-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
谷禾健康
你是否有过这样的经历:明明没有剧烈运动,却总是感到莫名的疲惫;或者明明控制了饮食,体重却依然居高不下,体检报告上的血糖、血脂指标也总是亮起红灯?我们习惯将这些问题归咎于“吃多了”或“动少了”,却往往忽视了一个潜伏在身体内部、肉眼不可见的“隐形杀手”——脂多糖(LPS)。
脂多糖并非外来入侵的毒素,而是我们肠道中数以万亿计细菌的细胞壁成分。在正常情况下,它安分守己地待在肠道内,甚至对免疫系统有益。然而,当肠道屏障受损或饮食结构失衡时,这些本不该进入血液的分子便会“越狱”,引发一场悄无声息却破坏力巨大的全身性慢性炎症。
这种持续存在的低水平炎症,被认为是代谢综合征、2型糖尿病、非酒精性脂肪性肝病以及动脉粥样硬化等多种慢性疾病的重要共同发病基础。脂多糖(LPS)在这一过程中发挥关键作用,可诱导并放大炎症反应,从而促进相关疾病的发生与发展。
那么,脂多糖是如何由肠道内的正常成分转变为影响机体健康的重要因素的?机体又可以通过哪些机制对其进行调控,以维持生理稳态?本文将从机制层面系统分析LPS的作用路径及其对健康的影响。
♥
▸ 脂多糖的定义与特性
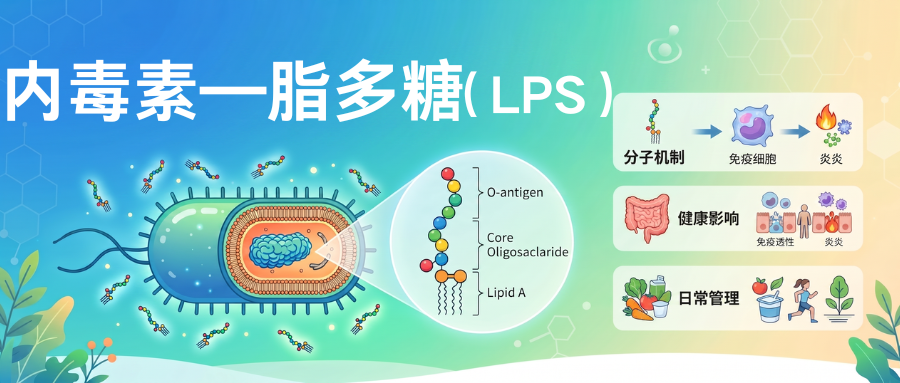
脂多糖(Lipopolysaccharide,简称LPS)是革兰氏阴性菌细胞壁外膜特有的结构大分子复合物,由三个功能区域共价连接组成的异源多糖-脂质复合物,三个区域分别承担不同生理功能。
革兰氏阴性菌膜由两层细菌膜组成,分别是外层和内层膜,外膜富含脂多糖(LPS)。
脂多糖(LPS)是两性分子,类脂A部分疏水,多糖部分亲水,所以LPS能够在细菌表面形成稳定的双亲结构,维持外膜完整性,同时作为屏障抵抗抗菌物质进入细菌体内。
↘ 脂多糖与内毒素的关系
脂多糖之所以被称为”内毒素”,是因为它是革兰氏阴性菌细胞外膜的重要结构成分,通常不以游离形式主动分泌。在细菌死亡或裂解时会大量释放出来,这一特点与由活菌主动分泌的“外毒素”形成鲜明对比,因此得名“内毒素”。
↘ 内毒素与外毒素的主要区别
编辑
需要注意的是,在感染过程中,细菌不断生长和死亡,会持续释放脂多糖;而在严重感染或抗生素治疗后,大量细菌同步裂解,可能在短时间内释放大量LPS,从而加重炎症反应,严重时可引发内毒素相关的全身炎症反应综合征。
▸ 脂多糖的分子结构
LPS分子从内到外分为三个部分:最内层是类脂A,中间是核心多糖,最外层是O抗原(O特异性多糖),三个部分共价连接形成完整LPS分子。
编辑
doi.org/10.3389/fimmu.2025.1588129
★ 每个部分结构和功能都不同:
•类脂A:嵌入外膜磷脂双层,是LPS的毒性中心和生物活性中心,人体对LPS的免疫识别主要识别类脂A结构;
•核心多糖:连接类脂A和O抗原,结构相对保守,是革兰氏阴性菌共同抗原成分;
•O抗原:最外层,是多糖重复单位,结构变异性最大,决定细菌血清型特异性,是细菌逃避免疫识别的重要方式。
编辑
doi.org/10.5217/IR.2014.12.2.90
这种三层结构设计是革兰氏阴性菌长期进化结果,它既保护细菌本身,也决定了它和人体免疫系统相互作用方式,类脂A是毒性中心,O抗原是免疫逃逸,这种结构对应功能的关系便于更好理解它对健康影响。
★ 脂多糖的结构多样性决定其生物活性差异
不同革兰氏阴性菌的脂多糖(LPS)在结构上存在显著差异,尤其是类脂A部分在酰基链数量、长度以及磷酸化修饰上的变化,会直接影响其免疫激活能力。因此,并非所有革兰氏阴性菌的LPS都具有相同毒性,共生菌来源的LPS通常显著低于典型致病菌。
很多人将“革兰氏阴性菌”与“强内毒素”直接等同,但实际上,不同细菌来源的LPS在毒性上差异很大。理解这种差异,有助于我们更准确地区分共生菌与致病菌的生物学影响。
↘ 类脂A:脂多糖的毒性中心和生物活性中心
类脂A嵌入外膜磷脂双层,是脂多糖的毒性中心和生物活性中心,人体对LPS的免疫识别主要识别类脂A结构。LPS的大部分生物学活性,包括毒性和免疫刺激性,都由类脂A部分决定,类脂A结构变异直接改变LPS毒性强弱,六酰化饱和类脂A毒性最强,酰基链减少毒性显著下降。
编辑
doi.org/10.5217/IR.2014.12.2.90
不同来源的类脂A结构与毒性差异:
•致病菌(如大肠杆菌、沙门氏菌):类脂A多为六酰化,且具有完整的双磷酸结构,与TLR4-MD2亲和力高,能够强烈激活免疫反应。
•肠道共生菌(如拟杆菌属):类脂A通常为五酰化或四酰化,并伴随去磷酸化或其他修饰,与受体结合能力较弱,炎症活性显著降低(在部分实验中可低几个数量级)。
•环境调控型细菌(如耶尔森氏菌):其类脂A结构可随温度变化而调整,在环境温度下多为低酰化形式,而在37℃宿主体内转为高酰化形式,从而增强毒性。
编辑
doi: 10.1128/mSystems.00046-17.
如上图所示,拟杆菌门(Bacteroidetes)是人类肠道微生物群中脂多糖(LPS)生物合成能力的主要贡献者。
↘ 核心多糖:连接类脂A和O抗原的保守结构
核心多糖位于类脂A和O抗原之间,主要作用是连接两个部分,它的结构相对保守,在同一属的不同细菌之间结构大多相似,含有特殊的糖分子庚糖和KDO。
核心多糖可以分为两个部分:
•内核心:靠近类脂A,含有特殊的糖分子:3-脱氧-D-甘露-辛酮糖酸(简称KDO)和L-甘油-D-甘露庚糖,这两种糖在其他地方很少见,是LPS核心多糖特征性成分。内核心结构非常保守,不同革兰氏阴性菌之间变化很小。
•外核心:连接内核心和O抗原,由己糖组成,结构比内核心变异大一些,但仍然比O抗原保守很多,同一属细菌外核心结构相似。
核心多糖带有多个负电荷,能够结合二价阳离子比如镁离子钙离子,这些阳离子中和负电荷,稳定LPS在细菌外膜的结构,如果去掉阳离子,LPS结构将变得不稳定,外膜通透性增加,细菌容易被抗菌物质杀死。
↘ O抗原:决定细菌特异性的高度变异部分
O抗原是LPS分子最外层的多糖重复单位,是LPS中变异性最大的部分,不同血清型革兰氏阴性菌O抗原结构不同,这种高度变异是细菌逃避免疫识别的重要进化策略。O抗原高度变异直接关系到细菌致病性和免疫识别,但是O抗原本身基本没有毒性,主要影响细菌免疫原性和致病性,不直接影响LPS毒性强弱。
O抗原由多个相同的寡糖重复单位聚合而成,每个重复单位通常含有2-6个糖分子,不同细菌重复单位的糖组成、排列顺序、连接方式都可以不同,所以变异非常大,理论上可以产生成千上万种不同O抗原结构。
实例:E. coli O157:H7(常见的致病性肠出血性大肠杆菌)与E. coli O26、O111等其他血清型的区别,本质上就是O抗原多糖重复单位的结构不同。有的重复单位含有不同种类的糖(葡萄糖、半乳糖等),有的糖连接方式不同(α-1,3还是β-1,4),排列顺序也不同。因此针对O157的抗体一般难以有效识别O26等其他血清型,但在个别情况下可能存在有限的交叉反应。
▸ 光滑型脂多糖和粗糙型脂多糖的差异
完整脂多糖(LPS)含有O抗原叫做光滑型LPS,缺失O抗原只剩下类脂A和核心多糖叫做粗糙型LPS,也叫脂寡糖(LOS),两者在稳定性、免疫原性和致病性上都有显著差异。
编辑
编辑
doi.org/10.3390/ijms24098395
LPS(脂多糖)结构示意图:图中亮红色的LPS锚定在革兰氏阴性(G-)细菌的外膜上。根据O抗原和核心区(core)的长度,可将LPS分为光滑型(S)、半粗糙型(SR)、粗糙型(R)和深度粗糙型(DR)。从LPS最外层到最内侧疏水部分,其结构变异性逐渐降低。
♥
▸ 脂多糖对革兰氏阴性菌生存的影响
脂多糖(LPS)不仅仅是致病因子,它对革兰氏阴性菌自身生存具有非常重要的生理功能,维持外膜结构完整性,抵抗抗菌物质入侵,保护细菌生存,虽然部分菌株即使缺失或改变LPS结构仍可存活,但是其生理功能和适应性会显著受损。
脂多糖对革兰氏阴性菌的主要生理功能:
①维持外膜结构完整性:脂多糖是革兰氏阴性菌外膜主要成分,占外膜干重三分之一以上,LPS分子通过二价阳离子交联形成稳定网状结构,维持外膜通透性和结构完整性,如果缺失LPS,外膜结构破坏,细菌存活能力下降。
②形成通透屏障抵抗抗菌物质:LPS外层亲水内层疏水,形成选择性通透屏障,能够阻挡疏水抗菌物质、胆汁盐、补体、溶菌酶进入细菌体内,保护细菌免受宿主防御物质和抗生素攻击,增加细菌耐药性。
③抵抗宿主免疫攻击:O抗原能够保护细菌不被补体激活和吞噬细胞吞噬,帮助细菌逃避免疫清除,增加细菌致病性,有O抗原的菌株更容易引起全身性感染。
④介导细菌粘附:LPS能够介导细菌粘附到宿主黏膜上皮细胞,帮助细菌定植,这是感染第一步,所以LPS对细菌定植也很重要。
因为脂多糖对细菌生存这么重要,所以几乎所有革兰氏阴性菌都必须合成LPS,只有极少数例外,比如某些衣原体可以缺失。
▸ 肠道中的脂多糖与人体屏障系统
据估计,人类结肠中含有约10¹³–10¹⁴个细菌,总生物量约为数百克(通常约0.2–0.5 kg)。这些细菌在生命活动过程中可产生脂多糖(LPS)。研究表明,以大肠杆菌为代表的革兰氏阴性菌,其单个细胞所含LPS约为几飞克至几十飞克(约2–50fg)。基于此进行粗略估算,肠道中LPS的总量大致处于几十毫克至数百毫克的范围,具体取决于菌群组成及生理状态。
然而,静脉注射LPS的致死剂量仅为1–2微克。也就是说,肠道内的LPS总量,可能是致死剂量的千倍甚至百万倍。那么为什么我们不会被”毒死”?答案在于——人体的屏障系统。
↘ 肠道屏障:把”危险”关在门外
人体的上皮组织(比如肠道内壁)在进化过程中,形成了极其高效的防御结构,能够将有害物质隔离在外。
在正常情况下,肠道细菌及其产生的LPS主要存在于肠腔内部,它们被粘液层和上皮细胞牢牢阻挡,不会接触到体内的深层组织。
研究发现,只要这些细菌停留在肠腔一侧,就不会对肠道细胞造成伤害。真正的危险发生在当LPS接触到上皮细胞”背面”(基底侧)时,毒性才会显现。
↘ 肠道屏障破坏的风险因素
当肠道屏障被破坏时,情况就会发生改变:细菌可能穿过粘液层,从细胞之间的缝隙渗透,甚至直接穿过细胞进入更深层组织。一些致病菌本身就具备”入侵能力”,可以主动突破防线。一旦细菌或大量LPS进入血液,免疫系统会迅速响应,但如果负荷过高,可能引发严重后果,比如全身炎症反应、败血症甚至败血性休克。
以下人群风险更高:
-新生儿(免疫系统尚未成熟);
-老年人或重症患者;
-免疫功能受损的人群;
-长期使用导尿管等侵入性器械者;
-肠道存在炎症、损伤或肿瘤的人。
▸ 肠道暴露导致脂多糖转移到循环中
在正常生理条件下,血浆中LPS水平极低。这是因为健康的肠道限制了LPS分子进入血液的途径。尽管持续暴露于LPS,肠细胞和结肠细胞对这些刺激表现出低反应状态,部分原因在于这些细胞中Toll样受体4(TLR4)表达较低。这种肠道耐受机制最大限度地减少了肠腔内的炎症反应。
此外,肠道微生物群中LPS的暴露在免疫系统的发育和成熟中起着关键作用,训练肠道相关淋巴组织(GALT)中的调控T细胞。个体的LPS谱系取决于肠道微生物群的组成,而肠道微生物群的组成因地理和生活方式等因素而有所变化。
▸ 脂多糖在肠道中”常态存在”的生理意义
很多人一听说脂多糖(LPS)有害,就想把肠道里的LPS彻底清除。实际上,这既不现实,也没有必要。相反,维持生理范围内的LPS暴露,对健康是有意义的。
在健康婴儿、成年人和老年人肠道菌群中,革兰氏阴性菌占有一定比例(如拟杆菌门)。这些细菌在更新过程中会不断死亡、裂解,从而释放LPS到肠腔中。因此,肠道内持续存在低水平LPS是一种正常的生理现象,而不是异常状态。生理水平的脂多糖具有以下作用:
•促进免疫系统正常发育:无菌动物由于缺乏包括LPS在内的微生物刺激,免疫系统发育不成熟,免疫应答能力不足,更容易出现过敏和自身免疫问题。适度的LPS等刺激有助于免疫系统建立“耐受”和”分辨能力”,这是”卫生假说”的重要机制基础之一。
•维持先天免疫的基础警戒状态:低水平的微生物信号可以让机体免疫系统保持”准备状态”。如果完全缺乏这类刺激,机体在真正遇到病原体时,可能反应迟缓,反而更容易感染。
•有助于睡眠:2024年发表的一项研究表明当LPS注入门静脉循环时,会引起睡眠增加和体温升高。门静脉循环负责将来自胃、小肠和大肠(这些器官中存在肠道微生物群)的血液输送到肝脏。像LPS这样的微生物分子可以通过“转位”过程进入门静脉,从而进入宿主体内环境。
编辑
事实上,在健康的人类、大鼠和小鼠体内,脂多糖天然存在于门静脉和全身循环血液中,是一种来源于原核生物的生理性血浆成分。
研究进一步暗示日常生活中的一些刺激会进一步促进LPS的转位,从而提高其在血液中的水平。例如,一顿高脂餐(如三片涂有50克黄油的吐司)可使人体血浆LPS水平升高约50%。此外,少量饮酒、急性或慢性睡眠不足以及轻度心理压力等常见情况,也都会增加循环中的LPS水平。
!
关键观点
问题不在”有没有”,而在”是否过量”。真正有害的是脂多糖异常升高并进入循环(如肠屏障受损导致的”内毒素血症”),这会诱导慢性低度炎症,进而与代谢性疾病等相关。因此,我们关注的重点不是”全部消灭LPS”,而是避免其异常升高和大量入血。
♥
▸ 肠道是人体内脂多糖的主要来源
在健康成年人中,肠道是体内LPS(脂多糖)最主要的产生和储存场所。健康成年人肠道内约有10¹³–10¹⁴数量级的微生物,其中约20%–40%为革兰氏阴性菌(以拟杆菌门为主),这些细菌在持续更新过程中不断裂解并释放LPS。
相比之下,口腔(如牙周炎时)虽也可产生LPS但总体规模较小,呼吸道和皮肤等部位菌量更低,对全身LPS贡献有限,但是注意在严重局部感染或全身感染时,这些部位也可能成为重要来源。
▸ 关键分界:是否进入循环
一个非常重要但常被忽略的点是:肠道中LPS很多≠血液中LPS一定高。
↘ 在健康状态下:
•肠道屏障(上皮细胞+紧密连接+黏液层+IgA)有效阻挡LPS进入体内;
•只有极少量LPS可进入循环;
•血浆LPS通常处于低水平(常见报道为pg/ml级别);
•这个水平属于生理暴露,不会引发系统性炎症。
↘ 何时变成问题?当出现以下情况时:
•肠道屏障受损(如”肠漏”);
•肠道菌群失衡(如革兰阴性菌过度增殖);
•高脂饮食促进LPS转运(经乳糜微粒)
就会导致进入循环的LPS增加(而不是肠道LPS本身增加才是关键),这就是所谓的代谢性内毒素血症,其特点是:
•血中LPS轻度但持续升高;
•激活TLR4等炎症通路;
•诱导慢性低度炎症;
•并与胰岛素抵抗、肥胖、非酒精性脂肪肝、动脉粥样硬化相关。
代谢性内毒素血症的经典模型认为,LPS可能进入全身循环并促进慢性炎症。该经典模型指出:
(1) 西式饮食可能增加产生LPS的细菌数量并增加脂肪吸收。
(2) LPS可在局部发挥作用并诱导炎症反应,也可通过刷状缘的移位转运以及在乳糜微粒内进入全身循环。
(3) 主要来源于肝脏的LBP与循环中LPS的脂质A部分结合。
▸ 脂多糖进入血液循环的主要途径
LPS从肠道进入血液循环主要通过两条途径:一是肠道通透性增加(肠漏)导致经细胞间隙进入循环,二是在脂肪吸收过程中结合乳糜微粒,经淋巴系统进入血液。这两条途径共同影响体内LPS的总体水平。
↘ 第一条途径:肠漏(细胞间隙通透增加)
当肠道上皮紧密连接受损,细胞间隙通透性增加,LPS可以从肠腔进入黏膜下组织,并进一步进入门静脉循环。这一过程不依赖脂肪摄入,而主要取决于肠屏障完整性。长期通透性升高,是循环LPS持续升高的重要原因之一。
↘ 第二条途径:乳糜微粒转运
LPS具有一定亲脂性。在摄入脂肪后,肠道形成乳糜微粒以吸收和运输脂类物质,部分LPS可结合在乳糜微粒上,随其进入淋巴系统,最终进入体循环。这一过程与脂肪摄入量相关:脂肪摄入越多,乳糜微粒生成越多,LPS转运量也可能相应增加。这也是高脂餐后出现”餐后内毒素血症“的一个重要机制。
这两条途径通常同时存在:肠道通透性决定基础LPS水平,而餐后乳糜微粒可造成短时升高,两者叠加,可能导致整体水平上移。
★ 短时过量脂肪摄入可能导致脂多糖过多的危害
当长期高脂摄入、频繁产生大量乳糜微粒,或肝脏清除能力下降时,可能出现LPS清除不及、循环水平升高的情况,从而与慢性低度炎症相关。
因此,并不需要避免脂肪摄入本身,而是应关注总量与分配:避免单次摄入过多脂肪,有助于降低一次性LPS负荷。只要控制每餐脂肪量,不要一次摄入太多脂肪,减少单次转运LPS量,就不会对健康造成危害,完全禁止脂肪摄入也不利于健康,关键是量和频率。
这个也解释了为什么中国人说“饱食伤身”,一次吃太多脂肪不仅热量过剩,还带来大量LPS进入循环,导致一次炎症峰,反复多次就是慢性炎症,所以吃七八分饱不仅减少热量,也减少LPS,对健康更有利。
♥
▸ 脂多糖的半衰期与清除动力学
LPS在人体内的半衰期取决于其浓度和人体解毒能力。生理低剂量LPS进入循环后,半衰期大约是几分钟到十几分钟,能够被肝脏快速清除,但是高剂量LPS超过肝脏清除能力,半衰期显著延长,能够在循环中停留几个小时,持续激活炎症。
了解半衰期能够帮助我们理解为什么低剂量持续暴露比一次性高剂量更有害,为什么慢性持续LPS升高导致慢性病。
▸ 肝脏:清除循环LPS的主要器官
进入门静脉的脂多糖约有80–90%在肝脏被清除,主要由Kupffer细胞介导,肝脏因此成为清除循环LPS的关键器官。
正常肝功能有助于维持低水平LPS,而功能受损时清除能力下降,易导致其升高。除直接吞噬外,LPS还可与LDL、HDL等血浆脂蛋白结合,并被肝细胞和Kupffer细胞摄取,从而进一步促进清除。
编辑
↘ 肝脏功能能够影响脂多糖的清除能力
因此,肝脏构成防止LPS进入体循环的重要屏障。肝功能正常,清除能力强,能够快速清除LPS,肝功能下降,清除能力下降,相同剂量LPS半衰期延长,炎症更明显,所以肝功能不好的人更容易发生内毒素血症。
编辑
▸ 肠道碱性磷酸酶对LPS的解毒作用
肠道上皮分泌的碱性磷酸酶(intestinal alkaline phosphatase, IAP)可通过去除LPS脂质A上的磷酸基团,将其转化为低毒形式,显著降低其激活TLR4-MD2受体的能力,从而减弱炎症反应。
↘ 碱性磷酸酶能够减弱脂多糖导致的炎症反应
这一过程发生于LPS进入体内之前,是肠道局部解毒的重要机制和防止系统性炎症的第一道屏障。IAP由小肠上皮刷状缘表达,直接与肠腔内LPS接触;其去磷酸化作用可使LPS炎症活性降低数十至数百倍。
动物研究表明,缺失IAP会增加对LPS的敏感性并加重炎症反应。IAP为含锌酶,锌对其结构稳定和催化活性至关重要,缺锌可降低其功能并增加活性LPS负荷。部分研究还提示,高脂饮食可能抑制IAP表达或活性,而低脂或特定膳食模式有助于维持其功能,但相关结论尚不一致。
▸ 低密度脂蛋白与脂多糖的相互作用
脂多糖(LPS)进入循环后,一部分游离存在,另一部分与低密度脂蛋白(LDL)结合。结合后,LPS的疏水结构被包埋于LDL颗粒内,减少与免疫细胞表面TLR4受体的接触,从而降低其炎症活性,这是一种机体的保护性机制。
↘ LDL与LPS的结合具有双重效应
然而,携带LPS的LDL更易被巨噬细胞通过清道夫受体摄取,促使其转化为富含胆固醇的泡沫细胞,进而在血管壁沉积形成脂纹,推动动脉粥样硬化的发生与发展。
因此,LDL与LPS的结合具有双重效应:既可降低游离LPS的炎症活性,又促进泡沫细胞形成和动脉粥样硬化进展。当LPS与LDL同时升高时,心血管风险显著增加,且两者具有协同放大效应。循环LPS水平升高会增强LDL的致动脉粥样硬化作用,这是其促进心血管疾病的重要机制之一。
▸ LPS清除的多器官协作(肝-脾-肾)
除肝脏外,脾脏和肾脏亦参与循环LPS的清除与代谢:肝脏为主要清除器官,脾脏提供辅助清除,肾脏负责排泄降解产物。整体上,网状内皮系统(单核-巨噬细胞系统)协同维持LPS稳态。
↘ 肝脏主要清除、脾脏辅助清除
进入循环的LPS大多由肝脏Kupffer细胞清除,其余部分可被脾脏巨噬细胞摄取。肾脏对完整LPS的直接清除有限,主要排泄其降解后的小分子片段,参与代谢末端处理。
LPS清除依赖单核-巨噬细胞系统的吞噬功能;当疾病、炎症或药物导致该系统功能下降时,清除效率降低、半衰期延长,循环水平升高并加重炎症反应。
总体而言,LPS清除体现多器官协同的分层防御:肝脏为核心屏障,脾脏补充清除,肾脏负责排泄。该体系功能正常时可有效控制LPS水平;若输入增加(如肠屏障受损或饮食因素)且清除能力下降(如肝功能或免疫功能减弱)并存,则可能突破防线,导致慢性低水平内毒素血症。
♥
▸ 肠道菌群失调
当肠道菌群失调时,菌群结构和代谢功能发生改变,可能导致脂多糖负荷及其炎症活性升高。肠腔中LPS水平上升,同时在饮食和屏障因素作用下进入循环的LPS增加。因此,通过改善菌群结构与代谢环境,可以从源头降低高炎症活性的LPS负荷。
健康成年人肠道中本就富含革兰氏阴性菌(如拟杆菌门)。但菌群失调(如高脂、低纤维饮食)会改变其结构,使部分菌群(尤其肠杆菌科)增加,同时短链脂肪酸等代谢产物减少,从而共同影响LPS相关效应。
不同来源的LPS免疫活性差异显著:拟杆菌来源LPS炎症活性较低,而大肠杆菌来源则更易诱导炎症。因此,关键不在于阴性菌数量多少,而在于高炎症活性LPS的比例是否上升。
↘ 在吸收方面,LPS进入循环主要通过:
•与脂质结合,经乳糜微粒转运(高脂饮食关键机制);
•肠屏障功能改变(紧密连接受损)。
因此,高脂低纤维饮食的作用是双重的:一方面改变菌群结构,另一方面显著促进LPS进入血液循环,共同提高循环LPS水平。
↘ 膳食纤维则通过不同路径发挥作用:
•促进产短链脂肪酸菌生长(如产丁酸菌);
•增强肠道屏障功能;
•调节菌群整体生态。
从而降低脂多糖相关炎症负荷。
这一机制把”菌群失调”和”慢性炎症”连接起来,但更准确的路径应理解为:
菌群失调 → LPS结构与负荷改变+代谢环境改变 → LPS更容易进入循环 → 诱导低度慢性炎症 → 胰岛素抵抗、脂肪肝、动脉硬化等问题 → 参与多种慢性疾病发生发展。
这里的关键,不是单一指标(比如阴性菌比例),而是菌群整体生态与宿主相互作用。所以,调节肠道菌群的核心,不是简单”降低革兰氏阴性菌”,而是恢复菌群结构与功能的平衡,从而降低高炎症活性的LPS负荷。
▸ 小肠细菌过度生长
正常情况下,小肠受胃酸和蠕动影响,细菌浓度远低于结肠(约10¹¹–10¹² CFU/ml的万分之一),因此LPS水平较低,即使全部吸收,总量也有限。
↘ 小肠细菌过度生长使脂多糖水平显著升高
发生小肠细菌过度生长(SIBO)时,小肠细菌增至10⁵–10⁸ CFU/ml,且多为革兰氏阴性菌,使腔内LPS浓度升高数十至上百倍。由于小肠吸收面积大、通透性高,更多LPS进入门静脉循环,导致循环水平升高。同时,SIBO还可损伤黏膜、破坏紧密连接,进一步增强LPS吸收,形成叠加效应。因此,不明原因的慢性LPS升高应考虑SIBO。
LPS的影响不仅取决于“数量”,还取决于“产生部位”:结肠产量高但吸收受限,小肠产量较低却更易入血。研究显示,SIBO患者循环LPS显著升高,根除后可下降并伴随症状改善,提示其为重要来源之一。这也解释了SIBO与多种肠外疾病的关联,即通过增加LPS吸收诱发全身慢性炎症。
▸ 牙周炎:被低估的”持续性LPS输入源”
牙周炎中,牙周袋内聚集大量革兰氏阴性厌氧菌(如牙龈卟啉单胞菌),持续释放脂多糖。在局部炎症和血管通透性增加的情况下,这些LPS可以进入血液循环,成为全身LPS负荷的长期来源之一。
很多人把牙周炎当作”口腔问题”,但忽略了一点:牙龈是一个高度血管化、长期暴露炎症的组织,这意味着,口腔里的炎症信号,是可以”进入全身”的。
↘ 牙周炎的核心环境是牙周袋:
•厌氧环境;
•大量革兰氏阴性菌;
•持续细菌更新与裂解释放脂多糖。
▸ 脂多糖与肠道屏障损伤的正反馈循环
脂多糖不仅可通过肠道进入循环,还可反过来破坏肠道屏障结构,增加通透性(”肠漏”),从而进一步促进LPS吸收,形成正反馈循环。
研究表明脂多糖可激活肠上皮细胞中的NF-κB通路,诱导炎症因子释放。这些炎症信号可导致紧密连接蛋白(如occludin、ZO-1)发生内吞和降解,从而破坏紧密连接结构。
结果是肠道通透性增加,使更多LPS从肠腔进入门静脉及全身循环。升高的LPS又进一步加重屏障损伤,形成循环:
LPS升高 → 屏障受损 → 通透性增加 → LPS吸收增加 → LPS进一步升高。
这一循环可持续放大低度慢性炎症状态。这表明一旦肠道屏障受损,单一干预往往难以逆转进程;同时降低LPS来源与修复屏障结构,可能更有助于打破这一循环。
▸ 其他升高脂多糖水平的风险因素
↘ 时间维度:停留越久→吸收越多(便秘)
便秘本质是”肠道内容物滞留”。停留时间延长→细菌裂解释放更多LPS + 与肠壁接触更久→吸收增加。同时伴随菌群失衡→进一步增加LPS来源。
结果:脂多糖”暴露时间+浓度”双升高。
↘ 屏障维度:肠道越”漏”→进入越多(压力/睡眠不足)
慢性压力通过皮质醇破坏紧密连接→肠道通透性增加。原本被阻挡的LPS更容易进入血液。同时压力还会改变菌群→增加LPS产生。
结果:LPS”通行能力”增强。
↘ 储存维度:储得越多→释放越久(肥胖)
肥胖不是单纯”多脂肪”,而是一个LPS放大系统:脂肪组织储存LPS并持续释放;菌群失调→产生更多LPS;肠道屏障变差→吸收更多LPS。
编辑
结果:LPS”长期维持在高水平”。
↘ 生活方式和饮食因素
多种生活方式和饮食因素通过”增加LPS产生+增加LPS吸收“两条路径,提高体内LPS水平:
•高脂饮食:一方面促进乳糜微粒把LPS带入血液,另一方面改变菌群增加革兰阴性菌→LPS显著升高。
•过量饮酒:破坏肠道屏障(肠漏)+ 改变菌群→更多LPS进入血液,加重肝脏炎症。
•吸烟:影响口腔和肠道菌群+ 破坏屏障→多来源增加LPS。
•膳食纤维不足:有益菌减少、短链脂肪酸下降→菌群失衡+ 屏障变差→LPS升高。
•长期抗生素:杀死有益菌、促使阴性菌占优势+ 损伤屏障→LPS增加。
♥
▸ LPS与TLR4的识别与信号传导
LPS通过与细胞膜上的Toll样受体4(TLR4)复合物结合,激活先天免疫细胞,其生物学效应由此启动。作为典型的病原体相关分子模式(PAMP),LPS被TLR4这一模式识别受体特异性识别。
↘ 脂多糖的识别过程需要多种蛋白质协同作用:
•LPS结合蛋白(LBP):首先结合循环中的游离LPS,从聚集体中提取LPS分子,并将其运输至CD14;
•CD14:将LPS转移至TLR4-MD2复合物;
•TLR4-MD2复合物:MD2蛋白结合LPS的脂肪酸A部分。结合后,MD2发生构象变化,导致TLR4二聚化并启动下游信号转导。
▸ TLR4多态性影响LPS敏感性与疾病风险
TLR4基因多态性(如Asp299Gly、Thr399Ile)可改变受体结构,降低其与LPS的结合亲和力和敏感性,从而减弱炎症反应,并影响慢性疾病的易感性。
因此,在相同脂多糖水平下,不同个体的炎症程度和疾病风险可存在显著差异。携带这些变异的个体炎症反应较低,动脉粥样硬化风险可能下降,但对革兰氏阴性菌感染的易感性及严重脓毒症风险增加,体现出权衡效应。相反,无变异者对LPS更敏感,更易发生强烈炎症反应及相关慢性疾病,需要更积极地控制LPS水平。
除上述多态性外,其他遗传变异亦参与调控LPS敏感性,共同决定个体差异。这也反映了遗传与环境的相互作用:环境决定暴露水平,遗传决定反应强度,两者共同影响疾病风险。
编辑
doi.org/10.3390/ijms24098395
▸ 脂多糖的免疫识别与炎症激活
↘ 识别与递呈(胞外阶段)
LPS在循环中先由脂多糖结合蛋白(LBP)结合并单体化,随后转移至CD14(mCD14或sCD14),再递交给TLR4-MD2复合物。MD2结合脂质A后诱导TLR4二聚化,启动信号转导。LBP和CD14提高了系统对低浓度LPS的敏感性。
↘ 两条主要信号通路(TLR4下游)
MyD88依赖通路(主要炎症通路):通过IRAK–TRAF6–TAK1–IKK级联,激活NF-κB,诱导TNF-α、IL-1β、IL-6等促炎细胞因子产生,是LPS炎症反应的核心机制。
TRIF依赖通路:在内体中激活TBK1–IRF3,诱导I型干扰素(IFN-α/β)产生,并促进共刺激分子表达,连接先天免疫与适应性免疫。
↘ 细胞内识别与炎症放大
NOD样受体(Nod1/Nod2)识别细菌肽聚糖成分(不是直接识别LPS),通过RIP2激活NF-κB,与TLR4信号协同放大炎症反应。
↘ 炎症小体激活(第二放大机制)
LPS作为”第一信号”诱导NLRP3和pro-IL-1β表达;ATP等作为”第二信号”激活炎症小体,caspase-1剪切成熟IL-1β,进一步放大炎症级联反应。
↘ 核心转录调控节点
NF-κB是关键汇聚点,几乎所有LPS诱导的促炎基因均依赖其激活,是慢性炎症持续存在的核心分子基础。
↘ 主要效应分子
TNF-α、IL-1β、IL-6共同介导LPS的炎症效应;CRP为IL-6驱动的临床替代指标,常用于炎症监测。
↘ 剂量决定结局
低剂量长期暴露:持续激活NF-κB → 慢性低级别炎症 → 代谢性疾病等。
高剂量急性暴露:细胞因子风暴 → SIRS/脓毒性休克。
♥
▸ 脂多糖与动脉粥样硬化
通常认为胆固醇是动脉粥样硬化的主要原因,但往往忽视了炎症因素,尤其是LPS对内皮细胞的直接激活作用。
血管内皮细胞表达TLR4和CD14。LPS与TLR4结合后激活NF-κB信号通路,诱导细胞间黏附分子-1(ICAM-1)和血管细胞黏附分子-1(VCAM-1)的表达。这些黏附分子为循环中的单核细胞和淋巴细胞提供黏附位点。
↘ 脂多糖作用于内皮细胞促进动脉粥样硬化
黏附的白细胞随后穿越内皮进入血管壁,单核细胞分化为巨噬细胞,摄取氧化低密度脂蛋白(oxLDL),形成泡沫细胞。泡沫细胞的聚集构成脂肪纹,这是动脉粥样硬化的早期病变。
此外,LPS还可诱导内皮细胞产生促炎细胞因子(如TNF-α、IL-6),进一步促进炎症反应、白细胞浸润及血管平滑肌细胞增殖,加速病变进展。
同时,LPS可通过增加活性氧生成、减少一氧化氮(NO)生物利用度,引发内皮功能障碍,表现为血管舒张能力下降及促凝状态增强。
因此,持续升高的LPS水平可通过直接作用于内皮细胞,促进动脉粥样硬化的发生和发展。
▸ 脂多糖与2型糖尿病及胰岛素抵抗
长期升高的循环LPS可激活脂肪组织、肝脏等处的巨噬细胞,促使其分泌TNF-α、IL-6等炎症因子。这些因子通过激活丝氨酸/苏氨酸激酶,促进IRS-1发生丝氨酸磷酸化,从而抑制其正常的酪氨酸磷酸化,阻断胰岛素信号传导,导致胰岛素抵抗。
↘ 降低脂多糖可改善胰岛素敏感性并降低血糖
在此状态下,β细胞需代偿性分泌更多胰岛素以维持血糖,但长期负担会导致功能衰竭,最终发展为2型糖尿病。研究表明,2型糖尿病患者循环LPS水平显著升高,而降低LPS可改善胰岛素敏感性并降低血糖,支持其在发病中的关键作用。
因此,糖尿病管理不应仅关注血糖,还应调节肠道菌群,从源头降低LPS水平。该机制也解释了部分体重正常者患病的原因:即使无肥胖,若存在菌群失调和LPS升高,仍可诱发炎症和胰岛素抵抗。对肠道菌群及LPS风险的评估,有助于实现更早期干预与预防。
▸ 脂多糖与非酒精性脂肪性肝病
肠道来源的脂多糖经门静脉直接进入肝脏。肝脏中的库普弗细胞高表达Toll样受体4(TLR4),可识别LPS并激活NF-κB信号通路,进而释放TNF-α、IL-1β等促炎细胞因子。
这些炎症因子一方面抑制肝脏胰岛素信号通路,降低脂肪酸氧化能力,促进甘油三酯在肝细胞内沉积,从而加重脂肪变性;另一方面可激活肝星状细胞,促进胶原合成和细胞外基质沉积,推动肝纤维化进展。
随着炎症持续存在,疾病可由单纯性脂肪肝逐步发展为非酒精性脂肪性肝炎(NASH),甚至进一步进展为肝纤维化和肝硬化。
↘ NAFLD患者存在脂多糖水平显著升高
研究显示,非酒精性脂肪性肝病(NAFLD)患者的门静脉及循环LPS水平显著升高;而降低LPS水平可减轻肝脏炎症反应,改善脂肪变性及纤维化程度,进一步支持LPS在NAFLD中的关键作用。
这一机制同样解释了为什么部分不超重、无饮酒史的人群仍会发生NAFLD:即使没有传统代谢风险因素,肠道菌群失调导致的LPS升高,依然可以驱动肝脏炎症和疾病进展。
▸ 脂多糖与高血压
↘ 脂多糖通过减少NO生成提高血管张力并升高血压
脂多糖可激活血管内皮细胞NF-κB通路,诱导促炎因子释放,并抑制内皮型一氧化氮合酶(eNOS)表达与活性,减少NO生成。NO减少会削弱血管舒张、增强收缩,进而提高血管张力并升高血压。
同时,LPS可损伤内皮结构、增加血管壁僵硬度和外周阻力;长期炎症还促进血管重构,使高血压逐渐固定化。动物研究表明,慢性低剂量LPS可升高血压,而降低LPS水平有助于改善血压,支持其在血压调控中的作用。
▸ 脂多糖与肥胖的相互促进
肥胖可促进脂肪组织炎症和脂多糖蓄积,而LPS介导的炎症又进一步干扰能量代谢、促进脂肪储存,二者形成相互促进的正向循环。
编辑
↘ 脂多糖干扰能量代谢、肥胖又促进脂多糖积累
此外,LPS还可能通过影响下丘脑能量调节,干扰食欲和能量摄入,进一步推动体重增加。
研究表明,减重可降低脂肪组织炎症和LPS负荷,改善胰岛素敏感性,从而有助于打破这一循环。
这一循环提示,单纯依赖热量限制可能不足以优化代谢状态;若同时改善肠道菌群、降低LPS水平,可能更有助于提高减重效果和代谢改善程度。
▸ 脂多糖与神经系统疾病
循环LPS能够通过多种途径穿过血脑屏障进入脑实质,激活小胶质细胞产生炎症,所以外周LPS升高能够影响中枢神经系统功能,参与神经精神疾病发生发展。
LPS分子量大约10-20 kDa,属于大分子,但是研究证实,循环LPS能够穿过血脑屏障进入脑内,主要通过几个途径:
•血脑屏障上有特定转运系统:能够转运LPS进入脑内,低浓度LPS就能够转运;
•炎症能够破坏血脑屏障完整性:LPS本身引发外周炎症,炎症介质破坏血脑屏障,更多LPS能够进入脑内,形成恶性循环;
•绕过血脑屏障:LPS能够通过血脑屏障薄弱部位比如延髓最后区进入脑内,然后扩散到其他脑区。
↘ 脂多糖入脑激活小胶质细胞致炎
进入脑内的脂多糖能够激活小胶质细胞,小胶质细胞是脑内巨噬细胞,表达TLR4,激活后产生促炎细胞因子,引发神经炎症,损伤神经元,所以外周LPS升高能够增加中枢神经炎症,参与抑郁症、帕金森病、阿尔茨海默病发生发展。
LPS能够穿过血脑屏障进入中枢这个事实,把外周肠道LPS和中枢神经疾病连接起来了,肠道菌群失调→LPS升高→进入中枢→激活小胶质细胞→神经炎症→神经精神疾病,这个完整通路现在已经被越来越多研究证实,这就是”肠脑轴”重要分子机制,所以改善肠道菌群降低LPS,能够帮助预防和改善神经精神疾病。
这一机制也解释了为什么肠道菌群失调能够影响情绪和认知,脂多糖进入中枢激活炎症就是重要介导。
编辑
doi.org/10.1021/acschemneuro.8b00733
♥
1
质谱检测
脂质A中的脂肪酸为3-羟基脂肪酸(3OH-FAs),其链长和酰化模式具有多样性,并可能影响LPS的免疫原性。
由于酯化的3OH-FAs仅来源于脂质A,因此其水平可作为循环中LPS的替代指标。早期采用GC/MS对其进行分离和定量;近年来发展出HPLC/MS/MS方法,通过计算总3OH-FAs与游离3OH-FAs的差值来定量酯化部分,从而实现更高灵敏度和准确性的LPS检测。
2
血浆脂多糖定量
血浆LPS定量检测是评估内毒素血症的重要方法之一,常用鲎试验(LAL)可用于检测血浆中LPS水平,反映循环内毒素负荷。
LAL试验基于鲎变形细胞溶解物对LPS的反应,可通过比浊或显色方法进行定量,检测灵敏度可达皮克级别。
↘ 但需要注意:
血浆中存在多种干扰因素(如脂蛋白、蛋白结合等),可能影响检测结果,因此不同实验条件下数值存在一定变异。
不同研究中报告的参考范围有所差异,临床上更常用于相对变化和趋势评估,而非绝对诊断标准。血浆LPS检测可用于研究或特定情况下评估内毒素水平变化,但在实际应用中通常需要结合其他指标综合判断。
3
炎症标志物间接反映
脂多糖可激活免疫系统,诱导IL-6等炎症因子释放,进而促进肝脏合成CRP,因此CRP和IL-6可以反映炎症反应水平。但需要强调的是CRP和IL-6是非特异性炎症指标,会受到感染、损伤、慢性疾病等多种因素影响。
在排除急性感染等因素的前提下,CRP(尤其hs-CRP)可以作为低度慢性炎症的实用监测指标,用于跟踪干预效果,但不能单独用于判断LPS水平。
4
肠道菌群检测推断LPS产生潜力
通过16S rRNA或宏基因组测序分析肠道菌群组成,可以从菌群结构角度推断LPS产生潜力,用于评估风险趋势。
LPS来源于革兰氏阴性菌,因此其相对丰度与LPS产生潜力相关,但这种关系是间接的、受多因素影响。
例如:
拟杆菌门虽为革兰阴性,但其LPS炎症活性差异较大,并非全部强致炎;
菌群功能、宿主状态、屏障功能等都会影响最终LPS水平;
菌群检测更适合用于风险评估和干预方向指导,而不是直接判断LPS实际水平。
5
谷禾健康肠道菌群检测评估LPS相关风险
谷禾报告对很多菌群多指标整合分析,可以更系统地评估LPS相关风险趋势,用于指导个体化干预。我们方法的价值在于从菌群结构和功能角度提供参考信息,但仍需结合临床指标(如炎症指标、代谢指标)综合判断。
↘ 高LPS风险的菌群特征
谷禾报告中高LPS风险通常表现为革兰氏阴性菌相对丰度偏高,伴随促炎菌增加、短链脂肪酸产生菌减少及综合评分升高,提示较高的LPS产生与炎症潜力。
典型特征包括:
•革兰氏阴性菌相对丰度升高→LPS来源增加(潜力增加);
•B/F比值升高→菌群结构向阴性菌倾斜;
•促炎革兰氏阴性菌增加→LPS炎症活性增强;
•短链脂肪酸产生菌减少→抗炎能力下降+屏障支持减弱;
•综合风险评分偏高→多因素叠加的结果。
这些特征反映的是LPS相关风险升高的趋势,提示需要从菌群和生活方式层面进行干预。
↘ 低LPS风险的菌群特征
低LPS风险通常表现为菌群结构相对平衡、短链脂肪酸产生能力良好、综合评分较低,提示LPS产生和炎症风险较低。
主要特征包括:
•革兰氏阴性菌比例在合理范围→LPS来源稳定;
•B/F比值平衡→菌群结构稳定;
•促炎致病菌未见明显富集→炎症驱动较低;
•短链脂肪酸产生菌充足→有助于抗炎和维持屏障;
•综合评分低→整体风险较低
低风险代表当前状态良好,但仍需维持健康饮食和生活方式以保持稳定。
6
结合肠道通透性综合评估LPS
LPS风险评估需要同时考虑“产生”和”吸收”两方面,即菌群结构与肠道通透性共同决定循环LPS水平。
关键理解三种情况:
•屏障受损(肠漏)时→即使LPS产生不高,也可能吸收增加;
•屏障正常时→即使LPS产生略高,进入血液的量可能有限;
•两者同时异常→LPS升高风险最大。
综合评估可以区分问题来源(“产多了”还是”漏多了”),从而更有针对性地干预,例如侧重调菌或修复屏障。
♥
1
修复肠道屏障—减少LPS吸收的根本措施
无论LPS升高的原因如何,修复肠道屏障是降低其进入循环的根本措施。完整的肠道屏障通过维持上皮细胞间的紧密连接,阻止LPS经细胞间隙进入血液;当屏障受损时,紧密连接蛋白减少、通透性增加,LPS更易入血。因此,恢复紧密连接完整性可有效减少LPS吸收并降低其循环水平。
①补充乳铁蛋白
乳铁蛋白是一种铁结合糖蛋白,可促进肠上皮细胞增殖与更新,维持紧密连接蛋白表达和屏障完整性,从而降低肠道通透性,减少LPS进入循环并降低其水平。
此外,乳铁蛋白具有特异性LPS结合结构域,能高亲和力结合并中和LPS,阻断其与免疫细胞膜TLR4受体结合,抑制下游炎症信号通路,减少促炎因子产生,从而直接抑制炎症反应。
②补充谷氨酰胺
肠上皮细胞更新迅速,需充足谷氨酰胺供能;其充足时可维持上皮更新和紧密连接蛋白合成,保持屏障完整与正常通透性,减少LPS经细胞间隙入血。
肠漏或屏障受损时,补充谷氨酰胺可促进上皮修复、增强紧密连接、降低通透性,从而减少LPS吸收,对此类人群尤为有益。
2
其他减少LPS吸收与转运的措施
①维生素D
可以上调紧密连接蛋白,降低肠道通透性,减少LPS跨上皮转运。
②改善便秘
缩短肠道转运时间,减少LPS在肠腔内累积及与肠壁接触时间,从而降低吸收机会。
③减轻压力+改善睡眠
通过降低HPA轴激活和皮质醇水平,改善紧密连接与肠屏障功能,减少LPS进入循环。
3
减少其他LPS产生来源
①治疗SIBO
小肠细菌过度生长增加潜在LPS来源,在肠屏障受损时更易转化为循环LPS,抗生素等针对性治疗可降低细菌负荷,从源头减少LPS产生。
②牙周治疗
牙周感染中革兰阴性菌持续释放LPS,可通过受损牙龈进入循环,系统治疗减少这一持续输入源。
4
控制每餐脂肪摄入
我们之前介绍过,吃完脂肪后,肠道合成乳糜微粒运输脂肪,LPS亲脂,结合到乳糜微粒上,随乳糜微粒进入淋巴循环,然后进入体循环,因此每餐脂肪越多,合成的乳糜微粒越多,进入循环的LPS越多,所以控制每餐脂肪量减少乳糜微粒,进而减少进入循环的LPS,降低循环LPS水平。
简单饮食调整:一餐不要吃太多脂肪,全天均匀分布脂肪,可以减少餐后LPS升高,减少累积炎症。
5
增加膳食纤维,黄酮、多酚等摄入
①芹菜素(apigenin)
芹菜素是一种广泛存在于温热带蔬果中的黄酮类化合物,在芹菜、洋甘菊和欧芹中含量较高,具有肠道靶向的抗炎作用。
其可调节肠道菌群、强化肠道屏障,从而减少LPS吸收;在高脂饮食诱导的肥胖模型中,补充芹菜素可显著降低全身LPS水平及相关低度炎症。
②菊粉(Inulin)
益生元纤维如菊粉可促进抑制LPS的共生菌生长,并增加短链脂肪酸(SCFAs)生成,从而修复肠黏膜。
在一项针对2型糖尿病女性的随机对照试验中,补充菊粉8周使血浆LPS较安慰剂下降27.9%,同时炎症因子和胰岛素抵抗显著改善。
这些效应主要归因于其对肠道菌群的调节和对紧密连接蛋白的增强,从而抑制LPS易位,表明膳食纤维是降低内毒素血症和炎症的可行策略。
③利福昔明
利福昔明是一种肠道特异性抗生素,针对革兰氏阳性和革兰氏阴性菌,且不进行全身吸收。通过重塑肠道微生物群,利福昔明可以降低内源性内毒素的产生。
在非酒精性脂肪性肝炎(NASH)患者中,28天的利福昔明治疗显著降低了血清内毒素水平(内毒素活性下降,P=0.03),同时肝酶和炎症标志物也有所改善。
④姜黄素
姜黄素可通过强化肠道屏障并解毒内毒素,减少LPS介导的炎症。在饮食诱导的代谢综合征啮齿动物模型中,姜黄素提高肠道碱性磷酸酶活性(去磷酸化并中和LPS),维持紧密连接结构,从而防止血浆LPS升高。
在高脂饮食的糖尿病大鼠中,姜黄素可使升高的血清LPS和TNF-α恢复至正常水平。这些作用与其重塑肠道菌群(增加有益菌)及直接抑制LPS诱导的肠道信号通路有关。初步人体研究亦支持其作为辅助干预,用于降低代谢疾病中的内毒素血症和炎症。
⑤膳食纤维的综合作用
膳食纤维不被人体消化,进入结肠后作为双歧杆菌、乳杆菌等革兰阳性有益菌的发酵底物,促进其增殖并相对降低革兰阴性菌比例,从而减少LPS生成。
有益菌发酵产生的短链脂肪酸具有抗炎作用并增强肠道屏障,降低LPS吸收,形成双重降LPS效应。其中丁酸盐既是能量来源,又可强化屏障功能,是膳食纤维降低LPS的重要机制之一。
6
调节肠道菌群,选择合适的益生菌
①从源头调整肠道菌群减少脂多糖产生
LPS由肠道革兰氏阴性菌产生;其比例升高会增加LPS生成。通过饮食调节促进革兰氏阳性有益菌生长、降低阴性菌比例,可从源头减少LPS产生并降低其循环水平,这种改变具有长期稳定性。
益生菌和益生元均有助于调节菌群,因此饮食干预是更为根本且可持续的策略。相比单纯依赖抗炎药物,从源头控制菌群更能长期维持LPS在正常水平,促进整体健康。
②施用以革兰氏阳性菌为主的益生菌
选择的益生菌应该以双歧杆菌、乳杆菌等革兰氏阳性菌为主,这些本身没有LPS细胞壁,不会给肠道额外增加LPS,同时抑制革兰氏阴性菌过度生长,减少总LPS产生。
避免选择含有大量革兰氏阴性菌的益生菌产品,这些会给肠道额外增加LPS,增加LPS负担,反而达不到降低LPS的效果。
7
抑制LPS诱导的炎症反应
①Omega-3脂肪酸
改变细胞膜结构与脂筏环境,影响TLR4信号复合体激活,并通过resolvins等介质促进炎症消退,从而减轻LPS炎症反应。
②小剂量阿司匹林
通过COX抑制及NF-κB通路调节发挥整体抗炎作用,同时抑制血小板聚集,降低LPS相关心血管风险(主要为系统性抗炎效应的一部分)。
8
减少脂肪组织LPS蓄积
LPS亲脂容易蓄积在脂肪组织中,脂肪组织质量越大,蓄积LPS总量越多,脂肪组织不断缓慢释放LPS进入循环,维持循环LPS持续低水平升高,减肥减少脂肪组织质量,LPS蓄积总量减少,释放进入循环的LPS减少,循环LPS水平下降。
减肥同时改善肠道菌群,减少革兰氏阴性菌比例,进一步减少LPS产生,多重机制共同降低循环LPS水平,因此肥胖症减少体重可以显著降低LPS水平。
9
不同LPS风险分层个体化干预策略
不同LPS风险程度需要不同强度干预策略,低风险维持健康生活方式,中风险饮食调整加针对性营养补充,高风险先排查继发性疾病针对性治疗,个体化干预更高效。
•低LPS风险:不需要特殊药物干预,维持低LPS饮食模式和健康生活方式,几个月复查一次即可,长期维持可以稳定保持低LPS。
•中度LPS风险:饮食调整基础上,针对性补充营养补充剂,比如益生元、益生菌、乳铁蛋白,根据是否存在屏障损伤选择修复屏障营养,3-6个月复查调整方案。
•显著LPS风险:先排查继发性疾病,常见继发性疾病是SIBO、牙周病、便秘,针对性治疗继发性疾病,治疗后继续生活方式维持。
根据风险分层选择不同干预强度,避免低风险过度干预浪费金钱扰乱菌群,高风险干预不足达不到效果,个体化分层干预平衡效果和安全性,更合理高效。
♥
很多人从未关注过脂多糖(LPS),也很少意识到它与常见慢性疾病之间的联系。通过本文的系统梳理,我们从结构、机制到疾病,再到干预路径,已经建立起一个相对完整的认知框架。
⑴脂多糖的基本特性
脂多糖由脂质A、核心多糖和O抗原构成,其中脂质A是主要的免疫活性结构。其物理化学性质较为稳定,一般灭菌手段难以完全去除其活性。
⑵代谢与清除路径
脂多糖主要来源于肠道菌群。正常情况下,肠道屏障可以限制其大量进入体内;进入循环后的LPS主要由肝脏清除。部分研究提示,脂肪组织在一定程度上也可参与LPS相关过程。
⑶致病机制
LPS可通过TLR4被机体识别,激活先天免疫系统,诱导TNF-α、IL-6等促炎因子释放。当这种激活处于长期低水平状态时,可对多组织产生慢性损伤。
⑷疾病关联
脂多糖升高与多种慢性疾病存在相关性,包括代谢综合征、2型糖尿病、非酒精性脂肪肝(NAFLD)、动脉粥样硬化、高血压、肥胖,以及部分神经系统疾病和自身免疫疾病等。但其作用通常是多因素共同参与的一部分。
⑸脂多糖升高的常见因素
高脂饮食、过量饮酒、吸烟、膳食纤维摄入不足、长期使用广谱抗生素,以及SIBO、牙周炎、便秘、慢性压力、睡眠不足和肥胖等,都可能通过不同路径影响脂多糖水平。
⑹检测与评估方式
脂多糖可以通过血浆检测进行评估,同时CRP等炎症指标可作为间接参考。结合肠道菌群检测,有助于从“产生—吸收—炎症反应”多个维度综合评估风险。
⑺干预基本原则
围绕三个核心方向展开:
•减少产生(调节菌群);
•减少吸收(修复肠道屏障);
•减少转运与炎症反应(饮食与系统状态调节)。
同时,对于SIBO、牙周炎等明确来源,应进行针对性处理,并结合个体情况进行分层干预。
脂多糖(LPS)是革兰氏阴性菌细胞壁的特有成分。在正常生理状态下,少量脂多糖进入循环,有助于维持基础免疫激活。
但当多种因素叠加,导致循环中LPS长期处于偏高水平时,机体会进入一种“慢性低度炎症状态”,这一状态被认为与多种慢性疾病的发生发展密切相关。因此,长期维持LPS在合理范围内,是慢性疾病预防与管理中一个值得关注的重要环节。
主要参考文献
Wassenaar TM, Zimmermann K. Lipopolysaccharides in Food, Food Supplements, and Probiotics: Should We be Worried? Eur J Microbiol Immunol (Bp). 2018 Aug 21;8(3):63-69.
Kim HS, Kim S, Shin SJ, Park YH, Nam Y, Kim CW, Lee KW, Kim SM, Jung ID, Yang HD, Park YM, Moon M. Gram-negative bacteria and their lipopolysaccharides in Alzheimer’s disease: pathologic roles and therapeutic implications. Transl Neurodegener. 2021 Dec 7;10(1):49.
Magdaleno F, Blajszczak CC, Nieto N. Key events participating in the pathogenesis of alcoholic liver disease. Biomolecules. 2017;7:pii:E9.28134813.
Gomes JM, Costa JA, Alfenas RC. Metabolic endotoxemia and diabetes mellitus: A systematic review. Metabolism. 2017;68:133–44.
Roth J, Blatteis CM. Mechanisms of fever production and lysis: lessons from experimental LPS fever. Compr Physiol. 2014;4:1563–604.
Tume R, El Sherbiny S, Bono R, Gautier T, Pais de Barros JP and Meroño T (2025) The balance between proinflammatory, “bad”, and immunomodulatory, “good”, lipopolysaccharide for understanding gut-derived systemic inflammation. Front. Immunol. 16:1588129.
Catorce MN, Gevorkian G. LPS-induced murine neuroinflammation model: main features and suitability for pre-clinical assessment of nutraceuticals. Curr Neuropharmacol. 2016
Laugerette F, Vors C, Peretti N, Michalski MC. Complex links between dietary lipids, endogenous endotoxins and metabolic inflammation. Biochimie. 2011;93:39–45
Neyen C, Lemaitre B. Sensing Gram-negative bacteria: a phylogenetic perspective. Curr Opin Immunol. 2016;38:8–17.
Candelli, M.; Franza, L.; Pignataro, G.; Ojetti, V.; Covino, M.; Piccioni, A.; Gasbarrini, A.; Franceschi, F. Interaction between Lipopolysaccharide and Gut Microbiota in Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2021, 22, 6242.
Szentirmai, É., Buckley, K., Massie, A.R. et al. Lipopolysaccharide-mediated effects of the microbiota on sleep and body temperature. Sci Rep 14, 27378 (2024).
Citronberg JS, Curtis KR, White E, Newcomb PA, Newton K, Atkinson C, Song X, Lampe JW, Hullar MA. Association of gut microbial communities with plasma lipopolysaccharide-binding protein (LBP) in premenopausal women. ISME J. 2018 Jun;12(7):1631-1641.
Lin TL, Shu CC, Chen YM, Lu JJ, Wu TS, Lai WF, Tzeng CM, Lai HC, Lu CC. Like Cures Like: Pharmacological Activity of Anti-Inflammatory Lipopolysaccharides From Gut Microbiome. Front Pharmacol. 2020 Apr 30;11:554.
d’Hennezel E, Abubucker S, Murphy LO, Cullen TW. Total Lipopolysaccharide from the Human Gut Microbiome Silences Toll-Like Receptor Signaling. mSystems. 2017 Nov 14;2(6):e00046-17.
Apigenin Alleviates Obesity-Associated Metabolic Syndrome by Regulating the Composition of the Gut Microbiome.. Qiao Y, Zhang Z, Zhai Y, Yan X, Zhou W, Liu H, Guan L and Peng L. (Front. Microbiol. 12:805827. (2022))
Orange juice neutralizes the proinflammatory effect of a high-fat, high-carbohydrate meal and prevents endotoxin increase and Toll-like receptor expression.. Ghanim H, Sia CL, Upadhyay M, Korzeniewski K, Viswanathan P, Abuaysheh S, Mohanty P, Dandona P.. (Am J Clin Nutr. 2010 Apr;91(4):940-9.)
Oral spore-based probiotic supplementation was associated with reduced incidence of post-prandial dietary endotoxin, triglycerides, and disease risk biomarkers.. McFarlin BK, Henning AL, Bowman EM, Gary MA, Carbajal KM.. (World Journal of Gastrointestinal Pathophysiology. 2017;8(3):117-117.)
Effect of Probiotic Supplementation on Intestinal Permeability in Overweight and Obesity: A Systematic Review of Randomized Controlled Trials and Animal Studies.. DiMattia Z, Damani JJ, Van Syoc E, Rogers CJ.. (Adv Nutr. 2024 Jan;15(1):100162.)
Inulin controls inflammation and metabolic endotoxemia in women with type 2 diabetes mellitus: a randomized-controlled clinical trial.. Dehghan P, Gargari BP, Jafar-Abadi MA, Aliasgharzadeh A.. (Int J Food Sci Nutr. 2014 Feb;65(1):117-23.)
Efficacy of rifaximin on circulating endotoxins and cytokines in patients with nonalcoholic fatty liver disease.. Gangarapu, Venkatanarayanaa; Ince, Ali Tüzüna; Baysal, Birola; Kayar, Yusufa; Kiliç, Ulkanb; Gök, Özlemb; Uysal, Ömerc; Şenturk, Hakana.. (European Journal of Gastroenterology & Hepatology 27(7):p 840-845, July 2015.)
Rifaximin: beyond the traditional antibiotic activity.. Calanni, F., Renzulli, C., Barbanti, M. et al.. (J Antibiot 67, 667–670 (2014).)
Improvement of intestinal barrier function, gut microbiota, and metabolic endotoxemia in type 2 diabetes rats by curcumin.. Huang J, Guan B, Lin L, Wang Y.. (Bioengineered. 2021 Dec;12(2):11947-11958.)