-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康
编辑
在全球范围内,乳腺癌已成为女性发病率最高的恶性肿瘤,同时也是导致全球癌症相关死亡的第四大原因之一,严重威胁着女性的生命健康。为了最大程度降低复发风险、延长生存期,辅助化疗几乎是绝大多数乳腺癌患者的必经之路。然而,化疗在带来生存希望的同时,也常常伴随着一系列令人痛苦的副作用。其中,癌症相关疲劳便是最为常见、也最容易被忽视的问题之一。
对于许多乳腺癌患者来说,化疗的艰辛不仅在于治疗本身,相比于脱发、恶心等常见副作用,更在于一种挥之不去的、深入骨髓的疲惫感。这种疲惫并非休息一下就能缓解,它有一个专业名称——癌症相关疲劳(Cancer-Related Fatigue,简称 CRF)。它正悄然影响着无数患者的生活质量,甚至可能比疾病本身更令人困扰。
很多人会觉得,“化疗累一点不是很正常吗?休息休息就好了。” 但事实上,癌症相关疲劳绝非简单的身体劳累,它有着明确的医学定义。
美国国家综合癌症网络(NCCN)将其描述为一种持续存在、令人痛苦的身体、情感和 / 或认知层面的疲惫感,这种疲惫与近期的活动量不成比,会严重干扰正常生活,比如无法完成家务、难以专注交流、甚至连起床都觉得费力。
更关键的是,这种疲劳很难通过常规休息缓解。哪怕患者睡足 10 小时,依然会觉得浑身无力、精神涣散,这种持续性的消耗,比身体上的疼痛更易击垮患者的心理防线。
研究数据显示,超过90%的化疗患者会经历癌症相关疲劳,而在乳腺癌患者群体中,由于常需要接受多种治疗手段(如手术、放化疗、内分泌治疗等),疲劳的发生率和严重程度更高。高达30%的乳腺癌幸存者在治疗结束后数年,仍被重度疲劳所困扰。
这种长期疲惫不仅限制了她们的身体活动能力,还阻碍了重返工作岗位的步伐,对心理和社交生活造成了深远影响。癌症相关疲劳的危害远不止于主观感受上的不适。研究证实,疲劳的严重程度与患者的生活质量呈显著负相关,甚至可能影响生存结局。
尽管如此,目前医学界对癌症相关疲劳的内在发生机制仍知之甚少,这也导致临床上缺乏足够有效的、基于循证医学的干预手段。传统的治疗手段,如睡眠指导、运动疗法、心理干预等,虽然有一定帮助,但对于中重度疲劳的患者往往效果有限。这也使得寻找有效的治疗靶点、开发针对性干预措施变得尤为迫切。
随着人体“第二基因组”——肠道菌群研究的深入,科学家们开始将目光投向了我们的肠道。肠道中庞大的微生物生态系统不仅在消化吸收中发挥重要作用,更是人体最大的免疫器官,并通过复杂的神经-免疫-内分泌网络与大脑进行双向沟通,形成所谓的”肠-脑轴”。越来越多证据表明,肠道菌群可能在癌因性疲劳的发生发展中扮演关键角色。
化疗药物在杀死癌细胞的同时,往往也会攻击肠道中的有益菌群,导致菌群结构失衡。当肠道菌群失去平衡,肠道这道天然屏障就会受损出现“肠漏”,原本被“关”在肠道内的微生物产物和某些神经活性代谢物质,就可能进入血液循环。这些物质会沿着“肠-脑轴”激活神经、免疫和内分泌等多条通路,促使体内释放出一系列炎性因子和活性物质(比如IL-6、脂多糖等)。这些物质可能引发神经炎症,对中枢神经系统产生影响,最终表现为一种难以缓解的疲惫感。与此同时,这种内在变化还可能影响患者的行为,比如让人不自觉地减少活动、不愿社交,从而进一步加重疲劳的恶性循环。
2026年,发表在国际知名肿瘤学期刊《Frontiers in Oncology》上的一项最新前瞻性队列研究,该研究不仅证实了肠道菌群在化疗疲劳中的核心作用,最新的研究发现,我们或许可以通过分析患者化疗前的肠道菌群特征,提前预测她们在治疗过程中可能出现疲劳的风险。这为早期干预、预防严重疲劳的发生开辟了全新的思路。通过该研究深入探讨肠道菌群如何成为预测化疗疲劳的”生物密码”。
编辑
为了探究肠道菌群与癌因性疲劳之间的真实因果与预测关系,苏州大学的研究团队设计了一项严谨的前瞻性队列研究。与以往只能观察“当前状态”的横断面研究不同,前瞻性研究的特点在于它能预见未来,通过在事件发生前就开始收集数据,而不是回顾过去发生的事情。这样的设计能够避免回忆偏差,更准确地揭示因果关系和预测模型。
研究团队在2021年期间,从两家三甲医院招募了100名即将首次接受辅助化疗(共四个周期)的女性乳腺癌患者。为了确保研究的纯粹性,采用严格的入组标准:患者在入组前均没有精神疾病、胃肠道疾病、血液或内分泌系统疾病,且在过去三个月内未使用过抗生素或益生菌。这些严格的排除标准,旨在最大程度地保证所采集到的肠道菌群样本能反映其“原生态”,排除其他疾病或药物的干扰。所有患者均在开始辅助化疗之前入组,确保可以获得她们的”基线数据“——也就是在化疗前还未对肠道菌群产生影响的状态。采用配对粪便和血液样本,以探索潜在的神经免疫-内分泌途径。
为什么特别关注“第三个化疗周期”?
临床经验和既往纵向研究表明,接受四个周期化疗的乳腺癌患者,其疲劳感往往在第三个周期达到顶峰。此时,化疗药物的累积毒性最大,患者的生理和心理承受力往往处于临界点。
如果在这个阶段出现中重度疲劳,极易导致化疗减量甚至中断,直接影响患者的生存预后。因此,精准预测并干预第三周期的疲劳,具有极高的临床价值。
研究人员在患者化疗前(基线)收集了她们的粪便样本、血液样本,并进行了详细的疲劳量表评估(包括癌症疲劳量表CFS和视觉模拟疲劳量表VAFS)。
1
癌症疲劳量表
癌症疲劳量表(Cancer Fatigue Scale, CFS)由日本学者Okuyama于2000年开发的,广泛用于评估癌症患者疲劳症状,该量表包括躯体疲劳、情绪疲劳、认知疲劳三个维度,共15个条目。每个项目均采用5分制李克特量表(1-5)进行评分,总分范围为0-60。分数越高,表示疲劳越严重。
2
视觉模拟疲劳量表
视觉模拟疲劳量表(Visual Analog Fatigue Scale, VAFS)用于使用具有11个等级的标尺评估患者的疲劳水平,从“我不觉得累”到“我觉得筋疲力尽”。VAFS采用0-10的数值评分。
0表示无疲劳,1-3表示轻度疲劳,4-6表示中度疲劳,7-10表示重度疲劳。
随后,在患者进行到第三个化疗周期时,再次进行随访评估。根据第三周期的VAFS评分,患者被分为两组:
16S rDNA测序和微生物多样性数据分析
该项目所有患者的粪便样本,使用杭州谷禾提供的专用常温保存管进行采集和储存,并在谷禾实验室完成了16S扩增子测序及生物信息分析。
样本采用专用的DNA提取试剂盒(GHFDE100 DNA isolation kit)从粪便样本中提取细菌总基因组DNA,对16S rDNA基因的V4高变区进行PCR扩增。采用Illumina NovaSeq 6000高通量测序平台测序,结合生物信息学分析流程(如VSearch聚类、SILVA数据库注释等),计算得到从门到属各个分类层级的微生物相对丰度。
16S扩增子测序的临床转化价值
在众多微生物组学技术中,16S扩增子测序在临床队列探索中具有不可替代的价值。其优势主要体现在:
研究结果发现:
基线时,Y0组和Y1组在躯体疲劳、认知疲劳、疲劳总评分(CFS评分)和疲劳程度(VAFS评分)方面无显著差异(P >0.05)。
然而,在第三个化疗周期,观察到两组之间在身体疲劳、情绪疲劳、疲劳总评分和疲劳程度方面存在显著差异(P< 0.05),Y1组的所有评分均高于Y0组。而在化疗开始前,那些后来在第三周期发展为中重度疲劳(Y1组)的患者,其肠道菌群结构已经与轻度疲劳(Y0组)患者出现了显著差异。这表明,肠道菌群并非在化疗后才被动改变,而是在治疗开始前就可能决定了个体对化疗毒副反应的易感性。
1. 菌群失衡的早期信号:
B/F比值升高与变形菌门增殖
在门水平上,肠道菌群主要由拟杆菌门 、厚壁菌门 、变形菌门、梭杆菌门 (Fusobacteria) 构成,这四大菌门的总和占比均超过了菌群总数的95%。进一步研究发现,中重度疲劳组患者在化疗前的拟杆菌门/厚壁菌门比值(B/F比值)显著高于轻度疲劳组(1.15 vs 1.03, P = 0.043)。
注:B/F比值长期以来被视为衡量肠道菌群健康状况的宏观指标之一,其升高通常与肥胖、炎症性肠病等多种代谢和炎症性疾病相关,暗示着肠道微生态的失衡。
更值得注意的是,中重度疲劳组患者肠道内的变形菌门丰度显著增加(8.03% vs 5.01%, P = 0.039)。变形菌门被誉为“微生物界的杂草”,其在肠道中的过度扩张是菌群失调的典型标志。变形菌门下包含了大量潜在的致病菌,在大肠杆菌、沙门氏菌、幽门螺杆菌等常见致病菌中都占主导地位,它们大多属于革兰氏阴性菌,其细胞壁外膜富含脂多糖(LPS,即内毒素)。
注:LPS是引发人体全身性炎症反应的强烈触发器,其水平的升高意味着肠道内存在一个潜在的“炎症火药库”。
在菌群多样性方面,两组之间的α多样性无显著差异(Shannon指数P = 0.505,Simpson指数P = 0.820,Chao1指数P = 0.326);β多样性分析也显示两组菌群结构整体相似(ANOSIM及PERMANOVA分析P值均大于0.05),主坐标分析结果见图1。
图1:两组患者肠道菌群的α多样性与β多样性分析
编辑
虽然整体多样性指数(如Shannon, Simpson, Chao1)无显著差异,但PCoA图显示两组样本在空间分布上略有分离趋势,预示着特定菌群的丰度已悄然改变。
2. 产短链脂肪酸菌匮乏
通过LEfSe(线性判别分析效应量)深度挖掘,研究人员发现,轻度疲劳组(Y0组)患者肠道内富含考拉杆菌属(Phascolarctobacterium)和瘤胃菌科(Ruminococcaceae)等有益菌。这些细菌是肠道内著名的产短链脂肪酸菌(尤其是丁酸)。
注:丁酸不仅是肠道黏膜细胞的主要能量来源,能维护肠道屏障的完整性,还能穿透血脑屏障,发挥强大的抗炎和神经保护作用。
相反,中重度疲劳组(Y1组)患者体内这些“护城河”细菌的丰度显著降低,取而代之的是韦荣球菌属(Veillonella)、巨球型菌属(Megasphaera)以及肠杆菌目(Enterobacteriales)等条件致病菌的增多。这些菌群的富集,进一步加剧了肠道微环境的促炎状态(图2)。
图2:LEfSe分析揭示了两组患者在化疗前肠道菌群的特征性差异
编辑
红色代表中重度疲劳组(Y1)富集的菌群(如变形菌门下的γ-变形菌纲、肠杆菌目、肠杆菌科),绿色代表轻度疲劳组(Y0)富集的菌群(如瘤胃菌科下的RFN20、考拉杆菌属)。
3. 构建高精度预测模型:AUC高达0.82
基于上述发现,研究团队将基线肠道菌群特征与临床协变量结合,构建了一个多因素逻辑回归预测模型。这个模型的强大之处在于,它不仅仅依赖于单一指标,而是综合了多个维度的信息,模拟了临床决策的复杂性。
模型变量的协同解读
最终进入模型的变量包括:
◐ 菌 群 变 量
◐ 临 床 变 量
◐ 其 他 临 床 协 变 量
包括年龄、BMI、癌症分期等,尽管这些因素在单变量分析中可能与疲劳有关,但在加入了强大的菌群变量后,它们的独立预测能力被削弱。这反过来证明,肠道菌群可能是连接这些临床因素与疲劳结果的更底层的生物学中介。
结果显示,该模型预测患者在第三周期是否会发生中重度疲劳的准确率(AUC值)达到了0.82(95% CI: 0.73 – 0.90, P < 0.001)。 AUC值是衡量模型预测能力的标准,0.82意味着该模型具有“良好”到“优秀”的区分能力。
在最佳截断值下,该模型的敏感性为82.8%(能正确识别出82.8%的未来高疲劳患者),特异性为71.4%(能正确识别出71.4%的未来低疲劳患者)。这意味着,通过化疗前的一次粪便16S测序,结合简单的疼痛评估,医生就能以较高的准确率找出那些容易在化疗中严重疲劳症的高危患者,从而提前进行干预。
为了弄清肠道里的细菌究竟是如何影响大脑产生疲劳感,研究人员从100人的大队列中随机抽取了13名患者组成子队列:CRF无显著变化组(n=6)和CRF增加组(n=7)。
先导队列(n = 13)与整体队列(n = 100)在社会人口学或临床特征上无显著差异(均为 P > 0.05),验证了随机抽样子样本的代表性。对比了她们化疗前后肠道菌群和血液中神经免疫内分泌指标的动态变化。结果揭示了一条清晰的“肠-脑轴”致病链。
1
屏障破损:产丁酸菌Blautia减少与LPS泄漏
在化疗过程中,随着疲劳感加剧的患者,其肠道内的厚壁菌门,特别是经黏液真杆菌属(Blautia)的丰度出现了显著下降(P = 0.011)。Blautia是肠道内极其重要的产丁酸菌。当它减少时,肠道内短链脂肪酸(SCFAs)的产量锐减。这不仅导致肠道黏膜细胞失去营养支持,发生肠漏,还使得变形菌门产生的内毒素(LPS)趁机大量进入血液循环。相关性分析也证实,在化疗前,Blautia的丰度与血清LPS浓度呈显著负相关(图3)。
图3:肠道菌群与神经免疫内分泌指标
及疲劳评分的相关性热图
编辑
化疗前(a),Blautia与BDNF呈显著正相关,与疲劳评分(CRF)和LPS呈显著负相关。而巨球型菌属(Megasphaera)则与CRF、IL-6和NGF呈正相关,清晰地展示了“好菌”与“坏菌”的不同角色。
2
神经炎症:LPS如何抑制“大脑肥料”BDNF?
——LPS引发的免疫反应与神经炎症
进入血液的LPS会激活免疫系统(如TLR4受体),引发全身性的炎症反应,导致白细胞介素-6(IL-6)等促炎细胞因子释放。这些炎症因子和LPS能够穿透或破坏血脑屏障,引起中枢神经系统的神经炎症。
已有研究表明,在神经炎症模型中,LPS会激活大脑中的小胶质细胞。被激活的小胶质细胞会释放大量炎症介质,同时上调组蛋白去乙酰化酶2(HDAC2)的表达。HDAC2的过度活跃会抑制BDNF基因的转录,从而导致BDNF蛋白合成减少。
简而言之,LPS就像一个信号,告诉大脑的免疫系统进入战备状态,而这种“战备状态”的代价之一,就是抑制了用于日常维护和修复大脑的“肥料”(BDNF)的生产。
——BDNF浓度下降与疲劳的关系
研究还发现:疲劳加剧的患者,血清中的脑源性神经营养因子(BDNF)浓度显著下降(P = 0.035)。BDNF被誉为“大脑的肥料”,是调节神经元能量代谢、促进神经可塑性和维持情绪稳定的核心物质。其水平的下降,直接与抑郁、认知障碍和疲劳感相关。这一发现也与以往“肠道菌群影响神经营养信号”的研究结论一致。
——肠道菌群失调对疲劳的影响
结合研究结果,为我们可以清晰理解肠道菌群通过肠道-脑轴影响癌因性疲乏的机制提供了线索。
本研究中观察到的血清脑源性神经营养因子BDNF水平下降,形成炎症与神经营养支持不足之间的恶性循环。很可能是短链脂肪酸减少与全身炎症增强共同作用的关键结果,而BNDF作为神经元能量代谢与可塑性的核心调控因子,其水平降低可能直接导致中枢性疲劳的发生(图4)。
图4:肠道菌群通过“肠-脑轴”影响癌因性疲劳的机制图
编辑
化疗导致菌群失调(产SCFA菌↓,革兰氏阴性菌↑) → 肠道屏障受损,LPS入血 → 激活TLR4等通路,引发全身炎症(IL-6↑) → 炎症信号和LPS穿过血脑屏障,抑制大脑BDNF表达(BDNF↓) → 最终导致中枢性疲劳(CRF↑)。
✍ 科普延伸:与“慢性疲劳综合征”异曲同工
其实,不仅是癌症化疗患者,在普通的肌痛性脑脊髓炎/慢性疲劳综合征(ME/CFS)患者中,科学家也观察到了极其相似的菌群特征。
一篇于2025年发表在《Journal of Translational Medicine》的综述指出,ME/CFS患者普遍存在肠道菌群失调,其核心特征同样是促炎类群(如变形菌门)增加,而产短链脂肪酸的有益菌(如粪杆菌、罗斯氏菌)减少。
这种跨疾病的共性,强有力地印证了本研究的结论:化疗作为一种强烈的外部冲击,诱导或加剧了与慢性疲劳疾病相似的肠道微生态失衡模式,最终通过“肠-脑轴”的神经炎症通路,在大脑中“投射”出了难以克服的疲劳感。
这项研究的重要价值在于,癌因性疲劳本是一种主观的、难以评估的症状,通过早期肠道菌群检测结合疲劳量表评估和相关血液标志物检测,可以为临床医生和患者提供了早期干预和防控的可能性。
总体而言,苏州大学的这项研究不仅为我们描绘了肠道菌群、神经免疫内分泌系统与化疗疲劳之间的复杂网络,更进一步证实并拓展了以往的研究结论:化疗前的肠道菌群特征,有望成为预测乳腺癌患者化疗期间CRF的生物标志物,例如,通过化疗前肠道菌群检测,评估其B/F比值、变形菌门丰度及产丁酸菌的比例,将可能成为由化疗引起的癌因性疲劳的重要评估预测指标。医生可以提前介入,给予更积极、更具针对性的支持性辅助干预。
通过监测血液标志物,来动态评估干预效果。血清BDNF水平的下降与疲劳加重密切相关。在临床实践中,血清BDNF可以作为一种便捷的血液生物标志物,用于动态追踪患者的疲劳进展,为干预措施的有效性提供有力证据。
我们可以从以下几个维度构建抗疲劳防线:
肠道菌群,作为连接化疗药物、宿主免疫和中枢神经系统的关键枢纽,未来有望成为肿瘤支持治疗的核心关注点。随着16S rDNA测序等高通量微生物组学技术的普及与成本降低,个体化肠道菌群检测或将成为肿瘤精准治疗不可或缺的一环。
注:本账号内容仅供学习和交流,不构成任何形式的医疗建议。
谷禾健康
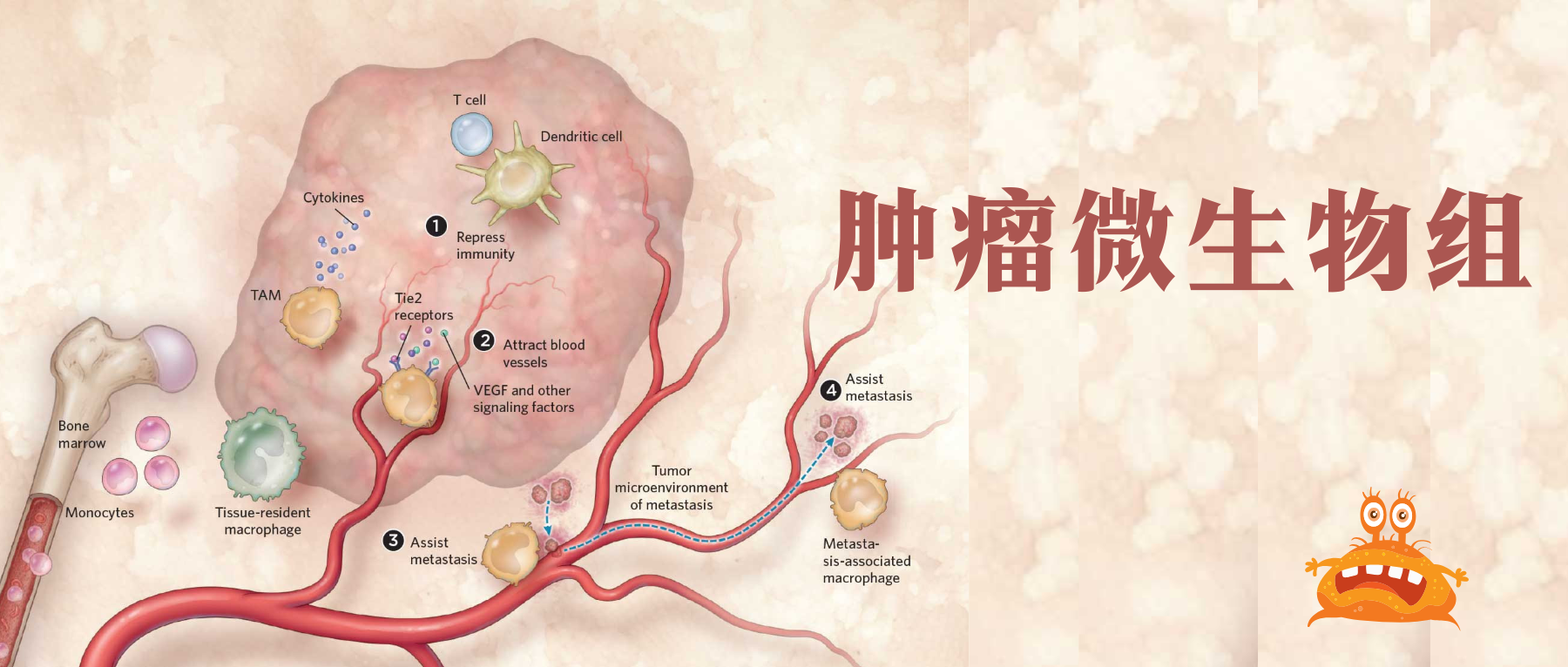
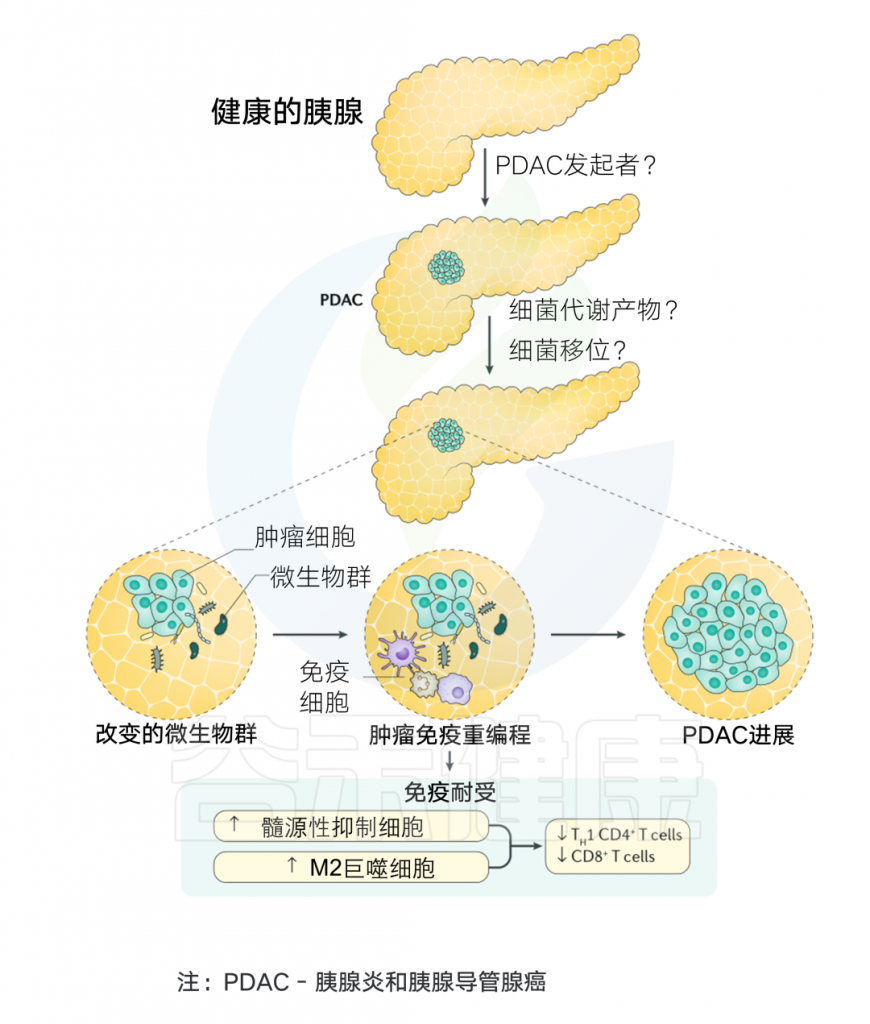
通常认为人类体内微生物群存在于与外部环境接触的体腔和器官中,例如胃肠道、皮肤、口腔、上呼吸道(尤其是鼻咽)和泌尿生殖道。然而在过去十年中,越来越多的研究确定了肿瘤内存在着低生物量而独特的微生物群落(瘤内微生物群)。
在肿瘤内,细菌、真菌、病毒和古菌等微生物形成了肿瘤微生物群,主要存在于肿瘤细胞、免疫细胞和细胞间基质中。这些微生物群落构成了肿瘤微环境(TME)的一部分,影响免疫调节、炎症和代谢控制等过程,这些过程与肿瘤的发生和进展密切相关。迄今为止报道的证据表明,肿瘤微生物组存在于33种主要癌症类型中。
大量证据表明,多达20%的癌症与微生物感染有着错综复杂的联系。最初在伯基特淋巴瘤中发现了EB 病毒(EBV)。从那时起,病毒感染就被认为是导致人类癌症的重要因素,包括确定人乳头瘤病毒(HPV)是导致宫颈癌的原因,乙型肝炎病毒(HBV)是导致肝癌的原因,以及人T细胞嗜淋巴细胞病毒是导致白血病和淋巴瘤的主要原因。
伯基特淋巴瘤(Burkitt淋巴瘤)是儿童恶性淋巴瘤中最常见的类型,起病急,进展快,侵袭强和恶性度高。1964年首先从非洲儿童Burkitt淋巴组织中分离出EB病毒。
1982年,就在胃中发现幽门螺杆菌(Helicobacter pylori)证实了其作为消化性溃疡和胃癌病原体的作用。2020年,对来自七种常见实体瘤(包括乳腺癌、肺癌、卵巢癌、胰腺癌、黑色素瘤、骨癌和脑肿瘤)的超过1526个样本进行了分析,发现大多数实体瘤含有细菌,其中许多是存活在癌细胞内的细胞内细菌。随后,在各种人类癌症标本中也发现了真菌,包括胰腺癌、肺腺癌和结直肠癌。
本文介绍了肿瘤微生物组的相关概念、来源和特征及检测方法。然后,我们描述了常见肿瘤中肿瘤微生物组的组成及其在塑造肿瘤微环境中的作用。肿瘤微生物组和免疫系统之间的相互作用可以调节宿主的抗肿瘤免疫,影响肿瘤治疗的疗效。理解肿瘤微生物组与肿瘤之间的相互关系,有助于为肿瘤的早期诊断和治疗带来新的突破。
▸ 肿瘤微生物的定义
“肿瘤微生物”,其定义为在肿瘤组织中发现的可影响癌症易感性和治疗反应的微生物。国际癌症登记协会(IARC)将11种微生物归类为人类致癌物。
其中包括7种病毒,即乙型肝炎病毒(HBV)、丙型肝炎病毒、人类嗜T细胞病毒、人乳头瘤病毒(HPV)、EB病毒、卡波西肉瘤相关疱疹病毒(KSHV)和默克尔细胞多瘤病毒(MCV);三种寄生虫,即华支睾吸虫、埃及血吸虫和Clonorchis mukusicus;和一种细菌,幽门螺杆菌(Helicobacter pylori)。
术语“瘤内微生物群 ”和 “瘤内微生物组 ”经常被人们混用。在本文中,我们将前者定义为肿瘤组织内所有微生物(包括细菌、病毒、真菌、原生动物和古细菌)的集合,而后者包括肿瘤组织内所有微生物的基因组和产物,包括所有分泌毒素(如来自脆弱拟杆菌和具核梭杆菌的毒素)、代谢物(如短链脂肪酸)和氧化还原活性小分子(活性氧、活性氮)。
并且在本文的术语中,“肿瘤”主要是指癌症(恶性肿瘤)。尽管良性肿瘤(如子宫肌瘤)也含有微生物群,但关于良性肿瘤的微生物群和微生物组的研究较少。此外,良性肿瘤不具有转移特性,限制了对微生物组在转移中作用的探索。
▸ 肿瘤微生物的来源
肿瘤微生物群的来源可分为两类。第一类是“常驻”生物,它们栖息在人类生态位中,在正常情况下通常不会引起疾病。然而,在微生物组的整体扰动导致生态失衡(生态失调)之后,共生微生物群的比例被破坏,导致共生微生物易位,从而诱导肿瘤形成。
第二类由“外来入侵者”组成,例如致癌微生物和其他病原微生物。入侵的肿瘤微生物群可以根据入侵途径进一步分为三种类型。
(一)微生物通过粘膜屏障侵入
微生物群落栖息在人体的各种粘膜表面,形成不同的微生物组生态位。在健康状态下,微生物群保持动态平衡,调节免疫系统以抵抗外部病原体的定植,并抑制内源性微生物的致病潜力。
然而,微生物组成的破坏或这些群落的位移会导致生态位间微生物串扰,从而可能影响癌症进展。在肿瘤发生过程中,粘膜屏障的损伤(无论是由细菌还是其他因素引起)可能使粘膜微生物有机会侵入肿瘤组织,从而导致结直肠癌、胰腺癌、肺癌和宫颈癌等癌症的发展。
(二)源自邻近健康组织的微生物
一些研究发现,肿瘤组织的微生物群组成与邻近正常组织的组成非常相似。胰腺癌患者肿瘤组织和十二指肠组织之间的细菌DNA谱相似性表明胰腺组织中的细菌可能起源于十二指肠。
起源于口腔的具核梭杆菌(Fusobacterium nucleatum)可进入肠道并在结直肠癌内增殖,从而在肿瘤微环境(TME)内诱导免疫细胞激活。小鼠模型显示,具核梭杆菌通过将其粘附素Fap2与结直肠癌细胞上表达的糖分子Gal-GalNAc结合来定植结直肠癌组织,而Fap2缺陷细菌在这些小鼠中的定植受损。
同样,发现口服荧光标记的粪肠球菌(Enterococcus faecalis)从肠道转移到肿瘤外围,改变了肿瘤微生物组的基因组特征并调节免疫功能。
此外,与正常组织相比,肿瘤微环境(TME)的特点是免疫抑制和缺氧,这些条件更有利于微生物定植,因此假设肿瘤微生物群可能起源于正常组织。
(三)微生物通过血液迁移到肿瘤
血液是肿瘤微生物组的另一个潜在来源。由于肿瘤组织的丰富血管化,它们为微生物的生存和传播提供了合适的环境。红细胞在细菌易位中起着至关重要的作用,有助于免疫逃避。使用 16S rDNA 定量聚合酶链反应测定和 MiSeq 测序(16S 靶向宏基因组测序)证实了健康人血液中存在细菌,并且在红细胞中检测到的细菌 DNA 浓度高于血浆。
活细菌,如肺炎链球菌和金黄色葡萄球菌,已被证明存在于红细胞中。
源自口腔、肠道、呼吸道和其他区域的微生物可以通过血流运输到肿瘤部位,通过受损的血管浸润肿瘤。坏死肿瘤细胞碎片的趋化梯度可能引导从身体不同部位进入血液的微生物迁移到肿瘤。
例如,金黄色葡萄球菌(Staphylococcus aureus) 的肺部感染已被证明会影响乳腺癌转移。结肠中的大肠杆菌(Escherichia coli)在结直肠癌期间破坏肠道血管屏障,进入血液,随后定植于肝脏,诱导转移前生态位的形成并促进肝转移。
★ 不同类型的癌症微生物群组成不同
许多研究已经确定了肿瘤组织内微生物组的存在,并强调了癌症微生物群的异质性。癌症微生物群是多种多样的,癌症微生物群的组成在不同类型的癌症之间,甚至在同一癌症的不同病理亚型之间也不同。
例如厚壁菌门(Firmicutes)和拟杆菌门(Bacteroidetes)是结直肠癌中最丰富的物种,而变形菌门(Proteobacteria)在胰腺癌的微生物群中占主导地位。肝细胞癌富含变形菌门、拟杆菌门和Patescibacteria。
不同的癌症类型承载着不同的微环境,具有不同的氧分压、血管生成能力和周围组织的微生物群,这会影响肿瘤微生物群的组成并产生肿瘤类型的异质性。
不同肿瘤类型中微生物群的组成



Ma Y,et al.Microb Pathog.2024
★ 癌症不同阶段的微生物群也存在变化
除了类型异质性外,癌症微生物群还表现出位点异质性和分期异质性。前者是指肿瘤组织和非肿瘤组织之间微生物群组成的差异,主要包括特定个体微生物的存在或生态失调。肿瘤组织中的微生物群多样性通常低于非肿瘤组织,这可能与肿瘤微环境中特定微生物的选择性扩增有关。
后者是指癌症发展不同阶段肿瘤微生物群的变化。在口腔鳞状细胞癌的进化过程中,癌前阶段(癌前病变)显示链球菌(Streptococcus)和罗氏菌(Rothia)的高富集。而在晚期癌症中,Capnocytophaga在肿瘤组织中富集。
★ 癌症微生物组与宿主之间存在双向作用
肿瘤微生物组和宿主之间的相互作用是双向的。
这种影响主要体现在两个方面:
首先,细胞内和细胞外肿瘤环境之间的生化和生物差异与细胞内和细胞外肿瘤微生物组之间的功能变化有关。例如,某些细菌可以在癌细胞侵袭过程中调节RhoA-GTP酶-Rock-肌动蛋白细胞骨架重塑途径,从而促进携带细菌的癌细胞远处转移——这是细胞内肿瘤微生物组特有的作用。
其次,肿瘤微环境对肿瘤微生物组产生选择性影响。不同微环境中血管生成、氧水平、微生物来源、内吞作用和胞饮作用活性的变化导致肿瘤内的微生物组成不同。
让我们一起来看下不同癌症中的肿瘤微生物组与宿主之间可能存在的相互作用。
结直肠癌(CRC)是全球最常见的肿瘤之一,也是与肿瘤微生物组密切相关的肿瘤之一。
菌群失调可能是结直肠癌的诱因之一
动态平衡的微生物群对人类健康有益,但菌群失调可能导致结直肠炎症、炎症性肠病,甚至结直肠癌。
一项对423名I-IV期结直肠癌患者的肿瘤组织和正常粘膜进行 16S rRNA 测序的研究发现,与正常肠粘膜相比,结直肠癌组织的α多样性降低,β多样性增加。
变形菌门、梭菌门、弯曲杆菌门在肿瘤组织中的含量增加,而拟杆菌门、厚壁菌门、疣微菌门、放线菌门和古细菌减少。
有益菌和有害菌在结直肠癌环境中的作用
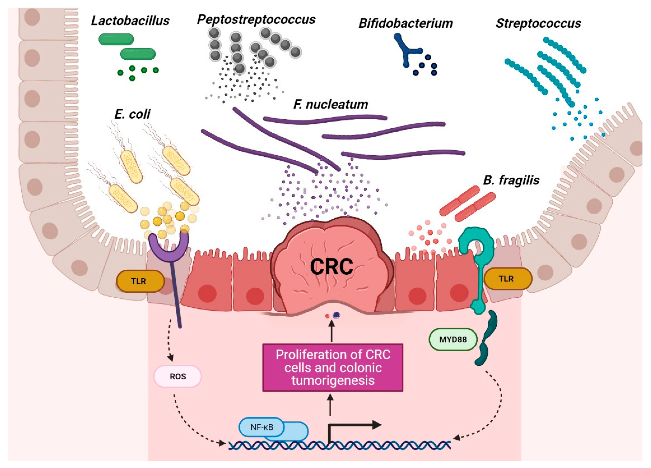
Torres-Maravilla,et al.Microorganisms.2021
产生毒素的特定细菌诱导肿瘤的进展
结直肠癌患者富含特定菌群,包括产生肠毒素的脆弱拟杆菌(B.fragilis)、肝螺杆菌(Helicobacter hepaticus)、败血梭菌(Clostridium septicum)、粪肠球菌(Enterococcus faecalis)、具核梭杆菌(F.nucleatum)、大肠杆菌、无乳链球菌和幽门螺杆菌。
这些细菌可通过释放破坏宿主细胞基因组的毒素,激活STAT3、NF-κB、Wnt和SREBP-2途径、诱导COX-2表达、与TRL2和TRL4相互作用、刺激促炎细胞因子(IL-1β、IL-6、IL-8、IL-17、TNF-α和IFN-γ)产生、调节NLRP3炎症体活性,通过氧化应激活性氧(ROS)和活性氮(RNS)DNA损伤来促进肿瘤中炎性微环境的形成并导致免疫逃逸来影响肿瘤的发展。
由于肿瘤粘膜微生态中的病原体网络与肿瘤突变和代谢特征相关,我们预计肿瘤微生物群组成的分析可以预测接受切除的结直肠癌患者的预后。
肺癌是常见的恶性肿瘤之一,肺癌的发生与下呼吸道以及口腔、鼻腔、胃肠道的微生物群密切相关。
肺癌患者中Gemmiger、Blautia等菌增加
普雷沃氏菌属(Prevotella)、链球菌属(Streptococcus)和韦荣氏球菌属(Veillonella)常见于口腔中,在肺癌中的含量增加。
并且与良性肺病患者相比,肺癌患者发现的菌群之间存在高度差异,Capnocytophaga、Sediminibacterium、吉米菌(Gemmiger)、Blautia和颤螺菌属(Oscillospira)这几个细菌相对丰富。
与肺癌可能相关的微生物
从全球角度来看,假单胞菌、链球菌、葡萄球菌、韦荣球菌属和莫拉克斯氏菌属经常被报道为与肺癌最相关的微生物群。

Liu NN,et al.NPJ Precis Oncol.2020
通过qPCR确定,二氧化碳嗜纤维菌(capnocytophaga)和韦荣氏球菌在肺癌患者的唾液样本中含量更高,而奈瑟菌属的丰度相对较低,它们有可能用作肺癌早期检测的生物标志物和微生物组治疗的靶标。
对30项相关研究的荟萃分析发现,肺部感染结核分枝杆菌、非结核分枝杆菌和其他病原体会增加患肺癌的风险。
胰腺癌是一种主要起源于胰腺导管上皮及腺泡细胞的恶性肿瘤,起病隐匿,早期诊断困难,进展迅速,生存时间短,是预后最差的恶性肿瘤之一,被称为“癌中之王”。
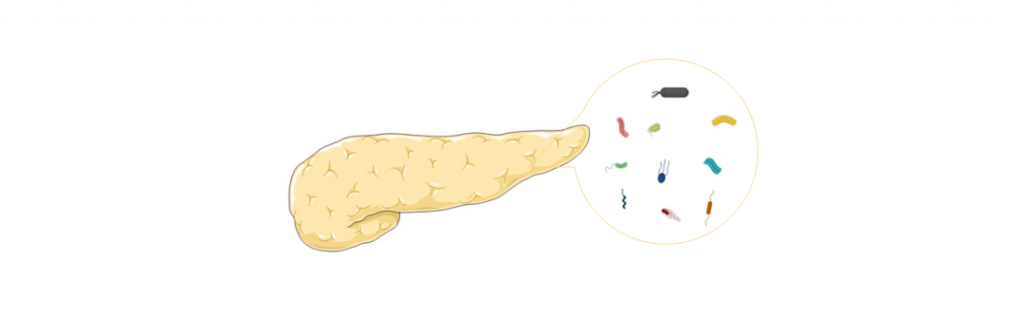
胰腺中存在细菌,胰腺癌患者细菌更多
胰腺以前被认为是一个完全无菌的器官。然而,在2017年,在人胰腺癌组织中发现了γ-变形杆菌。随后,其他研究通过 16S rRNA 测序、NGS 和免疫荧光鉴定了胰腺癌中的肠杆菌科、假单胞菌属和变形杆菌科等细菌。
用16S rRNA基因特异性PCR对胰腺囊肿标本进行分析,发现优势菌:氨基酸球菌属(Acidaminococcus),埃希氏菌属(Escherichia),拟杆菌属(Bacteroides),志贺氏菌(Shigella)。
最近,对胰腺癌样本进行的16S rRNA荧光探针和qPCR实验都证实,与正常人相比,胰腺癌患者中存在的细菌约为正常人的1000倍。
肿瘤微生物可引发炎症,促进胰腺癌变
此外,胰腺组织中的真菌组相比正常组织富集度增加了3000倍,主要成分为马拉色菌(Malassezia)。研究表明,马拉色菌属激活并结合甘露糖结合凝集素(MBL)蛋白,触发诱导胰腺癌的补体级联反应。
此外,肿瘤微生物组中的代谢物可以引发炎症和免疫抑制反应,并产生有利于肿瘤进展的免疫抑制微环境,促进胰腺癌变。
微生物组与胰腺癌发生的关系
Thomas R M, et al.Nature Reviews Gastroenterology & Hepatology.2019
乳腺癌是常见的癌症之一,也是全球女性癌症相关死亡的主要原因。
乳腺癌组织中的微生物稳态受到破坏
与正常乳腺组织相比,乳腺癌组织中的细菌 DNA 总水平较低,并且这些水平随着癌症的进展而持续下降,这表明癌症可能会破坏微生物组稳态。此外,已经确定肠道微生物群中预先存在的干扰增加了乳腺癌细胞转移,但还需要更多的研究来确定这些发现在临床环境中的相关性。
进一步表征乳腺组织中的正常微生物群和乳腺癌组织中的群落结构变化,可能会确定乳腺癌预防和诊断的新靶点。
阴道微生物组影响卵巢癌、子宫内膜癌和宫颈癌的发生和进展。研究表明,阴道细菌多样性的增加和乳杆菌丰度的降低可能导致持续的HPV感染。
阴道微生物组与HPV感染、宫颈上皮内瘤变相关
评估了250名女性的阴道菌群,证明了阴道微生物组、HPV感染与宫颈上皮内瘤变之间存在联系。阴道微生物群以阴道加德纳菌为主,其次是 Lactobacillus iners、Lactobacillus crispatus、Lactobacillus taiwanensis。
与健康女性相比,子宫肌瘤(UF)患者的宫颈和阴道微生物群相互作用和相对微生物丰度发生了改变。Erysipelatoclostridium、Mucispirillum、Finegoldia相对丰富,而Finegoldia的丰度降低,这表明子宫肌瘤患者可能存在宫颈和阴道微生物群的生态失调。
促炎微生物群与前列腺癌有关
前列腺癌是男性人群中常见的癌症,前列腺癌和非前列腺癌患者的尿液测序显示,促炎微生物群与泌尿生殖系统感染和前列腺癌有关。
研究报告了前列腺癌中促炎拟杆菌和链球菌丰度的显著差异,叶酸和精氨酸途径显著改变。对前列腺肿瘤微环境的分析显示,与非肿瘤组织相比,肿瘤/肿瘤周围组织中的葡萄球菌(Staphylococcus)明显更多,而丙酸菌属(Propionibacterium)在所有测试的肿瘤/肿瘤周围和非肿瘤组织中最为丰富。
病例对照研究还发现,良性对照受试者和前列腺癌男性的肠道微生物组的组成有很大不同,这可能适应前列腺癌的发病机制和对其危险因素的进一步研究。特别是,与对照组相比,前列腺癌病例中Bacteroides massiliensis的相对丰度较高,而对照组中普氏栖粪杆菌(Faecalibacterium prausnitzii)和直肠真杆菌(Eubacterium rectalie)的相对丰度较高。
超过700种不同的微生物定植于人类口腔,健康人的口腔微生物群保持相对稳定。而口腔癌患者的口腔微生物群发生了变化。
口腔癌患者丰度和多样性比正常人群更高
对121例患者的分析显示,与健康个体相比,口腔癌患者的戴阿利斯特杆菌属(Dialister)含量显著增加,放线菌属、乳酸菌属和链球菌属丰度显著降低。牙龈卟啉单胞菌和核镰刀菌等主要牙周病原菌的慢性感染可增强IL-6-STAT3 轴信号传导并诱导口腔鳞状细胞癌。
使用16S rDNA测序来表征口腔鳞状细胞癌(OSCC)组织的微生物群,肿瘤部位细菌的丰度和多样性显著高于来自同一患者的正常组织样本。在OSCC样本中检测到梭杆菌属、卟啉单胞菌属、消化链球菌科、Flavobacteriaceae、Prevotellaceae和Campylobacteraceae,可能是诊断标志物和治疗靶点。
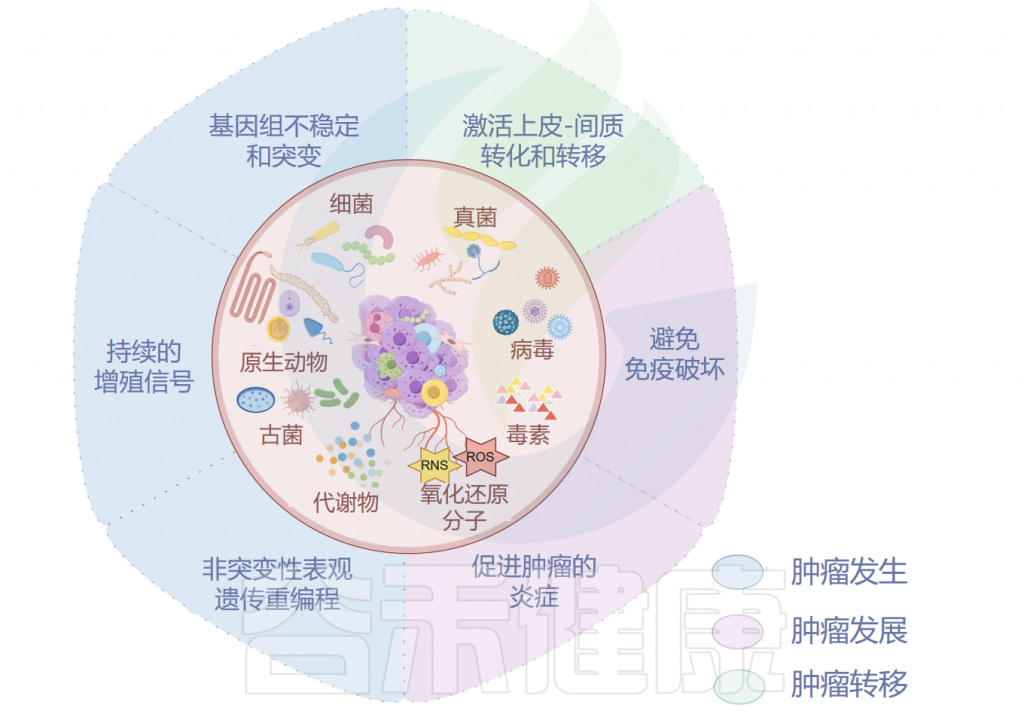
肿瘤微生物组与肿瘤发生、发展和转移密切相关,研究人员认为可以将多态性微生物组列为肿瘤的标志。接下来,我们将讲述肿瘤微生物组与肿瘤之间关系的代表性机制。
肿瘤微生物组与肿瘤密切相关
Ma Y,et al.Microb Pathog.2024
▸ 与肿瘤发生之间的关系
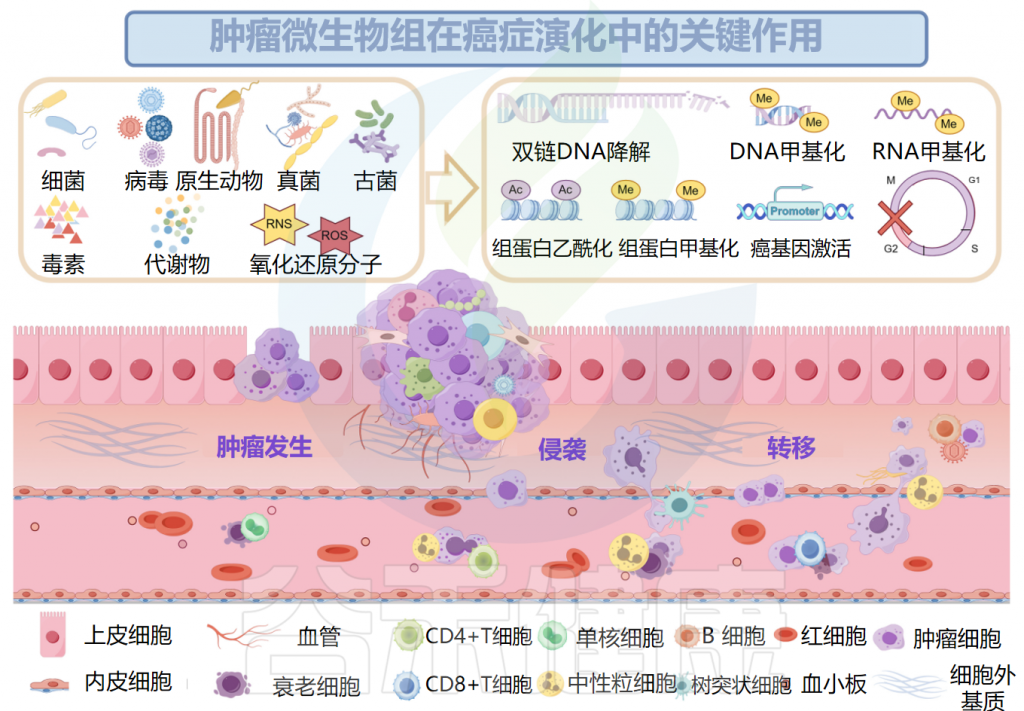
已被证明有助于肿瘤发生的肿瘤微生物组成分包括毒素、代谢物、酶和触发氧化应激的反应性小分子。这些物质可通过诱导基因组不稳定、表观遗传修饰和刺激宿主细胞的持续增殖而导致癌症。
细菌产生的毒素破坏基因稳定性并导致氧化应激
例如,由聚酮合酶阳性(pks+)大肠杆菌产生的大肠杆菌素和空肠弯曲菌分泌的细胞致死性膨胀毒素(Cdt)具有DNA酶活性,可以直接降解双链DNA,也可以导致DNA烷基化,从而在表观遗传水平上破坏基因组的稳定。
毒素介导的对DNA结构的间接损伤也可能是由反应性小分子的失衡引起的,例如脆弱拟杆菌毒素(BFT),它通过增加活性氧(ROS)的水平来诱导宿主细胞中的氧化应激。
此外,许多研究证实了肿瘤发生与癌微生物群产生的代谢物之间的联系。黄曲霉毒素B1(AFB1)是黄曲霉的代谢产物,可与DNA中的鸟嘌呤残基形成DNA加合物并诱导DNA损伤,最终致癌。在肝脏中,细胞色素P450酶参与AFB的代谢,它经历环氧化并转化为具有遗传毒性的8,9-环氧化物。8,9-环氧化物随后形成DNA加合物并促进突变,导致肝细胞癌的发展。
丁酸表达异常可能导致上皮细胞过度增殖
丁酸盐是一种短链脂肪酸,由结肠中的细菌发酵可溶性纤维衍生而来,是首选的宿主能量底物,可抑制结肠癌的发展。
丁酸抑制组蛋白脱乙酰酶(HDACs)和 DNA 修复蛋白的表达,在表观遗传水平上调节细胞增殖和凋亡,并减轻淋巴瘤。然而,在错配修复缺陷(dMMR)小鼠模型中,丁酸与结肠上皮细胞的过度增殖有关。
除了遗传起源外,表观遗传,包括由DNA甲基化、组蛋白甲基化和组蛋白乙酰化驱动的表观遗传,可以驱动宿主细胞恶性转化为癌细胞。
致癌微生物会驱动肿瘤的发生
EB病毒(EBV),也称为人类疱疹病毒4型(HHV-4),是一种致癌病毒,可诱导表观遗传变化以驱动肿瘤发生。溶原性EBV可诱导基因组不稳定并改变免疫逃逸,而潜伏的EBV有助于肿瘤细胞获得干性。
注:肿瘤干性被认为是肿瘤发展的关键组成部分
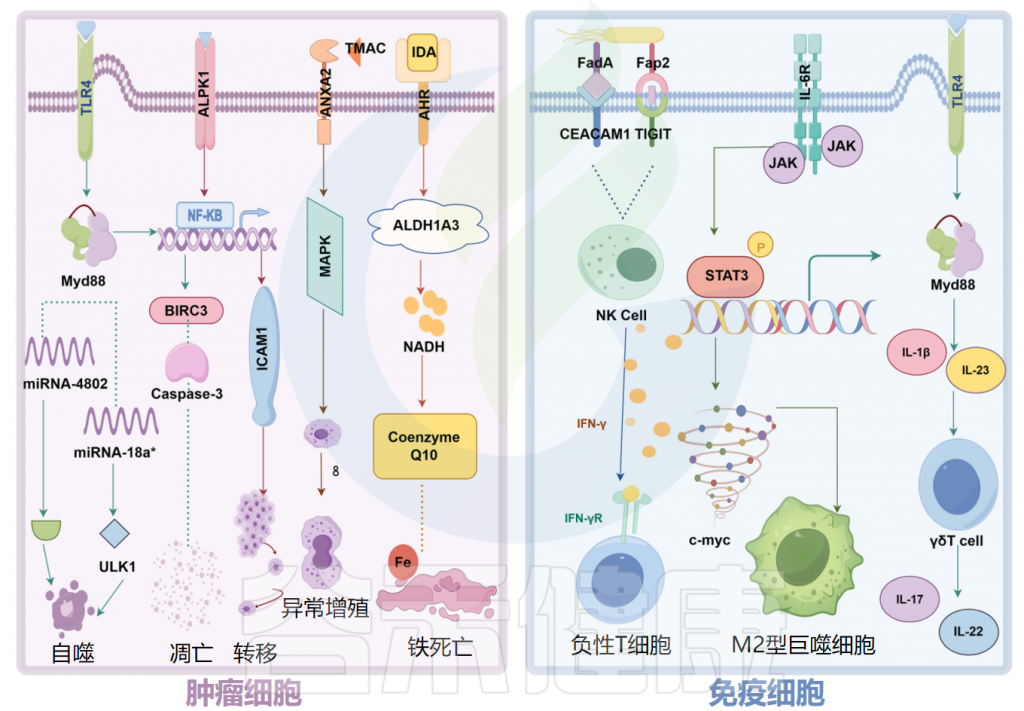
具核梭杆菌(F.nucleatum)通过钙黏附蛋白(E-cadherin)和粘附A蛋白(FadA)与癌细胞或免疫细胞结合,并激活β-catenin和Wnt信号通路以促进癌细胞增殖。
微生物组产生的炎症反应会增加对肿瘤的易感性
肿瘤微生物组产生炎症反应,以增加宿主对肿瘤的易感性。肿瘤微生物群的抗原表位被肿瘤微环境(TME)中的模式识别受体(如Toll样受体)识别,并导致活性氧、细胞因子和趋化因子等炎症介质的诱导,触发促进血管生成、癌细胞增殖以及肿瘤生长和进展的信号级联反应。
口腔牙龈卟啉单胞菌在癌症和胰腺导管腺癌中定植,促进CXCR2介导的中性粒细胞浸润,增强中性粒细胞弹性蛋白酶分泌,重塑肿瘤的炎症微环境,从而导致癌症进展。结直肠癌中,牙龈卟啉单胞菌激活造血NLRP3炎症小体,招募髓细胞,创造促炎微环境,与结直肠癌的发展和不良预后相关。
Ma Y,et al.Microb Pathog.2024
!
代谢物可能促癌也可能抑制癌症
重要的是要注意,代谢物的活性取决于肿瘤类型。色氨酸是变形杆菌和放线菌的能量来源,其代谢衍生物吲哚-3-醛激活肿瘤相关巨噬细胞中的芳烃受体(AHR),改变炎症基因的表达,随后可能诱发结直肠癌。
相反,在黑色素瘤中,罗伊氏乳杆菌代谢产生的吲哚-3-醛通过IFN-γ激活CD8+T 细胞中的AHR并增强免疫检查点抑制剂的疗效,从而促进肿瘤治疗。
▸ 与肿瘤发展之间的关系
肿瘤微生物群及其结构成分调节宿主代谢和免疫力,从而影响肿瘤的发展。
抑制自然杀伤细胞并促进肿瘤发展
除了激活长链非编码RNA ENO1-IT1的转录并影响组蛋白修饰外,具核梭杆菌(F.nucleatum)还调节结直肠癌细胞中的糖酵解途径,抑制自然杀伤细胞的活性并促进肿瘤发展。
肿瘤微生物的作用在不同肿瘤中可能有所不同,而不同生物体也可能在肿瘤中产生相同的生理效应。
例如,食管癌中的具核梭杆菌(F.nucleatum)以趋化因子(CCL20)依赖性方式促进Treg淋巴细胞浸润到肿瘤中,从而增强了肿瘤的侵袭性。在结直肠癌中,具核梭杆菌通过CCL20诱导巨噬细胞M2型极化参与肿瘤转移。
口腔鳞状细胞癌(OSCC)是一种起源于口腔粘膜复杂鳞状上皮的恶性肿瘤。具核梭杆菌激活自噬途径可促进体内癌细胞的迁移和侵袭。
同样,产气荚膜梭菌肠毒素诱导紧密连接蛋白Claudin 4(CLDN4)的核转位,增强OSCC细胞的增殖、迁移和侵袭,抑制YAP1磷酸化,促进YAP1表达以驱动肿瘤进展。
▸ 与肿瘤转移之间的关系
癌症转移是指肿瘤从原发部位迁移到远端器官的复杂过程,形成继发性肿瘤,是恶性肿瘤的一个重要标志。在发现癌微生物组作为肿瘤成分后,多项研究表征了癌微生物组在肿瘤转移中的作用。
调节上皮-间充质转化的激活
上皮-间充质转化(EMT)是将上皮细胞转化为间充质细胞的过程,其特征是细胞间极性和粘附丧失,运动和迁移增加。EMT的激活是肿瘤适应恶劣环境以促进侵袭和转移的关键策略,也可以由肿瘤微生物组调节。
在口腔癌小鼠模型中,具核梭杆菌(F.nucleatum)分泌的外膜囊泡调节EMT相关蛋白的表达,上调波形蛋白和神经钙网蛋白(N-cadherin)的表达,下调E-钙网蛋白(E-cadherin)的表达,从而促进口腔癌转移。
白色念珠菌是口腔癌菌群的主要组成部分,分泌蛋白酶诱导上皮细胞整合素的变化,增加E-钙粘蛋白表达,增强口腔癌的EMT表型,并促进侵袭和转移。
定植于乳腺导管腺癌的产肠毒素脆弱拟杆菌(ETBF)分泌的毒素可影响 Slug 和 Twist(EMT 标志物)的表达,并激活 β-catenin 和 Notch1 信号通路,促进肿瘤转移。
驱动细胞因子产生刺激癌细胞增殖和迁移
在胰腺癌中,具核梭杆菌(F.nucleatum)通过Fap-2依赖性途径靶向胰腺癌细胞紧密连接。这种相互作用驱动细胞因子的产生,这些细胞因子刺激癌细胞增殖并促进通过自分泌和旁分泌途径的迁移,最终驱动恶性肿瘤进展。
此外,紧密连接的破坏会激活YAP信号转导,从而抑制FOXD3的表达,从而降低m6A甲基转移酶甲基转移酶样3(METTL3)的表达。这随后降低了m6A甲基化,并促进了靶驱动蛋白家族成员26B的表达,从而驱动结直肠癌转移。
分泌细胞外囊泡,促进胰腺癌转移
最后,胰腺肿瘤微生物群可以分泌小的细胞外囊泡(sEVs),这些囊泡重塑细胞外基质,促进血管生成,并形成转移前生态位,促进胰腺癌转移。
与转移相关的肿瘤微生物群的研究

doi.org/10.1016/j.tcb.2022.11.007
对肿瘤微生物组及其异质性的研究发现了其在肿瘤诊断、预防和治疗方面的重要作用。
★ 有助于作为早期癌症的诊断标志物
迄今为止的研究表明,肿瘤微生物组可用作早期癌症诊断的标志物。幽门螺杆菌、梭杆菌、肠球菌属、沙门氏菌属、假单胞菌属和双歧杆菌属在特定的肿瘤部位富集,它们的检出可能用于诊断。
例如在原发性人类结肠癌和远处转移中检测到梭杆菌门(Fusobacteria),而使用甲硝唑抗生素治疗可以消除梭杆菌,并减缓肿瘤生长速度。
肝脏中Stenotrophomonas maltophilia丰度的增加诱导了肝星状细胞的细胞衰老相关的分泌表型(SASP),从而促进了肝癌发生。
微生物组对肿瘤细胞和免疫细胞的特异性机制
Ma Y,et al.Microb Pathog.2024
测序技术的广泛使用大大提高了我们研究肿瘤微生物组的能力。这些测序技术包括16S rRNA测序、DNA测序、下一代测序技术(NGS)、表观遗传学测序(例如染色质免疫沉淀测序和DNA/RNA甲基化测序)和三维(3D)基因组技术。
肿瘤内微生物的生物量相对较低,这导致了多路复用16S rRNA 测序方案的开发,以最大限度地减少污染并准确表征癌微生物组。
例如,利用qPCR和16S测序技术从肿瘤组织获取测序数据,并构建了一个表征肿瘤微生物组的数据库,实现了检测灵敏度103-104每克组织对应的细菌数。
宏基因组是一种针对样本中所有DNA的非靶向测序方法,包括微生物群落的全基因组序列,广泛应用于复杂微生物组的分析。宏基因组的分辨率更高,可以达到物种甚至菌株水平。此外,宏基因组学可以提供功能信息。最近的研究表明,宏基因组数据涵盖了更多类型的癌症,这可能促进肿瘤内微生物群领域的新进展。
代谢组学,特别是质谱法,可以检测和表征人类微生物群产生的小分子,并了解这些微生物代谢物的功能作用。这提高了我们研究肿瘤微生物群的能力,并可能开发出非侵入性的诊断性肿瘤生物标志物。
微生物产生的多种细胞毒性代谢物在肿瘤发生和发展中起着至关重要的作用。单一的实验技术可能不足以完全解释肿瘤和微生物之间的复杂相互作用,需要基因组学、转录组学、蛋白质组学和代谢组学的组合来了解各种代谢物在调节肿瘤发生和发展中的作用。
肿瘤微生物组检测技术

Ma Y,et al.Microb Pathog.2024
▸ 微生物组在癌症治疗中的作用
现有的癌症疗法很多,化疗、放疗等方式虽然有效,但副作用较多;免疫疗法有其局限性,有效率低且针对癌症类型有限。
靶向肿瘤微生物组的治疗方法可能通过增强宿主抗肿瘤免疫、诱导肿瘤细胞焦亡、促进CD8+ T 细胞活性以及避免干扰健康组织及其相关微生物群来改善肿瘤的治疗。
肿瘤微生物组在肿瘤治疗中的应用
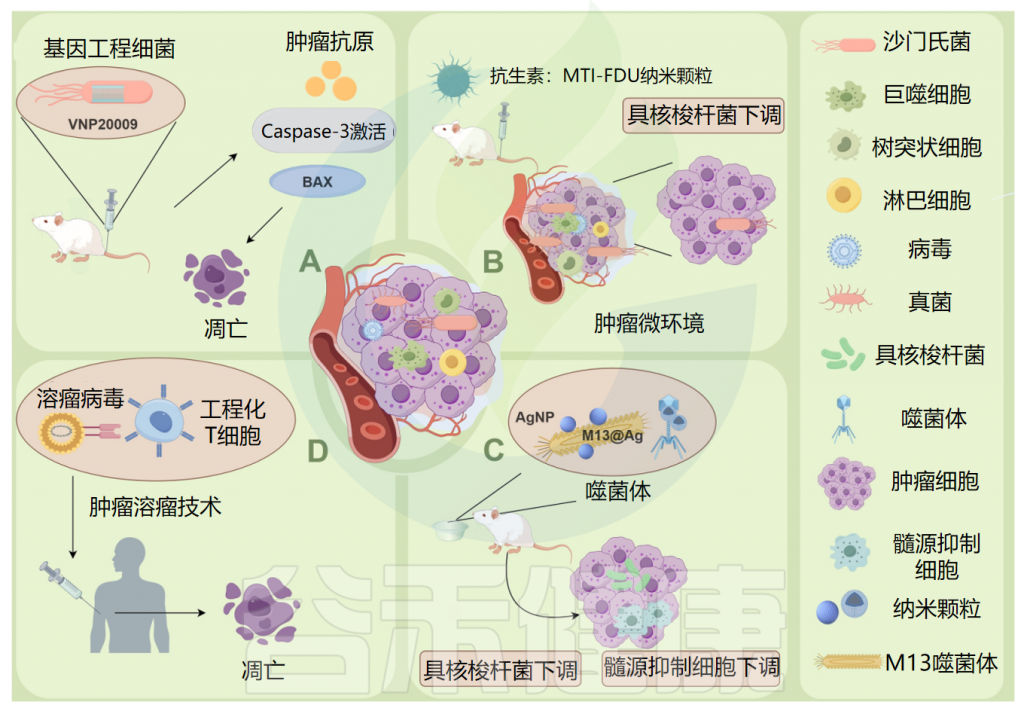
Ma Y,et al.Microb Pathog.2024
例如:(A)将基因工程减毒细菌菌株VNP20009直接递送到肿瘤发生部位,以及caspase-3凋亡酶活性的增加和促凋亡蛋白 Bax 的表达,显著诱导小鼠胰腺肿瘤细胞坏死,可作为杀死胰腺肿瘤的有效药物。
(B)甲硝唑-氟尿嘧啶利用增强的渗透性和保留作用,靶向肿瘤中的微生物群和肿瘤细胞,可以有效地从结直肠癌癌症组织中去除具核梭杆菌(F.nucleatum),具有低毒性和副作用。
(C)ONCOTECH(溶瘤病毒T细胞嵌合体)递送技术不仅增强了溶酶体病毒的靶向递送,而且改善了肿瘤微环境,能够在肿瘤中诱导长期免疫记忆。
(D)M13噬菌体特异性结合具核梭杆菌,在其表面外壳蛋白上静电组装银纳米粒子(AgNP)(M13@Ag),从肿瘤中去除具核梭杆菌,导致肿瘤部位髓系衍生抑制细胞(MDSC)减少。
除此之外,许多研究和谷禾的检测数据发现肠道微生物群可以调节癌症治疗,针对性地提高治疗效果并预防不良反应。
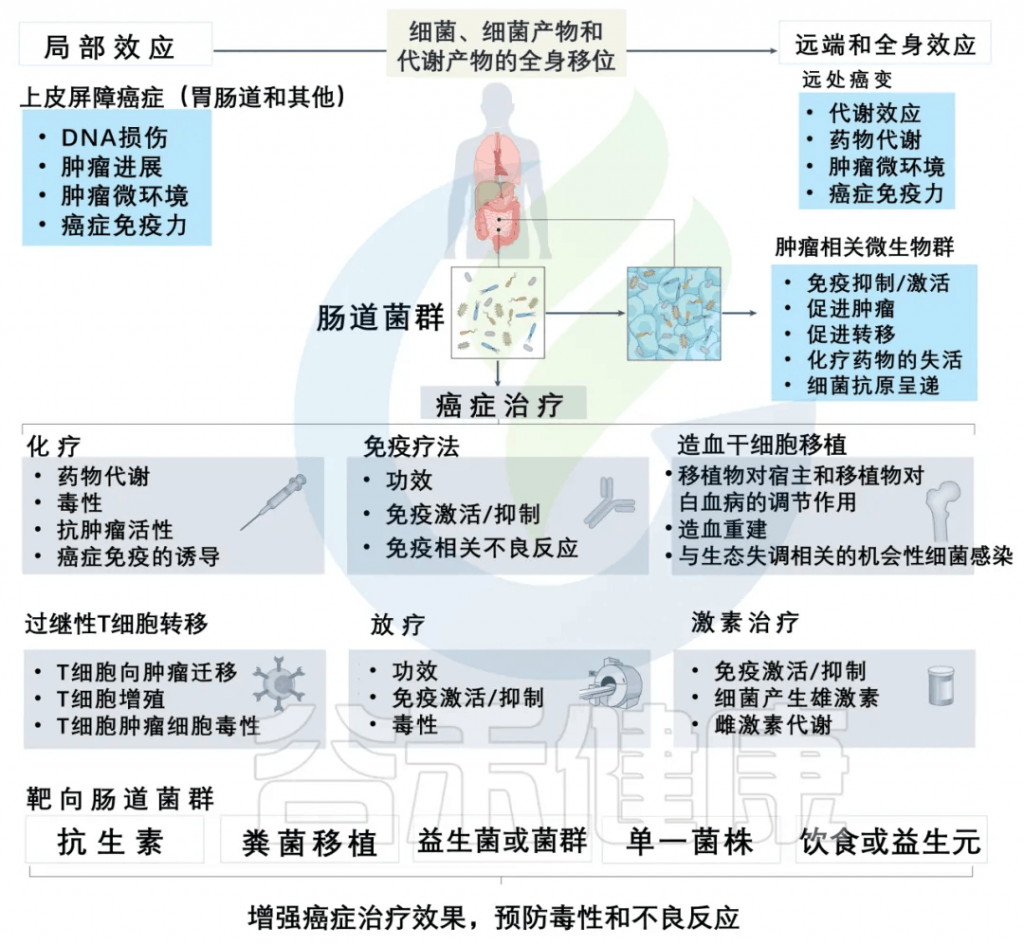
减轻化疗副作用,提高治疗效果
一些证据清楚地表明,调节肠道菌群可以减轻化疗药物的副作用,提高治疗效果。

减轻放疗毒性
在进行异基因造血干细胞移植预处理过程中,观察到患有白血病的患者的粪便样本中存在着毛螺菌科(Lachnospiraceae)和肠球菌科(Enterococcaceae)。这些患者在接受全身放疗治疗时出现的肠道毒性较轻。
几项研究表明,产生短链脂肪酸的益生菌,如乳酸杆菌和双歧杆菌,可以预防全身放疗治疗毒性。可能对癌症免疫和治疗毒性产生复杂影响,包括粘膜保护作用,部分由ANGPTL4、IL-18和IL-22的诱导介导,以及通过Treg细胞诱导和抑制树突状细胞功能介导的对立的免疫调节效应。
影响免疫疗法的效果
当前的癌症免疫疗法集中于利用特异性抗体来自我调节癌症免疫周期,这确保了应答的传播而没有生物中断。
微生态的改变会中断和削弱化学信号,导致致病状态,包括与炎症相关的疾病和癌症。
肠道微生物群对抗癌免疫反应的调节活性也与通过微生物群影响PD-L1和CTLA-4抑制剂的疗效有关。当与双歧杆菌的口服给药相结合时,PD-L1特异性抗体疗法的给药可以显著调节肿瘤的发展,在小鼠模型中肿瘤的生长几乎被消除。
我们预计肿瘤微生物组的研究将继续引起关注。然而,肿瘤微生物组在肿瘤预防和诊断中的应用,还存在着一些挑战。包括以下几点:
(一)人类微生物组在健康人群中的差异
同一个体在不同年龄的微生物组组成是可变的,更不用说不同个体中的微生物组成差异。部分原因是微生物组受饮食习惯、生活环境、药物暴露、生活方式和其他因素的影响。
因此,一个重大的挑战是根据微生物组的变化来确定个体的健康状况和癌症风险。未来,研究应探索肿瘤微生物组与环境、饮食和个人因素之间的相互作用。
(二)肿瘤微生物组的内容非常多样化
迄今为止已发表的大多数研究主要集中在细菌组上,而分析真菌组或病毒组的研究较少。此外,缺乏关于微生物组在非粘膜器官来源的肿瘤发展中的作用的数据。对肿瘤微生物组中微生物之间的关系也了解有限。例如,目前尚不清楚肿瘤微生物组中不同种类的细菌和真菌以及不同的病毒家族是相互独立的、协同的还是拮抗的。
(三)缺乏微生物代谢物等机制细节
尽管一些研究已经阐明了微生物改变肿瘤发生和发展的机制,但大多数仅具有相关性,缺乏与肿瘤发生因果关系的直接证据。特别是,其中许多研究无法确定微生物在肿瘤中定植的时间点。并且缺乏机制细节,主要集中在微生物种类的检测上,而忽略了分泌的毒素、代谢物和其他产物的功能。
未来的临床进展需要更精确地鉴定细菌分类单元对癌症早期诊断和治疗的因果关系,并了解其作用机制,因此需要进行更广泛的试验。改进的细菌可能作为抗癌药物,甚至可被改造成“微型机器人”用于药物传递。
总体而言,这一领域的发展为癌症患者提供了新的治疗选择和希望,但也需谨慎评估和监测以确保安全性和有效性。
微生物群在开发癌症诊断和抗癌策略中的重要性和潜力值得强调,将微生物调节疗法纳入癌症管理的整体方法很有必要。未来,我们有望实现更精准和个性化的菌群调节策略,为癌症治疗带来更大突破。
主要参考文献
Ma Y, Chen T, Sun T, Dilimulati D, Xiao Y. The oncomicrobiome: New insights into microorganisms in cancer. Microb Pathog. 2024 Oct 29;197:107091.
Azevedo MM, Pina-Vaz C, Baltazar F. Microbes and Cancer: Friends or Faux? Int J Mol Sci. 2020 Apr 28;21(9):3115.
Wong-Rolle A, Wei HK, Zhao C, Jin C. Unexpected guests in the tumor microenvironment: microbiome in cancer. Protein Cell. 2021 May;12(5):426-435.
Gagliani N, Hu B, Huber S, Elinav E, Flavell RA. The fire within: microbes inflame tumors. Cell. 2014 May 8;157(4):776-83.
Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, Rotter-Maskowitz A, Weiser R, Mallel G, Gigi E, Meltser A, Douglas GM, Kamer I, Gopalakrishnan V, Dadosh T, Levin-Zaidman S, Avnet S, Atlan T, Cooper ZA, Arora R, Cogdill AP, Khan MAW, Ologun G, Bussi Y, Weinberger A, Lotan-Pompan M, Golani O, Perry G, Rokah M, Bahar-Shany K, Rozeman EA, Blank CU, Ronai A, Shaoul R, Amit A, Dorfman T, Kremer R, Cohen ZR, Harnof S, Siegal T, Yehuda-Shnaidman E, Gal-Yam EN, Shapira H, Baldini N, Langille MGI, Ben-Nun A, Kaufman B, Nissan A, Golan T, Dadiani M, Levanon K, Bar J, Yust-Katz S, Barshack I, Peeper DS, Raz DJ, Segal E, Wargo JA, Sandbank J, Shental N, Straussman R. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science. 2020 May 29;368(6494):973-980.
Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, Kosciolek T, Janssen S, Metcalf J, Song SJ, Kanbar J, Miller-Montgomery S, Heaton R, Mckay R, Patel SP, Swafford AD, Knight R. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature. 2020 Mar;579(7800):567-574.
Narunsky-Haziza L, Sepich-Poore GD, Livyatan I, Asraf O, Martino C, Nejman D, Gavert N, Stajich JE, Amit G, González A, Wandro S, Perry G, Ariel R, Meltser A, Shaffer JP, Zhu Q, Balint-Lahat N, Barshack I, Dadiani M, Gal-Yam EN, Patel SP, Bashan A, Swafford AD, Pilpel Y, Knight R, Straussman R. Pan-cancer analyses reveal cancer-type-specific fungal ecologies and bacteriome interactions. Cell. 2022 Sep 29;185(20):3789-3806.e17.
El Tekle G, Garrett WS. Bacteria in cancer initiation, promotion and progression. Nat Rev Cancer. 2023 Sep;23(9):600-618.

谷禾健康
肠道微生物群在癌症中发挥免疫调节和抗肿瘤作用,肠道微生物失调可诱导有毒代谢物的释放,并在宿主体内表现出促肿瘤作用。肠道微生物群也能调节标准化疗药物和天然抗癌药物的疗效。
本文列举5种常见的癌症(结直肠癌、肺癌、乳腺癌、前列腺癌、胃癌),以及肠道微生物群在癌症中的复杂作用。
肠道微生物群与癌症发病的关系概览
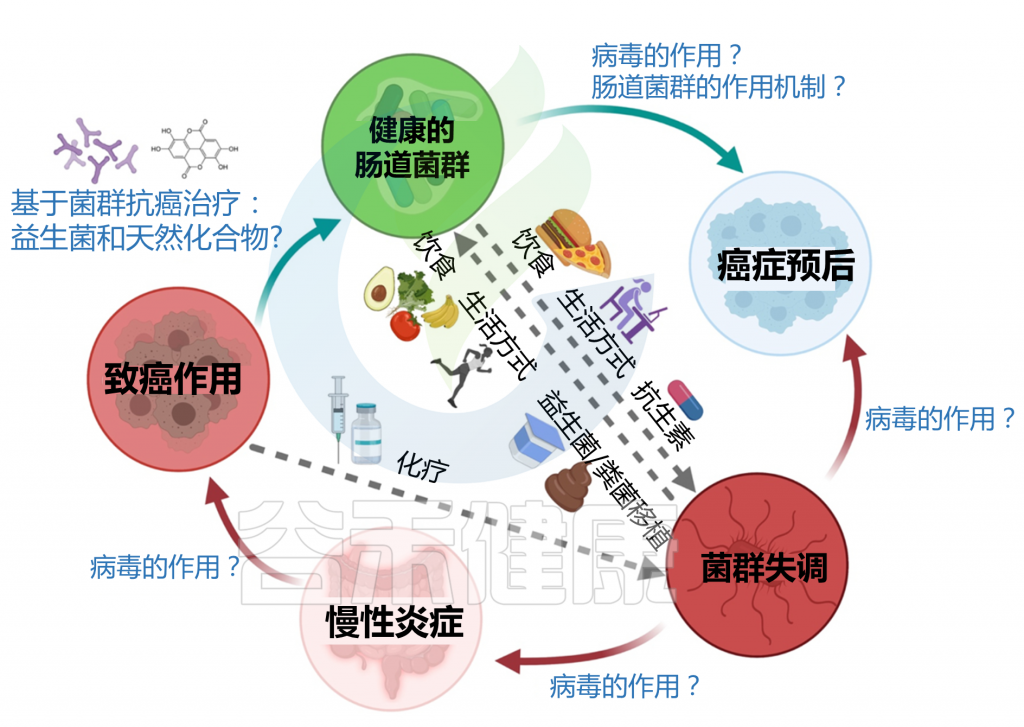

在进入具体的5种癌症章节之前,我们先来了解一下,微生物群与癌症的关系。有研究人员将微生物群和癌症之间的关系分为三个层次: 一级、二级和三级相互作用。
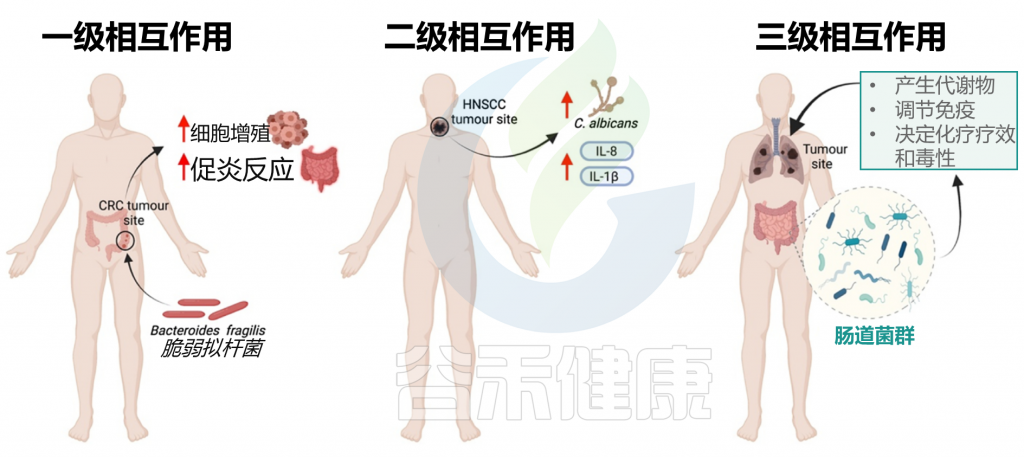

一级相互作用(主要)
主要的相互作用考虑了肿瘤微环境和微生物群之间的直接联系。几项体内和体外研究主要从两个方面支持了这种关系:
a) 肠道微生物群可通过生物失调导致致癌
b) 肠道微生物可通过调节肿瘤活性干扰化疗药物的疗效
二级相互作用(次要)
次要的相互作用考虑了组织或器官系统的微生物群和同一大体分区内的肿瘤之间的联系。这种相互作用水平有助于识别用于筛选不同癌症类型的潜在生物标志物。特别地,来自局部组织或器官环境的次级微生物群可包含来自肿瘤微环境和初级微生物群落的痕迹,其可用作癌症的生物标志物;但这些诊断过程往往很复杂。
三级相互作用
肠道微生物群和肿瘤之间的三级相互作用解释了位于体内不同部位的肿瘤上的微生物群的影响。对这种相互作用水平的研究对于确定生理上遥远的微生物种类和感兴趣的肿瘤之间的关系具有重要意义,这对于确定癌症患者中潜在治疗选择的功效也具有临床相关性。
这些三级相互作用可以通过以下方式影响癌症:
肠道微生物群可以通过启动代谢过程(包括水解和还原)来调节口服药物代谢,这直接影响药物毒性,并可以增强或抑制药物活性。微生物群与肿瘤之间的三级相互作用也可以帮助诊断不同类型的癌症。
肠道微生物群的促肿瘤、抗肿瘤和免疫调节作用
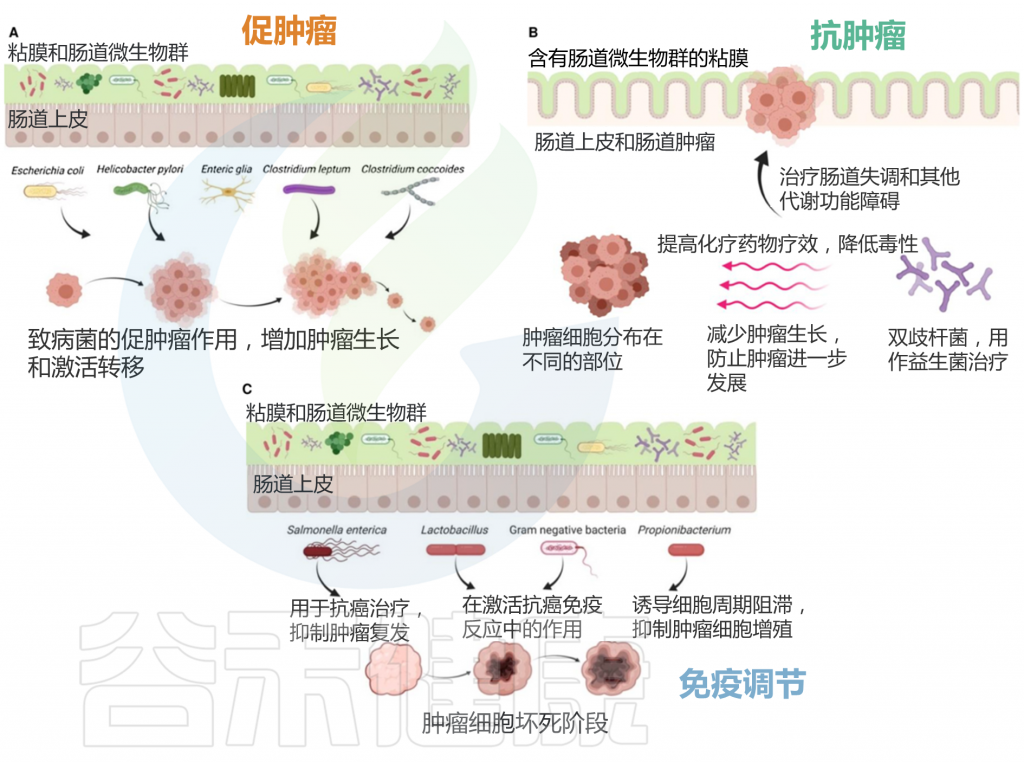


谷禾健康
辅助化疗可导致约三分之一的女性体重增加,葡萄糖耐量下降和高血压。这些事件的潜在机制尚未定义。这项研究评估了乳腺癌和妇科癌症辅助化疗患者的微生物组与体重增加之间的关联。近日发表在《BMC Medicine》上题为“The intestinal microbiome, weight, and metabolic changes in women treated by adjuvant chemotherapy for breast and gynecological malignancies”的一项小样本研究评估了乳腺癌和妇科癌症辅助化疗患者的微生物组与体重增加之间的关联。
方法:在开始辅助治疗之前招募患者。前瞻性收集有关肿瘤治疗,更年期状态和抗生素使用的数据。如果在研究期间接受抗生素治疗,则将患者排除在外。在治疗前和治疗结束后4-6周测量体重和身高。体重增加定义为体重增加3%或更多。治疗前收集粪便样本。并进行16S rRNA基因可变V4区扩增测序。数据使用QIIME 2进行处理和分析,并使用DADA2通过q2-dada2对读取进行了去噪和聚类。每个样品的读数均> 9300。使用Swiss Webster无菌小鼠进行了患者的粪便移植实验。
结果:招募了33名患者;其中9人增加了基线体重的3.5–10.6%。在治疗后体重增加的妇女的治疗前微生物组的多样性和分类学与对照妇女明显不同。与使用对照妇女的粪便样本进行移植的小鼠相比,从体重增加的患者的样本中进行粪便微生物菌群的移植诱导了无菌小鼠的代谢变化。
结论:肠道化学组的组成及其多样性与乳腺癌和妇科恶性肿瘤辅助化疗后体重增加有关。小鼠FMT实验表明,微生物组介导了化学疗法的不良代谢作用。值得进一步研究微生物组的预测价值,以及其对化疗后体重和代谢变化的作用机理。

图一:化疗后体重增重的妇女的肠道微生物组与对照组妇女的肠道微生物组成不同

图二:对GF小鼠使用化疗后体重会增加的妇女的预处理样本进行FMT,与不会增加体重的妇女的FMT相比,FMT会引起显著体重等指标的变化。

图三 对GF小鼠使用化疗后体重会增加的女性预处理样本的FMT,与不会增加体重的女性(对照)相比,会引起显著的微生物变化。
参考文献:Uzan-Yulzari, A., Morr, M., Tareef-Nabwani, H. et al. The intestinal microbiome, weight, and metabolic changes in women treated by adjuvant chemotherapy for breast and gynecological malignancies. BMC Med 18, 281 (2020). https://doi.org/10.1186/s12916-020-01751-2