-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康
婴儿早期机会之窗
生命早期,特别是从胎儿期到出生后的前几年,是人类免疫系统和肠道微生物组(菌群)协同发育的关键时期。这一时期被称为机会之窗,期间的微生物暴露、营养摄入和环境因素对个体长期的免疫健康具有深远且不可逆转的影响。
如果说菌群的组成和多样性是描述其状态的快照,那么菌群的成熟度则是一个动态的、纵向的衡量标准,它反映了菌群随时间演替的轨迹是否符合健康的模式。
一个健康的婴儿,其菌群年龄应与其生理年龄大致相符。如果菌群年龄显著低于生理年龄,则意味着其菌群的发育轨迹偏离了正常轨道,表现为“不成熟”或“延迟成熟”。
越来越多的证据表明,婴儿肠道菌群的成熟延迟,并非特定于某一种过敏疾病,而是临床表现各异的过敏性疾病一个普遍的、共同的上游风险生物标志。
近年来,随着工业化国家过敏性疾病发病率的持续攀升,卫生假说指出,现代社会生活方式导致的早期微生物暴露减少可能是关键诱因。
本文基于多项大规模前瞻性队列研究和多组学分析的最新文献,系统性地阐述了婴儿肠道菌群的初始定植过程、动态演替模式、关键微生物驱动因素及其功能代谢特征。深入探讨了早期肠道菌群图谱的构建(如ELi-CSTs)、菌群成熟度(如微生物组年龄)与儿童未来健康(特别是过敏性疾病)的预测关系。
此外,详细论述了菌群与宿主免疫系统之间的复杂互作机制,包括母体因素的跨代影响、菌群如何训练新生儿免疫系统建立耐受,以及菌群成熟延迟如何通过共享的功能代谢通路障碍和过敏性疾病的发生发展相关。旨在整合当前领域的关键证据,为理解和预防儿童过敏性疾病提供科学依据。
写在前面
得益于高通量测序、代谢组学等多组学技术的飞速发展,以及大规模、长周期的前瞻性出生队列研究(如加拿大的CHILD研究)的开展,我们现在能够以前所未有的深度和广度来描绘婴儿肠道菌群的动态发育图谱,并探究其与宿主免疫系统之间的分子对话。这些研究不仅验证了菌群组成和多样性的改变与过敏风险相关,更进一步揭示了菌群的成熟度或微生物组年龄可能是一个更为普适和关键的预测指标。
谷禾团队发表在《Gut》上的针对自闭症儿童菌群发育的研究显示,自闭症儿童的菌群发育要滞后于健康儿童。发育迟缓儿童的肠道菌群呈现出明显的年龄滞后特征,这种现象反映在菌群的多样性、组成结构和功能等多个方面。研究表明,这些儿童的肠道菌群发育水平往往落后于其实际年龄,这种滞后可能是导致发育迟缓的重要因素之一。
婴儿肠道微生物组的建立是一个高度动态且遵循特定生态学规律的过程。从无菌的子宫环境(尽管关于胎盘微生物组的存在仍有争议)到暴露于微生物丰富的外部世界,新生儿的肠道迅速被各种微生物定植,开启了一场深刻的生态演替。这一过程受到一系列内外因素的强烈影响,并为宿主与微生物的终身共生关系奠定基础。
初始定植:从出生开始的旅程
出生是微生物入住身体的开幕式时刻。分娩方式是决定新生儿初始菌群构成的首要和最强烈的因素。经阴道分娩的婴儿,在通过产道时会接触并获得大量来自母亲阴道和肠道的微生物,其早期肠道菌群主要以乳杆菌属、普雷沃氏菌、拟杆菌等为特征。
母婴间的微生物垂直传播
一项研究发现,阴道分娩婴儿的肠道菌群与母体粪便菌群的重叠率高达72%。这种母婴间的垂直传播被认为是自然选择的结果,为婴儿提供了适应性的初始微生物群落。
剖宫产对初始菌群的影响
相比之下,通过剖宫产出生的婴儿绕过了产道的微生物接种过程,其初始菌群更多地来源于母体皮肤、口腔以及医院环境中的微生物,如葡萄球菌(Staphylococcus)、棒状杆菌(Corynebacterium)和丙酸杆菌(Propionibacterium)。
剖宫产婴儿的菌群与母体菌群的共享率显著降低,特别是关键的共生菌如拟杆菌属和双歧杆菌属的传递受阻。这种初始定植模式的差异不仅是暂时的,其影响可以持续数月甚至更长时间,并与日后更高的过敏和代谢性疾病风险相关。
初生婴儿肠道菌群的特点
在出生后的最初24小时内,婴儿肠道菌群的特点是复杂性低、个体间差异大。由于肠道内尚存的氧气,首批成功定植的通常是兼性厌氧菌,特别是肠杆菌科(Enterobacteriaceae)的成员,如大肠杆菌。
这些“先锋物种”通过消耗肠道内的氧气,为后续严格厌氧菌的生长创造了必要的无氧环境,从而启动了微生物群落的生态演替。
演替模式
随着先锋物种创造出厌氧环境,肠道菌群的演替进入第二阶段。
在出生后的几周内,严格厌氧菌开始占据主导地位,其中最引人注目的是双歧杆菌属的爆发式增长,尤其是在母乳喂养的婴儿中。
母乳低聚糖与双歧杆菌的优势
母乳中富含的母乳低聚糖(HMOs)是婴儿自身无法消化的复杂碳水化合物,但却是双歧杆菌,特别是长双歧杆菌婴儿亚种(Bifidobacterium longum subsp. infantis)的优质益生元。
这些细菌拥有高效利用HMOs的特殊基因和酶系统,使其在母乳喂养婴儿的肠道中获得巨大的竞争优势,形成所谓的双歧杆菌峰值。这一时期的菌群特征是多样性相对较低,但结构稳定,由双歧杆菌主导(见下图)。
早期肠道菌群成熟过程及伴随的免疫表型
doi.org/10.1038/s41577-023-00874-w
出生后的前3-6个月被认为是机会之窗,在此期间肠道菌群训练着发育中的免疫系统。
此图展示了在此期间发生的主要肠道菌群建立模式。图中显示了健康、母乳喂养新生儿在婴儿期四个主要细菌门的相对丰度。
ILA:吲哚乳酸;SCFAs:短链脂肪酸;TH:辅助T细胞;Treg:调节性T细胞。
免疫系统的机会之窗
生命早期的前几个月,特别是3-6个月,被定义为免疫印记的“机会之窗”。在此期间,婴儿的免疫系统和肠道菌群都具有极高的可塑性,两者之间的相互作用对建立终身免疫耐受至关重要。例如,在小鼠模型中,乳汁中的表皮生长因子(EGF)在哺乳早期抑制了肠道上皮杯状细胞相关抗原通道(GAPs)的开放,限制了微生物抗原的跨上皮转运。
微生物抗原的刺激与免疫耐受
随着哺乳时间的推移,EGF水平下降,GAPs逐渐开放,允许微生物抗原进入并刺激T细胞反应,从而在新生儿免疫系统偏向调节性表型的背景下,诱导对共生菌的长期耐受。
辅食引入与菌群演替的新阶段
随着辅食的引入和断奶,婴儿的饮食结构发生根本性改变,从以母乳/配方奶为基础转变为更加复杂和多样的固体食物。这一转变标志着菌群演替进入一个新的阶段。以HMOs为食的双歧杆菌优势逐渐减弱,而能够降解复杂植物多糖的菌群,如厚壁菌门中的梭菌纲(Clostridia)和拟杆菌门的成员,开始增殖并占据生态位。
菌群多样性与免疫适应反应
这一时期,菌群的多样性迅速增加,并逐渐向成人化的菌群结构过渡。在小鼠中,断奶会引发一场断奶反应,表现为IFN-γ和TNF水平的短暂飙升,这被认为是免疫系统对营养和菌群组成突然变化的适应性反应。
这场由梭菌介导的反应对于建立对后期免疫病理的抵抗力至关重要,其核心机制在于诱导了由短链脂肪酸(SCFAs)和微生物抗原驱动的RORγt+调节性T细胞(Treg)。尽管断奶反应在人类中的确切机制尚待证实,但这一发现强调了在机会之窗内,特定时间点的特定微生物-宿主互作对于塑造长期免疫健康的关键性。
为了系统地理解婴儿肠道菌群的复杂性和动态性,研究人员通过整合全球范围内的大规模微生物测序数据,构建了早期人类肠道微生物组的参考图谱。
Tarracchini等人的一项综合性研究,分析了来自全球不同地区37个独立研究的5288份健康足月婴儿的粪便宏基因组样本数据为我们描绘了一幅前所未有的高清画卷。
早期生命群落状态类型 (ELi-CSTs)
通过对庞大的数据集进行聚类分析,该研究识别出六种反复出现的、具有代表性的微生物群落结构,并将其命名为早期生命群落状态类型(Early-Life Community State Types, ELi-CSTs)。
这些ELi-CSTs不仅捕捉了婴儿肠道菌群的组成特征和异质性,也记录了其在生命第一年内所经历的深刻转变。
编辑
doi.org/10.1038/s41522-025-00868-7
这六种ELi-CSTs的具体特征如下:
Ba
ELi-CST1
以拟杆菌科成员(如脆弱拟杆菌 Bacteroides fragilis、多形拟杆菌 Bacteroides uniformis)的高度富集为特征。该类型菌群结构较为复杂,多样性较高,类似于成人菌群,在断奶后婴儿中最为普遍。
Ec
ELi-CST2
表现为大肠杆菌(Escherichia coli)的单一物种绝对优势,其平均相对丰度可超过60%。这是一种典型的“先锋群落”,主要出现在新生儿期(0-1个月)。
mix
ELi-CST3
这是一个混合型群落,没有明显的优势菌种,由韦荣球菌(Veillonella)、克雷伯氏菌(Klebsiella)、链球菌(Streptococcus)和肠球菌(Enterococcus)等多种细菌组成,多样性中等。
Blo
ELi-CST4
以长双歧杆菌(Bifidobacterium longum)为主导,同时常伴有双歧双歧杆菌(Bifidobacterium bifidum)。这是最常见的双歧杆菌主导类型之一,在断奶前期的婴儿中尤为普遍。
Bbr
ELi-CST5
以短双歧杆菌(Bifidobacterium breve)为主导。这种类型与ELi-CST4清晰地分离开来,反映了双歧杆菌内部不同物种间的竞争和生态位分化,可能与HMOs中岩藻糖的利用等优先效应有关。
mix
ELi-CST6
这是另一种复杂的混合型群落,富含多种与成人肠道健康相关的产丁酸盐菌,如普拉梭菌(Faecalibacterium prausnitzii)、直肠真杆菌(Agathobacter rectalis)和罗氏菌属(Roseburia)等。该类型代表了向成熟菌群过渡的后期阶段,在断奶后婴儿中占主导。
结合其他大型队列和多组学研究:
双歧杆菌主导、菌群成熟度良好的模式(与 ELi‑CST4/5 具有相似特征)在多项研究中与较低的儿童期过敏风险相关。
菌群成熟延迟、长期依赖早期先锋菌/混乱菌群结构(可类比持续处于 CST2 或不稳定混合型)在多研究中与5 岁时多种过敏疾病的风险升高相关。
但真正要比较不同ELi‑CST婴儿后续过敏/疾病差异,必须在新的前瞻性队列里做结合临床随访的验证,目前尚无直接证据。
ELi-CSTs动态演变:年龄与地理的影响
ELi-CSTs的动态分布
ELi-CSTs的分布并非一成不变,而是与婴儿的发育阶段密切相关。研究显示,ELi-CST2 (Ec) 显著富集于新生儿期,随着年龄增长急剧下降。取而代之的是以双歧杆菌为主的ELi-CST4(Blo) 和ELi-CST5 (Bbr),它们在1-6个月的断奶前期占据主导。
辅食引入与菌群演化
随着辅食的引入,菌群结构进一步演化,ELi-CST1 (Ba) 和ELi-CST6(mix) 的比例显著上升,成为断奶后期的主要群落类型。这一清晰的年龄依赖性演替轨迹,反映了从依赖母乳到消化复杂固体食物的饮食转变,以及宿主免疫系统与肠道环境的共同成熟过程。
地理位置对ELi-CST分布的影响
除了年龄这一主要驱动力,地理位置也对ELi-CST的分布产生了显著影响。随机森林模型分析表明,虽然年龄是预测ELi-CST归属的最重要因素,但地理位置的预测能力也不容忽视。例如:
这种地理差异可能反映了不同地区的饮食文化、生活环境、母婴垂直传播的菌株特性以及遗传背景等因素的综合影响。
关键婴儿微生物调节剂 (KIMMs) 的识别
为了从物种层面精确解析驱动ELi-CST形成的关键成员,研究者结合了随机森林算法和指示物种分析两种机器学习方法,识别出了25个在塑造婴儿肠道菌群结构中起决定性作用的物种,并将其命名为关键婴儿微生物调节剂(Key Infant Microbial Modulators, KIMMs)。这些KIMMs是各自ELi-CST的标志性物种,具有高度的特异性和保真度。
婴儿肠道微生物群的关键调节因子和共变物种群(CSCs)
编辑
doi.org/10.1038/s41522-025-00868-7
通过构建物种间的共现网络,研究进一步发现这些KIMMs并非孤立存在,而是形成了多个共变物种簇(CSCs)。这些CSCs内部物种间表现出强烈的正相关,而与簇外物种则多为负相关,表明它们是功能上紧密联系的生态单元。
例如包含B. longum、B. bifidum、B. breve的CSC5,与ELi-CST4和ELi-CST5高度关联,这与它们在利用HMOs过程中已知的协同代谢和交叉喂养行为相符,这种互利共生关系增强了双歧杆菌主导群落的稳定性。
编辑
相反,一些由成人型菌种(如 F. prausnitzii 和 A. rectalis)组成的CSCs则与断奶后期的ELi-CST6紧密相关。这些发现揭示了KIMMs作为生态系统的基石菌或关键菌,通过复杂的种间相互作用,驱动和稳定了婴儿肠道菌群在不同发育阶段的特定构型。
新生儿的免疫系统并非一个不成熟的成人缩小版,而是一个经过精心设计、具有独特功能和偏好的系统,其主要任务是在保护新生儿免受病原体侵害的同时,对大量无害的环境抗原(包括食物和共生微生物)建立耐受。
肠道菌群的定植是这一训练过程的核心环节,菌群及其代谢产物通过多种途径与宿主免疫细胞直接对话,深刻地塑造着免疫系统的发育轨迹。
母体因素与胎儿免疫的“预编程”
免疫系统的训练甚至在出生前就已经开始。尽管胎儿肠道被认为是低菌的,但母体的微生物信号还可以通过胎盘传递给胎儿,对其免疫系统进行“预编程”。
母体菌群的代谢物与免疫健康
研究表明,母体菌群产生的代谢物,如短链脂肪酸(SCFAs)、芳香烃受体(AhR)配体和类维生素A,能够穿过胎盘,影响胎儿免疫细胞的发育。例如,在小鼠中,母体肠道菌群产生的乙酸盐能够促进胎儿肺部调节性T细胞(Treg)的发育。
在人类研究中也发现,孕期携带普雷沃氏菌物种(Prevotella copri,一种产乙酸盐的细菌)的母亲,其后代发生食物过敏的风险较低。
抗共生菌IgG抗体
此外,母体的IgG抗体在妊娠晚期通过胎盘主动转运给胎儿,其中不仅包含针对病原体的保护性抗体,也包含了针对母体自身共生菌群的抗体。这些“抗共生菌”IgG抗体能够在婴儿出生后,抑制其对新生定植菌群的过度免疫反应,从而促进和平共处的建立。
免疫细胞的产生和扩散波
编辑
doi.org/10.1038/s41577-023-00874-w
免疫细胞在产前生命中分三个阶段出现。卵黄囊、胎肝和骨髓都贡献了不同类型的细胞,它们在出生前的不同时间点开始出现。出生时及出生后的头几个月,依赖于微生物刺激,免疫系统不同部分会继续发展和成熟。
新生儿免疫系统的独特性与菌群的训练作用
新生儿免疫系统的独特特征
新生儿的免疫系统在细胞组成和功能上都表现出独特的偏好。其免疫反应通常是减弱的或偏向于调节性/抗炎性,以适应出生时接触到的大量新抗原。例如,新生儿的树突状细胞(DCs)在受到脂多糖(LPS)等微生物刺激时,倾向于产生免疫调节细胞因子IL-10,而不是促进TH1型炎症反应的关键细胞因子IL-12。
新生儿的CD4+ T细胞也天然地偏向于分化为TH2型细胞和Treg细胞,而非TH1型细胞。这种固有的TH2偏向,一方面使婴儿易受某些细胞内病原体的感染,另一方面也避免了对共生菌群和食物抗原产生剧烈的、破坏性的炎症反应。
编辑
肠道菌群的定植与免疫平衡
肠道菌群的定植是打破这种TH2偏向、促进免疫系统向平衡状态成熟的关键驱动力。无菌小鼠(Germ-free, GF)的免疫系统存在严重缺陷,包括肠道相关淋巴组织(GALT)发育不良、TH1和TH17细胞缺失、Treg细胞数量减少,以及全身性的TH2型免疫反应过度和IgE水平升高,这些都使其极易发生过敏反应。
将正常菌群定植到GF小鼠体内可以逆转大部分免疫缺陷,但许多关键的免疫训练过程必须在生命早期的机会之窗内完成。例如,只有在新生期而非成年期定植菌群,才能有效抑制肺部和结肠中促炎的iNKT细胞的积累,并纠正血清中高水平的IgE。
菌群如何塑造免疫耐受与TH1/TH2平衡
肠道菌群与免疫耐受的建立
肠道菌群通过多种机制促进免疫耐受和平衡的建立。其中,诱导Treg细胞是核心环节之一。Treg细胞是维持免疫稳态、抑制过度免疫反应的刹车。
特定细菌与短链脂肪酸的作用
特定种类的肠道细菌,特别是梭菌纲的成员,被证明是诱导结肠RORγt+ Treg细胞扩增的强大诱导剂。这些细菌通过发酵膳食纤维产生大量的短链脂肪酸(SCFAs),如丁酸盐、丙酸盐和乙酸盐。
注:SCFAs不仅是肠道上皮细胞的能量来源,也是重要的免疫信号分子。丁酸盐可以直接作用于T细胞和DCs,通过抑制组蛋白去乙酰化酶(HDACs)等表观遗传学修饰,促进Foxp3(Treg细胞的关键转录因子)的表达,从而驱动Treg细胞的分化。丁酸盐的减少与过敏风险的增加密切相关。
菌群对TH1和TH17细胞的影响
除了诱导Treg,菌群还能促进TH1和TH17细胞的发育,从而平衡新生儿固有的TH2偏向。
TH1细胞对于抵抗细胞内病原体至关重要,而TH17细胞则在维持肠道屏障完整性和抵抗胞外菌和真菌感染中发挥作用。
例如,脆弱拟杆菌产生的多糖A(Polysaccharide A, PSA)能够通过TLR2信号通路,诱导产生IL-12,从而促进TH1细胞分化,纠正GF小鼠的TH2偏向。
编辑
而分节丝状菌(Segmented Filamentous Bacteria, SFB)的定植则能强烈诱导小肠固有层中TH17细胞的产生。通过促进TH1和TH17反应,肠道菌群有效地“抑制”了TH2细胞的过度活化,从而降低了过敏反应的风险。这一机制是“卫生假说”的核心生物学基础之一。
如果说菌群的组成和多样性是描述其状态的“快照”,那么菌群的“成熟度”则是一个动态的、纵向的衡量标准,它反映了菌群随时间演替的轨迹是否符合健康的模式。越来越多的证据表明,婴儿肠道菌群的成熟延迟,是多种儿童过敏性疾病共同的、发生在临床诊断之前的核心生物学特征。
微生物组年龄:一个预测健康的新指标
婴儿肠道菌群的演替过程具有高度的可预测性,如果有足够样本仅凭菌群的物种组成,多项研究以及谷禾健康均可以通过机器学习模型相当准确地预测婴儿的实际生理年龄。这种基于菌群的预测年龄被称为“微生物组预测年龄”(microbiota-predicted age)或“菌群年龄”(microbiota age)。
一个健康的婴儿,其菌群年龄应与其生理年龄大致相符。如果菌群年龄显著低于生理年龄,则意味着其菌群的发育轨迹偏离了正常轨道,表现为“不成熟”或“延迟成熟”。
编辑
<来源:谷禾健康肠道菌群检测数据库>
利用CHILD队列的数据,研究也建立了一个强大的菌群年龄预测模型。
该模型基于婴儿从出生到1岁的粪便菌群物种丰度数据,通过嵌套交叉验证的随机森林算法进行训练,其预测的菌群年龄与婴儿的实际生理年龄高度相关(Pearson R = 0.89, p < 2.2e-16)。这一模型的建立,为量化菌群成熟度并将其与远期健康结局相关联提供了有力的工具。
婴儿肠道的微生物多样性和微生物组衍生年龄
编辑
doi: 10.1038/s41467-023-40336-4
(a, b) 分别展示了3个月和1岁样本中,健康对照组(HC)与一个或多个过敏诊断组(1+),以及各独立临床诊断组的Shannon多样性指数。
(c) 微生物组衍生年龄与实际生理年龄的散点图。
(d, e) 分别比较了各诊断组在1岁时的微生物组预测年龄。星号(*)表示与HC组相比有显著差异(p < 0.05)。结果显示,在1岁时,所有过敏组的菌群多样性和预测年龄均显著低于健康对照组。
菌群成熟延迟是儿童过敏性疾病的共同标志
利用上述菌群年龄模型,Hoskinson等研究者得出了一个较一致的结论:无论是在5岁时被诊断为特应性皮炎(AD)、哮喘(As)、食物过敏(FA)还是过敏性鼻炎(AR),这些患儿在1岁时的肠道菌群年龄均显著低于同龄的健康儿童(AD p = 0.000014; As p = 0.0073; FA p = 0.00083; AR p = 0.0021)。健康儿童在1岁时的平均菌群年龄为11.53个月,而所有四种过敏疾病组的儿童其菌群年龄均显著偏小(见上图e)。这一发现具有极其重要的意义。
菌群成熟延迟的临床意义与预测价值
首先,它表明菌群成熟延迟并非特定于某一种过敏疾病,而是这四种临床表现各异的过敏性疾病一个普遍的、共同的上游风险标志。
其次,这种菌群成熟的延迟发生在过敏性疾病的临床诊断之前,具有预测价值。调整了性别、分娩方式、母乳喂养、抗生素使用等多种混杂因素后,1岁时菌群年龄的增加仍然是5岁时患过敏性疾病的保护性因素,也就是说菌群发育得越好,孩子在5岁时患过敏性疾病的风险就越低。
编辑
菌群成熟延迟的微生物学特征
菌群成熟延迟的具体微生物学特征表现为:
促进健康免疫发育的好细菌丰度降低,而与不良健康结局相关的坏细菌丰度增加。
在过敏风险高的婴儿中,多种产短链脂肪酸的细菌(特别是丁酸盐),如 Anaerostipes hadrus、Fusicatenibacter saccharivorans、Eubacterium hallii、Blautia wexlerae 的丰度显著降低。
一些潜在的致病菌或促炎菌,如 Eggerthella lenta、Clostridium innocuum、Enterococcus faecalis、Escherichia coli、Tyzzerella nexilis的丰度则显著升高。这种此消彼长的模式,共同构成了不成熟菌群的物种画像。
共享通路功能障碍:菌群失调与过敏的连接
菌群的物种组成变化最终通过其功能和代谢产出来影响宿主。多组学分析进一步揭示了菌群成熟延迟背后的功能性后果,这些功能障碍构成了连接菌群失调与过敏性疾病发展的桥梁。研究发现,在1岁时菌群不成熟的婴儿肠道中,存在一系列共同的功能和代谢失衡特征。
肠道黏液屏障完整性受损
表现为与黏蛋白降解相关的通路上调,而与硫氧化(有助于维持屏障)相关的通路下调。这可能导致肠道屏障通透性增加,使更多过敏原和微生物产物进入体内,触发免疫反应。
氧化应激水平升高
表现为与氧化呼吸相关的通路(如NAD(P)/NADPH相互转换)上调。氧化应激环境会促进炎症,并产生氧化的单糖,这些单糖又可以作为某些病原菌的营养源,进一步加剧菌群失调。
次级发酵能力下降
表现为产丁酸盐等有益SCFAs的能力减弱,这直接削弱了对Treg细胞的诱导和对免疫系统的调节作用。代谢组学分析也证实,过敏风险高的婴儿粪便中丁酸盐水平有降低的趋势。
痕量胺水平升高
代谢组学分析发现,三种生物胺——苯乙胺(phenylethylamine)、色胺(tryptamine)、酪胺(tyramine)的水平在菌群不成熟的婴儿中显著升高。这些痕量胺能够与肠道和免疫细胞上的特定受体(TAARs)结合,诱导肠道细胞的氧化应激和免疫细胞的活化,并促进细菌黏附,可能形成一个促炎的恶性循环。
连接预测年龄和过敏性疾病的特定微生物和代谢组学特征
编辑
doi: 10.1038/s41467-023-40336-4
结构方程模型图显示了预测年龄对特应性和过敏性疾病的直接和间接影响,这种影响由1岁时的微生物组和代谢组特征所介导。
为了整合这些多维度的发现,研究者构建了一个结构方程模型来检验“菌群成熟延迟通过功能和代谢失调导致过敏”这一核心假说。
结果证实,菌群年龄对5岁过敏风险的影响,绝大部分是通过这些功能和代谢失衡(作为一个潜在变量)介导的,而直接效应不显著。
这些证据暗示一条从“菌群成熟延迟”到“功能代谢失调”再到“过敏性疾病”的病理生理通路,为将菌群成熟度作为预测和干预过敏性疾病的焦点提供了的理论基础。
婴儿肠道菌群的成熟延迟是多种儿童过敏性疾病(包括特应性皮炎、哮喘、食物过敏和过敏性鼻炎)共同的、发生在临床诊断之前的重要风险标志。
这种不成熟状态不仅表现为物种组成的失调(有益菌减少,有害菌增多),更关键的是导致了一系列共享的功能代谢通路障碍,包括肠道屏障功能受损、氧化应激增加、有益代谢物(如丁酸盐)产生不足以及有害代谢物(如痕量胺)的积累。这些功能性后果共同构成了连接早期菌群失调与远期过敏性疾病的生物学桥梁。
这些发现为我们理解和应对日益流行的儿童过敏问题提供了深刻的启示和新的方向。
从“静态组成”到“动态成熟”的视角转变
未来的研究和临床评估不应仅关注特定好或坏细菌的存在与否,而应更多地采用如“微生物组年龄”等纵向、动态的指标来评估菌群的健康状态。这为开发新的过敏风险早期筛查工具提供了可能。
精准干预的潜力
既然菌群成熟延迟是共同的上游风险因素,那么针对性地促进菌群健康成熟可能成为一种广谱预防多种过敏性疾病的策略。这包括优化分娩和喂养方式(如推广阴道分娩和母乳喂养)、审慎使用抗生素,以及开发基于证据的新一代益生菌、益生元或合生元制剂。这些干预措施的目标应是恢复健康的菌群演替轨迹,补充缺失的关键功能菌群(如产丁酸盐菌),并纠正代谢失衡。
机制研究的深化
尽管宏观通路已经明确,但微观的分子机制仍有待深入探索。例如,不同双歧杆菌亚种间的竞争与合作如何影响菌群的整体稳定性,母体因素如何通过表观遗传等方式影响后代的菌群-免疫轴,解答这些问题将为开发更有效的干预靶点提供依据。
总之,肠道菌群在生命早期的健康发育,是构建强大而平衡免疫系统的基石。将菌群成熟度纳入考虑,可能意味着一个新的起点,未来有望通过调节这个微小的内在生态系统,来预防和管理影响儿童的过敏性疾病。未来的挑战在于如何将这些基础研究的深刻见解,转化为安全、有效、个性化的临床实践。
主要参考文献:
Donald K, Finlay BB. Early-life interactions between the microbiota and immune system: impact on immune system development and atopic disease. Nat Rev Immunol. 2023 Nov;23(11):735-748.
Barker-Tejeda TC, Zubeldia-Varela E, Macías-Camero A, Alonso L, Martín-Antoniano IA, Rey-Stolle MF, Mera-Berriatua L, Bazire R, Cabrera-Freitag P, Shanmuganathan M, Britz-McKibbin P, Ubeda C, Francino MP, Barber D, Ibáñez-Sandín MD, Barbas C, Pérez-Gordo M, Villaseñor A. Comparative characterization of the infant gut microbiome and their maternal lineage by a multi-omics approach. Nat Commun. 2024 Apr 8;15(1):3004.
Tarracchini C, Longhi G, Gennaioli E, Muscò A, Rizzo SM, Viappiani A, Vitale SG, Mancabelli L, Lugli GA, Angioni S, Turroni F, van Sinderen D, Milani C, Ventura M. Compiling an early life human gut microbiome atlas and identification of key microbial drivers. NPJ Biofilms Microbiomes. 2025 Dec 5;12(1):4.
Hoskinson C, Dai DLY, Del Bel KL, Becker AB, Moraes TJ, Mandhane PJ, Finlay BB, Simons E, Kozyrskyj AL, Azad MB, Subbarao P, Petersen C, Turvey SE. Delayed gut microbiota maturation in the first year of life is a hallmark of pediatric allergic disease. Nat Commun. 2023 Aug 29;14(1):4785.

谷禾健康
下一代益生菌
传统益生菌在维持肠道稳定性方面具有一定优势,但其作用效果受宿主遗传背景、菌株生物学特性以及个体肠道微生态环境等因素的制约,这也使得其在临床应用中呈现出多样化的效果,且在调控全身性疾病方面仍需进一步探索其潜在机制。
与传统益生菌不同,下一代益生菌(NGPs)采用基因工程和下一代测序技术来识别具有特定功能特性的细菌菌株。一些研究者认为,下一代益生菌可能在微生物治疗领域具有发展前景,为某些健康问题的管理提供新的思路。
下一代益生菌(NGPs)通过筛选具备特定功能的菌株,不仅在肠道内发挥作用,还通过其他途径影响全身的免疫系统、代谢过程和神经调控机制。相较于传统益生菌,NGPs在以下方面具有显著优势:
①多系统靶向性——不仅修复肠道屏障,还可通过肠脑轴、口腔、皮肤、呼吸道等通道调控局部与全身免疫反应;
②精准性与个体化潜力——结合基因组学与代谢组学,可实现更精准的宿主-微生物互动干预;
③作用机制的扩展性——不仅抑制病原体、抗炎,还能通过代谢产物和免疫调控网络实现更广泛的治疗效应。
当前,NGPs的主要种类包括但不限于:
嗜黏蛋白阿克曼菌(Akkermansia muciniphila)、普拉梭菌(Faecalibacterium prausnitzii)、丁酸梭菌(Clostridium butyricum)、Eubacterium hallii及若干经基因改造或筛选的菌株,它们在慢性炎症、胃肠道疾病、代谢综合征、皮肤健康、口腔健康、免疫相关疾病以及精神疾病等多种疾病的治疗与缓解中展现出潜在价值。
随着基因组学、代谢组学、表观遗传学及循证临床研究的迅速发展,新型益生菌有望成为个体化干预的新载体,将疾病管理从“治疗反应”转向“精准预测与预防”。
什么是益生菌?
“益生菌(probiotics)”一词源自希腊语/拉丁词根
oro(意为’为了’)与bios(意为’生命”),这主要体现了这些生物体在确保生命健康方面的作用。
益生菌是具有活性的微生物,主要是细菌和酵母,在给予足够剂量时具有健康益处, 益生菌已被证明能改善消化健康、免疫系统功能,甚至心理健康。
有效益生菌微生物的特征
doi: 10.1007/s11033-024-09398-5.
◮ 益生菌发挥效果的条件
同时益生菌要发挥效果,必须:
-忍受胃的酸性环境;
-活着抵达肠道;
-附着在肠道细胞上以发挥其益处;
-与有害细菌竞争;
-以及在肠道内定植到一定程度。
益生菌的发展历史
益生菌的概念源远流长,我们在这里简要梳理益生菌的起源及其随时间的演变。以下为历代发展要点的回顾。
◮ 古代起源
中东和中亚:两千多年来,发酵乳制品已被证明是一个值得关注的问题。中东和中亚被视为大约1万年前处于原始发酵状态的文明的摇篮。
亚洲:在古代中国和日本,也有纳豆和味噌等富含益生菌的食物,分别是发酵大豆和大豆酱。
◮ 19世纪及20世纪初:科学发现
益生菌一词直到19世纪末才进入科学界。随着微生物学的兴起,巴斯德揭示了发酵由微生物所驱动,从而解释了为何某些菌类存在是为了发酵。
1907年,梅契尼科夫被誉为“益生菌之父”,指出保加利亚农民的长寿与牛奶发酵有关,乳酸菌能阻止细菌损害宿主肠道,防止自毒与衰老。1908年,梅契尼科夫因在免疫学领域的研究荣获诺贝尔奖,进一步提升了公众对乳酸菌的认知。
◮ 20世纪中叶:益生菌产品的发展
在20世纪40–50年代,仿制食品成为益生菌引入的关键因素。自1935年起,日本率先开展商业益生菌饮料“养乐多”的生产;1939年,通过干酪乳杆菌(Lactobacillus casei)展示了益生菌在胃肠治疗中的潜力。
随后,研究扩展至其他菌株,甚至酵母菌也被探索,其中Saccharomyces boulardii已用于腹泻治疗。
◮ 现代科学中的益生菌
益生菌研究广泛开展,尤其关注其在医学上的潜在益处。自1970年代初起,益生菌的活性逐步增强,医学与微生物学的结合揭示了其积极影响的机制。此间,益生菌不再仅是传统发酵食品的成分,而是逐渐被视为可用于治疗特定健康问题的药物。
▸ 益生菌如何与微生物群相互作用
益生菌的作用机制错综复杂,涵盖众多对消化系统有益并增强免疫力的作用过程:
益生菌的作用机制
编辑
doi: 10.1007/s12602-025-10606-2.
1) 病原体的竞争排斥
益生菌通过竞争排斥来抑制病原微生物的生长:利用可用资源并争夺肠道受体位点,限制入侵生物的扩张。
此外,某些益生菌还能分泌抑制物(如细菌素),抑制致病菌增殖,从而提升整个细菌群落的健康水平。
2) 免疫反应的调节
益生菌还能通过诱导调节性T细胞来调控免疫反应,这些T细胞维持免疫平衡,避免对无害物产生过度反应。因此,益生菌在炎症并发症如炎症性肠病和自身免疫性过敏的管理中具有关键作用。
它们还支持健康的免疫反应,帮助抵抗感染、在必要时抑制过度炎症,并整体促进免疫健康。
3) 强化肠道屏障
益生菌的重要性在于提升黏膜质量与细胞间紧密连接的完整性,从而预防肠漏综合征。肠漏指肠壁的病理生理改变,导致有毒物质进入循环并引发炎症。Faecalibacterium prausnitzii和Akkermansia muciniphila这两种菌株被公认有助于维持肠道屏障功能,前者对黏膜覆盖也有保护作用。
4) 有益代谢产物的生成
益生菌能合成重要代谢产物,如短链脂肪酸(SCFAs)—丁酸、乙酸、丙酸等。证据显示SCFAs具有抗炎作用,促进肠道稳态、保护细胞完整性并缓解炎症,滋养结肠上皮,帮助维持健康的肠道黏膜。此外,SCFAs在调节与健康微生物群相关的免疫与代谢方面也发挥重要作用。
益生菌的作用机制
编辑
doi: 10.1007/s12602-025-10582-7.
5) 对肠-脑轴的影响
近期研究显示,益生菌通过肠脑轴影响心理状态的潜力。某些菌株能产生神经递质,如血清素与GABA,调节情绪与压力水平。
瑞士乳杆菌和长双歧杆菌已被证实可缓解焦虑、改善情绪,从而将健康微生物群与心理健康联系起来。
随着对人类微生物群落的深入了解,益生菌及其产品开发的研究日益广泛。借助现代微生物学工具,如PCR的16S rRNA检测、下一代测序(NGS)和生物信息学,我们能够更精准地识别和检测肠道细菌菌株。
最近,基因组测序与培养技术的进步促使从人类微生物组中分离出多种新型微生物,显示出良好的健康益处和益生菌潜力,因而有望发展成为下一代益生菌(NGPs)。
下一代益生菌(NGPs)的定义
下一代益生菌(NGPs)是指基于宏基因组学或16S rRNA测序等比较微生物组研究,遴选或工程改造的特定菌株。这些菌株在适当摄入量下,通过调节宿主肠道微生物群落结构或功能,对特定疾病状态的改善具有潜在的生物学效应和科学证据支持。与传统益生菌相比,NGPs针对性更强,而且有的已经累积了更多相关临床研究数据。
这类非传统益生菌包括嗜黏蛋白阿克曼菌(Akkermansia muciniphila)和普拉梭菌(Faecalibacterium prausnitzii)等,它们可改善肠道健康、降低炎症并增强肠道屏障。
下表总结了传统益生菌与下一代益生菌(NGPs)之间的主要区别。
▸ 对下一代益生菌(NGPs)的需求
益生菌的修复与预防作用已广泛被认可和应用,不仅有助于消化健康,也提升免疫与整体健康。传统定义多聚焦于含发酵菌株的产品,如乳杆菌和双歧杆菌,在大量应用中发挥作用。然而,随着人类微生物组的日益复杂与深入了解,经典益生菌未必能满足所有健康需求,因此出现了新型益生菌(NGPs),旨在更精准地解决更多健康问题。
下一代益生菌可能的治疗作用
编辑
doi: 10.1007/s11033-024-09398-5.
1) 针对复杂疾病
全球慢性疾病如肥胖、糖尿病、自身免疫性疾病和精神健康障碍的患病率持续上升,促使科学界逐步揭示微生物组在这些疾病中的作用。这为非处方的下一代益生菌(NGPs)提供了更有针对性的治疗潜力。
例如,Akkermansia muciniphila与健康个体的优良新陈代谢活性相关,并有助于维持黏膜屏障;而屏障的降解与代谢性疾病密切相关。Faecalibacterium prausnitzii 被视为抗炎菌株,突显了其在炎症性肠病(IBD)治疗中的潜力。
2) 个性化医疗与精准健康
下一代益生菌(NGPs)正与日益增长的个性化医疗需求融合:根据患者的基因、代谢或微生物组特征来治疗个体疾病。
与此同时,益生菌研究持续推进,致力于深入理解肠道微生物组,以实现精准、定制的益生菌处方。
3) 通过调节微生物群落提高疗效
下一代益生菌(NGPs)更接近于益生元,研究显示,一些NGPs能释放代谢产物,调控群落结构与活性,长期有助于健康。例如,它们可产生短链脂肪酸(SCFAs)如丁酸,具有显著的抗炎作用,对肠道环境至关重要。相比之下,传统益生菌尽管可提高SCFA水平,但由于功能特性,其贡献可能不及NGPs。
4) 心理健康与肠-脑轴
近期肠-脑轴研究显示,肠道微生物组的状态显著影响心理健康、情绪、应激反应与高级认知功能。传统益生菌在此领域的作用有限,新型益生菌则具更大治疗潜力。
例如,脆弱拟杆菌与植物乳杆菌可能影响脑功能。新型益生菌能合成多种功能性代谢产物,用于调节脑活动,治疗焦虑、抑郁及其他精神障碍等问题。
5) 肠道屏障完整性与免疫调节
对于慢性疾病与自身免疫过程,肠道病变引发的免疫过度激活会使有毒物质从肠道扩散入血。新型益生菌,如嗜粘蛋白阿克曼菌,通过增强肠道黏液层提升屏障防御,进而减轻全身炎症。
此外,NGPs 还可调控免疫细胞功能与促炎/抗炎反应的平衡,可能有助于治疗免疫相关疾病。
6) 在癌症治疗中的潜在应用
下一代益生菌(NGPs)在癌症管理领域的应用备受关注。研究表明,某些微生物组变体可增强免疫疗法的效果。通过使用特定菌株,可提升宿主免疫系统对肿瘤细胞的杀伤力,从而为癌症治疗中的益生菌辅助治疗提供新的可能性。
▸ 下一代益生菌的标准
与传统益生菌相比,下一代益生菌(NGPs)能更有效、更精确地解决复杂的健康问题。考虑到功能性、特异性和安全性,已提出了一些标准来界定新型益生菌。关键标准如下:
1) 菌株特异性和选择
下一代益生菌(NGPs)通常源自非传统菌株,相较于乳杆菌和双歧杆菌等传统益生菌,它们在特定健康领域具更大潜力。每种菌株的选择都以实现精确的健康目标为导向,如代谢、免疫或认知健康,以提升针对性治疗效果。
2) 作用理解
就NGPs而言,核心在于菌株对免疫反应的调控、益生代谢物的产生、肠道通透性的改变,以及与宿主微生物组的相互作用机制。多数NGP已被筛选或改造以产生代谢物(如SCFA、神经活性物质)并执行支持其健康益处的功能。
3) 临床疗效和针对性健康益处
应通过一致的前临床与临床研究证明NGPs在健康状况下的病情调节功效。在某些情境下,可能表现为提升胰岛素敏感性以改善代谢健康,或通过调控炎症来治疗自身免疫性疾病。这种功效使NGPs从一般肠道健康支持,扩展到糖尿病、炎症性肠病和精神疾病等特定疾病的治疗。
4) 生存能力和定植潜力
要让NGPs菌株成功定植,关键在于其能耐受胃肠道恶劣环境,并在可能的情况下实现内腔幼体的稳定定植,以确保长期的遗留效应。其在宿主微生物组中的建设性相互作用以及在胃酸环境中的存活能力,对疗效至关重要。
5) 安全性和最小不良反应
安全性备受关注,部分原因在于许多新型益生菌来自非传统来源,且人类使用的历史证据稀少。因此需进行严格的综合安全评估,包括毒性和长期安全性研究。应尽量减少并密切监测可能引发不良反应的机制,如免疫紊乱或对自然微生物群的破坏。
6) 个性化应用的潜力
随着个体微生物组的多样性,开发下一代益生菌(NGPs)时已将个性化纳入考量。这意味着选取或设计符合特定微生物组特征或个人健康状况的菌株。
个性化的NGP应用通过识别微生物失衡与健康风险,提供更定制化的干预,因此产生了在健康益处上更具针对性的NGP,成为个性化与精准医疗领域的宝贵资产。
小结
传统益生菌和新型益生菌之间的主要区别
编辑
总得来说,新一代益生菌(NGPs)相对于传统益生菌的科学、具体优势主要有以下几点:
•更广的靶向健康作用谱;
•更强的肠道微生态调控能力;
•更精准的作用机制与靶点;
•先进的制备与设计策略;
•改善药品监管与可控性前景
•增强个体化干预潜力;
•安全性与耐受性改进方向。
传统益生菌和新型益生菌的特征比较
编辑
doi: 10.1007/s12602-025-10582-7.
下一代益生菌(NGPs)中具有潜力的候选株正在逐步浮出水面,其中一些已在代谢调控、炎症抑制和跨系统健康影响方面显示出特别的潜力。如Akkermansia muciniphila、Faecalibacterium prausnitzii、Clostridium butyricum、Christensenella minuta等,初步研究显示它们在代谢调控、炎症抑制和肠-全身轴的潜在影响方面具有积极信号。
但这类证据尚处于早期阶段,且个体差异、长期安全性、给药策略与潜在不良反应尚需进一步验证。此外,NGP 需要经过严格的毒理学评估、长期随访与可重复的生产质量控制,才能避免潜在风险并确保可追溯性。因此,在扩展NGP候选池时,必须遵循安全优先、证据驱动、标准化与伦理法规合规的原则,强调并非所有物种都具备成为下一代益生菌的潜力,也不应过早推广到临床应用。
Akkermansia muciniphila
极具潜力
嗜黏蛋白阿克曼菌(Akkermansia muciniphila,Akk)是一种定居肠道黏膜的共生菌,作为下一代益生菌的有力候选。它在增强代谢与免疫反应方面发挥关键作用,并可能改善癌症治疗效果。
◮ 能利用黏蛋白,促进肠道屏障完整性
A. muciniphila能够利用肠道黏膜层的黏蛋白与糖蛋白作为唯一的氮碳来源,促进肠道屏障完整性并附着于黏膜,是一种有益的益生菌特征。
肠道黏膜中该菌含量下降会削弱屏障,增加毒素侵袭风险。它不仅参与葡萄糖、脂质和蛋白质代谢,还影响黏膜层的完整性与黏膜免疫反应,因此对肠道健康具有支持作用。
◮ 在肥胖、炎症性肠病、自闭症中都发挥作用
近年研究表明A.muciniphila在机体稳态与疾病中具有重要作用。其丰度与多种疾病相关,水平下降与肥胖、炎症性肠病、自闭症和2型糖尿病等相关。
它通过调节神经系统控制葡萄糖代谢,帮助抵御肥胖与糖尿病。研究还发现经巴氏杀菌的A.muciniphila可显著延长结肠长度、增强对肥胖与胰岛素抵抗的抵抗力,且炎症性肠病与代谢疾病患者常见其水平降低,提示潜在抗炎作用。此外,A.muciniphila还能增强某些抗癌治疗在动物模型中的效果。
总体而言,益生菌领域的最新证据支持A.muciniphila作为可行治疗靶点,其水平变化可作为疾病进展的潜在生物标志物。因此,A.muciniphila在癌症、代谢综合征、炎症性肠病及免疫相关疾病等领域具有广阔的临床潜力。
肠道重要菌属——Akkermansia Muciniphila,它如何保护肠道健康
Faecalibacterium prausnitzii
具有较高潜力
普拉梭菌(Faecalibacterium prausnitzii)是一种极具前景的下一代益生菌。
F.prausnitzii在肠道中的下降与微生物群失调及多种代谢疾病和慢性免疫介导疾病(如炎症性疾病和肥胖)相关,并常作为衡量年轻肥胖者是否患溃疡性结肠炎的生物标志物,这可能源于其促进黏蛋白与紧密连接蛋白合成、修复受损肠黏膜的能力。此外,它还能调控粘液分泌、肠道杯状细胞分化和糖基化,并维持黏液屏障完整性,因此被视为有益的益生菌,对肠道具有关键保护作用,其枯竭会削弱肠道免疫调控与抗炎能力。
◮ 重要的产丁酸菌、具有抗炎活性
F.prausnitzii被认定为肠道中最重要的丁酸生产者之一。丁酸是肠道上皮细胞的主要能量来源,调节肠道T细胞的活性,抑制病原体入侵,促进结肠癌细胞凋亡,预防肠道炎症,调节免疫系统,并帮助代谢综合征恢复。
此外,F. prausnitzii还会产生来自微生物抗炎分子(MAM)的肽,抑制宿主激活NF-κB通路。这些活性代谢物的优点包括抗炎活性、维持肠道屏障功能、肠道免疫稳态以及在结直肠癌细胞中诱导凋亡。
◮ 可作为癌症免疫治疗中的调节因子
目前,从健康人群中分离出的F. prausnitzii已被证明具有体外抗炎和免疫调节活性。此外,Faecalibacterium属候选物种已被研究为癌症免疫治疗中的调节因子。研究人员发现肠道中Faecalibacterium的浓度与癌症患者的存活率之间存在关联。
此外,科学家们发现,黑色素瘤转移期间,调控T细胞数量和促炎细胞因子IL-2、IL-8和IL-6的血液水平与肠道中Faecalibacterium的数量呈负相关。因此,F. prausnitzii被认为是黑色素瘤患者的关键治疗靶点和预后标志物。
肠道核心菌属——普拉梭菌(Faecalibacterium Prausnitzii),预防炎症的下一代益生菌
Clostridium butyricum
具有较高潜力
丁酸梭菌(Clostridium butyricum)是一种嗜氧、形成孢子的革兰氏阳性细菌,因其发酵非碳水化合物并高产丁酸而得名。
◮ 对肠道健康、免疫调节有益,甚至改善抑郁
丁酸梭菌,被认为对炎症性肠病有益,并起到免疫调节作用。它已被用于肠道健康支持、免疫调节和感染预防。丁酸梭菌还对肠上皮增殖和维持结肠健康至关重要。
有研究报道了丁酸梭菌的抗癌潜力,观察到其可显著抑制小鼠肠道肿瘤形成、减少肠道癌细胞增殖并诱导凋亡。此外,丁酸梭菌的益生菌株与抗抑郁药联合使用,可显著改善抑郁症状。
Christensenella minuta
具有较高潜力
Christensenella minuta已成为下一代益生菌(NGPs)候选者中的重要一员,显示出在代谢调控与炎症抑制方面的潜力。
◮ 改善能量平衡、降低炎症水平
C. minuta在肠道微环境中能够促进有益代谢产物如乙酸和丁酸的产生,改善能量平衡与脂质代谢,同时通过调节炎性介质和免疫途径,降低慢性炎症的水平。这些作用不仅局限于肠道,还通过肠-全身轴、代谢网络和免疫调控网络对全身健康产生潜在影响。
C.minuta可能通过以下途径发挥作用:增强肠道屏障功能、改变宿主基因表达与信号传导、促进短链脂肪酸等有益代谢产物的产生、以及调节脂肪组织与肝脏的炎症反应。
◮ 临床层面
研究表明,Christensenella minuta在2型糖尿病和肥胖等代谢紊乱以及炎症性肠病中的丰度显著下降。其相对丰度与低 BMI 指数相关的瘦表型呈正相关。
除此之外,支气管哮喘和过敏性疾病、肾结石、情感障碍、甲状腺癌、粘膜类天疱疮、多囊卵巢综合征和复发性口疮性口炎等疾病中C. minuta的丰度也较低。
总体而言,C. minuta 的证据虽仍处于初步阶段,但其在代谢健康与炎症控制方面展现的多维潜力使其成为有希望的下一代益生菌候选株。未来需要更多高质量的人体研究来确认其安全性、最优给药策略、长期效果及与其他干预的协同作用,以便将这一候选菌株尽快转化为可用于临床的干预工具。
Christensenella minuta——下一代益生菌候选者:改善代谢、减轻炎症
Eubacterium hallii
有潜力
Eubacterium hallii是另一种潜在的益生菌生物治疗制剂(NGP)。能利用多种碳源(包括糖和有机酸)并产生两种关键的短链脂肪酸(SCFAs):丙酸和丁酸。SCFAs对肠道健康至关重要,促进黏液生成、刺激肠上皮细胞增殖分化并维持上皮健康;SCFA不足会引发炎症。
注:据报道,E.hallii是新生儿肠道中丁酸的主要来源之一。
◮ 能与肠道其他细菌协作产生短链脂肪酸
某些E.hallii能发酵复杂碳水化合物,但也有依赖其他肠道微生物产物的情况。多项研究证实了这种交叉摄食机制在Eubacterium属SCFA生成中的重要性。
相关研究多在含复杂碳水化合物的培养基中将Eubacterium属与双歧杆菌共培养,能降解复杂碳水化合物的双歧杆菌株可产生1,2-丙二醇、乙酸酯与乳酸盐,随后被Eubacterium属吸收并转化为丁酸与丙酸。
这一交叉摄食模式明确揭示了肠道微生物与复杂碳水化合物的丁酸化协同及其在肠道生态系统中的作用。
◮ 可能改善肥胖和糖尿病
研究了E.hallii在肥胖和糖尿病中的作用。发现E.hallii能够代谢丁酸以激活G蛋白联结受体信号通路,改善GLP1和GLP2的产生,强化肠道屏障功能,同时不影响体重或食物摄入,并提升胰岛素敏感性和能量代谢率。因此,它在胰岛素敏感性方面可能安全且有效。
◮ 在体内对一些物质进行关键转化
最新研究表明,Eubacterium属在肠道中进行关键的代谢转化,对人体健康有积极影响。在这些效果中,有毒化合物解毒为更良性的形态似乎具有价值。
例如,观察到E.hallii将极为丰富的食品来源杂环芳香胺致癌物——2-氨基-1-甲基-6-苯基咪达唑(4,5-b)吡啶(PhIP)转化为生物学上无法获得的7-羟基-5-甲基-3-苯基-6,7,8,9-四氢吡啶(3′,2′:4,5 咪唑(1,2-α)嘧啶-5-m氯化物(PhIP-M1)。此外,PhIP在模拟远端和近端结肠微生物群存在下被E.hallii转化,导致其丰度分别增加了120倍和300倍。这表明它作为一种保护性治疗具有极佳的视角。
此外,在同一研究中,检测到了E.hallii的抗菌活性。作者证明,E.hallii能够将甘油分解为3-羟基丙烯醛(3-HPA),这种甘油以水溶液中的榘蛋白形式存在。
研究还发现Eubacterium属通过调节胆汁酸代谢谱,有助于肠道和肝脏健康。近年来,胆汁酸代谢或肠道微生物群的调节正被研究为肝细胞癌(HCC)和结直肠癌(CRC)的创新治疗策略。
Roseburia
有潜力
罗氏菌属(Roseburia)是一种革兰氏阳性、厌氧、弯曲杆状细菌。Roseburia物种能使用复杂多糖,产生短链脂肪酸(丙酸盐、丁酸盐、醋酸盐)。它对炎症、帕金森病和炎症性肠病(IBD)有益,并且被充分考虑为下一代益生菌(NGPs)。
◮ 降低动脉粥样硬化、改善肠道、肝脏健康
令人瞩目的是,最新研究显示,食用Roseburia和富含纤维的饮食的小鼠,其动脉粥样硬化的发生率降低了。这归因于高纤维饮食,介导了Roseburia丁酸的形成以减少动脉粥样硬化。
此外,最新研究显示 Roseburia intestinalis(细菌鞭毛的关键结构成分)在治疗酒精性脂肪肝和溃疡性结肠炎方面具有潜力。口服 Roseburia 肠源性鞭毛蛋白在酒精性脂肪肝模型中显著改善了肠道上皮完整性并抑制了肠道损伤风险。然而,关于 Roseburia 的临床证据仍然有限,需要进一步研究以明确其在多种人类疾病中的应用价值。
Bacteroides fragilis
有潜力,但同时存在安全隐患
脆弱拟杆菌(Bacteroides fragilis)是机会性病原体,然而,近期研究证实了非致毒性脆弱拟杆菌的益生菌特性。脆弱拟杆菌可以刺激宿主适应性免疫,抑制炎症,激活免疫系统的成熟,调节肠道微生物群,并通过多糖A(PsA)及该NGP的其他外膜囊泡维持肠道健康和稳态。
◮ 抑制其他致病微生物
先前研究显示,脆弱拟杆菌通过抑制其他致病微生物的生长与定居来抑制它们。在动物模型中,该菌能抑制艰难梭菌感染,且治疗后肠道微生物多样性提升、AKK菌丰度增加。
研究还表明,B.fragilis通过抑制凋亡、维持ZO-1和MUC-2完整性,从而提高肠道屏障功能,降低艰难梭菌的黏附性和定植性。因此,脆弱拟杆菌有助于维护肠道屏障的完整性。
其他研究还发现脆弱拟杆菌对沙门氏菌的易位具有竞争性抑制作用,机制包括抗菌蛋白 BSAP-1 的生成。BSAP-1 含有 MACPF 结构域,能裂解侵染宿主细胞的细菌。除了 BSAP-1 外,泛素样蛋白(BfUbb)及 VI 型分泌系统(T6SSs)等因素可能也在该竞争中发挥重要作用。
◮ 产生的荚膜多糖具有免疫调节作用
最新研究显示脆弱拟杆菌在结肠内代谢多种碳水化合物,生成八种荚膜多糖,其中多糖A(PSA)是一种独特的两性多糖,具免疫调节作用。
PSA被抗原呈递细胞内吞并处理后被T细胞识别,持续增强宿主免疫。PSA通过调节树突状细胞,促使初始T细胞分化为Treg,提升Foxp3和CD39表达,抑制IL-17并诱导IL-10产生,帮助治疗肠道炎症性疾病。
近期研究还报道脆弱拟杆菌通过增强免疫功能、抑制脂多糖信号、改善肠道微生物群活性并维持肠道屏障稳态来预防肠漏,对癌症患者有潜在益处。
SK08活细菌粉末是一种以脆弱拟杆菌为基础的活体生物药,已获中国食品药品监督管理局认可,目前处于临床试验阶段,属于治疗性生物药范畴。
Prevotella copri
有潜力
P. copri是拟杆菌门中的另一种新型益生菌,据报道可改善葡萄糖耐量和肝糖原水平。它被视为2型糖尿病、肥胖等代谢疾病的潜在靶点。
◮ 微生物组学基础
P. copri在健康人群和特定疾病患者(如类风湿关节炎、2型糖尿病)的肠道微生物群落中存在明显差异,已通过宏基因组学研究识别。
◮ 功能特性研究
•能产生短链脂肪酸(SCFAs),特别是丙酸;
•参与多糖代谢,具有特定的代谢功能;
•与肠屏障完整性和免疫调节相关;
◮ 临床相关性
•在类风湿关节炎(RA)中丰度降低,补充可能改善症状;
•与代谢相关疾病的关联有一定研究基础。
广泛存在于人群的双面使者——Prevotella copri与疾病和健康
Parabacteroides goldsteinii
有潜力
研究表明,P.goldsteinii也有望成为下一代益生菌。中山大学的程芳/陈红波教授团队探讨了肠道共生细菌P.goldsteinii衍生的外膜囊泡(OMVs)通过传递关键的抗炎分子(例如十五烷酸)来调节宿主的免疫系统。这些OMVs能够在炎症皮肤区域积聚,有效抑制IL-23/Th17轴,改善全身免疫稳态,从而显著缓解银屑病症状。
编辑
Su D,et al.J Control Release.2025
P.goldsteinii已被证实能够显著改善肠道和肺部炎症。强烈推荐用于肥胖改善。此外,P.goldsteinii也表现出显著的抗炎和胰岛素刺激特性。
Propionibacterium freudenreichii
有争议
费氏丙酸杆菌(Propionibacterium freudenreichii)主要用于乳制品行业,作为生产瑞士奶酪的食品级微生物。研究者正探索其特定的益生菌特性以实现新的健康益处。该菌已能产生丙酸,具有抗菌活性并可降低胆固醇。
另一项研究则显示,P.freudenreichii 对肠道屏障完整性与炎症状态的影响提示其在胃肠道健康方面的潜在应用。
工程菌株
具有较高潜力
工程菌株是尖端的下一代益生菌代表。经过改造的乳酸乳球菌和大肠杆菌等菌株通过产生外源蛋白或分子,可用于治疗结直肠癌、炎症性肠病甚至精神健康问题等疾病。
注:益生菌还通过CRISPR技术进行基因改造,这扩大了接种疫苗的治疗选择,并增强了免疫反应。
编辑
doi: 10.1007/s12602-025-10606-2.
小结
无论来自天然还是合成来源,这些新一代菌株都代表益生菌的未来前沿。特定有益代谢物如丁酸、抗菌肽和免疫调节化合物可作为复杂疾病的治疗干预,提供传统益生菌难以实现的疾病特异性与个性化策略。因此,研究将聚焦新一代益生菌的关键点,推动医学在预防与治疗上的新前景。
下一代益生菌(NGP)在治疗领域的前景日益广阔,标志着其不再局限于传统的肠道健康维护,而是在疾病预防、诊断与治疗中发挥更为精准和多样的作用。通过基因改造或天然/合成菌株的多样化开发,NGP 能在人体内定向表达有益代谢物、抗微生物肽以及免疫调节因子,为难治性炎症、代谢疾病、免疫相关疾病等提供新的干预路径。
临床应用将持续扩展,覆盖免疫、神经、泌尿生殖、心脏、代谢、呼吸、皮肤、口腔及体重管理等领域。下面是当前新型益生菌已涉足的方向:
下一代益生菌在各种健康疾病中的应用
编辑
doi: 10.1007/s12602-025-10606-2.
胃肠道健康
下一代益生菌可能聚焦于已被证实对特定胃肠道问题有益的单一菌株,而非传统益生菌组合的多菌株混合,从而实现更个性化、针对性的治疗。
随着DNA测序技术的发展,下一代测序能够对肠道微生物群进行更为细致的分析,帮助理解胃肠道疾病患者的微生物组结构,并识别潜在的治疗重点菌群或菌株。
•炎症性肠病(IBD):包括Akkermansia muciniphila和Faecalibacterium prausnitzii,两者都与减轻肠道炎症和促进粘膜修复有关。抗炎分解代谢物(如SCFA)的合成有助于调节与IBD相关的炎症,包括克罗恩病和溃疡性结肠炎。
•肠易激综合征(IBS):某些益生菌可以缓解腹胀、疼痛和排便异常等症状。其中一些包括婴儿双歧杆菌和丁酸梭菌(Clostridium butyricum),它们可以对抗炎症并提高肠道内的健康运动能力,在这种情况下,它们在治疗IBS相关疾病方面非常有用。
•抗生素相关性腹泻:非致病性细菌大肠杆菌Nissle 1917通过引入有益细菌来缓解抗生素相关性腹泻和其他胃肠道问题,从而恢复肠道健康。
代谢健康
代谢综合征(MS)是一组特征性疾病,包含胰岛素抵抗、血脂异常、肝肾功能异常、高血压、血糖稳态紊乱等。如今,益生菌正被视为潜在的代谢疾病生物治疗药物,作为具有健康促进作用的功能性食品成分,并具备对抗特定疾病的潜力。
小鼠研究显示,A. muciniphila 与体重、脂肪量变化相关,新型益生菌菌株能够有效调节葡萄糖、脂质和体重的平衡。其改变必需代谢物谱,包括短链脂肪酸、维生素、 多种脂肪酸、氨基酸及胆汁酸代谢物等。
•肥胖和体重管理:肠道菌群的一些调节剂会影响促进脂肪储存、脂肪燃烧和饱腹感的因素。如前所述,使用Akkermansia muciniphila已被证明可以改善胰岛素反应,缓解脂肪堆积,从而治疗肥胖。
•2型糖尿病:新型益生菌可能有助于血糖控制,因为它们会产生SCFA,SCFA已被证明可以降低血糖水平并增强胰岛素敏感性。两种有益的微生物,Akkermansia muciniphila和鼠李糖乳杆菌,抑制胰岛素抵抗,已被推荐用于控制2型糖尿病。
免疫系统调节
健康的微生物群通过迷走神经直接或间接调节免疫系统。由于肠道微态的稳定性,益生菌可调节免疫环境,有助于自身免疫疾病的治疗。
•自身免疫调节:如脆弱拟杆菌通过产生多糖A调节促炎与抗炎反应,从而为治疗类风湿性关节炎和多发性硬化症等自身免疫疾病提供干预,目标是调节免疫与减轻自身免疫。
•过敏与哮喘:部分新型益生菌能调节免疫耐受,降低过敏反应。鼠李糖乳杆菌在给药后表现出抗过敏、抗哮喘特性,透过增强调节性T细胞并降低过度免疫反应来发挥作用。
•感染:部分新型益生菌具抗感染作用,如丁酸梭菌具有抗菌特性,能够增强对感染的免疫防御,但需在宿主体补偿机制恢复后方能实现。
心理健康
•抑郁与焦虑:仍有被称为“心理益生菌”(psychobiotics)的新型益生菌(NGP)与情绪和认知改善相关”。瑞士乳杆菌和长双歧杆菌已显示抗焦虑与抗抑郁效应,可能通过产生血清素和GABA等神经递质、以及降低全身炎症来实现。
•压力管理:与GBA(脑-肠-轴)相互作用的NGP 能通过维持微生物群平衡、调节 HPA 轴来减轻生理压力对身心的影响,关联情绪与压力相关的精神状态。
新型益生菌的治疗益处
编辑
doi: 10.1007/s12602-025-10582-7.
皮肤健康
健康的皮肤微生物群通过调节免疫细胞与炎症维持体内平衡。湿度、pH、温度、脂质、营养、运动、药物、手术以及身心压力等变量共同影响皮肤微生物组的日常波动。
表皮屏障缺陷与免疫失调与特应性皮炎(AD)密切相关,且具有遗传易感性和环境影响的个体更易发病,体现了 AD 的复杂病理生理。最新研究显示局部治疗可改善 AD 患者的微生物群,帮助清除有害菌、促进有益菌生长,从而平衡微生物群。
•湿疹与皮炎:益生菌已逐步用于炎症性皮肤病的治疗。鼠李糖乳杆菌与长双歧杆菌能改善屏障功能、降低炎症,可能对湿疹与皮炎患者有益。
•痤疮与牛皮癣:部分新型益生菌具抑炎特性、可抑制免疫活动,有助于治疗痤疮及银屑病等疾病;有助于控炎、保护屏障功能,对皮肤健康有益,减少炎症发作。
口腔健康
根据既有文献,口腔内约有700多种细菌构成口腔微生物群。免疫抑制、激素治疗及不良饮食等因素可引发牙釉质侵蚀与牙周疾病。
益生菌作为一种新兴且具成本效益的微生物疗法,能抑制口腔病原体并降低粘附,延长假体装置的使用寿命。
•牙龈疾病与龋齿:最新口腔研究显示,某些益生菌通过重新定植口腔菌群来预防口腔疾病,抑制有害生物。例如,唾液链球菌与罗伊氏乳杆菌通过抗炎、维持口腔菌群平衡及抑制致病生物膜,对牙龈疾病与龋齿具有积极作用。
变形链球菌在龋齿形成中扮演重要角色,能产生胞外多糖并在酸性条件下存活。研究了鼠李糖乳杆菌GG与发酵乳杆菌KU200060对变形链球菌KCTC 5316的最小抑菌浓度(MIC),抗菌效果分别为25%与12.5%。唾液链球菌DB-B5为从健康成年女性龈上牙菌斑分离的菌株,具备产生新型细菌素等益生菌特性。
•口臭:部分益生菌能降低挥发性硫化合物,这是口臭的主要成因。通过平衡口腔微生物群,可维持口腔健康环境,减少口臭发生。
呼吸健康
•上呼吸道感染:鼠李糖乳杆菌GG及其他新型益生菌在预防肺部感染方面已显示有效。它们可通过调节免疫反应来增强上皮黏膜对呼吸道病原体的防御,具备免疫抑制潜力。这些益生菌已用于预防普通感冒与流感等疾病。
•哮喘管理:一些NGP可通过降低全身炎症、促进免疫耐受来缓解哮喘,并帮助调节在寒冷天气下的异常免疫反应,通常用于慢性呼吸道疾病的治疗。
肝脏健康
•非酒精性脂肪肝(NAFLD):新型益生菌(NGPs)通过调整肠道脂质代谢相关微生物群,促进肝脏健康,抑制脂肪积聚。像Akkermansia muciniphila等菌株能减少肝脏氧化应激与炎症的证据正在积累,但仍需更多临床试验验证。
•肝脏排毒与健康维护:某些菌株通过改善肠道环境、降低进入肝脏的微生物内毒素,提升肝脏排毒能力,从而增强肝功能并降低全身炎症。
癌症预防与治疗
•癌症预防:特异性NGPs可通过产生抗氧化与抗炎应激因子来抑制癌症诱导效应,具有抗癌潜力。研究显示,干酪乳杆菌通过调节肠道菌群和免疫监测,减少结直肠癌风险。
•癌症治疗支持:NGPs可减轻化疗相关副作用(如黏膜炎)并支持免疫功能。据报道,益生菌通过增强免疫反应与改善肠道健康,抑制肿瘤细胞增殖,或作为标准肿瘤治疗的辅助手段。
新型益生菌(NGPs)已从单纯的肠道守卫者发展为多系统、多靶点的免疫与代谢调控工具。基于对肠道微生物组及其代谢产物的深入理解,NGPs显示出在慢性炎症、代谢疾病、免疫性疾病、皮肤与神经系统疾病等领域的广阔应用前景。它们不仅能够改善局部屏障功能与炎症状态,还可能通过肠脑轴、肠-皮肤轴等通路实现对远端器官的调控,从而带来更全面、个体化的健康管理方案。
尽管前景光明,NGPs的临床转化仍面临若干挑战:菌株的稳定性与可控性、个体微生环境差异、长期安全性评估、以及标准化的制备、给药剂量与疗效评估体系尚需完善。
未来发展的关键方向包括:
总之,NGPs具备成为下一代个体化健康管理核心元素的潜力。通过持续的基础研究与循证临床探索,未来的益生菌干预有望实现更高的治疗精准度、更广的适应证和更持久的健康收益。
主要参考文献
Gupta MK, Srivastava R. Gut Microbiome Interventions: From Dysbiosis to Next-Generation Probiotics (NGPs) for Disease Management. Probiotics Antimicrob Proteins. 2025 Aug;17(4):2629-2652.
Vijayaganapathi A, Mohanasrinivasan V. A Review of Next-Generation Probiotics-As a Gateway to Biotherapeutics. Probiotics Antimicrob Proteins. 2025 Aug;17(4):1985-1997.
Al-Fakhrany OM, Elekhnawy E. Next-generation probiotics: the upcoming biotherapeutics. Mol Biol Rep. 2024 Apr 15;51(1):505.
Maftei NM, Raileanu CR, Balta AA, Ambrose L, Boev M, Marin DB, Lisa EL. The Potential Impact of Probiotics on Human Health: An Update on Their Health-Promoting Properties. Microorganisms. 2024 Jan 23;12(2):234.
Santacroce L, Charitos IA, Bottalico L. A successful history: probiotics and their potential as antimicrobials. Expert Rev Anti Infect Ther. 2019 Aug;17(8):635-645.
Meng N, Liu Q, Dong Q, Gu J, Yang Y. Effects of probiotics on preventing caries in preschool children: a systematic review and meta-analysis. J Clin Pediatr Dent. 2023 Mar;47(2):85-100.
Evivie SE, Huo GC, Igene JO, Bian X. Some current applications, limitations and future perspectives of lactic acid bacteria as probiotics. Food Nutr Res. 2017 May 3;61(1):1318034.
Haghshenas B, Kiani A, Mansoori S, Mohammadi-Noori E, Nami Y. Probiotic properties and antimicrobial evaluation of silymarin-enriched Lactobacillus bacteria isolated from traditional curd. Sci Rep. 2023 Jul 5;13(1):10916.
Silva DR, Sardi JdCO, de Souza Pitangui N, Roque SM, da Silva ACB, Rosalen PL (2020) Probiotics as an alternative antimicrobial therapy: current reality and future directions. J Funct Foods 73:104080.
Mejía-Caballero A, Salas-Villagrán VA, Jiménez-Serna A, Farrés A (2021) Challenges in the production and use of probiotics as therapeuticals in cancer treatment or prevention. J Ind Microbiol Biotechnol 48(9–10):kuab052.

谷禾健康
随着全球人口老龄化加速,老年人多病共存与多重用药已成为常态,这给药物疗效和安全性带来了巨大挑战。
多药联用本身在控制多重危险因素、降低心脑血管事件方面具有明确获益;很多研究指出,多种药同时使用,住院、药物不良反应与全因死亡等风险往往显著上升。更棘手的是,在老年多病共存背景下,传统以单药证据为核心的处方逻辑,在面对真实世界的系统扰动时发现,同样方案,有人获益明显,有人疗效不足,甚至在疗效不佳的同时不良反应频发。
过去我们多从肝肾功能衰退、体成分变化、血浆蛋白结合率下降、受体敏感性改变,以及药物—药物相互作用来解释这种不确定性。但近十年的研究逐渐揭示一个被低估的关键变量:肠道菌群。
肠道菌群不仅能直接参与药物的转化、活化或灭活,还可通过代谢物谱改变、炎性衰老与屏障变化,沿着“代谢—免疫—屏障”三轴形成相互强化的级联反应,从而在药代动力学与药效学层面共同放大疗效波动与毒性风险。
基于此,本文围绕肠道菌群失调这一关键因素,阐述其通过”代谢—免疫—屏障“三轴相互作用,在老年多病共存与多重用药的临床背景下,对药物吸收、代谢和疗效的影响机制,并从代表性药物和治疗场景出发,阐明临床防治要点,为后续高质量试验设计与标准化评价体系的建立提供一些思考。
我们正处在一个不可逆转的全球老龄化时代。根据联合国的预测,到2050年,全球65岁以上的人口将占总人口的16.3%。
老年人普遍多病共存
世界卫生组织(WHO)将多病共存定义为”同一人体内存在两种或两种以上慢性疾病“。这听起来像是一个简单的数学叠加,实则是复杂的生物学重构。
流行病学数据显示,老年人群多病共存的患病率从15%-43%不等,且呈指数级增长。在我们国内,随着疾病谱从传染性疾病向慢性非传染性疾病转型,”高血压+糖尿病+冠心病+脑卒中“的组合已成为老年内科门诊的标准配置。
多药联用的益处和风险
为了管理多种慢性病,患者往往需要服用多种处方药。多药联用本身并非坏事。对于控制多重危险因素、预防心脑血管事件具有明确获益。但当药物数量超过5种时,风险陡然上升,例如住院率增加、药物不良反应增加、全因死亡风险上升。
在老年多病背景下,传统以单药证据为核心的处方逻辑,面对真实世界的系统扰动时,疗效与安全性的方差显著增大。
当多病共存遇上多重用药,药物的疗效和安全性变得极不确定。药物之间可能相互作用,而复杂的生理状况也让药物反应变得难以预测。这正是当前老年医学面临的核心难题之一。
传统观点认为,老年药代动力学改变主要源于肝肾功能衰退、脂肪/肌肉比例变化、血浆蛋白结合率下降。但近十年的研究揭示,肠道菌群构成了药物代谢的第三维度,在多药联用的老年人群中,这一维度的变异可能是疗效不确定性的最大来源。
肠道菌群:老年患者用药难题的关键一环
近年来的研究逐渐明确,肠道菌群不仅与消化相关,还深度参与免疫调控、代谢稳态与屏障维护。在老年人群中,其临床意义被进一步放大,原因主要有三点:
-个体差异更大
老年人因咀嚼/吞咽能力、胃肠动力、饮食结构与营养摄入能力变化,微生物群组成在个体间波动更明显。
-易受药物与疾病共同扰动
多病共存导致慢性炎症与器官轴(如肠-肝、肠-心、肠-肺)改变;多重用药(尤其抗生素、PPI、免疫抑制/抗肿瘤药物等)可改变群落结构与代谢功能。
特定药物和药物类别对肠道菌群影响的总结
编辑
doi: 10.1080/19490976.2025.2604867
-对药物治疗的影响:机制叠加而非单点效应
既往研究常把菌群影响分为三类来讨论。
问题在于:对老年多病共存患者而言,上述三类效应往往不是并列发生,而是形成相互强化的级联反应。也就是说,微生态失衡并非仅改变某一个药物的代谢,而是通过“代谢—免疫—屏障”三系统的协同失代偿,塑造一个对药物治疗不利的内环境:疗效更易不足,毒性更易放大,且波动更难预测。
下一章节,我们来详细了解代谢—免疫—屏障,它如何形成自我维持的恶性循环,并如何在药代/药效层面制造难以预测的真实世界差异。
在老年多重慢病与多重用药背景下,肠道微生态的紊乱正成为影响药物反应变异的新兴决定因素。菌群的异常不再局限于单一微生物水平的失衡,而会通过代谢-免疫-屏障三大系统的协同失代偿,引发药物动力学(PK)和药效学(PD)的广泛扰动。理清这种多轴联动机制,是实现精准干预的前提。
编辑
doi.org/10.1016/j.arr.2026.103023
代谢轴:药物转化的失控开关
很多口服药的结局,不只由肝肾功能决定——肠道菌群也在参与转化、激活、灭活与清除。
菌群主要从两条路影响用药
1) 直接改造药物分子
部分细菌能还原/脱羧/去甲基等,把药改形态,导致疗效或毒性变化。
2) 间接改造宿主代谢能力
健康菌群产生的短链脂肪酸、胆汁酸等代谢物,可影响肝脏药物代谢酶(如CYP、UGT等)的表达与活性;当菌群紊乱时,这类代谢物谱改变,可能让药物暴露升高或波动更大。
编辑
doi: 10.1080/19490976.2025.2604867
临床例子
注:老年人/多病共存/多药并用时,即使肝肾化验正常,也可能出现半衰期延长、血药浓度异常或ADR风险上升。
当遇到解释不清的疗效波动/不良反应,除了依从性、相互作用、肝肾功能,也可以把菌群状态纳入评估。
免疫轴:慢性低度炎症,正悄悄改写药物反应
老年人常见的菌群失调会提高机体对微生物相关分子(如LPS等PAMPs)的系统性暴露(其中一种来源是肠道通透性改变,下一个轴会详细了解),从而持续激活TLR4/NF-κB等通路,形成炎性衰老背景。
这会怎样影响用药?
1) 改变药代
慢性炎症可下调或扰动肝脏/肠道的药物处置系统(如部分CYP酶、转运体P-gp等),导致同样剂量出现血药浓度更高、波动更大,不良反应风险上升。
2) 改变药效
炎症会重塑靶器官微环境与免疫状态,使药物作用靶点是否可用、反应是否到位发生变化;在免疫相关治疗(如肿瘤免疫治疗、免疫抑制治疗、部分抗炎/免疫调节药)中尤其明显。
临床上你可能看到的矛盾现象
屏障轴:肠道屏障变薄,用药为什么不稳定?
在衰老、慢病与长期用药的叠加下,肠上皮修复能力下降,紧密连接与黏液层保护减弱,“肠漏”更常见——这不只是消化问题,也会直接影响药物反应。
两个核心后果
1) 吸收变得不确定
屏障受损与局部炎症会让肠道的通透性/转运发生改变:有的药吸收偏多,血药浓度升高;有的药吸收不稳,导致同剂量下疗效忽高忽低。
2) 全身稳态被扰动(并放大前两个轴)
屏障一旦松动,细菌成分与代谢产物更容易进入循环,促进低度炎症,进而影响肝药酶/转运体与靶器官微环境,从而形成肠漏 → 炎症 → 用药更波动的恶性循环。
临床上常见的表现
三轴不是并列问题,而是一个协同加剧的闭环
代谢-免疫-屏障三轴协同恶化的最终结果,是老年多病患者体内药物的药代动力学(PK,身体对药物的作用)和药效学(PD,药物对身体的作用)发生不可预测的改变。
PK改变
肠道屏障受损影响药物吸收;菌群代谢和肝脏酶活性改变影响药物的代谢和清除。这导致血药浓度要么过高(增加毒性风险),要么过低(导致治疗失败)。
PD改变
衰老本身就可能改变药物靶点(如受体)的敏感性。菌群失调引发的慢性炎症环境可以进一步改变靶组织的反应性,使得药物效果增强或减弱,或产生非预期的不良反应。
因此,理解这个三轴互作的复杂网络,对于在老年多病患者中实现安全有效的药物治疗至关重要。
老年多病状态下肠道菌群调节药物疗效的核心机制
编辑
doi.org/10.1016/j.arr.2026.103023
在处理患有多种疾病且服用多种不同药物的老年人时,肠道细菌与药物之间的关系变得至关重要。
老年多病患者的菌群失调与药物相互作用
编辑
doi.org/10.1016/j.arr.2026.103023
氯吡格雷 Clopidogrel
氯吡格雷是动脉粥样硬化性心血管疾病(ASCVD)二级预防中最常用的抗血小板药之一。但它有一个关键前提:需要在肝脏经CYP450酶系(尤其CYP2C、CYP3A相关通路)生物活化,才能产生真正抑制血小板的活性代谢物。
在多病共存的老年人群里,我们常看到“同样用药、抑制不够、血栓风险仍高”的情况。越来越多证据提示:肠道菌群失调及其引发的代谢—免疫—屏障轴紊乱,可能系统性地削弱氯吡格雷的有效反应。
下面我们来看这到底是怎么从肠道一路影响到血小板和肝脏的。
先决定药能不能起效——肝脏活化是门槛
氯吡格雷是典型前药(它最怕的是活化环节被压住):
老年人常合并慢性炎症、代谢异常、肝脏基础疾病、肠屏障受损与菌群失调——这些因素并不是彼此独立,而是会相互放大。
再看药效端:TMAO抬高血小板反应性,出现P2Y12旁路
肠道菌群可把膳食中的左旋肉碱、胆碱代谢成TMA,再由肝脏转化为TMAO进入循环。
TMAO升高不仅是风险相关指标,也可能参与机制:它可诱导TLR4介导的信号通路,机制研究提示可能促炎/促血栓,但尚需临床因果验证。
更关键的一点是“旁路效应”:即便P2Y12被抑制,血小板仍可能通过不依赖P2Y12的激活通路被推起来,于是临床上就表现为抗血小板效果不够/耐受倾向。
是谁在调CYP开关:T细胞与炎症信号如何改写药物活化?
CYP不是单纯的代谢酶,它受免疫—炎症信号调控。T细胞参与调控肝脏CYP2C与CYP3A表达,因此不同免疫状态下,氯吡格雷活化可能走向不同方向:
肠屏障受损经肠-肝轴放大炎症,压低CYP活化
多病共存老年人常伴慢性炎症、慢性肝病与心血管疾病,这些因素可破坏肠道紧密连接,增加通透性(也就是肠漏),促使炎症信号升级。系统性炎症常见TNF-α、IL-6升高,而这类促炎因子可能下调肝脏CYP酶,让氯吡格雷更难被充分活化。
菌群失调还可能伴随FXR–FGF15信号下降,扰动胆汁酸代谢,进一步削弱屏障与代谢稳态,让炎症-屏障受损-代谢紊乱更难刹车。
另一个值得关注的线索是黏膜免疫:肠黏膜免疫受损与致病共生菌(如AIEC)定植相关,提示IL-22信号受损;这会加重菌群失调与屏障障碍,经肠-肝轴放大代谢紊乱,并间接压低氯吡格雷的生物活化能力。
小 结
衰老肠道中的菌群失调触发代谢—免疫—屏障恶性循环,削弱氯吡格雷效应。
编辑
doi.org/10.1016/j.arr.2026.103023
三轴相互放大,最终削弱氯吡格雷的抗血小板作用。
地高辛Digoxin
地高辛是经典强心苷类药物,治疗窗很窄:剂量稍低可能无效,稍高就可能出现毒性与心律失常。因此,它对任何能改变肠道吸收、菌群代谢、炎症背景与转运蛋白(P-gp)的因素都格外敏感。
在多病共存的老年人群中,我们更容易看到“同样用药,却出现疗效不足或毒性增高、波动难控”的情况。由于其药代动力学特性较为敏感,容易受到患者肠道细菌影响。
菌群代谢:Eggerthella lenta如何把药变钝?
肠道共生菌 Eggerthella lenta 可通过其相关还原酶系统(如 强心苷还原酶)将地高辛转化为药理活性更弱/无活性的代谢物(二氢地高辛,dihydrodigoxin)。微生物失调导致该菌富集,从而使血清地高辛浓度降低至治疗阈值以下,最终因对心肌的正性肌力作用不足而导致心力衰竭治疗失败。
为什么炎症状态会把“窄治疗窗”变得更危险?
菌群失调不仅影响代谢,还可能通过免疫耐受破坏(如树突状细胞相关的免疫耐受下降)推动系统性炎症。与此同时,肠道屏障受损会促进LPS移位入血,激活TLR4通路,诱导TNF-α、IL-6等促炎因子升高。
对地高辛而言,这种炎症背景的风险在于:地高辛本就与心律失常风险相关,且其毒性更容易在电解质紊乱(如低钾)等情况下被放大。炎症信号进入心脏微环境后,可能增加心肌细胞的电不稳定性,从而提高地高辛相关心律失常的风险。
P-gp 如何决定地高辛的口服吸收上限?
地高辛是经典的 P-glycoprotein(P-gp)底物。P-gp 在肠上皮细胞腔面表达丰富,可把已吸收进入细胞的地高辛外排回肠腔,从而显著限制其通过肠壁进入血液的量,影响口服生物利用度。
动物证据也支持这一点:在保留人源 P-gp、缺失小鼠 P-gp 的模型中,口服后地高辛血浆浓度显著升高,提示P-gp外排是限制暴露的重要因素。因此从理论上说,抑制肠道P-gp活性,可能改善地高辛吸收。
老年多病共存为什么使地高辛疗效更不可预测?
多种因素叠加,使多因素健康问题老年人群中的药物相互作用更具变异性与不可预测性。
小 结
微生物可通过代谢—免疫—屏障三联机制调控地高辛的药代动力学与毒性。肠道菌群失调通过三条轴影响地高辛反应。
编辑
doi.org/10.1016/j.arr.2026.103023
二甲双胍Metformin
二甲双胍(metformin)作为2型糖尿病一线用药,其疗效部分依赖健康的肠道微生态环境;相反,菌群失调可能抑制二甲双胍的治疗效果。
代谢干扰:信号通路抑制与药物代谢加速
菌群失调(特别是拟杆菌属的丰度变化)可通过特定的酶学途径干扰二甲双胍的药代动力学和药效学。
免疫调节失衡:老龄化背景下的炎症干扰
在老龄化人群中,菌群失调与免疫系统的相互作用更为复杂。
屏障功能受损:系统性代谢紊乱的恶性循环
菌群失调导致的紧密连接蛋白降解和肠道通透性增加(即肠漏),不仅影响肠道局部环境,更具有全身性影响。
临床:多病共存老年患者适应不良
在多病共存老年人群中,肠道菌群失调会驱动一种不适应性的循环,形成代谢、炎症与免疫调控异常相互交织的三联网络。
肠道菌群在该循环三条轴中的作用,对于影响二甲双胍疗效至关重要。二甲双胍可作用于肠道微生物群以帮助调节代谢过程、调控免疫反应并维持肠屏障功能;然而,老年人中由衰老与多种慢性病共同导致的重度菌群失调也被证明会对上述结局产生负面影响。
在重度菌群失调状态下,二甲双胍对各轴的有益作用被削弱,最终导致其在多病共存老年患者治疗中的有效性出现极大异质性,甚至可能治疗失败。
小 结
肠道微生物群的代谢-免疫-屏障轴相互作用调控二甲双胍反应的变异性。在老年多病共存中,菌群失调通过相互关联的机制损害二甲双胍疗效。
编辑
doi.org/10.1016/j.arr.2026.103023
免疫检查点抑制剂ICIs
免疫检查点抑制剂(如抗PD-1/PD-L1抗体)的疗效高度依赖于机体的免疫基调(Immune Tone),而肠道菌群是这一基调的关键调节者。菌群失调通过干扰代谢重编程、加剧免疫衰老及破坏屏障功能,削弱T细胞的抗肿瘤应答。
代谢:短链脂肪酸匮乏,阻碍T细胞代谢重编程
在菌群失调期间,代谢轴的异常直接抑制了免疫细胞的抗肿瘤活性。
免疫衰老与分化失衡
老龄化背景下的菌群失调会加速免疫系统的无能化和肿瘤的免疫逃逸。
屏障破坏:系统性炎症干扰
菌群失调导致的物理屏障破坏是系统性干扰的源头。
总而言之,在老年多病患者中,一个失调的肠道菌群会从能量供应、免疫细胞平衡和全身炎症状态等多个维度,全面削弱ICIs的抗肿瘤效果。
小 结
肠道微生物群紊乱会破坏肿瘤微环境中的代谢—免疫—屏障网络,从而降低免疫检查点抑制剂(ICI)的疗效。菌群失调通过三轴失衡削弱ICI效应。
编辑
doi.org/10.1016/j.arr.2026.103023
全球人口老龄化与老年人多种慢性病患病人数增加并存。当前研究持续关注肠道微生物组失调与药物疗效之间的相互作用。
为有效应对这一问题,需要对更广泛且更具效能的治疗策略开展系统性研究与实施设计,并在三个层面的相互作用中统筹考量:代谢、免疫稳态与肠道屏障完整性。采用这一多维模型,可建立全面的调控框架,使研究者能够在系统层面借助网络药理学,精准靶向微生物组与药物治疗之间的相互作用。
益生菌
益生菌是调控肠道微生物组的多种方式之一,也是最常用的干预手段之一;其通常包含多种菌株,不同菌株因独特的代谢特征而产生不同治疗效应。
某些益生菌代谢产生的常见代谢物为短链脂肪酸(SCFAs)。研究表明,SCFAs可与CYP450酶(药物代谢酶家族的重要组成部分)直接相互作用;同时,SCFAs还可通过激活TLR/NF-κB通路增强机体免疫反应,并促进肠上皮细胞中紧密连接相关蛋白的生成。
与代谢类药物联用
多项研究提示,与不同种类的乳酸杆菌属和双歧杆菌属益生菌联用,可显著增强胰岛素、二甲双胍等降糖治疗的获益,其核心可归结为两条菌群介导的代谢调节路径:
不过总体来说仍然缺乏足够的预临床/小规模临床研究,高质量 RCT仍然不足。
-临床证据
胰岛素 + 乳酸杆菌
在1型糖尿病以及未使用其他降糖药的2型糖尿病人群中,联用可降低HbA1c;且这种改善与有益菌群上升(如 Bifidobacterium animalis、Akkermansia muciniphila)相关联。
二甲双胍 + 复合益生菌
例如以 L. rhamnosus Probio-M9、L. casei、L. plantarum P-8、以及 B. animalis subsp. lactis V9/M8(Probio-M8)等组成的组合,在与二甲双胍联用时可提升SCFAs水平,并通过改善胰岛素敏感性增强血糖调控。
抗感染与抗菌生态位竞争
乳杆菌补充剂具有一定的抗微生物效应,可在感染的预防与辅助治疗中发挥作用。其可能机制包括产生多种代谢物并改变局部理化环境,例如:
-代表性联合策略
Lacticaseibacillus paracasei LC11 + 蔓越莓 + D-甘露糖
可降低泌尿道感染复发(特定人群研究)。
乳酸杆菌/双歧杆菌 + 幽门螺杆菌三联疗法
提高根除率、并有望改善疗程耐受性。
肿瘤治疗支持
化疗与放疗常诱发显著的肠道菌群紊乱与黏膜损伤,进而表现为腹泻、黏膜炎、营养不良、炎症升高等一系列问题;在老年肿瘤患者中,这些问题更可能放大为剂量下降、延迟治疗甚至停药。
益生菌在这一场景下的价值更多体现为支持治疗。其有效性可归因于其能够稳定受损的肠道屏障并调节炎症和代谢。
鼠李糖乳杆菌GG+长双歧杆菌+嗜酸乳杆菌+粪肠球菌的组合
该组合已被证明能抵消这些影响。它们通过重建肠道紧密连接和减轻炎症来实现这一目标。
临床证据支持这一屏障恢复的重要性,它已与宫颈癌患者放疗引起的腹泻减少28%相关,从而减少了抗腹泻药洛哌丁胺的使用。
Bifidobacterium BB-536
可通过增加CD8⁺ T细胞向肿瘤浸润,增强抗PD-1免疫检查点抑制剂的疗效。
此外,益生菌通过减少体重增加、低密度脂蛋白(LDL)水平和与多西他赛治疗相关的菌群失调,有助于稳定代谢紊乱。
这些干预措施提高了患者的功能评分并改善了整体生活质量。总之,将益生菌纳入癌症治疗方案中可作为一项重要的支持性措施,有效减少肿瘤治疗相关的附带损伤,并提高患者对治疗方案的耐受性和依从性。
益生菌来源BEVs的跨器官信号:增强抗PD‑1免疫治疗的潜力
发表在《Nature Communications》的一项研究显示,肠道共生益生菌——双歧杆菌(Bifidobacterium)可释放细菌来源的细胞外囊泡(BEVs),这些BEVs能穿越肠屏障被机体吸收,并远程富集于肺癌肿瘤组织。
研究发现,BEVs可被肿瘤细胞摄取,通过TLR4–NF-κB信号上调肿瘤细胞PD-L1,并增强肿瘤微环境中免疫细胞的功能。
编辑
doi.org/10.1038/s41467-025-58553-4
动物实验中,联合给予BEVs和抗PD-1抗体可显著抑制肿瘤生长,较单用抗PD-1效果更佳。
编辑
doi.org/10.1038/s41467-025-58553-4
该机制提示,益生菌可能通过囊泡信号跨器官调节作用,帮助提升免疫治疗(如PD-1抑制剂)疗效。这为老年群体通过益生菌进行辅助抗肿瘤免疫调节提供了全新理论依据。
益生元
益生元(如菊粉与抗性淀粉)可作为有益菌群(如Faecalibacterium、Roseburia)的选择性底物,通过对有益菌的间接作用优化代谢—免疫串扰。
相较活菌制剂,益生元通常具有制剂稳定性更好、质量一致性更高、免疫安全性风险更低等优势,因此在体弱老年人或多病共存人群中,更适合作为低风险的长期辅助干预。
益生元的价值不止于改善菌,还可以作为饮食-微生物-药物三者之间的桥梁。通过改变菌群结构与代谢谱,进而影响宿主对药物的反应与耐受。
茯苓多糖 + 5-FU
动物研究提示其具有一定降毒/增效潜力——例如茯苓多糖可减轻5-FU相关体重下降与肠道损伤,可能与降低促炎因子、增强屏障功能及纠正菌群失调有关。
低聚果糖 + 二甲双胍
在饮食诱导肥胖动物中,低聚果糖与二甲双胍联用较单用进一步改善血糖与体重,并降低内毒素及炎症标志物水平,同时改善菌群。
多酚类既可被菌群转化为生物活性代谢物,也可反向塑造菌群组成,因此非常适合与药物形成代谢协同的联合策略。
多酚类+药物联合治疗非酒精性脂肪性肝病
编辑
doi.org/10.1016/j.jare.2024.03.004
粪菌移植(FMT)
粪菌移植(FMT)是一种独特疗法,通过递送健康供体的功能性菌群联合体来重建肠道微生态平衡。
FMT可调节胆汁酸代谢网络,并恢复产丁酸菌(如Faecalibacterium prausnitzii)的活性,以纠正菌群失调状态。
传统FMT给药方式包括结肠镜或灌肠;而口服胶囊递送与经内镜肠内管递送等新方式显著改善了患者耐受性与依从性。
与药物联用的证据示例
肿瘤免疫治疗(PD‑1/PD‑L1)
FMT可通过免疫重塑提高ICI疗效。
炎症性肠病(克罗恩病)
一项随机试点研究提示,FMT可能提高泼尼松龙治疗患者的临床缓解率,可能与屏障修复与SCFAs恢复相关。
高血压/心血管药物反应
在动物研究中,将“氨氯地平治疗后自发性高血压大鼠”的菌群移植给未治疗大鼠,可降低血压并改善血管舒张、氧化应激与Th17浸润等系统指标;而来自氢氯噻嗪处理动物的FMT未显示类似获益,提示FMT可能转移一种“代谢编程后的微生物表型”,但关键菌种与分子通路仍待明确。
目前FMT临床研究尚未形成标准化给药方案,因此需基于个体患者及其具体健康状态制定标准化剂量策略。迄今FMT研究主要聚焦于单次或短期治疗方案,尤其是在复发性艰难梭菌感染患者中;但对与衰老相关的虚弱等慢性状态的管理,可能需要重复和/或维持剂量方案。当前研究所收集的数据表明,老龄小鼠模型可产生与人类衰老相关的参考数据。
噬菌体疗法
噬菌体治疗是一种新型策略,利用噬菌体(感染细菌的病毒)对抗耐药菌感染,具有改变抗生素耐药性感染治疗范式的潜力。
通过靶向特定微生物,噬菌体可清除耐药病原体及相关菌群(如产生β-葡萄糖醛酸苷酶的E. coli),从而降低因抗生素相关毒素导致的药物再活化和/或代谢紊乱。例如,噬菌体能够从接受伊立替康治疗患者的肠道中清除产生β-葡萄糖醛酸苷酶的E. coli,从而阻止SN-38再活化所致的肠黏膜炎。
此外,这些噬菌体还可通过IL-22依赖的再生促进杯状细胞恢复,为黏膜屏障重建提供机制基础。病原清除与黏膜屏障重建的结合,对治疗多重耐药感染具有重要临床意义,尤其适用于老年人或免疫功能受损人群。
小鼠研究显示,经饮水口服给药(每日4×10⁸ PFU,连续31天)具有安全性证据;但目前尚未报道明确的人体剂量。
通过饮水口服噬菌体以改变肠道微生物组(并具良好安全性)具有前景,但仍需进一步研究以明确长期安全性与有效性,目前也缺乏既定的临床应用时间尺度。
饮食营养干预
营养饮食策略同样是调控肠道微生物群的主要手段之一,且具有多靶点特征。通过改变饮食结构(如增加高纤维食物摄入),可提升肠道微生物多样性并改善其功能,从而构建综合防御体系。
例如,摄入抗性淀粉或富含多酚的食物可改变肠道微生物对营养物质的代谢方式,并特异性触发有利于ICI发挥作用的不同通路。
锌与N-乙酰氨基葡萄糖可通过促进紧密连接相关蛋白(如ZO-2)生成以维持肠细胞间的致密连接,同时促进MUC2产生以利于损伤后修复,从而增强肠道保护层。
对于症状多样、且常合并多重用药的老年患者,单一疗法不足以应对复杂挑战。因此,有必要建立分层、整合的管理策略,以协调上述干预之间的相互作用。
例如,将益生元与工程化噬菌体联用,可在稳定代谢、可控调节细菌群落、以及具有抗炎特点的饮食干预之间形成协同,并与免疫功能与屏障功能改善相结合。
此外,可在全面评估个体肠型、用药史及动态微生物组特征的基础上,为患者制定个体化干预方案。通过组合这些治疗策略,或可提高特定药物或药物组合的治疗效应,降低多药并用导致不良事件的风险,并提升老年人健康管理的整体照护质量。
在老龄化与多病共存叠加的现实中,肠道微生物群已从背景因素走到台前:它通过代谢、免疫与屏障三条主轴,重塑药物疗效与不良反应的边界。现有从益生菌/益生元到FMT、噬菌体与饮食等多种工具,更多是可用的起点。
面向未来,关键在于把经验性调菌推进为可计算、可验证、可迭代的精准体系。
编辑
doi.org/10.1016/j.arr.2026.103023
一方面,以单细胞测序、空间转录组与代谢通量等多组学,在老年多病模型中绘制“宿主—微生物群—药物”互作图谱,解析微生物代谢物对CYP3A4、PD-1、Claudin-18等关键节点的时空调控;
另一方面,建立多中心、纵向随访队列,将标准化菌群检测纳入常规流程(基线分型与重复采样监测),持续记录菌群谱、用药史与结局,捕捉疗效波动及耐受/耐药的演化轨迹。
与之配套的是统一标准的微生物组—药物数据库与临床级算法:让AI/机器学习在临床试验中被严格验证,把高维数据转化为可预测的反应评估和可执行的干预建议。
这些数据的验证将推动针对多病老年人的个性化“菌群-药物”管理策略,提升多重用药安全性并促进健康老龄化。
更进一步,合成生物学有望带来可递送/可响应的智能工程菌、工程化噬菌体—纳米颗粒偶联体,并以连续菌群监测提供反馈,形成“检测—建模—干预—再检测”的闭环,使干预与个体衰老轨迹同频;同时也需以长期人群研究审慎评估气候与生态变化对微生物群及健康老龄化的潜在影响。
当然,前路仍有挑战,个体差异带来的可重复性问题、长期安全性与质控标准、以及高质量随机对照证据的缺口,决定了转化必须稳扎稳打——慢即是快。但方向已清晰:在多病共存与多重用药的时代,微生物群正在成为决定疗效与风险的重要变量。
主要参考文献
Yang D, Ren D, Zhang Y, Hao Y, Yue Y, Li Q, Fan Q, Sun C, Cui M, Zhang M. The gut microbiota dysbiosis in geriatric multimorbidity: Pharmacotherapeutic implications, pathophysiological mechanisms, and precision modulation strategies. Ageing Res Rev. 2026 Jan 13;115:103023.
Preet, R., Islam, M.A., Shim, J. et al. Gut commensal Bifidobacterium-derived extracellular vesicles modulate the therapeutic effects of anti-PD-1 in lung cancer. Nat Commun 16, 3500 (2025).
Li H, Liang J, Han M, Gao Z. Polyphenols synergistic drugs to ameliorate non-alcoholic fatty liver disease via signal pathway and gut microbiota: A review. J Adv Res. 2025 Feb;68:43-62.
Al-Btoosh S, Donnelly RF, Kelly SA. Microbes and medicines: interrelationships between pharmaceuticals and the gut microbiome. Gut Microbes. 2026 Dec 31;18(1):2604867.
de Ciutiis I, Djakovic S, Cagigas ML, Masedunskas A, Smith L, Franceschi C, Fontana L. Long-term fasting and its influence on inflammatory biomarkers: A comprehensive scoping review. Ageing Res Rev. 2025 Aug;110:102797.
Herisson FM, Cluzel GL, Llopis-Grimalt MA, O’Donovan AN, Koc F, Karnik K, Laurie I, Canene-Adams K, Ross RP, Stanton C, Caplice NM. Targeting the Gut-Heart Axis Improves Cardiac Remodeling in a Clinical Scale Model of Cardiometabolic Syndrome. JACC Basic Transl Sci. 2024 Nov 20;10(1):1-15..
谷禾健康
色氨酸是连接“吃什么”“肠道菌群状态”与“大脑和情绪”的关键枢纽。它不仅是蛋白质合成必需氨基酸,也是血清素与褪黑素的重要前体,并可通过犬尿氨酸通路参与NAD+的从头合成。
近年来,大量研究发现:色氨酸在不同代谢通路之间的“分流”,很大程度上受肠道微生物群及饮食结构(如多酚、膳食纤维、碳水化合物和脂肪比例)共同调控。这种调控一旦失衡,不仅会改变肠道屏障和免疫状态,还可通过肠–脑轴深刻影响情绪、认知以及多种神经精神和代谢性疾病的发生发展。
在这一背景下,围绕“肠道微生物–色氨酸代谢–肠脑轴”的研究迅速升温:一方面,人们逐步解析拟杆菌、梭菌、乳杆菌、双歧杆菌等代表菌属如何通过多样的色氨酸代谢途径生成吲哚类、犬尿氨酸类及血清素等关键代谢物;另一方面,肠易激综合征、自闭症谱系障碍、抑郁症、多发性硬化、阿尔茨海默病、帕金森病以及代谢综合征等疾病中,色氨酸代谢及其微生物调控异常的证据也不断被报道。
基于此情况,饮食干预、益生菌、粪菌移植乃至纳米技术等多种策略,正被尝试用于重塑肠道微生态、调整色氨酸代谢稳态,以期为肠脑轴相关疾病提供新的防治思路。
本文将系统梳理色氨酸的膳食来源和主要代谢通路,重点介绍肠道微生物中特定菌属参与色氨酸代谢的机制,阐述其在肠脑轴中的作用及与多类神经、免疫和代谢性疾病的关联,并总结目前通过饮食、益生菌、抗生素及新兴技术调控色氨酸稳态的研究进展和潜在应用价值。
▸ 色氨酸的重要性
编辑
色氨酸(Trp)是一种人体无法合成、需从饮食获取的必需氨基酸,也是血清素、褪黑素和NAD+等多种人体重要化合物的前体。
血清素:一种神经递质,在情绪调节和肠道蠕动中发挥作用;
褪黑素:一种调节睡眠-觉醒周期的激素;
NAD+:是500多种酶促反应所必需的,并且在几乎所有主要生物过程的调节中起着关键作用。
总得来说,可以概括为五个关键词:
•情绪与睡眠:是血清素和褪黑素的唯一氨基酸前体,直接影响情绪稳定、抗压能力、睡眠节律和食欲控制。
•免疫与神经保护:通过犬尿氨酸通路调节免疫反应与神经系统状态,在炎症和精神疾病中扮演关键角色。
•能量与修复:可转化为烟酸,支持 NAD⁺ 合成,保障细胞能量代谢、抗氧化、防老化和 DNA 修复。
•生长与组织结构:参与全身蛋白质合成,对儿童生长发育、肌肉维持和器官功能至关重要。
•肠道与肠—脑轴:影响肠道屏障功能和菌群代谢,通过肠—脑轴联系情绪、免疫和代谢健康。
▸ 色氨酸的主要膳食来源
①肉类
禽肉:鸡肉、鸭肉、火鸡肉(尤其是去皮鸡胸肉);
猪肉、牛肉、羊肉:瘦肉部分色氨酸含量相对更高。
②鱼类和其他水产
深海鱼:三文鱼(金枪鱼、鲭鱼等)、鳕鱼;
其他常见鱼类:草鱼、鲤鱼、罗非鱼等;
甲壳类:虾、蟹;
软体类:鱿鱼、贝类等。
③蛋类和乳制品
鸡蛋:尤其是蛋白和蛋黄整体算上;
奶及奶制品:牛奶、酸奶、奶酪(尤其是硬质奶酪,蛋白浓缩后单位重量色氨酸含量更高)。
④豆类及豆制品
豆类:黄豆、黑豆、毛豆、扁豆、豌豆、鹰嘴豆、红豆、芸豆等;
豆制品:豆腐、豆干、豆皮、素鸡、豆浆(色氨酸有一定稀释,但作为日常饮品仍有贡献)。
⑤坚果与种子
南瓜籽、葵花籽;
芝麻(黑白芝麻);
花生;
腰果、杏仁、核桃、开心果等坚果。
⑥谷物与伪谷物
全谷物:燕麦、糙米、荞麦、全麦面包/面条;
其他:藜麦(伪谷物,氨基酸组成较优)、玉米、小米等。
⑦水果(含量较低)
香蕉、牛油果(鳄梨)、榴莲;
一些干果:葡萄干、枣干等(因为脱水后营养被“浓缩”);
大多数鲜果(苹果、梨、橙子、葡萄、西瓜、草莓等)蛋白含量本身就低,色氨酸自然也少,更多作用是提供维生素、矿物质和膳食纤维。
注:咖啡豆中也含有较高含量的色氨酸。
▸ 色氨酸推荐摄入量
世界卫生组织建议最佳摄入量为4毫克/千克/天,主要用于成年人维持日常需求。
★ 不同年龄和阶段的人需求有所不同
婴儿需要更高的色氨酸摄入量,推荐剂量为13毫克/千克/天;
儿童推荐摄入量为6毫克/千克/天;
孕妇为7毫克/千克/天。
为保证获得足够氨基酸及其健康效应,研究还评估了色氨酸的可耐受最高摄入量(UL),结果为4.5g/天。不过迄今尚无因膳食色氨酸过量而致不良反应的报道。
注:高碳饮食会影响脑干和下丘脑的5-羟色胺(5-HT,也称为血清素)。膳食纤维通过调节肠道菌群也改变色氨酸代谢。微量营养素亦调控色氨酸/犬尿氨酸(KYN)通路,如维生素B6缺乏损害犬尿氨酸酶并可能降低某些脑区5‑HT合成。
食物中约90%的色氨酸由小肠吸收,吸收后,主要经犬尿氨酸(KYN)途径和血清素途径代谢。
体内约95%的色氨酸(Trp)在Trp 2,3‑二加氧酶(TDO)、吲哚胺 2,3‑二加氧酶1(IDO1)和吲哚胺 2,3-双加氧酶2(IDO2)作用下分解为喹啉酸(QA)、烟酸等产物;仅约 1%–2% 的色氨酸在色氨酸羟化酶(TpH)和芳香族氨基酸脱羧酶催化下转化为5‑羟色胺(5‑HT)。
通过5-HT、KYN和吲哚途径进行的色氨酸代谢
doi.org/10.1016/j.jfutfo.2024.09.006.
★ 肠道菌群参与重要的色氨酸代谢
若进入结肠,肠道微生物群也可以通过各种代谢途径产生多种色氨酸代谢产物。
注:早在1897年,即发现大肠杆菌和霍乱弧菌可将色氨酸转化为吲哚,提示色氨酸代谢在微生物过程及其对宿主环境影响中的重要性。
微生物群可以通过在宿主体内产生代谢物直接调节色氨酸代谢,或通过参与犬尿氨酸和血清素的产生间接影响这种代谢,而色氨酸代谢的变化会影响微生物的增殖和微生物群落的多样性。目前已鉴定出许多微生物色氨酸代谢途径,包括拟杆菌属、梭菌属、双歧杆菌、链球菌属和大肠杆菌的代谢途径。
虽然某些细菌色氨酸代谢产物的转化可以在分子层面上易于定义,但识别产生的具体代谢物类型却十分复杂。这种复杂性源于不同微生物催化酶的差异。在某些情况下,两个或多个细菌之间的协作可能是色氨酸代谢物生成的必要条件。
拟杆菌属(Bacteroides)
吲哚是通过色氨酸酶(Trpase)的酶促活性产生的,这种酶广泛存在于多种肠道细菌中,包括拟杆菌属、梭菌属和大肠杆菌。
√卵型拟杆菌、多形拟杆菌等可代谢产生吲哚乙酸
拟杆菌是人体和动物肠道中的一种主要菌群,一些拟杆菌属物种,如脆弱拟杆菌(Bacteroides fragilis)、卵形拟杆菌(Bacteroides ovatus)和多形拟杆菌(Bacteroides thetaiotaomicron),可以产生吲哚乳酸(ILA)和吲哚乙酸(IAA)。
√不同拟杆菌代谢色氨酸的产物影响不同(有的减轻肠道炎症、有的影响精神状态)
研究发现,肠道中不同拟杆菌的相关代谢物的影响不同。例如B.thetaiotaomicron可提高芳烃受体(AHR)配体吲哚乙酸(IAA)和吲哚丙酸(IPA)水平,增强 AHR 激活及相关T细胞转录因子表达。在结肠炎模型中,该菌通过激活AHR并调控CD4⁺T 细胞分化,减轻炎症性肠病。
微生物色氨酸分解物对宿主生理的作用
编辑
doi: 10.1038/s41467-018-05470-4.
通过分析无菌和单菌定植小鼠盲肠及粪便样本证实,B.ovatus可提高体内IAA浓度,重塑免疫细胞群,减轻炎症,并在三硝基苯磺酸诱导性结肠炎模型中上调结肠 IL‑22 表达;吲哚代谢物本身也能刺激免疫细胞分泌 IL‑22。研究人员认为,B.ovatus 产生的 IAA 通过促进 IL‑22 生成而对结肠炎具有保护作用。
还有研究表明,B.thetaiotaomicron和B.fragilis可诱发小鼠压力相关的抑郁样行为(如焦虑和绝望),而B.ovatus无此效应。
√同属不同菌种产生的代谢物可能差异显著
亲缘关系密切的属的不同菌株之间也存在显著的代谢差异,这很可能是由于微生物的遗传代谢和化学环境存在差异。使用四种不同的拟杆菌属菌株重建的小鼠回肠中色氨酸/犬尿喹啉酸(KYNA)途径的多种变化,支持了代谢调控的这种变异性。拟杆菌属还可能诱导中枢5-羟色胺代谢紊乱,使小鼠对应激诱导的行为障碍敏感。
√膳食缺乏色氨酸还会降低拟杆菌丰度
膳食中摄入的色氨酸也会显著影响拟杆菌属的生长状态和数量变化。研究表明,当饮食中实施严格的 色氨酸限制时,不仅会明显降低粪便中拟杆菌的丰度,还会同步重塑和改变整个肠道微生物群的组成与结构。
梭菌属(Clostridium)
梭菌属中的某些物种在吲哚衍生物的合成中发挥着重要作用。Clostridium drakei是一种能够产生乙酸的细菌,与C.scatologenes类似,可通过色氨酸的分解代谢转化3-甲基吲哚。3-甲基吲哚是一种常见的肠道代谢产物,又称粪臭素,被认为会导致猪肉产生异味。它可由脆弱拟杆菌(B.fragilis)和梭菌属(Clostridium)产生。
√丁酸梭菌抗肥胖作用还源于色氨酸代谢调节信号通路
丁酸梭菌(Clostridium butyricum)可以在肠道中产生丁酸盐,具有缓解多种疾病的潜力,包括炎症性肠病和肥胖症。
几种丁酸梭菌菌株具有抗肥胖作用,但这些作用并非完全通过提高肠道中丁酸盐的浓度来实现,还主要通过改变肠道微生物群和色氨酸(Trp)代谢来实现。丁酸梭菌可能通过产生吲哚和吲哚衍生物来激活芳香烃受体(AHR)信号通路,并调节宿主的代谢和免疫系统,从而改善肥胖。
√梭菌可能富含多种色氨酸代谢途径
最近的体外实验发现,B.thetaiotaomicron通过向大肠杆菌提供单糖,抑制其产生吲哚的能力,从而增加了对生孢梭菌(Clostridium sporogenes)的色氨酸供应,进而促进了吲哚乳酸和吲哚丙酸的产生。通过在小鼠饮食中补充富含色氨酸的食物,并增加梭菌属、乳杆菌属和双歧杆菌属的数量,重塑了小鼠肠道微生物的结构和组成,从而改善了类似抑郁的行为。
据报道,梭菌属(Clostridium)可通过色氨酸(Trp)的代谢产生吲哚、吲哚衍生物和色胺。研究发现,该属的菌株具有产生喹啉酸(QA)的潜力,且预测Clostridium hathewayi具有QA途径。然而,该菌株中缺失了负责合成QA途径前体犬尿氨酸(KYN)的基因,这表明QA途径具有代谢共享性,且包括梭菌属在内的部分生物以KYN为底物,而KYN是由包括芽孢杆菌属(Bacillus)、伯克霍尔德氏菌属(Burkholderia)和假单胞菌属(Pseudomonas)在内的另一组细菌产生的。
肠道微生物群通过肠脑轴影响色氨酸代谢
编辑
doi.org/10.1016/j.jfutfo.2024.09.006.
总体来看,梭状芽孢杆菌富含多种色氨酸代谢途径,相关分析表明其在肠道中具有更强的色氨酸代谢潜力。
乳杆菌属(Lactobacillus)
乳杆菌是正常肠道微生物群的重要成员,主要包括植物乳杆菌(Lactiplantibacillus plantarum)、干酪乳杆菌(Lactobacillus casei)、嗜酸乳杆菌(Lactobacillus acidophilus)、鼠李糖乳杆菌(Lactobacillus rhamnosus)、罗伊氏乳杆菌(Lactobacillus reuteri)、清酒乳杆菌(Lactobacillus sakei)和弯曲乳杆菌(Lactobacillus curvatus)。
√乳杆菌通过上调吲哚乙酸水平可以缓解结肠炎
乳杆菌可通过芳香族氨基酸氨基转移酶和吲哚乳酸脱氢酶(ILDH)将色氨酸转化为吲哚-3-醛(IAld)和吲哚乳酸(ILA)。研究人员筛选出色氨酸代谢活性最高的 L. plantarum,并联合 Trp 观察其在 DSS 诱导溃疡性结肠炎中的作用。
结果显示,该菌上调了吲哚乙酸(IAA)水平及 AHR mRNA 表达,进而激活 IL-22/STAT3 信号通路。通过比较不同 L. plantarum 菌株的基因组和代谢组,认为 IAA 可能是导致菌株间结肠炎缓解效果差异的关键代谢物。
√鼠李糖乳杆菌通过代谢色氨酸增强肠道屏障
筛选出可在肠道定植并在体外代谢色氨酸生成吲哚衍生物的L. rhamnosus MN-431。该菌同时上调孕烷 X 受体(PXR)和芳烃受体(AHR)的表达:大量 ILA 和 IAld 作为 AHR 配体,而共享配体 IA 主要结合 PXR,通过上调 PXR、AHR 并下调核因子 κB(NF-κB),增强肠道屏障功能。
尽管其具体吲哚代谢产物及体内代谢情况尚未完全明确,但多项证据提示L.rhamnosus对肠道益生健康具有潜在益处。
√罗伊氏乳杆菌产生的吲哚-3-醛有助于抑制结直肠癌
研究表明,吲哚-3-醛(I3A)是罗伊氏乳杆菌来源的膳食色氨酸(Trp)分解代谢产物,可诱导芳香烃受体依赖的环磷酸腺苷反应元件结合蛋白(CREB)活性,从而增强 Tc1 细胞(分泌干扰素-γ(IFN-γ)的 CD8⁺ T 细胞)的效应功能,并提高免疫检查点抑制剂的抗肿瘤效果。
当 I3A 自小肠转移至肿瘤微环境后,罗伊氏乳杆菌可激活肿瘤中的 I3A–AHR–CD8⁺ T 细胞轴,促进分泌 IFN-γ 的细胞毒性 CD8⁺ T 细胞活化。有研究发现,他汀类药物也可降低结直肠癌(CRC)风险,且该作用依赖肠道微生物群。分析显示,给药后肠上皮细胞 IDO1 基因表达受抑制,肠道色氨酸水平升高,罗伊氏乳杆菌丰度显著增加,更多 Trp 被其代谢为吲哚-3-乳酸(ILA),且二者呈正相关。ILA 通过调节 TH17 反应和维持屏障完整性抑制 结直肠癌,其中视黄酸受体相关孤儿核受体 γt 和 AHR 共同参与。
双歧杆菌属(Bifidobacterium)
双歧杆菌是经顺产和母乳喂养婴儿肠道微生物群的关键成员。母乳中色氨酸(Trp)含量高于大多数婴儿配方奶粉,且富含益生元,有助于双歧杆菌等有益菌在婴儿肠道定殖。
双歧杆菌是新生儿最早定殖的微生物之一,在其生理发育中发挥重要作用。一项研究显示,约90%新生儿在出生后肠道中检出双歧杆菌,该属在成人肠道菌群中仍占约 3%–5%。常见种类包括长双歧杆菌、短双歧杆菌、青春双歧杆菌和假长双歧杆菌等。
√双歧杆菌产生吲哚乳酸有助于抑制条件致病菌
检测从人类和动物胃肠道分离的双歧杆菌,发现吲哚乳酸(ILA)是其培养上清液中的色氨酸代谢物。来自婴儿粪便的长双歧杆菌和短双歧杆菌产生的 ILA 多于其他来源分离的菌株,因此婴儿肠道中双歧杆菌定殖所形成的 ILA 可能为其在结肠菌群中提供竞争优势。ILA 具有抗菌活性,可抑制大肠杆菌在肠道中的定植。
其他肠道微生物群
√大肠杆菌、霍乱弧菌
研究发现,大肠杆菌和霍乱弧菌(V. cholerae)可将色氨酸转化为吲哚。大肠杆菌具有活跃的色氨酸和吲哚转运蛋白,色氨酸酶可将色氨酸转化为吲哚,在大肠杆菌和乳酸菌中均有表达。然而,负责吲哚后续代谢的具体微生物酶途径,以及吲哚在其他共生菌中的分布和活性仍不明确。
√肠球菌、假单胞菌
多项研究表明,部分细菌可产生色胺(tryptamine)等神经活性分子,或通过影响肠嗜铬细胞/炎症状态间接调控宿主5-HT合成。
肠道微生物将 Trp 作为吲哚、血清素和褪黑素合成前体加以代谢,从而减少宿主可利用的 Trp。假单胞菌可利用现有色氨酸合成血清素,并通过其毒性及细胞间信号传导,使肠道菌群诱导循环 Trp 水平下降,进而影响 5-羟色胺能神经传递,以及中枢神经系统和肠神经系统(ENS)的功能。
√消化链球菌
消化链球菌(Peptostreptococcus)能够代谢色氨酸(Trp),从而产生吲哚-3-丙烯酸(IA,indole-3-acrylicacid)等吲哚类代谢物。IA对肠上皮屏障的功能有积极作用,并有助于减少免疫细胞中的炎症反应。Peptostreptococcus russellii具有苯丙氨酸脱水酶基因簇(fldAIBC),该基因簇与生孢梭菌(C. sporogenes)中的fldAIBC基因簇同源,且对苯丙氨酸的代谢至关重要。
同样,厌氧消化链球菌编码完整的fldAIBC基因簇,并产生色氨酸代谢产物吲哚丙酸(IPA)和异亮氨酸(IA),而口腔消化链球菌则缺乏初始因子fldI,导致色氨酸代谢产物水平较低。
小结
围绕特定肠道菌属或菌种的色氨酸代谢开展研究,有望为微生物群靶向调控色氨酸代谢提供新思路,例如通过基因序列同源性筛选具色氨酸代谢能力的菌株。
此类研究有助于构建基于肠道微生物群的精准干预策略,以改善 Trp 代谢失衡相关疾病。聚焦特定菌种或菌株与色氨酸的相互作用,科学家可开发更精细的调控手段,优化这一关键代谢通路,从而促进整体健康并应对多种代谢性疾病。
肠道微生物群通过产生激素、免疫因子和代谢物,在大脑行为和认知发育中发挥重要作用。出生后,肠道细菌的早期定殖可促进中枢神经系统发育。肠道微生物群影响中枢神经系统的结构和功能,并可调节情绪。
★ 肠道微生物群通过产生激素、信号分子等影响神经系统功能
动物研究表明,微生物群失衡常导致焦虑样行为;相反,特定益生菌干预或重建微生物群则有助于改善焦虑相关行为:缺乏正常肠道微生物群会严重干扰中枢神经传递,而肠道微生物群恢复后,小鼠的焦虑行为可恢复正常。
肠道微生物群可通过产生神经信号分子或调节内分泌细胞激素影响中枢神经系统功能。研究证实,口服婴儿双歧杆菌后,大鼠脑内 5-HTP、5-HIAA 和苯乙酸水平显著高于对照组。肠道微生物群还能通过影响内分泌细胞皮质酮水平调节 HPA 轴反应强度,在应激反应中发挥关键作用,并调控促肾上腺皮质激素释放因子和脑肠肽等激素的分泌。
注:脑肠肽是一类同时存在于大脑和肠道的活性肽类物质,通过脑-肠轴双向调节消化、代谢、情绪等生理功能,是神经、内分泌、免疫系统的重要信号分子。
编辑
★ 色氨酸代谢是肠脑轴中的重要组成部分
肠脑轴(GBA)描述了大脑与肠道之间的双向交叉。GBA拥有复杂的沟通系统,包括中枢神经系统(CNS)、肠神经系统(ENS)和胃肠道(GI)之间的双向相互作用。
脑肠相互作用主要有三个机制:神经通路、内分泌通路和免疫通路,微生物积极参与GBA内的代谢途径,影响微生物-肠-脑轴(MGBA)相关代谢产物。这些包括微生物产生的神经递质、短链脂肪酸,以及色氨酸等的直接或间接代谢。这些过程调节下丘脑-垂体-肾上腺(HPA)轴和细胞因子的产生,以调节免疫力。
色氨酸代谢及其相关代谢物对肠脑轴至关重要。微生物肠脑轴直接影响情绪和心理状态,还与免疫系统密切相关,并且与胃肠道慢性疾病相关。
▸ 血清素通路与肠脑轴
色氨酸(Trp)及其代谢物的三条关键代谢途径在大脑与肠道间的沟通中发挥重要作用。约 90% 的体内血清素由肠嗜铬细胞(EC)合成:Trp 先经色氨酸羟化酶1(TPH1)转化为5-羟色氨酸(5-HTP),随后进一步生成血清素(5-HT)。血清素作为核心胃肠信号分子,在肠道内外神经元之间传递信息,调节肠道蠕动与运动、上皮分泌、血管扩张及营养吸收,从而影响整体消化功能。
√色氨酸代谢调节血清素水平进而影响多种组织
在中枢神经系统与胃肠道的双向通讯中,血清素在中枢和外周均作为神经递质发挥作用。外周血清素不能通过血脑屏障,而色氨酸可跨越血脑屏障并在中枢合成血清素。因此,色氨酸代谢的改变会影响外周色氨酸的利用和中枢色氨酸水平,从而调节中枢血清素代谢。
经肠脑轴介导的胃肠神经调控可通过改变血清素信号传递,影响中枢神经系统的神经传递。早期神经元具有类似内分泌器官的功能,可从大脑释放多种生物活性物质(如血清素),既参与脑发育调控,又调节内脏靶器官(如胃肠道)及全身多种组织的发育。
例如,血清素(5-HT)通过其受体参与心血管调节,这些受体在调控心血管反射以及交感、副交感神经活动中具有重要生理作用。血清素受体分布于多种心血管组织及参与心血管调节的中枢区域,其激活可对心血管功能产生多方面影响。中枢血清素缺乏会导致 5-HT 能通路异常、血清素能支配减弱,并因血清素下降而诱导海马脑源性神经营养因子表达升高,引起过度神经支配。血清素水平改变也被认为与自闭症、精神分裂症等神经精神疾病的发生密切相关。
√血清素是肠神经系统的关键信号分子
肠神经系统(ENS)由胃肠道壁内的神经成分构成,是一个独立而复杂的神经网络,可在相对独立于中枢神经系统的情况下调节肠功能。
但 ENS 又通过交感和副交感神经与中枢神经纤维相连,对肠蠕动和局部血流等基本胃肠功能至关重要。5-HT 能神经元是 ENS 中最早出现的神经元之一,而 5-HT 是 ENS 的关键信号分子。
5-HT 受体的激活与成人期神经发生和神经保护相关;成人 ENS 的正常发育依赖5-HT受体4特异性信号,神经元来源的 5-HT 与黏膜来源的 5-HT 通过互补作用共同促进肠神经系统成熟。
▸ 犬尿氨酸路径与肠脑轴
将色氨酸代谢为犬尿氨酸(KYN)的关键酶包括色氨酸 2,3‑二加氧酶(TDO)、吲哚胺 2,3‑二加氧酶1(IDO1)和吲哚胺 2,3-双加氧酶2(IDO2)。其中,TDO以肝脏表达为主;肠道黏膜及免疫相关组织中IDO1更为关键。
√犬尿氨酸代谢影响中枢神经系统,并与神经疾病相关
约95%的色氨酸经犬尿氨酸通路(KP)代谢为烟酰胺和NAD+。TDO 负责启动 KP,是该通路的主要限速酶;肝外 KP 分支则由两种 IDO 酶调控,IDO 主要在黏膜组织(如免疫系统和肠道)中活跃。KYN 通常先被羟基化为 3-HK,再转化为 3-HAA;HAA 通过非酶促中间反应迅速生成喹啉酸(QA),并进一步转化为 NAD+,为 KP 的主要终产物。另一支 KP 则在 KAT 作用下生成犬尿氨酸(KYN),参与神经调节。
KYN 及其代谢物可影响中枢神经系统,并与多种神经系统疾病相关。约 60% 的外周 KYN 可通过血脑屏障进入中枢,在星形胶质细胞中转化为犬尿喹啉酸(KYNA),其水平与神经保护相关;而喹啉酸(QA)由小胶质细胞产生,其升高与神经元兴奋性毒性有关。KYNA 与 QA 之间的失衡被认为与抑郁症、精神分裂症和多发性硬化等神经系统疾病的发生密切相关。
注:在肠神经系统(ENS)和中枢神经系统(CNS) 中,犬尿喹啉酸(KYNA)作为 N-甲基-D-天冬氨酸受体和 α7 烟碱型受体的拮抗剂,同时又是G蛋白偶联受体 GPR35 的激动剂。
▸ 色氨酸代谢物与肠脑轴
肠道微生物群可直接利用色氨酸(Trp),将约 4%–6% 转化为吲哚、色胺及吲哚酸衍生物。
肠道菌群将色氨酸转化为色胺和吲哚-3-丙酮酸(IPYA),后者进一步生成吲哚、吲哚-3-乙醛(IAAld)和吲哚乳酸(ILA)。IAAld 可转化为吲哚乙酸(IAA)和色氨酸,IAA 还可继续代谢为粪臭素;ILA 则可转化为 IA,随后生成 IPA。
√色氨酸经肠道微生物代谢产生的吲哚和色胺影响神经系统
吲哚是调节肠道的重要信号分子。吲哚及其衍生物(如 IAA)可作为 AHR 配体,通过激活小胶质细胞和星形胶质细胞介导中枢神经系统炎症。AHR 配体可能通过干扰素α受体1相关通路,发挥抗炎和神经保护作用,从而调控中枢神经系统炎症。
吲哚作为 AHR 配体,还能通过影响电压门控钾通道和线粒体 NADH 脱氢酶,调节小鼠结肠 L 细胞中胰高血糖素样肽-1(GLP-1)的分泌。研究证实,肠源性吲哚代谢物可能作用于扩展奖赏网络,尤其是杏仁核–伏隔核以及杏仁核–前岛叶皮层通路。
微生物代谢产物扩散穿过肠道屏障后,可经迷走神经将信号由胃肠道传递至中枢神经系统。在大鼠中鞘内常规注射吲哚会升高脑内吲哚和色胺水平,激活迷走神经,降低运动活动,并诱导 c-Fos 过度表达。c-Fos 是迷走神经激活的重要标志,提示吲哚可激活肠黏膜中的迷走神经传入纤维。
色氨酸(Trp)作为必需氨基酸,不仅是蛋白质合成的基本原料,也是连接肠道微生物群与中枢神经系统的重要代谢枢纽。色氨酸代谢一旦在通路比例或关键代谢物水平上发生失衡,既可影响情绪与认知等神经精神功能,又与抑郁症、精神分裂症、多发性硬化等中枢神经系统疾病,以及多种慢性胃肠道疾病的发生和进展密切相关,因此成为近年来肠脑轴相关疾病研究与干预的核心焦点之一。
肠脑轴相关疾病及治疗策略
编辑
doi.org/10.1016/j.jfutfo.2024.09.006.
▸ 肠易激综合征(IBS)
肠易激综合征(IBS)是一种常见的肠道功能紊乱性疾病,可表现为腹痛、腹泻以及伴发的焦虑和抑郁。
√肠脑轴失衡是IBS的重要特征
肠脑轴(GBA)失衡是 IBS 发病机制的突出特征之一,而肠道微生物群及其代谢产物在其中起关键调控作用。多项针对 IBS 患者粪便样本的研究发现,在门水平上,Bacillota和Bacteroidota比例升高,而双歧杆菌(Bifidobacterium)和乳杆菌(Lactobacillus)比例降低;链球菌(Streptococcus)和瘤胃球菌(Ruminococcus)数量亦有所增加。
肠道微生物群结构和组成的改变与多种胃肠道疾病相关,包括 IBS。据估计,约10%的IBS发生于胃肠炎之后,此类患者的微生物特征与腹泻型 IBS(IBS-D)及单纯腹泻患者高度相似。胃肠蠕动异常是 IBS 的重要表现之一,而肠道微生物群及其代谢物可通过影响肠道神经元、胶质细胞和肠壁肌层巨噬细胞等多种途径调节胃肠动力。
√肠易激综合征患者存在血清素合成及功能受损
目前关于 IBS 机制的研究部分聚焦于色氨酸向血清素(5-HT)的转化通路,该通路同时调节胃肠功能和情绪。据报道,IBS 患者结肠和直肠中色氨酸羟化酶1(TPH1)表达降低,导致 5-HT 合成及其功能受损。
还观察到 IBS 患者的犬尿氨酸(KYN)水平升高,IDO 活性显著增强,并且与症状严重程度呈正相关;与此同时,KYN 代谢产物犬尿喹啉酸(KYNA)水平下降,KYNA/KYN 比值降低,提示 KYNA 与 IBS 发病呈负相关。KYNA 对中枢神经系统和肠道具有抗炎、镇痛及情绪保护作用,因此其代谢失衡可能构成 GBA 功能紊乱的重要基础。
▸ 自闭症谱系障碍(ASD)
自闭症谱系障碍(ASD)是一种复杂的神经发育障碍,主要影响沟通、社交能力和行为。许多儿童和成人 ASD 患者伴有明显的胃肠道症状,如便秘和腹泻,且这些症状与 ASD 的严重程度相关。
√自闭症患者肠道多样性降低
研究表明,这些胃肠道表现与肠道微生物群及其代谢物失衡有关,可影响胃肠功能和神经生物学状态。ASD 及其他神经发育障碍患者的粪便微生物群α多样性降低,且与健康对照组相比,自闭症谱系障碍患者的Bacillota与Bacteroidota比值升高。
√某些微生物可能通过影响色氨酸可用性参与自闭症的发病机制
几项评估自闭症患者肠道微生物丰度差异的研究将自闭症症状与Prevotella、Coprococcus、Veillonellaceae丰度较低联系起来。
脆弱拟杆菌(一种胰蛋白酶合成细菌),可能会降低自闭症患者的色氨酸可用性。非色氨酸衍生的微生物代谢产物也可能起到因果作用,一项观察自闭症小鼠母体免疫激活(MIA)模型中肠道微生物代谢产物的研究显示,微生物代谢产物4-乙基苯基硫酸盐增加了46倍,如果小鼠被脆弱拟杆菌定殖,则其正常化。
√自闭症患者存在色氨酸代谢异常
多项研究提示ASD患者存在色氨酸(Trp)代谢异常:血浆Trp水平降低,KYN/Trp比值升高,说明 Trp 代谢由血清素合成通路偏向犬尿氨酸通路(KP)。
研究还发现,ASD 患者细胞利用 Trp 作为能量底物的能力低于对照组,提示 Trp 代谢减弱可能通过影响大脑早期发育、线粒体稳态及脑内免疫代谢通路而发挥作用。进一步证明,ASD 儿童与对照儿童在尿代谢物上的主要差异集中于色氨酸和嘌呤代谢通路,ASD 儿童更倾向于将 Trp 代谢为黄尿酸和犬尿喹啉酸,同时减少 KYNA 生成。对这些代谢异常的深入认识有望促进 ASD 的早期和更可靠的诊断。
▸ 抑郁症
抑郁症是一种常见的精神障碍,也是当代社会致残的主要原因之一。患者常表现为情绪低落、绝望、快感缺失、疲乏及睡眠障碍。
√肠道微生物群可能通过多条通路影响抑郁
有研究发现,抑郁症患者的肠道微生物群组成和结构与健康人明显不同,提示“肠–脑”调控具有双向性。肠道微生物群可能通过影响多条在抑郁中被扰乱的通路(包括色氨酸代谢和免疫系统)参与其发病。血清素可用性下降是抑郁症的重要特征,而其他色氨酸代谢产物(如犬尿氨酸)同样与抑郁密切相关,这些代谢改变很可能由肠道微生物群驱动。
√色氨酸及血清素水平紊乱影响抑郁症状
作为关键神经递质,血清素(5-HT)参与调控中枢神经系统的应激与适应反应,其紊乱与抑郁发作密切相关。突触间隙 5-HT 含量下降可诱发抑郁,且抑郁患者突触后膜中 5-HT 受体密度与敏感性均降低。降低吲哚胺 2,3‑二加氧酶(IDO)表达、提高脑内5-HT水平被认为是潜在治疗靶点。
血浆色氨酸降低及犬尿喹啉酸(KYNA)/色氨酸(Trp)比值升高与抑郁相关;同时,抑郁患者血浆 KYNA/喹啉酸(QA)比值下降:KYNA 通过激活 GPR35 调节 cAMP 生成,并抑制交感神经元和星形胶质细胞的Ca2+通道,降低炎症转录因子 NF-κB 表达,抑制炎症相关通路,具有神经保护和抗炎作用;相反,喹啉酸(QA)具神经毒性,参与抑郁发生。肠上皮 GPR35 还能通过微生物群调控 IAld 与 ILA 等代谢产物的平衡;GPR35 缺失可诱导类抑郁行为,而在此背景下补充吲哚-3-醛(IAld)具有潜在抗抑郁效应。
√肠道微生物群通过调节色氨酸代谢影响抑郁
色氨酸代谢的紊乱是抑郁症病理的重要因素,肠道微生物群在影响这一代谢过程中起着关键作用。研究发现褪黑素以微生物群依赖性的方式改善色氨酸代谢,缓解卵巢切除小鼠的抑郁样行为。
细菌通过多种机制在抑郁症中发挥作用,益生菌与其他细菌的结合可以通过多种机制缓解抑郁症。探索多种益生菌疗法、益生元和共生菌对于进一步了解它们恢复色氨酸代谢和缓解抑郁行为的潜力至关重要。
▸ 多发性硬化症
多发性硬化症(MS)是一种常见的中枢神经系统自身免疫性疾病,也是青年人致残的主要原因之一。研究发现,MS 患者的色氨酸(Trp)水平显著低于健康对照,且 Trp 代谢关键产物犬尿喹啉酸(KYNA)与喹啉酸(QA)的平衡变化会通过调节犬尿氨酸通路(KP)影响疾病进程,从而有望缓解 MS 症状。
√丁酸梭菌调节色氨代谢减轻多发性硬化症
实验性自身免疫性脑脊髓炎(EAE)是研究 MS 的经典动物模型。研究显示,该模型中肠道微生物群可产生犬尿喹啉酸(KYNA),进而招募并激活 GPR35 阳性巨噬细胞,促进 EAE 发展。特别值得注意的是,在无菌小鼠中,只有能够产生 KYNA 的大肠杆菌菌株才能恢复 EAE 表型,而野生型大肠杆菌不足以诱导相同效应,突出了 KYNA 在该过程中的特异作用。
鉴于肠道微生物群在其中发挥关键调控作用,通过益生菌(如丁酸梭菌)调节色氨酸代谢通路,有望减轻神经退行性疾病。肠源性色氨酸代谢物还能作为 AHR 配体调控星形胶质细胞,从而影响中枢神经系统炎症。
▸ 神经退行性疾病
神经退行性疾病包括阿尔茨海默病(AD)和帕金森病(PD)等,其中 PD 的特征为多巴胺能神经元逐渐丧失。研究表明,与对照相比,PD 患者血浆和血清色氨酸浓度明显降低,血清色氨酸的下降可能与其常见的心理症状相关。AD 患者外周血中色氨酸降解增加,同样导致血清色氨酸水平下降。
√调节色氨酸代谢影响中枢神经从而减轻帕金森病
在帕金森病(PD)中,3-羟基犬尿氨酸(3‑HK)水平升高,可能通过诱导氧化应激损伤神经元;而 KYNA 水平降低,外源性 KYNA 注射可减轻 PD 症状。
进一步研究发现,通过调节 TDO 活性和色氨酸代谢,可影响中枢神经系统内外多种KYN通路神经活性代谢物的水平;抑制 TDO 既能增强神经保护、减少蛋白毒性、预防记忆减退,又有助于缓解胃肠功能障碍,有望成为帕金森病治疗的新靶点。
√色氨酸代谢产物调控阿尔茨海默病的Aβ沉积
阿尔茨海默病(AD)的主要病理特征为神经原纤维缠结和β淀粉样蛋白(Aβ)沉积。小胶质细胞和星形胶质细胞在 AD 的进展中起关键作用,色氨酸及其代谢物可调控这两类细胞的活化,从而改善认知功能并抑制 Aβ 沉积。
阿尔茨海默病患者体内3-羟基犬尿氨酸(3‑HK)和喹啉酸(QA)水平均升高,其中 QA 在多种神经系统疾病中已被证实具有神经毒性。相比之下,犬尿喹啉酸(KYNA)具有神经保护作用;部分研究发现,KYNA 和5-羟基哚烯乙酸(5-HIAA)能诱导蛋白酶 Neprilysin 的表达,进一步支持色氨酸代谢产物在脑内调控 Aβ 水平的作用。
注:Neprilysin(脑啡肽酶,NEP)是一种锌依赖性金属蛋白酶,在人体内具有多种重要的生理功能,主要涉及肽类物质的降解。
▸ 代谢综合征
代谢综合征(MetS)包括腹型肥胖、高血压、血脂异常和胰岛素抵抗等心血管危险因素。
√代谢综合征患者的色氨酸代谢紊乱
在肥胖和 MetS 人群中,犬尿氨酸(KYN)、犬尿喹啉酸(KYNA)和喹啉酸(QA)水平升高,犬尿氨酸/色氨酸比值上升,且尿酸、甘油三酯与色氨酸(Trp)向 KYN 的转化率呈正相关。受炎症状态调节的吲哚胺2,3-二氧化酶1(IDO1)将色氨酸代谢为犬尿氨酸。犬尿氨酸途径的抑制剂可能用于治疗代谢综合征。
外周血清素(5‑HT)水平升高及色氨酸羟化酶1(TPH1)多态性与肥胖相关。研究表明,缺失 TPH1 的小鼠在高脂饮食下可避免肥胖、胰岛素抵抗和非酒精性脂肪肝,提示外周 5‑HT 抑制能量消耗,而肥胖可反过来提高外周 5‑HT 水平。不过,也有研究发现 5‑HT 与肥胖呈负相关,说明其调控机制复杂且尚未完全阐明。此外,降低 5‑HT 活性的 TPH2 变异会增加抑郁症患者发生MetS的风险。
√色氨酸转化产物吲哚衍生物调控代谢
微生物群通过色氨酸转化产生的几种吲哚衍生物可能在代谢综合征的发病机制中起作用。
吲哚本身已被证明可以刺激肠内分泌 L 细胞产生胰高血糖素样肽-1 (GLP-1),这是一种刺激胰腺 β 细胞分泌胰岛素的肠降血糖素。这种机制涉及快速抑制刺激 GLP-1 分泌的电压门控 K+ 通道,但受 ATP 合成抑制的长期影响控制,减少GLP-1分泌。
除神经系统异常外,肠脑轴(GBA)相关疾病患者还常伴有便秘、腹泻等胃肠道症状,其中肠道微生物群失调及其代谢物改变是重要原因。
因此,越来越多研究尝试通过靶向肠道微生物群治疗此类疾病。愈来愈多证据将胃肠功能障碍与肠脑轴相关疾病联系起来,肠道极可能成为这类患者的潜在治疗靶点。
饮食
饮食被认为是影响微生物色氨酸代谢的重要因素。
√多酚、膳食纤维调节肠道菌群及色氨酸代谢
最新研究发现,小米中的多酚化合物奎宁酸通过改善肠道微生物群的组成并增加微生物代谢物吲哚乙酸和犬尿氨酸发挥抗炎作用,为预防阿尔茨海默病(AD)提供了潜在靶点。
膳食纤维可以减少吲哚的产生,同时促进其他有益健康的色氨酸代谢产物的生成。
√富含麦麸的饮食调节色氨酸代谢物的合成和转化
富含麦麸的饮食有效地抑制了色氨酸向犬尿氨酸途径代谢物的转化,同时增加了褪黑激素和微生物分解代谢物,即吲哚-3-丙酸、吲哚-3-乙醛和 5-羟基-吲哚-3-乙酸。
麦麸增加了促进健康的细菌(例如,Akkermansia和Lactobacillus),它们与色氨酸衍生的吲哚类代谢物显著相关。
富含麦麸的饮食还可有效调节与免疫功能相关的微生物转化和色氨酸合成(即增加 AhR 和 IL-22 的结肠表达),同时改善葡萄糖和脂质稳态,以及增加肠道健康促进菌的丰度。
√碳水化合物:影响色氨酸代谢速率
微生物色氨酸代谢速率可能受肠腔内营养物质(如碳水化合物)可利用性变化的影响。
体外研究显示,从仔猪粪便中分离的一株利用色氨酸的细菌,在可消化碳水化合物(葡萄糖)存在时,主要将色氨酸用于细菌蛋白质合成;而在不可消化碳水化合物(低聚果糖)存在时,则以其为底物产生吲哚。
进一步研究表明,通过添加低聚果糖、抗性淀粉等不可消化碳水化合物,可提高碳水化合物可利用性,促进其代谢并增加短链脂肪酸生成,同时减少色氨酸降解及吲哚类化合物的产生。
这些结果说明,提高碳水化合物可利用性可抑制肠道微生物对色氨酸的降解,进而影响循环色氨酸库。相反,碳水化合物供应增加还能促进肠道血清素(5‑HT)合成,并与胃肠道运动增强相关——这一点已在口服多糖的小鼠实验中得到证实。微生物产生的短链脂肪酸增加也可能参与其中。
√高脂肪饮食:抑制色氨酸向吲哚代谢物的转化
研究表明,高脂饮食会耗尽小鼠盲肠中的微生物代谢产物吲哚乙酸和色胺,提示在高脂状态下微生物色氨酸降解途径减弱。
同时,高脂饮食显著提高小鼠肠道 IDO 活性,促进色氨酸向犬尿氨酸的分解代谢。肠道微环境在高脂暴露下发生改变,抑制了微生物将色氨酸转化为吲哚类代谢物,尤其是吲哚‑3‑丙酸、吲哚‑3‑乳酸和吲哚乙酸盐。这些代谢物是 AhR 激动剂,在免疫调节中具有关键作用。
益生菌
益生菌,如属于乳杆菌属和双歧杆菌属的细菌,对色氨酸代谢产生有益影响。
√益生菌促进血清素合成
一方面,益生菌,如乳杆菌和双歧杆菌中的物种,可以直接将色氨酸转化为血清素。
另一方面,一些益生菌乳杆菌菌株,如干酪乳杆菌,可以通过增加TPH1表达间接促进结肠血清素合成。
√益生菌与犬尿氨酸途径的调节密切相关
与血清素水平升高相一致,大鼠口服约氏乳杆菌(Lactobacillus johnsonii)无细胞上清液后,血清犬尿氨酸水平及肠道 IDO 活性均下降。连续口服约氏乳杆菌 8 周的人体试验亦观察到血清犬尿氨酸降低,且色氨酸含量呈上升趋势。
此前研究还发现,大鼠补充益生菌婴儿双歧杆菌(Bifidobacterium infantis)可升高色氨酸水平,并降低血循环中犬尿氨酸/色氨酸比值。
上述结果提示,部分乳杆菌和双歧杆菌益生菌可能通过抑制犬尿氨酸途径来调节宿主色氨酸代谢。
慎用抗生素
√抗生素会调节肠道微生物群对色氨酸的代谢
已有研究报道,抗生素所致的微生物变化会影响犬尿氨酸通路:微生物群耗竭可提高小鼠和猪体循环中色氨酸的可用性,并减弱其沿犬尿氨酸通路的代谢。同时,一些研究也发现,抗生素诱导的菌群改变影响猪体内微生物色氨酸降解途径的激活。随着循环色氨酸升高,口服抗生素减少了空肠中色氨酸的可用性,并降低猪大肠中微生物色氨酸脱羧活性,却增加了大肠中吲哚及其衍生物含量。
有趣的是,最新研究发现,将主要作用于大肠菌群的广谱抗生素输注至回肠末端,反而使血循环中色氨酸水平下降,并增强微生物对色氨酸的降解,从而提高大肠吲哚水平。与既往结果相对,这提示肠道微生物群在应对不同抗生素干预时,对色氨酸代谢具有复杂而独特的调节作用。
其他方法
√粪菌移植通过影响肠道菌群调节色氨酸代谢
粪便微生物群移植(FMT)也被证明对受体结肠内菌群的色氨酸代谢具有调节作用,通过设计合成菌群,可以改善肠道微生物群的功能障碍,包括色氨酸代谢。
√纳米技术、增加多糖利用位点
一些能够代谢色氨酸的稀有细菌,如B.thetaiotaomicron,可以通过多个多糖利用位点来增加其丰度。纳米技术是一种在微观层面操纵肠道微生物群相互作用的新方法。该技术在诊断和治疗肠脑轴(GBA)相关疾病方面不断取得进展
还有研究人员设计了一种口服活生物治疗剂,利用乳酸乳球菌(Lactococcus lactis),该治疗剂能增强小肠靶向性并促进外源性乳酸乳球菌的产生,从而实现大脑功能的精确调控。
下表总结了相关疾病患者中益生菌、抗生素及饮食干预等的临床试验,显示这些干预可更有效缓解神经系统疾病相关的胃肠症状。
编辑
编辑
doi.org/10.1016/j.jfutfo.2024.09.006.
注:然而,目前尚无确切证据证明这些干预能缓解神经系统问题,难以直接肯定或否定其疗效,因为它们可能对由不同因素导致的神经系统疾病产生不同影响。
肠道微生物群、肠脑轴与色氨酸代谢共同构成了贯穿局部肠道环境与全身神经功能的核心网络。不同菌群通过竞争性利用色氨酸、分泌多种吲哚及其衍生物、调节血清素及犬尿氨酸等关键代谢通路,不仅重塑肠黏膜屏障和局部免疫微环境,也通过迷走神经、循环代谢物和炎症信号将“肠道事件”转译为情绪、认知及行为的改变。可以说,肠道微生物群对色氨酸代谢“走向”的精细调控,是肠脑轴发挥双向调节功能的重要生物学基础。
在此背景下,许多神经精神及肠道相关疾病可被重新理解为“微生物群–色氨酸代谢–肠脑轴”失衡的不同表现:一端是菌群多样性下降、关键代谢菌减少或致病菌增殖,另一端则是色氨酸流向偏移、保护性代谢物不足或神经毒性代谢物累积。
饮食模式变化、抗生素使用、感染、压力等多种因素,都可能通过扰动肠道微生态和色氨酸代谢,推动疾病的发生与进展。这一视角为从源头干预肠道微生物群、重构肠脑轴稳态提供了理论支撑。
未来,围绕这三者间的关系仍有大量问题有待解答。需要进一步解析不同菌群在色氨酸代谢网络中的分工与协同,明确哪些代谢通路、哪些受体信号是真正决定肠脑轴“方向”的关键节点;需要通过严谨设计的临床试验评估饮食干预、益生菌制剂、粪菌移植及新型靶向技术在神经精神结局上的真实获益与适用人群;更需要在此基础上构建以肠道菌群特征和色氨酸代谢谱为核心的分层管理与预测体系,推动从“经验调节微生态”走向“精准重塑肠脑轴”。
随着基础研究与转化医学的不断推进,以肠道微生物群‑肠脑轴‑色氨酸代谢为靶点,有望成为未来干预多类神经及代谢相关疾病的关键突破口。
注:本账号内容仅供学习和交流,不构成任何形式的医疗建议。
主要参考文献
Yuxuan Xia, Chuan Zhang, Leilei Yu, Qingsong Zhang, Arjan Narbad, Wei Chen, Qixiao Zhai, Fengwei Tian,Tryptophan metabolism and the gut-brain axis: focus on specific gut microbial genera,Journal of Future Foods,Volume 6, Issue 5,2026,Pages 740-752,ISSN 2772-5669,https://doi.org/10.1016/j.jfutfo.2024.09.006.
Seo SK, Kwon B. Immune regulation through tryptophan metabolism. Exp Mol Med. 2023 Jul;55(7):1371-1379.
Roager HM, Licht TR. Microbial tryptophan catabolites in health and disease. Nat Commun. 2018 Aug 17;9(1):3294.
Agus A, Planchais J, Sokol H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe. 2018 Jun 13;23(6):716-724.
Kundi ZM, Lee JC, Pihlajamäki J, Chan CB, Leung KS, So SSY, Nordlund E, Kolehmainen M, El-Nezami H. Dietary Fiber from Oat and Rye Brans Ameliorate Western Diet-Induced Body Weight Gain and Hepatic Inflammation by the Modulation of Short-Chain Fatty Acids, Bile Acids, and Tryptophan Metabolism. Mol Nutr Food Res. 2021 Jan;65(1):e1900580.
Mörkl S, Butler MI, Holl A, Cryan JF, Dinan TG. Probiotics and the Microbiota-Gut-Brain Axis: Focus on Psychiatry. Curr Nutr Rep. 2020 Sep;9(3):171-182. doi: 10.1007/s13668-020-00313-5. Erratum in: Curr Nutr Rep. 2020 Sep;9(3):183.
Yano JM, Yu K, Donaldson GP, Shastri GG, Ann P, Ma L, Nagler CR, Ismagilov RF, Mazmanian SK, Hsiao EY. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell. 2015 Apr 9;161(2):264-76.
Zhang Y, Fan Q, Hou Y, Zhang X, Yin Z, Cai X, Wei W, Wang J, He D, Wang G, Yuan Y, Hao H, Zheng X. Bacteroides species differentially modulate depression-like behavior via gut-brain metabolic signaling. Brain Behav Immun. 2022 May;102:11-22.

谷禾健康
明明滴酒未沾,却突然出现醉酒症状:说话含糊、步态不稳、意识模糊,严重者甚至被交警查出酒驾。
这听起来像天方夜谭,但在医学上,这是一种真实存在的罕见疾病——自酿综合征(Auto-Brewery Syndrome, ABS),也称为肠道发酵综合征。
其核心特征是:在未摄入外源性酒精的情况下,肠道微生物异常发酵碳水化合物,产生并吸收过量乙醇,使血液/呼气酒精水平可达到甚至超过法定阈值,从而出现类似醉酒的一系列神经与胃肠道症状。由于临床表现与饮酒高度相似,自酿综合征在现实中常被误解为酒精依赖、精神问题或刻意隐瞒饮酒史,患者往往经历漫长的误诊与社会压力,甚至常面临法律纠纷。
研究显示,70%的自酿综合征患者报告曾被朋友或家人怀疑撒谎,40%因此失去工作。提高公众和医疗界对ABS的认知,是改善患者生活质量的关键。
近期发表在《Nature Microbiology》的一项研究,通过对22名ABS患者和21名健康家庭成员的系统分析,首次揭示了这种罕见病的微生物学机制,为我们打开了一扇理解肠道菌群与人体健康关系的新窗口。
编辑
本文从自酿综合征的临床表现切入,结合最新《Nature Microbiology》上发表的研究队列从“菌群组成—功能通路—代谢物—体外发酵验证”四个层面系统呈现 ABS 的微生物学机制:发作期以变形菌门(肠杆菌科)异常富集为特征,血液酒精浓度与其相对丰度正相关;宏基因组功能注释显示混合酸发酵、异型乳酸发酵及乙醇胺利用等产醇相关通路显著增强,代谢组学提示乙酸显著升高并与醉酒程度关联。
在此基础上,本文进一步梳理易感人群与诱因、诊断思路与鉴别要点,并结合宏基因组与代谢证据说明产乙醇菌群的形成与维持机制;鉴于常规干预在部分患者中疗效有限且复发风险较高,本文还引入 FMT 个案,作为微生态“重置”策略的临床补充证据,为理解菌群干预与症状逆转之间的关联提供线索。
编辑
从未喝酒,为何会醉酒?
自酿综合征(简称ABS)是一种由于肠道微生物异常发酵碳水化合物,产生过量乙醇(也就是酒精)而导致的罕见代谢紊乱疾病。
患者在没有摄入任何酒精的情况下,血液酒精浓度可达到甚至超过法定酒驾标准(80 mg/dL),出现典型的醉酒症状。这种疾病最早于1948年由Ladkin和Davies首次报道,当时一名5岁非洲男孩因肠道异常发酵导致严重腹胀和穿孔死亡。
在日本,这种疾病被称为”meitei-sho”(肠内酒精发酵综合征),并在20世纪70年代有过系列报道。而在现代医学中,自酿综合征仍然常常被误诊为酒精依赖或精神疾病,许多患者在确诊前往往经历数年的误诊和社会误解。
患者的真实困境:从酒鬼到患者
Malik等人在2019年报道了一个典型案例:一名46岁男性在接受头孢类抗生素治疗拇指外伤后,出现性格改变、抑郁和脑雾症状。一次清晨,他因疑似酒驾被捕,血液酒精浓度高达200 mg/dL,但他坚称未饮酒。经过多次辗转,最终通过粪便检测发现大量酿酒酵母(Saccharomyces cerevisiae),确诊为自酿综合征(ABS)。
“最令人心碎的是,连医生和警察都不相信我没有喝酒。我被贴上了酒鬼的标签,失去了工作和朋友的信任。直到确诊ABS,我才终于洗清了冤屈。”
—— ABS患者自述
这种疾病不仅带来身体上的痛苦,更带来严重的社会和法律问题。据统计,已有多起ABS患者因酒驾被起诉后,通过医学证据最终被判无罪的案例。
如果说自酿综合征(ABS)是一个没喝酒也会醉的怪病,那么它的本质,其实就是肠道微生态,被重塑成了一座高效的小型酿酒厂。这一章节,我们就沿着最新《Nature Microbiology》队列研究的证据,看看这座酿酒厂是如何一步步搭建起来的。
自酿综合征:从正常发酵到病理酿酒
正常情况下,肠道菌群处于动态平衡状态,少量乙醇的产生会被肝脏迅速代谢。但在ABS患者中,这种平衡被打破,特定微生物过度生长,将摄入的碳水化合物大量转化为乙醇。
而在 ABS 患者身上,发生了三件关键的事:
1
菌群结构改变
原本占主导的共生菌减少,某些能高效产乙醇的细菌占位。
2
代谢通路重编程
多条能把糖和乙醇胺酿成酒的通路被全面激活。
3
代谢产物堆积,并进入血液
乙醇与相关代谢物(尤其是乙酸)在肠道内大量生成、被吸收,超出肝脏清除能力,如果超过一定量,甚至会诱发系统性中毒。
下面我们分步骤拆解这三层变化。
菌群失衡:谁在主导酿酒?
变形菌门(Proteobacteria)显著富集
在这项纳入 22 名经严格口服葡萄糖激发试验证实的 ABS 患者、21 名家庭伴侣和一组健康对照的研究中,宏基因组测序有一个醒目的共同点:
编辑
编辑
⭐️ 三个关键的酿酒菌(产乙醇)
多变量分析(校正家庭环境影响)指出了三种重要的菌群:
大肠杆菌(Escherichia coli)
肺炎克雷伯菌(Klebsiella pneumoniae)
Ruminococcus gnavus
⭐️ 有益菌下降
编辑
总的来说,ABS患者整个微生态从多样而稳定,滑向“低多样性 + 病理性富集”的状态。
产乙醇代谢通路拉满
如果说上述菌群失衡回答了谁在干,那代谢通路分析则是回答了他们在干什么。
研究利用宏基因组功能注释,对代谢通路进行比对,发现:
其中,与乙醇生产直接相关、在发作期明显富集的关键通路包括:
编辑
⭐️ 混合酸发酵通路(Mixed‑acid fermentation)
编辑
⭐️ 异型乳酸发酵通路(Heterolactic fermentation)
⭐️ 乙醇胺利用通路(Ethanolamine utilization)
乙醇胺来自于肠道上皮细胞膜磷脂的分解,是一种内源性氮源和碳源。一些细菌可以通过专门的乙醇胺利用操纵子,把乙醇胺转化为乙醛、乙酰‑CoA,最终还原为乙醇。
在 ABS 发作期:
编辑
⭐️ 基因水平与血液酒精浓度高度相关
研究者发现,所有与酒精脱氢酶活性相关基因总丰度,与患者的血液酒精浓度高度相关(Spearman R = 0.72,P=2.7×10⁻⁶)。
编辑
这就把微生物基因层面与临床醉酒程度直接关联起来,不是只有菌在那儿摆着,而是在真正参与乙醇的大量生产。
体外实验证实:这些菌真的在酿酒
为了避免一切只是推测,研究团队还做了一个非常关键的实验——体外粪便厌氧培养测乙醇。
取 100 mg 粪便,在含葡萄糖的厌氧培养基中培养 24 小时,并在 0、6、24 小时测定培养液中的乙醇浓度(HPLC/酶法)。结果显示:
ABS 发作期样本:中位乙醇浓度约 14.47 mg/dL;
同一患者缓解期样本:约 8.76 mg/dL;
家庭伴侣样本:约 5.00 mg/dL。
ABS 发作期(flare)样本的体外产乙醇能力更强,并且与采样时的血液酒精浓度相关。
关键菌群:细菌而不是真菌
自 ABS 被提出以来,很多早期病例把矛头指向肠道酵母过度生长,临床上因此常常首选抗真菌药物治疗。但这项队列研究给出了更细致的答案。
在体外培养体系中加入不同药物:
广谱抗生素——氯霉素
广谱抗真菌药——两性霉素B
代谢产物:乙酸是ABS的生物标志物
除乙醇外,ABS患者的粪便代谢组学分析显示,ABS 发作期患者的粪便乙酸水平显著高于家庭伴侣(P=1.2×10⁻⁵),且与血液酒精浓度呈正相关(R=0.6, P=0.00018)。
乙酸是乙醇代谢的中间产物,同时也可作为底物被某些细菌用于乙醇合成,形成乙醇-乙酸循环,这可能是ABS患者乙醇持续产生的另一重要原因。
编辑
在菌群–代谢物相关性分析中:
乙酸水平与 Escherichia、Blautia 等菌属呈正相关;与 Akkermansia 等典型有益菌负相关。
也就是说,比起频繁做口服葡萄糖激发试验,监测粪便乙酸或许在随访中更具可行性。
抗生素
多项研究表明,抗生素使用是ABS常见的诱因。抗生素会破坏肠道正常菌群平衡,导致产乙醇菌过度生长。
在Malik报道的案例中,患者在使用头孢氨苄(cephalexin)3周后出现ABS症状。
另一项研究发现,长期使用阿莫西林-克拉维酸、甲硝唑等广谱抗生素的患者,肠道变形菌门比例显著增加,ABS风险提。
基础疾病:肠道动力障碍与吸收不良
ABS常与以下基础疾病相关:
短肠综合征
肠道切除后,食物通过加快,碳水化合物未充分吸收即进入结肠,成为产乙醇菌的底物。
克罗恩病
肠道炎症和狭窄导致食物滞留,促进细菌过度生长。
糖尿病
高血糖环境促进酵母和某些细菌生长,研究显示糖尿病患者内源性乙醇水平显著高于健康人。
胃轻瘫
胃排空延迟导致食物在胃肠道停留时间延长,增加发酵机会。
饮食因素:吃太多碳水化合物
饮食中的碳水化合物是肠道菌群产乙醇的原料。研究发现,ABS患者在摄入高碳水化合物食物(如面包、 pasta、含糖饮料)后,症状明显加重,血液酒精浓度可在2-8小时内达到峰值。
一项病例报告显示,一名ABS患者在食用披萨和苏打水后,血液酒精浓度迅速升至400 mg/dL,出现严重醉酒症状。
遗传因素:酒精代谢酶的个体差异
酒精脱氢酶(ADH)和乙醛脱氢酶(ALDH)的基因多态性可能影响ABS的临床表现。
亚洲人群中常见的ALDH2*2突变(导致乙醛脱氢酶活性降低),可能使ABS患者更容易出现乙醛蓄积,加重醉酒症状。
警惕这些不寻常的醉酒症状
ABS的临床表现与普通醉酒相似,但具有以下特点:
关键诊断试验:碳水化合物激发试验
碳水化合物激发试验具体步骤如下:
1. 患者需禁酒48小时,空腹8小时
2. 基线血液酒精浓度和尿酒精代谢物(乙基葡萄糖醛酸苷、乙基硫酸酯)检测
3. 口服100-200克葡萄糖(或标准化碳水化合物餐)
4. 在0.5、1、2、4、8、12、24小时监测血液酒精浓度。
5. 若任何时间点血液酒精浓度≥0.01 g/dL(10 mg/dL),且排除外源性酒精摄入,则可诊断ABS。
注意:该试验需在严格医疗监督下进行,因为部分患者可能出现严重醉酒,甚至酒精中毒。
微生物检测
通过肠道菌群检测,可以从产乙醇相关菌群(如大肠杆菌、肺炎克雷伯菌)是否超标,以及相关代谢通路是否异常,来辅助判别是否存在自酿综合征的可能。
排除其他疾病
ABS需与以下疾病鉴别:
-酒精依赖
患者通常隐瞒饮酒史,尿酒精代谢物检测阳性
-肝性脑病
肝功能异常,血氨升高,无肠道产乙醇证据
-糖尿病酮症酸中毒
血糖显著升高,尿酮体阳性
-药物中毒
如苯二氮䓬类、阿片类药物过量
-罕见神经系统疾病
如发作性共济失调、卟啉病
饮食干预:减少酿酒原料
低碳水化合物饮食是ABS的基础治疗。具体建议包括:
一项病例系列研究显示,80%的ABS患者在严格低碳水饮食(每日碳水化合物<50克)6周后,症状显著改善,血液酒精浓度恢复正常。
抗生素治疗
根据微生物检测结果选择针对性药物,例如利福昔明、甲硝唑等。需注意,抗生素可能进一步破坏肠道菌群平衡,因此通常仅在严重病例中短期使用,可以考虑同时联合益生菌治疗。
益生菌调节
益生菌可通过竞争营养和黏附位点,抑制产乙醇菌生长。研究显示,Lactobacillus acidophilus、Bifidobacterium infantis等菌株可降低ABS患者肠道pH值,减少乙醇产生。
一项病例报告显示,一名ABS患者在使用多菌株益生菌(含12种细菌)1.5年后,症状缓解,可正常饮食。
长期管理:预防复发的关键
ABS治疗后复发率较高,长期管理需注意:
虽然前述多种常规治疗手段(包括抗生素、限制碳水、益生菌等)能在部分病例短期内缓解 ABS 症状,但对病情较重或反复发作的病例而言,疗效往往不够理想,难以在短时间内达到预期的控制目标。
对于难治性ABS,粪菌移植(FMT)可能是一种值得考虑的选择。该研究报道了一例ABS患者接受多轮FMT并长期随访的案例。这不仅丰富了我们对ABS发生机制的认识,也为探索菌群干预在临床治疗中的应用潜力提供了重要参考。
因此,我们将在下一章对 FMT 的这个案例进行专门介绍。
这个FMT案例是围绕单个患者的多轮 FMT、长期随访与宏基因组分型,系统性地提供了微生态干预下 ABS 逆转的因果证据。
粪菌移植干预过程和结果
患者背景
第一次 FMT:暂时好转
在 FDA 单例扩展使用批准下,接受:
结果:
第二次FMT
在首次FMT后9个月接受了第二次FMT。
采样时间线
编辑
这次口服万古霉素、甲硝唑和复方新诺明预处理3天,随后进行肠道准备。
注:
文中对该方案的解释:第一次用利福昔明与新霉素主要为肠腔内抗生素,全身吸收很少;
而第二次加入的甲硝唑与复方新诺明为可全身吸收抗生素,旨在不仅作用于肠腔内微生物,也可覆盖肠腔外/黏膜相关部位,包括绒毛刷状缘与肠隐窝等微生物,可能以生物膜或低代谢状态持续存在的部位。
给药与维持:预处理后先给 3 次、每次 15 粒胶囊;随后在不再预处理的情况下,用同一供者来源胶囊进行每月维持 15 粒,持续 6 个月。
辅助措施(促进定植):
同时嘱咐每日服用一大汤匙马铃薯淀粉,约 60% 抗性淀粉,溶于水;
复合益生菌每日 1 粒,含 Bifidobacterium infantis、Clostridium butyricum、Clostridium beijerinckii、Anaerobutyricum hallii、Akkermansia muciniphila,以促进移植菌群的持续存在/定植。
结果:
肠道菌群变化与临床关联
粪菌移植后菌群结构变化
第一次 FMT 后的缓解期样本,在主成分图上与供体的菌群组成聚在一起;
症状复发时,样本又跑回类似初始发作期的菌群结构。
第二次 FMT 后,菌群持续稳定且不同于术前/家庭成员菌群。
编辑
主成分贡献最大的菌
编辑
发作期 E. coli 丰度高;K. pneumoniae、R. gnavus 未显著增加。
编辑
FMT后大肠杆菌丰度、血液酒精浓度和发酵通路基因显著降低。
代谢和临床指标相关性
菌群应答、血乙醇浓度、AST/ALT、产醇相关通路丰度紧密相关。
编辑
同一时间轴上同时给出 BAC(血/呼气酒精)、AST/ALT、以及 fermentation pathway enrichment(混合酸发酵/异乳酸发酵/乙醇胺利用等的富集指标),并与菌群变化/发作缓解标注对应。
症状与供者菌株植入率正相关;与术前菌株保留(post-FMT 与 pre-FMT 共享比例)负相关。
编辑
总的来说,对这位患者而言,ABS 的发生与一个特定高产乙醇耐乙醇的大肠杆菌菌株群落密切相关;通过 FMT + 系统性抗生素预处理,成功完成菌群置换,症状得以长期缓解。
自酿综合征(ABS)表面上是一种罕见而离奇的疾病,却在机制、诊断与干预的层层展开中,清晰指向同一个核心事实:肠道菌群具备强大的代谢能力,其结构与功能的失衡,足以在人体内酿出可被检测、可致症状的乙醇水平。
面向未来,ABS 的意义并不止于罕见病本身。研究提示,即便在非 ABS 人群中也可能存在低水平内源性乙醇产生,这一现象或与非酒精性脂肪肝、肥胖等代谢异常相关;例如,肺炎克雷伯菌产生的乙醇可诱导动物出现肝脏脂肪变性。由此,ABS 也可能成为理解“菌群—代谢疾病”连接机制的窗口,并为常见代谢病提供新的干预靶点。
治疗层面,ABS 有望走向更具针对性的精准医疗路径:
与此同时,提高诊断率同样关键——ABS 的真实患病率可能被低估(有研究估计每 10 万人中或有 1–2 例),推动临床认知、完善流程并探索更便捷的检测手段(如肠道菌群检测),将有助于患者更早被识别、更少陷入误解与法律困境。
当肠道菌群喝醉,我们需要做的不仅是控制血液酒精和缓解症状,更是把 ABS 放回宿主—微生物共生系统的框架中重新理解:这并非人体的例外,而是微生物代谢能力在特定条件下的极端呈现。
通过提高认知、规范诊断与发展靶向菌群治疗,我们或许不仅能更好地帮助 ABS 患者,也能借此进一步理解菌群在代谢健康中的位置与边界。
在这个由 100 万亿微生物组成的“超级器官”面前,我们对生命的理解或许才刚刚开始。
主要参考文献
Hsu CL, Shukla S, Freund L, Chou AC, Yang Y, Bruellman R, Raya Tonetti F, Cabré N, Mayo S, Lim HG, Magallan V, Cordell BJ, Lang S, Demir M, Stärkel P, Llorente C, Palsson BO, Mandyam C, Boland BS, Hohmann E, Schnabl B. Gut microbial ethanol metabolism contributes to auto-brewery syndrome in an observational cohort. Nat Microbiol. 2026 Jan 8.
Dinis-Oliveira RJ. The Auto-Brewery Syndrome: A Perfect Metabolic “Storm” with Clinical and Forensic Implications. J Clin Med. 2021 Oct 10;10(20):4637.
Malik F, Wickremesinghe P, Saverimuttu J. Case report and literature review of auto-brewery syndrome: probably an underdiagnosed medical condition. BMJ Open Gastroenterol. 2019 Aug 5;6(1):e000325.
Xue G, Feng J, Zhang R, Du B, Sun Y, Liu S, Yan C, Liu X, Du S, Feng Y, Cui J, Gan L, Zhao H, Fan Z, Cui X, Xu Z, Fu T, Li C, Huang L, Zhang T, Wang J, Yang R, Yuan J. Three Klebsiella species as potential pathobionts generating endogenous ethanol in a clinical cohort of patients with auto-brewery syndrome: a case control study. EBioMedicine. 2023 May;91:104560.
Tameez Ud Din A, Alam F, Tameez-Ud-Din A, Chaudhary FMD. Auto-Brewery Syndrome: A Clinical Dilemma. Cureus. 2020 Oct 16;12(10):e10983.

谷禾健康
Akkermansia muciniphila(AKK菌)因其独特的黏蛋白降解能力和与宿主健康的复杂关系,目前已成为微生物学和医学研究的前沿热点。谷禾此前已对该菌进行过系统性介绍。
肠道重要菌属——Akkermansia Muciniphila,它如何保护肠道健康
本文将继续深入探讨AKK菌的研究前沿。
2004年,当这株微小的厌氧菌首次从人类粪便中被分离出来时,没人预料到它会凭借独特的黏蛋白降解能力(当然目前又陆续发现了几个黏蛋白降解菌),在此后近二十年间掀起一波又一波的研究热潮。
更引人关注的是AKK菌呈现出耐人寻味的双面性:它既是下一代益生菌的希望之星,与代谢健康、长寿紧密相连;又在某些特定情境下显现出促进疾病的潜在风险。这种复杂的功能属性,正是当前科研亟待厘清的关键议题。
三篇发表于顶级期刊的AKK菌研究为该领域带来了重要突破:
这三篇文献从不同维度构建了迄今为止最为全面和深入的AKK菌研究图景,涵盖了其基本生物学特性、复杂的基因组结构与菌株多样性、与宿主免疫系统的精密互作机制,以及在不同疾病模型中呈现的矛盾表型,系统解析了这些现象背后的科学逻辑。
因此,本文谷禾整合了这三篇文献的核心观点,以科研从业者的视角带领大家深入解构AKK菌的研究进展。我们希望与各位共同探讨一个关键问题:如何理性认知并合理应用AKK菌这个双面细菌,从而真正实现精准调控肠道健康的目标。
嗜黏蛋白阿克曼氏菌(Akkermansia muciniphila),简称为AKK菌,属于疣微菌门。这个名字是为了纪念荷兰微生物生态学家Antoon Akkermans博士,而“muciniphila”则源自拉丁语,意为偏爱黏蛋白。
注:Antoon Akkermans:荷兰瓦赫宁根大学(Wageningen University)微生物生态学家,对土壤和肠道微生物研究有重要贡献。
AKK菌的发现和命名具有里程碑意义
2004年,仅使用黏蛋白(mucin)作为唯一的碳源和氮源进行富集培养了该菌。AKK菌具备独特的酶系统,能够降解并利用黏蛋白作为其生存和生长的主要能量来源,这种自给自足的能力使其在竞争激烈的肠道环境中占据了稳定的一席之地。
其模式菌株为Akkermansia muciniphila MucT(亦写作Muc5),拥有多个菌株编号,如ATCC BAA-835、DSM 22959等,是目前研究中应用最广泛的菌株。
生态位
AKK菌在人体肠道中占据着一个非凡的生态位——肠道黏液层。这一层由宿主杯状细胞分泌的黏蛋白糖蛋白构成,是隔开肠道上皮细胞与肠腔内大量微生物的第一道物理和化学屏障。
从分布来看,AKK菌不仅存在于人类肠道,也广泛栖息于包括小鼠、牛、猪、兔等多种脊椎动物的胃肠道。
形态与基本特性
在显微镜下,AKK菌呈现为一种椭圆形、不运动、不产芽孢的革兰氏阴性严格厌氧菌。
AKK菌并非绝对厌氧
研究发现,在只有微量氧气的环境中,它不仅能存活,甚至可能生长得更好。这主要得益于其基因组编码的细胞色素 bd 氧化酶复合物等耐氧机制。
也正因为具备这种有限耐氧能力,AKK菌才能适应并定植于结肠黏液层——这里处在厌氧的肠腔与相对更富氧的上皮细胞之间,存在明显的氧梯度。
结构特征,驱动互作
AKK菌约2.66Mb的基因组,是其特殊生活方式的说明书。其中最引人注目的,就是黏蛋白利用基因簇(Mucin Utilization Loci, MULs)。
黏蛋白转运系统:把食物带回家再吃的策略
Grant等人的研究指出,这些基因编码了一套精密的黏蛋白转运系统,能将黏蛋白大分子吞入菌体内部一个叫黏蛋白体的特殊结构中进行降解。这种偏自私的代谢策略,有点像把食物带回家再吃,从而最大限度地减少了与肠道中其他微生物的营养竞争,进而巩固自身的生存优势。
编辑
此外,它的细胞表面也布满了武器。电镜观察到的菌毛样结构,特别是重要的外膜蛋白Amuc_1100,不仅参与黏附,更是与宿主免疫系统直接对话的关键分子。
其细胞壁的肽聚糖层含有非乙酰化的葡糖胺残基,这在革兰氏阴性菌中相当罕见,使其能被宿主的NOD1和NOD2受体识别,从而触发免疫应答。这些独特的结构,都为它与宿主之间复杂的相互作用埋下了伏笔。
生态分布:影响因素和趋势
AKK菌在人群中的分布呈现出鲜明的特征
根据人类肠道微生物组图谱对来自20个国家3268名健康人的数据分析,AKK菌在约40.3%的健康供体中被检测到,平均相对丰度为1.24%。这个数字看似不高,但其分布的倾向性却极具信息量。
AKK菌的分布图谱
编辑
Grant et al., Nature Microbiology
a) 在健康人群中,其丰度和患病率存在地理和性别差异。
b) 在不同疾病状态下,其丰度呈现显著变化,例如在IBD中减少,而在帕金森病中富集。
c) 其相对丰度随生命周期动态变化,在百岁老人中再次出现高峰。
d) 进化树揭示了AKK菌属内部复杂的系统发育关系。
从图1中我们可以清晰地看到:
-地域与生活方式
工业化人群中的丰度更高,这暗示着饮食或生活方式可能是其丰度的重要调节因素。
-年龄
AKK菌在婴儿期迅速定植,成年后逐渐下降,但在百岁老人中却意外地再次富集,被认为是健康长寿的潜在标志物。Luo等人的文章也系统总结了这一现象,指出AKK菌在人类中的年龄依赖性定植模式与在小鼠中的模式恰好相反,这提醒我们在选择动物模型时需格外谨慎。
-疾病状态
这是最能体现其双面性的一点。在炎症性肠病(IBD)患者中,AKK菌丰度显著降低(UC患者中为9.9%,克罗恩病患者中为14.3%);然而,在帕金森病(90.3%)、某些癌症(如黑色素瘤,65.9%)和动脉粥样硬化(83.3%)患者中,其丰度却异常增高。这种看似矛盾的分布模式,强烈暗示AKK菌的角色并非简单的好或坏,而是深度依赖于宿主的病理生理背景。
Tips:看到这些分布数据,首先想到的不是简单的因果关系,而是一个复杂的生态反馈回路。例如,在IBD中,AKK菌的减少可能是肠道炎症环境恶化、黏液层破坏的结果,而非原因。反之,在帕金森病中,其增多也可能是一种代偿性反应,或是神经退行性疾病引发的肠道环境改变所致。将AKK菌丰度作为疾病的生物标志物时,我们必须高度审慎,通过系统证据加以判别:该因素究竟是事件升级的关键驱动(促进因素),是对事态的响应性干预(缓解因素),还是与结果无显著因果关联的伴随出现(旁观变量)。
所以构建真实世界的特定疾病样本人群数据才能更有助于解析复杂的肠道微生态,类似的菌还有如活泼瘤胃球菌和普雷沃氏菌等相互矛盾的研究。
三篇文献都不约而同地强调了菌株水平多样性的重要性。
长久以来,大多数研究都围绕着模式菌株MucT(ATCC BAA-835)展开。然而,随着基因组学技术的发展,AKK菌家族的内部复杂性逐渐浮出水面。基于全基因组测序,研究人员已将AKK菌划分为至少四个系统发育群(phylogroups, AmI-AmIV)。
这些不同发育群的菌株虽然16S rRNA基因序列高度相似(>;99%),但其全基因组的平均核苷酸同一性(ANI)却可能低于95%,这已经达到了物种划分的界限。这意味着,我们过去所称的“A. muciniphila”很可能是一个包含多个物种或亚种的复合体。这种基因组上的差异,直接导致了功能上的多样性:
代谢能力差异
Loannou等人提到,AmII发育群的菌株拥有合成维生素B12的能力,而AmI发育群则不具备,这直接影响了它们的代谢产物谱。不同菌株对人类母乳寡糖(HMOs)的利用效率也存在显著差异。
对宿主影响的差异
在DSS诱导的结肠炎小鼠模型中,研究人员发现不同的人源AKK菌株对肠道炎症的影响截然不同:一个菌株表现出保护作用,两个菌株没有效果,而第四个菌株甚至有加剧炎症的趋势。这为我们敲响了警钟:随意使用一种AKK菌株来治疗IBD,可能不仅无效,甚至有害。
抗生素抗性差异
模式菌株MucT携带多种抗生素抗性基因(如blaA, dfrA, sul, tetM, van)。但从健康人中分离的菌株,其抗性谱各不相同。考虑到AKK菌具有通过水平基因转移获取新基因的能力,将一个未经充分安全性评估的活菌株作为益生菌推向市场,存在传播抗生素抗性的潜在风险。
对于产业界而言,开发AKK菌产品时,可能要对菌株进行全面的功能和安全性评估(包括代谢能力、免疫调节特性、抗生素抗性谱、基因转移能力等)是不可或缺的关键步骤。
AKK菌能在竞争激烈的肠道环境中占据一席之地,并与宿主展开如此复杂的对话,其背后是一套精妙的分子机制。这不仅是它生存的智慧,也是我们理解其双面性的钥匙。
黏蛋白降解
AKK菌对黏蛋白的降解,远非简单的啃食。它拥有一整套工具箱——即种类繁多的碳水化合物活性酶(CAZymes)。这个过程极具策略性:
-外层突破
首先,通过表面的唾液酸酶和岩藻糖苷酶,切除黏蛋白聚糖链最外层的唾液酸和岩藻糖残基。
-核心瓦解
接着,动用半乳糖苷酶、己糖胺酶和硫酸酯酶等,逐步分解聚糖核心结构。
-内部消化
降解产物通过糖转运系统高效内化。AKK菌虽不具备拟杆菌式的典型PULs/MULs基因簇,但其膜转运蛋白能有效摄取降解后的单糖和寡糖,进行发酵,最终产生乙酸、丙酸等短链脂肪酸(SCFAs)。
注:
编辑
其中,丙酸的产生尤为重要。它不仅是其他肠道菌的能量来源,还能通过激活G蛋白偶联受体GPR41和GPR43,刺激肠道L细胞分泌胰高血糖素样肽-1(GLP-1),从而参与调节宿主的血糖稳态和食欲。这部分解释了AKK菌在代谢性疾病中的有益作用。
与免疫系统的多渠道对话
AKK菌与宿主免疫系统的互作,是一场多层次、多渠道的复杂对话。Grant等人的文章中这个信息量大的图,为我们生动地展示了这一过程。
AKK菌与宿主结肠黏膜的相互作用机制示意图
编辑
Grant et al., Nature Microbiology
有几个关键的互作途径:
编辑
图源:doi.org/10.1038/s41579-024-01106-1
AKK菌并非孤立地存在于肠道中,而是作为复杂微生物网络的一部分,与其他细菌、古菌甚至病毒发生着密切的相互作用。这些相互作用包括协同合作、竞争排斥和营养交换(交叉喂养),共同决定了肠道微生态的结构、功能以及对宿主健康的影响。
交叉喂养:生态系统中的资源分享者
尽管AKK菌被描述为自私的黏蛋白降解者,因为它倾向于将黏蛋白内化代谢,但这一过程实际上为其他微生物创造了丰富的资源。
为产丁酸菌提供底物
AKK菌降解黏蛋白释放的单糖(如岩藻糖)和产生的代谢产物(如乙酸盐、丙酸盐),可以被许多重要的产丁酸菌利用。
例如,在共培养实验中,AKK菌能支持产丁酸菌如Anaerostipes caccae、Anaerobutyricum hallii、Faecalibacterium prausnitzii、Roseburia等细菌的生长,并促进有益代谢物丁酸盐的产生。丁酸盐是结肠上皮细胞的主要能源,具有强大的抗炎和维持肠道屏障完整性的功能。
编辑
图源:doi.org/10.1038/s41579-024-01106-1
双向营养交换
AKK菌与其他细菌的相互作用可以是双向的。例如,它与A. hallii之间存在互惠共生关系:AKK菌为A. hallii提供黏蛋白降解产物和乙酸用于丁酸合成,而A. hallii产生的丁酸等代谢物有助于维持适宜AKK菌生长的肠道微环境。
某些研究还提示可能存在更直接的代谢物交换,如维生素或辅因子的共享,但具体机制仍在探索中。
协同作用:共同抵御疾病
A. muciniphila 可能需要其他微生物的存在来预防某些疾病。
1+1>2:微生物协同互作的抗病潜力
例如,在结直肠癌(CRC)小鼠模型 Apc 突变小鼠中,单独定植 A. muciniphila 或幽门螺杆菌导致肿瘤负担增加,而两者共定植则减少了肠道肿瘤数量。
A. muciniphila Muc 和狄氏副拟杆菌(Parabacteroides distasonis)表现出协同的抗结肠炎关系。
在癫痫 Kcna1⁻/⁻ 小鼠中,A. muciniphila Muc和 狄氏副拟杆菌的联合给药通过降低细菌交叉代谢产生的γ-谷氨酰转肽酶活性,在控制饮食条件下预防了癫痫发作。
这些发现强调了一个核心观点:微生物之间的相互作用网络,比单一菌株的存在与否更能决定疾病的结局。
编辑
图源:doi.org/10.1038/s41579-024-01106-1
不单单是AKK菌,而应评估菌群的整体效应
这种复杂的互作关系提示我们在应用层面需要更宏观的视野。
通过菌株脱除试验发现, A. muciniphila 的引入,与抗炎共生菌普拉梭菌(F. prausnitzii)丰度降低相关(在疾病诱导之前),对柠檬酸杆菌(C. rodentium)感染的易感性部分归因于这种细菌网络的变化。这意味着,即使 AKK 菌无法长期定植,它作为过客仍可能重塑肠道菌群的功能结构。
因此,未来评估益生菌应用时,不能仅盯着 AKK 菌本身,而应将其视为生态系统的一个扰动因子,充分重视个体间菌群基线的差异,深入评估其对肠道微生态结构和功能的长期重塑作用,以及宿主原有菌群对其行为的反向调控。
竞争与拮抗
在肠道有限的生态位中,竞争是不可避免的。
与黏液降解菌的竞争
在以MUC2为唯一碳源的体外共培养体系中,当AKK菌与其他的黏液降解菌如普通拟杆菌(Bacteroides vulgatus)、活泼瘤胃球菌(Ruminococcus gnavus)等一起培养时,AKK菌的生长会受到抑制,而其他细菌的生长则被促进。这表明在黏液降解这一功能上存在激烈的竞争。
对其他菌群的负向影响
在某些情况下,AKK菌的存在可能对其他有益菌产生负面影响。
例如,在一个定义的微生物群落中,AKK菌的存在与抗炎共生菌Faecalibacterium prausnitzii丰度的降低相关。但这种相关性并不一定代表因果关系——可能是:环境条件改变导致两者同时变化,或其他因素的间接影响特定疾病状态的反映(如炎症性肠病)。
与普雷沃氏菌的负相关
在人类肠型(enterotype)研究中,AKK菌通常在以Ruminococcus属为主的肠型中富集,而与以普雷沃氏菌为主的肠型呈负相关,这反映的是菌群组成的自然差异,不同肠型由遗传、饮食、地理位置等多因素决定,不是AKK菌排斥普雷沃氏菌,而是不同的生态位和代谢特征。
目前最核心、也最令人困惑的部分:AKK菌在不同疾病背景下的双面角色。三篇文献都花费了大量篇幅,尽力为我们揭示了这种环境依赖性。
AKK菌与结肠炎:保护、致病与宿主互作的复杂博弈
关于AKK菌与结肠炎的关系,研究结论充满了矛盾。
保护作用
在常规的DSS化学诱导结肠炎模型中,灌胃AKK菌或其蛋白Amuc_1100,通常能观察到保护效果。其机制包括上调紧密连接蛋白(如ZO-1、Occludin)以修复屏障、减轻内质网应激、促进Treg细胞应答等。Luo等人的综述也证实,活菌能够增加紧密连接蛋白的表达。
致病作用
然而,一旦宿主背景改变,情况就可能逆转。在遗传易感的Il10-/-小鼠(一种自发性结肠炎模型)中,单独定植AKK菌反而会加剧炎症,导致黏液层变薄和促炎细胞因子上调。这表明,在宿主免疫调节能力受损(如缺乏关键的抗炎因子IL-10)的情况下,AKK菌的黏蛋白降解活性可能弊大于利。
宿主蛋白的策反
Grant等人的研究揭示了一个更为精妙的机制。在溃疡性结肠炎患者中,一种名为Intelectin-1(ITLN1)的宿主蛋白会过度表达。这种蛋白会特异性地结合AKK菌,将其拉到更靠近上皮细胞的位置。这种亲密接触在炎症背景下,反而加剧了免疫反应和组织损伤。
感染:启动防御,还是放大风险?
在面对外来病原体入侵时,AKK菌的角色同样摇摆不定。
有益面:抗感染
AKK菌的某些组分或代谢物显示出抗感染的潜力。例如,其产生的三肽RKH能通过阻断TLR4信号通路,保护小鼠免于致死性脓毒症。Amuc_1100蛋白则能预防沙门氏菌引起的肝损伤。
有害面:缺膳食纤维就吃粘液层,病原体侵入
AKK菌的关键功能——降解黏蛋白,在特定条件下会为病原体开门。
在一项设计精巧的无菌小鼠实验中,研究者发现,缺乏膳食纤维的饮食会导致肠道中的AKK菌饥不择食,转而大量消耗黏液层。这层被削弱的物理屏障,使得致病菌Citrobacter rodentium能够轻易入侵,导致致命感染。
更具说服力的是,当研究者从这个菌群中移除AKK菌后,即使在无纤维饮食下,小鼠也能免于感染。这证明了AKK菌在这种情境下的内鬼角色。有趣的是,一旦恢复富含纤维的饮食,AKK菌的存在反而与较低的病原体载量相关,显示出保护作用。
编辑
Tips:同一种菌,两副面孔:有膳食纤维时,AKK菌是肠道卫士;没有纤维时,它却会啃食肠道黏膜。这个营养开关,让它在天使与魔鬼之间切换。这个发现具有极其重要的实践意义。告诉我们,补充AKK菌益生菌的同时,如果忽略了饮食管理(特别是保证充足的膳食纤维摄入),可能不仅无法获益,甚至可能损害肠道屏障,增加感染风险。这为精准营养与精准菌群干预的结合提供了强有力的理论依据。
癌症:抗肿瘤反应与促肿瘤微环境的动态博弈
在癌症领域,尤其是结直肠癌,AKK菌的形象同样复杂。
促癌风险
部分研究在结直肠癌患者的肿瘤组织和相应的小鼠模型中,都观察到了AKK菌的富集。有观点认为,它通过降解黏液屏障,可能为肿瘤的发生发展创造了条件。
抑癌潜力
然而,另一些研究则得出了相反的结论。AKK菌的EVs、乙酰转移酶Amuc_2172等组分,在小鼠模型中显示出抑制肿瘤生长的效果。
在免疫治疗中的助攻
AKK菌在癌症研究中最高光的时刻,莫过于其在免疫检查点抑制剂(如PD-1抗体)治疗中的作用。
多项研究一致发现,对PD-1治疗有反应的癌症患者(包括肺癌、肾癌等),其肠道中AKK菌的基线水平显著更高。更关键的是,在小鼠模型中,将无反应者粪菌移植给小鼠后,再补充AKK菌,能够重新恢复小鼠对PD-1治疗的敏感性。这表明AKK菌可能通过调节全身免疫状态,增强了抗肿瘤免疫应答,从而成为免疫治疗的增效剂。
代谢与神经系统疾病:更偏向有益的角色
相较于在炎症和感染中的摇摆不定,AKK菌在代谢性疾病中的有益作用,是目前证据最为一致、也最具转化潜力的领域。无论是动物模型还是初步的人体研究,补充活菌或巴氏杀菌的AKK菌,都被证明能够改善胰岛素敏感性、降低胆固醇、减少脂肪堆积。其机制与促进GLP-1分泌、调节脂肪酸代谢、减轻低度炎症等密切相关。
在神经系统疾病中,AKK菌的关联性再次变得复杂。它在帕金森病和多发性硬化症患者中常常富集,体外实验也显示它可能诱导α-突触核蛋白聚集或促炎反应。然而,也有研究将多发性硬化症患者中AKK菌的增多与较低的残疾程度联系起来,提示这可能是一种有益的代偿反应。这种矛盾性再次凸显了菌株差异和宿主背景的重要性。
面对AKK菌如此复杂,我们该如何从科学研究走向临床应用?有幸的是三篇文献为我们提供了一些思考和前瞻性的指导。
编辑
Grant et al., Nature Microbiology
影响AKK菌双重性的因素及潜在治疗策略。AKK菌的最终效应受到菌株差异、饮食、微生物互作和宿主状态的共同调节。理解这些变量,有助于我们选择最优的治疗应用方案,例如是使用活菌、灭活菌,还是分离的生物活性组分。
精准应用的策略选择
未来的AKK菌疗法,绝非一招鲜,吃遍天,而应是量体裁衣的精准策略。
活菌 vs. 灭活菌
巴氏杀菌的AKK菌是一个极具吸引力的选择。它在欧盟已被批准为新食品原料,安全性更高,避免了活菌定植和基因转移的风险。
多项研究证实,灭活菌依然保留了大部分有益的代谢调节功能,这可能归功于其热稳定的细胞壁成分(如Amuc_1100)。
整体 vs. 组分
直接使用分离的生物活性组分,是更为精准和安全的策略。例如,将Amuc_1100蛋白作为药物开发,或利用其EVs作为治疗载体。
Ioannou 等人的综述强调了 AKK 菌作为“酶学底盘”的巨大潜力。例如利用AKK菌的糖苷酶来改造血细胞表面的ABO血型抗原,展示了从基础研究到生物技术应用的转化潜力。
补充 vs. 内源调节
除了直接补充AKK菌,通过饮食干预(如补充富含多酚的食物、膳食纤维)来扶持宿主内源AKK菌的生长,也是一种温和而有效的方法。
关 键 挑 战
要把 AKK从研究热点真正推进到可精准应用的干预手段,还需要把菌群检测作为贯穿研发—临床—产业化全链条的基础设施,融入以下关键环节:
菌株筛选与鉴定
建立一个标准化的 AKK 菌株功能评价体系,涵盖其代谢谱、免疫调节能力、安全性等多个维度,是实现精准应用的前提。与此同时,需要配套规范化的菌群检测(如 16S/宏基因组与定量检测),用于界定不同菌株在不同微生态背景中的适配性与可重复性,避免同名不同效。
情境依赖性的机制阐明
需要更深入地研究,在不同宿主遗传背景、饮食模式和共存微生物群落的影响下,AKK 菌的功能会发生怎样的改变。这需要更复杂的动物模型(如人源化小鼠模型)和多组学技术的结合;其中,纵向菌群检测是识别“谁在场、谁在协同/拮抗、何时发生生态位迁移”的关键手段,也是将机制与真实个体差异对齐的必要条件。
安全性评估
必须对活菌制剂的抗生素抗性传播风险进行严格评估。开发不含抗性基因的工程菌株,或优先使用灭活菌及组分,是未来的方向。同时,应将菌群检测用于追踪干预后菌群结构扰动、耐药基因负荷变化以及潜在机会致病菌扩增等风险信号,实现从前期评估到使用中/使用后监测的闭环。
递送与生产工艺
作为严格厌氧菌,如何实现 AKK 菌的大规模、低成本培养,并开发出能保护其在通过胃肠道时保持活性的口服制剂,是产业化面临的技术瓶颈。
在这一过程中,菌群检测同样不可或缺:一方面用于生产端的质量控制(纯度、污染菌与批间一致性),另一方面用于应用端的效果评估与分层(基线菌群与 AKK 定植/丰度变化、关键功能菌群响应),从而把工艺参数—活性保持—体内生态学结果真正连接起来,提升可复制性与可监管性。
编辑
图源:doi.org/10.1038/s41579-024-01106-1
通过对这三篇力作的梳理,我们对Akkermansia muciniphila的认知,从一个模糊的有益菌形象,变得立体、丰满,也更加敬畏。它不再是一个简单的标签,而是一个充满动态和变数的生命体。
毒理学有一句名言:剂量决定毒性。在微生物学领域,我们或许可以引申为:情境决定属性。对于AKK菌而言,这个情境包含了我们吃下的每一口食物,我们基因中编码的每一个蛋白,以及我们肠道中与之共存的亿万菌群。
作为科研和从业工作者,我们的核心任务在于阐明这些情境依赖性效应的内在机制。未来研究需要超越简单的相关性分析,通过严谨的实验设计和先进的技术手段,明确AKK菌发挥特定生物学效应的必要条件、充分条件及其剂量-效应关系,解析宿主遗传背景、肠道微生态结构、代谢状态等因素对其功能的调控作用等。
探索之路,道阻且长,但充满希望。与各位同仁共勉。
主要参考文献
Grant ET, Monzel E, Desai MS. Navigating the duality of Akkermansia muciniphila. Nat Microbiol. 2026 Jan;11(1):20-30.
Ioannou A, Berkhout MD, Geerlings SY, Belzer C. Akkermansia muciniphila: biology, microbial ecology, host interactions and therapeutic potential. Nat Rev Microbiol. 2025 Mar;23(3):162-177.
Luo Y, Lan C, Li H, Ouyang Q, Kong F, Wu A, Ren Z, Tian G, Cai J, Yu B, He J, Wright AG. Rational consideration of Akkermansia muciniphila targeting intestinal health: advantages and challenges. NPJ Biofilms Microbiomes. 2022 Oct 17;8(1):81.
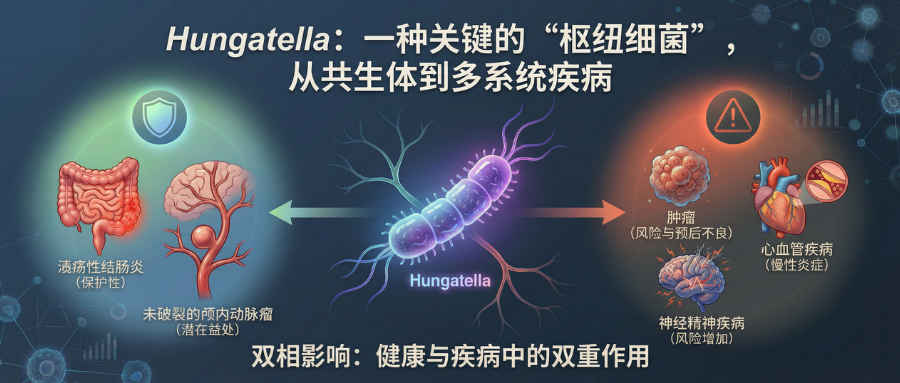
谷禾健康
Hungatella属隶属于厚壁菌门梭菌目梭菌科,是一种具运动性、严格厌氧、能形成内生孢子的杆菌,代表种包括 Hungatella hathewayi 和 Hungatella effluvii。是人类肠道中常见的细菌之一,谷禾菌群数据库显示,约90%的人群肠道中可检出该细菌。
这类细菌的不同菌株对革兰氏染色表现存在变异,例如Hungatella effluvii染色呈革兰氏阳性,而Hungatella hathewayi染色则为革兰氏阴性。提示同属细菌之间在细胞壁结构和染色特性上并不完全一致,菌株间差异较大。
它是人类肠道“重要”共生菌之一,具有较强的糖类和多糖(特别是糖胺聚糖,GAG)降解能力,能产生乙酸、丙酸等短链脂肪酸,同时在牛磺酸和氧化三甲胺(TMAO)等代谢通路中扮演重要角色。还有研究发现其与某些肿瘤化疗药物的敏感性相关,但证据仍属初步,需进一步验证因果性。
从目前的人群与动物研究来看,Hungatella在多种疾病中的丰度变化呈“双相”特点:一方面,在部分炎症性肠病(溃疡性结肠炎)和脑血管疾病(未破裂颅内动脉瘤)中表现出潜在保护作用;另一方面,在一些肿瘤、心血管及精神神经疾病中则与风险升高、预后不良或慢性炎症相关。
Hungatella是属于厚壁菌门、梭菌目、梭菌科下的一个属。Hungatella是人类肠道微生物群的成员之一,并具有重要的临床意义。
1
细胞形态
Hungatella属是一种能运动、严格厌氧、内生孢子(孢子形状为卵形至椭圆形,位于中央或近末端)的杆状细菌,长度为2-5μm,宽度为0.7-1.5μm。部分菌体还可产生粘稠胶囊。
固体琼脂培养基生长的Hungatella
编辑
doi: 10.3201/eid1011.040006.
▸ 革兰氏染色可变,取决于具体菌株
这类细菌的革兰氏染色结果因菌株而异,例如Hungatella effluvii染色呈革兰氏阳性,而Hungatella hathewayi染色则为革兰氏阴性。
一般而言,Hungatella菌落呈圆形,颜色为奶油色至灰白色,边缘稍不规则,凸起。通常,它们看起来有光泽且不透明。
注:37∘C 培养3天后,H.hathewayi 在哥伦比亚羊血琼脂培养基上形成灰白色、不透明、凸圆形、有光泽、边缘稍不规则的菌落。然而,在RCA培养基上,30°C 孵育24小时后,H.effluvii 形成奶油色、圆形、凸面和光滑的菌落,且边缘完整。
H.hathewayi经48小时培养后的菌落形态
编辑
doi: 10.3201/eid1011.040006.
2
营养和生长条件
▸ 生长环境与人体环境相近
菌株生长温度范围为10–45∘C(最佳温度为30–37∘C)、pH为5.0至8.5(最佳pH7.0)。生长可以发生在0–2.0%(w/v)NaCl 条件下,最佳条件是 0.5% NaCl,但在 3% (w/v) NaCl 条件下没有观察到生长。上述条件与人体内环境相近,说明 Hungatella可能适宜在人体肠道内定植与生长。厌氧培养的菌株通常需要24-72小时才能在RCA上形成菌落。
3
生态分布
Hungatella属分布广泛,构成正常人类微生物群的一部分,视为常见肠道成员之一。H.hathewayi(Clostridium hathewayi)最初从无胃肠道疾病的健康人粪便中分离并报道,随后也在术后血流感染患者中被检出。
编辑
另一种H.effluvii 菌株 DSM 24995T 是从用于处理益生菌制造业废发酵培养基的厌氧消化器获得的废水样品中分离出来的。
4
主要代谢与功能
▸ 糖类和多糖代谢
Hungatella属成员为糖分解菌,可发酵多种碳水化合物,包括苦杏仁苷、阿拉伯糖、纤维二糖、半乳糖、葡萄糖、乳糖、甘露糖、松三糖、蜜二糖、棉子糖、鼠李糖、核糖、水杨苷、山梨糖醇、淀粉、蔗糖和海藻糖,但不能将硝酸盐还原为亚硝酸盐。
葡萄糖发酵的主要终产物为乙酸盐和丙酸盐,并少量产生CO₂和H₂。
注:最新的功能组学研究表明,H.hathewayi在糖胺聚糖(GAG)代谢上具有非常突出的能力。
此外,Hungatella的过氧化氢酶、氧化酶和吲哚试验均为阴性,甲基红试验为阳性。其对七叶苷、淀粉和尿素的利用具有物种特异性,而所有已研究菌株均不水解明胶和酪蛋白。
▸ 与牛磺酸和氧化三甲胺含量相关
宏基因组关联研究及多个中国队列和相关动物模型的证据显示,Hungatella hathewayi 与血液中牛磺酸水平相关。但该关联尚不足以证明因果性,其在牛磺酸调控中的作用机制仍需进一步纵向研究和干预性试验加以证实。
而在另一项研究中,H.Hatheway产生氧化三甲胺(TMAO)的能力,与包括抑郁症在内的某些神经系统疾病有关。
▸ 可能会代谢5‑氟尿嘧啶
在结直肠癌队列中,H.hathewayi的肿瘤组织丰度与5-FU疗效下降及总体生存率下降呈相关关系。机制方面,研究提示可能通过CDX2/B-catenin通路的调控影响药物敏感性,但尚需因果性证据与纵向研究来证实。
5
耐药性
Hungatella属对青霉素、克林达霉素和莫西沙星的耐药性存在差异,但所有分离株均对美洛培南、β‑内酰胺/β‑内酰胺酶抑制剂及甲硝唑敏感。还发现利福昔明可抑制Hungatella hathewayi的生长。
▸ 抗生素体外敏感和实际临床药敏
•甲硝唑:迄今唯一在所有报道病例中均表现为敏感的药物。MIC 通常极低(如 0.032 μg/mL),无耐药报道,是治疗经验性厌氧覆盖首选。
•克林霉素:7/9例报道中Hungatella对其敏感。
•碳青霉烯类:约6例中表现为敏感,临床多作为重症败血症经验用药。
•β‑内酰胺/β‑内酰胺酶抑制剂:阿莫西林/克拉维酸、哌拉西林‑他唑巴坦在部分病例中敏感(4–5 例)。
•青霉素:多数早期H.hathewayi 菌株对其敏感,但不作为唯一经验药物。
•万古霉素:个别株报道为敏感,但Hungatella通常为厌氧 G+ 芽孢杆菌谱系,用万古霉素根除有限。
▸ 用药建议(基于有限病例):
•经验治疗严重感染:甲硝唑±第三/四代头孢或哌拉西林‑他唑巴坦或碳青霉烯。
•一旦确定为Hungatella:保持/调整为含甲硝唑方案通常可靠,再根据E‑test/MIC可逐步去除不必要的广谱药。
6
Hungatella属的重要物种
▸ Hungatella effluvii
细胞具运动性,杆状,长约2.0–4.0μm、宽1.0–1.5μm,革兰氏阳性。在pH6.0–8.0范围内可生长,最适pH为 7.0;适生温度为10–45℃,以30℃ 为最适。
菌株 UB-B.2ᵀ 的过氧化氢酶、氧化酶和吲哚试验为阴性,甲基红和脲酶试验为阳性;七叶苷、酪蛋白、淀粉和明胶水解阴性,不能还原硝酸盐,柠檬酸盐利用及 H₂S 产生均为阴性。
其对 L-吡咯烷酰芳香酰胺酶、脲酶和 β-吡喃半乳糖苷酶呈阳性,而对亮氨酸芳香酰胺酶、苯丙氨酸芳香酰胺酶、L-脯氨酸芳香酰胺酶、酪氨酸芳香酰胺酶、α-阿拉伯糖苷酶、β-甘露糖苷酶、七叶苷水解、β-D-岩藻糖苷酶、α-L-岩藻糖苷酶和碱性磷酸酶均为阴性。可利用半乳糖、蔗糖、葡萄糖、麦芽糖和甘露糖产酸。葡萄糖发酵的主要终产物为乙酸和丙酸。
DNA G+C 含量为 51.4 mol%。
遗传分析显示,Hungatella effluvii 与 Clostridium hathewayi(DSM 13479ᵀ)在 16S rRNA 基因水平的相似度为 97.84%,但全基因组亲缘性仅为 38.4%。在表型上,二者在七叶苷、淀粉和尿素水解等多项特征上存在显著差异。
▸ Hungatella hathewayi
细胞为严格厌氧、革兰氏阴性、具运动性的杆菌,宽0.7–1.0μm、长2.0–5.0μm,常成链排列,最长可达30个细胞。
生长pH范围为5.0–8.5,最适约7.0;温度范围15–45℃,最适约 37 ℃。菌株在含0–2.5%NaCl的培养基中生长最佳。
过氧化氢酶、氧化酶、吲哚、硝酸盐还原及 VP 试验均为阴性。葡萄糖发酵的主要终产物为乙酸、乙醇、H₂和CO₂。
能利用苦杏仁苷、阿拉伯糖、纤维二糖、果糖、半乳糖、葡萄糖、乳糖、麦芽糖、甘露糖、蜜二糖、松三糖、棉子糖、鼠李糖、核糖、水杨苷、山梨糖醇、淀粉、蔗糖、海藻糖和木糖产酸;不能利用麦芽三糖、糖原、肌醇、菊粉或甘露醇产酸。七叶苷和淀粉水解阳性,而酪蛋白、尿素、纤维素、明胶不被水解。
DNA G+C 含量为 50.7–50.9 mol%。
7
与人体及其他菌群的互作
▸ 与宿主的互作与潜在致病机制
(1)中性或有益方面
•作为肠道共生菌,可能通过产SCFA,维持结肠上皮能量与黏液层完整性;
•外泌GAG裂解酶,利用黏液层及 ECM 中的 GAG,为自身及共生菌提供碳源;在一定范围内维持黏膜更新与免疫稳态。
•牛磺酸产生:可能通过影响胆盐/抗氧化/血管平衡,对远端器官(如脑血管)产生系统性效应。
(2)有害或潜在有害方面
•当肠道屏障受损(阑尾炎、缺血、肿瘤、术后创面等),Hungatella 可由肠腔进入血流/腹腔,引起:菌血症、脓毒症、肝脓肿、胆囊炎、坏死性筋膜炎等;
•在合并其他菌(如 E. coli、Prevotella、Streptococcus constellatus)的多菌种感染中,则常与严重组织坏死、长期住院相关。
▸ 与其他肠道菌群的互作
•与Bacteroides的互补/竞争:
H.hathewayi拥有与B.thetaiotaomicron接近甚至更强的GAG降解能力,但使用的是完全不同的输运与调控网络(PTS/ABC vs SusC/D)。意味着在利用宿主黏液多糖这一生态位上,Hungatella可能与 Bacteroides形成功能冗余或在特定饮食/炎症条件下出现竞争/互补替代。
•向其他菌群“输出”寡糖与代谢中间体:
将大分子GAG裂解为寡糖/二糖,这些小分子自身能摄取利用,也可被缺乏初级外泌酶的共生菌进一步利用——这在微生态学上类似“初级降解者+次级利用者”的食物网结构。
1
影响炎症性肠病的风险
编辑
研究发现,Hungatella、Acidaminococcaceae及另外15个菌群是多种克罗恩病(CD)和溃疡性结肠炎(UC)亚型的保护因素,而Terrisporobacter、Anaerostipes及其他23个菌群与不同CD和UC亚型的风险增加相关。
▸ Hungatella有助于降低溃疡性结肠炎风险
分析显示,有9个肠道菌群对溃疡性结肠炎各亚型及肠外表现具有保护作用。其中,Lachnospiraceae 科 ND3007 组可降低溃疡性直肠乙状结肠炎风险;Ruminococcaceae UCG011和Hungatella属可降低溃疡性全结肠炎风险;Lachnospiraceae 科 ND3007 组和 Hungatella 属共同降低左侧溃疡性结肠炎风险。Prevotellaceae 科、Eubacterium fissicatena 属和 Ruminococcus gnavus 组则降低合并原发性硬化性胆管炎(PSC)的溃疡性结肠炎患者的风险。
▸ 与其他益生菌组合使用作为结肠炎的保护因子
Hungatella属和Lactobacillus属还可降低克罗恩病(CD)相关关节病变风险,而Barnesiella属则增加其发生风险。
在对比溃疡性直肠乙状结肠炎与左侧结肠炎时,Lachnospiraceae 科 ND3007 组在两者中均为保护因子,而 Hungatella 仅与左侧结肠炎相关,据此推测 Hungatella 可能参与降结肠炎症的起始。实验研究显示,克罗恩病术后吻合口处 Hungatella 丰度升高,提示其可能参与常见术后并发症的发生。
若肠道菌群在术后感染的发病机制中发挥促成作用,则针对肠道微生物群的靶向治疗或预防干预有望降低克罗恩病患者术后并发症风险。
2
丰度过高可能增加结直肠癌风险
编辑
近几十年来,早发结直肠癌(yCRC)发病率不断升高,但其肠道微生物特征仍知之甚少。既往研究多聚焦于老年发病型CRC(oCRC),尚不清楚老年患者的微生物特征能否外推至年轻患者。为此,有研究整合了迄今规模最大的 yCRC 肠道宏基因组数据,涵盖两个独立队列,发现CRC相关微生物组。
▸ H.hathewayi在结直肠癌患者中富集
分析显示,如Clostridium symbiosum、Peptostreptococcus stomatis、Parvimonas micra和Hungatella hathewayi在老年和年轻患者中均显著富集。并且在老年发病结直肠癌(oCRC)和早发结直肠癌(yCRC)中,观察到具核梭杆菌(Fusobacterium nucleatum)、脆弱拟杆菌(Bacteroides fragilis)和大肠杆菌(Escherichia coli)的菌株水平模式相似。
几乎所有与oCRC相关的宏基因组通路在年轻患者中均有方向一致的变化。值得注意的是,与对照组相比,oCRC和yCRC的相关毒力因子(fadA、bft)均为富集。此外,基于微生物组的分类模型在老年和年轻发病患者中对结肠癌状态的预测准确性相似,强调了不同年龄组微生物特征的一致性。
▸ H.hathewayi影响5‑氟尿嘧啶代谢进而干扰治疗效果
此外,刚才前文中也有讲到过的一项研究发现H.hathewayi在结直肠癌组织中的丰度显著增加,其高水平与整体生存率降低相关。
并且H.hathewayi处理明显减弱了5-氟尿嘧啶(5-FU)对结直肠癌(CRC)细胞系HCT116和HT29的增殖抑制和凋亡诱导作用,并增强了耐药细胞系 HCT116/5‑FU 和 HT29/5‑FU 对 5‑FU 的耐药性。机制上,H.hathewayi下调CDX2表达并促进 β‑catenin 在细胞核内的积累。CDX2 过表达可逆转H.hathewayi介导的细胞生长增强和对 HCT116/5‑FU、HT29/5‑FU 细胞凋亡的抑制,同时抑制 β‑catenin 的表达及其核内富集。
综上所述,结直肠癌组织中H.hathewayi高丰度不仅与较差生存预后相关,还可通过调控 CDX2/β‑catenin 信号通路影响结直肠癌细胞对5‑FU的耐药性。
3
未破裂动脉瘤患者中丰度较低
未破裂颅内动脉瘤(UIA)是一种危及生命的脑血管疾病。在两组中国UIA患者及接受人类供体粪便移植的对照个体和小鼠中进行了病例对照宏基因组全域关联研究。
注:非破裂颅内动脉瘤的全球患病率为3.2%,中国的社区调查表明中国成年人非破裂颅内动脉瘤患病率高达7%。颅内动脉瘤是一种极为凶险的脑血管疾病,一旦破裂其早期致死率为40-50%。
▸ 颅内动脉瘤患者中H.hathewayi减少,且与牛磺酸正相关
研究团队发现一群Clostridium属的物种包括C.bartlettii, C.nexile和C.bolteae在颅内动脉瘤组显著富集。然而,C.hathewayi(在2014年被更名为Hungatella hathewayi)却是唯一一个在颅内动脉瘤肠道中丰度降低的Clostridium属成员。
UIA患者的肠道微生物改变
编辑
doi: 10.1038/s41467-020-16990-3.
具体而言,在未破裂颅内动脉瘤(UIA)患者中Hungatella hathewayi 的丰度显著下降,并与人及小鼠体内的牛磺酸水平呈正相关。灌胃补充H.hathewayi可使小鼠血清牛磺酸水平恢复正常,并保护其免于颅内动脉瘤的形成与破裂;补充牛磺酸同样可逆转颅内动脉瘤的进展。上述结果提示,H.hathewayi相关的牛磺酸耗竭可能是UIAs发病机制中的关键因素。
此外,与单纯移植动脉瘤患者粪便的小鼠相比,补充H.hathewayi可显著降低颅内动脉瘤的总体发生率。Kaplan–Meier分析进一步证实,H.hathewayi 补充降低了动脉瘤破裂风险。同时,H.hathewayi 还能抑制脑血管炎症反应、细胞外基质重塑、MMP‑9 活性以及脑血管平滑肌细胞凋亡。
综上,H.hathewayi参与牛磺酸合成并提高血浆牛磺酸水平,从而降低颅内动脉瘤的发生率和破裂率。
4
在一些精神疾病中丰度较高
▸ 慢性精神分裂症患者中Hungatella丰度增加
编辑
研究共纳入115例处于阳性和紊乱症状缓解期的精神分裂症患者及119名对照者,评估了32项外周血标志物。
研究发现:在慢性精神分裂症患者中,Hungatella 的丰度显著增加,相比健康对照组其相对丰度更高。患者的肠道菌群多样性整体下降,而 Hungatella 等部分优势菌属呈富集状态。
差异丰度属与免疫炎症标志物、肠毒素、脂质谱成分和胰岛素抵抗水平的变化相关。此外,还观察到差异丰度属与认知障碍、阴性症状严重程度增加以及社交功能恶化存在若干相关性。甲烷短杆菌属(Methanobrevibacter)丰度与阴性症状、认知和社会功能水平的相关性似乎受白细胞介素-6和RANTES水平介导。
而Hungatella与注意力表现的相关性则受肠毒素水平介导。研究结果表明,精神分裂症患者肠道微生物群组成的改变与临床表现、肠道通透性、亚临床炎症、脂质谱改变以及葡萄糖稳态受损相关。亚临床炎症和肠道通透性受损可能介导了肠道微生物群改变与精神病理症状和认知障碍之间的关联。
▸ 产后抑郁的女性中H.hathewayi的含量较高
编辑
在另一项研究中,发现产后抑郁症状女性中 Hungatella hathewayi含量较高。在不同组学和横断面分析中观察到多种菌种丰度差异:中度且加重抑郁症状组中 Hungatella hathewayi丰度较高(FDR<0.25);高度且减轻抑郁症状组中 Bacteroides clarus 丰度较高(FDR<0.25)。
在妊娠晚期,与“低且稳定”组相比,“爱丁堡产后抑郁量表(EPDS)高且下降”组中 Bacteroides clarus、B. faecis 和 B. xylanisolvens 的相对丰度较高;而“EPDS 中且上升”组中 Hungatella hathewayi、Lachnospira SGB5076 和 Streptococcus thermophilus 的相对丰度较高。
既往研究也发现,H.hathewayi在非妊娠重度抑郁障碍人群中水平升高。其可能机制与产生三甲胺-N-氧化物(TMAO)的能力有关;TMAO 与包括抑郁在内的多种神经系统疾病有关,循环 TMAO 可激活促炎信号通路,从而诱发神经炎症。
5
冠状动脉病变中丰度升高
▸ 川崎病急性期合并冠状动脉病变儿童中富集
编辑
在川崎病急性期合并冠状动脉病变(AKDCAL)的儿童中,LEfSe 分析显示Hungatella显著富集,而在无冠状动脉病变的 AKDNCAL 组未见此现象。
注:川崎病(KD)是一种急性全身性免疫血管炎,主要影响中小动脉,常见于儿科患者。
虽然总体α多样性在川崎病急性期合并冠状动脉病变与无冠状动脉病变川崎病间无显著差异,但Hungatella、Burkholderia-Caballeronia-Paraburkholderia和Clostridium_innocuum富集,被鉴定为与冠状动脉病变(CALs)相关的特征菌属。
6
其他疾病中的丰度变化
⑴丰度增加可能与婴儿湿疹风险升高相关
在一项香港队列研究中,肠道Hungatella hathewayi丰度随时间增加与1岁前发生湿疹的风险升高显著相关。
⑵有时可能引起菌血症或严重感染
Hungatella 在多数情况下为肠道共栖菌,但在特定易感人群或肠道屏障受损的情况下,也可能导致菌血症或其他严重感染。
H.effluvii和H.hathewayi偶见引起败血症、坏死性筋膜炎或阑尾炎相关菌血症等重症感染。并且比较基因组学提示Hungatella属内部遗传差异大,不同“种/亚种”在毒力和耐药基因上差异明显。
⑶伴便秘的帕金森病患者中增多
与健康对照相比,伴有便秘的帕金森病(PD)患者粪便中Hungatella显著增多,同时伴随Collinsella升高、Lachnospira和Fusicatenibacter降低,提示整体向“促炎型菌群”转变。
反映便秘严重程度的 Wexner 评分与 Hungatella 丰度呈正相关,即Hungatella越高,便秘越重。
在机制层面,目前证据支持其作为参与PD相关肠道菌群失调和便秘,可能通过影响 SCFA 平衡、肠道炎症与肠‑脑轴功能发挥作用,是帕金森病合并便秘中值得关注的潜在干预靶点。
⑷乳腺癌患者中Hungatella丰度增加
乳腺癌(BCa)患者微生物多样性减少,但从BCa患者采集的粪便样本中,三种特定微生物属——Acidaminococus、Tyzzerella和Hungatella的丰度得到了富集。
⑸新冠后遗症/康复患者中增加
在新冠后遗症或康复期人群中,有研究发现,其肠道中Hungatella的总体丰度持续处于升高状态。进一步的分析显示,这种Hungatella丰度的增加与多种炎症因子水平呈显著正相关关系,尤其是与 IL‑8、IL‑1β 等经典促炎细胞因子相关联,因此提示 Hungatella 的升高可能反映或参与了机体长期存在的慢性炎症过程,与新冠感染后的持续性炎症状态具有一定联系。
综合目前多学科证据,Hungatella 在疾病中的作用大致通过以下几点体现:
1.黏膜与多糖代谢:强糖胺聚糖降解能力一方面为自身及其他菌群提供碳源,支持黏膜更新和生态食物网;另一方面,若失衡或过度活跃,可能削弱肠黏液屏障,增加内毒素和炎症因子易位,驱动全身慢性炎症。
2.小分子代谢(SCFA、牛磺酸、TMAO 等):
-通过产乙酸/丙酸等 SCFA 参与能量代谢和局部抗炎;
-通过促进牛磺酸合成或维持其水平,对脑血管等远端器官产生保护作用(如预防颅内动脉瘤);
-通过产生氧化三甲胺(TMAO),参与动脉粥样硬化和神经精神疾病相关的促炎与代谢异常。
3.免疫与炎症信号:与多种炎性细胞因子(IL‑6、IL‑8、IL‑1β 等)水平相关,可能通过诱导肠炎和系统性低度炎症,连接肠道微生态与代谢综合征、心血管疾病及精神神经障碍。
4.表观遗传与肿瘤信号通路:在结直肠癌中,通过调控 DNA 甲基转移酶活性,影响抑癌基因甲基化,以及通过 CDX2/β‑catenin 通路干预细胞增殖和 5‑FU 化疗敏感性,表现出典型的“促肿瘤菌”特征。
5.肠‑脑/肠‑血管轴:在帕金森病、抑郁、精神分裂症、川崎病和颅内动脉瘤等疾病中的一系列关联,提示 Hungatella不仅是肠腔共栖者,更可能通过代谢物与炎症通路参与中枢神经和血管系统的病理过程。
总体而言,Hungatella 是一个功能高度复杂且菌株异质性很大的肠道菌属:
•在某些场景中(如未破裂颅内动脉瘤、部分溃疡性结肠炎亚型),它可能具有保护性作用;
•在另一些场景中(如结直肠癌、乳腺癌、部分心血管和精神神经疾病、婴儿湿疹、新冠后遗症),其丰度升高则更多与风险增加、预后不良或慢性炎症相关;
•而在免疫低下或屏障破坏条件下,还可作为机会致病菌引发严重感染。
现有研究多为关联性和部分机制探索,因物种鉴定精度、株间差异及干预研究仍有限,Hungatella 在各类疾病中的真正“因果角色”和可干预潜力仍需更多纵向、功能和临床试验加以澄清。
注:本账号内容仅供学习和交流,不构成任何形式的医疗建议。
主要参考文献
Rawat PS, Li Y, Zhang W, Meng X, Liu W. Hungatella hathewayi, an Efficient Glycosaminoglycan-Degrading Firmicutes from Human Gut and Its Chondroitin ABC Exolyase with High Activity and Broad Substrate Specificity. Appl Environ Microbiol. 2022 Nov 22;88(22):e0154622.
Yang Q, Tang W, Ren J, Li M, Zhao C. Unveiling the gut-heart potential connection: microbiota’s role in kawasaki disease and coronary artery lesions. Front Cell Infect Microbiol. 2025 May 29;15:1560083.
https://doi.org/10.1002/9781118960608.gbm01640Digital Object Identifier (DOI)
Li F, Yu C, Zhao Q, Wang Z, Wang Z, Chang Y, Xu Z, Han X, Li H, Liu Y, Hu S, Chang S, Tang T, Li Y. Exploring the intestinal ecosystem: from gut microbiota to associations with subtypes of inflammatory bowel disease. Front Cell Infect Microbiol. 2024 Jan 4;13:1304858.
Qin Y, Tong X, Mei WJ, Cheng Y, Zou Y, Han K, Yu J, Jie Z, Zhang T, Zhu S, Jin X, Wang J, Yang H, Xu X, Zhong H, Xiao L, Ding PR. Consistent signatures in the human gut microbiome of old- and young-onset colorectal cancer. Nat Commun. 2024 Apr 22;15(1):3396.
Huang Z, Wang C, Huang Q, Yan Z, Yin Z. Hungatella hathewayi impairs the sensitivity of colorectal cancer cells to 5-FU through decreasing CDX2 expression. Hum Cell. 2023 Nov;36(6):2055-2065.
Li H, Xu H, Li Y, Jiang Y, Hu Y, Liu T, Tian X, Zhao X, Zhu Y, Wang S, Zhang C, Ge J, Wang X, Wen H, Bai C, Sun Y, Song L, Zhang Y, Hui R, Cai J, Chen J. Alterations of gut microbiota contribute to the progression of unruptured intracranial aneurysms. Nat Commun. 2020 Jun 25;11(1):3218.
Hieta J, Benchraka C, Pärnänen K, Houttu N, Mokkala K, Lotankar M, Kataja EL, Lahti L, Laitinen K. Perinatal depressive and anxiety symptoms are associated with gut microbiota in pregnant women with overweight and obesity. Brain Behav Immun Health. 2025 Jun 19;47:101042.
Misiak B, Pawlak E, Rembacz K, Kotas M, Żebrowska-Różańska P, Kujawa D, Łaczmański Ł, Piotrowski P, Bielawski T, Samochowiec J, Samochowiec A, Karpiński P. Associations of gut microbiota alterations with clinical, metabolic, and immune-inflammatory characteristics of chronic schizophrenia. J Psychiatr Res. 2024 Mar;171:152-160.
Elsayed S, Zhang K. Human infection caused by Clostridium hathewayi. Emerg Infect Dis. 2004 Nov;10(11):1950-2.

谷禾健康
准确的饮食评估是营养流行病学、临床营养学及公共卫生计划的基石,对于减轻全球饮食相关慢性疾病(如肥胖、三高)至关重要。
然而,传统的评估工具(如食物频率问卷、24小时回忆法和称重食物记录)长期面临着参与者负担重、数据偏差以及可扩展性有限等问题,这些局限性降低了营养数据的可用性和准确性。
人工智能与大型语言模型的兴起
近年来,人工智能(AI),特别是自然语言处理(NLP)和大型语言模型(LLMs)的飞速发展,为自动化营养评估提供了新的契机。以GPT、Claude、Gemini为代表的先进模型,具备了处理复杂自由文本、进行高通量食品分类及提供个性化营养建议的潜力。
编辑
然而,目前的验证研究主要集中在英语数据集上,遇到像波兰语这样语法复杂的语言,它们还能不能这么灵光? 此外,现实生活远比实验室复杂。比如在养老院里,饮食数据往往包含复杂的菜单外食品,要让AI准确识别这些带有强烈文化特色、又没写在标准食谱上的东西,对它的语言天赋和应变能力绝对是个巨大的考验。
分类框架的整合:NOVA与WHO指南
饮食质量的评估通常依赖于互补的框架:既要看食品是如何加工的(NOVA系统),又要看具体的营养成分是否超标(如糖、脂肪和钠),即参照世界卫生组织(WHO)的指南。
编辑
虽然这两大标准是全球饮食指南的基础,但直接让AI照搬却很困难,因为两者在定义上时有模糊,甚至相互打架。尤其是NOVA系统有时过于简单粗暴(非黑即白),无法完全反映食品的复杂性。因此,我们需要一种能同时考量加工深度和营养密度的混合系统,从而让AI做出更精准的判断。
为了填补这一空白,来自波兰的华沙医科大学Aia等人的研究团队利用一份包含1992种食品的波兰语纵向数据集,对目前市面上三款顶流AI模型——Claude Opus 4.5、Gemini 3 Pro 和 GPT-5.1-chat-latest,设了两场考试:一场是死磕标准的硬核测试(结合NOVA和WHO双重标准),一场是凭直觉的快速测试。
编辑
结果很明确,虽然AI面对复杂的波兰语还不能完全独立行医,但只要有人类专家的把关,AI完全有能力成为临床和公共卫生营养评估中的得力助手,让大规模的健康饮食管理变得既快又好。
本文我们一起来详细了解一下这项研究的主要设计、AI模型的真实表现、专家如何配合AI实现人机协作的最佳效果。无论你是营养师、AI从业者,还是对健康饮食感兴趣的科研工作者,相信都能从中获得新的启发。
该研究基于华沙医科大学2017–2021年开展的长期护理机构(LTCF)纵向项目数据,已获伦理批准(AK-BE/212/2017)。
原队列约1000名居民,收集人体测量、生物阻抗、体力活动、临床诊断与膳食信息。研究聚焦居民个人橱柜中菜单外食品(由本人购买或家属带入,非机构配餐),共纳入1992条食品记录,数据以波兰语原文处理。
选取三种大型语言模型(Claude Opus 4.5、Gemini 3 Pro、GPT-5.1-chat-latest),均通过API调用,统一温度参数为1.0,以提高可比性并贴近默认使用场景。
设置两种提示策略
① 结构化双步提示
1
按NOVA识别超加工食品(UPF),一经判定即归为不健康;
2
仅对第1步判为健康的条目,按WHO阈值评估游离糖(>总能量10%)、饱和脂肪(>10%)或钠(>2 g/日),超标则改判不健康。
模型需输出每个产品的名称、重量(g)、热量与二分类结果,并分别汇总两类总热量。
② 简化单步提示
仅要求对每个产品给出健康/不健康判断及简短理由(两列:evaluation与description)。
人工参照标准由两位专家完成:
统计分析以Pearson卡方检验(α=0.05)进行多组两两比较(模型-专家、模型间、提示策略间及“多模型共识”主导类别)。同时以不健康为阳性,基于混淆矩阵计算Accuracy、Precision、Recall、F1与Specificity,评估不同提示与模型的错误模式。
编辑
文中会出现的一个词:Dominant,指的是多模型共识(consensus response across LLMs),你可以理解为对多个模型的结果采用了投票机制,如果大多数模型对某一项食品都给出了相同的分类,那么这个分类就被认为是“Dominant”分类,即多模型共识的分类结果,有助于减少单个LLM的偏见,同时保持保守的风险评估。
1. 结构化双步提示结果
★ 核心结论
三种模型与专家一致性都很高(约90–91%);同时呈现更保守的倾向,也就是说更擅长抓出不健康(Recall很高),但更容易把健康误判为不健康(Specificity下降)。
双步提示下 LLM vs 专家的一致性分布
(占总样本1992的百分比)
编辑
双步提示下的混淆矩阵指标
(UNHEALTHY为阳性)
编辑
怎么看这组结果?
双步提示把规则写死(先NOVA判UPF直接不健康,再用WHO阈值复筛),所以模型更倾向于宁可判不健康也不漏掉,表现为 Recall极高(0.963–0.982)。
代价是 Specificity偏低(0.798–0.844),也就是说有一部分其实健康的条目被判成不健康(假阳性更多)。
2. 简化单步提示结果
★ 核心结论
简化提示下,总体一致率更高(约92.5–94.2%),并且 Specificity明显提升,Recall仍保持较强,整体更像专家的整体判断风格。
同时,作者指出:在简化提示下,模型整体会把更多条目判为健康(相比双步提示),显示规则约束变少后,模型不再那么保守。
简化提示下 LLM vs 专家的一致性分布
(占总样本1992的百分比)
编辑
简化提示下的混淆矩阵指标
(UNHEALTHY为阳性)
编辑
怎么看这组结果?
相比双步提示,简化提示的 Accuracy更高(0.927–0.942),并且 Specificity显著提高(0.897–0.951):更少把健康误判为不健康。
Recall略有回落但仍高(0.909–0.964),整体更均衡。
Dominant在简化提示下表现最好/接近最好(Accuracy 0.942,F1 0.949)。
这提示简化单步策略在召回率和特异性之间找到了一个更好的平衡点,使得模型的分类结果既能较好地识别不健康食品,也能较好地识别健康食品,减少了误报或漏报的情况。
3. 跨模型、跨提示词的整体差异(卡方检验)
为了评估提示结构对不同LLM及人类专家之间的一致性和差异性,使用Pearson’s Chi-square检验,对所有可能的成对比较(36对)进行统计显著性测试。
关键发现
所有两两比较均显著不同:
χ2=1174.5 –1897.1,p < 0.001
编辑
说明即使总体一致率很高,不同模型/不同提示词仍会导致系统性的分类分布差异(不只是随机波动)。
文中还给了两个极值例子:
各模型与专家总一致率(双步 vs 简化)
编辑
尽管多模型共识(Dominant)在一些指标上接近或略优于最佳单一模型,但与专家的判断仍存在显著差异,提示共识并不能完全替代专家判断,尤其在边缘案例上。这些差异也体现出提示词设计的重要性。
当前人工智能所展示的膳食分类的能力,虽然接近人类专家的水平,但无法完全替代专家,适合做初筛和前处理,可以用不确定性阈值触发强制人工复核。
未来的优化方向在于提示词设计、多语言本地化、多模态数据融合(例如包装、配料表的图片等)、混合工作流开发(AI+人类专家)、纵向验证等。
主要参考文献
Ase, A.; Borowicz, J.; Rakocy, K.; Piekarska, B. Large Language Models for Real-World Nutrition Assessment: Structured Prompts, Multi-Model Validation and Expert Oversight. Nutrients 2026, 18, 23. doi.org/10.3390/nu18010023

谷禾健康
健康的肠道菌群呈现出物种多样性高、结构稳定、功能良好等的特征,有益菌、中性菌和有害菌之间维持着动态平衡。然而,当这种平衡被打破,即发生菌群失衡,便可能成为多种急、慢性疾病的诱因或加剧因素。
目前谷禾积累了大量样本数据,通过对这些数据的深度分析,我们观察到多种多样的菌群状态,其中不乏一些表现出极端特征的案例。这些案例为我们理解菌群失衡的具体模式及其对健康的潜在影响提供了宝贵的窗口。
本文我们选取谷禾数据库中的一些检测报告,深入探讨几种典型的肠道菌群极端失衡状态,包括:菌群多样性严重低下、特定菌门/菌属的极端优势、益生菌缺乏、益生菌补充过量引发的新失衡等。
通过对这些案例的具体数据进行解读,我们希望能为相关研究人员和临床工作者及后端干预方提供更直观的参考,从而共同推动肠道微生态健康管理的发展。
编辑
( – 衡量微生态稳定的指标 – )
菌群多样性是评估肠道微生态健康状况的首要指标。它包含两个维度:一是物种丰富度,即菌种的数量;二是物种均匀度,即各种菌在群落中数量分布的均衡性。
一般情况下,一个高多样性的菌群生态系统更具弹性和稳定性,能够有效抵御外界干扰(如短期饮食改变、药物影响、病原体入侵),并维持正常的生理功能。相反,多样性低的菌群则显得脆弱,更容易在外界压力下崩溃,导致功能紊乱和疾病风险的增加。
案例一:
多样性荒漠化与病原菌疯长的恶性循环
这份报告显示,菌群多样性评分仅为3分(满分100),属非常低的水平。
编辑
<来源:谷禾健康肠道菌群检测数据库>
香农多样性指数仅为2.29(正常中位数约4,5左右)。
注:香农多样性(Shannon Diversity Index)是生态学中用于衡量生物群落多样性的一种常用指标。H′=−∑(pi⋅lnpi).
香浓多样性指数一个是反映了物种丰富度,一个是反映物种均匀度,综合考虑上述两者,数值越高越复杂。
这表明其肠道微生态环境极其单一,稳定性极差。这种荒漠化的肠道环境,为条件致病菌的过度繁殖提供了绝佳条件。
1)菌属构成分析
编辑
<来源:谷禾健康肠道菌群检测数据库>
可以看到,肠道内沙门氏菌属(Salmonella)的丰度非常高,达到了的68.365%,这其中主要是肠炎鼠伤寒沙门氏菌。
这是一种典型的致病菌,由于它的超高丰度,直接导致了变形菌门(Proteobacteria)的占比飙升至71.436%(正常人群中位数一般不超过10%,甚至低于5%)。
变形菌门被广泛认为是促炎菌门,其成员多为条件致病菌。沙门氏菌的肆虐,如同一场生态灾难,严重挤压了其他共生菌的生存空间,直接导致了有益菌(如双歧杆菌、乳杆菌)和大量核心功能菌属的缺失,这是其菌群多样性低下的直接原因。
编辑
<来源:谷禾健康肠道菌群检测数据库>
可以看到有益菌、核心菌属严重缺乏。
2)生理后果与炎症水平
这种极端失衡带来了严重的生理后果。报告显示,其肠道炎症水平高达95(重度炎症),肠道产气严重。这与沙门氏菌感染的典型症状(腹痛、消化不良、炎症反应)相吻合。
编辑
<来源:谷禾健康肠道菌群检测数据库>
同时,丁酸盐(1分,缺乏)、吲哚(2分,缺乏)等关键抗炎和维持肠道屏障功能的代谢物严重不足,进一步加剧了肠道损伤。
3)恶性循环的形成
这是一个典型的“病原菌疯长 → 多样性锐减 → 肠道功能崩溃 → 炎症加剧”的恶性循环。
此案例中我们看到了菌群多样性的重要性。当多样性降至冰点,肠道微生态的防御体系形同虚设,单一病原菌便可一家独大,主导整个群落,并引发严重的健康问题。
对于此类情况,干预策略不仅要考虑补充益生菌,更关键的是要针对性地清除优势病原菌,并创造条件帮助多样性恢复,当然这种较为复杂的情况是需要一个专业干预指导的过程。
上述是多样性很低,大量被病原菌占据的情况下,那么同样是多样性低的时候,有没有可能有一些相对“好”的菌群在竞争中脱颖而出呢?如果有的话,会发生什么呢?我们来看下一个案例。
案例二:
多样性低,大量普雷沃氏菌占据
接下来我们要看的这份报告是多样性低的另一种典型代表。其肠道菌群被普雷沃氏菌属(Prevotella)以61.025%的绝对优势所统治。
编辑
<来源:谷禾健康肠道菌群检测数据库>
普雷沃氏菌属通常与富含碳水化合物和植物纤维的饮食模式相关,常见于素食或以植物性饮食为主的人群。虽然在某些情况下,普雷沃氏菌属被认为是健康植物性饮食的标志,但当其丰度过高,形成单一优势时,问题便随之而来。
1)菌群多样性降低带来的影响
首先,普雷沃氏菌属的过度增殖同样挤压了其他核心菌属的生存空间,导致了菌群多样性的降低。
编辑
<来源:谷禾健康肠道菌群检测数据库>
报告显示,多个产丁酸的关键菌属如瘤胃球菌属(Ruminococcus)、罗氏菌属(Roseburia)、经黏液真杆菌属(Blautia)均处于缺乏状态。
丁酸是结肠上皮细胞的主要能量来源,具有强大的抗炎作用。这些菌的缺失意味着肠道抗炎能力的下降。
编辑
<来源:谷禾健康肠道菌群检测数据库>
2)促炎反应的风险
其次,高丰度的普雷沃氏菌属本身也与促炎反应相关。部分普雷沃氏菌菌株能够产生脂多糖(LPS),这是一种强烈的免疫刺激物,可引发炎症。
更值得警惕的是,该案例中同时伴随着变形菌门中病原菌埃希氏菌属(25.553%)严重超标,影响肠道菌群平衡,同时诱发高炎症风险。
其II型糖尿病风险评估为0.83(高风险),这与菌群的长期失衡和促炎状态可能存在关联,因为慢性低度炎症是胰岛素抵抗和II型糖尿病的重要病理生理基础。
编辑
<来源:谷禾健康肠道菌群检测数据库>
3)生态平衡的重要性
此案例说明,即使是与健康饮食模式相关的菌属,其过度增殖也可能打破生态平衡,导致核心功能性菌群的缺失和炎症风险的升高。肠道健康的关键在于平衡而非某种超级细菌的绝对优势。
普雷沃氏菌属(Prevotella)和拟杆菌属(Bacteroides)的相对丰度则与长期饮食模式(植物性饮食vs.动物蛋白/脂肪饮食)紧密关联。当某一菌群异常增殖,打破了原有的平衡,就会导致一系列代谢和免疫问题。
这提示我们,饮食干预需要精细化,在保证膳食纤维摄入的同时,也需关注蛋白质、脂肪的均衡配比,避免长期单一的饮食结构导致菌群结构的极化。
#########################################
关于 多样性过低 的分析思路:
谷禾报告中,多样性分值<15 提示 “单一菌群”(菌群数量极少,功能单一);15-30 提示 “多样性较低”(需结合临床症状干预) 。
◆ 核心判断
若多样性低伴随核心菌属缺失、 有害菌比例升高,则提示菌群结构失衡,需重点干预。
◆ 原因分析
——外部因素
饮食单一化:长期高糖 / 高脂 / 精粮饮食(如仅吃精制米面),缺乏膳食纤维(<15g / 日),导致菌群可利用底物不足。
药物滥用:广谱抗生素(如头孢类)、化疗药物直接杀灭有益菌,破坏菌群平衡。
生活方式:久坐少动(肠道蠕动减慢,菌群代谢产物不足)、熬夜(皮质醇升高抑制厌氧菌生长)。
——内部因素
慢性疾病:溃疡性结肠炎、糖尿病、肥胖等疾病直接损伤肠道黏膜,或通过代谢紊乱抑制菌群增殖。
衰老:70 岁后肠道菌群多样性自然下降,核心菌丰度都会有所降低。
◆ 潜在危害与关联疾病
肠道功能异常
腹泻 / 便秘交替、腹胀、肠易激综合征(IBS)风险升高(核心菌减少导致肠道屏障功能下降)。
代谢与免疫风险
短链脂肪酸合成不足,增加肥胖、胰岛素抵抗、心血管疾病风险。
免疫细胞(如T细胞)活化不足,易引发反复感染(如呼吸道 / 肠道感染)。
疾病恶性循环
菌群失衡→消化吸收差→营养缺乏→免疫力进一步下降,形成 “菌群 – 疾病” 循环。
多样性过低的本质是菌群结构失衡,需结合数值、阈值、核心菌 / 有害菌比例及代谢功能综合判断,重点关注核心菌缺失和功能受损,而非单纯追求菌群数量。
#########################################
上述我们看到了多样性低引发的单一菌群大量占据的现象,这是不利的情况,那么,是否意味着多样性越高就越好呢?接下来,我们将通过一个数据库中的高多样性例子来探讨这个问题。
案例三:
多样性很高,是否是更健康的菌群结构?
通常情况下,我们认为多样性越高,肠道菌群结构就越趋向于稳定。然而,当多样性超出一定范围时,情况是否仍然如此呢?
这份报告,在数据库中属于多样性高的情况。
编辑
<来源:谷禾健康肠道菌群检测数据库>
从表面指标看,本案例的菌群呈现“高度多样”的特征:α多样性水平高。
一般成年人能测到的在500-1800种菌之间,平均香浓多样性指数就4-5的样子,这个多样性是显著超过平均值,这类结构通常意味着菌种丰富、生态位充足,理论上更具稳定性与抗干扰能力。
1)菌多但不够好的结构
然而,进一步结合平衡指数、有益菌、有害菌水平、核心菌属占比等指标,会发现这并不是一个典型的“稳态高多样性”,而更像一种“菌多但不够好”的结构。
我们来看一下菌群结构:
编辑
<来源:谷禾健康肠道菌群检测数据库>
结合整个报告来看,虽然多样性高但结构质量一般:核心/有益菌偏少 + 有害菌略偏多 + 还有几项机会型/胆汁相关菌偏高。可以说是一种看起来很热闹,但关键功能菌没站稳的生态结构。
2)核心菌属的缺乏
首先核心菌属占比水平 46(参考80–100):核心菌属不足是最核心的结构信号,报告也明确提示可能存在不常见菌增多或生态紊乱。
编辑
核心菌属累积占比不足,常被解读为常见稳定菌群缺乏,而由更多非核心菌或波动性菌群填充生态位,导致群落看似丰富,却可能更容易在饮食、压力、作息或抗生素等扰动下发生漂移。
3)有益菌与有害菌的失衡
有益菌水平 11(偏低,建议≥50) + 有害菌水平 52(略超参考上限50):说明功能性倾向不理想,“平衡指数29”也与此一致。
编辑
<来源:谷禾健康肠道菌群检测数据库>
在具体菌属层面:
关键产丁酸/抗炎相关核心属,粪杆菌属(Faecalibacterium)、罗氏菌属(Roseburia) 偏低的情况。这些菌常与丁酸盐生成、肠道屏障支持和抗炎稳态相关,缺乏时往往意味着肠道自我修复与抗炎能力的底盘不足。
4)机会致病菌的增多
机会型/胆汁相关菌脱硫弧菌属(Desulfovibrio)、嗜胆菌属(Bilophila) 偏高,且检出的沃氏嗜胆菌(Bilophila wadsworthia) 超过人群90%。这类菌群多常见于饮食结构(高脂/胆汁酸压力)、或近期扰动后的生态位变化。
综上,这是一个典型的多样性高≠更健康的案例:与案例一“病原菌极端优势导致崩盘”、案例二“单一优势菌占据生态位”不同,本案例更像“群落过于分散、关键功能菌不稳”的隐性失衡——需要把关注点从多不多转向核心功能菌是否到位、结构是否稳态。
#########################################
关于 “多样性是否过高” 的分析思路:
多样性过高的情况数据库中并不常见,且相对来说缺乏明确的关于过高的定义及统一的临床问题描述。但从菌群生态平衡的一般原理及现有知识推断,若假设存在 “多样性过高”(如菌种数量异常庞大、均匀性严重失衡),可能存在以下潜在问题:
1. 菌群结构稳定性下降
菌群多样性高通常伴随菌群间竞争激烈,若某类菌种因环境波动(如饮食、药物)出现适应性下降,可能导致整体结构趋于失衡,反而降低稳定性。
如同生态系统中物种过多但缺乏优势种,易因单一物种消失引发连锁反应,例如 “过度分散的菌群” 可能无法有效抵御外界致病菌入侵。
2. 有益菌丰度被稀释
若多样性过高伴随大量非优势菌种(如潜在致病菌、条件致病菌)过度繁殖,可能导致核心有益菌(如双歧杆菌、乳杆菌)的相对丰度被稀释,削弱其生理功能(如短链脂肪酸合成、免疫调节)。
3. 与特定疾病的潜在关联
现有研究多关注多样性低与疾病的关联(如炎症性肠病、代谢综合征),而多样性过高的临床数据较少。但部分罕见病或特殊生理状态下,可能出现菌群异常丰富伴随功能紊乱,例如:自身免疫病、慢性炎症等。
建议:若报告中多样性评分异常偏高,需关注是否存在检测误差或特殊生理状态(如长期摄入广谱益生菌导致菌群过度分散),并结合临床症状(如频繁腹泻、过敏)综合判断。
#########################################
多样性高并不天然等于更健康。当菌群看起来很丰富,但核心菌属占比不足、有益菌被稀释、机会型菌群趁势上升时,这种热闹但不稳的结构同样可能带来功能层面的隐患。
那么,较好的菌群结构应该长什么样?接下来,我们将通过案例四展示一份多样性相对较好的报告,看看多样性恰到好处时,菌群结构在平衡性、核心菌群与功能表现上会有哪些共同特征。
案例四:
多样性较好,核心菌群到位的相对稳态结构
在大多数情况下,它并不追求极端的高或低,而是呈现适度且稳定的多样性:核心菌属占比高、有益菌占优势、有害菌受控,整体更接近一种可持续的稳态。
1)多样性水平稳定
首先从整体生态稳定性来看,多样性水平稳定,说明菌群并未出现荒漠化的低多样性,也没有走向极端的异常发散,多样性在合理区间内往往意味着菌群具备更好的弹性,能更从容地应对饮食波动、作息变化等外界扰动。
编辑
编辑
<来源:谷禾健康肠道菌群检测数据库>
2)核心菌属充足:健康结构的标志
其次,本案例的菌群结构质量也较为理想。核心菌属85,达到报告建议的核心菌属累积占比最好超过80。核心菌属相对充足通常提示群落中以常见、稳定的共生菌为主体,生态位更稳,不容易被少数波动性菌群或机会型菌群抢位,因此也更接近健康人群的结构特征。
编辑
3)有益菌占优:功能性倾向良好
从功能倾向来看,本案例呈现出更符合健康菌群原则的比例:有益菌水平整体已处在建议线以上,既有足够的有益菌群支撑稳定性,也没有出现有害菌明显失控的信号。
最后看综合平衡指标,菌群平衡54。该值位于“中等”区间的偏高段,提示整体已较接近良好状态,但仍存在进一步优化空间。
编辑
<来源:谷禾健康肠道菌群检测数据库>
4)产丁酸菌正常:核心功能菌的支撑
具体菌属层面来看:
重要的核心菌属,包括产丁酸菌等,都处于正常范围(未标红)。
编辑
<来源:谷禾健康肠道菌群检测数据库>
拟杆菌属(Bacteroides)(处于人群79%):典型“基石菌/主导菌属”,在此报告里占比高且仍在报告正常范围内。一般代表复杂多糖降解与交叉喂养能力较强,是核心菌属占比的重要支撑点。
粪杆菌属(Faecalibacterium) (处于人群83%):关键产丁酸、抗炎相关核心菌,在参考范围内,属于加分项。
瘤胃球菌属(Ruminococcus)(人群80%):参与抗性淀粉/纤维素利用,处于正常范围。
总的来说,本案例展示了一个多样性良好且核心菌群充足的稳态结构,反映了肠道相对健康的特征。适度的多样性增强了菌群对外界变化的适应能力,而有益菌的优势地位则支持了整体稳定性。
案例五:
厚壁菌门过度占优势的结构
健康的肠道菌群中,各大菌门、菌属之间维持着相对稳定的比例。厚壁菌门(Firmicutes)和拟杆菌门(Bacteroidetes)是成年人肠道中最主要的两个菌门,它们的比例(F/B ratio)与能量代谢和体重密切相关。
接下来我们要看的是一个典型的门水平比例明显偏移的菌群失衡案例,其最突出的结构特征是——厚壁菌门显著过多,拟杆菌门被严重压缩。
1)门水平的异常信号:厚壁菌门一家独大
报告显示,该个体:F/B 比值 = 12.98(在谷禾数据库中,大多数人群参考范围:0.3–1.0)
这已经不属于轻微偏移,而是一个明确的门层面的生态极化结构。
编辑
<来源:谷禾健康肠道菌群检测数据库>
需要强调的是,厚壁菌门本身并非坏菌门,其中也包含多种产丁酸、支持肠道屏障的有益菌;但当其整体比例被放大到压倒性优势时,往往意味着特定饮食或代谢环境长期偏向,使菌群结构失去弹性。
2)结构层面的真实情况:不是厚壁菌多=结构好
如果继续看整体健康评估,会发现厚壁菌门水平的强大优势,并没有转化为结构质量优势:
编辑
<来源:谷禾健康肠道菌群检测数据库>
可以看到菌群平衡指数、菌群多样性、有益菌水平都偏低,有害菌较多,核心菌属严重缺乏。
这说明一个关键事实:
👉 厚壁菌门虽然数量巨大,但该个体并不是由高质量核心功能菌主导,而更像是由部分机会型或代谢偏向特定方向的菌反复扩增,形成的伪优势。
3)菌属层面:韦荣菌属异常升高
进一步看关键菌属,问题更加清晰:
厚壁菌优势并非由典型肠道核心功能菌主导,而是被高度富集的口腔 / 上呼吸道来源菌属明显拉高,其中以韦荣菌属(Veillonella)异常增多最为突出。
编辑
<来源:谷禾健康肠道菌群检测数据库>
韦荣菌属经典生态位在口腔、咽部、上呼吸道,常与牙菌斑、龋齿、口腔炎症、呼吸道感染高度相关,属于乳酸利用菌,常与链球菌属形成稳定的共生复合体。
该样本中并非单一菌属异常,而是出现了口腔—上呼吸道菌群组合特征:
链球菌属、嗜血杆菌属都偏高,这些菌群通常在口腔、鼻咽部常见的条件致病或共居菌,这些菌属理论上在肠道中正常情况下应是低丰度。
编辑
编辑
<来源:谷禾健康肠道菌群检测数据库>
它们的同步扩增,提示很可能存在:
换句话说,这是一个上消化道菌群下行 + 肠道生态位防御能力不足共同作用的结果。
4)为何这些上消化道菌群下行还能定植?核心菌、有益菌让位
多个产丁酸、抗炎核心菌属显著偏低或未检出:
编辑
<来源:谷禾健康肠道菌群检测数据库>
也就是说,厚壁菌门虽然多,但是真正负责肠道屏障修复、免疫调节的高价值成员并没有站稳生态位。
当这些核心菌群不足时,肠道就会变成一个空位过多、防御薄弱的生态系统,使原本不应成为优势菌的口腔 / 呼吸道菌有机会向下迁移并异常扩增。
5)功能与代谢结果:炎症与内毒素压力升高
这种结构性失衡,在功能指标中已经有所体现:
编辑
<来源:谷禾健康肠道菌群检测数据库>
当菌群结构过于偏移时,内毒素压力、慢性低度炎症和代谢负担往往同步上升。
###################################################
关于厚壁菌偏高属于哪种类型,判断思路:
厚壁菌门/拟杆菌门(F/B)比值是一个简单但粗糙的门水平线索,这在我们之前的文章有写过,详见:
当报告出现厚壁菌门明显占优时,可能需要追问:
这种偏高是短期饮食塑形造成的可逆偏移,还是已经与宿主代谢环境绑定,形成更稳定的结构性失衡? 实践中可按以下路径判断。
首先,先看结构是否还完整
我们上述展示的是一个相对极端的案例,若厚壁菌门只是偏高,但菌群多样性、平衡指数相对尚可,核心菌属并未明显减少,说明生态位仍然多元,系统并未被单一菌群占死。这种情况更常见于饮食诱导型厚壁菌偏高,即在长期高能量、高精制或膳食结构单一的环境中,厚壁菌因能量获取效率较高而被动扩增,但其优势具有一定可逆性。
进一步,拆解厚壁菌门内部的组成质量
厚壁菌门中既包含大量产丁酸、维护肠道屏障与免疫稳态的关键菌属,也可能包含机会型扩增、功能贡献有限甚至与炎症相关的成员。
若厚壁菌门偏高同时,经典产丁酸核心菌(如粪杆菌属、罗氏菌属、部分Blautia 等)仍有存在,提示结构功能尚未完全崩塌;
若厚壁菌门明显占优,但上述核心功能菌反而显著偏低或未检出,则说明这种数量优势并未转化为功能优势,更接近一种生态失衡型扩张。
还需要警惕另一种情况:
若厚壁菌门内部主要由韦荣菌属、链球菌属等非典型肠源菌占据(例如案例五报告中),而典型肠道核心菌普遍缺位,则这一厚壁菌优势更可能是一种来源错位导致的假性结构优势,而非真正的肠道功能性强化。
功能与代谢指标进行交叉验证
饮食诱导型厚壁菌偏高,往往仅伴随轻度产气、能量吸收效率升高等变化,炎症与内毒素状态未必明显异常。
而代谢型厚壁菌偏高,则更常见于炎症水平升高、胆汁酸代谢异常及肠道屏障薄弱等背景之下,提示菌群结构已与宿主的代谢环境形成相互强化的状态。
###################################################
案例六:
拟杆菌过多 ≠ 结构更健康
接下来我们来看一份非常典型的拟杆菌优势型的肠道菌群报告:
拟杆菌的丰度高达 85.613%,菌群整体倾向拟杆菌型。
编辑
<来源:谷禾健康肠道菌群检测数据库>
F/B 比值 0.08(显著低于谷禾数据库中的人群常见范围 0.3–1.0)
编辑
<来源:谷禾健康肠道菌群检测数据库>
仅从这个结果看,容易给人一种偏向植物性、有利代谢的直觉印象。但和厚壁菌偏高一样,拟杆菌偏高本身也只是结构线索,而非结论,同样需要进一步拆解其结构质量与功能背景。
1)整体生态:并非严重失衡型
菌群多样性自然是低的,有害菌也较多,但核心菌属处于正常水平。
编辑
<来源:谷禾健康肠道菌群检测数据库>
进一步看属水平,就会发现问题的关键并不在于有没有拟杆菌,而在于谁在过度主导生态位。
2)拟杆菌属极度集中,占据绝对生态位
其中多株典型拟杆菌(如 B. fragilis、B. vulgatus、B. uniformis 等)同时出现高丰度。
这意味着:碳源利用、蛋白/脂类代谢路径高度集中,其他功能型菌群(尤其是厚壁菌中的发酵—丁酸路径)参与度极低。
编辑
<来源:谷禾健康肠道菌群检测数据库>
图中可以看到,尤其是脆弱拟杆菌(Bacteroides fragilis)特别多,脆弱拟杆菌是人类肠道中的一种重要共生菌,具有复杂的双重角色。它分为两种亚型:不产肠毒素脆弱拟杆菌(NTBF)和产肠毒素脆弱拟杆菌(ETBF)。NTBF通常被认为是有益的,能够增强肠道屏障、调节免疫系统并抑制病原菌感染;而ETBF则通过分泌脆弱拟杆菌毒素(BFT)导致肠道上皮损伤,促进炎症和慢病发展,包括结肠炎、阿尔兹海默病、结直肠癌等。
3)厚壁菌门过低,丁酸通路成为结构短板
与拟杆菌高度集中相对应的是:
这类结构通常意味着:
功能指标很好地解释了这种结构的真实状态。
编辑
<来源:谷禾健康肠道菌群检测数据库>
这类组合非常符合拟杆菌垄断型结构的典型表现:
于是就形成了:
👉 虽不至于严重炎症,但长期胀气、消化负担偏重、肠腔压力较大 的状态。
4)拟杆菌过多,通常出于什么原因?
综合结构与功能特征,可能是以下背景之一或叠加:
此外也有其他非饮食因素的可能:
因此,这种时候,如果强行引入新菌、猛加益生菌或突然大幅调整底物结构,结果有可能不是很理想,拟杆菌可能会再次快速适应,继续占住生态位,新来者被挤走。
总的来说,需谨慎评估拟杆菌过高的情况,避免简单地将其丰度与健康状态等同。
益生菌补充是改善肠道健康的常用手段,能够有效促进消化、增强免疫力和维持微生态平衡。我们的数据库中也显示出两种极端情况的风险:一方面,益生菌的严重缺乏可能导致肠道菌群失衡,增加消化不良和感染的风险;另一方面,过量补充益生菌则可能引发肠道不适,甚至导致菌群的进一步失调。
案例七:
益生菌严重缺乏
接下来我们要看的这份报告最突出的特征,在于典型益生菌的系统性缺失。
从整体结构看,菌群尚未完全崩盘,但支持肠道屏障、免疫调节和代谢稳态的关键益生菌未能有效定植,是该案例的核心问题。
1)核心表现:双歧杆菌、乳杆菌几乎全部检不出
在益生菌模块中,几乎所有常见且功能明确的益生菌,比如双歧杆菌、乳杆菌等均显示为 ND.(ND为未检出)
编辑
<来源:谷禾健康肠道菌群检测数据库>
单个益生菌未检出其实在成年人中并不少见,但当整个双歧杆菌、乳杆菌体系几乎集体缺位时,已不太能用正常个体差异来解释,而更像是一个明确的益生菌底盘不足的结构。
2)指标层面的对应信号:有益菌偏低、核心菌属不足
这一菌属层面的缺失,也体现在综合指标上:
编辑
<来源:谷禾健康肠道菌群检测数据库>
这种组合特征十分典型,不是单一病原菌爆表,而是益生菌长期缺席后,被其他机会型或功能偏向不利的菌群逐步填补生态位,使结构看似还能运转,但稳定性和健康收益明显下降。
3)功能后果:屏障与代谢压力开始显现
在功能与代谢相关指标中,也能看到益生菌缺乏可能带来的连锁反应。
炎症信号偏高,如 LPS、肠道炎症水平超标;肠道屏障不足。
编辑
<来源:谷禾健康肠道菌群检测数据库>
蛋白腐败代谢相关产物(如对甲酚、苯酚)偏高。
编辑
<来源:谷禾健康肠道菌群检测数据库>
这些情况往往与双歧杆菌、乳杆菌不足导致的短链脂肪酸合成下降、屏障支持能力减弱有关。
总的来说,这类情况的干预重点,通常不在单纯追求多样性,而在于重建益生菌生态位,并为其提供长期可持续的生存环境。
如果说这个案例的问题在于——益生菌缺位,生态位长期空置,那么接下来这个案例呈现的,则是另一个方向的偏移——益生菌占位过强,反而挤压了其他核心菌群的生存空间。
案例八:
益生菌补充过犹不及
此案例的报告中,乳杆菌属丰度达到了19.076%,远超正常人群平均水平。
编辑
<来源:谷禾健康肠道菌群检测数据库>
这很有可能是长期或大剂量补充特定乳杆菌制剂的结果。虽然其有益菌高达57,看似健康,但细究之下却隐藏着新的失衡。
首先,单一菌种的极高丰度本身就是一种多样性失衡的表现。尽管其整体多样性评分为39(中等),但这种由外源补充菌株主导的伪多样性可能并不稳定。一旦停止补充,菌群结构可能迅速改变。
编辑
其次,功能上的失衡已经显现。报告显示其肠道产气严重(91,超标)。乳杆菌是主要的产乳酸菌,在发酵碳水化合物时会产生气体。当其数量过多时,过量的气体产生会导致腹胀、消化不良等症状,这与报告中的评估结果相符。
编辑
<来源:谷禾健康肠道菌群检测数据库>
此外,其对甲酚(p-Cresol)水平过高(95,过多)。对甲酚是蛋白质发酵产生的有害代谢物,被认为是尿毒症毒素之一,与慢性肾病、结直肠癌等风险相关。虽然乳杆菌不直接产生对甲酚,但其过度增殖可能改变了整个肠道环境的pH值和代谢网络,为其他能够产生对甲酚的菌群(如部分梭菌)创造了更有利的生长条件。
总的来说,益生菌并非多多益善。盲目、过量地补充单一或少数几种益生菌,可能会人为地制造出新的菌群失衡,导致产气过多、有害代谢物积累等问题。益生菌的补充应更具针对性,最好在检测评估的基础上,根据自身缺乏的菌种进行精准补充,并注意适时调整剂量,同时配合益生元和多样化饮食,以支持整个微生态系统的健康与平衡。
通过对以上极端案例的分析,我们可以清晰地看到,肠道菌群的健康远不止有益菌与有害菌的简单二元对立。它是一个关乎多样性、平衡性、功能性的复杂多维系统。无论是病原菌入侵导致的多样性崩溃,还是长期单一饮食或不当补充导致的比例失衡,亦或是关键功能菌群的缺失,最终都会导向肠道功能的紊乱和疾病风险的升高。
这些案例也为我们提供了重要的启示:
肠道菌群检测的意义在于评估微生态健康。菌群多样性是关键指标,关注物种丰富度和均匀度可及时识别失衡状态。此外,结合关键代谢物(如丁酸盐、短链脂肪酸)的检测,能够全面评估肠道健康。
在临床和机构干预中,针对性策略至关重要。干预不仅应补充益生菌,还需清除优势病原菌,并创造条件以恢复菌群多样性,避免一刀切的方法。饮食干预应精细化,关注膳食纤维、蛋白质和脂肪的均衡,以防止长期单一饮食导致的菌群极化。此外,干预后需进行长期监测与评估,以确保肠道菌群的稳定性和健康,及时调整干预措施,确保最佳效果。
在研究与发展方向上,深入探索微生物与宿主之间的相互作用机制,将有助于更好地利用这些微生物促进健康。
随着研究的深入,我们对肠道菌群的认识仍在不断拓展。未来,结合宏基因组学、代谢组学等多组学技术,我们将能更全面、更深入地描绘个体的肠道健康图景。作为行业的先行者,我们的使命是持续优化检测技术和模型,提升准确度。同时利用大样本数据库将这些复杂的生物信息转化为更加清晰、可执行的健康指导,帮助更多人通过管理好自己的内在肠道微生态,构筑起坚实的健康防线。
注:本文所引用的案例均为谷禾肠道菌群检测数据库中的真实检测数据,但为保护用户隐私,个人信息已做脱敏处理。所有分析和结论仅供参考和学术探讨,不构成医疗诊断或治疗建议。任何健康问题,请务必咨询专业医生。
谷禾健康
肠道微生物组作为人体“第二基因组”,在癌症的发生、发展及治疗反应中扮演着日益重要的角色。这些微生物不仅在局部和全身层面调节免疫应答,还深刻影响着炎症、免疫监视和耐受性。
平衡良好的微生物群是有效免疫功能的基石,但由饮食,环境因素,生活方式,感染或药物驱动的干扰可能会促进慢性炎症并损害抗肿瘤免疫力。越来越多的证据表明,这些干扰不仅有助于肿瘤的发生和发展,而且还对治疗产生耐药性。
微生物及其代谢产物能够通过局部及全身性机制,直接影响药物代谢、塑造免疫微环境,并改变肠道、全身乃至肿瘤微环境(TME)中的转录和表观遗传程序。
大量新兴数据支持,微生物组作为预后和预测性生物标志物、癌症防治中可调控因素以及治疗靶点的巨大潜力。以微生物组为靶点的干预措施(如粪菌移植、饮食调整、益生元和益生菌)可作为传统癌症疗法的辅助手段,增强疗效并降低毒性。
本文探讨了微生物组驱动癌症发生发展的多维机制,包括基因毒性、慢性炎症和表观遗传调控。详细分析微生物组如何调节化疗、放疗及免疫治疗的疗效与不良反应,还系统梳理了当前基于微生物组的癌症治疗策略,如粪菌移植、饮食干预、益生菌和后生元等,并总结了相关临床试验的进展。
历史上,微生物与癌症的关联常采用科赫假设(Koch’s postulates)等框架,将疾病归因于具有确定潜伏期的单一病原体。然而,现在人们认识到,微生物作为复杂的群落发挥作用,对宿主产生集体效应,而不仅仅是孤立的病原体。这些效应包括调节炎症、免疫、代谢和表观遗传,从而深刻影响致癌过程。
微生物驱动的致癌机制
微生物群通过多种复杂的机制促进肿瘤的启动和进展,主要包括直接的基因毒性效应、诱导慢性炎症和肠道屏障功能障碍,以及对宿主细胞的表观遗传调控。这些机制相互交织,共同构成了一个促癌的微环境。
肠道微生物组可通过诱导基因组损伤、表观遗传调控或破坏肠道屏障功能参与癌症的启动和/或进展。这些机制共同作用,促进了从正常上皮到恶性肿瘤的转化。
病原菌的致癌策略:信号传导与毒素的双重威胁
定植于上皮表面的病原菌或机会性细菌可通过直接的宿主细胞信号传导、微生物间通讯或分泌毒素发挥致癌作用。其中,细菌基因毒素是研究最为深入的直接致癌因素之一。这些毒素包括tilimycin、indolimines、tilivalline、colibactin和脆弱拟杆菌毒素(Bacteroides fragilis toxin, BFT)等。
编辑
-Colibactin
在这些毒素中,Colibactin的机制研究最为透彻。Colibactin由存在于变形菌门(如特定大肠杆菌菌株)中的聚酮合酶(pks)基因岛编码。这种基因毒素能够诱导DNA双链断裂和烷基化,从而引发基因组不稳定性,促进癌症的发生。
编辑
图源:doi.org/10.1038/s41579-025-01268-6
例如,一项研究发现,在结直肠癌中,由产colibactin的大肠杆菌(pks+ E. coli)引起的突变特征与早发性CRC相关。此外,产BFT的脆弱拟杆菌(ETBF)能够协同pks+ E. coli,其产生的BFT和黏液溶解活性使得colibactin能够穿透黏液层,到达上皮细胞,从而加剧DNA损伤。
微生物代谢物:潜在的DNA损伤元凶?
除了直接的基因毒素,微生物代谢物也可能具有DNA损伤效应。例如,有研究指出微生物将初级胆汁酸转化为次级胆汁酸,后者过量可通过诱导DNA损伤、调节炎症通路和抑制抗肿瘤免疫反应(包括抑制在癌症免疫监视中至关重要的细胞毒性CD8+ T细胞)来促进结直肠肿瘤的发生。
同样,肠道微生物将管腔中的乙醇转化为乙醛,或将某些亚硝胺类致癌物(如N-丁基-N-(4-羟丁基)亚硝胺)转化为更具活性的形式,这些过程均可导致基因组损伤。
雌激素与微生物:远程影响
此外,微生物的β-葡萄糖醛酸酶(GUS)能够重新激活在肝脏中被结合失活的雌激素,提高循环雌激素水平,这可能对乳腺癌等雌激素敏感性肿瘤的发生构成远距离影响。
慢性炎症与肠道屏障功能障碍
慢性微生物感染是癌症发生的重要驱动力
例如,幽门螺杆菌(Helicobacter pylori)感染通过诱导持续性胃部炎症,促进胃癌的发生,而根除幽门螺杆菌已被证实可以预防胃癌。同样,伤寒沙门氏菌(Salmonella typhi)与胆囊癌的发生相关。
编辑
图源:doi.org/10.1038/s41579-025-01268-6
微生物组失衡促癌变
即使在没有明显感染的情况下,微生物组失衡也能通过调节局部炎症反应促进癌变。
例如,通常存在于口腔的具核梭杆菌(Fusobacterium nucleatum)在特定条件下可定植于肠道,并在CRC肿瘤中诱导肿瘤微环境的炎症。
炎症与癌症之间的复杂信号网络
微生物诱导的炎症部分通过模式识别受体(如Toll样受体,TLRs)信号和炎症小体激活介导,导致细胞因子和活性氧(ROS)的产生,这些分子刺激上皮增殖、免疫逃逸以及癌细胞在远端部位的播散和植入。
肠道炎症与肠道屏障功能障碍密切相关
屏障功能受损,即黏液层和上皮细胞无法在管腔微生物群落与宿主上皮之间提供足够的空间,增加了促癌微生物和分泌性基因毒素造成损害的易感性。受损的屏障完整性促进了微生物产物(如脂多糖,LPS)和代谢物(如胆汁酸)的易位,进入体循环。这些因素可驱动远端组织的全身性炎症或免疫抑制,可能导致远端上皮和非上皮癌症及转移的发生。
编辑
保护性微生物代谢物的作用
一些微生物代谢物,如膳食纤维在结肠中发酵产生的短链脂肪酸(SCFAs),通过促进紧密连接形成和刺激黏液产生,对肠道屏障具有保护作用。
表观遗传调控
肠道微生物组是一个强大的表观遗传调节器,能够通过代谢、炎症和饮食-微生物相互作用的组合来塑造基因表达和肿瘤行为。
图源:doi.org/10.1038/s41579-025-01268-6
丁酸盐的双重角色:从能量源到表观遗传调控
最典型的微生物驱动的表观遗传调控机制是SCFA中的丁酸盐对组蛋白去乙酰化酶(HDAC)的抑制。在健康的上皮细胞中,丁酸盐是支持增殖和分化的能量来源。然而,随着肿瘤细胞代谢转向有氧糖酵解(瓦博格效应),丁酸盐不再被有效消耗。其积累导致过度HDAC抑制,改变染色质构象,促进抑癌基因的转录激活,从而抑制癌变。
膳食成分与微生物的影响
其他微生物或膳食成分也能影响表观基因组。例如,叶酸、亚硝胺和致肥胖饮食在小鼠模型中影响DNA甲基化和组蛋白乙酰化,促进肠道肿瘤发生。
产毒脆弱拟杆菌的致癌机制
有趣的是,在小鼠模型中,产肠毒素的脆弱拟杆菌(ETBF)定植通过破坏DNA修复机制促进结直肠肿瘤发生,并诱导广泛的CpG岛高甲基化,这是肿瘤组织中表观遗传变化的标志。
不同类型的癌症与特定的微生物组特征相关联,这些特征不仅存在于胃肠道肿瘤中,也见于乳腺癌、前列腺癌等远端肿瘤,揭示了微生物组在肿瘤生态系统中的广泛影响。
结 直 肠 癌
结直肠癌(CRC)的发展与多种微生物、其代谢物和结肠上皮之间的相互作用密切相关。在动物模型和细胞培养研究中,特定微生物如ETBF(产肠毒素的脆弱拟杆菌)、具核梭杆菌(F. nucleatum)和携带pks基因岛的大肠杆菌(pks+ E. coli)被证明有助于肿瘤发生。
将结直肠癌患者的粪便微生物群移植到无菌小鼠体内,会导致细胞增殖增加、息肉数量增多、异型增生更严重以及炎症标志物水平升高,这强烈表明肠道微生物组失衡可能是结直肠癌发展的致病因素。
驱动-乘客模型:微生物的协同作用
“驱动-乘客”模型提出,像ETBF这样的驱动菌通过引发慢性炎症和破坏上皮屏障,创造一个允许性环境,让像具核梭杆菌这样的共生乘客菌得以定植并加剧肿瘤进展。具核梭杆菌通过其细胞表面的FadA黏附素与上皮E-钙黏蛋白直接相互作用,激活β-连环蛋白信号通路,促进肿瘤细胞增殖和存活。
编辑
具核梭杆菌与化疗耐药性
此外,具核梭杆菌还通过刺激肿瘤细胞中的自噬相关蛋白,与化疗药物的细胞毒性作用相抗衡,从而与化疗耐药性相关。临床上,结直肠癌肿瘤组织中的具核梭杆菌DNA水平较高与结直肠癌特异性死亡率增加相关,表明其可作为潜在的预后生物标志物。
胃 癌
在胃癌中,幽门螺杆菌(H. pylori)感染是公认的关键致病因素。它通过其毒力因子(如CagA和VacA,在谷禾16S Plus检测实践中,也会检出个别受检者携带 CagA和VacA毒力基因)引发炎症和免疫反应,进而可能有诱发癌症风险。然而,随着胃癌进展,胃内环境发生萎缩,幽门螺杆菌数量反而减少,而其他非幽门螺杆菌物种(如乳酸杆菌属、链球菌属)开始增多。
肝 细 胞 癌
在肝细胞癌(HCC)中,肠-肝轴起着关键作用。肠道屏障功能受损导致细菌及其产物(如LPS)易位至肝脏,诱发炎症、纤维化和肝细胞损伤,最终促进癌变。
肝细胞癌患者的肠道微生物组通常表现为产SCFA的细菌多形拟杆菌(Bacteroides thetaiotaomicron)减少,而促炎细菌(如大肠杆菌)增多。
胰 腺 导 管 腺 癌
胰腺导管腺癌(PDAC)同样与肠道微生物组失调相关。肠道屏障受损允许某些肠道微生物(如变形菌门)易位至胰腺,在局部创造一个免疫抑制的微环境,促进肿瘤进展。此外,微生物对胆汁酸的代谢异常也被认为是PDAC的风险因素之一。
激素依赖性癌症
肠道微生物组在激素代谢中发挥着重要作用,从而影响激素依赖性肿瘤的进展。所谓的雌激素菌基因组(estrobolome)是指能够代谢雌激素的细菌基因集合。肠道中的细菌β-葡萄糖醛酸酶可以解偶联在肝脏中结合的雌激素,使其重新被吸收入血,提高循环中活性雌激素的水平,从而促进乳腺癌等雌激素驱动肿瘤的生长。
在乳腺癌患者中,具有β-葡萄糖醛酸酶活性的菌种(如梭菌属)丰度增加可能与更严重的临床分期有关。同样,在前列腺癌中,肠道菌群能够从头合成雄激素或将皮质醇代谢物转化为雄激素,促进对内分泌治疗的抵抗。
癌症的多步发生模型认为,遗传和表观遗传的序贯改变是恶性转化的驱动力。新兴证据表明,微生物组的失调可能是这一模型中一个额外的、相互关联的因素。
生命早期的微生物组发育
微生物组的发育始于生命早期,受分娩方式、喂养习惯和饮食转变的影响。在这一关键窗口期的干扰可能导致微生物多样性永久性降低和对免疫与代谢稳态至关重要的关键菌种的缺失。
生活方式对微生物组的影响
在整个生命周期中,累积的损害(包括抗生素滥用、慢性压力和低纤维、超加工饮食)会进一步侵蚀微生物的多样性和功能。这种微生物恢复力的逐步丧失可能会损害上皮完整性、增加炎症、削弱代谢信号和免疫监视。
多个研究和科学家提出,微生物组的这种纵向退化可能会降低癌症启动和进展的阈值,与经典致癌过程中的逐步遗传事件相平行。
微生物组健康随生命周期的逐步退化可能促进癌症发展
编辑
图源:doi.org/10.1038/s41579-025-01268-6
图中显示了肠道微生物组健康和恢复力的两种对比轨迹
每次急性干扰后恢复不完全,导致微生物组健康和恢复力逐步退化。
持续的压力源可能将个体推向恶化轨迹,直至达到癌症发展概率更高的阈值。
癌症治疗反应的变异性受到多种宿主和肿瘤相关因素的影响,而肠道微生物组是导致这种变异性的一个重要贡献者。
微生物组主要通过两大机制调节癌症治疗结果:
一是通过微生物对治疗药物的生物转化以及药物对微生物组的反作用;
二是通过微生物介导的宿主免疫反应调节。
微生物组与化疗、放疗及靶向治疗
肠道微生物组编码着庞大的基因库,能够代谢多种肿瘤学和非肿瘤学药物,从而影响药物的生物利用度、疗效和毒性。
微生物介导的影响癌症治疗结果的机制
编辑
图源:doi.org/10.1038/s41579-025-01268-6
药物的微生物生物转化
一个典型的例子是化疗药物5-氟尿嘧啶(5-FU)及其口服前药卡培他滨。某些大肠杆菌和厌氧棒状菌属(Anaerostipes)表达细菌二氢嘧啶脱氢酶(DPD),能将5-FU转化为其非活性形式,从而可能降低其疗效。
类似地,用于治疗胰腺癌的吉西他滨可被某些肿瘤相关γ-变形菌纲细菌表达的胞苷脱氨酶(CDA)脱氨而失活,导致耐药。
微生物也能激活药物
拓扑异构酶I抑制剂伊立替康在肝脏中被代谢为活性形式SN-38,随后经葡萄糖醛酸化为无活性的SN-38G,通过胆汁排入肠道。肠道中的细菌β-葡萄糖醛酸酶(GUS)能将SN-38G重新激活为SN-38,后者会引起严重的上皮损伤、黏膜炎和腹泻等剂量限制性毒性。虽然理论上肿瘤内的再激活可能增强疗效,但肠道内的再激活主要导致毒性增加。
微生物影响疗效与毒性
除了直接代谢药物,微生物组还能通过调节免疫系统间接影响化疗效果。例如,化疗药物环磷酰胺能促进特定肠道细菌(如Enterococcus hirae)易位至次级淋巴器官,诱导产生有助于其治疗效果的TH17细胞(T辅助17细胞)反应。
同样,奥沙利铂的疗效被发现依赖于肠道微生物组,肠道菌群通过激活免疫系统来增强抗肿瘤效果。
放疗与微生物组之间的双向互动
一方面,放疗(尤其是盆腔放疗)会显著改变肠道微生物组的组成,导致多样性下降,并可能增加机会性病原菌。
另一方面,微生物组的组成可以预测患者对放疗的反应和毒性。例如,放疗前肠道菌群多样性较低与晚期放射性肠病相关。在小鼠模型中,粪菌移植(FMT)被证明可以保护机体免受辐射引起的毒性。
靶向治疗
对于靶向治疗,研究尚处早期。一项针对转移性CRC的研究发现,对抗EGFR(西妥昔单抗)和抗VEGF(贝伐珠单抗)治疗的疾病进展组患者,其肠道微生物组中具核梭杆菌的丰度显著高于部分缓解组。
在HER2阳性乳腺癌中,抗生素使用或移植来自抗生素处理供体的小鼠粪菌会削弱曲妥珠单抗的抗肿瘤效果。
微生物组与免疫治疗
免疫检查点抑制剂(ICIs)通过阻断T细胞上的抑制性信号(如PD-1、CTLA-4),重新激活细胞毒性CD8+ T细胞以攻击肿瘤,彻底改变了癌症治疗。然而,只有部分患者对ICIs有反应,而肠道微生物组是决定疗效的关键因素之一。
►▷ 微生物组如何调节免疫反应与治疗效果
多项早期研究独立证实,肠道微生物组的组成与抗PD-1/PD-L1治疗的反应相关。
尽管与疗效相关的具体菌种在不同研究中有所差异(如Akkermansia、Bifidobacterium、Ruminococcus),但其免疫调节机制常常趋同于增强CD8+ T细胞的数量或活性。
例如,在临床前模型中,口服双歧杆菌可增强树突状细胞(DC)的激活和CD8+ T细胞的启动,从而增强对PD-L1阻断的反应。同样,Akkermansia muciniphila通过促进CD8+ T细胞招募至肿瘤,来增强对PD-1抑制的反应。
这些发现表明,与ICI疗效相关的菌种可能共享一种能力,即在肠系膜淋巴结或肠黏膜中诱导依赖于DC的CD8+ T细胞启动,随后通过趋化因子介导浸润至TME。
肠道菌群的影响:U型效应
值得注意的是,微生物组对ICI疗效的影响可能具有“U型”或“倒U型”特征。
编辑
例如,一项针对晚期非小细胞肺癌(NSCLC)患者的研究发现,肠道中A. muciniphila的相对丰度处于中等水平(而非低或高水平)的患者,其临床反应最佳。这提示微生物的影响并非简单的“越多越好”,而是需要一个平衡的生态系统。
►▷ 微生物代谢物:抗肿瘤免疫的关键因素
微生物的功能冗余性可能比分类学组成更为关键,即不同的微生物群落可能产生相似的代谢或免疫输出。多种微生物代谢物被证明在调节免疫治疗反应中发挥核心作用。
肠道微生物代谢物与免疫
编辑
图源:doi.org/10.1038/s41579-025-01268-6
该图描绘了肠道微生物组与免疫系统之间的相互作用,说明了膳食成分(如纤维)如何影响SCFAs、胆汁酸和色氨酸衍生物的产生。这些代谢物调节多种免疫细胞,并通过调节炎症细胞因子和增强癌细胞凋亡来促进抗肿瘤免疫和ICI治疗效果。
编辑
图源:doi.org/10.1038/s42255-025-01287-w
代谢物的作用是情境依赖的
例如,丁酸盐在某些情况下能增强细胞毒性CD8+ T细胞功能,支持抗肿瘤免疫,但在其他条件下也可能促进免疫抑制性的Treg细胞扩增。
同样,具核梭杆菌在不同研究中显示出截然相反的作用:一项研究表明其产生的丁酸能敏化MSS型CRC对ICIs的反应,而另一项研究则发现其产生的琥珀酸会抑制免疫信号通路,导致耐药。
这凸显了菌株、肿瘤微环境和代谢产物背景的复杂性。
►▷ 肠道菌群预测免疫相关不良事件
ICIs激活的免疫系统也可能攻击正常组织,导致免疫相关不良事件(irAEs)。多项报告将基线肠道微生物组组成与irAEs的发生风险联系起来。
然而,由于患者群体异质性、队列规模小以及irAEs定义不一致等原因,目前尚未发现一致的预测性微生物标志物。一些研究提示,特定的菌群(如拟杆菌门丰度较高)可能与结肠炎风险增加有关,但仍需更多机制性研究来阐明因果关系。
鉴于微生物组与癌症疗效和毒性的密切联系,操纵微生物组成员为增强治疗效果提供了新的契机。一系列策略正在研究中,包括饮食干预、益生菌/益生元、粪菌移植(FMT)以及更前沿的工程菌和后生元等。
这些干预措施旨在通过重塑微生物组生态系统,来调节药物代谢、增强抗肿瘤免疫或减轻治疗毒性。
编辑
图源:doi.org/10.1038/s42255-025-01287-w
粪菌移植 (FMT)
粪菌移植是目前研究最深入的微生物组干预手段之一。两项开创性的I期临床试验表明,将来自对ICI有反应的黑色素瘤患者的粪便微生物群移植给耐药患者,可以使一部分患者转为应答者,并伴随着CD8+ T细胞活化增强或TME中免疫细胞浸润的有利变化。
最近,一项在晚期黑色素瘤患者中进行的 I期试验进一步证实,接受健康供体FMT后,患者对一线抗PD-1治疗的反应得到改善,其粪便样本能够在小鼠模型中恢复抗PD-1疗效。
此外,FMT也被用于治疗难治性ICI相关性结肠炎,初步研究显示其能有效缓解症状,并伴随免疫细胞谱的改变。目前,大量临床试验正在探索FMT联合ICI在不同癌种(如肾细胞癌、NSCLC)中的应用。
饮食、益生元与益生菌
饮食干预是一种极具性价比的策略
一项观察性研究发现,高纤维饮食(不使用益生菌)与改善的ICI结局相关。
BE GONE试验(NCT02843425)等临床研究正在评估高纤维食物(如豆类)对微生物组和临床结局的影响,初步结果显示其能增加有益菌(如Faecalibacterium)并改善炎症标志物。
注:zhuBE GONE试验(NCT02843425)是一项由MD安德森癌症中心开展的临床研究,旨在探索食用 canned navy beans对肠道微生物组的影响,以及这种饮食干预是否能减轻肥胖对结直肠癌风险的负面影响。
传统益生菌
益生菌的应用也备受关注。一些传统益生菌(如某些乳杆菌和双歧杆菌)被发现能通过产生特定的代谢物(如I3A、吲哚-3-羧酸)或直接作用来增强抗肿瘤免疫。
下一代益生菌 和 工程菌
例如,A. muciniphila被多项研究证实与ICI的良好反应相关,其巴氏杀菌形式(一种后生元)或其膜蛋白Amuc_1100在临床前模型中显示出抗肿瘤效果。
工程化的大肠杆菌Nissle 1917菌株被改造用于在肿瘤局部递送免疫刺激分子(如将氨转化为L-精氨酸)或免疫检查点抑制剂纳米抗体,从而在不产生全身毒性的情况下增强抗肿瘤免疫。
微生物组干预策略面临的挑战
– 机制不确定性
许多干预措施(如高纤维饮食、传统益生菌)缺乏对特定菌种或功能如何调节疗效的深入机制理解,导致策略普适化,而非个体化。
– 情境依赖性和个体差异
微生物组的效果受宿主遗传、肿瘤类型、治疗方案和生活方式等多种因素影响,使得干预效果难以一概而论。
– 缺乏个体化生物标志物
目前缺少可靠的生物标志物来指导患者分层,或确定应靶向微生物的哪个方面(如药物代谢、屏障功能或免疫调节)。
– 临床试验设计的局限性
癌症患者常伴有免疫抑制、恶病质或胃肠道毒性,难以依从严格的饮食方案或口服疗法。此外,抗生素的普遍使用也干扰了微生物组的评估和干预。
除了作为治疗靶点,微生物组在癌症的早期检测和风险分层方面也展现出巨大潜力。
早期检测与风险分层
肠道微生物组已被探索作为早期癌症检测的工具,尤其是在结直肠癌领域。多项研究通过比较癌症患者与健康对照的微生物组谱,试图识别可用于诊断的微生物标志物。
谷禾健康通过上万例的结直肠癌和结直肠息肉患者及健康对照构建的肠癌识别模型,在样本量、病例类型覆盖与真实世界差异性方面显著优于多数以“数十到数百例”为主的研究型队列。更大的队列意味着模型能够学习到更稳定、可重复的微生物特征组合,降低因地域、饮食、年龄结构、用药史等因素带来的偶然偏差,从而在实际应用中具备更强的泛化能力与可识别性。
同时,许多研究仅做“癌症 vs 健康”的二分类;谷禾模型纳入“结直肠息肉”等癌前人群,更贴合早筛需求,能在症状出现前识别风险,并捕捉从健康到息肉/腺瘤再到癌变的连续变化信号,从而支持早期风险分层与更有针对性的随访与干预。
谷禾结直肠癌模型示意图
编辑
<来源:谷禾肠道菌群检测数据库>
但是不管是谷禾的模型还是科学研究的结果直接引用于临床诊断还需要有一段路走。
首先,横断面研究难以区分因果关系和相关性,且易受混杂因素(如并存病、用药)的干扰。
其次,与现有筛查手段(如 FIT/粪便免疫化学、结肠镜、影像与血液指标)进行对照评估,确定其增益价值与适用场景;并建立标准化的采样、测序/检测、质控与解读体系,控制抗生素、饮食、肠道准备等混杂因素影响。
伴 随 诊 断
微生物组作为伴随诊断工具的前景可能更为广阔,可用于指导治疗决策、预测疗效或毒性风险。例如,通过检测治疗前的肠道或肿瘤微生物组,可以识别出对特定疗法(如ICI)反应可能性较高的患者,或发生irAEs风险较高的患者。
基于对瘤胃球菌(Ruminococcus)等丰度的检测,可以对接受ICI治疗的患者进行分层。理想的伴随诊断应植根于机制理解,而不仅仅是关联数据,从而指导个性化干预,例如决定是否需要将FMT作为一线辅助治疗,或是否需要先通过菌群移植恢复患者对膳食纤维的降解能力再进行饮食干预,而不是盲目的粪菌移植。
人工智能(AI)有望通过实现高级数据整合、模式识别和预测建模,彻底改变微生物组研究。
编辑
图源:doi.org/10.1038/s41579-025-01268-6
面对微生物组多组学数据维度灾难(即特征数量远超样本数量)的挑战,AI提供了解决方案。例如,通过降维、合成数据增强或迁移学习等策略,可以提高模型的稳定性和泛化能力。
新兴的AI方法,如变分贝叶斯神经网络和卷积神经网络,能够整合配对的微生物组和代谢组数据,或将多组学数据转换为图像格式进行分析,从而发掘深层生物学关联。
编辑
数字孪生是另一个令人兴奋的方向。这是一个动态的、虚拟的患者复制品,整合了静态数据(如基因信息)和实时数据(如临床记录、组学数据),以模拟、监测和预测健康结果。将微生物组数据纳入数字孪生模型,可以模拟其与宿主和治疗的复杂相互作用,支持假设驱动的干预措施和个性化治疗调整。
为不同生命阶段(如婴儿期和成年期)开发量身定制的数字孪生模型,将为整个生命周期的微生物组靶向干预提供宝贵的指导。
当前临床试验概览
近年来,旨在利用微生物组改善癌症治疗的临床试验数量激增。
FMT试验主要集中于免疫治疗效果良好的癌种(如黑色素瘤、RCC、NSCLC),旨在逆转或改善对ICIs的耐药性(如NCT05286294, NCT04988841)。其他试验则探索FMT在肝癌(NCT05690048)、胰腺癌(NCT06393400)等其他癌种中的应用,或用于减轻治疗相关的毒副反应,如免疫相关性结肠炎(NCT04038619)。
关于益生元、益生菌和后生元的临床试验同样蓬勃发展,多数旨在增强抗癌免疫治疗的效果(如NCT05303493, NCT06250335)。
A.muciniphila(NCT05865730)和C.butyricum等下一代益生菌的专项试验正在进行中。
这些研究不仅关注疗效,也探索其在围手术期管理、预防癌前病变复发以及减轻治疗副作用方面的潜力。
越来越多的证据表明,肠道微生物组是癌症治疗结果的关键调节器,其通过免疫调节、药物代谢和维持上皮屏障完整性等多种机制发挥作用。这一认知推动了新型诊断和治疗策略的开发,但临床转化仍受制于机制不确定性、缺乏个性化生物标志物以及在癌症人群中实施的挑战。
微生物组研究揭示,宿主与微生物群作为一个整合系统运作,治疗结果源于宿主-微生物-药物三者间的动态相互作用。
功能冗余、微生物可塑性和强烈的情境依赖性使得因果推断变得困难,也可能解释了为何微生物标志物在不同研究中难以重复。当前的许多干预措施基于普适性策略而非深刻的机制理解,而个体差异进一步加大了个性化治疗的复杂性。
为了推动该领域的发展,未来的研究必须转向由AI赋能、以机制为导向的精准干预,这需要以可靠的伴随生物标志物为支撑,并通过大规模、纵向的实用性临床试验证实。至关重要的是,临床试验必须考虑癌症患者的特殊限制,并在真实世界环境中关注有临床意义的功能性终点。随着微生物组科学日益融入肿瘤学,它有望通过将机制精度与临床相关性相结合,重新定义癌症治疗,实现更有效、更耐受的疗法。
注:本账号内容仅供参考和学术探讨,不构成医疗诊断或治疗建议。任何健康问题,请务必咨询专业医生。
主要参考文献
Hajjar R, Mars RAT, Kashyap PC. Harnessing the microbiome for cancer therapy. Nat Rev Microbiol. 2026 Jan 5.
Nobels A, van Marcke C, Jordan BF, Van Hul M, Cani PD. The gut microbiome and cancer: from tumorigenesis to therapy. Nat Metab. 2025 May;7(5):895-917.
de Vos, W. M., Tilg, H., Van Hul, M. & Cani, P. D. Gut microbiome and health: mechanistic insights. Gut 71, 1020–1032 (2022).
Domzaridou, E. et al. The impact of oral antibiotics prior to cancer diagnosis on overall patient survival: findings from an English population-based cohort study. Curr. Oncol. 30, 8434–8443 (2023).
Yuan, L. et al. The influence of gut microbiota dysbiosis to the efficacy of 5-fluorouracil treatment on colorectal cancer. Biomed. Pharmacother. 108, 184–193 (2018).
Routy, B. et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359, 91–97 (2018).
Elkrief, A. et al. Antibiotics are associated with worse outcomes in lung cancer patients treated with chemotherapy and immunotherapy. NPJ Precis. Oncol. 8, 143 (2024).
Ransohoff, J. D. et al. Antimicrobial exposure is associated with decreased survival in triple-negative breast cancer. Nat. Commun. 14, 2053 (2023).
Cong, J. et al. Bile acids modified by the intestinal microbiota promote colorectal cancer growth by suppressing CD8+ T cell effector functions. Immunity 57, 876–889.e11 (2024).
Tintelnot, J. et al. Microbiota-derived 3-IAA influences chemotherapy efficacy in pancreatic cancer. Nature 615, 168–174 (2023).
Bender, M. J. et al. Dietary tryptophan metabolite released by intratumoral Lactobacillus reuteri facilitates immune checkpoint inhibitor treatment. Cell 186, 1846–1862.e26 (2023).
Salminen, S. et al. The International Scientific Association of Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of postbiotics. Nat. Rev. Gastroenterol. Hepatol. 18, 649–667 (2021).