-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康

随着大家陆续“阳康”,大家逐渐恢复以往的生活,城市的烟火气回来了。
然而阳康后真的万事大吉了吗?
还是有很多朋友处于这样的状态:感觉恢复了,又好像没有完全恢复,身体多少有点不适,开始关心:
这种新冠肺炎后遗症,也就是临床上常说的“长新冠”,已经成为部分人不得不面临的事实。
“长新冠”是一种多系统疾病,据估计,全球至少有 6500 万人患有长新冠,病例每天都在增加。由于许多未记录在案,这个数字实际可能要高得多。
“长新冠”与所有年龄段和急性期疾病严重程度相关,许多患者在多个器官系统中经历了数十种疾病,包括心血管、血栓、脑血管疾病、2型糖尿病、自主神经功能障碍等。基于对其超过 2 年的研究,如果不采取行动任其发展,很大一部分患有长新冠的人可能会有严重后果。
新出现的临床前和临床研究表明,肠道微生物群可能有助于理解 COVID-19 的发病机制和疾病结果;SARS-CoV-2 感染与肠道微生物群的改变有关,并与炎症和免疫反应相关。
本文基于多篇文献报道,主要介绍关于“二次感染”以及“长新冠”问题的最新研究进展,同时也着重介绍了多种与新冠恢复期调理相关的干预措施。
本文由于篇幅较长,主要分为上、中、下三大篇章,主要包括以下内容:
上篇: 关于“二次感染”
中篇: 关于“长新冠”
下篇: 关于“干预措施”
上 篇
关于 “二次感染”
已经阳康的朋友,可能关心会不会马上又来一波,出现“二次感染”?
我们先了解一下,二次感染指的是什么?
“二次感染” 也叫“再感染”或者“重复感染”,是指一个人感染了新冠之后,经过足够的时间,然后再次感染。一般而言,如果一个人在第一次阳性检测后 90 天或更长时间再次检测呈阳性,则被视为二次感染。
注:也有部分研究定义间隔>30天。
再感染后可能出现临床症状,患者因核酸载量高而具有传染性。
与没感染过的人群相比,已经感染过的人群再感染风险低87%
先看一组数据:
➪
一项Meta分析综合了先前感染SARS-CoV-2人群再感染风险的全球证据:先前感染SARS-CoV-2的人可能会再次感染,他们的感染风险比之前没有感染的人低。
SARS-CoV-2再感染发病率为:
0.7 / 10,000人日(标准差0.33).
以前感染过的人比从未感染过的人再次感染的可能性低87%(HR = 0.12)。
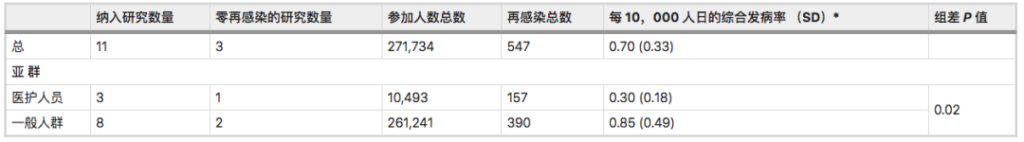
注:该研究荟萃分析和系统审查分别包括11项研究和11份病例报告(样本量都不小于100)。
看到这里是不是觉得可以放心了,再次感染的概率很低?
并不一定。需要注意的是,该研究虽在近期发表,但研究的数据是基于出现在2021年4月及之前的毒株,当时奥密克戎变种尚未出现。
研究表明,奥密克戎变异株的免疫逃逸,导致比其他变异株具有更高的传播。那么,有没有关于奥密克戎毒株的数据?
有。
与感染过德尔塔毒株的人群相比,在奥密克戎阶段,再感染风险显著高于德尔塔时期
➪
一项回顾性观察性研究,分析来自意大利北部利古里亚地区的感染者,在研究期间(2021年9月至2022年5月),利古里亚记录了335117例SARS-CoV-2感染病例,其中15715例再次感染。在奥密克戎阶段(从2022年1月3日开始占主导地位),再感染的风险是德尔塔阶段的4.89倍(p<0.001)。
其他也有来自意大利的研究(2021年8月至2022年3月)认为,奥密克戎时期的再感染风险比德尔塔时期高18倍。
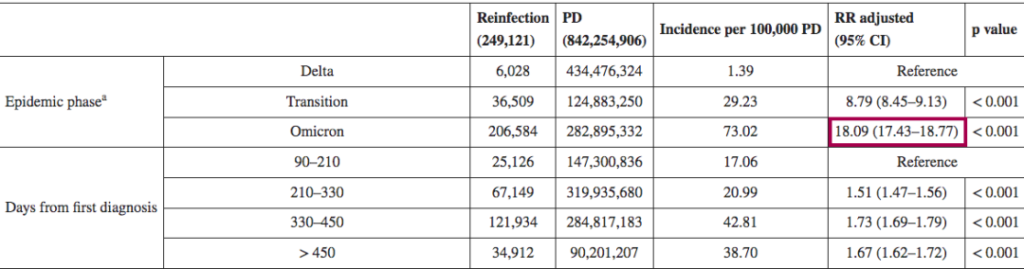
DOI: 10.2807/1560-7917.ES.2022.27.20.2200372
➪
英国爱丁堡大学的研究人员发现,奥密克戎变异株的可能再感染率约为德尔塔变异株的10倍。
➪
来自圣保罗大学医院281名医护人员的再感染率(2020年3月10日至2022年3月10日):
奥密克戎时期再感染率显著增加(0.8%到4.3%;相对风险5.45 [95%IC 3.80–7.81];p < 0.001)
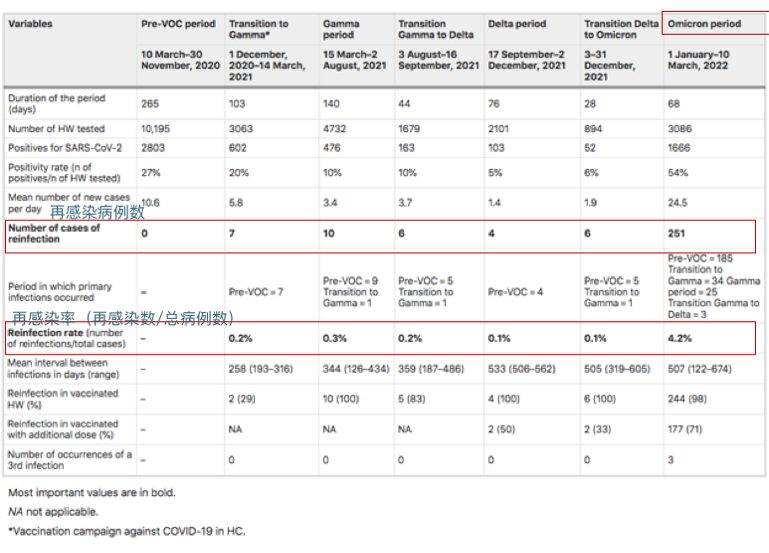
Guedes AR, et al., Sci Rep. 2023
关于奥密克戎再感染率上升的证据相当多,此处就不一一列举了。既往感染虽然可以在一定程度上防止新冠的再次感染,但对于奥密克戎来说,之前的抗体保护作用较弱,并且在一定期限后迅速减弱,接下来我们来看一下,既往感染的保护期限大概能维持多久。
队列研究证实,既往感染对奥密克戎变异株(BA.1、BA.2、BA.4 和 BA.5)再感染的保护作用低于对德尔塔和较旧变异株再感染的保护作用。
➪
50项研究共纳入118名再感染患者,从首次感染到再感染的最短时间为19日,最长为293日。
● 针对奥密克戎BA.1和BA.2毒株:
先前感染德尔塔变异株可将症状性感染的风险降低50%至67%.
➪
在丹麦的一项队列研究中,如果先前感染发生在3至6个月前,则对奥密克戎BA.1或BA.2的保护率为43.1%,如果先前感染至少发生在6个月前,则为22.2%.
● 针对奥密克戎BA.4和BA.5毒株:
BA.4 和 BA.5 这两个变体与 BA.2 的相似性高于 BA.1 菌株,携带着它们自己独特的突变,包括病毒刺突蛋白中 L452R 和 F486V 的变化,这些变化可能会调整其锁定宿主细胞并避开某些免疫反应的能力。
对卡塔尔人群的分析提供了关于奥密克戎BA.4和BA.5防护的详细信息:
在未接种疫苗的人群中,既往感染奥密克戎BA.1或BA.2可使任何感染奥密克戎BA.4或BA.5的风险降低至少68.7%(CI,64.0%至72.9%),而如果先前感染发生在奥密克戎变异株出现之前,则仅为27.7%(CI,19.3%至35.2%).
针对BA.4或BA.5免受BA.1或BA.2感染的保护作用在4个月内是强大的,但这种保护可能会迅速减弱。
关于再感染的风险和保护期限的研究
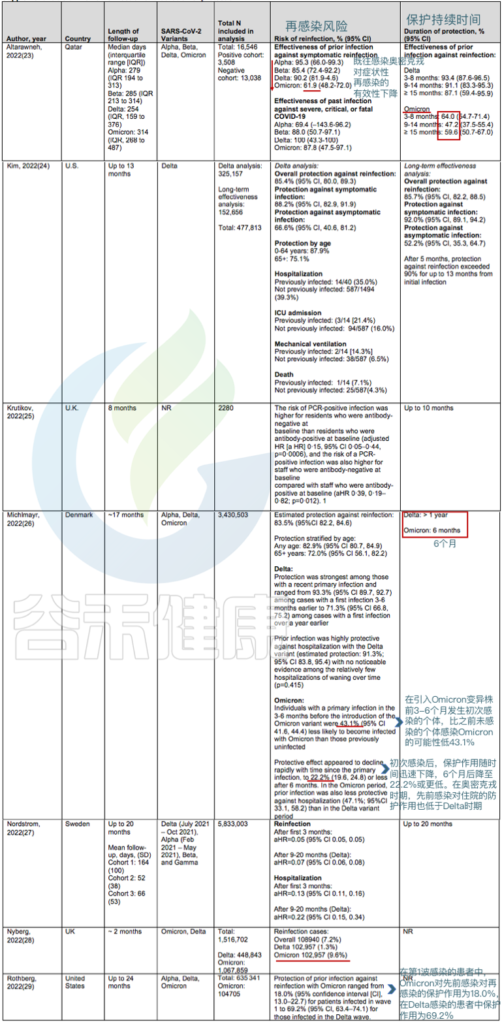
doi:10.7326/M22-1745
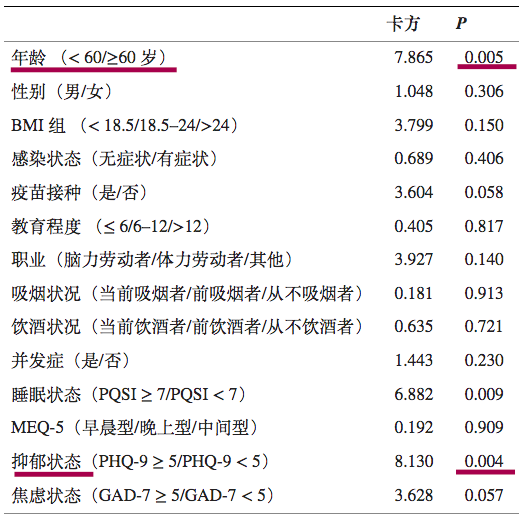
高龄和抑郁状态是奥密克戎再感染的危险因素
一项前瞻性队列研究,纳入了 933 名被诊断为 Omicron BA.2.2 感染且治疗后检测呈阴性的成年患者。
最终,683例符合标准,进行研究分析。
注:数据来源以及筛选标准:
患者来自四川省临水县,数据由四川省疾病预防控制中心提供。如果患者符合以下任何标准,则被排除在外:
感染前诊断为精神疾病,伴随需要住院治疗的严重疾病,以及交流障碍或拒绝参与。
在683名奥密克戎感染患者中,出院后30天内有116例再阳性,总体再阳性率为16.4%.
预测再阳性风险变量的单变量逻辑回归
DOI:10.3389/fpubh.2022.1014470
注:PHQ-9,患者健康问卷九。PHQ-9量表的总分范围为0至27分,其中0至5分表示没有抑郁,得分>5表示抑郁状态;评分越高,抑郁症状越严重。
比较分析显示:
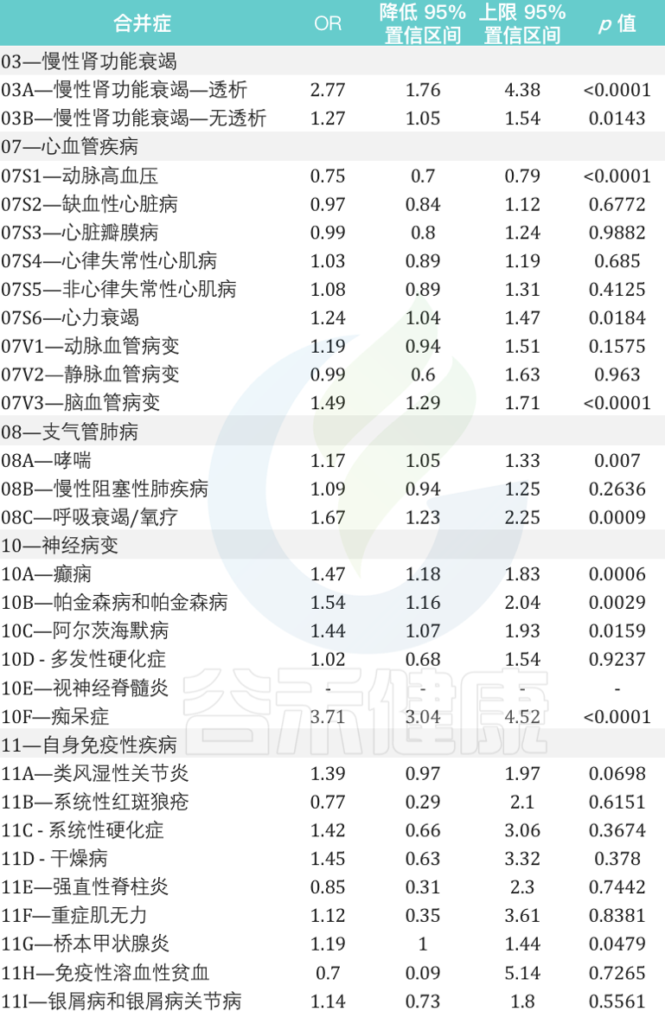
慢性肾功能衰竭、心血管疾病、支气管肺病、神经病变和自身免疫性等疾病患者再感染的风险相对增加
来自意大利北部地区的数据显示:
女性个体的再感染风险比男性高17%(OR为1.17,95%CI为1.13-1.21,p <0.0001)
在再感染者中,60岁及以上患至少一种潜在慢性病人群的风险比其他年龄组高7倍。
在慢性心血管疾病中,心力衰竭和脑血管病变与再感染风险的相关性最高,风险为1.24(95%CI为1.04-1.47,p = 0.0184)和1.49(95%CI为1.29-1.71,p <0.0001)是 未再感染个体的1.24倍(95%CI为1.29-1.71,p 0.0001).
在慢性肾衰竭患者中,接受透析的患者再感染风险几乎高出 3 倍(OR 为 2.77,95% CI 为 1.76–4.38,p < 0.0001).
与未再感染者相比,哮喘和呼吸衰竭/氧疗患者的再感染风险分别增加 1.17 倍(95% CI 为 1.05 –1.33,p = 0.0070)和 1.67 倍(95% CI 为 1.23–2.25,p = 0.0009).
在神经病变患者中,癫痫患者、帕金森病和阿尔茨海默病的再感染风险几乎是未再感染个体的两倍。
痴呆患者的风险大约高出四倍(OR为3.71,95%CI为3.04-4.52,p <0.0001).
在患有自身免疫性疾病的个体中,桥本甲状腺炎是再感染个体中最相关的疾病(OR为1.19,95%CI为1.00-1.44,p = 0.0479)
2021年9月至2022年5月期间与SARS-CoV-2再次感染患者有关的主要合并症的详细信息:
doi: 10.3390/vaccines10111885
此外,2022年发表的一篇系统评价报告了2019年12月1日至2021年9月1日的数据,发现高血压和肥胖是再感染患者中最常见的,其次是终末期肾衰竭、哮喘、慢性阻塞性肺病、痴呆、血脂异常和2型糖尿病。
其他研究也报道,终末期肾衰竭、高血压、糖尿病、慢性呼吸系统疾病、肝病和心血管疾病病史患者的再感染风险更高。
二次感染依然有症状
大多数再感染患者表现出临床症状,只有少数研究报告患者在第一次和继发感染时均无症状。
其他包括50项研究的数据显示再感染患者特征:


注:以上数据是2019年12月1日至2021年9月1日期间的研究
➪
二次感染相对首次感染严重程度有所减轻
韩国疾病控制和预防局于2020年1月20日至2022年5月7日在流行病学调查中通过综合系统报告的新冠肺炎病例数据库与健康保险审查和评估服务系统合并。使用具有二项分布的广义线性模型估计二次感染发作时与一次感染时的严重性比值比(SOR)。
结果发现,在所有患者中,再次感染的SOR为0.89(95%置信区间[CI]:0.82–0.95),与首次感染发作相比,严重程度有所减轻。
➪
一项研究显示,截至 2022 年 11 月 9 日,估计 94%(95% CrI,79%-99%)的美国人口至少感染过一次 SARS-CoV-2。
2022年11月针对SARS-CoV-2感染和重症的有效保护大大高于2021年12月
结果发现,2022年11月9日,在美国,对奥密克戎变异株SARS-CoV-2感染的保护估计为63%(51%-75%),对重症的保护率为89%(83%-92%).
11 月的人口免疫力高于最初奥密克戎激增后立即出现的情况。
对二次感染后的重症情况,不同研究结果不一致。
➪
发表在《Nature Medicine》的一项研究建立了一个包括5819264人的队列,其中SARS-CoV-2感染者(n = 443588)、再感染(两次或多次感染,n = 40947)和非感染对照(n = 5334729). 使用逆概率加权生存模型来估计死亡、住院和事件后遗症的风险和6个月的负担。
结果发现,与无再感染相比,再感染增加了死亡风险(危险比(HR) = 2.17,95%置信区间(CI)1.93–2.45),住院风险(HR = 3.32,95%可信区间3.13-3.51)和后遗症风险,包括肺部、心血管、血液学、糖尿病、胃肠道、肾脏、精神健康、肌肉骨骼和神经系统疾病。无论疫苗接种状况如何,风险都很明显。
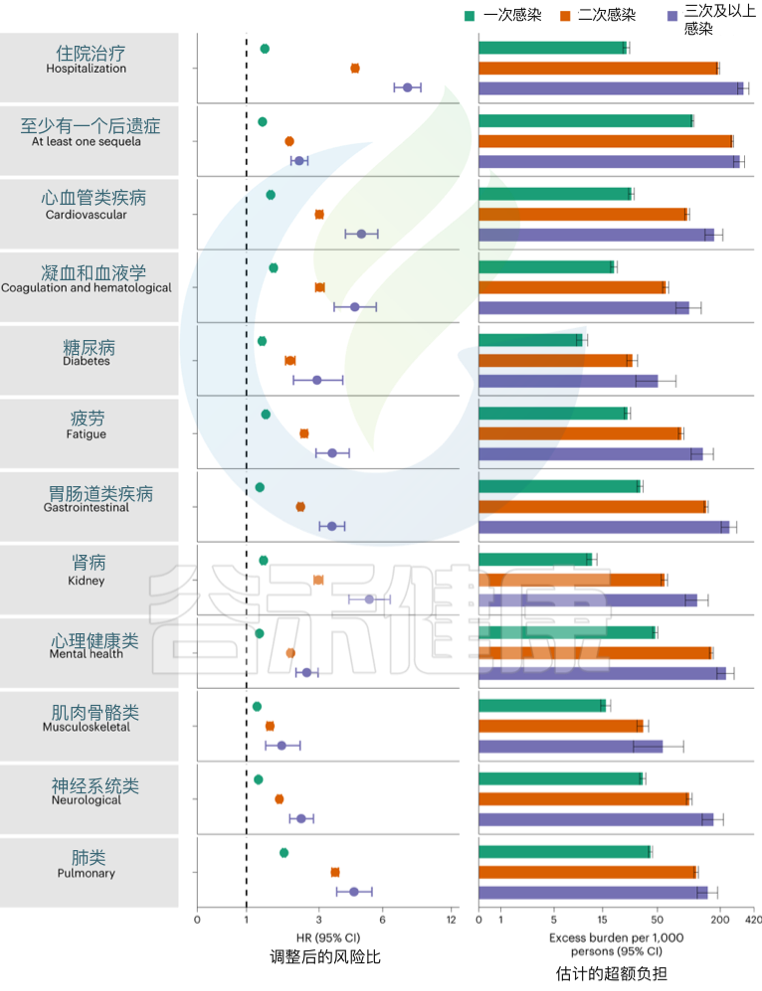
Bowe B, et al., Nat Med. 2022
风险在急性期最为明显,但在6个月后持续存在。与未感染的对照组相比,反复感染的累积风险和负担随着感染次数的增加而增加。
注:该研究数据主要来自美国退伍军人事务部国家医疗保健数据库,选取时间在2020年3月1日至2022年4月6日期间。退伍军人事务部的人口大多由老年人和男性组成,该队列也包括 10.3% 的女性,12% 在 38.8 岁以下(2021 年美国人口的中位年龄)。
关于未来:
英国牛津大学研究病毒进化的科学家认为,奥密克戎及其分支继续占据主导地位的时间越长,慢性感染产生全新变种的可能性就越小。
流行病学研究表明,连续的 COVID-19 浪潮正在变得温和。但这种趋势不应该被视为理所当然。
随着全球对反复接种疫苗和感染后免疫力的增强,研究人员预计 SARS-CoV-2 浪潮的频率将会放缓。
也有研究人员认为,SARS-CoV-2 的一个可能未来是,随季节起伏,通常在冬季达到顶峰,通常每三年左右重新感染一次。
关于其带来的症状是否会越来越轻等问题,研究人员还将持续关注。
总的来说,COVID-19疫情是一个高度动态的全球事件,各种流行病学驱动因素随着时间的推移而变化(包括新变异株的出现、疫苗吸收的增加和免疫力的减弱等各种因素),再感染的流行病学及其健康后果也可能随着时间的推移而改变。
对于已经感染过一次的人来说,继续保持警惕,降低再感染的风险,对于整体健康来说可能很重要。
以上是针对阳康后担心自己是否会二次感染的朋友的问题,然而也有一些朋友到现在,仍然觉得身体没有完全恢复,那就要考虑新冠可能带来的后遗症,也就是临床上说的“长新冠”,接下来章节,我们来了解一下关于“长新冠”及其持续时间,什么人群容易发生,会影响哪些器官功能,具体形成机制,与肠道菌群的关联等。
中 篇
关于“长新冠”
▾◆▽◆▽◆▾
►
“长新冠”是如何定义的?
2021年10月,世界卫生组织将其定义为:
有疑似或确诊新冠感染史,通常发生于起病三个月后,症状通常至少持续两个月,且不能被其他诊断所解释的症状。
►
“长新冠”会有哪些表现?
常见症状包括:
也包括其他一些症状,通常或多或少会影响日常生活。
症状可能是:
从新冠急性发作初步恢复后新出现的症状;
也可能从最初的疾病中持续存在;
也可能随着时间的推移而波动或复发。
►
“长新冠”什么时候发生发展?持续多长时间?
症状的发作和时间过程因个体和症状类型而异。
神经系统症状通常延迟发作数周至数月:在有认知症状的参与者中,43%的人报告在COVID-19至少1个月后才出现认知症状,延迟发作与年龄较轻有关。一些神经认知症状会随着时间的推移而恶化,并且往往会持续更长时间。而胃肠道和呼吸道症状更容易解决。
关节、骨骼、耳朵、脖子和背部的疼痛在1年时比2个月时更常见,感觉异常、脱发、视力模糊以及腿、手和脚肿胀也是如此。
麻痹症在初次感染后平均3个月发病;与其他神经认知症状不同,它通常会随着时间的推移而减少。
很少有长新冠患者能够完全康复,一项研究发现,在最初感染2个月后出现症状的患者中,85%在症状出现1年后出现症状。尽管ME/CFS和自主神经障碍的诊断通常是终身的,但未来的预后仍不确定。
►
有多少人会患“长新冠”?
据保守估计,全世界至少有 6500万 人患有长新冠,病例每天都在增加。
注:有许多未登记病例,这个数字实际上可能要高的多。
据估计,非住院病例的发病率为10-30%,住院病例为50-70%,接种疫苗病例为10-12%。
长冠肺炎与所有年龄段和急性期疾病严重程度相关,36岁至50岁之间的确诊率最高,大多数长冠肺炎病例发生在患有轻度急性疾病的非住院患者中,因为该人群占新冠肺炎总病例的大多数。
►
哪些人群更有可能患“长新冠”?
风险因素可能包括:
注:三分之一的长期 COVID 患者没有确定的原有疾病。
COVID-19 可能对任何患者产生长期影响,包括无症状或轻症患者。相比儿童和青少年,“长新冠”看起来在成人中更为常见。
►
“长新冠”的发病机制?
简单来说,潜在机制包括:
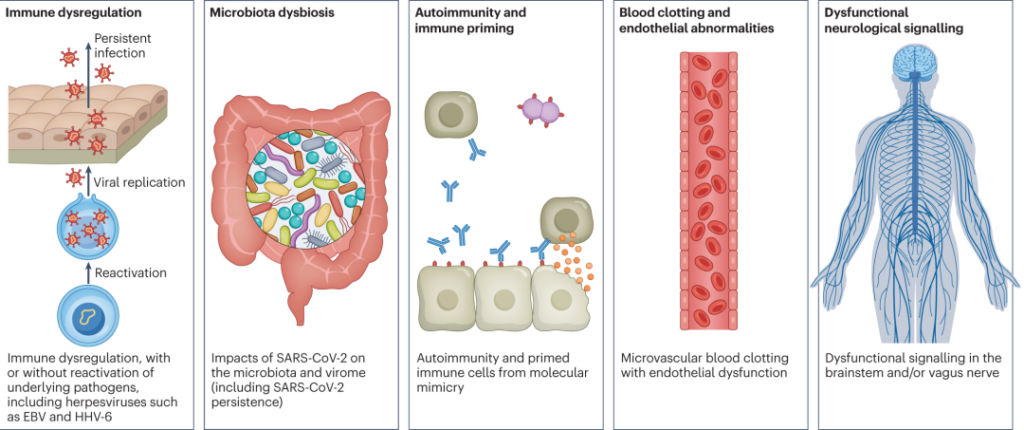
DOI: 10.1038/s41579-022-00846-2
以上是关于“长新冠”的一些基本科普,具体关于“长新冠”带来的多种不良后果,需要关注的疾病,形成机制,与菌群的关联等,我们将在接下来的小节详细介绍。
▾◆▽◆▽◆▾
长新冠包括多种不良后果,常见的新发疾病包括:心血管、血栓和脑血管疾病, 2型糖尿病、肌痛性脑脊髓炎/慢性疲劳综合征(ME/CFS)和自主神经功能障碍,体位性心动过速综合征 (POTS).
长新冠症状及其对多种不同病理器官的影响
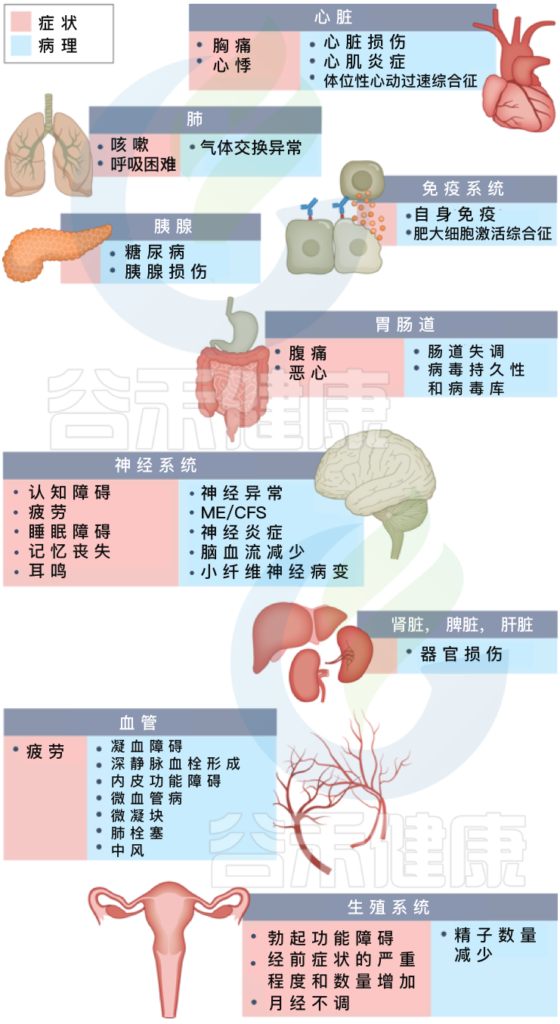
Davis HE, et al., Nat Rev Microbiol. 2023
★
血管问题和器官损伤
SARS-CoV-2会损害许多器官系统。在不同组织中已经证明的损伤主要归因于免疫介导的反应和炎症,而不是病毒对细胞的直接感染。
循环系统破坏包括内皮功能障碍和随后的下游影响,以及深静脉血栓形成、肺栓塞和出血事件的风险增加。
微凝块-> 促进血栓形成
在急性新冠肺炎和长冠肺炎中检测到的微凝块有助于血栓形成,在长新冠中也发现了血细胞大小和硬度的长期变化,有可能影响氧气输送。感染18个月后,长新冠患者的血管密度长期降低,特别影响小毛细血管。
心血管疾病风险增加
对美国退伍军人事务部数据库的分析显示,SARS-CoV-2感染1年后超过150000人患多种心血管疾病的风险显著增加,包括心力衰竭、心律失常和中风,与最初出现新冠肺炎的严重程度无关。
持久的心脏异常
心脏MRI研究显示,在100名既往有新冠肺炎发作史的患者中,78%的患者(感染后平均71天进行调查)和58%的长期冠状病毒感染者(感染后12个月进行研究)存在心脏损害,这增强了心脏异常的持久性。
新冠与多器官损害有关
除了心脏之外,新冠还与其他多种器官受损相关。
一项针对低风险个体的前瞻性研究,观察了心脏、肺、肝脏、肾脏、胰腺和脾脏,发现201名患者中70%至少有一个器官受损,29%有多个器官受损。
在同一研究小组对536名参与者进行的一项为期一年的随访研究中,研究作者发现,59%的人有单器官损伤,27%的人有多器官损伤。
一项针对VA数据的专门肾脏研究,包括89000多名患有新冠肺炎的患者,发现许多肾脏不良结果的风险增加。
另一项VA数据分析,包括181000多名患有新冠肺炎的患者,发现感染也会增加2型糖尿病的风险。
长新冠患者的心血管疾病风险增加,此外,器官损伤似乎是持久的,长期影响仍然未知。
★
神经和认知系统
神经和认知系统的症状是长新冠的主要特征,包括感觉运动症状,记忆丧失、认知障碍、感觉异常、头晕和平衡问题、对光和噪音的敏感性、嗅觉或味觉丧失(或幻觉)以及自主神经功能障碍,影响日常生活。长新冠的前庭听觉表现包括耳鸣、听力损失和眩晕。
认知障碍程度,类似酒驾上限
一项荟萃分析中,在感染后12周,32%的新冠肺炎患者出现疲劳,22%的患者出现认知障碍。长新冠患者的认知障碍会使人衰弱,与英国酒后驾驶限制的醉酒程度或认知衰老10年的程度相同,并且可能会随着时间的推移而增加。
犬尿氨酸途径的激活可能与认知障碍相关
一项研究发现,16%的患者在感染后2个月出现,26%的患者在感染后12个月出现。犬尿氨酸途径的激活,特别是代谢产物喹啉酸、3-羟基邻氨基苯甲酸和犬尿氨素的存在,已在长新冠中确认,并与认知障碍相关。
部分患者可能没有意识到认知障碍
在新冠康复的个体中也发现了认知障碍,当使用客观与主观测量时,认知障碍的比率更高,这表明有一部分认知障碍患者可能没有意识到和/或报告他们的障碍。
认知障碍是一种独立于焦虑和抑郁等心理健康状况的特征
认知障碍在住院和非住院患者中发生率相似。一份有130多万新冠肺炎患者参与的报告显示,随着时间的推移,焦虑和抑郁等心理健康状况恢复正常,但认知障碍(脑雾)、癫痫、痴呆、精神病和其他神经认知疾病的风险增加至少持续了2年。
神经病理学的可能机制包括:神经炎症、凝血病和内皮功能障碍对血管的损伤以及神经损伤
长新冠患者脑脊液异常,年龄较小可能延迟发作
研究发现,患有长新冠的患者存在阿尔茨海默病样信号,脑和脑干代谢低下与特异性症状和长新冠患者中的异常脑脊液发现相关,且年龄较小与神经症状延迟发作相关。
长新冠患者——“化疗脑”
在最近的一份预印本中,轻度感染的长新冠患者的多线细胞失调和髓磷脂丢失,其小胶质细胞反应性类似于化疗,即“化疗脑”。
即使非住院患者,认知能力也可能下降
英国生物银行(UK Biobank)的一项研究,包括新冠流行前后相同患者以及对照组的大脑成像,显示眶额皮层和海马旁回(初级嗅觉皮层相关区域的组织损伤标志物)的灰质厚度减少,与对照组相比,即使是非住院患者,新冠后患者的大脑整体缩小,认知能力下降更大。
尽管该研究将新冠感染者与对照组进行了比较,但并不是特别长的新冠患者,这可能对长新冠的认知成分有影响。在中枢神经系统中发现了线粒体蛋白以及SARS-CoV-2刺突蛋白和核衣壳蛋白的异常水平。在长新冠中也发现了四氢生物蝶呤缺乏症和氧化应激。
眼睛不适?可能与病毒在视网膜感染和复制有关
在眼睛中,在长新冠患者中发现角膜小神经纤维丢失和树突细胞密度增加,瞳孔光反应显著改变,视网膜微循环受损。SARS-CoV-2可以在视网膜和大脑类器官中感染和复制。长新冠的其他表现包括视网膜出血、棉絮斑和视网膜静脉闭塞。
焦虑和抑郁人群比例上升
在爱尔兰,研究报告了20-28%的普通人群出现焦虑和抑郁症状,多达4%的人有自残或自杀的想法。而在2018年爱尔兰健康调查(一项全国性的代表性调查),结果显示,在新冠大流行之前,自我报告的抑郁和焦虑发生率为6%.
仓鼠模型:持续炎症与焦虑和抑郁行为相关
轻度感染的小鼠模型显示小胶质细胞反应性和CCL11水平升高,这与认知功能障碍和受损神经发生有关。
仓鼠模型表现出持续的炎症状态,包括T细胞和髓细胞活化、产生促炎细胞因子和干扰素反应,与仓鼠的焦虑和抑郁行为相关,在新冠肺炎患者的组织中发现了类似的转录特征。轻度疾病感染的非人灵长类动物表现出神经炎症、神经元损伤和凋亡、脑微出血、慢性低氧血症和脑缺氧。
血液皮质醇水平较低,持续症状超过1年
最近的报告表明,与对照组相比,长新冠患者的血液皮质醇水平较低,持续症状超过1年。肾上腺产生的低皮质醇应通过垂体产生的促肾上腺皮质激素(ACTH)来补偿,但事实并非如此,这支持下丘脑-垂体-肾上腺轴功能失调。这也可能反映了潜在的神经炎症过程。此前,ME/CFS患者的皮质醇水平较低。
注:ME/CFS——肌痛性脑脊髓炎/慢性疲劳综合征,是一种多系统神经免疫性疾病。将在下一小节详细阐述。
★
ME/CFS、自主神经功能障碍和相关疾病
ME/CFS通常在病毒或细菌感染后发病。标准包括至少6个月的“疾病前从事职业、教育、社会或个人活动的能力大幅降低或受损”,伴随着无法通过休息缓解的严重疲劳,以及运动后不适、睡眠不足和认知障碍或直立不耐受(或两者兼有)。
高达75%的ME/CFS患者不能全职工作,25%的人患有重度ME/CFS,这通常意味着他们卧床,对感官输入极度敏感,并依赖他人照顾。
大约一半的长新冠患者符合ME/CFS标准
许多研究人员评论了ME/CFS与长新冠之间的相似性;据估计,大约一半的长新冠患者符合ME/CFS标准,在测量运动后不适的主要ME/CFS症状的研究中,大多数长新冠患者报告经历了运动后不舒服。
一项对长新冠患者和ME/CFS患者的直立压力的研究发现,与健康个体相比,两组患者的血流动力学、症状和认知异常相似。
• ME/CFS中一致的异常发现
包括自然杀伤细胞功能减弱、T细胞衰竭和其他T细胞异常、线粒体功能障碍以及血管和内皮异常,包括红细胞变形和血容量减少。
其他异常包括运动不耐受、耗氧量受损和无氧阈值降低,以及代谢异常,包括脂肪酸和氨基酸的使用改变。还观察到神经功能改变,包括神经炎症、脑血流减少、脑干异常和心室乳酸水平升高,以及眼睛和视力异常。反应性疱疹病毒(包括EBV、HHV-6、HHV-7和人巨细胞病毒)也与ME/CFS相关。
• 长新冠患者中观察到上述这些发现
长新冠研究发现,线粒体功能障碍包括线粒体膜电位丧失和可能的线粒体代谢失调、脂肪酸代谢改变和线粒体依赖性脂质分解代谢失调,与运动不耐受、氧化还原失衡、运动不耐受和氧提取受损的线粒体功能障碍一致。
研究还发现了内皮功能障碍、脑血流异常和代谢变化(即使是POTS症状减轻的长新冠患者)、广泛的神经炎症、疱疹病毒重新激活、红细胞变形以及其他地方讨论的许多发现。不仅在长新冠患者中,而且在ME/CFS患者中也发现了微裂纹和过度活化的血小板。
自主神经障碍,特别是POTS,通常与ME/CFS共病,也常伴有病毒性发作
POTS与G蛋白偶联的肾上腺素能受体和毒蕈碱乙酰胆碱受体自身抗体、血小板储存库缺陷、小纤维神经病变和其他神经病变有关。POTS和小纤维神经病变通常在长新冠中发现,一项研究发现67%的长新冠队列中存在POTS。
注:POTS——体位性心动过速综合征,一种随着姿势的改变而心率增加的情况,例如躺着坐起来或站着。这会导致头晕或昏厥。
肥大细胞激活综合征也通常与ME/CFS共病
与新冠前患者和对照组相比,长新冠患者肥大细胞激活综合征症状的数量和严重程度显著增加,组胺受体拮抗剂可改善大多数患者的症状。
注:肥大细胞活化综合征(MCAS)是一种由肥大细胞异常活化导致的慢性多系统性疾病.肥大细胞广泛分布于胃肠道,因此MCAS易累及胃肠道并出现相应症状。
其他可能共病的疾病
其他通常与ME/CFS共病的疾病包括结缔组织疾病,包括Ehlers–Danlos综合征和高移动性、神经矫形脊柱和颅骨疾病以及子宫内膜异位症。
长新冠中观察到与ME/CFS,自主神经障碍,肥大细胞激活综合征等疾病类似的发现,其他疾病也可能与长新冠合并。应进一步探讨病毒后条件与这些条件的重叠。
★
生 殖 系 统
长新冠中经常报告对生殖系统的影响。与无冠状病毒病史的女性以及患有新冠肺炎但不长时间冠状病毒的女性患者相比↓↓↓
患长新冠的女性更有可能发生月经改变
月经和月经前一周已被患者确定为长新冠症状复发的诱因。
在新冠肺炎患者中观察到卵巢储备下降和生殖内分泌紊乱,初步理论表明SARS-CoV-2感染会影响卵巢激素的产生和/或子宫内膜反应,因为卵巢和子宫内膜组织中ACE2受体丰富。
与那些没有月经变化的人相比,同时患有新冠和月经改变的人更容易出现疲劳、头痛、身体疼痛和气短,最常见的月经变化是月经不规律、经前症状增加和月经不频繁。
ME/CFS与多种妇科疾病存在关联
对ME/CFS的研究表明,ME/CFS与经前焦虑障碍、多囊卵巢综合征、月经周期异常、卵巢囊肿、绝经早期和子宫内膜异位症之间存在关联。妊娠、产后变化、围绝经期和月经周期波动会影响ME/CFS,并影响代谢和免疫系统变化。长新冠的研究应该关注这些关系,以更好地理解病理生理学。
病毒在阴茎组织中的持续存在,勃起功能障碍的风险也增加
这可能是由内皮功能障碍引起的。在一项研究中,与对照组相比,长新冠患者的精子计数、精液体积、活力、精子形态和精子浓度受损,并与细胞因子水平升高以及精液中胱天蛋白酶8、胱天蛋白酶9和胱天蛋白酶3的存在相关。
长新冠患者女性月经变化(月经不规律等),更容易出现疲劳,头痛等症状,男性精子质量变化,长期影响仍需进一步研究。
★
呼吸系统
呼吸系统疾病是长新冠的常见表现型,在一项研究中,新冠肺炎幸存者的发病率是普通人群的两倍。
呼吸短促和咳嗽是最常见的呼吸道症状,分别在40%和20%的长新冠患者中持续至少7个月。
几项包括长新冠患者的非住院患者的影像学研究显示了肺部异常,包括空气潴留和肺部灌注。
对感染后3-6个月的患者进行的免疫学和蛋白质组学研究表明,气道中的细胞凋亡和上皮损伤,但血液样本中没有。
进一步的免疫学特征比较了长新冠肺炎患者和新冠肺康复者,发现肺功能下降、全身炎症和SARS-CoV-2特异性T细胞之间存在相关性。



doi: 10.3389/fimmu.2021.686029
★
胃肠道系统
长新冠的胃肠道症状包括恶心、腹痛、食欲不振、胃灼热、便秘等。
持续的呼吸道和神经系统症状都与特定的肠道病原体有关。
SARS-CoV-2 RNA存在于新冠肺炎患者的粪便样本中,一项研究表明,12.7%的参与者在新冠肺炎确诊4个月后粪便中持续存在,3.8%的参与者在确诊7个月后持续存在。大多数感染7个月后出现长新冠症状和炎症性肠病的患者在肠粘膜中存在抗原持久性。
与没有长新冠或SARS-CoV-2阴性对照的患者相比,长新冠肺炎患者的血浆中发现了来自肠道和/或肺上皮的较高水平的真菌易位,可能会诱导细胞因子的产生。
长新冠患者的胃肠道症状可能与肠道菌群变化相关
与非新型冠状病毒肺炎对照组(疫情爆发前)相比,长新冠感染者中发现了较高水平的Ruminococcus gnavus和普通拟杆菌(Bacteroides vulgatus)和较低水平的普氏粪杆菌(Faecalibacterium prausnitzii),肠道失调持续至少14个月;低水平的产丁酸盐细菌与6个月时的长新冠密切相关。
将长新冠患者的肠道细菌转移到健康小鼠体内,导致小鼠认知功能丧失,肺部防御受损。
为什么长新冠患者的肠道菌群会发生变化?
胃肠道和呼吸道症状怎样将微生物群与SARS-CoV-2感染联系起来?
病毒是如何感染肠道的?
感染后的肠道菌群及宿主免疫会受到什么样的影响?
……
我们将在下一小节详细介绍。
▾◆▽◆▽◆▾
要了解病毒是如何感染,为什么会与肠道菌群相关联,我们来先从病毒的结构说起:
SARS-CoV-2是新冠肺炎的病原体。它是一种正向单链RNA病毒。它编码膜蛋白(M蛋白)、核衣壳蛋白(N蛋白)、刺突蛋白(S蛋白)和包膜结构蛋白(E蛋白)和多种非结构蛋白。
呼吸道胃肠道是人类微生物群的主要栖息地,也是SARS-CoV-2感染的目标
⇓⇓⇓
SARS-CoV-2病毒如何实现感染?
病毒利用表面的刺突蛋白和人体细胞上的ACE2(血管紧张素转化酶2)进行结合,从而实现感染。
在病毒体上,刺突蛋白(S蛋白)是包含S1和S2亚基的同源三聚体:
病毒劫持宿主细胞表面蛋白酶,如跨膜丝氨酸蛋白酶2(TMPRSS2),TMPRSS2反过来激活病毒S蛋白,切割ACE2受体,并促进病毒与宿主细胞膜的结合。
除了ACE2和TMPRSS2介导的进入外,SARS-Cov-2还可以利用宿主细胞的吞噬作用或内吞作用侵入某些免疫细胞类型,如巨噬细胞。
为什么SARS-CoV-2可感染肠道?
ACE2和TMPRSS2在呼吸道和胃肠道中强烈表达。由于后者与外部环境沟通,它们是SARS-CoV-2入侵的主要目标。
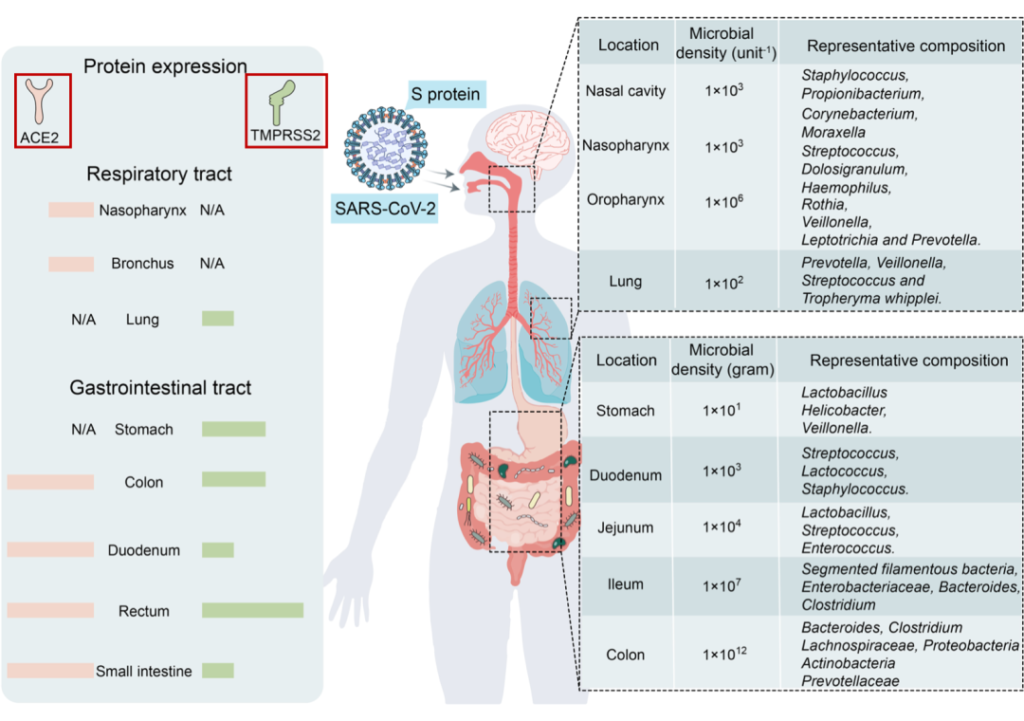
Wang B, et al., Signal Transduct Target Ther. 2022
SARS-CoV-2受体ACE2和TMPRSS2主要在呼吸道和胃肠道中表达,为微生物提供了许多合适的栖息地。
由于ACE2和TMPRSS2在胃肠道中高度表达,SARS-CoV-2也以肠道为靶点。一些研究报告称,新冠肺炎患者粪便样本中SARS-CoV-2病毒RNA呈阳性。
对一名新冠肺炎患者进行的活组织检查显示,SARS-CoV-2蛋白涂层存在于胃、十二指肠和直肠中。因此,SARS-CoV-2可感染肠道。
COVID-19的胃肠道和呼吸道症状将微生物群与SARS-CoV-2感染联系起来
⇓⇓⇓
几项临床研究报告,11-39% 的 COVID-19 患者有胃肠道症状,包括恶心、呕吐、腹泻和腹痛。
智利进行的一项研究报告称,在 7016 名 COVID-19 患者中,有 11% 表现出胃肠道症状。
在浙江的651例COVID-19患者中,8.6%表现出腹泻,4.15%表现出恶心或呕吐。
胃肠道症状与相对较高的住院风险和/或更高的疾病严重程度相关。在重症和/或危重患者中,疾病进展并引起并发症,例如急性呼吸窘迫综合征(ARDS),败血症,继发性病原体肺炎和终末期器官衰竭。
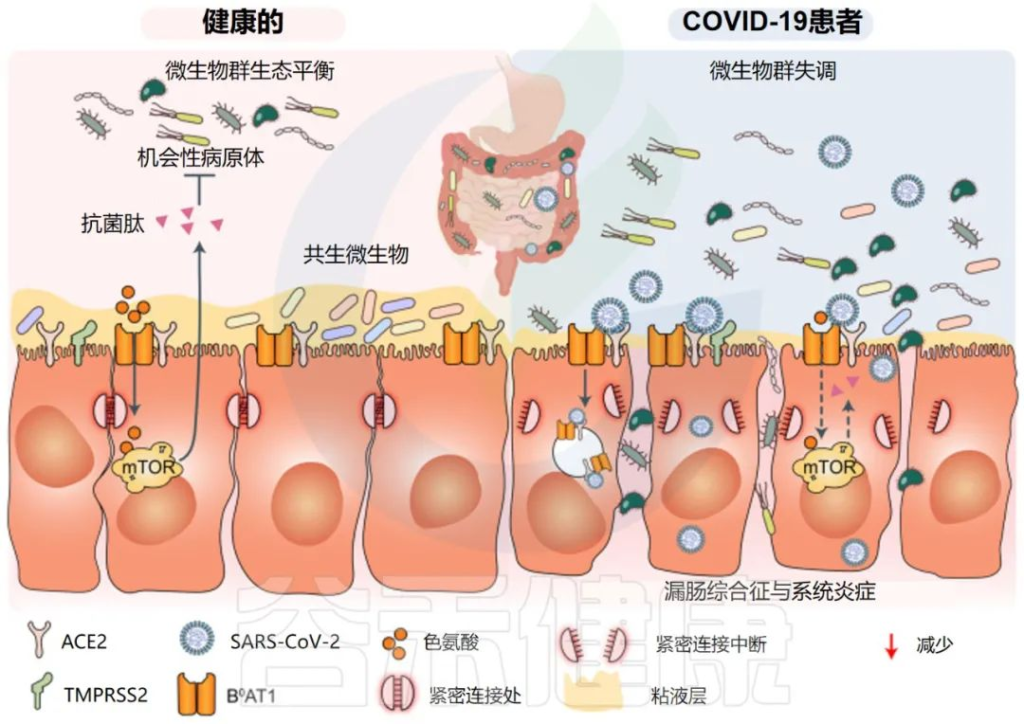
由于微生物群维持呼吸道和胃肠道稳态和健康,上述COVID-19相关症状可能将微生物群与SARS-CoV-2感染联系起来。
一些研究证实,肠道菌群有助于调节肠道免疫稳态和病原体感染。因此,肠道细菌可能对SARS-CoV-2感染的宿主免疫反应至关重要。
一些研究小组报告称,与新冠完全康复的患者相比,长新冠患者的肠道微生物群存在差异。在诊断时检测到微生物差异,但在6个月后被夸大。特别是,长新冠患者出院后,微生物群的丰富度没有恢复到正常状态。
持续的症状可能与免疫隔离组织中存在少量残余病毒有关,特别是身体中不受抗体直接保护的区域,如肠道。
一个重要的观察结果显示,病毒清除后,肠道失调持续了数月。与作为对照的健康个体相比,从新冠肺炎中恢复的患者在3个月时细菌多样性和丰富度降低,同时有益共生菌丰富度较低,机会性病原体丰富度较高。在随访6个月时,与对照组相比,COVID-19患者中双歧杆菌和瘤胃球菌的相对丰度显著降低(P < 0.001).
一项前瞻性研究追踪了香港106名新冠肺炎住院患者肠道微生物群的纵向动态,大约四分之三的患者在感染后6个月出现”长新冠”症状(通常是疲劳、记忆力差和焦虑)。
粪便样本的Shotgun宏基因组分析显示,与没有长新冠的个体和作为对照的健康个体相比,长新冠患者微生物多样性显著降低,细菌类型减少。
长新冠患者的Bacteroides vulgatus和Ruminococcus gnavus 的丰度增加,而P.prausnitzii的丰度减少。
有趣的是,与健康个体(n = 11)相比,PI-IBS患者(n = 11)的粪便样本中Bacteroides vulgatus也显示出6倍的升高;这一发现表明,Bacteroides vulgatus可能与长新冠和PI-IBS的发病机制有关。
注:因感染而导致的IBS被称为感染后肠易激综合征(PI-IBS)
doi: 10.1038/s41392-022-00986-0
此外,SARS-CoV-2感染后6个月出现呼吸道症状与Streptococcus vestibularis和Streptococcus anginosus等机会性致病物种的水平升高有关。
而疲劳和神经精神症状与医院内病原体有关,如:Clostridium innocuum 和 Actinomyces naeslundii。
在脱发患者中,产丁酸盐细菌显著减少,某些细菌,如Bifidobacterium pseudocatenulatum和F. prausnitzii,与长新冠的发展具有最大的负相关。入院时的细菌种类,包括长双歧杆菌(B.longum)和Blautia wexlerae,与6个月时长新冠的发展呈负相关,这意味着这些细菌在恢复期具有潜在的保护作用。
相比之下,在长新冠患者中富集的菌有:
Atopobium parvulum
Actinoomyces johnsonii
Actinomyces sp. S6 Spd3
这些发现表明,一个人在感染时的肠道微生物组组成能会影响其对新冠长期并发症的敏感性。尽管如此,这些变化可能代表长新冠的反应性变化,未来的研究需要包括从感染到症状发展的非住院患者的前瞻性纵向研究,以描述肠道菌群失调对长新冠症状的确切影响。
持久战——免疫细胞数量变化
针对患有轻度急性新冠肺炎的长冠肺炎患者的免疫失调进行的研究发现,T细胞改变,包括耗尽的T细胞、CD4+和CD8+效应记忆细胞数量减少以及中央记忆细胞PD1表达升高,持续至少13个月。
研究还报道了高度活化的先天免疫细胞,缺乏初始T细胞和B细胞,I型和III型干扰素(IFN-β和IFN-λ1)的表达升高,持续至少8个月。
一项综合研究将长新冠患者与未感染者和无长新冠的感染者进行了比较发现,在感染后中位数14个月,长新冠患者的非经典单核细胞、活化B细胞、双阴性B细胞以及分泌IL-4和IL -6的CD4+ T细胞数量增加,常规树突状细胞和T细胞数量减少,皮质醇水平降低。
疲劳和神经认知功能障碍可能与细胞因子变化和EBV再激活相关
细胞毒性T细胞的扩增已被发现与长冠的胃肠道表现有关。更多的研究发现细胞因子水平升高,特别是IL-1β、IL-6、TNF和IP10,最近的预印本报道了CCL11水平的持续升高,这与认知功能障碍有关。
在长新冠患者中发现了包括EBV和HHV-6在内的反应性病毒(已在ME/CFS45中发现),并导致线粒体断裂,严重影响能量代谢。最近的一份预印本报告,在长新冠患者中,EBV再激活与疲劳和神经认知功能障碍相关。
抗体生成量低,可预测长新冠
几项研究表明,在新冠肺炎急性期,无论是住院患者还是非住院患者,SARS-CoV-2抗体生成量低或无,以及其他免疫反应不足,都可以预测6至7个月后长新冠。
这些不足的免疫反应包括IgG的低基线水平、受体结合域和刺突特异性记忆B细胞的低水平、核衣壳IgG的低水平和刺突特异性IgG的低峰值。
在最近的一份预印本中,发现严重长新冠患者的CD4+T细胞和CD8+T细胞应答较低或缺失,另一项研究发现,与无长新冠的感染对照组相比,长新冠患者的CD8+T淋巴细胞表达CD107a的水平较低,核衣壳特异性干扰素-γ-产生的CD8+T细胞下降。
二次感染可能与抗体水平低相关
长新冠患者体内高水平的自身抗体与保护性新冠肺炎抗体呈负相关,这表明高水平自身抗体的患者更有可能发生突破性感染。
SARS-CoV-2病毒在肠道内反弹,可能是由于病毒持续存在,也与受体结合域IgA和IgG抗体的水平较低和产生较慢有关。
长新冠驱动因素:病毒持续存在
一些报告指出,病毒持续存在可能是长新冠的驱动因素;在生殖系统、心血管系统、大脑、肌肉、眼睛、淋巴结、阑尾、乳腺组织、肝组织、肺组织、血浆、粪便和尿液中发现了病毒蛋白和/或RNA。胃肠道活检后的多份报告表明存在病毒,提示某些患者体内存在持久性病毒库。
ACE2——病毒感染人体细胞的“钥匙”
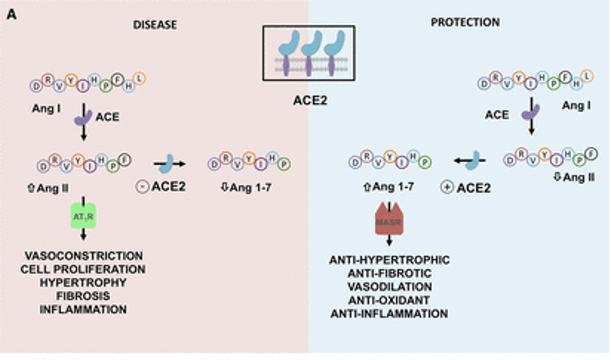
前面我们已经知道,病毒利用表面的刺突蛋白和人体细胞上的ACE2(血管紧张素转化酶2)进行结合,从而实现感染,可见,ACE2作为病毒进入的一个入口点,扮演者重要的角色。
ACE2能做什么?
ACE2在肾素-血管紧张素系统(RAS)中起主要作用,除此之外,其活性在肺部也有作用,因为它通过抑制des-Arg9-缓激肽调节缓激肽1受体信号传导,从而减少血管舒张和血管通透性。
ACE也在肠道肠细胞中表达,它可以调节微生物生态、先天免疫和饮食氨基酸稳态。
ACE2亦敌亦友
ACE2在肺部的保护作用可能是COVID-19中的一把双刃剑:
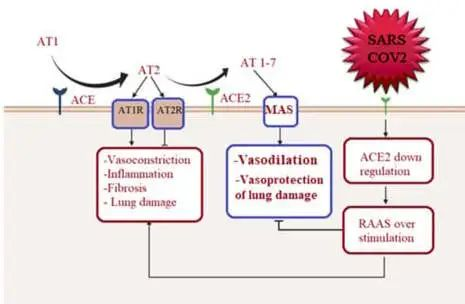
ACE2在肾素-血管紧张素系统(RAS)中的作用以及严重急性呼吸综合征SARS-CoV-2诱导的细胞表面ACE2表达下调的机制如下:
doi: 10.1161/CIRCULATIONAHA.120.047049
ACE2平衡肾素-血管紧张素系统的2个轴:
➭ 增加的ACE2促进保护性ACE2/Ang 1-7/MASR
➭ ACE2的丢失导致向以ACE/血管紧张素II(Ang II)/AngII受体1型(AT1)受体轴(AT1R)过度活性为特征的疾病状态转变。
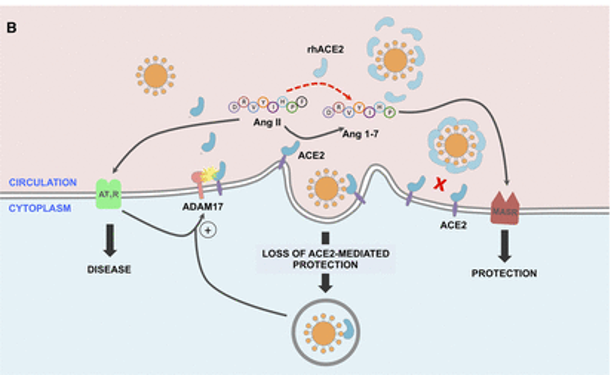
doi: 10.1161/CIRCULATIONAHA.120.047049
SARS-CoV-2的病毒刺突糖蛋白与细胞表面ACE2相互作用,并通过内吞作用一起内化,导致表面ACE2表达降低。ACE2的丢失导致Ang II的积累,Ang II通过AT1受体也上调ADAM17,导致细胞表面ACE2的进一步裂解。
肠道微生物群对肠道ACE2表达的影响?
动物研究中,无菌小鼠的肠道和肺部ACE2表达水平明显高于常规小鼠。以不同微生物群定殖的促性腺激素小鼠显示出肠道ACE2表达的可变性,这可能部分归因于微生物组编码的蛋白酶和肽酶类型的差异。
2021发表的一项研究确定了调节肠道ACE2表达的转录因子,包括已知受肠道微生物群调节的GATA4。这些数据表明,肠道微生物群可能在调节ACE2表达中发挥作用。
特定肠道菌群抑制ACE2表达
在小鼠模型中,特定的细菌种类,如多氏拟杆菌(Bacteroides dorei)和长双歧杆菌,可以抑制结肠ACE2的表达。
与小鼠结肠ACE2表达下调相关的四种拟杆菌显示出显著性差异(P < 0.05),与粪便SARS-CoV-2载量呈负相关。
“阳”之前肠道菌群已经异常,促进易感性
死于新冠肺炎的高危人群通常是那些患有糖尿病、心血管疾病和肥胖症等共病的人群,这些疾病也与微生物异常有关,其特征是细菌多样性减少。
这一发现表明,感染前的肠道微生物群可能促进宿主对SARS-CoV-2和ACE表达的易感性。
拟议的感染前、中、后肠道菌群变化模型及新冠措施如何影响人一生中的菌群多样性
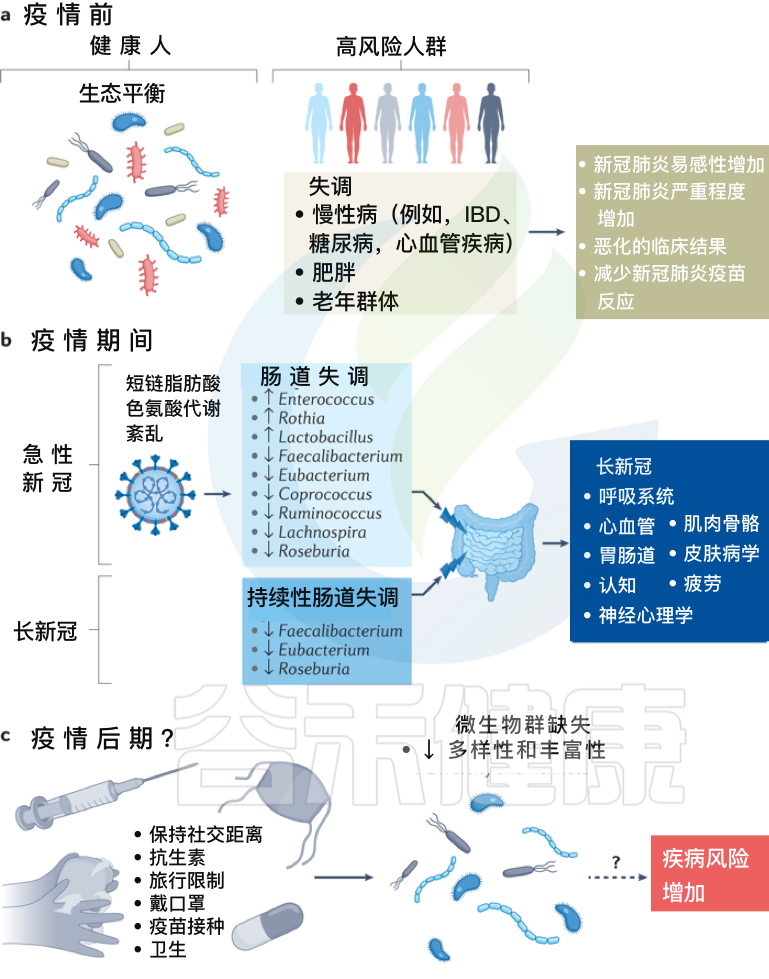
Zhang F, et al., Nat Rev Gastroenterol Hepatol. 2022 Oct
a、在新冠疫情之前,健康个体的肠道微生物群以“生态平衡”为特征,这是一个具有丰富微生物多样性的平衡肠道生态系统。
而某些个体,包括老年群体和患有炎症性肠病、糖尿病、心血管疾病和肥胖等慢性病的个体,肠道生态系统发生了变化,微生物多样性降低,肠道微生物组成失调可能导致严重急性呼吸综合征SARS-CoV-2感染的易感性增加,新冠肺炎严重程度增加,临床结局恶化和/或新冠肺炎疫苗反应降低。
b、在疫情期间,急性新冠感染与持续的肠道微生物群组成变化、短链脂肪酸生物合成受损和色氨酸代谢紊乱有关。
最初感染中出现的功能障碍也与急性后新冠肺炎综合征有关,包括慢性呼吸道症状(例如咳嗽或气短)、心血管症状(例如胸痛或心悸)、胃肠道症状(例如食欲不振或腹泻)、神经精神症状(例如焦虑或失眠)、肌肉骨骼症状(例如关节疼痛或肌肉无力)和皮肤病症状(例如皮疹或脱发)。
在急性新冠肺炎疫情后阶段,肠道微生物群持续受到破坏,其特征是产短链脂肪酸的细菌粪杆菌、真杆菌和Roseburia持续耗竭。急性期肠道菌群组成的改变也与多器官急性后新冠肺炎综合征有关。
c、除了大流行之外,现有的疫情控制做法,包括严格执行社交距离、广泛的卫生消毒措施、定期接种疫苗和限制旅行,可能会对婴儿的微生物组多样性产生负面影响,并对肠道中的早期细菌定植产生重大影响,对疾病风险产生未知后果。
需要做更多的工作来调查和确认新冠、微生物群丧失和未来疾病风险之间的潜在联系。未来导致微生物损失的感染控制措施需要与促进微生物多样性的战略相平衡,以确保子孙后代的健康益处。
迄今为止,大多数临床研究表明 SARS-CoV-2 感染与肠道菌群改变之间存在关联,但尚不清楚肠道菌群改变是感染的原因还是影响。
为此研究人员提出了两种假设,并推断了相应的机制。
SARS-CoV-2感染 ▬► 肠道菌群失调
那么研究人员假设了 SARS-CoV-2 可导致肠道菌群失调,推断了几种可能的机制:
1) SARS-CoV-2侵入肺部可导致组织损伤,可以激活以NF-κB和TNF途径上调为特征的强促炎途径,激活模式识别受体(TLR、RLR、NLR),这些受体被先天免疫细胞识别,导致各种促炎细胞因子的释放。
或者肠道感染可导致肠道结构的直接损伤和肠上皮屏障的破坏,可能损害肠道通透性,并促进肠道炎症。
激活的全身炎症和肠道炎症可能导致破坏肠道微生物群平衡,导致:
• 机会病原体(例如肠杆菌科和肠球菌)的丰度增加
• 共生菌(例如Faecaliberium、Eubacterium和Roseburia)的丰度降低
关于Faecaliberium详见:
肠道核心菌属——普拉梭菌(F. Prausnitzii),预防炎症的下一代益生菌
关于Eubacterium 详见:
肠道核心菌属——优/真杆菌属(Eubacterium),你为什么要关心它?
关于Roseburia 详见:
2) SARS-CoV-2感染可下调肠上皮细胞管腔表面ACE2和B0AT1(分子ACE2伴侣)的表达,这可能促进病原菌的生长。
3) 一项体外研究发现SARS-CoV-2可能直接感染细菌。揭示了SARS-CoV 2 影响肠道微生物群的另一种可能机制。
肠道菌群特征 ▬► 严重程度和免疫反应
动物研究提供了证据,SARS-CoV-2感染可能在驱动肠道微生物群生态变化中发挥作用。
➪
当用SARS-CoV-2攻击恒河猴和食蟹猴的非人灵长类动物模型时:
➪
仓鼠模型能够重现人类严重新冠的一些特征:
研究人员提出SARS-CoV-2感染点的特定内在“微生物组特征”,可以通过几种假设机制影响感染的严重程度和宿主免疫反应:
i) 增加的机会性病原体可能会被先天性淋巴细胞进一步识别,并强化肠道促炎反应。
ii)机会性病原体和毒素可能转移到循环系统中,导致菌血症,加剧系统炎症和疾病严重程度。
iii)有益菌逐渐减少耗尽,可能会对免疫细胞的募集产生负面影响,如激活的粘膜相关不变性T细胞(MAIT),从而影响呼吸道感染的易感性和严重性。
肠道菌群可能导致免疫反应功能失调和 COVID-19严重程度的潜在机制
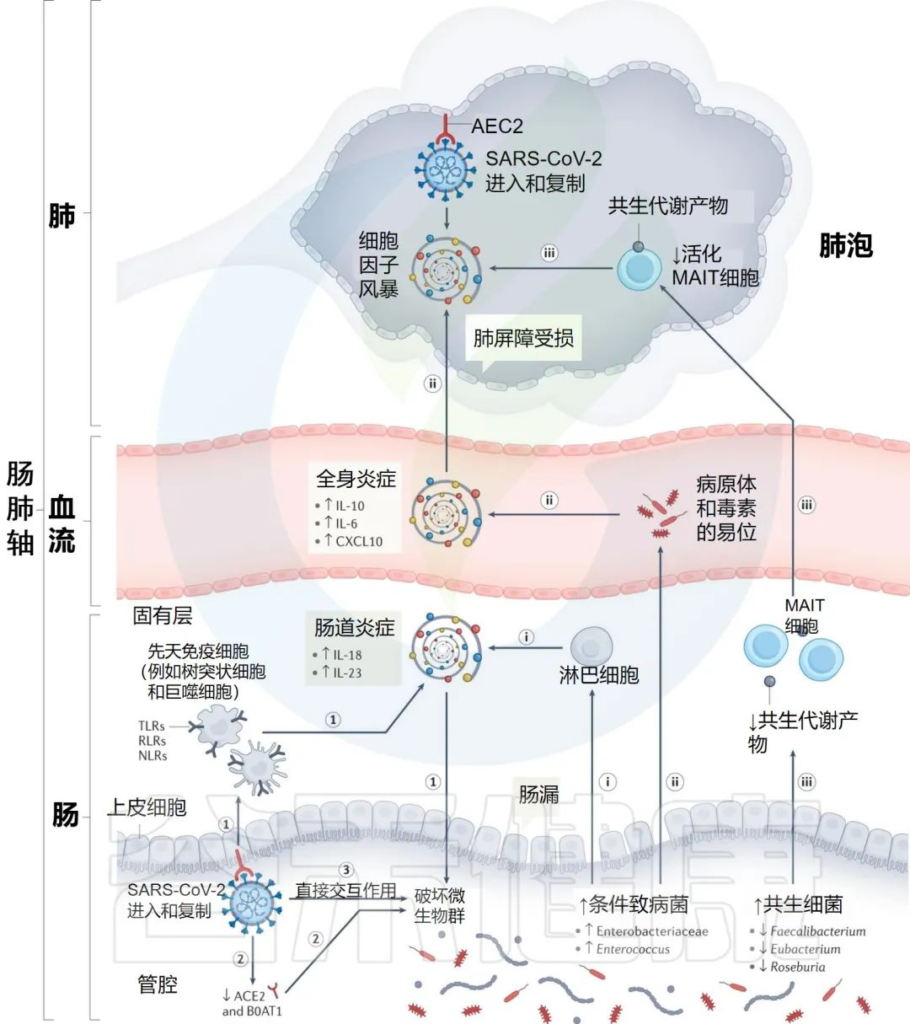
Zhang F, et al., Nat Rev Gastroenterol Hepatol. 2022 Oct
肠道菌群代谢产物
SARS-CoV-2感染与肠道微生物群的碳水化合物、脂质和氨基酸代谢改变有关。
——短链脂肪酸:合成受损
多项研究表明,COVID-19 患者的粪便样本中的短链脂肪酸生物合成受损。
在对66名新冠肺炎抗生素无效患者和70名未感染新冠的患者进行的宏基因组分析中,SARS-CoV-2感染患者的肠道微生物群合成短链脂肪酸的能力降低,这与疾病严重程度和血浆中促炎细胞因子IL-10和趋化因子CXCL10浓度升高呈负相关。
通过粪便代谢物的测量,19名与新冠相关的严重和/或危重疾病患者的粪便中短链脂肪酸(包括乙酸、丙酸、丁酸、戊酸和己酸)浓度持续下降。
短链脂肪酸可以激活免疫细胞的抗炎反应,抑制炎症信号通路,并保持肠道屏障的完整性,以防止肠道内毒素和细菌进入循环,从而减轻局部和全身炎症反应。
鉴于短链脂肪酸在调节宿主免疫反应中的重要性,新冠肺炎中短链脂肪酸生物合成不足可能与疾病发病机制和严重程度有关。然而,短链脂肪酸缺失是否是新冠感染的原因或后果尚待阐明。
——色氨酸代谢:受到干扰
一些测量新冠患者血浆代谢物的研究表明,与健康人作为对照组相比,色氨酸代谢受到干扰,与色氨酸新陈代谢有关的犬尿氨酸途径的激活增强。
色氨酸代谢通过调节调节性T细胞与TH17细胞的比率和B细胞活性与自身免疫、病毒感染和肠道健康相关。
在人类和动物研究中,犬尿氨酸途径代谢产物进入大脑的增加可能会引发疲劳、记忆力差和抑郁等症状,这是“长新冠”的常见症状。重要的是,色氨酸代谢产物是宿主-微生物群界面的关键介质。
根据人类和动物研究的证据,肠道微生物群可以直接使用色氨酸作为底物,并影响宿主色氨酸的吸收和代谢,以调节宿主的生理和免疫反应。内源性宿主色氨酸代谢产物可以深刻影响肠道微生物群的组成和功能,如阿克曼氏菌和乳杆菌。
综上所述,这些数据表明色氨酸代谢是肠道微生物群参与新冠肺炎的一种可能机制。
——胆汁酸代谢:菌群失调影响胆汁酸代谢,胆汁酸浓度升高破坏肠道屏障,引起炎症
初级胆汁酸由胆固醇、胆酸和鹅去氧胆酸通过与甘氨酸或牛磺酸结合在肝脏中合成。然后,它们被分泌到小肠,在那里它们被肠道菌群转化为次级胆汁酸,主要是脱氧胆酸(DCA)和石胆酸(LCA)。
次级胆汁酸在上皮细胞和内皮细胞以及肝细胞中充当核受体FXR、VDR和PXR的配体。它们还与TGR5相互作用。
注:TGR5是一种在肠、胰腺、淋巴组织和大脑中表达的膜结合受体。DCA和LCA都能够通过上述受体调节免疫系统。
研究人员根据疾病严重程度将新冠肺炎患者从无症状患者到处于关键阶段的患者进行分组。随着疾病严重程度的增加,来自患者的肠道菌群中厚壁菌/拟杆菌的比例逐渐受到更大的影响。
来自无症状患者的肠道菌群保留了有益菌种类,这些细菌与新冠肺炎的不良后果呈负相关:
此外,与危重症患者的微生物群不同,他们的微生物群的特征在于参与次级胆汁酸生物合成代谢途径的基因的高表达。
Harry Sokol等人观察了SARS-CoV-2感染对灵长类微生物群的影响,并发现总胆汁酸的数量随疾病严重程度而增加。值得注意的是,初级/次级胆汁酸的比率也明显较高。
这些数据表明,SARS-CoV-2感染对肠道菌群的破坏随着疾病的严重程度而加剧。随着菌群失调程度的增加,回肠的内在功能进一步改变,导致肠内转运增加,从而阻止胆汁酸的完全重吸收,从而增加其在结肠中的浓度。此外,重症新冠肺炎患者的肠道菌群功能有限,因此胆汁酸集中在这些患者的粪便中。
血清胆汁酸谱显示,急性呼吸窘迫综合征(ARDS)患者符合这些观察结果,因此可以推断,在严重的新冠肺炎中,胆汁酸浓度升高可能会破坏肠道屏障,并通过血流到达包括肺、心脏、肾和内皮在内的外周组织。它们的细胞毒性活性可能会损伤外周组织的细胞膜,导致局部和全身炎症反应,并在临床上表现出来。
——鞘脂:肠道微生物鞘脂代谢改变
据报道,新冠肺炎患者血清和粪便中鞘脂浓度降低,肠道微生物鞘脂代谢改变。鞘磷脂是生物膜的组成部分,介导信号转导和免疫激活。
拟杆菌产生的鞘脂可以增加外源鞘脂,从而增强体外或体内研究中观察到的调节性T细胞的分化,这可能抑制冠状病毒的复制。
这一观察支持肠道微生物群衍生的鞘脂可能调节宿主对SARS-CoV-2感染的防御的假设。
——蔗糖、葡萄糖:异常
与47名健康人作为对照组相比,56名新冠肺炎患者的粪便蔗糖水平升高,粪便葡萄糖水平降低。蔗糖和葡萄糖的异常水平可能与蔗糖酶-异麦芽糖酶活性受损有关。
这种变化可能与新冠肺炎常见的肠道症状有关,如腹泻、呕吐、肠胃气胀和腹痛。胀气通常是由细菌在肠道中发酵未吸收的碳水化合物引起的。
蔗糖水平的增加与放线菌和Streptococcus parasanguinis水平的增加有关,这意味着COVID-19 中的生态失调,可能会破坏肠道发酵并导致胃肠道症状。
▾◆▽◆▽◆▾
累积的证据表明,肠道菌群失调与 COVID-19 感染的严重程度和疾病恢复后的长期多系统并发症有关。
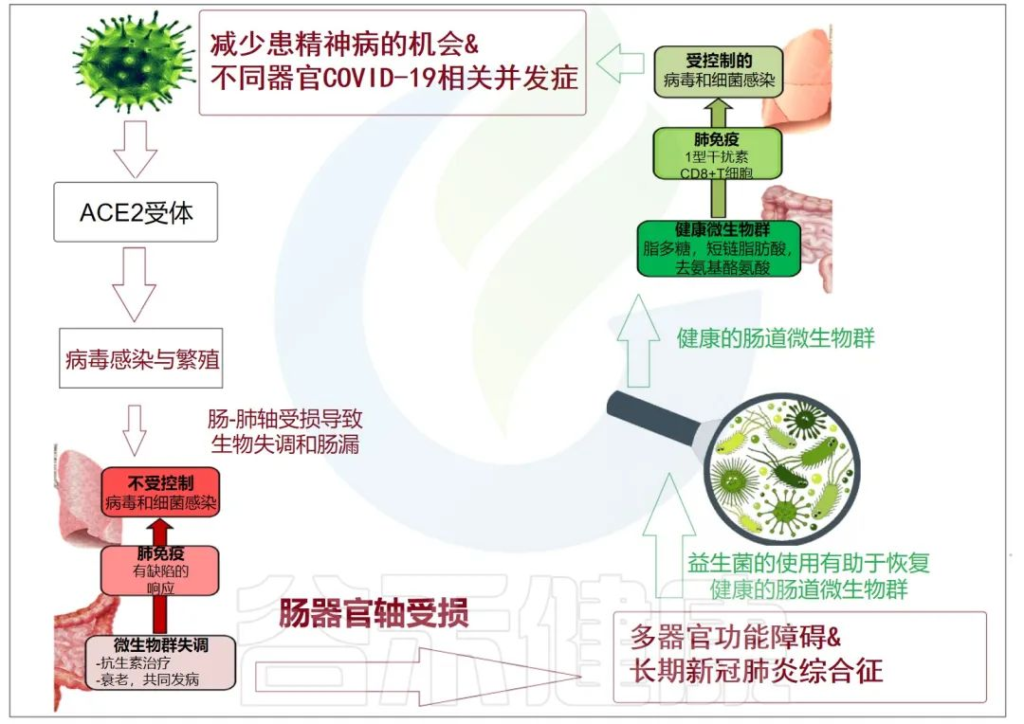
doi: 10.3390/metabo12100912
肠-肝轴是指肠道及其微生物群和肝脏之间的双向通路。这种相互作用是由门静脉建立的,通过门静脉,肠道菌群产物直接运输到肝脏,肝脏将胆汁和抗体反馈到肠道。
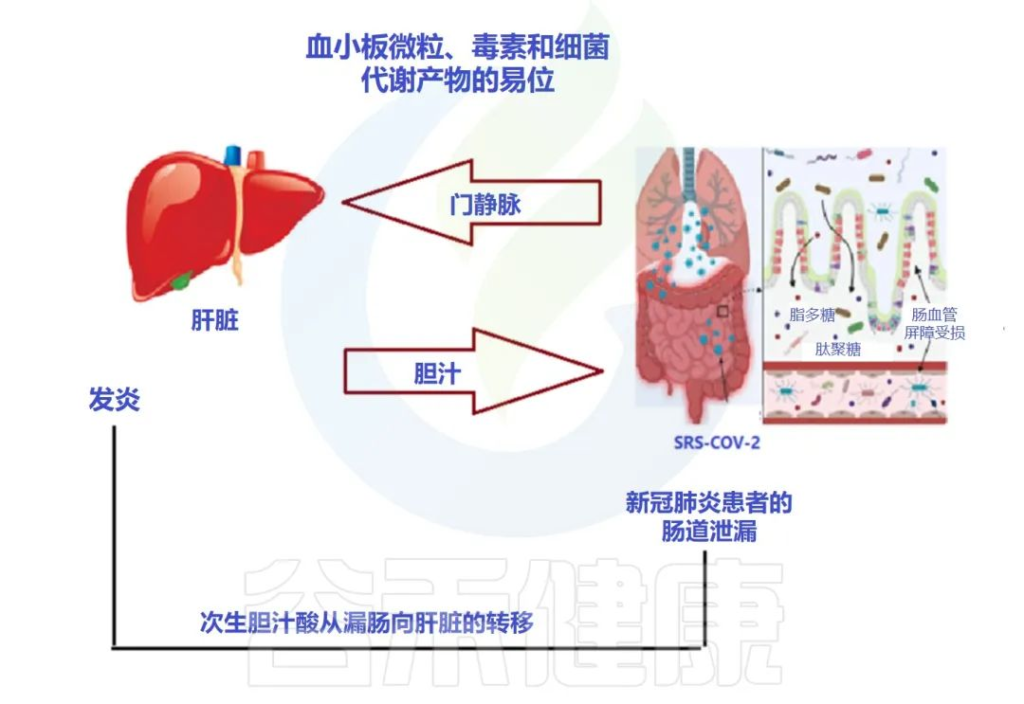
doi: 10.3390/metabo12100912
最近的两项研究表明:
代谢功能障碍相关性脂肪肝(MAFLD)是进展为严重和长期COVID-19的主要危险因素。
MAFLD征象的患者风险高
两项研究均证明,有MAFLD征象的患者发生呼吸系统疾病进展的风险高于无MAFLD的患者,年轻COVID-19患者的风险远高于老年COVID-19患者。
风险增加与病毒影响肠道通透性有关
研究人员认为,在MAFLD患者中观察到的风险增加可能与SARS CoV-2感染对肠道的影响有关,SARS CoV-2感染使肠道通透性和粘膜炎症恶化,从而加剧全身免疫功能障碍,这是严重COVID-19的特征。
当然,这个过程也可以阐明肥胖、2型糖尿病甚至炎症性肠病中 COVID-19 进展的较高风险,这与肠道微生物群改变、粘膜炎症和肠道通透性增加有关。
大量研究表明,腹泻、呕吐和腹痛等胃肠道症状在COVID-19患者中很常见,胃肠道症状的严重程度与呼吸系统疾病和肝功能障碍同时增加。
胃肠道中病毒进入受体表达的增加
已发现ACE-2 SARS CoV-2受体在肠细胞细胞上表达,因为粪便中高水平的SARS CoV-2病毒表明肠道是病毒感染和炎症的合理部位。
用于SARS-CoV-2进入的跨膜丝氨酸蛋白酶2在肠道细胞中也广泛表达。基于此,胃肠道中病毒进入受体表达的增加以及胃肠道症状的早期发作,意味着胃肠道异常可能是由病毒的直接恶化肠漏引起的,而不是对上呼吸道感染的继发性免疫致病反应的结果。
胃肠道症状的临床表现与肝功能不全的生物标志物呈正相关,支持了PAMPs向肝脏传播增加的观点。
SARS-CoV-2 感染会破坏肠道屏障,导致全身细菌脂多糖和肽聚糖升高,并有助于增强全身炎症。因此,肠漏和肠道菌群失调可能导致COVID-19重症患者发生细胞因子风暴。
基于此,已经开发的用于治疗肠漏的治疗方法,例如用于肠道粘膜保护/再生的益生菌和益生元,可以最大限度地减少进展为严重和长新冠的 MAFLD/肥胖/T2D 患者的数量。此外,在SARS CoV-2病毒感染期间,应避免使用干扰肠道微生物群的药物,例如抗生素。
充分证据表明,肠道微生物群改变和肠道细菌多样性减少,在心力衰竭合并冠状动脉疾病患者中很常见。
肠漏导致炎症,和心血管疾病相关
功能失调的肠道屏障会延缓菌群产物的被动泄漏,其中包括促炎脂多糖(LPS)进入血液,这可以通过炎症小体激活导致全身炎症。LPS结合蛋白(LBP)作为老年男性心血管风险高预测生物标志物的血浆水平显著升高,证明了这一点。
有趣的是已经发现,肠漏和炎症小体激活,与肌钙蛋白作为心肌损伤的标志物呈正相关。
肠道菌群-心轴在长新冠综合征中的作用
相当大比例的COVID-19住院患者有心脏问题。早期心血管疾病和心血管疾病危险因素(如肥胖)似乎是发生严重且长新冠并发症的关键危险因素。然而,高比例的COVID-19患者既往无心血管疾病的心脏受累。
在COVID-19患者中,心脏问题也被视为一个危及生命的实质性问题,从MI和心肌炎到伴有心脏应激的肺动脉高压。这种心脏受累的机制尚不清楚。
心肌感染可能与ACE2受体相关
ACE2在多个器官中表达,除肺、心脏和肾脏组织外,ACE2也在肠道中表达,肠细胞中的ACE2表达区可作为SARS-CoV-2进入和提示肠道感染的位点。继发于下调ACE2(SARSCoV-2受体)的抗炎和心脏保护性血管紧张素(AT)-1-7通路下调,通过表达ACE2的心脏细胞引导心肌感染,导致心脏炎症。
doi: 10.3390/metabo12100912
心脏成像的长期随访,结合肠道菌群分析,是进一步测试长冠状病毒肺炎患者肠道-心脏轴潜在影响的必要后续步骤。
肠道微生物群与肾脏疾病之间的致病性相互关联
肠道菌群参与广泛的临床表现,如慢性肾脏病(CKD),急性肾损伤(AKI)和高血压。
在肠漏的情况下,活细菌经常从肠道转移到其他肠外位置,例如肾脏。这种细菌易位可能伴有菌群失调、病原菌过度生长和宿主免疫系统低下。
在慢性肾病的情况下,肠道微生物群会产生许多毒素和尿毒症溶质,例如对甲酚硫酸盐 (PCS)、硫酸吲哚酯和三甲胺 (TMA) N-氧化物。另一方面,尿素水平升高可能导致肠道微生物群的改变(图3)。
尿毒症毒素可能导致慢性肾病患者出现疲乏、矿物质骨疾病、神经系统疾病和心血管损害。
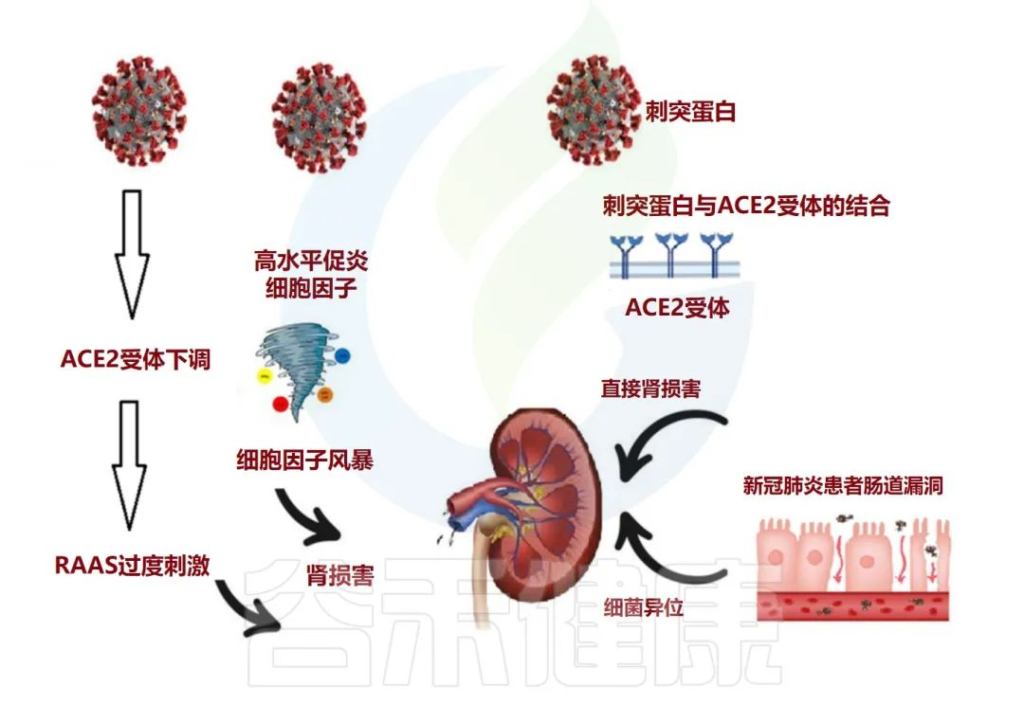
doi: 10.3390/metabo12100912
肠道生态失调在与 COVID-19 相关的长期肾脏问题中的作用
急性肾损伤 (AKI) 通常被视为 COVID-19 患者的并发症。除了先前存在的慢性肾病与COVID-19中的重症或死亡有关外,值得注意的是,解决SARS CoV-2通过AEC2受体进入肾脏,并诱导临床表现的不同途径。
人们普遍认为,该病毒可直接进入肾脏并复制,导致功能障碍,并且通过肾素-血管紧张素-醛固酮系统(RAAS)稳态的局部紊乱影响肾脏。
研究显示,既存慢性肾病患病率较高的群体可能更容易发生急性肾损伤。新出现的证据还表明,COVID-19的肾脏表现与长期严重COVID-19相关肾脏并发症的风险增加有关。
在菌群失调型COVID-19患者中,有益菌(主要是双歧杆菌和乳酸杆菌)逐渐消失,并且由于微生物群改变和病原体优势,观察到短链脂肪酸和胆汁酸水平下降。
短链脂肪酸,特别是丁酸盐是结肠细胞的重要能量来源,在上皮完整性中也起着重要作用。此外,短链脂肪酸受体GPR109A的激活与几种促炎介质的抑制有关。这可能解释了在 COVID-19 患者中出现的显著长期并发症。
作者报告说,慢性肾病患者的厌氧菌群减少,而有氧菌群增加,以肠杆菌科为主。所有这些机制都可以解释一些COVID-19患者的长期肾脏并发症。
在COVID-19住院期间监测肾功能,有助于识别后果更严重风险的患者,有助于早期和更有效的干预。
尽管 COVID-19 的主要临床表现与呼吸系统有关,但也会带来脑相关的问题,引发急性脑血管问题和颅内感染。
约35%的患者和高达85%的重症患者报告神经系统症状,包括头痛、头晕、肌痛或味觉和嗅觉丧失。
COVID-19感染可能导致神经系统疾病以及大脑结构和功能改变的机制有很多。
部分感染后的患者存在一致的记忆缺陷模式
认知问题是最常报告的症状之一,影响10%-25%的COVID-19患者,表现为SARS-CoV感染后的慢性疾病。作者发现,经历过COVID-19感染的人存在一致的记忆缺陷模式,随着自我报告的持续症状的严重程度,记忆缺陷也在增加。
此外,他们报告说,最初疾病期间的疲劳/混合症状和持续的神经系统症状可以预测认知能力。
doi: 10.3390/metabo12100912
COVID-19与神经损伤有关,主要见于神经系统症状
对COVID-19死亡患者的尸检显示,有缺血性损伤的指征和神经炎症的证据是病因机制。许多研究记录了不同脑区的功能和结构畸形,如出血性损伤和癫痫样放电。
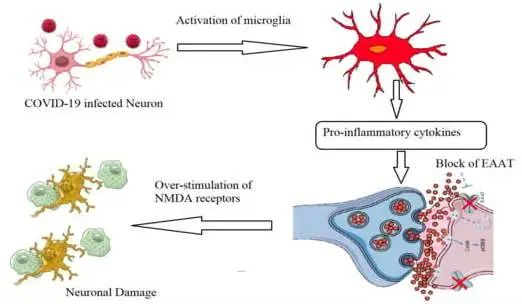
谷氨酸兴奋性毒性的作用:诱导促炎细胞因子产生
关于神经炎症,谷氨酸兴奋性毒性的作用应考虑为导致长新冠相关神经系统症状的原因。Ahmed等人(2020)报道,由于谷氨酸兴奋性毒性,SARS-CoV感染可诱导促炎细胞因子的产生和神经元变性显著增加。
简单地说,谷氨酸作为神经系统中的主要兴奋性神经递质,主要由神经元产生并在突触间隙中排出,之后它与配体依赖性AMPA受体(α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体)结合。这有助于钠离子的进入和神经冲动通过突触后神经元,导致N-甲基-D-天冬氨酸受体(NMDA)的激活,从而诱导钙离子进入。
新冠引起的神经元感染,扰乱谷氨酸稳态的控制
在新冠病毒引起的神经元感染过程中,小胶质细胞产生促炎细胞因子(TNF-α、IL-1β和IL-6),下调星形胶质细胞和突触前神经元上的谷氨酸转运蛋白1(GLT-1)。这将降低谷氨酸有效再摄取的速率,并导致谷氨酸 / GABA神经递质的不平衡和NMDA受体的过度刺激。这些事件扰乱了谷氨酸稳态的控制,突触间隙中谷氨酸的过量产生诱导神经元兴奋性毒性,钙显著进入,最终导致神经细胞变性和损失。
关于谷氨酸与大脑健康以及肠道菌群之间的关联,我们在之前的文章也有写过,详见:
新冠引起的肠道菌群失衡➔影响肠道屏障和血脑屏障通透性 ➔ 菌群代谢产物进入大脑➔脑功能障碍
肠道菌群维持肠上皮屏障、免疫稳态和防止病原体入侵。此外,肠道菌群可以通过增加紧密连接蛋白的表达来影响血脑屏障 (BBB) 的完整性和通透性。ACE2与肠道微生物稳态密切相关。
SARS-CoV-2 感染引起的肠道感染和 ACE2 表达下调可导致肠道菌群组成异常,包括乳酸杆菌和双歧杆菌等菌群水平降低。微生物失衡和肠道炎症势必会影响肠道屏障功能和血脑屏障的完整性和通透性,从而导致肠道细菌、毒素等肠道微生物代谢产物易位,通过血液循环进入大脑,最终导致脑功能障碍。
SARS-CoV-2侵入肠神经系统,出现异常,通过迷走神经影响大脑
迷走神经与肠神经系统中的神经元形成突触连接,并将肠道信息传输到NTS,在那里信息被整合并传递到大脑。
肠神经系统的异常不仅会导致胃肠功能障碍,还会通过肠-脑轴影响大脑功能。先前的研究表明,嗜神经病毒持续感染肠神经系统,并导致肠功能障碍。
SARS-CoV-2进入肠道后,通过与ACE2结合侵入肠神经系统,然后通过迷走神经进入大脑,影响中枢神经系统功能。
更重要的是,肠神经系统损伤导致肠道运动障碍、肠道血流异常和上皮屏障功能障碍,从而进一步促进肠道微生物和细菌代谢产物的毒素进入血液,加重大脑损伤。
越来越多的证据表明,肠道菌群失调也被发现是增加骨质流失的关键因素,而骨质流失又反过来促进了几种骨相关疾病的发展,例如类风湿性关节炎和骨质疏松症。
一系列报告显示,肠道菌群通过改变骨组织的质量来影响骨强度。肠道菌群调节骨骼健康的机制是通过促进调节代谢产物的生成,如吲哚衍生物、三甲基胺N-氧化物(TMAO:氧化胺)、短链脂肪酸和气体递质,如硫化氢(H2S)。
在卵巢切除术(ovx)诱导的绝经后骨质疏松小鼠模型中,H2S供体化合物GYY4137通过激活Wnt10b生成,从而增加骨形成,减少小梁骨损失,从而增强骨健康。
短链脂肪酸如乙酸、丙酸和丁酸诱导骨吸收细胞的代谢重编程,导致糖酵解增强,从而降低破骨细胞的特异性基因,如NFATc1和TRAF6,这是骨骼稳态的有效调节因子。
肠道菌群失调会加剧COVID-19疾病的严重程度和骨质流失
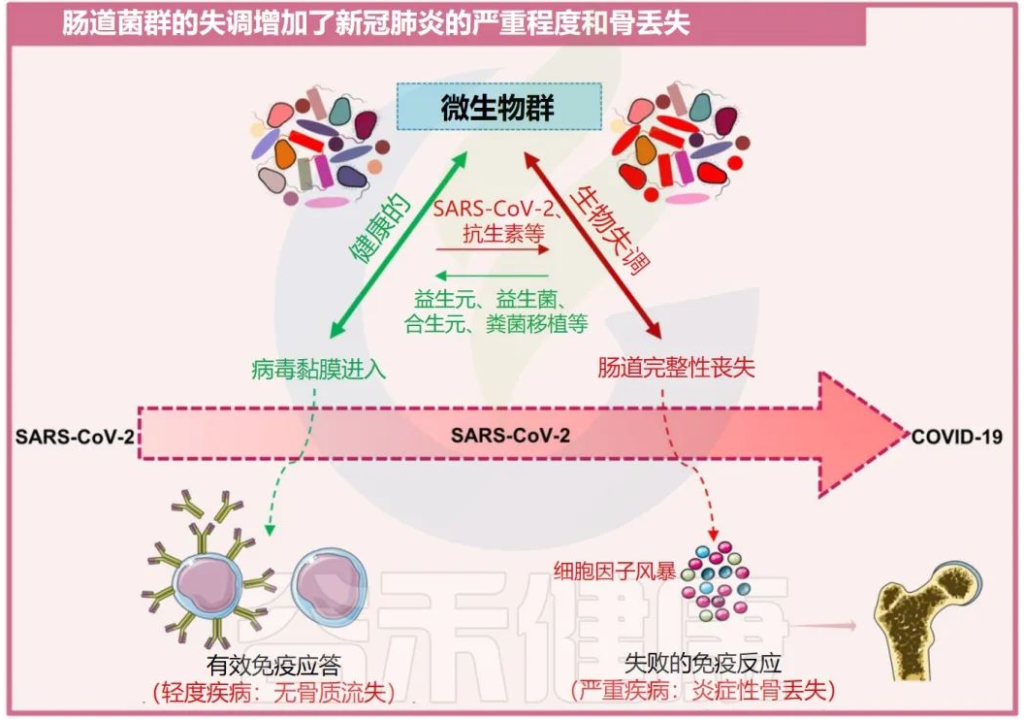
Sapra L, et al., Inflamm Res. 2022
还需要在该领域进行更多研究,并对康复的 COVID-19 感染患者进行长期随访,以确定 COVID-19 引起的骨病变的机制。
★ 肠-脾轴
SARS-CoV-2 被证明可能通过 ACE-2 受体诱导脾脏的特定嗜性。人们认为脾功能障碍与其他机制一起导致B细胞和T细胞淋巴细胞减少,这是感染后COVID-19的典型特征。
与健康对照组相比,脾切除术或脾功能障碍患者的革兰氏阴性菌产物LPSs丰度较高,因此肠道微生物群组成改变是血浆LPS升高的主要原因,可能与长期COVID-19并发症有关。
下 篇
干 预 措 施
饮 食 模 式
坚持高质量的饮食模式与较低的COVID-19感染和住院风险相关
一项Meta分析纳入5项研究,包括4023663名受试者(3149784名高质量饮食者和873881名对照组)。
高质量饮食模式对SARS-CoV-2感染和住院的有效性分别为28%(95% CI 19% ~ 36%)和62% (95% CI 25% ~ 80%)。
基于不同类型的高质量饮食和COVID-19感染风险的亚组分析显示:
一项病例对照研究调查了来自六个国家(法国、德国、意大利、西班牙、英国、美国)的2884名一线医护人员的饮食模式与新冠肺炎之间的关系。
注:这些医护人员是根据与新冠肺炎患者的大量接触情况进行筛选的。这项研究依赖于主要由男性医生组成的自我报告人群,不包括受最严重COVID-19病例影响的个体。
报告食用植物性饮食或鱼素饮食(包括益生元食品在内的)的人,中度至重度 COVID-19 的几率分别降低了 73% 和 59%。
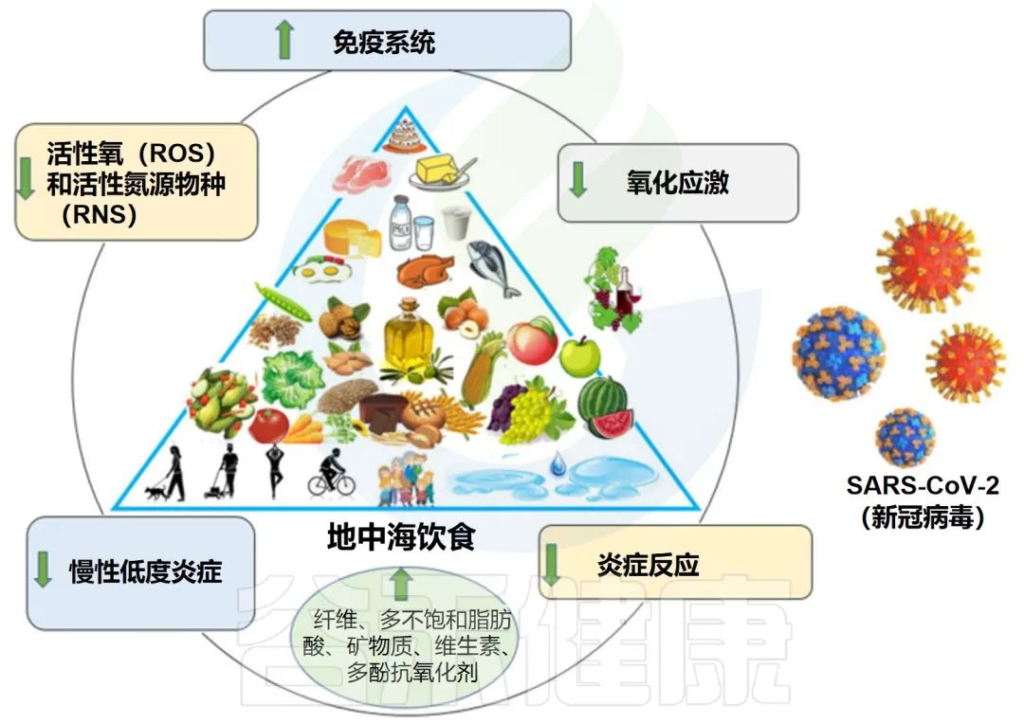
地中海饮食特征:
地中海饮食的依从性高,COVID-19严重程度和症状的可能性降低
一项横断面研究中,对 250 名年龄在 18 至 65 岁之间的 COVID-19 患者进行了检查。结果发现,对地中海饮食的依从性越高,COVID-19严重程度和症状的可能性降低,住院和康复时间缩短,炎症生物标志物也越短。
地中海饮食和COVID-19风险及相关死亡呈负相关
一项仅针对欧洲国家的生态学研究表明,地中海饮食与COVID-19相关死亡之间存在显着的负相关(r2= 0.771, p = 0.030).
一项观察性病例对照研究探讨了不同饮食模式与 COVID-19 事件和结局之间的可能关联。结果显示,病例的地中海饮食评分平均值(p=0.009)低于对照组,表明地中海饮食与COVID-19风险呈负相关。
地中海饮食有益地调节肠道微生物群和免疫系统
地中海饮食富含益生元物质,如半乳聚糖、果聚糖、纤维和菊粉。大量报道表明,这些化合物被宿主微生物使用,支持有利细菌的生长并促进有益代谢物的产生。
膳食纤维是影响复合碳水化合物对炎症影响的重要因素。研究表明,纤维消耗量的增加(约30g/d)与hs-CRP浓度的显着降低有关。膳食纤维摄入的另一个优点是对肠道微生物组组成更有利,可降低肠道和全身炎症。
多酚是地中海饮食中最丰富的次生植物化合物或植物化学物质之一,可能通过抑制NF-κB和AP-1以及激活Nrf2发挥许多抗氧化和抗炎作用。对肠道微生物群具有益生元作用。
地中海饮食:对抗冠状病毒感染的潜在策略
DOI:10.3390/medicina57121389
植物性饮食并不完全等于素食。在少量摄入动物源性食物的基础上,健康植物性饮食更倾向于新鲜蔬菜、坚果等健康的植物源性食物;而不健康的植物性饮食则更倾向于腌制蔬菜、糖等不健康的植物源性食物。
研究报告了植物性饮食是营养丰富的,包括高浓度的多酚,类胡萝卜素,纤维,维生素A,C和E,叶酸,铁,钾和镁。植物性饮食在预防高血压和心血管等疾病方面有益处。素食植物性饮食还可以增强免疫系统,减少炎症和氧化应激,并可能有助于预防慢性肾脏疾病和保持肾功能。
一项流行病学前瞻性队列研究表明,植物性食物与较低的COVID-19严重程度有关。
对来自美国和英国的五十多万参与者进行了一项研究,其中记录了 31815 例 COVID-19 病例。那些食用健康植物性饮食的人患 COVID-19 的风险降低了 10%,COVID-19 的严重程度降低了 40%.
对于年龄较大的COVID-19患者,研究人员发现,非素食饮食与COVID-19严重程度的风险较高有关。
对 2021 年 5 月至 2021 年 8 月期间在一家医疗中心被诊断为 COVID-19 的 509 名患者进行了回顾性评估。患者根据疾病严重程度分为三组。对于 ≥65 岁的患者,COVID-19 症状严重程度与坚持素食有统计学意义且呈负相关(p = 0.013).
水果、蔬菜摄入的重要性
一项针对COVID-19住院患者的横断面研究表明,水果、蔬菜和纤维的摄入量增加与 COVID-19 的严重程度、临床症状、住院和康复持续时间以及炎症标志物的浓度呈负相关。
水果和蔬菜富含纤维,是抗炎和增强免疫力的维生素,矿物质和抗氧化剂的良好来源。
为了确定饮食习惯对全球大流行期间COVID-19风险和严重程度的影响,需要进行进一步的研究。
间歇性禁食是一种潜在的补充疗法,不仅影响慢性病风险,而且有充分证据表明对传染病有影响。
SARS-CoV-2可能诱发肠道微生物群失调,导致致病菌的增殖增强,并导致有害的继发性病原体从肠道进入血流。
没有研究直接评估禁食对SARS-CoV-2感染者微生物组的影响,但由于其他观察结果,禁食塑造微生物组以支持与最佳代谢健康和低肥胖风险相关的物种,因此可以预期在预防不良变化方面产生影响。禁食期间,肠道微生物生长及其副产物(例如三甲胺N-氧化物)的产生受到抑制。
间接性禁食改变了微生物群丰度
间歇性禁食改变了各种微生物的丰度,例如脱硫弧菌科、Hydrogenoanaerobacterium、阿克曼氏菌、瘤胃球菌科等。
一项针对两种间歇性禁食(即限时进食和隔日禁食)的系统评价报道,禁食对微生物组有明显影响,例如改变厚壁菌/拟杆菌的比例,两种方案均增加了代谢保护微生物(如乳杆菌属和 Akkermansia municiphila)的丰度。
周期性禁食患者的死亡、住院和新诊断心力衰竭的风险也较低。特别是,禁食会在禁食期间急剧大幅降低葡萄糖水平,并在长期内降低基础葡萄糖水平,这在两种情况下都会使葡萄糖在感染期间减少使用,因此应该抵消SARS-CoV-2对糖酵解的刺激。
注意:禁食的安全问题包括轻微的潜在副作用,如饥饿、疲劳、头晕、便秘、头痛等,对健康的人是安全的。禁食会降低血糖,并可能导致低血糖。也可能导致脱水。2型糖尿病患者应谨慎禁食。其他安全问题可能包括身体过度紧张和营养缺乏等。
然而不可忽视的是间歇性禁食的一系列机制,使受损的人体免疫系统自我修复。在感染之前开始间歇性禁食方案可能对预防 COVID-19 等疾病的严重后果最有利。
总的来说,整体健康的饮食可能在预防SARS-CoV-2感染和降低感染严重程度方面发挥作用。需要更多的研究来证实这些发现,未来的研究应确定饮食质量与COVID-19感染风险之间关系的生物学机制。
生 活 方 式
注意,这里的运动并不是剧烈运动,特别是爬山、徒步等运动量大的活动更要注意量力而行。仍要加强健康监测。
刚阳康后需要进行一个休息阶段,尤其是老年“阳康”者,建议可循序渐进,先进行低等强度的温和运动,如适当散步、拉伸等。可以配合一些温和的养生方法,例如广播操,太极拳,八段锦等。
⇘⇘
太极拳作为中国传统运动之一,其特点是动作连贯、柔和,既能增强体质,调和身心,又能改善肺功能,避免运动时的呼吸短促。太极拳作为一种慢性病辅助治疗方法,已被临床证实能改善和缓解慢性阻塞性肺病。慢性阻塞性肺病,是一种常见的导致呼吸困难的慢性肺部疾病,被认为是COVID-19患者病情恶化的一个风险因素。
一项Meta分析共纳入11项随机对照实验,共纳入708名慢性阻塞性肺疾病患者。中期(3-6个月)或长期(12个月)太极拳运动对改善肺通气功能有效,且对改善慢性阻塞性肺疾病患者肺通气功能效果而言,干预时间在12个月以上的太极拳运动相较于中期运动更明显。
太极拳的简单易操作更容易让疫情期间老年人群体做出选择,同时也因为负荷量小更能够保证老年人的锻炼需求,从而达到免疫力提升的效果。
⇘⇘
八段锦具有运动强度小、动作幅度舒缓等特征,符合新冠肺炎患者年龄偏大、身体乏力、行动不便的需求,对于预防期新冠肺炎的发生以及预后呼吸功能的回复具有很好的预防和恢复作用。
90名亚健康状态学生为研究对象,随机分为对照组、散步组、八段锦组,30例/组。结果:八段锦组亚健康恢复率为50.00%(15/30),高于对照组的13.33%(4/30)和散步组的30.00%(9/30),差异有统计学意义(P<0.01). 八段锦练习能够显著改善亚健康状态,提高亚健康恢复率,且效果优于日常散步活动。
吸烟被认为与不良疾病预后有关,因为大量证据强调了吸烟对肺部健康的负面影响。吸烟会增加肺部感染的风险和严重程度,因为它会损害上呼吸道并降低肺部免疫功能。
吸烟已被证实是 COVID-19 负面进展的一个危险因素,尤其是在疾病严重程度和死亡方面。
Meta分析纳入 40 项研究,目前吸烟和以前吸烟都会显著增加疾病严重程度的风险(分别为:OR=1.58;95%CI:1.16–2.15,p=0.004;OR=2.48;95%CI:1.64–3.77,p<0.001),表现出中度异质性。
前吸烟者患疾病严重程度的几率是从不吸烟者的 1.58 倍。前吸烟者患疾病严重程度的几率是从不吸烟者的 2.48 倍。
同样,目前吸烟和以前吸烟也会显著增加死亡风险(分别为OR=1.35;95%CI:1.12–1.62,p=0.002;OR=2.58;95%CI:2.15–3.09,p<0.001;),并出现中度异质性。
对于死亡结果,当前和以前吸烟也分别使死亡风险显著增加 1.35 倍和 2.58 倍。
冥想练习调节注意力和情绪,向外关注特定的身体和感官刺激,向内转向精神体验和身体体验的躯体感觉。
由于冥想对包括抑郁症、焦虑症、慢性疼痛和药物滥用在内的精神病理学的特定领域产生积极影响,以及它与注意力障碍、创伤性压力、饮食失调和严重精神疾病的关联,冥想越来越多地被纳入心理健康干预措施。
与当地招募的对照组相比,长期冥想的佛教僧侣的肠道微生物群组成发生了显著变化。在属水平上,普氏菌属和拟杆菌属在冥想组中显着丰富。根据 LEfSe 分析,两个有益细菌属(巨型单胞菌属和粪杆菌属)在冥想组中显着增加。功能预测分析进一步表明,包括聚糖生物合成、新陈代谢和脂多糖生物合成在内的几种途径在冥想组中显着丰富。
这种改变的肠道微生物群组成可以降低焦虑和抑郁的风险,并改善身体的免疫功能。生化标志物概况表明冥想可以降低心身医学中心血管疾病的风险。这些结果表明,长期深度冥想可能对肠道微生物群产生有益影响,使身体保持最佳健康状态。
来自麻省理工学院、加州大学圣地亚哥分校、乔普拉综合研究图书馆和哈佛大学的研究人员探索后认为,某些冥想、瑜伽体式(姿势)和调息(呼吸)练习可能是治疗和/或预防SARS-CoV-2感染的有效辅助手段。
来自 13 个国家的 44 项研究,共 4023 人,时间范围从 20 到 4800 分钟不等。主要研究结果表明,瑜伽、冥想和调息,无论是单独使用还是组合使用,都可以通过调节抗炎和促炎生物标志物来有效提高健康和临床人群的免疫力,可能有效降低 IL-6、皮质醇和 TNF-α 患者的水平。
⇘⇘
晒太阳
时间可以选择在11点-15点,晒5-30分钟,尽量不隔着玻璃,夏季应注意避开紫外线最强的时候。
2022 年 1 月发表在《韩国家庭医学杂志》上的一项研究着眼于气候如何影响 COVID-19。研究人员发现,当涉及较高的湿度、气温和暴露在阳光下时,COVID-19 病例较少。
⇘⇘
卫生措施
阳康后依然有必要进行日常的防护措施,包括出门戴口罩,回家后勤洗手。
衣服上的病毒会传染吗?
2020 年 11 月发表的一项研究发现,虽然活病毒在皮肤上存在长达四天,但在衣服上,病毒存活不到八小时。
美国疾病控制与预防中心指出,COVID-19 主要通过三种方式传播:
受污染的衣服(或其他材料)不被视为主要传播方式。
马里兰州约翰霍普金斯健康安全中心资深学者、传染病专家认为,病毒的生长和存活在很大程度上取决于环境条件——温度和湿度。总的来说,衣服不是 SARS-CoV-2 的“主要传播媒介”。
衣服上的病毒可以被洗掉吗?
莱斯特德蒙福特大学的研究表明,使用洗涤剂在洗衣机中用热水洗衣服可以完全消除病毒。
⇘⇘
保暖
此外,天气寒冷时去户外应注意防寒保暖,天气寒冷时可适当减少外出。
以上是针对饮食、生活方式的调整,是所有调理方式的基石。当然可能还有许多没有列举出来的其他健康的生活方式,可以慢慢探索,找到一种适合自己的健康的生活方式,“病毒”就难以伤害你。
肠道菌群对免疫调节至关重要。针对肠道菌群的干预措施可能对COVID-19患者产生全身性有益作用。
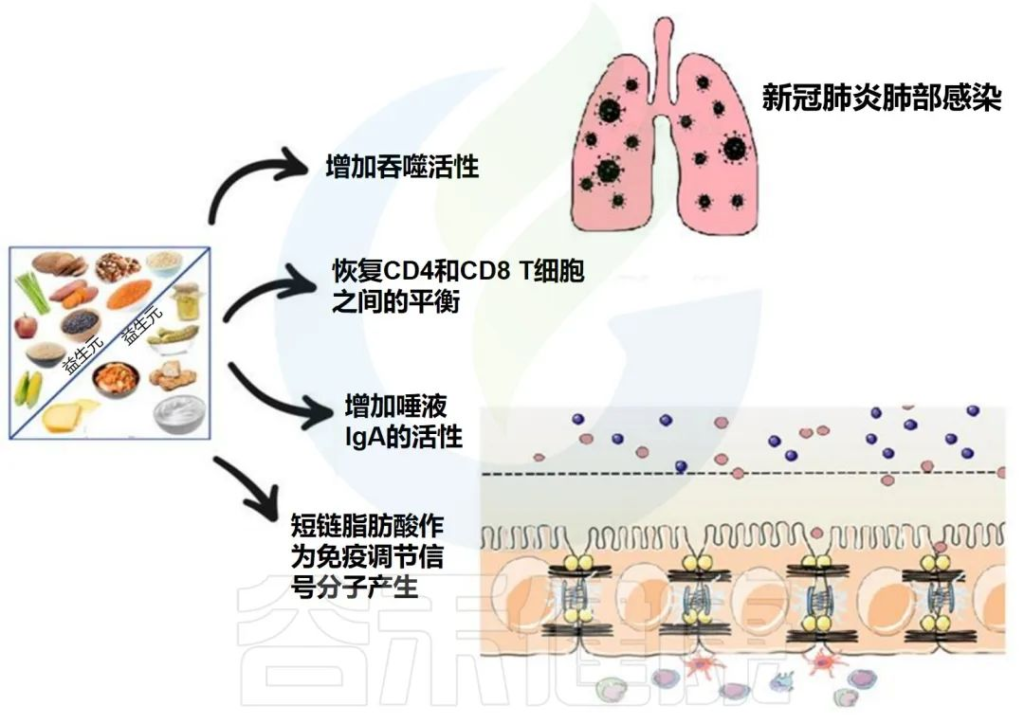
益生菌和益生元是我们饮食中会影响微生物组的两个组成部分。营养状况和饮食在COVID-19中起着至关重要的作用,主要是由于肺和肠道微生物群之间的双向相互作用。
下图描述了人体肠道和肺部之间的相互作用以及益生菌和益生元引发的潜在阳性免疫反应。
doi.org/10.3390/metabo12100912
益生菌和益生元都可以增强巨噬细胞的吞噬活性,平衡T细胞免疫以有利于更多的调节状态,增加唾液IgA的活性,并通过产生短链脂肪酸作为重要的信号分子,来发挥免疫调节细胞外和细胞内功能。
在临床研究中,使用益生元和益生菌操纵肠道微生物群是治疗肺部疾病的一种有前途的方法。
国家卫生健康委员会和国家中医药管理局指南建议重症COVID-19感染患者在常规治疗的同时食用益生菌,以改善肠道菌群平衡并预防继发细菌感染。
益生菌可能会改变肠道微生物群的组成,并在维持肠道微生物群的生态系统中发挥至关重要的作用。
益生菌通过调节肠道菌群有助于COVID-19治疗
益生菌有利于增强上皮屏障功能和改善肠道微生物多样性。此外,益生菌可对抗和阻断肠道中的有害细菌菌株或增强有益的信号通路。
尽管细菌引起的免疫反应与病毒引起的免疫反应相对不同,但许多临床研究得出结论,益生菌有助于治疗COVID-19。
注:已有超过 25 项注册临床试验旨在调查益生菌给药对 COVID-19 管理的生物学和治疗作用。
在迄今为止发表的有限试验中,关于益生菌给药的主要发现是:症状的更快改善,疲劳减少,并可能解决胃肠道问题。
已发表的研究:服用益生菌对COVID-19及其相关后遗症的影响

编辑
doi: 10.1016/j.clnesp.2022.08.023
益生菌通过调节宿主免疫系统带来益处
除了改善肠道微生物平衡外,最近的证据表明,益生菌还可以通过调节宿主免疫功能对宿主产生有益作用。一些研究报告了益生菌与ACE2相互作用的潜力,ACE2是SARS-COV2 进入宿主的受体。
例如,据报道,几种益生菌(主要是益生菌乳酸菌)在牛奶发酵过程中释放出对ACE2具有高亲和力的肽。同样,益生菌也可能通过ACE2途径改善呼吸道感染。
益生菌还可以提高肺免疫系统中自然杀伤细胞(NK细胞)、I.型干扰素、T和B淋巴细胞以及APC的水平。
注:NK细胞在针对病毒感染的早期免疫反应中起重要作用,主要是通过清除病毒感染。
先前的一项研究表明,益生菌会改变IL-10的表达并降低炎性细胞因子的表达。
益生菌还抑制其他促炎细胞因子,如TNF-a、CRP、IL-1b、IL2、IL-6、IL7、MCP1和LDH等。
益生菌通过细菌素抑制病毒与其受体的结合
除了抗炎作用、免疫调节和调节微生物组的机制外,益生菌还可以通过细菌素抑制 SARS-CoV-2 与其受体的结合。
益生菌植物乳杆菌(LPG)的独特菌株可以通过增强干扰素信号传导和抑制凋亡和炎症途径,在有效阶段和记忆阶段促进SARS-CoV-2特异性免疫反应。
由植物乳杆菌等益生菌分泌的植物素等细菌素参与抑制SARS-CoV-2的进入和复制
doi: 10.1016/j.clnesp.2022.08.023
分子对接研究预测,植物素结构可能通过靶向S蛋白或结合RNA依赖RNA聚合酶(RdRP)来阻碍病毒的进入,从而阻碍基因组的转录。
植物乳杆菌的PlnE和PlnF可以通过在解旋酶的ssRNA或ATP结合位点结合来抑制SARS CoV-2复制。
益生菌并不是治疗COVID-19的灵丹妙药,益生菌的功效和安全性在文献中存在争议,例如在接受Bacillus clausii 治疗的免疫功能低下患者中出现了菌血症等现象。因此,对一个病人有益的配方可能对另一个病人有害。还需要进行更多的研究。
此外,在推断临床试验结果时,重要的是要意识到混淆因素,如不同年龄组,不同免疫系统状态,不同季节,益生菌的成分,剂量,营养状况,正在服用的其他补充剂等。因此,也可能需要根据症状进行调整。
更有针对性的方法,包括肠道菌群检测等措施,以充分了解微生物群的作用及其在饮食和外部应激源之间的相互作用,以对抗SARS-CoV-2感染,可能会带来个性化治疗,以便益生菌能给每个需要的人带来真正益处。
益生元包括多不饱和脂肪酸、抗性淀粉、阿拉伯寡糖、低聚糖、果聚糖、低聚糖、半乳甘露聚糖、车前子、蔗糖乳糖、乳糖酸、多酚等。
大多数益生元是从植物多糖中合成或分离的,是低聚糖,例如:
含有益生元的食物,如纤维、低聚糖和多酚,可以改善细菌的生长。例如,富含菊粉的饮食刺激双歧杆菌和拟杆菌的生长;全麦谷物可以改变细菌谱,增加双歧杆菌和乳酸杆菌的相对数量。
益生元以与益生菌类似的方式调节肠道微生物群,从而抑制病原体并刺激免疫系统。同样,益生元通过直接和间接机制,对免疫系统和宿主的健康产生有益的改变。
益生元为益生菌的生长提供能量
此外,益生元选择性地刺激益生菌的有利生长并增强益生菌的活性。益生元通过增强益生菌的生长和生存能力,对 COVID-19 感染具有潜在作用。益生元也可能通过阻断ACE2对COVID-19引起的胃肠道症状产生潜在影响。
益生元明显降低了促炎IL-6的水平,这似乎是迄今为止描述的COVID-19严重预后的主要原因,并改善了抗炎IL-10的水平。
母乳低聚糖:在COVID-19中的潜在应用
母乳低聚糖在母乳中固体成分的比例排名第三。母乳低聚糖可以发挥多种功能,即抗感染(针对细菌和病毒),信号传导,抗炎/免疫调节和益生元作用。
母乳低聚糖对抗SARS-CoV-2的潜在作用模式
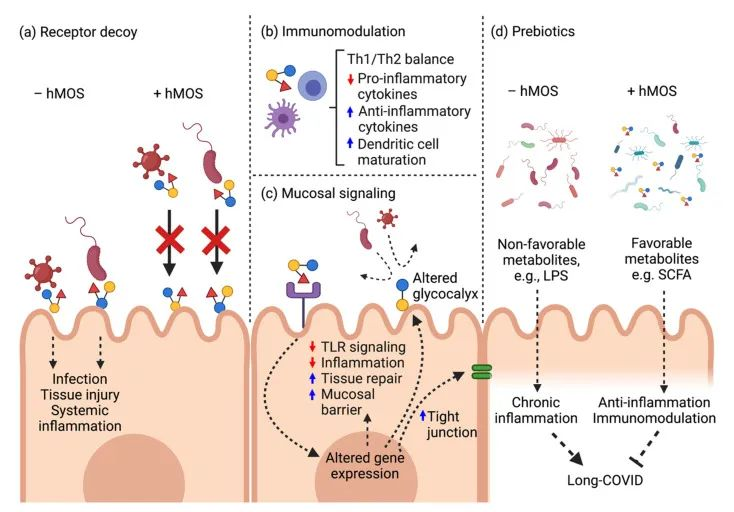
doi: 10.3390/biomedicines10020346
a) 母乳低聚糖分子结构类似于HBGA,并充当受体诱饵以阻止病毒进入。
b) 母乳低聚糖诱导局部防御和免疫调节。
c) 母乳低聚糖减弱TLR4介导的信号通路以维持粘膜稳态。
d) 母乳低聚糖在长新冠中缓解肠道菌群失调并恢复健康的肠道微生物群。
地中海饮食多酚:对 COVID-19 引起的炎症的潜在用途
研究人员发现,地中海饮食中存在的主要酚类化合物作为COVID-19预防/治疗剂的潜在用途,基于其抗氧化和抗炎作用。
目前的证据支持羟基酪醇、白藜芦醇、黄酮醇(如槲皮素)、黄烷醇(如儿茶素)和黄烷酮可能对 COVID-19 产生的潜在益处。
茶多酚:具有抗病毒固体和抗氧化特性,可能有助于降低出现严重COVID-19症状的风险
肠肺轴在SARS-CoV-2感染中起着重要作用,因此靶向肠肺轴治疗COVI-19尤为重要。茶多酚被认为是多功能生物活性分子,除了抗菌和调节肠道菌群以增强免疫功能外,还具有抗病毒作用。因此,茶多酚对COVID-19具有潜在的预防和治疗作用。
茶多酚降低 COVID-19 合并症风险

DOI:10.3389/fnut.2022.899842
茶多酚可以促进肠道中有益细菌的生长,并抑制肠道中病原微生物的生长,从而调节肠道菌群的组成。
研究人员研究了茶多酚对回肠损伤和肠道菌群紊乱的治疗和预防作用。结果表明,茶多酚可以减少炎症和氧化应激标志物,提高抗氧化酶和紧密连接蛋白的水平,有效改善肠道菌群失衡,减少对肠粘膜的损害,增强机体免疫力。
使用茶多酚预防和治疗COVID-19并发症
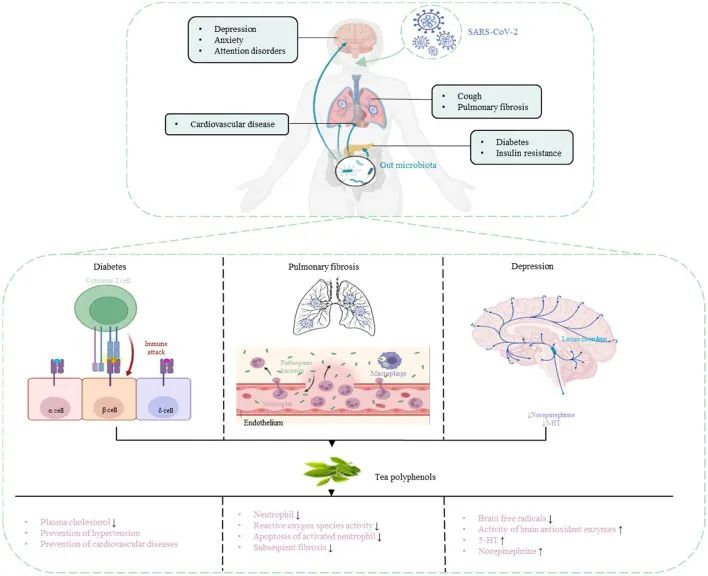
DOI:10.3389/fnut.2022.899842
在一项针对200名医护人员的随机双盲试验中,每天六粒胶囊(包括378毫克儿茶素和270毫克EGCG)持续5个月,在预防流感病毒方面优于安慰剂。
注意:在确定使用茶多酚治疗COVID-19之前,仍然需要大量实验来确认具体的药物给药(绿茶饮料、粉状绿茶提取物、儿茶素混合物、单独儿茶素)、剂量方案(不同剂量、不同治疗持续时间)和给药途径管理(饮食口服、饮料口服)。
针对COVID-19住院患者研究中,接受合生元患者临床症状缓解的比例更高
在一项针对55名COVID-19住院患者的开放标签研究中,与标准治疗组相比,接受双歧杆菌菌株和益生元的合生元配方(SIM01)4周的患者的临床症状得到缓解的比例更高(88%对63.3%),抗SARS-CoV-2的IgG抗体增加,IL-6、CCL2、M-CSF、TNF和IL-1RA等血液促炎标志物减少。
在接受SIM01的个体肠道菌群中,共生菌(如双歧杆菌、真杆菌和粪杆菌)的丰度也有所增加,而机会致病菌(如大肠杆菌和拟杆菌)的丰度则有所下降。
合生元配方 SIM01 可加速针对 SARS-CoV-2 的抗体形成,降低鼻咽病毒载量,减少促炎免疫标志物,并恢复住院 COVID-19 患者的肠道生态失调。
合生元辅助治疗两周可以有效调节针对COVID-19感染的炎症反应
一项随机安慰剂对照试验招募了 78 名确诊 COVID-19 感染的住院患者。干预组和对照组分别每天两次接受合生元或安慰剂胶囊,持续两周。
注:合生元胶囊含有多种菌株益生菌,如鼠李糖乳杆菌 、瑞士乳杆菌、干酪乳杆菌、乳双歧杆菌、嗜酸乳杆菌、短双歧杆菌、保加利亚乳杆菌、长双歧杆菌、植物乳杆菌、双歧双歧杆菌、格氏乳杆菌和嗜热链球菌,以及低聚果糖益生元剂。
结果发现:
后生元:在宿主中具有生物活性的微生物的非活细菌产物或代谢产物。也有研究人员称之为“幽灵益生菌”、“灭活益生菌”、“非活性益生菌”等。
短链脂肪酸、微生物细胞组分、功能蛋白、细胞外多糖(EPS)、细胞裂解物、替胆酸、肽聚糖衍生的多肽和毛状结构等多种代谢产物都算后生元。
后生元对抗 COVID-19 的抗病毒机制可以与以下作用相关联:
(a) 抗病毒抑制代谢物的产生
(b) 改善肠上皮衬里屏障功能
(c) 调节先天性和适应性免疫系统
(d) 对肠脑轴的影响
(e) 缓解继发性真菌感染
各种后生元对肠道屏障完整性的调节
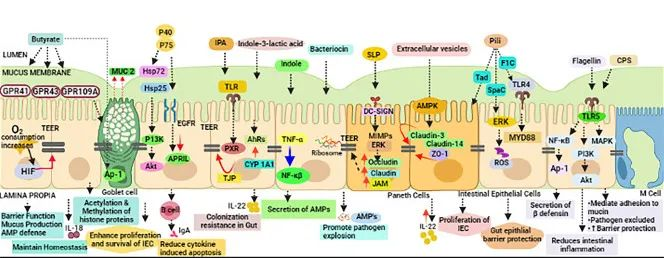
doi: 10.1007/s12602-021-09875-4
人们越来越热衷于利用益生菌,尽管由于宿主微生物组和侵入性病毒之间的固有复杂性和串扰,其详细的作用机制仍在研究中。后生元在降低致命的SARS-CoV-2感染严重程度方面可能比益生菌具有显着优势。
然而从治疗和调节的角度来看,还有一些关键问题需要回答,例如:最适合提取它们的方法,它们在宿主内的有效传递方法,它们的稳定性和保质期,后生元商业化的生物处理策略等。
未来,根据病原体变异的流行情况,将后生元用作个性化疗法或许是一种可行的选择。基于后生元在增强或调节个体免疫力方面的作用,也可以探索降低对其他病毒感染的易感性或严重程度。
在粪菌移植中,来自健康供体的体外培养或粪便物质纯化的粪便或复杂微生物群落被接种到患者的肠道中。粪菌移植已证明对结肠炎、糖尿病和复发性艰难梭菌感染有效。
11名COVID-19患者中有5名报告胃肠道症状有所改善,血液免疫标志物和肠道微生物群组成有良好的改善,双歧杆菌和粪杆菌的丰度增加。
在两名合并复发性艰难梭菌感染的 COVID-19 患者中,FMT 治疗似乎是安全的,并且 COVID-19 相关呼吸道症状在 FMT 后 1 个月内迅速消退。
一项注册临床试验(ClinicalTrials.gov 标识符号 NCT04824222),试图验证粪菌移植作为免疫调节风险降低剂,在与细胞因子风暴和炎症升级相关的COVID-19疾病进展中的功效。
对照组接受标准的药物治疗,而实验组也接受口服FMT,剂量为30–50,双层,耐胃酸,肠溶性冷冻60-g胶囊。
➳ 主要的结果指标是:
给药后第30天安全试验组的不良事件发生率。
➳ 另一个结果指标是:
研究组和对照组中需要升级无创氧疗方式的患者百分比
如增加FiO2、给予高流量鼻插管氧治疗(HFNOT)、持续气道正压(CPAP)或有创通气、呼吸机和/或ICU住院治疗,对应于新冠肺炎表现状态量表中5-7级疾病恶化。
这个试验仍在进行中。然而,考虑到肠道微生物群在免疫调节中的重要作用,研究人员认为FMT是抑制新冠肺炎诱导的细胞因子风暴和炎症的一种可能的治疗选择。
益生菌、益生元和FMT旨在增加有益细菌的丰度,而抗生素则用于抑制有害微生物群的丰度。世卫组织建议,患有可能患有严重急性呼吸道感染(SARI)和败血症的COVID-19患者可能需要广谱抗生素,这可以覆盖尽可能多的致病菌。
密歇根州38家医院新冠肺炎患者的随机数据显示,至少50%的患者接受了早期经验性抗生素治疗。抗生素在危重症患者中的应用也占很大比例,并在抑制患者继发感染方面发挥关键作用。
然而,抗生素治疗可以不加区别地消除正常的共生微生物群,同时消除病原体,导致肠道菌群失调。
与健康对照组相比,新冠肺炎患者的抗生素治疗对肠道微生物群有显著影响,有益于宿主免疫的共生菌较少,包括粪杆菌、粪球菌和直肠真杆菌,而铜绿假单胞菌和分枝杆菌,这已被证实与新冠肺炎的严重程度增加呈正相关。
特别是在疫情期间,抗生素的使用面临巨大挑战。
在早期大流行中,抗生素被普遍使用。一项荟萃分析估计,全球四分之三的新冠肺炎患者曾服用过抗生素,这一比例明显高于新冠肺炎中细菌合并感染的估计发生率,后者仅为8.6%。
45名中度新冠肺炎患者的临床结果显示,服用和未服用抗生素的患者之间没有差异,这表明抗生素对改善新冠肺炎的临床结局并无益处。
据报道,在住院期间接受抗生素治疗的新冠肺炎患者出现明显的肠道失调,抗生素诱导的肠道失调损害了人类对季节性流感疫苗的免疫反应。
一项对200名新冠肺炎患者的纵向研究表明,新冠肺炎爆发前一年抗生素摄入量的减少与疾病严重程度的减轻和SARS-CoV-2的快速清除有关。
因此,抗菌药物管理对于预防抗生素诱导的失调、严重的新冠肺炎和新冠肺炎患者的抗菌药物耐药性风险至关重要。抗菌治疗必须有明确严格的适应症,应谨慎选择病原学检测后敏感的抗生素,并根据患者的具体情况适当调整用药时间。
➤ 多 酚
多酚是源自植物的酚类化合物,富含抗氧化和抗炎特性。膳食多酚大致分为四类,包括酚酸、木脂素、二苯乙烯和类黄酮。
有大量证据强调了多酚对肠道的益生元作用。这可能有助于纠正据报道由SARS-CoV-2感染引发的肠道微生物群的生态失调。
——槲皮素
槲皮素被认为有助于预防严重的COVID-19症状,因为它具有已知的抗炎、抗氧化和免疫调节特性。分子对接和体外研究通过多种机制揭示了强大的抗病毒潜力,包括阻止与ACE2受体的附着和阻断病毒复制。
槲皮素的临床试验结果很有希望。在一项随机、开放标签试验中,研究人员测试了用向日葵磷脂配制的槲皮素提高吸收的有效性。
与标准护理相比,槲皮素组的住院风险降低了68.2%,住院时间缩短了76.8%,对氧气治疗的需求减少了93.3%。此外,槲皮素组没有患者入住ICU或死亡,对照组分别有10.5%和3.9%的患者入住ICU或死亡。同一组的进一步随访研究证实,槲皮素显著改善了病毒清除率,缩短了症状时间,改善了炎症标志物,与对照组的19%相比,槲皮素组的57%在7天后完全康复。
最近的一项小型研究证实了这些结果。槲皮素组的炎症标志物和住院时间显著减少。此外,槲皮素组入住ICU的患者数量减少,ICU天数减少,无死亡,而对照组有3例死亡。在所有情况下,这些结果都接近显著性,但很可能由于样本量小而没有统计学意义。
虽然缺乏大型临床试验,存在局限性,上述研究仍显示了症状减轻、进展至严重疾病和死亡率方面的益处。鉴于槲皮素在短期服用时具有良好的安全性,以及其广泛的可用性和众多其他健康益处,可以与患者讨论使用。
槲皮素可以从几种水果和蔬菜中获得,如浆果、芦笋,红叶生菜、洋葱、苹果、莳萝、萝卜、刺山柑、香菜、银杏叶、葡萄、葱、西红柿、西兰花、青椒、豌豆等。也可以从含有槲皮素或其一些合成衍生物的补充片剂中补充。
——姜黄素
姜黄素是姜黄中存在的生物活性化合物,具有多机制作用模式。
它可以抑制病毒进入细胞,包裹病毒和病毒蛋白酶。它调节各种信号通路。
姜黄素可能在治疗 COVID-19 感染中发挥有益作用,因为它能够调节负责 SARS-CoV-2 在许多器官(如肾脏、肝脏和心血管系统)中的附着和内化的各种靶点。它还可以抑制COVID-19感染时触发的纤维化相关通路和肺水肿。
研究表明,口服姜黄素可降低死亡率、恢复时间、对氧气的需求、机械通气、住院时间以及存在几种炎症标志物。
在三项安慰剂对照试验中,每天服用160 mg纳米姜黄素或1050 mg姜黄素和胡椒碱的患者的死亡率分别降低了80%、50%和82%。最近的一项荟萃分析表明,姜黄素治疗患者的死亡率总体降低了77%.
六项临床试验中,有五项也发现轻度、中度和重度新冠肺炎患者的症状持续时间显著缩短。
虽然还需要进行更大规模的试验,但现有数据表明姜黄素对降低新冠肺炎的严重程度非常有效。
鉴于几乎所有先前的临床试验都表明姜黄素补充剂是安全且耐受性良好的,即使剂量高达8000 mg/d,建议在症状出现时或首次阳性试验时每天服用1000 mg 较为合适。
虽然补充剂是达到试验中同等剂量的最佳方法,但用于烹饪的姜黄粉含有约3%的姜黄素。因此,一茶匙(5克)的姜黄粉含有大约150毫克。虽然姜黄素的低生物利用度一直存在问题,但胡椒碱(一种存在于黑胡椒中的化合物)已被证明可将生物利用度提高20倍,因此想要提高饮食中姜黄素摄入量的做法,可以在加姜黄粉的同时加入黑胡椒。
——白藜芦醇
白藜芦醇是一种多酚,白藜芦醇可能与SARS-CoV-2相互作用,至少部分是通过触发Nrf2,而Nrf2作为宿主防御机制对某些呼吸道病毒疾病(如呼吸道合胞病毒病)具有重要的调节作用。
白藜芦醇可以降低氧化应激,通过谷胱甘肽过氧化物酶的调节。这增强了谷胱甘肽的产生,并抵消了氧化应激介导的组织损伤。
白藜芦醇通过抗氧化和抗炎机制对SARS-CoV-2诱导的损害的主要潜在保护作用
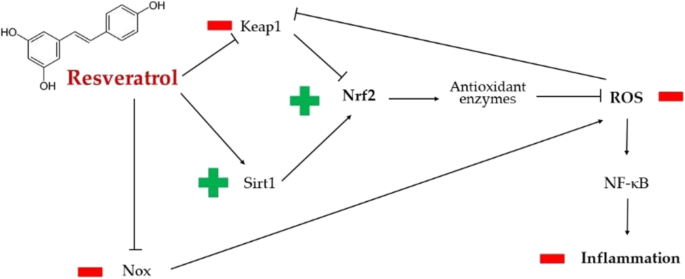
doi: 10.1007/s13105-022-00926-0.
白藜芦醇通过eIF2α和NADPH氧化酶途径降低氧化应激水平。
高血压和动脉粥样硬化是新冠病毒感染的两个危险因素。在这方面,白藜芦醇调节SIRT1和Nrf2通路的能力,以及ROS的产生,导致更大的一氧化氮(NO)生物利用度。因此,白藜芦醇介导的NO增加很可能是多酚的血管扩张剂和抗血小板作用的基础,这反过来又可以减轻许多患者的COVID-19严重程度。
白藜芦醇在内皮细胞中积累,由于其潜在的抗血栓作用,能够保护内皮屏障。
由于其抗氧化活性,白藜芦醇可以减轻与ROS介导的氧化应激相关的炎症反应。较低的ROS水平导致NF-κB和细胞外信号调节激酶/丝裂原活化蛋白激酶(ERK/MAPK)的抑制。
对随机临床试验的各种荟萃分析强调了白藜芦醇的抗炎作用,这可能有助于缓解新冠肺炎特有的所谓“炎症形式”。
项研究利用网络药理学方法和生物信息学基因分析探讨了白藜芦醇对新冠肺炎患者作用的潜在机制。该研究表明,白藜芦醇可以通过抑制IL-17、TNF和NF-κB信号通路来减轻SARS-CoV-2产生的过度炎症。
白藜芦醇,自然存在于不同的食物来源中,浓度较低,典型的地中海饮食,包括葡萄、红酒、浆果和坚果。
关于白藜芦醇与肠道菌群的关联详见:
——羟基酪醇
羟基酪醇,存在于橄榄中,在橄榄成熟过程中由于橄榄苦苷水解而增加。它是从橄榄叶和果实中提取出来的,在特级初榨橄榄油中含量尤其丰富。
注:橄榄油是地中海饮食中最具特色的食物之一。
羟基酪醇的抗病毒能力是众所周知的,部分原因是其抗炎作用。不同的研究表明,这种酚类化合物抑制MMP-9和COX-2酶的活性。
MMP-9循环水平升高被认为是COVID-19患者呼吸衰竭的早期指标。在急性肺损伤(例如COVID-19中发生的肺损伤)中,MMP-9从中性粒细胞中释放出来,从而产生炎症和肺泡毛细血管屏障的退化,进而促进炎症细胞的迁移,导致肺组织的进一步破坏。
羟基酪醇诱导的MMP-9抑制可以通过该途径减轻和/或部分预防COVID-19产生的肺损伤。
羟基酪醇通过抗炎机制对 SARS-CoV-2 诱导的肺泡组织损伤的潜在保护作用
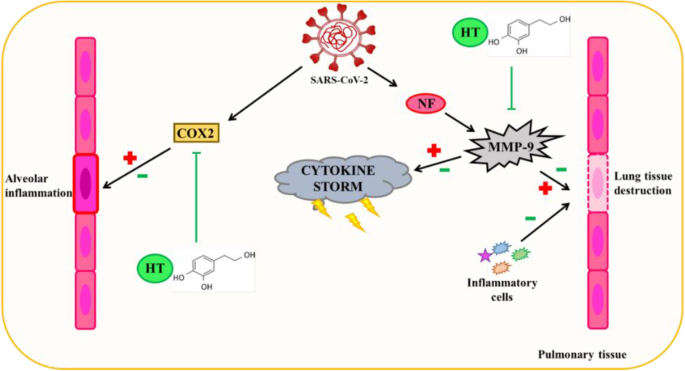
doi: 10.1007/s13105-022-00926-0.
➤ 类胡萝卜素
类胡萝卜素是四萜类。它们包括由植物、藻类和细菌产生的橙色、红色和黄色有机色素。一些常见的类胡萝卜素是α-和β-胡萝卜素、叶黄素、玉米黄质和番茄红素。类胡萝卜素以其抗氧化特性和抑制ROS而闻名。
因此,低水平的类胡萝卜素与氧化应激有关。叶黄素、胡萝卜素和玉米黄质的抗病毒作用已被报道。
一些类胡萝卜素可作为维生素A的前体,与免疫调节功能直接相关。据报道,类胡萝卜素可缓解COVID-19感染期间导致肺损伤的炎症反应。
当通过饮食提供必要的微量营养素(如维生素 A、B 和 D)以及硒、锌和铜等必要质量和数量时,可以达到最佳健康状况。
随着新冠的持续,很明显,最容易感染的人是那些失去生理营养状况和免疫系统平衡的人。这种不平衡使SARS-CoV-2病毒得以发展并导致疾病的不同临床形式(无症状,轻度,中度和重度)。
SARS-CoV-2感染中个体反应的微量营养素调节
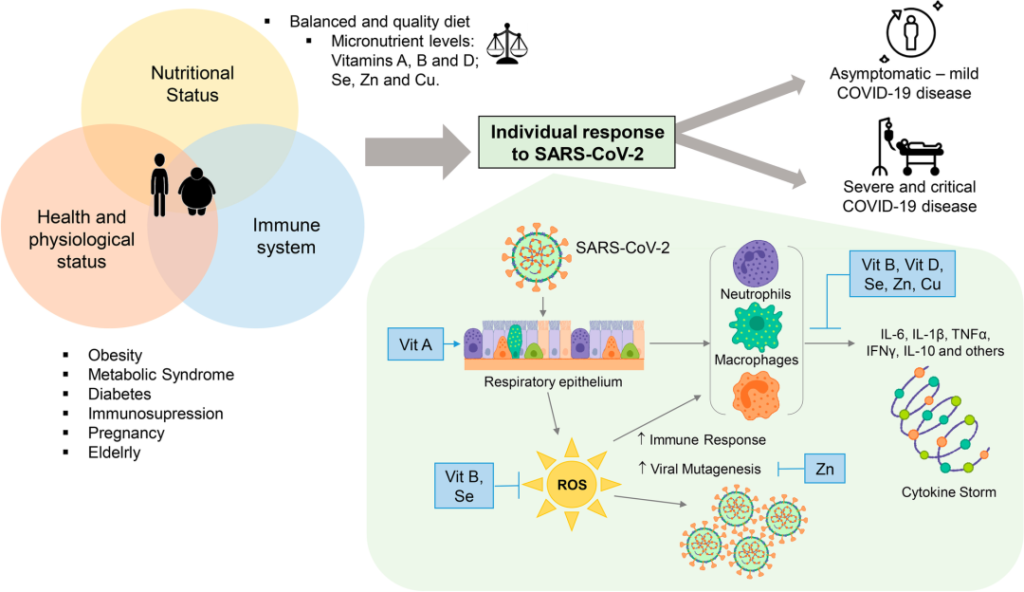
DOI:10.1007/s12011-022-03290-8
维生素A缺乏导致第一道防御屏障缺乏先天免疫的重要成分,从而增强了病毒的反应。
维生素B和硒在感染期间抵抗氧化应激的免疫和抗氧化反应中发挥作用。维生素B、D、硒、锌和铜以非常重要的方式参与,促进抑制促炎细胞因子的合成,促炎细胞素引发细胞因子风暴,从而通过抑制Th1反应和促进Th2细胞产生细胞因子来调节适应性免疫应答。
锌本身参与细胞信号转导,因此参与细胞和病毒基因表达模式,从而避免病毒突变。
➤ 维 生 素 A
维生素A,一种脂溶性维生素。是身体许多部位正常生长和功能所必需的,包括眼睛、皮肤和免疫系统。
维生素A或视黄酸在全身水平上作为一种激素,并通过核视黄酸受体(RAR和RXR)信号调节免疫系统中的I型干扰素(IFN)合成。此外,它还通过维甲酸诱导基因I(RIG-I)信号通路负责对病毒感染的永久性免疫系统反应。另外调节NF-kB的活化。
德国进行的一项多中心、观察性、横断面、前瞻性分析调查中,研究人员发现维生素A缺乏与ARDS的发展和死亡率之间存在很强的关联(OR 5.21 [1.06–25.5],p = 0.042)。
注:ARDS,呼吸窘迫综合征
该研究纳入了40名SARS-CoV-2感染住院患者,并被诊断为中度、重度和危重度ARDS。对照组由47例症状较轻且不需要住院治疗的康复患者组成,结果显示,随着ARDS严重程度的增加,维生素A缺乏症更高,康复组的维生素A缺乏症明显降低(p <0.01至p <0.001)。
最近一项针对155名老年患者(18-95岁)的研究显示,36.5%的患者缺乏维生素A (< 0.343 mg/L); 作者认为,COVID-19疾病患者耗尽了血清维生素A储存。
免疫反应机制从先天转变为适应性,阻止了维甲酸的使用,这表明炎症平衡了COVID-19严重程度和维生素A水平之间的关系。
维生素A缺乏会降低影响上皮机械屏障功能的先天免疫应答,并增强呼吸道和肠道感染。这些上皮中的粘蛋白产生受视黄酸调节;因此,中等剂量维生素A补充剂通过调节上皮生长因子和相关细胞因子的基因表达来改善屏障完整性。
食物来源:肝脏,鱼类,鸡蛋,乳制品等,还包括橙黄色蔬菜、绿叶蔬菜、西红柿、水果等。
注意:维生素 A 补充剂可能与某些类型的避孕、抗癌药物、痤疮治疗和血液稀释剂相互作用,服用任何这些类型药物的人在服用维生素A补充剂之前应咨询医生。
➤ B 族 维 生 素
B族维生素是多种细胞反应的辅助因子,介导氨基酸的合成。包括维生素 B1、B2、B3、B5、B6、B7、B9、B12等,对免疫系统对抗感染反应至关重要。
B族维生素在激活先天性和适应性免疫应答中起关键作用,下调促炎细胞因子和炎症的产生,并显著改善呼吸功能。B族维生素还可以减少胃肠道问题,预防高凝状态,并缩短COVID-19患者的住院时间。
一项研究的311名法国受试者中,与7455名健康受试者相比,SARS-CoV-2阳性患者的维生素B9摄入量较低(OR = 0.84 (0.72,0.98),p = 0.02)。
在以色列,162例诊断为重症COVID-19的患者叶酸水平低于中轻度病例(分别为9.6ng/mL vs 12.9ng/mL vs 18.2ng/mL,p=0.005),其中12%为免疫抑制,9%需要无创氧合,15%为插管。
一组来自新加坡的患者纳入了43例50岁以上的COVID-19患者,发现联合补充维生素B12、维生素D和镁入院后与疾病严重程度的降低有关。在补充的患者中,与未接受这些微量营养素补充剂的患者相比,对氧疗的需求减少(17.6 vs 61.5%,p = 0.006)和重症监护需求减少有关。
——维生素B12
维生素B12对DNA合成和调节至关重要。转钴胺素发挥其抗氧化机制,促进还原性谷胱甘肽在细胞质中的生物利用度,从而促进氧化性谷胱甘肽的合成。钴胺素由肠道微生物群产生,有助于调节肠-脑轴,防止肠道生态失调,并有利于产生适当比例的微生物代谢物。这些过程对DNA合成、细胞稳态、造血和免疫至关重要。
在生理条件下,VB12调节抗炎细胞因子和生长因子的表达,减轻全身炎症。此外,通过促进NK细胞和CD8 + T细胞的增加,提高免疫反应,调节抗病毒反应。
当维生素B12缺乏时,感染的风险更大,严重程度也会增加。
一些因素如年龄和某些药物的使用与维生素B12缺乏的风险较高有关。在老年人中,内因子(intrinsic factor)的产生减少导致B12吸收不良、营养不良或尿和肠损失增加。
二甲双胍作为2型糖尿病的治疗也与VB12吸收不良引发的缺乏有关。因此,这可能会使这些患者更容易受到感染。
➤ 维 生 素 C
维生素C在免疫功能和伤口愈合中很重要,并具有抗氧化,抗病毒和抗炎的特性。维生素C已被证明可以增加中性粒细胞向感染部位的迁移,引发活性氧(ROS)和吞噬作用的产生。
据报道,饮食中的抗坏血酸可以降低c反应蛋白的浓度。维生素C被认为通过增加干扰素蛋白的产生而表现出抗病毒活性。
维生素C被认为是严重疾病的潜在治疗方法,也是降低感染风险的预防措施,因为它能够调节免疫细胞活性并减少炎症。
小规模研究显示,在对ICU入院后24小时内循环维生素C水平的分析中发现,高达82%的COVID-19患者缺乏维生素C,许多患者的维生素C水平仅为0.1mg/dL(正常值为0.4-2mg/dL),18%的维生素C水平检测不到(低于0.1mg/dL)。
在 50 例 COVID 病例中进行了一项临床试验,其中给予了高剂量维生素C 静脉干预,结果显示 COVID 患者的氧合指数发生了积极变化。
有研究表明,高剂量(8000mg/d)口服维生素补充剂可使COVID-19恢复率提高70%.
仍需要进一步的临床试验来充分描述维生素 C 对 COVID-19 感染的影响。
维生素 C 静脉注射被 FDA 归类为药物,只有口服剂量才能被评估并用作膳食补充剂,儿童(取决于年龄)每天 400 至 1800 毫克,成人每天 2000 毫克被认为是安全的。
富含维生素C的食物包括:猕猴桃,橙子,辣椒,草莓,樱桃,苹果,西兰花,菠菜,青椒,甜菜,花椰菜等。
➤ 维 生 素 D
除了对钙的吸收和骨骼强度的强大影响外,维生素D对于维持免疫力至关重要。它还减慢病毒复制,降低炎症活动,并增加体内 T 调节细胞的数量。
维生素 D 还可以缓解传染病、感染性休克和 ARDS 疾病(由于维生素 D 水平低)等并发症,这可能有助于预防新型冠状病毒作为辅助补充剂。
维生素D通过中性粒细胞和巨噬细胞合成组织蛋白酶和防御素来提高先天免疫力。这些作用控制病毒复制,并下调肿瘤坏死因子TNFα和IFN-γ的表达。
维生素D在增强免疫力中的作用
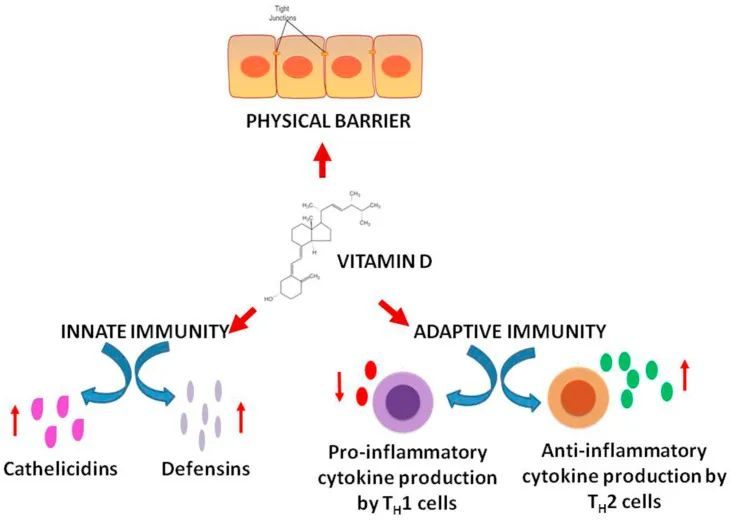
DOI:10.1016/j.clnesp.2022.04.007
低水平的维生素D(<20 ng/mL)与慢性疾病的发展和进展有关,如心血管疾病、2型糖尿病、癌症和抑郁症,也与骨骼健康不良有关。一些研究认为,维生素 D 水平低的人更容易患上 COVID-19 并患有更严重的疾病。维生素 D 缺乏会导致宿主体内的 SARS-CoV-2 病毒存活和复制。
与维生素D充足(感染率8.1%)或最高(感染率5.9%)的患者相比,缺乏维生素D的患者更有可能感染COVID-19(感染率12.5%)。
一项研究比较了中国335名COVID-19患者和560名健康志愿者的血液25(OH)D水平,发现COVID-19患者的25(OH)D浓度(中位数为26.5 nmol·L−1 [10.6 ng·mL−1])明显低于健康志愿者(中位数为32.5 nmol·L−1 [13 ng·mL−1])。
维生素D不足[具体定义为血清25(OH) D低于30 nmol·L−1 (12 ng·mL−1)]在COVID-19患者中比健康志愿者更常见。
与轻度COVID-19相比,重度COVID-19症状与最显著的维生素D缺乏相关。
土耳其的95名住院COVID-19患者,向他们补充了25-羟基维生素D,显示纤维蛋白原浓度降低,补充维生素D减少了住院时间,即使存在合并症。
在对13项研究的荟萃分析中,Pal等人计算出补充维生素D的人患严重疾病的风险降低了73%。在COVID-19发病后进行补充的研究中,风险降低最为显著(降低88%,OR 0.12)。
对SARS-CoV-2感染确诊病例的回顾性分析表明,维生素D水平低与较高的死亡风险显着相关,特别是在既往患病的老年人和男性个体中。
补充维生素D可以帮助25(OH) D水平低于25 nmol·L−1 (10 ng·mL−1) 的患者避免呼吸道感染。
补充维生素D,可以通过多晒太阳;饮食补充富含脂肪的鱼类如鲑鱼,鳟鱼,鲫鱼,金枪鱼,鳗鱼等,其他包括蘑菇,肝脏,蛋黄,新鲜水果蔬菜等。
注:冬季日照不够的情况可以通过维生素D补充剂,推荐最低剂量是每日400IU.
➤ 维 生 素 E
维生素E是一种脂溶性维生素,包括生育酚和生育三烯酚。是一种有效的抗氧化剂。可以影响免疫系统细胞,因为它具有抗氧化活性、蛋白激酶C(PKC)抑制和通过酶调节的信号转导。
在巨噬细胞中,维生素E修饰环氧化酶活性,从而控制过氧亚硝酸盐的合成。这导致前列腺素E2的产生降低,T细胞介导的T淋巴细胞反应上调。此外,它通过一氧化氮调节提高自然杀伤(NK)细胞活性。
与健康对照组相比,COVID-19患者的维生素E水平较低。
补充维生素E既往已被证明可增加免疫活性,特别是T细胞增殖,改善老年患者对疫苗接种的反应,并将老年肺炎患者的再住院风险降低63%.
维生素E来源:如小麦胚芽、葵花籽、杏仁、榛子、松子、鳄梨和甜红辣椒等。
扩展阅读:
➤ 铁
先天免疫反应精心控制铁代谢。铁稳态的破坏与感染,癌症,心血管疾病,肾脏疾病和血液疾病密切相关。
铁在病毒感染中也起着重要作用。在病毒复制过程中,需要ATP,而ATP合成需要铁。此外,铁调节素铁调素的表达增加几种细胞因子(例如,IL-6、IL-1),已知铁调素水平升高与血浆铁水平低有关(一种基于铁剥夺入侵病原体的非特异性宿主防御机制)。
注:铁调素通过结合和介导铁转运蛋白的降解来发挥其作用,铁转运蛋白是唯一已知的细胞铁输出剂,存在于肠细胞和巨噬细胞的细胞膜中,从而防止铁从这些细胞外排到血浆中。
铁缺乏在预测从轻度到重度疾病的转变方面很有价值,低血清铁水平是COVID-19患者死亡的独立危险因素。
汇总研究结果显示,COVID-19患者、严重程度状态和非幸存者的铁水平分别明显低于对照组、非严重程度状态和幸存者。
最近的研究中,与对照组(1.87±0.66mg/L)相比,COVID-19患者的血清铁(重症患者为1.33±0.7mg/L)显著降低。
此外,铁稳态的紊乱可在疾病发作后持续数月,并与肺部病变未消退和体能受损密切相关。
细胞内铁水平的升高和降低都是危险信号,分别通过NF-κB和HIF-1激活炎症和抗菌途径。
与中度病例相比,重症病例的血红蛋白、红细胞计数较低,铁蛋白水平较高,而非幸存者的铁蛋白水平高于幸存者。
铁过量也被认为是COVID-19发病机制的一个因素,因为它在活性氧的产生中发挥了作用,但更重要的可能是由于诱导非凋亡细胞死亡的铁死亡。
富含铁的食物包括:肝脏、动物血、红肉、扇贝、干、木耳、紫菜、菠菜、海带、黑芝麻、李子、桃、杏、苹果等。
扩展阅读
➤ 锌
锌是最普遍和最重要的微量元素,它参与细胞代谢的许多方面和近 100 种酶的催化活性。
它刺激适应性和固有免疫力,以及有助于维持呼吸上皮等组织屏障的抗病毒和抗炎特性。初步证据表明,ACE-2表达受Sirtuin 1(SIRT1)调节。由于锌能够下调SIRT1活性,这可能导致ACE-2表达降低,并减少SARS-CoV 2 进入细胞。
锌缺乏会阻碍淋巴细胞的产生、刺激和成熟,导致免疫功能下降。它还影响免疫介质,抑制 T 细胞、白细胞介素-2 合成以及抑制自然杀伤和细胞毒性 T 细胞活性。此外,它的缺失与更多的促炎介质有关,这可能会增加对疾病和炎症性疾病的易感性,尤其是那些影响肺部的疾病和炎症性疾病。
补充锌可能减轻COVID-19症状,因为这种金属抑制细胞内SARS-CoV 2复制的pH依赖性步骤,增加细胞内小泡的pH.
COVID-19典型的细胞因子过度产生(即“细胞因子风暴”),能够影响多个器官,似乎与短暂缺锌显著相关。
在2020年报告的一项研究显示,COVID-19患者的高剂量锌盐(每天<200毫克)改善了治疗后24小时的氧合并减少了发烧。尽管评估的病例数量很少,但这项工作确定了锌在SARS-CoV-2病毒控制和COVID-19并发症中可能产生的有益作用。
一项横断面对照研究报道,血清锌水平相对于疾病严重程度下降(中位数56.61μg/dL,n=200),重度COVID-19患者的降幅最大。
锌的典型每日剂量为15-30毫克锭剂,直接保护上呼吸道。补锌前检测确定锌水平至关重要。
➤ 硒
硒(Se)具有广泛的作用,如抗氧化和抗炎。硒在病毒感染中的氧化还原信号传导、氧化还原稳态和抗氧化防御中具有重要作用。
硒在保护呼吸系统,特别是抵御病毒感染方面发挥着重要作用。大量证据表明,硒缺乏与RNA病毒感染的易感性和不良结局有关。
硒与辅酶Q10联合使用可降低非特异性炎症反应和心血管疾病的死亡率。当与乙酰半胱氨酸一起服用时,硒有助于实现细胞内GSH(还原型谷胱甘肽)的正常水平,这是GPX(谷胱甘肽过氧化物酶是最强的抗氧化硒酶之一)的最佳水平的原因。GPX模拟依布硒(ebselen,一种合成硒化合物)是主要SARS-CoV-2蛋白酶的强抑制剂。
硒在正常免疫系统功能中的作用与其抗氧化特性和增加白细胞介素IL-2产生的能力有关,白细胞介介素IL-2具有免疫调节特性;例如,它根据当前需求刺激或抑制免疫反应。
适当剂量的硒对SARS-Cov-2感染患者的免疫系统的有益作用很重要,因为它调节IL-6的分泌,IL-6在疾病的病理生理学中起关键作用。硒的剂量建议为每天100至200µg。
➤ 铜
铜(Cu)是一种必需的微量元素,具有两种主要的生物学功能,第一种是酶的结构/催化辅因子,第二种是关键转录因子的辅激活因子。
铜转运基因参与巨噬细胞介导的宿主防御,其缺乏会降低IL-2和T细胞的增殖,并降低循环中性粒细胞的数量及其产生超氧阴离子的能力。在病毒感染和ROS启动氧化反应时,铜下调NF-κB表达,导致炎症细胞因子、趋化因子和粘附分子受到抑制。
一项对铜消费的荟萃分析显示,中国儿童反复呼吸道感染的发生与免疫功能、遗传因素和营养状况有关。在这项研究的结果中,锌和铜缺乏可能是儿童易患呼吸道感染的因素。

编辑
DOI:10.1007/s12011-022-03290-8
微量元素锌和铜之间存在拮抗作用和竞争吸收关系。当锌摄入量长期超标时,会导致铜缺乏,反之亦然。
自行服用锌补充剂的COVID-19患者,极有可能缺铜。作为一种必需的微量元素,铜是维持生物机制和细胞稳态所必需的;因此,有必要注意保持平衡。
关于微量元素的补充,应考虑锌与其他微量金属和维生素的相互作用。锌、铜、铁的吸收和生物利用度取决于元素之间的竞争,当以等比例给予时,铁和铜的吸收约减少40%。因此,过量补充锌可能导致铜或铁不足。
从目前的角度来看,除临床试验外,COVID-19中的维生素和微量元素补充剂不应超过一般人群和年龄组的推荐剂量。
药用植物,作为抗病毒、抗感染、抗炎、抗氧化、退热和肺肠道免疫增强剂,已被用于治疗与新冠肺炎相关的症状。
药用植物,在免疫刺激和维持平衡肠道微生物组中具有支持作用,可能是管理COVID-19的有效策略。
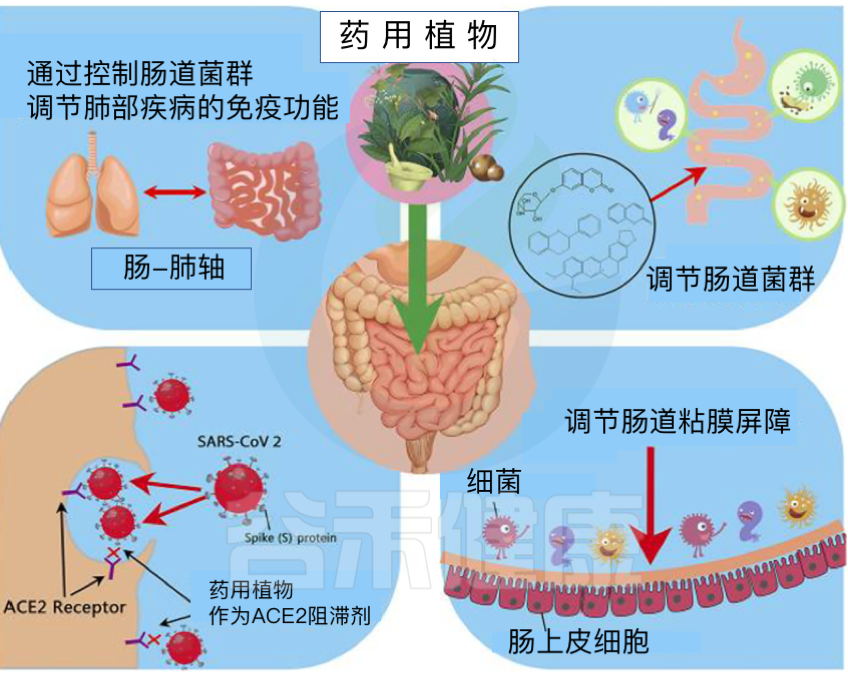
doi: 10.1186/s43088-022-00277-1.
➦ 吉 洛 伊 (Giloy,Guduchi)
由于其免疫调节特性,粉末状吉洛伊植物在前病毒期和后病毒期被彻底使用。此外,吉洛伊植物也可用作抗氧化剂,因此可以降低体内的整体氧化应激。这进一步防止了体内重要生物分子的氧化分解。
在慢性新冠肺炎感染期间,吉洛伊表现出抗炎作用,这在冠状病毒诱导的肺炎和肺部炎症中有很大的作用。血液中的氧饱和度低于正常范围,导致缺氧、疲劳,甚至死亡。
临床试验评估其对新冠肺炎确诊的无症状至轻度症状患者的疗效。91名年龄在18岁至75岁之间的患者,其中11.7%的对照组在平均1.8天后出现轻微症状,试验组没有任何其他症状。研究人员从研究中建议,Guduchi Ghan Vati可以作为无症状新冠肺炎患者的预防和治疗药物(NCT04480398)。
➦ 甘 草 (Mulethi)
由豆科甘草的根和细根组成,其活性化学成分是甘草酸。甘草是一种阿育吠陀药,以其外围和中枢作用的抗咳特性而闻名。
研究表明,甘草酸苷具有止咳作用,可以抑制咳嗽和相关的呼吸短促;也就是新冠肺炎感染的主要症状。它已被证明可以阻止被SARS感染的细胞释放HMGB1核蛋白(高运动性基盒蛋白1)。这使巨噬细胞失效,巨噬细胞在激活免疫中发挥积极作用,并负责产生促炎细胞因子。因此,避免了“细胞因子风暴”的开始及其对健康的不利影响。
在伊朗进行了一项单中心、开放标签、随机、平行组临床试验,以评估甘草对60名年龄≥18岁(体重≥35 kg)的新冠肺炎患者的抗炎作用(IRCT20200506047323N2)。
➦ 茯 苓 (Poria cocos)
其活性成分为茯苓酸。蛋白酶负责生物在宿主细胞中的复制。在冠状病毒中发现的一种蛋白酶是Mpro酶。Mpro是SARS-CoV-2的关键酶,参与介导病毒复制和转录。茯苓的主要活性成分茯苓酸与Mpro酶结合并抑制,从而阻止生物体在宿主细胞中的进一步复制和生长。
在COVID-19重症病例中,很少患者可能出现肺炎并发症,65岁或以上人群的并发症甚至更高。在这种情况下,茯苓酸的抗炎特性可以被解释为提供缓解。在一项研究中,用茯苓酸治疗肺炎大鼠肺部,结果显示通过NF-κB通路抑制炎症因子的表达。
➦ 蒙古紫云英(Mongolian milkvetch)
它由豆科黄芪的根、茎和叶组成。活性植物化学物质为黄芪甲苷、熊竹素和毛蕊异黄酮。黄芪甲苷是黄芪根、茎和叶中发现的多糖,可调节促炎因子如细胞因子、TNFα、IL-1β和NFATc4的表达,从而发挥免疫调节作用。它已被发现有助于避免细胞因子风暴的状况。
熊竹素在一定程度上也能成功抑制Mpro酶。作为一种免疫调节剂,在COVID-19的情况下,作为一种预防措施,可以使用蒙古紫云英来增强免疫力。
研究人员对小鼠血浆进行了代谢组学研究。结果表明,蒙古紫云英与人参同时食用可提高大鼠的脾、胸腺指数、脾淋巴细胞增殖和NK细胞毒活性。
➦ 其 他
doi: 10.1016/j.ccmp.2022.100021.
改变的肠道微生物群或菌群失调可以作为全身炎症活动的调节剂,并可以通过多个肠道器官轴影响不同的器官。肠道通透性增加或者说肠漏,使细菌代谢物和毒素进入循环系统,并进一步恶化全身炎症反应,导致不同的新冠肺炎并发症。
肠道微生物群不仅显着影响COVID-19的发展和疾病的严重程度,而且还反映了COVID-19患者对长期并发症的易感性。
初步临床研究揭示了益生菌对 SARS-CoV-2 感染及恢复期的潜在调节作用。鉴于针对COVID-19的特定药物仍然是个谜,疫苗是有效的预防和控制策略;然而,病毒的持续突变加剧了这一难题。因此,通过益生菌、益生元、膳食补充剂、药用植物、FMT等多种方式,进行肠道菌群干预,是未来治疗COVID-19的有前途的补充策略。
尽管大规模临床试验有限,但相信这个领域正在发展,并蕴藏着巨大的机遇,一个个新诞生的研究可以在疫情的不同浪潮中推动微生物群的发现走向临床应用。
在此,也向一直奋斗在前线的医务工作者、在该领域辛勤耕耘的科研人员、其他相关工作者致敬。
对于个人而言,通过多种方式增强体质提高自身免疫力,是抵抗病毒侵袭的最好选择。
在新开启的2023里,希望大家都能找到属于自己的健康生活。
注:本账号内容仅作交流参考,不作为诊断及医疗依据。
主要参考文献:
Li M, Peng H, Duan G, Wang J, Yu Z, Zhang Z, Wu L, Du M, Zhou S. Older age and depressive state are risk factors for re-positivity with SARS-CoV-2 Omicron variant. Front Public Health. 2022 Oct 4;10:1014470. doi: 10.3389/fpubh.2022.1014470. PMID: 36268004; PMCID: PMC9576942.
Sacco C, Petrone D, Del Manso M, Mateo-Urdiales A, Fabiani M, Bressi M, Bella A, Pezzotti P, Rota MC, Riccardo F; Italian Integrated Surveillance of COVID-19 study group. Risk and protective factors for SARS-CoV-2 reinfections, surveillance data, Italy, August 2021 to March 2022. Euro Surveill. 2022 May;27(20):2200372. doi: 10.2807/1560-7917.ES.2022.27.20.2200372. PMID: 35593164; PMCID: PMC9121659.
Deng L, Li P, Zhang X, Jiang Q, Turner D, Zhou C, Gao Y, Qian F, Zhang C, Lu H, Zou H, Vermund SH, Qian HZ. Risk of SARS-CoV-2 reinfection: a systematic review and meta-analysis. Sci Rep. 2022 Dec 1;12(1):20763. doi: 10.1038/s41598-022-24220-7. PMID: 36456577; PMCID: PMC9714387.
Hansen CH, Friis NU, Bager P, Stegger M, Fonager J, Fomsgaard A, Gram MA, Christiansen LE, Ethelberg S, Legarth R, Krause TG, Ullum H, Valentiner-Branth P. Risk of reinfection, vaccine protection, and severity of infection with the BA.5 omicron subvariant: a nation-wide population-based study in Denmark. Lancet Infect Dis. 2022 Oct 18:S1473-3099(22)00595-3. doi: 10.1016/S1473-3099(22)00595-3. Epub ahead of print. PMID: 36270311; PMCID: PMC9578720.
Guedes AR, Oliveira MS, Tavares BM, Luna-Muschi A, Lazari CDS, Montal AC, de Faria E, Maia FL, Barboza ADS, Leme MD, Tomazini FM, Costa SF, Levin AS. Reinfection rate in a cohort of healthcare workers over 2 years of the COVID-19 pandemic. Sci Rep. 2023 Jan 13;13(1):712. doi: 10.1038/s41598-022-25908-6. PMID: 36639411; PMCID: PMC9837751.
Holmer HK, Mackey K, Fiordalisi CV, Helfand M. Major Update 2: Antibody Response and Risk for Reinfection After SARS-CoV-2 Infection-Final Update of a Living, Rapid Review. Ann Intern Med. 2023 Jan;176(1):85-91. doi: 10.7326/M22-1745. Epub 2022 Nov 29. PMID: 36442059; PMCID: PMC9707440.
Bowe B, Xie Y, Al-Aly Z. Acute and postacute sequelae associated with SARS-CoV-2 reinfection. Nat Med. 2022 Nov;28(11):2398-2405. doi: 10.1038/s41591-022-02051-3. Epub 2022 Nov 10. PMID: 36357676; PMCID: PMC9671810.
Davis HE, McCorkell L, Vogel JM, Topol EJ. Long COVID: major findings, mechanisms and recommendations. Nat Rev Microbiol. 2023 Jan 13:1–14. doi: 10.1038/s41579-022-00846-2. Epub ahead of print. PMID: 36639608; PMCID: PMC9839201.
O’ Mahony L, Buwalda T, Blair M, Forde B, Lunjani N, Ambikan A, Neogi U, Barrett P, Geary E, O’Connor N, Dineen J, Clarke G, Kelleher E, Horgan M, Jackson A, Sadlier C. Impact of Long COVID on health and quality of life. HRB Open Res. 2022 Apr 22;5:31. doi: 10.12688/hrbopenres.13516.1. PMID: 36101871; PMCID: PMC9440374.
Zhang F, Lau RI, Liu Q, Su Q, Chan FKL, Ng SC. Gut microbiota in COVID-19: key microbial changes, potential mechanisms and clinical applications. Nat Rev Gastroenterol Hepatol. 2022 Oct 21:1–15. doi: 10.1038/s41575-022-00698-4. Epub ahead of print. Erratum in: Nat Rev Gastroenterol Hepatol. 2023 Jan 12;: PMID: 36271144; PMCID: PMC9589856.
Sun K, Tempia S, Kleynhans J, von Gottberg A, McMorrow ML, Wolter N, Bhiman JN, Moyes J, Carrim M, Martinson NA, Kahn K, Lebina L, du Toit JD, Mkhencele T, Viboud C, Cohen C; PHIRST-C group. Rapidly shifting immunologic landscape and severity of SARS-CoV-2 in the Omicron era in South Africa. Nat Commun. 2023 Jan 16;14(1):246. doi: 10.1038/s41467-022-35652-0. PMID: 36646700; PMCID: PMC9842214.
Wang B, Zhang L, Wang Y, Dai T, Qin Z, Zhou F, Zhang L. Alterations in microbiota of patients with COVID-19: potential mechanisms and therapeutic interventions. Signal Transduct Target Ther. 2022 Apr 29;7(1):143. doi: 10.1038/s41392-022-00986-0. PMID: 35487886; PMCID: PMC9052735.
Vaezi M, Ravanshad S, Rad MA, Zarrinfar H, Kabiri M. The effect of synbiotic adjunct therapy on clinical and paraclinical outcomes in hospitalized COVID-19 patients: A randomized placebo-controlled trial. J Med Virol. 2023 Jan 5. doi: 10.1002/jmv.28463. Epub ahead of print. PMID: 36602047.
Chutipongtanate S, Morrow AL, Newburg DS. Human Milk Oligosaccharides: Potential Applications in COVID-19. Biomedicines. 2022 Feb 1;10(2):346. doi: 10.3390/biomedicines10020346. PMID: 35203555; PMCID: PMC8961778.
Nasir Ahmed M, Hughes K. Role of ethno-phytomedicine knowledge in healthcare of COVID-19: advances in traditional phytomedicine perspective. Beni Suef Univ J Basic Appl Sci. 2022;11(1):96. doi: 10.1186/s43088-022-00277-1. Epub 2022 Aug 4. PMID: 35966214; PMCID: PMC9362587.
Gang J, Wang H, Xue X, Zhang S. Microbiota and COVID-19: Long-term and complex influencing factors. Front Microbiol. 2022 Aug 12;13:963488. doi: 10.3389/fmicb.2022.963488. PMID: 36033885; PMCID: PMC9417543.
Xu L, Ho CT, Liu Y, Wu Z, Zhang X. Potential Application of Tea Polyphenols to the Prevention of COVID-19 Infection: Based on the Gut-Lung Axis. Front Nutr. 2022 Apr 14;9:899842. doi: 10.3389/fnut.2022.899842. PMID: 35495940; PMCID: PMC9046984.
Xu L, Yang CS, Liu Y, Zhang X. Effective Regulation of Gut Microbiota With Probiotics and Prebiotics May Prevent or Alleviate COVID-19 Through the Gut-Lung Axis. Front Pharmacol. 2022 Apr 25;13:895193. doi: 10.3389/fphar.2022.895193. PMID: 35548347; PMCID: PMC9081431.
Rahmati M, Fatemi R, Yon DK, Lee SW, Koyanagi A, Il Shin J, Smith L. The effect of adherence to high-quality dietary pattern on COVID-19 outcomes: A systematic review and meta-analysis. J Med Virol. 2023 Jan;95(1):e28298. doi: 10.1002/jmv.28298. Epub 2022 Nov 18. PMID: 36367218.
Pandey M, Bhati A, Priya K, Sharma KK, Singhal B. Precision Postbiotics and Mental Health: the Management of Post-COVID-19 Complications. Probiotics Antimicrob Proteins. 2022 Jun;14(3):426-448. doi: 10.1007/s12602-021-09875-4. Epub 2021 Nov 22. PMID: 34806151; PMCID: PMC8606251.
Horne BD, Bunker T. Pathogenic Mechanisms of the Severe Acute Respiratory Syndrome Coronavirus 2 and Potential Direct and Indirect Counteractions by Intermittent Fasting. Nutrients. 2022 Dec 21;15(1):20. doi: 10.3390/nu15010020. PMID: 36615679; PMCID: PMC9823718.
Hou YC, Su WL, Chao YC. COVID-19 Illness Severity in the Elderly in Relation to Vegetarian and Non-vegetarian Diets: A Single-Center Experience. Front Nutr. 2022 Apr 29;9:837458. doi: 10.3389/fnut.2022.837458. PMID: 35571931; PMCID: PMC9101048.
Karupaiah T, Lu KC. Editorial: Nutraceuticals for the recovery of COVID-19 patients. Front Nutr. 2022 Nov 14;9:1054632. doi: 10.3389/fnut.2022.1054632. PMID: 36451742; PMCID: PMC9703639.
Itsiopoulos C, Mayr HL, Thomas CJ. The anti-inflammatory effects of a Mediterranean diet: a review. Curr Opin Clin Nutr Metab Care. 2022 Nov 1;25(6):415-422. doi: 10.1097/MCO.0000000000000872. Epub 2022 Aug 30. PMID: 36039924.
Yue Y, Ma W, Accorsi EK, Ding M, Hu F, Willett WC, Chan AT, Sun Q, Rich-Edwards J, Smith-Warner SA, Bhupathiraju SN. Long-term diet and risk of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) infection and Coronavirus Disease 2019 (COVID-19) severity. Am J Clin Nutr. 2022 Dec 19;116(6):1672-1681. doi: 10.1093/ajcn/nqac219. PMID: 35945354; PMCID: PMC9384672.
Zargarzadeh N, Tadbir Vajargah K, Ebrahimzadeh A, Mousavi SM, Khodaveisi H, Akhgarjand C, Toyos FMP, Cerqueira HS, Santos HO, Taghizadeh M, Milajerdi A. Higher Adherence to the Mediterranean Dietary Pattern Is Inversely Associated With Severity of COVID-19 and Related Symptoms: A Cross-Sectional Study. Front Med (Lausanne). 2022 Jul 19;9:911273. doi: 10.3389/fmed.2022.911273. PMID: 35928288; PMCID: PMC9343686.
Xu J, Ren Z, Cao K, Li X, Yang J, Luo X, Zhu L, Wang X, Ding L, Liang J, Jin D, Yuan T, Li L, Xu J. Boosting Vaccine-Elicited Respiratory Mucosal and Systemic COVID-19 Immunity in Mice With the Oral Lactobacillus plantarum. Front Nutr. 2021 Dec 22;8:789242. doi: 10.3389/fnut.2021.789242. PMID: 35004816; PMCID: PMC8733898.
Gualtieri P, Marchetti M, Frank G, Cianci R, Bigioni G, Colica C, Soldati L, Moia A, De Lorenzo A, Di Renzo L. Exploring the Sustainable Benefits of Adherence to the Mediterranean Diet during the COVID-19 Pandemic in Italy. Nutrients. 2022 Dec 26;15(1):110. doi: 10.3390/nu15010110. PMID: 36615768; PMCID: PMC9824251.
Ferro Y, Pujia R, Maurotti S, Boragina G, Mirarchi A, Gnagnarella P, Mazza E. Mediterranean Diet a Potential Strategy against SARS-CoV-2 Infection: A Narrative Review. Medicina (Kaunas). 2021 Dec 20;57(12):1389. doi: 10.3390/medicina57121389. PMID: 34946334; PMCID: PMC8704657.
Chavda VP, Patel AB, Vihol D, Vaghasiya DD, Ahmed KMSB, Trivedi KU, Dave DJ. Herbal Remedies, Nutraceuticals, and Dietary Supplements for COVID-19 Management: An Update. Clin Complement Med Pharmacol. 2022 Mar;2(1):100021. doi: 10.1016/j.ccmp.2022.100021. Epub 2022 Feb 5. PMID: 36620357; PMCID: PMC8816850.
Nasir Ahmed M, Hughes K. Role of ethno-phytomedicine knowledge in healthcare of COVID-19: advances in traditional phytomedicine perspective. Beni Suef Univ J Basic Appl Sci. 2022;11(1):96. doi: 10.1186/s43088-022-00277-1. Epub 2022 Aug 4. PMID: 35966214; PMCID: PMC9362587.
Renata RN, Arely GA, Gabriela LA, Esther MM. Immunomodulatory Role of Microelements in COVID-19 Outcome: a Relationship with Nutritional Status. Biol Trace Elem Res. 2022 Jun 6:1–19. doi: 10.1007/s12011-022-03290-8. Epub ahead of print. PMID: 35668151; PMCID: PMC9170122.
Li Y, Luo W, Liang B. Circulating trace elements status in COVID-19 disease: A meta-analysis. Front Nutr. 2022 Aug 12;9:982032. doi: 10.3389/fnut.2022.982032. PMID: 36034929; PMCID: PMC9411985.
Bego T, Meseldžić N, Prnjavorac B, Prnjavorac L, Marjanović D, Azevedo R, Pinto E, Duro M, Couto C, Almeida A. Association of trace element status in COVID-19 patients with disease severity. J Trace Elem Med Biol. 2022 Dec;74:127055. doi: 10.1016/j.jtemb.2022.127055. Epub 2022 Aug 4. PMID: 35985069; PMCID: PMC9349050.
Milton-Laskibar I, Trepiana J, Macarulla MT, Gómez-Zorita S, Arellano-García L, Fernández-Quintela A, Portillo MP. Potential usefulness of Mediterranean diet polyphenols against COVID-19-induced inflammation: a review of the current knowledge. J Physiol Biochem. 2022 Nov 8:1–12. doi: 10.1007/s13105-022-00926-0. Epub ahead of print. PMID: 36346507; PMCID: PMC9641689.
Sun Y, Ju P, Xue T, et al., Alteration of faecal microbiota balance related to long-term deep meditation. General Psychiatry 2023;36:e100893. doi: 10.1136/gpsych-2022-100893
Callaway E. What Omicron’s BA.4 and BA.5 variants mean for the pandemic. Nature. 2022 Jun;606(7916):848-849. doi: 10.1038/d41586-022-01730-y. PMID: 35750920.
Wang K, Gheblawi M, Oudit GY. Angiotensin Converting Enzyme 2: A Double-Edged Sword. Circulation. 2020 Aug 4;142(5):426-428. doi: 10.1161/CIRCULATIONAHA.120.047049. Epub 2020 Mar 26. PMID: 32213097.
Sapra L, Saini C, Garg B, Gupta R, Verma B, Mishra PK, Srivastava RK. Long-term implications of COVID-19 on bone health: pathophysiology and therapeutics. Inflamm Res. 2022 Sep;71(9):1025-1040. doi: 10.1007/s00011-022-01616-9. Epub 2022 Jul 28. PMID: 35900380; PMCID: PMC9330992.
Alenazy MF, Aljohar HI, Alruwaili AR, Daghestani MH, Alonazi MA, Labban RS, El-Ansary AK, Balto HA. Gut Microbiota Dynamics in Relation to Long-COVID-19 Syndrome: Role of Probiotics to Combat Psychiatric Complications. Metabolites. 2022 Sep 27;12(10):912. doi: 10.3390/metabo12100912. PMID: 36295814; PMCID: PMC9611210.
Shi Y, Li Z, Yang C, Liu C. The role of gut-brain axis in SARA-CoV-2 neuroinvasion: Culprit or innocent bystander? Brain Behav Immun. 2021 May;94:476-477. doi: 10.1016/j.bbi.2021.01.024. Epub 2021 Feb 15. PMID: 33600935; PMCID: PMC7883713.
De R, Dutta S. Role of the Microbiome in the Pathogenesis of COVID-19. Front Cell Infect Microbiol. 2022 Mar 31;12:736397. doi: 10.3389/fcimb.2022.736397. PMID: 35433495; PMCID: PMC9009446.
谷禾健康
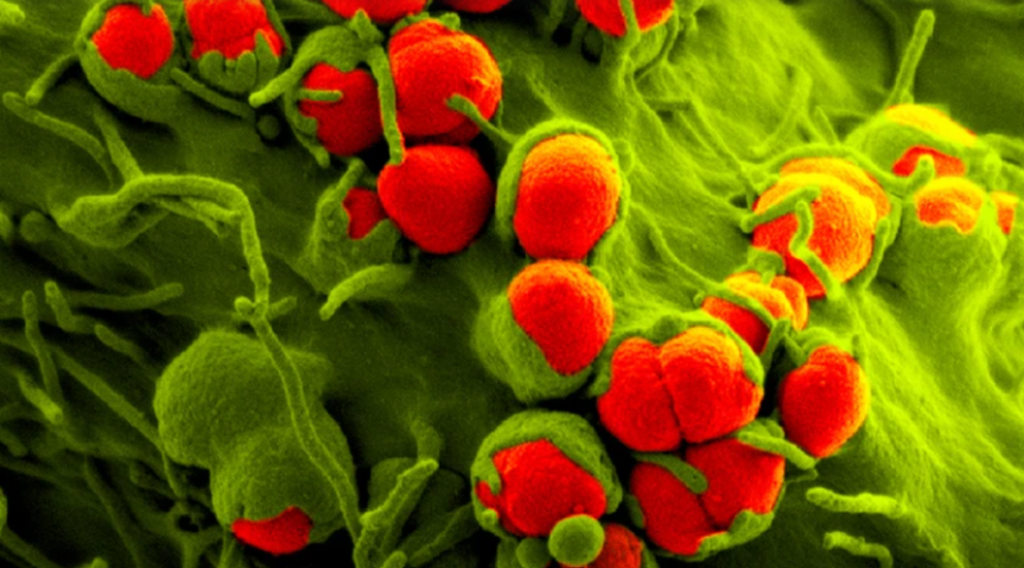
迄今为止最全面的全球抗菌素耐药性 (AMR) 研究发现,由耐药菌引起的感染是所有年龄段人群死亡的主要原因之一。
22年发表在《柳叶刀》杂志上的分析估计,2019 年有 495 万人死于细菌性 AMR 发挥作用的疾病。其中,127 万人死亡是 AMR 的直接结果——这意味着耐药性感染导致的死亡人数超过了艾滋病毒/艾滋病(864,000 人死亡)或疟疾(643,000 人死亡)。
细菌性 AMR 感染最常见的三个部位是胸部、血液和腹部——身体这些部位的感染占直接归因于 AMR 死亡的 78.8%。六种最致命的细菌病原体导致了将近四分之三的所有因耐药性导致的死亡。2019 年,仅耐抗生素大肠杆菌就导致约 20 万人死亡。
“AMR 确实是一个全球性问题,需要决策者和全球卫生界采取紧急行动,以避免可预防的死亡,”
对抗生素有耐药性的细菌比例正在上升。在一个抗生素使用变得如此普遍的世界里,耐药细菌比那些被药物杀死的细菌更胜一筹。
伤口敷料的微观纤维上的金黄色葡萄球菌
图片来源: Science Photo Library
预计到 2050 年,每年可能有多达 1000 万人死于抗菌素耐药性。如果任其发展,以前可以用几天抗生素治愈的感染可能变得无法治愈。
如果细菌感染无法控制会导致严重的健康问题,比如梅毒会引起失明和神经损伤。女性感染衣原体会导致不孕。梅毒可以传染给未出生的孩子,导致出生时患有这种疾病的婴儿流产、死产或大脑和器官受损。
淋病细菌(如图)对某些抗生素产生了耐药性
图片来源: SPL
数据显示,低收入国家的 AMR 相关死亡率最高。在 21 个 GBD 地理区域中,西撒哈拉以南非洲直接归因于 AMR 的死亡率最高,每 10 万人中有 27.3 人死亡。大洋洲最低,每 10 万人中有 6.5 人死亡。
低收入地区的耐药率和耐药菌感染数量均高于富裕国家。造成这种情况的原因包括环境卫生和卫生条件差、用于治疗的检测设施不足以及无法获得最新的抗生素和疫苗。
但是注意,许多低收入和中等收入国家的抗微生物药物耐药性数据很少,这表示实际的AMR比调查的数据更严重,这凸显了在这些地区大幅扩大实验室能力的必要性。
抗生素药物旨在消灭细菌,但它们不一定会杀死所有的细菌,它们只是将细菌减少到安全水平。但是不幸的是,这个过程选择留下了最强壮的细菌个体,然后可以通过基因传递产生耐药性。几十年后,就会有这么多种细菌进化出对一系列抗生素的抗性。
对抗生素产生耐药性的致病细菌的出现,往往归因于人类和牲畜过度和不合理使用抗生素。虽然这是问题背后的主要机制,但这不是唯一的因素。昆士兰大学的研究人员希望测试其他药物,即使是那些没有抗生素特性的药物,是否可以在超级细菌的出现中发挥作用。
最近昆士兰大学的研究人员已经将注意力集中在另一个潜在的抵抗力驱动因素上:抗抑郁药。通过研究在实验室中生长的细菌,已追踪到抗抑郁药如何引发耐药性。
“即使接触几天后,细菌也会产生耐药性,不仅针对一种抗生素,还针对多种抗生素”,布里斯班昆士兰大学澳大利亚水与环境生物技术中心的资深作者郭建华(英译)说。
早期线索
2014 年,郭开始对非抗生素药物对抗生素耐药性的可能贡献产生兴趣,因为他的实验室发现生活废水样本中循环的抗生素耐药基因比抗生素使用率更高的医院废水样本中循环的抗生素耐药基因更多。
郭的团队和其他团队还观察到,抗抑郁药——世界上使用最广泛的药物之一——可以杀死或阻碍某些细菌的生长。郭解释说,它们会引发“SOS 反应”,从而触发细胞防御机制,使细菌能够更好地在随后的抗生素治疗中存活下来。
在 2018 年的一篇论文中,该小组报告说,大肠杆菌在接触氟西汀后就对多种抗生素产生了耐药性,氟西汀通常以百忧解的形式出售。最新研究检查了6种此类药物中的5种其他抗抑郁药和13种抗生素,并调查了大肠杆菌的耐药性是如何产生的。
备注:百优解(盐酸氟西汀胶囊)是一种选择性血清素(5-羟色胺,5-HT)再吸收抑制剂(SSRI)型的抗忧郁药,其药物形态为盐酸氟西汀(Fluoxetinehydrochloride),商品名为“百优解”或“百忧解”(Prozac)。在临床上用于成人忧郁症、强迫症等。
在氧气充足的实验室条件下生长的细菌中,抗抑郁药会导致细胞产生活性氧:一种激活微生物防御机制的有毒分子。最重要的是,这激活了细菌的外排泵系统,这是许多细菌用来消除各种分子(包括抗生素)的通用排出系统。这可能解释了细菌如何在没有特定抗性基因的情况下抵抗抗生素。
在抗抑郁药存在的情况下,革兰氏阴性细菌大肠杆菌可以抵御抗生素
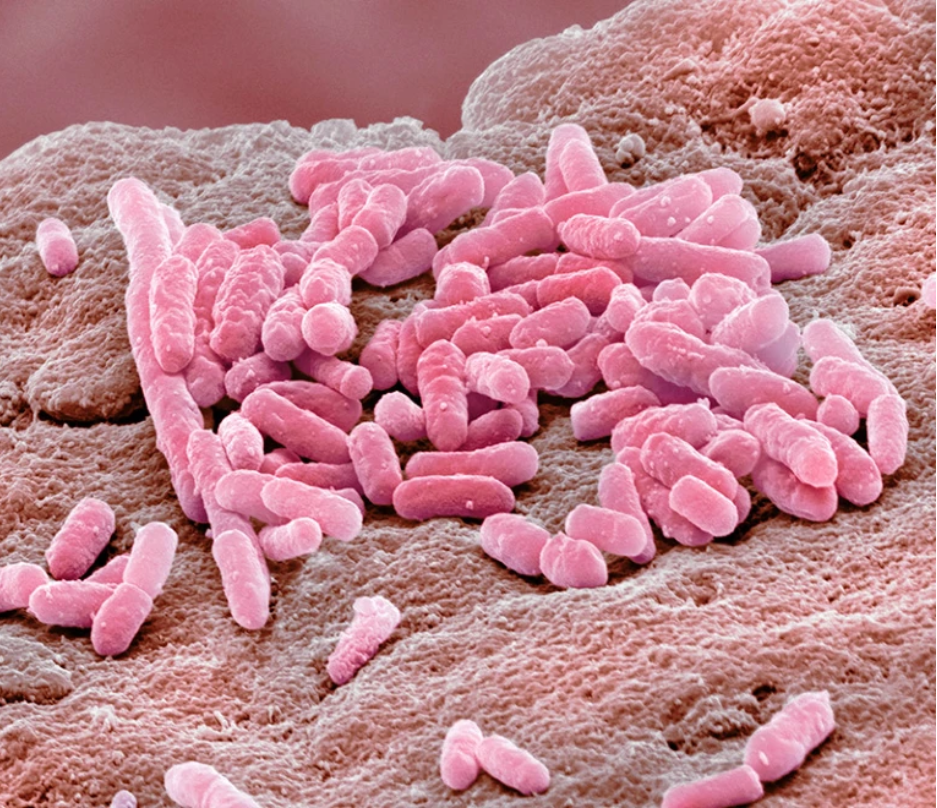
图片来源:Steve Gschmeissner/Science Photo Library
但是大肠杆菌暴露于抗抑郁药也导致微生物突变率增加,以及随后选择各种抗性基因。然而,在厌氧条件下生长的细菌中,活性氧水平要低得多,抗生素耐药性的发展要慢得多。
此外,至少一种抗抑郁药舍曲林(舍曲林是5-羟色氨再摄取抑制剂,是一种新型的抗抑郁剂,具有较强抗抑郁,抗焦虑作用,既可用于抑郁症,焦虑症、双相障碍的抑郁相,强迫障碍的治疗)促进了细菌细胞之间的基因转移,这一过程可以加速耐药性在人群中的传播。这种转移可以发生在不同类型的细菌之间,从而使耐药性在物种之间转移——包括从无害细菌到致病细菌。
越来越多的认可
英国剑桥大学研究微生物组-化学相互作用的 Kiran Patil 说,在过去五年中,人们越来越认识到许多针对人体细胞的非抗生素药物也会影响细菌并导致抗生素耐药性。
德国图宾根大学的 Lisa Maier 指出,研究药物与微生物组之间的相互作用,要了解抗抑郁药如何驱动抗生素耐药性,研究人员需要确定药物针对细菌中的哪些分子并评估药物对更广泛的临床相关细菌种类的影响。
2018 年,Maier 和她的同事调查了 835 种不针对微生物的药物,发现 24% 的药物抑制了至少一种人类肠道细菌菌株的生长。
Patil 和 Maier 表示,重要的是要收集证据来评估抗抑郁药在现实世界中对耐药性的影响,例如抗抑郁药是否正在推动抗生素耐药细菌的积累,尤其是致病细菌在人、动物或环境中的积累。
尽管在废水中发现了大量的抗抑郁药,但报告的水平往往低于郭的小组在大肠杆菌中看到显着效果的浓度。不过,预计在服用这些药物的人的大肠中会达到在这项研究中具有强烈作用的一些抗抑郁药的浓度。
后续研究
Maier 说,现在有几项研究将抗抑郁药和其他非抗生素药物与细菌的变化联系起来,并且初步研究已经给出了关于这些药物如何影响服用它们的人的微生物组的“初步提示”。
但在健康人类中,大肠杆菌主要存在于大肠中,那里的条件是厌氧的,这意味着论文中描述的过程可能不会以相同的速度发生在人身上,Maier 说,未来的研究应该使用细菌生长条件来模拟抗抑郁药可能起作用的部位。
郭说他的实验室现在正在研究给予抗抑郁药的小鼠的微生物组。早期未发表的数据表明,这些药物可以改变动物的肠道微生物群并促进基因转移。
但是郭和相关研究人员告诫人们不要根据这项研究停止服用抗抑郁药。“如果您患有抑郁症,则需要以最好的方式进行治疗,其次是关注肠道细菌”。
但是研究人员和制药公司需要量化非抗生素药物对抗生素耐药性的贡献。非抗生素药物是我们不应忽视的一个大问题。
寻找新抗生素的需求迫在眉睫,但许多科学家和政策制定者正在从其他角度解决抗菌素耐药性问题。《Nature》期刊详细地介绍了三种方法。
➤ 等离子清洗
富含化学不稳定形式的氧和氮(也称为自由基和活性物质)的等离子活化水被认为是潜在的新型消毒剂。“如果细菌被自由基淹没,它们最终就会死亡,” 澳大利亚阿德莱德大学的生物医学研究员 Katharina Richter 说。
Richter 和她的同事正在研究与未经治疗的伤口相比,血浆活化水清除感染耐甲氧西林金黄色葡萄球菌(MRSA) 的伤口的速度有多快。他们还将这项技术与通过静脉滴注施用抗生素进行比较。
她说,尚未公布的初步结果还不错。“治疗改善了伤口并比没有治疗更快地清除了感染,” 她说,“它不如抗生素治疗有效,但 Richter 说实验设计可能是罪魁祸首。将其与局部给予的抗生素进行比较会更公平。我们的下一项研究将有更好的控制。”
➤ 金属奇迹
细菌虽然在自然界中是单细胞,但确实聚集在一起并互相帮助以逃避药物和防腐剂。他们这样做的一种方法是形成生物膜——生活在它们自己制造的粘液环境中的细菌群。
生物膜保护居住在其中的单个细胞;据认为,大约 80% 的慢性人类感染是由生物膜引起的。金属元素镓会干扰细菌对铁的吸收,最终导致微生物缺乏营养。
正因为如此,含镓药物是一种正在探索的破坏生物膜的途径。英国曼彻斯特大学的科学家发现,镓化合物可以减少多达 87% 的细菌生长(JM Baker et al. Life Sci . 305 , 120794; 2022)。
这项工作建立在中国上海交通大学研究人员的研究结果之上,该研究表明镓可以有效溶解 MRSA 生物膜的结构,使细菌可以用常用抗生素剂量的十分之一杀死(W. Xia et al., ACS Infect. 2021, 2565–2582)。现在的研究重点是如何最好地输送镓以及输送多少剂量。
➤ 分子签证
抗生素应具有三个关键特性:溶解性,易于与细菌结合的能力,以及穿透细胞膜的能力。这使得设计一种新的抗生素成为一项艰巨的任务。
因此,一些研究人员并没有创造全新的抗生素,而是通过“大型数字图书馆”进行筛选,以预测哪些现有化合物可能已经具备所需的条件。
新加坡生物技术初创公司 BIOptimize 的计算化学家 Javad Deylami 说:“已经有既定的方法可以在几分钟内评估前两个特性,但渗透是这个难题中缺失的一块。”
Deylami 说,这意味着科学家们经常评估理论上可以很好地杀死细菌的化合物,但在进入细菌内部的第一个障碍上却达不到要求。“这就像他们到达边境但没有签证可以通过。”
Deylami 构建了一个细菌外细胞膜的计算机化版本,在上面进行模拟,测试分子穿透细胞膜的能力。他的模型确定了影响潜在药物通过或失败的力,从而使 Deylami 能够计算化合物的渗透性。
通过该程序运行已知的分子结构,Deylami 的团队能够在人工智能的帮助下了解化合物需要哪些“品质”来提高其渗透性。这些知识应该可以帮助他们在“庞大的图书馆”中寻找具有这些特性的现有药物。
主要参考文献
Wang, Y. et al. Proc. Natl Acad. Sci. USA 120, e2208344120 (2023).
Drew L. How antidepressants help bacteria resist antibiotics. Nature. 2023 Jan 24. doi: 10.1038/d41586-023-00186-y. Epub ahead of print. PMID: 36693968.
Antimicrobial Resistance Collaborators Lancet 399, 629–655 (2022).
Jin, M. et al. Environ. Int. 120, 421–430 (2018).
Maier, L. et al. Nature 555, 623–628 (2018).

谷禾健康

与人类密切相关的微生物
我们的世界大到浩瀚宇宙,小到微观下的生物分子。我们总说漫天繁星,其实身边微生物数量可能更多。动物、植物、真菌、细菌、病毒等,共同构成了丰富多彩的生命世界。
细菌、真菌、病毒是其中的三个大类,虽然它们都体型微小,但是相互之间可以说是天差地别。并且它们与我们的生活以及健康息息相关,有对人体有益的益生菌,也有对人体危害极大的病毒。

谷禾在本文中介绍了细菌、真菌、病毒的一些特征,它们的分类及繁殖方式,以及有致病性的微生物和感染后的一些症状与免疫过程。
在最后,针对一些病原体的感染。我们提出了一些预防的方法,以及目前技术条件下的治疗手段。微生物检测技术能够帮助人们更好地了解就在我们体内或身边的微生物,有助于塑造更健康的身体及生活。
本文主要从以下几个方面讲述
●什么是细菌、真菌、病毒?
●细菌、真菌、病毒的分类与繁殖方式
●细菌、真菌、病毒的致病性与感染症状
●细菌、真菌、病毒在体内的免疫反应
●病原体感染的预防及治疗方法
细菌
细菌(Bacteria)是生物的主要类群之一,属于细菌域。广义的细菌即为原核生物, 是指一大类细胞核无核膜包裹,只存在拟核区(或拟核)的裸露DNA的原始单细胞生物,包括真细菌(eubacteria)和古细菌(archaea)两大类群。
注:其中除少数属古细菌外,多数的原核生物都是真细菌。
✦结构简单、个体小
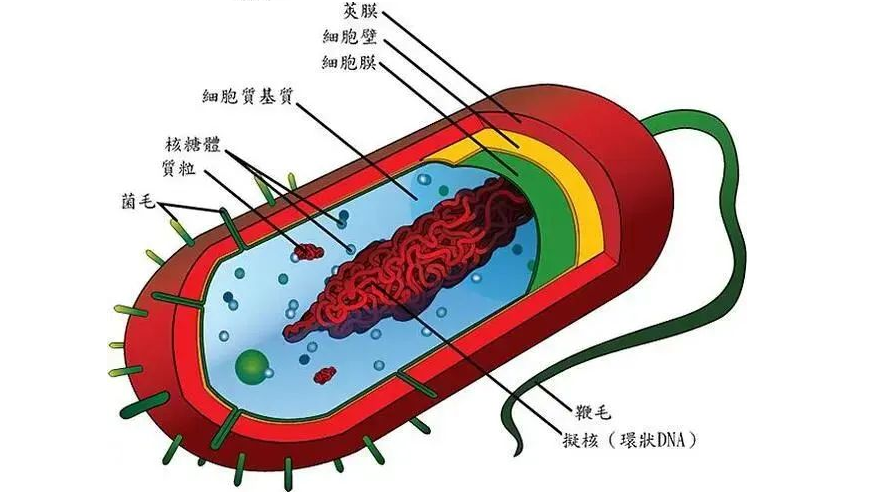
图片来源:百度
细菌为原核微生物的一类,是一类形状细短,结构简单,多以二分裂方式进行繁殖的原核生物。细菌一般是单细胞,主要由细胞壁、细胞膜、细胞质、核质体等部分构成,有的细菌还有荚膜、鞭毛、菌毛等特殊结构, 缺乏细胞核、细胞骨架以及膜状胞器,例如粒线体和叶绿体。
细菌的个体非常小,绝大多数细菌的直径大小在0.5~5μm之间。目前已知最小的细菌只有0.2微米长,因此大多情况只能在显微镜下看到它们。但处于有利环境中时,细菌可以形成肉眼可见的集合体,例如菌簇。
✦数量众多、分布广泛
细菌是所有生物中数量最多的一类,据估计,其总数约有5×10的三十次方个。
细菌广泛分布于土壤和水中,或者与其他生物共生。人体是大量细菌的栖息地;可以在皮肤表面、肠道、口腔、鼻子和其他身体部位找到。据估计,人体内及表皮上的细菌细胞总数约是人体细胞总数的十倍。
此外,也有部分种类分布在极端的环境中,例如温泉,甚至是放射性废弃物中,它们被归类为嗜极生物,其中最著名的种类之一是海栖热袍菌(Thermotoga maritima),科学家是在意大利的一座海底火山中发现这种细菌的。
★ 常见的细菌
大肠埃希氏菌(Escherichia coli)
大肠杆菌是短杆菌,两端呈钝圆形,属革兰氏阴性菌,于1885年首次被发现。
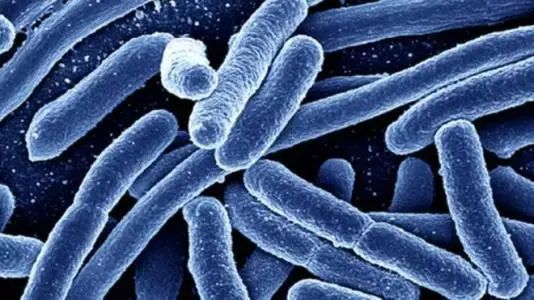
图片来源:百度百科
大肠杆菌是条件致病菌,在一定条件下可以引起多种疾病,如腹泻,肠炎,尿路感染,呼吸道感染、菌血症和其他临床感染(如新生儿脑膜炎)。
金黄色葡萄球菌(Staphylococcus aureus)
金黄色葡萄球菌也称“金葡菌”,隶属于葡萄球菌属,是革兰氏阳性菌代表,为一种常见的食源性致病微生物。
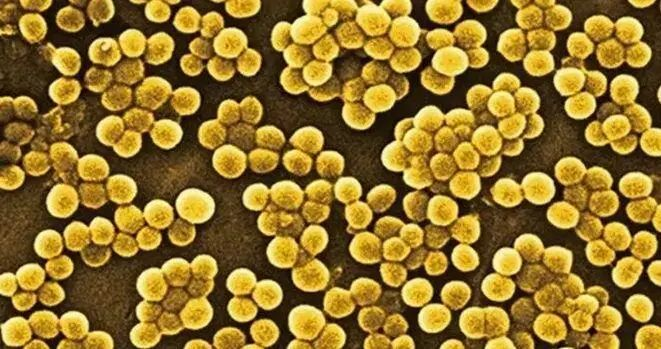
图片来源:百度
该菌最适宜生长温度为37℃,pH为7.4,耐高盐,可在盐浓度接近10%的环境中生长。金黄色葡萄球菌常寄生于人和动物的皮肤、鼻腔、咽喉、肠胃、痈、化脓疮口中,空气、污水等环境中也无处不在。
金黄色葡萄球菌是最臭名昭著、分布最广的细菌病原体之一。这种病原体可引起多种疾病,从中度严重的皮肤感染到致命的肺炎和败血症。
每年在全球范围内造成难以估计数量的无并发症皮肤感染,并可能导致数十万至数百万更严重的侵入性感染。
双歧杆菌属(Bifidobacterium)
双歧杆菌是一种革兰氏阳性、不运动、细胞呈杆状、一端有时呈分叉状、严格厌氧的细菌属,广泛存在于人和动物的消化道、阴道和口腔等生境中。双歧杆菌属的细菌是人和动物肠道菌群的重要组成成员之一。
双歧杆菌是一种重要的肠道有益微生物。双歧杆菌作为一种生理性有益菌,对人体健康具有生物屏障、营养作用、抗肿瘤作用、免疫增强作用、改善胃肠道功能、抗衰老等多种重要的生理功能。
★ 细菌对人类有利有弊
细菌也对人类活动有很大的影响。一方面,细菌是许多疾病的病原体,可以通过各种方式,如接触、消化道、呼吸道、昆虫叮咬等在正常人体间传播疾病,具有较强的传染性,对社会危害极大。
另一方面,人类也时常利用细菌,例如乳酪及酸奶和酒酿的制作、部分抗生素的制造、废水的处理等,都与细菌有关。在生物科技领域中,细菌也有着广泛的运用。
真菌
真菌(Fungus)是一种真核生物。在生物学分类上属于藻菌植物中真菌超纲。微生物中只有真菌具有真正的细胞核和完整的细胞器,故又称真核细胞型微生物。
最常见的真菌是各类蕈类,另外真菌也包括霉菌和酵母。现在已经发现了七万多种真菌,估计只是所有存在的一小半。大多真菌原先被分入动物或植物,现在成为自己的界,分为四门。
✦结构
菌体由菌丝组成,无根、茎、叶的分化,无叶绿素,不能自己制造养料,以寄生或腐生方式生活的低等生物。
真菌菌丝呈管状,多数菌丝有隔膜,此类菌丝为多细胞,隔膜中央有小孔,使细胞质、细胞核得以通过。有些真菌的菌丝无隔膜,为多核细胞。
✦分布
真菌广泛分布于全球各带的土壤、水体、动植物及其残骸和空气中,营腐生、寄生和共生生活。
★ 常见的真菌
蘑菇(Agaricus campestris)

图片来源:百度
蘑菇属于腐生真菌中的一种,其体内并没有叶绿素的存在,因此不能直接在光照下进行光合作用。蘑菇生长过程中,主要是将培养料中的各类营养物质作为营养来源,从而实现生长发育。
酵母(Saccharomyces)
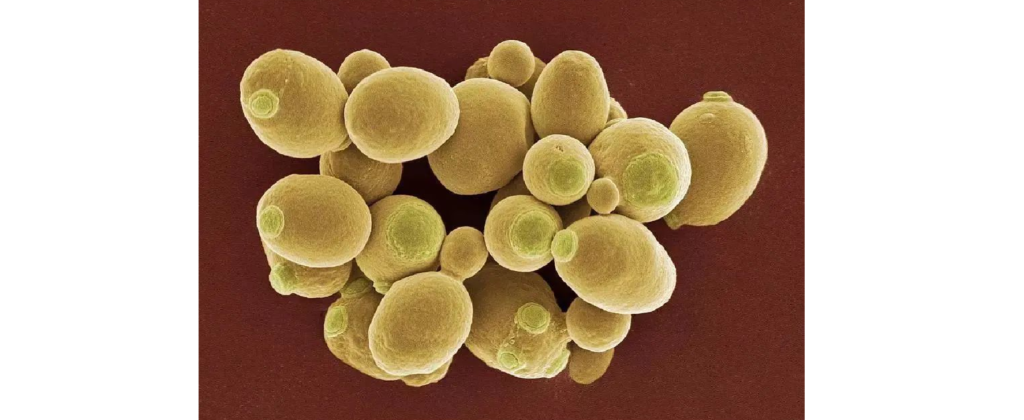
图片来源:百度
酵母菌是单细胞微生物。它属于高等微生物的真菌类。有细胞核、细胞膜、细胞壁、线粒体、相同的酶和代谢途径。酵母无害,容易生长,空气中、土壤中、水中、动物体内都存在酵母。有氧气或者无氧气都能生存。
酵母菌的作用
1、在面包、蛋糕、饼干和烤饼等这种一般的粮食制品掺入酵母菌可以提高食品的营养价值,酵母菌还在婴儿食品以及健康食品中作为食品营养的强化剂。
2、利用酵母菌可以将糖类发酵成酒精,在白酒、啤酒、果酒、黄酒等的酿造过程中都会加入酵母菌。
3、此外,大量的酵母菌还能令酒免受外界杂菌的侵害,在酵母菌发酵完成之后有澄清酒体的作用。
4、酵母菌还有入药价值,入药后的酵母菌不仅在治疗克山病和大骨节病起到辅助作用,还可以具有一定的防衰老作用。
酵母菌的危害
1、个别的酵母菌会危害生物或家庭用具,例如红酵母菌会生长在浴帘或者一些潮湿的家具上,会慢慢地腐蚀这些家具。
2、酵母菌种类中的白色假丝酵母菌,也就是俗称的白色念珠菌,主要出现在口腔、肠道、尿道和阴道等部位的粘膜上,会引起鹅口疮或尿道炎等感染疾病。
病毒
病毒(virus)是一种可以利用宿主细胞系统进行复制的微小, 无完整细胞结构的亚显微粒子。病毒不具细胞结构,无法独立生长和复制, 但病毒可以感染所有的具有细胞的生命体, 具有遗传、复制等生命特征。
✦无细胞结构
病毒主要由核酸和蛋白质外壳组成。有些病毒有囊膜和刺突,如流感病毒。病毒基因同其他生物的基因一样,也可以发生突变和重组,因此也是可以演化的。
对于病毒到底是一种生命形式,还是仅仅是一种能够与生物体作用的有机结构,人们的观点各不相同。
病毒有高度的寄生性,完全依赖宿主细胞的能量和代谢系统,获取生命活动所需的物质和能量,离开宿主细胞,它只是一个大化学分子,停止活动,可制成蛋白质结晶,为一个非生命体,遇到宿主细胞它会通过吸附,进入、复制、装配、释放子代病毒而显示典型的生命体特征,所以病毒是介于生物与非生物之间的, 一种处于“生命边缘的生物体”。
★ 常见的病毒
狂犬病毒(Rabies virus)
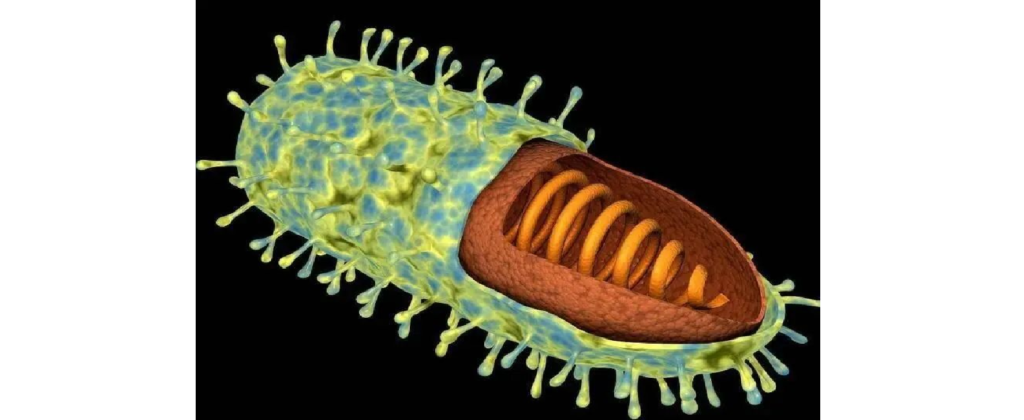
图片来源:百度
狂犬病病毒为弹状病毒,其头部为半球形,末端常为平端,形态呈典型的子弹状,长约130-240nm,直径65~80nm,内含有单链RNA。
狂犬病病毒是引起狂犬病的病原体。狂犬病毒具有两种主要抗原:一种是病毒外膜上的糖蛋白抗原,能与乙酰胆碱受体结合使病毒具有神经毒性,并使体内产生中和抗体及血凝抑制抗体,中和抗体具有保护作用;另一种为内层的核蛋白抗原,可使体内产生补体结合抗体和沉淀素,无保护作用。
冠状病毒
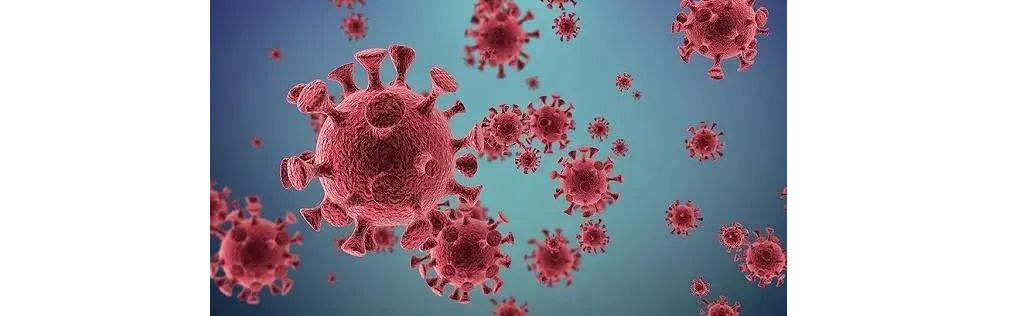
图片来源:百度
冠状病毒属的病毒是具囊膜、基因组为线性单股正链的RNA病毒,是自然界广泛存在的一大类病毒。
冠状病毒直径约80~120nm,基因组全长约27-32kb,是已知RNA病毒中基因组最大的病毒。
冠状病毒是成人普通感冒的主要病原之一,在儿童可以引起上呼吸道感染,一般很少波及下呼吸道。
冠状病毒还可以引起婴儿、新生儿急性肠胃炎,主要症状是水样大便、发热、呕吐,每天10余次,严重者可以出现血水样便。
2019新型冠状病毒(2019-nCoV ,引发新型冠状病毒肺炎COVID-19)是已知的第7种可以感染人的冠状病毒。
注:第一个已知的病毒是烟草花叶病毒,由马丁乌斯·贝杰林克于1899年发现并命名,如今已有超过5000种类型的病毒得到鉴定。研究病毒的科学被称为病毒学,是微生物学的一个分支。
细菌和真菌的名称中均有一个“菌”字,同属微生物,但两者在生物类型、结构、大小、增殖方式上却有着诸多不同。比较如下:
✦生物类型不同
一是就有无成形的细胞核来看:细菌没有核膜包围形成的细胞核,属于原核生物;真菌有核膜包围形成的细胞核,属于真核生物。
二是就组成生物的细胞数目来看:细菌全部是由单个细胞构成,为单细胞型生物;真菌既有由单个细胞构成的单细胞型生物(如酵母菌),也有由多个细胞构成的多细胞型生物(如食用菌、霉菌等)。
✦细胞结构不同
细菌和真菌都具有细胞结构,属于细胞型生物,在它们的细胞结构中都具有细胞壁、细胞膜、细胞质,但却存在诸多不同,具体表现在:一是细胞壁的成分不同:细菌细胞壁的主要成分是肽聚糖,而真菌细胞壁的主要成分是几丁质。
二是细胞质中的细胞器组成不同:细菌只有核糖体一种细胞器;而真菌除具有核糖体外,还有内质网、高尔基体、线粒体、中心体等多种细胞器。
三是细菌没有成形的细胞核,只有拟核;真菌具有。细菌没有染色体,其DNA分子单独存在;真菌细胞核中的DNA与蛋白质结合在一起形成染色体。
✦细胞大小差异
细胞大小:原核细胞一般较小,直径一般为1μm-10μm;真核细胞较大,直径一般为10μm-100μm。
✦增殖方式不同
细菌是原核生物,为单细胞型生物,通过细胞分裂而增殖,具有原核生物增殖的特有方式(二分裂);真菌为真核生物,细胞的增殖主要通过有丝分裂进行,因真菌种类的不同其个体增殖方式主要有出芽生殖(如酵母菌)和孢子生殖(食用菌)等方式。
注:尽管在细菌和真菌的名称中都有一个菌字,但细菌的名称中一般含有:球、杆、弧、螺旋等描述细菌形态的字眼,只有乳酸菌例外(实为乳酸杆菌);而真菌名称中则不含有。
✦体积差异大
细菌和病毒同属于微生物,只有在显微镜下才能看到。但两者是截然不同的东西。
细菌和病毒均属于微生物。在一定的环境条件下,细菌和病毒都可以在人体中增殖,并可能导致疾病发生。细菌较大,用普通光学显微镜就可看到,它们的生长条件也不高。病毒则较小,一般要用放大倍数超过万倍的电子显微镜才能看到。
注意:有一点值得指出的是,在人们身体的许多部位都有细菌的增殖。医学上称之为正常菌群,它们与我们和平相处,互惠互利。而在任何情况下从机体中发现病毒都非正常状况。因为只有侵入我们的活组织细胞中这些病毒才能存活。
✦结构不同
细菌是和植物一样,有细胞壁,而人的细胞是没有细胞壁的,这就是很多抗生素杀菌的原理。比如破坏它的细胞壁或者阻止合成细胞壁,细菌就死掉了,而人没有这个结构,所以对人无影响。
病毒与细菌不同之处是,病毒没有细胞结构,可以说是最低等的生物,但是它的能耐可不小,人类的疾病从小的感冒到大的癌症都和它有关系。
病毒构造很简单,外面是一层蛋白质,称为病毒外壳。蛋白质外壳内部包裹着病毒的遗传物质,可以是DNA,也可以是RNA。病毒自己不能完成新陈代谢,也不能完成繁殖,需要寄生在其它细胞内完成。
病毒没有自己的生长代谢系统,它的生存靠寄生在宿主(如人)和细胞中依赖他人的代谢系统。也是因为如此,目前抗病毒的特殊药物不多。
✦按形状分类
细菌具有不同的形状,并可根据形状分为三类,即:球菌、杆菌和螺旋菌(包括弧菌、螺菌、螺杆菌)。
✦不同生存条件
按细菌的生活方式来分类,分为两大类:自养菌和异养菌,其中异养菌包括腐生菌和寄生菌。
按细菌对氧气的需求来分类,可分为需氧(完全需氧和微需氧)和厌氧(不完全厌氧、有氧耐受和完全厌氧)细菌。
按细菌生存温度分类,可分为喜冷、常温和喜高温三类。
✦按细胞壁组成分类
细菌的结构十分简单,原核生物,没有成形的细胞核,没有膜结构的细胞器例如线粒体和叶绿体,但是有细胞壁,有的细菌还有鞭毛和荚膜,根据细胞壁的组成成分,细菌分为革兰氏阳性菌和革兰氏阴性菌。
“革兰氏”来源于丹麦细菌学家革兰,他发明了革兰氏染色。
革兰氏阳性菌具有较厚的肽聚糖细胞壁结构,在革兰氏染色试验中呈紫色/蓝色;
革兰氏阴性菌的细胞壁较薄,在革兰氏染色试验中呈红色至粉红色。
革兰氏阳性菌特征
革兰氏阳性细菌的主要特征是它们的结构。一般有以下特点:
●没有外膜。革兰氏阳性细菌没有外膜,但革兰氏阴性细菌有。
●复杂的细胞壁。包围细胞质膜的细胞壁由肽聚糖、多糖、磷壁和蛋白质组成。它很容易吸收外来物质。
●厚的肽聚糖层。在革兰氏阳性细菌中,肽聚糖有40到80层厚。
●某些表面附属物。革兰氏阳性细菌可能有鞭毛,可以帮助它们移动。它们很少有被称为菌毛的毛发状结构。
革兰氏阴性菌特征
革兰氏阳性菌和革兰氏阴性菌具有不同的结构。通常,革兰氏阴性菌具有以下特征:
●外脂质膜
●肽聚糖薄层(2-3纳米)
●通常不含磷壁酸
●可以有鞭毛或毛
小结
主要的区别是外脂膜。它很难渗透,这给了革兰氏阴性细菌额外的保护。革兰氏阳性细菌没有这种特征。
由于这种差异,革兰氏阴性细菌更难杀死。这意味着革兰氏阳性菌和革兰氏阴性菌需要不同的处理方法。
真菌较高层级的分类系统仍有很大争议,新理论不断被提出,各个分类阶层的名称均常有变动。且同一种真菌还可能在生活史的不同阶段,例如无性与有性世代拥有数种不同的学名,使真菌分类更加复杂。目前将真菌界分为4门和1类。
✦壶菌门
壶菌门(Chytridiomycota)壶菌门是游动细胞具有“9+2”结构的鞭毛,并能在水中游动的一类真菌,游动孢子具有一根后生尾鞭式鞭毛。
壶菌多水生,大多腐生在动植物残体上或寄生于水生植物、藻类、小动物和其他真菌上,少数寄生于高等种子植物上。大多数种类能分解纤维素和几丁质。
✦接合菌门
接合菌门(Zygomycota)是由低等的水生真菌发展到陆生种类,由游动的带鞭毛的孢囊孢子发展为不游动的孢囊孢子——静孢子或单孢孢子囊的分生孢子。
接合菌门菌物共同特征是有性生殖产生接合孢子。接合菌营养体为单倍体,大多是很发达的无隔菌丝体,少数菌丝体不发达,较高等的种类菌丝体有隔膜。有的种类菌丝体可以分化形成假根和匍匐丝。细胞壁的主要成分为几丁质。
✦子囊菌门
子囊菌门(Ascomycota)是真菌中最大的类群,与担子菌被称为高等真菌,生殖菌丝细胞出现较短双核阶段,其区别于其他真菌的一个特征是产生子囊。
子囊菌大都陆生,营养方式有腐生、寄生和共生。腐生的子囊菌可以引起木材、食品、布匹和皮革的霉烂以及动植物残体的分解。
✦担子菌门
担子菌门(Basidiomycota)是一类高等真菌,构成双核亚界,包含2万多种,包括蘑菇、木耳等主要食用菌。
担子菌门包括以下组:蘑菇,马勃,鬼笔科,和人体致病酵母隐球菌属等等。
担子菌门的真菌基本全为陆生品种,主要特征是由多细胞,有横隔膜的菌丝体组成,菌丝分为两种,初生菌丝体的细胞只有一个细胞核,次生菌丝体的细胞有两个核,两个核的次生菌丝体可以形成一种子实体,称为担子果,经过有性繁殖过程,在担子上生成担孢子;也可以经过无性繁殖过程生成无性孢子或出芽繁殖。
✦半知菌类
半知菌类(Deuteromycota)是一种已废止的生物分类,指在子囊菌、担子菌的同伴之中,还未发现有性繁殖阶段而在分类学上位置不明的一种临时分类。
只进行无性繁殖的菌类被称作不完全型,这一阶段被称为无性阶段。进行有性繁殖的被称为完全型,该阶段被称作有性阶段,通常有性阶段的菌类也是同时进行无性生殖的。
从遗传物质分类:DNA病毒、RNA病毒、蛋白质病毒(如:朊病毒)
RNA病毒和DNA病毒在结构、成分、复制能力和致病力等方面都不同。RNA病毒就是遗传物质是RNA的一种病毒。DNA病毒也称为脱氧核苷酸病毒,是一种生物病毒,属于原发病毒。
✦DNA病毒和RNA病毒的区别
1、结构:RNA病毒是单链病毒,比较容易发生变异。DNA病毒是双链结构的病毒,不容易发生变异,更稳定。
2、组成成分:RNA病毒是由核糖和磷酸组成,一般只有数百个或者数千个核苷酸。DNA病毒是由去氧核糖和磷酸组成,通常有上百万个核苷酸单位。
3、复制能力:RNA病毒感染人体后,在人体细胞中复制非常活跃,而复制过程中发生变异后也不修复,而是继续复制下去。这样就使RNA病毒变异非常快,不容易被攻破。比如甲型流感H1N1病毒和SARS病毒以及新冠病毒等,都是RNA病毒。
4、致病力:DNA病毒相对不容易变异,致病比较单一。由于RNA病毒相对比较容易变异,因此较容易致病。
从病毒结构分类:真病毒(Euvirus,简称病毒)和亚病毒(Subvirus,包括类病毒、拟病毒、朊病毒)
从寄主类型分类:噬菌体(细菌病毒)、植物病毒(如烟草花叶病毒)、动物病毒(如禽流感病毒、天花病毒、HIV等)
从性质来分:温和病毒(例如HIV)、烈性病毒(例如狂犬病毒)。
✦无性二分裂方式
细菌主要以无性二分裂方式繁殖,即细菌生长到一定时期,在细胞中间逐渐形成横隔,由一个母细胞分裂为两个大小相等的子细胞。
细胞分裂是连续的过程,分裂中的两个子细胞形成的同时,在子细胞的中间又形成横隔,开始细菌的第二次分裂。有些细菌分裂后的子细胞分开,形成单个的菌体,有的则不分开,形成一定的排列方式,如链球菌、链杆菌等。
分裂过程
采用电子显微镜研究细菌的分裂过程表明:细菌细胞分裂大致可经过核物质与细胞质分裂、横隔壁形成和子细胞分离等过程。
细菌细胞分裂时,核质DNA与中介体或细胞膜相连,首先DNA复制并向细胞两端移动,与此同时,细菌细胞膜向内凹陷并形成一垂直于细胞长轴的细胞质隔膜,使细胞质和核质均匀分配到两个子细胞中。
其次细胞形成横隔壁,在细胞膜不断内陷,形成子细胞各自的细胞质膜同时,母细胞的细胞壁也从四周向中心逐渐延伸。最后,逐渐形成子细胞各自完整的细胞壁。接着,子细胞分裂,形成两个大小基本相等的子细胞。
✦繁殖速度快
细菌繁殖速度快,一般细菌约20-30min便分裂一次,即为一代。接种子肉汤培养中的细菌在适宜的温度下迅速生长繁殖,肉汤很快即可变浑浊,表明有细菌的大量生长。不过也有些细菌,如结核分枝杆菌(M.tuberculosis)的繁殖速度较慢,需要15-18小时才能繁殖一代。
真菌的繁殖方式分为无性繁殖和有性繁殖两种。
✦无性繁殖
无性繁殖是指营养体不经过核配和减数分裂产生后代个体的繁殖。它的基本特征是营养繁殖通常直接由菌丝分化产生无性孢子。
常见的无性孢子有三种类型:
(1)游动孢子:形成于游动孢子囊内。游动孢子囊由菌丝或孢囊梗顶端膨大而成。游动孢子无细胞壁,具1-2根鞭毛,释放后能在水中游动。
(2)孢囊孢子:形成于孢囊孢子囊内。孢子囊由孢囊梗的顶端膨大而成。孢囊孢子有细胞壁,水生型有鞭毛,释放后可随风飞散。
(3)分生孢子(conidium)产生于由菌丝分化而形成的分生孢子梗(conidiophore)上,顶生、侧生或串生,形状、大小多种多样,单胞或多胞,无色或有色,成熟后从孢子梗上脱落。有些真菌的分生孢子和分生孢子梗还着生在分生孢子果内。孢子果主要有两种类型,即近球形的具孔口的分生孢子器(pycnidium)和杯状或盘状的分生孢子盘(acervulus)。
✦有性繁殖
真菌生长发育到一定时期(一般到后期)就进行有性生殖。有性生殖是经过两个性细胞结合后细胞核产生减数分裂产生孢子的繁殖方式。
多数真菌由菌丝分化产生性器官即配子囊,通过雌、雄配子囊结合形成有性孢子。其整个过程可分为质配、核配和减数分裂三个阶段。
第一阶段:质配阶段
即经过两个性细胞的融合,两者的细胞质和细胞核(N)合并在同一细胞中,形成双核期(N+N)。
第二阶段:核配阶段
核配阶段,就是在融合的细胞内两个单倍体的细胞核结合成一个双倍体的核(2N)。
第三阶段:减数分裂阶段
双倍体细胞核经过两次连续的分裂,形成四个单倍体的核(N),从而回到原来的单倍体阶段。
经过有性生殖,真菌可产生四种类型的有性孢子。
(1)卵孢子(oospore):卵菌的有性孢子。是由两个异型配子囊——雄器和藏卵器接触后,雄器的细胞质和细胞核经授精管进入藏卵器,与卵球核配,最后受精的卵球发育成厚壁的、双倍体的卵孢子。
(2)接合孢子(zygospore):接合菌的有性孢子。是由两个配子囊以配子囊结合的方式融合成1个细胞,并在这个细胞中进行质配和核配后形成的厚壁孢子。
(3)子囊孢子(ascospore):子囊菌的有性孢子。通常是由两个异型配子囊——雄器和产囊体相结合,经质配、核配和减数分裂而形成的单倍体孢子。子囊孢子着生在无色透明、棒状或卵圆形的囊状结构即子囊内。
每个子囊中一般形成8个子囊孢子。子囊通常产生在具包被的子囊果内。子囊果一般有四种类型,即球状而无孔口的闭囊壳,瓶状或球状且有真正壳壁和固定孔口的子囊壳,由于座溶解而成的、无真正壳壁和固定孔口的子囊腔,以及盘状或杯状的子囊盘。
(4)担孢子(basidiospore):担子菌的有性孢子。通常是直接由“+”、“-”菌丝结合形成双核菌丝,以后双核菌丝的顶端细胞膨大成棒状的担子。在担子内的双核经过核配和减数分裂,最后在担子上产生4个外生的单倍体的担孢子。
此外,有些低等真菌如根肿菌和壶菌产生的有性孢子是一种由游动配子结合成合子,再由合子发育而成的厚壁的休眠孢子(restingspore)。
✦自我复制
病毒繁殖借助宿主细胞为其提供的原料、能量和酶等必要条件,以自我复制的方式进行增殖,利用宿主细胞的核苷酸和氨基酸来自主地合成自身的一些组件,装配下一代个体。
流感病毒自我复制过程
1.病毒体附着到宿主细胞表面并通过胞吞进入细胞;2.衣壳分解后,病毒核糖核蛋白转运入核;3a.病毒基因组转录;3b.病毒基因组复制;4.新合成的病毒mRNA出核并完成翻译;5a.合成的核蛋白入核与新复制的核酸结合;5b.合成的病毒表面蛋白进入高尔基体完成翻译后修饰并转运上膜;6.新形成的核衣壳进入细胞质并与插有病毒表面蛋白的细胞膜结合;7.新生成的病毒体通过出泡方式离开宿主细胞。
✦细菌因素
主要与病原菌的毒力和数量有关。毒力强或数量多的致病菌进入机体,引起败血症的可能性较大。
注:败血症是指各种致病菌侵入血液循环,并在血中生长繁殖,产生毒素而发生的急性全身性感染。
✦人体因素
细菌侵入人体后是否引起感染,与人的防御、免疫功能有关。
•皮肤和黏膜是抵御细菌的有效武器
完整的皮肤和粘膜是防止细菌侵入人体的天然屏障,破损后细菌易于从此处侵入体内,挤压皮肤炎症部位或脓肿时细菌侵入的可能性更大。
严重烧伤时,创面为细菌敞开门户,皮肤坏死、血浆渗出又为细菌繁殖提供了良好环境,故极易发生感染。尿路、胆道、胃肠道、呼吸道粘膜受破坏后,若同时有内容物积滞、压力增高,细菌更易进入血中。保留导尿管、静脉等血管内留置导管、人工辅助呼吸时插管等,也使细菌易于侵入。
•免疫细胞能清除细菌
人体免疫功能正常时,进入血中的细菌迅速被血中免疫细胞如单核细胞、嗜中性粒细胞等所清除,而患肝硬变、糖尿病、血液病、结缔组织病等慢性病者,可因代谢紊乱、体液免疫及细胞免疫功能减低,易导致细菌感染发生;各种免疫抑制药物的使用、放射治疗亦是导致细菌感染发病率高的原因。
注:广谱抗菌药物使用后,对药物敏感的细菌虽被抑制或杀灭,而一些耐药菌乘机繁殖,亦可酿成细菌感染。
★细菌病
由细菌引起的疾病有许多,如:伤寒和副伤寒、细菌性食物中毒、 细菌感染性腹泻、霍乱、弯曲菌感染(弯曲菌肠炎、幽门螺杆菌感染) 细菌性痢疾、鼠疫、炭疽、白喉、百日咳、猩红热、流行性脑脊髓膜炎、结核病、人感染猪链球菌病、破伤风、败血症等。
✦症状
原发炎症:各种病原菌所引起的原发炎症与其在人体的分布部位有关。原发炎症的特点是局部的红、肿、热、痛和功能障碍。
皮疹:见于部分患者,以瘀点最为多见,多分布于躯干、四肢、眼结膜、口腔粘膜等处,为数不多。
关节症状:可出现大关节红、肿、热、痛和活动受限,甚至并发关节腔积液、积脓,多见于革兰阳性球菌、脑膜炎球菌、产碱杆菌等败血症的病程中。
感染性休克:约见于1/5~1/3败血症患者,表现为烦燥不安,脉搏细速,四肢厥冷,皮肤花斑,尿量减少及血压下降等,是严重败血症所致。
除外伤性、手术后、挤压疮疖后发生的败血症有较明显的潜伏期外,大多发病急骤。
注意
由于新生儿及老年患者具有不同的生理特点,其败血症亦各有特征。
•新生儿免疫力弱,发病率高
新生儿的皮肤粘膜屏障功能、淋巴及单核吞噬细胞系统功能尚不健全,补体尚缺乏,体液免疫水平低,细胞免疫也未完善;脐带残端为细菌入侵创造了有利条件,孕母泌尿生殖道感染或全身感染等均可使新生儿败血症发病率高、表现复杂、并发症多。
•老年人发病往往比较严重
老年人败血症的发病率有增高的趋势,由于机体反应性差,早期临床表现较隐蔽,热型往往不规则;又因免疫功能低下,病情常较严重,进展迅速且不易控制;老年人脏器功能多有减退或原有慢性病,败血症极易诱发脏器功能衰竭。
故新生儿及老年败血症预后差、死亡率亦高。及时发现新生儿和老年败血症,主要依据年龄特点和提高警惕。
真菌感染性疾病根据真菌侵犯人体的部位分为4类:浅表真菌病、皮肤真菌病、皮下组织真菌病和系统性真菌病;前二者合称为浅部真菌病,后二者又称为深部真菌病。
真菌感染多为继发性感染,由机会致病性真菌引起,特别是深部真菌感染多是由于各种诱因使机体免疫功能显著下降所致。
某些真菌如白假丝酵母菌、烟曲霉中可产生高分子强毒素或低分子毒素,这些毒素也会在治病中起到一定作用。另外,真菌的黏附能力,对免疫系统功能的抑制及胞壁中的酶类也与致病性有关。
诱发因素:发烧、创伤、肿瘤、严重其他微生物感染等。
✦浅部真菌感染
主要是由于人体接触所致,如皮肤廯菌、角层癣菌等皮肤感染真菌,多具有嗜角质性,可分解细胞的角蛋白和脂质,还可通过机械刺激和代谢产物作用,引起局部病变。
✦深部真菌感染
多发生于人体抵抗力下降或菌群失调时,为继发性感染,常见菌属有白色念珠菌、新生隐球菌、肺孢子菌、曲霉及毛菌等,因患者抵抗力较差,治疗效果往往不佳。
✦系统性真菌感染
在机体抵抗力低下时致病,如患有白血病、淋巴瘤、糖尿病等疾病或有长期大量广谱抗生素、激素使用史时,许多条件致病菌、如念珠菌、曲霉、毛霉等感染后在体内发展繁殖,从而引起系统性真菌感染。
✦症状
•浅表真菌病
感染仅仅局限于皮肤角质层的最外层,极少甚至完全没有组织反应,感染毛发时也只累及毛发表面,很少损伤毛发。
主要包括:花斑癣、掌黑癣和毛结节菌病。
•皮肤真菌病
感染累及皮肤角质层和皮肤附属器,如毛发、甲板等,能广泛破坏这些组织的结构并伴有不同程度的宿主免疫反应;这类真菌感染中最常见的是皮肤癣菌病,其他真菌引起的感染还包括皮肤念珠菌病等。
皮肤癣菌病根据不同的发病部位可以分为足癣(俗称”脚气”)、手癣、体癣、股癣、甲癣以及头癣等各类癣病;在世界范围内广泛发生,是最常见的真菌性疾病,发病率高。
•皮下真菌病
感染皮肤、皮下组织,包括肌肉和结缔组织,一般不会经血液流向重要脏器播散;但有些感染可以由病灶向周围组织缓慢扩散蔓延,如足菌肿等;也有些则沿淋巴管扩散,如孢子丝菌病、着色芽生菌病。免疫受损患者的皮下真菌具有潜在的播散全身的危险。
•系统性真菌病
除侵犯皮肤和皮下组织外,还累及组织和器官,甚至引起播散性感染,又称为侵袭性真菌感染。
近年来,随着高效广谱抗生素、免疫抑制剂、抗恶性肿瘤药物的广泛应用,器官移植、导管技术以及外科其他介入性治疗的深入开展,条件致病性真菌引起的系统性真菌病日益增多,新的致病菌不断出现,病情也日趋严重。
主要包括念珠菌病、曲霉病、隐球菌病、接合菌病和马内菲青霉病等。
病毒通过多种途径侵入机体,并在易感的宿主细胞中增殖。
✦水平传播
水平传播是指病毒在人群中不同个体之间的传播,包括病毒从动物到人的传播。常见的水平传播方式有以下几种。
(1)经呼吸道传播:病毒经空气、飞沫等吸入感染,如流感病毒、风疹病毒等。
(2)经消化道传播:病毒污染了食物和水源,经口食入而感染。如甲型肝炎病毒、脊髓灰质炎病毒等。
(3)经泌尿生殖道传播:由直接性接触而感染,如人类免疫缺陷病毒、单纯疱疹病毒等。
(4)经皮肤伤口传播:经昆虫媒介的叮咬、动物咬伤或皮肤伤口直接接触病毒而感染。如流行性乙型脑炎病毒、狂犬病病毒等。
(5)经血液传播:经输血或血液制品,包括经注射、器官移植等途径引起的感染,如乙型肝炎病毒、人类免疫缺陷病毒等。
✦垂直传播
病毒经胎盘、产道、哺乳由母亲传给胎儿或新生儿的方式,称为垂直传播。可经垂直传播的病毒有风疹病毒、人类免疫缺陷病毒、乙型肝炎病毒等。
✦症状
机体感染病毒后,可表现出不同的临床类型。依据有无症状,可分为显性感染和隐性感染;依据病毒滞留时间及症状持续时间长短,又可分为急性感染和持续性感染。
•隐性感染
由于侵入机体的病毒数量较少、毒力较弱或机体的抵抗力较强,病毒在宿主细胞内增殖,但机体不出现明显的临床症状,称为隐性感染。
隐性感染可使机体获得对该病毒的特异性免疫,保护机体免受该病毒的再次感染。隐性感染虽不出现临床症状,但病毒仍在体内增殖并向外界传播病毒,成为重要的传染源。
•显性感染
由于侵入机体的病毒数量较多、毒力较强或是机体的抵抗力较弱,病毒在宿主细胞内大量增殖,出现明显的临床症状,称为显性感染。显性感染根据感染持续时间长短。分为急性感染和持续性感染。
急性感染:病毒侵入机体后,其潜伏期短、发病急、病程数日至数周,病后常可获得特异性免疫力,机体可通过自身的免疫机制把病毒完全清除出体外,如甲型肝炎病毒。
持续性感染:病毒侵入机体后,在体内持续存在数月、数年,甚至数十年,机体可出现临床症状,也可不出现临床症状而长期带有病毒,成为重要的传染源。持续感染按病程、致病机制的不同,可分为三种。
①慢性感染:病毒侵入机体后,长期存在于血液或组织中,机体可出现症状,也可不出现症状。在整个病程病毒均可被查出,如乙型肝炎病毒引起的慢性肝炎。
②潜伏感染:原发感染后,病毒基因潜伏在机体一定的组织或细胞中,但不复制增殖出具有感染性的病毒,此时机体既没有临床症状,也不会向体外排出病毒。在某些条件下病毒可被激活而急性发作,并可检测出病毒,如单纯疱疹病毒。
③慢发病毒感染:经显性或隐性感染后,病毒长时间潜伏在机体内,潜伏期可长达数月至数年,此时机体一般无症状,一般也检测不出病毒。一旦发病,则呈亚急性进行性加重直至死亡,如人类免疫缺陷病毒的感染。
抗细菌感染的免疫是指机体抵御细菌感染的能力,是由机体的非特异性免疫和特异性免疫共同协调来完成的。
先天具有的非特异性免疫包括机体的屏障结构,吞噬细胞的吞噬功能和正常组织及体液中的抗菌物质;后天获得的特异性免疫包括以抗体作用为中心的体液免疫和致敏淋巴细胞及其产生的淋巴因子为中心的细胞免疫。
病原菌侵入机体后,由于其生物学特性的不同,致病物质的不同。机体对它们的免疫反应也各有差别。
✦宿主体表的防御功能
(一)机械的阻挡和排除作用
健康和完整的皮肤与粘膜能有效地阻挡细菌的侵入。
呼吸道粘膜上皮细胞的纤毛向上颤动,可将细菌咳出或咽下;随粪便每日约排菌1012个;小便可清除尿道上皮的细菌。
(二)分泌液中化学物质的局部抗菌作用
汗腺分泌的乳酸,皮脂腺分泌的脂肪酸均有一定的抗菌作用。
胃酸能杀死寒杆菌、痢疾杆菌和霍乱弧菌。阴道分泌物中的酸类亦有抗菌作用。前列腺分泌的精素是正常精液中存在的对革兰氏阳性细菌有效的抑制物。泪液、唾液、乳汗和呼吸道分泌物中广泛分布的溶菌酶能溶解革兰氏阳性细菌。
(三)正常菌群的拮抗作用
人体表以及与外界相通腔道中的正常菌群,可以通过它们的代谢产物对抗病原菌入侵。
例如皮肤上的痤疮丙酸菌(Propionibacterium acnes)能产生抗菌性脂类、抑制金黄色葡萄球菌和化脓性链球菌在皮肤上生长;肠道中的某些厌氧菌能产生脂肪酸阻止沙门氏菌在局部生存;肠道中大肠杆菌产生的大肠菌毒和酸性产物能抑制痢疾杆菌、金黄色葡萄球菌;咽部的草绿色链球菌(Viridans Streptococci)似能阻止肺炎球菌在局部生长;鼻腔的表皮葡萄球菌和类白喉杆菌能妨碍金黄色葡萄球菌定居等。当这种拮抗作用受影响时,则可发生菌群失调症。
✦机体抗毒性免疫
抗毒性免疫是一种以体液抗体为主的免疫应答。许多以外毒素致病的病原菌造成的感染,如白喉、破伤风、气性坏疽及内毒中毒等,机体的免疫应答,主要表现为抗毒素(lgG)中和毒素的作用。
由抗毒素与外毒素特异结合形成的复合物,可被吞噬细胞吞噬,并将其降解消除。抗毒素与毒素结合,可以通过空间阻碍使毒素不能吸附到敏感的宿主细胞(受体)上,或者使毒素生物学活性部位(酶)被封闭,从而使毒素不能发生毒性作用。
注意:抗毒素不能对已与组织结合的毒素起中和作用。
// 建议
根据外毒素的免疫特点,可应用类毒素进行预防接种,应用抗毒素血清进行早期治疗与紧急预防,使用时要保证“早期足量”。
✦机体的抗菌性免疫
病原侵入机体后,由于其生物学特征的不同,可分为胞外菌感染和胞内菌感染两类,机体对这两类感染的免疫反应是有差别的。
(一)胞外寄生菌的抗感染免疫
1.抗体对细菌繁殖的抑制作用:抗体与细菌结合,可以出现凝集和鞭毛制动现象,但一般而言,对细菌的活力只有微弱的影响,甚至没有影响。如果抗体的结合能抑制细菌的重要酶系统或代谢途径,则可能抑制细菌的生长。
例如,某些细菌(例如败血巴氏杆菌)从血清转铁蛋白摄取铁的能力可被特异性抗体封闭,从而导致细菌生长受抑制。
2.抗体对细菌吸附作用的抑制:病原菌吸附到粘膜上皮细胞是造成感染的先决条件。粘膜表面的抗体,在防止病原菌对粘膜的侵犯中具有更重要的作用。
在粘膜表面起这种作用的抗体主要是SlgA它是局部免疫的主要因素。SlgA抗细菌感染可有以下几种方式:在补体和溶菌酶的参与下溶解某些细菌;在肠道局部增强吞噬作用;防止细菌对粘膜上皮细胞的吸附。
例如SlgA能阻止链球菌、致病性大肠杆菌、霍乱弧菌、淋球菌、百日咳杆菌等对粘膜表面的吸附。
3.抗体和补体对细菌的溶解作用:在许多感染中,机体能产生相应抗体(lgG、lgM、lgA),当细菌表面抗原和lgG、lgM结合的免疫复合物一旦通过经典途径使补体活化或由分泌型 lgA或聚合的血清lgA通过替代途径活化补体,即可引起细胞膜的损伤,最终发生溶菌。
实验证明补体的溶菌作用仅对革兰氏阴性菌,其中包括霍乱弧菌、大肠杆菌、痢疾杆菌、伤寒杆菌等发挥作用。但这种作用往往并不彻底,仅使杆菌菌体膨大或变为球形,不引起溶解。但若于试验中系统中加入适量的溶菌酶,则可出现溶菌现象。
4.抗体和补体对吞噬作用的调理:抗体和补体单独能适当的靶细胞起调理吞噬作用,若两者联合作用效应更加强大。中性粒细胞和单核吞噬细胞表面具有lgG的Fc受体。当lgG通过其特异性抗原结合部位(Fab)与细菌表面相应抗原结合后,其Fc段可与吞噬细胞表面相应Fc受体结合,即可在细菌与吞噬细胞间形成抗体“桥梁”,这不仅能促进吞噬细胞对细菌的吞噬,而且有助于强化细胞内的杀菌作用。
注:中性粒细胞和单核细胞表面还有C3b 受体。因此,细菌与所有能结合补体的抗体(lgg 、lgM )形成的复合物,均可激活补体形成活化产物C3B,从而发挥调理吞噬作用。尤以lgM 的作用更强,此作用在感染的早期特别重要,因为此时lgM抗体占优势。
(二)胞内寄生菌的细胞免疫
凡侵入人体后大部分时间停留在宿主细胞内并繁殖的病原菌称胞内寄生菌。例如结核杆菌、麻风杆菌、布氏杆菌等均属此类。
由于抗体不能进入细胞内,所以体液免疫对这类细菌感染的作用受到限制,对胞内感染的防御功能主要靠细胞免疫。例如机体初次感染结核杆菌,由于细胞免疫尚未建立,吞噬细胞虽可将它们吞噬,但不能有效地消化杀灭,因此病原菌容易随吞噬细胞在体内扩散,蔓延,而造成全身感染。
但在传染过程中,机体在病原菌的刺激下逐渐形成细胞免疫,通过致敏淋巴细胞释放的各种淋巴因子,激活吞噬细胞,可增强其吞噬消化能力,抑制病原菌在吞噬细胞内生存,从而获得防御同种病种原菌再感染的免疫力。
✦非特异性免疫
人类对真菌感染有天然免疫力。包括皮肤分泌短链脂肪酸和乳酸的抗真菌作用,血液中转铁蛋白扩散至皮肤角质层的抑真菌作用;中性粒细胞和单核巨噬细胞的吞噬作用,以及正常菌群的拮抗作用。
注意:许多真菌病受生理状态影响,如婴儿对念珠菌病易感,学龄前儿童易患头癣。
✦特异性免疫
•细胞免疫排菌杀菌
真菌感染中细胞免疫是机体排菌杀菌及复原的关键,T细胞分泌的淋巴因子对加速表皮角化和皮屑形成,随皮屑脱落,将真菌排除;以T细胞为主导的迟发型变态反应引起免疫病理损伤能局限和消灭真菌,以终止感染。
一般反应强度与体内菌量呈反比,如阴性则菌量增加,病情严重,而经治疗又转阳性,说明治疗见效,预后良好。
•体液免疫具有保护作用
体液免疫对部分真菌感染有一定保护作用,如特异性抗体可阻止真菌转为菌丝相以提高吞噬细胞的吞噬率;抗白色念珠菌抗体与菌表面甘露醇蛋白质复合物结合,阻止本菌粘附宿主细胞;全身性白色念珠菌感染,尽管其迟发型变态反应阳性,或通过被动转移致敏淋巴细胞,还必须同时输入特异抗体才起保护作用。
注意:而DTH反应阴性者即使有抗体,不能引起保护作用,表明抗体须在具有良好的细胞免疫基础的机体内才发生保护作用。
DTH指的是迟发型超敏反应,主要是T细胞与相应抗原作用后,引起单个核细胞浸润以及组织细胞损伤为主的炎症反应。
✦先天性免疫
人体抵御病毒的第一道防线是先天性免疫系统。这一免疫系统由能够抵御非特异性病毒感染的细胞和其他机制组成,即以一种通用方式来对入侵的病原体做出识别和反应,但不同于获得性免疫系统,这一免疫系统并不产生持久的或保护性的免疫。
RNA干扰是对抗病毒的一种重要的先天性防御机制。
✦体液免疫
当人体的获得性免疫系统探测到病毒时,会产生特异性的抗体来与病毒结合并使其失去感染性,这种作用被称为体液免疫。
其中,有两类抗体非常重要。第一类被称为IgM(免疫球蛋白M),它能高效地使病毒去活,但免疫系统的细胞产生IgM的时间只有几个星期。第二类被称为IgG(免疫球蛋白G),它能够被免疫系统不停地制造出来。
IgM存在于宿主的血液中是用于急性感染的情况,而IgG的存在则表明过去曾经受到某种感染(用于防御以后的同类感染)。进行免疫性测试时,通常是对体内的IgG型抗体进行测量。
✦细胞免疫
人体对抗病毒的另一道防线是细胞免疫,包括了被称为T细胞的免疫细胞。人体中的细胞不断地将其内部蛋白质的片断展示在细胞表面(抗原呈递)供T细胞来进行检查,一旦T细胞识别出可能的病毒片断,那么对应的细胞就会被病毒特异性T细胞扩增所消灭。
诸如巨噬细胞在内的一些细胞专门负责抗原呈递。制造干扰素是一种重要的宿主防御机制。干扰素是病毒感染之后由机体所产生的一种激素,它在免疫中的作用较为复杂,可以确定的是它能够通过杀死受感染细胞及其邻近细胞来逐步阻止病毒的复制。
注:并非所有的病毒感染都会引起保护性免疫反应。例如,艾滋病毒可以通过不断地变换其病毒体表面蛋白的氨基酸序列来逃避免疫系统的打击。这些顽固的病毒采用多种方式来逃脱免疫系统的控制,如隔离、阻断抗原呈递、产生细胞因子抗性、逃避自然杀伤细胞的作用、逃脱细胞凋亡以及抗原转移。其他一些病毒,如向神经病毒,可以通过神经来传播,而在神经系统中免疫系统可能无法接触到它们。
✦预防
细菌感染的预防需要重点关注防止感染,对于具有传播性质的疾病,应从传染源、传播途径和易感人群三个基本环节中进行防控。
1、注意卫生:日常生活中注意饮食卫生,家庭居所最好日常进行消毒、灭菌,防止带入外界环境的致病菌。
2、接种疫苗:接种疫苗后可使机体产生免疫应答,产生特异性抗体,使机体获得针对病原疫苗的免疫力,应及时接种相关疫苗提高自身抗细菌感染能力。
3、体育锻炼:适当从事体育锻炼可以提高免疫力,可根据自身情况选择打太极拳、健身、跑步、郊游等,劳逸结合。
4、远离传染源:有传染性疾病的人群应做好隔离措施,其他人群也应做好防护措施,避免与其接触,避免到细菌感染流行病区。
小结
经常保持皮肤和粘膜的清洁和完整,避免创伤,控制慢性病,合理使用免疫抑制剂和抗生素类药物,烧伤病房应严格消毒等措施,均可预防细菌感染。
一切明显的或隐匿的化脓性病灶如能及早予以清除,感染的发生就可以减少。小儿时常见的传染病如麻疹、流行性感冒、百日咳等每易继发较重的呼吸道细菌感染,从而发生细菌感染。对这类病儿,必须加强保护。对不论多么细小的皮肤创伤必须予以重视,早作适当处理。
✦治疗
一般治疗:卧床休息,加强营养,补充适量维生素。维持水、电解质及酸碱平衡。必要时给予输血、血浆、白蛋白和丙种球蛋白。高热时可给予物理降温,烦躁者给予镇静剂等。
病原治疗:及时选用适当的抗菌药物是治疗的关键。应注意早期、足量并以杀菌剂为主;一般两种抗菌药物联合应用,多自静脉给药;首次剂量宜偏大,注意药物的半衰期,分次给药;疗程不宜过短,一般三周以上,或热退后7~10天方可酌情停药。
局部病灶的处理:化脓性病灶不论原发性或迁徙性,均应在使用适当、足量抗生素的基础上及时行穿刺或切开引流。化脓性胸膜炎、关节脓肿等可在穿刺引流后局部注入抗菌药物。胆道及泌尿道感染有梗阻时应考虑手术治疗。
建议
关键在于及时选用适当的抗菌药物,并予以休息及适量的营养。诊断基本肯定后应尽早治疗,在培养未获阳性结果前可根据细菌入侵途径及临床表现推测致病菌的种类给药,若获阳性培养而治疗效果欠佳时,则可按药物敏感试验选用适宜抗菌药物。
革兰氏阳性球菌感染者可选用青霉素、红霉素、头孢菌素等;革兰氏阴性杆菌感染则选用庆大霉素、丁胺卡那霉素、头孢菌素及半合成广谱青霉素;厌氧菌感染则首选甲硝唑,也可选用青霉素、氯霉素、氯洁霉素等;败血症确为真菌所致则应选用二性霉素。
此外,正确处理局部病灶及各阶段的突出矛盾(如感染性休克、弥漫性血管内凝血、心肾功能不全)亦很重要。
✦预防
1.保持皮肤干燥和清洁。
2.穿着宽松的衣服。
3.避免共用发刷,梳子和毛巾,因为它们可能含有与皮肤真菌菌落片段。
4.为避免脚气,应该使用备用鞋,每2、3天换洗。
5.尽量选择天然纤维制品的衣服,如棉花、蚕丝,使皮肤呼吸。
6.糖尿病患者应控制血糖水平。疾病有可能会导致免疫功能低下,增加了真菌感染的概率。
✦治疗
•大多真菌感染症状较轻微
除头癣和甲癣外,大多数真菌感染症状都较轻微,常用抗真菌霜剂治疗。一般不用抗真菌粉剂。抗真菌药物的活性成分有咪康唑、克霉唑、益康唑和酮康唑等。
一般霜剂每天涂敷两次,治疗持续到皮损消退后7~10天。如果霜剂停用太快,感染并未消除,皮疹又会复发。
抗真菌霜剂要在使用几天后才显效,其间可用皮质类固醇霜剂缓解瘙痒和疼痛。严重的或顽固性感染,可用灰黄霉素治疗几个月,有时同时用抗真菌霜剂。
•部分口服药物会引起副作用
口服灰黄霉素很有效,但可引起副作用,如头痛、胃肠道功能紊乱、光敏、水肿和白细胞减少等。停用灰黄霉素后,感染可能复发。皮肤真菌感染也可用酮康唑治疗。与灰黄霉素一样,口服酮康唑也有严重的副作用,包括肝脏损害。
保持感染部位清洁、干燥有助于抑制真菌繁殖,促进皮肤愈合。感染处应经常用肥皂和水清洗,擦干后扑撒滑石粉。避免使用含玉米粉的粉剂,因为它容易促进真菌生长。
注意
如果真菌感染有渗液,可能并发了细菌感染。需要用抗生素治疗。涂敷抗生素霜剂或口服抗生素。稀释醋酸铝溶液或怀特菲尔德软膏也可用来使渗液的皮肤干燥。
由于病毒使用了宿主细胞来进行复制并且寄居其内,因此很难用不破坏细胞的方法来杀灭病毒。现在最积极的对付病毒疾病的方法是疫苗接种来预防病毒感染或者使用抗病毒药物来降低病毒的活性以达到治疗的目的。
部分病毒感染以对症支持治疗为主,如普通感冒,甲型肝炎、病毒性胃肠炎等,可通过补液维持体内水分、电解质和酸碱平衡。
✦疫苗接种预防感染
疫苗接种是一种廉价而又有效的防止病毒感染的方法。早在病毒被发现之前,疫苗就已经为人们用于预防病毒感染。随着疫苗接种的普及,病毒感染相关的一些疾病(如小儿麻痹、痳疹、腮腺炎和风疹)的发病率和死亡率都大幅度下降,而曾经是致命疾病的天花已经绝迹。
目前各类疫苗可以预防超过30种对人体的病毒感染,而有更多的疫苗被用于防止动物受到的病毒感染。
疫苗的成分可以是活性降低或死亡的病毒,也可以是病毒蛋白质(抗原)。活疫苗包含了活性减弱的可致病的病毒,这样的病毒被称为“减毒”病毒。
注意
虽然活性减弱,但活疫苗对于那些免疫力较弱或免疫缺陷的人可能是危险的,对他们注射活疫苗可能反而会导致疾病。
生物技术和基因工程被用于改造病毒疫苗,改造后的疫苗(即亚单位疫苗)只含有病毒的衣壳蛋白,如乙肝疫苗。由于不含有病毒核酸,因此亚单位疫苗对于免疫缺陷的病人是安全的。
对于活疫苗的安全性也有一些例外,如黄热病毒疫苗,虽然是一种减毒病毒株(被称为17D),却可能是目前所有疫苗中最安全和最有效的。
✦治疗
•抗病毒药物
在过去的二十年间,抗病毒药物的发展非常迅速。艾滋病的不断蔓延推动了对抗病毒药物的需求。抗病毒药物常是核苷类似物,当病毒复制时如果将这些类似物当作核苷用于合成其基因组就会产生没有活性的病毒基因组(因为这些类似物缺少与磷相连能够相互连接形成DNA“骨架”的羟基,会造成DNA的链终止),从而抑制病毒的增殖。
核苷类似物作为抗病毒药物的例子包括阿昔洛韦,可用于抑制单纯疱疹病毒感染,和拉米夫定,可用于治疗艾滋病和乙型肝炎。阿昔洛韦是最早出现也是最经常被指定使用的抗病毒药物。其他使用中的抗病毒药物是针对病毒生活周期的不同阶段。艾滋病毒需要依赖一种被称为HIV-1蛋白酶的作用来获得完整的感染能力;而通过使用大量的蛋白酶抑制剂类的药物可以使这种酶失活。
•“吃病毒”生物
近日,美国的研究人员称,他所在的研究团队于近日首次发现了有生物会把“病毒”作为“食物”。他们研究后发现,两种浮游生物——“Halteria”和“Paramecium”可以主动食用病毒并茁壮成长。
研究发现,“Halteria”在两天内就有明显成长的迹象,纤毛的种群在两天内增长了约15倍,而氯病毒含量则下降了100倍以上。而在没有氯病毒的对照样本中,“Halteria”与初始状态相差无几。“Paramecium”也有类似表现,同样把氯病毒作为营养来源。并且,标记在氯病毒DNA的荧光绿移动痕迹证实病毒被“吃掉”了。
从科学上讲,这是人类第一次改变看待病毒的方式:病毒不仅是导致机体发生病变的“病原体”,还可以是自然界食物链中的一环。这可能对未来治疗病毒相关疾病具有重要作用。
细菌、真菌、病毒等生物共同构成了我们生活的世界。它们基本上无处不在,也时刻影响着我们的生命活动。而人体内的肠道菌群是其中数量最庞大的一类,其对人们的影响巨大。
微生物检测技术可以较为清晰地发现生活中的一些细菌和其他微生物,有助于我们判断健康状况,并根据此来做出一些调整。
随着测序技术和其他体外诊断技术的快速发展,新标志物的发现等新技术的发展将能更快更精准的区分和诊断感染病原,甚至大大提高用药的针对性和减少无效用药和耐药性,将有利于人类和微生物更好的共存。
主要参考文献:
Kwiecinski JM, Horswill AR. Staphylococcus aureus bloodstream infections: pathogenesis and regulatory mechanisms. Curr Opin Microbiol. 2020 Feb;53:51-60. doi: 10.1016/j.mib.2020.02.005. Epub 2020 Mar 12. PMID: 32172183; PMCID: PMC7244392.
Fisher CR, Streicker DG, Schnell MJ. The spread and evolution of rabies virus: conquering new frontiers. Nat Rev Microbiol. 2018 Apr;16(4):241-255. doi: 10.1038/nrmicro.2018.11. Epub 2018 Feb 26. PMID: 29479072; PMCID: PMC6899062.
Riley LW. Distinguishing Pathovars from Nonpathovars: Escherichia coli. Microbiol Spectr. 2020 Dec;8(4). doi: 10.1128/microbiolspec.AME-0014-2020. PMID: 33385193.
Cheung GYC, Bae JS, Otto M. Pathogenicity and virulence of Staphylococcus aureus. Virulence. 2021 Dec;12(1):547-569. doi:
10.1080/21505594.2021.1878688. PMID: 33522395; PMCID: PMC7872022.
Liu N, Pang X, Zhang H, Ji P. The cGAS-STING Pathway in Bacterial Infection and Bacterial Immunity. Front Immunol. 2022 Jan 13;12:814709. doi: 10.3389/fimmu.2021.814709. PMID: 35095914; PMCID: PMC8793285.
Klein EY, Monteforte B, Gupta A, Jiang W, May L, Hsieh YH, Dugas A. The frequency of influenza and bacterial coinfection: a systematic review and meta-analysis. Influenza Other Respir Viruses. 2016 Sep;10(5):394-403. doi: 10.1111/irv.12398. Epub 2016 Jun 24. PMID: 27232677; PMCID: PMC4947938.
Piepenbring M, Maciá-Vicente JG, Codjia JEI, Glatthorn C, Kirk P, Meswaet Y, Minter D, Olou BA, Reschke K, Schmidt M, Yorou NS. Mapping mycological ignorance – checklists and diversity patterns of fungi known for West Africa. IMA Fungus. 2020 Jul 7;11:13. doi: 10.1186/s43008-020-00034-y. PMID: 32699745; PMCID: PMC7341642.
Wu HY, Chang PH, Huang YS, Tsai CS, Chen KY, Lin IF, Hsih WH, Tsai WL, Chen JA, Yang TL, Lee CY, Ho TS, Wang HW, Huang SF, Wu AY, Chen HJ, Chen YC, Chen WC, Tseng CH, Lin PC, Yang CH, Hong PL, Lee SS, Chen YS, Liu YC, Wang FD; Infectious Disease Society of Taiwan; Medical Foundation in Memory of Dr. Deh-Lin Cheng; Foundation of Professor Wei-Chuan Hsieh for Infectious Diseases Research and Education; CY Lee’s Research Foundation for Pediatric Infectious Diseases and Vaccines,; 7th Guidelines Recommendations for Evidence-based Antimicrobial agents use in Taiwan (GREAT) working group; Members of the expert panel and board members of the IDST are listed in alphabetical order. Recommendations and guidelines for the diagnosis and management of Coronavirus Disease-19 (COVID-19) associated bacterial and fungal infections in Taiwan. J Microbiol Immunol Infect. 2022 Dec 21:S1684-1182(22)00284-5. doi: 10.1016/j.jmii.2022.12.003. Epub ahead of print. PMID: 36586743; PMCID: PMC9767873.
Dickson RP, Singer BH, Newstead MW, Falkowski NR, Erb-Downward JR, Standiford TJ, Huffnagle GB. Enrichment of the lung microbiome with gut bacteria in sepsis and the acute respiratory distress syndrome. Nat Microbiol. 2016 Jul 18;1(10):16113. doi: 10.1038/nmicrobiol.2016.113. PMID: 27670109; PMCID: PMC5076472.
de Wilde AH, Snijder EJ, Kikkert M, van Hemert MJ. Host Factors in Coronavirus Replication. Curr Top Microbiol Immunol. 2018;419:1-42. doi: 10.1007/82_2017_25. PMID: 28643204; PMCID: PMC7119980.
谷禾健康
癌症是一种复杂的疾病,归因于多因素变化,导致治疗策略困难。
90%的癌症患者死于复发或转移。癌症转移是恶性肿瘤进展的关键步骤,由癌细胞内在特性和外在环境因素决定。
一些微生物组通过诱导癌性上皮细胞和慢性炎症促进癌发生、癌症进展和调节癌症治疗。
关于微生物群在肿瘤发生和临床效率中的作用的大部分认知都与肠道微生物群有关。
然而,研究也证实了肿瘤内微生物群在癌症中的作用。近年来,肿瘤内微生物群已被确定为肿瘤的一个组成部分,并可能在功能上调节转移的各个方面。
肿瘤内微生物群与区分正常组织与癌组织、药物反应者与无反应者癌症、良好与不良预后、转移性与非转移性癌症有关。
肿瘤内微生物群的调节可以减少癌症转移,阻止癌症进展,并重新编程免疫反应。
本文主要集中于肿瘤内微生物群的发现和表征及其在肿瘤转移过程中的独特功能,并讨论了癌症治疗的挑战和意义。
癌症转移通常被定义为:
肿瘤从原始肿瘤部位转移到远端器官的多步骤过程。
这一过程涉及几个步骤,包括入侵、传播、血管内、外渗、定植。
转移的一个关键特征是其极低效率,这是由于癌细胞在成功到达并定居目的地之前,需要应对许多物理、化学和生物挑战。
转移级联期间的应激源包括:
• 细胞外基质(ECM)僵硬
注:肿瘤细胞外基质的硬度约为周围正常组织的1.5倍
• 失巢凋亡
注:失巢凋亡是由于细胞与细胞外基质和其他细胞失去接触而诱导的一种特殊的程序化细胞死亡形式,在机体发育、组织自身平衡、疾病发生和肿瘤转移等方面起重要作用。
• 流体剪切应力
注:压缩、拉伸、剪切力导致的组织变形导致组织液在细胞周围运动。
• 化疗
注:使用化学治疗药物杀灭癌细胞达到治疗目的。
• 免疫监视
注:免疫系统具有识别、杀伤并及时清除体内突变细胞,防止肿瘤发生的功能,称为免疫监视。
确定转移效率的关键是:
了解早期转移细胞如何能够抵抗这些挑战并增强其对不同环境的适应性,以及每种类型的压力对最终转移效率的影响程度。
转移是一个低效的多步骤易位过程
doi.org/10.1016/j.tcb.2022.11.007
新的研究扩大了我们对转移的认知。例如,研究表明转移开始发生在肿瘤进展的非常早期。
集体侵入相邻组织
在这些转移细胞到达远端器官之前,癌细胞甚至可以通过分泌成分远程准备转移前生态位(PMN)。当转移细胞开始迁移时,它们通常会集体侵入相邻组织,并作为寡克隆细胞簇在血流中传播,以增强其定植新生态位的能力。
doi.org/10.1016/j.canlet.2021.09.009
改变代谢程序,逃避免疫监视
这些先驱转移起始细胞改变它们的代谢程序以增强它们的转移潜能,并且可以逃避免疫监视并长时间保持休眠状态,直到开始分裂。
转移能力高度依赖于癌细胞内部细胞特性
这些研究使我们对转移细胞生存策略的理解更进一步,并证实了癌细胞转移能力高度依赖于癌细胞内部细胞特性的观点,例如 EMT 状态、干细胞可塑性、遗传学、表观遗传学、染色体不稳定性和代谢适应,以及环境因素,如机械压力、免疫反应、ECM、PMN 和肠道微生物组。
那么,癌细胞获得这些转移性状的驱动力是什么?

在实验上,肿瘤内微生物群已被确定为组织的一个组成部分。这些肿瘤内细菌是癌症进展不同阶段的新参与者,可以从外部相互作用和细胞内部影响癌细胞。
下面一个章节,我们来看肿瘤内微生物群是什么,有什么作用?
我们知道,已经有越来越多的文章阐述肠道微生物组在癌症进展中的作用,这方面我们的理解在迅速增长,然而我们对肿瘤内微生物群的理解仍处于初级阶段。
近期与转移相关的肿瘤内微生物群的研究

doi.org/10.1016/j.tcb.2022.11.007
人类组织,包括癌组织,通常被认为是无菌的,除了结肠、皮肤和口腔。
▸ 肿瘤内微生物群
癌症生物学的最新概念进展是,鉴定出癌症组织中存在微生物群。这些肿瘤组织驻留细菌被归类为“肿瘤内微生物群”。
我们知道,肠道微生物群可以通过代谢产物或通过与免疫细胞的相互作用,远距离影响肿瘤组织。
而肿瘤内微生物群与癌细胞密切接触,因此可能与肠道微生物群有不同的功能模式。
我们其他文章有对肠道微生物组在癌症诊断、预后和治疗反应中的作用进行详细介绍:
肠道微生物群与五种癌症的相互作用:致癌 -> 治疗 -> 预后
因此,本文主要集中于肿瘤内微生物群的发现和表征及其在肿瘤转移过程中的独特功能。
▸ 肿瘤内微生物群发现的证据:
-早前提出假设
一百多年前,威廉·科利发明了科利毒素(化脓性链球菌和粘质沙雷菌的混合物)来治疗一位癌症患者,并观察到肿瘤消退。
他假设“每一种恶性肿瘤都可能有外源性或微生物来源”。然而,在这个假设之后的几十年里,没有直接证据表明肿瘤内细菌的存在。
瘤内微生物群研究的重大突破包括发现、机制等成果
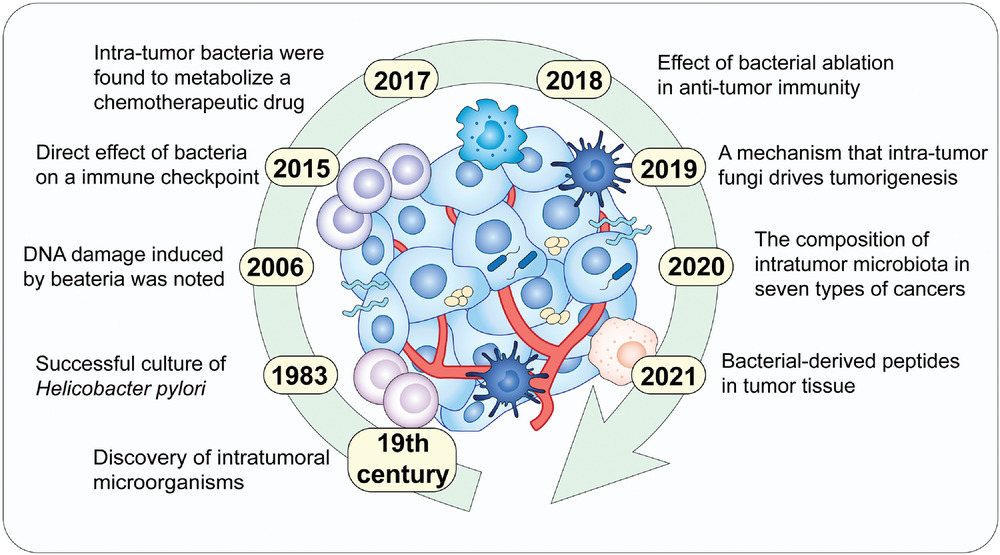
doi.org/10.1002/advs.202200470
-攻克瘤内微生物的检测技术挑战
到现在,下一代测序技术 (NGS) 能够使用 16S rDNA 测序将细菌 DNA 与肿瘤组织区分开来,然而,由于瘤内细菌丰度低和宿主基因组污染严重,从组织处理或试剂中引入的环境噪声信号使数据收集变得复杂,因为它们会掩盖组织的真实微生物概况并削弱结论的稳健性。
这些技术挑战在过去几年已被攻克,多个研究小组报告了大量数据,进一步支持瘤内微生物群的存在。此外,生物信息学微生物特征能够区分健康个体和癌症患者。
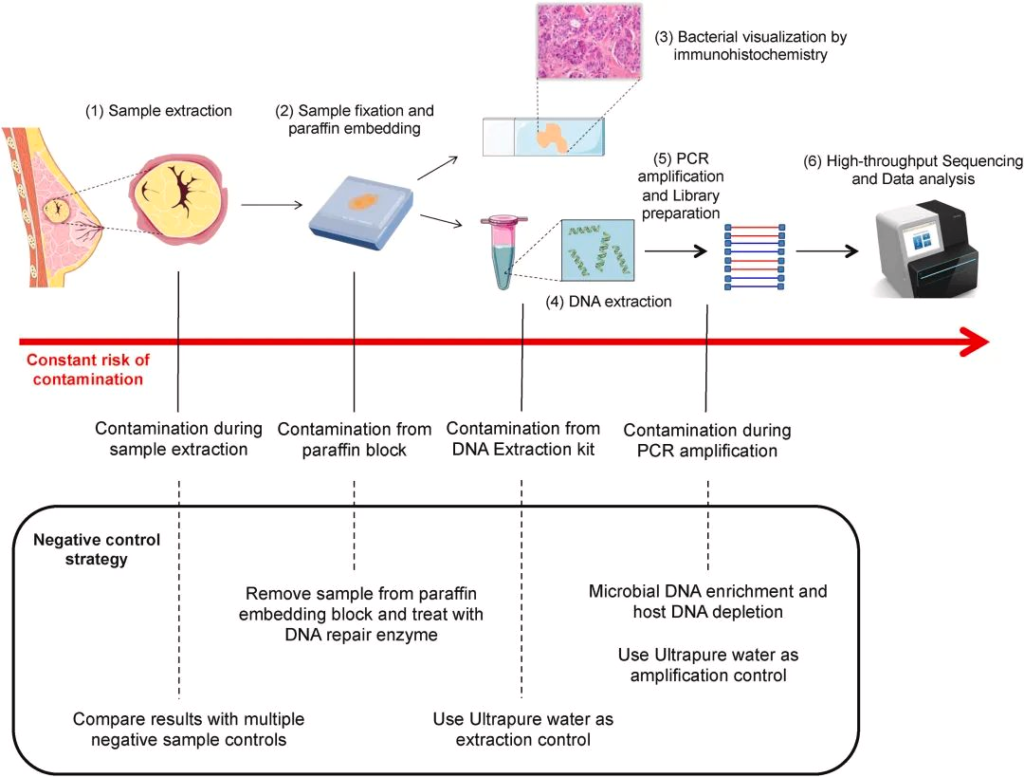
doi.org/10.1016/j.canlet.2021.09.009
识别肿瘤微生物组为癌症研究领域开辟了新的机遇。更好地表征肿瘤内微生物组可能会导致开发新的治疗方法,从而克服传统的癌症治疗方法。下一代测序方法,包括 16S 扩增子测序,可以在组织提取和石蜡固定后,将肿瘤内细菌精确地聚集在确定的细菌亚群中。
此外,宏基因组学对于肿瘤内微生物的鉴定也很重要。
宏基因组
宏基因组是一种针对样本中所有 DNA 的非靶向测序方法,包括微生物群落的全基因组序列,广泛应用于复杂微生物组的分析。宏基因组的分辨率更高,可以达到物种甚至菌株水平。此外,宏基因组学可以提供功能信息。
此外,宏基因组学可以与转录组分析结合使用,以消除死亡微生物和细胞外DNA造成的干扰。
最近的研究表明,最新的宏基因组数据涵盖了更多类型的癌症,这可能促进肿瘤内微生物群领域的新进展。
在瘤内微生物研究中,宿主DNA和环境微生物DNA的污染是最大的障碍。因此,需要开发从 TCGA 中丢弃不可信数据的方法。
在一项分析多种癌症的研究中,研究人员删除了总序列数据的 92.3%,以确保分析中数据的可靠性。2021 年,Dohlman 等人开发了一种去污染算法,可以去除 TCGA 数据中的污染。
随着这些方法的发展,宏基因组学可以为肿瘤内微生物群的研究提供更有力的支持。
▸ 细菌是各种癌症类型中肿瘤组织不可或缺的组成部分和活的居民
各种癌症类型有不同的微生物群。
肿瘤内微生物群的组成与许多类型的癌症有关。器官和组织包括食道、肺、乳腺、前列腺、膀胱、胃、肾、肝、胰腺等,以前被认为是无菌的。下一代测序显示这些器官含有低生物量微生物群。瘤内微生物组是肿瘤微环境的主要组成部分,影响肿瘤发生、疾病进展、耐药性和预后。
不同癌症类型的肿瘤内微生物群生态位
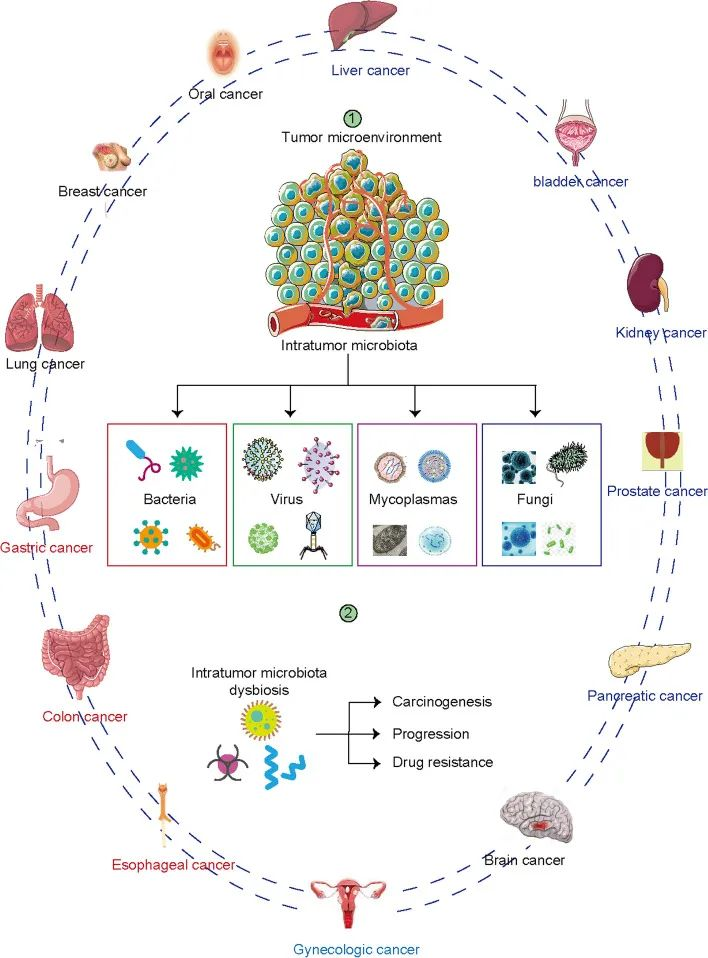
Liu J, et al., Biomark Res. 2022
在暴露于环境的组织(如肺癌和黑色素瘤)中并未发现微生物群丰度最高,而是在乳腺癌,骨癌,胰腺癌中。这表明肿瘤内微生物群的丰度是肿瘤特异性的。
作为癌症生态系统不可或缺的组成部分的肿瘤内微生物群
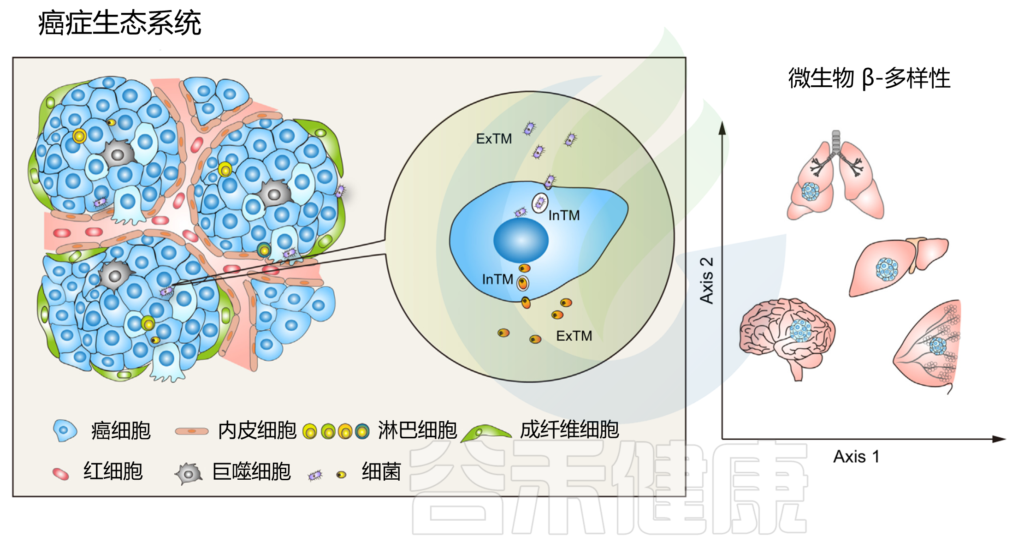
doi.org/10.1016/j.tcb.2022.11.007
如果肿瘤内微生物群存在于广泛的癌症类型中,那么它们来自哪里?
很少有研究专门去调查其原始来源。然而,对来自肿瘤组织的分离细菌菌株的分析提供了一些见解。
在小鼠乳腺肿瘤中,在正常组织对应物中检测到肿瘤内细菌菌株,这表明肿瘤组织从周围组织获得某些细菌。这些细菌菌株在体内的主要栖息地是多种多样的,有皮肤上的葡萄球菌、口腔中的链球菌和肠道中的肠球菌。
鉴于细菌具有在组织之间传播的能力,肿瘤内微生物群可能有多个起源。对鼻咽癌的分析表明,瘤内细菌主要来自鼻咽部,一小部分来自口腔和肠道。
* 也需要通过宏基因组比较和基因追踪分析来进一步加强。
肿瘤内微生物群的来源
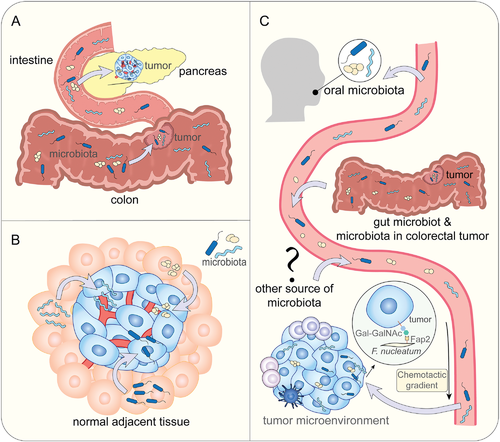
doi.org/10.1002/advs.202200470
A) 通过粘膜屏障从粘膜部位产生的肿瘤内微生物
B) 从正常邻近组织产生的肿瘤内微生物
C) 肿瘤内微生物是血行传播的结果
瘤内细菌的共同特征
1- 丰度低
它们在癌组织中的丰度远低于肠道中的丰度,根据 qPCR 定量和成像定量,0.1-10% 的癌细胞携带细菌,不同的量化方法和/或细菌 DNA 的提取效率引入了差异。
2- 多样性低
癌组织中微生物群落的多样性通常低于正常组织,这表明肿瘤可能形成一个独特的环境,选择性地扩展某些细菌种类。
3- 活的
这些细菌是活的。主要是主要存在于细胞内空间的共生生物。癌组织中不同的细菌栖息地可能与其在与癌细胞相互作用时的多效性作用模式有关。
细胞内外微生物群功能不一
鉴于细胞内和细胞外空间之间存在巨大的分子、生物化学和生物物理学差异,在肿瘤起始、肿瘤进展过程中,与细胞外肿瘤驻留微生物群 (ExTM) 相比,细胞内肿瘤驻留微生物群 (InTM) 可能具有完全不同的功能和免疫相互作用。
长期以来,细胞内细菌一直被研究为参与病原体-宿主相互作用的致病菌菌株。致病菌通过“触发”或“拉链”模式侵入宿主细胞,并能够迅速破开核内体膜进入细胞质。
肿瘤内共生细菌是遵循相同的原则还是使用不同的机制来侵入癌细胞?这方面仍知之甚少。在特定的癌症类型中,如乳腺癌,肿瘤内微生物群落主要以革兰氏阳性和兼性厌氧细菌为主,这表明肿瘤微环境具有选择效应。
不同的肿瘤类型具有不同的血管生成和氧水平、内吞作用和微胞作用以及周围组织中的微生物来源。这些因素共同决定肿瘤内微生物群的组成,并形成肿瘤类型特异性特征。
肠道菌群刺激特定代谢物的产生,调节免疫系统,并重建远端器官的微环境。相比之下,专门研究肿瘤内微生物群在癌症转移中的作用的研究有限。
这个领域的研究还比较浅,缺乏合适的实验工具来准确和特异性地调节肿瘤内的微生物群,同时又不扰乱身体其他部位的共生细菌。这个问题可以通过使用各种抗生素给药方案、使用无菌小鼠和原位细菌再给药来部分解决。
越来越多的证据证实,瘤内细菌可以调节癌细胞的内在特性及其外部环境,从而增强癌细胞的能力并为癌症转移铺平道路。
为了克服转移过程中的物理、化学和生物学挑战,癌细胞通常会改变其内在程序以应对不利的环境。这些包括干细胞程序/可塑性(用于新位点的肿瘤起始)、EMT 程序(用于癌症侵袭和传播)、粘附程序(防止失巢凋亡诱导的细胞死亡)和机械应激反应程序(抵抗机械力诱导的损伤) 。
研究表明,这些程序也可以通过肿瘤内微生物群进行调节。
肿瘤内微生物群改变癌细胞的内在特性并重塑转移中的肿瘤微环境
doi.org/10.1016/j.tcb.2022.11.007
我们先来看看,EMT程序是什么?
EMT程序赋予癌细胞迁移性间充质特征,具有松散的细胞间粘附特性,可动员癌细胞进行侵袭和扩散。这是由 TGFβ 信号通路的激活和与 Zeb、Twist 和 Snail 相关的协调转录程序驱动的。
微生物群和EMT程序之间有关联吗?
答案是肯定的。多项研究表明微生物群与 EMT 之间存在相关性。
在人类乳腺癌细胞系中,肿瘤驻留脆弱拟杆菌分泌的毒素诱导迁移和侵袭表型,EMT 相关的 Slug 和 Twist 的表达升高。在位于乳腺导管的肿瘤细胞中,脆弱拟杆菌的定植刺激了远端器官转移的增强。
这种功能调节是否仅限于细胞外肿瘤驻留微生物群,还是也适用于细胞内肿瘤驻留微生物群,以及不同的肿瘤驻留细菌对 EMT 的影响有多普遍,仍然是一个悬而未决的问题。
然而,有证据表明,脂多糖能够在依赖于 TLR-NFκB 通路的正常人肝内胆管上皮细胞中诱导 EMT.
在 EMT 驱动的小鼠结肠癌模型中,微生物群的存在对于肿瘤的发展至关重要。
这些研究支持组织驻留微生物群与 EMT 计划之间存在联系。
癌细胞的可塑性和干性是转移启动的另一个重要因素。
研究发现,脆弱拟杆菌毒素可以裂解 E-cadherin,触发下游 β-catenin 核定位,伴随 Notch 效应子 NICD 在乳腺癌中的核聚集。
在小鼠移植肿瘤模型中,Wnt 和 Notch 信号通路的后续激活,导致干性和肿瘤生长以及转移进展。
在自发性 MMTV-PyMT 乳腺肿瘤模型 [具有多瘤病毒中间 T 抗原 (PyMT) 的小鼠乳腺肿瘤模型在小鼠乳腺肿瘤病毒 (MMTV) 长末端重复序列下表达],各种肿瘤驻留细菌物种侵入 PyMT 癌症细胞触发了乳腺干细胞程序的富集。 由于与细菌侵入的癌细胞的体内分离相关的挑战,尚不清楚干细胞程序是否可以在生理细胞环境中被肿瘤内细菌激活。
癌细胞渗入血流引发细胞死亡程序
癌细胞渗入血流伴随着粘附丧失,这通常引发失巢凋亡,或其他形式的细胞凋亡的细胞死亡程序。癌细胞表面粘附分子的表达增强了它们的存活,并防止了转移失败。
在人类结直肠癌细胞系中,结直肠癌中常见的具核梭杆菌通过上调粘附分子 ICAM1 显着增强癌细胞对内皮细胞的粘附。这种增强的粘附力使癌细胞能够在尾静脉注射测定中外渗并引发新的转移灶。ICAM1 的上调部分是通过细菌依赖性激活 Alpk1-NFκB 通路实现的。
循环癌细胞受机械应力的影响导致细胞损伤
除了失巢凋亡依赖性细胞死亡外,循环癌细胞还会受到血液中各种机械应力的影响,从而导致细胞损伤,例如流体剪切应力,并在远端器官中,导致结构限制。
这些应激源部分被粘附分子(如整合素)感知,由 RhoGTPase 信号级联传递,并由 Yap/Taz 转录因子协调。
小鼠肿瘤模型的新发现表明,InTM 在侵入宿主癌细胞时会触发流体剪切应力反应,并且这种反应与细菌物种促进转移的能力相关。
被细菌侵入的癌细胞可以携带细菌,游走至远端器官,促进癌细胞的存活。这种表型是 InTM 特有的,因为通过调节 RhoAGTPase-Rock-actin 细胞骨架重组途径,癌细胞变得更能抵抗机械应力。引发这种反应的细菌机制仍不清楚。
然而,从肉毒梭状芽胞杆菌中分离出来并被多种细菌共享的 ADP-核糖基转移酶 C3 胞外酶是一个潜在的候选者,因为 C3 对细胞是不可渗透的,并且与膜穿透肽融合的 C3 经常被细胞生物学家用来解离肌动蛋白应力纤维并增强细胞扩散。
除了直接调节癌细胞外,瘤内细菌是重要的炎症介质,可以在癌细胞周围形成特定的微环境,从而间接促进癌症转移。
调节 PMN 的关键因素之一是细菌本身
结直肠癌研究表明,肿瘤驻留细菌能够通过毒力因子 VirF 调节肠道血管屏障。PV-1 表达升高的血管屏障受损,促进了细菌从原发性结直肠肿瘤传播到肝脏,并在癌细胞到达之前建立了 PMN.
注:PMN-迁移前生态位
患者体内较高的 PV-1 水平与较高的细菌负荷和较远的转移有关。这种依赖于细菌的 PMN 远程控制是一个新概念,可能对癌症以外的疾病有影响。
肿瘤外泌体可以调节 PMN 并决定转移器官的趋向性
肿瘤外泌体含有多种功能性脂类、蛋白质、RNA和DNA,释放到细胞外环境中调节靶细胞,重塑微环境。
源自具核梭杆菌侵入的人结直肠癌细胞,分离出含有 miR-1246/92b-3p/27a-3p 和 Cxcl16 的外泌体。这些外泌体在调节结直肠癌细胞迁移方面发挥作用,并通过靶向 GSK3β 激活 Wnt-β-catenin 信号通路显著增加肺转移。
这意味着邻近的癌细胞不一定需要被细菌侵入才能转移;相反,它们也可以通过旁分泌外泌体信号来动员以启动转移。
瘤内细菌最显着的特征之一是它们可以被免疫系统识别,从而触发特定的免疫反应
有许多关于肠道菌群失调与异常免疫反应之间关联的报道,但肿瘤内微生物群在调节免疫系统中的作用仍不清楚。
一方面,抗生素治疗和细菌再给药试验显示肿瘤内细菌抑制免疫反应的证据
在乳腺癌中,瘤内具核梭菌以免疫介导的方式加速肿瘤进展和肺转移,瘤内给药具核梭菌减少浸润的 CD4+ 和 CD8+ T 细胞。
在小鼠黑色素瘤癌症模型中,肺组织的抗生素治疗降低了细菌负荷,显示出调节性 T 细胞减少,T 细胞和自然杀伤 (NK) 细胞活化增强,同时肺转移显着减少。
在转基因小鼠肺癌模型中,肺部共生细菌激活了 γδT 细胞,这是一种 T 细胞亚群,通过刺激骨髓来源的 IL1β 和 IL23 并引发肿瘤炎症来促进淋巴和骨髓谱系的炎症反应。
另一方面,肿瘤内细菌可以触发抗肿瘤免疫。
例如,益生菌(鼠李糖乳杆菌)的施用强烈促进了针对小鼠黑色素瘤肺转移的肿瘤免疫。
此外,瘤内注射双歧杆菌可刺激 STING 通路,增加树突状细胞数量,并促进基于抗 Cd47 的免疫治疗。
因此,肿瘤内细菌的免疫调节作用是复杂的,并且依赖于环境,并且可能是细菌物种特异性的和/或受其细胞内/细胞外居住状态的高度影响。
传统癌症疗法的限制
迄今为止,主要的癌症疗法基于手术、放疗和化疗。尽管对大多数确定的肿瘤有效,但它们都有缺点,依赖于冗长、乏味的程序,非特异性地对抗肿瘤,通常无法区分恶性组织和健康组织。
由于缺乏对肿瘤样区域的特异性,某些癌细胞得以存活并定植在附近的组织中,从而导致潜在的癌症复发。靶向健康组织可能会产生意想不到的副作用,从而导致严重的致癌 DNA 损伤。
所有这些缺点,加上对治疗产生耐药性的持续风险,与癌症死亡率和发病率的增加有关。
90%的癌症患者死于复发或转移。
肿瘤内微生物群的作用可以通过具有肿瘤内微生物群信息的癌症患者的生存数据来评估。
在胰腺癌患者中,与短期幸存者相比,长期幸存者往往具有更高的微生物群落多样性。
此外,肿瘤内微生物群特征(假黄单胞菌Pseudoxanthomonas–链霉菌Streptomyces–糖多孢菌Saccharopolyspora –克劳氏芽孢杆菌Bacillus clausii)被确定与生存相关。
在其他癌症类型中,尽管样本量有限,但据报道特定的肿瘤内微生物组特征也与转移有关。
在对 800 多个患者样本进行分析的鼻咽癌临床研究中,肿瘤内细菌载量被确定为一种强有力的预后工具,可以区分恶性进展的风险。这些研究证实了肿瘤内微生物群的预后价值,并支持其在临床肿瘤进展中的作用。
然而,在临床上特异性调节肿瘤内微生物群具有挑战性。
有几项关于抗生素治疗和癌症风险、癌症反应和生存的回顾性研究,但它们很少专门设计用于剖析肿瘤内微生物群的消除和患者预后。
这些广泛的抗生素治疗数据分析报告了癌症发病率的增加和对免疫疗法的一般反应受损。鉴于已经确定肠道微生物组与免疫检查点抑制剂治疗密切相关,目前尚不清楚肠道肿瘤微生物组在调节癌症进展方面是否具有相似或不同的作用。
相比之下,一项胰腺腺瘤研究表明,抗生素治疗与晚期转移性胰腺导管腺癌的更好预后相关。
鉴于抗生素在效力、吸收效率、细胞渗透性以及给药途径和时间窗的可变性方面存在巨大差异,所有这些变量都可能导致肠道微生物组和细胞内/细胞外肿瘤微生物组概况的根本差异。因此,迫切需要精心定义的肿瘤内微生物群调节临床研究集。
肿瘤内微生物群数据在癌症筛查和治疗中的应用
Liu J, et al., Biomark Res. 2022
A) 来自临床样本的数据可能有助于开发新的癌症筛查和预后,包括来自肿瘤部位和易于获取的样本的微生物群模式。
B) 肿瘤内微生物群可用于癌症治疗,包括工程菌、饮食调节、粪便微生物组移植、抗生素和肿瘤内微生物组注射等。
新兴研究揭示了肿瘤内微生物群在癌症转移的各个步骤中的生物学功能。这些肿瘤内微生物群不仅是肿瘤环境的传感器、肿瘤病理类型、药物反应和预后的指标,而且在功能上也参与肿瘤进展。
肠道细菌的宿主内进化会导致共生菌株变成致病。因此,需要进一步的研究来测试肿瘤内细菌促进癌症转移的能力是否源于细菌进化。这或许可以解释不同的细菌种群及其在正常组织和癌组织中的各种功能,以及为什么某些肿瘤类型比其他肿瘤发展得更快。
未来,肿瘤内微生物领域将受到更多关注,该领域有四个方面可能成为未来研究的重点:
肿瘤内微生物群可以作为癌症筛查的生物标志物。
包括肿瘤内微生物组衍生的个性化数据,这些数据可以将食管癌、胰腺癌、肺癌和口腔癌患者与健康人区分开来。分析肿瘤内微生物群特征,可能为患者的预后提供潜在的生物标志物。
此外,肿瘤内微生物群为癌症治疗带来新的机遇。
考虑到肿瘤内微生物群的异质性,个性化治疗策略因其高效和靶向作用而具有吸引力。
肿瘤内细菌的细胞外和细胞内定位使它们成为药物载体的完美候选者,可以在肿瘤细胞内外递送,以倒带细胞间和细胞内信号网络。
与其他抗肿瘤疗法一样,细菌疗法和抗生素也可以与其他疗法结合使用,例如免疫疗法和化学疗法。
使肿瘤内微生物群正常化和移植某些微生物也是提高抗肿瘤治疗效率的潜在策略。
癌症疗法正面临着巨大的转变:传统疗法正逐渐被更精确和复杂的疗法所取代。了解肿瘤内微生物群对癌症发生和发展的不同贡献,将有助于制定癌症预防和治疗策略。
主要参考文献:
Fu A, Yao B, Dong T, Cai S. Emerging roles of intratumor microbiota in cancer metastasis. Trends Cell Biol. 2022 Dec 13:S0962-8924(22)00258-6. doi: 10.1016/j.tcb.2022.11.007. Epub ahead of print. PMID: 36522234.
Liu J, Zhang Y. Intratumor microbiome in cancer progression: current developments, challenges and future trends. Biomark Res. 2022 May 31;10(1):37. doi: 10.1186/s40364-022-00381-5. PMID: 35642013; PMCID: PMC9153132.
An Y, Zhang W, Liu T, Wang B, Cao H. The intratumoural microbiota in cancer: new insights from inside. Biochim Biophys Acta Rev Cancer. 2021 Dec;1876(2):188626. doi: 10.1016/j.bbcan.2021.188626. Epub 2021 Sep 11. PMID: 34520804.
Heymann CJF, Bard JM, Heymann MF, Heymann D, Bobin-Dubigeon C. The intratumoral microbiome: Characterization methods and functional impact. Cancer Lett. 2021 Dec 1;522:63-79. doi: 10.1016/j.canlet.2021.09.009. Epub 2021 Sep 10. PMID: 34517085.
Wang Y, Guo H, Gao X, Wang J. The Intratumor Microbiota Signatures Associate With Subtype, Tumor Stage, and Survival Status of Esophageal Carcinoma. Front Oncol. 2021 Oct 27;11:754788. doi: 10.3389/fonc.2021.754788. PMID: 34778069; PMCID: PMC8578860.
Xie Y, Xie F, Zhou X, Zhang L, Yang B, Huang J, Wang F, Yan H, Zeng L, Zhang L, Zhou F. Microbiota in Tumors: From Understanding to Application. Adv Sci (Weinh). 2022 Jul;9(21):e2200470. doi: 10.1002/advs.202200470. Epub 2022 May 23. PMID: 35603968; PMCID: PMC9313476.
Huang Y, Zhu N, Zheng X, Liu Y, Lu H, Yin X, Hao H, Tan Y, Wang D, Hu H, Liang Y, Li X, Hu Z, Yin Y. Intratumor Microbiome Analysis Identifies Positive Association Between Megasphaera and Survival of Chinese Patients With Pancreatic Ductal Adenocarcinomas. Front Immunol. 2022 Jan 25;13:785422. doi: 10.3389/fimmu.2022.785422. PMID: 35145519; PMCID: PMC8821101.

谷禾健康

色氨酸(Tryptophan,简称 Try)是人体必需氨基酸,也是唯一含有吲哚结构的氨基酸,由食物尤其膳食蛋白质提供,是正常细胞稳态所必需的,是维持细胞生长和协调机体对环境和饮食线索的反应(其中色氨酸代谢物充当神经递质和信号分子)。
不同组织内的色氨酸代谢与许多生理功能有关:
在哺乳动物中,色氨酸是代谢物的生化前体,显著影响哺乳动物的生理机能,包括胃肠道功能、免疫力、新陈代谢和神经系统。色氨酸及其代谢物水平的失衡与广泛的人类病理学相关,包括抑郁症、精神分裂症、自身免疫、神经退化和癌症。同时它也是自身免疫、癌症、神经退行性或肠道疾病的一个非常有吸引力的治疗靶点。
本文将总结和讨论色氨酸及色氨酸代谢的生理和病理学作用,肠道中色氨酸代谢物的产生和调控、肠道菌群衍生的色氨酸代谢物在全身健康稳态中的作用、以及基于色氨酸代谢药物开发的巨大机遇和挑战。
/
/
▼
色氨酸是一种必需氨基酸,是体内许多重要分子的前体。
如果您读过我们很多文章,可能读到比较多是短链脂肪酸。短链脂肪酸(SCFA),尤其是丁酸盐,通常会在肠道菌群失调和慢性疾病的状态下耗尽。
但短链脂肪酸只是肠道代谢物的一大类之一。其他两类——色氨酸代谢物和胆汁酸,在维持肠道健康方面发挥着同样重要的作用。所以我们也将逐步关注和分享它们。
● 什么是色氨酸?
色氨酸是一种氨基酸——我们体内蛋白质的众多组成部分之一。
色氨酸是一种必需的芳香族氨基酸,由连接到吲哚基团 3 位的 β 碳组成。在 20 种常见的经典氨基酸中,色氨酸的分子量最大。
虽然色氨酸是蛋白质和细胞中含量最少的氨基酸,但它是大量微生物的生物合成前体和宿主代谢物。
大多数游离色氨酸通过犬尿氨酸 (Kyn) 途径 (KP) 或血清素途径降解为具有生物活性的化合物。
色氨酸分解代谢途径
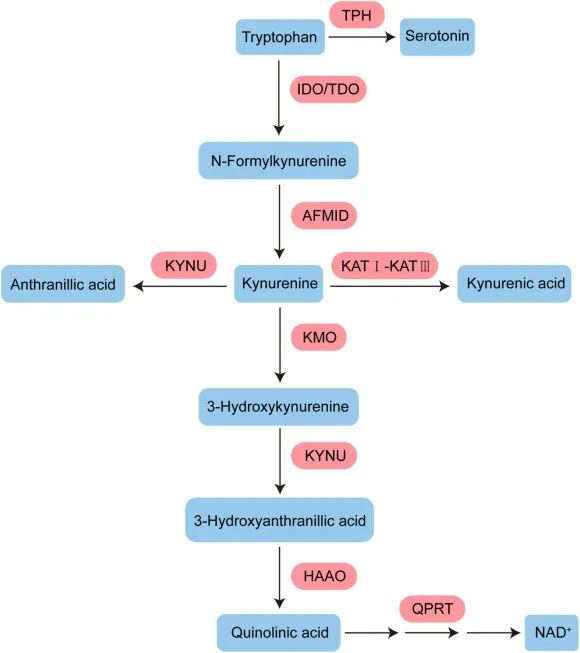
血清素途径产生血清素,可进一步转化为 N-乙酰血清素 (NAS) 和褪黑激素,后者对于昼夜节律调节和抗衰老至关重要。
肠道菌群对色氨酸的吸收很重要,限制和调节宿主细胞的使用。在此过程中生成吲哚衍生物,扩大了色氨酸分解代谢在不同组织器官中中的通讯作用。
色氨酸是体内许多其他化合物的重要前体,包括:
● 色氨酸 / 起源与生产
色氨酸是人体无法产生的必需氨基酸,必须通过饮食获取,主要来自动物或植物性蛋白质来源。
起源:酪蛋白分离
色氨酸是在 1900 年代初期从酪蛋白(一种在牛奶中发现的蛋白质)中分离出来后被发现的。几年后确定了它的分子结构。
释放:进入血液循环
小肠中膳食蛋白质的消化导致色氨酸的释放,色氨酸可以通过肠上皮细胞吸收并进入血液。色氨酸在血液中循环,主要与白蛋白结合,而在血液循环中只有 10-20% 的色氨酸是游离态。被吸收的色氨酸以其游离形式循环或与外周血流中的白蛋白结合。
据报道,健康献血者的总色氨酸平均血清水平为 73 ± 14.9 μmol/l 。
游离色氨酸的一个重要生理功能是对宿主蛋白质合成的贡献。
除了作为蛋白质合成的成分外,色氨酸还是生产多种重要生物活性物质的必需底物。例如,色氨酸是血清素合成(情绪相关)以及褪黑激素(睡眠相关)合成的底物。
全身和细胞色氨酸水平由食物摄入量、生物转化以及降解色氨酸的途径酶活性共同决定。
● 色氨酸的常见天然食物来源
乳制品、燕麦、香蕉、豆类、黑芝麻、李子干、金枪鱼、奶酪、面包、家禽、花生、黑巧克力、鱼肉、三文鱼、杏仁、南瓜和南瓜子等。
世界卫生组织将推荐的色氨酸摄入量设定为 4 毫克/千克/天,迄今为止,没有关于饮食中色氨酸过量的不良影响的报道。
注:含有色氨酸的食物对于制造激素血清素至关重要。但不应高估其影响。
一般来说,动物蛋白中的色氨酸含量往往高于植物蛋白。虽然色氨酸可以补充形式服用,但最好将其作为全食物中完整蛋白质的一部分。
● 为什么需要色氨酸?
色氨酸在体内的浓度是所有氨基酸中最低的,然而,色氨酸摄入量低与抑郁、焦虑、情绪低落、睡眠质量差、视觉认知能力下降以及学习和记忆受损有关。它还可能改变肠道微生物组并削弱肠道免疫力。
另一方面人们普遍认为色氨酸过量会导致困倦。比如在美国的感恩节食用大量火鸡,火鸡中的色氨酸含量很高,进食大餐会刺激胰岛素的产生,而胰岛素会清除血液中除色氨酸以外的所有氨基酸,会导致困倦。
/
/
▼
大量的数据表明色氨酸代谢的调节对环境条件很敏感,并且会影响生理和行为过程。
它因物种、细胞类型、诱导剂而异,并且可以通过组织之间的相互作用进行调节。
● 宿主色氨酸代谢
这里我们对色氨酸代谢先有个整体的认识:
色氨酸分解代谢主要两条通路:
▸犬尿氨酸通路占整体色氨酸降解的约95%
色氨酸 (TRP) 通过犬尿氨酸 (KYN) 通路 (KP) 的分解代谢,该通路占整体色氨酸降解的约 95%,形成主要最终产物 NAD+。
注:犬尿氨酸通路是炎症和免疫反应的重要参与者。
首先,色氨酸被转化为N-甲酰基-L-犬尿氨酸
该反应由三种限速酶之一催化:
注:这三种酶都是血红蛋白,并使用分子 O2作为共底物,这也使它们能够利用活性氧 (ROS) 并调节细胞内的氧化还原平衡。
IDO 和 TDO 酶在不同的组织中表达,暴露于不同的刺激物时被诱导,表明它们在健康和疾病中具有不同的功能。
TDO 在基础条件下催化色氨酸分解,而在免疫调节中具有关键作用的 IDO-1 受到多种刺激物的诱导和调节,例如炎症信号 。
进一步的,N-甲酰基-犬尿氨酸形式酰胺酶将 N-甲酰基-L-犬尿氨酸水解为 L-犬尿氨酸
成为三种具有不同氧化应激和器官毒性特性的替代代谢物:
最终在3-羟基邻氨基苯甲酸3,4-双加氧酶(3-HAAO)的催化下,进一步分解为喹啉酸、烟酰胺腺嘌呤二核苷酸(NAD+).
肝外色氨酸犬尿氨酸通路不提供所有必需的酶;因此,其中间代谢物及其特性在这些病症的发病机制和调节中变得至关重要(下图)。
注:在生理条件下,肝外通路仅占整体色氨酸降解的 5-10%.
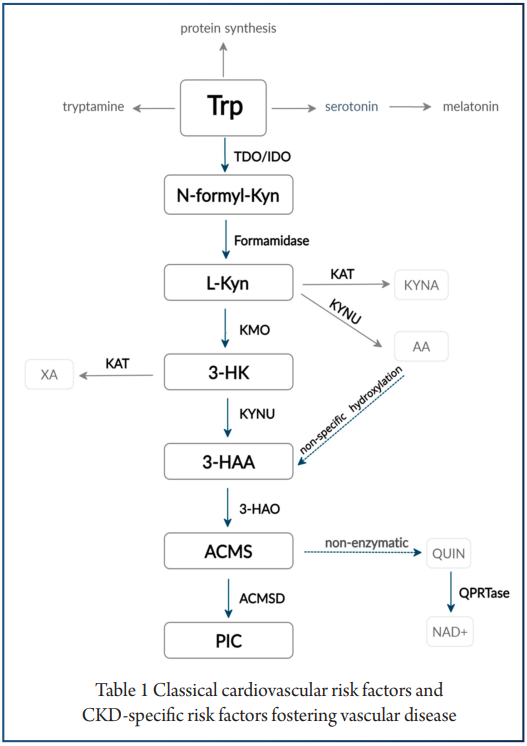
编辑
B 族维生素作为辅因子起着至关重要作用
KP 对B族维生素浓度的变化很敏感。维生素 B6(5′-磷酸吡哆醛,PLP)的活性形式影响犬尿氨酸酶 (KYNU) 和犬尿氨酸氨基转移酶 (KAT)。
维生素B6缺乏后,会影响色氨酸代谢。
▸ 大约不到5%的色氨酸会转化为5-羟色胺
通过色氨酸羟化酶(TPH)催化生成5-羟色胺(5-HT),也就是血清素。
doi.org/10.3389/fendo.2019.00158
以上是色氨酸代谢的两种主要途径。感染、压力和肠道菌群的变化都可以将色氨酸代谢从 5-HT的产生分流到犬尿氨酸途径,因此如抑郁症之类的病理变化,与人类的营养因素、压力和免疫功能有关。
简化的人类色氨酸的主要代谢途径
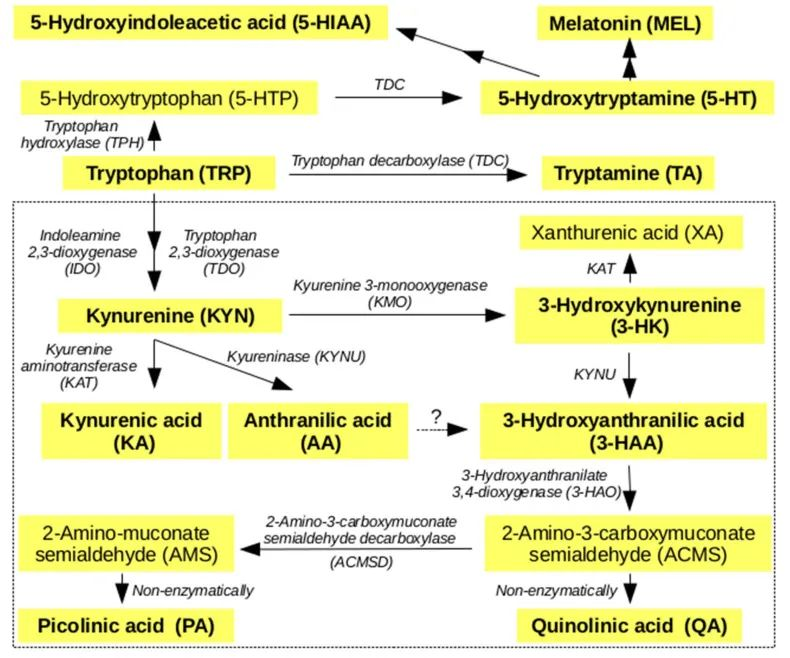
doi: 10.3390/metabo10050208.
● 色氨酸微生物代谢
肠道微生物将未吸收的 L-色氨酸 代谢成几个分子,如吲哚衍生物 [indole-3-aldehyde (IAld)、indole-3-acetic acid (IAA)、indole-3-propionic acid (IPA)、indole-3-acetaldehyde (IAAld)、吲哚-3-乳酸 (ILA) 和吲哚丙烯酸],还有色胺和粪臭素。
最近表明,其中一些分子不仅由微生物群合成,而且还通过 L-氨基酸氧化酶 (IL-4I1) 的作用由肿瘤细胞合成,代谢 L -色氨酸 转化为吲哚-3-丙酮酸,随后转化为 IAA、IAld 和 ILA,从而以 AhR 依赖性方式逃避免疫系统、存活和肿瘤运动。
AhR 信号是免疫反应屏障位点的重要组成部分。它通过作用于上皮更新、屏障完整性和许多免疫细胞类型(如上皮内淋巴细胞、T 辅助 (Th)17 细胞、先天性淋巴样细胞、巨噬细胞树突状细胞和中。
肠道环境的细菌色氨酸代谢
由于不同的微生物拥有不同的催化酶,需要两种以上的细菌相互合作才能从色氨酸中产生一种代谢物。与动物内源性色氨酸代谢相对简单的背景不同,人类肠道环境在细菌色氨酸代谢方面相对复杂。
肠道菌群通过各种代谢途径产生多种色氨酸代谢产物,例如:
肠道中微生物群相关的色氨酸代谢
不同菌种可能存在相同代谢能力
比如:消化链球菌属的相同代谢功能可能是基于这些菌种拥有苯乳酸脱水酶基因簇,在下列菌群中也发现与它们产生IPA能力一致的同源基因簇:
不同菌种之间也存在一定代谢能力差异
比如:几种拟杆菌属和梭菌(Clostridium bartlettii)可以产生ILA和吲哚乙酸(IAA),而双歧杆菌属(Bifidobacterium spp.) 产生ILA 。
通过 5-HT、Kyn 和吲哚/AhR 途径的色氨酸代谢途径
doi.org/10.1016/j.chom.2018.05.003
吲哚也是一种种间信号分子,能够控制细菌生理学的各个方面,例如抗生素抗性、孢子形成和生物膜形成。
在不产生吲哚的细菌中,吲哚及其衍生物显着抑制群体感应并调节毒力因子。然而,这些复杂现象在肠道生态系统中的重要性尚未得到具体解决。
微生物代谢的作用在肠道 AhR 活性中占主导地位。事实上,无菌或失调小鼠的肠道内容物缺乏 AhR 激动剂。只有少数共生物种能够产生 AhR 配体,例如Peptostreptococcus russellii罗氏消化链球菌和乳杆菌属已被表征,许多可能仍有待发现。
● 肠道色氨酸代谢平衡
虽然大部分色氨酸被小肠吸收,但其中一些会继续进入大肠,在那里它可以被微生物和宿主细胞作用。从上一小节的阐述,我们可以看到色氨酸在肠道内的三个主要归宿:
1) 吲哚/AhR 通路
肠道细菌直接将色氨酸转化为吲哚和相关分子。就像锁和钥匙一样,其中一些吲哚分子与整个肠道和其他器官细胞表面的芳烃受体 (AhR)结合。这会引发广泛的反应,促进肠道稳态。该途径的活性取决于饮食和微生物群的组成。
2) 犬尿氨酸(KP)通路
一些色氨酸被肠道上皮细胞和免疫细胞吸收,在那里它被 IDO1 酶转化为犬尿氨酸。犬尿氨酸可以进一步代谢为其他分子,例如具有神经毒性作用的喹啉酸。应激、炎症或感染会增加该通路的活性。
3) 血清素途径
色氨酸也被吸收到肠道肠内分泌细胞中,然后通过酶 TpH1转化为神经递质血清素。肠道中的血清素调节肠道运动、分泌和吸收,并在肠-脑信号传导中发挥作用。该通路的活性受禁食、饮食、肠道感染和某些微生物的影响。
当然,关键是平衡。在健康的肠道中,这三种途径是平衡的,从而产生最佳的肠道屏障功能、动力、免疫力和神经功能。
宿主生理学中肠道菌群控制下的综合色氨酸代谢
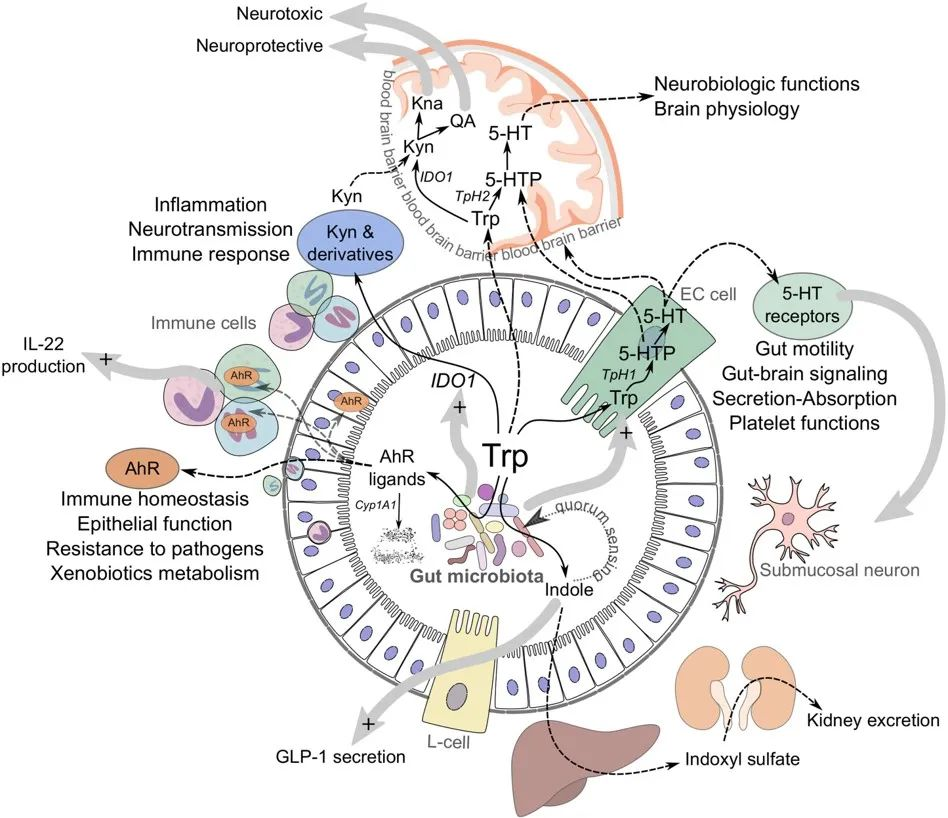
doi.org/10.1016/j.chom.2018.05.003
▼
在慢性疾病中,上述三种途径的平衡似乎出现了偏差,导致肠道功能受损和系统性影响。
/
/
▼
色氨酸及其代谢物水平的失衡与广泛的人类病理学相关,包括肠道疾病、抑郁症、精神分裂症、自身免疫、神经退化、癌症、心血管疾病、过敏、代谢综合征、肥胖、衰老等。
本章节我们对这些疾病中的色氨酸代谢先做个大致了解,后面章节会对各类疾病一一展开阐述。
由于许多 KP 代谢物具有神经活性,因此通常由炎症损伤引起的 KP 酶功能障碍可引发或促进中枢神经系统 (CNS) 疾病。
对于中枢神经系统疾病,人们越来越关注通过靶向特定 KP 酶来纠正 KP 代谢物变阻器的变化以实现净神经保护作用,以及色氨酸及其代谢物在调节肠道微生物组和大脑之间的相互作用中的作用。
肠道微生物组对膳食色氨酸吸收和代谢的影响也越来越受到关注,并且与中枢神经系统疾病以及肠易激综合征、胰腺炎和糖尿病具有潜在相关性。
降低维生素 B2 浓度会导致依赖于黄素腺嘌呤二核苷酸的犬尿氨酸 3-单加氧酶 (KMO) 的活性降低。B 族维生素,包括核黄素 (RBF) 和吡哆醇 ,在预防中风和中风后恢复中发挥作用。据报道,异常 KP 与神经系统疾病、癌症、心血管疾病和中风有关。
色氨酸分解代谢——涉及的关键器官
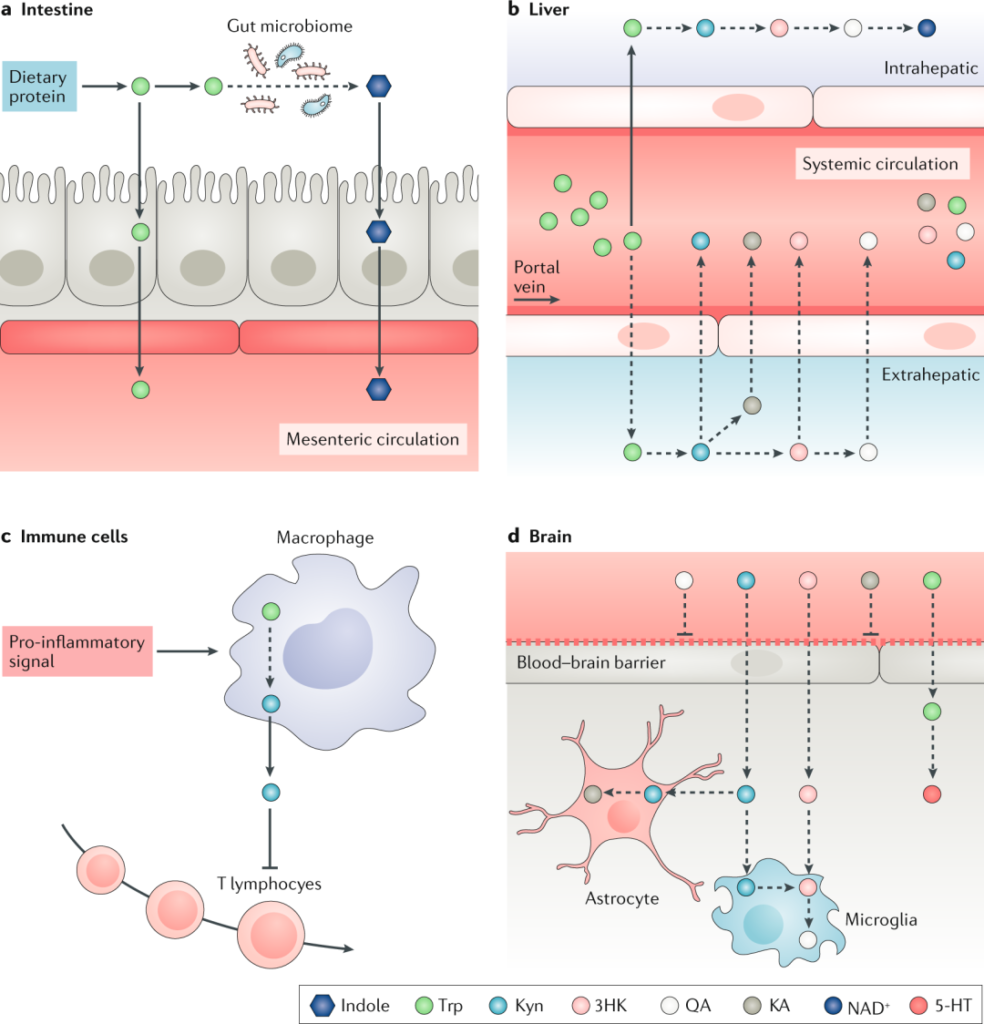
doi.org/10.1038/s41573-019-0016-5
a | 摄入膳食蛋白质后,肠上皮细胞将L-色氨酸转运穿过顶膜进入间质和肠系膜循环。或者,肠道微生物群合成色氨酸并将其代谢为吲哚并将其释放到体循环中。
b | 然后色氨进入肝脏,其中大部分被氧化为乙酰乙酰辅酶 A 并用于合成 NAD+。沿着犬尿氨酸 (Kyn) 途径 (KP) 代谢色氨酸 的肝外器官,包括肾脏、脾脏和免疫细胞,对 Kyn 和 KP 代谢物的循环水平贡献最大。
c | 在促炎性刺激后由骨髓细胞释放的 KP 代谢物抑制 T 细胞反应。
d | 色氨酸、Kyn 和 3-羟基犬尿氨酸 (3HK) 被转运穿过血脑屏障并被星形胶质细胞、小胶质细胞和神经元吸收。星形胶质细胞主要产生具有神经保护作用的犬尿酸 (KA),而小胶质细胞产生具有神经毒性的 KP 代谢物,例如喹啉酸 (QA)。
大约 5% 的色氨酸被代谢为血清素 (5-HT)、5-羟基吲哚乙酸 (5-HIAA)、褪黑激素 (MEL) 和色胺 (TA)。最近研究表明,5-HT除了在神经传递、血管收缩或血管舒张、止血控制和血小板功能中的作用外,还参与调节人体的能量平衡、食欲、肠道蠕动、免疫力、肝脏修复以及心血管和肺部生理学。
疾病中色氨酸代谢的扰动
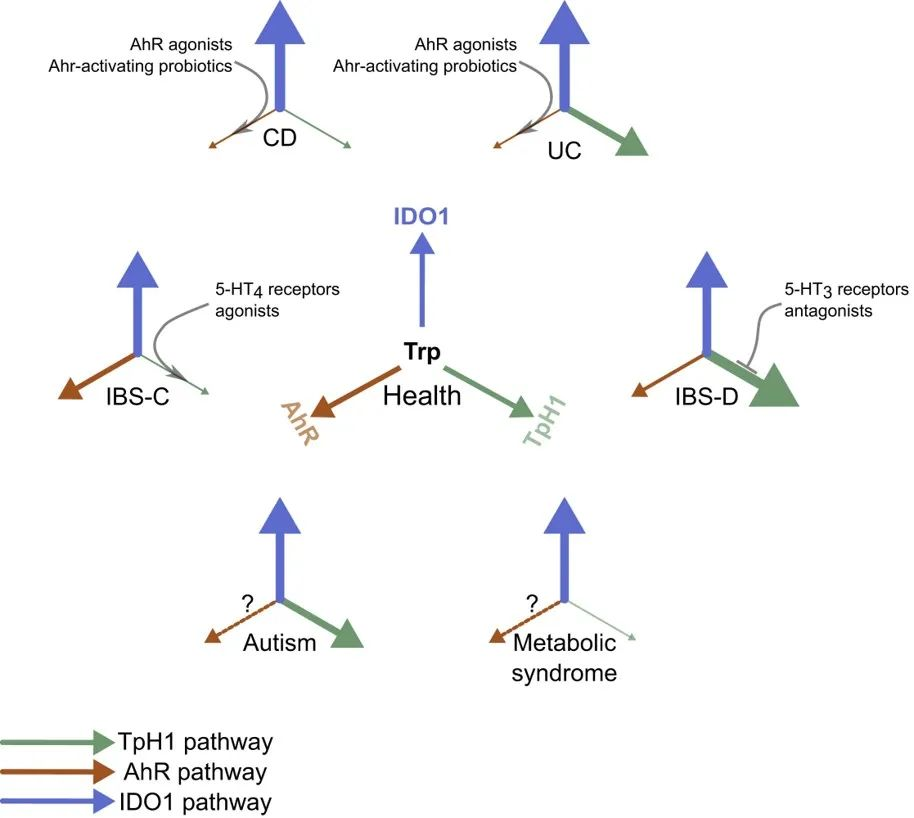
doi.org/10.1016/j.chom.2018.05.003
使用免疫组织化学监测 KP 代谢物的抗体的最新发展使得能够确定组织中 KP 代谢物的积累。
这些工具表明,犬尿氨酸通路在 IDO1 阳性癌症中积累,兴奋性毒性代谢物喹啉酸在脑肿瘤和神经退行性疾病的神经元中积累,而黄嘌呤酸 (XA) 是谷氨酸能突触传递的调节剂,定位于躯体和健康大脑中神经元的树突。
色氨酸代谢物与一系列疾病的联系导致人们在治疗上调节 KP 方面付出了巨大努力,特别是通过抑制所涉及的关键酶,包括 IDO1、TDO 和 KMO。
在癌症中,IDO1 和 TDO 的异常激活导致抗肿瘤免疫抑制。近年来 IDO1 抑制剂在癌症免疫治疗中得到了深入研究。
临床试验中有多种化合物,通常与免疫检查点抑制剂等其他药物联合使用。人们普遍预计领先的 IDO1 抑制剂将接近监管批准,但最近的 III 期试验终止引发了对该方法可行性的质疑,并强调需要更深入地了解 KP。
/
/
▼
● 神经退行性疾病中的色氨酸代谢
色氨酸代谢与多种神经退行性疾病有关,包括亨廷顿病 (HD)、阿尔茨海默病(AD)、肌萎缩侧索硬化 (ALS) 和帕金森病 (PD)。
尽管病理生理触发因素各不相同,但所有这些疾病的共同点是容易聚集的蛋白质引起神经元退化,从而导致细胞应激和有害的先天免疫反应。
基于人群的研究表明,就这些病理学特征而言,衰老和神经退行性疾病之间存在相当大的重叠,个体间差异很大。
虽然遗传和环境对色氨酸代谢的影响尚不完全清楚,但据信色氨酸代谢会导致衰老和神经退行性变,并且所涉及的机制即使不完全相同。这一观察得到了小鼠研究的支持,其中 TDO 的缺失已被证明会导致海马体和脑室下区的神经发生增强,可能抵消神经变性。
尽管生物标志物研究表明色氨酸代谢在神经退行性疾病患者中的活性不同,目前尚不清楚这是原发性倾向的结果还是神经变性或附带的先天免疫激活的结果。流行病学研究表明,KP 的激活与痴呆症风险增加有关。
然而,很难与生理老化明确区分。KP 对感染性和炎症性损伤的敏感性明显损害了其作为神经变性标志物的稳健性。另一方面,炎症对 KP 的激活可能在多发性硬化症等疾病中的神经炎症和神经变性之间建立联系。
由色氨酸代谢介导的神经变性的潜在机制包括:
➤ 阿尔茨海默氏病
色氨酸代谢物、肠道微生物和相关的神经炎症变化对阿尔茨海默病的病理生理学有显着影响。
阿尔茨海默患者的肠道微生物分类有显着差异,厚壁菌门和放线菌门减少,拟杆菌门增加。CSF 生物标志物升高与某些属的丰度相关,尤其是拟杆菌属和Blautia。
一项研究中,发现阿尔茨海默患者的循环色氨酸显著减少,犬尿氨酸/色氨酸比率升高,这反过来又与认知能力较差和促炎细胞因子升高相关。
几项临床前研究表明,在阿尔茨海默动物模型中具有保护作用。色氨酸代谢产物以芳基烃受体依赖的方式调节小胶质细胞和星形胶质细胞的活化。
此外,延缓阿尔茨海默进展的药物治疗的研究表明,肠道微生物和色氨酸代谢产物在阿尔茨海默的发展中可能发挥作用。
研究人员提供了犬尿氨酸代谢物在阿尔茨海默中的潜在毒性作用的间接证据,因为持续向小鼠腔内灌注犬尿氨酸会导致小鼠后代的学习和记忆缺陷。
吲哚途径代谢产物也可能介导阿尔茨海默病的发病。吲哚途径代谢物IPA在体外可抑制淀粉样蛋白-β诱导的神经毒性,并已被开发为治疗阿尔茨海默的神经保护剂。
➤ 帕金森病
帕金森病是一种进行性神经退行性疾病,其中α-突触核蛋白的聚集导致黑质神经毒性,导致多巴胺能神经传递不足。
大量数据表明肠道微生物组通过诱导炎性神经毒性参与帕金森发病机制。这些患者肠道拟杆菌属的丰度与运动症状严重程度和促炎性TNFα和IFNγ水平相关。尤其是疣微菌门(Verrucomicrobia)与循环中较高水平的IFNγ相关,说明了与IDO和色氨酸代谢产物的可能相互作用。
最近的研究还表明,帕金森病患者的色氨酸代谢紊乱,是潜在的治疗目标。帕金森患者血浆中3-HK显著升高,3-HANA降低。在这些患者中,犬尿酸显著降低,喹啉酸水平与疾病严重程度相关,表明色氨酸代谢产物在加重兴奋性毒性损伤中的潜在致病作用,尽管因果作用仍有待确定。
帕金森患者脑脊液和血浆中的犬尿氨酸/色氨酸比率升高,犬尿氨酸转氨酶活性降低。因此,犬尿酸合成类似物已成为治疗帕金森、亨廷顿病和阿尔茨海默病的神经保护药物。
➤ 其他神经退行性疾病
已知 NMDA 受体过度激活和随之而来的神经元兴奋性毒性在几种神经退行性疾病的发病机制中发挥作用。
——肌萎缩侧索硬化症
犬尿酸可能作为一种内源性神经保护剂发挥其拮抗 NMDA 受体过度激活的作用。临床数据表明了潜在的作用。晚期和延髓起病的肌萎缩侧索硬化症患者的 CSF 中 KA 水平显着升高。
——亨廷顿病
在亨廷顿病患者中,产生自由基的 3-HK 在早发性疾病中高度升高,同时纹状体和皮质喹啉酸也升高。然而,对于更晚期的疾病,这些浓度会降低。
虽然亨廷顿病患者的 CSF 中犬尿酸水平升高,对亨廷顿病大脑的尸检分析显示,与对照组相比,犬尿酸浓度降低,血清犬尿氨酸/色氨酸 比率升高。有趣的是,谷氨酰胺重复次数和疾病严重程度与循环色氨酸水平呈负相关。
亨廷顿病大鼠模型表明 3-HK 增强了神经兴奋性毒性,而自由基清除剂抑制了这种作用。
犬尿酸的合成类似物在原位产生神经保护和抗癫痫作用。鉴于IPA的抗氧化作用,研究人员还提出使用这种吲哚衍生物对亨廷顿病患者进行神经保护。
➤ 多发性硬化症
多发性硬化症是一种慢性、进行性和复发性中枢神经系统炎性脱髓鞘疾病。许多证据表明,这主要是由B和T细胞驱动的过程。最近,许多靶向B细胞和T细胞活化的药物被证明在预防复发方面具有临床疗效。
多发性硬化患者肠道菌群变化
多发性硬化患者粪便样本的微生物组分析显示,与对照组相比,多发性硬化患者中的Methanobrevibacter和Akkermansia增加,Butyricimonas 减少。
肠道微生物代谢产物参与多发性硬化发病
色氨酸代谢产物和I型IFN信号已显示在多发性硬化的实验性变态反应性脑脊髓炎(EAE)模型中激活星形胶质细胞AHR,从而抑制中枢神经系统炎症。
评估这种疾病中的犬尿氨酸途径的研究取得了有趣的结果。复发患者犬尿酸水平升高,而尸检样本显示犬尿氨酸转氨酶活性降低。
此外,喹啉酸可能诱导少突胶质细胞凋亡,导致脱髓鞘损伤。在其他EAE模型中,数据显示了有毒的犬尿氨酸代谢产物的集中聚集。
在这些模型中,肠道微生物也会影响中枢免疫,因为继发于微生物变化的免疫过度激活会加剧炎症损伤。
● 神经精神疾病中的色氨酸代谢
KP 的不平衡导致具有特定神经活性特性的代谢物过多,被认为是导致多种神经精神疾病的原因。
➤ 焦虑和抑郁
色氨酸代谢产物,尤其是血清素,与焦虑和抑郁的发病机制密切相关。
促进中枢5-羟色胺可用性的药物,特别是选择性5-羟色胺再摄取抑制剂(SSRIs)、MAO抑制剂(MAOIs)和三环类抗抑郁药(TCAs)已经彻底改变了这些疾病的治疗。
重度抑郁症、自杀倾向与喹啉酸水平升高相关
例如,重度抑郁症与 KP 的 3-羟基犬尿氨酸 (3HK) 分支下的新陈代谢增加有因果关系,导致大脑神经毒性喹啉酸水平高于神经保护性 KA。
同样,与 KA 和吡啶甲酸相比,喹啉酸水平升高也与自杀倾向相关。社会心理压力、感染或细胞因子治疗引起的免疫激活会导致抑郁症状。
系统性 IDO1 激活与抑郁症中 3HK 分支的激活有关
小鼠中的 IDO1 抑制或敲除可减轻抑郁样行为,细胞因子诱导的抑郁症易感性与 IDO1 基因的多态性有关。因此,系统性 IDO1 激活被认为与抑郁症中 3HK 分支的激活有关,但目前尚不清楚为什么 KA 和喹啉酸在对 IDO1 诱导的反应中没有同样上调。
随着对不同疾病过程中肠道微生物组组成的理解的增加,很明显,肠道微生物可能在这些疾病的起源和临床表型中发挥关键作用。
无菌小鼠表现出比常规饲养小鼠更焦虑的行为,这种行为在宿主断奶后不易随着微生物的重新繁殖而逆转,这表明肠道微生物组可能支持心理发育的关键时期。
一些益生菌在动物模型和人类中显示出减少焦虑和抑郁的功效。
补充色氨酸减少焦虑?仍然存在争议
瑞士乳杆菌R0052和长双歧杆菌R0175等物种的益生菌分别降低了小鼠和人类的焦虑、增强了情绪幸福感和抑郁症状。
患有抑郁症患者粪便微生物移植的小鼠表现出更严重的焦虑,这与更高的循环犬尿氨酸和犬尿氨酸/色氨酸比率有关。
有趣的是,已知慢性应激会增加循环色氨酸和皮质醇,由于糖皮质激素诱导的TDO表达增强,导致5-羟色胺代谢向犬尿氨酸及其代谢产物分流。
小鼠应激诱导的结果表明,外源性丁酸盐调节应激诱导的抑郁行为,降低海马血清素,增加海马脑源性神经营养因子(BDNF)。
肠道微生物群被抗生素耗尽的小鼠表现出类似焦虑的行为,循环的犬尿氨酸升高。在喂食高脂肪饮食的肥胖大鼠中,花青素可以防止神经炎症,并且循环色氨酸降低,犬尿酸增加。
➤ 精神分裂症
色氨酸向血清素的转换可能在精神分裂症的发病机制中受损,因为某些TPH1多态性增加了对精神分裂症和自杀的易感性。
精神分裂症患者 KA 的水平升高,与认知缺陷相关
精神分裂症和精神病似乎是由 NMDA 受体拮抗剂 KA 的形成增加引起的。在精神分裂症患者的死后大脑和脑脊髓液中测量到 KA 水平升高。KA 水平升高与在精神分裂症中观察到的认知缺陷相关、而KA 形成减少与认知功能改善相关。
同样,特别是在发育中的大脑中的神经炎症与精神分裂症的认知缺陷特征有关。KMO中的单核苷酸多态性基因与精神分裂症和双相情感障碍相关,这表明 KP 3HK分支下的流量减少可能使 Kyn 转向 KA 形成,KA 的积累与这些疾病有关。
脑脊液中低5-HIAA水平与自杀和攻击行为相关
由于精神分裂症患者皮质犬尿酸水平升高,犬尿氨酸代谢产物也可能起到致病作用。动物模型研究表明,色氨酸抑制攻击性行为,可能与增加中枢血清素的可用性有关。
小胶质细胞突触修剪过度激活
Sekar及其同事发表了一项具有里程碑意义的全基因组关联研究,该研究确定了与精神分裂症发病机制有关的基因位点,这涉及补体C4介导的小胶质细胞突触修剪过度激活。
肠道失调与免疫失调联系起来
鉴于肠道微生物组在介导中枢免疫中的既定作用,以及病例对照研究在精神分裂症患者中的优势,研究人员试图将肠道失调与免疫失调联系起来,导致大脑发育关键时期突触修剪过度活跃。
流行病学研究也支持这样的假设,即全身感染诱导的母体免疫激活是后代患精神分裂症的独立危险因素。
最近的一项研究表明,产前免疫暴露导致额叶皮质C4活性上调。母体微生物组向后代的垂直转移也可能导致持续的免疫功能障碍,增加突触过度修剪的风险。
需要进一步的机制研究来了解肠道微生物、色氨酸代谢产物和宿主免疫在精神分裂症和其他神经发育障碍发病机制中的相互作用。
➤ 自闭症
根据循环色氨酸的临床研究和排泄的犬尿氨酸代谢产物的检查,自闭症谱系障碍患者可能缺乏色氨酸。
自闭症患者来源的淋巴母细胞的代谢组学分析显示,当色氨酸是唯一可用的能量来源时,NADH生成减少,表明这些患者的喹啉酸降解途径可能受损。
某些微生物物种可能参与了自闭症的发病机制
几项评估自闭症患者肠道微生物丰度差异的研究将自闭症症状与Prevotella、Coprococcus、Veillonellaceae丰度较低联系起来。
脆弱拟杆菌(一种胰蛋白酶合成细菌),可能会降低自闭症患者的色氨酸可用性。非色氨酸衍生的微生物代谢产物也可能起到因果作用,一项观察自闭症小鼠母体免疫激活(MIA)模型中肠道微生物代谢产物的研究显示,微生物代谢产物4-乙基苯基硫酸盐增加了46倍,如果小鼠被脆弱拟杆菌定殖,则其正常化。
色氨酸代谢在肠道菌群-脑轴中的潜在作用
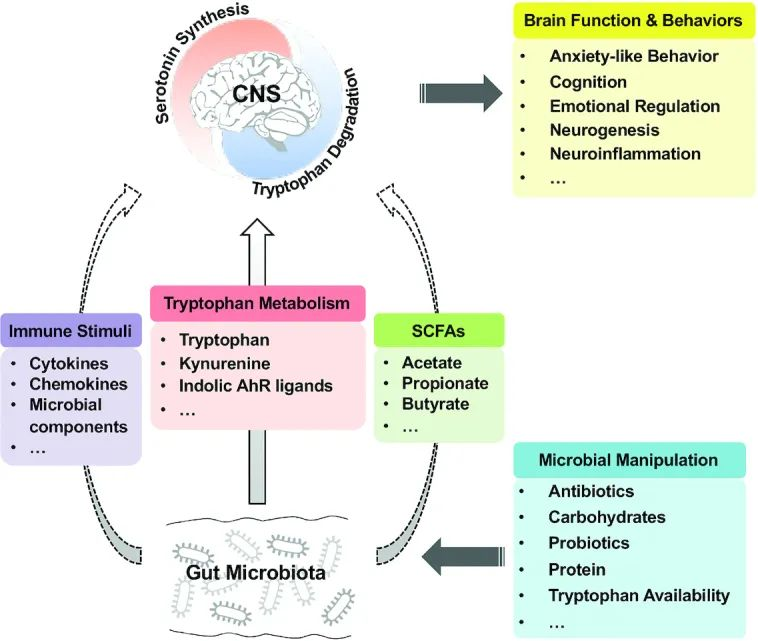
doi: 10.1093/advances/nmz127
通过各种方式(例如,抗生素和益生菌)对肠道微生物群组成和代谢的调控有助于在 5-羟色胺合成和色氨酸降解途径之间改变中枢色氨酸代谢,从而影响大脑功能和行为。
● 中枢神经系统疾病中靶向 KP 酶
尽管临床试验的重点(部分仍然是)补充或剥夺色氨酸或其代谢物以治疗神经精神疾病,但目前神经退行性和神经精神疾病药物开发的临床前工作主要集中在通过改变神经活性 KP 代谢物的变阻器抑制参与 喹啉酸或 KA 形成的酶。
从概念上讲,所有 KP 酶都代表潜在的治疗靶点,并且有几项研究调查了药理学抑制的影响。
例如,IDO1 抑制剂黄连碱已被证明可以减缓阿尔茨海默小鼠模型的认知障碍,尽管其对 IDO1 的特异性尚不清楚。
有趣的是,环氧合酶抑制通过抑制海马 TDO 表达,来防止类似阿尔茨海默模型中的行为下降。当使用 TDO 的药理学抑制剂时,观察到类似的神经保护作用。这些研究连同阿尔茨海默病和亨廷顿病患者中 KP 激活的证据, 表明抑制色氨酸降解中限速的第一个酶促步骤是一种潜在可行的治疗方法,可以抵消由淀粉样蛋白形成蛋白的积累引起的神经毒性。
尽管 IDO1 和 TDO 的抑制剂阻止了 KP 代谢物的产生,但这不会直接影响 KA/喹啉酸 喹啉酸变阻器,但会阻止两者的产生。这种治疗方法是可行的,因为它可以防止色氨酸的消耗,这可以减少在临床前模型中观察到的蛋白质毒性。
KAT 在辅助因子 pyridoxal-5-phosphate (PLP) 的帮助下催化 Kyn 转化为 KA。KATII 是哺乳动物大脑中最普遍的 KAT,并且正在寻求作为精神分裂症和认知障碍疾病的药物靶点。由于最近显示 KATII 也能催化 3HK 105形成 XA ,因此之前归因于 KA 的基于抑制 KATII 的效应也可能涉及 XA。
▸ KATII 抑制剂
KATII 的可逆抑制剂已经开发出来,包括 Kyn 类似物 (S)-4-(ethylsulfonyl)benzoylalalanine ( S -ESBA) ,它被证明可以降低大鼠大脑中的 KA 水平。
高效和选择性脑渗透不可逆抑制剂 PF-04859989 也报道了相同的抑制模式。然而,这些化合物都没有进入临床研究,这可能是由于它们与 KAT 同工酶和所有其他 PLP 依赖性酶所需的 PLP 辅因子发生不可逆相互作用而引起的毒性。
将 KATII 抑制剂推进临床试验的主要挑战包括由脑 KA 水平降低引起的潜在毒性、获得足够的效力和选择性以及 KATII 抑制剂效力的种间差异的发生。
▸ KMO 抑制剂
为了抑制 KP 的 喹啉酸分支和增加拮抗 KA 水平,KMO 抑制剂正在积极开发中。有关 KMO 晶体结构的信息有助于生成特异性更高的 KMO 抑制剂。众所周知的 KMO 抑制剂 Ro 61-8048已用于大量临床前研究,证明其作用范围从改善神经变性到减少大麻素滥用。
另一种广泛使用的工具化合物,UPF-648, 是一种不含氨基的 Kyn 类似物,在构象上受到环丙基环的限制。这种化合物,以及高效的恶唑烷酮 GSK180(在胰腺炎的背景下研究),是所谓的 I 型 KMO 抑制剂,它模仿 Kyn 并刺激有害的过氧化氢产生。
在一项基于结构的药物化学合作研究中,开发并评估了一种新的芳基嘧啶先导化合物 CHDI-340246,用于治疗 HD。然而,这种选择性 KMO 抑制剂的长期治疗并未显着改变 HD 小鼠模型的行为表型或自然进展,尽管它恢复了电生理学改变。
结构研究最近破译了 I 型和 II 型KMO抑制剂112、121之间的区别。II 型 KMO 抑制剂 GSK065 和 GSK366 显示出比 I 型 KMO 抑制剂更好的类药特性,因为它们具有皮摩尔亲和力、增加的停留时间和不产生过氧化物。
GSK065以GSK3335065(NCT03245619)的名称进入治疗胰腺炎的I期临床试验。有趣的是,KMO 抑制剂的外周给药足以影响 CNS KP 。然而,KMO 抑制剂是否需要穿透血脑屏障才能发挥作用,这仍然是一个有争议的问题。
最后,抑制初始限速 KP 酶 IDO1 和 TDO,它们分别在炎症条件或慢性社会心理压力下诱导,在神经退行性疾病和精神疾病中也可能值得探索。由于这些酶的抑制剂目前正在开发用于癌症治疗,因此可以使用多种化合物在临床环境中测试这些方法。
/
/
▼
● 感染中的色氨酸代谢
几条证据最近揭示了色氨酸代谢作为宿主-病原体相互作用和塑造宿主微生物群中免疫反应的重要调节因子的关键作用。
通过特定的色氨酸代谢酶,色氨酸代谢在细菌、病毒、真菌和寄生虫感染部位增加。通常以低基础水平表达,在抗原呈递细胞 (APC)中观察到 IDO1 增加,例如树突细胞 (DC) 和巨噬细胞,以响应多种微生物刺激,包括 Toll 样受体 (TLR) 配体(例如,脂多糖 (LPS) , CpG 寡核苷酸和聚肌胞苷酸 。
炎症刺激物诱导IDO1,IDO1 会耗尽色氨酸
此外,据报道,I 型和 II 型干扰素 、肿瘤坏死因子 (TNF)、前列腺素 和膜结合分子 等炎症刺激物可在特定APC类型中诱导IDO1。
在传染病中,IDO1 活性具有多效性,是一把双刃剑。实际上,IDO1 会耗尽色氨酸以饿死和重新编程营养缺陷型入侵者,同时有助于对在急性感染期间未清除的微生物产生 Kyn 依赖性免疫抑制状态或那些已经能够重新激活色氨酸生物合成的。
因此,已经表明色氨酸营养缺陷型病原体对 CD4 + T 细胞激活的巨噬细胞高度敏感。 在特定的环境条件下,色氨酸的微生物营养缺陷型可能会消失。
特定条件微生物重新获得合成必须氨基酸的能力
某些微生物可以在特定的胁迫条件下重新获得合成这种必需氨基酸的能力。此外,天然能够合成色氨酸的微生物群菌株可以在特定感染期间扩大,从而在色氨酸缺乏的条件下提供额外的这种必需氨基酸供应。
最近的研究结果表明,结核分枝杆菌等特定病原体可以在压力条件下重新获得合成色氨酸的能力,从而抵消 IDO1 饥饿驱动的抗菌作用。
此外,衣原体在由局部色氨酸剥夺引起的应激条件下进入非复制的持久状态。同样,IDO1 依赖性持久性已被记录在其他细菌物种中,包括肺炎积瘤。
除了调节病原体负荷外,通过 IDO1 活性进行的色氨酸代谢对于抑制最终阻止病原体根除的免疫病理也至关重要。
在这方面,最近对肠道微生物群的研究发现:
色氨酸代谢与通过充当特定 AHR 配体的微生物或细菌毒力因子在粘膜屏障表达的 AHR 激活之间存在重要联系。
值得注意的是,AHR +由于产生色氨酸代谢物(即吲哚 3 醛)的乳酸杆菌的选择性扩增,即使在 IDO1 缺乏的情况下,也会诱导产生IL-22 的第 3 组先天淋巴样细胞 (ILC3s)能够激活 AHR,从而在真菌感染模型中诱导保护性耐受状态。
TDO 在感染过程中的潜在作用
用 LPS 攻击的小鼠肝脏中 TDO 表达增加,而 TDO 缺陷小鼠更容易受到内毒素攻击。因此,在对弓形虫和金黄色葡萄球菌感染进行的体外研究中,已经报道了 TDO 依赖性抗菌和免疫调节作用。此外,代谢组分析揭示了原发性登革热感染患者 TDO 激活的变化。
因此,在宿主细胞中的三种不同色氨酸分解代谢酶中,IDO1 的影响已在几种临床前感染模型中得到解决。具体而言,据报道,IDO1 在体内抑制某些细胞内寄生虫和细菌的复制,例如弓形虫、衣原体和杜氏利什曼原虫。另一方面,弱 IDO1 抑制剂 1-甲基- L – Trp (L-1-MT) 增强了沙眼衣原体抗生素清除的功效,尽管可能涉及额外的 IDO1 独立机制。
IDO1活性还可以在体外抑制特定病毒的复制
例如人巨细胞病毒 (CMV)、2 型单纯疱疹病毒和痘苗病毒。然而,体内情况可能有所不同,因为病毒感染可能会诱导 IDO1 和 KP 逃避宿主免疫反应。
由于它们具有诱导 Treg细胞的能力 ,因此 IDO1 消耗色氨酸并产生 Kyn 是抑制抗菌 TH17 和 TH1 驱动的炎症的重要手段。
因此,病原体可能会劫持 IDO1 的免疫抑制作用,并利用它们来促进自身的生命周期。在这方面,尿道致病性大肠杆菌(UPEC) 在泌尿道的上皮细胞中诱导 IDO1 ,并且色氨酸分解代谢的免疫反应减弱使得 UPEC 能够成功定植。
HIV-1 等病毒利用 IDO1 的免疫抑制活性建立 HIV 慢性感染
KP 活性的增加也与丙型肝炎病毒感染患者的进行性肝硬化有关。
同样,小鼠感染甲型流感/PR/8/34 (PR8) 会刺激肺部和肺引流纵隔淋巴结中 IDO1 活性的快速升高,导致发病率增加、恢复减慢和肺部效应 T 细胞反应降低,尽管在原发性甲型流感病毒感染期间,IDO1 诱导不会影响病毒清除。在其他情况下,例如在真菌感染中,IDO1 可用作建立共生或慢性感染的逃避机制。
● 在传染病中靶向 KP 酶
在选定的微生物物种中调节特定的色氨酸生物合成途径并靶向宿主细胞中的 IDO1-AHR-微生物群轴可能代表了抗生素开发或补充抗病毒疗法的新颖有吸引力的策略。有必要更全面地了解特定感染期间色氨酸分解代谢酶或下游酶的作用,以便了解旨在调节色氨酸分解代谢以根除病原体同时保持与微生物群平衡的疗法的效用。
基于以上总结的证据,可以假设特定的 IDO1 阻断剂可能会发现潜在的应用作为辅助疗法来提高抗病毒药物的疗效,但可能证明对真菌感染有害,其中色氨酸分解代谢主要通过 IDO1 作用于维持免疫稳态和保护性耐受。
然而,这种作用可能构成使用 IDO1 抑制剂作为抗肿瘤药物的潜在缺点(下面讲)。实际上,在使用 IDO1 通路调节剂 1-甲基-D -Trp (D-1-MT)对转移性实体瘤患者进行的 I 期试验中,感染是最常见的不良事件。
有趣的是,最近的一项研究表明,KYNU 的靶向抑制会影响铜绿假单胞菌基因表达和群体感应,这表明一种新的潜在抗毒策略。具体而言,与 Kyn 具有结构相似性的S-苯基-L-半胱氨酸亚砜可抑制对铜绿假单胞菌毒力至关重要的邻氨基苯甲酸盐的产生。
/
/
▼
● 自身免疫中的色氨酸代谢
自身免疫是未能发展出对自身的中枢(胸腺)耐受性和外周耐受性维持不足的结果。免疫区室中的色氨酸代谢主要由 IDO1 启动,它代表主要促炎刺激的靶基因。
在这方面,IDO1 介导的色氨酸降解可被视为调节过度活跃的免疫反应的关键反馈机制,这是自身免疫性疾病的标志。
IDO1 在发炎组织中转录激活抑制适应性免疫反应的作用已经从最初在胎盘中观察到的维持胎儿耐受性扩展到多种自身免疫性疾病。
尽管 IDO1 缺陷不会导致与 Treg细胞重要检查点缺陷相关的整体自身免疫表型,但它与更微妙的炎症表型相关。这种关联可能部分是由于与其他双加氧酶共享的酶功能冗余。越来越多的证据表明,人类自身免疫性疾病是由免疫和/或基质细胞未能上调 IDO1 以响应炎症刺激驱动的。
然而,与自身免疫相关的上调 IDO 的结构性缺陷的潜在原因尚未阐明。连锁分析将IDO1和IDO2基因的多态性分别与克罗恩病的严重程度和风险相关联。需要进一步的研究来确定上调组织中色氨酸代谢的本构或诱导缺陷是否会导致组织特异性自身免疫。
许多针对多发性硬化症、类风湿性关节炎、狼疮和自身免疫性糖尿病的自身免疫性疾病小鼠模型的研究已经证明色氨酸代谢在调节疾病活动中的相关性。
综上所述,这些研究表明 IDO1 在组织驻留的骨髓细胞中表达,并限制对自身抗原和炎症病理学的先天性和适应性免疫。
然而,自相矛盾的是,在自发性类风湿性关节炎的动物模型中,使用 D/L-1-MT 对 IDO1 的药理学抑制减轻了疾病的严重程度,这可能是自身反应性 B 细胞活化减少的结果。
这一发现说明了 IDO1 在自身免疫中的复杂免疫调节功能,这取决于细胞区室。例如,B 细胞中免疫抑制细胞因子 IL-10 的表达依赖于 IDO1,这表明 IDO1 不仅会触发免疫抑制机制,还会协调对炎症的复杂免疫调节反应。
重要的是,转录激活和蛋白质表达不一定转化为人类 B 细胞中描述的酶活性。 在这方面,需要更多的研究来阐明 IDO1 的非酶功能。
此外,需要谨慎看待使用 D-1-MT 得出关于 IDO1 功能的关键结论的研究,因为 D-1-MT 不会抑制 IDO1 并显示出相当大的脱靶效应,从而导致 p38 MAPK 通路的激活。
对自身免疫性疾病模型的研究表明,与 IDO1 相比,IDO2 充当自身免疫的促进者,特别是由于体液免疫反应的调节。由于致病性自身抗体和抗体分泌细胞的减少,IDO2 缺陷小鼠表现出关节炎症减少。尽管与人类疾病的相关性仍不清楚, 这些研究突出了双加氧酶对色氨酸代谢的复杂和高度分隔的调节。
尽管 IDO1 介导的免疫调节的主要作用被认为是由组织炎症的局部微环境中的活动驱动的,但在患有自身免疫性疾病的患者中观察到色氨酸代谢的全身激活。在干燥综合征患者中,血清中色氨酸的降解增加,并与循环 Treg细胞频率增加有关。
相比之下,在多发性硬化症中,血清中的 IDO1 活性与健康对照相比没有显着差异,但抗炎治疗降低了 IDO1 活性。由于全身性 IDO1 活性会受到大量非特异性和难以控制的刺激的影响,包括感染、压力和营养,尝试通过循环色氨酸代谢物监测组织特异性自身免疫将具有挑战性。然而,对血清中 KP 代谢组的更详细分析不仅揭示了 KP 在多发性硬化症患者中的激活,而且还将 KP 活性的程度与疾病严重程度相关联。因此,KP 活性可以作为能够指导多发性硬化症治疗的预测性生物标志物。
● 靶向自身免疫性疾病中的色氨酸代谢
在治疗上针对色氨酸代谢的努力主要集中在开发具有 Kyn 样特性的药物上。
曲尼司特
曲尼司特是一种具有 AHR 激动特性的 AA 衍生物,能够在多发性硬化症和类风湿性关节炎的临床前模型中诱导免疫耐受和改善疾病活动。然而,一项针对类风湿性关节炎患者的 II 期临床试验(NCT00882024)因肝毒性而终止。
拉喹莫德
拉喹莫德是一种喹啉甲酰胺,在开发治疗多发性硬化症时显示出与 KA 的结构相似性,它以 AHR 依赖性方式抑制多发性硬化症临床前模型中的自身反应性 T 细胞免疫和疾病活动。
在针对复发和进行性多发性硬化症患者的一系列 II/III 期临床试验中,拉喹莫德未达到预先指定的主要终点,包括减少复发率和残疾进展,因此被终止 (NCT01707992)。
AHR 配体改善自身免疫神经炎症
AHR 的特定内源性配体足够稳定,可以在临床前疾病模型中进行肠胃外给药。2-(1’H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) 通过以 AHR 依赖性方式诱导致耐受性 DC,在实验性自身免疫性脑脊髓炎 (EAE) 模型中诱导 Treg细胞并改善自身免疫性神经炎症。
AHR 激活配体也可以与自身抗原偶联,从而导致 APC 的特异性靶向,然后耐受性抑制自身反应性 T 细胞反应,从而抑制系统性自身免疫。
从概念上讲,色氨酸代谢也可以通过全身给药色氨酸来增强,色氨酸在口服灌胃后会迅速代谢成 Kyn。尽管这种方法导致 TH17 免疫力的不同抑制,但这并不转化为实验性自身免疫性神经炎症的改善。
阻断IDO1降解,维持外周耐受性
认识到蛋白酶体降解是调节自身免疫中色氨酸代谢的免疫抑制活性的重要机制后,另一种治疗途径是阻断IDO1降解,从而维持外周耐受性。
硼替佐米是一种批准用于治疗多发性骨髓瘤的蛋白酶体抑制剂,可防止 IDO1 降解并以 IDO1 依赖性方式在临床前动物模型中改善自身免疫性糖尿病。
基于 IDO1 感受态细胞的疗法
尽管间充质干细胞的过继转移已被证明可以独立于IDO抑制自身免疫性神经炎症的临床疾病活动,但 IDO1 已被证明在其他自身免疫性体内疾病模型中与间充质干细胞的免疫抑制特性密切相关。
增强或诱导宿主 IDO 表达的另一种方法是通过局部基因治疗。例如,腺病毒将 IDO1 递送至移植器官可诱导免疫耐受并防止大鼠发生移植排斥反应。
IDO2 直到最近才成为潜在的治疗靶点
迄今为止,还没有对 IDO2 具有足够特异性的小分子。在自身免疫性关节炎的临床前模型中,一种通过内化靶向 IDO2 的抗体通过抑制自身反应性 T 细胞和 B 细胞减轻了疾病。
新开发的 IDO2 特异性测定系统和基于计算结构的研究可能有助于开发对 IDO1 没有交叉反应的 IDO2 抑制剂。
/
/
▼
● 肠 病
最近研究强调了肠道色氨酸代谢的改变与肠道微生物的潜在联系。发现 IBD 患者微生物群产生的 AhR 配体减少,这是受遗传因素的影响。与健康受试者相比, 肠道组织中 AhR 的表达降低。
IBD还与宿主和肠道细菌色氨酸代谢物的改变有关。IBD 患者的犬尿氨酸和 KA 血浆水平升高,血浆色氨酸浓度降低。
几种特定的肠道细菌色氨酸代谢物也参与 IBD 的病理生理学
在患有 IBD 的狗中,被认为在肠道中具有抗炎功能的细菌色氨酸代谢物(吲哚乙酸盐和吲哚丙酸盐)显着减少。在 IBD 患者中,粪便中 IAA(肠道抗炎功能)水平降低,表明细菌色氨酸代谢减少可能是 IBD 的病因。
此外,在 IBD 患者中,可利用 α-L-岩藻糖苷酶从肠粘蛋白中切割末端岩藻糖残基的细菌数量显着减少,这与来自色氨酸的吲哚丙烯酸和吲哚-3-丙酸产量减少有关。
IBD 患者的 IDO1 活性更高
据报道,IBD 患者外周血和结肠细胞中的 IDO1 活性增加。在 IBD 中,增加的促炎细胞因子,包括 IFN-γ、IL-1 和 IL-6,已被建议诱导色氨酸分解代谢途径以降低血浆色氨酸水平,并增加色氨酸分解代谢物水平。
此外,UC 患者血清中的 IPA 降低。在肠道局部观察到 IDO1 的过度激活和免疫系统过渡激活。与非活动性 IBD 患者相比,活动性 IBD 患者的 IDO1 活性更高,并且色氨酸和 C 反应蛋白血清水平呈负相关,这一假设得到了支持,C 反应蛋白是一种常用的生物标志物,对炎症反应增加。
IBD 中 5-HT 通路激活的状态存在争议。限速酶TpH1的表达增加在克罗恩病中已报道。
小鼠研究表明AhR 缺乏会增加实验性结肠炎的严重程度
这种结肠炎是由 T 细胞转移或通过施用葡聚糖硫酸钠 (DSS) 以化学方式驱动的。在这些模型中,AhR 缺陷部分通过改变白细胞介素 (IL)-22 的产生来驱动结肠炎,白细胞介素 (IL)-22 是一种对肠道稳态具有众所周知影响的细胞因子。
缺乏 caspase 募集域 9 (Card9)(一种 IBD 易感基因)的小鼠的肠道菌群失调无法将色氨酸催化成 AhR 配体,导致 IL-22 释放减少并最终导致Card9的易感性更高-/-小鼠对 DSS 诱导的结肠炎 。
在人类中也发现了一些功能相关性,因为 AhR 的药理学激活减少了促炎细胞因子干扰素 (IFN) γ 的产生,并增加了 IBD 患者固有层单核细胞中 IL-22 的产生。
此外,正如在患有 UC 的人类中观察到的那样,IPA 和吲哚在 DSS 诱导的结肠炎小鼠血清中减少,另外的证据表明口服 IPA 在该模型系统中具有保护特性。
KP 的改变也可能在机制上参与 IBD 发病机制
IDO1 -/-小鼠更易患结肠炎,表明 IDO1 是肠道炎症的负调节剂。与 IDO1 缺乏相关的病理损伤部分是由于促炎细胞因子的激活和结肠中 CD4+ Foxp3+ 调节性 T 细胞数量的减少。然而,所涉及的确切机制和代谢产物仍然未知。
众所周知,Kyn 是一种 AhR 激动剂,但在肝癌细胞系中引发报告基因 AhR 活性所需的浓度让人怀疑它在生理条件下作为 AhR 激活剂的相关性,可能涉及导致抗炎代谢物(如 Kna)缺乏的下游代谢途径的改变,但这仍有待证明。在 IBD 的背景下,来自失调微生物群的异常信号可能是 KP 的驱动因素。
5-HT 加重肠道炎症
化学诱导的结肠炎的严重程度在TpH1 −/−小鼠和用 5-HT 合成抑制剂对氯苯丙氨酸处理的小鼠中减弱,表明 5-HT 加重肠道炎症。此外,删除 SERT 会导致 5-HT 可用性增加,从而导致实验性结肠炎恶化。
这些促炎作用可能部分是由DC上 5-HT 7受体的激活驱动的。然而,新的线索表明 5-HT 还通过作用于 5-HT 4 发挥抗炎作用对肠上皮细胞屏障功能产生积极影响。
总之,这些数据表明在 IBD 中观察到的色氨酸代谢改变可能在疾病发病机制中发挥积极作用。就这些微生物产生 AhR 激动剂的能力受损而言,微生物群的参与是显而易见的,但也可能解释了在生理条件下微生物群的直接影响下发生的 IDO 和 TpH1 的局部激活加剧。
● 肠易激综合症
IBS 的病因在很大程度上是未知的,但可能与色氨酸代谢受损有关。IBS 患者血清中 Kyn 升高,外周 IDO1 活性与 IBS 严重程度呈正相关。
IBS 与通过 KP 增加的色氨酸代谢有关
犬尿氨酸:色氨酸比率与 IBS 症状严重程度呈正相关,IFN-γ 激活和随后的色氨酸 IDO1 氧化可能是 IBS 的致病机制。
此外,血清素能系统的功能障碍与 IBS 的病理生理学有关。与急性色氨酸耗竭治疗相比,IBS 患者通过急性色氨酸增加治疗进行的 5-羟色胺能调节导致更严重的胃肠道症状。
肠道运动的改变是 IBS 的关键特征之一,与 5-HT 代谢障碍有关
与健康对照组相比,IBS 患者的直肠活检组织中发现 TpH1 和 SERT 表达水平降低。
此外,5-HT 结肠内容物在便秘型和腹泻型 IBS 中分别减少和增加。5-HT 的多效性与其受体的多样性有关,这些受体能够触发特定器官的特定功能。
在胃肠道中表达最多的5-HT 3和 5-HT 4亚型将 5-HT 与内脏伤害感受和运动障碍联系起来。5-HT 的作用已经被开发为治疗靶点,使用 5-HT 3受体拮抗剂和 5-HT 4受体激动剂分别显示出对腹泻和便秘为主的 IBS 的一些疗效。
然而,受肠道微生物群调节的中枢血清素作用紊乱也可能参与 IBS 发病机制。肠道菌群对 5-HT 产生和肠道运动的影响已在小鼠身上得到证实,并表明 IBS 发病机制部分与微生物群对 5-HT 产生的功能失调控制有关。
● 与年龄有关的胃肠功能障碍
高龄增加了个体对胃肠道功能障碍的易感性,这归因于与年龄相关的神经元丢失。
5-HT4 激动剂刺激发育中的肠道中的神经突生长和网络形成,也已被证明可以防止神经元凋亡和炎症诱导的轴突变性和自噬。
此外,5-HT4 受体激动作用可促进成人肠道神经发生。相应地,其他方面健康的高龄个体表现出循环色氨酸减少,可能会限制血清素的可用性。
/
/
▼
● 衰 老
衰老与肠道微生物群的变化有关,这通常与胃肠道的生理变化有关,同时免疫系统功能下降可能导致感染、营养不良和其他功能缺陷的风险增加。
老年人菌群特征
老年人的肠道微生物群通常以细菌多样性降低、优势菌种改变、有益微生物减少和兼性厌氧菌增加为特征,所有这些都表明与衰老相关的微生物变化具有潜在的不利影响。微生物群组成的变化与老年人的免疫衰老和炎症有关。
色氨酸代谢受衰老影响
色氨酸在诱导免疫耐受和维持肠道菌群方面起着至关重要的作用。
对老年人和年轻人肠道核心微生物组直系同源基因的分析表明,丰度增加的年龄相关基因参与了色氨酸代谢途径 (ko00380),这与在百岁老人血清中发现的与年龄相关的色氨酸浓度降低一致。研究表明,肠道微生物群对色氨酸消耗的潜在增加可能会影响宿主对色氨酸的生物利用度。
最近的一项研究报告了血清色氨酸水平降低与免疫激活增加之间的关系。还推测微生物群依赖性色氨酸减少会增强百岁老人的炎症。
食物传感信号通路调节寿命,与色氨酸关联
几种食物传感信号通路,包括胰岛素/胰岛素样生长因子 (IIS) 通路和哺乳动物雷帕霉素靶标 (mTOR) 通路,已被证明可以调节模式生物的寿命,并且已经提出了类似的关联对于KP途径。
在人类中,表示该通路活性的 Kyn:色氨酸 比率随着年龄的增长而增加。这种增加与 65 岁以上人群的虚弱有关,并预示着 90 多岁人群的死亡率。
KP 的活性与衰老之间存在因果关系
此外,对成人个体外周血中年龄相关基因表达变化的荟萃分析将 KYNU 酶(犬尿氨酸酶,色氨酸降解途径关键酶)鉴定为表达差异最大的基因之一。在对秀丽隐杆线虫的后续研究中,通过 RNA 干扰 (RNAi) 敲低 KYNU 比敲低任何其他差异表达基因所达到的寿命更长,这表明 KYNU 对衰老有重要贡献。
连同独立发现,线虫和黑腹果蝇中 TDO 活性的遗传减少, 导致 Trp:Kyn 比率显着增加,延长寿命,这些研究表明 KP 的活性与衰老之间存在因果关系。
Kyn/Trp 分流在炎症中的后果
色氨酸代谢向肝外 Kyn 产生的炎症相关分流,可能影响衰老过程中一系列器官中色氨酸代谢物的功能。
编辑
doi.org/10.3389/fimmu.2019.02565
与年龄相关的组织稳态下降会导致生理上的低度慢性炎症表型,称为炎症。我们假设色氨酸向Kyn途径代谢,以控制与年龄相关的炎症。色氨酸和Kyn代谢产物的相应紊乱可能与年龄相关疾病和寿命缩短有关。
色氨酸参与调节寿命机制
KP 调节衰老的机制尚不清楚。已经针对不同的无脊椎动物和脊椎动物模型描述了氨基酸(包括色氨酸)在调节寿命方面的作用。在大多数情况下,色氨酸可用性的降低或细胞摄取的阻断可延长寿命。然而,这种机制与 TDO 抑制(增加色氨酸)延长寿命的发现相悖,除非这与细胞摄取减少有关。
此外,用 Kyn 喂养果蝇会缩短寿命,这表明该通路下游的代谢物水平也可能参与寿命的调节。TDO 耗尽对秀丽隐杆线虫延长寿命的影响取决于 FOXO 转录因子 DAF-16,它是寿命调节通路的介质,例如驱动细胞防御通路表达的 IIS 通路,表明它具有保护细胞免受细胞侵害的作用伤害。
有趣的是,防止与年龄相关的蛋白质毒性(这也是由秀丽隐杆线虫中的 TDO 耗尽引起的)不依赖于 DAF-16,并且独立于 KP 63中的下游酶。这一观察结果表明,延长寿命的效果要么是这种保护的结果,要么是由一种独立的机制引起的。
由于 NAD +正在成为一种潜在的延长寿命分子,KP 的改变可能通过 NAD +产生延长寿命的效果。然而,无脊椎动物的寿命更长是 KP 活性降低的结果,而通过外部供应其他 NAD +前体来延长寿命则表明 KP 活性的增加也是有益的。
需要更多的研究来理解这些看似矛盾的发现。由于 IDO1 或 TDO 的敲除小鼠是可行的,这些模型对于进一步研究 KP 中的寿命调节机制和潜在治疗靶点可能很有价值。KP 调节的寿命延长效应可能源于一般健康益处,而不是疾病特异性效应。
● 代谢综合征和肥胖
在患有代谢综合征的人类患者中,据报道 IDO1 过度激活会导致血清 Kyn 水平升高以及 Kyn/Trp 比率与肥胖、代谢综合征、BMI 和血液甘油三酯之间的相关性。
肥胖患者IDO1 的局部激活
IDO1和 KP 下游酶(如犬尿氨酸酶 (KYNU)、犬尿氨酸氨基转移酶 (KAT) 和犬尿氨酸 3-单加氧酶 (KMO))的基因表达增加已在肥胖患者的脂肪组织中观察到,表明 IDO1 的局部激活。
然而,循环 5-HT 水平在代谢综合征中降低,并且与 BMI 和体脂呈负相关。
色氨酸转化产物吲哚衍生物起作用
微生物群通过色氨酸转化产生的几种吲哚衍生物可能在代谢综合征的发病机制中起作用。
吲哚本身已被证明可以刺激肠内分泌 L 细胞产生胰高血糖素样肽-1 (GLP-1),这是一种刺激胰腺 β 细胞分泌胰岛素的肠降血糖素。这种机制涉及快速抑制刺激 GLP-1 分泌的电压门控 K+ 通道,但受 ATP 合成抑制的长期影响控制,减少 GLP-1 分泌。
硫酸吲哚酚促炎和氧化作用,与心血管和肾病等相关
吲哚也在肝脏中被吸收并代谢为硫酸吲哚酚。在肾衰竭期间,这种代谢物会积累,其促炎和氧化作用与动脉粥样硬化、动脉硬化、充血性心力衰竭和其他心血管并发症的发病机制有关,这些并发症在慢性肾衰竭患者中尤为突出。
硫酸吲哚酚在肾功能正常的受试者中的作用仍有待确定。KP 也与动脉粥样硬化有关。在小鼠模型中,IDO1 缺乏症通过 IL-10 产生失调减少了动脉粥样硬化病变的发展,这是一种通过施用 Kna 逆转的表型。在人类中,高 Kna 水平与不稳定的斑块表型相关。
低度慢性炎症可能有助于 IDO1 激活
KP 的过度激活也可能参与低度炎症情况下胰岛素抵抗的发生,例如肥胖、抑郁、丙型肝炎病毒感染和心血管疾病。人体和实验数据表明,黄嘌呤酸和 KP 的其他产物对胰岛素的产生和释放以及对靶组织的影响具有有害影响。
大脑中产生的血清素会引起饱腹感,但在微生物群的直接影响下产生的肠道来源的 5-HT 不会穿过血脑屏障。然而,色氨酸和直接 5-HT 前体 5-HTP 确实可以穿过血脑屏障,从而间接调节中枢 5-HT 的产生和功能。
外周 5-HT 独立于任何中枢效应影响宿主代谢
肠道来源的 5-HT 能够诱导食欲减退和饱腹感,其水平在禁食期间增加并刺激脂肪组织中的脂肪分解和肝细胞中的糖异生,有利于血糖控制。随后,通过一种涉及产热棕色脂肪组织消耗更多能量的机制,给予高脂肪饮食的 TpH1 基因或化学消融小鼠可免于肥胖、胰岛素抵抗和非酒精性脂肪肝疾病 (NAFLD)。
然而,这些结果可能不适用于棕色脂肪组织含量低且随年龄增长而减少的成年人。此外,人类肥胖与外周 5-HT 减少有关,表明其在发病机制中的复杂作用。
已经使用小鼠模型研究了 AhR 在代谢综合征中的作用,但尚未得出明确的结论。这可能与 AhR 的多重作用有关,AhR 在参与代谢综合征发病机制的各种细胞类型(肠细胞、肝细胞和免疫细胞)中表达。
/
/
▼
● 癌症中的色氨酸代谢
多项证据表明色氨酸代谢在癌症中具有重要作用,通过抑制抗肿瘤免疫反应和增加癌细胞的恶性特性来促进肿瘤进展。
首先,色氨酸降解酶在多种癌症中表达
IDO1 在大约 58% 的人类肿瘤中表达,其表达与多种癌症的不良临床结果相关,包括黑色素瘤、妇科癌症、结肠癌和血液系统恶性肿瘤。
IDO1 表达要么作为一种反调节机制被诱导,以响应从肿瘤浸润性免疫细胞释放的细胞因子,要么它的表达通过肿瘤固有的致癌信号传导维持。
TDO 催化与 IDO1 相同的反应,在神经胶质瘤、黑色素瘤、卵巢癌、肝癌、乳腺癌、非小细胞肺癌、肾细胞癌和膀胱癌中表达,并已被证明可促进肿瘤进展。
其次,各类癌症患者中全身色氨酸水平降低
已在成人 T 细胞白血病、结直肠癌 、妇科癌症、恶性黑色素瘤、肺癌和恶性神经胶质瘤患者中测量到全身色氨酸水平降低。在患有这些癌症的患者的血液中很少观察到 KP 代谢物浓度升高,这可能表明肿瘤微环境中 Kyn 和下游代谢物的局部变化受到更多限制。
第三,色氨酸降解在调节 Treg细胞和癌症中的免疫细胞浸润中发挥作用
FOXP3 + T reg细胞与宫颈癌引流淋巴结中表达 IDO1 的 DC 直接接触,IDO1 表达与转移性胰腺导管腺癌患者CD4 + CD25 + FOXP3 + T reg细胞增加有关,急性髓性白血病 (AML) 33和非霍奇金淋巴瘤。
此外,IDO1 表达与 CD3 + T 细胞、CD8的低肿瘤浸润相关+ T 细胞和 CD3 +和 CD8 + T 细胞以及 CD57 +自然杀伤细胞分别存在于结直肠癌、卵巢癌和子宫内膜癌患者中。
最近的一项研究表明,肿瘤再生细胞将 Kyn 转移到 CD8 + T 细胞,这反过来又以 AHR 依赖性方式上调程序性细胞死亡蛋白 1 (PD-1)。总而言之,这些观察结果为色氨酸代谢在肿瘤细胞免疫逃逸中的作用提供了机制解释。
第四,色氨酸代谢物可以有效促进癌细胞的运动和转移
例如,体外研究表明,TDO 在胶质母细胞瘤或乳腺癌细胞中的表达可促进肿瘤细胞迁移和侵袭。类似地,IDO1 的过表达增强了肺癌细胞的运动性,而敲除则降低了运动性。
这种促迁移表型也反映在临床前模型中由色氨酸降解引起的转移形成促进。药理学 TDO 抑制减少了肺癌小鼠模型肺部肿瘤结节的数量。
植入小鼠体内的人肺癌细胞中的 IDO1 过表达增加了大脑、肝脏和骨骼中的转移形成,而 IDO1 缺乏减少了转移负担并提高了乳腺癌衍生肺转移小鼠模型的存活率。
此外,TDO-AHR 信号轴促进了对贴壁依赖性细胞从周围细胞外基质分离时发生的程序性细胞死亡的抵抗,这是转移的关键步骤。最后,肿瘤内 IDO1 表达已被证明与结直肠癌肝转移、肝细胞癌远处转移和子宫内膜癌淋巴结转移的频率相关。
第五,NAD+ 在癌症生物学中通过色氨酸 de novo 途径产生的作用
在小鼠中,色氨酸代谢受损导致肝脏中从头合成 NAD+ 受到抑制,从而通过 DNA 损伤促进肝肿瘤发生。
在人类神经胶质瘤中,从色氨酸重新产生的 NAD+ 赋予对放化疗诱导的氧化应激的抗性。有趣的是,胶质瘤细胞和小胶质细胞合作产生 NAD+。
此外,在人类癌细胞中,IDO1 与通过产生 NAD +改善 DNA 修复和介导对治疗的抗性有关,例如 PARP 抑制剂奥拉帕尼、γ-辐射和化疗剂顺铂。因此,抑制色氨安代谢也可能通过从头形成 NAD +来防止治疗耐药性;然而,根据 NAD +合成所必需的 KP 酶的表达,这种效应可能是组织特异性或细胞特异性的,因此需要进一步研究。
● 靶向癌症中的 IDO1 和 TDO
基于 IDO1 和 TDO 的肿瘤促进功能,已经研究了这些酶的小分子抑制剂用于癌症治疗。临床阶段 IDO1 抑制剂 epacadostat (INCB024360)、navoximod (NLG-919/GDC919)等化学结构已被公开。未公开结构的化合物KHK2455、LY3381916和MK-7162也作为IDO1抑制剂进入临床评估。
TDO 抑制剂(最初被开发为抗抑郁药以提高全身色氨酸水平,从而提高大脑血清素浓度)也正在探索用于癌症治疗,但尚未进入临床试验阶段。
此外,indoximod 正在临床试验中进行研究,但与 L-1-MT 237不同,它们不是 IDO1抑制剂及其作用机制,尽管它似乎与 IDO1 表达有关,但仍存在争议。
然而,IDO1 抑制的最大治疗潜力预计是它与其他疗法的联合使用,这一直是大多数 II 期和 III 期研究的重点。
● 与免疫检查点抑制剂联合
IDO1 抑制剂的临床评估最先进的是它们与针对免疫系统检查点的单克隆抗体的组合,例如细胞毒性 T 淋巴细胞相关蛋白 4 (CTLA4)、PD-1 或其配体 (PD-L1),其中一些已被批准用于近年来治疗多种癌症的基础上部分患者出现前所未有的反应。
然而,由于相当大比例的患者无法从检查点抑制剂中获益,因此人们非常有兴趣确定缺乏治疗反应和治疗耐药性的分子基础,因为这些知识可能表明潜在的联合疗法可以改善反应。
有趣的是,在使用 PD-1 受体阻断剂 pembrolizumab 治疗期间,肉瘤患者的 Kyn:Trp 血浆比率增加,表明 IDO1 可能由免疫检查点封锁诱导。最有可能的是,这种 IDO1 的诱导,预计会抵消免疫检查点抑制的免疫刺激作用,是通过活化的 T 细胞产生的 IFNγ 介导的。
一项临床前研究表明,抑制 IDO1 会略微增强抗 CTLA4、抗 PD-1–PD-L1 和抗 GITR(糖皮质激素诱导的 TNFR 相关蛋白)疗法的疗效。
这些发现虽然不大,但引发了对 IDO1 抑制剂与免疫检查点抑制剂联合治疗的广泛临床研究。在 epacadostat 与 pembrolizumab 联合治疗的 I/II 期单臂试验获得令人鼓舞的数据后,在无法切除或转移性黑色素瘤患者中进行了 III 期试验。
尽管 ECHO-301 试验的阴性结果明显代表了 IDO1 抑制剂在癌症免疫治疗中的开发受挫,但它也激励人们利用临床试验来更多地了解 IDO1 抑制剂在癌症中的作用机制,以开发更复杂的生物标志物用于患者选择和治疗监测,并利用该途径中的新靶点,例如 AhR。
● 与免疫检查点抑制剂联合
目前已经计划在验证试验中继续研究 IDO1 抑制剂在联合免疫疗法中的潜力,包括不同于与 PD-1 和 PD-L1 拮抗剂组合的策略。
几项测试 IDO1 抑制剂 epacadostat 与抗肿瘤疫苗联合应用的临床试验正在进行中,可能会显示阻断 IDO1 是否会提高抗肿瘤疫苗接种的功效。这背后的基本原理是干扰素信号对 IDO1 的上调涉及多种免疫相关途径。例如,TLR 的激活通过干扰素诱导 IDO1 表达。
抑制抗肿瘤免疫反应的其他几种途径也与驱动色氨酸降解酶的肿瘤表达有关,包括 AhR 信号、TGFβ 信号和信号转导和转录激活因子 3 (STAT3)。
因此可以设想两种情况:
• 如果这些途径的抑制剂非常有效并且同时完全消除了色氨酸降解酶的表达,那么它们可能会使 IDO1 或 TDO 抑制剂在这种情况下变得可有可无。
• 相反,如果这些药物不能完全减轻 IDO1 和/或 TDO 的表达,它们可能与色氨酸代谢抑制剂协同作用。相比之下,其他治疗方法可能会诱导 IDO1 作为一种不良影响,这表明这些疗法与 IDO1 抑制剂的组合可能是有益的。
/
/
▼
前面概述了色氨酸及其在肠道中的三种命运。在这里主要谈论吲哚/AhR 通路,将深入探讨 AhR 信号的好处、为什么大多数人都缺乏这种途径、增加肠道中 AhR 活性的潜在策略,以及 AhR 可能被过度刺激的一些例外情况。
● 什么是AhR?
芳烃受体 (AhR)是一种转录因子——一种调节基因表达的蛋白质。结合并激活受体的分子称为激动剂。
AhR 的激动剂(即激活剂)主要分为三类分子:
AhR 最初因其在对二恶英和其他芳基碳氢化合物等环境毒素作出反应中的作用而被发现。这些污染物是 AhR 的非常强的激活剂。它们的结合增加了有助于促进其解毒的酶的表达。
肠道细菌,包括各种梭菌属、拟杆菌属、真杆菌属、乳杆菌属和双歧杆菌属,可以直接将色氨酸转化为称为吲哚的化合物,其中许多结合并激活 AhR。
近年来,膳食化合物也被证明可以激活 AhR。Indole-3-carbinol (I3C) 是一种源自十字花科蔬菜分解的化合物,可以结合并激活 AhR。
虽然环境污染物对 AhR 的慢性激活可能对健康产生负面影响,但肠道代谢物和膳食化合物对 AhR 的瞬时激活具有许多积极的下游影响。
● 肠道 AhR 激活的诸多好处
规律的、短暂的 AhR 信号在肠道和整体健康中起着许多重要作用。
1) 维持肠道屏障功能
AhR 刺激肠道中的先天免疫细胞产生细胞因子 IL-22,这是一种促进粘液产生和抗菌肽分泌的信号分子。在称为隐窝的肠道屏障口袋中,AhR 还支持干细胞增殖,这对于正常的肠道更新和修复至关重要。
2) 调节肠道菌群的组成
缺乏 AhR 刺激会导致促炎性肠杆菌科的扩张和产丁酸梭菌的减少,这是肠道菌群失调的常见特征。
3) 维持肠道免疫细胞群并减少炎症
AhR 支持肠道上皮细胞内足够数量的淋巴细胞。它还在将调节性 T 细胞引导至肠道并支持其抑制炎症的能力方面发挥关键作用。
4) 调节肠神经系统和肠蠕动
AhR 已被证明在调节蠕动方面发挥作用,蠕动是沿着胃肠道移动食物的肌肉收缩。AhR 也可能与损伤后肠神经的再生有关。
5) 防止念珠菌和其他肠道感染
AhR 激活通过支持 IL-22 信号传导在维持对酵母白色念珠菌和细菌病原体的定植抗性方面发挥重要作用。
6) 支持肺部的免疫防御
肠道 AhR 在防止其他粘膜表面(如肺)感染方面也起着重要作用。2019 年的一项研究发现,抗生素治疗后提高肠道中的 AhR 活性可显着减少肺部致病菌数量。
7) 促进健康的皮肤屏障功能
肠道 AhR 对于维持皮肤屏障的完整性至关重要。2016 年的一项研究发现,从饮食中去除 AhR 配体会损害皮肤屏障功能,而重新添加 AhR 激活剂吲哚-3-甲醇可挽救屏障缺陷,即使在老年小鼠中也是如此。
8) 激活解毒途径
AhR 在许多物质的解毒中发挥作用,包括多环芳烃、霉菌毒素、重金属和雌激素,激活整个身体的 解毒途径。
9) 保护肝肾功能
来自肠道色氨酸代谢的 AhR 信号也被证明可以预防非酒精性脂肪肝、酒精性肝损伤和肾纤维化。
10) 支持神经系统健康
星形胶质细胞中通过 AhR 发出的膳食色氨酸代谢物信号已被证明可以限制中枢神经系统的炎症。肠道 AhR 活性还促进成人神经发生,即新神经元的形成。
AhR 信号减少:许多慢性病的一个特征
在多种慢性疾病中观察到肠道 AhR 活性降低,包括炎症性肠病、肠易激综合征、结直肠癌、肥胖、代谢综合征、高血压、动脉粥样硬化、抑郁症、炎症性皮肤病、乳糜泻和多发性硬化症等疾病。
● 是什么导致 AhR 信号减少?
影响因素很多,包括:
改变的肠道微生物群组成通常无法产生已知可激活 AhR 的化合物,包括色氨酸衍生的吲哚和短链脂肪酸丁酸盐。
色氨酸(细菌吲哚形成的底物)的摄入量减少和/或植物性食物中 AhR 激动剂的摄入量减少都会减少 AhR 激动剂的总量。人造甜味剂的消费也被证明会减少 AhR 信号。
压力、炎症和某些类型的感染可以将色氨酸代谢从 AhR 转移到其他途径(后面我们会持续分享)。
解决这些根本原因始终是恢复 AhR 活动的第一步。
● 增加 AhR 活性的其他策略
以下是已知会暂时增加 AhR 活性的其他干预措施的总结。但是需要注意 AhR 激动剂具有物种特异性和组织特异性作用。它们的效果还取决于浓度,在存在多种化合物的情况下,它们甚至可能相互竞争——因此虽然这里列出了很多可能性,但“厨房水槽”方法并不一定是理想的。
希望在接下来的几年里,我们将看到更多的人体临床试验,以阐明这些疗法中的哪些可能对以 AhR 缺陷为特征的疾病状态最有帮助。以下信息不能视为医疗建议。
▸Indole-3-carbinol (I3C)
这种化合物由球芽甘蓝、卷心菜、西兰花、花椰菜和芥菜等十字花科蔬菜中的葡糖甘蓝素分解产生,是一种有效的 AhR 激活剂。
在动物模型中,I3C 已被证明可以诱导调节性 T 细胞的形成、抑制 Th17、保护粘液层、增加丁酸盐的产生、上调 PPAR-γ 并防止结肠炎。还对其潜在的抗癌和抗氧化作用进行了研究。虽然 I3C 或其衍生物二吲哚基甲烷 (DIM) 以补充剂形式提供,但较高剂量可能存在风险且人体研究有限,因此最好以整个食物形式食用 I3C。
注意:为了最大限度地提高膳食 I3C 的生物利用度,食用酸菜等生发酵形式的十字花科蔬菜,或在烹饪后加入芥末籽粉(含有黑芥子酶)。
▸丁酸盐
一种短链脂肪酸,是人肠上皮细胞中 AhR 的直接激活剂。在健康的肠道中,丁酸盐是由膳食纤维的发酵产生的,在较小程度上是蛋白质的发酵。它也以补充形式提供。一定情况下对结肠需要更有针对性。
▸尿石素 A
这种化合物由石榴、覆盆子和黑莓中的鞣花单宁分解产生,已被证明可以通过 AhR 增强肠道屏障功能。然而,估计只有 30-40% 的人拥有可以进行这种转化的细菌。尿石素 A也可以作为补充剂服用,并且作为食品成分已获得 FDA 公认的安全状态。
▸阳光照射
2019 年的一项研究发现,在小鼠身上,仅 15 分钟的 UV-B 照射就会诱导 AhR 靶基因在血液和外周组织(包括肠道)中的表达。
▸婴儿双歧杆菌(Bifidobacterium infantis)
该菌株在人乳低聚糖上生长后产生吲哚-3-乳酸,一种 AhR 的激活剂。它已在婴儿中得到充分研究,可用作婴儿益生菌。该菌株尚未在成人中进行研究。该配方确实含有大量的乳糖和残留量的大豆。
▸鼠李糖乳杆菌
某些乳杆菌菌株已被证明在色氨酸丰富时自然产生 AhR 激动剂。发现唯一已知可增加 AhR 活性且可商购的菌株是鼠李糖乳杆菌GG。
但是注意,不建议在使用抗生素期间或之后立即使用基于乳酸杆菌的益生菌,也不建议患有组胺不耐受/肥大细胞活化综合症的人服用。
▸Akkermansia muciniphila
至少在一项动物研究中,这种细菌或其外膜上的一种蛋白质增加了循环中的吲哚化合物并上调了 AhR 靶基因。
▸美沙拉嗪 (5-氨基水杨酸,5-ASA)
该药物是炎症性肠病 (IBD) 的一线治疗药物。这种药物上调 PPARgamma 和促细胞凋亡和抗增殖作用的能力。有趣的是,美沙拉嗪似乎也能激活 AhR。
▸咖啡
咖啡提取物,尤其是过滤较少的咖啡,如土耳其咖啡,已被证明可诱导肠上皮细胞中的 AhR 表达,并在啮齿动物模型中预防结肠炎。
▸萝卜硫素
虽然这种化合物的作用通常归因于 Nrf2 通路,但 AhR 似乎介导了它的许多保护作用。在喂食西方饮食的小鼠中,萝卜硫素增加了肠道中吲哚乙酸的产生,从而上调了 AhR 活性。
▸多酚
槲皮素、白藜芦醇和姜黄素都可以通过抑制控制 AhR 激动剂分解的 CYP1A1 酶来间接激活 AhR。其中,槲皮素在增强 AhR 信号传导方面最有效。
▸血清素
这种神经递质及其副产物 (5-HIAA) 也可以通过部分抑制 AhR 配体的清除来间接激活 AhR。这种效果取决于功能正常的血清素运输。
关于反馈调节而不是长期过度刺激 AhR 的重要性的说明:
这里不能忽略AhR 过度刺激的问题。虽然大多数慢性炎症性疾病都以 AhR 缺陷为特征,但在少数情况下,AhR 可能会过度激活,从而产生负面后果。这通常是由于污染物或霉菌暴露、严重病毒感染、或慢性肾病等引起的显着环境毒性。
这些条件的特点是持续的 AhR 激活,其对基因表达的影响与短暂的 AhR 激活截然不同。相反,由于某些解毒酶的上调,结合 AhR 的天然化合物在结合 AhR 时会被有效代谢。这种负反馈回路确保 AhR 信号是短暂的。
拥有足够的维生素,尤其B12 和叶酸可以防止慢性 AhR 过度刺激。尽管如此,在某些极端毒性或感染的情况下,短暂的 AhR 刺激是不提倡的。
/
/
▼
● 益 生 菌
益生菌,如属于乳杆菌属和双歧杆菌属的细菌,对色氨酸代谢产生有益影响。
益生菌促进血清素合成
一方面,益生菌,如乳杆菌和双歧杆菌中的物种,可以直接将色氨酸转化为血清素。
另一方面,一些益生菌乳杆菌菌株,如干酪乳杆菌327,可以通过增加TPH1表达间接促进结肠血清素合成。
益生菌与犬尿氨酸途径的调节密切相关
与血清5-羟色胺水平升高一致,大鼠口服约氏乳杆菌(Lactobacillus johnsonii) 无细胞上清液,也会导致血清中的犬尿氨酸水平降低,同时肠道IDO活性降低。口服约氏乳杆菌8周后,观察到人血清犬尿氨酸水平下降,色氨酸含量增加的明显趋势。
此前的一项研究还表明,大鼠服用益生菌婴儿双歧杆菌( Bifidobacteria infantis)会导致色氨酸水平升高,血液循环中的犬尿氨酸与色氨酸比率降低。
这些研究表明,一些属于乳杆菌和双歧杆菌的益生菌物种可能通过抑制犬尿氨酸途径改变宿主色氨酸代谢。
益生菌将色氨酸降解为吲哚化合物
此外,据报道,一些属于乳杆菌的细菌能够将色氨酸降解为吲哚化合物,如IAld、ILA和IAA。
向结肠炎易感小鼠口服3种色氨酸代谢乳杆菌菌株可促进微生物色氨酸新陈代谢芳基烃受体(Ah)依赖性信号传导,从而影响外周色氨酸的有效性。
尽管操纵肠道微生物群影响色氨酸代谢途径的机制尚未完全了解,但以肠道微生物群为靶点可能是调节色氨酸新陈代谢的一种有前途的方法。
● 抗 生 素
口服抗生素能够重塑肠道微生物群的组成和代谢。口服广谱抗生素会导致肠道微生物群耗竭,降低结肠血清素水平,进而延缓小鼠结肠运动。
正如结肠中关键合酶TPH1的下调所证明的,这项研究指出了共生微生物群在调节肠道血清素合成中的可能作用。
据报道,抗生素的微生物操纵会影响犬尿氨酸途径,因为抗生素诱导的微生物群耗竭会导致小鼠和猪的循环色氨酸可用性增加,并降低沿犬尿氨素途径的代谢。
此外,一些研究表明,抗生素诱导的肠道微生物改变也有利于猪体内的微生物色氨酸降解途径。随着循环色氨酸水平的增加,口服抗生素降低了空肠中色氨酸的可用性,并降低了猪大肠中的微生物色氨酸脱羧活性。
此外,口服抗生素会增加猪大肠中吲哚和吲哚化合物的含量。
有趣的是,最近的一项研究表明,回肠末端输注专门针对大肠微生物群的广谱抗生素会导致血液循环中的色氨酸水平降低,并增强微生物色氨酸降解,从而增加大肠中的吲哚水平。与之前的研究结果相反,该研究的发现表明,肠道微生物群在响应抗生素操作而调节色氨酸代谢方面发挥了独特的作用。
● 饮 食
饮食被认为是影响微生物色氨酸代谢的重要因素。
高脂肪饮食:抑制微生物从色氨酸向吲哚代谢物的转化,从而影响免疫调节
例如,最近的一项研究表明,高脂肪饮食会耗尽小鼠盲肠中的微生物代谢产物IAA和色胺,这表明在高脂肪饮食下,微生物色氨酸降解途径可以减弱。
高脂肪饮食增加了Alistipes和Bacteroides的丰度,同时减少了 Faecalibacterium。高脂肪饮食增加了致病菌属Alistipes,同时减少了有益菌 Parabacteroides distasonis ,导致小鼠肠道屏障功能受损。
高脂饮食显著增加小鼠肠道的IDO活性,促进色氨酸分解代谢为犬尿氨酸。
在暴露于高脂饮食的情况下,肠道微环境受到影响,随后抑制微生物从色氨酸向吲哚代谢物的转化,特别是吲哚-3-丙酸、吲哚-3-乳酸和吲哚乙酸盐。这些代谢物被认为是 AhR 激动剂,在免疫调节中起着关键作用。
配方奶:影响新生猪色氨酸代谢
母乳不仅是早期营养的唯一来源,而且有助于宿主肠道微生物群的成熟。有趣的是,之前的一项研究发现,配方奶引起的肠道微生物群的改变使新生猪结肠中的色氨酸代谢从血清素转变为色胺。
碳水化合物:影响色氨酸代谢速率
微生物色氨酸代谢的速率可能会受到管腔内营养物质(如碳水化合物)可用性变化的影响。
正如先前的体外研究所证明的,从仔猪粪便中分离出的一株利用色氨酸的细菌使用色氨酸进行细菌蛋白质合成,以可消化碳水化合物(葡萄糖)为底物,而不可消化碳水化合物(低聚果糖)是吲哚产生的底物。
此外,通过添加不可消化的碳水化合物,如低聚果糖和抗性淀粉,增加碳水化合物的可用性,促进碳水化合物代谢,从而增加短链脂肪酸的产量,同时减少色氨酸降解和仔猪大肠中的吲哚化合物。
事实证明,通过盲肠淀粉输注增加大肠碳水化合物的可用性可以抑制微生物色氨酸降解,从而导致大肠和血清中色氨酸水平的增加。
这些研究表明,增加碳水化合物的可用性抑制了肠道中的微生物色氨酸降解,这将进一步影响循环色氨酸库。
相比之下,增加碳水化合物的可用性促进了肠道血清素的合成,这与增加胃肠道传输有关,正如先前在口服多糖的小鼠中进行的一项研究所报告的那样。微生物短链脂肪酸的产生增强可以参与这一过程,因为它们已经被证明可以刺激结肠EC中的血清素释放。
腔内色氨酸可用性是影响微生物色氨酸代谢的另一个直接因素
IDO激活或饮食限制导致的宿主色氨酸耗竭可减少微生物增殖,尤其是乳酸杆菌中的细菌,据报道,其中一些细菌是利用色氨酸的细菌。通过饮食喂养选择性地恢复色氨酸水平导致乳杆菌的扩张,这进一步导致微生物色氨酸代谢的增强,IAld增加。
鉴于色氨酸也会被宿主直接吸收,肠道微生物群、管腔色氨酸可用性和宿主色氨酸代谢之间的复杂串扰需要进一步研究。
富含麦麸的饮食在调节色氨酸代谢物的合成和生物转化中的作用
富含麦麸的饮食有效地抑制了色氨酸向犬尿氨酸途径代谢物的转化,同时增加了褪黑激素和微生物分解代谢物,即吲哚-3-丙酸、吲哚-3-乙醛和 5-羟基-吲哚-3-乙酸。
麦麸增加了促进健康的细菌(例如,Akkermansia和Lactobacillus),它们与色氨酸衍生的吲哚类代谢物显著相关。
富含麦麸的饮食可有效调节与免疫功能相关的微生物转化和色氨酸合成(即增加 AhR 和 IL-22 的结肠表达),同时改善葡萄糖和脂质稳态,以及增加肠道健康促进菌的丰度。
/
/
色氨酸代谢调节炎症、肠道稳态和大脑功能等。色氨酸可用性和代谢的微生物调节对许多肠脑轴疾病具有重要意义,包括伴有精神疾病的胃肠道疾病,如IBS 、IBD,其他具有胃肠道功能障碍的中枢神经系统疾病,如自闭症等。
由于宿主色氨酸代谢直接或间接受肠道菌群调节,许多因素会影响肠道微生物群的组成和代谢,包括饮食、抗生素、益生菌等可以调节肠道微生物群,调节色氨酸的可用性,因此靶向肠道菌群干预是治疗肠脑轴疾病的有前途的方法。
色氨酸代谢可以作为与年龄相关的病理和寿命的调节剂。犬尿氨酸通路及其代谢产物可能成为预测衰老相关疾病的潜在风险标记物。
色氨酸主要经犬尿氨酸代谢,既促进肿瘤细胞固有的恶性特性,又限制肿瘤免疫,因此它是癌症免疫治疗的重要药物开发靶点。肿瘤中色氨酸代谢的改变常伴随色氨酸相关酶基因表达的异常,基于此,IDO抑制剂、TDO抑制剂及联合治疗被应用于大量的临床试验中。
随着这方面研究的不断深入,我们会持续关注和更新色氨酸代谢和全身健康稳态以及药物进展等。
主要参考文献
Le Floc’h N, Otten W, Merlot E. Tryptophan metabolism, from nutrition to potential therapeutic applications. Amino Acids. 2011 Nov;41(5):1195-205. doi: 10.1007/s00726-010-0752-7. Epub 2010 Sep 25. PMID: 20872026.
Gao K, Mu CL, Farzi A, Zhu WY. Tryptophan Metabolism: A Link Between the Gut Microbiota and Brain. Adv Nutr. 2020 May 1;11(3):709-723. doi: 10.1093/advances/nmz127. PMID: 31825083; PMCID: PMC7231603.
Sorgdrager FJH, Naudé PJW, Kema IP, Nollen EA, Deyn PP. Tryptophan Metabolism in Inflammaging: From Biomarker to Therapeutic Target. Front Immunol. 2019 Oct 30;10:2565. doi: 10.3389/fimmu.2019.02565. PMID: 31736978; PMCID: PMC6833926.
Richard DM, Dawes MA, Mathias CW, Acheson A, Hill-Kapturczak N, Dougherty DM. L-Tryptophan: Basic Metabolic Functions, Behavioral Research and Therapeutic Indications. Int J Tryptophan Res. 2009 Mar 23;2:45-60. doi: 10.4137/ijtr.s2129. PMID: 20651948; PMCID: PMC2908021.
Platten M, Nollen EAA, Röhrig UF, Fallarino F, Opitz CA. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat Rev Drug Discov. 2019 May;18(5):379-401. doi: 10.1038/s41573-019-0016-5. PMID: 30760888.
Chen G, Zhou S, Chen Q, Liu M, Dong M, Hou J, Zhou B. Tryptophan-5-HT pathway disorder was uncovered in the olfactory bulb of a depression mice model by metabolomic analysis. Front Mol Neurosci. 2022 Oct 10;15:965697. doi: 10.3389/fnmol.2022.965697. PMID: 36299862; PMCID: PMC9589483.
Roth W, Zadeh K, Vekariya R, Ge Y, Mohamadzadeh M. Tryptophan Metabolism and Gut-Brain Homeostasis. Int J Mol Sci. 2021 Mar 15;22(6):2973. doi: 10.3390/ijms22062973. PMID: 33804088; PMCID: PMC8000752.
van der Goot AT, Nollen EA. Tryptophan metabolism: entering the field of aging and age-related pathologies. Trends Mol Med. 2013 Jun;19(6):336-44. doi: 10.1016/j.molmed.2013.02.007. Epub 2013 Apr 2. PMID: 23562344.
Yao K, Fang J, Yin YL, Feng ZM, Tang ZR, Wu G. Tryptophan metabolism in animals: important roles in nutrition and health. Front Biosci (Schol Ed). 2011 Jan 1;3(1):286-97. doi: 10.2741/s152. PMID: 21196377.
Höglund E, Øverli Ø, Winberg S. Tryptophan Metabolic Pathways and Brain Serotonergic Activity: A Comparative Review. Front Endocrinol (Lausanne). 2019 Apr 8;10:158. doi: 10.3389/fendo.2019.00158. PMID: 31024440; PMCID: PMC6463810.
Gibson EL. Tryptophan supplementation and serotonin function: genetic variations in behavioural effects. Proc Nutr Soc. 2018 May;77(2):174-188. doi: 10.1017/S0029665117004451. Epub 2018 Jan 25. PMID: 29368666.
Gostner JM, Becker K, Kofler H, Strasser B, Fuchs D. Tryptophan Metabolism in Allergic Disorders. Int Arch Allergy Immunol. 2016;169(4):203-15. doi: 10.1159/000445500. Epub 2016 May 4. PMID: 27161289; PMCID: PMC5433561.
Liu XH, Zhai XY. Role of tryptophan metabolism in cancers and therapeutic implications. Biochimie. 2021 Mar;182:131-139. doi: 10.1016/j.biochi.2021.01.005. Epub 2021 Jan 16. PMID: 33460767.
Yan T, Shi L, Liu T, Zhang X, Yang M, Peng W, Sun X, Yan L, Dai X, Yang X. Diet-rich in wheat bran modulates tryptophan metabolism and AhR/IL-22 signalling mediated metabolic health and gut dysbacteriosis: A novel prebiotic-like activity of wheat bran. Food Res Int. 2023 Jan;163:112179. doi: 10.1016/j.foodres.2022.112179. Epub 2022 Nov 19. PMID: 36596122.

谷禾健康

我们知道,肠道微生物群对人类健康和福祉很重要,调节宿主代谢,塑造免疫系统并防止病原体定植。
通过粪便微生物群移植(FMT)恢复平衡多样的微生物群,已成为研究疾病发病机制中微生物群因果关系的潜在治疗策略和有前途的工具。
然而,FMT 带来了后勤方面的挑战和潜在的安全风险,如病原微生物的转移、不期望的表型(如肥胖)的潜在转移,或在生命后期发展疾病的风险增加。
因此,一种更可控、更个性化的培养有益微生物混合物可能是更好的选择。
这些有益微生物中的大多数将是宿主的内源性共生体,没有长期安全有益的使用历史,因此通常被称为下一代益生菌(NGP)或活生物治疗产品(LBP)。
植物乳杆菌菌株,其益生菌和功能特性及其促进健康的作用脱颖而出,可以很好地调节肠道菌群组成。
一项FMT研究发现共生产丁酸菌Anaerobutyricum spp.(以前称为Eubacterium hallii)与代谢综合征受试者胰岛素敏感性的提高有关。因此,着手进一步研究和开发这种潜在的有益微生物,并将重点放在Anaerobutyricum soehngenii L2-7等,因为它的特征最好。
在小鼠模型中使用Anaerobutyricum soehngenii 完成临床前试验后,在受控条件下生产菌株,并进行了几项临床研究,以评估其在人体中的安全性和有效性。
本文将以植物乳杆菌为例,介绍其益生菌特性;以A.soehingeii为例,介绍用于临床的的开发,为下一代益生菌的开发和测试提供了实践指导。
传统的益生菌被定义为“活的微生物,当给予足够的量时,会给宿主带来健康益处”。这些微生物使用历史悠久,被认为是安全的。
注:在美国具有公认安全(GRAS)状态,在欧盟具有合格安全推定(QPS)状态。
益生菌的使用可能代表一种调节肠道微生物群和改善人类疾病的治疗策略。
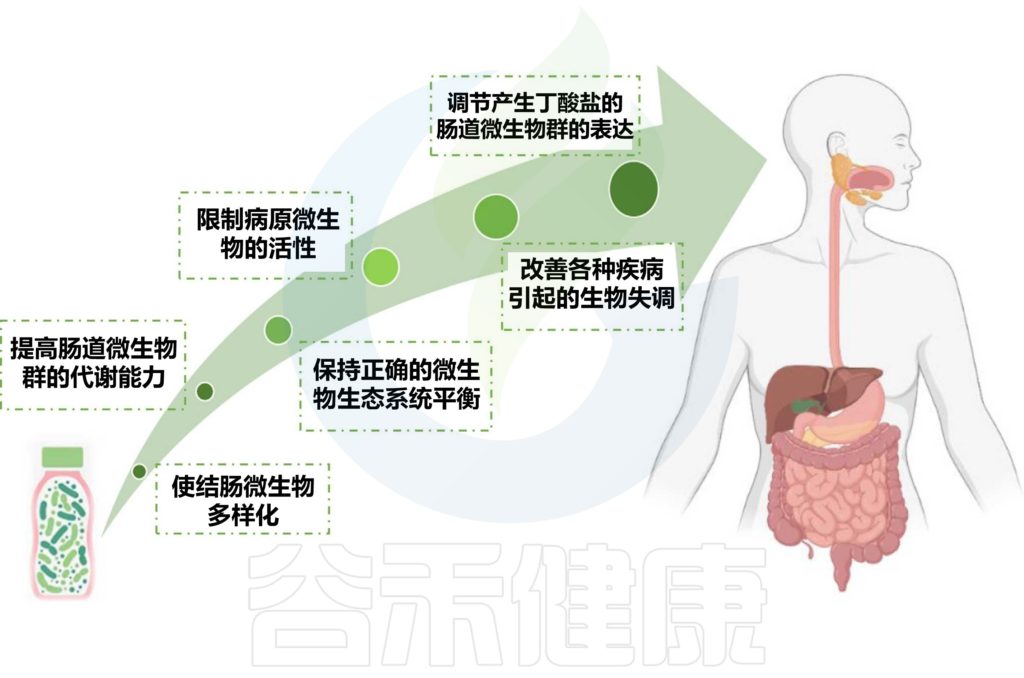
doi.org/10.1016/j.micres.2022.127289
相比之下,下一代益生菌(NGP)是一种没有长期安全有益使用历史的微生物,与传统益生菌一样,当以足够的量给药时,下一代益生菌对宿主健康有益。
2012年,美国食品和药物管理局引入了活生物治疗产品(LBP)一词,定义为“一种生物产品”,其:
(1)含有活生物体,如细菌;
(2)适用于预防、治疗或治愈人类疾病或病症;
(3)不是疫苗。
LBP在《欧洲药典》(Ph.Eur.)中被定义为“含有活微生物(细菌或酵母)的供人类使用的医药产品”。然而,由于LBP除了微生物外还包括最终产品的配方,并且被定义为药物产品,因此不应系统地使用该术语来替代NGP。
NGP一词更为广泛,包括LBP中存在的微生物和目前正在研究的、尚未在最终产品中配制的微生物。此外,NGPs既可以用作传统益生菌等食品补充剂,也可以用作预防、治疗或治愈疾病的医药产品。最后,转基因微生物也可以被视为NGP,尽管最有可能作为LBP上市。
下图示意性地描述了各种定义。
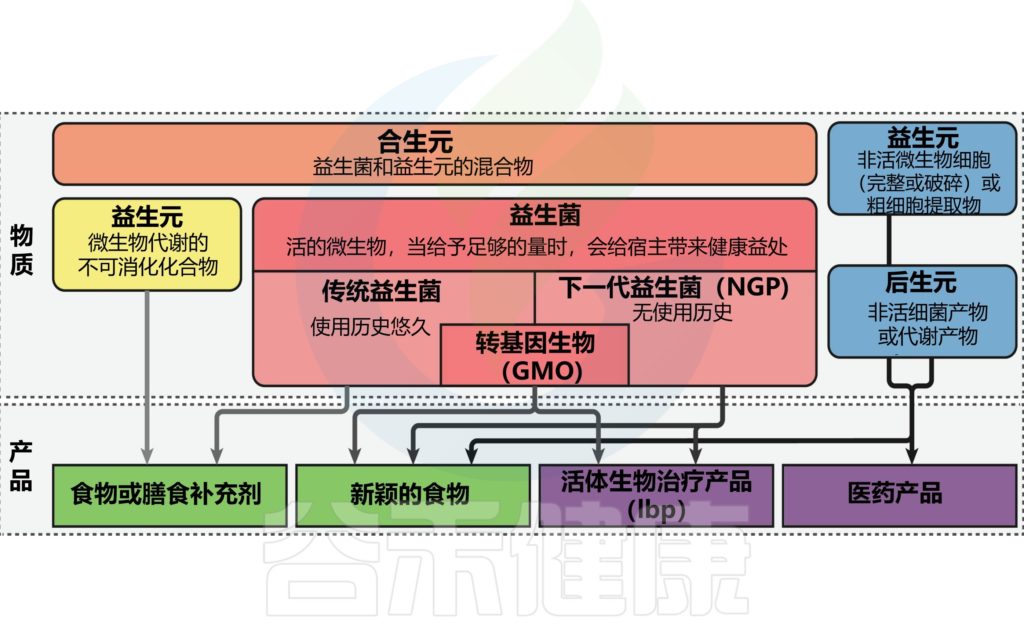
doi.org/10.3389/fmed.2022.1077275
植物乳杆菌是乳杆菌中最重要的成员之一,由于其出色的益生菌特性(良好的 GI 耐受性、粘附性、抗氧化性和抗菌性),它通常被用作益生菌。
✔ 抵抗胃肠道疾病
将微生物视为益生菌的一个基本特征是能够在人类胃肠道的恶劣条件下存活。
植物乳杆菌MA2菌株和B23菌株表现出良好的耐受性,可以在低pH值(2.5-3)下存活。植物乳杆菌KU15149 具有胃和胆汁盐耐受性。
✔ 对肠粘膜和/或细胞外基质成分的粘附能力
粘附到上皮细胞的粘膜或粘附到肠道细胞外基质的成分是益生菌微生物的理想特征,因为它们将有利于益生菌在宿主中的定植和持久存在。
两种植物乳杆菌菌株DKL3 和 JGR2 分别显示出 82.8% 和 79.6% 的粘附程度。
植物乳杆菌菌株 KACC11451 和 Wikim0112 的肠上皮粘附率约为 60–62%.
✔ 抗氧化活性
一些益生菌已被证明具有抗氧化活性,可减少氧化反应造成的损害。
✔ 细菌素生产
细菌素可以在食物和宿主中发挥各种益处,因为它们可以分别延长保质期和防止不必要的定植。许多植物乳杆菌菌株已被证明能够产生细菌素,赋予这种微生物益生菌特性。
植物乳杆菌产生通常称为 plantaricin 的细菌素。
KLDS1.0391、ZJ5、TN635、B23 和 AA135 菌株分别是细菌素 Plantaricin MG、Plantaricin ZJ5、细菌素 ST28MS 和 ST26MS、细菌素 BacTN635、细菌素 Lac-B23 和 Plantaricin AA135 的生产者,它们具有对几种革兰氏阴性菌的抗菌作用。
✔ 抗菌活性
益生菌的特点是抑制病原微生物的生长、发育和定植。
在植物乳杆菌的发酵代谢过程中,它会产生多种抗菌化合物(除细菌素外),其中可能包括有机酸,例如乳酸、柠檬酸、异丁酸和乙酸、乙醇、双乙酰和 H2O2. 植物乳杆菌还可以产生具有天然抗真菌活性的胞外多糖。
✔ 本土肠道调节
构成肠道微生物组的不同物种的生态平衡,对于预防传染性和非传染性疾病以及阻止微生物群平衡的紊乱至关重要。益生菌具有调整肠道菌群组成和纠正免疫系统异常反应的能力,从而对宿主产生不同的有益作用。
植物乳杆菌ZJ316 在体外肠道模型中发挥了微生物群的调节作用,增加了Veillonella的生长,这可以提高人体呼吸系统和消化系统的免疫力。同时减少了 Blautia 的存在。
注:Blautia与肥胖儿童的肠道炎症有关。
此外,ZJ316 菌株减少了肠杆菌科,包括共生生物和原发性和机会性病原体。
注:这些病原体很容易在发炎的肠道中繁殖,从而导致微生物群失衡。
不同植物乳杆菌菌株对肠道菌群的影响
doi.org/10.1016/j.micres.2022.127289
更多关于植物乳杆菌的介绍详见:客观认识植物乳杆菌 (L. plantarum) 及其健康益处
随着全球肥胖流行病的恶化,代谢综合征的发病率急剧增加,比较容易患上心血管疾病和2型糖尿病。肠道微生物群的动态变化与代谢综合征的出现相关。
进一步研究肠道微生物群在代谢综合征中的因果作用 ↓↓↓
研究人员先前向患有代谢综合征的男性受试者输注了来自瘦健康供体的粪便微生物群。输注供体微生物群6周后,与自体FMT组相比,外周胰岛素敏感性随着丁酸产生菌的水平而增加。
在这些产生丁酸的细菌中,厌氧产丁酸菌在小肠中更为丰富,这表明其在通过丁酸产生调节胰岛素敏感性方面具有潜在作用。
由于胰岛素抵抗代谢综合征受试者的特点是产短链脂肪酸菌水平降低,口服丁酸盐可改善饮食诱导的肥胖小鼠的胰岛素抵抗和血脂异常。
因此研究人员得出结论,A.soehingenii可能是一种有前途的下一代益生菌,可改善胰岛素抵抗。
//
1996年,从婴儿粪便中分离出的A.soehngenii菌株L2-7,以前被命名为E.hallii,是一种严格厌氧、革兰氏阳性、过氧化氢酶阴性的Lachnospiracae科细菌。A.soehngenii菌是人类胃肠道核心菌群的一部分。与其他已知的丁酸盐生产物种(如Roseburia和Faecalibacterium spp.)不同,A.soehingenii有能力在乙酸盐存在下利用D-和L-乳酸盐。此外,基因组中含有胆汁酸钠共转运蛋白和胆碱水解酶基因,表明A.soehngenii 可以影响宿主胆汁酸代谢。
学习要点和方向
下一代益生菌的开发通常采用两种策略。
第一种方法是将特定菌株的存在与健康表型相关联,并探讨该菌株是否对疾病表型有因果影响。
迄今为止,已经使用测序技术确定了许多NGP候选株,以选择患病受试者中丰度耗尽的菌株或与FMT治疗成功相关的菌株。
第二种策略是采用具有良好特征的益生菌菌株,并对该菌株进行基因修饰,例如通过生物活性分子的生产和递送,从而赋予健康益处。
后一种方法将导致转基因生物(GMO)在世界各地受到特定法规的约束,如欧盟。
无论用于识别或生成NGP的策略如何,在体内研究任何健康益处之前,需要在体外充分表征候选菌株。
下图总结了除了菌株的基因分型和表型外,必须评估的最重要特征。
doi.org/10.3389/fmed.2022.1077275
此外,必须记录菌株的起源和随后的操纵或基因修饰。如果存在任何抗微生物耐药基因或毒力基因,则应评估人体微生物群向其他微生物传播的可能性,并采取措施减轻这种风险。
当下一代益生菌用于患有免疫抑制的上皮屏障损伤的患者时,应确定细菌易位的风险。彻底的菌株特征评估对于在健康或患病人群中使用NGP的潜在安全问题至关重要。
在对A.soehngenii进行体外测试后,研究人员转向动物模型,以评估该菌株对胰岛素敏感性的安全性和有效性。
首先,在厌氧条件下生产了一批临床前的A.soehngenii。
简而言之,培养物在厌氧条件下生长至指数阶段结束,通过厌氧离心浓缩,用磷酸盐缓冲盐水(PBS)洗涤,最后用10%甘油稀释至100μl的106、108和1010菌落形成单位(CFU)浓度。
通过16S rRNA测序和细胞形态学的显微评估来评估纯度。
通过最可能数(MPN)分析评估生存能力,并通过显微分析确认。样品直接储存在−80°C下,并在生产6个月内使用,在此期间生存能力稳定。
此外,其中一些样品在2年内进行了稳定性测试,以支持临床试验的产品开发。
接下来,研究人员在雄性糖尿病(db/db)小鼠中进行了一项剂量发现研究,以测试口服A.soehingeii对胰岛素敏感性和脂质代谢的安全性和有效性。
每天用A.soehingeii或安慰剂(10%甘油)治疗小鼠达4周,期间未观察到不良事件(正常生命体征)。在胰岛素耐受试验期间观察到胰岛素敏感性的显著改善,这在108CFU剂量下最强。这伴随着肝脏脂肪的减少和Fasn和Acc1基因的表达减少,两者都参与脂肪生成。
为了证实这些发现并进一步剖析A.soehingeii的治疗机制,Bäckhed教授的实验室对db/db小鼠进行了第二项研究。
用108 CFU的A.soehingeii或热灭活A.soehingeii处理小鼠4周。当体重保持不变时,在活跃的A.soehingeii治疗后观察到静息能量消耗增加。此外,与热灭活的A.soehingeii相比,活性A.Soehingeii增加了粪便丁酸水平,并改变了胆汁酸代谢。
这两项小鼠研究表明,用 A.soehingeii 进行治疗是安全的,并对代谢产生有益影响,这可能由丁酸盐的产生和胆汁酸代谢的变化介导。这些数据用于获得我们在人类中进行的临床研究的伦理批准。
最近,对A.soehingeii CH106(一种来自A.soehingeii 菌株L2-7T的四环素敏感衍生物)进行了毒理学安全性评估,表明以推荐剂量摄入是安全的。
根据欧洲食品安全局(EFSA)和美国食品和药物管理局(FDA)对新的不可吸收食品成分进行安全评估的要求,对A.soehingeii进行了遗传毒性和亚慢性毒性评估。细菌反向突变和体外哺乳动物细胞微核试验均未显示出遗传毒性作用。
此外,大鼠的90天亚慢性毒性没有发现与A.soehingeii 喂养相关的任何不良事件,即使在最高剂量(5×1011 CFU/kg体重/天)下,也没有发现超过人类建议每日摄入量100倍以上的不良事件。
这些研究结果支持口服A.soehingeii 作为食物补充剂是安全的。
学习要点和方向
在临床前开发期间,应提供足够的药理学和毒理学信息,以支持拟议的临床试验。
NGP的安全性和毒性研究具有挑战性↓↓
由于该产品通常不会到达全身循环,但其代谢产物或其活性可能直接或间接影响身体的生理功能,因此疗效和毒性不一定与剂量有关。
人体生理学和微生物群组成等其他因素可能会影响安全性和疗效。
由于大多数NGP与人类宿主(全生物概念)共同进化,很难将动物研究的结果转化为人类环境。
因此,强烈建议将体外、离体和体内模型结合起来,以建立适应预期人群风险的全球安全性概况。
通常根据经济合作与发展组织(OECD)的良好实验室规范(GLP)原则进行安全性和毒性研究。然而,由于需要创新的方法和模型(例如,人类胃肠道的人工模型),而这些方法和模型可能既无法验证,也无法在GLP水平上验证,因此这可能很困难。
对于食品成分和膳食补充剂,EFSA建议采用毒理学研究的分级方法。
该分级方法评估NGP的毒代动力学、遗传毒性、亚慢性和慢性毒性、致癌性和致畸性,平衡数据要求和风险。该方法也用于A.soehingeii CH106的毒理学安全性评估。如果NGP打算用作患病人群中的药物产品,则必须证明目标人群的安全性。
前面的图总结了必须解决的最重要的问题,例如治疗剂量和持续时间对毒性反应的影响,以及致畸、致癌和遗传毒性的可能性。
在能够口服 A. soehngenii 给人类之前,必须制造出适合临床试验的产品。
在独立伦理委员会批准时(2014年),A.soehngenii被视为益生菌,必须遵守荷兰“Warenwet”,这符合欧盟膳食补充剂法规。这意味着必须根据危害分析和关键控制点(HACCP)标准进行生产。可根据HACCP标准进行临床干预研究。
首先,为了大规模生产食品级产品,进一步优化了生长培养基。该组合物基于先前的经验:
(1)实验室化学物质转化为食品级来源
(2)仅使用无动物成分(无血红素或肉蛋白胨)
(3)复杂性降低(微量矿物质、维生素、碳源和有机酸的去除/减少)
(4)生物量产量进一步提高。原材料来源于经过审计的可靠供应商,以确保高质量。发酵前,在大型发酵罐系统中制备并消毒生长培养基,通过氮气(N2)冲洗使其完全厌氧。
发酵分四个连续步骤进行,如下图所示:
doi.org/10.3389/fmed.2022.1077275
首先,用精心准备的A.soehngenii 冷冻种子储备接种少量食品级培养基。动物研究中使用了相同的菌株,因此,该菌株具有良好的特性,是可行的、纯净的,没有任何细菌或病毒污染物。在37°C下发酵24小时后,使用培养物接种1 L培养基,再次发酵18小时。
然后,使用该二级种子培养物在小型发酵罐中接种30 L培养基。该发酵罐发酵17小时,作为大规模发酵的试车。
最后,用10L小发酵罐的接种物接种大发酵罐中的290L培养基。控制小型和大型发酵罐的温度、pH和氧气水平,并使用培养物的光密度(OD)确定发酵时间(14至18小时)。在大型发酵罐中发酵16小时后,A.soehngenii生长至OD约为10.
使用中空纤维膜(Koch膜系统;HF3043-25-43-PM500;HF3043-16-106-PM500)和PBS渗滤,浓缩并洗涤细胞。将发酵液冷却至10°C,泵送通过厌氧膜装置,并在3小时内浓缩至40–50 L。
在第二阶段进行渗滤以降低培养基成分和发酵产物的水平。使用超高温对洗涤缓冲液进行灭菌、脱气并直接添加到返回的细胞流中进入发酵罐。6小时后,将细胞浓缩约20倍至15升,99.8%的培养基化合物被丢弃成废物,最终浓缩物中只剩下2.9%的培养基组分。
最后,可将9L产品从系统中收获到10L的无菌N2冲洗容器中。
为临床研究生产了四个不同的批次,包括600个试管和一个安慰剂批次,其中PBS中含有浓度为106、108和1010 CFU/mL的10mL A.soehngenii,PBS+10%甘油,PBS中只有10%甘油。
对于每一批,用甘油和PBS制备7L瓶用于进一步稀释,将其高压灭菌、冷却并用N2冲洗。从9L收获的浓缩液中,向这些瓶中加入必要的体积以获得正确的浓度。在连续搅拌和N2冲洗下,将瓶子置于冰上。
首先用N2填充10mL管,然后用定量管泵填充10mL产品。立即关闭试管,贴上标签,并在灌装后10分钟内将其置于−30°C的冰箱中。所有填充均在消毒层流柜内进行。
在制造过程中,持续监测温度、pH值和氧气水平。此外,在过程中的每一步都测定了细胞计数和OD,以及是否存在任何污染物。由于厌氧菌很难在琼脂平板上定量计数,因此在厌氧条件下进行MPN分析以获得活细胞的数量,并用显微镜评估细胞形态。所有上述质量控制均针对符合人类消费标准的包装小瓶进行。
Anaerobutyricum soehingeii 中间体和最终产品规范

doi.org/10.3389/fmed.2022.1077275
随后,每6个月测试生产的小瓶的稳定性。生产完成后,这些小瓶的“保质期”为6个月,这是荷兰法律要求的食品。如果满足生存能力和纯度标准,有机会延长小瓶的有效期。
下表显示了在3年时间段内具有最高剂量 A. soehngenii 的小瓶的效力和纯度。
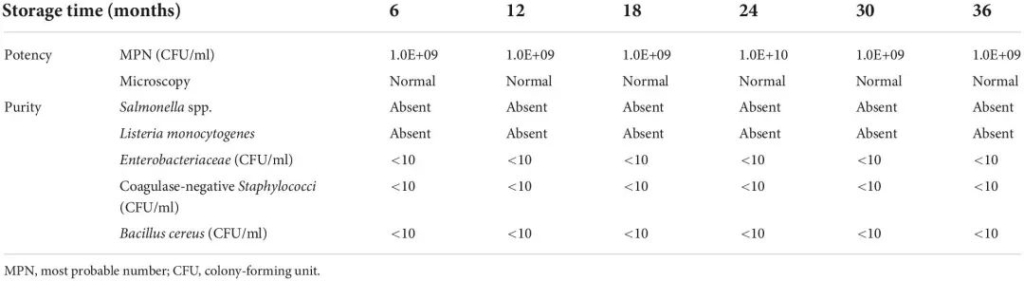
doi.org/10.3389/fmed.2022.1077275
学习要点和方向
▸ 工业规模生产是技术挑战
与实验室规模培养相比,以工业规模生产菌株对菌株和培养基的要求不同。因此,当一种菌株符合潜在NGP的条件时,应采取措施,看看该菌株是否可以在工业规模上培养。
培养NGPs所需的严格条件是技术挑战之一,例如需要特定的营养、缺氧、稳定的温度和合适的pH。此外,更长的保持时间、泵送的绝对压力、下游净化过程和储存可能会对细菌细胞的生存能力产生负面影响。
▸ 将菌株加入产品后需要有效策略,将其输送到作用部位
接下来,必须将菌株加入产品中,如胶囊、粉末或液体悬浮液。由于大多数NGP是严格的厌氧菌或兼性厌氧菌,因此应尽量减少接触氧气。为此,应降低容器中的氧气渗透性,并可添加抗氧化剂以降低氧化还原电位。
摄入产品后,NGP必须在胃肠道的恶劣环境中生存。肠溶胶囊和微胶囊是保护细菌并将其运送至其作用部位的有效策略。
▸ 有效期之前,足够量的递送剂量
最终,制造需要产生一种强健、稳定的产品,该产品将允许在有效期之前以足够数量的NGP递送有效剂量。
▸ 质量控制和质量保证计划需要到位
对于医药产品或LBP,需要按照良好生产规范(GMP)进行生产。对于食品和膳食补充剂,HACCP认证工厂的生产是标准。无论如何,质量控制和质量保证计划需要到位,以确保成分和最终产品的一致质量,并确保可靠的生产过程。
应从所用原材料、细胞库系统、细胞生长和收获、纯化和下游加工到过程中测试,应清楚记录菌株的制造过程。
▸ 彻底描述最终产品的制造
同样,必须彻底描述最终产品的制造,包括生产记录和配方、填充、标签和包装说明。对于菌株和产品制造,必须评估与同一房间或同一接触设备生产的其他产品交叉污染的风险。
▸ 必须描述菌株和产品的规格
包括采样程序和验证测试方法的说明。这些规范应描述身份、效力、纯度、污染、外观,如果适用,还应描述活细胞百分比、颗粒物、热原、pH和残留水分的附加测试。
▸ 必须生成稳定性数据
证明产品在计划的使用期限内,在效力和污染方面是稳定的。
对于冷冻产品,应评估多次冻融循环的影响,而对于冻干产品,应探讨重构后的保质期。
▸ 需要评估该产品对环境的影响
特别是当该菌株经过基因修饰、致病、生态上比野生型更适合或难以根除时。
为了验证人类环境中的小鼠数据,研究人员建立了一项单盲、I期/II期剂量递增试验,以确定Anaerobutyricum soehingeii 在肥胖、胰岛素抵抗受试者中的安全性和有效性。
在这项研究中,27名患有代谢综合征的肥胖高加索男性被纳入并分配接受soehngenii,剂量为107、109或1011个细胞/天,持续28天。
当受试者对其各自的治疗剂量进行盲测时,前9名受试者必须在剂量增加到更高浓度之前成功完成最低剂量的研究方案。
受试者在家中的−20°C温度下储存冷冻瓶,每天解冻一个10mL瓶,与100mL牛奶混合并口服。添加牛奶以增加胃中的pH值,从而在胃肠道通过期间保护活细胞。主要结果是安全性,此外,治疗4周后评估了对胰岛素敏感性和脂解的影响。
使用高达1011个细胞/天的A.soehngenii治疗耐受性良好,无任何严重不良事件。
当所有治疗组合并时,A.soehngenii 的粪便丰度与改善的外周胰岛素敏感性相关,并伴有胆汁酸分布的有益变化。
出乎意料的是,没有观察到粪便丁酸盐水平的增加,这可以通过短链脂肪酸的挥发性和化验的检测限来解释,这使得丁酸盐难以测量。
A.soehngenii 丰度的增加是短暂的,大多数在停止后2周消失。给药菌株的生存能力受到胃酸和氧气的负面影响。
然而,如接受最高剂量的受试者粪便中的最高复制信号所示,A.soehngenii部分能够在胃肠道中存活。通过包封和/或冷冻干燥更好地保护菌株免受酸性和含氧环境的影响,可以进一步提高生存能力(和治疗效果)。
为了进一步阐明A.soehngenii在人体中的作用模式,进行了一项随机安慰剂对照交叉试验,在该试验中,直接在十二指肠中施用该菌株,从而避免胃酸并减少氧气暴露。
由于小肠在葡萄糖增敏、调节胰岛素敏感性/分泌和葡萄糖稳态中起着核心作用,因此假设十二指肠直接输注A.soehngenii 可以进一步提高治疗效果。
同样,患有代谢综合征(N=12)的肥胖受试者被纳入并随机接受单次鼻十二指肠输注,输注最高剂量的A.soehngenii(1011个细胞)或安慰剂(PBS中的10%甘油)。6小时后,进行十二指肠活检和混合膳食试验。
此外,受试者监测了24小时血糖,并收集了一些粪便样本。经过4周的冲洗期后,受试者转为另一个治疗组,在第一次试验中,该治疗组被确定为足够长的时间来减轻压力。
再次,这项研究表明,A.soehngenii 的给药安全且耐受性良好。该菌株的治疗增加了促胰岛素激素胰高血糖素样肽1(GLP-1)的餐后漂移,伴随着葡萄糖变异性的降低。
鉴于A.soehngenii 具有产生丁酸盐的能力,并且在A.soehngenii处理后,粪便中的丁酸盐水平趋于较高,GLP-1分泌增加可能是丁酸盐激活肠L细胞上的G蛋白偶联受体43(GPR43)的结果。
由于A.soehngenii表达胆汁酸钠共转运蛋白和胆汁酸水解酶,并且二级胆汁酸的血浆水平升高,GLP-1表达增加也可能是TGR5被二级胆汁酸类激活的结果。
注:TGR5是G蛋白偶联受体超家族成员,TGR5不仅是胆汁酸受体,也是多种选择性合成激动剂的受体,调节不同信号通路的衍生物。参与能量稳态、胆汁酸平衡及葡萄糖代谢。
此外,用A.soehingeii治疗导致十二指肠核法尼素X受体(FXR)及其靶基因OSTa的表达降低,这也可能是GLP-1可用性增加的原因。
最后,葡萄糖变异性的改善可以通过GLP-1和丁酸盐的胰岛素增敏作用来解释。
此外,A.soehingeii改变了73个基因的十二指肠转录,最显著的是诱导REG1B和REG1A的表达,后者编码生成胰岛衍生蛋白1A/B。
注:Reg1A和Reg1B在肠隐窝底部的Paneth细胞中强烈表达,在管腔中分泌,可能通过诱导祖细胞或L细胞增生局部发挥作用。
此外,发现REG1B的诱导与施用A.soehngenii后24小时GLP-1分泌增加和葡萄糖变异性降低相关。单剂量A.soehingeii 的治疗不会影响微生物群的组成或多样性,正如之前的研究中所见。
此外,粪便A.soehngenii的丰度没有随时间变化,排除了交叉时微生物群介导的遗留效应。
学习要点和方向
第一次临床研究的主要目的是确定安全性,并根据产品的耐受性确定适当的剂量范围和方案。这包括确定最小有效剂量或最佳有效剂量范围,如果可能,还包括最大安全剂量。
除给药外,重点应是获取安全数据,以识别常见的产品相关不良事件。这些早期临床研究通常在健康志愿者中进行,但纳入患者可能更合适,例如当NGP应纠正生物失调时。应考虑确保研究参与者安全的风险缓解措施,如连续入组、剂量递增和独立数据监测委员会的监测。
此外,监测易位、炎症和感染以及确定NGP的持久性及其在最终给药后的作用是有利的。
重要的是考虑影响微生物群功能或组成的其他混杂因素,如年龄、饮食、生活方式和环境因素。在这方面,采用安慰剂对照交叉设计的研究非常有用,因为它们可以限制这种外在和内在混杂因素的影响,从而允许更小的样本量。不用说,盲板非常重要,应仔细考虑冲洗期。
越来越多地,基线微生物群组成也被纳入筛选标准,例如寻找特定肠道类型中特定细菌群的存在或集群。这将导致更具可比性的研究组,并且当特定的菌群参与作用机制时,可以优化干预的效果。
根据粮农组织和世界卫生组织对益生菌的定义,益生菌可分为膳食补充剂和药物,但两者在监管上存在着巨大的差异。同样,含下一代益生菌的产品可以作为食品、膳食补充剂或药物进入市场,具体取决于预期用途。
在欧盟,食品由欧洲食品安全局监管,药品由欧洲药品管理局监管,而在美国,食品和药物管理局负责这两类产品。当预期用途与预防、缓解或治疗疾病有关时,该产品将被视为医疗产品或医疗器械。
与增强生理功能或降低疾病风险因素有关的口服摄入产品可被归类为功能性食品或食品补充剂。此外,具有纯美容功能的局部应用产品可被评定为化妆品。为了确保法规遵从性,在临床前研究和制造之前,必须决定缩进的用途和随后的法规分类。
在欧盟,“食品” 被定义为“任何物质或产品,无论是加工的、部分加工的还是未加工的,旨在或合理预期被人类摄入”。根据标签、展示和广告的一般要求和规定,对每一类进行了相应的管理。
当NGP被用作食品或膳食补充剂时,它们很可能被认为是一种新型食品。然而,如果NGP经过了基因改造,它将作为转基因食品受到监管。为了使NGP作为一种新型食品进入市场,它需要获得授权并列入欧盟名单。
最重要的条件之一是NGP不会对人类健康造成风险,这必须得到科学证据的支持。这包括一项综合风险评估,结合预期人类接触的生物和毒理学研究,评估对人类健康的潜在风险。此外,申请应包含NGP、制造工艺、产品成分、使用的分析方法、标签和预期用途条件的详细说明。
除安全性外,该产品不得促进食品链或环境中抗微生物耐药性的传播,需要对抗微生物耐药性进行表型和基因型评估。
即使“含有益生菌/益生元”的声明在欧盟也被视为健康声明。为了接受健康声明,需要对NGP进行适当的描述,并通过高质量的研究证明其对健康有益的影响和因果关系。
自2012年和2019年以来,FDA和EDQM明确了LBP的质量要求,其中LBP被描述为含有供人类使用的活微生物的医药产品。除这些质量要求外,目前没有具体的LBP法规。
然而,由于LBP含有活微生物,它们被视为生物医药产品,因此必须遵守立法和监管框架。如果没有特定的LBP子类别,开发商将不得不依赖其他子类别生物医药产品的监管概念。
其中一个概念是基于从临床前和临床研究中获得的质量、安全性和疗效数据进行彻底的风险效益分析。
临床前和临床研究设计的其他相关指南包括:
迄今为止,没有LBP进入欧盟市场,这部分是由于缺乏明确的监管框架。在缺乏明确指导方针的情况下,尽早与主管当局进行互动,以讨论不确定性和减少风险的重要性。
随着对我们肠道微生物群的了解越来越多,将发现和开发越来越多的潜在下一代益生菌。本文以A.soehingeii为例,介绍了将其开发为下一代益生菌的经验。
重要的是,这些新菌株具有良好的特性、高质量和安全性。对NGPs进行彻底的安全评估非常重要(虽然很复杂),特别是因为疗效和毒性不一定与剂量有关。
由于这个领域相对稚嫩,目前还没有具体的LBP监管,因此在开发的早期阶段与监管机构进行沟通有助于降低风险并澄清任何不确定性。这需要在开发初期对市场(食品或药品)的路线有明确的看法。
在FMT干预后,A.soehingeii 这种微生物被确定为潜在的有益微生物,在临床前的体外和体内研究以及人类研究中都显示出很有前景的结果。它在改善胰岛素敏感性、增加GLP-1分泌和降低葡萄糖变异性方面显示出良好的效果。
这些效应可能通过丁酸和次生胆汁酸的产生介导。通过更好地保护菌株免受酸性和含氧环境的影响,例如通过冷冻干燥和封装,可以潜在地提高生存能力,从而提高治疗效果。
主要参考文献:Wortelboer K, Koopen AM, Herrema H, de Vos WM, Nieuwdorp M, Kemper EM. From fecal microbiota transplantation toward next-generation beneficial microbes: The case of Anaerobutyricum soehngenii. Front Med (Lausanne). 2022 Dec 5;9:1077275.
Kumari M, Singh P, Nataraj BH, Kokkiligadda A, Naithani H, Azmal Ali S, Behare PV, Nagpal R. Fostering next-generation probiotics in human gut by targeted dietary modulation: An emerging perspective. Food Res Int. 2021 Dec;150(Pt A):110716.
Echegaray N, Yilmaz B, Sharma H, Kumar M, Pateiro M, Ozogul F, Lorenzo JM. A novel approach to Lactiplantibacillus plantarum: From probiotic properties to the omics insights. Microbiol Res. 2022 Dec 22;268:127289.
Garcia-Gonzalez N, Battista N, Prete R, Corsetti A. Health-Promoting Role of Lactiplantibacillus plantarum Isolated from Fermented Foods. Microorganisms. 2021 Feb 10;9(2):349.
Seegers JFML, Gül IS, Hofkens S, Brosel S, Schreib G, Brenke J, Donath C, de Vos WM. Toxicological safety evaluation of live Anaerobutyricum soehngenii strain CH106. J Appl Toxicol. 2022 Feb;42(2):244-257.

谷禾健康

戴阿利斯特杆菌属 (Dialister)
✦
Dialister(戴阿利斯特杆菌属)是小的、厌氧或微需氧的革兰氏阴性球状或杆状菌,因次也被翻译成小杆菌属。
Dialister菌是人体肠道菌群中的一种常见菌种。该菌属物种被发现出现在人体全身各个部位,包括骨骼和血液,但是主要从人体粪便,口腔以及上呼吸道,阴道等部位分离或发现,属于人体肠道核心菌。
肠道菌群中Dialister菌属在96.15%的人群中检出,但平均丰度属于核心菌属中较低的,平均人群丰度为0.9%左右。
其中60.58%的人群中检出的是Dialister invisus,其次是55.77%的人检出Dialister sp.000434475,15.33%的Dialister propionicifaciens和12.98%的Dialister succinatiphilus(根据谷禾肠道菌群人群队列数据库)。
Dialister菌已在有症状和无症状的个体中被鉴定,因此被认为在正常微生物组中发挥了一定作用。目前尚不清楚这是如何发生的,但可以推断该菌所在身体环境与其致病或有益属性相关。
当在肠道中检出“Dialister invisus”与疾病无关,但是当在尿液中发现时,可能与尿路感染有关;当在口腔中检出“Dialister invisus”,它通常与冠周炎、边缘和根尖周炎、龋齿、口臭和牙髓感染有关。特别是“Dialister pneumosintes”被认为是一种新的牙周病病原体。
该菌属菌株难以与微小的革兰氏阴性厌氧球菌区分开来,所以在临床上比较难鉴定,一般需要分子方法,例如 16S rRNA 或宏基因组来鉴定。
Dialister 属于厚壁菌门,韦荣氏球菌科,代谢碳水化合物,产生琥珀酸和乙酸,丙酸,丁酸,产生组胺,过氧化氢酶。与抑郁症,自闭症、情绪控制、口腔疾病、减肥、强直性脊柱炎疾病,不同组织部位的感染,肾病等相关。
酸奶、胡桃、芽孢杆菌补充、双歧杆菌补充、菊粉以及运动可增加肠道Dialister 属的丰度。
Dialister(戴阿利斯特杆菌属)是厚壁菌门革兰氏阴性、厌氧杆菌。大部分菌种不形成孢子、不运动。产生琥珀酸和乙酸,丙酸,丁酸,产生组胺,过氧化氢酶。
已鉴定物种:
其中,D. pneumosintes和D. micraerophilus最常从临D. pneumosintes很难在常规培养基中生长,基于 16s rRNA 的 PCR 测定已开发用于检测这种病原体。这种微生物已从牙周炎、牙龈炎、根管感染、龈下菌斑 、人咬伤伤口感染 、呼吸道、头颈部感染 和阴道感染中分离出来。已报告严重的感染性并发症,包括脑脓肿 和肝脓肿,疑似牙源性感染。
Dialister 物种在人类感染中的作用已经明确,尽管真正的临床意义仍然未知。D. pneumosintes已从肺、血液、脑和上颌窦中分离出来和D. micraerophilus菌株已从多微生物培养物中的几个临床样本中得到表征。
Dialister 物种被认为是口腔、鼻咽、肠道和阴道菌群的共生生物。细菌可以从这些位置传播到各个器官,并可能导致严重的疾病,例如菌血症。患者的感染源可能是阴道菌群,尤其是当她经历过数次前庭大腺炎发作并接受过多种抗菌药物治疗时。正如先前报道的那样,应注意这些疾病,以避免传播到血液中。
Dialister菌属下的许多种都可能导致感染。例如,Dialister pneumosintes是一种常见的致病菌,可以导致呼吸道感染、皮肤感染和肠道感染。Dialister invisus也是一种常见的致病菌,可以导致呼吸道感染、皮肤感染和肠道感染。
其他常见的致病菌包括:
这些菌都可以导致许多不同类型的感染,包括呼吸道感染、皮肤感染和肠道感染。
应该注意的是,Dialister菌属下的所有种都不是总是致病的。在某些情况下,这些菌是人体的自然共生菌,并不会导致感染。然而,在免疫功能下降或者某些其他情况下,这些菌可能会导致感染。因此,应该根据临床症状和诊断结果来判断Dialister菌是否是致病的。
由于难以识别病原体, Dialister物种的抗菌药物敏感性数据仍然相对稀缺。Dialister分离株对根据 CLSI 指南测试的所有抗菌药物敏感,而 33 株菌株对一种或几种抗生素的敏感性降低,包括甲硝唑、红霉素、原始霉素、利福平、哌拉西林、左氧氟沙星和环丙沙星。
Dialister 已经从人类临床标本中分离出来,尤其是肺息肉,并且与人类临床感染有关,其中大多数是牙源性感染。
Dialister pneumosintes是一种不形成孢子、不运动、不发酵、革兰氏阴性厌氧杆菌。据报道,它作为正常菌群出现在鼻咽、口腔、肠道和阴道中。这种细菌于 1921 年首次在 1918-1919 年流感流行期间从患者的鼻咽分泌物中检测到,最初被命名为Bacterium pneumosintes.
Dialister pneumosintes 是一种与口腔感染相关的专性厌氧革兰氏阴性杆菌。研究报告了一名既往健康的 51 岁女性,她因 Dialister积气引起的肝脓肿作为牙脓肿的并发症就诊。通过在肝脏渗出液中使用广谱细菌 16S rRNA 基因 PCR 鉴定微生物。脓肿引流和 4 周抗生素治疗后,患者痊愈。
Dialister pneumosintes 是一种可疑的牙周病原体。它可以通过血行传播或区域传播影响身体的不同部位。这种微生物引起的牙周感染可能会引发潜在的危及生命的并发症。
D. invisus 物种于 2003 年由 J. Downes 首次发现,并通过 16S rRNA 测序从牙髓感染患者的根管中分离出来。D. invisus 主要从深牙周袋中回收,发现于龈缘以下。
由于 D. invisus 与边缘牙周炎、龋齿、口臭和根尖周炎有关,并且通常从牙髓感染中分离出来,因此它被认为是一种重要的人类病原体。了解持久存在的牙髓微生物(例如D. invisus)有助于确定牙髓感染患者的最佳治疗方案。为了控制或消除与牙髓病例相关的病原微生物,需要对这些病原体有透彻的了解。
由比利时鲁汶天主教大学的微生物学家 Jeroen Raes 领导的研究小组发现,被诊断患有抑郁症的人的肠道中缺乏的两种细菌。抑郁症患者的粪球菌 Coprococcus和Dialister菌也已耗尽。
虽然较低水平的Dialister与抑郁症有关,但最近的一篇论文将较高水平的Dialister与关节炎联系起来。Raes 说,可能是一种 Dialister 的流行增加了患关节炎的风险,而另一种的流行降低了患抑郁症的风险,但要确定这些细节还需要更多的研究。
安大略省圭尔夫大学微生物学教授 Emma Allen-Vercoe 表示,Coprococcus和Dialister可能是用作精神益生菌或针对心理健康的益生菌的理想候选者。
癌症是复杂的多因素疾病,被认为是一个全球性问题。Dialister 的终产物,如乙酸盐、乳酸盐和丙酸盐,似乎在致癌机制中起着重要作用。一项Meta调查人类癌症研究中微生物组与 Dialister 成分变化之间的关联。结果:荟萃分析包括 26 项研究,包含 1649 个对照样本和 1961 个癌症样本。与健康对照相比,Dialister 在癌症患者样本中显着升高。表明不同癌症类型与 Dialister 微生物组组成之间存在关系。
Lindefeldt 等人报告了 12 名儿童为治疗难治性癫痫开了生酮饮食,发现饮食处方的 alpha 多样性没有变化。双歧杆菌、直肠真杆菌和Dialister随着生酮饮食而减少。
Joossens 等人发现克罗恩病患者中有五种细菌具有生态失调的特征,即Dialister invisus减少,梭状芽胞杆菌簇 XIVa 的非特征性物种,Faecalibacterium prausnitzii和青春双歧杆菌,以及Ruminococcus gnavus增加。
食用油炸肉降低了肠道菌群的丰富度,以及毛螺菌科(Lachnospiraceae)和黄曲霉属(Flavonifractor)的丰度,同时增加了Dialister、多尔氏菌属(Dorea)和韦荣球菌属(Veillonella)的丰度(P FDR<0.05)。
通过比较3 个月后饮食转变 (DS) 从严重依赖肉食到素食和体育锻炼 (EX) 对肠道微生物组组成的影响发现,Dialister succinatiphilus的丰度被体育锻炼上调。
在一项先导性研究中,26名受试者采用了低热量、富含蔬果的饮食习惯,而其中有些人减重的幅度不如其他人高。对受试者肠道菌群的分析显示,有两类特定细菌的含量会影响减重速度,其中有一种为Dialister。
研究发现,比较容易减肥的人体肠道内考拉杆菌属(Phascolarctobacterium)水平较高,因此该菌也用来预测肥胖指标。而难以减肥的人体内则小类杆菌属(Dialister)水平较高。在难以减重的那部分人体内,这种细菌能够分解碳水化合物,更高效地利用其中的能量。
接受嗜酸乳杆菌和纤维二糖的健康志愿者表现出乳酸杆菌、双歧杆菌、柯林氏菌和真杆菌的水平升高,而Dialister降低了。
还有一些研究报告说,在1周龄时,非共生肠道细菌(如克雷伯氏菌和肠球菌)的相对丰度较高与1岁时的呼吸道感染有关;3 个月时链球菌的相对丰度与 5 岁时的特应性喘息有关;1岁时Rothia或Dialister的高相对丰度与4-5岁时哮喘相关。但是具体作用机制目前还不清楚。
发现 Dialister 属的丰度与强直性脊柱炎疾病活动评分呈正相关(Spearman 的 rho = 0.62,错误发现率 – 校正 q < 0.01)。在 SpA 患者和健康对照者的非发炎回肠和结肠活检组织中观察到的低频率 Dialister 进一步支持了这一发现。
Dialister 属会增加下列菌群的丰度:
Dialister 属会抑制下列菌群的丰度:
主要参考文献:
Morio F, Jean-Pierre H, Dubreuil L, et al. Antimicrobial susceptibilities and clinical sources of Dialister species. Antimicrobial Agents and Chemotherapy. 2007 Dec;51(12):4498-4501.
Wendy J. Dahl, … Jason M. Lambert, in Progress in Molecular Biology and Translational Science, 2020
Markus F. Neurath, in Mucosal Immunology (Fourth Edition), 2015
The Association of Fried Meat Consumption With the Gut Microbiota and Fecal Metabolites and Its Impact on Glucose Homoeostasis, Intestinal Endotoxin Levels, and Systemic Inflammation: A Randomized Controlled-Feeding Trial
Lkhagva E, Chung HJ, Ahn JS, Hong ST. Host Factors Affect the Gut Microbiome More Significantly than Diet Shift. Microorganisms. 2021;9(12):2520. Published 2021 Dec 6. doi:10.3390/microorganisms9122520
人体消化系统包含大约几百到几千种不同的细菌种类,其丰度构成因人而异。
其中少数益生菌乳杆菌属,即嗜酸乳杆菌、植物乳杆菌、短乳杆菌、乳酸乳杆菌、干酪乳杆菌、保加利亚乳杆菌、发酵乳杆菌、鼠李糖乳杆菌特异性产生细胞外蛋白、胞外多糖、细菌素和脂磷壁酸,通过与上皮细胞相互作用影响宿主的健康和生理,增强宿主免疫系统。
在乳杆菌菌种中,植物乳杆菌(L. plantarum)是革兰氏阳性、短杆状、微需氧、耐酸、不形成孢子、不呼吸、低 G + C 含量、异型发酵的乳杆菌群,具有一系列作为发酵剂和防腐剂在食品工业中的应用。
它是一种非孢子形成细菌,可产生有机酸,例如乙酸、琥珀酸和乳酸作为主要代谢物。植物乳杆菌在人类和其他哺乳动物的胃和其他复杂的胆汁盐分泌物中的低缓冲能力下生长。
除了在食品工业中的应用外,肠道微生物植物乳杆菌是一种很有前途的益生菌,可治疗腹泻、高胆固醇和特应性皮炎等。它是如何工作的,它还有哪些其他好处?本文带您了解更多。
植物乳杆菌是一种分布广泛、用途广泛的乳酸菌。它代表了许多食物和饲料的微生物群的一部分,包括乳制品、肉类、鱼类、蔬菜发酵产品(例如,葡萄汁、酸菜、泡菜、酸面团)和青贮饲料;它也是人和动物粘膜(口腔、胃肠道、阴道等)的天然居民。
植物乳杆菌是一种具有抗癌、抗炎、抗肥胖和抗糖尿病特性的抗氧化剂 [ 1 ] .
植物乳杆菌菌株的微观形态图像
DOI:10.1099/ijs.0.65319-0
植物乳杆菌耐受不同范围的盐,尤其是 NaCl 和胆汁盐,pH 值为 4.0-8.0,温度为 28-45°C,并且分别在 37°C 和 pH 7.0 的温度下具有最佳细胞生长。鉴定出的菌株在上消化道中经受了各种生物障碍,例如低 pH 值、裂解酶和胆汁盐。能够利用广泛的糖类,尤其单糖和双糖。此外,淀粉酶和蛋白酶等细胞外酶的产生对其有利。
B族维生素
从生牛奶中分离出的植物乳杆菌能够产生 B 族维生素核黄素( B2 ) 和叶酸(B9 ) [ 2 ].
铁吸收
植物乳杆菌可使健康女性从果汁饮料中吸收的铁增加约 50% [ 3 ].
植物乳杆菌可以使女性对燕麦中铁的吸收提高 100% 以上 [ 4 ].
钙吸收
含有植物乳杆菌的发酵乳表现出更高的钙保留摄取 [ 5 ].
植物乳杆菌是体内的短暂居民。它可以轻松抵御胃酸,并可以完成从补充品进入口腔,到肠道,到结肠,到粪便的完整旅程。植物乳杆菌生长的最佳温度非常接近体温。
植物乳杆菌也是一种强大益生菌,可猛烈攻击体内的致病性有害细菌。通过杀死坏菌,它帮助我们自己的本地细菌变得更强壮,并帮助我们人体更能抵抗外来病原体的入侵。
除了作为增强免疫系统的重要方式之外,人类在所有有记录的人类历史中都食用了大量的植物乳杆菌。
该菌株用于许多食品中,例如:
酵母面包;酸菜;泡菜;发酵食物
注意
植物乳杆菌益生菌补充剂尚未获得国家药监局和FDA的医疗用途批准,可能缺乏可靠的临床研究。为防止罕见的副作用,请在使用益生菌之前咨询权威的医疗建议。
可能有效
1) 腹泻
乳酸菌益生菌在改善与各种疾病相关的腹泻方面表现出很大的希望,包括旅行者腹泻和抗生素相关性腹泻 [ 6、7 ] .
在一项针对 438 名患有抗生素相关性腹泻的儿童的临床试验中,植物乳杆菌益生菌减少了稀便或水样便和腹痛的发生率,而且没有产生不良副作用 [ 7 ].
2) 皮肤健康
在临床试验中,植物乳杆菌显着增加了面部和手部的皮肤水分含量。益生菌组志愿者在第 12 周时皱纹深度明显减少,皮肤光泽度在第 12 周时也有显着改善。益生菌组的皮肤弹性在4 周后改善了 13.17%,在 12 周后改善了 21.73% [ 8 ] .
当作为益生菌服用时,植物乳杆菌改善皮肤水合作用,对人体皮肤具有抗光老化作用[ 9、10 ] .
植物乳杆菌抑制胶原蛋白的降解并促进其合成,减少活性氧 ( ROS ) 的产生 [ 11 ] .
在无毛小鼠中,植物乳杆菌降低了 UVB 诱导的表皮厚度,抑制了水分流失并增加了神经酰胺水平 [ 12 , 13 ] .
特应性皮炎
每天摄入含有热灭活植物乳杆菌的柑橘汁可减轻人类特应性皮炎的症状[ 14 ] .
从泡菜中分离出的植物乳杆菌改善小鼠特应性皮炎[ 15 ] .
3) 溃疡性结肠炎
在多项临床试验中,乳酸菌益生菌已显示出减轻溃疡性结肠炎症状的希望。含有植物乳杆菌的合生元混合物在8周后显着改善了 73 名患者的 UC 症状 [ 16、17 ] .
4) 胆固醇
在许多临床试验中,乳酸菌益生菌降低了胆固醇。在一项针对 60 名高胆固醇志愿者的研究中,含有植物乳杆菌的益生菌在 12 周后将总胆固醇降低了 13.6% [ 18 ] .
在患有糖尿病的大鼠中,植物乳杆菌降低血液甘油三酯和“坏”低密度脂蛋白胆固醇的比率,同时增加“好”高密度脂蛋白胆固醇的水平 [ 19 ] .
摄入植物乳杆菌后,胆固醇升高的小鼠的总血清胆固醇和甘油三酯显着降低 [ 20 ] .
双涂层植物乳杆菌可降低高脂肪饮食小鼠的胆固醇水平 [ 21 ] .
证据不足
研究人员目前正在调查植物乳杆菌是否具有其他健康益处。本节中的潜在益处至少在一项临床试验中产生了积极的结果,但这些研究规模小、相互矛盾或存在其他局限性。出于任何原因补充植物乳杆菌之前,请谨慎。
5) 肥胖
辅以含有植物乳杆菌的富含益生菌的奶酪的低热量饮食降低了患有肥胖症和高血压的俄罗斯成年人的 BMI 和血压[ 22 ].
植物乳杆菌还保护小鼠免受饮食引起的肥胖。这种细菌可降低肥胖小鼠的体重、脂肪量、空腹血糖、血清胰岛素、瘦素水平和促炎标志物 [ 23、24、25 ] .
植物乳杆菌发酵大麦逆转了高脂肪饮食大鼠的葡萄糖耐受不良,改善了升高的胰岛素,降低了甘油三酯和总胆固醇水平 [ 26 ].
植物乳杆菌通过诱导丙氨酸氨基转移酶( ALT )、γ-谷氨酰转移酶 ( GGT )、血浆甘油三酯、总胆固醇浓度、肌酐、尿素和体重的降低,来改善肥胖大鼠的肝功能和泌尿功能 [ 27 ].
6) 血糖
植物乳杆菌降低了绝经后妇女的血糖水平[ 22 ].
含有植物乳杆菌的豆浆具有抗氧化特性,可减少 2 型糖尿病患者的 DNA 损伤[ 22 ].
植物乳杆菌降低小鼠的食物摄入量、血糖水平、糖化血红蛋白水平和瘦素水平。这种细菌还有利于调节胰岛素水平并增加“好”(HDL) 胆固醇 [ 28 ].
植物乳杆菌导致高脂肪饮食小鼠对胰岛素的血糖水平显着降低 [ 29 ].
用植物乳杆菌治疗可有效调节糖尿病大鼠的血糖、激素和脂质代谢 [ 30 ].
植物乳杆菌显着改善糖尿病大鼠的免疫学参数并保护胰腺组织。此外,这种益生菌治疗显着降低了胰腺和血浆脂肪酶活性以及血清甘油三酯和低密度脂蛋白胆固醇率,并增加了高密度脂蛋白胆固醇水平。它还对肝肾功能发挥有效的保护作用 [ 19 ] .
7) 伤口愈合
在一项针对 34 名腿部溃疡患者的小型临床研究中,局部应用植物乳杆菌减少了糖尿病和非糖尿病患者的感染性慢性静脉溃疡伤口细菌负荷、中性粒细胞、凋亡和坏死细胞,并诱导伤口愈合[ 31 ].
8) 牙齿健康
热灭活的植物乳杆菌减少了接受牙周支持治疗的患者的牙周袋深度 [ 32 ].
9) 免疫力
在一项对 171 名成年人进行的临床研究中,植物乳杆菌提高了免疫活性并降低了应激标记物 [ 33 ].
即使是热灭活的植物乳杆菌也会激活人类的先天性和获得性免疫力 [ 32 ].
植物乳杆菌增强免疫抑制小鼠小肠的免疫力[ 34 ].
抗病毒
在感染单纯疱疹病毒 1 型 (HSV-1) 的小鼠中,口服植物乳杆菌显着延缓了感染早期皮肤损伤的发展,并减少了大脑中的病毒数量 [ 35 ].
同样在小鼠中,从发酵的韩国卷心菜中分离出的植物乳杆菌赋予了 100% 的保护作用,防止致命的甲型流感病毒感染,防止显着的体重减轻并降低肺部病毒载量 [ 36 ].
10) 过敏
在一项针对 42 名成年人的临床研究中,植物乳杆菌发酵的柑橘汁改善了日本柳杉花粉症的症状[ 37 ].
在一项细胞研究中,植物乳杆菌降低了大豆粉的过敏性 [ 38 ].
口服植物乳杆菌可减轻小鼠的气道高反应性和过敏反应 [ 39 ].
11) 念珠菌病
在外阴阴道念珠菌病 (VVC) 患者中,植物乳杆菌减少了常规治疗后的阴道不适,并改善了阴道细菌含量和阴道 pH 值 [ 40 ].
在一项临床试验中,使用植物乳杆菌可使外阴阴道念珠菌病复发风险降低三倍 [ 41 ].
植物乳杆菌还在实验室中杀死念珠菌 [ 42 ].
动物和细胞研究(缺乏证据)
没有临床证据支持将植物乳杆菌用于本节所列的任何病症。以下是对现有动物和细胞研究的总结,应指导进一步的研究工作。但是,不应将下列研究解释为支持任何健康益处。
12) 排毒
植物乳杆菌在实验室中减轻镉 (Cd) 在人类肠道细胞和小鼠中诱导的细胞毒性[ 43、44 ].
植物乳杆菌通过减少肠道铝吸收和组织积累,改善肝损伤、肾脏和大脑氧化应激,从而防止小鼠受到铝中毒[ 45 ].
用植物乳杆菌处理可通过增加铜排泄和减少铜在组织中的积累来减轻铜毒性。植物乳杆菌还逆转了铜暴露引起的氧化应激,恢复了 ALT 和AST血液水平并改善了小鼠的空间记忆 [ 46 ].
13) 血管性痴呆
植物乳杆菌发酵豆浆提取物可作为降血压剂和神经保护剂,改善血管性痴呆大鼠的学习和记忆 [ 47 ].
14) 焦虑
长期摄入植物乳杆菌可增加运动活性、多巴胺和血清素水平,同时减少小鼠的焦虑样行为。它还减少了抑郁样行为和炎性细胞因子水平,并增加了遭受早期生活压力的小鼠血清中的抗炎细胞因子水平[ 48、49 ].
15) 心血管健康
血压
植物乳杆菌发酵豆浆提取物可作为大鼠的降血压剂 [ 47 ].
同样在大鼠中,用植物乳杆菌发酵的蓝莓降低了血压并改善了可能指示心血管疾病风险的标志物 [ 50 ].
动脉粥样硬化
来自植物乳杆菌的脂磷壁酸 (LTA)可抑制小鼠促炎细胞因子的产生,并抑制动脉粥样硬化斑块炎症 [ 51 ].
16) 炎症
植物乳杆菌显着降低小鼠和大鼠促炎细胞因子的产生 [ 52 , 53 ]. 它还减轻了氧化应激和肾上腺素水平 [ 52 ].
17) 肝脏健康
用植物乳杆菌治疗大鼠阻塞性黄疸可恢复活跃的肝屏障功能 [ 54 ].
植物乳杆菌可保护小鼠免受氧化应激和肝脏炎症损伤 [ 55 ].
植物乳杆菌减轻了高脂血症小鼠的肝损伤[ 56 ].
补充植物乳杆菌 5 周可恢复患有非酒精性脂肪性肝病 (NAFLD) 大鼠的肝功能,并降低肝脏中的脂肪堆积水平。此外,该细菌显着减少了促炎细胞因子 [ 53 ].
18) 肠道健康
植物乳杆菌减少肠上皮细胞的炎症 [ 57 ].
摄入植物乳杆菌可以抵消肠道中不需要的细菌 [ 3 ].
植物乳杆菌增强了洛哌丁胺诱导的便秘小鼠的胃肠道转运[ 58 ].
溃疡
口服植物乳杆菌可通过抗炎和免疫调节活性改善小鼠的溃疡性结肠炎 [ 59 ].
肠易激综合症
植物乳杆菌可减少肠易激综合征患者的胀气问题和疼痛 [ 60 ].
幽门螺杆菌感染
植物乳杆菌可预防小鼠幽门螺杆菌感染引起的胃粘膜炎症和胃微生物群改变[ 61 ].
植物乳杆菌延缓幽门螺杆菌在大鼠胃中的定植,减轻胃炎症并改善胃组织病理学 [ 62 ].
19) 婴儿成长
植物乳杆菌菌株在慢性营养不良期间维持幼鼠的生长 [ 63 ].
20) 身体耐力
植物乳杆菌显着降低小鼠的体重并增加相对肌肉重量、握力和耐力游泳时间 [ 64 ].
21) 女性生育能力
植物乳杆菌改善了小鼠炎症诱导的不孕症 [ 65 ].
植物乳杆菌增强了自然微生物群落,并导致感染大肠杆菌的小鼠的生育能力恢复[ 66 ].
22) 组胺不耐受
植物乳杆菌可以在实验室环境中降解生物胺。在组胺不耐受的人群中可能值得研究 [ 67 ].
癌症研究
植物乳杆菌增强了肠腺癌小鼠的抗肿瘤免疫反应并延缓了肿瘤形成 [ 68 ].
长期服用植物乳杆菌可预防大鼠患乳腺癌 [ 69、70 ].
植物乳杆菌抑制大鼠结肠癌发生的发展 [ 71 ].
纳米尺寸的植物乳杆菌还在小鼠中表现出抗结直肠癌活性 [ 72 ].
植物乳杆菌显着抑制肝癌细胞、胃癌细胞和结直肠腺癌细胞的增殖[ 73 ].
这些影响尚未在人类受试者中进行过研究。
在大鼠身上没有观察到任何类型的不良反应,即使在大量食用后也是如此。然而,与其他益生菌一样,在器官衰竭、免疫功能低下和肠道屏障机制功能失调的患者中使用可能会导致感染 [ 74 ].
为了避免不良事件,请在使用益生菌之前咨询医生。
尽管这种情况可能很少见,但如果出现以下任何可能与非常严重的副作用相关的体征或症状,请立即寻求医疗帮助:
参考文献:
[1] Arasu MV, Al-Dhabi NA, Ilavenil S, Choi KC, Srigopalram S. In vitro importance of probiotic Lactobacillus plantarum related to medical field. Saudi J Biol Sci. 2016 Jan;23(1):S6-S10.
[2] Li P, Zhou Q, Gu Q. Complete genome sequence of Lactobacillus plantarum LZ227, a potential probiotic strain producing B-group vitamins. J Biotechnol. 2016 Sep 20;234:66-70.
[3] Hoppe M, Önning G, Berggren A, Hulthén L. Probiotic strain Lactobacillus plantarum 299v increases iron absorption from an iron-supplemented fruit drink: a double-isotope cross-over single-blind study in women of reproductive age. Br J Nutr. 2015 Oct 28;114(8):1195-202.
[4] Bering S, Suchdev S, Sjøltov L, Berggren A, Tetens I, Bukhave K. A lactic acid-fermented oat gruel increases non-haem iron absorption from a phytate-rich meal in healthy women of childbearing age. Br J Nutr. 2006 Jul;96(1):80-5.
[5] ergillos-Meca T, Cabrera-Vique C, Artacho R, Moreno-Montoro M, Navarro-Alarcón M, Olalla M, Giménez R, Seiquer I, Ruiz-López MD. Does Lactobacillus plantarum or ultrafiltration process improve Ca, Mg, Zn and P bioavailability from fermented goats’ milk? Food Chem. 2015 Nov 15;187:314-21.
[6] Olek A, Woynarowski M, Ahrén IL, Kierkuś J, Socha P, Larsson N, Önning G. Efficacy and Safety of Lactobacillus plantarum DSM 9843 (LP299V) in the Prevention of Antibiotic-Associated Gastrointestinal Symptoms in Children-Randomized, Double-Blind, Placebo-Controlled Study. J Pediatr. 2017 Jul;186:82-86.
[7] Hilton E, Kolakowski P, Singer C, Smith M. Efficacy of Lactobacillus GG as a Diarrheal Preventive in Travelers. J Travel Med. 1997 Mar 1;4(1):41-43.
[8] Lee DE, Huh CS, Ra J, Choi ID, Jeong JW, Kim SH, Ryu JH, Seo YK, Koh JS, Lee JH, Sim JH, Ahn YT. Clinical Evidence of Effects of Lactobacillus plantarum HY7714 on Skin Aging: A Randomized, Double Blind, Placebo-Controlled Study. J Microbiol Biotechnol. 2015 Dec 28;25(12):2160-8.
[9] Jeong JH, Lee CY, Chung DK. Probiotic Lactic Acid Bacteria and Skin Health. Crit Rev Food Sci Nutr. 2016 Oct 25;56(14):2331-7.
[10] Kim H, Kim HR, Jeong BJ, Lee SS, Kim TR, Jeong JH, Lee M, Lee S, Lee JS, Chung DK. Effects of oral intake of kimchi-derived Lactobacillus plantarum K8 lysates on skin moisturizing. J Microbiol Biotechnol. 2015 Jan;25(1):74-80.
[11] Hong YF, Lee Hy, Jung BJ, Jang S, Chung DK, Kim H. Lipoteichoic acid isolated from Lactobacillus plantarum down-regulates UV-induced MMP-1 expression and up-regulates type I procollagen through the inhibition of reactive oxygen species generation. Mol Immunol. 2015 Oct;67(2 Pt B):248-55.
[12] Ra J, Lee DE, Kim SH, Jeong JW, Ku HK, Kim TY, Choi ID, Jeung W, Sim JH, Ahn YT. Effect of oral administration of Lactobacillus plantarum HY7714 on epidermal hydration in ultraviolet B-irradiated hairless mice. J Microbiol Biotechnol. 2014 Dec 28;24(12):1736-43.
[13] Kim HM, Lee DE, Park SD, Kim YT, Kim YJ, Jeong JW, Jang SS, Ahn YT, Sim JH, Huh CS, Chung DK, Lee JH. Oral administration of Lactobacillus plantarum HY7714 protects hairless mouse against ultraviolet B-induced photoaging. J Microbiol Biotechnol. 2014 Nov 28;24(11):1583-91.
[14] Harima-Mizusawa N, Kamachi K, Kano M, Nozaki D, Uetake T, Yokomizo Y, Nagino T, Tanaka A, Miyazaki K, Nakamura S. Beneficial effects of citrus juice fermented with Lactobacillus plantarum YIT 0132 on atopic dermatitis: results of daily intake by adult patients in two open trials. Biosci Microbiota Food Health. 2016;35(1):29-39.
[15] Won TJ, Kim B, Lee Y, Bang JS, Oh ES, Yoo JS, Hyung KE, Yoon J, Hwang S, Park ES, Park SY, Hwang KW. Therapeutic potential of Lactobacillus plantarum CJLP133 for house-dust mite-induced dermatitis in NC/Nga mice. Cell Immunol. 2012 May-Jun;277(1-2):49-57.
[18] Fuentes MC, Lajo T, Carrión JM, Cuñé J. Cholesterol-lowering efficacy of Lactobacillus plantarum CECT 7527, 7528 and 7529 in hypercholesterolaemic adults. Br J Nutr. 2013 May 28;109(10):1866-72.
[19] Bejar W, Hamden K, Ben Salah R, Chouayekh H. Lactobacillus plantarum TN627 significantly reduces complications of alloxan-induced diabetes in rats. Anaerobe. 2013 Dec;24:4-11.
[20] Yoo JY, Kim SS. Probiotics and Prebiotics: Present Status and Future Perspectives on Metabolic Disorders. Nutrients. 2016 Mar 18;8(3):173.
[21] Jeun J, Kim S, Cho SY, Jun HJ, Park HJ, Seo JG, Chung MJ, Lee SJ. Hypocholesterolemic effects of Lactobacillus plantarum KCTC3928 by increased bile acid excretion in C57BL/6 mice. Nutrition. 2010 Mar;26(3):321-30.
[22] Sáez-Lara MJ, Robles-Sanchez C, Ruiz-Ojeda FJ, Plaza-Diaz J, Gil A. Effects of Probiotics and Synbiotics on Obesity, Insulin Resistance Syndrome, Type 2 Diabetes and Non-Alcoholic Fatty Liver Disease: A Review of Human Clinical Trials. Int J Mol Sci. 2016 Jun 13;17(6):928.
[23] Pothuraju R, Sharma RK, Kavadi PK, Chagalamarri J, Jangra S, Bhakri G, De S. Anti-obesity effect of milk fermented by Lactobacillus plantarum NCDC 625 alone and in combination with herbs on high fat diet fed C57BL/6J mice. Benef Microbes. 2016 Jun;7(3):375-85.
[24] Park JE, Oh SH, Cha YS. Lactobacillus plantarum LG42 isolated from gajami sik-hae decreases body and fat pad weights in diet-induced obese mice. J Appl Microbiol. 2014 Jan;116(1):145-56.
[25] Wu CC, Weng WL, Lai WL, Tsai HP, Liu WH, Lee MH, Tsai YC. Effect of Lactobacillus plantarum Strain K21 on High-Fat Diet-Fed Obese Mice. Evid Based Complement Alternat Med. 2015;2015:391767.
[26] Zhang J, Xiao X, Dong Y, Xu T, Wu F. Dietary supplementation with Lactobacillus plantarum dy-1 fermented barley suppresses body weight gain in high-fat diet-induced obese rats. J Sci Food Agric. 2016 Dec;96(15):4907-4917.
[27] Ben Salah R, Trabelsi I, Hamden K, Chouayekh H, Bejar S. Lactobacillus plantarum TN8 exhibits protective effects on lipid, hepatic and renal profiles in obese rat. Anaerobe. 2013 Oct;23:55-61.
[28] Li X, Wang N, Yin B, Fang D, Jiang T, Fang S, Zhao J, Zhang H, Wang G, Chen W. Effects of Lactobacillus plantarum CCFM0236 on hyperglycaemia and insulin resistance in high-fat and streptozotocin-induced type 2 diabetic mice. J Appl Microbiol. 2016 Dec;121(6):1727-1736.
[29] Sakai T, Taki T, Nakamoto A, Shuto E, Tsutsumi R, Toshimitsu T, Makino S, Ikegami S. Lactobacillus plantarum OLL2712 regulates glucose metabolism in C57BL/6 mice fed a high-fat diet. J Nutr Sci Vitaminol (Tokyo). 2013;59(2):144-7.
[30] Li C, Ding Q, Nie SP, Zhang YS, Xiong T, Xie MY. Carrot juice fermented with Lactobacillus plantarum NCU116 ameliorates type 2 diabetes in rats. J Agric Food Chem. 2014 Dec 10;62(49):11884-91.
[31] Peral MC, Rachid MM, Gobbato NM, Huaman Martinez MA, Valdez JC. Interleukin-8 production by polymorphonuclear leukocytes from patients with chronic infected leg ulcers treated with Lactobacillus plantarum. Clin Microbiol Infect. 2010 Mar;16(3):281-6.
[32] Iwasaki K, Maeda K, Hidaka K, Nemoto K, Hirose Y, Deguchi S. Daily Intake of Heat-killed Lactobacillus plantarum L-137 Decreases the Probing Depth in Patients Undergoing Supportive Periodontal Therapy. Oral Health Prev Dent. 2016;14(3):207-14.
[33] Nishimura M, Ohkawara T, Tetsuka K, Kawasaki Y, Nakagawa R, Satoh H, Sato Y, Nishihira J. Effects of yogurt containing Lactobacillus plantarum HOKKAIDO on immune function and stress markers. J Tradit Complement Med. 2015 Aug 21;6(3):275-80.
[34] Xie J, Yu Q, Nie S, Fan S, Xiong T, Xie M. Effects of Lactobacillus plantarum NCU116 on Intestine Mucosal Immunity in Immunosuppressed Mice. J Agric Food Chem. 2015 Dec 30;63(51):10914-20.
[35] Matsusaki T, Takeda S, Takeshita M, Arima Y, Tsend-Ayush C, Oyunsuren T, Sugita C, Yoshida H, Watanabe W, Kurokawa M. Augmentation of T helper type 1 immune response through intestinal immunity in murine cutaneous herpes simplex virus type 1 infection by probiotic Lactobacillus plantarum strain 06CC2. Int Immunopharmacol. 2016 Oct;39:320-327.
[36] Park MK, Ngo V, Kwon YM, Lee YT, Yoo S, Cho YH, Hong SM, Hwang HS, Ko EJ, Jung YJ, Moon DW, Jeong EJ, Kim MC, Lee YN, Jang JH, Oh JS, Kim CH, Kang SM. Lactobacillus plantarum DK119 as a probiotic confers protection against influenza virus by modulating innate immunity. PLoS One. 2013 Oct 4;8(10):e75368.
[37] Harima-Mizusawa N, Iino T, Onodera-Masuoka N, Kato-Nagaoka N, Kiyoshima-Shibata J, Gomi A, Shibahara-Sone H, Kano M, Shida K, Sakai M, Miyazaki K, Ishikawa F. Beneficial Effects of Citrus Juice Fermented with Lactobacillus plantarum YIT 0132 on Japanese Cedar Pollinosis. Biosci Microbiota Food Health. 2014;33(4):147-55.
[38] Frias J, Song YS, Martínez-Villaluenga C, González de Mejia E, Vidal-Valverde C. Immunoreactivity and amino acid content of fermented soybean products. J Agric Food Chem. 2008 Jan 9;56(1):99-105.
[39] Liu YW, Liao TW, Chen YH, Chiang YC, Tsai YC. Oral administration of heat-inactivated Lactobacillus plantarum K37 modulated airway hyperresponsiveness in ovalbumin-sensitized BALB/c mice. PLoS One. 2014 Jun 17;9(6):e100105.
[40] De Seta F, Parazzini F, De Leo R, Banco R, Maso GP, De Santo D, Sartore A, Stabile G, Inglese S, Tonon M, Restaino S. Lactobacillus plantarum P17630 for preventing Candida vaginitis recurrence: a retrospective comparative study. Eur J Obstet Gynecol Reprod Biol. 2014 Nov;182:136-9.
[41] Palacios S, Espadaler J, Fernández-Moya JM, Prieto C, Salas N. Is it possible to prevent recurrent vulvovaginitis? The role of Lactobacillus plantarum I1001 (CECT7504). Eur J Clin Microbiol Infect Dis. 2016 Oct;35(10):1701-8.
[42] Sharma A, Srivastava S. Anti-Candida activity of spent culture filtrate of Lactobacillus plantarum strain LR/14. J Mycol Med. 2014 Jun;24(2):e25-34.
[43] Zhai Q, Tian F, Zhao J, Zhang H, Narbad A, Chen W. Oral Administration of Probiotics Inhibits Absorption of the Heavy Metal Cadmium by Protecting the Intestinal Barrier. Appl Environ Microbiol. 2016 Jun 30;82(14):4429-40.
[44] Zhai Q, Wang G, Zhao J, Liu X, Narbad A, Chen YQ, Zhang H, Tian F, Chen W. Protective effects of Lactobacillus plantarum CCFM8610 against chronic cadmium toxicity in mice indicate routes of protection besides intestinal sequestration. Appl Environ Microbiol. 2014 Jul;80(13):4063-71.
[45] Yu L, Zhai Q, Liu X, Wang G, Zhang Q, Zhao J, Narbad A, Zhang H, Tian F, Chen W. Lactobacillus plantarum CCFM639 alleviates aluminium toxicity. Appl Microbiol Biotechnol. 2016 Feb;100(4):1891-1900.
[46] Tian F, Xiao Y, Li X, Zhai Q, Wang G, Zhang Q, Zhang H, Chen W. Protective Effects of Lactobacillus plantarum CCFM8246 against Copper Toxicity in Mice. PLoS One. 2015 Nov 25;10(11):e0143318.
[47] Liu TH, Chiou J, Tsai TY. Effects of Lactobacillus plantarum TWK10-Fermented Soymilk on Deoxycorticosterone Acetate-Salt-Induced Hypertension and Associated Dementia in Rats. Nutrients. 2016 May 2;8(5):260.
[48] Liu YW, Liu WH, Wu CC, Juan YC, Wu YC, Tsai HP, Wang S, Tsai YC. Psychotropic effects of Lactobacillus plantarum PS128 in early life-stressed and naïve adult mice. Brain Res. 2016 Jan 15;1631:1-12.
[49] Liu WH, Chuang HL, Huang YT, Wu CC, Chou GT, Wang S, Tsai YC. Alteration of behavior and monoamine levels attributable to Lactobacillus plantarum PS128 in germ-free mice. Behav Brain Res. 2016 Feb 1;298(Pt B):202-9.
[50] Ahrén IL, Xu J, Önning G, Olsson C, Ahrné S, Molin G. Antihypertensive activity of blueberries fermented by Lactobacillus plantarum DSM 15313 and effects on the gut microbiota in healthy rats. Clin Nutr. 2015 Aug;34(4):719-26.
[51] Kim JY, Kim H, Jung BJ, Kim NR, Park JE, Chung DK. Lipoteichoic acid isolated from Lactobacillus plantarum suppresses LPS-mediated atherosclerotic plaque inflammation. Mol Cells. 2013 Feb;35(2):115-24.
[52] Toshimitsu T, Mochizuki J, Ikegami S, Itou H. Identification of a Lactobacillus plantarum strain that ameliorates chronic inflammation and metabolic disorders in obese and type 2 diabetic mice. J Dairy Sci. 2016 Feb;99(2):933-946.
[53] Li C, Nie SP, Zhu KX, Ding Q, Li C, Xiong T, Xie MY. Lactobacillus plantarum NCU116 improves liver function, oxidative stress and lipid metabolism in rats with high fat diet induced non-alcoholic fatty liver disease. Food Funct. 2014 Dec;5(12):3216-23.
[54] Zhang M, Wang XQ, Zhou YK, Ma YL, Shen TY, Chen HQ, Chu ZX, Qin HL. Effects of oral Lactobacillus plantarum on hepatocyte tight junction structure and function in rats with obstructive jaundice. Mol Biol Rep. 2010 Jul;37(6):2989-99.
[55] Peng X, Jiang Y. Protective effects of Lactobacillus plantarum NDC 75017 against lipopolysaccharide-induced liver injury in mice. Inflammation. 2014 Oct;37(5):1599-607.
[56] Wang LX, Liu K, Gao DW, Hao JK. Protective effects of two Lactobacillus plantarum strains in hyperlipidemic mice. World J Gastroenterol. 2013 May 28;19(20):3150-6.
[57] Murofushi Y, Villena J, Morie K, Kanmani P, Tohno M, Shimazu T, Aso H, Suda Y, Hashiguchi K, Saito T, Kitazawa H. The toll-like receptor family protein RP105/MD1 complex is involved in the immunoregulatory effect of exopolysaccharides from Lactobacillus plantarum N14. Mol Immunol. 2015 Mar;64(1):63-75.
[58] Li C, Nie SP, Zhu KX, Xiong T, Li C, Gong J, Xie MY. Effect of Lactobacillus plantarum NCU116 on loperamide-induced constipation in mice. Int J Food Sci Nutr. 2015;66(5):533-8.
[59] Liu YW, Su YW, Ong WK, Cheng TH, Tsai YC. Oral administration of Lactobacillus plantarum K68 ameliorates DSS-induced ulcerative colitis in BALB
/c mice via the anti-inflammatory and immunomodulatory activities. Int Immunopharmacol. 2011 Dec;11(12):2159-66.
[60] Ducrotté P, Sawant P, Jayanthi V. Clinical trial: Lactobacillus plantarum 299v (DSM 9843) improves symptoms of irritable bowel syndrome. World J Gastroenterol. 2012 Aug 14;18(30):4012-8.
[61] Pan M, Wan C, Xie Q, Huang R, Tao X, Shah NP, Wei H. Changes in gastric microbiota induced by Helicobacter pylori infection and preventive effects of Lactobacillus plantarum ZDY 2013 against such infection. J Dairy Sci. 2016 Feb;99(2):970-981.
[62] Thiraworawong T, Spinler JK, Werawatganon D, Klaikeaw N, Venable SF, Versalovic J, Tumwasorn S. Anti-inflammatory properties of gastric-derived Lactobacillus plantarum XB7 in the context of Helicobacter pylori infection. Helicobacter. 2014 Apr;19(2):144-55.
[63] ME, Balmand S, Hudcovic T, Heddi A, Rieusset J, Kozakova H, Vidal H, Leulier F. Lactobacillus plantarum strain maintains growth of infant mice during chronic undernutrition. Science. 2016 Feb 19;351(6275):854-7.
[64] Chen YM, Wei L, Chiu YS, Hsu YJ, Tsai TY, Wang MF, Huang CC. Lactobacillus plantarum TWK10 Supplementation Improves Exercise Performance and Increases Muscle Mass in Mice. Nutrients. 2016 Apr 7;8(4):205.
[65] Bhandari P, Rishi P, Prabha V. Positive effect of probiotic Lactobacillus plantarum in reversing LPS-induced infertility in a mouse model. J Med Microbiol. 2016 May;65(5):345-350.
[66] Bhandari P, Prabha V. Evaluation of profertility effect of probiotic Lactobacillus plantarum 2621 in a murine model. Indian J Med Res. 2015 Jul;142(1):79-84.
[67] Capozzi V, Russo P, Ladero V, Fernández M, Fiocco D, Alvarez MA, Grieco F, Spano G. Biogenic Amines Degradation by Lactobacillus plantarum: Toward a Potential Application in Wine. Front Microbiol. 2012 Apr 2;3:122.
[68] Hu J, Wang C, Ye L, Yang W, Huang H, Meng F, Shi S, Ding Z. Anti-tumour immune effect of oral administration of Lactobacillus plantarum to CT26 tumour-bearing mice. J Biosci. 2015 Jun;40(2):269-79.
[69] Kassayová M, Bobrov N, Strojný L, Kisková T, Mikeš J, Demečková V, Orendáš P, Bojková B, Péč M, Kubatka P, Bomba A. Preventive effects of probiotic bacteria Lactobacillus plantarum and dietary fiber in chemically-induced mammary carcinogenesis. Anticancer Res. 2014 Sep;34(9):4969-75.
[70] Kassayová M, Bobrov N, Strojný L, Orendáš P, Demečková V, Jendželovský R, Kubatka P, Kisková T, Kružliak P, Adamkov M, Bomba A, Fedoročko P. Anticancer and Immunomodulatory Effects of Lactobacillus plantarum LS/07, Inulin and Melatonin in NMU-induced Rat Model of Breast Cancer. Anticancer Res. 2016 Jun;36(6):2719-28.
[71] Kumar RS, Kanmani P, Yuvaraj N, Paari KA, Pattukumar V, Thirunavukkarasu C, Arul V. Lactobacillus plantarum AS1 isolated from south Indian fermented food Kallappam suppress 1,2-dimethyl hydrazine (DMH)-induced colorectal cancer in male Wistar rats. Appl Biochem Biotechnol. 2012 Feb;166(3):620-31.
[72] Lee HA, Kim H, Lee KW, Park KY. Dead Nano-Sized Lactobacillus plantarum Inhibits Azoxymethane/Dextran Sulfate Sodium-Induced Colon Cancer in Balb/c Mice. J Med Food. 2015 Dec;18(12):1400-5.
[73] Wang K, Li W, Rui X, Chen X, Jiang M, Dong M. Characterization of a novel exopolysaccharide with antitumor activity from Lactobacillus plantarum 70810. Int J Biol Macromol. 2014 Feb;63:133-9.
[74] Biljana Novkovic, 11+ Health Benefits of Lactobacillus plantarum (L. plantarum). September 20, 2021. selfhacked
谷禾健康
阴道微生物组是一个复杂而动态的微生态系统,在女性月经周期和女性的一生中不断发生波动。
在过去几年中,对阴道微生物群关注随着测序技术的发展和应用逐渐广泛和突出,有关以往传统正常和异常阴道微生物组的知识也发生了变化。培养技术可能不再适用于确定正常或异常的阴道微生物群。以非培养为基础的分析生物技术揭示了一个动态且主要由乳酸杆菌主导的复杂动态系统。
该生态系统受基因、种族背景、发育以及环境和行为因素的影响。每个个体都有几种乳酸杆菌在健康的阴道中占主导地位。它们与抗菌物质、细胞因子、防御素和其他物质一起支持防御系统,以对抗菌群失调、感染和早产以及不孕不育等问题。
在这里,我们主要讨论和介绍女性生命周期不同阶段阴道区域微生物群落的变化,哪些因素会影响阴道菌群,及阴道菌群对性健康和病理状况的影响。
育龄女性产生约 1 至 4 mL 的阴道液,每 mL 含有 108至 109 个细菌细胞。阴道微生物从女性出生的最初几个小时开始就开始定居,并伴随她的一生一直存在到死亡。
这些细菌群落在个体之间和随着时间的推移可能会有产生差异。
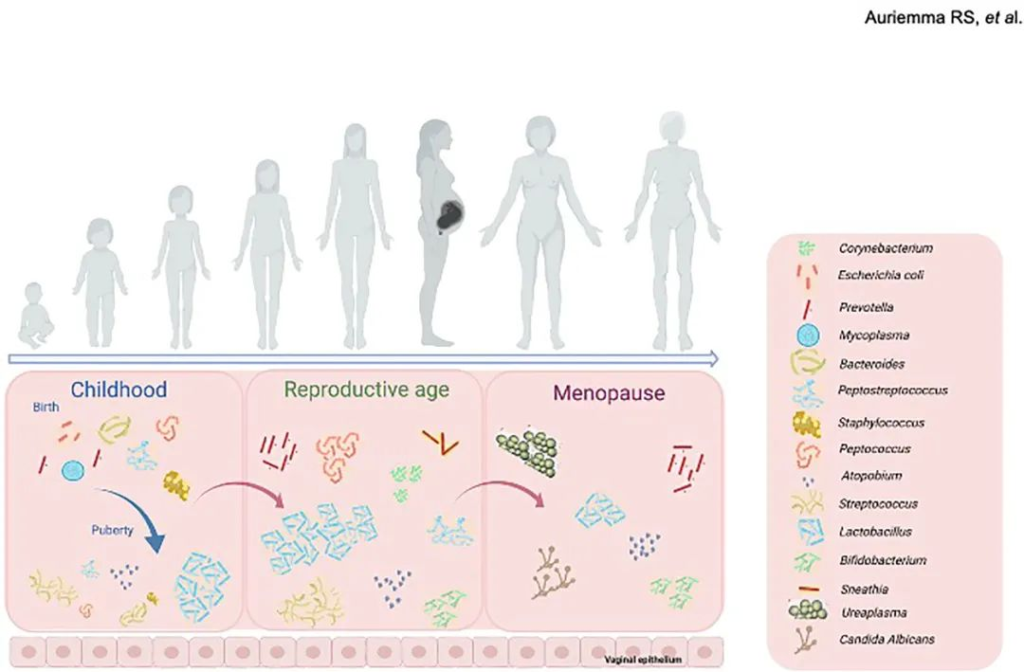
• 阴道微生物群被定义为在阴道内定殖的共生和病原微生物群;
• 阴道微生物群从女性出生的最初几个小时开始就开始定居,伴宿女性一生;
• 随着年龄的变化,从青春期前的厌氧微生物过渡到生殖年龄时富含乳酸杆菌的阴道。
• 在大多数健康育龄妇女中,阴道微生物群以乳酸杆菌为主。
+
阴道菌群中 70%是乳杆菌
阴道拥有自己的原生微生物群落,这是抵御病原体的主要防线。
最主要的阴道种群是乳酸菌,占细菌群落的近 70%。这些包括下列菌群等:
此外,其他细菌种类如下列菌群等也存在于健康个体的阴道中:
研究人员通过16s技术等在研究中将阴道微生物群分为编号为 I-V 的进化枝。其中,Clades I、II、III、V以乳酸杆菌为主,Clade IV无优势群,是微生物的混合体。
注:CST I、II、III 和 V 以L. crispatus 、L. gasseri、L. iners、L. jensenii 为主,而 CST IV 是指以专性厌氧菌为特征的微生物群落的高度多样性。
每个女性体内的原生微生物群数量因种族、地理、生活方式、卫生和冲洗方法而异。
注:已经发现,在疾病进展期间,乳杆菌种群发生了从L. crispatus 到 L. iners 的转变。这种转变的原因尚不清楚。有人提出,与L. iners 产生的L-乳酸相比, L . crispatus 产生的d-乳酸可能提供更好的保护,防止病原体定植。
因此,许多其他细菌在健康的阴道菌群中的浓度较低,例如消化链球菌(Peptostreptococcus)、拟杆菌(Bacteroides)、棒状杆菌(Corynebacterium)、链球菌(Streptococcus)和消化球菌(Peptococcus)。
阴道微生物群的组成在女性的一生中不断演变。在女性生命的这些不同阶段,阴道菌群会发生各种生理和荷尔蒙变化(下图)。
健康的阴道微生物群通常由乳杆菌属控制,乳杆菌属对阴道失调或感染具有保护作用。尽管这种保护作用的确切机制尚不清楚,但新出现的研究表明,阴道菌群产生多种分子来维持阴道内稳态并防止感染。
例如,健康的阴道环境的特征是阴道乳杆菌产生乳酸,这在消除入侵病原体方面发挥着重要作用。此外,Lactobacilius还产生许多化合物,如细菌素和H2O2,以选择性地抑制入侵病原体并保持阴道内稳态。
阴道微生物失调的特点是乳酸杆菌属的丰度较低,而CST IV微生物群的水平较高,如厌氧链球菌、阴道加德纳菌、阴道阿托波菌、莫比伦氏菌属、斯内氏菌属(Sneathia)、普雷沃氏菌属、支原体,这会对阴道和生殖健康产生不利影响,并导致对各种阴道感染(如细菌性阴道病和念珠菌病)的易感性,甚至可能导致不良妊娠结局,如早产、先兆子痫、妊娠高血压等。
世界范围内进行的大量研究已确定乳酸杆菌是育龄健康女性阴道微生物群的主要成分。阴道微生物组因种族和地区而异。目前尚不清楚种族因素是否单独或结合行为和环境变量可以解释这些差异。
▸ 不同地区的阴道菌群存在差异:
来自不同国家的女性在其阴道微生物群中有一定程度的差异,不过差异不大。
在中国育龄健康女性的生殖微生物群落中以 L. gasseri, L. crispatus 和 L. iners为主要。
欧洲女性有一个独特的阴道微生物群,包括L. crispatus, L. jensenii, L. gasseri, L. iners.
而非洲研究表明,植物乳杆菌是阴道微生物群中的主要物种,其次是:
L. gasseri
Lactobacillus rhamnosus
L. crispatus
Lactobacillus plantarum
上述发现表明,来自世界各地的健康女性的阴道微生物组存在物种水平的差异。这些差异也可以用所研究社区内的种族差异来解释。
▸ 同一地区不同种族的阴道菌群存在差异:
育龄妇女的阴道细菌群落在不同地区的妇女之间可能存在差异,但在生活在同一地理区域的不同种族的妇女之间也可能存在差异。
2011 年的一项研究对无症状北美女性的阴道微生物群进行表征,表明亚裔和美国白人女性的阴道菌群以乳酸杆菌为主,这与西班牙裔和非裔美国女性不同,后者只有 60% 的阴道菌群以乳酸菌为主。
此外,与非洲女性相比,白种人和亚洲女性的L. crispatus含量较高,而 L. iners 含量较低。
在另一项使用 16S rRNA 基因测序的研究中,表明欧洲血统女性的阴道微生物群以乳酸杆菌为主,而非裔美国女性则相反,后者呈现出混合的阴道群落,其中包括人型支原体、气球菌、惰性乳杆菌和许多严格的厌氧菌,包括:革兰氏-阳性厌氧球菌、细菌性阴道炎相关细菌、Sneathia、Prevotella amnii、Megasphaera、Atopobium、Gardnerella vaginalis。
阴道 pH 值也因种族而异。非裔美国人和西班牙裔女性的阴道 pH 值(分别为 4.7 和 5.0)高于标准值(<4.5)。
阴道及其微生物群形成了一个生态系统,随着时间的推移,从婴儿期到儿童期再到青春期和成年期,影响这些变化的主要力量是雌激素水平的波动和性活动的出现,卫生实践和药物,包括口服避孕药和抗菌剂,也会影响阴道中存在的各种菌群之间的复杂相互作用。
下面我们来看每个时期阴道菌群的特点及相关影响因素:
出生后或出生后不久,婴儿的阴道微生物群从母亲身上吸收细菌时开始繁殖。
许多研究表明,怀孕期间的母体微生物组对妊娠结局和分娩方式(阴道或剖宫产)有很大影响,这会显著影响婴儿的微生物组。
出生后,微生物群需要数年的时间才能充分发展和多样化,这是一个动态过程,并受到多种外部因素的严重影响。例如,早期喂养方式或接触抗生素会影响婴儿的微生物群。
▸分娩方式不同影响菌群定植
在阴道分娩时,新生儿暴露于母亲的阴道和粪便微生物群,这些微生物群中含有大量乳酸杆菌、普雷沃氏菌、斯奈氏菌和双歧杆菌;
然而,剖腹产婴儿从母亲的皮肤或周围医院环境中接受了不同的微生物接种物,这些接种物通常主要由棒状杆菌、葡萄球菌和丙酸杆菌居住,有时还含有传染性微生物制剂,如梭状芽孢杆菌、肠球菌和克雷伯菌。
胎盘和羊水的微生物组在成功分娩和新生儿微生物组发育中起着重要作用。最初的胎粪显示胎盘和羊水中的细菌占优势。
▸阴道菌群与激素调节相关
在怀孕和产后阶段,身体经历了许多变化,通常由雌激素等各种激素调节。
阴道雌激素水平高,有助于维持较高水平的阴道糖原,乳酸杆菌通常将其用作维持阴道酸性环境的食物。
此外,新生儿还通过母乳接触母体雌激素,这有助于维持健康的新生儿菌群。相反,阴道雌激素水平随着年龄的增长而降低,这反过来导致阴道乳杆菌和中性阴道pH值的降低。
▸乳酸杆菌是大多数女孩的主要菌群
在完成如厕训练之前,婴儿和学步儿童通常会遇到革兰氏阴性肠道细菌和肠球菌,但之后会较少遇到。
在儿童中,乳酸杆菌在 2 岁以下的女孩中比在青春期前年龄较大的女孩中更常见,并且可能在限制其他菌群过度生长方面发挥保护作用。
在儿童早期,阴道的 pH 值是中性或微碱性。
几个内在和外在因素可以显著影响女性一生中的阴道微生物群。
青少年时期的厌氧菌流行率更高。从青春期开始,有氧定植随着年龄、性活动的开始和胎次而增加。
在月经初潮前,低雌激素水平通常与由需氧、厌氧和肠道微生物组成的多种阴道微生物群落有关,微生物多样性增加的梭杆菌属、普雷沃氏菌属和Ezakiella属(普雷沃氏菌为主);在生命的这一阶段,阴道pH值呈微碱性(pH 7.5–8)或中性(pH 7.0)(下图)。
给父母的重要提示:
青春期前的儿童可能会出现阴道感染,这通常是由于高香味的沐浴产品造成的。鼓励孩子在上厕所时从前到后擦拭,洗澡时不要使用香皂等产品洗,尤其是直接进入阴道区域。
青少年时期对年轻人来说是一个充满挑战的时期,他们的身体在进入青春期时会发生变化。此时阴道微生物组也发生了很大变化。
从青春期初期到生殖阶段,阴道微生物组成不断变化:
青春期早期通常由Finegoldia、Anaerococcus、Prevotella、Dialister、Peptoniphilus和乳杆菌属组成,而后期主要由乳杆菌属和较低丰度的Sneathia、Prevotela、嗜血杆菌属、Atopobium、Gardnerella组成。
不断增加的雌激素水平导致高水平的糖原存在于阴道内膜中。乳酸杆菌家族的细菌发酵糖原,从而产生乳酸。这有助于保护阴道。
在这个阶段,乳酸杆菌的丰度降低会导致阴道生物失调和细菌性阴道病。
在生殖阶段的初始阶段,一系列因素有助于阴道微生物组的动态。青春期初期女性性腺激素水平的增加导致阴道微生物多样性的增加,这是通过诱导阴道壁中糖原沉积的增加,较低的阴道性腺水平和激素刺激的糖原支持乳酸杆菌的生长。
月经期的阴道微生物组成似乎与青春期早期观察到的相似:两个属(消化链球菌Peptostreptococcus和链球菌属Streptococcus)通常占主导地位,而不是乳杆菌属,这可以通过测定期间阴道pH值的变化来解释。
新的研究表明,在月经期间,月经液与阴道壁的相互作用中和了酸性阴道微环境,导致pH值升高(7.2–7.4)。乳杆菌属的下降导致许多其他厌氧菌上升。
尽管在月经周期中阴道菌群发生这些实质性变化的确切原因尚不清楚,但过去的结果表明,这一阶段的一些生理事件可能会影响阴道微环境。
例如,黄体期(月经前)被认为以子宫壁增厚为特征,特别是子宫内膜和阴道上皮增厚,导致阴道区域糖原沉积增加,而缺乏受精导致性激素水平突然下降,以及子宫内膜衬里脱落和中性pH,为许多微生物的增殖创造了理想的栖息地。
在随后的卵泡期,月经流量的减少再次降低了阴道pH值,增强了糖原降解和乳酸生产(通过乳酸杆菌),从而抑制了其他厌氧微生物的生长。
月经与阴道微生物群落的动态变化有关。一些女性有强健的阴道微生物群落,在月经期间群落状态之间有简单和可预测的过渡,而其他女性在整个月经期间表现出相对稳定的微生物群落。
在生殖阶段,阴道微生物群落通常以脆乳杆菌为主,而在月经期间观察到阴道微生物群失衡,其特征是脆乳杆菌减少,混合厌氧菌增加,如Gardnerella、Atopobium、Prevotella、Megasphaera sp.、L.iners、链球菌等。因此,CST从CST I转移到CST III或CST IV,有时在整个月经周期中,社区保持稳定和相同。
新的研究表明,阴道微生物环境是由宿主因素(种族、遗传和免疫介质)和微生物生物学在整个生殖周期中的微妙相互作用维持的。虽然在月经期间已描述了改变的阴道细胞因子模式,但其与阴道微生物群和生殖健康的关系尚未完全确定。此外,女性阴道微生物群落的个体间差异也很高。
一旦我们度过了青春期,我们就达到了生育年龄。性活动可能会随之而来,并伴随着怀孕的机会。在怀孕期间,阴道菌群会发生变化,并且在乳酸杆菌中变得更加占优势。人们认为,由于荷尔蒙的变化,乳酸杆菌的这种增加的优势是为了防止感染,这种感染可能在这个脆弱的时期被触发。
怀孕的特点是各种生理变化,这些变化有助于胎儿适应母亲身体的微生物群,反之亦然。这种多样化的状态受到激素和生理变化的调节,这些变化导致免疫调节、行为、粘膜理化、代谢和生殖道变化,从而导致微生物组的结构和功能被调节,这与非怀孕女性的不同。
怀孕是一个“形成期”,由各种相互关联的生理和分子过程控制,以支持胎儿的生长和发育。
在怀孕过程中,身体经历了大量的激素、免疫和微生物变化,以促进母体内稳态并支持胎儿生长。阴道菌群在怀孕过程中也会发生实质性变化,并随着怀孕的进行而变得更加均匀。在妊娠早期,胎盘为支持胎儿而增加的雌激素和孕激素分泌。
实际上触发了总体微生物多样性的丰富;然而,怀孕期间持续升高的激素水平和阴道乳杆菌的拮抗反应导致稳定和健康的阴道菌群。另一方面,不断变化的阴道环境也会导致几种氨基酸(例如,苯丙氨酸、丝氨酸、天冬氨酸、异亮氨酸、色氨酸、苏氨酸、甘氨酸、谷氨酸、亮氨酸)和乳酸水平升高,同时耗尽有机酸(乙酸盐、丙酸盐)、葡萄糖和生物胺(腐胺、酪胺、甲胺)以支持胎儿。
与怀孕期间阴道微生物的变化类似,母体免疫反应也发生了从促炎反应到抗炎细胞因子的实质性变化,以接受胎儿同种异体移植,同时性腺激素水平升高。阴道免疫系统在稳定阴道微生物群和防止上行生殖道感染中起着至关重要的作用。
产后阶段的阴道菌群显著不同,其特征通常是乳酸杆菌的丰度较低,而其他菌群的丰度相对较高,如阴道加德纳菌、无乳链球菌和普雷沃氏菌,导致阴道微生物群落更加多样。
怀孕后雌激素(雌二醇和雌二醇)水平的降低是一个显著影响阴道微环境的因素,包括菌群,这对于非怀孕阶段、恢复自然生理状态和为连续怀孕准备阴道微环境非常重要。未能在1年内恢复自然生理和免疫阴道微环境或随后的妊娠可能会严重影响妊娠结局。
产后抗生素预防是决定产后阴道微生物组成的另一个重要因素。与未经治疗的女性相比,分娩时使用抗生素预防的女性。
与雌激素水平降低同时,与分娩和分娩相关的自然生理变化会刺激炎症反应,以促进子宫收缩、宫颈扩张和胎膜破裂,这可能会显著导致阴道微生物成分的变化。
除雌激素外,其他分泌化合物如透明质酸和Hsp70在重塑阴道微环境中发挥着重要作用,因为它们在分娩期间的水平调节子宫颈并为分娩做好准备,而分娩后它们有助于重塑子宫颈,包括阴道上皮衬里和阴道液体成分。
更年期是女性停止月经周期的时候。在这个阶段,卵巢停止释放雌激素和卵子,这导致生殖周期结束,性腺激素和阴道壁糖原水平降低。随后,雌激素水平的降低也导致阴道微环境的变化,包括乳酸杆菌和乳酸的丰度降低以及中性pH值,这促进了潜在致病微生物的定植,并增加了对泌尿生殖道感染的易感性。
绝经后妇女不仅阴道感染风险增加,而且还会出现其他血管舒缩症状,如潮热、失眠、抑郁等。这些症状是更年期雌激素缺乏的结果,对女性的健康和生活质量产生负面影响。
阴道微生物组极其重要,对绝经后妇女的整体生活质量有重大影响,包括性健康、阴道干燥和外阴阴道萎缩等。不幸的是,新出现的结果表明,大约25-50%的女性在绝经后阶段经历了外阴阴道萎缩,这包括外阴阴道症状,如疼痛的性活动、性交后出血、小便时灼热、疼痛、瘙痒和带有难闻气味的阴道分泌物。
尽管医生已经尝试了不同的治疗方法来改善妇女的绝经后生活,包括使用雌激素的激素替代疗法,这种疗法导致乳酸杆菌数量和糖原水平的增加,从而导致泌尿生殖道感染的减少。
然而,这种疗法未能在不同人群中显示出一致的效果,特别是在有肝病、冠心病、心脏病或乳腺癌病史的患者中。在这些患者中,治疗会导致严重的不良反应,如乳房疼痛或持续阴道出血。
用益生菌鼠李糖乳杆菌GR-1和罗伊氏乳杆菌RC-14 进行的另一种阴道治疗已经证明,这显示出对阴道菌群的有益影响,有助于避免绝经后女性的阴道感染。有限的科学证据和不适当的数据阻止了此类益生菌的使用。
阴道微生物群在保护阴道上皮免受病原微生物污染方面发挥着重要作用。这种保护机制基于三种机制:
1)天然微生物群与病原体的竞争
乳酸杆菌粘附到阴道上皮,通过竞争性占领过程形成抵抗病原微生物的保护层。
2)针对这些不良微生物的抗微生物物质的生产
生产三种不同类型的物质:乳酸、过氧化氢和细菌素。
乳酸:维持阴道的 pH 值在酸性,抑制病原微生物的生长。
过氧化氢:由于其氧化能力而具有抗菌作用。
细菌素:是蛋白质来源的毒素,具有抗菌功能,因此它们可以抑制其他可能致病的微生物的生长。它们的作用是通过在细菌的细胞质膜中产生孔来溶解或破坏细菌的细胞质膜。
3)病原物种的共聚集能力以提高抗微生物能力
病原微生物被来自天然阴道微生物群的细菌包围的机制。
这些形成阴道菌群的微生物与其他微生物群在共生或稳态的情况下生活在一起,也就是说,在阴道中存在的所有物种之间保持平衡。
然而,当由于某种原因阴道中乳酸杆菌的浓度降低时,我们就会进入一种生态失调的情况,其中体内平衡被破坏,因此对粘膜的保护作用降低。
导致乳酸菌减少的原因是多种多样的:
在这种低保护的情况下,病原体会引起感染,包括细菌性阴道病、念珠菌属阴道炎(念珠菌病)、滴虫病或尿路感染等,其他包括早产,不孕不育等也与阴道菌群变化相关,接下来章节我们来详细了解一下相关妇科疾病。
早产(PTL)是指怀孕37周之前的分娩,影响15-30%的妊娠,是导致产前死亡的主要原因。尽管早产的确切病因仍不明确,但胎膜早破、阴道上升感染、羊膜内感染、压力、宫颈功能不全和血管疾病是关键的促成因素;其中,某些细菌引起的宫内感染或上行尿路感染是不良妊娠结局或PTL的影响关键因素。
越来越多的证据表明阴道微生物组与自发性早产风险有关。
阴道微生物群在妊娠健康和结局中起着重要作用,阴道失调的增加(通常以CST IV菌群的丰度较高和乳酸杆菌的丰度较低为特征)导致妊娠并发症和早产风险增加。
足月分娩中,阴道菌群以厚壁菌为主
足月分娩妇女的阴道微生物群落通常是稳定的,在怀孕早期以乳杆菌为主;而经历早产的女性通常阴道菌群以厌氧菌为主。
在大多数足月分娩中,阴道微生物群的特点是厚壁菌门成员占优势,放线菌门、变形菌门、拟杆菌门和细杆菌门成员的丰度较低,而在早产的情况下,厚壁菌的数量减少。
饮食对阴道微生物群有重要影响
在怀孕期间,几种维生素和小分子是必不可少的,每一种都在不同的代谢和生理变化以及胎儿的整体发育中发挥作用。
据指出,女性的饮食中缺乏铁、钙、叶酸、核黄素、钾和维生素D,更容易早产。
维生素D在怀孕期间会对阴道微生物群产生影响,因为它可以加强、保护和维持阴道的上皮衬里;诱导抗微生物肽(LL-37)的表达、增加胰岛素合成;并且通过抑制糖原合成酶激酶最显著地模拟糖原合成。
低丰度的乳酸菌与早产风险最高相关
对至少三种不同CST中的阴道微生物组进行分类以评估早产风险的纵向研究。所有17项研究均在2014年至2021期间发表,包括38-539例妊娠和8-107例早产。与脆乳杆菌占优势的女性相比,具有“低乳杆菌”阴道微生物组的女性早产风险增加(OR 1.69,95%CI 1.15–2.49).
网络荟萃分析支持微生物组可以预测早产,其中低丰度的乳酸菌与最高的风险相关,而L. crispatus优势菌群的早产风险最低。
不孕症对社会、经济以及夫妻双方的心理健康都有严重的不良影响。全球育龄夫妇的患病率为 8–12%.导致不孕的因素复杂且范围广泛。
年龄是导致女性生育力下降的关键影响因素,其他例如卵巢早衰、多囊卵巢综合征和子宫内膜异位症都是公认的导致女性不孕的原因。
越来越多的证据表明,每个女性独有的阴道微生物群在决定生殖健康许多方面起着重要作用。
之前的研究还表明, L. iners 、L . crispatus、L. gasseri可以区分特发性不孕女性与健康女性或阴道病患者。
乳杆菌主导的阴道菌群通常被视为正常的标志。然而,许多研究表明,并非所有类型的乳酸菌都是有益菌,例如,L . crispatus似乎具有有益特性,而L. iners 则没有。
患有特发性不孕的女性似乎更容易出现阴道菌群失调。
研究人员将阴道微生物群分为两类:低乳酸杆菌阴道微生物群 (LL-VMB) 和高乳酸杆菌阴道微生物群 (HL-VMB)。研究人员开始评估女性不育与阴道微生物群之间的统计关联,结果如下:
高乳酸菌阴道微生物群与不孕症之间呈负相关
DOI: 10.1007/s00404-020-05675-3
数据显示,细菌性阴道炎与女性不孕症呈正相关,并且细菌性阴道炎阳性者的影响大于细菌性阴道炎中间值者。
女性不孕症可根据不同的标准分为不同的类型,其中与阴道微生物群的关联可能有所不同。从病因学的角度来看,输卵管性不孕症是与阴道微生物群相关的最常见疾病。
由细菌性阴道炎引起的慢性炎症反应也可能是输卵管粘连的原因,至少是部分原因。
低乳酸杆菌阴道微生物群 或 细菌性阴道病 如何对受精过程产生影响?
通过回顾文献,研究人员确定了三种可能的途径。
第一个:慢性炎症假说
已知一部分女性不孕症可归因于亚临床盆腔炎。此外,细菌性阴道病常伴有 pH 值升高、粘膜细胞损伤和局部炎症反应。虽然阴道炎症不会直接影响卵子,但微生物群仍有可能发挥作用。
最近的一项研究还表明,女性生殖道存在微生物群连续体,包括宫颈管、子宫、输卵管和腹膜液。由于一些盆腔炎是慢性的,没有临床症状,许多女性在诊断出不孕症后才意识到这些问题。
第二个:对性传播感染 (STI) 的易感性
最近的一项荟萃分析提供了高乳酸杆菌阴道微生物群对 HPV 和沙眼衣原体的保护作用的证据。 此外,许多研究表明,细菌性阴道病是感染 STI/HIV 的危险因素。
第三个:指非因果关联
多囊卵巢综合征是女性不孕症的一个非常常见的原因,代表了以高雄激素血症、少排卵/无排卵和卵巢囊肿为特征的内分泌疾病综合症。
已知雌激素或下丘脑-垂体-卵巢轴的变化与 多囊卵巢综合征和阴道微生物群相关,但所涉及的机制仍不清楚。有一些证据表明肠道微生物群与多囊卵巢综合征有关。
需要进一步的研究来系统地探索阴道微生物群、不孕症和其他混杂/中介因素之间的因果关系。
总的来说研究结果表明,健康的阴道微生物群与较低的不孕风险相关。
健康的阴道微生物组以产生各种抗菌化合物的乳杆菌为主。细菌性阴道病(BV)的特征是乳酸杆菌总数的减少或急剧下降,以及相应的厌氧微生物浓度显着增加。
细菌性阴道病是一种在全球育龄妇女中非常普遍的阴道微生物群疾病。全世界23%–29%的女性患有此病。细菌性阴道病已被证实与妇科和产科不良结局有关,例如性传播感染、盆腔炎和早产。
BV的特征是:
乳酸杆菌总数的减少或急剧下降,同时兼性或专性厌氧微生物的浓度相应增加100–1000倍,如Gardnerella、Prevotella、Atopobium、Mobiluncus、双歧杆菌、Sneahia、Leptotrichia,以及梭状芽胞杆菌目中的一些新细菌,称为BV相关细菌(BVAB)1–3.
加德纳菌是从患有 BV 的女性阴道样本中鉴定出的最常见的微生物。
健康或无症状的女性也可能携带G. vaginalis. 这表明阴道中存在G. vaginalis并不一定导致 BV。因此,正确理解G. vaginalis的作用非常重要。
G. vaginalis 含有多种与致病潜力相关的毒力因子,其中唾液酸酶和阴道溶血素是研究最广泛的因子。唾液酸酶A基因与BV和生物膜的存在有关。
G. vaginalis 利用唾液酸酶水解阴道内粘液唾液聚糖中的唾液酸残基,然后分解代谢游离碳水化合物,从而促进阴道粘液屏障的降解。
至于阴道溶解素,它是一种属于胆固醇依赖性溶细胞素家族的成孔毒性化合物,有助于靶细胞(如阴道上皮细胞)的裂解。
细菌性阴道病的发病机制和益生菌对抗BV的作用机制
doi: 10.3390/antibiotics10060719
生物膜是一个附着在非生物或生物表面的结构化微生物群落,镶嵌在其自身分泌的聚合物基质中,包括碳水化合物、蛋白质和核酸。生物膜的形成是一个复杂、动态和相互作用的过程,与活动的浮游微生物和微生物聚集体有关。
多种细菌和真菌微生物,如加德纳菌属和念珠菌属,可以形成生物膜。
阴道上皮上形成的多微生物生物膜在BV的发病机制中起着关键作用。G. vaginalis是主要的定植体,它可以为其他BV相关微生物的附着建立支架,从而能够开发多微生物生物膜。
Atopobium vaginae 是多微生物生物膜的第二个定殖者之一,是一种严格的厌氧微生物,对BV具有很大的预测性。
G. vaginalis 生物膜对两种常见的健康阴道分泌物(即乳酸和H2O2)的耐受性高于浮游细胞。这可以保护阴道毛滴虫和其他BV相关微生物免受不利环境的影响。
BV被认为是一种生物失调,通常表现出临床症状,可由大量具有促炎特征的微生物以及宿主免疫反应引起。
据报道,BV女性的阴道样本中含有高水平的免疫介质,如白介素(IL)-8、IL-6、IL-1α、IL-1β、IL-12p70和TNFα。
不同物种可能采用不同的免疫因子。例如,脆乳杆菌与γ诱导蛋白10(IP-10)的显著增加以及IL-12(p70)、IL-8、IL-1β和IL-1α的显著下降相关。
然而,根据阴道拭子的分析,G. vaginalis与IP-10的下降和IL-12(p70)、IL-8、IL-1β和IL-1α的增加相关。G. vaginalis也与相同因素的增加和减少有关。
在携带大量Prevotella spp.的女性中观察到了更高水平的IL-1β、IL-8和干扰素(IFN-γ)。
IL-36G对BV女性的关键作用得到了验证。因此,BV患者阴道样本中IL-36G水平升高。IL-36G可能在BV和其他疾病的免疫应答中发挥重要作用。
通过阴道菌群的检测,可以了解更多相关疾病风险。
谷禾阴道菌群检测数据库
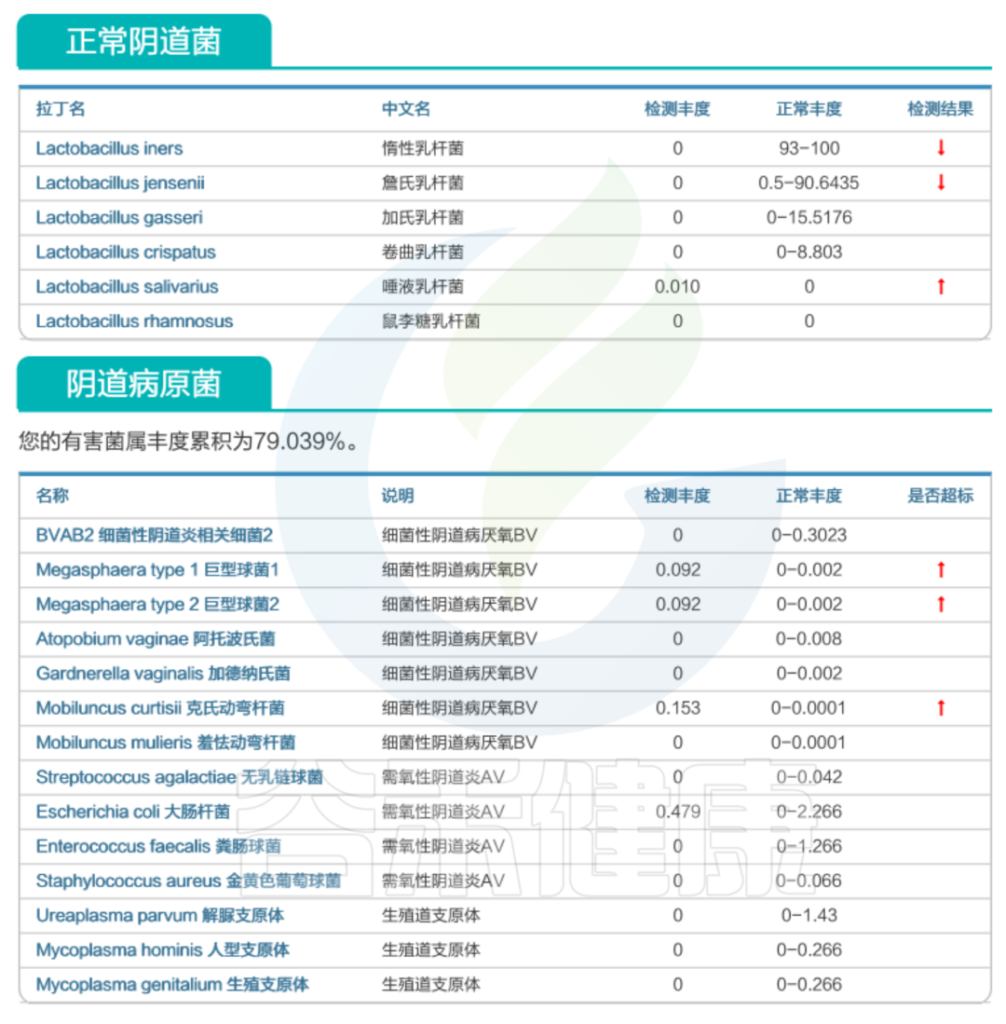
该案例选自谷禾阴道菌群检测数据库,相关菌群检测结果的异常对细菌性阴道病的风险具有提示作用。
盆腔炎是由上生殖道炎症引起的感染。细菌性阴道病被认为是盆腔炎的一个危险因素,可导致不良的生殖后遗症,如不孕、慢性骨盆疼痛和异位妊娠。
据报道,细菌性阴道病相关微生物与盆腔炎发病风险升高有关,而非细菌性阴道病相关微生物对盆腔炎发病风险没有影响。
急性子宫内膜炎患者更有可能细菌性阴道病,而携带乳酸杆菌的可能性较小。与阴道微生物群正常的女性相比,细菌性阴道病患者亚临床盆腔炎的检出率是正常女性的2.7倍。
A. vaginae, S. amnionii, BVAB1, S. sanguinegens的存在与盆腔炎及其后遗症相关,包括复发性盆腔炎和不孕。
在最近一项针对性传播感染高危女性的研究中,阴道中存在BV相关微生物,如A. vaginae, Megasphaera spp., Sneathia spp., Prevotella amnii, Eggerthella-like bacterium,可增加盆腔炎发生的可能性。
此外,BV相关微生物的更大细菌负荷预测了盆腔炎。盆腔炎中BV相关微生物的鉴定表明,生殖道上升的程度从低到高。
这一发现可能是由于BV相关微生物产生的酶。这些酶,如粘蛋白酶和唾液酸酶,可以降解粘蛋白屏障,促进上升感染,从而导致盆腔炎。
其他相关妇科疾病与阴道菌群详见:
阴道中有益和有害细菌之间的平衡非常脆弱,如果阴道 pH 值不够酸性,就会出现不平衡。阴道酸碱度应该在 3.8 到 4.5 之间,以保持健康的阴道酸度。
如果由于缺乏乳酸杆菌而导致阴道酸度不足,那么真菌和“坏”细菌的繁殖能力就会比平时更多。
可能增加阴道菌群失衡并因此感染的生理或外部风险因素包括:
帮助恢复阴道菌群平衡的措施:
益生菌是体内天然存在的细菌,通常被推荐用于治疗阴道菌群失衡。益生菌有助于恢复健康的乳酸杆菌水平。通过保持阴道菌群的健康平衡,身体对感染的防御能力得以恢复。
益生菌的开发和临床应用的最新进展为治疗开辟了新途径。极大地克服了因抗生素的适宜性、用量、给药方式等使用不当而导致病原体耐药性增强的现象。
有些益生菌例如鼠李糖乳杆菌、罗伊氏乳杆菌、植物乳杆菌、长双歧杆菌等,已被证明可以改善整体阴道健康。
每天口服一次或两次胶囊化鼠李糖乳杆菌GR-1和发酵乳杆菌RC-14菌株可将细菌性阴道病相关微生物群转化为以乳杆菌优势为标志的正常微生物群。
益生菌在外阴阴道念珠菌病(VVC)的阴道微生物组改变的治疗中也很有效。研究人员调查了7918名VVC孕妇阴道菌群的变化,发现外阴阴道念珠菌病阴道菌群可以在正常(以乳酸菌为主)和异常(乳酸菌减少)之间振荡。
临床试验表明,嗜酸乳杆菌、鼠李糖乳杆菌GR-1和发酵乳杆菌RC-14在恢复以乳酸杆菌为主的阴道菌群方面具有有效性。
它会对阴道健康产生负面影响。对于更容易受到阴道酵母菌感染的女性,建议采用均衡且高纤维的低糖饮食。摄入大量糖会促进肠道中致病性酵母菌的生长。如果这些在上厕所时进入阴道,可能会导致阴道酵母菌感染。
高热量和高脂肪消耗的女性患细菌性阴道病的风险增加。
一些研究已经确定微量营养素摄入不足,尤其是维生素 A、C、E 和 D 以及 β-胡萝卜素、叶酸、钙的不足,会增加细菌性阴道病的风险。
各种疾病所需的抗生素疗程会扰乱健康的阴道菌群。这使得病原体和酵母菌很容易传播。
有香味的卫生用品或其他物品,如用含有合成香料的洗衣粉洗涤的内衣,会破坏阴道微生物组。阴道有一种自然的气味。如果闻到比平时更刺鼻的气味,则可能表明阴道微生物组失衡。
冲洗会破坏阴道的天然微生物组,使其容易受到感染。肥皂和化学女性卫生用品会改变 pH 值的酸度,也可能杀死或去除有益细菌,还会引入新的细菌,从而降低抵御潜在感染的防御屏障。
有些女性认为所有的内衣都是一样的。但是紧贴生殖器区域皮肤穿的衣服会有所不同。“穿白色棉质内衣,不要染色”,研究人员表示,“而且阴道需要呼吸空间,所以不要穿紧身牛仔裤。”
膳食糖会滋养阴道中的酵母菌和其他有害细菌。“避免在饮食中加糖——不要吃小麦,不要吃甜食,”一些研究人员提倡“绿色”饮食,富含健康脂肪和低糖食物、富含微量营养素的植物性食物,如鳄梨、坚果、种子和十字花科蔬菜。
吸烟可以改变阴道内的雌激素和 pH 值;同时研究发现宫颈/阴道分泌物中含有微量尼古丁,可能会促进炎症。
如果低雌激素或阴道干燥影响阴道 pH 值,外用雌激素乳膏可能会有所帮助。 外用雌激素乳膏仅供处方使用,因此如果怀疑低雌激素(或雌激素波动不稳定)导致阴道生态系统失衡,请咨询医生。“如果女性处于围绝经期或绝经后,阴道雌激素治疗是降低阴道 pH 值的最有效方法。”
拥有健康的阴道菌群对孕妇尤为重要。怀孕对身体来说是一个非常特殊的条件。它还会改变荷尔蒙平衡。伴随的高水平雌激素也会影响阴道菌群。由于免疫系统更敏感,孕妇也更容易受到感染。
孕妇阴道菌群紊乱可能导致病原体传播。在细菌性阴道病的情况下,早产或流产的风险会增加。注意到阴道分泌物(外观和气味)发生变化的孕妇应咨询妇科医生。
阴道微生物组与人类宿主形成稳态和互惠关系,并在阴道健康和疾病中发挥重要作用。内部和/或外部因素的变化导致平衡的生态系统崩溃,这也称为生态失调。
乳酸杆菌似乎在维持健康的阴道微环境中起着核心作用。阴道菌群失调涉及微生物多样性或丰度的变化,特别是乳酸杆菌或某些厌氧细菌,这会导致炎症。这些相关微生物会影响免疫介质,而免疫介质可作为阴道环境中生态失调的预测性生物标志物。
基于调节阴道菌群平衡的研究,可用于了解阴道微生物组如何影响怀孕和分娩;同时可能成为细菌性阴道病、早产、不孕等疾病的新的靶点,深入了解相关作用机制,有助于更多改进的和准确的诊断和治疗策略的开发。

主要参考文献:
Gudnadottir U, Debelius JW, Du J, Hugerth LW, Danielsson H, Schuppe-Koistinen I, Fransson E, Brusselaers N. The vaginal microbiome and the risk of preterm birth: a systematic review and network meta-analysis. Sci Rep. 2022 May 13;12(1):7926. doi: 10.1038/s41598-022-12007-9. PMID: 35562576; PMCID: PMC9106729.
Hong X, Ma J, Yin J, Fang S, Geng J, Zhao H, Zhu M, Ye M, Zhu X, Xuan Y, Wang B. The association between vaginal microbiota and female infertility: a systematic review and meta-analysis. Arch Gynecol Obstet. 2020 Sep;302(3):569-578. doi: 10.1007/s00404-020-05675-3. Epub 2020 Jul 8. PMID: 32638096.
Joseph RJ, Ser HL, Kuai YH, Tan LT, Arasoo VJT, Letchumanan V, Wang L, Pusparajah P, Goh BH, Ab Mutalib NS, Chan KG, Lee LH. Finding a Balance in the Vaginal Microbiome: How Do We Treat and Prevent the Occurrence of Bacterial Vaginosis? Antibiotics (Basel). 2021 Jun 15;10(6):719. doi: 10.3390/antibiotics10060719. PMID: 34203908; PMCID: PMC8232816.
Chen X, Lu Y, Chen T, Li R. The Female Vaginal Microbiome in Health and Bacterial Vaginosis. Front Cell Infect Microbiol. 2021 Apr 7;11:631972. doi: 10.3389/fcimb.2021.631972. PMID: 33898328; PMCID: PMC8058480.
Zheng N, Guo R, Wang J, Zhou W, Ling Z. Contribution of Lactobacillus iners to Vaginal Health and Diseases: A Systematic Review. Front Cell Infect Microbiol. 2021 Nov 22;11:792787. doi: 10.3389/fcimb.2021.792787. PMID: 34881196; PMCID: PMC8645935.
Bastianelli C, Farris M, Bianchi P, Benagiano G. The effect of different contraceptive methods on the vaginal microbiome. Expert Rev Clin Pharmacol. 2021 Jul;14(7):821-836. doi: 10.1080/17512433.2021.1917373. Epub 2021 Apr 23. PMID: 33863265.
Saraf VS, Sheikh SA, Ahmad A, Gillevet PM, Bokhari H, Javed S. Vaginal microbiome: normalcy vs dysbiosis. Arch Microbiol. 2021 Sep;203(7):3793-3802. doi: 10.1007/s00203-021-02414-3. Epub 2021 Jun 13. PMID: 34120200.
Auriemma RS, Scairati R, Del Vecchio G, Liccardi A, Verde N, Pirchio R, Pivonello R, Ercolini D, Colao A. The Vaginal Microbiome: A Long Urogenital Colonization Throughout Woman Life. Front Cell Infect Microbiol. 2021 Jul 6;11:686167. doi: 10.3389/fcimb.2021.686167. PMID: 34295836; PMCID: PMC8290858.

谷禾健康
久坐不动的生活方式已逐渐成为现代社会很多人的一种常态,因此导致2型糖尿病 、肥胖、心血管疾病和非酒精性脂肪肝等代谢性疾病的发病率上升。
★ 代谢性疾病严重危害人体健康
根据世界卫生组织数据库,2019年,代谢风险(即高体重指数 (BMI)、高血糖、高血压和高胆固醇)占全球总健康损失的近 20%。调查发现,2019年,高血压导致了近五分之一的死亡(近1100万人),其次是高血糖(650万人死亡)、高BMI(500万人)和高胆固醇(440万人)。
这些疾病对人们的健康造成了巨大影响,不过定期和适当水平的体育锻炼可以起到预防作用。
最新的研究发现,运动与饮食结合:通过肠道微生物群的相互作用能够更好地预防和调节代谢性疾病。
根据世界卫生组织和美国疾病控制与预防中心的数据,定期进行体育锻炼和饮食干预可以将妊娠糖尿病的患病率降低30%,将死亡风险降低20%至30%。
•运动与肠道微生物
肠道微生物在宿主的整个生命周期中参与影响健康的各种相互作用。
运动促进的微生物群结构和状态的变化在促进有益代谢物的产生、刺激/调节免疫系统、保护宿主免受病原体定植以及控制脂质积累和胰岛素信号。
规律的运动是对肠道的刺激性应激源,可促进有益反应并改善肠道屏障的完整性。
•饮食与肠道微生物
饮食对于塑造微生物群落或代谢物很重要。
微生物群暴露于健康的膳食成分,如膳食碳水化合物、蛋白质、维生素、矿物质和多酚,它们可以产生有益的代谢物,特别是短链脂肪酸和色氨酸代谢物。
这些代谢物参与维持肠粘膜完整性,还介导宿主免疫和稳态反应。相反,不健康的饮食,如高脂饮食,会增加促炎细胞因子的产生,从而导致全身慢性炎症和脂多糖易位,从而增加代谢疾病的风险。
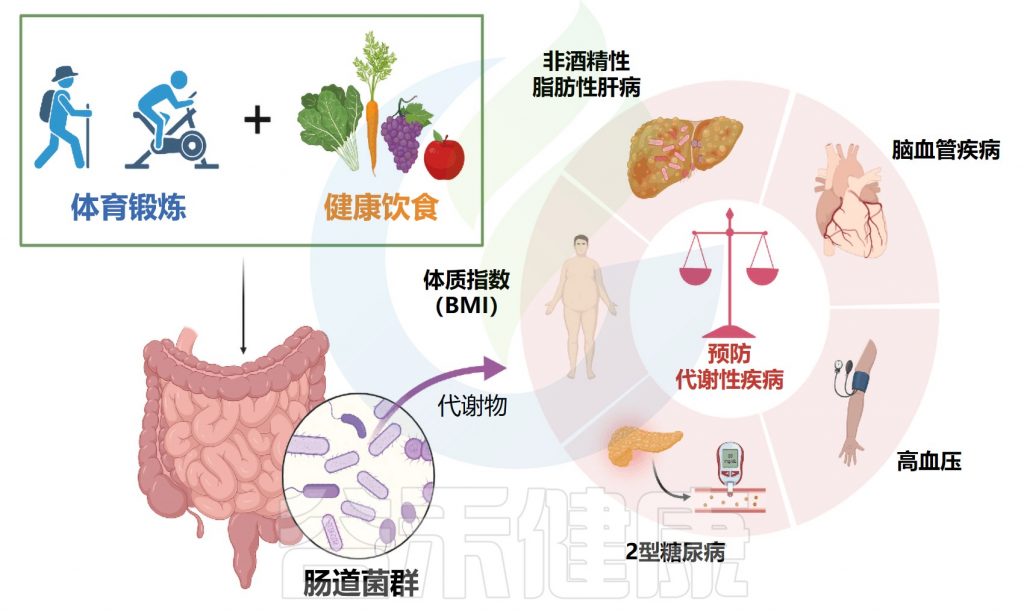

本文讲述了肠道微生物与代谢性疾病的关联,主要包括肥胖、2型糖尿病、心血管疾病和非酒精性脂肪肝。
我们还提到了体育锻炼、饮食成分和饮食模式对肠道微生物的影响。并介绍了通过体育锻炼和饮食相结合来预防代谢性疾病的一些研究和相关机制,这可能为预防代谢性疾病提供一条新途径。
▸ 体育锻炼
体育锻炼被定义为有计划、结构化和重复的体育活动的一个子集,旨在改善或保持身体健康。
注意:定期锻炼是指每周5天,每次至少30分钟的中等强度体育锻炼,或每周3天,至少20分钟的高强度体育锻炼。
★运动与炎症及代谢疾病有关
研究表明,习惯性运动会抑制基础促炎细胞因子的表达,但过度运动会引发多种促炎介质的产生。合理和适度的体育锻炼可以减少代谢性疾病的风险,只有在极端情况下,才会增加体育锻炼相关并发症的风险。
事实上,定期运动会独立影响肠道功能和微生物组特征,进而对预防代谢疾病具有有益作用。
体育锻炼对肠道菌群和宿主健康的影响
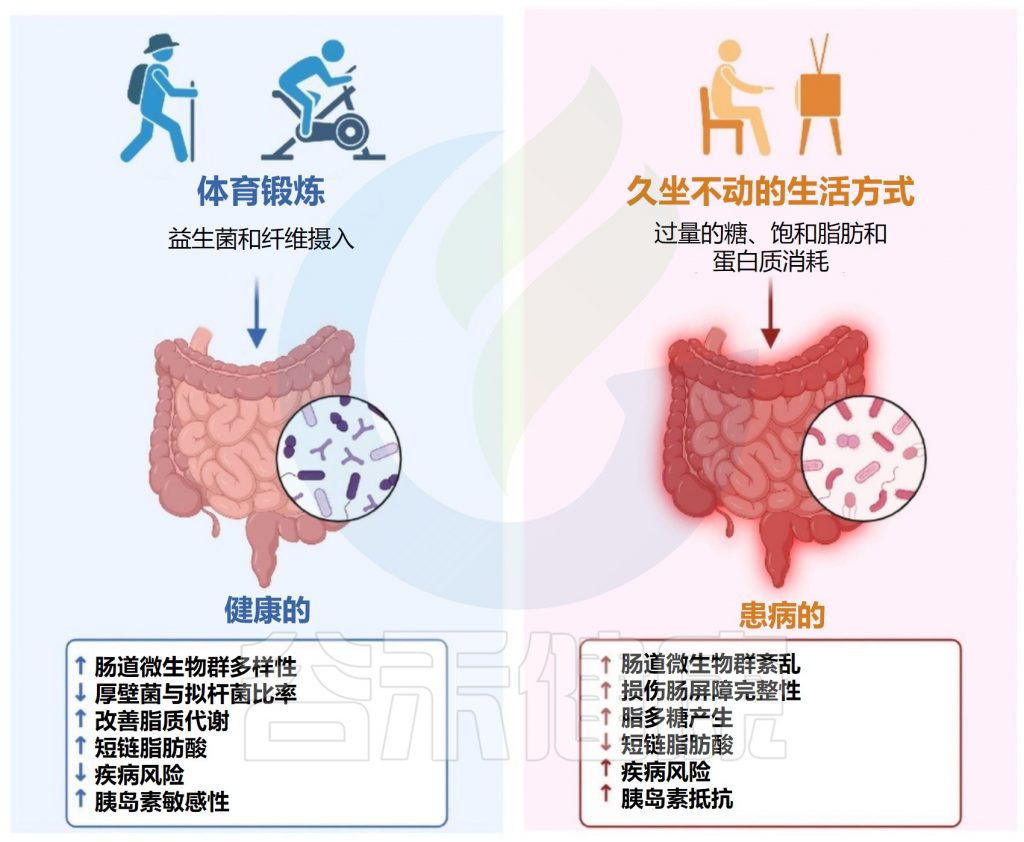

编辑
Zhang L,et al.Nutrients.2022
肠道菌群受性别、遗传、年龄和种族(即不可改变的因素)和可改变的因素(如宿主健康、身体活动、饮食和最终的抗生素治疗)的调节。研究表明,运动对微生物群有独特的影响。
体育锻炼与肠道菌群生物多样性的积极调节有关;体育锻炼在塑造肠道微生物多样性和调节其分布方面的作用已经得到证明。如下表所示:
运动对微生物群与代谢的影响








Donati Zeppa S,et al.Nutrients.2019
•改变原因
体育锻炼引起的肠道菌群改变是由于肠道转运时间 、胆汁酸谱的改变、通过AMPK激活产生短链脂肪酸 、Toll样受体 (TLRs) 信号通路、免疫球蛋白 A (IgA)、B和CD4+T细胞的数量,最后到体重减轻。
AMPK即AMP依赖的蛋白激酶,是生物能量代谢调节的关键分子。它表达于各种代谢相关的器官中,能被机体各种刺激激活,包括细胞压力、运动和很多激素及能影响细胞代谢的物质。
•体重与菌群变化显著相关
在一项分析运动活跃和久坐的40岁以下女性的研究中,几种细菌类群的变化与体重指数 (BMI) 显著相关。即使所有参与者的微生物群组成在运动后的一段时间内发生变化,具有已知抗炎特性和产生短链脂肪酸能力的物种在瘦受试者中更高。
✦运动促进新陈代谢
在运动条件下,肠道微生物的变化会影响营养物质的吸收,进而影响宿主的新陈代谢。来自美国肠道计划的数据表明,进行适度运动(从不运动到每天运动)重塑了微生物组成和功能的变化,促进了老年人尤其是超重老年人更健康的肠道环境。
在动物身上也得到了类似的结果。进行体育锻炼的小鼠通常表现出双歧杆菌(Bifidobacterium)、乳酸杆菌和嗜黏蛋白阿克曼菌(Akkermansia muciniphila)丰度增加。
这些结果可能反映了运动更高的新陈代谢,因为阿克曼菌比例增加通常与更健康的新陈代谢特征相关。
✦对肠道屏障、免疫系统有积极作用
此外,运动可能会对肠道粘液层产生积极影响,肠道粘液层是粘膜相关细菌(如嗜粘蛋白-阿克曼氏菌)的重要基质。适度运动还可以减轻慢性应激诱导的小鼠肠道屏障损伤,减少细菌移位并维持肠道通透性。
罗氏菌属(R.hominis)和普拉梭菌(F.prausnitzii)产生的丁酸盐对健康有益,对肠道功能和脂质代谢有积极影响。普拉梭菌还产生具有抗炎作用的代谢物。
粪球菌属(Coprococcus)属是一种产丁酸盐的属,在经常运动的女性中更为丰富,促进了一些与运动相关的健康影响。
•瘦的人群产丁酸盐菌群丰度较高
在另一项比较瘦和肥胖成年人在饮食控制下参加为期六周的监督耐力运动计划的研究中,仅在瘦受试者中发现产生丁酸盐的分类群增加。
此外,瘦成人的普拉梭菌(Faecalibacterium Prausnitzii)增加,而肥胖成人则减少,而拟杆菌属(Bacteroides)有相反的趋势,证实了体重的影响。
注意:在体重、饮食和年龄正常化后,有氧适能水平较高的个体中产丁酸盐类群的丰度更高。
研究表明,这些微生物是已知的丁酸盐生产者,对促进肠道屏障完整性、调节宿主免疫系统和脂质代谢具有有益作用。
✦肠道微生物影响运动表现
在运动期间和之后,大量的乳酸会释放到血液中。乳酸在耐力表现中具有重要作用,因为它被用作多种器官和组织的燃料。这些器官和组织“学习”使用乳酸作为底物的次数越多,性能提高的越多。
最近证明,全身性乳酸可以穿过肠道屏障进入肠腔,然后可以被韦荣氏球菌属(Veillonella)转化为丙酸。
有报道说,肠道微生物群中的韦荣球菌丰度增加,其甲基丙二酰辅酶A在运动后过度表达。
•提高抗氧化活性
此外,他们证明在老鼠身上,韦荣氏球菌属(Veillonella)接种改善了跑步性能,通过结肠内输注给予丙酸盐也改善了这种性能。
在一项关于小鼠耐力游泳时间的研究中,证明了肠道微生物群的表现和抗氧化活性之间的关系,表明“肠道微生物群的状态可能对运动表现及其与运动员抗氧化酶系统相关的潜在作用至关重要”。
因此,这些研究表明,肠道微生物群对短链脂肪酸产生的调节会影响运动过程中的能量代谢,从而有助于运动诱导的适应。这些微生物群发酵产物也可用作肝脏和肌肉细胞的能量来源,通过长期维持血糖来提高耐力表现。
✦运动频率不同体内菌群不同
已发现运动员微生物组包含不同的微生物组成,这些微生物主要由韦荣氏球菌(Veillonella)、拟杆菌属(Bacteroides)、普雷沃氏菌属(Prevotella)、甲烷杆菌(Methanobacteriaceae)和嗜黏蛋白阿克曼菌(Akkermansia muciniphila)所组成。
参与能量和碳水化合物代谢的分类群的丰度,如普雷沃氏菌和史密森甲烷杆菌,被发现在职业自行车手中明显高于业余自行车手,并且与训练频率相关。
在超重的成年人中,遵循富含纤维和全谷物的饮食六周后,普雷沃氏菌的丰度可预测体重减轻,这表明应在个性化营养策略中考虑肠型以对抗肥胖。短链脂肪酸的产生,尤其是丁酸,是肠道健康的重要标志,在人类运动后会增加。
✦经常运动肠道菌群多样有助于促进健康
职业橄榄球运动员的肠道微生物表现出更大的α多样性和厚壁菌门与拟杆菌门比率的下降. 与久坐不动的女性相比,进行常规运动量的女性显示出更多的促进健康的分类群,例如
普拉梭菌(Faecalibacterium Prausnitzii)↑↑↑
罗氏菌属(Roseburia hominis)↑↑↑
嗜黏蛋白阿克曼菌(Akkermansia muciniphila) ↑↑↑
这些物种与促进健康的作用有关。先前已在运动员的微生物群中描述了高丰度的阿克曼菌,而低水平与炎症性肠病患者的代谢紊乱(肥胖、代谢综合征和 II 型糖尿病)有关。
•增加有益菌丰度预防疾病
检查了20名业余跑步者在半程马拉松比赛前后的粪便代谢物和微生物群。
根据α多样性分析,多样性几乎没有差异,但是,某些微生物群成员的丰度在跑步前后显示出差异。在门水平上,跑步后检测到在人体肠道中的功能未知的Lentisphaerae和Acidobacteria。
在物种水平上,Coriobacteriaceae和Succinivibrionaceae显著增加。
放线菌门(Actinobacteria)参与胆汁盐和类固醇激素的代谢以及人体肠道中膳食多酚的激活。Coriobacteriaceae与15种代谢物呈正相关,表明Coriobacteriaceae的代谢可能是运动预防疾病和改善健康结果的潜在机制。
这些增加的代谢物表明,跑步促进了微生物群衍生的新陈代谢。
•减少致病菌,具有抗炎作用
在属水平上,半程马拉松跑减少了粪便中Ezakiella、Romboutsia和放线杆菌(Actinobacillus)的丰度,但增加了粪球菌(Coprococcus)和Ruminococcus bicirculans。
放线杆菌属会导致几种不同的动物疾病,例如牛的放线菌病、新生马驹的烈性败血症和人类牙周病。
因此,对这种潜在病原体的抑制表明运动具有抗炎作用。还需注意,戊糖磷酸途径是一种与糖酵解平行并涉及葡萄糖氧化的代谢途径,是半程马拉松跑后最丰富的途径。这些发现强调了运动促进健康益处的微生物群衍生机制。
✦不同运动类型菌群组成不同
为了研究特定运动类型和运动员饮食对肠道微生物群的长期影响。比较了15名久坐不动的健康男性(作为对照组)、15名健美运动员和15名长跑运动员的粪便微生物群特征、膳食摄入量和身体成分。
运动类型与运动员饮食模式相关(即,健美运动员:高蛋白、高脂肪和低碳水化合物/膳食纤维饮食;长跑运动员:低碳水化合物和低膳食纤维饮食)。
虽然运动员类型在肠道微生物群α和β多样性方面没有差异,但它与几种细菌的相对丰度显著相关。例如,在属水平上,普拉梭菌(Faecalibacterium)、萨特氏菌(Sutterella)、Clostridium、嗜血杆菌、艾森氏菌属最高,而双歧杆菌和副双歧杆菌在健美运动员中最低。
在物种水平上,广泛用作益生菌的肠道有益菌(青春双歧杆菌、长双歧杆菌、清酒乳杆菌)和产生短链脂肪酸的有益菌(经黏液真杆菌属、霍氏真杆菌(Eubacterium hallii))在健美运动员中最低,在对照组中最高。
在长跑运动员中,蛋白质摄入量与多样性呈负相关,而在健美运动员中,脂肪摄入量与双歧杆菌呈负相关。这些差异可能与运动中的的营养状况有关。
✦不同生理状态下运动效果不同
此外,体育锻炼所产生的变化似乎取决于个人的生理状态。例如,无论是肥胖-高血压大鼠还是正常大鼠,规律的强迫运动都会对微生物群丰富度产生不同的影响。高脂肪饮食后运动对大鼠微生物群的改变与正常饮食的大鼠不同,糖尿病小鼠产生的改变也不同于对照小鼠。
•幼年运动对微生物群影响更显著
最后,据观察,与成年大鼠相比,运动对幼年大鼠的微生物群产生更有效的改变。在这些研究运动训练对肠道微生物组影响的小鼠研究中,一个共同发现是α多样性增加。使用基于小鼠的模型的其他几项研究也表明,与久坐不动的动物相比,运动的动物的α多样性增加。
高强度运动对肠道微生物不利
需要注意的是,高强度运动可能会对肠道功能产生有害影响。总共70%的运动员在剧烈运动后可能会出现腹痛、恶心和腹泻。
长时间运动还会导致微生物多样性减少,幽门螺杆菌(Helicobacter pylori)数量增加。过度运动会诱发增加肠道通透性的压力,这可能导致细菌及其有毒产物(包括微生物群衍生的脂多糖)进入血液并激活全身炎症。易位的脂多糖激活 TLR,促进NF-kB通路激活和炎性细胞因子的产生,最终导致内毒素血症。
运动强度是一个有争议的问题;我们必须考虑到各种运动形式,以及运动的持续时间。同时,要针对不同人群制定不同的干预方案;目的在于激励久坐不动的人摆脱不健康的生活方式。
小结
运动会改变参与代谢模式的分子的转换,并刺激神经内分泌激素的释放,这些激素直接或通过免疫系统间接与肠道相互作用。
总之,运动的强度、时间和类型会影响肠道微生物群的组成,因为它还与受试者的性别、年龄、健康状况和训练状态有关。已经证明,低水平但持续进行的身体活动可以增加微生物群的多样性,改善受试者的代谢特征和免疫反应,而急性剧烈运动可能会对运动员的微生物群及其总体健康造成有害影响。
碳水化合物是由碳、氢和氧三种元素组成,自然界存在最多、具有广谱化学结构和生物功能的有机化合物。
不同种类的水果、蔬菜和全麦谷物是膳食碳水化合物的主要来源。在人类基因组中,只有不到20种糖苷酶被鉴定为参与消化膳食碳水化合物的酶。
唾液淀粉酶首先在口腔内将复杂的碳水化合物分解为单糖,易消化的碳水化合物可通过胰淀粉酶、蔗糖酶、麦芽糖酶、半乳糖酶和乳糖酶等降解消化。复杂的不易消化的膳食碳水化合物驱使我们的肠道微生物进化出碳水化合物活性酶库,以便有效地竞争营养。
✦不同的碳水化合物对肠道影响不同
宿主的肠道不断被动态排列的碳水化合物淹没。而不同的碳水化合物对肠道的影响各不相同。
•简单的碳水化合物导致宿主代谢紊乱
已经注意到,简单的碳水化合物(例如蔗糖、果糖)会引起微生物群快速重塑,从而导致宿主代谢紊乱。
✦复杂的碳水化合物对健康有利
复杂的碳水化合物,特别是某些微生物群可接触的多糖和膳食纤维,为在该栖息地竞争的密集微生物群提供食物,对肠道微生物生态学和健康产生重大影响。
多糖含量高的饮食与上调的肠道微生物群落多样性有关,并促进有益微生物的生长,例如阿克曼氏菌、双歧杆菌和乳杆菌。同时,肠道微生物可以使用中间寡糖来生成对宿主有益的短链脂肪酸。
•增强肠道屏障
例如,铁皮石斛多糖 (DOPs) 不易消化和吸收,但会促进肠道微生物产生更多的丁酸,主要由Parabacteroides sp. HGS0025产生,从而介导肠道健康和免疫功能的改善。
铁皮石斛多糖干预还可以通过作用于嗜黏蛋白阿克曼菌(Akkermansia muciniphila)来促进粘蛋白合成,从而增强肠道屏障功能。
•参与抗炎保护
五味子的其他多糖还逆转了肠道微生物生态失调并上调了丁酸和丙酸的产生,这可能参与了抗炎保护机制。
膳食蛋白质是另一种关键的常量营养素,人们每天必须摄入一定量蛋白质,以获得氨基酸和一定量的氮元素,用于合成组织蛋白质。
★ 蛋白质摄入过高或过低都不健康
它还可以调节微生物组成和代谢产物的产生。蛋白质摄入量与健康之间的关系遵循U形曲线,其中较低的蛋白质摄入量与营养不良状态相关,而高于可耐受限度的摄入量与营养过剩疾病相关。
注:世界卫生组织建议普通成人每日蛋白质摄入量为0.83g/kg。
✦影响肠道环境
膳食蛋白质消化的产物是氨基酸。肠道微生物降解的氨基酸代谢物包括短链脂肪酸、支链脂肪酸、吲哚、酚、硫醇、硫化物、氨和胺。这些代谢产物参与与宿主健康和疾病相关的各种生理功能。
一方面,蛋白质降解提供必需的游离氨基酸作为结肠细胞的替代能源. 另一方面,这个过程也会释放出有毒的代谢副产物,如氨、硫化物和酚类,它们对局部肠道环境有害。
研究表明,适度限制日粮蛋白质可以塑造微生物群组成和多样性的和谐平衡,并改善成年猪的肠道屏障功能。
▸ 高蛋白饮食
•高蛋白饮食导致菌群减少和一些疾病
高蛋白饮食者毛螺菌科(Lachnospiraceae)、瘤胃球菌科(Ruminococcaceae)和嗜黏蛋白阿克曼菌(Akkermansia)的丰度减少。
此外,蛋白质,尤其是红肉和加工肉类中的蛋白质,是左旋肉碱和胆碱的来源,可被肠道微生物代谢并产生三甲胺 (TMA),随后被氧化为三甲胺N-氧化物 (TMAO)。高TMAO浓度与心血管疾病或死亡风险增加相关。
•经常运动蛋白质需求大
值得注意的是,运动员可能需要更多的蛋白质来支持骨代谢,保持足够的蛋白质合成和能量代谢,以及在强化/长时间的运动程序中保持足够的免疫功能和肠道完整性。
研究建议接受过耐力和力量训练的运动员的蛋白质摄入量为1.2-1.7克每公斤体重/天。
注:缺乏蛋白质可能导致女运动员月经失调。
膳食脂肪是指我们每日所吃各种食物含油脂的总和。来自植物和动物的膳食脂肪是人类生长发育的能量储备来源。
▸ 消化过程
脂肪首先被口腔中的舌脂肪酶和胃脂肪酶消化。接下来被胰脂肪酶水解成游离脂肪酸(FFA);大部分游离脂肪酸被小肠吸收,少数会通过胃肠道并直接改变肠道微生物成分。
✦膳食脂肪导致肠道微生物改变
与橄榄油或红花油相比,以棕榈油为基础的饮食可能会导致体重增加,对微生物群多样性产生负面影响,并增加厚壁菌门与拟杆菌门的比例。
•高脂饮食减少了有益菌和短链脂肪酸
饱和脂肪酸降低拟杆菌属、普雷沃氏菌属、乳酸菌属和双歧杆菌属。与低脂饮食相比,食用高脂饮食也显著减少了短链脂肪酸的释放。
✦高脂饮食不利于健康
•高脂饮食易导致结肠癌
膳食脂肪引起的肠道微生物群成分变化也可以调节微生物衍生的次级胆汁酸 (BA) 的产生。高脂饮食引发增强的胆汁酸放电,导致初级胆汁酸的结肠浓度增加。然而,5%到10%的胆汁酸没有被重吸收,而是被大肠中的微生物转化为次级胆汁酸,这对人体有害并会促进结肠癌发生。
•高脂饮食易导致炎症
此外,在高脂饮食小鼠中观察到的微生物群失调引起脂多糖从肠腔进入体循环,从而激活宿主促炎信号通路,然后引发低度全身炎症。
▸ 定义
膳食纤维的定义一直存在争议,一般将膳食纤维定义为具有三个或三个以上单元的可食用碳水化合物聚合物,对内源性消化酶有抵抗力,因此在小肠中既不水解也不吸收。
✦膳食纤维的作用
•重要能量来源
膳食纤维是盲肠和结肠微生物群的重要能量来源。特定肠道条件下的厌氧菌会激活其由关键酶和代谢途径组成的机制,这些机制可以代谢复杂的碳水化合物,从而导致产生短链脂肪酸等代谢物。
•影响微生物多样性
值得注意的是,限制膳食纤维不仅会导致微生物多样性的减少和短链脂肪酸的产生,还会改变肠道微生物的代谢以利用不太有利的底物,这可能对宿主有害。
Q1
什么是短链脂肪酸?
短链脂肪酸是主要由乙酸盐、丙酸盐和丁酸盐组成的有机产物。短链脂肪酸在调节宿主代谢、免疫系统和细胞增殖方面具有关键作用。
短链脂肪酸在盲肠和近端结肠中浓度很高,它们被用作结肠细胞的能量来源(尤其是丁酸盐),但也可以通过门静脉输送到外周循环,作用于肝脏和外周组织。尽管短链脂肪酸在外周循环中的水平很低,但现在人们普遍认为它们在宿主体内充当信号分子并调节不同的生物过程。
✦高纤维饮食有助于降低危害
为人类志愿者提供高蛋白、低碳水化合物的饮食不仅显著减少了总短链脂肪酸和丁酸盐的产生,还导致氨基酸发酵产生的潜在有害代谢物增加,包括支链脂肪酸、氨、胺、N-亚硝基化合物、酚类化合物、硫化物、吲哚化合物和氢气硫化物。这些代谢物的细胞毒性和促炎特性导致慢性疾病的发展,尤其是结直肠癌。
考虑到糖酵解发酵和蛋白水解发酵之间的权衡,高纤维饮食可能会抑制蛋白质发酵,抵消肉类和脂肪的许多不利影响,从而降低这些食物成分的危害。
稳定的肠道微生物群落受多种必需成分的影响,例如维生素、矿物质和多酚。
✦维生素
维生素是维持正常生理功能所需的少量辅助因子。人类无法合成大多数维生素来满足我们的日常需求,因此必须从外部获取。
•改变肠道微生物丰度和多样性
值得注意的是,肠道微生物有能力调节各种维生素的合成和代谢输出。随后,维生素还可以显著改变肠道微生物的丰度和多样性。
例如,维生素A可以上调对健康有益的微生物群,包括双歧杆菌、乳酸杆菌和阿克曼氏菌。
✦矿物质
与维生素一样,矿物质是微量营养素,它们在宿主新陈代谢和与肠道微生物群进行积极互动方面发挥着重要作用。
•影响肠道菌群和慢性疾病
已经证明,镁缺乏与慢性病发病率增加有关,并且镁缺乏小鼠体内的双歧杆菌含量会降低四天。不过,如果长期缺镁(21 天),双歧杆菌和乳酸杆菌的丰度会增加。
需要进行更多临床试验来确定缺镁和补充镁对避免不良反应的影响。
✦多酚
多酚是广泛存在于植物性食物中的一大类化合物,其中一些与肠道健康有关。
•抑制有害菌,促进益生菌
例如茶多酚可以抑制幽门螺杆菌和金黄色葡萄球菌等有害细菌的生长,并刺激或促进双歧杆菌和嗜黏蛋白阿克曼菌(Akkermansia muciniphila)等肠道有益菌的生长。
•调节肠道微生物
类黄酮可以影响和重塑肠道菌群的组成,发挥益生元和杀菌作用,尽管证据尚不确凿,它们的全身抗炎作用可能至少部分与微生物群的调节有关。
多酚的“益生元样”作用已经通过对人类肠道微生物群的体外研究以及临床前和临床试验中的体内观察到,在这些试验中,补充多酚和富含多酚的食物被证明可以调节肠道微生物群。
多酚有利于生长的其他有益物种包括:
嗜黏蛋白阿克曼菌(Akkermansia muciniphila)、
普拉梭菌(Faecalibacterium Prausnitzii)
罗氏菌属(Roseburia spp)
✦多酚人体利用度较低
不幸的是,许多天然多酚,如浓缩或可水解的单宁和糖基化多酚衍生物(与葡萄糖、半乳糖、鼠李糖、核酮糖、阿拉伯吡喃糖等糖结合)的特点是人体肠道吸收率低。口服生物利用度的降低严重限制了这些化合物的潜在有益作用。
•经肠道微生物作用更容易吸收
有趣的是,这些通常在饮食中保持无活性的多酚在肠道微生物群去除糖部分后被生物转化为活性化合物。这些代谢物可以保留母体化合物的抗氧化和多效活性,同时还表现出增加的肠道吸收和更好的生物利用度。
因此,类黄酮通过微生物群的生物转化,可以更容易地到达血液并在全身水平发挥其生物学相关作用。
总的来说,微生物群和多酚之间的相互积极的相互作用可能会促进人们的健康。
除了个别营养素,饮食模式对肠道微生物群的代谢活动也有显著影响。
世界范围内的饮食习惯多种多样,包括西式饮食、地中海饮食、生酮饮食、间歇性禁食等。
饮食模式对肠道微生物群介导的健康的影响


Zhang L,et al.Nutrients.2022
▸ 西式饮食
西式饮食,是一种以高含量精加工糖和碳水化合物、高含量饱和脂肪酸、高含量动物蛋白以及低含量膳食纤维为特征的一种现代饮食方式。
•西式饮食影响肠道微生物群稳态
在西方饮食中,大部分能量由非细胞营养素提供,这些营养素更容易被微生物和人体细胞消化。易于获取的非细胞营养素的数量增加会影响pH值、肠道微生物群成分和新陈代谢的变化,从而影响肠道微生物群稳态的调节和维持。
•易导致炎症
另一方面,高脂饮食的消耗也增加了促炎细胞因子的产生,从而导致全身性慢性炎症和脂多糖易位。
不同饮食对肠道菌群和宿主生理功能的影响
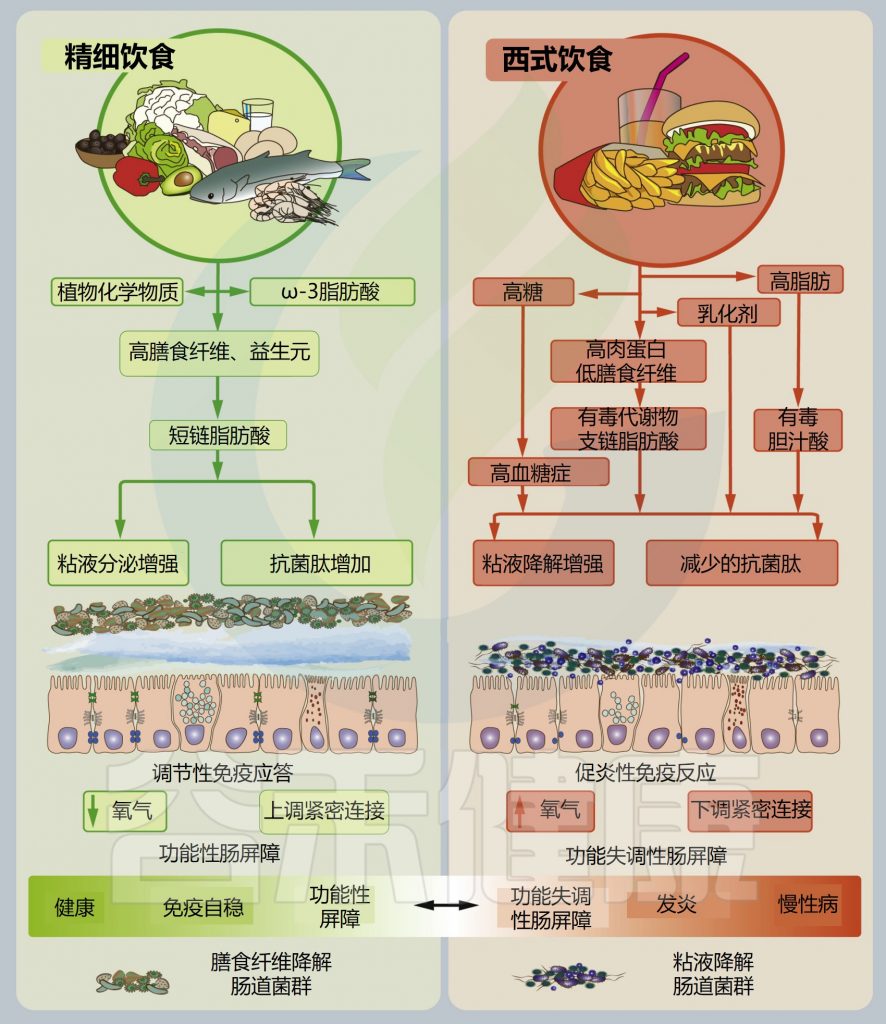

Makki K,et al.Cell Host Microbe.2018
▸ 地中海饮食
“地中海式饮食”是指有利于健康的,简单、清淡以及富含营养的饮食。这种特殊的饮食结构强调多吃蔬菜、水果、鱼、海鲜、豆类、坚果类食物,其次才是谷类,并且烹饪时要用植物油(含不饱和脂肪酸)来代替动物油(含饱和脂肪酸)。
•降低免疫性疾病风险
与西方饮食不同,地中海饮食被认为是全球最健康的饮食模式之一。更好地坚持地中海饮食与总死亡率的显著降低以及免疫系统失调、心血管疾病、认知能力下降和癌症的风险降低有关 。
•改善微生物群组成
此外,地中海饮食改变了微生物群的组成,有利于有益细菌,例如狄氏副拟杆菌(Parabacteroides distasonis)、多形拟杆菌(Bacteroides thetaiotaomicron)和青春双歧杆菌(Bifidobacterium adolescentis),并抑制病原体的生长,恢复可能有益的微生物。
▸ 生酮饮食
生酮饮食是一种高脂肪、充足蛋白质和低碳水化合物的饮食。
身体通过限制碳水化合物的可用性来燃烧脂肪而不是碳水化合物来获取卡路里。研究表明,生酮饮食对肠道微生物群的影响有好有坏。
•有营养不足的风险
一方面,生酮饮食营养不足的风险更大,并且由于缺乏纤维、必需的维生素、矿物质和铁,可能无法维持健康的微生物群。
•缓解结肠炎
另一方面,研究表明,随着阿克曼氏菌丰度的急剧增加,生酮饮食在DSS诱导的接受者中赋予微生物群益处并缓解结肠炎和产丁酸的罗氏菌属(Roseburia) ; 此外,在喂食生酮饮食的小鼠中发现大肠杆菌(Escherichia)/志贺氏菌(Shigella)的丰度减少。
▸ 间歇性禁食
间歇性禁食是一种类似于热量限制的饮食干预,包括各种操纵进餐时间以改善身体成分和整体健康的计划。
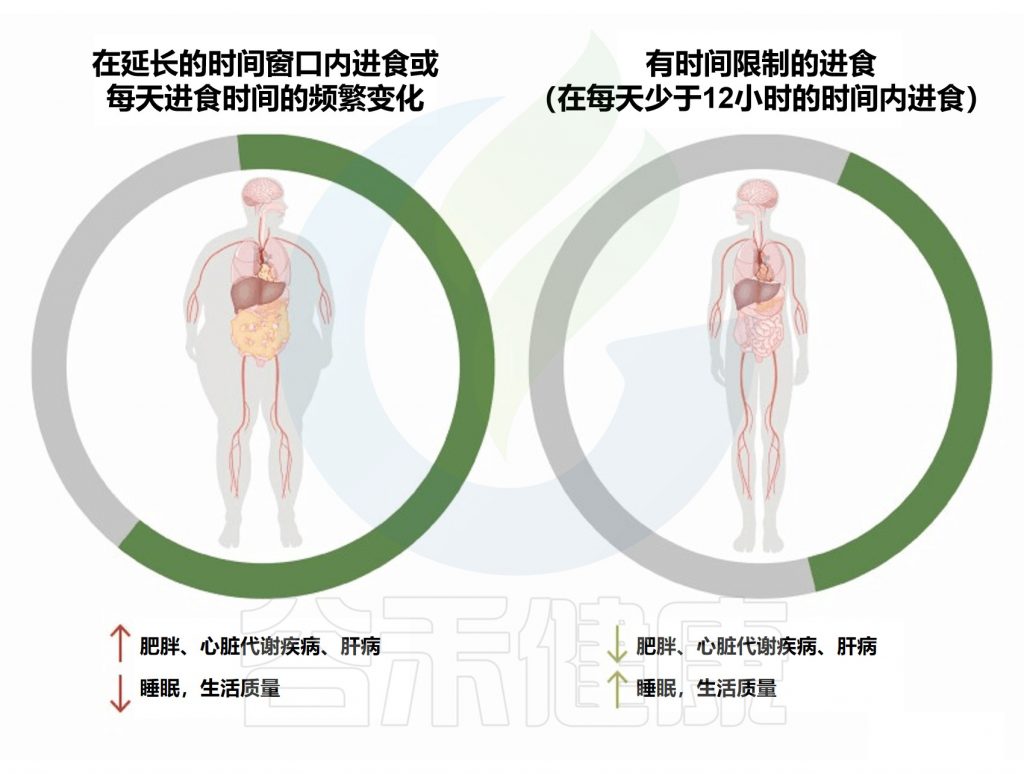

Chow LS,et al.Endocr Rev.2022
•缓解慢性疾病
研究发现间歇性禁食在动物模型中对广泛的慢性疾病(包括肝病、2型糖尿病、心血管疾病和脑功能)以及体重减轻具有强大的疾病缓解功效。
•增加肠道微生物丰富度
间歇性禁食似乎对肠道微生物群有积极影响。临床前研究一致表明,间歇性禁食有助于增加肠道微生物的丰富度,丰富嗜黏蛋白阿克曼菌(Akkermansia muciniphila)和乳杆菌(Lactobacillus),减少假定的促炎类群脱硫弧菌属(Desulfovibrio)和Turicibacter,并增强抗氧化微生物代谢途径。
建议
我们应该合理搭配膳食,尽量做到高纤维低脂肪的摄入,并保证一定量的碳水和蛋白质,以及不可缺少的微量元素。有助于降低代谢性疾病风险,恢复肠道环境,提升健康水平。
遗传变异被认为是代谢疾病的主要驱动因素,但这些变异的遗传概率相当有限。最近肠道微生物群被怀疑是驱动代谢疾病的另一个因素。
与健康个体相比,大多数患有肥胖、2型糖尿病、心血管疾病和非酒精性脂肪肝的人群肠道微生物多样性降低。肠道微生物的组成如果被外部因素改变,会导致肠道微生物与宿主之间的共生关系发生巨大变化,这对于代谢疾病的发展至关重要。
肥胖是指一定程度的明显超重与脂肪层过厚,是体内脂肪,尤其是甘油三酯积聚过多而导致的一种状态。由于食物摄入过多或机体代谢的改变而导致体内脂肪积聚过多造成体重过度增长并引起人体病理、生理改变或潜伏。
✦肥胖个体的微生物能量获取显著增加
通过行为改变(例如高脂饮食和抗生素的使用)改变肠道微生物可能是肥胖大流行的强大驱动力。
关于肠道微生物群在介导肥胖发病机制中作用,基于动物模型的发现。肥胖的微生物群导致从饮食中获取的能量显著增加。
据观察,与接受瘦捐赠者微生物群的小鼠相比,将肥胖捐赠者的微生物群引入无菌 (GF) 小鼠会导致能量获取能力增加。同样,可转移的肥胖相关微生物群比“瘦微生物群”定植更有助于全身脂肪的积累。
✦菌群丰度发生变化
肠道微生物成分在肥胖和瘦弱个体之间存在差异。人体研究观察到,与非超重个体相比,超重个体的微生物群的特征是拟杆菌(Bacteroides)的丰度较低,而厚壁菌门(Phylum Firmicutes)的丰度较高 。
在属水平上,一项宏基因组关联研究揭示了多形拟杆菌(Bacteroides thetaiotaomicron)在肥胖个体中的不足。
有趣的是,用B. thetaiotaomicron灌胃可以减轻饮食引起的小鼠体重增加和肥胖,这意味着益生菌或微生物化合物可能是未来潜在的抗肥胖方式。
2型糖尿病也被认为受到肠道微生物成分和功能失调的影响。
✦肠道微生物丰度与2型糖尿病相关
临床报告表明,双歧杆菌(Bifidobacterium)、乳杆菌(Lactobacillus)和产丁酸细菌 (例如 Akkermansia muciniphila) 的相对丰度与2型糖尿病呈负相关,而梭菌属(Clostridium spp)、瘤胃球菌属(Ruminococcus)、梭杆菌属(Fusobacterium)和经黏液真杆菌属(Blautia)与2型糖尿病呈正相关。
•肠道屏障受损影响2型糖尿病
肠道微生物的失调可能通过破坏紧密连接蛋白 (TJP) 损害肠道屏障,随后导致粘膜渗漏和代谢性内毒素血症,这是胰岛素抵抗和2型糖尿病发展的主要因素之一。
此外,肠道微生物可能参与葡萄糖调节。一项研究表明,与未接受结肠切除术的患者相比,接受全结肠切除术的患者患2型糖尿病的风险增加。
因此,营造一个良好的肠道稳态有助于预防2型糖尿病。
心脑血管疾病是心脏血管和脑血管疾病的统称,泛指由于高脂血症、血液黏稠、动脉粥样硬化、高血压等所导致的心脏、大脑及全身组织发生的缺血性或出血性疾病。
心脑血管疾病是一种严重威胁人类,特别是50岁以上中老年人健康的常见病,具有高患病率、高致残率和高死亡率的特点。
肥胖、2型糖尿病、血脂异常、高血压和不健康的生活方式,如吸烟、缺乏运动和不良饮食习惯等,都涉及心血管疾病的病理过程和危险因素。
✦肠道微生物影响心血管疾病
值得注意的是,这些因素中的大多数都与肠道微生物有关,基因组测序和宏基因组分析也揭示了心血管疾病表型与特定微生物类群变化或肠道微生物丰富度和多样性之间的关联。
早期研究表明,在动脉粥样硬化斑块中检测到细菌 DNA(主要是Chryseomonas),其特征与疾病状态相关的分类群相匹配.
•肠道菌群丰度发生变化
此外,宏基因组分析表明,心血管疾病患者的肠道微生物组与健康个体不同,这主要表现为链球菌属(Streptococcus spp.)和肠杆菌属(Enterobacteriaceae spp.)的丰度升高。以及拟杆菌属(Bacteroides spp.)、普氏菌属(Prevotella copri)和Alistipes shahii的丰度下降。
•肠道微生物作用机制
在机制层面,肠道微生物对心血管疾病的影响与炎症、肠道屏障功能和代谢物的调节有关。肠道微生物群中与生态失调相关的变化会损害肠道屏障,导致循环脂多糖水平升高,而脂多糖可通过 Toll 样受体 (TLR)-MyD88信号通路激活炎症信号,从而释放促炎细胞因子,从而在宿主中协调炎症状态。
先前的研究表明,心力衰竭患者的肠道完整性受损,血液中促炎细胞因子水平升高与症状严重程度和较差的预后相关。在依赖代谢的途径中,肠道微生物裂解一些含三甲胺的化合物产生三甲胺,三甲胺可被黄素单加氧酶进一步氧化成氧化三甲胺。氧化三甲胺激活 MAPK、NF-κB 信号通路,促进炎症基因表达,从而影响心血管疾病患者的脂质代谢并增加甘油三酯,降低高密度脂蛋白。
非酒精性脂肪肝是一种与肥胖有关的疾病,通常被认为是代谢综合征的肝脏表现。
多项临床前和临床研究强调了肠道微生物群在非酒精性脂肪肝发病机制中的作用,尽管对因果关系还不确定。
✦微生物多样性较低
简而言之,与健康受试者相比,非酒精性脂肪肝患者的肠道微生物群多样性较低,Anaerobacter、链球菌(Streptococcus)、大肠杆菌(Escherichia)和乳酸杆菌(Lactobacillus)的物种丰度增加,普雷沃氏菌(Prevotella)、颤螺菌属(Oscillibacter)和Alistipes spp的丰度较低。
注:肠道微生物群影响非酒精性脂肪肝的机制可能是在肠-肝轴方面。
✦影响其他疾病
除了肠道微生物群失调外,非酒精性脂肪肝还与胆汁酸的肠肝循环、肠道微生物群介导的肠粘膜炎症和相关的粘膜免疫功能损伤有关。
高热量饮食和久坐不动的生活方式导致肥胖的发病率上升,这在很大程度上是由能量摄入超过能量消耗造成的。
大量流行病学证据表明,肥胖是诱发其他代谢性疾病(包括2型糖尿病、心血管疾病和非酒精性脂肪肝)的危险因素。
代谢性疾病严重影响人们的健康,这一疾病主要是日积月累的不良习惯引起的,那么有什么可以预防或是降低这类疾病发病率的方法呢?
✦运动加饮食效果更好
确定有效的干预措施是改善代谢性疾病的重要途径。前文已有讲到运动和饮食都会调节肠道微生物并改善代谢性疾病。
事实上,当一项计划包括饮食和体育锻炼时,与单独锻炼或饮食相比,会有更有效的改变。肠道微生物的多样性和功能也受到饮食和体育锻炼的影响。
锻炼与饮食结合通过调节肠道菌群来预防代谢性疾病
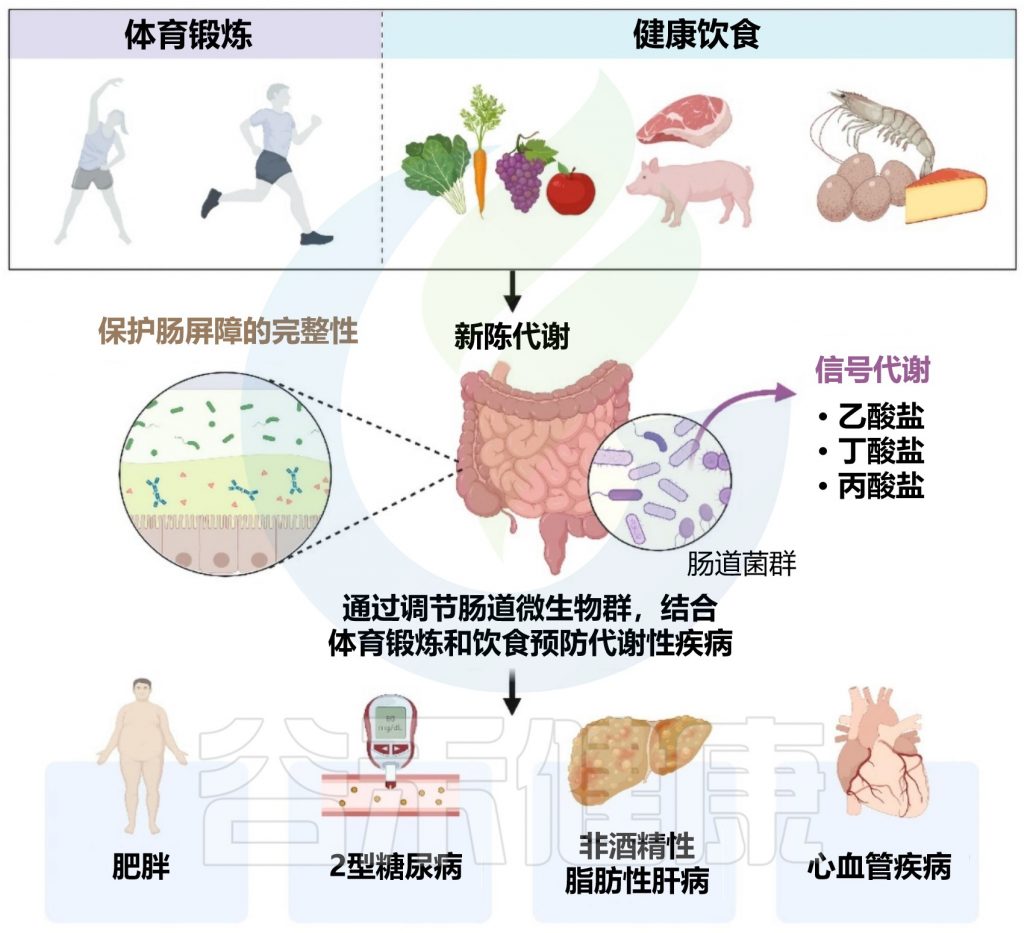

Zhang L,et al.Nutrients.2022
在这里,我们总结了一些动物和人类通过饮食加运动干预改善代谢性疾病的研究。
饮食诱导期间重复运动增加了免疫和代谢能力
在饮食诱导的肥胖期间,重复运动增加了小鼠远端肠道微生物群的α多样性和代谢能力。
适度运动和低脂饮食对高脂饮食诱导的肥胖小鼠的体重减轻和巨噬细胞免疫能力具有有益影响。
运动搭配低碳饮食减少了脂肪以及预防糖尿病
此外,一项为期6个月的随机干预计划表明,有氧运动和低碳水饮食提供了一种更有效的方法,可以通过改变肠道微生物群成分来减少肝脏脂肪和预防糖尿病。
低碳饮食加运动改善了心脏代谢
一项针对超重/肥胖中国女性表明,低碳水化合物饮食与运动训练相结合会增加产生短链脂肪酸的经黏液真杆菌属(Blautia)并减少与2型糖尿病相关的Alistipes属,导致显著的体重减轻,并改善血压、胰岛素敏感性和心肺健康,这表明低碳水饮食和运动干预可能通过调节肠道微生物在心脏代谢健康中发挥作用。
✦降低肝脏脂肪含量
最近的一项随机对照试验表明,与运动或单独饮食干预相比,饮食加运动干预可以显著降低肝脏脂肪含量并增加关键微生物的多样性和稳定性,这为制定饮食加运动干预策略提供了更有效的途径用于预防非酒精性脂肪肝。
✦有效控制血糖
在禁食状态下锻炼会产生有利的代谢适应,伴随着稳定的血糖浓度和升高的血液游离脂肪酸浓度,这可能更有效地改善胰岛素抵抗个体的胰岛素敏感性和控制血糖。
✦保护肠道屏障
肠道屏障是一种选择性的物理和免疫屏障,可促进营养、水和电解质吸收进入循环,同时阻止有害病原体和有毒管腔物质的转移。
如前所述,代谢疾病可能长期存在和加重的病理生理状态之一是肠道稳态失调释放内毒素,造成肠道渗漏,从而在宿主中诱发慢性低度炎症状态。
饮食和运动可以调节参与维持上皮膜完整性的紧密连接蛋白的表达,从而改善肠道通透性并降低慢性病风险。
从肠道微生物的角度来看,体育锻炼和饮食相结合可以缓和肠道屏障功能障碍,保持粘液厚度和肠道通透性。
✦影响代谢物的利用
饮食和运动的结合也会影响肠道微生物如何利用和合成代谢物。
肠道微生物和相应的代谢物以不同的方式与宿主协同作用,影响肠道稳态并为代谢性疾病提供保护性干预。具体而言,短链脂肪酸是微生物发酵或肠道中膳食多糖转化的主要终产物之一。而运动是短链脂肪酸的有效调节剂,对丁酸盐浓度具有特殊影响。
短链脂肪酸是肠上皮细胞的主要能量来源,参与维持肠粘膜完整性,改善糖脂代谢,控制能量消耗,调节免疫系统和炎症反应。在动物模型中,补充短链脂肪酸已被证明可以通过增加能量消耗和葡萄糖耐量来改善代谢,并且可能有助于延迟或减轻糖尿病并导致体重减轻。
益生菌、益生元等已被提议作为预防代谢性疾病的有效手段。
益生菌、益生元等对代谢性疾病的作用



编辑


Li HY, et al.Nutrients.2021
✦调节肠道菌群
含有Bifidobacterium lactis LMG P-28149和Lactobacillus rhamnosus LMG S-28148的益生菌混合物可以调节肥胖相关肠道菌群的组成,恢复嗜黏蛋白阿克曼菌(Akkermansia muciniphila)和Rikenellaceae的丰度,同时降低乳杆菌科的丰度。
✦改善代谢功能、减轻炎症
肠道微生物群被认为是肥胖和2型糖尿病代谢炎症的触发因素,罗伊氏乳杆菌(Lactobacillus reuteri)的给药可以通过抑制有害细菌(如小肠结肠炎耶尔森氏菌)的生长和改善TLR1-中的连四硫酸盐代谢来改善代谢功能有肠道炎症的缺陷小鼠。
此外,摄入干酪乳杆菌(Lactobacillus casei)可通过减少厚壁菌门和拟杆菌门来预防围产期大鼠代谢相关性高血压比率和血管紧张素转换酶 (ACE) 的表达,同时增加阿克曼菌和乳杆菌的丰度。
✦合生元有效改善肥胖
合生元被认为是预防肥胖的新领域,与单独的益生菌相比,omega-3脂肪酸与含有双歧杆菌、乳杆菌、乳球菌和丙酸杆菌的活益生菌混合物显示出更显著的肝脂肪变性和脂质积累减少。
此外,结合地衣芽孢杆菌和低聚木糖的口服补充剂可以更有效地改善肥胖大鼠的体重增加和脂质代谢,同时降低脱硫弧菌科和瘤胃球菌科的丰度。
Lactobacillus plantarum PMO 08与奇亚籽的混合物显示出对肥胖小鼠的协同抗肥胖作用,并为植物乳杆菌的生长创造了更有利的肠道微环境。
小结
益生菌(如双歧杆菌和乳酸杆菌)、益生元(如菊粉低聚果糖和其他多糖)、合生元(由益生菌菌株和益生元食品组成)等的干预可以使对代谢功能有重要影响。
主要通过调节肠道菌群组成、调节肠道微生物代谢物、改善肠道屏障功能这三个机制。
健康的饮食与体育锻炼相结合,可促进有益代谢物的产生并缓和肠屏障功能障碍,从而保护宿主免受入侵微生物的侵害,有助于维持体内平衡和预防代谢性疾病。
然而,虽然传统上这两种干预措施都被接受和实施,但很少有深入研究关注基于微生物群的策略。还需要更多的研究来确定肠道微生物是否可以作为对饮食和运动干预做出反应的代谢疾病的重要预测因子。
主要参考文献
Zhang L, Liu Y, Sun Y, Zhang X. Combined Physical Exercise and Diet: Regulation of Gut Microbiota to Prevent and Treat of Metabolic Disease: A Review. Nutrients. 2022 Nov 11;14(22):4774. doi:10.3390/nu14224774. PMID: 36432462; PMCID: PMC9699229.
Donati Zeppa S, Agostini D, Gervasi M, Annibalini G, Amatori S, Ferrini F, Sisti D, Piccoli G, Barbieri E, Sestili P, Stocchi V. Mutual Interactions among Exercise, Sport Supplements and Microbiota. Nutrients. 2019 Dec 20;12(1):17. doi: 10.3390/nu12010017. PMID: 31861755; PMCID: PMC7019274.
Makki K, Deehan EC, Walter J, Bäckhed F. The Impact of Dietary Fiber on Gut Microbiota in Host Health and Disease. Cell Host Microbe. 2018 Jun 13;23(6):705-715. doi: 10.1016/j.chom.2018.05.012. PMID: 29902436.
Allen JM, Mailing LJ, Niemiro GM, Moore R, Cook MD, White BA, Holscher HD, Woods JA. Exercise Alters Gut Microbiota Composition and Function in Lean and Obese Humans. Med Sci Sports Exerc. 2018 Apr;50(4):747-757. doi: 10.1249/MSS.0000000000001495. PMID: 29166320.
Mohr AE, Jäger R, Carpenter KC, Kerksick CM, Purpura M, Townsend JR, West NP, Black K, Gleeson M, Pyne DB, Wells SD, Arent SM, Kreider RB, Campbell BI, Bannock L, Scheiman J, Wissent CJ, Pane M, Kalman DS, Pugh JN, Ortega-Santos CP, Ter Haar JA, Arciero PJ, Antonio J. The athletic gut microbiota. J Int Soc Sports Nutr. 2020 May 12;17(1):24. doi: 10.1186/s12970-020-00353-w. PMID: 32398103; PMCID: PMC7218537.
Manoogian ENC, Chow LS, Taub PR, Laferrère B, Panda S. Time-restricted Eating for the Prevention and Management of Metabolic Diseases. Endocr Rev. 2022 Mar 9;43(2):405-436. doi: 10.1210/endrev/bnab027. PMID: 34550357; PMCID: PMC8905332.
Li HY, Zhou DD, Gan RY, Huang SY, Zhao CN, Shang A, Xu XY, Li HB. Effects and Mechanisms of Probiotics, Prebiotics, Synbiotics, and Postbiotics on Metabolic Diseases Targeting Gut Microbiota: A Narrative Review. Nutrients. 2021 Sep 15;13(9):3211. doi: 10.3390/nu13093211. PMID: 34579087; PMCID: PMC8470858.