-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业

谷禾健康
如果你正为宏基因组数据的组装和注释而忙于“拼工具、调环境、转格式”,那么annoSnake或许能让你从繁琐中解放。
它是一个基于Snakemake的自动化工作流程,从clean reads组装到物种分类、功能注释,再到MAGs的装配和注释。
作为开源工具,annoSnake具备良好的可重复性、可扩展性和可移植性,非常适合HPC集群环境。
本文将带你了解它的工作流程、在白蚁肠道宏基因组数据上的验证结果,以及它的优势与局限,帮助你快速判断是否值得上手。
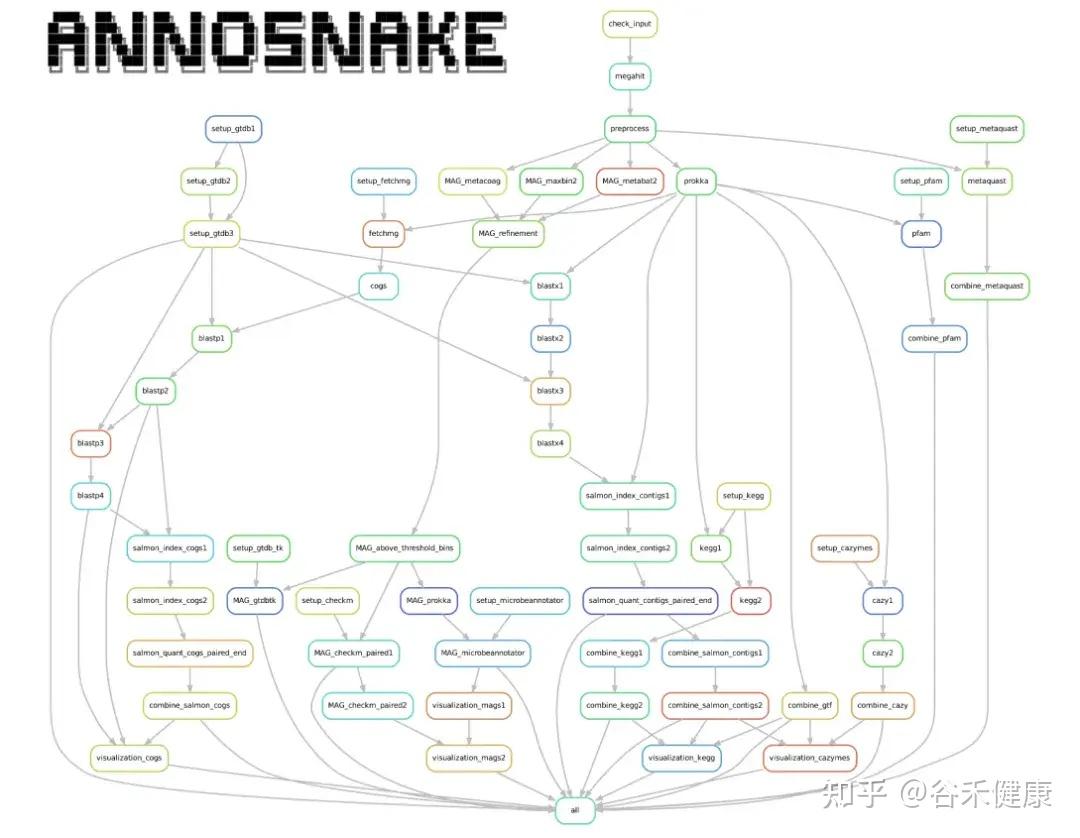
annoSnake以“自动化+模块化”为核心:输入clean reads → 组装 → 注释 → 分箱 → 结果汇总与可视化。
每个标准化步骤里,annoSnake使用的都是主流工具,如果你事先没有任何准备,也无需担心,它会自动创建独立的虚拟环境,并安装所需的分析工具和注释用的数据库。
分析前的准备
1. Mac OS或linux系统设备,磁盘空间推荐>100GB。如果运行单个宏基因组样本,只要有32 GB 内存和 8 核 CPU 基本就能跑完。
若要在集群上批量运行十几个样本或进行MAG分析,最好准备 ≥128 GB 内存和多核服务器。annoSnake 可批量化处理。
2. 安装mamba或conda用于管理环境,然后安装snakemake。
3.克隆Github仓库到本地(git clone https://github.com/bheimbu/annoSnake.git)。
4. 清洗后的测序数据,可以是双端,也可以是交错合并的fastq.gz文件,注意要是gzip格式。
5.编辑./profile/params.yaml和./profile/config.yaml文件。config.yaml 决定“要做什么”与“怎么做”,config.yaml 决定“在哪跑、分配多少资源”。
首次分析,annotSnake会自动下载并设置GTDB、dbCAN、Pfam、KEGG等数据库,总量约100GB。
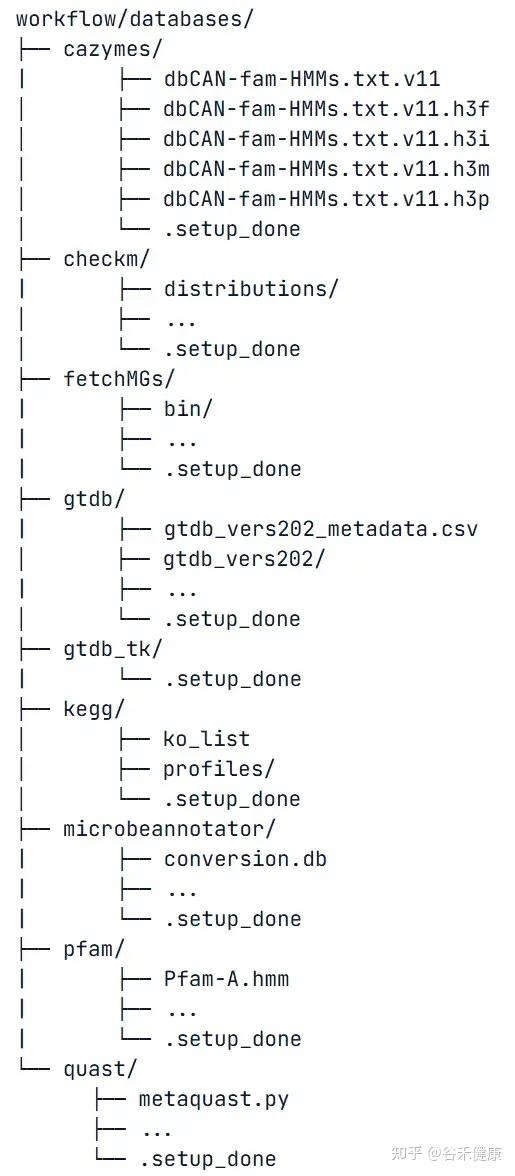
开始分析
1. 组装
MEGAHIT v1.2.9工具进行宏基因组组装,默认–presets meta-sensitive模式组装,保留≥1500 bp的contigs,并以metaQuast评估组装质量。
2. 物种分类注释
Prokka v1.14.6工具识别CDS、rRNA、tRNA;fetchMG v1.2提取40个单拷贝标记基因;结合GTDB(v202, (Parks et al.2022)数据库进行blastp和blastx注释;自定义R脚本gtdb_diamondlca.R进行LCA分类整合。
3. 功能注释
对细菌/古菌的contig执行注释,可以选择的功能数据库有:CAZy(dbCAN version 11)、Pfam(version 35)和KEGG。
针对Pfam搜索结果,可以自行借助在线工具HydDB进一步分类。针对KEGG结果,借助KofamScan工具重建以KEGG为基础的代谢通路。E-values阈值在params.yaml中设定。
4. 基因丰度量化与归一化
Salmon v1.10.2对CDS进行TPM定量,对于TPM>1的,予以保留,然后对剩余TPM做CLR对数转换(默认,log(TPM+0.65))。
5. 分箱与注释(可选)
同是采用三种分箱算法:MetaBAT v2.10.2、MetaCoAG v1.1.1、MaxBin v2.2.7,最后用metaWRAP v1.3的bin_refinement整合最优集合,CheckM 评估MAGs质量,默认阈值是完整性≥50%且污染≤10%。
对优质的MAGs使用GTDB-Tk v2.3.2进行物种分类(数据库v214),Prokka做基因预测,然后用MicrobeAnnotator进行功能注释,该工具使用DIAMOND和KofamScan,并以通路基因存在/缺失评估完整性。
6. 输出与可视化
输出包括CSV表格和ggplot2/plotly生成的PDF/HTML图表。
!
Tips
annoSnake已经内置了“KEGG条目(KO编号)→基因名称/通路名称”的映射表文件。如果你想重点关注某些特定的KEGG基因或通路(比如只看甲烷生成、乙酸生成或硫酸盐还原等),可以在/workflow/rules/scripts目录下,直接编辑这类映射文件,把你关心的KO编号及其基因名、通路名加入或调整。
管道运行时会按你改过的清单去批量检索与汇总这些目标基因/通路的注释与丰度,并在输出图表中优先呈现,从而实现“按课题定制”的结果视图。
作者用来自澳大利亚Amitermes组(AAG)的白蚁肠道宏基因组作为测试数据,与已知发现进行一致性检验。
▸在测序深度不高时仍能识别主要细菌谱系和代谢通路
31个群体,Illumina NextSeq双端,平均每样本约700万条reads。尽管测序深度不高,但annoSnake仍有效识别了主要的细菌谱系,以及大量与木质纤维素消化相关的代谢通路和基因。
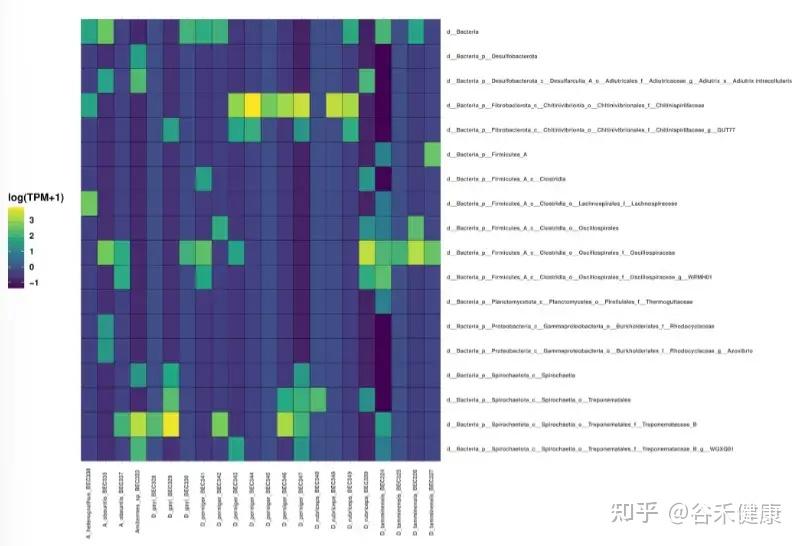
图中是annoSnake识别出的主要菌群,结果显示不同取食类型白蚁肠道的优势类群模式,这与已知发现相符,通常,食草和食木的白蚁其肠道群落以螺旋体为主,而食腐殖质和土壤的白蚁则富含梭菌(clostridia)。
但也有与已知发现不符的结果,D. tamminensis物种的肠道群落以梭菌为主,几乎不见螺旋体或Fibrobacterota,部分D. gayi群体也表现出类似模式,这与“草/木料取食类型的白蚁常以螺旋体占优”的普遍模式不一致,作者解释这是数据特性使然,低覆盖度数据只能恢复高丰度群落成员,而非物种生态学的结论。
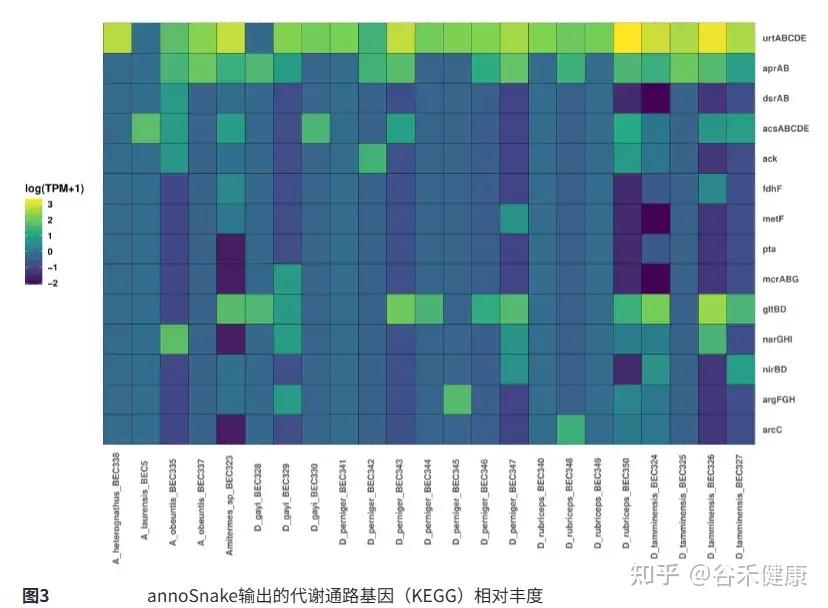
▸ 识别出硫酸盐还原等重要通路的基因
在所有样本中,annoSnake识别出硫酸盐还原通路的关键基因,如aprA、aprB和dsrAB,这符合白蚁肠道微生物组中硫酸盐还原过程的常见模式。还有许多与木质纤维素消化相关的KEGG代谢通路基因,这支持白蚁肠道微生物组在碳循环和能量代谢方面的普遍功能特征。
仅检测到少量与甲烷生成相关的基因,如mcrABG。已知甲烷生成主要局限于厌氧甲烷生成古菌,而本次分析中未有样本被检测到古菌,所以甲烷生成基因稀少是符合预期的。这与低覆盖度数据仅恢复高丰度群落成员的特性一致,古菌可能被低估,而不是KEGG注释的偏差。
识别出fdhF和acsABCDE等基因预测还原性乙酸生成的存在,这一点由分箱得到的15个Bacillota和6个螺旋体MAGs所支持,这两类群包含潜在的乙酸生成菌。这与其他白蚁和千足虫研究中已知的乙酸生成潜力一致。
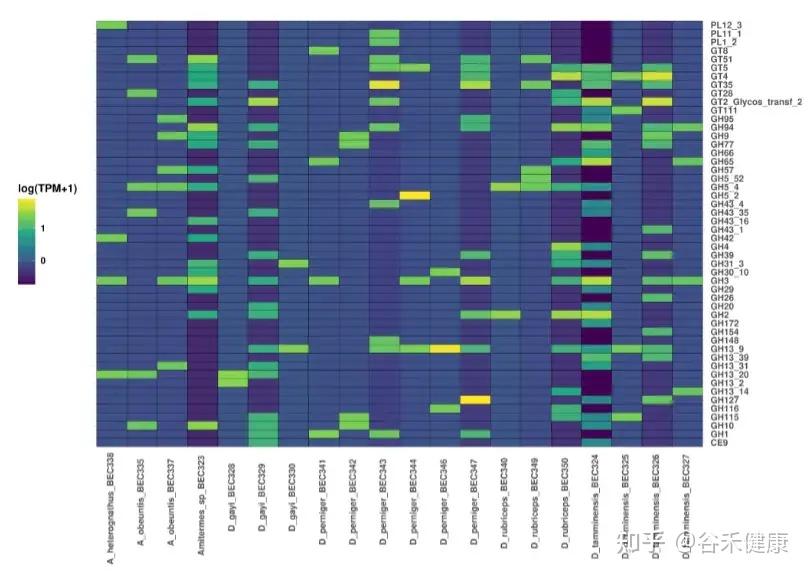
▸ 能够检测到大量碳水化合物活性酶
annoSnake检测到大量CAZymes(碳水化合物活性酶)。D. tamminensis在不同群体间的GHs丰度差异再次暗示饮食灵活性,且部分群体GHs模式与腐殖/土壤取食物种一致;螺旋体主导的D_gayi_BEC329中GHs丰富,符合凋落物取食物种的特性,而在梭菌主导的D. gayi群体中GHs较低。
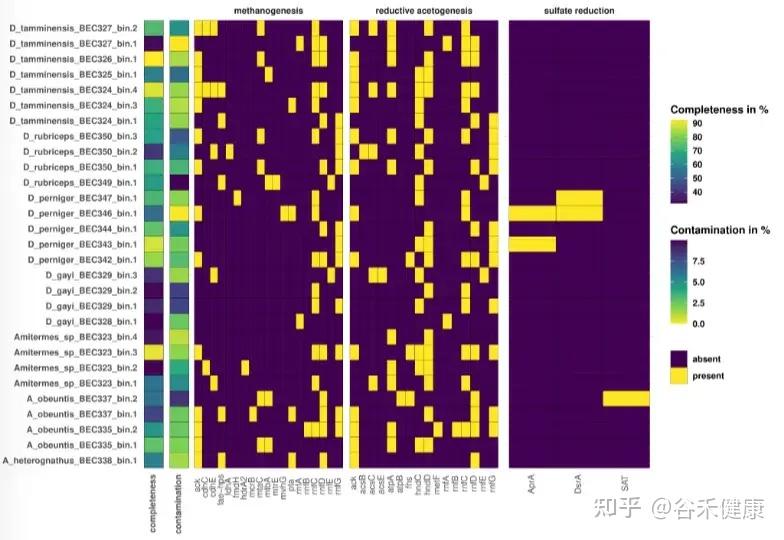
annoSnake从低覆盖度数据中获得30个MAGs,其中包括15个Bacillota(内含大量梭菌纲)、1个Desulfobacterota、7个Fibrobacterota、1个Pseudomonadota、6个Spirochaetota。
图中展示了MAGs中木质纤维素消化相关代谢途径(甲烷生成、还原性乙酸生成、硫酸盐还原)基因的存在/缺失。左侧给出MAG的完整性和污染分值,颜色越浅表示完整性越高,污染越少。 紫色方块表示基因缺失,黄色方块表示基因存在。
优势
• 覆盖全流程的一站式自动化:从输入reads→组装→物种注释→功能注释→丰度定量→分箱(可选)→可视化,节省操作时间。
• 数据库自动下载和配置。
• 兼具一些灵活性,比如可以自定义数据库,也能调整分析参数。
• 可重复、可扩展、可移植。Snakemake内核+HPC优化,可以在不同HPC环境中高效执行。
劣势
• 资源占用较高,数据库体量约100GB,完整流程在大规模数据上更适合HPC环境;本地轻量设备可能受限于存储、内存与时长。
• 需要有一定代码基础,掌握Snakemake、Conda与YAML配置,能调试环境配置时可能出现的错误。
• 范围聚焦细菌/古菌,真核生物未被纳入默认流程,氢化酶精细亚型分类需借助HydDB等外部工具,未在管道集成。
• 低覆盖度数据的固有限制:对稀有类群的恢复能力受限,更偏向于恢复高丰度成员,需要结合研究设计与深度规划权衡。但作者也没有发表对高覆盖度数据的测试结果,所以工具对高覆盖度数据的表现不明确。
• 工具较新,容易出现环境/兼容性问题或边缘情况未覆盖;第三方依赖更新也可能引入不稳定性。数据库管理灵活性受限,版本固定且无更新管道。虽支持自定义数据库,但需自行调整文件格式。
o 输出的图像不够美观,可视化类型单一。
annoSnake适合具备中级生信技能,需快速产出的微生物组学研究者。如样本量大,需批量分析,则需要配备高性能设备。研究范围在细菌/古菌的宏基因组与MAGs。
下面这个网址可访问 annoSnake 文档:
https://annosnake.readthedocs.io/en/latest/index.html
参考文献:
Bastian Heimburger, Rebecca Clement, Tamara R. Hartke
bioRxiv 2025.11.03.686227; doi: https://doi.org/10.1101/2025.11
谷禾健康

导语
微生物在在地球上无处不在,适应了几乎所有可用的生态栖息。 微生物在不同物种和个体之间差异性很大,存在着广泛的微生物多样性。
野生动物之所以能够耐受病原菌的感染和有毒食物的威胁以及抵御多种疾病,可能与其体内或体表生存的微生物密切关联。然而,与已被广泛研究的人类微生物群相比,野生动物的微生物群受到的关注较少。
当宿主有着共同的饮食或共同祖先,尤其是哺乳动物,通常肠道菌群构成也更为相似,不过这种相关性在鱼类、两栖类、鸟类和非脊椎动物中较弱。在许多情况下,肠道微生物都参与宿主的关键生理过程,包括代谢特殊的饮食化合物。
近日,以色列魏茨曼科学研究院Eran Segal团队采用一致的方法从全球四大洲采集了406份动物粪便样品,包括121份养殖样品和285份野生样品。共涉及184个动物物种,包括哺乳类、禽类、两栖类、硬骨鱼类等的物种。这些物种在分类单元、觅食/取食行为、地理分布、性状等方面具有较高的多样性。
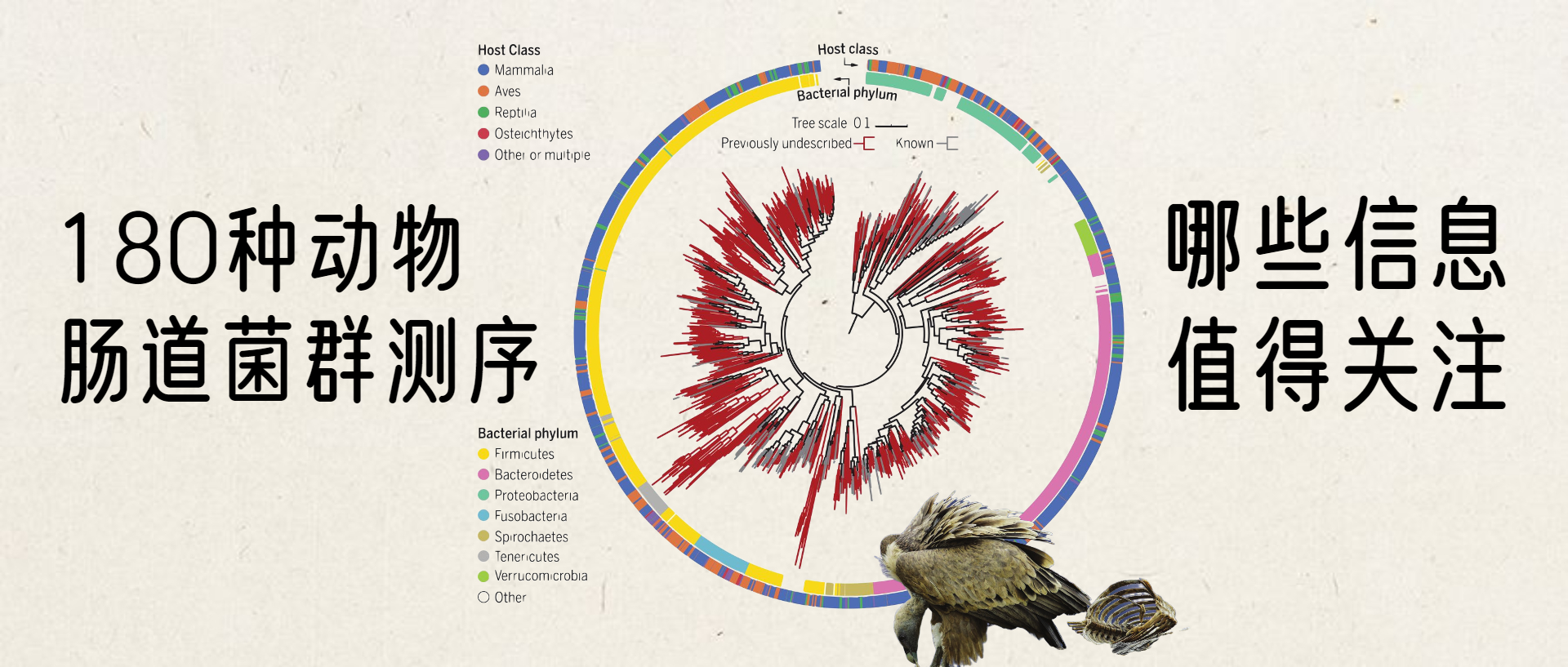
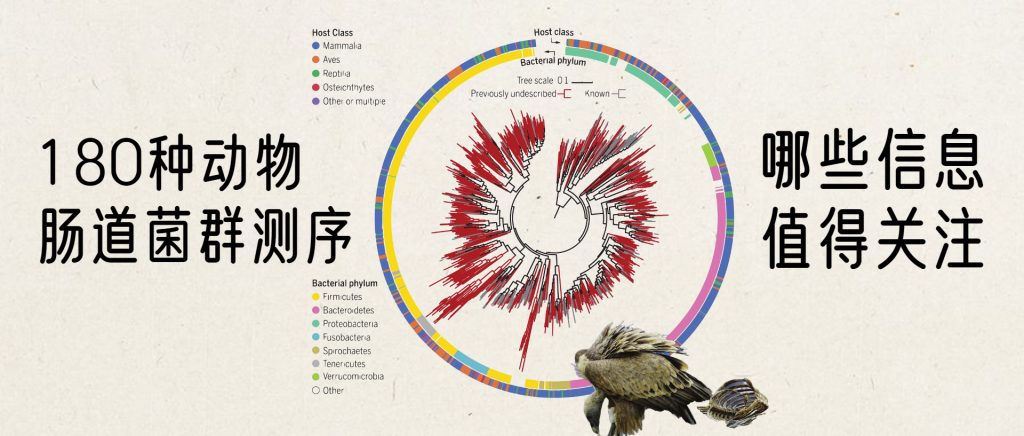
使用宏基因组学来分析这180多个物种的肠道菌群,使用从头基因组组装,构建并在功能上注释了5000多个基因组的数据库,其中包括1209种细菌,但是其中75%未知。
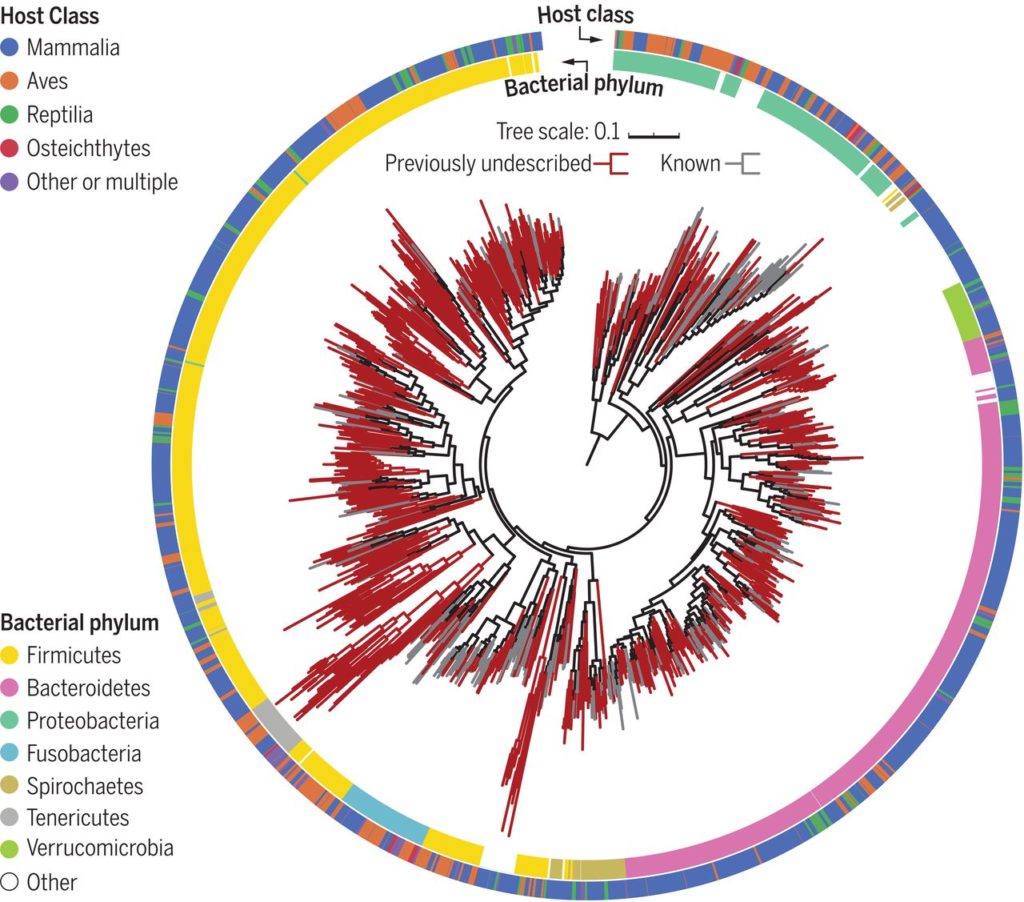
在这项研究中组装的1209个基因组的最大似然比的系统发育树。
内有色环和外有色环分别表示细菌门和宿主类别。先前未描述的基因组进化枝为深红色。
一,坚持野外采样。野外采样,尤其对于动物采样存在很多挑战,但是证据表明圈养动物会改变微生物组,而且,过往大量有关哺乳动物的研究大部分来来自于是圈养动物,包括大小鼠。这次大规模全球野外采样,可以扩大和了解动物宿主栖息微生物的机会。
二,要获得广泛的野生动物代表性,需要在全球不同的生态环境采样,并从具有不同特征和喂养方式的多种动物中取样。此外,该研究为每个物种手工制定了特质,包括饮食适应性,活动时间和社会结构,使我们能够系统地研究微生物群组成与宿主表型之间的关系。
三,为未知物种的大规模注释细菌基因组数据库,并确定了与这些动物的性状和分类相关的多种微生物模式,并强调了其潜力作为发现新的工业酶和治疗剂的主要未开发资源。
微生物的组成,多样性和功能含量与动物分类,饮食,活动,社会结构和寿命相关。动物微生物群系是生物功能的丰富来源,可能会对生物技术产生影响,包括抗生素,工业酶和免疫调节剂。
此外,野外动物表现出适应性,例如安全食用腐烂,感染病原体的肉类和有毒植物,可以产生强效毒素、生物发光以及各种疾病和微生物病原体具有特异性免疫力、再生能力并且在某些物种中具有极长的寿命。这些适应性中的某些,例如毒素产生和生物发光至少部分是由生活在动物体内/上的微生物共生体赋予的。该研究构建和功能注释从自然栖息地的野生动物中提取的微生物群的综合数据库可以对动物性状与其微生物群之间联系的进行全面了解。
例如,为了证明在动物微生物群中可以发现新的细菌功能,作者在实验中验证了细菌毒素——食用腐肉的欧亚兀鹫(Gyps fulvus)的MAG中发现的代谢蛋白酶。这些蛋白酶可用作抗菌化合物,具有抗菌活性潜在的应用包括对抗人类食物中毒。
欧亚兀鹫(学名:Gyps fulvus):体长95-105厘米,尾长24-29 厘米,翼展240-280厘米,体重6-11千克。是一种大型的褐色鹫。栖息在海拔高达2,500米的范围内。主要以山羊、鹿和瞪羚等野生动物,以及人类养殖的绵羊、山羊、牛和马为食。靠灵敏的嗅觉来找寻腐烂的动物尸体,并常常为抢一块肉而争个不停。而且习惯把头伸进动物尸体的腹腔内,啄食内脏和肌肉。分布范围非常广泛,遍布欧洲、中东和北非,也分布于印度、喜马拉雅山脉。在地中海沿岸国家最常见。
这项研究的最大贡献是其丰富的、系统生成的数据集。很容易想象,微生物保护和新出现的抗生素耐药性等不同领域的突破是由这些亚基因组的发现推动的。在欧亚兀鹫微生物群中发现的蛋白酶证明了从野生动物微生物群中进行生物勘探的原理,尽管尚不清楚该案例研究是否应被视为例外或预期的发现。


新发现的食腐肉的欧亚兀鹫(Gyps fulvus)的肠道细菌中的毒素代谢蛋白酶可能在抵抗食物中毒方面有应用。
这项研究仅仅触及了可以用这个数据集检验的假设的表面。未来方向包括:
01 微生物群如何帮助动物降解有毒的植物化学物质。
02 抵御食物中的病原体。
03 从多种食物来源中提取营养的问题。
四,丰富了许多未知物种的细菌门,并发现某些细菌进化枝相对于同一门中的其他细菌具有独特的功能特性。动物种类和已发现的动物种类(共存细菌的特定簇)之间的细菌状况有所不同。动物中这种未被探索的微生物多样性与被充分研究的人类微生物组形成了对比,而人类微生物组在参考数据库中表现得更好。未描述的物种中的富集度最高的是疣状菌属(Verrucomicrobia),这是一个存在于水、土壤和人类肠道中的门,但培养物种相对较少。
确定了多种途径和直系同源物,这些途径和直系同源物在特定的动物性状中显着丰富,并表明功能性景观与这些性状相关。这些功能中的一些功能提示了野生生物微生物群的新角色和特性。绘制野生动物的微生物群落图也可能有助于野生微生物的保护工作。
五,重述了首次通过扩增子测序发现的结果,包括食草动物微生物组比食肉动物微生物组更加多样化。这是一个比较重要的发现,是否说明植物性饮食可以提高微生物多样性?此外,不同动物群体的微生物组编码的遗传途径因宿主饮食、体型和其他特征而不同。
六,野生动物的微生物区系也是动物和人类病原体的天然库,如当前的COVID-19大流行一样,通过对野生生物微生物景观的广泛的基因组集合可以阐明其传播到人群中的时间和途径。
一,使用MAGs(宏基因组组装的基因组)限制了研究其中一些目标的拓展,即保护医学和生态上重要的细菌菌株。有些微生物物种将需要分析未组装的读取、培养或富集技术,而不是本研究中使用的全基因组测序。
二,即使是那些基因组被MAG组装捕获的细菌,这些组装体也倾向于排除“辅助”基因组,即在不同菌株间存在的基因。然而,这些基因往往在适应特定的宿主和环境中发挥作用。例如,抗菌素抗性、致病性和能量收集是经常由移动元件或其他辅助基因编码的性状。
三,Levin等人检测到的大多数非特征微生物在宿主体内的活动,以及它们是稳定地定殖在动物体内还是短暂地通过它们的胃肠道的问题仍然存在。在这个庞大的数据集上测试每个假设需要大量的计算、解释和实验验证。
参考文献:
Levin D, Raab N, Pinto Y, et al. Diversity and functional landscapes in the microbiota of animals in the wild[J]. Science, 2021, 372(6539).
Coleman M. Diagnosing nutritional stress in the oceans[J]. Science, 2021, 372(6539): 239-240.
L.-X. Chen, K. Anantharaman, A. Shaiber, A. M. Eren, J. F. Banfield, Genome Res. 30, 315 (2020)
E. C. Lindsay, N. B. Metcalfe, M. S. Llewellyn, J. Anim. Ecol. 89, 2415 (2020)