-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
谷禾健康
记忆力下降、反应变慢、注意力不集中、认知功能减退等问题,正在受到越来越多关注。
尤其是在新冠疫情之后,脑雾、记忆力下降、疲劳和注意力减退等认知相关症状影响了更多人群,也让大脑健康不再只是老年人的话题。
大脑衰老不仅会带来记忆和反应能力下降,还会增加阿尔茨海默病、帕金森病等神经退行性疾病的发生风险。随着认知功能逐渐受损,一个人的学习能力、工作效率、生活质量,甚至独立生活能力都可能受到影响。
以往谷禾写过:大脑退行疾病的两个重要诱因:氧化应激和肠道失衡,人的脑部大约有860亿个神经元和数万亿个突触连接,由250-300亿的神经胶质细胞支持,消耗基础氧气中约20%的比例来维持ATP驱动的活动。
这种高代谢特征,也让大脑更容易产生大量活性氧,也就是ROS。同时相比其他组织,大脑的抗氧化防御系统相对较弱。一旦ROS水平长期超过抗氧化系统的清除能力,就可能出现氧化还原稳态紊乱,形成氧化应激。
氧化应激会损伤细胞结构,干扰能量代谢和神经信号传递,也是多种神经系统疾病的重要病理基础。
可以说,大脑是一个高度耗能、也高度脆弱的器官。
过去,人们更多把神经退行性疾病理解为神经系统内部的问题,而相对忽略了外周器官,尤其是肠道,对大脑功能的长期影响。现在,越来越多证据显示,肠道和大脑之间存在一条复杂的双向通讯网络,也就是微生物群-肠-脑轴。
这条通路并不是单一机制,而是通过神经、代谢、免疫和内分泌等多种方式,把肠道微生态状态与大脑功能紧密连接起来。
肠道菌群可以通过自身代谢产物或次生代谢产物,影响氧化应激、系统性炎症、能量代谢、血脑屏障、神经递质和应激反应,进而参与大脑衰老和认知功能变化。
本文将从机制到干预,系统梳理肠道菌群如何影响大脑衰老:包括微生物群-肠-脑轴的核心通路、可能加速认知衰退的风险菌、支持认知健康的有益菌,全面总结包括饮食、生活方式和精准微生态管理等干预策略,希望为关注脑健康的专业人士和大众提供科学、前沿、实用的知识框架。
如果说过去对大脑衰老的理解更多集中在中枢神经系统内部,例如神经元损伤、突触丢失、小胶质细胞活化、Aβ相关蛋白病理,那么近年的研究正在把视角进一步前移:大脑衰老并不只是脑内事件,它与肠道微生态、外周免疫、代谢信号和神经内分泌网络密切相关。
肠道微生物群与中枢神经系统之间的双向通讯网络是理解肠道菌群如何影响大脑衰老的基础。
编辑
doi.org/10.1007/978-981-99-8803-7
接下来,我们将从神经通路、代谢通路、免疫通路和内分泌通路四个层面,梳理肠道菌群如何影响血脑屏障、神经炎症、海马功能和认知表现。
神 经 通 路
迷走神经把肠道状态传给大脑
在微生物群-肠-脑轴中,迷走神经是一条非常重要的神经通路。
迷走神经如何把肠道信号传给大脑?
迷走神经是人体最长的脑神经之一,其中约80%的纤维属于传入纤维。
它可以把来自胃肠道的机械刺激、化学信号、免疫状态和代谢变化,传递到脑干孤束核,再进一步影响海马、杏仁核、下丘脑、前额叶皮层等脑区。
编辑
doi.org/10.1007/978-981-99-8803-7
这些脑区分别与记忆、情绪、压力、执行功能有关。因此,当肠道菌群失衡影响迷走神经信号时,最终可能反映为认知表现、睡眠状态、情绪稳定性和压力应答能力的变化。
肠道菌群不是直接发消息,而是先改变肠道信号环境
肠道菌群产生或调节的多种代谢物,包括:短链脂肪酸、胆汁酸、色氨酸代谢物和微量胺类物质等,首先影响的是肠道局部环境,包括肠上皮细胞、肠嗜铬细胞、肠神经系统和免疫细胞。随后,这些局部变化再通过迷走神经、内分泌信号和免疫炎症信号传递到中枢神经系统。
也就是说,肠道菌群先改变肠道内部环境,再通过迷走神经把这些变化翻译成大脑能够识别的信号。
★ 5-羟色胺
——肠道产生,但主要通过信号通路影响大脑
人体外周约90%的5-羟色胺(简写5-HT)由肠道肠嗜铬细胞合成,但肠源性5-HT通常不能大量直接穿过血脑屏障。
因此,肠道5-HT对脑功能的影响,更多是通过调节肠神经系统、迷走神经活性、免疫细胞和内分泌信号间接实现。
已有研究显示,部分共生菌能够促进肠嗜铬细胞释放5-HT,并改变肠道神经活动和胃肠动力。
肠道产生的5-羟色胺通常不能大量直接穿过血脑屏障。因此,它对脑功能的影响,并不等同于“肠道5-羟色胺直接进入大脑”。它主要通过激活肠神经系统、迷走神经传入纤维、免疫细胞和内分泌信号,间接影响脑内神经活动、情绪调节和认知过程。
★ GABA和多巴胺
——更多是间接影响神经信号
除了5-HT,肠道菌群还可能影响GABA、多巴胺等神经递质相关通路。
GABA 能帮助大脑放松。部分乳酸菌和双歧杆菌具有谷氨酸脱羧酶系统,可能参与GABA相关代谢。
比如植物乳杆菌 PS128,被认为是一种有潜力的精神益生菌,能改善情绪和运动功能。
注:目前仍然缺乏脑脊液检测等严格的临床数据来证明这种直接的转化关系。
多巴胺,是大脑的快乐与动力分子,一些肠道细菌也可以影响酪氨酸、苯丙氨酸等芳香族氨基酸代谢,并产生酪胺、苯乙胺等微量胺类物质。
这些代谢物可以作用于微量胺相关受体,例如TAAR家族受体,从而影响肠道神经、免疫和内分泌信号。
不过,这里也需要避免一个常见误解:不能简单说“酪胺进入大脑后,会被多巴胺β-羟化酶转化为多巴胺”。事实上,多巴胺β-羟化酶的经典作用,是将多巴胺转化为去甲肾上腺素。
肠道菌群影响多巴胺系统,并不是直接往大脑里输送多巴胺,而是通过间接方式,比如:
衰老后,这条肠-脑神经通路可能变迟钝
在衰老过程中,肠道菌群组成、肠屏障功能和色氨酸代谢都会发生变化。这些变化可能影响5-羟色胺合成相关酶,例如色氨酸羟化酶1(TPH1)的表达或活性,并进一步改变肠—脑信号传递。
与此同时,衰老相关的肠道菌群失调,还可能削弱迷走传入神经元功能。也就是说,肠道里的信号还在发生,但大脑对这些信号的接收和响应能力可能下降。
而海马与学习、记忆和空间认知密切相关。因此,当迷走神经介导的肠—脑通讯减弱时,海马神经元活动和记忆编码过程也可能受到影响。
相关动物研究显示,恢复较年轻的肠道菌群组成,或改善肠道代谢环境,可能有助于增强迷走神经介导的肠—脑通讯,并改善部分与衰老相关的认知功能下降。
最新 Nature 研究:神经通路在认知老化中具有因果作用
近期,Cox等人在《Nature》发表的研究进一步证实:
衰老相关的肠道菌群失调,会通过抑制迷走传入神经元功能,削弱大脑接收肠道内感受信号的能力。
这种信号传递受阻,最终会导致海马功能下降和记忆编码受损。
这一发现的重要意义在于,它不仅说明肠道菌群与大脑衰老有关,更进一步证明了:神经通路在认知老化过程中具有直接的因果作用。
▼
编辑
代 谢 通 路
肠道菌群释放的化学信号,如何影响大脑衰老?
如果说迷走神经像是信息高速路,那么微生物代谢产物就像是肠道菌群释放出来的化学信号。
这些信号可作用于肠道、免疫系统、血管内皮细胞,甚至影响中枢神经系统的炎症和屏障功能。其中研究最深入的代谢物,就是短链脂肪酸。
★ 短链脂肪酸:连接饮食、菌群和大脑
短链脂肪酸,简称SCFAs,是膳食纤维经肠道菌群发酵后产生的主要代谢物,主要包括:乙酸、丙酸、丁酸。
编辑
doi.org/10.1007/978-981-99-8803-7
它们不仅作用于肠上皮细胞、免疫细胞和肠神经系统,也可以通过血液循环、迷走神经和免疫调节等途径,影响中枢神经系统功能。
短链脂肪酸在体内存在明显的浓度梯度
一般来说,肠腔和粪便中的浓度最高,门静脉中降低,进入外周血后进一步下降,而进入脑组织或脑脊液中的浓度通常更低。
编辑
因此,短链脂肪酸影响大脑,更可能是先作用于肠道和外周免疫系统,再通过炎症调节、迷走神经信号、血脑屏障和胶质细胞功能,间接影响脑功能。
★ GPR受体:短链脂肪酸发挥作用的关键开关
短链脂肪酸可以通过多种受体发挥作用,其中较重要的包括:GPR43、GPR41、GPR109A。
GPR43(FFAR2)主要对乙酸和丙酸敏感,可以调节免疫细胞活动、炎症因子释放和代谢状态。
在神经炎症模型中,SCFAs可抑制NF-κB相关促炎信号,降低IL-1β、TNF-α、IL-6等炎症因子表达,并影响小胶质细胞成熟、活化状态和吞噬功能。
GPR41(FFAR3)更多参与肠神经系统、外周神经节和肠内分泌细胞相关信号。丙酸等短链脂肪酸可能通过这一通路影响神经元兴奋性、肠激素释放和自主神经活动。
GPR109A(HCA2)是丁酸和烟酸相关的受体之一。
丁酸除了可以作用于受体,还能作为HDAC抑制剂,通过表观遗传机制调节炎症、神经发生、髓鞘形成和突触可塑性相关基因表达。因此,丁酸对脑功能的影响是受体信号、免疫调节、表观遗传和代谢调控共同作用的结果。
★ 血脑屏障:守住大脑稳定环境的关键屏障
代谢通路影响大脑衰老,一个非常关键的环节是血脑屏障。
血脑屏障由脑微血管内皮细胞、紧密连接蛋白、基底膜、周细胞和星形胶质细胞终足共同构成。它就像大脑的安全门,负责控制哪些物质可以进入脑内,哪些物质应该被阻挡在外。
当血脑屏障完整时,大脑可以维持相对稳定的内部环境;但如果血脑屏障变得薄弱,外周炎症因子、微生物相关分子和其他有害信号就更容易影响中枢神经系统,进而推动神经炎症和认知功能下降。
肠道菌群可以影响血脑屏障的成熟和通透性
研究显示,无菌小鼠的血脑屏障通透性增加,紧密连接蛋白表达下降;而定植正常菌群或补充丁酸钠后,血脑屏障完整性可以得到改善,并伴随occludin、claudin-5等紧密连接相关蛋白上调。
丁酸盐对血脑屏障的保护作用,主要体现在几个方面:
在部分衰老或神经炎症动物模型中,丁酸盐补充可降低血脑屏障通透性和脑内炎症负荷。不过,这些效果仍会受到实验模型、剂量、干预时间和宿主状态影响。
★ 色氨酸:连接菌群、情绪和大脑炎症的通路
色氨酸主要有三条代谢方向:
当衰老、慢性炎症、肠道菌群失调时,色氨酸代谢方向可能发生偏移。也就是说,同样是色氨酸,最终走向不同代谢分支,对大脑产生的影响可能完全不同。
编辑
吲哚丙酸:具有神经保护潜力的菌群代谢物
在色氨酸代谢产物中,吲哚丙酸(IPA)是较受关注的一种菌群来源代谢物。
部分细菌,例如Clostridium sporogenes,可以通过与fldAIBC基因簇相关的芳香族氨基酸代谢通路参与吲哚丙酸生成。
吲哚丙酸具有抗氧化、抗炎、屏障保护作用,可能通过清除自由基、减轻线粒体损伤、调节AhR/PXR相关信号,以及维持肠屏障和血脑屏障功能发挥保护作用。
在阿尔茨海默病和认知障碍相关研究中,较低的吲哚丙酸水平可能与认知下降、代谢异常或神经退行性改变有关。
注:这类发现仍需要更多队列研究和干预研究验证。
IPA受到关注,主要有几个原因:
★ TMAO:放大炎症与认知风险的信号
TMAO,也就是三甲胺-N-氧化物,就是近年来受到关注的一类潜在风险代谢物。
TMAO的形成通常需要两个步骤:
TMAO最早主要在心血管疾病和肾脏疾病中受到关注,近年来也被用于研究肠道菌群与认知障碍、脑血管损伤和阿尔茨海默病之间的关系。
人体研究显示,轻度认知障碍或阿尔茨海默病患者脑脊液中的TMAO水平可能高于认知正常个体,并且较高TMAO水平可能与阿尔茨海默病相关病理、神经退行性标志物或认知表现存在相关性。
注:这些结果主要是观察性关联,不能直接证明TMAO就是认知下降的因果驱动因素。TMAO水平还会受到饮食、肾功能、年龄、心血管代谢状态、药物和检测方法等多种因素影响。
★ 细菌结构成分:也会影响炎症
除了小分子代谢物,细菌来源的结构成分也可能参与肠-脑轴调节,例如脂多糖LPS、肽聚糖PGN、脂壁酸LTA,以及细菌淀粉样蛋白。
这些分子本身不一定需要大量进入脑实质,也可以通过外周免疫激活、血脑屏障损伤、脑血管内皮反应、迷走神经信号和脑室周围器官等途径,影响中枢炎症状态。
LPS是研究最充分的细菌结构成分之一
当肠屏障受损、外周LPS水平升高时,它可以激活免疫系统,诱导IL-1β、TNF-α、IL-6等炎症因子释放;也可以作用于脑血管内皮细胞,破坏紧密连接结构。
若血脑屏障通透性本身已经升高,这些炎症信号就更容易影响中枢神经系统,进一步推动神经炎症和认知功能下降。
细菌淀粉样蛋白curli/CsgA与神经退行性疾病的关系近年来也受到关注。部分研究认为,curli可能通过交叉播种或免疫预激活机制,促进宿主淀粉样蛋白病理,例如Aβ或α-syn聚集。
注:不过,目前关于细菌淀粉样蛋白在人体脑内直接转运和共定位的证据仍然有限。
机制补充:LPS如何通过TLR4/NF-κB通路放大炎症反应?
LPS引发炎症反应的核心通路之一,是TLR4/NF-κB信号轴。简单来说,LPS会先被免疫识别分子捕获,并激活TLR4受体,随后启动MyD88依赖性炎症通路,最终促使NF-κB进入细胞核,诱导IL-1β、TNF-α、IL-6等炎症因子表达。
编辑
这一过程在细胞模型中启动很快,近端信号可在数分钟至十余分钟内发生,炎症基因表达则通常在数小时内明显升高。因此,如果LPS刺激持续存在,就可能不断放大外周炎症,并通过血脑屏障、脑血管内皮细胞和小胶质细胞等途径,推动中枢神经炎症。
血脑屏障渗漏:炎症信号如何更容易影响大脑
衰老、慢性炎症、肠道菌群失调和代谢异常,都可能削弱血脑屏障完整性。
其核心变化包括:紧密连接蛋白下降、内皮细胞炎症增强、周细胞覆盖减少、基底膜重塑、星形胶质细胞终足功能异常,以及转胞吞作用增加。
在衰老动物中,海马、皮层和脑室周围区域可出现不同程度的血脑屏障通透性升高。常见分子改变包括occludin、claudin-5、ZO-1等紧密连接蛋白表达下降,内皮细胞黏附分子增加,MMP-2/MMP-9活性增强,以及小胶质细胞和星形胶质细胞炎症反应增强。
编辑
doi.org/10.1007/978-981-99-8803-7
LPS和其他细菌来源分子可通过TLR4激活脑血管内皮细胞炎症通路,诱导NF-κB、MLCK和RhoA/ROCK等信号,导致细胞骨架收缩、紧密连接重排,最终增加血脑屏障通透性。
其中,MLCK激活后可促进肌球蛋白轻链磷酸化,引起细胞骨架收缩和紧密连接蛋白重新分布。同时,炎症信号还可促进occludin、claudin-5、ZO-1的内吞或降解,使屏障结构进一步变弱。
▸▸
总体来看,代谢通路是肠道菌群影响大脑的一套化学语言。短链脂肪酸和吲哚丙酸等代谢物,可能通过抗炎、抗氧化和保护血脑屏障支持脑健康;而TMAO、LPS及部分细菌结构成分,则可能在菌群失衡和慢性炎症背景下,增加神经炎症和认知下降风险。
免 疫 & 内 分 泌 通 路
炎症和压力轴如何共同影响大脑?
很多时候,肠道菌群失调会先影响肠屏障和外周炎症状态,而炎症信号又会进一步影响血脑屏障、胶质细胞以及HPA轴,也就是下丘脑—垂体—肾上腺轴。
因此,理解免疫与内分泌通路,要看到它们如何共同参与“肠道菌群—系统性炎症—神经炎症—压力反应—认知功能下降”这一连续过程。
肠屏障受损:系统性炎症的起点
肠道本身就是人体最大的免疫相关器官之一。正常情况下,肠道菌群与肠黏膜免疫系统之间维持着一种动态平衡:既要允许共生菌存在,又要防止有害微生物和炎症信号过度扩散。
但在衰老、慢性应激、感染、膳食结构改变或菌群失调等情况下,这种平衡可能被打破。此时,肠上皮屏障功能下降,紧密连接蛋白减少,黏液层变薄,抗菌肽分泌降低,肠道就可能进入通常所说的肠漏状态。
编辑
doi.org/10.1007/978-981-99-8803-7
一旦肠屏障变得不够严密,LPS、肽聚糖、细菌DNA、鞭毛蛋白等微生物相关分子就更容易进入肠道固有层和循环系统,激活单核细胞、巨噬细胞、树突状细胞和中性粒细胞。
随后,TNF-α、IL-1β、IL-6、IL-17、IFN-γ、CCL2、CXCL10等炎症因子释放增加,形成持续的低度系统性炎症。
这种炎症并不一定非常剧烈,却可能长期存在。对大脑来说,长期低度炎症比短期强烈炎症更隐蔽,也更容易在衰老过程中逐渐累积影响。
外周炎症如何传到大脑?
外周炎症影响大脑,并不一定意味着炎症因子必须大量直接进入脑实质。外周炎症可以通过多条路径把信号传递给中枢神经系统。
编辑
doi.org/10.1007/978-981-99-8803-7
因此,外周炎症与神经炎症之间,长期系统性炎症会让大脑处于更容易被激活的状态,降低神经元对代谢压力、氧化应激和炎症刺激的耐受性,进而影响突触修剪、神经递质平衡和认知功能。
小胶质细胞:从巡逻者变成高敏状态
在中枢神经系统中,小胶质细胞是最重要的固有免疫细胞之一。
在年轻健康状态下,小胶质细胞主要处于稳态监视状态,像大脑的巡逻者,负责识别病原信号、清除细胞碎片、参与突触修剪,并与神经元、星形胶质细胞和血管细胞共同维持脑内稳态。
这类稳态小胶质细胞通常表达P2RY12、TMEM119、CX3CR1等标志物。
编辑
但随着年龄增长,小胶质细胞会逐渐发生变化。它们可能出现促炎因子表达升高、吞噬功能异常、脂质代谢紊乱、补体通路激活,以及抗原呈递相关基因增强等特征。如果同时存在肠道菌群失调和系统性炎症,小胶质细胞就更容易进入一种预激活或疾病相关状态。
预激活,可理解为:小胶质细胞还没有完全进入强烈炎症反应,但已经变得更敏感。一旦再次遇到LPS、IL-1β、TNF-α、Aβ、损伤相关分子或氧化应激信号,就可能产生更强烈的炎症反应。
此时,IL-1β、TNF-α、IL-6、C1q、C3、iNOS等分子可能升高,进而影响突触功能和神经网络稳定性。
编辑
过去常用M1/M2模型描述小胶质细胞状态,认为M1偏促炎,M2偏抗炎和修复。但近年来单细胞测序研究显示,小胶质细胞远比这种二分法复杂。
在不同疾病和衰老背景下,小胶质细胞可能呈现疾病相关型、干扰素响应型、增殖型、吞噬型和衰老相关状态等多种亚群。
总的来说,衰老和菌群失调会让小胶质细胞从稳态监视状态,逐渐偏向促炎、吞噬异常、补体激活和疾病相关转录状态。
星形胶质细胞:保护反应也可能变成损伤因素
星形胶质细胞是大脑中非常重要的支持细胞。它们参与维持离子稳态、清除谷氨酸、支持血脑屏障、调节能量代谢和维持突触功能。
在健康状态下,星形胶质细胞更多是在帮助大脑维持稳定环境。但在神经退行性疾病、长期慢性炎症或血脑屏障受损的背景下,星形胶质细胞会从维持稳定的状态,转变为更加活跃的反应性状态。
可以把它理解为:当大脑受到炎症或损伤刺激后,星形胶质细胞会被激活,开始参与防御、修复和炎症调节。
编辑
这种反应性星形胶质细胞通常会出现一些分子标志物升高,例如:GFAP、Vimentin、C3、S100B、CXCL10、SERPINA3和LCN2。
同时,它们的形态也会发生变化,比如细胞体变大、突起重新排列,并分泌更多炎症因子、趋化因子或补体蛋白。
在肠道菌群失调和全身炎症背景下,来自外周的LPS、TNF-α、IL-1α、C1q和IL-1β等信号,都可能进一步激活星形胶质细胞。
短期来看,这种激活有一定保护意义,可以帮助隔离损伤、限制炎症扩散。
但如果激活长期持续,就可能从保护反应转变为损伤因素。长期反应性星形胶质细胞会释放更多炎症介质、趋化因子和补体蛋白,进而促进突触损伤、血脑屏障功能下降,并与认知功能受损有关。
因此,GFAP、S100B、CXCL10等指标,可以作为观察星形胶质细胞反应性和神经炎症程度的辅助参考。
HPA轴:菌群也会影响压力反应
HPA轴是人体应对压力的重要系统。简单来说,当身体感受到压力时,下丘脑会释放相关信号,促使垂体释放ACTH,最终刺激肾上腺分泌糖皮质激素。
编辑
经典无菌动物研究显示,无菌小鼠在急性应激后会表现出更强的ACTH和皮质酮反应。这提示,正常肠道菌群可能参与HPA轴发育和应激敏感性的设定。
早期定植某些共生菌,例如Bifidobacterium infantis,可以部分逆转无菌小鼠夸大的HPA轴应激反应。这说明,特定菌群可能在一定程度上帮助调节压力轴的敏感性。
压力轴失衡:为什么慢性压力会影响记忆?
当慢性应激持续存在时,HPA轴会长期处于激活状态,导致糖皮质激素持续升高。过量糖皮质激素可以通过糖皮质激素受体影响海马、杏仁核和前额叶皮层功能。
在海马中,长期高糖皮质激素可能抑制BDNF表达,削弱神经发生和突触可塑性,并增加神经元对谷氨酸兴奋性毒性、氧化应激和炎症损伤的敏感性。
这对认知功能尤其重要。因为海马不仅参与学习和记忆,还参与HPA轴的负反馈调节。
于是,可能形成一个恶性循环:
慢性应激 → HPA轴过度激活 → 海马功能下降 → 负反馈减弱 → 应激反应进一步放大。
编辑
菌群如何调节HPA轴?
肠道菌群可以通过多种方式调节HPA轴敏感性。
也就是说,肠道菌群对HPA轴的影响是神经、免疫、代谢、内分泌信号共同参与的结果。
随着肠道菌群-肠-脑轴研究的深入,越来越多证据显示,特定菌群变化可能参与认知老化、神经炎症、睡眠障碍和神经退行性疾病进展。
需要强调的是,不同菌种的证据强度并不相同。有些已经在动物模型中获得较强的因果验证,有些主要来自人群相关性研究,还有一些机制仍停留在体外实验或推测阶段。
1、Parabacteroides goldsteinii
动物模型中证据较强的促认知衰退候选菌
在年龄相关认知下降研究中,Parabacteroides goldsteinii 是近年来受到高度关注的菌种之一。
Cox 等人在 2026 年发表于《Nature》的研究中,通过多组学筛选、菌群移植、单菌定殖和靶向干预等方法,提示该菌可能参与小鼠年龄相关记忆下降过程。
研究显示,随着小鼠年龄增长,P. goldsteinii 的丰度升高,并与认知表现下降相关。
更重要的是,在功能验证中,该菌及其相关代谢产物能够削弱肠-脑信号,影响外周免疫状态,并最终损害学习和记忆功能。
目前较明确的机制,集中在中链脂肪酸产生上。
P. goldsteinii 可以产生己酸、庚酸、辛酸等中链脂肪酸。这些代谢物可能通过影响外周髓系细胞和肠道传入神经信号,进一步影响脑功能。
相比之下,胆汁酸代谢、黏液层降解和色氨酸代谢是否构成该菌促认知衰退的主要机制,目前仍需更多直接证据支持。
编辑
P. goldsteinii 可以被视为“小鼠年龄相关认知下降中具有较强因果证据的候选风险菌”。但还不能直接等同于人类认知衰退的核心病因。
2、 Enterobacteriaceae
内毒素和细菌淀粉样蛋白相关的炎症风险
肠杆菌科细菌,包括 Escherichia coli 、 Klebsiella pneumoniae 等,是肠道炎症和系统性低度炎症研究中常见的风险菌群。
它们的一个共同特点是含有脂多糖,也就是 LPS。
LPS 是一种典型的内毒素,可以通过TLR4/NF-κB等通路诱导促炎因子释放,增加肠屏障和血脑屏障通透性,并促进小胶质细胞活化。这一过程可能参与神经炎症、突触功能损害和认知下降。
编辑
其中,部分 E. coli 菌株还能产生curli菌毛淀粉样蛋白。Curli的主要结构蛋白CsgA具有淀粉样特征,在实验模型中可能促进宿主淀粉样蛋白聚集,并增强神经炎症反应。
也就是说,E. coli 的潜在风险不只来自LPS诱导炎症,也可能与细菌淀粉样蛋白影响Aβ或α-syn相关病理有关。
Klebsiella pneumoniae 在部分炎症性疾病、代谢异常和神经退行性疾病患者中也可见富集。
它的潜在风险主要来自LPS、荚膜多糖和促炎免疫激活。
若要进一步声称其特定代谢物可直接穿过血脑屏障并促进 α-syn 磷酸化,则需要明确的原始研究支持。
3、Veillonella、Citrobacter
可能通过睡眠和炎症影响认知
睡眠质量下降,是认知衰退的重要风险因素。因为睡眠不仅关系到精神状态,也参与记忆巩固、突触可塑性维持,以及大脑代谢废物清除。
在一些临床研究中,Veillonella 和 Citrobacter 与睡眠障碍和潜在认知损害有关。
例如,在卒中后睡眠障碍患者(PSSD)的粪便中,这两类细菌被观察到显著富集,并且其丰度与匹兹堡睡眠质量指数评分呈正相关。
▾
Veillonella是革兰氏阴性厌氧球菌,主要通过发酵乳酸产生丙酸和乙酸。
在睡眠障碍背景下,如果Veillonella过度生长,可能通过影响GABA能传递、促进神经炎症或改变肠—脑轴信号,间接影响睡眠质量。而睡眠紊乱本身,又会进一步影响海马突触可塑性、记忆巩固和执行功能。
▾
Citrobacter属于肠杆菌科,其富集常提示肠道生态失调、肠屏障功能障碍和炎症状态。该属细菌产生的内毒素可能触发系统性炎症,进而影响脑功能。
这两种细菌通过干扰睡眠质量和昼夜节律调节,形成”菌群失调-睡眠障碍-认知下降“的恶性循环。
4、脱硫弧菌属 Desulfovibrio
帕金森和硫化氢代谢相关的风险菌
脱硫弧菌属(Desulfovibrio属)是典型的硫酸盐还原菌,可以产生硫化氢,也就是H₂S。
已有研究报道,脱硫弧菌属在帕金森病患者肠道中可能升高,并且部分菌株可在模型系统中促进α-突触核蛋白聚集。这提示它可能参与帕金森病相关的肠-脑轴病理过程。
编辑
潜在机制主要包括几个方面。
不过,H₂S并不是单纯的有害分子。在生理浓度下,它也是一种重要的气体信号分子,参与血管舒张、线粒体调节和神经信号转导。在特定病理背景下,脱硫弧菌属可能通过增加硫化氢、LPS和炎症负荷,加重神经退行性病理风险。
编辑
5、Prevotella copri
高度依赖饮食背景的双向菌
Prevotella copri 不能简单归类为“有害菌”或“有益菌”。它的功能很大程度上取决于饮食结构、宿主代谢状态和菌株差异。
这个例子也提醒我们:
判断一种菌是风险还是保护,不能脱离饮食结构、菌株差异和宿主状态。同一种菌,在不同生态背景下,可能产生完全不同的健康效应。
广泛存在于人群的双面使者——Prevotella copri与疾病和健康
风险菌如何共同推动认知衰退?
总体来看,可能加速认知衰退的菌群并非通过单一路径发挥作用,而是通过多个相互连接的机制节点共同影响脑功能。
代谢节点
部分菌群可增加 LPS、H₂S、中链脂肪酸、TMAO 等促炎或应激相关代谢物;
同时减少短链脂肪酸、吲哚丙酸等具有屏障保护和神经保护潜力的代谢物。
屏障节点
菌群失衡可能削弱肠道黏液层和紧密连接结构,增加肠道通透性,使 LPS 和其他微生物相关分子更容易进入循环。
系统性炎症进一步影响血脑屏障,使外周炎症信号更容易传递至中枢神经系统。
免疫节点
LPS、细菌淀粉样蛋白和异常代谢物可激活 TLR4/NF-κB、NLRP3 炎症小体等通路,促使 TNF-α、IL-1β、IL-6 等促炎因子升高,并诱导小胶质细胞活化。
神经信号节点
菌群变化可能影响 5-HT、GABA、多巴胺等神经递质相关通路,也可能通过迷走神经、肠神经系统和内分泌信号改变脑内网络活动。
蛋白病理节点
部分细菌产物,如 LPS、curli 淀粉样蛋白或硫化氢相关应激信号,可能促进 Aβ、α-syn 或 tau 相关病理过程,但不同蛋白病理对应的证据强度并不一致,需要分别讨论。
这些节点之间存在正反馈关系:菌群失衡可诱导炎症,炎症进一步破坏屏障和改变肠道生态,屏障损伤又放大微生物产物进入循环的机会,最终形成:
菌群失衡—屏障破坏—系统性炎症—神经炎症—认知下降
编辑
因此,针对认知衰退的菌群干预不应只盯着某个坏菌,而应同时考虑饮食结构、炎症状态、屏障功能、代谢物、宿主本身是否存在神经退行性病理背景。
与促炎或促衰老菌群相对应,肠道中也存在一批可能支持认知健康的有益菌。这些菌群可通过产生短链脂肪酸、调节色氨酸代谢、降低系统性炎症、维护肠道屏障和影响肠—脑轴神经信号,对脑功能产生保护作用。
需要注意的是,认知保护作用往往具有明显的菌株特异性和宿主背景依赖性。
因此,下述菌种应按照证据强度分层理解:部分已有小样本人群试验支持,部分主要来自动物实验,还有一些仍属于机制推测或观察性关联。
1、双 歧 杆 菌 属
认知和压力调节研究中证据较多的益生菌
双歧杆菌属是认知健康研究中较受关注的一类有益菌。它们的潜在作用包括改善肠道屏障、降低低度炎症、调节色氨酸代谢,以及影响肠-脑轴神经信号。
其中,Bifidobacterium longum 1714 是研究较多的精神益生菌菌株之一。已有随机、双盲、安慰剂对照研究显示,该菌株可能影响健康成人的压力反应、脑电活动和记忆相关表现。
研究提示,补充 B. longum 1714 后,受试者在急性压力任务中的生理和心理反应有所减弱,部分记忆任务表现改善,并伴随脑活动模式变化。
从机制上看,B. longum 1714 可能通过调节压力轴、肠-脑神经信号和神经递质相关通路,对认知和情绪状态产生影响。
在轻度认知障碍人群中,Bifidobacterium breveMCC1274/A1 比 B. breve M-16V 有更直接的证据。
已有研究提示,B. breve MCC1274/A1 补充可能改善部分认知测验表现,并可能减缓脑萎缩进展。
不过,这类结果目前仍需要更大样本、更长随访和独立重复研究来确认。
总体而言,双歧杆菌属可以被看作认知健康干预中较有潜力的一类益生菌,但实际效果仍要看具体菌株、受试者基线菌群状态,以及是否存在认知障碍、炎症或代谢异常等背景。
2、乳 酸 杆 菌 属
通过神经递质、炎症和屏障功能影响脑健康
乳酸杆菌属包含多个被研究较多的益生菌菌株。鼠李糖乳杆菌Lacticaseibacillus rhamnosus GG(LGG) 是研究最充分的益生菌之一。
动物研究显示,LGG 可在炎症、应激或代谢异常相关模型中改善认知表现,并与海马 BDNF/TrkB 信号、肠屏障改善和系统性炎症下降有关。
在人群研究中,LGG 也被观察到可能与中老年人认知表现改善相关,但具体效果受人群基础状态、干预时间和联合饮食因素影响。
从机制上看,LGG 可能通过改善肠屏障、减少外周炎症、调节 GABA 受体表达和影响迷走神经信号发挥作用。经典动物研究曾显示,某些乳酸杆菌对 GABA 受体表达和行为表型的影响依赖迷走神经完整性。
Lactiplantibacillus plantarum PS128是另一个受到关注的精神益生菌菌株。
动物研究显示,PS128 可在阿尔茨海默病模型中改善认知功能,并可能通过调节丙酸水平、GSK3β 活性和神经炎症发挥作用。
该菌株也被用于抑郁、焦虑、睡眠和神经发育相关研究,但目前关于其在人类轻度认知障碍中显著提高脑脊液 GABA 或直接改善执行功能的证据仍需进一步核实。
乳杆菌属Lactobacillus——维持肠道和阴道健康不可忽缺的角色
3、Akkermansia muciniphila
以屏障和代谢改善为核心的下一代益生菌
Akkermansia muciniphila 是下一代益生菌研究中的代表菌种。它能利用肠道黏液作为营养来源,并与黏液层更新、肠屏障完整性和代谢健康密切相关。
编辑
其潜在有益机制主要包括三方面。
人体研究方面,已有试验显示,A. muciniphila,尤其是巴氏灭活形式,在超重、肥胖或胰岛素抵抗人群中具有良好的安全性,并可能改善部分代谢指标和炎症相关指标。
在动物模型中,A. muciniphila 已被报道可改善睡眠剥夺、肝病相关脑功能异常或阿尔茨海默病模型中的认知损害,并与海马炎症降低、BDNF/5-HT 信号改善或 Aβ 病理减轻相关。
不过,关于 A. muciniphila 直接改善老年人认知、提高 BDNF 或改善处理速度的临床证据,目前仍不充分。
双面明星菌:Akkermansia muciniphila——从肠道微生态到情景治疗的探索
肠道重要菌属——Akkermansia Muciniphila,它如何保护肠道健康
4、产 丁 酸 菌
Faecalibacterium 与 Roseburia 的屏障和抗炎作用
产丁酸菌是认知健康相关菌群中最值得关注的一类菌。丁酸不仅是结肠上皮细胞的重要能量来源,还具有组蛋白去乙酰化酶抑制、抗炎、促进紧密连接和调节小胶质细胞功能等作用。
▾
Faecalibacterium prausnitzii是人体肠道中重要的丁酸产生菌,也是健康成人肠道内较丰富的厌氧菌之一。它主要通过丁酰辅酶A:乙酸辅酶A转移酶等通路产生丁酸,并具有较强的抗炎潜力。
在阿尔茨海默病和认知障碍相关研究中,Faecalibacterium 等产丁酸菌减少常被认为是菌群失衡的重要特征。
丁酸可能通过多条路径支持认知健康:
肠道核心菌属——普拉梭菌(Faecalibacterium Prausnitzii),预防炎症的下一代益生菌
▾
Roseburia intestinalis 同样是重要的产丁酸菌。Roseburia属丰度降低常见于炎症性肠病、代谢异常和部分神经退行性疾病相关菌群失衡状态。其潜在保护作用主要来自丁酸产生、肠屏障维护和炎症调节。
关于 Roseburia 是否通过共轭亚油酸、胆汁酸或吲哚丙酸直接发挥神经保护作用。
Faecalibacterium 和 Roseburia 共同维持肠道厌氧生态、丁酸供给、屏障稳定和低炎症状态。这些基础环境对于延缓脑老化和维持认知功能可能具有重要意义。
5、其 他 菌
与认知保护相关的候选菌
▾
Clostridium sporogenes是色氨酸代谢研究中的重要菌种,可通过相关代谢通路产生吲哚丙酸。吲哚丙酸是一种具有抗氧化和屏障保护作用的微生物来源代谢物,也被认为可能抑制 Aβ 聚集和神经炎症。
动物研究显示,通过 C. sporogenes 与膳食底物联合提高吲哚丙酸水平,可能改善阿尔茨海默病模型中的认知表现。不过,人类临床证据仍有限。
▾
Lachnospiraceae 中包含多种产短链脂肪酸菌。部分成员可能参与膳食多酚、坚果成分和复杂碳水化合物代谢,并与较好的炎症状态和代谢健康相关。
若某些队列研究显示 Lachnospiraceae UCG-004 与坚果摄入和认知表现相关,是一个观察性发现,不能直接推断为因果保护菌。
▾
Flavonifractor plautii 可参与黄酮类化合物代谢,但其健康意义具有情境依赖性。它可能参与多酚转化,提高部分代谢物的生物利用度;但在不同疾病状态下,其丰度变化和功能解释并不一致。
有益菌如何共同支持认知健康?
认知保护相关菌群可能通过多个互补层面共同发挥作用。
编辑
——代谢层面
有益菌群可产生丁酸、丙酸、乙酸等短链脂肪酸,激活 GPR41、GPR43、GPR109A 等受体,并通过表观遗传机制调节炎症和神经营养相关基因表达。
——在屏障层面
双歧杆菌、乳酸杆菌、Akkermansia 和产丁酸菌均可能增强肠道紧密连接、促进黏液层稳定、减少 LPS 易位,并间接降低血脑屏障受到系统性炎症冲击的风险。
——在免疫层面
这些菌群和代谢物可能促进 Treg 分化、增加 IL-10 等抗炎信号,抑制 NF-κB 和 NLRP3 炎症小体过度激活,并调节小胶质细胞和星形胶质细胞反应状态。
——在神经信号层面
部分菌株可能影响 GABA、5-HT、多巴胺和色氨酸代谢相关通路,也可能通过迷走神经和肠神经系统改变脑内应激反应和情绪—认知网络活动。
——在结构和功能层面
有益菌群的长期影响可能表现为海马炎症减轻、BDNF 等神经营养因子上调、突触可塑性改善和记忆巩固能力增强。
不过,这些结构层面的证据目前更多来自动物实验,人类中仍需更多影像学、脑脊液标志物和长期认知随访研究支持。
编辑
总体来看,维持认知健康的菌群依赖一个稳定、多样、低炎症、富含产短链脂肪酸和色氨酸有益代谢能力的肠道生态系统。益生菌干预若要真正影响认知健康,通常也需要与高纤维饮食、多酚摄入、规律运动、睡眠管理和代谢风险控制共同发挥作用。
除了直接靶向肠道微生物本身,通过调节菌群下游的免疫、代谢和神经通路来干预认知衰老,也是一个重要方向。
这类策略的优势在于,它们不完全依赖于复杂且高度个体化的菌群组成,而是瞄准更接近病理效应的分子和通路,例如 GPR84、IL-1β、短链脂肪酸、胆汁酸受体和迷走神经信号。
不过,这些策略的证据强度并不相同。
因此,在讨论这些策略时,应区分机制证据、动物实验结果、临床可用疗法。
GPR84 抑制:阻断菌群代谢物引发的炎症信号
GPR84 抑制是连接菌群代谢物与免疫炎症反应的一个新兴干预方向。
GPR84 是一种主要在髓系免疫细胞中表达的 G 蛋白偶联受体,可被中链脂肪酸激活。这就让 GPR84 成为一个重要接口:
Cox 等人在 2026 年的研究中发现,在小鼠模型中,Parabacteroides goldsteinii 可以产生中链脂肪酸,并通过激活髓系细胞上的 GPR84 信号,引发外周炎症。
这种炎症反应进一步削弱迷走神经介导的肠—脑信号传递,最终影响学习和记忆功能。
更重要的是,研究显示,使用小分子 GPR84 拮抗剂后,可以减轻 P. goldsteinii 或中链脂肪酸诱导的记忆损害,并抑制相关髓系炎症反应。
编辑
这一发现的意义在于,它把 GPR84 从一个机制节点推进到了潜在药物靶点的位置。
相比直接清除某一种细菌,靶向 GPR84 的理论优势是:即使不同人的菌群组成不一样,只要最终都汇聚到“中链脂肪酸—GPR84—髓系炎症”这条通路,就有可能通过阻断下游炎症效应来发挥作用。
不过,目前 GPR84 抑制用于认知老化仍处于早期转化阶段。现有 GPR84 拮抗剂主要围绕炎症性肠病、纤维化、慢性炎症疾病开发。
未来还需要进一步验证它在老年人、轻度认知障碍或神经退行性疾病中的疗效和安全性。同时,也要评估长期抑制 GPR84 是否会影响正常免疫防御和组织修复。
IL-1β 通路阻断:能否恢复被炎症削弱的肠—脑信号?
IL-1β 是连接外周炎症与神经功能改变的重要促炎细胞因子。它不仅参与系统性炎症和神经炎症,也可能通过作用于外周感觉神经元,改变肠-脑轴信号传递。
Cox 等人的机制研究提示,在小鼠中,P. goldsteinii 和中链脂肪酸诱导的髓系炎症可以增加 IL-1β 信号。随后,IL-1β 作用于表达 PHOX2B 的迷走感觉神经元,抑制其正常传入功能,从而削弱肠道向大脑传递的保护性内感受信号。
换句话说,炎症并不只是让身体处于炎症状态,它还可能直接干扰肠道向大脑传递信息的神经通路。
动物实验显示,阻断 IL-1β 相关信号可以部分逆转这一过程,恢复迷走神经功能,并改善认知表现。
目前,临床上已经有多种 IL-1 通路药物。
例如,Anakinra 是 IL-1 受体拮抗剂,可以同时阻断 IL-1α 和 IL-1β 信号;
canakinumab 是靶向 IL-1β 的单克隆抗体;
rilonacept 则是可以结合 IL-1 的融合蛋白。
这些药物主要用于自身炎症性疾病、部分风湿免疫疾病和复发性心包炎等场景。
CANTOS 研究显示,canakinumab 可以降低既往心肌梗死且炎症水平升高患者的心血管事件风险,支持了靶向炎症可以改善慢性疾病结局这一思路。
但需要注意的是,CANTOS 不能直接证明 IL-1β 阻断能够改善认知功能。IL-1β 抑制剂是否能用于认知老化或阿尔茨海默病,还需要专门设计的临床试验验证。
此外,IL-1β 并不是单纯有害的信号。它也参与宿主防御、感染控制和组织修复。长期或全身性阻断 IL-1 通路,可能增加感染风险。
因此,未来如果将 IL-1β 阻断用于认知老化,更可能适合特定人群,例如存在明显外周炎症、肠屏障损伤或 IL-1β 活化特征的亚型患者,而不是广泛用于所有老年人。
短链脂肪酸补充:与其直接补充,不如促进自身产生
在几类短链脂肪酸中,丁酸最受认知健康研究关注。
动物实验显示,丁酸钠可以通过抑制组蛋白去乙酰化酶,也就是 HDAC,增加 BDNF 等神经营养因子的表达,促进突触可塑性,并减轻小胶质细胞过度活化和神经炎症。
这些作用使丁酸成为连接“肠道菌群—代谢物—脑功能”的重要分子。
乙酸和丙酸也可通过 GPR41、GPR43 等受体调节免疫反应、能量代谢和肠道屏障功能。
但需要注意的是,短链脂肪酸并不是越多越好,也不能把乙酸、丙酸和丁酸简单看成完全相同的保护分子。
尤其是丙酸,在不同模型中可能呈现不同效应,具有明显的剂量和背景依赖性。
编辑
直接补充短链脂肪酸也面临一些现实问题。
因此,相比直接大量补充短链脂肪酸,更现实的策略可能是:
也就是说,短链脂肪酸干预的关键,不是简单补多少,而是如何让身体在合适的位置、合适的时间,产生合适水平的代谢信号。
编辑
胆汁酸代谢:一个新兴但还不成熟的方向
胆汁酸不仅参与脂肪消化吸收,也是重要的信号分子。
肠道菌群可通过胆盐水解、脱羟基化等反应改变胆汁酸谱,而不同胆汁酸又可通过 FXR、TGR5 等受体影响糖脂代谢、炎症反应和屏障功能。
在脑健康研究中,TGR5 尤其受到关注。TGR5 在小胶质细胞和星形胶质细胞等细胞中均有表达。
动物和细胞实验提示,激活 TGR5 可能抑制神经炎症、调节小胶质细胞活化状态,并在某些模型中改善认知表现。
FXR 主要在肝脏和肠道发挥代谢调节作用,但也可能通过改善外周代谢、炎症状态和胆汁酸稳态间接影响脑功能。
奥贝胆酸是 FXR 激动剂,已获批用于原发性胆汁性胆管炎。它在代谢性肝病中的研究提示,胆汁酸受体调节可影响全身代谢和炎症状态。但奥贝胆酸也存在瘙痒、血脂变化等安全性和耐受性问题,因此不能简单外推到认知老化干预。
目前,胆汁酸受体调节剂在神经退行性疾病和认知老化中的应用仍处于早期阶段。更稳妥的方向可能包括:通过饮食、益生元、胆汁酸代谢菌群调节和选择性 TGR5/FXR 调节剂,改善胆汁酸谱和慢性炎症状态。未来需要更多人体研究来确认其认知获益和长期安全性。
神经通路辅助干预:恢复肠—脑轴信号传递
迷走神经是最值得关注的神经通路。它连接肠道和脑干,并进一步影响海马、下丘脑和边缘系统等与记忆、情绪和应激反应相关的脑区。
在衰老和菌群失调状态下,肠道炎症增加、屏障功能下降以及异常微生物代谢物升高,可能削弱迷走神经传入信号,使大脑对肠道内感受信息的响应下降,进而影响海马神经活动和记忆功能。
因此,恢复迷走神经功能被认为是潜在的辅助干预方向。传统迷走神经刺激和经皮耳迷走神经刺激等方式,已在情绪、炎症和认知相关研究中受到关注,可能通过增强副交感神经活性、降低炎症反应和调节胆碱能信号影响脑功能。
不过,目前这类方法仍属于探索性干预,尚不能作为认知衰老的标准治疗。
此外,肠道菌群还可能通过影响 5-羟色胺、GABA、多巴胺和胆碱能系统参与脑功能调节。但这些作用多是间接发生的,通常依赖前体代谢、肠内分泌细胞、免疫炎症状态、迷走神经活动和屏障功能,而不是肠道神经递质直接进入大脑。
总体来看,神经通路干预的价值在于为菌群代谢、外周炎症和脑功能变化提供一个可调节的连接点。
高膳食纤维饮食:菌群健康的基础
如果要从一个最基础的饮食策略开始,那一定是增加膳食纤维。膳食纤维是肠道菌群的重要能量来源。充足的膳食纤维摄入,可以帮助维持菌群多样性和代谢活性,并促进产短链脂肪酸菌生长。
一般建议每日摄入约25–30克膳食纤维,来源可以包括:全谷物、豆类、蔬菜、水果、坚果和种子。
不同类型的纤维,会被不同菌群利用。例如:
燕麦、香蕉、洋葱、大蒜、芦笋等食物富含低聚果糖和菊粉,是双歧杆菌和乳酸杆菌喜欢利用的底物。
编辑
全谷物中的β-葡聚糖可以促进产丁酸细菌生长。
豆类中的抗性淀粉经过肠道菌群发酵后,也可以产生较多短链脂肪酸。
一些干预研究显示,增加膳食纤维摄入数周后,肠道菌群组成就可能发生明显变化,产短链脂肪酸细菌丰度增加,系统性炎症标志物下降,认知测试表现也可能出现改善。
从长期来看,坚持高纤维饮食的老年人,认知衰退速度可能更慢。
但需要注意的是,增加纤维要循序渐进。突然大量增加豆类、全谷物或菊粉类食物,可能引起腹胀、排气增多或肠胃不适。可以逐步增加,比如先把部分精米白面换成全谷物,再增加豆类、蔬菜和坚果摄入,让肠道菌群有适应过程。
地中海饮食:证据较充分的饮食模式
在认知健康相关饮食模式中,地中海饮食的证据相对最充分。
多项大型前瞻性队列研究显示,坚持地中海饮食模式的人群,认知衰退速度更慢,痴呆风险降低30%-50%。
地中海饮食的核心并不复杂,主要包括:
编辑
这种饮食模式富含膳食纤维、多酚、不饱和脂肪酸和抗氧化成分。
从肠道菌群角度看,地中海饮食可以增加一些与短链脂肪酸产生和抗炎作用相关的有益菌,例如 Roseburia、Faecalibacterium 等,并提高粪便和循环中的短链脂肪酸水平,从而减轻系统性炎症。多酚类化合物,是地中海饮食发挥作用的重要成分之一。
橄榄油中的橄榄苦苷、浆果中的花青素、姜黄中的姜黄素、葡萄中的白藜芦醇等,都具有抗氧化和抗炎潜力。
坚果:小份量也可能带来认知保护
坚果是饮食干预中比较容易执行的一类食物,也有较多流行病学和机制研究支持。
Ni et al.(2025)在747名社区老年人中进行的前瞻性队列研究提供了高质量证据,研究显示,每周食用3–7份坚果的老年人,在长期随访中整体认知功能下降速度更慢。
与不食用坚果的人相比,每周食用3份及以上坚果者,认知衰退风险明显降低。
这种认知益处可能与肠道菌群变化有关。
研究中,坚果消费组Lachnospiraceae UCG-004、Roseburia 和 Flavonifractor等菌群富集,整体微生物多样性更高,粪便短链脂肪酸水平也更高。
降解多酚黄酮的肠道重要菌——Flavonifractor,能促进痛风、抑郁也能改善便秘肥胖
机制研究提示,坚果中的多酚和不饱和脂肪酸共同作用,塑造了有益于大脑健康的肠道微环境。坚果中的精氨酸还可以转化为一氧化氮,改善脑血流。维生素E和硒等抗氧化成分则直接保护神经元免受氧化损伤。
推荐每日食用一小把坚果(约25-30克),种类多样化更佳:核桃富含ω-3脂肪酸,杏仁和腰果富含多酚,开心果富含叶黄素,花生富含白藜芦醇,各有侧重。
需要注意的是,坚果能量密度较高,最好选择原味、少盐、少糖、不过度油炸的类型。如果已经有肥胖、脂代谢异常或肾脏疾病等问题,摄入量应结合个人情况调整。
生酮饮食:有潜力,但争议也很明显
生酮饮食的证据相对复杂,存在明显的争议。该饮食通过极低碳水化合物、高脂肪摄入诱导酮症,使大脑主要依靠酮体供能。
在癫痫治疗中,生酮饮食已有近百年的应用历史。
从菌群角度看,生酮饮食显著改变肠道菌群组成,增加Bacteroides丰度,减少Firmicutes。这种菌群变化与免疫反应调节相关,可能部分解释生酮饮食的抗癫痫作用。
在认知领域,小型临床试验显示生酮饮食可以轻度改善轻度认知障碍和早期阿尔茨海默病患者的认知功能。酮体可能为受损的神经元提供替代能源,同时减少氧化应激和炎症。
但生酮饮食的高脂肪特性也可能加剧肠道菌群失调,长期使用的安全性尚未得到充分验证。可能的风险包括营养不均衡、血脂异常、肾结石风险增加等。对于老年人,还需要考虑吞咽困难和消化功能下降等问题。
目前来看,生酮饮食可能适用于特定人群的短期干预,但作为长期的认知保护策略仍需更多研究支持。个性化的生酮方案,如周期性生酮或改良生酮,可能平衡收益与风险。
注:如果要执行需要在专业指导下进行,而不是自行长期严格执行。
其他饮食模式与认知健康
▾
MIND饮食:可以理解为DASH饮食(停止高血压饮食法)与地中海饮食的结合,专门为大脑健康设计,已被证明可以降低阿尔茨海默病风险。
DASH饮食原本用于帮助控制高血压,而MIND饮食则进一步强调与认知保护相关的食物,例如绿叶蔬菜、浆果、坚果、全谷物、豆类、鱼类和橄榄油。它的优势在于执行逻辑清楚,既关注心血管代谢健康,也强调大脑友好的植物性食物和抗氧化成分。
▾
间歇性禁食作为近年来热门的饮食模式,在动物实验中显示出显著的认知改善作用。
隔日禁食、限时进食等模式可以调节肠道菌群组成,增加菌群节律性,促进短链脂肪酸产生。
注:如果存在低血糖风险、营养不良、消瘦、胃肠疾病、糖尿病用药或进食障碍史,就不宜自行尝试严格禁食。
更温和的方式,可能是避免夜间过晚进食,保持相对规律的进餐时间,而不是追求极端断食。
▾
发酵食品也是值得关注的一类食物。酸奶、泡菜、纳豆、味噌等传统发酵食品,是天然微生物和发酵代谢产物的来源。发酵食品在人类饮食历史中占有重要地位,它们不仅延长了食物保存时间,也可能提高营养价值和生物活性成分。
定期摄入发酵食品,可能有助于增加肠道菌群多样性,补充部分有益微生物,并与更好的免疫调节状态有关。
不过,发酵食品也要注意选择。优先选择低盐、低糖、少添加的产品。尤其是泡菜、咸菜、味噌等高盐发酵食品,不能因为发酵就不限制摄入。
总的来说,饮食是调节肠道菌群和支持认知健康最基础、最安全、也最容易长期坚持的方式。高纤维饮食、地中海饮食、坚果摄入、MIND饮食和适量发酵食品,都是相对更值得优先考虑的方向。需要长期建立一个高纤维、多植物、低炎症、低加工、可持续的饮食模式。
生活方式是影响肠道菌群和大脑健康的另一个重要维度。运动、睡眠和压力管理不仅直接影响大脑功能,也通过肠道菌群发挥作用。
运动:菌群重塑与认知增强
有氧运动可以显著改变肠道菌群组成,增加菌群多样性,提高产短链脂肪酸细菌的丰度。无论是耐力运动还是力量训练,都对肠道菌群产生积极影响。
运动促进肠道蠕动,改变肠道环境的物理和化学特性,为不同菌群创造适宜的生存条件。运动还通过增加血流和调节免疫反应间接影响菌群。这些变化共同促进了菌群向更健康的方向发展。
动物实验显示,运动可以增加Lachnospiraceae、Ruminococcaceae、Bacteroidetes的丰度,这些变化与认知功能改善相关。
在人体研究中,长期坚持运动的人群菌群多样性更高,产丁酸菌丰度显著增加。
推荐每周至少150分钟中等强度有氧运动,配合2-3次力量训练。运动方式应多样化,包括快走、跑步、游泳、骑行等。即使是轻度运动,如每日散步30分钟,也能对菌群产生积极影响。
睡眠:菌群节律与认知巩固
睡眠与肠道菌群之间存在双向调节关系。睡眠质量影响菌群组成和代谢活性,而菌群代谢产物反过来调节睡眠质量和昼夜节律。
睡眠剥夺导致菌群多样性下降,促炎细菌增加,产短链脂肪酸细菌减少。这些变化可能通过炎症途径影响认知功能。反过来,菌群失调产生的代谢信号也可能干扰睡眠调节,形成恶性循环。
肠道菌群具有自身的昼夜节律,这种节律与宿主的睡眠-觉醒周期同步。菌群节律的破坏可能影响褪黑素等睡眠调节激素的合成和释放,进而影响睡眠质量和认知功能。
改善睡眠质量的策略包括:
睡眠质量的改善不仅直接有益于认知功能,也通过菌群途径发挥保护作用。
压力:打破脑-肠恶性循环
慢性压力是肠道菌群和大脑健康的重要威胁。压力通过HPA轴释放皮质醇,改变肠道通透性和菌群组成,而菌群失调反过来加剧压力反应和焦虑症状。
压力-菌群失调-炎症-认知损害
压力导致的菌群特征包括:菌群多样性降低,Lactobacillus和Bifidobacterium等有益菌减少,促炎细菌增加。这些变化增强了肠道的炎症反应,削弱了屏障功能。
编辑
正念冥想、瑜伽、深呼吸训练、渐进性肌肉放松等压力管理技术可以有效降低压力水平,改善菌群状态。这些技术通过调节自主神经系统和HPA轴活动,减轻压力对肠道和大脑的负面影响。
社会支持也是压力管理的重要组成部分。良好的人际关系和社会联系可以缓冲压力的影响,保护菌群多样性和认知功能。孤独和社会隔离则会加剧压力反应,对菌群和大脑产生不利影响。
每个人的肠道菌群并不一样。有些人本身产短链脂肪酸菌较少,可能对高纤维饮食反应更明显;有些人促炎菌群较多,可能需要先降低炎症;还有些人菌群整体状态不错,可能只需要调整生活方式,不一定需要复杂干预。
这也是精准微生态医学出现的原因。核心是根据个体菌群特征、遗传背景、代谢状态、炎症水平和生活方式,制定更适合个人的干预策略。
基线菌群分层:先看属于哪一类
精准干预的第一步,是了解一个人的基线菌群。
所谓基线菌群,就是在干预开始前,个体肠道菌群原本的组成和功能状态。
这一步非常重要。比如说,基线菌群中产短链脂肪酸细菌丰度较低的个体,可能从高膳食纤维饮食中获得最大益处。而Parabacteroides丰度较高的个体,可能更适合噬菌体靶向治疗、GPR84抑制剂,或者其他针对下游炎症通路的方式。
编辑
当然,这些方法目前很多还处于研究阶段,并不是常规临床方案。
基线菌群的炎症潜能也是重要考量因素。如果一个人的菌群整体偏促炎,肠屏障受损明显,LPS等炎症相关信号较高,可能需要优先控制炎症、改善屏障功能,再考虑菌群重建。而如果菌群状态相对稳定,炎症水平不高,可能通过饮食、运动、睡眠和压力管理,就能获得较好的改善。
未来,随着机器学习和微生物组数据积累增加,研究者可能会把人群分成不同菌群亚型,并为不同亚型匹配更可能有效的干预方案。这种思路已经在肿瘤免疫治疗、代谢疾病等领域显示出价值,在认知健康领域也值得期待。
多组学整合的精准诊断
单看菌群组成,往往不足以指导精准干预。因为知道有哪些菌,并不一定等于知道这些菌正在做什么。因此,未来更有价值的方向,是把菌群和多组学数据结合起来。
例如,宏基因组测序可以告诉我们,菌群具备哪些功能潜能:它们可能合成哪些代谢物,具有哪些酶活性,参与哪些信号通路。
代谢组学则可以进一步反映菌群实际产生了什么,以及宿主身体如何响应这些代谢变化。
除此之外,还可以结合血液炎症标志物、神经递质相关指标、激素水平、认知测试结果、脑影像数据和遗传风险评分。
这些信息放在一起,可以更完整地判别一个人的肠-脑轴状态。
例如,一个人可能菌群多样性不算差,但炎症标志物偏高、TMAO水平较高、睡眠差、认知测试表现下降。另一个人可能菌群多样性较低,但炎症不明显,主要问题是膳食纤维摄入不足。
这两类人显然不应该采用完全相同的干预方案。
因此,精准微生态医学未来的方向,是建立一个多维度模型。不过,多组学整合也不是一件简单的事。它需要生物信息学分析、机器学习算法和足够大的数据库支持。随着测序成本降低和分析工具成熟,未来5–10年,多组学整合诊断有望逐步进入更多临床和健康管理场景。
动态监测与适应性调整
肠道菌群不是固定不变的。它会受到饮食、睡眠、压力、药物、感染、运动和年龄变化的影响。因此,单一时间点的检测,很难代表一个人的长期菌群状态。
这也是为什么精准微生态医学强调动态监测。通过定期采样和连续评估,可以观察菌群是否真的对干预产生反应,也可以及时调整方案。
比如,一个人开始补充益生菌后,4周过去了,菌群组成和相关代谢物并没有明显变化。这时就可能需要重新评估:是不是剂量不合适?是否缺少膳食纤维作为底物?是否存在长期睡眠不足或压力过大,抵消了干预效果?
如果只做一次检测,就很难发现这些问题。肠道菌群检测是非侵入性的,操作相对方便,受检者可以在家完成粪便采样,再寄回实验室进行分析。这样是适合后续做阶段性复测,观察菌群变化趋势。
未来,随着居家检测和可穿戴设备发展,动态监测可能会变得更容易,可以帮助人们在家中完成部分监测,降低检测成本和负担。
可穿戴设备还可以记录睡眠、心率变异性、活动量和压力相关指标,与菌群和代谢数据结合后,形成更完整的健康画像。
闭环干预
基于实时监测数据的闭环干预系统是未来的发展方向。该系统可以自动分析监测数据,预测认知变化趋势,推荐个性化的饮食、运动和补充方案,形成”监测-分析-干预-再监测“的闭环管理。
未来,人工智能和机器学习可能会参与这类闭环系统,自动分析监测数据,预测认知风险变化趋势,并给出更个体化的干预建议。当然,这仍然是未来方向,目前还不能替代专业医疗判断。
精准干预更适合用于例如认知下降风险较高、已有轻度认知障碍、存在明显炎症或代谢异常、肠道症状较多,或者普通生活方式调整效果不明显的人。它的意义是减少盲目干预。
注:本账号内容仅供学习和交流,不构成任何形式的医疗建议。
主要参考文献
Chen, Z., Bao, W., Su, J., Wu, X., Tan, Y., & Yang, X. (2026). Research on gut microbiota and metabolic characteristics in patients with sleep disorders after ischemic stroke. Metabolic Brain Disease, 41, 114.
Cox, T. D., Devason, A. S., de Araujo, A., et al. (2026). Intestinal interoceptive dysfunction drives age-associated cognitive decline. Nature, 652, 442-450.
Ma, X., Liu, J., Jiang, L., et al. (2025). Dynamic changes in the gut microbiota play a critical role in age-associated cognitive dysfunction via SCFAs and LPS synthesis metabolic pathways during brain aging. International Journal of Biological Macromolecules, 304, 140945.
Surajit Pathak, Antara Banerjee, et al.(2024). Gut Microbiome and Brain Ageing,Brain Aging, April.
Ni, J., et al. (2025). Nut consumption, gut microbiota and cognitive function in older adults at high cardiovascular risk. Age and Ageing, afaf208.
Shen, H., Wang, S. Y., Zhao, Y. Y., Zhou, J. L., Zhao, J., & Zhu, W. K. (2026). Brain-gut-microbiota axis: a review on the bidirectional regulatory mechanisms between gut microbiota and brain and their disease interactions. Frontiers in Microbiology, 17, 1768891.
谷禾健康
你是否有过这样的经历:明明没有剧烈运动,却总是感到莫名的疲惫;或者明明控制了饮食,体重却依然居高不下,体检报告上的血糖、血脂指标也总是亮起红灯?我们习惯将这些问题归咎于“吃多了”或“动少了”,却往往忽视了一个潜伏在身体内部、肉眼不可见的“隐形杀手”——脂多糖(LPS)。
脂多糖并非外来入侵的毒素,而是我们肠道中数以万亿计细菌的细胞壁成分。在正常情况下,它安分守己地待在肠道内,甚至对免疫系统有益。然而,当肠道屏障受损或饮食结构失衡时,这些本不该进入血液的分子便会“越狱”,引发一场悄无声息却破坏力巨大的全身性慢性炎症。
这种持续存在的低水平炎症,被认为是代谢综合征、2型糖尿病、非酒精性脂肪性肝病以及动脉粥样硬化等多种慢性疾病的重要共同发病基础。脂多糖(LPS)在这一过程中发挥关键作用,可诱导并放大炎症反应,从而促进相关疾病的发生与发展。
那么,脂多糖是如何由肠道内的正常成分转变为影响机体健康的重要因素的?机体又可以通过哪些机制对其进行调控,以维持生理稳态?本文将从机制层面系统分析LPS的作用路径及其对健康的影响。
♥
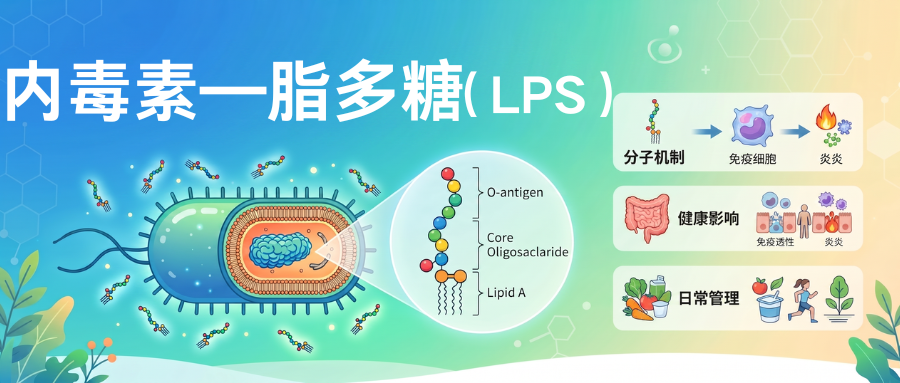
▸ 脂多糖的定义与特性
脂多糖(Lipopolysaccharide,简称LPS)是革兰氏阴性菌细胞壁外膜特有的结构大分子复合物,由三个功能区域共价连接组成的异源多糖-脂质复合物,三个区域分别承担不同生理功能。
革兰氏阴性菌膜由两层细菌膜组成,分别是外层和内层膜,外膜富含脂多糖(LPS)。
脂多糖(LPS)是两性分子,类脂A部分疏水,多糖部分亲水,所以LPS能够在细菌表面形成稳定的双亲结构,维持外膜完整性,同时作为屏障抵抗抗菌物质进入细菌体内。
↘ 脂多糖与内毒素的关系
脂多糖之所以被称为”内毒素”,是因为它是革兰氏阴性菌细胞外膜的重要结构成分,通常不以游离形式主动分泌。在细菌死亡或裂解时会大量释放出来,这一特点与由活菌主动分泌的“外毒素”形成鲜明对比,因此得名“内毒素”。
↘ 内毒素与外毒素的主要区别
编辑
需要注意的是,在感染过程中,细菌不断生长和死亡,会持续释放脂多糖;而在严重感染或抗生素治疗后,大量细菌同步裂解,可能在短时间内释放大量LPS,从而加重炎症反应,严重时可引发内毒素相关的全身炎症反应综合征。
▸ 脂多糖的分子结构
LPS分子从内到外分为三个部分:最内层是类脂A,中间是核心多糖,最外层是O抗原(O特异性多糖),三个部分共价连接形成完整LPS分子。
编辑
doi.org/10.3389/fimmu.2025.1588129
★ 每个部分结构和功能都不同:
•类脂A:嵌入外膜磷脂双层,是LPS的毒性中心和生物活性中心,人体对LPS的免疫识别主要识别类脂A结构;
•核心多糖:连接类脂A和O抗原,结构相对保守,是革兰氏阴性菌共同抗原成分;
•O抗原:最外层,是多糖重复单位,结构变异性最大,决定细菌血清型特异性,是细菌逃避免疫识别的重要方式。
编辑
doi.org/10.5217/IR.2014.12.2.90
这种三层结构设计是革兰氏阴性菌长期进化结果,它既保护细菌本身,也决定了它和人体免疫系统相互作用方式,类脂A是毒性中心,O抗原是免疫逃逸,这种结构对应功能的关系便于更好理解它对健康影响。
★ 脂多糖的结构多样性决定其生物活性差异
不同革兰氏阴性菌的脂多糖(LPS)在结构上存在显著差异,尤其是类脂A部分在酰基链数量、长度以及磷酸化修饰上的变化,会直接影响其免疫激活能力。因此,并非所有革兰氏阴性菌的LPS都具有相同毒性,共生菌来源的LPS通常显著低于典型致病菌。
很多人将“革兰氏阴性菌”与“强内毒素”直接等同,但实际上,不同细菌来源的LPS在毒性上差异很大。理解这种差异,有助于我们更准确地区分共生菌与致病菌的生物学影响。
↘ 类脂A:脂多糖的毒性中心和生物活性中心
类脂A嵌入外膜磷脂双层,是脂多糖的毒性中心和生物活性中心,人体对LPS的免疫识别主要识别类脂A结构。LPS的大部分生物学活性,包括毒性和免疫刺激性,都由类脂A部分决定,类脂A结构变异直接改变LPS毒性强弱,六酰化饱和类脂A毒性最强,酰基链减少毒性显著下降。
编辑
doi.org/10.5217/IR.2014.12.2.90
不同来源的类脂A结构与毒性差异:
•致病菌(如大肠杆菌、沙门氏菌):类脂A多为六酰化,且具有完整的双磷酸结构,与TLR4-MD2亲和力高,能够强烈激活免疫反应。
•肠道共生菌(如拟杆菌属):类脂A通常为五酰化或四酰化,并伴随去磷酸化或其他修饰,与受体结合能力较弱,炎症活性显著降低(在部分实验中可低几个数量级)。
•环境调控型细菌(如耶尔森氏菌):其类脂A结构可随温度变化而调整,在环境温度下多为低酰化形式,而在37℃宿主体内转为高酰化形式,从而增强毒性。
编辑
doi: 10.1128/mSystems.00046-17.
如上图所示,拟杆菌门(Bacteroidetes)是人类肠道微生物群中脂多糖(LPS)生物合成能力的主要贡献者。
↘ 核心多糖:连接类脂A和O抗原的保守结构
核心多糖位于类脂A和O抗原之间,主要作用是连接两个部分,它的结构相对保守,在同一属的不同细菌之间结构大多相似,含有特殊的糖分子庚糖和KDO。
核心多糖可以分为两个部分:
•内核心:靠近类脂A,含有特殊的糖分子:3-脱氧-D-甘露-辛酮糖酸(简称KDO)和L-甘油-D-甘露庚糖,这两种糖在其他地方很少见,是LPS核心多糖特征性成分。内核心结构非常保守,不同革兰氏阴性菌之间变化很小。
•外核心:连接内核心和O抗原,由己糖组成,结构比内核心变异大一些,但仍然比O抗原保守很多,同一属细菌外核心结构相似。
核心多糖带有多个负电荷,能够结合二价阳离子比如镁离子钙离子,这些阳离子中和负电荷,稳定LPS在细菌外膜的结构,如果去掉阳离子,LPS结构将变得不稳定,外膜通透性增加,细菌容易被抗菌物质杀死。
↘ O抗原:决定细菌特异性的高度变异部分
O抗原是LPS分子最外层的多糖重复单位,是LPS中变异性最大的部分,不同血清型革兰氏阴性菌O抗原结构不同,这种高度变异是细菌逃避免疫识别的重要进化策略。O抗原高度变异直接关系到细菌致病性和免疫识别,但是O抗原本身基本没有毒性,主要影响细菌免疫原性和致病性,不直接影响LPS毒性强弱。
O抗原由多个相同的寡糖重复单位聚合而成,每个重复单位通常含有2-6个糖分子,不同细菌重复单位的糖组成、排列顺序、连接方式都可以不同,所以变异非常大,理论上可以产生成千上万种不同O抗原结构。
实例:E. coli O157:H7(常见的致病性肠出血性大肠杆菌)与E. coli O26、O111等其他血清型的区别,本质上就是O抗原多糖重复单位的结构不同。有的重复单位含有不同种类的糖(葡萄糖、半乳糖等),有的糖连接方式不同(α-1,3还是β-1,4),排列顺序也不同。因此针对O157的抗体一般难以有效识别O26等其他血清型,但在个别情况下可能存在有限的交叉反应。
▸ 光滑型脂多糖和粗糙型脂多糖的差异
完整脂多糖(LPS)含有O抗原叫做光滑型LPS,缺失O抗原只剩下类脂A和核心多糖叫做粗糙型LPS,也叫脂寡糖(LOS),两者在稳定性、免疫原性和致病性上都有显著差异。
编辑
编辑
doi.org/10.3390/ijms24098395
LPS(脂多糖)结构示意图:图中亮红色的LPS锚定在革兰氏阴性(G-)细菌的外膜上。根据O抗原和核心区(core)的长度,可将LPS分为光滑型(S)、半粗糙型(SR)、粗糙型(R)和深度粗糙型(DR)。从LPS最外层到最内侧疏水部分,其结构变异性逐渐降低。
♥
▸ 脂多糖对革兰氏阴性菌生存的影响
脂多糖(LPS)不仅仅是致病因子,它对革兰氏阴性菌自身生存具有非常重要的生理功能,维持外膜结构完整性,抵抗抗菌物质入侵,保护细菌生存,虽然部分菌株即使缺失或改变LPS结构仍可存活,但是其生理功能和适应性会显著受损。
脂多糖对革兰氏阴性菌的主要生理功能:
①维持外膜结构完整性:脂多糖是革兰氏阴性菌外膜主要成分,占外膜干重三分之一以上,LPS分子通过二价阳离子交联形成稳定网状结构,维持外膜通透性和结构完整性,如果缺失LPS,外膜结构破坏,细菌存活能力下降。
②形成通透屏障抵抗抗菌物质:LPS外层亲水内层疏水,形成选择性通透屏障,能够阻挡疏水抗菌物质、胆汁盐、补体、溶菌酶进入细菌体内,保护细菌免受宿主防御物质和抗生素攻击,增加细菌耐药性。
③抵抗宿主免疫攻击:O抗原能够保护细菌不被补体激活和吞噬细胞吞噬,帮助细菌逃避免疫清除,增加细菌致病性,有O抗原的菌株更容易引起全身性感染。
④介导细菌粘附:LPS能够介导细菌粘附到宿主黏膜上皮细胞,帮助细菌定植,这是感染第一步,所以LPS对细菌定植也很重要。
因为脂多糖对细菌生存这么重要,所以几乎所有革兰氏阴性菌都必须合成LPS,只有极少数例外,比如某些衣原体可以缺失。
▸ 肠道中的脂多糖与人体屏障系统
据估计,人类结肠中含有约10¹³–10¹⁴个细菌,总生物量约为数百克(通常约0.2–0.5 kg)。这些细菌在生命活动过程中可产生脂多糖(LPS)。研究表明,以大肠杆菌为代表的革兰氏阴性菌,其单个细胞所含LPS约为几飞克至几十飞克(约2–50fg)。基于此进行粗略估算,肠道中LPS的总量大致处于几十毫克至数百毫克的范围,具体取决于菌群组成及生理状态。
然而,静脉注射LPS的致死剂量仅为1–2微克。也就是说,肠道内的LPS总量,可能是致死剂量的千倍甚至百万倍。那么为什么我们不会被”毒死”?答案在于——人体的屏障系统。
↘ 肠道屏障:把”危险”关在门外
人体的上皮组织(比如肠道内壁)在进化过程中,形成了极其高效的防御结构,能够将有害物质隔离在外。
在正常情况下,肠道细菌及其产生的LPS主要存在于肠腔内部,它们被粘液层和上皮细胞牢牢阻挡,不会接触到体内的深层组织。
研究发现,只要这些细菌停留在肠腔一侧,就不会对肠道细胞造成伤害。真正的危险发生在当LPS接触到上皮细胞”背面”(基底侧)时,毒性才会显现。
↘ 肠道屏障破坏的风险因素
当肠道屏障被破坏时,情况就会发生改变:细菌可能穿过粘液层,从细胞之间的缝隙渗透,甚至直接穿过细胞进入更深层组织。一些致病菌本身就具备”入侵能力”,可以主动突破防线。一旦细菌或大量LPS进入血液,免疫系统会迅速响应,但如果负荷过高,可能引发严重后果,比如全身炎症反应、败血症甚至败血性休克。
以下人群风险更高:
-新生儿(免疫系统尚未成熟);
-老年人或重症患者;
-免疫功能受损的人群;
-长期使用导尿管等侵入性器械者;
-肠道存在炎症、损伤或肿瘤的人。
▸ 肠道暴露导致脂多糖转移到循环中
在正常生理条件下,血浆中LPS水平极低。这是因为健康的肠道限制了LPS分子进入血液的途径。尽管持续暴露于LPS,肠细胞和结肠细胞对这些刺激表现出低反应状态,部分原因在于这些细胞中Toll样受体4(TLR4)表达较低。这种肠道耐受机制最大限度地减少了肠腔内的炎症反应。
此外,肠道微生物群中LPS的暴露在免疫系统的发育和成熟中起着关键作用,训练肠道相关淋巴组织(GALT)中的调控T细胞。个体的LPS谱系取决于肠道微生物群的组成,而肠道微生物群的组成因地理和生活方式等因素而有所变化。
▸ 脂多糖在肠道中”常态存在”的生理意义
很多人一听说脂多糖(LPS)有害,就想把肠道里的LPS彻底清除。实际上,这既不现实,也没有必要。相反,维持生理范围内的LPS暴露,对健康是有意义的。
在健康婴儿、成年人和老年人肠道菌群中,革兰氏阴性菌占有一定比例(如拟杆菌门)。这些细菌在更新过程中会不断死亡、裂解,从而释放LPS到肠腔中。因此,肠道内持续存在低水平LPS是一种正常的生理现象,而不是异常状态。生理水平的脂多糖具有以下作用:
•促进免疫系统正常发育:无菌动物由于缺乏包括LPS在内的微生物刺激,免疫系统发育不成熟,免疫应答能力不足,更容易出现过敏和自身免疫问题。适度的LPS等刺激有助于免疫系统建立“耐受”和”分辨能力”,这是”卫生假说”的重要机制基础之一。
•维持先天免疫的基础警戒状态:低水平的微生物信号可以让机体免疫系统保持”准备状态”。如果完全缺乏这类刺激,机体在真正遇到病原体时,可能反应迟缓,反而更容易感染。
•有助于睡眠:2024年发表的一项研究表明当LPS注入门静脉循环时,会引起睡眠增加和体温升高。门静脉循环负责将来自胃、小肠和大肠(这些器官中存在肠道微生物群)的血液输送到肝脏。像LPS这样的微生物分子可以通过“转位”过程进入门静脉,从而进入宿主体内环境。
编辑
事实上,在健康的人类、大鼠和小鼠体内,脂多糖天然存在于门静脉和全身循环血液中,是一种来源于原核生物的生理性血浆成分。
研究进一步暗示日常生活中的一些刺激会进一步促进LPS的转位,从而提高其在血液中的水平。例如,一顿高脂餐(如三片涂有50克黄油的吐司)可使人体血浆LPS水平升高约50%。此外,少量饮酒、急性或慢性睡眠不足以及轻度心理压力等常见情况,也都会增加循环中的LPS水平。
!
关键观点
问题不在”有没有”,而在”是否过量”。真正有害的是脂多糖异常升高并进入循环(如肠屏障受损导致的”内毒素血症”),这会诱导慢性低度炎症,进而与代谢性疾病等相关。因此,我们关注的重点不是”全部消灭LPS”,而是避免其异常升高和大量入血。
♥
▸ 肠道是人体内脂多糖的主要来源
在健康成年人中,肠道是体内LPS(脂多糖)最主要的产生和储存场所。健康成年人肠道内约有10¹³–10¹⁴数量级的微生物,其中约20%–40%为革兰氏阴性菌(以拟杆菌门为主),这些细菌在持续更新过程中不断裂解并释放LPS。
相比之下,口腔(如牙周炎时)虽也可产生LPS但总体规模较小,呼吸道和皮肤等部位菌量更低,对全身LPS贡献有限,但是注意在严重局部感染或全身感染时,这些部位也可能成为重要来源。
▸ 关键分界:是否进入循环
一个非常重要但常被忽略的点是:肠道中LPS很多≠血液中LPS一定高。
↘ 在健康状态下:
•肠道屏障(上皮细胞+紧密连接+黏液层+IgA)有效阻挡LPS进入体内;
•只有极少量LPS可进入循环;
•血浆LPS通常处于低水平(常见报道为pg/ml级别);
•这个水平属于生理暴露,不会引发系统性炎症。
↘ 何时变成问题?当出现以下情况时:
•肠道屏障受损(如”肠漏”);
•肠道菌群失衡(如革兰阴性菌过度增殖);
•高脂饮食促进LPS转运(经乳糜微粒)
就会导致进入循环的LPS增加(而不是肠道LPS本身增加才是关键),这就是所谓的代谢性内毒素血症,其特点是:
•血中LPS轻度但持续升高;
•激活TLR4等炎症通路;
•诱导慢性低度炎症;
•并与胰岛素抵抗、肥胖、非酒精性脂肪肝、动脉粥样硬化相关。
代谢性内毒素血症的经典模型认为,LPS可能进入全身循环并促进慢性炎症。该经典模型指出:
(1) 西式饮食可能增加产生LPS的细菌数量并增加脂肪吸收。
(2) LPS可在局部发挥作用并诱导炎症反应,也可通过刷状缘的移位转运以及在乳糜微粒内进入全身循环。
(3) 主要来源于肝脏的LBP与循环中LPS的脂质A部分结合。
▸ 脂多糖进入血液循环的主要途径
LPS从肠道进入血液循环主要通过两条途径:一是肠道通透性增加(肠漏)导致经细胞间隙进入循环,二是在脂肪吸收过程中结合乳糜微粒,经淋巴系统进入血液。这两条途径共同影响体内LPS的总体水平。
↘ 第一条途径:肠漏(细胞间隙通透增加)
当肠道上皮紧密连接受损,细胞间隙通透性增加,LPS可以从肠腔进入黏膜下组织,并进一步进入门静脉循环。这一过程不依赖脂肪摄入,而主要取决于肠屏障完整性。长期通透性升高,是循环LPS持续升高的重要原因之一。
↘ 第二条途径:乳糜微粒转运
LPS具有一定亲脂性。在摄入脂肪后,肠道形成乳糜微粒以吸收和运输脂类物质,部分LPS可结合在乳糜微粒上,随其进入淋巴系统,最终进入体循环。这一过程与脂肪摄入量相关:脂肪摄入越多,乳糜微粒生成越多,LPS转运量也可能相应增加。这也是高脂餐后出现”餐后内毒素血症“的一个重要机制。
这两条途径通常同时存在:肠道通透性决定基础LPS水平,而餐后乳糜微粒可造成短时升高,两者叠加,可能导致整体水平上移。
★ 短时过量脂肪摄入可能导致脂多糖过多的危害
当长期高脂摄入、频繁产生大量乳糜微粒,或肝脏清除能力下降时,可能出现LPS清除不及、循环水平升高的情况,从而与慢性低度炎症相关。
因此,并不需要避免脂肪摄入本身,而是应关注总量与分配:避免单次摄入过多脂肪,有助于降低一次性LPS负荷。只要控制每餐脂肪量,不要一次摄入太多脂肪,减少单次转运LPS量,就不会对健康造成危害,完全禁止脂肪摄入也不利于健康,关键是量和频率。
这个也解释了为什么中国人说“饱食伤身”,一次吃太多脂肪不仅热量过剩,还带来大量LPS进入循环,导致一次炎症峰,反复多次就是慢性炎症,所以吃七八分饱不仅减少热量,也减少LPS,对健康更有利。
♥
▸ 脂多糖的半衰期与清除动力学
LPS在人体内的半衰期取决于其浓度和人体解毒能力。生理低剂量LPS进入循环后,半衰期大约是几分钟到十几分钟,能够被肝脏快速清除,但是高剂量LPS超过肝脏清除能力,半衰期显著延长,能够在循环中停留几个小时,持续激活炎症。
了解半衰期能够帮助我们理解为什么低剂量持续暴露比一次性高剂量更有害,为什么慢性持续LPS升高导致慢性病。
▸ 肝脏:清除循环LPS的主要器官
进入门静脉的脂多糖约有80–90%在肝脏被清除,主要由Kupffer细胞介导,肝脏因此成为清除循环LPS的关键器官。
正常肝功能有助于维持低水平LPS,而功能受损时清除能力下降,易导致其升高。除直接吞噬外,LPS还可与LDL、HDL等血浆脂蛋白结合,并被肝细胞和Kupffer细胞摄取,从而进一步促进清除。
编辑
↘ 肝脏功能能够影响脂多糖的清除能力
因此,肝脏构成防止LPS进入体循环的重要屏障。肝功能正常,清除能力强,能够快速清除LPS,肝功能下降,清除能力下降,相同剂量LPS半衰期延长,炎症更明显,所以肝功能不好的人更容易发生内毒素血症。
编辑
▸ 肠道碱性磷酸酶对LPS的解毒作用
肠道上皮分泌的碱性磷酸酶(intestinal alkaline phosphatase, IAP)可通过去除LPS脂质A上的磷酸基团,将其转化为低毒形式,显著降低其激活TLR4-MD2受体的能力,从而减弱炎症反应。
↘ 碱性磷酸酶能够减弱脂多糖导致的炎症反应
这一过程发生于LPS进入体内之前,是肠道局部解毒的重要机制和防止系统性炎症的第一道屏障。IAP由小肠上皮刷状缘表达,直接与肠腔内LPS接触;其去磷酸化作用可使LPS炎症活性降低数十至数百倍。
动物研究表明,缺失IAP会增加对LPS的敏感性并加重炎症反应。IAP为含锌酶,锌对其结构稳定和催化活性至关重要,缺锌可降低其功能并增加活性LPS负荷。部分研究还提示,高脂饮食可能抑制IAP表达或活性,而低脂或特定膳食模式有助于维持其功能,但相关结论尚不一致。
▸ 低密度脂蛋白与脂多糖的相互作用
脂多糖(LPS)进入循环后,一部分游离存在,另一部分与低密度脂蛋白(LDL)结合。结合后,LPS的疏水结构被包埋于LDL颗粒内,减少与免疫细胞表面TLR4受体的接触,从而降低其炎症活性,这是一种机体的保护性机制。
↘ LDL与LPS的结合具有双重效应
然而,携带LPS的LDL更易被巨噬细胞通过清道夫受体摄取,促使其转化为富含胆固醇的泡沫细胞,进而在血管壁沉积形成脂纹,推动动脉粥样硬化的发生与发展。
因此,LDL与LPS的结合具有双重效应:既可降低游离LPS的炎症活性,又促进泡沫细胞形成和动脉粥样硬化进展。当LPS与LDL同时升高时,心血管风险显著增加,且两者具有协同放大效应。循环LPS水平升高会增强LDL的致动脉粥样硬化作用,这是其促进心血管疾病的重要机制之一。
▸ LPS清除的多器官协作(肝-脾-肾)
除肝脏外,脾脏和肾脏亦参与循环LPS的清除与代谢:肝脏为主要清除器官,脾脏提供辅助清除,肾脏负责排泄降解产物。整体上,网状内皮系统(单核-巨噬细胞系统)协同维持LPS稳态。
↘ 肝脏主要清除、脾脏辅助清除
进入循环的LPS大多由肝脏Kupffer细胞清除,其余部分可被脾脏巨噬细胞摄取。肾脏对完整LPS的直接清除有限,主要排泄其降解后的小分子片段,参与代谢末端处理。
LPS清除依赖单核-巨噬细胞系统的吞噬功能;当疾病、炎症或药物导致该系统功能下降时,清除效率降低、半衰期延长,循环水平升高并加重炎症反应。
总体而言,LPS清除体现多器官协同的分层防御:肝脏为核心屏障,脾脏补充清除,肾脏负责排泄。该体系功能正常时可有效控制LPS水平;若输入增加(如肠屏障受损或饮食因素)且清除能力下降(如肝功能或免疫功能减弱)并存,则可能突破防线,导致慢性低水平内毒素血症。
♥
▸ 肠道菌群失调
当肠道菌群失调时,菌群结构和代谢功能发生改变,可能导致脂多糖负荷及其炎症活性升高。肠腔中LPS水平上升,同时在饮食和屏障因素作用下进入循环的LPS增加。因此,通过改善菌群结构与代谢环境,可以从源头降低高炎症活性的LPS负荷。
健康成年人肠道中本就富含革兰氏阴性菌(如拟杆菌门)。但菌群失调(如高脂、低纤维饮食)会改变其结构,使部分菌群(尤其肠杆菌科)增加,同时短链脂肪酸等代谢产物减少,从而共同影响LPS相关效应。
不同来源的LPS免疫活性差异显著:拟杆菌来源LPS炎症活性较低,而大肠杆菌来源则更易诱导炎症。因此,关键不在于阴性菌数量多少,而在于高炎症活性LPS的比例是否上升。
↘ 在吸收方面,LPS进入循环主要通过:
•与脂质结合,经乳糜微粒转运(高脂饮食关键机制);
•肠屏障功能改变(紧密连接受损)。
因此,高脂低纤维饮食的作用是双重的:一方面改变菌群结构,另一方面显著促进LPS进入血液循环,共同提高循环LPS水平。
↘ 膳食纤维则通过不同路径发挥作用:
•促进产短链脂肪酸菌生长(如产丁酸菌);
•增强肠道屏障功能;
•调节菌群整体生态。
从而降低脂多糖相关炎症负荷。
这一机制把”菌群失调”和”慢性炎症”连接起来,但更准确的路径应理解为:
菌群失调 → LPS结构与负荷改变+代谢环境改变 → LPS更容易进入循环 → 诱导低度慢性炎症 → 胰岛素抵抗、脂肪肝、动脉硬化等问题 → 参与多种慢性疾病发生发展。
这里的关键,不是单一指标(比如阴性菌比例),而是菌群整体生态与宿主相互作用。所以,调节肠道菌群的核心,不是简单”降低革兰氏阴性菌”,而是恢复菌群结构与功能的平衡,从而降低高炎症活性的LPS负荷。
▸ 小肠细菌过度生长
正常情况下,小肠受胃酸和蠕动影响,细菌浓度远低于结肠(约10¹¹–10¹² CFU/ml的万分之一),因此LPS水平较低,即使全部吸收,总量也有限。
↘ 小肠细菌过度生长使脂多糖水平显著升高
发生小肠细菌过度生长(SIBO)时,小肠细菌增至10⁵–10⁸ CFU/ml,且多为革兰氏阴性菌,使腔内LPS浓度升高数十至上百倍。由于小肠吸收面积大、通透性高,更多LPS进入门静脉循环,导致循环水平升高。同时,SIBO还可损伤黏膜、破坏紧密连接,进一步增强LPS吸收,形成叠加效应。因此,不明原因的慢性LPS升高应考虑SIBO。
LPS的影响不仅取决于“数量”,还取决于“产生部位”:结肠产量高但吸收受限,小肠产量较低却更易入血。研究显示,SIBO患者循环LPS显著升高,根除后可下降并伴随症状改善,提示其为重要来源之一。这也解释了SIBO与多种肠外疾病的关联,即通过增加LPS吸收诱发全身慢性炎症。
▸ 牙周炎:被低估的”持续性LPS输入源”
牙周炎中,牙周袋内聚集大量革兰氏阴性厌氧菌(如牙龈卟啉单胞菌),持续释放脂多糖。在局部炎症和血管通透性增加的情况下,这些LPS可以进入血液循环,成为全身LPS负荷的长期来源之一。
很多人把牙周炎当作”口腔问题”,但忽略了一点:牙龈是一个高度血管化、长期暴露炎症的组织,这意味着,口腔里的炎症信号,是可以”进入全身”的。
↘ 牙周炎的核心环境是牙周袋:
•厌氧环境;
•大量革兰氏阴性菌;
•持续细菌更新与裂解释放脂多糖。
▸ 脂多糖与肠道屏障损伤的正反馈循环
脂多糖不仅可通过肠道进入循环,还可反过来破坏肠道屏障结构,增加通透性(”肠漏”),从而进一步促进LPS吸收,形成正反馈循环。
研究表明脂多糖可激活肠上皮细胞中的NF-κB通路,诱导炎症因子释放。这些炎症信号可导致紧密连接蛋白(如occludin、ZO-1)发生内吞和降解,从而破坏紧密连接结构。
结果是肠道通透性增加,使更多LPS从肠腔进入门静脉及全身循环。升高的LPS又进一步加重屏障损伤,形成循环:
LPS升高 → 屏障受损 → 通透性增加 → LPS吸收增加 → LPS进一步升高。
这一循环可持续放大低度慢性炎症状态。这表明一旦肠道屏障受损,单一干预往往难以逆转进程;同时降低LPS来源与修复屏障结构,可能更有助于打破这一循环。
▸ 其他升高脂多糖水平的风险因素
↘ 时间维度:停留越久→吸收越多(便秘)
便秘本质是”肠道内容物滞留”。停留时间延长→细菌裂解释放更多LPS + 与肠壁接触更久→吸收增加。同时伴随菌群失衡→进一步增加LPS来源。
结果:脂多糖”暴露时间+浓度”双升高。
↘ 屏障维度:肠道越”漏”→进入越多(压力/睡眠不足)
慢性压力通过皮质醇破坏紧密连接→肠道通透性增加。原本被阻挡的LPS更容易进入血液。同时压力还会改变菌群→增加LPS产生。
结果:LPS”通行能力”增强。
↘ 储存维度:储得越多→释放越久(肥胖)
肥胖不是单纯”多脂肪”,而是一个LPS放大系统:脂肪组织储存LPS并持续释放;菌群失调→产生更多LPS;肠道屏障变差→吸收更多LPS。
编辑
结果:LPS”长期维持在高水平”。
↘ 生活方式和饮食因素
多种生活方式和饮食因素通过”增加LPS产生+增加LPS吸收“两条路径,提高体内LPS水平:
•高脂饮食:一方面促进乳糜微粒把LPS带入血液,另一方面改变菌群增加革兰阴性菌→LPS显著升高。
•过量饮酒:破坏肠道屏障(肠漏)+ 改变菌群→更多LPS进入血液,加重肝脏炎症。
•吸烟:影响口腔和肠道菌群+ 破坏屏障→多来源增加LPS。
•膳食纤维不足:有益菌减少、短链脂肪酸下降→菌群失衡+ 屏障变差→LPS升高。
•长期抗生素:杀死有益菌、促使阴性菌占优势+ 损伤屏障→LPS增加。
♥
▸ LPS与TLR4的识别与信号传导
LPS通过与细胞膜上的Toll样受体4(TLR4)复合物结合,激活先天免疫细胞,其生物学效应由此启动。作为典型的病原体相关分子模式(PAMP),LPS被TLR4这一模式识别受体特异性识别。
↘ 脂多糖的识别过程需要多种蛋白质协同作用:
•LPS结合蛋白(LBP):首先结合循环中的游离LPS,从聚集体中提取LPS分子,并将其运输至CD14;
•CD14:将LPS转移至TLR4-MD2复合物;
•TLR4-MD2复合物:MD2蛋白结合LPS的脂肪酸A部分。结合后,MD2发生构象变化,导致TLR4二聚化并启动下游信号转导。
▸ TLR4多态性影响LPS敏感性与疾病风险
TLR4基因多态性(如Asp299Gly、Thr399Ile)可改变受体结构,降低其与LPS的结合亲和力和敏感性,从而减弱炎症反应,并影响慢性疾病的易感性。
因此,在相同脂多糖水平下,不同个体的炎症程度和疾病风险可存在显著差异。携带这些变异的个体炎症反应较低,动脉粥样硬化风险可能下降,但对革兰氏阴性菌感染的易感性及严重脓毒症风险增加,体现出权衡效应。相反,无变异者对LPS更敏感,更易发生强烈炎症反应及相关慢性疾病,需要更积极地控制LPS水平。
除上述多态性外,其他遗传变异亦参与调控LPS敏感性,共同决定个体差异。这也反映了遗传与环境的相互作用:环境决定暴露水平,遗传决定反应强度,两者共同影响疾病风险。
编辑
doi.org/10.3390/ijms24098395
▸ 脂多糖的免疫识别与炎症激活
↘ 识别与递呈(胞外阶段)
LPS在循环中先由脂多糖结合蛋白(LBP)结合并单体化,随后转移至CD14(mCD14或sCD14),再递交给TLR4-MD2复合物。MD2结合脂质A后诱导TLR4二聚化,启动信号转导。LBP和CD14提高了系统对低浓度LPS的敏感性。
↘ 两条主要信号通路(TLR4下游)
MyD88依赖通路(主要炎症通路):通过IRAK–TRAF6–TAK1–IKK级联,激活NF-κB,诱导TNF-α、IL-1β、IL-6等促炎细胞因子产生,是LPS炎症反应的核心机制。
TRIF依赖通路:在内体中激活TBK1–IRF3,诱导I型干扰素(IFN-α/β)产生,并促进共刺激分子表达,连接先天免疫与适应性免疫。
↘ 细胞内识别与炎症放大
NOD样受体(Nod1/Nod2)识别细菌肽聚糖成分(不是直接识别LPS),通过RIP2激活NF-κB,与TLR4信号协同放大炎症反应。
↘ 炎症小体激活(第二放大机制)
LPS作为”第一信号”诱导NLRP3和pro-IL-1β表达;ATP等作为”第二信号”激活炎症小体,caspase-1剪切成熟IL-1β,进一步放大炎症级联反应。
↘ 核心转录调控节点
NF-κB是关键汇聚点,几乎所有LPS诱导的促炎基因均依赖其激活,是慢性炎症持续存在的核心分子基础。
↘ 主要效应分子
TNF-α、IL-1β、IL-6共同介导LPS的炎症效应;CRP为IL-6驱动的临床替代指标,常用于炎症监测。
↘ 剂量决定结局
低剂量长期暴露:持续激活NF-κB → 慢性低级别炎症 → 代谢性疾病等。
高剂量急性暴露:细胞因子风暴 → SIRS/脓毒性休克。
♥
▸ 脂多糖与动脉粥样硬化
通常认为胆固醇是动脉粥样硬化的主要原因,但往往忽视了炎症因素,尤其是LPS对内皮细胞的直接激活作用。
血管内皮细胞表达TLR4和CD14。LPS与TLR4结合后激活NF-κB信号通路,诱导细胞间黏附分子-1(ICAM-1)和血管细胞黏附分子-1(VCAM-1)的表达。这些黏附分子为循环中的单核细胞和淋巴细胞提供黏附位点。
↘ 脂多糖作用于内皮细胞促进动脉粥样硬化
黏附的白细胞随后穿越内皮进入血管壁,单核细胞分化为巨噬细胞,摄取氧化低密度脂蛋白(oxLDL),形成泡沫细胞。泡沫细胞的聚集构成脂肪纹,这是动脉粥样硬化的早期病变。
此外,LPS还可诱导内皮细胞产生促炎细胞因子(如TNF-α、IL-6),进一步促进炎症反应、白细胞浸润及血管平滑肌细胞增殖,加速病变进展。
同时,LPS可通过增加活性氧生成、减少一氧化氮(NO)生物利用度,引发内皮功能障碍,表现为血管舒张能力下降及促凝状态增强。
因此,持续升高的LPS水平可通过直接作用于内皮细胞,促进动脉粥样硬化的发生和发展。
▸ 脂多糖与2型糖尿病及胰岛素抵抗
长期升高的循环LPS可激活脂肪组织、肝脏等处的巨噬细胞,促使其分泌TNF-α、IL-6等炎症因子。这些因子通过激活丝氨酸/苏氨酸激酶,促进IRS-1发生丝氨酸磷酸化,从而抑制其正常的酪氨酸磷酸化,阻断胰岛素信号传导,导致胰岛素抵抗。
↘ 降低脂多糖可改善胰岛素敏感性并降低血糖
在此状态下,β细胞需代偿性分泌更多胰岛素以维持血糖,但长期负担会导致功能衰竭,最终发展为2型糖尿病。研究表明,2型糖尿病患者循环LPS水平显著升高,而降低LPS可改善胰岛素敏感性并降低血糖,支持其在发病中的关键作用。
因此,糖尿病管理不应仅关注血糖,还应调节肠道菌群,从源头降低LPS水平。该机制也解释了部分体重正常者患病的原因:即使无肥胖,若存在菌群失调和LPS升高,仍可诱发炎症和胰岛素抵抗。对肠道菌群及LPS风险的评估,有助于实现更早期干预与预防。
▸ 脂多糖与非酒精性脂肪性肝病
肠道来源的脂多糖经门静脉直接进入肝脏。肝脏中的库普弗细胞高表达Toll样受体4(TLR4),可识别LPS并激活NF-κB信号通路,进而释放TNF-α、IL-1β等促炎细胞因子。
这些炎症因子一方面抑制肝脏胰岛素信号通路,降低脂肪酸氧化能力,促进甘油三酯在肝细胞内沉积,从而加重脂肪变性;另一方面可激活肝星状细胞,促进胶原合成和细胞外基质沉积,推动肝纤维化进展。
随着炎症持续存在,疾病可由单纯性脂肪肝逐步发展为非酒精性脂肪性肝炎(NASH),甚至进一步进展为肝纤维化和肝硬化。
↘ NAFLD患者存在脂多糖水平显著升高
研究显示,非酒精性脂肪性肝病(NAFLD)患者的门静脉及循环LPS水平显著升高;而降低LPS水平可减轻肝脏炎症反应,改善脂肪变性及纤维化程度,进一步支持LPS在NAFLD中的关键作用。
这一机制同样解释了为什么部分不超重、无饮酒史的人群仍会发生NAFLD:即使没有传统代谢风险因素,肠道菌群失调导致的LPS升高,依然可以驱动肝脏炎症和疾病进展。
▸ 脂多糖与高血压
↘ 脂多糖通过减少NO生成提高血管张力并升高血压
脂多糖可激活血管内皮细胞NF-κB通路,诱导促炎因子释放,并抑制内皮型一氧化氮合酶(eNOS)表达与活性,减少NO生成。NO减少会削弱血管舒张、增强收缩,进而提高血管张力并升高血压。
同时,LPS可损伤内皮结构、增加血管壁僵硬度和外周阻力;长期炎症还促进血管重构,使高血压逐渐固定化。动物研究表明,慢性低剂量LPS可升高血压,而降低LPS水平有助于改善血压,支持其在血压调控中的作用。
▸ 脂多糖与肥胖的相互促进
肥胖可促进脂肪组织炎症和脂多糖蓄积,而LPS介导的炎症又进一步干扰能量代谢、促进脂肪储存,二者形成相互促进的正向循环。
编辑
↘ 脂多糖干扰能量代谢、肥胖又促进脂多糖积累
此外,LPS还可能通过影响下丘脑能量调节,干扰食欲和能量摄入,进一步推动体重增加。
研究表明,减重可降低脂肪组织炎症和LPS负荷,改善胰岛素敏感性,从而有助于打破这一循环。
这一循环提示,单纯依赖热量限制可能不足以优化代谢状态;若同时改善肠道菌群、降低LPS水平,可能更有助于提高减重效果和代谢改善程度。
▸ 脂多糖与神经系统疾病
循环LPS能够通过多种途径穿过血脑屏障进入脑实质,激活小胶质细胞产生炎症,所以外周LPS升高能够影响中枢神经系统功能,参与神经精神疾病发生发展。
LPS分子量大约10-20 kDa,属于大分子,但是研究证实,循环LPS能够穿过血脑屏障进入脑内,主要通过几个途径:
•血脑屏障上有特定转运系统:能够转运LPS进入脑内,低浓度LPS就能够转运;
•炎症能够破坏血脑屏障完整性:LPS本身引发外周炎症,炎症介质破坏血脑屏障,更多LPS能够进入脑内,形成恶性循环;
•绕过血脑屏障:LPS能够通过血脑屏障薄弱部位比如延髓最后区进入脑内,然后扩散到其他脑区。
↘ 脂多糖入脑激活小胶质细胞致炎
进入脑内的脂多糖能够激活小胶质细胞,小胶质细胞是脑内巨噬细胞,表达TLR4,激活后产生促炎细胞因子,引发神经炎症,损伤神经元,所以外周LPS升高能够增加中枢神经炎症,参与抑郁症、帕金森病、阿尔茨海默病发生发展。
LPS能够穿过血脑屏障进入中枢这个事实,把外周肠道LPS和中枢神经疾病连接起来了,肠道菌群失调→LPS升高→进入中枢→激活小胶质细胞→神经炎症→神经精神疾病,这个完整通路现在已经被越来越多研究证实,这就是”肠脑轴”重要分子机制,所以改善肠道菌群降低LPS,能够帮助预防和改善神经精神疾病。
这一机制也解释了为什么肠道菌群失调能够影响情绪和认知,脂多糖进入中枢激活炎症就是重要介导。
编辑
doi.org/10.1021/acschemneuro.8b00733
♥
1
质谱检测
脂质A中的脂肪酸为3-羟基脂肪酸(3OH-FAs),其链长和酰化模式具有多样性,并可能影响LPS的免疫原性。
由于酯化的3OH-FAs仅来源于脂质A,因此其水平可作为循环中LPS的替代指标。早期采用GC/MS对其进行分离和定量;近年来发展出HPLC/MS/MS方法,通过计算总3OH-FAs与游离3OH-FAs的差值来定量酯化部分,从而实现更高灵敏度和准确性的LPS检测。
2
血浆脂多糖定量
血浆LPS定量检测是评估内毒素血症的重要方法之一,常用鲎试验(LAL)可用于检测血浆中LPS水平,反映循环内毒素负荷。
LAL试验基于鲎变形细胞溶解物对LPS的反应,可通过比浊或显色方法进行定量,检测灵敏度可达皮克级别。
↘ 但需要注意:
血浆中存在多种干扰因素(如脂蛋白、蛋白结合等),可能影响检测结果,因此不同实验条件下数值存在一定变异。
不同研究中报告的参考范围有所差异,临床上更常用于相对变化和趋势评估,而非绝对诊断标准。血浆LPS检测可用于研究或特定情况下评估内毒素水平变化,但在实际应用中通常需要结合其他指标综合判断。
3
炎症标志物间接反映
脂多糖可激活免疫系统,诱导IL-6等炎症因子释放,进而促进肝脏合成CRP,因此CRP和IL-6可以反映炎症反应水平。但需要强调的是CRP和IL-6是非特异性炎症指标,会受到感染、损伤、慢性疾病等多种因素影响。
在排除急性感染等因素的前提下,CRP(尤其hs-CRP)可以作为低度慢性炎症的实用监测指标,用于跟踪干预效果,但不能单独用于判断LPS水平。
4
肠道菌群检测推断LPS产生潜力
通过16S rRNA或宏基因组测序分析肠道菌群组成,可以从菌群结构角度推断LPS产生潜力,用于评估风险趋势。
LPS来源于革兰氏阴性菌,因此其相对丰度与LPS产生潜力相关,但这种关系是间接的、受多因素影响。
例如:
拟杆菌门虽为革兰阴性,但其LPS炎症活性差异较大,并非全部强致炎;
菌群功能、宿主状态、屏障功能等都会影响最终LPS水平;
菌群检测更适合用于风险评估和干预方向指导,而不是直接判断LPS实际水平。
5
谷禾健康肠道菌群检测评估LPS相关风险
谷禾报告对很多菌群多指标整合分析,可以更系统地评估LPS相关风险趋势,用于指导个体化干预。我们方法的价值在于从菌群结构和功能角度提供参考信息,但仍需结合临床指标(如炎症指标、代谢指标)综合判断。
↘ 高LPS风险的菌群特征
谷禾报告中高LPS风险通常表现为革兰氏阴性菌相对丰度偏高,伴随促炎菌增加、短链脂肪酸产生菌减少及综合评分升高,提示较高的LPS产生与炎症潜力。
典型特征包括:
•革兰氏阴性菌相对丰度升高→LPS来源增加(潜力增加);
•B/F比值升高→菌群结构向阴性菌倾斜;
•促炎革兰氏阴性菌增加→LPS炎症活性增强;
•短链脂肪酸产生菌减少→抗炎能力下降+屏障支持减弱;
•综合风险评分偏高→多因素叠加的结果。
这些特征反映的是LPS相关风险升高的趋势,提示需要从菌群和生活方式层面进行干预。
↘ 低LPS风险的菌群特征
低LPS风险通常表现为菌群结构相对平衡、短链脂肪酸产生能力良好、综合评分较低,提示LPS产生和炎症风险较低。
主要特征包括:
•革兰氏阴性菌比例在合理范围→LPS来源稳定;
•B/F比值平衡→菌群结构稳定;
•促炎致病菌未见明显富集→炎症驱动较低;
•短链脂肪酸产生菌充足→有助于抗炎和维持屏障;
•综合评分低→整体风险较低
低风险代表当前状态良好,但仍需维持健康饮食和生活方式以保持稳定。
6
结合肠道通透性综合评估LPS
LPS风险评估需要同时考虑“产生”和”吸收”两方面,即菌群结构与肠道通透性共同决定循环LPS水平。
关键理解三种情况:
•屏障受损(肠漏)时→即使LPS产生不高,也可能吸收增加;
•屏障正常时→即使LPS产生略高,进入血液的量可能有限;
•两者同时异常→LPS升高风险最大。
综合评估可以区分问题来源(“产多了”还是”漏多了”),从而更有针对性地干预,例如侧重调菌或修复屏障。
♥
1
修复肠道屏障—减少LPS吸收的根本措施
无论LPS升高的原因如何,修复肠道屏障是降低其进入循环的根本措施。完整的肠道屏障通过维持上皮细胞间的紧密连接,阻止LPS经细胞间隙进入血液;当屏障受损时,紧密连接蛋白减少、通透性增加,LPS更易入血。因此,恢复紧密连接完整性可有效减少LPS吸收并降低其循环水平。
①补充乳铁蛋白
乳铁蛋白是一种铁结合糖蛋白,可促进肠上皮细胞增殖与更新,维持紧密连接蛋白表达和屏障完整性,从而降低肠道通透性,减少LPS进入循环并降低其水平。
此外,乳铁蛋白具有特异性LPS结合结构域,能高亲和力结合并中和LPS,阻断其与免疫细胞膜TLR4受体结合,抑制下游炎症信号通路,减少促炎因子产生,从而直接抑制炎症反应。
②补充谷氨酰胺
肠上皮细胞更新迅速,需充足谷氨酰胺供能;其充足时可维持上皮更新和紧密连接蛋白合成,保持屏障完整与正常通透性,减少LPS经细胞间隙入血。
肠漏或屏障受损时,补充谷氨酰胺可促进上皮修复、增强紧密连接、降低通透性,从而减少LPS吸收,对此类人群尤为有益。
2
其他减少LPS吸收与转运的措施
①维生素D
可以上调紧密连接蛋白,降低肠道通透性,减少LPS跨上皮转运。
②改善便秘
缩短肠道转运时间,减少LPS在肠腔内累积及与肠壁接触时间,从而降低吸收机会。
③减轻压力+改善睡眠
通过降低HPA轴激活和皮质醇水平,改善紧密连接与肠屏障功能,减少LPS进入循环。
3
减少其他LPS产生来源
①治疗SIBO
小肠细菌过度生长增加潜在LPS来源,在肠屏障受损时更易转化为循环LPS,抗生素等针对性治疗可降低细菌负荷,从源头减少LPS产生。
②牙周治疗
牙周感染中革兰阴性菌持续释放LPS,可通过受损牙龈进入循环,系统治疗减少这一持续输入源。
4
控制每餐脂肪摄入
我们之前介绍过,吃完脂肪后,肠道合成乳糜微粒运输脂肪,LPS亲脂,结合到乳糜微粒上,随乳糜微粒进入淋巴循环,然后进入体循环,因此每餐脂肪越多,合成的乳糜微粒越多,进入循环的LPS越多,所以控制每餐脂肪量减少乳糜微粒,进而减少进入循环的LPS,降低循环LPS水平。
简单饮食调整:一餐不要吃太多脂肪,全天均匀分布脂肪,可以减少餐后LPS升高,减少累积炎症。
5
增加膳食纤维,黄酮、多酚等摄入
①芹菜素(apigenin)
芹菜素是一种广泛存在于温热带蔬果中的黄酮类化合物,在芹菜、洋甘菊和欧芹中含量较高,具有肠道靶向的抗炎作用。
其可调节肠道菌群、强化肠道屏障,从而减少LPS吸收;在高脂饮食诱导的肥胖模型中,补充芹菜素可显著降低全身LPS水平及相关低度炎症。
②菊粉(Inulin)
益生元纤维如菊粉可促进抑制LPS的共生菌生长,并增加短链脂肪酸(SCFAs)生成,从而修复肠黏膜。
在一项针对2型糖尿病女性的随机对照试验中,补充菊粉8周使血浆LPS较安慰剂下降27.9%,同时炎症因子和胰岛素抵抗显著改善。
这些效应主要归因于其对肠道菌群的调节和对紧密连接蛋白的增强,从而抑制LPS易位,表明膳食纤维是降低内毒素血症和炎症的可行策略。
③利福昔明
利福昔明是一种肠道特异性抗生素,针对革兰氏阳性和革兰氏阴性菌,且不进行全身吸收。通过重塑肠道微生物群,利福昔明可以降低内源性内毒素的产生。
在非酒精性脂肪性肝炎(NASH)患者中,28天的利福昔明治疗显著降低了血清内毒素水平(内毒素活性下降,P=0.03),同时肝酶和炎症标志物也有所改善。
④姜黄素
姜黄素可通过强化肠道屏障并解毒内毒素,减少LPS介导的炎症。在饮食诱导的代谢综合征啮齿动物模型中,姜黄素提高肠道碱性磷酸酶活性(去磷酸化并中和LPS),维持紧密连接结构,从而防止血浆LPS升高。
在高脂饮食的糖尿病大鼠中,姜黄素可使升高的血清LPS和TNF-α恢复至正常水平。这些作用与其重塑肠道菌群(增加有益菌)及直接抑制LPS诱导的肠道信号通路有关。初步人体研究亦支持其作为辅助干预,用于降低代谢疾病中的内毒素血症和炎症。
⑤膳食纤维的综合作用
膳食纤维不被人体消化,进入结肠后作为双歧杆菌、乳杆菌等革兰阳性有益菌的发酵底物,促进其增殖并相对降低革兰阴性菌比例,从而减少LPS生成。
有益菌发酵产生的短链脂肪酸具有抗炎作用并增强肠道屏障,降低LPS吸收,形成双重降LPS效应。其中丁酸盐既是能量来源,又可强化屏障功能,是膳食纤维降低LPS的重要机制之一。
6
调节肠道菌群,选择合适的益生菌
①从源头调整肠道菌群减少脂多糖产生
LPS由肠道革兰氏阴性菌产生;其比例升高会增加LPS生成。通过饮食调节促进革兰氏阳性有益菌生长、降低阴性菌比例,可从源头减少LPS产生并降低其循环水平,这种改变具有长期稳定性。
益生菌和益生元均有助于调节菌群,因此饮食干预是更为根本且可持续的策略。相比单纯依赖抗炎药物,从源头控制菌群更能长期维持LPS在正常水平,促进整体健康。
②施用以革兰氏阳性菌为主的益生菌
选择的益生菌应该以双歧杆菌、乳杆菌等革兰氏阳性菌为主,这些本身没有LPS细胞壁,不会给肠道额外增加LPS,同时抑制革兰氏阴性菌过度生长,减少总LPS产生。
避免选择含有大量革兰氏阴性菌的益生菌产品,这些会给肠道额外增加LPS,增加LPS负担,反而达不到降低LPS的效果。
7
抑制LPS诱导的炎症反应
①Omega-3脂肪酸
改变细胞膜结构与脂筏环境,影响TLR4信号复合体激活,并通过resolvins等介质促进炎症消退,从而减轻LPS炎症反应。
②小剂量阿司匹林
通过COX抑制及NF-κB通路调节发挥整体抗炎作用,同时抑制血小板聚集,降低LPS相关心血管风险(主要为系统性抗炎效应的一部分)。
8
减少脂肪组织LPS蓄积
LPS亲脂容易蓄积在脂肪组织中,脂肪组织质量越大,蓄积LPS总量越多,脂肪组织不断缓慢释放LPS进入循环,维持循环LPS持续低水平升高,减肥减少脂肪组织质量,LPS蓄积总量减少,释放进入循环的LPS减少,循环LPS水平下降。
减肥同时改善肠道菌群,减少革兰氏阴性菌比例,进一步减少LPS产生,多重机制共同降低循环LPS水平,因此肥胖症减少体重可以显著降低LPS水平。
9
不同LPS风险分层个体化干预策略
不同LPS风险程度需要不同强度干预策略,低风险维持健康生活方式,中风险饮食调整加针对性营养补充,高风险先排查继发性疾病针对性治疗,个体化干预更高效。
•低LPS风险:不需要特殊药物干预,维持低LPS饮食模式和健康生活方式,几个月复查一次即可,长期维持可以稳定保持低LPS。
•中度LPS风险:饮食调整基础上,针对性补充营养补充剂,比如益生元、益生菌、乳铁蛋白,根据是否存在屏障损伤选择修复屏障营养,3-6个月复查调整方案。
•显著LPS风险:先排查继发性疾病,常见继发性疾病是SIBO、牙周病、便秘,针对性治疗继发性疾病,治疗后继续生活方式维持。
根据风险分层选择不同干预强度,避免低风险过度干预浪费金钱扰乱菌群,高风险干预不足达不到效果,个体化分层干预平衡效果和安全性,更合理高效。
♥
很多人从未关注过脂多糖(LPS),也很少意识到它与常见慢性疾病之间的联系。通过本文的系统梳理,我们从结构、机制到疾病,再到干预路径,已经建立起一个相对完整的认知框架。
⑴脂多糖的基本特性
脂多糖由脂质A、核心多糖和O抗原构成,其中脂质A是主要的免疫活性结构。其物理化学性质较为稳定,一般灭菌手段难以完全去除其活性。
⑵代谢与清除路径
脂多糖主要来源于肠道菌群。正常情况下,肠道屏障可以限制其大量进入体内;进入循环后的LPS主要由肝脏清除。部分研究提示,脂肪组织在一定程度上也可参与LPS相关过程。
⑶致病机制
LPS可通过TLR4被机体识别,激活先天免疫系统,诱导TNF-α、IL-6等促炎因子释放。当这种激活处于长期低水平状态时,可对多组织产生慢性损伤。
⑷疾病关联
脂多糖升高与多种慢性疾病存在相关性,包括代谢综合征、2型糖尿病、非酒精性脂肪肝(NAFLD)、动脉粥样硬化、高血压、肥胖,以及部分神经系统疾病和自身免疫疾病等。但其作用通常是多因素共同参与的一部分。
⑸脂多糖升高的常见因素
高脂饮食、过量饮酒、吸烟、膳食纤维摄入不足、长期使用广谱抗生素,以及SIBO、牙周炎、便秘、慢性压力、睡眠不足和肥胖等,都可能通过不同路径影响脂多糖水平。
⑹检测与评估方式
脂多糖可以通过血浆检测进行评估,同时CRP等炎症指标可作为间接参考。结合肠道菌群检测,有助于从“产生—吸收—炎症反应”多个维度综合评估风险。
⑺干预基本原则
围绕三个核心方向展开:
•减少产生(调节菌群);
•减少吸收(修复肠道屏障);
•减少转运与炎症反应(饮食与系统状态调节)。
同时,对于SIBO、牙周炎等明确来源,应进行针对性处理,并结合个体情况进行分层干预。
脂多糖(LPS)是革兰氏阴性菌细胞壁的特有成分。在正常生理状态下,少量脂多糖进入循环,有助于维持基础免疫激活。
但当多种因素叠加,导致循环中LPS长期处于偏高水平时,机体会进入一种“慢性低度炎症状态”,这一状态被认为与多种慢性疾病的发生发展密切相关。因此,长期维持LPS在合理范围内,是慢性疾病预防与管理中一个值得关注的重要环节。
主要参考文献
Wassenaar TM, Zimmermann K. Lipopolysaccharides in Food, Food Supplements, and Probiotics: Should We be Worried? Eur J Microbiol Immunol (Bp). 2018 Aug 21;8(3):63-69.
Kim HS, Kim S, Shin SJ, Park YH, Nam Y, Kim CW, Lee KW, Kim SM, Jung ID, Yang HD, Park YM, Moon M. Gram-negative bacteria and their lipopolysaccharides in Alzheimer’s disease: pathologic roles and therapeutic implications. Transl Neurodegener. 2021 Dec 7;10(1):49.
Magdaleno F, Blajszczak CC, Nieto N. Key events participating in the pathogenesis of alcoholic liver disease. Biomolecules. 2017;7:pii:E9.28134813.
Gomes JM, Costa JA, Alfenas RC. Metabolic endotoxemia and diabetes mellitus: A systematic review. Metabolism. 2017;68:133–44.
Roth J, Blatteis CM. Mechanisms of fever production and lysis: lessons from experimental LPS fever. Compr Physiol. 2014;4:1563–604.
Tume R, El Sherbiny S, Bono R, Gautier T, Pais de Barros JP and Meroño T (2025) The balance between proinflammatory, “bad”, and immunomodulatory, “good”, lipopolysaccharide for understanding gut-derived systemic inflammation. Front. Immunol. 16:1588129.
Catorce MN, Gevorkian G. LPS-induced murine neuroinflammation model: main features and suitability for pre-clinical assessment of nutraceuticals. Curr Neuropharmacol. 2016
Laugerette F, Vors C, Peretti N, Michalski MC. Complex links between dietary lipids, endogenous endotoxins and metabolic inflammation. Biochimie. 2011;93:39–45
Neyen C, Lemaitre B. Sensing Gram-negative bacteria: a phylogenetic perspective. Curr Opin Immunol. 2016;38:8–17.
Candelli, M.; Franza, L.; Pignataro, G.; Ojetti, V.; Covino, M.; Piccioni, A.; Gasbarrini, A.; Franceschi, F. Interaction between Lipopolysaccharide and Gut Microbiota in Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 6242.
Szentirmai, É., Buckley, K., Massie, A.R. et al. Lipopolysaccharide-mediated effects of the microbiota on sleep and body temperature. Sci Rep 14, 27378 (2024).
Citronberg JS, Curtis KR, White E, Newcomb PA, Newton K, Atkinson C, Song X, Lampe JW, Hullar MA. Association of gut microbial communities with plasma lipopolysaccharide-binding protein (LBP) in premenopausal women. ISME J. 2018 Jun;12(7):1631-1641.
Lin TL, Shu CC, Chen YM, Lu JJ, Wu TS, Lai WF, Tzeng CM, Lai HC, Lu CC. Like Cures Like: Pharmacological Activity of Anti-Inflammatory Lipopolysaccharides From Gut Microbiome. Front Pharmacol. 2020 Apr 30;11:554.
d’Hennezel E, Abubucker S, Murphy LO, Cullen TW. Total Lipopolysaccharide from the Human Gut Microbiome Silences Toll-Like Receptor Signaling. mSystems. 2017 Nov 14;2(6):e00046-17.
Apigenin Alleviates Obesity-Associated Metabolic Syndrome by Regulating the Composition of the Gut Microbiome.. Qiao Y, Zhang Z, Zhai Y, Yan X, Zhou W, Liu H, Guan L and Peng L. (Front. Microbiol. 12:805827. (2022))
Orange juice neutralizes the proinflammatory effect of a high-fat, high-carbohydrate meal and prevents endotoxin increase and Toll-like receptor expression.. Ghanim H, Sia CL, Upadhyay M, Korzeniewski K, Viswanathan P, Abuaysheh S, Mohanty P, Dandona P.. (Am J Clin Nutr. 2010 Apr;91(4):940-9.)
Oral spore-based probiotic supplementation was associated with reduced incidence of post-prandial dietary endotoxin, triglycerides, and disease risk biomarkers.. McFarlin BK, Henning AL, Bowman EM, Gary MA, Carbajal KM.. (World Journal of Gastrointestinal Pathophysiology. 2017;8(3):117-117.)
Effect of Probiotic Supplementation on Intestinal Permeability in Overweight and Obesity: A Systematic Review of Randomized Controlled Trials and Animal Studies.. DiMattia Z, Damani JJ, Van Syoc E, Rogers CJ.. (Adv Nutr. 2024 Jan;15(1):100162.)
Inulin controls inflammation and metabolic endotoxemia in women with type 2 diabetes mellitus: a randomized-controlled clinical trial.. Dehghan P, Gargari BP, Jafar-Abadi MA, Aliasgharzadeh A.. (Int J Food Sci Nutr. 2014 Feb;65(1):117-23.)
Efficacy of rifaximin on circulating endotoxins and cytokines in patients with nonalcoholic fatty liver disease.. Gangarapu, Venkatanarayanaa; Ince, Ali Tüzüna; Baysal, Birola; Kayar, Yusufa; Kiliç, Ulkanb; Gök, Özlemb; Uysal, Ömerc; Şenturk, Hakana.. (European Journal of Gastroenterology & Hepatology 27(7):p 840-845, July 2015.)
Rifaximin: beyond the traditional antibiotic activity.. Calanni, F., Renzulli, C., Barbanti, M. et al.. (J Antibiot 67, 667–670 (2014).)
Improvement of intestinal barrier function, gut microbiota, and metabolic endotoxemia in type 2 diabetes rats by curcumin.. Huang J, Guan B, Lin L, Wang Y.. (Bioengineered. 2021 Dec;12(2):11947-11958.)

谷禾健康
肠道屏障是将宿主与外界隔离的主要防御,具有多项关键生理功能,包括营养消化、吸收以及防止潜在有害的膳食抗原和致病微生物的侵害。然而,饮食、药物、昼夜节律紊乱、年龄、肠道微生物群、微生物代谢物和遗传易感性等多种因素都可能破坏肠道屏障。这种破坏可能导致细菌易位,进而引发肠肝和全身性炎症。
目前,肠道屏障受损已被认为与多种疾病的发病机制有关,包括炎症性肠病(克罗恩病和溃疡性结肠炎)、肠易激综合征和结直肠癌等肠道疾病。此外,肝病(如代谢功能障碍相关脂肪性肝病、酒精性肝病和原发性硬化性胆管炎)和全身性代谢疾病(如糖尿病和肥胖)也与肠道屏障受损有关。
然而,目前大多数临床数据仅显示相关性,尚无法明确肠道屏障损伤是这些疾病的原因还是结果。目前,全世界药监督管理局尚未批准专门用于修复肠道屏障损伤的药物。现有疗法主要侧重于疾病的预防和管理,并严重依赖免疫抑制剂来控制炎症。
不幸的是,持续的屏障损伤和延迟愈合会降低这些治疗的疗效,并可能导致治疗耐受甚至复发。因此,开发直接靶向肠道上皮屏障的治疗策略至关重要。
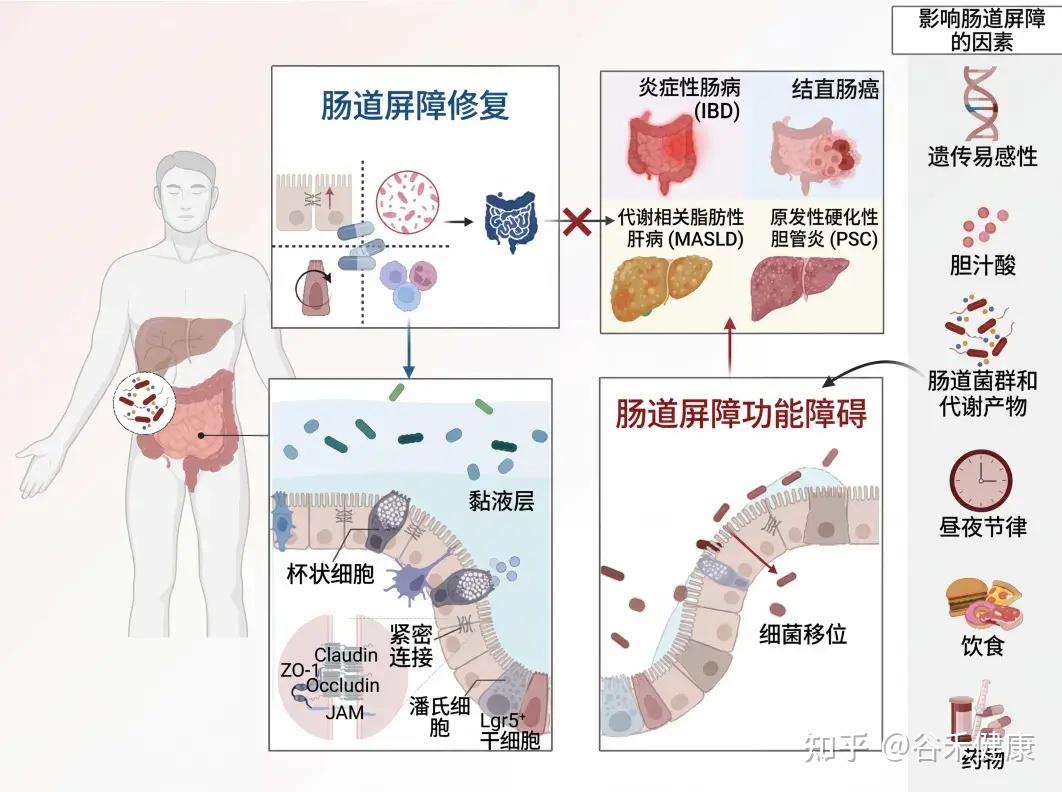
本文介绍肠道屏障的基本功能,生理结构和功能、影响其完整性的内外因素,重点介绍一些潜在的治疗策略,旨在恢复屏障完整性,改善和优化疾病管理。
肠道是一个独特的器官,在食物消化、营养吸收、动态的宿主与环境互动以及维持身体稳态方面发挥着关键作用。
为了保护宿主免受外部威胁,例如长期暴露于膳食抗原和病原微生物,肠道上皮细胞(IECs)形成了多种类型的屏障,包括机械屏障、富含共生微生物的粘液层,以及由免疫细胞及其活性物质组成的免疫屏障(下图)。

机械屏障:由紧密连接(TJs)形成的紧密排列的IECs层构成,确保肠道的结构完整性,调节肠道通透性,并控制水和大分子的运输。
粘液层:由杯状细胞分泌的粘蛋白组成,为共生细菌提供栖息地和营养。其独特的结构特征限制了病原体的渗透,进一步增强了TJs的物理隔离功能。
干细胞微环境:位于肠腺隐窝底部,由隐窝驻留肠道干细胞(ISCs)、间充质细胞、免疫细胞以及肠道分泌细胞(如杯状细胞和潘氏细胞)组成。这个微环境具有高度增殖性,因此负责组织更新和肠道屏障修复。它还介导抗原吞噬并释放抗菌肽(AMPs),通过清除潜在病原体来维持内部稳态。
这些元素共同构建了物理和生化屏障,保护宿主并调节内部和外部环境之间的交流。这种复杂的平衡对于胃肠系统的正常运作和维持身体整体平衡至关重要。
1
粘液层组成与免疫监测
粘液层的物理与生化屏障
粘液层是胃肠道的第一道防线,主要由90%-95%的水、1%-2%的脂质和1%-5%的粘蛋白组成。粘蛋白通过糖基转移酶高度糖基化,含有50%-80%的碳水化合物(w/w)。
粘蛋白聚糖多样而复杂的结构,为粘液相关细菌(如 R.torques、A.muciniphila、B.bifidum和R.gnavus)提供了理想的定植位点和营养来源。
动态防御:细菌即是住客,也是建筑师
MUC2是胃肠道中表达的主要粘蛋白。在细菌暴露后,杯状细胞通过meprin β介导的裂解机制分泌MUC2,形成保护性粘蛋白层。该结构允许共生微生物在外粘液层定植,并利用其多糖降解酶从粘蛋白O-聚糖中获取营养能量。这种宿主与微生物的相互作用,有助于调节近端结肠微生物群的结构和转录。
别让细菌吃肠壁:低纤维饮食的代价
研究表明,低纤维饮食会促进微生物群降解宿主粘蛋白,导致粘液层变薄,从而削弱屏障功能。Muc2缺陷小鼠表现出结肠组织学损伤增加、细菌易位至肝脏增多以及肠道紧密连接蛋白显著减少。此外,粘蛋白O-糖基化紊乱导致的粘液屏障完整性和功能受损与代谢疾病的发病机制有关。
粘液层的水龙头:谁在掌控肠道保护液的释放?
最近的研究发现,Gasdermin D (GSDMD) 是一种参与细胞凋亡的成孔效应蛋白,它通过scinderin介导的F-肌动蛋白解聚,促进钙依赖性胞吐作用,从而调节杯状细胞的粘蛋白分泌。GSDMD缺陷会破坏粘液屏障,使病原体粘附到上皮细胞,导致肠道疾病的发生。
严防细菌偷渡:一套保护肝脏的精密免疫系统
粘液层凭借其独特的粘弹性,能够有效滞留并扩散来自潘氏细胞和杯状细胞的抗菌物质及免疫细胞因子,形成化学屏障。
-抗菌肽 (AMPs) 与IgA的协同作用
潘氏细胞产生隐匿防御素、抗菌素和溶菌酶等AMPs。这些AMPs大量存在于肠道上皮表面,能够直接清除有害微生物。在新生非肥胖糖尿病小鼠模型中,生态失调导致的结肠AMPs缺乏会导致1型糖尿病中的胰腺自身免疫。
AMPs与微生物特异性免疫球蛋白IgA协调作用,在维持屏障稳定性和抑制炎症中发挥关键作用。
-IL-17和IL-22的调控
它们的调节受T辅助17(Th17)细胞和III型固有淋巴细胞(ILC3)产生的IL-17和IL-22的影响。
ILC3依赖于树突状细胞(DC)相关的Mincle信号通路,该通路与酪氨酸激酶偶联的C型凝集素受体有关。在Mincle缺失或酪氨酸激酶受损的情况下,肠道再生胰岛衍生III-γ(RegIIIγ)和IgA的合成会减少,从而导致肠道微生物群移位,进而引发肝脏炎症和脂质代谢失调。
这些发现表明粘液层完整性在维持肠肝稳态中的关键作用。
2
上皮连接:构筑肠壁防线的灰浆与砖块
微绒毛:不仅仅是吸收养分的地毯
功能:肠道上皮细胞顶端的微绒毛密集排列成刷状缘。它们既是营养吸收的高效界面(扩大表面积),又是阻止细菌附着的第一道物理防线。
脆弱性:在克罗恩病中,这些绒毛会变短、基因表达混乱。
破坏机制:就像拆除帐篷的支柱一样,肠出血性大肠杆菌的毒力因子通过CDK1-Formin信号轴,攻击支撑微绒毛的骨架蛋白(ACT-5),导致微绒毛坍塌消失,引发严重腹泻、出血性结肠炎等。
紧密连接(TJs):细胞间的拉链
在微绒毛下方,紧密连接(TJs)像拉链一样把相邻细胞的细胞膜紧紧锁死。
核心作用:这种吻合结构封堵了细胞间的空隙,相当于门控功能——只允许特定的物质通过,严防细菌和有害大分子渗透。
关键零件:谁在控制拉链的松紧?
病菌如何撬开防御?
紧密连接的稳定性高度依赖于细胞骨架的支撑,这成为了病原体的攻击目标:
3
动态防御机制:从干细胞再生到免疫感知
隐窝深处的生命源泉:肠道干细胞 (ISCs)
肠道屏障之所以能抵御消化道内持续的磨损与危害,归功于其惊人的自我更新能力。
核心机制: 位于肠道隐窝底部的肠道干细胞是这一过程的“总工程师”。它们通过持续增殖,不断分化补充受损的肠道上皮细胞(IECs),维持着组织的修复与动态平衡。
谁在调控修复?信号通路、压力与衰老
ISCs 的功能受到微环境信号的精密调控,同时也易受外部因素干扰:
-修复的加速器
当肠道受损时,IL-1R1 信号 和 Wnt 激动剂 RSPO3 会协同作用,强力促进 ISCs 的修复功能,加速伤口愈合。
-心理压力:让干细胞电量耗尽
心理压力不仅仅是情绪问题,它能产生实实在在的生理毒性。
心理压力会导致 ISCs 内部的 线粒体能量代谢 受损,进而干扰细胞分化。这种微观层面的能量危机削弱了宏观的肠道屏障,这科学地解释了为何精神疾病患者常伴有肠道问题。
-衰老的阻滞剂
随着年龄增长,ISCs 的数量和活性会下降,导致屏障完整性受损及菌群失衡。
关键原因:维持 ISCs 活性的关键信号——Wnt 信号通路随衰老而减弱,导致干细胞枯竭。
簇状细胞:不仅是免疫哨兵
簇状细胞是肠道屏障中一种重要的分泌型肠上皮细胞,与潘氏细胞和杯状细胞共同发挥作用。它们通过分泌IL-25来抵御病原体感染,激活2型免疫并清除病原体。簇状细胞还能感知病原体代谢物,并通过G蛋白偶联信号通路产生PGD2,从而刺激杯状细胞分泌粘液并促进自身增殖,进一步增强抗菌防御。此外,在肠道损伤时,簇状细胞可以充当储备肠道干细胞,协助屏障修复。
肠道屏障的健康受多种因素影响,包括遗传、饮食(如西式饮食)、药物(抗生素等)、疾病、生活习惯(昼夜节律)、心理状态(压力)和生理过程(衰老)。这些因素共同作用,可导致肠道微生物失衡,进而引发全身性代谢紊乱。这些代谢紊乱又会加剧炎症反应,进一步损害肠道屏障,形成一个恶性循环,持续破坏肠道健康。
下表总结了影响肠道和肝脏疾病的因素及机制与肠道屏障功能障碍相关的研究

1
遗传易感性
基因组关联研究(GWAS)的进展显著提升了疾病易感基因的识别和相关生物学通路的理解,这对临床转化具有重要价值。这些研究分析基因组中的遗传变异,以探索基因型和表型之间的关系。
免疫刹车失灵:IL-10 信号通路的遗传缺陷
尽管基因突变与肠道屏障功能障碍之间的直接联系有限,但GWAS已识别出许多与IBD(炎症性肠病)发展相关的基因座。例如,IL10基因突变是首批被发现能诱发IBD的突变之一。IL10基因敲除小鼠会自发发展结肠炎并增加肠道微生物易位。IL10受体(IL10R)的突变(由IL10RA和IL10RB基因编码的IL10R1和IL10R2蛋白组成)已与早发性小肠结肠炎相关联。这些突变损害了IL10诱导的信号传导,这可能增加TNF-α和其他促炎细胞因子的分泌,从而加剧炎症并削弱肠道耐受性。
离子转运障碍:SLC26A3 与物理屏障的松动
GWAS还显示SLC26A3基因(编码DRA蛋白,一种肠道氯离子转运蛋白)的显著下调。该基因在人类基因组中有害IBD变异中排名在前1%。SLC26A3表达的降低显著增加结肠旁细胞通透性,降低紧密连接(TJ)和黏附连接(AJ)蛋白的表达,从而增加对IBD的易感性。
黏液防线的溃败:ST6GALNAC1 与糖基化异常
糖蛋白组学分析揭示,先天性IBD(炎症性肠病)患者可能携带ST6GALNAC1(ST6)基因突变,该基因编码一种对维持黏液屏障稳态至关重要的唾液酸转移酶。该基因的突变会导致肠道黏膜层厚度减少,并破坏其保护功能。阐明这些调控机制对于理解先天性IBD的发病机制至关重要。此外,屏障功能还受到遗传易感性和环境因素相互作用的影响。DNA甲基转移酶编码基因DNMT3A的突变与IBD风险增加相关,已被证明能减少杯状细胞数量,缩短黏附连接(AJ)复合体,并增加肠道通透性。这些改变增加了对结肠炎的易感性,并阻碍了上皮再生和修复过程。
2
胆汁酸
胆汁酸(BAs)不仅是消化和脂质吸收的关键,还扮演着调节全身代谢和免疫的激素角色。它们通过激活FXR和TGR5等受体发挥作用。
修复的动力(次级胆汁酸)
最近研究发现,胆汁酸在维持肠道上皮屏障方面至关重要,能刺激Lgr5+肠道干细胞(ISCs)的自我更新,从而促进肠道修复。
修复的阻力(过量初级胆汁酸)
然而,过量的初级胆汁酸(如胆酸)会通过抑制脂肪酸氧化来减缓ISC的增殖,进而影响屏障修复。另一方面,TGR5在肠道代谢稳态中作用显著,其被次级胆汁酸(脱氧胆酸)激活后,会促进ISCs中YAP1和SRC等因子的转录,从而有效驱动肠道上皮的再生。
孕烷X受体 (PXR):抗炎与修复的化学感应器
孕烷X受体(PXR)是调节肠道上皮屏障稳态的关键核受体,它通过调控外源性物质代谢和先天免疫发挥作用。
溃疡性结肠炎患者中PXR及其靶基因的下调表明,PXR功能受损可能导致肠道损伤后屏障修复缺陷。PXR缺陷会加剧肠道上皮功能障碍,并增加对肠道损伤的易感性。
PXR的激活(例如通过石胆酸LCA)通过抑制NF-κB通路和炎症因子释放,以及维持紧密连接的完整性来发挥保护作用。
维生素 D 受体 (VDR):从抗菌防御到防癌屏障
胆汁酸也是维生素D受体(VDR)的内源性配体,对肠道稳态具有保护作用。VDR通过调节上皮分化和增强紧密连接表达来维护肠道屏障完整性,并支持潘氏细胞的抗菌功能。然而,在炎症性肠病(IBD)和结直肠癌(CRC)中,VDR的保护机制受损,其信号通路的失调与CRC的加速进展和不良预后密切相关。VDR激活不仅直接抑制肿瘤细胞增殖,还能通过增强黏膜屏障完整性来限制肿瘤进展,从而抑制CRC。
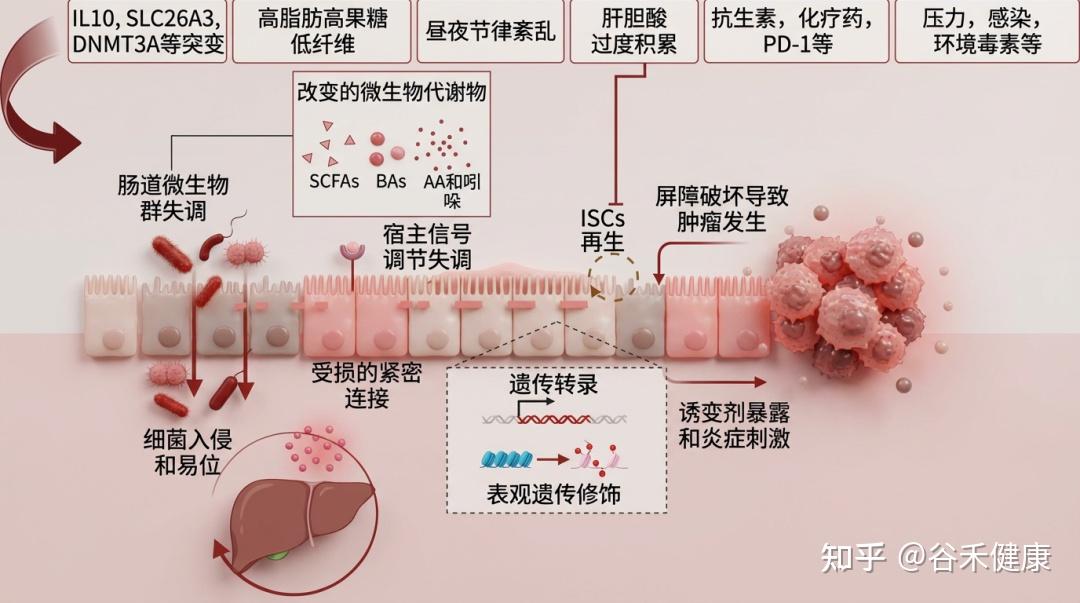
3
肠道微生物群及衍生代谢物
肠道微生物群通常与宿主共生,并被肠道上皮屏障限制在黏液层外。但有些微生物能突破屏障,损害胃肠道健康。抗生素虽看似能保护肠道,但实际上可能削弱屏障功能,增加新发炎症性肠病(IBD)的风险,这已在大量的研究中得到证实。与此相反,良好的肠道微生物结构对肠道屏障修复有积极作用。研究发现,肠道微生物群能通过激活巨噬细胞信号促进结肠上皮前体细胞生长,从而帮助修复受损的肠道上皮屏障。
色氨酸代谢物:PXR 与 AhR
肠道微生物群通过产生多种代谢物(如次级胆汁酸和色氨酸衍生物)来维护肠道屏障的完整性和整体健康。在IBD患者中,色氨酸水平显著降低,其代谢改变对疾病进程和预后有重要影响。
路径一:芳烃受体 (AhR) 的激活
色氨酸衍生物中的吲哚类物质是芳烃受体(AhR)的天然配体,AhR的激活能调节免疫细胞(如Tregs和Th17)及其细胞因子(特别是IL-22)的产生,从而促进肠道屏障的保护、修复和稳态。
路径二:孕烷 X 受体 (PXR) 的增强
除了AhR途径,这些代谢物还能通过孕烷X受体(PXR)增强屏障。例如,吲哚-3-丙酸(IPA)作为内源性PXR激动剂,通过TLR4信号通路减轻肠道通透性和炎症。PXR激活通过稳定紧密连接蛋白ZO-1、抑制MLCK表达和JNK1/2磷酸化来保护肠道屏障。
综上所述,色氨酸衍生的吲哚代谢物通过同时激活PXR和AhR信号通路,对肠道上皮发挥双重保护作用。
反面:犬尿氨酸 (Kyn) 途径
色氨酸代谢的犬尿氨酸(Kyn)途径与IBD进展密切相关,其限速酶IDO在炎症下促进Kyn途径,IDO的抑制或缺陷可减轻肠道炎症并增强屏障功能。
短链脂肪酸:能源与信号的结合
短链脂肪酸(SCFAs),包括丁酸、乙酸和丙酸,是肠道微生物分解膳食纤维等产生的关键代谢物,对肠道屏障功能至关重要。它们通过激活G蛋白偶联受体(如GPR43或FFAR2)来增强肠道屏障,并保护免疫细胞免受损伤,从而有助于预防结直肠癌(CRC)。
丁酸抑制组蛋白脱乙酰酶(HDACs)以调节基因转录,并通过调控紧密连接处的蛋白来促进屏障修复。GPCR信号和HDAC共同抑制维护上皮屏障完整性。此外,SCFAs还能刺激杯状细胞分泌黏蛋白,进而促进黏蛋白降解菌生长,通过消耗黏蛋白促进肠道干细胞分化,确保肠道上皮的再生能力。
4
昼夜节律
昼夜节律,常被称为生物钟,是代谢稳态的内部控制系统,旨在同步诸如光暗周期等周期性环境信号。该系统通过转录、转录后和翻译后修饰,在特定时间协调基因表达,以实现最佳代谢适应。
“什么时候吃”可能比“吃什么”更重要
多项研究表明,昼夜节律紊乱与代谢疾病之间存在密切关联。具体而言,不规律的进食时间会增加小肠暴露于膳食抗原和微生物刺激的风险,从而加重肠上皮细胞(IECs)主要组织相容性复合体II(MHC II)的负担。这种紊乱会削弱肠道微生态的调节功能,并减少IL-10的分泌。
神经免疫回路的失调:VIP 与 ILC3 的博弈
昼夜节律还会影响肠道中的神经免疫回路,这些回路受进食行为激活,并在饮食不规律时可能导致病理变化。食物摄入会触发肠道神经元分泌血管活性肠肽(VIP),该肽会上调与脂质吸收和转运相关的蛋白质。同时,VIP会降低IECs中的AMP水平,并减少ILC3产生的IL22。这种饮食节律的紊乱有助于病原体的肠道定植,尤其是在神经免疫回路的屏障功能受损时。
微生物振荡器:细菌也有生物钟
肠道微生物群自身也表现出丰度和功能的昼夜波动,被称为微生物振荡器,它们通过微生物代谢物或自身抗原影响宿主昼夜节律。肠道微生物群与宿主生物钟之间的这种相互作用显著影响屏障完整性和先天免疫反应。例如,短链脂肪酸通过抑制组蛋白脱乙酰酶(HDAC)活性,有助于调节小肠的昼夜节律相移。
肠道中的分节丝状细菌(SFB)驱动着与宿主节律同步的节律性ILC3回路振荡,从而通过时间依赖性地表达抗菌肽(AMPs)来介导感染抵抗力的昼夜变化。
5
饮食
高脂与快餐:胆汁酸的黑化与致癌风险
高脂肪和高糖、高加工饮食,与现代社会代谢疾病患病率的增加密切相关。饮食成分通过改变肠道微生物群以及主动参与宿主生理过程的次级代谢产物的产生,显著影响肠道微环境。
研究表明,现代快餐和西方饮食引起的肠道微生物群变化会提高脱氧胆酸(DCA)水平,这通过激活肠道FXR和I型干扰素(IFN)信号通路损害潘氏细胞。
动物研究进一步揭示,高脂肪饮食(HFD)在不同的结肠癌模型中,包括偶氮甲烷(AOM)-葡聚糖硫酸钠(DSS)诱导模型以及Apc突变诱导的自发模型,都会加剧肠道屏障损伤。HFD受损的肠道屏障允许更多病原体和衍生代谢物穿透上皮,从而加速结直肠癌(CRC)的发展。
糖衣炮弹:果糖与高血糖的双重打击
除了高脂肪摄入,过量的膳食果糖摄入也会损害上皮屏障。果糖水平升高会增加循环内毒素,从而激活巨噬细胞上的Toll样受体4(TLR4),引发全身性炎症反应。同时,葡萄糖代谢既对代谢综合征具有治疗潜力,也是肠道屏障功能的关键协调者。
在瘦素缺乏(ob/ob)和瘦素受体(LepR)缺乏(db/db)的2型糖尿病(T2DM)小鼠模型中,高血糖通过诱导肠上皮细胞(IECs)中葡萄糖转运蛋白2(GLUT2)依赖性转录重编程来破坏肠道屏障,这随后损害了紧密连接(TJ)和黏着连接(AJ)结构的完整性。
富含果糖的食物

6
药物
抗生素:精准打击与地毯式搜捕
抗菌素的使用对肠道微生物群落的丰度和结构有着深远的影响,进而影响肠道屏障的完整性。
例如,利福昔明-α (Rifaximin-α)常用来治疗小肠细菌过度生长:这种抗生素可以减少破坏粘膜的细菌,增加肠道内TNF-α和IL-17的水平,从而增强对病原体的抵抗力。它还能通过增加回肠中的乳杆菌水平来改善应激引起的肠道屏障功能障碍。
广谱抗生素会降低肠道微生物多样性,导致免疫失调,并增加感染的易感性。在健康成年小鼠中,广谱抗生素治疗会导致菌群失调,肠道上皮紧密连接(TJ)的完整性受损,表现为ZO-1表达减少和NLRP3炎症小体激活。
非抗生素药物:阿司匹林的隐形副作用
非抗生素药物也可能损害肠道屏障,例如阿司匹林,广泛用作消炎镇痛药,但会引起胃肠道损伤。
它会激活肠道FXR信号,并减少戈氏副拟杆菌(Parabacteroides goldsteinii)的数量。
戈氏副拟杆菌产生7-酮-LCA,这种物质能抑制肠道干细胞(ISC)的干性,从而减缓肠道屏障的修复。
化疗药物:再生能力的丧失
化疗药物:化疗是导致肠道屏障损伤的另一个主要原因。例如,5-氟尿嘧啶会加速黏膜细胞的死亡,而非再生。因此,超过40%的化疗患者会出现胃肠道损伤,表现为腹泻、便秘和消化不良等症状。
免疫检查点抑制剂:免疫激活的附带损伤
免疫检查点抑制剂(ICIs):靶向PD-1的ICIs彻底改变了抗肿瘤治疗。尽管这些抗体通过阻断PD-1通路来重建正常的免疫反应,但它们经常引起胃肠道毒性。
PD-1信号被发现对调节结肠淋巴组织诱导(LTi)细胞(ILC3的一个特定亚群,对维持免疫稳态至关重要)至关重要。
PD-1信号的缺失会导致LTi细胞中脂肪酸氧化过度激活,并反馈抑制LTi细胞的激活和IL-22的产生。这种失衡导致生态失调、肠道屏障损伤,并增加肠炎的易感性。
功能性测试:探针分子与 LMR 比值
乳果糖/甘露醇比值 (LMR) 是评估肠道通透性的重要生物标志物。
注:乳果糖(大分子)的尿液排泄量反映“肠漏”程度和上皮损伤。甘露醇(小分子)主要通过跨细胞途径吸收,代表非特异性上皮转运。
通过口服探针分子后检测尿液排泄来评估肠道通透性,这是最常用和直接的方法。
常用的探针分子包括不易代谢、吸收差的糖类(如甘露醇、鼠李糖、三氯蔗糖、乳果糖)或低分子量聚乙二醇(PEG)和乙二胺四乙酸(EDTA)。
为了避免饮食干扰,这些探针常使用同位素标记。
乳果糖-甘露醇测试 (LMT) 是临床广泛使用的评估方法。
在动物模型中,常用FITC标记的葡聚糖或铬-51标记的EDTA (51Cr-EDTA) 吸收到体循环的量来评估肠道屏障完整性。
其他方法:生物标志物的间接分析和多组学分析平台也是新兴的评估手段。
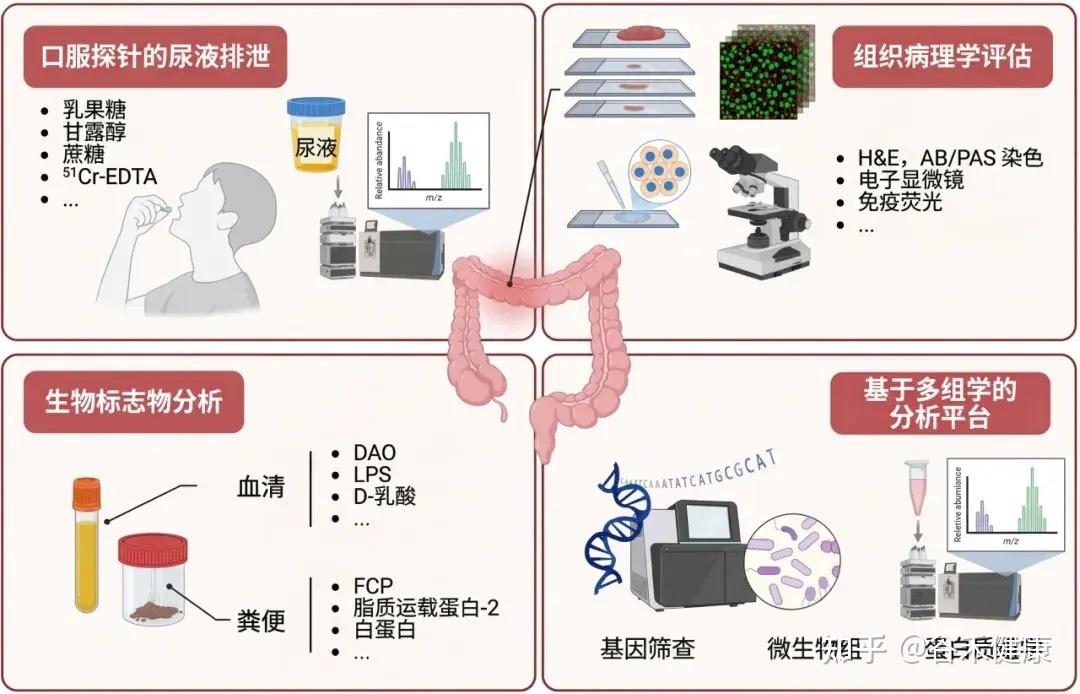
生物标志物:血液与粪便的线索
血清蛋白质组学和基因筛查技术:用于早期检测屏障功能障碍,并支持对其潜在原因的机制研究。
这些间接方法通过检测血液、粪便或分子水平上的特定物质,提供肠道屏障健康状况的重要信息。
组织病理学:屏障损伤的可视证据
内窥镜活检标本的组织病理学评估可以详细检查上皮绒毛形态和评估肠道屏障完整性。可以使用黏蛋白特异性阿尔辛蓝/过碘酸-希夫 (AB/PAS) 染色来评估黏液层,使用电子显微镜进行超微结构分析,以及使用免疫荧光技术精确定位和量化紧密连接 (TJ) 蛋白的表达,从而系统地分析黏膜成分的结构改变。
这些方法共同为评估肠道屏障完整性提供了全面的视角。
肠道上皮屏障的失调对多种肠道和肝脏疾病的发展和进展具有重要意义。肠道上皮的紧密连接功能障碍增加了通透性,促进细菌及其致病因子穿越屏障进入全身循环及其他组织。这种入侵会引发全身性炎症,破坏代谢稳态,而代谢稳态是肠道和肝脏疾病发生和进展的关键事件。虽然尚不确定肠道屏障的损伤是主要驱动因素还是偶然的病理特征,但越来越多的证据表明,肠道屏障的完整性在这些疾病的病因中起着关键作用。
1
炎症性肠病与结直肠癌
IBD 的病理核心:炎症因子的破坏机制
炎症性肠病 (IBD),包括克罗恩病 (CD) 和溃疡性结肠炎 (UC) 的特征是慢性胃肠道炎症。上皮屏障功能障碍是导致 IBD 发生和发展的关键因素。
临床研究表明,UC 和 CD 患者的肠道紧密连接 (TJ) 结构发生改变,导致肠道通透性增加。此外,黏膜免疫激活会引起TNF-α、IL-13、IL-17等细胞因子的产生。
屏障损伤是“因”而非“果”
进一步研究表明,由于 TJ 功能障碍和细胞骨架变化,细胞旁通透性增加可能在疾病发生前就已出现,例如 CD 患者亲属中早期肠屏障异常以及肠易激综合征 (IBS) 的较高患病率。同样,大规模队列研究已经发现 CD 诊断前三年内的屏障障碍标志物。这些发现支持屏障功能障碍是疾病进展的主要原因,而不是次要效应。
致癌风险:暴露于诱变环境
上皮屏障缺陷会使肠道内壁暴露于诱变化合物或长期炎症刺激,这可能通过氧化应激等机制启动并促进结直肠肿瘤的发生。在早期和晚期结直肠癌中,各种屏障相关蛋白的表达显著降低,这促进了微生物移位,引发炎症,并进一步加速肿瘤生长。然而,结直肠癌发生发展过程中上皮屏障成分异常表达的确切机制仍不清楚。
关键分子机制:GOLM1 与 NDRG2
事实上,实验模型表明,上皮屏障受损的小鼠更容易患结肠炎和炎症驱动的肿瘤。例如:
-GOLM1 缺失(细胞分化紊乱)
高尔基体膜蛋白 1 (GOLM1) 的缺失会过度激活 Notch 通路,从而破坏肠道稳态,改变肠上皮细胞 (IEC) 的分化和分泌细胞谱,并减少杯状细胞的数量,所有这些都会增加肠道通透性和促肿瘤炎症。机械屏障在预防结直肠癌发展中也至关重要。
-NDRG2(肿瘤抑制基因)
肿瘤抑制基因 N-myc 下游调节基因 2 (NDRG2) 已被证明可以增强 E3 连接酶 FBXO11 和 E-钙粘蛋白抑制剂 Snail 之间的相互作用,促进 Snail 泛素化。E-钙粘蛋白的这种稳定作用增强了黏附连接,从而限制了结肠炎相关肿瘤的发展。
2
慢性肝病
慢性肝病,包括 MASLD、ALD 和 PSC,常伴有肠道屏障受损和肠肝轴的失调。胃肠道物质,如营养素、分泌因子和微生物代谢物, 通过门静脉进入肝脏,并被代谢用于全身利用。这一过程使肝脏对病原体相关分子模式(PAMPs)极为敏感,成为肠道物质的主要靶点。
代谢功能障碍相关脂肪性肝病
代谢功能障碍相关脂肪性肝病 (MASLD, Metabolic dysfunction-associated steatotic liver disease):以前被称为NAFLD(非酒精性脂肪性肝病)。它是一种与代谢功能障碍相关的肝脏疾病,通常与肥胖、2型糖尿病、高血压和血脂异常有关。主要特征是肝脏中脂肪过度积聚,但与大量饮酒无关。
MASLD中,内毒素血症常见,提示肠屏障与肝炎症相关。DSS诱导小鼠肠屏障损伤加剧肝损伤。Il-10敲除鼠中,西方饮食+DSS处理降低BA水平,抑制肝脏FXR信号,加重MASLD。
肠道菌群失调是MASH早期驱动因素,宏基因组研究表明MASLD患者肝病发作前已存在肠道菌群失调。MASH患者粪外囊泡可能通过nmMLCK机制降低TJ表达,加剧肠屏障功能障碍。恢复肠屏障可逆转结肠炎引起的肝脂代谢失衡,特别是通过调节次级BA-TGR5/mTOR/氧化磷酸化通路。
酒精性肝病
酒精性肝病 (ALD, Alcoholic Liver Disease):是由于长期过度饮酒引起的肝脏损伤。 它可以表现为脂肪肝、酒精性肝炎和肝硬化等不同阶段。
酒精暴露破坏上皮连接,诱导肠屏障分解。ALDH2缺乏加剧酒精引起的肠道TJ/AJ蛋白降解,肠道ALDH2可能是酒精诱导的肠-肝轴损伤靶点。慢性酒精摄入减少肠道cDCs,导致AMP产量下降,保护性A. muciniphila丰度下降,AJs破坏,最终通过IL-12-IFNγ信号通路引起肝损伤。
原发性硬化性胆管炎 (PSC)
原发性硬化性胆管炎 (PSC, Primary Sclerosing Cholangitis):是一种慢性胆汁淤积性肝病,其特征是肝内外胆管的炎症和瘢痕形成。 最终可能导致胆管狭窄、肝硬化和肝功能衰竭。
PSC常与溃疡性结肠炎共病。PSC患者肠道微生物群与健康个体不同。门静脉中存在微生物提示肠屏障破坏,可能导致细菌易位。肺炎克雷伯菌等致病菌可通过接触依赖性细胞凋亡诱导上皮孔形成,加剧炎症和肝胆损伤。BDL诱导胆汁肝病小鼠模型中,肠道屏障破坏独立于菌群失调发生。敲除CHOP可缓解BDL小鼠肠屏障损伤、ISC干性丧失以及肝脏炎症和纤维化。
肠道屏障受损可刺激PSC中的保护性负反馈回路,LPS激活NF-κB通路抑制肝细胞BA代谢,减缓胆汁性肝病进展。抗生素或泛半胱天冬酶抑制剂能减弱菌群失调诱导的内毒素易位或NLRP3炎症小体激活,有望治疗PSC。
免疫靶向疗法,包括抗炎药、免疫抑制剂和生物制剂,已被证明在控制多种肠道疾病(如炎症性肠病)炎症方面有效。然而,这些治疗并不能直接解决肠道屏障的根本损伤。不幸的是,长期使用这些药物甚至可能增加感染、耐药性和疾病复发的风险,以及恶性肿瘤和死亡等严重不良反应。
确实,虽然减少炎症至关重要,但促进上皮屏障的愈合和恢复对于肠道疾病,尤其是炎症性肠病(IBD)的长期缓解至关重要。目前,没有临床批准的疗法专门针对肠道屏障的修复和维护。鉴于屏障功能障碍与肠道和肝脏疾病发病机制的密切联系,及时修复和恢复上皮屏障是一种有前景的治疗策略。与其仅关注症状管理,提升上皮愈合和健康完整性的治疗方法,可以减缓肠道和肝脏疾病的进展。
通过加强屏障,这类疗法还可能减少细菌易位、炎症以及肝损伤的加重。

1
紧密连接蛋白调节
靶向 MLCK:紧密连接稳态的核心调控
肠道屏障的维持在很大程度上依赖于多种 TJ 蛋白。肌球蛋白轻链激酶(MLCK)被视为开发炎症性肠病障碍疗法的有力候选目标,其在调节 TJs 及其分解中发挥核心作用,通过调节紧密连接(TJs)及其分解来保护屏障功能免受免疫诱导损伤。
对 MLCK 的缺失或抑制可以有效保护屏障功能免受免疫诱导损伤,但这些干预无法预防晚期结肠炎,后者涉及细胞凋亡和黏膜损伤,这些过程与 MLCK 介导的 TJ 分解无关。这些发现反映了 MLCK 抑制剂的治疗局限性。目前的 MLCK 抑制剂对上皮和平滑肌的催化域缺乏特异性,因此可能出现毒性副作用。
新机制发现:TCPTP 的双重保护
紧密连接 (TJ) 通过调节闭合蛋白 (occludin) 和封闭蛋白 (claudin) 来维持肠道完整性,而闭合蛋白和封闭蛋白是两种关键蛋白。
一项研究发现,上皮细胞中的蛋白酪氨酸磷酸酶 (TCPTP) 通过以下两种方式保护肠道屏障:
后者可以防止闭合蛋白 (occludin) 错位并降低上皮细胞的通透性。
目前尚无专门针对 TJ 蛋白的药物进入临床试验阶段,因此,需要在此领域进行深入研究和开发。
2
肠道微生物群的生态调控
益生菌疗法:从天然菌株到工程改造
益生菌已被发现通过多种机制调节屏障功能。例如,罗伊氏乳杆菌(Lactobacillus reuteri)激活Wnt/β-catenin信号通路,诱导Paneth细胞分化和AMP(抗菌肽)分泌,同时刺激Lgr5+肠道干细胞(ISCs)增殖以促进上皮修复。
此外,罗氏菌属(Roseburia)的鞭毛蛋白与Toll样受体5(TLR5)结合,可以上调occludin和MUC2,从而改善肠道屏障完整性,并增加IL-22和REG3γ水平,进一步调节肠道生态。
嗜黏蛋白阿克曼氏菌(AKK菌,Akkermansia muciniphila)也通过调节ISC程序来支持肠道上皮的修复、增殖、分化和稳定。因此,靶向肠道微生物失调成为恢复受损屏障功能的一种有前景的治疗策略。
最新的进展包括使用生物纳米材料包裹过表达人工酶的基因工程益生菌,这是一种用于黏膜修复和炎症治疗的新方法。长双歧杆菌(Bifidobacterium longum)经过修饰以表达过氧化氢酶和超氧化物歧化酶,表现出增强的肠道定植能力、强大的抗炎活性,促进肠道屏障重塑,并调节微生物平衡。这些进展有望减少传统抗炎药物的不良反应。
胆汁酸信号:FXR 激动剂的多重获益
胆汁酸在宿主与肠道菌群的交流以及肠道菌群的构成调节中起着关键作用。胆汁酸合成失调与多种疾病的发生密切相关。激活法尼酯X受体(FXR)能有效缓解胆汁酸过量带来的危害,它通过抑制胆固醇代谢、促进肝细胞将胆汁酸转运出去,从而减少胆汁淤积性肝损伤。
研究显示,FXR激动剂奥贝胆酸(OCA)能够:
此外,OCA治疗还能通过稳定内皮细胞内的β-连环蛋白来防止肠道血管屏障受损。
色氨酸代谢:AHR 通路与 IDO 的平衡
-AHR:屏障完整性的总开关
AHR(芳香烃受体)是一种色氨酸代谢物的受体,对维持肠道屏障的完整性至关重要。研究发现,Ahr−/−小鼠(即缺乏AHR的小鼠)的上皮屏障功能明显丧失,表现为肠道机械屏障受损以及细胞无法正常分化。缺乏AHR配体的小鼠也会出现类似的症状。
进一步研究表明,来自食物的AHR配体可以通过促进细胞内的锌离子(Zn2+)信号传导,从而提高紧密连接(TJ)蛋白的表达,有效预防损害肠道屏障功能的疾病。
-AHR 激动剂的治疗潜力:以尿石素 A 为例
其他AHR激动剂,例如微生物代谢产物尿石素A(UroA),也显示出保护肠道屏障功能的潜力。UroA具有抗炎作用,并且可以通过激活AhR–NrF2依赖性通路来上调TJ蛋白的表达,从而促进肠道屏障的修复。
这些研究表明,激活肠道AHR通路对于治疗酒精性肝病(ALD)具有重要意义,因为ALD与TJ屏障的丧失密切相关。因此,AHR可能成为修复肠道屏障的一个有价值的治疗靶点,尤其是在与肠道屏障功能障碍相关的慢性疾病中。
-IDO 抑制剂:代谢平衡与潜在毒性
正向:抑制 IDO -> 阻断色氨酸向犬尿氨酸转化 -> 迫使色氨酸转化为吲哚
抑制IDO(一种将色氨酸转化为犬尿氨酸的酶)可能有助于治疗肠道屏障功能障碍。IDO抑制剂通过促进色氨酸转化为吲哚,增加AHR配体的生成,从而改善肠道完整性。
虽然IDO抑制剂在癌症治疗中显示出潜力,但它们在治疗肠道屏障损伤方面的应用仍需更多研究。Indoximod是一种IDO抑制剂,已被证明能有效维持细胞间的紧密连接,并显著减轻DSS诱导的结肠损伤。
反向:过度抑制 IDO -> 减少犬尿酸生成 -> GPR35 失去激活 -> 削弱屏障保护
需要注意的是,IDO抑制也可能导致不良的肠道毒性反应。完全阻断犬尿氨酸通路可能会适得其反,损害上皮屏障的完整性。
研究表明,化疗药物引起的肠道损伤会激活色氨酸-犬尿氨酸-犬尿酸通路,增加肠道内犬尿酸的生成。犬尿酸随后激活GPR35,从而增强肠道完整性。这意味着,过度抑制犬尿氨酸通路可能会降低犬尿酸水平,削弱GPR35介导的保护作用,并可能延缓屏障修复。
综上所述,色氨酸代谢在肠道健康中扮演着复杂的角色。因此,在开发治疗肠道屏障功能障碍的新方法时,需要进行更深入的研究。
临床挑战:个体化差异
虽然大量研究表明直接调节肠道微生物群可以改善肠道屏障功能并缓解肠肝疾病,但基于微生物的疗法在临床应用上仍然受到限制。
肠道微生物群产生的有益代谢物,如吲哚-3-乳酸(ILA),其增强肠道屏障的特性取决于宿主特异性的微生物群调节。临床疗效主要取决于患者的初始肠道微生物群组成,这会影响微生物的定植情况。
此外,宿主的遗传变异也是治疗结果的关键因素。例如,携带CARD9风险基因的炎症性肠病(IBD)患者,其膳食色氨酸向芳香烃受体(AHR)激活代谢物的微生物转化能力受损。这些发现解释了微生物靶向疗法中个体差异显著的分子机制。
益生菌作为疾病状态下耗尽的共生微生物,需要稳定的宿主微环境才能定植和发挥作用。如果事先不恢复受损的肠道生态系统,微生物干预疗法往往无法建立持久的微生物平衡或实现有意义的临床结果。因此,未来的治疗策略应采用结合微生物调节和微环境恢复的双靶点方法。
3
肠道干细胞再生
Lgr5+ 肠道干细胞 (ISCs):修复的原动力
肠上皮细胞的更新、修复和再生在很大程度上依赖于位于隐窝底部的Lgr5+肠道干细胞(ISCs)。
Lgr5作为ISCs的特异性标记,编码一种受体,该受体能够响应Wnt等信号,从而触发ISCs重编程为上皮细胞谱系。ISC的活性受到细胞外信号和旁分泌信号的精密调控,以维持肠道稳态,并在损伤发生时启动适应性分化,从而保障肠道的基本生理功能。
调控胆汁酸水平,恢复干细胞活力
胆汁酸在介导肠肝轴通讯中扮演着关键角色,并整合了调控ISC功能的饮食和代谢信号。近期研究提示,通过减少病理条件下肠道内过量的胆汁酸累积,或可为治疗炎症性肠病(IBD)相关的肠道损伤提供新的干预策略。
例如,FXR激动剂治疗通过抑制肝脏中CYP8B1的表达,进而降低肠肝胆汁酸水平,从而减轻胆汁酸对Lgr5+ ISCs的相关毒性。上述发现提示,基于跨器官代谢调节靶向ISCs有望成为治疗IBD的新型目标。
4
免疫调节
传统抗炎疗法的局限性
传统的抗炎疗法,包括皮质类固醇、5-氨基水杨酸制剂以及新型TNF-α单克隆抗体,一直是炎症性肠病(IBD)和其他免疫介导疾病的标准治疗方法。
虽然这些疗法能有效减轻炎症并缓解症状,但它们在实现长期愈合方面往往力有不逮,尤其是在肠道屏障方面。许多患者随着时间推移出现疗效丧失和疾病复发。例如,尽管抗IL-6抗体疗法在临床试验中对中度至重度克罗恩病(CD)或溃疡性结肠炎(UC)有效,但在一些患者中仍持续发生脓肿和肠穿孔等严重不良反应。
这些疗法通常未能解决根本的上皮功能障碍问题,而这正是维持慢性肠道炎症的关键因素。
新兴策略:直接靶向屏障修复
为了解决这些局限性,人们对靶向更直接参与上皮屏障修复的免疫通路越来越感兴趣。IL-10是一种有效的抗炎和组织再生细胞因子,最近开发的IL-10制剂旨在通过靶向黏膜屏障来增强治疗效果。
另一个有前景的靶点是IL-22,其受体IL-22R在上皮细胞上表达,这使得IL-22和IL-22R成为旨在恢复上皮完整性和增强黏膜愈合疗法的潜在候选者。
IL-22通过刺激AMPs(抗菌肽)和粘蛋白的产生以及ISC(肠道干细胞)的再生,在维持肠道稳态中发挥关键作用。因此,IL-22被认为是屏障修复的潜在治疗方法。
IL-22 激动剂的临床与代谢获益
IL-22融合蛋白激动剂Efmarodocokin alfa (UTTR1147A) 目前正在研究中,用于活动性溃疡性结肠炎(UC)和克罗恩病(CD)的治疗(NCT02749630)。
临床试验表明,UTTR1147A在UC患者和健康个体中都能激活IL-22R信号通路,并改善UC相关的菌群失调。
此外,肠道中IL-22信号的特异性激活可以在代谢紊乱模型中以微生物依赖的方式增强肝脏和全身葡萄糖和脂质代谢稳态。此外,IL-22还对MASLD(代谢功能障碍相关脂肪性肝病)、ALD(酒精性肝病)和饮食诱导的肥胖表现出积极作用。
当外源性给药时,IL-22通过其在肠上皮细胞(IECs)而非肝细胞上的受体发挥治疗作用,然后激活STAT3并抑制WNT–β-catenin信号传导以减少吸收性肠上皮细胞的数量。
然而,IL-22在肠道屏障维持中的作用仍存在争议,人们担心可能产生致病性的免疫调节作用,例如在结肠组织中介导CXCR2+中性粒细胞的趋化作用,以及增加对IL-23单克隆抗体Ustekinumab的抵抗力。这些观察结果表明,IL-22靶向疗法可能并非总能达到预期的疗效,需要进一步研究以更好地了解IL-22激活的全部影响和潜在副作用。
5
肠道屏障功能增强相关的临床试验进展
前面介绍了目前针对肠道屏障完整性的多种创新疗法,包括药物、微生物疗法、吸附剂、饮食干预和工程益生菌,并同时也探讨了这些疗法在临床验证中的进展和未来面临的挑战。
其实核心要点如下:
创新疗法:多项创新疗法正在进行临床验证,以靶向肠道屏障完整性。
小分子创新 / 微生物疗法
ISM5411:一种新型肠道限制性选择性脯氨酰羟化酶结构域(PHD)抑制剂,通过AI平台开发,已完成I期临床试验。其在肠黏膜修复和免疫调节方面具有双重机制,在IBD模型中显示出显著疗效。
利福昔明-α (Rifaximin-α):通过上调粪便中的TNF-α和IL-17E来调节肠道微环境,增强抗菌防御,有效促进肠道屏屏障修复。
ZED1227:在乳糜泻中,作为转谷氨酰胺酶2抑制剂,显著改善十二指肠黏膜结构,减少上皮内淋巴细胞浸润,通过抑制免疫原性谷蛋白肽中谷氨酰胺残基的脱酰胺化来防止T细胞活化和黏膜损伤。
粪便菌群移植(FMT):健康供体FMT在恢复糖尿病远端对称性多发性神经病变(DSPN)患者肠道屏障功能和减轻全身炎症方面显示出治疗潜力。
工程益生菌:在恢复肠道屏障完整性和维持黏膜稳态方面具有显著治疗潜力,目前研究重点是结合其屏障增强和免疫调节作用,但仍在临床前阶段。
吸附剂:非吸收性、肠道限制性工程化碳珠吸附剂Yaq-001通过改善肠道屏障功能障碍和减少全身内毒素负荷,在肝硬化中显示出临床疗效。
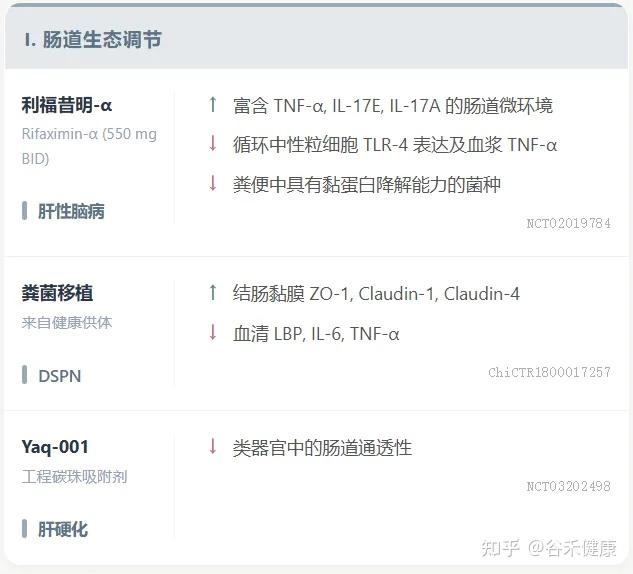

饮食干预
临床研究证实,膳食干预在多种胃肠道疾病中有效。
谷氨酰胺补充剂:显著恢复肠道通透性并缓解感染后肠易激综合征的腹泻 (NCT01414244)。
在肠易激综合征-腹泻、代谢紊乱和轻中度克罗恩病中均显示出益处。

未来挑战
尽管这些进展显示了靶向屏障修复策略的广阔前景,但仍需要在人体试验中全面评估长期安全性和有效性,以推进临床应用。
肠道屏障功能障碍是多种胃肠和肠外疾病(如IBD、MASLD)的关键因素。传统免疫抑制疗法虽能改善症状,但无法根治屏障问题,且副作用明显。新型疗法应结合屏障修复策略,如干细胞再生、微生物疗法、胆汁酸调节、TJ调节剂等。
肠道屏障功能障碍与疾病互为因果,受遗传、环境等因素影响。屏障破坏可引发炎症和多器官功能障碍,导致MASH、IBD、CRC等。同时,疾病微环境反过来又损害屏障,形成恶性循环。
免疫抑制疗法可能抑制黏膜愈合,加剧微生物失衡。需深入研究肠道细胞间通讯,以确定有效治疗靶点。
独立于免疫抑制的屏障防御和修复是治疗肝肠疾病的重要目标。个性化治疗策略可能更有效。增强肠道屏障完整性在MASLD预防和逆转中潜力巨大。未来人体研究至关重要。
恢复肠道屏障是治疗肝肠疾病的重要机遇。新生物技术、再生医学和微生物组研究有望重塑胃肠道治疗格局。
主要参考文献:
Zhang Y, Liu Y, Liang X, Wen Y, Zhao J, He Y, Xie Q, Xie C. Intestinal barrier in chronic gut and liver diseases: Pathogenesis and therapeutic targets. Acta Pharm Sin B. 2025 Nov;15(11):5515-5536.
Macura B, Kiecka A, Szczepanik M. Intestinal permeability disturbances: causes, diseases and therapy. Clin Exp Med. 2024 Sep 28;24(1):232.
Farré R, Vicario M. Abnormal Barrier Function in Gastrointestinal Disorders. Handb Exp Pharmacol. 2017;239:193-217.
Brandl C, Bucci L, Schett G, Zaiss MM. Crossing the barriers: Revisiting the gut feeling in rheumatoid arthritis. Eur J Immunol. 2021;51(4):798–810.
Ramakrishna BS. Role of the gut microbiota in human nutrition and metabolism. J Gastroenterol Hepatol. 2013;28(Suppl 4):9–17.
Bäumler AJ, Sperandio V. Interactions between the microbiota and pathogenic bacteria in the gut. Nature. 2016;535(7610):85–93
谷禾健康
当我们的皮肤被轻微割伤或烧伤时,伤口周围区域可能会变得红肿、发热,甚至伴有疼痛;感冒时,喉咙痛、肿胀;不小心扭伤后,可能会肿胀、疼痛和僵硬…这些都与炎症相关。
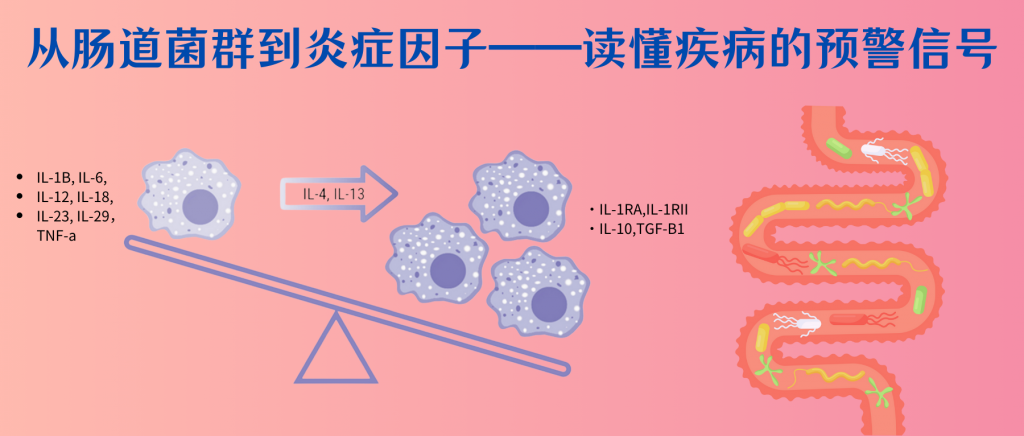
炎症,作为身体对损伤或感染的自然防御机制,是一种复杂的生物学过程,涉及到免疫细胞和多种分子介质的相互作用。它可以帮助身体对抗病原体、清除死亡细胞和促进组织修复。然而,当炎症反应过度或持续时间过长时,它也可能成为许多疾病的驱动因素,包括心脏病、糖尿病、某些类型的癌症,自身免疫疾病等。因此,了解炎症及其相关症状对于早期识别和治疗炎症相关疾病至关重要。
你是否想过,在身体出现炎症之前,其实已经有一些潜在的信号出现?炎症这个过程看似简单,但背后却涉及复杂的免疫系统调控,以及多种炎症因子的参与。值得注意的是,在这些可见的炎症症状出现之前,我们体内的炎症因子可能已经悄然发生了改变。
研究表明,肠道菌群的失调往往是最早的预警信号之一。当肠道微生物群的平衡被打破时,会引发一系列连锁反应:有益菌群(如双歧杆菌、乳酸杆菌)数量减少,条件致病菌和致病菌的比例升高。这种失衡会导致肠道屏障功能受损,使得细菌产物(如脂多糖LPS)更容易进入血液循环。
早期的肠道菌群改变会引起炎症因子水平的显著变化。比如说:血清中促炎因子如TNF-α、IL-1β和IL-6的水平开始升高,而抗炎因子如IL-10的水平则呈现下降趋势。同时,由于有益菌群减少,短链脂肪酸等具有抗炎作用的代谢产物的产生也会减少。这些变化都可能发生在明显的炎症症状出现之前。
这种早期的炎症因子改变往往具有预警作用。例如,在动脉粥样硬化的发展过程中,即使尚未出现明显的斑块形成,血液中的炎症因子水平就已经开始发生变化。
本文将带大家了解炎症因子,具体怎么看,它的高低代表着什么,发挥什么作用,探讨炎症因子作为早期诊断生物标志物的价值,以及肠道菌群与炎症因子之间的复杂联系。通过监测肠道菌群的变化和炎症因子水平的波动,我们可以更早地发现潜在的健康问题,为疾病的预防和早期干预提供重要的时间窗口。

◑ 炎症
炎症是身体对外界损伤、感染或内部损伤的一种自然防御反应,旨在清除有害刺激因子、清除死亡细胞和组织碎片,并启动修复过程。它是免疫系统的一部分,涉及多种细胞类型和分子介质,包括白细胞、血管系统、补体系统和各种炎症因子。
日常可见的炎症反应可分为两类:
不健康的生活方式,如吸烟、不良饮食、过量饮酒、久坐不动、压力、体重增加等,都可能导致慢性炎症。
慢性低度炎症——疾病之源
慢性低度炎症是在没有明显感染的情况下,体内炎症介质水平持续升高。这种炎症状态往往不会引起明显的临床症状,但会长期影响人体健康。
慢性炎症可以攻击全身,并在此过程中增加特定区域某些类型疾病和紊乱的风险,如心脏、大脑、关节、胃肠道等。
炎症是如何被触发的?
炎症的触发是一个复杂的过程,感染、损伤、应激、自身免疫反应、坏死细胞、代谢紊乱…这些都可能是炎症触发的途径。
随着损伤信号的识别,免疫细胞如巨噬细胞和树突状细胞迅速响应,受体的激活促使免疫细胞分泌炎症因子,这些炎症因子是炎症反应中的关键分子。
炎症因子的释放不仅放大了炎症信号,还促进了血管的扩张和通透性增加,使得免疫细胞和分子能够更有效地到达受损部位。这些炎症因子的相互作用和级联反应构成了炎症反应的基础,它们共同协调了机体对损伤和感染的防御机制。
接下来我们来深入认识炎症过程中的核心”信使”——炎症因子。
◑ 炎症因子
炎症因子在疾病发展过程中扮演着关键角色,其水平变化不仅反映了疾病的发展态势,更为疾病的早期诊断和预后评估提供了重要依据。研究表明,炎症因子的变化往往早于临床症状的出现,这种特性使其成为疾病发展的重要生物标志物。
我们先看一下,炎症因子是什么?
炎症因子是一类由免疫细胞和其他细胞产生的特殊蛋白质分子,它们在体内发挥着”信使“的作用。也就是说,炎症因子就像是我们身体内的一支特殊”信号部队“,它们负责在炎症发生时传递各种指令,协调免疫系统的行动。
无论是急性炎症还是慢性炎症,炎症因子都扮演着不可或缺的角色,它们决定着炎症反应的强度、持续时间和最终结果。
炎症因子与细胞因子有什么区别?
炎症因子主要是指那些能够引起或加剧炎症反应的分子,而细胞因子则是一类更广泛的信号分子,包括炎症因子在内的多种类型,比如生长因子、趋化因子等,它们参与调节免疫细胞的功能和相互作用。
炎症因子可以被视为细胞因子的一个子集,也就是那些具有促进炎症反应功能的细胞因子。
在疾病发展的早期阶段,即使尚未出现明显的临床表现,体内的炎症因子已经开始发生显著变化。
例如,促炎因子TNF-α和IL-6的水平会逐渐升高,而抗炎因子IL-10的水平则呈现下降趋势。这种变化具有明显的时序性和渐进性特征,为疾病的早期预警提供了可能。同时,C反应蛋白(CRP)等急性期蛋白的轻微升高,也常常预示着潜在的健康问题。
主要的促炎因子和抗炎因子有哪些?
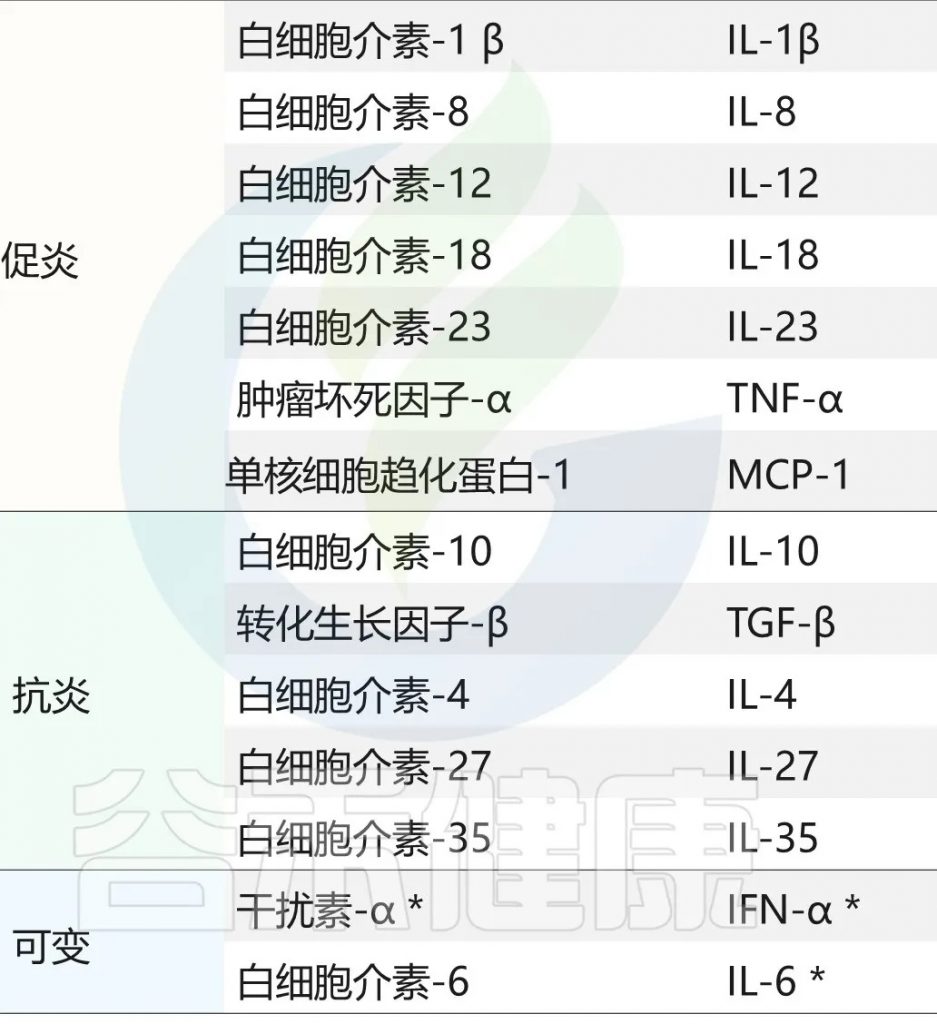
doi:10.3390/ijeph17207618
随着疾病的进展,炎症因子会呈现出不同的变化模式。在急性疾病中,IL-1β和TNF-α等促炎因子会快速升高,同时伴随着中性粒细胞趋化因子的显著增加。而在慢性疾病中,则常常表现为持续的低度炎症状态,多种炎症因子维持在较高水平,形成促炎和抗炎因子的失衡状态。
不同类型的疾病中,炎症因子往往表现出特征性的变化。以心血管疾病为例,患者体内的CRP和IL-6水平通常持续升高,同时伴有粘附分子表达的增加。在自身免疫性疾病中,TNF-α和IL-17的显著升高,以及特异性自身抗体的出现,往往是重要的诊断依据。而在代谢性疾病中,脂联素水平的改变和瘦素抵抗的出现,则与胰岛素抵抗密切相关。
炎症因子作为生物标志物在临床实践中有多重价值
在诊断方面,它可以用于疾病的早期筛查、鉴别诊断和病情评估;
在预后评估方面,能够预测疾病的进展趋势,评估并发症风险,并监测治疗效果;
在治疗指导方面,帮助医生选择合适的治疗方案,调整用药剂量,评价治疗效果。
了解炎症因子的作用机制不仅有助于我们更好地理解炎症过程,也为开发新的治疗策略提供了靶点,通过调节炎症因子的活性来治疗炎症性疾病,那么,如何有效地调节这些炎症因子的活性呢?近年来,越来越多的研究开始关注肠道菌群在调节炎症因子和免疫反应中的作用。
在疾病发展的早期阶段,即使在临床症状尚未显现时,身体内部可能已经发生了微妙的改变。肠道菌群作为人体最大的微生态系统,常常能够最早感知这些变化,并通过多种途径向身体发出预警信号。
我们以前的文章也有很多提及,肠道菌群不仅能影响局部的免疫反应,还能通过多种机制影响全身性的炎症状态,比如说:
代谢产物:
肠道菌群通过发酵膳食纤维产生短链脂肪酸(SCFAs),如乙酸、丙酸和丁酸,这些代谢产物能够调节免疫细胞的功能,特别是对调节性T细胞(Treg)的分化和功能具有重要作用,进而影响炎症因子的产生。
免疫细胞调节:
肠道菌群能够影响树突状细胞(DCs)和巨噬细胞的成熟和功能,这些免疫细胞在调节炎症因子的表达和释放中起着关键作用。
肠道屏障功能:
维持健康的肠道菌群有助于维持肠道屏障的完整性,防止病原体和有害物质的入侵,从而减少炎症因子的激活。
抗炎和促炎平衡:
某些肠道菌群成员能够促进抗炎细胞因子如IL-10的产生,而抑制促炎细胞因子如TNF-α和IFNγ的表达。
肠道菌群发出的预警信号,主要通过代谢产物、免疫细胞调节、肠道屏障功能维持以及抗炎促炎平衡等途径实现,而炎症因子则是传递这些预警信号的关键分子。
通过监测这些早期的分子标志物,我们可以在疾病发展的早期阶段进行干预,从而更有效地预防和控制炎症反应的发生发展。
接下来,我们将深入探讨常见的炎症因子(炎症标志物),包括C反应蛋白、粪便钙卫蛋白、TNF-α、IL-1、IL-6、IL-10、IL-17、IL-22等,这些炎症因子的具体功能,肠道菌群与这些炎症因子的关联等。
C反应蛋白(CRP)是一种重要的由肝脏产生的急性期反应蛋白,是临床上最常用的炎症标志物之一。比如,广泛使用于IBD筛查和评估疾病活动性、临床复发和治疗反应。
什么时候需要做CRP测试?
——急性
CRP水平在体内发生炎症或组织损伤时会急剧上升。比如说,感冒时身体的免疫系统会被激活,C反应蛋白的产生显著增加,反映了身体正在经历一种急性炎症反应,通常伴随着其他症状,如发热、 寒战、喉咙痛、全身乏力、呼吸急促、恶心呕吐等。一般出现这种情况的时候医生会要求抽血,看看CRP指标。
——慢性
除了感染性疾病的早期诊断外,CRP水平的升高还可用于判断疾病的严重程度和监测治疗效果。例如,在风湿性关节炎、狼疮、炎症性肠病、血管炎、哮喘等慢性疾病中,CRP常常作为一种重要的生物标志物,帮助医生评估病情进展及治疗反应。也可用于术后并发症监测。
例如,血液中高水平的hs-CRP与心脏病发作的风险增加有关。如果hs-CRP水平较高,心脏病发作的人更有可能再次心脏病发作。
CRP水平的高低意味着什么?
一般来说,健康人的血液中CRP含量很低。

CRP与肠道菌群有什么关联?
一项研究中,在CRP血浆水平升高的肥胖小鼠中,Akkermansia muciniphila的比例下降。
Phascolarctobacterium属成员的丰度与较低水平的CRP有关。这种关系可能会解释为什么该菌属比例的下降与结肠炎症有关:Phascolarctobacterium是丙酸的生产者,丙酸是一种短链脂肪酸,通过抑制促炎调节因子NFκB的活性来抑制促炎级联反应。同样,粪杆菌的丰度与CRP水平呈负相关。
因此,CRP是一种下游炎症标志物,可以通过特定肠道微生物的抗炎代谢产物的作用下调。
对BMI超过25的健康受试者的基线血清和微生物群数据的评估表明,CRP水平较高的受试者乳杆菌属和双歧杆菌属的细菌丰度明显较低,但大肠杆菌属和拟杆菌属的丰度较高。
钙卫蛋白是一种胞浆蛋白复合物,在中性粒细胞中组成性表达,并在肠道炎症期间迁移至肠粘膜时释放。
在健康状况下,钙卫蛋白具有免疫调节功能,对免疫防御至关重要;在慢性炎症性疾病中,钙卫蛋白可通过细胞因子受体结合和活性氧的产生来促进疾病进程。
钙卫蛋白——非侵入性生物标志物
自20世纪80年代钙卫蛋白的鉴定和表征以来,粪便钙卫蛋白成为一种经过显著验证的非侵入性生物标志物,可用于评估肠道炎症,是短期复发和IBD炎症活动的可靠预测指标。
举个例子:一个人去医院看病,主诉反复腹泻、腹痛、体重下降,有慢性腹泻的病史,过去几个月中症状有所加重,医生考虑他可能与肠道疾病相关,为了确诊,医生可能会开具粪便钙卫蛋白检测进行辅助诊断,帮助区分炎症性肠病和肠易激综合征。
钙卫蛋白水平高低意味着什么?
粪便钙卫蛋白与肠道菌群有什么关联?

一项来自TREND队列的大样本老年人研究发现,在高钙卫蛋白组与低钙卫蛋白组中,几种促炎肠道微生物属显著增加,而产短链脂肪酸菌减少。
在粪便钙卫蛋白升高的组中,几种产短链脂肪酸菌属(如梭菌属、Blautia、Turicibacter)的丰度降低与IBD、帕金森和心血管疾病中的许多发现一致。这些产生SCFA菌减少可能是几种疾病机制的关键,因为SCFA可以防止病原体,调节代谢,内分泌和免疫功能,并影响药物代谢和吸收。
发炎的肠道中较高水平的氧气可以允许肠杆菌科的有氧呼吸,同时抑制专性厌氧菌、拟杆菌和产生SCFA的梭菌的生长。此外,通过与富含脂肪的西方饮食的相互作用,含有促炎脂多糖作为膜成分的革兰氏阴性菌可能会引发炎症和粪便钙卫蛋白水平升高。
血清中,高粪便钙卫蛋白组IL-17 C、CCL 19和毒性代谢产物硫酸吲哚酚升高。这些变化部分由肠道微生物群介导。此外,高粪便钙卫蛋白组显示BMI增加,心脏病发作和肥胖的患病率较高。
在免疫疗法治疗肝细胞癌患者期间,粪便钙卫蛋白显示出与阿克曼氏菌与肠杆菌科比例和肠道微生物群α多样性相反的时间演变,但与zonulin-1和LBP相似。
肿瘤坏死因子-α(TNF-α)是驱动炎症的关键细胞因子。TNF-α在炎症级联反应的上游启动阶段,是Th1信号通路关键的细胞因子,在人体免疫系统中扮演着“紧急呼叫器”的角色。
该分子水平升高与胰岛素抵抗和葡萄糖耐受不良相关,因为TNF-α能够激活各种信号传导途径,包括mTOR途径,使其成为代谢疾病发展中的关键分子。
TNF的“好”
TNF诱导睡眠,并增加非快速眼动睡眠。因此,当我们晚上想入睡时,它提高一些是很好的。TNF在健康人中在夜间自然升高。
TNF是一种直接的脂肪克星,它导致脂肪细胞中的胰岛素抵抗,但也导致肌肉细胞中的胰岛素抵抗。这意味着葡萄糖不能进入这些细胞。
TNF通过抑制食欲素来抑制食欲。因此,高水平的TNF会使你吃得更少,并抑制葡萄糖进入脂肪细胞,从而使你变瘦。如果你减少TNF,会变得更饿,储存更多的脂肪。因此,抗TNF治疗导致体重增加并不奇怪。
TNF的“坏”
TNF通过抑制食欲素让人感到疲劳,降低情绪并降低认知和身体表现。
注:食欲素是一种非常重要的神经递质,对许多身体功能。食欲素在记忆获得和巩固以及长期记忆强化中起着许多关键作用。因此,如果你有炎症升高,它会损害你的认知能力。
TNF可以通过破坏线粒体造成持久的伤害。
TNF还可以降低甲状腺激素,导致“低T3综合征”。它也可以降低睾酮(趋势,但不显著)。
TNF会减缓伤口愈合,这意味着需要更多的时间从运动/受伤中恢复。
TNF可诱导“肠漏”。
长期升高的TNF-α也会扰乱昼夜节律并导致白天疲劳。
与TNF-α相关的疾病:炎症性肠病(IBD)、类风湿性关节炎(RA)、2型糖尿病、肥胖、系统性红斑狼疮、神经退行性疾病、精神分裂症等。
哪些肠道菌群与TNF-α呈负相关?
哪些肠道菌群与TNF-α呈正相关?
TNF-α在不同类型的感染中表现出不同的作用,例如在真菌感染中,TNF-α通过调节Th17/Th2和中性粒细胞/嗜酸性粒细胞的平衡来影响炎症反应。而在克罗恩病患者中,TNF-α的诱导活性与某些革兰阴性菌有关。
肠道菌群通过其代谢产物,如短链脂肪酸,可以影响TNF-α的水平。短链脂肪酸能激活AMPK,减少FIAF(诱导型脂肪因子)的产生,从而抑制脂多糖的活性,进而影响TNF-α的产生。
★ 如何减少TNF-α?
生活方式:运动(骑车)、瑜伽、太极拳、睡眠不足后的小睡、谈恋爱等。
饮食:沙丁鱼、蘑菇、大蒜、蜂蜜、大豆、苦瓜、十字花科蔬菜(西兰花、花菜)、水果(红树莓、蓝莓、黑醋栗果、李子、桃、荔枝、巴西莓)等。
其他补充剂:姜黄素、鱼油、肉桂、精氨酸、甘氨酸、组氨酸、铬、植物淄醇、黄芪、青蒿素、柠檬苷、辅酶Q10、紫锥菊、葡萄糖胺、小檗碱、黄芩、银杏、南非醉茄、槲皮素等。
IL-1是一种重要的促炎细胞因子。促进炎症细胞的招募和活化,增加血管通透性,吸引免疫细胞到达炎症部位。参与调节T细胞和B细胞的活化、增殖和分化,促进发热反应、疼痛、肿胀。同时也可以诱导某些细胞类型的凋亡,参与组织修复和再生。
与IL-1相关的疾病:自身免疫性疾病、感染性疾病、心血管疾病、代谢性疾病、神经退行性疾病等。
哪些肠道菌群与IL-1呈负相关?
哪些肠道菌群与IL-1呈正相关?
肠道菌群代谢产物,短链脂肪酸,通过激活AMPK和抑制PGC-1α,进而影响IL-1的产生。
◆ IL-1β
IL-1β是IL-1家族的重要成员之一,同属于促炎性细胞因子,IL-1β常作为特异性炎症标志物。
IL-1β的“好”
IL-1β是诱导睡眠机制的一部分,并增加非快速眼动睡眠,IL-1β具有昼夜节律,在睡觉前升高,但如果你是夜班工人,它会转移到白天。
IL-1在记忆功能中发挥作用,所以人需要一定水平的IL-1,但要“尽可能低”。
在啮齿动物中,IL-1β增加了催产素和加压素的释放。
IL-1β增加NGF。
NGF和NT-3在刺激神经突起生长方面有些独特,这是NGF、BDNF或NT-4无法单独完成的。IL-1β还会增加GDNF,这再次刺激神经突起的生长。
此外,它可以通过增加bFGF来增加多巴胺促进神经元。
IL-1β的“坏”
如果你的IL-1水平升高,那么这将影响你的情绪、认知功能、清醒程度和动力,需要关注降低慢性炎症。
IL-1与抑郁症有关,可能通过减少雌激素合成、增加黄体酮分解影响情绪,同时降低雄性激素水平。压力可通过IL-1b介导的途径导致抑郁,同时损害认知能力,影响学习和记忆,这可能与脑源性神经营养因子(BDNF)的减少有关。
IL-1通过抑制食欲素引起疲劳,并与焦虑、HPA轴激活、IBS、认知缺陷和多种慢性疾病相关。
IL-1β与低睾酮水平相关,可能通过影响甲状腺激素和皮质醇水平降低性能。它还抑制胰岛素释放,影响血糖水平,降低乙酰胆碱水平,并在肠道中抑制胃酸,可能导致幽门螺杆菌感染失控。
IL-1β与肠道菌群的关联
★ 如何减少IL-1β?
饮食:蔬菜和水果(花青素)、芹菜、生姜、十字花科蔬菜(西兰花、花菜)、燕麦鱼(虾青素)等。
益生菌:植物乳杆菌等
其他补充剂:维生素A、VB2、穿心莲、β-葡聚糖、葡萄籽提取物、水飞蓟素、红景天、黄连素、小檗碱、葡萄籽提取物、鱼油、茶多酚、蜂蜜、黄芩苷、迷迭香酸、丹参、锌(如果缺乏)、苜蓿、白术、紫苏等。
IL-6是与系统性炎症相关的炎症因子,调节免疫反应,参与激活和分化T细胞,促进B细胞分化和抗体产生,促进急性期蛋白的合成,促进中性粒细胞的募集,参与炎症性疾病的发生发展。
IL-6在中枢神经系统中也发挥作用,参与调节神经炎症和情绪反应。
IL-6可以通过两种方式激活细胞:
IL-6在你生病和运动后升高,特别是有氧运动。如果运动增加炎症标志物,那么怎么理解运动是健康的?
当你运动时,肌肉会释放IL-6,这是抗炎的。然而,当免疫细胞(巨噬细胞)释放它时,它是促炎性的。
IL-6还抑制Th1细胞,同时诱导Th2细胞,因此对Th2占主导地位的人来说情况更糟。它还增加了B细胞,这是产生抗体并导致过敏和自身免疫的原因。
不易患自身免疫性疾病的人也可能患有IL-6升高。这是与现代文明病有关的细胞因子。最常见的原因可能是肥胖。
IL-6升高的其他常见原因可能是慢性压力、睡眠太少、吃得太多(特别是吃太多糖或精制食品)、吸烟、过量酒精、运动过多。
IL-6的“坏”
IL-6水平在几乎所有疾病状态中都升高。
它减少了Treg细胞,这反过来又阻碍了我们对摄入的蛋白质产生耐受性的能力-引起过敏。它还增加了中性粒细胞的产生,这是炎症。
IL-6是中年后期认知能力下降的一个很好的预测因子。
IL-6可能会导致情绪恶化,绝望的感觉。IL-6还与暴力自杀、冲动和避免单调乏味相关。
IL-6导致血糖水平升高,增强了应激激素(CRH)对肠道粘膜的影响,导致IBS。IL-6可导致肠漏。
IL-6通过抑制(或超甲基化)对GABA正常工作重要的基因(GAD 67)而导致精神分裂症。
IL-6是CRP最有效的诱导剂,CRP就是我们前面讲的炎症标志物。
它可以通过增加IgG和IgM抗体产生和恶化食物敏感性和自身免疫问题。
IL-6也会导致皮肤问题。IL-6在患有皮肤真菌变色菌的人中升高。IL-6还增加了Th 22细胞,这破坏了皮肤微生物平衡。
IL-6的“好”
IL-6如果升高大多是不好的,但短暂的峰值可能是有益的。
TNF和IL-1β增加IL-6,但IL-6反过来抑制这两种细胞因子,其比IL-6本身更有害。在这种方式下,它是一种抗炎。抑制TNF,分解脂肪细胞并降低胰岛素抵抗。
IL-6在一些细菌、病毒和真菌感染中起保护作用。
IL-6的正常水平是什么?
在健康受试者中,IL-6血液水平几乎检测不到,范围在2-6 pg/ml之间。另一项研究提到,健康人的平均水平为0.5 pg/ml 。
抑郁症患者的IL-6水平比健康人高约1.78 pg/ml。
在患有风湿性关节炎的人中,水平可以增加到1000倍(不常见)。在败血症中,这是非常危险的,它可以增加到一百万倍。
IL-6与肠道菌群有什么关联?
高脂饮食会削弱粘液层的完整性,增加血液中LPS的水平,通过TLR-4传递,导致血液中TNF-α、IL-1、IL-6和PAI-1(纤溶酶原激活抑制剂-1)的水平升高,引起系统性炎症。
坏死的肠道粘膜细胞会刺激巨噬细胞产生IL-6。
★ 如何减少IL-6?
生活方式:情绪积极、睡眠不足后的小睡、听音乐等。
饮食:地中海饮食、燕麦、多酚、坚果(腰果)、豆类、橄榄油、蔬菜、蓝莓、红树莓、蜂蜜等。
其他补充剂:鱼油、植物甾醇、螺旋藻、维生素B2、VB12、VE、镁、铬、锌、精氨酸、组氨酸、甘氨酸、银杏、维生素E、黄芩苷、乳铁蛋白、葡萄籽提取物、黄芪、丹参、紫锥菊、迷迭香酸、牛至、鼠尾草等。
IL-10是一种重要的抗炎因子。
它能抑制促炎细胞因子的产生(如TNF-α、IL-1β、IL-6),降低抗原呈递细胞的活性,减少炎症细胞的募集,有助于炎症反应的消退。抑制Th1细胞的活化,从而调节Th1/Th2平衡。
IL-10的“好”
在自身免疫性疾病(如类风湿性关节炎、系统性红斑狼疮等)和移植免疫中,IL-10有助于防止自身反应性T细胞的活化,减少对移植器官的排斥反应。
IL-10抑制COX-2,这是参与偏头痛,疼痛和炎症。COX-2通常被非甾体抗炎药如阿司匹林和布洛芬阻断。
通过抑制肥大细胞,它抵消了这些细胞在过敏反应部位的炎症作用。
IL-10通过减少暴饮暴食并降低下丘脑(控制食欲的腺体)中的胰岛素和瘦素抵抗(通过抑制细胞因子、Nf-kB和ER应激)来减少肥胖。
IL-10的“坏”
IL-10可以阻断对病毒感染的反应,甚至直接增加病毒蛋白的产生,比如说,在慢性疲劳综合征中,IL-10增加;慢性感染的丙型肝炎患者在遗传上倾向于高IL-10产生,对治疗的积极反应较低。
平衡 IL-10 的“好”与“坏”
Th 1免疫系统,特别是CD 8 + T细胞和IFN γ,是人体对抗癌症的机制的一部分。阻断IL-10显示出作为癌症治疗的前景。然而,IL-10在某些情况下也通过促进细胞毒性T细胞活性和IFN-γ产生而发挥保护性抗癌作用。
重要的是认识到全身和癌组织中的IL-10水平之间存在差异。如果IL-10在正常组织中处于健康水平,在癌组织中处于低水平,那么这是比较理想健康的。
血液中IL-10的水平并不一定代表肠道或其他组织中的水平,但通常存在相关性。
在现代环境中,高IL-10水平可能比低IL-10水平更好,因为可以对抗细菌感染。
IL-10与肠道菌群有什么关联?
乳杆菌、双歧杆菌、普氏粪杆菌、某些产丁酸菌:能促进IL-10的产生,从而改善肠道炎症。
益生菌通过促进调节性T细胞(Treg)的分化来增加IL-10的产生。
★ 如何增加IL-10?
生活方式:运动、晒太阳、冥想等。
饮食:芝麻油、肉桂、大蒜、辣椒素、甘草、芥末等。
益生菌:植物乳杆菌、布拉氏酵母菌、干酪乳杆菌、枯草芽孢杆菌等。
益生元:阿拉伯半乳聚糖
其他补充剂:姜黄素、表儿茶素EGCG、Boswellia、橄榄苦苷、褪黑素、白藜芦醇、维生素D3、辅酶Q10、植物甾醇等。
IL-17(白细胞介素-17)是一种重要的促炎细胞因子,在自身免疫和炎症反应中发挥关键作用。IL-17能够促进多种细胞产生炎症因子,如IL-1β、TNF-α和IL-6,促进中性粒细胞募集,加剧炎症反应。
在肿瘤微环境中具有双重作用,既可以促进抗肿瘤免疫反应,也可能促进肿瘤的侵袭和转移。
与自身免疫性疾病、肿瘤、感染性疾病等相关。
IL-17水平的变化可以作为某些疾病预后的生物标志物。
IL-17 C属于IL-17细胞因子家族,由上皮细胞而不是免疫细胞产生。它作为对上皮损伤的快速局部自分泌反应,促进抗微生物保护反应和肠屏障维持。
分节丝状菌(SFB):通过其鞭毛蛋白促进其产生。
青春双歧杆菌(Bifidobacterium adolescentis):能够诱导肠道Th17细胞积累,从而促进IL-17的产生。
IL-22(白细胞介素-22)是IL-10家族的重要成员,主要参与组织修复和黏膜免疫。
具体来说,IL-22在组织损伤后的修复和再生过程中起着重要作用,特别是在肝脏和肠道等上皮组织中。
它是一种促炎细胞因子,但它也能发挥抗炎作用,特别是在抑制过度的炎症反应和促进组织稳态方面。
IL-22能够增强上皮细胞的抗菌肽表达,从而增强机体对细菌和病毒感染的防御能力。
在急性结肠炎中具有保护作用,但在慢性结肠炎中,IL-22与IL-17A协同介导致病性。
分节丝状菌(SFB):通过鞭毛蛋白参与调节其产生,诱导SAA产生来促进IL-22的分泌,刺激树突状细胞促进IL-22的产生。
IFNγ(干扰素-γ)是一种重要的细胞因子,它在免疫反应、抗感染、抗肿瘤和调节免疫应答中扮演着重要角色。
IFNγ是Th1细胞介导免疫反应的关键因子,它能够促进Th1细胞的分化,并抑制Th2细胞的分化,从而调节Th1/Th2平衡。
在某些自身免疫性疾病中,如多发性硬化症和类风湿性关节炎,IFNγ的过度产生与疾病的发生和发展有关。
肿瘤:IFNγ在肿瘤免疫监视中起关键作用,但其在肿瘤微环境中的作用可能更为复杂,包括促进肿瘤免疫逃逸。
TGF-β(转化生长因子-β)是一种多功能细胞因子,在调节免疫反应和促进Tregs细胞的增殖中起作用。
肠道菌群的变化可以影响TGF-β的产生,进而影响Tregs和Th17细胞的平衡。
丁酸梭菌,诱导树突状细胞中TGF-β信号传导,进而诱导调节性T细胞(Treg)的产生。
以上我们了解一些常见的炎症因子的功能,与肠道菌群的关联,基于这一认识,我们将进一步探讨这种异常变化如何表征不同疾病的风险,以及如何通过早期干预来预防和治疗相关疾病。
在探讨肠道菌群与炎症因子异常之间的关系时,我们不得不关注它们在多种慢性疾病中的作用,尤其是那些与炎症密切相关的疾病。
这里我们通过举例分析几种典型疾病(抑郁症、心血管疾病、炎症性皮肤病)中肠道菌群与炎症因子的动态变化特征,更深入地理解肠道菌群如何作为炎症反应的早期指标,为疾病的早期预防和个体化治疗提供科学依据。
抑郁症是一种常见的精神障碍,其特征为情绪低落、食欲不振和高自杀率。研究表明,炎症反应在抑郁症的发病中起着关键作用,炎症水平的紊乱可增加抑郁症的发病率。
调节炎症水平可能是肠道菌群影响宿主健康的途径之一。通过调节特异性肠道菌群和炎症反应,可以实现抑郁症的早期预测、预防和个性化治疗。
➤ 抑郁症
一项研究通过孟德尔随机化分析方法确定了15个与抑郁症相关的肠道菌群分类群和4种细胞因子,并证实了某些细胞因子在肠道菌群对抑郁症影响中的介导作用。
研究发现,Romboutsia、Intestinimonas、瘤胃球菌UCG 011等对抑郁症具有保护作用。
Romboutsia,Ruminococcaceae UCG 011,Intestinimonas都是产丁酸菌,临床研究发现,Romboutsia的丰度与帕金森病患者的抑郁状态呈负相关。同样,应激耐受性较差的小鼠含有较少的Romboutsia,导致对抑郁症的易感性更高。
Intestinimonas与IL-10水平呈正相关,与能够修复肠道损伤的促炎细胞因子DAO、D-LA呈负相关。
瘤胃球菌科UCG 011能够影响促炎细胞Th17的分化,减少促炎细胞因子IL-17的分泌,改善慢性炎症,缓解疼痛和焦虑抑郁症状。
毛螺菌FCS 020、链球菌、Marvinbryantia等被确定为抑郁症的危险因素。
毛螺菌科FCS 020能够诱导炎症反应,与血清脂多糖和细胞因子产生正相关,并且在自闭症儿童中丰度增加。
链球菌是一种与LPS密切相关的促炎性肠道菌群。
Marvinbryantia在慢性轻度应激大鼠中数量增加。
ADA 和 IL-18 R1是抗抑郁的保护性细胞因子。
VEGF_A和TNFSF 14是促进抑郁的危险因子。
抑郁症的严重程度与VEGF_A呈正相关。这可能与VEGF介导的血脑屏障功能障碍有关。许多抗抑郁药物通过调节VEGF_A发挥抗抑郁作用,因此VEGF_A对抑郁症的治疗效果具有预测作用。
TNFSF 14是TNF受体超家族的成员,其通过激活NF-κB信号通路促进促炎细胞因子的产生。TNF损害神经递质的合成并降低5-羟色胺的可用性,导致神经毒性代谢物的积累,这反过来会导致神经系统损伤并导致抑郁症。
循环细胞因子介导的肠道菌群对抑郁症的影响
doi.org/10.1007/s13167-024-00379-z
➤ 重度抑郁症(MDD)
一项研究探讨了首发未经治疗的重度抑郁症(MDD)患者的肠道菌群组成与炎症因子和认知功能之间的关系。
研究发现MDD患者的肠道菌群多样性显著降低,某些菌群如拟杆菌属、Alistipes增加;一些菌群如梭菌科、Turicibacter减少。这些肠道菌群的改变与炎症因子(如IL-6、CRP)水平升高以及认知功能障碍(如执行功能、记忆力下降)显著相关。
在MDD患者中,拟杆菌科和拟杆菌属均与hsCRP、CCT1、CCT2呈正相关。
拟杆菌属是嗜酸性和革兰氏阴性的,是存在于人类胃肠道系统中非常丰富的细菌。拟杆菌属的某些菌株由于其许多能力对人类健康有利。然而,当人们经历压力,如休克,虐待,失去家庭成员等。拟杆菌属物种的某些菌株如脆弱拟杆菌可分泌脂多糖(LPS)、细菌淀粉样蛋白、内毒素(如脆弱溶素)和外毒素。这些神经毒素刺激各种细胞类型中TNF-α、IL-1β、IL-8、IFN-γ、CXCL 8和其他炎性细胞因子和趋化因子的释放,导致对这些细菌分子病原体的炎症反应。
活化的炎症反应可以破坏肠粘膜屏障以及血脑屏障,并且进一步地,它们可以活化CNS的小胶质细胞。发现活化的小胶质细胞参与促炎细胞因子的分泌,包括IL-1β、IL-6、TNF-α和TGF-β,从而有助于患有神经障碍的个体中认知障碍的发展。
Alistipes与IL-6呈正相关,与延迟记忆、总分和标准化评分呈负相关。
Alistipes也是促炎细菌,有研究表明,Alistipes属以IL-6依赖性方式参与促进炎症和肿瘤发生。本研究中Alistipes与IL-1、IL-6无显著相关性,与TNF-α呈负相关。因此,Alistipes可以通过炎症反应以外的其他方式加重认知障碍。
扩展阅读:
肠道重要菌属——另枝菌属(Alistipes),调节炎症情绪等的潜力菌
Clostridiaceae、Turicibacterae、Turicibacter与IL-1β和IL-6均呈负相关。
➤ 伴有厌食症的重度抑郁症
与健康个体相比,患有厌食症重度抑郁症患者具有不同的肠道微生物群,具有更高的CRP水平。伴厌食症的抑郁症患者中Blautia含量更丰富,并与CRP、HAMD评分和厌食症呈正相关。肠道菌群可能通过炎症因子CRP影响MDD和厌食症。
粪杆菌、拟杆菌、Roseburia和副拟杆菌与厌食、HAMD评分和CRP水平呈负相关。
短链脂肪酸通过干扰NF-κB通路发挥抗炎作用,补充短链脂肪酸可以减少炎症并缓解抑郁症状。
因此,MDD患者中产短链脂肪酸菌的减少可能会通过炎症反应引起抑郁症。这些炎症分子干扰食欲并促进厌食。下丘脑的炎症通过上调5-羟色胺的可用性,并刺激其在下丘脑中的信号传导途径而引起厌食症。
总的来说,由于肠道生态失调引起的免疫失衡是抑郁症发病的早期风险指标。这为利用无创肠道菌群检测对抑郁症进行早期筛查、及时预防和个性化治疗提供了依据。通过将非侵入性肠道菌群检测与现有方法(如心理问卷)相结合,可以共同预测和评估患抑郁症的风险。
心血管疾病(CVD)对人类健康构成了巨大的威胁。炎症是心血管疾病病理过程的普遍原因,包括免疫细胞的激活、积聚和炎症因子的释放。
➤ 动脉粥样硬化(AS)
动脉粥样硬化是CVD的病因之一。脂质沉积和持续性血管炎症被认为是动脉粥样硬化斑块进展的两个核心因素。
一项基于粪便宏基因组学、临床测量和流行病学的研究表明,日常饮食通过影响肠道微生物群促进动脉粥样硬化的形成,而抗炎反应功能障碍可能是核心过程。
脆弱拟杆菌的增加减少了乳酸杆菌的丰度,增加了脱硫弧菌科的丰度,导致葡萄糖或脂质代谢功能障碍,加重炎症反应。
循环中低密度脂蛋白含量显著增加,斑块中CD 36、F4/80增加,促进主动脉斑块的形成和动脉粥样硬化的进展。
衰老是动脉粥样硬化发展的另一个关键危险因素。
在炎症反应加重的衰老个体中,参与花生四烯酸(AA)代谢途径的组分(如20-HETE、PGF 2 α、花生四烯酸和LTB 4)显著增加。因此,建议肠道微生物群与动脉粥样硬化可能通过“肠道微生物群-代谢物-局部炎症-动脉粥样硬化”轴联系起来。
花生皮提取物(PSE)可降低动脉粥样硬化小鼠的血清TC和LDL-C含量,增加HDL-C含量,从而减缓动脉粥样硬化斑块的形成。
天麻素可以调节肠道微生物种类和丰度,降低促炎细胞因子TNF-α和IL-6的水平,增加抗炎因子IL-10的水平。
通心络干预是一种传统中药,通过增加肠道中益生菌的水平来改变斑块稳定性,从而增加有益代谢产物的含量,如反式阿魏酸,这可以抑制斑块中NLRP 3相关的炎症通路并稳定斑块。
乳酸杆菌的管理减少了来自受干扰的肠道微生物群的毒素,并增加了SCFA的水平,抑制了动脉粥样硬化的进展。
黄酮类化合物通过抑制TMA裂解酶而具有治疗冠心病的作用。
燕麦纤维可以防止动脉粥样硬化的恶化,阻断了TLR 4信号通路,降低了NF-κB p65的表达,并通过影响肠道微生物源性异丁酰-L-肉碱、戊酰肉碱、1-甲基鸟苷和2-甲基鸟苷来维持肠粘膜屏障的完整性。
鱼油衍生的长链单不饱和脂肪酸(LCMUFA),通过降低厚壁菌门和拟杆菌门的比例,增加肠道中阿克曼氏菌的丰度,上调SCFA以及SCFA诱导的一些胰高血糖素样物质来维持肠道微环境的平衡,降低了血清中炎性细胞因子的水平,抑制了动脉粥样硬化病变的进展。
支链氨基酸(BCAA)补充剂可以通过调节炎症来减轻动脉粥样硬化,包括减少巨噬细胞浸润、降低炎症因子的血清水平和抑制炎症相关信号通路。
➤ 慢性心力衰竭(CHF)
对53名慢性心力衰竭患者和41名对照成员的粪便宏基因组分析显示,慢性心力衰竭患者的肠道微生物群组成和代谢特征与对照组有显著差异,提示肠道微生物群功能障碍与慢性心力衰竭密切相关。
当肠道微生物群被破坏时分泌的LPS,以TLR4依赖性方式减少ZO-1紧密连接(TJ),并诱导肠上皮TJ的明显变形,导致肠屏障完整性的破坏。
心力衰竭患者会发生慢性全身炎症反应,血浆中几种促炎细胞因子的水平与疾病的严重程度和预后相关。肠道微生物群破坏和细菌产物(如LPS)易位到血液中是炎症过度状态的主要因素。
LPS是最强的促炎介质之一,可诱导心力衰竭患者血清中TNF-α、IL-1和IL 6的释放。另一方面,LPS通过TLR 4直接诱导心肌细胞损伤。
研究表明,晚期心力衰竭患者心脏中TLR 4的表达增加,与心脏炎性损伤高度相关,而抑制TLR 4可缓解心力衰竭的进展。
然而,一些研究表明,通过适当调节肠道微生物群,从而减轻或逆转心室重塑,可以减轻炎症。
目前有报道称,一些中药,如芪荔强心(QL),可稳定心力衰竭后的肠道菌群,通过减少炎症因子(如NLRP 3、IL-1β和TNF-α)的产生来抑制心肌纤维化和心脏重塑。
严重的心力衰竭总是伴随着肠道功能障碍。因此,重视肠功能的维持,可能会改善心力衰竭患者的预后。
肠道微生物群失衡会加重心血管疾病
doi.org/10.3390/nu15030607
➤ 高血压
• 高盐饮食与肠道菌群失衡:
高盐饮食导致乳杆菌属减少,这些菌株能够产生肠道吲哚-3-乳酸,进而影响Th17细胞的活化。
• Th17细胞与炎症因子IL-17A:
Th17细胞的活化与IL-17A的产生增加有关,IL-17A通过上调肾脏中的钠转运蛋白(如钠氢交换蛋白3和氯化钠共转运蛋白)促进肾钠再吸收,这可能导致盐敏感性高血压。
• 益生菌治疗与血压调节:
用鼠乳杆菌(Lactobacillus murinus)进行的益生菌治疗可以预防小鼠的盐敏感性高血压,这表明通过调节肠道菌群可以影响血压。
• 高盐饮食与肠道菌群代谢物:
高盐饮食降低了肠道中脆弱拟杆菌和花生四烯酸的水平,这可能增加肠源性皮质酮的产生,进而提高血清和肠道中的皮质酮水平,促进血压升高。
• 饮食中钠的减少与SCFA:
减少饮食中的钠可以增加循环中的短链脂肪酸(SCFA),降低血压,这表明菌群代谢物SCFA可能在血压调节中发挥作用。
• 神经炎症与高血压:
肠道微生物群的紊乱及其代谢产物失调,会刺激巨噬细胞释放过量的炎症因子,加剧高血压的进展。
• 交感神经系统的激活与肠道菌群:
交感神经系统的激活增加了肠粘膜的渗透性,破坏了肠道微生物群的平衡,导致炎症细胞的动员和分化,这些炎性细胞返回大脑后加重神经炎症。
双歧杆菌能够增加eNOS的活性,降低血清过氧化氢酶的活性,这可能有助于维持血管的舒张和降低血压。
高纤维饮食增加了共生细菌的丰度,抑制了机会致病菌的繁殖,例如,产乙酸菌比例的增加有效降低了血压,减轻了心脏肥大和纤维化。
总的来说,维持肠道微生物群的稳定性对CVD的进展具有抑制作用。合适的饮食疗法,如益生菌和益生元补充剂,可以维持肠道微生物群的平衡,这已被证明可以有效降低血液中炎症因子的水平,改善心血管疾病的预后。
炎症性皮肤病代表了一组具有多种病因的疾病,包括遗传因素、感染和免疫失调,涉及先天性和适应性免疫系统中各种免疫细胞和炎症介质的激活。
一项研究首次使用双样本MR分析评估肠道微生物群与炎症性皮肤病之间的双向因果关系。
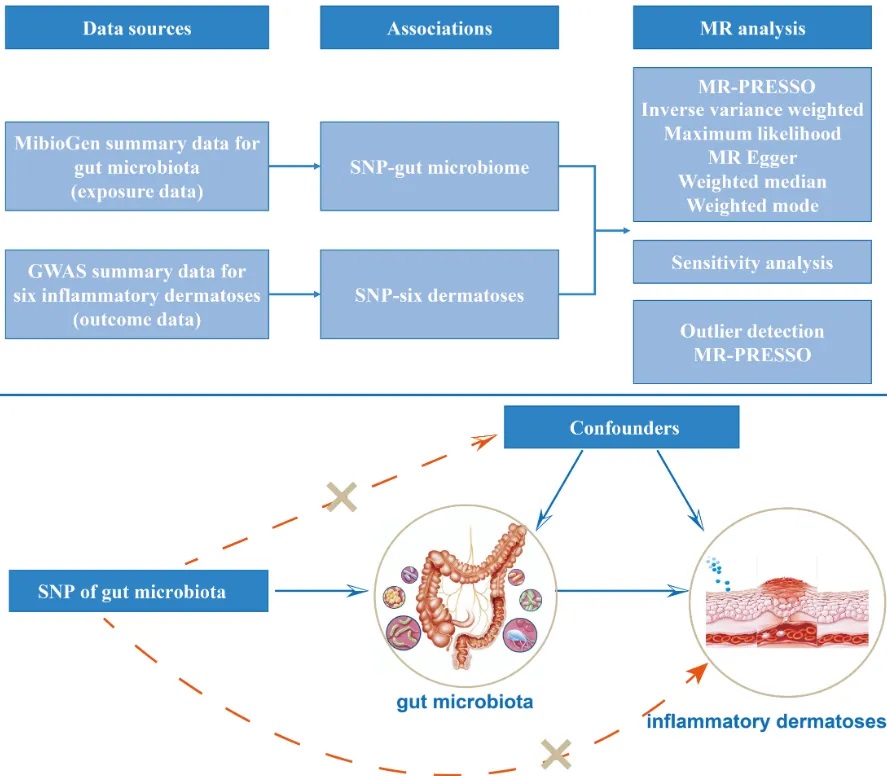
doi.org/10.3389/fimmu.2023.1231848
▸阿克曼氏菌属、瘤胃球菌属、双歧杆菌属、真杆菌属、粪球菌属等:
产生乙酸盐、丙酸盐和丁酸盐,通过GPCR和PPARγ受体调节免疫细胞,减少炎症因子释放,抑制组蛋白脱乙酰酶(HDAC),促进线粒体脂肪酸β-氧化。
▸乳酸杆菌和双歧杆菌:
增加色氨酸(Trp)和Trp代谢物,维持肠道屏障功能,减少痤疮炎症。
▸长双歧杆菌特殊作用:
将Trp代谢为吲哚-3-甲醛(I3C),通过AHR途径抑制Th2细胞,缓解特应性皮炎(AD)。
▸双歧杆菌、乳杆菌、Roseburia:
将多不饱和脂肪酸代谢为共轭亚油酸(CLA),抑制COX-2/5-LOX途径,抑制TLR4/NF-κB信号通路,减轻特应性皮炎皮肤病变。
▸硫酸盐还原菌(SRB):
如Desulfovibrionaceae,产生硫化氢(H2S),干扰丁酸盐氧化,损害肠道屏障,增加炎症因子释放。
以银屑病(牛皮癣)为例
银屑病是一种慢性复发性炎症性皮肤病,其特征是促炎细胞因子的释放增加。
Th-1、Th-17、Th-22细胞的扩增和活化, 一旦活化,这些细胞促进大量促炎介质的产生,包括但不限于来自角质形成细胞、淋巴细胞和其它免疫细胞的TNF-α、IL-6、IL-1、IL-17、IL-22、IL-23、VEGF、IFN-γ。此外,这些细胞促进皮肤病变的免疫发病机制,并在银屑病过程中驱动全身参与。
通过健康的饮食和运动获得的体重减轻能够改善银屑病的临床病程和治疗反应,甚至防止其发生,这种平衡的一个关键因素是肠道微生物群。(这在我们之前的文章中也详细阐述过)
肠道菌群在代谢综合征和银屑病中的多效性作用
doi.org/10.3390/ijms25158098
哪些菌群产生炎症因子,影响银屑病发生发展?
厚壁菌门中,金黄色葡萄球菌(Staphylococcus aureus)产生超抗原如TSST-1,其刺激角质形成细胞和DC分别产生促炎细胞因子和IL-23。这导致Th17细胞活化和IL-17产生,促进银屑病特征。
放线菌门中,纹状体棒状杆菌(Corynebacterium striatum)直接刺激角质形成细胞和DC产生IL-1β、IL-6和IL-23,增强Th17细胞分化。
在变形菌门中,大肠杆菌和粘膜奈瑟氏菌(Neisseria mucosa)通过LPS激活巨噬细胞和DC,触发IL-23、IL-6和IL-1β的产生,从而稳定Th17细胞并促进IL-17的产生。
孢子形成细菌如梭菌和脆弱拟杆菌通过诱导结肠T淋巴细胞和平衡Th1/Th2/Th17细胞来调节免疫应答.
分节丝状细菌(SFB)诱导Th17细胞分化。
在标准治疗的协同作用下,可以建议采用适当的饮食或其他干预措施来调节肠道菌群,从而改善银屑病的临床表现,并降低合并症的发生率。
婴儿双歧杆菌35624和1:1:1的益生菌混合物(即长双歧杆菌CECT 7347、乳双歧杆菌CECT8145、鼠李糖乳杆菌CECT 8361)在8至12周的治疗过程后,要么降低促炎TNF-α和血浆C-反应蛋白,要么降低患者的银屑病面积和严重程度指数。
在用咪喹莫特治疗的BALB/c小鼠中,戊糖乳杆菌GMNL-77显著降低了红斑鳞屑病变和促炎细胞因子如IL-23和IL-27的mRNA水平。
有趣的是,补充短双歧杆菌CCFM683有效地下调了角蛋白16/17、IL-17和TNF-α的表达,通过调节FXR/NF-κB通路和角质形成细胞增殖来改善银屑病。
以上我们了解了炎症因子在多种疾病中的作用机制,以及肠道菌群如何通过影响这些因子的水平和功能来调节炎症反应。炎症因子不仅反映了炎症的状态,还可以作为疾病进展和治疗效果的生物标志物。
为了有效地评估炎症因子的变化,临床上采用了多种检测方法。这些方法能够提供准确的炎症因子水平信息,辅助医生做出及时的诊断和治疗决策。接下来,我们将详细介绍几种常用的检测技术。
检测体内炎症通常需要特定的血液检查,以测量指示炎症反应的各种标志物。
◉ ELISA(酶联免疫吸附测定)
原理:抗原-抗体特异性结合,通过酶标记检测
优点:
灵敏度高、特异性强、可批量检测
缺点:
检测时间较长、操作步骤多、成本较高
适用:
IL-1β、IL-6、TNF-α等细胞因子的定量检测
◉ 化学发光免疫分析
原理:利用化学发光物质标记抗体或抗原
优点:
检测速度快、灵敏度高、自动化程度高
缺点:
仪器要求高、试剂成本高
适用:CRP、PCT等急性期蛋白的快速检测
◉ 免疫比浊法
原理:
抗原抗体形成免疫复合物产生浊度
优点:
操作简单、成本低、检测快速
缺点:
灵敏度较低、易受干扰
适用:CRP、血清淀粉样蛋白等的常规检测
◉ 流式细胞术
原理:通过荧光标记检测细胞因子
优点:
可同时检测多个指标、特异性强、定量准确
缺点:
设备昂贵、要求技术人员专业水平高
适用:细胞内细胞因子和膜表面标志物检测
◉ PCR技术
原理:检测炎症因子的基因表达水平
优点:
灵敏度极高、特异性强、可检测微量样本
缺点:
操作复杂、成本高、易污染
适用:炎症因子基因表达研究
◉ 蛋白质芯片
原理:多种抗原抗体反应的微阵列分析
优点:
可同时检测多个指标、样本用量少、高通量
缺点:
成本高、技术要求高、标准化难度大
适用:多种炎症因子的同时检测
◉ 肠道菌群健康检测
原理:通过分析肠道菌群及其代谢产物的变化情况,间接反映人体的炎症状态
优点:
非侵入性、可重复采样、早期预警、利于干预、反映整体状态、长期监测
缺点:
成本高、技术要求高、数据分析复杂
适用:多种炎症因子的同时检测
通过肠道菌群监测炎症因子的方法虽然存在一些局限性,但其独特的预警价值和非侵入性特点使其成为传统炎症监测方法的重要补充。
随着技术的进步和研究的深入,这种检测方法的局限性正通过多种创新手段得到克服:
这些技术创新显著提升了肠道菌群检测在临床应用中的价值,特别是在精准医疗和预防医学领域,使其成为疾病预警和健康管理的重要工具。
饮食方式
地中海饮食(MD)包括水果、蔬菜、全谷物、橄榄油、红酒等,可能对IBD有益。有证据表明,地中海饮食后可降低IBD和炎症的发生率。地中海饮食有助于抗炎菌群的存活,并防止肠道微生物群的失调发展。
通过调节肠道微生物群,改变肠道微生物组成,增加SCFA水平,降低尿TMAO水平,地中海饮食可以成为阿尔茨海默的潜在治疗干预措施。此外,更严格地遵守地中海饮食会延缓阿尔茨海默的进展,并提供1.5-3.5年的阿尔茨海默防护。
更多抗炎饮食,详见我们之前的文章:
益生菌
益生菌引入人体后,不仅会产生抗炎代谢产物,下调IL-6、IL-12、TNF-α等炎症因子和NF-κB通路等相关信号通路,还有助于抑制病原体的生长,修复肠道屏障,调节初始淋巴细胞的分化和增殖。
在BALB/c小鼠中,引入干酪乳杆菌可以预防肠道和关节炎症,不仅在膝盖,还在肠系膜和腘淋巴结中下调IL-1β、IL-6、IL-17、IL-23、TNF-α。
在一项涉及18名活动性强直性脊柱炎患者的试点研究中,补充嗜酸乳杆菌和唾液酸乳杆菌4周后,巴斯强直性脊柱炎疾病活动指数和视觉模拟量表有所改善。
此外,干酪乳杆菌、嗜酸乳杆菌、罗伊氏乳杆菌、双歧杆菌和嗜热链球菌的组合成功降低了用光受体间类维生素A结合蛋白免疫的C57BL/6小鼠的视网膜组织学评分,这是一种自身免疫性葡萄膜炎的动物模型。
在牙周炎的背景下,引入乳双歧杆菌HN019或罗伊氏乳杆菌显著改善了牙周炎的临床指标,包括减少探诊深度、减少探诊出血和降低手术风险。上述临床试验或动物实验中没有报告严重不良事件。
下一代益生菌,如F.prausnitzii、A.muciniphila,它们对IBD的治疗效果已经得到证实。这些益生菌可以减少浸润的巨噬细胞,抑制NF-κB信号通路,减少IL-8的产生,最终降低结肠炎的严重程度。
考虑到IBD和常见肠易激综合征之间肠道生态失调的相似模式,下一代益生菌在肠易激综合症管理中的应用也可能很有前景。
益生元
益生元是指微生物选择性利用以带来健康益处的底物。菊粉和低聚果糖等益生元的微生物发酵会产生代谢物(如SCFA),进一步调节肠道微生态系统和免疫反应。
在SpA的背景下,据报道,口服长链菊粉和低聚果糖可显著降低HLA-B27转基因大鼠结肠炎和关节炎的发病率。
对于牙周炎的动物模型,口服甘露寡糖成功地防止了牙槽骨丢失,降低了IL-10和IFN-γ的表达,下调了TNF-α和IL-1β的水平,并显著恢复了肠绒毛和隐窝深度。
值得注意的是,益生元具有广泛可接受的安全性,报告的严重不良事件很少。
后生元
在国际益生菌和益生元科学协会的指导下,益生元是指死亡的微生物或其对宿主有益的成分,包括SCFA、SBA等。
在SpA的背景下,直接外源性补充短链脂肪酸可以减轻各种动物模型中的关节炎严重程度。此外,口服短链脂肪酸还可以防止效应T细胞的激活和免疫细胞向脾脏和颈部淋巴结的运输,最终降低C57BL/6J和Kaede转基因小鼠模型中葡萄膜炎的严重程度。
该领域对原发性硬化性胆管炎的研究相对丰富。已经启动了几项为期12-24周的II期临床试验,报告称,无论是去甲熊去氧胆酸(SBA的衍生物)、奥贝胆酸(FXR配体)还是西洛菲索(FXR激动剂),都能显著降低原发性硬化性胆管炎患者(有或没有IBD)血清中的碱性磷酸酶。
此外,在患有IBD相关肝损伤的C57BL/6J小鼠中,添加乳脂球膜(Milk Fat Globule Membrane)与促炎细胞因子减少、Faccalibacumum和Roseburia恢复、结肠炎和肝损伤减轻以及谷胱甘肽转移酶途径的重新激活有关。
对于牙周炎患者,口服热灭活植物乳杆菌L-137有效地减少了同时接受支持性牙周治疗且基线深度不小于4mm的患者的探诊深度。
此外,小檗碱促进丁酸盐的产生,改善肠道屏障,降低循环LPS和促炎细胞因子水平,下调牙槽骨中的促炎细胞,最终改善牙周炎动物模型中的牙槽骨损失。同样,在后生元中也没有报告严重的不良事件。
抗生素
临床上,抗生素用于杀死致病菌或抑制其增殖。
在SpA的背景下,口服美罗培南和万古霉素有效地抑制了BALB/c和SKG小鼠脾脏中Th1和Th17细胞减少的外周附着点炎的发展。
对于葡萄膜炎,甲硝唑或万古霉素已被证明可以缓解葡萄膜炎,并增加用光受体间维甲酸结合蛋白预处理的B10.RII小鼠肠外淋巴组织中Tregs的丰度。
对于原发性硬化性胆管炎患者,应用万古霉素、甲硝唑和米诺环素可改善肝酶和梅奥风险评分。
此外,阿莫西林联合甲硝唑、单独甲硝唑和阿奇霉素可有效改善牙周炎患者的临床结果,其中阿莫西林联合甲硝唑在减少探诊袋深度、探诊出血和提高临床附着水平方面表现最佳。
粪菌移植 (FMT)
FMT是指将健康人类粪便中的微生物群移植到患者消化道中,使肠道微生态系统快速恢复的疗法。FMT的效果受到各种因素的干扰,包括供体的粪便质量、粪便的制备程序、给药方法和频率、FMT前的个体化肠道微生物组成(包括细菌、真菌和病毒)以及操作员技术。
一项试点随机对照研究表明,FMT组的内镜严重程度指数降低,C反应蛋白水平降低,这证明了FMT在维持克罗恩病缓解方面的疗效。
FMT还可以通过改善肠道微生物群的失调来减少肝脏脂肪积聚,从而减少NAFLD。
对FMT在阿尔茨海默病治疗中的作用的系统综述表明,FMT可以恢复SCFA和健康的微生物组,从而破坏阿尔茨海默患者的Aβ寡聚体,从而减少阿尔茨海默的发病机制。进一步机制研究表明,FMT降低了结肠、血清和SN中的LPS水平,抑制了TLR4/MyD88/NF-κB信号通路,使肠道微生物群和SCFA正常化,增加了突触素I的表达,并改善了阿尔茨海默模型小鼠的认知缺陷和Aβ沉积。
在许多慢性疾病中,例如肥胖症、动脉粥样硬化、2型糖尿病、炎性肠病、多发性硬化、类风湿性关节炎等,炎症是主要原因。肠道菌群的结构成分可能引发炎症,这可能引发一系列涉及白细胞介素和其他细胞因子的炎症反应。同样,某些短链脂肪酸和细菌代谢的其他代谢副产物可能有助于抑制炎症反应。因此,肠道菌群改变往往可能是炎症的预警信号,更早于临床症状的出现,其在疾病防治中的价值正逐渐被认识和挖掘。
特别是在当今精准医疗时代,结合人工智能深度学习算法、多组学整合分析等创新技术,肠道菌群检测的准确性和临床应用价值将得到进一步提升。随着检测技术的不断优化、数据分析方法的持续改进,以及临床验证研究的深入开展,肠道菌群检测将在疾病预警、健康管理、个体化治疗,响应监测以及预后评估中发挥越来越重要的作用。
未来,深入了解肠道微生物诱导的炎症因子的信号机制,有助于为肠道微生物诱导的炎症性疾病开发更精确、更有效的治疗方案。
本文内容仅供学习和交流目的,不构成任何形式的医疗建议。
主要参考文献
Chaudhary S, Kaur P, Singh TA, Bano KS, Vyas A, Mishra AK, Singh P, Mehdi MM. The dynamic crosslinking between gut microbiota and inflammation during aging: reviewing the nutritional and hormetic approaches against dysbiosis and inflammaging. Biogerontology. 2024 Oct 23;26(1):1.
Bai S, Bai H, Li D, Zhong Q, Xie J, Chen JJ. Gut Microbiota-Related Inflammation Factors as a Potential Biomarker for Diagnosing Major Depressive Disorder. Front Cell Infect Microbiol. 2022 Mar 15;12:831186.
Wu, J., Ou, G., Wang, S. et al. The predictive, preventive, and personalized medicine of depression: gut microbiota and inflammation. EPMA Journal (2024).
Liu P, Gao M, Liu Z, Zhang Y, Tu H, Lei L, Wu P, Zhang A, Yang C, Li G, Sun N, Zhang K. Gut Microbiome Composition Linked to Inflammatory Factors and Cognitive Functions in First-Episode, Drug-Naive Major Depressive Disorder Patients. Front Neurosci. 2022 Jan 28;15:800764.
Secchiero, P.; Rimondi, E.; Marcuzzi, A.; Longo, G.; Papi, C.; Manfredini, M.; Fields, M.; Caruso, L.; Di Caprio, R.; Balato, A. Metabolic Syndrome and Psoriasis: Pivotal Roles of Chronic Inflammation and Gut Microbiota. Int. J. Mol. Sci. 2024, 25, 8098.
Wang, W.; Zhu, L.-J.; Leng, Y.-Q.; Wang, Y.-W.; Shi, T.; Wang, W.-Z.; Sun, J.-C. Inflammatory Response: A Crucial Way for Gut Microbes to Regulate Cardiovascular Diseases. Nutrients 2023, 15, 607.
Mao R, Yu Q, Li J. The causal relationship between gut microbiota and inflammatory dermatoses: a Mendelian randomization study. Front Immunol. 2023 Sep 27;14:1231848.
Heinzel, S., Jureczek, J., Kainulainen, V. et al. Elevated fecal calprotectin is associated with gut microbial dysbiosis, altered serum markers and clinical outcomes in older individuals. Sci Rep 14, 13513 (2024)
Saedi, S., Derakhshan, S., Hasani, A. et al. Recent Advances in Gut Microbiome Modulation: Effect of Probiotics, Prebiotics, Synbiotics, and Postbiotics in Inflammatory Bowel Disease Prevention and Treatment. Curr Microbiol 82, 12 (2025).
Jukic A, Bakiri L, Wagner EF, Tilg H, Adolph TE. Calprotectin: from biomarker to biological function. Gut. 2021 Oct;70(10):1978-1988.
Zhao M, Chu J, Feng S, Guo C, Xue B, He K, Li L. Immunological mechanisms of inflammatory diseases caused by gut microbiota dysbiosis: A review. Biomed Pharmacother. 2023 Aug;164:114985.
Tie Y, Huang Y, Chen R, Li L, Chen M, Zhang S. Current insights on the roles of gut microbiota in inflammatory bowel disease-associated extra-intestinal manifestations: pathophysiology and therapeutic targets. Gut Microbes. 2023 Dec;15(2):2265028.
Nigam M, Devi K, Coutinho HDM, Mishra AP. Exploration of gut microbiome and inflammation: A review on key signalling pathways. Cell Signal. 2024 Jun;118:111140.
Soares CLR, Wilairatana P, Silva LR, Moreira PS, Vilar Barbosa NMM, da Silva PR, Coutinho HDM, de Menezes IRA, Felipe CFB. Biochemical aspects of the inflammatory process: A narrative review. Biomed Pharmacother. 2023 Dec;168:115764.