-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康
肠道菌群检测的第一步是取样,取样的重要程度不言而喻,实验人员只有在拿到合格样本后才能开展后续实验。
如果储存和运输不当菌群结构就会发生变化,进而导致菌群测序不准确,因此,便捷可靠是关键。
谷禾经过多年肠道菌群检测实践和研发,开发出适用于肠道菌群取样和常温储存的取样管,可以采集并稳定DNA,用于定量肠道菌群组成分析。

整个取样盒包括:
取样管(内含裂解液和稳定液);
无菌棉签;
回寄袋;
每个取样管上均有唯一条码。
主要特点:
在家中轻松自行采样高质量样品
起始样品需要量低至0.01g,快速且稳定
常温保存运输
标准样品适合手动或高通量自动处理
获得适用于16S ,qPCR,宏基因组的高质量DNA
条形码化全样本可追溯性
谷禾取样管的独特特点使得取样变的异常简便,下面是取样演示:
仅需使用棉签从厕纸上沾取粪便,然后洗脱到取样管的保存液中即可,使保存液可见粪便颜色即表示取样量足够。

适用于-20°C至65°C下保持DNA完整性
室温下有效存储长达60天
与新鲜样本一致的菌群构成特征
低成本
下面来看一下取样管在不同条件下的保存效果,我们使用凝胶电泳来检测不同保存处理条件下提取菌群DNA的状态:
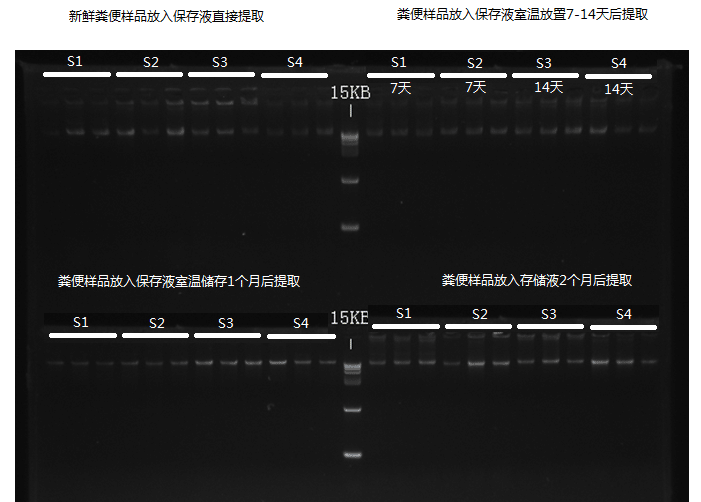
可以看到,使用谷禾保存管的DNA样品即便在存储至60天仍然没有出现明显的DNA降解情况。
专利号:ZL201511009389.7
配合谷禾肠道菌群取样保存管
适用于提取极低当量菌群DNA
具备以下特点:
磁珠法-适用于自动化高通量提取
起始量限制低
与MoBio试剂盒一致性高
现有样本处理量450例/天

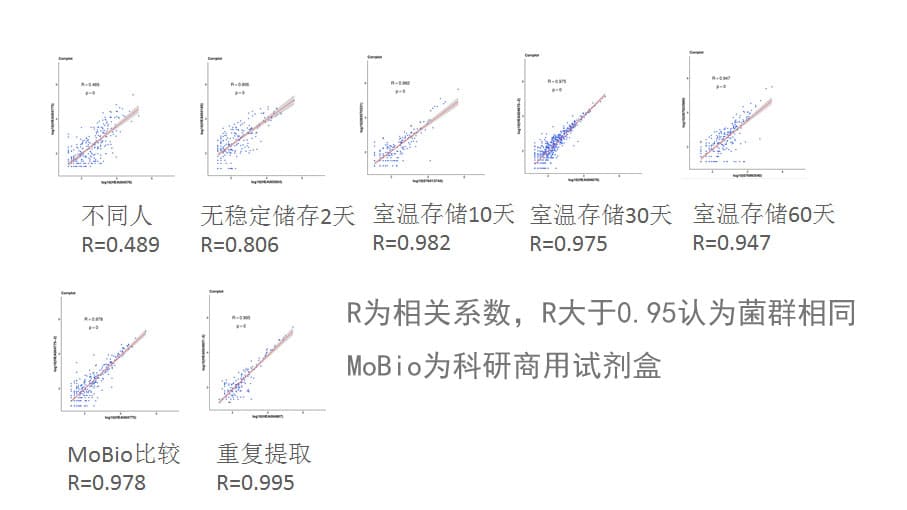
下图可以看到我们使用谷禾提取方法与MoBio试剂盒比较以及重复提取的菌群相关性。另外同时比较了使用谷禾取样管保存不同天数后的提取菌群结果。
专业的实验环境
让整个实验操作
得以高效可靠运行
二级生物安全实验室

注:生物安全实验室的分级

生物安全实验室一般实施两级隔离。一级隔离通过生物安全柜、负压隔离器、正压防护服、手套、眼罩等实现;二级隔离通过实验室的建筑、空调净化和电气控制系统来实现。

谷禾健康
一个多世纪前,埃利·梅奇尼科夫提出了人类健康和预期寿命可以通过操纵肠道菌群来改善的概念。
现在,科学家们已经将目光投向了如何利用微生物来促进人类健康。
来自美国国立卫生研究院国家过敏症和传染病研究所的Stacy及其同事的最新研究表明,有感染史的肠道菌群能够为宿主提供更强的抗感染力。也就是说急性感染后,共生微生物群也可以被“训练”以增强对异源感染的定植抗性。
定植抗性:微生物群阻碍病原体入侵肠道生态系统,这种现象称为定植抗性。定植抗性是肠道微生物群的原始功能之一,为宿主提供了明显的益处。
训练免疫:感染或免疫可以重新编程先天免疫细胞,产生对后续感染具有广泛保护作用的记忆反应,这一过程被称为“训练免疫”。
肠道微生物群是宿主防御网络的基础。除了塑造免疫系统的发展和维护,共生微生物通过战胜病原体获得必需的营养物质和分泌抗生素来确保宿主(和它们自己)的生存,这些因素共同使肠道成为入侵的病原菌不可生存的环境。
肠道微生物群的训练总览
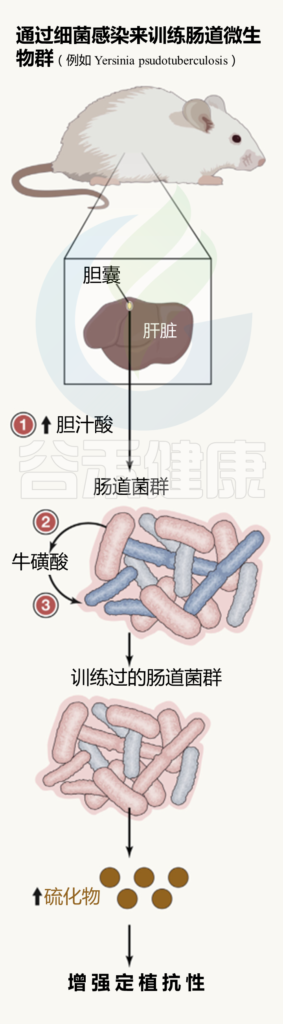
最初的肠道感染后:
( 如Y. pseudotuberculosis )
(1) 肝脏中与牛磺酸结合的胆汁酸产生增加,储存在胆囊,并释放到肠道;
(2) 在肠道中,胆汁酸被特定的微生物群化学解共轭;
(3)牛磺酸变得可利用,导致产生硫化氢的细菌大量繁殖,从而阻止肠道病原体的有氧生长。
下面我们来看Stacy等人是如何一步步进行的实验探究过程。
Stacy等人在研究中发现,与实验室特有的无病原体(SPF)微生物群的小鼠相比,拥有野生小鼠(即WildR)微生物群的自交系小鼠表现出对致命病毒感染的增强保护。
在这里,作者还发现野生小鼠比SPF小鼠对肠道肺炎克雷伯菌感染更具抵抗力。考虑到WildR小鼠的未知感染史,作者用肺炎克雷伯菌对以前感染过急性Yersinia pseudotuberculosis ( post-△yopM) 的SPF小鼠进行了挑战。
值得注意的是,post-△yopM 的小鼠在初次感染后的至少15周内对肺炎克雷伯菌感染的抵抗力也更强。与SPF重建的对照组相比,用post-△yopM小鼠的粪便重建无菌小鼠也增强了对肺炎克雷伯菌感染的保护,这证实了微生物群在抗性中的作用。
这些结果表明,感染史可以改变肠道微生物群,从而诱导定植抗性,因此称为“微生物群训练”。
Stacy等人使用代谢组学方法记录了清除了假结核杆菌Y. pseudotuberculosis△yopM的小鼠肠道中胆汁酸水平的升高。微生物群对初级胆汁酸的解偶联释放了偶联组分,如牛磺酸。
注:牛磺酸在胰腺中合成,对肌肉和大脑发育至关重要,是哺乳动物中最丰富的游离氨基酸。最近发现牛磺酸可以激活杯状细胞中的NLRP6炎症小体,并通过肠道上皮细胞调节稳定状态的抗微生物肽的产生。
在短暂的假结核杆菌感染后牛磺酸水平升高。之前的研究表明,牛磺酸支持解决肠道炎症和清除肠道病原体的柠檬酸杆菌感染。
他们发现,补充牛磺酸会损害柠檬酸杆菌C. rodentium和肺炎克雷伯菌的肠道定植。而当补充给无菌小鼠时,牛磺酸对肠道感染没有影响,这表明它通过调节共栖群落而不是直接影响病原体。
从机制上讲,牛磺酸可增强微生物群中硫化物的产生,硫化物是细胞呼吸的抑制剂,这是宿主被多种病原体入侵的关键。
Stacy等人用16S rDNA和宏基因组测序来解决这个问题。肠道炎症通常与肠杆菌科(Enterobacteriaceae)的大量繁殖有关。
参与对抗感染菌– δ 变形菌 Deltaproteobacteria
他们发现,与对照组小鼠相比,清除假结核杆菌△yopM感染的小鼠微生物群中变形菌门增加,特别是δ 变形菌纲(Deltaproteobacteria)。
宏基因组学揭示,δ 变形菌纲的这种繁殖导致感染小鼠的硫代谢途径增加。虽然在感染后的微生物群中没有发现代谢硫的菌(δ 变形菌纲Bilophila wadsworthia),但当补充到无菌小鼠中时,足以提高对肺炎克雷伯菌的定殖抗性。
牛磺酸的施用同样影响了微生物群;然而,这些变化与post-Yersinia状态相当不一致,包括梭状芽孢杆菌而不是δ变形菌的增殖。这些不一致的分类变化表明,仅补充牛磺酸并不能完全重现共生微生物群的感染后状态,多种因素和微生物群可能能够增强对继发感染的定殖抗性。
Stacy等人通过宏基因组学检测牛磺酸处理过的小鼠微生物组,发现与post-Yersinia菌感染后的微生物组相似,编码异化亚硫酸盐还原酶(dsr)的基因得到了富集。这种蛋白质参与牛磺酸酶转化为硫化物的最后一步。
当牛磺酸调节的微生物群在体外培养中暴露于牛磺酸时,会产生更多的硫化氢,这与肠道中牛磺酸水平较高支持能将这种代谢物转化为硫化物的物种生长的观点一致。
重要的是,硫化氢通过抑制细胞色素氧化酶来限制有氧呼吸,而细胞色素氧化酶是病原体在不可发酵的底物上进行有氧呼吸和生长的常用物质。因此,牛磺酸依赖的硫化氢的产生可能通过阻断有氧呼吸来提高定植抗性。
原发感染后牛磺酸代谢菌群的扩张增强了对继发感染的定植抗性
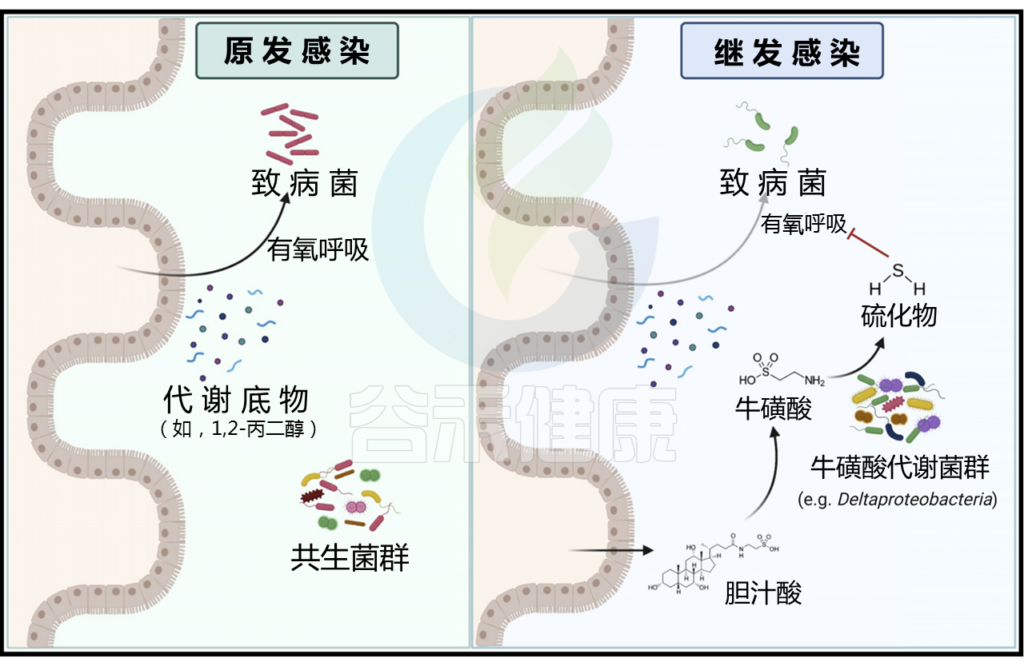
在原发感染(左)期间,病原体通过有氧发酵利用代谢底物,如1,2-丙二醇。清除感染后,宿主产生的胆汁酸水平增加,可以通过共生微生物群与牛磺酸解偶联。牛磺酸反过来被代谢成硫化物,抑制有氧呼吸,从而增强对继发感染的定植抗性(右)。
Stacy等人用两种方法验证:
(1) 使病原体不能在不可发酵的物质上生长
Stacy等人对肺炎克雷伯菌进行了高通量转座子测序。他们让12000个转座子突变体通过小鼠,然后测序,以确定与输入库相比丢失的突变体。
在未感染的小鼠中,导致细菌适应的因素包括与1,2-丙二醇利用有关的基因以及与细胞色素氧化酶bd-II同源的假定氧化酶。因此,肺炎克雷伯菌在低氧环境下对1,2丙二醇作用最为有利,低氧环境受硫化物抑制。
此外,缺乏在1,2-丙二醇或细胞色素氧化酶II上生长能力的肺炎克雷伯菌突变体在体内被它们的野生型对应物所取代,但当牛磺酸被补充到肠道生态系统中时却没有这种情况,表明牛磺酸破坏了病原体在1,2-丙二醇上的呼吸和生长能力。
(2) 隔离硫化氢
Stacy等人使用铋来隔离硫化物。铋对小鼠的处理阻止了微生物群产生硫化氢,导致了具有有氧呼吸功能的细菌数量的增加。
重要的是,铋处理还增加了对肺炎克雷伯菌感染的易感性,突出了硫化物隔离对定植抗性的影响。
这种对继发感染的增强抗性似乎是高度非特异性的,因为弱毒假结核分枝杆菌的初次感染可以防止肺炎克雷伯氏菌或柠檬酸杆菌的继发感染。
事实上,Stacy等人使用“wildR”小鼠的微生物群发现了相同的效果,这些小鼠是无菌小鼠,与野生小鼠的微生物群一起定居,野生小鼠可能有许多肠道感染的历史。
野生小鼠的肠道也显示出δ变形菌纲的扩张,但牛磺酸的水平降低而不是升高,这表明感染后牛磺酸可能会随着时间的推移而消耗,或者是不同的机制导致了这些小鼠中δ变形菌纲的大量繁殖。
Stacy等人没有用相同的病原体进行原发性和继发性感染,因此尚不清楚保护机制是否也适用于这种情况,以及增强定殖抗性的进化目的论是否是为了避免用相同的病原体进行二次感染。同样,确定感染后微生物群是否对不依赖硫化物敏感有氧呼吸的病原体无效也很重要。
Stacy等人的发现也存在一些问题。
首先,为什么在成功清除感染后胆汁酸分泌持续升高?
作者在post–Yersinia感染后的小鼠中发现了增大的胆囊,但这可能不是主要原因。
二、这种增强的殖民抵抗的状态持续多久?
Stacy等人在post-Yersinia感染后15天观察到,接受来自小鼠体内微生物群的无菌小鼠的定殖减少,但尚不清楚肠道牛磺酸水平是否表现相似,以及保护作用是否在超过15天的时间内减弱。
同样,确定多轮感染是否会进一步增强最终的定植抗性能力也很有意思。
Stacy等人的工作使用了生物化学、微生物学和生物信息学方法等,不仅形成了一个关于感染史如何导致肠道-肝脏保护轴的新概念框架,使“微生物群记忆”的概念得到推广,还提供了对宿主及其微生物群之间存在的互利途径的新见解。
这可能是通过精确调节微生物群来提高抗病原体入侵能力的重要一步。
相关阅读:
主要参考文献:
Stacy, A., Andrade-Oliveira, V., McCulloch, J.A.,Hild, B., Oh, J.H., Perez-Chaparro, P.J., Sim, C.K., Lim, A.I., Link, V.M., Enamorado, M., et al. (2021). Infection trains the host for microbiota-enhanced resistance to pathogens. Cell 184, this issue, 615–627.e18
Wong Andrea C, Levy Maayan,Microbial memories.[J] .Immunity, 2021, 54: 201-204.
King Irah L,Divangahi Maziar,Training the metaorganism: the microbial counterpart.[J] .Cell, 2021, 184: 574-576.
Rosshart, S.P., Vassallo, B.G., Angeletti, D.,Hutchinson, D.S., Morgan, A.P., Takeda, K., Hickman, H.D., McCulloch, J.A., Badger, J.H., Ajami, N.J., et al. (2017). Wild Mouse Gut Microbiota Promotes Host Fitness and Improves Disease Resistance. Cell 171, 1015–1028.

谷禾健康
当生命逐渐衰老时,肠道微生物群的脆弱性和随之而来的疾病易感性显得更加突出。
老年人肠道菌群中双歧杆菌、普氏栖粪杆菌(Faecalibacterium prausnitzii)和XIVa梭菌的丰度减少,而产气荚膜梭菌(Clostridium perfringens)、大肠杆菌、肠球菌、链球菌、葡萄球菌、肠杆菌的丰度增加。
因此,衰老的人类肠道微生物群显示出与短链脂肪酸(SCFAs)生产相关的基因缺失和糖分解能力下降,淀粉、蔗糖、半乳糖、糖酵解和糖异生代谢途径的表达降低;伴随着纤维分解微生物的损失;以及蛋白水解功能的全面增强。
益生元低聚半乳糖(GOS)对肠道健康有广泛的有益影响。近日,来自美国北卡罗莱纳大学医学院的研究人员在《Microbiome》上发表文章,题为《益生元低聚半乳糖对衰老肠道的多效性作用》。

在这项研究中,研究人员确定了GOS饮食对肠道老化特征的影响。
在艰难梭状芽胞杆菌感染的小鼠模型中,研究人员还评估了短期喂养低聚半乳糖是否会影响衰老的肠道对抗生素挑战的反应。最后,评估结肠类器官是否能复制体内观察到的低聚半乳糖应答-非应答表型。
老年动物有一个独特的微生物组,其特征是非糖化细菌与糖化细菌的比例增加,相应地,β-半乳糖苷酶的丰度较低。
低聚半乳糖降低了总体多样性,增加了特定的糖化细菌(拟杆菌和乳酸杆菌的种类)的丰度。
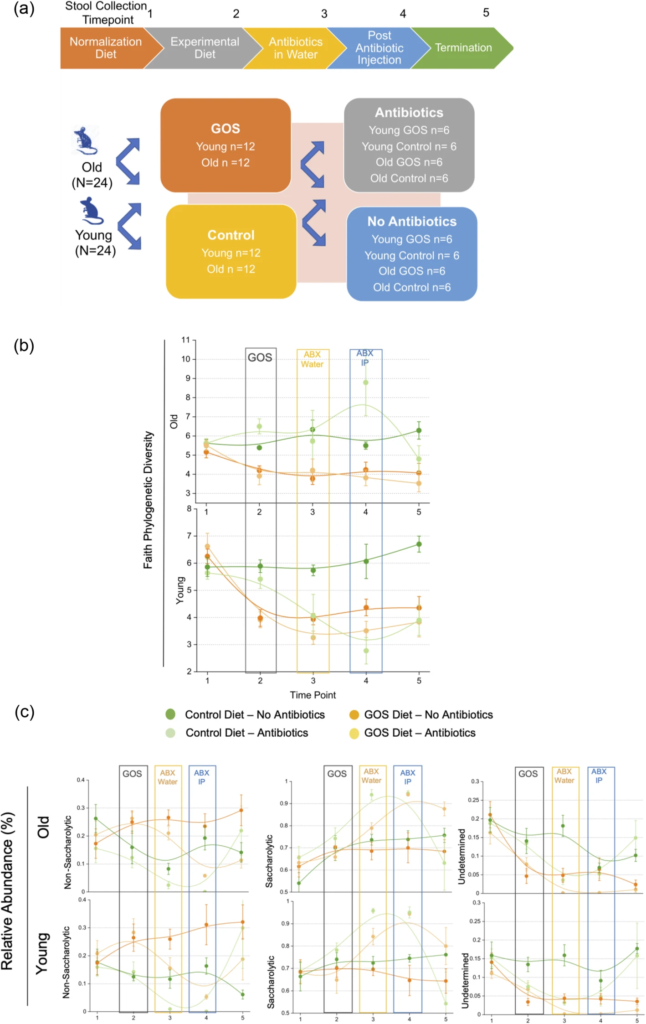
增加了幼年和老年动物中β-半乳糖苷酶的丰度,并增加了非糖化生物;但是,没有观察到强有力的、均匀的双歧作用。
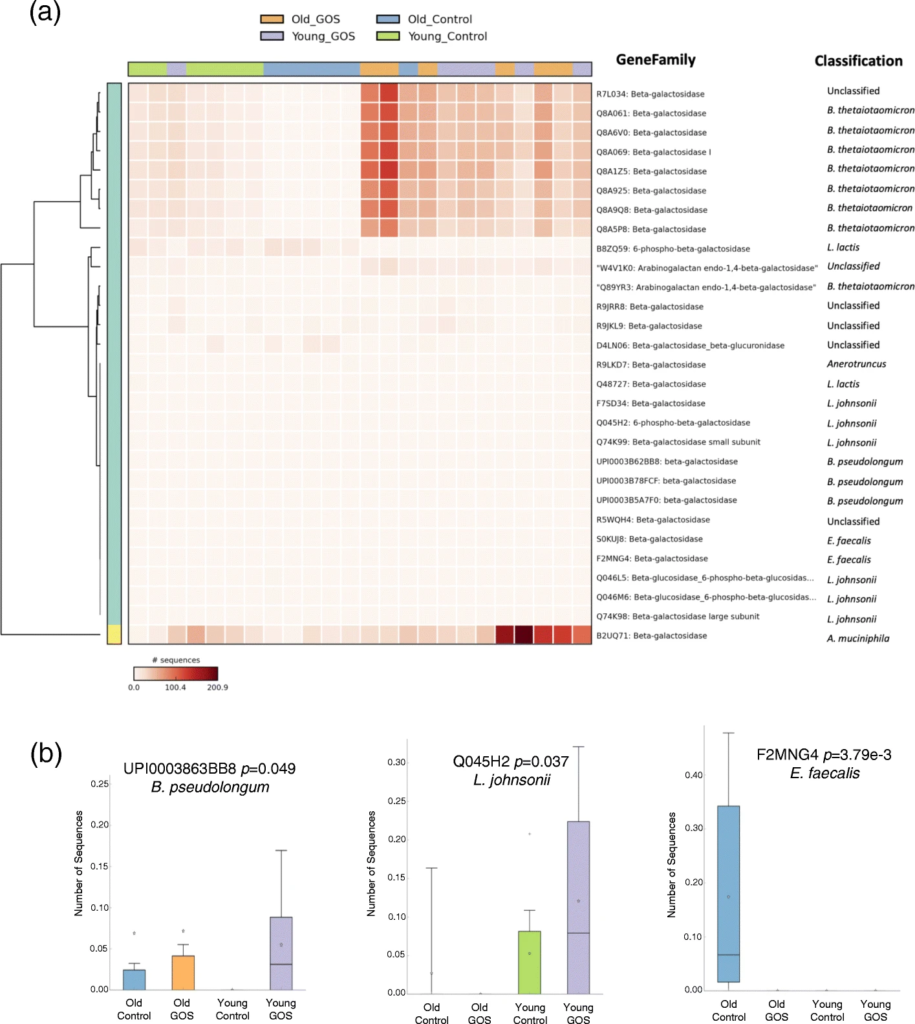
低聚半乳糖降低了老年小鼠与年龄相关的肠通透性增加,增加了MUC2 的表达和黏液厚度。
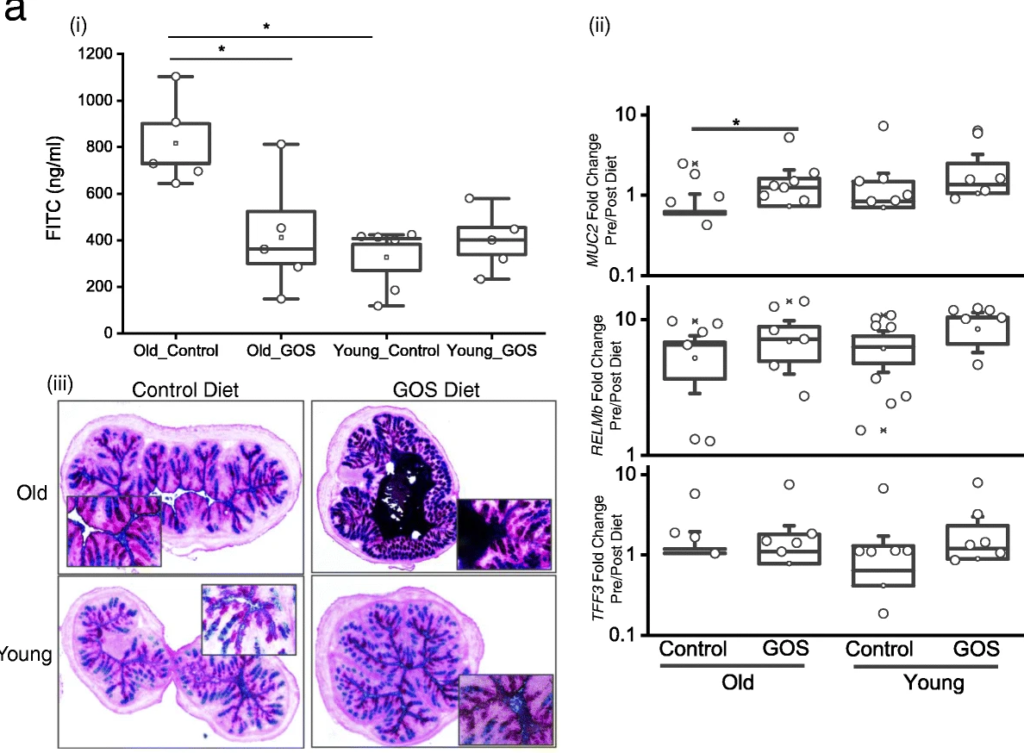
抗生素降低了老年小鼠双歧杆菌的丰度,同时增加了阿克曼菌、梭状芽孢杆菌、粪球菌、芽孢杆菌、拟杆菌和瘤胃球菌。
抗生素在调节血清炎症标志物方面比低聚半乳糖更有效。在抗生素组的对照组和低聚半乳糖饮食中观察到较高的血清IL-17和IL-6水平,并且在这些组中,无论年龄大小,低聚半乳糖组的IL-6水平较高,并且与对照饮食组的幼年动物相比,老年动物的IL-6水平较高。
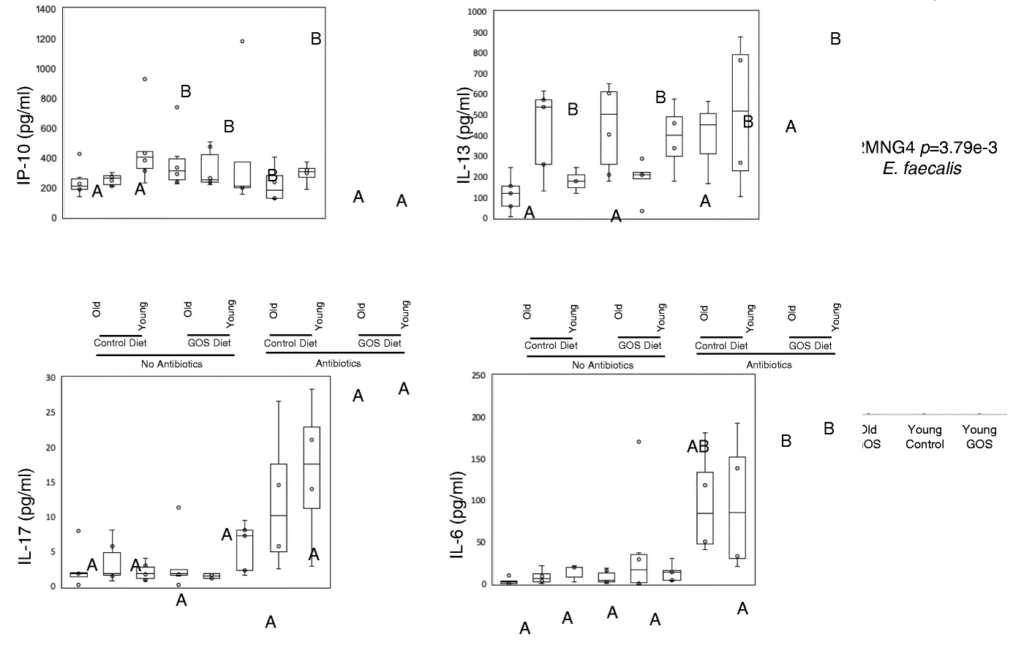
RTqPCR显示老年小鼠远端结肠组织中TNFα的基因表达显著增加,低聚半乳糖饮食降低了这种表达。
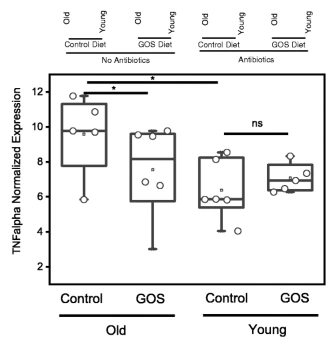
结肠转录组学分析显示,在年老的动物中,小分子代谢过程,特别是呼吸体相关基因的表达增加,这可能表明氧化代谢和能量效率增加。
在幼龄小鼠中,低聚半乳糖诱导了结合相关基因的表达。半乳糖凝集素基因Lgals1是一种β-半乳糖结合凝集素,通过其糖基连接分子,是免疫应答的重要调节剂,PI3K-Akt和ecm受体相互作用通路也在年轻小鼠中被诱导。
在益生元存在的情况下,将GOS注射到结肠类有机物中,小鼠粪便显示出不同的双歧作用,重现了体内观察到的应答和非应答表型,这表明微生物群的组成和功能是表型的主要贡献者。
膳食GOS通过促进微生物组分和宿主基因表达的变化来调节老化肠道的内稳态,这种变化转化为降低肠道通透性和增加粘液生成。
年龄是益生元如何影响肠道上皮细胞的微生物组和表达的决定因素,尤其是在年轻而非老年小鼠中诱导半乳糖凝集素-1时更为明显。
相关阅读:
参考文献:
Arnold Jason W,Roach Jeffery,Fabela Salvador et al. The pleiotropic effects of prebiotic galacto-oligosaccharides on the aging gut.[J] .Microbiome, 2021, 9: 31.0人点赞日记本

谷禾健康
今日,两条关于睡眠问题登上热搜。
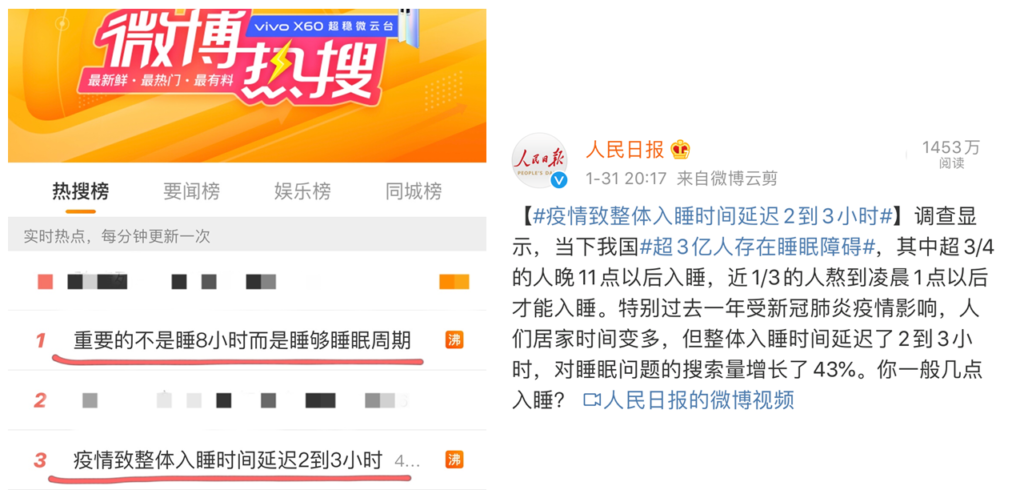
我国有超3亿人存在睡眠障碍,尤其过去这一年,人们整体入睡时间延迟2-3小时,对睡眠搜索量增长43%,看来睡眠问题正在影响越来越多人。
疫情致使整体入睡时间晚2-3小时_腾讯视频
睡眠是由人脑控制的一种复杂的生理行为过程,与免疫功能同为正常生活所必要的生理机能。睡眠是在漫长的一天之后舒缓和恢复的良好方式,睡眠可以让身体和大脑补充能量,良好的睡眠对于巩固记忆、处理信息、生长身体、修复肌肉,增强免疫,抵御疾病至关重要的。
睡眠障碍与各种疾病的发生和发展有关,例如肥胖,II型糖尿病,心血管疾病,抑郁症,癌症等。睡眠不足也会影响判断力和智力。
本文我们来详细了解下,睡眠障碍——这个大多数人都有可能遇到的难题。
首先,关于热搜第一条“睡够睡眠周期”到底什么意思呢?
要了解睡眠障碍之前,我们的先看下,正常生理性睡眠。
正常睡眠结构的特征是轻度睡眠,更深的慢波睡眠和快速眼动(REM)睡眠周期。
第一阶段睡眠(清醒和睡眠的过渡期)
第一阶段睡眠是睡眠周期的开始,被视为清醒和睡眠之间的过渡期。这段睡眠时间仅持续5-10分钟,其特征是混合频率的theta波(非常慢的脑波)。
第二阶段睡眠(体温下降,心率减慢)
第2阶段持续约20分钟,涉及混合频率的脑电波,具有快速的节奏性脑电波活动。在第2阶段,体温开始下降,心率开始减慢。
第三阶段睡眠(从轻度到深度过渡期)
第3阶段睡眠的特征是20%-50%的缓慢脑电波(称为δ波)。这是从轻度睡眠到深度睡眠的过渡时期。
第四阶段睡眠(缓慢脑电波)
阶段4的δ波大于50%,在此期间发生了缓慢的脑电波。阶段4持续约30分钟。
第五阶段睡眠(快速眼动睡眠)
睡眠的第5个阶段,即快速眼动(REM)睡眠,是大多数做梦的时候。第五阶段的特征是呼吸频率增加,大脑活动增加,体内各种代谢功能都显著增加。REM睡眠具有混合频率的EEG和theta波。成年人大约每90分钟出现一次REM睡眠。
睡眠以正常顺序开始,但随后以不规则的顺序循环进行。它开始于阶段1,然后进入阶段2、3和4。在阶段4睡眠之后,在开始REM(阶段5)睡眠之前,重复阶段3和2。REM睡眠结束后,身体通常会返回第2阶段睡眠。REM睡眠的第一个周期是入睡后约90分钟,并且只能持续很短的时间。每个周期,REM睡眠持续时间更长。


失眠是最普遍的睡眠障碍。判断失眠的标准:

标准一: 3个30分钟
入睡时间 [ 入睡时间超过30分钟 ]
睡眠维持困难 [ 醒后再入睡超过30分钟 ]
早醒 [ 比平时提前醒来超过30分钟 ]
标准二:
以上情况 一周超过三天
标准三:
社会功能受损,第二天身体不适
如何判断失眠?权威专家来解答_腾讯视频
影响睡眠质量和持续时间的因素如下,多种内部和外部因素都会对其进行干扰。
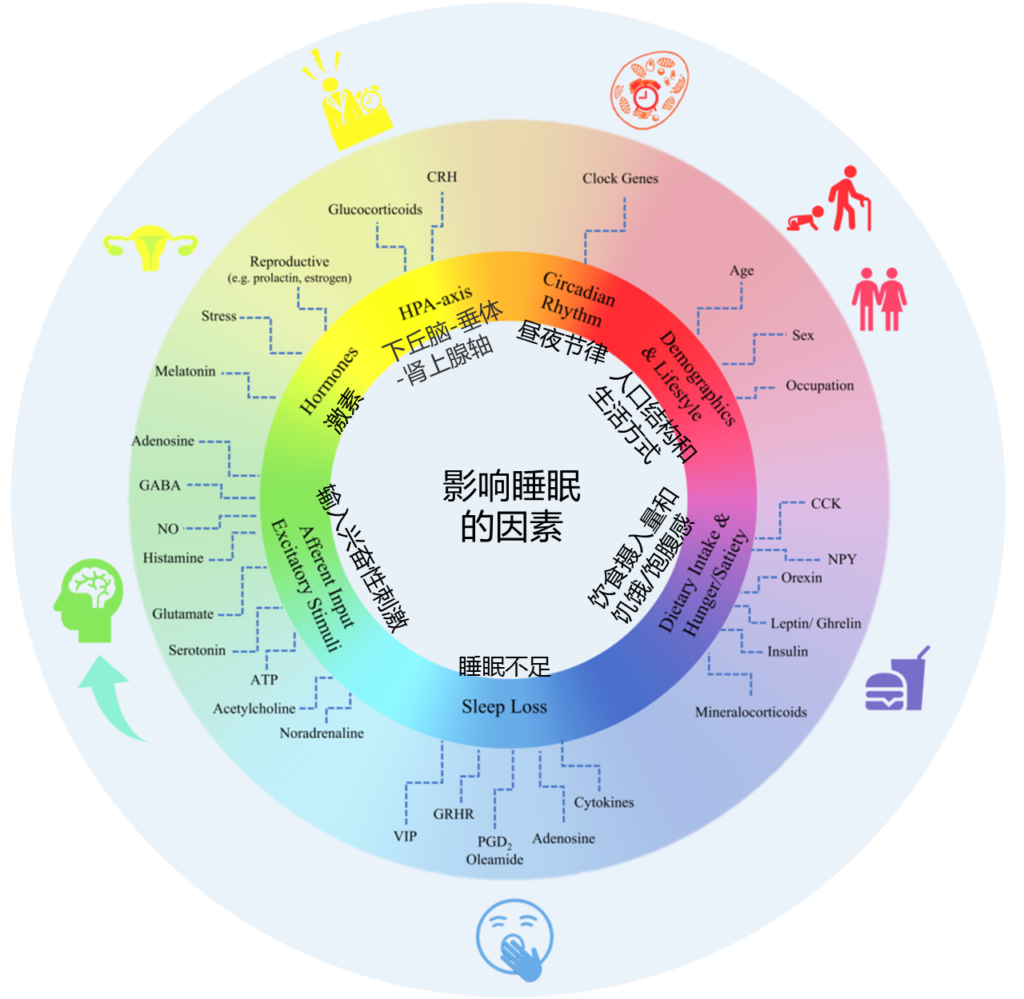
Matenchuk Brittany A,et al., Sleep Med Rev, 2020
睡眠障碍与多种原因有关,通常与不良饮食以及饮食习惯、昼夜节律、压力情绪、生活方式、疼痛炎症、以及慢性疾病等有关。
引起睡眠障碍的原因有很多,但有一个容易被忽略,那就是肠道菌群。
人类微生物群是体内复杂,动态的生态系统。越来越多的研究表明它似乎以许多重要的方式与睡眠相互沟通,相互作用。
菌群改变与睡眠密切相关
研究表明失眠症患者和健康人群肠道微生物的组成、多样性和代谢功能发生了显著变化。随机森林结合交叉验证确定了两种标志性细菌,可用于区分失眠患者和健康人群——拟杆菌属,梭菌属。
对微生物组组成的分析表明,拟杆菌门(Bacteroidetes)和厚壁菌门(Firmicutes)的丰度与睡眠质量呈正相关,而Lachnospiraceae、棒状杆菌(Corynebacterium)、Blautia等几种菌与睡眠质量测量值呈负相关。
Faecalibacterium是肠道微生物群中产丁酸菌,可能有助于双相患者减轻疾病负担和改善睡眠质量。其潜在机制可能是产生促进睡眠的丁酸盐。
高质量的睡眠与肠道菌群相关,包括Verrucomicrobia菌和Lentisphaerae菌 ,占比偏高,与认知功能改善相关。
乳酸菌数量与睡眠呈负相关。干酪乳杆菌对健康成年人的应激性睡眠障碍有有益作用。短乳杆菌对小鼠的睡眠节律有好处。
微生物组多样性(丰度,香农多样性和辛普森多样性)与睡眠质量和总睡眠时间增加呈正相关。
研究发现,睡眠不足与肠道微生物的多样性降低有关,睡眠越好,微生物组的多样性就越丰富。
一项2019年的研究发现,睡前60分钟(这是衡量睡眠量和睡眠质量的指标)与肠道微生物多样性降低26%有关。这是在控制了可能影响微生物组成的其他因素之后,包括饮食中纤维和脂肪的摄入量,体力活动和身体质量指数。
肠道微生物的多样性高有助于减轻压力和改善睡眠。除了睡眠不足之外,微生物组多样性的降低还与一系列健康问题有关,包括情绪障碍,焦虑,抑郁,免疫系统功能障碍和自身免疫性疾病。
失眠患者肠道菌群的α和β多样性发生了显著改变。睡眠时间减少可能会导致肠道菌群失调。
肠道菌群是如何影响睡眠的呢?
可以通过肠道菌群与大脑之间的持续不断的相互作用来影响。主要有以下途径:
· 免疫系统途径
大脑和肠道微生物组都影响免疫细胞的活性,并依次相互影响。
肠道细菌被吞噬细胞(如巨噬细胞或中性粒细胞)吞噬并被消化;消化产物(如MPs、LPS)被释放到周围的细胞间液中。MPs和LPS反过来激活吞噬细胞(如锯齿状细胞膜所示),然后释放细胞因子。全身性细胞因子通过至少两种途径(迷走神经和血脑屏障)进入大脑。
免疫细胞在保持肠道微生物组健康方面发挥了重要作用,并且帮助免疫系统发挥最佳功能。这些细胞执行许多关键功能,包括:
帮助调控微生物组的组成
调节新陈代谢
限制炎症
保护肠道不受感染
保持肠壁坚固(并避免所谓的“漏肠”)
——细菌细胞壁结构成分影响睡眠
微生物细胞壁的结构成分不断刺激先天免疫系统产生细胞因子,产生一种免疫激活的基本状态,从肠粘膜表面开始,影响全身。
当细菌分裂、生长或死亡时,肽聚糖、脂多糖和其他成分被细菌酶降解或改变。宿主吞噬细胞如巨噬细胞和中性粒细胞也可以消化肽聚糖产生胞壁肽(小糖肽)。从革兰氏阳性或革兰氏阴性细菌中分离出来的肽聚糖,诱导睡眠反应,例如,非快速眼动睡眠的持续时间和强度会增强几个小时。如果给吞噬细胞喂养细菌,它们就会释放出具有生物活性的胞壁酰肽;其中一些胞壁酰肽诱导睡眠反应与完整的肽聚糖和热杀死的整个细菌所诱导的睡眠反应相似。
细菌肽诱导肠巨噬细胞和T细胞产生细胞因子白细胞介素-1β(IL-1β)和肿瘤坏死因子α(TNFα);细菌细胞壁脂多糖(LPS)诱导IL-18的合成。
IL-1β,TNFa22,IL-18是非快速眼动睡眠的诱导因子。
其他微生物,如病毒及其组分也通过内源性受体(识别病原体相关分子模式,如Toll样受体)促进细胞因子的产生,从而影响睡眠。
· 神经内分泌途径
肠道内有20多种肠内内分泌细胞,构成最大的内分泌器官。
肠道菌群直接参与多种神经递质,细胞因子和代谢产物的产生,例如5-HT,多巴胺,γ-氨基丁酸(GABA),SCFA和褪黑激素等。
某些乳酸杆菌和双歧杆菌可以产生GABA。在失眠患者中经常观察到GABA mRNA的异常表达。
大肠杆菌产生去甲肾上腺素、5-羟色胺和多巴胺;
链球菌和肠球菌产生5-羟色胺;
芽孢杆菌产生去甲肾上腺素和多巴胺。
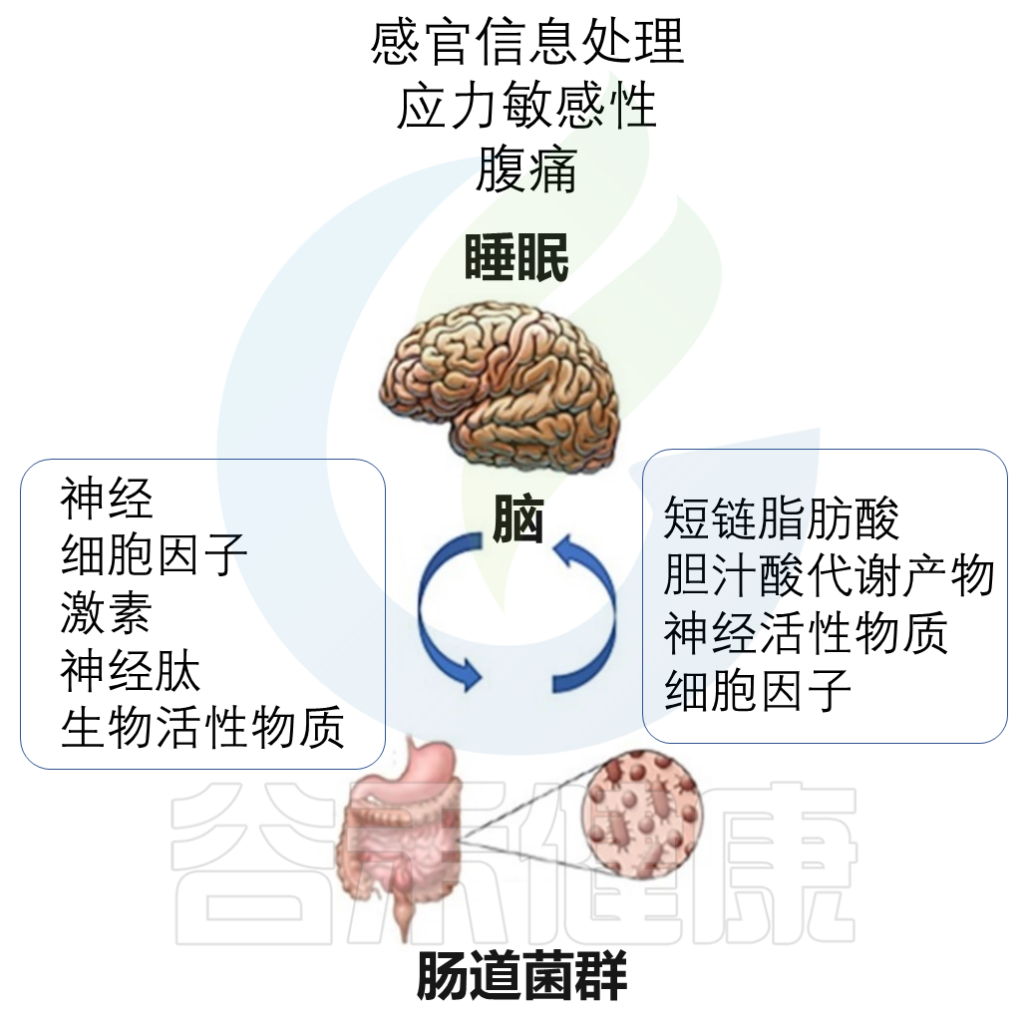
Vernia F,et al., Int. J. Med. Sci.2021
这些代谢物直接作用于肠神经系统和迷走神经,并影响中枢神经系统的活性。
此外,肠道菌群还影响下丘脑-垂体-肾上腺(HPA)轴。
HPA轴参与稳态,参与对新刺激的反应。HPA轴是一种自适应系统,目的是在不断变化的环境中保持体内动态平衡。越来越多的研究表明,睡眠与HPA轴活动之间存在相互关系。
HPA轴亢进会对睡眠产生负面影响,导致睡眠碎片化,深度慢波睡眠减少和睡眠时间缩短。反过来,包括失眠和阻塞性睡眠呼吸暂停在内的睡眠障碍会进一步加剧HPA轴功能障碍。
干预以使HPA轴异常正常化,减少夜间CRH亢进和降低皮质醇可能对治疗失眠和其他睡眠障碍有益。详见本文后面改善睡眠章节。
说起HPA轴,就不得不提到皮质醇。它的作用不容小觑。
皮质醇如何产生?
HPA轴被激活,下丘脑促肾上腺皮质激素释放激素(CRH)的分泌,然后刺激垂体前叶释放促肾上腺皮质激素。然后促肾上腺皮质激素刺激肾上腺释放皮质醇,导致交感神经系统的各种生理反应(如肾上腺素的释放、心率加快和血压升高)。
皮质醇升高可能是睡眠障碍的主要原因
HPA轴障碍可能导致皮质醇升高,当皮质醇水平较高时,会激活糖皮质激素受体。在压力时期去甲肾上腺素和糖皮质激素受体可以优先激活,从而增加促肾上腺皮质激素释放激素。这种升高的促肾上腺皮质激素释放激素会增加睡眠脑电波频率,减少短波睡眠,并增加轻度睡眠和频繁醒来。
皮质醇还与昼夜节律相关,这部分我们在下一章节昼夜节律篇讨论。
· 迷走神经途径
肠肌层神经丛的感觉神经元通过调节肠蠕动和肠激素分泌而接触肠道菌群。肠神经系统也与迷走神经形成突触连接,迷走神经将肠道与大脑连接起来。
细胞因子通过迷走神经传入向大脑发出信号,迷走神经的动作电位进一步诱导胶质细胞和神经元在大脑中产生细胞因子。细胞因子浓度高低与睡眠有关。
低浓度的脑细胞因子能促进睡眠,而高浓度的脑细胞因子则不利于睡眠。
睡眠障碍与肠道菌群失调存在循环关系
前面我们知道,肠道菌群会通过多种途径影响睡眠。
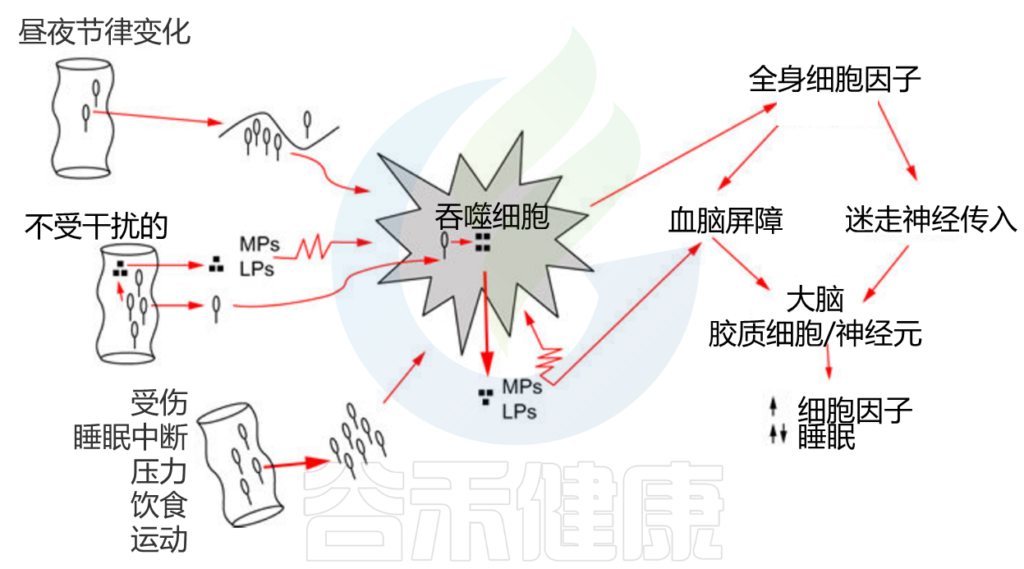
Krueger JM,et al .,Int Rev Neurobiol. 2016
反过来睡眠也会影响肠道菌群。
睡眠不足或者其他因素如受伤、食物摄入、压力、昼夜节律和运动等,可致肠屏障损伤和细菌移位,增加感染易感性,激活HPA轴从而影响菌群。
大多数人(和其他哺乳动物)都存在昼夜节律–控制进食和睡眠等过程的代谢时钟。最常见的昼夜节律周期是控制睡眠的周期,科学家们已发现存在着多种控制着不同生物系统的昼夜节律。
过去的研究已表明如果昼夜节律紊乱,人们可能会遇到健康问题。比如,改变工作时间的轮班工人更容易患睡眠障碍、肥胖、糖尿病等。
昼夜节律——皮质醇
前面提到的皮质醇分泌就有昼夜节律。皮质醇的最低点出现在午夜左右。睡眠开始后约2-3小时,皮质醇水平开始上升,并一直持续到清晨。
早晨醒来时,皮质醇开始迅速升高,并持续升高约60分钟。皮质醇的峰值大约是上午9点。随着一天的继续,水平逐渐下降。随着睡眠的开始,皮质醇持续下降直至最低点。
此外,越来越多的研究都表明,机体的昼夜节律能够调节肠道的免疫反应。
昼夜节律——免疫系统
昼夜节律调节免疫系统,并随之调节炎症水平。
第3组先天淋巴细胞(ILC3s)是昼夜脑-肠信号转导的关键介质。ILC3s表达高水平的昼夜节律基因,光-暗周期的反转导致ILC3s主要的昼夜节律振荡。这种作用依赖于中枢神经系统(CNS)和下丘脑SCN中ARNTL的存在,并进一步与肠道菌群组成的变化有关,特别是变形菌门和拟杆菌门丰度的改变。
注:ARNTL——芳香烃受体核转位因子样蛋白
当昼夜节律被破坏时,正常的免疫功能也会被破坏。这样的情况下,人更容易患上各种疾病。
昼夜节律——肠道菌群
研究发现肠道菌群的两个主要组成部分拟杆菌门(Bacteroidetes)和厚壁菌门(Firmicutes)的丰度从白天到晚上呈周期性变化。
肠道菌群受昼夜节律信号的影响,同时也对生物钟基因的表达产生交互作用。
来自美国德克萨斯大学西南医学中心的研究人员发现小鼠小肠中的微生物参与肠道昼夜节律。该研究发现改变受试小鼠中组蛋白乙酰化的过程,即在组蛋白末端添加乙酰基的过程,细菌便可开启HDAC3在位于小肠内壁的上皮细胞中的表达。这进而导致了参与基因表达的同步振荡,这些基因表达与脂质代谢和营养物运输有关。相比之下,肠道无菌的小鼠没有表现出这种节律性调节。
肠道微生物的昼夜节律振荡导致血清代谢产物的振荡,并与周围组织的转录和表观遗传波动有关。
昼夜节律——肠道菌群代谢产物
短链脂肪酸影响生物钟基因表达和睡眠模式
肠道微生物代谢产物,短链脂肪酸乙酸、丙酸、丁酸在一天中会发生变化,粪便样本中的最高浓度出现得较早,并且在一天中不断降低。短链脂肪酸可能会影响生物钟基因的表达。
研究发现,肠道微生物群的缺乏,以及微生物代谢物的缺乏,导致中枢和肝脏生物钟基因表达明显受损,这表明肠道微生物群在分子水平上传播生物钟的可能性。
在体外,发现在给予乙酸钠和丁酸钠后,小鼠肝细胞中时钟基因Bmal1和Per2的表达发生了显著变化。
在不同的光照-暗期和摄食周期下,添加乙酸后Per2表达量较高,添加丁酸后Per2表达量较低;短链脂肪酸处理后Bmal1表达持续升高,尤其是丁酸处理。
在无菌小鼠体内,关灯两小时后用丁酸盐治疗5天(小鼠处于活跃期),导致肝细胞中Per2:Bmal1 mRNA比值显著增加。此外,同样的处理也导致了中基底下丘脑细胞中Per2:Bmal1 mRNA比值的非显著增加(p=0.053)。Bmal1和Per2等时钟基因在分子水平上调控昼夜节律;它们的比率是肝脏代谢调节网络的标志。
丁酸盐在肠道菌群与大脑产生睡眠的机制之间提供重要联系。
进一步的研究表明,门静脉注射丁酸盐可导致小鼠非快速眼动睡眠增加70%;全身皮下和腹腔注射丁酸盐对睡眠无影响。这些结果表明,丁酸盐的睡眠诱导作用是由肝脏感觉机制介导的。
昼夜节律——肠上皮屏障
肠道菌群通过肠上皮细胞昼夜节律因子调节。
肠上皮细胞协调消化、免疫和神经内分泌功能,是人体最重要的屏障之一。胞壁肽(MPs)或脂多糖(LPS),通过肠上皮屏障转运。
通过受损的伪反应调节器(PRR)信号,导致过氧化物酶体增殖物激活受体α(PPARα)的永久表达,肠道微生物群的消失会破坏肠上皮细胞中Bmal1和Cry1时钟基因的表达,导致肠上皮细胞活动的完全丧失。
此外,肠道菌群也受饮食周期调控,我们将在下一章节详细了解它们之间的关联。
睡眠与昼夜节律、食物摄入、运动和压力源密切相关;这些变量还相互影响,使它们在睡眠中的行为复杂化。饮食、进餐时间和睡眠之间的联系是相互的,因为昼夜节律驱动着代谢模式的变化,而代谢和营养状况的改变则影响着昼夜节律。
我们常听说健康的饮食,生活方式以及合理的饮食习惯有助于心理和身体健康。
辛辣食物、兴奋剂和不良食物反应(不耐受和食物过敏)影响睡眠可以理解。然而,为什么说不吃饭,吃得太快或吃得过饱,吃饭时间不规律,食物质量差,这些也都是导致睡眠障碍的饮食原因?
从本质上讲,饮食摄入与肠道菌群组成有关,因为我们摄入的食物是微生物生长的主要基质。我们饮食的改变可以在几天内导致我们肠道菌群重塑。
摄食节律和昼夜节律的破坏会导致肠道细菌的时间特异性变化。昼夜节律紊乱也会增加肠上皮屏障的通透性。
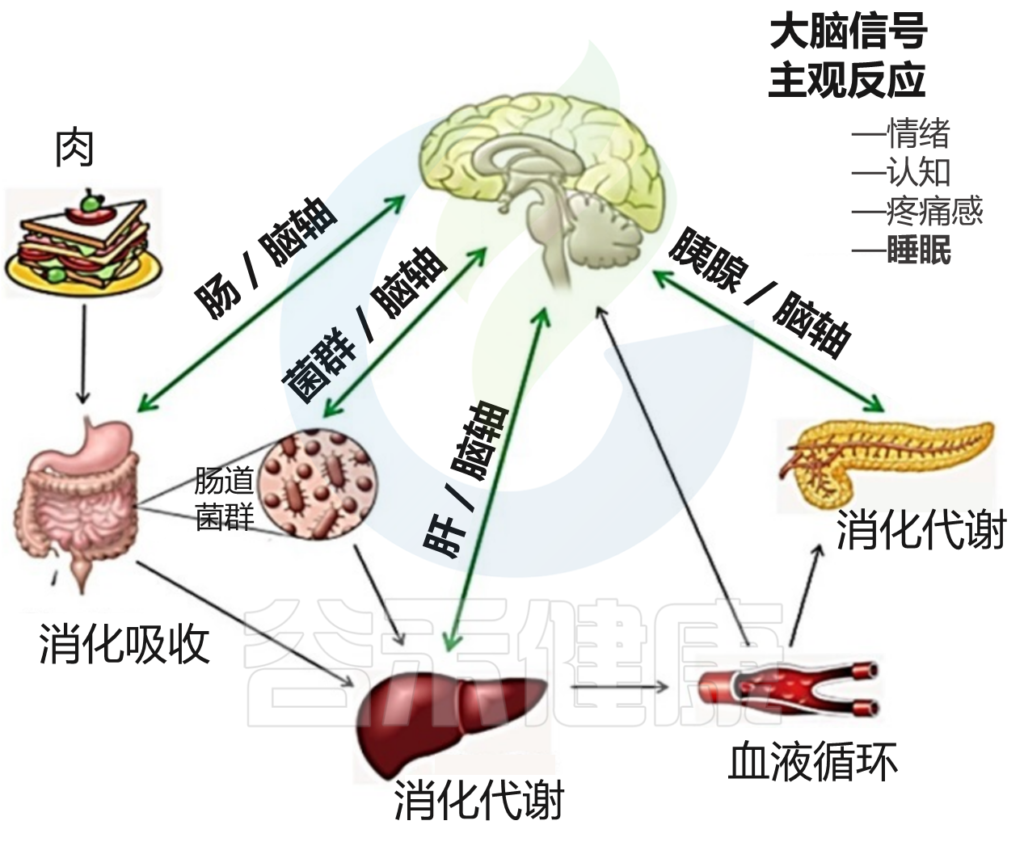
Vernia F,et al., Int. J. Med. Sci.2021
饮食行为影响人类睡眠的时间和质量。睡眠时间短和高能量摄入之间有一致联系。
食物中营养物质影响睡眠
营养物质影响激素的产生,包括生长激素、催乳素、睾酮、褪黑素和血清素,所有这些都在调节生物钟中发挥作用。
食物中存在的氨基酸,如苯丙氨酸、组胺和酪氨酸,促进肾上腺素、去甲肾上腺素和其他刺激性神经递质的产生和释放,可能损害睡眠。
影响色氨酸供应或血清素和褪黑素合成的食物则促进睡眠。一些维生素(B1和B6)也能诱导褪黑素和血清素的产生和释放。
饮食习惯影响睡眠
进餐的时间,特别是零食的频率,使昼夜节律失去同步,影响新陈代谢,并促进肥胖。这与生物钟在调节激素和神经递质释放中的作用是一致的。
不吃饭、或者晚餐十分丰盛的现象越来越普遍。然而将主要热量摄入转移到一天结束时会对消化产生不利影响,并使睡眠困难;如果膳食丰富且脂肪丰富,则更是如此。
相反,碳水化合物对睡眠模式的作用仍有争议,碳水化合物的重量与热量负荷的关系也有争议。
为什么很想吃垃圾食品?
压力在影响饮食模式方面很重要,可能是通过改变下丘脑-垂体-肾上腺轴,让人对垃圾食品(高脂肪和精制糖)产生强烈的渴望。
久坐的生活方式,睡眠时间短同样会让人想吃高能量食物。
为什么睡眠不足与想吃高能量食物有关?
下丘脑外侧神经元通过不同的回路表达神经肽,如黑色素浓缩激素和食欲素/下视黄醇,在调节食物摄取、觉醒、运动行为和自主神经功能方面发挥重要作用。
睡眠限制与饱食因子瘦素浓度降低、促饥饿激素ghrelin浓度增加有关,从而改变了它们发出正确热量需求信号的能力。于是又会促进代谢综合征和肥胖,并再次对生物钟产生不利影响。
注:Ghrelin是一种神经肽,参与睡眠-觉醒调节。
此外,食欲素Orexins在能量稳态和警觉状态之间提供联系,并参与多巴胺能奖赏系统。在动物模型中,产生食欲素的基因突变导致了睡眠表型的改变。有假设说,在清醒时,产生食欲素的细胞的高活性,而在睡眠时几乎没有这种活性,也会影响睡眠。
越来越多的证据也表明睡眠会影响饮食选择。睡眠较少的人更可能喜欢高能量的食物(如脂肪和精制碳水化合物),吃较少的蔬菜,并选择不规律的饮食模式。
糖摄入与睡眠
糖会对肠道健康产生特定作用。有大量证据表明,标准的西方饮食(加工糖和高脂)会导致肠道微生物群的组成发生变化。
上一小节提到的多巴胺奖赏系统与糖摄入也有关系。研究表明,糖是一种有力的触发剂,含糖的食物足以刺激大脑的奖赏系统,从而对食物产生更多的渴望,
糖还有其他间接影响我们肠道健康的方法。高糖饮食会加剧慢性炎症,而炎症则会损害肠道菌群的多样性和功能。经常食用添加糖的饮食可能导致体重增加。
另外添加糖还会升高胆固醇,这与炎症增加有关。关于炎症和睡眠的关系将在下一章节详述。
所有含糖食物(例如水果)都会影响睡眠吗?
不是的。水果之类的天然含糖的食物提升人血糖的速度,远没有含添加糖的食物快。天然食品中纤维含量很高,人体吸收糖的速度变慢,阻止血糖水平飙升。
炎症和睡眠障碍也是双向联系的。
炎症是免疫系统的一种天然的,保护性的生物反应,可以抵抗有害的外来病原体(细菌,病毒,毒素),并帮助身体从受伤中恢复健康。急性炎症的症状包括肿胀和发红,发烧,发冷,疼痛和僵硬以及疲劳,这些迹象表明人体的免疫系统处于“战斗模式”。
睡眠障碍会加剧慢性低度炎症,这是导致疾病的重要因素。不需要几年或者几个月,哪怕只是一晚上的完全睡眠不足就足以提高促炎生物标志物、肿瘤坏死因子α(TNFα)和C反应蛋白(CRP)的循环水平;血清CRP水平随着4天的完全睡眠不足而逐渐升高。
有研究发现,一晚上完全睡眠不足,白细胞介素(IL-6)细胞因子升高,一周失眠不足(每晚4-6小时),IL-6和TNFα的24小时分泌量也会增加。
全身性炎症也会破坏健康的睡眠。通过触发生理和心理变化,让人难以获得良好的睡眠。
细胞因子升高与睡眠困难有关。炎症会在体内造成疼痛和僵硬,使人难以入睡。身体上的疼痛是失眠和其他睡眠问题的常见因素。关于慢性疼痛将在下一章节详细介绍。
炎症涉及较高水平的皮质醇,皮质醇前面了解过,可刺激机敏并导致心理压力。压力是健康睡眠的最重要的常见障碍之一。
7.1 压力,抑郁与睡眠障碍
压力与睡眠
2017年进行的一项研究,压力对大鼠睡眠和肠道健康的影响。通过对小鼠尾部冲击睡眠模式中断。结果发现肠道菌群失去了多样性。少数菌群控制着肠道微生物,失去平衡是不健康的。当他们给小鼠服用益生元时,肠道菌群变得更加多样化,并包含了更多有益菌,如鼠李糖乳杆菌,睡眠变得更好,包括REM和非REM睡眠。
昼夜节律引发情绪波动和睡眠障碍
临床经验表明,扰乱昼夜节律挑起时差综合征或减少睡眠可以触发情绪波动和睡眠障碍。
核心时钟基因突变会引起肠道菌群失调。多种时钟基因变异易患精神疾病,例如重度抑郁症(MDD),双相情感障碍(BD),注意力缺陷多动障碍(ADHD),精神分裂症等。
微生物GABA产生(这是中枢神经系统的主要抑制性神经递质,已证实GABA受体的激活有利于睡眠)对抑郁症和肠道微生物多巴胺代谢物的能力的潜在贡献。
3,4-二羟基苯乙酸(一种主要包含在浆果、水果和蔬菜中的膳食多酚)的合成,与较高的心理生活质量感知相关。
7.2 慢性疼痛与睡眠障碍
慢性疼痛可以对睡眠有不同的影响并取决于疼痛的性质。
疼痛可能在夜间无法缓解,导致睡眠不足。除了缩短总体睡眠时间外,最常见的,慢性疼痛还会导致夜间频繁起床。我们会在轻度睡眠,慢波睡眠和快速眼动(REM)睡眠之间循环。破坏该周期会干扰睡眠阶段的进展,并导致睡眠不足和第二天的疲倦。
疼痛带来的情绪不佳
疼痛也可能伴有焦虑,压力或抑郁。据估计,三分之一的慢性疼痛患者也符合临床抑郁症。这些状况本身会导致睡眠问题。
慢性疼痛间接影响睡眠
患有慢性疼痛的人白天可能会感到疲劳。那么他们不太能做到锻炼或遵循健康饮食,然而这两者对于获得良好的睡眠很重要。
慢性疼痛导致的不稳定睡眠也会打扰夫妻同床,对他们的睡眠质量和健康产生相应的影响。
睡眠对疼痛的影响
新的研究表明,睡眠对疼痛的影响甚至可能比疼痛对睡眠的影响还要强。
睡眠不好导致对疼痛敏感性增强
研究人员发现,睡眠时间短,睡眠分散和睡眠质量差等问题通常会导致第二天对疼痛的敏感性增强,诸如类风湿关节炎。患有睡眠问题的人似乎更有可能最终患上诸如肌痛和偏头痛等疾病。当失眠引起的疼痛加剧时,女性比男性更敏感,年轻人比老年人更有弹性。
慢性疼痛与睡眠障碍的不良循环
患有慢性疼痛的人可能患有自我延续的周期,疼痛,失眠,抑郁或焦虑。例如,遭受痛苦的人在无法入睡时可能会感到焦虑,睡眠不好,醒来时会感到沮丧,这增加了他们对疼痛的敏感性。第二天晚上又开始疼痛,无法入睡,周期一直循环。久而久之,状况可能更加恶化。
前面提到的褪黑素,除了它在调节昼夜节律中的作用,新的研究开始发现褪黑激素在我们对疼痛的感知中产生作用。维生素D、多巴胺也似乎在睡眠和疼痛中都起着作用。
7.3 消化系统疾病与睡眠障碍
胃食管反流性疾病
胃食管反流病以病理性酸或非酸反流为特征,并与多种可能影响上消化道(反流、烧心、疼痛)和/或诱发呼吸道症状(声音嘶哑、发音困难、慢性喉炎、咳嗽、哮喘和慢性支气管炎)的紊乱有关。
有强有力的证据表明胃食管反流病与睡眠障碍之间存在双向关系,因为胃食管反流病的症状会导致入睡困难、睡眠分裂和清晨醒来,而睡眠障碍又会诱发食管痛觉过敏。
因此,有睡眠障碍的胃食管反流病患者比没有睡眠障碍的患者有更严重的症状和更差的生活质量。据报道,在这些患者中,焦虑和抑郁的患病率很高,在某种程度上是由睡眠障碍直接介导的。
IBS
IBS患者的睡眠障碍是有据可查的,入睡困难、睡眠时间短、频繁觉醒等。最近的一项荟萃分析有63620名参与者,结果显示IBS患者睡眠障碍的患病率为37.6%。
IBD
前面章节我们已经知道,炎性细胞因子如肿瘤坏死因子-α(TNF-α)、IL-1和IL-6可引起睡眠障碍,而睡眠障碍可上调细胞因子,尤其是IL-1和TNF-α。(IL-1参与生理性睡眠调节和睡眠对微生物的反应)
临床研究发现睡眠障碍、亚临床炎症和IBD复发风险之间存在关联。最近的一项研究报道,使用匹兹堡睡眠质量指数评估睡眠质量差与粘膜愈合不良有关(P<0.05)。
7.4 肝病与睡眠障碍
睡眠障碍可能发生在急性和慢性肝炎,但更常见于肝硬化患者。相当一部分肝硬化和急慢性肝衰竭患者患有失眠、睡眠延迟和白天过度嗜睡。
肝硬化
最近一项对341名病毒性肝硬化患者的研究证实了这种关联,报告称匹兹堡睡眠质量指数显著升高。多导睡眠图异常也存在。
肝性脑病
睡眠障碍通常是肝性脑病的早期症状,导致日常嗜睡,增加受伤风险,降低生活质量。
肝脏和大脑之间的神经和体液通讯途径尚不完全清楚,但炎症细胞因子如TNF-α、IL-1和IL-6发挥了作用,它们改变了中枢神经递质(血清素和促肾上腺皮质激素释放激素)的浓度。
60%的慢性丙型肝炎患者存在睡眠障碍。
脂肪性肝炎
脂肪性肝炎患者的睡眠障碍可能与肝细胞活性受损和多余脂质处理受损有关。酒精对肝脏和中枢神经系统有直接毒性作用。
最近的分析(2272名参与者)表明,阻塞性睡眠呼吸暂停与脂肪变性、小叶炎症、气球样变性和纤维化显著相关。
瘙痒在慢性肝病患者中很常见,在原发性胆管炎等胆汁淤积性肝病患者中更常见。随之而来的往往是睡眠障碍和生活质量低下。
肝病中瘙痒的患病率从慢性丙型肝炎的5%到原发性胆汁性肝硬化的70%不等。胆汁盐、组胺、5-羟色胺、孕酮代谢物浓度的增加可能与此有关。
7.5 肥胖与睡眠障碍
前面饮食章节我们已经知道,睡眠不足会使身体发出错误信号导致饮食过量,对高热量食物难以抗拒,吃过多自然容易肥胖。
当然,肥胖也会导致睡眠障碍。
超重和肥胖通过胃食管反流病和非酒精性脂肪肝以及阻塞性睡眠呼吸暂停患病率的增加而导致睡眠障碍。
肥胖与阻塞性睡眠呼吸暂停综合征之间存在着相互关系。阻塞性睡眠呼吸暂停会促进行为、代谢和/或激素的变化,促使体重增加和/或减肥困难。阻塞性睡眠呼吸暂停综合征(OSA)与激素水平有关,其特点是瘦素和胃饥饿素水平高,进而促使能量摄入过高。
体重增加10%与患阻塞性睡眠呼吸暂停综合征的概率增加50%有关。当然,体重减轻会减少严重的阻塞性睡眠呼吸暂停,改善睡眠,进一步减轻体重。
因此,阻塞性睡眠呼吸暂停、睡眠时间短和体重增加之间存在关系。一些证据表明,嗜睡与肥胖有关,在没有睡眠呼吸暂停的情况下也是如此。
营养物质改善睡眠
维生素B6
在失眠研究中分析失眠患者中肠道菌群中的维生素B6分解代谢(ko00750)显着增强,导致宿主体内维生素B6缺乏。据报道,维生素B6是失眠症的一种常见治疗方法,维生素B6缺乏会导致疲劳和抑郁。因此,补充维生素B6可以改善失眠症状。
维生素B6食物来源:麦麸、葵花子、大豆、糙米、香蕉、动物肝脏及肾脏、鱼类、瘦肉、坚果等。
叶 酸
叶酸参与髓鞘的形成,在脑脊液和细胞外液中分布较多,可缓解因抑郁导致的失眠,对于人体精神和情绪方面的健康起到重要性的作用。
叶酸食物来源:芦笋,西兰花,胡萝卜,燕麦,奇异果等。
镁
镁补充剂有时用于治疗睡眠障碍,改善睡眠质量并减少睡眠潜伏期(即入睡时间)。一项研究发现,每天服用500mg可以改善老年人的失眠症状。
同时,补充镁也有助于减轻抑郁症症状。
镁食物来源:南瓜子,煮熟的菠菜,黑豆,藜麦,杏仁,腰果,鳄梨,三文鱼等。
锌
除了镁,锌也有促进睡眠的作用,可以改善大脑神经细胞的代谢,平时可以适当多吃一些海鲜、坚果类食物以及全谷类食物,都有助于为身体补充锌元素。
L-茶氨酸
L-茶氨酸:一种氨基酸,L-茶氨酸可以改善放松和睡眠。
益生菌干预
益生菌是一种活的微生物,当其存在的量足够时,可以为宿主带来健康益处,例如发酵食品,如酸奶,开菲尔,豆豉,泡菜,康普茶等。
很少有研究测试通过控制肠道微生物群来改善睡眠的有效性。在一项32名医科学生参加的临床试验中,发现益生菌加氏乳酸杆菌CP2305能显著改善睡眠质量,这可以通过PSQI评分的变化来衡量。在服用了益生菌的男性参与者中,这种改善更为明显,在床上入睡时间的减少。
注:匹兹堡睡眠质量指数(Pittsburgh sleep quality index,PSQI)是美国匹兹堡大学精神科医生Buysse博士等人于1989年编制的。该量表适用于睡眠障碍患者、精神障碍患者评价睡眠质量,同时也适用于一般人睡眠质量的评估。
同时,15种肠道微生物的相对丰度在对照组和益生菌组之间有所不同,包括Bact. Vulgatus的减少,在使用益生菌后增加了Dorea Longicatena.
额外的双盲随机对照试验发现,补充益生菌混合物(含Lactobacillus fermentum LF16, L.rhamnosus LR06, L.plantarum LP01,长双歧杆菌 Bifidobacterium longum BL04 ),在年轻健康的参与者中,随着时间的推移,导致PSQI得分下降。
注:PSQI得分越高,表示睡眠质量越差。
高皮质醇诱发的睡眠问题的替代方法
解决慢性皮质醇水平升高的有效方法是确保肾上腺得到适当的营养支持。维生素B6,维生素B5(泛酸)和维生素C通常会由于肾上腺活动时间过长和皮质醇的产生而耗尽。这些营养物质在肾上腺的最佳功能和肾上腺激素的最佳制造中起关键作用。在压力时期,这些营养素的水平可以降低 。
改善睡眠的另一种方法是针对GABA(γ-氨基丁酸)活性。增加GABA活性将降低蓝斑,下丘脑室旁核和HPA轴活性。支持GABA功能的一种方法是减少谷氨酸信号。谷氨酸和GABA活性彼此相反。因此,降低谷氨酸的活性将支持健康的HPA轴活性。
Tips
1 不要在深夜吃东西,破坏微生物生物钟,还会促进胃反流。
2 多吃纤维。纤维有助于有益菌生长。纤维食物包括朝鲜蓟,芦笋,洋葱,豆类,绿叶蔬菜和大多数非淀粉类蔬菜。

3 尝试睡前禁食,禁食会使身体处于“待机”状态,可以自我修复。身体在睡眠过程中会继续燃烧卡路里。睡前禁食,早晨更有可能感到饥饿。可能会促使早起。
4 如果一定要吃,尽量吃易消化食物。消化过程让人清醒睡不着,因此最好在睡前避免食用难消化的食物。包括:脂肪或油炸食品、辛辣食物、酸性食品、碳酸饮料等。
5 多吃各种食物,有益于维持人体健康的微生物群。均衡饮食,食物中的营养素在产生褪黑素以及其他有助于调节睡眠的重要神经递质中起着巨大作用。

6 尝试补充益生元。已显示许多益生元可在人类受试者中发挥作用。如低聚果糖和低聚半乳糖等。
7 创建理想睡眠环境。
关闭电子产品(就寝前30分钟至1小时),保持卧室适宜温度(在16至19°C之间)等

8 调整灯光。晚上关掉灯或调暗灯,黑暗下人体会分泌更多褪黑素,有助于睡眠,当然,早上拉开窗帘享受阳光,可以帮你清醒。
9 舒适的床是最佳睡眠环境。旧的床垫和枕头会引起疼痛和酸痛,难以获得优质的睡眠。通常,专家建议每10年更换一次床垫,每两年更换一次枕头。当然也取决于床垫枕头质量。

10 保持规律作息。最好每天在同一时间上床睡觉,早上同一时间起床,确保人体昼夜节律时钟正常运作。即使在周末或休息日最好也是如此。

11 避免白天睡过多。如果已经出现睡眠障碍,那么白天尽量不要睡觉。如果有午睡习惯,尽量控制在30分钟之内,且在下午3点之前完成。
12 睡前放松,可以进行温水浴,泡脚,深呼吸,做些伸展运动,适量阅读,听听舒缓的音乐等,这些准备工作都有助于良好的睡眠。当有压力或焦虑时,身体会产生更多的皮质醇,皮质醇过高可能导致夜间频繁醒来。

13 如果实在在20分钟或更长时间内无法入睡,请起床并做一些容易累的事情。最重要的是离开床。
14 运动是帮助睡眠的良好方式,如果可以的话,每天至少20-30分钟锻炼,每周五次左右,但不要在睡前剧烈运动。

15 随着年龄的增长,褪黑素水平会下降。可以购买褪黑激素补充剂,该补充剂已被证明可以帮助55岁以上的人们更快入睡和更长的睡眠。睡前一个小时服用。褪黑激素还可以增强肠道微生物的健康多样性。如长期服用需咨询医生。

【附录】
需要多少睡眠时间取决于年龄,并且因人而异。大多数成年人每晚至少需要七个或七个以上的睡眠时间。
新生儿(0到3个月):睡眠14到17个小时
婴儿(4至11个月):睡眠12至15小时
幼儿(1至2岁):睡眠11至14小时
学龄前儿童(3至5岁):睡眠10至13小时
学龄儿童(6至13岁):睡眠9至11小时
青少年(14至17岁):睡眠8至10小时
年轻人(18至25岁):睡眠7至9小时
成人(26至64岁):睡眠7至9小时
老年人(65岁或以上):睡眠7至8小时
当然以上只是参考,并不是所有人必须达到的标准,少数人的需要的睡眠时间本来就不多,且没有睡眠困扰或不适症状,则无需参考以上标准。
相关阅读:
主要参考文献:
Vernia F, Di Ruscio M, Ciccone A, Viscido A, Frieri G, Stefanelli G, Latella G. Sleep disorders related to nutrition and digestive diseases: a neglected clinical condition. Int J Med Sci. 2021 Jan 1;18(3):593-603. doi: 10.7150/ijms.45512.
Krueger JM, Opp MR. Sleep and Microbes. Int Rev Neurobiol. 2016;131:207-225. doi: 10.1016/bs.irn.2016.07.003. Epub 2016 Aug 31.
Matenchuk Brittany A,Mandhane Piush J,Kozyrskyj Anita L,Sleep, circadian rhythm, and gut microbiota.[J] .Sleep Med Rev, 2020, 53: 101340.
Hertenstein E., Feige B., Gmeiner T., Kienzler C., Spiegelhalder K., Johann A., Jansson-Frojmark M., Palagini L., Rucker G., Riemann D., et al. Insomnia as a Predictor of Mental Disorders: A Systematic Review and Meta-Analysis. Sleep Med. Rev. 2019;43:96–105.
Poroyko V.A., Carreras A., Khalyfa A., Khalyfa A.A., Leone V., Peris E., Almendros I., Gileles-Hillel A., Qiao Z., Hubert N., et al. Chronic Sleep Disruption Alters Gut Microbiota, Induces Systemic and Adipose Tissue Inflammation and Insulin Resistance in Mice. Sci. Rep. 2016;6:35405.
Kinnucan J.A., Rubin D.T., Ali T. Sleep and Inflammatory Bowel Disease: Exploring the Relationship between Sleep Disturbances and Inflammation. Gastroenterol. Hepatol. (N.Y.) 2013;9:718–727.
Bowers S.J., Vargas F., Gonzalez A., He S., Jiang P., Dorrestein P.C., Knight R., Wright K.P., Jr., Lowry C.A., Fleshner M., et al. Repeated Sleep Disruption in Mice Leads to Persistent Shifts in the Fecal Microbiome and Metabolome. PLoS ONE. 2020;15
Smith R.P., Easson C., Lyle S.M., Kapoor R., Donnelly C.P., Davidson E.J., Parikh E., Lopez J.V., Tartar J.L. Gut Microbiome Diversity is Associated with Sleep Physiology in Humans. PLoS ONE. 2019;14:e0222394.
Durgan DJ. Obstructive sleep apnea-induced hypertension: role of the gutmicrobiota. Curr Hypertens Rep. 2017; 19: 35
Reynolds AC, Paterson JL, Ferguson SA, Stanley D, Wright KP Jr, Dawson D.The shift work and health research agenda: considering changes in gutmicrobiota as a pathway linking shift work, sleep loss and circadianmisalignment, and metabolic disease. Sleep Med Rev. 2016; 34: 3-9.
Parisi P, Pietropaoli N, Ferretti A, Nenna R, Mastrogiorgio G, Del Pozzo M, etal. Role of the gluten-free diet on neurological-EEG findings and sleepdisordered breathing in children with celiac disease. Seizure. 2015; 25: 181-183
Michalopoulos G, Vrakas S, Makris K, Tzathas C. Association of sleep qualityand mucosal healing in patients with inflammatory bowel disease in clinicalremission. Ann Gastroenterol. 2018; 31: 211-216.
Wang B, Duan R, Duan L. Prevalence of sleep disorder in irritable bowelsyndrome: A systematic review with meta-analysis. Saudi J Gastroenterol.2018; 24: 141-150.

谷禾健康
基因组规模代谢网络模型(Genome-scale metabolic model,GEM),是一种包含了某种特定生物或者是细胞基因组范围代谢反应,及其酶及基因关联的数学模型。
这里,我们基于文章的描述,介绍一款新软件——MetaGEM。
研究者认为,目前代谢建模的工作流程仍然是倾向于依赖参考基因组作为重建和模拟GEMs的起点,这忽略了微生物群落中存在的物种内和物种之间的多样性。也限制了对已知参考基因组空间中的代谢网络的分析和解释。
可能导致假阳性(即在参考基因组中存在但在群落中的变量中缺失的通路)或假阴性(即在参考基因组中缺失但在群落变量中存在的通路)结果,最终导致对个别物种代谢通路以及交互营养共生(cross-feeding)相互作用的不准确预测。
也就是说当前的代谢建模方法很可能无法捕捉特定物种在不同环境中的特定代谢特征,例如具有不同疾病状况的个体的微生物群。为了克服这一局限,研究者们开发了MetaGEM。
MetaGEM可以不依赖参考基因组,直接从短读的宏基因组数据中重建样本特定的代谢模型。
下图是该软件的流程图,图中蓝底白字的部分是该流程中所使用到的软件,都是已经由他人开发完成的。
研究者们自己开发的部分有两个:
一是end-to-end的框架,能够进行群落水平的代谢交互模拟;
二是一个来自宏基因组生物群落的14,000多个MAGs,包括3750份高质量的MAGs,以及来自人类肠道微生物组研究和全球微生物组项目的相应的随时可用的GEMs。
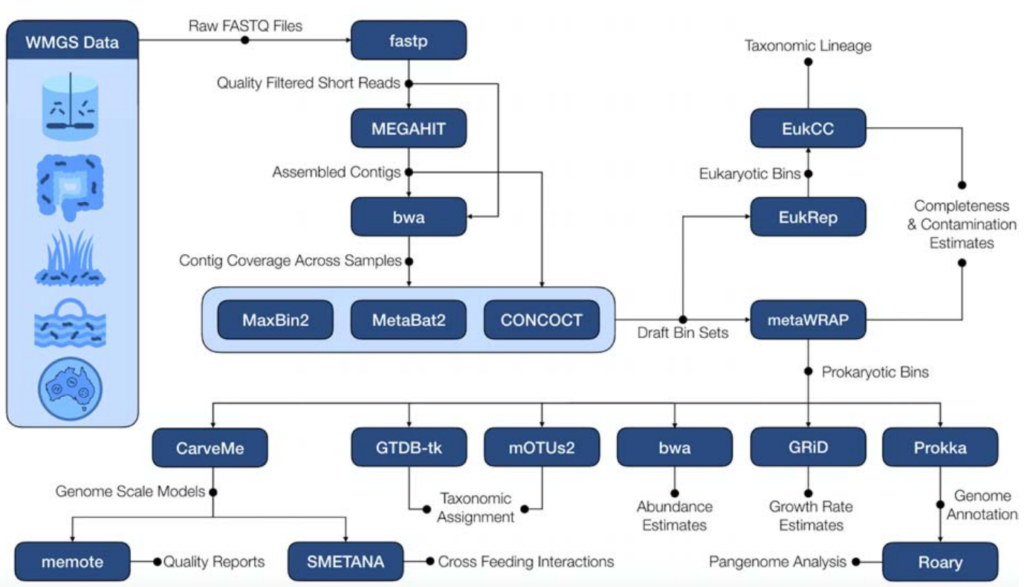
整个流程使用Snakemake实现,从原始的宏基因组的fastq文件开始,质控、组装、估计contig覆盖率、binning、Bin的改进和重组、MAG丰度定量和物种分类、CarveMe进行基因组规模代谢模型重建及质量报告,Smetana模拟重建的基因组规模代谢模型的肠道微生物群落。
(这里只简单介绍了处理步骤,文章中的“Methods”部分有给出使用的参数)
除了以上的必备选项,该流程还有一些附加功能可供用户选择。可以使用GRID估计中和高覆盖率的MAGs的增长率。
Prokka可以对MAGs做功能注释,并且其结果可以提供给Roary,获得一组MAGs的核心MAG和泛基因组的可视化结果。
EukRep可以用于寻找真核生物的MAGs。
EukCC可以对真核生物的bins做后续的分析。
MetaGEM流程具有两个特点:
一是直接从宏基因组获得高质量的代谢重建;
二是可以为个性化的人类肠道群落建模,研究者通过两个实验进行了描述:
MetaGEM模型与EMBL、AGORA、KBase和Bigg模型相比较
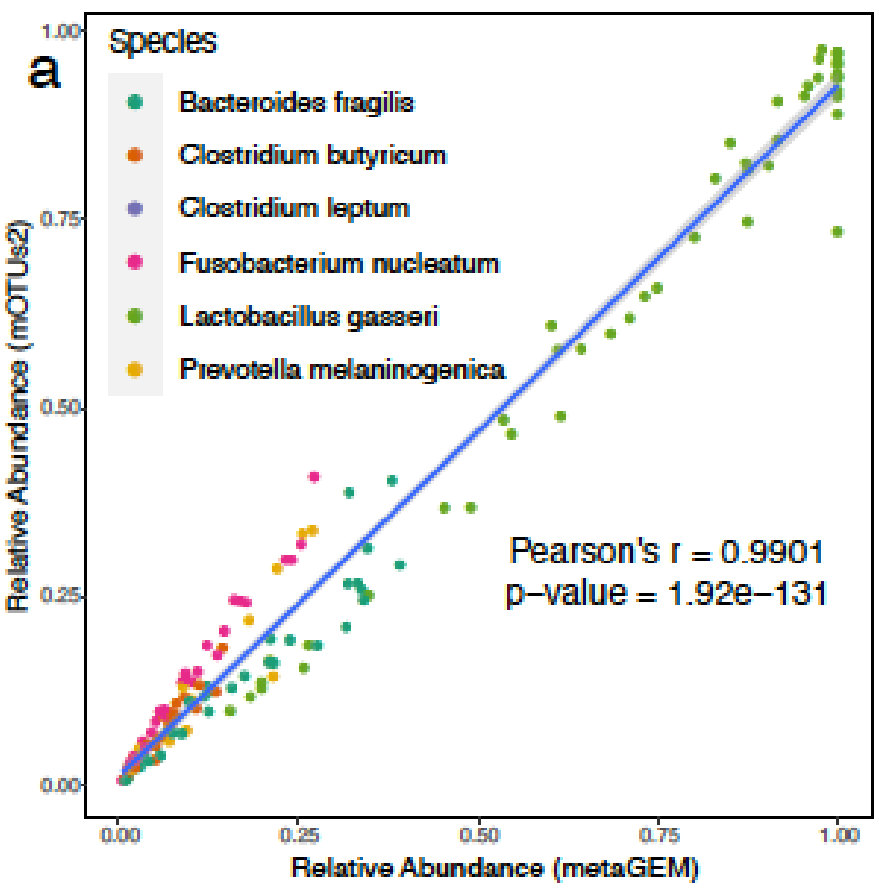
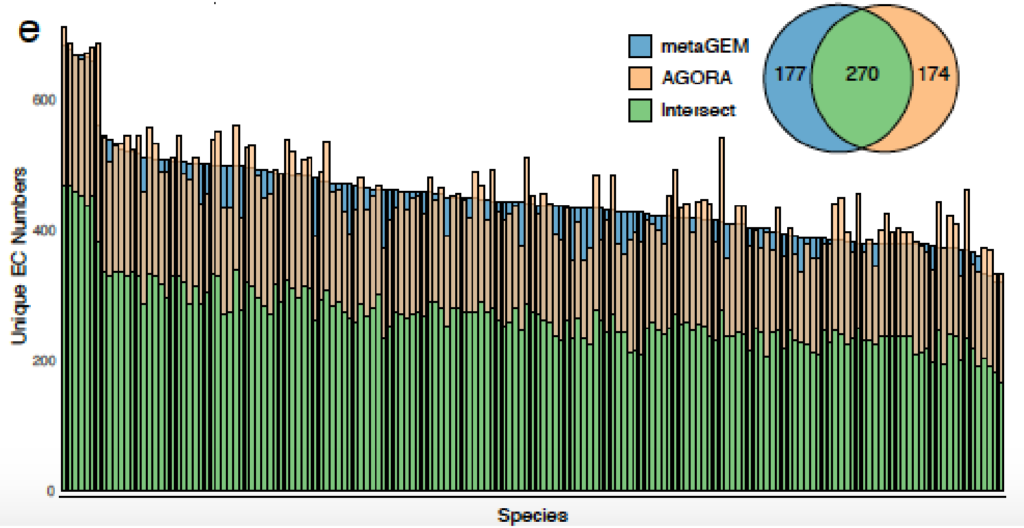
用MetaGEM基于宏基因组短读序列构建MAGs,分为HQ(高质量的),MQ(中等质量的),并以此进行代谢重建,总共获得14087个GEMs,然后将它们与高度精选的基于参考基因组的BIGG模型、AGORA、EMBL和KBase模型进行了比较。
利用基于定位的方法(方法)生成的丰度估计值与基于标记基因的丰度估计值完全相关;
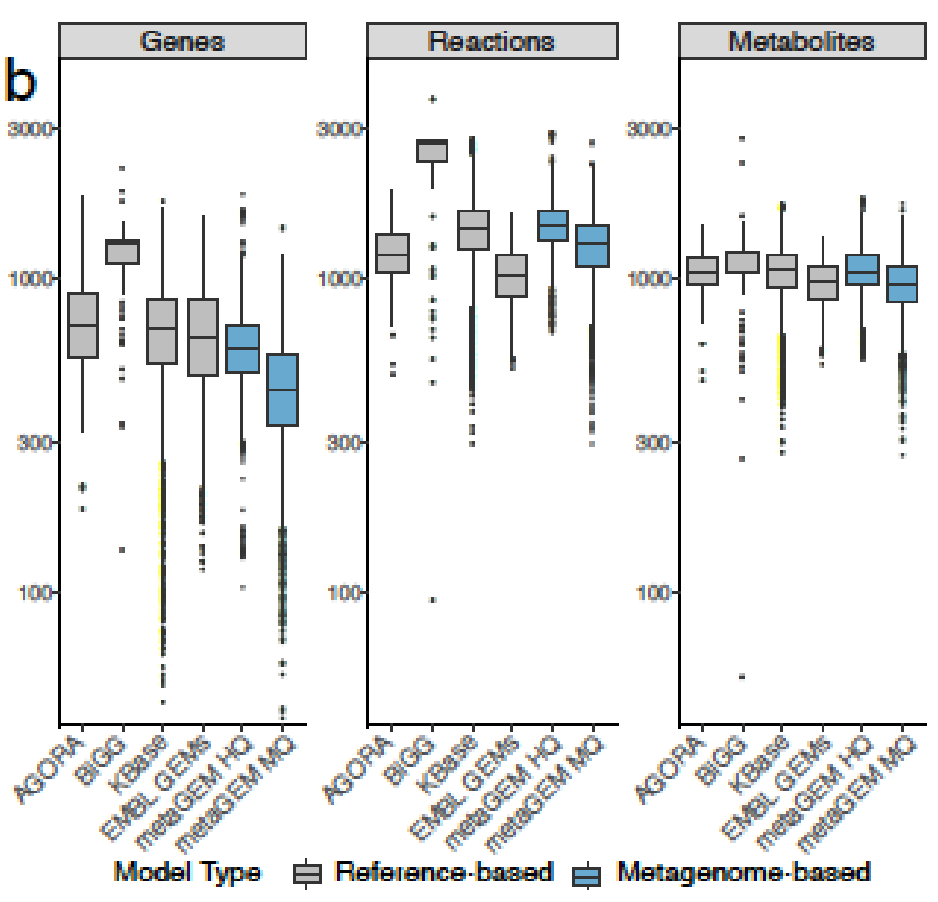
MetaGEM和其他模型都具有类似数量的反应和代谢物,但基因数量相比较少;
通过计算模型之间成对的代谢之间的距离,发现MetaGEM具有相似的酶多样性分布;
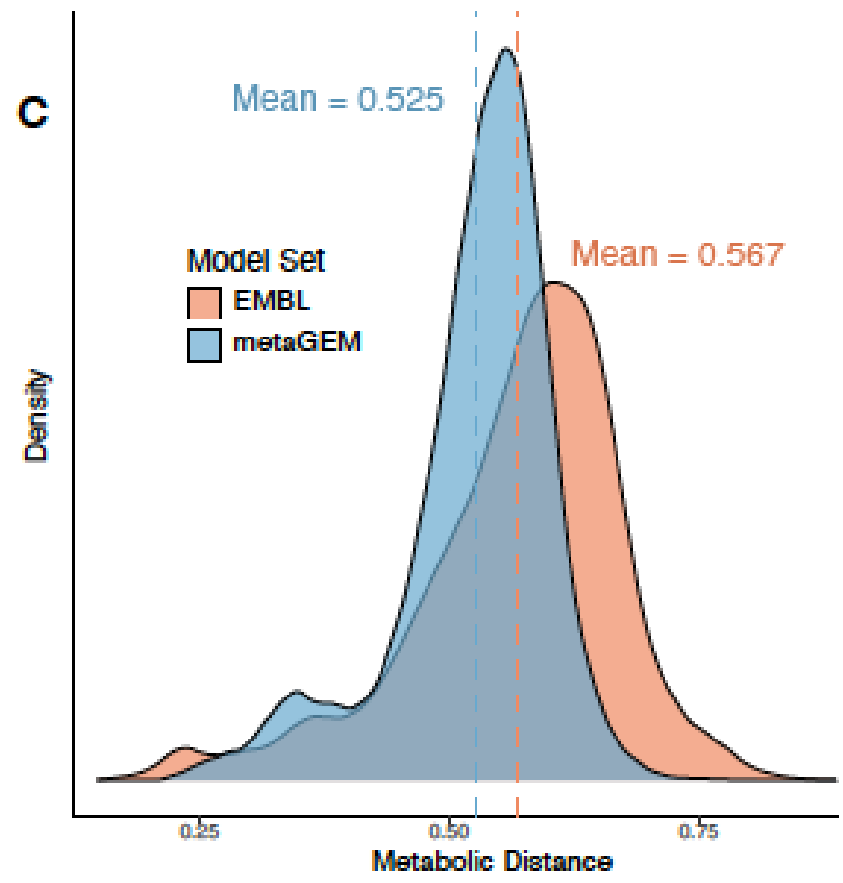
可以捕捉到种水平物种间的显著的代谢差异。高达60%的代谢多样性存在于物种泛基因组中,metaGEM模型捕获的物种内代谢变异程度显著
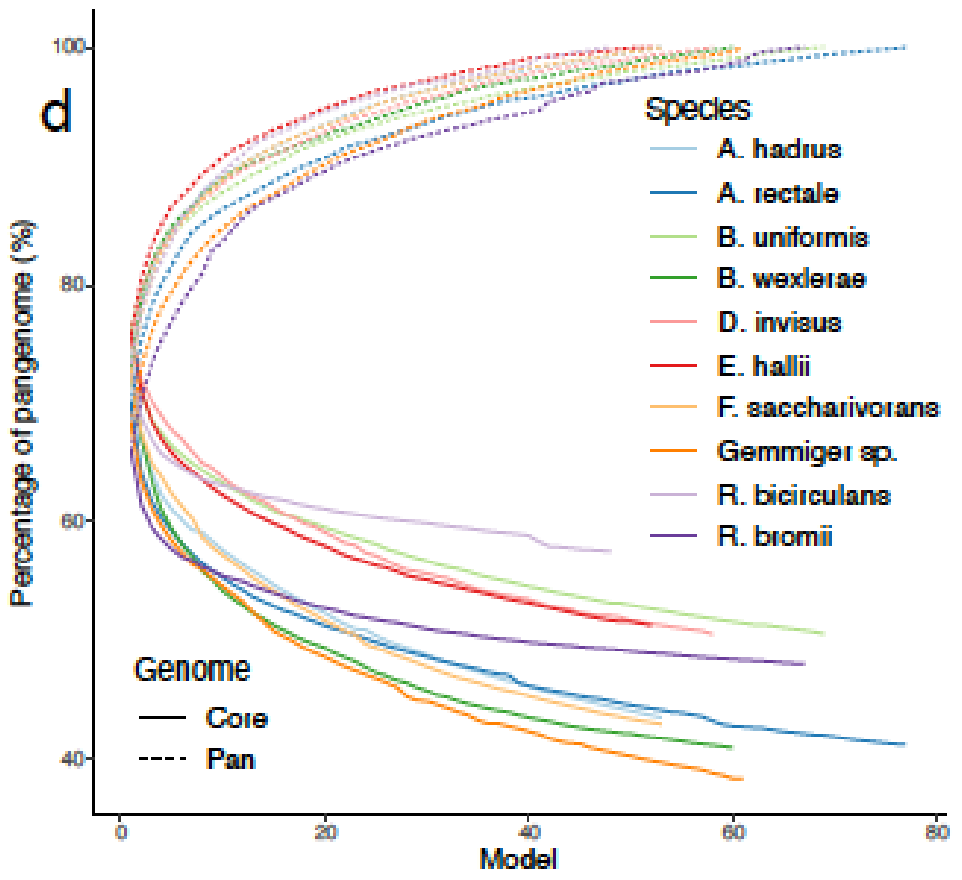
与基于参考基因组的肠道物种代谢模型AGORA比较,发现基于参考的模型引入的代谢反应不一定存在于每个宏基因组环境中,而MetaGEM模型是完全基于实际的宏基因组在特定环境下重建的代谢模型。
AGORA和MetaGEM模型的EC数的交集在48.9%到69%之间,其中53.9%的情况下MetaGEM模型比相应的AGORA模型包含更多的EC数。
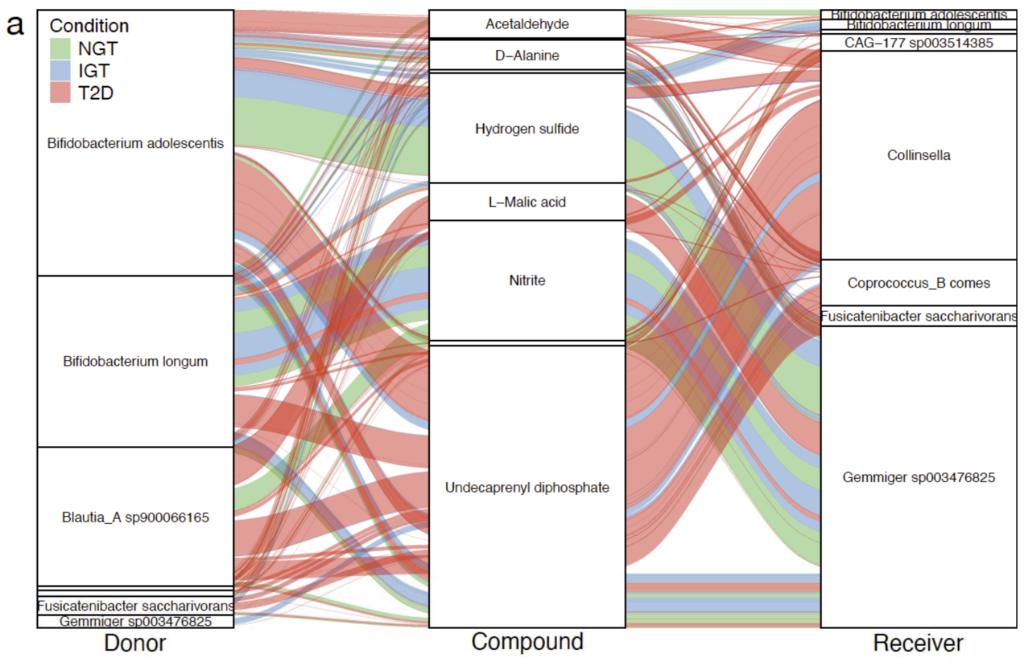
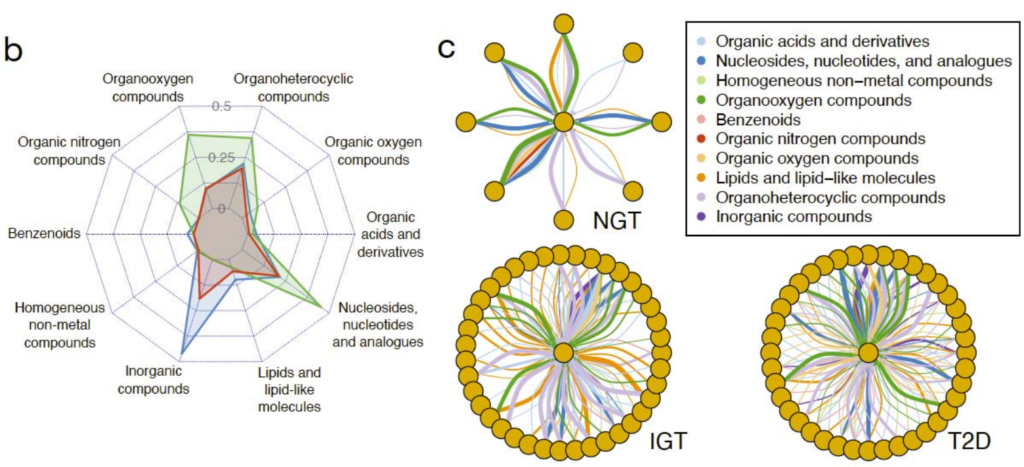
研究健康和代谢受损的2型糖尿病患者肠道微生物群落中潜在的微生物代谢相互作用。
使用metaGEMs通过137个宏基因组数据重建了4127个个性化的GEMs。
根据疾病状况分类,即正常糖耐量(NGT,n=42)、糖耐量受损(IGT,n=42)、 2型糖尿病(T2D,n=53),然后应用Smetana软件模拟微生物群落中的物种间依赖关系,Smetana为每个群落输出一个分数表,对应于在给定条件下为支持群落成员的成长而应发生的交叉喂养相互作用强度的度量,即物种A生长的可能性取决于物种B的代谢物X。
不同的2型糖尿病疾病组(NGT、IGT、T2D)相对应的肠道代谢基因组产生具有不同代谢结构的群落。
MetaGEM具有完善的流程,搭载的工具也是生物信息分析中常用的处理工具,下载很方便,用conda就能完成。无需参考基因组,这也意味着不需要下载动辄几十Gb的文件。使用Snakemake做流程的自动化管理,运行命令简单,也可以分步骤运行。
总体而言,MetaGEM可以直接从宏基因组数据中研究复杂微生物群落中特定样本(sample-specific)的新陈代谢。
【附录】
关于文中MetaGEM流程搭建所应用到的宏基因组分析软件,这其中也有我们常用的软件,比如fastp、MEGAHIT、bwa、SAMtools、metaWRAP,它们在处理数据时非常的方便也易于上手。
参考文献:
Zorrilla F, Patil K R, Zelezniak A. metaGEM: reconstruction of genome scale metabolic models directly from metagenomes[J]. bioRxiv, 2021: 2020.12. 31.424982.
相关阅读:
谷禾健康
介绍三种菌:
大肠埃希氏菌、血链球菌、李斯特菌。
Escherichia coli
大肠杆菌是短杆菌,两端呈钝圆形,属革兰氏阴性菌,于 1885 年首次被发现。

大肠杆菌是条件致病菌,在一定条件下可以引起多种疾病,如腹泻,肠炎,尿路感染,呼吸道感染、菌血症和其他临床感染(如新生儿脑膜炎)。
生化代谢活跃
大肠杆菌的生化代谢非常活跃。大肠杆菌可以发酵葡萄糖产酸、产气,个别菌株不产气,大肠杆菌还能发酵多种碳水化合物,也可以利用多种有机酸盐。
大肠杆菌具有三种硝酸盐还原酶和三种一氧化氮还原酶。因此,大肠杆菌菌株能够将不可发酵的营养物/硝酸盐转化为可发酵的硝酸盐。
传染性
致病性的大肠杆菌具有高度的传染性,会严重危害健康。大肠杆菌的肠道传染具有比较广泛的特性,而食品在生产、包装及运输过程中极易感染此菌,进而引发传染性疾病。
大肠杆菌病的主要传染源是因为在胃肠道感染患者的粪便中有大量大肠杆菌病原菌排至体外构成的。大肠杆菌在人之间的传播途径多是通过粪—口这一传播途径,在一定的条件下可引起大肠杆菌病散发或流行。
致病型、症状及毒力因子

Allocati N, et al., Int J Environ Res Public Health. 2013
致病机制
克罗恩病中,粘附侵袭性大肠杆菌对宿主细胞的侵袭作用(下图)。回肠粘膜的异常定植是由粘附侵袭性大肠杆菌与肠上皮细胞相互作用引起的。
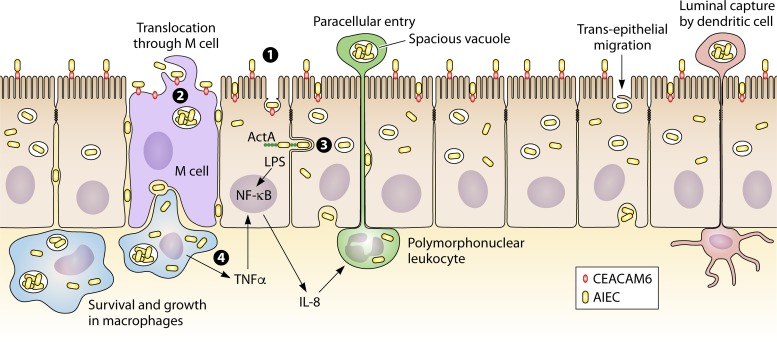
Mirsepasi-Lauridsen HC, et al., Clin Microbiol Rev. 2019
溃疡性结肠炎中,弥散粘附性大肠杆菌感染(下图)。弥散粘附性大肠杆菌通过细菌识别衰变/加速因子(DAF),癌胚抗原相关细胞黏附分子CEACAM1或CEACAM6(通过Afa / Dr CEA粘附素)来启动其与完全分化的上皮细胞的相互作用。
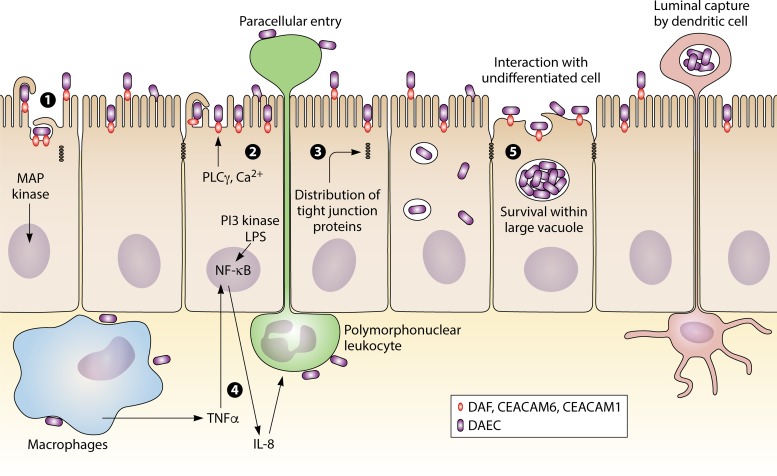
Mirsepasi-Lauridsen HC, et al., Clin Microbiol Rev. 2019
预 防
益生菌可能是预防几种大肠杆菌感染的方法。益生菌是可行且安全的微生物,主要是乳杆菌属、双歧杆菌属等,能够在肠道中定植,与致病菌竞争。
短链脂肪酸的产生速率取决于结肠中微生物群的种类和数量。短链脂肪酸具有抗炎作用,并有助于抑制肠内大肠杆菌的生长。饮食中的碳水化合物,淀粉和纤维是发酵的底物,可产生短链脂肪酸。
高脂/高糖饮食会导致微生物失调,黏液层厚度减少,通透性增加,增加对致病性大肠杆菌定植的敏感性。
参考文献:
Allocati N, Masulli M, Alexeyev MF, Di Ilio C. Escherichia coli in Europe: an overview. Int J Environ Res Public Health. 2013;10(12):6235-6254. Published 2013 Nov 25. doi:10.3390/ijerph10126235
殷泽禄, 万虎. 大肠杆菌的研究综述[J]. 甘肃畜牧兽医, 2019, 049(005):33-35.
Mirsepasi-Lauridsen HC, Vallance BA, Krogfelt KA, Petersen AM. Escherichia coli Pathobionts Associated with Inflammatory Bowel Disease. Clin Microbiol Rev. 2019;32(2):e00060-18. Published 2019 Jan 30. doi:10.1128/CMR.00060-18
Streptococcus sanguinis
血链球菌属革兰氏阳性,无孢子形成的兼性厌氧菌。像其他链球菌一样,血红链球菌的细胞分裂沿单个轴发生,从而形成链球或成对链球菌。
血红链球菌是一种共生细菌,广泛分布在口腔中,主要是牙齿表面,口腔粘膜的表面和人唾液。
血链球菌在不同的牙齿位置,血红链球菌的生物量可能存在显着差异。它以较高的比例存在于牙齿的下门齿/犬齿位置,但以较低的比例存在于牙齿的上门齿。
生物膜的形成对细菌适应环境和侵袭宿主都起到了重要作用。血链球菌是口腔生物膜发育的开拓者和关键角色。
机制
血链球菌最初的附着是由它的毛和粘附素促成的,葡聚糖和eDNA的产生促进血链球菌生物膜的成熟。流行病学研究表明,血链球菌可能抑制龋齿的产生。体外研究表明血链球菌和变形链球菌之间存在竞争,变形链球菌是最常见的致龋物种。
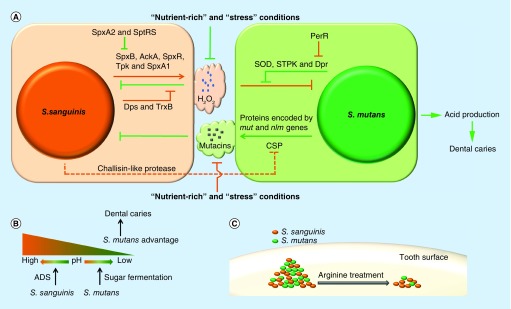
Zhu B,et al., Future Microbiol. 2018
16S rRNA测序结果表明,血链球菌可能与牙周健康有关。与患病的龈下微生物组相比,健康人的血链球菌的丰度显著增加。
然而,体外研究表明,血链球菌也可能促进后续与牙周炎相关的病原体附着。
血链球菌与牙周炎相关病原体的相互作用
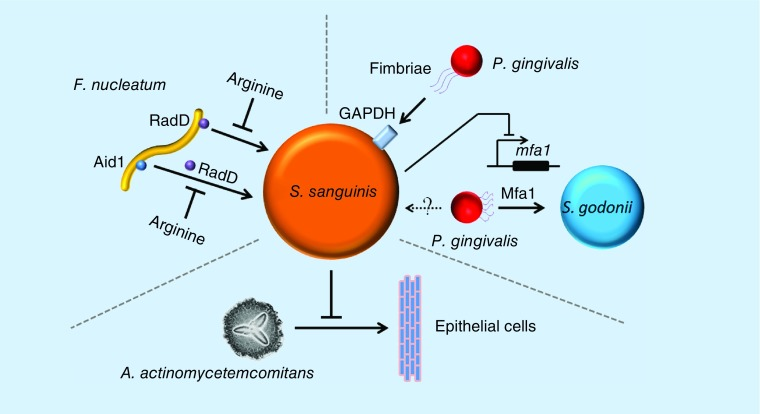
Zhu B,et al., Future Microbiol. 2018
除了在口腔中作为主要定居者的作用外,血链球菌还被广泛认为是感染性心内膜炎,心脏瓣膜或心内膜感染的病因。在一般条件下血链球菌定植于牙菌斑中,对人体无害,但当其通过口腔内的微小创口进入血液循环引起菌血症,就有可能引发感染性心内膜炎。
参考文献:
Haffajee AD, Teles RP, Patel MR, Song X, Yaskell T, Socransky SS, Factors affecting human supragingival biofilm composition. II. Tooth position. J Periodontal Res. 2009 Aug; 44(4):520-8.
Zhu B, Macleod LC, Kitten T, Xu P. Streptococcus sanguinis biofilm formation & interaction with oral pathogens. Future Microbiol. 2018 Jun 1;13(8):915-932. doi: 10.2217/fmb-2018-0043. Epub 2018 Jun 8.
Listeria monocytogenes
单核细胞增生杆菌(单核细胞增生李斯特菌)是革兰氏阳性,兼性厌氧,无芽孢的细胞内杆状细菌,在血琼脂上生长时为过氧化氢酶阳性和β-溶血性。
注:李斯特菌属有六个种:单核增生李斯特菌,英诺克李斯特菌,绵羊(伊氏)李斯特菌,威尔氏李斯特菌,西式李斯特菌和格氏李斯特菌,只有单核细胞增生李斯特氏菌对人具有致病性。
该菌生命力顽强,能适应各种环境 (例如低温、高 pH 值、高盐等),在冰箱的冷藏温度下仍可生长,这也是它不同于其他食源性致病菌的重要特征。
单核细胞增生李斯特菌能发酵多种糖类,产酸不产气。该菌产生生物膜的能力增强了其在恶劣环境中生存的能力。它还在较低温度下利用鞭毛。这种机制使细菌能够在感染早期自我驱动并附着在肠上皮细胞上,但最终细菌暴露在更高温度下的时间越长,鞭毛就会消失。
传播途径
该菌已被列为20世纪90年代四大食源性疾病致病菌之一。传播是通过粪-口途径发生的,最常见的是食物,与单核细胞增生李斯特菌相关感染率最高的食物包括:
· 生豆芽
· 未经巴氏消毒的乳制品
· 软奶酪
· 冷熟食肉
· 冷热狗
· 预制或贮存的沙拉
此外,食品加工、储存设备(设备表面,管道,生产环境和冰箱) ,自然环境(土壤,污水)和动物宿主(哺乳动物,非哺乳动物)细胞中均有分离获得该菌的报道 。
感染后症状
感染后3天~70天出现症状(潜伏期长),平均潜伏时间为3周,一旦发生感染,主要临床表现为发热、上呼吸道感染、腹泻、呕吐等。
该细菌具有穿过肠屏障,胎盘和血脑屏障的能力,会引起羊膜炎,败血症,孕妇自然流产,肺炎、急性胃肠炎、肺炎、婴儿败血性肉芽肿和脑膜炎等。
其中感染风险最高的人群包括孕妇,婴儿,免疫功能低下的人和老年人(65岁及以上)。孕妇感染该菌后可能将其传给未出生的胎儿。
发病率
李斯特菌病与高发病率和高死亡率相关。
根据疾病控制中心(CDC)的数据,每年约有1600人患有李斯特菌病,其中约260人死于该病。确诊的单核细胞增多性乳杆菌感染的死亡率约为15%,但根据患者状况,具有更多合并症的患者死亡率增加。
早期识别是增加生存机会的关键。
致病机制
该菌能够借助肠道内皮细胞自身的内吞作用通过肠道黏膜进入机体。在它入侵宿主细胞的过程中,涉及众多蛋白分子,包括毒力因子、黏附分子等,构成了一系列复杂的过程。
主要毒力因子:
内化素(InlA和InlB):
用于宿主细胞附着的细菌表面蛋白
李斯特菌溶血素O(LLO):
帮助细菌逃离宿主细胞液泡
肌动蛋白聚合(ActA):
帮助细菌在细胞内和细胞间移动
磷脂酰肌醇特异性磷脂酶C(PI-PLC):
帮助细菌逃脱宿主细胞液泡并引起细胞膜破裂
单核细胞增生李斯特氏菌具有细胞表面半乳糖残基,脂蛋白酸和内化素,它们主要通过宿主蛋白钙黏着蛋白与胃肠道上皮细胞结合,从而进入细胞。侵入宿主细胞后,细菌倾向于在宿主中启动细胞介导的免疫反应。
吞噬的单核细胞增生李斯特菌可以通过“李斯特菌溶血素O”(LLO)的成孔细胞毒性蛋白和其他非成孔的磷脂酶蛋白裂解内化液泡。一旦没有液泡,细菌就能通过肌动蛋白聚合作用穿过细胞,从而破坏正常的细胞过程。
预防
随着饮食习惯逐渐变化,食用生冷及半熟食的人群增多,致使李斯特菌感染率增高。
高温烹饪食物可杀灭李斯特菌。
另外有研究表明,短期高脂饮食会增加宿主对单核增生李斯特菌的易感性。
以下是一些预防李斯特菌感染的小建议:
食品洗涤和处理:
1在进食,切割或烹饪之前,用自来水彻底冲洗生的农产品。即使产品脱落,也必须先清洗
2用干净的工具擦洗瓜类等坚硬的食物
保持厨房及其周围环境清洁安全:
1处理和准备未煮熟的食物后,请洗手,清洁刀具和切菜板
2冰箱中的所有溢出物需要及时清理,尤其是香肠汁,生肉,生禽肉等
彻底煮熟肉类和家禽:
1彻底烹饪动物生食,例如牛肉,猪肉或家禽,使其内部温度达到安全
2尽快食用预煮或即食食品。请勿将食物放在冰箱放到过期
3尽量少吃剩菜,尤其不要超过3天
选购安全食品:
1选购时查看食品包装是否完整以及是否过期
2不要喝未经消毒的牛奶及其他未消毒乳制品
针对李斯特菌病高风险人群的饮食建议(孕妇,免疫低下者,老年人):
1吃热狗,熟食肉等要注意吃热食(74℃以上),不要吃冷菜,冷盘,冷的热狗等。
2避免使液体从香肠和食品包装中流到其他食品、器皿和食物制备表面上,处理冷熟食等要洗手。
3不要直接食用超市柜台或商店的冷藏室里冷藏的肉酱或肉类产品。不需要冷藏的食品都可以安全食用,例如罐头或黄油稳定的馅饼和肉类。
·芝士
– 吃奶酪或其他乳制品要注意看是否标有用巴氏杀菌牛奶制成的标签。
·鱼
– 请勿食用冷冻的熏制海鲜,除非它是在煮熟的食物中,例如砂锅,或者是罐装或耐久的产品
– 金枪鱼罐头,鲑鱼和其他腌制,货架稳定的海鲜可以安全食用
·甜瓜
– 处理甜瓜前后,请用温水至少洗净双手20秒钟
– 在切割之前,用干净的刷子在流水下擦洗瓜子的表面,并用干净的布或纸巾拍干。每次使用后请确保刷子已消毒
– 立即食用切好的瓜或迅速冷藏。将切好的瓜冷藏但不能超过7天
– 丢弃在室温下超过4小时的切开的瓜
这个菌看起来十分可怕,那如果粪便中检出该菌,意味着什么?
健康人的粪便也可能检测到。
有研究表明,在900名健康无症状捐赠者的独立队列中,10%的人类粪便样本中也检测到该菌。
肠道微生物群组成影响该菌的粪便携带。
无症状粪便携带对该菌“毒力基因”进行的是纯化选择,而不是疾病。
单增李斯特菌进入人体是否得病与菌量和宿主的年龄、免疫状态等有关,健康成人可能无症状或出现轻微类似流感症状,但易感人群可能会引发前面所述多种疾病,如出现症状需引起重视并立即就医。
参考文献:
Rogalla D, Bomar PA. Listeria Monocytogenes. 2020 Jul 10. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan–. PMID: 30521259.
Rodríguez-Auad JP. Panorama de la infección por Listeria monocytogenes [Overview of Listeria monocytogenes infection]. Rev Chilena Infectol. 2018;35(6):649-657. Spanish.
Las Heras V, Clooney AG, Ryan FJ, Cabrera-Rubio R, Casey PG, Hueston CM, Pinheiro J, Rudkin JK, Melgar S, Cotter PD, Hill C, Gahan CGM. Short-term consumption of a high-fat diet increases host susceptibility to Listeria monocytogenes infection. Microbiome. 2019 Jan 18;7(1):7.
周继福,王娉等人,单增李斯特菌进化家系的研究进展[J]. 食品科学,2021. DOI:10.7506/spkx1002-6630-20201009-053
焦颖, 张巍. 李斯特菌生物学特征与临床相关性[J]. 中国感染与化疗杂志, 2015.

谷禾健康
迄今为止,已经有了许多对呼吸道微生物组通过16S rRNA高通量测序的研究。这其中所有基于扩增子的研究的共同之处就是PCR的应用:
一是扩增待测序的目标标记基因,
二是为多样本的混合测序添加必要的索引序列。
这些步骤可以通过一步PCR或两步PCR完成,但没有研究说明两步PCR方案相关的实验室处理步骤是否会使样品比一步PCR方案更容易受到来自实验室的细菌DNA污染的影响。
本文
试图确定对16S rRNA V3V4与V4基因区域的一步或两步PCR的建库方案对上呼吸道和下呼吸道微生物组的影响
对收集的样本进行了三个设置下的lllumina MiSeq测序
设置1(两步PCR,V3V4区域)
设置2(两步PCR,V4区域)
设置3(一步PCR,V4区域)
分别对这三个设置产生的测序数据进行分析
结论
PCR步骤数量的差异会影响对呼吸道微生物群落的物种组成分析,且对上呼吸道(高细菌载量)的影响小于下呼吸道(低细菌载量),这表明PCR设置的偏差与样本生物量有关。
通过三个实验,即对模拟群落样品HM-783D、NCS样品、呼吸道样品采用三种PCR方案进行建库后的测序结果分析,研究这三种建库方案对其菌群描述的影响。
模拟群落样品HM-783D,来自20种不同细菌物种(17个属)的基因组DNA。
阴性对照样本NCS
呼吸道样品,从Bergen COPD微生物组研究中选择了23名研究对象,其中9名健康,4名患哮喘,10名患COPD(慢阻肺)。上呼吸道样本以漱口水(OW)为代表,下呼吸道样本以标本刷(PSB)和支气管肺泡灌洗液(PBAL)为代表。
三种PCR方案:
细菌DNA提取后通过三种不同的建库设置进行MiSeq测序,分别为
Setup1(两步PCR,V3V4区域);
Setup2(两步PCR,V4区域);
Setup3(一步PCR,V4区域)
下图完整的展示了三个PCR设置下的使用呼吸道样本的生物信息学过滤步骤:
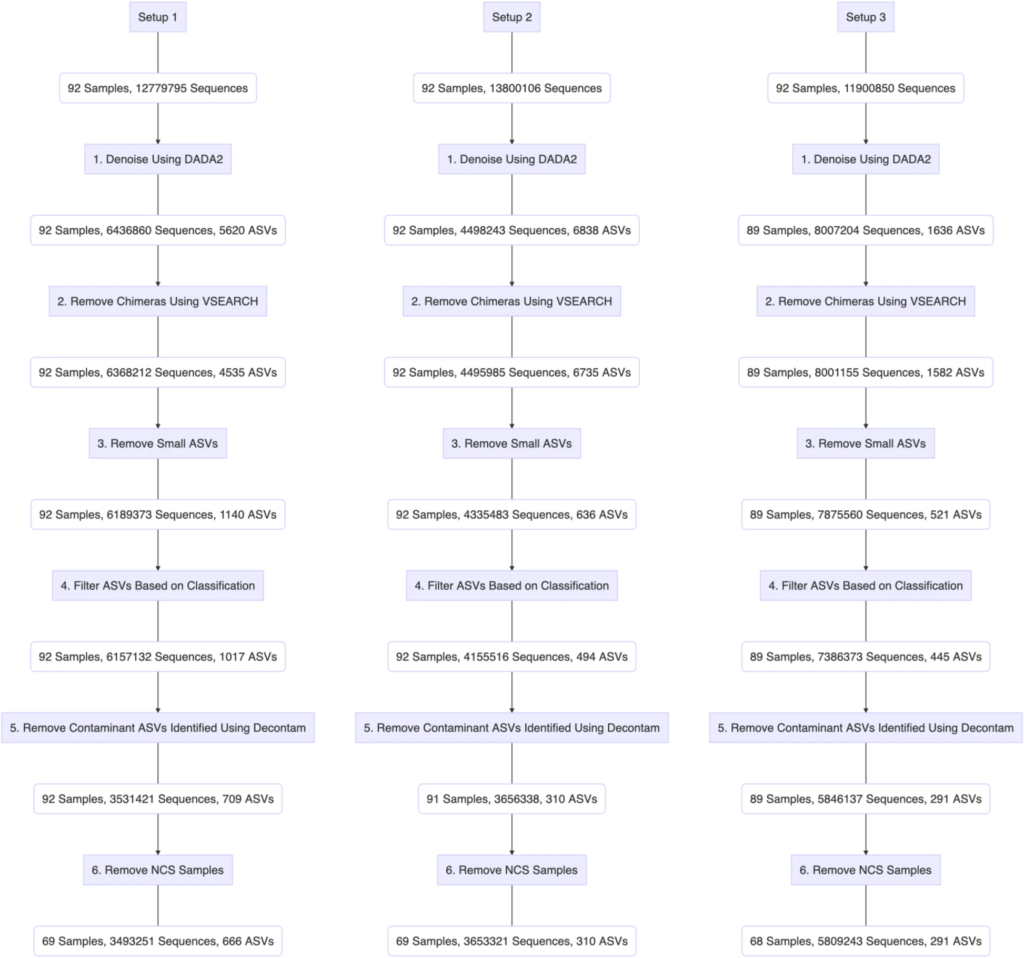
最终:
设置1:得到了666个ASVs
设置2:得到了310个ASVs
设置3:得到了291个ASVs
1. 对模拟群落样品HM-783D的分析
在设置1中进行了四次测序,设置2和设置3分别进行了一次。
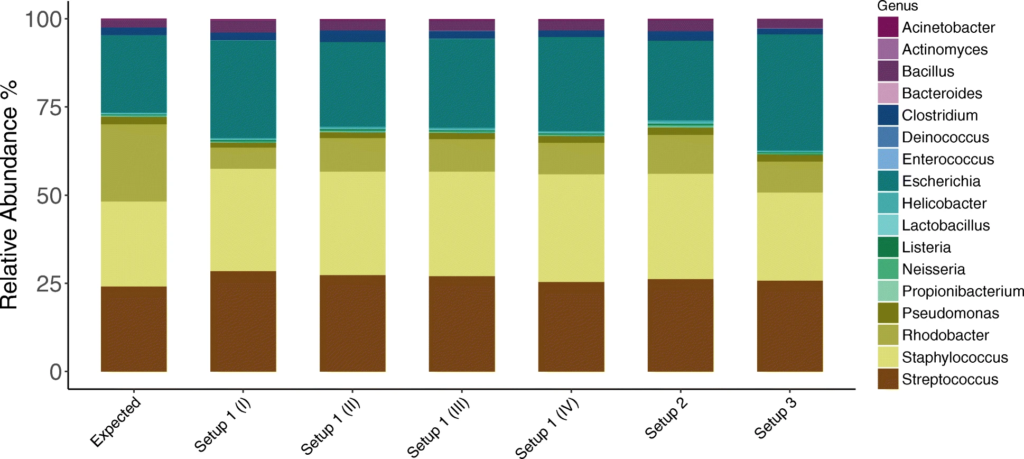
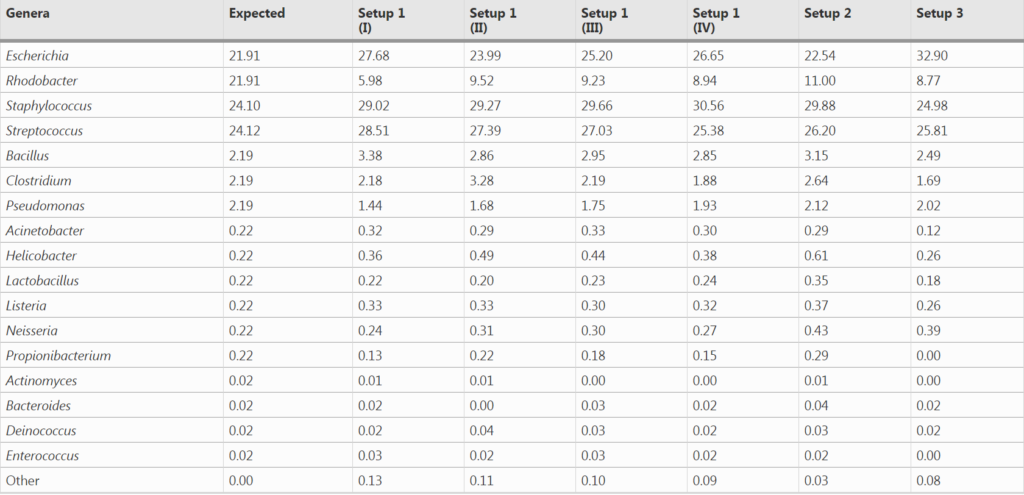
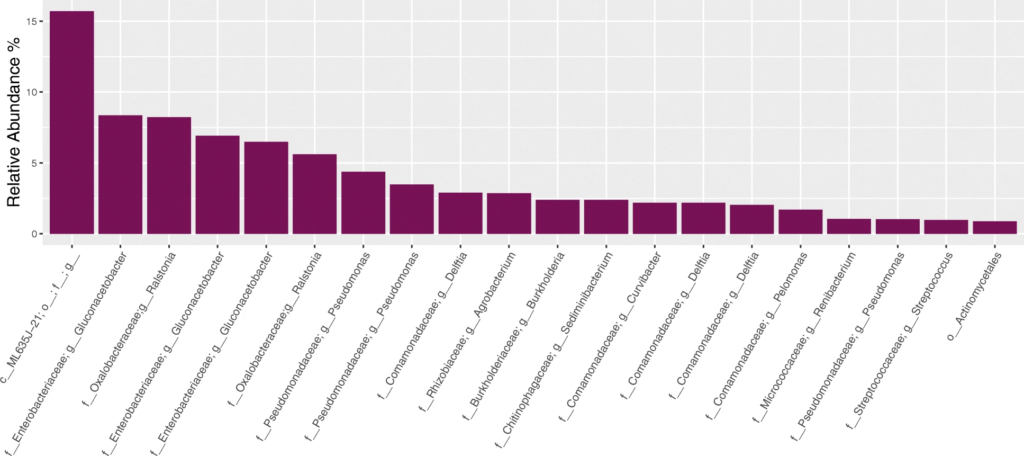
与预期丰度(Expected)相比,柱状图中观察到三种PCR设置下的各菌属的相对丰度与预期丰度相差不大,表中数据显示,三种PCR设置都在恢复高丰度物种方面具有最高的效率,但设置3在回收低丰度物种时的效率最低。
2. 对阴性对照样品的分析
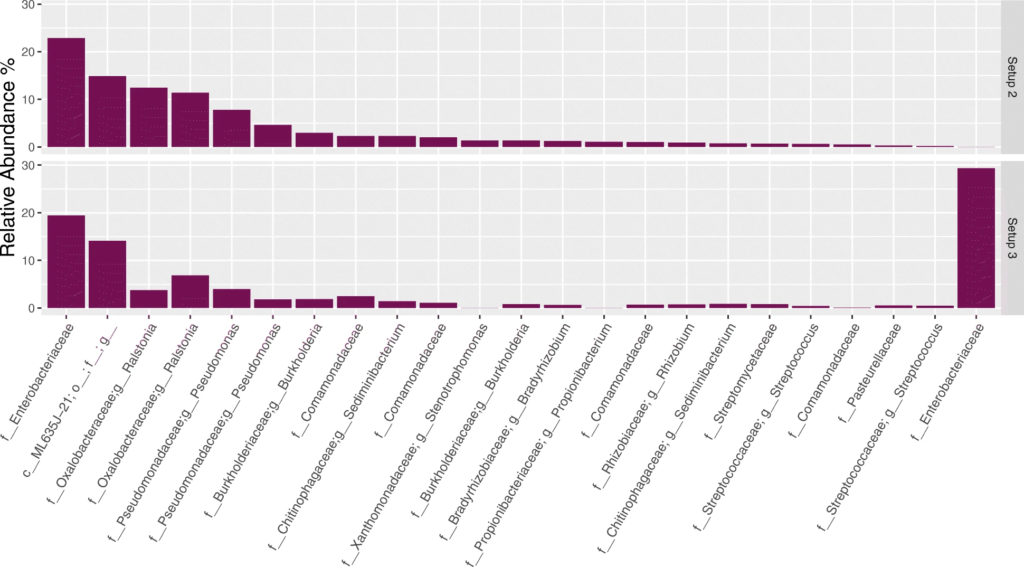
从上至下分别为设置123测序后,在NCS样品中观察到的20种最丰富的ASV。通过R包Decontam去除污染物,在设置23之间差异最大的是属于肠杆菌科的ASV,与后续的对水样品进行设置23下的测序分析结果相比较,发现大肠杆菌ASV就是在建库步骤中使用设置3的试剂时引入的污染物。
3. 对采集的呼吸道样品的分析
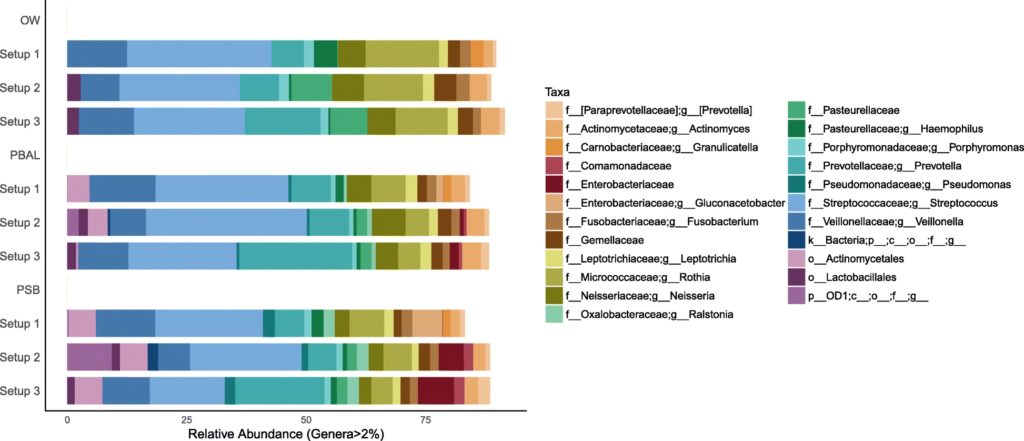
在去除污染物前后,代表为呼吸道菌群的链球菌,普雷伏氏菌,Veillonella和Rothia属的相对丰度变化不大,而去除污染物后,预测作为污染物代表的数量较少的物种被滤出。基于主坐标分析,发现高细菌载量的OW样品聚集在一起,低细菌载量PBAL,PSB一句设置23分离开。
去除污染物之前的三种类型样品的三种PCR设置下的物种分类
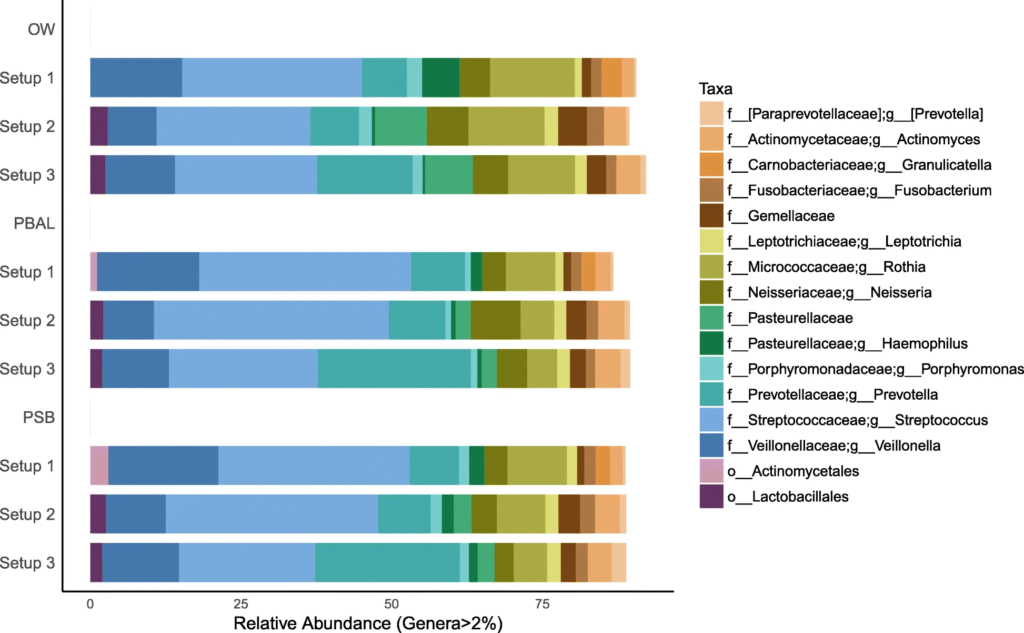
去除污染物后的
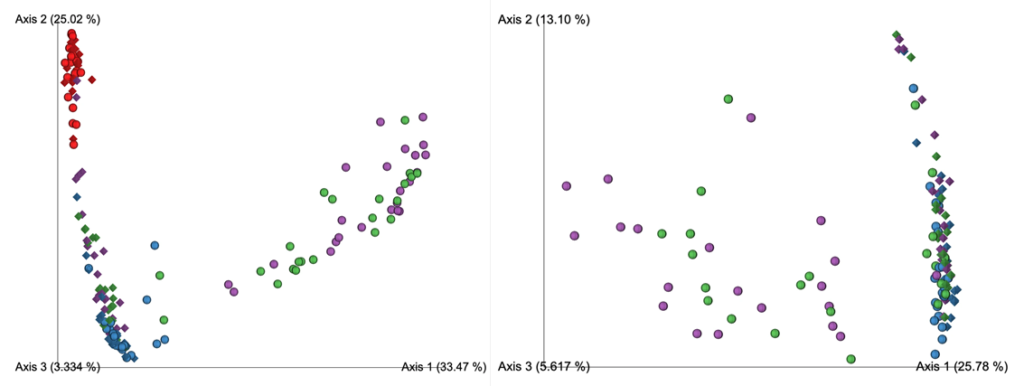
从左至右分别为去除污染物前后的未加权UniFrac距离的主坐标分析
OW:蓝色; PBAL:绿色;PSB:紫色;NCS:红色。
设置2(球形),3(菱形)
文章作者给出的结论是文库制备和测序方法的选择会对呼吸道微生物组的分析产生影响,且对上呼吸道的影响小于下呼吸道。靶向扩增子区域的差异(16S rRNA基因V3 V4与V4)并未表现出对细菌群落描述的重大影响。对于整篇研究存在的主要的局限性在于仅研究了DNA提取后的PCR步骤,污染或影响也可能来自于更前期的处理。
编者按
在使用测序技术进行的微生物研究中,测序偏差和污染物是一直存在的问题,也因此诞生了许多工具和计算方法用于尽可能的消除或降低这方面的影响。这篇研究也提醒了我们,在呼吸道微生物组的研究中,要注意上呼吸道与下呼吸道的菌群差异或相似可能不仅仅来源于样本自身,还可能掺杂着PCR方法选择上的影响。
参考文献:
Drengenes C, Eagan TML, Haaland I, Wiker HG, Nielsen R. Exploring protocol bias in airway microbiome studies: one versus two PCR steps and 16S rRNA gene region V3 V4 versus V4. BMC Genomics. 2021 Jan 4;22(1):3.
相关阅读:

谷禾健康
大部分人都遭受过头皮屑的困扰。头皮屑虽然看起来不是严重的疾病,但有时候足以令人头疼和尴尬,影响整体生活质量。

头皮屑是一种非常常见的皮肤病,无论年龄,性别等,任何人都可能会有。
头皮屑通常看起来像油腻的皮肤片,如果飘落在深色衣服上更加明显。在头皮上,它也可能表现为皮肤的鳞片状斑点,有时伴有发红、发痒或发炎。
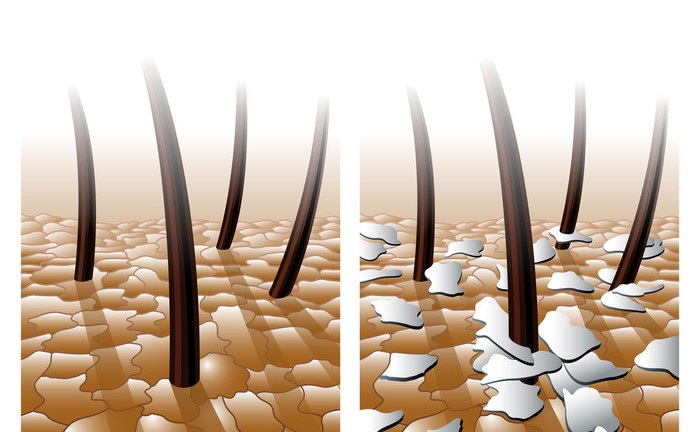
头皮表面为微生物提供了独特的微环境,主要来自宿主的生理条件,包括皮脂含量,水分,pH值等。
头皮屑与多种可能的原因有关,包括卫生差,压力大和饮食不当,当然头皮屑的产生也离不开微生物群。
微生物包括真菌和细菌。
真菌
少量脱落实际上是正常的,因为皮肤细胞会死亡,以便形成新的皮肤。然而,在某些情况下,由于一种或多种潜在的问题可能会使头皮真菌的过量生长,导致剥落异常大量。
头皮表面发现的最多的真菌是马拉色菌(malassezia)。这种真菌生活在头皮上,以皮肤油脂为食。
马拉色菌里有限制性马拉色菌、球形马拉色菌等,不同种类的马拉色菌的致病性不同,并适应人体皮肤的不同部位,这主要归因于宿主的生理条件(皮脂,免疫反应,汗液等),环境(温度,湿度,紫外线暴露)以及其他微生物的存在。
其中限制性马拉色菌与球形马拉色菌的比例较低与健康的头皮有关。
当球形马拉色菌利用脂肪酶代谢其消耗的油脂时,会产生一种副产物——油酸,该副产物渗透到头皮并引起头皮屑。
功能分析表明,真菌菌群主要通过与头皮屑头皮细胞-宿主粘附有关的途径富集。马拉色菌的粘附与头皮角质层屏障功能受损有关,可能进一步有助于头皮屑区马拉色菌的发育。
与健康头皮相比,头皮屑头皮中还发现了大量与一般功能和遗传信息处理相关的通路(硫中继系统、蛋白酶体通路、酵母细胞周期和减数分裂等),且与限制性马拉色菌呈显著正相关。
“酵母细胞周期”和“减数分裂”是参与真菌细胞周期和增殖的重要通路,这两通路在头皮屑头皮上的较高数量表明,它们在头皮屑头皮上的马拉色菌的增殖中起作用。
“蛋白酶体通路”与包膜的形成有关,包膜是真菌的主要致病决定因素。

细菌
头皮表面发现的最丰富的两个细菌属是Cutibacterium菌(健康头皮)和葡萄球菌(头皮屑头皮)。
研究表明,与健康人群相比,头皮屑患者的限制性马拉色菌和葡萄球菌的丰度较高(p <0.05)。
不同的葡萄球菌丰度变化也不同。
与健康人群相比,头皮屑患者头皮上表皮葡萄球菌的丰度减少,而同时头状葡萄球菌的丰度显著增加。头状葡萄球菌丰度的增加足以驱动头皮屑损伤部位葡萄球菌绝对物种丰度的总体增加。
细菌菌群的功能分析显示出,健康头皮与以下代谢相关通路明显高于头皮屑头皮:
氨基酸:丙氨酸、天冬氨酸、谷氨酸、精氨酸、脯氨酸、赖氨酸、组氨酸、半胱氨酸、蛋氨酸代谢;
维生素:维生素B6,B7
其他辅助因子:卟啉,叶绿素,烟酸和烟酰胺代谢,泛醌和其他萜醌的生物合成。
这些头皮微生物群合成的维生素、生物素、氨基酸等被头皮角质形成细胞吸收,为头皮提供营养,在保持头发和头皮健康以及控制头皮屑方面起着有益的作用。
可能引起头皮屑的一些潜在潜在问题包括:
•皮肤状况-例如脂溢性皮炎,湿疹和牛皮癣,会增加头皮屑的风险。
•神经系统疾病-发现患有帕金森氏症等神经系统疾病的人更容易患头皮屑。
•洗护用品的刺激-购买的洗护品含有许多化学物质,可能会刺激头皮并导致发炎,发痒和脱落。
•不良的饮食习惯-摄入含锌和B族维生素的食物不足,可能会导致头皮屑。
•年龄-虽然说任何年龄的任何人都可能患有头皮屑,但往往在年轻人和中年人中更为普遍。
•气候-冬季的寒冷天气可能促进头皮中酵母样真菌的生长。
•激素-体内激素的变化可能会影响头皮的天然皮脂分泌。激素失调的人更容易有头皮屑。

为了想要尽快清除头皮屑,有人会选择含药的洗发水。但是,药用洗发水真的可以治愈头皮问题吗?
首先,含药的洗发香波只有在您持续使用时才有效-它们不能彻底治愈头皮屑。这就意味着,必须要每周使用几次这种洗发水才能看到效果,一旦停止使用,头皮屑很可能卷土重来。

这些洗发剂大多数都含有抗真菌成分,例如酮康唑。虽然可以有效控制头皮屑,但连续使用会刺激头皮,并使头发变得蓬松干燥。
其他药用去屑洗发水也可能含有煤焦油,煤焦油是通过延迟皮肤细胞再生的过程来控制头皮屑。但是,煤焦油是已知的致癌物质,因此最好避免含有这种成分的洗发水。
皮肤会吸收施加的任何物质,自然疗法是最佳选择。
盐
快速去除头皮屑且不使用任何有害化学物质的最安全方法之一是:用盐来去除片状头皮屑。
在洗澡之前,将盐轻轻按摩到干头皮上,使头发在漂洗时更容易去除。
椰子油
椰子油可以帮助改善水合作用,减少刺激感,并防止真菌在头皮上生长。恢复头皮的水分有助于止痒。

根据一项临床试验的结果,在117名1-13岁的儿童中,将纯净的椰子油涂在皮肤上可使特应性皮炎症状减少68.23%。当然也需要进行更多研究,以评估椰子油在治疗头皮屑中的作用。
椰子油的保湿和抗菌特性,使其成为抵抗干燥的绝佳自然选择。
茶树油
使用茶树油治疗头皮屑也是比较快速简便的。有研究人员认为茶树油中的化合物可以有效控制表皮葡萄球菌。

只需在头皮上按摩几滴茶树油,可减少瘙痒和多余油脂的产生。也可以在洗发水中混合几滴茶树油。
芦荟凝胶
芦荟是一种多汁植物,以其愈合特性而闻名。叶片中的凝胶含有几种生物活性化合物,例如氨基酸和抗氧化剂,可减少头皮屑。
2019年一项研究检查了23项涉及芦荟的临床试验。这些研究的结果表明,芦荟凝胶可以改善皮肤中的水分保留并促进伤口愈合。芦荟还可以减轻炎症,这可以帮助有头皮屑症状的人减轻症状,例如发痒。
2015年的研究表明,芦荟的抗菌性能可预防头皮屑。

饮 食
头皮屑通常与不良饮食有关,首先请开始调整饮食习惯。
Omega-3脂肪酸
Omega-3脂肪酸可以降低血压,增加“良好的” HDL胆固醇水平,并支持心脏和大脑健康。这种脂肪酸的缺乏会导致不良症状,例如头皮屑,指甲变脆和皮肤干燥。
锌
锌是支持人体免疫系统并促进细胞生长的矿物质。可以从动物蛋白,坚果等获取锌。

根据NIH研究表明,严重的锌缺乏与脱发、腹泻、皮肤损伤等有关。
2016年的一研究将缺锌列为脂溢性皮炎和头皮屑的潜在诱因。
减少糖和谷物
应该减少糖和谷物的摄入,因为已知这些会促进酵母或真菌的过度生长。
水果
水果和蔬菜含有许多必需的维生素,矿物质和抗氧化剂,可以帮助减轻炎症。

一项涉及4379人的观察性研究称,多吃水果的人患脂溢性皮炎的可能性较小。
研究结果还表明,典型的西方饮食可能会增加女性脂溢性皮炎的风险。
生物素
2018年的一项审查显示,生物素缺乏症可能导致几种皮肤疾病,包括脂溢性皮炎。生物素,也称为维生素B7,在支持健康的头发,指甲和皮肤方面发挥作用。富含生物素的食物包括:肝、蛋黄、坚果、三文鱼等。

晒太阳
还有一种非常简单的方法是:经常晒晒太阳。阳光可以防止真菌在头皮上繁衍。同时可以增加维生素D的含量,从而有助于改善皮肤状况。

解 压
此外,压力是头皮屑的最常见诱因之一。学习如何正确处理压力也很重要。适当培养一些兴趣爱好,定期放松解压,尽可能远离压力源。

其 他
最后,要保持良好的个人卫生习惯,避免过多抓挠,发痒最初是由头皮屑引起的刺激引起的,抓挠会增加刺激性并导致恶性循环。
避免使用过多的护发产品,防止头皮发炎。
摆脱头皮屑需要时间,精力和耐心,才能获得持久的效果。
Q & A
如何区分头皮屑和虱子?
如果白色颗粒在吹动或梳理头发时掉落,痒的程度相对较轻,则很可能是头皮屑。头虱在头皮部叮咬吸血,主要症状头皮部剧烈瘙痒,斑点粘在发干上,则可能是虱子或虱子空卵。
头皮屑可以治愈吗?
没有永久治愈的方法,但合理的方式可以控制住头皮屑。一些自然疗法包括茶树油、椰子油等。
头皮屑会导致脱发吗?
头皮屑本身不会直接导致脱发。然而,因为头皮屑抓挠发痒的头皮,久而久之可能会导致发丝脱落。
相关阅读:
主要参考文献:
Grimshaw SG, Smith AM, Arnold DS, Xu E, Hoptroff M, Murphy B. The diversity and abundance of fungi and bacteria on the healthy and dandruff affected human scalp. PLoS One. 2019;14(12):e0225796. Published 2019 Dec 18.
Saxena R, Mittal P, Clavaud C, et al. Comparison of Healthy and Dandruff Scalp Microbiome Reveals the Role of Commensals in Scalp Health. Front Cell Infect Microbiol. 2018;8:346. Published 2018 Oct 4.
National Health Service, Dandruff
Lorch JMP J.M.; Vanderwolf K.J.; Schmidt K.Z.; Verant M.L.; Weller T.J.; Blehert D.S. Malassezia vespertilionis sp. nov.: a new cold-tolerant species of yeast isolated from bats. Persoonia. 2018; 41:56–70. 10.3767/persoonia.2018.41.04
Symptom Find, Dandruff
Lin Qingbin,Panchamukhi Ananth,Li Pan et al. Malassezia and Staphylococcus dominate scalp microbiome for seborrheic dermatitis.[J] .Bioprocess Biosyst Eng, 2020
Zinc, Fact Sheet for Health Professionals
Borda, L. J., & Wikramanayake, T. C. (2016). Seborrheic dermatitis and dandruff: A comprehensive review.
Hekmatpou, D., et al. (2019). The effect of aloe vera clinical trials on prevention and healing of skin wound: A systematic review.
Kim, S., et al. (2017). Enhanced barrier functions and anti-inflammatory effect of cultured coconut extract on human skin.
Omega-3 fatty acids fact sheet for consumers. (2018).
Sanders, M. G. H., et al. (2019). Association between diet and seborrheic dermatitis: A cross-sectional study.
Varma, S. R., et al. (2019). In vitro anti-inflammatory and skin protective properties of virgin coconut oil.
谷禾健康
收样安排
春节前实验室的收样时间截止2月7日,客户的样本必须在2月5日前送到实验室会安排节前上机。春节期间2月8日至2月18日实验室无法收样。
报告交付
2月1到5日收到样本交付期限为节后2月19日;
2月19号起恢复正常上机和报告出具。

杭州谷禾信息技术有限公司
2021年1月14日
谷禾健康

现在经济飞速发展,随着生活条件改善,人们的寿命开始变长,对健康长寿的研究也逐渐开始增多。
然而寿命变长却不一定健康,越来越多人开始患上各种慢性疾病。
慢性疾病怎么来的?
首先从炎症开始。炎症其实是身体在与自身有害的物质(例如感染,毒素)作斗争来自愈的过程。当细胞要被破坏时,身体就会释放化学物质,从而触发免疫系统的反应。
当这种反应持续存在时,就会发生慢性炎症,身体处于持续的警觉状态。随着时间的流逝,慢性炎症可能会对组织和器官造成负面影响。于是各种疾病就开始了。
那慢性疾病为什么与肠道健康有关呢?
看过我们文章的朋友,大概已经开始有了这样的概念:许多疾病始于肠道。
因为免疫系统有很大一部分在肠道,具体来讲,这要涉及到肠道通透性的问题。
来自麻省总医院儿童医院腹腔研究和治疗中心主任Fasano博士和他的团队发现了zonulin蛋白(连蛋白),这为研究肠道通透性功能的新方法打开了大门,不仅因为它影响肠道,而且还影响了整个过程中炎症和自身免疫的作用。
除了基因组成和暴露于环境诱因外,还有三个引起慢性炎症性疾病的额外因素:
肠道通透性的不适当增加(可能受肠道菌群组成的影响);
负责耐受性免疫应答平衡的“超好战”免疫系统;
肠道菌群的组成及其对免疫系统的表观遗传影响宿主基因组的表达。
近十年来,人们开始越来越多关注到人类遗传学、肠道微生物组学和蛋白质组学,表明粘膜屏障功能的丧失,特别是胃肠道粘膜屏障功能的丧失,可能会严重影响抗原的运输,最终影响肠道微生物组和免疫系统之间密切的双向相互作用。
这种相互作用对宿主肠道免疫系统功能的形成有很大影响,并最终将遗传易感性转化为临床结果。这一观察导致了对慢性炎症性疾病流行的可能原因的重新审视,表明肠道通透性的关键致病作用。
临床前和临床研究表明,连蛋白家族是调节肠通透性的一组蛋白质,与多种慢性炎症性疾病有关,包括自身免疫性,感染性,代谢性和肿瘤性疾病。这些数据为多种慢性炎症性疾病提供了新的治疗靶点,其中连蛋白途径与它们的发病机理有关。
Fasano指出,根本没有足够的基因来解释众多慢性疾病,基因也不能解释疾病发作的时间。他说,要解决这些谜团,我们必须关注微生物组,因为“决定个人临床命运的是个体之间的相互作用和我们所生活的环境。”
除了微生物本身,肠粘膜的状况也起着重要作用。Fasano解释说:“尽管这种巨大的粘膜界面(200 m2)看不见,但它通过与周围环境中各种因素的动态相互作用而起着关键作用,这些因素包括微生物,营养素,污染物和其他物质。”
虽然过去人们认为细胞内紧密连接是静态且不可渗透的,但我们现在知道并非如此。正如Fasano所解释的,连蛋白是肠道渗透性的强大调节剂。然而,尽管连蛋白是肠道通透性的生物标志物,并在许多慢性炎性疾病中起着致病作用,但并非所有慢性炎症性疾病都是由肠道渗漏引起的。
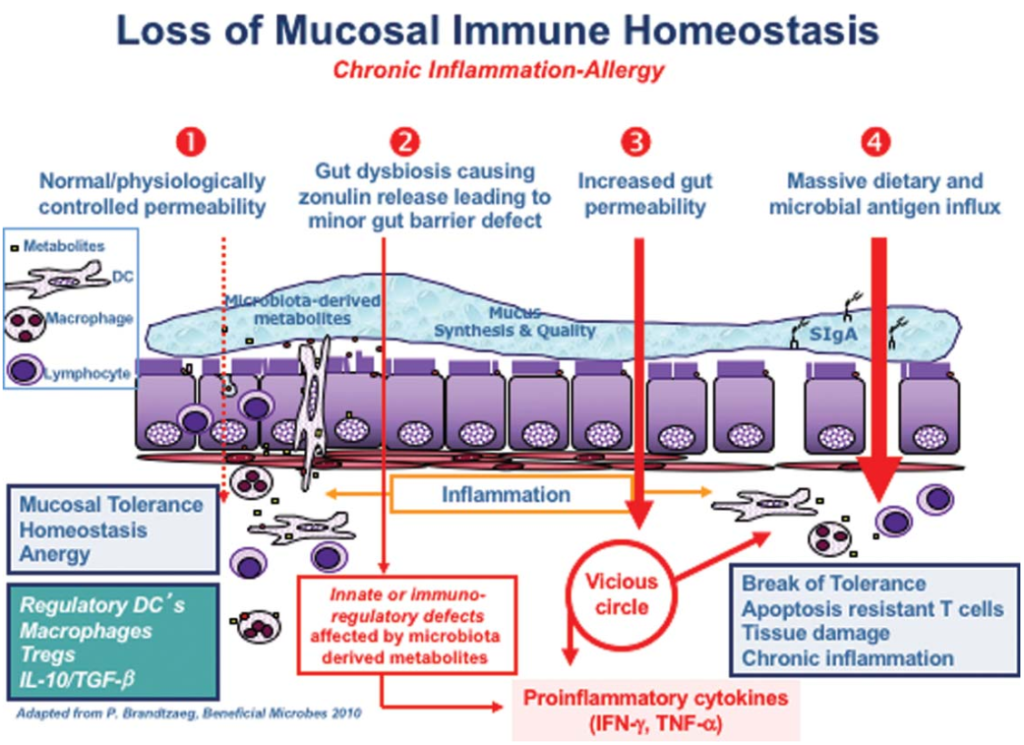
在他的综述中,一篇题为“Zonulin,一种上皮和内皮屏障功能的调节因子,及其在慢性炎症疾病中的作用”的文章,详细描述了“导致慢性炎症疾病的连锁反应”。
在正常情况下,你的肠道会保持健康的内稳态,当遇到抗原时,不会发生过度的免疫反应。在图中第2点,肠道菌群失调(即肠道菌群的数量和多样性不平衡)正在形成,导致连蛋白的过量生产,从而使肠道内壁更容易渗透。
Sturgeon C et al., Tissue Barriers, 2016
两个最强大的触发连蛋白释放是细菌过度生长和谷蛋白。连蛋白是对坏细菌的反应产生的——它通过打开紧密连接帮助细菌排出体外,所以细菌过度生长是有意义的。但是为什么它对谷蛋白有反应呢?
有趣的是,连蛋白途径将谷蛋白误解为微生物的潜在有害成分。这就是为什么谷蛋白会触发连蛋白的释放。虽然Fasano没有提到,除草剂草甘膦也触发连蛋白,而且是谷蛋白10倍的效力!
随后的通透性允许微生物群衍生的抗原和内毒素从管腔迁移到固有层(肠粘膜的结缔组织),从而引发炎症。
随着过程的继续恶化(上图中第3阶段),适应性免疫反应开始,触发促炎性细胞因子的产生,包括干扰素γ(IFN-γ)和肿瘤坏死因子α(TNF-α)。这些细胞因子使通透性进一步恶化,从而形成恶性循环。
最终(第4阶段),粘膜耐受性被完全破坏,导致慢性炎症性疾病的发作。
最终出现的特定的慢性炎症性疾病,部分取决于你的基因组成,部分取决于你所接触的类型以及部分取决于肠道菌群组成。
除了遗传易感性和环境触发因素外,各种慢性炎症性疾病的发病机理还涉及到相互影响的肠道通透性/ Ag转运,免疫激活以及肠道菌群的组成/功能的变化。
连蛋白是上皮和内皮屏障功能的调节剂,肠营养不良可能导致连蛋白的释放,从而导致腔内物质穿过上皮屏障的释放,导致促炎性细胞因子的释放,而促炎性细胞因子本身会导致通透性增加,形成恶性循环,从而导致大量的饮食和微生物Ag大量涌入,触发了T细胞的活化。
根据宿主的遗传组成,活化的T细胞可能保留在胃肠道内,导致肠道慢性炎症性疾病或迁移到几个不同的器官以引起全身性慢性炎症性疾病。”
与zonulin通路失调相关的慢性炎症疾病包括:
自身免疫性疾病如腹腔疾病、1型糖尿病、炎症性肠病、多发性硬化症和强直性脊柱炎
代谢紊乱如肥胖、胰岛素抵抗、非酒精性脂肪肝、妊娠期糖尿病、高脂血症和2型糖尿病
肠道疾病如肠易激综合征、非腹腔麸质敏感性和环境肠道功能障碍
神经炎症性疾病如自闭症谱系障碍、精神分裂症、重度抑郁症和慢性疲劳/肌痛性脑脊髓炎
癌症脑癌和肝癌
2018年,发现的肠道菌群实际控制肝脏中的抗肿瘤免疫应答,并且抗生素可以改变免疫细胞的组成在肝脏中触发肿瘤生长。
哈佛医学院的研究人员已经确定了肠道微生物的特定种群,可以调节局部和系统的免疫反应来抵御病毒入侵。
某些肠道细菌也会促进炎症,炎症是几乎所有癌症的潜在因素,而其他细菌则会抑制炎症。某些肠道细菌的存在甚至可以增强患者对抗癌药物的反应。
肠道菌群提高癌症治疗效果的一种方法:
激活你的免疫系统,让它更有效地发挥作用。
研究人员发现,当这些特定的微生物缺失时,某些抗癌药物可能根本不起作用。
最近的研究表明,肠道细菌也参与了抗病毒防御。
哈佛医学院的研究人员第一次确定了特定的肠道微生物群,这些菌群调节局部和全身免疫反应,抵御病毒侵略者。这项工作确定了一组肠道微生物,以及其中的一个特定物种,它能使免疫细胞释放出抗病毒化学物质——1型干扰素。
研究人员进一步确定了许多肠道细菌共有的确切分子,它开启了免疫保护级联反应。研究人员指出,这种分子并不难分离,可能成为增强人类抗病毒免疫的药物的基础。”
虽然这些发现还需要重复和证实,但它们指出了一种可能性:你也许可以通过在肠道中重新播种脆弱拟杆菌和拟杆菌科的其他细菌,来增强你的抗病毒免疫。
这些细菌启动一个信号级联,诱导干扰素的释放,通过刺激免疫细胞攻击病毒,并导致病毒感染的细胞自我毁灭来保护免受病毒入侵。
具体来说,驻留在细菌表面的一个分子通过激活所谓的TLR4-TRIF信号通路触发干扰素的释放,这种细菌分子刺激免疫信号通路,该通路由9种toll样受体(TLR)之一启动,TLR是先天免疫系统的一部分。
最近的研究还强调了维生素D在肠道健康和全身自身免疫中的作用。一篇综述文章发表于《免疫学前沿》中:
自身免疫性疾病往往会导致维生素D缺乏症,这会改变微生物组和肠道上皮屏障的完整性。
这篇综述总结了肠道细菌对免疫系统的影响,探讨了自身免疫疾病研究中出现的微生物模式,并讨论了维生素D缺乏症如何通过其对肠道屏障功能,菌群组成的影响而有助于自身免疫,和/或对免疫反应的直接影响。
维生素D对免疫系统具有多种直接和间接的调节作用,包括促进调节性T细胞(Tregs),抑制Th1和Th17细胞的分化,损害B细胞的发育和功能,减少单核细胞的活化和刺激来自免疫细胞的抗菌肽。
也就是说,维生素D与自身免疫之间的关系很复杂。除了免疫抑制,维生素D还通过影响菌群组成和肠道屏障的方式改善自身免疫性疾病。
该文章引用了一些研究,这些研究表明维生素D会改变肠道微生物组的组成。一般而言,维生素D缺乏倾向于增加拟杆菌和变形杆菌,而更高的维生素D摄入量则倾向于增加普氏杆菌并减少某些类型的变形杆菌和厚壁菌。
虽然关于维生素D对肠道细菌的影响的研究仍很薄弱,尤其是在患有自身免疫性疾病的患者中,但已知维生素D缺乏症和自身免疫性疾病是合并症,通常建议这些患者补充维生素D。
众所周知,维生素D支持肠道和免疫细胞的防御。维生素D是维持紧密连接所需的关键成分之一。
肠上皮与外部环境不断相互作用。上皮表面适当的屏障完整性和抗菌功能对于维持内稳态和防止特定微生物物种的入侵或过度定殖至关重要。
健康的肠上皮和完整的粘液层对于防止病原性生物入侵至关重要,而维生素D有助于维持这种屏障功能。多项研究发现,维生素D3 / VDR信号调节紧密连接蛋白的数量和分布。
作为一种可使离子进入肠腔的“泄漏”蛋白,在功能性维生素D缺乏症的情况下,claudin-2表达可能会导致结肠炎。
维生素D上调抗菌肽的mRNA和蛋白质表达,包括抗菌肽,防御素和溶菌酶。
抗菌肽主要由肠道Paneth细胞分泌,是微生物组组成的重要介质。
防御素由上皮细胞,Paneth细胞和免疫细胞分泌,并且是肠道固有免疫反应的重要组成部分。
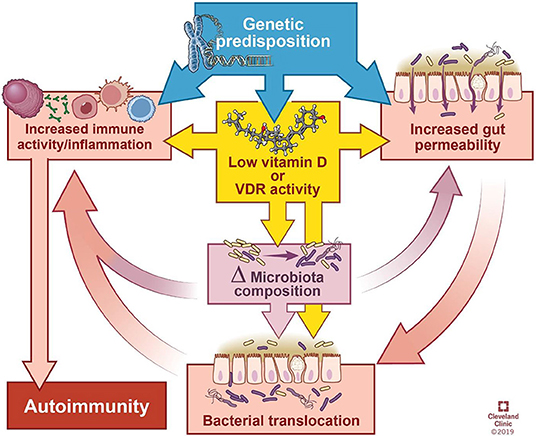
维生素D缺乏症可能通过以下方式影响微生物组和免疫系统,从而导致自身免疫疾病:
1 维生素D缺乏或补充会改变微生物组,细菌丰度或组成的操纵会影响疾病的表现。
2 由于饮食不足而缺乏维生素D信号传导会损害肠道的物理和功能屏障完整性,从而使细菌之间的相互作用刺激或抑制免疫反应。
3 如果缺乏维生素D,先天免疫防御能力可能会受到损害。
Yamamoto Erin A et al.,Front Immunol, 2019
以上所有,我们可以看到,优化肠道菌群和维生素D水平对于保持健康至关重要。通过肠道菌群检测,查看自己的肠道菌群的构成,适当补充益生菌,维生素D将有助于避免肠道泄漏。
对肠道微生物组产生重大影响的最简单,最有效和最便宜的方法:定期食用发酵食品。
健康的选择包括酸奶,纳豆和各种发酵蔬菜。
避免破坏或杀死微生物组,其中包括:
如果可以的话,尽量避免抗生素。抗生素杀菌一视同仁,不管好坏。
尽量少吃常规饲养的肉类和其他动物产品,因为这些可能会被喂食低剂量的抗生素。
尽量避免经基因工程处理和/或草甘膦处理的谷物。
少吃加工食品(由于过量的糖会滋生病原菌)
相关阅读:
参考文献:
Krautkramer KA, Kreznar JH, Romano KA, Vivas EI, Barrett-Wilt GA, Rabaglia ME, Keller MP, Attie AD, Rey FE, Denu JM. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol Cell. 2016 Dec 1;64(5):982-992. doi: 10.1016/j.molcel.2016.10.025. Epub 2016 Nov 23. PMID: 27889451; PMCID: PMC5227652.
Guglielmi Giorgia,How gut microbes are joining the fight against cancer.[J] .Nature, 2018, 557: 482-484.
Larsen Nadja,Vogensen Finn K,van den Berg Frans W J et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults.[J] .PLoS One, 2010, 5: e9085.
Sturgeon Craig,Fasano Alessio,Zonulin, a regulator of epithelial and endothelial barrier functions, and its involvement in chronic inflammatory diseases.[J] .Tissue Barriers, 2016, 4: e1251384.
Yamamoto Erin A,Jørgensen Trine N,Relationships Between Vitamin D, Gut Microbiome, and Systemic Autoimmunity.[J] .Front Immunol, 2019, 10: 3141.