-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康

人们逐渐发现数以万亿计的共生微生物不仅占据人体各处,而且往往是人体健康生理不可或缺的参与者。这是自 19 世纪细菌理论首次彻底改变生物医学领域以来,生物医学领域最重大的进步之一。事实上,早在人类出现之前,我们可能就一直在与微生物一起进化和共同进化,我们必须将人类(以及大多数多细胞生命)重新设想为“全生物”,即超有机体,其进化轨迹只能根据宿主和微生物基因组赋予的复合表型来考虑。
人体是多种多样且功能重要的共生微生物群落的家园,这些微生物群落在不同的空间尺度上变化可预测,无论是在身体部位内还是跨身体部位。这些空间上不同的微生物群落的组成可能受到各种随机性和确定性因素的影响,包括来自不同来源群落的扩散,以及区域特定宿主过程对生理重要类群富集的选择。
尤其在过去十年中,科学家已经积累了大量证据,健康身体功能的许多方面都与我们携带的特定微生物组合及其各自的基因组或我们的“微生物组”密切相关。因此,了解哪些微生物出现在身体的哪个部位以及它们在做什么具有极其重要的临床意义。这一直是微生物组领域许多基础探索性工作的重点,特别是随着不依赖培养的细菌定量方法的出现,我们极大地扩展了我们对各种身体部位的微生物组组成、功能、稳定性和健康和疾病恢复力的认识。
本文我们将初步了解和认识有关健康人类胃肠道、皮肤、阴道和呼吸道微生物组的组成和功能的当前知识状态,特别是胃肠道内微生物的区域特异性,以及可能有助于每个栖息地群落聚集的生态过程。
微生物组研究已经从单纯的相关性调查发展到探讨微生物群落组装的机制,以期能够操纵这些过程,实现治疗目的。
微生物群落组装受宿主基因型、微生物间相互作用和环境因素(如饮食)的复杂动态影响。这些过程可以概括为四个基本过程:传播、选择、多样化和漂移。了解这些基本过程有助于更好地理解和操纵微生物群落的组装。(下图)
影响群落聚集的生态过程总结
★ 扩散
扩散是指微生物从一个局部栖息地迁入和移出到另一个局部栖息地(上图1A)。
想象一下,我们每个人的身体就像一个小岛。这个小岛上住着很多微生物。这些微生物可能来自周围的环境,比如其他人或者我们周围的空气、物品等。
我们体内能有哪些微生物,取决于两点:
举个例子,刚出生的婴儿体内最初的微生物,主要是从母亲身上或医院环境中”搬家”过来的。如果是顺产,婴儿会接触到母亲阴道里的微生物;如果是剖腹产,婴儿接触到的可能更多是手术室里的微生物。这就决定了婴儿一开始能获得哪些微生物。
★ 选择
适者生存,不适者淘汰
选择是一种确定性的进化力量,那些更适应环境的生物,能够很好地利用这里的资源(比如食物)的微生物,能活得更好,生更多的后代。而那些不太适应环境的生物,可能就会慢慢消失(上图1B)。
这些选择压力通常以“栖息地过滤器”或环境条件的形式出现(例如代谢资源的可用性、pH 值或粘附位点),限制哪些分类群能够在特定地点生存和生长。比如说,在一个特别酸的地方,只有能耐酸的微生物才能活下来。
有时候,微生物还要和其他微生物、我们身体的细胞,甚至是一些病毒竞争。
在微生物群落中,选择压力也可以通过与群落中的其他微生物、宿主细胞和噬菌体的相互作用来施加,从而改变可用的生态位空间。
即使一种细菌菌株理论上可以利用给定环境中的可用资源生存,但为了在该群落中生存,它必须能够胜过同样争夺这些有限资源的其他微生物。宿主细胞和噬菌体也可以对特定微生物的存在作出反应,并直接或通过改变环境来抑制或促进某些群体的发展。
★ 漂移
生态系统像一个随机彩票抽奖,持有彩票少的生物(丰度低的物种) 更容易在这个抽奖中“输光”
生态漂变是指由于出生率和死亡率的随机变化而导致物种丰富度的随机波动(上图1C)。从实际角度来说,这意味着低丰度物种更有可能从群落中灭绝,尤其是在受到干扰之后,即使它们可能比其他丰度较高的类群更适应该群落。在受到强烈干扰(如抗生素疗程)后,漂变在群落重组中可以发挥特别突出的作用。
★ 多样化
多样化是微生物不断地改写自己的
“生存说明书”,以适应不断变化的环境。
多样化描述了特定种群中新遗传变异的产生(上图 1D)可能通过突变、重组或细菌中常见的水平基因转移发生。
如果想象成一本书的话,“多样化” 就是不断地给这本书添加或修改内容:
值得注意的是,与较大规模的生物体相比,微生物的多样化发生的速度要快得多,因为微生物种群规模庞大、繁殖率高、突变率高。
最近的数据显示,常见的与人类相关的细菌,例如,脆弱拟杆菌,它能在一个人的一生中不断”改写”自己,更好地适应这个人的身体环境。
药物失效?可能和水平基因转移有关
当我们使用抗生素时,一些细菌可能会很快”学会”如何抵抗这种抗生素,这就是通过”复制”其他细菌的”抗生素抵抗“内容实现的。这就是为什么有时候药物会失效,因为细菌已经”学会”了如何对抗它们。
因此,多样化为群落中已经存在的细菌提供了一种适应不断变化的选择压力的方法。
在人体中,许多这些力量可能都在发挥作用。毕竟,不同的身体部位构成了截然不同的生态位。例如:
在每个部位,微生物还面临着来自宿主免疫系统的不同压力、暴露于外来微生物的程度以及资源可用性模式。
我的肠道微生物群与你的肠道的相似性,
比我的肠道与我的肘部的相似性更高
鉴于不同身体部位的环境差异,栖息地过滤和确定性选择过程在群落形成中发挥重要作用是合乎情理的,而且这在很大程度上得到了数据的支持。
人类微生物组计划的证据表明,人类微生物群在个体之间的相似性比在身体部位之间的相似性更高。也就是说,我的肠道微生物群与你的肠道的相似性比我的肠道与我的肘部的相似性更高,这表明肠道(或肘部)独有的选择过程以可预测的方式塑造了这些群落。
驱动因素是什么, 确定性力量 or 随机?
尤其是从生物医学和转化的角度来看,人们很容易认为选择等确定性力量是微生物组组成的主要驱动因素。
然而,微生物生态学家越来越多地致力于量化和解释更随机的过程(如扩散和漂移)在群落组装中的作用程度。
例如,寻求了解群落构成的微生物组研究人员必须考虑优先效应和历史偶然性,在这种情况下,群落可以通过在生命早期偶然接触有限的微生物子集而转向另一种稳定状态,然后占据和/或修改可用的生态位空间,影响后来到达的微生物的定殖能力。类似于“先到先得”,先到的微生物可能会占据最好的位置。
这凸显了这样一种可能性,即随机过程和确定性过程之间的反馈可能会放大群落构成的差异,从而使处于类似选择压力下的群落经历不同的组成轨迹。
这可能解释了健康个体体内微生物组成的一些显著差异。即使是健康的双胞胎,他们体内的微生物组成也可能很不一样。
就像是在玩桌游,游戏规则是固定的(环境的选择压力),但骰子的点数是随机的(微生物随机到达)。两个人玩相同的游戏,但因为骰子点数不同,最终结果可能大不相同。理解这一点对于研究人体微生物以及它们如何影响我们的健康非常重要。
胃肠道 (GIT) 非常与众不同,因为它要面对来自外界的大量挑战,还要在消化食物的同时做好防御。
每次我们吃东西,它都得识别并处理潜在的危险物质,同时把食物变成营养,便于吸收,还得向身体其他部位传递信息。
胃肠道不仅是消化的大本营,还有感觉、神经、内分泌和免疫系统,是丰富微生物群的家园。
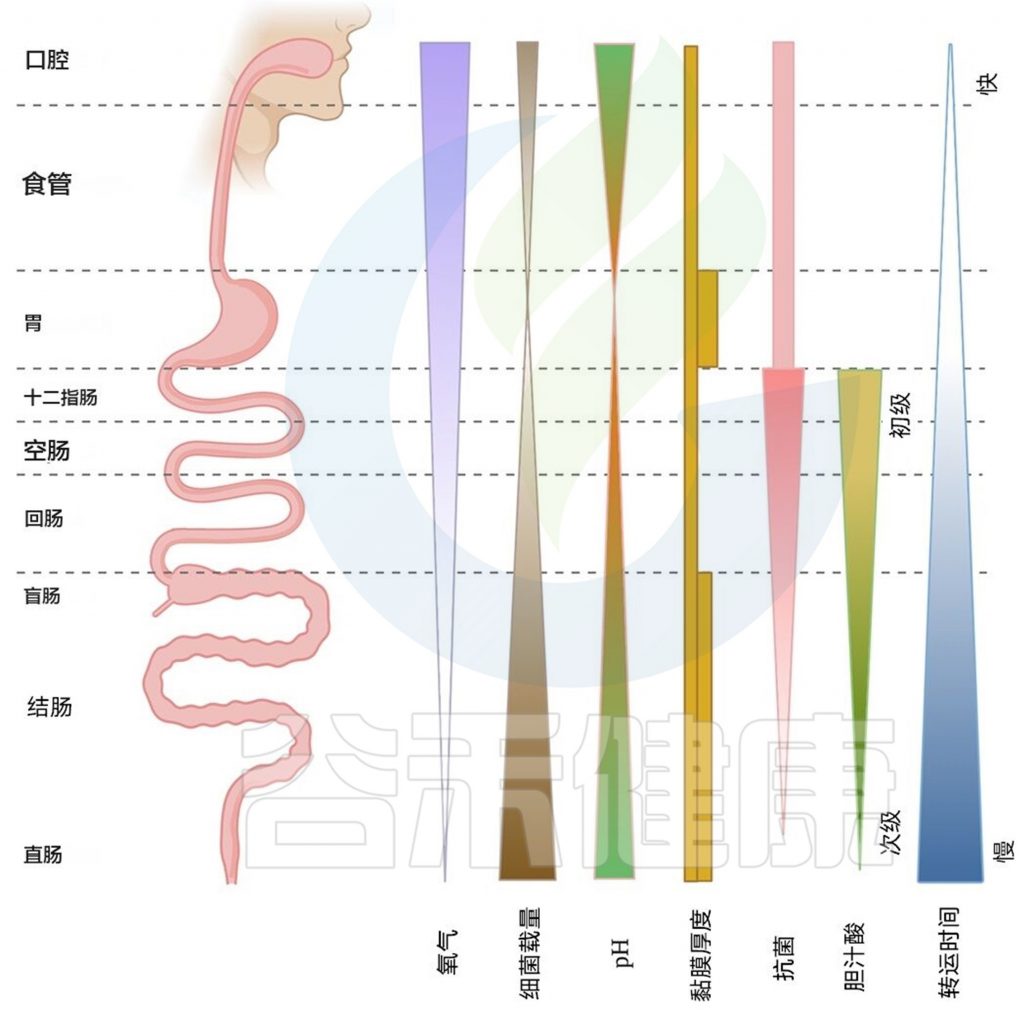
整个胃肠道的生态相关空间梯度
编辑
沿着这个轴线,有几个关键的梯度:
微生物负荷和多样性从喙端到尾端增加,氧气水平降低,粘液厚度发生变化,pH 值在胃中下降,并从那里向远端增加(上图)。
胃肠道的每个部分(我们将其分为口腔、食道、胃、小肠和结肠),都有自己的消化、代谢、免疫和内分泌功能,导致营养吸收和微生物环境有区域性差异。
这些区别以显著且通常可预测的方式影响局部微生物组成,而这些局部不同的微生物群落所执行的功能随后可以反馈到局部宿主过程中,形成互惠互利的共生关系,或者在疾病状态下,“菌群失调”的微生物群可能导致宿主功能受损。
尽管人们付出了大量努力来描述健康个体的微生物组,但对“健康”肠道微生物组的具体定义却一直难以确定。部分原因是,即使是在健康个体的同一身体部位,也存在相当大的微生物多样性,特别是在更高的分类分辨率下(即属、种或菌株水平)。
其中一些变化可能归因于起作用的随机和确定性构成力的特殊组合。在这里,我们描述了“健康”区域胃肠道微生物群的一些组成和功能特征。
口腔:防御与消化的前线
口腔是胃肠道和呼吸道的入口,承担着多种功能性作用,是重要的呼吸管道,也是许多消化过程的起始点。这些作用包括咀嚼、润湿、酶分解的初始阶段,以及味觉和其他感官接收,这些作用在防御有毒或致病因子方面发挥着重要作用。
作为入侵病原体与胃肠道首次接触的机会,口腔还发挥着关键的免疫学作用。口腔反映了这种功能作用的多样性,拥有一系列解剖和功能适应性,这些适应性创造了丰富多样的空间独特栖息地,这些栖息地的相对可达性为高分辨率的口腔微生物群空间采样铺平了道路。
口腔微生物群
最近的研究估计,大约有 200-500 种独特的细菌栖息在口腔中,数量达到约 2000 万个单细胞。
从解剖学角度来看,口腔包含各种不同的栖息地和拓扑特征,包括嘴唇、牙齿、牙龈、舌头、脸颊、硬腭和软腭以及扁桃体。这些特征之间的第一个关键区别是它们是否有脱落或不脱落的表面:口腔中唯一不脱落的表面是牙釉质,而口腔黏膜的其余部分则被定期更新和脱落的复层鳞状上皮细胞覆盖。
不脱落的牙釉质是口腔中更持久的结构,可导致牙菌斑或细菌生物膜的积聚。这些多种类结构是微生物合作和连续群落组装的迷人例子:丝状的棒状杆菌(Corynebacterium)触须提供了一个支架,组织良好的分类群(如链球菌、嗜血杆菌/聚集杆菌、卟啉单胞菌、口腔拟杆菌和梭状芽胞杆菌)根据微米级生化梯度附着在“刺猬”结构上。牙龈上方和下方的菌斑——即分别称为龈上菌斑和龈下菌斑——也包含不同的微生物群落,部分原因是营养物质和免疫分子在牙龈和牙齿之间的缝隙中被捕获和积累。
牙龈线上方和下方的牙菌斑(即龈上牙菌斑和龈下牙菌斑)也含有不同的微生物群,部分原因是牙龈和牙齿之间的缝隙中捕获和积累了营养物质和免疫分子。
尽管龈上菌斑的主要成员在整个口腔中仍然很丰富,但这些群落中较少丰富的成员因牙齿类别(门牙与臼齿)和牙齿方面(颊侧或舌侧)而异。
唾液流动是口腔中的主要选择力量
当唾液从各种主要和次要唾液腺从口腔后部流向前部时,它会产生水分和 pH 值的生物物理梯度,同时也起到物理运送细菌、释放或清除部分消化食物中的代谢物并刺激粘蛋白分泌的作用。
这些梯度选择富集臼齿上的韦荣球菌等分类单元,以及门牙上的链球菌和放线菌。牙周炎和龋齿等微生物介导的疾病通常与唾液分泌不足有关,这表明在缺乏唾液流动和口腔内相关微环境梯度的情况下,菌群失调群落可能会聚集在一起。
与不脱落的牙釉质相比,口腔黏膜的软组织具有脱落的上皮细胞,这些细胞大约每两到三周更新一次。据推测,角化(硬腭、舌背、角化牙龈)和非角化(颊唇黏膜、舌腹、软腭)组织上的微生物群落会根据粘附机制和与唾液腺的接近程度的差异而变化。
口腔微生物与免疫系统相互作用
作为摄入食物的初始部位,口腔也会接触到许多病原体,而这个群落聚集的主要选择力量是免疫系统的持续基础激活。因此,鉴于可分散到口腔中的微生物种类繁多,定期监测和基础免疫活动(例如通过 IL-17 途径)负责过滤掉潜在的致病因子。
口腔微生物群的共生菌,如链球菌(Streptococcus)、韦荣球菌(Veillonella)、Granulicatella,已被证明有助于这些防御过程,刺激免疫效应物(如抗菌肽 (AMP) 和促炎细胞因子)的产生增加,并增加上皮屏障功能和粘膜厚度。
与口腔相比,食管的解剖结构和功能相对统一。食管两侧为上、下食管括约肌,主要负责将食物从口腔运送到胃中。食管表面覆盖着非角化的复层鳞状上皮细胞,表面覆盖着粘膜下腺分泌的粘液。
尽管大量微生物会随摄入的食物经过食管进入胃部,但这些微生物被认为大多是短暂的。食管中更稳定的常驻菌群附着在粘膜表面,只能通过侵入性上消化道内镜检查(刷检或活检)进行取样,这严重限制了对该身体部位的特征描述。尽管如此,少数研究发现,食管粘膜主要被从口腔摄入的微生物定植,表现出高度的组成重叠,且均来自相同的主要类群,包括链球菌、普氏菌和韦荣球菌。
与口腔不同,食管中明显缺乏螺旋体。这些模式与分散受限的群落装配相一致,其中定植在食管的微生物是口腔中存在的元群落的一个子集。
胃在胃肠道中发挥着重要的消化、防御和内分泌作用。到达胃中的摄入物质通过蠕动搅拌进行机械分解,通过盐酸进行化学降解,并通过胃蛋白酶原等酶原蛋白酶进行酶促分解,胃蛋白酶原在酸性胃环境中被激活。胃还通过各种机制感知和调节食物摄入、运动和食欲,包括拉伸机械感受器和产生胃泌素和生长素释放肽等激素。这些功能既影响胃微生物群,又受其影响。
胃微生物:极端环境中的生存挑战
这些条件对微生物来说非常恶劣。胃部的低 pH 值(~1.5 – 2.5)构成了一个相当极端的栖息地过滤器,除了作为抵御摄入病原体的防御机制外,还显著限制了能够在那里生存的共生微生物的多样性。
从历史上看,这让科学家认为胃是完全无菌的,直到发现幽门螺杆菌在患病个体的胃粘膜中旺盛生长,这种传染性嗜酸细菌会导致消化性溃疡。然而,最近,不依赖培养的技术表明,虽然胃中的总体细菌负荷相对较低(~10 1 – 10 3 CFU/ml),但有多种门类的代表,包括链球菌、普氏菌、韦荣球菌和罗氏菌。
胃微生物群落:口腔与肠道的交汇点
在胃中发现的菌群中,超过 65% 属于口腔菌群,不过其中许多是在胃液中发现的,被认为主要是暂时性的。
胃中的群落聚集可以理解为口腔和十二指肠的扩散与主要受 pH 值(主要栖息地过滤器)驱动的选择之间的平衡。有证据表明,胃中 pH 值较高与可能来自口腔和肠道来源群落的分类单元的过度生长相对应。
幽门螺杆菌:敌人 or 朋友
酸度水平会受到质子泵抑制剂等药物或甚至常驻微生物的影响:幽门螺杆菌,据记录在美国无症状成人中的患病率约为 50%,它可以降低胃酸度。通过将尿素分解为氨(一种中和胃酸的基本物质),幽门螺杆菌可以潜入胃粘膜,在那里刺激免疫反应,导致胃壁细胞萎缩,进一步减少酸的分泌。
随着胃酸减少,它变得更适合从口腔或十二指肠扩散的微生物,最终导致“菌群失调”疾病状态。
尽管幽门螺杆菌在现代社会中被普遍视为一种地方性病原体,但有人推测,在工业化之前,这种普遍存在的微生物所引发的促炎性 IL-17 反应对其他传染性病原体具有保护作用。此后,细菌理论诞生,卫生习惯不断发展,抗生素也得到了开发,导致此类传染性病原体的流行率下降,并使幽门螺杆菌的促炎作用与我们“干净”的环境格格不入。因此,幽门螺杆菌现在最常与消化性溃疡病和胃癌有关。
小肠是胃肠道中营养吸收的主要部位
它具有单层吸收性柱状肠上皮细胞 (IEC),其间散布着各种特殊细胞,包括杯状细胞、潘氏细胞、微皱襞细胞和肠内分泌细胞,有助于消化、防御和分泌。这些 IEC 的顶端表面最大化了吸收表面积,具有隐窝、绒毛和微绒毛,并被一层松散附着的粘液包裹,其中基本没有微生物。
与大肠相比,小肠的需氧量更高、酸性更强、分泌的抗菌分子水平更高,并且运输时间更短。
小肠微生物:快速生长,抵抗抗生素
在这些区域定殖的微生物不仅必须通过这些栖息地过滤器,还必须与其他细菌和宿主细胞竞争营养。
因此,一般来说,小肠中含有大量快速生长的兼性厌氧细菌,这些细菌可以利用碳水化合物作为能量来源,具有强大的 AMP 耐受机制,并且尽管胃肠道运动迅速,但仍可以粘附在粘液、饮食底物或上皮组织上。
小肠的防御系统
为了促进营养物质进入肠内消化道,小肠的黏液层比胃肠道其他区域的黏液层薄得多。因此,小肠面临着在厚的次级黏液层可以提供的防御屏障功能与有效消化和吸收肠腔中营养物质的能力之间的权衡。为了对抗这种情况,小肠具有特殊的免疫适应性,例如隐窝底部的潘氏细胞,它们会将大量的 AMP(如溶菌酶和 α-防御素)分泌到黏液层中,以及丰富的浆细胞,它们会将分泌性 IgA 释放到黏液中,从而抑制微生物定植。
小肠上皮覆盖在黏膜和黏膜下层中特殊的肠道相关淋巴组织 (GALT) 和派尔集合淋巴结上,这些组织在抗原呈递和适应性免疫系统的激活中发挥作用。由于这种广泛的免疫活动,一些肠道共生微生物已被证明拥有独特的抗原修饰,以逃避先天免疫机制。
小肠微生物的重要性
小肠微生物群在健康宿主生理学中的独特功能作用的研究是医学微生物组研究中一个经常被忽视但新兴的子领域。虽然这主要是因为与粪便取样相比,小肠部位相对难以接近,但最近在动物模型中的研究已开始确定小肠微生物群在宿主代谢和营养吸收中的重要作用。例如,与已充分表征的结肠微生物群一样,小鼠的小肠微生物群受到西式高脂肪 (HF) 饮食的显著影响,事实上在甘油三酯的消化和吸收中起着不可或缺的作用。
这项研究表明,小肠微生物对于胰腺脂肪酶在饮食脂质反应中的释放是必不可少的,从而间接帮助宿主脂质消化。此外,作者还表明,在西式高脂肪条件下大量繁殖的梭菌科等菌类释放的代谢物会与十二指肠和空肠的吸收性肠细胞相互作用,从而增强游离脂肪酸的吸收。
因此,随着宿主消耗更多的脂肪,微生物群会发生变化,使宿主能够吸收这些脂肪。这是宿主和微生物之间双向串扰的一个显著例子,这对于维持健康的宿主生理至关重要,尤其是在饮食变化等快速变化的条件下。
从胃部向外,小肠可分为三段,每段都具有不同的功能作用、解剖特征和微生物栖息环境:十二指肠、空肠和回肠。
十二指肠——食物加工的第一站
十二指肠是小肠的前 10-15 英寸,就像是一个工厂的第一个车间。它是分泌和消化活动的重要部位,杯状细胞的粘液保护肠上皮免受酸性胃食糜的侵蚀,胰腺的碳酸氢盐将 pH 值恢复到中性区,胆汁和胰酶流入以帮助分解和吸收碳水化合物、脂肪和蛋白质。
十二指肠的特殊设计
从解剖学上讲,十二指肠上皮具有隐窝和长绒毛,微绒毛“刷状缘”充满了用于分泌的消化酶和用于吸收的营养转运蛋白。从免疫学上讲,十二指肠具有粘膜下 GALT,尽管派尔集合淋巴结比空肠和回肠少。
十二指肠的“工作环境”:氧气足,食物通过快
这些特征以特定的方式影响十二指肠微生物群。
因此,细菌负荷相当低,在 101 -103 CFU/ml 范围内,并且往往以厚壁菌门、放线菌门和变形菌门的细菌为主,这些细菌含有许多兼性厌氧菌。
胆汁酸:分解脂肪,耐受性强的细菌才能生存
初级胆汁酸 (BA) 由肝脏中的胆固醇合成,通过胆管直接沉积到十二指肠中。这些两亲性分子充当清洁剂,通过乳化脂肪球帮助脂质吸收。然而,初级胆汁酸已被证明具有很强的杀菌活性,因此可作为整个小肠中胆汁耐受性更强的微生物的选择压力。
胆汁盐水解酶 (BSH) 是一种分解初级 BA 的酶,它们广泛存在于肠道微生物中。有人推测这些 BSH 酶可增强对初级 BA 的耐受性,这可能在初级 BA 最丰富的近端小肠中尤为重要。
细菌版的”无线充电”
关于细胞外电子转移 (EET) 的令人兴奋的新研究还表明,十二指肠和空肠中特别丰富的铁可能是肠道细菌的重要代谢资源,肠道细菌可以利用 EET 产生能量。与哺乳动物细胞内的氧化磷酸化类似,这些细菌可以利用铁等细胞外矿物质作为电子受体,产生电能和 ATP,并支持细菌在不可发酵碳源上的生长。最近,在多种致病和共生厚壁菌种的基因组中发现了 EET 通路直系同源物,这表明 EET 可能是近端小肠资源有限环境中的重要竞争性适应。
总的来说,十二指肠就像一个高效、复杂的工厂车间,它不仅要处理食物,还要平衡各种化学物质,同时为特殊的微生物提供生存环境。这个”车间”的独特设计和工作方式,使它成为消化系统中非常重要的一部分。
空肠:吸收营养,高效的分拣中心
空肠具有与十二指肠相同的许多功能和解剖学特征,但含氧量较低、pH 值为中性、绒毛较长、派尔集合淋巴结数量增加、运输时间较慢,营养成分也发生了改变,这取决于十二指肠中已经消化或吸收的物质。与十二指肠相比,空肠的消化功能较少,更擅长吸收,是碳水化合物、小肽、维生素和矿物质(如叶酸、钙、镁和其他微量元素)的主要吸收部位。
微生物数量比十二指肠多
由于人类空肠只能通过内窥镜检查,因此研究健康个体空肠微生物组成的研究很少。早期与小肠细菌过度生长综合征相关的研究表明,健康个体空肠的典型细菌负荷约为 10 3 – 10 5 CFU/ml。
随后的独立培养分析证实了这一估计,并表明空肠在组成上主要由多种兼性厌氧菌属组成,包括链球菌、普氏菌、韦荣球菌、罗氏菌和梭杆菌,以及大肠杆菌和克雷伯菌。
虽然尚未从同一个体同时采集粘膜和腔内空肠样本进行直接比较,但对粘膜和腔内空肠微生物群的独立研究已确定了相似的分类群和丰度。
总的来说,空肠就像一个专门负责吸收的高效车间。它的环境比十二指肠更适合微生物生存,但仍然保持着良好的秩序。同时也为特定类型的微生物提供了生存环境。
回肠:最后的营养吸收,食物通过慢,粘液保护层加强防护
回肠继续沿着同样更广阔的环境梯度:氧气水平较低、运输时间较慢、绒毛较短、较宽且消化酶较少。到达回肠的食糜已基本耗尽宿主可利用的营养物质,其主要吸收作用是吸收维生素 B12和再吸收胆汁酸。
虽然回肠中的派尔集合淋巴结稀疏,但其隐窝中有大量分泌 AMP 的潘氏细胞,以及数量相对较高的分泌粘液的杯状细胞,这些细胞会形成厚度不一的斑块状粘液层。
在这里,向远端移动,肠道环境开始接近结肠中微生物生长的最佳条件:由于宿主已经吸收了其能够利用的大部分营养物质,因此栖息在远端回肠中的微生物不再与宿主细胞竞争剩余的资源。此外,吸收能力和防御之间的权衡已大大缓解,因为营养吸收在回肠中不那么重要,因此被认为可以降低近端小肠微生物密度的免疫机制也放松了。
回肠微生物:数量大幅增加,厌氧菌
与空肠相比,微生物负荷增加了几个数量级,估计范围为 103 – 10 8 CFU/ml。
由于如果不进行侵入性操作就很难对回肠进行取样,因此对其组成的了解大多来自接受回肠造口术或根治性膀胱切除术的患者群体。需要注意的是,这类患者群体通常具有潜在的健康状况,可能会或可能不会影响回肠微生物群的组成,这些研究表明,回肠是兼性和专性厌氧菌的家园,包括链球菌属、颗粒菌属、放线菌属、索洛杆菌属、罗氏菌属、孪生菌属和TM7(G-1)属的分类单元。
这与结肠微生物群有显著不同,严格厌氧菌科如梭菌科、消化链球菌科或真细菌科的丰度较低,而兼性厌氧菌科如链球菌科和乳酸杆菌科的丰度较高,尽管这可能部分归因于回肠造口患者的氧张力差异。
尽管免疫释放被认为是回肠中细菌负荷高于小肠其他部分的原因之一,但粘膜免疫在塑造回肠微生物组成方面仍然发挥着关键作用,反之亦然。
回肠微生物的重要性:帮助刺激免疫系统
潘氏细胞大量产生抗菌肽,如抗菌肽、C 型凝集素和防御素,在上皮细胞和微生物栖息者之间形成防御边界。然而,这些免疫效应分子的产生和分泌依赖于共生微生物的刺激:无菌小鼠的抗菌肽水平降低,如革兰氏阳性靶向 REGIII-γ,在与特定病原体无小鼠的盲肠内容物常规化后恢复。
共生微生物的免疫刺激也可以在抵抗某些病原生物的定植中发挥重要作用。例如,缺乏节段丝状细菌(一种回肠末端特有的微生物)的小鼠无法发育出Th17 细胞,因此极易受到柠檬酸杆菌和鼠伤寒沙门氏菌等病原体的感染。
回肠与生物钟:自动化系统,影响睡眠、进食和新陈代谢
回肠和结肠微生物在维持宿主昼夜节律方面也发挥着重要作用。昼夜节律是指全身组织基因表达的周期性 24 小时振荡,其作用是协调稳态功能,包括睡眠-觉醒周期、摄食行为以及葡萄糖和脂质代谢。
通过对各种分解代谢和合成代谢过程进行时间划分,昼夜节律能够减少身体资源利用效率低下,而这些节律的失调与糖尿病和肥胖症等慢性代谢疾病有关。
虽然中央昼夜节律时钟位于大脑的视交叉上核,主要受光刺激的影响,但肠道等外周组织中的振荡动力学也可以受摄食行为和微生物刺激的影响。
研究表明,在小肠中,Nfil3的节律性、昼夜节律性表达受肠道微生物群的调节,而抗生素引起的Nfil3节律性丧失与体重增加和脂肪储存有关。
总的来说,回肠就像一个复杂的、多功能的收尾车间。它不仅负责最后的营养吸收,还为大肠做准备,同时还是大量微生物的家园。这些微生物不仅帮助消化,还参与调节免疫系统和身体的生物钟。
结肠:巨大的发酵罐,数以万亿计的微生物
结肠是人体中微生物种类最丰富的场所,其微生物数量比身体其他部位加起来要多几个数量级,细菌密度为 1010 – 1012 CFU/ml。这是由该区域独有的解剖和功能特点所促成的。
厚厚的粘液层,高度厌氧,分解无法消化的纤维
与小肠一样,结肠具有单层极化的柱状肠上皮细胞,具有类似凹陷的隐窝,但与小肠不同的是,结肠上皮缺乏绒毛和微绒毛。结肠上皮中的杯状细胞分泌出厚厚的双层粘液层,内层致密且基本无菌,外层疏松且含有大量特殊微生物。
结肠是高度厌氧的,通过的消化物主要由宿主过程无法消化的复杂多糖和纤维以及微量营养素和回肠中未吸收的任何剩余胆汁酸组成。结肠中的运动要慢得多,通常运输时间长达 30 小时。
为什么微生物能在结肠大量繁衍?
首先,发酵代谢在细菌中广泛存在,大肠中的厌氧环境和丰富的纤维基质为这一过程创造了最佳条件。
其次,通过结肠的缓慢运输时间使微生物有足够的时间在物理空间中粘附、消耗、繁殖和扩张。因此,由于它们没有定期通过系统,结肠中的微生物可以积累到更高的水平。
第三,结肠提供了许多空间上不同的生态位,可以支持不同的微生物群落,从结肠隐窝到外粘液层,再到管腔和中央管腔的褶皱间区域。这种空间异质性允许对有限资源进行生态位划分,从而增加了可以支持的总体多样性水平。
最后,作为长期共同进化的共生体,结肠中的微生物已经发展出独特的免疫逃避和调节适应机制,以避免被宿主防御机制主动从肠道中清除。
微生物代谢产物——短链脂肪酸
微生物通过复杂多糖的发酵代谢合成短链脂肪酸 (SCFA) 是肠道微生物群的典型互利功能:这也许是第一个被发现完全依赖于肠道微生物“器官”的主要生理功能。在这个分解代谢过程中,纤维素、果胶、菊粉、高直链淀粉等纤维底物或宿主衍生的粘膜聚糖,被厚壁菌门和拟杆菌门的微生物共生菌厌氧发酵,释放出乙酸盐、丙酸盐和丁酸盐等 SCFA。
这些 SCFA 被宿主组织回收,估计占人类总热量需求的 5-15%。虽然所有这些 SCFA 都可以进入各种宿主和微生物代谢途径。
丁酸盐尤其是结肠细胞的首选能量来源
这些细胞将丁酸氧化成二氧化碳,从而消耗结肠中的氧气供应,并促进厌氧状态,这对于抗病原体、免疫稳态,当然还有产生丁酸的厌氧微生物种群的生长都很重要,这是一个典型的正反馈回路。
饮食对微生物的影响
宿主所摄入的饮食成分会显著影响结肠中的微生物组成,进而影响发酵和代谢。一项早期研究调查了在指定时间内从动物饮食转换为植物饮食的人类受试者,结果表明,微生物组成会随着饮食成分的变化而迅速且可逆地发生变化。
其他研究广泛记录了高脂和低脂饮食对粪便微生物群落的深远影响,高脂饮食使群落向厚壁菌门与拟杆菌门的比例转变。在稍高的营养分辨率下,最近的研究表明,施用特定的复合多糖可以促进特定拟杆菌种群的生长。
虽然结肠腔内的资源可用性因饮食摄入量而异,但许多结肠微生物都寄居在粘膜中并以宿主衍生的成分为食,而这些成分在不同的饮食行为模式下可能保持得更稳定。
结肠不同的”社区”:腔内生态位
构成结肠粘液大部分的糖蛋白粘蛋白 2 (MUC2) 被各种各样的 O-连接聚糖、寡糖所包裹,这些寡糖可以从 MUC2 上裂解并代谢以支持各种特殊微生物类群(如 Akkermansia muciniphila)的生长。
不同微生物在穿透和粘附粘液层的能力上有所不同,以及它们对从下层上皮细胞通过粘液向外扩散的 AMP 和氧气的耐受性方面也有所不同。
结肠不同的”社区”:粘膜生态位
与结肠中的腔内生态位相比,粘膜生态位经历了独特的选择压力,它支持来自放线菌门和变形菌门的耐氧、不进行糖分解的蛋白质代谢物种的富集。
在粘膜内,研究人员也记录了进一步的空间生态位划分。例如,不动杆菌属(Acinetobacter)物种特别擅长穿过粘液层及其相关的生化梯度,直接与结肠隐窝的 IEC 结合。
结肠不同的”社区”:褶间区域
与粘膜一样,管腔本身也具有显著的空间异质性,在大多数结肠微生物组研究中,由于使用粪便取样,因此忽略了这一点。小鼠的证据表明,结肠壁的特征性粘膜褶皱在肠腔内形成了不同的“褶间”区域,这些区域富含毛螺菌科和瘤胃球菌科的分类群。这些分类群被认为既受益于上皮粘液的局部积累,也受益于相对不受其他管腔内容物流动影响的环境。
结肠不同的”社区”:中央管腔
相比之下,中央管腔的消化物主要由来自拟杆菌科、肠球菌科(Enterococcaceae)、普氏菌科(Prevotellaceae) 、文肯菌科(Rikenellaceae)
从近端到远端,结肠中的微生物表现出局部特化功能,似乎广泛反映了资源的可用性。由于进入结肠的食糜具有较高浓度的复合多糖和胆汁酸,因此发酵和胆汁酸代谢主要位于盲肠和近端结肠。
胆汁酸代谢广泛影响结肠中的微生物组成
未被回肠吸收的初级胆汁酸进入盲肠和结肠,在那里,它们可以通过共生微生物中广泛存在的 BSH 酶进行去结合,和/或通过具有 7-α-脱羟酶的梭菌类(Clostridia)中的特定细菌物种转化为次级胆汁酸。
胆汁酸杀菌
胆汁酸对许多分类群具有杀菌作用,因此去结合和转化过程可以作为微生物群落成员的防御措施。次级胆汁酸通常对合成它们的细菌无害,但仍会对其他微生物分类群产生毒性作用。
一些细菌能把胆汁酸转化为对自己有利的形式
因此,在初级胆汁酸浓度较高的条件下,如在近端结肠中,我们预计将初级胆汁酸转化为次级胆汁酸的能力将带来物种特异性的生长优势。这一预测得到了文献的支持。一项针对大鼠的研究表明,胆汁酸库的大小可以对结肠微生物群落施加极强的选择压力:接受膳食胆汁酸补充剂的啮齿动物表现出微生物组成显著的门级变化,尤其有利于能够将初级胆汁酸转化为次级胆汁酸的梭菌种。
由于高脂饮食会诱导产生更多的胆汁盐以促进脂质吸收,这些胆汁盐反过来又会影响结肠微生物群。
胆汁酸与微生物的双向作用
胆汁酸池的组成和大小既影响整个结肠的微生物代谢,又受其影响,同样对宿主的健康和代谢有重大影响。例如,微生物 BSH 酶作用产生的去结合胆汁酸会破坏胶束形成,抑制胆固醇和其他脂质通过肠膜的吸收,这对心血管疾病有影响。
共生微生物产生的次级胆汁酸可以激活核受体,如法呢醇 X 受体 (FXR) 和 G 蛋白偶联受体,如 TGR5,在宿主组织之间进行通讯,调节初级胆汁酸的合成和降解、葡萄糖和脂质代谢以及能量稳态。最后,微生物合成的胆汁酸作为炎症疾病介质的作用正受到越来越多的研究。
炎症与微生物
宿主的炎症状态本身在结肠微生物群的组成和功能中起着重要作用,反映了免疫系统和胃肠道这个密集区域中的微生物群之间微妙的稳态平衡。在健康肠道中定植的微生物已经进化出多种机制来避免引发免疫反应。例如,许多共生菌株,特别是拟杆菌属的菌株,已经进化出一种对其外膜脂多糖 (LPS) 的修饰,使其对宿主衍生的 AMP 不可见。
其他共生类群已经进化出更明确的免疫调节策略。例如,脆弱拟杆菌多糖荚膜的一种成分可刺激调节性T细胞产生免疫抑制性白细胞介素10,从而使脆弱拟杆菌能够定植于粘膜微环境。虽然尚不清楚菌群失调是否会引发病理性免疫反应或反之亦然,但许多研究已证实,在炎症条件下,微生物组组成截然不同,其主要特征是厚壁菌门和拟杆菌门的含量减少,放线菌门和变形菌门的丰度增加。
皮肤是一个完全外在的巨大器官,其相关微生物群可能会暴露于任何身体部位最易变的条件。微生物可以从宿主遇到的任何源群落到达皮肤,扩散限制非常低,尽管皮肤在区域和局部尺度上表现出不同的生物地理学,但这些部位的条件往往因环境条件和宿主行为而异。
一般来说,皮肤是干燥、凉爽、有氧和酸性的
尽管在区域结构和褶皱中,这些物理化学参数以及微尺度拓扑结构的密度存在差异。某些区域,例如腋窝,往往更温暖、更潮湿,汗腺和毛囊密集。然而,即使是这个口袋也会受到时间和行为变化的影响——在炎热天气锻炼的人的腋窝环境与在寒冷天气坐着的人不同。
虽然毛囊或汗腺等微尺度拓扑特征往往可以充当更一致的栖息地过滤器,但即使是这些地点也会由于宿主不同的美容和卫生行为而频繁受到干扰。
因此,皮肤成为具有以下特征的场所:
这些标准是生态学文献中被最广泛引用的维持多样性的机制。这些特征为扩散和漂移等随机过程与皮肤的不同选择压力相互作用铺平了道路,从而在个体之间产生了各种可能的群落结果。
事实上,人类微生物组计划的数据表明,尽管胃肠道表现出可比的 alpha 多样性或站点内多样性,但皮肤在所有身体部位中具有最高的 beta 多样性,这意味着它在人与人之间变化最大。
此外,对皮肤微生物组的纵向跟踪表明,它也是时间上最不稳定的身体部位,随着时间的推移,个体内差异很大。
从结构上看,皮肤由脱落的层状角质化鳞状上皮细胞组成,其间散布着毛囊、毛孔和皮脂腺等结构。估计有 1011 个微生物细胞覆盖表皮表面,深入毛孔、腺体和毛囊。
这些结构不仅是物理空间异质性的重要来源,而且它们的生理功能会导致皮肤局部水分、酸度和盐度不均匀,从而形成独特的、通常具有选择性的生态位(下图) 。
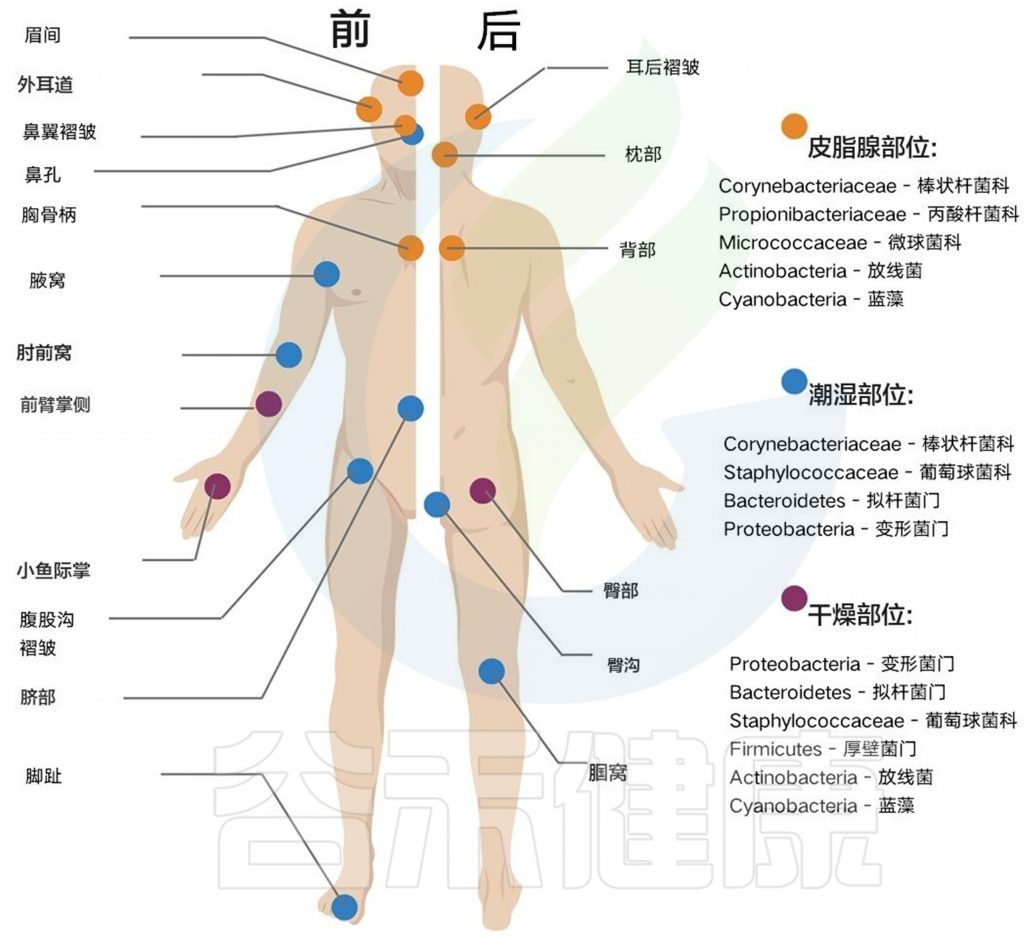
例如,顶泌汗腺会吸引棒状杆菌属(Corynebacteria)。在肾上腺素的作用下,这些腺体释放无味的类固醇、酸和其他挥发性分泌物,这些物质被棒状杆菌消耗并代谢成汗液特有的恶臭化合物。
皮脂腺与毛囊相连,会分泌一种富含脂质的物质——皮脂,以保护和润滑皮肤,皮脂腺对特定细菌也具有高度选择性。
皮脂腺密集区:吸引喜欢油脂的微生物
面部、胸部和背部等皮脂腺密集的皮肤区域与其他皮肤部位相比,其 β 多样性最低,并且不同个体的细菌类群始终相同。
这些腺体会招募亲脂性物种,如痤疮丙酸杆菌(Propionibacterium acnes),它会消耗和降解构成皮脂的脂质。这会释放出游离脂肪酸作为副产品,促进皮肤基线酸化,而皮肤是抵御病原体的主要屏障。皮脂腺部位也是真菌共生菌的家园,例如另一种亲脂性微生物马拉色菌(Malassezia)。

皮肤折叠区域:吸引大量喜湿细菌
此类拓扑结构的密度以及不同皮肤区域的更广泛解剖结构会影响这些位置的生理和环境。例如,温度和湿度在不同的解剖结构中存在巨大差异。较少暴露的皮肤折叠区域(如腋窝、腹股沟皱褶、臀皱或脐)往往比其他暴露的皮肤区域更温暖、更潮湿,因此会招募大量喜湿细菌,如棒状杆菌(Corynebacteria)和葡萄球菌。
较干燥、较暴露的部位(如四肢)会比身体其他部位经历更大的温度波动和更频繁的扰动,这可能是导致这些区域的细菌生物量相对较低的原因。尽管如此,这些暴露区域是人体中最具多样性的部位之一,代表了放线菌门、变形菌门、厚壁菌门和拟杆菌门。
从功能上看,皮肤微生物群对健康生理的完整性似乎不如其他身体部位的微生物群。大多数皮肤共生菌被认为是共生的,只有相对较少的专性互利共生的例子,即宿主依赖其微生物来实现特定功能。这在像皮肤这样多变的环境中可能并不奇怪——选择压力不一致,因此选择不能以定向的方式起作用。
尽管如此,有新证据表明皮肤微生物群在刺激和教育宿主免疫系统方面发挥着作用:分泌 AMP,如抗菌肽和 β-防御素,以及产生补体,都是由共生菌刺激宿主先天免疫受体引起的。
此外,在小鼠中,最近的证据表明皮肤共生菌可以通过改变 IL-1 产生的能力来调整和调节适应性免疫反应。最后,表皮葡萄球菌(S.epidermidis)等共生微生物已证实能够直接抑制金黄色葡萄球菌和 A 组链球菌(A Streptococcus)等致病菌的生长。
阴道微生物群是人体中多样性最低的部位之一,并且与宿主呈现出最紧密、最一致的互利共生关系。虽然阴道微生物物种会消耗宿主脱落的细胞和分泌物中的物质,但宿主被认为受益于阴道微生物可能发挥的保护作用,防止各种病原体的定植,包括那些可能导致酵母菌感染、性传播感染和尿路感染的病原体。
宿主与特定微生物类群之间的紧密联系以及机制上易于理解的互利共生表明,阴道微生物群可能是宿主-微生物共同进化的一个特殊例子。
阴道上皮由分层的、充满糖原的、非角化的鳞状细胞组成,这些细胞会迅速脱落,最上层大约每四小时更新一次。
厌氧、酸性、乳杆菌为主
它在育龄女性中主要是厌氧的,而且酸性相当强(pH 值 3.5 – 4.5)。虽然阴道上皮细胞可以产生乳酸,但大量证据表明,互利共生细菌(主要是乳酸杆菌)是这种酸性的主要来源,它们通过厌氧发酵过程将脱落上皮细胞中的糖原储存转化为乳酸。
在大多数健康育龄妇女中,该生态系统以乳酸杆菌为主导。从历史上看,大量乳酸杆菌和随之而来的酸性 pH 值被认为是“健康”阴道微生物群的标志,而 pH 值较高且更加多样化的微生物群与早产、细菌性阴道病 (BV) 等“菌群失调”情况以及加德纳菌或滴虫等阴道病原体的存在有关。
然而,在不同人口群体中进行的更广泛的不依赖培养的抽样显示,大约 20%-30% 的健康女性拥有更加多样化、非乳酸杆菌主导的微生物群,这再次混淆了我们对该生态系统中“健康”的确切定义的理解。
健康女性的五种阴道菌群状态类型(CST)
这项研究在健康女性中发现了五种不同的“群落状态类型”(CST),其中四种类型分别由不同种类的乳酸杆菌(L.crispatus、L.gasseri、L.jensenii、L.iners)主导,第五种(CST – IV)由多种兼性和严格厌氧菌群主导,包括来自Atopobium、棒状杆菌、厌氧球菌、Peptoniphilus、普氏菌和加德纳菌属的微生物。

从人口统计学上看,黑人和西班牙裔女性更有可能患上 CST-IV 群落,而白人和亚洲女性更有可能患上其他任何类型的群落。
阴道菌群五种分类详见我们之前的文章:
人们认为,阴道天然微生物群通过多种机制对阴道抵抗病原体具有重大贡献。
乳酸产生,降低pH值,增强保护
首先,常驻乳酸杆菌会产生大量乳酸,而宿主细胞和其他微生物栖息者会产生较少的乳酸,这种乳酸对许多入侵病原体来说是不适宜的,或者直接具有抑制作用,包括性传播病原体,如沙眼衣原体、淋病奈瑟菌(Neisseria gonorrhoeae)和 HIV。
不同乳酸杆菌种会产生不同数量和异构体的乳酸,从而不同程度地降低阴道 PH 值,从而降低其保护能力。
免疫调节
其次,乳酸已被证明具有保护性的免疫调节特性。尽管促炎环境与 BV 和 STI 的获得有关,但 Hearps等人的研究发现。
已经证明乳酸可以诱导抗炎细胞因子 IL-1RA 的产生,并抑制促炎细胞因子 IL-6 和 IL-8 的产生。
抗菌物质
第三,阴道乳酸杆菌可以产生抗菌细菌素,专门针对克雷伯氏菌、加德纳氏菌、大肠杆菌和粪肠球菌等病原体。
竞争排斥
最后,有人假设天然阴道微生物有助于通过竞争性排斥部分防止病原体定植:天然阴道微生物群更适应阴道环境,可以更有效地提取那里的有限资源,防止潜在入侵者生长和建立。
为什么某些健康女性的阴道菌群以乳酸杆菌为主导,而其他女性却没有,以及在有乳酸杆菌的女性中,为什么不同的乳酸杆菌种类会占主导地位,这是一个悬而未决的问题。
在胃肠道中,我们很容易推测选择压力和栖息地过滤器会影响群落的形成,但在阴道中尚未发现明确的栖息地过滤器。
许多人假设乳酸杆菌是因为它们具有代谢糖原衍生资源的能力而被选择,但这仍然无法解释为什么其他糖原代谢细菌在这些群落中并不更突出,或者为什么在已知的 130 多种乳酸杆菌中,只有 4 种在 70-80% 健康女性的阴道菌群中占主导地位。
旨在识别这四种乳酸杆菌特有的栖息地相关性状的功能基因组学研究未能成功。
关于单菌株优势的一种推测性解释可能是,优先效应使得单一、早期到达的乳酸杆菌种群得以建立,从而赋予其空间和数量的生长优势,限制了后来到达的种群的竞争能力,但这种可能性需要实验研究。
慢性干扰
鉴于生殖生理的周期性,阴道的生理学与其他身体部位相比具有独特的时间依赖性:激素水平随规律的月经周期而波动,怀孕会导致一系列剧烈的解剖和功能变化,这些变化随着时间的推移有序地展开。
急性干扰
除了这些对阴道微生物群的长期慢性干扰外,阴道还经常因性行为、使用避孕药、润滑剂和月经产品以及其他行为习惯而受到更剧烈的干扰。鉴于该群落慢性和急性干扰的规律性,阴道微生物群是否稳定或有多稳定一直是一个至关重要的问题。
集“稳定”与“弹性”于一身
有趣的是,健康的阴道既非常稳定,又具有很强的弹性。阴道群落的急性行为干扰以及每月规律的月经周期似乎只会引起微生物群落成员的轻微变化,大多数群落最终会恢复到其基线组成状态。
类似地,对整个妊娠期阴道微生物群的纵向研究表明,尽管 CST-IV 在孕妇中的代表性降低,表明妊娠导致乳酸杆菌占主导地位,但在整个妊娠过程中,阴道微生物群落与非孕妇相比保持异常稳定。
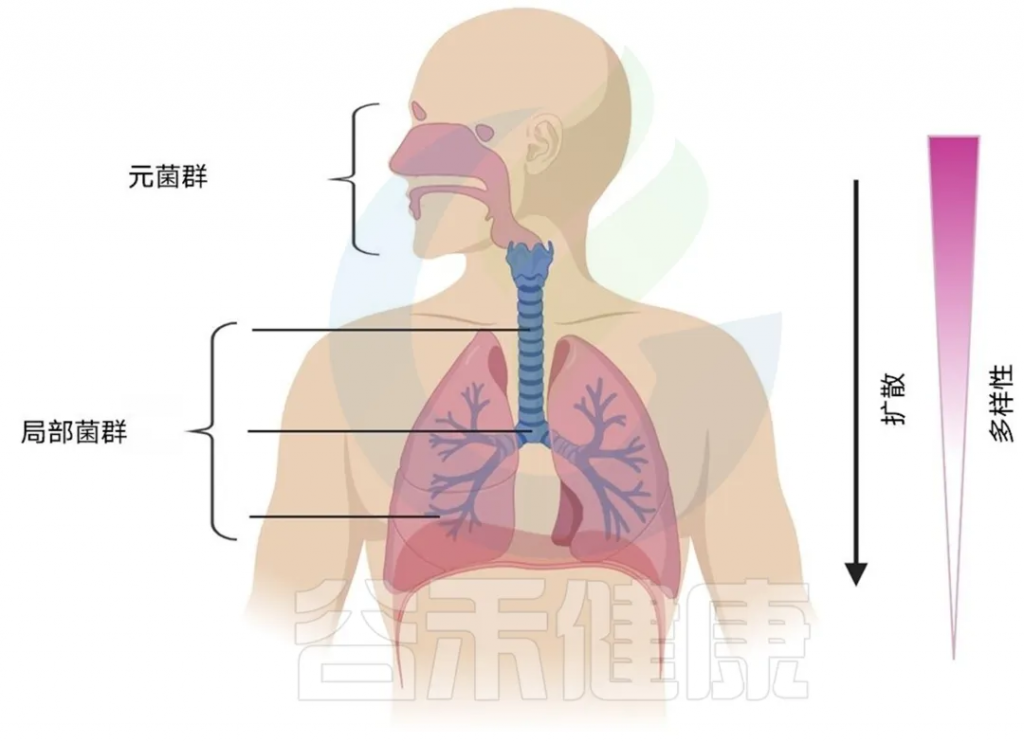
根据 Dickson等人提出的模型,上呼吸道 (URT),尤其是口腔,构成微生物源群落,微生物从该群落扩散到下呼吸道 (LRT)。因此,LRT 每个局部栖息地中存在的微生物是 URT 源群落中存在的微生物的一个子集。LRT 内较深的地点面临的扩散限制更大,因为它们在物理上距离源群落较远,因此多样性随着与 URT 距离的增加而减少。
适应健康肺微生物组组装的岛屿理论
疾病状态的肺部
有趣的是,随着患者从健康转变为疾病,中性和选择性过程的相对重要性似乎发生了变化:患有晚期肺病(如囊性纤维化或慢性阻塞性肺病)的患者的肺部表现出微生物群落结构明显的空间异质性,而健康人则没有这种现象。
这可能是由于在局部加剧的疾病条件下茁壮成长的本地或入侵微生物的增殖,或者由于现有微生物对这种局部条件的敏感性增加,并因此而灭绝。
为什么某些微生物在疾病状态下会突然增多?
通常,呼吸道损伤和炎症会导致温度升高和粘液产生增加,进而形成厌氧袋,可支持特定群落成员或病原体的生长。
局部严重的疾病可能会在不同地区造成这样的条件,从而使这些新的栖息地过滤器塑造那里的群落组成。
从历史上看,尽管众所周知上呼吸道(URT)藏有大量细菌,但人们认为下呼吸道(LRT)是无菌的,除非处于活动性疾病状态。
随着不依赖培养的测序技术的出现,这种误解已被纠正,我们现在知道实际上存在一个生物量低但相当健康的 LRT 微生物组。呼吸道微生物组的特征及其随时间和疾病状态的动态是一个新兴领域,这可能带来未来肺部疾病治疗方法的重大变革,比如开发针对特定微生物群落的靶向治疗,或者使用益生菌来恢复健康的微生物平衡。
上鼻腔由鼻孔、鼻腔和口腔、咽部和上喉部组成。有关口腔微生物群的描述,请参阅上文的胃肠道部分。
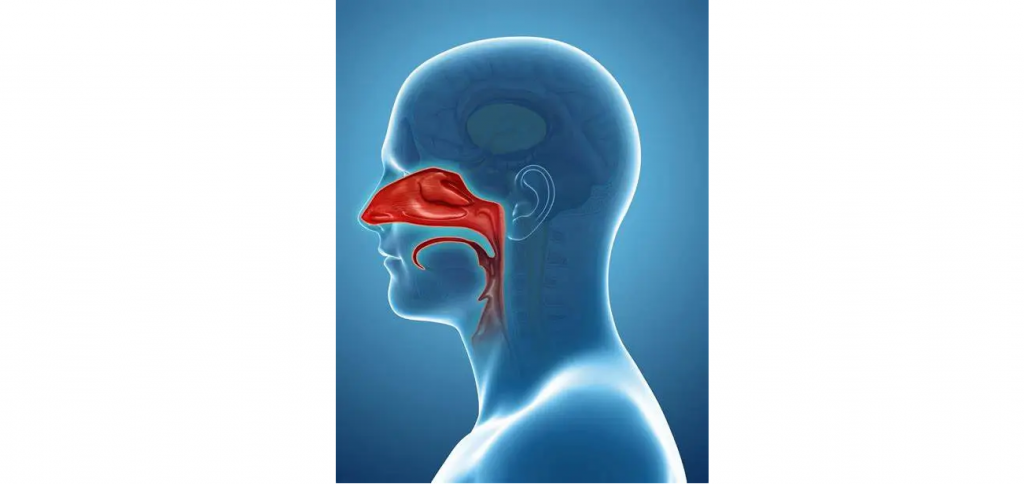
虽然咽喉的微生物群通常反映了口腔的微生物群,但鼻孔和鼻腔却拥有一组独特的微生物。
鼻孔(鼻前庭):类似皮肤、较凉爽、干燥、
棒状杆菌、葡萄球菌、丙酸杆菌
从解剖学上讲,鼻孔比鼻粘膜的其他部分具有更多类似皮肤的特征,包括角化、分层鳞状上皮细胞、汗腺和皮脂腺,以及毛囊,粗糙的特殊毛发(称为触须)从毛囊中长出。虽然空气在通过鼻腔时会变湿变暖,但进入鼻前庭的空气处于环境温度,因此这个栖息地比鼻腔的其他部分更凉爽、更干燥。栖息在鼻前庭的微生物群落高度反映了皮肤群落,以丰富的棒状杆菌(Corynebacteria)、葡萄球菌(Staphylococcus)和丙酸杆菌(Propionibacteria) 为特征。
鼻粘膜:鼻腔深处、更温暖湿润、清除颗粒物
放线菌、变形菌比例较高(与鼻孔的主要区别)
鼻粘膜位于鼻腔深处,具有几个重要的解剖学和生理学特征,使其与鼻前庭和口腔区分开来。与鼻前庭相比,鼻粘膜更温暖、更湿润。上皮从角化的复层鳞状细胞转变为假复层纤毛柱状细胞,并被一层流动的粘液覆盖。纤毛与粘液一起在整个呼吸道中进行上下向外的清扫运动,以清除颗粒物。这个区域主要由放线菌组成,包括棒状杆菌和丙酸杆菌,就像在前鼻孔中一样,但变形菌种类的代表性更高。这表明,鼻腔可能由从鼻前庭分散出来的微生物播种,但具有独特的栖息地过滤器,可以选择这些微生物的独特子集。
下呼吸道的结构与功能
下呼吸道 (LRT) 由气管、支气管、细支气管、肺泡组成。这些环境通常营养不良且需氧,尽管从近端到远端会发生许多重要的解剖和生理转变。
从鼻腔到细支气管,这些组织中的大多数构成呼吸道的“传导”部分,负责将空气带入肺部并清除所有其他颗粒物,而肺泡构成“呼吸”部分,负责气体交换。这些区域的上皮在很大程度上反映了这些不同的生理作用。
气管和支气管与鼻腔一样,具有纤毛假复层柱状上皮。在细支气管处,这种上皮转变为单层柱状细胞,然后扁平化为立方形上皮,最终发展为肺泡中薄薄的鳞状细胞内层。各种分泌细胞散布在整个下呼吸道中,释放粘液,粘液覆盖上皮表面并捕获分泌的抗菌肽和免疫调节分子。
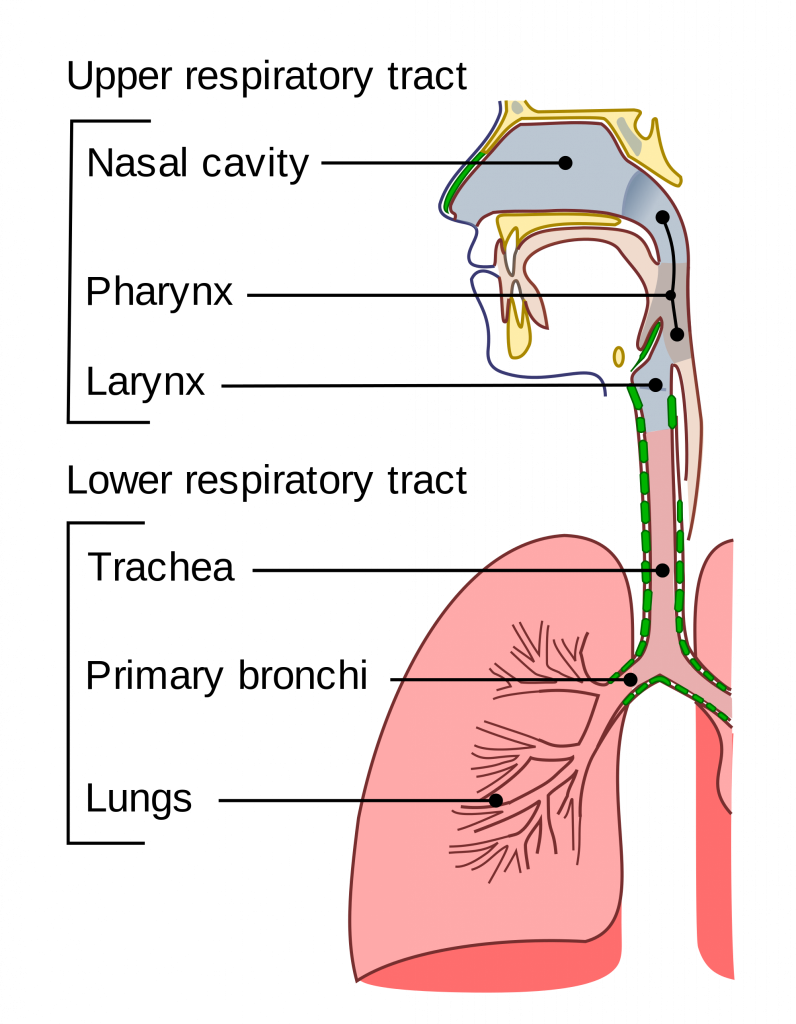
气道粘液在下呼吸道的近端区域最厚,向远端细支气管变薄,最终被肺泡中的表面活性剂取代。从近端到远端,下呼吸道还具有氧气张力、pH、温度和吸入颗粒密度的梯度。
采样技术与局限性
虽然这种空间异质性很可能导致整个下呼吸道中微生物组成的局部差异,但在人类中,目前的采样技术不具备研究这种可能性的空间分辨率。
下呼吸道微生物组的样本通常通过称为支气管肺泡灌洗(BAL)的过程收集,在此过程中,将支气管镜插入细支气管,并引入无菌盐水,然后重新收集被冲洗到其中的任何微生物群落成员。
上皮粘液不同层中的微生物,或可能出现在肺泡而不是细支气管中的微生物,都混合成单一的均质溶液,阻碍了跨微生境检测不同群落。
不同肺叶微生物群落有相似性,个体差异大于位置差异
然而,在更广阔的空间尺度上,从不同肺叶收集的 BAL 数据似乎表明,尽管在同一区域范围内观察到环境梯度,但微生物群落几乎难以区分。
对来自舌叶、右中叶、左上叶和右上叶以及气管上腔的样本进行测序分析,发现均存在大量耐氧普雷沃氏菌、韦荣球菌、链球菌。
每个个体的下呼吸道位置的样本与同一个体的 URT 源群落的相似性高于其他个体的同一下呼吸道位置的样本,这可能反映了下呼吸道群落组装的独特、基于个体的元群落。这些结果表明,栖息地过滤器在肺群落组装中发挥的作用可能不如中性的、基于分散的过程那么重要。
鉴于这些发现,有人提出了一种肺内微生物群落获得模型,该模型改编自岛屿生物地理学理论,这是一种经典的生态模型(上图)。
在这个模型中,肺的多样性是来自元群落物种库 (URT) 的随机移民与灭绝事件之间的平衡。一个地点离源群落越近,移民率就越高,该地点维持的多样性就越高。
微生物从元群落到达肺部的迁移过程
在这个模型下,当来自 URT 的微生物物理迁移到口咽部、被吸入气溶胶颗粒物或在睡眠期间被微吸入时,就会发生向 LRT 的移民。
灭绝事件如何影响肺部微生物的多样性
灭绝事件是由于咳嗽、粘液纤毛清扫和免疫清除等各种力量而发生的。由于不同地点的群落来自同一个元群落池,因此地点之间会存在显著的重叠——每个本地群落理论上都应该是同一源群落的一个子集——但预计物种丰富度会随着与源群落的距离而降低。
注:灭绝可能因为资源不足、环境变化,或者被岛上的”本地居民”(免疫系统)驱逐。
虽然 LRT 微生物组的高分辨率空间数据对于测试和改进这一假设非常有价值,但物种丰富度会随着与源群落的距离而降低。发现物种丰富度确实随着每个采样点与 URT 之间的距离而降低,支持了这一预期。
人体拥有如此多的特殊功能,因此在不同的解剖和功能部位拥有多样化、特殊的微生物群落是再合适不过的了。人类相关的微生物群落可以通过多种不同的过程形成,如扩散、选择和漂移,这些过程可以以不同的程度影响不同身体部位的组装和组成。
近年来,微生物组研究领域取得了显著进展。在菌群检测方面,无创采样技术的进步大大降低了研究的难度和患者的不适。高通量测序技术的不断进步,结合人工智能辅助的数据分析方法,使我们能够更快速、更准确地分析复杂的微生物组数据。
希望有一天,对这些更深入理解将使我们能够更有策略地操纵这些群落,以实现健康。
随着我们对微生物组的定义、框架和机制理解的不断发展和完善,我们在促进健康方面工程化这一复杂生态网络的能力也将不断扩大。不远的未来,个性化的微生物组干预策略可能成为常规医疗实践的一部分,为慢性疾病、自身免疫疾病,甚至是某些精神疾病提供新的治疗方案。
主要参考文献:
Kennedy MS, Chang EB. The microbiome: Composition and locations. Prog Mol Biol Transl Sci. 2020;176:1-42.
Vellend M Conceptual synthesis in community ecology. Q Rev Biol. 2010;85(2):183–206.
Costello EK, Stagaman K, Dethlefsen L, Bohannan BJM, Relman DA. The application of ecological theory toward an understanding of the human microbiome. Science. 2012;336(6086):1255–1262.
Dominguez-Bello MG, Costello EK, Contreras M, et al. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107(26):11971–11975.
Chibani-Chennoufi S, Bruttin A, Dillmann ML, Brüssow H. Phage-host interaction: An ecological perspective. J Bacteriol. 2004;186(12):3677–3686.
Sun CL, Relman DA. Microbiota’s “little helpers”: Bacteriophages and antibiotic-associated responses in the gut microbiome. Genome Biol. 2013;14(7):127.
Gilbert B, Levine JM. Ecological drift and the distribution of species diversity. Proc R Soc B Biol Sci. 2017;284(1855).
Zaura E, Brandt BW, de Mattos MJT, et al. Same Exposure but two radically different responses to antibiotics: Resilience of the salivary microbiome versus long-term microbial shifts in feces. MBio. 2015;6(6).
Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(SUPPL. 1):4554–4561.
Marais GAB, Calteau A, Tenaillon O. Mutation rate and genome reduction in endosymbiotic and free-living bacteria. Genetica. 2008;134(2):205–210.
Zhao S, Lieberman TD, Poyet M, et al. Adaptive Evolution within Gut Microbiomes of Healthy People. Cell Host Microbe. 2019;25(5):656–667.e8.
Huddleston JR. Horizontal gene transfer in the human gastrointestinal tract: Potential spread of antibiotic resistance genes. Infect Drug Resist. 2014;7:167–176.
Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214.
Sprockett D, Fukami T, Relman DA. Role of priority effects in the early-life assembly of the gut microbiota. Nat Rev Gastroenterol Hepatol. 2018;15(4):197–205.
Friedman ES, Bittinger K, Esipova TV., et al. Microbes vs. chemistry in the origin of the anaerobic gut lumen. Proc Natl Acad Sci U S A. 2018;115(16):4170–4175.
Donaldson GP, Lee SM, Mazmanian SK. Gut biogeography of the bacterial microbiota. Nat Rev Microbiol. 2016;14(1):20–32.
Johansson ME V, Sjövall H, Hansson GC. The gastrointestinal mucus system in health and disease. Nat Rev Gastroenterol Hepatol. 2013;10(6):352–361.
Tropini C, Earle KA, Huang KC, Sonnenburg JL. The Gut Microbiome: Connecting Spatial Organization to Function. Cell Host Microbe. 2017;21(4):433–442.

谷禾健康
微生物群代表宿主肠道中存在的整个微生物群。肠道内细菌界的“贫富差距”非常大,和人类社会创造的大部分的财富都流向少部分人口的现实类似,只有少数几十种的细菌分布在近乎90%的人群中。换句话说,大部分细菌都只能在特定的环境中生存,只有少数细菌适应能力超强,这可能也是我们需要重点关注的对象。
如果把不同细菌品种看作互相竞争的国家,那么细菌界的“超级大国”就属拟杆菌门和厚壁菌门了。当然它们都不是单独某一种细菌,而是一大类细菌的统称。
然而近年来随着患有肠内外疾病的人群越来庞大,变形菌门也逐渐被关注和研究,变形菌门是含有最丰富细菌的门,麾下包括多种“著名的”病原菌,如大肠杆菌、幽门螺杆菌、克雷伯氏菌、沙门氏菌、志贺氏菌、绿脓杆菌、霍乱弧菌、空肠弯曲菌、鼠疫杆菌、脑膜炎双球菌、淋球菌等,让其备受关注。
事实上,越来越多的数据将变形菌确定为疾病的可能微生物特征。目前主要证据涉及代谢紊乱和炎症甚至癌症。然而,最近的研究表明,在哮喘和慢性阻塞性肺病等肺部疾病中也有作用,有些疾病中变形菌不受控制扩张导致疾病易感和发生。
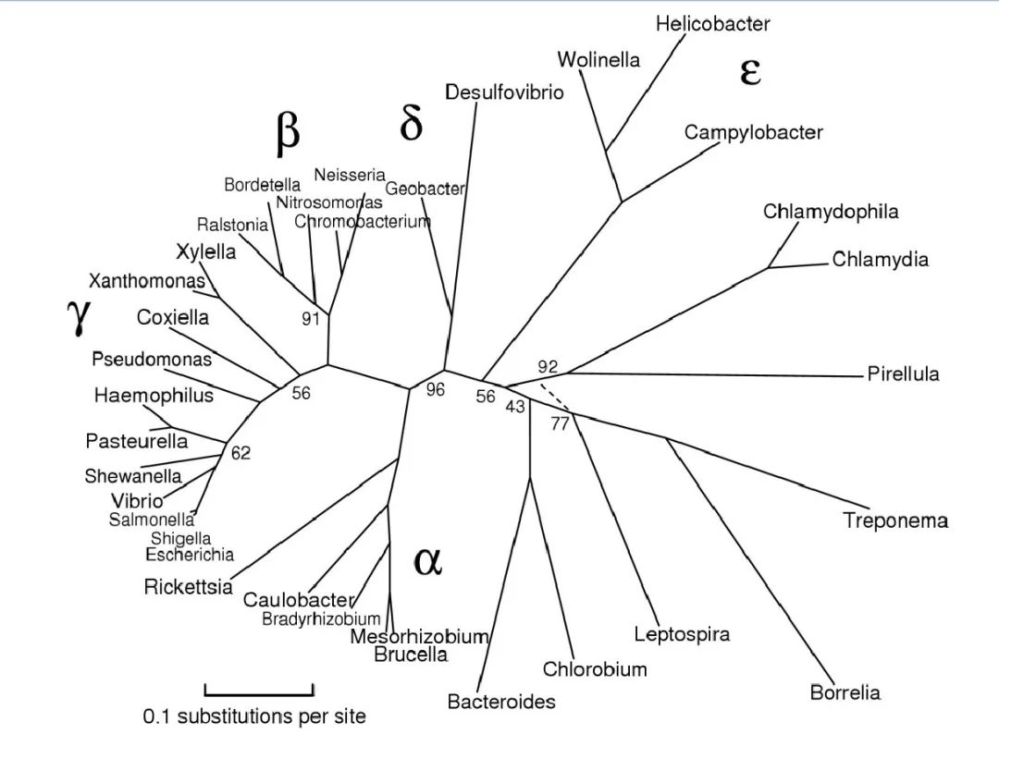
变形菌(proteobacteria)是细菌中最大、种类最多的一个门,它们在系统发育、生态和致病方面具有广泛的重要性。所有变形菌都是革兰氏阴性菌,外膜主要由脂多糖组成。
图源:esacademic
变形菌门主要是由核糖体RNA序列定义的,名称取自希腊神话中能够变形的神普罗透斯(这同时也是变形菌门中变形杆菌属的名字),因为该门细菌具有极为多样的形状,代谢特征等。
△ 形状:杆状和球菌、弯曲的、螺旋状的、环状的、丝状的和带鞘的细菌都有。
△ 新陈代谢:新陈代谢类型也多种多样,一系列代谢特征包括化学自养(从无机化合物的氧化中获取能量)、化学有机营养(从有机化合物的氧化中获取能量)和光养(从光中获取能量)。
△ 氧气利用:从严格厌氧菌和严格需氧菌到兼性厌氧菌和微需氧菌株的都有,但是大多数变形菌门的成员是兼性厌氧菌。
△ 运动:许多使用鞭毛移动,但有些不能移动或依赖细菌滑动,而一些细菌是不运动的。
△ 生态分布:变形菌门的成员具有极大的可变形态和多才多艺的生理学,这使它们在各种生态位中生存具有竞争优势。已观察到变形菌在不同生境中无处不在。
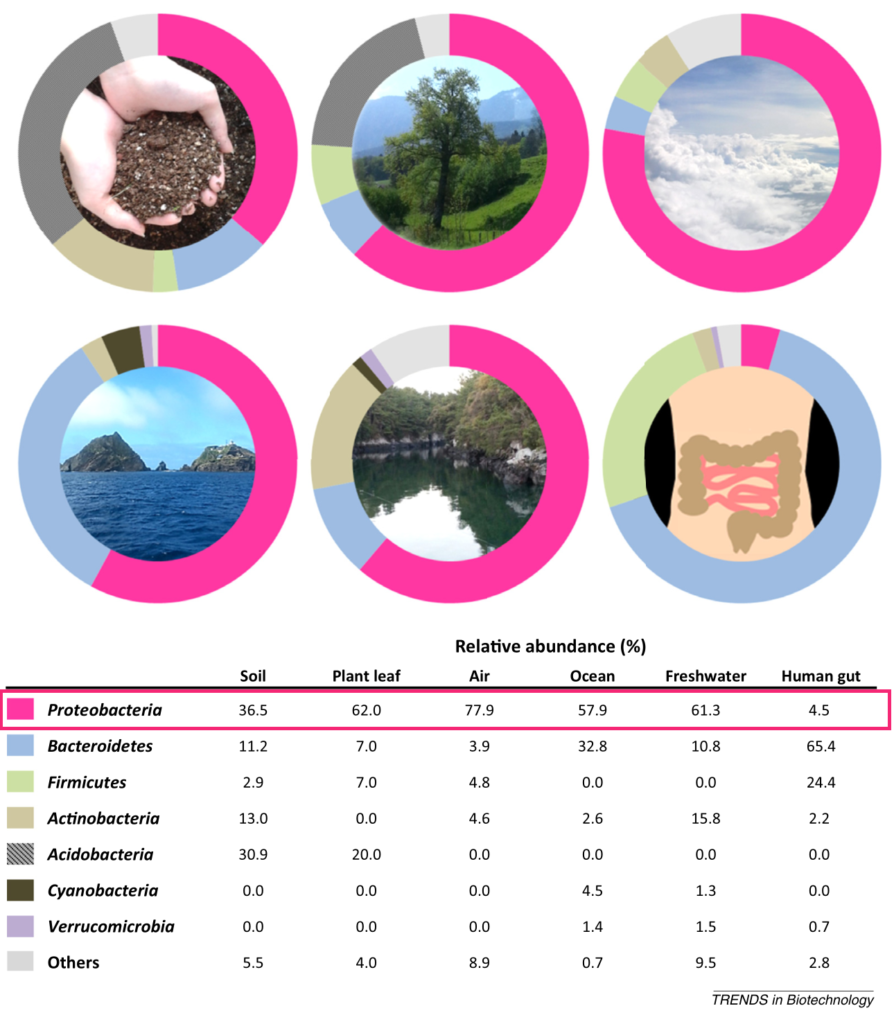
Shin NR, et al., Trends Biotechnol. 2015
植物 、海水、淡水 ,空气,以及人和动物的身体部位,包括肠道、口腔、皮肤、阴道。尽管存在研究间差异,但健康人口腔微生物群的变形菌相对丰度最高(17.2-36.8%),其次是皮肤(6.8-30.0%)、胃肠道(2.5-4.6%)和阴道(2.3%)。
在系统发育学上,变形菌是根据小核糖体亚单位RNA基因(16S rRNA)的测序定义的。这是一个巨大的革兰氏阴性原核生物门,原线粒体起源于此。
图片来源:Maria Lane,eportfolio
该门主要分为以下几大类:
最初,变形菌包括 α、β、γ 和 δ 四个亚类。ε变形菌 和 δ变形菌 通常被认为是最古老的变形菌群,因为它们包括利用硫化合物进行能量代谢的专性厌氧菌。
α变形菌(Alpha-proteobacteria)
第一类变形菌是α-变形菌。这一类的统一特征是它们是寡营养生物,能够生活在低营养环境中,如深海沉积物、冰川或深层地下土壤。同时α-变形菌是多样化的细菌分支之一,在生活方式、地理分布和基因组大小方面表现出极大的差异。
在 α-变形菌 中有两个重要分类群,衣原体和立克次体,它们是专性细胞内病原体,这意味着它们的部分生命周期必须发生宿主细胞内。由于它们无法合成自己的三磷酸腺苷 (ATP),因此,量需求依赖宿主于细胞。
立克次体属是人类很多严重疾病的病原体。例如,布鲁氏菌属、埃立克体属和立克次氏体。立克次氏杆菌会导致落基山斑疹热,这是一种威胁生命的脑膜炎(包裹大脑的膜发炎)。R. rickettsii 感染蜱,并可以通过被感染的蜱叮咬传播给人类。此外,布鲁氏菌科(Brucellaceae)和巴尔通氏菌科(Bartonellaceae)的细菌是人类病原体。
α-变形菌 还包括固氮细菌,例如固氮螺菌属和根瘤菌属。这两种细菌都使用一种称为固氮酶途径的复杂酶途径将大气中的氮 (N2) 转化为氨 (NH3)。此外,α变形菌还包括硝化细菌。这种类型的细菌将氨和铵 (NH4+) 还原为硝酸盐 (NO3–)。乙酸杆菌属和葡糖杆菌属的变形菌可用于生产乙酸。
β变形菌(Beta-proteobacteria)
与依靠最少量营养物质生存的 Alpha-proteobacteria 不同,Beta-proteobacteria 类是富营养生物,这意味着它们需要大量的有机营养物质。
Beta-proteobacteria 通常在需氧和厌氧区域之间生长(例如,在哺乳动物的肠道中)。一些属包括作为人类病原体的物种,能够引起严重的,甚至可能危及生命的疾病。例如,奈瑟球菌属包括淋病奈瑟菌( STI淋病的病原体)和脑膜炎奈瑟菌(细菌性脑膜炎的病原体)
β变形菌中的亚硝化单胞菌可以将亚硝酸盐还原为亚硝酸盐 (NO2–)。同时,硫杆菌属物种是将硫化氢 (H2S) 和元素硫氧化成硫酸盐 (SO42-) 的细菌,以及用于污水处理的菌胶团(Zoogloea)和Sphaerotilis 。
γ变形菌(Gamma-proteobacteria)
最多样化的革兰氏阴性细菌是γ-变形菌,它包括许多人类病原体。包括几个医学和科学上重要的细菌群,例如肠杆菌科、弧菌科和假单胞菌科。
此外,许多重要的病原体属于这一类,例如:
Richard B. Frankel
△ 铜绿假单胞菌
一个庞大而多样的科,假单胞菌科,包括假单胞菌属。铜绿假单胞菌在该属内,它是一种病原体,可以造成身体不同部位的各种感染。铜绿假单胞菌是一种严格需氧、不发酵、高度运动的细菌。
它通常可能造成伤口和烧伤感染,也可能是慢性尿路感染的原因,并且可能是囊性纤维化患者或机械呼吸机患者呼吸道感染的重要原因。
铜绿假单胞菌感染通常难以治疗,因为该细菌对许多抗生素具有抗性,并且具有形成生物膜的非凡能力。
△ 肠杆菌科
肠杆菌科是属于γ-变形菌 的一大类肠道细菌。它们是兼性厌氧菌,能够发酵碳水化合物。在这个家族中,微生物学家认识到两个不同的类别。
第一类,大肠杆菌,以其原型细菌种类大肠杆菌命名。大肠菌能够完全发酵乳糖(即产生酸和气体)。
第二类,非大肠杆菌,要么不能发酵乳糖,要么不能完全发酵(产生酸或气体,但两者不能同时产生)。
非大肠杆菌包括一些值得注意的人类病原体,例如沙门氏菌属,志贺氏菌,鼠疫耶尔森氏菌。
δ 变形菌(Delta-proteobacteria)
δ-变形菌(Delta-proteobacteria )包括基本好氧的形成子实体的粘细菌和严格厌氧的一些种类,如脱硫球菌属(Desulfococcus)、脱硫线菌属(Desulfonema)、硫酸盐还原菌(脱硫弧菌属(Desulfovibrio)、脱硫菌属(Desulfobacter)、和硫还原菌(如除硫单胞菌属Desulfuromonas),以及具有其它生理特征的厌氧细菌,如还原三价铁的Geobacter和互营菌属(Syntrophus)。
△ 蛭弧菌属:
δ-变形菌还包括蛭弧菌属,Bdellovibrio侵入宿主细菌的细胞,将自身定位在周质中,即质膜和细胞壁之间的空间,以宿主的蛋白质和多糖为食。这种感染对宿主细胞是致命的。
△粘细菌:
粘细菌(“粘液细菌”)是一组主要生活在土壤中并以不溶性有机物质为食的细菌。与其他细菌相比,粘细菌具有非常大的基因组,例如 9-1000 万个核苷酸。
Sorangium cellulosum 拥有最大的已知(截至 2008 年)细菌基因组,有 1300 万个核苷酸。
粘细菌产生许多在生物医学和工业上有用的化学品,例如抗生素。他们将这些化学物质输出到细胞外。
ε变形菌(Epsilon-proteobacteria )
ε-变形菌(Epsilon-proteobacteria) 是革兰氏阴性微需氧细菌(意味着它们在其环境中只需要少量氧气)。多数是弯曲或螺旋形的细菌,如沃林氏菌属(Wolinella)、螺杆菌属(Helicobacter)和弯曲菌属(Campylobacter)。它们都生活在动物或人的消化道中,为共生菌(沃林氏菌在牛中)或致病菌(螺杆菌在胃中或弯曲菌在十二指肠中)。
△ 弯曲杆菌:
变形菌门Epsilon-proteobacteria 中的两个临床相关属是弯曲杆菌属和螺杆菌属,它们都包括人类病原体。
弯曲杆菌可引起食物中毒,表现为严重的肠炎(小肠发炎)。这种由空肠弯曲杆菌引起的疾病在发达国家相当普遍,通常是因为食用了受污染的家禽产品。鸡通常携带空肠弯曲杆菌在胃肠道和粪便中,它们的肉在加工过程中可能会受到污染。
△螺杆菌:
螺杆菌是ε-变形菌的一个属,具有特征性的螺旋形状。它们最初被认为是弯曲杆菌属的成员,但自 1989 年以来,它们独立为自己的属。
螺杆菌属属于ε-变形菌,弯曲杆菌目,螺杆菌科,已经有超过 35 种。已经发现一些菌生活在上胃肠道的内壁,以及哺乳动物和一些鸟类的肝脏中。
该属中最广为人知的物种是幽门螺杆菌,它感染多达 50% 的人口。这种细菌的某些菌株对人类具有致病性,因为它与消化性溃疡、慢性胃炎、十二指肠炎和胃癌密切相关。它也作为该属的模式种。
幽门螺杆菌在胃的高酸性环境中存活的能力有些不同寻常。它产生脲酶和其他酶来改变其环境以降低其酸性。
幽门螺杆菌也有它存在的意义,可能抑制引起结核的细菌(结核分枝杆菌),预防哮喘,克罗恩病,食管反流,腹泻病以及食道癌。
❥ 识别微生物编码的基因,与特征相关联
栖息在哺乳动物肠道中的微生物编码了大量的蛋白质,这些蛋白质有助于广泛的生物功能,从调节免疫系统到参与新陈代谢。
我们从这些微生物中识别蛋白质编码基因并将基因水平与疾病、药物功效或副作用以及其他宿主特征相关联。
例如,与传统的高纤维农业饮食相关的人类肠道微生物群编码了参与纤维素和木聚糖水解的基因家族,而这些基因家族在吃典型西方饮食的人群(年龄匹配)中不存在。
一般编码适应肠道环境所必需的功能的微生物有很强的选择性,在不同宿主中具有大量冗余的基因库。然而,目前的研究和临床很容易忽略健康人类微生物组之间基因丰度的生理意义差异。
❥ 较少丰度的变形菌门,才是是跨宿主丰度变异性最大的基因的主要来源
人体肠道通常由拟杆菌门和厚壁菌门主宰,这些门内的进化枝(尤其是拟杆菌属、普氏菌属和瘤胃球菌科)是最常用于将个体聚集成“肠型”,因为它们解释了最多的分类变异。Bacteroidetes 与 Firmicutes 的比率也被推定为疾病或健康的潜在生物标志物。
有人提出,人类肠道微生物组中可能存在少量“肠型”,每一种都具有不同的分类组成。因此,虽然拟杆菌门和厚壁菌门可能对宿主之间的分类变异贡献最大,但变形菌门的丰度可能会捕获更多的功能变异。
与先前确定的肠型标记分类群相比,变形菌门的水平和可能的 Euryarchaeota 更好地解释了肠道微生物基因功能的人与人之间的差异。
在肠型研究中遗漏了这些不太丰富的门,可能是因为肠型是通过倾向于对高丰度分类群进行更多加权的方法鉴定的,并且肠型是从分类学而非功能数据中鉴定的。这对解释人类肠道微生物群的分类数据具有重要意义。
例如,变形菌门的过度生长与代谢综合征和炎症性肠病有关。通过 TLR5 敲除小鼠测试的肠道炎症关联到变形菌门(超过拟杆菌门和厚壁菌门),并且一些变形杆菌可以在这种背景下诱发结肠炎,可能导致反馈循环。因此,可变基因家族对解释人类肠道微生物群的分类数据具有重要意义。
备注:肠道受体蛋白TLR5参与积极地塑造新生小鼠肠道微生物群落的长期组成,敲除的Toll样受体(TLR5),是免疫系统识别鞭毛细菌(比如变形菌和梭状芽孢杆菌)的关键受体,缺乏它则机体可能不会在感知到细菌鞭毛时对细菌产生免疫应答。
肠道相关微生物群落组成的变化与许多人类疾病有关,但驱动这种不平衡(生态失调)的机制尚不完全清楚。
在肠道菌群失调期间观察到的最一致和最强大的生态模式是属于变形菌门的兼性厌氧细菌的扩张。
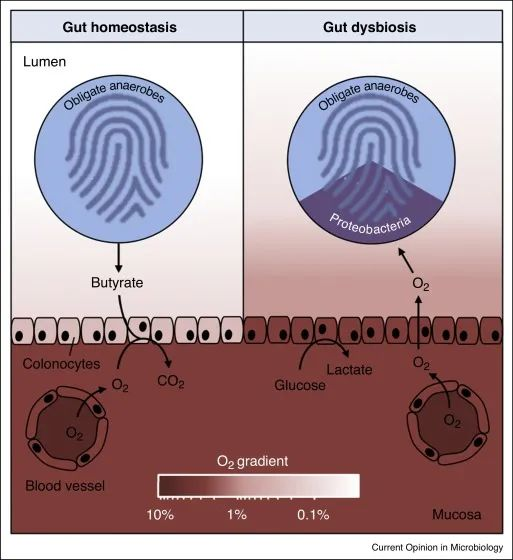
变形菌的菌群失调是上皮功能障碍的微生物特征
在肠道稳态期间(左),微生物群衍生的丁酸盐的 β 氧化导致上皮缺氧,从而维持大肠腔内的厌氧状态。反过来,腔内厌氧症导致肠道微生物群内专性厌氧菌占主导地位。
备注:丁酸(Butyrate acid,BA),俗称酪酸,是构成脂肪的一种脂肪酸,含有4个碳原子又称短链脂肪酸。人体的丁酸部分来自于食物中丁酸的吸收,主要的来自结肠厌氧菌的发酵产生。人体结肠产生的短链脂肪酸丁酸占比大部分)。
在肠道菌群失调期间(右),表面结肠细胞通过无氧糖酵解获得能量,从而导致上皮氧合增加,这种上皮功能障碍破坏了管腔中的厌氧菌,从而通过有氧呼吸推动兼性厌氧变形菌的扩张。
健康结肠的厌氧菌导致肠道微生物群的组成以专性厌氧菌为主,而菌群失调通常与兼性厌氧变形菌的丰度持续增加有关,这表明厌氧菌的破坏。
结肠上皮是缺氧的,但肠道炎症或抗生素治疗会增加结肠中的上皮氧合,从而破坏厌氧作用,通过有氧呼吸驱动兼性厌氧变形菌的菌群失调。
肠沙门氏菌(S. enterica)是一种食源性病原体,属于肠杆菌科,变形菌门,可引起小鼠结肠炎。在肠道沙门菌S. enterica诱导的结肠炎期间,肠腔内的氧气可用性增加,这表明结肠中病原体的氧气呼吸依赖性大量繁殖以及随之而来的专性厌氧梭状芽胞杆菌的丰度下降。
同样,结肠隐窝增生由鼠肠道病原体柠檬酸杆菌(肠杆菌科,变形菌门)引发,可提高肠腔内的氧气利用率,从而通过有氧呼吸推动变形菌病原体扩张。
这些观察结果表明,变形菌的菌群失调是上皮功能障碍的潜在诊断微生物特征,建议将变形菌负荷作为生态失调和疾病的潜在诊断标准,所以在谷禾即将更新的肠道菌群检测报告中,我们会加入变形菌门丰度和参考范围这一指标。
大肠中专性厌氧菌的优势可能是宿主环境的氧气限制严重的结果,这反过来又对用于营养物质的分解代谢途径产生重要影响。
避免被上消化道中的宿主酶降解的复合碳水化合物,可以被大肠中的专性厌氧细菌水解并发酵成更小的化合物。专性厌氧菌最终将许多发酵产物转化为短链脂肪酸,其中乙酸盐、丙酸盐和丁酸盐是最丰富的产物。宿主吸收了大约 95-99% 的微生物产生的短链脂肪酸,它到达血流以影响免疫发育。因此,大肠中专性厌氧菌的优势确保了维持肠道稳态的代谢物的产生。
变形菌是平衡的肠道相关微生物群落中的一个次要成分。然而,由遗传易感性、化学物质或肠道病原体感染引起的肠道炎症会导致小鼠模型中变形杆菌的管腔扩张不受控制。
同样,在患有严重肠道炎症的人类中,包括炎症性肠病、结直肠癌或坏死性小肠结肠炎的患者中观察到变形杆菌的丰度增加。此外,在包括肠易激综合征和代谢综合征在内的低水平肠道炎症条件下观察到大量变形菌。
肠道炎症增加了替代电子受体的可用性,这些电子受体通过厌氧呼吸支持兼性厌氧细菌的生长。肠道炎症过程中产生的活性氧可以将内源性硫化合物氧化为连四硫酸盐,这是一种电子受体,通过连四硫酸盐呼吸作用在鼠结肠中驱动类似肠沙门氏菌和Yersinia enterocolitica(一种属于肠杆菌科,变形菌门的病原体)的管腔扩张 。
一氧化氮由宿主酶产生化学诱导的结肠炎或由遗传易感性引发的结肠炎期间的诱导型一氧化氮合酶(iNOS) 。一氧化氮在肠腔内分解成硝酸盐,从而通过硝酸盐呼吸支持生长,从而增加小鼠结肠中共生大肠杆菌的丰度。类似,宿主衍生的硝酸盐的呼吸有助于在 S. enterica 诱导的小鼠结肠炎期间腔内病原体扩张。
有趣的是,即使在没有明显肠道炎症的情况下,例如在抗生素治疗期间,呼吸电子受体也有助于细菌群落从专性厌氧菌转变为兼性厌氧菌。为了支持这一观点,用链霉素治疗小鼠可将盲肠中的氧化还原电位提高到接近需氧培养液的水平。链霉素治疗通过硝酸盐呼吸和氧气呼吸的结合增加结肠中共生大肠杆菌或致病性肠杆菌的生长。
其他类似研究的结论也表明,氧气,单独或与其他呼吸电子受体结合,是广泛的胃肠道失衡中肠道菌群失调的常见驱动因素。因此,为了开发新的预防或治疗策略,必须了解在肠道菌群失调期间呼吸电子受体的可用性如何升高。
基于这些观察,有人提出变形菌的扩增是肠道菌群失调的微生物特征,而氧气、用药,遗传易感,肠炎驱动了变形菌的扩张,反过来加剧疾病的进展。
宿主遗传因素和外在环境因素,如饮食和生活环境,不断影响肠道微生物群的分类和功能组成。鉴于具有高度稳定性的平衡肠道微生物群与宿主的免疫系统具有共生相互作用,能够抑制变形杆菌失控的扩张,肠道中变形杆菌的大量繁殖可以反映肠道微生物群落结构的不稳定;这种不稳定的结构可以在非疾病状态下观察到(例如,新生儿期 和胃绕道手术后和疾病状态例如,代谢紊乱和肠道炎症)。
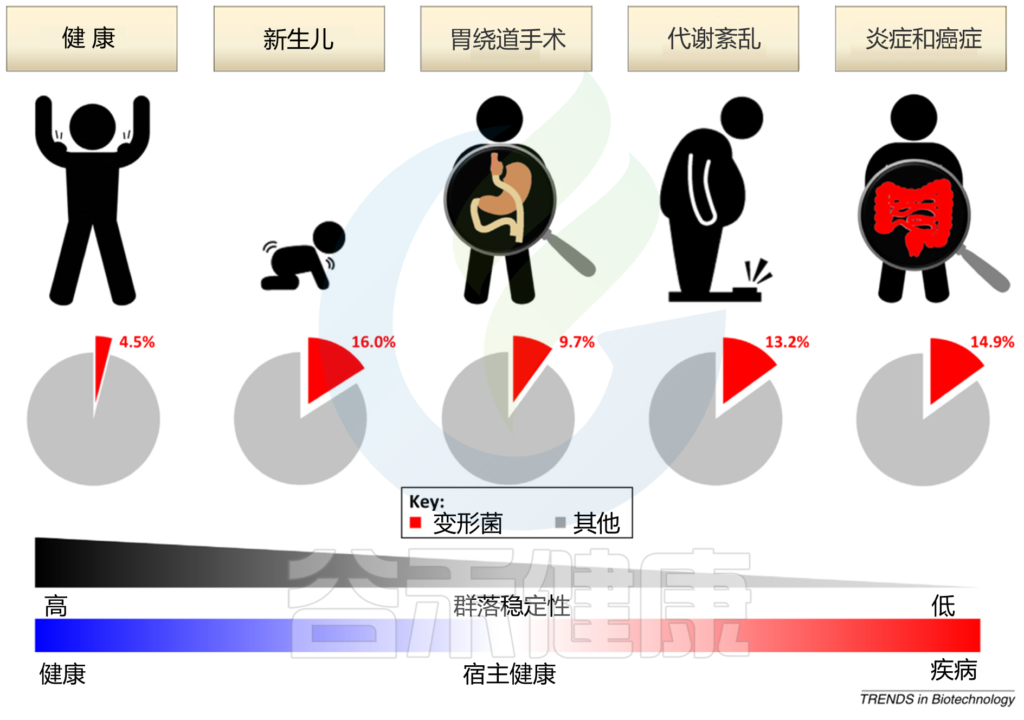
Shin NR, et al., Trends Biotechnol. 2015
在新生儿胃肠道的初始定植期间,兼性厌氧变形菌使肠道生态位有利于专性厌氧菌的定植;后者很快被专性厌氧的厚壁菌门和拟杆菌门所取代,它们在健康成年人的肠道微生物群中占主导地位。胃绕道手术导致的胃肠道重排可以改变 pH、胆汁流量和肠道激素,所有这些因素都会影响变形杆菌的丰度。
新生儿肠道中的变形菌
新生儿肠道中的微生物群备受关注,因为它不仅反映了细菌群落的脆弱结构,而且反映了哺乳动物肠道微生物群的真正起源。新生儿肠道中的细菌群落由于其快速的时间变化而不稳定。然而,这种脆弱性与更重要的肠道菌群定植有关,例如严格的厌氧菌。
具体来说,由于新生儿肠道中的氧气丰富,生命第一周的微生物群经常以兼性厌氧菌为主,主要是变形菌属(例如,埃希氏菌属、克雷伯氏菌属和肠杆菌属)。这些兼性厌氧菌通过消耗氧气、改变 pH 值、降低氧化还原电位并产生二氧化碳和营养物质,使栖息地适合严格的厌氧菌定殖。
因此,可以推测变形杆菌在为新生儿肠道准备好接受严格厌氧菌的连续定植方面发挥了作用,这些厌氧菌在健康成人的肠道中含量丰富。
最近对母体胎盘微生物组的一项研究描述了共生细菌群落的存在,其中大肠杆菌的丰度最高。尽管关于胎盘微生物群的活力和起源存在争议,但在母体胎盘中发现的这些有趣的细菌群落与来自母体羊水和新生儿胎粪的细菌群落重叠。
因此,新生儿肠道中的变形菌可能通过胎儿在子宫内吞咽羊水从母体胎盘传播。有趣的是,妊娠后期孕妇肠道中变形菌的比例增加。这意味着母亲微生物群中的这种特定细菌群转移到了新生儿身上。
在新生儿肠胃道中观察到的变形杆菌定植生长的持续时间很可能在母体控制之下。事实上,新生儿微生物群会受到各种母体因素的影响,例如分娩方式、饮食和怀孕期间接触抗生素。
最重要的是,新生儿肠道中变形菌的丰度受喂养类型的影响,这些细菌在配方奶喂养的婴儿中的频率更高,但在母乳喂养的婴儿中很少见。
人乳寡糖 和分泌型 IgA 的产生参与在最初的肠道定植过程中选择性抑制变形菌。因此,越来越多的人认为,及时减少变形菌的丰度是初始微生物定植的正常部分,而这种定植模式的紊乱与新生儿疾病的风险增加有关。
肠道中微生物群和宿主细胞之间的相互作用对于免疫系统的形成和调节至关重要,由于肠腔内有大量外源性抗原,免疫系统必须严格调节其反应以维持与共生菌的共生关系。共生体传递一种信号,诱导宿主免疫的耐受性反应。因此,宿主可以区分有益的本土微生物和有害病原体,并建立健康的微生物群。
变形杆菌的主要分类及其与IBD的关系
Mukhopadhya I, et al., Nat Rev Gastroenterol Hepatol. 2012
为了防止对共生细菌的炎症反应,肠道内的免疫细胞,如单核吞噬细胞(巨噬细胞和树突状细胞)和 CD4 + T 细胞,对微生物刺激反应迟钝或表现出共生反应。
同时,黏膜免疫系统负责清除病原体,这一过程需要积极的促炎信号级联反应。因此,不适当的免疫反应会破坏肠道稳态,引发生态失调,并导致局部和全身炎症和代谢功能障碍。
这种慢性进行性肠道炎症的状态在临床上被诊断为炎症性肠病 (IBD),其中包括溃疡性结肠炎 (UC) 和克罗恩病 (CD)。IBD 的确切病因仍然无法获得,但新出现的证据表明,肠道微生物群成为了这种疾病的主要嫌疑。
许多研究报告了动物和人类各种炎症持续条件下微生物群组成的改变。在这种情况下,通常发现变形菌在疾病中增加,变形菌在肠道炎症中的作用已在各种结肠炎小鼠模型中得到解决,与疾病呈正相关。
例如,使用易发炎症的小鼠模型,即鞭毛蛋白受体 TLR5 缺陷小鼠 (T5KO),发现,进展为结肠炎的小鼠表现出明确的微生物群特征,其特征是变形菌的水平增加,尤其是大肠杆菌属。并且一些作者已将其确定为微生物群不稳定性的潜在标志物,因此易诱发疾病发作。
与变形杆菌属大量繁殖的同时,结肠炎Tlr5-/- 小鼠表现出杂乱无章的结肠粘液层,与非结肠炎Tlr5-/- 同胞相比,感染性病原体的清除延迟。
这些结果表明,短暂不稳定的肠道微生物群,尤其是以变形菌为主的群落,会使遗传易感的小鼠易患慢性结肠炎。
先天免疫反应失调推动变形杆菌生长的假设这反过来又会促进肠道炎症,这一点得到了其他小鼠模型研究的支持,这些小鼠模型具有影响适应性免疫的突变,白细胞介素 (IL)-10 是对本地微生物群产生免疫耐受所需的主要免疫调节细胞因子。
IL-10 缺陷小鼠由于对肠道菌群不耐受而表现出自发性结肠炎。随着结肠炎症的发生和发展,在定植常规微生物群或缺乏特定病原体的微生物群的 IL-10-/- 小鼠中,变形杆菌和大肠杆菌比野生型小鼠多。
在另一项对 IL-10 缺陷小鼠的研究中,富含饱和乳脂的饮食扰乱了肠道微生物群,导致亚硫酸盐还原Delta-proteobacteriumBilophila wadsworthia 大量繁殖。这种病原菌在 IL-10 -/-小鼠中诱导促炎性黏膜免疫反应并促进自发性结肠炎的发生率和严重程度;它还在喂食高乳脂饮食的野生型小鼠中促进葡聚糖硫酸钠 (DSS) 诱导的结肠炎。
除了对结肠炎的易感性与肠道变形菌的相对丰度之间存在正相关性之外,对先天性和适应性免疫系统均缺陷的小鼠的研究提供了支持变形菌在肠道炎症中的致病作用的证据。
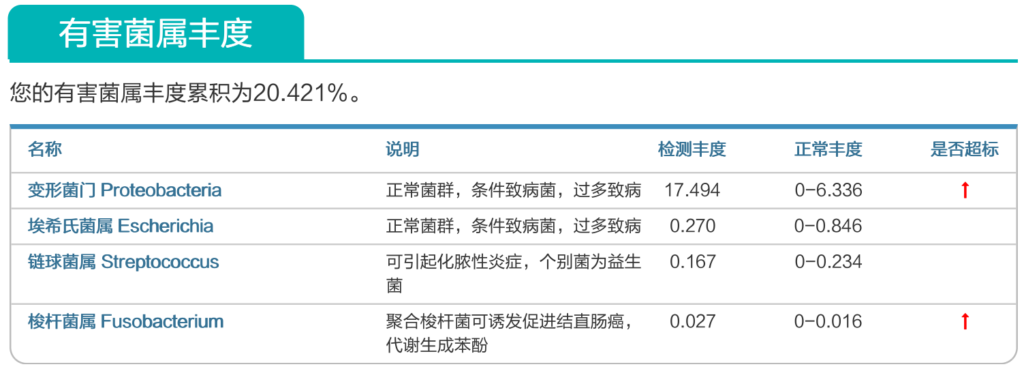
来源:谷禾健康肠道菌群数据库
谷禾健康肠道菌群检测大数据也显示,在炎症性肠病,结直肠癌等患者的肠道菌群检测报告中,85%以上的患者显示变形菌门超标或多项变形菌门病原菌超标或占比丰度偏高。
在最近的一项研究中重现了,结肠炎中变形杆菌的显着扩增,该研究比较了患有活动性结肠炎的 TRUC 小鼠的肠道微生物组与因庆大霉素、甲硝唑或抗肿瘤坏死因子 (TNF)-α 治疗而缓解的小鼠的肠道微生物组。
值得注意的是,从 TRUC 小鼠的粪便中分离出的两种肠杆菌科细菌(肺炎克雷伯菌和奇异变形杆菌)即使在没有任何遗传免疫缺陷的受体小鼠中也足以引发结肠炎。
然而,这两种微生物的致结肠潜力并未在无菌 TRUC 小鼠中复制,这表明结肠炎的发病机制需要其他共生成员。口服伤寒杆菌,另一种富含 TRUC 小鼠的变形菌,也会在非结肠炎 TRUC 小鼠中引发结肠炎,这些小鼠具有大量的促炎细胞因子(例如,TNF-α)。
遗传易患结肠炎的小鼠的生态失调与人类 IBD 特别相关,因为与 IBD 相关的风险等位基因或多态性与先天性和适应性免疫成分有关。与小鼠研究相似,两项人类研究表明,与健康受试者相比,IBD 患者肠道微生物群落的特点是微生物多样性低、变形菌门(尤其是肠杆菌科)的产物以及厚壁菌门的减少。
一项人类队列研究发现,核苷酸结合寡聚化结构域 (NOD)-2 风险等位基因剂量与 IBD 患者肠道标本中肠杆菌科的相对丰度呈正相关。
在 UC 患者中,与炎症的中度和轻度阶段相比,在严重阶段观察到的变形杆菌水平显着升高。
在新发 CD 的初治儿科患者和非 IBD 对照受试者之间,回肠和直肠活检(但不在粪便样本中)的粘膜相关微生物组存在明显差异。变形菌的相对丰度增加,包括肠杆菌科、巴氏杆菌科和奈瑟菌科,将 CD 相关细菌群落与健康对照组区分开来。与慢性炎症一致,伴随变形杆菌属优势的肠道微生物群落改变不仅见于传染性病原菌或原生动物寄生虫引起的急性炎症,而且见于实验性和人类结肠炎相关的结肠直肠癌。
最有趣的生物体,通过一个孤立的病例报告与 IBD 有关,该病例报告一名感染这种细菌的小男孩在放射成像上出现回肠增厚,这是克罗恩病的典型表现。
血清学研究表明,与健康对照相比,克罗恩病患者的大肠杆菌抗体数量增加。具体地说,已发现37-55 % 的克罗恩病患者、2-11% 的溃疡性结肠炎患者和 <5% 对照组患者的百分比。
此外,克罗恩病患者中这些抗体的存在与更严重的表型相关,其特征是小肠受累、疾病进展频繁、病程更长和对手术的需求更大,这表明它们可以用作克罗恩病的预后标志物。
饮食被认为是塑造肠道微生物结构的最关键的环境因素之一。
△ 肥胖:丰富的变形菌为特征
累积证据表明,人类和啮齿动物的健康和肥胖个体的肠道微生物群的分类和功能组成存在差异。
此外,肥胖表型通过粪便移植的传播能力表明肠道微生物群落的改变,作为主要触发因素,是因果关系而不是结果。
肠道微生物群的分类组成失衡,称为生态失调,在代谢紊乱中得到充分证明,并被视为厚壁菌门相对于拟杆菌门的相对丰度增加(F:B 比率)。尽管一致的研究结果普遍支持这一概念,但代谢紊乱期间的生态失调通常包括变形菌的患病率增加。
例如,一项对儿童肠道微生物群的研究发现,与低脂肪、高纤维饮食儿童相比,食用高热量、高脂肪、低纤维饮食的欧洲儿童中的变形杆菌数量更多。
这种差异揭示了肠道微生物群落对非洲儿童饮食的适应性,这可以提高他们从难消化的多糖中获取能量的能力。此外,一些导致有害代谢影响的因素,例如食用无热量的人造甜味剂和乳化剂(通常用作加工食品中的添加剂),也会损害血糖控制并诱发变形杆菌繁殖。
特别是,人造甜味剂介导的肠杆菌科和Delta-proteobacteria类相对丰度的升高与 2 型糖尿病 (T2DM) 患者的结果一致,表明葡萄糖稳态和肠道变形菌之间存在联系。相比之下,证明变形菌的丰度与糖尿病表型呈负相关,挑战代谢疾病患者中高丰度变形菌的概念。
为支持代谢紊乱与变形菌属的扩张之间的关系,变形杆菌属的致肥胖潜力已在无菌小鼠的单关联研究中被确定。
在对一名病态肥胖志愿者进行的减肥试验中,肠杆菌科的相对丰度逐渐减少,假设肠杆菌在代谢恶化中具有致病作用。用从肥胖的人类肠道中分离出来的阴沟肠杆菌B29对无菌小鼠进行单菌定植足以诱导肥胖和胰岛素抵抗。
这一发现支持了这样一个假设,即以丰富的变形菌为特征的不稳定的肠道微生物群落可能代表代谢紊乱的主动特征,而不是被动后果。
△ 营养不良儿童:变形菌成为优势菌
营养不良会导致其他健康问题,例如消瘦和夸希奥科病。在发展中国家,营养不良是威胁 5 岁以下儿童生命的疾病。
营养不良的主要病因是在孕期或产后头 3 年由于大量营养素缺乏和微量营养素缺乏导致的慢性能量负平衡。
然而,最近的研究表明,孟加拉国和马拉维营养不良儿童的肠道微生物群落结构和基因含量与营养良好的儿童不同。在这些研究中,在营养不良的儿童中普遍观察到变形菌的优势和肠道微生物群的低多样性,并被认为是肠道微生物群成熟的障碍。
此外,最近的一项研究揭示了肠杆菌科细菌与营养不良下的肠道黏膜免疫球蛋白 A (IgA) 反应之间存在机制上的相互关系,这会引发肠病并中断黏膜免疫的发展和健康微生物群的组装。
鉴于生态失调驱动的选择压力似乎干扰了微生物群的稳定性,变形菌随后借此机会增加了它们的适应性。微生物群落在异常代谢条件下的不稳定性已被解释为对定植的抵抗力受损。
当接种来自肥胖人类供体的培养细菌(“肥胖受体小鼠”)的无菌小鼠与携带来自瘦肉供体的细菌物种(低脂肪、高纤维饮食)的小鼠共同饲养时,它们被瘦肉有效定殖供体来源的细菌菌株及其肥胖表型得到改善。相比之下,瘦小鼠没有被来自肥胖小鼠的外源或外源细菌菌株定殖。
这一发现表明,生态失调的特点是传播能力减弱和对定植的抵抗力。鉴于 kwashiorkor 儿童的肠道微生物不成熟且富含肠道病原体营养不良被认为与对殖民化的抵抗力有缺陷有关。
总的来说,这一间接证据导致了这样一种观点,即肠道变形菌的扩张反映了宿主的能量不平衡和不稳定的微生物群。有趣的是,在非疾病状态下,如新生儿期和胃绕道手术后也观察到肠道微生物群落的不稳定结构和高丰度的变形菌。
与大多数细菌一样,在细胞外环境中对变形菌的初步识别是通过病原体识别受体 (PRRs) 发生的,PRRs 识别微生物相关分子模式 (MAMPs)——一个包括病原体相关分子模式 (PAMPs) 和危险相关分子模式的统称分子模式(DAMP)。
这些信号受体可分为三个家族:
尽管至关重要的是,只有 TLR 家族参与识别肠细胞表面的细菌配体。
存在于变形菌细胞表面的主要 MAMP 是脂多糖 (LPS) 和鞭毛蛋白,它们分别被 TLR4 和 TLR5 识别。其他参与细菌识别的TLR包括检测细菌脂蛋白的TLR2和检测未甲基化 CpG DNA 的细胞内受体 TLR9。
LPS 的产生和鞭毛组装是在原核生物中观察到的两个最动态的过程,这些结构组成的巨大差异反映在不同变形菌家族成员中观察到的先天免疫反应的强度和方向上。例如,弯曲杆菌和螺杆菌属LPS 与大肠杆菌LPS 的不同之处在于具有更长的酰基链和增加的链连接和脂质 A 磷酸基团的修饰。
在许多病原生物体(例如百日咳杆菌和幽门螺杆菌)中观察到脂质 A 锚中的一个或两个磷酸基团丢失,并且已被证明可提供对抗菌肽的抗性。
参与细菌识别的 TLR 的遗传变异与 IBD 相关。2010 年发表的一项荟萃分析表明,TLR4 Asp299Gly 和 Thr399Ile 变体都赋予白人患克罗恩病和溃疡性结肠炎的统计学显着风险。有趣的是,这两种变体都位于 LPS 结合域内 TLR4 的胞外域,并且被认为会影响蛋白质的二级结构。
这些功能变体的存在已被证明会影响 LPS 反应性,并使个体更容易受到革兰氏阴性菌的感染。证据还表明,这些遗传变异的存在可能会影响基础免疫状态。
因此,有理由推测,在 TLR4 基因变异的携带者中,在营养不良事件之前或期间发生的免疫反应改变,可能足以驱动 IBD 发生不可挽回的免疫反应改变。TLR9 中的遗传变异也与 IBD 易感性增加有关。证据不如TLR4那样令人信服,尽管这一警告可能反映了 TLR9 处理来自所有细菌的配体而 TLR4 反映革兰氏阴性菌易感性的事实。
变形菌门是肠道菌群中四个主要门(厚壁菌门、拟杆菌门、变形菌门和放线菌门)中最不稳定变化最快的门。变形菌门作为一线反应者,对环境因素(如饮食)反应敏感。
总的来说,迄今为止的许多研究都支持这样一个概念,即肠道中大量变形菌反映了生态失调或不稳定的肠道微生物群落结构。除了外源性肠致病性变形杆菌外,健康的哺乳动物肠道还含有数种属于该门的共生细菌,作为其天然肠道菌群。
这些细菌在比例较小时似乎是良性的,而在某些肠道环境下,它们会变成可引发炎症反应甚至代谢障碍。
然而,肠道中变形菌的长期富集可能代表不平衡的不稳定微生物群落结构或宿主的疾病状态。因此,时间顺序监测,而不是横断面研究,可能是根据肠道中变形菌的比例确定疾病风险的更好方法。
在健康肠道中,免疫系统严格调节其反应以维持与共生菌的共生关系。这种可能性表明存在正反馈循环。环境或宿主因素(例如低纤维饮食和急性或慢性炎症)破坏体内平衡,具有选择性并导致肠道内大量变形菌的生态失调。由于宿主无法保持共生的变形菌而导致变形菌的不受控制的扩张,在一小部分和微生物群落对定植的抵抗力降低的情况下,可以进一步促进炎症或外源性病原体的入侵。
因此,切断反馈回路的策略可能包括优化肠道微生物群和宿主之间的伙伴关系。鉴于大多数研究已经在与宿主生理学相关的背景下描述了微生物群落状态,因此对于未来的炎症和代谢干预治疗,首先需要判别变形菌的丰度以及是其是否不受控制扩张,另外需要确定变形杆菌大量繁殖的原因以开发有效的治疗方法。
主要参考文献:
Rizzatti G, Lopetuso LR, Gibiino G, Binda C, Gasbarrini A. Proteobacteria: A Common Factor in Human Diseases. Biomed Res Int. 2017;2017:9351507. doi: 10.1155/2017/9351507. Epub 2017 Nov 2. PMID: 29230419; PMCID: PMC5688358.
Mukhopadhya I, Hansen R, El-Omar EM, Hold GL. IBD-what role do Proteobacteria play? Nat Rev Gastroenterol Hepatol. 2012 Feb 21;9(4):219-30. doi: 10.1038/nrgastro.2012.14. PMID: 22349170.
Litvak Y, Byndloss MX, Tsolis RM, Bäumler AJ. Dysbiotic Proteobacteria expansion: a microbial signature of epithelial dysfunction. Curr Opin Microbiol. 2017 Oct;39:1-6. doi: 10.1016/j.mib.2017.07.003. Epub 2017 Aug 4. PMID: 28783509.
Shin NR, Whon TW, Bae JW. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015 Sep;33(9):496-503. doi: 10.1016/j.tibtech.2015.06.011. Epub 2015 Jul 22. PMID: 26210164.
Shin NR, Whon TW, Bae JW. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015 Sep;33(9):496-503. doi: 10.1016/j.tibtech.2015.06.011. Epub 2015 Jul 22. PMID: 26210164.
Rigottier-Gois L. Dysbiosis in inflammatory bowel diseases: the oxygen hypothesis. ISME J. 2013 Jul;7(7):1256-61. doi: 10.1038/ismej.2013.80. Epub 2013 May 16. PMID: 23677008; PMCID: PMC3695303.