-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康
前面的文章中,我们已经了解到,在肝病的发生发展中肠道菌群的变化,详见:
深度解析 | 肠道菌群与慢性肝病,肝癌
到目前为止,大多数研究都集中在细菌多样性及其代谢物与靶癌细胞表型的关联上,而没有考虑环境的微调。实际上生态系统中,个体在环境中与其他个体相互作用、相互影响和限制。
在肿瘤微环境中,特定的微生物会影响其他细胞(微生物或宿主细胞)并受其影响。
微生物群及其代谢物影响肠-肝轴的组织细胞。肠道微生物群以及肿瘤本身的细菌可以影响肿瘤微环境,包括通过调节癌症、基质和炎症/免疫细胞中的基因转录以及促进或抑制肿瘤进展。
同时,微生物群也受饮食、环境等因素影响,在环境扰动后会适应,从而影响宿主-微生物的相互作用。
本文主要阐述了微生物群在肝脏稳态中的作用,肠道菌群及其代谢物直接和间接地调节肝脏基因表达,导致肠-肝轴失衡,从而促使肝病的发生发展,甚至致癌,并对肿瘤微环境产生影响。
我们的健康和生存能力取决于共生微生物(微生物群)的存在,它们主要存在于上皮细胞界面上,在下消化道中特别丰富。
菌群维持肠道稳态
肠道微生物群中的平衡生态有助于食物加工和吸收,调节宿主新陈代谢,并通过防止病原体和病原菌的扩张或通过调节宿主免疫力和维持肠道上皮的完整性来防止感染。
肠道-肝脏-胰腺轴
在物理上,肝脏和胰腺通过胆管和胰管与肠道相通,而门静脉将肠道菌群产物输送到肝脏。因此,肠道菌群与肝脏和胰腺之间的串扰(肠道-肝脏-胰腺轴)可以将信号整合为一个相互关联的系统。
宿主与微生物群之间复杂而高度协调的相互作用代表了一个自然生态系统。共生相互作用,如互利共生、共栖、捕食、寄生和竞争,是微生物、微生物-宿主和宿主-宿主细胞之间相互作用的基础。
因此,肠-肝轴中的细胞处于稳态平衡,环境扰动会改变这种平衡,从而调节局部和全身的转录反应,并影响健康和疾病,例如癌症。
图1 微生物与宿主的相互作用调节体内平衡和疾病
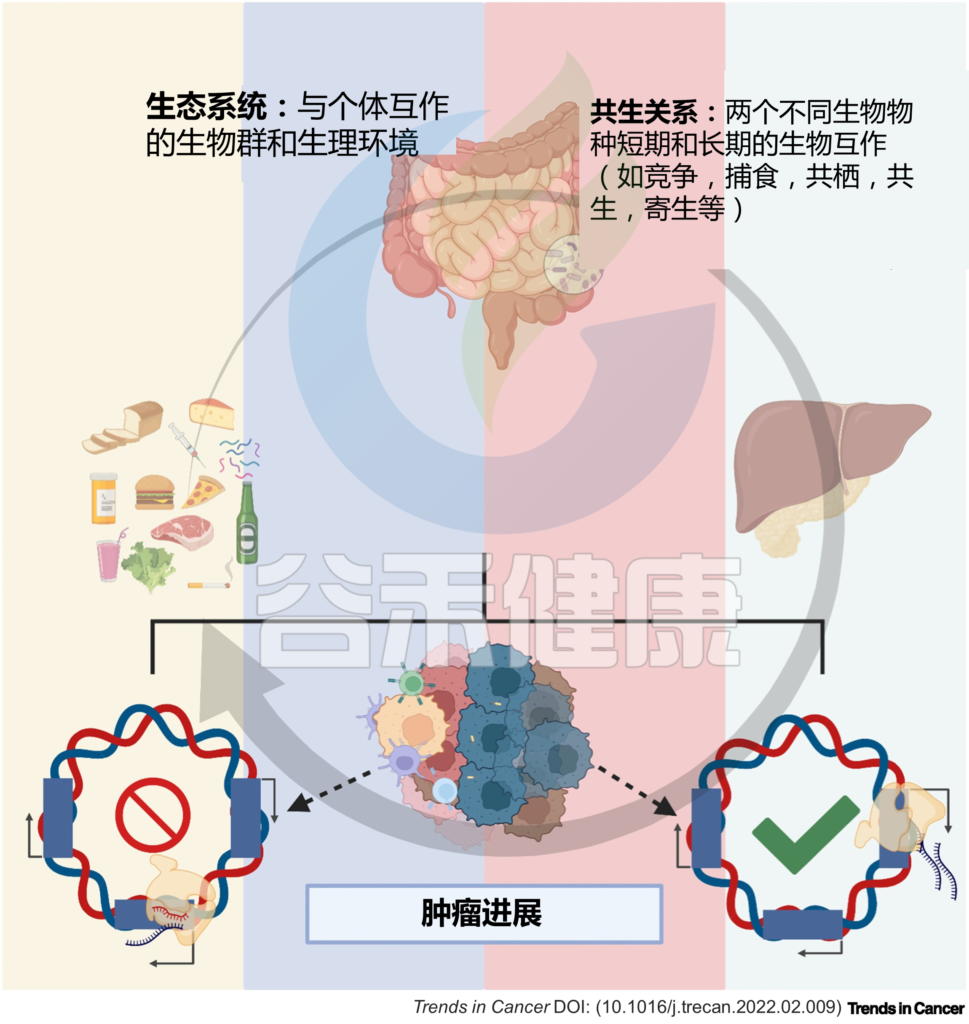
关于微生物群在致癌中的作用的研究,最初集中在上皮屏障界面的肿瘤上,如胃癌和结肠癌,但胰腺导管腺癌 (PDAC) 为口腔和肠道微生物群以及癌症相关微生物群的作用提供了很多证据。
微生物群对致癌作用的影响,及细菌调节肿瘤微环境的一些机制如下:
胰腺癌研究解决了肠道菌群失调、瘤内细菌和癌症之间的联系
人类胰腺癌前体病变显示被产生 IL-17 的Th17 细胞浸润,加速了癌症的发生和进展。
几项研究表明,胰腺导管腺癌与口腔微生物组的组成、口腔病原体(如牙龈卟啉单胞菌Porphyromonas gingivalis、伴放线聚合杆菌Aggregatibacter actinomycetemcomitans)的丰度增加或口腔微生物抗体的存在有关。胰腺癌中出现了较低的α多样性,产生LPS的细菌增加,产丁酸盐菌减少。
基于与胰腺导管腺癌相关的口腔和肠道微生物群落的概况,已经提出了将胰腺导管腺癌患者与健康个体区分开来的无创诊断模型。
此外,胰腺导管腺癌研究已经确定,胰腺含有与组织相关的细菌和真菌,这些细菌和真菌在胰腺癌中比在正常胰腺组织中的含量更高。胰腺导管腺癌中的细菌主要存在于免疫细胞和癌细胞内。
在分析的每种肿瘤类型中,肿瘤内细菌的组成是不同的,可用于预测肿瘤与正常组织和肿瘤类型。
尽管在大约三分之二的胰腺导管腺癌中观察到细菌,并且数量高于大多数其他肿瘤类型,但与人类细胞相比,它们在肿瘤中的绝对数量仅为约 1/40 – 1/400。
与其他肿瘤相比,胰腺导管腺癌中的肿瘤相关细菌 Gammaproteobacteria较多,尤其是肠杆菌Enterobacterales,与胃癌和结肠癌不同的是,它们的梭杆菌fusobacteria较少。
胰腺导管腺癌相关微生物群的组成与十二指肠微生物群相似,这一事实表明细菌可能是通过胆胰管逆行迁移。
肿瘤相关微生物通过各种机制参与胰腺癌的发生或对治疗产生抵抗
例如,表达胞苷脱氨酶长同工型的细菌(如 Gammaproteobacteria)将吉西他滨代谢成无活性形式,导致胰腺导管腺癌出现耐药性。
瘤内细菌可能通过诱导 MDSCs 和抑制 M1 巨噬细胞分化和 CD4 +和 CD8 + T 细胞活化来重新编程肿瘤微环境;通过抗生素治疗进行的细菌消融可重新编程胰腺肿瘤免疫微环境,防止癌变并使免疫检查点抑制治疗产生反应。
然而,肿瘤内细菌也可能是有益的,并且 胰腺导管腺癌的长期幸存者在肿瘤相关微生物群中表现出更高的微生物 α 多样性和独特的特征(假黄单胞菌属Pseudoxanthomonas、链霉菌属Streptomyces、糖多孢菌属Saccharopolyspora),这可能诱导有效的免疫细胞浸润和抗肿瘤免疫。
最近有多项综述回顾了微生物调节在胰腺导管腺癌中的作用。这些研究支持胰腺导管腺癌患者的肿瘤外和肿瘤内微生物群与发育和临床进展的易感性之间的直接联系。
作为一个相互关联的系统,肠-肝轴中的微生物-宿主串扰有望成为肝癌发生的一个重要因素,就像在胰腺导管腺癌中一样。
细菌通过微生物-宿主和宿主-宿主相互作用参与致癌作用
幽门螺杆菌,已被正式确定为胃癌的明确人类致癌物。然而,新出现的证据表明,细菌,无论是存在于上皮屏障界面上还是存在于肿瘤中,都与局部或远处组织的癌变和肿瘤进展有关。
细菌在肿瘤微环境中产生选择性压力以促进肿瘤发生,部分原因是引发 ROS 的产生,影响对 pH 变化的反应,竞争有限的营养物质,增加 DNA 损伤和诱变,调节癌基因途径,影响化学疗法的代谢药物,或调节免疫。
携带产生大肠杆菌素的聚酮化合物-非核糖体肽合酶操纵子 (pks) 的大肠杆菌菌株在结直肠癌中诱导了明显的突变特征,这为细菌在基因组突变中的作用提供了证据。
微生物群对致癌基因诱导的肿瘤进展的影响得到以下观察结果的支持:
突变的p53仅在远端结肠中致癌,因为存在微生物产生的没食子酸,通过破坏 WNT 通路阻止突变的 p53 作为肿瘤抑制因子。相反,肿瘤会对局部组织细胞施加的竞争压力可能会影响肿瘤微环境、周围组织和肠道中的细菌。
宿主-宿主相邻细胞之间的肿瘤微环境竞争动态是生态系统模型的基础,在结肠和肝脏中均发现有致癌作用,并且可以为细菌调节肿瘤微环境提供底物。
恶性干细胞分泌促进邻近干细胞分化为含有促癌突变的克隆的因子。微生物群的改变可能代表另一个触发因素,结合宿主细胞串扰中涉及的其他多个信号,不仅影响癌症前体靶细胞,而且通过调整局部组织环境影响所有细胞。
此外,肿瘤相关细菌大多存在于癌细胞和免疫细胞的细胞内,可能影响癌细胞的信号传导,并在抗原呈递细胞表面以 MHC 限制性肽的形式呈递,从而刺激宿主免疫。
尽管在改进低生物量正常和肿瘤组织样本中稀有细菌的鉴定和分析技术方面取得了很大进展,但这些结果仍需谨慎解读。不过,这支持肿瘤微环境选择居住的微生物群并反过来受微生物群影响的新概念。
此外,肝脏和胰腺之间的生理联系提出了一个问题,即:最近在胰腺导管腺癌中建立的模型是否可以应用于肝细胞癌 (HCC)?
在此阐述了微生物群在维持肠-肝轴稳态中的作用,并关注环境扰动如何直接(通过诱导微生物相对丰度/多样性的变化)或间接(通过微生物代谢物的作用)触发与肝癌发生相关的基因反应。
微生物群的组成是在婴儿早期建立的,并在成年后保持相对稳定。然而,由于生活方式、饮食、疾病、感染和抗生素的使用,细菌种类的相对丰度可能会迅速改变。
微生物群与宿主之间的串扰对健康和疾病产生关键影响
微生物群通过其扩张/收缩、占据不同的解剖生态位以及遗传物质的突变和交换来适应环境变化并调节宿主反应。
微生物群在宿主从出生开始的先天性和适应性免疫系统发育中发挥着重要作用,免疫系统也塑造了宿主-微生物的相互作用。这个过程依赖于肠道黏膜表面的分隔、微生物群感应和信号传递以及免疫细胞启动,以产生特定的反应并维持体内平衡。
结合起来,上皮屏障、其微生物群落和局部免疫系统不仅可以耐受环境中的共生细菌,还可以使免疫系统对条件致病菌或微生物产物做出反应。
当这些防御机制失败时,例如由于肠道通透性增加(肠道渗漏)或生态失调(与疾病相关或与疾病有因果关系的微生物群组成的变化),就会出现细菌代谢物的涌入或先前被分隔,并且有害微生物可能通过肠道血管屏障(GVB)并通过门静脉循环延伸到肝脏。
微生物群在肝脏稳态中的关键作用
在肝脏中,环境扰动会触发肝脏免疫反应,这种反应依赖于常驻免疫细胞以及来自肠道微生物群的循环抗原和内毒素。由与肠道相关淋巴组织 (GALT) 相关的免疫监视形成,证实了微生物群作为一个相互关联的系统在肝脏稳态中的关键作用。
为什么肠道屏障的紊乱会改变肝脏的微环境?
小肠细菌过度生长(SIBO)也与肠漏和细菌流入肝脏有关。
细菌产物或微生物易位的增加会刺激与慢性肝病相关的促炎反应。相反,肝脏通过胆道在肠道中释放胆汁酸和其他生物活性介质,这些介质可能会被肠道微生物群进行生物转化,然后被吸收并释放到体循环中。
生理组织稳态的改变可能导致癌症等疾病。此外,通过将饮食模式与微生物组对免疫和代谢状态的影响和癌症治疗反应联系起来,这种关系揭示了微生物群和肝脏之间的联系,这是由局部环境扰动引发的一系列相互关联的反应。
总的来说,肠-肝轴生态系统可以触发局部和远处的反应,并勾勒出肠道微生物群与肝脏之间的直接和间接相互作用(图 2)。
图2 肠肝轴的双向关系调节体内平衡
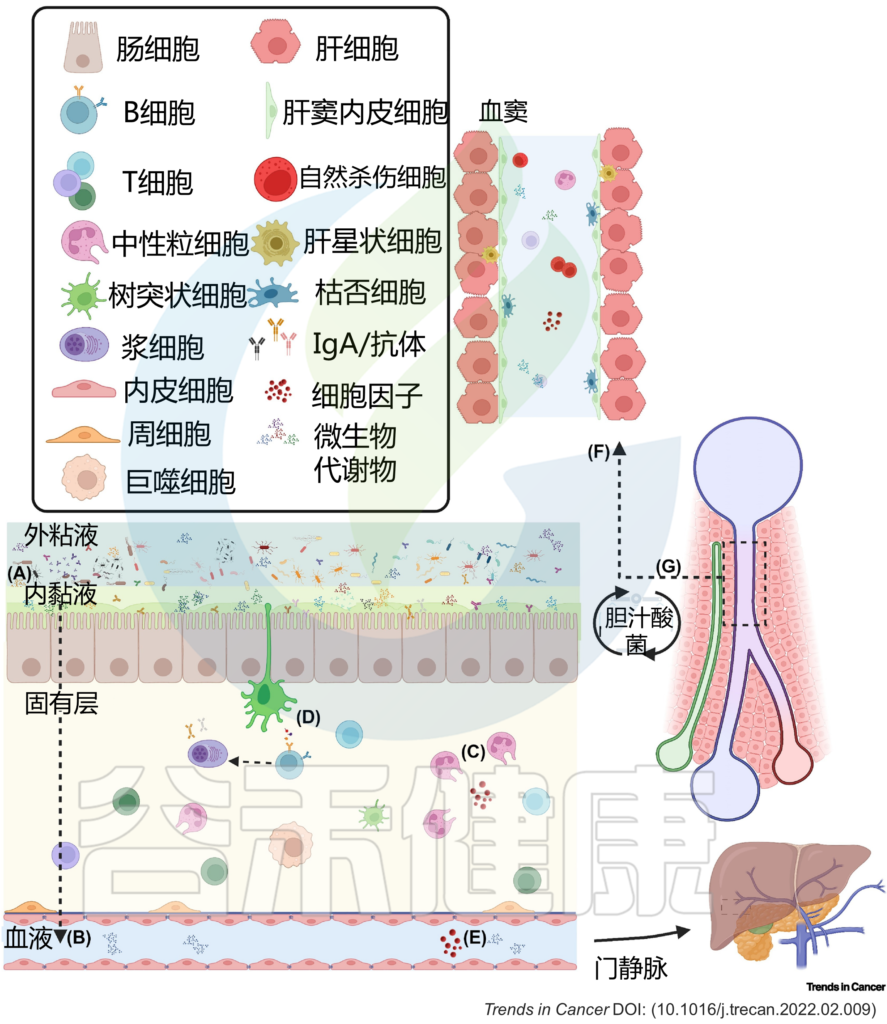
(A) 粘液从物理上将微生物群与上皮衬里分离,而抗菌肽使内部粘液几乎无菌。(B) 因此,微生物群与宿主之间的相互作用主要是间接的,并由代谢产物介导,这些代谢产物可能穿过粘液和上皮屏障,到达固有层中的免疫细胞和基质细胞,或通过淋巴和血管系统到达肝脏和体循环。(C) 免疫细胞通过产生生长因子和细胞因子来巡逻上皮细胞,并加强上皮和粘液屏障。这些产物对微生物产生选择性压力。(D) 树突状细胞感知环境并诱导T细胞和B细胞反应,从而产生IgA抗体,这些抗体转移到管腔,并通过调节微生物组成和多样性来促进粘膜免疫保护。代谢产物通过(E)肠血管屏障通过门静脉流入肝脏。(F) 在肝窦中,免疫细胞扫描异物。(G) 反过来,肝脏通过释放胆汁酸和其他生物活性介质进入胆道与小肠沟通;这些代谢物可被回肠和大肠末端丰富的微生物群生物转化,部分通过门静脉再循环到肝脏,从而可能影响局部和全身功能。
健康的肠道屏障对一些微生物代谢产物是可渗透的,但大多数完整的微生物却不能通过。
然而,饮食和其他环境因素的影响会迅速丰富或消耗特定的营养物质和细菌。这种效应会对微生物代谢物的产生和胆汁酸的转化产生影响,这有可能塑造局部微环境并与包括癌症在内的慢性肝病的发展相关。
例如,酒精会导致肠漏。与 SIBO 一起,它允许内毒素进入循环,导致肝脏疾病。SIBO 是肠道微生物菌群失调的一种表现,其特征是 α 多样性降低,在某些情况下,β 多样性增加,这可能导致全身性炎症。
增加的细菌易位会促进肝硬化进展(肝细胞癌的前体),包括:导致纤维化,通过增加肝细胞与微生物和细菌代谢物的接触,或通过在肝脏中创造转移前的小生境,改变环境以利于转移细胞的募集和增殖,从而促进肝硬化进展。
最后,在某些慢性肝病中,活细菌的易位可能导致 GALT 的免疫麻痹,表明微生物平衡的变化直接影响局部环境的机制。
下文将阐述肠道细菌如何通过扩散到肝脏、影响环境或改变其他细胞中的串扰直接影响肝癌发生。
饮食改变肠道微生物组,对免疫和新陈代谢状况、癌症风险和对癌症治疗的反应产生影响。因此,摄入食物的来源和类型在调节肠道微生物组中起着关键作用,并对宿主-微生物相互作用产生影响。
人类肠道中主要有三种类型(肠型)(拟杆菌属Bacteroides、普氏菌属Prevotella和瘤胃球菌属Ruminococcus)。
在工业化和非工业化人群中观察到的蛋白质和动物脂肪(拟杆菌属Bacteroides)与碳水化合物或植物性食物(普氏菌属Prevotella)的摄入比例不同。工业化与天然富含纤维食品的减少有关。
膳食纤维是饮食中的重要组成部分,分为不溶性(抗发酵)或可溶性(可被肠道微生物群代谢),例如益生元纤维菊粉,在结肠中它被肠道微生物群发酵成短链脂肪酸:乙酸盐、丁酸盐、和丙酸盐。
菊粉 是果糖聚合物的异质混合物。果糖摄入转化为乙酸盐,并通过微生物群衍生的乙酸盐经门静脉到达肝脏触发从头脂肪生成。在肠道中,菊粉对成年人微生物组组成的影响:
下列菌增加:
双歧杆菌Bifidobacterium
厌氧菌Anaerostipes
粪杆菌Faecalibacterium
乳杆菌属Lactobacillus
下列菌减少:
拟杆菌属Bacteroides
普氏菌Prevotella、密螺旋体Treponema、琥珀弧菌属Succinivibrio的丰度增加主要与蔬菜的纤维和碳水化合物发酵相关的饮食有关。
高纤维饮食增加了微生物组编码的聚糖降解碳水化合物活性酶,而不影响群落多样性。相比之下,高发酵食品饮食增加了微生物组的多样性并减少炎症。
从机制上讲,缺乏纤维的饮食可以使结肠粘液降解细菌增多,增强柠檬酸杆菌粘液层翻转和相关的结肠炎。这表明饮食模式可能影响肠-肝轴的多种机制。
健康的植物性食物会影响肠道微生物的多样性和组成,包括产丁酸菌的富集,例如:
Roseburia hominis
Agathobaculum butyriciproducens
普氏粪杆菌Faecalibacterium prausnitzii
厌氧菌Anaerostipes hadrus
丁酸盐是一种短链脂肪酸,通过肠道内膳食纤维的微生物发酵产生。丁酸盐有助于黏膜稳态和肠道内壁的完整性,从而提供肠细胞的大部分能量需求,并通过与几种 G 蛋白偶联受体结合并作为组蛋白脱乙酰酶抑制剂发挥抗炎作用。在小鼠模型中,增加的膳食纤维以微生物群和丁酸盐依赖的方式防止结直肠肿瘤发生。
总的来说,这些研究强调了饮食对微生物群落的深远影响及其对宿主的影响,具体取决于摄入的食物来源。
先前的研究表明,地中海饮食与拟杆菌门Bacteroidetes和某些有益梭菌群Clostridium的富集以及变形杆菌门Proteobacteria和芽孢杆菌门Bacillota的减少有关,可以减少肝脏脂肪,被推荐用于预防非酒精性脂肪性肝病 (NAFLD)。
调节与饮食相关的微生物组是预防肝癌的潜在途径
长期食用可发酵的富含纤维的食物(如可溶性纤维菊粉或富含菊粉的高脂肪饮食)容易导致生态失调的小鼠出现炎症、胆汁淤积和肝细胞癌。总细菌负荷增加,多样性减少,变形杆菌和纤维发酵细菌(如梭菌属)的特定增加。消除这些产生丁酸盐的细菌成功地预防了富含菊粉的肝细胞癌,这表明调节与饮食相关的微生物组是预防肝癌的潜在途径。
由于肠道微生物群失调,高胆固醇/高脂肪饮食依次导致小鼠阶段进展为脂肪变性、脂肪性肝炎、纤维化,最终导致 NAFLD-肝细胞癌。
在每个阶段都富集了不同的微生物群组成,因为在肝细胞癌患者中:
Mucispirillum、Desulfovibrio、Anaerotruncus和 Desulfovibrionaceae依次增加,
而Bifidobacterium、Bacteroides被耗尽。
综合这些发现,饮食模式会通过富集与健康或疾病相关的特定细菌来影响肠道微生物组,从而对肝脏产生潜在影响。
许多微生物群代谢物是肝癌发展的危险因素。考虑到环境塑造和选择特定的微生物群,推测某些物种可能获得哪些竞争优势以及特定微生物群的富集如何影响肝癌的进展。
相似或不同的微生物群组成是否可能参与肝细胞癌和胆管癌 (CCA) 的发展?现在下结论还为时过早,在这部分讨论这两种类型的肝癌。
肝脏中肿瘤和非肿瘤区域之间菌群不同
最近有报道称,病毒和非病毒病因的肝细胞癌中的 16S rRNA 基因测序确定了肝脏中肿瘤和非肿瘤区域之间不同的微生物组成,其中拟杆菌门Bacteroidetes、厚壁菌门Firmicutes、变形杆菌门Proteobacteria的物种占肿瘤相关菌群的主导地位。相关微生物群Ruminococcus gnavus被确定为感染肝炎病毒的 肝细胞癌患者的特征分类群。
肝硬化、肝细胞癌患者的肿瘤微生物群显示出更高丰度的嗜麦芽窄食单胞菌Stenotrophomonas maltophilia,这与肝星状细胞 (HSC) 中的衰老相关分泌表型 (SASP) 相关,证实了菌群失调与肝细胞调节之间的关联。
NAFLD肝硬化中,伴或不伴肝细胞癌的患者菌群不同
肝细胞癌患者的拟杆菌属Bacteroides和瘤胃球菌科Ruminococcaceae丰度增加,双歧杆菌Bifidobacterium丰度降低,这与粪便钙卫蛋白水平升高和全身炎症相关。同样,与肝硬化患者相比,早期肝细胞癌患者的肠道菌群中产生脂多糖 (LPS) 的菌群增加,而产生丁酸盐的菌群减少。
这些数据表明,在患有 NAFLD 和肝硬化的肝细胞癌患者中,肠道菌群组成与全身炎症相关,并可能促进肝癌的发生。
乙肝病毒感染进展的肝细胞癌的菌群特点
从乙型肝炎病毒 (HBV) 感染进展的肝细胞癌患者显示出丰富的抗炎细菌(例如,普氏菌Prevotella、乳酸杆菌Lactobacillus、双歧杆菌Bifidobacterium、粪杆菌Faecalibacterium)和减少的促炎细菌(例如,大肠杆菌-志贺氏菌Escherichia-Shigella、肠球菌Enterococcus),肠道微生物群可能与调节宿主免疫生物学途径的 HBV 感染有关的成分。
这些研究强调了在肝细胞癌中观察到的微生物多样性,这代表了已知风险因素与肝细胞癌发展之间的相关性。
作为饮食和其他环境因素影响微生物组的相互关联的系统,有必要在多个评估部位(肿瘤和非肿瘤)和组织中表征局部微生物群的组成和多样性,与单细胞分析相关并可能相关与环境因素。此外,突出特定细菌物种富集的潜在因果关系的功能分析是超越相关性的关键步骤。
胆管相关的微生物群
一些细菌科,如Dietziaceae、Pseudomonadaceae、Oxalobacteraceae主导了胆管相关微生物群,表明独特的微生物群落存在于这一解剖学定位中。
肝外胆管癌患者有大量肠球菌Enterococcus、链球菌Streptococcus、拟杆菌属Bacteroides、克雷伯氏菌属Klebsiella、锥体杆菌属Pyramidobacter。
此外,与胆总管结石患者相比,胆管癌患者的胆汁样本富含肠杆菌属Enterobacter、假单胞菌属Pseudomonas、窄食单胞菌属Stenotrophomonas。
最后,与肝细胞癌或肝硬化患者和健康个体相比,肝内胆管癌患者肠道菌群中的4个细菌属增加:
乳酸杆菌Lactobacillus、放线菌Actinomyces、消化链球菌Peptostreptococcus、异体卡多菌Alloscardovia。
肠道微生物群特征可以来区分胆管癌和胆石症
在胆管癌中富集的菌群:拟杆菌属Bacteroides、Muribaculaceae_unclassified、Muribaculum、Alistipes属的物种。
而不同的微生物物种在胆石症组中富集,这表明在从良性肝胆疾病到恶性肝胆疾病的演变过程中微生物关联发生了变化。
总的来说,这些过程可能解释了肠道细菌易位直接导致建立有利于肝癌发展和进展的发炎肝脏环境之间的联系。然而,很难根据与微生物相对丰度的相关性来唯一地假设因果关系,微生物相对丰度由于多种环境因素而迅速改变。
因此,全面的跨界网络分析比较肝细胞癌和胆管癌,并将局部肠道和组织微生物群的组成和多样性以及环境因素对代谢、免疫和转录改变的影响联系起来,对于剖析微生物群在肝癌的发生及其作用机制调节中的因果作用至关重要。
“
强调了微生物群变化对肝脏环境的间接影响(可能通过环境中其他细胞或微生物代谢物的串扰)及其与肝癌发生和进展的关系。
为了了解特定微生物群在肝肿瘤中的潜在影响,有必要将潜在机制以及微生物与其他细胞之间的串扰联系起来。
小鼠中肝细胞癌发展的演变
从这个意义上说,最近的一项研究描述了Mdr2 缺陷小鼠中肝细胞癌发展的时间演变,这些小鼠缺乏从肝脏将磷脂分泌到胆汁中的能力,从而经历胆汁淤积和肝细胞癌发展。
在这些代表炎症诱导肝细胞癌的有用模型的小鼠中,肠道菌群失调诱导肠道屏障功能障碍,先于 LPS 介导的肝脏转录改变,从而导致肝细胞癌发展。
此外,肝内炎症基因谱从肝损伤早期的促炎表型转变为肝细胞癌的免疫抑制表型。这种变化与通过微生物组功能从碳水化合物向氨基酸代谢的转变来重新调整能源利用有关。
菌群改变通过代谢和炎症影响肿瘤发生
微生物群的改变通过影响肝脏碳水化合物和脂质代谢调节炎症,从而导致 NAFLD 及其进展为非酒精性脂肪性肝炎 (NASH)。代谢和炎症的调节可能同样影响肝脏肿瘤发生。
肠道微生物组受饮食和其他环境因素的影响,微生物与营养物质的竞争是调节新陈代谢和免疫反应的关键步骤。例如,通过微生物群介导的膳食纤维发酵产生 SCFA 与胆汁淤积型肝细胞癌相关。
有人认为,细菌产物的易位可能会刺激炎症并释放 GALT 中的活性氧 (ROS),从而影响机械和分泌屏障以及局部微生物群。
这些研究强调需要继续进行系统和全球研究,将肠-肝轴中微生物物种的多样性和丰度作为一个生态系统进行表征,同时也需要开始剖析这些表型背后的机制。
最近有人提出,母亲在怀孕期间摄入丁酸盐和谷氨酰胺会影响新生小鼠的粪便微生物群和代谢物,这与拟杆菌和梭状芽胞杆菌的粪便特征有关。
此外,这些新生小鼠对肝脏免疫激活有抵抗力,导致胆管炎症和损伤。
从机制上讲,细菌代谢物在宿主细胞中触发基因反应的影响可能取决于环境中的转录改变。
对急性肝衰竭动物模型中不同的转录特征进行了检查,表明肠道微生物群和 Toll 样受体 (TLR) 信号激活肝星状细胞、枯否细胞和肝窦内皮细胞 (LSEC) 中的 MYC 依赖性转录程序,导致 Ly6C 阳性炎性单核细胞浸润和肝功能衰竭。
图3 微生物代谢产物和多样性是肝癌进展的触发因素


(A)一些细菌可能会穿透粘液屏障或参与其降解,在某些情况下导致上皮屏障的破坏或破坏,从而允许(B)微生物和微生物代谢物和免疫细胞之间的直接接触,诱导促炎细胞因子的产生和全身传播。粘液/上皮屏障的破坏也可能促进(C)细菌转移到肝脏(D),这为癌细胞的播散创造了一个有利的生态位。因此,先前划分的细菌和微生物产物的涌入影响了局部肝细胞的基因表达。例如:(E)肝细胞可能表达CXCR1并诱导CRCX2+多形核髓系衍生抑制细胞(PMN-MDSCs)的积累,创造免疫抑制环境促进胆管癌(CCA);(F)激活的肝星状细胞在肝细胞癌(HCC)和癌症转移中发挥多种功能,可能通过CXCL12-CXCR4相互作用破坏肝脏中自然杀伤细胞(NK)的功能,改变NK细胞介导的免疫,促进乳腺向肝脏转移;而(G)其他的肝脏免疫细胞可能通过脂多糖(LPS) – toll样受体4 (TLR4)或脱氧胆酸(DCA) -TLR2调节而被激活,并诱导促进肝细胞癌发生的炎症反应。相比之下,(H)被肠道菌群修饰后的肝脏产生的胆汁酸可能会激活肝脏自然杀伤T细胞(NKTs)在肝脏中的趋化因子依赖性积累,从而控制肿瘤的生长。
LPS调节影响肝细胞癌
循环水平的LPS通过TLR4 激活并诱导肝星状细胞分泌生长因子、调节肝脏慢性炎症状态和抑制细胞凋亡,这些过程与肝细胞癌促进有关。门静脉区域 LPS-TLR4 相互作用下游的转录调节因子 YAP1 的激活调节肝细胞的干性。
因为肝细胞周转的位点定位在激活基础稳态和再生的分子途径中很重要。推测 LPS 也可以通过调节局部微环境重编程来调节这些机制以影响肝细胞癌。
肠道微生物群调节肝细胞的基因表达程序,促进肝细胞癌和胆管癌
在胆管癌中,增加的肠道通透性诱导微生物 LPS 易位进入肝脏,通过 TLR4 依赖性机制诱导肝细胞中 CXCL1 的表达。这种表达反过来导致 CCR2 +多形核髓源性抑制细胞 (MDSCs) 的积累。
肝硬化肝细胞癌患者的瘤内S. maltophilia丰度更高。 通过激活 TLR4/NF-κB/NLRP3 通路诱导衰老肝星状细胞中衰老相关分泌表型SASP因子和促炎因子的表达,从而促进肝纤维化,随之而来的肝纤维化加重并发展为肝细胞癌。
肝星状细胞增殖是肝纤维化发展的关键事件。最后,胆汁酸通过激活表皮生长因子受体诱导肝星状细胞增殖。
饮食会迅速改变人体肠道微生物组。饮食衍生的微生物代谢物对甲酚硫酸盐、4-乙基苯基硫酸盐和 4-甲基儿茶酚会影响肝细胞癌亚型。
人类肠道微生物组编码的代谢途径通过众多生物活性分子不断与宿主基因产物相互作用。例如,营养过剩会增加 IL-17A,进而诱导白色脂肪组织中的中性粒细胞浸润和 NASH 诱导的肝细胞癌。
IL-17A 是一种促肿瘤细胞因子,通过调节Kupffer细胞和骨髓源性单核细胞的炎症反应和脂肪变性肝细胞的胆固醇合成,调节酒精诱导的肝脂肪变性、炎症、纤维化和肝细胞癌的进展。
地高辛,一种类视黄醇孤儿受体 γ t (RORγt) 拮抗剂,降低了 IL-17A 水平并稳定了体重。表明其在代谢紊乱中的关键作用。
此外,TNF和IL-17A 与骨髓来源细胞中NLRP3 炎性体激活诱导的肝脏炎症和纤维化的发展有关。
总的来说,这些研究表明饮食代谢物、细胞因子和肝癌疾病之间存在机制联系。
初级胆汁酸在肝细胞中合成,释放到十二指肠,大部分在小肠中重新吸收。一小部分初级胆汁酸逃逸到结肠,肠道共生细菌将其转化为次级胆汁酸,次级胆汁酸对新陈代谢和宿主先天免疫反应具有多种重要功能。
饮食和微生物胆汁酸代谢物均可调节 RORγt 阳性调节性 T 细胞(Treg) ,有助于维持宿主免疫稳态和改善肠道炎症。此外,胆汁酸代谢物可以通过调节 Th17 和 Treg 细胞的平衡来控制宿主免疫反应。
胆汁酸可以在高脂肪饮食中发挥积极作用
膳食胆固醇诱导肠道细菌代谢物改变,包括增加牛磺胆酸和减少 3-吲哚丙酸,从而在小鼠中驱动 NAFLD-肝细胞癌。因此,胆固醇抑制疗法和肠道菌群操作可能是预防 NAFLD-肝细胞癌的有效策略。
石胆酸 (LCA) 衍生物直接影响 CD4+ T 细胞(3-oxoLCA 和 isoalloLCA)
3-oxoLCA 通过直接结合转录因子 RORγt 抑制 Th17 细胞分化,而 isoalloLCA 增强 Treg 细胞分化,证实肠道微生物群可能控制宿主免疫反应。
对于肝内胆管癌,观察到甘熊去氧胆酸和牛磺脱氧胆酸血浆:粪便比率增加,血浆牛磺胆酸和 IL-4 呈正相关,表明肠道微生物群、代谢物、细胞因子和胆汁酸之间存在相互关系。
肥胖诱导的菌群失调促进肝癌发生
最近的一项综合组学研究揭示了一种胆汁酸代谢物升高、胆固醇代谢失调和与 BMI 增加相关的独特炎症反应的胆管癌亚型,这表明肥胖诱导的肠道微生物群失调促进肝癌发生的模型。
从机制上讲,胆汁酸/致癌轴涉及胆汁酸受体,例如法尼醇 X 受体 (FXR) 和 G 蛋白偶联胆汁酸受体 1,它们可能代表癌症的重要治疗靶点。胆汁酸如脱氧胆酸 (DCA) 被证明可阻断 FXR 的功能及其抑制肠癌干细胞增殖的能力,从而影响肠-肝轴稳态。
此外,胆汁酸传感器 FXR 或 G 蛋白偶联受体 TGR5 的激活通过抑制 NF-κB 依赖性信号通路和 NLRP3 依赖性炎症小体活性来抑制炎症信号传导。因此,饮食-肝脏-胆汁酸-微生物群的串扰在胃肠道炎症以及结直肠癌和肝癌的发生中起重要作用,可用于预防癌症的发生或进展。
菌群利用胆汁酸作为信使影响抗肿瘤免疫
肠道微生物组可以利用胆汁酸作为信使,来控制趋化因子依赖性肝脏自然杀伤 T 细胞 (NKT) 的积累,从而影响肝脏中的抗肿瘤免疫。这一过程是由共生肠道细菌的改变、初级和次级胆汁酸的平衡以及 LSEC 中的 CXCL16 表达介导的,LSEC 是最早暴露于肝脏中肠道衍生代谢物的细胞之一。
相反,NKTs 通过与肝细胞的相互作用与 CD8 T 细胞合作,促进 NASH 和 NASH相关的肝细胞癌。
NK 细胞和肝星状细胞之间的相互作用——癌症休眠和转移的主要开关
基质反应阻碍了 NK 细胞和干扰素 γ 介导的肿瘤细胞休眠的维持,并通过组织损伤和活化的肝星状细胞分泌 CXCL12 的过程诱导肝转移,CXCL12 通过 CXCR4 保留和使 NK 细胞静止,抑制免疫监视和促进转移性生长。
某些与肥胖相关的细菌具有增加次级胆汁酸脱氧胆酸的能力
脱氧胆酸的增加会导致肝星状细胞中的 DNA 损伤,从而诱导衰老相关分泌表型,导致炎症和肿瘤促进因子以及 COX2 诱导的免疫抑制性 PGE2 在肝脏中的产生,从而促进肝细胞癌的发展。
肥胖还与微生物群改变有关,导致 TLR2 激动剂脂磷壁酸 (LTA)这一革兰氏阳性菌的主要细胞壁成分的积累增加。脱氧胆酸增加肝星状细胞上 TLR2 的表达,并与 LTA 协同诱导衰老相关分泌表型因子和 COX2,后者通过 PGE2 诱导免疫抑制,并在化学致癌物暴露后促进肥胖相关的肝细胞癌。
总之,这些研究支持了一个模型,即平衡状态的改变会对环境中的连接细胞产生影响,证实微生物组及其代谢物是肠道和肝脏基因反应的关键影响因素,对肝癌发生具有影响。
本文主要介绍了影响肝细胞癌的微生物群的复杂相互作用,强调了微生物群多样性或肠-肝轴中微生物代谢物的改变触发局部细胞中的基因反应。
与肝细胞癌相比,微生物群和PDAC对患者预后的相关性已得到更彻底的研究,并已明确开始确定特定机制,通过这些机制,微生物群可以局部或系统地影响肿瘤微环境,以及肿瘤进展和对治疗的反应。因此,胰腺导管腺癌研究中使用的一些策略可以应用于肝细胞癌研究。
可以看到,目前的研究分析环境中局部细胞内微生物群介导的直接和间接变化,研究视角已逐渐从靶细胞转向微环境/生态系统。肠道微生物群以及肿瘤本身的细菌通过调节癌症、基质和炎症/免疫细胞中的基因转录程序以及促进或抑制肿瘤进展来影响肿瘤微环境。
癌症干细胞和免疫细胞之间的串扰在癌症进展中起着重要作用,那么癌症干细胞是否容易被微生物/代谢物的改变触发?这也是值得关注的问题。由于物理和生理上的联系,评估微生物群对癌症干细胞分化的影响以及作为一个综合系统对肠-肝轴生态系统的影响也非常重要。
主要参考文献:
Silveira MAD, Bilodeau S, Greten TF, Wang XW, Trinchieri G. The gut-liver axis: host microbiota interactions shape hepatocarcinogenesis. Trends Cancer. 2022 Mar 21:S2405-8033(22)00045-0. doi: 10.1016/j.trecan.2022.02.009. Epub ahead of print. PMID: 35331674.
Komiyama S, Yamada T, Takemura N, Kokudo N, Hase K, Kawamura YI. Profiling of tumour-associated microbiota in human hepatocellular carcinoma. Sci Rep. 2021 May 19;11(1):10589. doi: 10.1038/s41598-021-89963-1. PMID: 34012007; PMCID: PMC8134445.
Zhang T, Zhang S, Jin C, Lin Z, Deng T, Xie X, Deng L, Li X, Ma J, Ding X, Liu Y, Shan Y, Yu Z, Wang Y, Chen G, Li J. A Predictive Model Based on the Gut Microbiota Improves the Diagnostic Effect in Patients With Cholangiocarcinoma. Front Cell Infect Microbiol. 2021 Nov 23;11:751795. doi: 10.3389/fcimb.2021.751795. PMID: 34888258; PMCID: PMC8650695.
Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, Rotter-Maskowitz A, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science. 2020 May 29;368(6494):973-980. doi: 10.1126/science.aay9189. PMID: 32467386; PMCID: PMC7757858.

谷禾健康
人类肠道菌群复杂多样,在与人类长期的共同进化过程中,具备了调节人体免疫应答、影响疾病发展等作用。这种作用与肠道菌群本身的多样性和关键核心菌种的是否存在等具有紧密联系。
在前面的文章我们已经了解到,肠道菌群失调与很多疾病相关,详见:
肠道菌群失衡的症状、原因和自然改善
造成菌群失调的原因有很多,比如抗生素的使用,膳食营养不均衡,感染,重金属污染,疾病发生以及过渡清洁肠道等。
一般肠道菌群失衡可以通过一些明显的迹象表明肠道菌群失衡,如腹胀气、腹泻、便秘、间歇性或慢性腹泻、肠易激综合征、溃疡性结肠炎和克罗恩病,频繁呼吸道感染、过敏、神经问题、免疫低下或代谢异常等来判别。
说到对改善修复肠道菌群,一般是针对菌群存在异常或者偏离健康状态的特定情况才进行针对性调节,这些调节思路主要包括 “清除”,“补充”,“置换”,“塑造”。常用的手段或措施如,使用抗生素或抗菌剂,益生菌,益生元,膳食纤维,粪菌移植,饮食或天然补充物等。
以上改善措施单一或者组合对改善和调节宿主微生态平衡发挥重要作用。不过个体的菌群构成和状态差异很大,由此带来的干预对不同个体和状态的干预效果同样有很大差异,这也反映在很多菌群干预临床研究上。
所以盲目的补充益生菌,益生元等单纯的从菌量或功效来评价益生菌产品的好坏,都可能不利于有效的改善健康状况和调整微生态平衡。除了对比如益生菌菌株,益生元结构区分等进行更加精细化的功能分析外,还需要结合肠道菌群检测,基于不同肠道菌群特点进行精准化的匹配干预和临床研究。
今天我们主要简单讲下不同的一些益生菌、益生元、天然调节剂等对肠道菌群的调节以及对宿主健康的影响及其差异化。
益生菌的现代定义为“活的微生物,当给予足够的剂量时,会赋予宿主健康”。益生菌主要存在于人体肠道内,通过维持肠道微生物平衡,在宿主体内发挥有益作用。在日常生活中,常见的益生菌,如乳酸杆菌或双歧杆菌,通常作为活性菌制剂食用。
近年来,益生菌的研究取得了重大进展。例如,益生菌益生菌可以改善肠道菌群的组成缓解便秘,IBS,IBD,改善腹泻,修复多种与肠道相关的损伤等,此外,益生菌可以在慢性炎症性疾病的治疗中发挥作用,具有抗癌、抗肥胖和抗糖尿病等作用。
本章节我们列举一些常见的益生菌及其功效。
双歧杆菌
人体内双歧杆菌的数量实际上随着年龄的增长而下降。双歧杆菌在提高整体免疫力、减少和治疗胃肠道感染以及改善腹泻、便秘和湿疹等方面发挥作用。
双歧杆菌中常见的种类有:双歧双歧杆菌、长双歧杆菌、婴儿双歧杆菌、乳酸双歧杆菌、短双歧杆菌等。
双歧双歧杆菌B.bifidum是一种通常用于改善消化问题的益生菌。B.bifidum与健康饮食相结合还可以改善血糖控制、减轻压力并帮助对抗感染,有助于增强免疫系统并减少过敏。
双歧双歧杆菌是在母乳喂养婴儿中发现的第二大菌种。在成年期,双歧杆菌的水平显著下降,但保持相对稳定 (2-14%),在老年时再次开始下降。
对其他肠道菌群的影响
在一项针对 27 名健康志愿者的临床试验中,双歧杆菌的摄入量减少了普氏菌科和普氏菌属,并增加了瘤胃球菌科和Rikenellaceae。
在一项针对 53 名慢性肝病患者的临床试验中,双歧双歧杆菌是成功防止小肠细菌过度生长的益生菌之一。同样,在一项针对 66 名酒精性肝损伤患者的试验中,它与植物乳杆菌(后面会讲到)的组合恢复了肠道菌群。
在对 30 人进行的另一项试验中,双歧双歧杆菌与嗜酸乳杆菌(后面会讲到)结合也在抗生素治疗后恢复了肠道菌群。
健康益处


双歧杆菌除在以上列举的疾病发挥作用之外,还在压力、过敏等方面发挥作用(小规模研究或临床试验单一)。
安全性
B. bifidum一般都是安全的,但应避免在免疫功能低下的个体、器官衰竭和“肠漏”的人群中使用。在这些情况下,益生菌可能会导致感染。在具有自身免疫性甲状腺疾病遗传易感性的人群中,双歧杆菌可能导致其发展和恶化。
短双歧杆菌是一种有益细菌,可以在人类母乳以及婴儿和成人的胃肠道中找到。随着个体年龄的增长,其肠道内的短双歧杆菌减少。
对肠道菌群的影响
在一项对 30 名没有其他畸形、染色体异常或宫内感染的低出生体重婴儿的研究中,早期给予短双歧杆菌促进了双歧杆菌的定植和正常肠道菌群的形成。
B. breve还显著减少了 10 名极低出生体重婴儿的吸入空气量并改善了体重增加。
健康益处


短双歧杆菌除在以上列举的疾病发挥作用之外,还在肥胖、坏死性小肠结肠炎、乳糜泻、感染等疾病中发挥作用(小规模研究或临床试验单一)。
目前已有文献中,部分关于短双歧杆菌菌株的研究:
短双歧杆菌M -16V (B. breveM-16V) 显着抑制 Th2 和 Th17 淋巴细胞亚群。
同时,B. breve M-16V 可能激活 MyD88 表达并促进 Th1 相关细胞因子 IL-12 的产生。此外,B. breve M-16V 可能部分恢复肠道菌群失调。
B. breve CCFM1025 是一种很有前途的候选精神生物菌株,可减轻抑郁症和相关的胃肠道疾病。
B. breve FHNFQ23M3可以缓解腹泻症状。
母乳分离的益生菌菌株B. breve CECT7263 是一种安全有效的婴儿绞痛治疗方法。
B. breve UCC2003 在生命早期驱动肠上皮稳态发育中发挥着核心作用。
安全性
B. breve被证明是适合早产儿常规使用的益生菌。
与使用短双歧杆菌相关的不良事件发生率极低,且严重程度较轻。
长双歧杆菌是一种革兰氏阳性、杆状细菌,天然存在于人体胃肠道中。它可以改善人体免疫反应并帮助预防肠道疾病。早期证据表明,它还可以抑制过敏、降低胆固醇和改善皮肤健康。
我们之前这篇文章有详细介绍,详见:双歧杆菌:长双歧杆菌
乳酸杆菌
鼠李糖乳杆菌是一种革兰氏阳性乳酸菌,是人类正常肠道菌群的一部分。通常都是安全的,并已广泛用于食品和保健品中。
健康益处


鼠李糖乳杆菌除在以上列举的疾病发挥作用之外,还在体重管理、肝功能、牙齿健康、免疫、怀孕与分娩等方面发挥作用(小规模研究或临床试验单一,证据还不够充分)。
注意事项
鼠李糖乳杆菌在健康成人中是安全的并且耐受性良好,并且似乎不会对年轻或老年受试者造成不良影响。
但是,免疫功能低下的人不应服用它,因为它可能导致菌血症。在器官衰竭、免疫功能低下状态和肠道屏障功能失调的患者中使用益生菌可能导致感染。
短乳杆菌L. brevis是一种植物来源的乳酸菌,L. brevis可以在酸菜和泡菜等发酵食品中找到。它也是人体肠道微生物群的正常组成部分。
健康益处


短乳杆菌除在以上列举的疾病发挥作用之外,还在睡眠、口腔黏膜炎等方面发挥作用(小规模研究或临床试验单一,证据还不够充分)。
注意事项
L. brevis被认为对人类食用是安全的。
L. brevis可以产生生物胺,如酪胺和腐胺。
对于器官衰竭、免疫功能低下和肠道屏障机制功能障碍的患者,应避免使用益生菌,因为可能会导致感染。为避免任何不利影响或意外相互作用,请在服用短乳杆菌之前咨询医生。
干酪乳杆菌是一种革兰氏阳性、非致病性乳酸菌。它存在于发酵乳制品(例如奶酪)、植物材料(例如葡萄酒、泡菜)以及人类和动物的生殖和胃肠道中。
作为一种营养补充剂,干酪乳杆菌已被证明可以改善肠道微生物平衡、关节炎、2 型糖尿病,并具有潜在的抗癌特性。
干酪乳杆菌在动物消化道中的运输过程中增强了免疫系统,可以刺激一氧化氮、细胞因子和前列腺素的产生。
干酪乳杆菌通过激活自然杀伤 (NK) 细胞、细胞毒性 T 细胞和巨噬细胞来促进小鼠化疗药物引起的免疫抑制的恢复。这些都是识别和消除肿瘤细胞和感染细胞的白细胞。
对其他肠道菌群的影响
干酪乳杆菌与普氏菌属、乳酸杆菌属、粪杆菌属、丙酸杆菌属、双歧杆菌属和一些拟杆菌科和毛螺菌属呈正相关,与梭菌属、芽孢杆菌属、沙雷氏菌属、肠球菌属、志贺氏菌属和希瓦氏菌属的存在呈负相关。
在志愿者实验中,L. casei 抑制了潜在有害的假单胞菌和不动杆菌。
含有干酪乳杆菌的发酵乳保留了肠道微生物群多样性,缓解了腹部功能障碍,在学业压力的健康医学生中,它并防止了皮质醇水平升高。
健康益处


干酪乳杆菌除在以上列举的方面发挥作用之外,还在压力、免疫力、呼吸道和胃肠道感染、病毒感染、炎症、关节炎、过敏、牙齿健康、心血管疾病、糖尿病、吸烟的并发症等方面发挥作用(小规模研究或临床试验单一,证据还不够充分)。
注意事项
干酪乳杆菌通常具有良好的耐受性。应避免在器官衰竭、免疫功能低下和肠道屏障功能障碍的患者中使用益生菌。为避免不良反应,请在开始任何新的益生菌补充剂之前咨询医生。
格氏乳杆菌是一种乳酸菌,具有抗菌活性、产生细菌素以及调节先天和适应性免疫系统。
健康益处


格氏乳杆菌除在以上列举的疾病发挥作用之外,还在胆固醇、免疫力、肠道健康(腹泻、溃疡、幽门螺杆菌)、过敏、疲劳、子宫内膜异位症等方面发挥作用(小规模研究或临床试验单一,证据还不够充分)。
注意事项
一般认为是安全的。然而,应避免在器官衰竭、免疫功能低下状态和肠道屏障功能障碍的患者中使用益生菌。为防止不良副作用,请在开始服用新的益生菌补充剂之前咨询医生。
嗜酸乳杆菌是一种革兰氏阳性乳酸菌,传统上广泛用于乳制品行业,最近还用作益生菌。
嗜酸乳杆菌因其风味和益生菌作用而被添加到商业酸奶和乳制品配方中,并且是最常选择的用于饮食的乳酸菌之一。
对其他肠道菌群的影响
接受嗜酸乳杆菌和纤维二糖的健康志愿者表现出乳酸杆菌、双歧杆菌、柯林氏菌和真杆菌的水平升高,而Dialister降低了。
酸奶中的嗜酸乳杆菌可正向改变肥胖小鼠的肠道微生物群并增加肠道双歧杆菌。
嗜酸乳杆菌增加了大鼠中乳酸杆菌和双歧杆菌的数量,增加了乙酸、丁酸和丙酸的水平,并降低了人体微生物群模拟器中的铵盐。


嗜酸乳杆菌除在以上列举的疾病发挥作用之外,还在改善儿童叶酸和 B12 状态、糖尿病、轻微肝性脑病、老化、疲劳等方面发挥作用(小规模研究或临床试验单一,证据还不够充分)。
注意事项
嗜酸乳杆菌通常耐受性良好。然而,应避免在器官衰竭、免疫功能低下状态和肠道屏障功能障碍的患者中使用益生菌,因为它可能导致感染。
肠道微生物植物乳杆菌是一种很有前途的,用于治疗腹泻、高胆固醇和特应性皮炎的益生菌。植物乳杆菌是一种广泛分布的乳酸菌。它常见于许多发酵的植物产品中,例如酸菜、泡菜、卤橄榄和韩国泡菜。
植物乳杆菌是一种具有抗癌、抗炎、抗肥胖和抗糖尿病特性的抗氧化剂。植物乳杆菌可以减少促炎细胞因子(IL-6、IL-8和MCP-1)的产生,增加抗炎细胞因子 ( IL-10 ) 的产生,降低 ALT 和 AST,减少NF-κB.
营养益处
从生牛奶中分离出的植物乳杆菌能够产生 B 族维生素核黄素(B2) 和叶酸(B9)。
植物乳杆菌可使健康女性从果汁饮料中吸收的铁增加约 50%。
植物乳杆菌可以将女性从燕麦基质中的铁吸收提高 100% 以上。
含有植物乳杆菌的发酵乳表现出更高的钙保留摄取。


植物乳杆菌除在以上列举的疾病发挥作用之外,还在改善肥胖、血糖、伤口愈合、牙齿健康、免疫、过敏、念珠菌病等方面发挥作用(小规模研究或临床试验单一,证据还不够充分)。
注意事项
在大鼠中未观察到任何类型的不良反应,即使在大量食用后也是如此。然而,与其他益生菌一样,用于器官衰竭、免疫功能低下状态和功能失调的肠道屏障机制的患者可能会导致感染。为了避免不良事件,请在使用益生菌之前咨询医生。
菊粉(不要与胰岛素混淆,胰岛素是一种控制血糖水平的激素)是一种存在于多种植物中的可溶性纤维。纤维是不被人体肠道消化或吸收的化合物。可溶性纤维吸水并在消化过程中变成凝胶。
来源及用处
菊粉存在于 36,000 种植物中,包括我们日常饮食中食用的植物,如小麦、洋葱、香蕉、大蒜和芦笋。它们也存在于不太常见的食物中,例如菊芋,尤其是菊苣,菊粉是商业提取菊粉的主要来源。
菊粉的其他天然来源有:菊苣根、龙舌兰、雪莲果根、新鲜香草等。不太常见的菊粉来源是蒲公英根、松果菊、牛蒡根等。
含有菊粉的植物用它来储存能量和抵御低温。当暴露于低温时,菊粉起到防冻剂的作用。
菊粉的溶解度使其能够吸收大量水分。当它膨胀时,它会形成一种凝胶,沿途聚集脂肪颗粒并将它们排出体外。
此外,它通过充当有益菌的食物,来增加肠道中有益细菌的数量。
对其他肠道菌群的影响
前面我们知道,双歧杆菌是肠道中的有益菌。菊粉基本上是双歧杆菌的食物并刺激它们的生长和活动。
多项研究表明,菊粉可刺激双歧杆菌的生长。例如:
8 名健康受试者被给予低聚果糖 15 天,并监测他们的粪便。虽然粪便中的细菌总数没有变化,但双歧杆菌成为主要类型。
在另一项研究中,10 名便秘的老年患者服用菊粉 19 天,并监测他们的大便情况。这些患者还表现出双歧杆菌数量增加,同时有害细菌减少。
此外,其他菌群似乎也受到菊粉的影响。
在一项针对 165 人的临床试验中,这种纤维还增加了厌氧菌的丰度(可以通过产生丁酸改善消化,甚至预防结肠癌),并减少嗜胆菌(与大便和便秘有关)。
不一样的研究结果
对实验室培养的细菌进行的一些研究表明,菊粉还会增加沙门氏菌等有害细菌,以及那些不会在正常人中引起疾病但可能导致免疫系统较弱的人感染的细菌,例如克雷伯氏菌和大肠杆菌。然而,其他实验室研究表明,菊粉通过增加双歧杆菌的生长来抑制艰难梭菌等有害细菌的生长。


菊粉除在以上列举的疾病发挥作用之外,还在增加钙和镁的摄取、骨骼健康、炎症性肠病、预防结肠癌的发展等方面发挥作用(小规模研究或临床试验单一,证据还不够充分)。
注意事项
菊粉可能对敏感个体产生某些副作用,或者如果使用的剂量太大产生不良反应。这些包括:
肠道不适,包括胀气、腹胀、胃部噪音、嗳气和痉挛、结肠肿胀、腹泻等。
此外可能会发生严重的过敏反应,但很罕见。在一些孤立的案例中,它会导致过敏反应,可能与食物过敏反应有关。
此外,对于在怀孕和哺乳期间补充菊粉的效果知之甚少。因此,孕妇应避免服用菊粉补充剂。
对于肠易激综合征 (IBS) 患者,低剂量可能会调节肠道细菌并减轻症状,但大剂量可能会产生中性甚至负面影响。
果胶是一种复杂的碳水化合物(多糖),存在于植物细胞壁中,有助于维持其结构。它是一种粘性可溶性纤维,具有形成凝胶的能力。果胶由主要在大肠(结肠)中的有益菌群发酵,产生短链脂肪酸。
由于其凝胶状稠度,果胶是一种流行的食品添加剂,作为增稠剂和纤维的重要来源,具有许多潜在的健康益处。研究表明,它可能有助于治疗高胆固醇、反酸、减肥和糖尿病。
果胶含量高的水果和食物
果胶存在于水果、蔬菜、豆类和坚果中。柑橘皮中的果胶含量最高,如橙皮、柠檬皮和葡萄柚皮(30% 至 35%)和苹果果肉(15% 至 20%)。其他主要来源包括木瓜、李子、醋栗、樱桃、杏子、胡萝卜等 。


在食品工业中用作胶凝剂(用于果酱和果冻)或用作稳定剂(用于糖果、果汁和奶饮料)的果胶主要从苹果果肉或柑橘类水果的果皮中提取。


果胶除在以上列举的疾病发挥作用之外,还在改善糖尿病、减肥、辐射损伤、便秘、呕吐、降血压、溃疡性结肠炎、铅毒性等方面发挥作用(小规模研究或临床试验单一,证据还不够充分)。
对肠道菌群的影响
果胶在结肠中由不同的细菌属发酵,如双歧杆菌、乳酸杆菌、肠球菌、直肠真杆菌、普氏粪杆菌、梭菌、厌氧菌、Roseburia属,以促进其生长。
果胶的降解由不同的细菌衍生酶(如果胶酶、甲基酯酶、乙酰酯酶和裂解酶)促进,产生不同的POS,其取决于微生物群组成和果胶结构。
体外发酵系统报告的果胶效应


Blanco-Pérez F, et al., Curr Allergy Asthma Rep. 2021
果胶通过增加拟杆菌的丰度来改变肠道菌群的组成,并改善酒精诱导的肝损伤(在非酒精性脂肪肝中,拟杆菌的丰度降低)。
膳食纤维果胶可以改变肠道和肺微生物群中厚壁菌门与拟杆菌门的比例,增加粪便和血清中短链脂肪酸的浓度。
通过果胶产生的短链脂肪酸进行免疫调节
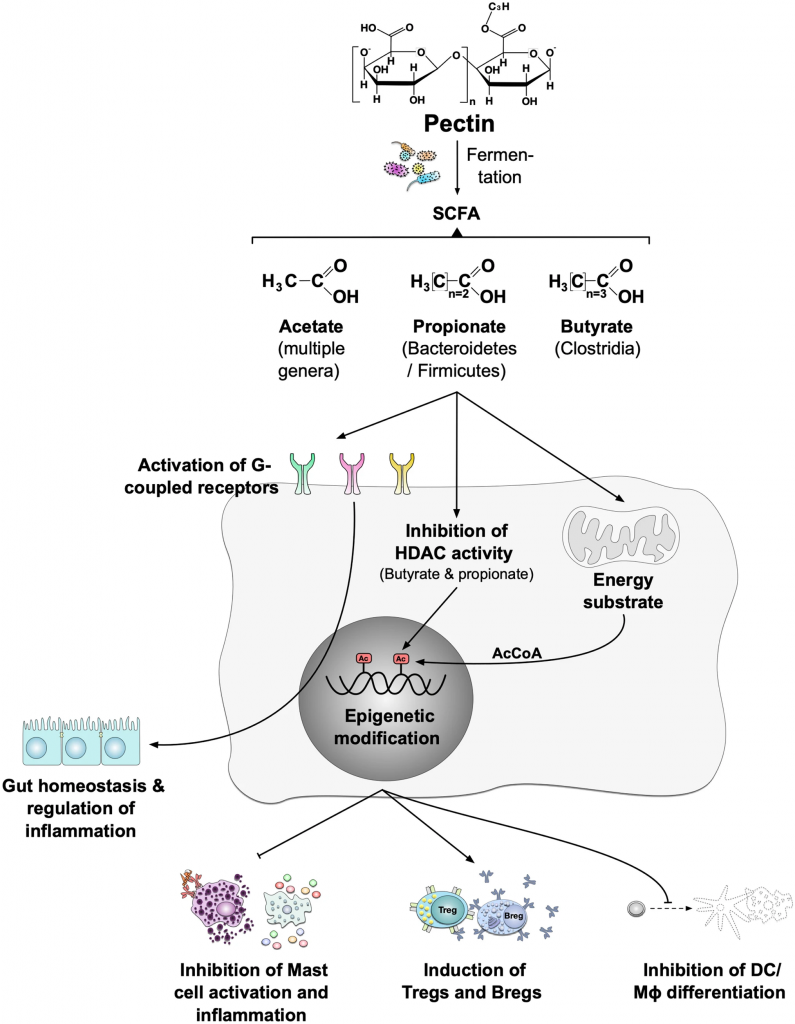

Blanco-Pérez F, et al., Curr Allergy Asthma Rep. 2021
果胶经肠道菌群发酵可产生短链脂肪酸。不同的属可以产生不同的短链脂肪酸。例如,乙酸盐可以由许多不同的属生产;丙酸主要由拟杆菌门和厚壁菌门产生,丁酸主要由梭状芽胞杆菌产生。短链脂肪酸结合“代谢感知”G蛋白偶联受体,如GPR41、GPR43、GPR109A和嗅觉受体(Olfr)-78。这些受体促进肠道内稳态和炎症反应的调节。GPRs及其代谢产物影响Treg活化、上皮完整性、肠道稳态、DC生物学和IgA抗体反应。通过抑制HDAC的表达或功能,短链脂肪酸还影响许多细胞和组织中的基因转录。
过敏:果胶在过敏反应中的作用存在争议
一些临床报告表明,食用果胶后出现过敏反应,这可能归因于果胶和过敏原之间的交叉反应。此外,果胶还被描述为防止胃中过敏原的消化,促进完整的过敏原分子到达肠道并诱发过敏反应。
然而,其他人则认为果胶有直接和间接免疫调节作用。已经提供了一系列广泛的证据,描述了应用果胶诱导有益微生物群的转变和SCFA水平的增加,这两者都与减少体内外的炎症和过敏反应有关。由于不同的果胶增加或减少了与人类健康相关的细菌数量,因此,施用果胶可能会对肠道中的菌群进行特异性调节。
果胶能够通过与TLR2的静电相互作用直接与免疫细胞(如DC和巨噬细胞)相互作用,从而抑制促炎症TLR2-TLR1途径,同时不影响TLR2-TLR6耐受途径。
此外,它能够结合LPS,影响其与TLR4的结合。其他类型的细胞,如T细胞、B细胞和NK细胞也被果胶激活,而腹腔巨噬细胞中的iNOS和COX-2表达则被IKK活性、MAPK磷酸化和NF-κB激活抑制,表明其具有抗炎特性。
炎症性肠病:果胶调节IBD相关菌群
在短链脂肪酸中,丁酸能滋养结肠细胞并抑制结肠肿瘤,因此在大肠中表现出促进局部健康的特性。
产丁酸菌(主要属于厚壁菌门)的流失被认为是IBD期间微生物失调的一个特征。果胶可以促进厚壁菌门中许多丁酸生产者的生长。
果胶物质在调节 IBD 相关肠道微生物群中的综合概况
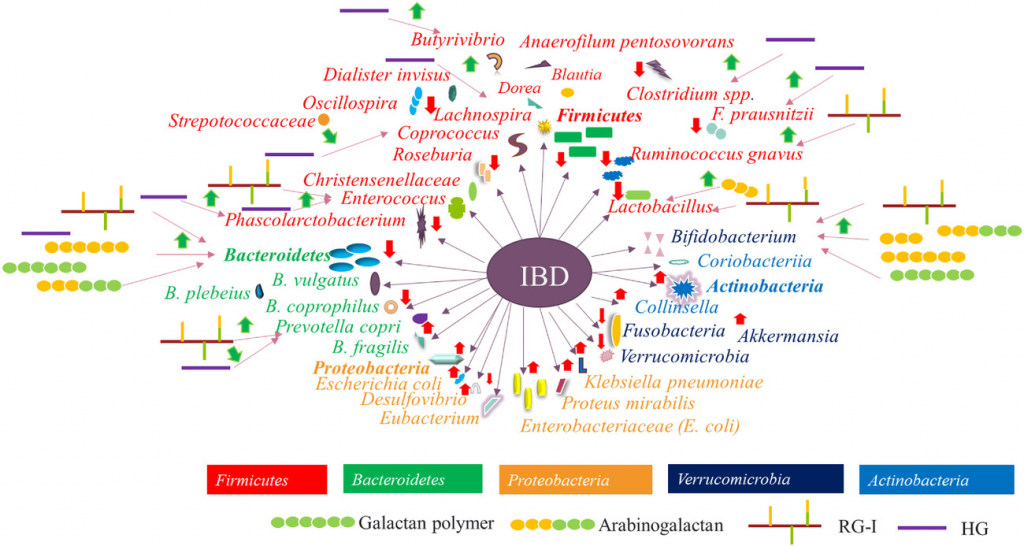

Wu D, et al., Compr Rev Food Sci Food Saf. 2021
注:不同颜色表示不同的细菌门。绿色箭头表示果胶物质对细菌的调节作用,红色箭头表示 IBD 与特定微生物群之间的正相关或负相关。
注意事项
果胶通常对人类食用是安全的。然而,在临床试验中,纤维与果胶的混合物(每天 20 克,持续 15 周)会引起一些与肠道相关的副作用,例如腹泻、肠胃胀气和稀便。
与药物相互作用:
在 3 名高胆固醇患者中,每天服用 15 克果胶和 80 毫克降胆固醇药物(洛伐他汀)会增加 LDL 水平。
建议把果胶和地高辛分开至少 2 小时。
在一项对 7 名健康受试者进行的研究中,他们服用 12 克柑橘果胶和 25 毫克 β-胡萝卜素,果胶将 β-胡萝卜素(维生素 A的前体)血液水平降低了 50% 以上。
姜黄是一种来自植物的香料,通常用于调味或着色咖喱粉、芥末和其他食物。姜黄根也用于制造替代药物。
姜黄已被用于替代医学中,作为降低血液胆固醇、减轻骨关节炎疼痛或缓解慢性肾病引起的瘙痒的一种可能有效的帮助。
姜黄含有几种被称为类姜黄素的主要成分,姜黄素是姜黄中最活跃的植物化学物质。它占类姜黄素的 77%.
姜黄素的健康益处
姜黄素和整个姜黄根茎在治疗慢性疾病如胃肠道、心血管和神经系统疾病、糖尿病和几种癌症方面具有一些有益作用。


姜黄素除在以上列举的疾病发挥作用之外,还在关节痛和关节炎、克罗恩病(肠蠕动、腹泻和胃痛)、狼疮、糖尿病、经前综合症等方面发挥作用(小规模研究或临床试验单一,证据还不够充分)。

姜黄素目前已被认为可以治疗许多疾病,肠道微生物群在姜黄素生物活性机制中可能产生的作用,是一个有趣且有吸引力的研究领域。下面我们来看它们之间有怎样的互作关系。
姜黄素与肠道菌群的相互作用
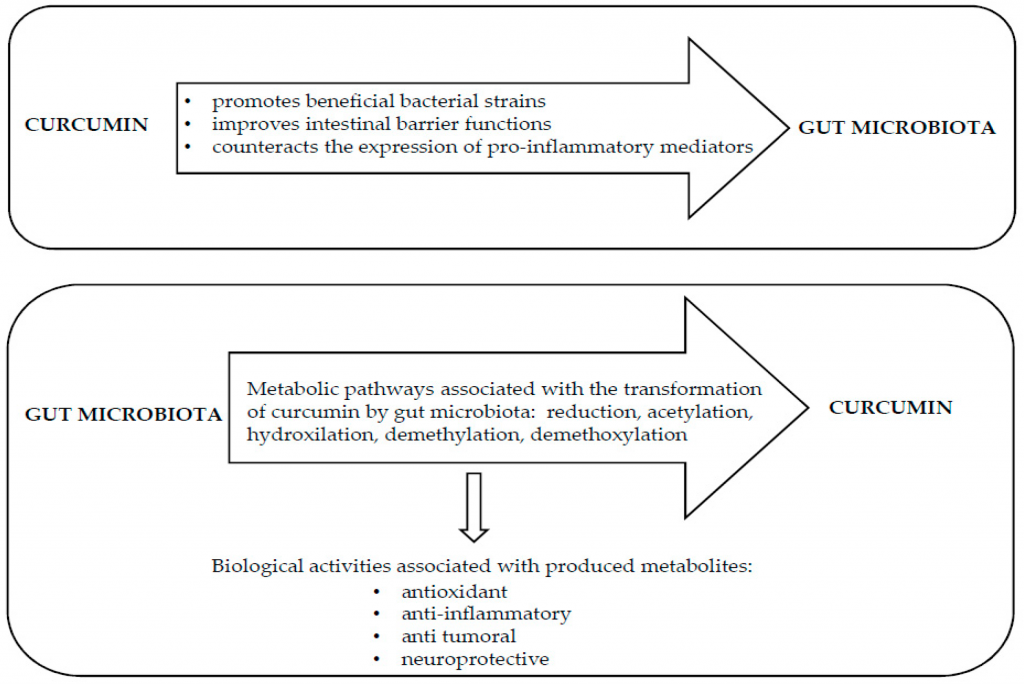
★ 姜黄素直接调节肠道菌群
食用姜黄素与梭状芽孢杆菌、拟杆菌属物种的增加以及Blautia、Ruminococcus的减少有关。
研究证实,口服姜黄素能够显著改变肠道微生物群落中有益细菌和有害细菌的比例,有利于有益细菌菌株的生长,如双歧杆菌、乳酸杆菌和产丁酸菌,并减少致病菌的丰度,如普雷沃氏菌科、Coriobacteries、肠杆菌、Rikenellaceae.
姜黄素治疗会物种的微生物丰度,例如发现结直肠癌患者粪便中的普雷沃氏菌多。患有结肠癌的小鼠被喂食不同的颗粒饲料,姜黄素的计算人体等效剂量为8/mg/kg/天-162 mg/kg/天。最高剂量的姜黄素给药可减少或消除结肠肿瘤负担,增加乳酸杆菌,减少Coriobacteries。
还清楚地证明,姜黄素治疗可减少几种瘤胃球菌;这是一个有趣的发现,因为瘤胃球菌种类的增加与大肠癌的发生有关。此外,在使用致突变化合物治疗的小鼠中,膳食姜黄素能够将乳酸杆菌的数量恢复到控制水平,这已被证明具有抗肿瘤功能。

Scazzocchio B, et al., Nutrients. 2020
★ 肠道菌群对姜黄素进行生物转化,产生活性代谢物
姜黄素的代谢转化不仅发生在肠细胞和肝细胞中,还通过肠道微生物群产生的酶进行,这些酶产生许多活性代谢物。姜黄素代谢产物的生物活性可能不同于天然姜黄素,姜黄素的特定生物学特性实际上取决于肠道微生物群消化产生的生物活性代谢产物。
已经鉴定出几种能够修饰姜黄素的肠道细菌:对从人类粪便中分离的微生物的分析表明,大肠杆菌代表了姜黄素代谢活性最高的细菌,通过NADPH依赖的姜黄素/二氢姜黄素还原酶。这种酶能够将姜黄素转化为二氢姜黄素,然后转化为四氢姜黄素。
其他菌群,如长双歧杆菌、假链状双歧杆菌、粪肠球菌、嗜酸乳杆菌和干酪乳杆菌,代表了能够代谢姜黄素的相关菌株,母体化合物的还原率高于50%.
姜黄素代谢物具有与姜黄素相似的特性和效力:四氢姜黄素表现出与母体化合物相同的生理和药理特性,可能是通过β-二酮部分以及酚羟基。此外,四氢姜黄素能够预防氧化应激和神经炎症,还表现出抗癌作用,这可能是由于抑制了显着的细胞因子释放,例如 IL-6 和 TNFα。因此,在对姜黄素的进一步研究中应考虑细菌分解产物,因为它们可能具有有益作用。
对肠道屏障的影响
体外研究表明,姜黄素是一种潜在的化合物,可以恢复被破坏的肠道通透性。在 CaCo2 细胞中,姜黄素能够减轻肠上皮屏障功能的破坏,抵消 LPS 诱导的 IL-1β 分泌并防止紧密连接蛋白破坏。此外,姜黄素还能够减少由 IL-1β 诱导的 p38 MAPK 活化,以及随后紧密连接蛋白磷酸化的升高。
对肠道炎症的影响
代谢组学分析显示姜黄素对氧化应激和炎症的生物标志物具有有益作用,作者认为,姜黄素治疗抵消了非酒精性脂肪肝进展过程中一些细菌菌株的增加。
一项活体动物研究报告,新开发的纳米姜黄素通过抑制促炎介质的表达和诱导Treg扩张(同时伴随粪便丁酸水平的增加)积极改善DSS-结肠炎小鼠的炎症。将姜黄素与正常啮齿类动物饮食的粉末形式(含有0.2%(w/w)纳米姜黄素)混合:该化合物能够抑制NF-κB的激活和治疗小鼠结肠上皮细胞中促炎症介质的表达。
或者,姜黄素可以通过抑制TLR4/MyD88/NF-κB信号通路的激活来减轻LPS诱导的炎症。此外,姜黄素能抑制NF-κB核转位,并减轻其他在癌症中过度激活的促炎症基因的表达。
在喂食添加姜黄素(300 mg/kg姜黄素,与正常饲料混合)28天的断奶仔猪中,Gan等人证明,这种多酚能够通过抑制大肠杆菌增殖来减轻炎症,下调TLR4的表达。
虽然姜黄素迄今为止在体内研究中描述了所有有益的作用,但这些结果必须通过更大的人体临床试验得到一致的支持。
潜在风险和副作用
姜黄素通常耐受性良好。
常见的副作用包括便秘、消化不良(消化不良)、腹泻、腹胀、胃食管反流(胃酸反流)、恶心、呕吐和其他肠道问题。
极少的情况下,姜黄素会引起瘙痒或凹陷性水肿。
姜黄涂在皮肤上可能会引起过敏性接触性皮炎。
在高剂量体外模型中,姜黄素可引起细胞毒性和 DNA 损伤。
协同效应
添加胡椒碱(来自黑胡椒)可能会增加姜黄素在血液中的吸收。研究人员估计它可能会将姜黄素的生物利用度提高多达 2000%.
白藜芦醇是一种多酚,主要存在于葡萄皮和红酒中。
白藜芦醇是一种小多酚,在 1990 年代引起了科学界的关注。这种化合物被戏称为“瓶中的法国悖论”,因为在红酒中发现了白藜芦醇,法国人喜欢在高饱和脂肪饮食的同时食用不太适量的白藜芦醇。然而,法国人的心脏病发病率非常低。
虽然红酒中的白藜芦醇不太可能完全解释这一悖论,但一些科学家表示它可能是一个促成因素。
葡萄皮中的白藜芦醇含量很高,因为葡萄会产生白藜芦醇来防御毒素和寄生虫。它也存在于各种浆果、花生、大豆中。
有限的研究探索了它的抗氧化、抗炎、抗衰老和植物雌激素活性。白藜芦醇确实具有改善慢性疾病的一些潜力。
白藜芦醇有一个主要缺陷:生物利用度差。
白藜芦醇比其他多酚(如槲皮素)更容易从肠道吸收到血液中。但它会很快分解,在血液中留下很少的游离白藜芦醇。
与肠道菌群之间的关系
白藜芦醇和肠道菌群之间的双向相互作用:肠道菌群进行白藜芦醇生物转化,白藜芦醇对肠道菌群进行相互靶向,从而维持肠道稳态。
白藜芦醇的长期摄入改变了DSS诱导小鼠的肠道菌群,厚壁菌门/拟杆菌门的比例显著提高,这反过来又改变了白藜芦醇的代谢。
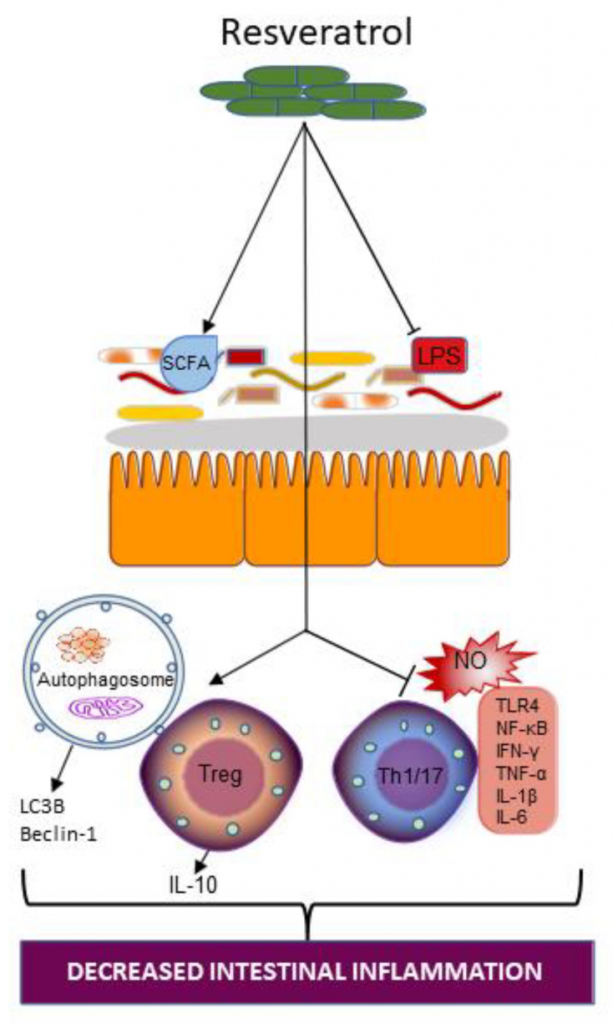
白藜芦醇补充对肠道生态系统的作用

Wellington VNA, et al., Int J Mol Sci. 2021
白藜芦醇添加增加了产短链脂肪酸菌,同时减少了产生LPS的肠道细菌。
补充白藜芦醇也可以通过增加自噬小体的数量和诱导微管相关蛋白1A/1B-light chain 3和Beclin-1的表达来恢复自噬,这两种蛋白在自噬中都是重要的蛋白。白藜芦醇补充也可能会中断Th1/17和细胞因子依赖的促炎通路,一氧化氮依赖的促氧化通路,并干扰toll样受体(TLR) 4信号转导。
白藜芦醇能减轻LPS对小鼠肠道和肝脏的炎症损伤。白藜芦醇减少了拟杆菌和Alistipes的相对丰度,增加了乳酸杆菌的相对丰度。白藜芦醇治疗降低了肝脏中TNF-α、IL-6、IFN-γ、髓过氧化物酶和丙氨酸转氨酶的水平。
此外,益生菌Ligilactobacillus salivarious Li01促进白藜芦醇大量代谢为二氢藜芦醇、硫酸白藜芦醇和白藜芦醇葡萄糖苷酸。在代谢产物中,二氢藜芦醇水平升高最为显著。
肠道菌群的存在促进了二氢藜芦醇的产生,同时促进了硫酸白藜芦醇和白藜芦醇葡萄糖苷酸的消除。

补充剂量
大多数补充剂含有 50 – 500 毫克白藜芦醇。有些含有更高的剂量,高达 1,200 毫克。
临床数据仍然有限。可用的临床研究使用典型的白藜芦醇剂量:
口服纯白藜芦醇的剂量在 150 – 500 毫克/天之间变化。每天喝 1 – 2 杯葡萄酒(100 – 300 毫升)可降低患心脏病的风险并改善血管健康。不含酒精的葡萄酒可能更有益,尤其是对于已经有患心脏病风险的人。
注意:对于有自身免疫和组胺问题的人来说,葡萄酒可能不合适。某些慢病患者如果不确定每天喝一杯葡萄酒是不是安全,请咨询医生。
在接受高剂量(每天 2.5 克或 5 克)白藜芦醇 29 天的人中观察到频繁的胃肠道不适/腹泻。根据 NOAEL 研究,使用 10 倍的安全系数,对于体重 60 公斤的个体,每日 450 毫克白藜芦醇的剂量被认为是安全的。
补充形式
白藜芦醇有多种形式作为口服补充剂:
大多数研究得出结论,反式白藜芦醇是更活跃的白藜芦醇形式。
增加生物利用度的方法
一些可能增加白藜芦醇生物利用度的方法包括:
协同效应
在研究中探索了以下白藜芦醇协同作用:


与药物相互作用
白藜芦醇可能与肠道或肝脏中的药物相互作用,尤其是那些被相同肝酶 (CYP450) 分解的药物。
白藜芦醇还可以减少血液凝固,增强抗凝血药物(抗凝血剂或抗血小板药物,如阿司匹林、氯吡格雷、达肝素、肝素和华法林)的活性。
一起来看下白藜芦醇可能与之相互作用的药物:
“
他汀类药物(Mevacor)
降低高血压的药物(如硝苯地平)
用于减少心律失常的药物(胺碘酮)
抗真菌剂(Sporanox)
抗组胺药(Allegra)
镇静剂/抗焦虑药(安定等苯二氮卓类药物)
抗抑郁药(Halcion)
抗病毒药物和 HIV 药物(蛋白酶抑制剂)
降低免疫反应的药物(免疫抑制剂)
勃起功能障碍(ED)药物
抗凝血药物
NSAID 止痛药/抗炎药,如双氯芬酸 (Voltaren)、布洛芬 (Advil, Motrin)、萘普生 (Anaprox)
……
来源
你知道葡萄酒中含有多少白藜芦醇吗?
通常,白藜芦醇的总浓度为:
红葡萄酒中 0.2 – 5.8 mg/L(平均约为 2 mg/L)
白葡萄酒中仅约 0.68 mg/L
红葡萄酒的反式白藜芦醇含量是白葡萄酒的六倍;白葡萄酒含有高水平的顺式白藜芦醇。红葡萄酒是在不去除葡萄皮的情况下提取的。
白藜芦醇的其他食物来源包括:
黑巧克力、各种浆果、大豆和生或煮花生。
1 杯煮花生含有 1.28 毫克白藜芦醇,大多数食物中的白藜芦醇含量可能太低,无法指望太多特定的健康益处。
副作用
在对健康人进行的临床研究中,每天服用 500 毫克的白藜芦醇具有良好的耐受性。
给予癌症患者高剂量的高生物利用度白藜芦醇(每天 5 克)不会引起任何严重的副作用,但一些患者会出现恶心和胃部不适。
由于缺乏适当的安全数据,儿童应避免使用白藜芦醇。
有人提出,补充白藜芦醇在怀孕期间有益于平衡新陈代谢和产前健康。不过,没有充分的临床研究调查孕妇中的白藜芦醇。
注意事项
白藜芦醇可减少铁吸收和/或血液水平,这可能会恶化贫血。
白藜芦醇转向代表涉及铁代谢(肝素)的重要蛋白质的基因,这可能降低铁吸收。另一方面,白藜芦醇对铁代谢的影响可能是有益的铁过载。
“
肠道菌群的维护和有益菌等的获得以及核心菌群的定植等,与每个人自身的饮食,遗传,生活环境等息息相关,尽量保持饮食多样化,每天食物应该包括蛋白,肉,全谷物,蔬菜,水果,坚果,鱼油/亚麻籽,发酵和多酚食物,食材选择新鲜,无过渡添加/烹饪同时清洁干净的食物,此外,不滥用抗生素或过渡清洁肠道,合理睡眠,适量运动,保持乐观,将有利于肠道和菌群健康。
主要参考文献:
Kato-Kataoka A, Nishida K, Takada M, et al., Fermented Milk Containing Lactobacillus casei Strain Shirota Preserves the Diversity of the Gut Microbiota and Relieves Abdominal Dysfunction in Healthy Medical Students Exposed to Academic Stress. Appl Environ Microbiol. 2016 May 31;82(12):3649-58. doi: 10.1128/AEM.04134-15. PMID: 27208120; PMCID: PMC4959178.
Segers ME, Lebeer S. Towards a better understanding of Lactobacillus rhamnosus GG–host interactions. Microb Cell Fact. 2014;13 Suppl 1(Suppl 1):S7. doi:10.1186/1475-2859-13-S1-S7
Ren JJ, Yu Z, Yang FL, et al. Effects of Bifidobacterium Breve Feeding Strategy and Delivery Modes on Experimental Allergic Rhinitis Mice. PLoS One. 2015;10(10):e0140018. Published 2015 Oct 7. doi:10.1371/journal.pone.0140018
Diaz Ferrer J, Parra V, Bendaño T, Montes P, Solorzano P. Utilidad del suplemento de probioticos (Lactobacillus acidophilus y bulgaricus) en el tratamiento del sindrome de intestino irritable [Probiotic supplement (Lactobacillus acidophilus and bulgaricus) utility in the treatment of irritable bowel syndrome]. Rev Gastroenterol Peru. 2012 Oct-Dec;32(4):387-93. Spanish. PMID: 23307089.
Jeun J, Kim S, Cho SY, Jun HJ, Park HJ, Seo JG, Chung MJ, Lee SJ. Hypocholesterolemic effects of Lactobacillus plantarum KCTC3928 by increased bile acid excretion in C57BL/6 mice. Nutrition. 2010 Mar;26(3):321-30. doi: 10.1016/j.nut.2009.04.011. Epub 2009 Aug 19. PMID: 19695834.
Yoo JY, Kim SS. Probiotics and Prebiotics: Present Status and Future Perspectives on Metabolic Disorders. Nutrients. 2016;8(3):173. Published 2016 Mar 18. doi:10.3390/nu8030173
Tursi A, Brandimarte G, Giorgetti GM, Forti G, Modeo ME, Gigliobianco A. Low-dose balsalazide plus a high-potency probiotic preparation is more effective than balsalazide alone or mesalazine in the treatment of acute mild-to-moderate ulcerative colitis. Med Sci Monit. 2004 Nov;10(11):PI126-31. Epub 2004 Oct 26. PMID: 15507864.
van Zanten GC, Krych L, Röytiö H, Forssten S, Lahtinen SJ, Abu Al-Soud W, Sørensen S, Svensson B, Jespersen L, Jakobsen M. Synbiotic Lactobacillus acidophilus NCFM and cellobiose does not affect human gut bacterial diversity but increases abundance of lactobacilli, bifidobacteria and branched-chain fatty acids: a randomized, double-blinded cross-over trial. FEMS Microbiol Ecol. 2014 Oct;90(1):225-36. doi: 10.1111/1574-6941.12397. Epub 2014 Sep 5. PMID: 25098489.
Kadooka Y, Sato M, Ogawa A, Miyoshi M, Uenishi H, Ogawa H, Ikuyama K, Kagoshima M, Tsuchida T. Effect of Lactobacillus gasseri SBT2055 in fermented milk on abdominal adiposity in adults in a randomised controlled trial. Br J Nutr. 2013 Nov 14;110(9):1696-703. doi: 10.1017/S0007114513001037. Epub 2013 Apr 25. PMID: 23614897.
Fujii T, Ohtsuka Y, Lee T, Kudo T, Shoji H, Sato H, Nagata S, Shimizu T, Yamashiro Y. Bifidobacterium breve enhances transforming growth factor beta1 signaling by regulating Smad7 expression in preterm infants. J Pediatr Gastroenterol Nutr. 2006 Jul;43(1):83-8. doi: 10.1097/01.mpg.0000228100.04702.f8. PMID: 16819382.
Kanjan P, Hongpattarakere T. Prebiotic efficacy and mechanism of inulin combined with inulin-degrading Lactobacillus paracasei I321 in competition with Salmonella. Carbohydr Polym. 2017 Aug 1;169:236-244. doi: 10.1016/j.carbpol.2017.03.072. Epub 2017 Apr 1. PMID: 28504142.
Micka A, Siepelmeyer A, Holz A, Theis S, Schön C. Effect of consumption of chicory inulin on bowel function in healthy subjects with constipation: a randomized, double-blind, placebo-controlled trial. Int J Food Sci Nutr. 2017 Feb;68(1):82-89. doi: 10.1080/09637486.2016.1212819. Epub 2016 Aug 5. PMID: 27492975.
Blanco-Pérez F, Steigerwald H, Schülke S, Vieths S, Toda M, Scheurer S. The Dietary Fiber Pectin: Health Benefits and Potential for the Treatment of Allergies by Modulation of Gut Microbiota. Curr Allergy Asthma Rep. 2021 Sep 10;21(10):43. doi: 10.1007/s11882-021-01020-z. PMID: 34505973; PMCID: PMC8433104.
Wu D, Ye X, Linhardt RJ, Liu X, Zhu K, Yu C, Ding T, Liu D, He Q, Chen S. Dietary pectic substances enhance gut health by its polycomponent: A review. Compr Rev Food Sci Food Saf. 2021 Mar;20(2):2015-2039. doi: 10.1111/1541-4337.12723. Epub 2021 Feb 16. PMID: 33594822.
Scazzocchio B, Minghetti L, D’Archivio M. Interaction between Gut Microbiota and Curcumin: A New Key of Understanding for the Health Effects of Curcumin. Nutrients. 2020;12(9):2499. Published 2020 Aug 19. doi:10.3390/nu12092499
Ding S, Jiang H, Fang J, Liu G. Regulatory Effect of Resveratrol on Inflammation Induced by Lipopolysaccharides via Reprograming Intestinal Microbes and Ameliorating Serum Metabolism Profiles. Front Immunol. 2021 Nov 15;12:777159. doi: 10.3389/fimmu.2021.777159. PMID: 34868045; PMCID: PMC8634337.
Wellington VNA, Sundaram VL, Singh S, Sundaram U. Dietary Supplementation with Vitamin D, Fish Oil or Resveratrol Modulates the Gut Microbiome in Inflammatory Bowel Disease. Int J Mol Sci. 2021;23(1):206. Published 2021 Dec 24. doi:10.3390/ijms23010206
Yao M, Fei Y, Zhang S, et al. Gut Microbiota Composition in Relation to the Metabolism of Oral Administrated Resveratrol. Nutrients. 2022;14(5):1013. Published 2022 Feb 28. doi:10.3390/nu14051013
Del Follo-Martinez A, Banerjee N, Li X, Safe S, Mertens-Talcott S. Resveratrol and quercetin in combination have anticancer activity in colon cancer cells and repress oncogenic microRNA-27a. Nutr Cancer. 2013;65(3):494-504. doi: 10.1080/01635581.2012.725194. PMID: 23530649.
Malhotra A, Nair P, Dhawan DK. Curcumin and resveratrol synergistically stimulate p21 and regulate cox-2 by maintaining adequate zinc levels during lung carcinogenesis. Eur J Cancer Prev. 2011 Sep;20(5):411-6. doi: 10.1097/CEJ.0b013e3283481d71. PMID: 21633290.

谷禾健康

随着高通量多组学技术的快速创新推动,微生物群,尤其是肠道菌群失调已被明确与许多人类疾病有关,包括 2 型糖尿病和炎症性肠病。
多组学数据的综合分析,包括宏基因组学和代谢组学以及宿主指标的检测和细菌物种的分类,已经确定了许多与疾病相关的细菌和细菌产物。然而,深入了解微生物影响肠道健康的机制需要从关联拓展到因果关系。
目前对肠道微生物群对疾病因果关系的贡献的理解仍然有限,这主要是由于微生物群落结构的异质性、疾病进化的个体差异以及对将微生物群衍生信号整合到宿主信号通路中的机制的不完全理解。
最近,德国慕尼黑工业大学从事肠道菌群和营养研究的Haller 教授团队在《Nature reviews gastroenterology & hepatology》 (自然评论胃肠病学和肝病学)发表评论文章,系统讨论了目前已知的炎症和代谢紊乱相关微生物组的特征和认知,并讨论提高对其作用机制理解的困难所在。
在这里我们将文章整理与大家分享。


关键信息
1、肠道菌群组成的改变和细菌衍生代谢物经宿主加工后的变化与 IBD 和 T2DM 相关,并提供了共同的潜在致病机制。
2、益生菌与 IBD 或 T2DM 之间的因果关系已通过无菌小鼠实验和综合多组学研究明确。
3、对于疾病特异性生物标志物发现的挑战,包括确定观察到的变化的因果关系,了解它们在疾病机制中的功能以及肠道微生物群的地理和种族差异。
4、特定细菌菌株、其编码基因和代谢副产物的大数据细化、测试和验证对于识别疾病生物标志物是必要的。
文章内容
人体消化道含有一系列复杂的微生物,包括细菌、古细菌、病毒和真菌。由于消化道及其微生物组被认为位于免疫和代谢过程的交叉点,本文重点关注炎症性肠病 (IBD) 和 2 型糖尿病 (T2DM) 作为微生物群相关疾病的范例。
IBD 和 T2DM 都被认为是多因素疾病,随着工业化的进展其发病率在全球范围内呈上升趋势。病因涉及遗传易感性、环境诱因和城市生活方式相关因素的复杂相互作用。
在这种共同的背景下,代谢疾病(如 T2DM)的另外特征是肝脏、脂肪组织、肌肉、胰腺和肠道的慢性亚临床炎症,而炎症性胃肠道疾病,如克罗恩病(CD)和溃疡性结肠炎(UC) ,也与炎症驱动的代谢改变有关。
环境触发因素的重要性(肠道菌群)
全基因组关联研究已经确定了大量的遗传变异与 T2DM (143 位点) 或 IBD (>240 位点)的易感性增加相关。然而,这些变异共同解释了这些疾病的一小部分遗传性:T2DM < 10 %,UC < 15 % 和 CD < 50 % 。这种情况表明环境触发因素的重要性,特别是肠道微生物组,作为这些疾病病因的主要贡献者。对大型人群研究和 IBD 或 T2DM 患者队列的多项分析已经确定了与特定疾病表型、复发风险和治疗反应相关的微生物组特征。
IBD 和 T2DM 都与特征性微生物改变有关,特别是随着有益微生物的减少和病原菌的增加而降低群落多样性。尽管它们的病理学不同,IBD 和 T2DM 有几个共同的机制特征。T2DM 的代谢特征伴随着慢性低度炎症和肠道屏障的破坏,IBD 患者的复发性炎症发作与细胞和全身水平的代谢改变共同发生。
这些复杂疾病的治疗仍然具有挑战性,但粪便微生物群移植(FMT) 的对照试验已显示出对T2DM和IBD的临床疗效,包括UC以及较轻的 CD。
FMT对炎症、免疫和代谢疾病有效果但存在差异
FMT 的临床试验还提供了肠道菌群与其他炎症、免疫或代谢疾病之间存在因果关系的证据。例如,FMT 在治疗大约 90% 的艰难梭菌(以前的艰难梭菌)感染患者方面非常有效并已被评估为治疗肥胖和移植物抗宿主病。
在四项随机临床试验中,FMT 在 28% 的 UC 患者中诱导了临床缓解。但很少有临床试验检查过 FMT 在 CD 患者中的疗效,而且结果相当不同。
在一项对 174 名接受 FMT 治疗的 CD 患者进行的研究中,20% 的患者获得了临床缓解,总体而言,43% 的患者获得了临床缓解。
一项单独的随机对照试验发现 FMT 对 CD 患者的临床缓解率没有影响,但供体微生物群的植入增加与维持缓解有关。相反(尽管大量研究表明,特定的菌群失调或特定的微生物群谱与代谢紊乱有关),FMT 对代谢性疾病患者有益的证据尚不明确。具有里程碑意义的研究表明,从较瘦、健康的捐赠者那里接受 FMT 的代谢综合征患者的代谢改善以及肠道微生物组的有益变化。然而,这些影响是不一致和短暂的,这可以通过供体微生物群的有限移植或基线时供体粪便微生物多样性的变化来解释。
有趣的是,口服 FMT 后补充低发酵性纤维可改善肥胖和代谢综合征患者的胰岛素敏感性、增加微生物多样性,并延长供体微生物定植。这些数据强调了微生物调节疗法在逆转代谢功能障碍中的价值。
与这些发现一致,来自代谢受损的肥胖供体的 FMT 会暂时恶化代谢综合征受体的胰岛素敏感性,而胃旁路术后健康供体的 FMT 会导致代谢综合征受体的胰岛素敏感性略有增加。
几项大型队列研究(表 1、表2)通过分析 IBD 患者的肠腔和黏膜微生物群落,研究了肠道微生物群的改变。
总体而言,活跃期IBD 与某些菌群的数量过多有关,如:
肠杆菌科Enterobacteriaceae
梭杆菌属Fusobacterium
咽峡炎链球菌Streptococcus anginosus
肠球菌Enterococcus
巨球菌Megasphaera
弯曲杆菌Campylobacter
Gammaproteobacteria
Deltaproteobacteria
相反,IBD 与有益菌群的缺失有关,例如:
Faecalibacterium prausnitzii
Christensenellaceae
Collinsella
Roseburia
Ruminococcus
其他产丁酸盐的细菌
在我们检测的炎症性肠病的菌群报告中也发现,炎症性肠病风险高的人群中,炎症水平很高,肠杆菌科Enterobacteriaceae,梭杆菌属Ruminococcus gnavus偏高,而Faecalibacterium prausnitzii和Roseburia丰度普遍降低或者缺乏。


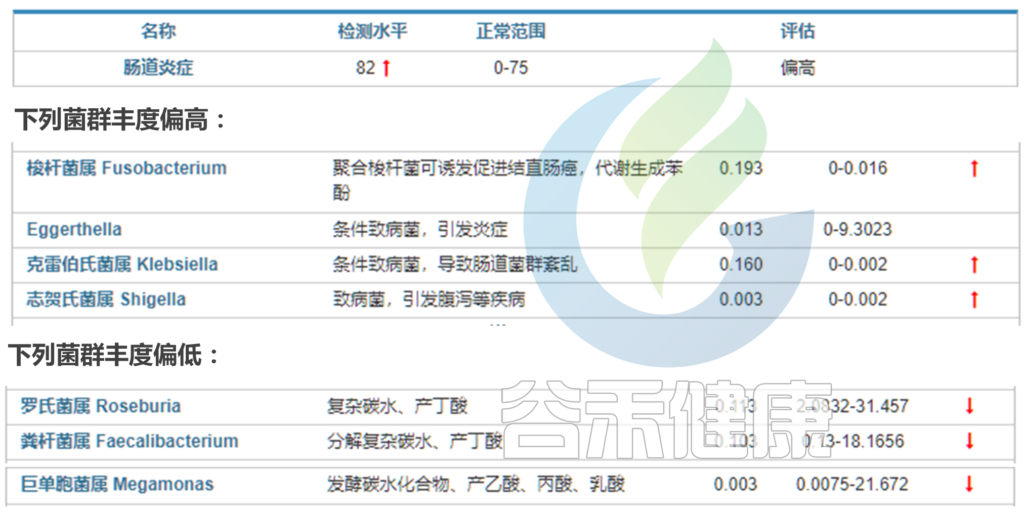
< 选自:谷禾肠道菌群健康检测报告 >
菌群代谢功能
粪便样本的宏基因组学为 IBD 中发生的功能失调和代谢途径的扰动提供了更全面的观点。这些研究表明,参与含硫氨基酸合成、核黄素代谢、谷胱甘肽转运蛋白、氧化应激和营养转运的代谢途径均被上调。
一项能够将微生物群落分解到物种内单个菌株水平的研究显示,与健康对照相比,IBD 或肠易激综合征 (IBS) 患者粪便样本中致病菌的菌株多样性增加,而有益微生物的菌株多样性降低。深入分析表明,219个类群(包括152种)与CD相关,102个类群(包括93种)与UC相关。
CD的主要特征是属于毛螺菌科和瘤胃球菌科的分类群减少和属于肠杆菌科的分类群增加,而对于UC观察到属于拟杆菌科的分类群减少和属于毛螺菌科的分类群增加。与这种异质性一致,在不同的 IBD 研究中仅存在少数物种的共同变化,这表明尽管疾病表型和病程相似,但在 CD 患者群体中仍存在个体间差异。
IBD和肠道菌群的因果关系探究
肠道微生物群在 IBD 中的致病作用的首批临床证据之一源于实验表明,从 CD 患者的小肠发炎段转移粪便流可改善疾病症状。粪便流的恢复和新末端回肠术后暴露于肠腔内容物会诱发炎症,这表明肠道微生物群会引发 CD的术后复发。此外,抗生素治疗对活动性 CD 患者亚群的疗效强调了肠道细菌和 IBD 之间的因果关系。
急性和慢性肠道炎症小鼠模型的机制研究为微生物失调与 IBD 之间因果关系的提供了进一步证据。例如,将IBD 患者的菌群移植到无菌受体小鼠,会将IBD 表型转移到无菌小鼠中。而具有 IBD 遗传易感性的小鼠在无菌条件下不会发生自发性炎症。
免疫响应
IBD 发病的遗传易感小鼠的失调菌群,能够将这种疾病症状转移到无菌受体小鼠。将 IBD 微生物群转移到无菌的野生型小鼠体内会导致肠道 T 细胞反应失衡,肠道 T 辅助 17 (TH17) 细胞和 TH2 细胞数量增加,RORγt +数量减少。同样,肠道微生物群的人类共生细菌脆弱拟杆菌对无菌小鼠的定植诱导CD4 + T 细胞转化为产生IL-10的FOXP3 + T reg细胞,这表明微生物群驱动的存在IBD 的发病机制。
在 T2DM 中也发现了几种细菌类群的丰度变化很大。
例如,据报道,2型糖尿病患者中下列菌相对丰度增加:
大肠杆菌E. coli、
韦荣氏菌属Veillonella、
布劳氏菌属Blautia、
厌氧菌属Anaerostipes、
乳杆菌属Lactobacillus、
粪杆菌属Faecalibacterium、
梭状芽胞杆菌属Clostridiales(等)
相反,下列菌丰度降低:
拟杆菌属Bacteroides
双歧杆菌属Bifidobacterium、
副拟杆菌属Parabacteroides、
颤螺菌属Oscillospira
可降解粘蛋白的阿克曼菌Akkermansia muciniphila
2019 年发表的一项宏基因组和宏蛋白质组学研究分析了来自 254 名中国个体的粪便样本中的肠道微生物群组成和功能,其中包括 77 名未接受治疗的 T2DM 患者、80 名糖尿病前期患者和 97 名葡萄糖耐量正常的对照个体。与代谢健康的对照组相比,T2DM 患者和前驱糖尿病患者的梭菌目Clostridiales细菌丰度较低,而埃氏巨球形菌Megasphaera elsdenii的丰度较高。
菌群代谢功能
在 T2DM 患者和糖尿病前期患者的微生物组中观察到功能差异。与对照个体相比,糖尿病前期个体的肠道微生物群显示出与糖磷酸转移酶系统、细菌分泌系统和氨基酸的 ATP 结合盒 (ABC) 转运蛋白有关的途径富集。这些发现表明,在糖尿病前期患者转变为 T2DM 之前,可以检测到肠道微生物组的疾病特异性变化。
环境因素影响
细菌种类和代谢基因簇谱的差异已被用于确定一组具有正常葡萄糖耐量或 T2DM 的个体的糖尿病状态。然而,包括地理位置、种族、健康状况和用药史在内的混杂因素导致在识别与 T2DM 相关的微生物变化方面不同研究存在不一致。
因果关系研究
几项研究提供了肠道微生物群特定成员与 T2DM 发病机制之间因果关系的证据。例如,A. muciniphila属于在人类和小鼠研究中显示对代谢紊乱具有保护作用的分类群。有趣的是,补充益生元使A. muciniphila的丰度正常化并改善了人类的代谢健康。同样,对喂食高脂肪饮食的小鼠施用A. muciniphila可逆转其增加的脂肪量、代谢性内毒素血症、脂肪组织炎症和胰岛素抵抗。
此外,产生丁酸盐的细菌Anaerobutyricum soehngenii(以前称为Eubacterium hallii菌株 L2-7)显示出丰度增加,这与来自瘦供体的FMT受体的外周胰岛素敏感性改善相关。
对T2DM 患者A. soehngenii菌水平进行管理,在治疗 4 周后改善了外周胰岛素敏感性,这些益处伴随着微生物群组成的改变和胆汁酸代谢的变化。
奇怪的是,特定的细菌分类群在 IBD 和 T2DM 中表现出相似的变化,这表明免疫介导和代谢疾病的共同特征导致微生物群的相似适应。
下列菌丰度下降:
梭状芽孢杆菌属 Clostridium spp.
粪杆菌属 Faecalibacterium
瘤胃球菌属 Ruminococcus
阿克曼氏菌属 Akkermansia
柯林斯氏菌属 Collinsella
罗斯氏菌属 Roseburia
下列菌丰度增加:
肠杆菌科 Enterobacteriaceae
大肠杆菌 E. coli
具核梭杆菌 Fusobacterium nucleatum spp.
这为定义疾病特异性标志物提出了挑战(下图)


Metwaly et al.,Nat Rev Gastroenterol Hepatol. 2022.
例如,一项针对 2,045 名 IBD 患者的研究的作者确定了 8 个分类群的特征,包括克里斯滕森菌科Christensenellaceae和梭杆菌属Fusobacterium的未知成员,它们可以区分 CD 患者和健康个体。
然而,Christensenellaceae 的丰度增加与低 BMI 和体重减轻有关,这是一种在 IBD 患者中经常观察到的分解代谢状况。同样,梭杆菌的富集被认为是转移性结直肠癌患者预后不良的标志。鉴于 IBD 患者患结直肠癌的风险增加,这一提议的微生物组特征可能是一种附带现象,与潜在的疾病机制没有因果关系。
对来自 132 名个体的微生物组、代谢组和转录组数据集的综合网络分析确定了连接关键细菌分类群(即F. prausnitzii 、未分类的Subdoligranulum、Alistipes、大肠杆菌、Roseburia)的某些代谢物(短链脂肪酸、辛酰肉碱和几种脂质)。有趣的是,有和没有 IBD 的研究参与者之间的差异在粪便代谢组中,比在粪便宏基因组、粪便宏转录组或粪便蛋白质组中更明显。
在综合个人组学分析研究 (iPOP) 中,血浆代谢物与来自 106 名个体的纵向样本中的胰岛素抵抗密切相关,这表明宿主代谢组和肠道微生物组之间的相互作用在胰岛素抵抗个体中受到干扰。
许多研究调查了微生物改变作为疾病生物标志物的效用,特别是在 CD 或 UC 患者中。最初试图定义可以作为疾病活动指标的单一细菌分类群。
例如,F. prausnitzii (一种产生丁酸盐的厚壁菌)在 CD患者中被耗尽。CD 患者回肠黏膜活检样本中这种细菌丰度的降低与回肠切除术后内镜下复发风险的增加密切相关。相反,粘附的侵袭性大肠杆菌丰度增加与回肠 CD相关。
然而,由于大多数细菌物种由许多个体菌株组成,这些菌株可以表现出相当大的基因组和蛋白质组变异,因此菌株多样性具有重要的功能,特别是在确定致病性方面。
例如,R. gnavus和大肠杆菌的亚种都与 IBD 的严重程度增加有关。此外, R. gnavus的一个特定亚种表明,来自 IBD 患者的粪便样本中丰度增加含有菌株特异性基因(与改善的细菌定植有关)。这些基因涉及诸如氧化应激反应、细菌粘附、铁获取和宿主粘液利用等功能。同样,不同的脆弱拟杆菌菌株表现出功能差异,导致 IBD 相关小鼠模型中 IgA 诱导水平不同。这些遗传上不同的脆弱拟杆菌菌株在接种到受体小鼠时也表现出不同的致结肠和免疫调节作用。在一项旨在定义用于监测 IBD 患者疾病活动的关键菌群失调的研究中,使用定量 PCR 计算了F. prausnitzii和大肠杆菌的绝对丰度比(也称为 F-E 指数)。F-E 指数的使用提高了 UC 和 IBS 患者与 CD 患者的区分,并有助于区分结直肠癌与其他肠道疾病。然而, F-E 指数无法区分 IBD亚型,这表明单一分类群指标在分类疾病亚型方面的效用有限。
大规模生物标志物分析
基于微生物特征的判别模型
几项研究已经使用机器学习算法来验证横截面和纵向患者队列中复杂的微生物组特征。
例如,2017 年发表的一项研究使用 16S rRNA 微生物群来分析来自大量 IBD 患者和没有 IBD 的对照个体的粪便样本。研究人员使用序列聚类算法根据八种细菌类群的丰度来识别 CD 特异性微生物特征。
此外,另一组研究表明,基线肠道微生物组组成的特征可以预测 IBD 患者在治疗开始后14周对抗整合素治疗的反应。由深度神经网络生成的微生物组特征的受试者工作特征曲线 (AUC) 下面积为 0.87,而基于临床协变量的模型的 AUC 为 0.62。研究小组还评估了微生物组特征作为 IBD 和 T2DM 生物标志物的效用。
在一项研究中,检查了 29 名接受过自体造血干细胞移植的 CD 患者的独特队列中的疾病活动性和对治疗的反应。来自人类供体和人源化小鼠的微生物组和代谢组风险概况的整合将疾病结果的预测模型的性能从 AUC 0.79 提高到 0.96,并确定了与硫代谢相关的疾病相关细菌和代谢物网络。
这些发现听起来很有前景,但重要的是要承认微生物组风险概况是基于来自前瞻性队列研究中的人群或患者组的预测模型,因此比起对于个人的预测结果,对于相似患者组(人群或队列)可能更准确。重要的是记住,预测的风险可能不会直接转化为个体患者,这可能是由于在异质环境中风险概况的普遍性有限。
不忽略混杂变量
另一项研究调查了以德国人群为基础的 1,976 人队列中的代谢健康和肠道微生物群的昼夜节律性。粪便微生物群分析确定了 13 个微生物分类群的风险特征,这些分类群显示 T2DM 患者的昼夜节律性受到破坏。基于这种心律失常风险特征的预测模型成功识别出有患 T2DM 风险的个体,当模型中包含 BMI 时,AUC 为 0.78。
这些例子为微生物组特征在用于诊断和治疗目的的生物标志物发现中的作用提供了证据。然而要注意,生态失调指数不是独立的测量值,需要整合到额外的宿主衍生数据和临床数据中。这些指标的标准化和验证需要大规模究研,包括对潜在生物标志物的纵向评估,并考虑可能的混杂变量,例如饮食、年龄、种族、病史和最后的排便时间,所有这些因素都会影响微生物组的改变。
在寻找疾病生物标志物时,代谢物作为疾病活动的最接近指标,并且与作为疾病机制基础和调节疾病机制的调节信号密切相关。事实上,代谢组和微生物组都随着饮食、环境、衰老和整体健康状况等内源性和外源性因素而波动。
许多研究报告了 IBD或 T2DM患者的肠道代谢物谱的显着变化。
例如,已在 IBD 患者的粪便代谢组中发现中链脂肪酸(如戊酸和己酸水平降低)和 B 族维生素水平降低。相反,据报道,成人和儿童 IBD 患者的粪便和血清中氨基酸、胺和肉碱的含量分别增加。
一项具有里程碑意义的研究结果表明,代谢物分析可以区分 IBD 患者和健康个体。该报告之后有许多其他人一致表明 IBD 患者的代谢物表型与健康个体的不同。有趣的是,代谢物分析还可以区分不同形式的 IBD,例如 CD 和 UC,并且可以进一步将 CD 患者分类为患有回肠或结肠炎症。同样,T2DM 患者的代谢物分析表明代谢途径的活性发生了改变。
在多份报告中,支链和芳香族氨基酸(如亮氨酸、异亮氨酸、缬氨酸、苯丙氨酸、酪氨酸和色氨酸)的血清水平与胰岛素抵抗、肥胖和发生 T2DM的风险相关。
T2DM 患者的代谢物分析还揭示了特定细菌代谢物水平与疾病发作之间的强关联。
例如,色氨酸代谢途径包括几个候选代谢物生物标志物,这些生物标志物由于与人类和小鼠研究中炎症和代谢疾病的发展相关而引起了研究关注。色氨酸是一种从饮食中获得的必需氨基酸,主要在小肠中吸收,尽管一小部分在结肠中分解代谢为吲哚代谢物。
一项纵向队列研究证实,在 213 名中国个体(包括 51 名继续发展为 2 型糖尿病和 162 名保持代谢健康的个体)中,高基线空腹血清色氨酸浓度与患 T2DM 的风险增加有关。此外,色氨酸水平作为生物标志物的预测能力与五种现有氨基酸在区分患有和未患 T2DM 的个体方面的预测能力相当 。
值得注意的是,先前的报告表明,几种氨基酸的血清水平可以以不同的准确性识别来自不同人群的 T2DM 患者。例如,苯丙氨酸和缬氨酸在美国人群中表现最好,而酪氨酸在南亚人群中最准确。这些发现指出了识别区域特异性生物标志物在实现最佳诊断准确性方面的重要性。
有价值的生物标志物必须为从临床信息中获得的分类能力提供额外的分类能力。因此,粪便生物标志物是粘膜疾病诊断标志物的明显来源,因为粪便流与肠粘膜直接接触。
钙卫蛋白
粪便钙卫蛋白是一种可在粪便中检测到的粒细胞衍生的细胞溶质蛋白,由于炎症严重程度与粪便钙卫蛋白水平之间的相关性强,它是最广泛使用的用于炎症性疾病的粪便生物标志物。
两份报告证实了粪便钙卫蛋白水平检测内窥镜炎症的能力,报告的敏感性为70-100% ,特异性为44-100%。这些值的广泛范围可以通过每项研究中应用的截止阈值的变化来解释。
然而,粪便钙卫蛋白水平升高并不是 IBD 特有的,而是反映了肠道炎症状况,这也与其他肠道和代谢疾病(包括 IBS、胃肠道恶性肿瘤、肥胖和 T2DM)有关。
例如,对来自 1,792 个人的粪便样本中肠道微生物群的鸟枪宏基因组分析能够区分 IBD 和 IBS,与单独的粪便钙卫蛋白水平 (AUC) 相比,机器学习算法显着提高了这些预测模型的准确性 (AUC 0.91>0.80)。
重要的是,具有最高预测准确度 (AUC 0.93) 的模型包括粪便钙卫蛋白水平以及前 20 个选定分类群的宏基因组分析。这些结果表明,临床和微生物生物标志物的整合提高了诊断的准确性。这种综合方法已被用于预测 IBD 患者对治疗的反应。
在这项研究中,基线临床数据(包括血清学、内窥镜和临床生物标志物)不足以预测缓解(AUC 0.62),而添加分类学和代谢谱将诊断能力分别提高到 AUC 0.72 和 AUC 0.74。
此外,仅粪便钙卫蛋白水平就能够区分储袋炎患者和无储袋炎患者 (AUC 0.63)。相比之下,微生物组物种模型(有或没有粪便钙卫蛋白水平作为额外的预测因子)实现了 0.78 的 AUC,证实微生物分析在识别储袋炎方面具有优于仅粪便钙卫蛋白水平的诊断性能。
用于诊断 T2DM 的葡萄糖代谢受损的血清学生物标志物包括空腹血糖水平、75g口服葡萄糖激发后 2 小时血糖水平(口服葡萄糖耐量试验)和糖化血红蛋白水平。
一项使用来自两个瑞典队列136数据的研究确定了预测 T2DM 进展的血清学和微生物组生物标志物的组合。
在这项研究中,多变量分析表明胰岛素抵抗程度与微生物组变异之间存在很强的相关性。
有趣的是,使用基于微生物组的机器学习模型来区分验证队列中胰岛素抵抗程度最低和最高的个体的 AUC 为 0.78,这表明肠道微生物群是 T2DM 进展的重要调节因子。
事实上,尽管已经为 T2DM 提出了广泛的诊断生物标志物,但它们中的大多数未能捕捉到这种疾病的复杂性或掌握微生物和代谢的变化。在这方面,已将代谢物生物标志物与已确定的临床风险因素结合使用,以显着改善疾病分类。
微生物特征
了解单个细菌类群(病原菌)和/或复杂微生物群落(生态失调)变化的功能作用和特异性对于解决IBD或T2DM中微生物-宿主相互作用的发病机制至关重要。在这种情况下,肠道微生物组的功能改变可能代表宿主适应的结果。

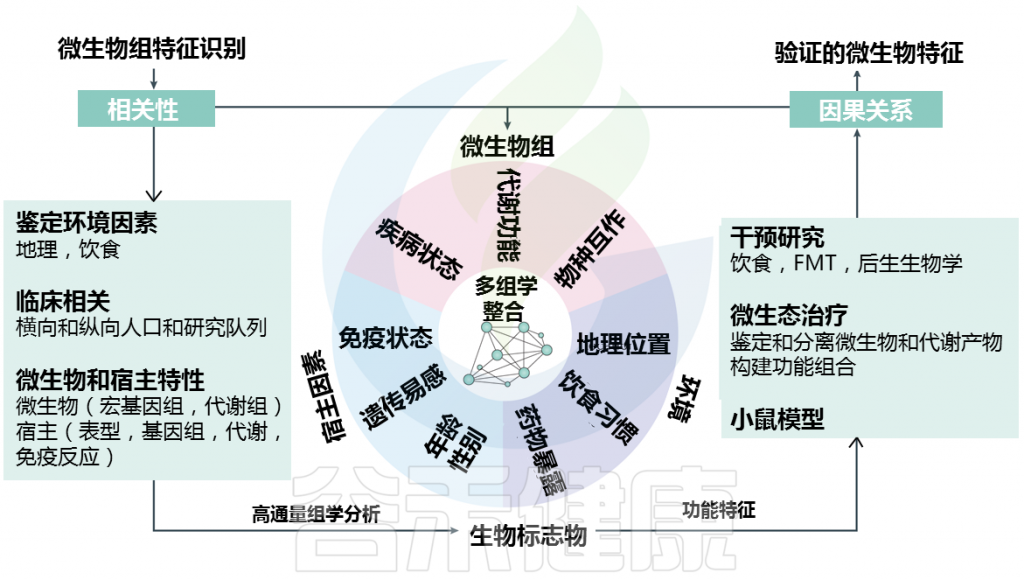
Metwaly et al.,Nat Rev Gastroenterol Hepatol. 2022.
肠道微生物群与多种疾病之间存在因果关系,已在小鼠实验中得到证实。无菌小鼠模型可以选择性地用单一细菌菌株、最小细菌聚生体或来自人类粪便或其他供体材料的定义复杂的肠道微生物生态系统进行定植,以研究它们对宿主表型的影响。在 IBD 中,无菌小鼠模型的单菌株定植有多种共生细菌,包括大肠杆菌、粪肠球菌、普通拟杆菌和Bilophila wadsworthia使我们能够了解疾病引发或保护的一些潜在机制。已有的研究工作表明,肠道菌群是驱动结肠炎小鼠模型炎症所必需的,而这些细菌与 CD患者的复发风险相关。肠道微生物群移植到无菌小鼠体内导致了几种疾病状态,从而揭示了与炎症有关的共享功能代谢途径。同样,以前的工作表明葡萄糖耐量和胰岛素抵抗受肠道微生物组组成的影响,已通过一系列 FMT 研究得到证实。
在过去的二十年里,人类和小鼠研究的证据揭示了肠道微生物组在炎症和代谢疾病(如 IBD 和 T2DM)的发病机制中的基本作用。肠道微生物生态系统结构和功能的变化(失调),与这些疾病患者的疾病活动、复发风险或对治疗的反应有关。然而,大多数这些疾病的复杂性和多因素发病机制,以及人类研究中存在多种混杂因素,依然对微生物组特征在诊断、预测预后和治疗决策中的临床应用提出重大挑战。
当前的微生物组研究不仅仅局限于描述微生物群落结构和疾病关联,还在了解肠道菌群在复杂慢性病发病机制中的致病作用方面取得进展。预计这些努力将增强微生物组建模,并推进可用于临床环境的基于菌群特征和/或疾病风险的模型开发。
参考文献:
Metwaly A, Reitmeier S, Haller D. Microbiome risk profiles as biomarkers for inflammatory and metabolic disorders. Nat Rev Gastroenterol Hepatol. 2022 Feb 21. doi: 10.1038/s41575-022-00581-2. Epub ahead of print. PMID: 35190727.

谷禾健康

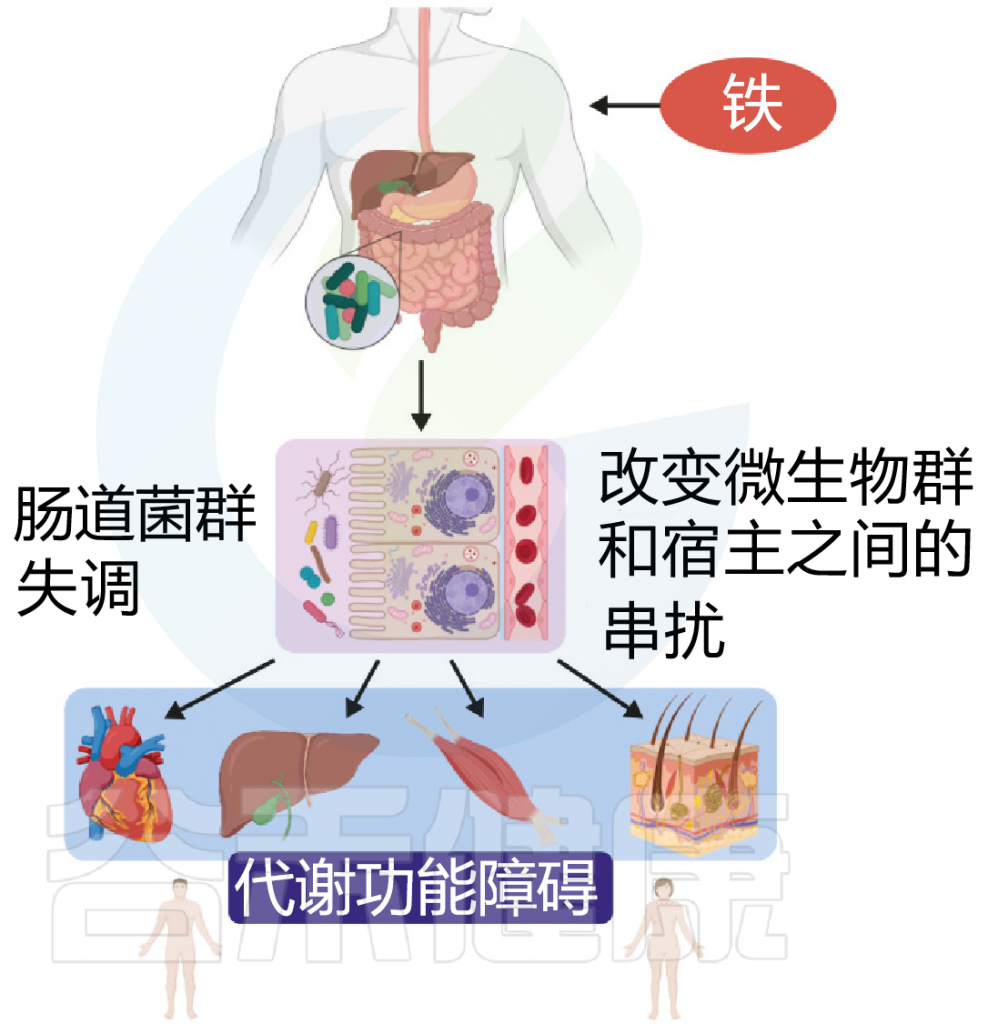
铁 (Iron)作为人体不可或缺的元素之一,在氧气运输,新陈代谢和免疫防御中起着基本作用。铁元素的良好调控是人体健康的保障,过多过少都会对人体产生不利影响。铁稳态的失调与各种疾病的发生和发展息息相关:铁缺乏会导致宿主发育迟缓,免疫低下,而铁过载更易引发炎性反应和代谢问题, 还可能与癌症的发生发展密切相关。
在人类中,小肠作为宿主体内铁的主要吸收场所,大约每天吸收2-3mg铁,但是在一些病理条件下,小肠并不能完全将食物中的铁吸收,而相当一部分铁进入结肠中与肠道菌群发生密切的交互作用,并影响肠道菌群的“生态系统”,从而进一步调节宿主的健康和代谢。
自法国国家农业研究院与法国国家健康与医学研究院(INSERM)的研究人员首次发现肠道菌群如何影响肠道内铁元素的转运与储备成果后,多项研究表明,铁失衡会导致肠道菌群的改变,进而改变微生物多样性,增加病原体丰度并诱导肠道炎症的发生发展。
谷禾在多年的肠道菌群研究和检测实践中,也发现饮食中的铁水平是导致肠道菌群改变的重要因素。我们基于机器学习算法,建立了预测模型,可以预测评估膳食摄入和铁的菌群代谢的总体水平,这有助于避免缺铁和铁过量引起的肠道菌群失调及其可能导致的宿主健康损害和疾病风险。
铁和微生物组之间相互作用
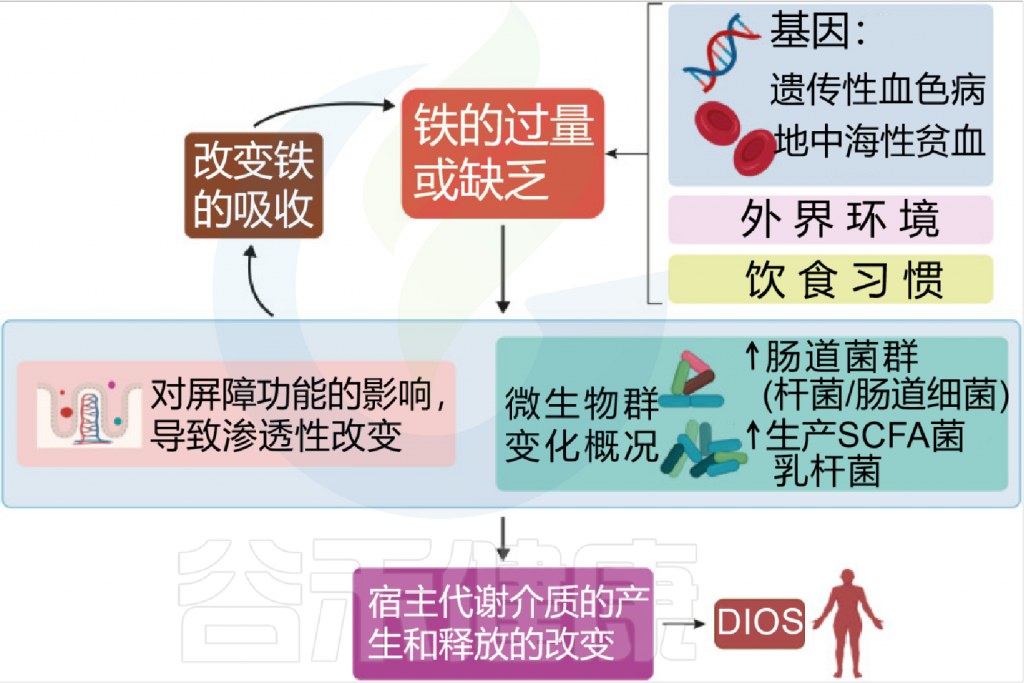

Botta A, et al., J Lipid Atheroscler. 2021
铁是人类必需的营养素,在环境中含量非常丰富,并参与了许多生物过程,如氢气的产生、呼吸和DNA 生物合成。它还在宿主细胞内的许多代谢途径中起到辅助因子的作用。
在人类中,由于没有排泄铁的途径,小肠上皮细胞对铁的吸收是一个非常严格的调节过程。
吸收效率取决于饮食中铁的潜在可用性,并受生理铁需求的调节,包括体内铁储存,铁调素在控制吸收中起核心作用。
膳食铁大致分为两种类型,非血红素铁和血红素铁。两种形式的膳食铁都有单独的肠细胞吸收途径:


这里主要关注的是非血红素铁(口服铁通常以非血红素铁的形式给予)。
肠上皮细胞对非血红素铁的吸收
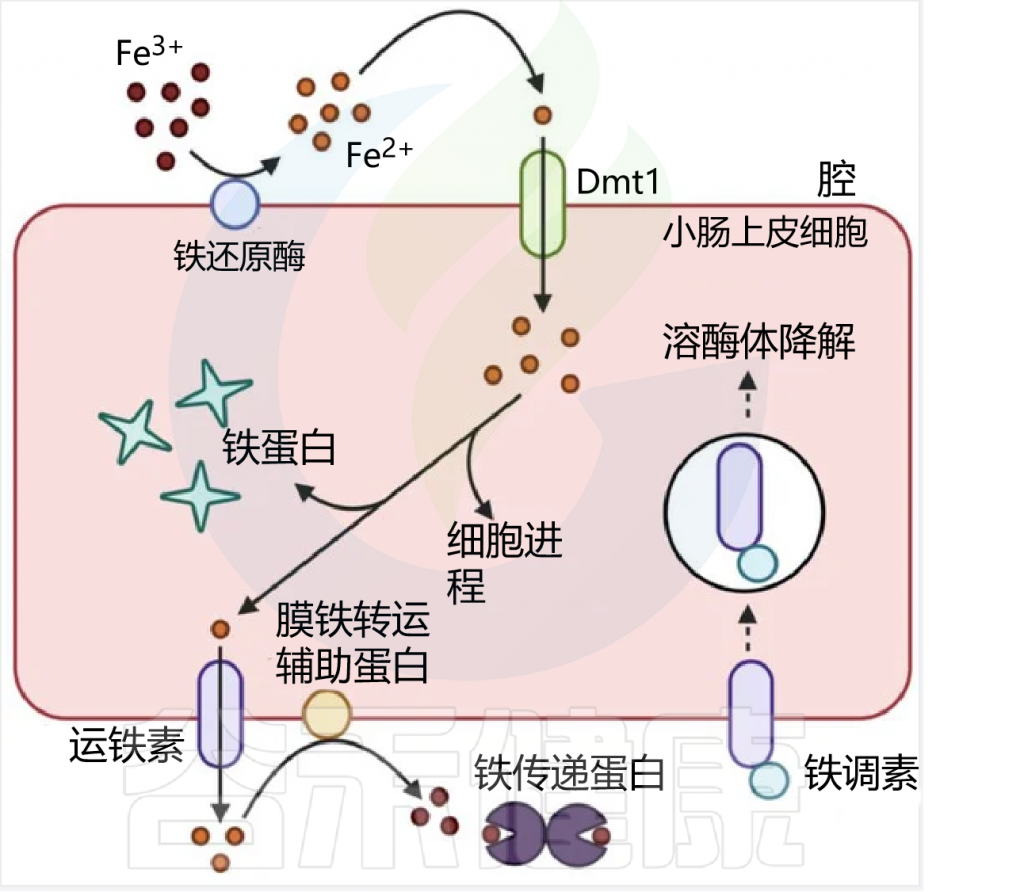

日常生活中,缺铁的常见症状包括:
一般我们常见的缺铁,主要是以下几个原因:
这很好理解。我们的身体不能制造铁,需要从食物中获取。如果你每天摄入的铁量没有达到身体需要的量,就会出现缺铁。
快速生长会增加铁需求,因此儿童、孕妇和哺乳期女性更可能缺铁。此外,进行耐力锻炼的人更容易患低铁症。
出血和失血会增加铁的流失,包括:月经出血、分娩、溃疡、痔疮、因受伤或手术而出血、献血等。长期使用消炎药(布洛芬、萘普生、双氯芬酸)会增加肠道出血的可能性。
需要注意的是,缺铁性贫血的一个常见且常被忽视的原因是月经过多。随着时间的推移,这种缓慢的血液流失通常会导致铁流失过多。
在慢性炎症性疾病中也会出现缺铁,例如自身免疫性疾病、慢性感染、慢性肾病或癌症。这被称为慢性病性贫血。
因为铁对病原体和癌细胞的生长很重要,所以当发生感染或炎症时,身体会试图通过锁定铁来抑制病原体或恶性细胞的生长。
受感染或炎症影响的组织会释放降低铁血水平的细胞因子,从而导致贫血的发展。
消化系统的疾病会降低铁的吸收,包括:
乳糜泻、炎症性肠病、胃炎、幽门螺杆菌感染、小肠细菌生长过度等,此外,减肥手术也会减少营养吸收,包括铁。
铁吸收不足的其他原因包括大量摄入抑制铁吸收的食物或药物,包括:植酸盐(全谷物、豆类)、多酚(茶、咖啡、葡萄酒)、抗酸药、H2受体阻滞剂、四环素或消胆胺等药物等。还有一个很重要的我们可能会忽略的环节,就是菌群。
细胞铁浓度是感染性的关键决定因素之一。肠道病原体在宿主细胞中的存活可能部分取决于宿主铁的状态。铁也可以促进肠道病原体的复制和毒力,例如沙门氏菌属、志贺氏菌属和弯曲杆菌属。
利用铁载体获取铁的一些细菌还包括如:
大肠杆菌、铜绿假单胞菌、肺炎克雷伯菌、金黄色葡萄球菌和结核分枝杆菌。
这些病原菌的定植会与宿主进行铁的竞争吸收,同时诱发肠道炎症,改变肠腔环境,影响菌群构成,进一步导致缺铁加剧。


大多数细菌都依赖于铁的存在来进行呼吸和各种代谢过程。在细菌内部,铁在生长和增殖中起着至关重要的作用,例如,某些细菌蛋白质和酶的正常功能需要铁。此外,铁还可以调节某些毒力因子的表达。
铁浓度的波动会产生病理影响,对肠道微生物群组成产生负面影响。铁的波动有两种情况,过多或者缺乏,接下来我们逐一来看铁过量和缺乏会对菌群造成什么影响。
宿主铁稳态的改变可能会影响肠道的管腔铁含量,从而影响肠道菌群的组成。在无菌小鼠中,铁转运蛋白表达减少两倍,细胞铁含量较低,在移植肠道细菌定植后,上皮细胞利于铁的能力增加,细胞内铁含量增加。
铁过量
肠道中富含铁的环境有利于变形菌。对儿童的研究表明,过量的铁会导致炎症和病原菌的生长。这些病原菌可能会诱发炎症性肠病或结直肠癌。铁的强化增加了粪便钙卫蛋白的水平,显示了肠道的炎症。
患有铁过载综合征(包括血色素沉着症和难治性贫血)的人更容易受到细菌感染,包括:
耶尔森菌属Yersinia
单核细胞增生李斯特菌Listeria monocytogenes
创伤弧菌Vibrio vulnificus
膳食铁过量摄入可影响大鼠正常的生长发育,并引起小肠粘膜炎性损伤。过量铁摄入可引起大鼠肠道菌群失调,肠道菌群丰度有所降低,其中乳酸杆菌、双歧杆菌降低,血清中肿瘤坏死因子和内毒素水平升高可能与肠道菌群紊乱有关。
铁缺乏
当铁水平下降时,也会对肠道微生物群组成产生影响,诱发肠道感染。缺铁会抑制细菌细胞繁殖过程,从而损害细菌生长。
在啮齿类动物模型中,缺铁导致微生物群组成的显著重组,微生物多样性降低。
在铁含量非常低的条件下(0.9 mg Fe/L),Roseburia,肠杆菌减少,丁酸盐水平也降低,而Lactobacillus增加。此外,低铁条件下生长的Roseburia gutis优先产生乳酸而不是丁酸盐。
图 铁和炎症之间相互作用
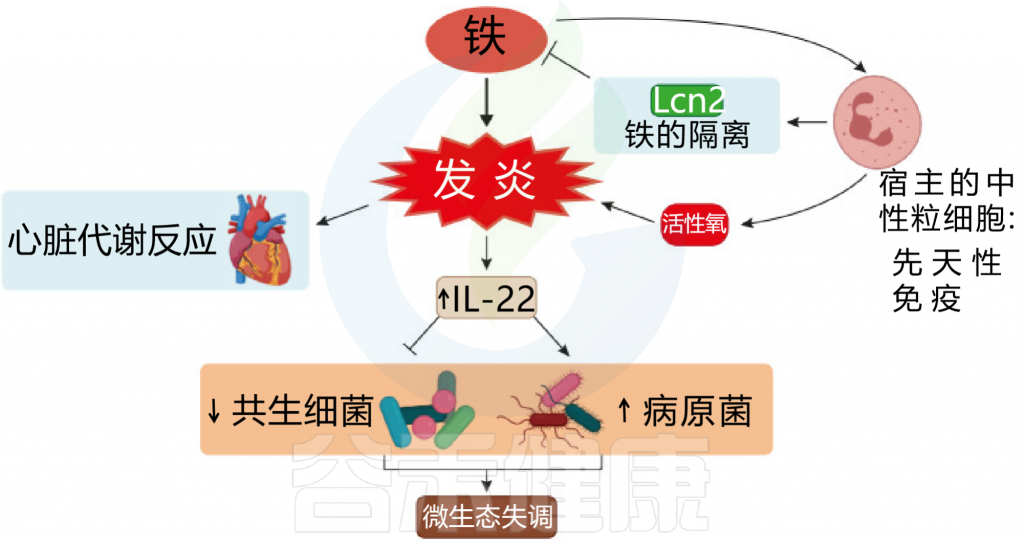

Botta A, et al., J Lipid Atheroscler. 2021
铁补充
铁的补充和强化对人体肠道细菌组成有不同的影响。
接受低剂量(0-10 mg Fe/天)或高剂量(大于60 mg Fe/天)铁补充剂的孕妇在肠道菌群任何分类水平上均没有显著差异。
接受含铁微量营养素粉(12.5 mg/天)的肯尼亚婴儿的病原体丰度增加。在健康、非贫血的瑞典婴儿中,食用高铁配方奶粉(6.6 mg Fe/天)45天不会增加病原菌的生长;然而,双歧杆菌的相对丰度降低。
但是,值得注意的是在同一项研究中,与服用高铁配方奶粉的婴儿相比,服用铁滴剂(6.6 mg Fe/天)的婴儿的乳酸菌种类相对丰度较低。尽管剂量相当,但这项研究表明,给药形式(即配方奶粉与滴剂)对肠道微生物组成的影响存在差异。
此外,由于铁滴剂会导致乳酸杆菌的减少,乳酸杆菌是重要的共生细菌,因此铁滴剂可能会增加感染的易感性。
在疟疾流行地区补充铁被证明会增加严重不良事件的发生率,包括因疟疾和其他感染而住院。疟疾感染恶化的潜在机制被认为是过量铁抑制铁转运蛋白(ferroportin,防止红细胞中铁过量,防止感染)。
口服铁补充剂和强化对 4-59 个月儿童腹泻发病率的影响。在19项研究中发现,12项研究中铁不会影响腹泻发病率,在其余的研究中,四项记录的腹泻发病率显着增加,三项记录在特定亚群中的增加。
为什么有些研究表明补铁导致腹泻增加?
有两个主要假设可以解释有时观察到的效果。
首先,铁可以在肠道内产生活性氧,从而导致肠道损伤,并导致炎症性腹泻。这一假设得到了体外实验的支持,在体外实验中,铁暴露后,肠上皮细胞失去了完整性。
其次,铁可以改变肠道细菌的组成,创造一个更具炎症性的环境。
口服铁剂后结肠腔内的微生物和代谢变化
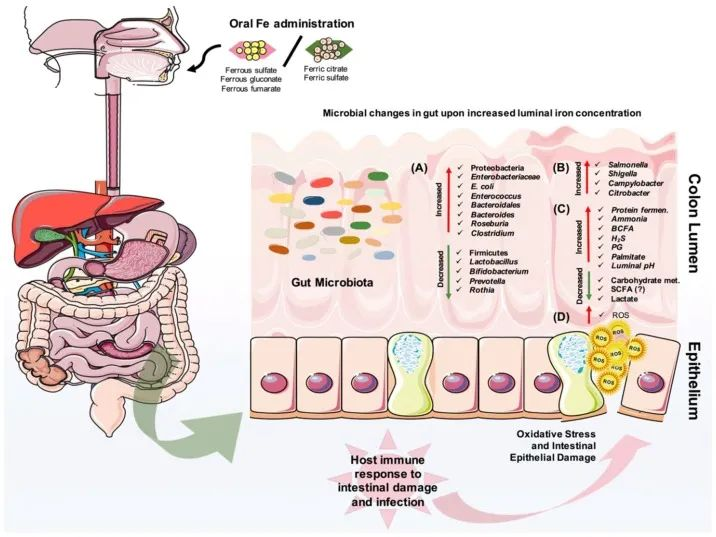
Yilmaz B, Li H. Pharmaceuticals (Basel). 2018
口服铁对肠道微生物组成的改变有直接影响。
(A)它可以导致有益微生物群的减少和致病菌的扩张,
(B)也可以为肠道病原体的扩张提供机会。
(C)此外,蛋白质发酵的增加和碳水化合物代谢的减少也会影响宿主的代谢。
(D)重要的是,铁可以在肠道中诱导活性氧(ROS)的产生,从而导致氧化应激,从而导致肠上皮损伤。
· 反过来,宿主的肠道免疫系统会对炎症、肠道损伤和可能的感染做出反应。
对于缺铁个体的补铁,可以改善肠道微生物的组成,降低致病菌的数量。但是在一些研究中,缺铁性贫血的个体口服补铁常会伴随胃肠道症状和肠道感染等副作用。在非洲的研究中,接受了铁强化剂饼干的儿童,铁补充并未改善机体贫血状况,反而增加了肠道致病菌肠杆菌数量,减少了乳酸菌和双歧杆菌数量,这些现象的发生可能与宿主的肠道高炎症水平密切相关,受到肠道内铁含量和微生物的影响。
当人体存在低肠道病原体负担时,补充铁剂对于肠道优势菌群或肠道炎症没有明显的影响;当人体不存在肠道病原负担的时候,补充铁剂可恢复肠道菌群,显著增加粪便中抗炎短链脂肪酸浓度并且降低肠道炎症,改善肠道微环境。
铁与病原菌
在哺乳动物中,大多数铁在血红素的卟啉结构中被螯合。由于病原菌生长需要铁,有的病原菌如霍乱包含的基因使霍乱弧菌能够从血红素中获取铁。霍乱毒素通过堵塞末端的毛细血管增加管腔血红素的生物利用度,导致宿主铁利用降低。
图 铁和代谢功能之间联系

Botta A, et al., J Lipid Atheroscler. 2021
此外,霍乱弧菌产生一种称为弧菌素的铁载体。与肠杆菌素等其他儿茶酚酸酯铁载体不同,这种独特的协同作用有助于逃避宿主免疫系统。霍乱毒素还会增加管腔内的长链脂肪酸和L-乳酸代谢产物,从而导致编码TCA循环含铁硫簇酶的霍乱弧菌基因上调。
因此,霍乱和霍乱毒素的产生在肠道中创造了一个缺铁代谢生态位,通过获得宿主来源的血红素和脂肪酸,选择性地促进霍乱弧菌的生长。
空肠弯曲菌也能捕获宿主铁并在宿主内引起感染。空肠弯曲菌感染是通过食用生的或未煮熟的家禽、海鲜、肉类和未经处理的饮用水发生的,当空肠弯曲菌通过胃时,它必须首先在极端酸性环境中存活。铁的存在增强了它在酸胁迫环境中的生存能力,因此它含有与铁介导的酸保护有关的基因,包括鞭毛生物发生基因、细胞膜生物发生基因、热休克蛋白(GroEL、GroES),这些基因有助于它的生存。
膳食铁可抑制肠道病原体柠檬酸杆菌的生长,并促使选择无症状的柠檬酸杆菌菌株;这些反应与胰岛素抵抗和抑制病原体毒力的葡萄糖水平升高有关。
除了促进胰岛素抵抗外,膳食铁还增加了肠道葡萄糖水平,这是抑制病原体毒力的关键肠道环境变化,并推动了无症状柠檬酸杆菌菌株的选择。然而,相比之下,其他研究表明,铁可用性的降低是有益的,因为它可以减少潜在致病性肠道细菌的生长。
铁对促、健康的 SCFAs 产生的影响
细菌代谢对我们的肠道健康很重要。短链脂肪酸是结肠中的主要代谢物,对肠道健康非常有益,并且是肠细胞和更远距离组织的能量来源。
铁与短链脂肪酸
两项研究首次描述了铁和短链脂肪酸产生之间的联系。
具体来说,体内大鼠管腔缺铁期间丁酸盐和丙酸盐水平较低,并通过补铁恢复。尽管不能排除饮食干预会改变肠道对短链脂肪酸的摄取,但这些结果表明,补铁可以通过增加短链脂肪酸的产生对肠道健康产生有益的影响。
然而,相比之下,高铁条件下似乎没有太大刺激体外短链脂肪酸的产生,而在极低铁条件下丁酸盐和丙酸盐的产生最明显受到损害。这种产量下降伴随着产生短链脂肪酸的菌Roseburia spp./ E. rectale和Clostridium Cluster IV 成员的减少。
体外发酵研究表明,补充铁会略微增加丙酸盐水平,但总短链脂肪酸水平没有显着变化。
与补充铁饮食的大鼠相比,缺铁饮食大鼠的盲肠乙酸盐、丙酸盐和丁酸盐水平降低。他们还表明,与对照组饮食(并且没有首先耗尽铁)的大鼠相比,补充铁饮食的大鼠的丙酸盐和丁酸盐水平更高。因此可以假设,缺铁饮食(与缺铁相结合)可能对肠道健康无益,因为在这些条件下,腔内短链脂肪酸水平会降低。相反,铁补充剂可能会增加促进健康的肠腔短链脂肪酸水平。
值得注意的是,短链脂肪酸可能会影响肠道病原体的毒力。丁酸盐的减少也可能会降低肠内 AMP 导管素的表达,从而削弱宿主的防御能力。
铁与支链氨基酸(BCFA)
与碳水化合物发酵相比,肠道细菌的蛋白质发酵会产生有毒或潜在有毒的代谢物,例如氨、H2S、BCFA(例如异丁酸和异戊酸)、吲哚和酚类化合物。
在体外,低铁条件下 BCFAs 的产生减少,也就是说:铁增加了成人粪便微生物群的 BCFA 的产生以及有毒氨的产生。
值得注意的是,BCFAs 和氨被认为是蛋白质发酵的指标。研究表明铁会刺激蛋白质发酵,这可能会导致更腐败、潜在有毒或致癌的环境。相比之下,在体外发酵研究中发现乳酸(主要来自碳水化合物)水平会随着铁的反应而降低。
虽然在多项体外研究中显示了源自蛋白质发酵的产品毒性的证据,但体内毒性是有限的,最近的一项试验不支持蛋白质发酵在人体肠道毒性中的作用。
另一方面,蛋白质发酵通常与病原菌的生长有关。因此,有必要在体内研究铁对蛋白质发酵和毒性的影响。
重要的是要认识到微生物代谢物的影响不仅限于肠道,因为它们被吸收,并且可能对远处部位和全身宿主代谢产生影响。目前我们还不知道这如何影响人类健康和疾病,但研究暗示铁诱导的肠道微生物活性变化也可能具有全身性影响。
在稳态条件下,肠道内的微生物必须相互竞争,并与宿主竞争可用铁。因此,细菌发展了不同的吸收系统,如铁运输系统和铁载体(铁螯合分子),以便在铁有限的环境中更有利地竞争。
前面我们知道,铁对于细菌的生长繁殖至关重要,但过量的铁也会产生毒性,因此,细菌对铁离子的摄取必须受到精确严格的调控。
细菌已经进化出了铁源的摄取系统。
细菌获取铁的机制
细菌可以通过分泌铁载体摄取Fe3+。铁载体是一种Fe3+特异性的螯合剂,对Fe3+具有超强的络合力。
铁载体能够与宿主体内的转铁蛋白、乳铁蛋白等铁结合蛋白竞争Fe3+,从而形成可溶性的Fe3+-铁载体复合体,这种复合体可以特异性地与细菌细胞外膜上的铁载体受体蛋白(OMRs)相结合,最终被转运至细胞周质中,转运过程通过TonB系统提供能量。
细胞周质中的Fe3+-铁载体复合体与周质结合蛋白(PBPs)相结合,形成Fe3+-铁载体-PBPs复合物。最后Fe3+-铁载体-PBPs复合物由ABC转运蛋白介导,通过内膜进入胞浆。进入细胞后,Fe3+-铁载体-PBPs复合物中的Fe3+被铁还原酶还原为Fe2+,Fe2+与铁载体的亲和力低,从而被释放。
除了分泌高亲和力的铁载体竞争Fe3+外,一些细菌还进化出直接利用转铁蛋白或乳铁蛋白中Fe3+的机制。
大多数革兰氏阳性菌存在直接吸收血红素而获得铁元素的转运系统。
细菌外膜上的血红素受体可以直接与血红素或血红蛋白结合,并将血红素或血红蛋白转运至周质,通过ABC转运蛋白转运至胞质降解或利用,整个过程由TonB系统提供能量。
金黄色葡萄球菌铁依赖性表面决定系统(Isd)可从血红蛋白中获取Fe2+。
除直接的血红素转运系统外,还存在间接的血红素转运系统。如革兰氏阴性菌中存在的Hemophore蛋白介导的血红素转运系统。
除此之外,细菌也可以产生分泌的或位于膜上的铁还原酶,将Fe3+还原成更容易溶解的Fe2+形式,并通过Feo、Yfe、Efe等转运系统来摄取Fe2+。其中Feo转运系统最为重要,大约80%的革兰氏阴性菌都存在Feo转运系统。
调节铁稳态
铁储存蛋白通过以可溶且无毒的形式储存细胞内游离铁来降低其浓度。在铁缺乏时,例如当存在于哺乳动物宿主细胞内或血液中时,铁可以从这些细菌储存中释放出来。
例如,大肠杆菌可以通过FieF输出铁,而且已经描述了一种用于伤寒杆菌的柠檬酸铁外排转运体(IceT);这些铁外流系统的目的是防止细胞内高水平的游离铁造成的压力。此外,血红素输出机制(HrtAB或同源蛋白)已被证明可以缓解某些细菌中血红素的铁胁迫。
最后,双歧杆菌科能够将铁结合到其表面,从而减少周围环境中自由基的形成,并可能在结肠腔中起到铁螯合的作用,以防止病原菌吸收铁。
铁的形态、可用性和结肠腔中的铁之争

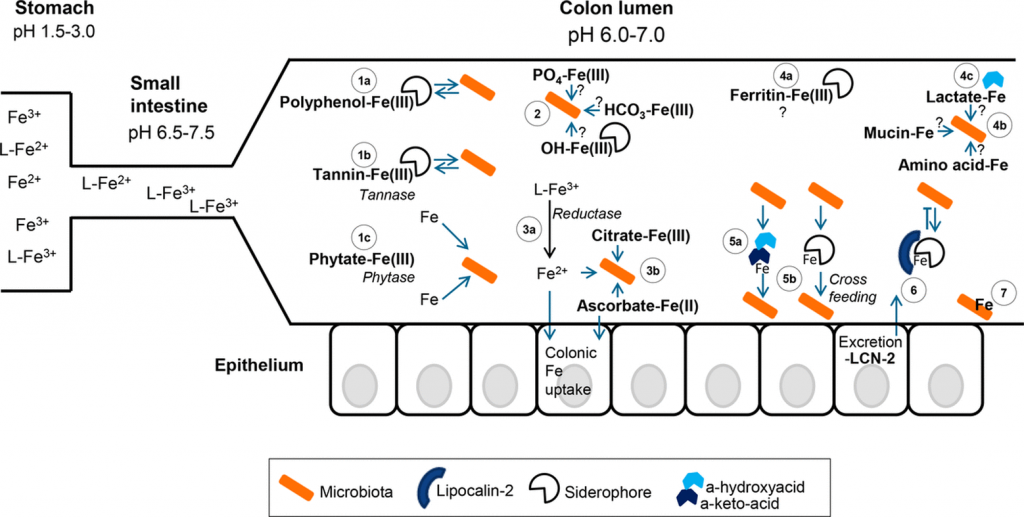
Kortman GA, et al., FEMS Microbiol Rev. 2014
胃肠道中的铁形态可能对肠道微生物群获取铁起着重要作用。
低pH值有利于铁和亚铁的溶解性,不一定需要配体(L)来溶解。
当小肠内的pH值升高时,主要是三价铁的溶解度降低,并与食物成分和宿主排泄物形成更多的络合物。
在结肠内,由于微生物群产生乳酸和短链脂肪酸等,pH值略有下降。图中结肠部分的微生物群以橙色表示(有益的)常驻物种和致病物种。
铁调素(Hepcidin)是一种由肝脏产生的肽激素,是全身铁稳态的主要调节剂。铁调素结合并降解铁转运蛋白,从而影响铁被巨噬细胞回收、被肠上皮细胞吸收以及被肝细胞储存的过程。
当体内缺铁时,铁调素浓度较低,从而有利于铁的吸收和从储存部位输送到血浆;
但当体内铁含量充足时,较高的铁调素浓度会降低铁的吸收,并损害铁的释放。
体内“铁稳态”
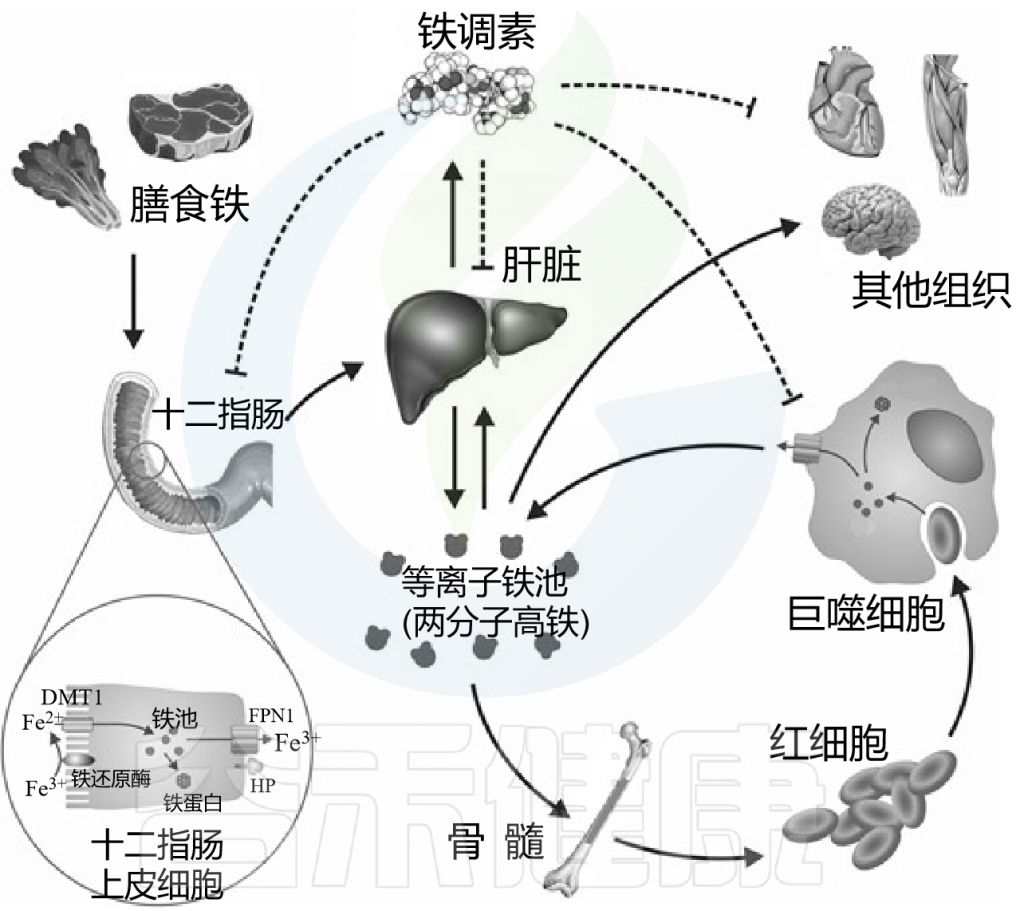

Anderson GJ, et al., Am J Clin Nutr. 2017
铁在饮食中以血红素和非血红素形式存在。非血红素铁通过DMT1穿过肠上皮细胞的根尖膜,通过FPN1穿过基底外侧膜后进入血液循环。铁与血浆TF结合,分布到全身组织。
菌群会通过代谢物信号传导途径调控系统铁稳态平衡。美国密西根大学研究人员在《Cell-Metabolism》上发表的研究证明了,细菌具有铁依赖性机制,可以抑制宿主铁的运输和储存。肠道菌群产生的代谢物能抑制肠道铁吸收主要转录因子低氧诱导因子 2α (HIF-2α),并增加铁存储蛋白铁蛋白 (Ferritin),从而抑制宿主的铁吸收。
两种菌群代谢物——1,3-二氨基丙烷 (DAP) 和 Reuterin,通过抑制异二聚化作用作为 HIF-2α 抑制剂,可以有效缓解全身铁超负荷。
与铁摄取相关的抗菌治疗
针对细菌生存繁殖对铁稳态的严重依赖,可将病原菌的铁获取系统作为抗菌治疗的靶点,开发针对病原菌铁稳态的化合物治疗细菌感染,同时可基于该系统研发疫苗。
大鼠和家兔服用抗生素后,铁的吸收也减少了。然而,小鼠研究发现,抗生素治疗后,铁的吸收增加了。这些发现表明,抗生素的使用可能改善铁缺乏症患者的铁吸收。
铁是饮食变化、微生物组改变和代谢功能障碍之间串扰的关键节点。
代谢综合征指的是一组异常,包括肥胖、血脂异常、胰岛素抵抗和2型糖尿病,这些疾病共同增加了心血管疾病的风险,包括心力衰竭(HF)和非酒精性脂肪性肝病。
通过血清中非转铁蛋白结合铁的存在、高铁蛋白血症、肝铁超载与胰岛素抵抗的相关性,明确了代谢综合征患者中轻度铁过量的患病率。
铁过量与胰岛素抵抗的结合通常被称为代谢异常铁过量综合征,15%-30%的代谢综合征患者会出现这种情况。因此,目前铁过量与代谢当量的关系已被充分认识,但导致代谢功能障碍的机制尚不完全清楚。
降低代谢疾病中铁的效果
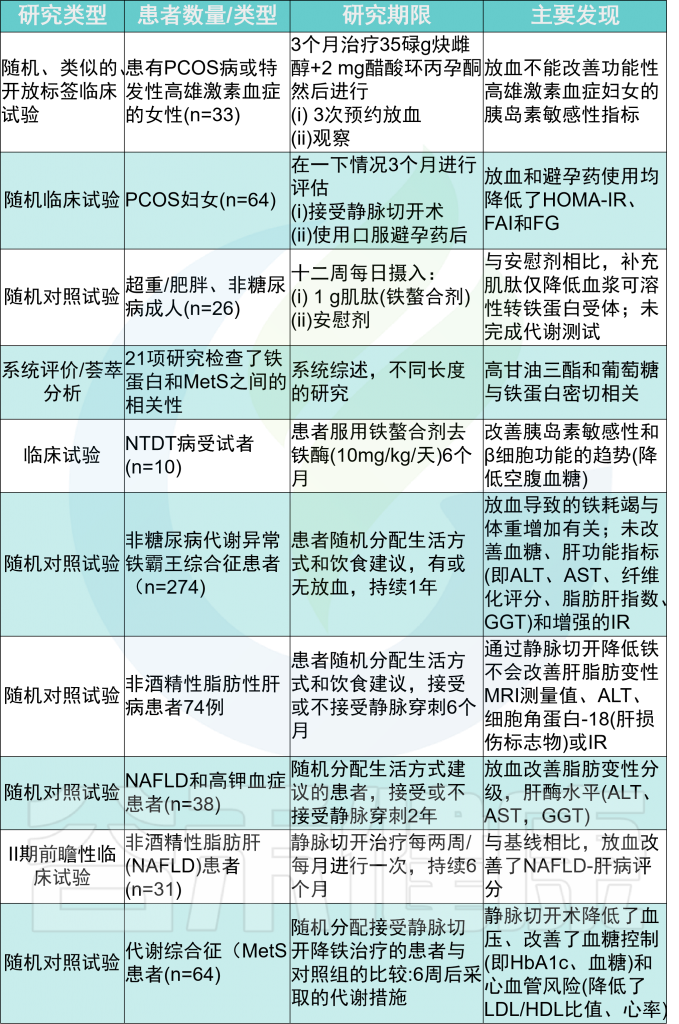

在铁储量较高的MetS患者中,男性对心脏和肝脏疾病的易感性较高。
通过静脉切开或使用螯合剂等降低铁含量的干预措施,在某些情况下可以提高胰岛素敏感性,延缓2型糖尿病(T2DM)和心力衰竭的发病,但并不总是成功的。
在长期病态肥胖后,缺铁是一种常见现象,同样也可导致2型糖尿病和心力衰竭。因此,之前的研究表明铁和葡萄糖稳态或心肌病之间存在双向关系,表明最佳铁水平的平衡至关重要。
可能铁在代谢综合征发病机制及其并发症中的作用仍未得到充分重视,而微生物群的修饰是铁代谢影响的一种重要且相对未被探索的中介物。特别是,肠道内饮食中的铁水平改变了微生物群的组成。预计随后会影响微生物组的代谢组谱功能,包括短链脂肪酸和支链氨基酸。这种改变的后果将是宿主的外周胰岛素抵抗和代谢功能障碍。
炎症性肠病 (IBD) 的特征是胃肠道的慢性炎症。炎症与溃疡性结肠炎 (UC) 和克罗恩病 (CD) 中的肠道溃疡有关。IBD 也可能出现出血和吸收不良,三分之一的患者会出现缺铁性贫血。
一项IBD小鼠模型的研究发现,铁含量的改变显着影响 DSS 在小鼠中诱导的结肠炎的严重程度,铁含量的过多或过少都会加剧结肠炎的严重程度。
DSS治疗的高铁饮食小鼠的体重减轻程度不如低铁饮食小鼠,但粪便钙卫蛋白测定的肠道炎症更严重。这些喂食高铁饮食的小鼠经历了变形菌的增加,同时厚壁菌和拟杆菌的减少。
膳食铁水平与结肠炎症的 DSS 治疗和粪便钙卫蛋白水平之间似乎存在协同作用。
摄入两倍于标准水平铁(400 ppm)的饮食会导致微生物组的关键变化,这意味着观察到的这些变化不仅仅是由炎症的严重程度驱动的,还有管腔游离铁也会导致导致IBD中经常观察到的异源状态发展的复杂因素相互作用。
还需要更多了解的是,管腔铁如何影响IBD。此外,还需要研究管腔铁的增加对肠道微生物群的生理影响,以及这可能如何影响菌群多样性。未来也需要更多人体干预研究,进一步确定不同剂量的治疗性口服铁对人体肠道微生物群的复杂影响,尤其是代谢后果。
研究表明,缺铁和铁过量都与结直肠癌的发病机制有关,这表明必须谨慎平衡最佳铁摄入量。
在 965 名 50-75 岁的人群研究中,发现铁摄入量与结直肠息肉之间存在 U 型关系,铁摄入量高(>27.3 毫克/天)或低量(<11.6 毫克/天)的人更容易患上结直肠息肉,这是结直肠癌的前兆病变。
缺铁
由于铁在维持免疫功能中至关重要,铁的可用性不足可能会通过削弱对肿瘤变化的免疫监测,并潜在地改变肿瘤免疫微环境而增强致癌性。来自临床研究的数据表明,在结直肠癌患者中,缺铁与较差的预后和较低的治疗反应相关。
铁过量
大多数强有力的研究证实,膳食铁和铁储存过量都会增加结直肠癌的风险。
五项前瞻性人类队列研究,包括566607名个体和4734例结肠癌患者的数据,表明高血红素铁摄入量与结肠癌风险增加有关(虽然有一个队列没有发现任何关联)。
在防御方面,长双歧杆菌和嗜酸乳杆菌是肠道保护性共生菌。它们形成了一道保护屏障,防止病原菌定植,并产生丁酸盐,作为一种抗癌剂。双歧杆菌科可通过将铁结合到其表面来影响自由基的形成,并促进结肠上皮的日常更新,而乳酸杆菌菌株可降低胆汁酸的诱变效应。
大多数致病菌都具有增强的铁获取机制,因此往往比保护性细菌更容易获得游离铁。也就是说,铁可以促进致病菌和肠道共生菌之间比例的变化,增加肠道中的特定代谢物和炎症。
研究显示,拟杆菌/普氏杆菌、梭状芽孢杆菌、牛链球菌和粪肠球菌可产生遗传毒性代谢物,如硫化氢和次生胆盐,这可能会促进炎症和致癌。
基于抗生素的肠道致病菌清除降低了结肠癌的发病率,并改变了小鼠的肠道微生物群。这些发现得到了人体研究的支持。
可见,针对肠道微生物群的饮食干预有望治疗结直肠癌,但这些方法仍需要进一步研究。
过量的铁对身体会带来不良影响,因此如果体内铁过量则需要采取相应措施。
多酚-铁:结合牢固,防止铁被吸收
饮食中通常含有单宁和儿茶酚等多酚,它们大量存在于茶和咖啡中。这些化合物可以非常牢固地结合铁,从而防止宿主吸收铁,但也防止细菌吸收铁。
然而,在铁缺乏的环境中,致病菌可以产生和/或占用含铁细胞可能受益于铁多酚提取机制,很可能通过清除铁绑定到多酚。
目前尚不清楚这种机制是否在肠腔中发挥重要作用,但可能与此有关,因为肠环境中可能同时存在多酚和铁载体。
此外,某些细菌,如甘蓝链球菌或卢格敦葡萄球菌,可以降解多酚鞣酸盐,并通过这种方式暂时将铁从这种有效的铁粘合剂中释放出来。
可以通过在用餐后一小时内饮用以下饮料来减少铁的吸收:


植酸盐-铁:结合物通过菌群降解,释放铁
另一种具有强铁结合活性的化合物是植酸盐,在食用谷物和豆类为主的饮食后,其肠道可利用性很高。
与单宁酸类似,某些肠道微生物(如大肠菌和双歧杆菌科)可以降解植酸盐,这可能是一种特殊的释放铁的方式,铁可能被降解生物体或其他细菌物种利用。因此,与植酸盐结合的铁可能是结肠肠道微生物群的相关铁源。
然而,应该注意的是,铁与植酸盐的复合物到达结肠时大多是不溶性的,因此不易被降解。然而,之前在常规大鼠的结肠中发现了只能通过微生物作用产生的植酸盐降解产物,但在无菌大鼠中却没有发现。这些发现表明微生物降解的植酸发生在结肠。
在每餐 2-10mg 植酸盐的极低浓度下,植酸盐会降低铁的吸收。
大豆蛋白(存在于豆腐、组织化植物蛋白和一些加工肉制品中)可以减少铁的吸收,因为它含有植酸盐。
钙
钙对骨骼很重要,但它也可能抑制铁的吸收。为了更好地吸收铁,避免在吃富含铁的食物的同时服用钙补充剂。
牛奶和鸡蛋中的蛋白质
虽然动物肉中的蛋白质会增加铁的吸收,但牛奶和鸡蛋中的蛋白质(卵清蛋白除外)在与低铁食物一起食用时会减少铁的吸收。
锌
过量摄入锌(由于过度使用锌补充剂)也会损害铁的吸收。
考虑到缺铁对人体健康影响重大,因此实施适当的策略来解决这一问题至关重要。
最常见的策略是食物中的铁补充、益生菌、益生元、铁药物补充剂等方法。
富含铁的食物
前面我们知道,膳食铁有两种形式:血红素和非血红素。血红素铁具有较高的生物利用度,膳食因素对其吸收的影响最小,而非血红素铁的吸收要低得多,并受到其他食物类型的强烈影响。
大多数对年轻女性的研究发现铁状态(铁蛋白和铁)与肉类和其他血红素铁的消耗量之间存在正相关关系。
增加富含铁的食物的摄入量,以补充铁储备,确保饮食健康均衡。
其中常见的富含铁元素的食物包括:
红肉、家禽(包括肝脏类)、鱼、豆类、扁豆、豆腐、豆豉、坚果、种子等
非血红素形式的铁含量高的植物性食物(但也可能含有高含量的阻止铁吸收的物质)包括:
● 豆类,包括鹰嘴豆、豆类、豌豆和小扁豆
● 种子,包括芝麻和南瓜子
● 绿叶蔬菜,包括西兰花和羽衣甘蓝
以上是富含铁元素的食物,同时其他富含维生素的食物也能辅助铁的吸收,例如,维生素A,维生素C.
维生素C:促进铁的吸收
如果膳食中含有大量维生素C,那么蔬菜膳食的吸收量可能会增加六倍。维生素C和柠檬酸以剂量依赖的方式促进铁的吸收,部分是通过充当弱螯合剂来帮助溶解小肠中的铁。
一项研究发现,维生素 C 的摄入量与女性的铁含量呈正相关。
在存在抑制铁吸收的物质(包括植酸盐、多酚、钙和蛋白质)的情况下,维生素 C 还有助于铁的吸收。
如果同时服用,维生素 C可将植物性食物中的非血红素铁的吸收提高 2 至 3 倍。因此,为了提高铁的摄入量,可以将富含铁的植物性食物与富含维生素 C 的食物结合起来。
水果或蔬菜中都会包含维生素 C,包括:
● 柑橘类水果
● 奇异果
● 草莓
● 番茄
● 辣椒
● 西兰花
● 卷心菜
● 菠菜


维生素A:克服植酸盐的影响促进铁的吸收
维生素 A直接影响铁转运和红细胞生成。
全谷物和豆类中的植酸盐会降低铁的吸收,但如果加入富含维生素A和β-胡萝卜素的食物可以增加铁的吸收,并可以克服植酸盐的影响。
维生素A(视黄醇)有助于治疗缺铁性贫血,并能改善儿童和孕妇的铁状况。
常见的富含维生素A和β-胡萝卜素的食物:
● 胡萝卜
● 红薯
● 鱼
● 哈密瓜
● 甜椒
● 南瓜
● 葡萄柚
益生菌
大多数益生菌产生乳酸,这可能会降低 pH 值,从而增加铁的溶解度,帮助其吸收。
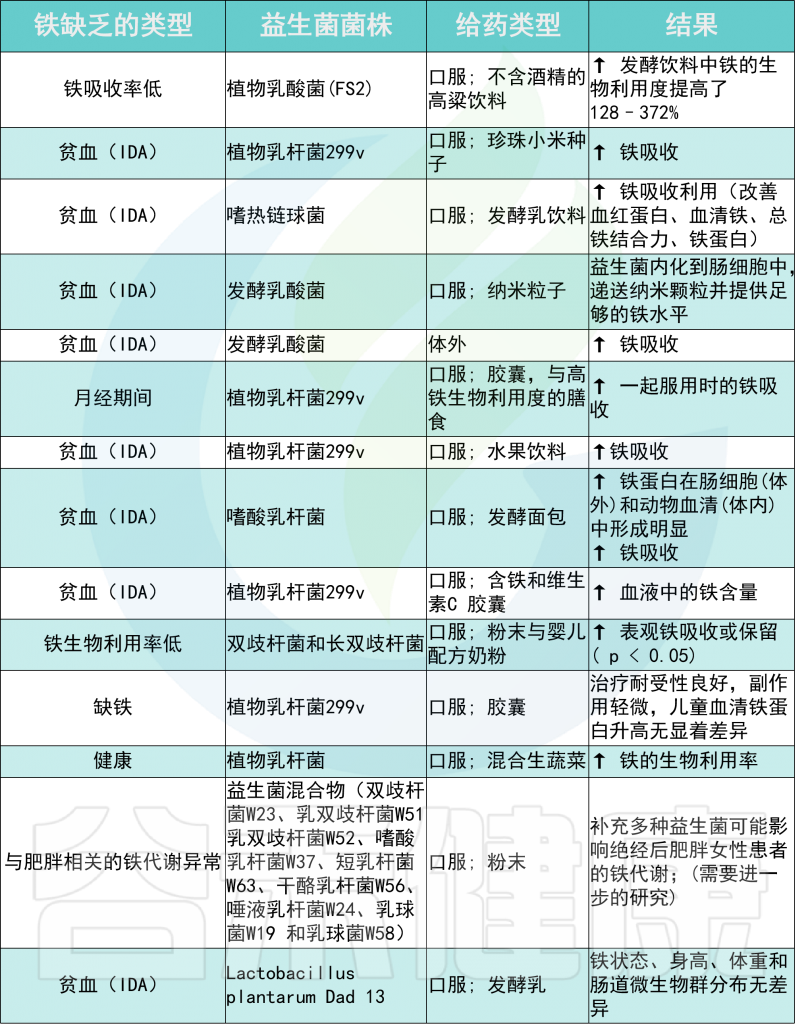
例如,植物乳杆菌 299v 有助于预防缺铁性贫血。这种益生菌可以改善活跃的高加索欧洲人的膳食非血红素铁吸收。
发酵乳杆菌是人类微生物群中的一种主要益生菌,具有显著的铁还原活性。对羟基苯乳酸是该菌株产生的代谢物,通过DMT1转运体将Fe3+还原为Fe2+来增加肠细胞对铁的吸收。
与益生菌菌株给药相关的铁吸收

Rusu IG,et al., Nutrients. 2020
益生元
益生元是功能性食品成分,可刺激肠道中有益细菌的生长和定植,最终改善身体健康。肠道微生物群定植在肠道生理学中起着重要作用。
几项研究将益生元和/或合生元的摄入与铁可用性的增加联系起来,主要是通过将 Fe 3+转化为 Fe 2+(由于它们的铁还原活性),并促进肠细胞对铁的吸收。
例如,在肯尼亚儿童中进行的一项补铁试验报告称,在服用益生元期间铁吸收更高半乳糖寡糖 (GOS)底物。
不同类型铁缺乏症的益生元和合生元摄入量及其对机体铁水平的影响
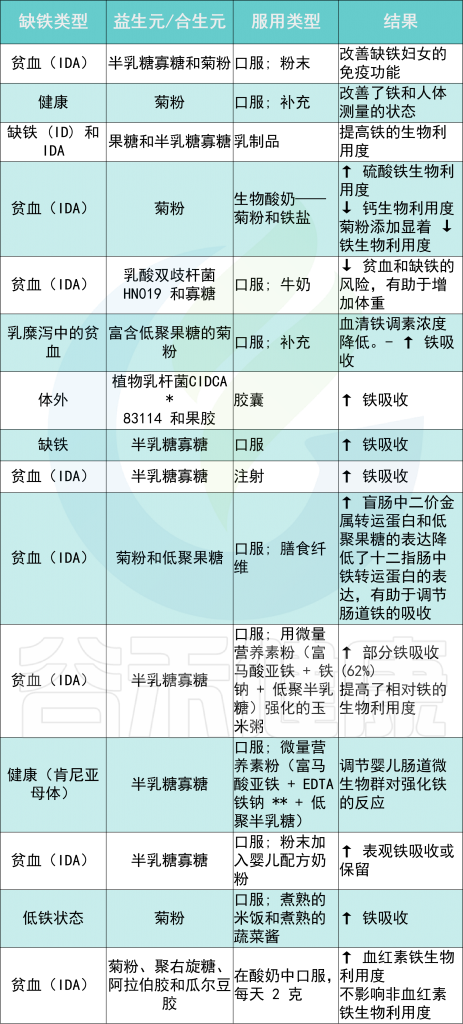

Rusu IG,et al., Nutrients. 2020
* CIDCA——食品冷冻技术研发中心
** EDTA——乙二胺四乙酸
补充剂
不同形式的铁补充剂补充铁以亚铁(+2)和铁(+3)的形式存在。由于铁形态必须在体内转化为亚铁形态以供吸收,因此亚铁形态更具生物利用度。
常用的口服亚铁补充剂包括:
● 柠檬酸亚铁
● 硫酸亚铁
● 葡萄糖酸亚铁
● 铁琥珀酸亚铁
● 氨基酸螯合物(如双甘氨酸铁、天冬氨酸铁)
● 血红素铁
虽然最常研究的铁补充剂是硫酸亚铁,但食品强化和补充剂研究表明,氨基酸螯合形式的铁(如甘氨酸)更好或同样好地被吸收。
例如,在一项针对孕妇的研究中,25mg甘氨酸亚铁能够将铁水平提高到与50mg硫酸亚铁相同的水平。
较新配方的铁补充剂可能比亚铁盐更容易耐受,胃肠道副作用更少,如:
然而,在提高产品螯合率、安全性,降低产品成本等方面有待进一步研究。
铁注射需要医疗监督。当口服铁补充剂不能耐受时,可以注射,这通常适用于患有腹腔疾病和炎症性肠病的患者。在手术或输血后血红蛋白必须迅速增加的情况下,也建议使用。注射铁剂比口服疗法更昂贵,而且不能在怀孕的前三个月用。
建议注射羧麦芽糖铁和异麦芽糖铁,因为它们能够以更大剂量给药,且安全性好。


另外,特殊人群对铁的需求量不同:
健身人群补铁
运动会加快铁在机体中的代谢,长期的运动使组织内储存铁的含量明显下降,是红细胞的更新速度加快,运动还导致机体对铁的吸收率降低,这些都增加了健身人群对铁的需要量。
我国对健身人群每日膳食的推荐的摄入量为:男性20毫克/天,女性25毫克/天。
妊娠期补铁
孕妇对铁的需求量比一般人群更高,每天 27 毫克。
妊娠期间,受母体铁状态调节的铁调素血清浓度极低。由于铁调素是肠道铁吸收的负调节剂,低水平表明对铁的需求高。
妊娠早期和中期缺铁与孕产妇发病率增加和不良妊娠结局风险增加有关,包括低出生体重、早产或宫内生长受限。
大多数产前维生素都含有足够的铁来弥补这种增加,但由于对血液产生的需求增加,缺铁性贫血在怀孕期间很常见。因此需要合理补铁。
具体补铁方式可参考上面列出的形式。
铁对于维持宿主肠道菌群稳态和肠道微生物的生长定植有重要作用。一些菌群,尤其是致病菌的生存,必须依靠铁的存在,因此形成了多种摄取铁的机制,并且参与调节宿主的肠道铁吸收。同时,铁与宿主的肠道微生物和微生物代谢产物共同作用,可对肠道乃至整体健康产生影响。
在补铁的同时,需要防止补铁带来的代谢紊乱和炎症损伤,也就是说铁的补充要在允许的条件下适度补充。那么怎么知道是否在合理范围内?目前对铁的监测以血液检测较常见,但血液检测波动较大。
此外,既已发现了铁与肠道菌群相关性,肠道菌群检测也是一个可行方向。谷禾肠道菌群健康检测报告中包含铁的水平,这是基于菌群代谢计算得到的,与血液检测有所不同,肠道菌群检测反映的是一段时间(2周左右)的长期状态。
未来需要更多的研究来证明铁、宿主与肠道菌群的相互作用机制,以及其与肠道炎症疾病等多种慢性疾病发生发展之间的因果关系,为铁补充的个性化策略提供更多支持。
各类人群的铁需求量


►►►
铁补充的副作用及相关禁忌
► 可能出现的副作用
► 禁忌症
铁补充剂不应用于以下人群:
声明:本账号发表的内容用于信息的分享,仅供学习参考使用。在采取任何预防、治疗措施之前,请先咨询临床医生。
主要参考文献:
Yilmaz B, Li H. Gut Microbiota and Iron: The Crucial Actors in Health and Disease. Pharmaceuticals (Basel). 2018;11(4):98. Published 2018 Oct 5. doi:10.3390/ph11040098
Botta A, Barra NG, Lam NH, et al. Iron Reshapes the Gut Microbiome and Host Metabolism. J Lipid Atheroscler. 2021;10(2):160-183. doi:10.12997/jla.2021.10.2.160
Finlaysontrick E C , Fischer J A , Goldfarb D M , et al. The Effects of Iron Supplementation and Fortification on the Gut Microbiota: A Review[J]. Gastrointestinal Disorders, 2020, 2(4):327-340.
Yilmaz B, Li H. Gut Microbiota and Iron: The Crucial Actors in Health and Disease. Pharmaceuticals (Basel). 2018 Oct 5;11(4):98. doi: 10.3390/ph11040098. PMID: 30301142; PMCID: PMC6315993.
Kortman GA, Raffatellu M, Swinkels DW, Tjalsma H. Nutritional iron turned inside out: intestinal stress from a gut microbial perspective. FEMS Microbiol Rev. 2014 Nov;38(6):1202-34. doi: 10.1111/1574-6976.12086. Epub 2014 Sep 29. PMID: 25205464.
Georgieff MK. Iron deficiency in pregnancy. Am J Obstet Gynecol. 2020;223(4):516-524. doi:10.1016/j.ajog.2020.03.006
Rusu IG, Suharoschi R, Vodnar DC, et al. Iron Supplementation Influence on the Gut Microbiota and Probiotic Intake Effect in Iron Deficiency-A Literature-Based Review. Nutrients. 2020;12(7):1993. Published 2020 Jul 4. doi:10.3390/nu12071993
Anderson GJ, Frazer DM. Current understanding of iron homeostasis. Am J Clin Nutr. 2017 Dec;106(Suppl 6):1559S-1566S. doi: 10.3945/ajcn.117.155804. Epub 2017 Oct 25. PMID: 29070551; PMCID: PMC5701707.
LIU Fang-Tong, FAN Hao-Nan, SHEN Li-Xin, LI Bo. Iron acquisition by bacterial and adaptive immune responses[J]. Microbiology China, 2019, 46(12): 3432-3439.
Tolkien Z, Stecher L, Mander AP, Pereira DI, Powell JJ. Ferrous sulfate supplementation causes significant gastrointestinal side-effects in adults: a systematic review and meta-analysis. PLoS One. 2015;10(2):e0117383. Published 2015 Feb 20. doi:10.1371/journal.pone.0117383
GUAN Lingjuan,CAO Congcong,TU Piaohan,et al. Research progress of the effect of iron deficiency on intestinal immune function and new iron supplements[J]. Food and Fermentation Industries,2020,46(19):264 -270
高鹤, 杨浕滢, 应晓玲,等. 铁,宿主和肠道菌群相互作用的研究进展[J]. 现代预防医学, 2020, 47(20):4.

谷禾健康

尽管地球上微生物类群的繁多,但只有一小部分得到了培养和有效命名。因为大多数菌无法在非常特定的条件下培养分离鉴定。
在过去十年中,宏基因组研究的重要性已经凸显,因为它能够评估细菌基因库并发现当前实验室培养技术无法掌握的新细菌基因组。这些数据对于扩大我们对地球上微生物多样性的理解至关重要。
由于宏基因组测序数据由来自多个物种和菌株的 DNA 序列片段组成,通常有数千个来自不同生命领域,因此此类分析的主要挑战是正确确定每个 DNA 序列片段的真实来源。不幸的是,这些步骤容易出错,因此必须对结果进行严格审查,以避免发布不完整和低质量的基因组。
最近,比利时研究人员新开发MAGISTA,这是一种评估宏基因组基因组组装质量的新方法,基于随机森林的方法估计MAGs的完整性和污染度,解决了当前基于参考基因的方法经常被忽视的一些缺陷。
MAGISTA是基于宏基因组bins内的contig片段之间的无对齐距离分布,而不是一组参考基因。该方法利用了来自整个 bin 的信息。为了正确评估此方法,并说明基于参考的工具的缺点,最近,比利时研究人员构建了一个高度复杂的 DNA 模拟群落,由 227 个细菌菌株组成,并且具有不同程度的相似性。

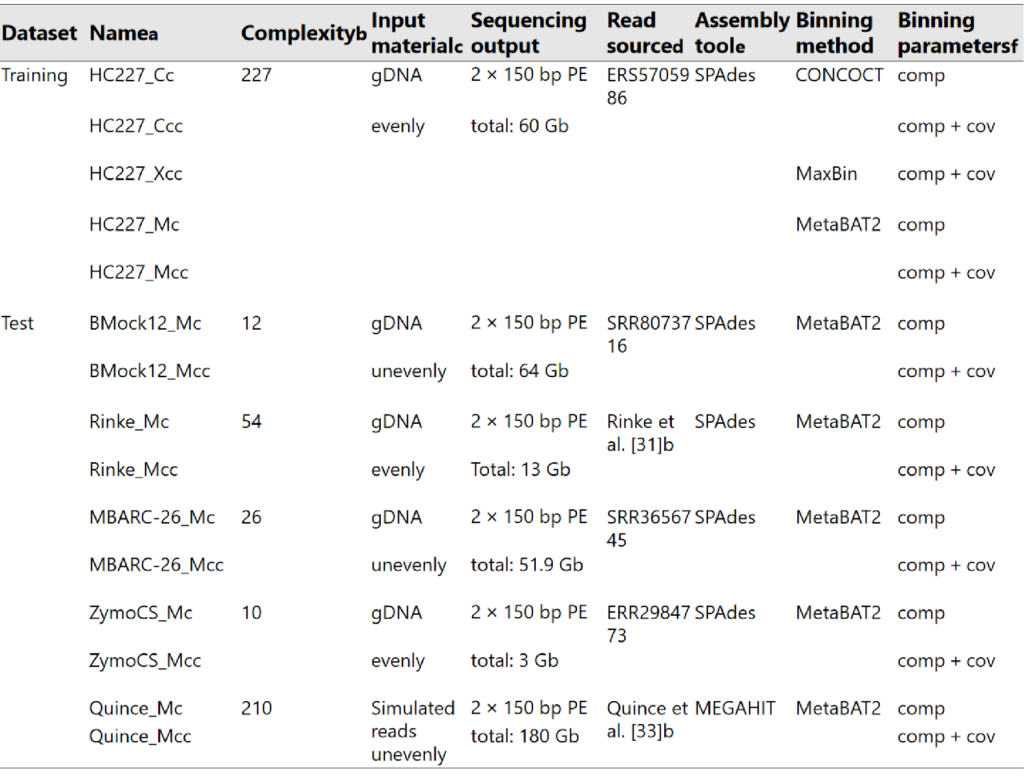
方 法
训练集来(HC227)自 227 个细菌菌株,测试数据集由五个公开可用的短读(short reads)子集构成,其中四个含有来自复杂度相对较低的基因组 DNA 模拟群落的reads。具体情况如下图所示。
Complexity列指示菌株数;Assembly tool列表示所使用的用于组装的软件;Binning method列表示所使用的用于分箱的工具;Binning parameters列表示所使用的用于评估分箱质量的指标,comp为完整度,cov为覆盖率。
MAGISTA计算步骤:
输入binning后的每个bins
-●-
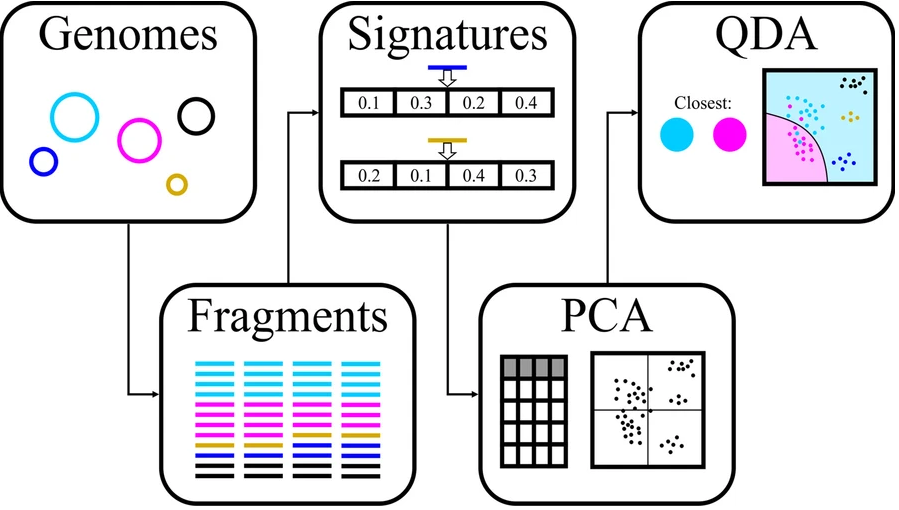
第 1 步:选择适合的片段大小与距离计算方法
-●-
首先将每个 bin 中的每个 contig 拆分为固定长度的片段,然后使用四种不同的方法(即 PaSiT4、MMZ3、MMZ4 和 Freq4)计算一个 bin 中的片段之间的所有距离。对于每种方法,都选择了特定的片段长度,以便为不同的生物产生不同的特征分布。
每种方法的最终片段长度的选择是通过不同方法分析整合决定的,方法如下图所示。每组的设计中至少两个基因组来自同一个家族,两个基因组来自相同的顺序但来自不同的家族。这些基因组被人为地分成所需长度的片段,并为每个片段计算目标特征。
对于每组五个基因组,混合所有片段并根据它们的特征进行主成分分析(PCA),然后进行二次判别分析,用于生成分类器,旨在区分每组中重叠最多的两个基因组。对该分类器的准确度取平均值,结果用于选择方法和片段长度的最终组合。
-●-
第 2 步:模型中特征变量的选择
-●-
为每种方法选择片段长度后,使用平均值、标准差、偏度、峰度和中位数以及 2.5%、5%、10%、90%、95% 和 97.5% 百分位数计算距离分布。此外,还计算了 1 kb 片段的 GC含量分布。以及每个bin的大小,共计66个特征变量。
-●-
第3步:模型构建
-●-
使用 R (v 4.0.3) 包“RandomForest”中的“RandomForest”函数和默认参数训练随机森林模型。同时使用R包lm再建立一个线性模型执行线性回归,输入经对数转换后的特征变量值,用于交叉验证分析。
主 要 结 果
一个高度复杂的基因组DNA模拟群落
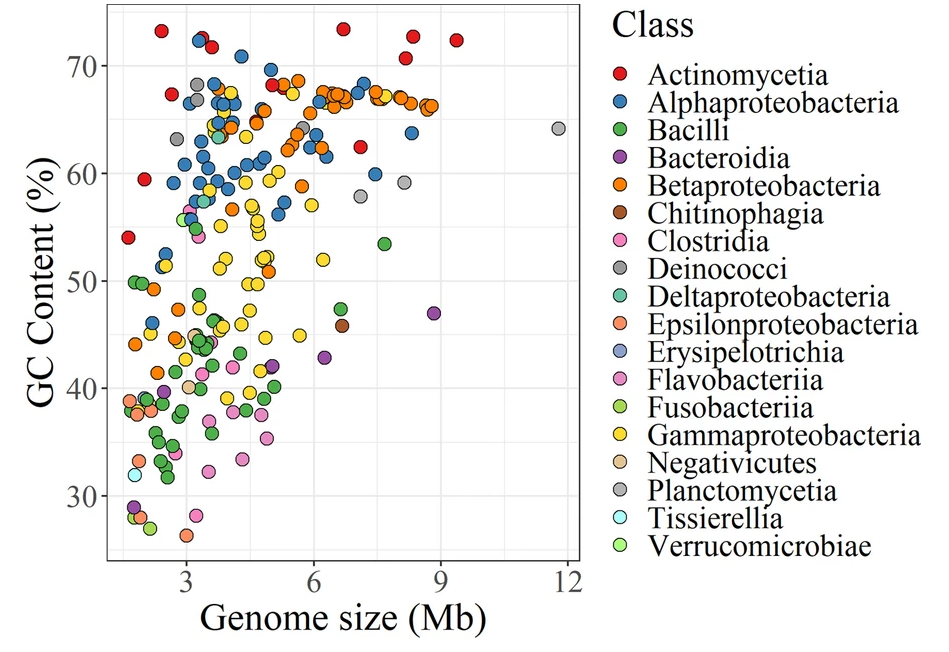
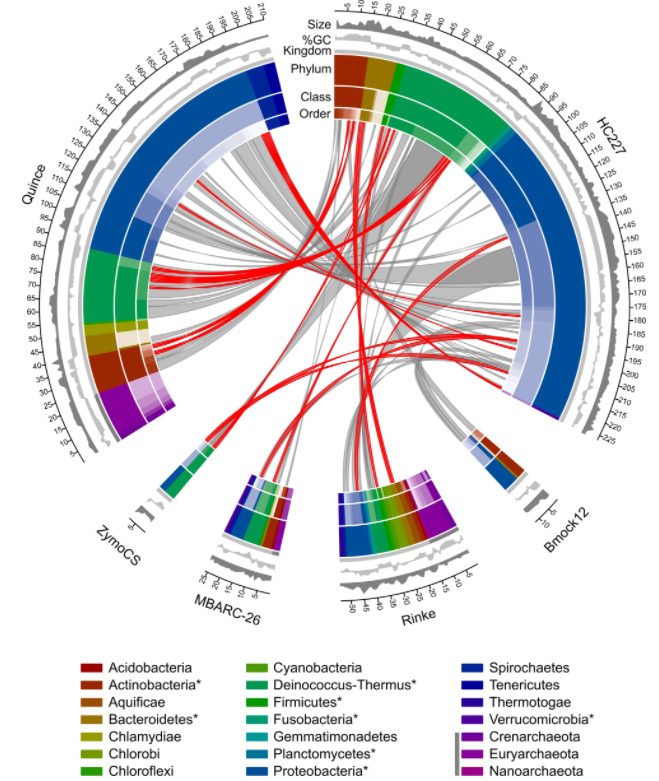
由来自 227 个细菌菌株的基因组 DNA 组成,这些菌株属于8 个门(Actinobacteria, Bacteroidetes,Deinococcus-Thermus, Firmicutes,Fusobacteria,Planctomycetes, Proteobacteria和Verrucomicrobia),18 类,47目,85科,175属,197种。
编辑
上图为模拟群落中的细菌菌株的基因组大小和GC含量(从26.3%到73.4%)散点图;
编辑
图为训练集与测试集中物种之间的关系图。红色线条表示在训练集中存在的菌种,灰色线条表示在训练集中存在的菌属。环状图中的不同颜色代表不同分类水平。图例中存在于训练集中的菌门用*标记,存在于古生菌的菌门用深灰色色带标记。
CheckM中基于单拷贝标记基因(SCMG)来评估 bin 质量的存在的缺陷
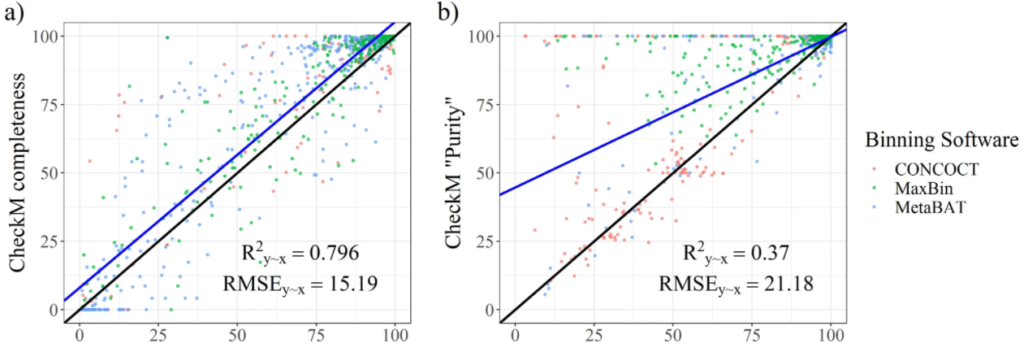
图a和b分别为从CheckM中输出的完整性指标和污染度。使用R^2y∼x(解释方差的百分比),RMSE(相对于实际值的均方根误差)两个参数评估结果。结果表示CheckM高估了bin的质量。许多受污染的bins被预测为接近未受污染。
使用MAGISTA分析模拟群落中的bins
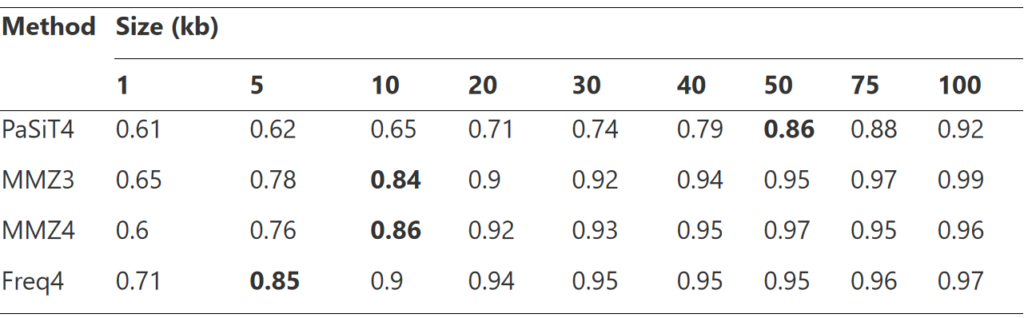
首先选择最佳片段大小用于计算距离分布,如上图所示,考虑了 1、5、10、20、30、40、50、75 和 100 kb 的片段,最终选择了粗体所示的片段大小。
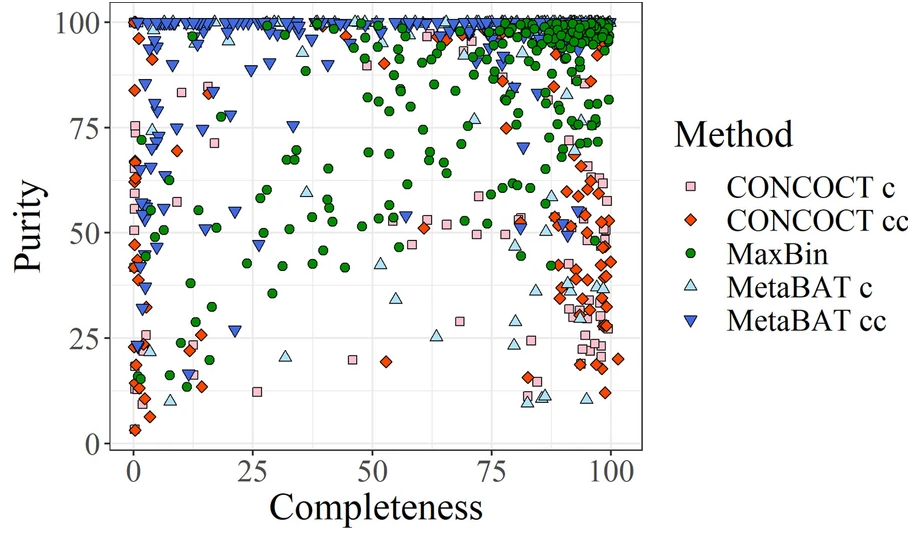
图为concont、MetaBAT和MaxBin产生的bins的完整性和污染度信息。
由于通过模拟生成这样的数据集并不能准确地表示真实的结果,所以使用了binning软件的结果,提供了一组不同质量的真实的bins。训练数据集的完整性和未污染度均在90%以上。
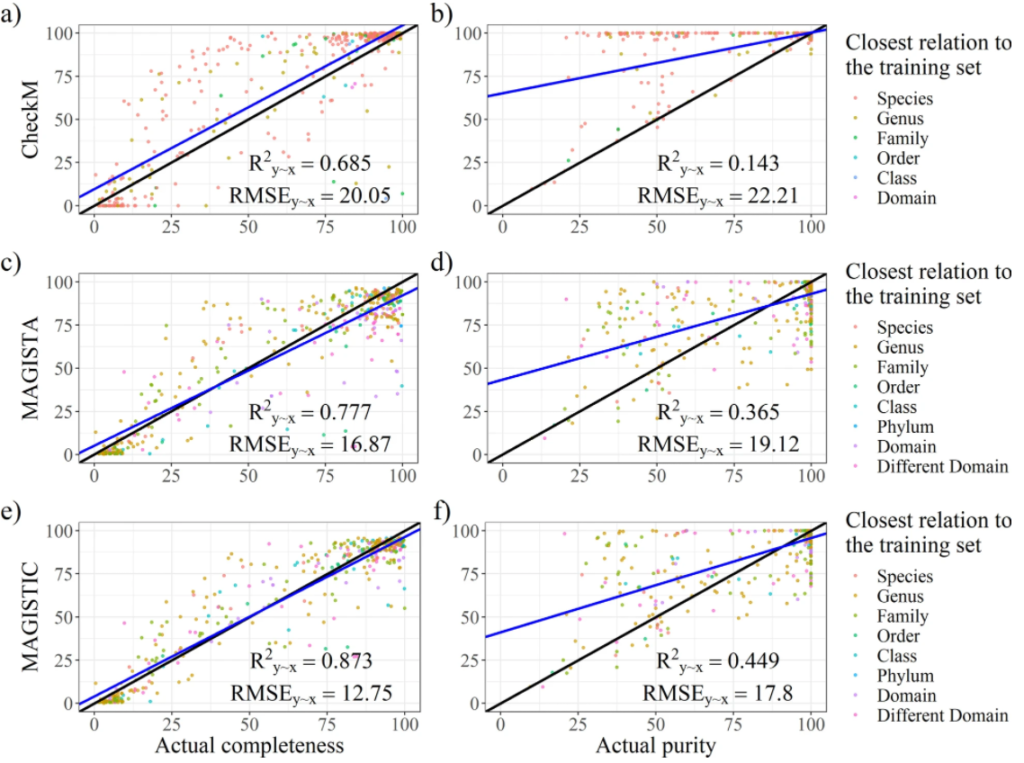
最后是模型构建,建立完整性和污染度的预测模型。并进行了模型评估,如图所示。分别对CheckM、MAGISTA 和 MAGISTIC测试了其性能。CheckM是现在主流的一款评估bin质量的工具。MAGISTIC是一款结合了CheckM和MAGISTA 的工具。使用解释方差的分数(R2y∼x)和均方根误差(RMSE)作为评估性能的指标。对于完整性的预测,MAGISTA 优于 CheckM。对于污染度的预测,MAGISTA 的表现优于 CheckM。
结 论
研究人员开发了一种新的用于预测高度复杂的宏基因组组装基因组bin的质量的方法,MAGISTA。是基于 SCMG 的低复杂性宏基因组方法的一个同样好的替代方法。除了MAGISTA之外,还通过结合CheckM的结果,使用MAGISTIC生成了一个更准确的预测。
研究人员在文章中指出MAGISTA 和 CheckM 都没有达到足够的准确度来被认为是可靠的。MAGISTIC 产生了比 MAGISTA 更好的结果。
在附加分析中,将测试集分为了两个子集,从真实和模拟reads中获得的bins,对此再进行分析,结果表示,CheckM 对于“真实”子集表现良好(但相比MAGISTA 和 MAGISTIC还是较差),对于“模拟”子集部分表现较差。而MAGISTIC相比MAGISTA会更准确些。但是文章中并没有详细说明MAGISTIC的工作流程。
查看作者在github上公开的软件说明,地址如下。但是没有说明和给出输出文件的内容。个人认为还不太成熟。
https://github.com/LM-UGent/MAGISTA
参考文献:
Goussarov G, Claesen J, Mysara M, Cleenwerck I, Leys N, Vandamme P, Van Houdt R. Accurate prediction of metagenome-assembled genome completeness by MAGISTA, a random forest model built on alignment-free intra-bin statistics. Environ Microbiome. 2022 Mar 5;17(1):9. doi: 10.1186/s40793-022-00403-7. PMID: 35248155; PMCID: PMC8898458.

谷禾健康
人的身体拥有数千种细菌,这些细菌在维持健康方面发挥着重要作用。当这些细菌失控繁殖并侵入身体的其他部位或将有害细菌引入身体的系统时,可能会发生细菌感染。
细菌感染的严重程度取决于所涉及的细菌类型和所感染的部位等。细菌最常感染肠道、皮肤和呼吸系统,包括肺、泌尿道和阴道。
目前应对细菌感染的主要治疗方法还是使用抗生素,但是抗生素管理,或改进抗生素的处方和使用方式,对于优化感染患者的治疗、保护患者免受伤害和对抗抗生素耐药性至关重要。
今天我们重点了解下什么是细菌感染,感染的症状、原因和类型,包括诊断测试和治疗方案,抗生素什么时候使用,什么时候不用,抗生素的耐药性如何产生以及使用抗生素的注意事项等。
了解什么是细菌感染以及如何治疗细菌感染,我们应该先了解细菌的概念。
来源:MedicineNet
地球上所有的生物都是由两种基本类型的细胞中的一种组成:
真核细胞,其遗传物质被包裹在核膜内;
原核细胞,其遗传物质与细胞的其他部分不分离。
传统上,所有的原核细胞都被称为细菌,被归为原核生物界。
1970年代后期,美国微生物学家卡尔·沃斯(Carl Woese)率先在分类上进行了重大变革,将所有生物分为真核生物、细菌(原名真细菌)和古细菌(原名古细菌)三个领域,以反映三条古老的进化路线。原核生物以前被称为细菌,然后被分为两个领域,细菌和古细菌。
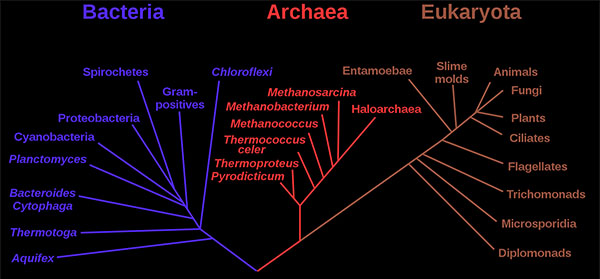

published by Woese et al.
细菌和古细菌在表面上是相似的,例如,它们没有细胞内的细胞器,它们有环状DNA。但是,它们在本质上是截然不同的,它们的分离是基于其古老而又独立的进化谱系的遗传证据,以及其化学和生理学的根本差异。这两个原核域的成员彼此之间的区别与它们和真核细胞中的区别一样。
细菌细胞在几个方面不同于动物细胞和植物细胞。一个根本的区别是细菌细胞缺乏动物细胞和植物细胞中都存在的细胞内细胞器,例如线粒体,叶绿体和细胞核。细菌体积小,设计简单,代谢能力强,使它们能够迅速生长和分裂,并在几乎任何环境中生存和繁衍。
细菌,动物和植物细胞的比较

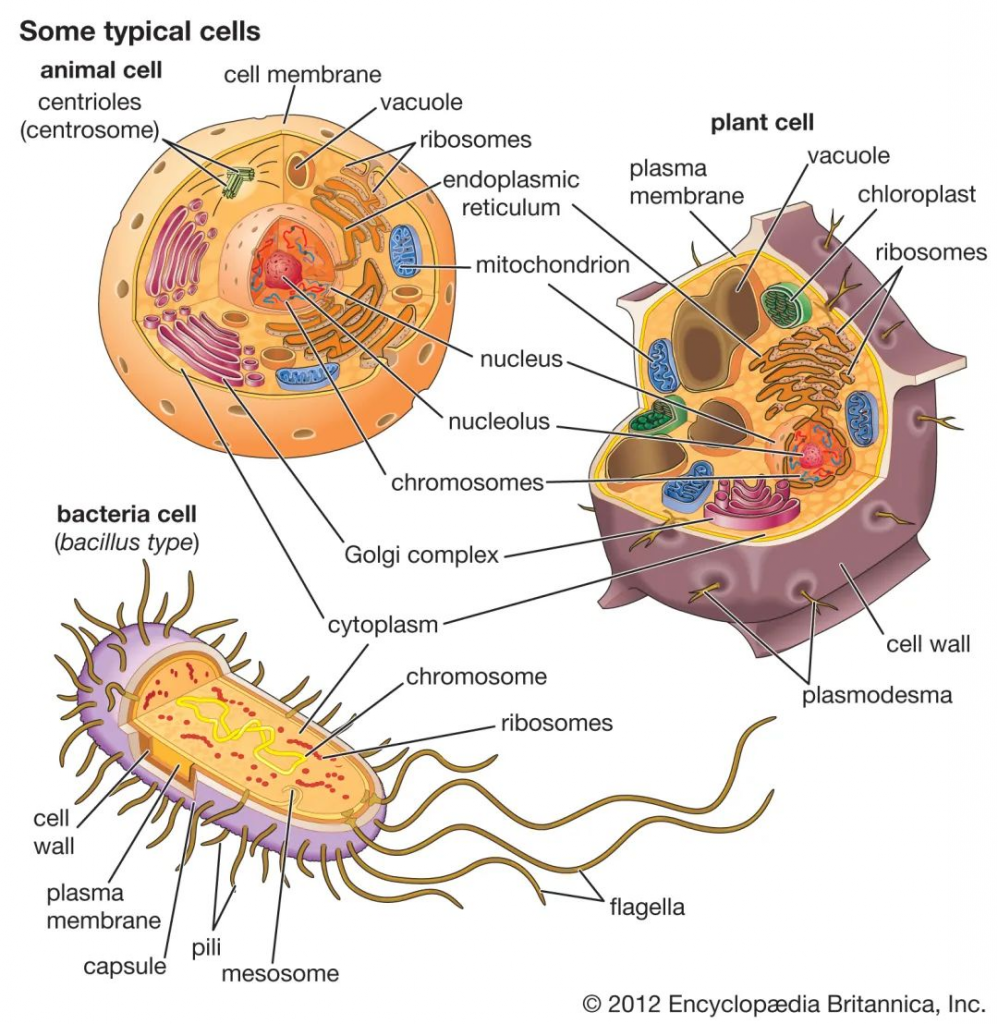
除此之外,原核和真核细胞在许多其他方面有所不同,包括脂质组成,关键代谢酶的结构,对抗生素和毒素的反应以及遗传信息的表达机制。
真核生物包含多个线性染色体,这些染色体的基因比编码蛋白质合成所需的基因大得多。遗传信息的核糖核酸(RNA)副本(脱氧核糖核酸或DNA)的大部分被丢弃,剩余的信使RNA(mRNA)在被翻译成蛋白质之前已被充分修饰。相反,细菌具有一个包含所有遗传信息的环形染色体,它们的mRNA是其基因的精确副本,不会被修饰。
细菌细胞在结构上比真核细胞小得多,也简单得多,但细菌是一个在大小、形状、生境和代谢上都有差异的极其多样化的有机体群体。
许多关于细菌的知识来自对致病细菌的研究,这些细菌比许多自由生活的细菌更容易在纯培养中分离出来,也更容易被研究。必须注意的是,许多自由生活的细菌与适应作为动物寄生虫或共生体生活的细菌有很大的不同。因此,关于细菌的组成或结构没有绝对的规则,任何说法都会有许多例外。
大多数细菌对地球上的生命都是有益的,甚至是生命所必需的,但少数细菌却对人类有害。目前,没有一种古细菌被认为是病原体,但是包括人类在内的动物不断遭到大量细菌的“轰击和居住”。
口腔,肠道和皮肤被大量特定类型的细菌定殖,这些细菌适应这些栖息地的生活。这些微生物在正常情况下是无害的,并且仅在它们以某种方式穿过身体的屏障并引起感染时才变得危险。
一些细菌擅长入侵宿主,被称为病原体或疾病产生者。一些病原体作用于人体的特定部位,例如:
在食物中传播的许多致病菌的毒素在摄入时会引起食物中毒。
由金黄色葡萄球菌(Staphylococcus aureus)产生的毒素,能引起迅速、严重但有限的胃肠道不适。
肉毒梭菌(Clostridium botulinum)的毒素,通常是致命的。在密封前未完全煮熟的罐装非酸性食品中可能产生肉毒杆菌毒素。肉毒梭菌形成耐热孢子,可以发芽为营养细菌细胞,在厌氧环境中茁壮成长,这有利于产生其极强的毒素。
其他食源性感染实际上是由受感染的食物处理者传播的,包括伤寒、沙门氏菌病(沙门氏菌属)和志贺氏菌病(痢疾志贺氏菌属)。
关于食物中毒相关细菌详见:正值夏季,警惕食源性疾病,常见的食物中毒的病原菌介绍
细菌感染是指病原菌侵入宿主组织。当有害细菌进入人体或伤口并繁殖,导致疾病、器官损伤、组织损伤或疾病时,就会发生细菌感染。细菌可以感染身体的任何部位。
细菌感染很常见,但它们并不完全相同。细菌有很多种,每种对身体都有不同的影响。广义上来讲,细菌感染也可以是由任何细菌引起的任何内部或外部疾病的广义术语。
细菌存在于与人体的各种关系中。它们在体表内定殖并提供益处,正常情况下细菌可以与人类身体共存不会产生有害的影响(在共生关系中)。但是有时候,通常无害地存在于我们体内或皮肤上,而不会造成任何异常问题的细菌会失控并导致感染。这种情况通常发生在我们的免疫系统不够强大,无法保持它们的平衡,或者我们的微生物区系的组成发生了一些变化,对某些细菌生存繁殖更有利。
细菌感染是由体内的坏细菌引起的。一些细菌感染可能危及生命。标准的医学治疗通常涉及一剂抗生素。细菌感染很容易与病毒感染混淆。但是他们并不一样,后面我们会单独解释。
当皮肤粘膜有破损或发生化脓性炎症时,细菌则容易侵入体内。
人体的免疫反应可分为非特异性免疫反应及特异性免疫反应两种,后者又可分为细胞免疫与体液免疫两方面。当机体免疫功能下降时,不能充分发挥其吞噬杀灭细菌的作用时,即使入侵的细菌量较少,致病力不强也能引起感染;条件致病菌所引起的医源性感染也逐渐增多。
人体免疫功能正常时,进入血中的细菌迅速被血中防御细胞如单核细胞、嗜中性粒细胞等所清除,而患肝硬变、糖尿病、血液病、结缔组织病等慢性病者,可因代谢紊乱、体液免疫及细胞免疫功能减低,易招致败血症发生;各种免疫抑制药物的使用、放射治疗亦是导致败血症发病率高的原因。广谱抗菌药物使用后,对药物敏感的细菌虽被抑制或杀灭,而一些耐药菌乘机繁殖,亦可酿成败血症。
细菌侵入人体后是否引起感染,除了与人的防御、免疫功能,还与细菌的毒力及数量有关。毒力强或数量多的致病菌进入机体,引起感染的可能性较大。
严重烧伤时,创面为细菌敞开门户,皮肤坏死、血浆渗出又为细菌繁殖提供了良好环境,故极易发生感染。
尿路、胆道、胃肠道、呼吸道粘膜受破坏后,若同时有内容物积滞、压力增高,细菌更易进入血中,保留导尿管、静脉等血管内留置导管、人工辅助呼吸时插管等,也使细菌易于侵入。
细菌是微观的,通常是单细胞生物,到处都可以找到(例如,在我们体内、皮肤上、空气中、水里、土壤中或我们吃的食物里)。细菌具有传染性,因此,它可以通过与患者的密切接触在人与人之间传播。接触受污染的表面、食物、水以及打喷嚏和咳嗽就是例子。
发生感染的几个条件:
此外,细菌还可以导致急性感染(快递治疗)和慢性感染(持续很长时间甚至终生),以及潜伏感染(最初可能没有任何明显迹象或症状但可以过段时间自然发展)。症状可以从轻度、中度到重度不等。在严重的情况下,导致数百万人死亡;例如,许多人死于黑死病或鼠疫。
细菌感染的常见特征:
常见的各部位细菌感染的症状

细菌性感冒
细菌性感冒(Bacterial cold)是指继发细菌感染引起的感冒。感冒在习惯上分为病毒性感冒和细菌性感冒。
病毒性感冒有:普通感冒、流行性感冒和病毒性咽炎等。其主要不同是致病因素不同,病毒性感冒是由于病毒所致,而细菌性感冒是由于细菌所致。
注:流行性感冒(流感通过病毒传播,不是细菌性感冒,注意区分),是由流感病毒引起的急性呼吸道传染病。
细菌的感染还取决于受感染细菌的类型,常见的细菌感染例如:
细菌感染具有高度传染性,因此需要特别注意避免通过洗手、打喷嚏和咳嗽时遮盖以及不共用杯子和饮料瓶来传播感染。
细菌有多种传播方式,包括:

当你忍不住咳嗽或打喷嚏时,用纸巾捂住口鼻或用手肘内侧。
在下述情况下需要洗手,至少洗20秒:
如果没有肥皂和水,可以使用酒精含量至少为 60% 的酒精类洗手液。
除此之外,其他需要注意的:
接种疫苗。如轮状病毒是一种导致婴儿和幼儿严重肠胃炎的病毒。所有婴儿都应在 6 周和 4 个月大时接种轮状病毒疫苗。
流感疫苗。当患上流感时,免疫系统会被削弱。这使细菌有机会侵入肺部并引起感染。每年注射一次流感疫苗可以防止感染肺部细菌。
肺炎球菌多糖疫苗。美国疾病控制与预防中心 (CDC) 推荐 65 岁及以上的人接种肺炎球菌多糖疫苗 (PPSV)。它也适用于免疫系统较弱或患肺炎风险较高的成年人。
治疗取决于感染的严重程度、患者的年龄、免疫系统有多强、是否存在任何并存症状以及现有的医疗条件。
治疗可能包括:
扩展阅读:
细菌素——对抗感染、保存食品、重塑肠道菌群
还在滥用抗生素?15种天然抗生素助你调节肠道菌群
这里我们主要来了解一下,用于细菌感染的药物——抗生素。
目前抗生素是对抗细菌感染的常用药物。它们通过破坏细菌细胞生长和增殖所需的过程来发挥作用。
抗生素根据抗菌谱可以分为:
广谱抗生素,中谱抗生素和窄谱抗生素。
✓ 广谱抗生素
广谱抗生素可对抗多种细菌。广谱抗生素可治疗革兰氏阳性菌和阴性菌,因此如果医生不确定有哪些细菌,他或她可能会开出其中一种抗生素。
阿莫西林、奥格门汀、头孢菌素(第 4 代和第 5 代)、四环素氨基糖苷类和氟喹诺酮类(环丙沙星)是广谱抗生素的例子。
✓ 中谱抗生素
中谱抗生素针对一组细菌。青霉素和杆菌肽是流行的中谱抗生素。
✓ 窄谱抗生素
窄谱抗生素用于治疗一种特定的细菌。多粘菌素属于这一小类抗生素。当确定患有哪种细菌感染时,治疗会更容易和更有效。
抗菌活性通常分为五种机制:
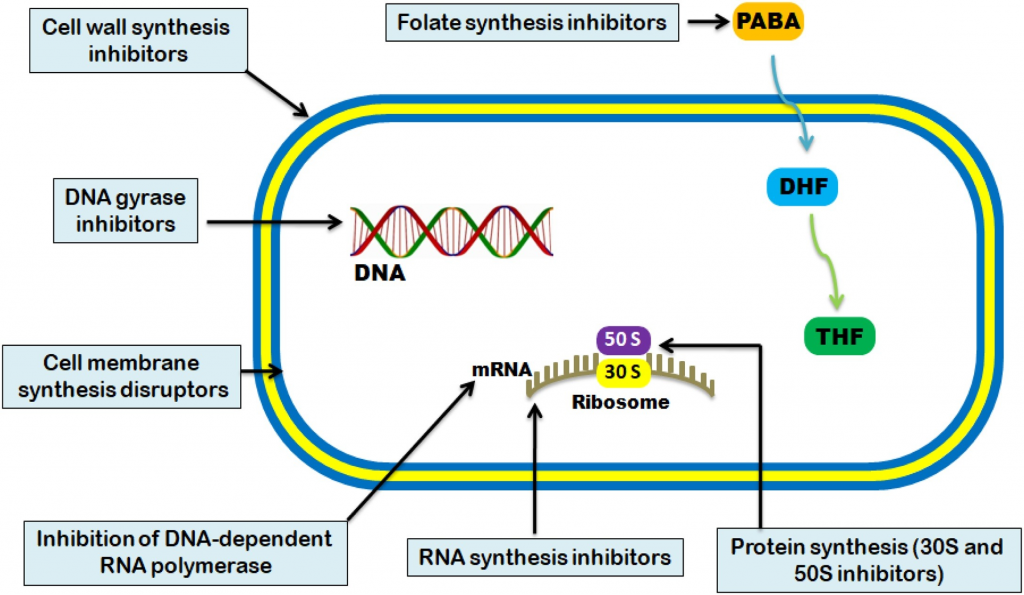

Uddin TM, et al., J Infect Public Health. 2021
不同种类抗生素作用的方式


Uddin TM, et al., J Infect Public Health. 2021
青霉素
最初的青霉素仅用于革兰阳性菌感染的治疗,由于金黄色葡萄球菌很快产生一种青霉素酶对青霉素产生了耐药,人们研制出了以甲氧西林、苯唑西林为代表的耐酶青霉素,用于产青霉素酶的葡萄球菌( 甲氧西林耐药者除外) 感染。
之后,以氨苄西林、阿莫西林为代表的广谱青霉素由仅对革兰阳性(G+) 菌有效扩展到对革兰阴性(G-) 菌( 主要为肠杆菌科细菌) 也具有抗菌活性。在广谱青霉素的基础上又发展为对铜绿假单胞菌等非发酵菌也具抗菌活性者,当前应用主要为脲基类青霉素,包括: 哌拉西林、阿洛西林、美洛西林。
代表药物: 阿莫西林(amoxicillin)
阿莫西林是一种抗细菌的青霉素抗生素。
阿莫西林用于治疗由细菌引起的多种不同类型的感染,例如扁桃体炎、支气管炎、肺炎以及耳、鼻、喉、皮肤或泌尿道感染。
阿莫西林有时还与另一种称为克拉霉素( Biaxin ) 的抗生素一起用于治疗由幽门螺杆菌感染引起的胃溃疡。这种组合有时与一种称为兰索拉唑(Prevacid)的胃酸减少剂一起使用。
四环素
四环素是一类抗生素,可用于治疗由易感微生物引起的感染,例如革兰氏阳性菌和革兰氏阴性菌、衣原体、支原体、原生动物或立克次体。
它们是在 1940 年代发现的,第一个四环素是从链霉菌中获得或衍生的。
四环素抑制微生物 RNA 中的蛋白质合成(一种重要的分子,作为 DNA 的信使)。它们主要是抑菌剂,这意味着它们可以防止细菌繁殖,但不一定会杀死它们。
四环素类药物现今应用的品种除四环素外,更常用有多西环素( 强力霉素) 和米诺环素( 二甲胺四环素) 。由于常见病原菌对本类药物耐药性普遍升高及其不良反应多见,当前本类药物临床应用已受到很大限制。
代表药物:强力霉素(doxycycline)
强力霉素是一种四环素抗生素,可以对抗体内的细菌。
强力霉素用于治疗许多不同的细菌感染,例如痤疮、尿路感染、肠道感染、呼吸道感染、眼部感染、淋病、衣原体、梅毒、牙周炎(牙龈疾病)等。
强力霉素也用于治疗由红斑痤疮引起的瑕疵、肿块和痤疮样病变。
某些形式的强力霉素用于预防疟疾、治疗炭疽或治疗由螨虫、蜱虫或虱子引起的感染。
什么是林可霉素衍生物?
林可霉素衍生物是一小类抗生素,可抑制细菌蛋白质的合成,而细菌蛋白质对细菌的生存至关重要。林可霉素衍生物保留用于治疗由肺炎球菌、葡萄球菌和链球菌的敏感菌株引起的感染。一种衍生物也可用于治疗恶性疟原虫(疟疾)。
林可霉素类包括林可霉素及克林霉素。克林霉素的体外抗菌活性优于林可霉素,主要可应用于敏感肺炎链球菌、其他链球菌属( 肠球菌属除外) 及甲氧西林敏感金葡菌所致的各种感染。该类药物对厌氧菌有良好的抗菌活性常与其他抗菌药物联合用于腹腔感染及盆腔感染,也由于其骨组织浓度较高适用于骨和关节的G + 菌感染。使用本类药物时,应注意假膜性肠炎的发生,如有可疑应及时停药。本类药物有神经肌肉阻滞作用,应避免与其他神经肌肉阻滞剂合用,应注意静脉滴注速度宜缓慢滴注,不可静脉推注。
代表药物:克林霉素(Clindamycin)
克林霉素是一种抗生素,可以对抗体内的细菌。
克林霉素用于治疗由细菌引起的严重感染。
服用此药前 如果对克林霉素或林可霉素过敏,则不应使用该药。
为确保克林霉素的安全,请告诉医生是否曾经有过:结肠炎、克罗恩病或其他肠道疾病; 湿疹或皮肤过敏反应; 肝病; 哮喘或对阿司匹林的严重过敏反应; 对黄色食用色素过敏。
什么是喹诺酮类药物?
喹诺酮类是一种抗生素。抗生素杀死或抑制细菌的生长。
有五种不同的喹诺酮类。此外,另一类抗生素,称为氟喹诺酮类,是从喹诺酮类通过氟修饰其结构衍生而来的。氟喹诺酮类抗生素可导致严重或致残的副作用,这些副作用可能不可逆。
喹诺酮类药物和氟喹诺酮类药物有许多共同点,但也有一些区别,例如它们对哪些生物有效。有些人互换使用喹诺酮和氟喹诺酮这两个词。
喹诺酮类和氟喹诺酮类对细菌产生的两种酶(拓扑异构酶 IV 和 DNA 促旋酶)的功能产生不利影响,因此它们不能再修复 DNA 或帮助其制造。
现今临床主要应用为氟喹诺酮类,常用有诺氟沙星、环丙沙星等主要用于单纯性下尿路感染或肠道感染。但应注意,现今国内尿路感染的主要病原菌大肠埃希菌中,耐药株已达半数以上。环丙沙星尚与β-内酰胺类抗生素联合用于治疗中、重度革兰阴性杆菌感染包括铜绿假单胞菌的感染。
近年来研制的新品种左氧氟沙星、莫西沙星等对肺炎链球菌、化脓性链球菌等G + 球菌的抗菌作用增强,对衣原体属、支原体属、军团菌等细胞内病原或厌氧菌的作用亦有增强,但对G-菌活性增强不明显,对铜绿假单胞菌的抗菌活性仍以环丙沙星为最强。左氧氟沙星、莫西沙星除可用于尿路感染或肠道感染外也可用于呼吸道感染,特别适合于肺炎链球菌( 包括耐青霉素肺炎链球菌) 、支原体、衣原体、军团菌等所致社区获得性肺炎的治疗,此外亦可用于皮肤软组织感染。
在治疗腹腔、胆道感染及盆腔感染时除莫西沙星外需与甲硝唑等抗厌氧菌药物合用。部分品种可与其他药物联合应用作为治疗耐药结核分枝杆菌和其他分枝杆菌感染的二线用药。
代表药物:环丙沙星(Ciprofloxacin)和左氧氟沙星(Levofloxacin)
环丙沙星和左氧氟沙星是一种氟喹诺酮抗生素,用于治疗不同类型的细菌感染。也用于治疗接触过炭疽或某些类型鼠疫的人。环丙沙星和左氧氟沙星应仅用于无法用更安全的抗生素治疗的感染。
左氧氟沙星、莫西沙星:对肺炎链球菌、A 组溶血性链球菌等革兰阳性球菌、衣原体属、支原体属、军团菌等细胞内病原或厌氧菌的作用强。
环丙沙星、左氧氟沙星:主要适用于肺炎克雷伯菌、肠杆菌属、假单胞菌属等革兰阴性杆菌所致的下呼吸道感染。
头孢霉素
头孢菌素是一大类从霉菌顶孢菌(以前称为头孢菌素)中提取的抗生素。头孢菌素具有杀菌作用(杀死细菌),其作用方式与青霉素相似。它们结合并阻断负责制造肽聚糖的酶的活性,肽聚糖是细菌细胞壁的重要组成部分。它们被称为广谱抗生素,因为它们对多种细菌有效。
自 1945 年发现第一个头孢菌素以来,科学家们一直在改进头孢菌素的结构,以使其对更广泛的细菌更有效。每次结构发生变化,都会产生新的“一代”头孢菌素。迄今为止,头孢菌素已有五代。所有头孢菌素均以 cef、ceph 或 kef 开头。请注意,该分类系统在不同国家/地区的使用并不一致。
第一代头孢菌素是指发现的第一组头孢菌素。它们的最佳活性是对抗革兰氏阳性细菌,如葡萄球菌和链球菌。它们对革兰氏阴性菌几乎没有活性。
第二代头孢菌素尚可应用于由流感嗜血杆菌、大肠埃希菌、奇异变形杆菌等中的敏感株所致的尿路感染、皮肤软组织感染、败血症、骨及关节感染和腹腔、盆腔等感染。但如用于腹腔感染和盆腔感染时需与抗厌氧菌药合用。头孢呋辛尚可用于脑膜炎球菌、流感嗜血杆菌所致脑膜炎的治疗,也可作为围术期预防用药。
第三代头孢菌素:对肠杆菌科细菌等革兰阴性杆菌具有强大抗菌作用,头孢他啶和头孢哌酮对铜绿假单胞菌亦具较强抗菌活性;注射品种有头孢噻肟、头孢曲松、头孢他啶、头孢哌酮等,口服品种有头孢克肟和头孢泊肟酯等,口服品种对铜绿假单胞菌均无作用。应注意的是,不是所有的三代头孢菌素都可用于非发酵菌( 如铜绿假单胞菌、不动杆菌等) 感染的治疗,除了头孢哌酮和头孢他啶;外,如头孢曲松和头孢噻肟不可以用于非发酵菌感染的治疗。
第四代头孢菌素:常用者为头孢吡肟,对肠杆菌科细菌作用与第三代头孢菌素大致相仿,对铜绿假单胞菌的作用与头孢他啶相仿,对革兰阳性球菌的作用较第三代头孢菌素略强。其不同于三代头孢菌素之处为对产头孢菌素酶( AmpC) 的肠杆菌属有效,其次是增强了对作用靶位细菌青霉素结合蛋白( PBP) 的亲和力以及其抗革兰阳性菌的抗菌活性亦较三代头孢菌素增强。
第五代(或下一代)头孢菌素头孢洛林( Teflaro ) 对耐甲氧西林金黄色葡萄球菌(MRSA)具有活性。Avycaz 含有β-内酰胺酶抑制剂avibactam。
代表药物:头孢氨苄(Cephalexin)
头孢氨苄是一种头孢菌素(SEF 一种低孢子)抗生素。它通过对抗体内的细菌起作用。
头孢氨苄用于治疗由细菌引起的感染,包括上呼吸道感染、耳部感染、皮肤感染、尿路感染和骨骼感染。
头孢氨苄用于治疗成人和至少 1 岁儿童的感染。
注意事项:
如果对头孢氨苄或类似抗生素(如Ceftin、Cefzil、Omnicef等)过敏,则不应使用该药。如果对任何药物(尤其是青霉素或其他抗生素)过敏,请告诉医生。
对任何药物(尤其是青霉素)过敏;
肝脏或肾脏疾病;要么肠道问题,例如结肠炎,请告诉医生。
头孢氨苄可以进入母乳。如果正在哺乳婴儿,请咨询医生。
β-内酰胺酶抑制剂
β-内酰胺酶抑制剂是一类阻断 β-内酰胺酶(也称为 β-内酰胺酶)活性,防止 β-内酰胺类抗生素降解的药物。本类药物适用于因产β-内酰胺酶的细菌感染,其抗菌谱主要依据原有抗生素的活性而酶抑制剂仅有对抗细菌产酶的作用,一般不增加抗菌活性。
β-内酰胺酶由以下细菌的某些菌株产生:拟杆菌属、肠球菌属、流感嗜血杆菌、卡他莫拉菌、淋病奈瑟菌和葡萄球菌属,无论是组成型还是暴露于抗微生物剂。
β-内酰胺酶裂解易感青霉素和头孢菌素的 β-内酰胺环,使抗生素失活。一些抗微生物药物(如头孢唑啉和氯唑西林)对某些 β-内酰胺酶具有天然耐药性。β-内酰胺类:阿莫西林、氨苄青霉素、哌拉西林和替卡西林,可以通过与β-内酰胺酶抑制剂组合来恢复和扩大其活性。
克拉维酸、舒巴坦和他唑巴坦都是β-内酰胺酶抑制剂。
大环内酯类
大环内酯类药物主要作为青霉素过敏患者的替代药物,用于治疗β-溶血性链球菌、肺炎链球菌中的敏感菌株所致的上、下呼吸道感染、敏感β-溶血性链球菌引起的猩红热及蜂窝织炎等感染。
大环内酯类是从红糖多孢菌(原名红链霉菌)中提取的一类抗生素,一种土壤传播的细菌。
红霉素是第一个发现的大环内酯类;其他大环内酯类包括阿奇霉素、克拉霉素和罗红霉素。
它们的作用主要是抑菌,但在高浓度时可能具有杀菌作用,或取决于微生物的类型。
代表药物:红霉素
红霉素为大环内酯类原型代表药物,由于其胃肠道反应大、口服吸收差使其疗效受到影响。当下在临床应用的主要为大环内酯类新品种罗红霉素、阿奇霉素和克拉霉素等,其他大环内酯类疗效多不如该三种药物。
罗红霉素主要是改善了其药代动力学,口服生物利用度明显提高、给药剂量减小、不良反应明显减少,但其抗菌谱没有明显的改善,是红霉素的替代药物。
氨基糖苷类
临床常用的氨基糖苷类药物主要有: 链霉素、卡那霉素、丁胺卡那霉素、庆大霉素、妥布霉素、奈替米星、依替米星。
其中链霉素当前主要用抗结核治疗,其次可用于治疗鼠疫及布鲁菌病; 庆大霉素、妥布霉素、奈替米星、依替米星肠杆菌科细菌和铜绿假单胞菌等G - 杆菌具强大抗菌活性,对葡萄球菌属亦有良好作用者。所有氨基糖苷类药物对肺炎链球菌、溶血性链球菌的抗菌作用均差。
链霉素、卡那霉素:对肠杆菌科和葡萄球菌属细菌有良好抗菌作用,但对铜绿假单胞菌无作用者。
庆大霉素、妥布霉素、奈替米星、阿米卡星、异帕米星、小诺米星、依替米星:对肠杆菌科细菌和铜绿假单胞菌等革兰阴性杆菌具强大抗菌活性,对葡萄球菌属亦有良好作用。
碳青霉烯类
碳青霉烯类药物是当下抗菌药物中抗菌谱最广的药物,对各种革兰氏阳性球菌、革兰氏阴性杆菌( 包括铜绿假单胞菌的非发酵菌) 和多数厌氧菌具强大抗菌活性,对大多数β-内酰胺酶高度稳定,但对甲氧西林耐药葡萄球菌和嗜麦芽窄食单胞菌等抗菌作用差。
当下在国内应用的碳青霉烯类抗生素有亚胺培南-西司他丁、美罗培南、帕尼培南-倍他米隆、比阿培南。厄他培南与上述品种不同对非发酵菌缺乏抗菌活性,而其他抗菌作用与上述药品基本相同,适合于治疗社区与医院早期特别是G-杆菌感染的重症患者。
碳青霉烯类对各种革兰阳性球菌、革兰阴性杆菌(包括铜绿假单胞菌、不动杆菌属)和多数厌氧菌具强大抗菌活性,对多数β-内酰胺酶高度稳定,但对甲氧西林耐药葡萄球菌和嗜麦芽窄食单胞菌等抗菌作用差。
现用于细菌感染的抗生素被认为是医学史上最重要的突破之一。不幸的是,细菌的适应性很强,抗生素的过度使用让许多人对抗生素产生了耐药性,造成了严重的问题,尤其是在医院的环境里。
抗生素耐药性是细菌抵抗抗生素杀伤力的能力。换句话说,以前治疗感染的抗生素没有效果了,或者说根本无法杀死细菌。
由于过度使用抗生素,抗生素耐药率持续上升,新的抗菌药物开发缓慢。感染耐药细菌可能导致住院时间更长、费用更高,并增加死于感染的风险。
抗生素耐药性是对全球健康的紧迫威胁,包括后来逐渐出现危险耐药细菌——“超级细菌”。
疾病控制和预防中心 (CDC)发布了对美国的前 18 种耐药性威胁的清单,至少包括:
注:以上这些并未全面列出所有耐药菌。抗生素耐药性模式不断演变,细菌可能并不总是对每位患者的选择抗生素表现出耐药性。在正常情况下,抗生素的选择应基于感染部位和由医疗保健专业人员评估的临床表现、培养/敏感性和其他所需的实验室结果、局部耐药性/敏感性模式以及患者的特定特征。
细菌通过以某种方式调整其结构或功能作为防御机制,从而对抗生素产生抗药性。
当细菌以某种方式发生变化时,细菌会抵抗药物。这种变化可以保护细菌免受药物的影响或限制药物接触细菌。或者这种变化可能导致细菌改变药物或破坏药物。
细菌可以用以下方式与抗生素抗争:
如果说以上列举的是抗生素产生耐药性的内因,那么除细菌本身外的因素也会导致抗生素耐药性的出现。
早前,医生有时会依赖不可靠或不准确的知识,开具抗生素“以防万一”,或在特定窄谱抗生素可能更合适时却开出广谱抗生素。这些情况加剧了选择压力并加速了抗菌素耐药性。
当医生不清楚是细菌或病毒加剧了感染时,他们可能会开抗生素。抗生素对病毒感染不起作用,可能会产生耐药性。
抗生素自我治疗(SMA)与药物使用不当的可能性有关,这会使患者面临药物不良反应的风险,掩盖潜在疾病的迹象,并在微生物中产生耐药性。
过度使用抗生素,尤其是在不正确治疗的情况下服用抗生素,会促进抗生素耐药性。根据疾病控制和预防中心的数据,人类使用大约三分之一的抗生素既不需要也不合适。
抗生素可以治疗由细菌引起的感染,但不能治疗由病毒引起的感染(病毒感染)。例如,抗生素是可以治疗由细菌引起的链球菌性咽喉炎。但对于大多数由病毒引起的喉咙痛,并不是正确的治疗方法。
每天都有成千上万的患者、工作人员和访客到达医院,每个人的衣服上和身体上/体内都有自己的一套微生物组。如果医院没有适当的程序和规程来帮助保持空间清洁,细菌就会传播。
抗生素被用作动物的生长补充剂和生长促进剂。在家畜中发现的耐抗生素细菌可能对人类致病,很容易通过食物链传播给人类,并通过动物粪便在生态系统中广泛传播。在人类中,这可能会导致复杂的、无法治疗的和长期的感染。
由于技术挑战、缺乏知识、对抗细菌生理学的重大困难,制药业对新抗生素的发明在很大程度上放缓了,当新抗生素普及时,耐药性的发展(在相对较短的时间内)几乎是不可避免的。
世界范围内抗生素的过度使用和滥用正在导致抗生素耐药性的全球医疗问题。可能会发生抗生素耐药性感染,在最坏的情况下,可能会没有有效的抗生素。这种情况在严重感染时可能会危及生命。
大多数病毒性疾病不需要特殊药物并且是“自限性”的,也就是说患者自身的免疫系统可以抵抗疾病。病毒性疾病的患者可以休息、多喝水并使用对症治疗。
由病毒引起的疾病包括:
有时,在复杂或长期的病毒感染中,细菌也可能侵入,并导致所谓的“继发感染”。在这些情况下,如果需要,可以使用抗生素。
在感染由病毒引起的疾病中,例如咳嗽、感冒或流感,患者不应要求医生开抗生素。抗生素不能治愈病毒感染,患者可能会因不必要的药物而产生副作用。医生可以用其他方法来帮助患有病毒性疾病的患者进行治疗。
如果感染没有得到正确治疗,抗生素耐药细菌也可能传播给其他人。
——有以下几个原因:
它可能不是治疗感染的正确抗生素
它可能已过时且无效
整个疗程可能没有足够的药物
如果新疾病是病毒感染,则不需要抗生素
一些疫苗可以预防细菌性疾病。抗病毒疫苗,例如流感疫苗或COVID-19 疫苗,可以帮助预防可能与继发性细菌感染(如严重的肺部肺炎)相关的原发性疾病。
在治疗的最初几天感觉好些了的时候,仍应完成整个抗生素疗程。
抗生素使用注意事项
✓ 按照指示使用抗生素
按照医生的指示治疗感染。医生会选择最能对抗导致感染的特定细菌的抗生素类型。目前市面上有许多不同种类的抗生素,大部分需要医生开出的非处方抗生素。
✓ 了解抗生素
有的抗生素需要进行皮试,比如青霉素,以防过敏,不同的抗生素杀菌的作用原理不尽相同,比如青霉素是通过能破坏细菌的细胞壁并在细菌细胞的繁殖期起杀菌作用的一类窄普抗生素,杀菌作用强,可用于敏感菌所致的严重感染,由于该类药物作用的靶位为细菌细胞壁,而人体的细胞没有细胞壁,因此毒性低,用于老年人,新生儿和孕妇时安全性相对较高。
✓ 确保清楚抗生素的使用方式
确保明确知道应该服用多少抗生素,以及何时服用。有些抗生素需要与食物一起服用,有些需要在晚上服用等。特别注意要用完整的病征疗程抗生素,有些人担心抗生素对身体有副作用,病情稍微好转就会停止服用或注射抗生素,但是这可能导致体内的致病细菌未完全清楚,再次发生感染,或引起形成抗生素抗药性,使得以后的感染治疗变得困难。
✓ 清洁伤口以防止细菌感染
通过立即正确清洁和包扎伤口来预防皮肤感染。适当的急救治疗对于帮助预防细菌感染至关重要,但不应尝试自己治疗严重的皮肉伤口。如果伤口很深、很宽或出血很多,应该立即寻求医疗帮助。
治疗伤口前先洗手。如果你用脏手治疗伤口,会增加细菌感染的机会。用温水和抗菌肥皂洗手 20 秒,然后擦干。如果有条件可以戴上手套。
细菌感染性疾病起病急、进展快,重症疑难患者病原细菌复杂,诊断及治疗困难,可危及生命。早期、准确地明确病原微生物对感染判定至关重要,有助于改善预后。
准确诊断细菌感染对于避免不必要的抗生素使用和集中适当的治疗至关重要。
细菌感染是细菌的存在与炎症或全身功能障碍的结合;因此,通常需要不止一种诊断方式进行确认,确定患者是否符合临床病例定义的病史和检查。
不同的病原微生物检测技术对感染的判定价值不同,临床上也会根据患者的病情,可能感染的部位,可供采集的标本等选择适宜的检测技术。
当前,病原微生物检测技术层出不穷,能识别的微生物种类越来越多,但如何正确利用和分析微生物检测结果仍是临床关注的难题。值得注意的是,感染首先是个临床诊断,所有的判定必须基于临床。
定植容易对感染判定产生混淆。正常生理状态下人体口腔、胃肠道、呼吸道及体表等部位均有细菌、真菌存在,患者无感染症状时称为定植菌。当定植菌的致病力改变或机体防御能力下降时,可大量繁殖或被带入机体深部引起感染成为致病菌。
临床医生应如何判断所获取的病原体为定植还是感染,是长久以来的难题,且未形成成熟统一的解决方案。
呼吸道定植菌的综合分析:
就呼吸道定植菌而言,既往认为可根据菌落数量或菌种拷贝数来判定定植与感染,但结果存在偏差。越来越多的学者认为,临床上判定是定植还是感染,需结合患者症状、体征及其他检查结果综合分析。
若患者存在与检测阳性结果相匹配的临床症状和体征,如发热、咳嗽、咳痰等,感染指标升高,肺部出现新发病灶,应考虑感染并给予相应的抗感染治疗。如仅培养阳性,患者无任何感染相关临床表现,则倾向于定植菌。
念珠菌定植的综合分析:
念珠菌可广泛定植于呼吸道、胃肠道及泌尿生殖道,正常屏障破坏、免疫功能受损或局部菌群失调时,定植的念珠菌可生长繁殖引起感染。痰和尿标本中分离出的念珠菌亦应根据临床症状和体征鉴别是定植还是感染,念珠菌血培养阳性时要高度警惕念珠菌血症。
此外,病原微生物检测结果阳性,并不意味着患者一定存在感染或感染一定由检出的病原体所致,需结合标本质量、采集部位、病史及其他检测结果综合分析。
不同部位标本检测出同一种病原体,其代表的临床意义可能并不相同。
以肺炎克雷伯氏菌为例,从肠道粪便样本中检出肺炎克雷伯氏菌,其中丰度占比超过1%的人群有3765例,占比28.2%【谷禾健康数据库】。但是当肺炎克雷伯菌进入血液并感染时,通常会发生细菌血症。症状可能包括发冷,发烧,发抖,肌肉酸痛,疲劳和嗜睡。当肺炎克雷伯菌感染肝脏时,会引起脓性病变,也被称为化脓性肝脓肿。这种类型的感染更常见于患有糖尿病一段时间或已经服用抗生素多年的人。
因病原微生物种类不同,微生物检测方法的选择也不相同;即便检测同一种微生物,因采集部位不同所选择的检测方法也有所差别。
如大肠埃希菌,血培养阳性时需考虑血流感染,怀疑致病性大肠埃希菌感染肠道时需特殊培养或者血清学、PCR,16s测序检测等。因普通大肠埃希菌可寄居在肠道中,粪便普通培养有大肠埃希菌生长并不能直接考虑大肠埃希菌肠道感染。
病原微生物检测技术快速发展,能识别的微生物种类越来越多,但感染首先是个临床诊断,所有的判定必须基于临床。随着检测技术革新带来的是临床辅助手段的进步和多样化,更快、更准确地判定感染一直都是病原学检测的挑战,不同方法各有优劣。当前,并没有一项技术可通过从标本中识别微生物来直接判定机体是否感染该病原体,甚至依靠检测的阴性结果来排除感染都很难实现。
在面对细菌感染诊疗过程中始终需要思考3个问题:患者是否存在感染?如果有感染,致病病原体是什么?如何治疗干预?
充分利用现有的病原微生物检测方法和其他实验室检查手段,综合判定,给出合适的抗菌治疗或其他综合治疗方法,是科学可持续的手段。
主要参考文献:
Uddin TM, Chakraborty AJ, Khusro A, Zidan BRM, Mitra S, Emran TB, Dhama K, Ripon MKH, Gajdács M, Sahibzada MUK, Hossain MJ, Koirala N. Antibiotic resistance in microbes: History, mechanisms, therapeutic strategies and future prospects. J Infect Public Health. 2021 Dec;14(12):1750-1766. doi: 10.1016/j.jiph.2021.10.020. Epub 2021 Oct 23. PMID: 34756812.
Cheung GYC, Bae JS, Otto M. Pathogenicity and virulence of Staphylococcus aureus. Virulence. 2021 Dec;12(1):547-569. doi: 10.1080/21505594.2021.1878688. PMID: 33522395; PMCID: PMC7872022.
Tshibangu-Kabamba E, Yamaoka Y. Helicobacter pylori infection and antibiotic resistance – from biology to clinical implications. Nat Rev Gastroenterol Hepatol. 2021 Sep;18(9):613-629. doi: 10.1038/s41575-021-00449-x. Epub 2021 May 17. PMID: 34002081.
BRM, Mitra S, Emran TB, Dhama K, Ripon MKH, Gajdács M, Sahibzada MUK, Hossain MJ, Koirala N. Antibiotic resistance in microbes: History, mechanisms, therapeutic strategies and future prospects. J Infect Public Health. 2021 Dec;14(12):1750-1766. doi: 10.1016/j.jiph.2021.10.020. Epub 2021 Oct 23. PMID: 34756812.
Carmen Fookes, BPharm, Bacterial Infection. January 13, 2020 Medicine
Spagnolo F, Trujillo M, Dennehy JJ. Why Do Antibiotics Exist? mBio. 2021 Dec 21;12(6):e0196621. doi: 10.1128/mBio.01966-21. Epub 2021 Dec 7. PMID: 34872345; PMCID: PMC8649755
Baquero F, Coque TM, Cantón R. Counteracting antibiotic resistance: breaking barriers among antibacterial strategies. Expert Opin Ther Targets. 2014 Aug;18(8):851-61. doi: 10.1517/14728222.2014.925881. Epub 2014 May 31. PMID: 24881465.
武洁, 王荃. 病原微生物检测在感染判定的意义[J]. 中国小儿急救医学, 2020, 27(3):6.
李丹鹤, 荣爱国, 马瑞芝,等. 病原微生物检验在抗感染经验治疗中的临床意义[J]. 医学理论与实践, 2019, 32(09):127-129.