-

 CNAS L23010
CNAS L23010

 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业 二级病原微生物安全实验室

谷禾健康
基因组规模代谢网络模型(Genome-scale metabolic model,GEM),是一种包含了某种特定生物或者是细胞基因组范围代谢反应,及其酶及基因关联的数学模型。
这里,我们基于文章的描述,介绍一款新软件——MetaGEM。
研究者认为,目前代谢建模的工作流程仍然是倾向于依赖参考基因组作为重建和模拟GEMs的起点,这忽略了微生物群落中存在的物种内和物种之间的多样性。也限制了对已知参考基因组空间中的代谢网络的分析和解释。
可能导致假阳性(即在参考基因组中存在但在群落中的变量中缺失的通路)或假阴性(即在参考基因组中缺失但在群落变量中存在的通路)结果,最终导致对个别物种代谢通路以及交互营养共生(cross-feeding)相互作用的不准确预测。
也就是说当前的代谢建模方法很可能无法捕捉特定物种在不同环境中的特定代谢特征,例如具有不同疾病状况的个体的微生物群。为了克服这一局限,研究者们开发了MetaGEM。
MetaGEM可以不依赖参考基因组,直接从短读的宏基因组数据中重建样本特定的代谢模型。
下图是该软件的流程图,图中蓝底白字的部分是该流程中所使用到的软件,都是已经由他人开发完成的。
研究者们自己开发的部分有两个:
一是end-to-end的框架,能够进行群落水平的代谢交互模拟;
二是一个来自宏基因组生物群落的14,000多个MAGs,包括3750份高质量的MAGs,以及来自人类肠道微生物组研究和全球微生物组项目的相应的随时可用的GEMs。
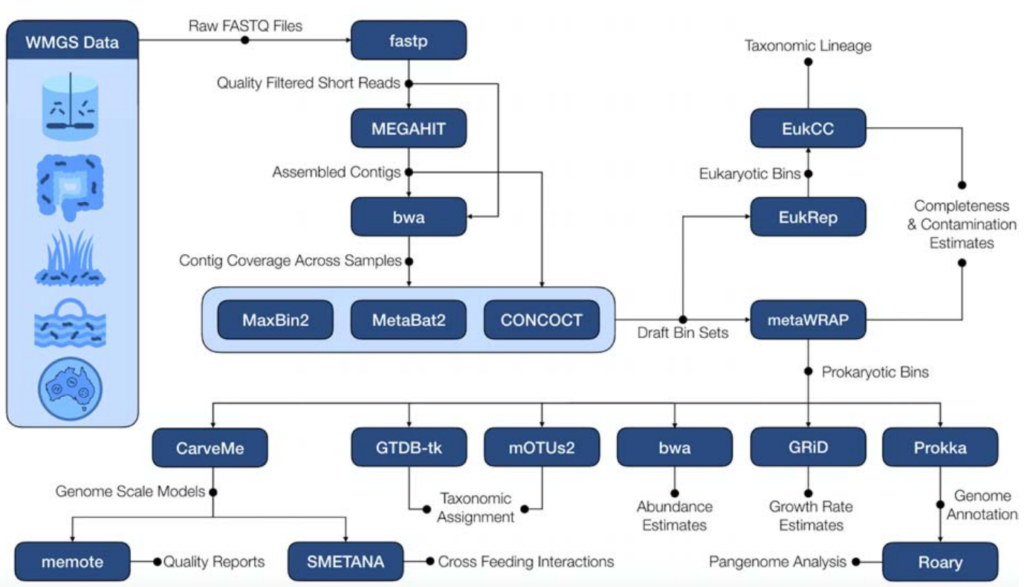
整个流程使用Snakemake实现,从原始的宏基因组的fastq文件开始,质控、组装、估计contig覆盖率、binning、Bin的改进和重组、MAG丰度定量和物种分类、CarveMe进行基因组规模代谢模型重建及质量报告,Smetana模拟重建的基因组规模代谢模型的肠道微生物群落。
(这里只简单介绍了处理步骤,文章中的“Methods”部分有给出使用的参数)
除了以上的必备选项,该流程还有一些附加功能可供用户选择。可以使用GRID估计中和高覆盖率的MAGs的增长率。
Prokka可以对MAGs做功能注释,并且其结果可以提供给Roary,获得一组MAGs的核心MAG和泛基因组的可视化结果。
EukRep可以用于寻找真核生物的MAGs。
EukCC可以对真核生物的bins做后续的分析。
MetaGEM流程具有两个特点:
一是直接从宏基因组获得高质量的代谢重建;
二是可以为个性化的人类肠道群落建模,研究者通过两个实验进行了描述:
MetaGEM模型与EMBL、AGORA、KBase和Bigg模型相比较
用MetaGEM基于宏基因组短读序列构建MAGs,分为HQ(高质量的),MQ(中等质量的),并以此进行代谢重建,总共获得14087个GEMs,然后将它们与高度精选的基于参考基因组的BIGG模型、AGORA、EMBL和KBase模型进行了比较。
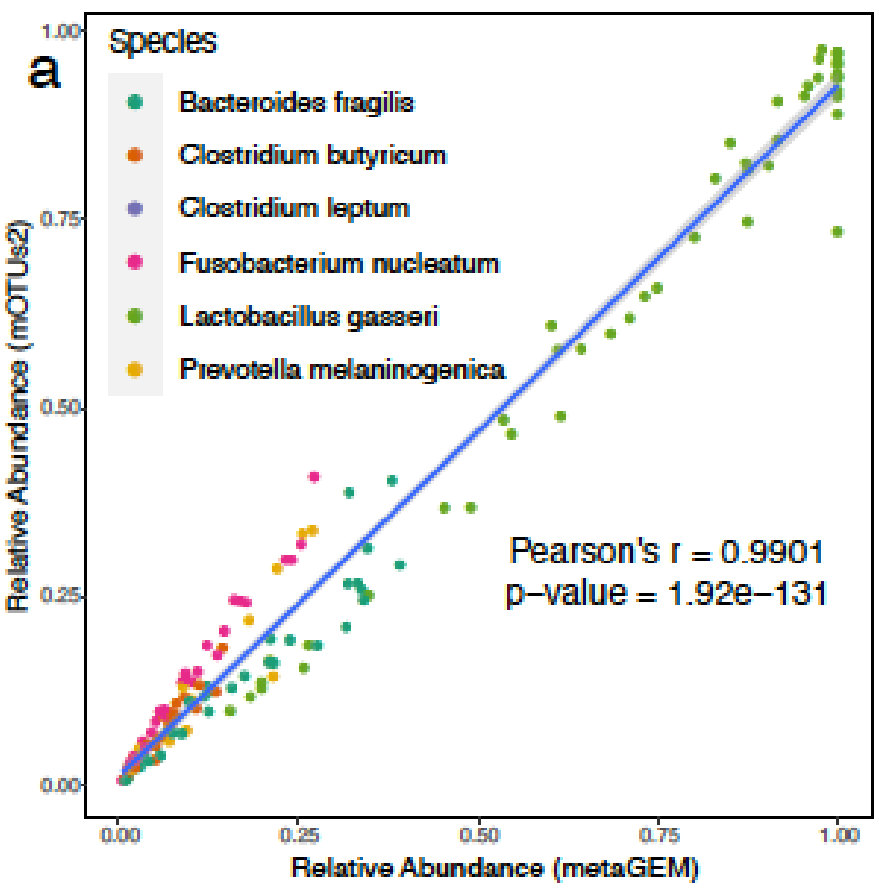
利用基于定位的方法(方法)生成的丰度估计值与基于标记基因的丰度估计值完全相关;
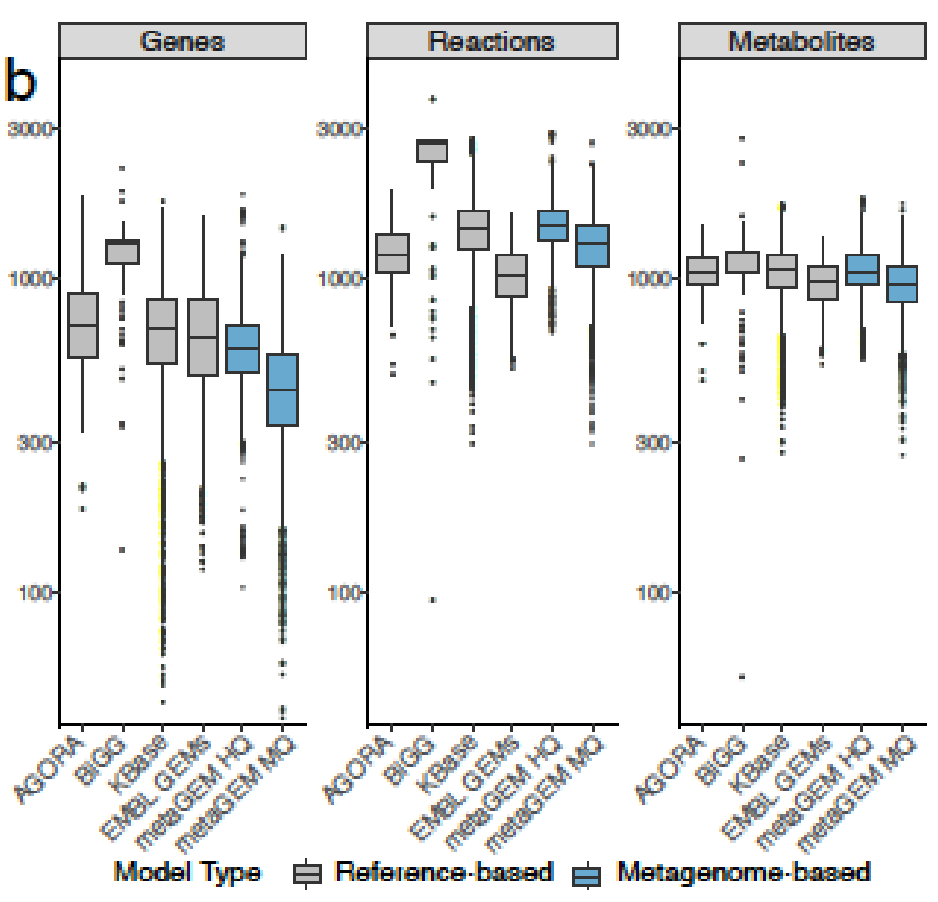
MetaGEM和其他模型都具有类似数量的反应和代谢物,但基因数量相比较少;
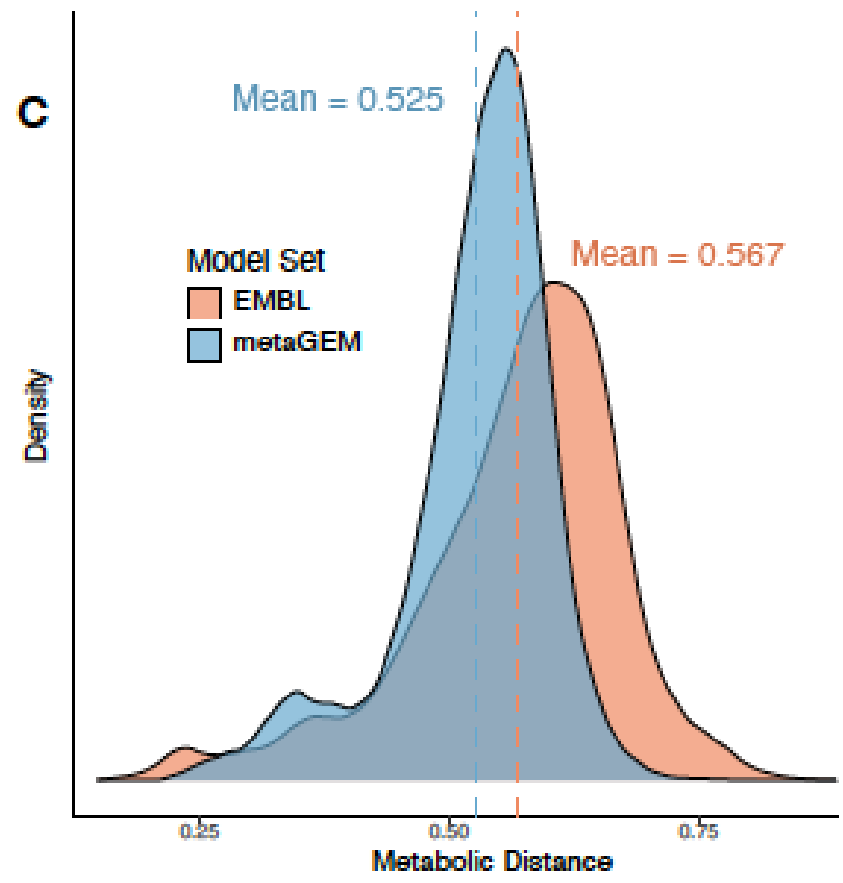
通过计算模型之间成对的代谢之间的距离,发现MetaGEM具有相似的酶多样性分布;
可以捕捉到种水平物种间的显著的代谢差异。高达60%的代谢多样性存在于物种泛基因组中,metaGEM模型捕获的物种内代谢变异程度显著
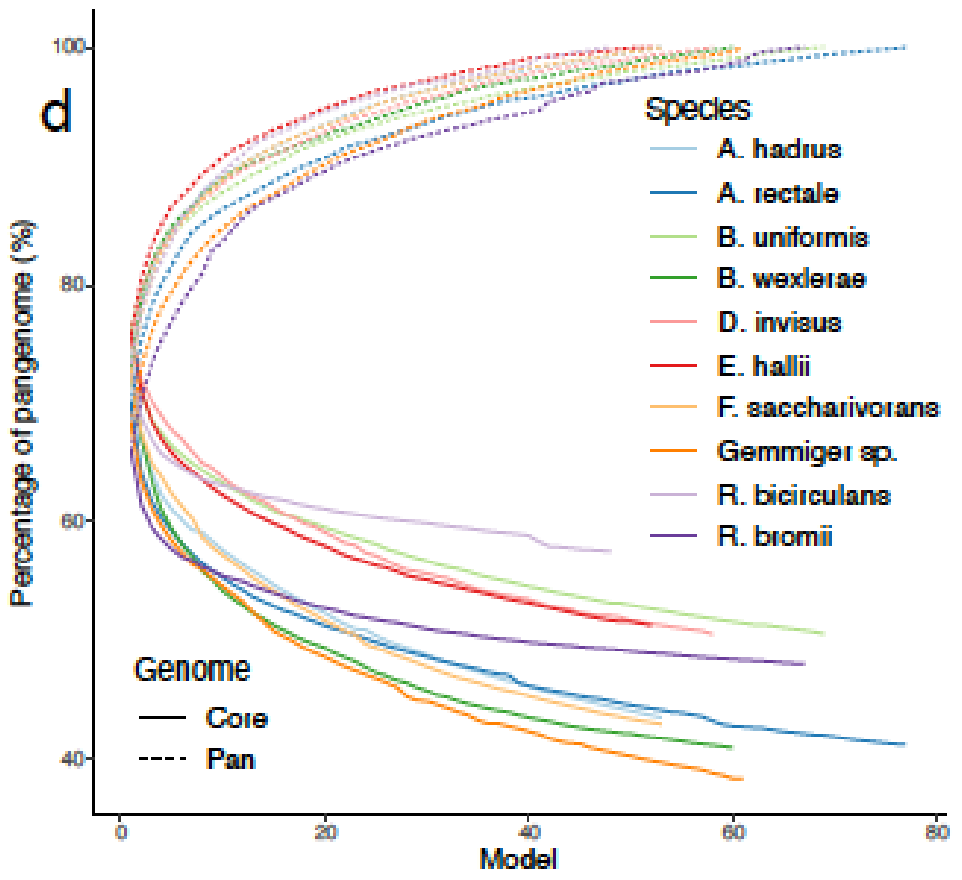
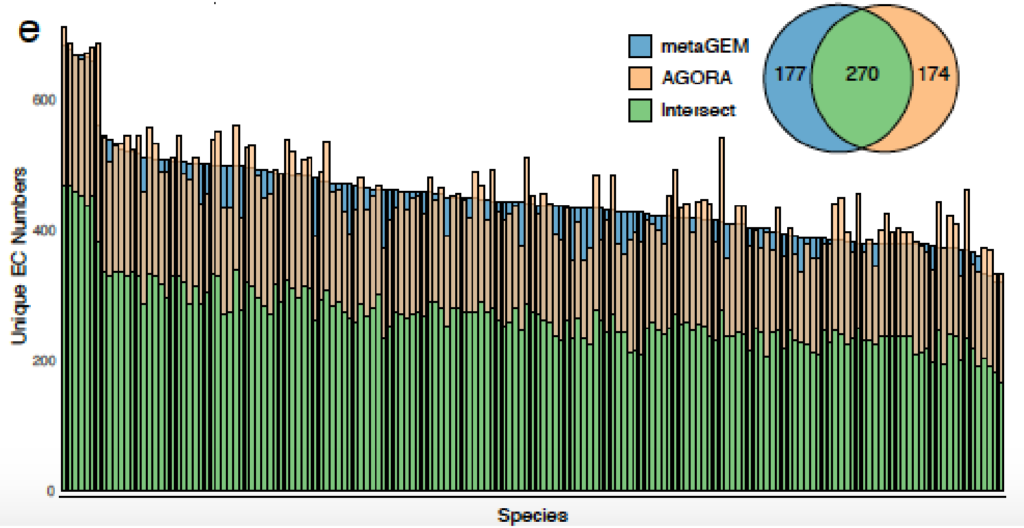
与基于参考基因组的肠道物种代谢模型AGORA比较,发现基于参考的模型引入的代谢反应不一定存在于每个宏基因组环境中,而MetaGEM模型是完全基于实际的宏基因组在特定环境下重建的代谢模型。
AGORA和MetaGEM模型的EC数的交集在48.9%到69%之间,其中53.9%的情况下MetaGEM模型比相应的AGORA模型包含更多的EC数。
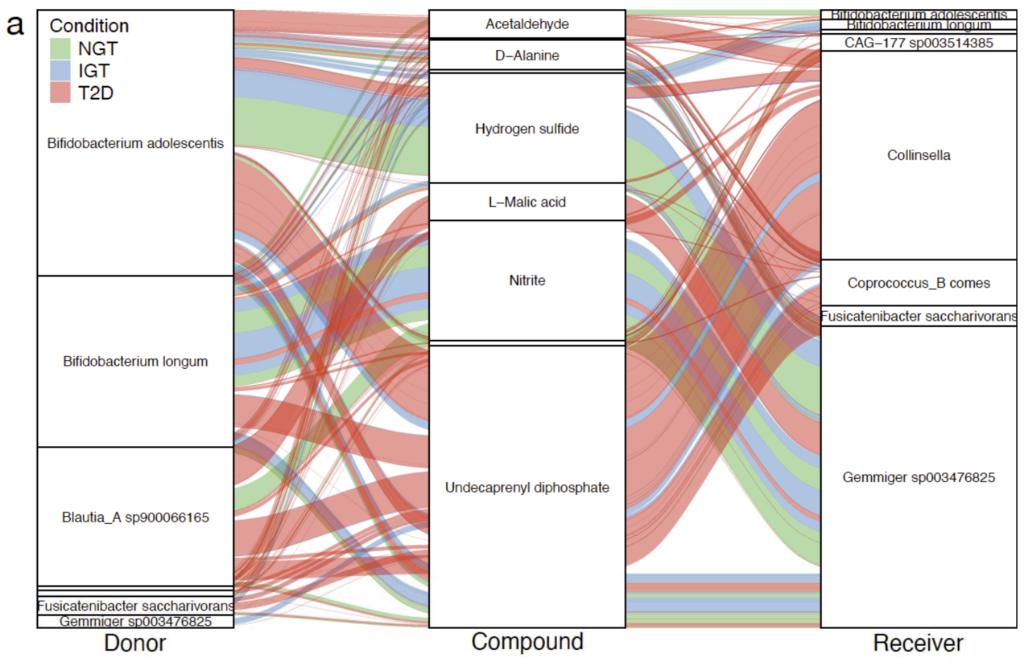
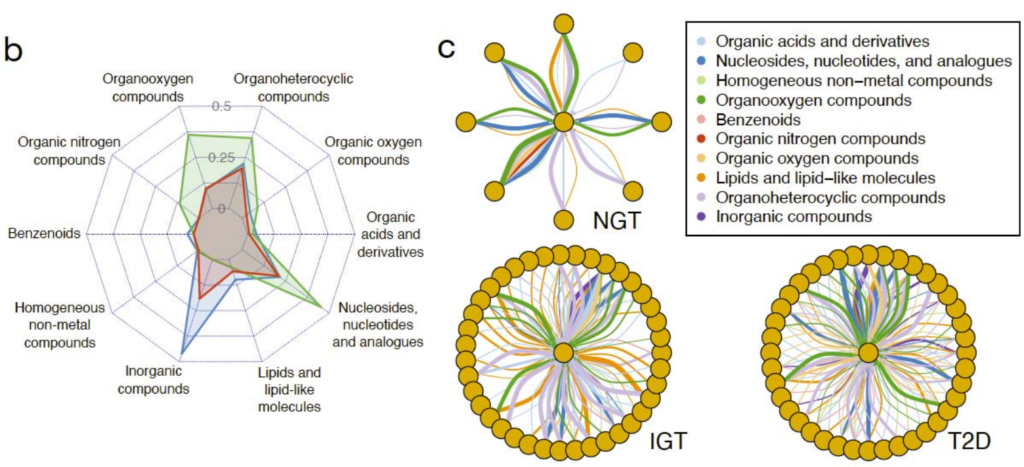
研究健康和代谢受损的2型糖尿病患者肠道微生物群落中潜在的微生物代谢相互作用。
使用metaGEMs通过137个宏基因组数据重建了4127个个性化的GEMs。
根据疾病状况分类,即正常糖耐量(NGT,n=42)、糖耐量受损(IGT,n=42)、 2型糖尿病(T2D,n=53),然后应用Smetana软件模拟微生物群落中的物种间依赖关系,Smetana为每个群落输出一个分数表,对应于在给定条件下为支持群落成员的成长而应发生的交叉喂养相互作用强度的度量,即物种A生长的可能性取决于物种B的代谢物X。
不同的2型糖尿病疾病组(NGT、IGT、T2D)相对应的肠道代谢基因组产生具有不同代谢结构的群落。
MetaGEM具有完善的流程,搭载的工具也是生物信息分析中常用的处理工具,下载很方便,用conda就能完成。无需参考基因组,这也意味着不需要下载动辄几十Gb的文件。使用Snakemake做流程的自动化管理,运行命令简单,也可以分步骤运行。
总体而言,MetaGEM可以直接从宏基因组数据中研究复杂微生物群落中特定样本(sample-specific)的新陈代谢。
【附录】
关于文中MetaGEM流程搭建所应用到的宏基因组分析软件,这其中也有我们常用的软件,比如fastp、MEGAHIT、bwa、SAMtools、metaWRAP,它们在处理数据时非常的方便也易于上手。
参考文献:
Zorrilla F, Patil K R, Zelezniak A. metaGEM: reconstruction of genome scale metabolic models directly from metagenomes[J]. bioRxiv, 2021: 2020.12. 31.424982.
相关阅读: