-

 国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子
国家认可委 CNAS L23010 认可项目:微生物宏基因组 | 16S rRNA扩增子

 二级病原微生物安全实验室
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
二级病原微生物安全实验室 国家高新企业 | ISO9001认证 | 肠道健康精准检测高新技术研发中心 | 专精特新企业
谷禾健康
肾脏的主要功能是保持体内平衡,包括酸碱平衡,水平衡,血压调节和葡萄糖稳态在内的功能需要肾脏进行精确的协调和调节。 全球大约9%的人口正在遭受慢性肾脏疾病。近年来,研究表明,肠道菌群的变化(通常是各种细菌的相对比例的变化)与多种疾病和病症相关,包括肥胖,糖尿病,躁郁症和抑郁症,人们也越来越关注肠道菌群在调节肾脏疾病结局中的潜在作用。但是这些菌群和疾病研究中有许多是相关的,通常不清楚为什么会发生这些变化,或者是否是因果关系以及这些变化是否以某种方式促进了相关的表型?
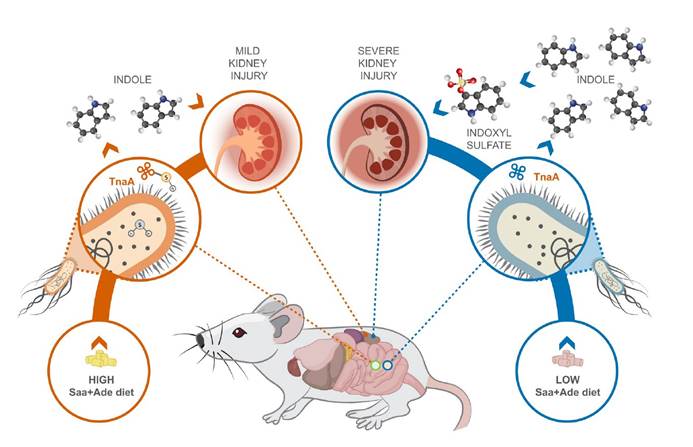
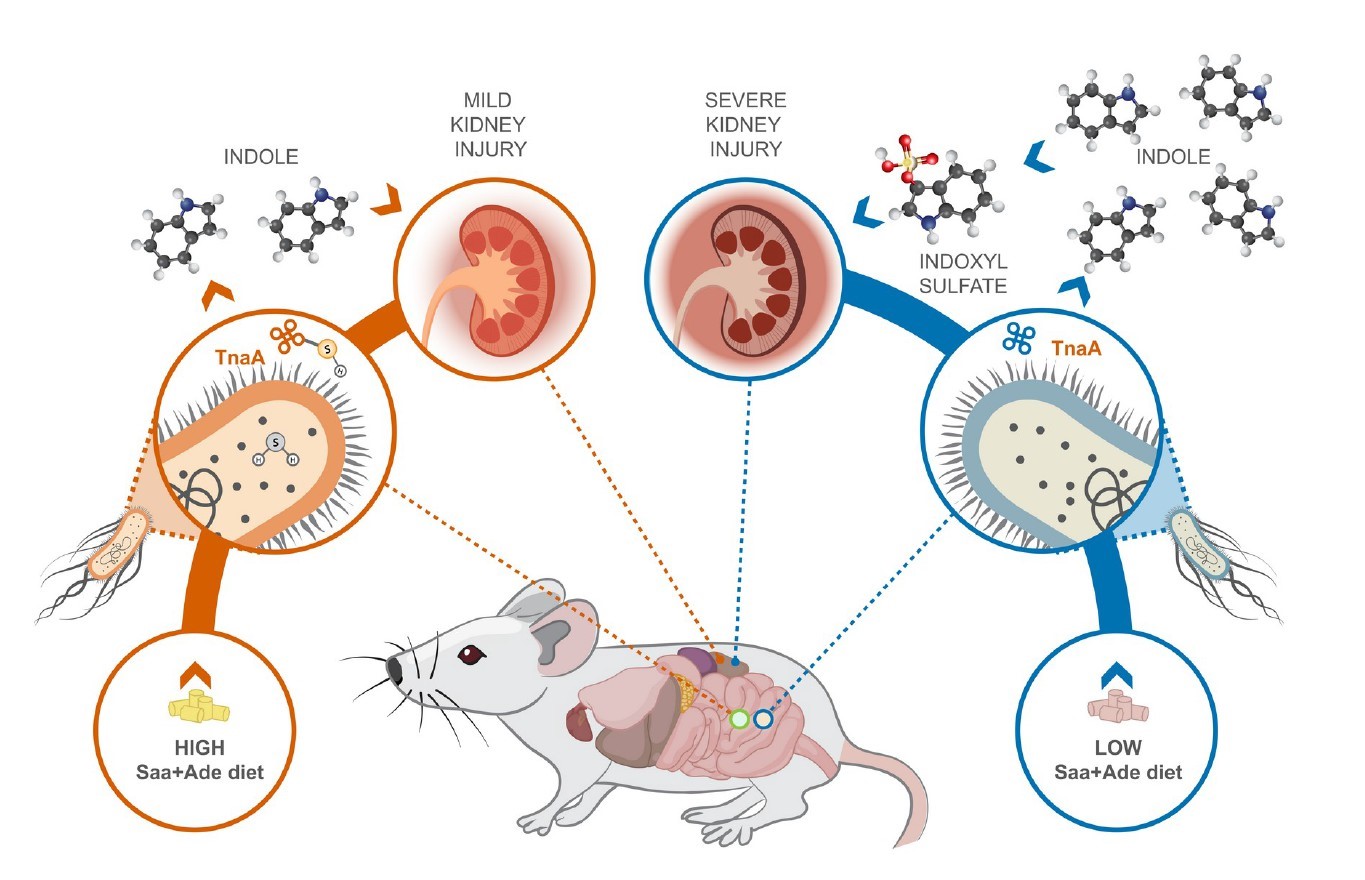
近日,发在在《Science》上的一篇来自哈佛大学公共卫生学院和医学院的教授Wendy S. Garrett团队的关于慢性肾脏病与肠道菌群的研究(Diet posttranslationally modifies the mouse gut microbial proteome to modulate renal function)在这个范例上引入了全新的思路。文章指出饮食变化可以触发微生物蛋白的翻译后修饰,从而改变尿毒症毒素的形成,从而影响慢性肾脏疾病的进展。这些发现可能有助于慢性肾脏病的治疗,并为改善人类健康提供了针对微生物群及其蛋白质组酶活性的临床方法。

缩略词:
Saa:含硫氨基酸
Ade:腺嘌呤
SPF:无特殊病原体
GF:无菌
SAF: 无菌小鼠肠道中接种ASF
研究背景:
尽管饮食调整是慢性肾脏病治疗的基石,但饮食与微生物群相互作用在慢性肾脏病发病机理和治疗中的机制作用尚未得到充分研究。 数十年来,改变患者饮食中蛋白质的摄入量一直是治疗慢性肾脏病的临床策略,更具体地说,是饮食中含硫氨基酸(Saa)的摄入量会影响患者和疾病模型的慢性肾脏病进展。尽管膳食氨基酸中有5%到10%到达了大多数肠道细菌代谢发生的结肠。 在人类中,膳食蛋白质的增加会增加肠道细菌产生硫化氢(H2S),吲哚和吲哚酚硫酸盐的产量。 吲哚和吲哚酚硫酸盐是尿毒症毒素,H2S具有多种生理功能,其中一些功能是由翻译后修饰S-硫酸化介导的。尿毒症毒素是肾功能受损时残留在血液中的化合物,尿毒症毒素的增加会导致多种疾病,包括内皮功能障碍。尽管已在哺乳动物系统中进行了大量研究,但仍未充分研究H2S在调节宿主内肠道细菌功能方面的生理作用。
实验方法:
⑴ 组织学观察:小鼠肾脏石蜡包埋切片观察,包括肾皮质和髓质的定向切片。
⑵ 血清肌酐测定,血清肌酸酐比色测定试剂盒(Cayman Chemical)测量肌酐水平。
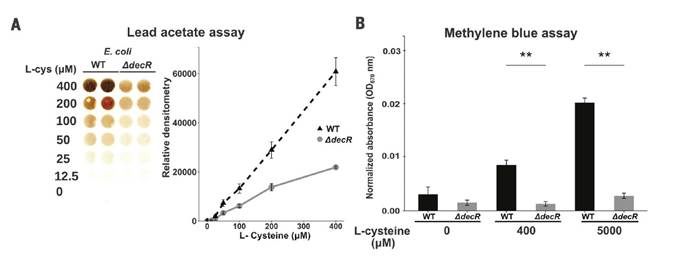
⑶ H2S测定:ASF小鼠盲肠硫化物的检测,采用上述醋酸铅硫化物分析法进行分析。
⑷ 宿主肾基因表达分析:石蜡块中提取小鼠肾RNA,然后进行RT-PCR定量分析。
⑸ 慢性肾病患者粪便微生物组学公共数据集收集和Meta分析
⑹ 菌种培养和试验:E. coli K-12 BW25113菌株自己克隆培养,E. coli MG1655,
BW25113, W3110 和tnaA739::kan来自耶鲁大学大肠杆菌遗传储备中心(CGSC)。
⑺ 盲肠DNA提取及RT-qPCR分析:
⑻ 细菌16srRNA基因扩增子序列:盲肠内容物16s,V4可变区测序分析
⑼ 蛋白质实验:
硫水合蛋白质的降解
蛋白质印迹分析实验
蛋白质消化和标记实验
⑽ 质谱分析,吲哚和硫酸吲哚的LC-MS/MS分析
⑾用Kovac试剂比色法测定吲哚
⑿ 体外色氨酸酶测定
⒀大肠杆菌的基因插入与克隆
实验结论:
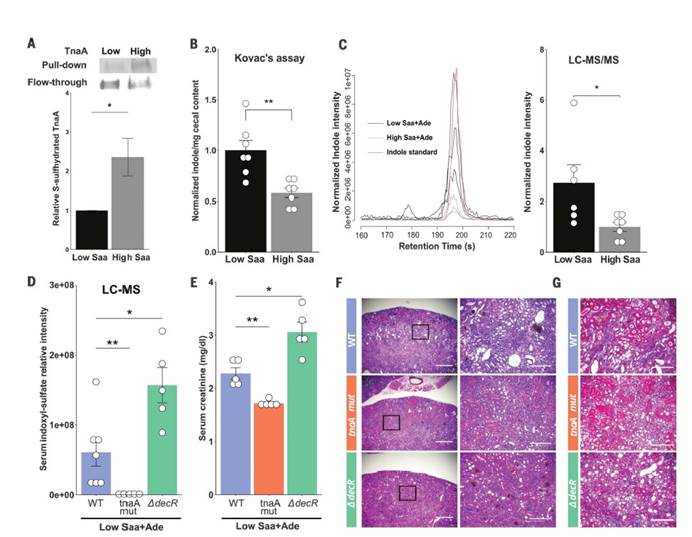
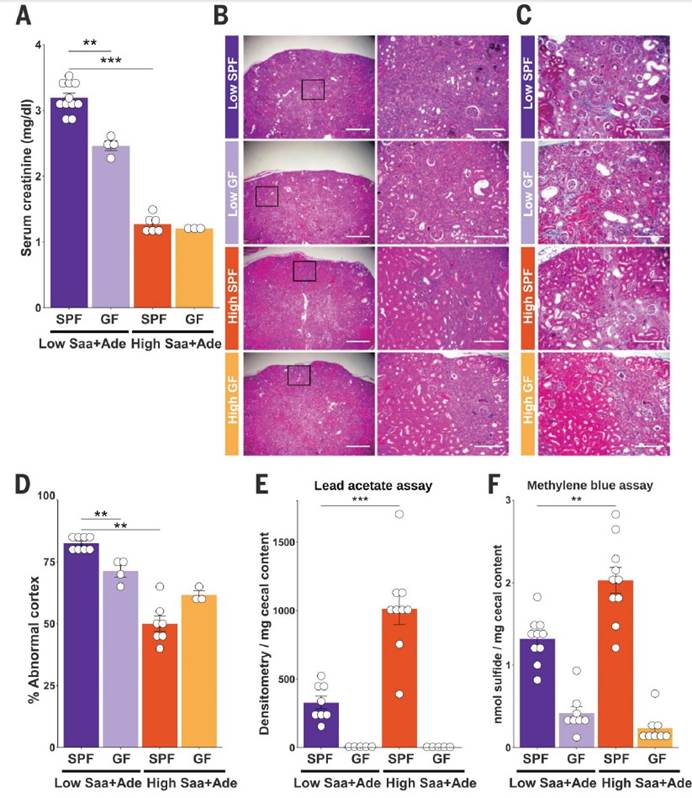
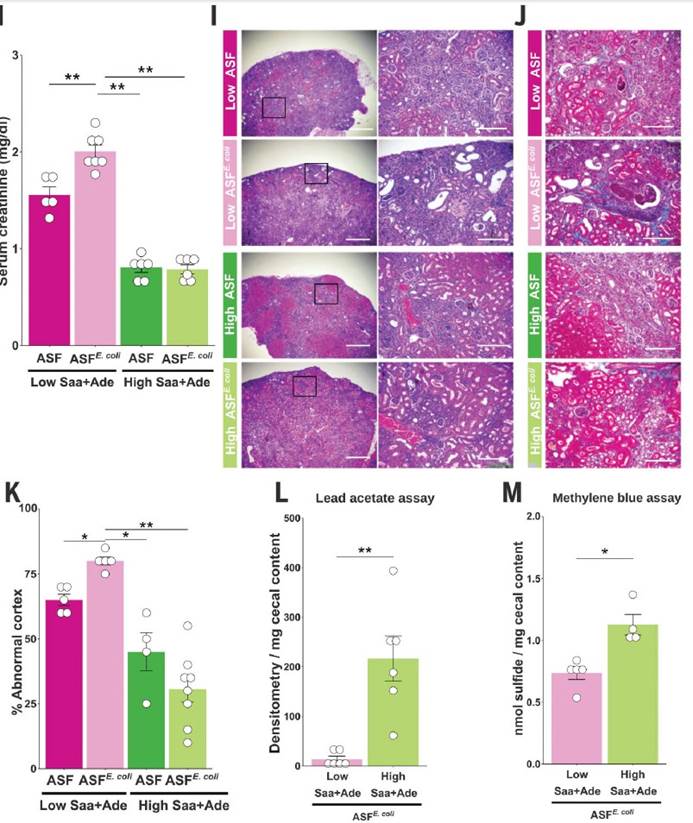
一,与高Saa + Ade(腺嘌呤)的小鼠相比,低Saa + Ade的饮食常规饲养,无特定病原(SPF)小鼠的血清肌酐水平显着升高,而且范围更广以及严重的肾皮质组织病理学改变,包括肾小管扩张和脱落,肾小管炎伴肾小管周围纤维化以及皮质晶体沉积。与低Saa + Ade饮食的SPF小鼠相比,GF小鼠的血清肌酐和肾脏损害明显减少,并且在高Saa + Ade饮食的GF和SPF小鼠中存在相似的表型。
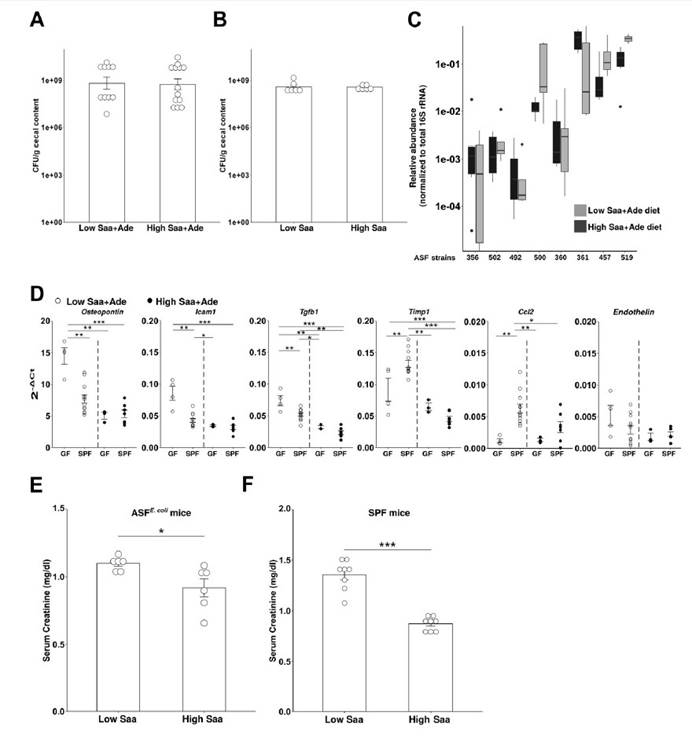
二,微生物群可以缓冲某些宿主基因的表达,同时刺激其他宿主基因的表达。低Saa饮食会加剧观察到的慢性肾脏病表型,而肠道菌群的存在会进一步放大这些影响。
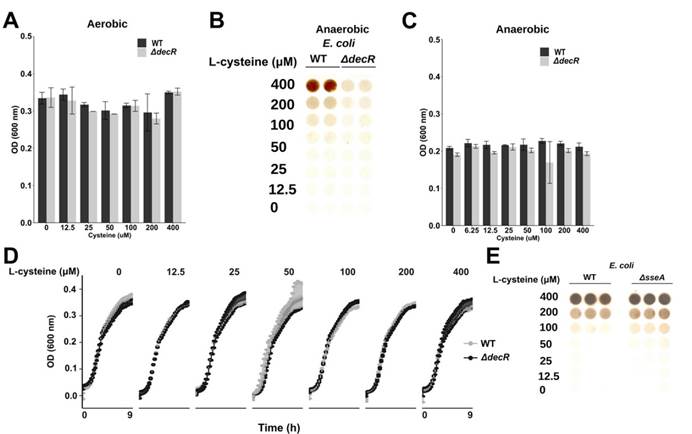
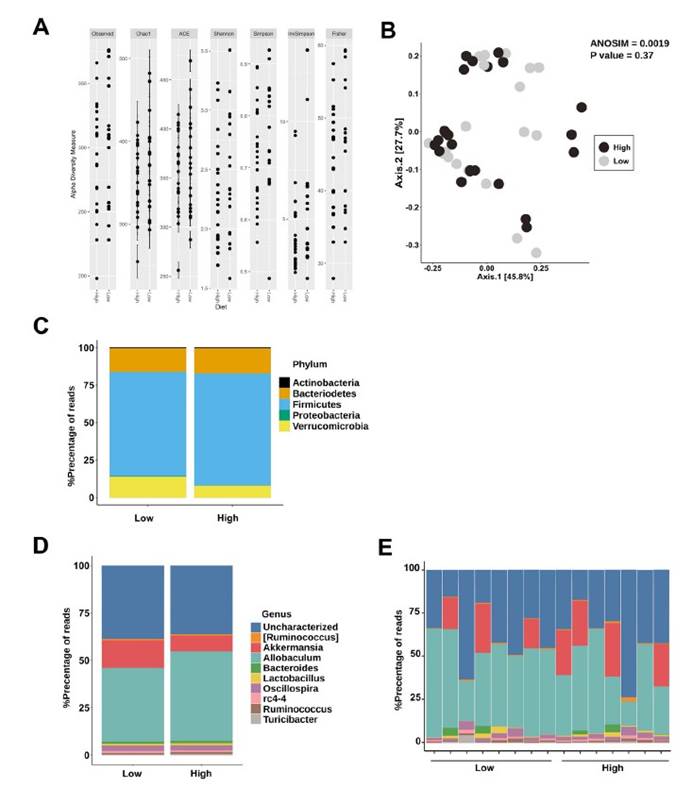
三,未观察到低Saa和高Saa饮食的SPF小鼠肠道微生物群成员的分类丰度存在任何显著差异,支持健康小鼠盲肠硫化物的差异可能是通过改变微生物功能而不是通过改变微生物功能来实现的微生物群落结构的变化。
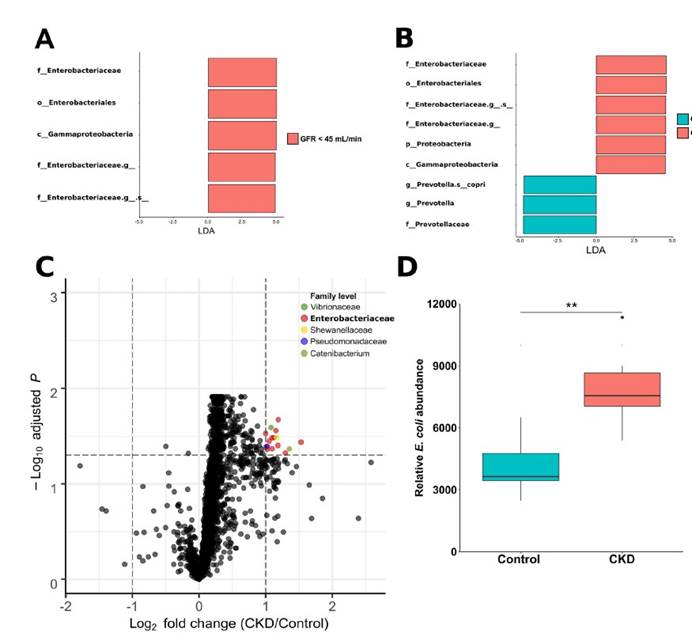
四,在患有终末期肾脏疾病的慢性肾脏病患者的粪便样本中测得的7种大肠杆菌的总平均丰度显着增加。
五,在低Saa + Ade饮食下,定植于大肠杆菌(ASFE. coli)的ASF小鼠比ASF小鼠具有更高的血清肌酐和更广泛的肾小管炎,肾小管萎缩和脱落,肾小管周围纤维化和皮质晶体。在低Saa+Ade饮食中,ASF小鼠感染了大肠杆菌(ASFE. coli)比ASF小鼠血清肌酐更高,更广泛的小管炎,肾小管萎缩和脱落,肾小管周围纤维化和皮质结晶。
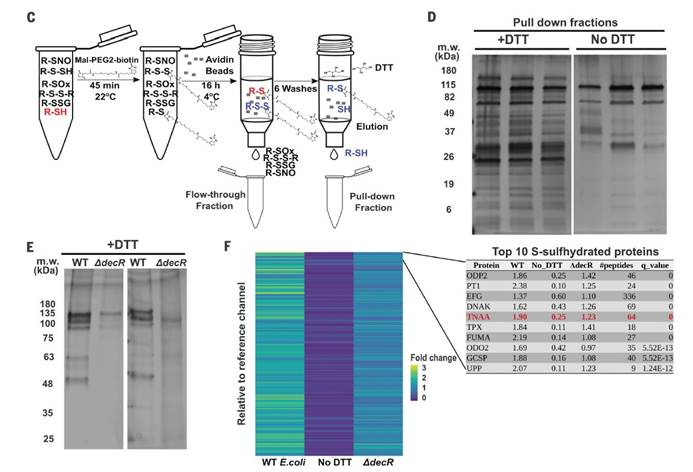
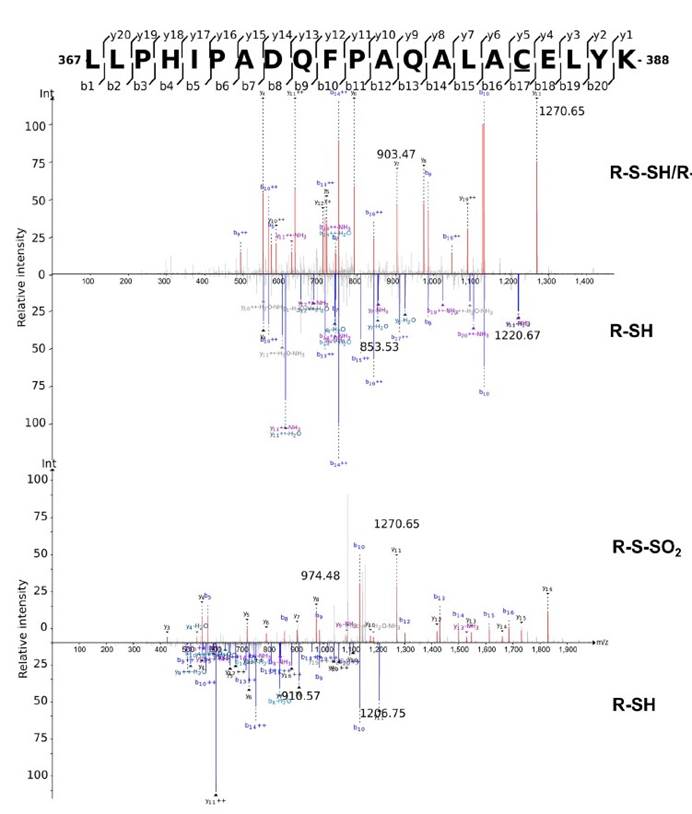
六,用定量串联质谱(TMT)和液相色谱-多阶段质谱(LC-MS3)分析表明大多数在大肠杆菌裂解物中富集的蛋白质确实是S-硫水合物洗脱的。
七,SPF和ASFE.coli中的血清肌酐水平结果支持大肠杆菌与饮食性Saa相互作用以调节肾脏功能。
八,SPF和ASFE.coli小鼠中盲肠H2S的观察结果显示需氧或厌氧生长的大肠杆菌以剂量依赖性方式从半胱氨酸产生硫化物,而对生长没有任何影响。
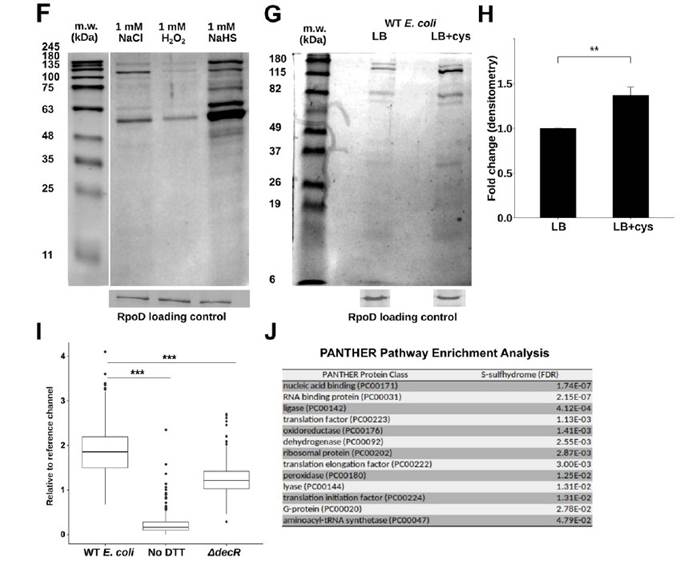
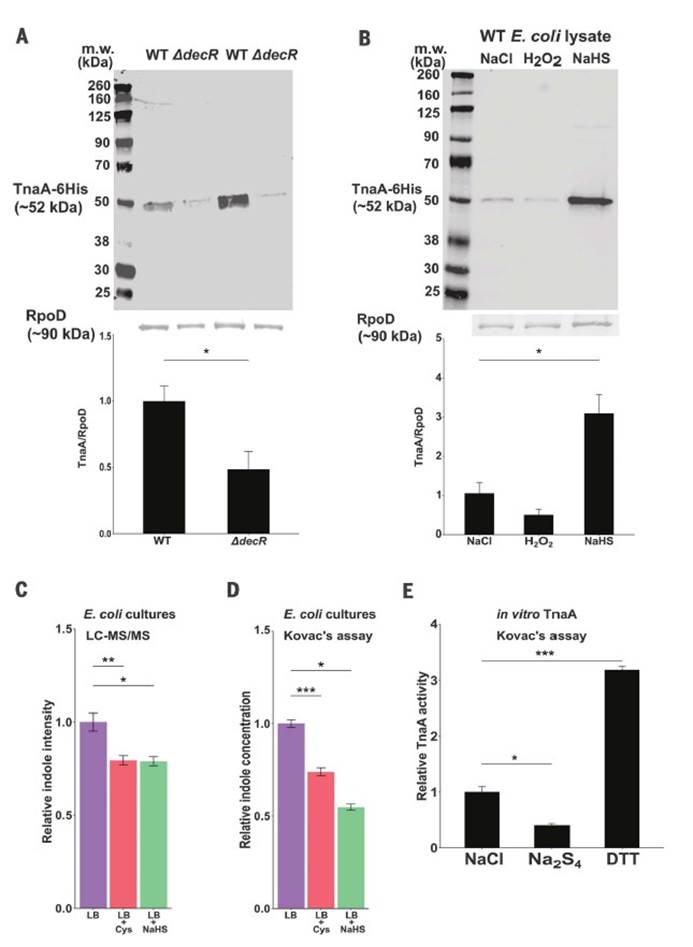
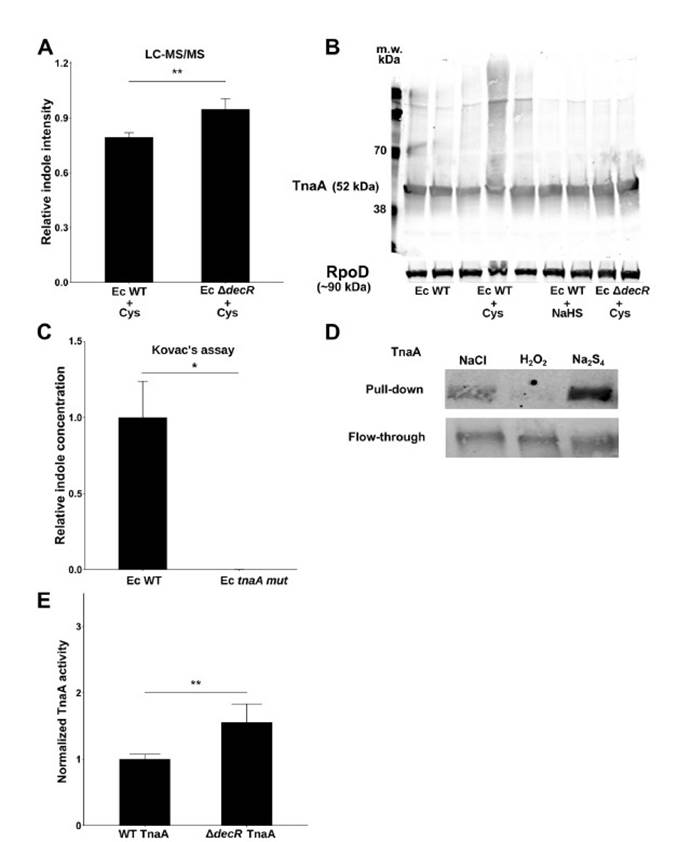
九,Western印迹分析法分析WT与DdecR大肠杆菌裂解物中的TnaA S-硫酸化反应验证了S-巯基水解结果,并发现DdecR裂解物中TnaA S-硫酸化反应降低。 用H2O2和NaHS处理的大肠杆菌裂解物分别显示出减少和增加的TnaA S硫酸盐化作用。
十,外从细菌细胞裂解物中检测到纯化的蛋白质和TnaA的TnaA S硫酸盐化反应,证明了这种修饰抑制了其活性。
十一, 发现采用高Saa饮食的小鼠的吲哚水平显着降低,这表明高饮食Saa不仅增加了TnaA S的硫酸化作用,而且这种修饰足以影响体内TnaA的活性。
研究总结:
研究发现,源自饮食Saa细菌代谢的硫化物通过抑制S-巯基化色氨酸酶来调节大肠杆菌的吲哚生产。表明饮食成分可以被微生物群代谢,从而产生微生物蛋白的翻译后修饰,从而影响宿主的生理,并为宿主-饮食-微生物群之间的相互作用如何促进或阻止诸如慢性肾脏病的疾病状态提供了框架。小鼠慢性肾脏病模型中,饮食的细微变化不会导致微生物成分的变化,这揭示了大肠杆菌产生的吲哚受到肠道细菌内源性产生的硫化物水平的差异性影响。表明细菌代谢不仅可能对宿主生理产生直接影响,而且还可能影响宿主饮食介导的细菌翻译后修饰驱动的微生物相互作用。
局限性:
研究观察表明,GF小鼠在腺嘌呤模型中表现出肾衰竭表型,尽管比SPF小鼠轻,但表明宿主因素以及氧化还原状态(例如,谷胱甘肽水平改变)在饮食Saa中也起作用调节肾功能。
饮食中Saa和细菌TnaA吲哚生产对人慢性肾脏病肾衰竭的贡献值得进一步研究。 在小鼠慢性肾脏病模型中盲肠吲哚的2倍增加最终导致血清吲哚酚硫酸盐增加10倍,这是值得注意的。
研究意义和启示:
研究报道了一种饮食干预措施,该干预措施诱导了微生物酶的翻译后修饰(S-巯基化),导致尿毒症毒素产生的减少,这对肾脏疾病的小鼠模型具有保护作用。尽管这样的研究很困难,但这篇研究提供如何实现它们的示例蓝图。此外,这些研究暗示可以通过改变肠道微生物群和/或肠道微生物蛋白质组的酶活性的新策略来改变慢性肾脏疾病的进展。
这些类型的方法不仅可以为慢性肾脏疾病带来好处,而且可以为与微生物群有关的许多其他疾病和状况带来好处。根据定义,这些类型的研究需要采取多学科的方法,因为不仅必须认识微生物生物学和翻译后修饰方式,还必须了解疾病的整个动物生理学和病理生理学。 因此,对于来自不同领域的科学家们来说,这是一个巨大的机会。通过汇集不同领域的知识和方法,可以增进对宿主-微生物组相互作用和疾病进展的了解,也许可以发现新的疾病治疗和预防方法。
主要图表:


谷禾健康
肠道微生物组与多种人类慢性胃肠道(GI)疾病有关。肠易激综合症(IBS)是一种普遍存在的疾病,其特征是反复出现腹痛或不适。IBS主要见于女性,并与粪便形式或频率变化有关,并基于主要便秘形式(便秘为主(IBS-C),腹泻为主(IBS-D)或混合型(IBS-M))。 由于动物和人类研究之间明显的脱节以及缺乏针对疾病特异性生理变化的综合多组学观点,因此很难确定肠道菌群在疾病中的作用机理。
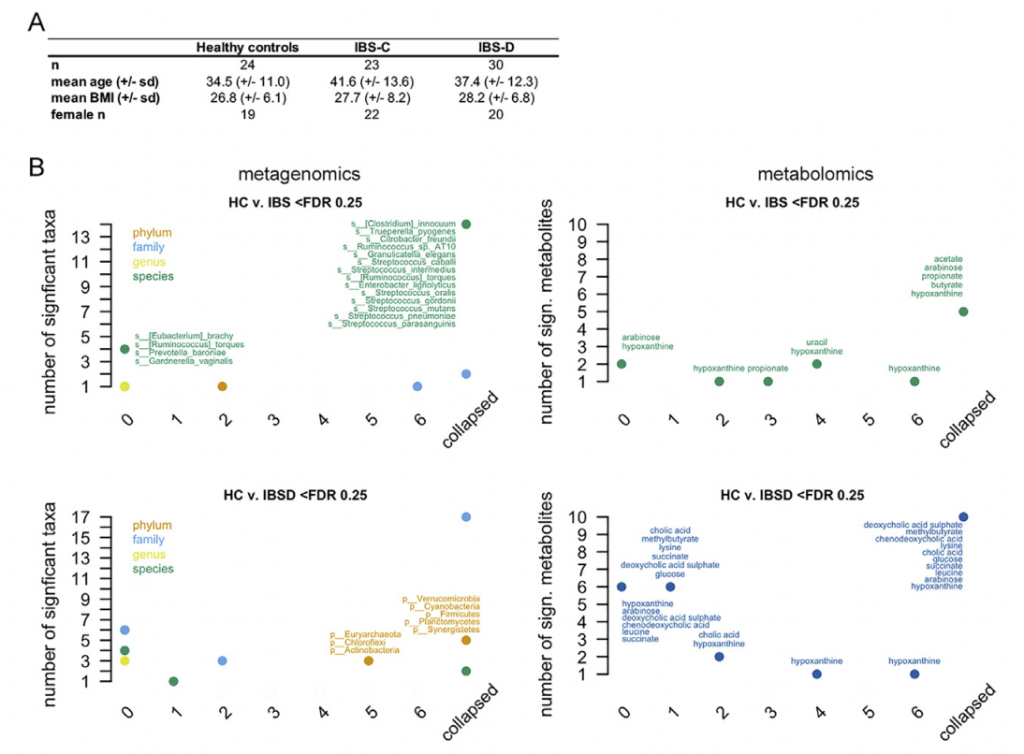

近日,美国梅奥诊所消化内科和肝病科 Purna C. Kashyap研究团队和明尼苏达大学生物科学学院 Dan Knights团队合作在 Cell上发表了题为 Longitudinal Multi-omics Reveals Subset-Specific Mechanisms Underlying Irritable Bowel Syndrome 的文章,研究团队对IBS宿主生理进行了多组学测量的纵向研究 (gun弹枪浅宏基因组测序、16S rRNA基因测序、代谢组学研究,细胞因子测量,转录组和甲基化组分析)最终确定了IBS亚类型特异性、症状相关的微生物组成和功能变异,其中一组已确定的微生物代谢产物变异子集与IBS有关的宿主生理机制相关。 通过Lasso回归机器学习方法整合多个数据层,鉴定出的微生物代谢物变化的子集对应于与IBS相关的宿主生理机制。研究团队将嘌呤代谢确定为一种新型IBS宿主-微生物代谢途径,同时嘌呤饥饿被确认为潜在的IBS治疗靶标。
这项研究主要采用了纵向多时间点采样,针对IBS这类症状波动较为明显的疾病研究纵向多时间点采样可以减少横断面研究的采样偏差,减少入组病人的数量但提高统计检验的效力。可以看到研究纳入的病人并不很多,但采用平均值之后统计效果明显。
另外研究同时进行了多组学的检测,包括代谢组和黏膜转录组等,通过对宏基因组功能差异的分析以及代谢组的差异分析同时锁定了多项代谢异常,尤其是次黄嘌呤的水平异常。进一步对次黄嘌呤水平与宏基因组的菌种进行关联分析锁定了重要的相关菌,通过SV关联分析,在更细尺度上筛选出了可能的功能基因区段。
这种组合方式提供了完整的研究思路,从人群尺度寻找疾病可能的代谢和生理机制,并经由多组学的关联分析锁定可能的菌种标志,再到精细化筛选出可能的功能基因。
在菌群方面研究采用了宏基因组和16S,对粪便样本采用宏基因组,对黏膜样本采用16S,因为黏膜样本含有较高的人体DNA,16S更为合适。粪便样本的宏基因组直接采用和RefSeq89版本进行比对注释,基因部分同时结合了序列比对和利用基因组数据直接提取注释相结合。宏基因组测序能够提供菌株层面的分辨率,同时也是后续结构变异关联分析的必要条件,随着参考基因集的完善,中等测序深度的浅宏基因组将可以大量应用于这类研究中(浅宏基因组文末详细介绍和福利活动 )。
全文缩略词整理:
肠易激综合征(IBS)
便秘为主的肠易激综合征(IBS-C)
腹泻为主的肠易激综合征(IBS-D)
混合型为主的肠易激综合征(IBS-M)
健康对照(HCs)
症状严重程度评分(SSS)
短链脂肪酸(SCFA)
靶向液相色谱-质谱(LC-MS)
5-羟色胺受体4(5-HT4R)
胆汁酸(BAs)
胆酸(CA)
鹅去氧胆酸(CDCA)
核磁共振氢谱(1H-NMR)
黄嘌呤磷酸核糖基转移酶(XPRT)
嘌呤核苷磷酸化酶(PNP)
缺失区(DR)
可变区(VR)
Bray-Curtis差异(BCD)
不规则性(BCDI)
差异甲基化区域(DMR)
本文用到的实验方法汇总:
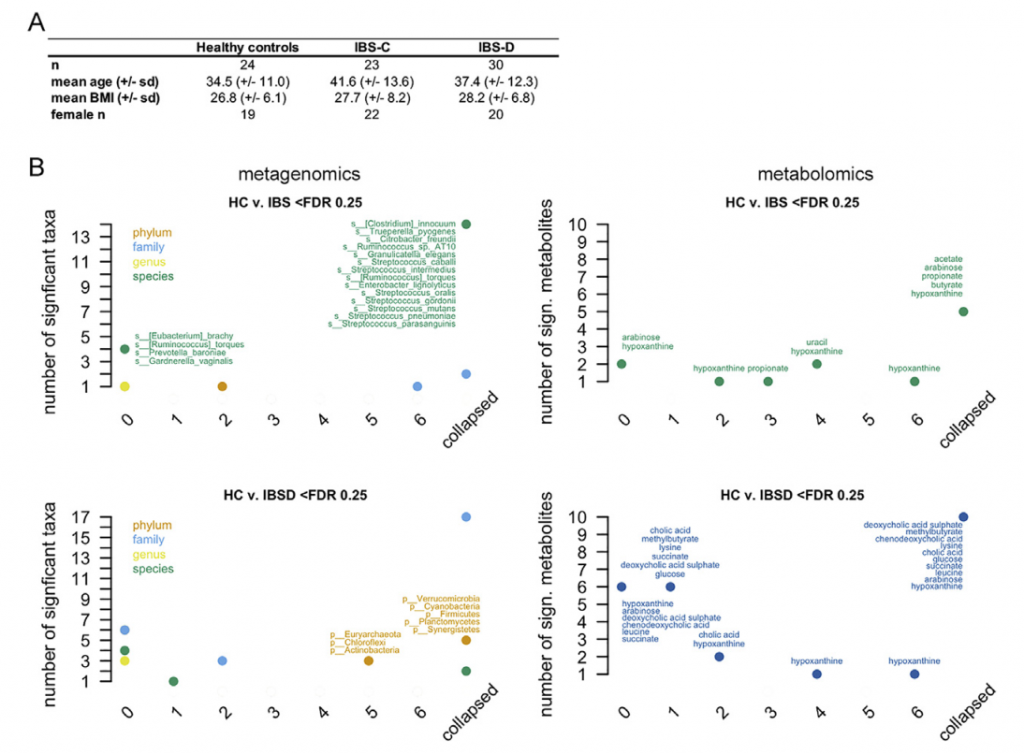
样本人群和数据生成
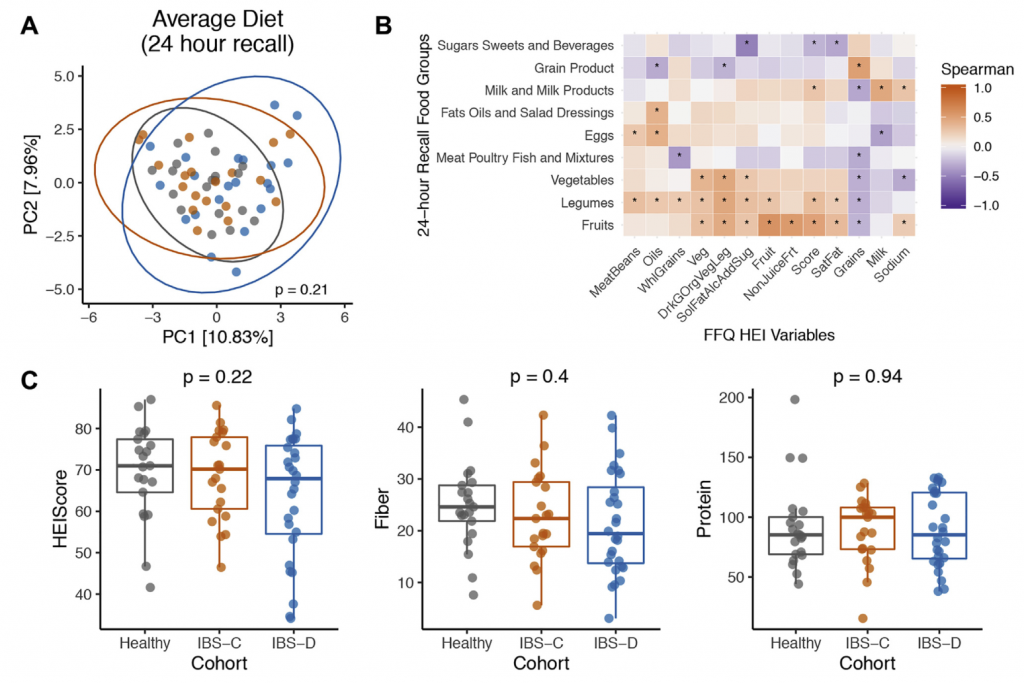
18-65岁的IBS-C和IBS-D患者经过严格的筛查确诊患者入组。IBS-C和IBS-D受试者均符合罗马III级标准。有腹部手术史(阑尾切除和胆囊切除除外)、被诊断为炎症性肠道疾病、显微镜下结肠炎、腹腔疾病或其他炎症性疾病、在过去4周内使用抗生素、出血风险或服用增加出血风险的药物、过去一周准备接受结肠镜检查、怀孕、计划在研究期间怀孕、是易受感染的成年人以及年龄在18岁以下或65岁以上的志愿者被排除在外。
食物频率问卷(FFQ)和24小时饮食回想问卷
动物实验
小鼠实验
Ussing chamber 试验
Ussing Chamber,尤斯灌流室,离是研究跨上皮转运的工具,可用于包括离子转运、营养物质转运及药物转运等的研究。通过跨上皮转运的研究,可以了解上皮的离子通道机制、营养成分及药物透过上皮的吸收、影响上皮屏障功能以及通透性的因素等。本文的Ussing Chamber实验研究了5-羟色胺(5-HT)对结肠上皮短路电流(ISC;一种反映肠道分泌物的跨上皮离子流量的测量)的变化。
微生物组测序
QIAGEN PowerSoil 提取粪便和组织DNA
浅宏基因组使用HiSeq 2500(快速模式)单端读数为100 bp(1×100)和NextSeq 150 bp单端读数(1×150)进行测序。
扩增子测序,对核糖体RNA基因的V4区进行测序,扩增子序列与来自同一细菌基因组的16S rRNA基因在shotgun测序法中使用BURST。
微生物组数据分析
宏基因组部分使用的是浅宏基因组,使用1×100或1×150单端,使用BURST以97%相似度和RefSeq(v86版本)进行比对,过滤比对测序深度低于1万reads的样本。KEGG正射图也可以从参考基因组中进行预测,并利用SHOGUN对预测的图谱进行扩充,以改进对低丰度基因的估计
基因丰度部分是直接比对从refseq基因组中提取的KEGG注释序列,并利用SHOGUN改进低丰度基因的预测)
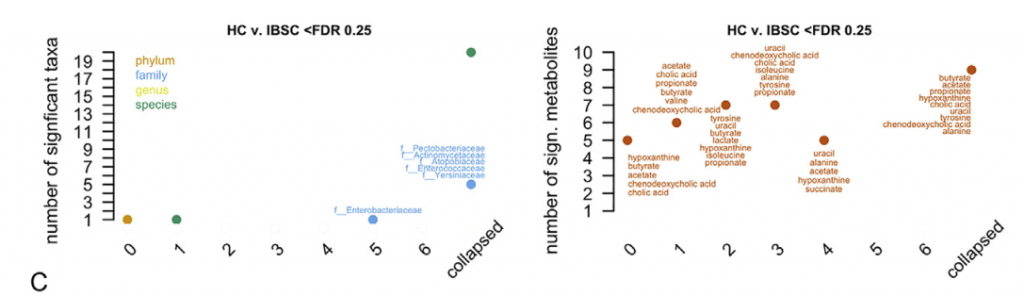
在测试亚组之间的分类单元差异之前,删除了90%的受试者中不存在的分类单元。 为了识别差异丰富的特征,使用FDR截止值<0.25。
通过提取所有健康对照(HC)与HC或IBS样本之间的成对差异来计算基于Bray-Curtis差异(BCD)的不规则性(BCDI),并存储这些差异的中位数。HC值的第90个百分位数用作鉴定与HC样品不同的微生物组样品的临界值。通过随机抽取每个HC对象一个样本并在这些样本中识别BCDI 500次,对第90个百分位数的临界值进行了敏感性分析。 此外,还计算了平均HC微生物组丰度的第90个百分位数临界值。 使用平均值不会改变第90个百分位数的临界值(0.63)。
代谢组学
血清和粪便样品的核磁共振代谢(1H-NMR)谱分析,测定粪便样品中丙酸、丁酸和醋酸的相对丰度,非靶向1H-NMR谱判别分析(PLS-DA)模型确定了IBS亚组和HC粪便样本之间的代谢变异
通过LC-MS /MS进行胆汁酸分析
使用ACQUITY超高效液相色谱(UPLC)系统和Xevo G2-S四极飞行时间(Q-TOF)质谱仪上对样品进行了分析。
通过GC-MS / MS进行SCFA定量
使用7000D 三重四极杆 GC/MS((Agilent Technologies Ltd.)对SCFA进行定量分析。7000D 三重四极杆气质联用系统是 GC/MS/MS 发展史上迄今为止最为成功的最新型号。使用真正的SCFA标准品绘制了11点校准曲线和合并的QC样品。
通过LC-MS / MS进行色氨酸定量
色氨酸定量使用LC-MS/MS在Waters Acquity UPLC上进行,色谱柱为T3 C18柱(1 ×50 mm,1.7 uM),联用Waters Xevo TQ-S三重四极杆质谱仪。
细胞因子测量
使用多路Luminex定量分析IL-8,IFNγ,IL-10,II-18,IL-22,瘦素,血管内皮生长因子(VEGF),膜结合免疫球蛋白(MIG),IL-1β,IL-17A,IL-1RA,IL-6和 TNFα。使用酶联免疫吸附测定(ELISA)对TGFb-1进行了定量。
RNA测序和分析
提取mRNA, 使用Illumina High Seq-2000测序,使用MAP-RSeq v.2.0.0、edgeR 2.6.2、R包RITAN(https://www.bioconductor.org/packages/release/bioc/html/RITAN.html)进行基因机损,差异表达和路径富集分析。
甲基基因组测序和分析
使用Illumina Infinium甲基化EPIC BeadChip评估基因组DNA中的全基因组甲基化,使用R软件包ChAMP 2.9.10、Combat方法、limma函数和Benjamini-Hochberg(BH)得到甲基化的CpG位点,使用R包RITAN,将与DMC或DMR相关的基因用于途径富集分析。
多组学数据整合
使用R中的Maaslin2软件包(http://huttenhower.sph.harvard.edu/maaslin)研究了粪便微生物特征与粪便代谢产物之间的关联。 使用最小丰度运行Maaslin2,将微生物特征的最小患病率分别设置为0.0001和0.5。 经FDR校正的q值的阈值设置为0.25。 将线性混合效应模型应用于与被设置为随机效应的受试者的关联。
用Lasso回归机器学习方法拟合,鉴定基因-微生物组和基因-代谢物关联,该基因模型使用每个基因的基因表达作为响应,并使用微生物组丰度或代谢物浓度作为预测因子。回归方法是一种对数值型连续随机变量进行预测和建模的监督学习算法,回归分为Linear Regression线性回归,Logistic Regression逻辑回归,Polynomial Regression多项式回归,Stepwise Regression逐步回归,Ridge Regression岭回归,Lasso Regression套索回归,ElasticNet回归。其中,Lasso回归方法是在统计学和机器学习中同时进行特征选择和正则化(数学)的回归分析方法,旨在增强统计模型的预测准确性和可解释性,在实践中,岭回归与套索回归首先岭回归。但是,如果特征特别多,而某些特征更重要,具有选择性,那就选择Lasso可能更好。
体内和体外次黄嘌呤消耗实验
Mega培养基中培养细菌,使用LC-MS测定培养上清液中的次黄嘌呤水平,在6周龄的无菌Swiss Webster小鼠上进行了单菌落实验。
次黄嘌呤(Sigma-Aldrich)管饲喂养后第4天,处死小鼠并收集盲肠内容物,使用Amplex Red黄嘌呤/黄嘌呤氧化酶测定试剂盒(Thermo Fisher)测定盲肠内次黄嘌呤和黄嘌呤的总浓度
一 纵向采样克服了跨部门微生物组研究中的异质性
慢性胃肠道疾病中肠道微生物组的横断面研究提供了高度动态的生态系统的快照。 除了饮食,药物使用,生活方式和其他环境因素的影响外,随着时间的流逝微生物群的变化也可能反映疾病活动的变化。
该文通过对纵向数据进行二次采样,测试显着的分类单元,将单个时间点的结果与受试者平均所有数据所获得的结果进行比较,来评估纵向采样与横截面采样相比对识别成分变化的影响时间点。
比较不同时间点时,在各个时间点观察到的HC和疾病组之间的分类单元丰度差异非常不一致,并且与平均数据中观察到的变化不重叠。 当使用平均数据而不是单时间点数据时,发现多个链球菌属物种的丰度明显更高。与HC相比,IBS-D中新近鉴定出的门合生植物的丰度要低得多。 我们还发现,个体内部差异高于个体内部差异,这支持了我们对每个个体的纵向数据进行平均的方法(STAR方法,t检验p <0.0001)。
这些发现凸显了在慢性疾病中进行纵向采样的必要性,以可靠地识别使用横截面采样可能遗漏的微生物群变化。 因此,我们主要报告时间平均数据的发现。 最近的一项研究进一步证明了这一点,该研究表明,在多个采样时间点使用平均值时,常用的“组学”方法更为准确。


二 纵向采样揭示了随着时间的流逝,IBS-C微生物群具有更大的可变性
与HC和IBS-D受试者相比,IBS-C患者的粪便微生物群组成随时间表现出更大的变异性。 此外,与IBS-D样本相比,平均IBS-C粪便样本的香农多样性更高(ANOVA,Tukey HSD p值为0.016)。
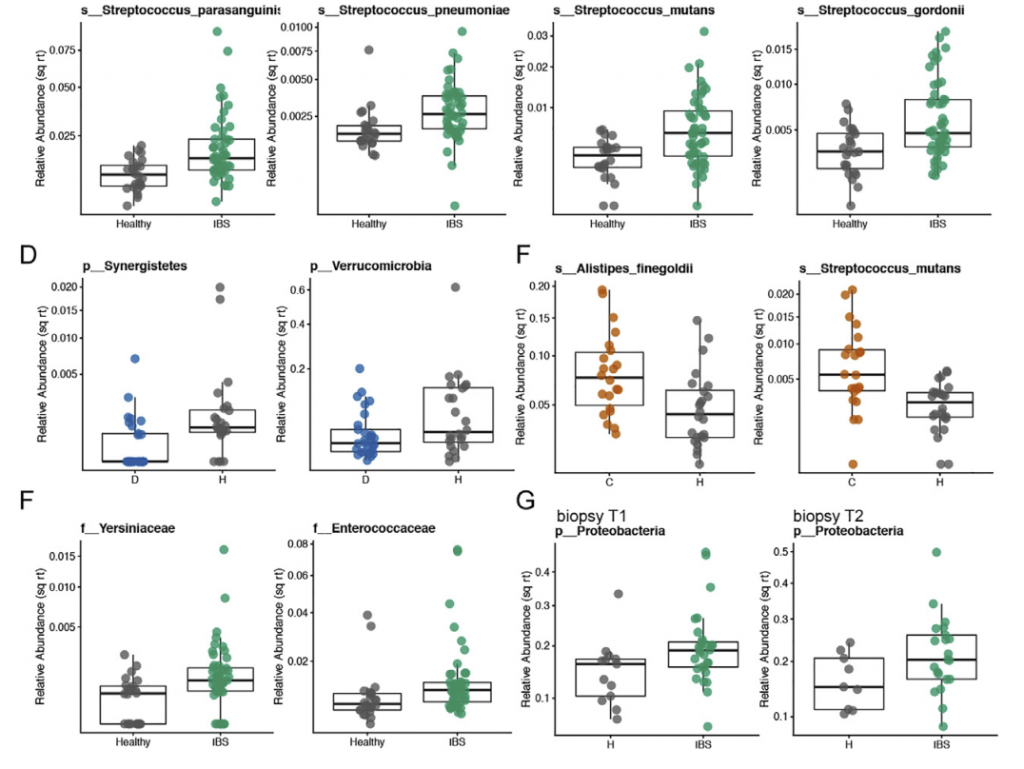

粪便样品中结肠黏膜的微生物组成与腔微生物群有显着差异。 与HC相比,IBS患者的粘膜相关菌群的特征是变形杆菌水平明显较高。 与IBS-D或HC相比,IBS-C患者的与黏膜相关的微生物菌群与其各自的腔内微生物菌群相似性较低。 这可能反映出IBS-C患者菌群的迁移时间较长,而社区之间有更多的时间分化。 此外,IBS-C患者的粘膜相关菌群的个体内变异性随时间变化更大,这与我们在腔菌群中观察到的相似。

三IBS症状严重程度与肠道菌群的功能变化有关
在特定采样点使用IBS SSS(0-500)报告IBS的严重程度,IBS SSS是腹痛强度、频率、肿胀、对排便习惯的不满以及IBS对一般生活的影响的累积度量。我们观察到重度IBS-D(SSS>300)中超过20种乳杆菌的相对丰度高于轻中度IBS-D(SSS<300)。而且这与受试者食用益生菌或乳制品无关。
在粪便宏基因组学功能富集分析时,我们发现在FDR中74个KO与严重的IBS-C相关,而44个与严重的IBS-D相关。 重度IBS-C和IBS-D中均发现了乙醇脱氢酶(ADH)的KO途径,且在重度IBS-C和IBS-D比轻中度IBS中高。 乙醇脱氢酶(ADH)显示与双歧杆菌和链球菌属呈正相关。这些数据表明ADH活性可能与腹痛有关,腹痛是IBS-C和IBS-D共同的主要症状。
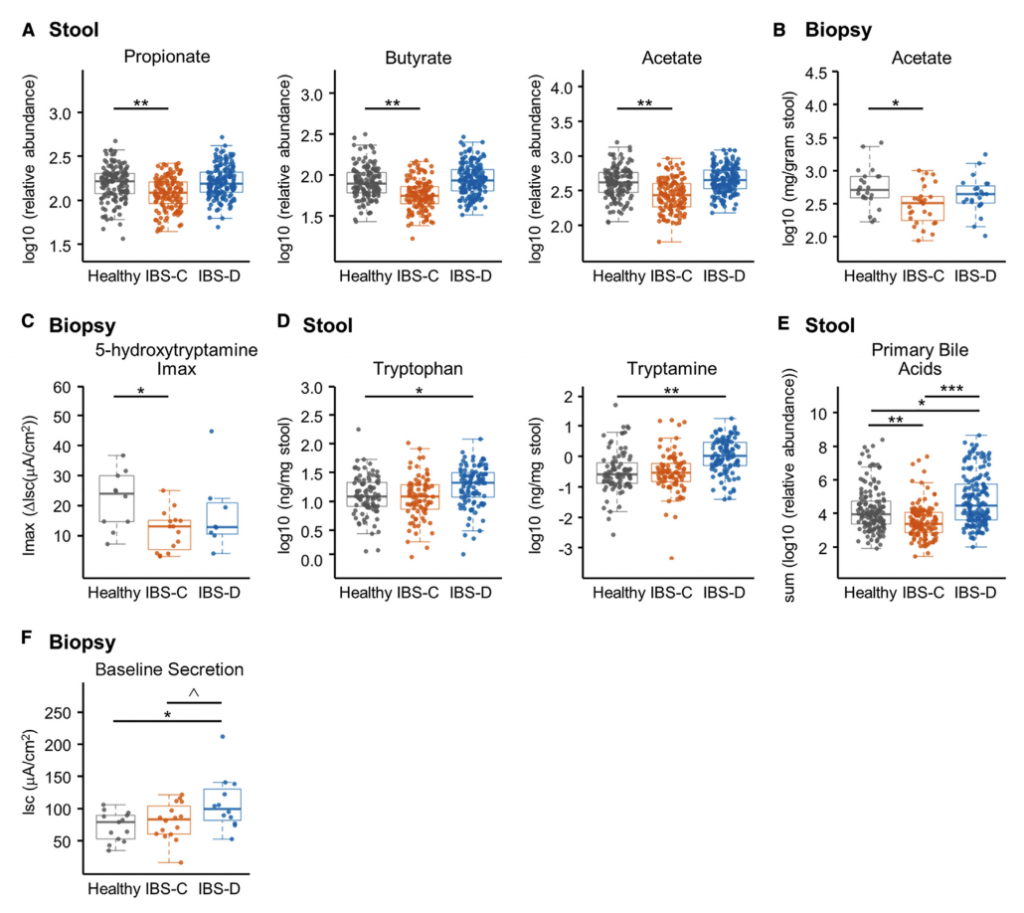

四 代谢组学与生理学结果阐明了肠道微生物群代谢对胃肠功能的影响
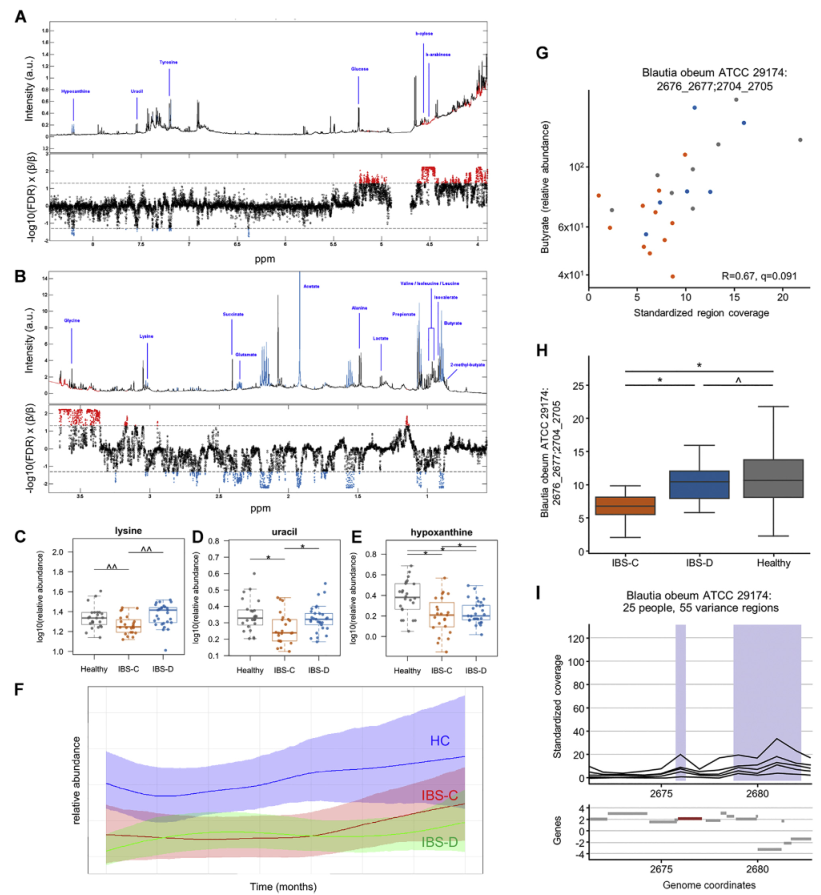
H1核磁共振(NMR)光谱显示,与HC相比,IBS-C患者粪便样本中的短链脂肪酸(SCFA)丙酸酯,丁酸酯和乙酸酯显着降低。与腔内代谢产物一致,与HC组相比,IBS-C组的结肠黏膜活检样品中的乙酸盐(通过气相色谱-质谱[GC-MS]测定)也显着减少。,SCFA的这些差异与膳食纤维的总摄入量无关,因为这在各组之间没有显着差异。
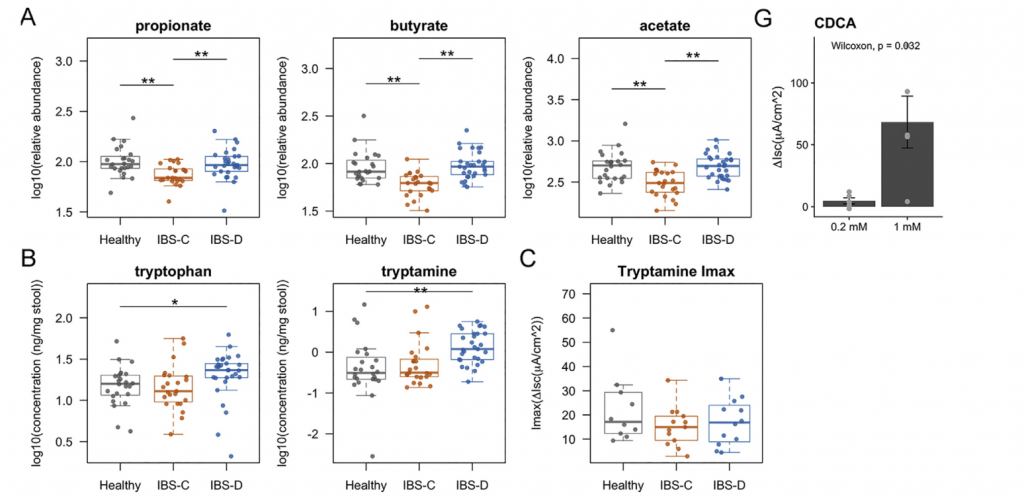

Ussing chamber 试验结果表明水分流伴随离子通量,并且分泌减少会导致便秘中粪便含水量降低。 相反,增加的离子通量可导致分泌更多的水,导致腹泻。色胺均显着增加了结肠分泌且在各组之间没有显着差异,表明,IBS患者和HCs的结肠上皮能够由色胺引起的液体分泌,因此观察到的变化可能是由于色胺的丰度变化导致。
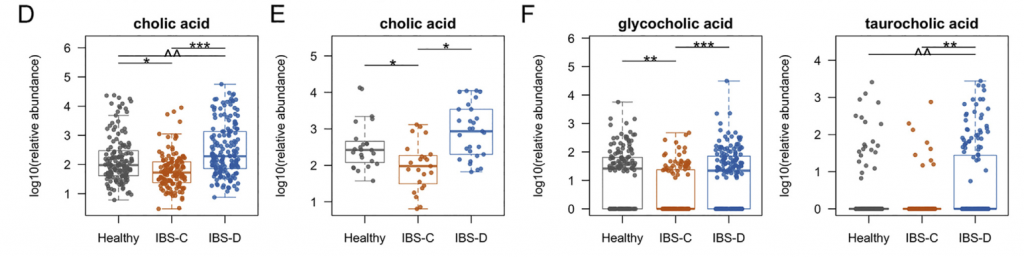

进一步使用靶向液相色谱-质谱(LC-MS)方法研究了粪便样品中色胺和其他色氨酸代谢物的变化。 发现,IBS-D患者粪便样品中的色氨酸和色胺都显着增加,因此可能部分归因于IBS-D粪便中水分的增加。 我们证实这些代谢物的变化与蛋白质摄入的饮食差异无关。
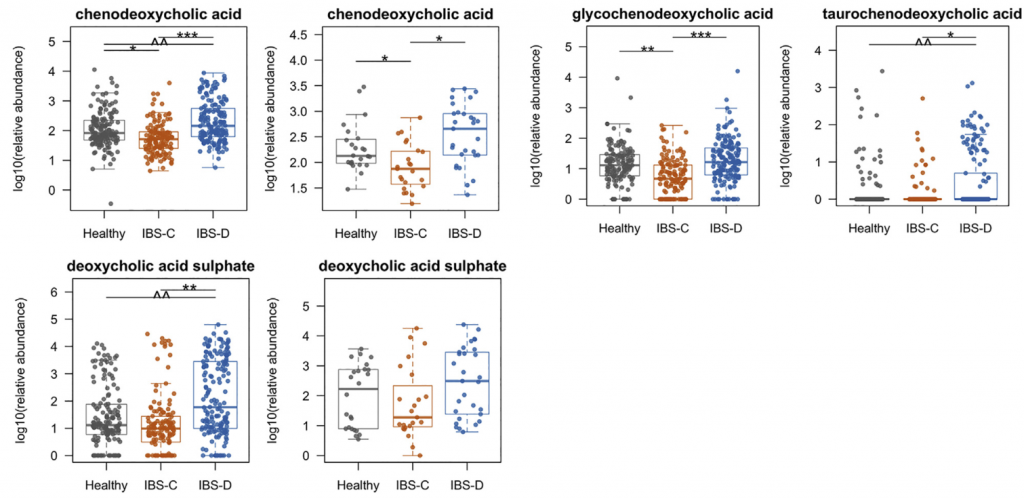

使用LC-MS / MS我们IBS-D患者粪便样品中未结合的初级BA含量明显较高,而IBS-D患者粪便样品中未结合的初级BAs含量明显较低。与HC和IBS-C受试者相比,IBSD中个体初级共轭和非共轭BA和DCA-S的量更高。 由于像CDCA这样的羟基化初级BA可能会增加结肠分泌,因此运用Ussing chamber测试了CDCA在无菌小鼠结肠黏膜下黏膜下层制剂中的作用,结论支持了CDCA水平升高在增加IBS-D患者粪便中水分含量方面的生理作用。
五 联合多组学分析确定了IBS中的新型微生物代谢途径
采用了非靶向代谢组学方法来鉴定可能导致IBS病理生理变化的新型微生物途径。基于无目标1H-NMR光谱图的潜在结构判别分析(PLS-DA)模型的投影确定了IBS亚组和HC粪便样品之间的代谢变化。 与HC相比,IBS-C患者粪便样品中的赖氨酸,尿嘧啶和次黄嘌呤均显着降低。 IBS-D患者的次黄嘌呤含量也较低,尽管与IBS-C的意义不同。

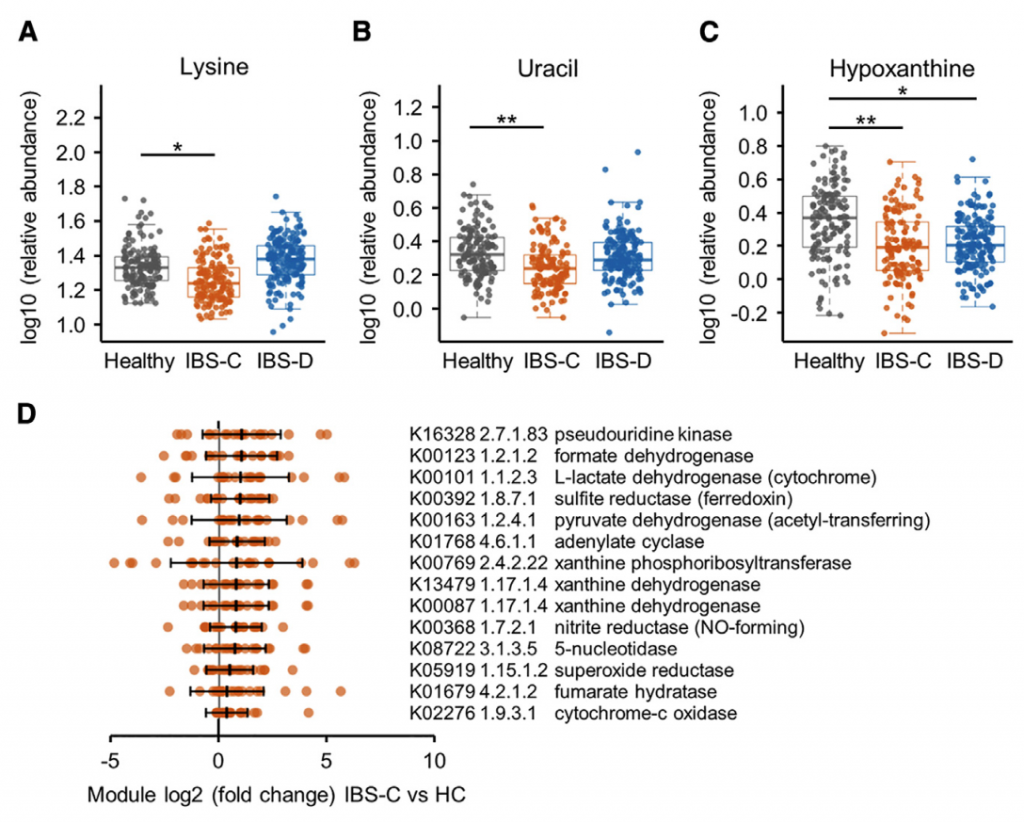
分析IBS和HC患者粪便样本中次黄嘌呤相关的宏基因组学功能,发现IBS-C中的黄嘌呤脱氢酶/氧化酶(XO; 1.17.1.4)和黄嘌呤磷酸核糖基转移酶(XPRT; 2.4.2.22)途径相对于HC有所升高。 XPRT从黄嘌呤单磷酸中释放出黄嘌呤,这是抢救嘌呤的第一步。 在下游,XO是一种具有低底物特异性的酶,可作用于黄嘌呤或次黄嘌呤以产生尿酸。 这些较高水平的XPRT和XO模块表明,IBS患者中肠道菌群对嘌呤的分解作用增加。


进一步检查了宏基因组的KO分析,以探索次黄嘌呤代谢的两个方面,即其在调节上皮能量状态中的作用以及在假定的氧化剂作用下生成H2O2和超氧阴离子。 与HC相比,IBS-C粪便中C氧化酶的丰度明显更高。 有趣的是,IBS-C中的超氧化物还原酶(1.15.1.2)升高,这可能反映了IBS-C肠道微生物组应对氧化应激的能力增强。 在XO活跃的情况下,这可能是必要的。

综上所述,这表明IBS患者的微生物组对次黄嘌呤的利用和分解能力增强,这与IBS-C粪便中次黄嘌呤水平的降低是一致的。
六 胆汁酸,丁酸和次黄嘌呤代谢有关
为了进一步阐明微生物对IBS中鉴定的代谢物丰度的贡献,首先基于线性模型(Maaslin2; http://huttenhower.sph.harvard.edu/maaslin)进行了直接多元相关分析。 这确定了HC样品有60种重要的代谢物种类相关性,IBS-C有28种,IBS-D有46种。 所有组均无相关性。 HC和IBSC或IBS-D中存在12个。 IBS-C和IBS-D子组都存在两种相关性。

尽管以上相关方法使我们能够识别粪便代谢物差异的潜在微生物驱动因素,但无法识别可能与检测到的代谢物差异相关的特定微生物基因。 因此,我们使用最近描述的将结构可变的基因组区域与代谢物丰度相关联的方法(SV关联),测试了可能导致各组之间代谢输出变化的特定细菌基因组区域。
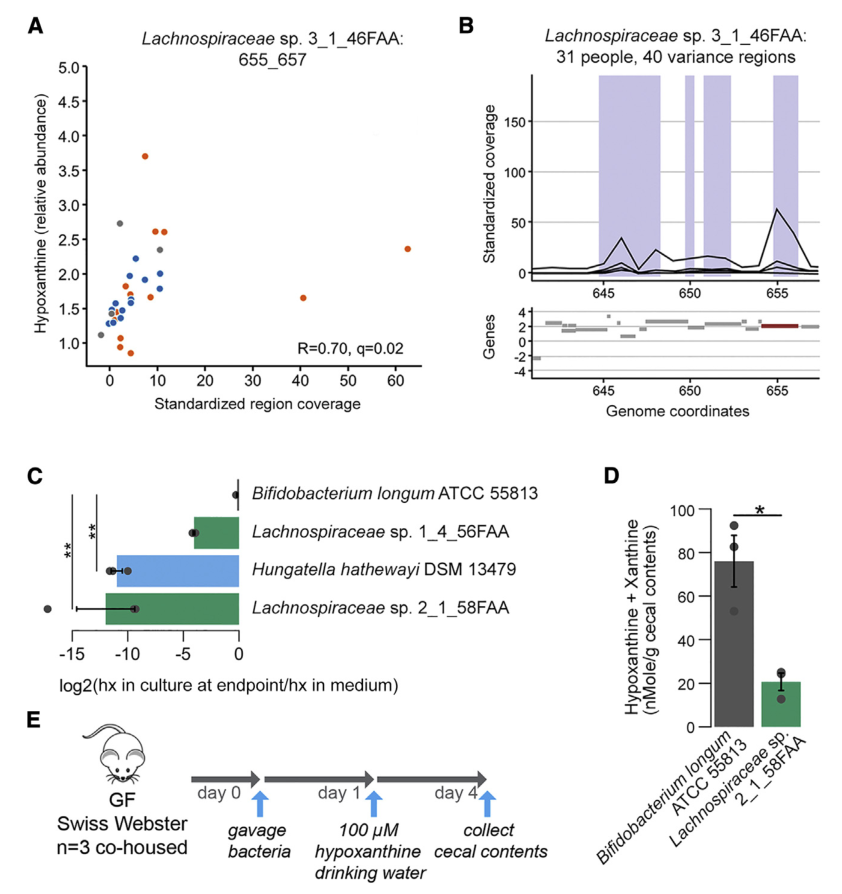
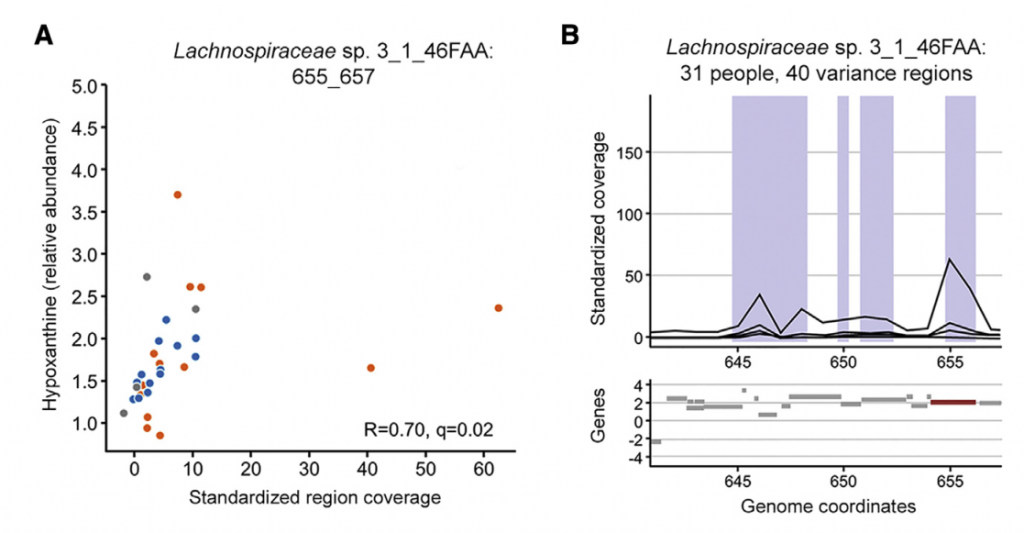
七 微生物代谢有助于次黄嘌呤水平
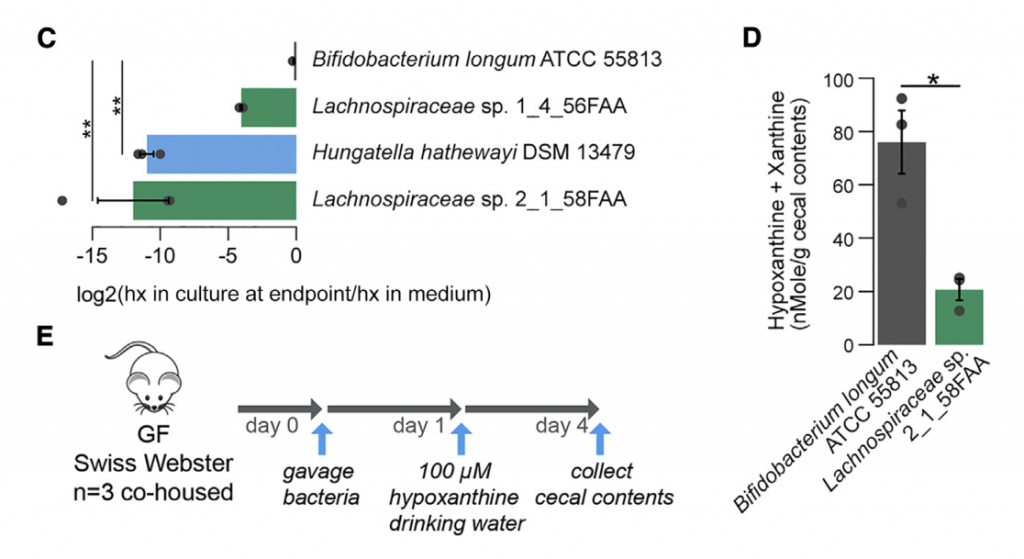
为了更深入地了解微生物组在降低次黄嘌呤水平中的作用,选择了与Lachnospiraceae sp的基因组相似性选择了2个Lachnospiraceae菌株进行无菌小鼠实验,结果表明现定植了Lachnospiraceae sp的小鼠的盲肠内次黄嘌呤水平明显降低。 与长双歧杆菌定植的小鼠相比为2_1_58FAA(图4E)。 由于常规使用次黄嘌呤水平会增加,这表明微生物确定的次黄嘌呤水平是微生物生产与消耗之间平衡的结果。
八 IBS患者爆发时肠道微生物组和微生物代谢产物的改变
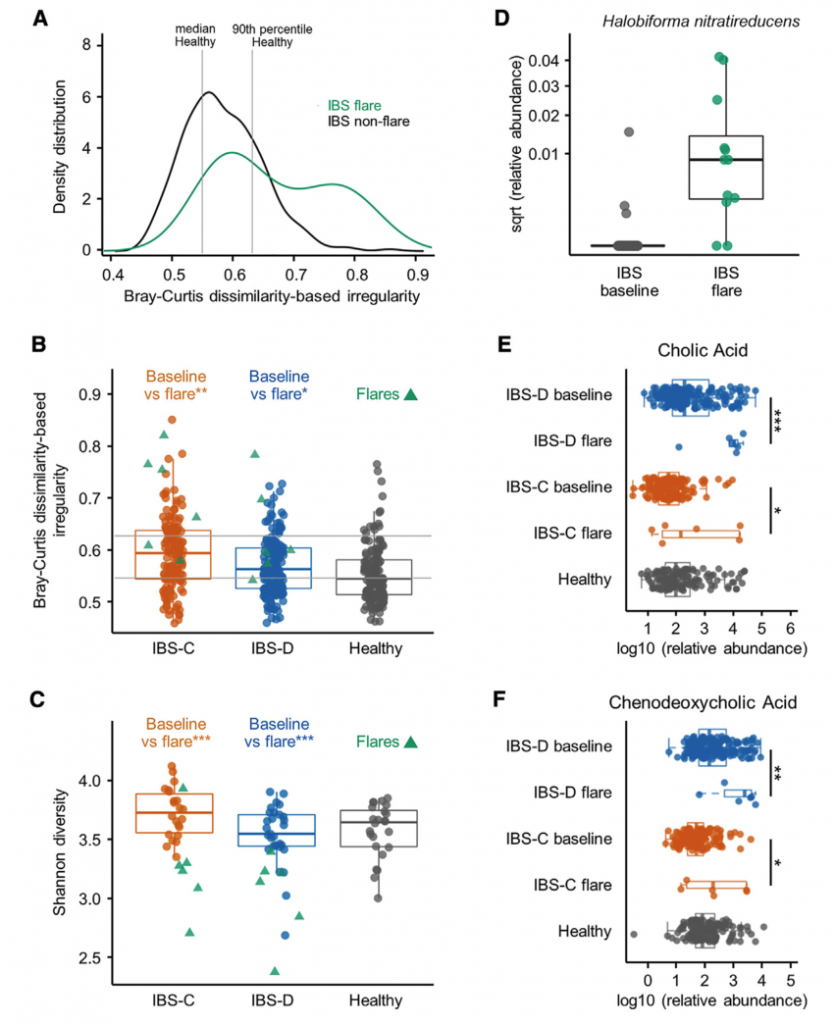
IBS是一种症状严重程度随时间变化的慢性疾病,大多数患者会出现短暂的症状恶化。前面纵向分析确定了肠微生物组与IBS患者症状严重程度之间的潜在联系。对个别报告中显示恶化时收集的粪便样本进行分析,与非爆发基线组合IBS样本相比,爆发期样本显示出更高的BCDI),与各个IBS子组的平均样本相比,爆发期样本的Shannonα多样性更低(图5C)。 在将IBS患者作为一组时以及在IBS-D和IBS-C患者中,特定细菌类群都与耀斑显着相关(IBS-D和IBS-C患者为168种,IBS-C为40种,IBS-D为7种) 与各自平均基线样本相比,来自Mann-Whitney U检验的q <0.1(表S2)。 这些重要物种在爆发期间几乎普遍减少。发现包括色胺,CA和CDCA在内的分泌代谢物在亚组中升高爆发时IBS患者的比例(BA为6/11,色胺为4/11)。 这些观察结果表明,不同患者的症状恶化可能是独特的微生物和代谢特征。


九 微生物组和代谢组学数据与转录组学和表观遗传学差异的整合揭示了IBS中新型的宿主-微生物组相互作用
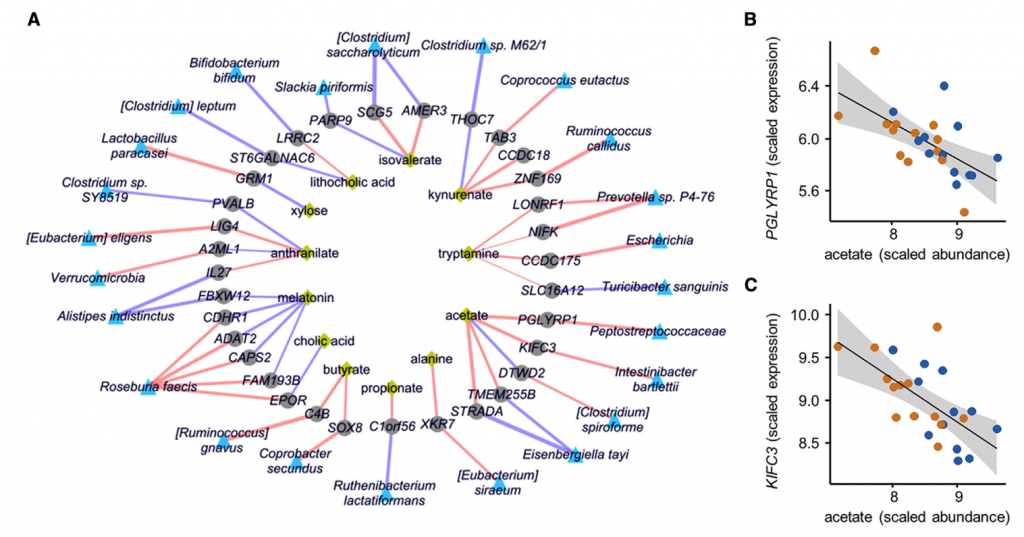
对于大多数慢性疾病,IBS的病理生理是多因素的,其来自宿主途径,微生物途径和宿主-微生物共代谢。 为了确定微生物代谢对宿主功能的影响,我们首先比较了在结肠活检组织中观察到的转录和表观遗传学变化。 还通过构建跨组学相关网络,将转录组数据与代谢物和微生物群的丰度相集合,从而以无针对性的方式,使用这些数据来确定推定的宿主-微生物-代谢相互作用。
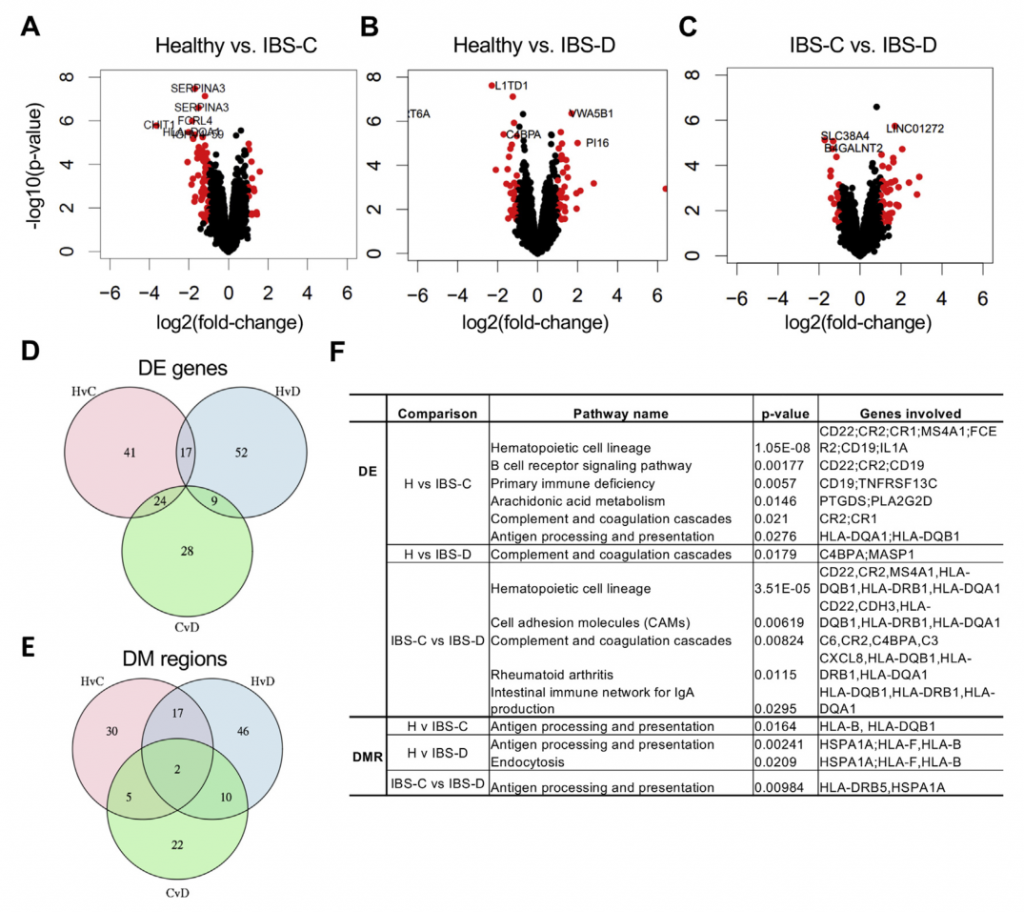

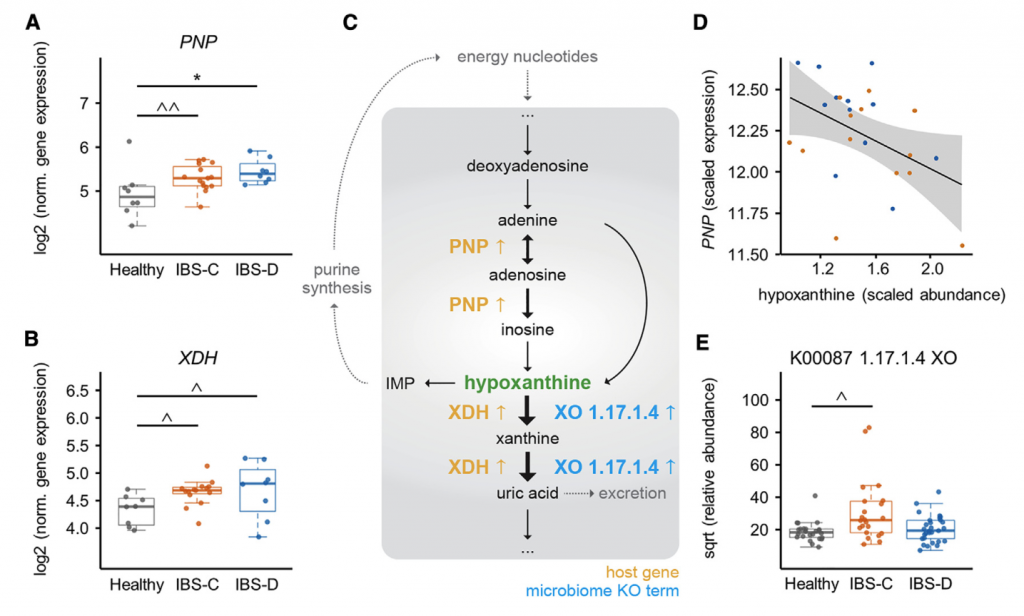

十一 多组学集成确定结肠上皮中的嘌呤饥饿是潜在的新机制IBS
前面确定了IBS-C和IBS-D中粪便次黄嘌呤的含量显着降低,确定了微生物次黄嘌呤的降解导致肠道次黄嘌呤的水平降低,并确定了功能性变化,表明IBS-C粪便中微生物组导致嘌呤降解的增加。 然而,由于次黄嘌呤是宿主-微生物共代谢物,因此其库可同时受到微生物和宿主代谢的影响。
其实,肠上皮细胞的嘌呤从头合成能力有限,而是主要依靠挽救途径进行腺苷酸的生物合成,因此,为了确定由于次黄嘌呤池的消耗而导致宿主的继发效应,需要进一步确认嘌呤挽救途径中可能的转录变化。嘌呤挽救途径中的第一个基因嘌呤核苷磷酸化酶(PNP)在IBS-C和IBS-D中均表达高2倍。在IBS患者中PNP表达呈与次黄嘌呤水平呈负相关。 重要的是,宿主遗传学的变异不负责基因表达的这些差异,因为Illumina全局筛选阵列显示XDH和PNP中的单核苷酸多态性(SNP)在IBS亚组和HC之间没有差异分布。总之,这些发现提出了一种模型,其中微生物群和宿主的嘌呤核苷酸在结肠组织中诱导代谢应激。 反过来,这可能会通过增加嘌呤挽救而导致代偿性反应。 使用这种多组学观点,我们建议低水平的嘌呤核苷酸可能导致较低的上皮能量状态和粘膜修复能力,这可能部分是IBS的病理生理基础。

研究结论
在这项研究中,我们描述了在不同IBS亚型患者的宿主生理情况下,对肠道微生物组,代谢组,宿主表观基因组和转录组进行综合纵向多组学分析的结果。IBS-D患者活检中基线结肠分泌增加,这表明上皮运输的固有变化或促进液体分泌的代谢产物增加。 观察到的促分泌素(例如主要的BA CDCA和细菌代谢物类胰蛋白酶)的增加表明,较高水平的微生物群相关分泌化合物可能会导致IBS-D分泌增加。 三组结肠活检样本之间对色胺的分泌反应缺乏显着差异,这进一步得到了支持,如果结肠上皮存在固有缺陷,这是可以预期的。
先前的研究表明,BA吸收不良驱动了IBS-D的肠道分泌增加,但是在该研究中,继发性BA与原发性BA并存的增加并没有增加,这表明原发性BA的微生物生物转化减少可能至少部分地驱动了这种作用。通过进一步有针对性地整合多个宿主和微生物组数据层,确定了嘌呤代谢的宿主-微生物途径,这可能在IBS的病理生理中起重要作用。
研究意义
这是首次将次黄嘌呤与IBS发病机制联系起来,包括先前对动物模型进行的致生菌研究。 这说明了在人类中采用多组学测量以鉴定可能依赖于基因表达中人类特异性反应的潜在疾病机制的相关性。由于黄嘌呤氧化酶抑制剂别嘌呤醇(用于治疗痛风)和硫嘌呤(用于优化炎症性肠病治疗)的可用性,次黄嘌呤是有吸引力的药物靶标。
该研究为将来的研究提供了多个新的治疗靶标。IBS-D患者中BA明显减少的微生物生物转化可以使用确定的微生物菌群进行治疗,同样,在IBS-C患者中,细菌SCFA和/或色胺的产生增加可能是可行的治疗策略。
最后,在肠道内局部刺激微生物次黄嘌呤的产生或抑制黄嘌呤氧化酶将是一种增加腔次黄嘌呤的量而没有全身作用的新方法,并且可能对独立于疾病亚型的IBS有益。
研究的局限性
研究存在一些局限性。本文主要关注结肠微生物组,但我们知道小肠可能在IBS症状的产生中起重要作用。 需要专门针对小肠微生物组的纵向研究来补充发现并增进对IBS的理解。
福利活动和技术推广
本文的浅宏基因组测序方案是针对16s分辨率和宏基因组高成本之间的一个折中方案,通过降低测序深度,但是物种的分辨率并没有低于一般宏基因组(普遍5~10G数据量)。
经过几个月的研发的测试,谷禾推出浅宏基因组测序分析服务,每个样本数据量不低于100万reads,不通过拼接组装,直接基于kraken2等kmer,或MetaPhlAn2等标记基因的参考基因组方法进行种属丰度分类。结合其到菌株的物种分类和丰度数据可较16s方案下的PICRUST更加准确的预测基因构成。周期在:2-3周左右,尤其适合粪便样本, 特别推荐,目前在推广,价格非常实惠,之比谷禾16s测序价格稍高一点,欢迎咨询和合作。
浅宏基因组分析内容

科研路漫漫,谷禾伴左右!

目前,关于肠道菌群的研究越来越多,相关科研文献数量每年递增。早期对微生物组的研究重点在于对微生物进行分类,使用临床前动物模型来了解表型等,而现有的研究更多地展示了肠道菌群的功能及其作用机制,推进基于菌群治疗技术的发展。
我们也有很多文章介绍了目前肠道菌群的最新研究进展,在向大家展示目前研究进展的同时,我们也一直着力于微生物组在临床上的应用。
这次,我们整理了一些谷禾数据库中关于菌群检测应用于多疾病临床辅助诊断的案例。
我们从常见的儿童疾病中选取了4例较常见的病例,分别是便秘,睡眠障碍,腹泻,自闭症。这些病例从临床症状诊断开始,结合肠道菌群检测结果进行有针对性的临床干预,取得了明显的康复效果。
孩子不吃饭,孩子不排便…这些对于家长来说都是比较头疼的事。宝宝排便情况一定程度上反映着身体健康状况。
我们知道,良好的肠道微生态环境有助于排便,改善结肠传输时间,改善便秘症状。因此,我们需要帮助孩子创建良好的肠道微生态环境。
家长也不用过于焦虑,只要通过合理调理,基本可以改善。我们选取了一个4岁患者的案例,来详细看下:

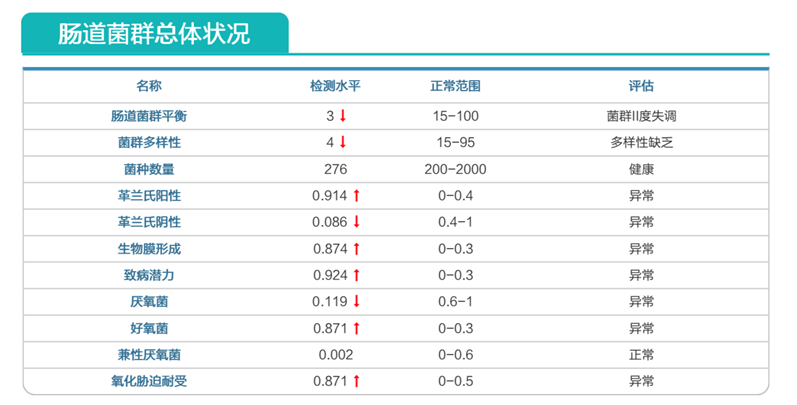
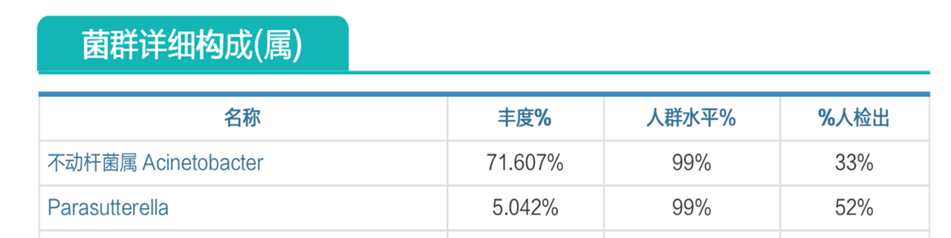
肠道菌群明显失衡,变形菌门(Proteobacteria)扩张,不动杆菌属(Acinetobacter)中约式不动杆菌滋生严重,比例达71.607%。
营养评估里面有明显缺乏项,如下:
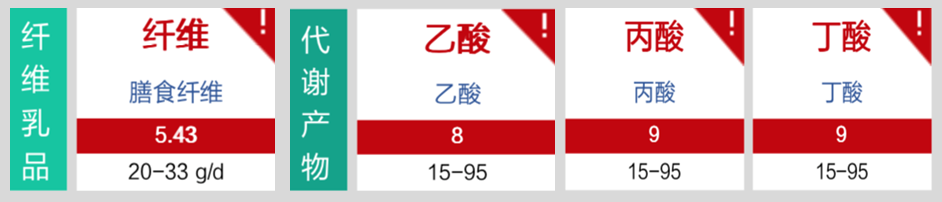
优先解决不动杆菌感染问题;
临床用抗生素治疗。
增补膳食纤维;
混合膳食纤维10g/天;
特定益生菌补充;
清淡饮食,控制饮食中的油脂、肉类总量, 引入粗纤维食材、蔬果等。
2个月后复查菌群,结果如下:
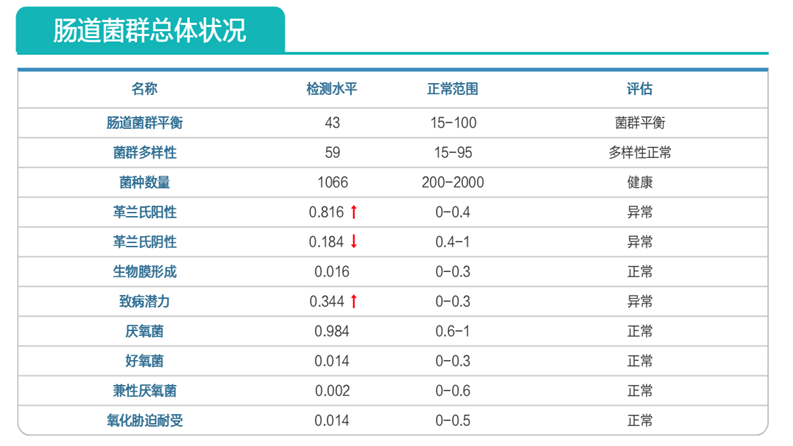
约式不动杆菌感染问题已解决;
菌群结构明显改善。
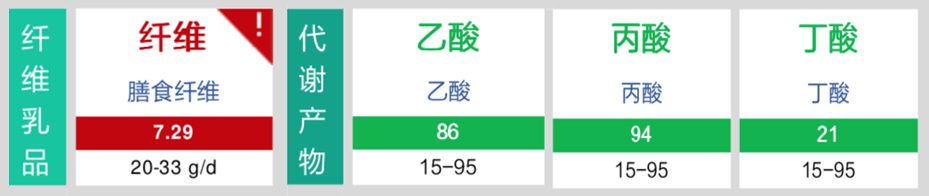
膳食纤维总量提升
短链脂肪酸合成正常

便秘问题明显改善,目前1-2天一便,且形态气味正常。
情绪改善,注意力更集中。
体格发育明显,身上明显更结实。
目前再未发现腹胀、腹痛的情况。
对于宝宝来说,睡眠质量直接影响其身体健康和发育。有些睡眠障碍不仅给孩子身高、免疫力等发育带来影响,还会影响记忆力、智力等潜在发育,更是让家长深受困扰。
以下这个案例是一个11个月宝宝的睡眠障碍问题,进行菌群检测后发现营养缺乏严重,我们来看下具体情况:

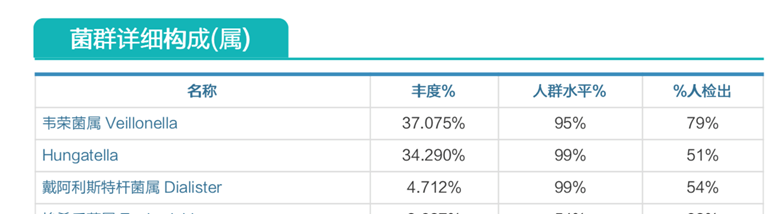
肠道菌群明显失衡,韦荣菌属、Hungatella、戴阿利斯特菌属病理性滋生;

酵母菌感染。
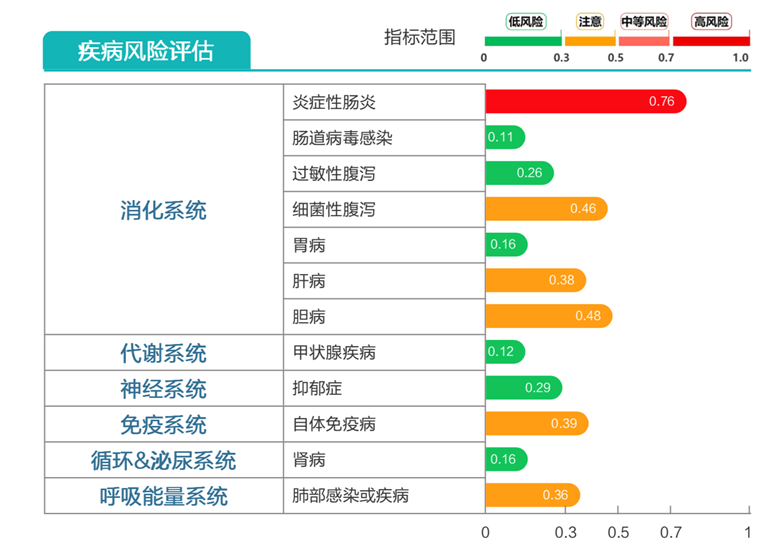
炎症性肠病高风险。
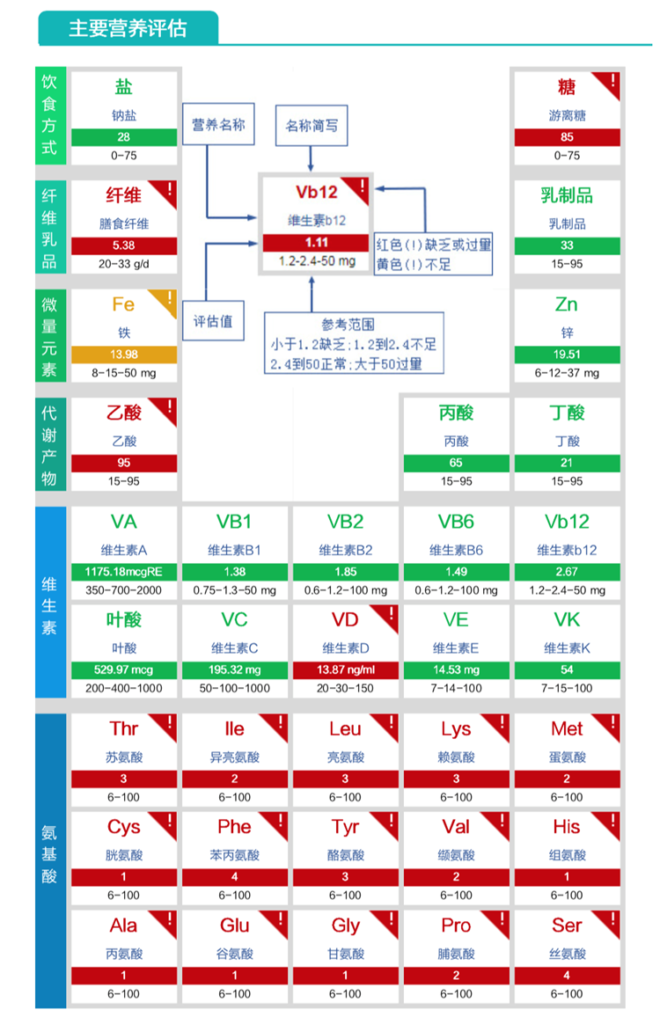
膳食纤维缺乏、VD缺乏、氨基酸成分严重缺乏。
经菌群检测后,补测血液免疫指标,见抗链O超标,提示感染存在。
考虑到目前情况,未见肝功异常,且同时存在细菌、真菌异常。
处理微生物问题;
抗生素治疗
益生菌补充;
由于患儿小龄,特定的益生菌组合;
维生素D补充;
氨基酸奶粉补充。
肠炎备选治疗方案。

在用抗生素的当晚,近2月的睡眠障碍直接改善。
睡眠稳,仅见腹部咕噜声;无夜醒、无夜哭。且后无复发。
益生菌、VD、氨基酸奶粉补充后,大便正常、残渣消失;目前处香蕉软便状。
未到复查期,但身体已基本恢复正常。
在医院每天都会有家长咨询关于宝宝腹泻的问题,宝宝的不正常便便让家长十分紧张,比如说突然开始腹泻,反反复复……
这时候有些家长情急之下根据以往的经验就开始给宝宝吃药,这是比较冒险的选择,如果不对症,容易引起宝宝肠道损伤。
有些家长怕宝宝接受各种检查不舒服,其实随着现代化医疗水平的发展,有很多检测项目可以选择。例如肠道菌群检测,下面这个就是关于4个月婴儿反复腹泻的临床案例。

初次肠道菌群检测结果:
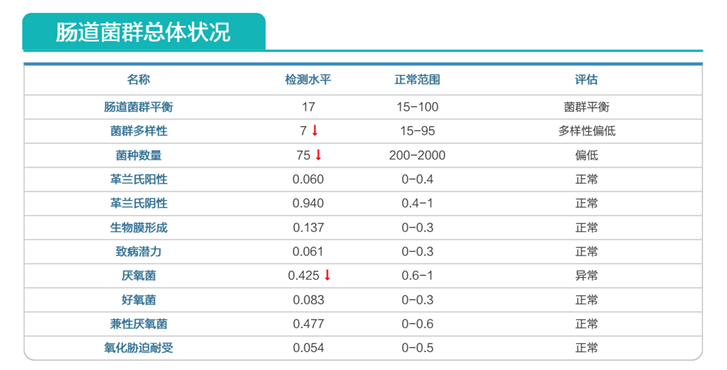
肠道菌群多样性降低


链球菌感染、艰难梭菌感染;
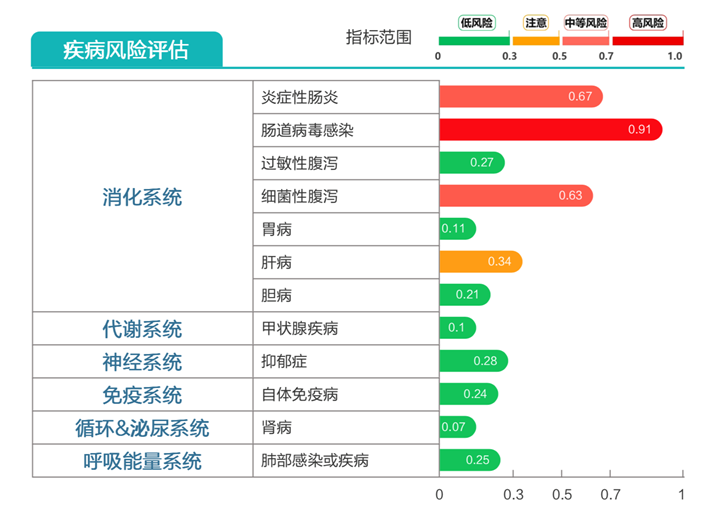
炎症性肠病高风险;
病毒性腹泻、细菌性腹泻高风险。
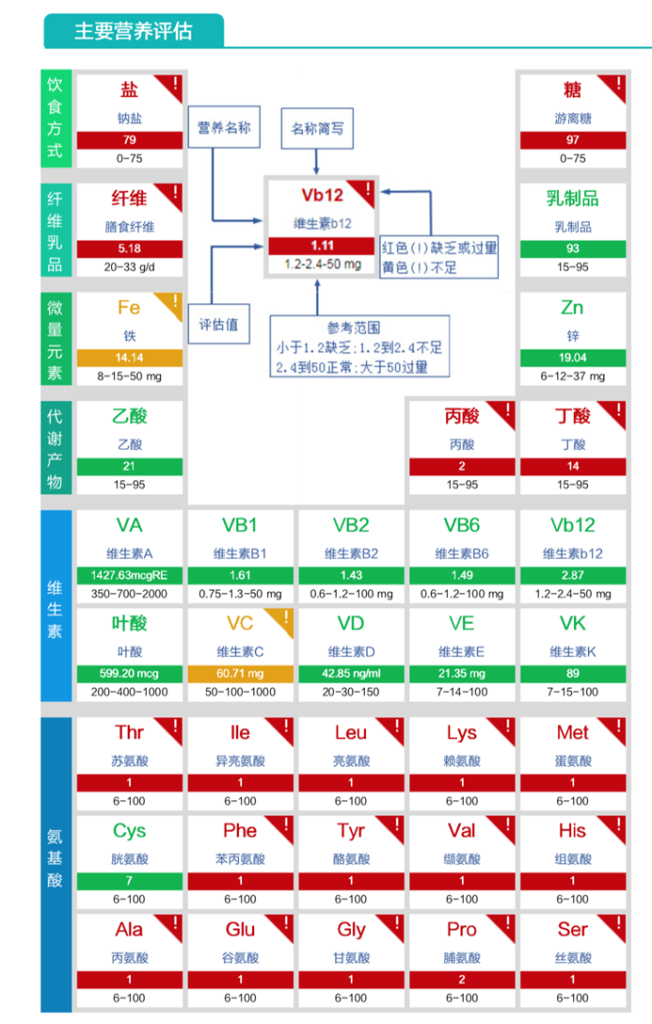
膳食纤维缺乏、丙酸与丁酸缺乏、氨基酸成分严重缺乏。
处理微生物问题;
抗生素治疗
益生菌补充;
由于患儿小龄,特定的益生菌组合;
氨基酸奶粉补充。
营养以及特定维生素补充。
治疗后1个月后复查菌群,结果如下:

链球菌感染已解决,但艰难梭菌仍超标。

菌群结构明显改善;营养状态改善。
考虑到肠道症状的明显改善,与生长发育的改善。认定先前的肠道问题以链球菌感染为主,艰难梭菌为辅。
目前未见肠道问题且患儿明显改善,决定暂时对艰难梭菌姑息治疗。待患儿身体更佳时再行复查治疗。

抗生素治疗4天腹泻止,一周后复查便隐血无。
抗生素治疗与益生菌补充后,消化正常,无腹泻,大便成形,但微臭。
益生菌、VD、氨基酸奶粉补充后,大便正常、残渣消失;目前处香蕉软便状。
孤独症谱系障碍又称为自闭症,自闭症其实并不算罕见,《中国自闭症教育康复行业发展状况报告III》中的数据显示,在我国,基于比较保守的推算,自闭症发生率为1/100,其中儿童约占300万左右。
看起来1%的发病率,落到一个孩子,一个家庭上,就是巨大的阴影,多少父母在孩子确诊后崩溃地痛哭……
对于自闭症,虽然目前尚未有完全治愈自闭症的方法,但是早发现、早干预对于预后发展是非常重要的作用。自闭症治疗的黄金时段是1-3岁,一旦错过最佳时期,难度就加大了。
接下来我们这个案例是一个3岁的患者,在被诊断为自闭症的同时,也伴有一些其他症状。

初次肠道菌群检测结果:
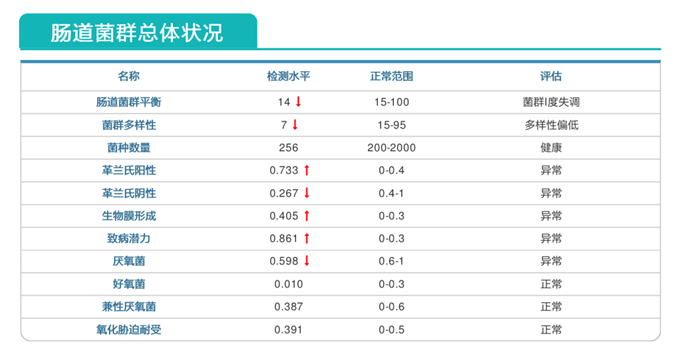
报告中可以看到,肠道菌群失衡明显。


存在肺炎克雷伯式菌感染问题;戴阿斯特菌属病理性滋生。
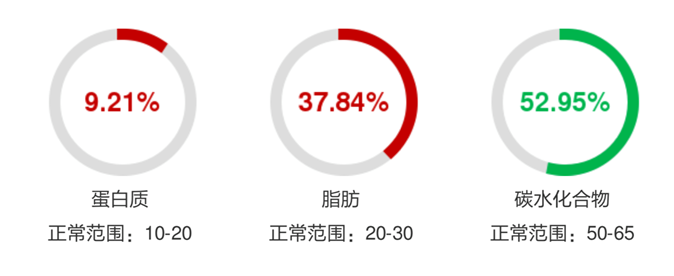
营养状况差
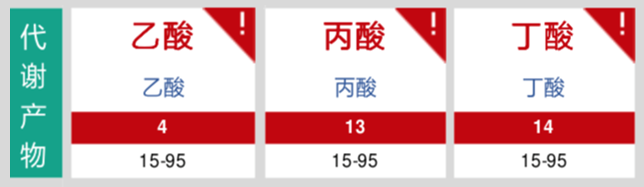
短链脂肪酸合成异常。


意识明显清醒,注意力集中。
能坐住,情绪好,无尖叫情况。
康复机构反应配合度高,进步大。
益生菌、膳食纤维等补充后,大便正常。目前处香蕉软便状。
氨基酸奶粉补充后,面色正常。
目前治疗后3个月,整体稳定。计划复查持续治疗。
肠道菌群临床应用治疗一般路径:

精准检测+精准干预应该是未来肠道菌群临床应用和发展的趋势,目前国家正在加大在肠道菌群研究上的科研资金和人才投入,肠道菌群真正应用于临床并造福病人需要扎实的临床数据和更多的临床积累。我们会积极探索和与更多临床科室合作,将谷禾多年的菌群检测与临床需求相结合,科学和精准的推进临床诊疗的应用落地。
本期分享的多例儿童临床案例展现了肠道菌群与临床相结合的一些应用和实践,未来随着数据和案例的积累,我们还会在更多科室和应用场景见到肠道菌群为临床提供多种视角和更精确化的干预方案。
感谢湖南省妇幼保健院和齐鲁医院提供的病例。
相关阅读:
国庆前实验室的收样时间截止9月30日,客户的样本必须在9月29日前送到实验室会安排节前上机。国庆期间10月1日至10月8日实验室无法收样。
原定国庆期间出具报告的样本报告将延后到10月9号后出具。

杭州谷禾信息技术有限公司
2020年9月14日

谷禾健康
最近,欧洲心脏病学杂志《European Heart Journal》发表了荷兰由阿姆斯特丹大学研究的最新成果:“Associations between gutmicrobiota,faecal short-chain fatty acids, and blood pressure across ethnic groups: the HELIUS study(不同民族群体间肠道菌群、粪便短链脂肪酸和血压之间的关系)”,这是首个评估不同种族肠道菌群组成与血压关系的研究。研究人员发现肠道菌群组成和血压之间存在一致的联系,而年龄和种族之间的解释差异很大。

摘要
文章主要调查了不同种族人群肠道菌群、粪便SCFA水平和血压之间的关系。研究人员纳入了来自6个不同种族的4672名受试者(平均年龄49.8±11.7岁,女性占52%)。使用16S rRNA基因扩增子测序对肠道菌群进行分析。利用机器学习预测模型评估菌群组成和室血压之间的关系。在相关性最大的亚组中,比较了200名收缩压较低或较高的受试者的粪便SCFA水平。结果表示,粪便微生物群组成可解释总收缩压方差的4.4%。对收缩压最好的预测因子有Roseburia spp.、梭状芽孢杆菌属、Romboutsia spp.和瘤胃球菌科。微生物群落组成的解释方差在荷兰人中最高(4.8%),但在南亚苏里南人、非洲苏里南人、加纳人、摩洛哥人和土耳其人后裔中很低(解释方差<0.8%)。在低收缩压的年轻的荷兰受试者中,粪便SCFA水平,包括醋酸(P<0.05)和丙酸(P<0.01)水平较低。
背景
高血压是心血管疾病发病率和死亡率的主要可改变危险因素,因此是全世界可预防死亡的最重要危险因素。原发性高血压的发病机制尚不完全清楚,目前被认为是遗传和心血管危险因素复杂的相互作用造成的。来自动物和人类研究的初步证据表明,肠道菌群组成和菌群衍生代谢物的水平,包括短链脂肪酸(SCFAs),都与血压(BP)有关。肠道菌群产生的主要代谢物是短链脂肪酸(SCFAs),这是膳食纤维在肠道发酵的最终产物。动物研究指出,通过肾脏和血管中的SCFAs受体介导,粪便中的SCFAs与血压之间存在直接联系。在人类研究中,证明粪便中SCFA水平与血压之间的关系的证据很少且粪便SCFAs的高低都与较高的血压有关。假设肠道菌群和SCFAs确实与高血压有关,这将为高血压的发病机制和治疗提供新的视角。
实验设计
受试者研究:
随机抽样年龄在18岁到70岁之间的人,按种族分层(荷兰人、南亚苏里南人、非洲苏里南人、加纳人、土耳其人或摩洛哥人)。只选择有可用的血压测量、体重指数(BMI)和粪便样本的参与者。所有参与者都被要求戒烟。体重指数根据身高和体重计算。在仰卧位休息至少5分钟后测量血压,使用经过验证的半自动振动装置连续测量的两次平均值。在静脉血标本中测定空腹血糖和肌酐水平,并使用CKD-EPI公式计算肾小球滤过率(eGFR)。从清晨尿样中测定尿白蛋白/肌酐比值,蛋白尿的定义为大于等于30 mg/mmol。糖尿病的定义是空腹血糖水平升高(≥7 mmol/L)或服用降糖药物。高血压定义为收缩压SBP >140 mmHg或舒张压(DBP)>90 mmHg或使用了降血压药物。
样本采集及16S rRNA扩增子测序
粪便样本采集后需在6小时内处理,如若不能,-20℃最多存放一天,在这期间必须转移至-80℃存放。在采集前一周内出现腹泻或在采集前3周内使用抗生素的参与者的样本不予使用。使用Illumina MiSeq测序16S rRNA基因的V4区来确定粪便微生物群的组成。
粪便短链脂肪酸测定方法
采用高效液相色谱(HPLC)紫外检测粪便中SCFA的含量。对于所有样品,在均质粪便冷冻干燥24小时后测定干重。由HPLC测量得到的所有浓度都根据每个样品的湿重和干重的差异进行了校正。
统计分析
使用机器学习模型来评估肠道菌群组成和血压之间的关系。对全部研究人群和按年龄(<50岁,>50岁)、性别和种族的亚组进行了分析。模型是使用迭代流构建的。在每次迭代中,数据集被随机分成包含20%的参与者的测试集和包含剩余80%的训练集。之后,严格在训练集内进行五次交叉验证,以拟合和优化模型超参数。最后在测试集上对得到的模型进行了评估。在每次迭代期间,两个随机变量被添加到预测器数据中作为基准。Mann-Whitney U检验比较高血压组和低血压组之间粪便SCFAs浓度和丰度。
主要结果
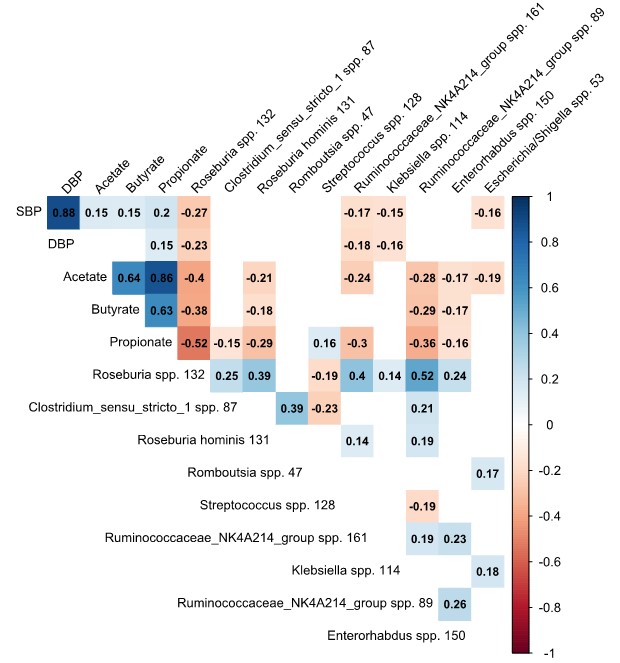
1.肠道菌群的组成与血压有关,而血压在不同种族之间存在很大差异。
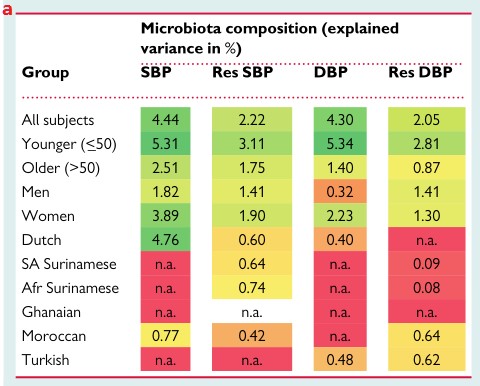
a.解释了不同亚组中的肠道微生物组成百分比的变化对血压的影响。在整个队列中,基于肠道菌群组成的机器学习模型可以解释SBP和DBP的4.4%和4.3%的方差,而在年轻人、女性和荷兰人亚组中解释的差异最高。颜色表示解释方差的水平。Afr Surinamese:非洲苏里南人;DBP:舒张压;n.a.:解释这些模型的方差为负;Res:根据年龄,性别,BMI调整的残差;SA Surinamese:南亚苏里南人;SBP:收缩压。
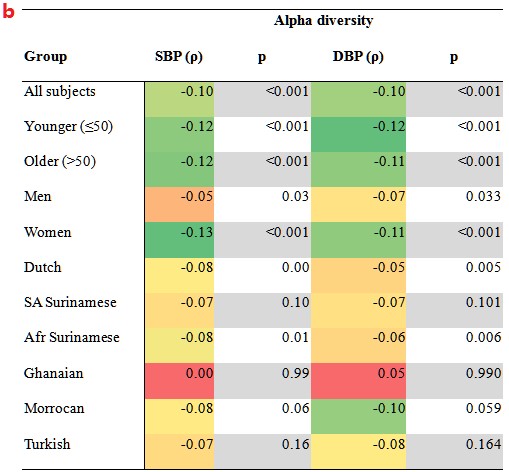
b.不同亚群中微生物群落的香农指数与血压的相关性。(p):相关系数;p:相关性的显著性。在年轻人、女性和荷兰人亚组中显示出与血压的相关性更强。
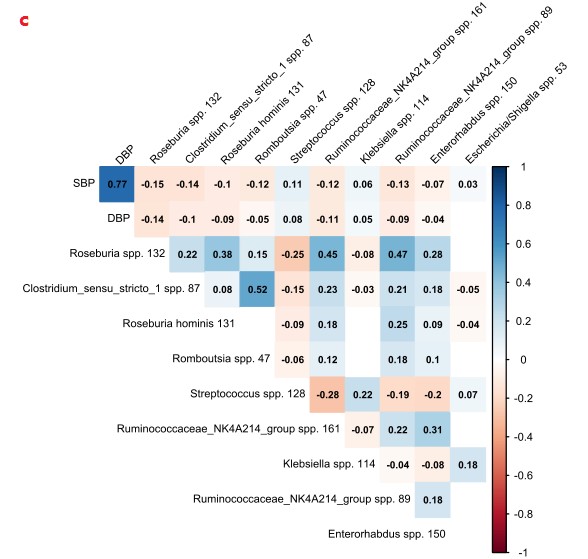
c.预测到的前10个物种与收缩压的关联图(P<0.0 5,Spearman),除链球菌和克雷伯菌外,其余物种均与SBP和DBP均呈负相关。格子中的数值表示每个物种的相对丰度与收缩压(SBP)和舒张压(DBP)之间的相关系数。红色为负相关,蓝色为正相关,颜色越深,相关性越强。
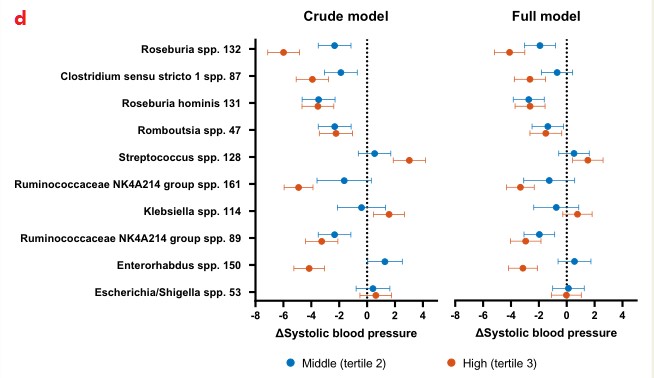
d.在回归分析中,这些物种的影响效应介于SBP降低6.0 mmHg和升高3.0 mmHg之间,大多数物种的效应大小随着丰度的增加而增加。Roseburia spp对血压的绝对影响最大:中/最高值与收缩压分别为2.3 mmHg (95%CI 1.2-3.5)和6.0 mmHg (95%CI 4.9-7.1)。
Crude model:粗略模型(根据年龄和性别进行校正);Full model:对体重指数、吸烟、使用降压药和糖尿病病史进行额外校正,校正BMI和其他协变量后,效应影响减弱到2-4 mmHg,这表明BMI只是造成这种影响的部分原因。
2.虽然产生SCFA的微生物与较低的血压相关,但粪便中SCFA水平的增加与较高的血压相关。产生SCFA的微生物与粪便SCFA水平呈负相关。因此推测,较高的SCFA上调了肠道对SCFA的吸收,从而导致粪便中SCFAs排泄水平相对较低。
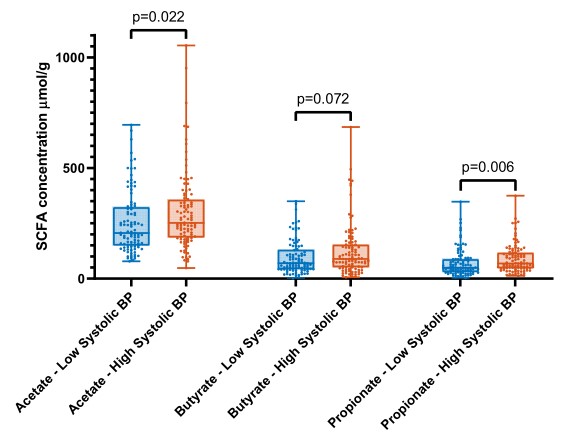
将荷兰参与者的年龄、性别和BMI进行配对,结果有100名参与者的收缩压较低,100名参与者的收缩压较高,对这200名受试者进行高、低血压(BP)与短链脂肪酸(SCFA)水平的比较(盒形图),Mann-WhitneyU统计差异(P<0.0 5)。低收缩压受试者粪便中醋酸(P=0.022)和丙酸(P=0.006)水平显著降低,丁酸水平也有降低趋势(P=0.072)。此外,粪便SCFA水平与前10位物种呈负相关,与SBP、DBP呈正相关(关联图)。
结论
这是首个评估不同种族肠道菌群组成与血压关系的研究。研究人员发现肠道菌群组成和血压之间存在一致的联系,而年龄和种族之间的解释差异很大。观察到的产生SCFA的微生物与血压之间的关联,为SCFAs在血压调节中发挥作用的假设提供了进一步的证据。研究人员认为未来在研究肠道微生物区系与血压的关系时应该考虑到种族差异。用SCFAs进行干预研究,可以更深入地了解这些代谢物对血压的潜在机制。
欢饮关注谷禾健康,专注做肠道菌群检测

世界卫生组织(WHO)已将癌症确立为一种全球威胁,每年夺去大量生命。 仅在2018年,它就造成960万人死亡,是全球第二大死亡原因。癌症仍然是主要的健康灾难。
最近的研究已指出人类共生肠道微生物群参与调节化学疗法和免疫疗法的结果。 它主要是通过调节药物的代谢和宿主免疫反应来实现的。
在过去的十年中,在诸如外科手术,化学疗法,放射疗法,免疫疗法和激素疗法之类的针对癌症的治疗方式的进步上的不懈而巨大的努力已经成功地改善了大多数患有该疾病的个体的临床结果。
然而依然会发现许多不利因素,例如,肿瘤复发和转移的频率增加以及新兴的癌症耐药性,此外,大多数抗癌药物对正常细胞仍具有攻击性。这些都影响了生活质量,且癌症患者的死亡人数仍然居高不下。

我们需要对肿瘤发生基础的多因素病因有更清楚的理解,这有助于增强抗癌治疗的有效性。值得关注的是,一些研究表明,人体中存在的微生物是致癌作用的关键决定因素,可以影响癌症的发生,进展,最重要的是对治疗的反应。
人体是数万亿种微生物的生态家园,它们与宿主健康的维护密切相关。诺贝尔奖获得者约书亚·莱德伯格(Joshua Lederberg)于2001年将这样的微生物群落称为“微生物组”。
微生物遍布全身,其中肠道微生物群是最受欢迎和研究最深入的系统。人的肠道微生物主要包含细菌,病毒,真菌,古细菌和小型原生动物的异质等。
最新研究强调了肠道菌群对宿主健康的不同作用。 肠道内的微生物通过调节大量的基本生物学过程,在宿主系统内提供保护并维持体内平衡。 这些可能包括监测上皮的发育,营养吸收,代谢功能和先天免疫反应,包括免疫细胞的活化和成熟,防止全身性渗透和肠道病原体排出。
相反,对这种自然存在的原有菌群的扰动,被称为微生物失调,与各种病理状况有关,例如糖尿病,肥胖症,炎性疾病,代谢综合症,肝硬化甚至各种癌症。
宿主和肠道微生物群共同努力的结果是,全身功能(如能量平衡,营养,代谢,认知功能,心血管功能,昼夜节律,炎症,先天性和适应性免疫)之间的微妙平衡。
肠道菌群的各种成员,包括拟杆菌属,乳杆菌属和双歧杆菌属,与某些食物的消化密切相关。肠道微生物群除了影响脂类和蛋白质的稳态以及必需营养维生素的合成外,还有助于维持能量平衡。
此外,肠道体液和细胞黏膜免疫系统的正常发育是由肠道菌群分泌的代谢物和信号分子介导的。
肠道微生物群可调节多种生理功能
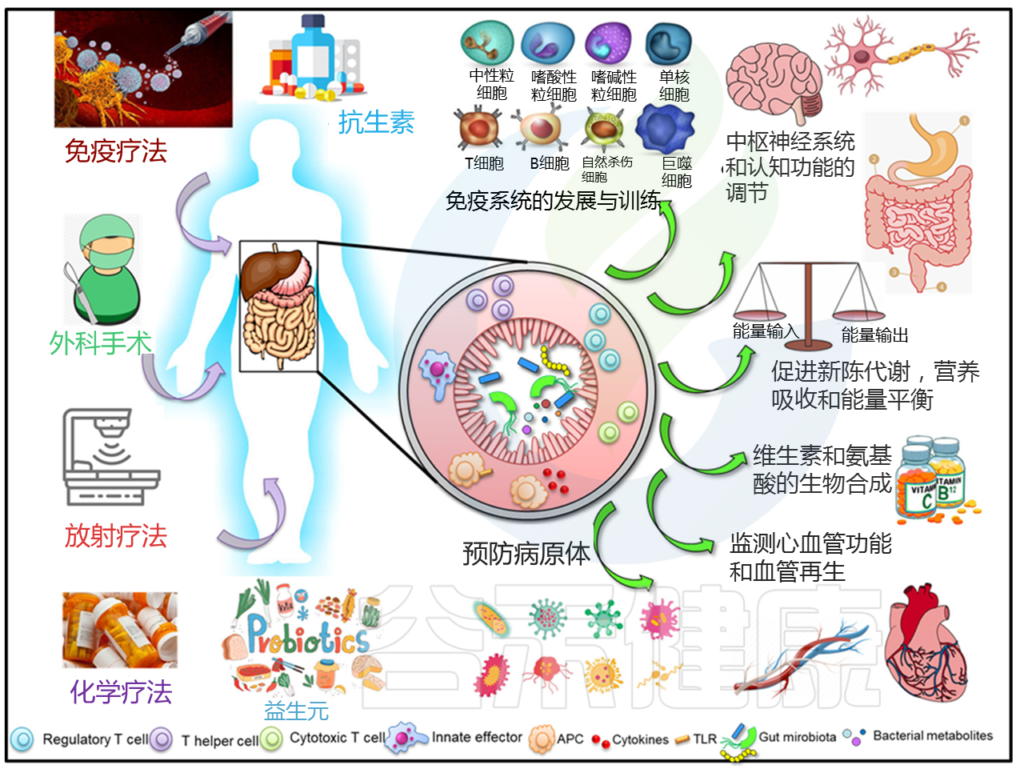
肠道微生物通过调节大量的系统功能,在维持宿主正常健康方面起着关键作用。它对中枢免疫系统、消化系统和其他系统有很大的贡献。肠道微生物群本身会受到多种外部因素的影响,如饮食、抗生素的使用和不同的治疗模式。这种修改反过来又会影响其监管功能。
随着宏基因组学以及涉及下一代测序(NGS)和16S rRNA扩增子计算分析的多学科方法的出现,人们对肠道微生物组多样性和丰富度有了更多的了解,宏基因组测序已揭示癌症患者中微生物群落的显著改变。
胃癌:
从胃癌患者提取的组织中检测到厚壁菌、变形菌、放线杆菌和梭杆菌。此外,胃癌症患者表现出21个细菌类群的显著富集,包括核梭杆菌、微小微胞菌、血管紧张链球菌和消化链球菌口炎,并减少了10个分类群。
胰腺癌:
一项宏基因组研究还表明,口腔中存在嗜血杆菌、卟啉单胞菌、瘦肉杆菌和梭杆菌属物种与胰腺癌的风险增加有关。
16srrna基因测序预测卟啉单胞菌、放线菌、奈瑟菌、链球菌、类双歧杆菌和梭杆菌在胰腺癌的发生发展中起重要作用。
结直肠癌:
研究表明,大肠埃希菌、脆弱类杆菌和厌氧消化链球菌具有致癌潜力,可诱导遗传毒性应激、胆固醇生物合成和激活Th1免疫反应。宏基因组分析发现,在结直肠肿瘤患者中,除了明显的病毒组特征外,微小单胞菌、消化链球菌、梭杆菌和卟啉菌都有明显的积累。
另有更详细的研究表明,CRC相关菌群可分为三种不同的模式。

肝细胞癌:
肝癌发生发展涉及慢性肝细胞死亡、炎症和肝组织修复诱导的纤维化等多个阶段。肠道菌群的变化趋势如下:
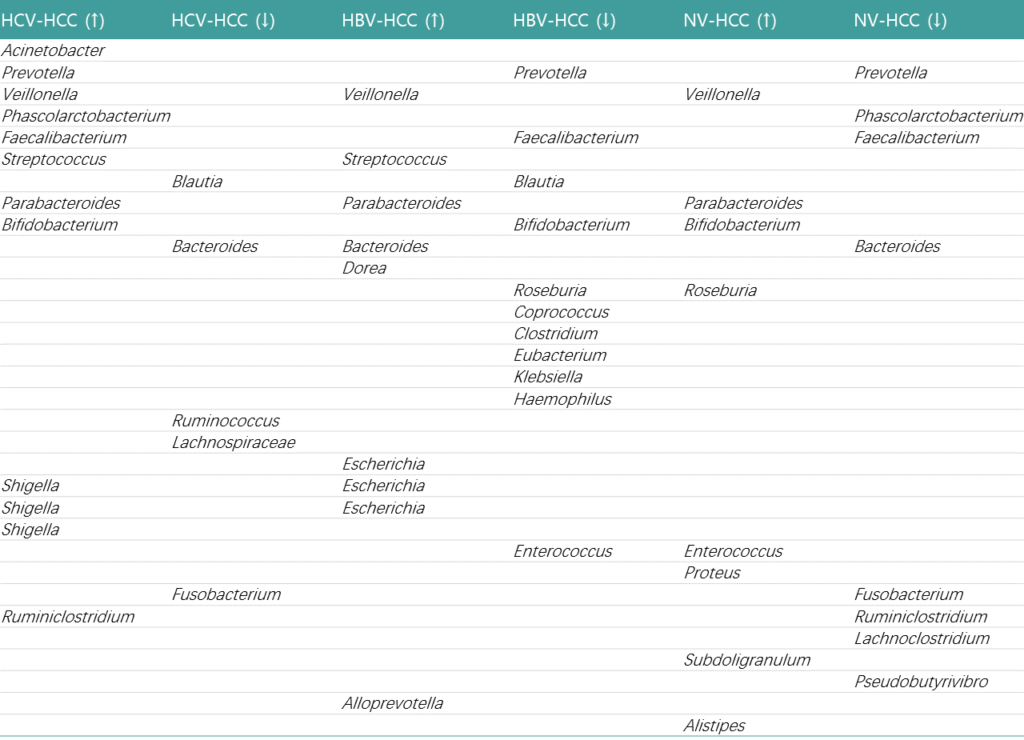
注:HCC-肝细胞癌;HBV-HCC: 乙肝感染引起肝细胞癌;HCV-HCC: 丙肝感染引起肝细胞癌;NV-HCC: 非病毒性肝细胞癌
癌变起因于肠道微生物群的生态失调
良好平衡的肠道微生物群对健康生活十分重要。而肠道微生物群的失调会加剧不同癌症的致癌发展。
几项使用无菌小鼠模型的临床前研究揭示了肠道微生物组通过不同机制在癌症发生和进展中的作用。
肠道微生物群积极参与肿瘤的发生和发展

癌症的发生是一个动态的过程,许多内在因素促成了它的发生。肠道微生物群在决定转化细胞的命运方面起着至关重要的作用。微生物失调可能会促进肿瘤的发生,并导致肿瘤转移。
促癌微生物最突出的例子是幽门螺杆菌,它会感染胃并刺激胃癌的形成。幽门螺杆菌是唯一获得IARC认可的I类人类致癌物的细菌。 其主要作用方式包括分泌毒力因子,主要是空泡细胞毒素A(vacA)和细胞毒素相关基因A(CagA).
这些已被证明可以激活致癌途径,从而异常转化细胞增殖,细胞周期转变和细胞死亡。 可以诱发癌变的其他间接机制包括氧化应激的产生。
这有助于促进突变的基因组不稳定性和宿主炎症和免疫反应的操控,从而帮助肿瘤细胞逃避免疫监视. 例如,幽门螺杆菌和粪肠球菌成功诱导氧化应激,从而导致基因组损伤并最终导致肿瘤发生。
参与肿瘤发生的肠道菌群列表

其他毒素,例如大肠杆菌产生的大肠杆菌杆菌毒素和细胞致死性扩张毒素(CDT)表现出DNase活性,为DNA损伤铺平了道路,最终导致异常的细胞周期,基因组攻击和致瘤性进展。肠杆菌科的Colibactin类似地在宿主细胞中诱导DNA双链断裂。
致病性弗氏志贺氏菌是肠道微生物的另一个例子,其酶肌醇磷酸磷酸酶D(IpgD)和半胱氨酸蛋白酶样毒力基因A(VirA)介导宿主p53降解。
另一方面,梭菌梭状芽胞杆菌(Clostridium scindens)参与胆酸向脱氧胆酸(DCA)的转化,所述脱氧胆酸起肿瘤诱导剂的作用。DCA还参与了花生四烯酸的释放,花生四烯酸被COX-2和脂氧合酶转化为炎症分子前列腺素和DNA损伤剂活性氧(ROS).
粪肠球菌可促进细胞外超氧化物(O2-)的产生,从而引入DNA双链断裂和点突变。
某些炎性细菌,例如沃兹沃氏菌(Bilophilia wadsworthia)参与了肿瘤诱导剂的生成,例如牛磺胆酸。
核梭状芽孢杆菌的FadA通过凝集素和宿主上皮细胞的E-钙粘着蛋白的结合诱导β-连环蛋白。β-catenin的核易位通过激活c-MYC癌基因诱导细胞增殖。
具核梭杆菌的另一种细菌致病因子Fap2抑制T淋巴细胞和自然杀伤(NK)细胞的作用,从而阻止了髓样抑制细胞在肿瘤部位的募集。
肠道菌群通常有助于产生短链脂肪酸(SCFA),例如甲酸,乙酸,丙酸和丁酸。这些SCFA通常参与多种生理功能,例如激活G偶联受体,抑制组蛋白脱乙酰基酶(HDAC)以及将T细胞分化为效应T细胞(例如Th1和Th17细胞),从而赋予整体抗肿瘤功能。
肠道菌群负责循环中性粒细胞的成熟以及循环单核细胞的昼夜波动。通过分泌白介素(IL)-10和转化生长因子(TGF)-β参与促炎反应下调的调节性T细胞(Tregs)也被某些肠道菌群激活。例如,脆弱的共生拟杆菌(Bacteroides fragilis)有助于Treg细胞的成熟和抗炎细胞因子IL-10的分泌。
重要的是,丁酸和丙酸参与了CD8+ 细胞毒性T淋巴细胞的调节表达,而CD8+ 细胞毒性T淋巴细胞是抵抗肿瘤活性的主要哨兵。已知微生物配体的产生会诱导核因子κB(NF-κB)的活化,从而驱动促炎性细胞因子,例如肿瘤坏死因子(TNF)-α或IL-1的分泌。共生菌在肠道内的定植还导致了来自Paneth细胞的重要抗菌肽的表达。
脂多糖(LPS)和肽聚糖是革兰氏阴性细菌外膜的重要组成部分,它们通过激活宿主Toll样受体(TLR)导致肠道免疫调节,而Toll样受体主要由肠上皮细胞和树突状细胞表达(DC)。这些TLR积极参与介导针对肿瘤细胞的T细胞反应。
除此之外,细菌产物诱导IFN-γ的表达,从而影响中性粒细胞的存活和成熟。低水平的细菌LPS可能会对髓样细胞的活化产生巨大影响,从而引发增强的炎症反应。
更具体地说,细菌产生的吡哆醇可用于刺激宿主免疫监视。几种活细菌,如果摄入足够的量,它们能够给宿主带来健康益处,因此可能发挥益生菌的作用。此类益生菌微生物有助于保护宿主肠道稳态,并在很大程度上调节宿主生理和免疫力。这些结果揭示了健康肠道微生物群的日常功能,这是维持宿主内正常内环境平衡的必要条件。
一些证据清楚地表明,调节肠道菌群可以提高治疗效果,减轻化疗药物的副作用。
肠道菌群对化疗药物疗效和毒性的调节作用

上表总结了几种常用的治疗各种恶性肿瘤的化疗药物及其与肠道微生物的关系。
细菌通过多种机制影响化疗药物和免疫检查点抑制剂的疗效,如下图:
肠道细菌调节抗癌药物疗效的机制
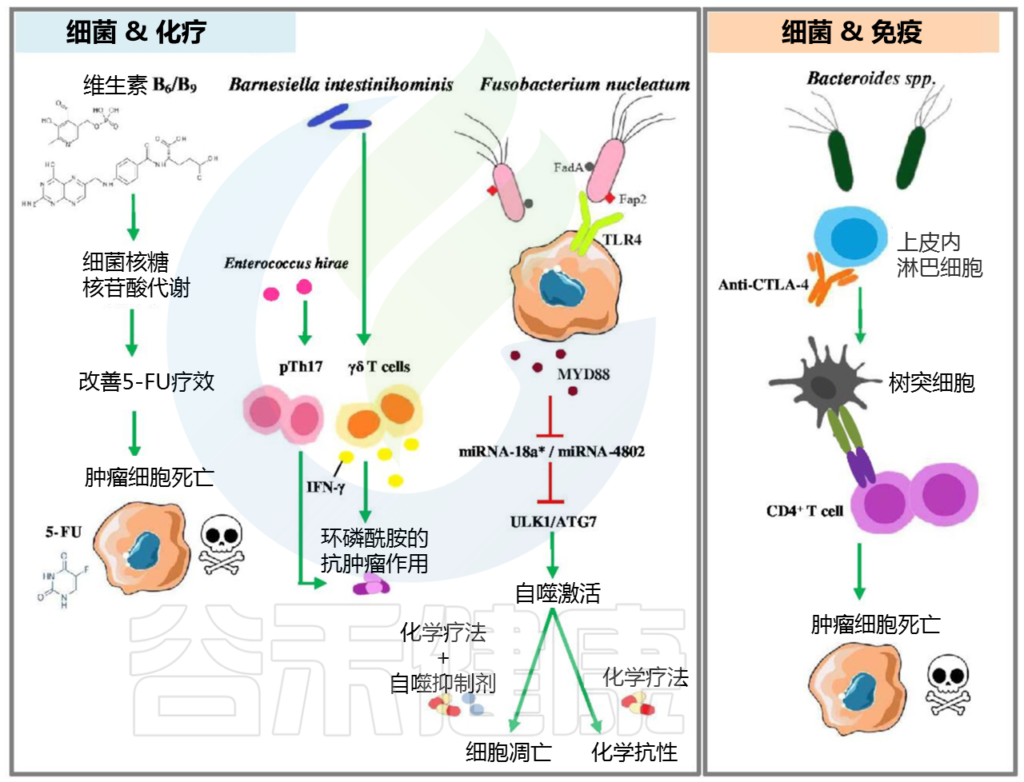
微生物转化的代谢物一旦进入循环,就可以到达人体内遥远的器官,影响那里的癌症发生。例如,在肝癌中,肠道定植者代谢胆汁酸,胆汁酸经过再循环,调节免疫细胞向癌区募集。
肠道微生物的雌激素代谢也改变了绝经后妇女患乳腺癌的风险。肠道失调加上分泌β-葡萄糖醛酸酶的微生物水平增加,如瘦肉梭菌和球菌,显著激活雌激素受体,刺激靶细胞(主要是乳腺和子宫内膜)的细胞增殖。雌激素摄取的增加与乳腺癌的发生直接相关。
肠道微生物群的变化是否会导致癌症的发生,或者致癌转化是否会刺激微生物组的这些变化,仍然是个模棱两可的问题,因此需要进行深入的研究。
细菌素是主要由所有细菌核糖体合成的阳离子肽。癌细胞膜的主要负电荷使细菌素优先结合到癌细胞而不是健康细胞上。
革兰氏阴性菌的细菌素分为微球菌素、大肠杆菌素和泰洛霉素。牛链球菌HC5的牛维素HC5抑制MCF-7等乳腺癌细胞的生长。
由大肠杆菌和其他肠杆菌科合成的大肠杆菌素,特别是大肠杆菌素E1和A,可以抑制乳腺癌细胞系MCF7、ZR75、BT549、BT474、MDAMB231、SKBR3和T47D等多种乳腺癌细胞株的生长,对结肠、骨和子宫等恶性肿瘤也有抗肿瘤作用。
肺炎克雷伯菌分泌的大肠杆菌素microcin也具有很好的肿瘤抑制和凋亡功能,在Jurkat、HeLa和结直肠癌细胞系中都有观察到。然而,它对正常细胞没有任何毒性。
对HeLa和HT29细胞株特异性的细胞毒性作用与从Pediococcus acidlicati中提取的pediocin有关。
细胞溶血素A是一种细菌毒素,可引起胱天蛋白酶介导的细胞死亡。用大肠杆菌或鼠伤寒沙门氏菌治疗的小鼠,可以产生细胞溶血素A,已经显示出很有前景的抗癌机制。
同样,革兰氏阳性菌的侧凸蛋白10(LS10)对MCF-7乳腺癌细胞的抑制活性最高。
此外,Nisin是世卫组织批准的乳酸乳杆菌Ⅰ类细菌素,可显著抑制人肝细胞癌、头颈癌和乳腺癌细胞的侵袭和转移。
铜绿假单胞菌产生的一种含铜金属蛋白Azurin(14kda,128个氨基酸),通过抑制肿瘤细胞中cop1介导的泛素化和蛋白酶体降解,增强p53的细胞内稳定性。Azurin对乳腺癌细胞株MCF7、ZR-75-1、T47D、MDA-MB-157、MDD2和MDA-MB-231也具有潜在的抗癌活性。
此外,肺炎链球菌分泌的Pep27anal2可诱导胱天蛋白酶非依赖性和细胞色素依赖性凋亡,从而显著阻断白血病、胃癌和乳腺癌细胞系的细胞增殖。
肠球菌产生的Entap对三阴性乳腺癌细胞株MDA-MB-231具有良好的抗增殖活性。
白喉毒素(DT)由白喉棒状杆菌产生。交叉反应物质197(CRM197)是白喉毒素的无毒突变体,通过与肝素结合的表皮生长因子结合促进细胞凋亡和抑制血管生成,从而抑制人肾上腺皮质癌的增殖。CRM197和顺铂的组合方法触发胶质瘤细胞凋亡介导的死亡。
肉毒梭菌的A型肉毒毒素(BoNT-A)通过激活程序性细胞死亡阻止前列腺增生的生长和增殖。这些研究代表了一些在肿瘤发生中具有独特抑制作用的微生物代谢物的显著例子。
临床前小鼠模型和癌症患者的测序数据是微生物组学研究中有价值的工具,有助于癌症患者新疗法的开发。
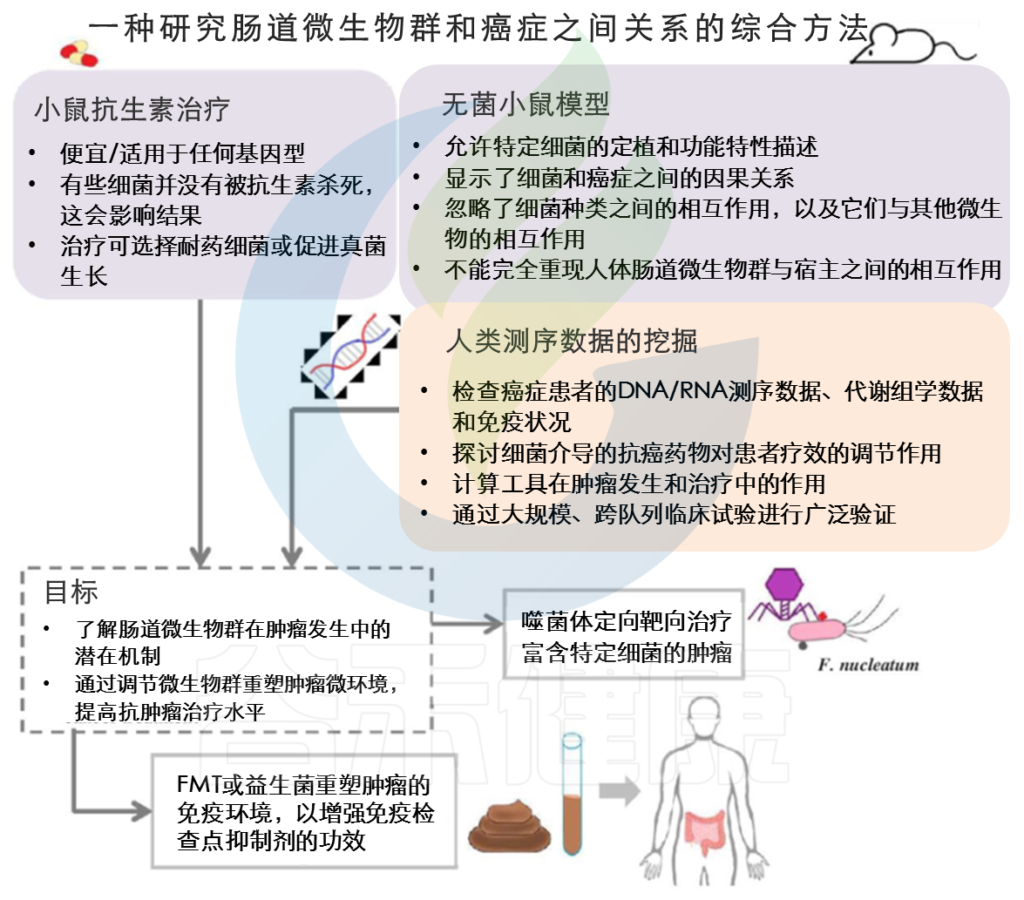
临床前小鼠模型是在没有微生物(无菌小鼠)或存在有限数量的微生物(抗生素治疗)的情况下剖析特定细菌机制的有价值工具。
然而,由于选择偏差等局限性,这些临床前模型无法完全重现人类肠道微生物群与癌细胞之间的相互作用。抗生素治疗可能选择耐药菌或促进真菌生长,这与实验结果相混淆。
其他因素,包括居住环境、饮食和遗传背景,也会影响小鼠体内的微生物群落及其与肿瘤细胞或抗癌药物的相互作用。为了补充这些临床前模型,癌症患者的测序数据在抗癌治疗前后根据宿主基因组、代谢组和免疫谱进行检查。
总之,小鼠模型和人类测序数据的META分析为肠道微生物群和癌症治疗之间的关系提供了见解。通过选择性地用噬菌体靶向癌相关细菌、通过施用益生菌或进行FMT来调节肠道微生物群,可以重塑肿瘤微环境和宿主免疫反应,从而增强抗癌药物的疗效,改善癌症患者的预后。
抗生素用于治疗多种疾病,从常见感染到更严重和更严重的疾病,清楚地说明了抗生素在我们日常生活中的重要性。
抗生素对于预防术后或免疫功能低下患者的致病微生物的生长极为有用,然而,不加控制地使用抗生素可能会在不同程度上改变我们的肠道共生菌群。这有时可能会对健康造成有害影响。
多项研究表明,由于抗生素的使用,免疫治疗的益处可能会大大中断。艾哈迈德等人研究表明在抗PD-1治疗期间使用抗生素会对患有各种癌症的个体的治疗结果产生不利影响,包括黑色素瘤、头颈部癌、肺癌、肾癌、肝癌和尿路上皮癌。
事实上,没有接受任何广谱抗生素治疗的病人完全受益于免疫治疗。根据Huemer等人的观点抗生素和免疫检查点抑制疗法的引入削弱了免疫治疗的益处。然而,也有一些相互矛盾的报道,表明在这方面计划周密的临床试验对于阐明抗生素和免疫治疗之间的确切联系是非常重要的。
益生菌参与改变肠道微生物群,增强肠道屏障的完整性,抑制致病菌的生长,并降低原癌物质的代谢。
一些临床前研究和临床试验已经确定了益生菌在改善传统肿瘤抑制模块的风险、严重程度和相关毒性方面的整体功能。给癌症患者服用益生菌的主要目的是重新填充受损的肠道微生物群,恢复微生物群落的正常功能。
一项针对CRC患者的前瞻性干预研究显示,嗜酸乳杆菌NCFM和乳酸双歧杆菌Bl-04的使用改变了患者的微生物特征。益生菌增加了产生丁酸盐的细菌(例如粪杆菌属和梭状芽孢杆菌)的数量,同时降低了CRC相关属(包括梭菌属和消化链球菌)的数量。
除了改变微生物特征外,研究表明益生菌还可以抑制癌症的发展。
益生菌在抗癌反应和治疗中的作用

尽管益生菌通常是安全的,但在免疫功能受损的癌症患者中引入益生菌,有时可能会促进机会性感染的潜在风险和抗生素耐药性的转移。
大多数益生菌产品是从乳酸产生菌(LAB)中获得的,它们属于乳酸杆菌和双歧杆菌。酪乳杆菌和嗜酸乳杆菌已被证明能增强肿瘤细胞的凋亡。鼠李糖菌GG株还具有抗胃癌和结肠癌细胞的增殖作用。
人类肠道微生物组是遗传多样性不可缺少的资源来源,是宿主免疫系统的重要组成部分,是调节代谢、影响药物相互作用和疾病结果的功能实体。
肠道微生物的双重作用体现在其诱人的益生菌和有益的特性上,它们可以预防或治疗某些疾病,而肠道微生物的失调似乎是癌症等疾病发展的关键决定因素。
目前正在进行的临床试验主要关注肠道菌群在治疗不同癌症中的作用,如下表:

这些数据清楚地强调了癌症患者在接受不同的抗癌治疗策略时所遇到的各种临床挑战。同时也指出了共生微生物在避免这些并发症方面的潜在用途。
应用微生物组学诱导抗肿瘤反应在肿瘤治疗中具有潜在的应用前景。益生菌/抗生素与手术、放疗和化疗的组合方法可能被认为是未来新的抗肿瘤策略。
目前,大多数与微生物组效应有关的研究都是在小鼠肿瘤模型上进行的,在人类身上的重复性是一个需要深入研究的领域。
了解微生物组特征和个体微生物特征可能有助于个性化治疗。微生物组学领域的研究进展为宿主和微生物群之间复杂的相互作用,调节癌症发生带来大量信息,可以说是挖掘出了一座金矿。
控制肠道微生物群可以推动癌症系统朝着理想的方向发展。确定化疗和免疫治疗药物、肠道微生物和宿主之间复杂的相互作用途径,以及我们对系统性微生物致癌作用的进一步理解,可能会为癌症的管理和控制带来前所未有的机遇。
最近的研究结果支持微生物标志物或肠型在癌症诊断和预后方面的潜力,噬菌体疗法在靶向输送癌症药物方面的潜力,以及FMT或益生菌在重塑肿瘤微环境或增强抗癌免疫方面的潜力。
而延长患者寿命,改善患者生活质量的合理解决方案可能在于我们能够利用微生物群对癌症进行控制及长期管理。
相关阅读:
菌群最新资讯 | COVID-19 消毒剂正在改变微生物群;利用微生物群来提高癌症免疫治疗的疗效?六位科学家分享见解
【 参考文献 】
[1] Chattopadhyay I, Nandi D, Nag A, The pint- sized powerhouse:Illuminating the mighty role of the gut microbiome in improving the outcome of anti- cancertherapy, Seminars in Cancer Biology (2020)
[2] Cheng, W. Y., Wu, C.-Y., & Yu, J. (2020). The role of gut microbiota in cancer treatment: friend or foe? Gut, gutjnl–2020–321153.
[3] Wei Jia, et al. Gut microbiota alterations are distinct for primary colorectal cancer and hepatocellular carcinoma. Protein & Cell, August 14, 2020.


谷禾健康
在世界各地,包括亚洲,特应性疾病的患病率已经上升。这一增长与快速的城市化和相关的生活方式变化相吻合,如抗生素过度使用、超卫生的生活条件、气候变化和空气污染。从传统饮食向西化饮食的转变会引起肠道微生物群的组成和功能变化,并被认为是发展中国家“西方生活方式”疾病发病机制的基础。一项涉及从东南亚移民到美国的亚裔少数民族的大型队列研究表明,生活在西化环境中与肠道微生物多样性、本土肠道菌群物种和植物纤维降解功能的丧失有关。这些变化可能与亚洲传统饮食中普遍存在的主要植物性食物的摄入减少有关,或者与西化饮食中的基质纤维总体缺乏有关。

亚洲传统饮食的特点和降低特应性风险的潜在机制
特应性疾病(atopic diseases)主要包括特应性皮炎、变应性鼻炎以及哮喘,属于遗传过敏性疾病,即过敏性体质对环境中常见抗原产生IgE类抗体应答的倾向性,是特应性疾病的发病基础。
传统饮食与西化饮食对肠道微生物群的影响及其与特应性皮炎的关系在非洲已有报道儿童。流行病学有证据表明,坚持传统的地中海饮食是以大量食用水果、蔬菜为基础的,怀孕和儿童时期的鱼和橄榄油对儿童哮喘和过敏性疾病有有益的作用,但结果是矛盾。肠道微生物群和代谢组学研究证实,高水平食用符合地中海饮食的植物性食品与肠道中纤维降解微生物和短链脂肪酸(SCFA)生成有关。有趣的是,生活在英国的亚裔移民儿童,如果他们保持一种独特的亚裔饮食,他们患支气管高反应性的风险比那些采用西化饮食的儿童要低。这个保护作用呈剂量依赖性,与地中海饮食或饮食类型的SCFA产量和依从性水平之间的正相关一致。
与地中海和西方饮食相比,传统亚洲饮食的另一个显著特点是大豆和豆制品(如豆腐、味噌和纳豆)的摄入量较高。大豆食品富含益生元、益生菌、非动物蛋白、不饱和脂肪和高含量的多酚,被认为具有抗氧化、抗炎和抗过敏特性。在人类和动物的研究中,食用大豆增加了双歧杆菌和乳酸杆菌的水平,并抑制了肠道中潜在的致病肠道菌群。大豆异黄酮-染料木素和大豆苷元在小鼠模型中有预防花生过敏和过敏的作用。功能性食品也是传统亚洲饮食不可或缺的一部分。泡菜(泡菜)、大黄(凝乳或酸奶)以及豆豉(发酵大豆)分别是中国、韩国、印度和马来群岛的本土发酵食品,它们有助于丰富多样的、富含益生菌的饮食。
关于亚洲国家儿童肠道微生物群和饮食模式的研究数据很少,主要来自东亚、东南亚和南亚人群。一般来说,城市化或西化饮食中动物蛋白、糖和脂肪含量高与拟杆菌的优势有关。相比之下,传统的农村饮食与普雷沃氏菌(Prevotella)的增加有关,普雷沃氏是一种祖先发酵的细菌,擅长复杂的碳水化合物代谢和SCFA的产生。食物过敏,特别是树坚果和花生过敏与类杆菌过度生长相关,与普雷沃氏菌呈负相关,而IgE介导的牛奶过敏儿童拟杆菌相对丰富。
微生物源性SCFAs,特别是丁酸盐,对特应性有内在的抗炎和免疫调节作用。正如儿童和牧场队列研究所证明的,高丁酸水平的婴儿不太可能发生特应性敏化,而在斑贴和糖尿病免疫研究中,患有湿疹的婴儿丁酸盐水平降低。饮食干预旨在增加SCFA产生菌群和丁酸盐浓度,这是一种有吸引力的减少过敏的策略风险。这个可包括高消耗的丁酸基料,如抗性淀粉、含丁酸盐的食品(如南亚烹饪中使用的酥油或澄清黄油),以及活的生物疗法。在牧场研究中,食用酸奶(每100克酸奶中含有0.1克丁酸盐)且一岁时粪便中丁酸盐含量高的婴儿在6岁时被发现不会对食物和吸入性过敏原过敏。
目前有些国家新的过敏预防护理标准正在形成,其基础是提倡有意识地接触过敏原以诱导免疫耐受。建议包括在4-6个月大时及时引入潜在致敏蛋白,并为所有婴儿(包括有可能患上特应性疾病的婴儿)提供饮食多样性。后者促进肠道微生物多样性,并增加代谢底物的可用性,以支持SCFA生产物种在婴儿肠道中的优先生长。当补充喂养期间引入新的食物时,婴儿肠道微生物群迅速扩张,进而引发强烈的T调节细胞生成或“断奶反应”。在这一关键时期,如果错过断奶反应,可能会导致免疫系统的病理印记,并增加宿主对日后过敏性炎症的易感性。
目前从观察性研究中获得的证据提供了有说服力的推论来支持“饮食-微生物群-疾病”假说,但在指导临床实践方面存在固有的方法论局限性。需要设计良好的人体干预试验来确定饮食和过敏风险之间的因果关系,并解决饮食干预的剂量、时间、持续时间和频率。由于具有不同微生物组学特征和危险表型的个体对饮食干预的反应可能不同,还需要使用集成多组学方法进行的未来研究,因为具有不同微生物组特征和风险表型的个体对饮食干预的反应可能不同。
更好地了解祖先肠道微生物群的特征及其与营养成分和宿主特应性状况的关系,可能会为提供新颖的干预措施,其中包括新一代的益生菌和益生菌食物来重新填充或保存亚洲微生物组。 在精准营养研究方面取得足够进展之前,采用文化适应饮食策略来调节儿童肠道菌群可能是减轻特应性风险的合理方法。
主要参考文献:
Ismail IH, Lay C, Majid NH, et al. Dietary patterns in childhood and their effect on gut microbiota – an Asian perspective on atopy risk [published online ahead of print, 2020 Aug 26]. J Allergy Clin Immunol. 2020;S0091-6749(20)31175-1. doi:10.1016/j.jaci.2020.05.057
Mahdavinia M, Rasmussen HE, Botha M, et al. Effects of diet on the childhood gut microbiome and its implications for atopic dermatitis. J Allergy Clin Immunol. 2019;143(4):1636-1637.e5. doi:10.1016/j.jaci.2018.11.034
Masilamani M, Wei J, Bhatt S, Paul M, Yakir S, Sampson HA. Soybean isoflavones regulate dendritic cell function and suppress allergic sensitization to peanut. J Allergy Clin Immunol. 2011;128(6):1242-1250.e1. doi:10.1016/j.jaci.2011.05.009
De Filippis F, Pellegrini N, Vannini L, et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut. 2016;65(11):1812-1821. doi:10.1136/gutjnl-2015-309957

宏基因组测序方案是针对16s分辨率和宏基因组高成本之间的一个折中方案,通过降低测序深度,每个样本100万reads左右,但是物种的分辨率并没有一般宏基因组(普遍5~10G数据量)差很多。 不通过拼接组装,直接基于kraken2等kmer,或MetaPhlAn2等标记基因的参考基因组方法进行种属丰度分类。结合其到菌株的物种分类和丰度数据可较16s方案下的PICRUST更加准确的预测基因构成。


比较适合想要获得更全和更精细分类精度而同时不需要获得完整基因组序列和重建菌群基因的,浅宏基因组测序就可以成为很好的选择,其成本低,分析简便快速,同样能获得宏基因组的基本丰度数据。
不过浅宏基因组也有其适用范围,根据样品类型的不同,一些样品可能包含 >99%的人类宿主DNA,这不仅增加了序列成本,而且给测量带来了不确定性。
在许多研究中也会采取在进行宏基因组测序文库的准备之前去除宿主DNA的方法。但是,在去除宿主DNA后,可能没有足够的微生物基因组DNA用于宏基因组测序,这通常需要最少50ng的输入。
浅宏基因组较适合于宿主DNA含量较低的样本如:人类粪便、水体、土壤等;
不太适合:口腔唾液、肺泡灌洗液、血液等人体体液类样本。
我们可以免费提供针对粪便及环境样本助力临床/科研取样。
人体口腔、痰液、腹水、脑脊液、尿液、皮肤、阴道分泌物等高寄主细胞含量样本可根据我们的处理方案简单处理后大幅降低宿主DNA比例。
测序平台:Illumina Novaseq,PE150,默认:100万reads/样
2-3周左右出报告


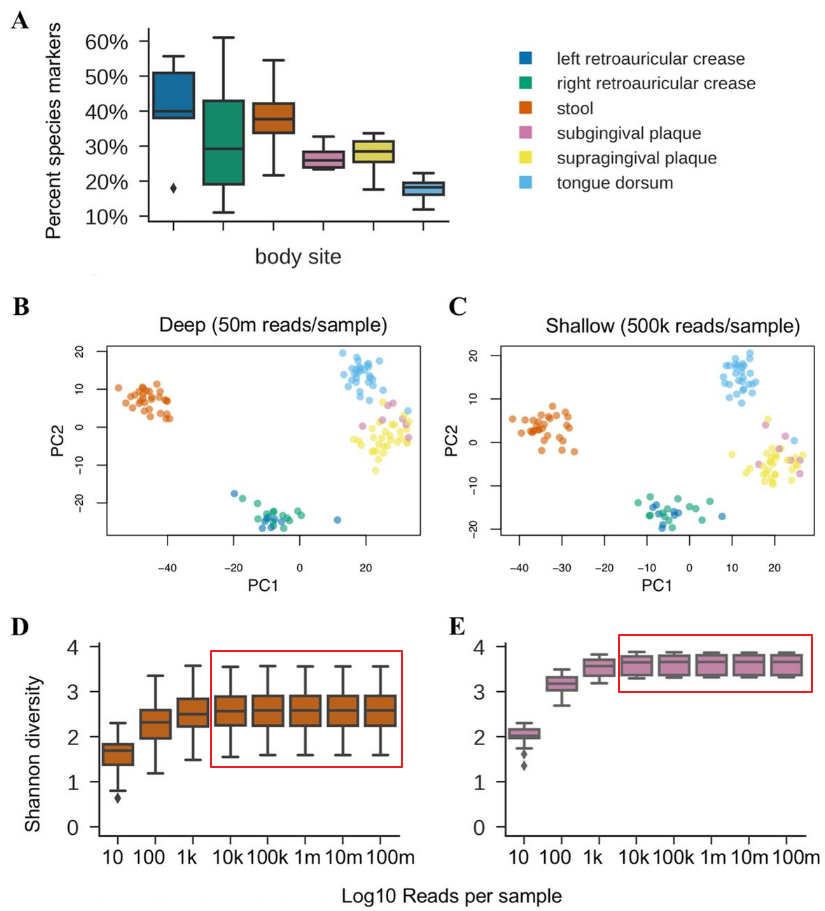
当测序深度达到50万reads以上,浅宏基因组与深度测序宏基因组在主要物种构成的丰度已经基本一致。如下图所示:

样本需求量低:公司研发实现了低当量微生物样本提取和建库,保证提取丰度以及片段完整性同时,样本量需求低于同行其他公司要求;对于样本获取困难的样本,也可以选择微量建库,样本量可低至10ng。
免费取样盒和针对性取样建议:粪便及环境样本提供取样盒助力临床/科研取样,人体口腔、痰液、腹水、脑脊液、尿液、皮肤等高寄主细胞含量样本可根据我们的处理方案简单处理后大幅降低宿主DNA比例。
严格标准的实验流程:自动化样品处理平台辅助,每轮设置阳性对照,上轮检测样本对照,阴性对照。评估污染,轮次比对,最大化减少误差,保证样本重复性和稳定性
Illumina测序平台:宏基因组测序(PE150)采用先进的Illumina Novaseq测序平台,快速、高效地读取高质量的测序数据、结合样品特点和数据的产出,充分挖掘环境样品中的微生物菌群和功能基因
大数据分析流程质量流程控制严格:优化的数据质量控制,包括过滤比对质量低、非特异性扩增、覆盖度低、低复杂度的序列,从而快速准确获得样本中微生物信息及其丰度信息,最大化提高质量数据
分析内容丰富全面:物种分析,基因预测与分析,多样性和相似性分析,功能分析,网路互作分析,代谢网络,关联分析等
完整详细的报告:提供质检实验报告,分析统计报告,分析报告解读,原始数据
高效个性化服务:在线项目系统方便您及时查看项目动态和下载报告以及与分析人员高效交流,免费支持个性化图表修改以及重新分组出报告。
价格低,周期快:包括提取,测序到分析,最快2周出报告。
大数据分析团队和多中心大项目分析经验(团队主要源自浙江大学,包括生物信息学,计算机,微生物以及统计分析等专业,积累了多年的大健康项目多中心项目分析经验,有助于多样本,多表型,多组学联合分析
若需要进一步了解咨询,可以致电联系我们

谷禾健康


假设你能看到人体内所有的基因,你应该会感到震惊。他们中只有1%是人类。另外99%属于细菌、酵母、病毒和微小的原生动物。这些微生物被称为微生物群,它覆盖着你的全身内外。
可以说,潮湿温暖的肠道环境是地球上最拥挤的空间之一。它容纳了大约上千种不同的微生物物种,其所含基因远超过我们的人类基因。

神奇的是,这些微生物及其基因会影响我们的爱情生活,帮助我们像应用程序一样挑选相配的伴侣。取决于你的微生物群状态,这可能是好事,也可能是坏事。
人类可能需要花费数千年的时间才能改变基因,但是微生物可以在20分钟左右的时间内产生新一代的基因泵送的同胞细胞。然后微生物围绕着我们发展进化圈,古老的防御体系,我们的身体要不断为这些变化的微生物创造环境。
因此,在原始时代的某个时候,我们募集了有益的细菌来保护我们免受致病菌的侵害。在潮湿和温暖的环境中,微生物通过抵抗病原体来保护我们,这种关系使我们在充满病原体的世界中成长[1]。
这种共生有多重要?所有的动物都不遗余力地将选定的微生物传给它们的孩子,例如小马会去吃马妈妈的便便,因为它需要摄取母亲粪便中的微生物。
在人类中,产道为早期微生物的形成提供了的途径。母乳中含有的细菌会进一步建立婴儿的初始肠道微生物群。母乳甚至含有专门为喂养这些微生物而生产的益生元。

然而,我们的肠道微生物(统称为微生物群)也有自己的需求-并且他们知道如何满足。
令人惊讶的是,微生物可以产生人类神经递质,包括多巴胺和5-羟色胺,这是目前抗抑郁药物最流行的两个目标。微生物也会产生激素和脂肪酸,所有这些都是有效的情绪操纵器。
再加上细菌毒素和微生物,对他们的宿主有着强大的控制能力。你是不是认为自己有意识地决定爱吃甜食?不,很可能你的微生物才是真正的甜食渴望者,并且你的欲望已经被塑造成可以满足它们的欲望。
事实是,肠道微生物并不是我们的“朋友”,它们是我们共同用餐的伙伴,但随时都可以帮助我们。
它们都有自己的食欲:酵母渴望糖;拟杆菌属(Bacteroidetes)会喜欢脂肪;普雷沃特菌属则比较喜欢碳水化合物;双歧杆菌是纤维爱好者。
它们都有自己的方法来讨好自己喜欢的,使用两种基本技术的变化来影响我们的食物选择[3]。
如果我们不给它们想要的东西,微生物就会产生毒素,使我们感到不舒服,因为他们知道如何使我们痛苦。
微生物通过改变我们的味蕾,增加阿片和大麻素受体以及产生神经递质(如多巴胺和5-羟色胺)来增加我们对食物的渴望。也是因此他们也知道如何使我们快乐[4]。

如果微生物能够指导我们的行为,那么他们还能做什么?从微生物的角度考虑,为了扩大其领土,它们可能会驱使我们更多地社交。从微生物的角度来看,有什么比亲吻,握手或拥抱更好的传播途径呢?
以色列2010年的一项研究将一批果蝇分成了两组。他们给一组喂以糖蜜为食的饮食,另一组为淀粉。每组都建立了合适的微生物群,经过精心调整以消化各自的饮食。
然后,他们在交配室中把这四个混合在一起(每组取一对)。一个交配室实际上只是一个很小的塑料碗,上面有透明的盖子,这样研究人员就可以算出性行为。
具有相同微生物群的果蝇更有交配意向
尽管所有果蝇之间都可以完全通行,但他们还是希望与具有相同微生物群的果蝇交配[5] 。
从几十代来看,果蝇一直生活在自己的群体中,基本上是两个独立的物种。那么有人问,怎么知道这是由于微生物引起的,而不仅仅是呼吸中有糖蜜的气味?答案很简单:当研究人员给果蝇使用抗生素杀死其微生物群时,他们的偏好就消失了。
细菌不会产生性信息素,但会调节其信息素
首席科学家 尤金·罗森伯格(Eugene Rosenberg)说:“共生细菌可通过改变性信息素(sex pheromones,在哺乳类动物中通过唾液,汗液和尿液释放)水平来影响果蝇交配偏好。细菌不会产生性信息素,但会调节其信息素。”
信息素是一种神秘的化学物质,可以起性吸引剂的作用。昆虫会嗅到其中的单个分子而飞走数英里。它显示了微生物群对于交配选择的重要性。
罗森伯格在后续论文中提到,蝇类并不是唯一被微生物操纵配偶选择的动物:“细菌对人类和其他动物的嗅觉有影响,而嗅觉是选择性伴侣的重要输入。”
可以这样理解,你的细菌将皮肤油脂转化为属于你自己的脂肪酸混合物,从而决定了你的气味。这是你的“特定味道”。
罗森伯格及其小组进一步推测,婴儿的抗生素治疗可能会影响这种情况,并可能导致菌群的流失和产生不同的气味[6] 。
早期使用抗生素会影响未来的爱情生活?这种猜测令人担忧,但罗森伯格认为,通过谨慎地在抗生素使用后重新繁殖微生物群,这种情况可能会逆转。
从客观上看,性有时让人满头大汗,但我们还是设法克服了这一点。细菌也可能通过操纵我们的激素发挥作用。
就像动物一样,微生物利用激素彼此交流[7] 。这些化学物质可以帮助我们改善性生活。例如催产素,参与保持身体健康,也和快速愈合有关,但它也在社会关系中发挥作用。催产素是“荷尔蒙激素”,它可以帮助我们忽略性的不便,得以繁衍后代。
这对微生物来说,也是一个巨大的胜利,因为性可以帮助它们找到新的人类领土。接吻可以使它们的生活更美好:亲密接吻每秒可在参与者之间转移800万细菌[8]。

摄入益生菌如罗伊氏乳杆菌可提高体内催产素的水平[9]。看到这,你是不是想赶紧去买益生菌补充剂了?别急,高纤维食物也可以提高自己的罗伊氏乳杆菌水平。
伴侣的选择还受微生物自身免疫系统的影响。主要的组织相容性复合体(MHC)由免疫蛋白组成,这些免疫蛋白为我们每个人提供了独特的气味特征。MHC蛋白是通过与微生物接触而产生的,代表了与病原体战斗的浓缩版本。
从理论上讲,我们无意识地选择了拥有完全不同的MHC的伴侣。这意味着将补充我们自身,有效地使我们对病原体的抵抗力加倍。
当你选择一个具有兼容的MHC特性的伴侣时,它们会让你闻起来很友好。如果你有孩子,他们就会在健康的微生物群上有一个良好的开端。
已婚或同居伴侣可以在很大程度上影响彼此的压力水平,情绪和健康行为方式。这意味着配偶不快乐或不健康,或敌对婚姻会让你更不健康。
当然另一方面,你的伴侣可以起到积极的作用,让你更活跃,减轻体重或减少饮酒,从而有可能改善您的身体健康。
一个最近的评论文章俄亥俄州立大学的Janice Kiecolt-Glaser及其同事的研究表明,婚姻互动,配偶情绪和生活方式习惯对健康的影响可能是通过肠道来介导的[10]。
最近,研究人员已更加意识到人体中,肠道是与大脑,免疫系统和心血管系统(通过迷走神经)进行通讯的中心枢纽。

研究表明,抑郁,饮食,睡眠,压力和敌对的婚姻相互作用会减少肠细菌的生物多样性,或使肠屏障更具渗透性,并更有可能使炎性因子渗入血液(称为“渗漏性肠”)。
肠道菌群多样性较低或肠道泄漏较多,会增加您对慢性炎症,肥胖症和糖尿病或心脏病等慢性疾病的抵抗力。因此,与伴侣生活在一起可能会以更多的你想象不到的方式影响肠道和整体健康[11]。
在一起生活为什么会影响这些因素?事实证明,身体互动,触摸,亲吻和性行为促进了微生物群的共享。肠道菌群与心血管疾病或糖尿病的许多危险因素有关,包括葡萄糖代谢,体重指数,腰围和高密度脂蛋白。
共享的压力源,情绪和健康习惯
促进已婚夫妇健康风险相似性的另一个因素是,他们可能共享共同的压力源,或者受到彼此情绪和压力水平的影响。无论是面对共同的压力源(例如经济压力或生病的孩子),还是伴侣将工作压力带回家,压力都具有传染性。
婚姻中的冲突,压力或敌意也可能影响您的情绪并增加皮质醇水平。伴侣也容易受到彼此健康或不健康行为和睡眠方式的影响。

研究表明:
拥有沮丧的伴侣会使你患抑郁的风险加倍。
不幸福的夫妻比幸福的夫妻更难消除负面情绪和压力。
伴侣有慢性睡眠问题的人有的炎症水平更高。如果你的伴侣在晚上醒着,可能会打扰你的睡眠。
肠道菌群,肠道渗漏和慢性炎症
一个健康的肠道具有多种细菌或病毒,它们分布均匀,没有一个物种能占主导地位。研究表明,菌群多样性低的人比菌群多样性高的人更容易发生慢性炎症。一些研究表明,饮食习惯也会影响微生物群的多样性。
与西方饮食相比,传统西方饮食富含红肉,精制糖和饱和脂肪,而微生物多样性较低,而地中海饮食则更多地依赖植物和健康脂肪。高饱和脂肪饮食还可以增加肠道的通透性,使毒素和炎性物质更有可能渗入您的血液中。
从婚姻敌意到全身性炎症的途径
一项研究表明,对婚姻的看法更加敌对的夫妇减少了肠道微生物的多样性,并增加了不健康的饮食习惯。在这项研究中,研究人员通过对脂多糖结合蛋白(LBP)进行评估,对婚姻状况进行了录像和编码,并测量了微生物的多样性。具有更多敌对性的夫妻的LBP较高,这又与C-反应蛋白(炎症的标志物)的水平较高有关。
因此,这项研究显示了通过减少肠道生物多样性,从婚姻敌意到全身性炎症的途径。此外,较敌对的夫妇的饮食中饱和脂肪含量较高,这可能会影响肠道微生物并增加炎症[11]。
配偶比兄弟姐妹具有更相似的微生物群
一项美国的研究,针对威斯康星州人群的研究,对配偶(N = 94)和兄弟姐妹对(N = 83)的分析进一步表明,配偶比兄弟姐妹具有更相似的微生物群和更多共同的细菌类群,在兄弟姐妹和不相关配偶之间没有观察到差异。即使考虑到饮食因素,这些差异仍然存在。
结果表明,人与人之间的互动,特别是持续的密切婚姻关系,会影响肠道菌群。已婚个体所拥有的微生物群落相对于独居者具有更大的多样性和丰富性,具有亲密关系的夫妻具有最大的菌群多样性,鉴于数十年来的研究记录了婚姻对健康的益处,这点值得注意[12]。
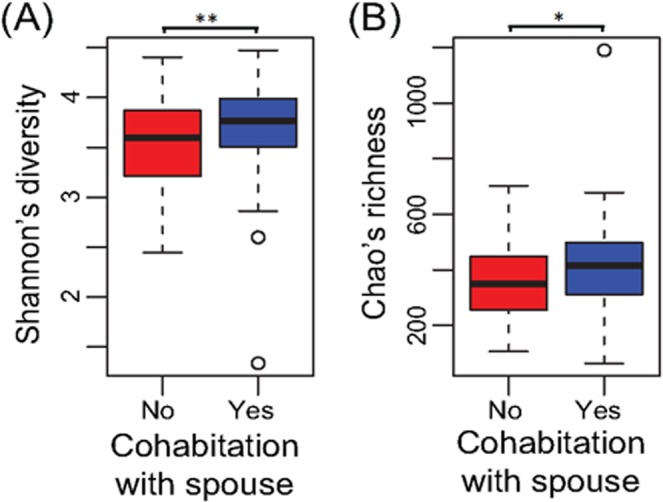
可以看到,具有亲密关系的夫妻其菌群多样性更高,而且存在显著差异。
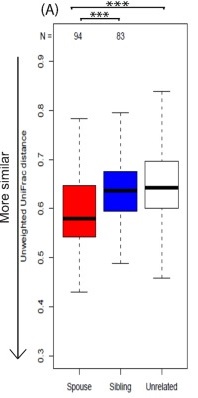
夫妻之间的肠道菌群相似度也超过兄弟姐妹和不相关人群。
虽然饮食通常与胃肠道微生物群相关,但亲密关系表明人类互动和共同行为的影响较少得到理解。紧靠和频繁的身体接触与促进相似个人之间直接微生物共享灵长类动物中微生物的相似性是相关的。
在这项研究中,亲密关系可能代表了在一起度过的时间,身体情感以及其他可能导致微生物共享的人类互动的总和。事实上,有证据表明唾液微生物群影响肠道微生物群,而唾液微生物群可能受接吻的影响。
高质量婚姻可能有助于微生物
通过进一步分析共享的菌发现,有趣的是,这些潜在共享的OTU中的大多数都来自严格厌氧的分类单元,这表明在宿主之间的富氧环境中持续存在可能不是密切人际关系的传播限制因素。
我们进一步发现,不仅已婚夫妇具有更多相似的肠道菌群,而且已婚个体所拥有的微生物群落相对于独居者更具多样性和丰富性。亲密的婚姻关系比兄弟姐妹之间共享的遗传因素和早期生活环境影响更大。
这一发现之所以有趣,部分是因为它与大量证据相吻合,证明了高质量婚姻与发病率和死亡率之间的牢固联系。未来可能会试图弄清将密切关系与微生物成分联系起来的机制。例如,尽管我们没有发现证据表明饮食共享是造成这些发现的主要原因,但我们无法测试精确的身体接触和亲密关系频率作为替代性解释机制。
菌群预测家庭关系
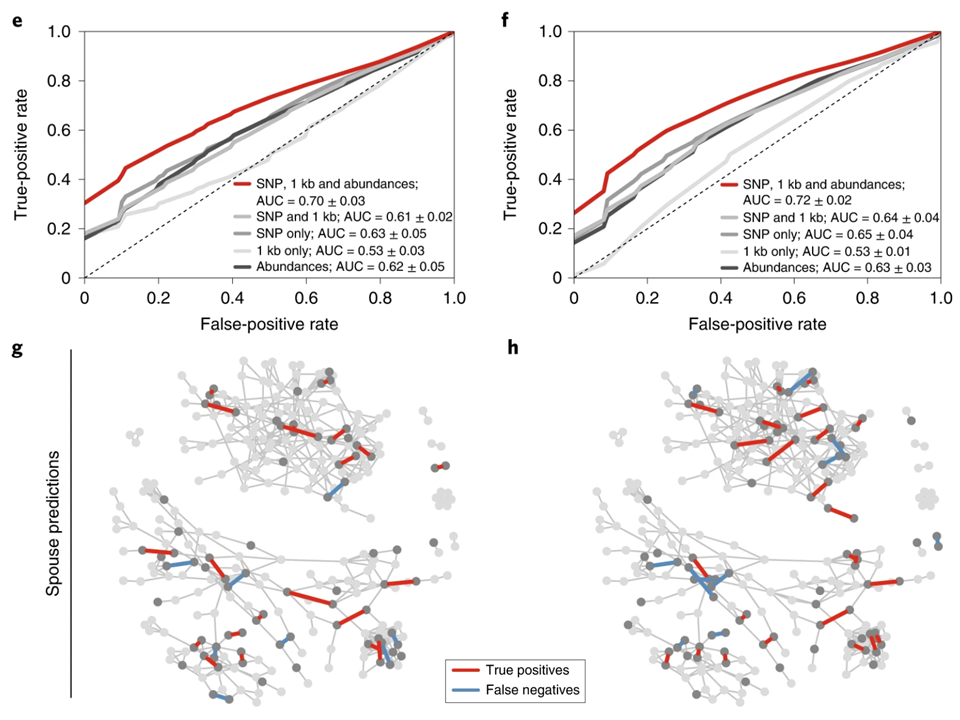
通过评估共有菌群来预测区分家庭和夫妻配偶,预测家庭模型的整体性能较差,但这些预测似乎仍依赖于网络结构,因为在肠道和口腔样本中,某些家庭内部的所有关系都得到了准确的预测。
值得注意的是,模型表明,近25%的配偶非常容易以高置信度进行预测。为什么某些夫妻比其他夫妻更容易预测,这可能反映出共同的敏感性,特定行为或婚姻关系的相对重要性[13]。

1. 共同努力,让生活更健康
当你和伴侣同时工作以减轻体重,多睡,多运动,少喝酒时,更有可能互助成长。看到伴侣变得更健康可以激励你去做同样的事情。
2. 更加富有同情心和相互尊重,而不是敌对地进行沟通
记住,你们是站在同一边的。请注意,对话如果朝消极方向进行,请稍作休息。尝试降低防御力,保持耐心,减少批评。
3. 如果你的伴侣在工作,家庭或育儿方面有压力,请尝试提供帮助并给予支持
最终,压力较小的配偶对你们俩来说都意味着更好的健康。如果尽管您尝试帮助您的伴侣仍然感到压力,则应集中精力分散自己,锻炼,冥想或进行愉快的活动,以减轻压力。
4. 另外,请确保了解您的肠道菌群和拥有良好的菌群
这样可以帮助你拥有美丽的皮肤,光亮的头发,毕竟健康的人自带魅力。

要调整您的微生物群,以下简单步骤供参考:
有了健康的微生物群,你会处于健康状态,并选择了与微生物相容的伴侣,它们可能会比任何约会应用程序更能帮助你找到良好合适的伴侣。
相关阅读:
肠道微生物组如何影响运动能力,所谓的“精英肠道微生物组”真的存在吗?
欢迎关注:谷禾健康
——让你和你的家人更健康
参考文献
1. Scott C. Anderson,Microbes and the Mind. Psychologytoday., 2019,07
2. Scott C. Anderson,The Shocking Source of Your Cravings. Psychologytoday, 2019, 07
3. Alcock, Joe, Carlo C Maley, and C Athena Aktipis. “Is Eating Behavior Manipulated by the Gastrointestinal Microbiota? Evolutionary Pressures and Potential Mechanisms.” Bioessays 36, no. 10 (October 2014): 940–49.
4. Temko, Jamie E., Sofia Bouhlal, Mehdi Farokhnia, Mary R. Lee, John F. Cryan, and Lorenzo Leggio. “The Microbiota, the Gut and the Brain in Eating and Alcohol Use Disorders: A ‘Ménage à Trois’?” Alcohol and Alcoholism 52, no. 4 (July 1, 2017): 403–13.
5. Sharon, Gil, Daniel Segal, John M. Ringo, Abraham Hefetz, Ilana Zilber-Rosenberg, and Eugene Rosenberg. “Commensal Bacteria Play a Role in Mating Preference of Drosophila Melanogaster.” Proceedings of the National Academy of Sciences of the United States of America 107, no. 46 (November 16, 2010): 20051–56.
6. Sharon, Gil, Daniel Segal, Ilana Zilber-Rosenberg, and Eugene Rosenberg. “Symbiotic Bacteria Are Responsible for Diet-Induced Mating Preference in Drosophila Melanogaster, Providing Support for the Hologenome Concept of Evolution.” Gut Microbes 2, no. 3 (June 2011): 190–92.
7. Stabb, Eric V. “Could Positive Feedback Enable Bacterial Pheromone Signaling To Coordinate Behaviors in Response to Heterogeneous Environmental Cues?” MBio 9, no. 3 (July 5, 2018): e00098-18.
8. Kort, Remco, Martien P. M. Caspers, Astrid van de Graaf, Wim van Egmond, Bart J. F. Keijser, and Guus Roeselers. “Shaping the Oral Microbiota through Intimate Kissing.” In Microbiome, 2014.
9. Varian, Bernard J., Theofilos Poutahidis, Brett T. DiBenedictis, Tatiana Levkovich, Yassin Ibrahim, Eliska Didyk, Lana Shikhman, et al. “Microbial Lysate Upregulates Host Oxytocin.” Brain, Behavior, and Immunity 61 (March 2017): 36–49.
10. Kiecolt-Glaser JK, Gouin JP, & Hantsoo LV (2010). Close relationships, inflammation, and health. Neuroscience and Biobehavioral Reviews. Close relationships, inflammation, and health. Neuroscience and Biobehavioral Reviews. 35, 33-38. PMC2891342
11. Melanie Greenberg Ph.D. How Married Couples Influence Each Other’s Physical Health. psychologytoday. 2019.10
12. Dill-McFarland KA, Tang ZZ, Kemis JH, et al. Close social relationships correlate with human gut microbiota composition. Sci Rep. 2019;9(1):703. Published 2019 Jan 24. doi:10.1038/s41598-018-37298-9
13. Brito IL, Gurry T, Zhao S, et al. Transmission of human-associated microbiota along family and social networks. Nat Microbiol. 2019;4(6):964-971. doi:10.1038/s41564-019-0409-6









