-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
谷禾健康

最近的Nature 和 Nature Medicine 连发表了好几篇关于肠道菌群的文章,包括肠道菌群与神经互作,和基于这个原理的针对自闭症的临床治疗方案。心血管疾病的微生物组和代谢特征等。
今天我们主要介绍心血管疾病中冠状动脉疾病的相关重要研究发现和意义。
复杂的疾病,如冠状动脉疾病(CAD),往往是多因素的,由多种潜在的病理机制引起。尽管冠状动脉疾病在预防、诊断和治疗方面取得了巨大进展,但仍然是世界范围内发病率和死亡率的主要原因。目前对冠状动脉疾病的治疗基于传统的和可控制的冠状动脉疾病风险因素,只能取得部分成功。
冠状动脉疾病的发展包括血管壁上动脉粥样硬化斑块的逐渐生长,这通常与代谢状态受损有关。人体接触环境分子的主要部位是胃肠道,其中膳食成分被微生物群转化,利用产生代谢物传播到全身器官。
血液充当体内分子的液体输送器, 特别是数以千计的循环代谢小分子,它们可以帮助我们了解体内生物过程状况,并且是研究冠状动脉疾病多因素性质疾病的宝贵来源。肠道微生物组积极参与血液代谢物的代谢。
几种肠道微生物群衍生的循环代谢物与心血管疾病相关:
三甲胺 N-氧化物
三甲胺 N-氧化物被确定为人类心血管疾病的标志物,进一步的证据表明在小鼠模型中具有促动脉粥样硬化性和促血栓形成。
硫酸吲哚酚
硫酸吲哚酚在细菌色氨酸酶降解色氨酸后在肝脏中产生,并被证明与动脉僵硬和外周血管疾病有关。
对甲酚
对甲酚是苯丙氨酸和酪氨酸的结肠细菌发酵产物,显示与心血管事件增加相关。
近期,以色列科学家招募了下列人群,采集其粪便和血清样本进行了全面的多组学分析,同时调查详细的医疗、生活方式和营养问卷等。
通过对粪便样本宏基因组测序(每个样本1000万 reads,约3G/样本)和对血清样本的进行非靶向质谱LC-MS测量了 961 种代谢物的水平,包括脂质、氨基酸、异生物质、碳水化合物、肽、核苷酸和大约 30% 的未命名化合物。
通过 Nightingale Health 的质子核磁共振 ( 1 H-NMR) 平台测量了另外 228 种血浆代谢物和比率,并使用了一个独立宏基因组数据集MetaCardis进行验证(该数据集样本来自于北欧血统队列,在地里区域上与该研究样本来源不同,这样可以分析遗传,饮食差异变量)。
MetaCardis数据集主要由四个主要群体组成:缺血性心脏病、健康对照组、代谢匹配的对照组和未经治疗的代谢受损对照组(详细数据集描述可以参看原文)
一、ACS的肠道微生物组特征
1. ACS 患者的变形杆菌丰度更高
这与之前的大多数研究结果一致,变形菌增多会导致处于炎症状态,是生态失调的标志。
20个在 ACS 或对照个体中显着富集的细菌,包括产丁酸盐的细菌如:梭菌属(Clostridium)、Anaerostipes hadrus嗜热链球菌(Streptococcus thermophilus)和Blautia菌属,以及Odoribacter splanchnicus 和大肠杆菌。
2. ACS患者队列中一种梭菌科的细菌物种 SGB 4712缺乏
在20 个显着富集的基因组中,鉴定到了一种以前未知的梭菌科细菌物种,索引为 SGB 4712。为了进一步验证该结果稳定和实用性,使用另外一个来自北欧血统地理上分布不同的队列,MetaCardis宏基因组数据集进行验证,与该研究结果一致,该物种的相对丰度随着具有 CAD 传统风险因素的种群逐渐减少。
3. SGB 4712关联15种显著差异的代谢物,其中包括降低心血管疾病风险的独立标志物——麦角硫因(ergothioneine,天然氨基酸)
对照组相比, 鉴定到SGB 4712 菌种与15 种循环代谢物的水平显着相关,在 MetaCardis 研究中,所有 15 种代谢物与 SGB 4712 的相关系数均可以重复,其中 10 种相关性仍然显著。
值得注意的是,SGB 4712与麦角硫因呈正相关,麦角硫因是一种天然存在的氨基酸,在体外显示对细胞应激源具有抗氧化和细胞保护能力,最近被证明是降低心血管疾病和人类死亡率风险的独立标志物。
此外,SGB 4712 与七种化学结构未知的化合物有关。其中包括 X-11315 和 X-24473,预测它们来自饮食,并与 SGB 4712 呈正相关。
图一 ACS 的微生物组和血清代谢组学特征
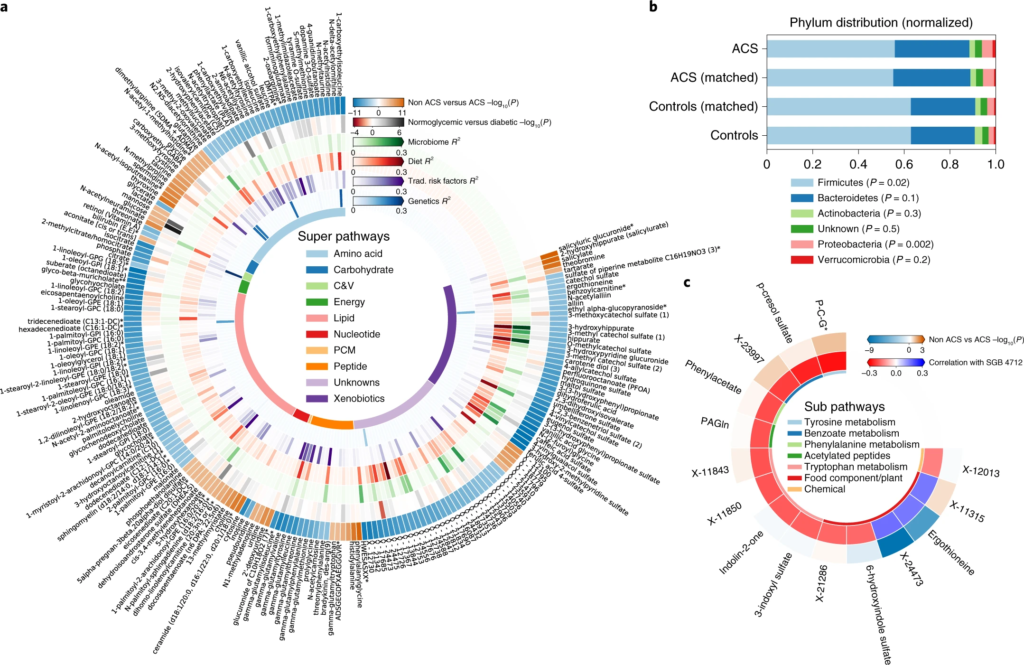
圆形热图显示 ACS 和非 ACS 对照组之间显着差异的前 200 种代谢物,与年龄、性别、BMI、吸烟状况和 DM 相匹配(方法)。每个切片代表一个代谢物,其名称显示在图表的外层周围。
这些结果突出了SGB 4712菌种在 CAD 发展中具有潜在的保护作用,由一系列循环血液代谢物介导,其中一些以前被证明在元生物途径中发挥核心作用,而另一些则未知。
因此,在实验研究中进一步验证后,这些代谢物可能会形成降低 CAD 风险的新目标。
二、ACS 的代谢特征因人而异
1. ACS 患者的血清代谢物水平个体化差异较大
虽然 CAD 患者具有共同的内表型,但他们通常表现出生物学上不同的疾病特征。为了更好地了解 ACS 的个体水平变异性,作者试图检查与非 ACS 对照的代谢偏差,并询问它们是否是个体特异性的。
计算了他们的个体偏差,并根据之前根据饮食、微生物组、传统风险因素和遗传学估计的 EV 对每个个体的前 100 个偏差代谢物进行加权。最后发现ACS 患者与其匹配对照的代谢偏差是因人而异的。
急性冠脉综合征患者的血清谱在血清代谢物水平上表现出广泛的扰动,包括533种显著改变的代谢物。
ACS的血清代谢组遵循一种主要的消耗模式,因为在对照组参与者中,358种代谢物(67%)的平均测量值较高。然而,这一趋势在主要的生物途径中并不一致。但是,与富含 ACS 的代谢物相比,饮食和微生物组在与 ACS 耗尽代谢物的偏差相关联方面更为显着(双尾 Mann–Whitney U-检验,P-value小于10 -20),这表明微生物组对 CAD 起保护作用。
值得注意的是,超过 90% 的显着扰动的代谢物无法用血糖状态来解释,这表明这种变化背后还有其他机制。所以进一步分析了其他系列综合因素(包括宿主遗传学、微生物组和饮食),得到一个重要发现就是:饮食和微生物组可以更好地解释 ACS 缺乏或含量低的代谢物,而传统的风险因素可以更好地解释 ACS 富集的代谢物。
图2 代谢偏差由潜在决定因素解释,并与临床参数相关
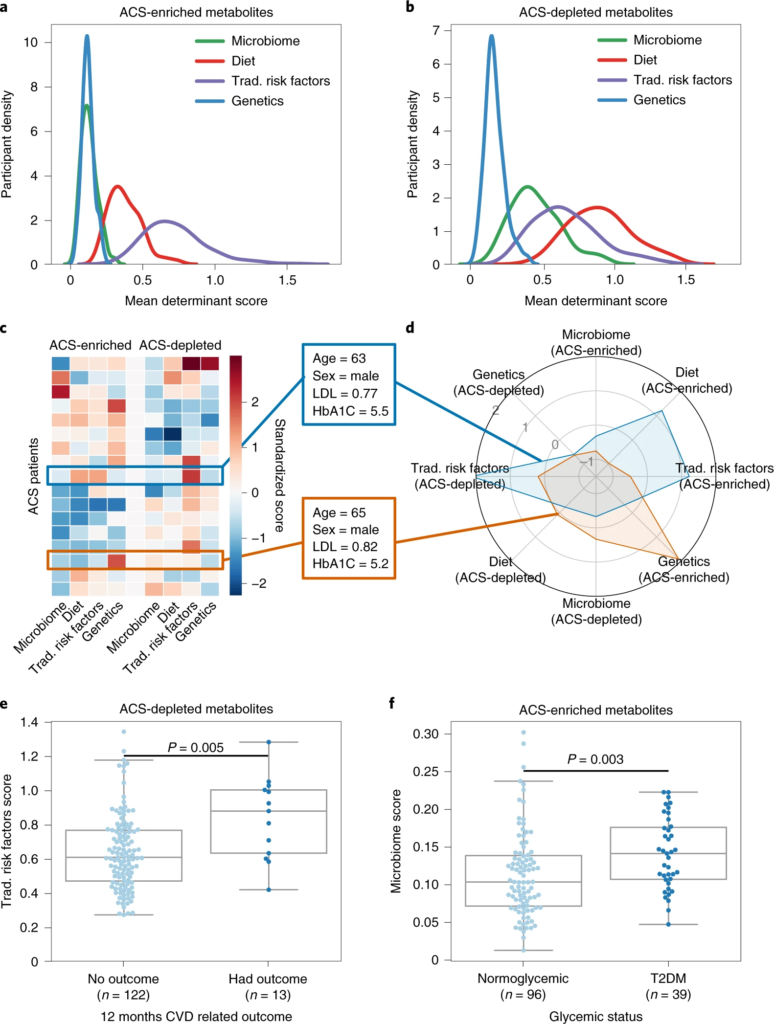
a、b、密度图显示 ACS 参与者的分布(y轴)与代谢物的潜在决定因素(微生物组、饮食、传统风险因素或遗传学)的平均加权R 2 ( x轴);富含 ACS 的代谢物。
2. 相似的临床特征,但其动脉粥样硬化负担的代谢机制却不同
虽然一些患者可能具有相似的临床特征,但他们的潜在生理状态和疾病轨迹可能不同。为了强调这种 CAD 患者的变异性,作者选择了 ACS 患者的常规危险因素的同质亚组。其中包括 17 名 60 至 70 岁的男性患者,低密度脂蛋白 (LDL) 在 0.70–1.30 mg ml -1范围内,糖化血红蛋白 (HbA1C) 低于 6%。尽管具有相似的临床特征,但该 ACS 患者亚组在代谢偏差方面表现出异质性。
三、微生物组在CAD早期阶段发挥作用
动脉粥样硬化是一种经过多年发展的进行性疾病,其中动脉粥样硬化斑块形成的每个阶段的特点是不同的病理过程。在早期阶段,血管壁上的动脉粥样硬化斑块的生长通常与代谢状态的损害有关。
为了解释每个代谢成分在 CAD 发展的时间轴上的参与,作者将个体代谢偏差的分析应用于代谢受损的对照(定义为 T2DM、高血压或血脂异常的诊断,或 BMI > 35),以及到非 ACS 个体的随机子集。
在比较这三组的分数时,我们发现分数分布存在一致的差异。与微生物组和饮食相关的代谢异常呈现出渐进的趋势,与对照组的随机子集相比,代谢受损的对照参与者的代谢物存在显着偏差。
这表明,微生物组和饮食对ACS的贡献可能是通过受损的代谢状态介导的,而不是代谢受损个体中尚未表现出的与传统风险因素和遗传学相关的代谢物异常。
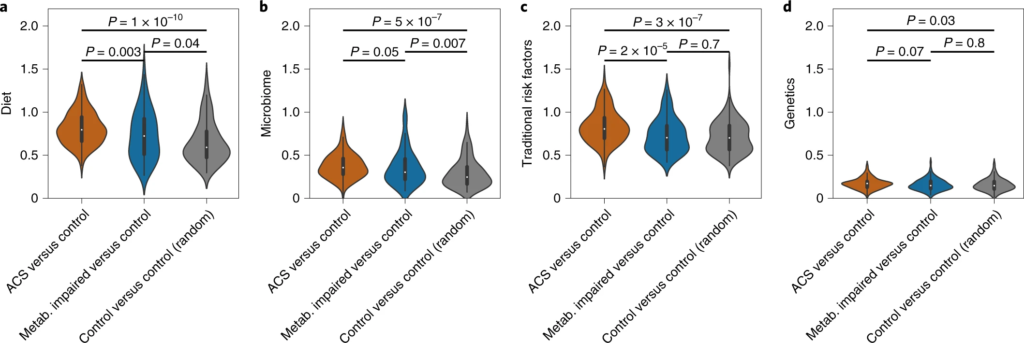
a – d,归因于饮食 ( a )、微生物组 ( b )、传统风险因素 ( c ) 和遗传学 ( d ) 的代谢偏差分数计算三个亚组:(1) ACS 个体 ( n = 135) 与非 ACS 对照与年龄、性别和 BMI 相匹配(橙色);(2) 患有代谢障碍的非 ACS 对照(定义为:诊断为 T2DM、高血压或血脂异常,或 BMI > 35;n = 102)与其他年龄、性别和 BMI 匹配的非 ACS 对照(蓝色);(3) 一组随机的非 ACS 个体 ( n = 132) 与其他匹配年龄、性别和 BMI(灰色)的非 ACS 对照。
四、血清代谢组学预测ACS患者 BMI 更高
肥胖是 CAD 的主要独立危险因素,影响已知的危险因素,如血脂异常、高血压、葡萄糖耐受不良和炎症状态,以及可能尚未认识到的机制。BMI 测量被用作肥胖的标志和代谢健康的指标。
为了研究肥胖作为 CAD 的独立危险因素,该研究设计并彻底验证了基于血清代谢组学的 BMI 模型,并表明较高的预测 ΔBMI 对应于更广泛的动脉粥样硬化疾病。
作者分析了CAD 患者的 BMI-代谢组平衡是否以及如何被破坏。使用了梯度提升决策树 (GBDT) 算法预测 BMI,结果表明在非ACS受试者中发现的代谢组-BMI模式在ACS患者中受到干扰。
为了研究这些扰动,作者测试了对照组和 ACS 测试集中预测和测量 BMI 之间的差异,这里称为 ΔBMI。结果发现,与非 ACS 受试者相比,该研究的模型预测 ACS 的 ΔBMI 更高。
为了验证这些结果的稳健性,作者试图根据其他类型的代谢组学数据和独立队列来复制这些发现。将相同的预测程序应用于基于 NMR 的代谢组学数据,并观察到ACS 和对照之间 ΔBMI 的更大差异,应用于为发表的MetaCardis 队列数据中得出在所有 BMI 范围内,与血糖正常的缺血性心脏病患者相比,患有糖尿病的缺血性心脏病患者的 ΔBMI 显着更高。
进一步分析推断哪些特定代谢物是 ACS 患者高 ΔBMI 的主要驱动因素,发现两种脂质在对照组中与 BMI 呈负相关,后者在患有更广泛疾病的患者中也显着减少,这两种脂质分别是:
1-(1-enyl-palmitoyl)-2-oleoyl-GPC (P-16:0/18:1)
1-(1-enyl-palmitoyl)-2-linoleoyl-GPC (P-16:0/18:2)
最近的研究表明,脂质1-linoleoyl-GPC (18:2) 与肥胖和 T2DM呈负相关,并且脂质水平的增加显着降低了T2DM的风险。该研究发现 1-linoleoyl-GPC (18:2) 和 1-(1-enyl-palmitoyl)-2-linoleoyl-GPC (P-16:0/18:2) 在对照组中与 BMI 呈负相关,并且在患有更广泛 CAD 的患者中显着耗尽,这表明这些代谢物可能作为降低 CAD 风险的潜在靶点。
此外,两种代谢物都含有一条亚油酸链,一种必需脂肪酸,与 T2DM 风险呈负相关。然而,这些假设应在干预性研究中进一步检验。
迄今为止,大多数研究都集中在寻找在 CAD 患者中增加的新代谢物,而该研究对 199 名 ACS 患者进行了全面的多组学分析结果强调, ACS 的代谢组学特征是缺乏多种血清代谢物,其中许多与饮食和微生物组有关。
其中一个重要的发现是以前未知的细菌物种 SGB 4712,它在 ACS 患者和独立验证队列中都显着缺乏或偏低。通过进一步将这种细菌与心脏毒性和心脏保护代谢物的水平联系起来,证明了特定细菌基因组的缺失可能与 CAD 风险增加相对应,并提出在后续干预研究中评估的具体目标。总体而言,这些发现因此为 CAD 患者的预测甚至治疗提供了一种新方法。
迄今为止,大多数研究都对 CAD 患者进行了批量分析,寻找人群水平的风险因素,而不是关注个体水平的生物变异性。在这项研究中,作者使用全面的代谢组学和微生物组分析,呈现了 CAD 内部变异性的深度映射。总之,结果揭示了新的范式和治疗方向。
参考文献:Talmor-Barkan Y, Bar N, Shaul AA, Shahaf N, Godneva A, Bussi Y, Lotan-Pompan M, Weinberger A, Shechter A, Chezar-Azerrad C, Arow Z, Hammer Y, Chechi K, Forslund SK, Fromentin S, Dumas ME, Ehrlich SD, Pedersen O, Kornowski R, Segal E. Metabolomic and microbiome profiling reveals personalized risk factors for coronary artery disease. Nat Med. 2022 Feb;28(2):295-302. doi: 10.1038/s41591-022-01686-6. Epub 2022 Feb 17. PMID: 35177859.
谷禾健康
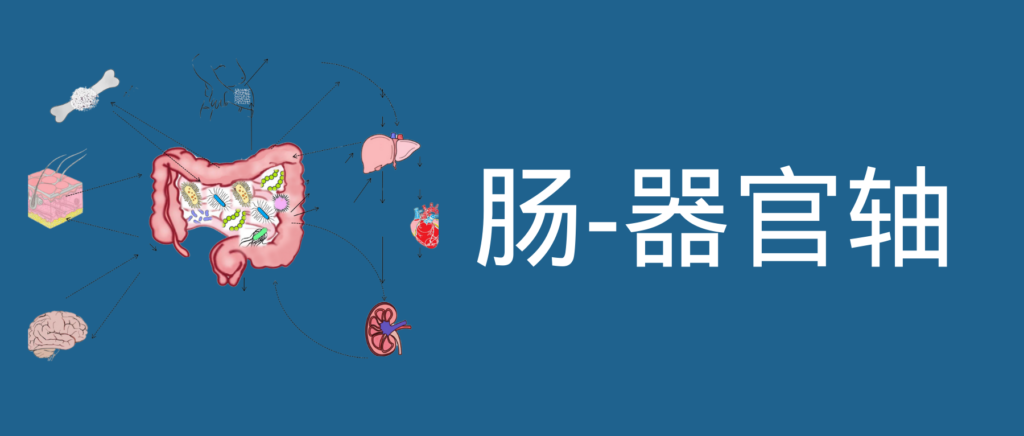
肠道菌群和宿主免疫代谢系统之间的复杂相互作用会影响与其他器官相关的身体功能,它们之间形成“轴”。
这种串扰通过宿主和微生物及其代谢物之间的直接或间接相互作用发生。宿主-微生物免疫代谢轴是宿主细胞途径和各种微生物群之间的多向通信系统。在这个轴内,不同的微生物通过产生胆汁酸、胆碱、短链脂肪酸、神经递质、小分子、有毒物,炎症因子等来调节生理代谢过程,从而对健康和疾病发生产生重要的影响。
图1 在肠道、相关微生物群和各种器官之间的双向或多向通信连接(轴)
Ahlawat S,et al.,Lett Appl Microbiol. 2021
肠道菌群在个体之间或在同一个人的一生中是不同的,并且受到各种因素的影响,包括饮食、年龄、生活方式、药物、疾病状态等。受这些因素影响的动态的肠道菌群成分可能会通过改变多样性或组成来影响健康或疾病风险。
因此,胃肠道和相关微生物群的一个复杂综合的对话机制在越来越多的研究中被证实,肠道以及肠道菌群与各个器官的对话交流机制,即“肠道-器官轴”,在维持各个器官的健康方面变得越来越重要。
本文主要讨论了肠道和人体重要器官之间的双向关系,以及在各类疾病中肠道菌群发挥的作用。
本文主要介绍的各类肠轴如下:
01 肠-大脑轴
02 肠-肾脏轴
03 肠-肝脏轴
04 肠-骨骼轴
05 肠-皮肤轴
06 肠-脂肪轴
07 肠-心脏轴
“肠道作为“第二大脑”影响情绪和行为
大脑和肠道的双向沟通,构成了“肠-脑轴”的基础。肠-脑轴相互作用的证据来自各种研究,涉及无菌动物模型、抗生素、益生菌、中枢神经系统疾病和功能性胃肠道疾病的生态失调。
通过建立肠-脑轴,肠道菌群可以影响大脑,如行为、食欲调节、肠道糖异生和5-羟色胺代谢。
肠道菌群的改变与焦虑、多发性硬化、自闭症谱系障碍、帕金森病等多种神经系统疾病有关。
在了解肠道菌群是如何影响肠脑轴之前,我们先了解一下肠-脑轴是怎么回事。
肠道-大脑轴将大脑的认知和情感中心与外周肠道功能(即免疫激活、肠道反射、肠道通透性和肠道内分泌信号)联系起来。
肠-脑轴涉及:
中枢神经系统(CNS),包括大脑和脊髓;
自主神经系统(ANS)及其交感和副交感肢体;
肠神经系统(ENS);
下丘脑-垂体-肾上腺轴(HPA)
自主神经系统驱动从管腔到中枢神经系统的传入信号,通过脊髓、肠道和迷走神经通路传输,以及从中枢神经系统到肠壁的传出信号。
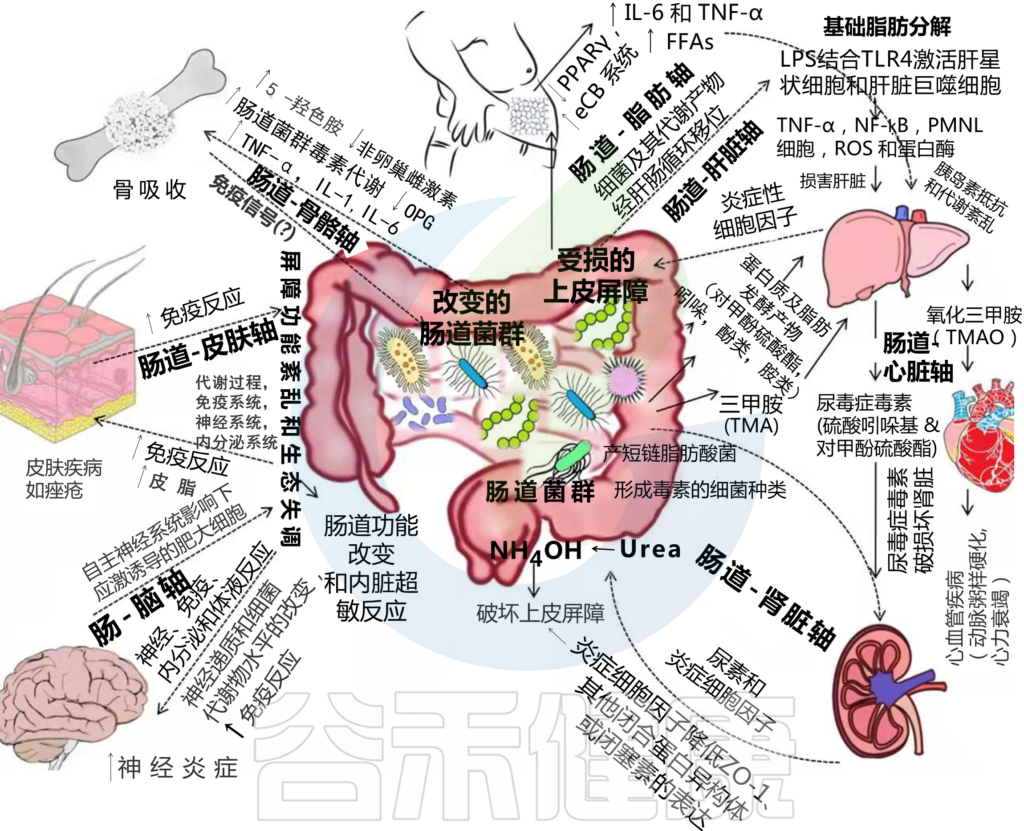
HPA轴涉及记忆和情绪反应,协调生物体对压力源的适应性反应。促炎细胞因子增加的环境应激通过下丘脑分泌促肾上腺皮质激素释放因子(CRF)触发该系统。CRF刺激垂体分泌促肾上腺皮质激素(ACTH),进而导致肾上腺分泌皮质醇,影响包括大脑在内的各种器官(下图)。
图2 “肠-脑轴”(GBA)结构示意图
Ahlawat S,et al.,Lett Appl Microbiol. 2021
因此,不同的通讯线路一起允许大脑控制肠道效应细胞的活动,这些细胞也受肠道菌群的影响。
关于肠脑轴,在我们之前的文章中也有详细介绍:深度解读 | 肠道菌群和中枢神经系统的关系
肠道菌群通过代谢和神经内分泌途径与肠道细胞和肠神经系统以及中枢神经系统进行沟通。
肠-脑轴的破坏控制着肠道功能的变化,如分泌和运动,导致内脏过敏,从而导致肠内分泌和免疫系统的细胞变化。
如果没有微生物定植会发生什么?
改变神经递质的表达(包括5-羟色胺、褪黑素、乙酰胆碱、GABA和组胺)和肠道感觉运动功能,如减少肠道运输、迁移性运动复合体循环复发、远端繁殖、延迟胃排空、扩大盲肠大小…
可能还有各种想象不到的后果,总之,肠道菌群在调节肠道和大脑功能方面起着至关重要的作用。
以细菌物种特异性的方式对动物进行定植可以恢复所有这些异常。
微生物群通过在肠腔中产生局部神经递质和儿茶酚胺的生物活性形式来影响肠神经系统活性。短链脂肪酸等细菌代谢物通过刺激交感神经系统、粘膜5-羟色胺释放、记忆和学习过程影响肠神经系统。
肠神经系统是什么?
肠神经系统主要由肠胶质细胞(EGCs)组成,类似于中枢神经系统中的星形胶质细胞。肠上皮细胞分布于肠壁,包括粘膜固有层。
肠神经系统有什么功能?
——肠神经系统自主调节胃肠道的生理和功能
肠神经系统可以自主调节胃肠道的生理和功能,并通过迷走神经通路与中枢神经系统进行双向沟通,从而形成“肠-脑轴”。
随着肠神经胶质网络的发展,肠道菌群在肠神经系统的调节中起着关键作用。肠神经胶质网络通过钙依赖性信号传导在调节胃肠功能方面发挥作用(如血流、肠道运动、免疫炎症反应和外分泌/内分泌)。
所以说,如果肠神经胶质细胞出了问题,就会导致胃肠道疾病,如炎症性肠病、运动障碍、PD等神经退行性疾病和感染引起的肠道炎症。
通过Toll样受体(TLR)发出的LPS信号在肠道菌群和肠神经系统发育之间起着中介作用。
——肠神经系统是肠内神经肽合成的关键来源
通过信号传递到远端器官,如大脑,产生肠外作用,而远端器官有迷走神经上的神经肽受体。
这些神经肽可以通过影响肠道菌群成分来调节肠道内稳态。它们水平的改变导致各种肠道失调和肠道炎症相关的神经精神障碍。例如,自闭症和重度抑郁症分别与降钙素基因相关肽(CGRP)、神经肽Y(NPY)和P物质(SP)(即神经传递介质)的循环水平改变有关。
微生物群调节血清素能系统,因为在无菌动物的边缘系统中发现血清素和相关代谢物水平的改变。
此外,肠道菌群具有多种氧化还原酶,可以调节神经递质的水平。例如,漆酶(一种多铜氧化酶或MCO)调节肠道中合成的血清素的数量,参与其代谢,在肠-脑轴中起重要作用。漆酶可将儿茶酚胺氧化为活性氧(ROS)和多巴胺奎宁(DAQ)。
研究发现,发现生成的 ROS 受损细胞和 DAQ, 与 帕金森患者的线粒体功能障碍和痴呆有关。微生物群通过迷走神经与大脑沟通,迷走神经将信息从管腔传输到中枢神经系统(图2)。
肠道细菌具有神经递质的表面受体,使肠道和中枢神经系统之间的联系更加有效。
例如,Pseudomonas fluorescence 具有GABA受体,而肾上腺素和去甲肾上腺素受体存在于大肠杆菌O157:H7上(图2)
肠道菌群调节肠道屏障和传入感觉神经,增强其兴奋性,从而调节肠道运动和疼痛感知。
肠道菌群通过激素调节饱腹感
肠道菌群也可能通过从肠内分泌细胞释放各种生物活性肽与肠-脑轴相互作用。例如,甘丙肽触发HPA轴的活动;因此,释放CRF和ACTH可增强肾上腺皮质的糖皮质激素释放,或直接刺激肾上腺髓质的去甲肾上腺素和肾上腺皮质细胞的皮质醇分泌。
几种外围使食欲减退的激素,如肽YY(PYY)、胰高血糖素样肽-1(GLP-1)、胰岛素、瘦素和阿片黑皮素原,以及含有可卡因和安非他明调节转录物的神经元,可诱导饱足感。然而,含有NPY和刺鼠相关肽(AGRP)的神经元的ghrelin(胃饥饿素)会增加饥饿感。
肠道菌群影响粘膜免疫激活
这可能部分由蛋白酶介导。蛋白酶是粘膜和肠神经损伤的终末期效应物,在几种肠道免疫介导的疾病中上调。
大脑调节来自免疫细胞、神经元和胃肠道嗜铬细胞的信号分子的分泌,这可能会影响微生物群的组成。
肠-脑轴的任何失调都会通过扰乱正常粘膜栖息地,影响肠道菌群。胃肠道转运的变化对营养物质输送到肠道微生物群有着深远的影响。
由于自主神经系统影响下应激诱导的肥大细胞导致肠道通透性增强,因此微生物群组成有所不同。这导致类胰蛋白酶和组胺失衡。微生物群的改变也是由于胃肠道Paneth细胞释放抗微生物肽,如α-防御素。
总之,肠道菌群对中枢神经系统的发育和功能有重大影响。同样,中枢神经系统调节胃肠道的生理,最终调节肠道环境。

Ahlawat S,et al.,Lett Appl Microbiol. 2021
许多神经系统疾病包括多发性硬化、帕金森病、阿尔茨海默病、癫痫症、中风与脑损伤等在这两篇文章中有详细介绍:
肠道微生物组在人类神经系统疾病中的作用
最新研究速递 | 柳叶刀:肠道微生物群在神经系统疾病中的作用
“调节体内废物的水平
肠道和肾脏也具有双向协同关系。
在一个方向上,尿毒症毒素如三甲胺-N-氧化物(TMAO)、对甲酚硫酸盐和吲哚硫酸盐从微生物代谢中产生;
而在另一个方向上,尿毒症会破坏肠道菌群组成和代谢。
这种双向交流之间的任何干扰都会导致各种严重并发症,如慢性肾病(CKD)、终末期肾病(ESRD)和脓毒性急性肾损伤(AKI)。
膳食纤维:通过菌群发酵成短链脂肪酸,修复肾上皮细胞
膳食纤维是菌群的碳水化合物来源。结肠微生物群的组成受到饮食及其在小肠中的同化作用的显著影响。膳食纤维逃避小肠的消化过程,成为结肠菌群碳水化合物的主要来源。
膳食纤维被结肠微生物群发酵成短链脂肪酸。短链脂肪酸在保持肠道上皮完整性和能量稳态方面发挥着重要作用。它们通过改善线粒体生物发生来修复肾上皮细胞的缺氧损伤。
膳食蛋白质:通过菌群发酵,形成尿毒症毒素,加剧肾病
抵抗上消化道消化的膳食蛋白质是结肠微生物群的氮源。这些蛋白质在结肠中的命运主要取决于结肠菌群生长和发育所需能量的可用性,而结肠菌群主要来自碳水化合物发酵。
如果碳水化合物的利用率较高,蛋白质及其中间产物要么被同化为细菌生物量,要么在碳水化合物缺乏的情况下被梭菌和拟杆菌发酵为对甲酚、吲哚、酚和胺。
蛋白质发酵产物经过进一步加工,形成尿毒症毒素,如对甲酚硫酸盐和硫酸吲哚酚。
——尿毒症毒素如何导致肾病发展?
这些毒素由于与白蛋白有较高的亲和力(非共价相互作用)而在血液中循环,并由肾小管分泌物释放出来。如果尿毒症滞留溶质在体内积聚,则会增加肾小球硬化的发生率和肾脏疾病的进展。因此,它们在血液中的浓度可以用来衡量肾脏的功能效率。
硫酸吲哚酚和对甲酚硫酸盐的其他毒性作用包括炎症反应增加、内皮功能障碍、血管钙化、氧化应激增强、红细胞生成减少、细胞衰老增加、血栓形成、动脉粥样硬化形成,左心室肥厚、胰岛素抵抗、肾小管-间质纤维化和肾素-血管紧张素-醛固酮系统激活。
膳食脂肪:高脂饮食导致TMAO升高,与慢性肾病进展直接相关
胆碱、肉碱和卵磷脂是膳食脂肪的主要成分。哺乳动物缺乏打破这些脂肪成分的氰化物键所需的酶。
然而,结肠微生物群有TMA裂解酶,可以破坏氰化物键。胆汁中的TMA裂解酶和肝酶(即含黄素的单加氧酶)的联合作用导致肉碱和胆碱形成TMAO。
2021年发表在Science期刊上的一篇文章指出,高脂饮食会损害结肠上皮细胞线粒体的功能,使肠道氧气和硝酸盐的浓度增加,促进大肠杆菌的生长以及对胆碱的分解,导致TMA水平增加,最终导致循环中有害代谢物TMAO水平的升高。
——TMAO的升高会发生什么?
TMAO与其他尿毒症毒素一样,进入身体循环,并由肾脏释放。TMAO增加与慢性肾病进展直接相关。与健康对照组相比,终末期肾病患者体内的TMAO浓度可高出20倍。
TMAO高导致有害后果,如血小板活性增加、血栓形成潜能、肾小管间质纤维化和动脉粥样硬化的发展。
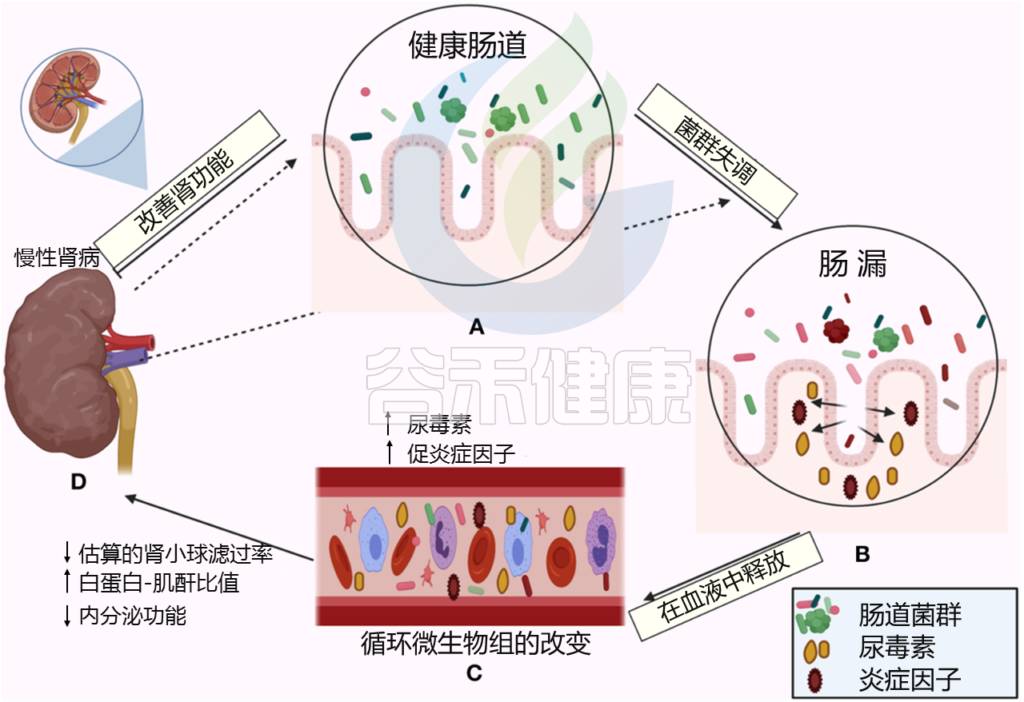
Al Khodor D, et al., Frontiers in Medicine,2022
← 在一个方向上,肠道菌群影响肾脏:
(A)健康的肠道
(B)肠道微生物失调和破坏粘膜层
(C)释放血液中炎性因子和炎症级联的开始,尿毒症毒素积累
(D)估计的肾小球滤过率下降(eGFR),白蛋白肌酐比值(ACR)升高,肾脏内分泌功能丧失
→ 在另一个方向,慢性肾病驱动肠道内的生态失调(虚线箭头所示),并引发炎症级联
尿毒症患者的微生物群组成与健康人不同。据观察,在接受血液透析的尿毒症患者中,需氧菌如肠杆菌和肠球菌增加很多倍,而厌氧菌如双歧杆菌则减少。
5期慢性肾病患者体内含有短链脂肪酸形成酶的细菌较少;然而,其中含有更多的脲酶、尿酸酶、吲哚和对甲酚产生菌。
另一项研究发现,在区分慢性肾病患者和健康对照组方面表现最好的两种菌:
Lachnospira 和 Ruminocococus gnavus
慢性肾病进展和血液透析和Holdemanella、巨单胞菌、普雷沃氏菌属Prevotella和Scardovia有关。
关于慢性肾病的进一步报告表明,慢性肾病是一种蛋白质发酵增加的状态,与肾功能恶化有关。发现这些细菌蛋白发酵产生的代谢物(对甲酚硫酸盐、吲哚硫酸盐和苯乙酰谷氨酰胺)与肾功能呈负相关。
结肠微生物群通过其代谢产生尿毒症毒素,其失衡可能导致上皮屏障损伤。尿毒症毒素的增加会降低紧密连接蛋白的表达,如紧密连接蛋白(ZO-1)、闭合蛋白、claudin-1。
急性肾损伤也有各种原因引起的,有感染性的,败血症性的,脓毒性的等。
感染性急性肾损伤:
肾功能受损->肠道通透性增高->全身炎症->肾功能进一步损伤
患者的炎性细胞因子增加,肾功能受损,导致肠道损伤。细胞因子水平的增强作用于胃肠道壁的连接复合体;因此,通过改变紧密连接蛋白的表达,导致其通透性增高。此外,增加的肠道通透性会以正反馈方式放大全身炎症反应。增强的全身炎症进一步促进肾功能障碍。
败血症性急性肾损伤:
水分潴留->肠壁水肿->尿素代谢->破坏屏障->细菌移位->炎症->衰竭
该类患者中,钠、尿素、尿毒症溶质和水的潴留是由于其肾功能障碍引起的。过多的水分滞留导致肠壁水肿显著增加。来自体循环的尿素扩散到胃肠道腔,肠道细菌脲酶在那里代谢尿素。
尿素转化为氨[CO(NH2)2+H2O→ CO2+2NH3]
进一步代谢为氢氧化铵(NH3+H2O→ NH4OH)
此后,肠道菌群产生的氢氧化铵(NH4OH)作用于连接蛋白,破坏上皮屏障(图1)。
受损的上皮屏障导致肠腔毒素流入,同时细菌从肠腔转移到肠系膜淋巴系统和体循环。这会促进局部和全身炎症,从而导致多器官衰竭和死亡。
脓毒性急性肾损伤:
外部+内部因素->菌群变化
患者的微生物组成发生改变,这可能是由于炎性细胞因子或上皮屏障受损。肠道菌群及其代谢产物能够改变胃肠道细菌细胞受体的表达,从而改变微生物组成。
肠道微生物组成也受到脓毒症患者肠道生理学各种变化的影响,这些变化可能是由于肠外营养和抗生素等外部因素,也可能是肠道渗漏和全身炎症等内部因素。
当使用微生物源性短链脂肪酸治疗时,败血症性AKI患者的肾功能得到改善。这种改善与低水平的细胞浸润/激活、炎症、氧化性细胞应激和凋亡有关。
一项确定蛋白质发酵代谢物对肠道微生物特征的作用的研究显示,梭菌有56个成员,在早期肾脏疾病中,以Christensenellae、Ruminococaceae和Lachnospiraceae为代表。
对终末期肾病患者的类似研究表明,从普雷沃氏菌向拟杆菌转变,产丁酸菌包括 Roseburia、粪球菌、梭菌、粪杆菌、普雷沃氏菌减少。
此外,他们还增加了来自下列菌的OTU:Brachybacterium, Catenibacterium,
Enterobacteriaceae, Moraxellaceae,
Nesterenkonia, Halomonadaceae,
Pseudomonadaceae, Polyangiaceae, Thiothrix
据报道,钙肾结石患者粪便微生物多样性降低,粪便杆菌、大肠杆菌和肠杆菌的代表性显著降低。
此外,在泌尿系结石病中,抗生素引起的泌尿道微生物群的长期变化从乳杆菌(健康保护)转变为肠杆菌科(促结石)。

Ahlawat S,et al.,Lett Appl Microbiol. 2021
扩展阅读:慢性肾脏病中的人类微生物组:一把双刃剑
“酒精不一定是导致肝脏退化的罪魁祸首
肠道和肝脏之间的双向通讯网络涉及这些器官之间的相互关系,肝脏在这些器官中产生有益物质并被肠道吸收。
肠道菌群变化使肠道通透性增加,内毒素移位,肝毒素进入肝脏。
“肠道-肝脏轴”对于理解各种肝脏疾病的病理生理学至关重要。
肠道菌群如何与肝脏产生联系?
肝脏通过门静脉从肠道的静脉流出接收近70%的血液供应,因此,它持续暴露在肠道菌群及其代谢产物。
在健康个体中,进入肝脏的微生物代谢产物,如氨、乙醛和乙醇,由肝脏巨噬细胞(库普弗细胞)代谢。因此,肠道菌群对肝脏生理有很大影响。
肠道菌群变化如何导致肝脏受损?
炎症和门脉高压或肠道微生物组成变化导致的肠道上皮改变增加了肠道通透性。肠道通透性增加导致内毒素移位,导致肝脏中各种促炎基因和细胞因子的转录激活。由于肠道屏障受损,大量细菌及其代谢物如脂多糖(LPS)通过肝肠循环进入肝脏。
在激活LPS时,发生了一系列级联事件,通过NF-kβ介导的机制产生促炎症细胞因子,如TNF-α,这些细胞因子与肝损伤有关(图1)。
LPS是一种肝毒素;因此,暴露于肝脏会导致形态和功能改变。诱导的变化导致急性炎症反应和多形核细胞的积聚,通过从颗粒中释放蛋白酶、活性氧代谢物和其他酶,进一步加剧肝脏损伤。
肝硬化合并门脉高压导致肠道运动受损,促炎细胞因子释放增加,上皮通透性增加,从而影响肝脏。
对于理解各种肝脏疾病的病理生理学至关重要,如非酒精性脂肪肝(NAFLD)、脂肪性肝炎(NASH)、酒精性肝病(ALD)、肝癌发生和肝性脑病(HE)、急性或慢性肝衰竭、肝硬化的进展和并发症。
非酒精性脂肪肝是代谢综合征的一种肝脏表现,其原因是肥胖的普遍存在。
原发性脂肪性肝炎与肥胖、高脂血症和2型糖尿病(T2D)等代谢表现相关。而继发性脂肪性肝炎则是由于空肠回肠旁路手术、全肠外营养、快速减肥、脂肪营养不良或威尔逊氏病以及药物摄入所致。
肠道菌群在能量摄取中的作用
有趣的是,瘦肉型和肥胖型个体的肠道菌群成分不同,肥胖型个体具有的厚壁菌、普雷沃菌、卟啉单胞菌和更少的拟杆菌,这表明肠道菌群在肠道内容物的能量摄取中的作用。
此外,肠道菌群对能量收集和脂肪储存途径有影响,表明它们在胰岛素抵抗和相关代谢疾病的发展中起直接作用。在肥胖个体中,双歧杆菌与血清丙氨酸转氨酶水平呈负相关,丙氨酸转氨酶可作为非酒精性脂肪肝的标志物。
肠道通透性增加、小肠细菌过度生长
肠细胞释放乳糜微粒是脂肪代谢(代谢性内毒素血症)的结果,支持含LPS细菌的生长和移位。它会导致肠道失调和促炎细胞因子的释放(即由于LPS、乙醇和内毒素等细菌产物与TLR之间的相互作用),以应对肥胖。因此,非酒精性脂肪肝的发展主要取决于TLR-4或TLR-9和TNF-α受体的流行。
然而,非酒精性脂肪肝导致脂肪性肝炎的发生过程尚不清楚,但研究表明,产乙醇的肠道菌群的改变导致脂肪性肝炎的发生。肥胖患者的肠道菌群改变可诱导瘦素产生,通过STAT3信号上调CD14。这会导致对低剂量LPS的更高反应性,导致脂肪性肝炎中的肝脏炎症和纤维化。
非酒精性脂肪肝中的肠道菌群变化
在一项对86名经活检证实的非酒精性脂肪肝患者(72名患者患有轻度/中度非酒精性脂肪肝,14名患者患有晚期纤维化)的研究中,在轻度/中度非酒精性脂肪肝患者中,厚壁菌、Eubacterium rectale、普通拟杆菌Bacteroides vulgatus 数量丰富,而变形菌门,晚期(3-4期)纤维化患者中大肠杆菌和普通拟杆菌的比例过高。与轻度/中度非酒精性脂肪肝相比,晚期纤维化患者下列菌群显著减少:
Eu. rectale, Ruminococcus obeum , R. obeum
另一项针对非酒精性脂肪肝患者的研究显示,Alistipes和Prevotella的丰度较低。厌氧菌属、大肠杆菌属、链球菌属和乳酸杆菌属在这些患者中更为丰富。
紧密连接变宽,微绒毛排列异常,T淋巴细胞减少,TNF-α、IFN-γ和IL-6升高,揭示了肠道菌群介导的炎症在非酒精性脂肪肝发病机制中的重要性。
其他各类非酒精性脂肪肝中肠道菌群变化
一项针对100名患有2型糖尿病的非酒精性脂肪肝患者的研究中,50名糖代谢正常的非酒精性脂肪肝患者和60名对照组报告,患有2型糖尿病的非酒精性脂肪肝患者的双歧杆菌显著减少。然而,这两组患者的多形拟杆菌丰度均较低,而Eu. rectale ,Lactobacillus 较高。
在一项研究中,使用定量实时PCR进行肠道菌群肠道菌群结构分析,涉及11名单纯性脂肪变性(SS)患者、22名非酒精性脂肪性肝炎(NASH)患者和17名健康对照者(HC)。与单纯性脂肪变性患者和HC患者相比,脂肪性肝炎患者中的拟杆菌显著减少,而与SS患者相比,脂肪性肝炎患者中的Clostridium coccoides 更丰富。
另一项针对22名脂肪性肝炎患者、25名肥胖者和16名对照者的研究显示,肥胖和脂肪性肝炎患者的拟杆菌数量增加,厚壁菌数量减少。与对照组相比,脂肪性肝炎患者的放线菌丰度较低;然而,肥胖组和脂肪性肝炎组之间的变形菌存在显著差异(表1)。
肠道通透性增加、内毒素进入肝脏、促炎
酒精性肝病(ALD)是一系列肝脏疾病,包括脂肪肝、脂肪变性、急性酒精性脂肪性肝炎,酒精性肝纤维化和肝硬化是由成瘾性饮酒引起的,酒精及其代谢物(如乙醛)通过产生ROS导致肝损伤。
它们能够破坏上皮细胞紧密连接,从而导致肠道通透性增加。这会导致细菌易位,增加内毒素、LPS、细菌DNA和其他代谢物通过门静脉进入肝脏。库普弗细胞激活在ALD的发病机制中起着核心作用,LPS通过TLR-4或TLR-9激活库普弗细胞,导致促炎细胞因子的释放。
LPS、TLR-4和炎症细胞因子通过生长因子-β信号激活星状细胞,导致纤维化,这是一个的渐进过程。
酗酒者:
人类酗酒者的肠道菌群与健康对照组不同,前者的拟杆菌科减少,而后者的拟杆菌科增加。
酒精依赖综合征和酒精性肝硬化:
一项关于酒精依赖综合征(ADS)和酒精性肝硬化(ALC)的研究报告,肠杆菌科、拟杆菌、普氏杆菌、粪杆菌、克雷伯菌、乳球菌增加。然而,通过对肠道群落结构的比较,可以确定这两个群体之间差异丰富的分类群。尤其是,ADS患者的阿克曼病、粪球菌、未分类梭状芽胞杆菌显著减少。然而,ALC表现为拟杆菌、Blautia、双歧杆菌、链球菌、乳酸杆菌增加,普雷沃氏菌属、Paraprevotella、Alistipes 减少(表1)。
肝硬化:
肝硬化患者低水平的胆汁酸分泌和门脉高压会影响肠道菌群的组成和生长。肝硬化患者肠道内的病原体(链球菌科和肠杆菌科)增加,有益细菌(双歧杆菌和乳酸菌科)减少,从而导致生态失调。进一步的报道表明,拟杆菌门、变形菌门和毛螺菌科的比例降低;然而,肝硬化患者中梭杆菌类、肠杆菌科、韦荣球菌科和链球菌科的比例增加。
在另一项研究中,在肝硬化患者中观察到厚壁菌门比例降低,链球菌和韦荣球菌数量增加。在这些患者中,生物失调导致其他严重并发症,如菌血症、HE伴SIBO和肠道通透性增加。他极大地影响着生活质量;由于器官损伤和微生物产生的有毒物质(主要是氨,其他还有酚类、硫醇、苯二氮卓、短链和中链脂肪酸等)的综合作用,以认知功能受损为特征。
TH17与肠道菌群相互作用
肠道菌群在肝癌发生中的作用也很明显,研究表明,肠道菌群可以减少无菌小鼠的肝癌发生,丙酸盐可以抑制肝癌细胞的增殖。产生肿瘤内IL-17的T辅助细胞(Th17)通过与肠道菌群相互作用在肠道中生成。发现它们与肝细胞癌患者的不良预后相关,可能是由于血管生成和肠道菌群肿瘤生长的进展。
因此,从上述讨论中可以看出,不是单个微生物而是微生物失调会导致几种肝脏相关疾病。因此,可以得出结论,肠道菌群在通过“肠道-肝脏轴”维持个体健康方面起着关键作用。

Ahlawat S,et al.,Lett Appl Microbiol. 2021
扩展阅读:深度解析 | 肠道菌群与慢性肝病,肝癌
“微生物代谢产物和骨骼健康
肠道菌群及其衍生分子对骨骼健康有影响。
肠道菌群对骨密度、骨强度、养分吸收产生影响。
饮食影响肠道菌群,从而影响骨骼健康。
骨骼:身体的支撑框架,细胞因子的仓库
人体骨骼对身体的整体功能起着至关重要的作用。除了作为身体的特殊支撑框架,骨骼还保护重要器官,充当钙稳态的矿物质储库,为骨髓(脂肪储存和血液形成)提供环境,并且是细胞因子和生长因子的仓库。
有趣的是,肠道菌群及其衍生分子对骨骼健康有影响。
最近的报告揭示了肠道和骨骼健康之间通过“肠道-骨骼轴”的复杂关联。在宫内和产后早期,暴露或限制环境因素可调节生长迟缓、骨矿化以及身体和肠道微生物组成。
肠道菌群对骨密度和骨强度的影响
各种临床前试验显示,乳酸杆菌是主要影响因素。骨丢失和肠道细菌过度生长验证了肠道、其微生物群和骨骼健康之间的沟通。肠道菌群通过各种潜在机制调节骨骼生长,如营养吸收、免疫系统成熟、释放各种代谢产物、改变胃肠道通透性、肠源性血清素和LPS诱导的全身炎症。
肠道菌群对养分吸收有重大影响
例如,胃肠道中长双歧杆菌和罗伊氏乳杆菌水平的增加通过钙、磷和镁等矿物质吸收水平的提高导致骨密度增加。肠道菌群在维生素B和K的合成中起着重要作用,维生素B和K对骨骼健康调节和胆汁酸代谢至关重要。
胆汁酸调节钙的吸收过程,就像脱氧胆酸抑制钙的吸收一样;然而,熊去氧胆酸会提高钙吸收过程。
饮食->肠道菌群->营养吸收->进一步影响骨骼
饮食对肠道菌群(碳水化合物作为主要能源)的组成有很大影响;因此,对营养吸收过程的影响进一步影响骨骼健康。有效的蛋白质含量对骨骼生长至关重要,而高蛋白饮食会导致胃肠道中甲烷和硫化氢等毒素的产生增加。
因此,饮食中适当的碳水化合物与蛋白质比例至关重要,任何偏差都会导致肠道菌群成分的破坏,从而可能导致骨代谢过程的故障。骨干细胞分化依赖于细胞因子等全身因素;此外,微生物群组成破坏导致的免疫改变会影响骨骼。
多种免疫因子参与骨代谢的调节
核因子-κB受体激活剂(RANK)、RANK配体(RANKL)和骨保护素(OPG)等多种免疫因子参与骨代谢的调节。
有两种类型的细胞,间充质干细胞来源的成骨细胞和单核细胞系来源的破骨细胞。单核细胞系细胞通过RANKL途径分化为巨噬细胞/树突状细胞或破骨细胞,具体取决于细胞周围的微环境。巨噬细胞集落刺激因子(M-CSF)的存在导致RANK表达增加,导致RANKL信号的刺激,最终通过形成破骨细胞导致骨吸收。
TNF-α、IL-6和IL-1等细胞因子也通过RANKL途径直接或间接放大骨吸收,因为细胞因子增加髓系细胞上RANK受体的表达。然而,OPG(RANKL的天然受体)阻止它与RANK结合,从而减少破骨细胞的生成过程。OPG的主要来源是B细胞,炎症状态下B细胞的失调导致B细胞RANKL表达升高,OPG表达降低,从而导致更高水平的骨吸收和破骨细胞过度生成。
血清素水平的增加与骨量的减少有关
据报道,肠道菌群通过维持代谢激素5-羟色胺或5-羟色胺(5-HT)的水平来影响骨代谢,5-羟色胺是由肠道嗜铬细胞在色氨酸羟化酶-1(Tph1)酶的帮助下合成的。由于这两种细胞类型,即骨细胞和成骨细胞都有5-HT受体,它通过5-HT信号转导途径在骨发育和维持的调节中起主要作用。
短链脂肪酸等微生物产物在骨密度调节中发挥作用
短链脂肪酸通过OPG和Runx信号通路参与骨矿化和骨形成;然而,丁酸通过抑制RANKL信号通路减少破骨细胞的生成过程。研究表明,短链脂肪酸通过潜在影响宿主内分泌因子(如胰高血糖素样肽1(GLP-1)和肽YY(PYY))的功能,在维持骨密度方面发挥间接作用,这些内分泌因子与骨代谢有关。
PYY和GLP-1均由胃肠道内分泌L细胞分泌,其中GLP-1通过破坏成骨细胞和脂肪细胞之间的平衡,从骨髓间充质干细胞分化为骨代谢调节器。然而,在绝经前妇女中,PYY与全身和髋部骨密度之间存在矛盾的联系。
肠道菌群改变激素水平影响骨骼健康
肠道菌群还通过改变肠道来源的非卵巢雌激素(如己烯雌酚和类黄酮)水平影响骨骼健康。雌激素水平低是绝经后骨质疏松风险的主要因素。
其他疾病中,肠道菌群和骨骼健康的联系
克罗恩病(CD)和肥胖会增加骨折风险,因此,将骨密度和肠道菌群联系起来。
早些时候,一项用于测定类风湿性关节炎(一种关节慢性炎症性疾病)中肠道菌群改变的研究显示乳酸杆菌显著增加。
另一份报告显示拟杆菌、普雷沃菌,Porphyromonas的数量较低。然而,最近的一项研究表明,以下菌群比例过高:Porphyromonadaceae, Carnobacterium, Parabacteroides, Phascolarctobacterium, Bacteroides, Paraprevotella. 此外,据报道,产生丁酸盐的粪杆菌、Roseburia、Subdoligranulum、瘤胃球菌、Pseudobutyrivibrio 数量减少。
类似地,强直性脊柱炎(关节炎的一种形式)患者在回肠末端有离散的微生物特征,属于毛螺菌科、卟啉单胞菌科、瘤胃球菌科、拟杆菌科、Rikenellaceae的细菌数量增加。Prevotellaceae和Veillonellaceae科细菌的丰度降低(表1)。

Ahlawat S,et al.,Lett Appl Microbiol. 2021
扩展阅读:肠道微生物组:肌肉骨骼研究的新领域
“肠道健康对容光焕发的皮肤的重要性
皮肤内稳态和外稳态与胃肠道有关,皮肤和肠道之间存在双向沟通。
胃肠道疾病和饮食都会影响皮肤的病理生理学。
肠道菌群通过产生短链脂肪酸、免疫系统修饰等影响皮肤健康。
各类皮肤病中肠道菌群的作用。
皮肤和肠道对维持生理内环境平衡至关重要。它们具有多种共同特征,如大量微生物群的定植、高度神经支配、大量血管化,并提供与外部环境的接口。皮肤再生过程对于维持其内环境稳定状态非常重要,这是通过持续更新和有效的表皮更新来实现的。
处于稳态的皮肤可以执行各种基本功能,如温度调节、保护和保水。皮肤内稳态和外稳态与胃肠道有关;因此,考虑到皮肤和肠道之间的双向沟通。尽管肠道和皮肤关系的完整机制尚不清楚;然而,研究表明,它涉及代谢系统、免疫系统、神经系统和内分泌系统之间的复杂通信网络。
胃肠道疾病和饮食都会影响皮肤的病理生理学,皮肤表现与某些胃肠道疾病有关。
皮肤通常由四个细菌门控制,包括拟杆菌门、变形菌门、放线菌门和厚壁菌门。
肠道菌群通过产生短链脂肪酸影响皮肤微生物群
短链脂肪酸在决定皮肤微生物群物种的流行程度方面具有重要作用,从而影响皮肤免疫反应机制。通过抑制炎症细胞的粘附、迁移、增殖和细胞因子的产生,短链脂肪酸(尤其是丁酸)减缓了免疫反应。短链脂肪酸还通过组蛋白去乙酰化酶(HDAC)抑制和NF-kB信号通路失活在调节免疫细胞凋亡和激活中发挥作用。
许多皮肤生理功能,如伤口愈合和毛囊干细胞分化的调节,都受皮肤调节细胞的控制,其增殖受到HDAC抑制的刺激。
重要的皮肤菌——表皮葡萄球菌和痤疮丙酸杆菌
有趣的是,据报道,两种最显著的皮肤共生菌,即表皮葡萄球菌和痤疮丙酸杆菌能够耐受短链脂肪酸的显著变化。丙酸杆菌本身产生类似丙酸和乙酸盐的短链脂肪酸。丙酸对条件致病菌耐甲氧西林金黄色葡萄球菌具有抗菌作用。
补充副干酪乳杆菌NCC2461降低皮肤敏感性
在皮肤的功能中,屏障功能可以防止病原体入侵以及皮肤水分、电解质和蛋白质的流失。经表皮失水(TEWL)是一种测量稳态水蒸气穿过皮肤进入环境的通量,已被用作皮肤屏障功能的标志。人类临床研究表明,服用短乳杆菌SBC8803口服补充剂12周后,角膜水合作用显著增加,TEWL降低。同样,补充副干酪乳杆菌NCC2461 2个月的个体TEWL和皮肤敏感性降低。
肠道微生物组主要通过免疫系统修饰影响皮肤健康
它通过影响T细胞对各种免疫刺激的反应,从而增强皮肤的异质性。Th-17细胞数量异常或更高,以及促炎细胞因子与白塞病(BD)等多种炎症表现有关。这验证了肠道菌群在通过免疫系统修饰维持皮肤健康方面的关键作用。
免疫改变、皮肤屏障功能障碍
特应性皮炎(AD)是一种皮肤炎症性疾病,其发病机制主要由免疫反应改变和皮肤屏障功能障碍控制。由于Th1/Th2比率中断,以细胞因子产生增加(IL-4、IL-5和IL-13)为标志的免疫改变导致IgE水平升高,并增加金黄色葡萄球菌与特应性皮炎患者皮肤的结合。这种免疫失衡是由于存在特定类型的微生物群时,炎症微环境导致肠道菌群及其代谢物发生改变的结果。
特应性皮炎患者肠道菌群增加了金黄色葡萄球菌、大肠杆菌、艰难梭菌的数量,其中拟杆菌和双歧杆菌减少。
另一项针对特应性皮炎患者的研究显示,Faecalibacterium prausnitzii在亚种水平上存在失调,丙酸盐和丁酸盐的产生量较低,包括与菌株A2-165相关的产生量。它导致皮肤对过敏原产生异常的Th2型免疫反应(表1)。此外,还发现大肠杆菌和梭菌通过嗜酸性炎症与特应性皮炎相关。
然而,屏障功能障碍的主要遗传原因是丝聚蛋白(filaggrin)基因突变导致的功能丧失,这对维持表皮内环境稳定至关重要,因为丝聚蛋白基因有助于屏障功能和保水。因此,对环境抗原的易感性增加和TEWL增加可能是该特定基因突变的结果。
逐渐地,已确定的肠道菌群失调以及免疫系统失衡持续到成年,从而导致疾病的自然病程。肠道菌群破坏降低了其调节宿主免疫系统的能力。这会导致局部和系统性炎症,如牛皮癣(银屑病)。
扩展阅读:微生物群对三大过敏性疾病发展的影响
生命早期微生物接触和过敏风险:如何预防
菌群变化、肠道炎症
据报道,牛皮癣患者的肠道菌群多样性较低,粪球菌数量减少。
此外,一项针对15名银屑病患者、16名银屑病关节炎(PsA)患者和17名对照受试者的研究显示,银屑病和PsA患者中的粪球菌数量均减少。同时,PsA患者的Akkermansia、瘤胃球菌和假丁酸菌数量减少(表1)。
银屑病可导致身体其他器官发炎。据报道,7–11%的炎症性肠病患者患有银屑病,银屑病进一步将皮肤与胃肠道连接起来。
发现银屑病患者的血浆中含有肠道细菌的DNA。其他皮肤表现如酒渣鼻与幽门螺杆菌感染有关。这些患者的SIBO发病率更高,通过产生有毒代谢物导致肠道通透性、肠细胞损伤和全身炎症。
扩展阅读:牛皮癣看似皮肤病,实则关系到肠道
阻塞、炎症、痤疮杆菌介导、mTOR途径
痤疮,一种较常见的皮肤异常,是由于皮脂分泌过多、导管阻塞和炎症引起的,由痤疮杆菌介导。对寻常痤疮患者粪便的高通量测序显示,变形菌数量增加,放线菌、双歧杆菌、丁酸杆菌、共细菌、乳酸杆菌和异杆菌的数量减少(表1)。
据报道,痤疮的病理生理学受到mTOR途径和肠道菌群之间双向通讯的影响。
在一个方向上,肠道代谢物对mTOR途径介导的代谢过程具有调节作用,如脂质代谢和细胞增殖。或者,mTOR途径通过调节肠道屏障的完整性来影响肠道菌群成分。
扩展阅读:痘痘?粉刺?皮肤问题很可能是肠道问题
这7种类型的食物可能引起 “痘痘”
菌群变化
湿疹的标志是双歧杆菌、巨球菌、嗜血杆菌、脆弱拟杆菌和唾液链球菌的丰度降低。此外,这些患者以下菌群数量增加:
Escherichia/Shigella, Veillonella, Clostridium XlVa,
Lachnospiraceae incertae sedis, F. prausnitzii,
Ruminococcus gnavus, A. muciniphila
免疫功能障碍、菌群变化
研究表明,肠道菌群的改变与白塞病患者的免疫功能障碍有关。这些患者双歧杆菌和埃格特菌的数量增加,而巨单胞菌和普氏杆菌的数量减少(表1)。
总之,这些发现为皮肤和肠道之间的功能互动机制提供了强有力的支持性证据。

Ahlawat S,et al.,Lett Appl Microbiol. 2021
“肠道菌群和能量平衡
脂肪介导的小肠效应可能有助于理解脂肪介导的代谢紊乱的病因。
肠道脂肪吸收可以根据饮食中的脂肪含量进行调整。
肠道菌群通过与脂肪组织的通讯轴影响代谢
膳食脂肪的消化吸收过程
在我们的饮食中,甘油三酯(TAG)几乎占膳食脂质的95%。在吸收之前,它在胃中被酸性稳定的胃脂肪酶部分水解成二酰甘油(DAG)和游离脂肪酸(FFA)。
它在小肠中继续消化,在小肠中,依赖于脂肪酶的胰脂肪酶释放2-单酰甘油(MAG)和长链脂肪酸(LCFA)。长链脂肪酸具有可能对细胞完整性有害的清洁剂特性。因此,它们分散在肠腔中的胶束中,与肠吸收细胞中的脂质结合蛋白结合,并作为富含甘油三酯的脂蛋白(乳糜微粒)分泌到淋巴中,这些脂蛋白被内皮脂蛋白脂肪酶进一步水解,为外周组织提供长链脂肪酸。
小脂蛋白中剩余的甘油三酯和残余物被肝脏脂肪酶进一步水解,并被肝脏从血液中清除。因此,膳食脂肪的高效消化和吸收确保了长链脂肪酸正确供应到发挥各种基本细胞功能的身体。
脂肪吸收可以根据饮食中的脂肪含量进行调整
小肠是一种选择性屏障,可有效吸收膳食脂肪并负责其处置。新出现的数据表明,肠道的甘油三酯的高生物利用度是后天获得的特性。
这表明肠道脂肪吸收可以根据饮食中的脂肪含量进行调整。有趣的是,这些脂质介导的肠道适应防止了高脂肪饮食期间粪便中脂质的过度清除。
因此,在食物匮乏的环境中提供生存优势。相反,在食物充足的时期,它们会增加肥胖和相关疾病的患病率。
总之,这些数据强调,脂肪对小肠的影响可能是促成脂肪介导的代谢紊乱的病因。
过度摄入脂肪的肥胖,引起系列代谢紊乱
过度摄入脂肪会增加肥胖的风险,肥胖涉及一系列代谢改变,如葡萄糖稳态紊乱(胰岛素抵抗和2型糖尿病或T2D)、心血管疾病(CVD)或风险因素(如高血压)和非酒精性脂肪肝。它涉及器官间(肠-脂肪组织和肠-大脑)通讯网络的严重紊乱,这有助于能量消耗、脂肪组织发育和胰岛素抵抗的改变。然而,并非所有肥胖者都有胰岛素抵抗和糖尿病。
它与两个主要细菌分类的相对丰度的改变有关,即拟杆菌减少,厚壁菌增加。因此,微生物组从饮食中提取能量的能力更高。
肠道菌群不仅是代谢紊乱的结果,也可能是原因
有几项研究将肠道双歧杆菌数量的减少与肥胖和/或糖尿病的发病联系起来。然而,现有证据表明,肠道菌群成分的改变不仅仅是结果,还可能导致肥胖。
高脂饮食改变菌群引发炎症反应,导致代谢综合征
高脂饮食也与肠内拟杆菌相对丰度降低、含LPS细菌丰度增加以及血浆LPS水平升高(称为代谢性内毒素血症)有关,通过增加LPS-TLR4结合增强局部炎症反应。
其次是脂肪/肠系膜脂肪组织积聚和胰岛素抵抗。胰岛素敏感性受绕过LPS诱导的CD95介导的髓样细胞炎症的影响。然而,在生理学上,局部肠道炎症是一种调节肠道屏障功能和促进乳糜微粒转运的适应,以在脂质摄入增加的情况下存活。但长期高脂肪摄入和肠道菌群改变会在肠道引发持续/慢性低度局部炎症反应,进而导致代谢综合征(图1)。
代谢异常也受脂肪分布的影响。中枢性肥胖的个体,脂肪主要集中在上胸廓和腹腔内沉积,更容易发生这种异常。
肠道菌群通过与脂肪组织的通讯轴影响代谢
由生物活性脂质组成的内源性大麻素(eCB)系统可以调节肠道和脂肪组织之间的联系。它通过与大麻素受体结合而引发细胞信号。此外,它的严格调控依赖于特定酶的合成和降解之间的平衡。它在控制能量平衡的组织中表达,因此,其失调会导致各种代谢状况,包括肥胖和2型糖尿病。
免疫反应诱导胰岛素抵抗,维持平衡,但长时间会引起代谢异常
免疫细胞尤其是巨噬细胞(具有LPS受体)对代谢内毒素血症的反应,增加白色脂肪组织浸润,产生炎症免疫反应,增强IL-6和TNF-α,从而诱导胰岛素抵抗,以维持饥饿和感染条件下的体内平衡。
然而,在肥胖患者中观察到的,如果它持续很长时间,就会产生有害影响,并导致代谢异常。
类似地,肠屏障功能受损和肠系膜脂肪大量积聚的克罗恩病患者中,细菌向肠系膜脂肪的移位增加。
脂肪细胞肥大->游离脂肪酸升高->代谢紊乱
LPS可以降低前脂肪细胞中过氧化物酶体增殖物激活受体γ(PPARγ)的表达,即脂肪生成(脂肪细胞分化和脂肪生成)的关键调节因子,并激活eCB的产生。这会对前脂肪细胞分化产生不利影响,并导致脂肪细胞肥大。
肥大脂肪细胞沉积游离脂肪酸的能力受损,从而导致循环游离脂肪酸水平升高,从而导致基础脂肪分解(图1)。
相反,它们会释放更多的游离脂肪酸和炎性细胞因子,包括脂肪因子,也就是不好的代谢特征。
总之,肝脏代谢受内脏脂肪和肠道分泌特征的影响。在肥胖个体中,内脏脂肪沉积物中的FFA和促炎细胞因子以及肠道进入门静脉循环的内毒素(LPS)的释放增加对肝脏胰岛素敏感性产生负面影响,这可能导致代谢综合征的发生。这证实了“肠道脂肪组织”和肝脏之间存在一个网络。
扩展阅读:体重增长:目前为止我们所知道的一切(更新你的减肥工具箱)
2型糖尿病如何做到可防可控?肠道菌群发挥重要作用
“双向通信网络
大多数心血管疾病风险因素,都会导致与肠道炎症和肠道屏障完整性降低相关的生态失调。
肠道和心脏之间双向沟通,“肠道-心脏轴”
肠道菌群参与各类心血管疾病的进展。
心血管疾病由多种风险因素引起,分为可改变的(饮食和生活方式)和不可改变的(年龄和遗传学)。
几项研究表明,肠道菌群与宿主生理过程保持着复杂的关系,因此表明它是心血管疾病风险的基因外因素。
大多数心血管疾病风险因素都会导致与肠道炎症和肠道屏障完整性降低相关的生态失调,提高肠道细菌结构成分和循环中微生物代谢产物的水平,从而加快了心血管疾病的发展。
一些影响心脏的疾病,如代谢综合征或肥胖症,与出生后微生物组的获取受到干扰或不足有关,或与儿童时期肠道微生物组中存在特定细菌或细菌家族的环境微生物的早期接触有关。
此外,患有肠易激综合征等肠道疾病的患者患冠心病的风险增加。这表明肠道和心脏之间存在额外的连接。
以上证据共同表明,肠道和心脏之间存在双向通信网络,即“肠道-心脏轴”。
越来越多的数据表明,肠道在心力衰竭(HF)的病理生理学中起着至关重要的作用。
心力衰竭患者损害肠道,导致疾病发展
心力衰竭患者会出现外周血管收缩、心输出量减少和组织充血等障碍,这些障碍会在结构和功能上损害肠道,导致肠道血流量减少、肠壁增厚(结肠和回肠末端),增加(小肠中)胶原蛋白的积累和血流动力学改变。由于缺氧诱导的肠缺血,这些微循环障碍在功能上损害了肠上皮细胞,从而损害营养吸收,导致疾病发展和营养不良。
肠道细菌及其产物转移到循环中,诱发炎症反应
屏障功能障碍导致肠道细菌及其产物转移到循环中。当进入循环系统时,肠道细菌源性内毒素(如LPS)结合其受体,即心肌细胞上的Toll样受体4(TLR-4)。这种结合与循环细胞因子(TNF-α)增加、结构组织损伤、收缩力下降和心功能受损等炎症反应的诱导有关。LPS还会触发吞噬细胞和粒细胞释放儿茶酚胺,从而对肠道灌注产生额外的不利影响。
心力衰竭与菌群失调有关
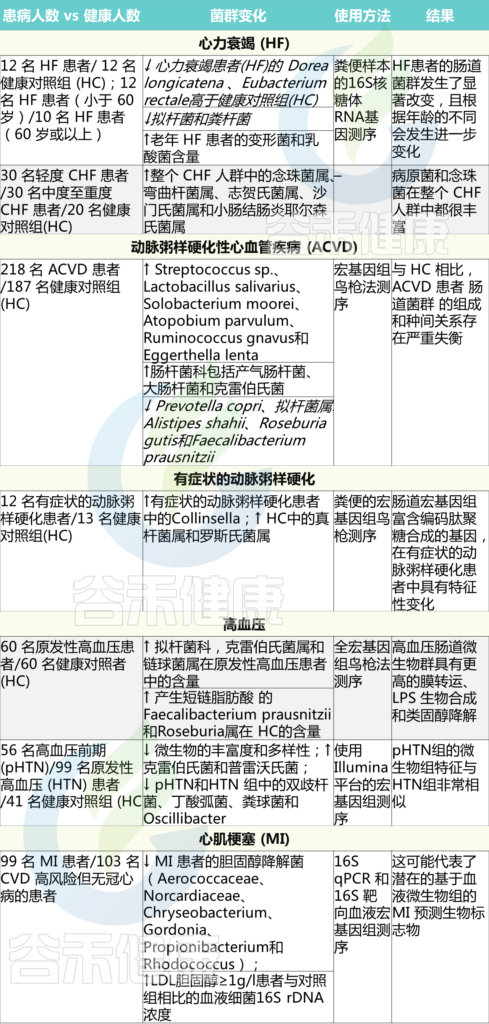
最近,心衰患者的肠道菌群分析表明,肠道菌群发生了显著改变, 以下菌群较少:
Dorea longicatena 、 Eu. rectale
菌群随着年龄的不同而进一步变化,因此老年心力衰竭患者(60岁或以上)的拟杆菌和粪杆菌数量较少,变形菌和乳酸杆菌比例较大。
另一项针对慢性心力衰竭(CHF)患者的研究显示,患者的肠壁厚度增加,三氯蔗糖和乳果糖/甘露醇的通透性增加,D-木糖吸收减少,同时乙状结肠粘膜生物膜中的粘附细菌水平升高。这些变化共同导致肠道缺血、慢性炎症和营养不良。
此外,与健康对照组相比,CHF患者肠道内念珠菌和沙门氏菌、弯曲菌、志贺氏菌和小肠结肠炎耶尔森氏菌等病原菌过度生长(表1)。
心力衰竭和代谢产物的异常产生有关
肠道菌群衍生的代谢物也会促进疾病进程。尿毒症毒素,如TMAO、对甲酚硫酸盐和吲哚氧基硫酸盐,是从饮食摄入的微生物发酵中产生的。硫酸吲哚氧基对心脏有促肥大和促纤维化作用,而TMAO是预测心血管疾病风险的一个有前途的生物标志物。
一项大型队列研究表明,在接受选择性冠状动脉造影的患者中,血浆TMAO水平升高与心肌梗死、中风和死亡风险增加有关。此外,心力衰竭患者的TMAO血浆水平也明显高于健康对照组。
动脉粥样硬化涉及代谢和炎症成分,受肠道菌群变化的影响
新出现的报告提出了一种新的途径,将膳食脂质摄入、肠道菌群和动脉粥样硬化联系起来。从膳食磷脂酰胆碱(卵磷脂)中产生甜菜碱、胆碱和TMAO代谢物取决于肠道菌群的代谢,TMAO与心血管疾病风险呈最强正相关。
根据一项研究,TMAO可减少胆汁酸的合成,并抑制胆固醇的逆向转运,而胆固醇的逆向转运与动脉粥样硬化的增加有关。然而,确切的机制仍然难以捉摸。
此外,人们认为慢性肺炎衣原体和幽门螺杆菌感染以及随后的免疫反应对动脉粥样硬化的发展至关重要。
LPS升高与动脉粥样硬化有什么样的关联?
各种研究支持血清LPS(内毒素)水平升高与动脉粥样硬化之间的关联。IBD或肝硬化患者的肠道屏障功能受损,导致血清LPS水平升高,动脉粥样硬化发生率增加。
LPS通过与低密度脂蛋白(LDL)相互作用影响脂蛋白代谢,诱导内皮细胞损伤,刺激超氧阴离子释放和低密度脂蛋白氧化。氧化低密度脂蛋白有利于巨噬细胞释放细胞因子(IL-1和TNF-α),刺激巨噬细胞转化为泡沫细胞。这些特征共同促进动脉粥样硬化的发展和进展。
肠道菌群参与动脉粥样硬化性心血管疾病的发展
动脉粥样硬化斑块和同一个体的肠道中存在不同种类的细菌DNA,表明肠道菌群是动脉粥样硬化细菌的潜在来源。因此,肠道菌群可能参与冠心病的发病和进展。
在早期的研究中,动脉粥样硬化性心血管疾病患者中链球菌属和肠杆菌科的比例高于健康对照组。另一项针对症状性动脉粥样硬化患者的研究显示,与健康对照组相比,症状性动脉粥样硬化组的Collinsella数量增加,Eubacterium ,Roseburia比例降低(表1)。
此外,肠道菌群是多种疾病的风险因素,如代谢综合征、肥胖、糖尿病和动脉粥样硬化,这些疾病与高血压有关。
高血压的发病机制是复杂的、多因素的。由细菌诱导的膳食纤维厌氧发酵在肠道产生的短链脂肪酸在调节血压(BP)方面具有生理功能。短链脂肪酸的高血压和降压作用分别通过与受体Olfr78和GPR41结合来介导(图3)。
图3 “肠道-心脏”轴
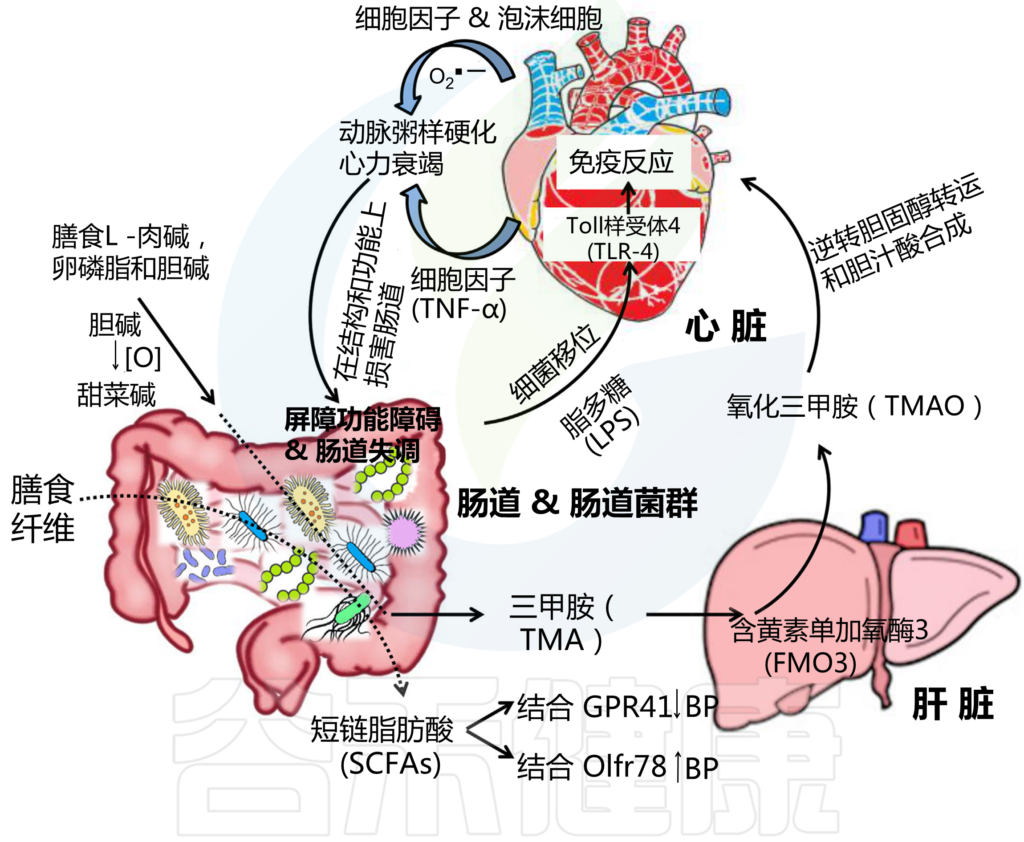
Ahlawat S,et al.,Lett Appl Microbiol. 2021
最近一项针对56名高血压前期(pHTN)、99名原发性高血压(HTN)患者和41名健康对照(HC)的研究表明,在pHTN和HTN两组中,产生短链脂肪酸的F. prausnitzii 和 Roseburia 均减少。
另一份报告提出了条件致病菌(Parabacteroides merdae, Klebsiella, Streptococcus)在高血压发病机制中的作用(表1)。
扩展阅读:认识肠道微生物及其与高血压的关系
总之,血压与肠道菌群的多样性、丰富度和均匀度密切相关,并受厚壁菌/拟杆菌比率的影响。
Ahlawat S,et al.,Lett Appl Microbiol. 2021
扩展阅读:与心血管疾病相关的肠道菌群代谢产物或毒素
最新 | 肠道微生物群与心血管疾病:机遇与挑战
微生物群与宿主之间的相互作用对维持内稳态很重要,但这种相互作用一旦受到干扰,就会成为许多慢性疾病的核心驱动因素。
目前随着对肠道菌群的了解逐步深入,我们开始了解它们的信号以及与人类健康和相关疾病的相关性。当然还有更多需要探索的问题,肠道菌群的改变是导致疾病的原因还是仅仅反映疾病状态,应该如何针对菌群作出精准干预等。
当我们真正开始理解微生物彼此之间的关系,及其与宿主之间复杂多变的进化和生态关系时,对疾病的机制理解就会越来越清晰,从而在菌群的基础上进行有效的干预措施。
主要参考文献:
Ahlawat S, Asha, Sharma KK. Gut-organ axis: a microbial outreach and networking. Lett Appl Microbiol. 2021 Jun;72(6):636-668. doi: 10.1111/lam.13333. Epub 2020 Jul 16. PMID: 32472555.
Yoo W, Zieba JK, Foegeding NJ, Torres TP, Shelton CD, Shealy NG, Byndloss AJ, Cevallos SA, Gertz E, Tiffany CR, Thomas JD, Litvak Y, Nguyen H, Olsan EE, Bennett BJ, Rathmell JC, Major AS, Bäumler AJ, Byndloss MX. High-fat diet-induced colonocyte dysfunction escalates microbiota-derived trimethylamine N-oxide. Science. 2021 Aug 13;373(6556):813-818. doi: 10.1126/science.aba3683. PMID: 34385401; PMCID: PMC8506909.
Rinninella E, Raoul P, Cintoni M, Franceschi F, Miggiano GAD, Gasbarrini A, Mele MC. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms. 2019 Jan 10;7(1):14. doi: 10.3390/microorganisms7010014. PMID: 30634578; PMCID: PMC6351938.
Parikh K, Antanaviciute A, Fawkner-Corbett D, Jagielowicz M, Aulicino A, Lagerholm C, Davis S, Kinchen J, Chen HH, Alham NK, Ashley N, Johnson E, Hublitz P, Bao L, Lukomska J, Andev RS, Björklund E, Kessler BM, Fischer R, Goldin R, Koohy H, Simmons A. Colonic epithelial cell diversity in health and inflammatory bowel disease. Nature. 2019 Mar;567(7746):49-55. doi: 10.1038/s41586-019-0992-y. Epub 2019 Feb 27. PMID: 30814735.
Lun H, Yang W, Zhao S, Jiang M, Xu M, Liu F, Wang Y. Altered gut microbiota and microbial biomarkers associated with chronic kidney disease. Microbiologyopen. 2019 Apr;8(4):e00678. doi: 10.1002/mbo3.678. Epub 2018 Aug 7. PMID: 30088332; PMCID: PMC6460263.
谷禾健康
现状
全球肥胖患病率的上升是一个主要的社会经济负担,肥胖与许多疾病的风险增加有关,包括糖尿病、心血管疾病和癌症。
尽管人们努力改善生活方式选择,提高对潜在病因的认识,但在预防和治疗肥胖方面的长期成功似乎有限,因为饮食诱导的体重减轻在5年随访后仅维持约25%。
近年来,在了解肠道微生物群作为宿主能量和底物代谢调节器参与肥胖和相关心脏代谢并发症方面取得了进展。因此,通过肠道微生物群靶向宿主代谢可能是饮食干预减轻体重的一项重要策略。
过去十年中,关于肠道微生物组对宿主代谢影响的研究数量呈指数增长,研究的数量和质量都在迅速发展,这些研究表明,基线微生物组成可以预测包括肥胖在内的代谢综合征。然而,研究同时表明微生物群组成的调节不可能会在所有条件下对人体代谢产生重要积极的影响,而这种影响取决于个体的特征,例如年龄、习惯性饮食、代谢表型和基线肠道微生物谱。
肠道微生物群的组成由可遗传、人口统计和环境因素决定,包括出生时的分娩方式、年龄、性别、胃肠道转运时间和药物使用。但是诸多因素中,饮食已成为塑造和定义肠道微生物组的关键因素。
饮食尤其是膳食纤维等引起的肠道微生物群组成和功能变化与肥胖和相关疾病的发展有关。这些研究结果发现肠道微生物群的个体间差异可以作为对抗肥胖代谢疾病的更精确饮食方法的基础。
本文将介绍有关饮食成分、肠道微生物组和宿主代谢之间相互作用的知识和研究成果,以及如何整合这些知识来制定基于精确的营养策略,以改善人类的体重控制和代谢健康。
厚壁菌/拟杆菌门
肠道微生物群影响免疫功能和上皮完整性、能量和底物代谢以及葡萄糖稳态。初步研究表明,与瘦个体相比,肥胖的人类和啮齿动物的厚壁菌门与拟杆菌门的比例增加,但也有个别研究未能观察到这种差异,甚至报告了比例下降。
多样性和微生物基因丰富度
在代谢健康与不健康个体的比较中,代谢不健康组的α多样性较低。而且重度肥胖症患者的低微生物基因丰富度比例高达75%,而瘦或超重/中度肥胖症患者的低微生物基因丰富度比例为23%-40%。
(小编推测可能是由于中重度肥胖人群其饮食比较丰富且量大,微生物不需要太多多余的基因就可以代谢获得生存繁殖的食物,而较瘦的个体食物不太丰富,那么菌需要更多的基因才获取生存的食物和繁殖生存)
具体菌属
具体而言,颤螺菌属(Oscillospira)和 红蝽菌科(Coriobacteriaceae)的细菌与良好的代谢健康相关。 在一项包含正常体重和超重/肥胖人群的研究人群中,特定菌属的丰度与代谢特征相关。 例如,产气柯林氏菌、Dorea formicigenans 和 Dorea longicatena 在超重/肥胖人群中的丰度更高。
Akkermansia属的细菌是最有说服力的证据,它与患肥胖症和代谢综合征的风险呈负相关。在超重/肥胖患者中,为期 3 个月的 Akkermansia muciniphila 补充剂可改善胰岛素敏感性并降低肝功能障碍和炎症的血液标志物。
基线菌属
另一项研究表明,在瘦肉型个体中,嗜粘菌A.muciniphila和Alistipes obesi显著富集,而在肥胖型个体中,Ruminococcus gnavus显著富集。该研究还确定,当在基线检查时高丰度存在的菌,如Blautia wexlerae 和 Bacteroides dorei 减肥前以高丰度存在时将有助于减肥。此外,基线普雷沃菌属 (Prevotella)普氏菌丰度可以预测肥胖人群在膳食纤维干预减肥中是否可以成功。
此外,与健康个体相比,II型糖尿病患者和代谢受损个体表现出微生物功能改变和发酵能力降低,尤其是产丁酸盐细菌丰度较低的个体。此外,胰岛素抵抗个体的肠道微生物组可能具有增加的生物合成潜力,并减少了支链氨基酸(BCAA,主要由Prevotella copri,B. vulgatus驱动)的吸收和分解代谢,这与有害代谢效果有关。
总之,代谢受损个体的微生物基因丰富度和多样性降低。肠道微生物群组成和功能的个体差异与饮食干预的反应变化有关。
在当前的西方世界,习惯性饮食结构已转向高能量密集型食物,包括相对较高的饱和脂肪和简单碳水化合物含量,以及较低的膳食纤维含量。尤其是膳食纤维的消耗,以及大量营养素的质量和消耗量都会强烈影响肠道微生物群的组成和功能。基于人群的宏基因组分析揭示了微生物组成和多样性与60多种饮食因素的习惯饮食之间的关联。这些因素包括能量和大量营养素的摄入,以及面包和软饮料等特定食品的消耗。这些数据证实了饮食对塑造肠道微生物群的重要性。
饮食塑造肠型
在一项纵向单卵双生子研究中,粪便微生物群分析表明,能量的习惯性摄入、不饱和脂肪酸(FA)的类型和可溶性纤维会影响微生物群的组成,尤其是拟杆菌属和双歧杆菌的丰度。微生物肠道类型与长期习惯性饮食密切相关,尤其是蛋白质和动物脂肪(拟杆菌属)与碳水化合物摄入(普雷沃氏菌属)相比。
与此一致,长期坚持地中海饮食与特定分类群以及肠道微生物谱的功能有关。肠道微生物组的组成是地中海饮食与心脏代谢疾病风险之间保护性关联的调节因素。当比较习惯性高脂肪饮食和高碳水化合物饮食时,高脂肪饮食的微生物多样性似乎较低。此外,与高(饱和)脂肪饮食和高碳水化合物/纤维饮食相比,微生物多样性似乎更低。这种饮食诱导的失调被认为是肥胖症代谢障碍的诱因。
饮食干预菌群变化较快,但是整齐菌群结构稳定
虽然主要在动物模型中得到证实,但数量有限的人体研究表明,饮食干预引起的微生物组成和功能改变可能已经在饮食摄入改变后的几周甚至几天内发生。在人类中,在严格转向完全以植物或动物为基础的饮食后,发现了适度的微生物变化。这些相当极端的饮食干预形式提供了对饮食-肠道微生物组相互作用的潜在机制的见解,并表明饮食干预引起的微生物变化可能会非常迅速地发生。
与此一致,一项小型控制喂养研究显示,在开始高脂肪/低纤维或低脂肪/高纤维饮食后 24 小时内微生物组组成发生了变化,尽管在整个为期 10 天的研究中肠型特征保持稳定。这些研究结果表明,成年人存在微生物复原力的趋势,这可能与长期习惯性饮食摄入有关。然而,由于缺乏对肠型动力学和复原力的理解,细菌肠型的概念受到了其他几项研究的质疑。
一项为期 1 年的干预研究比较了限制能量的地中海饮食和增加体力活动与等热量地中海饮食对超重/肥胖成年人的影响,结果显示两组之间肠道菌群组成的变化存在显著差异。尽管如此,两种饮食的微生物转移趋势是相同的。这表明饮食模式对于肠道微生物的整齐迁移起关键作用。
饮食与肠道和宿主代谢中的糖酵解和蛋白水解发酵之间的相互作用
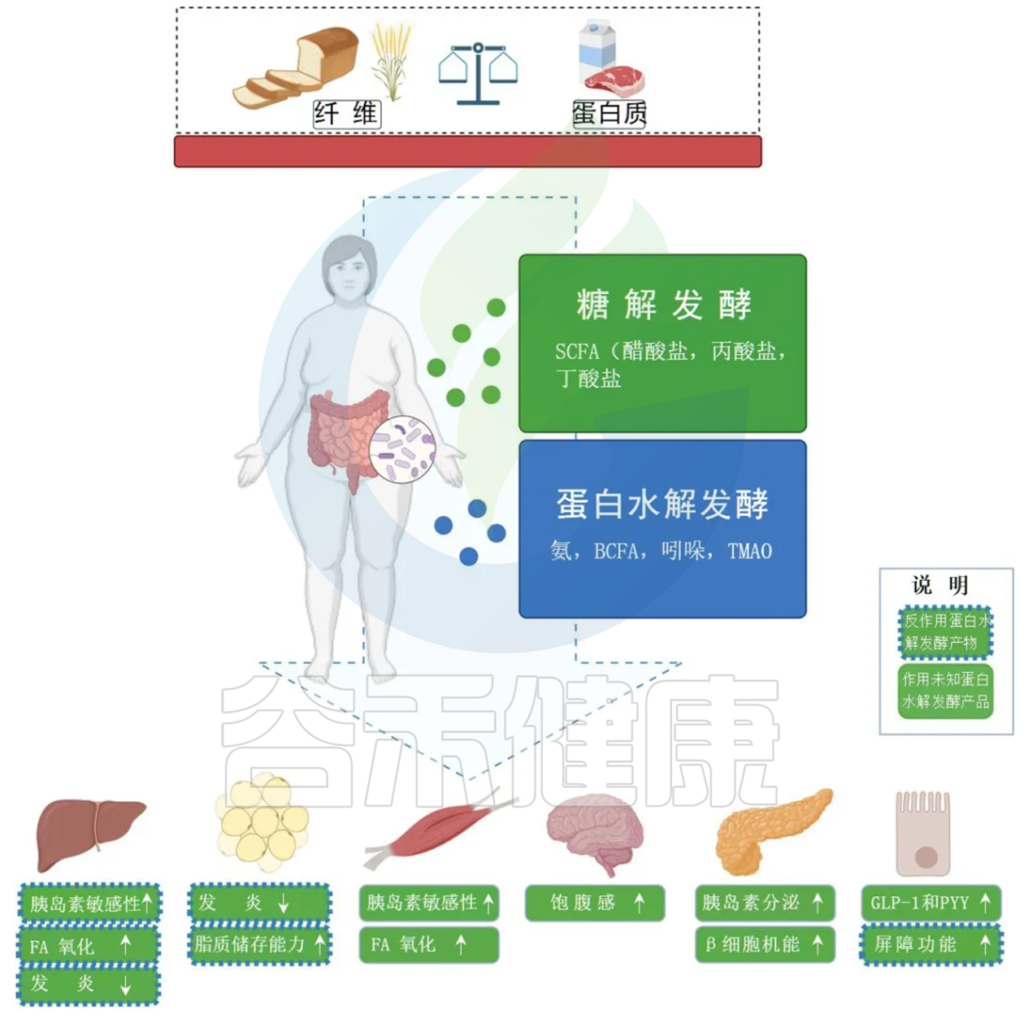
Jardon KM et al., Gut. 2022
膳食纤维的发酵主要发生在近端结肠并产生 SCFA,既可以用作肠细胞的燃料,也可以充当外周信号分子。SCFA 通过影响 GLP-1 和 PYY 的分泌,参与集中调节食物摄入和能量消耗。
蛋白质发酵主要发生在远端结肠并产生更多样化的代谢物,包括与肠道和代谢健康有害影响的 BCFA。
绿框表示 SCFAs 对周围器官代谢过程的影响。
蓝色边框表示蛋白水解发酵产物的相反方向位点方向(虚线)或未知方向(无线)的影响。
BCFA,支链脂肪酸;FA,脂肪酸;GLP-1,胰高血糖素样肽 1;PYY,肽YY;SCFA,短链脂肪酸;TMAO,三甲胺 N-氧化物。
成人肠道微生物组的塑造在生命早期就已经开始,这取决于诸如暴露于母体微生物组、分娩方式和早期暴露于膳食成分等因素。在所有生命阶段影响肠道微生物群组成和功能的众多因素中,饮食是调节特定细菌种类及其功能的丰度的关键。反之亦然,个人对某种饮食或饮食成分的反应可能在很大程度上受肠道微生物群特征的影响。
肠道微生物群能够发酵宿主无法获得的食物成分。小肠中不能被酶分解的膳食纤维和其他复杂碳水化合物可以(部分)被大肠中的细菌发酵,这一般是细菌作为首选能源,发酵后产生微生物产品,如短链脂肪酸(主要是乙酸盐、丙酸盐和丁酸盐)。
主要的产丁酸菌属于厚壁菌门,尤其是:
Faecalibacterium prausnitzii、Clostridium leptum、Eubacterium rectale 、Roseburia.
其他短链脂肪酸的产生由双歧杆菌等细菌介导,双歧杆菌在碳水化合物发酵过程中产生乙酸盐和乳酸。此外,A. muciniphila 物种同时产生丙酸盐和乙酸盐。
稳定同位素技术与13 C标记的短链脂肪酸可根据呼吸、尿液和血液分析对体内结肠产生的短链脂肪酸进行量化。短链脂肪酸主要在结肠中形成,其中约95%随后被吸收。
短链脂肪酸的作用
丁酸盐主要用作结肠细胞的主要能量来源,而丙酸盐和乙酸盐则通过门静脉进入肝脏。特别是,乙酸盐在进入体循环后也能到达外周组织,引起多种代谢和饱腹感相关效应。
短链脂肪酸可与G蛋白偶联受体(GPRs)结合。研究最好的受体包括GPR41、GPR43、GPR109a和GPR164,它们在大量细胞中表达,包括结肠上皮、胰腺β细胞、免疫细胞和周围组织,如脂肪组织。
短链脂肪酸对外周组织的影响包括脂肪生成、抑制脂肪组织脂肪分解(尤其是通过乙酸盐)和减轻脂肪细胞炎症、骨骼肌脂质氧化能力增加、胰腺胰岛素分泌和β细胞功能增加,肝脏的胰岛素敏感性和脂质氧化增加并改变肠-脑相互作用。但是注意这些数据主要来自体外和啮齿动物研究。
短链脂肪酸减脂(人类研究)
在人类研究中发现,长期结肠丙酸盐输送可防止体重增加,减少腹部肥胖和肝细胞内脂质含量,并防止超重成年人胰岛素敏感性的恶化。与这些发现一致,人体内数据表明,在超重或肥胖的成年人中,饮食诱导微生物短链脂肪酸产生变化或直接结肠短链脂肪酸输注后,空腹脂质氧化和静息能量消耗增加。
碳水化合物的消化是一个复杂的过程,涉及摄入的碳水化合物类型的特定酶。大多数可消化的膳食碳水化合物在小肠中被消化和吸收,而某些不可消化的碳水化合物,包括抗性淀粉和膳食纤维,很容易被结肠中含量最高的肠道微生物发酵。
膳食纤维对肠道菌群的有益影响
膳食纤维已被证明对与健康益处相关的肠道微生物群的组成和功能具有显著影响。这些因膳食纤维的结构、物理和化学特性可能会有所不同,例如水溶性、粘度、粘合和膨胀能力以及发酵性。高度可发酵的纤维,如 β-葡聚糖、菊粉和低聚半乳糖,在对微生物群组成和肠道代谢物产生的影响方面得到了很好的定义,而不溶性纤维虽然部分发酵,但大多数人都知道它们对粪便的有益作用一致性和结肠传输时间。

摄入高纤维饮食有益地影响宿主的健康,其中包括影响葡萄糖和脂质代谢。重要的机制包括调节营养吸收或产生短链脂肪酸,但有关膳食纤维对健康影响的数据存在争议。
对于膳食纤维研究中不一致发现的解释:
首先,在大多数人体研究中,只补充了一种特定的可发酵纤维,因此只刺激了一种或几种个体(潜在有益的)细菌属。后者的后果可能是其他必需细菌或核心菌属的丰度减少,这可能导致微生物生态系统的不平衡。因此,结合刺激多种不同细菌属的不同纤维可能对维持微生物丰富度以及对免疫状态和代谢健康产生更显著的(相加或协同)影响很重要,所以多样化膳食纤维和饮食摄入对于健康益处的微生物调节更有用。
有趣的是,一项研究表明,结肠中产生短链脂肪酸的部位可能是代谢健康的决定因素。急性远端结肠乙酸盐给药增加了超重男性的循环乙酸盐浓度,增加了脂肪氧化和刺激饱腹感激素 PYY,并降低了血浆肿瘤坏死因子-α。与远端输注相比,近端结肠中的乙酸盐给药不影响代谢特征。因此,通过结合不同的膳食纤维和/或更复杂的膳食纤维,针对远端结肠中微生物物种的膳食纤维可用性和短链脂肪酸形成,可能是改善免疫和代谢健康的有前景的策略。
TIPs
短链脂肪酸在一定范围内是越高越好,但是超过一定范围,也会产生害处。例如,高纤维饮食增加丁酸盐,诱导Stx受体球形三酰神经酰胺表达从而促进致病大肠杆菌定植。
此外,有益的短链脂肪酸一般需要通过结肠部位的菌群发酵产生,如果外源性的补充摄入,例如,丙酸盐有助于防止食物上霉菌,被广泛使用于烘焙食物、动物饲料和人造调味品中。如果长期摄入过量含有丙酸盐的食物,可能会增加人类患糖尿病和肥胖症的风险。
其次,到目前为止,大多数膳食纤维干预研究都没有考虑基线微生物组或代谢表型。基线肠道微生物组的特征可能与饮食干预结果密切相关。例如,已经表明肠道微生物群对膳食纤维(抗性淀粉与非淀粉多糖)的反应可以根据肥胖男性的基线微生物多样性来预测。高微生物多样性与微生物群的较低膳食反应性相关,这可能支持肠道微生物的更高多样性与微生物生态系统的稳定性有关的假设。
与此一致,与基因计数低的个体相比,基线时的高微生物基因计数与对减肥饮食的不太明显的反应有关。在低基因计数组中,基因丰富度和临床参数有所改善,尽管在基因丰富度低的个体中炎症标志物的变化不太明显。
一项针对肥胖个体的研究表明,不是基线微生物多样性而是厚壁菌门的基线丰度预测了个体微生物群的饮食反应。总之,这些发现表明微生物多样性并不总是饮食反应性的预测指标,这意味着需要进一步研究以更好地了解复杂的饮食-微生物组-宿主代谢相互作用。
作为对菊粉型果聚糖益生元的反应,具有高习惯性膳食纤维摄入量的健康个体的肠道菌群组成发生了更大的变化,而习惯性纤维摄入量低的人肠道菌群似乎更能适应变化。在II型糖尿病患者中进行的一项研究表明,膳食纤维促进了一组精选的产生短链脂肪酸的菌株,而许多其他微生物,包括蛋白水解发酵中的微生物,要么减少要么不变,表明微生物基因丰富度总体下降。粪便短链脂肪酸增加,尤其是丁酸盐,伴随着葡萄糖稳态的改善。因此,如几项人类纤维膳食干预研究所示,更高的微生物基因丰富度本身可能无益,但生理结果可能更依赖于微生物网络的功能。
在一项调查 6 周全麦饮食对体重变化影响的研究中,普雷沃氏菌属的高基线丰度与超重、健康成年人的体重减轻程度较高相关。这些发现表明,作为对特定饮食干预的反应,肠道微生物群的影响调节剂具有预测能力。
此外,发现超重、前驱糖尿病个体与瘦个体相比,对短期施用长链菊粉和抗性淀粉的微生物多样性和餐后胰岛素敏感性的变化的反应降低。与此一致,最近的研究表明,基线肠道微生物特征可以预测补充 3 个月长链菊粉后 BMI 的变化,这种效应在不同个体的粪便微生物群定植的小鼠中得到了复制。
有趣的是,可溶性菊粉纤维已被证明可以降低空腹血糖受损人群的胰岛素抵抗,但不能降低葡萄糖耐量受损的人群。鉴于空腹血糖受损与肝脏胰岛素抗性密切相关的发现,后一发现可能表明纤维 – 肠道微生物群 – 宿主代谢串扰中的组织特异性。
总体而言,益生元膳食纤维对代谢健康结果的有效性可能取决于几个参数,包括基线微生物组成以及微生物发酵的部位。
在低膳食纤维的西方饮食人群中,结肠远端的微生物群更擅长于利用剩余肽和蛋白质的发酵,因为首选的燃料,可发酵碳水化合物,已经在近端结肠中被人体大量使用。这种蛋白水解发酵过程的产物包括气体产物,如氢、甲烷、二氧化碳和硫化氢;BCFAs异丁酸酯、2-甲基丁酸酯和异戊酸酯(源自BCAAs发酵)、酚类和吲哚类化合物(源自芳香族氨基酸微生物发酵)以及较小的、未知的短链脂肪酸。
与糖解发酵产物相比,大多数蛋白水解发酵产物被认为对宿主肠道和代谢健康有害,尽管一些动物数据表明吲哚和硫化氢对肠道和外周组织功能有益。
例如,一些只能由肠道细菌(吲哚)或哺乳动物宿主(酪胺、色胺和短链脂肪酸)产生的氨基酸衍生化合物通过影响GLP-1和肠内分泌细胞血清素的分泌,直接影响哺乳动物的饱腹感和肠道运动。
然而,大多数这些化合物对宿主肠道和周围组织的生理作用仍不清楚。许多此类化合物的人类来源和细菌来源之间的区别尚未完全确定,需要进一步的体内研究来验证此类效应。
结肠中糖酵解和蛋白水解发酵之间的平衡,以及对宿主生理的假定有益和有害调节之间的平衡,可能对制定饮食干预策略很有意义。
一些研究表明,增加膳食纤维的摄入量,特别是缓慢发酵纤维的摄入量,会减少肠道微生物群仅产生有害的蛋白水解代谢物,使得整体发酵平衡向更有益的糖酵解发酵转变。
摄入的膳食蛋白质首先在小肠中被胰酶和来自肠细胞的肽酶消化。然后,大量的寡肽和氨基酸通过肠细胞转运蛋白被转运到门静脉血流中,在那里它们被用作蛋白质合成的氨基酸前体或被代谢为燃料或肠粘膜代谢物必需的前体。
由于远端小肠和近端结肠中的大多数细菌优先使用可发酵碳水化合物而不是蛋白质,因此大多数氨基酸作为能量来源的发酵发生在碳水化合物被耗尽的远端结肠。
摄入的蛋白质到达大肠的百分比也可能取决于蛋白质质量,估计约为 10%。由于植物的细胞壁不易消化,源自植物的蛋白质的消化率较低,而源自动物的蛋白质更容易在大肠中消化,这表明功能结果存在潜在差异。
酪蛋白是一种从动物产品中提取的相对缓慢消化的蛋白质,是防止高脂肪/高蛋白饮食小鼠体重增加和脂肪量增加的最有效蛋白质来源。
蛋白水解和糖酵解发酵之间的平衡可能决定对生活方式干预的反应情况,因此应在未来的研究中加以考虑。


流行病学研究还表明,摄入乳制品和素食蛋白质来源与预防肥胖有关,而大量摄入肉类(尤其是红肉)则预示着体重增加会更高。
尽管研究较少,但蛋白质摄入已被证明会影响微生物群的组成和功能。效果取决于蛋白质的氨基酸组成和消化率,而蛋白质的来源和摄入量会影响它们。
蛋白质摄入影响微生物组成
在大鼠研究中,高蛋白饮食与C. coccoides, C. leptum, F. prausnitzii 减少有关,而超重或肥胖雄性中Roseburia, E. rectale, C. aerofaciens, Bacteroides, Oscillibacter 减少。
值得注意的是,以等热量的方式比较高脂肪/高蛋白饮食与中等蛋白质或低蛋白质饮食会导致饮食之间碳水化合物或脂肪含量的差异。因此,对于所有的等热量膳食宏量营养素交换研究,很难确定导致肠道微生物群组成变化的主要膳食因素,这可能归因于一种(宏量)营养素的增加或另一种营养素的减少。
膳食脂肪已被广泛研究与饮食相关的代谢疾病(如肥胖)相关,但其对人类肠道微生物群的影响尚不明确,而且研究通常会得出相反的结果。
不同类型的脂肪酸(饱和、单不饱和、多不饱和脂肪酸)、碳链长度和饱和度可能对肠道微生物群组成有明显影响。
横断面研究表明,食用富含动物蛋白和脂肪的饮食与拟杆菌属肠型有关,而高纤维、水果和蔬菜的摄入与健康成年人的普氏菌肠型有关。

此外,主要饱和脂肪酸(SFA)的高摄入量与成人和婴儿肠道微生物丰富度和多样性的降低有关。在超重和肥胖人群中, 主要饱和脂肪酸与肠单胞菌属呈负相关,而主要饱和脂肪酸与Roseburia呈正相关,后者在体重正常的个体中也非常丰富。在这项研究中,根据 BMI,习惯性 主要饱和脂肪酸摄入量与产丁酸菌表现出相反的关联特征。
总体而言,应该注意的是,与膳食纤维相比,膳食脂肪-微生物组-宿主生理学相互作用的研究较少,而且其机理知识主要基于动物研究。根据人类生理学比较难解释这些发现,应进一步研究。
多酚主要作为酚类化合物存在于水果和蔬菜中,以其作为抗氧化、抗炎、心脏保护、癌症预防和神经保护剂的有益作用而闻名。
补充天然存在于茶中的表没食子儿茶素-3-没食子酸酯(epigallocatechin-3-gallate) 2个月,对肥胖小鼠胆汁酸代谢和疣微菌科Verrucomicrobiaceae丰度均有影响,促进了A. muciniphila丰度的增加。在其他研究中,后者与有益的代谢作用有关。
此外,虽然也在动物模型中,但 8 周的多酚补充剂可防止饮食引起的肥胖和肠道炎症,这与Akkermansia的丰度增加有关。在健康、超重或肥胖的个体中,12 周的白藜芦醇和表没食子儿茶素-3-没食子酸酯联合补充剂改善了男性的代谢参数并减少了拟杆菌门,但女性没有。
以上两项研究都表明存在性别特异性微生物反应,在评估干预反应时应考虑这一点。

总体而言,在饮食中添加膳食多酚似乎可以促进肠道和代谢健康,尽管仍然需要对人体研究的机制见解。
基于微生物组的精准营养预测代谢健康参数,如血糖反应和变异性,或用于抵消代谢紊乱,目前已受到很大关注。
该领域的一项具有里程碑意义的研究表明,尽管餐后血糖反应的人际差异很大,但在机器学习算法的帮助下创建的个性化饮食(基于习惯性饮食、身体活动和肠道微生物群)可能会成功降低血糖反应和不良代谢健康,还有助于减肥。
研究测试在对不同类型面包的血糖反应中发现了显著的人际差异,并且这种血糖反应可以通过基线微生物组特征来预测。值得注意的是,这些研究主要基于他们对急性膳食挑战和短期干预的反应,而不是长期干预反应。
肠道微生物组的预测能力正变得越来越明显,特别是在检查纤维和粪便微生物群移植效果的研究中。在长期的肠道菌群检测经验实践中也证实,基线微生物特征是对饮食干预(例如,膳食纤维或复合蛋白质)的反应性的有趣生物标志物,也是个性化健康管理的应该纳入的指标基础。
微生物组-宿主代谢轴可能对胰岛素抵抗患者的饮食干预存在抗性,这表明干预可能需要更长的时间,或者需要摄入的功能性膳食成分(如膳食纤维)来诱导有益的效果。特定功能微生物群的特点是对膳食成分的不同消化能力,导致微生物代谢物(如 短链脂肪酸)的不同产生,随后影响宿主代谢的调节。
总的来说,在评估饮食模式和常量营养素组成不同的饮食时,重要的是要同时考虑饮食成分的数量和质量,由于与宿主的微生物和代谢表型的不同相互作用,在整体饮食方法中要考虑到微量营养素和生物活性成分,如多酚。
对饮食干预的反应不仅取决于肠道微生物群的特征,还取决于饮食、生活方式和环境因素以及代谢表型等临床特征之间复杂的多因素相互作用。
Jardon KM, et al., Gut. 2022
为了将基于精确的策略转化为医疗保健实践或指南,我们需要彻底了解为什么人们对饮食的反应不同,差异反应和相关表型是否长期保持,以及开发的算法在多大程度上是可重复的。
在饮食干预研究中通过最先进的方法进行详细的微生物和代谢表型分析至关重要。显然,鉴于复杂性,除了生活方式和环境因素的详细信息外,还需要详细的信息,包括出生方式、病史、药物使用情况(尤其是抗生素)、身体活动、心理压力和睡眠质量等。这也意味着需要先进的统计和建模方法来梳理不同因素的重要性。

主要参考文献:
Jardon KM, Canfora EE, Goossens GH, Blaak EE. Dietary macronutrients and the gut microbiome: a precision nutrition approach to improve cardiometabolic health. Gut. 2022 Feb 8:gutjnl-2020-323715. doi: 10.1136/gutjnl-2020-323715. Epub ahead of print. PMID: 35135841.
Agus A, Clément K, Sokol H. Gut microbiota-derived metabolites as central regulators in metabolic disorders. Gut. 2021 Jun;70(6):1174-1182. doi: 10.1136/gutjnl-2020-323071. Epub 2020 Dec 3. PMID: 33272977; PMCID: PMC8108286.
Jie Zhuye,Yu Xinlei,Liu Yinghua et al. The Baseline Gut Microbiota Directs Dieting-Induced Weight Loss Trajectories.[J] .Gastroenterology, 2021
Jie Z, Yu X, Liu Y, Sun L, Chen P, Ding Q, Gao Y, Zhang X, Yu M, Liu Y, Zhang Y, Kristiansen K, Jia H, Brix S, Cai K. The Baseline Gut Microbiota Directs Dieting-Induced Weight Loss Trajectories. Gastroenterology. 2021 May;160(6):2029-2042.e16. doi: 10.1053/j.gastro.2021.01.029. Epub 2021 Jan 20. PMID: 33482223.
谷禾健康
Phascolarctobacterium,考拉杆菌属,专性厌氧和革兰氏阴性细菌,可产生短链脂肪酸,包括乙酸盐和丙酸盐,并可能与宿主的代谢状态和情绪有关,在人体胃肠道大量定植。
每个人都有可能具有与其他人不同的个体特异性微生物遗传成分,并且可能从童年到成年持续存在。Phascolarctobacterium属物种在较长的时间内显示出相对较高的个体内稳定性。一项调查中国南方1-80 岁健康个体研究发现,随着年龄的增长(1至60岁),该菌数量逐渐增加,维持在高水平,但随着年龄继续增加,老年人(> 60岁)的其数量反而减少)。
来自梅奥临床医学中心的研究人员研究发现比较容易减肥的人体肠道内考拉杆菌属水平较高。该菌除了减肥同时也是肠道菌群动态平衡的关键调节因素。
Phascolarctobacterium 多形棒状杆菌,0.5 × 2.0 µm 至 0.5 × 5 –20 µm。在琥珀酸的存在下,小棒会变成带有多个分支的细长和碎片状的棒。革兰氏染色阴性、不运动、不形成孢子的化学有机营养物。丙酸是琥珀酸发酵的主要终产物。富马酸盐抑制其生长。在 30–37°C 时生长最快。
系统发育上属于韦荣菌科,厚壁菌门。到目前为止, Succiniclasticum ruminis是最近的系统发育邻居。
DNA G + C 含量(mol %):41.4–42.3 ( Tm )
主要存在于人体肠道内,我们根据谷禾数据库认为其是基石核心菌。
目前报告的3个物种如下:
Phascolarctobacterium faecium
Phascolarctobacterium succinatutens
Phascolarctobacterium wakonense
其中,Phascolarctobacterium faecium (P. faecium)最早是从以有毒桉树叶为食的动物考拉中分离出来的,它可能与肠道菌群的解毒有关。因此,P. faecium可能在人体胃肠道中发挥有益作用。该菌种是一种专性厌氧和革兰氏阴性细菌,不形成孢子、不运动、分解酶,属于厚壁菌门。
它在普通琼脂上生长不良,但在培养基中添加琥珀酸盐可促进其生长。表明其利用生长需要琥珀酸。
虽然在人类胃肠道的样本中经常检测到与P. faecium密切相关的未培养菌落,但文献中尚未描述从人类胃肠道中分离出Phascolarctobacterium和扩大培养物,从而限制了Phascolarctobacterium faecium相关的功能研究和临床应用。
Phascolarctobacterium wakonense从普通狨猴 (Callithrixjacchus) 粪便中分离出,他们不仅利用了琥珀酸,还利用了丙酮酸。补充丙酮酸后,他们同时产生丙酸和乙酸,而琥珀酸仅产生丙酸。
肠道菌群的遗传特异性可用来做宿主的“微生物指纹”。
研究发现人体肠道菌群的遗传特征比其相对丰度更具有个体特异性。其中Phascolarctobacterium succinatutens 的鉴别准确率达到了88%。
饮食模式
一项调查中国南方1-80 岁健康个体研究发现,随着年龄的增长(1至60岁),该菌数量逐渐增加,维持在高水平,但随着年龄继续增加,老年人(> 60岁)的其数量反而减少。作者指出这种现象可能与饮食习惯有关。
考拉杆菌属专注于利用其他细菌产生的琥珀酸盐,同时,琥珀酸的主要生产者拟杆菌属和副拟杆菌属的丰度因高脂饮食而增加,并且与体重呈正相关。
老年人和1岁以下个体消耗的脂肪相对较少,体重相对较低,这可能导致拟杆菌属和副拟杆菌属减少,可用于考拉杆菌属的琥珀酸盐减少。
已发现体重和脂肪量与Phascolarctobacterium丰度呈负相关,因此可以帮助预测肥胖风险。
与年轻人相比,老年人群的体育锻炼较少,这可能是Phascolarctobacterium减少的另一个原因。
高脂饮食组富含拟杆菌属和Phascolarctobacterium,在人体肠道微生物群中,拟杆菌属产生乙酸和琥珀酸作为主要代谢产物。琥珀酸在肠道中的过量积累会导致腹泻,而利用琥珀酸的细菌的存在可能对人类有益。因此,Phascolarctobacterium可能和拟杆菌,尤其Bacteroides thetaiotaomicron(常栖息在人类肠道中,能够消化多糖)等菌存在共生。
此外,除了Phascolarctobacterium,研究发现高脂肪饮食更有可能导致大量产生丙酸和乙酸的细菌物种,如奇异变形杆菌(Proteus mirabilis)和韦荣氏球菌 (Veillonellaceae)。
含淀粉类食物、谷物和奶制品,与较高的Phascolarctobacterium 相对丰度。
一些小样本证据显示:
菊粉、岩藻多糖、中等剂量木糖醇可以增加Phascolarctobacterium 的丰度,但是低聚果糖的补充会降低Phascolarctobacterium。
此外,小檗碱和二甲双胍可以显着增加这种菌,这反过来可能有助于这两种药物对宿主的有益作用。
与其他菌互作
人体肠道中存在多种微生物,其中一些被认为是相互作用的。大多数这些相互作用涉及细菌代谢物。考拉杆菌属Phascolarctobacterium几乎不用碳水化合物进行生长,而是使用琥珀酸盐作为底物。研究发现Bacteroides thetaiotaomicron产生的琥珀酸支持Phascolarctobacterium菌的生长和伴随的通过琥珀酸途径产生丙酸。
丙酸生产的三种不同生化途径包括琥珀酸、丙烯酸酯和丙二醇途径。拟杆菌属拥有琥珀酸途径,该途径也存在于Phascolarctobacterium。然而,由于缺乏延胡索酸还原酶,推测P. faecium JCM 30894 无法产生琥珀酸,这是琥珀酸途径的关键代谢物。因此,与产生琥珀酸的细菌(如拟杆菌属)共存对于考拉杆菌属是必不可少的。
此外,从琥珀酸到丙酸的转化反应之一涉及甲基丙二酰辅酶 A 变位酶,它需要维生素 B12 。Bacteroides thetaiotaomicron 一些菌株缺乏维生素 B族 所需的上游基因12生物合成。
此外,发现Phascolarctobacterium与颤螺菌属一般呈正相关。
在一项对 314 名中国健康青年样本的队列研究中,9 个核心属中的 8 个,包括Blautia、Clostridium、Ruminococcus、Faecalibacterium、Subdoligranulum、Roseburia、Coprococcus、Bacteroides,彼此之间呈显著正相关,而核心属Phascolarctobacterium与其他八个核心属呈负相关。综合谷禾数据库和相关研究结果:

环境条件
考拉杆菌属(Phascolarctobacterium),这些细菌与他们的种族/地理和生活方式有关。通过对来自9个省份与自治区、7个民族的20个健康年轻人群的314名居民粪便进行16SrRNA测序,发现厚壁菌门、拟杆菌门、变形杆菌门和放线菌门是4种最主要的细菌门,其中,来自厚壁菌门的考拉杆菌属在人群中丰度占比较高。
人体肠道微生物群是可塑的,与周围环境密切相关。医务人员在日常工作中不断与患者接触,并暴露于医院环境中,这种高风险的接触与暴露使很多病原微生物成为医务人员手部微生物群的一部分而被携带。
万献尧教授团队2021年发表在《Clin Microbiol Infect》上的研究评估了医务人员与非医疗人员肠道微生物组的变化。与非ICU工作人员相比,ICU工作人员肠道内Phascolarctobacterium丰度显著增加。
疾病状态
而与健康人相比,早期肝癌患者中,考拉杆菌属(Phascolarctobacterium)和瘤胃球菌属(Ruminococcus) 明显减少。
在重度抑郁、阿尔茨海默病(AD)、自闭症等疾病中发现Phascolarctobacterium高富集,尽管疾病组内异质性也较高,因此,有必要开展大人群队列和临床验证该菌对于神经类疾病的发生和发展贡献情况。
在参与一项小型研究的复发缓解型多发性硬化症(RRMS) 患者中确定了肉类消费与其如何影响肠道细菌、免疫细胞谱和新陈代谢之间的关系。发现许多与多发性硬化症和多发性硬化症患者残疾严重程度与肠道4种细菌产气柯林氏菌、Coprococcus come、Phascolarctobacterium succinatutens、Sutterella wadsworthensis呈正相关。
Phascolarctobacterium属的减少与结肠炎症的存在有关。
阻止艰难梭菌定植
宿主免疫在肠道微生物群介导的对艰难梭菌感染 (CDI) 的定植抗性中发挥重要作用。研究发现人类微生物群相关小鼠中的 IL-22 信号传导调节宿主糖基化,这使得消耗琥珀酸的细菌Phascolarctobacterium 能够生长。在肠道微生物群中,Phascolarctobacterium降低了琥珀酸的可用性,这是艰难梭菌生长的关键代谢物,因此阻止了艰难梭菌的生长。
Phascolarctobacterium有助于肺癌的免疫治疗
免疫检查点阻断(ICB),特别是PD1/PDL1轴的阻断,为非小细胞肺癌(NSCLC)的治疗开辟了新的标准。然而,尽管临床护理取得了重大进展,但许多患者仍然对这些疗法无效。PD-L1 表达和肿瘤突变负荷等生物标志物与 ICB 疗效相关。Phascolarctobacterium在具有临床益处的患者中富集,并与延长的无进展生存期相关,而Dialister在进展性疾病患者中的代表性更高,其较高的相对丰度与无进展生存期和总生存期降低相关,具有独立的预后价值多变量分析。
有助于减肥
研究发现,比较容易减肥的人体肠道内考拉杆菌属(Phascolarctobacterium)水平较高,因此该菌也用来预测肥胖指标。而难以减肥的人体内则小类杆菌属(Dialister)水平较高。此外,在代谢综合征女性中观察到的Phascolarctobacterium属的丰度高于代谢综合征男性。
无论健康的核心细菌是如何定义的,以及群体研究中的鉴定结果有多么不同,可以肯定的是,普遍和优势的核心细菌对于宿主肠道稳态和健康至关重要。因此,重要的是发现一个全面的核心微生物群概况,以定义健康的肠道微生物群并指导它们对宿主健康的干预。
考拉杆菌属作为我们东方人肠道的核心菌属 ,其丰度高低对于维持健康和情绪等非常重要,后续期待更多关于该菌的深入研究信息。
主要参考文献:
Wu F, Guo X, Zhang J, Zhang M, Ou Z, Peng Y. Phascolarctobacterium faecium abundant colonization in human gastrointestinal tract. Exp Ther Med. 2017;14(4):3122-3126. doi:10.3892/etm.2017.4878
Watanabe Y, Nagai F, Morotomi M. Characterization of Phascolarctobacterium succinatutens sp. nov., an asaccharolytic, succinate-utilizing bacterium isolated from human feces. Appl Environ Microbiol. 2012 Jan;78(2):511-8. doi: 10.1128/AEM.06035-11. Epub 2011 Nov 11. PMID: 22081579; PMCID: PMC3255759.
Ogata Y, Suda W, Ikeyama N, Hattori M, Ohkuma M, Sakamoto M. Complete Genome Sequence of Phascolarctobacterium faecium JCM 30894, a Succinate-Utilizing Bacterium Isolated from Human Feces. Microbiol Resour Announc. 2019;8(3):e01487-18. Published 2019 Jan 17. doi:10.1128/MRA.01487-18
Bhandarkar NS, Mouatt P, Majzoub ME, Thomas T, Brown L, Panchal SK. Coffee Pulp, a By-Product of Coffee Production, Modulates Gut Microbiota and Improves Metabolic Syndrome in High-Carbohydrate, High-Fat Diet-Fed Rats. Pathogens. 2021 Oct 22;10(11):1369. doi: 10.3390/pathogens10111369. PMID: 34832525; PMCID: PMC8624503.
Zheng YH, Xu Y, Ma HX, Liang CJ, Yang T. Effect of High-Fat Diet on the Intestinal Flora in Letrozole-Induced Polycystic Ovary Syndrome Rats. Evid Based Complement Alternat Med. 2021 Jun 25;2021:6674965. doi: 10.1155/2021/6674965. PMID: 34257691; PMCID: PMC8257354.

谷禾健康

有没有觉得无论你一天刷了多少次牙,仍然需要吃几颗口香糖?
或者外出与人交谈时对自己的呼吸感到不自在?
……
你不是一个人。
事实上,现代人们饮食不规律,加上高油高脂高盐以及辛辣等饮食习惯,存在口臭问题的人群越来越多,口臭是目前消化科门诊较为常见的主诉之一。口臭给人们自身健康和心理问题都会带来很大困扰和担忧。

口臭与多方面因素有关,大多数口臭可能是由各种口腔问题引起的,但同时,口臭也可能是其他健康问题的征兆。例如消化系统的失调可能是重要原因。消化系统不仅包括口腔,还包括食道、胃、小肠和大肠等,因此这些地方出现问题都可能导致口臭。这其中,被扰乱的微生物群平衡是造成口臭的不可忽视的因素之一。
Zanetti F, et al., Front Oral Health. 2021
本文结合对口臭研究的最新进展,从更广泛的角度来了解与口臭相关的原因,包括口腔病理因素、口腔菌群的作用、细菌代谢途径、消化道疾病,肠道菌群失调等,同时也包括一些口臭相关干预措施。
口臭分为几种:真口臭、假口臭、口臭恐惧症。
这其中真正的口臭又分为生理性和病理性两种。
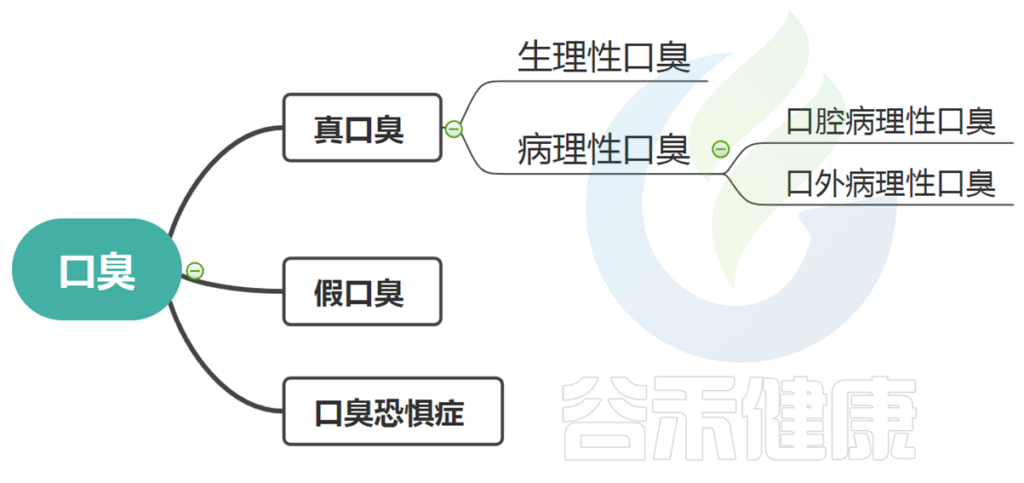
什么叫真口臭和假口臭?口臭恐惧症又是什么?
我们一条条来看。
——“ 没有口臭觉得有口臭 ”
这种分类是指患者没有实际的呼吸气味问题(嗅觉或科学测试无法检测到),但他们仍然确定自己有口臭。
假性口臭约占15%。
——“ 总觉得口臭被别人嫌弃,尴尬,不敢正常交流 ”
此类别是指尽管患者的真正口臭状况得到成功治疗,或者在接受咨询后假性口臭的情况下,患者仍然对呼吸问题有感知,其他人的行为(例如,打开窗户、嗅嗅、摸鼻子等)被误解为口臭的证据。并且在许多情况下,朋友和家人无法说服ta。其症状包括害怕呼气、抑郁和社会孤立,甚至会产生离职、离婚或自杀念头。
直接原因在于心理问题:
在许多情况下,患者受到有关口臭的评论或戏弄,存在心理创伤。这通常发生在童年时期,伴随着被拒绝或嘲笑的感觉一直到成年。其他也可能包括疑病症或强迫症等疾病。
后果可能加剧心理问题:
这种情况可能导致妄想的心理疾病,例如精神分裂症或双相情感障碍,也可能导致口臭恐惧症的发展。
此时,需要将患者病情的治疗转到心理医生处。
这一类别指的是口腔腐败(有机物的腐烂)导致的臭味,通常出现在舌头后部(最后端)的白色涂层内。
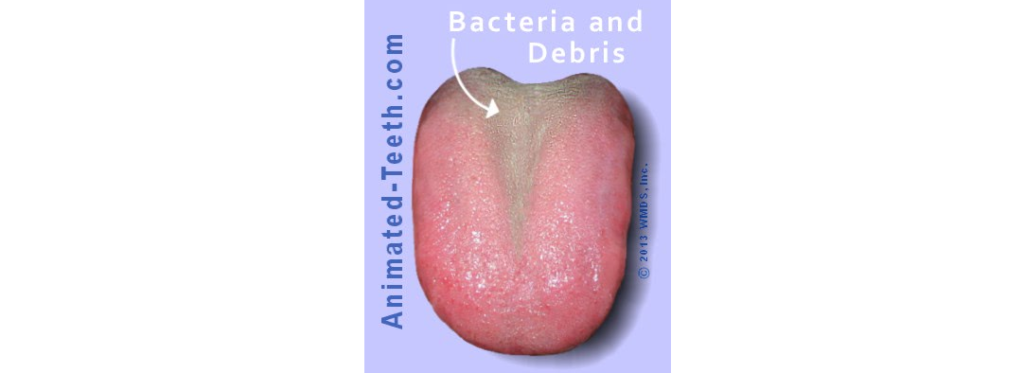
这是最常见的口臭形式,涉及约90%的真口臭病例。
不过这种口臭一般被认为是短暂的,因为它时有时无,由口腔中的临时局部条件决定。
患者通常可以通过改善口腔护理来解决生理性口臭,尤其是舌头清洁。
病理性口臭又分为口腔病理性口臭和口外病理性口臭。
口腔病理性口臭:
这一类别包括由口腔内组织相关疾病或其他病理条件引起或加重个人气味问题的情况。
举个例子,一个人口臭可能是由于牙龈疾病的存在,或因其加重。此外,相关因素(如口干和吸烟)也可能在病理性口臭中起作用。
口外病理性口臭:
在这些情况下,口臭源于涉及口腔以外的身体组织的疾病或病理状况。气味可能来自:
• 鼻、鼻旁或喉区域(上呼吸道)
可能的相关病症:鼻后滴漏、慢性鼻窦炎、急性病毒或细菌感染、扁桃体炎、扁桃体结石、深扁桃体隐窝。
• 下呼吸道(肺)或上消化道
可能的相关病症:慢性支气管炎、支气管扩张、裂孔疝、幽门螺杆菌感染、吸收不良病症。
• 身体其他部位的疾病。在这些情况下,疾病过程产生的化合物是血液传播的,当它们从肺部呼出时,会产生一种呼吸恶臭的状态。
可能的相关疾病:糖尿病、肝硬化、尿毒症、肾功能不全、月经周期、内出血。
由此可见,口臭的原因包括口腔和消化道等其他因素,下面我们主要从口腔和消化道两方面对口臭的形成进行深入了解。
前面我们了解到,在这几种口臭类型中,口源性口臭占比较大。其病因有龋齿、牙周病、口腔感染、种植体周围炎、冠周炎、黏膜溃疡、潴留的食物或残渣、舌苔等。
虽然牙周健康的人也可能出现口臭,但牙周炎是口臭的原因之一。如果不及时清洁,口腔中残留的食物颗粒会腐烂并产生异味。牙齿护理不到位可能会导致口腔中的菌斑积聚,从而产生气味。牙菌斑积聚在牙齿上也会导致牙周病。当斑块变硬时,变成牙垢。牙垢中的细菌会刺激牙龈,导致牙龈疾病。轻微的牙龈疾病称为牙龈炎;如果不治疗牙龈炎,可能会发展为牙周炎。
这些患者的口臭主要是由口腔环境中基质腐败引起的,这其中微生物扮演了重要角色。研究发现,口腔内的多种细菌与口臭的产生紧密相关。
目前认为,口源性口臭源于口腔微生态的平衡被打破,口腔内菌群失调造成产臭厌氧菌比例的增加,致臭菌含硫蛋白代谢产物释放增多,从而引起口臭。
硫氨基酸降解的一般模式和口臭的机制
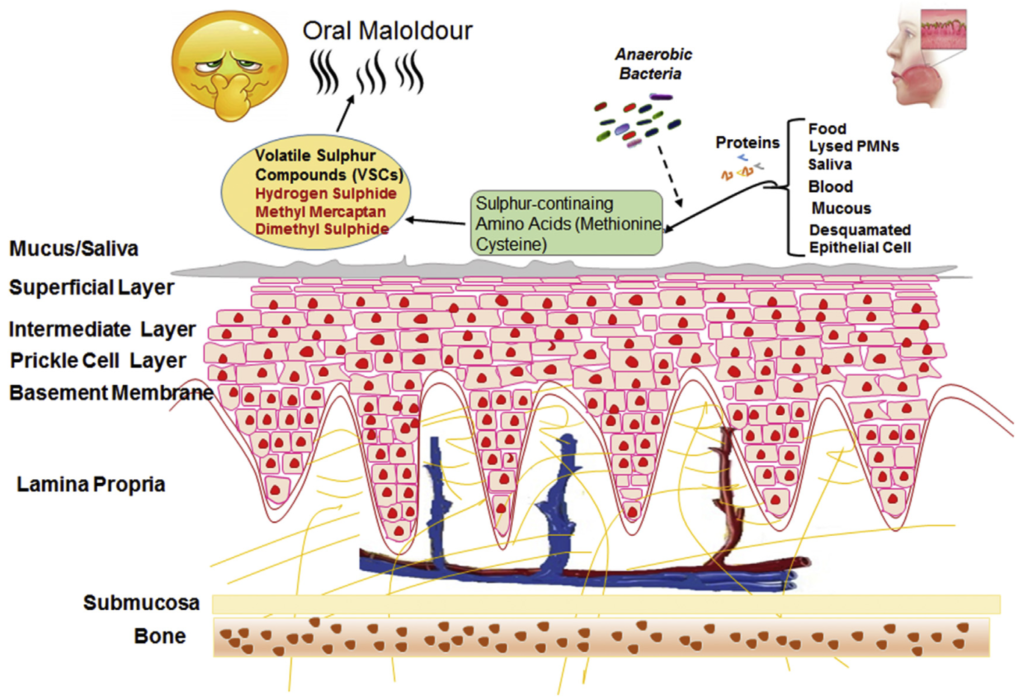
Karbalaei M, et al., New Microbes New Infect. 2021
接下来,我们将从与口臭有关的化合物、菌群、发酵底物、代谢途径、环境这些个方面来阐述它们是如何导致口臭的发生。
与口臭有关的化合物
口腔中基质腐烂会产生各种代谢物,包括挥发性硫化合物 (VSC) 和其他有机化合物,这些代谢物挥发气味就出现了口臭。
口臭主要归因于挥发性硫化合物 (VSC)。在患有口内口臭和牙周炎的受试者中,还报告了挥发性芳香族化合物水平升高,例如吲哚、多胺、粪臭素、吡啶和甲基吡啶,而这些在健康受试者中没有检测到。
呼吸气体中腐败化合物列表
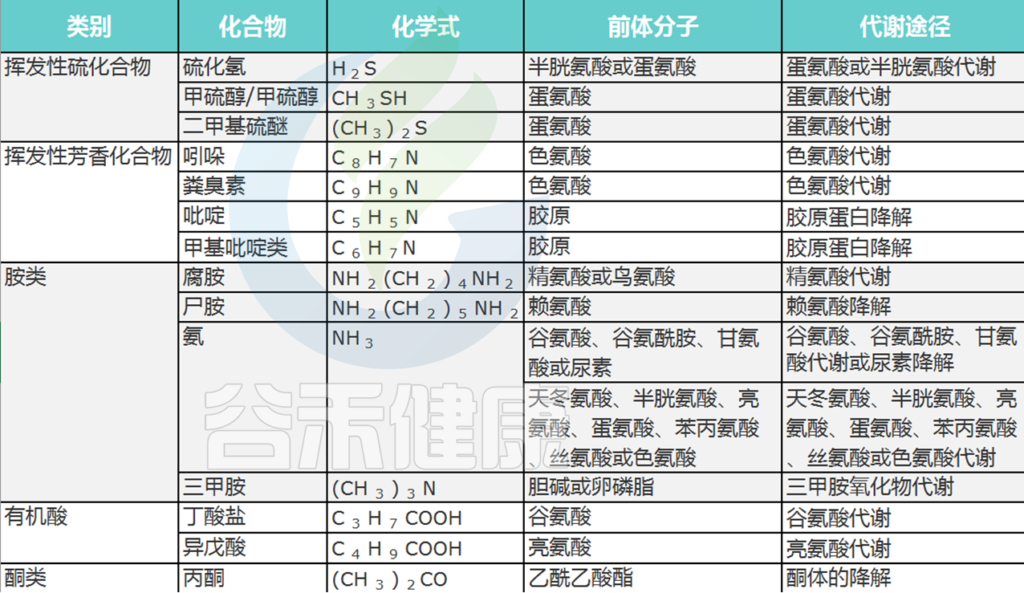
Foo LH,et al., Crit Rev Microbiol. 2021
在VSC中,与口臭有关的最重要的化合物是:
硫化氢 (H2S)
甲硫醇 (CH3SH)
二甲硫醚 [(CH3)2S]
其中,硫化氢和甲硫醇占口腔内VSC的90%.
口臭患者的口腔空气中可能含有H2S和CH3SH或两者。这两者浓度在口内口臭中可能会有所不同。这是因为高H2S和高CH3SH患者的菌群结构在系统发育上是不同的。
接下来我们来看看具体哪些菌与口臭相关。
与口臭有关的口腔菌群
研究表明,产VSC的细菌很多,如Solobacterium moorei,牙龈卟啉单胞菌,密螺旋体Treponema denticola,中间普雷沃氏菌Prevotella intermedia,口腔链球菌Streptococcus oralis,Tannerella forsythia 等。
挥发性硫化合物(VSCs)与口腔细菌的相关性
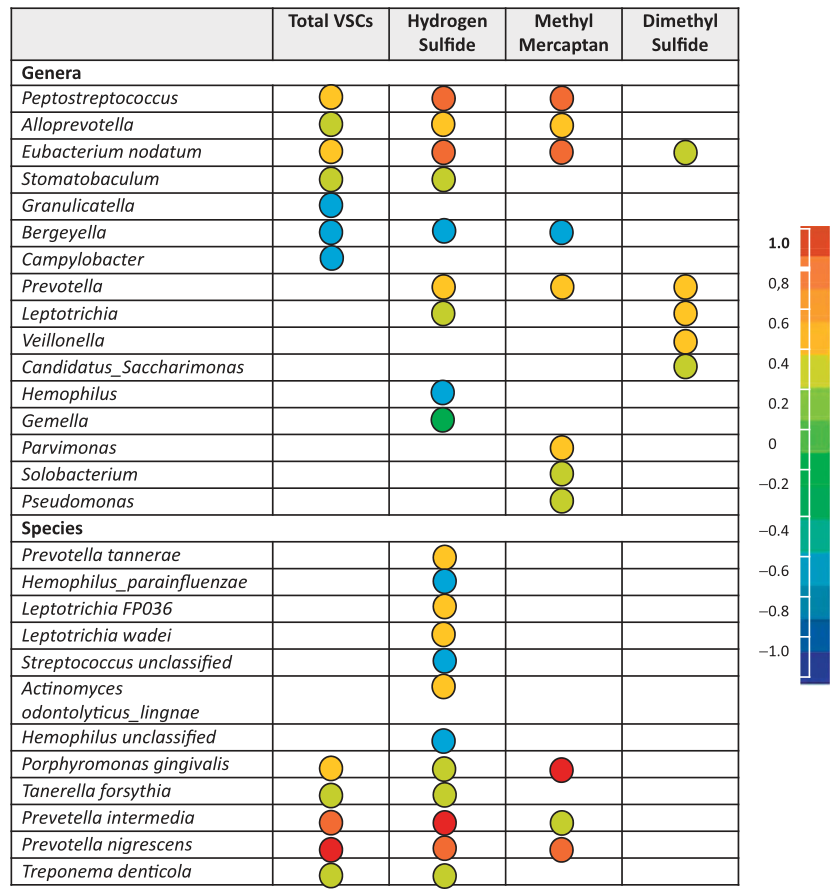
Foo LH,et al., Crit Rev Microbiol. 2021
前面我们知道,在口腔空气中H2S和CH3SH含量高与口臭相关。
在H2S较高的患者中,奈瑟菌属、梭杆菌属、卟啉单胞菌属和SR1属的比例更高。
相比之下,高CH3SH患者的普雷沃菌属、韦荣球菌属、阿托波姆属Atopobium、巨球菌属Megasphaera、硒单胞菌属Selenomonas的比例较高。
在体内研究发现,嗜血杆菌Hemophilus和孪生球菌属Gemella与口内口臭呈负相关。
那么这也说明了,气味化合物的产生是一个非常复杂的现象,任何单一细菌物种的存在都不能解释口臭的原因。不同菌群之间会产生相互作用,口臭可能是许多不同细菌的活动共同导致的。
这些细菌为生存形成了代谢网络。
不同口腔生态位中口臭和健康的微生物群结构
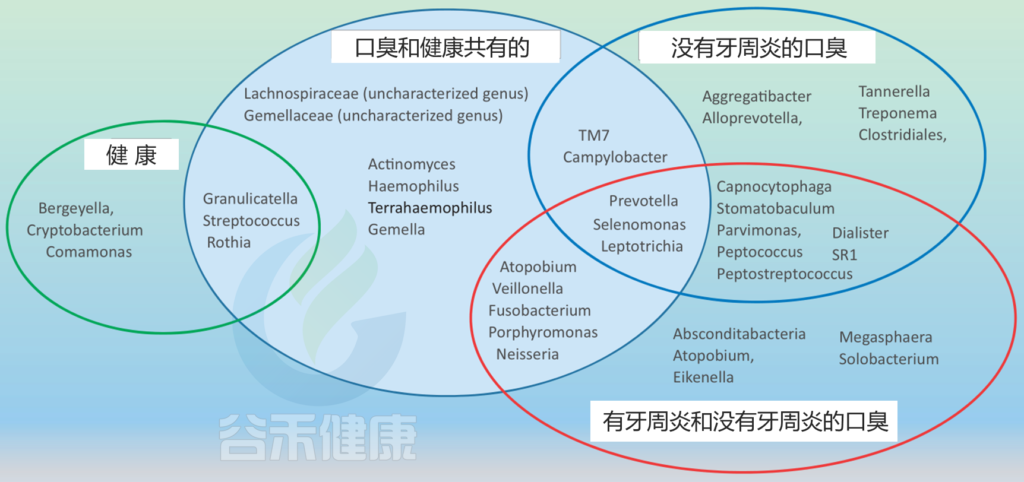
Foo LH,et al., Crit Rev Microbiol. 2021
健康组包括在没有口臭的受试者中存在的属;非牙周炎组的口臭包括牙周健康者伴有口臭的属;口臭和健康共有组包括健康和口臭的属,不考虑牙周炎;有牙周炎和没有牙周炎的口臭组包括存在于口臭的属,这些属的研究没有明确排除或提到牙周炎的影响。
与无口臭者相比,牙周健康者的微生物群中下列菌较多:
普雷沃菌属Prevotella、Alloprevotella、
Aggregatibacter、Campylobacter、
嗜二氧化碳噬细胞菌Capnocytophaga、
梭菌目Clostridiales、 小类杆菌属Dialister、
Parvimonas、消化球菌属Peptococcus、
Selenomonas、SR1 genera、
坦纳菌属Tannerella、TM7、Treponema、
纤毛菌属Leptotrichia、 Stomatobaculum、
消化链球菌属Peptostreptococcus 等。
有趣的是,在物种水平上,牙周炎相关的物种在牙周健康受试者舌背的微生物谱中均未检测到,如:
牙龈卟啉单胞菌P. gingivalis、Treponema denticola、T. forsythia、Capnocytophaga.
在一项基于PCR的研究中,81.1%的口臭患者样本中未检出牙龈卟啉单胞菌,86.5%的样本中未检出F. nucleatum和48.6%的样本中未检出P. intermedia。
由此可以推断,健康相关性口臭的口腔微生物群与牙周炎引起的口臭不同。
细菌发酵底物
吃东西的过程中,有些食物可能会卡在牙齿里,成为细菌的发酵底物,促进细菌和牙菌斑的生长,从而导致口臭。
口腔细菌的主要营养素来自唾液、龈沟液和脱落的上皮,含有糖蛋白、蛋白质、肽和氨基酸。
蛋白水解细菌可以通过细胞膜结合或细胞外分泌的蛋白酶将这些化合物降解为小肽和氨基酸,以便随后用作代谢底物。
那么具体哪些细菌,如何将这些底物发酵,从而导致口臭?
接下来的小节,我们来详细看看它们的代谢途径。
口臭相关细菌的代谢途径
牙龈卟啉单胞菌有牙龈蛋白酶(类胰蛋白酶半胱氨酸蛋白酶)和二肽基肽酶,而中间普氏菌有几种降解白蛋白和免疫球蛋白的蛋白酶。
革兰氏阳性菌,如唾液链球菌S.salivarius和 S. moorei,可以通过使唾液糖蛋白去糖基化,使蛋白核心可供革兰氏阴性菌进一步降解,从而间接促进口内口臭。
革兰氏阳性菌产生的酶和蛋白水解细菌的协同作用可以促进VSC的产生。虽然关于口腔中氨基酸降解的信息不多,但最常见的口臭代谢途径如下:
蛋氨酸和半胱氨酸代谢
蛋氨酸和半胱氨酸是主要的含硫氨基酸,属于正常发育所需的必需氨基酸。蛋氨酸-半胱氨酸代谢的第一步涉及天冬氨酸末端羧基的两次连续减少,以形成高丝氨酸(下图)。
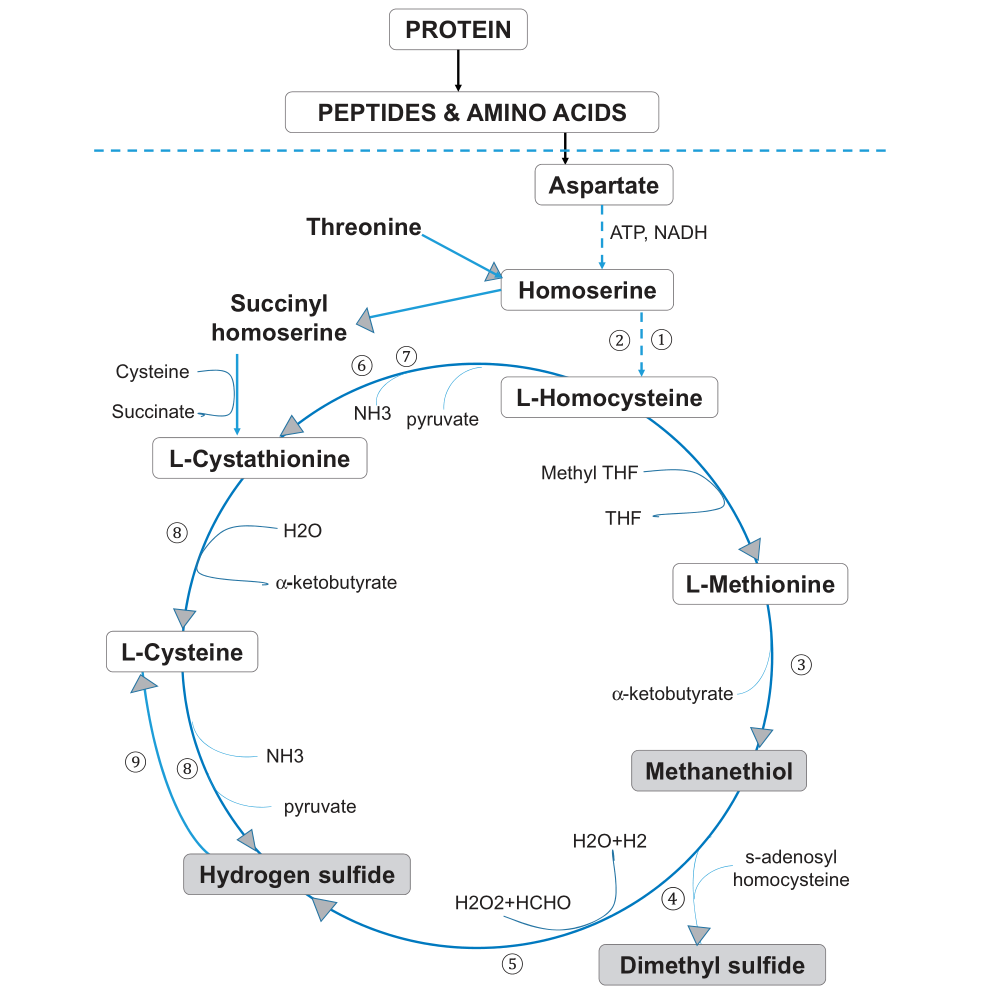
Foo LH,et al., Crit Rev Microbiol. 2021
口腔厌氧菌利用谷氨酸和/或天冬氨酸或其肽作为能量底物。
例如,牙龈卟啉单胞菌P. gingivalis、 中间普氏菌P. intermedia、P. nigrescens可以使用aspartyl aspartate,而牙龈卟啉单胞菌、具核梭杆菌F. nucleatum也可以使用谷氨酰谷氨酸glutamylglutamate作为能源。
高丝氨酸的激活允许发生反式磺酰化,形成同型半胱氨酸。同型半胱氨酸可以重新甲基化形成蛋氨酸,也可以通过反式硫化途径转化为半胱氨酸。通过METase酶促作用,同型半胱氨酸再甲基化为蛋氨酸导致VSC、甲硫醇或CH3SH的形成。
牙龈卟啉单胞菌、具核梭杆菌、齿垢密螺旋体T. denticola是通过转化酶活性产生甲硫醇的关键口腔微生物群。METase存在于细菌的细胞内和真菌的细胞外,但在哺乳动物中不存在。因此,抑制这种酶对人类的影响应该很小,使其成为治疗口腔异味的潜在药物靶点。
在蛋氨酸代谢的后续步骤中,甲硫醇通过酶、硫醇S-甲基转移酶和CH3SH氧化酶进一步甲基化,生成其他VSC,如(CH3)2S和H2S.
色氨酸代谢
经典的牙周病原体牙龈卟啉单胞菌、中间普氏菌和核梭杆菌是通过色氨酸代谢途径产生吲哚和粪臭素的主要细菌。
色氨酸也是一种必需氨基酸,由膳食蛋白质提供。它被色氨酸酶降解,色氨酸酶在氧化和脱氨基后产生中间产物吲哚-3-丙酮酸和吲哚-3-乙酸,最终形成粪臭素(下图)。
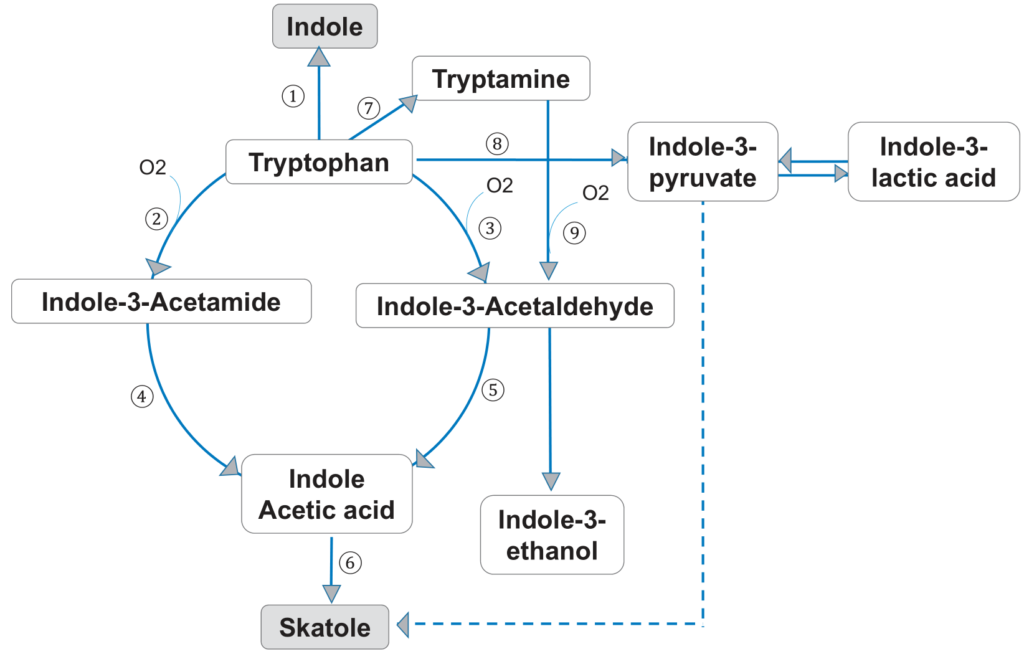
Foo LH,et al., Crit Rev Microbiol. 2021
另一方面,吲哚可以通过色氨酸酶的一步催化作用产生。吲哚和粪臭素是色氨酸的两种主要代谢物,与口内口臭有关。据报道,中间普氏菌、牙龈卟啉单胞菌、具核梭杆菌等革兰氏阴性蛋白水解细菌在口腔内产生大部分吲哚和粪臭素。
然而,纯革兰氏阳性细菌培养物,如S. salivarius, S. mutans, A. naeslundii, L. acidophilus,并不产生这些代谢物。
相比之下,最近在肠道微生物组学方面的研究表明,色氨酸的降解可能不仅限于蛋白水解细菌,革兰氏阳性乳酸杆菌也表现出分解色氨酸的能力。
影响口臭的环境因素
舌苔
舌苔的形成是与口腔卫生不良、牙周组织发炎、戴假牙、吸烟和饮食摄入等因素有关。舌背不规则且深度裂开,难以进入该区域进行清洁,这会促进脱皮的上皮细胞和食物残渣保留在舌背表面。这种位置为微生物在舌头上粘附和增殖创造了理想的环境,并能很好地防止唾液的冲洗作用。
在戒除舌头卫生的情况下,平均细菌总数往往会显著增加,而在清洁舌头后,VSC减少75%。
舌头生态位中的低氧水平会使微生物群向厌氧菌群倾斜。
在一项实验研究中,当受试者停止舌头清洁七天时,牙龈卟啉单胞菌P. gingivalis、齿垢密螺旋体T.denticola、T. forsythensis、中间普氏菌P. intermedia、P. nigrescens的数量显著增加。虽然口臭的强度与不同的细菌定植模式显著相关,但这些关联与舌苔评分无关。
其他研究也报告了患有和不患有口臭的牙周健康受试者在可观察到的舌苔和厚度得分方面没有显著差异。
因此,可以推断,VSC和口内口臭的原因不是舌苔厚度,而是高细菌密度。
唾液流
平均未受刺激的唾液流速范围为0.3至0.4 毫升/分钟,进餐时可以增加到4-5.0 毫升/分钟。但在唾液流量减少的受试者中,静息唾液流量降至0.1毫升/分钟以下,并被指定为唾液分泌不足。
药物治疗(如抗抑郁药、抗利尿药和抗高血压药)、唾液腺疾病(如Sjögren综合症)、不受控制的糖尿病、病毒感染和其他医疗(如化疗和放射疗法)都可能导致唾液分泌减少。唾液流量减少使口腔的正常清洁机制效率低下,增加了细菌腐败的基质。
此外,唾液本身具有抗菌活性。晨间口臭是一种非病理性口臭,由夜间唾液分泌减少引起,具有短暂特征。
需要注意的是,口呼吸还可以促进硬腭、舌背和口腔粘膜周围的干燥,间接促进VSC的产生。
在当前的SARS-CoV-2大流行中,长时间使用口罩,以及戴口罩时的口腔呼吸习惯,往往会让人们更加注意自己的口气(“口罩口臭”)。
牙周袋
在牙周炎患者中,VSC水平和CH3CH/H2S比率与出血指数和探测深度成比例增加。这主要是由于牙周袋深处的氧气压力较低,使其成为能够产生VSC的厌氧细菌的理想环境。在可培养的口腔细菌中,与牙周炎相关的厌氧革兰氏阴性菌(P. gingivalis, T. denticola, T. forsythia)是体外H2S最活跃的产生菌。
由于牙齿和舌头周围的清洁不足,牙周炎患者的口腔卫生状况通常较差。此外,它们的唾液中含有脱落的上皮细胞和白细胞,这些细胞可能沉积在舌头表面,并间接为VSC的形成提供二硫化物和硫醇基团。这解释了牙周炎患者产生的舌苔是非牙周炎患者的四倍。
然而,它并不一定代表牙周炎是口内口臭的原因,因为舌苔的厚度不一定等同于微生物量。事实上,如前所述,已知与VSC产生有关的是舌苔中的微生物密度,而不是舌苔溃疡。因此,可观察到的舌苔数量与舌苔上的微生物数量关系不大。
同样,也可以说,像脱落的上皮细胞一样,来自牙周袋深处的牙周病原体也可能会移位,并在舌头表面定居,在那里它们可以生长和繁殖。因此,牙周微生物群可能会影响舌头微生物群的定植,并加剧VSC的生成。
牙列(包括牙齿修复中的问题)
口腔内任何部位的细菌积聚、食物嵌塞和随后的腐败都可能有利于口内口臭。因此,口内口臭最常见的牙齿来源包括深龋病变、牙间食物嵌塞和牙齿拥挤。
固定正畸支架和悬垂修复体也可能是食物滞留和腐败的潜在场所。悬垂修复可以改变微环境,有利于增加间接导致口内口臭的厌氧细菌(V.alcolescens、V.parvula和产黑色素拟杆菌)。
丙烯酸假牙,尤其是在夜间存放在口腔中或不定期清洁时,也会产生典型的口腔气味。面向牙龈的义齿表面是多孔的,对细菌、酵母、碎屑和所有导致腐败的因素都有保持力。
暴露在外的牙髓营养丰富,是细菌和口内口臭的另一种合适基质。已从坏死牙髓中分离出混合厌氧菌(普雷沃氏菌、Furobacterium、梭菌、消化链球菌Peptostreptococcus、链球菌、乳酸杆菌)和常见的牙周病原体(牙龈卟啉单胞菌、齿垢密螺旋体),表明它们在口内口臭中的作用。
牙齿修复可以改善口腔微生物生态,这取决于所用修复材料的性质。例如,修复过程中金属或氟离子的泄漏可能会干扰细菌粘附并防止其积聚。另一方面,当修复材料深入牙龈时,可能会导致出血,并增加拟杆菌和螺旋体等厌氧菌的比例。
物理化学条件
除了存在细菌和基质外,物理化学条件对于产生有气味的挥发物至关重要。口腔卫生不良会导致菌斑厚度增加,以及菌斑外层细菌对氧气的利用率增加。这导致大面积的厌氧和低氧化还原条件,促进口内口臭的形成。
此外,在口腔中,温度在34-37℃之间变化,经口呼气时湿度在91%-96%之间变化。这些条件也可能为口内口臭相关细菌的生长提供合适的环境。
一般来说,温度会影响分子本身的挥发性,气体在较低温度下的挥发性较小。要使化合物有气味,它必须是挥发性的。口内口臭的强度与吲哚或粪便的浓度之间缺乏相关性,这可能是因为吲哚在室温和37℃下的挥发性较低 。
pH值也是产生口臭的关键因素之一。上述pH值对于口腔异味的形成至关重要,而酸性pH值可以起到抑制作用。由于口腔粘膜表面的pH值主要由粘附细菌的发酵和腐败活动决定,因此了解pH值是有必要的。
以上是与口臭有关的口腔原因,当然口臭的原因不仅限于此。
消化系统疾病导致许多口臭。这类口臭主要是由气味挥发物从肠胃通过食道泄漏到口鼻。然而,由肠道引起的口臭通常是消化系统总体失衡的标志。
消化道疾病
胃食管反流病,胃酸/胃灼热/胃痛的情况都可能导致气味。
腹胀、胀气和打嗝
任何让你打嗝的消化系统疾病都可能导致口臭。这些包括消化系统的不平衡,如肠易激综合征、食物不耐受或高糖摄入。
肠梗阻或便秘
当你的身体不能正常消化食物时,一个不幸的副作用可能是,出现类似粪便的口臭。
肠道屏障薄弱
当肠道屏障被削弱时,它会被损坏,也就是出现“肠漏”,不能再作为屏障发挥最佳作用,于是一下麸质、有害细菌、未消化的食物颗粒、化合物和毒素等有害物质都开始“泄漏”出来。这些物质会进入身体系统,可能出现发炎,对健康造成损害,也包括出现口臭问题。
导致肠漏的因素有很多,例如药物的使用,过多摄入糖分,食物过敏刺激肠壁,缺乏维生素 A 和 D 等营养素,过量饮酒,慢性压力,自身免疫性疾病等。
菌群失调
除了这些明显的疾病或症状之外,有些容易被忽略的问题也可能导致口臭,比如肠道菌群失衡。
前面我们知道,挥发性亚硫酸盐化合物是罪魁祸首——硫化氢、甲硫醇、二甲硫醚。
这些也会由结肠细菌通过食物发酵产生的。
这些气体可能会从结肠中被重新吸收,然后出现在呼吸中。
结合谷禾肠道菌群数据库,我们整理了与口臭相关的常见肠道菌属如下:
口臭相关肠道菌属

幽门螺杆菌:
幽门螺杆菌感染造成口臭存在争议。幽门螺杆菌具有菌株依赖的能力,可以在体外从结合的半胱氨酸-蛋氨酸底物中合成硫化氢和甲硫醇。
与健康对照组相比,感染幽门螺旋杆菌的患者呼吸中氰化氢和硝酸氢水平都有所升高;然而,这是否代表这类型的口臭尚不清楚。也可能是由于幽门螺杆菌在口腔的定植引起口内口臭。
一些研究报告了幽门螺旋杆菌和口臭之间的正相关关系,然而,其中一些研究可能因为基于自我报告的口臭标准,而不是呼吸分析。其他的报告没有统计上显著的相关性。
饮食
食物是口腔异味的主要来源。有些食物,如大蒜、洋葱、辛辣食物、一些奶酪、鱼、酸性饮料,如咖啡,可能会留下挥之不去的气味。大多数时候,这种气味是短暂的。
然而,饮食与肠道菌群密切相关,糖的摄入过多,蛋白质摄入缺乏等都会导致肠道菌群紊乱,对人体有害菌群大量生长,有益菌生存空间不足,身体受到损害,当然也包括可能出现的口臭。
低碳水化合物饮食也可能导致“酮呼吸”。这些饮食使身体燃烧脂肪作为能量来源。产生这种能量的最终产物是酮,当呼出时会在呼吸中产生口臭,类似丙酮的果味。
吸烟和咀嚼烟草会在口腔中留下化学物质。吸烟还可能导致口臭,如牙龈疾病或口腔癌。
口臭的相关因素有很多,除了上述常见的原因之外,一些身体疾病也会出现口臭症状,例如:
鼻窦感染、肺炎、喉咙痛(咽炎)和其他喉咙感染、普通感冒、流感、扁桃体结石、鹅口疮、支气管炎、鼻后滴漏、糖尿病、胃酸反流、乳糖不耐症、其他肠胃或消化等问题,一些肝脏疾病或肾脏疾病可能与口臭有关。
物体卡在鼻子里(通常是儿童)、酗酒和大剂量维生素补充剂也可能引起口臭。
扁桃体炎
与扁桃体炎有关的口臭可发生在急性、慢性和复发性扁桃体炎中。
扁桃体是位于喉部两侧的两小块腺体组织。它们构成你免疫系统的一部分,制造抗体和白细胞来攻击口腔内的细菌。它们是抵御食物或空气中细菌的第一道防线的一部分。
扁桃体像蜂窝一样,在它的表面和里面有一些孔隙,这些孔隙被称为隐窝。在这些隐窝里,藏有一些灰色或者灰白色的、质地比较硬的块状物质。这些块状物质就是扁桃体结石。
扁桃体结石最常见的症状是咽痛和口臭。如果结石比较小可能感受不到明显的症状,但是多数情况下会有明显的口臭。
鼻窦炎
鼻窦炎可能是一种长期的慢性疾病,伴有过敏和/或鼻子结构问题。长期鼻窦炎会严重影响生活质量。
鼻窦位于脸颊内、鼻子周围和后面。它们的作用是加热、湿润和过滤进入鼻腔的空气。它们还帮助发声。
鼻窦炎的症状因严重程度和涉及的鼻窦而异。它们可能伴随着口臭。鼻窦炎患者窦腔内的分泌物不能排出来。伴有细菌感染的话就会发生脓性分泌物,久而久之就可能发生腐败,通过鼻咽部流向口内,就会造成口臭。
其他呼吸系统疾病
如支气管扩张、肺脓肿或坏死性肺肿瘤都会产生难闻的气味。
过敏
许多用于治疗过敏的药物都会导致口干,这是导致口臭的另一个原因。此外,鼻后滴漏是一种常见的过敏症状,可导致口臭。过敏引起的鼻窦充血也会导致人们用口呼吸,导致口干,从而引起口臭。
药物
许多药物,包括治疗过敏的抗组胺药和利尿剂,可能会导致口干(如前所述),从而导致口臭。其他可能导致口臭的药物可能包括氨苯蝶啶(Dyrenium)和多醛。
糖尿病
糖尿病患者的胰岛素产量不足,导致他们燃烧脂肪并产生酮。这意味着他们容易出现酮呼吸。糖尿病患者的另一个口臭原因可能是慢性肾功能衰竭。
慢性肾功能衰竭
这可能会导致闻起来有鱼腥味或像氨水的气味,唾液中的大量尿素及其分解为氨会导致口臭。
怀孕
怀孕本身不会引起口臭,但怀孕期间常见的恶心和晨吐可能会引起口臭。此外,激素变化、脱水以及因渴望而吃不同的食物也可能导致怀孕期间的口臭。
首先,要排除是否是一些特殊疾病引起的口臭,例如糖尿病、肝功能衰竭等,如果是这类原因,则需要进行相应的疾病治疗。
其次,需要检查是否存在其他不太严重的疾病,如口腔疾病、鼻窦炎、胃食管反流、肠道菌群失调等问题。
如果存在较为明显的疾病症状,例如牙周炎,则配合口腔科医生进行相应治疗;如果症状不是特别明显,则可进行口腔菌群或者肠道菌群检测,看看是否由于菌群失调引起的口臭。
益生菌
研究表明,益生菌可以解决部分口腔问题,包括口臭。
唾液链球菌是一种非致病性的口腔优势菌,是最重要的共生益生菌之一,最常从无口臭的人群中分离出来。唾液链球菌K12可产生两种抗生素——唾液霉素A2 (SalA2)和唾液霉素B (SboB)。临床试验表明,含唾液链球菌K12的抗菌漱口水显著降低了VSC产生菌的水平。唾液链球菌K12作为一种益生菌,最初来源于口腔共生菌,在口臭的治疗中可以发挥关键作用。
益生菌预防龋齿
研究表明,食用含有鼠李糖乳杆菌GG (LGG) 和罗伊氏乳杆菌的牛奶可显着减少变形链球菌和远缘链球菌的数量,这是龋齿的两种主要致病菌。链球菌属发酵饮食中的碳水化合物。牙菌斑的 pH 值降低(从 7.0 到 4.0)会导致牙釉质脱矿。因此,益生菌可用作乳制品中的预防性细菌。
最近,基于体外和体内实验证明,魏斯氏菌Weissella cibaria(原属乳酸杆菌属)作为一种新型益生菌菌株可以预防龋齿,并显着抑制变形链球菌形成生物膜。这种细菌会产生大量的过氧化氢,并且可以与具核梭菌聚集并抑制这些病原体在口腔中产生VSCs(下图)。
益生菌预防口臭的可能机制
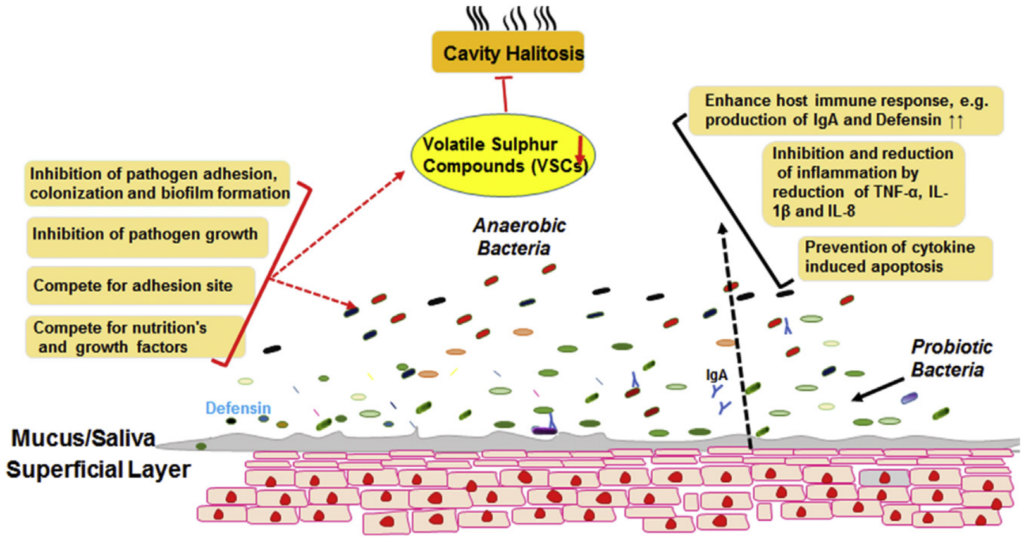
Karbalaei M, et al., New Microbes New Infect. 2021
应用于食品和健康领域的常用益生菌

*a 安全微生物的合格推定
Bustamante M, et al., Probiotics Antimicrob Proteins. 2020
另外需要注意的是,口臭并没有想象中的可怕。排除各类疾病的可能性,其余口臭原因都是生理性的,也就是短暂的,可能是吃了味道比较大的食物造成的,那么这类人群只要注意饮食和养成良好的口腔卫生习惯,问题不大。
注意饮食
吃的东西会影响呼吸的味道。需要注意的一些食物和饮料包括:
咖啡。除了喝完后留在嘴里的刺鼻气味外,咖啡中的咖啡因也会导致口干。
糖。高糖和精制碳水化合物(即白面包)的饮食与口臭有关。含糖食物会导致酸产生,不仅会增加蛀牙的风险,还会促进念珠菌的过度生长。
大蒜、洋葱和辛辣食物等味道浓郁的食物会在口腔中留下难闻的气味。虽然大蒜通常属于健康的食物,但不可否认,这些食物会导致暂时的口臭。
避免发炎的食物
炎症性食物会增加体内炎症,往往会加剧疾病症状,并可能导致口腔健康恶化。这些食物包括加工肉类、苏打水和含糖饮料、咸味零食、包装糖果和精制碳水化合物。特别是盐分过多会加剧口干并导致口臭。
抗炎饮食
另一方面,抗炎食物有助于减少体内炎症,并可能减轻症状,包括减少口臭。有许多抗炎食物,包括姜黄、生姜、绿叶蔬菜、浆果、亚麻和奇异籽、核桃、杏仁、橄榄油和鲑鱼等。
试试这些健康的饮食秘诀,包括吃新鲜蔬菜和尽量减少零食。还有一些更具体的食物已被经验证明可以改善口臭:
肉桂是一种抗病原菌的抗菌剂。为了缓解口臭,咀嚼肉桂棒、喝肉桂香料茶、咀嚼含有肉桂油的口香糖或用肉桂味漱口水漱口。
绿茶具有抗菌特性,可以减少异味。这是因为它含有抗氧化剂和多酚。

研究表明,绿茶可以减少与S. moorei 相关的口臭,并且它可能会降低龋齿和牙周病的发病率/严重程度。喝绿茶以促进整体口腔健康和治疗口臭。也可以制作冰绿茶,只需将绿茶叶放入罐中,加水并冷藏即可。
多喝水,少喝咖啡因和酒精
多喝水,但是避免苏打水、含糖饮料和酒精是个好办法。
如果患有口干症,那么饮用的饮料就显得尤为重要。全天饮水,以保持口腔中的液体持续流动。但是,避免气泡水,它会使口干变得更糟糕。含咖啡因的饮料和酒精都会使口腔干燥。尝试改用不含咖啡因的咖啡或茶来增加能量。
虽然有些食物可能会导致口臭,但还有其他食物实际上可以帮助改善口臭。某些食物可以通过它们的酶作用、食物中抗氧化剂的活性和/或食物的 pH 值来除臭。
例如,菠菜,生苹果,香菜,薄荷,绿茶等。
此外,发酵、富含益生菌的食物,如酸奶、泡菜、康普茶、开菲尔、酸菜等,可以帮助促进口腔中“好”细菌的平衡,并阻止念珠菌的生长。
多喝水不仅几乎可以立即冲走细菌,还有助于防止口干。目标是每天 6-8 杯。
椰子油
椰子油具有天然的抗炎和抗菌特性,每天用椰子油漱口或在嘴边刷几分钟,可以预防蛀牙和牙龈炎。
柠檬
由于其抗菌和抗氧化作用,柠檬可以帮助控制口臭。在吃过大量洋葱或大蒜后,用柠檬水漱口可能特别有用。此外,在舌尖滴一滴柠檬有助于刺激唾液分泌,从而缓解口干。
补充剂
锌
体内有 300 多种不同的细胞功能使用锌,包括与肠道菌群、口腔和微生物组有关的细胞功能。研究表明,锌可有效减少导致口臭的含硫化合物的积累。可以使用含锌漱口水来控制口臭。
维生素D
获得足够的维生素 D很重要,原因有很多,包括口腔健康。维生素 D 的缺乏会导致灼口综合征,它会使口腔中出现金属味或苦味,同时还会导致口臭。
良好的口腔卫生习惯
每天刷牙(和舌头)两次,每天使用牙线,每六个月去看一次牙医,但口腔健康不仅仅是最基本的要求。以下是维持长期口腔健康的 3 个不太明显的技巧。
正确的刷牙
刷牙有助于清除口腔中的菌斑和细菌,防止蛀牙、蛀牙甚至牙周病。虽然刷牙是我们每天都会做的事,但刷牙也是个技术活,刷得不到位可能会出现口腔问题。
这里再次强调一下正确的刷牙方式。
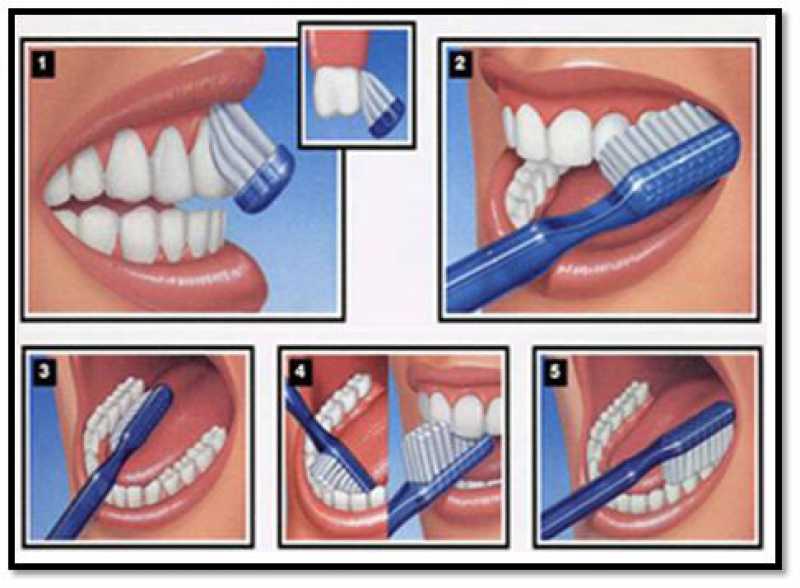
首先,刷牙齿的外表面。轻轻地刷你牙齿的上下部分,可以帮助你减缓这个过程,防止你遗漏任何斑点。
然后,倾斜你的刷子45度来清洁牙龈线。它可以帮助清除牙龈上滞留的菌斑和食物残渣。
牙齿的内表面是不可见的。因此,以45度的角度前后刷牙对清洁牙齿内部至关重要。
此外,来回刷你的后牙顶部。很多食物残渣都会在那个地方积聚。
最后,刷一下舌头,也就是下面提到的使用刮舌器,去除异味,呼吸清新。
使用刮舌器
口臭可能是由残留在舌头上的病原菌堆积引起的。使用刮舌器,或者牙刷背面刮舌。小心不要对舌头施加太大压力,因为重度刮擦会割伤组织。
治疗口干
口臭的另一个常见罪魁祸首是口干。如果觉得自己的口气需要清新,请记得多喝水有助于清除细菌并保持口腔湿润。为了长期增加唾液量,也可尝试使用益生菌等产品。
尝试盐水冲洗
将盐倒入温水中搅拌至溶解,然后在嘴巴和喉咙周围漱口。盐水的温和酸度会阻止有害微生物的生长。
每天使用漱口水
我们的嘴里含有平衡的好细菌和坏细菌。使用漱口水也是一种良好的习惯,不过尽量避免使用含酒精的漱口水,也可以试试专为口干设计的非酒精漱口水。
适度运动
新的研究表明,适度运动有益于口腔健康。锻炼时,可随身携带一瓶水(可能含有镁或钾)以保持水分。

主要参考文献:
Foo LH, Balan P, Pang LM, Laine ML, Seneviratne CJ. Role of the oral microbiome, metabolic pathways, and novel diagnostic tools in intra-oral halitosis: a comprehensive update. Crit Rev Microbiol. 2021 May;47(3):359-375. doi: 10.1080/1040841X.2021.1888867. Epub 2021 Mar 3. PMID: 33653206.
Mogilnicka I, Bogucki P, Ufnal M. Microbiota and Malodor-Etiology and Management. Int J Mol Sci. 2020;21(8):2886. Published 2020 Apr 20. doi:10.3390/ijms21082886
Bustamante M, Oomah BD, Mosi-Roa Y, Rubilar M, Burgos-Díaz C. Probiotics as an Adjunct Therapy for the Treatment of Halitosis, Dental Caries and Periodontitis. Probiotics Antimicrob Proteins. 2020 Jun;12(2):325-334. doi: 10.1007/s12602-019-9521-4. PMID: 30729452.
Zanetti F, Zivkovic Semren T, Battey JND, Guy PA, Ivanov NV, van der Plas A, Hoeng J. A Literature Review and Framework Proposal for Halitosis Assessment in Cigarette Smokers and Alternative Nicotine-Delivery Products Users. Front Oral Health. 2021 Dec 10;2:777442. doi: 10.3389/froh.2021.777442. PMID: 35048075; PMCID: PMC8757736.
Karbalaei M, Keikha M, Kobyliak NM, Khatib Zadeh Z, Yousefi B, Eslami M. Alleviation of halitosis by use of probiotics and their protective mechanisms in the oral cavity. New Microbes New Infect. 2021 Apr 23;42:100887. doi: 10.1016/j.nmni.2021.100887. PMID: 34123388; PMCID: PMC8173312.
Jo JK, Seo SH, Park SE, et al. Identification of Salivary Microorganisms and Metabolites Associated with Halitosis. Metabolites. 2021;11(6):362. Published 2021 Jun 7. doi:10.3390/metabo11060362

谷禾健康

口服酶可以对肠道微生物群的组成产生深远的影响,并且可以作为一种有吸引力的替代调节剂。
本文总结了酶影响肠道微生物群的三种方式,并讨论了选择合适的酶来调节肠道微生物群的挑战。
酶:一种被忽视的调节肠道微生物群的因子
人类肠道容纳多达100万亿微生物,包括细菌、古细菌、真菌等。
最近的一项研究发现,有178种肠道微生物可产生400多种高度丰富的代谢物,其中许多可通过肠道屏障迁移,进入血液循环,并对宿主进行免疫、代谢和神经元调节。
目前用于调节肠道微生物群的大多数策略都集中在化学品和整个微生物细胞上。而酶,作为具有催化功能的特殊蛋白质,却没有被广泛考虑。
几乎在所有生化反应中都需要酶。在畜牧业中,各种酶被广泛添加到饲料中,以促进动物的生长。它们的有益作用与肠道菌群的变化有关,并可能部分归因于肠道菌群的变化,这强烈表明可以有意选择酶来调节肠道菌群。
事实上,在小鼠和其他实验动物中,酶无疑已被证明对肠道微生物群有潜在影响。
酶可以通过三种方式影响肠道微生物群。
1
// 一些酶可以杀死肠道微生物
溶菌酶、类溶菌酶糖苷水解酶和细菌噬菌体溶菌酶直接降解细菌细胞壁的主要成分肽聚糖。

Su X, Yao B. Trends Microbiol. 2022
在小鼠中,口服赖氨酸-防御素嵌合蛋白可显著减少粪便艰难梭菌孢子,从而降低死亡率。酶可以产生对肠道微生物有害的反应产物。
在肠道中,管腔葡萄糖(浓度为几十毫摩尔)、L-氨基酸(回肠中的几毫摩尔),甚至细菌衍生的游离D-氨基酸(盲肠内容物中的约200-500 nmol/g)都可以被酶氧化,释放过氧化氢杀菌。在小鼠体内,D-氨基酸氧化酶导致霍乱弧菌(Vibrio cholera)在小肠中的定植显著减少。
2
// 酶可以刺激肠道微生物生长
在人类中,食物中约一半的木聚糖被肠道微生物木聚糖酶降解为低聚木糖,低聚木糖支持某些肠道微生物(如拟杆菌、双歧杆菌和乳酸杆菌属)的生长。

Su X, Yao B. Trends Microbiol. 2022
木聚糖酶的刺激谱可以通过肠道微生物的生态网络进一步扩展。
酶还可以通过催化去除有害化学物质来刺激肠道微生物生长。
肠道碱性磷酸酶(IAP)是一种由肠上皮细胞分泌的内源性酶,通过降低抑制细菌的管腔核苷酸三磷酸的浓度来促进特定肠道微生物的生长。这与减轻肠道屏障损伤和减轻酒精诱导的肝脂肪变性等疾病密切相关。
3
// 酶能通过干扰微生物网络影响肠道菌群
群体感应(QS)是微生物群形成网络的一种方式。通过感应信号分子,微生物同步生成生物膜并排出有毒分子。

Su X, Yao B. Trends Microbiol. 2022
通过N-酰基高丝氨酸乳糖等酶去除QS分子(群体猝灭)可以调节金鱼的肠道微生物群,这可以通过增加变形菌的丰度和减少肠道中致病性嗜水气单胞菌(Aeromonas hydrophila)来证明。
分散素B也证明了微生物网络的干扰,它水解生物膜稳定剂1,6-N-乙酰-D-葡萄糖胺,从而显著减少铜绿假单胞菌(Pseudomonas aeruginosa)在肠道中的定植。
研究表明,有充分的机会利用酶来调节肠道微生物群,进而促进宿主的健康。
这其中最大的挑战可能是该使用什么酶。
以治疗慢性疾病为例。由于整合多组学分析的能力不断发展,慢性病中的致病菌群正逐渐被识别出来。
因此,酶可以作为有选择地富集或减少有害致病微生物的宝贵工具。
因为酶能够通过将致病微生物与微生物结合模块(如抗体)连接起来,从而瞄准致病微生物。
为了制造这种具有精确调节功能的酶,可以从拟杆菌的“自私”策略中获取经验。这种细菌编码细胞表面附着的甘露聚糖酶,以帮助它在竞争激烈的肠道生态位中靠甘露聚糖繁衍生息。
此外,成功地将赖氨酸与细菌素融合,使酶能够杀死革兰氏阴性细菌,可以减少不必要的致病菌。
酶可以作为已知化学物质或微生物的增强剂,影响致病微生物。
例如,2型糖尿病患者(50%)对两种精心设计的高纤维饮食没有积极的反应。在那些从饮食中受益的人中,利用低聚糖的细菌,如双歧杆菌属,属于少数“公会”致病微生物。在这些饮食中添加纤维特异性酶可以释放更多低聚糖,从而改善细菌的生长,从而有助于缓解更多患者的疾病。
然而,许多其他疾病与肠道微生物群变化之间的机制联系目前仅限于关联而非因果关系。因为肠道中几乎所有的成分都是酶的潜在底物,除了上述几种酶之外,还有大量其他候选酶。
从理论上讲,直接受酶影响的肠道微生物可以从酶的作用模式中推断出来,根据已知的机械关联,可以使用该模式选择酶作为进一步测试的候选酶。
酶可以用来帮助释放真正的效应剂
这在使用化学药品和微生物来调节肠道微生物群的组成时是不容易做到的。例如,溶菌酶通过杀死乳酸乳球菌(Lactococcus lactis)和释放细胞内超氧化物歧化酶来缓解结肠炎。
总的来说,这些结果表明,通过询问与上述三种酶以及其他类似的候选酶,并对其进行系统地测试,可以发现在有效调节肠道微生物群方面的隐藏酶。或者,将有关酶、饮食和宿主基因型的累积信息集成到现有数据库中,如Amadis (http://gift2disease.net/GIFTED/)将肠道微生物群与疾病联系起来,可能会有更合理的选择,并减少需要筛选的酶的数量。
尽管生产、储存和口服都很容易,但在选择合适的酶时仍有重要的考虑因素。
1、考虑酶的耐受条件,相应增加剂量
口服给药时,酶必须能够耐受肠道内的恶劣条件,包括酸性pH值和蛋白酶消化,这表明应选择候选酶来满足抵抗力,最初计算的剂量应在实验验证的基础上相应增加,以补偿储存和使用过程中任何可能的活性损失。
2、考虑酶的多效作用甚至危害影响
虽然酶不具有水平基因转移和引入多药耐药生物体甚至与基因工程益生菌和粪便微生物群移植相关的病原体的安全风险,但酶可以发挥不必要的多效作用,甚至产生有害影响。例如,磷脂酶将磷脂酰胆碱水解为胆碱,可被肠道细菌进一步转化为代谢产物三甲胺,三甲胺与不良疾病相关。
3、同一家族中不能调节菌群的酶需要排除
同一家族中的一些酶略有不同。那些不能调节目标微生物的也应该被排除在外。例如,木聚糖酶释放大量不同的低聚木糖,肠道细菌如Roseburia intestinalis和Bacteroides ovatus对具有不同糖链长度的低聚木糖的反应非常不同。
酶影响肠道微生物群,进而影响宿主健康,再加上大量候选者的可用性和可感知的优势,使其成为调节肠道微生物群的一种不错的方式。了解酶的功能和催化机制可以更好地调节肠道菌群,指导药物使用,治疗人类疾病。
然而,由于酶与其底物、肠道微生物群和宿主之间的复杂相互作用,开发酶等新试剂并非易事。需要针对不同情况选择合适的酶。此外,酶并非排他性的,它们可以与化学物质和微生物合作,改变肠道微生物群的组成,进一步促进宿主的健康。
主要参考文献:
Su X, Yao B. Exploiting enzymes as a powerful tool to modulate the gut microbiota. Trends Microbiol. 2022 Feb 1:S0966-842X(22)00003-8. doi: 10.1016/j.tim.2022.01.003. Epub ahead of print. PMID: 35120774.
Jia B, Han X, Kim KH, Jeon CO. Discovery and mining of enzymes from the human gut microbiome. Trends Biotechnol. 2022 Feb;40(2):240-254. doi: 10.1016/j.tibtech.2021.06.008. Epub 2021 Jul 22. PMID: 34304905.
Neves ALA, Yu J, Suzuki Y, Baez-Magana M, Arutyunova E, O’Hara E, McAllister T, Ominski KH, Lemieux MJ, Guan LL. Accelerated discovery of novel glycoside hydrolases using targeted functional profiling and selective pressure on the rumen microbiome. Microbiome. 2021 Nov 23;9(1):229. doi: 10.1186/s40168-021-01147-1. PMID: 34814938; PMCID: PMC8609826.

谷禾健康

维生素对人体健康至关重要;它们是无数酶的辅助因子,包括促进脂肪和碳水化合物代谢的酶,并具有直接和间接的抗氧化特性。
由于人类自身无法产生足够量的维生素(维生素 D 除外),因此要从食物中获取维生素。然而,许多维生素对温度敏感,在食品加工和储存过程中容易降解。因此,维生素缺乏症在某些人群(例如,素食主义者或老年人)中很常见。
例如口角炎,脚气病,腿部容易抽筋,皮肤容易红肿,容易烦躁和疲倦等,都有可能与缺乏B族维生素有关。
B族维生素从哪里来?
曾经有人认为B族维生素只从饮食中获得;然而并非如此,肠道微生物群也是维生素的重要来源。
B 族维生素包括哪些?
常见的有维生素 B1 (硫胺素)、B2(核黄素)、B3(烟酸/烟酰胺)、B5(泛酸)、B6(吡哆醇)、B7(生物素)、B9(叶酸)和B12 (钴胺素)等。

B族维生素的吸收情况
B族维生素主要通过结肠产生和吸收,大多数B族维生素的吸收情况类似:在低浓度时,主动运输系统促进吸收,而在较高浓度时,被动扩散主要发生在小肠。过量摄入B族维生素会使B族维生素到达大肠。

B族维生素是多种代谢途径中普遍必需的辅助因子的生物合成前体,对宿主和肠道微生物群都是不可或缺的,它们在维持免疫稳态中也发挥着重要作用。
本文来详细了解一下B族维生素(8类)和肠道微生物群之间有什么联系,它们在免疫代谢中有哪些作用,缺乏会导致什么症状,如何补充B族维生素。
维生素B1是维持神经、心脏及消化系统正常机能的重要生物活性物质。
维生素B1参与肠道粘膜免疫系统的免疫防御,调节免疫细胞在肠道内发挥作用。维生素B1对巨噬细胞有调控作用,能够介导巨噬细胞的生长及其细胞因子的分泌,间接促进免疫细胞的增殖和分化。
维生素 B1(硫胺素)是几种酶的辅助因子,包括丙酮酸脱氢酶和 α-酮戊二酸脱氢酶,它们都参与三羧酸 (TCA) 循环,从而为人体提供能量。
维生素B1在体内是如何参与代谢的?
前面我们已经知道,维生素B的两种来源:
摄入食物和细菌代谢生成。
膳食和细菌维生素B1在宿主中有不同的作用。因此我们从这两个方面分别了解其吸收转化过程。
膳食维生素B1
维生素 B1 在肉类(尤其是猪肉和鸡肉)、鸡蛋、谷芽、米糠、豆类中以高浓度的焦磷酸硫胺素 (TPP) 形式存在。
膳食TPP被碱性磷酸酶水解,并在小肠内转化为游离硫胺素。游离硫胺素通过硫胺素转运体(例如,THTR-1、THTR-2)被肠上皮吸收,并被输送到血液中以分布在全身。游离硫胺被转化回TPP,用于TCA循环中的能量代谢。
细菌维生素B1
结肠中各种类型的肠道细菌也会产生维生素B1作为游离硫胺素和TPP。在结肠中,游离细菌硫胺素主要被硫胺素转运体吸收,运输到血液中,并分布在全身;这一机制与小肠吸收游离膳食硫胺素的方式相似。
然而,肠道细菌产生的TPP不会转化为游离硫胺素,因为结肠中不会分泌碱性磷酸酶。相反,TPP通过在结肠顶膜上高度表达的TPP转运体(如TPPT-1)直接被结肠吸收。被吸收的TPP通过MTPP-1进入线粒体,MTPP-1是一种TPP转运体,在线粒体内膜中表达,用作ATP生成的辅助因子。这表明细菌TPP对结肠中的能量生成很重要。
这两者之间的区别在于:肠道细菌产生TPP这条途径,并不是通过游离硫氨酸,而是直接被结肠吸收。
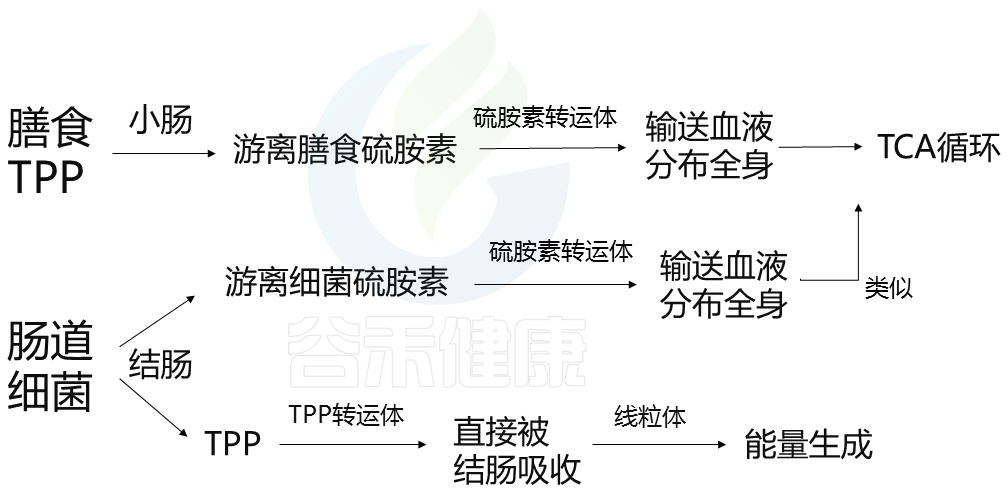
哪些菌会产生维生素B1呢?
对人类肠道微生物群研究和谷禾数据库预测总结以下主要菌群:脆弱拟杆菌和普雷沃氏菌、艰难梭菌(Clostridium difficile)、一些乳杆菌属、Ruminococcus lactaris、双歧杆菌属、可变梭杆菌(Fusobacterium varium)是维生素B1的产生菌。

此外,比如干酪乳杆菌(Lactobacillus casei)在发酵乳饮料的生产过程中产生硫胺素,婴儿双歧杆菌和双歧双歧杆菌在培养上清液中产生硫胺素。
许多肠道细菌拥有完整的维生素B1合成途径,包括噻唑和嘧啶的合成途径。
维生素B1的结构由嘧啶环和噻唑环通过亚结合而成。
细菌从甘氨酸或酪氨酸和1-deoxy-d-xylulose-5-phosphate中获得噻唑部分;
嘧啶部分来自5-氨基咪唑核糖核苷酸,这是嘌呤途径的中间产物。
宿主和某些菌存在VB1竞争
在肠道微生物群中,并非所有细菌都产生 B 族维生素,一些细菌利用膳食 B 族维生素或其他肠道细菌产生的 B 族维生素来满足自己的需要;因此,宿主和肠道微生物群之间可能存在对 B 族维生素的竞争。
比如,粪杆菌属(Faecalibacterium)的生长虽然需要维生素B1,但它却缺乏维生素B1合成途径。因此,必须通过硫胺素转运体从其他细菌或宿主饮食中获得维生素B1,这表明宿主和某些肠道细菌之间存在维生素B1竞争。
建议摄入量
世界卫生组织 (WHO)/粮食及农业组织 (FAO) 建议成人每日维生素 B1 摄入量为 1.1-1.2 毫克。
缺乏导致
维生素 B1 缺乏会导致嗜睡,如果不及时治疗,可能会发展成脚气病,这是一种影响周围神经系统和心血管系统的疾病。
含量较高的食物
维生素B1 存在于全麦谷物、酵母、豆类、坚果和肉类(尤其是猪肉和鸡肉)等食物中。

维生素 B2(核黄素)及其活性形式(黄素腺嘌呤二核苷酸 [FAD] 和黄素单核苷酸 [FMN])是 TCA 循环和脂肪酸氧化(也称为 β-氧化)中酶促反应的辅助因子。
维生素B2通过调节脂肪酸氧化来控制免疫细胞的分化和功能。
除了产生能量外,维生素B2通过启动NADPH氧化酶2与免疫细胞中活性氧(ROS)的产生有关,ROS是炎症和免疫过程中重要的效应分子和信号分子。
维生素B1和B2介导肠道B细胞分化的免疫代谢
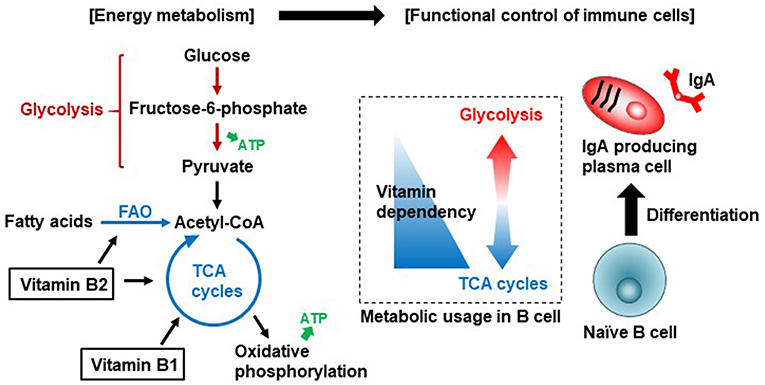
Yoshii K, et al., Front Nutr. 2019
维生素B1是参与TCA循环的丙酮酸脱氢酶和α-酮戊二酸脱氢酶等酶的辅助因子。维生素B2是TCA循环中琥珀酸脱氢酶和脂肪酸氧化(FAO,也称为β-氧化)中酰基辅酶a脱氢酶等酶的辅助因子。初始B细胞优先使用TCA循环有效产生能量。一旦B细胞被激活,分化为产生IgA的浆细胞,它们就会利用糖酵解产生IgA抗体。
膳食维生素B2
膳食维生素 B2 以 FAD 或 FMN 形式存在,并在小肠中通过 FAD 焦磷酸酶和 FMN 磷酸酶转化为游离核黄素。游离核黄素通过小肠上皮上表达的核黄素转运蛋白被吸收,然后释放到血液中。在血液中,游离核黄素转化回 FAD 或 FMN 并分布在全身。
细菌维生素B2
细菌维生素B2是由三磷酸鸟苷(GTP)和5-磷酸核酮糖合成的。细菌维生素B2以游离核黄素的形式存在,可直接在大肠中吸收,转化为FAD或FMN,并如上所述分布在全身。
哪些菌会产生维生素B2呢?
对人类肠道微生物群研究和谷禾数据库预测总结以下主要菌群:脆弱拟杆菌和普雷沃特菌属;艰难梭菌(Clostridium difficile)、植物乳杆菌(Lactobacillus plantarum)、发酵乳杆菌(L. fermentum)、Ruminococcus lacttaris表达合成维生素B2所必需的因子,表明这些细菌是大肠中维生素B2的重要来源。
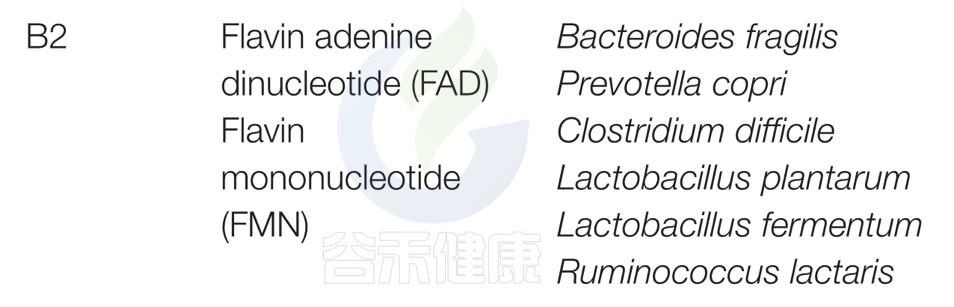
从酸面团中分离出的发酵乳杆菌可以在体外合成维生素B2。
双歧杆菌和Collinsella缺乏维生素B2途径。
在缺乏维生素B2的小鼠中,添加含有植物乳杆菌的发酵豆浆可以促进维生素B2的产生,防止维生素B2缺乏。
宿主和共生细菌之间对VB2的竞争
拟杆菌门中的某些物种比放线菌门和厚壁菌门产生更多的VB2。然而,放线菌和厚壁菌门仍然表达核黄素转运蛋白以及从游离核黄素生成 FAD 和 FMN 所必需的酶(即 FAD 合酶和黄素激酶),这表明所有细菌,包括那些不能合成维生素的细菌B2 本身需要 FAD 和 FMN 才能生长和生存。因此,与维生素 B1 一样,宿主和共生细菌之间可能存在对维生素 B2的竞争。
某些菌产生维生素B2中间体,有助于防御病原体
除了能够产生维生素 B2 之外,一些细菌(例如嗜酸乳杆菌等共生菌,结核分枝杆菌和鼠伤寒沙门氏菌等病原体)还产生维生素 B2 中间体(6-hydroxymethyl-8-d-ribityllumazine ),该中间体与抗原呈递细胞上的主要组织相容性复合物I类相关基因蛋白(MR1)结合;这会导致粘膜相关不变T细胞(MAIT)产生细胞因子,如干扰素γ和IL-17,这有助于宿主防御病原体(下图)。
来自维生素B2和B9的微生物代谢产物对MAIT细胞的调节
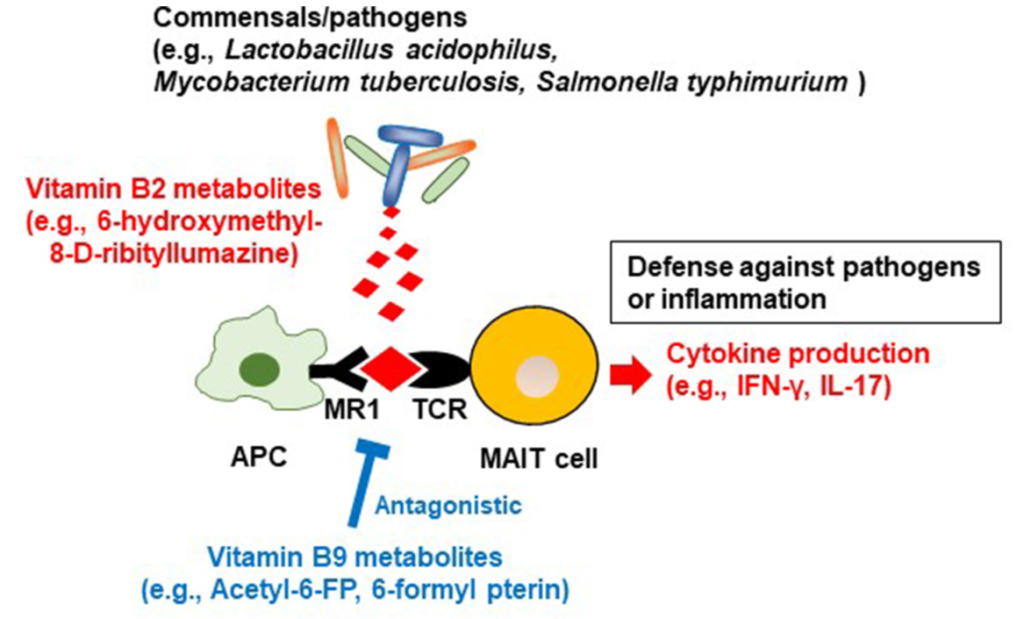
Yoshii K, et al., Front Nutr. 2019
有人认为,共生细菌的刺激有助于MAIT细胞的发育和激活,以便对病原体进行免疫监测。MAIT细胞还产生炎性细胞因子并具有组织归巢特性,表明这些细胞也参与自身免疫和炎症疾病的发展。
建议摄入量
世卫组织/粮农组织建议成人每日维生素 B2 摄入量为 1.0–1.3 毫克。
缺乏会导致
维生素 B2 缺乏会抑制参与脂肪酸氧化生成乙酰辅酶 A 的酰基辅酶 A 脱氢酶的活性,而乙酰辅酶 A 被线粒体用来通过 TCA 循环产生 ATP。
含量较高的食物
绿叶蔬菜、肝脏和鸡蛋中的维生素 B2 含量很高。

维生素 B3(烟酸)通常被称为烟酸和烟酰胺。这些化合物是烟酰胺腺嘌呤二核苷酸 (NAD) 的前体,NAD 是细胞氧化还原反应中的一种辅酶,在有氧呼吸中起核心作用。
免疫功能:抗炎,抑制结肠炎
维生素 B3 也是 GPR109a 的配体,GPR109a 是一种 G 蛋白偶联受体,在包括免疫细胞在内的多种细胞上表达。维生素 B3-GPR109a 信号传导诱导调节性 T 细胞分化并以 GPR109a 依赖性方式抑制结肠炎。维生素 B3 还抑制巨噬细胞和单核细胞产生促炎细胞因子 IL-1、IL-6 和肿瘤坏死因子 α (TNF-α)。
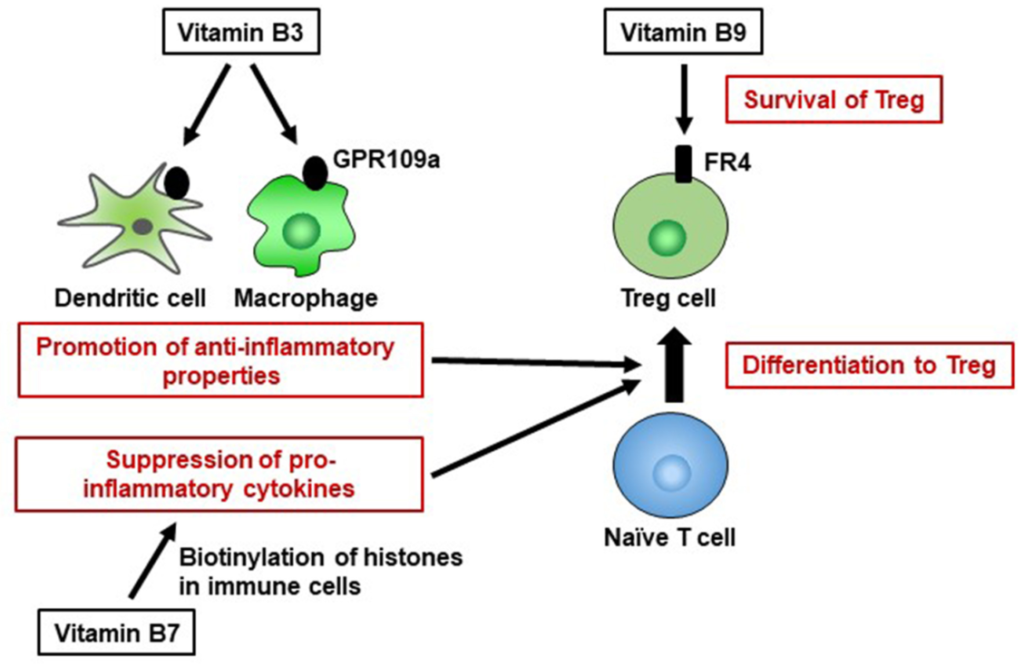
Yoshii K, et al., Front Nutr. 2019
因此,维生素B3通过调节宿主免疫细胞而具有抗炎特性,并在维持免疫稳态中发挥重要作用。
与其他B族维生素不同,维生素B3可由哺乳动物通过色氨酸的内源性酶途径生成,并储存在肝脏中,也可从饮食中获得。
膳食维生素B3
动物性食品如鱼和肉含有维生素B3作为烟酰胺,而植物性食品如豆类含有维生素B3作为烟酸。烟酰胺和烟酸都通过小肠直接吸收,烟酸在小肠转化为烟酰胺。
哪些菌会产生维生素B3呢?

维生素B3也是由肠道细菌从色氨酸合成的。可能参与B12合成和代谢的菌如:脆弱拟杆菌和普雷沃氏菌属、乳瘤胃球菌、艰难梭菌、婴儿双歧杆菌、幽门螺杆菌、可变梭杆菌。
因此,来自不同属的许多肠道细菌可以产生维生素B3,这表明饮食和共生细菌来源的维生素B3对宿主免疫都很重要。
缺乏会导致
在人类中,维生素B3缺乏会导致糙皮病,这是一种以肠道炎症、腹泻、皮炎和痴呆为特征的疾病。
建议摄入量
世卫组织/粮农组织建议成人每日维生素 B3 摄入量为 11-12 毫克。
含量较高的食物
全麦制品、糙米、绿豆、芝麻、花生、香菇、紫菜、无花果、乳制品、蛋、鸡肉、肝、瘦肉、鱼等。

维生素B5(泛酸)是辅酶a(CoA)的前体,辅酶a是TCA循环和脂肪酸氧化的必要辅助因子。
与维生素B1和B2一样,维生素B5通过免疫细胞的能量生成参与宿主免疫的控制。
缺乏导致
维生素B5缺乏会导致皮炎等免疫疾病,以及疲劳和失眠等非免疫相关疾病。
维生素B5缺乏如何引起炎症?
在一项针对成年人的随机、双盲、安慰剂对照研究中,膳食中补充维生素B5可改善面部痤疮,表明上皮屏障功能通过促进角质形成细胞增殖和分化为成纤维细胞而改善。
为了在缺乏维生素B5的时候保持维生素B5水平,辅酶A通过泛碱转化回维生素B5或半胱胺。然而,半胱胺抑制过氧化物酶体增殖物激活受体γ(PPARγ)信号,导致炎症。在泛碱产生酶基因敲除小鼠中,结肠炎已得到改善。因此,维生素B5缺乏通过上皮屏障功能障碍和促炎分子的产生引起炎症。
维生素B5通过激活免疫反应来促进宿主防御
在免疫反应方面,维生素B5通过促进先天免疫和适应性免疫,增强抗结核分枝杆菌感染的保护活性。在小鼠中,补充维生素B5可激活巨噬细胞的吞噬作用和细胞因子(包括IL-6和TNF-α)的产生,并随后促进Th1和Th17反应,以清除肺结核分枝杆菌。因此,维生素B5通过激活免疫反应来促进宿主防御,表明这种维生素作为一种天然佐剂具有重要作用。
膳食维生素B5
维生素B5在肝脏、鸡蛋、鸡肉和发酵大豆中以辅酶A或磷酸泛酰巯基乙胺pantetheinase的形式存在,浓度很高。辅酶A和磷酸泛乙烯通过小肠内的内源性酶(如磷酸酶和泛乙烯酶)转化为游离泛酸。游离泛酸通过小肠上皮上表达的钠依赖性复合维生素转运体(SMVT)被吸收,然后释放到血液中。最后,游离泛酸被转化回辅酶A并分布在全身,尤其是肝脏和肾脏。
细菌维生素B5
细菌维生素B5由2-二氢泛酸盐和β-丙氨酸通过从头合成途径合成。细菌维生素B5以游离泛酸的形式存在,其在大肠中直接吸收,转化为辅酶A,并以与膳食维生素B5相同的方式分布。
哪些菌会产生维生素B5呢?

对人类肠道微生物群研究和谷禾数据库预测总结:脆弱拟杆菌、普雷沃氏菌属、一些瘤胃球菌属(乳链球菌和扭链球菌)、肠道沙门氏菌、幽门螺杆菌具有维生素B5生物合成途径,表明肠道共生菌可以产生维生素B5。
细菌与宿主竞争维生素B5
相比之下,大多数梭杆菌和双歧杆菌属、艰难梭菌、粪杆菌属和乳酸杆菌属(厚壁菌属)的一些菌株缺乏这种途径,尽管其中一些菌株确实表达泛酸转运体以利用维生素B5产生能量,表明这些细菌与宿主竞争维生素B5。
摄入量
世卫组织/粮农组织建议成年人每天摄入5.0毫克维生素B5。
含量较高的食物
蔬菜中的卷心菜类,例如西兰花羽衣甘蓝,以及鳄梨、全麦谷物、土豆、乳制品、内脏等食物。

维生素B6以多种形式存在,包括吡哆醇、吡哆醛和吡哆胺。这些形式的维生素B6是辅酶磷酸吡哆醛(PLP)和磷酸吡哆胺(PMP)的前体,参与多种细胞代谢过程,包括氨基酸、脂质和碳水化合物代谢。
维生素B6在肠道免疫监测中起着重要作用
维生素B6调节炎症的机制目前尚不清楚。维生素B6通过脂质介质1-磷酸鞘氨醇(S1P)的代谢促进肠道免疫调节。
S1P调节淋巴细胞进入肠道,尤其是大肠。淋巴细胞转运依赖于S1P的产生及其降解产生的S1P梯度。S1P降解由S1P裂解酶介导,该裂解酶需要维生素B6作为辅助因子。服用维生素B6拮抗剂会损害S1P裂解酶活性,并产生不适当的S1P梯度,导致淋巴细胞从淋巴组织迁移受损,并减少肠道中淋巴细胞的数量。位于肠道上皮细胞之间的淋巴细胞被称为上皮内细胞(IEL),参与抵抗病原体的保护。
膳食维生素B6
膳食维生素B6以PLP或PMP的形式存在;它通过内源性酶(如吡哆醛磷酸酶)转化为游离维生素B6,然后被小肠吸收。虽然已经证明维生素B6通过酸性pH依赖和载体介导的转运被吸收,但尚未在任何哺乳动物物种中鉴定出肠吡哆醇转运体。在吸收游离维生素B6后,它进入血液,并转化回PLP或PMP。
细菌维生素B6
细菌维生素B6是由脱氧基果糖5-磷酸和4-磷酸羟基-L-苏氨酸或甘油醛-3-磷酸和d-核酮糖5-磷酸合成的PLP。在大肠中,细菌衍生的PLP转化为游离维生素B6,通过被动运输被吸收,运输到血液中,并分布在全身。
哪些菌会产生维生素B6呢?

可能参与B6合成和代谢的菌如:脆弱拟杆菌、普雷沃氏菌属、长双歧杆菌、产气柯林斯菌、幽门螺杆菌。
拟杆菌和变形杆菌可能从5-磷酸脱氧纤维素和4-磷酸羟基-l-苏氨酸开始产生维生素B6,而放线菌可能从甘油醛-3-磷酸和d-核酮糖5-磷酸开始产生。
相比之下,大多数厚壁菌属(韦荣球菌、瘤胃球菌、粪杆菌和乳杆菌属)都缺乏维生素B6生物合成途径,除了一些梭菌(C. bartlettii、C. leptum、C.methylpentosum和C. sporogenes)和乳杆菌(短乳杆菌和瘤胃乳酸杆菌)外。
摄入量
世卫组织/粮农组织建议成年人每天摄入1.3-1.7毫克维生素B6。青少年和怀孕或哺乳的女性每天需要更多的维生素 B6,大约 2 毫克。
缺乏导致
维生素B6缺乏与过敏和类风湿性关节炎等炎症性疾病的发展以及神经元功能障碍有关。维生素B6缺乏会破坏Th1-Th2平衡,导致过度的Th2反应,导致过敏。此外,在类风湿性关节炎患者中观察到血浆维生素B6水平较低,以及促炎细胞因子(如TNF-α和IL-6)水平升高。
其他还包括导致肌肉无力、抑郁、易怒、短期记忆丧失、紧张和注意力不集中等问题。
含量高的食物
鱼类、鸡肉、豆腐、红薯、鳄梨、扁豆、牛奶、三文鱼、虾、菠菜、葵花籽、金枪鱼、糙米等。

维生素B7(生物素)是葡萄糖、氨基酸和脂肪酸代谢所必需的几种羧化酶的辅助因子。例如,维生素B7是参与脂肪酸生物合成的乙酰辅酶A羧化酶和脂肪酸合成酶的必要辅助因子。因此,维生素B7可能会影响免疫代谢。
维生素B7的抗炎作用
维生素B7通过结合(生物素化)组蛋白抑制基因表达;这些基因包括编码NF-κB的基因,NF-κB是产生几种促炎细胞因子(例如,肿瘤坏死因子α、IL-1、IL-6、IL-8)的主要信号分子。NF-κB的核转录在维生素B7缺乏时被激活,表明组蛋白的生物素化抑制NF-κB信号中编码促炎细胞因子的基因的表达。
因此,维生素B7通过抑制NF-κB活化而具有抗炎作用,膳食维生素B7缺乏通过促进促炎细胞因子的分泌而引起炎症反应。
膳食维生素B7
膳食生物素以游离蛋白质结合形式或生物素形式存在。在小肠中,生物素酶从结合蛋白中释放游离生物素,游离生物素通过生物素转运体SMVT被吸收。
细菌维生素B7
维生素B7也是由肠道细菌产生的游离生物素,由丙二酰辅酶A或吡美乐酸通过吡美乐酰辅酶A合成。细菌游离生物素被结肠中表达的SMVT吸收。
哪些菌会产生维生素B7呢?

可能参与B7合成和代谢的菌如:脆弱拟杆菌、普雷沃氏菌属可变梭杆菌、大肠弯曲杆菌(campylobacteric coli)。
维生素B7的产生似乎在不同的肠道细菌之间以合作的方式进行;肠道中的长双歧杆菌产生吡美拉酸,它是维生素B7的前体,可增强其他肠道细菌产生维生素B7。
细菌与宿主竞争维生素B7
相比之下,普雷沃氏菌属、双歧杆菌属、梭菌属、瘤胃球菌属、粪杆菌属和乳酸杆菌属缺乏这种途径;然而,它们确实表达游离生物素转运体,这表明这些细菌也利用饮食和细菌维生素B7,因此可能与宿主竞争。
因此,游离生物素可能会影响肠道微生物群的组成,因为生物素是微生物群生长和存活所必需的。
缺乏导致
生物素缺乏会导致肠道失调和鼠乳杆菌过度生长,从而导致脱发。
什么情况会缺乏?
生蛋清中含有大量的抗生物素,它与维生素B7紧密结合,阻止维生素B7在肠道中被吸收。因此,维生素B7缺乏症不仅可能由维生素B7摄入不足引起,还可能由过量摄入生蛋清引起。
摄入量
世卫组织/粮农组织建议成年人每天摄入30微克。
含量较高的食物
维生素B7在坚果、豆类和油籽等食物中含量丰富。

维生素B9(叶酸)以四氢叶酸的形式存在,是包括DNA和氨基酸合成在内的多种代谢反应的辅助因子。
缺乏导致
由于红细胞对维生素B9的高需求,维生素B9缺乏会导致巨幼细胞性贫血。维生素B9缺乏症还通过将细胞周期阻滞在S期并增加DNA损伤的频率,在体外抑制人类CD8+T细胞的增殖。
维生素B9有助于维持免疫稳态
调节性T细胞(Treg)表达高水平的维生素B9受体(叶酸受体4[FR4])。服用抗FR4抗体会导致Treg细胞群的特异性减少,这表明维持Treg细胞需要维生素B9–FR4轴。
在维生素B9减少的条件下体外培养Treg细胞会导致细胞存活受损,抗凋亡Bcl2分子的表达减少,尽管幼稚T细胞仍保留分化为Treg细胞的能力;这表明维生素B9是Treg细胞的生存因子。
与这些发现一致,膳食维生素B9缺乏导致小肠中Treg细胞数量减少。由于Treg细胞在预防过度免疫反应中起着重要作用,喂食缺乏维生素B9饮食的小鼠对肠道炎症的易感性增加。
膳食维生素B9
维生素B9以单谷氨酸和多谷氨酸叶酸的形式存在于饮食中。叶酸-聚谷氨酸酯与谷氨酸单酯脱结合,然后通过叶酸转运体(如质子耦合叶酸转运体(PCFT))在小肠吸收。在肠上皮中,叶酸单谷氨酸盐在被输送到血液之前转化为四氢叶酸(THF),四氢叶酸是一种活性形式和辅助因子。
细菌维生素B9
肠道细菌从GTP、4-磷酸红糖和磷酸烯醇式丙酮酸合成维生素B9作为THF。细菌性THF通过PCFT在结肠中直接吸收,并通过血液分布到全身。
哪些菌会产生维生素B9呢?

对人类肠道微生物群研究和谷禾数据库预测总结:脆弱拟杆菌、普雷沃氏菌属、艰难梭菌、植物乳杆菌、罗氏乳杆菌、德氏乳杆菌、保加利亚和嗜热链球菌、双歧杆菌属中的一些物种、可变梭杆菌、肠道沙门氏菌具有维生素B9生物合成途径。这表明几乎所有门中的所有物种都会产生叶酸。
膳食中添加双歧杆菌益生菌菌株(青春期双歧杆菌和假链双歧杆菌)可提高叶酸缺乏大鼠和无叶酸培养基大肠中叶酸的生成。此外,植物乳杆菌、德氏乳杆菌、保加利亚乳杆菌和罗氏乳杆菌可提高缺乏叶酸合成所需成分的细菌培养上清液中叶酸的生成。
维生素B9代谢物防止过敏和炎症
在共生细菌中,维生素B9代谢产物6-甲酰蝶呤(6-FP)是通过光降解产生的。与维生素B2代谢物6-羟甲基-8-d-三嗪一样,6-FP与MR1结合,但与B2代谢物不同,它不能激活MAIT细胞。6-FP的类似物乙酰-6-FP是MR1的拮抗剂,可抑制MAIT细胞活化。
如关于维生素B2的章节所述,6-羟甲基-8-d-三硝基脲嗪可激活MAIT细胞,从而抵御病原体,因此维生素B9代谢物可抑制过度的MAIT细胞反应,并防止过度过敏和炎症反应。
膳食维生素B2和B9与微生物群的组成及其代谢这些维生素的能力之间的定量平衡,可能是理解肠内MAIT细胞介导的稳态的关键。
摄入量
世卫组织/粮农组织建议成年人每天摄入400微克维生素B9。美国国立卫生研究院建议母乳喂养的母亲每天需要 500 微克,青少年和怀孕女性每天应该摄入 600 微克。
较高含量的食物
牛肉肝、深绿叶蔬菜、芦笋、抱子甘蓝、橙子、坚果、豆类等食物含有高水平的维生素B9。

维生素B12(钴胺素)是一种含钴的维生素,以甲钴胺素和腺苷钴胺素的活性形式催化甲硫氨酸的合成。
维生素B12促进免疫反应
在宿主免疫方面,膳食维生素B12缺乏会减少小鼠体内CD8+T细胞的数量,并抑制自然杀伤T细胞的活性;补充甲钴胺可以改善这些状况,这表明维生素B12通过CD8+T细胞和自然杀伤T细胞促进免疫反应。
膳食维生素B12
膳食维生素B12与膳食蛋白质复合存在,胃蛋白酶将其分解为游离维生素B12。游离维生素B12通过胃糖蛋白内源性因子(IF)被小肠上皮细胞吸收。在上皮细胞内,如果维生素B12复合物被溶酶体分解为游离维生素B12,然后释放到血液中,在血液中转化为活性形式并分布在全身。
细菌维生素B12
细菌维生素B12由前甲胎蛋白-2合成,产生腺苷钴胺素,直接被大肠吸收并分布在全身;这种吸收的机制目前尚不清楚。
哪些菌会产生维生素B12呢?

可能参与B12合成和代谢的菌如:脆弱拟杆菌和普雷沃氏菌属、艰难梭菌、粪杆菌和乳瘤胃球菌、动物双歧杆菌、婴儿双歧杆菌和长双歧杆菌、可变梭杆菌。
从发酵食品中分离出的植物乳杆菌和棒状乳杆菌产生维生素B12,而动物双歧杆菌在牛奶发酵过程中合成维生素B12。
摄入量
世卫组织/粮农组织建议成年人每天摄入2.4μg维生素B12。青少年和怀孕或哺乳的女性需要更多:每天 2.6 至 2.8 μg。
缺乏导致
与维生素B6和B9一起,维生素B12在红细胞形成和核酸合成中起着重要作用,尤其是在神经元中。因此,维生素B12缺乏会导致巨幼细胞性贫血和神经系统症状,如手脚麻木,刺痛。
其他还会导致包括贫血、失智、沮丧、难以保持平衡、疲劳、虚弱、体重减轻、肠道问题、肌肉无力、口腔或舌头酸痛等症状,缺乏也会损害神经系统,并可能导致记忆力差、情绪障碍、抑郁、精神错乱和痴呆。
什么情况会缺乏?
除了饮食摄入量不足的原因之外,维生素 B-12 缺乏还可能是由于恶性贫血(一种影响壁细胞和内因子释放的自身免疫性疾病,维生素 B-12 吸收所必需的)导致的生物利用度低或吸收受损;随着年龄的增长,药物(例如质子泵抑制剂)的使用,胃肠道疾病(例如炎症性肠病)或胃肠道感染(例如幽门螺杆菌、肠道蠕虫)的发生,萎缩性胃炎、吸收不良和恶性贫血的风险增加。
含量较高的食物
牛肝、双壳类、鱼类、鸡肉和鸡蛋含有高水平的维生素B12。

微生物群失调和维生素缺乏是相互关联的,这种关系可能直接影响宿主健康。
对哺乳期女性进行的一项关联研究发现,增加维生素 B2、B5、B6 和 B12 的摄入量与120个粪便样本中普氏菌的相对丰度增加和拟杆菌的相对丰度降低有关。
炎症性肠病(IBD)患者会出现血浆维生素B2浓度较低。肠道慢性炎症包括促炎细胞因子浓度升高,已被证明会导致上皮吸收功能的改变。肠上皮细胞暴露于促炎细胞因子肿瘤坏死因子-α中会显著抑制维生素B2的摄取,这可能解释了IBD患者中维生素B2浓度显著较低的原因。
维生素B2和B3浓度可能是疾病状态的标志物。
克罗恩病恶化与参与抗炎的维生素B2、B1、B9生物合成相关的微生物基因减少有关。
此外,患有 2 型糖尿病的受试者在与微生物介导的维生素代谢相关的基因丰度方面表现出显着变化。
营养不良儿童的微生物组显示参与B 族维生素代谢的多种途径显着减少,包括烟酸/NADP 生物合成。
所有这些观察结果表明,微量营养素的缺乏会改变肠道微生物群。
因此,在某些条件下,针对肠道微生物群的维生素补充剂(直接和间接)可能对健康有益。
越来越多的证据表明,当大量服用以逃避完全吸收或以结肠靶向递送系统 (CTDS) 的形式服用时,维生素可以直接调节肠道微生物组。此外,维生素似乎通过体循环的间接机制影响肠道微生物群,与人类健康直接相关。
结肠靶向维生素补充剂可能通过三种相互关联的途径影响宿主健康
1) 对肠道免疫系统的直接影响(上左图)
2) 对肠道上皮屏障的直接影响(上右图)
3) 通过微生物代谢产物对肠道微生物群以及随后对肠道免疫和上皮屏障的影响(中)
在一个小型成人志愿者组中进行了一项试点研究,该组补充了过量的维生素B2(100 毫克),为期 14 天。他们发现,在补充过程中,每克粪便中的Faecalibacterium prausnitziip数量增加。作者还注意到厌氧罗氏菌属的增加和大肠杆菌减少。
关于维生素的结肠靶向性,最近的一项研究表明,通过缓释微囊结肠靶向补充烟碱酸有益于影响微生物组成和胰岛素敏感性。有趣的是,作者还观察到肥胖受试者群体中α多样性和拟杆菌丰度降低,并指出这种变化与膳食烟酸摄入量降低有关。
针对克罗恩病的研究发现发现,补充维生素 B2 可减少全身氧化应激、一些抗炎作用,降低 C 反应蛋白、红细胞沉降率、血小板和 IL-2,并减轻临床症状。粪便微生物群的荧光原位杂交分析显示 克罗恩病患者中粪便肠杆菌科细菌减少、粪便钙卫蛋白水平降低。
维生素可以调节微生物组、改变氧化还原电位,并且就维生素 B3而言,对全身胰岛素敏感性和代谢性炎症的生物标志物产生积极影响。虽然需要进行额外的临床研究以充分了解结肠靶向递送维生素的优势和潜在副作用,但在菌群失调的情况下,这种方法可能成为预防和治疗微生物组相关疾病的新的菌群调节途径。
该领域的研究很复杂,因为不仅肠道菌群的组成因人而异,饮食也会改变肠道菌群的组成和功能,维生素介导的免疫维持也因人而异。
补充结肠靶向维生素可能与基于个人健康状况和微生物组组成的个性化营养建议相关。
目前,对肠道微生物群的研究及其与多种 B族维生素的相互作用机制的理解正逐步深入,精准健康和营养新时代正在开启。
主要参考文献
Beane, K.E., Redding, M.C., Wang, X. et al. Effects of dietary fibers, micronutrients, and phytonutrients on gut microbiome: a review. Appl Biol Chem 64, 36 (2021). doi.org/10.1186/s13765-021-00605-6
Uebanso T, Shimohata T, Mawatari K, Takahashi A. Functional Roles of B-Vitamins in the Gut and Gut Microbiome. Mol Nutr Food Res. 2020 Sep;64(18):e2000426. doi: 10.1002/mnfr.202000426. Epub 2020 Aug 19. PMID: 32761878.
Yoshii K, Hosomi K, Sawane K, Kunisawa J. Metabolism of Dietary and Microbial Vitamin B Family in the Regulation of Host Immunity. Front Nutr. 2019 Apr 17;6:48. doi: 10.3389/fnut.2019.00048. PMID: 31058161; PMCID: PMC6478888.
Steinert RE, Lee YK, Sybesma W. Vitamins for the Gut Microbiome. Trends Mol Med. 2020 Feb;26(2):137-140. doi: 10.1016/j.molmed.2019.11.005. Epub 2019 Dec 17. PMID: 31862244.
Heather M Guetterman, Samantha L Huey, Rob Knight, Allison M Fox, Saurabh Mehta, Julia L Finkelstein, Vitamin B-12 and the Gastrointestinal Microbiome: A Systematic Review, Advances in Nutrition, 2021;nmab123, doi.org/10.1093/advances/nmab123
Putnam EE, Goodman AL. B vitamin acquisition by gut commensal bacteria. PLoS Pathog. 2020 Jan 23;16(1):e1008208. doi: 10.1371/journal.ppat.1008208. PMID: 31971969; PMCID: PMC6977713.

谷禾健康

Alistipes是拟杆菌门的一种革兰氏阴性细菌,也是相对新的细菌属,主要从医学临床样本中分离出来。该菌的生态失调,可能是有益的也可能是有害的。
Alistipes可能对某些疾病有保护作用,包括肝纤维化、癌症免疫治疗和心血管疾病。相比之下,其他研究表明Alistipes在结直肠癌中具有致病性,并且与抑郁症有关。
该菌避开富含植物性食物的饮食,可以在高脂肪饮食中茁壮成长,并且在肥胖患者的肠道微生物群中生长得特别好,表明与肥胖相关。
Alistipes 是拟杆菌门中的一个属,革兰氏阴性,专性厌氧,是肠道共生的细菌,G+C 含量为55–58%,直径为 0.2–0.9 µm,长度为 0.5–4 µm 的直或略微弯曲的棒状,末端为圆形。不会形成孢子。细胞通常单独或成对出现,偶尔以较长的细丝出现。
不运动,可产生吲哚。不能还原硝酸盐,不水解精氨酸和尿素,葡萄糖代谢终产物是琥珀酸和少量的乙酸,丙酸。
microbiomology
在分类学上,Alistipes 是在 2003 年在患有阑尾炎的儿童的组织样本中发现后描述的一个属。第一个被发现的Alistipes物种是Alistipes Finegoldii,它是以美国厌氧细菌学和传染病临床研究学家Sydney M. Finegold 的名字命名,极大地促进了我们对厌氧菌的理解。
根据NCBI分类数据库,目前Alistipes由13个物种组成:
2017 年从肠易激综合征患者的结肠中分离出来一种名为Tidjanibacter massiliensis Marseille-P3084 的新物种,与A. putredinis具有 92.1% 的序列同源性,不过该菌株尚未被官方认可或存放在公共菌株生物储存库中。
此外,三个较新的亚种:
从生态学角度来看,Alistipes主要存在于健康人的肠道中。然而,Alistipes也从其他液体中分离出来、如尿液、阑尾、腹部、直肠周围和脑脓肿中分离出来,突出了它们在人类疾病中潜在的机会致病作用。
A. putredinis(ATCC 29800 T型菌株)已从各种标本中分离出来,例如粪便、急性阑尾炎患者的阑尾组织、腹部和直肠脓肿、甚至农场土壤。该菌株对克林霉素、头孢西丁、氯霉素、红霉素和甲硝唑敏感,对四环素和强力霉素有中度耐药。
Alistipes finegoldii 已被认为是肉鸡的生长促进剂,并且已观察到在人类中,A. putredinis随着十字花科蔬菜摄入量的增加而增加,因此可以合理假设不同的Alistipes物种在营养和健康方面可能具有不同的作用。
A. onderdonkii和 A. shahii 分别从腹部脓肿和阑尾组织以及尿液中分离出来。它们具有圆形菌落。这两个种都对 20% 的胆汁具有抗性,并将色氨酸水解为吲哚。它们是过氧化氢酶、氮还原酶和脲酶阴性。琥珀酸是主要的代谢终产物,少量产生乙酸和丙酸。
其他两个种,A. senegalensis 和 A. timonensis 最初是从健康患者的粪便菌群中分离出来的。它们有圆形菌落并产生色素,可以将色氨酸水解为吲哚。A. senegalensis会发酵甘露糖,而A. timonensis 不会。这些细菌菌株对青霉素、阿莫西林加克拉维酸、亚胺培南和克林霉素敏感。此外,A. senegalensis菌株对甲硝唑耐药,A. timonensis 菌株对甲硝唑敏感。
Alistipes inops从人类粪便中分离出物种。它对吲哚产生呈阳性,对过氧化氢酶、硝酸还原酶和脲酶呈阴性。这种细菌是非发酵的,主要代谢终产物是琥珀酸和乙酸。
A. megaguti (Marseille-P5997 T型菌株)是从一名年轻健康女性的新鲜粪便样本中分离出来的。
A. megaguti是过氧化氢酶、脲酶和氧化酶阴性。
A. provencensis (Marseille-P2431 T型菌株) 分离自一名患有高血压和糖尿病的男性患者。该种属氧化酶和脲酶阴性,而过氧化氢酶阳性。
A.ihumii 已从患有神经性厌食症的患者的粪便中分离出来,而A.obesi则从患有病态肥胖症的患者中分离出来。
该菌属大部分耐胆汁,因此,有必要确定对胆汁酸的抗性是否确实决定了Alistipes胃肠道内的区域性富集,或临床上以胆汁改变为特征的疾病,其与Alistipes菌的关系。
肝病和短链脂肪酸中的 Alistipes
肝细胞癌 (HCC) 是全球第二大致命癌症。HCC 通常是由肝硬化、非酒精性脂肪肝病 (NAFLD) 或非酒精性脂肪性肝炎 (NASH) 引起的晚期肝纤维化发展而来的。这些肝脏疾病与“微生物-肝轴”有关,表明生态失调是潜在原因之一。
肝纤维化中Alistipes减少
在对微生物群组成和肝纤维化进行的研究中可以看出,在纤维化的整个进展过程中,Alistipes减少。
例如,在代偿性和失代偿性肝硬化 (LC) 患者中,来自健康志愿者和各种类型 LC 患者的新鲜粪便宏基因组序列表明,与健康对照组相比,A. shahii和A. putredinis菌减少。
针对 LC 患者粪便和活检的研究中,与 LC 患者相比,健康对照患者的Alistipes丰度增加。
代偿发展到失代偿,Alistipes减少
随着疾病从代偿发展到失代偿,观察到Alistipes indistinctus 的减少。一旦个体出现失代偿性肝硬化,患者就会开始产生多种严重并发症,例如肝性脑病。
另一项研究表明,在比较失代偿性肝硬化和急性肝性脑病患者的粪便微生物群时,Alistipes具有保护作用,其丰度的降低与肝性脑病复发的增加相关。因此,Alistipes的减少与肝硬化进展为失代偿状态有关。
NASH 和 NAFLD中 Alistipes 降低
在NASH 和 NAFLD 等其他纤维化疾病中可以看到肝纤维化患者的Alistipes丰度降低。有实质性纤维化的 NAFLD 患者粪便中乙酸盐和丙酸盐浓度降低,丁酸盐浓度无显着差异。当将健康对照与 NASH 患者进行比较时,A. finegoldii 显着减少,标准化计数平均值从 542 减少到 19。在具有显着纤维化的 NAFLD 患者中,观察到 A. onderdonkii 的显着减少从 285 到 31。
Alistipes生产短链脂肪酸
值得注意的是,这些晚期纤维化患者的粪便乙酸盐和丙酸盐水平降低。
Alistipes是一种丙酸生产者,表达甲基丙二酰辅酶A差向异构酶,其中该酶的基因位于具有乙酰辅酶A羧化酶基因的操纵子上。
此外,Alistipes是乙酸盐生产者,由于先前的研究表明短链脂肪酸具有抗炎机制,因此可以表明Alistipes 减少促使短链脂肪酸减少,可能加剧这些 NAFLD 患者中的晚期纤维化。
心血管疾病、高血压和上皮中的 Alistipes
心血管疾病 (CVD) 是发展中国家和发达国家死亡和发病的主要原因。随着全球人口老龄化,预计心血管疾病在未来会上升,因此对与肠道微生物群关系的评估进行了更广泛的研究。
Alistipes与高血压等心血管疾病风险因素以及心房颤动 (AF)、充血性心力衰竭 (CHF) 和动脉粥样硬化性心血管疾病 (ACVD) 等多种心血管疾病相关。
各种研究表明,Alistipes在心血管疾病中起保护作用。
心房颤动患者Alistipes减少
Alistipes与心房颤动 (AF) 等心血管疾病直接相关。心房颤动是最常见的心律失常,在高血压、心力衰竭和肥胖患者中普遍存在。
为了量化肠道微生物组和心房颤动之间的关系,对来自中国参与者的 100 个粪便样本全基因组鸟枪序列进行分析,显示在心房颤动患者肠道中Alistipes急剧减少。
Alistipes和链球菌的拮抗作用
然而,研究中提出在心房颤动期间急剧增加的细菌,如链球菌,可能是导致Alistipes下降的原因,这表明Alistipes和链球菌之间存在潜在的拮抗作用。这种趋势在其他心脏病中很常见,例如 ACVD和 CHF。
但是Alistipes在心血管疾病中证据是矛盾的,尚不清楚关联是保护性的、有益的还是致病的。因为大多数心血管疾病具有共同的病理生理特征,例如内皮功能障碍 ,Alistipes的作用可能取决于多个心血管疾病共享的疾病机制。因此,更多关于肠心轴的研究可能会导致未来对微生物组相关疾病和潜在疗法的理解。
高血压中Alistipes有助于炎症和上皮细胞的改变
宏基因组分析研究了 22 名高血压患者和 18 名对照患者的粪便样本。数据显示A. finegoldii和A. indistinctus的增加与收缩压呈正相关,表明该物种是高血压患者肠道屏障功能障碍和炎症的潜在驱动因素,但是该研究样本量较少,证据力度不大。
肠道炎症和结直肠癌中不一样的 Alistipes
由于胃肠道中微生物群的多样性,菌群失调与炎症性肠病(IBD)之间存在很强的相关性。
人群中最常见的炎症性肠病是克罗恩病 (CD) 和溃疡性结肠炎 (UC)。溃疡性结肠炎是一种主要针对结肠的慢性炎症性疾病。有人提出A. Finegoldii是一种针对结肠炎的保护性物种,因为A. Finegoldii在患有结肠炎的小鼠中减少。
基于这个事实,进行了一项研究,其中微生物群耗竭的小鼠用口服 DSS 治疗以诱发结肠炎。当用 A. finegoldii 灌胃时,结肠炎的严重程度与野生小鼠相似。当给患上结肠炎小鼠添加了A. finegoldii和 Bacteroides Eggerthii (一种结肠炎诱导细菌)时,与单独添加B. Eggerthii 或添加其他细菌(如Parabacteroides distasonis或Prevotella falsenii)的小鼠相比,结肠炎的严重程度显着降低,这进一步表明A. Finegoldii是一种减轻结肠炎的细菌。
具有对比临床意义的是,已从严重克罗恩病的肠壁海绵状瘘管 (CavFT) 微病变中分离出与其他拟杆菌有关的 A. finegoldii. 对自发性克罗恩病回肠炎小鼠模型粪便样本的宏基因组研究显示,与亲代 AKR/J 小鼠群体相比,Alistipes 富集。
8 周龄 NOD2 敲除小鼠富含Alistipes、抗炎细胞因子(TGF-β 和 IL-10)和 CD4 + LAP + FoxP3 –调节性 T 细胞。这些观察结果的一个可能联系来自对姜黄素的研究,姜黄素是一种已被证明可通过 IL-10增加 CD4 + LAP + FoxP3 -细胞来调节肠道炎症的香料。
有趣的是,有炎症的患者服用益生菌后,Alistipes增加了。迄今为止,尚不清楚该属与肠道中的其他微生物(包括食物和益生菌菌株)之间存在哪些相互作用机制。
诱发结直肠癌
已发现Alistipes作为潜在的病原体可能会诱发结直肠癌。A. Finegoldii 通过 IL-6/STAT 3 途径促进右侧结直肠癌。报告指出Alistipes能产生磺脂,已知磺脂类药物可作为血管性血友病因子受体的拮抗剂并抑制肿瘤坏死因子-α (TNF-a),这些物质与微炎症和血管内皮功能障碍有关。
Lipocalin 2 (LCN 2) 是一种与铁载体结合的抗微生物蛋白,最终会降低铁的利用率。
在 IBD 患者中,LCN 2 在黏膜和粪便样本中的浓度很高。从本质上讲,这可以减少Alistipes的繁殖,因为铁是A. finegoldii 生长的调节因子。
小鼠研究表明A. finegoldii在 WT、LCN 2 KO 和 IL-10 KO C57BL/6J 小鼠中口服给药 1 周后引起肠道炎症。因此,该论文得出结论,Alistipes在缺乏 LCN 2 并促进炎症和肿瘤形成的发炎环境中茁壮成长。此外,他们发现在盲肠中发现Alistipes finegoldii的丰度高于大肠内的其他位置。
癌症免疫疗法中的Alistipes
该属已被证明可以通过调节肿瘤微环境在癌症免疫治疗中发挥有益作用。癌症的主要标志之一是逃避免疫系统。因此,一种形式的抗癌治疗是操纵肿瘤微环境。
免疫疗法的一个例子是通过诱导肿瘤相关骨髓细胞产生肿瘤坏死因子 (TNF) 来操纵微环境,最终导致肿瘤根除。
一种方法是使用肿瘤内 CpG-寡脱氧核苷酸 (ODN) 的组合来激活 TLR9 和抑制性 IL-10R 抗体。这种免疫疗法通常会阻止肿瘤生长并通过肿瘤相关的骨髓细胞诱导肿瘤坏死因子依赖性出血性坏死,从而导致肿瘤抑制。
免疫疗法效果依赖微生物群的存在
由于 TNF 产生减少,抗生素导致肿瘤根除效率降低。然后,研究人员怀疑这是否取决于肠道中的细菌负荷。因此,具有 MC38 肿瘤的无菌 (GF) 小鼠接受了抗 IL-10R/CpG-ODN 治疗。与无特定病原体 (SPF) 小鼠相比,经处理的 GF 小鼠产生的 TNF 量显着降低。
这表明肿瘤相关的先天性骨髓细胞由微生物群引发,以响应抗 IL-10R/CpG-ODN 产生炎性细胞因子,并且抗生素治疗或无菌状态导致的细菌负荷减少会降低这种反应,并且TNF依赖的早期肿瘤坏死。
为了更好地了解抗生素的作用和肠道微生物群的作用,用 LPS 灌胃 MC38 荷瘤小鼠并重建 TNF 表达。Alistipes属和 TLR4 引发/TNF 产生的作用,两者之间存在正相关。
TNF 恢复是由于促炎性革兰氏阴性菌A. shahii与 TLR4 结合,启动 TNF 产生表达的作用。
为了进一步证明他们的假设,作者随后显示了抗生素治疗后A. shahii恢复的延迟,这也与抗生素给药后约 4 周的 TNF 恢复阶段平行。
此外,他们表明,当用抗生素预处理并用A. shahii灌胃的小鼠时,与肿瘤相关的骨髓细胞产生 TNF 的功能得到恢复。临床相关,该研究表明,当Alistipes减少,对癌症免疫疗法的最佳反应平行减少 。陆续其他人也确定了Alistipes在癌症免疫治疗中的作用。
精神健康中的 Alistipes
虽然Alistipes常见于肠道,但它已被证明对定位于肠道外的疾病有显着影响,例如抑郁症、焦虑症、慢性疲劳综合征、自闭症、肝硬化和衰老。肠道内的生态失调会影响肠脑轴,并用于解释肠道微生物群、抑郁症和其他情绪障碍(如焦虑)之间的关系。
抑郁患者中Alistipes增加
与健康对照组相比,重度抑郁症患者的粪便中肠杆菌科和Alistipes水平升高,粪杆菌水平降低。Alistipes属吲哚阳性,影响血清素前体色氨酸,而Faecalibacterium具有抗炎特性。
在一项对 BALB/c 小鼠进行的研究中,该小鼠置于压力环境中,Alistipes丰度显着增加。此外,还发现挪威患有慢性疲劳综合症的患者的Alistipes浓度增加了近 4 倍。这些发现与抑郁症患者Alistipes增加的证据相关,因为抑郁症患者通常与疲劳和压力作斗争 。
Aistipes丰度的增加可能与GABA增加有关
研究指出,Alistipes的增加扰乱了肠-脑轴,因为Alistipes是一种吲哚阳性生物体,因此会降低血清素的可用性。色氨酸是血清素的前体,血清素的减少与抑郁症有关。此外,Alistipes已被证明可表达谷氨酸脱羧酶,这是一种在鸡体内将谷氨酸代谢为 γ-氨基丁酸 (GABA) 的酶。因此Alistipes丰度的增加也可能与 GABA 的增加有关。然而,应该进行研究以证明 GABA 是否被分泌到肠腔中。
自闭症患者中Alistipes研究不一致
此外,自闭症谱系障碍患者的大脑和肠道之间存在关联。研究发现自闭症谱系障碍患者的Alistipes减少。然而,另一项针对不同形式的自闭症 PDD-NOS 的研究表明,儿童中存在大量Alistipes。据推测,这可能来自丙酸的产生,丙酸已被证明对大鼠具有神经生物学作用。需要对Alistipes进行更多研究及其对肠脑轴的影响,因为关于其在两个系统中的保护/致病作用存在矛盾的证据。
磺胺类和生化标志物
Alistipes是一种细菌,具有许多与上述疾病相关的免疫和生化途径。一个重要的意义是Alistipes通过 IL-6/STAT 3 途径促进结直肠癌。
因此,未来的研究可以考虑使用Alistipes物种作为结直肠癌的潜在生物标志物,利用我们基于微生物组 DNA 数据的理解以及对疾病发病机制的生化概念的整合。
实现这一目标的一种潜在方法是寻找磺基脂,这是一类独特的鞘脂,在鞘氨醇碱基中具有磺酸基团。
Alistipes产生磺基脂
研究表明,当 C57BL/6N 小鼠喂食含有红花油或猪油脂肪的高脂肪饮食 3 周时,与喂食正常食物的小鼠相比,磺胺脂以及体重都会增加。进行了宏基因组分析并筛选了这些小鼠盲肠中参与磺胺脂生物合成的细菌基因。发现除A. inops外的所有Alistipes物种都产生磺基脂(关于最新物种 A. megaguti、A.provencensis和磺基脂生产的信息仍然未知)。
为了进一步证明磺基脂是细菌的产物和Alistipes的标志物,科学家们对带有Alistipes的无菌 (GF) 小鼠进行了单定殖研究并检测到单定殖小鼠盲肠中明显出现了以前在无菌小鼠中不存在的磺基脂。
因此,由于结直肠癌的常见风险因素是高脂肪饮食、肥胖和年龄,除了这些结肠疾病中Alistipes的丰度增加外,还有一个有趣的提议是使用磺基脂类作为结直肠癌患者风险的标志物。此外,应进行研究以验证Alistipes丰度是否在息肉中增加,癌前与癌性,息肉是结直肠癌的另一个危险因素。
有害代谢产物与结直肠癌相关
此外,Alistipes在共生细菌中具有最多的腐败途径。腐败是肠道微生物群在胃肠道中发酵未消化的蛋白质,通常会导致细菌产生有害代谢物。据报道,这些产物有害并与结直肠癌相关。此类产品包括氨、H2S、甲酚、吲哚和苯酚。
在一项旨在确定肠道细菌使用的主要腐败途径的研究中,发现Alistipes有助于组氨酸降解/四氢呋喃产生、吲哚产生和苯酚产生。已发现组氨酸降解/THF 产生会释放过量的氨,当吸收时会损害结肠细胞。还发现氨会增加肠细胞增殖并有助于结直肠癌中癌细胞的生长。当患者有结直肠癌风险时,发现过量氨和其他Alistipes产生的腐败产物可能对临床医生有用。
Alistipes是一种从临床样本中分离出来的相对较新的细菌属,尽管与拟杆菌门中的其他细菌相比,其分离率较低。全基因组蛋白质系统发育分析表明,与拟杆菌门中的其他成员相比,该属可能具有独特的功能特性。
一般,超过一定量的Alistipes在临床和临床前研究中已被认为是导致疾病的原因。有趣的是,其他研究表明它们的存在与促进健康表型相关,例如Alistipes在结肠炎、自闭症谱系障碍以及各种肝脏和心血管纤维化疾病等疾病中的保护作用。
尽管Alistipes在健康表型中发挥作用,但与之形成鲜明对比的是,Alistipes在焦虑、肌痛性脑脊髓炎/慢性疲劳综合征、抑郁症、PDD-NOS 和 CRC 等疾病中具有致病作用。
根据相关研究的结论,该属可能在疾病的调节中起主导作用,或者可能只是具有辅助作用或共同诱导作用。将进一步需要动物研究来破译其复杂多模式疾病机制,以及亚型表型的有针对性研究。
使用无菌动物和模型将有助于了解该属在疾病和健康中的作用以及与宿主免疫防御耐受性的相互作用,例如应该有研究调查Alistipes及其生产的 SCFAs 的作用对各种肝病的影响和Alistipes对T细胞分化的直接作用。
主要参考文献
Parker BJ, Wearsch PA, Veloo ACM, Rodriguez-Palacios A. The Genus Alistipes: Gut Bacteria With Emerging Implications to Inflammation, Cancer, and Mental Health. Front Immunol. 2020 Jun 9;11:906. doi: 10.3389/fimmu.2020.00906. PMID: 32582143; PMCID: PMC7296073.
Sakamoto M, Ikeyama N, Ogata Y, Suda W, Iino T, Hattori M, et al. . Alistipes communis sp. nov., Alistipes dispar sp. nov. and Alistipes onderdonkii subsp. vulgaris subsp. nov., isolated from human faeces, and creation of Alistipes onderdonkii subsp. onderdonkii subsp. nov. Int J Syst Evol Microbiol. (2020) 70:473–80.
Rau M, Rehman A, Dittrich M, Groen AK, Hermanns HM, Seyfried F, et al. . Fecal SCFAs and SCFA-producing bacteria in gut microbiome of human NAFLD as a putative link to systemic T-cell activation and advanced disease. United Euro. Gastroenterol J. (2018) 6:1496–1507. 10.1177/2050640618804444
Song Y, Könönen E, Rautio M, Liu C, Bryk A, Eerola E, et al. . Alistipes onderdonkii sp. nov. and Alistipes shahii sp. nov., of human origin. Int J Syst Evol Microbiol. (2006) 56:1985–90. 10.1099/ijs.0.64318-0
Kim S, Goel R, Kumar A, Qi Y, Lobaton G, Hosaka K, et al. . Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin Sci. (2018) 132:701–18. 10.1042/CS20180087
Shen L, Liu L, Ji HF. Regulative effects of curcumin spice administration on gut microbiota and its pharmacological implications. Food Nutr Res. (2017) 61:1361780. 10.1080/16546628.2017.1361780
Merrill AH. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem Rev. (2011) 111:6387–422. 10.1021/cr2002917
Parker BJ, Wearsch PA, Veloo ACM, Rodriguez-Palacios A. The Genus Alistipes: Gut Bacteria With Emerging Implications to Inflammation, Cancer, and Mental Health. Front Immunol. 2020 Jun 9;11:906. doi: 10.3389/fimmu.2020.00906. PMID: 32582143; PMCID: PMC7296073.
Sakamoto M, Ikeyama N, Ogata Y, Suda W, Iino T, Hattori M, et al. . Alistipes communis sp. nov., Alistipes dispar sp. nov. and Alistipes onderdonkii subsp. vulgaris subsp. nov., isolated from human faeces, and creation of Alistipes onderdonkii subsp. onderdonkii subsp. nov. Int J Syst Evol Microbiol. (2020) 70:473–80.
Rau M, Rehman A, Dittrich M, Groen AK, Hermanns HM, Seyfried F, et al. . Fecal SCFAs and SCFA-producing bacteria in gut microbiome of human NAFLD as a putative link to systemic T-cell activation and advanced disease. United Euro. Gastroenterol J. (2018) 6:1496–1507. 10.1177/2050640618804444
Song Y, Könönen E, Rautio M, Liu C, Bryk A, Eerola E, et al. . Alistipes onderdonkii sp. nov. and Alistipes shahii sp. nov., of human origin. Int J Syst Evol Microbiol. (2006) 56:1985–90. 10.1099/ijs.0.64318-0
Kim S, Goel R, Kumar A, Qi Y, Lobaton G, Hosaka K, et al. . Imbalance of gut microbiome and intestinal epithelial barrier dysfunction in patients with high blood pressure. Clin Sci. (2018) 132:701–18. 10.1042/CS20180087
Shen L, Liu L, Ji HF. Regulative effects of curcumin spice administration on gut microbiota and its pharmacological implications. Food Nutr Res. (2017) 61:1361780. 10.1080/16546628.2017.1361780
Merrill AH. Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem Rev. (2011) 111:6387–422. 10.1021/cr2002917
谷禾健康
慢性肾病 (CKD) 影响着全球约 13.4% 的人口,是一个日益严重的全球健康负担。成人中,高血压和糖尿病是慢性肾病的主要原因,而先天性肾脏和泌尿生殖道异常占儿童慢性肾病病因的大部分。慢性肾病与心血管疾病、神经系统并发症、不良妊娠结局和高钾血症等严重健康状况的发展有关。在儿童中,慢性肾病会影响神经认知能力、学校表现、成长、生活质量等。
而许多研究已证明,慢性疾病过程与人类肠道微生物群及其代谢物之间存在关联。
那么慢性肾病与肠道微生物群之间存在什么关系?
肠道菌群代谢产物在慢性肾病中起着什么样的作用?
肠道失调是如何启动炎症过程并导致菌群代谢产物泄漏到血液中的?
哪些饮食方式可以对其进行干预?
本文一起来了解一下。
概 要
· 慢性肾病与菌群关系是双向的;肠源性代谢物和毒素影响慢性肾病的进展,尿毒症环境影响微生物群。
· 微生物代谢物和毒素的积累与肾功能丧失和死亡风险增加有关,但短链脂肪酸和胆汁酸等肾脏保护代谢物有助于恢复肾功能和提高慢性肾病患者的存活率。
· 改变肠道微生物组的特定饮食干预可改善慢性肾病患者的临床结果。
·低蛋白和高纤维饮食增加了产生短链脂肪酸和抗炎菌的丰度。
· 尿液微生物组的波动与感染易感性和抗生素耐药性的增加有关。
01
慢性肾病是什么
肾脏的存在好处多多。肾可以帮助调控血液稳态,维持电解质平衡,调控全身水平衡,甚至可以产生激素。
慢性肾病是一个泛指,包含多种肾功能轻微下降的症状,且肾功能下降和微结构改变持续时间超过3个月,而小于3个月的肾功能恶化则是急性肾损伤。
慢性肾病的病理生理学
慢性肾病包括一系列与肾功能异常和肾小球滤过率(GFR)逐渐下降相关的病理生理过程。
注:单位时间内两肾生成滤液的量称为肾小球滤过率,正常成人为100-125ml/min/1.73㎡。
但是呢,由于GFR的测量麻烦、经济与时间成本较高,临床实践中相对较少使用。于是就出现了eGFR(Estimated Glomerular filtering Rate,即估算的肾小球滤过率),临床上一般用这个指标来衡量肾脏的工作情况。可以对慢性肾病患者的疾病严重程度进行分级,分级越高,滤过率越低,病情越严重。

慢性肾病的潜在病因因年龄、合并症、急性肾损伤反复发生和蛋白尿水平而异。
无论潜在的病因如何,剩余肾单位的过度过滤和肥大、肾小管间质纤维化、肾素-血管紧张素-醛固酮系统的激活以及内皮屏障的破坏都很常见,并导致肾排泄功效和eGFR下降。
从一个等级到下一个等级的转变通常伴随着肾脏内分泌功能的丧失。特别是,患有心血管病的慢性肾病患者表现出肾功能恶化和严重炎症。
肾小管间质间隙中免疫细胞的浸润和免疫衍生成分的积累促进慢性肾病的进展。
慢性肾病治疗的一个关键目标是防止患者进展到疾病的下一阶段。
02
慢性肾病中的肠道菌群失调
最近的研究表明,肠道微生物群失调在慢性肾病的病理生理学中起着关键作用,并导致严重的慢性肾病。
慢性肾病中的菌群变化
双歧杆菌和乳酸杆菌与慢性肾病进展和长期生存率呈负相关。
一项对223名终末期肾病患者的研究表明,与对照组相比,慢性肾病患者的次级胆汁酸和尿毒症毒素水平升高与Eggerthella lenta、Fusobacterium nucleatum、Alistipes shahii呈正相关。在这项研究中,作者表明,Faecalibacterium prausnitzii(普拉梭菌)、Roseburia、Prevotella(产短链脂肪酸菌)的存在与疾病进展和尿毒症毒素积累呈负相关。
另一项对92例慢性肾病患者的研究报告称:
慢性肾病队列中的Paraprevotella,Pseudobutyrivibrio(假丁酸弧菌属),Collinsella数量增加;这一发现使作者提出,这个特征可以用来区分慢性肾病患者(甚至是处于疾病早期的患者)和健康人。
肠道菌群失调引发慢性肾病的两种机制
其一:影响肠道屏障
微生物群组成的变化增强了肠道氨的产生,从而提高了肠腔的生理 pH 值,导致粘膜刺激并破坏了结肠上皮屏障。这导致肠道通透性增加,通常称为“肠漏”。
因此,内毒素和细菌产物易位进入循环并诱导局部炎症,由免疫细胞激活和促炎细胞因子和趋化因子的释放引起,以及慢性全身炎症,加剧肾功能的恶化。
其二:影响血压变化
肠道菌群失调可能促进慢性肾病进展的另一个机制是通过肠道生态失调在内皮功能障碍、血管收缩反应和随后的高血压发展中的作用。
肠道中乳酸杆菌的较低丰度与高血压和肾脏疾病的发生有关。与正常饮食的小鼠相比,高盐饮食的小鼠具有异常的微生物群;这些变化与T淋巴细胞活化和血压升高有关。
肠道微生物群的变化可能是慢性肾病通过一系列免疫反应改变、血压改变、代谢变化和长期炎症进展的起点。
上述是菌群失调影响慢性肾病,反过来,慢性肾病也影响菌群失调。
肠道菌群 ⇋ 慢性肾病
菌群失调与慢性肾病的发病机制之间存在双向关系。
吃进去的营养物质被分解代谢最终产物中的氨,通过肝脏代谢转化为尿素,并释放到循环中。尿素主要通过肾脏排出,部分通过结肠排出。
肾功能的恶化将主要排泄部位从肾脏转移到结肠。结肠中尿素的持续存在会触发产脲酶菌的增殖,导致肠道生态失调。
肠道微生物组与慢性肾脏病之间的关系是双向的
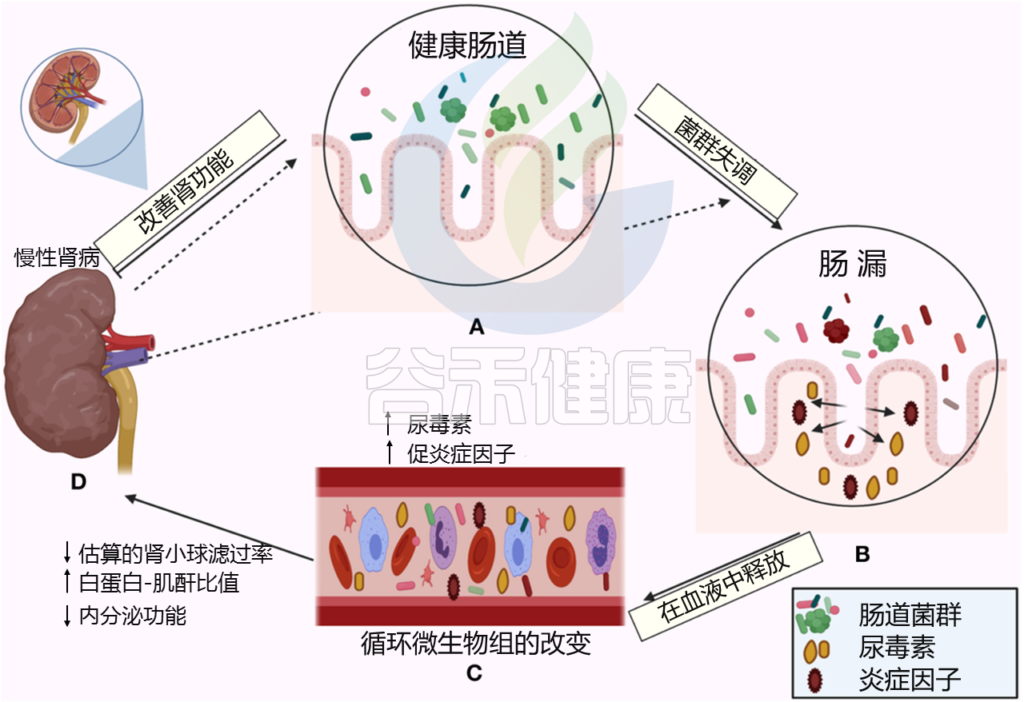
Al Khodor D, et al., Frontiers in Medicine,2022
← 在一个方向上,肠道菌群影响肾脏:
(A)健康的肠道
(B)肠道微生物失调和破坏粘膜层
(C)释放血液中炎性因子和炎症级联的开始,尿毒症毒素积累
(D)估计的肾小球滤过率下降(eGFR),白蛋白肌酐比值(ACR)升高,肾脏内分泌功能丧失
→ 在另一个方向,慢性肾病驱动肠道内的生态失调(虚线箭头所示),并引发炎症级联
03
慢性肾病中的微生物代谢物
一般来说,与慢性肾病相关的微生物代谢物分为两类;有害和肾脏保护代谢产物。
一方面,有害代谢物的水平增加,包括三甲胺 N-氧化物 (TAMO)、硫酸吲哚酚和对甲酚硫酸盐与肾纤维化、内皮功能障碍、估计肾小球滤过率 (eGFR) 下降、心血管并发症以及慢性肾病死亡率和发病率增加有关。此外,5-甲氧基色氨酸和硫酸吲哚酚的血清水平与慢性肾病进展呈正相关。
另一方面,包括短链脂肪酸在内的肾脏保护代谢物通过抑制上皮屏障的破坏和调节抗炎反应来预防慢性肾病的进展。来自肠道菌群的吲哚丙酸水平与慢性肾病患者的对甲酚硫酸酯和吲哚硫酸酯浓度呈负相关。
菌群代谢产物和慢性肾病的关系
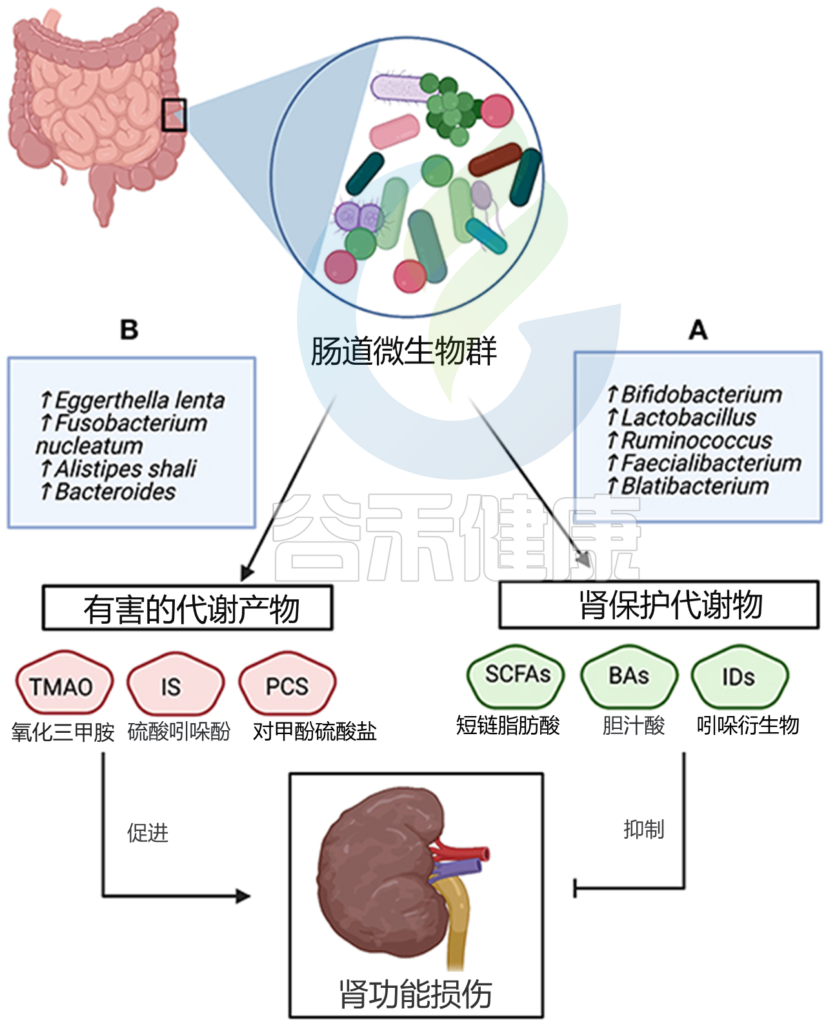
Al Khodor D, et al., Frontiers in Medicine,2022
一些人类和动物研究已经证明了TAMO对肾脏的有害影响,表现为肾间质纤维化、eGFR下降、内皮功能障碍和心血管疾病风险增加。
我们知道,肾脏的功能就是代谢身体废物,它的功能类似一个“清洁工”,肾脏生病也就是清洁工罢工,那么代谢废物就清除不出去了,于是在血浆中累积。
慢性肾病患者死亡率和发病率的增加归因于毒素的积累:硫酸吲哚酚和硫酸对甲酚。这些毒素与血浆蛋白具有很高的亲和力,从而减轻了它们通过透析膜的清除。
TAMO、硫酸吲哚氧基和硫酸对甲酚分别参与SMAD信号传导、色氨酸代谢和酪氨酸途径。
广泛的尿毒症毒素和其他微生物代谢物积聚在慢性肾病患者的生物样本中,包括血浆、粪便和尿液等常见生物样本中的毒素和其他微生物代谢物,也包括呼出气中的挥发性代谢物和粪便培养物中收集的气体。例如,慢性肾病患者体内会积聚气体代谢物,包括异戊二烯、醛、二甲基二硫、二甲基三硫和硫酯。
04
慢性肾病中的饮食干预
慢性肾病患者存在微生物失调和肠道代谢物积累。
益生菌
对慢性肾病患者进行的随机对照临床试验表明,益生元和益生菌治疗后肠道微生物群组成的变化改善了疾病结果,并降低了尿毒症毒素水平。
双歧杆菌和乳酸杆菌含量高的患者血清中尿毒症毒素水平较低,炎症环境减轻,肾功能改善。
益生元
益生元是不易消化的膳食成分,如膳食纤维和耐消化淀粉。它们存在于谷物、水果、牛奶、蜂蜜和蔬菜中,或者可以作为膳食补充剂。益生元发酵通过增加双歧杆菌和乳酸杆菌的丰度,降低类杆菌、梭状芽孢杆菌和肠杆菌的水平,有益地改善肠道细菌。
不利肾脏的食物:
摄入富含胆碱和 L-claritin的食物(TMAO的前体),如蛋黄、内脏、肉类和牛奶,与尿毒症毒素的大量积累和肾小球滤过率的下降相关。
低蛋白饮食减少炎症菌
一项前瞻性交叉临床试验将60例慢性肾病患者随机分为不同的饮食干预组;与常规饮食组相比,极低蛋白饮食组的肠道放线菌丰度增加,炎性变形菌减少。
膳食纤维降低慢性肾病风险
在慢性肾病患者中,膳食纤维摄入可降低循环中促炎细胞因子的水平,减缓eGFR的下降,降低尿毒症毒素的血浆水平,并将慢性肾病相关的心血管风险降至最低。
抗性淀粉降低血浆毒素
研究人员研究了补充抗性淀粉(16克/天)对慢性肾病患者的影响;他们观察到尿毒症毒素(硫酸吲哚氧基和硫酸对甲酚)、IL-6和硫代巴比妥酸反应物质的血浆水平降低。
乳果糖糖浆降低血清肌酐
这些结果与另一项将32例慢性肾病患者随机分为两组的研究一致;接受乳果糖糖浆治疗8周的组,肠道微生物群中双歧杆菌和乳酸杆菌的含量更高,血清肌酐水平降低。
虽然这些研究表明益生菌和益生元对慢性肾病有有益的作用,但也有其他研究表明循环肠道菌群代谢物或慢性肾病结果没有显著变化。
重要的是要指出,现有的研究是异质的;他们使用不同的膳食补充剂,有不同的干预持续时间,并对其他共病患者、肾病严重程度和潜在病因不同的患者进行管理。这种异质性使得从这些研究中得出结论非常困难。这就是说,在儿童饮食干预研究中可能会获得更好的结果,因为其他共同因素是最小的。
总之,这些研究表明,饮食干预疗法有可能调节微生物组组成及其代谢产物,从而改善慢性肾病并发症和慢性肾病进展率。然而,需要进一步设计良好的前瞻性研究来明确证明营养疗法对慢性肾病的益处。
05
尿和血液微生物群在慢性肾病中的作用
微生物组学领域的大部分注意力集中在肠道微生物组及其代谢产物上;然而,尿液微生物组正受到更多的关注。
新一代测序技术的发展使研究表明,健康个体的尿路由不同种类的微生物控制,这些微生物的分布模式影响尿路健康。
泌尿系微生物组的波动发生在泌尿系感染中,并与抗生素耐药性有关。肾移植后,尿液微生物组发生变化,这些改变被认为是导致同种异体移植功能障碍和增加感染易感性的原因。此外,慢性肾病患者尿液微生物多样性与eGFR值相关。
健康个体的循环微生物群包含多种细菌类群,其中以变形菌门为主。血液中循环的肠源性内毒素可改变血液微生物组。
一项研究调查了399名参与者的血液代谢组与肠道微生物群α多样性之间的相关性,结果表明,对甲酚和TAMO等肠道菌群代谢物反映了肠道细菌的香农多样性,可能是反映肠道健康的生物标记物。
使用16S rRNA靶序列对血液样本进行的病例对照研究表明,与对照组相比,慢性肾病患者的肠杆菌科和假单胞菌科的多样性较高,这也与较低的eGFR相关。
因此,肠道微生物群通过不同途径对慢性肾病的结局产生最终影响。
06
结 语
双歧杆菌、乳酸杆菌和胆汁酸成分水平较低与慢性肾病患者的不良后果有关。TAMO、硫酸吲哚氧基、硫酸对甲酚和其他有害微生物代谢产物在慢性肾病患者体内积累,这些代谢产物的水平与疾病进展相关。
肠道、泌尿道和血液微生物群以及相关代谢物之间的复杂相互作用可能协调慢性肾病发病机制中的亚临床变化,并促进疾病的发生。
通过饮食干预调节肠道微生物群可以改善慢性肾病患者的临床结果。
随着肠道微生物群的深入研究,可为慢性肾病的病因、代谢途径和潜在治疗提供线索。
未来可在以下方面深入开展研究:
主要参考文献:
Al Khodor D, Wehedy E, Shatat I F. The human microbiome in chronic kidney disease: a double-edged sword[J]. Frontiers in Medicine, 2986.
Mertowska P, Mertowski S, Wojnicka J, et al. A Link between Chronic Kidney Disease and Gut Microbiota in Immunological and Nutritional Aspects. Nutrients. 2021;13(10):3637. Published 2021 Oct 17. doi:10.3390/nu13103637
Feng Z, Wang T, Dong S, et al. Association between gut dysbiosis and chronic kidney disease: a narrative review of the literature. J Int Med Res. 2021;49(10):3000605211053276.
Giordano L, Mihaila SM, Eslami Amirabadi H, Masereeuw R. Microphysiological Systems to Recapitulate the Gut-Kidney Axis. Trends Biotechnol. 2021 Aug;39(8):811-823. doi: 10.1016/j.tibtech.2020.12.001. Epub 2021 Jan 6. PMID: 33419585.
谷禾健康
你知道肠道和大脑之间的秘密吗?
大脑自闭了,为什么是肠道的锅?
肠道真的会影响大脑嘛,原理是啥?
……
已经有越来越多人开始好奇肠道和大脑之间的联系,关于这方面的前沿研究也在不断更新,人们开始逐渐深入了解相关机制。

中枢神经系统功能与肠道微生物之间存在关联,即大脑和肠道之间的串扰,与迷走神经、肠神经系统、免疫系统和循环相互作用。胃肠道微生物群可以影响神经系统,无论是通过迷走神经直接输入大脑,还是通过间接激活整个胃肠道的肠神经系统。
以下是我们整理过相对较全面的关于肠道和神经系统相关的文章:
深度解读 | 肠道菌群和中枢神经系统的关系
肠道微生物群在神经系统疾病中的作用
本文在这个基础上,结合最新研究进展,再次阐述人类肠道微生物组在神经系统疾病发病机制中的潜在作用,讨论了精神药物、益生菌、益生元、合生元、后生元、粪菌移植等方式治疗神经系统疾病的潜在作用。

大家越来越有这样的认知:胃肠道微生物群的不平衡会影响大脑的生理、认知和行为。
肠道微生物群通过神经、免疫、体液和内分泌联系参与肠-脑双向相互作用。我们先来了解一下以下它们之间几种“交流方式”:
肠道主要通过两条神经解剖学途径与大脑进行沟通。
首先,大脑和肠道直接通过迷走神经(VN)和脊髓中的自主神经系统(ANS)进行沟通。
其次,细菌通过迷走神经和肠神经系统传入神经元的刺激在大脑和胃肠道微生物群之间建立直接的神经联系。
此外,迷走神经激活表现出抗炎作用,迷走神经活动对肠道微生物群及有益菌的产生积极影响。
迷走神经可以将胃肠道中的内分泌、神经元和微生物改变转移到大脑。
几项临床前研究表明,肠道疾病的病理生理学和发病机制,包括炎症性肠病(IBD)和肠易激综合征(IBS),以及神经系统疾病和精神疾病,包括焦虑、抑郁、自闭症、阿尔茨海默、多发性硬化和帕金森病,与肠道微生物群失衡有关。
由于微生物群-肠-脑轴(MGBA)内存在多种相互作用机制,胃肠道微生物组主要通过免疫相关、神经、内分泌和代谢信号通路与中枢神经系统进行通信。
肠道微生物可通过在肠腔中产生大量代谢物与宿主交换感官信息,包括神经递质、GABA、血清素、多巴胺和去甲肾上腺素,激素(如下丘脑-垂体-肾上腺轴中促肾上腺皮质激素释放激素的分泌)、组胺、乙酰胆碱、儿茶酚胺,以及几种维生素和短链脂肪酸。其中一些分子可以通过血脑屏障进入大脑,并影响神经回路。在这些代谢物中,短链脂肪酸是结肠细菌发酵膳食纤维产生的主要代谢物,在调节神经免疫内分泌、代谢稳态、感染和炎症方面发挥着关键作用。
许多种类的乳酸杆菌和双歧杆菌产生γ-氨基丁酸(GABA),这是大脑中主要的抑制性神经递质。
念珠菌、大肠杆菌和肠球菌会产生神经递质5-羟色胺,而一些芽孢杆菌会产生多巴胺。
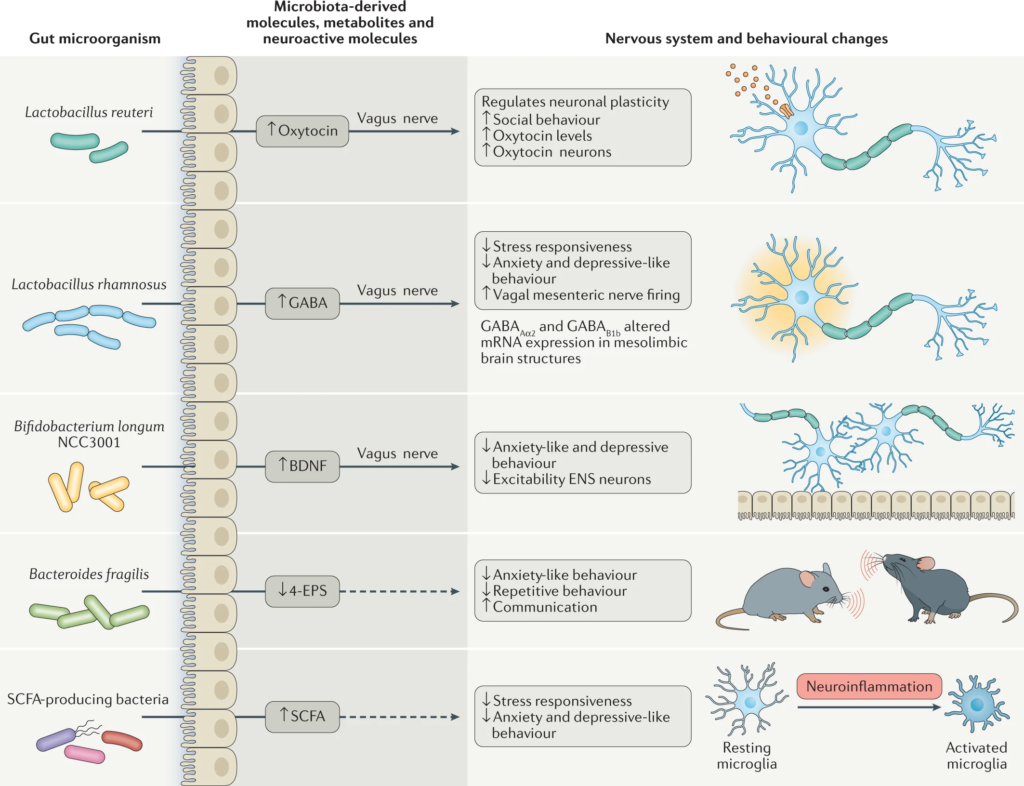
Morais LH, et al. Nat Rev Microbiol. 2021.
微生物群代谢产物,包括维生素、神经活性代谢物(如短链脂肪酸)和神经递质,介导双向微生物群-肠-脑轴相互作用以调节宿主神经生理学和免疫。
微生物代谢产物——短链脂肪酸,如乙酸、丙酸、丁酸,也可以通过进入体循环影响中枢神经系统。
短链脂肪酸能够刺激交感神经系统,粘膜血清素的释放,从而影响大脑的记忆或学习过程。
大约95%血清素(5-羟色胺)由肠粘膜嗜铬细胞产生。在外周,5-羟色胺参与Gl分泌、运动(平滑肌收缩和放松)和疼痛感知的调节,而在大脑中,5-羟色胺参与情绪和认知的调节。
肠道微生物群在色氨酸代谢中也起着重要作用,色氨酸代谢是产生5-羟色胺的前体。例如,婴儿双歧杆菌通过增加血浆色氨酸影响中枢5-羟色胺的传递。
我们知道了以上化学信使,那么它们通过什么途径去发挥作用?
细菌神经活性代谢物和饮食分子可以通过多种方式改变大脑和行为,例如影响上皮细胞以影响上皮屏障的功能,肠内分泌细胞释放激素,通过树突状细胞调节小胶质细胞和免疫细胞的功能。
代谢产物在通过血脑屏障运输后直接影响,或通过神经内分泌、免疫或迷走神经途径间接影响。
血脑屏障
细菌可以直接将因子释放到体循环中或可以转移到血液中。一旦进入血液,微生物组及其因子可以改变外周免疫细胞,促进与血脑屏障的相互作用,并最终与神经血管单元的其他元素相互作用。菌群代谢产物,例如短链脂肪酸,可穿过血脑屏障以影响脑功能。
肠道屏障
肠道微生物群还可以影响肠道屏障的完整性,控制信号分子从肠腔到固有层(包含免疫细胞和ENS神经元末端)或门静脉循环的通道。肠道屏障的完整性在某些神经精神疾病中会被破坏,如焦虑症、自闭症谱系障碍和抑郁症。
Li XJ,et al., CNS Neurosci Ther. 2020
肠道通透性
慢性应激可改变肠道通透性(肠漏综合征),这与低度炎症有关,在功能上与抑郁症等精神疾病有关。
在许多情况下,循环中细菌内毒素(脂多糖,LPS)的增加是导致疾病的基本危险因素。
肠道屏障和肠道通透性区别:
肠屏障
是将肠腔与内部宿主分开的功能实体,由机械元件(粘液、上皮层)、体液元件(防御素、IgA)、免疫元件(淋巴细胞、先天免疫细胞)、肌肉和神经元件组成
肠道通透性
指给定部位肠道屏障的功能特征,可通过分析整个肠壁或跨壁成分的定义分子的通量率进行测量
神经内分泌(HPA轴)
在神经系统内,应激是如何激活HPA轴反应?
该反应涉及下丘脑神经元,该神经元向大脑或门静脉循环分泌激素,如促肾上腺皮质激素受体激素(CRH),触发促肾上腺皮质激素(ACTH)的释放,然后启动皮质醇的合成和释放。
皮质醇调节神经免疫信号反应,进而影响肠屏障的完整性。
肠道渗漏导致促炎状态,循环中TNF-A、干扰素-y和IL-6水平升高。已知IL-6激活HPA轴。
随着时间的推移,它也下调糖皮质激素受体。这些受体是抑制HPA轴的反馈机制,然而,它们的下调导致HPA轴过度活跃和过度敏感。
有研究表明,这些变化导致海马5-羟色胺的减少以及BDNF表达的减少。BDNF表达降低是抑郁症发病的危险因素。
应激激素、免疫介质和CNS神经递质可激活肠神经系统的神经细胞和迷走神经的传入通路,从而改变肠道环境和微生物群组成。
目前,已经表明胃肠道微生物群在发展大脑免疫和神经发育中起着核心作用。
大脑并非免疫“特权”器官
免疫系统和中枢神经系统都是复杂而有组织的系统,在运作模式和发育过程中具有共同的特征。大脑中可以产生参与先天免疫的分子,如Toll样受体(TLR)、细胞因子、以及适应性免疫相关分子,如抗体受体和主要组织相容性复合体(MHC),这些分子在脑发育中起着关键的调节作用。
脑膜淋巴细胞和血脑屏障
尽管以前认为大脑是一个免疫特权器官,但它包含脑膜淋巴管。脑膜中淋巴管的存在使我们能够深入了解中枢神经外周免疫系统之间的可能联系,从而影响自身免疫。
此外,淋巴细胞和小胶质细胞可以调节认知,对神经元回路的正确连接也是必不可少的。小胶质细胞是巨噬细胞,占所有神经细胞的10%。它们负责中枢神经系统主动免疫防御的基本作用。而且大脑大部分区域的血管系统发展出选择性血脑屏障的组织特异性,允许所需分子进入大脑,并限制潜在有毒物质或细胞的渗透。
免疫影响大脑和神经
免疫细胞具有渗透大脑的能力。浸润性免疫细胞或小胶质细胞能够与中枢神经系统有效地相互作用,并影响大脑功能和病理学。
小胶质细胞从胚胎祖细胞中出现,并可在中枢神经系统中经历自我更新的过程。它们不仅参与典型的免疫功能,如吞噬和抗原递呈,还参与一些大脑生理活动。
免疫细胞如中性粒细胞、巨噬细胞、T细胞和自然杀伤(NK)细胞从大脑的外周循环血液进入。小胶质细胞对行为和某些神经系统疾病有着巨大的影响,如神经退行性疾病。
通常,成人大脑神经发生受宿主肠道微生物群的影响。成年小鼠的抗生素治疗影响肠道微生物群的多样性和海马的神经发生,而且益生菌具有重建肠道微生物群的能力并显示神经发生改善。
菌群与免疫
最近的许多研究表明肠道微生物组与大脑以及肠道微生物组与免疫系统调节之间存在联系。对无菌和对照啮齿动物的研究表明,肠道微生物组的缺失会加剧焦虑样行为。此外,如果肠道微生物群在生命早期恢复,这种行为完全可以治愈。微生物代谢产物可通过血液循环转移到大脑,并影响迷走神经或免疫系统和炎症反应,这表明由于生态失调引起的微生物代谢紊乱可对焦虑相关疾病产生巨大影响。
菌群招募免疫细胞
肠道微生物群落通过在各种免疫条件下招募不同的免疫细胞,直接或间接调节肠道内的免疫反应。胃肠道微生物的动态和异质性特征是宿主体内平衡的基础。据报道,与野生型小鼠相比,以胃肠道淋巴细胞水平降低为特征的免疫系统发育不平衡减少了无菌小鼠中免疫球蛋白a(IgA)、抗菌肽(AMP)和未成熟肠道相关淋巴组织(GALT)的数量,强调胃肠道微生物组在宿主免疫形成中的重要作用。
病原识别和抗体反应
此外,肠道免疫系统在区分共生动物和病原体以及确定导致免疫耐受的因素方面至关重要。因此,肠道菌群可以调节免疫系统的发育和功能,形成肠道微生物群落并调节肠道粘膜表面的病原体。例如,据报道,无菌小鼠中辅助性T细胞1(Th1)和Th17细胞数量的减少,以及IL-22和IL-17的减少,导致固有层数量减少。
影响T和B细胞发育和反应
肠道微生物组影响肠道T和B细胞反应的诱导和发展。肠道微生物群在肠道CD81 T淋巴细胞的激活中起着重要作用。
胃肠道微生物组影响固有层处肠道驻留B淋巴细胞的发育,因为无菌小鼠的固有层处B细胞计数较低。此外,这种类型的细胞也能够产生IgA,作为微生物成分的强调节因子。这表明在促进对共生微生物的免疫耐受以及在固有层中实现IgA的广泛多样化方面具有突出作用。
菌群代谢物参与免疫
对短链脂肪酸的研究表明,除了增强肠道系统中调节性T淋巴细胞的功能和数量外,它们还可以通过抑制转录因子NF-kB和HDAC活性来促进抗炎作用和肠道屏障功能。微生物肠道菌群与芳香烃受体(AhR)结合产生的色氨酸衍生物影响肠道免疫系统的功能。肠道菌群可产生精氨酸衍生物,包括二胺、精胺、亚精胺和多胺,通过增强常驻免疫细胞和肠粘膜的内环境平衡来调节免疫反应 。
微生物群与大脑之间通过肠脑轴的分子通讯途径
Sorboni SG, et al.,Clin Microbiol Rev. 2022
肠神经系统在人的一生中经历了一个巨大的发育变化,同时在病理生理功能方面保持了灵活性。因此,随着年龄的增长,肠神经系统开始衰弱,宿主微生物群、免疫系统和生理学也开始衰弱。
注:肠道神经系统是周围神经系统中最复杂、最重要的部分之一,由小神经节和神经元组成。这些神经元分布在整个胃肠道膜。
在了解菌群与神经衰退之前,首先看一下菌群与神经发育的关系。
大脑发育是一个复杂的过程,通常从妊娠第三周开始,一直持续到青春期晚期,生命的前3年被认为是胃肠道微生物群和脑突触形成的中心时期。
对无菌(GF)小鼠模型的研究表明,胃肠道微生物群、行为表现和大脑功能之间存在相关性。应激反应比无特定病原体(SPF)小鼠强烈得多。据报道,无菌小鼠模型中突触形成标记物水平的降低,包括分别负责突触成熟的突触素和PSD95,突出了共生细菌在大脑发育中的重要性。基于这些结果,肠道菌群似乎在大脑发育过程中对神经网络的形成起着核心作用。
部分自闭症患者表现出慢性便秘、肠道通透性增高、腹痛和肠道微生物群紊乱的症状,从而提供了生态失调和神经发育障碍之间的可能联系。微生物从母亲转移到胎儿、分娩方式、抗生素暴露和饮食习惯都会改变婴儿微生物群的定植和成熟。
怀孕期间补充抗生素会导致母体和新生儿肠道菌群的破坏,随后运动活动的减少,以及新生儿行为的改变。因此,临床证据支持抗生素诱导的失调与几种神经发育障碍的发展相关,包括精神分裂症、抑郁症和双相障碍。
尽管需要更多的研究来阐明上述因素与神经发育障碍之间的分子联系,但操纵早期生命微生物群可以被认为是预防自闭症和其他神经疾病的有益手段。
肠神经系统是否随年龄增长而变化?
一些研究认为随着年龄的增长,肌间神经元的数量和功能都会减少,而其他研究则没有报道这种后果。因此,肠神经系统是否会随着年龄的增长而发生变化仍在激烈争论中。
肠神经系统随年龄变化的研究表明,肠神经节的形态发生了变化,变性神经纤维的识别,α-突触核蛋白(α-syn)和脂褐素积累。这些报告表明老化和肠神经系统退化之间存在相关性。
肠神经系统在肠道细胞活动、营养吸收和肠道激素分泌中具有重要作用。针对肠神经系统的研究虽多,但机制不明确。根据以上研究证据,有理由假设肠神经系统发生了退化性变化,与宿主生理学、代谢、微生物群以及与衰老相关的免疫系统的变化相一致。
考虑到肠神经系统细胞不同的功能和形态特征,不同的细胞类型在年龄相关疾病的易感性中起主要作用。代谢活跃的神经细胞中氧化DNA损伤和活性氧(ROS)产生的负担可能是肠神经系统衰老的另一个潜在因素。
根据强调在老年动物模型中降低钙结合蛋白表达的重要性的研究,钙失调也可被认为与肠神经系统衰老有关。也有报道称,在衰老过程中,肠神经细胞中的钠通道基因表达显著改变。
对肠神经系统的年龄相关影响的额外研究是必要的,可能有助于对这些复杂的衰老和胃肠道关联的新理解,也可能带来新机会,发现治疗各种年龄相关神经疾病的新治疗方法,以及改善老年人的生活质量。
肠道微生物群与肠神经系统
考虑到胃肠道肠道神经元附近定植的微生物,肠神经系统似乎与肠道微生物群高度相关或可能受其影响。
几项研究表明,新生儿肠神经系统的发育是由早期暴露于肠道常驻微生物形成的。此外,肠道微生物群可以调节神经胶质细胞对固有层的初始定植以及稳态。
随着年龄的增长,拟杆菌和变形杆菌(尤其是Gammaproteobacteria)数量增加,厚壁菌和双歧杆菌数量显著减少。
与衰老过程中肠道微生物群的实质性变化类似,成人肠道神经元对年龄相关损伤的敏感性更高。肠道微生物群平衡的改变,包括条件致病菌的增加和有益菌或共生菌的减少,可导致胃肠道的不同微生物代谢产物谱。
因此,由肠道菌群的年龄相关改变引起的肠道炎症水平升高会影响肠神经系统,并导致肠道神经元不同生理和神经化学功能的损害或丧失,从而导致年龄相关疾病的发生。
肠神经系统还可以调节肠道微生物群落组成,维持和促进肠道健康。此外,肠道微生物群落的缺失可导致肠神经系统功能的异常和改变。
也有研究表明,肠道失调和肠道病理的发展与胃肠动力紊乱相关,表明肠神经系统在肠道微生物群维持和预防可导致宿主疾病的病原菌过度生长中的重要作用。
尽管确切的机制仍需澄清,但从肠-脑轴和老年人群神经病变的角度来看,肠道微生物群、粘膜免疫系统、肠道神经元和肠上皮细胞之间复杂的相互作用和相互作用强调了该领域研究的重要性,并强调了进一步研究的必要性。
胃肠道菌群和神经-肠内分泌系统中与年龄相关的变化可能通过肠-脑信号通路功能障碍影响大脑健康的分子通路图如下。
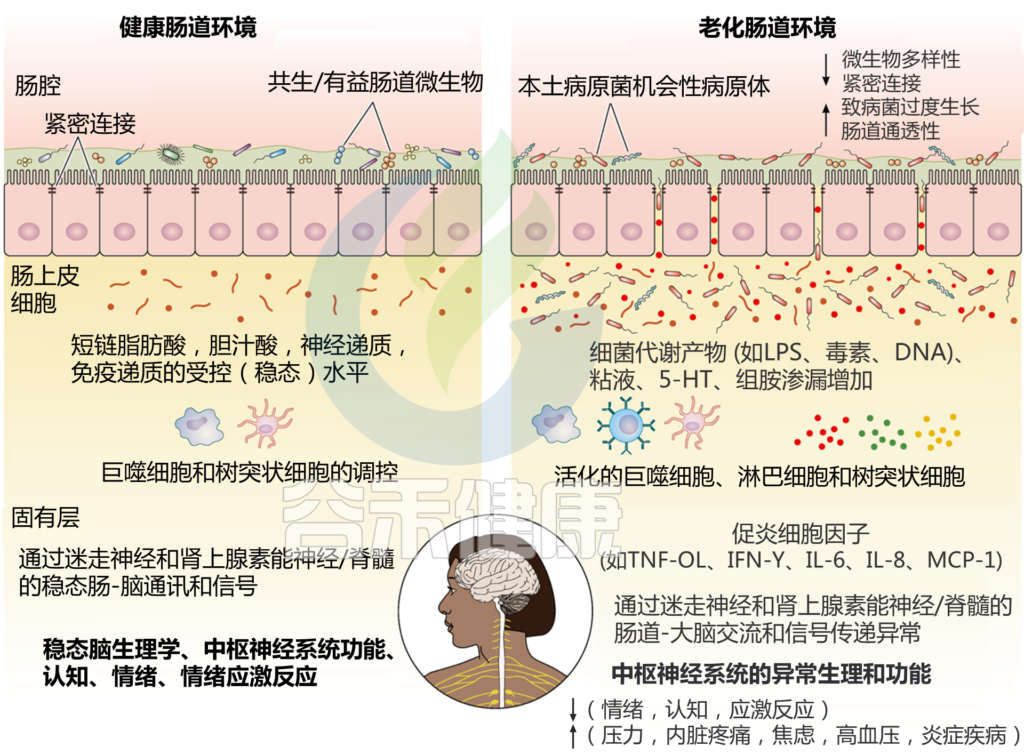
Sorboni SG, et al.,Clin Microbiol Rev. 2022
在健康成人中,平衡的肠道菌群和肠道屏障完整性有助于维持平衡的微生物群落及其代谢产物,包括短链脂肪酸。胃肠道中神经递质的适当产生有助于通过巨噬细胞和树突状细胞的平衡增殖维持受控的肠道炎症和免疫系统,最终导致受控的肠脑通讯和中枢神经系统的适当功能。
然而,在衰老宿主中,胃肠道微生物群落多样性的改变和肠道屏障完整性的破坏,通过短链脂肪酸、LPS、5-羟色胺、组胺、葡萄糖和葡萄糖水平的不平衡,导致胃肠道上皮细胞衬里的生化和微生物微环境发生扰动,分泌性免疫球蛋白(sIgA)等。因此,在肠道环境中诱导过度激活的炎症环境导致健康肠道-大脑沟通中断。
老化的微生物组本身足以导致认知障碍
微生物群移植研究表明,老化的肠道微生物群可导致年轻受体发病。从老年供体小鼠到无菌受体小鼠的肠道微生物群移植有助于促进肠道炎症和增加通透性,这表明与较高水平的变形菌和TM7细菌相关。
最近的一项研究表明,来自老年供体小鼠的粪便微生物群移植导致年轻受体小鼠的空间学习和记忆障碍,产短链脂肪酸菌显著减少,包括Faecalibaculum、毛螺菌科和瘤胃菌科。
从老龄小鼠模型到无菌小鼠的粪菌移植导致粪便短链脂肪酸生成减少、促进抑郁样行为和短期记忆障碍,表明老龄肠道微生物组能够降低宿主的短链脂肪酸水平和随后的认知能力下降。
从老龄供体到年轻受体大鼠的粪菌移植导致受体小鼠的认知行为损伤、突触结构改变、糖基化终产物水平升高以及炎症和氧化应激增加。
自闭症谱系障碍
精神分裂症
抑郁症
自闭症谱系障碍(ASD) 包括一系列复杂的神经发育障碍症状,包括社交和交流障碍,以及限制性和重复性行为模式。
谷禾参与组织的一项多中心合作的自闭症谱系发育障碍与肠道菌群研究,该项目共包括 773 名自闭症受试者(16 个月至 19 岁)和 429 名神经典型 (NT) 发育受试者(11 个月至 15 岁)。该研究已发表的在《GUT》。【实际上这个研究仍在继续,目前我们已经构建了超过3000例的自闭症样本队列】
这项研究详细分析了不同年龄发育阶段自闭症儿童的菌群与正常儿童的差异和变化,并揭示了临床症状相关的自闭症儿童肠道微生物组发育动态特征。
研究显示多种菌、菌群代谢功能的改变与自闭症儿童的行为、睡眠和胃肠道症状的严重程度有关。
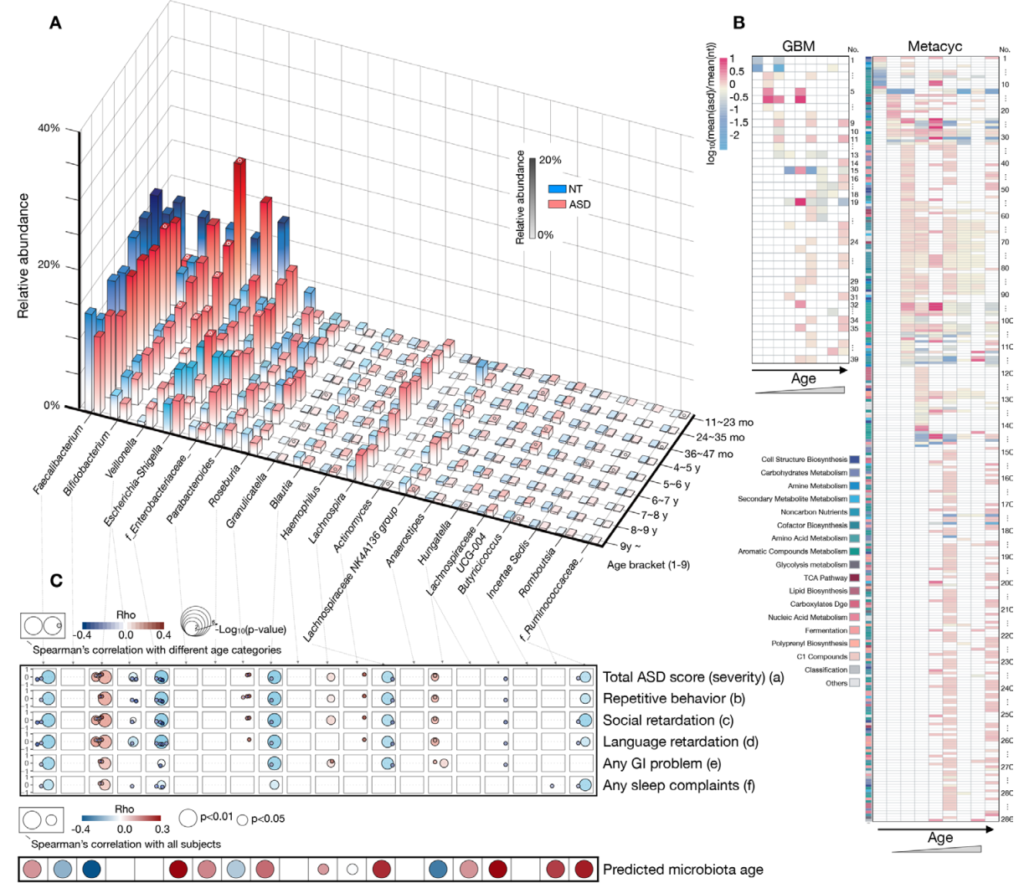
Lou M, et al., Gut. 2021
为了更好的应用于临床,我们尝试并给出了基于微生物群的疾病诊断模型,并在不同年龄和地区显示出很好的模型效果。
Lou M, et al., Gut. 2021
尤其是在早期,基于肠道菌群的模型对于临床鉴别和评估有更大价值。因为很难对低龄幼儿进行行为学评估,而错误的评估很容易错过早期干预的黄金时机,肠道菌群模型可以更加量化的评估,而不依赖问询或行为学,可以很好的对现有临床诊疗进行补充。
遗传和环境因素
自闭症的确切病因尚不确定。然而,有大量的临床证据表明,遗传和环境因素在该病的发病中起着至关重要的作用。已经确定了100多个影响中枢神经系统发育的基因和基因组区域,这些基因和基因组区域可能与自闭症的发展有关。
环境因素,如营养不良、病毒和婴儿期发育错误,特别是发育中大脑中七种蛋白质的母体自身抗体,也与自闭症有关。这些环境因素现在已经被证明对自闭症有着比以前认为的更重要的作用。
微生物群-大脑重要作用
大约40%的自闭症患者经历更多的胃肠功能障碍,包括肠功能改变和腹部痉挛(疼痛)、腹泻、反流和呕吐。
胃肠道症状与自闭症严重程度之间的相关性表明了肠道与大脑之间联系的重要性。
人们逐渐发现肠道中的微生物群和大脑相互作用在自闭症等神经精神疾病中起着关键作用。
肠道微生物群的组成与年龄有关。肠道微生物群正常组成的改变会增加致病菌的数量,从而导致感染。
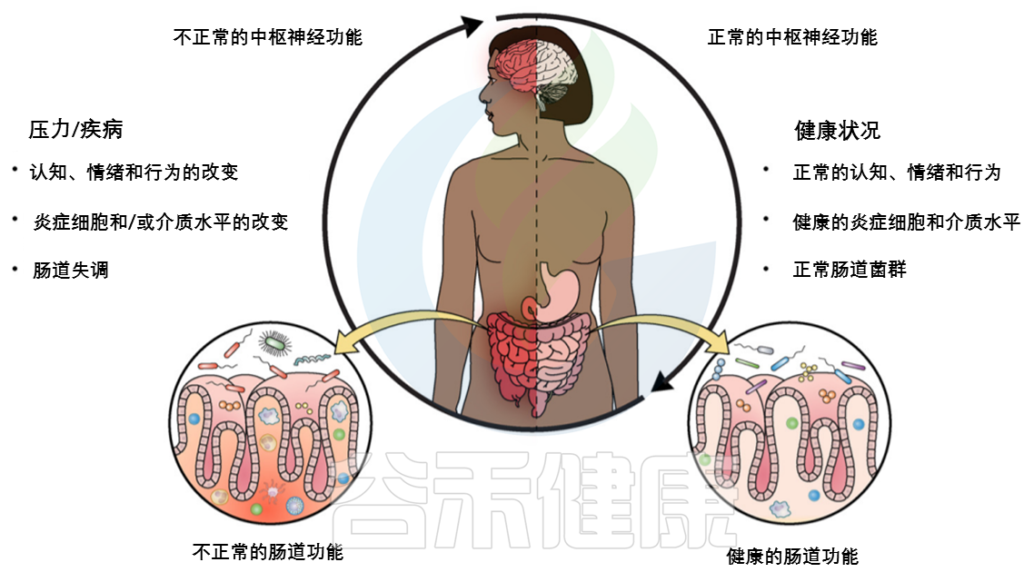
Sorboni SG, et al.,Clin Microbiol Rev. 2022
肠道失调 -> 炎症
自闭症患者的胃肠道紊乱和中枢神经系统症状可能与肠道失调引起的炎症状态有关。
根据最新研究,自闭症儿童的肠道微生物群组成发生了显著变化,胃肠道症状可能代表了炎症过程。炎症与肠粘膜屏障对细菌神经毒性肽(如脂多糖)的通透性增加和炎性细胞因子的产生有关。细菌代谢产物在肠-脑轴中起着至关重要的作用;因此,肠脑信号中断可能与自闭症和帕金森等神经精神疾病有关。
最近关于微生物干预预防和治疗自闭症的潜力的研究
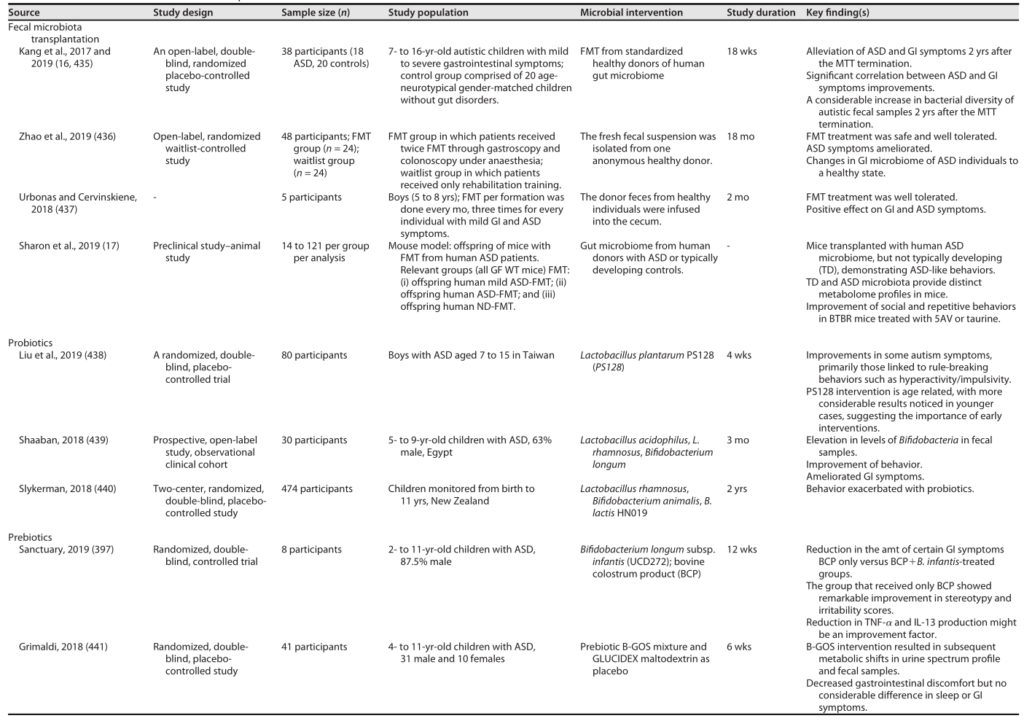
Sorboni SG, et al.,Clin Microbiol Rev. 2022
自闭症患者肠道菌群变化
自闭症儿童甚至成人的肠道微生物群与健康对照组完全不同。
对自闭症儿童粪便样本的调查表明,由于拟杆菌数量减少,拟杆菌/厚壁菌比例降低。
在患有自闭症的儿童中,乳酸杆菌、梭状芽孢杆菌、脱硫弧菌、Caloramator、Alistipes、Sarcina、Akkermansia、Sutterellaceae、肠杆菌科的水平升高。
许多研究人员已经评估了梭菌的丰度及其作为危险因素的作用。对自闭症儿童每周使用万古霉素治疗可显著改善神经行为和胃肠道症状。
发送信号 -> 控制肠通透性
除了肠道微生物群在免疫系统发育中的作用外,梭状芽孢杆菌通过肠上皮或迷走神经的传入纤维向大脑发送信号,并控制肠道通透性。
代谢
据报道,肠道微生物可通过产生酚类、短链脂肪酸和游离氨基酸等代谢物在肠道通透性中发挥重要作用。自闭症儿童丙酸和乙酸的比率较高,但丁酸的比率较低。非消化性碳水化合物的最终产物可能与自闭症发病机制有关。
详见:如何通过喂养菌群产生丁酸调节人体健康
自闭症儿童和正常发育儿童的粪便和血浆代谢组比较显示线粒体功能障碍;自闭症儿童中不同水平的酚类微生物代谢产物、脂质、氨基酸和外源性代谢可能被用作自闭症的分子生物标记物。
在另一项研究中对患有自闭症的儿童的血浆代谢物进行精确评估后,血浆代谢物的水平,包括烟酰胺核糖苷、IMP、亚氨基二乙酸、甲基琥珀酸、半乳酸、丙氨酸甘氨酸、肌氨酸和亮氨酸甘氨酸,明显较低。
然而,在微生物群转移疗法(MTT)后,这些代谢物发生了实质性变化,使其中一些与典型发育中儿童的代谢产物相似。
干预(饮食、微生物等)
已经证明,影响孕妇胎儿微生物群的高脂肪饮食也可能与自闭症有关。此外,母乳喂养6个月降低了自闭症表现的机会,而配方奶粉喂养与肠道艰难梭菌数量增加相关。
《cell》发表的关于自闭症的文章显示,与自闭症相关的行为与饮食多样性的减少有关。并提示在对精神疾病患者的微生物组分析时,应考虑饮食相关因素。
由于益生菌可以发挥抗炎作用并减轻IBD受试者的胃肠道症状,据报道微生物干预,如益生菌,可有助于减少自闭症患者的社会行为症状和炎症水平。
总的来说,自闭症患者肠道微生物群改变已得到证实。然而,考虑到参与患者的异质性和几个相互矛盾的结果,很难建立自闭症的独特特征。考虑到肠道功能障碍与自闭症患者社会行为障碍严重程度的不一致,这些数据提示我们应该考虑两种不同类型的自闭症,这些炎症类型与胃肠道并发症相关。
在各种治疗自闭症的方法中,益生菌治疗的结果很有潜力,同时也应考虑耐受性和安全性评估。鉴于微生物群分析方法的局限性,有必要进一步使用随机、安慰剂对照临床试验,以验证益生菌治疗自闭症的有效性。
精神分裂症(SCZ) 是一种严重的精神疾病,与幻听、妄想、思维和行为紊乱有关,损害日常功能和社会交往。
精神分裂症的生理病理学尚未得到解释,但最近的研究表明,环境因素增加了可能具有该疾病遗传易感性的个体发生精神分裂症的风险。
神经递质在多个系统中的功能障碍已被广泛研究,特别强调了信号异常的重要性,包括多巴胺、5-羟色胺、谷氨酸和GABA。
此外,炎症的重要性以及胃肠系统在精神分裂症病因中的可能作用正在考虑之中。
肠道菌群及其代谢物的影响
胃肠道微生物群在神经生成途径和肠道微生物组中起着至关重要的作用,微生物代谢物扰动已被证明会影响情绪和行为。
肠道微生物组的改变与几种神经发育和神经系统疾病相关。最近有研究表明,来自精神分裂症受试者的粪便移植到无菌受体小鼠,可诱发精神分裂症相关的行为症状。这与海马中GABA、谷氨酰胺和谷氨酸水平的改变有关。这表明精神分裂症患者的微生物群可能对神经化学产生影响,这可能与这些人类条件有关。
目前还没有报告可以促进受试小鼠模型行为改变的特定细菌功能。根据各种研究显示,放线菌、变形菌、拟杆菌和厚壁菌群在精神分裂症患者中的差异最大。
抗生素或许发挥作用
有趣的是,在一项体外研究中,补充抗生素减少了小胶质细胞对突触的吞噬。小胶质细胞降低了中枢神经系统突触的密度,这被认为是精神分裂症发育的一个重要步骤。
在对一组青少年的电子健康记录进行检查后,服用二甲胺四环素与精神分裂症的发病率略有降低有关,这意味着需要更多的研究来调查精神分裂症中微生物群的相关性。
益生菌缓解精神分裂症的消化障碍
根据SCZ的严重性和复杂性,尚未有研究证实精神分裂症患者通过补充益生菌来缓解任何行为症状。然而,一些研究认为,服用益生菌至少可以缓解与精神分裂症相关的消化障碍。
精神分裂症的微生物干预
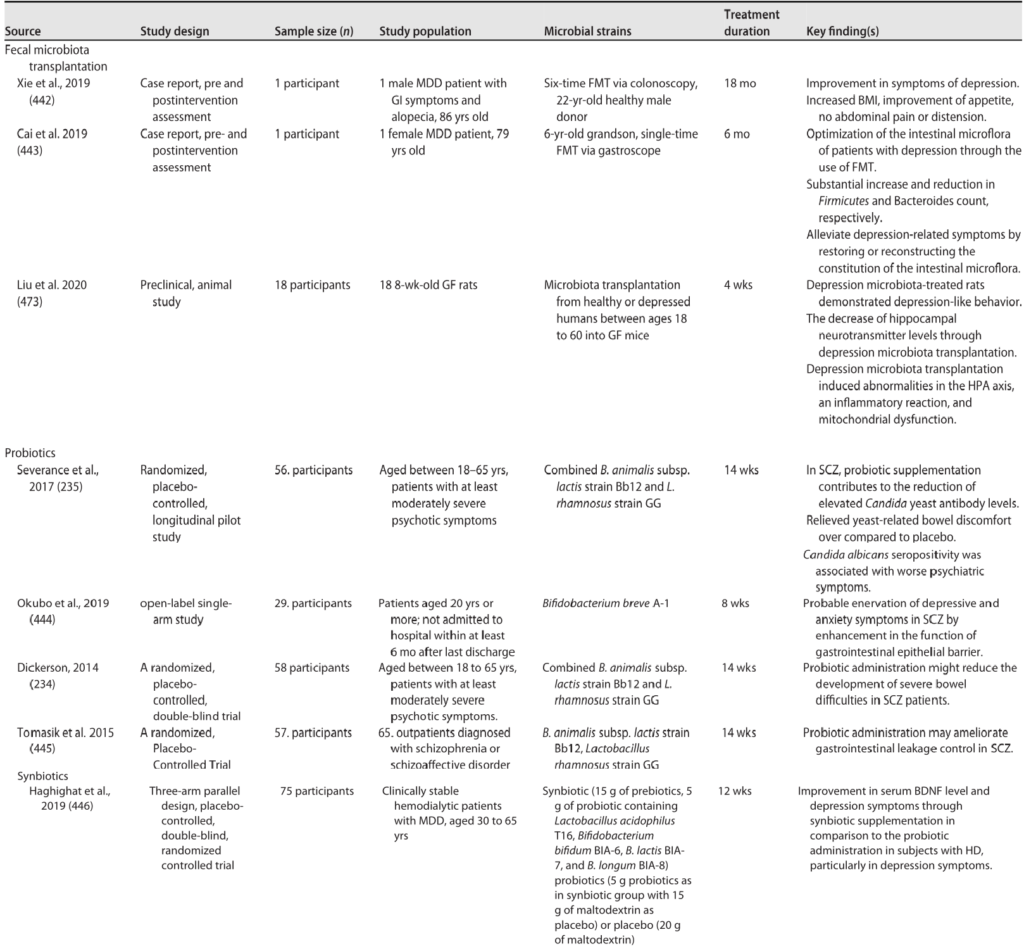
Sorboni SG, et al.,Clin Microbiol Rev. 2022
在一项人类临床试验中,严重的胃肠道问题减少了,精神分裂症患者的精神症状没有任何改变。
另一项人体试验证明了白色念珠菌与胃肠道问题之间的相关性,在服用特定益生菌补充剂(包括鼠李糖乳杆菌GG和动物乳双歧杆菌Bb12)的男性患者中,精神症状有所改善,且白色念珠菌血清阴性。
已经发现了一整套针对严重精神病性疾病的新颖、可能的治疗干预措施,包括考虑肠道舒适性。需要进行更多的研究,通过纵向数据分析和更大的样本量来提高我们对胃肠道微生物群参与精神分裂症的理解。胃肠道微生物的功能和分类对于全面精神分裂症至关重要。
抑郁症是世界范围内导致严重残疾的一种常见的异质性疾病,其特征是情绪低落,经常伴随着对个人通常认为是愉快的活动失去兴趣。其严重形式为重度抑郁症,被归类为心境障碍。抑郁症对人们的健康有着更大的负面影响,并且它带来了与吸烟、血压和饮酒类似的死亡风险。它是癫痫和主要神经退行性疾病(下节内容)的相关共病,其病因包括代谢、神经内分泌和神经免疫因子。
抑郁症主要是一种脑部疾病,但大脑并不是孤立存在的,它存在于包括肠道在内的整个身体生理系统中。如果说成年健康个体的肠道菌群主要由拟杆菌和厚壁菌门组成(90%),那么重度抑郁症患者的肠道菌群在拟杆菌门、厚壁菌门、变形菌门和放线菌门的不同属的丰度上表现出显著变化。
在人类和动物模型中研究发现,临床抑郁症与肠道微生物群丰富度和多样性降低有关。来自抑郁症患者或对照组的粪便微生物群样本移植到微生物群缺陷大鼠模型中,诱发了抑郁症的行为和生理特征,包括快感丧失和焦虑样行为。
慢性炎症可能在重度抑郁症(MDD)的发病机制中发挥重要作用,肠道菌群的内环境平衡失调可能导致此类炎症,这表明胃肠道菌群在影响大脑发育、情绪和行为方面起着中心作用。
这些作者得出结论,生理和情绪压力会影响肠道微生物组的组成。
抑郁症通常与肠易激综合征共存,肠易激综合征的特征是肠道功能的改变,从动物研究中获得的数据表明,肠道微生物群可能影响抑郁症的神经生物学特征。
利用小鼠双侧嗅球切除术(已知会诱发抑郁样行为)来研究其是否会导致微生物群组成的变化:球切除术诱导的慢性抑郁导致肠道微生物谱改变,同时结肠运动、c-Fos活性和5-羟色胺水平增加。
由于抑郁症是肥胖患者的常见症状,研究人员从肥胖小鼠(表现出抑郁症样行为)移植到非肥胖对照小鼠体内的微生物组,观察发现在体重无显著差异的情况下,对照组非肥胖小鼠中肥胖衍生的微生物群重新定植导致探索、认知和定型行为的中断。
抑郁样行为也可以通过应激模型在小鼠中诱导,例如慢性社会挫败应激(CSDS)范式,在该范式中,小鼠反复遭受更大、更具攻击性的小鼠的社会挫败。应激小鼠表现出抑郁样行为,并表现出微生物多样性的变化,其中脱硫弧菌科、Rikenellaceae、毛螺菌科的数量增加,Allobaculum、Mucispirillum的数量减少。
现有证据表明,肠道微生物群可能在抑郁症的发展中起到因果作用,并且可能被认为是治疗/预防这种疾病的一个有价值的靶点。
神经退行性疾病是一类复杂疾病,脑和脊髓的神经元随时间发展而损害逐渐加剧,以特异性神经元的大量丢失为主要特征。这里介绍几种常见的神经退行性疾病及其与肠道微生物群的关系:
多发性硬化
帕金森病
阿尔茨海默
癫痫症
中风和脑损伤
多发性硬化
多发性硬化症(MS)是一种免疫介导的慢性中枢神经系统疾病,涉及受损轴突和脱髓鞘,影响全球约230万人,女性发病率较高。
多发性硬化的致病特征
在中枢神经系统中形成炎性局灶性脱髓鞘斑块,包括脊髓和大脑的灰质或白质,并触发神经炎症反应,导致包括少突胶质细胞在内的特殊细胞脱髓鞘,并导致神经退行性变。
脱髓鞘如何形成?
由于血脑屏障的异常通透性,免疫系统的各种细胞渗入中枢神经系统,导致脱髓鞘的发生。髓鞘抗原特异性T细胞(CD81和CD41 T细胞)穿过这一屏障,导致一系列事件导致脱髓鞘病变的形成。
多发性硬化发病的免疫机制
最近对多发性硬化小鼠模型(包括实验性自身免疫性脑脊髓炎模型)的研究表明,CD41 T淋巴细胞在多发性硬化发病机制中起主要作用。尤其是,CD41Th17和Th1淋巴细胞在多发性硬化发病中具有最突出的作用。
Th1有助于分泌δ干扰素(IFN-d),在活化后促进巨噬细胞酶的产生。此外,IFN-d刺激活性氮和活性氧的产生,分别导致细胞结构的亚硝化和氧化损伤。Th1细胞还能够产生IL-12,从而诱导肿瘤坏死因子(TNF-a)和IFN-d的分泌,导致慢性炎症反应和进一步的组织损伤。
由Th17细胞介导的特定细胞因子(包括IL-22、IL-21和IL-17)的产生导致慢性炎症进展。识别中枢神经系统自身抗原(如Th1和Th17)的CD41 T淋巴细胞参与多发性硬化的病理生理学。
除了CD81和CD41细胞外,其他免疫细胞也与多发性硬化发病有关,包括NK细胞、小胶质细胞和巨噬细胞。这些细胞与其细胞因子之间的分子相互作用维持了中枢神经系统内的炎症级联反应。
多发性硬化的几种临床变异
包括最常见的复发缓解型多发性硬化和进行性复发型多发性硬化,以及原发性进行性多发性硬化和继发性进行性多发性硬化(SPMS)。
遗传易感性和环境因素在多发性硬化症的病因中都起着重要作用。
肠道微生物群参与免疫调节
最近的研究表明,肠道共生微生物群落也与多种免疫介导的疾病(如多发性硬化)有关,可以认为是一种新的环境风险因素。换句话说,肠道微生物群负责免疫调节,改变血脑屏障的完整性和功能,刺激自身免疫脱髓鞘过程,并与中枢神经系统中存在的各种细胞类型直接相互作用。
与肠道微生物群α或β多样性的广泛差异不同,横断面调查主要揭示了多发性硬化儿童与健康个体相比在分类上的明显改变。
一些研究评估了多发性硬化患者微生物移植到两种不同的实验性自身免疫性脑脊髓炎(EAE)模型中的效果;这些研究强调了产生IL-10的CD1 T细胞在胃肠道微生物群介导的免疫调节中的重要性。
此外,胃肠道中SFB的存在,可能在Th17细胞活化中起作用 ,显著影响EAE小鼠的多发性硬化样症状。根据多发性硬化作为脱髓鞘疾病的定义,临床前抗菌研究表明,在汇集来自无菌小鼠模型的数据后,胃肠道微生物群可以调节小鼠模型前额叶皮质髓鞘的生成。
肠道菌群在血脑屏障调节中的基本作用
无菌小鼠研究表明,作为多发性硬化的一个主要标志,微生物组与血脑屏障完整性的丧失之间可能存在关联。
研究还表明,在膳食中补充短链脂肪酸或产短链脂肪酸的菌,可以逆转血脑屏障完整性的丧失。此外,饮食诱导的肠道微生物群落结构变化也参与了EAE的表现。
有证据表明,肠道微生物群可以调节大量的神经炎症途径。然而,补充研究对于理解多发性硬化病因的确切作用机制至关重要。动物和人类研究表明,肠道菌群可能与多发性硬化生理病理学的许多方面有关。
干预措施
关于如何有效地控制肠道微生物组作为一种干预措施,以最大程度地阻止复发和缓解症状,问题仍然悬而未决。
在一项试点实验中,补充一种特定的益生菌制剂(含有双歧杆菌、乳酸杆菌和链球菌)可以逆转微生物群的改变并调节炎症反应,这表明这种微生物群靶向治疗是有希望的(下表),尽管需要进一步调查以确认这些结果。
多发性硬化的微生物干预
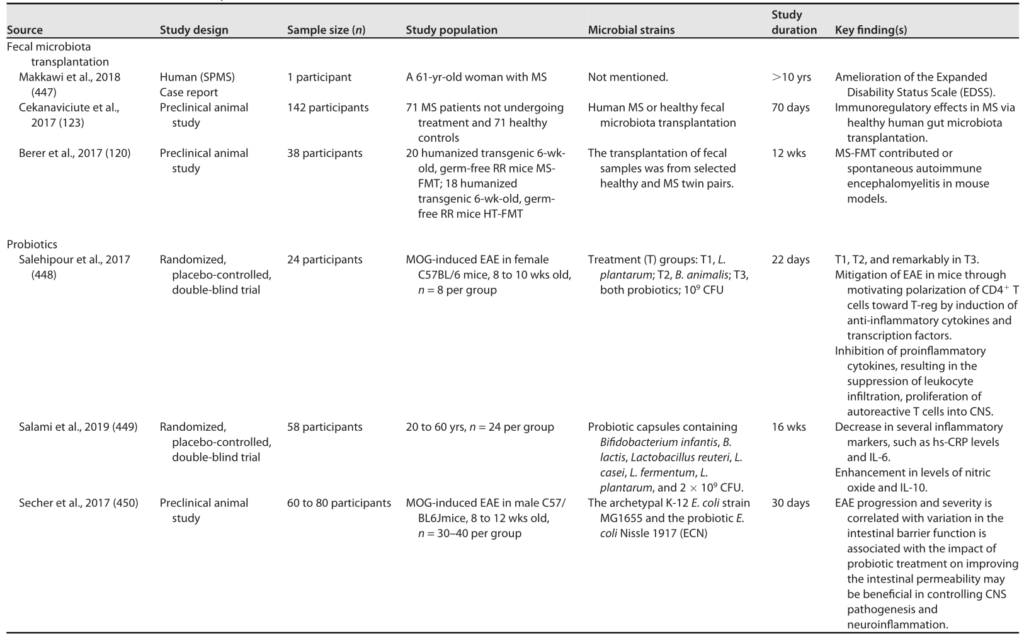
Sorboni SG, et al.,Clin Microbiol Rev. 2022
帕金森病
帕金森病(PD)是一种进行性多中心神经退行性疾病,由α-突触核蛋白(α-syn)沉积在部分大脑中心黑质的多巴胺能神经细胞中引起。这些过程促进了圆形片状嗜酸性细胞质内含物的逐渐聚集,称为Lewy小体。
然而,帕金森发病机制的确切机制仍不明确,它可能是一种多因素疾病,在这方面引入了各种理论。
衰老是帕金森发展和进展的重要风险因素
影响多种细胞途径,导致这些过程受损,并导致神经退行性变。可以想象,年轻神经元可以容忍的相同分子扰动在老年神经元中显示出一些灾难性后果。
帕金森病的临床症状主要表现为运动障碍症状,包括肌肉僵硬、静止性震颤、运动迟缓和姿势不稳 。帕金森病在50岁之前很少见,但随着年龄的增长,发病率会增加5到10倍。它主要发生在男性,每年每100000人中有5-35例新病例。
多巴胺能神经元逐渐退化,非运动和运动症状如抑郁症、痴呆症和胃肠道问题(包括便秘、唾液分泌异常、排便功能障碍、恶心和吞咽困难)之间存在着强相关。帕金森症状因个体而异。
几项研究表明,帕金森受试者的胃肠道异常与肠神经系统中的肠道失调和α-突触核蛋白沉积有关。
肠道菌群如何影响帕金森?
由于最初的胃肠道参与帕金森,并且宿主微生物组之间的生理相互作用潜力很大,因此有人认为胃肠道菌群可能影响帕金森。胃肠道功能异常,尤其是便秘,影响多达80%的帕金森病患者,并且可能在出现运动症状前几年发生。
特发性便秘是帕金森的主要相关因素,与肠神经系统的神经退行性改变有关。肠神经系统中的α-syn神经退行性变可能是帕金森的运动前临床症状之一。它与慢性便秘和胃肠道壁的生理改变有关。
肠道微生物群可能影响参与α-syn分泌的肠道神经元。这些变化在运动症状出现之前的帕金森开始时就已出现,可认为是运动前生物标记物。
——Prevotellaceae减少
对肠道微生物组与帕金森患者之间的相关性进行了不同的研究。一项研究观察到帕金森病患者粪便样本中Prevotellaceae种类显著减少。与对照组相比,Prevotellaceae的丰度显著降低(77.6%)。
Prevotellaceae通过膳食纤维发酵和肠道中的粘蛋白率先产生短链脂肪酸。Prevotellaceae减少引起的细菌内毒素全身暴露和肠道通透性的增强可触发α-syn结肠的不受控制的表达和错误折叠。
这种肠型负责硫胺素、叶酸和神经活性短链脂肪酸的生物合成。因此,补充这些维生素和短链脂肪酸可能有助于治疗帕金森。
最近的研究首次承认,帕金森病患者的机会性病原体数量显著增加。
——肠杆菌科丰度增加
步态困难姿势不稳的严重程度与肠道内肠杆菌科丰度呈正相关。肠杆菌科细菌在肠道中的过度生长导致作为血清中革兰氏阴性细菌细胞壁一部分的LPS滴定增强 。因此,研究表明,由于帕金森患者血液样本中LPS的吸收增加,LPS结合蛋白的全身浓度异常高。
——乳酸杆菌科的丰度增加
与Prevotellaceae一样,乳酸杆菌科与胃肠激素ghrelin有关。也有报道称帕金森患者的ghrelin分泌减少。
总的来说,研究结果揭示了胃肠道微生物群和帕金森作用之间的联系。进一步的微生物组学分析可能会提高准确性,澄清关系以及机制。
帕金森患者中菌群失衡可能会影响炎症,因为菌群失调会损害肠道屏障功能并触发免疫激活和全身炎症反应。
帕金森的持续存在会影响微生物群,肠道菌群可能在一些腹部症状中起作用,如便秘和炎症。
肠道屏障破坏
简而言之,LPS和其他细菌神经毒素在穿过肠壁后进入血液,导致肠上皮屏障的破坏。血液中细菌LPS的存在导致通过核因子kB(NF-kB)和TLR4产生炎症细胞因子,导致全身炎症。细菌LPS和炎性细胞因子(包括TNF-a、IL-1b和IL-6)诱导的血脑屏障破坏触发α-syn的积累。位于黑质的多巴胺能神经元丢失可能是血脑屏障分解的结果。由于帕金森的肠屏障破坏导致微生物易位升高和促炎症基因谱升高,结肠活检标本显示TLR4或细菌内毒素特异性配体、CD31 T 细胞和其他细胞因子的表达增强。
产短链脂肪酸的细菌减少
帕金森病患者在肠道失调期间产短链脂肪酸的细菌减少。TLR4介导的炎症在脑或肠道炎症中发挥重要作用,这可能是导致帕金森神经退行性变的重要因素之一。因此,增强肠道内肠杆菌科后LPS的相对增强与帕金森的发生相关。
闭塞和其他紧密连接蛋白对肠屏障结构至关重要。肠道生态失调,使闭塞素降解,导致肠道通透性增强。
促炎细胞因子升高
另一项针对帕金森病患者的研究调查了其粘膜中Ralstonia、肠球菌和变形菌浓度的增加,导致促炎细胞因子的升高。
帕金森受试者粪便样本中被认为具有抗炎作用的产丁酸菌(如布氏杆菌、粪球菌、粪杆菌和罗氏菌)数量显著减少。
此外,据报道,帕金森患者粪便样本的微生物群中LPS生物合成基因表达增加。有趣的是,幽门螺杆菌感染可被认为是帕金森发病机制中的一个重要触发因素。
小肠细菌过度生长(SIBO),与运动功能障碍有关,尤其是在帕金森患者中。
牙龈假单胞菌感染在帕金森的病因/危险因素中的重要作用
牙龈卟啉单胞菌的牙龈蛋白酶和LPS导致帕金森样本中出现异常血凝块。研究发现只有帕金森样本的凝块中观察到了牙龈蛋白酶抗体信号,这证实了这种细菌在帕金森病理学中的潜力。他们进一步指出,据报道,牙龈假单胞菌诱导的外周炎症导致肠道微生物群失衡,黑质多巴胺能神经元减少,肠道通透性增加,以及富含亮氨酸重复激酶2(LRRK2)相关帕金森病理生理学中小胶质细胞活化增强。
益生菌缓解症状
益生菌,包括乳酸杆菌和双歧杆菌,已被证明可以缓解帕金森样症状。
帕金森的微生物干预

Sorboni SG, et al.,Clin Microbiol Rev. 2022
芽孢杆菌作为一种益生菌,能够将L-酪氨酸转化为L-多巴,L-多巴是多巴胺的重要前体分子,其转化为多巴胺是通过多巴脱羧酶进行的。
据报道,定期服用含有干酪乳杆菌shirota的发酵乳饮料可通过减少帕金森患者粪便中葡萄球菌的数量促进排便。
肠道微生物群从二芳基黄烷醇中积极产生多酚,干扰α-突触核蛋白的错误折叠和毒性,是帕金森和其他α-突触核蛋白病的基本病理机制。
对口服富含拉法诺制剂(FRP)的异源性人源化侏儒小鼠的研究表明,FRP衍生代谢物的产生存在特殊差异,影响α-突触核蛋白的错误折叠或炎症。
对果蝇α-突触核蛋白病模型的研究表明,它对运动功能障碍有影响,从而导致其发病和进展的调节。
体外研究表明,在细菌发酵过程中,特定的细菌可以产生这些具有生物活性的酚酸。
总之,已经得出结论,个体间异质肠道微生物群诱导的二芳基黄烷醇的变化证明了益生菌、益生元和共生策略在调节帕金森和其他共核病变进展中的潜力。
目前,关于微生物组与帕金森病之间的相关性的不同研究结果并不一致。微生物组的结果产生了有价值的信息。帕金森患者使用的药物与肠道微生物之间存在联系,因为肠道微生物群在处方药物代谢中发挥作用,甚至药物对微生物组成产生影响。
对胃肠道微生物组和肠-脑轴的相互作用有一个完整的认识,可能阐明帕金森的病因和进展因素,以提供新的治疗视野和手段。例如,FMT和肠道微生物组作为帕金森临床诊断的新生物标记物的评估可能揭示传统治疗方法的替代治疗。
肠道不适可发生在帕金森的初始阶段;这有助于在出现震颤和强直等运动症状之前对该疾病进行早期诊断。微生物组学研究可以提供关于帕金森病的有用信息,但目前,我们不能依赖它们作为生物标志物。
阿尔茨海默病是一种慢性不可逆的大脑疾病,脑细胞的进行性退化导致记忆障碍、认知能力下降。它是老年人最常见的痴呆类型。阿尔茨海默患者表现出严重的学习、行为和记忆障碍,严重到足以影响日常活动。
阿尔茨海默的特征
阿尔茨海默患者的大脑中的神经元细胞死亡和进行性突触衰竭,伴随着神经元周围或外部的β淀粉样蛋白(amyloid-β,简称Aβ)沉积,伴随着皮质神经元树突和轴突中微管相关蛋白tau异常磷酸化的聚集。Aβ的积累和tau蛋白的聚集有助于微管稳定性的降低、突触失效和神经元钙稳态的紊乱,最终导致神经元凋亡。
尽管已经对阿尔茨海默的病因进行了大量研究,但阿尔茨海默的潜在机制尚不完全明确,目前的Aβ疗法对症状的缓解作用有限。据报道,淀粉样蛋白可能在大脑中充当AMP。
与中枢神经系统炎症有关
最近的研究发现阿尔茨海默的发病机制与周围感染引起的中枢神经系统炎症有关。在感染单纯疱疹病毒1型(HSV-1)的小鼠中,可以看到阿尔茨海默受试者中tau和Aβ沉积的共同特征。病毒感染诱导的高细胞内胆固醇25羟化酶(CH25H)水平对于调节Aβ产生和阿尔茨海默易感性至关重要。
阿尔茨海默 & 微生物群
此外,先前的研究已经证明阿尔茨海默与其他微生物感染(包括真菌、肺炎衣原体和螺旋体感染)之间存在潜在联系。阿尔茨海默患者脑脊液中肠道菌群微生物驱动代谢物的测定与阿尔茨海默生物标记物(如磷酸化tau和tau/Aβ42)相关,表明肠道微生物群在阿尔茨海默发病中的意义。
一项研究报告,根据Aβ前体蛋白转基因小鼠(APP)粪便样本的细菌16S rRNA序列分析,与野生型小鼠模型对照组相比,肠道微生物组成存在显著差异。研究还表明,具有阿尔茨海默表型的转基因小鼠模型具有多种肠道微生物。
对无菌小鼠的研究表明,在没有微生物的情况下,不会出现淀粉样斑块和神经炎症症状。
根据横断面研究结果,与健康对照组相比,阿尔茨海默患者粪便样本中参与炎症反应的两种细菌大肠杆菌和志贺菌的丰度显著增加。在患有认知障碍和脑淀粉样变性的患者中,可能与外周炎症状态有关的两种主要情况包括促炎性大肠杆菌和志贺菌的增加以及抗炎性直肠真杆菌的降低。
菌群失调 & 全身炎症
肠道微生物群失调和全身炎症之间存在联系,这可能是阿尔茨海默患者大脑中发生的神经退行性变的一个促成因素。
这些观察结果基于小规模研究,需要更多具有较大统计组的研究来评估肠道微生物群与阿尔茨海默进展的关系。一些科学家指出,在阿尔茨海默患者大脑中发现的感染因子可能与该疾病的发展有关,但在这方面,强有力的证据是必不可少的。
牙龈卟啉单胞菌在患者大脑中定植
在最近的一项阿尔茨海默治疗研究中,使用合成的神经毒性抑制剂是有益的。在这项研究中,牙龈卟啉单胞菌与慢性牙周炎有关,在阿尔茨海默患者的大脑中被发现。这些细菌在大脑中的定植导致Aβ1-42的产生增加。此外,神经毒性姜黄素对tau蛋白Aβ1-42有破坏性影响。
NLRP3炎症小体与阿尔茨海默
肠道微生物群失调与阿尔茨海默相关神经炎症之间的潜在关联。肠道NLRP3异常表达的增加与外周炎性体的激活呈正相关,后者随着阿尔茨海默的进展而增强神经炎症。因此,观察到,与年龄匹配的对照组小鼠相比,年轻和老年5xFAD小鼠模型的肠道微生物群组成发生了相当大的变化。
与非转基因小鼠相比,com 5xFAD小鼠由于粘附蛋白和紧密连接蛋白的丢失而表现出肠道屏障功能受损。此外,已经证明肠道微生物炎症体蛋白的高表达可能是激活下游细胞毒性和炎症介质的重要主导因素。因此,NLRP3炎症体介导的神经炎症可能通过胃肠道NLRP3促进。因此,肠道微生物群调节可能是治疗遗传易感个体阿尔茨海默相关神经系统疾病的一种可能策略。
将Tg2576小鼠阿尔茨海默模型(包括症状前和症状中转基因)与野生型进行比较,研究人员观察到肠上皮屏障(IEB)中血管A β肽的沉积破坏了IEB,并且吸收失调发生在其脑聚集之前。得出结论,肠-脑轴的改变与较高水平的炎症血浆细胞因子(如IL-9、IP-10和VEGF)相关。
考虑到阿尔茨海默患者的肠道功能障碍,阿尔茨海默治疗的未来治疗策略可能涉及肠道微生物群的早期调理。根据肠道微生物群参与阿尔茨海默Aβ病理学的发展,研究人员开发了一个新的框架,通过肠-脑轴确定阿尔茨海默的潜在机制,并将肠道微生物群的操作转化为临床实践。
抗生素疗法
用接受来自老年(16个月)APPSWE/PS1DE9小鼠的粪便供体移植的APPSWE/PS1DE9小鼠进行短期抗生素鸡尾酒疗法,收集粪便颗粒进行进一步分析。
抗生素治疗前小鼠的FMT重建主要归因于供者来源,如梭状芽孢杆菌和Coriobacteriae,有助于Aβ斑块的更高沉积。有趣的是,在微生物群植入后,Aβ斑块周围星形胶质细胞的激活受到抑制,而不是小胶质细胞。
在阿尔茨海默小鼠模型中,长期服用广谱抗生素也可以减少Aβ积累并调节影响Aβ淀粉样变的先天免疫反应。此外,在转基因小鼠中,通过定期使用抗生素鸡尾酒疗法,海马淀粉样斑块周围的小胶质细胞和星形胶质细胞聚集以及不溶性Aβ斑块减少 。
也有报道称,通过比较不同年龄的野生型和阿尔茨海默小鼠模型之间的粪便短链脂肪酸和微生物组成,观察到具有阿尔茨海默表型的小鼠中的丁酸球菌和瘤胃球菌数量显著减少,变形菌和疣状菌数量增加,提供改变的微生物组成和多样性。短链脂肪酸水平的下降表明至少有30条代谢途径受到干扰。
先前的一项研究也表明,小胶质细胞激活抑制Aβ清除和降解,Aβ的进一步积累导致阿尔茨海默的病理学。此外,Aβ沉积水平的升高有助于小胶质细胞内几种促炎介质的释放,如ROS、iNOS、NF-kB和COX2,从而促进阿尔茨海默患者的神经炎症。
阿尔茨海默的微生物干预
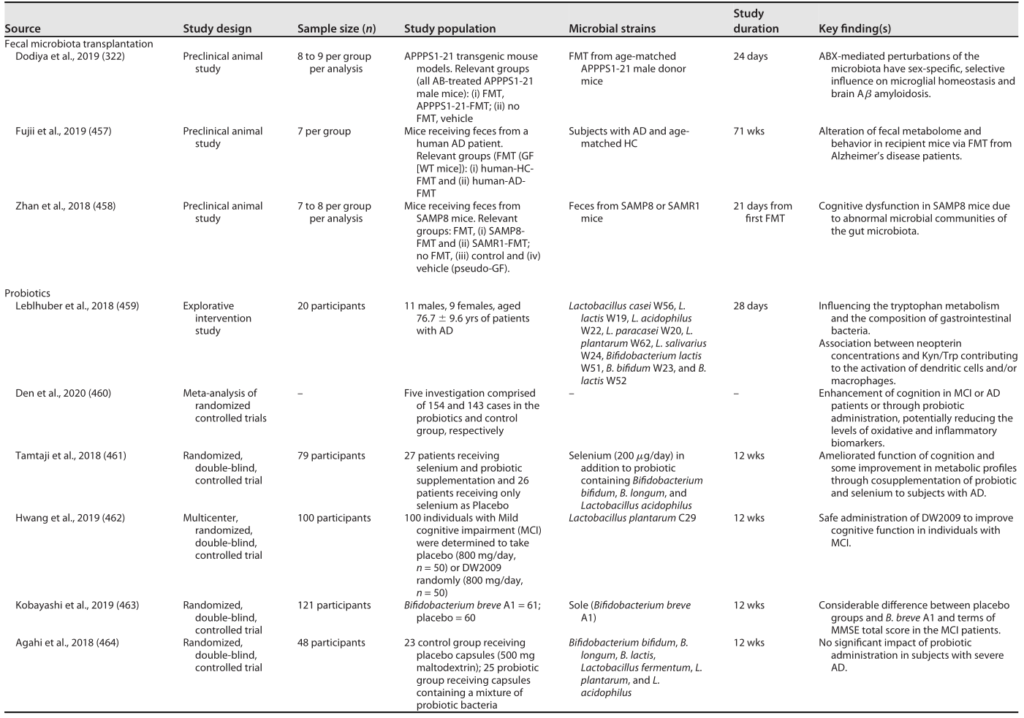
Sorboni SG, et al.,Clin Microbiol Rev. 2022
这些研究表明,某些种类的肠道微生物可激活Aβ信号通路,参与阿尔茨海默的发病机制,并在阿尔茨海默的分子调节中发挥关键作用。益生菌补充和营养干预可能成为阻碍阿尔茨海默进展的一种有希望的治疗方法。
癫痫是一种使人衰弱的神经系统疾病,影响全世界约6500万人。尽管医学上有许多新的进展,但确切的病因仍需完全阐明。大约一半的癫痫患者病因不明。据报道,癫痫患者的医疗费用是正常人的13倍。癫痫显著的社会经济影响是由于其死亡率和致残率高于正常人群。尽管正在使用抗癫痫药物(AEDs)进行药物治疗,但只有70%的癫痫患者能够完全控制癫痫发作。因此,大约三分之一的癫痫患者会出现难治性癫痫,影响他们的日常生活活动。
环境和遗传因素都决定癫痫的易感性。
此外,一些研究表明肠道细菌种类与癫痫的病理生理学之间存在关联。肠道微生物组失调与癫痫等神经精神疾病的发生有关。平衡的肠道微生物群与健康的大脑和免疫系统之间存在相关性。
最近的研究表明,慢性炎症在癫痫的发病和进展中起着重要作用。研究还表明,肠道菌群可以调节免疫和炎症反应。因此,操纵肠道微生物组作为癫痫的治疗策略具有潜力。
腹腔注射LPS诱导大鼠更易发生癫痫发作,同时增加血脑屏障的通透性和大脑中更高水平的促炎细胞因子。
未控制癫痫的替代治疗策略包括迷走神经刺激和生酮饮食。因此,控制肠道微生物群的多样性可以被认为是一种潜在的治疗方法。
癫痫患者肠道菌群变化
在几项研究中发现,与健康人群相比,采用各种治疗方法的癫痫患者的肠道微生物特征存在差异。
所有这些研究表明,在未控制的癫痫中,厚壁菌/拟杆菌比率增加。一些属于厚壁菌门的细菌能够调节神经递质水平。对肠道微生物群(包括α-多样性)的进一步分析表明,结果存在显著差异。在另一项研究中,与拟杆菌相比,厚壁菌的数量增加。
此外,与药物反应性患者相比,耐药患者的α-多样性测量结果与健康受试者相似。实质上,更高水平的α-多样性与罕见肠道细菌种类的异常增加有关。此外,在属水平上,报告了显著差异。根据这些结果,可以认为细菌在癫痫的有效治疗中起作用。
有趣的是,肠道微生物群可以调节唑尼沙胺代谢,唑尼沙胺是一种抗癫痫药物。此外,乳酸杆菌和双歧杆菌数量的增加与每年较少的癫痫发作相关。
在临床前和临床研究中,关于抗生素给药是否能诱导或预防癫痫发作,也有争议的发现。值得注意的是,潜在传染病在治疗过程中可能产生的促癫痫作用或抗生素直接引起的神经毒性副作用可能更为重要。
生酮饮食降低癫痫发作率
据报道,癫痫患者的生酮饮食可降低癫痫发作率,并与肠道菌群组成和功能的改变有关。
生酮饮食在颞叶癫痫的无菌小鼠模型中介导抗癫痫作用。事实上,这些研究人员发现,SPF小鼠在移植生酮饮食的微生物群或长期治疗细菌后癫痫发作阈值升高(长期治疗细菌包括Akkermansia muciniphila, Parabacteroides distasonis, Parabacteroides merdae)。
一些研究表明补充益生菌对癫痫有积极作用。
癫痫的微生物干预
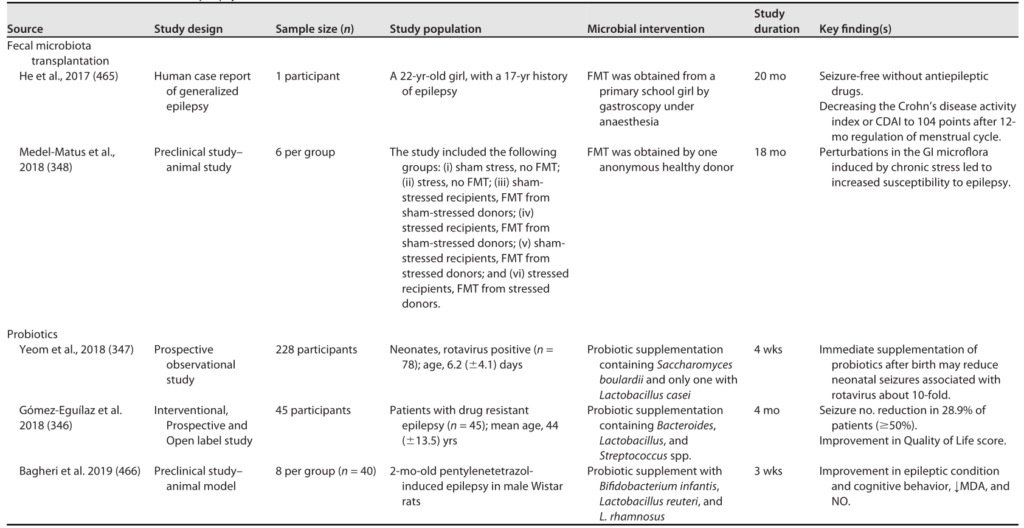
Sorboni SG, et al.,Clin Microbiol Rev. 2022
在全球范围内,中风和脑损伤是发病率和死亡率的重要原因。肠道菌群中的共生细菌可能通过调节多种脑血管疾病的危险因素(包括动脉粥样硬化、糖尿病、血脂异常和动脉高血压),与中风的发生有关。饮食也是一个重要的危险因素。
将动脉粥样硬化和失调联系起来会直接影响微生物组的组成和多样性。然而,越来越多的证据表明,肠道微生物群可能在脑血管疾病和中风中发挥更直接的作用。
三甲胺n-氧化物(TMAO)升高
三甲胺n-氧化物(TMAO)作为微生物群衍生的代谢物,可以从膳食胆碱中合成,可在体液和组织中检测到。最近的研究表明,TMAO与脑血管和心血管疾病的风险增加有关,这表明可能通过这种代谢物的治疗潜力调节肠道微生物群。
横断面调查表明,与健康对照组相比,中风患者的肠道微生物群组成不平衡。在一项对4000多例患者的纵向研究中,血浆样本中TMAO水平升高与卒中和心血管事件的高风险呈剂量依赖性相关。
抗生素补充引起的TMAO水平降低突出了肠道细菌在该化合物合成中的重要性。
同时,与无症状动脉粥样硬化患者相比,中风和短暂性脑缺血发作患者的TMAO水平相对较低。
磷脂酰胆碱代谢物
临床前研究表明,服用磷脂酰胆碱代谢物(如胆碱和TMAO)可上调参与动脉粥样硬化的巨噬细胞清除受体的表达,这可能是由于肠道中存在的细菌物种所致。
对无菌小鼠的研究表明,胆碱的服用与较高的动脉粥样硬化率无关,并且有助于减少主动脉斑块的体积。然而,关于饮食对TMAO和胆碱的影响以及肠道微生物群在动脉粥样硬化发病和进展中的有害和保护作用,重要的是不要过度解释临床前研究的结果。
健康的微生物组在动脉粥样硬化病变的恢复中起着重要作用。
大脑中动脉闭塞后补充广谱抗生素与小鼠存活率降低相关。抗生素诱导的胃肠道菌群改变也导致IL-17相关趋化因子表达下调和促炎性IL-17gd T细胞迁移减少。
因此,肠道细菌通过调节肠道T细胞向大脑的浸润来调节中风后的神经炎症。
卒中后含有短链脂肪酸产生菌的粪菌移植,包括发酵乳杆菌、长双歧杆菌、Faecalibacterium prausnitzii、Clostridium symbiosum,可缓解卒中后的认知障碍和炎症,还可增加血浆、肠道和大脑短链脂肪酸浓度,促进老年模型卒中后恢复。也有报道称,将中风患者粪菌移植到抗生素给药的小鼠,以及从中风模型转移到无菌小鼠,会增加缺血性脑损伤和相关功能损伤的大小。
中风和脑损伤的微生物干预
Sorboni SG, et al.,Clin Microbiol Rev. 2022
据报道,中风后拟杆菌的数量和多样性减少。另一项研究表明,短暂性脑缺血发作和中风患者的机会性病原体数量较多,包括脱硫弧菌、肠杆菌、巨球形杆菌和Osicillibacter,有益或共生菌属的数量较少,如拟杆菌、粪杆菌和Prevotella。此外,Prevotellaceae和Peptococcaceae的丰度增加与中风严重程度相关。
用一种特殊的细菌菌株,即丁酸梭菌进行治疗,可改善缺血/再灌注小鼠模型的认知功能,并减少神经元损伤。
根据这些解释,肠道微生物群在中风和脑损伤发病和进展中的作用尚不完全清楚。虽然临床前和临床研究提供了有趣的结果(表7),但还需要进一步的研究。有人建议在饮食中补充精神生物素,以减少创伤性脑损伤后的精神后果和共病。然而,需要更多的临床研究来阐明这种微生物治疗干预的潜力。
药物
益生元
益生菌
合生元
后生元
粪菌移植
其他
越来越多的证据表明,人们越来越认识到肠道微生物组在调节不同药物(如精神药物)的疗效和副作用方面的重要性。
抗生素
抗生素是影响胃肠道菌群的最有效和最直接的方法。
对1135名个体的肠道微生物组的深度测序表明,肠道微生物组与各种药物组之间存在关联。抗生素与胃肠道微生物组的改变密切相关。
值得注意的是,作者提供了其他几种治疗药物对胃肠道微生物群影响的证据,如二甲双胍、泻药、他汀类药物和质子泵抑制剂(PPI)。
多药疗法
同时使用多种药物治疗患者,也与肠道微生物群的改变有关。一项调查显示,服用药物的数量与微生物多样性之间存在显著的负相关。尤其是抗抑郁药、PPI和抗精神病药与分类单元丰度的相关性最大。
非抗生素药物
同时,新证据表明,除了药物药代动力学的调节外,非抗生素药物还可以改变肠道微生物组结构,对情绪和行为产生潜在影响。
另一方面,人们越来越重视肠道微生物组与药物之间的相互作用,这支持了肠道菌群可影响药物代谢和吸收的观点。在一项大规模队列研究发现,包括抗生素、抗抑郁剂、苯二氮卓类药物等在内的医疗干预可以改变肠道微生物组的组成。
精神药物
此外,研究了精神药物对40名焦虑症和/或重度抑郁症患者胃肠道菌群的影响。在其队列研究中,研究人员得出结论,抗精神病药物降低了胃肠道微生物组α多样性。这些研究人员确定抗精神病药物的剂量与这些患者的α-多样性呈负相关。
已经进行了多项体外研究,以评估非抗生素药物的抗菌活性,所有这些药物都具有抗菌活性,可能通过与特殊分子靶相互作用影响中枢神经系统功能。
其他报告显示抗抑郁药选择性5-羟色胺再摄取抑制剂(SSRIs)、氟西汀、舍曲林、西酞普兰和帕罗西汀对芽孢杆菌、梭状芽孢杆菌、肠球菌、假单胞菌和葡萄球菌菌株具有抗菌活性。
在最近的一项研究中,作者测试了不同类别的抗抑郁药对12种肠道菌群共生细菌菌株的抗菌活性。大多数被检查的抗抑郁药对被检查菌株的生长具有相当大的浓度依赖性抑制作用。此外,在对雄性BALB/c小鼠肠道微生物群的体内研究中,作者发现与对照组相比,地昔帕明可增加β-多样性并降低丰富度。
这些作者还发现,在补充了地昔帕明的小鼠模型中,Adlercreutzia、瘤胃球菌和未分类的α-变形杆菌的数量减少。三环类抗抑郁药,包括阿米替林,也被证明对致病细菌菌株,如芽孢杆菌属、葡萄球菌属和霍乱弧菌具有体外抗菌活性,而丙咪嗪分别对小肠结肠炎耶尔森菌和大肠杆菌具有生长抑制作用。
在一项老年住院患者队列研究中,研究了精神药物对肠道微生物群组成的影响。在受试药物中,与PPI和抗抑郁药相比,抗精神病药物与微生物群落α多样性的负相关性最高。
在另一项对双相情感疾病受试者的调查中,非典型抗精神病药物(APP)治疗与女性的微生物多样性降低相关,但与APP治疗的男性患者无关。在这一队列中,服用APPs的患者,其衣原体科和阿克曼氏菌的数量分别显著增加和减少。
在针对40种肠道共生菌代表的1000多种药物的大规模体外筛选研究中,据报道,24%的受试药物对至少一种细菌菌株表现出生长抑制。这些药物对相当相似的物种模式具有抗菌活性,表明直接抗菌活性可能是其药理作用的一部分,不应将其视为副作用。因此,迫切需要评估精神药物对胃肠道菌群的潜在影响。
阿片类药物
肠-脑轴的双向方面也反映在这样一个事实,即肠道神经胶质细胞的 GDNF 等分泌因子也通过粘膜免疫系统的成熟以及通过加强上皮紧密连接功能来调节微生物稳态。阿片类药物介导的 GDNF 表达降低也与肠道通透性增加和肠粘膜表面免疫监视改变有关。这些因素有助于维持阿片类药物治疗观察到的促炎环境,导致下游阿片类药物相关的合并症,如阿片类药物耐受、依赖和戒断。

Jalodia R, et al., J Neuroimmune Pharmacol. 2022
此外,来自肠神经元的神经元介质(例如,VIP、ACh、NO)的阿片类药物抑制释放会导致胃肠道分泌减少、水和电解质吸收增加、蠕动减少,从而导致阿片类药物诱导的便秘,进一步导致微生物群失调。总之,虽然 OUD 的药物治疗是可用的,但它们并非对所有患者都有效。
根据国际益生菌和益生元科学协会(ISAPP)的说法,“益生元是指宿主微生物群体专门利用的、对健康有益的非活性食品成分。”作为益生菌补充的替代品,益生元可用于调节肠道菌群。
这组化合物通过其影响胃肠道健康的能力进行鉴定,包括不可消化低聚糖(NDO)、母乳低聚糖(HMO)和可溶性可发酵纤维。尽管益生元疗法在增强有益细菌(如双歧杆菌和乳酸杆菌)方面具有潜力,但只有少数研究检测了这些化合物对人类和动物肠道菌群的有益影响。对低聚半乳糖和低聚果糖或其组合对雄性小鼠的作用的研究表明,这些化合物具有抗抑郁、抗焦虑作用,并逆转慢性应激的作用。
在一项安慰剂对照临床试验中,服用N-乙酰半胱氨酸8周后,自闭症婴儿的易怒性和重复行为有所下降。此外,补充商业益生元药物B GOS(Bimuno)和限制性饮食可改善自闭症儿童的行为,这可能是由于乳酸杆菌和双歧杆菌含量较高。
最近的一项研究还表明,益生元乳果糖可以通过自噬和抗炎途径改善阿尔茨海默小鼠模型的认知缺陷。因此,这些发现似乎表明益生菌和益生元是神经系统疾病的有效治疗选择。然而,考虑到仅仅是相关性并不一定表明因果关系,需要进行额外的调查以详细了解潜在的机制。
健康个体和患者肠道微生物群组成和功能的变化已被确定为各种神经系统疾病。人们已经认识到,饮食可以影响微生物组成,改变肠-脑轴的功能。多种治疗干预措施已用于治疗肠道微生物群落失调,恢复肠道微生物群落平衡,改善神经系统疾病的临床结果,包括使用益生菌。
益生菌在普通食品和药片中的应用越来越流行。益生菌主要由双歧杆菌和乳酸产生菌组成,例如乳酸杆菌。越来越多的证据表明,益生菌合成的代谢物是饮食诱导的宿主-微生物相互作用的基本介质。此外,一些肠道细菌种类,如拟杆菌、梭菌、双歧杆菌、消化链球菌、乳酸杆菌和瘤胃球菌,可产生多种色氨酸分解代谢产物,包括吲哚、3-甲基吲哚、吲哚乙酸(IAA)、色胺等。
新出现的数据表明,微生物组衍生的色氨酸分解代谢影响宿主健康。已经证明,这些代谢物可与AhR结合,从而激活免疫系统,改善肠道屏障功能,刺激胃运动活动(以及胃肠激素的分泌),发挥全身或局部抗氧化、抗炎作用,并可能调节肠道微生物组和代谢组。
据报道,由共生微生物群合成的色氨酸分解代谢物可诱导小胶质细胞AhR激活,抑制NF-kB信号、VEGF-B和TGF-α的激活。此外,AhR在树突状细胞中高度表达,控制分化和功能。树突状细胞中维甲酸、犬尿氨酸和AhR驱动的细胞因子的产生增强T-reg细胞的分化,抑制EAE作为多发性硬化动物模型的发展。
AhR信号在肠道和大脑中的作用
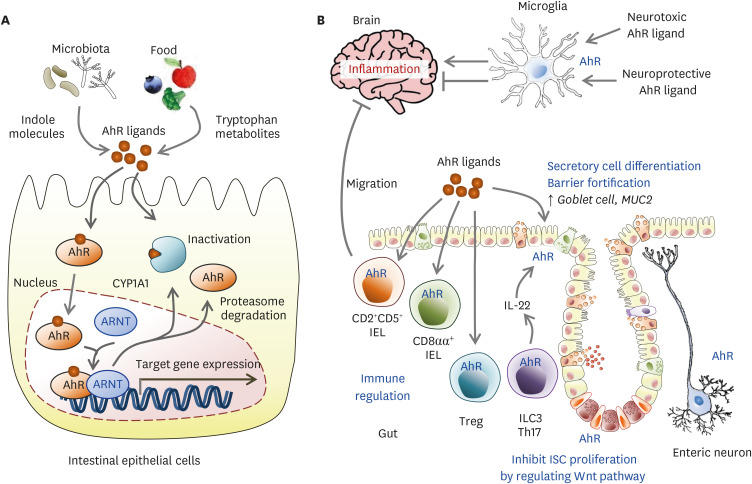
Gwak MG, et al., Immune Netw. 2021
星形胶质细胞在炎症介导的神经退行性变中发挥关键作用,发挥神经毒性作用,激活和招募与中枢神经系统发病机制有关的其他细胞。据报道,在EAE和多发性硬化动物模型中,星形胶质细胞的转录谱显示AhR表达上调。
最近的研究得到了几种神经系统疾病相关临床证据的支持,证明越来越重视使用益生菌和益生元来调节胃肠道微生物群。
使用小鼠模型进行的几项研究表明,益生菌的施用可有益于几种神经系统疾病(如自闭症、癫痫和阿尔茨海默),从而改善认知结果。然而,关于益生菌给药对人类神经功能障碍的有效性,临床证据仍然很少。
对几名患有焦虑和胃肠道症状的3-12岁自闭症儿童进行的调查发现,使用名为Visbome的特殊配方,包括八种不同的益生菌菌株,主要是乳酸杆菌,是安全的,并导致保留乳酸菌患者的自闭症和胃肠道症状的健康改善。
此外,当使用益生菌治疗人类神经退行性疾病(包括阿尔茨海默)时,已经发现了有希望的结果。首先,据报道,植物乳杆菌能够改善阿尔茨海默小鼠模型的认知能力并增加大脑中乙酰胆碱酯酶的水平。在患有阿尔茨海默的啮齿动物散发模型中,用嗜酸乳杆菌、发酵乳杆菌、乳酸双歧杆菌和长双歧杆菌进行Ab注射也发现类似结果。另一项随机临床研究表明,益生菌与鼠李糖乳杆菌GG(ATCC 53103)合用可能减少75名自闭症婴儿的多动症发展,并可能减少神经精神疾病的发展。
证据还表明益生菌对帕金森病患者的影响。最近的一项研究表明,长期服用由六种细菌组成的益生菌可减轻帕金森病遗传小鼠模型的运动损伤,并对多巴胺能神经元具有神经保护作用。
合生元是指益生元和益生菌的混合物,其中益生元有利于益生菌微生物的生长和代谢,提高其生存能力和效益,通过增加胃肠道中有益微生物的丰度来影响宿主。
合生元中使用的组合必须适当,以支持益生菌微生物在胃肠道中的存活。研究表明,使用合生元比单独使用益生菌或益生元更有效。结果表明,由GOS和包括瑞士乳杆菌和长双歧杆菌在内的多序列益生菌组成的合生元制剂可减少抑郁症症状,并改善重度抑郁症中的色氨酸信号。
随机对照试验中使用含合生元的多序列益生菌和益生元治疗的结果导致帕金森队列中功能性胃肠道症状的改善。婴儿双歧杆菌和低聚糖作为合生元被证明对缓解自闭症中的肠道相关疾病有效。然而,合生元对微生物群-肠-脑轴的影响还需要更多的研究 。
后生元,也称为代谢、生物原或CFSs(无细胞上清液),由细菌发酵代谢产物和从活细菌中获得或在细菌细胞裂解后释放的可溶性因子组成,如短链脂肪酸、酶、AMP、磷壁酸、胞内和胞外多糖、细胞表面蛋白、维生素、血浆素和有机酸。
非活性益生菌paraprobiotics被定义为不可存活或失活的微生物细胞,而一些研究人员将其作为后生物的一个亚组。非活性益生菌是一种结构成分,若给予适量,可能会触发宿主的生物活性。
灭活可通过各种方法实现,如物理(热灭活益生菌、紫外线照射或超声波)或化学方法。生物活性化合物,如肠道肽,是细菌与宿主相互作用的结果,被认为是益生元。热灭活副干酪乳杆菌PS23缓解了皮质酮诱导的焦虑样表型,改善了海马和前额叶皮质中的多巴胺水平。
关于大脑健康,对患有心理社会应激的小鼠进行短链脂肪酸组合(乙酸盐、丙酸盐和丁酸盐)治疗的研究显示了抗焦虑作用。
根据最近发现的微生物干预在调节肠道失调引起的神经系统疾病方面的潜力,粪便微生物群移植(FMT)似乎是一种有希望的治疗策略。
这种相对较新的治疗方法包括从健康粪便样本捐赠者及其微生物和代谢物转移到接受者。这种方法目前被用于治疗艰难梭菌感染,与抗生素治疗一起使用。
通过FMT,健康微生物群通过繁殖自我替换,并产生生物活性代谢物。口蹄疫是通过使用内窥镜、灌肠和冷冻干燥材料口服喂养来完成的。该方法的潜力已被用于治疗帕金森病、自闭症和多发性硬化症等神经系统疾病。这种方法的优点之一是没有明显的副作用报告,即使在高危患者中也被认为是安全的。
在最近一项关于自闭症小鼠的研究中,评估了体外培养的肠道微生物移植(GMT)的效果,该移植显著减轻了小鼠的焦虑样行为。在另一项研究中,对接受FMT的自闭症患者进行了结肠镜检查,结果证明自闭症相关症状显著改善,他们的肠道微生物群改变为健康状态。然而,需要更多的研究来进一步阐明FMT对自闭症患者的影响。
在动物模型中对阿尔茨海默进行了大量研究,但没有对人类患者进行具体研究。对阿尔茨海默小鼠模型的研究结果表明,认知功能障碍与肠道微生物群组成的变化有关;因此,通过FMT对该微生物群进行修饰被证明能有效缓解阿尔茨海默患者的认知功能障碍。许多关于FMT的神经系统疾病的研究已经完成,许多试验正在进行中。因此,很快就会有大量证据。
通过治疗性微生物干预调节肠道微生物群
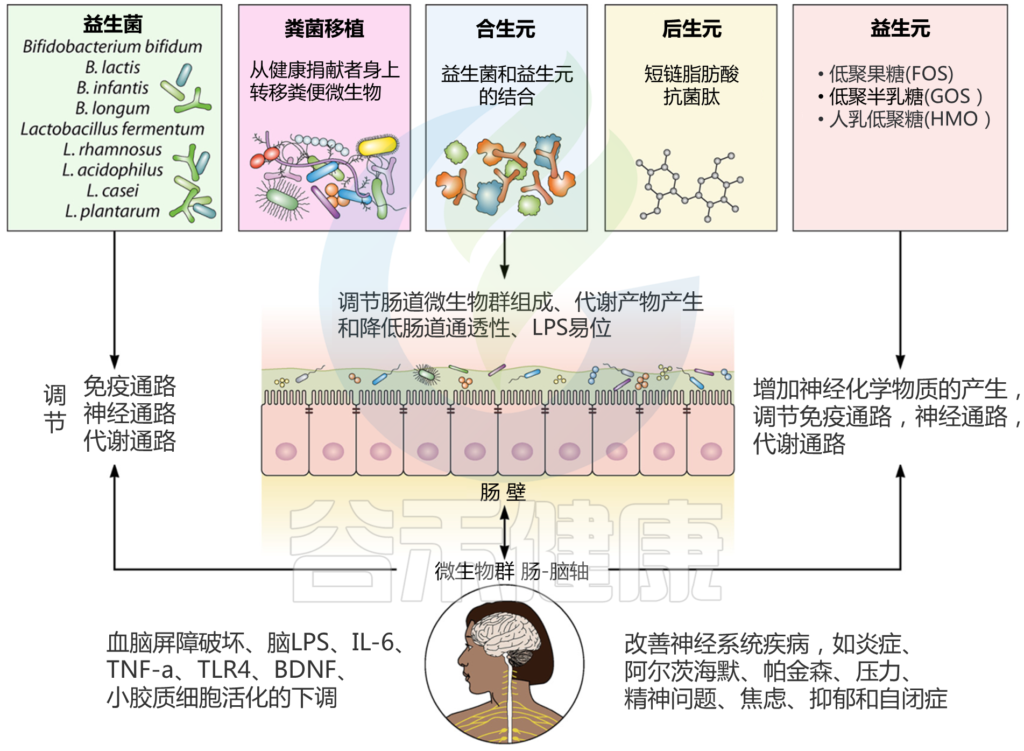
Sorboni SG, et al.,Clin Microbiol Rev. 2022
其他(食物及补充剂)
改善肠脑轴的食物
食物已被证明可以改善肠脑功能。其中许多含有精神生物化合物,包括:
Omega-3 脂肪
存在于油性鱼类和亚麻籽中,已被证明可以改善青春期和成年期的微生物群多样性。这可以降低患精神分裂症和抑郁症等脑部疾病的风险。
发酵乳制品
如酸奶和奶酪中发现的各种益生菌菌株。这些已被证明可以调节大脑活动。
富含纤维的食物
如水果和蔬菜、全谷物和坚果。这些含有益生元纤维,可降低皮质醇水平并改变情绪偏见。
富含多酚的食物
肠道菌群失调导致肠上皮黏膜屏障和血脑屏障通透性增高,并通过肠-脑轴的免疫、内分泌和肠神经途径影响阿尔茨海默的发生发展。多酚类化合物可能通过上述机制发挥防治阿尔茨海默的作用。
详见:肠道微生物群与膳食多酚互作对人体健康的影响
食用蘑菇类
香菇含有大量的维生素B6。因为维生素 B6 会影响血清素和神经递质的产生,所以健康的 B6 水平与积极的情绪和自然地减轻压力有关。
在动物研究中,它也被证明可以有效治疗抑郁症等情绪障碍。
坚果
如杏仁、腰果、核桃和巴西坚果。研究表明,食用这些会提高血清素的水平,血清素是一种让人感觉良好的化学物质,血清素降低,人会感到沮丧。
富含酪氨酸的食物
如杏仁、蛋鱼和鸡肉,富含酪氨酸,酪氨酸是一种能提高大脑多巴胺水平的氨基酸。
其他:
L-苏糖酸镁
镁对大脑至关重要,它是制造许多参与大脑功能的酶的必要辅助因子。它对于维持和发展突触之间的联系也至关重要,突触是学习和记忆的核心任务。大脑和脑脊液中高水平的镁与阿尔茨海默病和大脑衰老的发病率降低有关。补充剂 L-苏糖酸镁比其他镁形式更有效地通过血脑屏障,并且不会引起消化不良。
维生素 D3 和 维生素 K2
血清维生素 D 高水平对于维持大脑健康和降低阿尔茨海默病和其他神经退行性疾病导致的认知障碍风险极为重要。维生素 D 的抗炎和抗氧化特性也很重要。
相反,低维生素 D 会导致大脑中的钙含量增加,这与抑郁症有关,并且会导致表征痴呆的淀粉样蛋白斑块增加。
维生素 D 与维生素 K 协同作用以调节钙并防止其在软组织中积累,例如滋养大脑的血管。
补充剂应包括最易吸收的胆钙化醇(维生素 D3)形式的维生素 D,以及甲基萘醌(维生素 K2)形式的维生素 K,以帮助调理肠道。这种组合对预防血管钙化最有效。
姜黄素
姜黄根中的活性化合物姜黄素使咖喱粉呈现鲜艳的黄色。姜黄素激活 Nrf2 抗氧化信号通路,开启参与解毒和消除自由基的基因。
在大脑中,姜黄素补充剂具有强大的抗炎和抗氧化特性。姜黄素还通过抑制淀粉样蛋白的形成来帮助预防痴呆。姜黄素补充剂可以显著提高 BDNF(脑源性神经营养因子)的血清水平。BDNF 在保护现有神经元和刺激新神经元生长方面发挥着重要作用。
高水平可以帮助预防认知障碍,并有助于从脑震荡和脑损伤中恢复。
肠道微生物群不仅与消化吸收相关,还与大脑密切相关,从而影响各类神经系统疾病,如常见的自闭症、抑郁症、精神分裂症等。改善肠道健康后,通过肠脑轴的连接减少全身炎症,同时也改善心理健康。在现代社会普遍压力较大的情况下,注意减压的同时更不能忽视肠道健康。
主要参考文献:
Li XJ, You XY, Wang CY, et al. Bidirectional Brain-gut-microbiota Axis in increased intestinal permeability induced by central nervous system injury. CNS Neurosci Ther. 2020;26(8):783-790. doi:10.1111/cns.13401
Deidda G, Biazzo M. Gut and Brain: Investigating Physiological and Pathological Interactions Between Microbiota and Brain to Gain New Therapeutic Avenues for Brain Diseases. Front Neurosci. 2021;15:753915. Published 2021 Oct 12. doi:10.3389/fnins.2021.753915
Gwak MG, Chang SY. Gut-Brain Connection: Microbiome, Gut Barrier, and Environmental Sensors. Immune Netw. 2021;21(3):e20. Published 2021 Jun 16. doi:10.4110/in.2021.21.e20
Sorboni SG, Moghaddam HS, Jafarzadeh-Esfehani R, Soleimanpour S. A Comprehensive Review on the Role of the Gut Microbiome in Human Neurological Disorders. Clin Microbiol Rev. 2022 Jan 5;35(1):e0033820. doi: 10.1128/CMR.00338-20. Epub ahead of print. PMID: 34985325; PMCID: PMC8729913.
Suganya K, Koo BS. Gut-Brain Axis: Role of Gut Microbiota on Neurological Disorders and How Probiotics/Prebiotics Beneficially Modulate Microbial and Immune Pathways to Improve Brain Functions. Int J Mol Sci. 2020;21(20):7551. Published 2020 Oct 13. doi:10.3390/ijms21207551
Jalodia R, Abu YF, Oppenheimer MR, Herlihy B, Meng J, Chupikova I, Tao J, Ghosh N, Dutta RK, Kolli U, Yan Y, Valdes E, Sharma M, Sharma U, Moidunny S, Roy S. Opioid Use, Gut Dysbiosis, Inflammation, and the Nervous System. J Neuroimmune Pharmacol. 2022 Jan 7. doi: 10.1007/s11481-021-10046-z. Epub ahead of print. PMID: 34993905.
Morais LH, Schreiber HL 4th, Mazmanian SK. The gut microbiota-brain axis in behaviour and brain disorders. Nat Rev Microbiol. 2021 Apr;19(4):241-255. doi: 10.1038/s41579-020-00460-0. Epub 2020 Oct 22. PMID: 33093662.