-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康
用于生物数据可视化的方法不断改进,但是在一些可视化图形的着色方面仍然存在根本性的挑战。
生物学数据的视觉不应淹没,掩盖或偏倚结果,而应使其更易于理解。这是对于在创建可视化效果时如何有效使用颜色的挑战。
生物数据的可视化处理是计算机图形学,科学可视化和信息可视化在生命科学各个领域的应用。

本文将介绍10条简单的规则来对生物数据进行可视化着色。
总览
规则1:确定数据的性质
规则2:选择色彩空间
规则3:根据所选颜色空间创建调色板
规则4:将调色板应用于数据集以进行可视化
规则5:套用调色板后,检查数据中的颜色上下文
规则6:在数据可视化中评估颜色的交互作用
规则7:要了解特定学科的颜色约定和定义
规则8:评估色差
规则9:考虑网络内容的可访问性和打印实际情况
规则10:黑白分明
数据是有价值的信息记录。可视化数据是将这些数据中包含的想法、经历和故事联系起来的一种重要而有力的方式。
图形和数据可视化促进了生物信息在不同背景下的表达和交流,形成叙述、想法和经验。要使数据中包含的信息具有形状,了解数据的性质是重要的。借用描述性统计的领域知识,数据如性别、年龄、身高、体重和眼睛颜色等被称为变量。变量的类型与数据的性质有关。
区分变量类型的一种方法是依赖于分配给变量的值中的信息的性质。这个被称为测量的水平或尺度,将观察到的变量分为4个级别:名义、序数、区间和比率。这些数据也可以分为两种不同的数据类型:定性或分类(名义、序数)和定量(区间、比率)。
下面我们分别用一个例子来描述和解释:
名义描述了一个变量的属性,只通过名称(类别)来区分,没有顺序(等级、方向或位置)。
例如:性别、生物种类、眼睛颜色,血型(A、B、AB、O),细菌类型(球菌、芽孢杆菌、螺旋菌等)。它们是一个多值变量,没有明确的尺度来适应不同的值。
序数层次描述了按顺序(等级、规模或位置)区分的变量的分类属性,但没有关于它们之间差异相对程度的信息。要注意这种变量可能会用数字编码。
举例:热度(低、中、高);疾病的严重程度(轻度、中度、重度);一致量表,如李克特量表,(强烈不同意、不同意、无意见、同意或强烈同意)。
注:李克特量表是一种心理反应量表。
区间级别描述变量的属性,通过差异程度来区分,没有绝对零度,并且属性之间没有已知的比率。通常,该变量的数值为正、负或零。
例如:公制摄氏温标,温差(摄氏度和开尔文),1年的间隔。20℃和30℃之间的差异与25℃和35℃间的差异相同.。
比率级别描述变量的属性,这些属性通过它们之间的差异程度来区分,绝对为零,并且属性之间的比率是已知的。具有负值是不典型的。
例如:年龄、身高、体重、持续时间、开尔文温标。此外,假设数值的定量数据(区间或比率)可以进一步分为离散或连续。
离散(可计算的)变量仅假设整数和某种计数。
例如:年龄和日期是离散的。年龄在1年内保持不变,而日期在24小时内保持不变。它们都以“1”跳跃或增加。
连续(定义范围内的任何值)变量可以取某个值范围内的任何值。对这种测量的观察会受到测量仪器的限制。
例如:身高(厘米,英寸),体重(公斤,磅),温度(摄氏度,华氏度),时间(小时,分,秒)。温度逐渐升高,时间不断流逝。
当只有两个可能值时,二进制或二分变量类型是一种特殊类型。示例:是或否调查问卷和二进制数字(0或1)。
表1根据4个不同的测量相关类别介绍了4个测量级别,包括从最低到最高的测量分辨率。
表1 四个层次的测量


使用4个与测量相关的类别来比较等级:分辨率、属性、数学运算符和中心趋势。
颜色空间指的是颜色转化为数字的颜色模型。基于一组原色,颜色模型创建许多颜色。每个模型都有其可以产生的特定颜色范围,该范围定义了色彩空间。
通常,红、绿、蓝(RGB)和青色、洋红、黄色和黑色(CMYK)是最常见的系统,当然还有其他系统。例如,色调、饱和度和亮度/值(HSB/HSV)颜色空间是RGB颜色模型或标准红绿蓝(sRGB)颜色空间的替代表示。
注:关于这些维度的更多信息,大卫·布里格斯的网站名为《颜色的维度》是一个关于颜色理论和使用的信息宝库。
传统的颜色工具,如色轮,鼓励艺术/手工颜色选择。颜色或代码的数值是不同的,将颜色视为特定颜色空间中的数字。此外,由于我们选择的数字和输出颜色之间可能会出现差异,颜色空间应该在感知上是一致的。
在颜色科学领域,已经努力建立独立于特定颜色显示或复制设备的颜色空间。人们努力创造出感觉上统一的色彩空间。这些颜色空间背后的动机是使空间与人类视觉感知颜色属性的方式紧密一致。
下表是常用的颜色空间(表2),接下来将要讨论的是解决感知一致性问题的颜色空间。
表2 常用色彩空间的优缺点
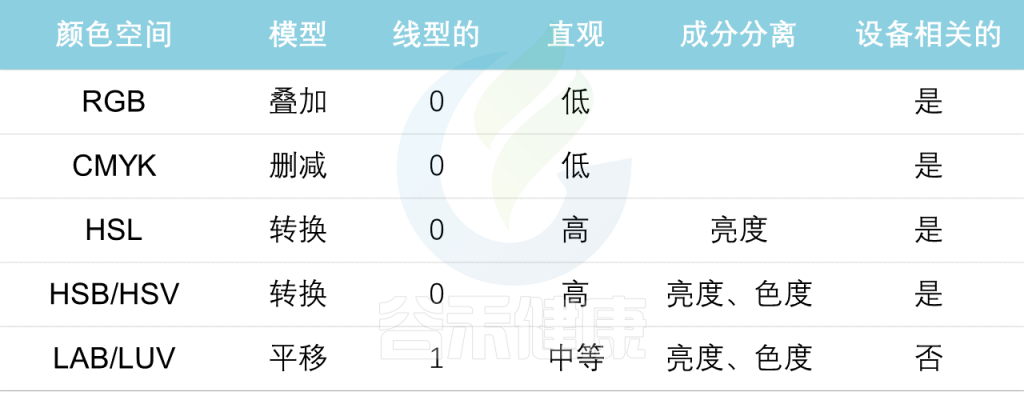

由于复杂的颜色转换,光线混合的维度反映了人类视觉的工作方式。下面几种是我们需要考虑的各种特征:模型,线性,直观,组件分离以及设备相关。
模型
一个有序的系统,用于从一小组原色中创建一个完整的颜色范围
线性
颜色值相同的变化应该会产生视觉重要性大致相同的变化
直观
指颜色维度易于重新映射到不同的颜色模型
组件分离
指相对于其他维度分离1个颜色维度。
例如,色调、饱和度和亮度(HSL)分离亮度分量(明度),在图像处理的领域知识中特别有用。
设备相关
颜色空间依赖于所使用的设备来设置、制作和渲染的情况。
亮度是光的可见能量或根据人类视觉系统的逐波长响应加权的物理光能。
色度是一个区域的颜色,它被判断为一个相似的被照亮的区域的亮度的一个比例,这个区域看起来是白色的或高度透射的。虽然色度描述了光的心理物理颜色,但它与光的强度(亮度)无关。
所列出的感知一致颜色空间优于RGB和CMYK颜色空间。RGB用来表示颜色,但它不足以进行颜色处理,并且不是行业标准。
由于CMYK主要用于印刷,它有许多缺点,将在下一篇规则9中进一步讨论。然而,它们并非没有混淆的效果,例如亮度随色调而急剧变化。
LUV和LAB都追求感知一致性。虽然两者都已被CIE(国际照明委员会)采用,但通过依靠三个组成部分并计算相邻颜色之间的椭圆距离,可以观察到空间中不同颜色的相对概念差异。
因为它们是独立的,我们建议使用它的颜色空间。如果选择了,就需要为数据创建一个合适的调色板。
创建一个调色板很像选择一套衣服。重要的是要了解允许选择颜色来给数据可视化着色的规则。
为了根据特定的颜色空间选择调色板,通常使用色轮。它是一种围绕一个圆圈组织不同颜色以显示颜色之间关系的工具。通常,色轮包含12种颜色。
创建色彩和谐是一个选择在图像合成中协同工作的色彩的过程。基于色轮上的颜色组合,有助于为色彩如何协同工作提供共同的指导方针。
我们可以区分有助于使用色轮创建配色方案的软件和/或网络工具,即Adobe color和配色方案设计器Paletton。
除了创造美学上令人愉悦的颜色组合,调和性还可以用来指导调色板的创建。它们包括单色、模拟和互补。
下图描绘了青色调中的三个调和示例。应该注意颜色的小点,以描绘出特定的色调排列(单色、相似和互补)。
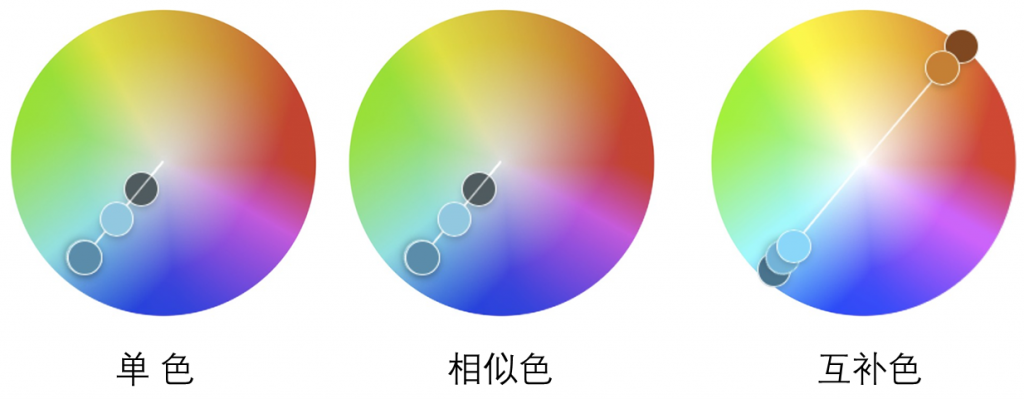

单色或单调色度
是一种单一色调,它在色调、色度和饱和度方面有所变化。一个特别的例子是单调方案,但非彩色(没有色调),仅由从黑色到白色的灰度值组成,即灰度。
相似色
是那些位于任何给定颜色的两边或被一个颜色分开的颜色。这些通常是自然界中的配色方案。
互补色
是色轮上彼此直接相对的颜色。他们经常形成对比,相互突出。当用作数据中的高亮颜色时,它们非常有用。
为了更好地将颜色的使用与数据类型联系起来,信息设计师和数据科学家将上述数据类型(规则1)简化为三种主要类型:连续的、发散的和定性的。
这些分类是在ColorBrewer工具中开发的,最初旨在为制图提供颜色建议。这一概念已经被数据可视化社区所采用,反映在蒙兹纳的可视化分析和设计教科书中。该网络工具可以在colorbrewer2.org找到。
下图展示出了每个数据类型的调色板的例子。
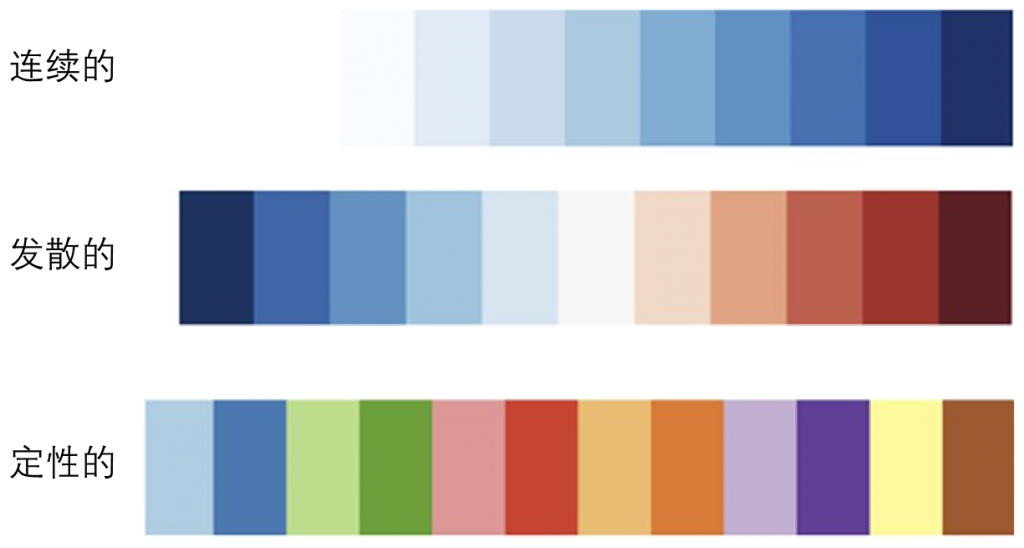

顺序调色板
适用于从低到高变化的有序数据。视哪一方对观察者来说最重要,视觉编码是两种颜色之间的变化,分别从白色或较亮的颜色到黑色或较暗的颜色。这种颜色使用是明度逐步变化,通常重要的数据值具有较暗的颜色。这些调色板对应于包含1种颜色变化的单色调色板。
发散调色板
显示两个方向的视觉变化。主要用于在区间数据范围的两端同等强调中间值和极值,它们通常是对称的。颜色在黑暗中增加,以表示断点(如零变化或平均值)周围与数据中特定有意义的中间值之间的差异。
定性调色板
不依赖或暗示类别之间的数量差异。通常,色调以一致的亮度来表示名义和分类数据。还有另外两种变体:成对和强调。处理无序数据时,成对调色板通过视觉关联类来处理成对数据,但强调调色板通过更饱和的颜色来强调相关类。
除了前面提到的工具ColorBrewer,还有两个调色板:一个用于连续数据,另一个用于定性数据。
对于连续数据,推荐 viridis调色板。它在感觉上是均匀的,并以多种色调显示单调增加的亮度。多亏viridis调色板和其他调色板,一个连续数据集的所有数据点都具有同等的视觉重要性。此外,我们将在下一篇的规则8中看到,这些调色板对色弱和色盲是友好的。
对于定性数据,Tableau 10调色板可以推荐给大家。它包含几个非常不同的色调,亮度值范围很广。虽然它是用10种颜色设计的,而且很适合三色异常,但它所有颜色的使用对其他颜色缺陷是一个挑战。我们将在规则4和下一篇规则8中讨论定性数据的颜色限制。
另外还有一个创建调色板的网络工具,即Colorgorical( http://vrl.cs.brown.edu/color)。
要应用选定的调色板,需要考虑将颜色映射到数据点的过程。基于规则3,我们考虑了3种不同的颜色映射调色板:连续的、发散的和定性的。
对于连续调色板,色调应该受到限制,只有亮度或饱和度应该变化。根据背景颜色、手头的任务和数据的性质,将较高的值映射到较暗或较亮的颜色非常重要。
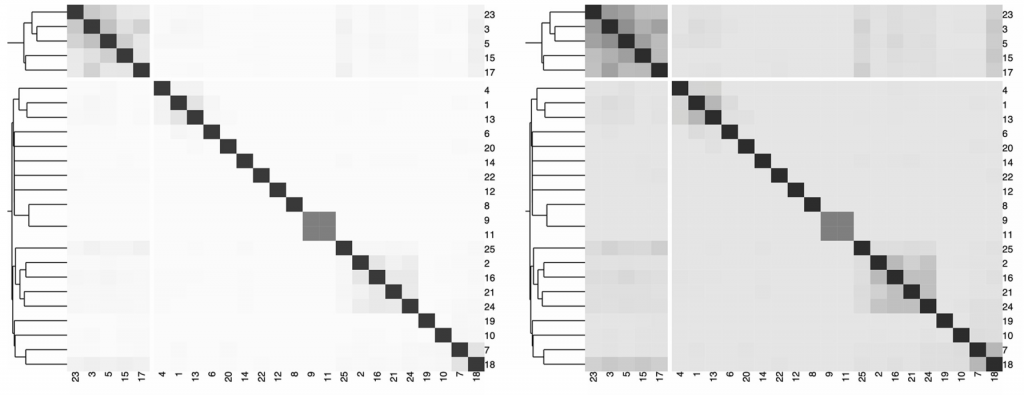
在下图中,我们展示了一个热图,描述了不同字符串之间的Jaccard索引和由分层聚类提供的背景信息。
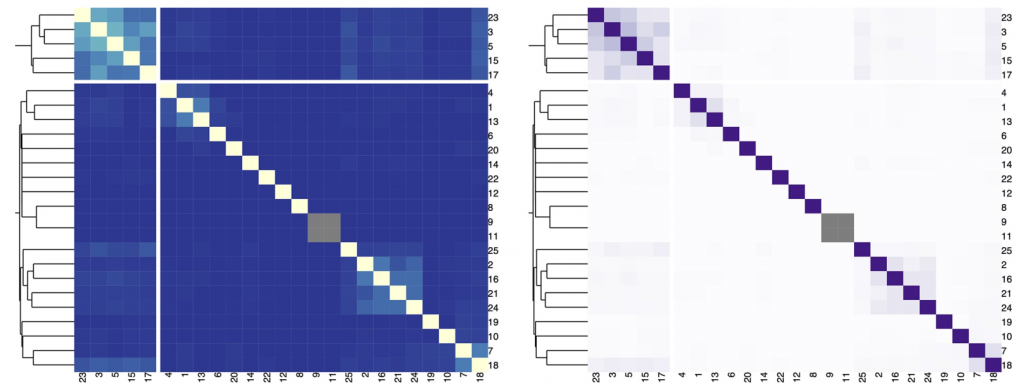

对于发散的调色板,当数据有有意义的或关键的中断时很重要。通常,关键断点应该采用中性颜色,如灰色,端点应该采用饱和颜色。一般来说是对称的,临界断点可以是平均值、中间值或零变化值。
在平均值或中位数的情况下,通常有低终点和高终点。在负值和正值有零值中断的情况下,端点应使用不同的色调。为了突出分歧,中断可以去饱和,端点可以饱和。
对于定性调色板,建议仅使用5 –6 种颜色,如果绝对需要,也可以使用更多颜色。实际上,当使用ColorBrewer时,限制被设置在3到9的范围内。
如果有理由的话,我们认为颜色是不变的,即使它们在不同的光线下。事实上,颜色恒常性是感知物体颜色的能力,不受光源颜色的影响。这主要是因为颜色是一种相对的媒介。
举个例子,比如我们可以看到一根香蕉在阳光充足的中午或光线微弱的黑暗房间里呈黄色。然而,在某些情况下,相邻的颜色会改变我们的感知和区分某种颜色影响的能力。
下图显示了一个数据视觉的例子,其中白色可以与灰色背景区分开来,比如在你的电脑屏幕上。然而,同样的白线在白色背景下很难区分,也许当打印在白纸上时。当白线变成黄线时,情况就解决了。

有个“Interaction of color”app可以进一步教会你如何意识到颜色的背景,它是约瑟夫·阿尔伯斯50年前写的《色彩交互》一书的数字化延伸。它为在不同的显示背景中学习颜色提供了练习。该应用还允许在interactionofcolor.com创建个性化的色彩研究和调色板。
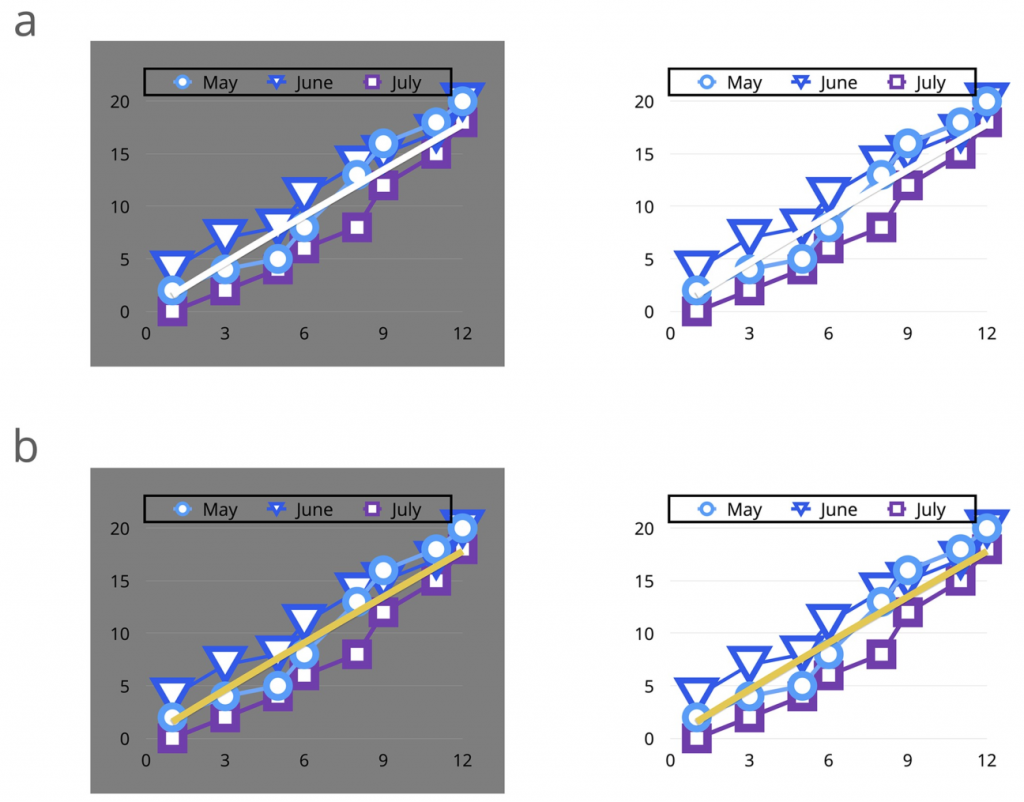
在生物数据可视化中,通常会看到红色/蓝色的数据可视化。由于同时对比,对红/蓝颜色组合的偏好是可以解释的。下图就是这种情况。
-1024x454.png)
-1-1024x454.png)
左:红色/蓝色组合。右图:绿色/紫色补色组合改善了数据可视性。
同时对比是指两种不同的颜色相互影响的方式。这也是蓝色背景下很难阅读红色文字的原因。理论是,当两种颜色并排放置时,一种颜色可以改变我们对另一种颜色的色调感知。实际的颜色本身不会改变,但我们认为它们已经改变了。
法国化学家米歇尔·欧仁·切夫勒发展了这种同时对比的规则。它坚持认为,如果两种颜色靠得很近,每种颜色都将呈现相邻颜色补色的色调。类似的结果也可以发生在数据可视化中。然而,同时使用对比色可能难以评估数据趋势的变化。
颜色的使用取决于大量数据和介质特性。除了某些颜色的不良相互作用之外,我们会看到颜色可能带有某种意义。
对于交互,存在红/蓝颜色或文本内容的交互。首先,文本看起来模糊不清,伤害眼睛。这是称为色差的现象的结果,色差对应于不能同时聚焦在两种颜色上。
对于下图中提供的例子,还需要解决色彩不足问题的补色组合。绿色/紫色方案提供了两个数据变量之间的转换组合,同时允许有色觉障碍的个人区分这两个变量。绿色和紫色是这种特殊情况下的最佳组合。
-2-1024x454.png)
-1-1024x454.png)
在某些情况下,感知的一致性至关重要。一个简单的例子是,在不规则的颜色空间中,选择一种随机的颜色以便在黑暗的背景下可读,这是很困难的,因为相同亮度或光度的颜色看起来非常不同(蓝色和黄色在HSV中都有100%的亮度,但是蓝色比黄色暗得多)。
为了解决这个问题,需要考虑所选色调的复杂计算,以使随机颜色看起来同样明亮。有一个更简单的方法,即选择更好的颜色空间。
基于jet或彩虹的调色板是最常用的调色板,因为它是在软件工具中作为标准提供的。它具有很高的对比度,这使得它能够突出手头数据的特征。
然而,当查看颜色图表时,色带或色块尤其出现在青色和黄色区域。这个看似不错的调色板在应用于描述同等重要的顺序数据时会导致急剧的转换,尽管底层数据变化均匀。事实上,由于非恒定的感知颜色变化,这是误导,甚至对色弱的个人更是如此。
下图描述了这些部分。
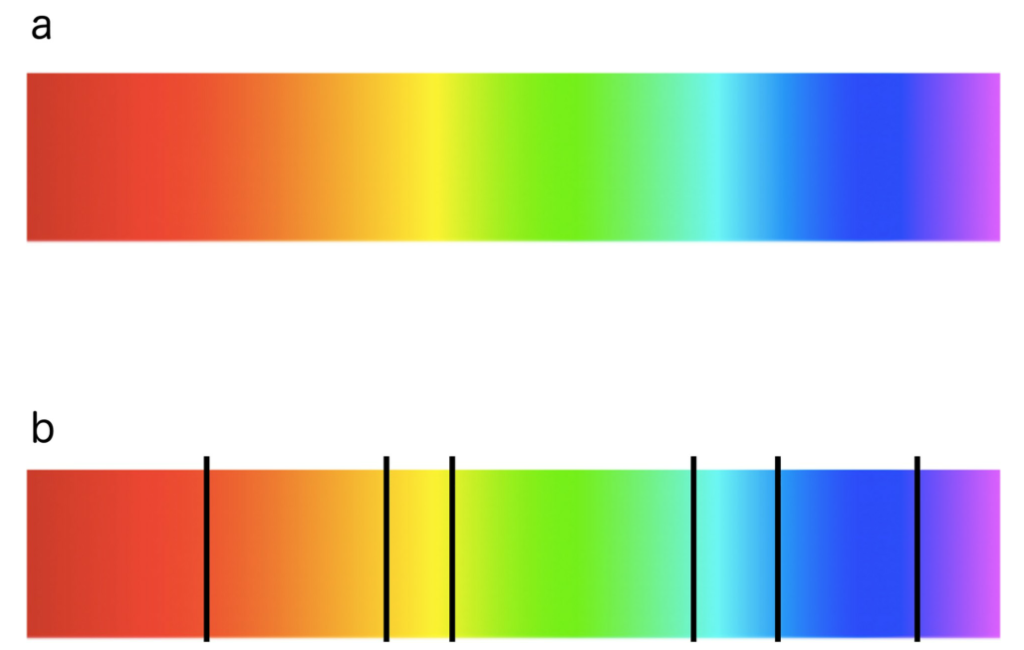
虽然许多研究人员都在抱怨它的误导,但在实际许多应用中,基于彩虹的调色板仍然在被使用,并且有可能对解决任务的准确性有潜在的负面影响。
不幸的是,由于人类通常将颜色分类,彩虹调色板的使用会给数据的解释带来偏差。此外,由于色调的自然顺序,这甚至可能被放大。然而,不同的方面可以被智能地集成。例如,不同的亮度强调某些标量值,而低亮度颜色(例如,蓝色)可能隐藏高频。
生物学描述了生物组织的不同层次(从分子到细胞、有机体到生态系统),整合了多种领域,如生物化学和生物物理学。这涉及到大量不同风格的数据,这些数据可能受特定领域惯例的约束。
我们简要讨论4个与生物化学、生物物理学、解剖学和细菌学相关的显著例子。
生物化学
在化学中,一个分子中不同原子的颜色遵循标准的科里·鲍林·科尔顿(CPK)规则。最重要的颜色是氢的白色(H),碳的黑色(C),氮的蓝色(N),氧的红色(O),硫的深黄色(S),磷的紫色(P)。其余的原子呈现出亮、中、暗、卤素组为深绿色,金属组为银色。生物化学遵循这些惯例,例如,给20种蛋白质氨基酸的生化结构着色。
生物物理学
在过去的几年里,已经开发了广泛的荧光蛋白遗传变异体,其特征是荧光发射光谱分布几乎跨越了整个可见光谱。借助这种分子和显微镜技术,科学家可以看到特定的细胞反应甚至亚细胞机制。例如,这种特定的分子可以在不同的光谱范围内(例如青色、绿色、黄色或红色)发出荧光。当然,最著名的分子是绿色荧光蛋白。如果数据集涉及荧光图谱,或包括光谱范围的信息,惯例是根据它们给数据着色。
解剖学
解剖学上,颜色约定从第一张解剖草图就已经存在。虽然第一幅彩色印刷的医学插图显示了文字颜色的用法,但现代颜色的用法是相当象征性的。事实上,颜色通常用于肤色、内脏、循环和神经系统,甚至是选定的身体组织(如肌肉或脂肪)。尽管在体内动脉和神经呈白色,静脉呈淡蓝色,但既定的颜色惯例是动脉为红色,静脉为蓝色,神经为黄色。
细菌学
在细菌学中,科学家对许多细菌特性和机制感兴趣,例如革兰氏染色、形态学、遗传学和抗生素抗性。前者通过细胞壁的化学和物理特性来区分细菌(革兰氏阳性细菌有厚厚的肽聚糖细胞壁,保留了结晶紫的主要染色)。后者发生在细菌和真菌等细菌有能力击败用来杀死它们的抗生素(如青霉素)时。
基于包含3种最流行的抗生素对16种细菌的性能的数据,我们在下图中展示了青霉素与新霉素的有效性的2个实例数据。
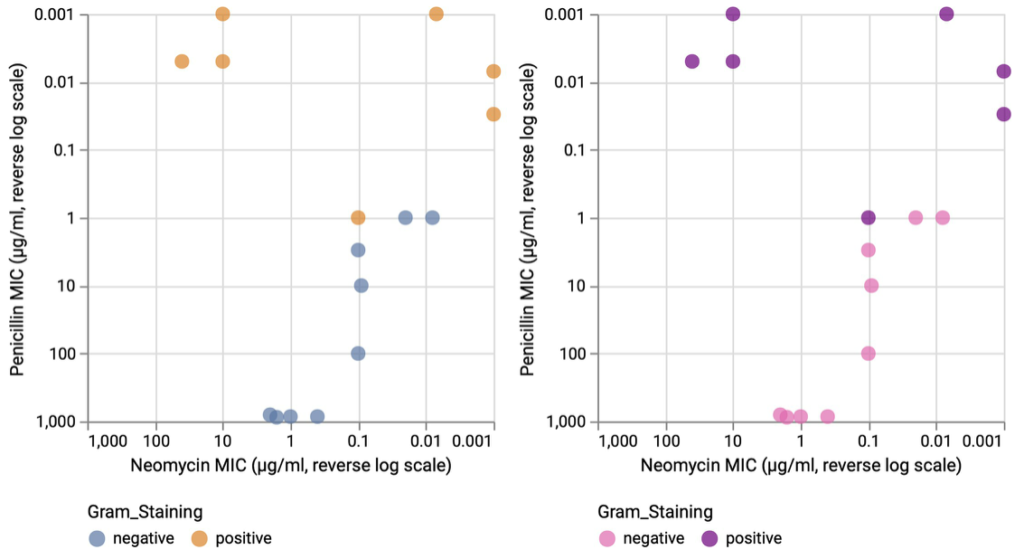
虽然选择蓝色/橙色的配色方案是为了为名义比较提供可感知的可分辨颜色(左),但采用革兰氏染色颜色惯例呈现出更适合具体问题的颜色用法(右)。
其他生物领域
其他实践存在于特定的生物研究领域。例如,在分子和进化生物学中,基因表达水平和基因保护的视觉编码依赖于红/蓝发散调色板。然而,我们不能谈论颜色惯例,因为在红/绿、红/蓝以及断点不是白色而是黄色的其他情况下,这个值会有很大差异。
值得一提的是要注意文化习俗。事实上,在不同的国家或文化中,颜色可能具有非常不同的象征意义,如果不是相反的话。一个很好的例子是红色,它在西方社会象征着危险和激情,在东方社会象征着幸福和繁荣。
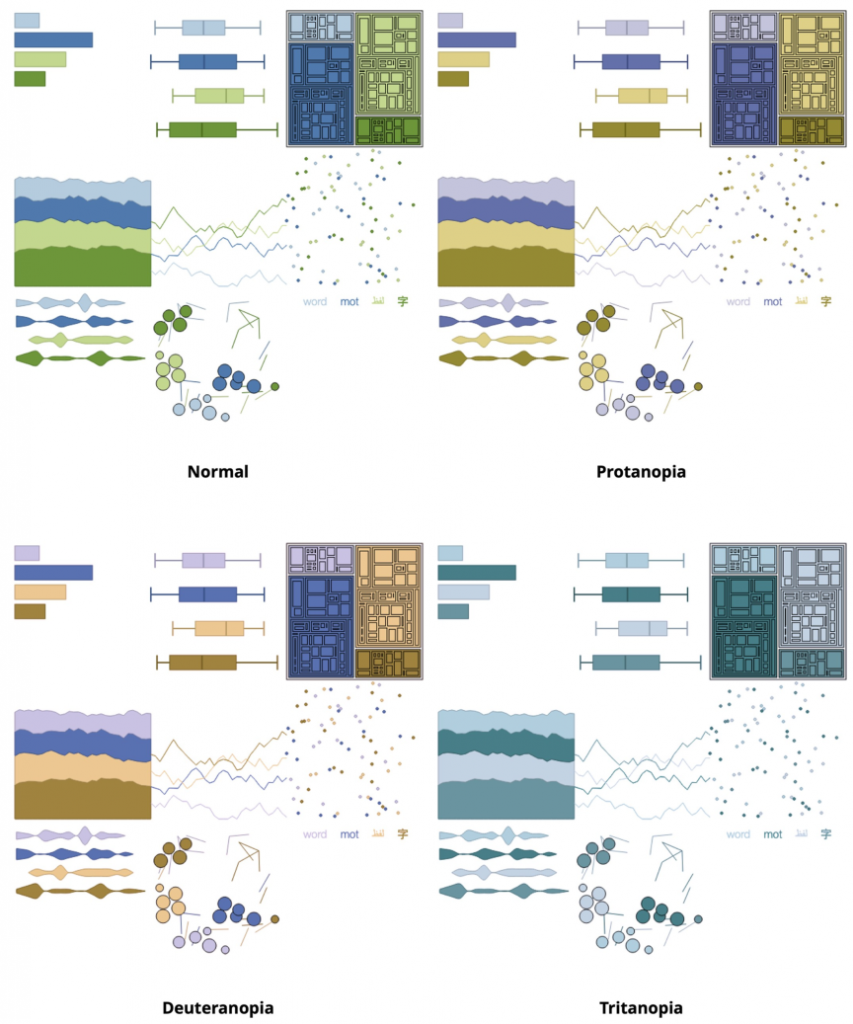
人类有3种感光细胞或视锥细胞,每一种都对视觉光谱的不同部分敏感,以促进丰富的彩色视觉。我们需要尊重一些人的颜色感知是不同的,并评估所选择的调色板是否适合有色弱或色盲的人。
如果一个或多个视锥细胞不能正常工作,就会导致色觉缺失。红色锥细胞缺乏症被归类为红色盲。绿色锥细胞缺陷被归类为绿色盲。蓝锥细胞缺陷被归类为蓝色盲。
当创建或选择调色板时,不同的网络工具允许测试色弱和色盲。一方面,可以使用Adobe Color web工具(color.adobe.com)或Paletton–配色方案设计师(paletton.com)来测试调色板的颜色缺陷。另一方面,网络工具Coblis (Matthew Wickline和人机交互资源网络)使我们能够评估数据可视化是否对更大的受众可用,包括颜色缺陷。
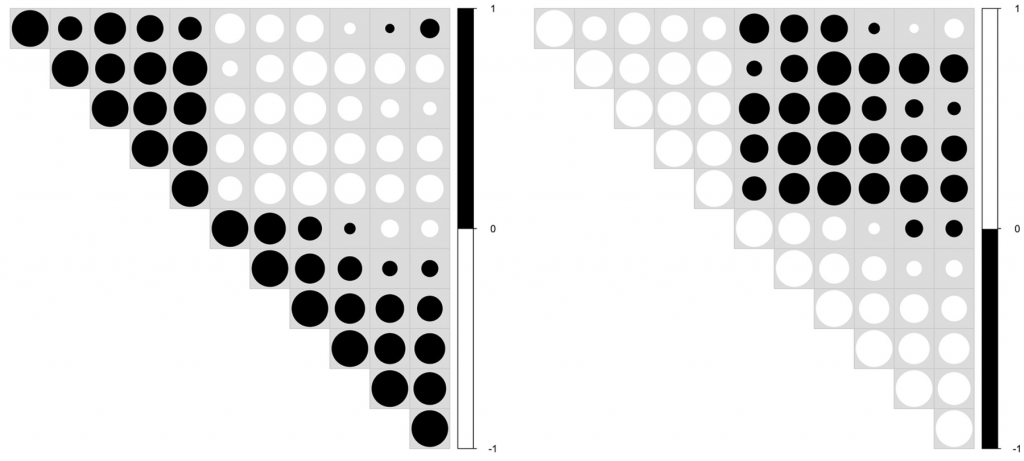
另一个值得注意的工具是Viz调色板。它允许通过模拟选定的信息可视化示例来测试特定调色板的颜色缺陷。下图结合了3个工具,ColorBrewer, Viz Palette和 Coblis,提供了一个用例的例子。

在许多情况下,生物数据可视化超越了研究工作,成为一般在线(如网站)和印刷(如期刊论文)出版物的一部分。对于这些情况,我们简要讨论了网络内容的可访问性和打印现实。
对于基于网络或桌面和移动设备,我们建议遵循万维网联盟(W3C)制定的网络内容可访问性指南(WCAG)。网站必须是可感知的、可操作的、可理解的和健壮的,根据4个原则组织有12个指南。
虽然有一些技术可以帮助作者满足指导方针和成功标准,但是这些技术会随着时间的推移而发展和适应。在列出的技术中,有8种与颜色有关。我们将范围限制在非交互式数据可视化,并借用有利于数据可视化可访问性的技术:
· 确保通过颜色差异传达的信息在文本中也可用
· 使用颜色和图案
·使用颜色提示时使用语义标记
·当使用文本颜色差异来传达信息时,确保附加的视觉提示可用
·对周围的文本使用3:1的对比度,并在链接或控件的焦点上提供额外的视觉提示,仅用颜色来识别它们
·包括用于彩色表单控件标签的文本提示
事实上,其中大部分是为了网页颜色的使用,但我们认为它们是相关的。所报道的技术解决了如何提高看不见颜色的用户的可访问性,因此可以寻找或倾听文本线索;使用盲文显示器或其他触觉界面的人可以通过触摸来检测文本提示。
此外,一些技术解决了实现文本信息和内容的更好对比度的问题。即G17:确保文本(和文本的图像)和文本后面的背景之间存在至少7:1的对比度。
事实上,这个想法是为了确保阅读文本时亮度有对比,而不是色调有对比。
网络工具 Colorable 允许使用十六进制格式的web十六进制代码测试两种颜色,并提供滑块来控制色调、饱和度和亮度。
观看和阅读生物数据可视化取决于目标受众使用的媒介。一方面,使用桌面和移动设备,其中光源用于在RGB颜色空间中混合不同强度的红色、绿色和蓝色。当所有颜色混合时,出现白色。
另一方面,使用纸张打印件,其中打印机将不同程度的CMYK颜色与物理墨水颜色(青色、品红色、黄色和黑色)相结合。当所有颜色混合在一起时,就会产生黑色。为了方便起见,我们可在以下工作时提出一项易于遵守的入围名单要求:
台式机和移动设备,最适合的色彩空间是RGB。关于在网页上应用颜色的指导性文件可以在这里找到:w3.org/TR/css-color-3/#rgb-color
小印刷件,如小册子,或期刊纸图,我们鼓励在300 DPI分辨率的CMYK色彩空间的图像。
非常大的图形并不总是控制它们的质量,我们建议从灰度、位图或RGB颜色空间转换到打印机友好的CMYK颜色空间。
在某些需要考虑印刷成本的情况下,黑白配色方案可能是首选。此外,黑白会增加那些色盲者以与您相同的方式看到和阅读数据可视化的机会。
在不同的领域,如图形和渲染,甚至摄影,这条规则通常被表述为“检查它在黑白和彩色中是否工作良好”。在数据可视化中,这通常与测试所呈现的故事是否仍然可见或可辨别有关。
黑白分明意味着两件事。当不确定调色板时,尝试用灰度显示数据,或者比较两个彩色版本的数据,当不确定哪个更易读时,用黑白打印出来。大多数情况下,后者是找到对比度更好的可视化。
此外,一个建议是关于影印友好的调色板。为了对抗影印过程的损耗,单色或连续调色板是最有弹性和最合适的。
着色并不容易。如果对颜色有需求,可以选择适合并使用少量的颜色,避免饱和的颜色,并且要符合阅读者的期望。


以上两图详细说明了根据示例任务的黑白数据可视化的变化。安全选择的一个例子是选择一种颜色和几种灰色阴影。
最后,总结一下这十种规则。

【参考文献】
Hattab G, Rhyne T-M, Heider D (2020) Ten simple rules to colorize biological data visualization. PLoS Comput Biol 16(10): e1008259.
Rhyne T. M. (2017). Applying Color Theory to Digital Media and Visualization., CRC Press, Boca Raton, Florida, ISBN 9781498765497
Smith N, van der Walt S. A better default colormap for Matplotlib. SciPy2015. 2015
Stokes D., Matthen M., & Biggs S. (Eds.). (2015). Perception and its modalities. Oxford University Press, USA
Gramazio CC, Laidlaw DH, Schloss KB. Colorgorical: Creating discriminable and preferable color palettes for information visualization. IEEE Trans Vis Comput Graph. 2016; 23(1):521–530.

谷禾健康


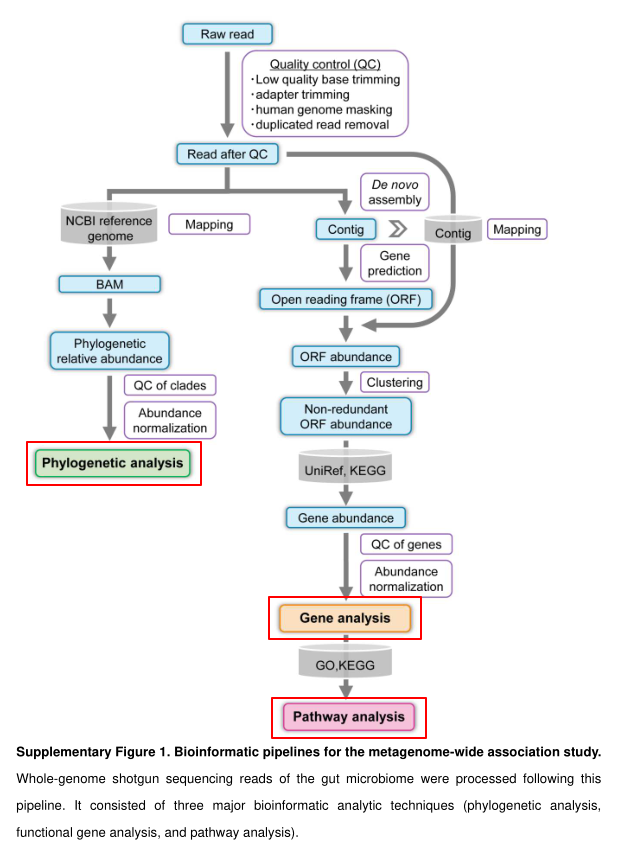
在现代测序技术的帮助下,微生物组研究的范围被扩大,通过16S rRNA测序或鸟枪法宏基因组测序可以生成大量的微生物组数据。而微生物群落研究中的一个重要问题是对这些微生物的归类,模拟和分析人类微生物群。
通常使用16S rRNA技术量化微生物群落的组成,但量化后的数据是偏斜的,带有过多的0。目前还缺乏对复杂的微生物群落测序数据的标准化的聚类分析方法。
近日,加拿大多伦多大学研究人员在《Microorganisms》上发表的一篇研究,针对上述问题构建了一个参数化的混合模型用于计算聚类分析的距离度量,模型根据观察到的OTU计数和估计的混合权重产生sample-specific的分布。这个方法可以准确的估计真实的0比例,从而构建一个精确的beta多样性度量。
大量的模拟研究表明,与一些被广泛使用的距离度量方法相比,当存在较大比例的0时,该方法取得了较好的聚类效果。
该研究人员提出了一种具有特定beta多样性度量的聚类算法,该算法可以解决稀疏计数数据遇到的有无偏差问题且能有效的度量样本距离,达到分层的目的。
微生物群落研究中的一个重要问题是对这些微生物的归类,它们是否能被划分为亚群。如果有,有多少组亚群,如何解释这个亚群。例如,这种分类是否区分了治疗方法、疾病或遗传类型。
为了回答这些问题,需要测量两个微生物群落之间的相似性。beta多样性是为了适应不同的目的而提出的,在评估群落之间的差异时提供不同的结果。对于微生物组成,beta多样性根据测量丰度来衡量群落之间的距离,丰度可以是观察到的计数,也可以是相对丰度,这些丰度是根据不同或距离度量计算出来的,以量化样本之间的相似性。
现如今,已经有许多非参数统计方法来量化距离度量。例如Euclidean和Manhattan距离是最常用的。其它beta多样性指标,例如Bray-Curtis距离、Jensen-Shannon距离、Jaccard指数、UniFrac距离(未加权的、加权的和广义的)也经常用于微生物组研究。
除了距离度量之外,还引入了用于生态关联推理的稀疏逆协方差估计(SPICE-EASI)的图形网络模型。然而这些方法都会有一定的局限性,例如SPIEC-EASI方法依赖于单一的方差-协方差矩阵,由于微生物群落结构复杂,可能无法完全恢复底层OTU网络。
于是,研究人员开发了一种创新的聚类方法,以混合模型而不是beta多样性度量作为距离度量,并将聚类算法应用于微生物群落数据来表征亚群。该算法还包括根据选择的内部指标选择最优聚类数,并将结果在几种距离度量和不同评估方法之间进行比较。通过全面的模拟研究和一个真实的帕金森病肠道微生物群数据集对该算法的性能进行了评估。
1. 构建混合模型
混合模型是一种概率模型,用于表示在无监督学习中经常使用的总体内的子群体。该模型关注单个OTU在种群中的分布,可以解决样本间的稀疏性问题。它参数化地模拟了计数的潜在分布,包括低计数OTU和极高计数。对于个体样本之间的成对距离,在L2范数距离中使用公式化的混合概率。
2. 距离度量
在确定混合模型分布后,使用概率分布通过样本之间的两两距离计算距离度量。为了进行比较,考虑了基于L2范数的三种距离度量(L2-PDF、L2CDF、L2-DCDF、L2-CCDF)。
除此之外还选择了一些其他的距离度量进行比较,即Manhattan距离和Euclidean距离,以及微生物组分析中特有的三个距离度量:Bray-Curtis距离、加权UniFrac距离和广义UniFrac距离。本研究不考虑未加权的UniFrac距离,因为它不包含类群丰度信息。
3. 聚类分析验证指数
这些指数用于衡量集群在集群内部和集群之间的可分离性表现很好。验证指标可以分为内部指标和外部评估,许多内部验证指标被用来选择最优聚类数。外部评估分数是在假设标签在建模阶段没有使用时,通过直接将划分结果与之前的标签进行比较来计算的。
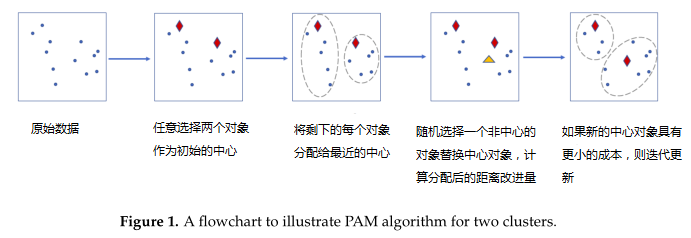
4. 用于聚类的分区算法(PAM)

使用混合分布的聚类过程的详细步骤:
为了测试该方法在聚类中的表现如何,研究人员推导了其准确性和Jaccard指数。
准确性是指聚类结果与真实的聚类指数的接近程度。它被定义为正确聚集的受试者所占的比例。
Jaccard指数衡量聚类结果与原始聚类标签之间的相似性,原始聚类标签定义为正确分类的主题数量(预测集与真实集的交集 )与两组总样本量(两集的并集)之比。
研究人员用类标签模拟数据来模拟OTU计数及其复杂的结构。研究人员考虑两个有两个子类和三个子类的场景,每个子类包含200个样本,总样本量分别为400和600。所有的结果被重复了100次。
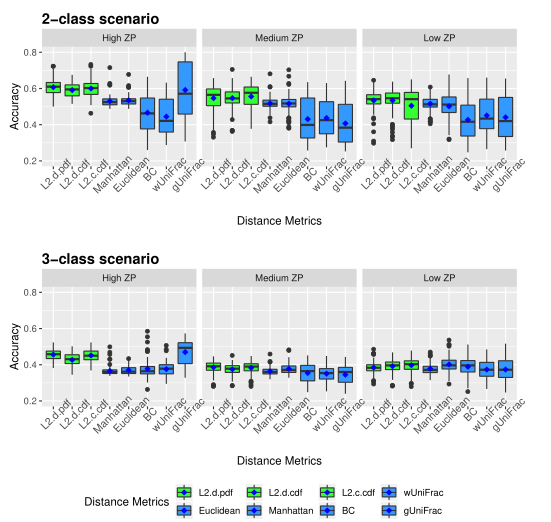
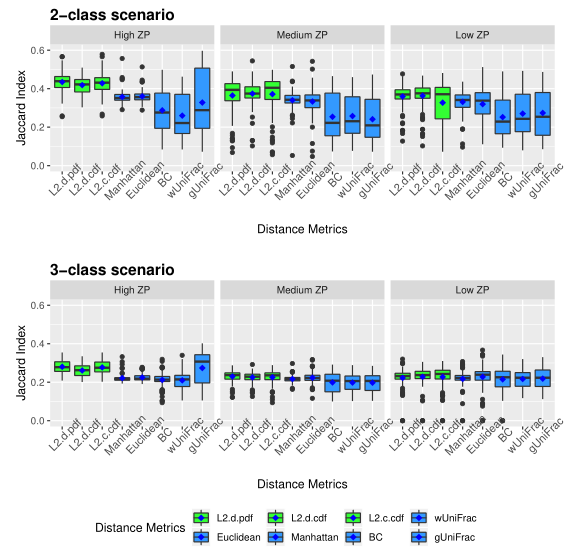
下图展示了模拟数据的准确性。评估了三种不同的0的比例(ZP)情况,左中右分别为高ZP、中等ZP、低ZP。
下图展示了模拟数据的Jaccard指数。同上图一样评估了三种不同的0的比例。
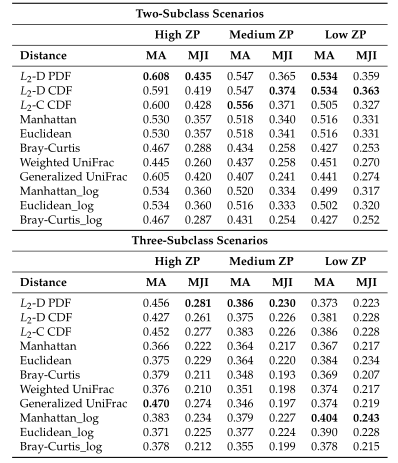
以上两图显示了具有不同ZP和子类数量的不同场景下模拟数据集的聚类结果。通过准确率和Jaccard指数对基于距离的算法性能进行了评估。填充颜色为绿色的箱形图为研究人员建议使用的距离度量。所有的距离都是根据相对丰度计算的。
Table1平均准确率(MA)和平均Jaccard指数(MJI)估计。粗体表示每个方案下的最佳情况。Log表示对输入的模拟数据进行了对数变换。
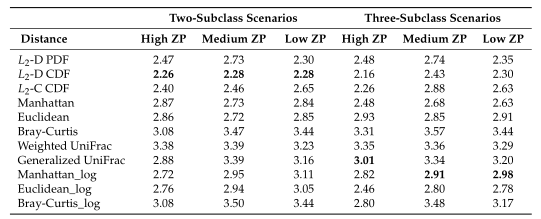
Table2所有模拟场景的平均集群数。根据Dunn内部指数计算出每次重复的最优聚类数。
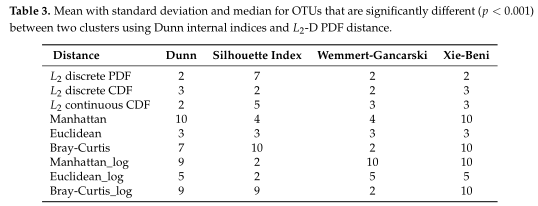
Table1 的结果是通过对每个场景中的100个重复结果求平均值计算得出的。观察得到在聚类算法中实现的距离度量(即绿色标识的箱形图)的准确率和Jaccard指数都优于其他距离度量,特别是在数据集中包含大量0时(高ZP)。在MA和MJI方面,L2范数也显示了其优势,在基于100次重复计算的L2范数在两个子类场景下的;平均准确率约为0.6秒,在三个子类场景下的平均准确率为0.45。而广义Unifrac距离(gUniFrac)在准确性估计中有很大的波动变化。
数据集为197名PD患者和130名对照的粪便样本的16S rRNA测序数据。首先过滤掉在80%的OTU中相对丰度都为0的OTU,因此,此次分析中共使用280个OTUs。将基于相对丰度计算的L2范数与其他三个距离度量进行了比较,包括对数变换和不变换的比较。
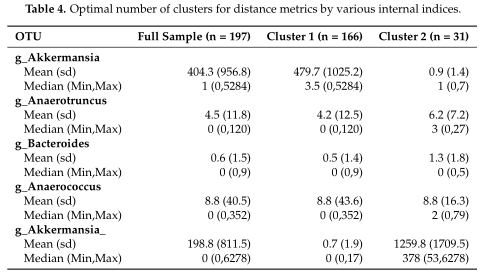
如Table3所示,距离度量采用各种内部验证指标(列名)进行灵敏度分析。对于Dunn和Xie-Beni指数,l2范数倾向于将数据聚类为两到三个亚群,而在有和没有对数变换的情况下,除了未变换的欧氏距离外,Manhattan、Euclidean和Bray-Curtis更倾向于聚类更多的亚群。(设置了最多聚类数目为10)
接着选择L2-D PDF范数作为进一步分析的例子。
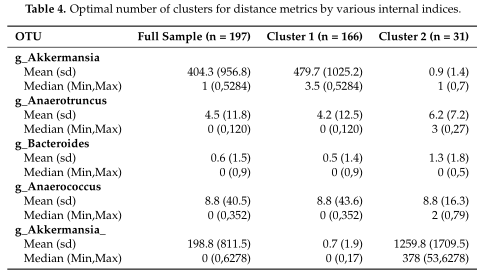
结果在Table4中展示,对数据集中的两个集群之间的OTUs进行了探索,得到了聚类之间差异最大的前5个OTU。
研究认为该聚类算法在高ZP和中等ZP的情况下表现最好,因此,当数据中出现大量的0时,建议使用。并且,在PAM框架下,文章中列出的那些距离度量都可以用作聚类的输入。
如模拟研究中显示的那样,在各种场景下,由混合模型计算的成对距离比其他距离度量表现的更好。但是与所有聚类方法一样,都有一个局限性,就是很难为每个新数据选择合适的内部指标,因此很难获得最优和最稳健的集群数。
此外,对于L2范数距离,在聚类中无法进行变量选择。但不可否认,该聚类算法结合了微生物测序数据的特殊距离,所介绍的聚类算法除了目前使用的方法之外,还可以被看作是分析微生物数据的一个很好的辅助工具。
研究人员指出,下一步会基于Dirichlet-Polyomial等模型进行分区,与文章中的方法进行比较,并努力将该方法扩展到其他微生物群落和疾病相关的数据上。
【参考文献】
Yang D, Xu W. Clustering on Human Microbiome Sequencing Data: A Distance-Based Unsupervised Learning Model. Microorganisms. 2020 Oct 20;8(10):E1612.

谷禾健康
宏观生态学中研究最多的模式是物种分度分布(SAD),它被定义为具有给定丰度的物种的比例。研究中虽然对SAD进行了高度的研究和表征,但往往忽略了三个不同而独立的变异源对其的影响:采样噪声、个体物种丰度的波动和物种间丰度的变化。最近,发表在《Nature Communications》的一篇文章使用来自许多不同环境的微生物群落的纵向和横截面数据从宏观生态学的角度研究微生物群落中物种的存在、丰度和多样性的模式。确定了三条普遍的,基本的宏观生态规律,这些规律定量地表征了单个物种在空间和时间上的丰度变化,以及不同物种之间典型丰度的差异。这三条宏观生态规律适用于整个微生物群落,也适用于横断面和剖面数据,因为它们足以在不拟合任何附加参数的情况下预测多样性的尺度和其他普遍研究的宏观生态模式,如SAD。利用这三条规律,人们可以预测物种的存在与否、群落的多样性和普遍研究的宏观生态模式。

背景
没有两个生态群落是相同的,因为物种组成和丰富度差异很大。环境波动、竞争、交叉喂养、环境改造、人口随机性、迁徙和许多其他生态力量在时间和空间上塑造着微生物群落。在数千种物种相互作用的群落中,要理清多种机制的作用是极其困难的。但最近的实验证明了几种推动体外多样性的生态机制的存在,并对其影响进行了量化。宏观生态学,即通过丰度、多样性和分布模式研究生态群落,是定量研究微生物群落的变异,并提供塑造微生物群落的机制的量化方法。
方法
数据集来自EBI宏基因组。只保留至少有50个样本的数据集,且这些样本的reads数大于10^4。为了研究(相对)丰度如何在群落和物种之间变化,需要消除采样噪声的影响,文章中通过建模抽样的方法进行。(由于方法均由数学公式推导得出,这里就不一一搬运了,有兴趣可以阅读原文,更为详细)
SLM模型
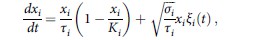
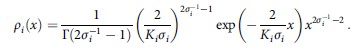
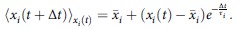
主要结果
1. 三个宏观生态学规律:丰度波动是伽马分布的;Taylor定律;平均丰度呈对数正态分布。
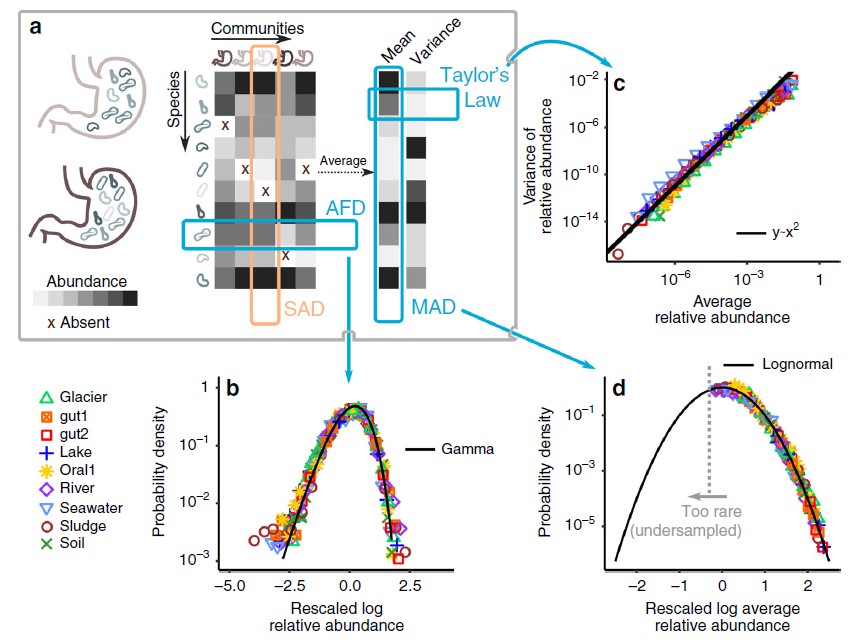
丰度波动分布(AFD),它被定义为物种丰度在群落之间的分布。这个数量受到抽样误差的强烈影响,特别是当一个物种因为波动而变得稀少的时候。对于最丰富的物种,这些抽样误差可以忽略不计。b图描述了物种丰度在群落间的分布。图例代表不同数据集。实心黑线为伽玛分布, 很好地描述了整个微生物群落中丰富物种的AFD。
Taylor定律,描述物种丰度的均值和方差之间的关系。丰度波动的均值和方差足以表征物种丰度在群落间的完全分布。Taylor定律适用于描述群落的组成,如c图。其中,方差尺度与均值成平方关系,这意味着丰度波动的变异系数是恒定的。因此,每个物种只需要一个参数,即它们的平均丰度来描述物种丰度的波动。
平均丰度分布(MAD),描述了平均丰度是如何在物种间分布的。d图表示MAD呈对数正态分布。由于在有限数量的样本中,稀有物种可能永远不会被采样,因此实证的MAD显示出一个由抽样确定的较低的截止值。通过对无限次重复取平均值,会发现,在中性模型中,MAD则呈Delta分布。
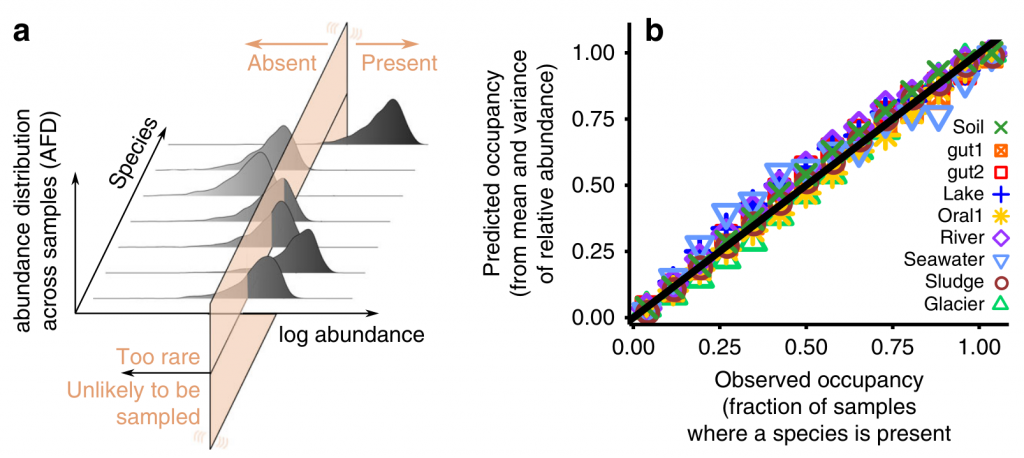

Gamma AFD规律表示所有物种缺失的情况都应该归因于抽样误差这一结果。a图展示了丰度波动与物种缺失之间的关系,通过丰度的波动来预测物种的存在与否。b图通过比较不同数据集中物种的占有率(一个物种在群落中所占的比例)与伽玛分布相对丰度的独立抽样所预测的结果,来检验这一预测。
2. 其他宏观生态模式的预测。
了解了这三个定律及其参数,并假设物种丰度波动是独立的,就可以为任意水平的抽样生成合成群落。通过比较它们的统计特性,将人造合成的群落与实证的群落进行对比。图中从a-d列举了四种宏观生态模式研究,结果表明这些宏观生态模式的预测与数据准确吻合。
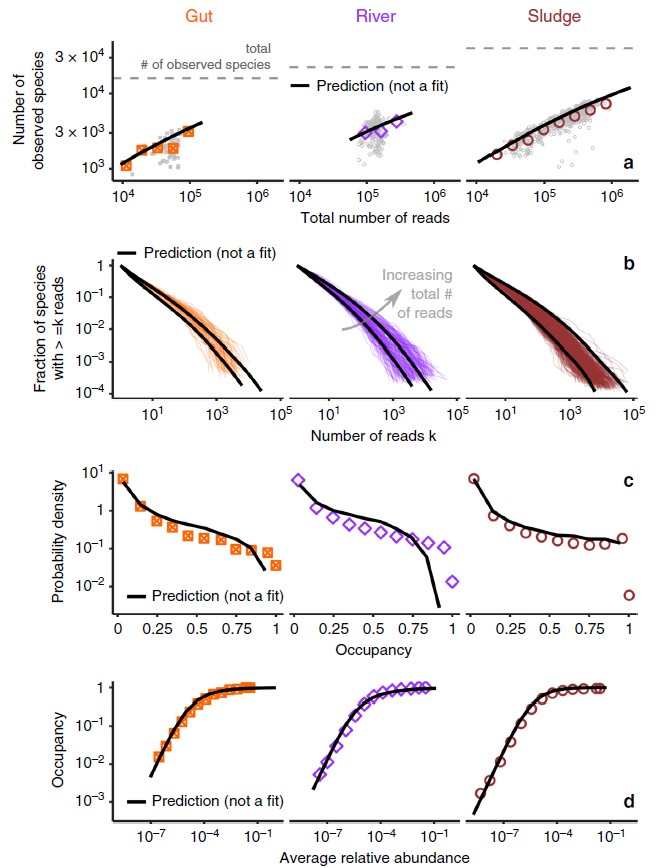
a. 多样性(以物种数量衡量)与读取总reads数的比例
b.物种丰度分布(SAD)。
c.占有率分布。
d.物种丰度与占有率间的关系。
不同颜色的图例代表不同数据集。(黑线)预测值是在不拟合任何附加参数的情况下
3. 宏观生态学规律适用于时态数据

a-c.基于纵向时态数据的群落中物种间丰度变化。结果表明这三条宏观生态规律也适用于纵向数据,而丰度的波动主要是由于时间的随机性。
4. 随机Logistic模型(SLM)再现了宏观生态规律
研究人员认为SLM是用来描述物种种群动态的。SLM假设种群数量呈逻辑增长,增长速率随时间的变化而变化,其波动速度快于平均增长率(即,与增长率波动相关的时间尺度比种群动态的典型时间尺度短得多)。
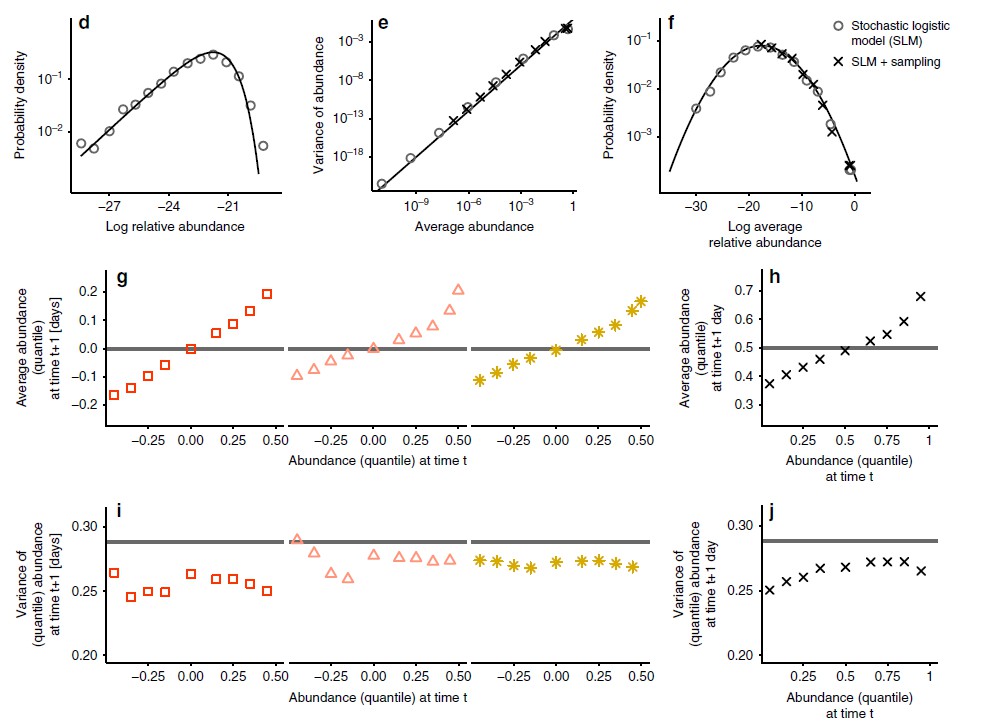
d-f. 随机Logistic模型(SLM)分别再现了三大宏观生态规律。灰色的圆圈是通过SLM得到的结果,黑色十字架是采用SLM和抽样得到的结果。
g.横轴描述了前一天的丰度的平均分位数,纵轴描述了当天的丰度的平均分位数。灰色实线表示在没有时间条件下的预期结果。
i.与g图类似,只是纵轴计算的是丰度的平均分位数的方差值。
h.j. SLM正确的预测了g和i图中展示的非平稳属性。
结论
宏观生态学的三个规律分别为丰度波动是伽马分布的;Taylor定律;平均丰度呈对数正态分布。SLM既描述了横截面数据中的平稳模式,又描述了时态(纵向)数据中的丰度动态。该模型指出,环境波动是微生物群落中物种存在和丰度变化的主要来源。Taylor定律和对数正态分布不是SLM能预测到的,但它们掣肘了SLM的参数化。微生物群落中SAD的对数正态分布只是明显的,并且是MAD对数正态分布的结果。在多个空间、时间和分类学尺度上对这些规律进行表征,将有助于理清和量化造成我们星球令人惊叹的(微生物)生物多样性的生态力。
TIPs:文章利用数学建模方面的知识验证了宏观生态学的三条规律,在验证的过程中点出环境波动是影响微生物群落中物种存在和丰度变化的主要来源。改文章扩宽了分析思维,在做宏观生态学研究时,不能只从物种丰度的变化得出整个群落的物种变化,在空间和时间上也要做分析。如果要直接使用文章中提出的三大规律做宏观生态学研究,目前主要问题是能否重现它的模型。

宏基因组测序方案是针对16s分辨率和宏基因组高成本之间的一个折中方案,通过降低测序深度,每个样本100万reads左右,但是物种的分辨率并没有一般宏基因组(普遍5~10G数据量)差很多。 不通过拼接组装,直接基于kraken2等kmer,或MetaPhlAn2等标记基因的参考基因组方法进行种属丰度分类。结合其到菌株的物种分类和丰度数据可较16s方案下的PICRUST更加准确的预测基因构成。


比较适合想要获得更全和更精细分类精度而同时不需要获得完整基因组序列和重建菌群基因的,浅宏基因组测序就可以成为很好的选择,其成本低,分析简便快速,同样能获得宏基因组的基本丰度数据。
不过浅宏基因组也有其适用范围,根据样品类型的不同,一些样品可能包含 >99%的人类宿主DNA,这不仅增加了序列成本,而且给测量带来了不确定性。
在许多研究中也会采取在进行宏基因组测序文库的准备之前去除宿主DNA的方法。但是,在去除宿主DNA后,可能没有足够的微生物基因组DNA用于宏基因组测序,这通常需要最少50ng的输入。
浅宏基因组较适合于宿主DNA含量较低的样本如:人类粪便、水体、土壤等;
不太适合:口腔唾液、肺泡灌洗液、血液等人体体液类样本。
我们可以免费提供针对粪便及环境样本助力临床/科研取样。
人体口腔、痰液、腹水、脑脊液、尿液、皮肤、阴道分泌物等高寄主细胞含量样本可根据我们的处理方案简单处理后大幅降低宿主DNA比例。
测序平台:Illumina Novaseq,PE150,默认:100万reads/样
2-3周左右出报告


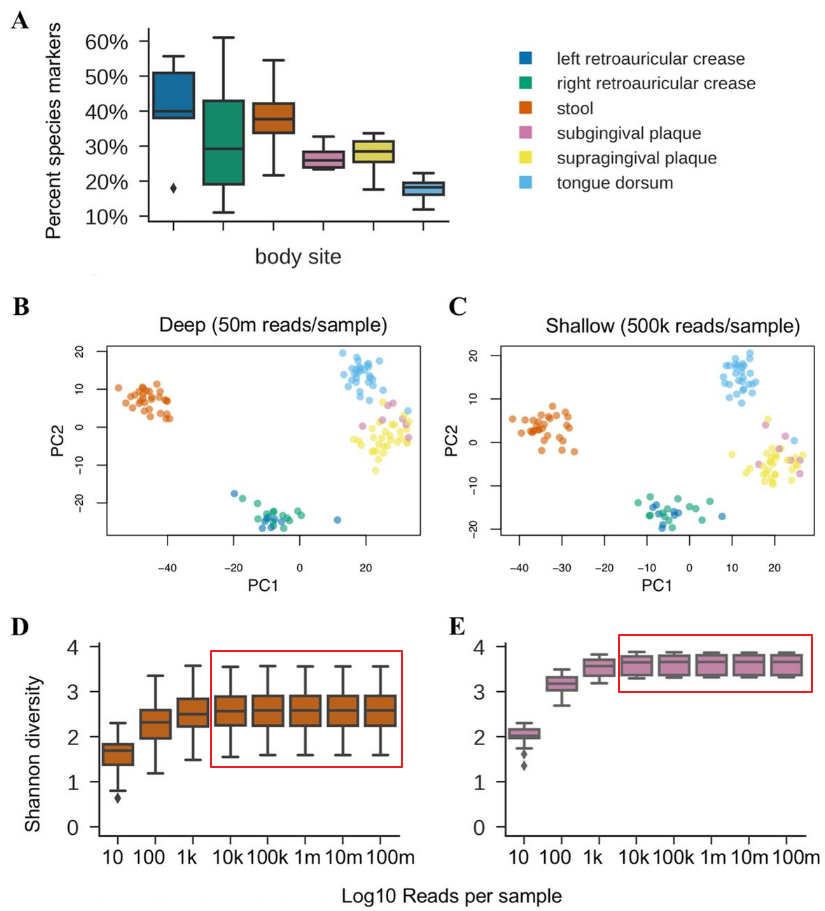
当测序深度达到50万reads以上,浅宏基因组与深度测序宏基因组在主要物种构成的丰度已经基本一致。如下图所示:

样本需求量低:公司研发实现了低当量微生物样本提取和建库,保证提取丰度以及片段完整性同时,样本量需求低于同行其他公司要求;对于样本获取困难的样本,也可以选择微量建库,样本量可低至10ng。
免费取样盒和针对性取样建议:粪便及环境样本提供取样盒助力临床/科研取样,人体口腔、痰液、腹水、脑脊液、尿液、皮肤等高寄主细胞含量样本可根据我们的处理方案简单处理后大幅降低宿主DNA比例。
严格标准的实验流程:自动化样品处理平台辅助,每轮设置阳性对照,上轮检测样本对照,阴性对照。评估污染,轮次比对,最大化减少误差,保证样本重复性和稳定性
Illumina测序平台:宏基因组测序(PE150)采用先进的Illumina Novaseq测序平台,快速、高效地读取高质量的测序数据、结合样品特点和数据的产出,充分挖掘环境样品中的微生物菌群和功能基因
大数据分析流程质量流程控制严格:优化的数据质量控制,包括过滤比对质量低、非特异性扩增、覆盖度低、低复杂度的序列,从而快速准确获得样本中微生物信息及其丰度信息,最大化提高质量数据
分析内容丰富全面:物种分析,基因预测与分析,多样性和相似性分析,功能分析,网路互作分析,代谢网络,关联分析等
完整详细的报告:提供质检实验报告,分析统计报告,分析报告解读,原始数据
高效个性化服务:在线项目系统方便您及时查看项目动态和下载报告以及与分析人员高效交流,免费支持个性化图表修改以及重新分组出报告。
价格低,周期快:包括提取,测序到分析,最快2周出报告。
大数据分析团队和多中心大项目分析经验(团队主要源自浙江大学,包括生物信息学,计算机,微生物以及统计分析等专业,积累了多年的大健康项目多中心项目分析经验,有助于多样本,多表型,多组学联合分析
若需要进一步了解咨询,可以致电联系我们

原创 谷禾健康


近年的研究热点集中于环境和生物体相互作用的微生物群体,而大量复杂的微生物群体存在培养困难,构成复杂(包括细菌、古菌、真菌、原生生物、病毒甚至小型真核生物)。因此如何用高通量精准的了解这些群体的构成,基因功能分布以及具体的表达活性和代谢状况成为首要问题。
高通量测序技术的发展,让我们可以不经过培养,一次性了解微生物群落构成甚至基因代谢组成。
随着技术的进步,检测方法也逐渐丰富,对应的分析手段和软件算法也逐步完善,使我们可以根据研究需要选择不同的检测和分析策略来获得海量的数据并进行相应的研究分析。


免于培养的微生物学研究方法主要基于测序,高通量测序使我们一次可以获得整个微生物群体的数据信息,简单来说包括两种策略:
1、基于特定标记基因的扩增测序方案(常见的16s,ITs,18s或特定功能基因)
2、对整个群落DNA进行测序,获取全部微生物基因组进而进行分类和功能分析的策略(鸟枪法宏基因组测序shotgun metagenomics)。
基于16s基因的分析方法
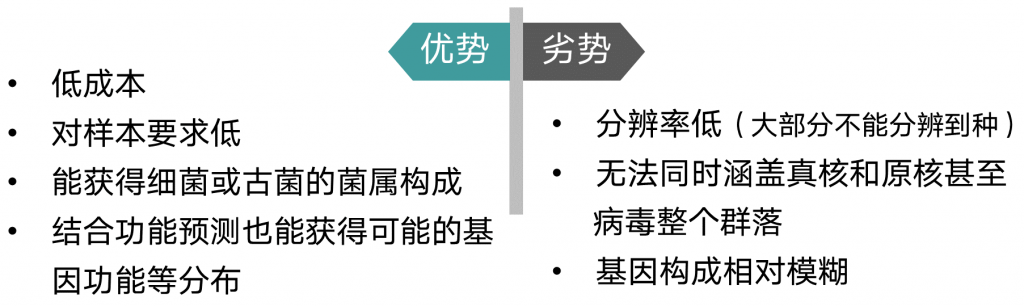

由于其极低的成本,对于样本DNA的低要求非常适合于大规模群体样本的调查和分析,随着DADA2等分析方法的改进,物种分类精度和准确度也有所提升,加上PICRUST等功能预测方法一定程度上弥补了基因信息的缺失,因此16s这类基于基因的微生物研究方法仍然是不可或缺的方案。
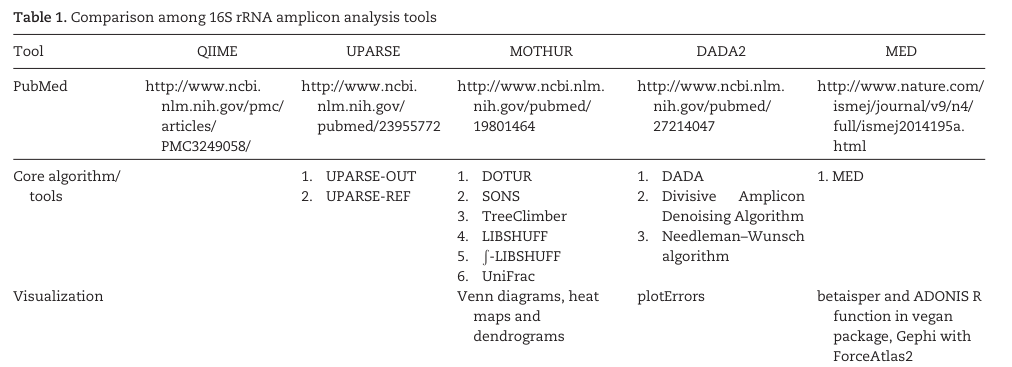
下表列了16s常见的分析软件,目前QIIME2作为整合包使用最为方便,VSEARCH也作为UPARSE的开源版本使用也非常广泛。
16s测序的分析流程如下图,获得序列经过聚类后获得OTU或ASV,并得到相对丰度。
经过PICRUSt可以得到预测的基因分类丰度,进而进行alpha多样性和Beta多样性以及组间差异和相关性分析。
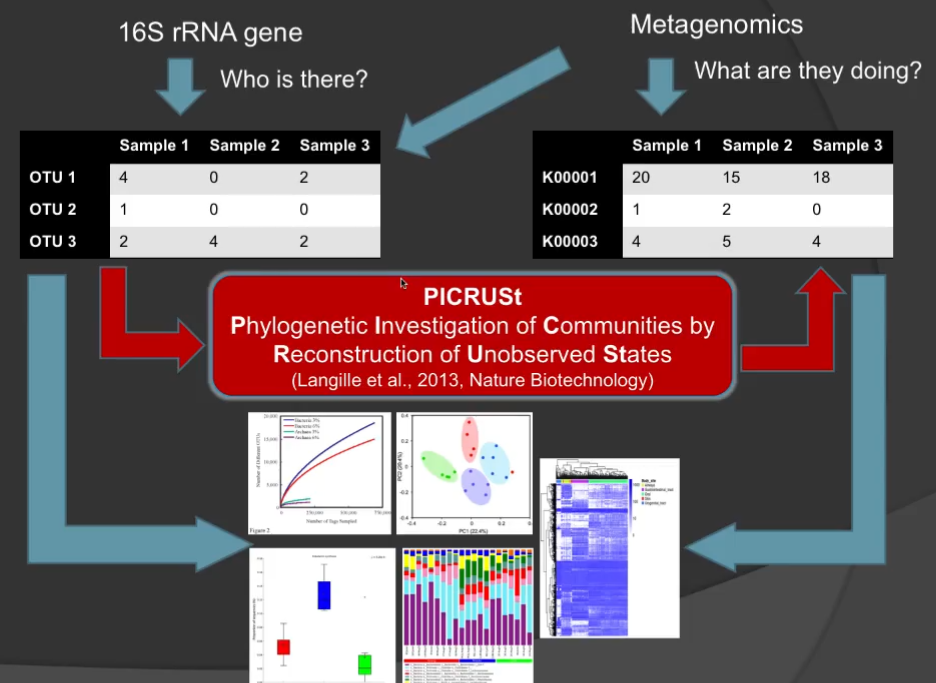
PICRSt的工作原理如下图,将OTU表内16s序列进行对应物种16s拷贝数标准化后,将物种丰度乘以已经整理好的物种的基因注释数表就获得基因的预测丰度。
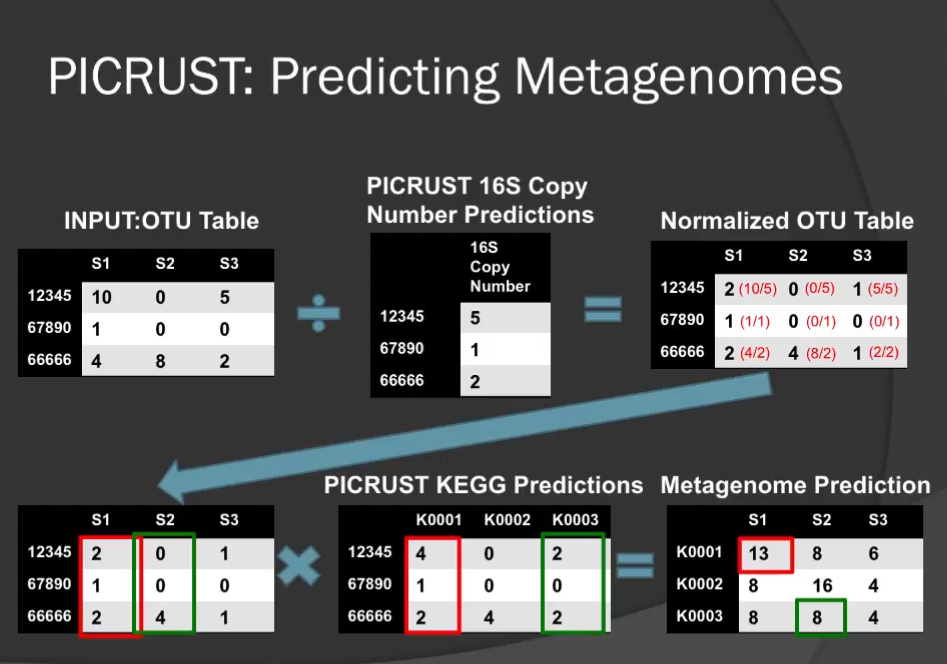
浅宏基因组测序方案是去年knights-lab在msystems上发表的针对16s分辨率和宏基因组高成本之间的一个折中方案,通过降低测序深度,每个样本50万reads,但是物种的分辨率并没有低于一般宏基因组(普遍5~10G数据量)。
不通过拼接组装,直接基于kraken2等kmer,或MetaPhlAn2等标记基因的参考基因组方法进行种属丰度分类。结合其到菌株的物种分类和丰度数据可较16s方案下的PICRUST更加准确的预测基因构成。

Hillmann B, Al-Ghalith GA, Shields-Cutler RR, Zhu Q, Gohl DM, Beckman KB, Knight R, Knights D. 2018. Evaluating the information content of shallow shotgun metagenomics. mSystems 3:e00069-18. https://doi.org/10.1128/mSystems.00069-18.
我们发现有些小伙伴的需求是:
想要获得更全和更精细分类精度同时不需要获得完整基因组序列和重建菌群基因的。
那么这时候,我们提供的浅宏基因组测序就可以成为很好的选择,其成本低(快要接近16s测序分析的价格了,文末有福利),分析简便快速,同样能获得宏基因组的基本丰度数据。不过浅宏基因组也有其适用范围,根据样品类型的不同,一些样品可能包含 >99%的人类宿主DNA,这不仅增加了序列成本,而且给测量带来了不确定性。
在许多研究中也会采取在进行宏基因组测序文库的准备之前去除宿主DNA的方法。但是,在去除宿主DNA后,可能没有足够的微生物基因组DNA用于宏基因组测序,这通常需要最少50ng的输入。因此浅宏基因组较适合于宿主DNA含量较低的样本,如人类粪便、水体、土壤等;而如口腔唾液、肺泡灌洗液、血液等人体体液类样本就不太适合。
下图是宏基因组测序数据中比对到人类基因组的序列比例,根据样本类型不同而不同。

我们可以免费提供针对粪便及环境样本助力临床/科研取样。
人体口腔、痰液、腹水、脑脊液、尿液、皮肤、阴道分泌物等高寄主细胞含量样本可根据我们的处理方案简单处理后大幅降低宿主DNA比例。
高宿主含量DNA样本(包括唾液、血液、肺泡灌洗液、腹水、阴道分泌物和黏膜类样品)的取样前处
将200微升唾液等体液样本以10,000g离心8分钟
弃去上清液,通过移液将细胞沉淀重悬于200μl无菌水中,短暂涡旋,然后在室温下静置5分钟,以渗透压裂解哺乳动物细胞
添加终浓度为10μm的PMA(叠氮溴化丙锭)(向200μl样品中添加10μl的0.2 mM PMA溶液),并将样品短暂涡旋,然后在黑暗中于室温温育5分钟
然后将样品从标准台式荧光灯放置在<20cm的冰上水平放置25分钟,短暂离心并每5分钟旋转一次
完成后,可将样品冷冻在−20°C或转移到取样管的储存液中
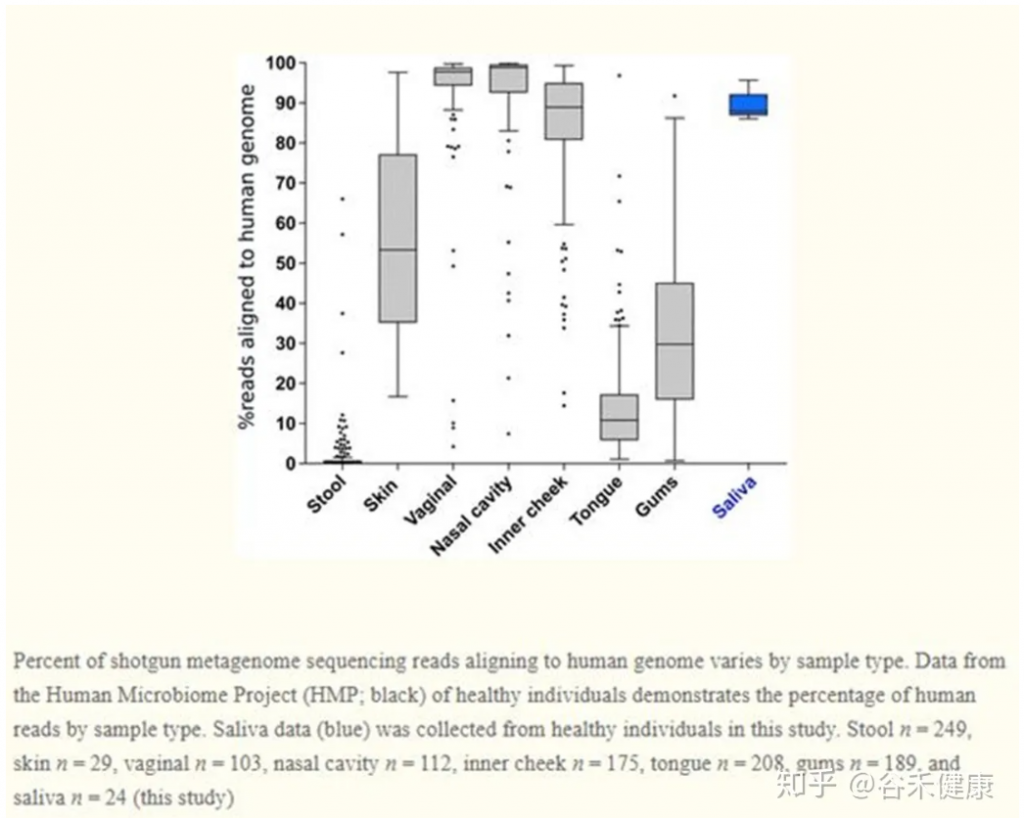
Marotz CA, Sanders JG, Zuniga C, Zaramela LS, Knight R, Zengler K. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome. 2018;6(1):42. Published 2018 Feb 27. doi:10.1186/s40168-018-0426-3
本处理方案以后宿主DNA可以降低8%以下。
说起宏基因组,对于熟悉宏基因组或者打算做宏基因组的同学可能已经迫不及待想知道这个怎么分析啊,怎么看结果啊之类的问题… 但在这之前,首先你应该了解的是宏基因组是什么,做宏基因组你能得到什么。


此外,对于缺乏深度研究和高质量参考基因组的样本,如土壤和特殊环境下的样本,宏基因组获得的较为完整的基因组不仅可以丰富参考基因组数据库,同时可以提供更加准确的物种分类。
因此,深度宏基因组测序是解析新环境样本的核心方法,不过从单一样本中重建出完整的菌株基因组有相当困难,一般需要较多样本或设置梯度样本从而利用更高深度和共同变化来获取分箱信息,当然对应测序和分析成本会更高。
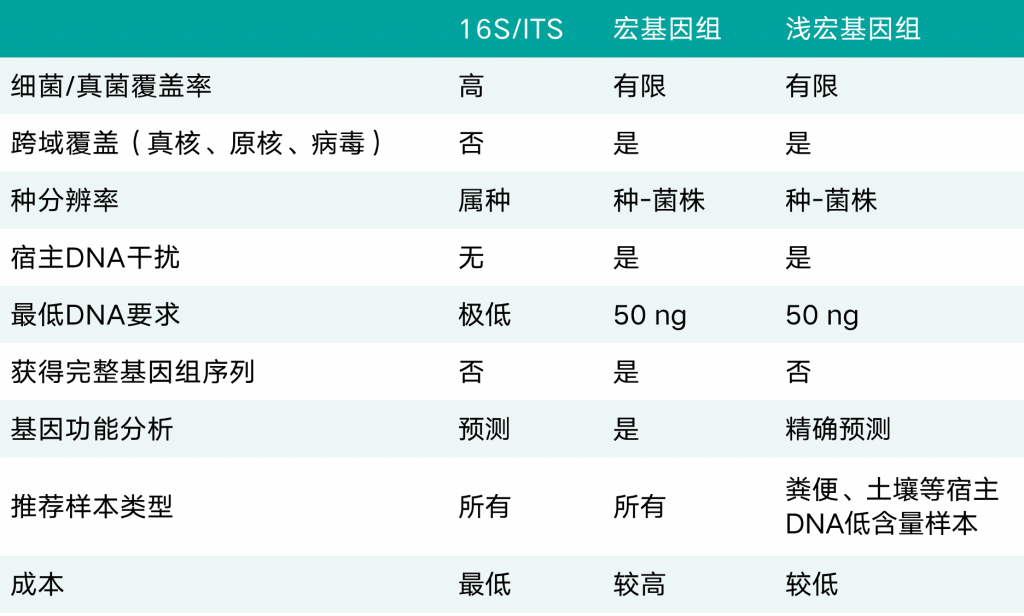
至此,我们了解了16s、浅宏基因组、宏基因组三种方式,我们将它们各自的特点总结如下表,便于你更直观地去了解(文末有福利~)。

宏基因组报告中有哪些分析内容?
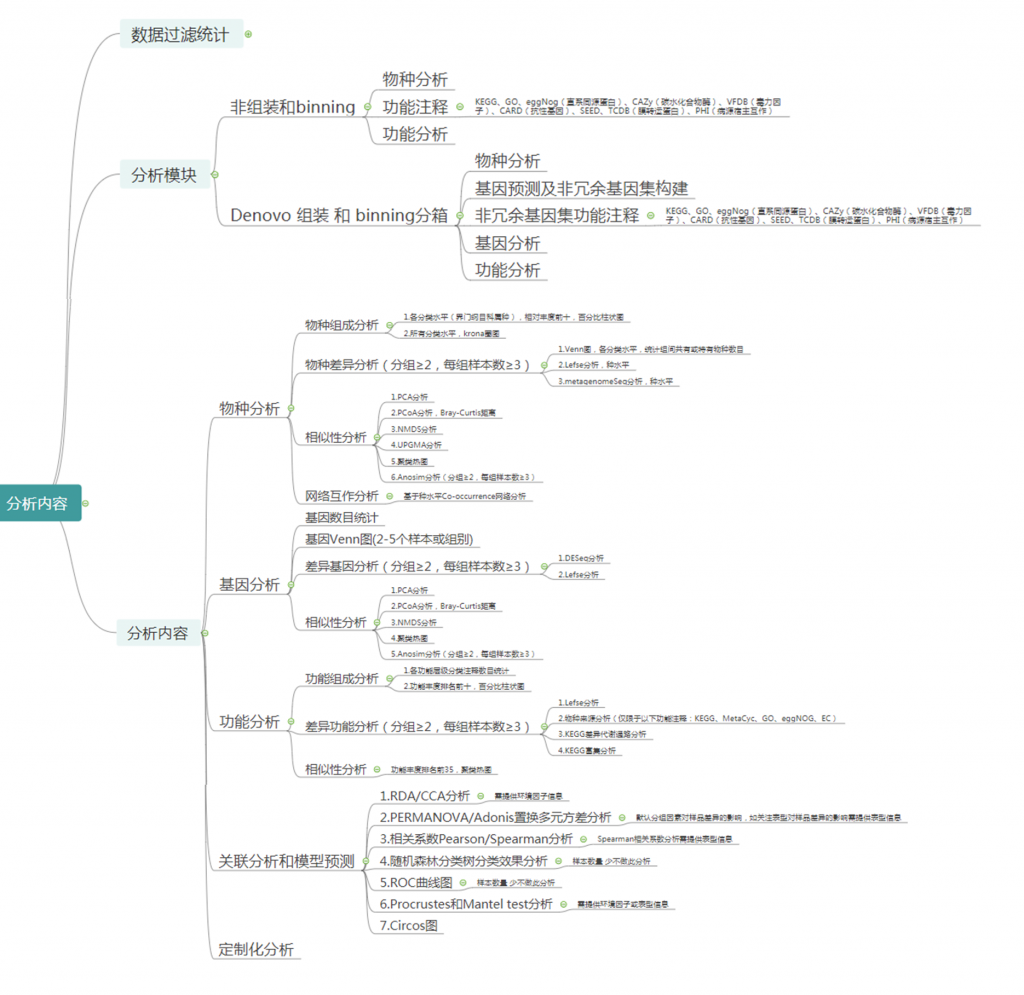

上图可以快速预览一下我们报告中的分析内容。
接下来,我们会详细介绍这些内容是如何从原始数据开始一步步实现的,同时也会选取一些文章案例来给大家做详细解读,希望给大家带来一些思路。
测序数据需要经过质检,去除接头和低质量序列,一般还会进行一步过滤人的基因组序列,然后分为两个路径,使用参考数据的比对方法和从头组装的方法,下图是一个完整的宏基因组分析流程:

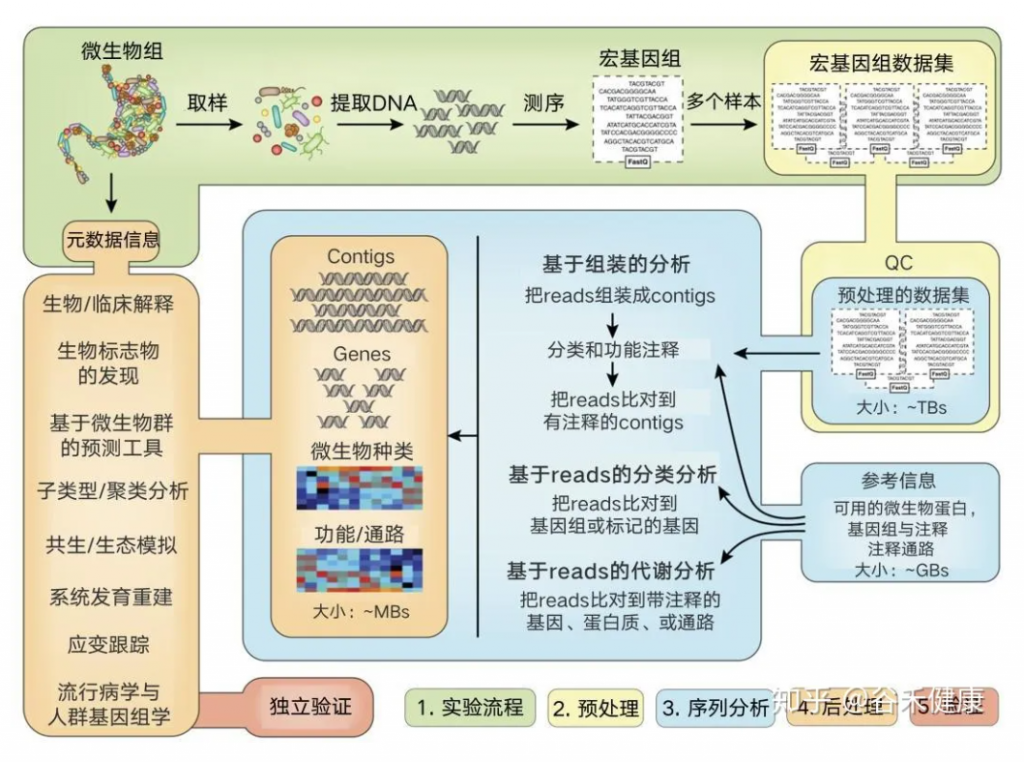
看完上图,可以对宏基因组测序的基本流程有个大致了解。
对于宏基因组测序而言,最重要的就是获得微生物群准确的物种构成及其丰度。
首先你需要了解的是无论16S测序还是宏基因组测序获得的均是相对丰度,即每种菌占所有菌属的比例。
要获得绝对的丰度需要在取样时做好取样量的计量,并在提取和建库中加入已知绝对量的参照DNA。
宏基因组测序获得物种构成及其丰度有以下两条路可以走:

我们先讲其中之一: 直接比对 。
直接比对是基于参考数据的,那么基于参考数据的物种构成分析主要有两类方法:
一类是基于Kmer和LCA比对特征来分析对应物种丰度,如kraken2等。
另一类是基于特征标记基因进行分析的,如MetaPhlAn2等。
基于参考基因组的分析工具如下表:
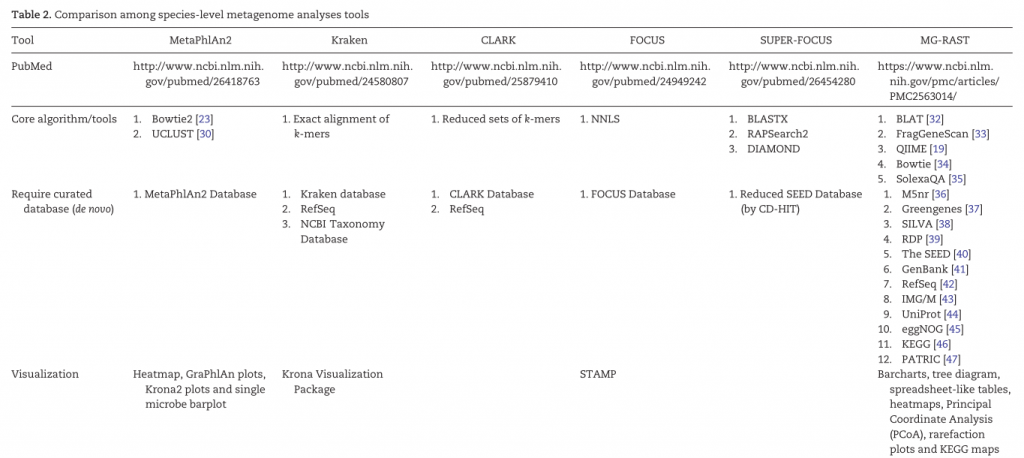

除了上面表中列出来的,另外还有
Centrifuge:比kraken2慢2x,内存使用少很多
Sourmash:类似CLARK,可以使用整个refseq作为数据库。
主流的kraken2——快速、准确度高、内存要求高
目前主要使用kraken2为主,因为快速,准确度也相当不错。不过,对于内存的要求较高,另外受数据库本身质量影响较大,默认kraken2的参考数据库只包括了细菌、古菌、病毒和人,还需要添加其他域的参考基因组。但涵盖的测序参考种仍然有限,对于菌株水平的鉴定受一定影响。后续使用Bracken可以针对kraken2的比对结果进行计算相对丰度。
MetaPhlAn2——物种跨度大、实用
MetaPhlAn2首先从全基因组数据库中找出clade-specific marker genes,然后利用这个marker genes的数据库对高通量测序得到的shotgun序列进行注释,目前主要用于后面直接使用reads获得基因和代谢通路丰度的HUMANn2的流程中,其物种跨度较大,速度也可以接受。
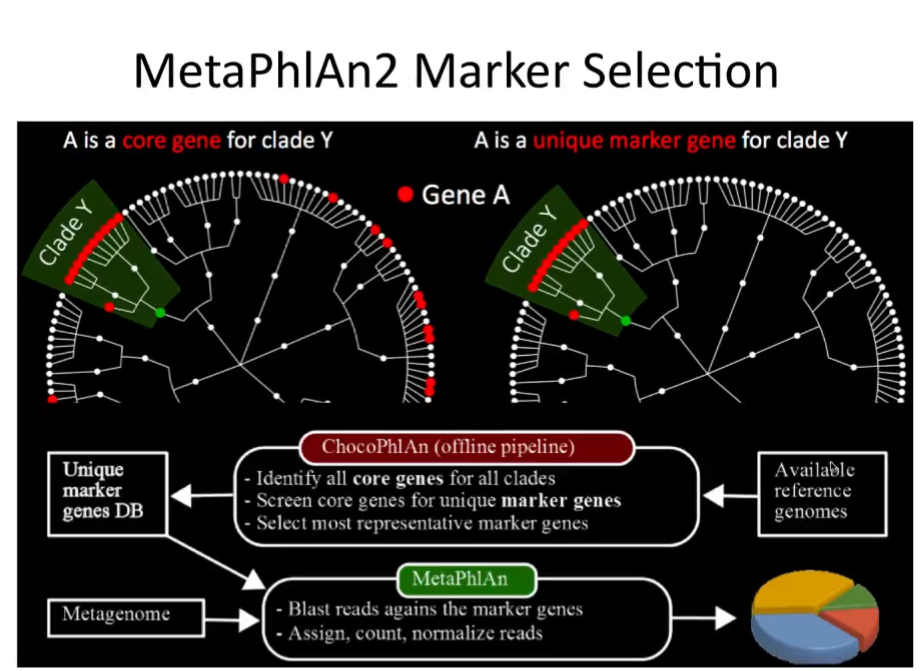

以上我们了解了直接使用reads获得丰度。
如果有足够测序深度和样本数量还可以通过组装出参考基因组来鉴定获得。该部分我们在下面的组装和分箱流程部分详细讲。
接下来,看一下我们报告中获得的结果和图:
使用Kraken2对其中的微生物进行物种注释。我们的Kraken2使用的数据库是由Refseq(2020.04.20)细菌,古细菌、真菌、原生动物和病毒库以及GRCh38人类基因组构建的。
通过查询数据库序列中的每个k-mer,然后使用所得的LCA分类单元集确定序列的适当标签,对序列进行分类。数据库中没有k-mers的序列不会被Kraken2分类。这里我们是在使用k-mer=35的条件下进行物种注释。
使用Bracken对物种注释结果计算相对丰度。Bracken是一种高度精确的统计方法,可从宏基因组学样本计算DNA序列中物种的丰度。Braken使用Kraken2分配的分类标签来估计源自样本中每种物种的读数数量。
对物种注释结果使用 KRONA 进行可视化展示。
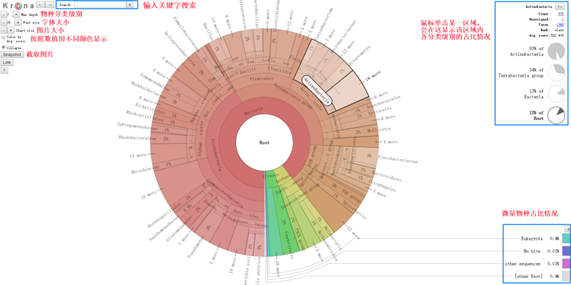

注:圆圈从内到外依次代表不同的分类级别(界门纲目科属种),扇形的大小代表不同注释结果的相对比例。
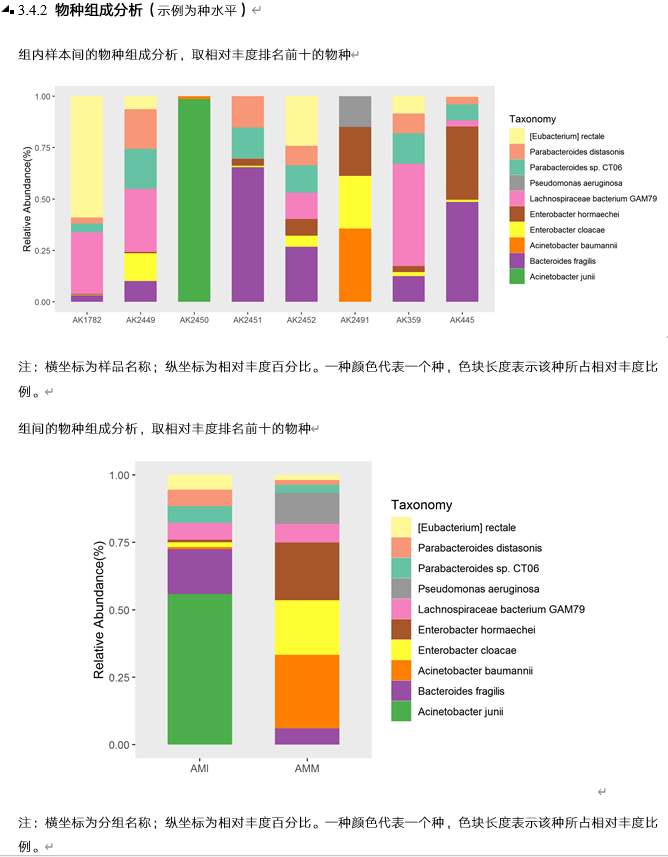
上面的是使用KRONA对单个样本的构成图形化,所有样本合并使用柱状图就可以了解具体的样本构成丰度,从门-纲-目-科-属-种-甚至菌株每个层次都可以进行显示(下面是截取我们报告中的相关图)。

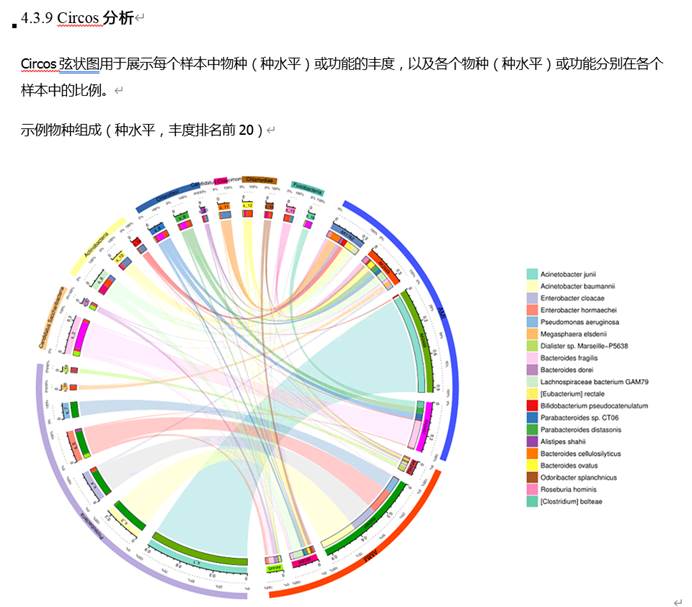
如果嫌柱状图的展示方式单一,当然也可以有别的选择。比如说以Circos的环图形式展现:

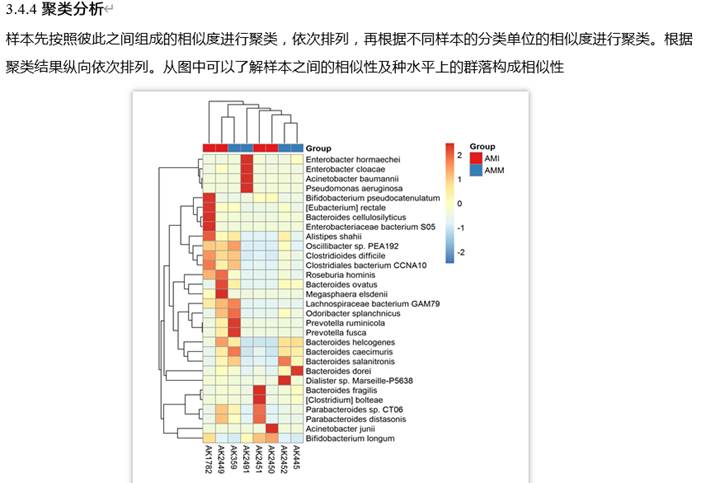
也可以进行聚类分析:

有了这些数据我们就可以进行alpha多样性(指每个样本内部菌群多样性)的分析了。
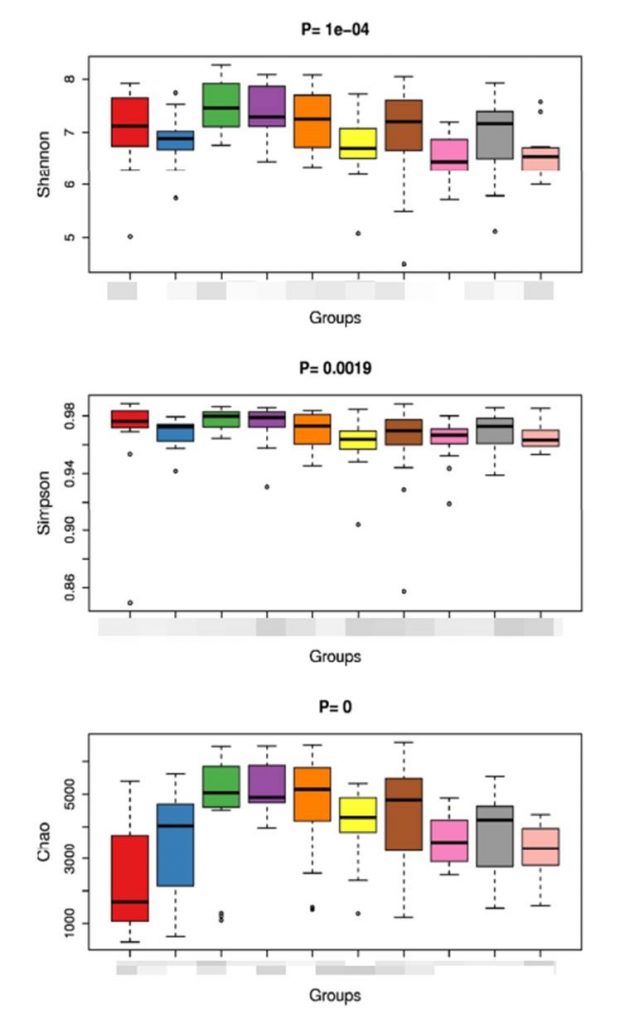

各样本和多组之间也可以进行Beta多样性的比较分析:
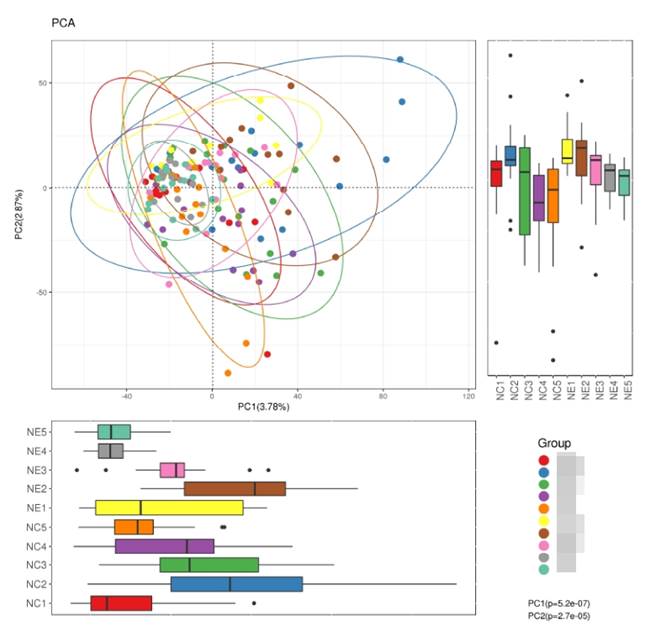

计算样本之间的菌属构成相似度:
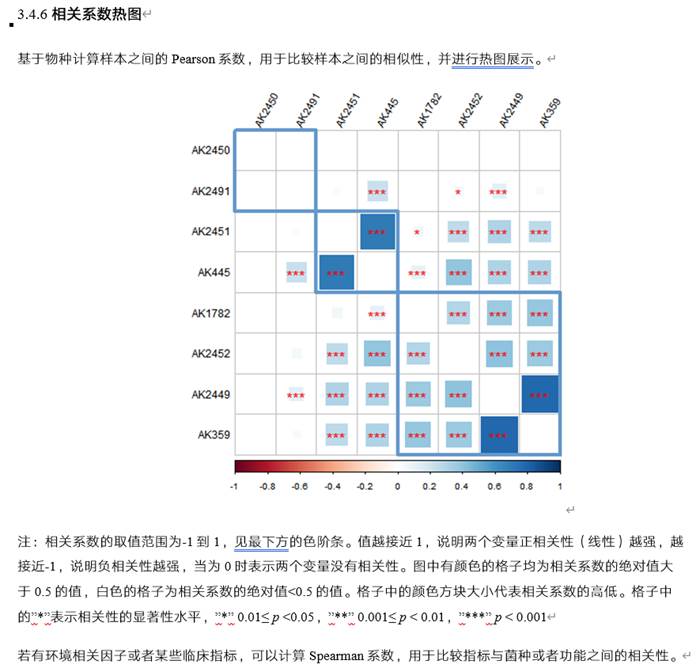

组间的差异分析:寻找差异或代表性菌属,如下:
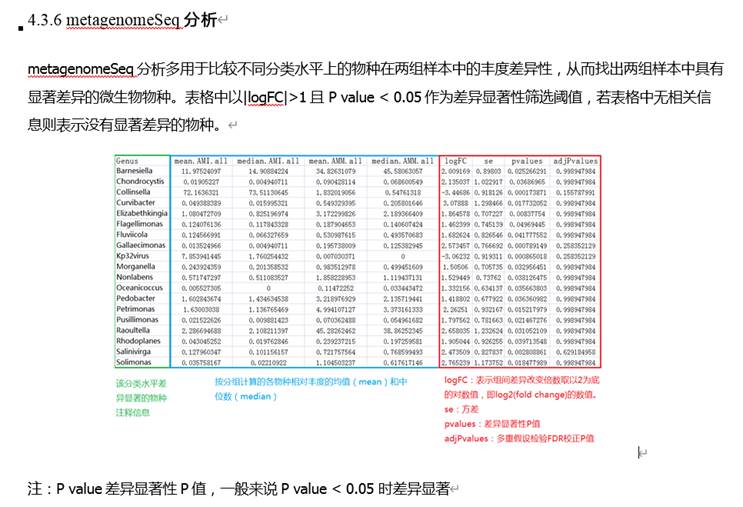

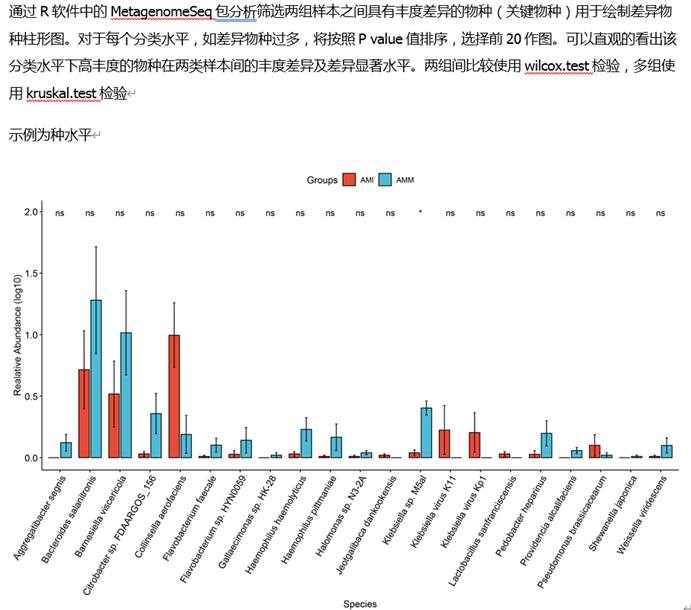

Trukey多组间检验
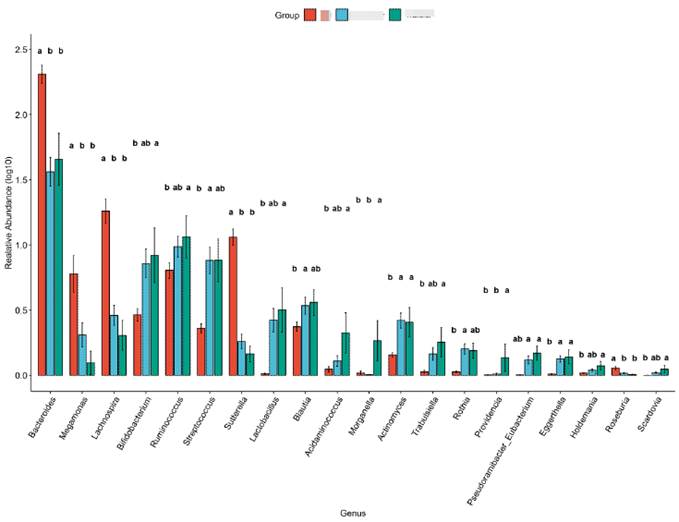

LefSe分析
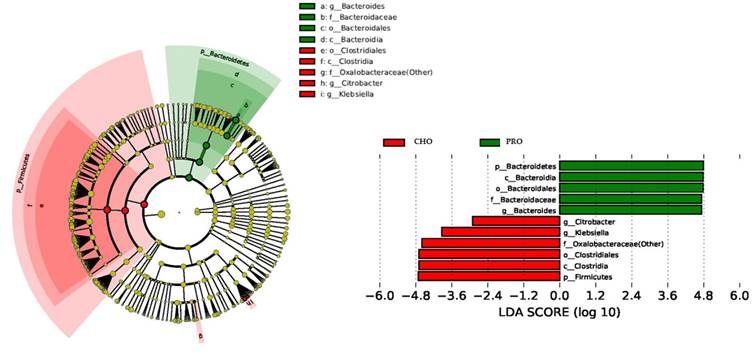

其中LEfSe基于线性判别分析(Linear discriminant analysis,LDA)的分析方法,筛选组与组之间生物标记物Biomarker(基因、通路和分类单元等),即组间差异显著物种或基因。当分组较多时较难获得每个分组独特的Biomarker。
以上是关于物种组成部分,但是有些小伙伴会有这样一些疑惑:物种构成变化很大怎么办?个体差异也很大?之类的诸多疑问。
是的,微生物群落一般对应特定的环境,其物种构成有时候变化迅速,而且个体或不同地点的构成差异极大。如人体的肠道菌群,个体之间的菌群构成差异很大,仅少量核心菌在绝大部分人的肠道内出现,个体特异性菌株也非常常见。那么如此多样性和复杂的构成如何应对相似的环境呢?
研究显示不同的菌属可能有着相似的基因或代谢能力,差异极大的种属在基因功能层面可能有着相似的构成。因此,获得微生物群的基因和功能代谢构成及分布对于解释和了解微生物如何响应和适应环境就尤为重要。
下图可以帮你更好地理解上面这段话。从图中我们可以看到,舌背样本和粪便样本虽然在种属上有很大差异,但它们在基因功能层面却有着相似的构成。
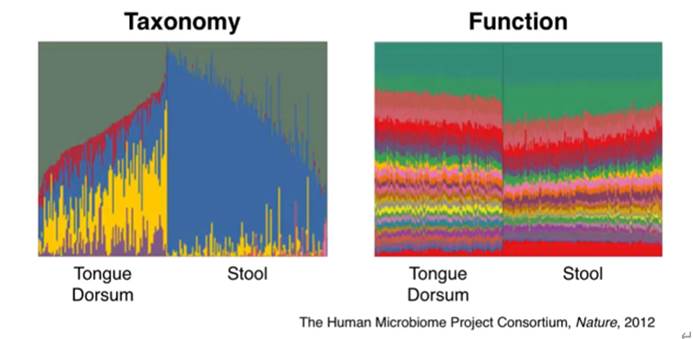

与物种构成丰度的分析类似,基因功能构成分析也同样可以包括两种方法:
方法一、通过直接基于reads的参考数据库方法获得
方法二、通过组装后预测注释基因并得到丰度
在具体展开方法之前,我们需要先了解关于基因功能的基本概念。
基因功能
每个菌的基因组中都包含大量的编码基因(ORF)以及非编码的RNA。这些基因之间又存在同源或序列相似性,达到一定相似程度的称为同源基因(一般通过CD-hit聚类为unigene,gltA这类基因名称,而数据库中一般聚类为如uniref90,eggNOG_ortholog等不同相似度的非冗余基因),这些同源基因除了序列相似同样也有着相似的功能,基于其功能或具备的蛋白功能域可以进一步分类为基因家族(Pfam),酶(EC 1.4.1.13),代谢通路(ko:K00266),更进一步层层分类为GO或顶层代谢通路Metacyc或COG等。
我们先来看方法一,具体是如何操作的?
主流的HUMAnN2——获得基因和代谢通路丰度的同时可直接进行下游分析
基于测序原始序列直接获得基因构成丰度的软件目前最主要的是HUMAnN2,其首先使用MetaPhlAn2进行物种分类(关于这个软件我们在前面物种组成部分已经讲过),并提取相应物种参考基因组用于比对,未比对上的用于进一步和uniref数据库进行蛋白质序列比对。原理见下图:
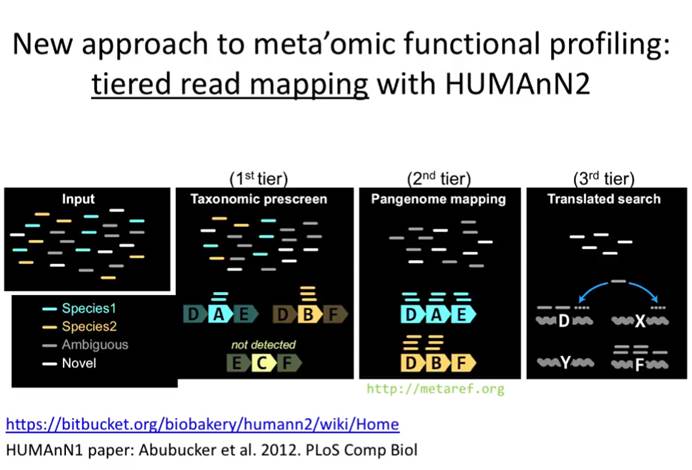

HUMAnN2的便利之处在于获得基因和代谢通路丰度的同时可以直接进行下游分析,将导出的表用于如LEFSE等差异分析,此外还可以反向给出不同样本中每个基因或代谢通路里的物种贡献。
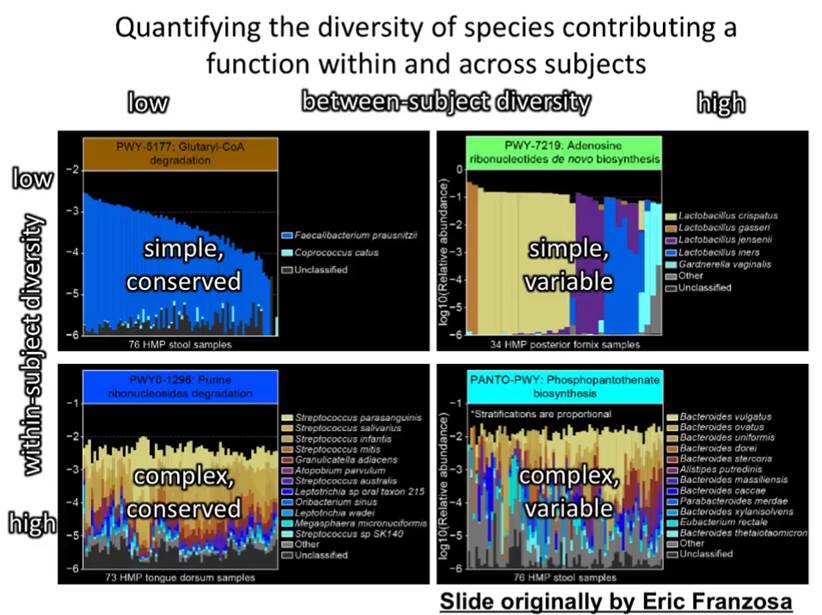
下图是基于HUMAnN2的不同代谢通路的菌贡献比例图:

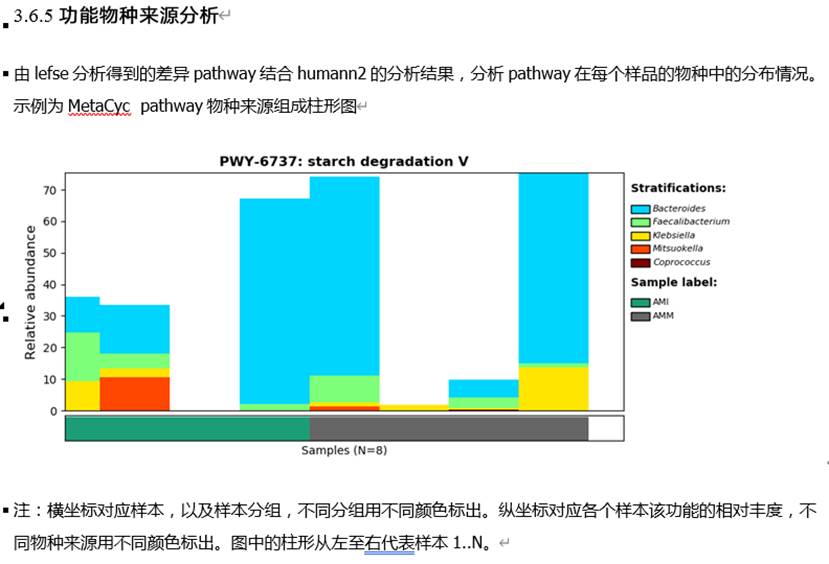
在我们的宏基因组报告中获得的是这样的:

而另外一种方法是通过组装获得,我们在前面物种构成小节也已经提到过组装分析,那么这里我们就组装拼接分析这部分展开讲解一下。
什么样的条件下可以进行组装分析?
当测序深度足够的情况下,目前illumina二代和Pacbio以及Nanopore等长片段测序技术已经足以组装出高质量的细菌基因组草图,结合Binning方法可以一次性获得大量物种的高质量接近完成基因组。此外还有Hi-C等手段可以进一步完成基因组以及对应质粒的完整拼接。
组装的流程是什么样的?
来看一下整个基于组装的流程:
① 提取、测序
首先从样本中提取基因组DNA,进行测序,可以使用Illumina的段片段深度测序也可以辅助三代长片段测序。
② 获得contig序列
接着对序列经过质检过滤处理后直接使用序列进行拼接,获得contig序列,这时通常每个菌的基因组会有几十到数千个contig片段,由于构成复杂,很多近缘菌之间的基因组存在大量相似序列,以及每种菌丰度都不一致,所以contig阶段的片段仍然较多。
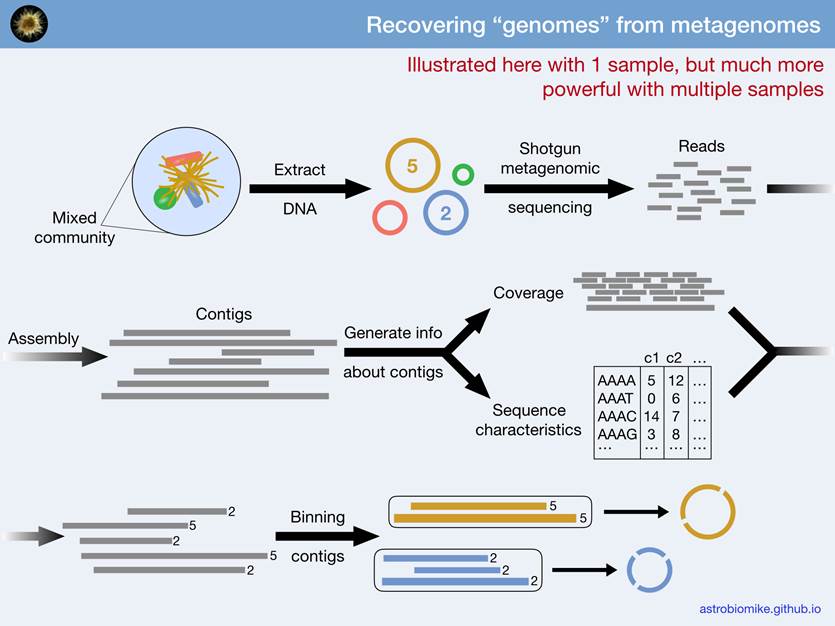

③ Binning分析
基于序列构成特征如GC含量、核苷酸多态性、覆盖度以及基因的物种相似度等多种数据,如果有多个样本或梯度可以同时结合样本丰度变化来进行分箱也就是Binning分析,将具有相同特征和变化的contig聚类归为同一个来源的箱,每个bins通常来自单一菌也就是一个菌株的基因组(我们的数据分析中包含这部分分析内容)。
④ 进一步质检评估
之后会进行进一步的质检,如checkM等评估每个Bin的完整度(核心基因以及rRNA等的完整性)和污染比例(如错误拼接,不同物种来源等)。一般要求50%以上的完整度以及10%以下的污染,当然样本数量越多,测序深度越高,测序读长越长理论上binning的质量也会更好,能获得更多高质量的单一菌完整基因组。
借用一张分箱的说明PPT:
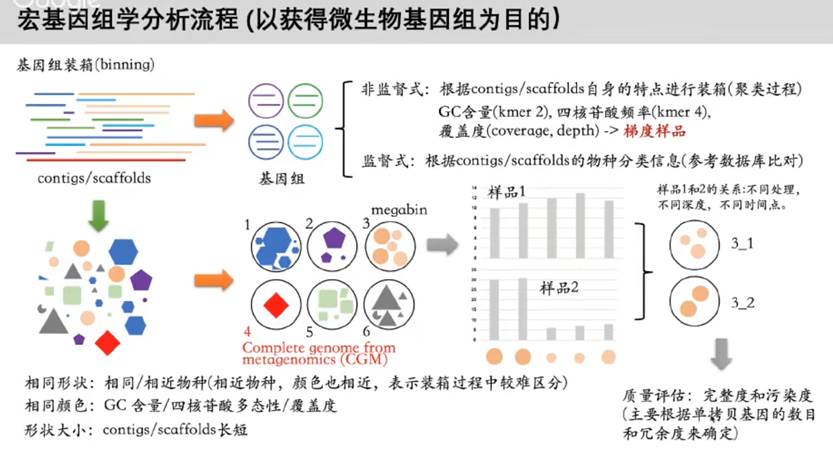

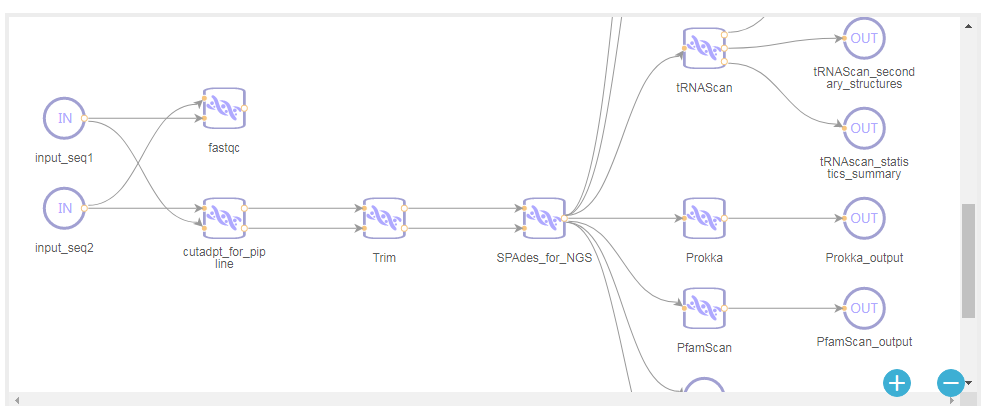
目前组装contig方面比较好的软件主要是SPAdes和MegaHIT。分箱方面MetaWRAP流程可以将整个组装和分箱优化全部完成,包括前期质检到组装以及使用三种分箱方法concoct, metabat2和maxbin2,并最终进行合并提纯优化,输出最终的分箱。
同时还可以对每个分箱bins进行物种鉴定和定量,这样我们就可以获得基于拼接组装后的物种丰度构成表,开展上述的物种多样性和样本差异统计分析。
⑤ 注释
最后使用PROKKA进行基因预测,获得的编码序列我们经过进一步CD-Hit聚类去冗余,然后使用eggNOG-mapper对其进行进一步的功能比对注释。使用salmon完成基因的定量,这样我们就得到基于组装注释的基因丰度数据了。之后就可以进行基因和功能层面的多样性、构成以及样本和组间差异分析。
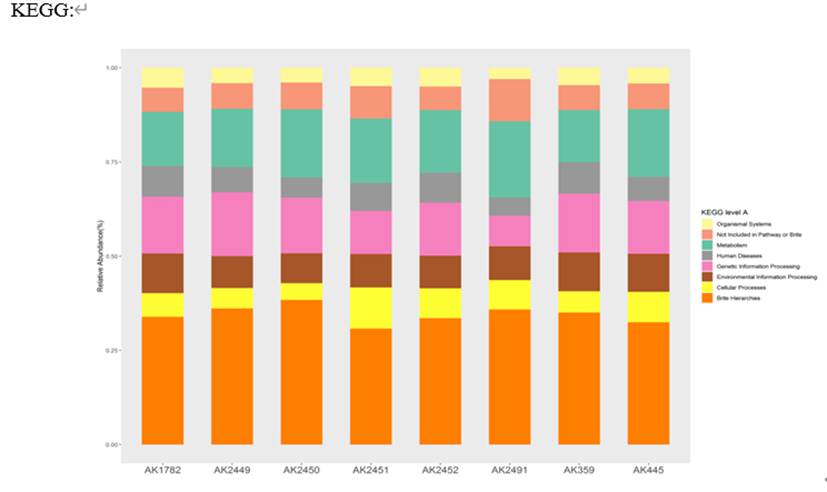
我们获得的最基础的uniref,eggNOG,KEGG和GO等注释如下:
KEGG

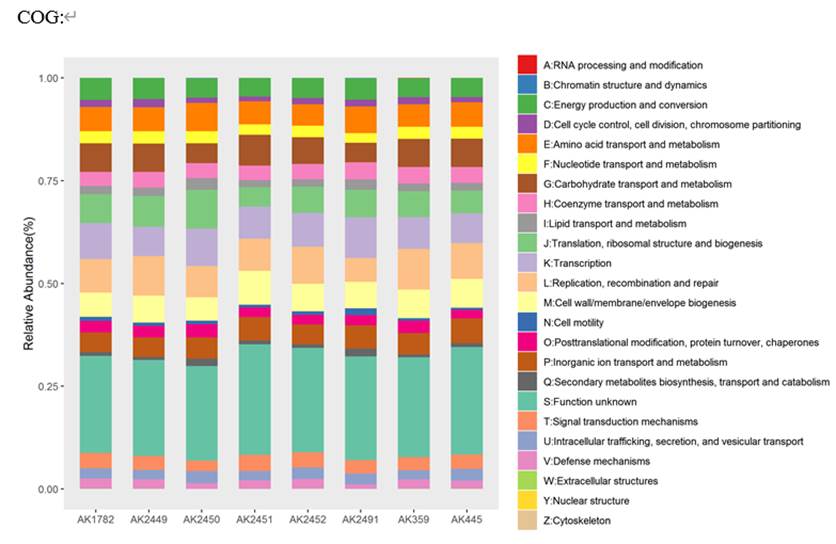
COG

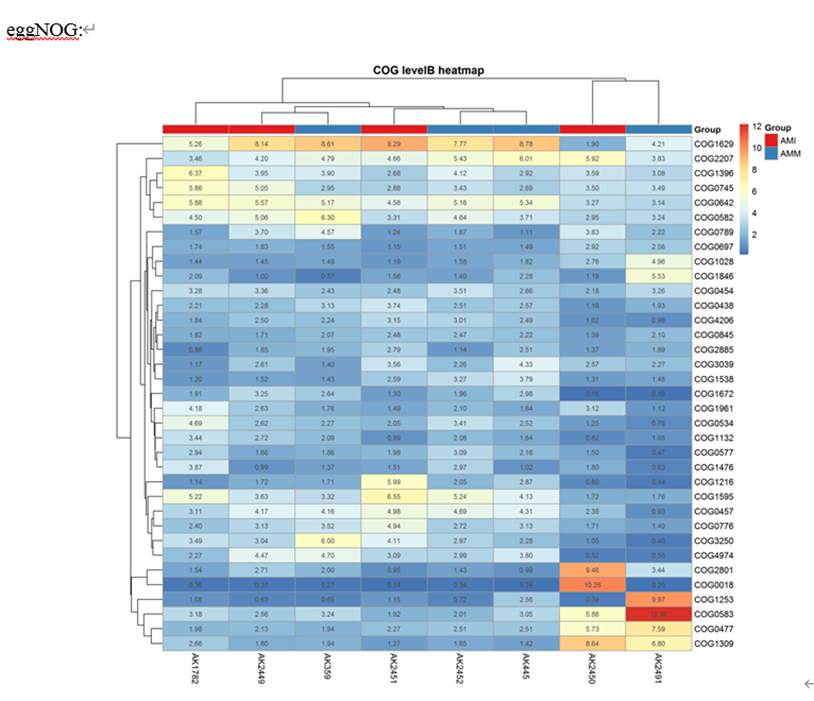
eggNOG

组间差异分析,如KEGG途径:

除此之外,还可以使用其他的功能基因数据库来进行进一步的基因注释和分析。比如:
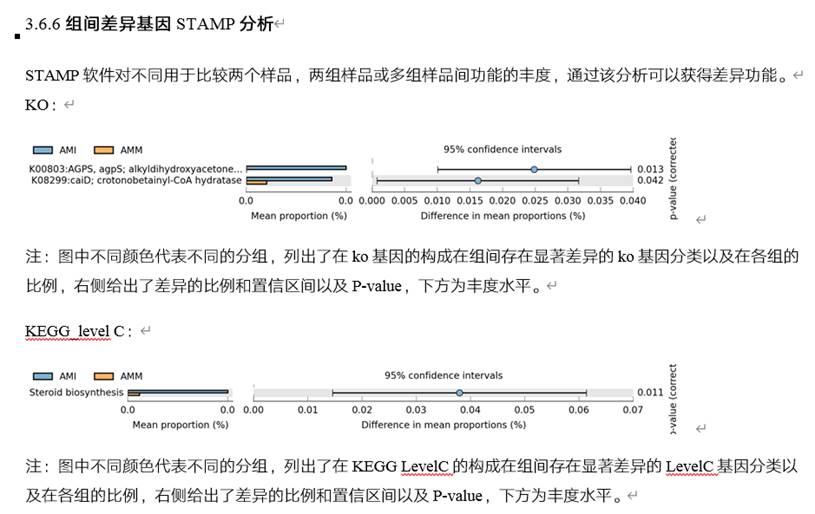
CAZy:

VFDB毒力因子注释:


抗性基因注释:




TCDB数据库注释:
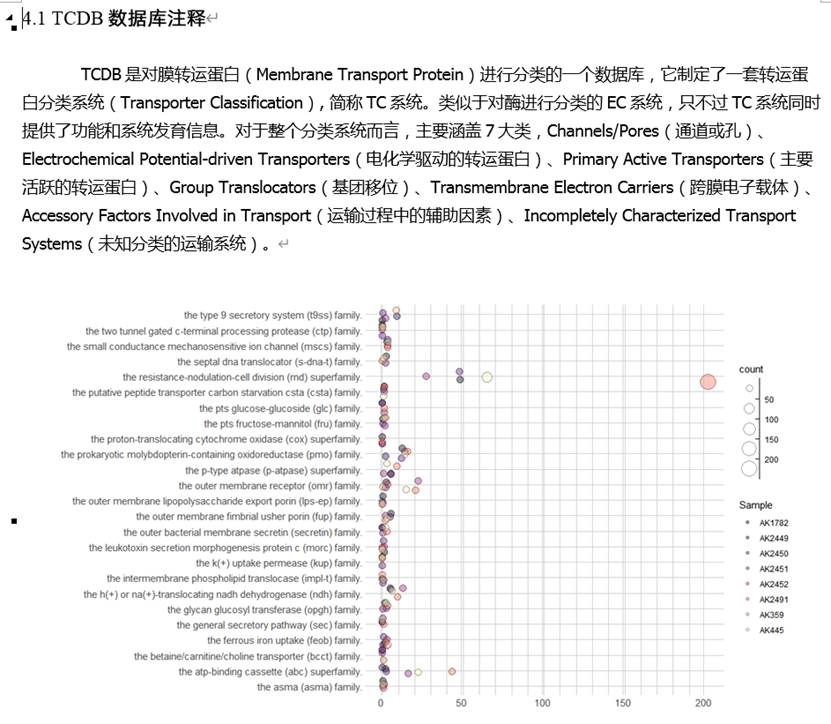

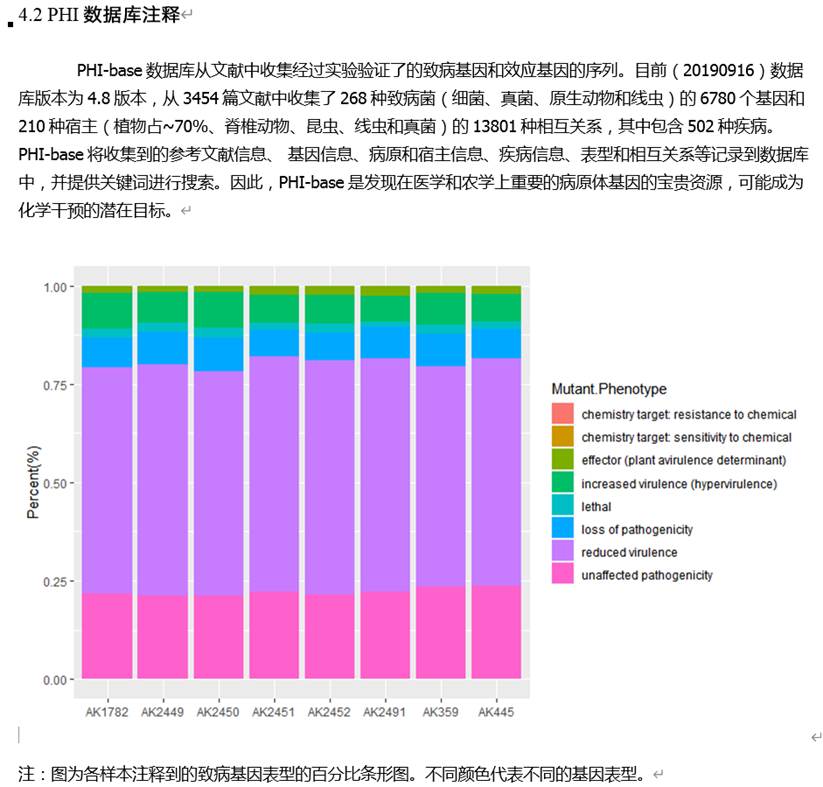
PHI数据库注释:

BCGs分析:
以及基于antiSMASH和BiG-SCAPE来对代谢物的合成生物基因簇BCGs进行分析。
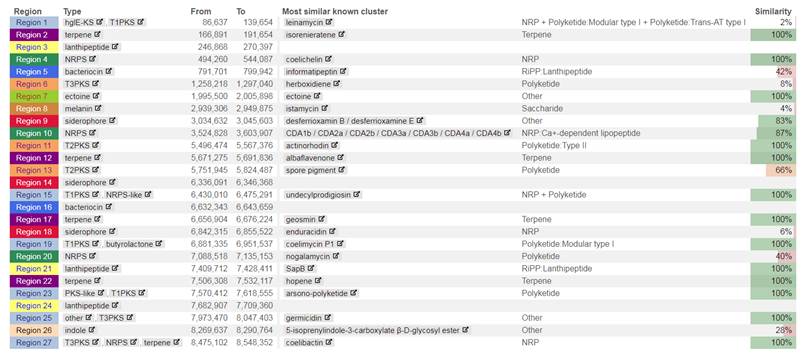

固定代谢能力评估:
或更聚焦于特定代谢的如下图中的氮、磷、硫和碳固定代谢能力和水平的评估:
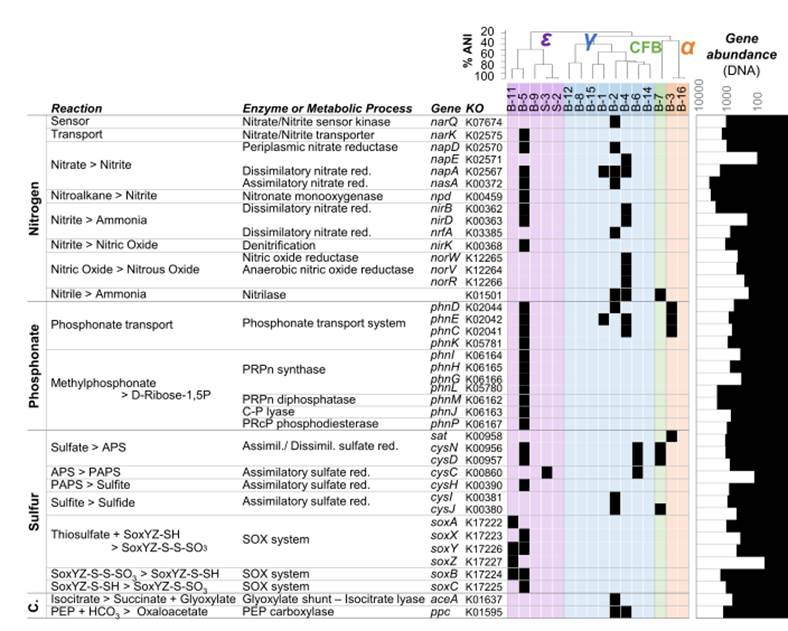

当有了大量样本的菌群构成丰度信息,以及各种基因和代谢丰度数据后,我们需要根据样本的meta信息,基于不同分组,时间或环境因子等数据进行统计分析和检验,进而发现和探索可能的关联以及背后的生物学意义。
那么在面对宏基因组这类数据时在进行统计检验分析时需要注意什么呢,应该采用哪些分析,并如何解读这些结果呢?
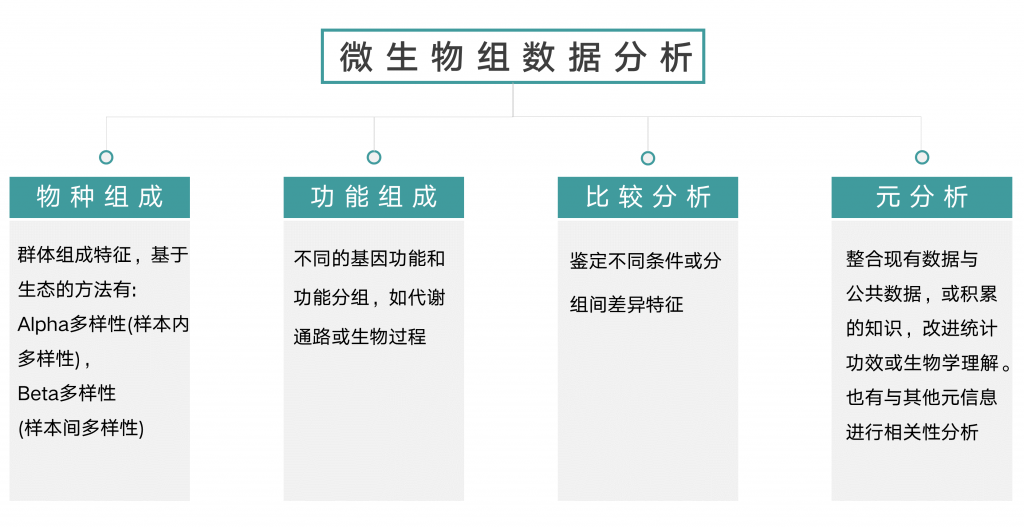
首先,微生物组数据分析分为四大类:

在对所有数据进行统计检验前一般建议对数据进行基本的质量过滤。一类是去除绝大部分样本都不存在的物种和基因,如Prevalence in samples (20%),还有一类是去除变异度过小的Percentage to remove (10%)基于Inter-quantile range。
为什么可以过滤这两类?
上述的两类由于其携带的信息量和变化过小在进行组间比较统计检验的时候都建议过滤,因为要么是污染,要么与差异无关。
宏基因组数据具有一些独特的特征,例如测序深度的巨大差异,稀疏性(包含许多零)和分布的巨大差异(过度分散)。在进行后续的统计检验之前建议针对不同的分析方法进行相应匹配的标准化处理。标准化包括:
Rarefaction和缩放方法:这些方法通过将样本放到相同的比例进行比较来处理不均匀的测序深度;
转换方法:包括处理稀疏性,组成性和数据中较大变化的方法。
那么各种标准化方法是什么,应该选择哪种方法?
参考MicrobiomeAnalyst网站提供的信息,以下是一个简短的介绍:


请注意,数据标准化主要用于可视数据探索,例如beta多样性和聚类分析。有时候不使用标准化也能获得最佳结果,比如:单变量统计和LEfSe。
同时,其他比较分析将使用其自己的特定标准化方法。例如,对metagenomeSeq使用累积总和定标(CSS)标准化,对edgeR应用M值的修剪均值(TMM)。
经常有小伙伴问,这个数据是用的什么标准化?没有做标准化怎么办?这类问题。
目前,尚无关于应使用标准化的共识性指南。建议大家可以探索不同的方法,然后目视检查分离模式(即PCoA图)以评估不同标准化程序对实验条件或其他感兴趣的宏基因组数据的影响。
有关这些方法的详细讨论,请参考使用者最近发表的两篇论文
① Paul J. McMurdie等
(https://doi.org/10.1371/journal.pcbi.1003531)
② Jonathan Thorsen等
(http://doi.org/10.1186/s40168-016-0208-8)
以上是关于标准化的这部分内容需要了解的知识,接下来我们来看具体如何操作,怎么得到那些图表?它们分别代表什么?
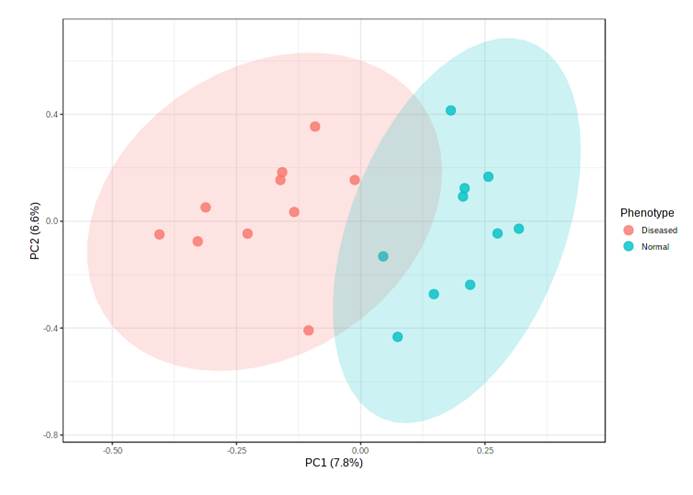
一般我们需要先进行探索性分析,也就是不设预订的假设,首先从主成分分析结果中了解样本的菌属和基因的大概分布。
主成分分析是根据不同距离算法计算样本之间的距离矩阵,然后进行降维,最终形成一个三维的空间分布。样本之间在空间上分隔越远表明样本之间的差异越大。
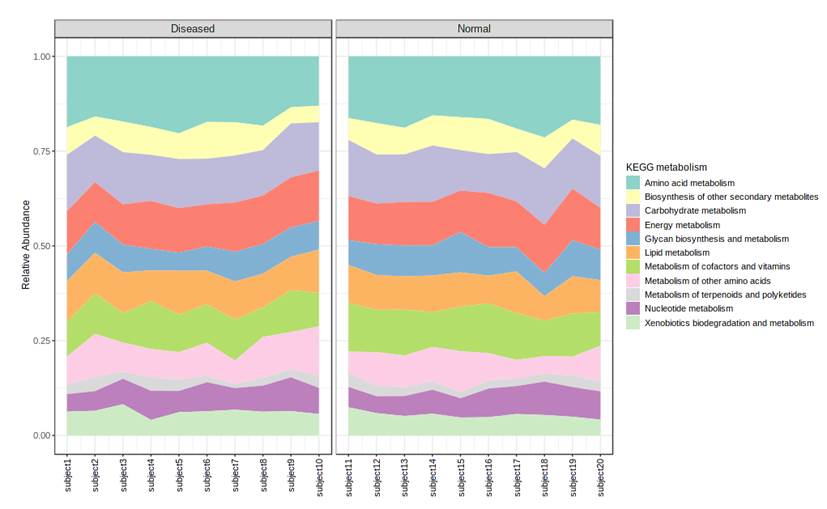
比如我们报告中的下图,疾病和正常样本可以较好的区分,一般此处我们还会进行一个统计检验,来判别PC1和PC2这几个维度上两组之间是否真的存在统计差异。

基于丰度图来评估各样本和分组的基本构成,如:
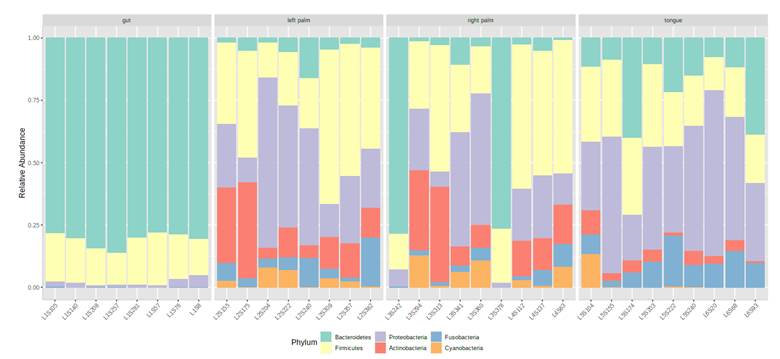

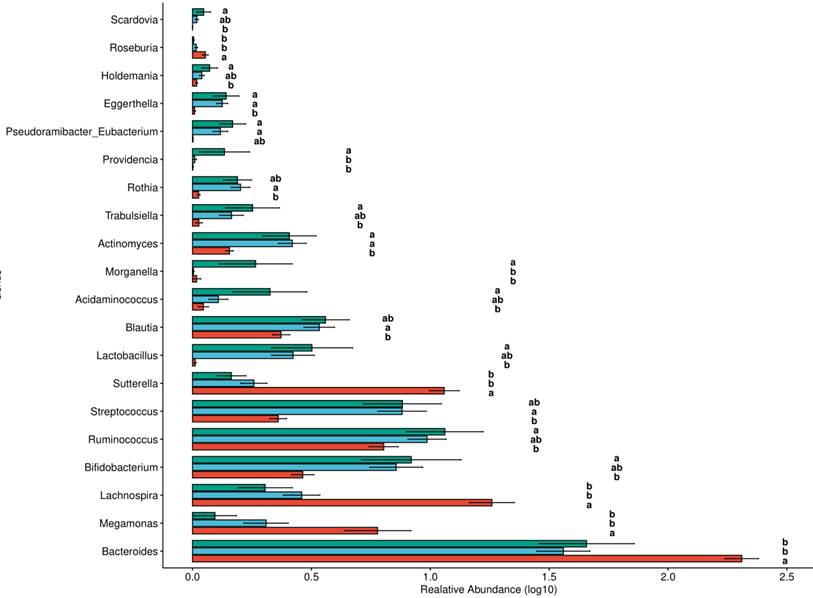
之后我们可以针对不同分组或处理之间的样本进行统计检验,可以使用的检验方法包括两组间的非参数统计检验T-test/ANOVA,3组以上组间统计检验可以使用Tukey test,其直接生成各组将的统计差异,并提供字母标注,直观简便,如:

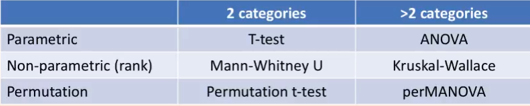
具体的统计方法选择可以参考下表:

除了常规的非参数检验外,包括metagenomeSeq和DEseq以及edgeR等统计方法包可以很好的分析组间差异特征。LEfSe则一般用于寻找特征标志物。
那么有了大量的差异特征菌属或基因之后,我们是否能基于这些差异菌属有效的区分不同的分组呢,或构件一个模型来预测或分类呢?
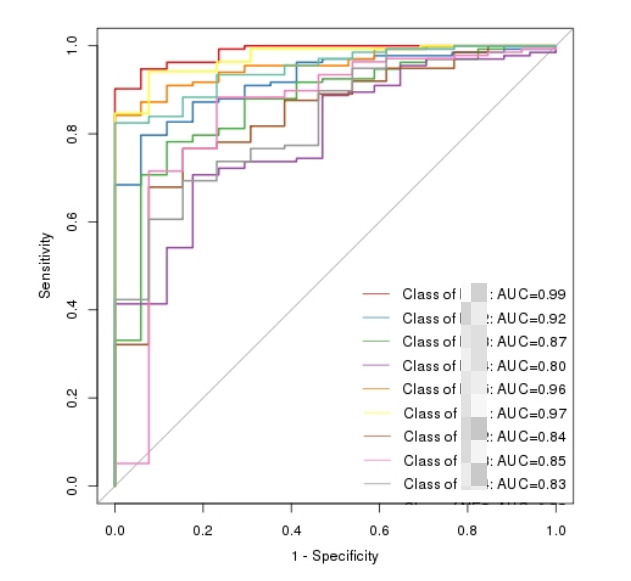
这时候可以使用随机森林(Random Forest)一类的决策树机器学习模型,来利用这些差异特征构建分类模型,并使用AUC等指标来评估基于这些模型的预测有效性和准确度(我们报告中如下图)。
当然也可以使用其他更复杂的如深度学习等方法来构建分类模型。

除了性别、疾病、地点等分类差异之外,我们通常还有很多元数据,包括临床指标或环境因子等信息,这些数据通常是连续型数值,对于这类数据我们可以进行相关性分析。
当然反过来,将菌群特征作为表型也可以和如基因组的基因型或SNP构成来进行相关性分析。
对于菌群数据的相关性分析比较推荐:
SparCC方法,可以构建菌种或菌属之间的相关性网络,相对稳定。
对于与疾病或环境变量进行相关性分析可以使用:
Sperman秩相关分析。
另外RDA/CCA分析也可以有效的反映菌属与环境因子等指标直接的关系(我们报告中如下图)。
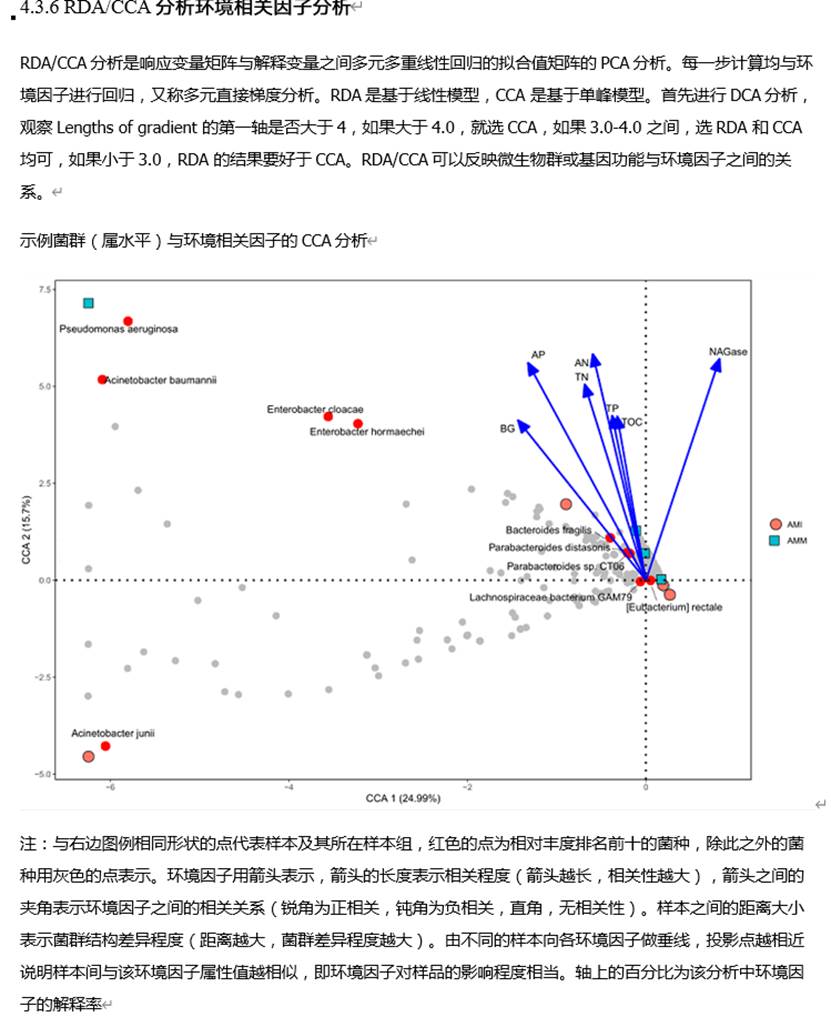

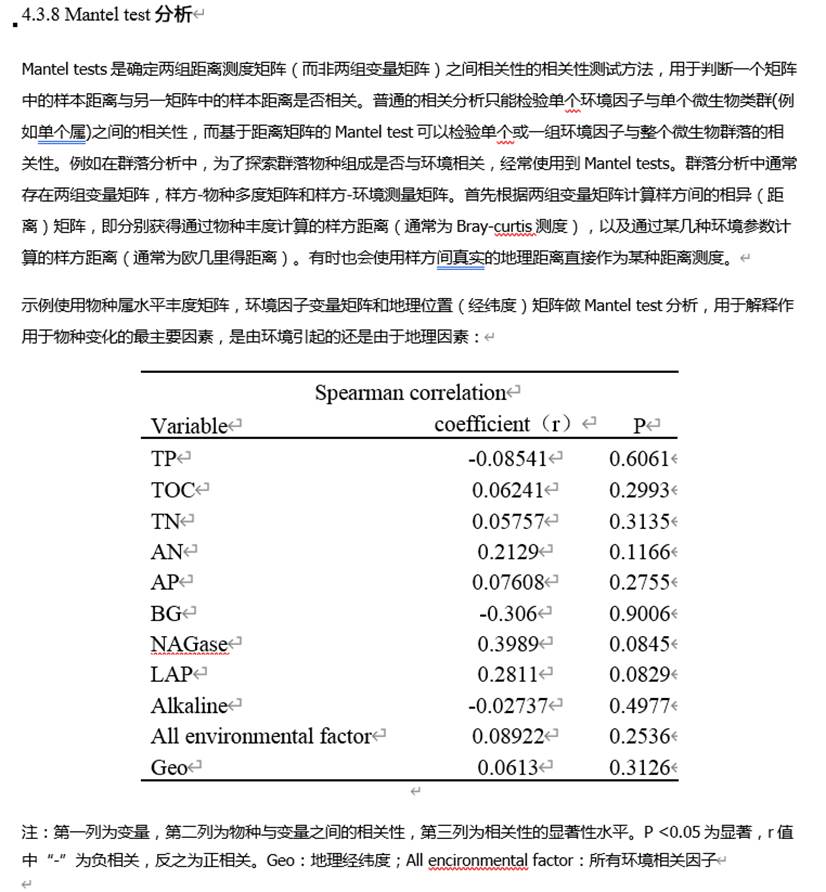
Mentel检验也可以用于判断菌群构成特征与单个或一组环境因子之间是否存在显著相关。

要 点
宏基因组从大量菌群和基因构成中寻找关联是需要足够的样本量才能达到有效的统计效力,因为一次性获得了大量的特征数据,样本量过少会带来统计结论的无效,越是组内差异大,组间差异小的研究足够大的样本量才能得到可靠的结论。
一般动物样本具有较好的背景可控,组内样本数量建议至少6个,而人群研究由于背景复杂,个体多样性高,一般建议组内50例以上较好。
以上看完后,你应该对宏基因组的数据分析流程有了整体的认识,也学会了相应的一些操作,但是不一定能直接从自己的这些数据、图表中真正探索到和实际生物学相关的有价值的研究成果。
所以,我们又选取了一些已发表的研究作为案例,结合实际问题来具体分析,从实验设计到具体分析流程方法和图表的展示,再到相应的结论,掌握这类文章的总体思路。
之后无论是刚开始的实验设计,还是后面的分析,都会更加得心应手。
建议想好整个实验思路再开始(或者也可以咨询我们,我们专业的数据分析团队会为你提供切实可行的项目方案)。
第一项研究是关于肥胖患者减肥手术后的宏基因组和代谢数据的分析研究。
文献来源:Aron-Wisnewsky J, Prifti E, Belda E, et al. Major microbiota dysbiosis in severe obesity: fate after bariatric surgery.Gut. 2019;68(1):70–82. doi:10.1136/gutjnl-2018-316103
研究纳入了61名严重肥胖的受试者,他们是可调节胃束带术(AGB,n = 20)或Roux-en-Y胃旁路术(RYGB,n = 41)的候选人。减肥手术后1、3和12个月随访24名受试者。使用宏基因组学测序和LC-MS分析肠道菌群和血清代谢组。另外纳入了10人和147人分别作为宏基因组和代谢检测的验证集。
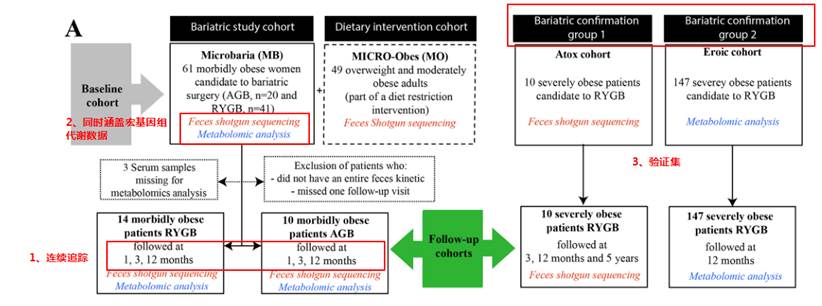

研究思路

这样的设计分别有什么作用?
第一点持续的动态采样可以获得持续变化情况,尤其是在一个特定变化后(减肥手术),持续的最终采样有助于确认菌群的变化出现和特定事件或生理病理变化的前后,尤其是在确定因果中有重要帮助。
第二点获得多维的数据有助于帮助我们全方位的了解菌群变化背后的带来的生理和代谢变化以及之间的关联。
第三点独立验证集的存在将大大增强研究的可信度,尤其是该研究纳入的样本量并不多,无法全面有效的控制无关因素,使得很多统计检验的效力无法显现。这也导致该研究仅在基因总量和多样性上获得较好的重复效果,而更多的菌群精细特征以及具体基因和代谢通路没有得到深入分析。但是独立验证集保证了核心结论的可靠性和重复性,这点在宏基因组研究中非常重要。
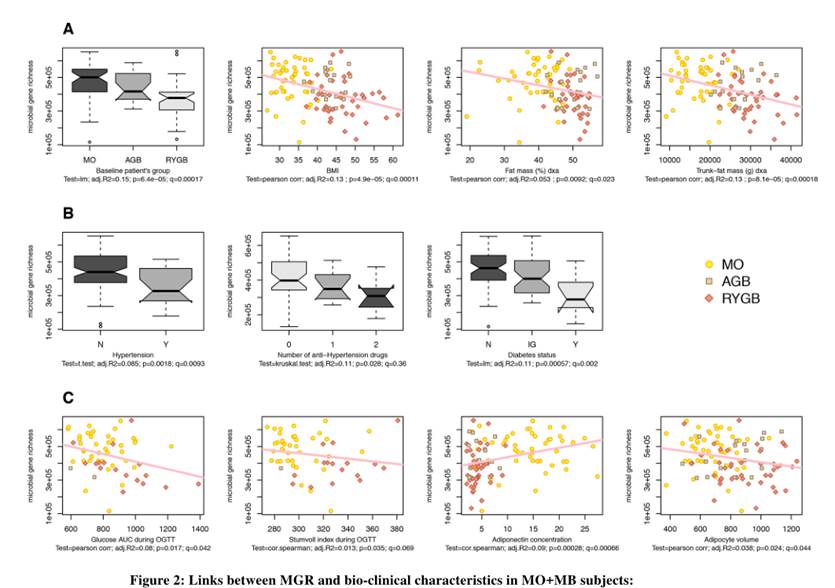
从下图可以看到研究针对样本的总基因多样性水平与生理指标和疾病状态进行相关性分析和组间差异分析,图中给出了显著相关和差异的指标。
使用的统计检验方法是pearson和sperman相关和t-test以及Kruskal-Wallis检验。
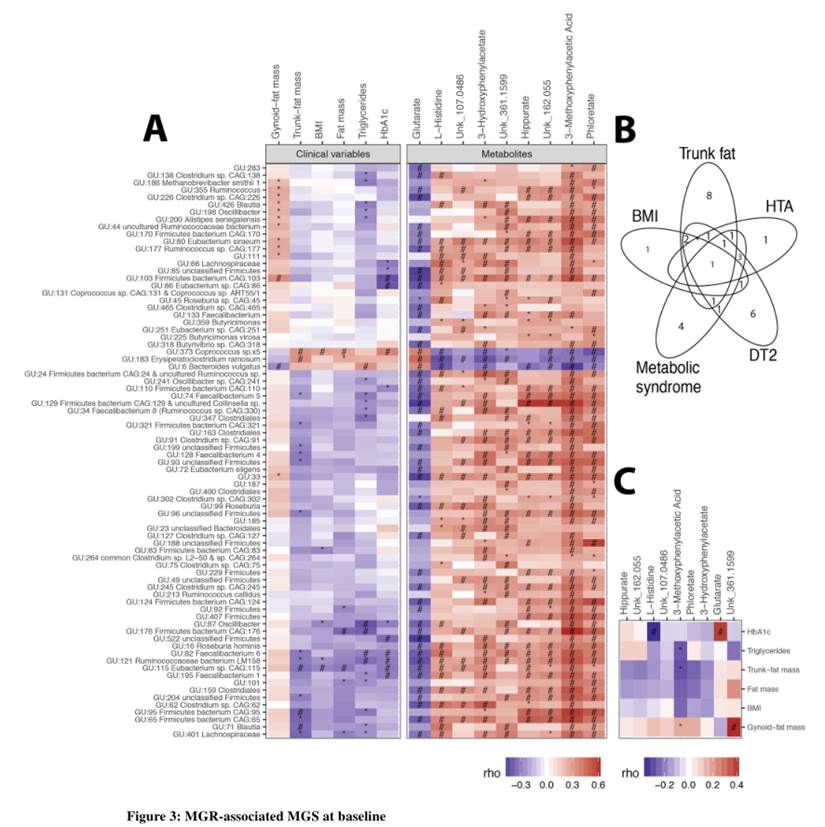

下图是研究将MAGs与各项生理和代谢值进行相关性分析后的热力图。该研究由于测序较早,并未独立拼接,而是直接使用了之前一项人类肠道菌群研究获得组装基因组参考序列。
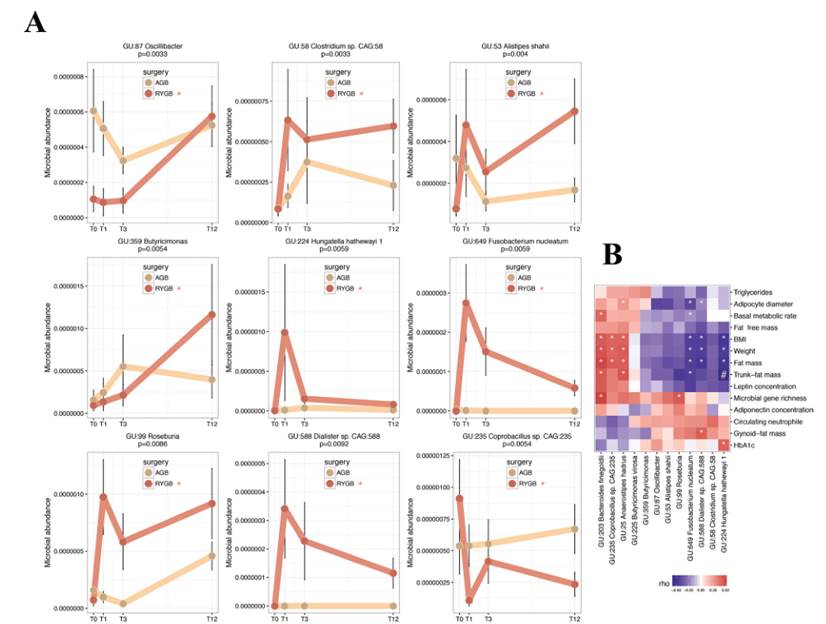

进一步研究分析了术后特定变化模式的MAGs以及它们与代谢生理指标的相关性,见下图:

上图的研究可以通过pattern search的方法寻找特定变化模式的菌种。
研究的主要结论发现是低基因丰富度(LGC)存在于75%的患者中,并且与躯干脂肪质量和合并症(2型糖尿病,高血压和严重程度)增加相关。LGC改变了78种宏基因组种(MGS),其中50%与不良的身体成分和代谢表型有关。九种血清代谢产物(包括谷氨酸盐,3-甲氧基苯基乙酸和L-组氨酸)和含有参与其代谢的蛋白质家族的功能模块与低MGR密切相关。术后一年,BS会增加MGR,但尽管RYGB患者的代谢改善比AGB患者大,但术后一年的MGR仍然很低。
总体而言该项研究可以使用浅宏基因组(在文章开头第二部分详细介绍过)来完成所有测序和分析,进一步扩大样本数量,如果能同时获得人的转录组数据甚至能更加明确的找到菌群变化与特定代谢通路的关联关系。
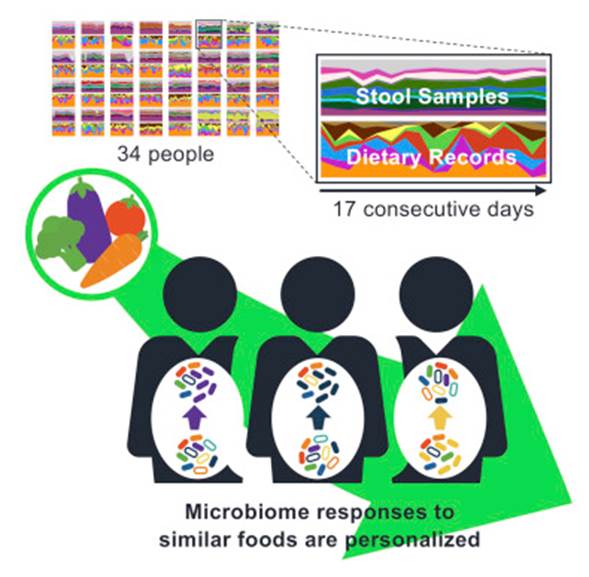
第二项研究是Dan Knights实验室发表在Cell Host & Microbe,2019的一篇针对34个人17天每日饮食和菌群变化的相关研究,试图揭示日常食物选择与人类肠道微生物组组成之间的精细关系。
文献来源:Johnson Abigail J,Vangay Pajau,Al-Ghalith Gabriel Aet al. Daily Sampling Reveals Personalized Diet-Microbiome Associations in Humans.[J].Cell Host Microbe, 2019, 25: 789-802.e5.
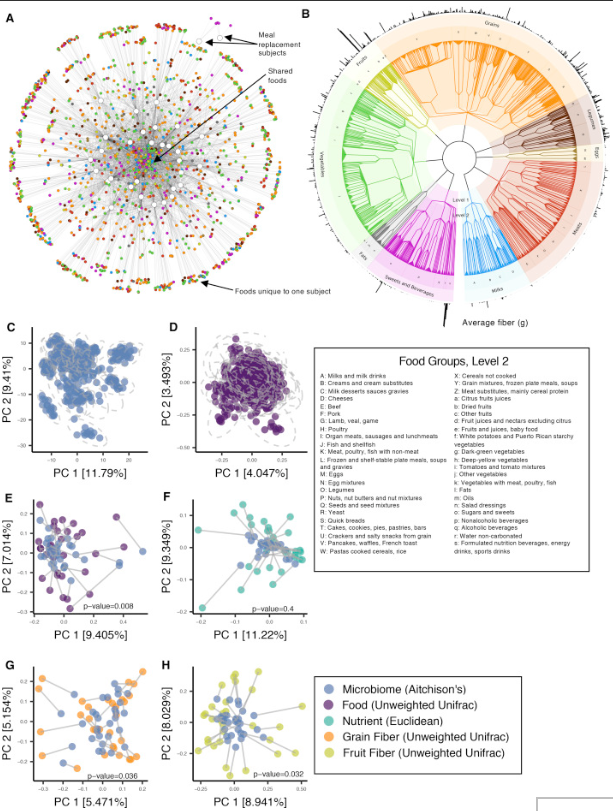

可以看到,研究同时记录了粪便样本的菌群宏基因组和每日的饮食记录。研究的核心在于将每日饮食的食物通过营养构成进行量化,并构建类似物种进化树的食物物候树。
此外由于有每日的数据,可以通过前一日的食物与第二日的菌群数据进行时间序列分析,构建食物与菌之间的关联以及时间相关性。
最后基于菌群数据和前一日饮食来构建模型预测判断后一日的菌群状态,帮助我们了解食物对于个体菌群的影响因素并实现定量和预测。
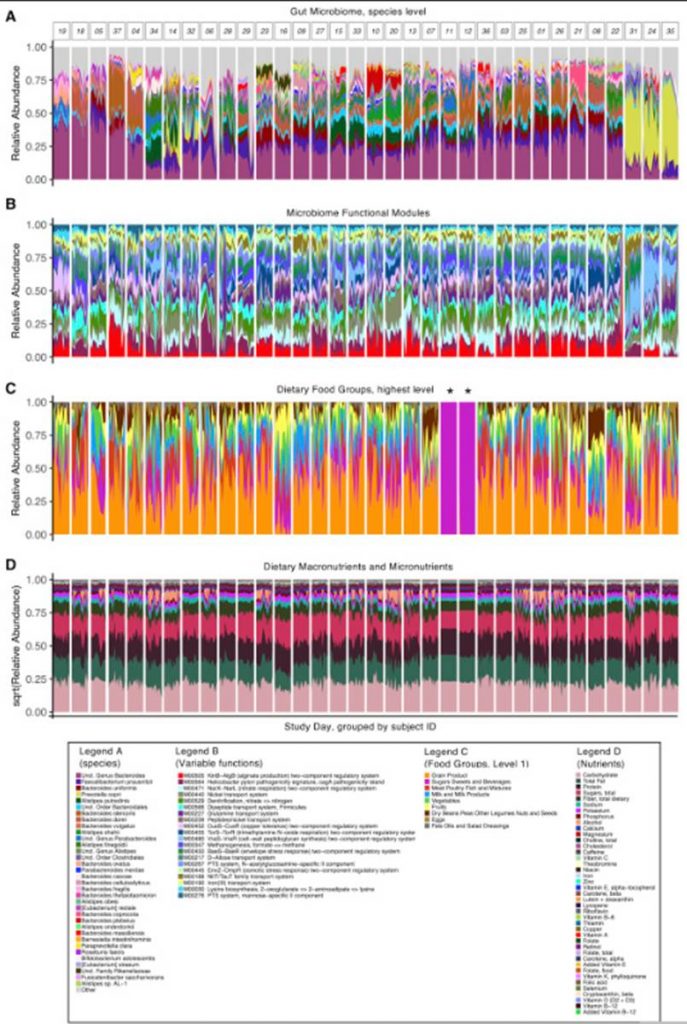

研究中对数据的处理过滤标准如下:删除所有具有低读取计数(每个样品<23,500个读取)的样品。物种级别的分类表仅限于研究对象中至少存在25%的研究日,且在10%的研究样本对象中发现的那些物种。
最后,相对丰度<0.01%的稀有物种被丢弃,将物种数量限制为290个注释。将得到的分类表汇总到较高的分类级别(即属,科,门等),以进行下游分析。
菌群和饮食以及营养构成的堆叠图很好展现了变化和对应。
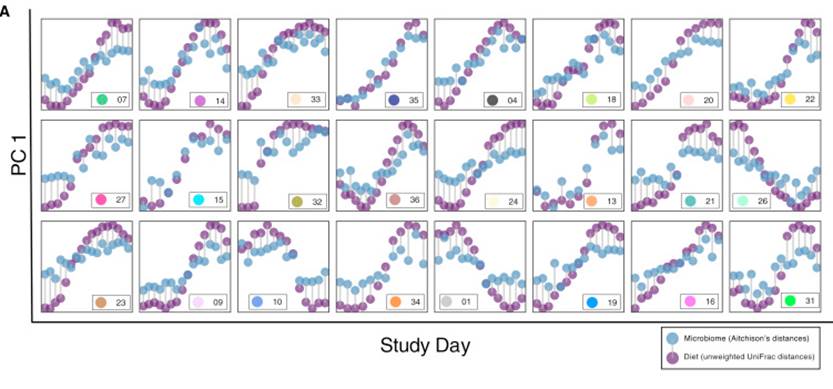

下面这张图很好的显示了饮食食物的变化与菌群变化之间的时间变化关系:
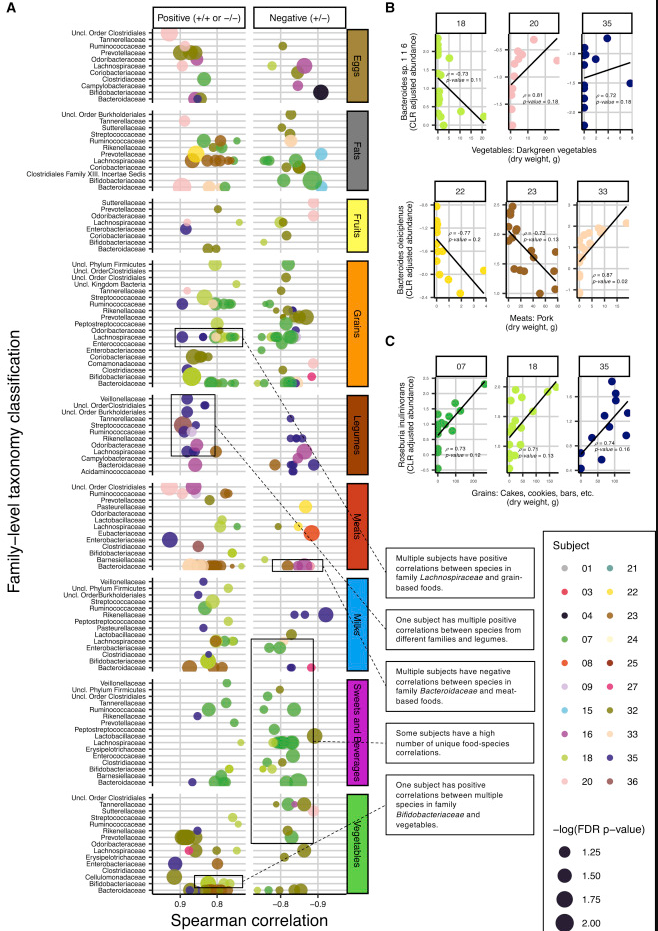

下面这张图通过对每个人单独进行菌属与食物的Spearman相关,展现了菌与食物之间的关联的个体化差异,在特定菌属对应相同食物不同人会出现完全不同方向的变化,这也正是这项研究所揭示的,这种关联关系的复杂性。

本研究虽然有大量样本,但并未进行组装,而是直接使用了Refseq的细菌完成基因组序列作为参考。研究由于样本数量众多,测序深度也很有限,类似研究也可以使用浅宏基因组方式完成。
接下来的一个研究是比较典型的宏基因组组装并与疾病进行关联分析的案例,研究的是日本人群类风湿关节炎的肠道微生物组的全基因组关联研究。
文献来源:Kishikawa Toshihiro, Maeda Yuichi,Nii Takuroet al. Metagenome-wide association study of gut microbiome revealed novel aetiology of rheumatoid arthritis in the Japanese population.[J] .Ann. Rheum. Dis., 2020, 79: 103-111.
研究使用较高深度的宏基因组shotgun测序(每个样本平均13 Gb)对日本人群(病例 = 82,对照 = 42)进行了RA肠道微生物组的MWAS分析。MWAS由三个主要的生物信息学分析渠道(系统发育分析、功能基因分析、途径分析)组成。
使用了之前研究中6139个完成拼接日本人肠道宏基因组作为参考序列以及其他几项研究的参考基因组,在过滤部分种过多的基因组之后,最后一共使用了7881个参考基因组。
将QC后的序列直接比对到参考基因组,并根据基因组长度计算对应物种的相对丰度。
基因方面选择denovo组装,使用MegaHIT,然后再contig上完成ORF预测和CD-HIT聚类去冗余,最后与UniRef和KEGG数据库比对。
最后使用bowtie2将测序序列比对到注释后的unigene序列上获得基因丰度,经过KEGG注释得到代谢途径的丰度。研究的数据处理流程图如下:
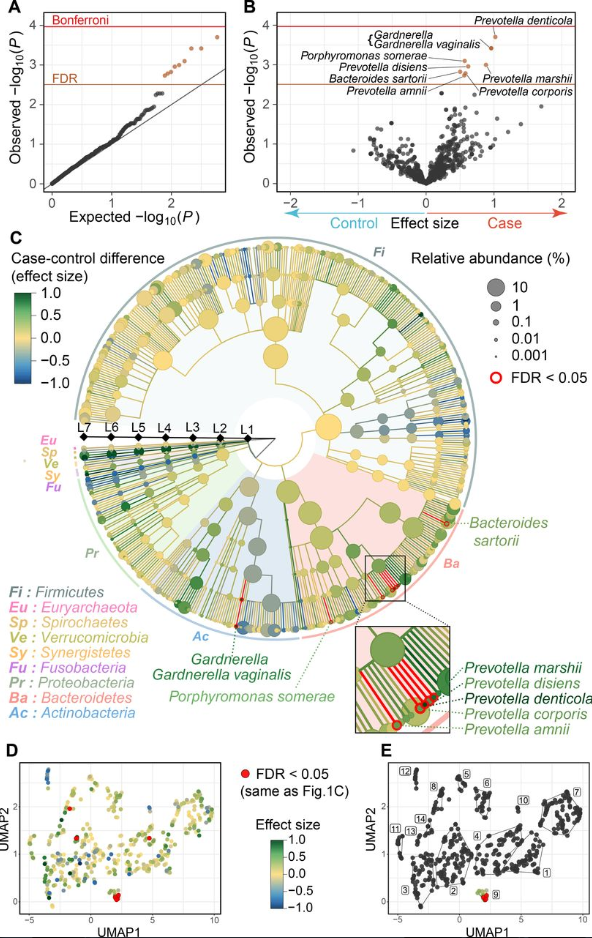

在数据分析流程和方案选择上人体肠道菌群由于研究众多,以及有多个深度测序拼接完成的Binning参考基因组数据集,确实可以直接使用参考基因组直接比对。
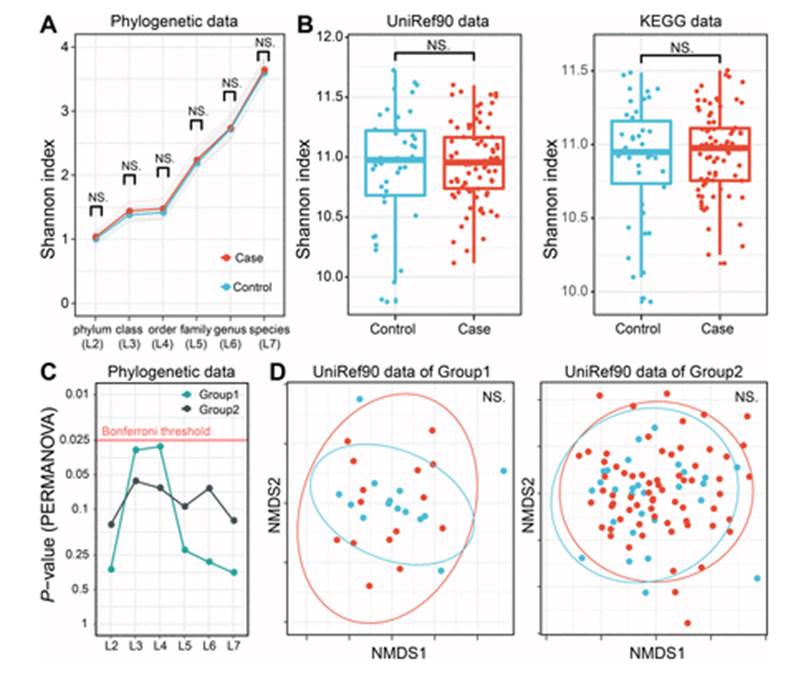
对于其他一些环境或来源的样本这个深度的数据量可以考虑独立拼接和分箱。该研究中使用已有参考基因组,大概88%的序列能比对到参考基因组,如果直接组装这个比例应该可以更高一些。另外在获得基因丰度是可以考虑使用Salmon,比对获得基因丰度更为方便。
获得相应数据后对相对丰度,该研究使用Box-Cox transformation对数据进行标准化,并过滤了一些低丰度的菌属。Case-control的相关性分析使用的R的glm2模块,将年龄、性别和测序上机分组作为协变量。
对于菌属的关联分析,最终将显著性结果以火山图和GraPhlAn图的形式展现如下:

上面其中D图使用每个菌的丰度进行UMAP分析,并映射关联效应的展示比较有意思。
不过在基因层面上并未找到相应的关联,可以看到下图UniRef90的NMDS分布图两组之间无法有效区分,多样性也没有显著差异。

这项研究在菌层面发现了多个普雷沃氏菌属的菌在日本人群中与类风湿性关节炎存在关联,不过除此之外其他方面的发现并不多,仅找到一个基因存在显著关联,涉及的临床调查也相对有限,且人群队列数量不算多,并无独立验证集,因此亮点并不多。如果能纳入免疫相应指标可能能研究的更细致一些。
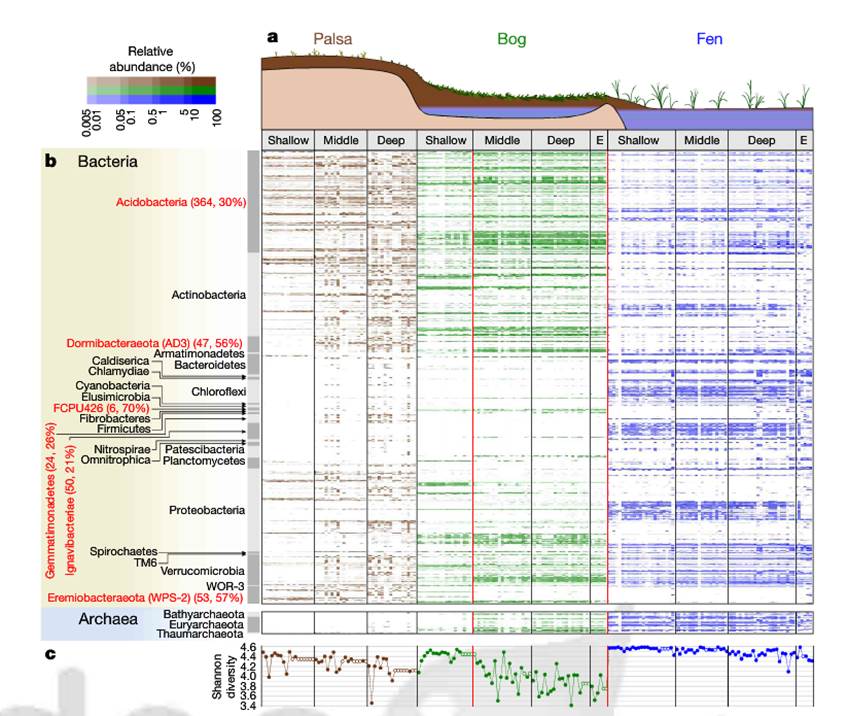
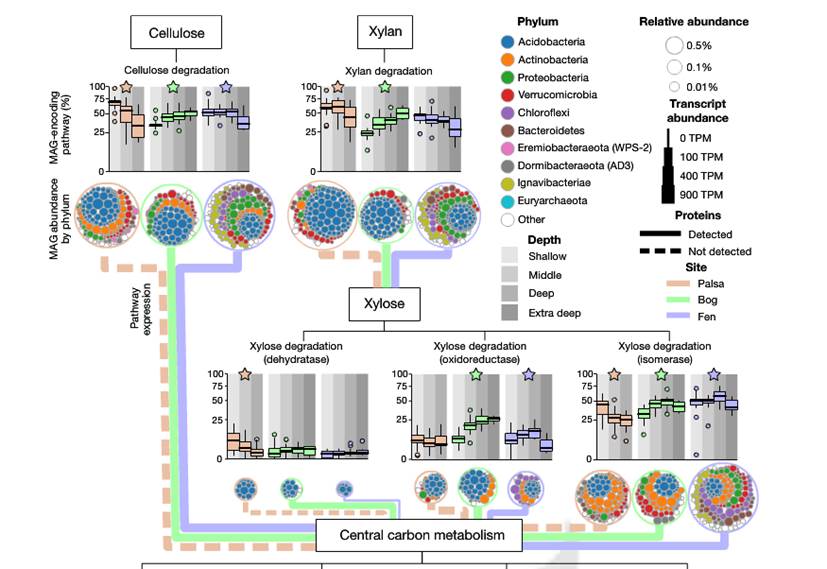
最后这项研究是对来自永冻土融化梯度的214个样品的宏基因组测序组装了1,529个基因组,揭示了参与有机物降解的关键种群,包括其基因组编码先前未描述的木糖降解真菌途径的细菌。
文献来源:Woodcroft Ben J,Singleton Caitlin M,Boyd Joel Aet al.Genome-centric view of carbon processing in thawing permafrost.[J] .Nature, 2018, 560: 49-54.
通过宏基因组denovo组装和分箱Binning,最终获得了1529个永冻土菌群基因组。基于这些数据描绘了永冻土融化梯度下的菌群构成特征,如下图。

论文是2018年发表的,测序是在2011和12年测的,使用的是CLC Genomics Workbench 较早的4.4版分单样本组装,然后使用MetaBAT进行分箱,最后的标准是70%完成度和低于10%的污染。
其中糖苷水解酶基因使用dbCAN数据库的HMM进行预测,碳代谢使用KEGG数据。
研究还同时选择了部分样本进行了宏转录组和宏蛋白组的测序,对碳代谢同时结合转录组和蛋白组的数据,展现了特定通路下不同永冻土的菌群构成和表达丰度差异。
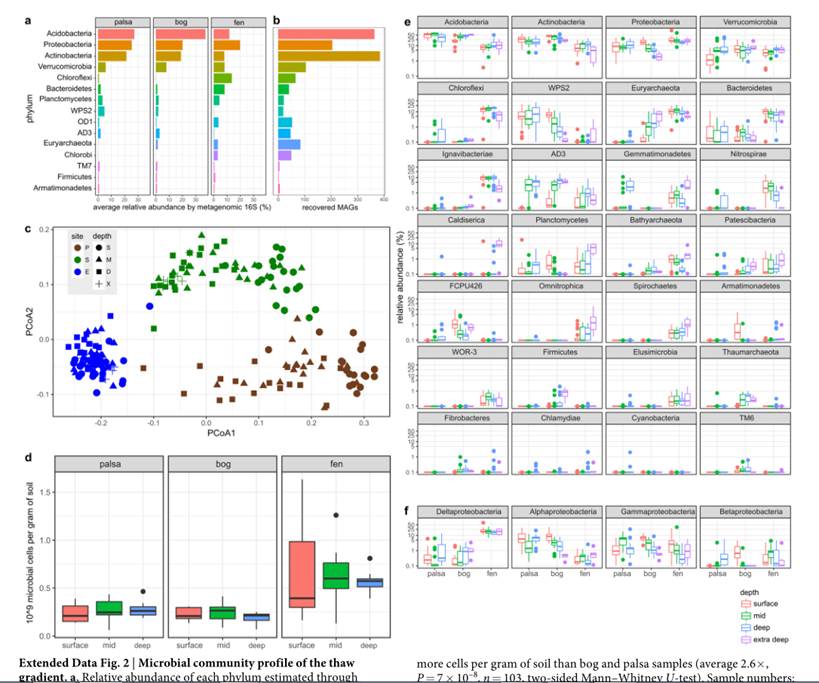

基因组拼接的分布情况,以及不同地域样本分布和菌属丰度情况如下:
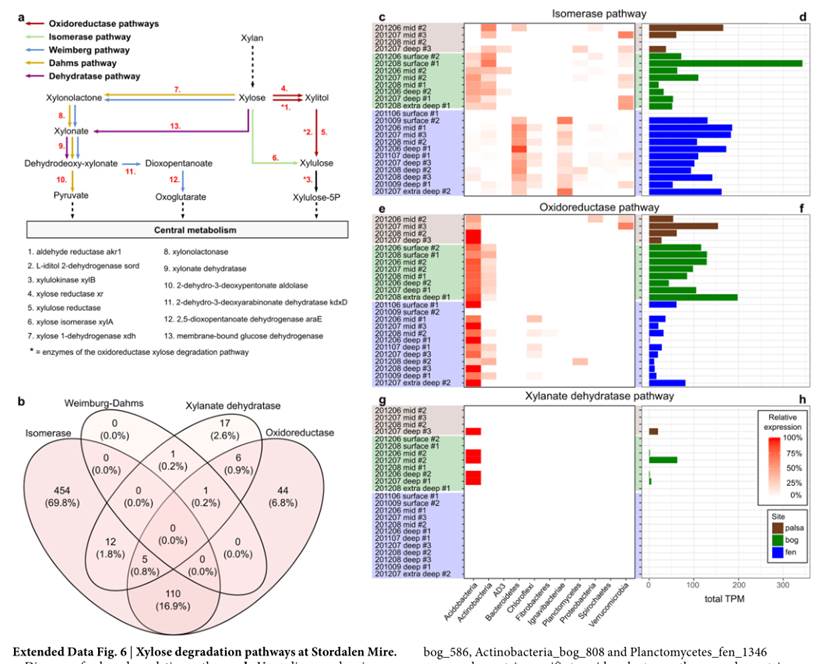

木糖降解途径在每个样本中的分布和维恩图,另外详细的展现了主要门对每个代谢途径的贡献和基因表达丰度,如下图:
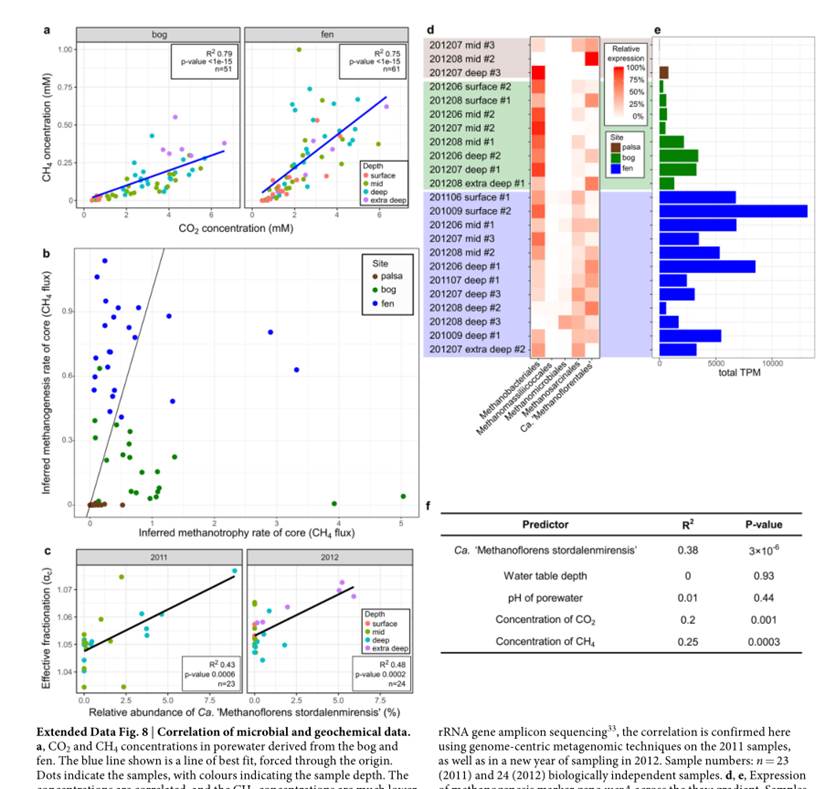

这张图分析了特定菌与地理位置和CO2以及甲烷的浓度的关联性,如下图:
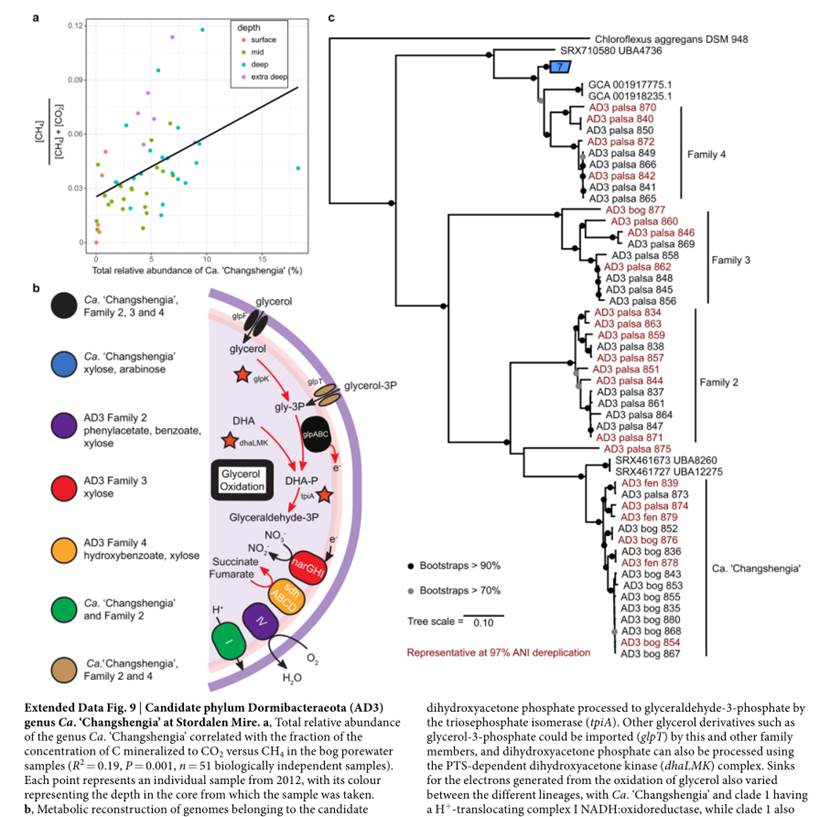

对关键物种的CH 4 :CO2浓度比相关代谢途径的重建,以及相应基因的基因家族分析。

总结一下这项研究,永冻土的菌群参考基因组数据缺乏,该研究从大量地点采集样本重建了1500多个参考基因组。
首先从物种构成特征上与永冻土融化阶段特征进行分析,并与重要环境因子进行分析,锁定重要的特征菌。
然后针对重要的代谢途径和关键基因结合宏转录组和宏蛋白组全面解析代谢途径的分布和差异变化。对关键的物种重建了相关代谢途径并对其相关基因家族进行分析。
研究基本上从头构建了一个生态环境下的菌群结构数据,并利用获得的基因组深度解析特定代谢途径和基因的构成和表达变化,应该说既宽又深。很多样本采集和测序是2011年和 2012年就开展的,虽然测序技术远不如现在成本低和成熟,但是其独特的研究对象和全面深入的分析仍然使整项研究和目前的一些研究相比完成的更加出色。
p.s. 以上展示的图表,我们都可以帮你实现~
网址:https://www.microbiomeanalyst.ca/,只需要biom文件或丰度表就可以进行绝大部分统计检验分析,而且生成图表完整,可以直接使用。偶尔会有服务器不稳定,上传提示错误的情况。
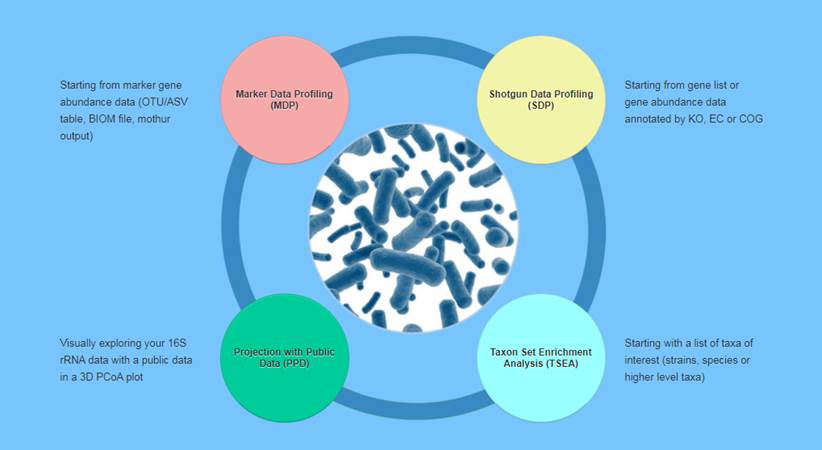

特别推荐其中的Taxon Set Enrichment Analysis模块,直接提交物种列表(一般是找到的差异物种列表),可以直接在各种已有的相关性(人体基因-菌属相关,生活方式-菌属相关,疾病-菌属相关)中进行富集分析,能很好的帮助判断和提供差异菌群的具体关联和证据支持。
完整的支持分析包括:


可以直接生成下面的图:


基本上常见的分析和图都能在线实现。
另一个是https://gcmeta.wdcm.org/,是中科院微生物研究所搞的平台,里面包括了宏基因组的样本数据和在线分析平台,可以直接上传原始数据,直接使用工具进行在线分析,大部分常见工具都有,也有一些流程。



对于缺乏计算资源或想自己动手分析的朋友挺友好的,非常推荐试试看。
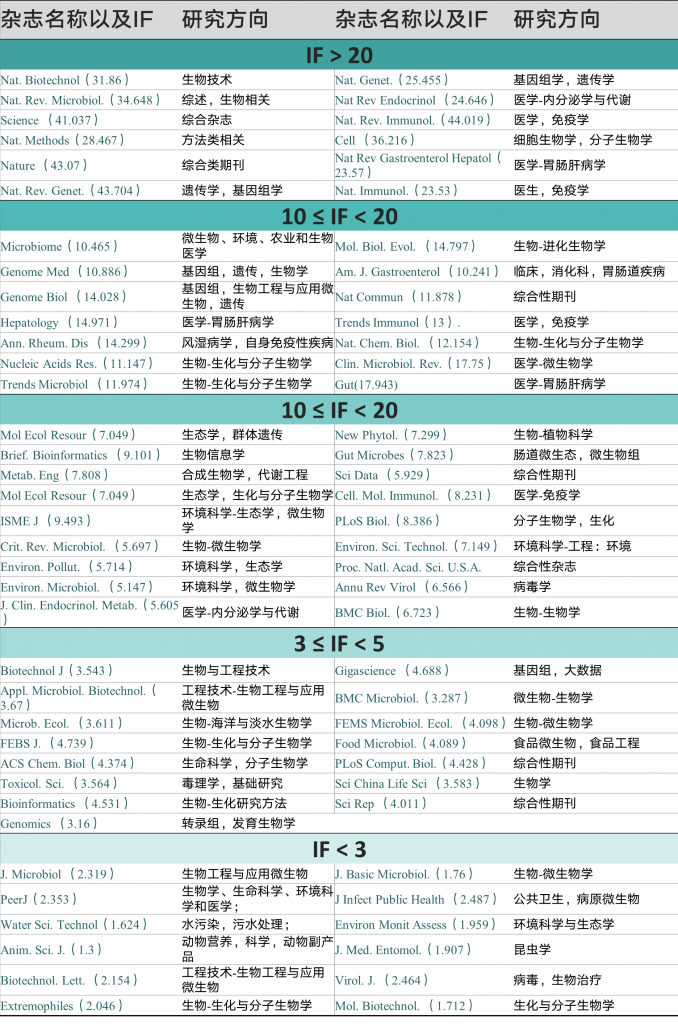
最后,帮大家整理了宏基因组可投稿的期刊,具体研究方向和影响因子见下表:



来源: 谷禾健康

谷禾健康 原创
最近, 国际顶级方法学期刊《Nature Methods》发表了由加利福尼亚大学圣地亚哥分校儿科、加州大学计算机科学与工程系 、加州大学合作质谱创新中心以及加利福尼亚大学圣地亚哥分校微生物群创新中心 多学科合作研究的最新成果:“Learning representations of microbe–metabolite
Interactions(可用于分析微生物与代谢产物之间相互作用的人工神经网络)”,此项研究恢复微生物与代谢物之间关系的能力,并证明了该方法如何发现微生物产生的代谢产物与炎症性肠病之间的关系。


摘要
研究人员表示,整合多组学数据集对于微生物组研究至关重要。但是,推断整个组学数据集之间的交互具有多种统计学上的挑战。文章中通过使用神经网络(https://github.com/biocore/mmvec)来解决了此问题,其能够在存在特定微生物的情况下估算每个分子存在的条件概率。研究人员以已知的环境(沙漠土壤湿润生物结壳)和临床(囊性纤维化肺)实例为例,展示了这一方法恢复微生物与代谢物之间关系的能力,并证明了该方法如何发现微生物产生的代谢产物与炎症性肠病之间的关系。
背景
虽然已经有广泛的努力来开发整合多组学数据的方法,但一些概念上的挑战限制了整合不同组学数据的技术,例如,将微生物测序和非靶向质谱联系起来。因此,需要新的方法来处理不同的数据类型。为此,研究人员提出了“mmvec”(微生物-代谢物载体),一种神经网络,可以从单个微生物序列预测整个代谢物丰度曲线。通过迭代训练,mmvec可以学习微生物和代谢物之间的共现概率。微生物-代谢物相互作用可以通过标准的降维界面进行排序和可视化,从而产生可解释的结果。
主要结果
1.使用模拟囊性纤维化生物膜的数据集,将mmvec与Pearson’s、Spearman’s、SPIEC-EASI、SparCC和proportionality方法进行基准比较。证明了mmvec优于所有旨在推断成对微生物-代谢物丰度数据集之间相互作用的现有工具。
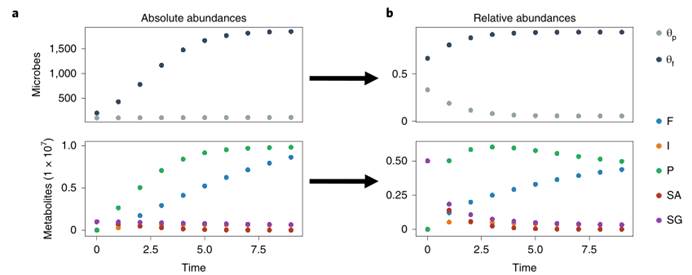
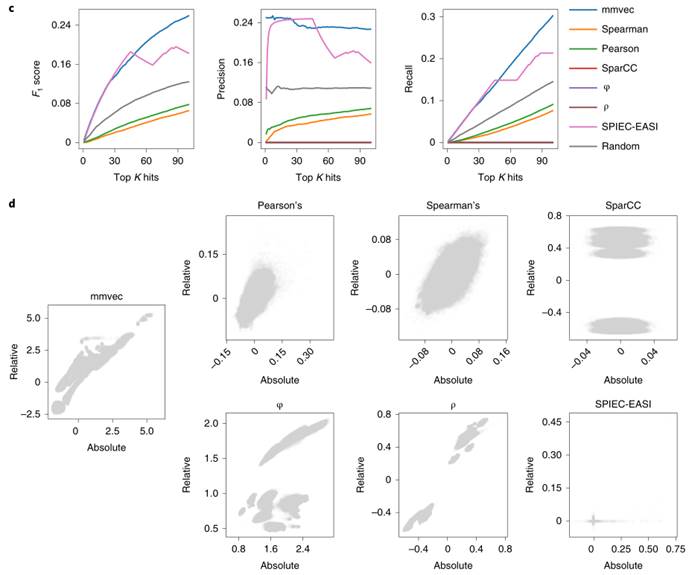
图a.两个微生物和多个分子之间的相互作用被模拟成单分子动力学和扩散过程,(发酵剂由θf表示,铜绿假单胞菌由θp表示)从推导的微分方程模拟的微生物和代谢物的绝对丰度,图b. 为图a.中绝对丰度的比例。这里模拟了五种代谢物,即糖(SG)、抑制剂(I)、酸(F)、铵(P)和氨基酸(SA)
图c. 在每种微生物的前100个代谢产物中,使用 F1 score、precision(精确率)和recall curves(召回率)比较了mmvec与Pearson’s、Spearman’s、SparCC、SPIEC-EASI(生态关联的稀疏逆协方差估计与统计推断)、比例度量(φ和ρ)。图中表示mmvec和SPIEC-EASI的随机表现(Random)优于其它所有工具,其中mmvec表现最好。
图d.从绝对丰度和从所有基准测试方法获得的相对丰度的系数比较。图中显示mmvec是唯一对比例偏差具有鲁棒性的方法。这对于保持绝对丰度和相对丰度之间的一致性至关重要,否则可能导致虚报假阳性和假阴性。
2.沙漠土壤生物润湿事件,测试mmvec是否可以解决微生物-代谢物相互作用中无法解释的差异。结果是mmvec解决了阴道分枝杆菌释放的体外验证代谢物与环境样品的测序和质谱分析之间的冲突发现。
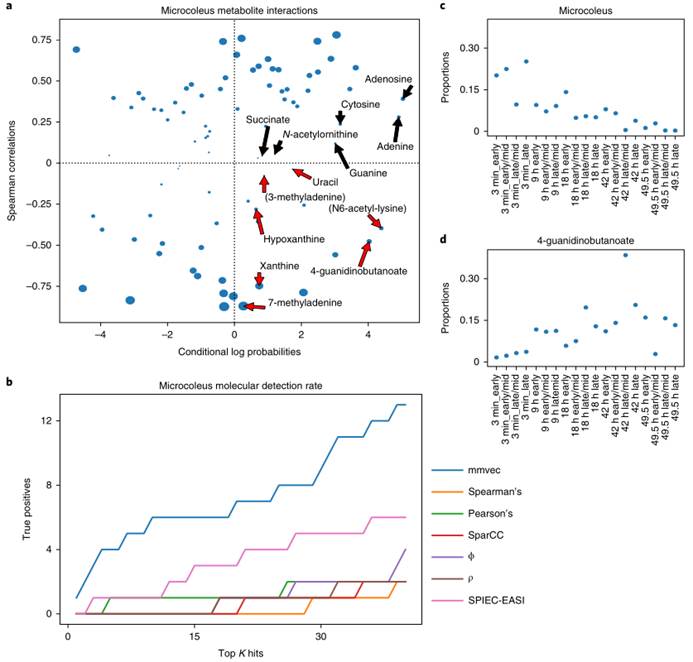
图a. 阴道分枝杆菌-代谢物相互作用的比较,根据Spearman‘s和mmvec估计(n = 19个样品)。由阴道分枝杆菌释放的所有经实验验证的代谢物都被标记。所有与生物润湿实验结果和体外实验结果相矛盾的代谢物都用红色突出显示。Spearman‘s标记的13个标签中有7个具有负相关性,表明这些分子被阴道分枝杆菌消耗而不是释放。
图b.经实验验证的分子在不同统计方法中的检测率的基准比较。mmvec具有相当高的真阳性率。
图cd. 阴道分枝杆菌(c)和4-胍丁酸(d)在生物润湿事件后的比例
MMVEC和Spearman‘s之间的冲突结果可以用生物润湿后微生物生物量的增长(c)和可用资源(d)的转移来解释。
3. 囊性纤维化患者的肺粘液微生物组研究,进一步验证mmvec是否可以检测已知的微生物-代谢物相互作用。结果表明mmvec可以可靠地识别由铜绿假单胞菌产生的所有经实验验证的感兴趣的分子
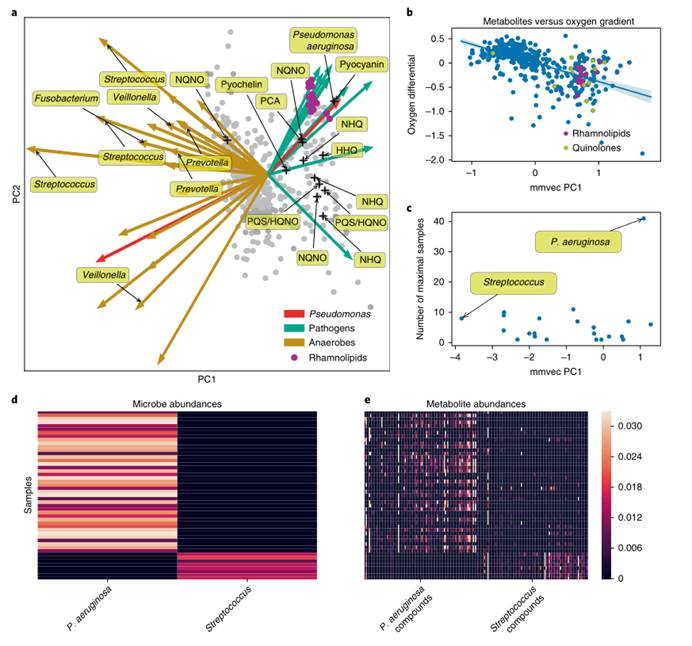
图a.依据mmvec在囊性纤维化数据集中估计的条件概率做的双标图。箭头代表微生物,圆点代表代谢物。x轴和y轴表示由mmvec (n = 138个样本)估计的微生物代谢产物的条件概率的奇异值分解(SVD)的主成分(PCs)。点之间的距离量化了代谢物之间的共现强度,较小的距离表明代谢物有很高的共现概率。箭头尖端之间的距离可以量化微生物之间的共现强度。箭头的方向性可以用来确定哪些微生物可以解释代谢产物的共现模式。绿色箭头表示推测的囊性纤维化病原体,黄色箭头表示已知的厌氧菌。只有铜绿假单胞菌产生的已知分子被标记。mmvec清楚地分离了厌氧菌和病原体,左侧是已知的厌氧微生物,右侧是显著的病原体。
图b.从mmvec学习到的第一主成分与代谢物在氧梯度上的对数倍数变化之间存在负相关 ( Pearson‘s r=−0.59,P=1.8×10−44,n=442个分子)。Pearson‘s法未发现氧梯度与第一微生物主成分之间的这种相关性(r=0.11,P=0.16,n=138个分子)。
图c. 第一主成分与样本数量的关系,其中分类群是该样本中最丰富的分类群。
图d. 铜绿假单胞菌和链球菌最丰富的样品的热图(log ratio t test = 6.51, P = 4.4 × 10−8, n = 49 个样本)。这提供了证据表明,在本研究的背景下,代谢组学特征在很大程度上受到最丰富的微生物的影响。
图e. 与铜绿假单胞菌和链球菌共生的前100个代谢物分子的热图。图中表示仅是预测铜绿假单胞菌代谢物谱就可以解释这些样品中10%的代谢物变异(r = 0.319, P = 1.18×10−11,n = 442个分子)。
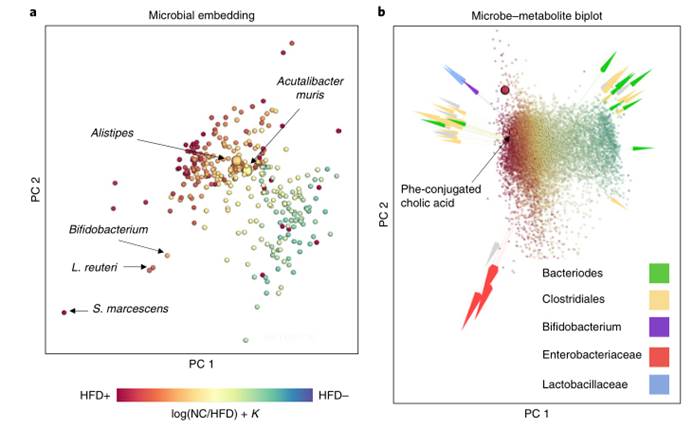
4. 胆汁酸研究。证明mmvec能够在复杂的生物系统中进行探索性分析,并简化特定代谢物的微生物来源的发现
图a. 微生物共生模式的可视化,其中点之间的距离近似于微生物之间的Aitchison距离,它量化了微生物发生的情况。较小的距离表明微生物具有很高的共生概率。微生物根据它们与HFD(高脂肪饮食)的关联被着色,这是通过多项式回归用差异丰度分析估计的。mmvec的使用显示了与HFD相关的不同微生物群
图b. 微生物-代谢物相互作用的双图,代谢物根据它们与HFD的关联而着色。HFD关联性通过多项式回归的差异丰度分析进行估计。点之间的距离近似代谢物之间的Aitchison距离,箭头间距近似微生物之间的Aitchison距离。表明mmvec根据饮食对质谱数据进行了清晰的分层。
5.炎症性肠病中微生物-代谢产物的相互作用。结果表示mmvec能够确定IBD研究中对代谢物丰度最强的微生物贡献,并发现了在最初的研究中被遗漏的一种微生物(Propionibacterium)
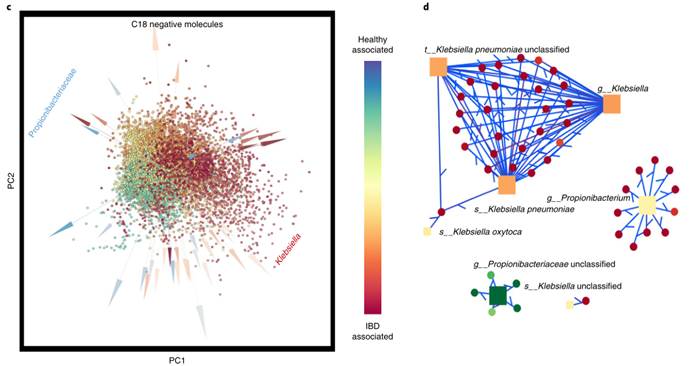
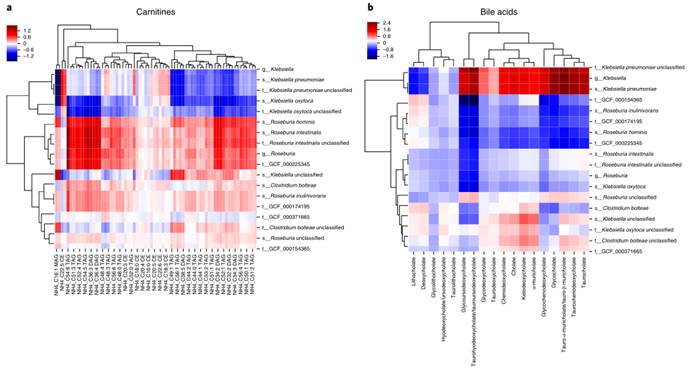


图a.和图b.分别为在三种菌属Klebsiella,Roseburia和Clostridium bolteae存在的情况下,推断各种胆汁酸(a)和肉碱(b)的条件概率的热图
图c.从宏基因组的概要文件和C18负离子模式LC-MS中学习到的微生物-代谢物相互作用的多组学双图。微生物(箭头)和代谢物(球体)根据多项式回归估计的差异着色。Klebsiella似乎与IBD密切相关,而Propionibacterium有强烈的负相关。
图d. 前300条边缘的网络,只有包含Klebsiella和Propionibacterium的边缘可见。
结论
作者表示,鉴于这些发现,目前的方法仍有局限性。目前还不清楚如何使用共现概率来获得相互作用的统计意义。同样,还不能计算每个微生物-代谢物相互作用强度的置信区间,还需要理论工作来处理连续值的输入。
Tips:文章末尾有关于mmvec算法的推导公式。
越来越多小伙伴开始关注我们产品的同时也会对产品的准确性,检测使用的技术等产生强烈的好奇日常提问:
检测后能得到什么有用的信息?
这个结果的准确性如何?
通过检测能直接区分患者和健康人吗?
同个人过一段时间做菌群变化大吗?
取样时间不同影响大吗?
疾病之间会不会相互干扰?
……各种问题层出不穷今天就为大家详细解答
以下是分享原文。

非常高兴有机会在这里跟大家分享我们这几年在肠道菌群检测应用于临床和健康管理方面的尝试,以及我们的一些经验。

谷禾成立于 2012年,是最早从事肠道菌群健康事业的公司,技术骨干源自浙大。建立了完整的pcr分子实验室。
我们比较专注,一直在做肠道菌群检测,从整个样本量来说,我们更关注来自于临床的有病理的信息,以及临床辅助诊断的数据样本。所以我们在这方面积累了相当大一部分的数据集。
而且这些临床样本相对来说,对于我们后续将肠道菌群检测应用于临床辅助诊断当中有很大的帮助。

我们其实不只是检测一下肠道菌群的构成,以及哪些菌有异常,我们是希望将肠道菌群检测直接做成一个临床辅助诊断的产品。
不只是告诉你可能有哪些疾病风险,我们希望可以直接给出包括结直肠癌、胃癌、甚至肝癌、抑郁症、自闭症的临床辅助诊断提示。
从这个角度上来说,可能跟目前已有的检测,包括这几天嘉宾分享的一些研究,可能就会有很大的不同。
你可以通过检测找到很多肠道菌群构成的显著性差异,比如自闭症患者跟健康人之间有相当大一部分的菌是存在丰度上的显著差异,但是:
能不能准确告知,哪个是自闭症患者,哪个是健康人,或者是他甚至有中间型的状态?
那么这个过程当中就需要解决几个问题。
第一个问题,首先是准确度
不仅仅是告诉你一个概率的问题,而且需要临床辅助诊断,那么就需要提高样品的处理本身,以及疾病诊断模型的准确度。
第二个是稳定性
大家都知道肠道菌群其实受的影响因素非常多,你的饮食方式、生活、地域、健康状态、甚至情绪状态都可能对菌群有巨大的影响。
这种巨大的影响的来源有这么多的情况,那么如何保证无论你什么时候检测,都能够是可靠稳定的?
假设一个结直肠癌患者,他今天做了,和隔了一个礼拜之后再去做,从病理的状态上来说,他还是个结直肠癌患者,但是他的菌群状态可能会产生巨大的变化。
那么这些变化本身是否会对我们的检测和临床结论产生巨大的影响,这种波动如何去消除,所以这个是个稳定性的问题。我等会也会讲到。
再一个是可解释性
因为菌群相对来说,实际算是一个大的数据。我们现在如果采用高通量测序的方式来做,一次性可以拿到大量的数据集。
这些数据集本身会有各种各样的菌的构成差异,我们的经验是差不多每个人,从婴幼儿开始到成年人,大概两岁以上的婴幼儿,菌群构成会从200到2000种菌不等,也就是说每个人会有这么大的菌的种类。
但是总的数据集有多大呢?我们自己有几万例的人群的样本,构建了一个人的肠道菌群的参考数据集,这个数据集里目前包括7500多种菌。但是我们自己的评估,全人类的肠道当中可能出现的定植菌应该会超过10万例。
那么这么大的数据量当中的异常菌如何去进行解释,如何给临床上提供更有意义的,病理上也好,或者机制上的一种解释,以及可以量化的干预方案,这就是可解释性。
最后一个是经济性
因为如果希望肠道菌群检测能够作为一个临床辅助诊断的项目,或者是针对具体的临床疾病的辅助诊断来说,它不仅仅要做到准确,它要具有足够大的经济性。
也就是说成本必须要得到控制,几千块钱的项目可能能做,但是它无法做到普及,也无法在临床当中被大量的应用,所以如何控制成本也是个巨大的一个问题。

我们做了很多的工作,尝试在上面提到的方向去努力实现在临床应用当中的可能,以下几方面我后面会逐一讲一下我们所做的工作。
第一是取样和储存运输,然后是如何大规模、低成本、高效准确的去处理样品。
再一个是参考数据库,完整的数据库的建立,其实也是非常重要的。
然后是大规模人群队列和临床数据。我们的核心经验,由于肠道菌群的多样性,以及受各种因素的影响比较多,那么大规模的人群队列就变成一个非常重要的点。如何去构建大规模的临床队列,以及从这个大规模的临床队列当中,我们能不能拿到一些信息和有用的经验。
再有是全方位的解析,我们等会儿会讲到不只是在菌群层面上,也不只是在代谢层面上,我们甚至可以基于肠道菌群,把代谢营养,生理生化指标,免疫指标,我们都是来自于临床的,包括血常规,尿常规,我们都能够进行解析。
还有重要的一点是如何使用人工智能的高可用性的模型,从这么大的数据当中精细化的提高检出率的同时,又能够保证它的特异性和敏感度,这是个巨大的一个挑战,这个我后面也会讲到。

第一个方面,可能我们现在采用的这个取样方式,应该相对来说最简便和最小的一个取样方式,我们直接可以用棉签从厕纸上蘸取,直接洗脱在取样管当中。
你可以看到取样颜色达到左侧第二个管子的这种颜色的粪便量,我们就可以完成整个检测,从使用体验上来说,会比较简便,而且需要量少。
样本保存也可以在室温下至少可以保存一个月,运输过程当中就不需要涉及到冷链,可以直接快递,便捷性也会大大提高。
有了这个取样管之后,实际上从临床和门诊当中可以快速的拿到大量的方便的样品,因为不需要采用非常复杂的取样和储存的方案。
我们讲第二个方面,刚才提到菌群的构成特点是很多样性的,而且跟很多因素包括取样时间有关,比如说早上取、晚上取,取的粪便的部位以及取样的量的多少,可能出来的菌群构成都会有一些区别。
如何再将这些区别和波动有效地控制,并且从中提取稳定准确的信息
这就涉及到一个我们能够从菌群数据当中能拿到一些什么结果,我主要从几个维度来讲。
——首先是菌群丰度和菌群结构
你首先可以知道每一种菌大概有多少的量,相对比例是多少。你还会知道菌群构成,也就是说都有些什么菌。
——然后是菌群结构。
所谓菌群结构就是说,有一些菌它会共同出现。甚至你会发现你检测到了有几种菌,并不代表其他的菌可能就没有出现。我们的肠道菌群总共可能会有七万多种菌,每个人差不多200到2000种,但是在99%的人当中都出现的菌,可能不超过30几种。你的肠道当中有这种菌,但是很多人当中都没有这种菌,那么很多的信息是稀疏的。但是通过构建菌群结构之后,你会发现这两种菌,可能一个在你这里有,一个在另外一个人当中有,但是这两种菌它其实代表的意义和内涵是类似的。
——再一个方面,我们通过数据的模型的挖掘,可以拿到更高维的特征。
这些特征反应的是生理的,比如说你的尿酸量,你的尿蛋白,你的血液当中的高密度脂肪酸,包括一些代谢的指标,这些指标的生理的特征和病理的特征,我们也可以通过菌群结构来进行提取。
那么从信息的维度上提升了之后,你可以看到数据的稳定性在不断提高。
最底层数据菌种的丰度波动性是非常大的。前一天的饮食如果有改变,跟你日常的饮食习惯有一些稍微的改变,第二天的检测,菌群的构成丰度上,波动甚至会达到30%。这种菌群丰度的变化,就代表如果你单纯依据少量的几种菌的丰度变化去检测它的异常,或者是这个疾病的状态的话,稳定性是很差的。那么你就需要控制各种场景,各种条件,才能拿到一个稳定的结果。但是菌种丰度又代表了一个非常高的信息量。
那我们尝试的更多的是从第二个维度开始,就菌群的构成,菌群的结构以及高维的菌群特征这个角度,因为它的稳定性更好。我们通过不断的去加入各种各样的临床病例的数据的方式来提取这些菌群的附加信息。
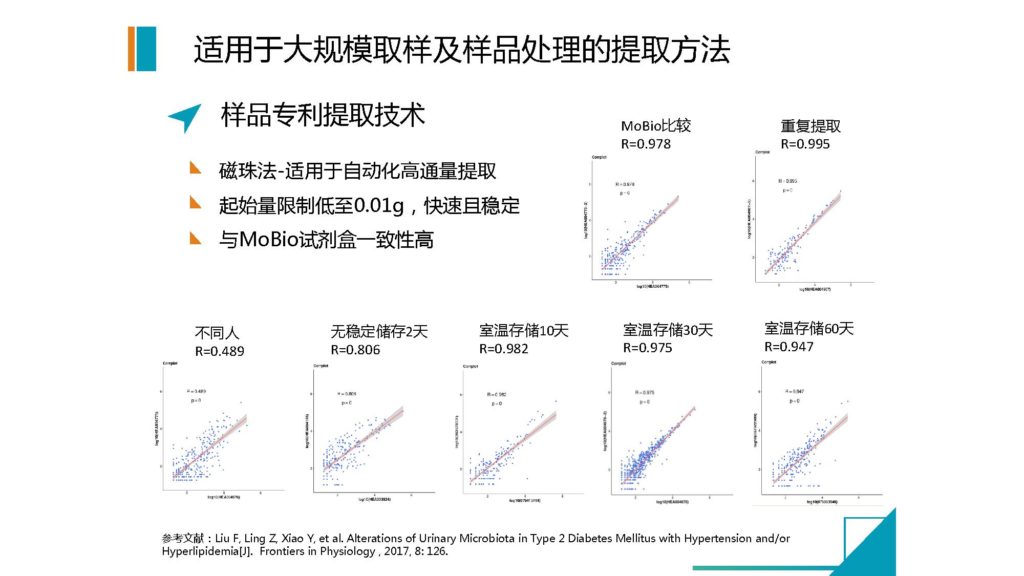
这就涉及到第二个问题,我们要把这更多维度的信息量能够提出来的话,你就必须要有涉及到非常大规模的样本人群,包括疾病状态、年龄、社会生活区域、饮食方式等各种因素的情况。
那么大量样本的话,我前面提到我们在取样盒上的改进,对应的我们还提供了一个快速的提取方式。就是通过磁珠法,来完全全自动化的来配合我们的取样盒,来做到大规模,自动化的,低成本的快速提取。
因为一般来说像MoBio这一类的试剂盒它对于样本的起始量有一个比较高的要求,并不适用于我们前面那种非常低当量的一个量。
我们自己改进之后的方法,稳定性和可靠性也是相当高的,这样是极大地降低了我们的整个实验处理过程当中的成本,同时又能够有效地保证这个检测结果的可靠性。我们的方法已经有文章发表。

那么当有了大量的样本之后,第二个问题就是
需要你更精准,更精细化的去提取这些数据
提取这些数据的过程当中我们自己也做过比较。用公共数据库包括Greengene、RDP或者HMP这些数据参考集,我们大概也就只能最多到95%的数据是能比对上去的,到种属的鉴定甚至会更低一些。
我们自己大概用了24000例的来自全球各地的样本,包括我们自己大概测了将近有17000多例的我们早期测的样本,还有各种来源的,特征的,包括疾病状态的,包括我们纳入了从各种基因组数据库拿到的肠道疾病和人体病原物的这些菌的数据,总共汇总之后,我们有24000多人的样本。
最后,我们构建了一个全新的人体肠道的一个参考集,这个参考集大概有75000多种OTU的菌,然后我们做了大量的注释,超过99.5%的菌是都能够完成比对的,这就大大的提高了对于菌属和样品当中菌构成的分辨率。
我们目前总的样本量已经接近快20万了,估计今年应该会超过20万例。
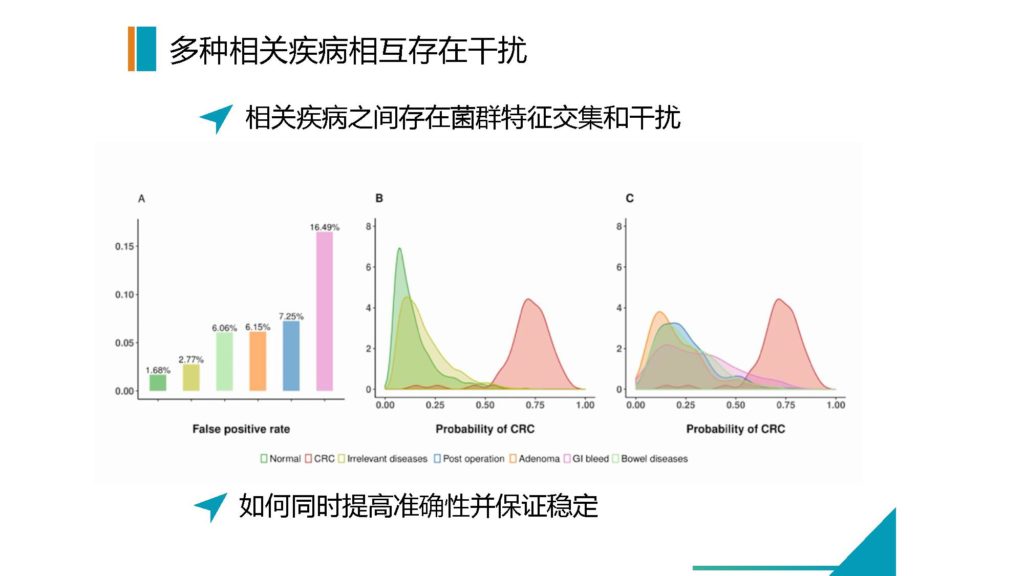
多种相关疾病互相存在干扰
这个是遇到的另外一个问题。
当我们解析了这些菌之后,我们尝试去做不同的疾病状态下菌的构成丰度和这些菌的特征信息,我们去尝试做疾病的分类。
前期做的时候效果相对还是非常不错的,因为它的特征菌比较明显。但是实际上面对临床的时候会遇到第二个问题,临床当中没有一个人的样品是非常干净,他可能会有结直肠癌,但是同时又会有高血压,或者是有消化道的疾病。
这些样本在你做检测之前你其实不知道他的状态,在试验或者研究型论文当中,你可能做的队列一个是健康人,再加一个某种疾病的患者,那么这两类的样本做出来,统计差异是非常明显的。但是如何在临床样本当中做到非常精准地将这两类人区分,而且不受任何中间因素的干扰,比如说阴性干扰样本的这个影响,这是需要面临的问题。
图解
上面图的左侧,我们自己做了一个结直肠癌的模型。结直肠癌我们现在检测的准确度可以达到非常高了。
但是,最开始做的时候,其实我们做了一下测试,单纯的模型去做预测分析的时候,会有其他中间疾病的大量干扰。尤其是消化道出血的情况下,会对整个菌群状态有非常大的影响。
包括腺瘤的阶段,刚才几位也都提到肠癌,肠癌其实是一个菌群变化要早于癌症发生的过程。但是菌群变化和癌症的阶段是有一些特征性的影响的,那么腺瘤的阶段跟肠癌是有大量的菌群特征是重叠的。
我们前期由于收集来自于各个来源的病例样本,所以可以大量的去寻找哪一些疾病是和我们要检测的目标疾病存在干扰因素的,我们可以挑选出这些疾病作为一个控制集,那么可以大大的减少假阳性和干扰的因素。
这也就是另外一个因素,就是我们在构建人群队列的时候,务必不能以一个相对干净的样本集去做。对于研究来说,它可能是很好的一个方式,你可以做前瞻性来寻找这是否可能以及效果。但是实际临床过程当中,你需要纳入大量的,可能影响你这个菌群,或者跟这个疾病有相互干扰和影响的相应的疾病来作为控制集,才能够提高它的可靠性和准确度。
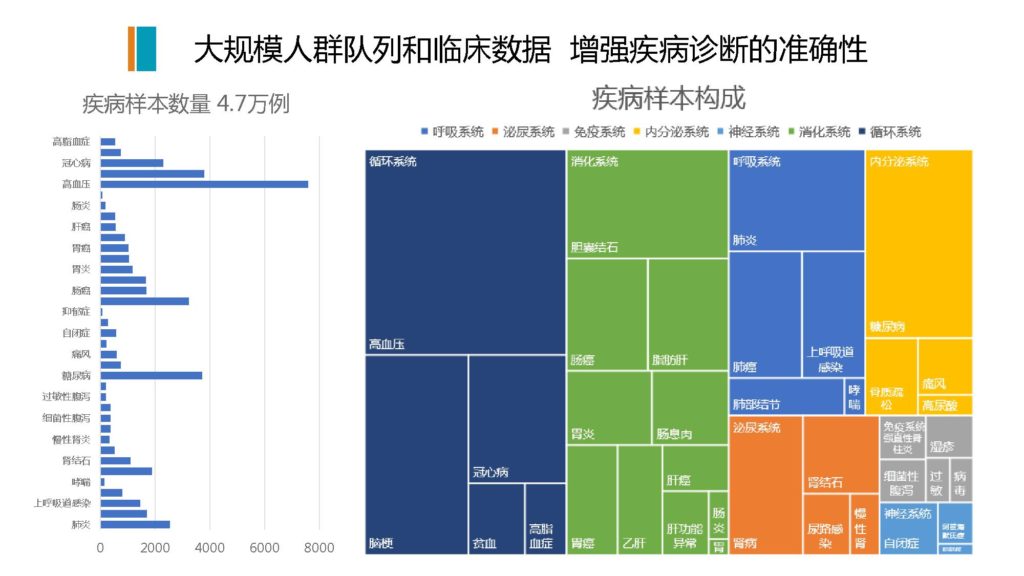
图解
这个图是我们自己有完整的临床病例,我们跟大量的医院和研究所在合作,我们自己构建了大量的人群队列规模,全部都是住院病人,有明确的临床的诊断和所有的病例信息,这个样本规模差不多有4.7万例。
图的左侧是各种疾病的类型,我们也通过各种疾病和菌群的模型构建,分析了七大类系统,包括呼吸系统,泌尿系统,免疫系统,内分泌系统,神经系统和消化系统,还有循环系统,跟肠道菌群能够有相对可靠的临床应用和检测,用于临床疾病的辅助诊断的可能性的。
右边这里是一个疾病的构成,其中有很多病跟菌群的关系目前甚至都没有发表过论文,就是说并不知道肠道菌群跟它有关。我们实际通过大规模的临床样本的实际筛查和模型构建之后,发现有很多病,通过肠道菌群可以做到非常精准。
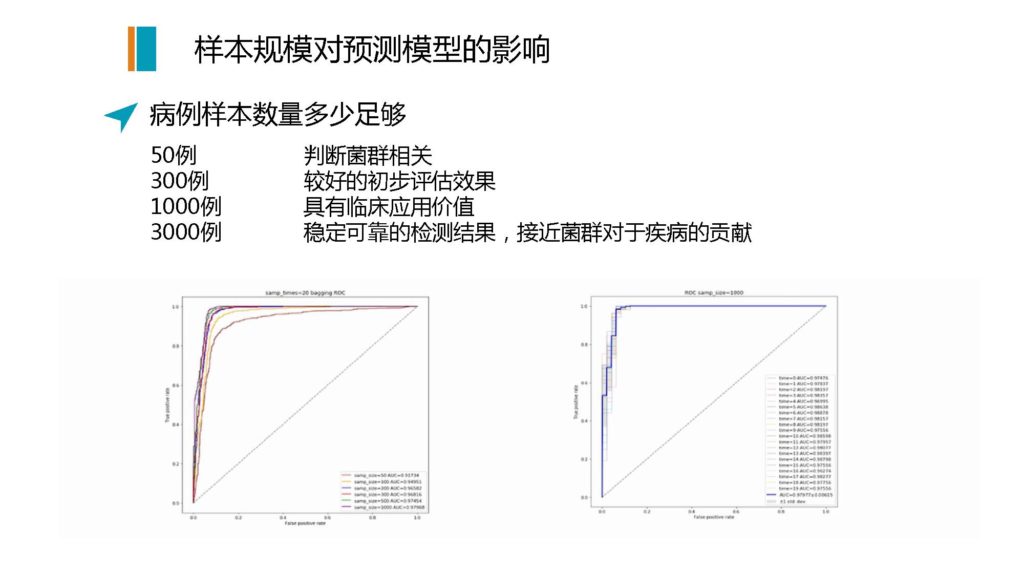
另外一个问题就是,我们对于一个病的预测也好,或者进行辅助诊断也好,基于肠道菌群
需要多大样本的量才能够做到足够的准确度
来看一个我们自己做的一个模型,是拿实际真实临床样本的数据来做的
图解
这个图实际上是拿二型糖尿病的患者的诊断来做的,可以看到不同的曲线丰度。
我们自己做了从50例、300例、1000例到3000例,这些都是病人的样本量,对照集的样本量一般会在两到三倍的量来进行构建模型。
从我们自己的这个模型数据来看,50例的样本,你可以有效地判断菌群到底能否对这个疾病进行一个相对较好的评估;那么如果是300例,你基本上可以拿到一个相对可用的模型,进行初步评估。
如果是要达到一个相对稳定的有临床应用价值的模型,我们认为差不多要1000例。因为你要纳入各种来自于不同临床疾病状态的样本,因为可能这个患者虽然有这个病,但是他同时还会有其他的疾病,包括不同的年龄和饮食习惯的这些背景因素要做控制,至少要1000例。
如果想要得到稳定可靠的检测结果,而且因为不是所有的病,菌群都是在其中起到绝对性的作用,有些是属于间接的,那么你希望菌群本身的检测,它需要有一个贡献度的上限,也就是说,菌群本身与这个疾病的参与度和关联性的上限。那么要达到这个上限,我们认为差不多要3000例的临床的阳性样本,就是病例患者的样本,可以达到上限的结果。
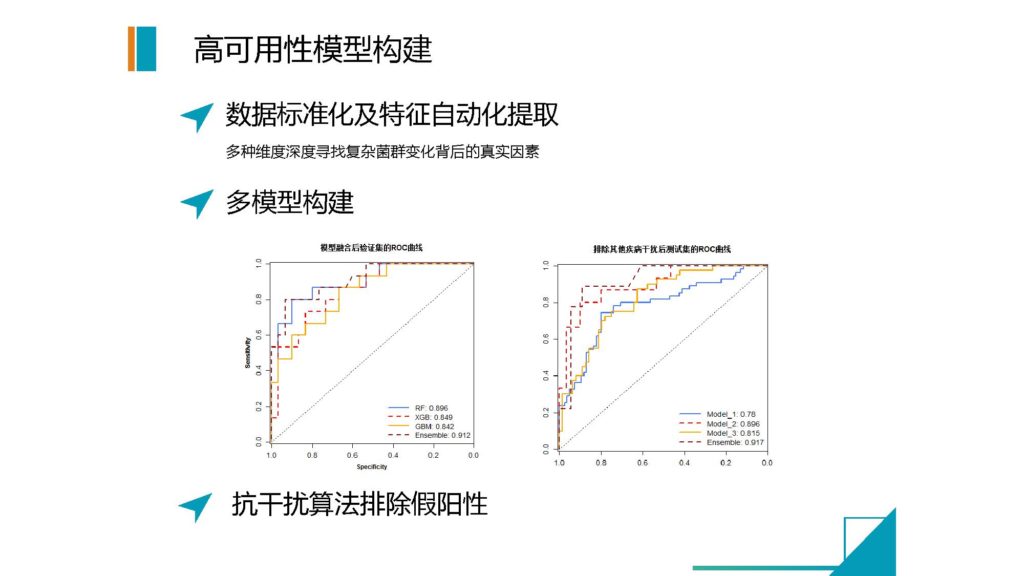
再下来,就是我们需要构建可靠的模型。
因为菌群是一个相对数据源,你的各种生活方式,疾病状态,营养健康的情况都会影响它。可能这个菌既在肠癌当中属于特征菌,同时也是一个炎症性疾病的特征菌,那么这些状态都会影响同一个菌的结果。
如何将这个菌的结果的变化反馈到去解释它到底是哪一个病的问题呢?
我们通过数据标准化和多维度的提升来构建一个判断的模型。
我们用人工智能和深度学习的方法,通过足够大的样本数据,来提取各种各样的菌群特征,并不直接用菌群自身的信息,而是用高维度自主学习的方式来提取这些菌群特征。
然后纳入各种各样数据,
比如有营养学的数据,有质谱的数据,
也有一些病理的数据,包括一些生理生化的数据,
都纳入之后,我们去解析它。
而且我们并不是用一个模型来做,我们现在是用三个模型来做。
我们第一轮是将所有的可能的干扰性疾病和有影响的疾病,全部作为一个病的类型,来进行模型的分析,筛出所有可能有问题的人。
然后第二轮我们需要精准化的去提取,到底哪些病是明确就是单一这种疾病的。
第三个模型,就是我们要对目标疾病与其他干扰的疾病进行区分。
通过多个模型之后,我们可以极大地提高菌群检测的稳定性,以及这个疾病当中的特异性程度。

图解
这个图是我们自己在做的一些疾病的检测结果。从目前来看,很多疾病的稳定性和效果都相当不错,这里每一个病至少都有将近500例病人的样本数据,来去做一个验证。每一个疾病的类型,我们差不多都有两到三个中心的检测结果数据去汇总。
通过多维度之后,我们就可以探寻不同的菌群变化背后,它可能真实驱动的因素。
我们还加入了营养的部分,这些营养其实我们是通过营养调查和一些质谱的数据,然后通过机器学习的方式来去把它解析出来。
我们也加入了像血常规,尿常规,生化,免疫组化,代谢组产物指标,肿瘤标志物,还有激素水平。我们将这些数据纳入之后,通过构建模型,可以将菌群的结果转换为这些生理生化相应的指标。也就是说,你如果只给我菌群的数据,我可以将这些生理生化的相应指标也能够给你体现出来。
甚至还有包括艾滋病的特征的,以及另外一些其它疾病,这里没有列出来,但是效果也是相当不错。
但是你可以看到,它的解释并不是指这个菌群直接的特征变化。我们通过菌群解析,像艾滋病,我们有免疫组化的数据解读了之后,我可以明确告诉你,CD4和CD8的比值会有特征性的差异。但它本身的菌群特征上并没有直接体现出这个东西,是通过生理生化指标的转换之后,我就可以告诉你,菌群特征的变化在具体哪些生理生化方面产生一些影响。
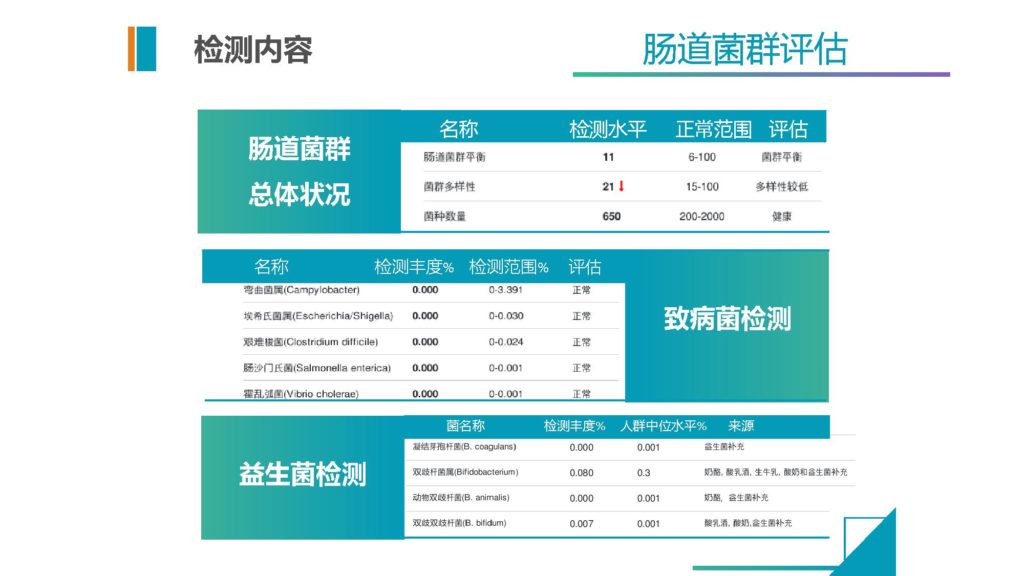
这个是我们现在提供给包括临床和健康检测的一些基本的内容。
可以提供菌群的总体状况,以及致病菌的情况,益生菌的情况。因为本身测的就是菌群,所以它直接就能提供这些最基本的一些信息。当然正常范围都是我们基于将近上万人的健康人群来做的正常范围的评估。
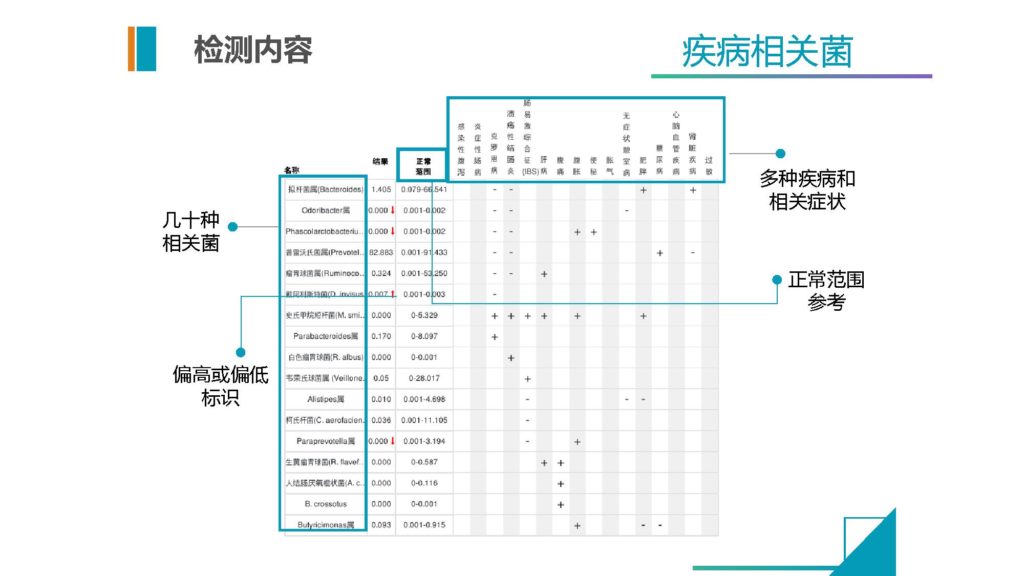
这个是除了来自文献之外,我们自己通过算法提取到的相关的菌,每一种菌在这个疾病内的相关性情况,以及它在正常人群的基本的正常范围是多少。然后我们通过检测这个是否异常,在临床当中给医生来做快速判断的一个内容和信息。
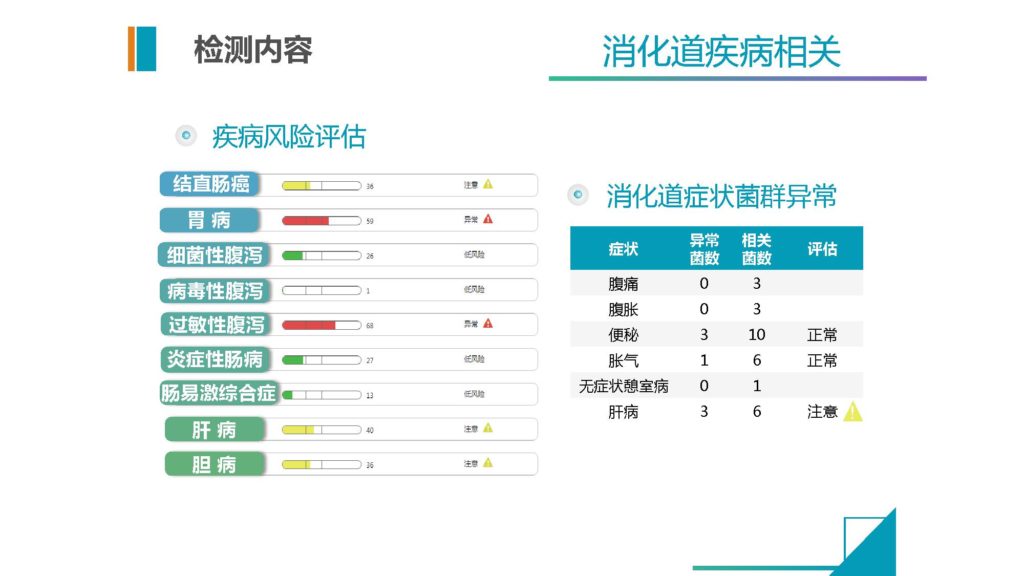
再有一个我们现在给一个疾病的辅助诊断,这些部分它可以相对有效的提供整个维度的不只是一个专科的信息,可以给到包括我们前面讲到七个系统的相关疾病的一个提示。
这些提示可以帮助我们来做一些专科性的疾病辅助诊断,能排除一些其他科室可能漏掉的一些疾病症状。另外包括消化道症状的解读,我们也会有一个菌群异常的评估提示。
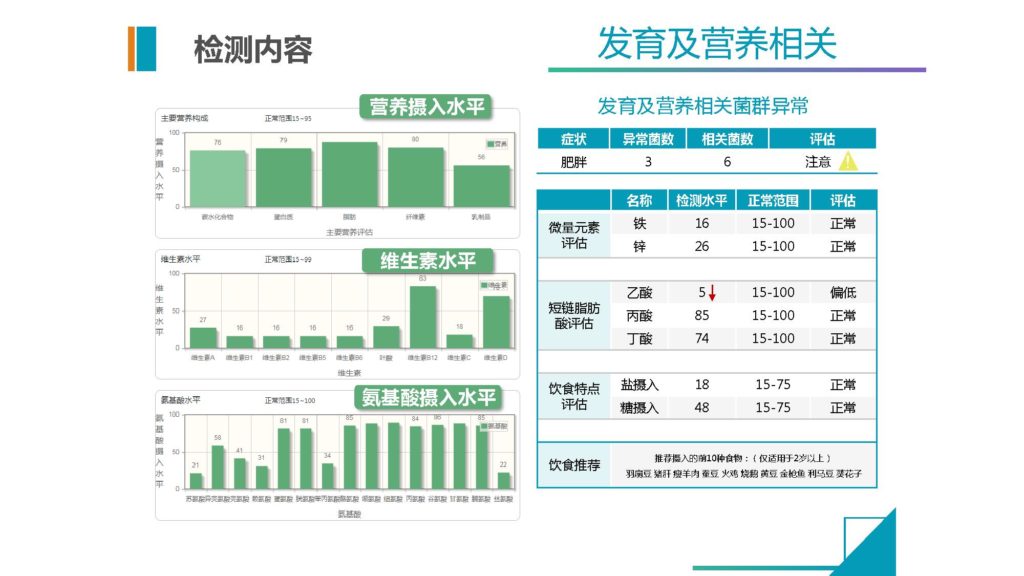
包括营养的部分,我们也单独加入营养摄入水平,维生素摄入水平,氨基酸摄入水平。
这个值目前来说我们是基于人群分布数据,就是说我们通过菌群来预测模型构建之后,评估出人群基础的水平,然后再基于人群的营养调查的水平,我们做拟合。现在来看,准确度还是相当的高。
那另外也包括微量元素,现在重金属的部分我们已经完成了,也很快会加入包括饮食特点、盐摄入、精制糖摄入对应的信息,还有短链脂肪酸的这些指标。

另外这里还有包括有心脑血管、神经系统的疾病的风险评估,包括过敏的一些问题。
过敏的话现在我们也在开展一个比较大的多中心的项目。关于过敏Broad Institute(博德研究所)去年还是今年有一篇文章,他们做的是一个大的欧洲的队列。
也就是通过菌群的数据,从刚出生开始持续采集,差不多到六岁,然后再去评估过敏以及过敏原。目前他那个数据我们做过测试,完全基于菌群的数据,对于过敏包括过敏体质的评估,我们差不多现在能到0.78左右。那如果你是做特异性的过敏原的检测的话,我们甚至也能够进行过敏源的评估,完全基于菌群数据。
所以我们自己的大量的数据检测完了之后,会发现,通过菌群数据本身,不止是能够告诉你菌群的结果,甚至能够反映非常非常多原来你意想不到的,需要用其他手段来进行检测的结果。
另外,我们现在的检测全部是基于16S,16S大家知道它本身的精细度可能还是有缺陷,就是它并不能到菌株;另外它只测细菌的部分,你的病毒和真菌是没有的。
但是我们这里可以看到有一项检测是病毒性腹泻,就是说我测的是肠道菌群,但是我们能够发现,这个病毒的感染,也会对整个肠道菌群产生一个巨大的影响。
所以我们通过整个菌群结构的特征变化,能够只检测细菌,也仍然能够发现病毒性的这种变化。

最后我说一下挑战。
第一个挑战,到目前为止我们做了这么多的数据和这么多样本量之后,竟然发现要真正去完整的解析整个菌群的特征,我们需要更大规模和全面多维度的数据集。不只是菌群检测本身的或者疾病的信息,我们需要纳入比如代谢组,以及其他的一些数据的情况,来构建更完整的数据集。
第二,我们发现不同的疾病,它的诊疗需求和特点是不一样的。虽然信息很多,但如何去跟临床辅助诊疗特定的疾病去做对接和结合是很重要的一点。
第三,我们现在的肠道菌群干预的手段其实也蛮多的,但是这些干预手段呢,现在缺乏量化,就是如何去评估每个人的干预手段,包括饮食的习惯,益生菌的菌株的水平的评估,益生元的效应,甚至粪菌移植的配体。这些量化的方面需要有大量的工作去做。
以上是我们这么多年做的实际临床大量样本的菌群检测的一些经验,跟大家分享一下,谢谢。

20251120
将以下菌列入病原菌列表,单独显示
解齿放线菌 Actinomyces odontolyticus
摩氏摩根氏菌 Morganella morganii
无乳链球菌 Streptococcus agalactiae
粘质沙雷菌 Serratia marcescens
索氏志贺氏菌 Shigella sonnei
化脓链球菌 Streptococcus pyogenes
解脲脲原体 Ureaplasma urealyticum
肺炎支原体 Mycoplasma pneumoniae
共生梭菌 Clostridium symbiosum
20250825
疾病模型有较大更新 , 胃 病 、 胆 病 、 肝 病 、 甲 状 腺 疾 病 、 I I 型 糖 尿 病 、 肺 部 疾病,大幅增加了临床病例样本数量,改善了检出率,部分疾病的假阳性也大幅改善了。
20250419
宏基因组报告采用新的版式
20240823检测报告
增加了【核心菌属】这个指标。
增加了【肠道炎症】和【肠道产气】两个指标
20240312
更新新增加了包括肠道屏障、菌群代谢物以及神经递质在内的19个指标。
20231116
对菌群多样性,菌群平衡,有益菌有害菌评估指标的分值评估标准进行了更新,以便更好地指导和区分状态
V20181010
修正苹果手机使用微信浏览器查看提示信息无法点开的问题
开始启用v3.3疾病预测模型,疾病之间干扰会更加少
修正认知障碍风险提示总览与详情部分分值不一致问题
V20180913
修正疾病总分计算中对于年龄的识别错误,原来部分低年龄段样本由于年龄识别判断bug会因为不符合年龄段的疾病风险导致疾病分值偏低。
修正后部分样本的疾病分值会提高,相应的健康总分也会更新。请以修正后的分值为准。
V20180808:
打印版增加个体食物推荐列表
雌激素水平评估修正完善
v20180801:
在线版更新
修正说明?点击不消失问题
修正侧边菜单栏窗口尺寸变化异常问题
加强报告主菜单栏阴影,便于快速识别
修正儿童总分计算偏低问题
修正管理版后台不同报告版本跳转问题,正确识别菌群版
v20180725:
打印版更新
修正建议文字清晰度
修正指标范围清晰度
在线版更新
增加40岁以上绝经女性非卵巢雌激素水平评估指标
更新健康人群队列数量,增加0~7岁健康儿童参照人群数量
v20180718:
更新在线绑定页面,减少问题填写
v20180714:
打印版更新
重新设计版式,全面更新以页面模式展示
增加厚壁菌、拟杆菌比例
增加香浓指数和菌种数量
更新疾病风险分值分布图,以实际验证人群检验模型和风险分值分布
在线版更新
以分栏式页面展示不同内容
新增分项健康评估分值
使用v20180710疾病模型,大幅增加结直肠癌、肝病等人群数量,部分疾病检出率大幅上升,总体分值相对统一
v20180615:
在线版更新
增加菌群详情部分,在页面中给出具体门、纲、目、科、属、种对应注释具体菌及丰度
修正部分文案错误
去除血清素缺乏建议中的药物建议
v20180610:
在线版更新
重新增加抗生素抗性中beta-内酰胺类药物的抗生素抗性风险评估
v20180510:
在线版更新
胃病调整,增加检测模型敏感度,增加胃癌样本
新增菌群检测版本,仅提供菌群信息
v20180420:
疾病模型更新,部分疾病使用深度神经网络模型,使用7万例样本进行基础网络构建。迁移学习提升少样本疾病预测可靠性。
v20180310:
在线版更新
增加个体化食物推荐
增加自闭症疾病风险评估
v20180114:
菌群注释大幅增加人体致病病原菌增加至340种,并全部注释到种,已去除25种分辨精度不足的菌
v20171210:
增加菌群详情,提供完整的菌群构成丰度和菌注释
v20171116:
更新疾病预测模型,提高疾病检出率,增加疾病风险预防提示,将潜在风险人群分值提升至0.3~0.5区段
v20170730:
更新疾病预测模型,使用新的机器学习方法,减少疾病之间相互干扰
v20170503:
暂时下线肥胖、慢性疲劳综合征以及季节性过敏和骨质疏松,因样本数量尚不足以使模型获得稳定预测,转入alpha测试阶段
调整肝病风险,精细化预测乙肝、肝功能异常、肝癌
增加营养部分,加入各种氨基酸、加入维生素A、B族维生素、D
v20170415:
君验专业版上线
完整提供菌群构成、疾病风险和营养构成三大模块
在线版更新
重新排版并增加说明和背景知识
增加疾病风险评估的准确度及人群分布图
v20161013:
增加血清素代谢及睡眠质量
增加人群分布,扩增为2.4万人
v20160715:
疾病增加胃病、肾病、肝病、胆病、心脑血管疾病、抑郁症、老年痴呆
v20160322:
增加疾病风险评估
首批疾病包括:IBS、IBD、结直肠癌、II型糖尿病以及自体免疫疾病
肥胖、慢性疲劳综合征以及季节性过敏
另外初步测试骨质疏松
v20151005:
增加代谢和6种营养成分分析内容
增加抗生素抗性评估内容
v20150712:
首个正式在线评估版上线
提供菌群多样性及菌群失调
健康人群分布
具体菌群情况
v20150510:
首批1万例临床病理样本检测完毕,疾病模型及人群菌群分布初步构建完成。
谷禾精准健康检测报告包含三个主要部分:肠道菌群、疾病风险、营养饮食
下面我们来详细解释报告是如何生成以及背后的技术和原理,以及如何解读报告。
我们首先使用24317例核心人群的肠道菌群基因测序数据构建了核心参考数据集,包括:
并对这些菌的特征序列进行详细物种注释。
这为我们对肠道菌群的构成和致病菌的检测奠定了基础,相较于目前的Greengene和SILVA132数据库的85%水平,我们的肠道菌群数据库涵盖了超过98%的人体肠道菌群。
基于这一标准化菌群特征参考数据集,我们进一步收集样本,并构建了如下样本人群队列
谷禾对全部样本和来自临床的病例进行了数据清洗和整理,并通过深度特征工程结合已有的基因组、药物、代谢等信息提取和构建深度菌群特征。
对每种疾病、营养指标都采用包括深度学习和基于决策树的人工智能模型进行预测和分析。

为了获得稳定可靠的预测效果,我们在模型构建和样本选择上经过多次迭代更新,针对肠道菌群数据开发了一系列优化方法,最终达到极高的准确度。
精准健康检测报告中首先给出了健康总分,总分100分,越高越好。
分值综合评估了菌群状况、疾病风险以及营养饮食的情况。存在疾病风险、有致病菌检出或饮食营养不合理都会降低健康评分。
健康总分的评价范围为:
>95:最健康
90~95:健康典范
80~90:很健康,针对性改善就好
70~80:健康但请注意生活方式和饮食
60~70:亚健康及营养饮食不合理
50~60:疾病高风险
40~50:疾病急需医疗关注
<40:多项疾病高风险,菌群严重破坏
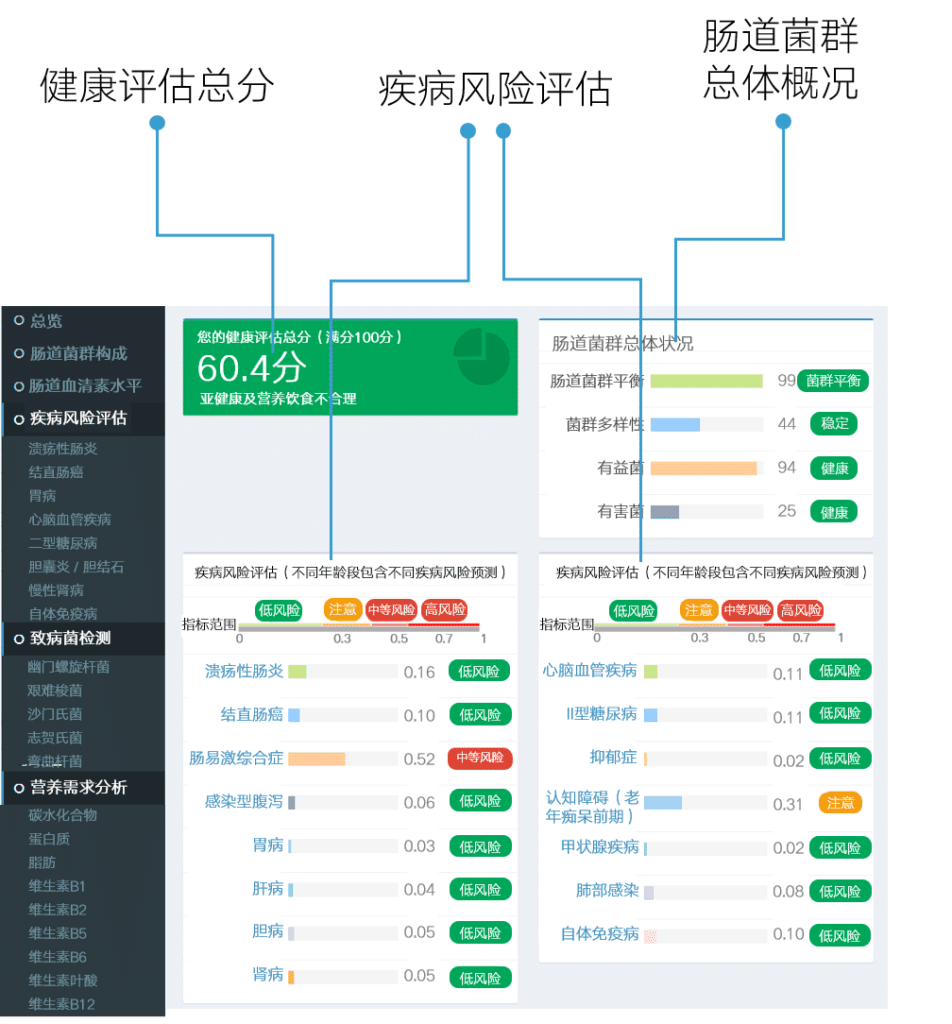
基因测序是直接对肠道菌群的16s进行测序,因而获得了极为准确和详尽的菌群构成特征。
通过对这些菌群数据的进一步分析,我们对肠道菌群部分给出如下结果:
下图给出了主要的菌群状况评估:

说明:报告中的分值包括两种数值类型,一类是0~100的分值,另一类是0~1的分值。
其中0~100表示的是在人群中的分布水平,比如70表示位于人群70%的水平。
肠道菌群平衡是根据有害菌和有益菌的比例分布确定的。
其中有益菌主要为乳杆菌和双歧杆菌。
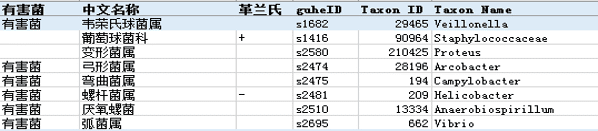
有害菌的定义如下:
目前的有害菌包括致病菌和条件致病菌,以及属内主要菌种为致病菌的属。为便于统计,我们在计算的时候统一按照属层级进行计算比例。下表是我们归属于有害菌的属。
另外报告中还会给出详细的主要菌属的丰度和人群分布情况。更加详细的数据表可以点击
下载完整菌群构成表
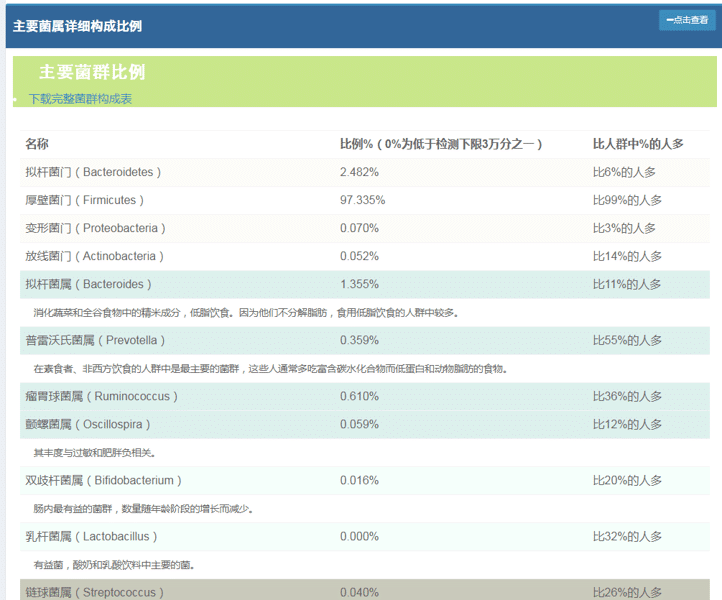
此外报告专门将常见益生菌和有益菌列出:
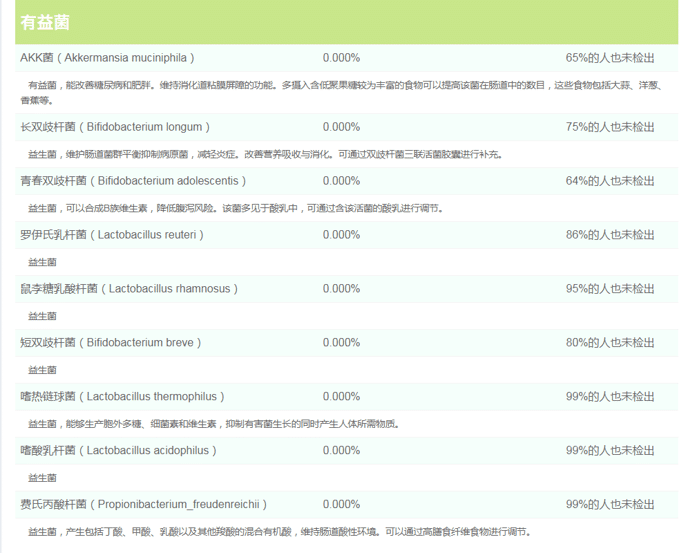
根据我们大量人群样本数据的统计和分析,我们从菌的层面提取了和不同疾病相关的菌,并监测其是否超标,超标标准为超出99%的人群或低于1%人群。
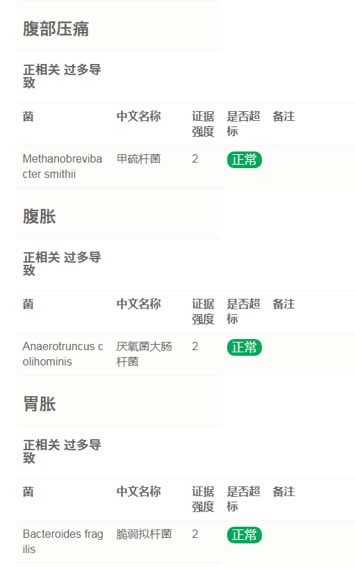
然后对每一种疾病分为病人和健康人两组队列,使用机器学习方法提取相关特征,使用深度神经网络进行模型训练,并在新样本人群中进行准确度的检验。
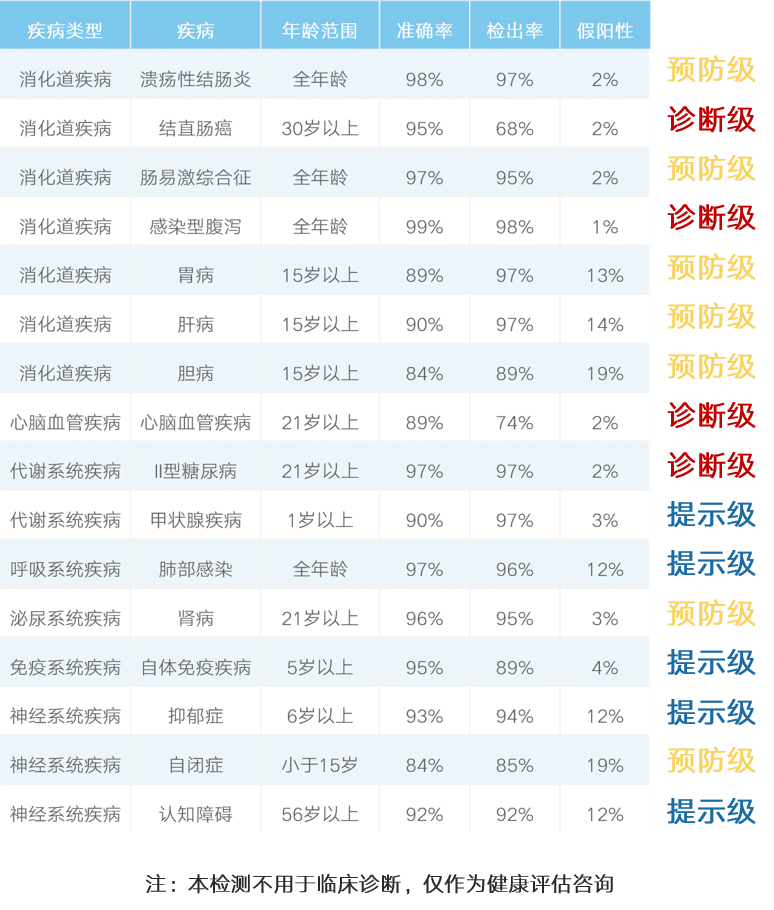
目前我们疾病风险检测部分包括16类主要疾病,根据疾病检测准确度和稳定性,我们将检测疾病的水平分为三个等级:诊断级、预防级和提示级。
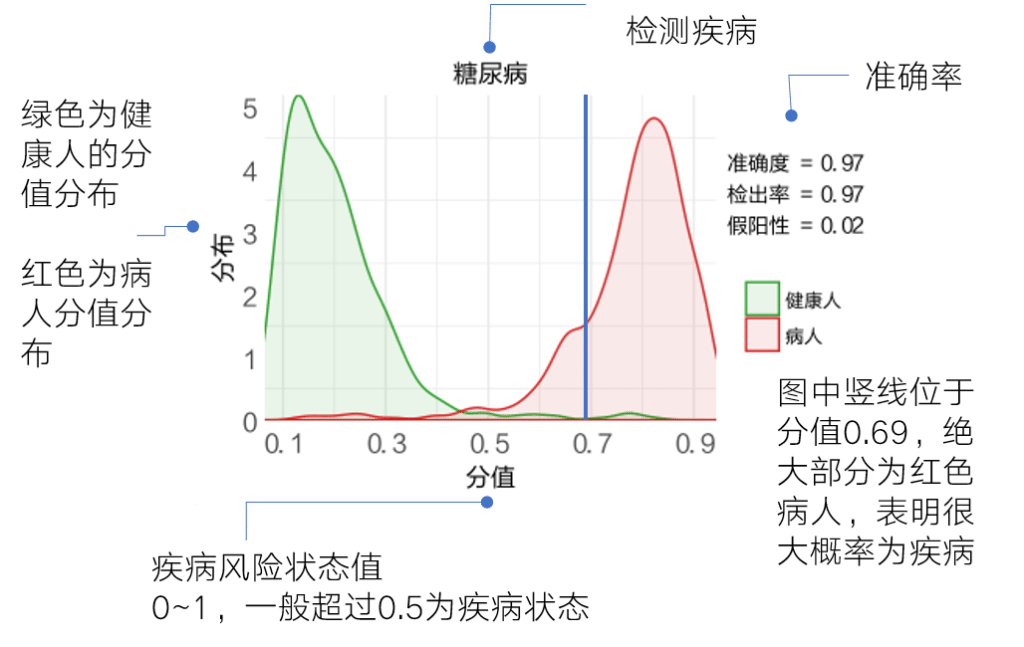
最终报告中,疾病风险以0~1的分值出现,并根据分值分为不同的提示级别,见下图:
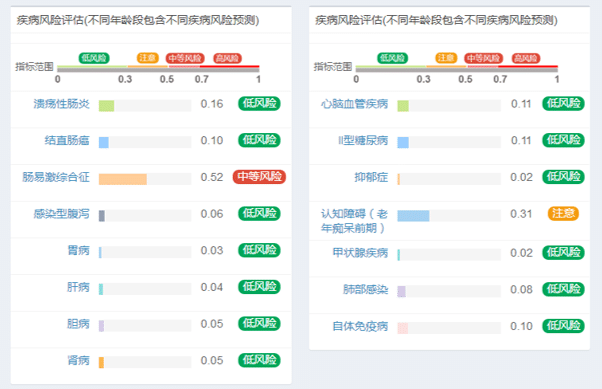
根据每种病的分值,0~0.3归为低风险,0.3~0.5评估为注意,0.5~0.7为中等风险,超过0.7为高风险。
目前报告中提供的疾病均经过大量病例样本检验并且准确率超过90%,虽然不作为疾病的诊断依据,但是其分值的高低仍然具有很强的指示作用。
如果您某种疾病的风险值低于0.3以下表明菌群状态提示疾病风险较低,不同身体条件和生活方式下会有0.05的波动。
如果您某种疾病的风险值位于0.3~0.5之间我们认为属于病前期阶段,通过饮食调理和相应的注意就可以降低风险。
如果您某种疾病的风险值位于0.5~0.7之间表明您可能患有该疾病或处于疾病风险阶段,我们建议您最好前往医院相关科室进行一下检查,如果不便前往医院也可根据建议先进行饮食调理和相应的注意,一般一个月后再进行一次检测查看疾病风险是否下降到正常范围,如果仍然较高甚至升高建议您最好前往医院复查。
如果您某种疾病的风险值超过0.7表明您有很大可能已患有该疾病,且分值越高表明风险越高。因此我们强烈建议您去医院进行相应检查并听从医生建议。
注意:本检测目前尚不属于医疗诊断,疾病分值作为提示,低分值不代表完全没有疾病,只表示风险较低,也可能存在一定的未检出。高分值只表示存在很大疾病风险,疾病的确诊和精确诊断需要通过进一步的医疗检查确认。
根据谷禾大规模人群饮食和营养元素调查的数据,通过机器学习模型构建基于肠道菌群的营养饮食和微量营养物质的水平评估模型。
报告中量化了包括主要饮食成分、主要氨基酸以及维生素和微量元素的水平。
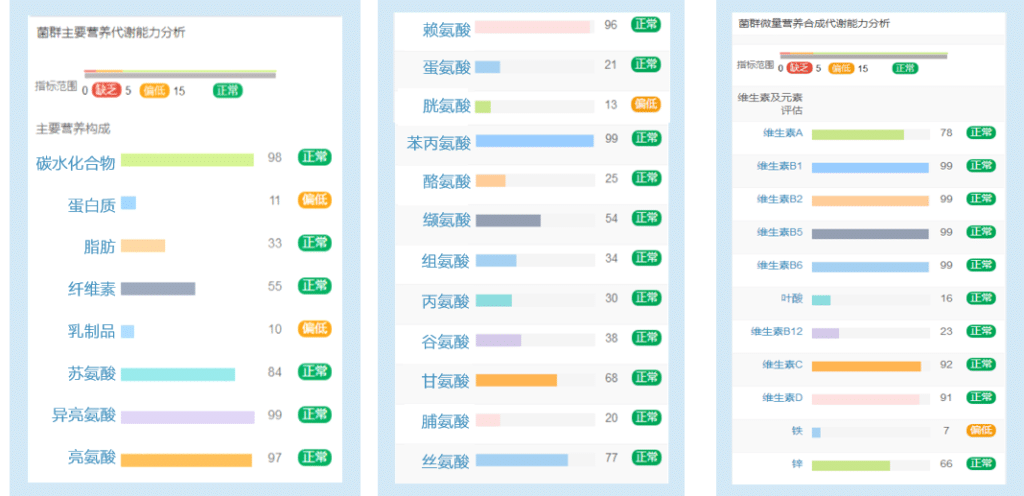
其中的分值为在人群中的分布水平,代表的是您的单项营养水平位于人群中的位置,一般最佳的营养分值为70左右,过高或过低都可能不均衡。
最佳的营养状况是各项营养水平相对一致,均衡是评判健康的主要标准。
上述营养指标根据我们对人群长达6个月的追踪发现,营养饮食的指标相对稳定,反应的是最近2周左右的一段时间平均的饮食摄入水平。
由于营养物质和微量元素随当日饮食会迅速变化,包括血液指标也会迅速改变,而肠道菌群反应的营养饮食状况受取样前一天的饮食的影响在15~30%左右,所以建议取样前一天尽量保持近期正常的饮食。
而营养指标的根本性改变通常需要改变饮食2周以上会有明显的变动,而维持该水准需要保持2个月以上的饮食习惯。
基于上述检测的营养饮食指标和疾病风险状况,我们结合不同食物的营养成分构成使用机器学习和统计方法计算了每种食物的推荐指数,从-100到+100。
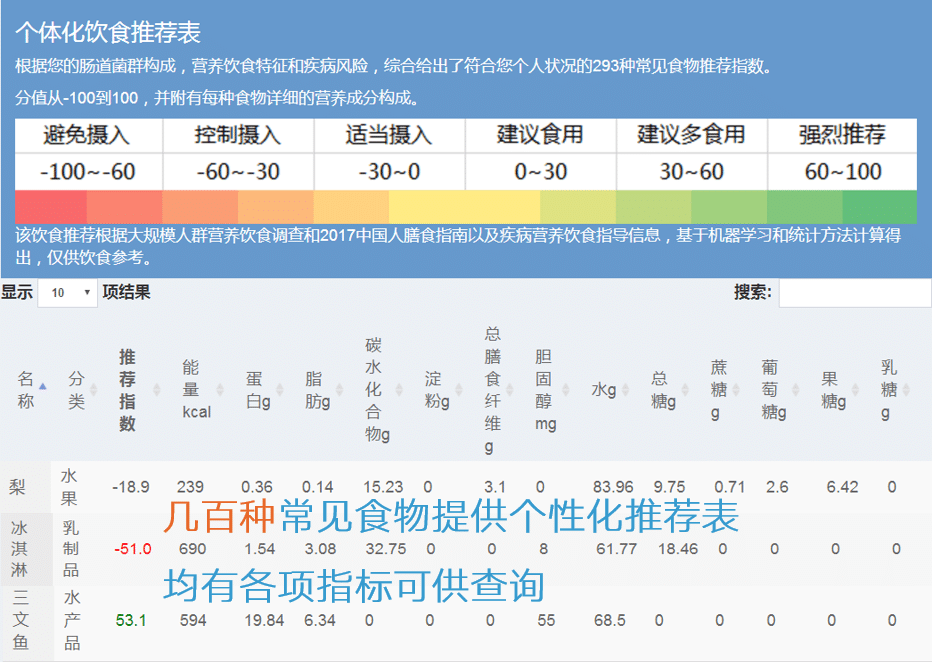
注:低于2岁以下婴儿,本食物推荐表仅做参考,也可作为母乳喂养妈妈的饮食参考。
以上报告版本为2018年3月v0.0.5版,疾病检测模型一般3个月左右会快速更新迭代一次以使用更大样本量来提升检测准确度和检出率。

谷禾菌群测序是通过对细菌的16S V4可变区域及部分病毒、真菌和寄生虫以及毒力和耐药基因进行靶向扩增测序,来对肠道菌群的种属和基因丰度进行检测。
肠道菌群DNA/RNA样本使用辅助自动化移液工作站自动提取和分液后进行PCR扩增。
再经过凝胶电泳和荧光定量PCR双重质检,最终进入上机基因测序。
严格的质量和扩增管控:
我们的测序平台使用二代高通量测序平台。

以下是谷禾测序检测的数据参数:
检测技术及方法:
自主粪便肠道菌群取样和提取方法
高通量测序
Q30质量大于93%
平均10万reads,最低5万reads
细菌16SDNA,V4区,引物:F515-R806
70%到种,致病菌95%特异性
最低质检标准1万reads