有益细菌的丰度,如双歧杆菌、Akkermansia、乳酸杆菌属。这些细菌在维持肠道屏障功能、调节免疫系统、促进肠道稳态和抑制病原菌生长方面起着至关重要的作用。
此外,多酚在肠道内表现出显著的抗炎和抗氧化作用。多酚代谢的副产物,缺乏酚类的代谢产物,在系统循环中被吸收,在那里它们发挥显著的免疫调节作用。例如,这些代谢产物已被证明可以通过减少炎症和氧化应激,以及改善内皮功能,从而改善肺部、大脑和心脏功能,增加周围血流。
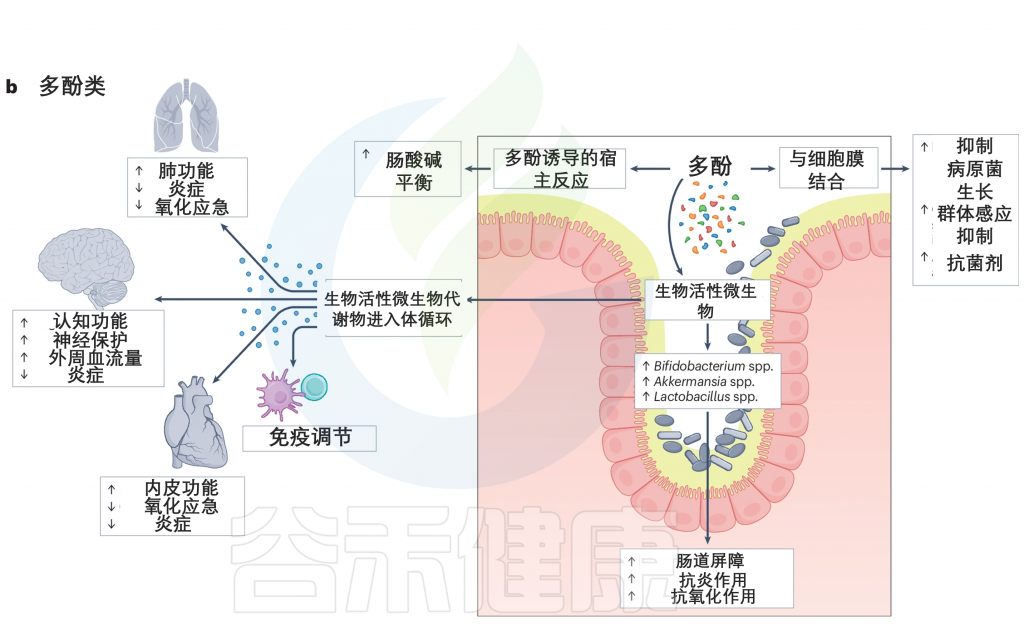
doi.org/10.1038/s41579-024-01068-4
肠道微生物组的差异性影响
不同肠道微生物群对宿主能量状态的贡献存在差异,与肥胖相关的肠道微生物组特征可能会加剧宿主的表型。
遗传性肥胖小鼠及其瘦弱的同窝小鼠在肠道微生物组成上存在差异,从ob/ob供体获得的肠道微生物群受体增加的体脂,比从遗传性瘦弱供体获得的微生物群受体多。
将适应高脂高糖(HFHS)饮食的小鼠肠道微生物群与适应低脂高植物多糖饮食的小鼠肠道微生物群进行移植,一致地增强了接受控制饲料的无菌受体小鼠的脂肪积累。
这些研究表明,无论是由遗传还是饮食驱动的肥胖表型,都可以通过肠道微生物群传播。
肠道微生物组与营养不良
患有夸希奥科病(kwashiorkor)的儿童的肠道微生物群表现出发育不良的特征,并通过在无菌小鼠中定植后与健康对照相比,损害了营养吸收,从而在因果上对营养不良有所贡献。
肠道微生物群的变化也已被证明有助于极低热量饮食(VLCDs)和Roux-en-Y胃旁路手术后的快速减重。
例如,对超重或肥胖的绝经后妇女进行每天800千卡的极低热量饮食,导致肠道微生物群的变化和改善的代谢表型,如体重减轻和减少的脂肪量,这些变化可以在接受了节食前后肠道微生物群的无菌小鼠受体中重现。
肠道微生物组的能量缓冲作用
与低消化性饮食相关的更高营养流入结肠可以以一种增强其对宿主能量状态贡献的方式改变肠道微生物群,表现为接受低消化性饮食条件的微生物群的无菌小鼠受体体重增加和脂肪量更多。
在这个宿主-微生物组生态共生的例子中,宿主的营养吸收较低被肠道微生物群衍生的代谢产物及其下游效应所部分缓冲,例如增加宿主的能量摄入。这样的能量缓冲在能量受限条件下可能有助于宿主的代谢健康,但在能量过剩条件下也可能妨碍体重管理。
肠道微生物组的环境和饮食依赖性
肠道微生物群对宿主能量平衡的贡献可能依赖于环境和饮食背景,即使不通过饮食操纵宿主能量平衡也是如此。
来自肥胖不一致的人类双胞胎的无菌小鼠受体通常模仿了它们供体的代谢表型,但是当差异性定植的受体动物共同饲养时,来自瘦弱供体的微生物群侵入了来自肥胖供体的微生物群,结果是两者都保持了瘦弱。
当共同饲养的受体动物被喂食高脂肪和低水果蔬菜的饮食时,与瘦弱相关的微生物群的传播性被破坏了。
这些复杂的相互作用强调了饮食对宿主-微生物组代谢相互作用的影响有时可能难以追踪。
肠道微生物通过其代谢产物影响健康
短链脂肪酸可以被各种宿主组织转化为ATP,其中:
SCFAs具有多样的信号功能,影响能量平衡。
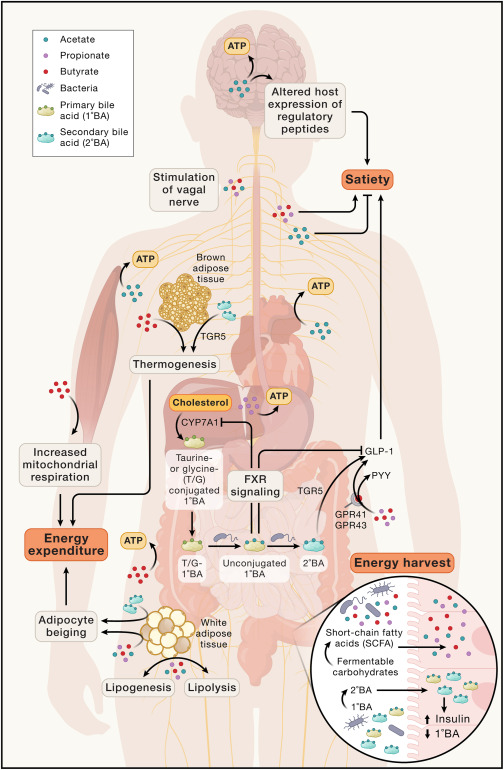
doi.org/10.1038/s41579-024-01068-4
SCFA通过各种方法影响能量摄入,包括乙酸盐穿过血脑屏障,介导调节性神经肽的表达,丙酸盐和丁酸盐结合肠内分泌L细胞中的GPR41和GPR43受体,刺激GLP-1和PYY的释放,以及通过迷走神经的肠脑信号传导,乙酸盐与SCFA混合物可能不同地介导这些信号传导。
SCFA通过促进棕色脂肪组织的产热、白色脂肪组织的米色和骨骼肌的线粒体呼吸来影响能量消耗。SCFA还可以影响脂肪生成和脂肪分解的动力学,据报道,丁酸盐促进脂肪分解,而乙酸盐和丙酸盐促进脂肪生成。
此外,肠道微生物组可以使宿主肝脏分泌的牛磺酸或甘氨酸结合的初级胆汁酸(T/G-1°BA)脱偶联和脱羟基,产生调节宿主能量代谢各个方面的非偶联初级胆汁酸和次级胆汁酸。未结合的初级胆汁酸通过法尼醇X受体(FXR)发出信号,抑制CYP7A1,CYP7A1是初级胆汁酸合成的限速步骤,对饮食脂肪吸收具有潜在的下游影响。次级胆汁酸激活TGR5,促进棕色脂肪组织的产热、白色脂肪组织的米色和胰腺β细胞的胰岛素产生。
肠道微生物胆汁酸代谢也可能通过对厌食素GLP-1的对比作用来影响能量摄入,2°BA激活的TGR5信号促进L细胞分泌GLP-1,1°BA活化的FXR信号在小鼠中显示出抑制GLP-1活性。这些多效性效应强调了对SCFA和胆汁酸的看法正在发生变化,从能量收获的载体转变为能够对宿主能量状态产生净积极和净消极影响的代谢调节因子.
地中海饮食(MD)强调摄入大量未加工的全植物性食品、橄榄油、乳制品、适量家禽和鱼类,以及少量红肉。
降低癌症死亡率及糖尿病风险
一项对美国25,315名女性的前瞻性研究显示,那些坚持地中海饮食模式的人在25年的随访期间全因死亡率降低了23%。这项研究还显示,较高的地中海饮食摄入量与20年随访期间未来2型糖尿病风险降低30%相关。地中海饮食模式可能还对癌症有保护作用。实际上,高度遵守这种饮食与普通人群中的癌症死亡率降低、癌症幸存者的全因死亡率降低,以及降低发展结直肠癌、头颈癌、呼吸、胃、肝和膀胱癌风险有关。
增加产丁酸菌
两项干预研究将地中海饮食与特定分类特征联系起来,增加Faecalibacterium prausnitzii、Roseburia丰度,减少Ruminococcus gnavus、Collinsella aerofaciens、Ruminococcus torques丰度。这些因饮食而导致的微生物组变化与短链脂肪酸产量的增加和代谢副产物(如乙醇、对甲酚和二氧化碳)产量的减少有关。
地中海饮食与特定功能途径有关
之前研究用宏基因组测序分析了307名男性长期饮食信息的微生物组数据。结果显示,地中海饮食与36条功能途径有关,这些途径大多类似于植物性饮食,具有丰富的微生物功能,用于SCFA发酵和膳食纤维降解。对地中海饮食的坚持显示出与特定功能途径的正相关,如用于果胶分解的d-果糖醛酸降解途径和用于半纤维素分解的甘露聚糖降解途径。地中海饮食的坚持和降低心血管疾病风险在P. copri水平较低的个体中更为明显。
地中海饮食plus版——更积极的变化相关
最近,DIRECT-PLUS研究包括294名肥胖或血脂异常的参与者,发现与地中海饮食相比,绿色地中海饮食与更显著的组成变化相关。绿色地中海饮食是地中海饮食的增强版,它增加了植物性食品的摄入量,减少了红肉的摄入,并且每天还摄入富含多酚的绿茶和Mankai水生植物。
这种饮食在微生物组成和多样性上产生了更大的变化,包括增加普雷沃特氏菌的丰度和支链氨基酸降解酶(异亮氨酸降解),减少双歧杆菌和支链氨基酸生物合成酶(缬氨酸和异亮氨酸生物合成)。这些变化与体重和心代谢指标的积极变化相关联。
膳食纤维对人类健康至关重要,它有助于降低长期体重增加,低纤维摄入量会增加患2型糖尿病和结肠癌的风险。
高纤维饮食会改变肠道微生物的组成,包括显著增加乳酸杆菌属和双歧杆菌属的丰度。
断奶后饮食变化,引起代谢复杂多糖的菌增加
不同的膳食纤维组分对肠道微生物的影响各不相同。例如,母乳喂养的婴儿表现出更高丰度的适应于利用人乳寡糖(HMOs——母乳中大量存在的不可消化的益生元糖类)的双歧杆菌。断奶后,肠道微生物组成会发生明显变化,这主要归因于饮食组成的改变。这导致能代谢更复杂多糖的拟杆菌门和厚壁菌门的扩张。
超重个体:改善菌群预防代谢疾病
在超重的个体中,阿拉伯木聚糖低聚糖的干预增加了普雷沃氏菌和直肠真杆菌(Eubacterium rectale)的丰度,伴随着代谢组学特征的有利变化,可能有助于预防代谢性疾病。
全谷物和小麦麸皮:双歧杆菌、乳杆菌↑↑
在31名志愿者中补充全谷物和小麦麸皮,导致双歧杆菌属和乳酸杆菌属的水平增加。全谷物消费者中的增加更为明显;两组都经历了总胆固醇的降低。
燕麦:厚壁菌门↑ 拟杆菌门↓ 心血管疾病风险↓
来自燕麦的高分子量β-葡聚糖减少了厚壁菌门,增加了拟杆菌门,并伴随着心血管疾病风险标志物的减少。
抗性淀粉:影响短链脂肪酸产生
以IV型抗性淀粉形式的膳食纤维对肠道微生物群的组成和功能以及丁酸盐或丙酸盐的产生了不同的影响。
简单碳水化合物在小肠中吸收,而复杂碳水化合物如膳食纤维则经历结肠微生物发酵,从而产生短链脂肪酸。人类只产生非常有限的用于碳水化合物降解的碳水化合物活性酶(CAZymes),因此依赖于肠道微生物群间接代谢几种膳食纤维。低纤维的饮食与肠道微生物群中减少的CAZyme储备相关。
短链脂肪酸的健康益处
包括前面文中提到过的,通过GPCRs传递信号,以及刺激肠道内分泌细胞分泌饱腹感激素(GLP-1和肽YY)。这影响了食欲调节,并调节了调节性T细胞的功能,以及脂质和葡萄糖代谢,在调节宿主能量代谢和结肠稳态中发挥关键作用。
丁酸盐作为结肠细胞的能量来源,通过肠细胞(巨噬细胞和树突状细胞)介导抗炎特性,并增强粘液产生,这突出了其在优化肠道吸收和肠道屏障功能中的作用。
短链脂肪酸与GPCRs及其他细胞的作用和互动不仅限于肠道,还扩展到外周组织、器官和免疫细胞。在小鼠模型中的报告表明,SCFAs和高纤维饮食可能在降低1型糖尿病、2型糖尿病、哮喘和压力的风险,减少脂肪酸合成和脂肪分解方面发挥作用,从而减轻体重并增强神经认知发展。SCFA的吸收导致肠腔pH值降低,这抑制了对pH敏感的病原体如梭菌纲和肠杆菌科的生长,并增加了营养素吸收。
全谷物中的不可溶纤维影响肠道传输速率和细菌发酵
两项随机对照交叉试验涉及50名超重或有代谢综合征风险的个体,表明全谷物饮食增加了粪便中的丁酸盐和己酸盐,改善了血脂水平,减少了炎症标志物,并与精制谷物饮食相比改善了体重减轻。产短链脂肪酸的菌与结肠传输时间显示出负相关关系。这进一步有助于调节肠道微生物组成和多样性,从而缓解各种肠道疾病,如肠易激综合症、炎症性肠病、结直肠癌和胃癌以及便秘。
微生物群与人类健康之间的相互作用强调了采取整体方法和更大规模的人类研究的必要性,以便深入认识饮食碳水化合物、肠道微生物群组成和疾病易感性之间复杂的关系。
植物性饮食富含多酚类、宿主可消化和不可消化的碳水化合物,并发挥益生元和后生元的双重效应。素食饮食导致形成独特的细菌环境,这一点从细菌功能能力的转变中得到证实。
素食者:拟杆菌↑ 普雷沃氏菌属↑
例如,素食者表现出低肉碱降解但增加氮同化。与杂食者饮食相比,这些饮食促进了拟杆菌门和普雷沃氏菌属的丰度,尽管由于微生物个体差异和研究方法的不一致性,研究结果有时会出现矛盾。
某些属或种的对比水平可以归因于饮食快速与逐渐转变对微生物造成的压力、健康与不健康饮食成分的存在,以及各种生物活性化合物的来源。例如:
植物性饮食的这些特性使其在预防和管理慢性疾病,如心血管疾病、2型糖尿病和某些癌症方面显示出潜力。然而,需要更多的研究来充分理解植物性饮食对肠道微生物组的具体影响,以及这些变化如何影响宿主的健康和疾病风险。
多酚类物质的吸收:少量在小肠,大量在结肠
多酚类物质,分为类黄酮和非类黄酮,是植物的次级代谢产物,存在于水果、蔬菜、谷物、葡萄酒、茶、咖啡等食物中。
少量的多酚类物质(5%~10%)在小肠中被吸收,主要是那些具有单体和二体结构的多酚。吸收后,苷元在肠细胞内经历生物转化,然后在肝细胞内继续转化。这些代谢产物通过循环系统运输到肾脏和肝脏等器官,并最终随尿液排出。
大部分多酚类物质(90%-95%)在回肠和结肠中与肠道微生物发生作用,它们促进双歧杆菌、Akkermansia、乳杆菌等物种的丰度,从而提供显著的抗炎和抗病原体特性,以及心血管保护作用。
最近一项涉及超过2万名成年人的随机对照试验表明,食用富含多酚的可可提取物减少了心血管疾病导致的死亡。然而,心血管疾病的发生并没有减少。
多酚类物质的抗菌和抗病原体特性
多酚类物质可以通过几种机制抑制细菌生长,包括结合并改变细胞膜的功能特性。它们还展现出对食源性病原体的抗菌活性,并以剂量依赖性方式作为群体感应抑制剂和抗菌剂。
肠道微生物群代谢多酚
肠道微生物群双向调节并代谢多酚类物质,将它们转化为更具生物活性的微生物代谢产物,并提高其相对于原始化合物的吸收。
代谢产物的健康益处
研究表明,食用生物活性微生物代谢产物对人类健康有益处。例如:
多酚类物质对肠道微生物群的调节
多酚类物质可以通过改变肠道微生物群的组成和影响各种微生物酶的功能,调节肠道微生物代谢产物,包括短链脂肪酸、TMAO、多巴胺、脂多糖、胆汁酸。
这最终可以通过多种方式引起多酚类物质诱导的宿主反应,例如,作为调节肠道酸碱平衡的调节器。多酚类物质对肠道微生物群的调节已被证明支持肺功能、中枢神经系统功能和肠道屏障完整性的稳态。
植物和动物源食物类型不同,对菌群影响有差异
植物和动物源性食物中蛋白质和脂肪类型的不同导致了肠道微生物组成和代谢组的差异。例如,基于动物的饮食导致耐胆汁细菌种类的丰度增加,如Alistipes、Bilophila,同时减少了厚壁菌门的丰度,降低了支链氨基酸(BCAAs)的水平,并增加了SCFAs和二甲基硫化物。
其他植物化合物,如纤维、萜类和类胡萝卜素,也已显示出健康益处。个体在从饮食多酚中产生酚类衍生代谢产物的量上的差异归因于每个人肠道微生物组的独特组成。
因此,分析多酚代谢产物可以作为一种有价值的方法,以更深入了解生物活性化合物效应,并为理解个体间的显著多样性提供全面的认识。
每日蛋白质摄入量超过1.5克/千克体重的饮食通常被认为是高蛋白饮食。这种饮食通常用于运动员或为超重人群减肥时所推荐。
蛋白质的消化和吸收
饮食中的蛋白质主要由宿主的蛋白酶分解,但每天有12-18克的蛋白质可到达大肠并被微生物群代谢。
不同类型的复杂蛋白质具有不同程度的可消化性,以及不同的氨基酸组成。
参与蛋白质分解的菌群
一些细菌物种参与蛋白质分解,并在高蛋白饮食者的肠道微生物群中富集,主要是拟杆菌属、芽孢杆菌属(Bacillus)、梭菌属(Clostridium)、Phocaeicola、丙酸杆菌属(Propionibacterium)、梭杆菌属(Fusobacterium)、乳杆菌属、链球菌属。
其他细菌可以直接利用氨基酸,并从蛋白质分解中受益,形成交叉喂养的相互作用。
蛋白质分解细菌使用多种酶
蛋白质分解细菌使用多种外肽酶、蛋白酶(包括金属、丝氨酸、半胱氨酸、天冬氨酸、苏氨酸、谷氨酸和天冬酰胺蛋白酶)和内肽酶来释放短肽和游离氨基酸。
氨基酸代谢产生短链脂肪酸
大多数氨基酸被发酵成短链脂肪酸:
部分发酵产物可能带来的健康危害
其他发酵产物包括可能的炎症化合物,如来自芳香族氨基酸(例如色氨酸)的吲哚和酚类化合物,以及氨、胺、有机酸和气体(即由含硫氨基酸半胱氨酸和甲硫氨酸产生的硫化氢,以及二氧化碳)。
值得注意的是,这些最终产物中的一些可能与疾病有关。吲哚和吲哚相关化合物可以到达肝脏并转化为硫酸吲哚酚,这是一种对肾脏有害的有毒代谢产物,并参与内皮功能障碍。此外,硫化氢可能具有致突变性,并可能在炎症中发挥作用,增加结肠癌的风险。
生酮饮食是一种极低碳水化合物、适量蛋白质和高脂肪的饮食模式,模拟了禁食期间的代谢反应,这种状态下循环酮体水平升高。
注:酮体是脂肪酸衍生的分子,当葡萄糖可用性受限时作为替代能量来源。这些酮体(KBs)包括β-羟基丁酸(βHB)、乙酰乙酸和丙酮,主要在肝脏中产生。
生酮饮食长期以来一直作为治疗癫痫的饮食疗法,并且越来越多的研究表明这种饮食在治疗阿尔茨海默症、肥胖症、癌症等各种疾病方面的益处。
注:传统的长链甘油三酯生酮饮食遵循脂肪(克)与蛋白质和碳水化合物总和的4:1比例。变体包括中链甘油三酯生酮饮食、改良阿特金斯饮食和低血糖指数治疗,每种方法都有稍微不同的宏观营养素比例。
在人类中,诱导生酮状态需要严格限制碳水化合物摄入(5%–10%千卡/天),适量蛋白质摄入(30%–35%),和高脂肪摄入(55%–60%)。
生酮饮食的潜在风险和副作用
生酮饮食(利于拟杆菌门) ≠ 高脂饮食(利于厚壁菌门)
典型的高脂饮食通常会增加厚壁菌门的丰度并减少拟杆菌门;然而,生酮饮食的效果不同。
——超重成年人
在涉及17名超重成年人的研究中,为期4周的生酮饮食显示在人肠道中放线菌门(Actinobacteria)和厚壁菌门的大量减少。具体来说,有益的双歧杆菌的19种物种减少了,而拟杆菌门丰度增加。这些变化部分是通过宿主产生酮体诱导的。
——癫痫儿童
在涉及12名严重癫痫儿童的为期3个月的研究中,遵循生酮饮食的儿童显示健康促进和消耗纤维的双歧杆菌属、直肠真杆菌(E. rectale)和Dialister属的丰度大幅减少。相反,儿童显示拟杆菌属和大肠杆菌属的丰度增加,后者部分归因于大肠杆菌(Escherichia coli)的增加。
生酮饮食对肠道微生物组的影响
临床前研究也表明,肠道微生物组的组成在响应生酮饮食时发生了显著变化,最明显的是:
酮体βHB↑ 双歧杆菌↓
一项分析生酮饮食对肠道微生物组组成的变化的潜在机制的研究报告了在人类和鼠类受试者中,双歧杆菌属和酮体β-羟基丁酸(βHB)之间的显著负相关,也就是说,随着βHB水平的增加,双歧杆菌属的水平会降低。
来自人类、啮齿动物和细胞培养的数据支持β-羟基丁酸抑制NLRP3炎症体的能力。高水平的酮体可以降低血压并增加血管功能。循环酮体水平的增加还可以减少心脏炎症和心力衰竭的可能性。酮体也可能通过刺激胰岛素受体,通过诱导AMP激活蛋白激酶(AMPK)和下调mTOR来改善胰岛素敏感性。高水平的酮体可能减少食欲,从而使体重减轻。
生酮饮食→双歧杆菌↓→减少诱导Th17→促炎降低
将生酮饮食者的粪便微生物群移植到无菌小鼠中,研究揭示了肠道TH17细胞的变化。
注:Th17细胞是一种辅助性T细胞亚群,其主要特征是能够产生多种促炎细胞因子,如IL-17、IL-21和IL-22等。
双歧杆菌属对肠道TH17细胞的有强烈诱导作用,而生酮饮食改变肠道菌群(双歧杆菌降低)也减少了诱导Th17的能力,可能导致这些细胞的促炎性降低,从而影响肠道和脂肪组织的炎症状态,
然而,由于有益的肠道微生物群的减少和促炎性及病原性肠道细菌的促进,需要进一步的研究来了解生酮饮食对宿主健康的长期影响。
西方饮食的特点是高热量含量,富含动物蛋白、饱和脂肪、简单糖和超加工食品,同时纤维、水果和蔬菜的摄入量不足。
西方饮食:多样性下降,拟杆菌为主
与其他饮食相比,西方饮食与肠道微生物组多样性的显著降低有关,其肠道特征转向以拟杆菌属为主的肠道特征。其他丰富的物种属于Ruminococcus、Faecalibacterium、双歧杆菌属、Alistipes、Blautia、Bilophila。
由于纤维摄入较少和不同的微生物组成,相关的微生物群产生的短链脂肪酸较少。
红肉中胆碱→TMAO→多种慢病相关
红肉中的特定化合物,如胆碱和肉碱,也可以被肠道微生物群转化为三甲胺,然后在肝脏中转化为与慢性疾病相关的三甲胺-N-氧化物(TMAO)。
加工食品和添加剂的影响
加工食品包含各种添加剂、防腐剂和乳化剂,能够直接或间接与肠道微生物群相互作用。
非营养性人造甜味剂,如低热量或饮食食品和饮料中的糖精、三氯蔗糖和阿斯巴甜,对微生物组多样性和组成的潜在长期影响尚不清楚。
其他添加剂,如卡拉胶(一种从红海藻中提取的增稠剂或凝胶剂,存在于许多加工食品中,如乳制品),已知会促进肠道炎症和破坏粘液层,导致肠道微生物组的变化。
人工食品色素,如糖果和烘焙产品中的Allura Red AC,赋予颜色并通过与肠道细菌的相互作用改变硫的稳态。
一些防腐剂,如加工肉类中的硝酸钠,也可以调节肠道微生物组的组成,而乳化剂,如羧甲基纤维素(一种存在于酱汁中的增稠剂)和聚山梨醇酯-80(一种存在于酱汁和烘焙食品中的乳化剂和稳定剂),直接冲击肠道微生物组的组成和功能。
详见我们之前的文章:
你的焦虑可能与食品添加剂有关,警惕食品添加剂引起的微生物群变化
糖,功能糖,代糖,如何从健康角度看这些肠道菌群的“甜蜜伙伴”
总体而言,西方饮食与慢性炎症的激增有关,导致与饮食相关的疾病,包括肥胖和其他非传染性疾病。
过去50年中,一种受到极大关注的饮食疗法是日常热量限制(CR),它被定义为在保持充足营养的同时,将饮食摄入量减少至低于维持体重所需的能量水平。观察性、临床前和临床试验的发现表明,CR可能将寿命延长1-5年,同时改善生活质量。
最严格的CR随机试验来自国家老龄化研究所资助的CALERIE(减少能量摄入长期效应综合评估)联盟。CALERIE研究包括CALERIE第一阶段(三项为期6至12个月的CR小规模试点研究)和CALERIE第二阶段(一项大型、多中心、为期2年的CR随机试验)。
注:这些研究招募了体重正常且健康状况良好的成年人。每项试验中实施的CR程度不同,但通常涉及日常能量摄入量减少10%至30%,同时确保其他关键营养素的充足摄入。
CR的健康益处
CALERIE研究的发现显示,短期和长期CR都可以减少体重、皮下脂肪、内脏脂肪和肝内脂肪含量。
CR减少了微生物表达的酶
这些酶能够使脂多糖A生物合成,从而限制了脂多糖(LPS)的产生,并以药理学上已知能刺激脂肪细胞褐化和减少内脏脂肪的方式抑制了LPS-TLR4途径。
将经过CR调节的与对照肠道微生物群移植到未经处理的无菌小鼠中,导致体重和体脂肪的增加减少,胰岛素敏感性提高,UCP1+(即褐/产热)脂肪细胞增加,这表明CR诱导的肠道微生物组变化在这些效应中起到了因果作用。
Dorea弱预测了CR诱导的体重减轻
人类的CR研究报道了肠道微生物组组成和功能的多种变化,但据所知,还没有研究表明这些变化是代谢益处的基础。
最近一项随机对照试验比较了147名超重或肥胖成年人中12周间歇性与持续性CR的效果,发现体重减轻与细菌相对丰度、群落α多样性或循环微生物代谢产物(例如短链脂肪酸)的变化之间没有关联。尽管如此,基线微生物组组成——特别是Dorea的相对丰度——弱预测了CR诱导的体重减轻。
超重人群日常热量限制后相关菌群变化
同样,一项涉及80名超重或肥胖成年人进行14周CR的前瞻性研究发现,体重减轻5%或以上与Collinsella和Christensenellaceae的丰度正相关,与大肠杆菌/志贺菌属、克雷伯菌属、巨球形菌属(Megasphaera)、Sellimonas、乳杆菌属的丰度负相关。
微生物组特征与特定代谢健康标志物之间的关联
如Akkermansia和Christensenellaceae与基于HOMA-IR的胰岛素敏感性之间的关系。需要额外的功能研究来测试这些微生物组特征与代谢反应之间的联系是因果关系还是其他生理状态的共线性结果。
解决开始和维持饮食模式重大转变挑战的一个潜在解决方案来自于一组数据,即间歇性禁食可以导致显著的体重减轻。
最常见的间歇性禁食形式是时间限制性进食(TRE),它涉及将进食窗口限制在4-10小时内,并在一天剩余的14-20小时内禁食。
TRE的做法
在进食窗口期间,个人不需要计算卡路里或以任何方式监测食物摄入,这种简单性可能解释了近期TRE受欢迎度的上升。在禁食窗口期间,个人被鼓励大量饮水,也可以消费无能量饮料,如不加添加剂的茶和咖啡。当肥胖成年人将进食窗口限制在每天4-10小时时,他们通常会将能量摄入减少200-550千卡/天,这种能量限制程度与日常CR(热量限制)相当。
TRE的减重效果
随机对照试验显示,TRE在降低体重和改善一些心血管健康标志物方面是有效的。体重通常在2-12个月的TRE后减少3%-5%,减少主要来自脂肪质量和内脏脂肪质量的减少,而不是瘦体重。
然而,并非所有关于人类TRE的研究都报告了体重减轻。有研究表明,3个月的8小时TRE(下午12点至晚上8点的进食窗口)对肥胖成年人的体重与无干预对照组相比没有影响。
注:然而,这项研究是在自由生活的参与者中进行的,他们在试验期间与研究团队的接触很少。
当进食窗口较早时,降血压效果才较为明显
即使实现了减重,也不是所有受试者都表现出代谢改善。血压通常在2-12个月的TRE后降低5-10毫米汞柱,但这些效果通常只有在进食窗口设在一天中较早的时候(即下午2点前)才会被注意到。早期进食窗口可能通过促进钠尿(通过肾脏在尿液中排泄钠)来降低血压,因为当盐分摄入转移到一天中较早的时候,由昼夜节律系统调节的钠排泄会增加。TRE似乎并不影响低密度脂蛋白胆固醇、高密度脂蛋白胆固醇或甘油三酯水平。循环炎症标志物,如C反应蛋白(CRP)、白细胞介素-6(IL-6)和肿瘤坏死因子-α(TNF-α),也不受TRE影响,尽管数据有限。
TRE改善血糖效果明显(早点吃,进食时间短)
临床试验发现,TRE在改善前驱糖尿病和肥胖个体的空腹胰岛素和胰岛素敏感性方面表现出相当一致的效果。TRE还改善了葡萄糖耐受性并减少了血清葡萄糖波动。这些改善更常见于早期进食窗口(即在下午3点前吃完所有食物)和较短的进食窗口(4-6小时)。
在2型糖尿病成人中,TRE改善了糖化血红蛋白水平,与每日CR相当,并且没有增加低血糖的风险。
TRE如何改善糖调节?
来自人类试验的数据显示,身体在TRE期间经历了代谢转换。
肠道微生物群发挥作用
在小鼠中,时间限制性喂养(TRF)通过恢复肠道细菌相对丰度的昼夜变化,减轻高脂高糖(HFHS)饮食的影响。
这些变化在远端小肠(回肠)最为明显,并与促胰高血糖素基因Gcg的表达增加和GLP-1的血浆水平升高相对应。
经抗生素处理和无菌小鼠的研究支持肠道微生物群在昼夜GLP-1释放中发挥因果作用,但具体的微生物效应因子仍不清楚。
一个概念验证来自于肠道共生菌Akkermansia muciniphila的研究,它分泌一种84kDa的蛋白质(P9),足以通过与细胞间粘附分子2(ICAM-2)相互作用诱导GLP-1的分泌。
需要更多的工作来理解参与TRE的糖调节和其他有益效应的微生物群的全范围,以及它们的临床相关性。
TREplus版:肠道菌群变化更显著
值得注意的是,最近的一项临床研究比较了CR与能量匹配的TRE加蛋白质plus(定义为每天四次均匀间隔的餐食;TRE-P)方案在超重或肥胖成年人中的效果,发现TRE-P与肠道微生物组组成的更显著变化相关,包括之前与减重和蛋白质消费有关的类群的丰富,如Christensenellaceae。此外,在TRE-P干预期间,体重减轻高与低的参与者之间观察到肠道微生物组组成和功能能力的差异,但这些微生物组变化是否对TRE-P诱导的代谢改善有因果贡献仍不清楚。
母乳是大量生物活性化合物的来源,包括人乳寡糖(HMOs)、免疫球蛋白G(IgGs)、免疫细胞和微小RNA(miRNA),其中一些可以影响婴儿的肠道微生物群。与配方奶相比,母乳喂养会导致粪便钙保护素和β-防御素2等炎症标志物水平更高,这反映了随着促炎血清细胞因子减少,免疫成熟的过程。
双歧杆菌和拟杆菌利用HMOs,因此占主导地位
HMOs被双歧杆菌属(包括Bifidobacterium breve、Bifidobacterium bifidum、B. longum、B. infantis、Bifidobacterium pseudocatenulatum)以及拟杆菌属物种利用,导致这些物种在母乳喂养的婴儿肠道中占主导地位。
这可能会改变宿主中微生物与代谢产物之间的关系,如降低的肌苷水平与长双歧杆菌丰度增加之间的相关性所证明的,这表明其可能在婴儿的免疫和神经发育中发挥作用。
HMOs作为益生元发挥作用
乳铁蛋白和溶菌酶具有抗菌特性,能够调节对感染的保护。
肠道中由HMO利用形成的SCFAs被宿主用作能量来源。
非母乳喂养的肠道菌群
非纯母乳喂养的配方奶喂养婴儿拥有更高丰度的链球菌属、肠球菌属、韦荣球菌、梭菌属,并表现出在更多碳水化合物代谢途径上的功能能力差异,这证明了饮食对肠道微生物组的重要性。
较短的母乳喂养时间,菌群多样化
较短的母乳喂养持续时间与早期生活中高度多样化且类似成人的微生物组成相关联。
母乳中的HMOs调节婴儿肠道微生物群,并提供若干健康益处,如长期保护免受过敏、特应性皮炎和肥胖的影响,以及增强肠道屏障功能。同样,引入辅食会导致肠道微生物群的变化,这些变化促进了碳水化合物的利用、维生素的合成和外源性物质的降解,结果是厚壁菌门和拟杆菌门中的微生物水平增加。
最近的研究报道,涉及脂肪和糖摄入的孕妇饮食干预改变了婴儿肠道微生物组的功能,而另一项研究则报告没有关联。
小鼠实验:母亲孕期低纤维饮食,幼鼠呼吸感染的严重程度增强
最近的研究显示,在怀孕期间接受低纤维饮食的小鼠在后代中经历了延迟的浆细胞样树突状细胞和调节性T细胞扩增的扰动,导致呼吸感染的严重程度增强。同样,在无纤维饮食的小鼠中,幼崽中的比例较低的Akkermansia muciniphila、固有淋巴细胞和TH17细胞,而缺乏AKK菌属且被喂食纤维的小鼠显示出减少的固有和适应性RORγt‐阳性免疫细胞亚群。
小鼠实验:富含发酵食品,减少新生儿结肠炎症
另一项在母猪和小鼠上进行的研究表明,富含发酵食品的母亲饮食影响了新生儿肠道微生物群的发展,并通过p38丝裂原激活蛋白激酶和c-Jun氨基末端激酶激活的caspase 3的磷酸化减少了结肠炎症。母亲饮食对婴儿长期健康影响的程度需要进一步研究。
肠道微生物群在调节宿主代谢方面发挥着关键作用,微生物组成的某些变化和多样性的减少与多种代谢性疾病发病率的上升有关。
肥胖与肠道菌群有关
利用无菌啮齿动物模型,研究人员已经建立了肠道微生物群与肥胖之间的联系。将肥胖小鼠的肠道微生物群定植到无菌小鼠体内,导致体重和胰岛素抵抗显著增加,而当无菌小鼠被喂食西式饮食时,肥胖的发展则不存在,这突显了肠道微生物群在肥胖中的作用。然而,其他几项同意微生物群在能量稳态中的作用的研究未能显示其在肥胖发展中的决定性作用,并指出需要更多的研究来探索这种复杂的关系。
2型糖尿病和肥胖的个体的肠道菌群特征
患有2型糖尿病和肥胖的个体通常表现出产丁酸菌减少,乙酸盐及促炎物种增加,这些与胰岛素抵抗性升高有关。在肥胖小鼠上进行的研究支持肠道微生物群在2型糖尿病中的作用。双歧杆菌属、拟杆菌属、Faecalibacterium、Akkermansia与2型糖尿病负相关,其中双歧杆菌增加了胰高血糖素样肽-2(GLP-2)的水平,从而改善肠道通透性并减少代谢性内毒素血症。
注:二甲双胍,一种常见的2型糖尿病药物,与肠道微生物群相互作用,可能通过调节葡萄糖稳态和短链脂肪酸的产生来介导其抗糖尿病效应。
饮食、肠道微生物组、代谢性疾病
doi.org/10.1038/s41579-024-01068-4
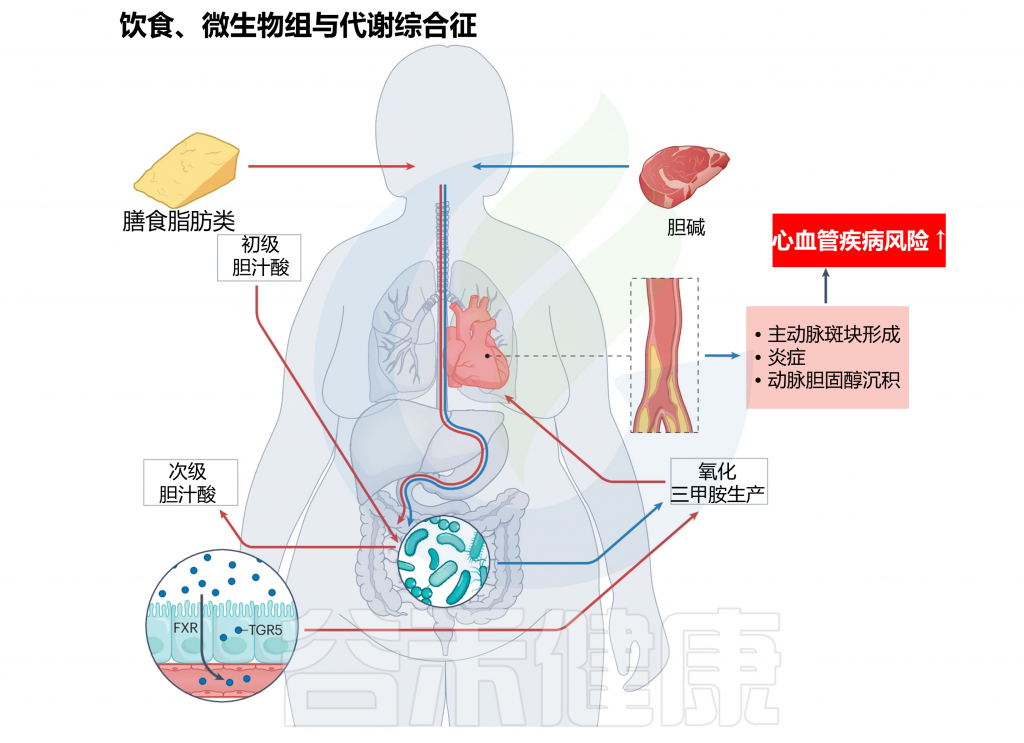
注:红色箭头表示饮食脂肪可以通过何种作用机制对宿主健康产生下游影响,最终导致CVD风险。此外,蓝色箭头显示了主要存在于动物产品中的胆碱如何引起CVD风险。
心血管代谢疾病的个体的肠道菌群变化
特征是增加的肠杆菌科(Enterobacteriaceae)物种和减少的拟杆菌属以及抗炎的F. prausnitzii。肠道微生物群的这些变化与更具炎症性和较少发酵性的肠道环境有关。
TMAO
三甲胺-N-氧化物(TMAO),一种由肠道细菌从饮食化合物产生的代谢产物,与动脉硬化、血小板聚集和血栓形成有关。
在小鼠和人类的研究表明,饮食因素影响TMAO水平,某些情况下抗生素降低了TMAO,而杂食饮食增加了它。TMAO水平升高与心力衰竭患者的高死亡率相关。然而,结果并不一致,一些研究表明某些饮食成分如左旋肉碱和富含TMAO的食物可能有助于预防动脉粥样硬化,这引发了关于饮食、微生物组和宿主遗传学在动脉粥样硬化发展中复杂相互作用的问题。
增加的饮食脂肪可以影响FXR和TGR5等胆汁酸受体的激活,它们在脂质和葡萄糖代谢中发挥重要作用。这些途径的调节失常可能导致心血管疾病的发展。
由于微生物组改变导致的能量稳态的微小变化可能具有长期效应,在代谢性疾病中发挥作用,既是因果因素也是促成因素。此外,它们可以作为使用微生物组靶向治疗改善这些状况的目标。
饮食在肠道疾病的病理生理学中起着关键作用,特别是炎症性肠病、肠易激综合症和结肠癌。
肠易激综合征
过敏、食物不耐受、微生物群组成的转变、轻度粘膜炎症和肠道通透性的增加可能促成了肠易激综合症的表现。
研究发现,类似于病原性肠易激综合症的人类微生物组表现出拟杆菌门的丰度减少,以及厚壁菌门和与氨基酸及碳水化合物代谢相关的基因丰度增加。
饮食成分与炎症性肠病风险
饮食也可以改变炎症性肠病(包括克罗恩病和溃疡性结肠炎)的肠道微生物群落组成,影响短链脂肪酸和纤维等物质的代谢,这反过来又可能促成疾病的发生。
动物蛋白、乳制品、碳水化合物和多不饱和脂肪酸等食物成分与发生炎症性肠病的风险有关。
动物蛋白与炎症性肠病的机制
一个将炎症性肠病与动物蛋白联系起来的机制涉及小肠中的氨基酸和血红素吸收不良,导致产生酚类和氢气等有害副产物。这通过抑制丁酸盐的产生和减少肠道屏障中的二硫键,促成了炎症性肠病的发病机制。
高脂肪饮食也与炎症性肠病强烈相关
在实验模型中,高脂肪饮食可以破坏肠细胞间的结合蛋白功能,从而改变粘液层的组成和肠道微生物群。
持续且控制不当的炎症性肠病,以及由于不良饮食模式(如西方饮食)导致的慢性胃肠道炎症,是影响结肠炎相关结直肠癌风险的主要外部因素。这些因素影响免疫反应、肠道组织平衡和肠道微生物组。
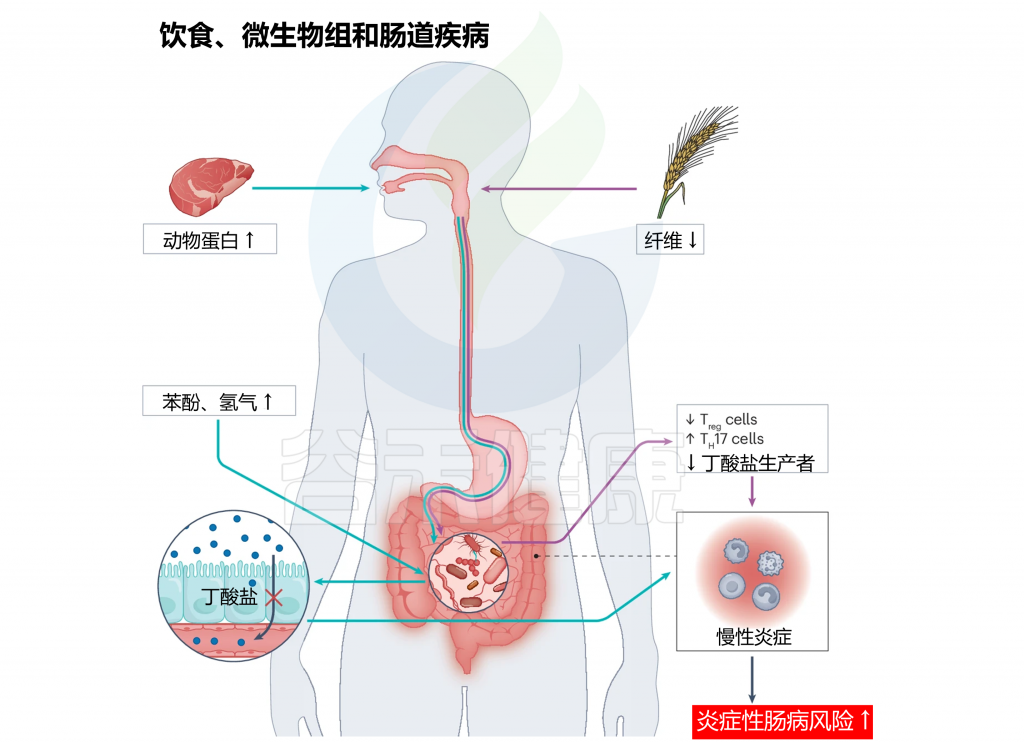
doi.org/10.1038/s41579-024-01068-4
注:增加的动物蛋白(绿色箭头)和低纤维(紫色箭头)饮食可能对生理功能和宿主健康产生下游影响。
增加红肉消费可导致胆碱水平升高,由于血红素吸收不良,在小肠中产生更多的氢气和苯酚。这反过来可以减少胃肠道中的丁酸盐生产,导致炎症增加。同样,饮食中纤维摄入减少可能通过增加TH17的产生,同时减少Treg和短链脂肪酸产生,对肠道健康产生负面影响。这种不平衡最终导致胃肠道内慢性炎症加剧。肠道内长期的慢性炎症可能大幅增加发展成炎症性肠病的风险。
饮食在散发性结直肠癌中的作用
研究发现,低纤维、高脂肪饮食与Fusobacterium nucleatum有关。拟杆菌属通过激活E-钙粘蛋白-β-链球蛋白信号、表观遗传变化和改变肿瘤微环境等机制与结直肠癌有关,从而促进恶性转变。同样,诸如产毒脆弱拟杆菌(Bacteroides fragilis)等致癌细菌被假设通过直接与结肠上皮细胞相互作用和改变局部微生物群组成来触发结直肠癌的发病。
人类肠道是真菌和病毒群的栖息地,分别称为肠道真菌组和病毒组。尽管这些群落只占肠道中总微生物的0.1%-1%,但它们都受到饮食的影响。
婴儿肠道真菌组中,酿酒酵母(Saccharomyces cerevisiae)是优势物种,断奶后被其他酵母属(丝孢酵母属Cystofilobasidium、曲霉属Ascomycota、单孢子酵母属Monographella)取代。
城市居民的肠道真菌组成包括酿酒酵母和较少的产短链脂肪酸菌,农村居民则有更多样化的真菌物种。
念珠菌属(Candida species)与富含碳水化合物的饮食相关,与富含蛋白质的饮食负相关。
母乳喂养和配方奶喂养婴儿的肠道病毒组组成差异由肠道微生物群变化和母乳垂直传递病毒引起。
高脂饮食与Siphoviridae病毒丰度减少和Microviridae噬菌体丰度增加有关。
无麸质饮食则与相反的变化有关,Siphoviridae在Microviridae之上,占主导地位。
肥胖和1型及2型糖尿病患者的病毒组成也发生变化,高脂饮食喂养小鼠的粪便病毒移植降低肥胖风险。
肠道耐药组,赋予微生物抗微生物药物耐药性的所有基因或遗传物质的集合,随着细菌微生物组和病毒组的变化而变化。
一些研究报告γ-变形菌纲(Gammaproteobacteria)属拥有丰富的抗生素抗性基因(ARG)储备。
配方奶喂养的婴儿ARG负荷更高,与细菌组成有关。
纯素和鱼素食饮食个体肠道中的微生物组成不同,但他们的耐药组档案并没有显著差异,表明耐药组主要由抗微生物药物暴露而非饮食塑造,可能的例外是含有特定防腐剂的食物。
需要进行详细的饮食干预研究,以了解饮食是否可以减少ARG的负担。
地中海饮食在缓解和管理多种疾病方面已被证明是有效的,包括心血管疾病、2型糖尿病、炎症性肠病、肠易激综合症、认知能力下降和抑郁症。此外,对这种饮食的调整,如MIND饮食,已成功降低阿尔茨海默病的风险并减缓认知能力下降。同样,DASH(阻止高血压的饮食方法)饮食已证明在治疗高血压方面有效。
特定的碳水化合物饮食在临床实践中用于治疗炎症性肠病的症状。特定的碳水化合物饮食在儿童和成人队列中已证明其有效性,并已与改善的临床参数和炎症标志物相关联。然而,使用这种饮食时必须保持营养控制,以避免营养不足和体重下降。
对于肠易激综合症的治疗,通常使用低发酵性低聚糖、二糖、单糖和多元醇(低FODMAP)饮食,有50%~80%的患者有积极的临床反应。
在41名患者中进行的为期四周的低FODMAP饮食研究显示,从类似病原性肠易激综合症的肠道微生物组向健康相关的肠道微生物组发生了组成和功能上的转变。
同样,研究表明,坚持低FODMAP饮食,双歧杆菌(Bifidobacterium adolescentis)方面表现出显著降低,这种细菌会破坏肠道屏障功能并改变紧密连接的完整性,从而支持低FODMAP饮食的积极效应是通过肠道微生物群介导的假设。
无麸质饮食目前是治疗乳糜泻的方法,研究已证实这种饮食在缓解胃肠道症状方面的有效性。采用这种饮食方案与肠道微生物组成和肠道微生物途径的改变有关。
最近一项研究分析了乳糜泻患者的小RNA和宏基因组测序数据,研究结果显示,采用无麸质饮食改变了miRNA和微生物群落的轮廓。该研究还揭示了乳糜泻患者中的miRNA-细菌关系和特定的分子模式,表明可能存在用于监测无麸质饮食依从性和评估肠道炎症状态的生物标志物。
对于慢性肾病的管理,推荐采用低蛋白饮食,目的是减缓进入终末期肾病的进展,并推迟对肾脏替代治疗的需求。
综述表明,极低蛋白饮食可能有效减少4期或5期肾病的发生。然而,仅采用低蛋白饮食并未影响终末期肾病的发展。
此外,五篇文章的系统综述和元分析发现,低蛋白饮食增加了拟杆菌科、乳酸菌科、咽峡链球菌Streptococcus anginosus的丰度,同时减少了Roseburia faecis和Bacteroides eggerthii的丰度。但是,在没有微生物多样性和丰富度的整体构成变化的情况下,这些主要在物种和科水平上的变化似乎不足以影响代谢或临床结果。
用于管理2型糖尿病的血糖指数饮食,因其对肠道微生物群的影响及其在影响疾病发展和严重程度方面的潜在作用而受到关注。
这种饮食包括消耗低血糖指数的碳水化合物(例如,豆类、燕麦和小麦),促进血糖水平逐渐且持续上升。尽管关于这种饮食对肠道微生物群影响的研究有限,但小鼠研究表明,它与因摄入大麦而增加的乳酸杆菌属、普雷沃特氏菌属和纤维降解S24-7细菌的丰度有关,或因摄入全谷物燕麦而增加的双歧杆菌属和乳酸杆菌-肠球菌属(Lactobacillus-Enterococcus)有关。
肠道微生物组在人体生理学中的中心作用彻底改变了我们对健康的看法,并日益渗透到营养研究和建议中。
目前,全球饮食指南普遍达成共识,但不幸的是,这种均质性也延伸到了微生物组,只有少数几个国家(例如美国和南非)明确考虑了饮食-微生物组相互作用。
很多文章已经讨论了肠道微生物组知识如何与当前的营养指南相结合,为包含微生物组的精准营养提供了机会,并广泛考虑了将微生物组科学纳入研究、教育、政策和公共卫生沟通的更广泛问题。
几乎所有方面的人类营养最终都需要根据饮食-微生物组相互作用对人类健康的直接和间接后果重新评估。
这里强调微生物组知识挑战营养科学的三个原则:
宿主卡路里≠宿主-微生物组卡路里
由美国化学家威尔伯·奥林·阿特沃特(Wilbur Olin Atwater)在19世纪末提出的阿特沃特系统,用于估算食物中各种营养成分的热量值,反映了食物中的平均化学能量减去粪便、尿液、分泌物和气体中排泄的平均分数。
阿特沃特系统估算热量含量的方法存在三个关键疏漏:
1、食物基质效应
没有捕捉到更广泛食物基质的效果,如植物性宏观营养素在细胞壁或亚细胞结构中的封装。
2、饮食诱导的热生成
没有捕捉到消化的代谢成本,这基于宏观营养素含量、餐食的可口性和食物加工而变化。
3、宿主与微生物组的卡路里区分
只在很小程度上区分了对人类可利用的卡路里和对肠道微生物组可利用的卡路里。
营养学领域长期以来一直合理地关注那些被吸收进入人体组织的饮食成分,因为这些成分有潜力直接影响健康。然而,大量证据表明肠道微生物组对饮食消化性很敏感,并且饮食引起的肠道微生物组的变化可以在不同情况下因果地塑造宿主的健康和疾病,这日益凸显了未吸收营养素的重要性。
未吸收营养素的重要性
与被吸收的营养素不同,未吸收的营养素可靠地到达结肠中最密集的微生物群落。此外,随着消化液在胃肠道内向下推进,未吸收的营养素会因为被吸收的营养素和水分的消失而浓缩。因此,可以预期,未吸收的营养素在塑造肠道微生物组及其对健康和疾病的下游影响方面,可能比被吸收的营养素具有更大的影响力。
饮食与肠道微生物组的相互作用
目前研究主要关注食物入口时的状态,而未充分考虑小肠末端的消化残余物。
虽然历史上对回肠消化性的描述依赖于体外模型或复杂的体内模型,例如插管动物、回肠造口术后的人类患者、健康人体中的侵入性鼻-回肠或结肠插管,以及在血浆中检测同位素标记的营养素,但受微生物组启发的新方法可能证明是有希望的。
深入理解饮食-微生物组相互作用的新视角
例如,基于DNA的饮食底物表征——一种称为DNA metabarcoding的技术,可能与基于DNA的微生物组分析相结合,研究特定排泄样本中直接的饮食-微生物组相互作用。可以在动物模型中或使用新的可吞咽装置在人体中执行对饮食和微生物组信号的双重表征,这些装置能够在由pH变化确定的胃肠道间隔处采样消化液。
许多食品物质已根据美国食品药品监督管理局(FDA)基于动物毒理学试验和/或过去在人类中广泛使用且未产生已知有害影响的基础上,被授予“通常认为安全”(GRAS)的认定。
潜在健康影响
然而,GRAS评估通常并未考虑这些物质对肠道微生物组的影响,或者通过微生物组介导的间接健康效应的潜力。
专注于宿主组织的危险通过发现乳化剂如卵磷脂和人造甜味剂如糖精等GRAS物质在饮食相关水平下可能通过影响肠道微生物组诱导肥胖和胰岛素抵抗的情况得到了说明。
牛磺胆酸可能通过菌群与肠道病理的关联
GRAS化合物牛磺胆酸及其化学成分,GRAS化合物牛磺酸和胆酸,可能与肠道微生物组相互作用,促进肠道病理。具体来说,由Bilophila wadsworthia细菌在牛磺胆酸的脱结合过程中释放的牛磺酸产生遗传毒性的硫化氢,同时释放的胆酸作为微生物产生促炎的次级胆汁酸脱氧胆酸的基质。因此,补充牛磺胆酸的饮食导致了B. wadsworthia的增长和易感基因型(IL-10−/−)小鼠中结肠炎的发展。
肠道微生物组可能转化为更有害的形式:杂环胺的肠肝循环
此外,肠道微生物组可能使用其广泛的酶库将饮食化合物或宿主代谢产物转化为更具有害的形式。例如,细菌β-葡萄糖醛酸酶有助于致癌的杂环胺(如IQ,2-氨基-3-甲基咪唑[4,5-F]喹啉)的肠肝循环,这些物质通过肝脏的葡萄糖醛酸化被解毒。
在暴露于IQ时,常规小鼠比无菌小鼠显示出更多的DNA加合物和DNA损伤。单核子大肠杆菌携带功能性与非功能性uidA基因(编码β-葡萄糖醛酸酶)的大鼠表现出增加的结肠遗传毒性,与这种化合物排泄的多个峰值相结合,这与肠肝循环一致。
三聚氰胺污染+肠道微生物组→肾脏病理
肠道微生物组还与由饮食污染物三聚氰胺引起的肾脏病理有关,三聚氰胺是一种用于许多食品制备工具的塑料添加剂。体外和体内实验表明,存在于一些婴儿肠道中的克雷伯菌可以将三聚氰胺转化为三聚氰酸,三聚氰酸现在已知与三聚氰胺形成不溶性的肾脏聚集体。
有益效应
另一方面,肠道微生物组对未吸收的饮食化合物的生物转化可能有助于有益效应,这些效应如果只关注饮食对宿主的直接影响则可能被忽视。
对抗乳腺癌的保护作用
例如,植物衍生的饮食木脂素(如全谷物、种子、豆类和坚果中发现的)的肠道微生物生物转化被认为是它们对抗乳腺癌的保护作用的基础。一组肠道细菌类群(例如,Eggerthella lenta、Blautia producta、Gordonibacter pamelaeae和Lactonifactor longoviformis)将饮食木脂素松香转化为具有抗癌作用的雌激素模拟物enterodiol和enterolactone。
因此,与无菌动物相比,在化学诱导乳腺癌时,能够从饮食木脂素前体产生enterodiol和enterolactone的细菌群落定植的无菌大鼠显示出较少的肿瘤数量和较小的肿瘤大小。
扩展阅读:
肠道菌群有助于饮食解毒改变疾病风险
例如,肠道细菌Oxalobacter formigenes参与草酸盐的分解,草酸盐是一种螯合饮食毒素,通过结合游离金属阳离子,有助于肾结石和肾衰竭。缺乏O. formigenes与高草酸尿症的风险增加有关,其在大鼠中的施用以剂量依赖性的方式减少了饮食诱导的高草酸尿症。
在探索肠道微生物群与饮食之间错综复杂的关系后,我们不难发现,这个微小的生态系统对我们的健康有着深远的影响。从调节能量平衡到影响免疫功能,从塑造情绪到预防疾病,肠道微生物群的作用远远超出了我们的想象。
当然,饮食也只是众多生活方式因素之一,例如身体活动、环境暴露和睡眠,这些因素都会影响宿主的能量平衡和肠道微生物群。此外,药物的广泛使用已经显著改变了饮食干预的背景。例如,GLP-1 激动剂延迟胃排空,这对消化有着深远的影响,包括肠道微生物代谢可用底物的变化。
即使仅考虑饮食,现在也非常清楚,肠道微生物影响宿主代谢的多种途径,加上关键的饮食和微生物组相关代谢物(如短链脂肪酸、次级胆汁酸等)的多效性作用,使预测特定饮食或微生物组特征的代谢影响变得复杂。
实现基于微生物组的精准营养方法需要对人类进行实验研究,以测量整个生物体水平的综合影响,涵盖地理、性别、种族和年龄等各种因素,以及更大规模的横断面研究,针对饮食成分、肠道微生物组结构和功能以及宿主健康之间的特定联系。
这些数据将受益于机器学习的快速发展并将人工智能与实施精准医疗方面的结合起来。随着技术的进步和数据的积累,肠道菌群检测有望成为精准营养和个性化医疗的重要组成部分,帮助我们更好地管理健康,预防疾病,并提升生活质量。
主要参考文献
Carmody RN, Varady K, Turnbaugh PJ. Digesting the complex metabolic effects of diet on the host and microbiome. Cell. 2024 Jul 25;187(15):3857-3876.
Ross FC, Patangia D, Grimaud G, Lavelle A, Dempsey EM, Ross RP, Stanton C. The interplay between diet and the gut microbiome: implications for health and disease. Nat Rev Microbiol. 2024 Jul 15.
Ahmad S, Moorthy MV, Lee IM, Ridker PM, Manson JE, Buring JE, Demler OV, Mora S. Mediterranean Diet Adherence and Risk of All-Cause Mortality in Women. JAMA Netw Open. 2024 May 1;7(5):e2414322.
McEvoy CT, Jennings A, Steves CJ, Macgregor A, Spector T, Cassidy A. Diet patterns and cognitive performance in a UK Female Twin Registry (TwinsUK). Alzheimers Res Ther. 2024 Jan 23;16(1):17.
Link VM, Subramanian P, Cheung F, Han KL, Stacy A, Chi L, Sellers BA, Koroleva G, Courville AB, Mistry S, Burns A, Apps R, Hall KD, Belkaid Y. Differential peripheral immune signatures elicited by vegan versus ketogenic diets in humans. Nat Med. 2024 Feb;30(2):560-572.
Staudacher HM, Mahoney S, Canale K, Opie RS, Loughman A, So D, Beswick L, Hair C, Jacka FN. Clinical trial: A Mediterranean diet is feasible and improves gastrointestinal and psychological symptoms in irritable bowel syndrome. Aliment Pharmacol Ther. 2024 Feb;59(4):492-503.

谷禾健康

丁酸弧菌属(Butyrivibrio)是人体的重要菌属,属于厚壁菌门毛螺菌科,毛螺菌科包括了人体很多重要的菌属,与健康紧密相关。该菌最早于1956年被发现,其革兰氏染色呈阴性,但具有非常薄的革兰氏阳性细胞壁结构。
丁酸弧菌属严格厌氧,不产生孢子。目前已知该菌属包含60多种菌株,溶纤维丁酸弧菌(Butyrivibrio fibrisolvens)为其的模式菌种,能够发酵多种糖和纤维。该菌对植物结构(主要是半纤维素)的降解具有特别重要的作用。因此该菌常见于牛和其他反刍动物(如绵羊、山羊、驯鹿)中,可能与经常摄入纤维有关。该菌也存在于人体的胃肠道中,并且研究发现其丰度与纤维摄入量相关。
丁酸弧菌是一种重要的丁酸生产者,其主要通过代谢可溶性多糖以及植物纤维来产生丁酸盐。而丁酸盐具有多种健康益处,会影响维生素的合成,有助于维持肠道上皮,发挥抗炎、抗氧化作用,以及对其他器官的正常功能也存在一定影响。此外,丁酸弧菌通过蛋白质分解(氨是其最主要的氮源)调节体内的氨水平,还能产生细菌素影响其他菌,例如可以抑制多种革兰氏阳性病原菌,以此来调节人体免疫。
由于丁酸弧菌能够产生丁酸盐,目前认为其是一种对人体有益的菌属。并且已经有一些研究发现在病原体感染、2型糖尿病、白塞氏病、婴儿胆汁淤积症、肺动脉高压、银屑病关节炎、肥胖和代谢紊乱、阿尔兹海默病患者中丁酸弧菌(Butyrivibrio)的丰度显著下降。
丁酸弧菌对抗生素比较敏感,因此在使用一些广谱抗生素时可能也会降低其丰度,但是对四环素具体耐药性。由于丁酸弧菌主要代谢纤维和多糖等物质,通过补充富含儿茶素、花青素、原花青素、咖啡酸、绿原酸和芦丁等多酚类物质的水果和蔬菜,或是使用合生元疗法如共同施用枯草芽孢杆菌和L-丙氨酰-L-谷氨酰胺可以提高丁酸弧菌的丰度。
▼1
丁酸弧菌(Butyrivibrio)一般为弯杆状,末端呈锥形或圆形,大小约为0.3~0.8μm×1.0~5.0μm,它们以单链或短链或长链的形式出现,呈螺旋状排列。成对的细胞可能以“S”形排列,有时呈丝状。
不同菌株之间存在相当大的差异。一些菌株的细胞几乎是纺锤形的,而另一些菌株的细胞非常弯曲,可以形成由2到4个细胞组成的螺旋。Butyrivibrio crossotus直径往往比Butyrivbrio fibrisolvens大。
溶纤维丁酸弧菌的电子显微镜照片
Sewell et al.2008.
✦ 革兰氏染色阴性,但细胞壁属革兰氏阳性类型
丁酸弧菌染色为革兰氏阴性,但缺乏与革兰氏阴性菌相关的三层外膜结构。一些菌株具有非典型的,非常薄的革兰氏阳性细胞壁结构。与正常革兰氏阳性菌壁(约30-50nm)相比,薄壁(约12-20nm)可能是菌株染色反应呈革兰氏阴性的原因。
丁酸弧菌菌株的肽聚糖含有乳酸、葡萄糖胺、丙氨酸、谷氨酸和中氨基戊酸。这些成分符合二氨基苯甲酸直接交联结构。此外,一些丁酸弧菌中还含有少量的甘氨酸和天冬氨酸,表明这些氨基酸可能存在反应。
✦ 菌落的特点
许多丁酸弧菌菌株在含复合碳水化合物琼脂培养基上生长时,表面菌落直径为2至4mm,完整,微凸,半透明,颜色为浅褐色至白色。
一些菌株会产生具有丝状边缘的粗糙菌落,可能代表菌株产生很少的细胞外多糖物质。相比之下,CF型菌株产生大量的胞外多糖,菌落呈粘液状,可见菌落中冒出气泡。
在含纤维素的培养基中,消化纤维素的菌株被纤维素降解区包围,降解区大小和消化程度(通常较弱)都是可变的。当使用含有天然木聚糖的培养基时,也会出现类似的观察结果。
▼2
丁酸弧菌属(Butyrivebrio spp.)是牛和其他反刍动物(如绵羊、山羊、驯鹿)瘤胃中的常见菌,在喂食饲料含量高的动物中数量可观,通常是数量较多的细菌物种之一。它们也存在于这些动物的盲肠和结肠中。
除了反刍动物,丁酸弧菌也存在于各种其他哺乳动物的胃肠道中,并已从兔子、马、人类和猪的粪便或盲肠内容物中分离出来。
还应该注意的是,丁酸弧菌可以在动物以外的环境中生存,并且已经从Napier草食厌氧消化器中分离出菌株。
▼3
通常只有少数细胞表现出运动性。表现出以快速或强烈的振动运动为特征的平移运动。运动是通亚极性鞭毛进行的,鞭毛附着在腋下。
✦ 运动性与生长条件相关
在分离菌株中缺乏运动性通常是由于所采用的培养条件。报道了15株菌株在瘤胃液-葡萄糖-纤维素二糖培养基中生长时不活动,但如果去除纤维素二糖并降低葡萄糖浓度,则所有菌株都可以活动。大多数菌株生长迅速(2小时或更短时间内翻倍),并产生大量酸。由于产酸造成的低pH值可能会抑制运动,也会抑制生长。
▼4
丁酸弧菌是一种严格的厌氧菌,存在于各种家畜和人类的胃肠道系统中。最佳生长温度是37°C,在30°C以下或45°C以上时很少生长或不生长。
它们具有发酵代谢的能力,能够代谢多种碳水化合物,能够利用各种半纤维素和木聚糖。大多数菌株可以降解淀粉和果胶,但很少有菌株可以在纤维素上生长。大多数菌株可以发酵各种可溶性糖、双糖或低聚糖,形成丁酸盐、乳酸盐和醋酸盐。
▸ 代谢多种糖类产生丁酸盐、乙酸盐等产物
几乎所有菌株都能发酵15到20种不同的可溶性碳水化合物,包括葡萄糖、麦芽糖、蔗糖、果糖、纤维素二糖、木糖和阿拉伯糖。
许多研究都集中在特定模式菌株的碳水化合物代谢上。在溶纤维丁酸弧菌ATCC 19171和CE 51中,葡萄糖培养的细胞干物质产量和生长速度高于木糖培养。在后一种培养中,更多的碳被转化为代谢物,而较少的碳被转化为细胞物质。
当同时提供葡萄糖和木糖时,溶纤维丁酸弧菌ATCC 19171和86同时使用两种糖,菌株X1和CE 51表现出典型的先使用葡萄糖生长,而菌株X2D62先缓慢使用木糖直到耗尽,然后快速使用葡萄糖。
此外,菌株CE 51也发生了磷酸烯醇丙酮酸依赖性的葡萄糖磷酸化。溶纤维丁酸弧菌菌株49能迅速水解淀粉,产生葡萄糖、麦芽糖、麦芽糖三糖、麦芽糖四糖和麦芽糖戊二糖。
溶纤维丁酸弧菌发酵葡萄糖的最终产物包括甲酸盐、丁酸盐、乙酸盐以及不同数量的乳酸盐和琥珀酸盐。
然而,菌株之间有相当大的差异:菌株D1和A38显示产生少量乳酸,而菌株49和菌株37产生大量乳酸,但这可以被抑制,而乙酸的存在会产生丁酸盐。
▸ 代谢和降解植物纤维
丁酸弧菌可以利用植物细胞壁的各种成分,丁酸弧菌在各种植物降解中的作用已被广泛研究。
溶纤维丁酸弧菌与其他瘤胃细菌共培养能够降解大麦秸秆、高粱秸秆、苜蓿干草和紫菜干草、棉花秸秆、小麦秸秆和苜蓿。
在不同来源果胶利用的比较研究中,溶纤丁酸弧菌对柑橘果胶的利用效果最好,对甜菜果胶的利用效果优于苹果果胶和苜蓿果胶。与生长在l-阿拉伯糖和d-葡萄糖上的培养物相比,生长在果胶上的溶纤维丁酸弧菌787产生了更多的乙酸和更少的丁酸、乳酸、琥珀酸和氢。
但丁酸弧菌的代谢也被其中一些化合物所抑制。例如,溶纤丁酸弧菌菌株49的生长受到酯连接的阿魏酰和对香豆醇基的限制。酚酸-碳水化合物复合物的生长限制因其水解碳水化合物键的能力而异。
▸ 氨为大多数丁酸弧菌的氮源
大多数丁酸弧菌能以氨为唯一氮源;许多菌株也可以使用尿素。一些菌株还可以使用氨基酸混合物或更复杂的氮源(胰酶、酪蛋白水解物、蛋白胨)。然而,当这些化合物以限制生长的浓度提供时,氨比等量的肽或氨基酸氮支持更多的生长。
多肽不是必需的,但在含氨培养基上经常刺激生长。并且不同的丁酸弧菌可能所需的营养物质不同,据报道菌株E14生长需要蛋氨酸,菌株S2生长需要脂肪酸。添加维生素/酪蛋白水解物混合物或酵母提取物可以刺激菌株TC33在含有葡萄糖的澄清瘤胃液中的生长。最快速的生长是通过添加叶酸、吡哆胺.2HCl和酪蛋白酶解产物的组合获得的。
▸ KEGG 通路
氨基酸的生物合成
抗生素的生物合成
次生代谢产物的生物合成
不饱和脂肪酸的生物合成
生物素代谢
丁酸代谢
2-氧代羧酸代谢
丙氨酸、天冬氨酸和谷氨酸代谢
氨基糖和核苷酸糖代谢
氨酰-tRNA生物合成
花生四烯酸代谢
精氨酸和脯氨酸代谢
精氨酸生物合成
抗坏血酸和醛糖酸代谢
C5-支链二元酸代谢
碳代谢
阳离子抗菌肽(CAMP)耐药性
柠檬酸循环(TCA循环)
氰氨基酸代谢
半胱氨酸和蛋氨酸代谢
D-丙氨酸代谢
D-谷氨酰胺和D-谷氨酸代谢
DNA复制
芳香族化合物的降解
脂肪酸生物合成
脂肪酸降解
脂肪酸代谢
叶酸生物合成
果糖和甘露糖代谢
半乳糖代谢
谷胱甘肽代谢
甘油脂代谢
甘油磷脂代谢
甘氨酸、丝氨酸和苏氨酸代谢
糖酵解/糖异生
乙醛酸和二羧酸代谢
组氨酸代谢
肌醇磷酸代谢
柠檬烯和蒎烯的降解
脂多糖生物合成
赖氨酸生物合成
赖氨酸降解
甲烷代谢
不同环境中的微生物代谢
单环内酰胺生物合成
烟酸和烟酰胺代谢
氮代谢
非核糖体肽结构
新生霉素的生物合成
泛酸和辅酶 A 的生物合成
戊糖和葡萄糖醛酸相互转化
磷酸戊糖途径
肽聚糖生物合成
苯丙氨酸代谢
磷酸转移酶系统(PTS)
聚酮糖单元生物合成
卟啉和叶绿素代谢
丙酸代谢
蛋白质输出
嘌呤代谢
嘧啶代谢
丙酮酸代谢
RNA降解
RNA聚合酶
核黄素代谢
硒化合物代谢
淀粉和蔗糖代谢
链霉素生物合成
硫代谢
硫磺中继系统
牛磺酸和亚牛磺酸代谢
萜类化合物主链的生物合成
硫胺素代谢
色氨酸代谢
酪氨酸代谢
缬氨酸、亮氨酸和异亮氨酸的生物合成
缬氨酸、亮氨酸和异亮氨酸降解
万古霉素耐药性
维生素B6代谢
β-丙氨酸代谢
▼5
丁酸弧菌于1956被首次发现,当时只提出了一个物种,即溶纤维丁酸弧菌(Butyrivibrio fibrisolvens),这也是丁酸弧菌的模式菌种。
目前主要的丁酸弧菌有三个菌种,溶纤维丁酸弧菌(Butyrivibrio fibrisolvens)、Butyrivibrio crossotus和Butyrivibrio hungatei。
▸ 溶纤维丁酸弧菌为模式菌种
溶纤维丁酸弧菌细胞呈革兰氏阴性,但细胞壁很薄,具有革兰氏阳性的超微结构。葡萄糖培养基中的生长变化从均匀的浑浊到絮状或粒状沉积物。37°C时生长良好,一般在45°C时生长,但在30°C时生长较慢。在22以下或50°C以上时没有生长。
溶纤维丁酸弧菌是正常瘤胃菌群中的一种非致病性成员,并且能够产生丁酸盐,被认为对人体健康有益。
DNA的G+C (摩尔%)是42
模式菌株:ATCC 19171
菌株将葡萄糖或纤维素二糖作为能量来源,氨基酸混合物和铵盐作为氮源,矿物质,B族维生素和半胱氨酸等营养物质也会影响生长。氨基酸混合物通常促进生长,醋酸酯也能刺激生长。
▸ Butyrivibrio crossotus
Butyrivibrio crossotus是从人类粪便或直肠内容物中分离出来的,目前还不知道是否具有致病性。
细胞形态与属描述中呈现的相同,只有少数菌株在厌氧培养的血琼脂板上生长。除非存在可发酵的碳水化合物,否则在蛋白胨酵母提取物肉汤中生长很差。
最佳生长温度是37°C,在30°C时生长缓慢,有些菌株可以在45°C时生长。
DNA G+C (摩尔%):37
模式菌株:ATCC 29175
▸ Butyrivibrio hungatei
Butyrivibrio hungatei,以美国微生物学家罗伯特·e·亨盖特(Robert E. Hungate)命名,他在20世纪60年代分离出了类似的菌株。
细胞形态与属描述相同,由单极或近极鞭毛运动的。可以在39°C无氧环境生长,但在25°C时无法生长,在45°C时生长受限。
色氨酸、尿素和明胶不被利用。不表现出显著的纤维水解或蛋白水解活性,主要利用寡糖和单糖作为生长底物。
可以产生α-半乳糖苷酶、α-阿拉伯糖苷酶、苯丙氨酸芳基酰胺酶和亮氨酸芳基酰胺酶。未检测到淀粉酶、木聚糖酶、β-内切葡聚糖酶、层粘连酶、果胶水解酶、蛋白酶或DNA酶活性。
DNA G+C (摩尔%):44.8
模式菌株:JK 615 (= DSM 14810 = ATCC BAA-456).
▸ 丁酸弧菌的其他菌种
对丁酸弧菌菌株16S rDNA基因的系统发育研究揭示了12种rRNA类型,形成了三个不同谱系。下图给出了这些组的详细视图。
不同丁酸弧菌群的详细组成
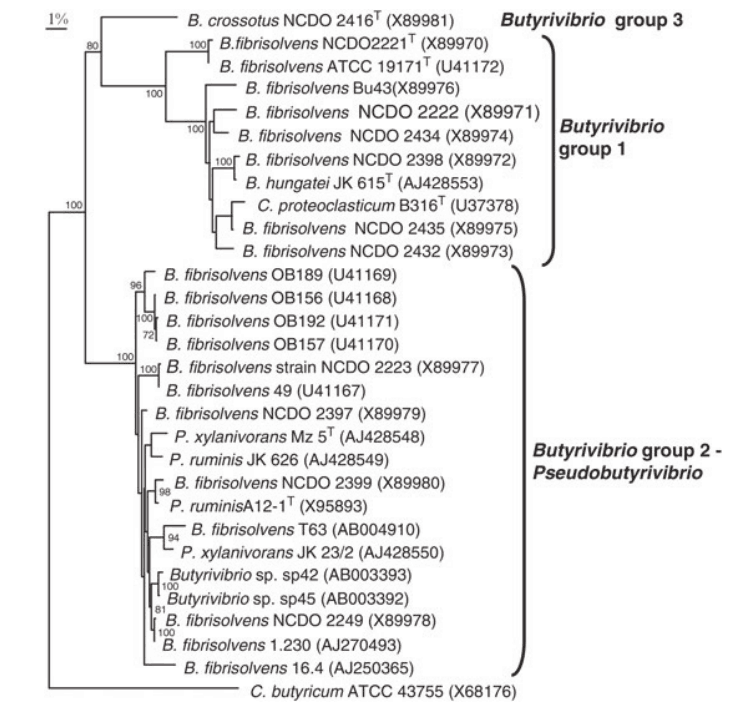
DOI: 10.1002/9781118960608.gbm00640.
核糖体RNA1 – 7型形成第一组,其中rRNA 1型(包括溶纤维丁酸弧菌型菌株)是最外围的。16S rDNA序列与2-7型rRNA序列相似性为93-94%,与2-7型rRNA序列相似性为96-98.5%。
除溶纤维丁酸弧菌外,至少报道了13个不同的丁酸弧菌基因型群,下面给出了属于这些组的菌株的概述。
根据遗传信息将菌株划分为不同的丁酸弧菌群
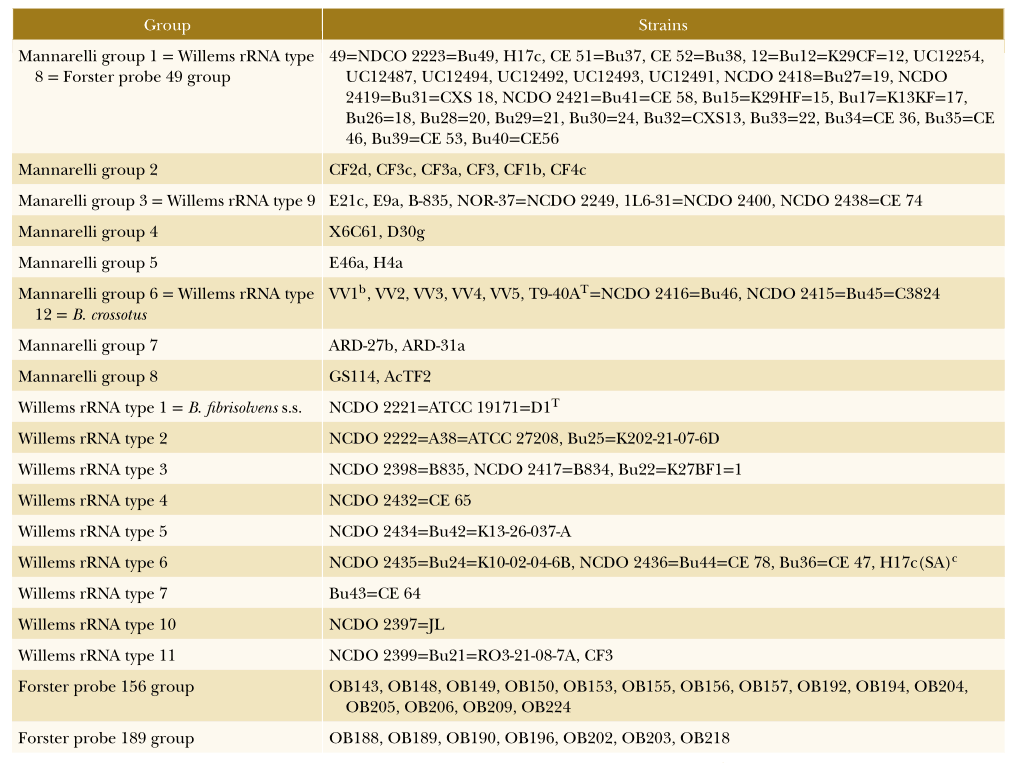
DOI: 10.1002/9781118960608.gbm00640.
目前已知的致病菌种有以下几种:
Butyrivibrio sp AE3009
Butyrivibrio sp XPD2006
Butyrivibrio sp NC2007
▼6
▸ 对大部分影响细胞壁合成的抗生素敏感
溶丁酸弧菌菌株对高水平的萘啶酸(一种DNA旋切酶抑制剂,30-500 μg/ml)具有耐药性。它们通常对许多其他抗生素敏感,特别是那些影响细胞壁合成的抗生素,如β-内酰胺类、头孢菌素和杆菌肽。5株瘤胃溶纤丁酸弧菌对离子载体和蛋白质合成抑制剂非常敏感,但对阿霉素不敏感。
它们对碳水化合物代谢抑制剂和解偶联剂相对不敏感,对盐碱霉素、金霉素和杆菌肽的敏感性各不相同。
▸ 对四环素具有耐药性
溶纤维丁酸弧菌中存在两种类型的四环素耐药基因:一种是非传染性的,其序列与肺炎链球菌tet(o)相同;另一种是传染性耐药基因tet(W)。该基因的DNA GC含量高于其他溶纤丁酸弧菌基因,类似的序列也存在于其他瘤胃厌氧菌中,表明最近在瘤胃细菌之间发生了属间转移。
▼7
许多溶纤维丁酸弧菌菌株(从羊、鹿和牛中分离出的菌株超过50%)具有抗菌活性,可能是由于它们产生不同的抑制化合物。
▸ 可以产生一些细菌素
来自牛瘤胃的溶纤维丁酸弧菌菌株JL5产生一种细菌素,这种细菌素可以抑制多种革兰氏阳性菌,如专门氨基酸发酵的粘连梭菌和嗜氨梭菌,并可能在体内调节氨的产生中发挥作用。
此外,菌株AR10的细菌素样活性是由于单个肽与先前报道的细菌素没有同源性。菌株OR79产生类似细菌素的活性,具有广谱活性。氨基酸序列比较表明,这些分子代表一种新型抗生素。
▼8
丁酸弧菌通过代谢植物纤维素和其他碳水化合物,产生丁酸等短链脂肪酸,为宿主提供能量。同时,丁酸弧菌还能影响其他菌株的生长,包括促进和抑制作用。以下是丁酸弧菌对其他菌株生长影响作用的总结:
▸ 可以增强以下菌属的生长:
Bacteroidales
Bacteroides
Odoribacter
Peptococcaceae
Bacteroidales
Bacteroides
Odoribacter
Peptococcaceae
Bacteroidales
Bacteroides
Odoribacter
Peptococcaceae
▸ 可以抑制以下菌属的生长:
向上滑动阅览
Bifidobacterium
Coriobacteriales
Adlercreutzia
Collinsella
Porphyromonas
Prevotella
Clostridium
Clostridiales incertae sedis
Clostridiales Family XIII. Incertae Sedis
Blautia
Coprococcus
Dorea
Lachnospiraceae
Ruminococcaceae
Ruminococcus
Dialister
Campylobacteraceae
Erysipelotrichaceae
Bifidobacterium
Coriobacteriales
Adlercreutzia
Collinsella
Porphyromonas
Prevotella
Clostridium
Clostridiales incertae sedis
Clostridiales Family XIII. Incertae Sedis
Blautia
Coprococcus
Dorea
Lachnospiraceae
Ruminococcaceae
Ruminococcus
Dialister
Campylobacteraceae
Erysipelotrichaceae
Bifidobacterium
Coriobacteriales
Adlercreutzia
Collinsella
Porphyromonas
Prevotella
Clostridium
Clostridiales incertae sedis
Clostridiales Family XIII. Incertae Sedis
Blautia
Coprococcus
Dorea
Lachnospiraceae
Ruminococcaceae
Ruminococcus
Dialister
Campylobacteraceae
Erysipelotrichaceae
▸ 受到以下菌属的抑制作用
Bifidobacterium
Coriobacteriales
Adlercreutzia
Collinsella
Bacteroidales
Bacteroides
Porphyromonadaceae
Odoribacter
Parabacteroides
Porphyromonas
Prevotella
Rikenellaceae
Alistipes
Turicibacter
Streptococcus
Clostridiales
Catabacteriaceae
Clostridium
Clostridiales incertae sedis
Peptoniphilus
Clostridiales Family XIII. Incertae Sedis
Lachnospiraceae
Blautia
Lachnospiraceae
Coprococcus
Dorea
Eubacterium
Lachnobacterium
Lachnospira
Roseburia
Lachnospiraceae
Peptococcaceae
Ruminococcaceae
Ruminiclostridium
Acetivibrio
Eubacterium
Faecalibacterium
Oscillospira
Ruminococcus
Acidaminococcus
Dialister
Phascolarctobacterium
Veillonella
Rubrivivax
Alcaligenaceae
Oxalobacter
Bilophila
Desulfovibrio
Campylobacteraceae
Enterobacteriaceae
Escherichia
Erysipelotrichaceae
Erysipelotrichaceae
Holdemania
Akkermansia
丁酸弧菌(Butyrivibrio)是一种重要的微生物,广泛分布于人体和反刍动物的胃肠道中,对胃肠道生态系统的健康和功能发挥着至关重要的作用。
在众多生理功能中,产生丁酸盐等短链脂肪酸和共轭亚油酸的能力尤为关键。以下是对丁酸弧菌产生丁酸盐进而对健康影响的总结性描述:
▼1
✦ 维持肠道厌氧环境,减少病原体的定值
结肠细胞利用丁酸产生能量,从而增加上皮氧消耗。因此,产丁酸细菌的存在有助于维持肠道的厌氧环境,从而进一步防止机会性需氧病原体(如沙门氏菌和大肠杆菌)的定植。
✦ 产生具有广谱抗菌活性的肽
丁酸还调节cathelicidins的产生,cathelicidins 是一种参与哺乳动物先天免疫并对潜在肠道病原体表现出广谱抗菌活性的多聚阳离子肽。
✦ 增强巨噬细胞的抗菌活性
丁酸通过抑制 HDAC 来调节固有层中的结肠巨噬细胞,并限制促炎性IL-12和IL-6的产生,以及脂多糖刺激的巨噬细胞产生的抗菌一氧化氮。
丁酸通过代谢物增强巨噬细胞的 GPCR 独立的抗菌活性,一项研究表明,在微生物丁酸存在下生长的巨噬细胞上调了抗菌蛋白钙卫蛋白的表达,但降低了抗炎 IL-10 的表达。此外,微生物丁酸可显著增强巨噬细胞消除潜在病原体(如肠道沙门氏菌和柠檬酸杆菌)的能力。
因此,丁酸可增强肠道对侵入性病原体的防御能力,而不会引起组织损伤性炎症或过度反应。
产丁酸的微生物群在肠道中的动态作用
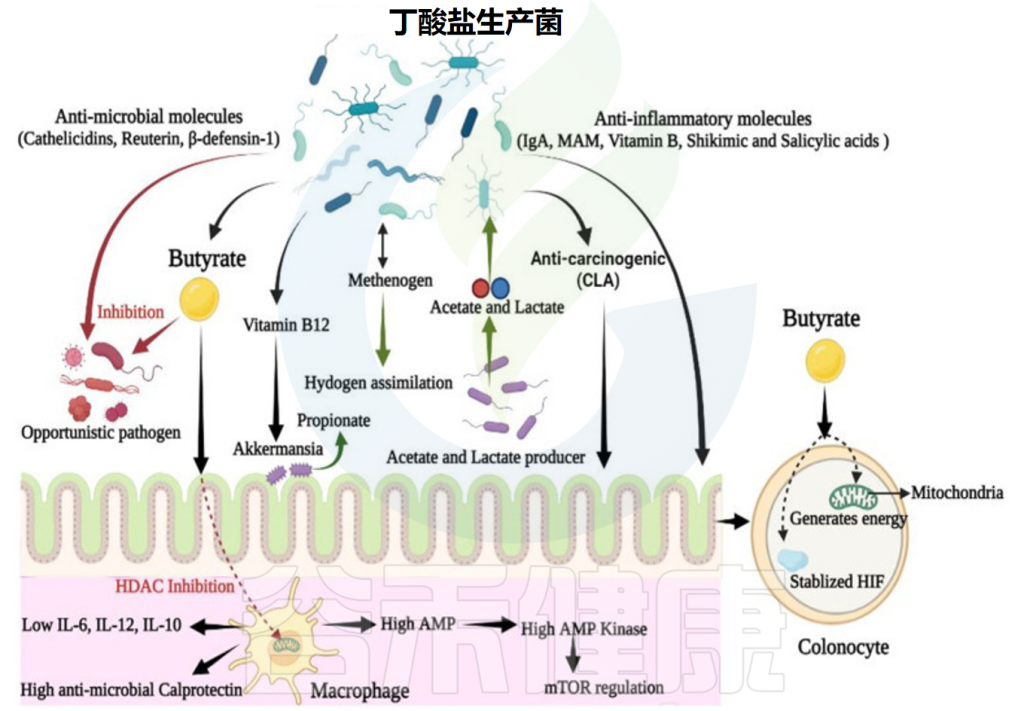
Singh V,et al.Front Microbiol.2023
▼2
体外和体内研究表明,丁酸生产者参与维生素生物合成,特别是维生素B复合物的生物合成。维生素B复合物是各种代谢活动中必需的辅助因子,也与调节宿主的免疫稳态有关。
健康的肠道屏障对于维生素的吸收至关重要,许多脂溶性维生素(如维生素A、D、E和K)需要通过肠道吸收才能进入血液循环。丁酸盐通过促进肠道健康,间接地支持了维生素的吸收过程。
▼3
肠上皮是一种单层结构,外覆黏液层,是抵御肠道病原体的第一道防线。肠上皮细胞通过紧密连接相互连接。肠上皮含有分泌黏液的杯状细胞,这些细胞通过分泌黏液提供屏障保护,黏液还充当免疫球蛋白IgA和抗菌肽的储存器。
✦ 增强粘液的形成
丁酸弧菌是人体肠道中的重要丁酸生产者,它通过增加杯状细胞分化与粘蛋白糖基化相关的基因表达来增强粘液形成。
临床研究表明,霍乱患者口服抗性淀粉(一种丁酸前体)后迅速康复。此外,肠道细菌产生的丁酸会加速肠道上皮细胞中线粒体依赖的氧消耗,从而稳定 缺氧诱导因子(HIF)。丁酸本身也会抑制降解 HIF 的 HIF-脯氨酰羟化酶。稳定化的HIF调节紧密连接蛋白claudin-1、MUC2表达和抗菌肽β防御素-1的生成。
✦ 调节肌动蛋白结合蛋白
丁酸还通过诱导抗炎细胞因子 IL-10RA 依赖性抑制 claudin-2 蛋白来加强肠上皮细胞屏障,而 claudin-2 蛋白在紧密连接处形成旁细胞通道并增加肠道通透性。
最近的一项研究还证明了丁酸在调节肌动蛋白结合蛋白突触蛋白(SYNPO)中的作用,该蛋白在肠上皮紧密连接中表达,对肠屏障完整性至关重要。
✦ 促进肠道蠕动,缓解便秘
另外,肠道中的丁酸一方面可以促进胃肠肌肉的收缩能力,增强胃肠的蠕动;另一方面还可以促进神经递质的传递,以此来促进胃肠蠕动,缓解便秘问题。
▼4
✦ 肠道炎症患者中的丁酸弧菌减少
在小鼠中开展的结肠炎研究证实,产生丁酸的丁酸弧菌群减少,肠道中丁酸水平降低,这导致了需氧肠杆菌科细菌的扩张,这是肠道菌群失调的常见标志。
研究表明,溃疡性结肠炎患者肠道中产丁酸的丁酸弧菌数量减少。另一方面,据报道, 丁酸弧菌的培养上清液对小鼠模型中的IBD(克罗恩病) 和结肠炎有效。
✦ 增强IL-10抗炎细胞因子,减少促炎细胞因子
微生物丁酸作为配体参与抗炎反应,通过芳烃受体(AhR)和各种G蛋白偶联受体(如GPR109a、GPR43 和 GPR41)来减轻炎症并维持肠道稳态。
AhR和GPR是转录因子,激活后可控制各种免疫调节剂的转录机制。AhR通过增强抗炎IL-10分泌B细胞和Th2细胞,同时减少促炎Th1和Th17细胞,发挥抗炎作用。
丁酸激活的GPR109a促进Treg细胞分化,增强产生抗炎IL-10的Th2细胞和血浆IL-10水平,进而抑制促炎性IL-17。GPR43降低CD4 T 细胞增殖并限制促炎性细胞因子(如IL-17和IL-22)的分泌。
▼5
✦ 提高谷胱甘肽的水平
生物体处于应激状态时,体内积累大量未能被消除的活性氧,破坏氧化还原平衡,导致DNA、脂质和蛋白质受到破坏,机体物理屏障受损。丁酸能够提高细胞内谷胱甘肽(一种内源性抗氧化剂清除剂)的水平。
一项首次利用人体研究丁酸盐对于氧化作用影响的实验开展,结果表明丁酸盐处理结肠后,促进了肠粘膜中还原性谷胱甘肽表达通路的进行,还原性谷胱甘肽的含量显著提高。
✦ 影响酶促抗氧化系统
丁酸还可以通过影响酶促抗氧化系统达到抗氧化作用。在日粮中添加适量丁酸钠,其血浆中超氧化物歧化酶和过氧化氢酶的含量显著高于对照组,并且增强了机体免疫能力。然而丁酸对于机体的抗氧化作用从根本上说是源自对基因和蛋白的调控。
Nrf-2是抗氧化反应基因的重要调节因子,肠道内的丁酸通过静脉门转移到肝脏,在肝脏中激活Nrf-2并通过AMPK激活或抑制HDAC诱导Nrf-2在细胞中的表达;另外,丁酸还可以抑制Keap1蛋白(Nrf-2的一种胞质抑制因子)的表达,以此维持氧化还原平衡。
▼6
丁酸生产者丁酸弧菌与各种肠器官轴相关,例如肠脑、肠肺、肠肝、肠道、肾脏和肠心脏轴。在这种复杂的关系中,丁酸弧菌充当微生物调节剂,并通过其代谢物丁酸盐发挥作用。
✦ 与精神状态如抑郁相关
如前所述,丁酸充当AhR的配体,使丁酸生产者成为肠脑轴中的相关群落。研究已经确定了产生丁酸的丁酸弧菌的抗抑郁作用以及它们在抑郁症个体中的减少。
✦ 丁酸盐的减少与肺部疾病风险增加相关
研究还证实了肠肺轴,因为研究发现肠道菌群失调与哮喘和肺部疾病的发生密切相关。据报道,婴儿肠道微生物多样性降低会增加哮喘和传染性呼吸道疾病的风险。特别是,肠道中产丁酸菌丰度的减少与过敏症和哮喘风险的增加密切相关。
此外,在流感等病毒感染期间,丁酸盐通过GPCR41受体增强肺中的Ly6C-单核细胞,这些单核细胞分化为替代激活的巨噬细胞(AAM),通过限制中性粒细胞进入呼吸道,减轻肺部的免疫病理反应。
✦ 产丁酸菌株可抑制肝脏炎症和氧化应激
肠道微生物群也参与肠肝轴,因为肝脏大约70%的血液供应来自肠道,甚至在慢性肝病期间门静脉和肝循环中存在较高的微生物脂多糖(LPS)水平。丁酸可维持肠道屏障的完整性,并抑制LPS的流入。在小鼠研究中,发现以三丁酸甘油酯形式的丁酸补充剂可有效缓解酒精引起的肝损伤。
酒精引起的菌群失调会显著减少厚壁菌门和毛螺菌科的成员,其中产丁酸的属(如Butyrivibrio)的数量较少。一项基于大量人类群体的研究(n =1148)也发现,产丁酸的丁酸弧菌菌株可抑制非酒精性脂肪肝病(NAFLD)中的氧化应激和肝脏炎症指标。
✦ 减少心血管疾病风险,降低血压
蛋白质发酵的代谢物,如胆碱、磷脂酰胆碱和肉碱,被肠道微生物代谢为三甲胺,三甲胺在肝脏中通过含黄素的单加氧酶(FMO)进一步转化为氧化三甲胺(TMAO)。已知 TMAO 会导致慢性肾病(CKD)并诱发心血管疾病,如动脉粥样硬化和冠心病。
而丁酸可以通过刺激含 apoA-IV 的脂蛋白的分泌,通过逆向胆固醇转运降低循环胆固醇。此外,丁酸还能增强肠道分泌胰高血糖素样肽-1(GLP-1),从而降低血压。
近年来的研究表明,肠道微生物群的平衡与疾病或健康状况密切相关,丁酸弧菌在不同疾病中的丰度会发生变化。
我们通过查阅和整理相关研究,将丁酸弧菌(Butyrivibrio)在不同疾病中的丰度变化总结在下面。
感染患者中的丰度较低

丁酸盐产生菌水平较低与人群严重感染风险增加存在关联。因感染住院的参与者中,Veillonella和链球菌的相对丰度更高,而丁弧菌相(Butyrivibrio)对丰度较低。未因传染病住院的受试者具有较高水平的专性厌氧菌,如Butyrivibrio。
2型糖尿病患者中的丰度较低

几种细菌(如Butyrivibrio和Faecalibacterium)和参与纤维降解的酶(如木聚糖酶EC3.2.1.156)与纤维摄入呈正相关,与流行的2型糖尿病(T2D)呈负相关。
通过对一项11394名参与者的队列研究发现,膳食纤维摄入量与丁酸弧菌(Butyrivibrio)和普拉梭菌(Faecalibacterium)等肠道细菌,以及吲哚丙酸和3-苯基丙酸等代谢产物相关,这些细菌及代谢产物又与较低的2型糖尿病风险关联。
在白塞氏病患者中丁酸弧菌减少

在患有白塞氏病(BD)的患者中,放线菌门、乳杆菌科和双歧杆菌属的肠道微生物群相对丰度增加。而超巨型巨单胞菌、丁酸弧菌、婴儿链球菌和Filifactor属的相对丰度显著减少。
Eggerthella lenta、Acidaminococcus物种、Lactobacillus mucosae、Bifidobacterium bifidum、Lactobacillus iners、Streptococcus物种和Lactobacillus salivarius的相对丰度在白塞氏病患者中显著增加。而与BD患者相比,正常个体中Megamonas hypermegale、Butyrivibrio、Streptococcus infantis和Filifactor物种的相对丰度显著增加。
在PICRUSt的功能注释分析中,研究发现白塞氏病患者中普遍存在戊糖磷酸途径和肌苷单磷酸生物合成的基因功能。数据表明白塞氏病患者中的肠道微生物改变了核酸和脂肪酸的合成。
婴儿胆汁淤积症中丰度降低

本研究结果显示,在属的水平上,婴儿胆汁淤积症(IC)组婴幼儿的瘤胃球菌属、丁酸弧菌属、产粪真杆菌属、粪杆菌属、毛螺菌科的丰度均显著降低。先前的研究表明,瘤胃球菌和丁酸弧菌参与胆汁酸羟基的氧化和熊去氧胆酸的生成,而产粪真杆菌属中含有胆盐水解酶(BSH),影响胆汁酸的代谢。
差异菌属与代谢物的相关性分析进一步显示,瘤胃球菌和丁酸弧菌与α-亚麻酸呈负相关,且研究发现IC患者的差异代谢物中α-亚麻酸代谢产物和亚油酸代谢产物含量丰富。亚麻酸的代谢产物二十碳五烯酸(EPA)和二十二碳六烯酸(DHA)通过提高ALT和AST活性,减轻肝细胞炎症反应和肝脏脂肪沉积,从而减轻儿童非酒精性脂肪性肝病的症状。
但婴儿胆汁淤积症(IC)患者肝细胞功能受损,影响亚麻酸在肝脏的代谢和DHA的合成,导致代谢产物蓄积,可能加重肝细胞异常炎症反应。
婴儿胆汁淤积症患者应早期加强肠内营养和氨基酸摄入
因此,婴儿胆汁淤积症(IC)患者应早期加强肠内营养和氨基酸摄入,减少长链脂肪酸摄入,增加短链和中链脂肪酸摄入比例,改善肝功能和营养状况,减轻肝脏负担,促进营养吸收和肠道菌群稳态的逆向调控。
肺动脉高压患者中减少
肺动脉高压(PAH)是一种以肺血管压力和阻力升高为特征的疾病,是由肺小动脉的增殖、外源性和纤维化重塑引起的。
在PAH患者肠道菌群中,产生三甲胺(TMA)/氧化三甲胺(TMAO)的细菌增加,但产生丁酸盐和丙酸盐的细菌减少,具体包括粪球菌(Coprococcus)、丁酸弧菌(Butyrivibrio) 、真杆菌(Eubacterium)、阿克曼氏菌(Akkermansia)、拟杆菌(Bacteroides)和毛螺菌(Lachnospiraceae)。
代谢紊乱和肥胖人群中减少
研究发现肥胖与宿主肠道菌群组成存在关联。肥胖患者肠道中微生物多样性降低、厚壁菌门与拟杆菌门比值(Firmicutes:Bacteroidetes)增加。
而较低的丁酸弧菌丰度与代谢紊乱和肥胖有关。这暗示了丁酸弧菌可能在体重管理和代谢健康中发挥作用。较高水平的丁酸弧菌可能有助于防止体重增加。
银屑病关节炎患者中减少
纽约大学的研究人员发现,银屑病关节炎患者的特定细菌类型水平较低,包括阿克曼氏菌、瘤胃球菌和丁酸弧菌。
阿尔兹海默症患者中减少
阿尔茨海默病除了其他清醒症的思维之外,海默组中丁酸合成细菌的种类(例如丁酸弧菌属Butyrivibrio和真细菌属 Eubacteria)有所减少。
✦ 补充多酚、儿茶素、花青素等益生元
补充益生元对丁酸弧菌丁酸生产者有积极影响,因为它们将益生元代谢为丁酸。不可消化的膳食纤维通常用作益生元,但其他生物活性分子,如多酚,也可以作为益生元产生丁酸。多酚干预显著增加了丁酸生产者如丁酸弧菌和瘤胃球菌科成员的丰度。
在其他多酚中,儿茶素、花青素和原花青素作为益生元的影响更为明显,因为它们增加了丁酸弧菌和粪杆菌属的丰度。
据报道,咖啡酸、绿原酸和芦丁等其他酚类化合物也能增加微生物丁酸的产生。此外,不同益生元的微生物可及性在丁酸生产者之间也存在差异;因此,施用不同的益生元可以选择性地丰富特定的丁酸生产者。
以下食物中含有较多的多酚等物质:
水果:颜色较深的水果当中多酚含量比较高,如樱桃、李子、蓝莓、草莓、李子等,平时可以多吃这些水果补充多酚类物质;
蔬菜:蔬菜中含有一定的多酚,比较常见的有卷心菜、香菜、胡萝卜、洋葱、菠菜等,紫甘蓝中的多酚含量偏高,适当吃能为机体补充多酚类物质,还能身体提供维生素等多种营养,维持身体健康;
全谷类:比较常见的粗粮食物,包括小麦、黑麦、燕麦等,里面都有多酚的成分,平时适当吃对身体健康有帮助,能快速补充多酚;
豆类:比如黄豆、豌豆等,里面含有天然的类黄酮成分以及多酚,都是对人体有益的营养物质;
其他食物:包括坚果类,如核桃、山核桃、栗子、亚麻籽等,里面的多酚化合物属于天然植物代谢物,进入人体之后能发挥一定的作用。黑巧克力中也含有多酚类物质,主要是可可豆中的天然多酚含量比较高。
✦ 补充合生元也能促进丁酸弧菌的丰度
除益生元外,还可以使用合生元疗法来促进肠道产生丁酸。合生元含有益生元和益生菌的组合,它们的协同作用比单独使用益生元和益生菌更为突出。
用枯草芽孢杆菌DSM 32315 和L-丙氨酰-L-谷氨酰胺进行合生元治疗可以增加主要丁酸生产菌,如普拉梭菌(F.prausnitzii)和丁酸弧菌(Butyrivibrio),从而增加丁酸水平。
另一项研究报道了在富含纤维的酸奶中使用合生元后,产生丁酸的丁酸弧菌丰度增加。
丁酸弧菌属(Butyrivibrio)是一类在人体肠道微生物群中占有重要地位的细菌,它们属于革兰氏阴性厌氧菌,能够产生丁酸,作为肠道细胞的主要能量来源,促进肠道屏障的完整性,调节免疫反应,以及抑制炎症。
在谷禾的大人群数据库中,丁酸弧菌属的检出率为35.58%,表明它在人群中的分布较为普遍。然而,在某些疾病状态下,该菌的丰度可能会降低,这可能与疾病的发生发展有关。例如,肠道炎症性疾病、肥胖、糖尿病等情况下,丁酸弧菌属的丰度可能会受到影响。
丁酸弧菌属的代谢特性使其能够利用纤维素和氨,这意味着富含纤维的食物(如蔬菜、水果、全谷物)和高蛋白食物(如肉类、豆类)可能有助于增加其丰度。此外,多酚类化合物(存在于许多植物性食物中)也可能对丁酸弧菌属的生长有积极影响。
在谷禾的检测报告中,如果能够看到丁酸弧菌属的丰度高低,结合相关的研究和文献,可以进一步了解其在健康中的作用和生理代谢机制。例如,高丰度的丁酸弧菌属可能与更好的肠道健康、更低的炎症水平和更稳定的肠道屏障功能相关。
总之,丁酸弧菌属作为人体重要的产丁酸菌之一,其丰度的变化可能与多种健康状况有关。通过饮食调整和生活方式的改变,可能有助于维持或增加其在肠道微生物群中的丰度,从而促进肠道健康。
主要参考文献
Wang Z, Peters BA, Yu B, Grove ML, Wang T, Xue X, Thyagarajan B, Daviglus ML, Boerwinkle E, Hu G, Mossavar-Rahmani Y, Isasi CR, Knight R, Burk RD, Kaplan RC, Qi Q. Gut Microbiota and Blood Metabolites Related to Fiber Intake and Type 2 Diabetes. Circ Res. 2024 Mar 29;134(7):842-854.
Singh V, Lee G, Son H, Koh H, Kim ES, Unno T, Shin JH. Butyrate producers, “The Sentinel of Gut”: Their intestinal significance with and beyond butyrate, and prospective use as microbial therapeutics. Front Microbiol. 2023 Jan 12;13:1103836.
Jin M, Cui J, Ning H, Wang M, Liu W, Yao K, Yuan J, Zhong X. Alterations in gut microbiota and metabolite profiles in patients with infantile cholestasis. BMC Microbiol. 2023 Nov 18;23(1):357.
Pidcock SE, Skvortsov T, Santos FG, Courtney SJ, Sui-Ting K, Creevey CJ, Huws SA. Phylogenetic systematics of Butyrivibrio and Pseudobutyrivibrio genomes illustrate vast taxonomic diversity, open genomes and an abundance of carbohydrate-active enzyme family isoforms. Microb Genom. 2021 Oct;7(10):000638.
Onohuean H, Agwu E, Nwodo UU. A Global Perspective of Vibrio Species and Associated Diseases: Three-Decade Meta-Synthesis of Research Advancement. Environ Health Insights. 2022 May 18;16:11786302221099406.
Rodríguez Hernáez J, Cerón Cucchi ME, Cravero S, Martinez MC, Gonzalez S, Puebla A, Dopazo J, Farber M, Paniego N, Rivarola M. The first complete genomic structure of Butyrivibrio fibrisolvens and its chromid. Microb Genom. 2018 Oct;4(10):e000216.
Sengupta K, Hivarkar SS, Palevich N, Chaudhary PP, Dhakephalkar PK, Dagar SS. Genomic architecture of three newly isolated unclassified Butyrivibrio species elucidate their potential role in the rumen ecosystem. Genomics. 2022 Mar;114(2):110281.
Kelly WJ, Leahy SC, Altermann E, Yeoman CJ, Dunne JC, Kong Z, Pacheco DM, Li D, Noel SJ, Moon CD, Cookson AL, Attwood GT. The glycobiome of the rumen bacterium Butyrivibrio proteoclasticus B316(T) highlights adaptation to a polysaccharide-rich environment. PLoS One. 2010 Aug 3;5(8):e11942.


16s 测序
几十年来,16S rRNA基因一直是基于序列的细菌分析的基石。
16S rRNA 基因是识别复杂群落中微生物的快速有效标记,因此已被用于人类微生物组计划 (HMP) 、地球微生物组计划 (EMP) 和人类肠道宏基因组学 (MetaHIT) 等研究。
该基因的一大优势在于,其分析方法不依赖于样本中细菌的可培养性,可以确定样本中所有细菌的相对丰度。此外,它能够同时对数百至数千样本进行并行测序,并在采样后最短两天内即可获得结果。
基于二代测序的16S可变区域和基于纳米孔测序的全长的16S核糖体RNA (rRNA)测序,是微生物基因组学的核心技术,广泛应用于针对16S rRNA基因特定区域的细菌和古细菌群落分析。
二代测序平台和华大DNBSEQ测序平台(特别关注V4或V3V4区域),以及基于纳米孔技术的全长16S测序,是该领域的两种主要方法。这些方法在功能、应用和局限性上存在显著差异,为微生物多样性和分类学提供了互补的见解。
二代测序平台以其高通量和高碱基准确性闻名,能够高效生成约300至500个碱基对(bp)的短读段,通常覆盖16S rRNA基因的1-2个区域。这种读段长度和分类分辨率之间的平衡使其成为土壤、水体及人类微生物组等多种环境和临床研究的可靠选择。
基于二代测序的16S 可变区具备更低的成本,更高的通量,但因其序列较短,在种水平的分辨率并不高。不过由于其已应用于大量的研究,因此有着广泛的研究样本数据和队列积累,对于基于16S菌群数据的疾病和代谢机器学习和模型更为适合。
同时,改善引物的通用性和减少单分子错误提升16S rRNA测序效率。以人体肠道菌群应用为例,谷禾结合宏基因组数据和已有的菌群组成数据,通过算法大幅提高了V4区数据的物种分辨率,达到约70%的种注释比例。此外,谷禾还通过增加靶向引物,针对病毒、真菌、寄生虫及部分难以区分的16S病原菌开发tNGS方法,从而显著扩展了二代测序在微生物多样性检测领域的应用范围。
基于纳米孔测序的全长16S rRNA基因测序,尤其是采用牛津纳米孔技术(Oxford Nanopore Technology, ONT)进行的测序,能够覆盖所有高变区(V1-V9)。该技术在物种层面上提供了更高的分类学分辨率,适用于病原体的快速诊断和鉴别。然而,这一技术同样面临序列准确性较低的挑战,并且需要为长读段数据分析量身定制先进的生物信息学工具。此外,由于全长扩增对样本质量的要求较高,该方法可能不适合某些低丰度或高度污染的样本。
当前,高通量测序数据的爆炸式增长,促使对新的生物信息学工具及数据管理和分析策略的开发。这种快速扩展推动了分类学方法和下游分析技术的改进,进一步巩固了测序技术在现代微生物学研究中的核心地位。随着人工智能(AI)技术的发展以及大量微生物测序数据的积累,结合特定领域的菌群数据与AI可为微生物群落外的疾病和代谢等问题提供新的见解。长期以来,通过二代测序积累的大量样本和数据,已成为其重要的应用优势。
细胞中有三种重要的 RNA:
核糖体RNA 是细胞中发现的分子,参与细胞器蛋白质的合成,扩散到细胞质中。这有助于将 mRNA中包含的信息翻译成蛋白质。
编辑
这些分子是在核仁中产生的,核仁看起来像是细胞核中一个密集的区域,其中包含编码 rRNA 的基因。这些编码的 rRNA 大小不一。至少每个核糖体包含一个大 rRNA 和一个小 rRNA。
核仁中的这两个 rRNA 与核糖体蛋白结合,形成核糖体的大亚基和小亚基。要知道,核糖体蛋白是在细胞质里合成的,然后一路移动到细胞核,就在核仁里进行亚组装。紧接着这些亚基又被送回细胞质去完成最终的组装。
注:-50S是核糖体中较大的亚基,-30S是核糖体中较小的亚基。S是用来描述核糖体亚基的相对大小和沉降速度的单位。
古细菌和细菌中发现的 rRNA 有所不同,因为古细菌和细菌在真核细胞发育之前,就已从共同的前体中分离出来。
16S rRNA 是编码细菌核糖体小亚基 RNA 的 DNA 序列。
16S rRNA 的功能
16S rRNA 是一种参与制造原核核糖体小亚基的 rRNA;它们是 30S 亚基(原核核糖体)的组成部分。
一般来说,它具有与较大亚基中的 rRNA 类似的结构功能,将核糖体的蛋白质保持在固定位置。它们还参与通过与较大亚基中的 23s rRNA 相互作用来促进小亚基与较大亚基的融合。古细菌、细菌、叶绿体和细菌中的小核糖体单元含有 16S。
关于 16S 中的“S”
16S 中的“S”是沉降系数,该指数表示大分子在离心场中的下沉速度。
16S rRNA的基因特性
16S rRNA 基因是与细菌基因组中看到的 rRNA 编码细菌相对应的 DNA 序列。
这些是特异性的,高度保守的;它们的基因序列足够长。
细菌包含大约 1 到 几十个16S rRNA 拷贝,因此检测极其敏感。16S rRNA编码基因的大小接近1500bp,包含50个功能结构域。
在原核生物中,核糖体 RNA 如下:
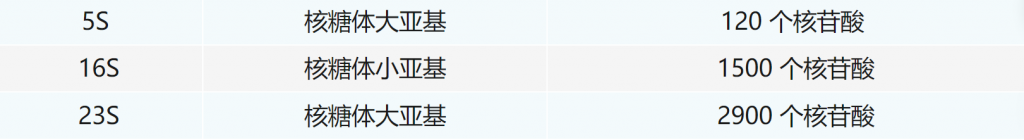
16S rRNA 基因的内部结构
16S rRNA基因内部结构包括保守区和可变区,不同细菌的通用引物可按保守区制定,特定细菌的特定引物可按可变区制定,16S rRNA不同区域的信息在种间存在变异,使得识别具有特异性。
16S rRNA 在蛋白质合成中的作用
它们与 23S 相互作用,帮助整合两个核糖体亚基 (50S+30S)。3’端包含一个反向 SD 序列,用于结合 mRNA 的 AUG 密码子(起始)。16S rRNA 的 3′ 末端与 S1 和 S21 的组合被认为与蛋白质合成的起始有关。
核糖体蛋白的固定可作为支架。因此,它们在确定核糖体蛋白的位置方面具有结构作用。
它稳定 A 位上准确的密码子-反密码子配对,从而在腺嘌呤残基的 N1 原子和 mRNA 骨架的 2′OH 基团之间形成氢键。
随着PCR技术的进步和核酸研究技术的持续进步,16S rRNA基因检测技术已成为最广泛使用的菌群丰度识别和检测工具。
该技术可用于更快更准确地识别、分类和发现病原体。
一般rRNA基因测序涉及以下步骤:
在分子技术中使用核糖体RNA的一些好处是:
16S rRNA 扩增子测序能够区分细菌或古细菌微生物物种,已成为研究微生物组结构和多样性的“指纹”。
海量数据集表明,不同类型的栖息地中微生物组结构存在很大差异,导致微生物分布不均。例如,厚壁菌门和拟杆菌门在人类肠道菌群中占主导地位,而变形菌门和蓝藻则在自然环境中普遍存在。不同的微生物在16S可变区上对扩增灵敏度和核苷酸序列可识别性有各自的偏好。因此可以通过优化变异区选择、测序策略和读长等配置来进一步改进基于16S的分析。
WGS、三代全长测序
近年来,鸟枪法全基因组测序(WGS)和第三代全长16S rRNA扩增子测序技术使得在物种和菌株水平上对微生物组进行注释成为可能,同时对生物量、测序成本、分析时间和存储空间的要求也提高了1到2个数量级。
更重要的是,由于大量的微生物组已通过16S扩增子测序进行调查(数千项研究产生了超过500,000个样本,存储在Qiita、MSE和NCBI等开放存储库中),其中一些难以重新取样或重新测序(例如纵向队列样本或从深海沉积物中收集的样本)。因此,对16S扩增子短读序列在分析中的应用进行全面评估以及不断更新和优化,对于先前数据的可重复使用具有重要意义。
16S rRNA扩增子在微生物组研究中的独特价值与再利用潜力
2023年,有研究模拟扩增子产生和密切参考分类注释的整个计算机模拟过程,以衡量使用 16S rRNA 基因进行微生物组分析的性能。研究超过 35,000 个物种的基因组和相应的 16S 片段,研究发现扩增后的短读测序可以接近全长 16S rRNA 基因,与参考数据库进行序列比对,在物种水平分类中达到 73% 的准确率。
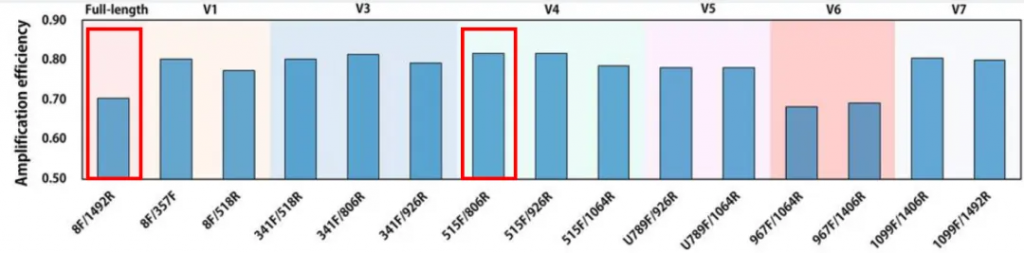
DOI: 10.1128/spectrum.00563-23
而且,发现16S V4区515F/806R引物组扩增效率最高,为81.72%(511,460个引物中有417,965个引物,如上)。
数据库方面
RefSeq表现出比其他两个数据库更好的比对率和精确度(在物种水平上)。而Greengenes和Silva的精度低主要是因为参考数据库中的注释不一致、不完整,例如一些分类单元与国际原核生物命名法规(ICNP)不匹配,大量序列缺少种甚至属级别的注释。此外,研究发现16S较长序列测序虽具有精度优势,但也存在因末端合并失败和测序错误率较高而导致的灵敏度损失。
最佳扩增区域
此外,以厚壁菌门和拟杆菌门为主的人类肠道微生物组的最佳扩增区域是V4,但对于富含变形菌门和酸杆菌门的河流样本,V3区域效果更好。下面列表还包含只考虑性能而不考虑每个生境测序成本的替代方案,例如对于土壤,V4-V5 PE 250 bp序列在物种水平上的精度比V4 SE 150 bp序列更高,但也使成本更高(下表)。
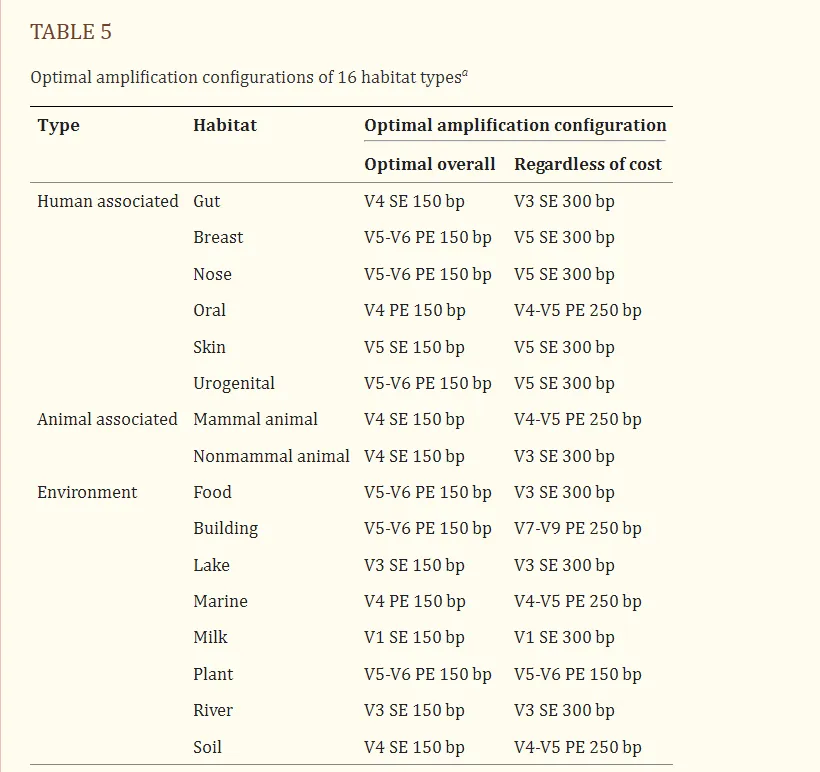
DOI: 10.1128/spectrum.00563-23
扩增子可变区对β多样性模式的影响最大
详细的 alpha 和 beta 多样性分析还展示了从多种方法中得出的分类学概况。16S V1、V3、V4 和 V5 区域的组成与实验设计和 WGS 相似,而 V6 和 V7 与其他配置表现出巨大的差异。
结果表明,扩增子可变区对β多样性模式的影响最大(Adonis R2 = 0.69,P < 0.01),其次是拷贝数校正(Adonis R2 = 0.15,P < 0.01),序列读长(Adonis R2 = 0.14,P < 0.01)和测序类型(Adonis R2 = 0.12,P < 0.01)。
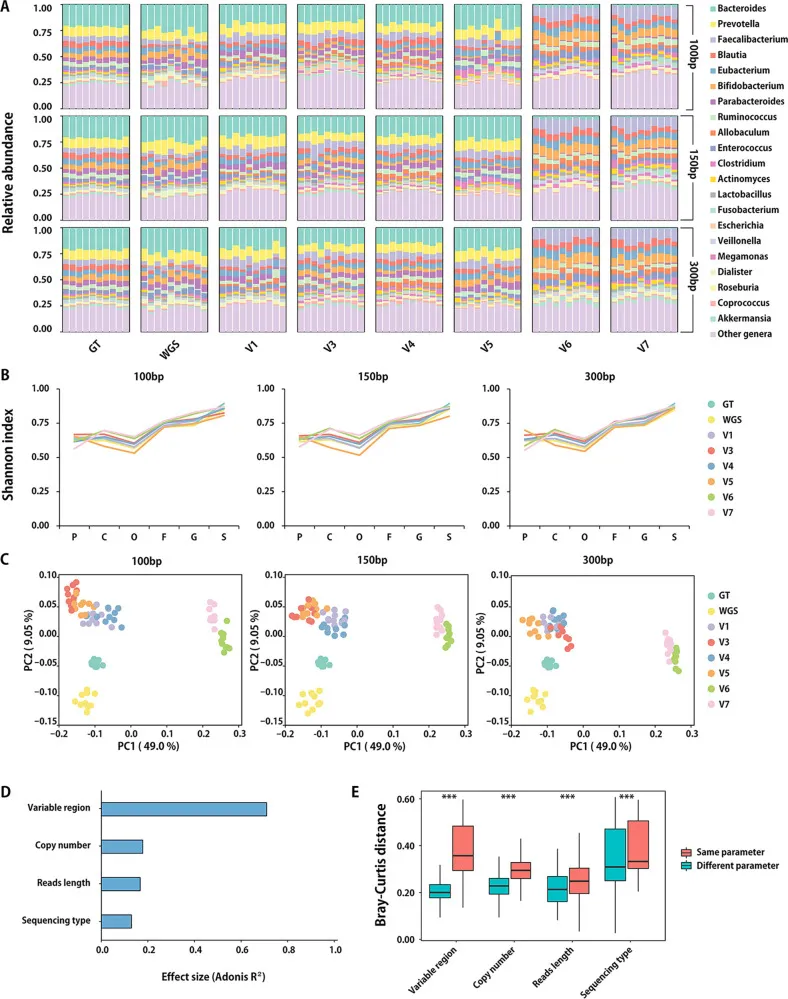
编辑
Zhang W et al.,2023 Microbiol Spectr.
因此,通过合适的测序策略,扩增子可以在物种水平上以低成本提供接近WGS的可靠微生物组分析。
扩增子二代测序
二代测序,尤其是使用 Illumina 的 Novaseq 或华大基因的DNBseq 系统,由于其高碱基调用准确性和吞吐量,已成为微生物分析的基石。
二代测序系统产生的300~500bp读长,足以覆盖 V4 区域的 515-806 片段,该区域以其可重复性和分类准确性而闻名。尽管有这些优势,V4 区域的长度有限也可能是一个缺点。
为了缓解这种情况,测序方法通常采用经过修改的定制引物或增加测序深度等方法来提高保真度,此外新的靶向测序方案也使得二代测序平台成为更具成本效益的方案。
优化和减少偏差
尽管有这些优点,二代测序和纳米孔测序技术都有其局限性,包括由不同的引物组和测序平台引入的偏差。例如,研究表明,纳米孔平台上的 V3-V4 区域引物可能表现出物种依赖性偏差,这可能并不适合所有类型的微生物群落。针对这些问题,研究人员通过调整PCR循环次数、引物组和生物信息学工作流程来优化测序方案,从而提高微生物群落分析的可靠性和准确性。
16S全长测序
相比之下,纳米孔测序,尤其是使用牛津纳米孔技术 Oxford Nanopore Technologies (ONT) 平台,可以对全长 16S rRNA 基因进行测序,涵盖所有高变区 (V1–V9)。虽然单碱基准确度较低,但综合来看物种分辨率更高。ONT MinION 等设备的便携性、成本效益和实时测序功能使其成为快速现场应用的理想选择,例如诊断细菌感染和监测传染病爆发。
尽管纳米孔测序具有这些优势,但它也面临挑战:相对测序和建库成本更高,通量较二代测序更低,尾端数据质量的下降以及缺乏专门用于分析纳米孔 16S 序列的生物信息学工具和方案。对样本要求更高,可能不适合部分低丰度或较多污染的样本。
多项比较研究表明,纳米孔技术可以在物种水平上识别微生物组成,其结果与二代测序等其他测序技术的结果非常相似,同时还可以识别出更多的细菌种类。
总体而言,在实际应用中,选择二代测序还是纳米孔测序取决于具体的研究和应用目标。
克服挑战和限制
16S rRNA 测序虽然功能强大,但也存在一些固有的挑战和局限性。一个主要问题是由于细菌和古细菌 DNA 从极少量扩增而导致的污染,使其容易受到有害环境细菌的影响。此外,与宏基因组测序方法相比,16S rRNA 测序的成本效益和可及性是以数据较少为代价的。为解决这些问题而做出的努力促成了新技术和新策略的开发。引入唯一分子标识符 (UMI) 为测序深度问题提供了潜在的解决方案。解决引物通用性和减少单分子错误也是提高 16S rRNA 测序效率的关键。
针对人体肠道菌群应用
谷禾结合宏基因组数据和已有的菌群构成数据,通过算法可以大大提高扩增子数据的物种分辨率。此外病毒、真菌、寄生虫和部分16S难区分的病原菌目前通过增加靶向引物进行tNGS的方式来实现,大大拓展了二代测序微生物多样性检测领域的涵盖范围。
高通量测序数据的爆炸式增长,要求开发新的生物信息学工具和数据管理和分析策略。这种快速扩展导致了分类学方法和测序数据下游分析的改进,进一步巩固了测序技术在现代微生物学研究中的作用。随着AI和大量微生物测序数据的积累,特定领域的菌群数据结合AI可以提供菌群之外的疾病和代谢等问题的答案,长久以来二代测序积累的大量样本和数据变成一种重要的应用优势。
主要参考文献:
Zhang W, Fan X, Shi H, Li J, Zhang M, Zhao J, Su X. Comprehensive Assessment of 16S rRNA Gene Amplicon Sequencing for Microbiome Profiling across Multiple Habitats. Microbiol Spectr. 2023 Jun 15;11(3):e0056323.
Bel Mokhtar N, Catalá-Oltra M, Stathopoulou P, Asimakis E, Remmal I, Remmas N, Maurady A, Britel MR, García de Oteyza J, Tsiamis G, Dembilio Ó. Dynamics of the Gut Bacteriome During a Laboratory Adaptation Process of the Mediterranean Fruit Fly, Ceratitis capitata. Front Microbiol. 2022 Jul 1;13:919760.
Pichler M, Coskun ÖK, Ortega-Arbulú AS, Conci N, Wörheide G, Vargas S, Orsi WD. A 16S rRNA gene sequencing and analysis protocol for the Illumina MiniSeq platform. Microbiologyopen. 2018 Dec;7(6):e00611.
Yeo K, Connell J, Bouras G, Smith E, Murphy W, Hodge JC, Krishnan S, Wormald PJ, Valentine R, Psaltis AJ, Vreugde S, Fenix KA. A comparison between full-length 16S rRNA Oxford nanopore sequencing and Illumina V3-V4 16S rRNA sequencing in head and neck cancer tissues. Arch Microbiol. 2024 May 7;206(6):248.

谷禾健康

质子泵抑制剂(PPI)是一种抑制胃酸分泌最有效和最常见的胃酸抑制药物, 于1989年被引入医学领域,可阻断胃壁细胞中的H+/K+ ATPase 酶系统,从而抑制壁细胞分泌氢,并抑制细胞外钾的吸收,这种机制抑制了胃壁细胞的酸分泌。
由于质子泵抑制剂抑制胃酸分泌的能力,已成为治疗和预防胃酸反流、胃食管反流病(GERD)、溃疡非糜烂性反流病(NERD)、非甾体抗炎药相关溃疡、食管炎、消化性溃疡病(PUD)和卓-艾氏综合症(ZES)等疾病的一线药物。此外,PPI还与抗生素一起用于幽门螺杆菌根除疗法。
质子泵抑制剂(PPI)通常被认为是安全的,因此经常被过度开具和长期使用(多项研究发现,在住院患者中,约有40%-71.4%在住院期间接受了PPI治疗)。然而,越来越多的研究表明,PPI的过度使用与多种健康状况有关,已知PPI引起的胃内pH值升高和随后的胃肠道生理改变会对整个胃肠道造成不良影响。
国际著名期刊《GUT》等多项期刊都有发表研究显示,质子泵抑制剂(PPI)治疗可能会破坏肠道微生物群,降低胃肠道微生物群的多样性和丰富度。检测了1827名患者的粪便样本。研究人员表示,质子泵抑制剂(PPI)会导致肠道共生菌的丰度降低,微生物多样性减少。具体来说,具有抗炎特性的Faecalibacterium减少,肠道重要基石菌瘤胃球菌、毛螺菌科也显示出减少。
此外,肠道感染病原体风险增加;一项荟萃分析了涉及近30万名患者的23项研究的结果。结果表明,65%的患者在使用PPI后出现了与艰难梭菌肠道定植相关的腹泻。使用PPI还可能导致小肠细菌过度生长;并且还会增加炎症性肠病患者的病情严重程度。2023年对45151名成年患者的数据研究发现,PPI会增加结肠炎的严重程度,并显示体重减轻和疾病活动指数评分显著增加。
质子泵抑制剂的副作用不仅限于肠道微生物群的破坏,长期使用PPI似乎还会影响营养吸收,包括钙、镁、维生素B12的吸收不良。此外,PPI可能会干扰骨代谢,从而增加骨折的发生率。胃液抗酸药已被证明能抑制肠道对磷酸盐的吸收。而这一过程又会导致低磷血症和骨矿化受损。
还有研究表明,PPI可通过影响脑-微生物轴诱发包括痴呆在内的神经退行性疾病。在小鼠模型中,PPI 已被证明会增加脑内淀粉样蛋白的沉积,从而诱发阿尔茨海默病。PPI使用者患痴呆症的风险增加了1.4倍。

此外,研究还显示,每日两次使用PPI的人更容易感染 COVID-19。使用PPI后的细菌易位可能与肝硬化腹水或隐源性肝脓肿患者发生自发性细菌性腹膜炎有关。因此,找到一种方法来减轻质子泵抑制剂(PPI)给药带来的副作用非常重要。
本文将在众多临床和实验研究的基础上客观的讨论下什么是质子泵抑制剂(PPI),包括哪些常见的药物,对应的治疗适用性,益处,副作用,尤其对肠道和神经系统等的副作用,以及有哪些改善副作用的措施和建议。
▼
质子泵抑制剂(PPI)是一类药物,它们能够抑制胃壁细胞中的质子泵,从而减少胃酸的分泌。
质子泵抑制剂(PPI)是苯并咪唑衍生物,由两个杂环基团组成,这些杂环基团包含吡啶和苯并咪唑基团,通过甲基亚磺酰基连接。
PPI通过与质子泵的活性位点形成共价结合,不可逆地抑制其活性,从而减少胃酸的生成。这种抑制作用是剂量依赖性的,意味着质子泵抑制剂的剂量越高,胃酸的分泌就越少。
▼
质子泵抑制剂可阻断产生胃酸的酶。这种酶被称为氢钾ATP酶泵,简称“质子泵”。质子泵是一种H+/K+-ATPase酶,主要存在于胃壁细胞的分泌小管膜上,负责将氢离子(H+)从细胞内部泵入胃腔,同时将钾离子(K+)泵回细胞内,这个过程是形成胃酸的关键步骤。
质子泵抑制剂的作用机制
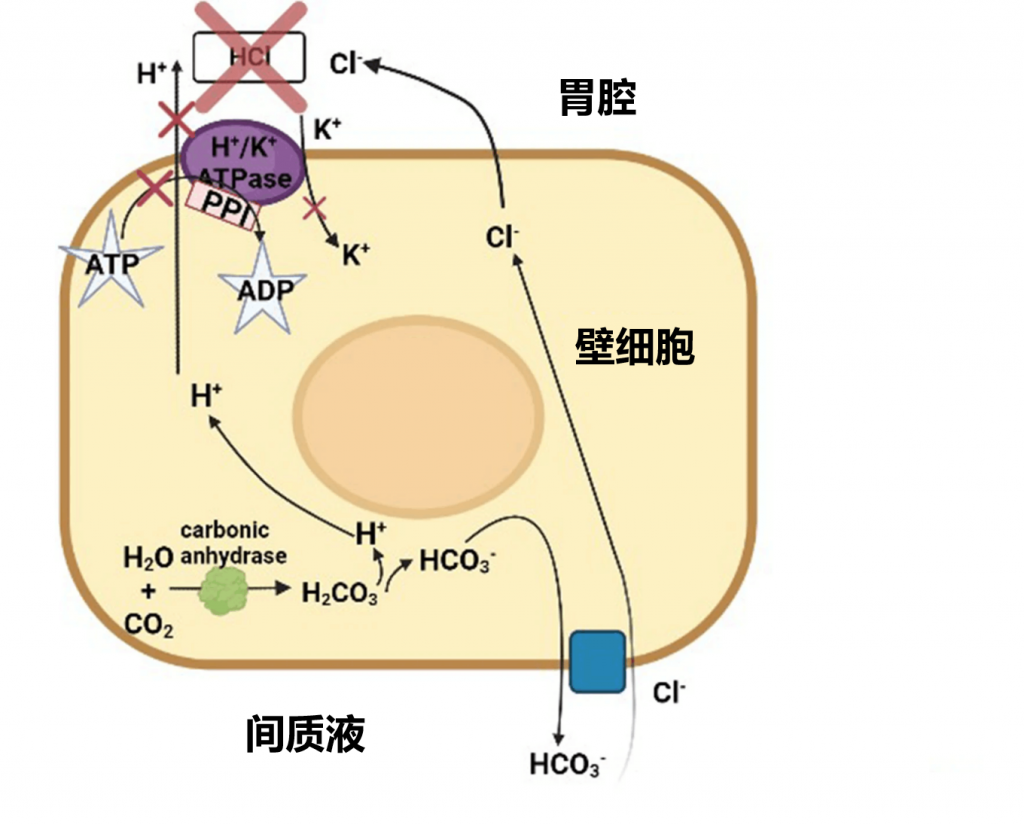
Morris N,et al.Natl Res Cent (2023)
可以这样想:质子泵抑制剂“抑制”(或阻止)质子泵完成产生胃酸所需的化学过程。
•使用质子泵抑制剂后仍有一部分胃酸供分解食物
但质子泵抑制剂(PPI)不会阻止所有胃酸的产生。你仍然有足够的胃酸来消化食物。如果你每天服用 PPI 约五天,这些药物会减少你约65%的胃酸。你仍然有剩余的35%供你的胃分解食物。
由于PPI是弱碱,它们会积聚在胃壁细胞活跃的酸性空间中。这种酸性很重要,因为PPI是前体药物,需要酸才能被激活。不同的质子泵抑制剂(PPI)与H+/K+ ATPase 的不同半胱氨酸残基结合,从而导致PPI的特性略有不同。
• 质子泵抑制剂的持续时间
由于质子泵抑制剂(PPI)通过不可逆抑制胃H+/K+ ATPase,因此它们能够产生较为持久的作用。但是,由于胃H+/K+ ATPase 的周转,并不是所有的H+/K+ ATPase 泵都能被阻断,并且可能需要几天的PPI给药才能达到最佳效果。
PPI从胃中排出后,会在小肠近端被吸收。血清半衰期很短,约为1-2小时。吸收后,血液循环将PPI输送到腹壁细胞,并在那里聚集在酸性分泌小管中。然后,PPI在酸催化下生成活性亚磺酸或磺酰胺,这些化合物与H+ /K+ ATPase中的半胱氨酸残基共价结合,并对酸分泌产生抑制作用,直到替代泵合成,也就是大约36小时。这些化合物与H+/K+ -ATPase 泵形成不可逆二硫键。
质子泵抑制剂(PPI)通常在餐前使用,因为它们需要小管中H+/K+ -ATPases 的活跃表达,才能在进餐后发生结合。建议胃内pH值高于4,胃腔内的pH值在2以下时,每天服用一片PPI可使胃液pH值维持在4以上达10-16小时。
注:PPI与蛋白质结合力强,可被肝细胞色素 P450 降解。奥美拉唑和埃索美拉唑经CYP2C19代谢,雷贝拉唑、兰索拉唑和右兰索拉唑也经CYP2C19代谢,但也对CYP3A4有亲和力。经肝脏代谢后,大多数苯并咪唑类药物最终通过肾脏排泄,但兰索拉唑和右兰索拉唑也经胆道排泄。
▼
质子泵抑制剂主要治疗因胃酸刺激或损害消化系统某些部位(如胃、十二指肠(小肠最靠近胃的部分)或食道)而引起的病症。例如以下几种疾病:
-胃食管反流病(GERD)
质子泵抑制剂(PPI)可缓解GERD症状,如胃灼热。GERD会导致胃酸渗入食道,刺激食道。胃酸会损害食道内壁(糜烂性食管炎)。PPI可减少胃酸,让食道有时间愈合和修复。
-糜烂性食管炎
质子泵抑制剂通过显著减少胃酸的分泌,可以帮助缓解症状,促进食管黏膜的愈合。质子泵抑制剂是治疗糜烂性食管炎的常用药物之一。
-胃溃疡和十二指肠溃疡
PPI可帮助治愈胃酸破坏胃的保护性内壁而形成的胃溃疡和小肠溃疡。PPI还可帮助治愈和预防因服用NSAID(非甾体抗炎药)而形成的溃疡。
定期服用NSAID的人中,多达30%的人会患上胃溃疡。
-幽门螺杆菌感染
质子泵抑制剂(PPI)可以与某些抗生素联合使用,用于根除幽门螺杆菌,从而使抗生素更有效地杀死细菌。
-佐林格-埃利森综合征
质子泵抑制剂(PPI)可抵消佐林格-埃利森综合征的影响。患有这种罕见疾病的人,肿瘤会释放一种促进胃酸分泌的激素。
-其他与质子泵抑制剂治疗或相关的疾病:
巴雷特食管
胃炎/十二指肠炎
胃肠道出血
食管裂孔疝
消化不良
多发性内分泌腺瘤
病理性高分泌情况
消化性溃疡
应激性溃疡预防
系统性肥大细胞增多症
▼
质子泵抑制剂是一类具有选择性抑制胃酸分泌作用的药物,临床常用的质子泵抑制剂包括奥美拉唑、泮托拉唑、雷贝拉唑、埃索美拉唑、兰索拉唑、右兰索拉唑等。一个简单好记的方法就是:一般带有“拉唑类”的药物,即质子泵抑制剂。
它们的名称听起来相似,作用相似,副作用也类似。但还是存在差异,在这里,我们简单介绍六种常用的质子泵抑制剂。
■ 奥美拉唑
奥美拉唑用于治疗非癌性胃溃疡、胃食管反流病、活动性十二指肠溃疡、佐林格-埃利森综合征和糜烂性食管炎等疾病引起的胃酸过多。奥美拉唑通过阻止胃酸产生起作用,属于质子泵抑制剂类药物。
奥美拉唑还可以与抗生素一起使用来治疗由幽门螺杆菌(H. pylori) 感染引起的胃溃疡。
奥美拉唑治疗胃食管反流病的剂量一般为每天一次,每次20毫克。应在每日第一餐前至少30分钟服用。每天服用效果最好,而不是仅在出现症状时服用。
非处方奥美拉唑只能连续服用14天。可能需要1至4天才能改善症状。
一些药物可能会影响奥美拉唑的效果:
■ 埃索美拉唑
埃索美拉唑(Nexium)是奥美拉唑的近亲。它具有相似的化学成分,也获准用于相同的用途。更重要的是,它既可以作为非处方药,也可以作为处方药。
埃索美拉唑治疗的剂量通常为每天一次20毫克,在一天的第一顿饭前至少30分钟服用。埃索美拉唑不能立即缓解胃灼热症状。
一些药物可能会影响埃索美拉唑的效果:
■ 泮托拉唑
泮托拉唑用于治疗成人和至少5岁儿童的糜烂性食管炎(胃食管反流病(GERD)引起的胃酸对食管的损害)。泮托拉唑通常一次给药长达8周,直至食管愈合。
与奥美拉唑和埃索美拉唑不同,它未被批准用于治疗溃疡或预防幽门螺杆菌溃疡。
泮托拉唑的剂量一般为每天一次,每次40毫克,在第一次进餐前至少30分钟服用。但是,体重低于40公斤的儿童的剂量会有所不同。
一些药物可能会影响泮托拉唑的效果:
■ 兰索拉唑
兰索拉唑(Prevacid)获准用于治疗与奥美拉唑和埃索美拉唑相同的疾病,以及其他一些疾病。它可以预防和治疗成人非甾体抗炎药(NSAID)相关胃溃疡。它还可以治疗1岁及以上儿童的胃食管反流和糜烂性食管炎。
治疗胃食管反流的兰索拉唑剂量通常为每天一次15毫克,在第一次进餐前至少30分钟服用。
硫酸铝(Carafate)会使身体更难吸收兰索拉唑。服用兰索拉唑后至少等待30分钟再服用硫酸铝。
■ 右兰索拉唑
右兰索拉唑(Dexilant)和泮托拉唑一样,只能凭处方购买。它是兰索拉唑的近亲,具有相似的化学结构。右兰索拉唑用于治疗胃食管反流病引起的胃灼热,以及糜烂性食管炎。
治疗胃食管反流病时,右兰索拉唑的剂量通常为每天一次30毫克。服用右兰索拉唑的好处是,它可以在一天中的任何时间服用,无需考虑是否进食。
一些药物可能会影响右兰索拉唑的效果:
■ 雷贝拉唑
雷贝拉唑(AcipHex)是另一种需凭处方购买的质子泵抑制剂 。雷贝拉唑用于成人治疗胃酸过多的疾病,例如Zollinger-Ellison 综合征。雷贝拉唑还用于成人促进十二指肠溃疡或糜烂性食管炎的愈合。
注:它是20毫克DR片剂,每天服用一次。
雷贝拉唑的第一剂可能比奥美拉唑的第一剂产生更大的效果。这是因为雷贝拉唑被认为在阻止酸产生方面比奥美拉唑起效稍快。
一些药物可能会影响雷贝拉唑的效果:
▼
质子泵抑制剂(PPI)通常耐受性良好且安全。它们是治疗各种胃肠道疾病的常用药物,包括胃十二指肠溃疡、糜烂性食管炎、胃食管反流病、胃酸分泌过多综合征,并用于治疗幽门螺杆菌感染。
如果按照推荐剂量和推荐时间服用,PPI通常是安全的。然而,它们可能与几种严重的不良反应有关,包括:
急性间质性肾炎(一种肾衰竭):可能在PPI治疗期间的任何时间发生;
艰难梭菌相关性腹泻:这是一种特别严重且持续性的腹泻;
骨质疏松症导致髋部、腕部或脊柱骨折的风险增加:接受高剂量治疗(通常每日多次服用)和PPI治疗持续时间超过一年的患者风险更高;
皮肤型红斑狼疮(CLE) 和系统性红斑狼疮(SLE);
抑制氯吡格雷的作用,氯吡格雷是一种用于降低心脏病患者血小板凝结能力的药物;
此外,使用PPI可能会掩盖胃癌症状。所有对PPI反应不佳或停用PPI后症状复发的患者都应接受额外的诊断检测,老年人在开始治疗前应考虑进行内镜检查。
质子泵抑制剂(PPI)通常耐受性良好。使用PPI后报告的较常见副作用包括:
临床治疗中还发现,长期过量使用PPI所引起的病症会影响各种身体系统。受影响的系统可能包括胃肠道、呼吸系统、骨骼肌肉系统、免疫系统、泌尿系统和神经系统。
这些系统受到PPI引起的不同机制的影响,例如营养缺乏、pH值变化、肠道菌群组成变化和胃肠道微生物过度生长等等。研究表明,长期过量使用PPI会损害肠粘膜。这些变化可能会增加胃肠道癌症的风险。长期或过量使用PPI的风险与受影响的身体系统的关系将在下面的章节详细讨论。
尽管通常情况下质子泵抑制剂(PPI)耐受性良好且安全。但是已经有许多研究发现质子泵抑制剂的长期使用可能会导致胃肠道菌群的失衡。
胃肠道微生物对于消化/分解膳食营养素以及抵御病原体至关重要。这些微生物群受pH值、其他微生物、环境因素和遗传因素等不同因素的调节。正常的微生物群彼此之间处于精确的平衡状态,这种平衡正是促进肠道健康的关键。如果微生物群的平衡被打破,就会引发肠道和肠外疾病。
质子泵抑制剂(PPI)可以降低胃酸度,PPI使用者的胃液pH值>4,因此口腔和胃中的许多细菌可以存活。

口腔中一些菌群的丰度增加
对1815名荷兰个体进行了研究。发现,PPI使用者口腔中的Rothia mucilaginosa、Rothia dentocariosa、细菌属Scardovia和放线菌以及微球菌科等物种更多。
在健康个体中,服用埃索美拉唑4周后,牙周袋中梭杆菌(Fusobacterium)和纤毛菌(Leptotrichia)增多,但唾液中奈瑟菌(Neisseria)和韦荣氏球菌(Veillonella)减少。
注:有记录显示,健康个体的食管中大多发现革兰氏阳性菌,而不健康个体的食管(如 Barrett 食管)中大多发现革兰氏阴性菌。这种细菌类型的变化可能导致脂多糖增加,从而引发炎症反应。
食管和胃中的菌群组成也发生变化
同时,食管微生物群中微球菌科(Micrococcaceae)、丹毒丝菌科(Erysipelotrichaceae)和肠杆菌科(Enterobacteriaceae)的数量有所增加,而丛毛单胞菌科(Comamonadaceae)减少。研究表明,使用质子泵抑制剂(PPI)会改变食管菌群,导致厚壁菌门增多、拟杆菌门和变形菌门减少。
而PPI使用者的胃液pH值 >4,因此许多口腔细菌可以在胃中存活。胃中链球菌科增多、普雷沃氏菌科减少。
pH 值的增加还可能导致幽门螺杆菌感染以及其他微生物的过度生长。
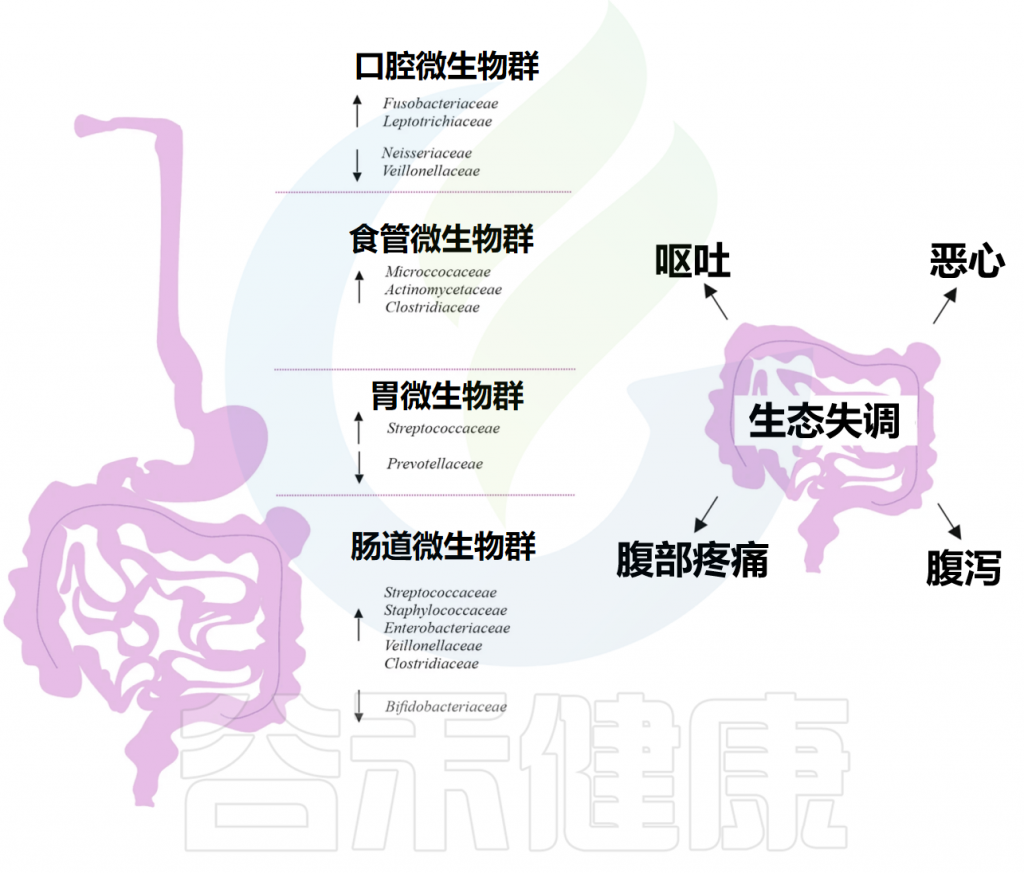
doi: 10.1007/s43440-023-00489-x.

国际著名期刊《GUT》发表的一项研究表示,检测了1827名患者的粪便样本。研究人员表示,质子泵抑制剂(PPI)会导致肠道共生菌的丰度降低,微生物多样性减少。
具有抗炎特性的Faecalibacterium减少
服用PPI的受试者粪便中Faecalibacterium属有所减少。众所周知,该属具有抗炎特性。此外,粪便微生物群中的 OTU 有所减少。
乳杆菌的数量有所增加
对20名反流性食管炎患者施用PPI(埃索美拉唑),持续 8 周。他们随后测定了粪便和血液中的细菌组成,此外还测定了粪便中的有机酸浓度和pH值。研究显示,使用PPIs治疗后,加氏乳杆菌、发酵乳杆菌、罗伊氏乳杆菌和瘤胃乳杆菌的数量增加。
乳杆菌被认为对人体健康有益,但有几份报道称乳酸杆菌在敏感的免疫功能低下患者中引起了菌血症和肝脓肿等严重感染。
胃食管反流病患者使用PPI后的菌群变化
使用质子泵抑制剂(PPI)治疗会导致整个胃肠道中的微生物群多样性显著降低。研究了胃食管反流病患者胃黏膜和粪便的微生物群变化,发现长期使用PPI的患者组粪便微生物群中链球菌科、韦荣氏球菌科、氨基酸球菌科(Acidaminococcaceae)、微球菌科和黄杆菌科(Flavobacteriaceae)的丰度较高,而Pelobacter、脱硫单胞菌属(Desulfuromonas)、Alkanindiges、Koridiimonas、Marinobacterium和Marinobacter的相对丰度降低。
而Luteimonas、Limonobacter、Herbaspirillum、Sphingobium、Phenylobacterium、Comamonas、Chryseobacterium、Duganella、Pedobacter的相对丰度也较高。
短期使用质子泵抑制剂(PPI)与细菌水平显著降低有关,例如Pelobacter、Desulfuromonas、Alcanlvorax、Kordiimonas、Desulfuromusa、Marinobacterium和Marinobacter丰度均有所下降。
使用质子泵抑制剂后发生改变的胃肠道细菌
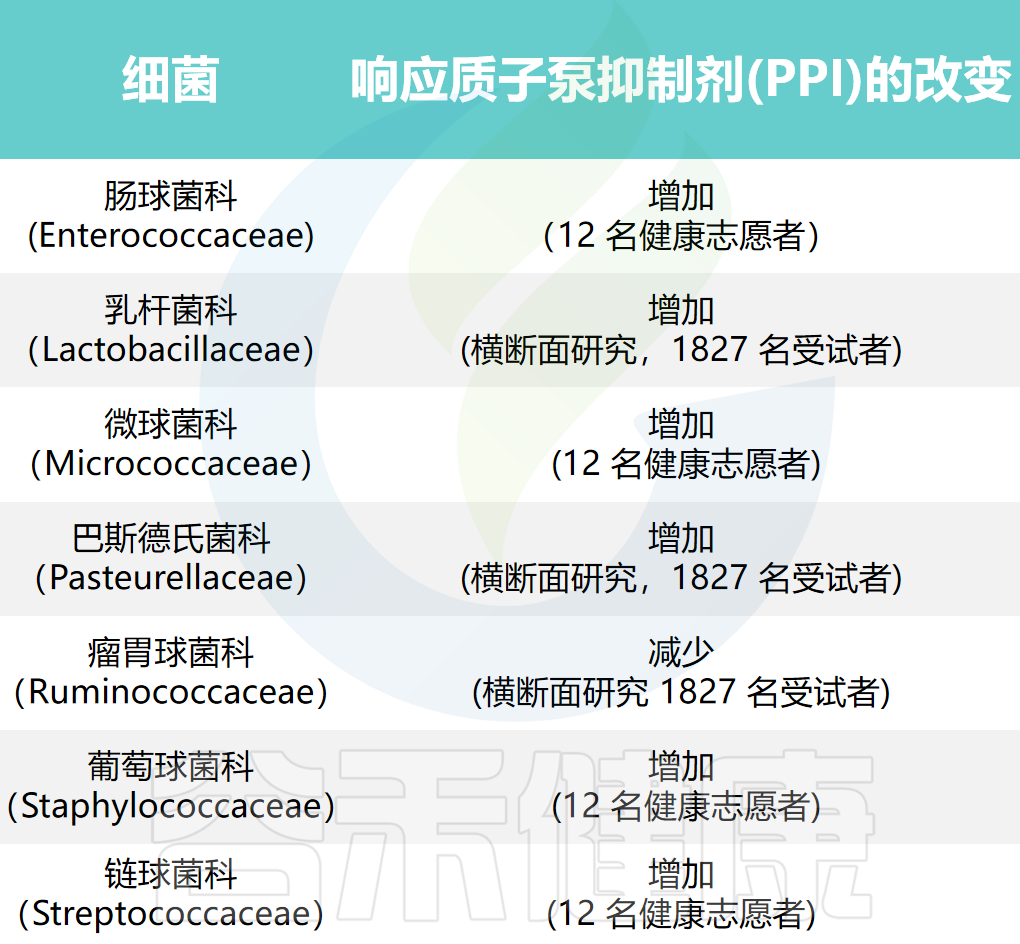
Morris N,et al.Natl Res Cent (2023)
使用质子泵抑制剂(PPI)还会增加致病微生物感染的风险。这可能是因为益生菌由于其他微生物过度生长而丢失,或者由于胃酸分泌受到抑制导致pH值变得更碱性,这也会允许通常被胃酸消灭的微生物存活,从而导致后续微生物环境的改变,使致病微生物更容易引发感染。
使用质子泵抑制剂的人艰难梭菌感染风险更高
艰难梭菌感染是最常见的感染之一。它会导致严重腹泻、结肠扩张甚至死亡。
人们怀疑肠道菌群失调和诱发的炎症可能是艰难梭菌感染的原因。胃酸是许多病原微生物的重要屏障,抑制胃酸产生可能促使肠道感染倾向增加。
研究了艰难梭菌感染相关腹泻的发生与质子泵抑制剂(PPI)使用之间的关系发现,使用PPI的患者发生艰难梭菌感染相关腹泻的风险更高。未服用PPI的患者艰难梭菌感染的发生率为4.4%,而服用PPI的患者为9.3%。

另一项研究也发现,服用质子泵抑制剂(PPI)的患者艰难梭菌感染的发生率从近37%上升到63%左右。此外,美国胃肠病学杂志进行的一项荟萃分析了涉及近30万名患者的23项研究的结果。结果表明,65%的患者在使用PPI后出现了与艰难梭菌肠道定植相关的腹泻。
PPI治疗后促炎细胞因子表达增加,易感性增强
用质子泵抑制剂(PPI)治疗的结肠炎小鼠表现出促炎细胞因子表达增加,例如IL-1β、IL-6、白细胞介素-17A(IL-17A)、TNF-α、干扰素-γ(IFN-γ)、巨噬细胞炎症蛋白-2(MIP-2)和单核细胞趋化蛋白-1 (MCP-1)。
研究还显示,PPI可导致瘤胃球菌科和双歧杆菌属细菌减少,而γ-变形菌纲、肠球菌科、肠杆菌科和乳酸杆菌科以及肠球菌属和韦荣球菌属细菌增加,这些细菌与艰难梭菌感染易感性增加有关。
产短链脂肪酸的菌群减少,炎症加重
与PPI使用相关的肠道微生物群的改变可能会改变产生短链脂肪酸的细菌种类的数量和比例。研究发现,PPI给药减少了拟杆菌属和普雷沃氏菌属。这种减少会改变短链脂肪酸的总体产生量,导致结肠调节性T细胞活化减少,从而可能导致炎症加重。
因此,PPI不仅可能增加艰难梭菌感染的风险,还可能增加相关死亡率。
质子泵抑制剂使用者沙门氏菌定植增加
沙门氏菌病是一种主要由沙门氏菌引起的胃肠道感染。研究表明,非伤寒沙门氏菌病患者更多包括质子泵抑制剂(PPI)使用者。
一项病例对照研究表明,沙门氏菌感染的原因之一是近期使用PPI。原因可能是PPI减少胃酸分泌作用可能抑制胃液的保护性抗菌作用,从而促进沙门氏菌感染。
报告还显示,使用PPI治疗降低了小肠和大肠各个部位的丁酸水平。丁酸显著影响对沙门氏菌的抑制。在沙门氏菌感染期间也观察到IL-1β和TNF-α表达增加。
长期使用质子泵抑制剂(PPI)治疗会影响小肠微生物群,由于失去胃酸这一“防御屏障”,可能引发小肠细菌过度生长。
小肠细菌过度生长是指每毫升上肠道抽吸物中的细菌数量超过10^5个,而正常值则为每毫升上肠道抽吸物中的细菌数量少于10^4个。
小肠细菌过度生长是一种以小肠中细菌增多或类型异常为特征的综合征。它可能没有症状,也可能出现非特异性症状,包括腹胀、腹部不适、腹痛或腹泻。更严重的病例会出现体重减轻、营养不良、肝脏变化、关节痛和贫血、维生素D3缺乏引起的低钙血症所致的手足搐溺症、代谢性骨病以及维生素B12缺乏引起的多发性神经病。
使用质子泵抑制剂可能导致小肠细菌过度生长风险增加
一些报告表明,质子泵抑制剂(PPI)的使用会影响小肠细菌过度生长(SIBO)的发生。给一组老年患者每天服用20毫克奥美拉唑。他们在试验前后采集了患者的十二指肠抽吸物。试验前,患者的细菌计数< 10^4 菌落形成单位(CFU)/mL(96%的患者),而在接受PPI治疗后,43%的患者细菌计数>10^5 CFU/mL。
对200例使用PPI36个月的胃食管反流病患者、未使用PPI的肠易激综合征(IBS)患者和健康对照者进行了葡萄糖-氢呼气试验(GHBT)以检测小肠细菌过度生长。
50%的PPI使用者、24.5%的IBS患者和6%的健康对照者患有小肠细菌过度生长。比较了使用埃索美拉唑20mg每日两次治疗糜烂性反流病和胃食管反流病的患者,持续6个月。在PPIs治疗8周后,患者主诉腹胀(43%)、气胀(17%)、腹痛(7%)和腹泻(2%)。经过6个月的治疗,肠道症状的发生率继续增加。
小肠细菌过度生长的患者肠道多样性降低
而小肠细菌过度生长(SIBO)有影响到肠道菌群的构成。与没有小肠细菌过度生长的患者相比,在诊断患有小肠细菌过度生长的患者中,肠道菌群的组成显示出α多样性显著降低。链球菌的相对丰度增加,拟杆菌的相对丰度降低。
此外,研究表明SIBO与炎症有关。在SIBO患者十二指肠中发现促炎细胞因子IL-1β、IL-6和TNF-α升高。
使用质子泵抑制剂会增加肠道炎症的严重程度

在研究质子泵抑制剂(PPI)使用及其导致的实验性结肠炎严重程度的小鼠中,发现PPI会增加结肠炎的严重程度,并显示体重减轻和疾病活动指数评分显著增加。
在同一研究中,长期使用质子泵抑制剂(PPI)导致炎症性肠病(IBD)患者的住院率增加,其中包括 45151 名匹配的成年患者。
这些实验数据表明,长期使用PPI会对与炎症性肠病相关的各种途径产生深远影响,从而导致更严重的并发症。此外,在一项分析了3个队列的研究中(n=82269),与非PPI使用者相比,PPI使用者患IBD的风险增加。
IBD患者同时使用PPI会影响肠炎的治疗效果
此外,证据还表明,正在接受治疗的 IBD 患者在同时接受 PPI 治疗时不太可能获得缓解。在一项使用生物药物英夫利昔单抗治疗 IBD 的研究中,缓解率下降与 PPI 的使用有显著关联。第30周 PPI 组的缓解率为30%,非PPI组的缓解率为49%。
注:这种显著性仅在克罗恩病中可见,而在溃疡性结肠炎中则不可见。与非PPI使用者相比,使用PPI的患者住院率增加。
研究观察到在克罗恩病和溃疡性结肠炎中,质子泵抑制剂(PPI)使用与炎症性肠病相关住院和手术的比值比增加。经过倾向评分匹配后,发现与不使用 PPI 相比,PPI使用与新生物制剂使用、IBD相关入院和手术均显著相关。此外,PPI剂量与新生物制剂使用和 IBD 相关住院也存在关联,但与手术无关。
使用质子泵抑制剂还可能增加儿童患IBD风险
质子泵抑制剂(PPI)还与儿童IBD有关,PPI的疾病风险评分为3.6,研究使用H2RA(H2受体阻滞剂)作为对照,将PPI给药与之比较,H2RA的疾病风险为 1.6。
然而,由于样本量小(对照组n=6,PPI组n=6)以及研究前可能对 IBD 进行误诊,因此有必要进一步调查。
这些发现表明,使用质子泵抑制剂(PPI)可能会增加患炎症性肠病(IBD)的风险,并且可能与某些 IBD 治疗的有效性降低有关。
不同人群的基础生理特征不同,年龄、体重、原有基础疾病、肠道微生物群的构成等都会影响质子泵抑制剂使用后的副作用。以下是更容易受到质子泵抑制剂(PPI)对肠道微生物群影响的人群:
-体质较弱或微生物群失调的群体
研究发现,体质虚弱或微生物群失调的群体更容易受到质子泵抑制剂(PPI)使用影响,从而导致肠道微生物群进一步紊乱。
-存在胃肠道疾病的人群
例如患有炎症性肠病(IBD)或肠易激综合征(IBS)的患者,他们的肠道微生物群可能已经因为疾病本身而受到影响,PPI 的使用可能会进一步改变其微生物群组成。
-肝硬化患者
两项正在进行的研究,提供了更多证据,说明PPI引起的微生物群改变与肝硬化患者存在临床关联。
-胃食管反流病(GERD)患者
同样,GERD患者也是质子泵抑制剂(PPI)使用后微生物群改变显著的群体之一。
-老年人
老年人可能因为使用质子泵抑制剂(PPI)而面临更高的风险,因为他们可能已经是易受感染和其他并发症影响的个体。
-长期使用PPI的人
研究表明长期使用PPI的人肠道微生物群会发生了一些变化,但这些变化是否会对健康产生不利影响仍有待证实。
-使用抗生素或其他常用药物的人
质子泵抑制剂(PPI)使用者中观察到的微生物组成变化不是由于腹泻或排便频率增加引起的,而是与抗生素和除PPI之外的其他常用药物类别有关。
-社区获得性肺炎的高风险群体
使用PPI后,社区获得性肺炎的风险显著增加,特别是链球菌源性肺炎,这表明胃肠道可能成为其他身体部位潜在病原体的储存器。
长期使用质子泵抑制剂(PPI)会使身体对多种重要营养素的吸收产生负面影响。营养素吸收受阻会导致多种健康并发症。
与长期使用PPI相关的营养异常包括低镁血症、低钙血症、维生素B12缺乏症和低/高钾血症。
值得注意的是,并非每个人都会出现与长期使用PPI 相关的异常,但使用 PPI 可能会增加此类缺陷的风险,例如在之前已经存在营养吸收问题的个体中。
长期使用质子泵抑制剂导致镁缺乏,低镁血症可导致严重的健康问题,如癫痫和心律失常。
研究发现长期使用PPI,尿液中Mg2+排泄量减少,表明肠道对Mg2+吸收减少的补偿,并且排除了肾脏 Mg2+丢失是缺乏的原因。
奥美拉唑会抑制镁吸收,受到剂量和时间影响
据估计,30-50%的Mg2+在肠道中吸收。奥美拉唑治疗 14 天后, Mg2+从顶端到基底外侧的转运受到抑制。此外,值得注意的是,在 PPI 治疗期间补充镁对镁水平没有影响。如果缺乏,则需要停止 PPI 治疗以恢复镁水平。
这种抑制可能呈剂量和时间依赖性,因为与14天奥美拉唑治疗相比, 21天奥美拉唑治疗进一步降低了 Mg2+从顶端到基底外侧的转运。瞬时受体电位黑色素瘤素(TRPM6/7)负责结肠对镁的吸收,PPI可抑制或降低其活性。
有研究提出,PPI对胃酸的抑制会阻碍从食物中溶解钙的能力,并由于pH值的变化而导致吸收减少。低钙血症会导致精神状态改变,并且由于骨骼变弱而导致骨折增加。
长期使用PPI导致胃泌素增多,钙流失增加
甲状旁腺激素(PTH)通过维持血清钙水平、刺激骨吸收、增加肾小管钙重吸收和骨化三醇生成(可增加上肠钙的主动转运)来调节钙和骨代谢。
质子泵抑制剂(PPI)通过抑制粘膜 D 细胞释放生长抑素而导致胃泌素显著增加,这已被证明对甲状旁腺有刺激作用,表现为高胃泌素血症导致大鼠甲状旁腺体积和重量增加。长期使用PPI会导致高胃泌素血症,这还会影响甲状旁腺,导致骨骼中钙质流失增加。
在人类中,八周的奥美拉唑疗法导致 PTH 水平增加 28%。如前所述,长期使用PPI会导致钙吸收降低。研究还发现,奥美拉唑会降低老年女性吸收碳酸钙的能力。
多吃牛奶和奶酪可以适当减轻钙缺乏
值得注意的是,牛奶和奶酪等钙源具有更高的生物利用度,因此如果遵循适当饮食的方案,可能能够减少钙缺乏症。
由于质子泵抑制剂(PPI)的使用,胃酸分泌受到抑制,维生素B12不能被正确分解,从而导致吸收减少。
在缺乏胃酸和不能被正确分解的情况下,维生素B12无法避免胰腺消化,导致吸收量降低。维生素B12缺乏会导致精神状态改变、虚弱、贫血和心悸。
维生素B12摄入受到阻碍的人群应谨慎使用PPI
由于维生素B12的生理储备,大部分人群不会出现维生素B12缺乏症,但在维生素B12摄入已经受到阻碍的人群中,应谨慎使用质子泵抑制剂(PPI)。
此外,还有研究表明,质子泵抑制剂(PPI)会抑制肾上腺皮质类固醇的合成,从而导致血清钾水平升高。高钾血症可导致心律失常和肌肉无力,从而引起严重的健康并发症。
也有患者在接受PPI治疗时出现低钾血症,尿钾排出量增加。因此,可能需要测量服用PPI的患者的钾水平。
使用质子泵抑制剂(PPI)对神经系统是否有影响?虽然罕见,但已有因质子泵抑制剂(PPI)治疗导致神经病变的病例报告,使用了奥美拉唑,也有兰索拉唑。
▼

与H2RA(H2受体拮抗剂)给药相比,PPI给药的记忆障碍不良反应结果显著增加。H2RA给药组没有关于阿尔茨海默病型痴呆的报告,而PPI给药组有80份。注:与PPI治疗相关的神经病变可能是维生素B12缺乏症的继发性因素。
记忆障碍包括阿尔茨海默病(AD)型痴呆、非AD型痴呆、记忆障碍和遗忘症。此外,研究还发现,与H2RA治疗相比,PPI治疗在神经病变、听力障碍、视力障碍和癫痫方面具有更高的风险关联。
神经病变症状出现的时间与使用PPI的时间重叠
在该案例研究中,神经病变症状的出现与开始使用兰索拉唑的时间点相重叠,并且停用兰索拉唑后这些临床和电生理症状的停止,因此我们推断质子泵抑制剂(PPI)与神经病变存在因果关系。
▼
一项使用德国老龄化、认知和痴呆数据库进行的研究发现,长期使用质子泵抑制剂(PPI)的个体与非长期使用PPI的个体相比,患痴呆症的风险增加。
质子泵抑制剂会导致β淀粉样蛋白增加
研究发现,质子泵抑制剂(PPI)可能会破坏清除酶及其活性,导致脑中β淀粉样蛋白的数量增加,这已被证明与痴呆进展有关。
在细胞培养和小鼠模型中,兰索拉唑、奥美拉唑、泮托拉唑和埃索美拉唑均显示出使淀粉样蛋白β浓度增加,特别是淀粉样蛋白β40和42以剂量依赖性方式增加(5µM-50µM),这在生理上是相关的。
注:γ-分泌酶具有多个淀粉样蛋白前体裂解位点,并且可以受到称为γ-分泌酶调节剂的小分子药物的影响。这些调节剂可以改变γ-分泌酶裂解位点,产生不同的淀粉样β蛋白生成模式,会产生多种淀粉样β蛋白,其中淀粉样β40是最常见的。
此外,PPI 可能对多种途径产生影响,导致淀粉样β蛋白生成异常,表现为PPI给药增加β位淀粉样蛋白前体蛋白裂解酶 (BACE1) 活性,并可能通过pH值变化影响蛋白酶(如meprinβ),导致淀粉样β蛋白生成增加。
▼
胆碱能神经元网络的退化是阿尔兹海默病、唐氏综合症、帕金森病痴呆和路易体痴呆的主要特征。质子泵抑制剂(PPI)会抑制人类胆碱乙酰转移酶(chAT)的活性,所有测试的PPI均几乎完全抑制 chat 的酶活性。奥美拉唑的效力与强效chAT抑制剂α-NETA相似,而埃索美拉唑和雷贝拉唑的效力分别强2倍和5倍。
使用质子泵抑制剂导致胆碱能神经元网络的退化可能是质子泵抑制剂对神经系统产生的另一个负面影响。
除了对胃肠道和神经系统的影响外,质子泵抑制剂还可能影响心血管系统、肾脏,甚至还可能与肺炎有关。
▼
长期使用质子泵抑制剂(PPI)与心肌梗死和中风等心血管事件有关。人们推测长期使用PPI会导致心血管事件的途径有多种,包括一氧化氮水平变化、镁缺乏、嗜铬粒蛋白A增加以及对细胞色素P450酶的影响。

缺血性中风和心肌梗死风险上升
质子泵抑制剂(PPI)引起的心血管事件大多与高剂量和长期使用有关。一项平均随访时间约为6年、涉及214998人的研究表明,长期使用PPI会增加缺血性中风和心肌梗死的风险。
短期和长期使用PPI均与缺血性中风和心肌梗死的风险比增加相关,风险比分别为1.13和1.31。而高剂量PPI的使用与上述风险显著相关。
在使用PPI时应考虑患者的心血管疾病风险
因此,在开具和使用质子泵抑制剂(PPI)时应考虑心血管疾病风险,尤其是在需要长期使用或高剂量使用的情况下。
此外,应密切评估和监测患有心血管疾病高风险的个体的长期PPI使用情况。
▼
质子泵抑制剂(PPI)治疗是否会导致肾脏损伤还存在争议。
正在使用质子泵抑制剂的人患急性间质性肾炎风险更高
一项研究调查了肾脏损伤是否与当前、近期或既往使用PPI有关,结果发现,当前使用PPI的人患急性间质性肾炎的风险高于既往或近期使用PPI的人。该研究还指出,随着年龄的增长,PPI与肾脏损伤之间的关联性增强。
另一项利用医疗健康记录的研究发现,质子泵抑制剂(PPI)的使用与急性肾损伤风险之间存在关联,还与慢性肾病风险之间存在关联。
然而,不同的慢性肾病分期和PPI的使用与死亡率无显著相关性。这些异质性结果源于不同研究人群的基础生理特征不同,需要进行更多随机研究来调查肾损伤与长期PPI使用之间的关联。
▼
还有一些研究评估了长期质子泵抑制剂(PPI)使用与肺炎的关联,但结果也相互矛盾。
使用质子泵抑制剂肺炎发病率会增加,但具体原因还不确定
多项研究表明,在使用PPI之前和之后,肺炎发病率都会增加。然而,解释有所不同,一些人将这种肺炎关联归因于在PPI给药前后存在的潜在肺炎风险,而与PPI的使用无关。但进一步的研究证明肺炎发病率的增加与第二年长期使用PPI有关。
此外,在台湾患有二型糖尿病的患者中,使用 PPI 与肺炎风险增加有关。研究发现,在二型糖尿病患者队列中,PPI 使用者的肺炎发病率比非使用者高 11.4% (30.3%vs.18.9%)。肺炎的相关风险也呈剂量依赖性增加,即PPI剂量越高,肺炎风险越高。
质子泵抑制剂(PPI)用于治疗胃酸相关疾病,如胃食管反流病(GERD)、胃溃疡和十二指肠溃疡等。
但是,长期或不当使用PPI可能会引起一些副作用,如导致肠道微生物群紊乱、骨折风险增加、维生素和矿物质缺乏、感染风险增加等。以下是一些详细的措施,可以帮助减轻或预防PPI的副作用:
▼
-医生评估
在使用PPI前,请务必咨询医生进行全面评估,确保有明确的使用指征。仅在医生建议的情况下使用PPI,避免不必要的长期使用。
-尽量使用最小有效剂量
使用最低有效剂量和最短疗程,以减少长期使用的风险,不要自行增加剂量或延长使用时间。
-定期评估PPI使用的必要性
定期与医生讨论PPI使用的效果和副作用,评估是否需要继续使用。如果症状得到缓解,考虑在医生的建议下逐渐减少PPI的剂量或停药。
注:PPI突然停药可能引起反弹性胃酸分泌。建议逐渐递减剂量,逐步停药,减少反弹风险。
-替代药物选择
对于需要长期减酸治疗的患者,可以在医生指导下尝试替代药物如H2受体拮抗剂。
-注意PPI与其他药物的相互作用
了解PPI可能与其他药物(如抗抑郁药、他汀类药物等)发生相互作用,影响药物的效果或增加副作用。
在使用其他药物时,告知医生你正在使用的PPI,以便医生调整药物剂量或更换药物。
-定期复查治疗和体检
定期复诊和进行体检,评估是否需要继续使用PPI。部分患者可能在症状缓解后可以逐渐停药,及时发现并处理健康问题。
-多学科协作,个性化治疗
将PPI作为综合治疗的一部分,包括生活方式改变、其他药物治疗和行为疗法等。
根据个体情况调整治疗方案,确保治疗的安全性和有效性。
▼
长期使用质子泵抑制剂(PPI)可导致肠道菌群失调,从而增加感染的易感性,并会导致肠道疾病以及其他疾病。最近有研究发现在PPI治疗期间额外补充益生菌,除了增强PPI治疗效果外,益生菌补充剂还能潜在地抑制肠道菌群失调和长期使用PPI的副作用。
减轻了肠道菌群失调

对患有胃食管反流病的儿童进行了12周的质子泵抑制剂(PPI)治疗,同时还给研究组施用了益生菌罗伊氏乳杆菌DSM 17938。结果显示,未服用益生菌的组中56.2%的儿童出现了肠道菌群失调,而服用益生菌的组中只有6.2%的儿童出现了肠道菌群失调。
减少病原菌的定值
益生菌在PPI治疗中发挥健康促进作用的潜在机制可能有很多。首先,益生菌的作用机制之一可能是竞争性排斥。通过与其他细菌竞争胃肠道中的受体位点,从而减少一些病原菌的定值。
益生菌充当限制致病菌增殖的“屏障”。研究表明,某些益生菌代谢物可能在调节细胞中的各种代谢和信号通路中发挥作用。此外,乳酸杆菌和双歧杆菌可产生抑制某些病原体增殖的细菌素。益生菌菌株能够通过影响肠道菌群来刺激短链脂肪酸(SCFA)的产生,这对于肠道健康也至关重要。
已有报道显示益生菌可有效减轻艰难梭菌感染,将含有艰难梭菌毒素的上清液与克劳氏芽孢杆菌一起可保护细胞免受毒素的影响,这与克劳氏芽孢杆菌产生丝氨酸蛋白酶有关。使用双歧杆菌和布拉氏酵母菌等益生菌也显示出对艰难梭菌的抑制。
嗜热双歧杆菌RBL67对沙门氏菌有抑制作用。其中,VSL#3 益生菌混合物中的婴儿双歧杆菌和短双歧杆菌被证实可改善细胞上皮完整性和对沙门氏菌侵袭的抵抗力。
缓解小肠细菌过度生长
研究发现,一些益生菌菌株可有效调节小肠菌群,这与小肠细菌过度生长的减少有关。
给使用质子泵抑制剂(PPI)超过12个月的患者施用了四种选定的益生菌,即鼠李糖乳杆菌LR06 (DSM 21981)、戊糖乳杆菌LPS01 (DSM 21980)、植物乳杆菌LP01 (LMG P-21021) 和德氏乳杆菌LDD01 (DSM 22106)。益生菌混合物显著减少长期使用PPI治疗的患者的细菌过度生长。
在128名接受PPI治疗的胃食管反流病(GERD)儿童中,服用额外的罗伊氏乳杆菌DSM 17938益生菌菌株降低了肠道菌群失调的发生率并抑制小肠细菌过度生长的风险。
调节宿主免疫反应
此外,益生菌还能调节宿主的免疫反应。超过70%的免疫细胞位于肠道,尤其是小肠,形成肠道相关淋巴组织 (GALT)。GALT 包含派尔集合淋巴结、淋巴小结、淋巴细胞和粘膜等结构。
益生菌已被证明能诱导树突状细胞(DC)成熟模式,其特征是释放少量TNF-α和白细胞介素12(IL-12),同时增加IL-10水平,从而抑制促炎性Th1淋巴细胞的产生。
因此,益生菌对小肠细菌过度生长、艰难梭菌感染、沙门氏菌感染有影响,即对质子泵抑制剂(PPI)治疗副作用的有一定的改善效果。
除了补充益生菌可以调节肠道微生物群并减轻使用质子泵抑制剂的副作用外,以下这些方法可能也有助于改善使用质子泵抑制剂后的副作用。
▼
益生元是一类不可消化的食物成分,它们可以促进益生菌的生长和活动。常见的益生元包括:
可溶性纤维:如菊粉、低聚果糖和低聚半乳糖。
不可溶性纤维:如全谷物中的纤维素。
益生元可以通过以下方式帮助减轻质子泵抑制剂的副作用:
促进益生菌生长:益生元为益生菌提供营养,促进其生长和活动。
改善肠道屏障功能:益生元可以帮助维持肠道屏障的完整性,减少病原体的入侵。
▼
-增加膳食纤维的摄入
膳食纤维可以促进肠道蠕动,帮助缓解便秘,这是某些质子泵抑制剂可能引起的副作用之一。建议多食用洋葱和大蒜等蔬菜、柑橘类水果和全谷类食品等富含膳食纤维的食物。
-均衡摄入各类营养素
长期使用PPI可能导致钙、镁、维生素B12等营养素的吸收障碍,增加骨折或其他营养不良风险。需要保证足够的维生素和矿物质摄入,可以通过食物或补充剂来确保足够的摄入量。
-适量摄入发酵食品
发酵食品如酸奶、酸菜等含有益生菌,可以改善肠道微生物群的平衡,有助于维持消化系统的健康,并可能减轻由于长期使用质子泵抑制剂可能引起的肠道菌群失衡问题。
-避免卧床进食
饭后至少待立或坐姿30分钟,避免立即卧床,以减少胃酸反流。
-避免刺激性食物
辛辣、油腻、烧烤等食物可能刺激胃肠道,加重消化不良症状。减少这类食物的摄入,选择易消化的食物如米粥、面条、煮熟的蔬菜等。
-少食多餐
每天分成4-5餐,每餐食量适中,避免暴饮暴食,这有助于减轻胃部负担,减少胃酸分泌,从而可能降低质子泵抑制剂的副作用。
-避免过度饮酒和吸烟
酒精和烟草会刺激胃肠道,加重消化不良症状,应尽量避免。
-注意饮食卫生
避免食用过期食品、生冷食物等容易引起胃肠道感染的食物,PPI使用可能增加肠道感染的风险,如艰难梭菌感染。保持良好的个人和食品卫生习惯,减少感染风险。
-喝足够的水
保持足够的水分摄入可以促进肠道蠕动,缓解便秘症状。
-限制饮料的摄入
咖啡是一种带有兴奋作用的饮料,可能会导致身体进入兴奋状态,增加胃酸分泌,从而加重症状或副作用。而碳酸饮料可能增加胃部气体,引起腹胀等不适,应尽量避免。
▼
定期检测肠道健康可以帮助及时发现和纠正肠道微生物群的失衡。这可能包括:
肠道微生物群分析:通过检测肠道内的微生物组成,了解益生菌和有害菌的比例以及相关代谢物的含量,并根据此来更有针对性、个性化的饮食及相关调整。
肠道屏障功能检测:评估肠道屏障的完整性,及时发现潜在的肠道屏障功能障碍。
骨密度检查:长期使用PPI可能增加骨折风险,尤其是老年人。建议定期进行骨密度检测,评估骨骼健康。
血液检查:定期进行血液常规、肝功能、肾功能、维生素和矿物质水平检测,及早发现和纠正异常。
▼
适量的运动可以帮助改善肠道健康,促进益生菌的生长。运动可以通过以下方式帮助减轻质子泵抑制剂的副作用:
促进肠道蠕动:运动可以增加肠道蠕动,帮助食物通过肠道,减少有害菌的生长。
降低压力水平:运动可以帮助降低压力水平,减少压力对肠道微生物群的负面影响。
小 结
通过补充益生菌和益生元、调整饮食、定期检测肠道健康以及适量运动,可以帮助改善肠道微生物群的平衡,从而减轻质子泵抑制剂的副作用。
然而,这些策略的有效性可能因个体差异而异,因此在实施这些策略时应考虑个人的健康背景和医生的建议。
质子泵抑制剂(PPI)包括奥美拉唑、埃索美拉唑、兰索拉唑、右兰索拉唑、泮托拉唑和雷贝拉唑,是治疗食管反流病(GERD)、溃疡非糜烂性反流病(NERD)、非甾体抗炎药相关溃疡、食管炎、消化性溃疡病等疾病的一线药物。
近年来,质子泵抑制剂对肠道微生物群的潜在影响及其对健康的长期后果引起了广泛关注。PPI治疗可能会带来一些副作用,如头晕、头痛、发热、腹痛、便秘、腹泻、胀气、恶心或呕吐等,科学界正在积极研究如何减轻这些影响。
PPI治疗的一些报道指出,其可能会对肠道微生物群产生影响,包括可能降低胃肠道微生物群的多样性和丰富度。这可能使得某些肠道感染风险增加,但这种情况各异。长期使用PPI还可能与小肠细菌过度生长及炎症性肠病病情波动有关。但这些关联在个体间的表现并不一致。
此外,PPI可能会影响某些营养的吸收,如钙、镁和维生素B12,并可能对骨代谢产生影响,但发生骨折的风险需个体风险因素综合考虑。PPI与神经退行性疾病(如痴呆)之间的关系也在研究中,但目前的证据并不确定。同样,有关PPI与心血管疾病、肾脏损伤、肺炎之间联系的证据也仍然在不断完善。
为了减轻PPI可能带来的长期影响,优化肠道微生物群平衡显得尤为重要。研究表明,益生菌(如罗伊氏乳杆菌DSM 17938)在接受PPI治疗的个体中显示出缓解某些副作用的潜力。
通过合理使用质子泵抑制剂(PPI),定期监测评估症状包括肠道,神经系统以及营养状况如维生素B12, 钙,镁等,针对性使用益生菌、益生元、改善饮食或选择替代药物,同时预防肠道感染或菌群失衡,从而综合减轻质子泵抑制剂药物(PPI)给个体带来的可能副作用。
注:本篇内容仅为科学知识的分享,无任何立场,仅供参考与学习。
主要参考文献
Kiecka A, Szczepanik M. Proton pump inhibitor-induced gut dysbiosis and immunomodulation: current knowledge and potential restoration by probiotics. Pharmacol Rep. 2023 Aug;75(4):791-804.
Morris, N., Nighot, M. Understanding the health risks and emerging concerns associated with the use of long-term proton pump inhibitors. Bull Natl Res Cent 47, 134 (2023).
Nighot M, Liao PL, Morris N, McCarthy D, Dharmaprakash V, Ullah Khan I, Dalessio S, Saha K, Ganapathy AS, Wang A, Ding W, Yochum G, Koltun W, Nighot P, Ma T. Long-Term Use of Proton Pump Inhibitors Disrupts Intestinal Tight Junction Barrier and Exaggerates Experimental Colitis. J Crohns Colitis. 2023 Apr 19;17(4):565-579.
Fossmark R, Olaisen M. Changes in the Gastrointestinal Microbiota Induced by Proton Pump Inhibitors-A Review of Findings from Experimental Trials. Microorganisms. 2024 May 30;12(6):1110.
Belei O, Olariu L, Dobrescu A, Marcovici T, Marginean O. Is It Useful to Administer Probiotics Together With Proton Pump Inhibitors in Children With Gastroesophageal Reflux? J Neurogastroenterol Motil. 2018 Jan 30;24(1):51-57.
Lespessailles E, Toumi H. Proton pump inhibitors and bone health: An update narrative review. Int J Mol Sci. 2022;18:10733.
Willems RPJ, Schut MC, Kaiser AM, Groot TH, Abu-Hanna A, Twisk JWR, et al. Association of proton pump inhibitor use with risk of acquiring drug-resistant Enterobacterales. JAMA Netw Open. 2023;6: e230470.
Su T, Lai S, Lee A, He X, Chen S. Meta-analysis: proton pump inhibitors moderately increase the risk of small intestinal bacterial overgrowth. J Gastroenterol. 2018;53:27–36.
谷禾健康
人的脑部大约有860亿个神经元和数万亿个突触连接,由250-300亿的神经胶质细胞支持,消耗基础氧气中约20%的比例来维持ATP驱动的活动。
与其他器官相比,大脑及其神经组织由于其高代谢率和能量活动而产生大量的 ROS(活性氧)。与此同时,与其他组织相比,大脑的抗氧化防御系统较弱,因此容易出现氧化还原稳态紊乱。
氧化应激是指由于 ROS 水平高于抗氧化剂水平而导致细胞内氧化还原信号通路中断。这种不平衡状态会产生有害影响,是许多神经系统疾病的主要原因。当大脑缺氧时,每分钟有大约190万个神经元和1400万个突触会开始丧失。
人们最初认为神经退行性疾病 (NDD) 是由神经系统缺陷引起的,而忽略了肠道和大脑之间的通讯涉及神经、代谢、内分泌和免疫途径。
大量研究观察到,肠道微生物组在通过其自身代谢产物或产生次生代谢产物减轻氧化应激、炎症和能量代谢方面发挥了不可忽视的作用,而通过使用具有抗氧化和抗炎活性的益生菌来调节肠道微生物组种群已显示出有希望的神经恢复能力。据报道从德氏乳杆菌(Lactobacillus delbrueckii)亚种Lactobacillus delbrueckii ssp. bulgaricus B3和 Lactobacillus plantarum GD2中分离出的胞外多糖可保护 SH-SY5Y 细胞免受 Aβ(1–42) 诱导的细胞凋亡,这表明它们有望成为药物治疗阿尔茨海默病 (AD)的有前途的天然化学成分。
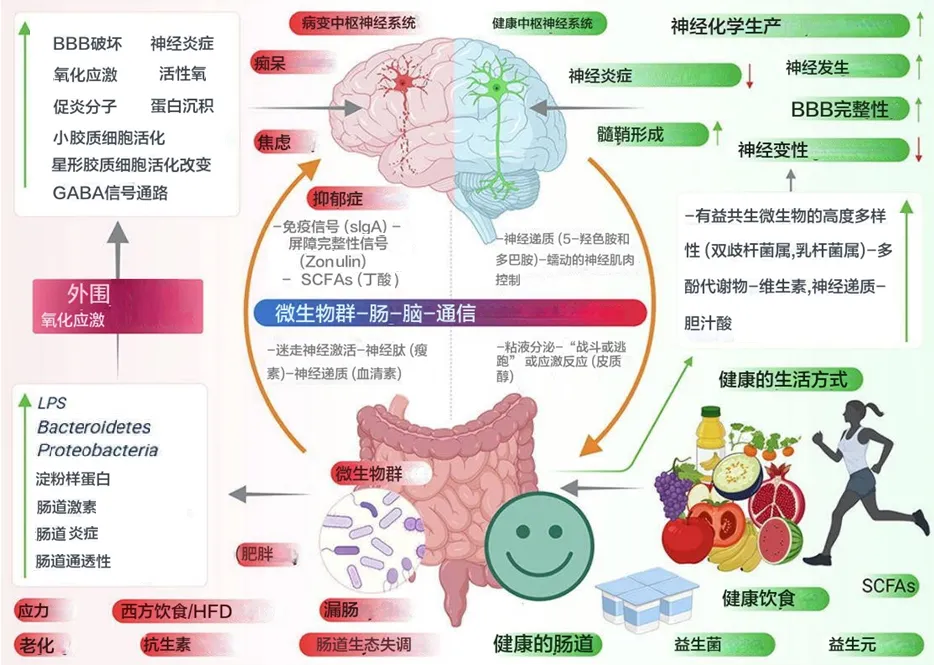
doi: 10.1016/j.jare.2021.09.005
肠道微生物分子,如神经递质、氨基酸、短链脂肪酸 (SCFA)、淀粉样蛋白、脂多糖 (LPS) 和微生物相关分子模式 (MAMP),通过循环与宿主免疫系统相互作用,影响宿主的代谢和神经系统,并通过肠道神经系统直接激活迷走神经影响大脑。压力等情况会导致下丘脑神经元分泌促肾上腺皮质激素(CRH),触发肾上腺皮质激素释放激素(ACTH) 的释放,随后激活皮质醇的释放,影响肠道屏障完整性,进而影响肠道健康。
当肠道菌群失调时,抗炎分子(如SCFAs、H2)的含量减少,而促炎分子(LPS、淀粉样蛋白)的含量增加,同时有益菌种数目减少,致病菌增加。这导致肠道和血脑屏障通透性增加,随之增加外周免疫反应,进而在中枢神经系统(CNS)中增加氧化应激。在神经元的细胞器(如线粒体、内质网(ER)和过氧化物体)中观察到反应性氧化物种(ROS)的产生增加,同时伴随神经毒素的聚集,导致神经退行性变化。
近期的研究提出了肠道微生物群与大脑之间的两个最重要的联系:
所以本文想和大家一起深入了解相关方面研究成果和进展,重点关注涉及神经退化的肠道微生物群介导的氧化应激,以及显示肠道微生物群及其代谢物参与神经保护的研究。
神经退行性疾病(NDD)是一类涉及神经元退化和功能损害的疾病,通常会导致神经细胞的死亡或损伤,进而引起神经系统功能障碍。随着时间的推移逐渐恶化,影响患者的日常生活和活动能力,目前的发病人群越来越大。
阿尔茨海默病:一种进行性痴呆疾病,主要表现为记忆丧失、认知功能下降和行为变化。
帕金森病:一种影响运动控制的疾病,其中运动功能受损,如震颤、肌肉僵硬和运动迟缓。
亨廷顿病(Huntington’s Disease):一种遗传性疾病,表现为神经元的逐渐死亡,导致运动障碍、认知功能下降和精神障碍。
路易体痴呆症(Amyotrophic Lateral Sclerosis, ALS):一种肌萎缩性疾病,导致运动神经元受损,最终导致肌肉无力和萎缩。
多发性硬化:这是一种影响中枢神经系统的慢性疾病,导致神经纤维的髓鞘损伤,影响神经信号的传递。
中风与脑损伤:中风是一种急性神经退行性疾病,由于脑部血流中断导致脑组织损伤。脑损伤也可以是慢性的,随着时间的推移导致神经细胞的损伤。
肠道的“第二大脑”:肠神经系统 (ENS)
肠道不仅仅是消化食物的地方,它还有一个聪明的“大脑”——肠神经系统 (ENS)。它在胃肠道 (GI) 和中枢神经系统 (CNS) 之间架起了一座沟通的桥梁。
肠道神经元通过迷走神经、内分泌和免疫途径与中枢神经系统进行对话,确保我们的肠道健康有序。
肠道微生物群:调节通讯的关键
上面说的“肠神经系统”并不是独自工作的,它有一个重要的合作伙伴——肠道微生物群。这些微生物不仅数量庞大,其代谢能力甚至可以与我们的肝脏相媲美。它们是调节肠道与大脑之间通讯的幕后英雄。
肠道微生物群的多重角色
该领域的持续研究表明,肠道微生物群不仅参与免疫系统的建设和代谢调节,还在我们身体各种器官的发育中扮演着重要角色。
饮食、药物等各种环境因素以及年龄和遗传等宿主因素,不仅会改变肠道菌群的组成,还会造成它们的信号活性发生变化,进而影响我们的健康。
IgA:肠道菌群的守护者
免疫球蛋白A (IgA) 是粘膜表面分泌的最丰富的抗体,她不仅能够包裹病原体以防止其侵入,还能包裹肠道中的共生菌,从而共同抵抗病原菌,维持着肠道菌群的稳定。
连蛋白:肠道屏障的调控者
肠道菌群还会影响连蛋白(zonulin),而连蛋白是调节肠道和血管内皮 (血脑屏障) 紧密连接所必需的。肠道菌群的变化直接影响连蛋白通路,我们常听说的“肠漏”就与此相关。
肠道菌群与情绪
肠道菌群还能影响我们的肠道蠕动,甚至与我们的内分泌系统有关。例如,压力和抑郁可能与皮质醇水平的升高有关,而这种激素的变化又与肠道菌群的平衡有关。
简而言之,肠道菌群通过感知和调节大量化学信号,直接影响我们的身心健康。
肠道细菌与神经系统的联系:从被忽视到被认可
肠道细菌与神经系统疾病之间的关系首次在20世纪提出,现在已被众多研究人员认可。
最初,人们认为神经退行性疾病(NDDs)仅由神经系统内部的缺陷引起,但现代研究揭示了一个事实:肠道微生物能产生和改变各种免疫、代谢和神经化学因子,它们实际上在调控我们的大脑健康方面发挥着关键作用。
肠道菌群:参与神经退行性疾病的双重角色
——失调带来的破坏
神经退行性疾病主要由氧化损伤、活性氧 (ROS) 生成增加、神经炎症和能量代谢紊乱引起,这些病理过程不仅侵袭大脑,也影响肠道微生物群的平衡。
肠道微生物组成会随着身体新陈代谢从健康状态到患病状态的变化而改变。这表明肠道菌群与宿主和环境之间存在交集,且与各种神经和心理疾病存在关联。
肠道菌群失调和神经炎症是各种神经系统疾病病理生理学中一致的因素。在本文中,我们重点展示了肠道微生物群介导的氧化应激在神经退行性疾病中的作用,包括 ROS 产生机制的解释、大脑更容易受到氧化应激的原因以及肠道微生物代谢物如何影响氧化应激引起的大脑损伤,重点关注阿尔茨海默病、帕金森病、创伤性脑损伤。
——保护作用
虽然说肠道菌群在神经退行性疾病的病理学中发挥着作用,但肠道菌群也具有保护大脑免受损伤的潜力,一些细菌能够释放代谢物,这些代谢物转化自膳食纤维、多酚或宿主分子,如胆汁酸、类固醇激素等。此外,通过益生元调节肠道菌群的组成,可以增强神经恢复力。
益生菌的神经保护作用:实验室到临床的探索
肠道菌群的神经保护作用在最近的研究中已得到充分证实,从发酵食品中分离的 Lactobacillus buchneri KU200793菌株表现出较高的抗氧化活性,并且能够保护 SH-SY5Y 细胞免受有害物质 1-甲基-4-苯基吡啶 (MPP + ) 的侵害,表明其具有益生菌和神经保护作用。
同样,从德氏乳杆菌亚种Lactobacillus delbrueckii ssp. bulgaricusB3和 Lactobacillus plantarum GD2 中分离出的胞外多糖,展现出保护神经细胞免受阿尔茨海默相关毒性的能力。
热灭活的Rumnicoccus albus菌株在细胞和小鼠模型中显示出降低ROS水平和提高抗氧化酶(SOD和GSH)水平的能力。此外,植物乳杆菌MTCC1325在动物模型中显示出抗阿尔茨海默病的潜力,预防记忆缺陷。
总之,这些研究反映了肠道微生物群的良好作用、它们的抗氧化作用以及随后的神经保护作用。
氧化应激的微妙平衡
氧化应激是一种生物学现象,当细胞内的活性氧(ROS)水平超过抗氧化剂的防御能力时,就会发生。这种失衡可能会导致细胞内的氧化还原信号通路中断,进而引发一系列有害的生物效应,成为众多神经系统疾病的潜在推手。
ROS是怎么产生的?
有氧代谢中涉及的每个化学反应都会形成不稳定且短暂的反应性中间产物,即 ROS。
注:生物分子氧 (O2) 具有两个不成对电子,不能被完全还原,因此其不完全还原会形成高度亲电且短寿命的 ROS,如:H2O2、超氧化物阴离子、一氧化氮、过氧亚硝酸根阴离子、羟基、过氧化物自由基等。
ROS要么在正常细胞过程中通过ROS生成酶作为中间体产生,要么在药物、毒素和辐射等外源性因素存在下产生。
神经组织更容易产生ROS
与其他器官相比,神经组织因其高代谢率而更容易产生ROS。
线粒体,作为细胞的能量工厂,同时也是ROS生成的主要场所。在ATP生成的过程中,线粒体可能会产生超氧化物,这些超氧化物随后被转化为H2O2和O2。O2的量越多,超氧化物的形成越多,这进一步导致更多的 ROS,如 H2O2和羟基自由基等。
线粒体与神经元活动的紧密联系
线粒体 ROS 的产生与神经元活动密切相关。强烈的突触传递不仅能促进超氧化物的产生,而且这种产生还受到细胞内钙(Ca2+)水平的调节,线粒体 ROS 产生的增加也与线粒体膜电位的增加有关。
单胺氧化酶(MAO)与神经健康
线粒体外膜上有一种酶,叫单胺氧化酶(MAO),它在神经细胞的发育和功能中起着关键作用。MAO通过催化单胺的氧化脱氨,产生H2O2作为副产物,这在神经退行性疾病中可能起到重要作用。
注:MAO-A 主要存在于儿茶酚胺能神经元中,参与去甲肾上腺素和血清素的氧化,而 MAO-B 则特别表达于血清素能神经元和神经胶质细胞中,并氧化 β-苯乙胺。
一氧化氮合酶(NOS):神经信号与氧化应激的交汇点
一氧化氮合酶(NOS)是神经元中产生ROS的酶,也是脑内ROS的来源之一。它通过催化L-精氨酸的氧化,产生一氧化氮(NO),NO在调节突触传递和干扰氧化还原稳态中起着双重作用。
NADPH氧化酶(NOX):神经元活动的响应者
NADPH氧化酶(NOX)是另一种在神经组织中产生ROS的酶,也是脑内ROS的来源之一,在脑皮质和海马区域丰富。它在神经元活动中的作用表明,NOX可能参与了多种中枢神经系统疾病的进展,包括AD、PD、肌萎缩侧索硬化症 (ALS) 和亨廷顿氏病 (HD)等。
注:Ca2+是 NOX 的主要激活剂,导致酶复合物在神经元中定位于突触后,从而表明NOX 参与神经元活动。已报道 7 种 NOX 的同源物,即 NOX (1-5)、双氧化酶 DUOX (1 和 2),它们的大小和结构域不同,但主要参与 ROS 的生成。
先前的报道还揭示了线粒体 ROS 和 NOX-ROS 之间的协同关系,从而支持彼此的 ROS 生成。
因此,开发异构体选择性 NOX 抑制剂可能是治疗急性和慢性中枢神经系统疾病的一种有前途的治疗方法。
其他产ROS的酶
在细胞质中,非血红素铁酶(如脂氧合酶)在分子O2存在下催化花生四烯酸的过氧化,并产生超氧化物和羟基自由基。
细胞质中的许多其他酶,如黄嘌呤氧化酶、细胞色素 P450 单加氧酶、环氧酶、D-氨基氧化酶也是重要的ROS 产生者。
氧化应激通过介导脂质过氧化、蛋白质氧化和核酸损伤三种主要反应导致细胞损伤。
事实上,氧化应激是衰老过程中正常生理过程的一部分,但已知它与大脑慢性疾病有关,如阿尔茨海默病 (AD)、帕金森病 (PD)、HD、缺血性中风、抑郁症和硬化症。此外,它在生活方式相关的代谢紊乱中也起着重要作用,如 2 型糖尿病 (T2D)、非酒精性脂肪肝、非酒精性脂肪性肝炎、肥胖、心血管疾病和癌症。
Tips:生理状态下正常浓度ROS在脑中的作用
虽然高浓度的ROS是有害的,会导致生物分子损伤,从而引起多种细胞功能障碍,但在安全稳定的水平上,ROS其实是细胞生理功能中不可或缺的助手。
详情展开如下:
免疫反应的哨兵
在正常生理条件下,细胞外的ROS是先天免疫系统的得力助手,它们能够引发免疫反应,帮助身体抵御外来的感染。
细胞信号的激活者
细胞内产生的ROS也是信号通路的激活者,它们参与刺激细胞凋亡和增强细胞对氧化应激的防御能力。
NF-κB的触发器
ROS在激活核转录因子NF-κB方面扮演着重要角色,这一过程会引发炎症反应,进而可能引发氧化应激。
病原体的克星
次氯酸(HOCl)这样的自由基,由髓过氧化物酶在溶酶体中作用产生,是一种对抗病原体的强大氧化剂。
中枢神经系统的调控者
对于中枢神经系统,生理条件下许多反应产生的副产物ROS,不仅是细胞内信号转导途径的调节者,还参与调控细胞的增殖、分化和成熟。
促进神经发生
研究表明,ROS的产生和氧化还原平衡有助于前体神经元祖细胞向神经元的分化,以及轴突的形成,促进神经元在其微环境中的扩增。
影响信号级联
氧化还原信号,包括ROS,能够调节多种转录因子和信号分子的功能,影响神经发生的信号级联。
神经元兴奋性的调节者
ROS,如H2O2,能通过增强细胞内钙(Ca2+)信号来调节皮质神经元的兴奋性。
影响神经系统信号级联
氧化还原信号(ROS和氧化状态)还调节转录因子(如NF-κB)、活化T细胞的核因子和活化蛋白1(AP-1)以及酪氨酸磷酸化蛋白PKC的氧化还原状态的功能。
研究人员观察到 H2O2可增加皮质神经元和 PC12 细胞中 ERK 和 cAMP 反应元件结合蛋白 (CREB) 的磷酸化。
参与学习和记忆
ROS 在大脑的不同部位(如海马、大脑皮层、下丘脑、杏仁核和脊髓)充当第二信使,海马中的长期增强(LTP)被证实是必不可少的,而海马与哺乳动物的学习和记忆有关,因此表明 ROS 参与了突触增强。
参与疼痛调节
ROS还通过参与增加杏仁核中央核的兴奋性来影响疼痛相关行为,杏仁核是大脑中负责疼痛调节情绪方面的区域。同样,在脊髓中,与神经性疼痛和炎症疼痛相关的神经可塑性过程也受 ROS 作为信号分子的控制。
ROS在细胞生理中的作用是复杂而精细的。它们既是细胞健康的守护者,也是潜在的挑战者。了解ROS如何在不同生理条件下发挥作用,对于我们理解健康和疾病的机制至关重要。
(为何大脑更容易积累过量ROS,清除机制vs产生机制)
我们将从以下方面来阐述大脑易受氧化应激影响的因素:
Ca2+在神经细胞中的作用:在神经细胞中,动作电位引起的Ca2+瞬变对于维持神经元之间的连接强度(即双向突触可塑性)至关重要。
Ca2+稳态失调的影响:当细胞膜上的Ca2+通道功能受损,导致Ca2+流入过多时,细胞内的游离Ca2+浓度会异常增加。
激活有害酶类:这种高浓度的Ca2+会激活一些有害的酶,如神经一氧化氮合酶(NOS)、磷脂酶A2和钙蛋白,这些酶的活性增加可能导致细胞骨架的破坏。
NO的产生及其影响:NOS的激活会产生一氧化氮(NO),NO是一种信号分子,但高浓度时会对细胞产生负面影响。NO会与线粒体内的细胞色素C氧化酶结合,抑制线粒体的呼吸作用,影响能量产生。
形成有害物质:NO还可以与超氧阴离子(O2.– )反应生成过氧亚硝酸盐(ONOO–),这是一种强氧化剂,可以对细胞造成进一步的损害。
线粒体功能障碍:线粒体内的Ca2+超载会导致线粒体通透性转换孔(MPTP)开放,使得Ca2+和H2O2流出,这可能导致细胞坏死。
大脑易受氧化应激:由于上述过程,Ca2+的稳态失调会使大脑细胞更容易受到氧化应激的影响,氧化应激是指细胞内氧化剂和抗氧化剂之间的不平衡,可能导致细胞损伤。
谷氨酸的兴奋性作用:谷氨酸是一种主要的兴奋性神经递质,它在神经元之间的信号传递中起着关键作用。然而,当谷氨酸在细胞外环境中大量积累时,它可能导致神经细胞的损伤甚至死亡。
谷氨酸的毒性作用:谷氨酸的过量积累可以激活神经元上的谷氨酸受体,尤其是AMPA和NMDA受体。这些受体的激活会导致细胞内Ca2+和Na+的持续流入,引发所谓的“兴奋性毒性”。
注:AMPA和NMDA是两种类型的谷氨酸受体,它们在神经元之间的信号传递中起着关键作用。这些受体是离子通道受体,意味着它们不仅能够结合神经递质谷氨酸,还能够控制特定离子的流动,从而影响神经细胞的电活动。
AMPA受体:AMPA受体(α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体)是快速兴奋性突触后电流的主要介质。当谷氨酸结合到AMPA受体时,受体通道打开,允许Na+和K+离子通过,导致突触后膜的去极化,这是神经信号传递的一个重要步骤。AMPA受体的激活通常与快速的、短暂的信号传递相关。
NMDA受体:NMDA受体(N-甲基-D-天冬氨酸受体)是另一种类型的谷氨酸受体,它们在学习和记忆中起着重要作用。NMDA受体通道的开放需要谷氨酸的结合以及突触后膜的去极化(通常由AMPA受体的激活引起)。NMDA受体通道开放时,允许Ca2+和Na+离子进入细胞,同时也允许K+离子流出。Ca2+的流入可以激活多种细胞内信号途径,包括那些涉及长期增强(LTP)和长期抑制(LTD)的途径,这些都是学习和记忆的关键机制。
这两种受体在神经传递中的协同作用对于正常的脑功能至关重要,包括感知、运动控制、学习和记忆。然而,当这些受体过度激活时,它们也可能参与神经退行性疾病和神经损伤的过程,如兴奋性毒性。
反应性物质的形成:细胞内Ca2+的增加可以激活NOS,产生一氧化氮(NO),NO与超氧阴离子(O2–)反应生成过氧亚硝酸盐(ONOO–)。ONOO–是一种强氧化剂,可以对细胞造成损害。
谷氨酸合成酶的抑制:ONOO–等反应性物质可以使谷氨酸合成酶失活,这种酶负责将谷氨酸转化为谷氨酰胺,从而抑制了谷氨酸的代谢。
谷氨酸与半胱氨酸的交换受阻:谷氨酸还可以通过Xc-载体抑制细胞内谷氨酸与半胱氨酸的交换。半胱氨酸是合成谷胱甘肽(GSH)的关键成分。
主编解读:在细胞内,氨基酸不仅是蛋白质的构建块,还参与多种生物化学反应。谷氨酸(Glutamate)和半胱氨酸(Cysteine)是两种重要的氨基酸,它们在细胞内的水平受到严格调控。
谷氨酸与半胱氨酸的交换通常通过特定的转运蛋白(载体)进行,这些转运蛋白位于细胞膜上,负责将氨基酸从细胞外环境转运到细胞内。这种交换是双向的,意味着谷氨酸可以从细胞内转运到细胞外,同时半胱氨酸可以从细胞外转运到细胞内。
半胱氨酸是合成谷胱甘肽(Glutathione, GSH)的关键前体。谷胱甘肽是一种重要的抗氧化剂,对于保护细胞免受氧化应激的损害至关重要。因此,半胱氨酸的供应对于维持细胞内GSH的水平和细胞的抗氧化能力非常重要。
当谷氨酸与半胱氨酸的交换受阻时,意味着这种双向转运过程受到了干扰。这可能是由于转运蛋白的功能障碍、细胞外谷氨酸水平的异常升高(如在兴奋性毒性情况下),或者其他因素导致的细胞膜通透性的改变。
这种交换受阻可能导致细胞内半胱氨酸的供应不足,进而影响GSH的合成。GSH的减少会使细胞更容易受到氧化应激的影响,可能导致细胞损伤或死亡。在某些情况下,这种交换受阻还可能与特定的疾病状态或病理过程相关。
谷胱甘肽的耗竭与铁死亡:由于谷氨酸与半胱氨酸的交换受阻,导致细胞内GSH的耗竭。GSH的减少会使细胞更容易受到氧化应激的影响,并可能导致一种称为“铁死亡”的细胞死亡形式,这是一种由铁依赖的脂质过氧化驱动的细胞死亡。
总结来说,谷氨酸的过量积累通过激活神经元上的受体,导致细胞内Ca2+和Na+的持续流入,进而引发一系列的生物化学反应,包括反应性物质的形成、谷氨酸代谢的抑制、GSH的耗竭,最终可能导致神经元的损伤和死亡。这些过程表明,谷氨酸的稳态对于维持神经细胞的健康至关重要。
扩展阅读:
过渡金属离子,如铁(Fe2+)和铜(Cu+),在大脑中扮演着多重角色。它们不仅是许多酶的辅因子,参与能量代谢、抗氧化防御和神经递质的合成等多种生物化学过程,而且还与神经退行性疾病和脑损伤有关。
酶的辅因子:Fe2+和Cu+等过渡金属离子是许多酶的必要组成部分,这些酶参与细胞内的各种代谢过程。例如,铁是细胞色素c氧化酶和铁硫蛋白的组成部分,而铜是细胞色素c氧化酶和超氧化物歧化酶的组成部分。
自由基反应的催化剂:在某些情况下,过渡金属离子可以催化自由基反应,这些反应可以产生有害的氧化剂,如羟自由基(·OH)。自由基是不稳定的分子,它们可以损害细胞结构,包括脂质、蛋白质和DNA。
铁的持久存在:大脑中的铁主要以Fe2+的形式存在,它在脑脊液中的结合能力较低,这意味着铁离子可以在脑脊液中相对自由地移动,并且可能积累在某些区域。铁的积累与氧化应激和神经退行性疾病(如阿尔茨海默病和帕金森病)有关。
脑损伤的应激因素:在脑损伤或疾病状态下,细胞可能会释放更多的过渡金属离子。这些离子在细胞外环境中可能催化自由基的产生,导致进一步的细胞损伤。
总结来说,过渡金属离子在大脑中的正常功能对于维持神经细胞的健康至关重要。然而,当这些离子在细胞外环境中过度积累或以不适当的形式存在时,它们可能成为氧化应激和细胞损伤的催化剂。因此,维持大脑中过渡金属离子的稳态对于预防神经退行性疾病和脑损伤具有重要意义。
神经递质的自动氧化:多巴胺、5-羟色胺和去甲肾上腺素等神经递质在有氧条件下可以发生自动氧化反应。这些反应涉及神经递质与氧气之间的化学反应,产生活性氧(ROS),如超氧阴离子(O2–)和羟自由基(·OH)。
多巴胺的自动氧化过程:多巴胺与氧气反应首先生成半醌(一种不稳定的中间产物)和超氧阴离子(O2–)。半醌可以进一步与氧气反应生成醌。醌是一种氧化产物,它可以被氧气重新氧化为醌醇和过氧化氢(H2O2)。
线粒体和溶酶体功能障碍:ROS的积累可以导致线粒体和溶酶体的功能障碍。线粒体是细胞的能量工厂,而溶酶体是细胞的“消化系统”,负责分解和回收细胞内的废物。这些细胞器的功能障碍可能导致细胞能量代谢的紊乱和细胞内废物的积累。
总结来说,神经递质的自动氧化是一个产生ROS的过程,这些ROS可以对细胞造成损害,特别是通过损害线粒体和溶酶体的功能。这种损害可能与多种神经退行性疾病有关,包括帕金森病和阿尔茨海默病。因此,控制神经递质的自动氧化和ROS的产生对于维持神经细胞的健康至关重要。
大脑对葡萄糖的依赖:大脑是身体中对葡萄糖依赖性最高的器官之一。葡萄糖不仅是大脑的主要能量来源,还参与多种代谢途径和信号传导过程。
糖醇磷酸途径:糖醇磷酸途径(Glycolysis)是葡萄糖分解代谢的第一步,产生能量和中间代谢产物。磷酸果糖激酶(Phosphofructokinase, PFK)是糖醇磷酸途径中的一个关键酶,它控制着糖酵解的速率。
神经元降解磷酸果糖激酶:在某些情况下,神经元可能会降解磷酸果糖激酶,这可能导致糖酵解速率的降低。糖酵解速率的降低意味着葡萄糖的利用效率下降。
蛋白质糖基化和AGE的形成:糖酵解速率的降低可能导致葡萄糖在细胞内的积累。过量的葡萄糖可以非酶促地与蛋白质发生反应,形成糖基化产物,最终生成高级糖基化终产物(Advanced Glycation End Products, AGE)。
AGE的损害作用:AGE可以与细胞内的AGE受体结合,引发炎症反应和氧化应激。这种氧化应激可以损害蛋白质和线粒体的功能。线粒体是细胞的能量工厂,其功能障碍可能导致细胞能量代谢的紊乱。
炎症性氧化应激:AGE诱导的炎症性氧化应激可能导致细胞内氧化剂和抗氧化剂之间的不平衡,进一步加剧细胞损伤。
Tips:以上总结就是葡萄糖诱导的氧化应激涉及糖酵解速率的降低、蛋白质糖基化和AGE的形成,以及由此引发的炎症性氧化应激。这些过程可能导致蛋白质和线粒体功能的损害,从而影响神经细胞的健康。因此,维持适当的糖酵解速率和控制AGE的形成对于预防神经退行性疾病和脑损伤具有重要意义。
大脑中的多不饱和脂肪酸:大脑富含多不饱和脂肪酸,特别是二十碳五烯酸(Docosahexaenoic Acid, DHA)。DHA是构成神经细胞膜的重要成分,对于维持神经细胞的结构和功能至关重要。
氧化应激的影响:多不饱和脂肪酸由于其化学结构中的多个双键,容易受到氧化应激的影响。氧化应激可以引发脂质过氧化反应,导致脂肪酸的氧化和损伤。
脂质过氧化和信号传导:脂质过氧化产物可以参与脑部的信号传导过程。然而,这些过氧化产物也可能对神经细胞产生负面影响,包括细胞膜的损伤和细胞功能的紊乱。
4-羟基壬酸醛的神经毒性:脂质过氧化产物之一,4-羟基壬酸醛(4-Hydroxynonenal, 4-HNE),可以通过提高细胞内Ca2+水平使谷氨酸转运体失活,从而具有神经毒性。谷氨酸转运体的失活可能导致谷氨酸的积累,引发兴奋性毒性。
α-酮戊二酸脱氢酶的失活:脂质过氧化物还可以使α-酮戊二酸脱氢酶(α-Ketoglutarate Dehydrogenase)失活,这是一种参与三羧酸循环(TCA循环)的酶。该酶的失活可能导致能量代谢的紊乱。
血管收缩作用和蛋白酶体的破坏:脂质过氧化物具有血管收缩作用,可能导致脑血流的减少。此外,它们还可以破坏蛋白酶体,这是一种负责蛋白质降解的细胞器,其功能障碍可能导致细胞内废物的积累。
脂质过氧化物在多种神经退行性疾病,如阿尔茨海默病中起着持续的作用。这些疾病的特点是神经细胞的进行性损伤和死亡。
大脑中多不饱和脂肪酸的易感性使得神经细胞更容易受到氧化应激的影响。脂质过氧化产物如4-HNE可以通过多种机制对神经细胞产生负面影响,包括神经毒性、能量代谢的紊乱、血管收缩和蛋白酶体的破坏。这些过程可能与神经退行性疾病的病理机制有关。
微胶质细胞的角色:微胶质细胞(Microglia)是大脑和脊髓中的常驻免疫细胞,它们在维持大脑的正常功能和发育中起着关键作用。微胶质细胞参与多种生理过程,包括神经保护、突触修剪、炎症反应和组织修复。
吞噬活动和反应性物质的产生:在正常的吞噬活动中,微胶质细胞可以清除死亡的细胞碎片和外来的病原体。在这个过程中,微胶质细胞产生超氧阴离子(O2–)和其他反应性物质,这些物质在清除病原体和受损细胞中起着重要作用。
NOX-2酶的作用:NOX-2(NADPH氧化酶2)是一种酶,它在微胶质细胞中产生O2–。NOX-2的激活可以增加O2–的产生,这是微胶质细胞应对损伤和感染的一种防御机制。
氧气生物可用性与微胶质细胞活性:微胶质细胞的活性取决于总的氧气生物可用性。在损伤或疾病状态下,微胶质细胞可能通过消耗更多的氧气来产生更多的O2–,这可能导致局部氧浓度的降低。
反应性物质与突触损伤:H2O2和NO等反应性物质在损伤部位吸引微胶质细胞,这些物质可以引发局部炎症反应。炎症反应可能导致突触的损伤,这是神经退行性疾病中的一个关键过程。
神经退行性的推动:微胶质细胞的过度激活和炎症反应可能推动神经退行性过程。在某些情况下,微胶质细胞的持续激活可能导致神经细胞的损伤和死亡,这是多种神经退行性疾病的共同特征。
一句话总结就是微胶质细胞在大脑中扮演着多重角色,包括免疫防御、吞噬活动和炎症反应。然而,微胶质细胞的过度激活和炎症反应可能导致神经细胞的损伤和神经退行性疾病的进展。因此,平衡微胶质细胞的活性对于维持大脑健康和预防神经退行性疾病至关重要。
大脑抗氧化防御系统的脆弱性:大脑是一个高代谢活跃的器官,对氧气的需求很高,因此容易受到氧化应激的影响。与其他组织相比,大脑的抗氧化防御系统相对较弱,这使得大脑更容易出现氧化还原稳态的紊乱。
过氧化氢酶的含量:过氧化氢酶(Catalase)是一种重要的抗氧化酶,它能够分解过氧化氢(H2O2)为水和氧气。神经元中的过氧化氢酶含量比肝细胞中的低得多,这可能是因为神经元对H2O2的处理能力较弱。
过氧化物酶体的限制:过氧化氢酶主要存在于过氧化物酶体中,这是一种细胞器,专门负责处理过氧化氢和其他有害物质。然而,过氧化物酶体的存在限制了过氧化氢酶的活性,使其无法作用于其他亚细胞区室(如线粒体和细胞质)产生的H2O2。
谷胱甘肽的含量:谷胱甘肽(Glutathione, GSH)是一种重要的细胞内抗氧化剂,它能够清除自由基并参与亲电子物质的代谢。神经元中的GSH含量非常低,这使得它们更容易受到氧化应激的影响,并且难以代谢亲电子物质。
铁死亡的风险:铁死亡是一种由铁依赖的脂质过氧化驱动的细胞死亡形式。神经元中GSH的低含量可能增加它们发生铁死亡的风险,这是一种与多种神经退行性疾病相关的细胞死亡机制。
总结来说,大脑的抗氧化防御系统相对脆弱,这使得神经元更容易受到氧化应激的影响。过氧化氢酶和谷胱甘肽的低含量限制了神经元处理氧化应激的能力,可能导致氧化还原稳态的紊乱和细胞损伤。
血红蛋白与H2O2的反应:血红蛋白(Hemoglobin)是红细胞中的一种蛋白质,负责携带氧气。当血红蛋白与过量的过氧化氢(H2O2)反应时,它可以释放出铁离子(Fe2+)和血红素(Heme)。这些产物可以参与进一步的化学反应,产生有害的过氧化物。
血红蛋白的氧化:在氧化应激条件下,血红蛋白可以被过氧化氢(H2O2)或其他氧化剂氧化。这种氧化过程可以破坏血红蛋白的结构,导致其携氧能力下降。
铁离子的释放:血红蛋白的氧化可以导致铁离子(Fe2+)从血红蛋白中释放出来。这些游离的铁离子可以参与Fenton反应,即铁离子与H2O2反应生成羟自由基(·OH)。羟自由基是一种强氧化剂,可以对细胞造成广泛损害。
血红素的释放:血红蛋白的氧化还可以导致血红素的释放。血红素是一种铁卟啉化合物,它是血红蛋白的活性部分。血红素可以催化脂质过氧化反应,导致细胞膜的损伤。
脂质过氧化:血红素可以催化不饱和脂肪酸的过氧化反应,生成脂质过氧化物。这些过氧化物可以进一步分解产生更多的自由基,加剧氧化应激。
血管收缩:血红素还可以与一氧化氮(NO)结合,形成复合物。这种复合物可能导致血管收缩,影响脑血流和氧气供应。
因此,控制血红蛋白与H2O2的反应对于预防神经毒性和维持大脑健康具有重要意义。
CYP2E1(Cytochrome P450 2E1)是一种属于细胞色素 P450 酶家族的酶。细胞色素 P450 酶是一组含有血红素(铁卟啉)的酶,它们在许多生物体内参与多种化合物的氧化代谢。CYP2E1 主要存在于肝脏中,但它也在其他组织中表达,包括大脑。
CYP2E1 的主要功能是催化小分子化合物的氧化反应,包括乙醇、某些药物、致癌物和其他外源性物质。这些反应通常涉及将分子中的氢原子移除,并添加一个氧原子,从而使化合物更容易被进一步代谢或排出体外。
CYP2E1 酶的角色:CYP2E1(Cytochrome P450 2E1)是一种存在于肝脏和其他组织中的酶,包括大脑。它参与多种代谢过程,包括小分子(如乙醇、某些药物和致癌物)的氧化代谢。
电子泄漏与氧化应激:CYP2E1 在催化反应时可能会发生电子泄漏,这意味着在代谢过程中,它可能会产生超氧阴离子(O2–)和其他活性氧(ROS)。这些 ROS 是高度反应性的分子,可以导致氧化应激,即细胞内氧化剂和抗氧化剂之间的不平衡。
大脑中的 CYP2E1:虽然大脑中含有低水平的细胞色素 P450 酶,但 CYP2E1 的存在使得大脑在代谢过程中容易受到氧化应激的影响。大脑中的 CYP2E1 活性可能导致神经细胞的损伤。
饮酒和吸烟的影响:研究表明,CYP2E1 的水平可能会因饮酒和吸烟而增加。这是因为乙醇和其他烟草中的化合物可以诱导 CYP2E1 的表达和活性。CYP2E1 的增加可能导致更多的 ROS 产生,从而加剧氧化应激。
总结来说,DNA 修复酶如 PARP-1 在修复 DNA 损伤中起着关键作用,但它们的过度激活可能导致 NAD+ 的消耗和能量产生的限制,以及可能通过 TRPM2 通道导致细胞内 Ca2+ 浓度的增加,从而导致神经元细胞死亡。
DNA 修复酶的角色:DNA 修复酶是一类酶,它们在细胞内负责修复 DNA 分子的损伤。这些损伤可能由多种因素引起,包括氧化应激、紫外线辐射、化学物质和复制错误。
PARP-1 的功能:聚 ADP 核糖聚合酶 (PARP-1) 是一种重要的 DNA 修复酶。当 DNA 发生单链断裂时,PARP-1 会被激活。它通过裂解烟酰胺腺嘌呤二核苷酸 (NAD+) 并将 ADP 核糖部分转移到核蛋白上,从而启动 DNA 修复过程。
PARP-1 的过度激活:在严重的 DNA 损伤或氧化应激条件下,PARP-1 可能会过度激活。这种过度激活会导致大量的 NAD+ 消耗,因为 NAD+ 是 PARP-1 活性所必需的底物。
因此,平衡 PARP-1 的活性对于维持细胞健康和预防神经退行性疾病具有重要意义。
RNA 的易感性:RNA 是单链分子,不像 DNA 那样有双螺旋结构和组蛋白的保护。因此,RNA 更容易受到氧化应激的影响。氧化应激可以导致 RNA 分子中的核糖和碱基发生氧化损伤。
氧化的 RNA 的影响:氧化的 RNA 可能会阻止核糖体合成蛋白质。核糖体是细胞内的蛋白质合成工厂,它们通过读取 RNA 分子上的遗传信息来合成蛋白质。如果 RNA 被氧化,其结构可能会改变,导致核糖体无法正确读取遗传信息,从而影响蛋白质的合成。
蛋白质合成异常:如果 RNA 的氧化损伤未得到修复,可能会导致合成的蛋白质未折叠或截短。未折叠或截短的蛋白质可能无法正常执行其功能,甚至可能形成有害的蛋白质聚集体,这在多种神经退行性疾病中是一个关键的病理特征。
氧化的 RNA 与氧化还原活性过渡金属(如铁和铜)一起可以催化 Fenton 反应。Fenton 反应是一种产生羟自由基(·OH)的化学反应,羟自由基是一种强氧化剂,可以对细胞造成广泛损害。
此外,超氧化物歧化酶(SOD)是一种重要的抗氧化酶,能够将超如果氧阴离子(O2–)转化为过氧化氢(H2O2)。铜锌超氧化物歧化酶(CuZn-SOD)的 mRNA 被氧化,可能是肌萎缩侧索硬化症(ALS)的临床前征兆。ALS 是一种神经退行性疾病,其特点是运动神经元的进行性退化和死亡。
注意,尽管神经退行性疾病中氧化的 RNA 在神经退行性疾病中的作用已经引起了关注,但其确切的作用机制和重要性仍需要进一步的研究。
胃肠道 (GI) 包含数万亿共生微生物和约 200-2000左右个物种,它们在维持膜屏障功能方面发挥着重要作用。这些微生物是肠道内的永久居民,参与宿主生物体内分子的不断流动,从而调节各种代谢功能。
人在出生后两年内,宿主胃肠道中的微生物群就会稳定下来,但它们的组成因个体而异,并且会根据年龄、健康、遗传和生活方式等外界因素而改变。胃肠道的腔侧暴露于饮食成分和肠道微生物群,此外,70% 的免疫细胞位于肠道组织,并由连接肠道和大脑的神经元支配,涉及肠道和大脑之间的持续沟通。
肠道微生物群与大脑之间的沟通主要涉及四条途径;
1、神经高速公路——迷走神经
第一种重要模式包括激活迷走神经,迷走神经连接胃肠道的肌肉层和粘膜层与脑干。最近研究显示,肠道病原体和益生菌通过激活迷走神经元改变大脑中的 γ-氨基丁酸 (GABA)、色氨酸、催产素和脑源性神经营养因子 (BDNF) 信号,从而调节宿主的焦虑、进食和抑郁等行为。
2、肠嗜铬细胞释放的血清素影响大脑
直接或间接影响大脑活动的通讯途径涉及通过肠道内壁的肠嗜铬细胞 (EC) 释放的血清素发出信号。一项研究表明,在用益生菌双歧杆菌治疗抑郁症小鼠模型时,其血清素和血清素前体水平会增加,从而改善其抑郁状态。同样,据报道,产芽孢细菌(梭菌属)的代谢物能够刺激 EC 产生血清素。
3、肠道菌群通过小胶质细胞介导对神经系统的调节
肠道菌群在小胶质细胞的发育、成熟和激活中起着至关重要的作用。在一项研究中,据报道,无菌 (GF) 小鼠携带的未成熟小胶质细胞数量比传统小鼠多,而且用双歧杆菌治疗时,可通过转录激活小胶质细胞。在行为和神经退行性疾病 (NDD) 中观察到小胶质细胞功能的变化,表明肠道微生物群通过小胶质细胞介导对 NDD 的影响。肠道微生物群还通过全身免疫系统(即细胞因子和趋化因子)影响神经系统。研究表明,与传统小鼠相比,GF 小鼠的血脑屏障 (BBB) 通透性更大,因此大脑更容易接触微生物产物,进而导致神经病理学状况。
4、肠道菌群传递化学信号
肠道微生物群通过直接向大脑传递化学信号进行交流。例如,肠道细菌发酵膳食纤维会产生短链脂肪酸 (SCFA),已被证明可以调节中枢神经系统的神经可塑性,还可以改善小鼠的抑郁行为。此外,肠道微生物群如拟杆菌、双歧杆菌、副拟杆菌、大肠杆菌属。能够产生神经递质 GABA,这表明肠道微生物群调节宿主生物体神经递质的浓度。
人体肠道内定植着四种主要的共生菌门,包括厚壁菌门、拟杆菌门、放线菌门、变形菌门。肠道中的共生菌和致病菌都能通过调节线粒体活性来改变细胞的 ROS。
共生菌产生甲酰化肽,这些肽与巨噬细胞和中性粒细胞上的 G 蛋白偶联受体 (GPCR) 结合,从而引发上皮细胞炎症。该过程导致 NOX-1 产生超氧化物,从而增加细胞的 ROS。
肠道乳酸杆菌和双歧杆菌具有将硝酸盐和亚硝酸盐转化为 NO 的能力,使肠道上皮成为 NO 的丰富来源。类似地,链球菌和杆菌利用 NOS 从 L-精氨酸产生 NO。
低浓度NO→保护;高浓度NO→有害
纳摩尔浓度的 NO 具有神经保护作用,是去甲肾上腺素能、非胆碱能肠道神经元的神经递质。而在较高浓度下,它会导致产生活性氧和氮物质 (RONS)(如超氧化物和 H2O2)而引起的有害影响,这进一步形成高活性羟基自由基,使其与神经炎症、轴突变性和神经退行性疾病相关。
肠道细菌产生的有益代谢物(如 SCFA)有助于通过影响线粒体活动来降低 ROS。这个后面章节详细讨论。
MAMPs维持微生物结构功能,影响大脑发育与炎症反应
膜相关分子模式 (MAMP) 维持着所有微生物类别的结构完整性和基本功能,甚至大脑也能检测到。这些是不同的化学基团,包括肽、核苷酸、碳水化合物和脂质。当宿主无法检测到此类分子模式时,它可能伴有急性到慢性炎症,并被发现会改变大脑的发育和功能。这些高度保守的结构基序与先天免疫系统细胞上的模式识别受体 (PRR) 结合,从而诱导线粒体 ROS 的产生和 NF-κB 通路的激活,导致炎症反应,引起神经元应激和细胞死亡。
在最近的一项研究中,据报道,细菌细胞壁成分肽聚糖易位到发育中的大脑,影响基因表达并导致社会行为的改变。同样,革兰氏阴性细菌细胞壁中的脂多糖被发现会损害小鼠的胎儿大脑发育、急性抑郁和认知障碍。此外,在 PD、自闭症谱系障碍 (ASD) 和突触核蛋白病模型中,急性和慢性暴露于 MAMP 是导致疾病症状的一个因素。
机会性病原体产生的细菌毒素,也对神经系统产生负面影响
发现梭菌属(Clostridium )产生的致命毒素,如毒素 B、肠毒素和ε 毒素,可降低神经元活力,并通过破坏的 BBB到达大脑,抑制神经递质的释放。
葡萄球菌属(Staphylococcus)和芽孢杆菌属(Bacillus)产生的肠毒素和麦芽孢杆菌素可通过刺激迷走神经引起呕吐和疾病行为。
沙门氏菌和大肠杆菌等致病菌能够降解含硫氨基酸,从而在肠道中产生硫化氢 (H2S) 。H2S水平的升高会引起各种代谢活动的变化,如乳酸增加和ATP生成减少、环氧合酶2 (COX-2)活性抑制、线粒体对O2的消耗减少、促炎细胞因子表达增加,并且已知会刺激高血压和神经炎症。
肠道菌群介导的氧化应激在神经退行性病变中的作用
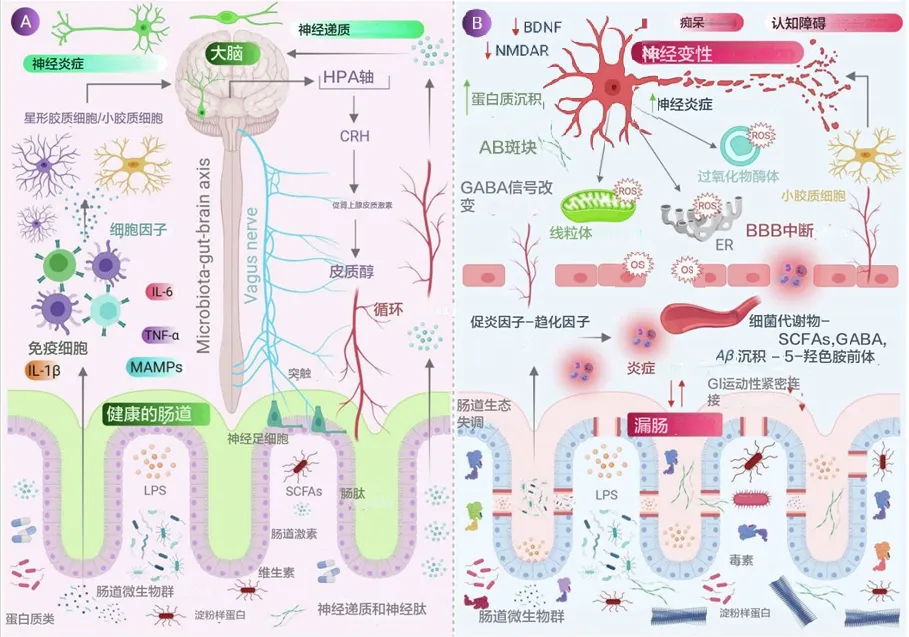
doi: 10.1016/j.jare.2021.09.005
神经退行性疾病(如阿尔茨海默病和帕金森病)的病因病理涉及神经细胞内蛋白质错误折叠及其聚集,氧化应激也被认为是其病理因素之一。发现帕金森患者的肠道中肌间神经丛(Auerbach丛)和粘膜下神经丛(Meissner丛)存在α-突触核蛋白的聚集,暗示了肠道菌群在启动肠道α-突触核蛋白聚集中的作用,然后通过跨突触途径向上影响中枢神经系统神经元导致神经退行性变化。
阿尔茨海默病:全球性的健康挑战
阿尔茨海默病 (AD) 是导致痴呆的主要原因,影响着全球超过 5000 万人口,其中老年人群的发病率更高,85 岁以上每 1000 人中约有 80 人患有该病。阿尔茨海默病的无症状病理学在记忆力减退和认知障碍等症状出现前约20年就开始了。
病理变化:蛋白质的异常积累
与阿尔茨海默相关的脑病理变化包括蛋白质淀粉样β蛋白 (Aβ-淀粉样斑块) 的细胞外积累和 tau 蛋白 (tau 缠结) 的细胞内积累。这种异常的蛋白质积累导致小胶质细胞被激活以清除 Aβ 和 tau 蛋白,但随着随后的衰老,会发生慢性炎症,引起神经元细胞死亡,从而导致萎缩。
氧化应激:阿尔茨海默的潜在危险因素
在可能的危险因素中,氧化应激和肠道微生物群的作用引起了科学界的关注,并被认为是神经退行性过程的直接可能后果。
许多研究表明,阿尔茨海默病患者大脑的抗氧化防御系统发生了变化,即超氧化物歧化酶和过氧化氢酶的活性和水平降低。同时,氧化应激生物标志物(如丙二醛、4-羟基壬烯醛和F2-异前列腺素、蛋白质羰基、3-硝基酪氨酸、8-羟基脱氧鸟苷)在血液和脑脊液中含量较高,这些物质的浓度与认知障碍和脑重量成正比。
阿尔茨海默症脑内ROS的产生还表现为细胞器功能障碍,如线粒体(细胞色素C氧化酶缺乏)、未折叠蛋白反应 (UPR) 引起的内质网、神经斑块中金属离子的积累以及小胶质细胞的过度活化和随后的NADPH氧化酶的过度表达。
Aβ沉积和氧化应激之间也存在相互关系
即Aβ聚集会诱导氧化应激(也存在于线粒体、内质网和高尔基体等细胞器中),而氧化应激会诱导Aβ积累。甚至,神经元中tau蛋白的聚集会导致NADH-泛醌还原酶活性降低,从而导致ROS生成增加和线粒体功能障碍。
肠道菌群失调→炎症→神经退行性病变
最近的事实和数据显示,阿尔茨海默不仅是局限性脑炎症的结果,也是外周炎症的结果。肠道菌群失调会导致炎症,而炎症会随着年龄的增长而增加,血脑屏障被破坏,免疫系统被激活,随后出现神经退行性病变,另一方面,健康均衡的肠道有助于减少 ROS 产生的有害影响。
共生菌减少,致病菌增加
已发现患有阿尔茨海默的个体体内共生菌如双歧杆菌和厚壁菌数量减少,大肠杆菌、志贺氏菌和拟杆菌数量增加,随后出现炎症和 Aβ 积累增加。
肠道菌群变化与阿尔茨海默病严重程度的关联
同样,在 APP/PS1 小鼠模型中,当用广谱抗生素联合治疗时,Aβ 斑块形成也减少。5xFAD 小鼠阿尔茨海默模型显示微生物群向促炎物种转变,同时氨基酸分解代谢也发生变化,相反,抗生素治疗可逆转这种影响,这表明疾病的严重程度和肠道菌群转化之间可能存在联系。
Toll样受体激活与肠道屏障紊乱
肠道中形成的微生物淀粉样蛋白激活 Toll 样受体 (TLR),分化簇 14 (CD14) 促进免疫反应,导致被忽视的错误折叠 Aβ 和 Aβ 清除受损,随后细胞因子产生增加,导致肠道和血脑屏障紊乱。此外,研究还表明,阿尔茨海默患者的肠道激素水平下降,相反,肠道微生物代谢物如 H2S 和三甲胺增加。
与年龄有关的肠道微生物多样性下降
研究表明,随着年龄的增长,双歧杆菌属减少,变形菌增加,痴呆症不是由于 SCFA 减少而是由于脂质代谢受到干扰。双歧杆菌在调节胆固醇水平方面发挥着重要作用,直接促进胆固醇通过粪便排出,间接通过增加血清瘦素水平,从而参与维持海马可塑性和记忆功能。
肠道菌群影响神经递质
肠道细菌如乳酸杆菌和双歧杆菌会代谢抑制性神经递质 GABA。一项研究发现,在 APPSwe/PSEN1DeltaE9 双基因 (人类早老素(DeltaE9)和人鼠淀粉样前蛋白(APPswe)融合体阿尔茨海默小鼠模型中,海马突触可塑性发生了改变,其中发现 GABA 生成减少,同时谷氨酸能神经传递增加。虽然有报道显示蓝藻门太高会产生导致认知障碍的神经毒素,但尚未观察到与阿尔茨海默的关系。
淀粉样蛋白的交叉播种机制
另一个可能的连接环节是脑淀粉样蛋白的积累和肠道微生物群,涉及微生物淀粉样蛋白的交叉播种机制,其方式类似于朊病毒的传播,因此形成的不同淀粉样蛋白构象体在其细胞靶标中诱导不同水平的毒性,推测存在阿尔茨海默表型。
口腔菌群与阿尔茨海默
除了肠道微生物群外,还研究了口腔共生菌与阿尔茨海默之间的联系。有趣的是,口腔卫生不良和牙齿脱落会增加阿尔茨海默早期发病的风险。
关于口腔菌群与神经系统疾病的关联,详见:
帕金森病 (PD) 的病理特征是多巴胺能神经元的进行性退化、磷酸化蛋白 α-突触核蛋白的聚集、过量 ROS 产生、线粒体功能障碍和小胶质细胞活化。其症状特征是患者无法控制自主运动(震颤、肌肉僵硬、行走困难和驼背姿势),这是由于大脑的黑质和纹状体区域受损所致。
它是第二常见的神经退行性疾病 (NDD),影响全球超过 1% 的老年人口。
关于肠道与帕金森病之间的关系的第一份报告出现在1817年詹姆斯·帕金森的一篇关于震颤麻痹的文章中。
注:詹姆斯·帕金森,我们所说的描述帕金森病的这位前辈,全名是“James William Keys Parkinson”。1817年,62岁的James Parkinson发表了论文“An Essay on the Shaking Palsy”,在文中,详细地描述了六例被其称为患有“震颤麻痹”的患者,在论文中他注意到了患者不适,像被无法逃脱的魔鬼所控制。他对这种疾病的描述,很多都被现在的医学观察所承认:不自主震颤的同时还有肌肉乏力,即使在被支撑时也不能缓解;身体前驱,不能刹车,越走越快甚至要小跑起来;感觉和智能几乎不受影响…..
肠道病理与帕金森病的联系
后来的研究支持了这一观点,表明了病理过程是从肠道开始,然后影响大脑。越来越多的证据显示,帕金森病患者常见的特征包括肠道炎症、磷酸化α-突触核的早期积累导致跨过迷走神经的腹背侧运动神经元、便秘问题和肠道通透性增加,这表明了肠道菌群与帕金森病发病机制之间的密切关系。
接受迷走神经切断手术的个体患帕金森病的风险降低,这进一步支持了肠道与PD之间的联系。
氧化应激与神经元损伤
较低水平的谷胱甘肽(GSH)和较高水平的铁和H2O2使黑质致密部 (SNc) 神经元易受氧化应激。而且,该区域的脂质过氧化和多巴胺氧化会导致神经元细胞死亡。研究还表明,线粒体呼吸链功能障碍会导致过量的 ROS 产生。
相关病理与细胞毒性
复合物 1 的抑制剂会对多巴胺神经元产生细胞毒性作用,这一事实也支持了这一观点,患有 α-突触核蛋白、磷酸酶和张力蛋白同源物 (PTEN) 诱导的假定激酶 1 (PINK1) 和 Parkin 病理的患者被检测到患有线粒体功能障碍和氧化应激增加。
异常蛋白聚集与肠道微生物群
与 AD 类似,异常蛋白质 α-突触核蛋白的聚集与氧化应激增加相关,反之亦然。谈到肠道微生物群的作用,一些致病菌会释放毒素,导致肠道和肠神经系统 (ENS)细胞的线粒体功能障碍,从而导致神经退行性。
肠道致病菌及其产物参与发病
帕金森病患者肠道中的致病菌增加及其微生物产物直接参与帕金森病的发病机制。为了支持这一事实,最近有报道称,大肠杆菌会产生一种名为 curli 的淀粉样蛋白,这种蛋白会促进 α-突触核蛋白在肠道和大脑中的聚集,会导致小鼠的运动障碍。
另一方面,当小鼠接受肠道限制性淀粉样蛋白抑制剂治疗时,小鼠的运动功能得到改善,便秘也得到改善,表明肠道与 PD 症状的病因有关。
克罗恩病与帕金森病共病风险高
肠道细菌引起的肠道炎症与 PD 中的进行性神经退行性病变直接相关。虽然 PD 患者的血清代谢谱和肠道成分发生了改变,但研究发现,在严重的 PD 情况下,肠道中的肠杆菌科细菌水平会增强,而抗炎细菌水平却非常低,这也表明它与克罗恩病的肠道炎症具有平行关联。这表明克罗恩病患者患 PD 的风险非常高。
同样,研究发现,在PINK1基因敲除小鼠模型中,柠檬酸杆菌(Citrobacter rodentium)感染会通过诱发肠道炎症加重 PD 症状。
肠道菌群代谢物影响PD药物疗效
除了诱发炎症之外,肠道菌群还会发挥代谢作用,例如,在 PD 患者中,其代谢物 β-葡萄糖醛酸、色氨酸和 SCFA 会发生改变。
肠道菌群的一个显著特性是,它能够通过降低抗 帕金森药物的生物利用度或增加药物失活来降低其疗效,就像在标准左旋多巴治疗中的那样。
H2饱和水对PD症状的改善
在这些研究中,还发现肠道细菌产生H2的减少是导致 PD 的因素之一。
当向患PD大鼠和 MPTP 小鼠模型提供 50% H2饱和水时,发现它能够成功减少黑质中的神经元丢失以及氧化应激标志物,当在人体中进行双盲随机试验时,PD患者的运动评分有所改善。
肠道菌群参与帕金森病的证据
此外,针对这一观点,最近对过表达人类 α-突触核蛋白基因的 GF 小鼠 PD 模型的研究表明,SCFA 水平降低,小胶质细胞活化减少,运动功能改善,这表明肠道菌群直接参与了增强 PD。
当将 PD 患者的肠道菌群移植到无菌α-突触核蛋白过表达的小鼠模型中时,运动症状恶化,表明肠道菌群功能失调在 PD 患者中起着作用。同样,用神经毒素治疗的小鼠模型显示肠道菌群组成发生改变,致病性肠杆菌科细菌水平升高。
一些特定的细菌物种,如奇异变形杆菌(Proteus mirabilis)发现能促进小鼠的神经退行性病变。综合起来,这些研究表明,肠道微生物群加剧了人类和动物帕金森病模型中的神经元功能障碍和神经炎症。
注:奇异变形杆菌(Proteus mirabilis, PM)是革兰阴性细菌,属于肠杆菌科。在自然界中广泛存在,具有特殊的群集运动能力,可促进结石形成,在导管、泌尿系上皮中形成结晶生物膜,是引起感染的重要病原。
创伤性脑损伤 (TBI) 是世界范围内最常见的损伤类型之一,是导致死亡和残疾的主要原因之一。
TBI的广泛影响
TBI 造成的残疾不仅包括脑的原发性机械损伤,还包括损伤后的继发性损伤,这种损伤发生在细胞和分子水平,可能导致代谢异常,如线粒体功能障碍、氧化应激、炎症、小胶质细胞活化、兴奋性毒性,从而造成暂时或终身的认知障碍。TBI 的严重程度不仅集中在大脑上,还可能造成多器官损伤,是一种异质性病理生物学状况。
TBI的治疗现状
由于脑损伤的异质性,针对 TBI 引起的神经病理学的治疗方法仍然缺乏,需要考虑新的治疗方案。针对这一问题,肠道益生疗法因其能够恢复肠道菌群失调和 TBI 之间的双面关系而受到广泛关注。
肠道微生物群与 TBI 损伤之间存在双向关系
TBI 的全身表现之一是肠道蠕动和通透性紊乱、粘膜损伤、肠绒毛组织病理学改变,这表明肠道微生物群组成受到干扰。
最近的报告显示,暴露于轻度反复性脑损伤 20 天的小鼠会逐渐出现白质损伤、认知能力下降和轻度、短暂性的肠道菌群失调。
肠道菌群失调也会影响创伤性中枢神经系统损伤的病理生理、血脑屏障通透性改变和小胶质细胞激活,导致严重后果。
研究发现,在脑损伤之前和之后,小鼠 TBI 模型中的肠道菌群耗尽会导致海马 CA1 区神经元密度增加、联想学习障碍减轻以及病变体积缩小。
TBI中肠道菌群的变化
最近的研究还表明,脊髓创伤后会出现肠道菌群失调,导致椎管内炎症和病变病理。
肠道菌群在TBI恢复中的作用
最近的一项研究指出,在 TBI 之前、期间和之后,广谱抗生素引起的肠道菌群失调会导致神经元丢失增加、神经发生受到抑制以及小胶质细胞和外周免疫反应发生改变,同时恐惧记忆反应也会发生调节。
因此,肠道菌群对 TBI 患者的影响具有至关重要的临床意义,因为 TBI 患者由于定期使用抗生素和长期住院,肠道菌群容易发生改变。此外,检测肠道菌群调节可能为识别 TBI 严重程度提供诊断工具,从而提供有针对性的治疗方法。
中枢神经系统与氧化应激
中枢神经系统极易受到氧化应激的影响,并导致神经系统疾病。由于高 O2需求和过氧化敏感脂质细胞的涌入,中枢神经系统运作期间会产生高水平的 ROS。这种氧化代谢产生活性物质,用于传递氧化还原信号以调节突触可塑性等关键功能。
抗氧化剂(无论是酶促还是非酶促、内源性还是外源性)通过阻止 ROS 的产生或清除自由基或使自由基产物失活来保护大脑免受氧化应激。
抗氧化防御机制
抗氧化酶的作用
抗氧化酶如:SOD通过催化超氧化物歧化为O2和H2O2来降低超氧化物阴离子的浓度;GPx还原H2O2和脂质过氧化物;硫醇特异性过氧化物酶如过氧化物酶减少羟基过氧化物的量,过氧化氢酶将H2O2转化为H2O和普通分子O2。
自由基激活参与抗氧化途径的基因转录,保护细胞免受不利影响。
谷胱甘肽不足可能会限制过氧化物酶的活性,从而使神经元更容易受到氧化应激的影响。
体内和体外研究表明:
重要途径与相关机制
Keap1-Nrf2-ARE 与神经退行性疾病相关的氧化应激防御机制有关,调节 SOD、硫氧还蛋白、过氧化物酶和 GPx 的活性。
NF-κB 是中枢神经系统 (CNS) 中的氧化还原传感器,可由 ROS 激活。
中枢神经系统抗氧化代谢的调节受到严格控制,而肠道微生物的作用是高度动态的。
肠道菌群之间、与宿主等相互作用产生代谢物
肠道菌与菌之间、菌与宿主之间的相互作用通过产生各种代谢物(如可吸收维生素、多酚、SCFA、BDNF、可扩散抗氧化剂和氧化剂气体等)来调节内源性和外源性 ROS 水平。
肠道菌群通过代谢物调节屏障、免疫系统、神经系统
肠道微生物还控制代谢物对血脑屏障的通透性、紧密连接完整性和肠道屏障,调节免疫系统,阻止病原体在肠道定植。副交感神经系统的迷走神经感知肠道代谢物,并将肠道信息传达给中枢神经系统,以产生特定的反应。
肠道菌群与神经系统疾病的关系
在应激条件下,迷走神经张力受到抑制,并由于菌群失调而表现出有害影响,如肠易激综合征 (IBS) 和炎症性肠病 (IBD)。参与 AD 发病机制的 Aβ 蛋白由肠道细菌(如ENS 中的大肠杆菌和肠道沙门氏菌)表达。
有益的肠道微生物还会产生多巴胺、血清素和 GABA。这些是调节 ENS 活性并可能相互关联的中枢神经递质。
肠道菌群管理小胶质细胞,失调引发神经炎症
一些研究表明,肠道微生物管理小胶质细胞的激活和成熟,而激活的小胶质细胞会释放大量诱导型一氧化氮合酶 (iNOS) 来调节 NO 的产生。菌群失调会引发炎症性 iNOS 并导致神经炎症。
我们已经看到肠道菌群的潜在作用以及氧化应激在介导神经元疾病中的重要作用,近年来,开发基于抗氧化剂的疗法来治疗氧化应激诱导的神经退行性疾病的需求日益增长,并且成为科学研究的重点。
抗氧化剂疗法与肠道微生物组的作用
抗氧化剂是能够抵消ROS/RNS诱导的氧化应激的化学或天然物质。尽管已经观察到抗氧化剂对糖尿病、关节炎、白内障和骨质疏松症等疾病的强大治疗作用,但用于中枢神经系统疾病的抗氧化疗法有限,仍然需要深入了解其机制。
肠道微生物组的双重作用
肠道微生物组的作用具有两面性,一方面它负责神经退行性(肠道菌群失调和神经炎症)的基本机制,另一方面,肠道微生物组及其代谢物调节许多相关通路,表明它们具有神经保护治疗作用。
蛋白质、维生素等微生物分子通过多步生物合成途径产生,可能对宿主系统产生有益或有害的影响。因此,通过包括益生元和益生菌在内的适当饮食来维持健康的肠道微生物群是维持神经元健康的先决条件。
肠道菌群代谢物的作用及其对肠道菌群的调节过程在神经退行性疾病(NDDs)中的作用
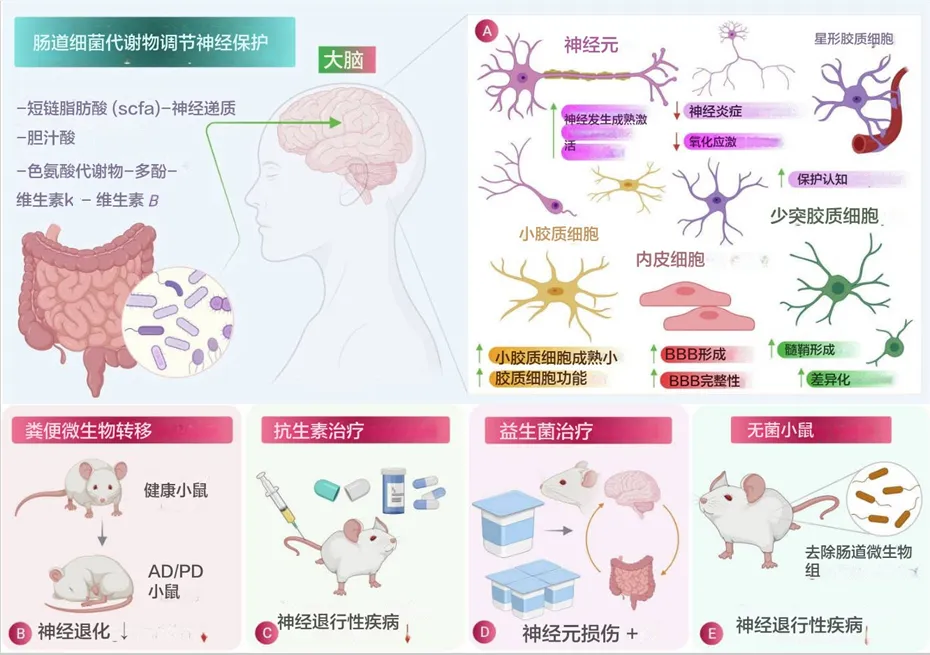
doi: 10.1016/j.jare.2021.09.005
肠道菌群在神经保护中的作用:
(A)肠道菌群释放的有益代谢物在减少炎症和氧化应激方面对脑细胞的特定影响示意图。
(B)粪菌移植 (FMT) 涉及将粪便细菌从健康个体转移到有病理状况的个体,被发现是一种有效的程序,可减少阿尔茨海默病、帕金森病、亨廷顿病和多发性硬化症等神经退行性疾病的病理生理。
(C)和(D)分别使用抗生素治疗和益生菌治疗,它们在减少神经退行性疾病的发病机制方面显示出相当大的效果。
(E)无菌小鼠(没有肠道微生物的小鼠)显示神经退行性疾病的减少,也用于研究肠道微生物对大脑生理的影响,从而显示肠道微生物参与神经退行性疾病。
肠道微生物群在神经保护中的作用

doi: 10.1016/j.jare.2021.09.005
肠道菌群与宿主分子的相互作用
胆汁酸的生成与功能
胆汁酸在肝脏中产生并在肠腔中释放,主要参与脂质和脂溶性维生素的溶解、能量代谢信号传导,并且在脑的生理学和病理生理学中发挥重要作用。
胆汁酸对神经系统的影响
胆汁酸通过直接结合穿过血脑屏障的脑内受体或通过与肠道受体结合间接诱导成纤维细胞生长因子 (FGF) 和胰高血糖素样肽 1 的释放,影响脑不同区域的神经元活动以及迷走神经活动。
胆汁酸的神经保护特性
胆汁酸如熊去氧胆酸 (UDCA) 和牛磺熊去氧胆酸 (TUDCA) 具有神经保护特性且无细胞毒性,这分别在其 III 期临床试验和动物研究中得到证实。最近的数据显示,TUDCA 有助于减弱慢性 PD 小鼠模型中的自噬、α-突触核蛋白聚集和蛋白质氧化。
此外,它还有助于通过Takeda G 蛋白偶联受体 5/sirtuin-3 (TGR5/SIRT-3) 通路防止蛛网膜下腔出血大鼠的神经元凋亡。同样,UDCA 在带电多泡体蛋白 2B (CHMP2B) 内含子 5 额颞叶痴呆模型中表现出神经保护作用。
肠道微生物对胆汁酸的影响
肠腔内的肠道微生物群在脱水酶的作用下将初级胆汁酸(胆酸和鹅去氧胆酸)转化为次级胆汁酸,包括氨基酸与胆汁盐水解酶和其他酶促过程的解离,从而改变它们的核受体结合、溶解度和循环。在人类和小鼠的 AD、PD、ASD多发性硬化症模型中发现了次级胆汁酸水平的变化。
此外,细菌修饰的胆汁酸对 ALS 和中风有神经保护作用。肠道微生物群落的调节可导致胆汁酸水平和性质的变化,这可能是神经退行性或神经保护性的。
胆汁酸在神经保护和疾病治疗中的潜在应用
肠道微生物介导的脱氧胆酸增加会诱导小鼠 EC 中神经递质血清素的释放。胆汁酸代谢物被发现可改善脱髓鞘和减少氧化应激,通过分别作用于少突胶质细胞和小胶质细胞增强其神经保护作用。然而,微生物操纵的胆汁酸的潜在作用和影响尚不清楚,仍有待明确定义。
类固醇激素在大脑发育和功能中的作用
类固醇激素发出的信号对大脑的发育和功能(记忆、决策和性行为)至关重要。类固醇激素在整个身体中循环时,会遇到肠腔内的微生物群。
肠道微生物对类固醇激素的修饰作用
肠道细菌在 β-葡萄糖醛酸酶和 β-葡萄糖苷酶介导的去结合反应中修饰类固醇激素,从而重新激活激素并阻止其排泄。因此,肠道微生物群通过降解和活化途径影响活性和非活性类固醇激素的水平。
肠道微生物与性激素代谢
研究发现,雄激素和雌激素受到肠道微生物群的影响。研究发现,大量肠道细菌能代谢雌激素,雌激素在粪便样本中也会发生氧化还原反应,这表明肠道微生物群发挥了作用。
研究发现,肠道微生物群还具有将睾酮和胆固醇转化为雄激素的能力。受微生物影响的雌激素具有神经保护作用,对小胶质细胞具有抗炎作用。
此外,肠道微生物群落改变会导致雌激素水平降低,从而导致慢性炎症和认知障碍。雌激素分子还会影响少突胶质细胞的分化和髓鞘形成。最近的报告还显示,即使在 MPTP 帕金森病小鼠模型中使用黄体酮治疗也显示出神经保护、抗炎和免疫调节作用,但神经保护作用是始于肠道还是大脑仍不清楚。
肠道菌群与饮食分子的相互作用
膳食氨基酸与肠道微生物代谢
膳食氨基酸也可以被肠道微生物代谢,它们对大脑的影响取决于膳食摄入的类型和频率。虽然肠道微生物编码的氨基酸在宿主体内循环,但影响中枢神经系统的氨基酸是由肠道细菌代谢的膳食氨基酸。
芳香族氨基酸
酪氨酸、色氨酸和苯丙氨酸等芳香族氨基酸被肠道细菌代谢为短链脂肪酸、吲哚衍生物、神经递质、有机酸、胺和氨。
酪氨酸
酪氨酸代谢的最终产物是由酪胺中间体形成两种儿茶酚胺、多巴胺和去甲肾上腺素。体外研究表明,大量肠道细菌可以产生毫摩尔范围内的去甲肾上腺素。最近的报告显示,非肾上腺素通过刺激 B-3 肾上腺素能受体增加星形胶质细胞的谷胱甘肽供应,从而保护神经元免受 H2O2诱导的神经元死亡。
研究发现,酪氨酸也被肠道微生物代谢为苯酚,如 4-乙基苯酚,随后在宿主体内被硫酸化为 4-乙基苯酚硫酸盐,在 ASD 小鼠模型中发现该物质升高,也是 ASD 儿童尿液的生物标志物。
色氨酸代谢产生的神经活性分子
色胺和犬尿氨酸等吲哚衍生物是肠道细菌代谢色氨酸的产物,是一种神经活性分子。吲哚丙酸是一种吲哚衍生物,可作为抗氧化剂减少神经炎症,并被观察到在减少 AD 病理方面具有潜在作用。
发现犬尿氨酸代谢物会影响焦虑、记忆和应激样行为。还观察到犬尿氨酸代谢途径的紊乱会促进炎症、兴奋毒性谷氨酸的产生和自由基攻击,表明平衡的犬尿氨酸具有神经保护作用,并在 AD、PD 和 HD 中具有抗炎作用。
色氨酸代谢物通过调节其芳烃受体有助于降低星形胶质细胞的炎症反应,同时也影响它们与小胶质细胞的相互作用。同样,还观察到吲哚酚-3-硫酸盐控制小胶质细胞的活化,随后控制其与星形胶质细胞的相互作用。
谷氨酸和精氨酸的神经保护
氨基酸谷氨酸也被肠道细菌谷氨酸脱羧酶转化为 GABA,一种抑制性神经递质,在小鼠模型中观察到它可以减轻抑郁和焦虑症状。
就氨基酸精氨酸而言,它被代谢成四种多胺,即胍丁胺、腐胺、亚精胺和精胺,它们的作用机理是谷氨酸受体并参与维持突触可塑性和记忆形成。
胍丁胺在作为脑中 α-2 肾上腺素能受体和咪唑受体的配体时,对中枢神经系统疾病有治疗作用。亚精胺也在 3-硝基丙酸 3-NP HD 模型中显示出神经保护作用。
此外,胍丁胺能刺激 Nrf-2 信号通路,改善脂多糖 (LPS) 诱导的 ROS 产生。体外和体内研究还表明,胍丁胺可保护星形胶质细胞和小胶质细胞免受氧化应激引起的损伤。这些研究表明肠道微生物内分泌学在神经科学中的潜在作用。
肠道菌群可以将膳食纤维代谢成短链脂肪酸
未消化的膳食纤维(如复合碳水化合物多糖)在肠道微生物酶糖苷水解酶和多糖裂解酶的作用下,通过厌氧发酵转化为短链脂肪酸(SCFA)。
丁酸、乙酸和丙酸组成短链脂肪酸,是结肠上皮细胞的能量来源。除此之外,它还进入体循环,随后直接或间接影响许多器官的生理功能,包括神经发育和功能。
短链脂肪酸在神经退行性疾病中的作用
有报道显示,当通过使用益生菌混合物或使用抗炎短链脂肪酸来调节 AD 小鼠模型的肠道菌群时,有助于抵消疾病的进展。同样,短链脂肪酸被发现能有效加剧 GF 小鼠 PD 模型中的运动症状。
SCFA对神经系统的直接影响
短链脂肪酸和乙酸穿过血脑屏障,激活神经元,调节神经递质和神经营养因子的水平。一项研究表明,丙酸和丁酸会影响神经元细胞内的钾水平。
丁酸是调节表观遗传基因活化(组蛋白去乙酰化酶)的酶的强效抑制剂,并被发现在 AD、PD、HD、中风和记忆障碍的小鼠模型中起到强效抗炎剂的作用。
有趣的是,SCFA 会干扰 Aβ 肽之间的相互作用,形成神经毒性低聚物,从而预防 AD 病理。据报道,将野生型小鼠的粪便微生物群移植到 PD 动物模型中,同时给予丁酸,可显著改善运动症状和多巴胺缺乏症。
SCFA对神经系统保护的多重机制
当我们观察细胞特异性反应时,丁酸已被证明可以在体外减少星形胶质细胞中的神经炎症和氧化,而乙酸盐被这些细胞用作能量来源。
一项研究发现,SCFA 通过与内皮细胞上的 SCFA 受体结合来增加紧密连接蛋白的表达,从而有助于降低 血脑屏障的通透性并预防 LPS 诱发的癫痫和中风。
SCFA 通过减少小胶质细胞活化来减少大脑中的氧化应激,从而对抗 AD 和 PD 中的神经炎症。总之,从膳食纤维中获得的 SCFA 有助于改善大脑健康,具体取决于个人的肠道健康状况。
多酚的分类及抗氧化特性
多酚是植物中存在的生物活性分子,在植物的生长、保护和繁殖中起着根本性的作用。多酚分为黄酮类化合物、酚酸类化合物和单宁类化合物。
多酚的分子结构,即羟基的位置和芳香环取代的性质,使其具有清除自由基的能力,并被广泛研究作为治疗NDD的抗氧化疗法。
多酚的代谢及其神经保护机制
未吸收的多酚在宿主体内通过水解和酯化作用,在肠道微生物群的作用下转化为生物可利用和生物活性的代谢物,随后在到达外周组织之前进行甲基化、硫酸化、羟基化等修饰。
研究表明,从葡萄和葡萄酒中提取的白藜芦醇、从绿茶中提取的姜黄素和表没食子儿茶素-3-没食子酸酯等不同多酚通过激活蛋白激酶通路(如 Keap1/Nrf-2/ARE)发挥神经保护作用,而这些通路是缓解内源性和外源性 ROS 的主要通路。
多酚代谢物在神经退行性疾病中的潜在作用
据报道,细菌多酚代谢物如 3-羟基苯甲酸和 3-(3′-羟基苯基)丙酸可抑制淀粉样蛋白聚集,从而有助于抑制 AD 的进展。
同样,据观察,黄酮类槲皮素可作为 BACE-1 抑制剂。此外,天然黄酮类原花青素可在体外 减轻鱼藤酮诱导的多巴胺能神经元的氧化应激。此外,肠道菌群产生的多酚(如阿魏酸)可促进神经发生,并在 AD和脑缺血的小鼠模型中显示出神经保护作用。
多酚与肠道菌群的相互作用及其对神经保护的影响
雌马酚和肠内酯也是肠道细菌通过代谢植物雌激素(多酚之一)产生的衍生物,可能对雌激素受体介导的经典神经保护途径产生影响。有趣的是,多酚还可以调节肠道菌群的组成,进一步将其转化为抗炎和神经保护代谢物。
扩展阅读:
肠道微生物与维生素合成
肠道微生物也是维生素的重要来源,特别是维生素B和K,它们不仅对肠道微生物代谢至关重要,而且还对宿主的生理途径产生影响。
维生素的吸收与生理作用
虽然膳食维生素通过小肠吸收,但微生物衍生的维生素的吸收发生在结肠中。为了预防新生儿出血性疾病,在肠道菌群建立之前,需要补充维生素K,这是血栓形成过程中必不可少的物质。
此外,维生素B和K对大脑发育和功能也很重要。就神经退行性疾病 (NDD)而言,许多研究表明维生素B和K在改善神经元健康方面发挥着有效作用。
维生素K在神经退行性疾病中的作用
研究发现,维生素K缺乏与AD的发病机制有关,增加膳食中维生素K的摄入有助于改善老年患者的记忆功能。
最近的报告显示,维生素K2(甲基萘醌-4)具有强大的抗氧化特性,能显著抑制鱼藤酮诱导的p38活化、ROS产生和caspase-1活性,进而恢复线粒体膜电位,显示出其在治疗神经炎症诱导的帕金森方面的潜力。
类似地,维生素 K2 可有效调节 PC12 小鼠神经母细胞瘤细胞中的 bax 和 caspase-3 活化,保护其免受 6-OHDA 诱导的细胞凋亡。
维生素缺乏与神经系统疾病的关系
最近的一份报告显示,帕金森病患者体内维生素 K2 水平低与炎症反应失调和凝血信号级联有关。
维生素 B 缺乏还与脚气病和多发性神经病等神经系统疾病有关。同样,叶酸缺乏与老年女性的认知障碍和进行性痴呆有关。而且,大量摄入 B6、B9 和 B12 可降低与 AD 认知能力下降有关的大脑区域萎缩的速度。此外,还需要更多建设性的研究来证明肠道微生物群产生的维生素在神经保护中的潜在联系。
扩展阅读:
自由基可能带来的危害
ROS 形式的自由基是细胞正常代谢过程的副产物。自由基有可能破坏遗传物质、使酶失活、使复合碳水化合物解聚以及使脂质过氧化。
抗氧化剂的保护机制
细胞内分子的这种破坏会导致细胞死亡。为了平衡自由基,身体具有特定的抗氧化剂,如谷胱甘肽。
一些产品也开始流入医药市场以应对氧化应激,如阿魏酸 (FA),因为它具有抗氧化和抗炎特性。它通过增强 BDNF 和神经生长因子 (NGF) 以及一些具有抗炎特性的神经肽的产生来帮助神经干细胞增殖。
益生菌在抗氧化应激中的作用
2017 年的研究,益生菌如植物乳杆菌 NCIMB 8826、发酵乳杆菌 NCIMB 5221和动物双歧杆菌也能通过细菌 FA 酯酶大量产生 FA。
由于其治疗效果,它在 AD 治疗中的应用越来越受到关注,用 FA 进行预处理已被证明可以减少 Aβ 原纤维并治疗 AD 小鼠的神经炎症。产生 FA 的益生菌通过清除 ROS 来抑制 β-淀粉样蛋白原纤维的形成和聚集。
SIRT1蛋白脱乙酰酶的抗氧化特性
其他益生菌蛋白,sirtuin-1 (SIRT1) 蛋白脱乙酰酶已被证明具有抗氧化特性。这种蛋白质调节宿主抗氧化途径的基因,并具有神经保护作用。
在接受治疗的转基因 3xTg-AD 小鼠中,观察到大脑中 SIRT1 蛋白的表达和活性恢复,Aβ 肽的形成减少。然而,在未经治疗的 AD 小鼠中,发现 SIRT1 的表达显著下降。
此外,SIRT1 活性增强可降低 p53 蛋白乙酰化,并通过抑制凋亡途径提高应激细胞的存活率。其他研究也表明,益生菌补充剂可激活 SIRT1 途径并激发抗氧化作用。
SLAB51益生菌配方的神经保护作用
最近的研究探究了一种名为 SLAB51(嗜热链球菌、嗜酸乳杆菌、植物乳酸杆菌、副干酪乳酸杆菌、德氏乳酸杆菌保加利亚亚种、短乳酸杆菌、长双歧杆菌、短双歧杆菌、婴儿双歧杆菌)的益生菌配方缓解氧化应激的能力,并发现了其作用的分子机制。
SLAB51 还能增强 GPx 和过氧化氢酶抗氧化酶的活性,从而减轻氧化应激引起的损伤。在人类中也观察到了类似的发现,AD 患者大脑中的 SIRT1 浓度显著降低,这与 AD 患者大脑皮层中淀粉样蛋白 β 和 tau 的积累密切相关。一项补充益生菌菌株干酪乳杆菌 01 的人体研究报告称,SIRT1 水平有所提高。
肠道菌群与免疫反应
肠道菌群与宿主免疫细胞之间的关系非常脆弱。免疫细胞专门区分宿主友好细菌和致病细菌。如果这种关系受到损害,则会导致不必要的免疫反应,引发慢性炎症。这种分化是由肠上皮细胞启动的,肠上皮细胞负责根据细菌细胞表面抗原(如 LPS、肽聚糖和鞭毛蛋白)产生受过训练的巨噬细胞表型。当上皮细胞变得容易受到致病攻击时,抗原会转移到脉管系统中,并产生促炎细胞因子,如 IL(白细胞介素)-1、IL-6 和肿瘤坏死因子-α (TNF-α),从而导致感染性休克以及肠道和脑部炎症。
肠道微生物与神经系统炎症的关联
一些细菌毒素也可以穿过血脑屏障。为了探究微生物在淀粉样变性中的作用,研究人员通过评估认知障碍患者几种肠道微生物的促炎(CXCL2、CXCL10、IL-1β、IL-6、IL-18、IL-8、炎症小体复合物 NLRP3、TNF-α)和抗炎(IL-4、IL-10、IL-13)细胞因子活性进行了研究,结果发现:
大肠杆菌/志贺氏菌的数量增加和直肠真杆菌的数量减少与认知障碍和淀粉样蛋白阳性患者的促炎和抗炎细胞因子浓度变化显著相关。同时,在 AD 患者中观察到 IL-6、CXCL2、NLRP3 和 IL-1β 水平升高,而 IL-10 水平降低。这项研究表明,肠道微生物群可能引发、加重或减轻神经系统疾病的外周炎症。
扩展阅读:
随着技术的进步,有关大脑的知识正在成倍增加。神经科学家从分子生物学、分子遗传学、脑成像和其他新技术中生成了大量数据,并且对共享神经科学数据以进行各种分析有着浓厚的兴趣。
神经成像在预测和检测神经退行性疾病和精神障碍方面也非常有帮助。为了收集和分析神经科学数据,正在开发各种生物信息学工具,以进一步开发基于大脑功能的设备。到目前为止,研究人员对因各种压力条件而导致神经退化的分子通路有了初步了解。
集成数据库将有助于从公开来源收集与神经退行性疾病相关的不同类型的数据。
当前与神经数据相关的活跃数据库
编辑
doi: 10.1016/j.jare.2021.09.005
随着高通量技术的加强,人们对微生物组的全面了解及其在人类健康中的重要性正在快速增长。
近期已证明肠道细菌可以改变中枢神经系统生理,而菌群失调可能是神经炎症的一个潜在因素。
微生物群影响左旋多巴治疗PD效果,数据库助力解析神经退行性疾病
据报道,在临床试验中,左旋多巴治疗 PD 的结果在招募受试者之间有所不同。这种差异是由于他们的微生物群造成的。L-多巴由不同肠道细菌物种(即粪肠球菌和Eggerthella lenta A2)的酪氨酸脱羧酶 (TDC) 和多巴胺脱羟酶 (Dadh) 代谢。
为了灭活细菌 L-多巴脱羧酶,人们发现了一种药物,即 (S)-α-氟甲基酪氨酸 (AFMT)。L-多巴和 AFMT 的组合被用于治疗帕金森病。与健康个体相比,AD 患者的肠道微生物群在分类学水平上发生了改变,例如拟杆菌、放线菌、瘤胃球菌、毛螺菌科。
宏基因组数据/16S RNA 测序数据支持菌群失调与 AD 之间存在明确关联。细菌产物(如 LPS 和 SCFA)与淀粉样蛋白病理有关。开发这些数据库的主要目的是将数据组织成一组结构化记录,这些记录有助于分析师检索信息进行不同的分析,从而为与神经退行性疾病相关的各种未解之谜提供解决方案。
肠道微生物群与大脑的相互联系已导致神经科学研究取得变革性进展。迄今为止,发现的大多数研究都与肠道细菌神经科学研究有关,但肠道微生物群非常庞大,需要科学家的关注,以确定和描述肠道微生物及其群落对肠脑信号传导途径的影响。
★ 为什么大脑容易发生氧化应激?
目前我们了解到大脑含有丰富的神经元,突触以及神经胶质细胞,相比于其他器官有更高的能量代谢活动和要求,大脑的抗氧化防御系统较弱,因此容易出现氧化还原稳态紊乱。
大脑Ca2+的稳态失调,神经元的兴奋性氨基酸在大量摄入导致谷胱甘肽(SH)的耗竭,从而引发神经元的铁死亡。
神经递质如多巴胺、5-羟色胺和去甲肾上腺素自动氧化生成ROS。大脑对由葡萄糖诱导的氧化应激敏感。大脑富含多不饱和脂肪酸,尤其是二十碳五烯酸(DHA),这使得其更容易受到氧化应激的影响,因为脂质过氧化和脑部利用过氧脂质进行信号传导。
脑部的微胶质细胞是大脑的常驻免疫细胞,对于大脑发育和功能至关重要。微胶质细胞活性取决于总的氧气生物可用性,并借此展示对突触的损伤,通过消耗更多氧气来产生O2–。H2O2和NO等反应性物质在损伤部位吸引微胶质细胞,引发局部炎症并推动神经退行性。
因此综合以上因素表面大脑容易发生氧化应激,这是很多神经退行疾病比如阿尔茨海默病,帕金森以及痴呆等的主要原因。
而大量的研究观察到肠道微生物组在通过其自身代谢产物或产生次生代谢产物减轻氧化应激、炎症和能量代谢方面发挥了不可忽视的作用。
★ 肠道菌群如何影响神经系统?
肠道微生物还控制代谢物对血脑屏障的通透性、紧密连接完整性和肠道屏障,调节免疫系统,阻止病原体在肠道定植。副交感神经系统的迷走神经感知肠道代谢物,并将肠道信息传达给中枢神经系统,以产生特定的反应。
在应激条件下,迷走神经张力受到抑制,并由于菌群失调而表现出有害影响,如肠易激综合征 (IBS) 和炎症性肠病 (IBD)。参与 AD 发病机制的 Aβ 蛋白由肠道细菌(如ENS 中的大肠杆菌和肠道沙门氏菌)表达。有益的肠道微生物还会产生多巴胺、血清素和 GABA。这些是调节 ENS 活性并可能相互关联的中枢神经递质。
肠道微生物管理小胶质细胞的激活和成熟,而激活的小胶质细胞会释放大量诱导型一氧化氮合酶 (iNOS) 调节 NO产生。菌群失调会引发炎症性 iNOS 并导致神经炎症。
★ 肠道菌群代谢物的作用
胆汁酸通过直接结合穿过血脑屏障的脑内受体或通过与肠道受体结合间接诱导成纤维细胞生长因子 (FGF) 和胰高血糖素样肽 1 的释放,影响脑不同区域的神经元活动以及迷走神经活动。
色胺和犬尿氨酸等吲哚衍生物是肠道细菌代谢色氨酸的产物,是一种神经活性分子。吲哚丙酸是一种吲哚衍生物,可作为抗氧化剂减少神经炎症,并被观察到在减少 AD 病理方面具有潜在作用。
短链脂肪酸和乙酸穿过血脑屏障,激活神经元,调节神经递质和神经营养因子的水平。一项研究表明,丙酸和丁酸会影响神经元细胞内的钾水平。
雌马酚和肠内酯也是肠道细菌通过代谢植物雌激素(多酚之一)产生的衍生物,可能对雌激素受体介导的经典神经保护途径产生影响。
而通过使用具有抗氧化和抗炎活性的益生菌来调节肠道微生物组已显示出有希望的神经恢复能力。
★ 益生菌帮助恢复神经系统
益生菌如植物乳杆菌 NCIMB 8826、发酵乳杆菌 NCIMB 5221和动物双歧杆菌也能通过细菌 FA 酯酶大量产生 FA。由于其治疗效果,它在 AD 治疗中的应用越来越受到关注,
最近的研究探究了一种名为 SLAB51(嗜热链球菌、嗜酸乳杆菌、植物乳酸杆菌、副干酪乳酸杆菌、德氏乳酸杆菌保加利亚亚种、短乳酸杆菌、长双歧杆菌、短双歧杆菌、婴儿双歧杆菌)的益生菌配方缓解氧化应激的能力,并发现了其作用的分子机制。
从德氏乳杆菌(Lactobacillus delbrueckii)亚种Lactobacillus delbrueckii ssp. bulgaricus B3 和 Lactobacillus plantarum GD2 中分离出的胞外多糖可保护 SH-SY5Y 细胞免受 Aβ(1–42) 诱导的细胞凋亡,这表明它们有望成为药物治疗阿尔茨海默病 (AD)的有前途的天然化学成分。
★ 饮食塑造肠道菌群缓解氧化应激助力大脑健康
氨基酸谷氨酸也被肠道细菌谷氨酸脱羧酶转化为 GABA,一种抑制性神经递质,在小鼠模型中观察到它可以减轻抑郁和焦虑症状。
就氨基酸精氨酸而言,它被代谢成四种多胺,即胍丁胺、腐胺、亚精胺和精胺,它们的作用机理是谷氨酸受体并参与维持突触可塑性和记忆形成。
从葡萄和葡萄酒中提取的白藜芦醇、从绿茶中提取的姜黄素和表没食子儿茶素-3-没食子酸酯等不同多酚通过激活蛋白激酶通路(如 Keap1/Nrf-2/ARE)发挥神经保护作用,而这些通路是缓解内源性和外源性 ROS 的主要通路。
主要参考文献
2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement 2020;16:391–460.
Akasaka Naoki, Fujiwara Shinsuke. The therapeutic and nutraceutical potential of agmatine, and its enhanced production using Aspergillus oryzae. Amino Acids. 2020;52(2):181–197.
Barua Sumit, Kim Jong Youl, Kim Jae Young, Kim Jae Hwan, Lee Jong Eun. Therapeutic Effect of Agmatine on Neurological Disease: Focus on Ion Channels and Receptors. Neurochem Res. 2019;44(4):735–750.
谷禾健康
2024年6月19日,广东省人民医院儿科杨敏团队取得了一项备受瞩目的成果。该团队关于“肠道菌群失调通过糖原贮积病中的 CCL4L2-VSIR 轴引发炎症性肠病”,在国际著名期刊《Advanced Science》(影响因子IF=15.1)发表,针对这一通路不仅可以为 GSD 患者提供有益的治疗,还可以为结肠炎和其他形式的 IBD 患者提供有益的治疗。
谷禾健康技术团队参与并支持本研究的菌群测序和分析部分。
这一突破性研究为糖原贮积病及其相关并发症的治疗提供了新的视角,这也是我国儿科在罕见病领域的研究中取得的重大突破。

糖原贮积病(GSD),是一类由于先天性酶缺陷所造成的糖原代谢障碍疾病,多数属常染色体隐性遗传,发病因种族而异。该病类型多样,其中GSD-Ib型患者常伴有炎症性肠病。
在GSD-Ib型患者中,肠道菌群失调通过CCL4L2-VSIR轴影响炎症性肠病(IBD)。研究发现,与典型IBD不同,GSD-Ib患者展现出独特的消化道症状和肠道菌群特征。
肠道菌群失调主要由特定病原菌引起,导致肠道巨噬细胞过度激活,CCL4L2-VSIR轴的过度激活促进上皮细胞特定基因的表达,从而推动IBD的进展。
研究结果表明,靶向肠道菌群失调或CCL4L2-VSIR轴可能成为治疗GSD相关IBD的潜在策略。
糖原是肝脏和骨骼肌中储存的葡萄糖形式,其分解提供稳定的葡萄糖供应。当特定酶的活性缺失时,会导致糖原积累,进而影响组织的功能,导致一系列健康问题,包括低血糖、肌肉痉挛、疲劳、肝脏肿大等。
糖原贮积病的类型多样,根据受影响的酶和组织不同,可以分为几种主要类型。例如:
糖原贮积病的治疗主要是通过调节血糖水平和预防低血糖来管理。对于GSD-Ia和GSD-Ib,过去十多年的治疗方法包括间歇性未煮玉米淀粉或通过胃管给予夜间葡萄糖输注。给GSD-III患者高蛋白饮食,以增加儿童的生长速率。
GSD不仅影响儿童,成人也可能受到影响。未经最佳终身饮食葡萄糖治疗的成人GSD患者的长期预后不佳,可能会出现多种并发症,成人GSD患者常见的并发症包括骨质疏松症和骨折、肾结石、肾盂肾炎等。
▼
正常情况下,人体摄入的碳水化合物会被转化为葡萄糖,一部分葡萄糖会被立即利用以提供能量,而多余的葡萄糖则会在肝脏和肌肉等组织中合成糖原储存起来。当身体需要能量时,糖原又会分解为葡萄糖以供使用。
然而,在糖原贮积病患者中,由于参与糖原合成或分解的某些酶存在缺陷,导致糖原的合成、分解或储存出现异常,从而引起一系列的健康问题,包括低血糖、肌肉痉挛、疲劳、肝脏肿大等。
这类疾病有一个共同的生化特征,即是糖原贮存异常,绝大多数是糖原在肝脏、肌肉、肾脏等组织中贮积量增加。
▼
糖原贮积病是一种罕见病,总体发病率相对较低,但具体的发病率因不同类型的糖原贮积病而有所差异。
《世界胃肠病学杂志》资料显示,发病率约为20000-43000名新生儿中的1人。
由于其症状的多样性和隐匿性,一些病例可能未被准确诊断,实际的发病率可能被低估。
为了调查中国大陆GSD的流行病学和临床特征,杨敏及其团队进行了两项大型多中心研究,并通过问卷调查报告了2020年10月至2021年6月的209例GSD病例(如图)。
关于患者的就医情况,其中最显著的一个特点是超半数家庭(52.4%)为“省级”外来病例,就医的医院主要来自相对发达地区。且就诊地主要集中在上海、北京、广东等医疗资源相对发达的城市。
▼
糖原合成和分解代谢中所必需的各种酶至少有8种,由于这些酶缺陷所造成的临床疾病有15型,其中:
doi: 10.3748/wjg.v29.i25.3932
杨敏团队的调查研究中,主要包括6种类型的GSD,即:
其中最常见的是I型(66.9%)。
▼
不同类型的糖原贮积病临床表现各异,但常见的症状包括:
低血糖、肝肿大、酸中毒(乳酸性酸中毒)、高脂血症、高尿酸血症、疲劳、肌肉无力等。
由于糖原无法正常分解为葡萄糖,患者在空腹或长时间运动后容易出现低血糖症状,如头晕、乏力、出汗、心慌等。
糖原在肝脏中过度积累,导致肝脏体积增大。
长期的低血糖和代谢紊乱可能影响儿童的生长发育,导致身高、体重增长缓慢。
某些类型的糖原贮积病会影响肌肉功能,表现为肌肉无力、疼痛、痉挛等。
如Ⅱ型糖原贮积病可能导致心脏肥大、呼吸肌无力等,严重影响心肺功能。
其中:
口疮性口炎、肛周病变、胃肠炎、呼吸道感染和皮肤感染等症状在I型GSD中更常见;
厌食、呕吐、腹泻、粘液/血便、腹痛和腹胀是 GSD 患者常见的胃肠道并发症,尤其是在 GSD-Ib 组(77.8%)。
造成患者家庭问诊困难的一个主要原因是该疾病表型的复杂,例如下图:
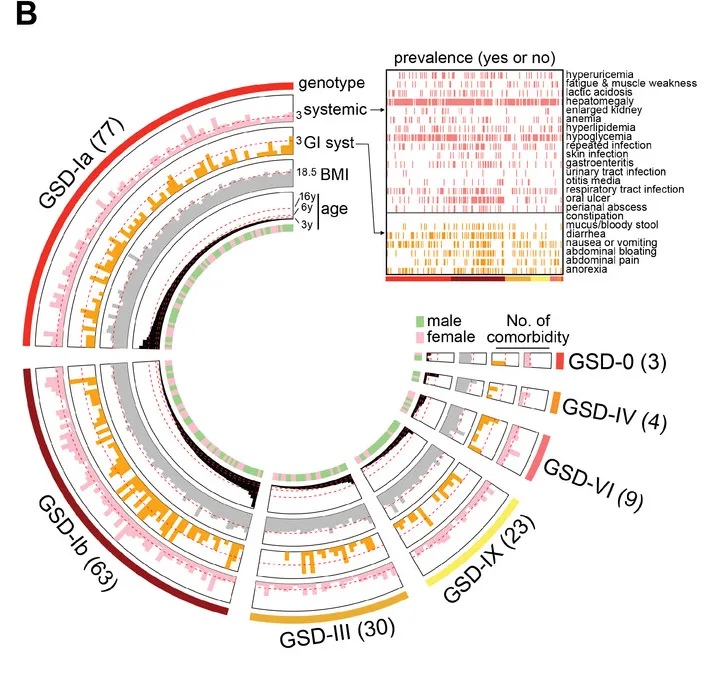
一种奇怪的糖原贮积病亚型:许多GSD-lb患者也患有慢性 IBD
杨敏教授表示,他们关注 GSD相关IBD 已有近10年了。多中心内镜监测研究中,他们招募了 32 名 GSD 患者。这些患者在内镜检查期间表现出活动性胃肠道症状。在这 32 名患者中,27 名患有 GSD-Ib,25 名最初诊断为 IBD。
其中GSD-Ib型患儿消化道症状比例最高(77.8%)且最为特殊,表现为单个或多发散在深圆形溃疡、炎性假息肉、梗阻和狭窄(如下图)。
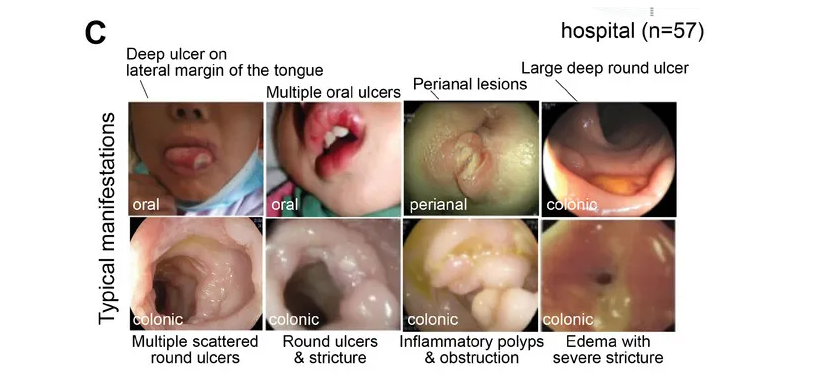
目前人们对 GSD-lb 中 IBD 的发病机制和发病原因知之甚少,超过 70% 的病例会出现严重的消化道症状,而这种症状在其他疾病亚型中偶尔才会出现。
如果不具体了解这些机制以及免疫系统与肠道微生物群之间的关系,科学家将无法开发出针对与 GSD 相关的 IBD 症状的有效治疗方法或干预措施。杨敏及其团队希望改变这一现状,开始了他们的实验探索过程。
▼
为了探究GSD群体消化系统异常,特别是IBD高发的原因,杨敏及其团队在全国23个省份招募了150名GSD患者(共涵盖Ia、Ib、III、IX、VI、IV及0型共7种类型)采集粪便样本, 327个健康对照者的粪便样本。
注:这327个对照样本包括:137 名家庭对照者和 190 名在山东、浙江、湖南等地收集的无关健康对照者。
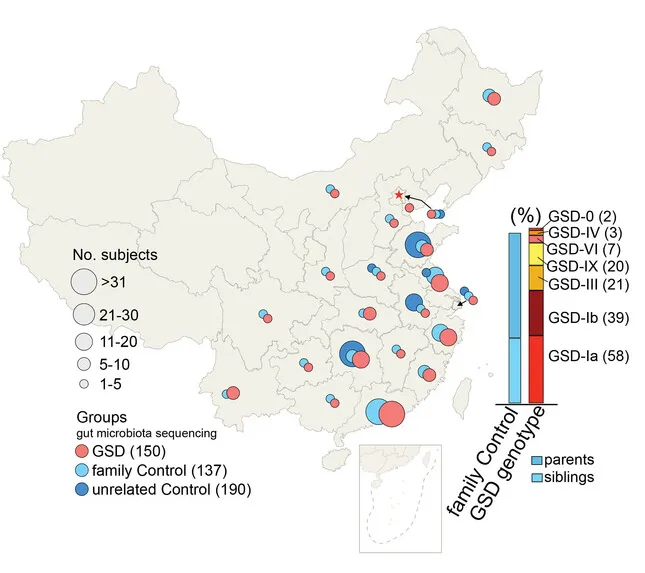
GSD基因型是肠道菌群的主要影响因素
肠道菌群受多种因素影响,包括饮食、地域和医疗条件等。
通过结合个人临床信息与肠道菌群数据的EnvFit分析发现(如下图),共有45个个人因素与肠道菌群的变异显著相关,如疾病情况(基因型)、家庭因素、年龄、身高体重、共患病等等。
值得注意的是,在这个队列中,GSD基因型主要影响肠道菌群组成。总体而言,家庭因素(家庭组配对比较)和居住地区是影响肠道菌群组成的第二和第三重要因素。
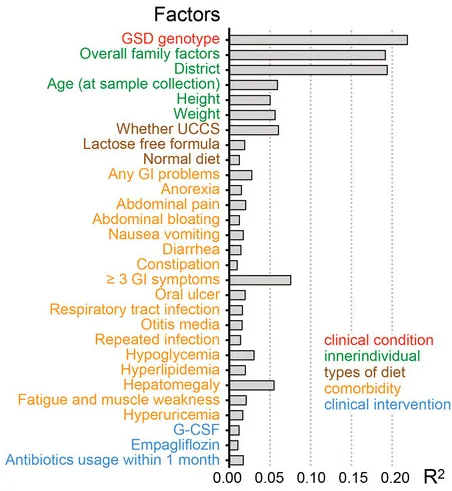
这在肠道菌群研究中颇为少见,因为多数研究认为地域因素极大且显著地影响个体菌群构成。这说明,特殊的基因型对于塑造个体肠道菌群构成起到决定性作用。
具体而言,研究人员将患者分为三组:
GSD-Ia、GSD-Ib、GSD-no IaIb
▼
GSD引起了个体肠道菌群多样性显著降低(下图H)及潜在致病菌显著升高(下图G),特别是GSD-Ia和GSD-Ib。这些肠道菌群的变化在校正性别、年龄、BMI和地域等因素后,仍然存在。
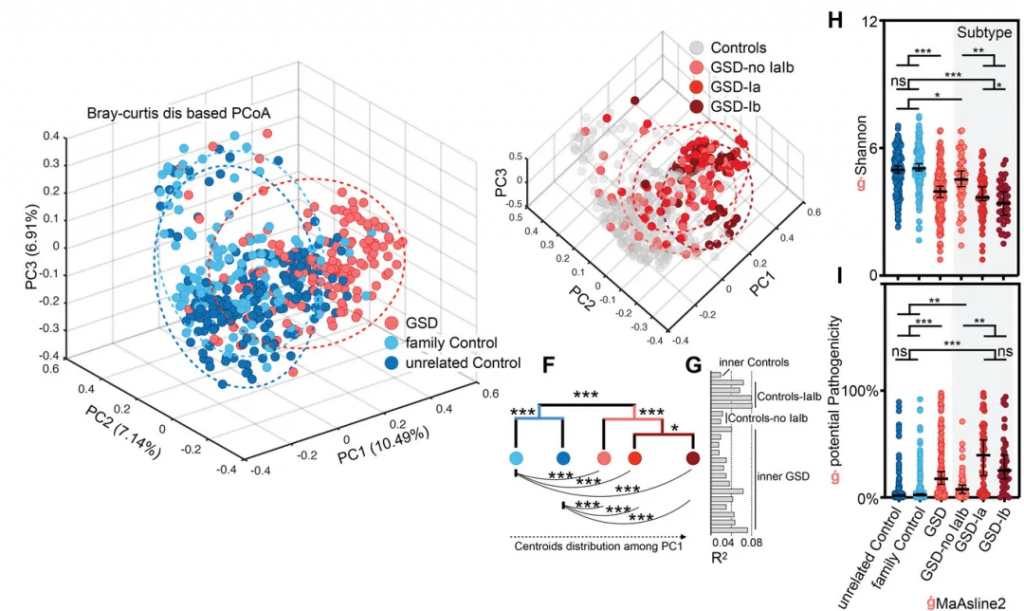
厚壁菌门和拟杆菌门仍是主要菌群;但变形菌门和放线菌门的比例显著增加。
GSD 患者共有31个属发生显著改变。其中18个属减少,而其余13个增加。这31个属中28个属在校正掉性别、年龄、BMI和地域等因素影响后,仍与GSD显著相关(下图D)。
有22物种的丰度和GSD显著关联(下图E),其中10 个增加和12个降低。
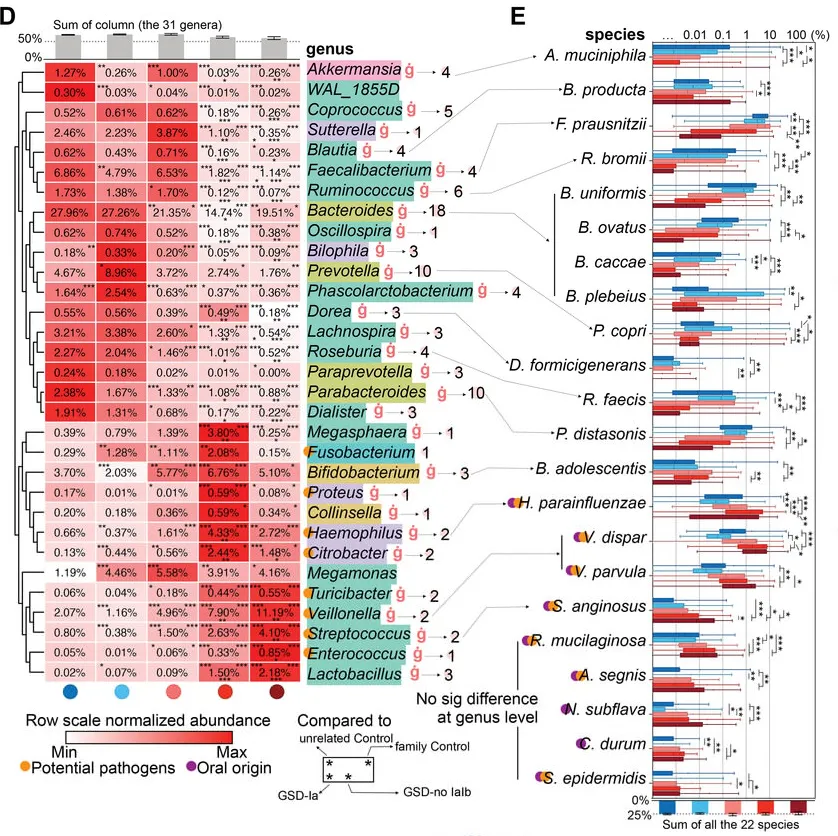
这31个显著改变的属的总丰度在不同分组人群中的总占比达到60-70%,种水平则为~25%,表明这些GSD相关的属种变化,是肠道菌群构成主体的紊乱。
▼
GSD-Ia和GSD-Ib患者表现出更严重的菌群紊乱,特别是那些最常见、功能性的微生物类群在其肠道内丢失,例如下列菌群在GSD-Ia和-Ib几乎完全耗尽:
丁酸可增强上皮氧合,有利于结肠中的低氧微环境并抑制病原体定植。然而,主要丁酸生产菌,如拟杆菌属、粪杆菌属和瘤胃球菌,以及相关代谢途径在GSD中显著降低,这可能导致兼性厌氧病原菌的延伸。
具体到每个菌来说:
AKK菌与肠道屏障的维持和抗炎作用有关,其减少可能会削弱肠道屏障,增加肠道炎症的风险,从而可能与IBD症状的出现有关。
粪杆菌参与肠道内的发酵过程,对维持肠道健康和免疫功能有重要作用。在糖原贮积病患者中,由于糖原代谢异常可能影响了肠道环境,进而影响了粪杆菌的定殖和功能,导致其数量减少。这可能会减少短链脂肪酸的产生,影响肠道健康。
拟杆菌在肠道内参与碳水化合物的分解,产生短链脂肪酸,糖原贮积病患者拟杆菌的减少可能导致肠道炎症反应加剧,与IBD症状的出现有关。
瘤胃球菌是产生短链脂肪酸的重要菌群,对肠道健康和宿主能量代谢有重要作用。瘤胃球菌的减少可能会导致短链脂肪酸的产生下降,影响肠道屏障功能和抗炎作用,从而可能与IBD症状的出现有关。以动物为基础的饮食会增加有害菌Ruminococcus gnavus。
GSD患者中存在口腔源的潜在病原体激增,例如:
口腔中的致病菌可以在肠道中定植和繁殖,说明肠道为这些菌群的生长繁殖提供了条件和环境。
链球菌,已被证明可通过GasderminA依赖性细胞焦亡诱导肠道损伤;可能通过产生超抗原等物质激活宿主T细胞免疫反应进而引发肠道炎症。
肠球菌的成员已显示出促炎作用;肠球菌可以通过分泌金属蛋白酶分解上皮钙黏蛋白破坏肠屏障。
韦荣氏球菌属在IBD中延伸,细小病毒通过产生硝酸盐诱导炎症。韦荣氏球菌属和链球菌的组合抑制了IL-12p70的生物合成,随后增强了IL-8、IL-6和TNFα的炎症反应。
菌群该定植时未定植,该退出时却长期占据
值得注意的是,双歧杆菌、韦荣球菌和乳杆菌这些本应该在健康儿童生命早期保持较高比例的细菌,在GSD患者的肠道中扩张,并且这些细菌在 GSD 患者的整个童年和成年期都会扩增,这可能抑制了其他正常菌群的定植。
▼
GSD患者肠道菌群发育落后于实际年龄
使用微生物组年龄Z分数(MAZ)评估菌群发育状态,随机森林回归和调整的深度神经网络分析均显示GSD患者的MAZ显著降低,特别是在GSD-Ia和Ib患者中。
这表明GSD患者的肠道菌群发育落后于实际年龄,呈现发育延迟状态。
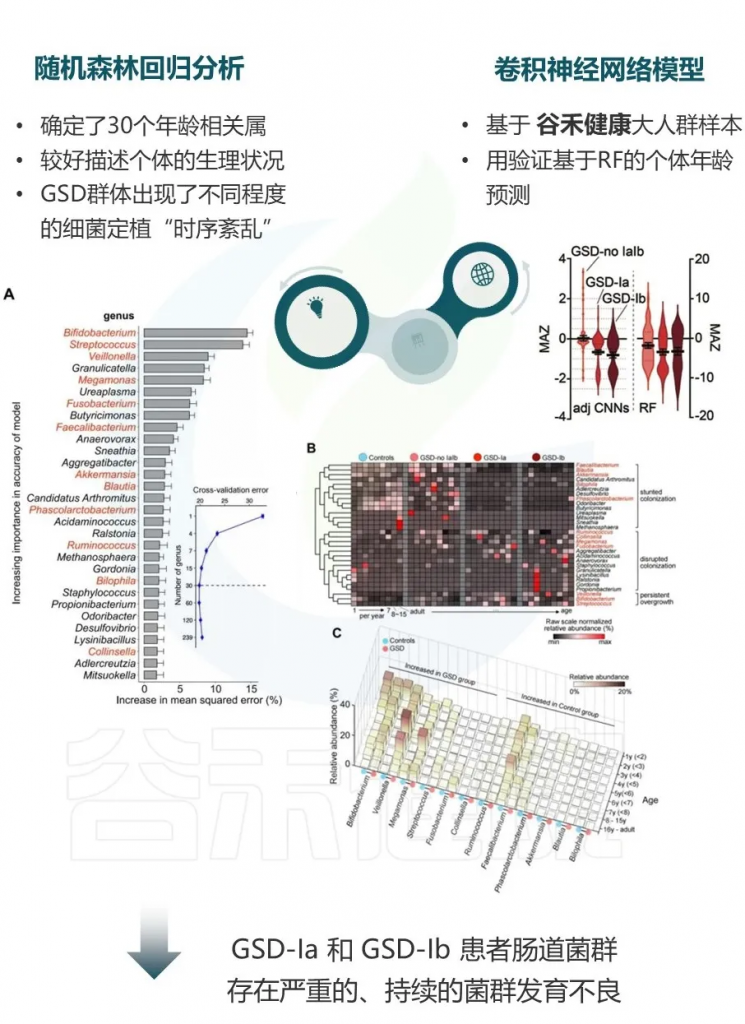
▼
从数据分析的角度,研究团队进一步分析了GSD患病情况、共患病与肠道菌群紊乱的中介关系。
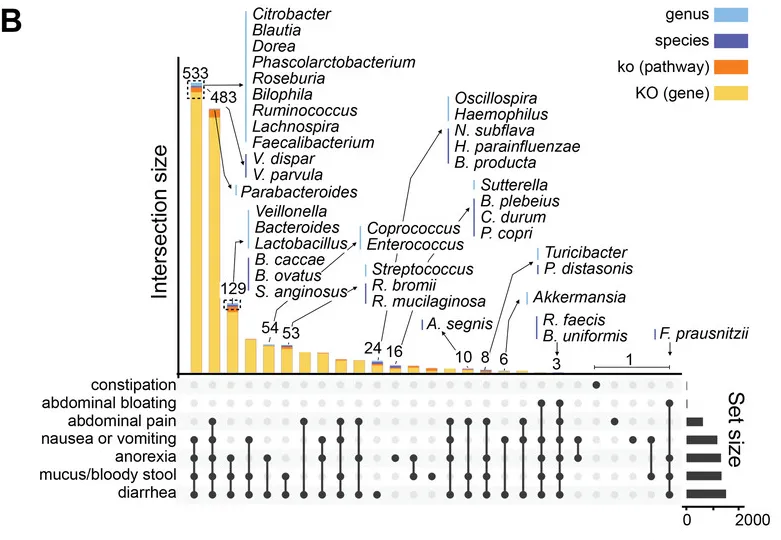
他们用最具代表性的肠道微生物特征和临床元数据构建了一个中介模型,在校正年龄、性别、BMI、地域及特殊治疗性饮食(生玉米淀粉)的影响后,肠道菌群仍然是GSD背景下,患者消化道异常的显著中介因子(下图)。说明GSD会通过影响肠道菌群构成最终造成消化道异常。
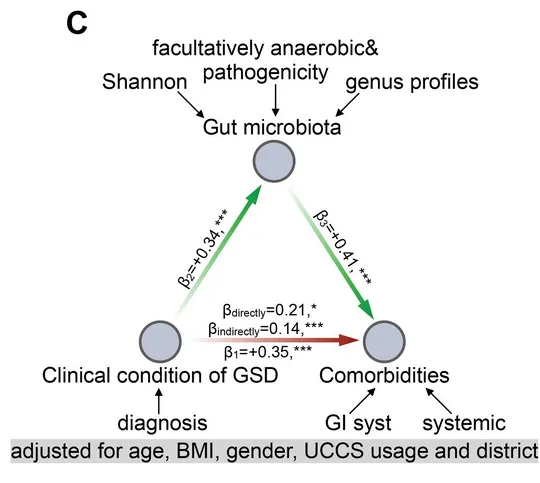
GSD 中这些细菌可能相互依赖资源和生存,形成一个复杂的相互支持网络,肠道菌群失调可能通过细菌的“群体犯罪”引起 GSD 患者的肠道上皮功能障碍。
下面我们来看一下该研究的另一个重要内容——肠道微生物群和免疫细胞之间的“串扰”。
▼
为了明确GSD相关的肠道菌群紊乱,如何引起个体IBD表性的产生。研究者们采集3名GSD-Ib患者的结肠粘膜组织(下图G),并使用单细胞测序技术分析患者结肠粘膜单细胞层面的转录水平变化。通过转录水平注释发现,相较于健康人、典型IBD和结肠炎的肠粘膜组织,GSD-Ib患者肠粘膜上有非常高的巨噬细胞聚集(下图H)。
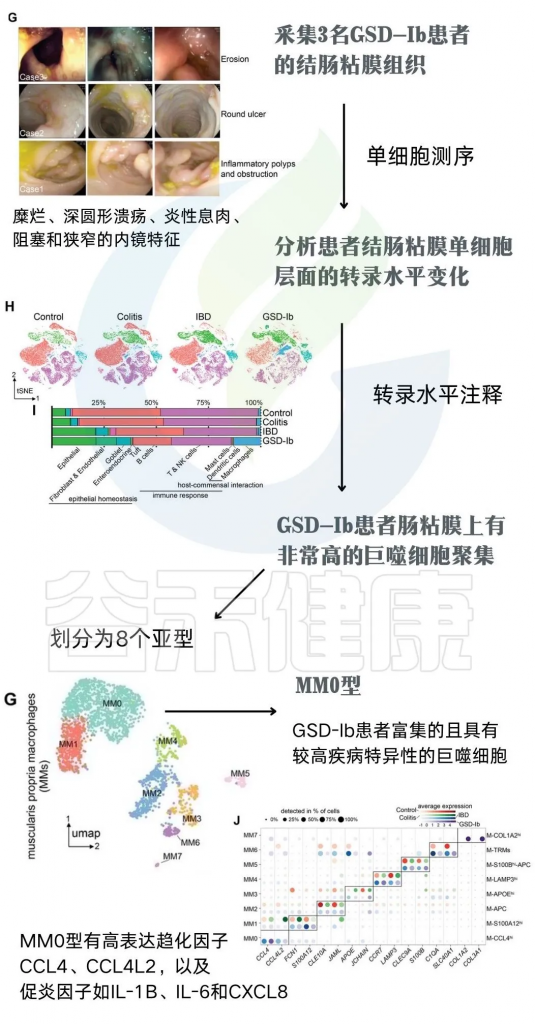
注:巨噬细胞中差异表达基因的KEGG通路注释显示,GSD-Ib中调节抗菌体液免疫反应和趋化因子介导的信号通路的基因集显著富集。GSD-Ib组中上调的基因主要参与环境信息处理和人类疾病。
“
更深入的分析确定了一种巨噬细胞亚型(MM0)的重要性,这种亚型产生一种名为CCL4L2的趋化因子,负责激活其他免疫细胞对抗有害细菌的生长。
虽然CCL4L2的自然功能通常是保护性的,但杨敏及团队怀疑,由于GSD-Ib患者相关肠道细菌的存在,CCL4L2的过度或失调刺激可能是他们出现IBD症状的可能原因。
这一点最初在实验室中得到了支持,其中用有害细菌感染的细胞获得的细胞培养基处理的巨噬细胞,比用对照培养基处理的巨噬细胞更加“激活”。
下面我们来看一下他们选用了哪些菌群进行相关实验,来刺激巨噬细胞:
▼
研究人员选择了三种GSD相关菌群来刺激巨噬细胞,即:
暴露于粪肠球菌、小弧菌和咽峡炎链球菌以及混合感染 (EVS) 的巨噬细胞中 CCL4L2 的表达在 mRNA 和蛋白质水平上显著增加,但不影响 IL-1 β和 IL-6 的表达。
来自EVS感染的条件培养基(CM)与对照组相比,诱导了显著的巨噬细胞迁移趋化性,而这种趋化性部分被CCL4L2中和所减弱。
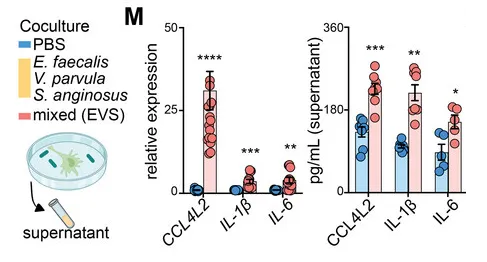
因此,是特定的肠道病原菌(如E. faecalis、V. parvula、S. anginosus)引发了CCL4L2的高表达,那么具体是如何影响炎症性肠病的进展?我们接着看。
“
然后,团队在从GSD-Ib患者取得的活检样本中寻找CCL4L2,并发现CCL4L2蛋白与一种叫做VSIR的受体结合,这种受体存在于肠道细胞壁中。
在IBD的小鼠模型中也发现了类似的结果,科学家们发现与健康对照组相比,巨噬细胞的水平更高,并且在用一种旨在抑制VSIR的抗体处理后,他们发现小鼠的症状加重了。
巨噬细胞通过CCL4L2-VSIR配体-受体信号与肠道上皮细胞相互作用,以促进损伤修复。
具体研究如下:
调节核糖体、氧化磷酸化途径和抗病原体反应的基因集在GSD-Ib上皮细胞中特异性富集。
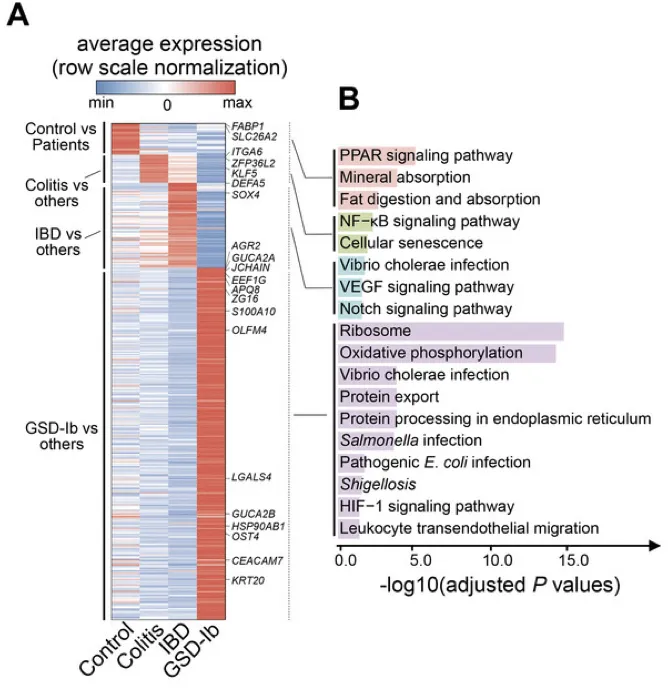
上皮细胞中典型的差异表达基因是AQP8、ZG16、GUCA2C、SLC26A3、AGR2,它们负责吸收、分泌、代谢、上皮屏障完整性、pH稳态、粘液屏障功能。
▼
免疫荧光染色显示GSD-Ib患者肠道组织中CCL4L2和VSIR的显著共定位,进一步验证了CCL4L2在巨噬细胞中的高表达及其与VSIR的相互作用。
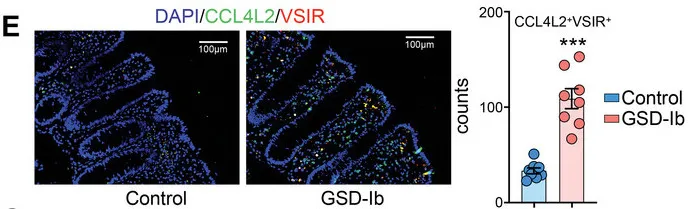
CCL4L2与其受体VSIR形成的CCL4L2-VSIR轴主导参与了MM0型巨噬细胞与其他类型细胞交互,特别是OLFM4+上皮细胞(这类细胞是GSD特异性增加的,同时也被报道在克罗恩病中较高)。
预测MM0巨噬细胞与其他细胞类型之间的相互作用
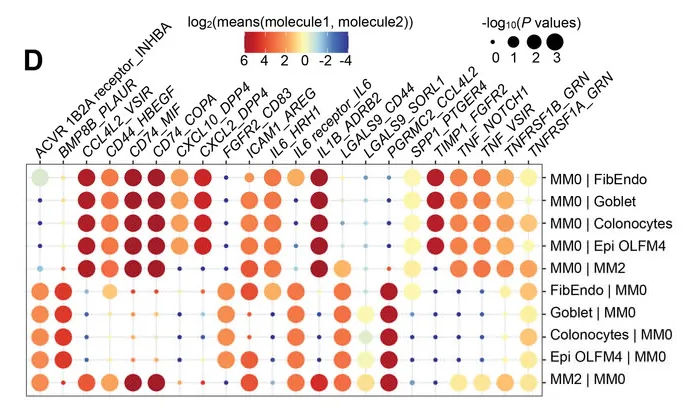
而这些受体细胞的基因表达情况,决定了患者肠道组织的疾病发展方向,如AGR2、ZG16、MUC2等。
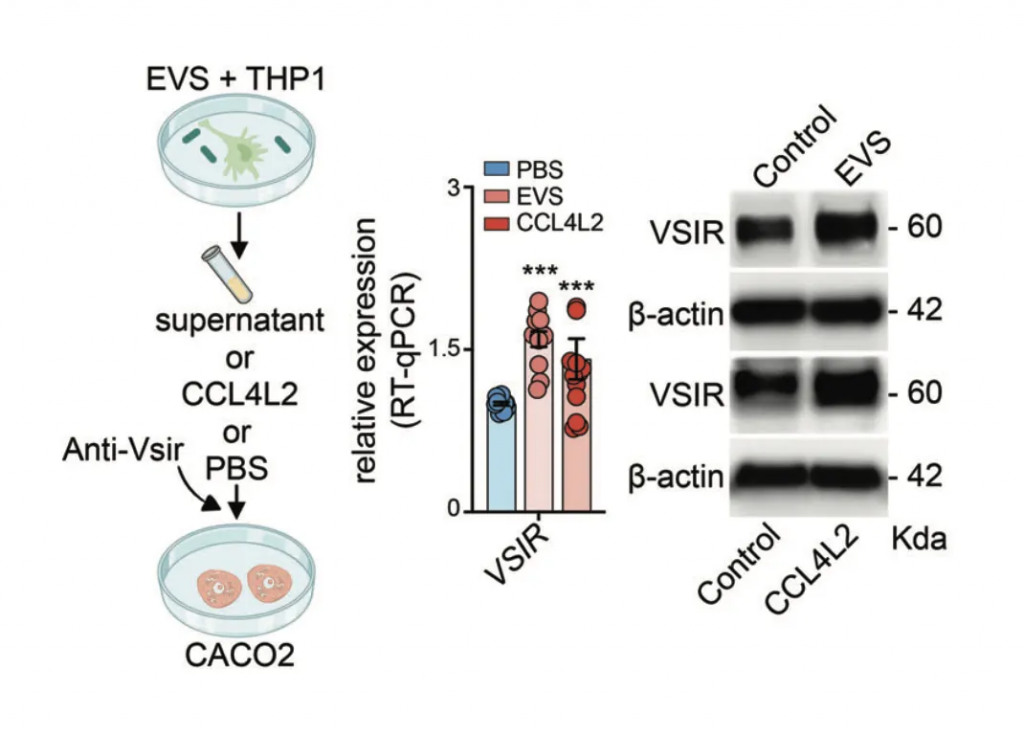
表明GSD相关的肠道菌群紊乱,通过引起肠上皮巨噬细胞高表达趋化因子CCL4L2,激活其他下游细胞膜蛋白VSIR表达,从而引起了GSD独有的消化系统IBD表型。这个现象进一步在C57BL/6小鼠中得到验证。
▼
研究人员在C57BL / 6小鼠中进行了选择性细菌移植,通过口服灌胃的方式将EVS移植到小鼠体内,并用DSS诱导结肠炎。研究发现,抗生素治疗显著降低了EVS灌胃诱导的小鼠血清CCL4L2水平,而抗VSIR则显示出完全相反的趋势。
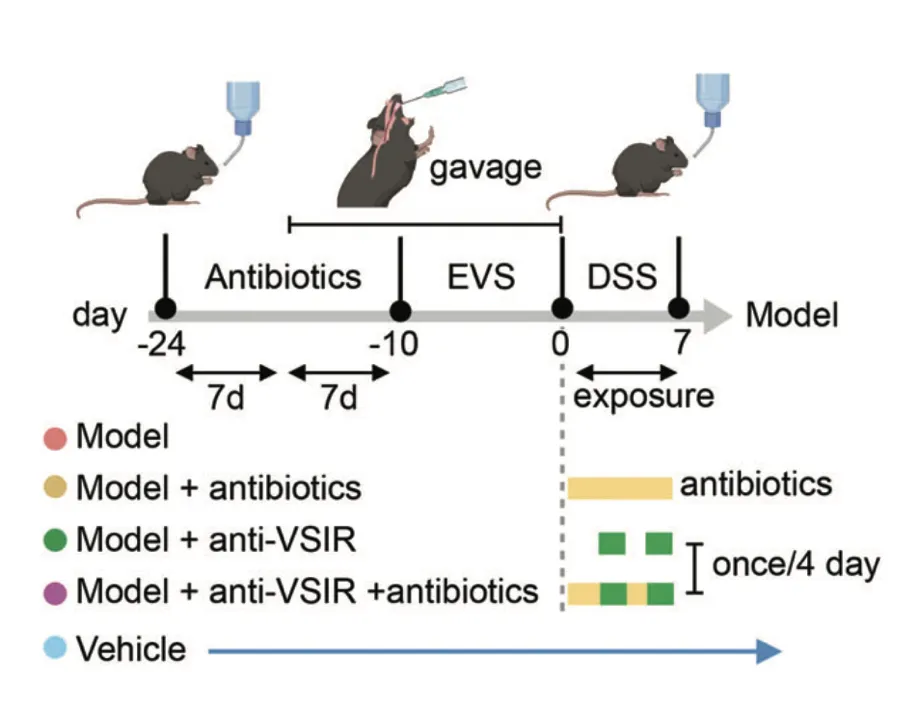
免疫荧光染色显示,模型组中MM0巨噬细胞增强,但在给予抗生素或抗VSIR注射后减少,这进一步导致模型组中肠上皮细胞的MUC2、AGR2和ZG16表达显著下调,引发粘膜损伤。
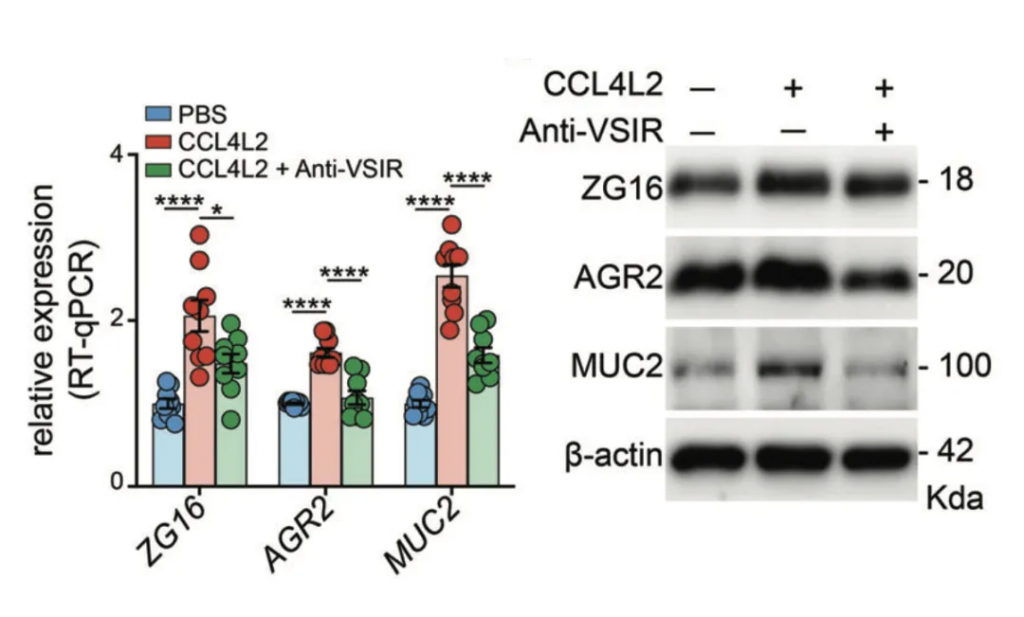
研究结果表明,EVS 增强的 MM0 巨噬细胞可以通过调节 CCL4L2-VSIR 轴来介导结肠肠上皮稳态。因此,靶向CCL4L2-VSIR通路可用于结肠炎和IBD的广泛应用。
肠道驻留免疫细胞和上皮之间的串扰,对于胃肠道稳态、抗原致敏调节、预防感染和 IBD 发展至关重要。在该研究中 GSD相关IBD 与典型的 IBD 相比具有不同的特征,特别是复杂的内镜表现及其特殊的解构:大量巨噬细胞积聚和上皮细胞增殖。
从病因上讲,肠道致病菌激活粘膜巨噬细胞导致胃肠道内形成炎症环境。在 GSD-Ib 中,一群独特的巨噬细胞由致病菌诱导,并表达高水平的趋化因子CCL4L2。
关于CCL4L2的有益方面:
潜在有害方面:
值得注意的是,CCL4L2-VIR通路的激活可能促进严重细胞因子风暴中的T细胞耗竭,这与新冠肺炎的不良预后有关。
靶向CCL4L2-VSIR可以抑制各种炎症性疾病、自身免疫性疾病和肿瘤转移。在这里,研究人员揭示了胃肠道中CCL4L2-VSIR轴的异常激活,最终驱动GSD-Ib中的非典型IBD。该发现为诊断和治疗GSD-Ib以及潜在的GSD相关IBD引入了一个新的靶点,并为VSIR相关研究提供了证据。
总的来说,这项研究证实了在糖原贮积病群体中独有的肠道菌群紊乱,即:
而这样的肠道菌群紊乱会引起个体结肠巨噬细胞高表达CCL4L2,并通过其受体蛋白VSIR介导上皮细胞的异常基因表达,最终造成GSD相关的IBD表型。
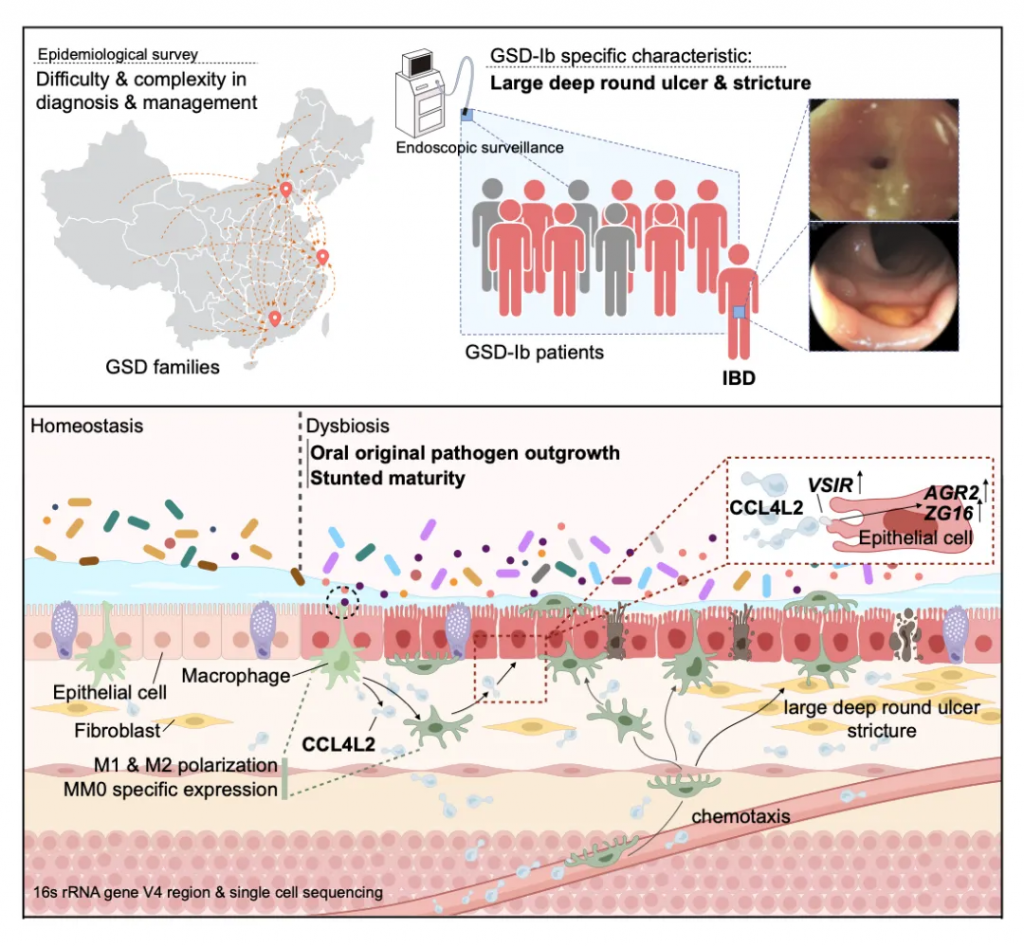
该研究的意义及未来方向:
该研究通过识别GSD-Ib与IBD之间的特定生物学联系,推动了精准医疗在代谢性疾病和消化系统疾病中的应用。
目前杨敏团队正在研究 CCL4L2-VSIR 通路的作用机制,并将探索其在患者治疗中的潜力。该团队预计,针对这一通路不仅可以为 GSD 患者提供有益的治疗,还可以为结肠炎和其他形式的 IBD 患者提供有益的治疗。
CCL4L2-VSIR轴的发现也可能促进新药开发,特别是针对GSD-Ib相关IBD的治疗。
这些研究结果也可应用于个性化治疗方案(基于肠道菌群特征),为患者提供更有效的治疗选择,有助于改善GSD-Ib患者及更广泛消化系统疾病患者的生活质量。
主要参考文献
Lan J, Zhang Y, Jin C, Yang M et al., Gut Dysbiosis Drives Inflammatory Bowel Disease Through the CCL4L2-VSIR Axis in Glycogen Storage Disease. Adv Sci (Weinh). 2024 Jun 18:e2309471.
Gümüş E, Özen H. Glycogen storage diseases: An update. World J Gastroenterol. 2023 Jul 7;29(25):3932-3963.
Hannah WB, Derks TGJ, Drumm ML, Grünert SC, Kishnani PS, Vissing J. Glycogen storage diseases. Nat Rev Dis Primers. 2023 Sep 7;9(1):46.
Zhong J, Gou Y, Zhao P, Dong X, Guo M, Li A, Hao A, Luu HH, He TC, Reid RR, Fan J. Glycogen storage disease type I: Genetic etiology, clinical manifestations, and conventional and gene therapies. Pediatr Discov. 2023;1(2):e3.
Wang Y, Liu H, Dong F, Xiao Y, Xiao F, Ge T, Li D, Yu G, Zhang T. Altered gut microbiota and microbial metabolism in children with hepatic glycogen storage disease: a case-control study. Transl Pediatr. 2023 Apr 29;12(4):572-586.

谷禾健康

小克里斯滕森氏菌(Christensenella minuta)是一种革兰氏阴性、不产生孢子、不运动的细菌:它属于厚壁菌门,这是细菌界中最大的门之一,包括多种对人类健康有重要影响的细菌。
在厚壁菌门下,C.minuta属于梭菌纲,这一纲的细菌多为厌氧菌,能在缺氧的环境中生长。梭菌目是梭菌纲下的一个目,包含了多种与消化道健康密切相关的细菌。
在梭菌目下,C.minuta属于Christensenellaceae属,这是一个与肠道健康紧密相关的科。由Christensenella minuta和少数其他菌种组成的属。
Christensenella minuta于2012年首次从健康人类粪便中发现,被认为是新一代益生菌。克里斯滕森菌科(Christensenellaceae)及其成员Christensenella minuta已被证明具有许多健康益处。
研究表明,Christensenella minuta在2型糖尿病和肥胖等代谢紊乱以及炎症性肠病中的丰度显著下降。其相对丰度与低 BMI 指数相关的瘦表型呈正相关。
除此之外,支气管哮喘和过敏性疾病、肾结石、情感障碍、甲状腺癌、粘膜类天疱疮、多囊卵巢综合征和复发性口疮性口炎等疾病中Christensenella minuta的丰度也较低。
Christensenella minuta能够代谢多种碳水化合物,例如纤维素、半纤维素和果胶,产生乙酸和丁酸等短链脂肪酸,这些物质不仅对肠道健康有益,还能调节宿主的代谢过程。
C.minuta通过几种机制影响代谢健康,包括使肠道微生物群重新正常化、产生功能性短链脂肪酸、抑制脂肪生成、维持肠道上皮完整性以及通过胆汁酸代谢调节能量代谢。
此外,C.minuta还通过抑制NF-κB信号通路和促炎细胞因子IL-8的分泌来缓解炎症性肠病。
C.minuta还可能对患有过敏性疾病的患者大有裨益,因为它可以改善肠道通透性并减轻全身炎症。而患有哮喘、湿疹和食物过敏的患者更容易出现“肠漏”,这被认为是导致疾病发病和诱发病情的一个因素。
鉴于其能够限制肠道干细胞增殖,未来C.minuta衍生的益生菌也可能对恶性肿瘤患者有益,尤其是结肠癌患者。
此外,Christensenella minuta显示出与其他潜在有益菌株的强烈相关性;由于C.minuta能够产生短链脂肪酸或塑造酸性环境,有利于其他有益菌(如Akkermansia muciniphila和Roseburia faecis)或与传统益生菌菌株(如双歧杆菌和乳酸杆菌)的生长,C.minuta还能够产生氢气,为M.smithii提供代谢底物(它利用H2和CO2产生甲烷),同时会抑制如克雷伯菌、大肠杆菌等机会性病原体的定值和增殖。
本文从基本属性、人群分布、Christensenella minuta的丰度与一些人体疾病(如肥胖、炎症性肠病和 2 型糖尿病)中的关联,以及与其他细菌的相关性,讲述了可能成为新一代益生菌的Christensenella munita。更好的了解Christensenella可以为基于肠道微生物群的个性化药物或疗法铺平道路。
▸ 发现历史
小克里斯滕森氏菌(Christensenella minuta)(DSM 22607) 于2012年通过 16S rRNA 测序发现,并首次从健康日本男性的粪便样本中培养出来。
2021年,发现了另一个C.minuta菌株DSM33407。其序列与菌株DSM22607有 99% 的一致性,并表现出相似的微生物学特性。次年,另一株菌株C.minuta DSM 33715 被公布并登记。
此外发现了两个新的细菌物种Christensenella massiliensis和Christensenella timonensis ,经 16S rRNA 测序,它们与C. minuta的序列相似性分别为 97.4% 和 97.5%。
而Caldicoprobacter oshimai JW/HY-331 T、Tindallia californiensis DSM 14871 T和Clostridium ganghwense JCM 13193 T是最近的亲属。
▸ 基本属性
从分类学上讲,Christensenella minuta属于厚壁菌门、梭菌纲和梭菌目。该细菌以丹麦微生物学家 Henrik Christensen 的名字命名,其种名反映其小巧的体型(Minuta在拉丁语中是“小”的意思)。该菌株的基因组相对较小,由大约150万个碱基对组成。
Christensenella minuta的细胞形态
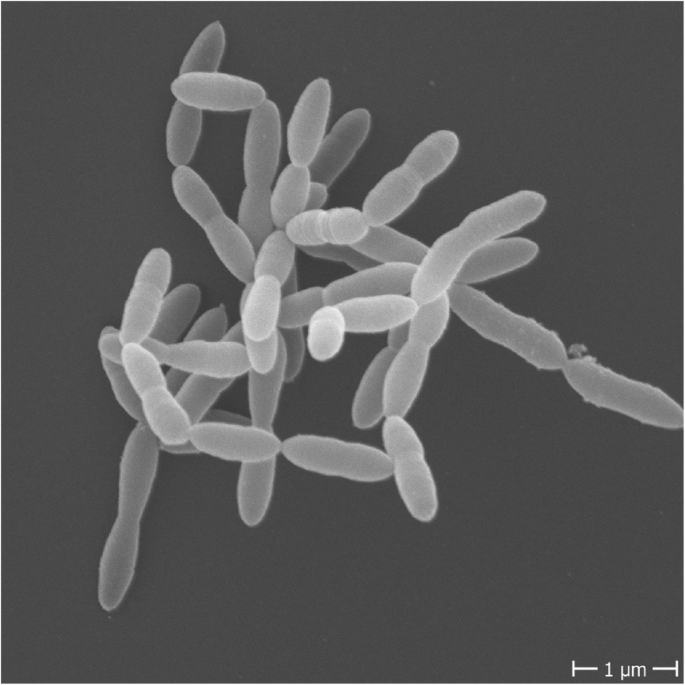
doi: 10.1186/s12915-019-0699-4.
Christensenella minuta是一种小型杆状细菌,末端呈锥形、大小从0.5毫米到1.9毫米不等、革兰氏阴性、不产生孢子、不运动的细菌,可形成圆形、几乎无色的菌落。菌落的平均尺寸为:宽度0.507±0.04μm、长度1.27±0.28μm、直径0.5–1.0μm,单独或成对出现。
Ignatyeva O,et al.Front Microbiol.2024
A)革兰氏染色;B)在Schaedler琼脂上生长的菌落
-暴露于空气中会显著降低活性
它最初被描述为严格厌氧的;然而,后来的研究表明,它可以耐受氧气数小时。与普遍看法相反,暴露在大气中不会立即杀死细菌,而是会降低其活力。Christensenella minuta在37–40°和pH7.5时生长最快。它不是特别挑剔,可以在各种培养基中生长。
Christensenella minuta具有边界清晰的细胞壁,由丙氨酸、谷氨酸、丝氨酸和LL-二氨基庚二酸组成,这些细胞壁与半乳糖、葡萄糖、鼠李糖和核糖作为全细胞糖连接。
-对氨苄西林和四环素有抗性
菌株DSM22607的胆汁抗性为20%,而菌株 DSM33715的胆汁抗性高达80%。
对氨苄西林和四环素有抗性,但对氯霉素、克林霉素、美罗培南、甲硝唑、莫西沙星和哌拉西林/他唑巴坦敏感。
-可以利用多种单糖,但无法代谢色氨酸
在碳源利用方面,Christensenella minuta显示出对多种碳水化合物的利用能力,包括纤维素、半纤维素和果胶等。这些碳水化合物在肠道中不易被宿主消化酶分解,但C. minuta能够通过其独特的酶系统进行发酵,从而获取能量和营养物质。
C.minuta可以利用多种单糖,如葡萄糖、D-木糖、L-阿拉伯糖、L-鼠李糖和D-甘露糖,进行糖酵解发酵。C. minuta对葡萄糖的主要发酵产物是短链脂肪酸(乙酸和丁酸)。此外,已证明它可以通过发酵转化有机底物以产生大量氢气。
C. minuta对过氧化氢酶、氧化酶和脲酶的检测结果均为阴性。它也不能还原硝酸盐,也不能代谢色氨酸。最近, C. minuta菌株之一 DSM 22607被证明能产生一种新型胆盐水解酶(BSH)。
克里斯滕森菌科(Christensenellaceae)成员遍布各大洲。它们生活在各种动物的微生物群中,从蟑螂和蜥蜴到鸟类和哺乳动物,包括人类。这些细菌主要存在于胃肠道中,但也存在于灵长类动物的呼吸道和泌尿生殖道。
▸ 不同种族和性别之间丰度差异显著
Christensenella minuta作为健康成人结肠中的亚优势共生微生物种群,约占细菌总种群的0.2%到2%。其流行率在个体之间差异很大。在人群中,与克里斯滕森菌科(Christensenellaceae)不同相对丰度相关的特征包括种族和性别。
例如,对居住在阿姆斯特丹的2000多名不同种族的个体进行了研究,报告称荷兰受试者的Christensenellaceae科相对丰度最高。同样,比较了1673名居住在美国的人种族间微生物组差异,报告称与其他种族相比,亚太岛民的粪便样本中Christensenellaceae科的总体代表性较低。

一项研究还发现,与男性相比,女性中Christensenellaceae的相对丰度更高,在动物中也报告了类似的观察结果。这些种族和性别差异的根本原因尚不清楚。
▸ 百岁老人和瘦体型的人群中丰度更高

还有研究表明,在中国、意大利和韩国等国,百岁老人和超百岁老人体内的Christensenellaceae相对丰度高于年轻人群,因此Christensenellaceae可能与人类长寿有关。
针对多个地理位置的相对年轻个体的研究也发现了Christensenellaceae与年龄的正相关的关系 。鉴于这些研究均未对同一个体进行长期跟踪,因此 Christensenellaceae 与年龄的关联可能反映的是队列效应,而非年龄效应。
例如,随年龄而变化的饮食模式可能会影响这种关联,或者较早出生的个体体内的Christensenellaceae含量可能一直高于较晚出生的个体。
基于 16S rRNA 基因的肠道微生物群检测,该菌群在瘦体型个体中较为丰富。M.smithii是最丰富的产甲烷菌,它利用H2和CO2(C.minuta细菌发酵膳食纤维的产物)产生甲烷,表明以H2为基础的共营养与瘦表型和健康状态相关。这表明C.minuta和M.smithii之间存在跨物种氢转移,并且这两个物种与瘦表型呈正相关。
▸ 炎症性肠病等疾病中丰度降低
研究发现,C. minuta在健康个体的肠道微生物群中普遍存在,但在患有炎症性肠病(IBD)等特定疾病的个体中,其丰度显著降低。
这种分布的差异性提示了C. minuta在维持肠道健康和预防疾病方面可能具有重要作用。
越来越多的研究揭示了Christensenella minuta在人体健康和疾病中的重要作用。Christensenellaceae与多种代谢过程密切相关。研究表明,这种细菌在调节宿主的能量平衡、脂质代谢、脂多糖代谢、抗炎作用以及维持肠道屏障功能方面发挥着关键作用。
进一步的研究还发现,Christensenellaceae的丰度与肥胖、糖尿病、炎症性肠病等多种疾病的风险呈负相关。它还与健康衰老有关。在短短10年内,已经积累了大量证据表明,Christensenellaceae在许多疾病中显著减少。
Christensenellaceae与人类健康之间的关联

Ignatyeva O,et al.Front Microbiol.2024
▸ 肥胖患者中减少 ↓
肥胖是一种复杂的疾病,由体内过多的脂肪堆积引起,会对身体健康产生不利影响。许多非传染性疾病,如心血管疾病、各种癌症、2 型糖尿病、高血压和中风,以及精神健康问题都与肥胖有关。
研究认为,肥胖的病因与肠道菌群失调和先天性瘦素缺乏有关。已提出了几种机制将肥胖的发生与肠道菌群组成联系起来,这些机制是通过代谢和炎症活动的功能障碍实现的。
肥胖的发生涉及肠道菌群和宿主,是通过与近端器官的直接相互作用或通过代谢物分泌与肝脏、脂肪组织和大脑等远处器官的间接相互作用介导的。
在许多研究中,C. minuta被反复与其治疗性抗肥胖潜力联系在一起,这表明C. minuta在肠道微生物生态系统中的作用与宿主代谢的调节之间存在很强的相关性。C. minuta发挥治疗性抗肥胖作用的机制包括调节肠道上皮完整性、产生短链脂肪酸、改善脂质代谢和胆汁酸代谢。
Christensenella minuta抗肥胖作用的潜在机制
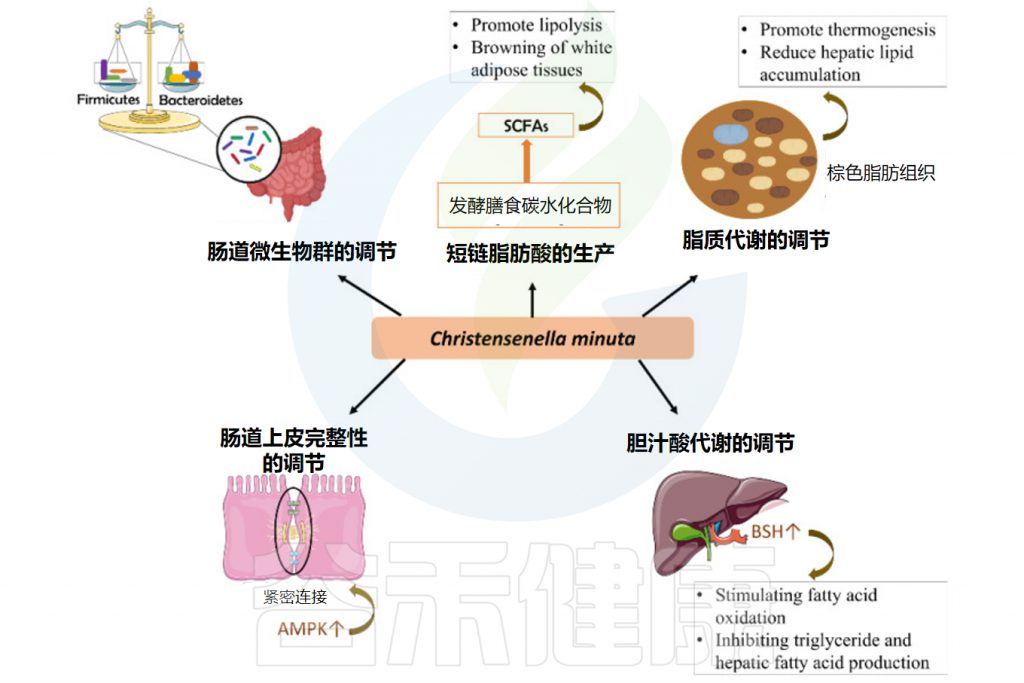
Ang WS,et al.Foods.2023
-产生短链脂肪酸来调节能量代谢和肠道稳态
研究发现C. minuta DSM 22607以5:1的比例产生高水平的乙酸盐和中等水平的丁酸盐,而不产生丙酸盐。
同时,在近端和远端结肠中均发现了低水平的支链脂肪酸,即异丁酸、异戊酸和异己酸,这表明C.minuta可降低细菌蛋白水解并刺激碳水化合物发酵。
肠道菌群通过短链脂肪酸(SCFA)产生参与调节能量代谢和肠道稳态。SCFA 被吸收并充当宿主体内葡萄糖和脂质代谢的能量来源或前体。SCFA可能通过G蛋白偶联受体,即游离脂肪酸受体2(FFAR2)和3(FFAR3)与结肠、肝脏、肌肉和脂肪组织相互作用。
-调节瘦素水平,减少脂肪的生成
此外,短链脂肪酸会上调抑制饥饿的瘦素合成,抑制脂肪生成并促进脂肪分解。研究评估了丁酸盐的施用可以通过促进脂肪细胞形成和脂肪组织褐变来减少能量摄入,增强脂肪氧化和能量消耗,从而治疗和预防肥胖。
这些由肠道微生物群产生的短链脂肪酸还与结肠粘膜中的肠内分泌细胞相互作用,诱导释放胰高血糖素样肽1(GLP-1)和肽YY(PYY)。这些激素进入体循环并对许多器官和组织发挥作用,最重要的是胃和胰腺。GLP-1和PYY共同防止胃排空过快,抑制酸分泌和运动,减缓胃肠道运输,从而导致食欲减少和食物摄入量减少。GLP-1还能刺激胰岛素分泌并防止胰腺β细胞衰竭。
–C.minuta改善了肠道上皮完整性,增强肠道屏障

此外,C. minuta合成的两种短链脂肪酸可能都参与维持肠上皮屏障。例如,乙酸和丁酸可通过与G蛋白偶联受体GPR43和GPR109A 结合来激活核苷酸结合寡聚化结构域3(NLRP3)炎症小体,这会增加IL-18的释放,促进上皮细胞的修复。
丁酸能稳定缺氧诱导因子(HIF),后者是屏障保护和组织再生的关键分子,它上调紧密连接蛋白,增加杯状细胞的黏蛋白生成,从而增强肠道屏障。
一项研究表明,C. minuta在体外和涉及高脂饮食组的动物研究中改善了肠道紧密连接蛋白(ZO-1)、闭合蛋白(OCLN)和紧密连接蛋白-1(CLDN1)的表达。
肠道通透性增加被认为是脂肪诱导性肥胖的一个驱动因素,与肠道菌群失调和肠道炎症有关。已证实肥胖小鼠的紧密连接减少,这表明肥胖是由肠道通透性增加和跨上皮阻力降低引起的。
肠道紧密连接蛋白下调可导致肠道渗漏,其中脂多糖细菌物质和其他炎症介质通过紧密连接扩散并与宿主免疫细胞相互作用,导致低度炎症、暴食并最终导致体重增加。
-调节甘油三酯和游离脂肪酸的积累
此外,与肥胖水平或体脂相关的身体质量指数(BMI)也与肠道中C.minuta的丰富程度密切相关。调节脂肪酸的合成和氧化以及抑制脂肪生成对体脂和体重有有利的影响。
早期的一项研究表明,肠道微生物群中C.minuta的存在与肥胖降低之间存在联系。一项针对C.minuta的干预研究表明,在饮食诱导的肥胖小鼠模型中,肝脏甘油三酯和游离脂肪酸的积累受到阻碍。这一发现与基因表达水平一致,在补充了C.minuta的动物模型中,编码肝脏葡萄糖激酶的gck基因受到强烈抑制。
葡萄糖激酶的过度表达促进了糖的过度吸收、肝脏脂质积累和棕色脂肪组织(BAT)中产热蛋白的下调,导致肥胖。从机制上讲,增强BAT中的脂肪组织产热作用并诱导白色脂肪组织(WAT)褐变可导致体重减轻。
-通过调节胆汁酸代谢来发挥抗肥胖能力
胆汁酸代谢对于调节葡萄糖和能量代谢、肠道完整性和免疫力至关重要。胆汁酸代谢的改变与肥胖密切相关。胆汁酸代谢通过刺激脂肪酸氧化和抑制甘油三酯和肝脏脂肪酸的产生。
C.minuta的抗肥胖潜力还通过结肠中的胆酸/牛磺胆酸(CA/TCA)比率来展现。这一发现意义重大,为抗肥胖与糖分解代谢以及初级胆汁酸的有效解离之间的关联提供了重要的理论基础。
C.minuta菌株 DSM33407和DSM22607在80%胆汁存在48小时的情况下均对胆汁酸具有高度耐受性。胆汁酸水解酶(BSH)基因已在两种C.minuta菌株中被鉴定,并且由于其水解结合胆汁酸的强能力而高度表达。
C.minuta还通过法尼醇x受体和G蛋白偶联胆汁酸受体(TGR5)促进胆汁酸代谢,而 TGR5 在肠道中高度表达。一些体内研究表明,肠道生态系统中BSH的高水平表达被认为是抗肥胖的关键调节因素,可显著降低体重、肥胖、循环低密度脂蛋白(LDL)胆固醇和甘油三酯。
注:BSH活性是肥胖控制的关键机制目标。利用具有高BSH活性的细菌菌株来丰富肠道微生物群可能是预防和控制肥胖的一种策略。
▸ 炎症性肠病中减少 ↓
炎症性肠病(IBD)是一种影响胃肠道的慢性炎症性疾病,主要有两种类型:克罗恩病和溃疡性结肠炎,每种类型都有不同的生理症状。研究发现,IBD患者的微生物组成会发生变化,其特征是厚壁菌门与拟杆菌门的比例下降。
-抑制NF-κB信号通路和IL-8来减轻结肠炎症
肠道菌群失调伴有短链脂肪酸组成的变化,随后是肠道屏障完整性的破坏,最终通过免疫系统调节引发炎症反应。尽管 IBD 的病因仍不太清楚,但研究表明,它们是由不受控制的炎症反应引发的,与白细胞介素8(IL-8)细胞因子和活性氧(ROS)的增加有关。
许多研究已经证明克罗恩病(CD)和溃疡性结肠炎(UC)患者的Christensenellaceae会减少。在发作前立即观察到静止期CD患者中Christensenellaceae的丰度显著下降,这可能表明它们在疾病进展中发挥了作用。在腹泻患者中也观察到了Christensenellaceae的丰度较低。
最近的体外和体内研究表明,C.minuta具有强效抗炎和免疫调节特性。C. minuta通过抑制 NF-κB 信号通路和促炎细胞因子IL-8的分泌来减轻结肠炎症。
-克罗恩病患者缺乏C.minuta

一项对C.minuta的介入研究表明,克罗恩病(CD)患者中缺乏Christensenella minuta(C.minuta),并且有记录证明它们会在人类上皮细胞中诱导抗炎作用,这支持了它们作为一种新型生物疗法的潜力。
在两种不同的急性结肠炎动物模型和一种人类肠道细胞系中,C.minuta限制结肠损伤、促进粘膜愈合并降低因炎症引起的中性粒细胞(特别是髓过氧化物酶和嗜酸性粒细胞过氧化物酶)的活化。
在C.minuta治疗动物模型中,肠道炎症的非侵入性生物标志人脂质运载蛋白-2(LCN-2)的浓度降低。在基因层面,携带克罗恩病风险基因 IL23R 的个体体内与C.minuta相关的微生物丰度降低,表明肠道微生物组受到宿主遗传学的影响。
据报道,当小鼠补充C.minuta时,IL23R 保护性编码变体会增加,从而预防克罗恩病。C.minuta还会产生丁酸,通过丁酸受体 GPR109a 来控制脂肪细胞、肠上皮细胞和免疫细胞中的炎症反应。
值得注意的是,C.minuta的抗炎功效已被证实与美沙拉嗪(也称为5-氨基水杨酸(5-ASA),一种用于治疗IBD的药物)相似。
▸ 2型糖尿病患者中减少 ↓
2型糖尿病(T2D)是一种复杂的代谢和内分泌功能障碍,其特征是胰岛素抵抗、胰腺β细胞功能障碍、低度全身炎症、肠道菌群失调、肥胖和其他内分泌疾病引起的高血糖。
中药是一种源自天然产物的补充药物,在治疗代谢综合征方面具有潜力。口服中药干预会影响肠道微生物群,但由于其活性成分(如黄酮类化合物)的亲脂性较差,因此生物利用度较低。肠道菌群的生物转化促进药物的吸收,这对药理学有重大影响。
-增加C.minuta有助于改善糖尿病
中药成分可能会调节宿主肠道菌群的数量。黄芪苓化散(HQLHS)由黄芪、灵芝、桦褐孔菌和苦瓜组成,是专门用于治疗2型糖尿病的中药复方。

最近的一项研究表明,在小鼠模型中,HQLHS抑制了致病菌并丰富了有益菌,特别是C.minuta和Christensenella timonensis。值得注意的是,该研究表明HQLHS显著增加了小鼠肠道菌群中Christensenella的相对丰度。该研究还描述了C.minuta对肝脏代谢的影响,为理解C.minuta在糖尿病治疗和控制中的药理机制奠定了基础。
-减少氧化应激、改善葡萄糖代谢
在同一项研究中,C. minuta DSM 22607 降低了糖尿病大鼠体内的氧化应激、色氨酸和酪氨酸等糖尿病诱因。抗氧化酶和脂质过氧化生物标志物 MDA 的水平也得到了控制。
C. minuta的抗糖尿病特性有多种机制,例如改善糖脂代谢、通过抑制肠道葡萄糖转运中SGLT1和GLUT2的表达来抑制葡萄糖吸收、促进GLP-1分泌以刺激胰岛素抵抗并调节葡萄糖稳态。
▸ 支气管哮喘和过敏性疾病中减少 ↓
-C.minuta丰度较高时患哮喘的可能性较低
小克里斯滕森氏菌(Christensenella minuta)还与支气管哮喘和过敏性疾病有关。研究发现,粪便微生物组中富含C. minuta的儿童患湿疹和对吸入性过敏原致敏的可能性较小。
值得注意的是,家庭环境中克里斯滕森菌科的丰富程度可能在支气管哮喘中发挥重要作用。分析了从健康儿童以及患有哮喘的儿童和成人家中收集的灰尘的宏基因组学谱。他们发现,克里斯滕森菌科在“健康”房屋的灰尘中显著过多,而在“哮喘”房屋的灰尘中却很少。
其他一些研究还发现:肾结石、情感障碍、甲状腺癌、粘膜类天疱疮、多囊卵巢综合征和复发性口疮性口炎也与Christensenellaceae科丰度较低有关。
▸ 过高在一些患者中可能有害 ↑
然而,有证据表明,Christensenellaceae科的丰度较高可能与一些病理之间存在联系。在一项研究中,Christensenellaceae科的丰度较高会增加重症监护病房中患有神经系统疾病的危重患者的死亡风险。
该分类单元在帕金森病患者中显著富集,尤其是在临床特征较差的患者中。在患有阿尔茨海默病、多发性硬化症和神经性厌食症的人中也观察到了Christensenellaceae科的丰富度增加。
Christensenellaceae科与健康表型或疾病之间关联




Ignatyeva O,et al.Front Microbiol.2024
C.minuta和Christensenellaceae科的有益作用可能归因于它们与肠道中许多其他细菌群落相互作用的特殊能力。除了直接作用外,C.minuta还可以通过促进或限制某些分类群的生长来间接影响宿主。
-与Christensenellaceae正相关的菌群
Christensenellaceae与许多菌群呈正相关,包括:
颤螺菌属(Oscillospira)
瘤胃球菌属(Ruminococcus)
粪球菌属(Coprococcus)
普雷沃氏菌属(Prevotella)
嗜粘蛋白-阿克曼氏菌(Akkermansia muciniphila)
罗氏菌属(Roseburia)
-与Christensenellaceae负相关的菌群
相反,几个属与Christensenellaceae呈负相关:
克雷伯氏菌属(Klebsiella)
链球菌属(Streptococcus)
梭杆菌属(Fusobacterium)
经黏液真杆菌属(Blautia)
Magamonas
▸ 能够促进一些有益菌的生长
此外, Christensenellaceae丰度越高,微生物丰富度和多样性就越高。值得注意的是,几种与之呈正相关的菌群已被提议作为新一代益生菌(Oscillospira、Roseburia)或目前已经是这种身份(Akkermansia)。
–C.minuta能够促进双歧杆菌生长
C.minuta能够促进双歧杆菌属的生长,这是通过产生代谢产物如短链脂肪酸(SCFAs),特别是乙酸和丁酸,这些SCFAs是双歧杆菌属的重要能量来源。这种交叉喂养关系有助于双歧杆菌属在肠道中的定植和增殖。
–C.minuta塑造适合乳酸杆菌生长的环境
C.minuta可能通过产生乳酸来降低肠道pH值,从而为乳酸杆菌属创造一个更适宜的生长环境。此外,C.minuta产生的代谢产物可能直接或间接地激活乳酸杆菌属的代谢途径,增强其在肠道中的竞争力。
–C.minuta促进普拉梭菌生长
Faecalibacterium prausnitzii是一种潜在的下一代益生菌,具有高丁酸生产、抗炎和预防肠道病原体的作用。
研究发现,Christensenella minuta通过产生外源乙酸、半胱氨酸、脯氨酸和赖氨酸来交叉喂养F.prausnitzii,这些都是F. prausnitzii发酵和繁殖所必需的。从而促进了Faecalibacterium prausnitzii的增殖。
–C.minuta与粪肠球菌
C.minuta与F.prausnitzii之间存在正向的相互作用。C.minuta产生的代谢产物可能作为F.prausnitzii的底物,促进其生长和SCFA的生产,特别是丁酸盐,这是一种对肠道健康至关重要的短链脂肪酸。
▸ 减少一些有害菌群
有趣的是,在存在Christensenellaceae的情况下通常会减少几种有害菌群,例如机会性病原体,已知会导致人类和动物感染的克雷伯氏菌和链球菌,以及可能导致癌症的微生物梭杆菌。
因此,我们假设C. minuta可以通过支持有益物种的生长和抑制潜在有害物种来调节肠道微生物群。
–C.minuta抑制克雷伯菌的定植
C.minuta可能通过竞争营养物质或产生抗菌物质来抑制K.pneumoniae等潜在的病原菌。这种竞争和抑制作用有助于维持肠道微生物群的平衡,防止病原菌的过度生长。
–C.minuta限制大肠杆菌的增殖
C.minuta可能通过调节肠道中的氧化还原电位来影响大肠杆菌的生长。由于C.minuta是严格的厌氧菌,它可能通过降低肠道的氧化还原电位来限制需氧菌如大肠杆菌的增殖。
▸ 与甲烷杆菌存在互作并可能影响体重
不同细菌种属很可能通过代谢物转移进行相互作用。Christensenellaceae科和甲烷杆菌科的相互作用可能影响体重指数并呈负相关。

对来自10项独立研究的1821个样本进行了荟萃分析,证实了在科水平(Christensenellaceae科和甲烷杆菌科)和种水平(C.minuta和Methanobrevibacter smithii)之间均存在很强的正相关性。
此外,发现了它们之间的物理和代谢相互作用。作为一种氢气生产者,C.minuta有效地支持了依赖氢气供应的M.smithii的生长。在共同培养时,C.minuta释放的氢气量足以确保M.smithii的生存力,与氢气过量的单一培养中相当。
反过来,M.smithii也能调节C.minuta的代谢,导致短链脂肪酸的产生从丁酸转向乙酸。根据观察到的乙酸盐产量的增加。除此之外,甲烷杆菌科的甲烷生产会导致碳损失和宿主可用能量减少,这可能部分解释了该细菌与体重减轻之间的关联。
Christensenellaceae可能是一种高效益生菌药物的来源,可使许多患者群体受益,尤其是那些患有代谢紊乱和炎症性胃肠道疾病的患者。我们在下面总结了C.minuta作为益生菌在人体健康中的一些作用。
▸ 抗炎作用
C.minuta益生菌活性的所有潜在机制尚未完全了解;不过,已经取得了许多积极的进展。在一系列体外和体内实验中测试了C. minuta DSM 22607。首先,该细菌及其上清液均表现出强大的抗炎潜力,因为它们能够限制HT-29细胞中的IL-8产生,此外发现上清液还能抑制NF-kB信号通路。
从9位捐献者身上分离并测试了 32 株新的C.minuta菌株,以确定最佳候选益生菌药物。他们在一系列实验中分析了这些菌株的抗炎和保护特性,并选出了5种主要候选菌株。五种候选菌株均在体外细胞模型中阻止TNF-α刺激后的NF-kB通路激活并诱导IL-10的产生。
在动物模型中,五种菌株中的两种显著改善了TNBC引起的炎症病变,并具有明显的局部抗炎作用。
此外,还证明了C.minuta菌株在体外模型中刺激人源 PBMC 产生 IL-10的能力。
▸ 保护肠道屏障
其次,C.minuta还显示出保护 TNF-α 受损的 Caco-2 细胞中肠道屏障的能力。这些结果在二硝基苯磺酸(DNBS)和三硝基苯磺酸(TNBS)诱发的结肠炎小鼠模型中得到了证实。
在这两项实验中,C.minuta表现出独特的抗炎特性,保护结肠组织的效果与5-氨基水杨酸(5-ASA)一样有效。该细菌减少了宏观和微观化学损伤,减少了结肠中的免疫细胞浸润(ICI),限制了氧化应激,并降低了促炎细胞因子的分泌和脂质运载蛋白-2的表达。
▸ 能够产生乙酸盐、丁酸盐
实验还揭示了C. minuta的代谢作用,特别是其产生大量乙酸盐和适量丁酸盐的能力。据报道, C.minuta可以同时产生短链脂肪酸(SCFA)中的乙酸盐和丁酸盐,而大多数微生物只能产生丁酸盐或乙酸盐其中一种。
▸ 抗肥胖能力
对C.minuta DSM22607 的抗肥胖能力进行了研究。他们发现,每天施用2×10^9个C. minuta菌落形成单位(CFU)可防止喂食高脂饮食(HFD)的小鼠体重增加和高血糖,但不影响它们的食物摄入量。
令人惊讶的是,食用益生菌菌株的动物和喂食正常食物的动物在体重增加方面没有统计学上的显著差异;然而,接受载体的HFD喂养小鼠体重增加明显且快速。这强烈表明C.minuta通过改变新陈代谢而不是影响进食行为来限制脂肪堆积。
这些发现与对血清代谢标志物的观察结果相关,即高脂饮食小鼠的瘦素和抵抗素水平下降。C.minuta可能破坏了肝脏脂肪生成,这通过编码葡萄糖激酶的Gck基因表达降低来证明。
此外,益生菌菌株通过上调编码主要紧密连接蛋白的Ocln和Zo1基因,对肠道通透性具有强大的保护作用。这也可能有助于C. minuta通过限制由肠漏引起的全身炎症而发挥抗肥胖作用。
▸ 改善糖尿病
进一步证实了C. minuta的有益作用及其在代谢过程中的关键作用的证据。使用两种Christensenella属菌株(C.minuta DSM 22607 和C.timonesis DSM 102800)治疗小鼠2型糖尿病,两种菌株均改善了许多代谢指标。
管饲益生菌可降低血糖水平、限制氧化应激、促进受损胰岛和肝细胞的修复,并抑制肝脏和结肠中几种促炎细胞因子和 TLR4 的表达。
重要的是,用C.minuta和C.timonesis治疗还上调结肠中的Zonula occludens-1和Claudin-1,从而加强肠道屏障。测试对象血清脂多糖水平下降支持了这一发现。这两种菌株还通过刺激胰高血糖素原的表达、增加血清胰高血糖素样肽-1(GLP-1)水平和限制肝糖异生,对代谢产生了重大影响。
总体而言,C.minuta和C.timonesis改善了2型糖尿病的代谢过程并减轻了炎症反应。
▸ 免疫调节作用
C.minuta能够通过调节肠道微环境影响宿主的免疫系统。研究表明,C. minuta能够促进Th17细胞的分化,这些细胞在维持肠道免疫耐受和防御病原体入侵方面发挥着关键作用。
此外,C.minuta还可能通过与其他肠道共生菌的复杂相互作用,间接影响宿主的免疫系统。肠道微生物群落之间的相互作用是复杂的,C.minuta通过其代谢产物和分泌物,可以影响其他菌群的生长和功能,进而调节宿主的免疫反应。这种间接调节机制为宿主免疫系统提供了额外的调节途径。
最后,C.minuta对宿主免疫系统的调节作用可能还与其在肠道中的定植能力有关。研究表明,C.minuta在人体肠道中具有较高的遗传性,这意味着它能够有效地定植并长期存在于宿主肠道中。这种定植能力可能是C. minuta发挥其免疫调节作用的基础。
▸ 增加有益菌丰度,改善肠道菌群组成
此外,Christensenella菌株通过增加许多有益微生物(如双歧杆菌和Phascolarctobacterium)的丰度来改变肠道微生物群的组成。
06
调节C.minuta的策略
调节C.minuta的策略可以通过多种途径实现,包括饮食调节、益生菌和益生元的使用、药物治疗以及功能性食品的摄入。
▸ 通过饮食调节C.minuta的策略
调节肠道共生菌C.minuta的一种有效方法是通过饮食。
研究表明,某些食物成分可以促进C.minuta的生长和活性。例如,高纤维食物,如全谷物、豆类、坚果和水果,可以作为益生元,为C.minuta提供必要的营养物质。这些纤维在肠道中被微生物发酵,产生短链脂肪酸,这些物质对C.minuta的生长至关重要。
此外,一些特定的益生元,如低聚果糖和菊粉,已被证明能够特异性地增加C.minuta的数量。
▸ 益生元和合生元的应用
益生元是指能够促进肠道内有益菌生长的非消化性食品成分,而合生元则是益生元和益生菌的组合。在调节C.minuta中,益生元和合生元的应用是两个重要的策略。
益生元如多糖、半纤维素、果胶等,能够通过刺激C.minuta的生长,增强其在肠道中的竞争力。合生元则结合了益生元和益生菌的双重优势,通过提供C.minuta所需的营养物质和直接补充C.minuta,更有效地调节肠道微生物群。
▸ 药物和功能性食品的作用
在某些情况下,药物和功能性食品也可以用于调节C.minuta。例如,某些抗生素可以在必要时用来减少有害菌的数量,为C.minuta提供更好的生长环境。
功能性食品,如含有特定益生菌的酸奶或补充剂,可以直接补充C.minuta,增加其在肠道中的数量。此外,一些植物提取物和天然化合物也被研究用于调节肠道微生物群,包括C.minuta。
主要参考文献
Ignatyeva O, Tolyneva D, Kovalyov A, Matkava L, Terekhov M, Kashtanova D, Zagainova A, Ivanov M, Yudin V, Makarov V, Keskinov A, Kraevoy S, Yudin S. Christensenella minuta, a new candidate next-generation probiotic: current evidence and future trajectories. Front Microbiol. 2024 Jan 11;14:1241259.
Ang WS, Law JW, Letchumanan V, Hong KW, Wong SH, Ab Mutalib NS, Chan KG, Lee LH, Tan LT. A Keystone Gut Bacterium Christensenella minuta-A Potential Biotherapeutic Agent for Obesity and Associated Metabolic Diseases. Foods. 2023 Jun 26;12(13):2485.
Pan T, Zheng S, Zheng W, Shi C, Ning K, Zhang Q, Xie Y, Xiang H, Xie Q. Christensenella regulated by Huang-Qi-Ling-Hua-San is a key factor by which to improve type 2 diabetes. Front Microbiol. 2022 Oct 12;13:1022403.
Mazier W, Le Corf K, Martinez C, Tudela H, Kissi D, Kropp C, Coubard C, Soto M, Elustondo F, Rawadi G, Claus SP. A New Strain of Christensenella minuta as a Potential Biotherapy for Obesity and Associated Metabolic Diseases. Cells. 2021 Apr 6;10(4):823.
Relizani K, Le Corf K, Kropp C, Martin-Rosique R, Kissi D, Déjean G, Bruno L, Martinez C, Rawadi G, Elustondo F, Mazier W, Claus SP. Selection of a novel strain of Christensenella minuta as a future biotherapy for Crohn’s disease. Sci Rep. 2022 Apr 11;12(1):6017.
Waters JL, Ley RE. The human gut bacteria Christensenellaceae are widespread, heritable, and associated with health. BMC Biol. 2019 Oct 28;17(1):83.
Pető, Á.; Kósa, D.; Szilvássy, Z.; Fehér, P.; Ujhelyi, Z.; Kovács, G.; Német, I.; Pócsi, I.; Bácskay, I. Scientific and Pharmaceutical Aspects of Christensenella minuta, a Promising Next-Generation Probiotic. Fermentation 2023, 9, 767.
Xu C, Jiang H, Feng LJ, Jiang MZ, Wang YL, Liu SJ. Christensenella minuta interacts with multiple gut bacteria. Front Microbiol. 2024 Feb 19;15:1301073.

谷禾健康

随着技术的发展,高通量测序技术已成为研究微生物群落的重要工具。这种技术使得科学家们能够解析巨量微生物DNA序列,从而获得丰富的微生物组数据,包括16S rRNA基因、ITS序列和宏基因组。然而,这些数据只是迈向揭示微生物群落复杂性的第一步。
通过对环境样本的可变区域如16S、18S、ITS序列进行高通量测序获得的原始序列数据,再对其进行聚类,数据分析,统计学差异比较等得到微生物多样性分析报告。那么,什么是微生物群落多样性?
微生物群落多样性(Microbial Community Diversity)是指在特定环境中存在的微生物种类的数量和分布情况,它不仅包含不同种类微生物的丰度,还包括它们之间的相互关系。多样性可以从不同角度进行评价,主要分为以下几种:
α多样性(Alpha Diversity): 这是衡量某一特定样本内部多样性的一种指标。常用的α多样性指标包括物种丰富度(Species Richness)、香农指数(Shannon Index)和辛普森指数(Simpson Index)。这些指标可以帮助我们了解样本内部的复杂性和均一性。
β多样性(Beta Diversity):不同样本之间的多样性比较被称为β多样性。常用的β多样性指标包括Bray-Curtis距离和Jaccard指数,通过这些指标可以探索样本之间的相似性和差异性,揭示不同环境或条件下微生物群落的变化模式。
γ多样性(Gamma Diversity): 这是指在一个更大尺度、多个样本的总体多样性,通常用以评估一个较大区域的整体多样性水平。
为理解这些多样性指标,我们可以借助一些简单的比喻来形象解释。例如,α多样性就像是在观察一个花园的花卉种类和数量;β多样性则是比较不同的花园之间的相似性或不同之处;而γ多样性则是对一个城市中所有花园的总览评价。
在接下来的部分中,我们将深入探讨这些多样性指标的详细内涵,以及从多个角度展示如何通过高通量测序技术解析微生物群落中的这些多样性规律。
下图是实验上机测序流程,提取的样本总DNA经过质检、PCR扩增、建库等步骤进行高通量测序得到测序原始数据。

原始数据经过Reads拼接、tags过滤、去嵌合体等步骤得到有效数据clean data。在特定的相似度下进行聚类得到OTU/ASV,报告中通过降噪方法得到ASV表,一切后续分析都围绕ASV表来进行。根据ASV表可以继续做物种分类注释、丰度计算、多样性分析、差异分析、功能预测等。所以ASV特征表是微生物多样性分析中关键数据结果。
OTU和ASV的区别
OTU和ASV是微生物组学中用来表示微生物多样性的两个不同概念。两者都是从环境样本中获得的DNA序列数据,通过一定的分析方法分类得到的用于表示微生物种类或种群的单位。它们之间的主要区别在于定义的精确度和建立的方法。
– OTU(Operational Taxonomic Units):
OTU是一种将序列通过相似度聚类的传统方式,来表示相似序列组成的种群。通常,这种聚类方法会将序列之间相似度达到97%(或其他设定的阈值)的序列分到同一个OTU。OTU聚类通常不考虑序列中的单个变异位点,而是基于整体相似度。
由于使用阈值聚类,OTU不能准确反映序列之间的实际差异,可能会将生态学意义上不同的微生物序列归为一个OTU。OTU分析可能过于简化,有时无法捕获低水平的微生物多样性。
– ASV(Amplicon Sequence Variants):
ASV采用较新的降噪方法,可以精确地解析序列中的每一个核苷酸差异,简单来说就是以100%相似度进行聚类,对低质量序列进行去除和校正,这种方法可以生成“零半径OTU”,即互不相同的基于序列的变体。
ASV通常使用误差校正算法来排除测序错误,从而提供更精确的序列变体识别。ASV方法对单一核苷酸变异敏感而能提供更细粒度的微生物多样性解析。ASV为每一种变异提供更一致、可复制的标识符,这在比较不同研究之间的微生物群落组成时非常有用。
简而言之,ASV方法提供了比OTU更高分辨率、更精准的序列变体检测。换句话说,ASV提供了一种微生物组多样性分析的“高清”视角,它更可能捕捉到微生物群落内变化的微妙差异,尤其是在不同环境或时间点间的比较中。
原始序列数据(raw tags)经过质控、过滤、去嵌合体,最终得到有效数据(effective tags)。再对有效数据进行UNOISE降噪处理,得到ASV特征表。数据处理过程中各步骤得到的序列进行途径统计,可以直观的反映每个样本的数据量和物种丰度。
文件目录:
01_pick_otu/summary/sumOTUPerSample.txt
raw-tags:每个样本的原始序列数据;
singleton :每个样本中无完全匹配的单条序列的数量。singleton ASV 是指只有单条代表序列的 ASV,可能由于测序错误,或者是来自于PCR过程中产生的嵌合体;
tagsmatchedASVs: 每个样本中比对到ASVs的最终有效序列数据 及其比例,聚类的同时vsearch会根据UCHIME算法将singleton ASV及嵌合体去除,得到最终的有效序列数据 Effective Tags;
ASVs:每个样本的ASVs数量。
一般文献中的测序原始数据量raw-tags 要求达到3万条以上,可以满足数据分析的基本要求。绝大多数文献数据量平均在5万条左右。世面上不同公司承诺的数据指标有所不同,谷禾测序得到的原始数据一般可以达到10万 reads左右,足够满足当前文章发表要求的参考数据量。
若原始数据量低于1万条,尤其是少于3000条reads以下,则很有可能受环境污染的杂带较多,建议重新上机补测数据。ASVS列可以反映每个样本的物种多样性,一般一个ASVs就代表一个物种。因此可以用ASV数量来代表物种数量。将每个样本的有效原始数量和ASVs数据可视化做成柱状图,可以更直观的观察每个样本/分组数据量的变化。
每个样本/分组可能会有一些共有的和独有的ASV,通常用韦恩图或花瓣图表示(样本数/分组数<=5个样本用维恩图,数量大于5出花瓣图)。除了用Venn图将几个数据集之间的交集进行可视化,还可以使用upset图表示。
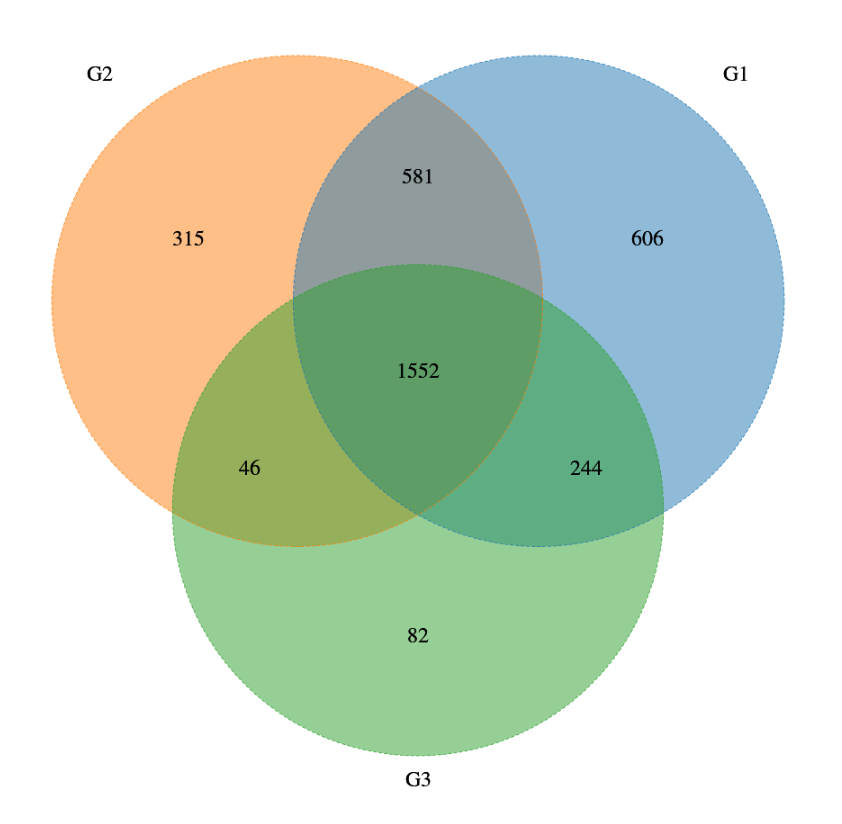
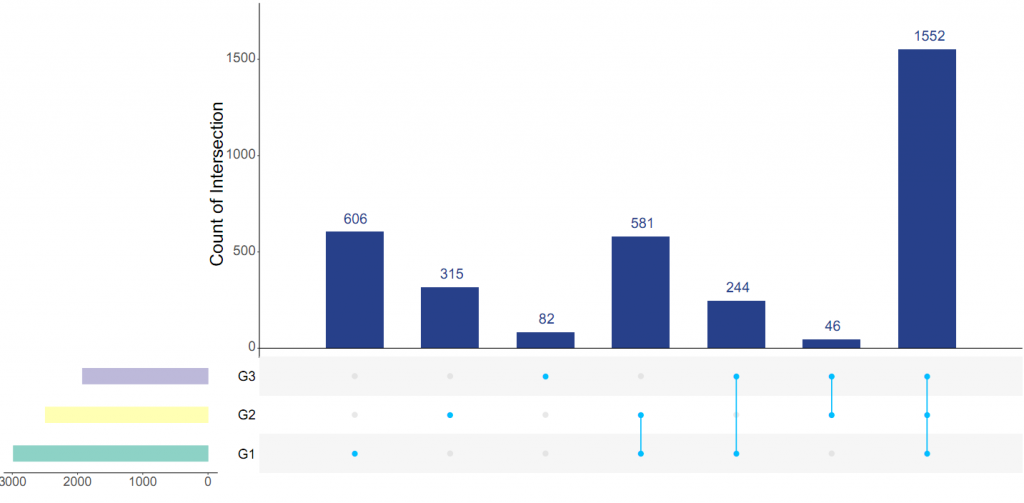
韦恩图中不同颜色的圆圈代表一个样本/分组,圈之间的重迭区域表示样本/分组间共有的ASVs,每个区域的数字大小表示该区域对应的ASVs数目。
UpSet图主要包含三个部分:上部分为各个分组独有和共有的ASV数量,下部分为各个分组独有和共有的分类情况,左部分每一个行代表一个分组。
alpha多样性主要用来衡量单个样本内的菌群多样性,不涉及样本之间的比较。alpha多样性与两个因素相关,分别是:一、丰富度(richness),二、多样性(diversity)。
丰富度指的是单个样本物种的种类数目;而多样性是指菌群在个体中分配的均匀度。样本的丰度高不一定就代表菌群的多样性丰富,丰度高如果是因为里边含有较多低丰度的杂带,这些可能是来源于环境的污染物导致的,这些低丰度的物种并不会使菌群的多样性增加。
alpha多样性有三类相关指数,其中包括菌群丰度指数(Chao1和ACE)、菌群多样性指数(shannon和simpson)和测序深度指数(Goods coverage和Observed spieces)。
Chao1:Chao1算法用于评估样本中所含ASV数目的指数,Chao1在生态学中常用来估计物种总数,由Chao(1984)最早提出。通过计算群落中只检测到1次和2次的ASV数估计群落中实际存在的物种数。chao1指数可以评估一个样本中的ASV数量,chao指数越大,ASV数目越多,说明该样本物种数越多。
计算公式如下:
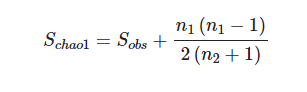
编辑
其中:
Schao1=估计的OTU数;
Sobs=观测到的OTU数;
n1=只有一条序列的OTU数目(如“singletons”);
n2=只有两条序列的OTU数目(如“doubletons”)。
ACE:用来估计群落中含有ASV数目的指数,由Chao提出,是生态学中估计物种总数的常用指数之一,与Chao1的算法不同。预设将序列量10以下的ASV都计算在内,从而估计群落中实际存在的物种数。
计算公式如下:
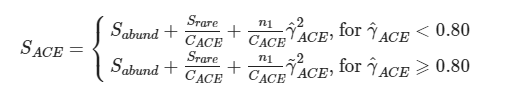
其中
ni=含有i条序列的ASV数目;
Srare=含有“abund”条序列或者少于“abund”的OTU数目;
Sabund=多于“abund”条序列的OTU数目;
abund=被视为“优势”的ASV的阈值,默认为10。
Shannon:香农-威纳指数综合考虑了群落的丰富度和均匀度,是用来评估样本中微生物多样性指数之一。Shannon指数值越高,表明群落的多样性越高。
计算公式如下:
其中:
Sobs=观测到的ASV数目;
ni=含有i条序列的ASV数目;
N=所有的序列数。
Simpson:辛普森多样性指数对菌群多样性评估,Simpson指数值越高,表明群落多样性越高。由EdwardHugh Simpson(1949)提出,在生态学中常用来定量描述一个区域的生物多样性。一般而言,Shannon指数侧重对群落的丰富度以及稀有ASV,而Simpson指数侧重均匀度和群落中的优势ASV。
计算公式一如下:
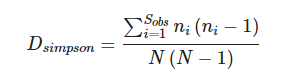
计算公式二如下:
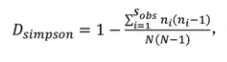
此时,Simpson指数越大,说明群落多样性越大。报告中用到的是计算公式二。
其中:
Sobs=观测到的ASV数目;
ni=含有i条序列的ASV数目;
N=所有的序列数。
Coverage:是指各样品克隆文库的覆盖率,其数值越高,则样品中序列被测出的概率越高,而没有被测出的概率越低。该指数反映本次测序结果是否代表了样品中微生物的真实情况。
计算公式如下:
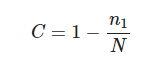
其中:
n1=只含有1条序列的ASV数目;
N=所有的序列数。
下表统计了每个样本的各项alpha多样性指标:
结果目录:
03_diversity-metrics/alpha/alpha_div.txt
可以选择不同的alpha多样性指数进行显著性差异比较,一般常用丰富度指数Chao1,多样性指数Shannon、simpson,比较不同组间指数是否有显著差异。Alpha多样性分析将样本的菌群群整体研究并转换为具体的指数与p值,来说明群落的变化与差异。
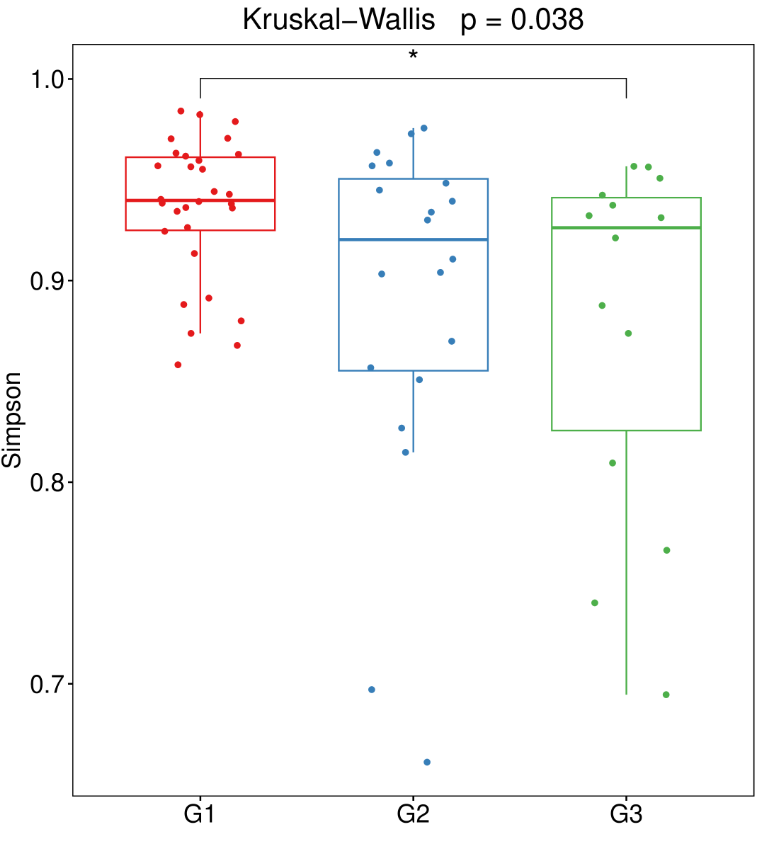
•稀释性曲线(Rarefaction curve)
稀释曲线是从每个样本中随机抽取一定数量的序列,统计这些序列所代表的ASV数目,以随机抽取的序列数与ASV数量来构建曲线。可以用来比较不同样本中的物种多样性,也可以用来说明样本出测序数据量是否足以反映环境中的物种多样性。
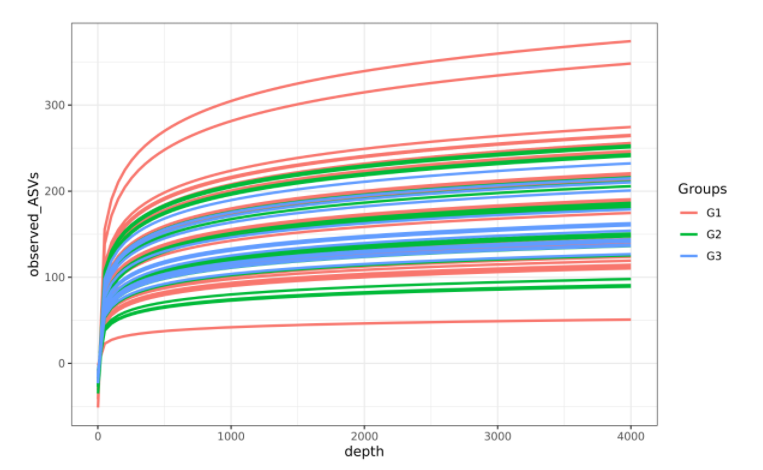
•
菌群多样性指数(shannon和simpson)
丰度等级曲线(Rank abundance curve)是分析多样性的一种方式。构建方法是统计单一样品中,每一个OTU所含的序列数,将OTU按丰度(所含有的序列条数)由大到小等级排序,再以OTU等级为横坐标,以每个OTU中所含的序列数(也可用OTU中序列数的相对百分含量)为纵坐标做图。
Rank-abundance曲线可用来解释多样性的两个方面,即物种丰度和物种均匀度。在水平方向,物种的丰度由曲线的宽度来反映,物种的丰度越高,曲线在横轴上的范围越大;曲线的形状(平滑程度)反映了样品中物种的均度,曲线越平缓,物种分布越均匀。
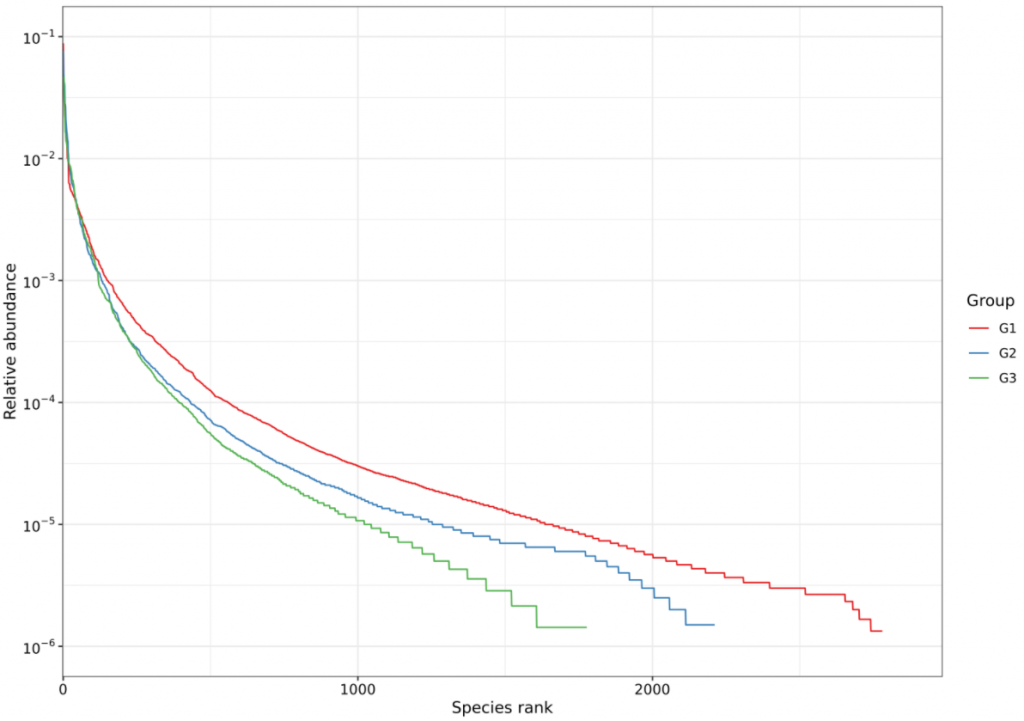
Beta多样性指的是样本间多样性,Beta多样性是衡量个体间菌落构成相似度的一个指标。通过计算样本间距离可以获得beta多样性距离矩阵,Beta多样性计算主要基于OTU的群落比较方法,有欧式距离、bray curtis距离等,这些方法优势在于算法简单,考虑物种丰度(有无)和均度(相对丰度),但其没有考虑OTUs之间的进化关系,认为OTU之间不存在进化上的联系,每个OTU间的关系平等。
另一种算法Unifrac距离法,是根据系统发生树进行比较,并根据16s的序列信息对OTU进行进化树分类,因此不同OTU之间的距离实际上有“远近”之分。而其他距离算法认为OTU之间的关系是平等的。Unifrac距离分为加权距离和非加权距离。
1
欧式距离(Euclidean distance):
欧几里得距离是空间中两点间“普通”(即直线)距离。
2
Bray-Curtis距离:
Bray-Curtis距离是生态学中用来衡量不同样地物种组成差异的测度。由J. Roger Bray and John T. Curtis 提出。其计算基于样本中不同物种组成的数量特征(多度,盖度,重要值等)。
计算公式为:
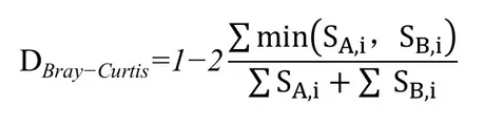
SA,i=表示A样本中第i个OTU所含的序列数;
SB,i=表示B样本中第i个OTU所含的序列数。
3
Unweighted UniFrac距离:
非加权距离包含特征之间的系统发育关系的群落差异定性度量。
4
Weighted UniFrac距离:
加权距离包含特征之间的系统发育关系的群落差异定量度量。
两者的区别在于:Weighted Unifrac 距离是一种同时考虑各样品中微生物的进化关系和物种的相对丰度,计算样品的距离,而Unweighted Unifrac则只考虑物种的有无,忽略物种间的相对丰度差异。
一般采用PCA、PCoA、NMDS等进行图像化展示,区分样本间的菌群组成差异。其原理是利用降维思想把样本平铺到二维平面上,使得相似的样品距离相近,相异的样品距离较远。
PCA图是基于ASV table的欧式距离,PCoA是基于两两样品之间的距离矩阵(有Bray-Curtis距离、加权距离、非加权距离),基于距离矩阵的统计检验方法有ANOSIM相似性分析和Adonis多元方差分析。
Anosim分析是一种非参数检验,用来检验组间差异是否显著大于组内差异,从而判断分组是否有意义。对 Anosim 的分析结果,基于两两样本之间的距离值排序获得的秩(组间的为 between,组内的为 within),这样任一两两组的比较可以获得三个分类的数据,并进行箱线图的展示(若两个箱的凹槽互不重迭,则表明它们的中位数有显著差异)。
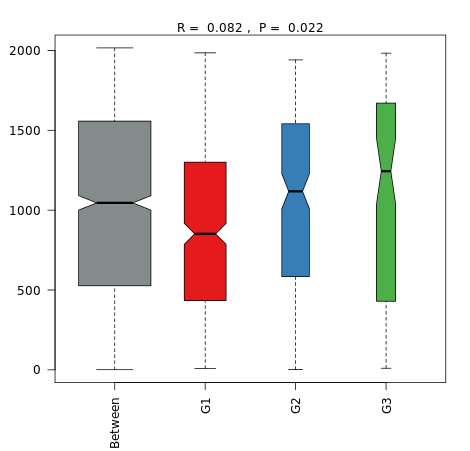
该方法主要有两个数值结果:R值,用于比较不同组间是否存在差异;P值,用于说明是否有显著差异。
R-value 介于(-1,1)之间,R-value > 0,说明组间差异大于组内差异。R-value < 0,说明组间差异小于组内差异, R只是组间是否有差异的数值表示,并不提供显著性说明。统计分析的可信度用 P-value 表示,P< 0.05 表示统计具有显著性。
Adonis检验,多元方差分析,其实就是PERMANOVA,亦可称为非参数多元方差分析。其原理是利用距离矩阵(比如基于Bray-Curtis距离、Unifrac距离)对总方差进行分解,分析不同分组因素对样品差异的解释度,并使用置换检验对其统计学意义进行显著性分析。它与Anosim的用途相似,也能够给出不同分组因素对样品差异的解释度(R值)与分组显著性(P值)。
PCA(Principal Components Analysis)即主成分分析,首先利用线性变换,将数据变换到一个新的坐标系统中;然后再利用降维的思想,使得任何数据投影的第一大方差在第一个坐标(称为第一主成分)上,第二大方差在第二个坐标(第二主成分)上。
这种降维的思想首先减少数据集的维数,同时还保持数据集的对方差贡献最大的特征,最终使数据直观呈现在二维坐标系。经过一系列的特征值和特征向量进行排序后,选取PCA分析得到的前三个主成分(PC1、PC2和PC3)中的任意两个数据作图。通过PCA 可以观察个体或群体间的差异。
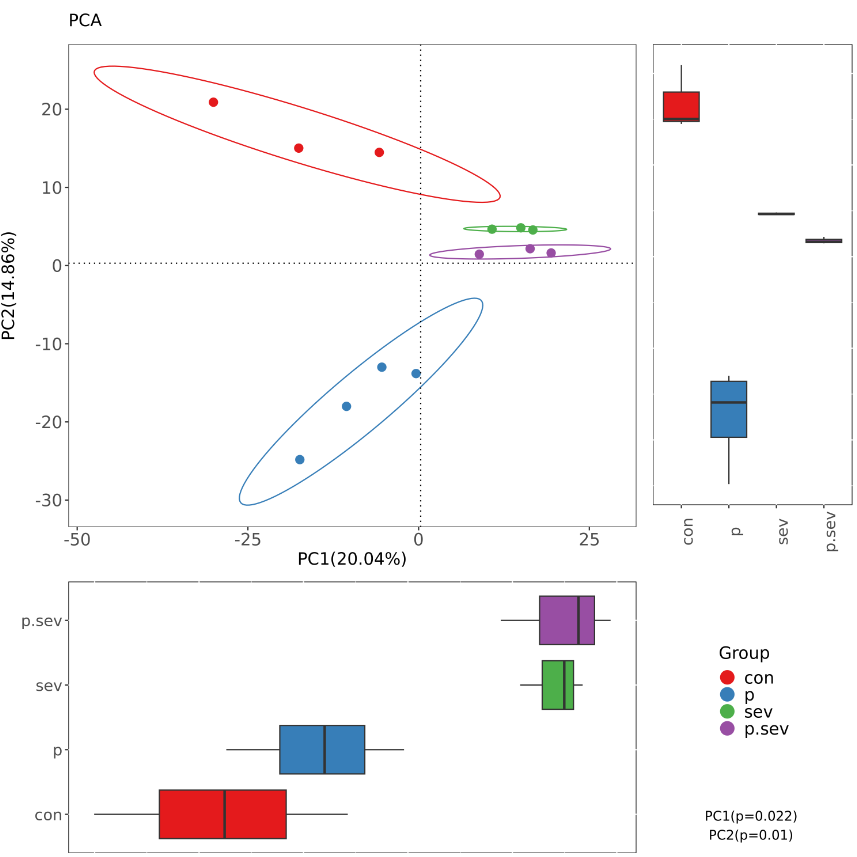
主坐标分析 PCoA (Principal component analysis)是一种非约束性的数据降维分析方法,可用来研究样本群落组成的相似性或相异性。通过 PCoA 可以观察个体>或群体间的差异。
它与PCA类似,两者的区别为PCA是基于样本的相似系数矩阵(如欧式距离)来寻找主成分,而PCoA是基于距离矩阵(欧式距离以外的其他距离)来寻找主坐标。我们基于Bray-Curtis 距离、 Weighted Unifrac 距离和Unweighted Unifrac 距离来进行 PCoA 分析。
该图是基于Bray-Curtis距离做的PCoA图,图中右下角的P值就是基于Adonis检验得到的结果:
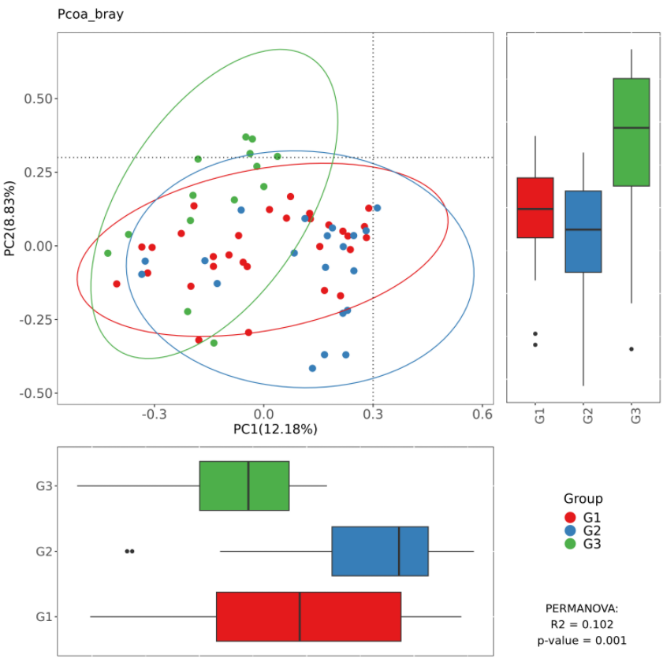
编辑
非度量多维尺度分析 NMDS 分析(Nonmetric Multidimensional Scaling)与上述 PcoA 分析类似,也是一种基于样本距离矩阵的分析方法,通过降维处理展现样本特定的距离分布。
与 PcoA 的区别是 NMDS 分析不依赖于特征根和特征向量的计算,而是通过对样本距离进行等级排序,使样本在低维空间中的排序尽可能符合彼此之间的距离远近关系(而非确切距离数值)。因此,NMDS 分析不受样本距离的数值影响,对于结构复杂的数据排序结果可能更稳定。