 国家高新企业 | ISO9001认证
国家高新企业 | ISO9001认证 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 二级病原微生物安全实验室

谷禾健康
呼吸道感染(RTIs)是全世界儿童发病和死亡的主要来源。在过去的十年中,已经清楚的是,呼吸道和肠道菌群与呼吸道感染的发病机理有关。宿主和环境因素可以促使呼吸微生物群在早期生活中走向成熟,这又与对呼吸道感染的持续易感性有关。近日,荷兰研究团队发表在《Cell host microbe》的一篇综述论文,详细阐述了微生物群在儿童呼吸道感染易感性中的作用,并回顾了呼吸道微生物群组成与呼吸道感染易感性联系起来的流行病学和机制证据,讨论了微生物在呼吸道感染急性期驱动感染和炎症中的作用机理。

背景:
呼吸道感染是全世界儿童发病和死亡的主要来源。 这些感染的范围从轻度的上呼吸道感染(U呼吸道感染)或普通感冒,到威胁生命的疾病,包括下呼吸道感染(L呼吸道感染)或肺炎。尽管全球死亡率总体上正在下降,但它仍然是5岁以下儿童死亡的主要原因,占该年龄组总死亡人数的15%(联合国儿童基金会和世界银行,2019年)。
重要的是,肺炎是导致肺功能下降和慢性后遗症的驱动因素,包括成年后哮喘和慢性阻塞性肺病(COPD)的发展。考虑到呼吸道感染在生命早期可能产生长期影响,因此越来越需要了解调节这些感染易感性和严重性的因素。
尽管揭露了许多这些因素,包括出生方式,进食类型,吸烟,早产,托儿所的出勤率和兄弟姐妹的存在,但潜在的生物学机制仍然难以捉摸。传统上,呼吸道感染被认为是由病毒或细菌引起的,由诸如流感嗜血杆菌和肺炎链球菌等“病原体”以及呼吸道合胞病毒(RSV)引起。但是,考虑到这些相同细菌和病毒的无症状“定殖”是普遍现象,这种关于呼吸道感染病因的以病原体为中心的观点似乎已经过时了。相反,最近对呼吸微生物群(即,由细菌,病毒,古细菌和位于粘膜表面上和表面的单细胞真核生物组成的群落)的研究表明,更广泛的微生物群落参与了呼吸道感染的病因。 除了局部微生物群的影响外,肠道微生物群还似乎通过其免疫调节特性促进了呼吸道感染的发病机理和严重程度。总之,这些新证据强调了我们对呼吸道感染病因学机制复杂性的历史性认识不足。
呼吸道微生物的早期来源和组装
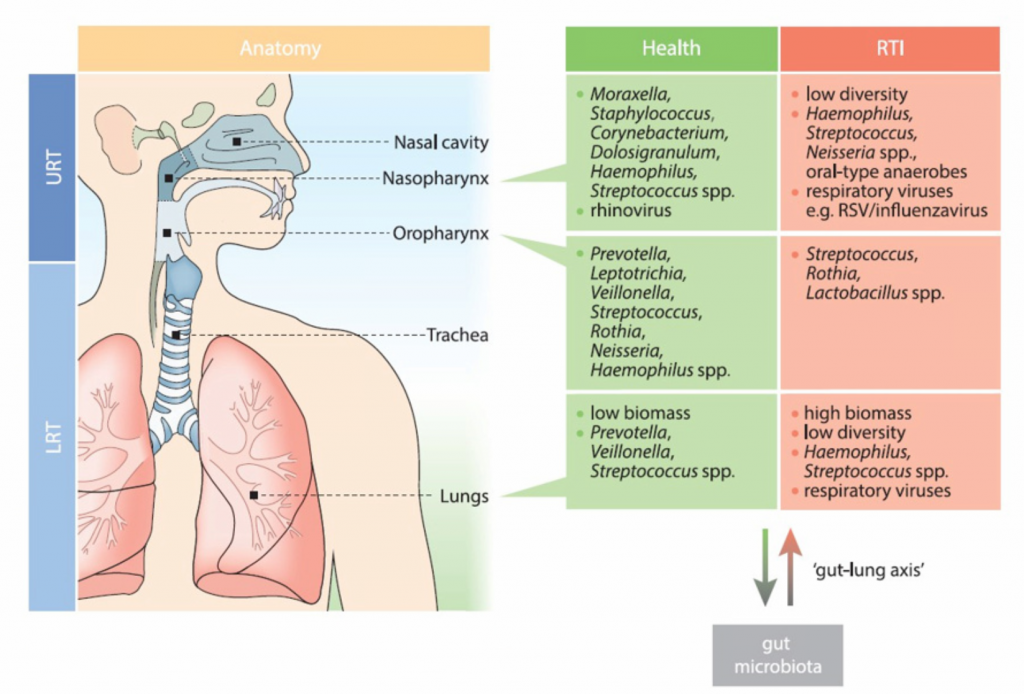
在健康方面,如图1所示,整个解剖龛的呼吸道细菌群落组成均不同。
尽管现在有几项研究表明,在怀孕期间胎儿会暴露于母体微生物产物中,而且甚至可能稀疏地存在活的微球菌属某些种。新生儿的大部分微生物组是在出生期间均匀混合微生物后获得的,因此主要来自母亲。一般来说,这种母体“初始试剂盒”通常由阴道、肛肠和皮肤微生物组成,尽管这与分娩方式不同。这种最初同质的微生物群在局部选择性压力下迅速在身体部位多样化。例如,鼻咽微生物群在生命的最初几周发展最为迅速,最初的金黄色葡萄球菌作为核心微生物出现,随后是革兰氏阳性共生菌棒状杆菌和Dolosigranulum菌(Dolosigranulum菌是1993年Aguirre等提议设立的一个菌属,该菌属细菌为兼性厌氧、触酶阴性或弱阳性的革兰阳性球菌,仅1个菌种)的定植和生长。从6周龄开始,莫拉菌属某些种迅速开始繁殖,并最终在3个月龄时超过该生态位。
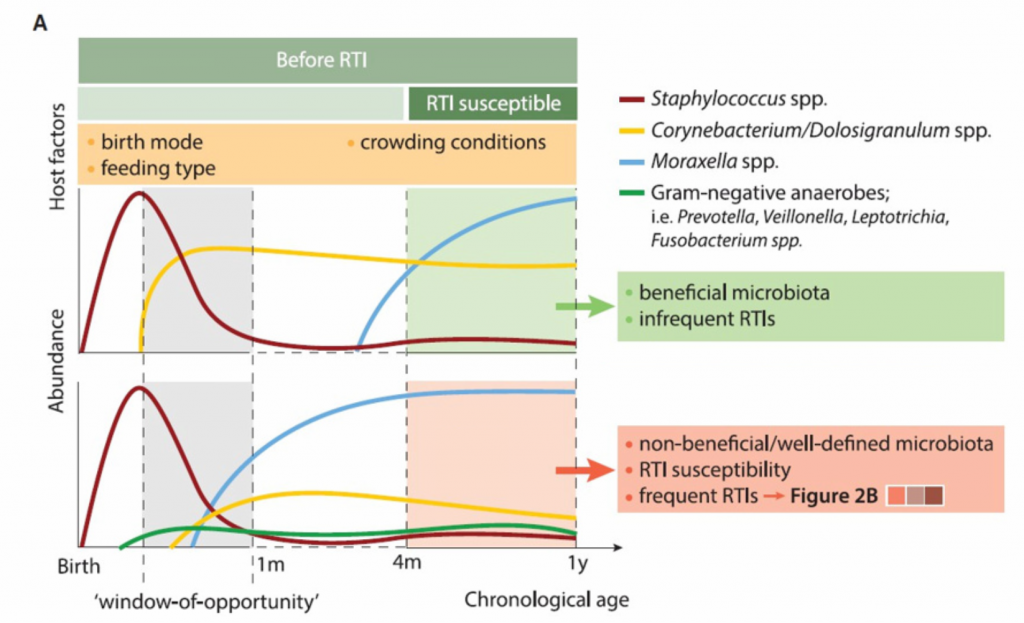

图1:健康和呼吸道感染期间各呼吸生态位的关键微生物群落成员健康和疾病期间上呼吸道(URT;鼻咽、口咽)和下呼吸道(LRT;肺)微生物群落组成概述。
至关重要的是,在围产期和新生儿期,这些微生物的成熟方式受到宿主和环境因素的严重影响。 与肠道相似,早期上呼吸道(URT)菌群的发育因出生方式而异,通常早期和长期的棒状杆菌/Dolosigranulum spp数量丰富。 在阴道分娩的婴儿中,金黄色葡萄球菌的持续存在和厌氧菌的出现,包括普雷沃氏菌,韦荣氏菌和卟啉单胞菌。在剖腹产(剖腹产)出生的孩子中。 喂养的方式类似地影响定居模式,除了金黄色葡萄球菌富集外,这表现出阴道分娩和母乳驱动的群落之间的相似性,这主要与皮肤-皮肤接触有关(即都是通过母乳和剖腹产获得的)。 与阴道分娩和母乳有关的微生物群组成的相似性可能是由母乳相关的免疫学因素驱动的,例如免疫球蛋白(Ig)A和生长因子,很可能自然地支持源自母亲的共生微生物。
最近的证据表明,母乳还含有自己的微生物,可能直接填充呼吸道粘膜。URT中微生物群结构和发育的其他重要决定因素包括暴露于拥挤条件下,例如托儿所的出勤和年幼的兄弟姐妹的存在。 这些因素也是呼吸道感染发生的众所周知的危险因素,可以通过观察到的相关微生物变化来解释,例如,普雷沃氏菌的早期富集和嗜血杆菌属的较高丰度。
解开影响微生物群组成的不同新生儿因素具有挑战性,因为许多因素之间有着千丝万缕的联系。例如,通过剖腹产分娩的母亲更可能早日停止母乳喂养。 此外,母乳菌群的组成似乎也受到分娩方式的高度影响,突显了研究其独立作用的难度。很少有研究调查下呼吸道(LRT)微生物群的驱动因素,这是一项极富挑战性的工作,因为这种利基的可获得性有限且细菌密度低,尤其是在健康婴儿中。但是,一项研究评估了健康早产儿和足月儿的气管抽吸物,结果表明,LRT菌群组成的差异最大,其胎龄不同,早产儿有葡萄球菌或脲原体,足月儿出现混合的成人型厌氧菌群,包括静脉内拉菌、普雷沃特菌和梭杆菌。与其他生态位类似,影响早期LRT微生物群的因素包括出生方式(仅早产儿)和产后年龄(仅足月儿)。
早期呼吸道微生物和儿童呼吸道感染
在呼吸道感染发展的各个阶段,有人提出了呼吸菌群组成的重要性(图2)。 多项纵向(出生)队列研究已证明了早期呼吸道微生物群发育与呼吸道感染敏感性和晚年呼吸道健康之间的关联。值得注意的是,在出生后的第一个月内,URT中过早从葡萄球菌过渡到以莫拉氏菌为主的分布,省略了棒状杆菌和Dolosigranulum占主导地位的早期阶段,这与出生后第一年发生呼吸道感染的频率更高有关。 尽管仍然需要证明其因果关系,但微生物群发育的第一个偏差和呼吸道感染发生之间的时间差异(通常从4个月开始)表明,早期微生物群暴露可能有助于先天和全身免疫,从而调节连续的呼吸道感染敏感性 。
或者,呼吸道感染敏感性增加至少可以部分归因于与微生物群落定殖事件的发生时间一致的其他事件,特别是由于母体IgG水平下降导致被动免疫的逐步下降。 值得注意的是,这些不是孤立的事件,也不是相互排斥的事件,可能会发生母体免疫力下降与婴儿特异性微生物和免疫介导作用之间的相互作用。
微生物群发育与呼吸道感染敏感性之间关系的关键似乎是定植事件的发生时间。
尽管到3-6个月时,几乎所有儿童都过渡到了以莫拉氏菌为主导的菌群,但莫拉氏菌的早期出现。 与气道粘膜的炎性免疫反应有关,以及第一次呼吸道感染和喘息的早期发作。 同时,链球菌早期富集(约9周)与早期L呼吸道感染的较高风险相关。 应当指出的是,除了定居的时间外,这些相关性可能是由物种或什至菌株水平的变化所驱动的,这可以通过卡他莫拉氏菌或林肯莫拉氏菌在早期定殖的婴儿的呼吸道感染风险差异来说明。
尽管奖赏可能引起免疫调节作用,但研究还暗示携带特定病原体(如肺炎链球菌)是呼吸道感染发生的直接危险因素。
传统上,特别是肺炎链球菌和流感嗜血杆菌是直接引起严重感染如细菌性肺炎的病原体。然而,尽管这些病原体在明显的细菌性呼吸道感染中已经确立并发挥了重要作用,但最近的证据表明,它们与更广泛的疾病表型有关,例如传统上认为是病毒起源的细支气管炎和并发性喘息。结合在一起,发现三种病原体(RSV,肺炎链球菌和流感嗜血杆菌)的存在和丰富程度可指示幼儿的疾病状况,而与感染表型(即肺炎,细支气管炎和混合感染)无关(Man 等,2019a)。 这与先前的研究一致,URR中以链球菌和嗜血杆菌为主的微生物群谱与RSV疾病的严重程度相关,而以葡萄球菌为主的谱菌似乎具有保护性。棒状杆菌和Dolosigranulum spp的丰度同时下降。 与更严重的预后相关,包括入住重症监护室。 因此,我们假设这些生态力量的总和最终将支撑呼吸道感染结果。
流感嗜血杆菌与肺炎链球菌和RSV之间的相互作用是有关病毒-细菌相互作用如何影响疾病严重程度的临床相关实例。重要的是要注意,尽管呼吸菌群领域取得了进步,但仍需要阐明支持这些关联数据的机制。 结果,越来越需要研究“混合感染”中的因果途径,重点研究共生微生物群可调节临床呼吸道感染结果的机制。
微生物群介导的呼吸道感染易感性调控机制
生命早期的微生物群落可能直接通过微生物与微生物的相互作用或通过宿主免疫系统间接预防呼吸道感染。 尽管现有的实验模型无法在真实,复杂的人类微生物群落中重建这些相互作用,但有关流行病学研究中突出的特定呼吸道法则已有相关证据。例如,尽管金黄色葡萄球菌因其致病潜力而广为人知,但由于其免疫调节特性,似乎是重要的早期鼻咽共鸣。 小鼠研究表明,URT中的金黄色葡萄球菌定植可通过诱导抗炎性M2肺泡巨噬细胞来抑制流感诱导的免疫介导的肺损伤。 这种共鸣的行为可能进一步取决于共同定殖的微生物物种,例如纹状棒状杆菌,抑制金黄色葡萄球菌的毒力基因的表达。葡萄球菌进化枝的其他成员,包括沙门氏菌和表皮葡萄球菌,也通过其抗菌肽诱导能力和生物膜形成能力在塑造鼻腔微生物群落中起着至关重要的作用。Dolosigranulum与棒状杆菌属共存的流行病学关联。 健康的部分原因可能是直接抑制了潜在的病原体。 实验性人类挑战研究支持了这一点,研究表明,直接挑战后,Dolosigranulum的高丰度与肺炎球菌的低丰度有关,并且与微生物群稳定性提高和黏膜细胞因子生成低有关。 体外研究已经证实,Dolosigranulum本身或与棒状杆菌属菌种协同作用可分别抑制金黄色葡萄球菌和肺炎链球菌的生长,这很可能是一系列抗菌肽产生的结果。棒状杆菌同时也是已证明通过Toll样受体(TLR)3介导的保护作用和体外直接代谢活性抑制潜在的病毒和细菌病原体。
早期微生物群发育与健康结果之间关联的潜在机制可能涉及其他免疫介导途径。 在早产儿和足月儿的早期气管样本中,宿主IgA基因表达被证明与预测的微生物IgA蛋白酶功能相关,这意味着微生物与宿主的串扰。此外,在小鼠身上,局部微生物群和树突状细胞之间的特定早期相互作用会在肺部产生调节性T(Treg)细胞亚群,防止气道高反应性。
当这些相互作用在以后的生活中发生时,这种保护就被取消了,再次强调了及时暴露于微生物对呼吸道粘膜免疫稳态发展的重要性。 除了对自然感染的免疫反应外,还建议肠道和鼻微生物会影响宿主对呼吸道疫苗的免疫反应,潜在地调节疫苗效力并影响宿主对后续呼吸道感染的防御。相反,肺炎球菌疫苗与呼吸道微生物群组成的变化有关,活减毒流感病毒疫苗的接种导致其他呼吸道病原体(如肺炎链球菌)的获得和密度的瞬时增加。 这些发现强调,应该做更多的工作来理解这些双边互动,并将其用于未来的量身定制的预防策略。
呼吸道感染中微生物介导的机制
除了在改变呼吸道感染敏感性中可能发挥作用外,呼吸道微生物群还与呼吸道感染发作的开始以及症状的严重程度有关。 尽管已知细菌在明显的细菌感染中具有致病作用,但特定的细菌群落成员,尤其是链球菌和嗜血杆菌属菌种,已牵涉到更广泛的疾病实体中,包括被认为是病毒起源的L呼吸道感染。 入侵这些病态生物可能会引起生态不稳定,并因此引起某种程度的炎症,尽管这本身可能尚未导致症状。 这种情况可能会促使包括环境刺激物(如香烟烟雾暴露)在内的触发因素进一步升级为有症状的感染(图2)。 确实,在小鼠中发现只有在也存在肺炎链球菌的情况下,烟雾暴露才可以改善微生物群落的平衡。 在这种情况下,并发病毒感染也可能成为“第二击”。 支持这一想法的是,在检测到病毒存在之前,患有富含莫拉氏菌的微生物群的儿童没有症状,从而导致L呼吸道感染。 在L呼吸道感染的细菌-病毒组合诊断模型中,病毒的存在最能将健康儿童与患有呼吸道感染的儿童区分开,无论其表型如何,其中包括推测的(细菌性)肺炎病例。这表明病毒感染可能会进一步扰乱已经受到挑战的宿主-微生物界面,从而导致疾病的表型和严重程度。 符合条件的是,同时感染了链球菌/嗜血杆菌为主的微生物群的RSV感染婴儿更有可能住院治疗,并显示出与TLR信号相关的基因表达更高以及嗜中性粒细胞和单核细胞募集。
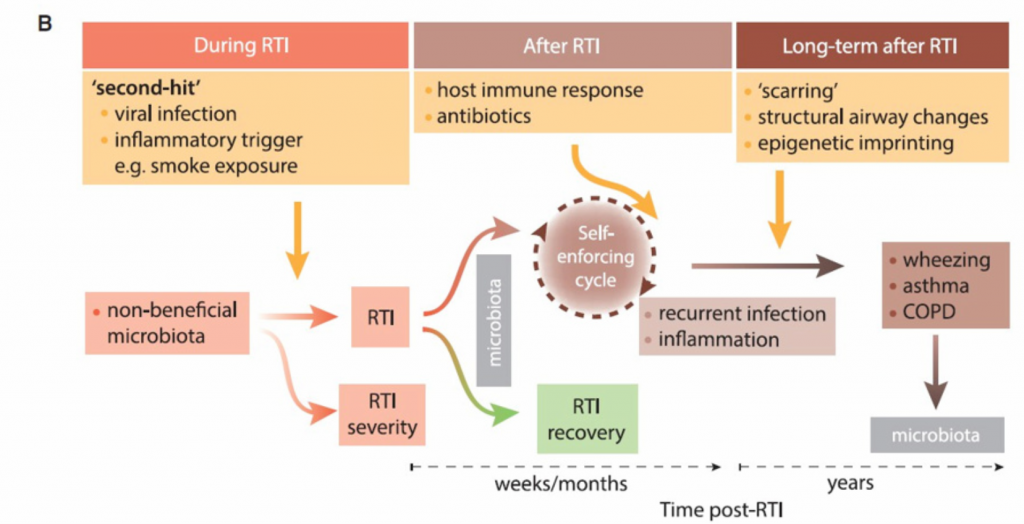


图2 呼吸道感染前、中、后呼吸道微生物区系的影响。
A.在4个月大的第一次RTI发作之前,呼吸菌群已经显示出与连续RTI敏感性相关的独特的微生物成熟模式。 这些早期的发育模式转化为有益的,平衡的微生物群,或转化为定义不明确和失衡的微生物群,后者通常与RTI的风险有关。
B. 一旦非有益的呼吸道微生物群建立起来,可能需要“第二次打击”来打破平衡,导致严重程度不同的感染。在RTIs之后,我们认为微生物群可以通过促进恢复状态而恢复到以前的状态,或者保持在扰动状态,这可能引发一个促炎症途径的自我强化循环。这种炎症状态反过来又使个体易受复发性rti的影响。长期来看,这种状态可能通过气道结构的改变永久改变呼吸道微环境,甚至可能导致长期后遗症,包括哮喘和慢性阻塞性肺病。
相反,对于以葡萄球菌为主的微生物群定殖的儿童,情况恰恰相反。 体外研究进一步证明了这一点,表明用不可分型的流感嗜血杆菌引发初级支气管上皮细胞可导致ICAM-1上调,并促进促炎性细胞因子(包括IL-6和IL- 8在受到额外的RSV攻击后。 除了这些通过宿主炎症引起的间接相互作用外,直接病毒细菌相互作用还可能影响L呼吸道感染发病的早期阶段,这项研究表明,预先与流感病毒一起孵育的肺炎球菌证明了在其他幼稚小鼠中细菌粘附的增加。
一旦达到平衡,感染和炎症就会并存,相互促进。该理论在L呼吸道感染的背景下得到了很好的描述。 在这些感染期间,正常的低密度和高多样性微生物区系会被一种推定的病原菌过度繁殖。随后的病原体诱导的宿主炎症可能作为炎症和微生物变化的自我强化循环的起点,从而促进了进一步的病原体生长。
呼吸道生态环境中的微生物与宿主的相互作用
尽管宿主-微生物相互作用对呼吸系统健康的重要性显而易见,但这些相互作用可能会因解剖位置而异。在健康的URT中,自然存在于鼻咽和口咽生态位的微生物群落之间存在明显的区别。 但是,这种地形上的区别在呼吸道感染发作之前和期间开始逐渐消失,尤其是口型细菌包括Prevotella,Neisseria,Veillonella,Fusobacterium和Porphyromonas出现在鼻咽中。
同样,在L呼吸道感染的背景下,微生物群落之间的区别在解剖边界变得模糊。 在患有这些感染的儿童中,在成对的鼻咽和气管样本中检测到的病毒和细菌群落组成大部分重叠,这表明URT微生物群(例如通过微生物过度生长和微量抽吸)可能是LRT微生物群的来源,从而导致了症状。急性L呼吸道感染。 或者,可以在不同的呼吸生态位中发挥类似的控制力(例如炎症),选择相似的微生物群落。 尽管本研究中URT和LRT微生物群落之间存在大量重叠,但仍选择了一个分类单元,包括葡萄球菌,棒状杆菌和Dolosigranulum spp。 在鼻咽以外很少发现,这说明这些物种具有生态位特异性的生态作用。
与不同生态位的微生物组成不同,某些物种确实在整个呼吸道中发生,但是它们的免疫调节功能可能会随它们所嵌入的生态位和特定生态位社区而变化。 例如,厌氧菌(如厌氧杆菌和Veillonella)被认为是口腔和口咽的正常标志,它们的丰度似乎表明呼吸系统健康。 然而,当在肺中鉴定出相同的微生物物种时,它们与IL17 + CD4 + T细胞和中性粒细胞的水平升高以及TLR4反应减弱有关(Segal等,2016)。这些发现都强调了呼吸微生物物种和健康状况之间的联系只能在其完整的生态环境中进行评估,而生态环境可能因生态位而异。
呼吸道感染术后微生物扰动的影响
如上所述,证据支持早期呼吸微生物群在呼吸道感染发展中的作用,并强调呼吸道感染期间发现的微生物群组成的扰动。我们假设,这种受干扰的微生物群组成不仅导致微生物群的定殖抵抗力和恢复力降低,并且在呼吸道感染期间增强症状,而且导致症状持续存在和未来复发的风险。最近的数据表明,婴儿呼吸道感染后三周内链球菌科的富集与随后一段时间内的持续症状有关。此外,抗生素治疗,通常为生殖道感染,可能会进一步扰乱已经受到挑战的微生物群落,有可能降低感染后对新获得病原体的抵抗力。
事实上,在抗生素治疗后,居住在呼吸道的促进健康的共栖体,包括棒状杆菌和白云雀属,以及肠道共生体,如双歧杆菌属,在抗生素治疗后被消除,从而可能增加感染复发的风险。这也许可以解释为什么抗生素治疗后急性中耳炎复发率更高,尽管这还没有在随机对照环境中进行研究。
尽管可能涉及一个自我执行的循环,在该机制中特定的微生物群落可以促进或维持促炎环境,从而促进病原体大量繁殖,但尚未阐明这种现象的确切机制。 当这些病原体引发进一步的促炎性级联反应时,恶性循环迫在眉睫(图2)。
据推测,当不能恢复到稳定状态时,这些变化可能会持续并转化为疾病的长期后遗症。对易患哮喘儿童的研究证据表明,下咽部微生物群的组成特征为Veillonella,Prevotella和Gemella spp。 出生1个月时,患者气道炎症与6岁时哮喘的诊断有关。 这项最新研究增加了先前的研究,将卡他莫拉菌和流感嗜血杆菌的早期存在与1个月大的哮喘多发新生儿气道中的促炎性免疫特征联系起来。此外,婴儿期急性感染期间莫拉菌属,嗜血杆菌和链球菌的富集与变态反应致敏或细支气管炎后嗜血杆菌和链球菌的富集有关。 或3至5岁时反复喘息,进一步支持了这一理论。 无论如何,需要进一步研究支持这些关联的机制,以了解确切的因果关系。
通过表观遗传的印记,早期生命中的微生物和免疫扰动可能在晚些时候改变气道的生理功能的一种可行方式是。
体外模型和分子流行病学研究表明,年轻时的RSV感染确实会在生活中涉及哮喘的各种免疫改变基因中诱导组蛋白和DNA甲基化修饰。
同样,有益的微生物可以通过产生短链脂肪酸(SCFA)来调节宿主表观基因组,如肠道营养证明的那样。 这些SCFA与呼吸道的生物学相关性已通过一项体外研究得到证明,该研究表明暴露于acc.accolens后从鼻上皮表面释放的SCFA抑制了肺炎链球菌的生长。
肠肺轴
除了呼吸道中局部微生物介导的作用外,越来越多的证据表明,肠道菌群在调节呼吸道免疫力中起着重要作用。这两个粘膜部位之间的连接通常称为肠肺轴。
除了肠道和肺微生物群之间的一般联系外,人们还发现,肠道微生物定植似乎与早期生命特别相关。 在小鼠中进行的一项研究表明,肠道定居,尤其是在新生儿窗口内,可以防止卵白蛋白诱导不变的自然杀伤性T(iNKT)细胞积聚到肺中,这一过程与过敏性哮喘的发展有关,并通过降低Cxcl16基因的甲基化程度来介导 。 肠道菌群似乎也促进了第3组先天性淋巴样细胞(ILC3s)向肺的动员,有助于防御肺炎。 新生儿时期肠道菌群的耗竭严重损害了这种防御,而成年小鼠的相同程序显示出对肺炎易感性的作用有限,强调了微生物菌群与宿主相互作用的时机很重要。一项最近的出生队列研究支持了这一点,该研究表明阴道分娩支持的双歧杆菌属早期丰度与生命的第一年间的呼吸道感染之间存在时间上的联系。 该研究表明肠道微生物组可以充当介体,将已知的危险因素(例如分娩方式与呼吸道感染易感性)联系起来。
此外,在呼吸道感染期间,肠道微生物群落可能会影响疾病的严重程度,这已经通过鼠类研究调查了细菌和病毒性呼吸道病原体。 符合条件的是,已提出了益生菌给药(主要是乳酸杆菌和双歧杆菌属)对U呼吸道感染的发生率和持续时间的影响,尽管这些研究的质量有限阻碍了结论,这主要是由于研究规模不足或随机方法不清楚以及盲目性。
此外,在呼吸道感染期间,肠道微生物群落可能会影响疾病的严重程度,这已经通过鼠类研究调查了细菌和病毒性呼吸道病原体。 符合条件的是,已提出了益生菌给药(主要是乳酸杆菌和双歧杆菌属)对U呼吸道感染的发生率和持续时间的影响,尽管这些研究的质量有限阻碍了结论,这主要是由于研究规模不足或随机方法不清楚以及盲目性。如前所述,肠道微生物群影响呼吸道感染的机制大概是免疫介导的,或者涉及微生物衍生产物向血流的释放。例如,微生物代谢物去氨基酪氨酸通过增强I型干扰素信号传导,消除肺部免疫病理学来保护小鼠免受流感的侵害。有趣的是,这些微生物群驱动的有益影响不仅限于细菌,因为共生真菌在这方面也被证明是重要的。
般而言,这些常驻社区成员似乎可以校准免疫语调并教育免疫系统如何控制病原体以预防症状性感染。 后者似乎与病毒感染最相关,但也与细菌感染有关,已经揭示了特定的机制。例如,已显示通过特定细菌群的Nod样受体(NLR)-可通过肺泡巨噬细胞中IL-17A依赖性的粒细胞-巨噬细胞集落刺激因子(GM-CSF)信号传导增强保护免受细菌感染。
肠道菌群可能影响呼吸道感染的一种不太直接的免疫介导途径可能是与针对呼吸道病原体的疫苗相互作用。 小鼠模型揭示了潜在的机制,肠道共生可能通过细胞或代谢物介导的与巨噬细胞或B或T细胞之间的相互作用来增强免疫记忆的形成,Lynn和Pulendran(2018)综述。例如,微生物鞭毛蛋白已显示以TLR5介导的方式诱导更高水平的浆细胞和三价流感疫苗(TIV)特异性IgG。 肠道菌群对人类疫苗接种反应的影响可能涉及复杂的途径,正如对抗生素改变的菌群患者进行的TIV应答的多组学分析所表明的那样。 尽管抗生素的耗竭仅对免疫力低的患者显示了对TIV特异性抗体的影响,但微生物菌群的畸变与TIV后转录组和代谢组学模式的变化相关,包括炎性体信号转导增加。
尽管仅是关联性的,但该证据表明肠道菌群可通过局部和全身相互作用介导疫苗效力。
肠与肺之间的相互影响是相互的。 研究表明,小鼠的RSV和流感感染都会影响肠道菌群的组,使这些小鼠更容易受到随后的肠感染的影响。流感引起的肠道微生物变化和SCFA产量减少也显示出增加了对继发性肺炎球菌感染的易感性,强调了肠道与肺之间的强烈串扰。
未来的展望
在过去的几年中,一般的“一种病原体,一种疾病”的范例已经开始向整个生态系统的呼吸道感染发病机制理论转移。 该理论既包括微生物相互作用,也包括微生物-宿主介导的免疫调节,它们是呼吸道感染的适应性和敏感性的基础。
最近的知识不仅加深了我们对呼吸道感染发病机理的了解,而且还有助于重新定义预防,诊断或治疗方式。例如,Langelier等结果表明,他们可以通过使用集成的微生物组和宿主转录组分析,将重症患者的肺炎与其他急性呼吸衰竭的其他原因准确区分(Langelier等,2018)。 此外,实时宏基因组测序已被用于诊断细菌性肺炎的微生物起源,同时能够检测微生物抗性基因,从而可以在感染的早期阶段进行量身定制的抗生素治疗。这种知识还可以导致新的生物治疗方法。
尽管这些实例很有前途,但我们应该加大对生物学机制的认识,以支持宿主,微生物和外界之间的健康平衡。 为此,作者建议并行使用模型系统和系统生物学方法。
尽管揭示微生物群组成和功能与呼吸道感染发病机理各个阶段的健康结果之间的关联非常重要,但重要的警告仍然是已证实的因果关系的稀疏性。微生物组研究的一个主要挑战是在模型系统中捕获代表人类有机体的整个生态系统范围的宿主-微生物相互作用的复杂性。 结合基因和组织工程学等其他领域的最新进展(例如人类微生物菌落定殖的[人源化]鼠模型),可以为这些问题提供一些解决方案。
通过对这些模型生物进行遗传操作获得的功能丧失或丧失功能的实验,再结合下一代测序技术,可用于评估微生物宿主相互作用或微生物基因组介导的表观遗传改。此外,先进的体外模型,包括芯片上气道模型或气道类器官可用于研究可控环境下的宿主-微生物相互作用,其中后者已被证明可用于对RSV或流感病毒感染进行建模。
体内动物模型和类器官系统都可以与基因编辑技术相结合,以调节宿主或组织侧(基因表达和代谢产物/蛋白质的产生)或改造微生物的基因组。
尽管取得了这些进展,但由于每个定义的体内和体外模型都缺乏人类生理学和环境变化的复杂性,因此人类研究具有希望。与观察性研究相反,这些研究使我们能够深入了解病原体获取周围的微生物群落动态,并允许对单个变量进行操作,例如共同定殖细菌或病毒,或测试疫苗相互作用。 与疫苗学领域类似,近年来,疫苗学领域采取了“系统科学”的方向,类似的多组学方法也可能有助于了解呼吸道感染的病因。 包括病毒,噬菌体和真菌在内的目前尚未深入研究的非细菌微生物群落,将使我们能够更详细地捕获整个生态系统范围内微生物群落的真实变化。这将使我们能够更广泛地研究环境,微生物和宿主之间与呼吸系统疾病的病因和表型之间的相互作用。
儿童呼吸道感染的长期后果以及微生物群在呼吸道感染发病机理中各个阶段的全面作用要求采取早期预防和治疗干预措施以促进呼吸系统健康。

谷禾健康


日常生活中,我们更多地认为心情是由思想控制的。这虽然是常识,但并非完全正确。
荷尔蒙也十分影响我们的情绪,同时我们的情绪也受到免疫系统和肠道微生物的影响。更复杂的是,在这个纠结的生物网络中,我们的生活承受着巨大的压力。
精神压力是身体对生活压力的反应。它设法使身体恢复平衡,正常发挥免疫力,激素和神经的作用。来自工作上的压力或生活的压力都可能会导致抑郁和焦虑。无论哪种情况,都可以从免疫系统开始。
抑郁症是全世界致残的主要原因之一。据估计,五分之一的人在一生中都会经历抑郁症。大约85%的人在经历第一次抑郁症发作后的10年内会复发。

今年,COVID-19大流行,更给全球人类造成了极大的焦虑和不安全感,持续的危机将导致很多心理和精神健康问题。
肠道是人体最大的器官,肠道中微生物包括细菌,病毒,真菌等。微生物的平衡对于身心健康至关重要。
而现代生活中的各个方面,例如压力大,加工食品饮食,抗生素,杀虫剂以及经过消毒的城市环境,都减少了健康肠道菌群的数量和丰富度,同时增加了有害菌群的数量。最新的抑郁理论表明,肠道菌群失衡可能在这种疾病中起主要作用。
一项2019年的研究将来自精神分裂症患者的粪便微生物移植物引入无菌小鼠中,发现这些小鼠随后表现出与精神分裂症有关的行为。
同样,另一项研究指出对患有抑郁样特征的啮齿动物施用益生菌可以减少这些特征,从而使啮齿动物恢复正常行为。所有这些表明,肠道菌群的外部操纵具有进行新的心理健康治疗的潜力。
抑郁症患者的肠道菌群与健康人不同。与健康个体相比,它们的肠道菌群多样性和丰富性较少。《自然》杂志上的一项新研究发现,肠道微生物对于消除小鼠的恐惧反应至关重要。
众多研究表明肠道微生物连接大脑至少有三种途径:免疫、神经系统,激素和代谢物。
病原体发作后,免疫系统就会立即按下紧急按钮。但是免疫系统也可以对心理压力做出反应,即所谓的“无菌压力”。与侵略者作斗争是一回事,但是让我们抵抗精神压力似乎是对免疫系统的一大要求。
有一种简单的方法来证明压力和免疫力之间的密切联系:每天将普通小鼠引入恶霸小鼠中几分钟。结果很神奇,受欺负的小鼠仅需一到两周即可产生炎症,这是通过其血液中的促炎化学物质测得的。其中,他们的淋巴细胞——白细胞发生了变化。
另外,有一种特别培育出来的淋巴细胞水平较低的老鼠比普通老鼠更焦虑,更不善于交际。但是,当您向这些害羞小鼠注入来自受欺负小鼠的淋巴细胞时,会发生一些有趣的事情:可能与你期望的相反,它们变得不那么焦虑,开始更多地社交。
在应激小鼠中,这些淋巴细胞正疯狂地试图减少恶霸引起的炎症。这是自然界最持久的回路之一:负反馈回路,可防止反应失控。威胁带来的心理压力会引发这些淋巴细胞抑制炎症。一旦转移,它们对宿主小鼠产生有益的“减轻压力”作用,导致促炎性化学物质减少并赋予其新宿主弹性。
这个有趣的实验表明,纯粹的心理压力可以改变免疫系统。这表明,通过简单地重新平衡免疫系统可以治疗抑郁症。
美国阿尔伯特·爱因斯坦医学院Paul S. Frenette小组发现,肠道微生物组可以调节心理压力引起的炎症。这一研究成果在线发表于2020年7月30日的 《免疫》。利用镰状细胞病的血管闭塞性发作(简称VOE)作为血管疾病模型,研究人员发现压力可通过引起糖皮质激素的激素反应增强VOE,这些激素反应可增强肠道壁的通透性,导致微生物依赖的IL-17A固有层辅助性T细胞17的分泌,随后引发VOE中性粒细胞循环池扩大。
我们已经看到反复的压力如何导致免疫反应,以及由此产生的炎症导致抑郁和恐惧。但是微生物扮演什么角色?
虽然认知疗法和增加5-羟色胺的抗抑郁药可以有效地治疗抑郁症,但这些治疗方法对超过三分之一的抑郁症患者无效。最近的抑郁理论表明,肠道微生物群的失衡和连接肠道和大脑的轴的功能障碍可能参与其中。
这个世界上布满了细菌,其中许多细菌都比吃我们的早餐更好。为了应对这种恶劣的现实,动物们很早就招募了自己的一组友好微生物来保护自己。这些有益细菌并没有真正站在我们这边,只是他们很乐意在温暖和潮湿的肠子中发酵,不断地吃自助餐。这些细菌被称为共生细菌,拉丁语是“共享餐桌”。
当病原体试图侵入这一长期建立的伙伴关系时,共生菌会手撕它们。从本质上讲,我们的微生物群是免疫的先锋。为了与人类宿主建立如此重要的关系,共栖者需要教会免疫系统容忍它们。这是在生命的早期,出生后一千天之内发生的,如果这个过程受到干扰,那么可能会终身受疾病困扰。
就像牧羊犬引导羊群一样,免疫细胞监视着共生菌,使它们在肠道内和血液循环外被包围。而细菌则分泌脂肪酸来安抚免疫细胞,保护它们免受攻击。通过这些分泌物,肠道细菌可以在远离肠道的地方发挥作用,甚至可以改变大脑的形状和功能。
这意味着微生物会改善情绪。
那些可以改善情绪的菌群,包括某些乳酸菌和双歧杆菌,甚至可以降低皮质醇水平。这些被称为精神药物,它们代表了另一种可能的抑郁症治疗方法。尽管这些都属于该领域的早期研究,但已证明它们可以减少负面想法,改善认知并降低IBS症状。
微生物可以影响你的海马体或杏仁核的大小,这些大脑中心与情绪、食欲和恐惧有关。海马体试图阻止下丘脑-垂体-肾上腺轴(HPA轴)偏离轨道,如果它的生长发育迟缓,就会影响应激反应。

如果说免疫细胞和微生物是生物乐队的主要参与者,那么HPA轴就是指挥。HPA轴是下丘脑-垂体-肾上腺的缩写,它协调着逐渐增加的激素释放与皮质醇的释放,皮质醇是战斗或逃避反应的药物。压力通路会通过交感神经系统(SNS)和/或下丘脑-垂体-肾上腺(HPA)轴从大脑传递信号。SNS的激活导致肾上腺素(从肾上腺髓质)和神经递质去甲肾上腺素(从各个器官的神经末梢)释放激素。
HPA轴激活最终导致肾上腺皮质分泌糖皮质激素。 糖皮质激素通常具有免疫抑制功能。 例如,在压力下升高的糖皮质激素浓度显示可通过B和T淋巴细胞的凋亡引起适应性免疫缺陷,并且糖皮质激素已在药理学上用作有效的抗炎药。 炎症被认为是导致与压力有关的疾病的常见途径。 考虑到糖皮质激素作为免疫抑制剂的作用,应激诱导炎症的机制仍然不清楚。
迷走神经是肠道和大脑之间的主要传输通路。
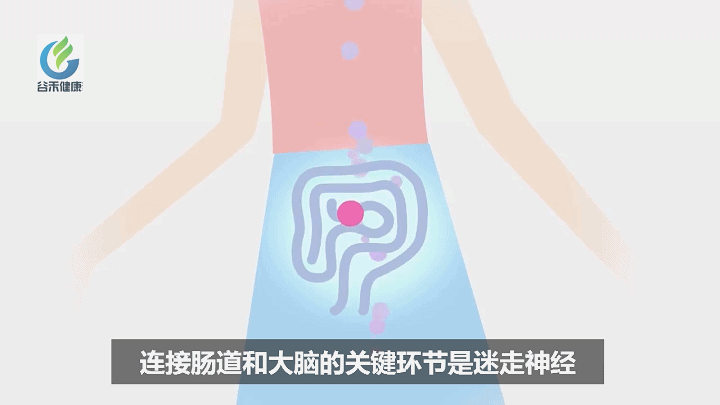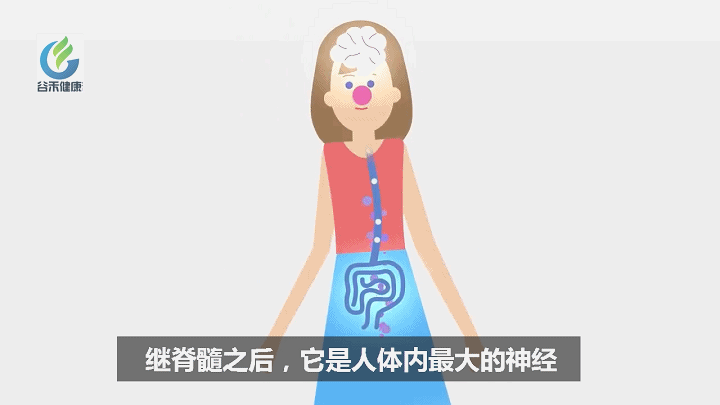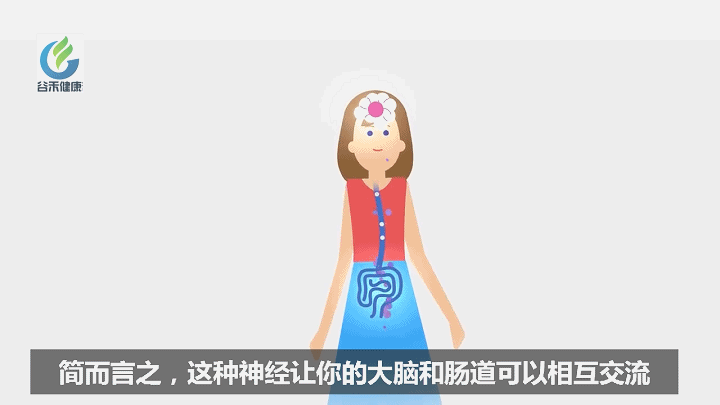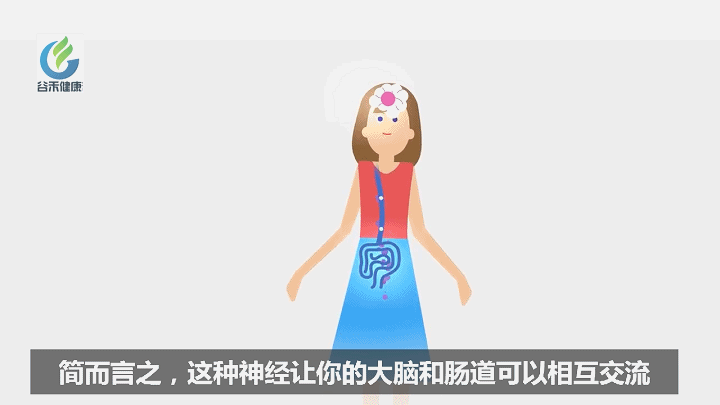
迷走神经也被称为“徘徊神经”,因为它是人体中最长的神经,并且有无数分支从脑干一直延伸到肠的最下部,并一路接触大部分主要器官。
迷走神经是副交感神经系统的主要组成部分,它调节“休息和消化”或“趋于友好”的反应。另一方面,为维持体内平衡,交感神经系统驱动“战斗或躲避”反应。
迷走神经通过引起所谓的“放松反应” 来抵消战斗或躲避中的应激反应。
迷走神经的早期解剖图
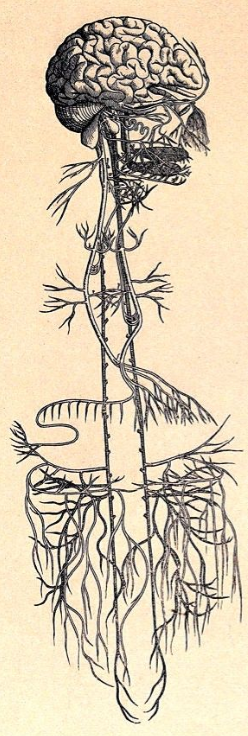
来源:Wellcome Library/Public Domain
炎症反应在许多疾病的发展和持续中起着核心作用,并可能导致慢性疼痛衰弱。在许多情况下,炎症是您身体对压力的反应。因此,减少神经系统中的“战斗或逃跑”反应并降低应激的生物标志物也可以减轻炎症。

瑜伽和冥想等日常习惯或可以对抗炎症
越来越多的证据表明,对抗炎症的另一种方法是通过接合迷走神经并改善“迷走神经张力”。这可以通过使用瑜伽和冥想等日常习惯来实现。
植入迷走神经刺激装置,改善类风湿关节炎
在更极端的炎症情况下,例如类风湿关节炎(RA),可以使用植入的迷走神经刺激装置(VNS)来实现。
最近,来自阿姆斯特丹和美国的国际研究人员团队进行了一项临床试验,该试验表明,使用小型植入装置刺激迷走神经可通过抑制细胞因子的产生显着减少炎症并改善类风湿关节炎患者的预后。
学会呼吸,缓解焦虑,辅助降压
健康的迷走神经表现为吸气时心率略有上升,呼气时心率略下降。
每次呼气时,迷走神经都会向心脏喷射一些乙酰胆碱。
横膈膜深呼吸(长时间缓慢呼气)是刺激迷走神经并减慢心率和血压的关键,特别是在表现焦虑时。
迷走神经张力指数与健康状况相关
较高的迷走神经张力指数与身心健康息息相关。相反,迷走神经张力指数低与发炎,抑郁,情绪低落,孤独,心脏病发作和中风有关。
2010年的一项研究发表在《心理学》杂志上:积极情绪如何建立身体健康:感知到的积极社会关系解释了积极情绪和迷走神经张力之间的螺旋上升。
在这项研究中,北卡罗来纳大学教堂山分校的Barbara Fredrickson和Bethany Kok研究了迷走神经,发现高迷走神经张力指数是积极情绪、身体健康和积极社会关系之间反馈回路的一部分。
慢性压力与胃溃疡有关
相信大家可能有过对公开演讲感到恐惧的经历,这种站在人前的演讲会导致消化不良。由于沿着肠脑轴的迷走神经通路是双向的,因此当人真正感到压力时,这些脑-肠连接创造了一条来回交流的高速公路,可能会失去控制。
脑岛接收来自肠道的压力信号,从而使大脑在胃部发出更多与压力有关的警钟。
注:脑岛为大脑的岛叶,控制很多感觉和情绪的产生。
Levinthal和Strick(2020)的最新研究表明,大脑会立即将压力信号从脑岛发送到肠道。随着时间的推移,慢性压力以一种可能导致胃溃疡的方式劫持了这些脑肠连接。
有趣的是,他们推测,促进“战斗或逃跑”反应的交感神经系统会沿着迷走神经通路发送大脑到肠道的信息,这些信息与大脑皮层的运动区域更紧密地联系在一起。
更好地理解脑-肠连接如何控制胃,将为肠胃科医生治疗肠道问题和胃肠道疾病带来新的更好的方法。
HPA轴通过几种不同的化学物质与微生物群和大脑进行通讯
细菌分泌物:代谢物如短链脂肪酸。
免疫化学物质:细胞因子,如干扰素,白介素和肿瘤坏死因子。
腺分泌物:肾上腺素和皮质醇等激素。
神经传递化学物质:神经递质,例如5-羟色胺,多巴胺和GABA。
更多关于肠道菌群与神经系统这部分内容详见之前的文章:肠道微生物(菌群)与脑神经(中枢神经)到底如何联系?
微生物代谢产物是细菌在进食和代谢食物时分泌的化学物质。
不同的细菌在不同的情况下会产生独特的代谢产物。其中一些代谢物是短链脂肪酸,例如丁酸,可以使其进入大脑,从而影响大脑的生长和功能。
从肠道内的微生物和细胞释放的化学物质可直接导致迷走神经和迷走神经支配的大脑区域的电活动变化。迷走神经的活动众所周知会影响人类行为。实际上,植入刺激该神经的电子设备(迷走神经刺激器)可以有效地治疗患有严重的,对治疗有抵抗力的抑郁症和癫痫症的患者。
越来越多的研究表明,与健康对照组相比,患有多种精神疾病的人倾向于具有不同的肠道菌群模式。例如,两项研究发现与健康对照组相比,精神分裂症患者的肠道菌群多样性要低得多。
其他研究表明,抑郁症患者和自闭症患者与健康对照组相比,肠道菌群的模式也不同。这与相关研究有关提示特定的肠道菌群在涉及精神疾病的神经递质和代谢产物的产生和调节中起重要作用。这包括5-羟色胺,多巴胺和γ-氨基丁酸(GABA)。
肠道微生物影响大脑和行为的其中一种方法是通过这些微生物或是受这些微生物影响的胃肠系统细胞释放化学物质。这些化学物质释放到血液中,然后穿过血脑屏障并影响神经细胞的生长,功能和连通性。
这些化学物质中的一部分也会影响大脑免疫系统的细胞。此外,人们早就知道,肠道中合成的某些化学物质也是由大脑细胞产生的(例如生长抑素和胆囊收缩素等),这些由大脑合成的肠道化学物质可有效调节大脑功能。
发现的第一种神经递质——乙酰胆碱
1921年,一位名叫奥托·洛维的德国生理学家发现,刺激迷走神经会触发释放Vagusstoff物质(德语为“ Vagus物质”),从而导致心率降低,“迷走神经物质”后来被识别为乙酰胆碱。并成为科学家发现的第一个神经递质。
乙酰胆碱就像一种镇静剂
Vagusstoff就像镇静剂一样,可以减缓心跳间隔并改善心率变异性(HRV)。稳健的迷走神经张力和更高的HRV齐头并进,是整体心理和身体健康的标志。
你可以通过做深呼吸并长时间呼气来简单地自我管理。有意识地利用迷走神经的力量可以在抑制炎症反射的同时建立一种内心平静的状态。
皮质醇
皮质醇是人体通过增加血压,心率和呼吸来增强压力反应的方式,以便逃脱或者逃离烦扰。 在此过程中,它还会降低免疫反应:首先,你逃脱烦扰,然后才处理流感或食物中毒。这是一个合理的回应方式,只是片刻的恐慌。 但是随着时间的流逝,防御能力下降可能导致肠漏,从而将心理压力转化为微生物压力。
5-羟色胺
在肠道中也发现了与抑郁和幸福有关的化学物质,例如血清素(5-羟色胺)。90%的5-羟色胺是在消化道而非大脑中产生的。许多抗抑郁药通过增加5-羟色胺发挥作用。
科学家发现肠道菌群会产生许多其他神经递质,例如多巴胺,去甲肾上腺素,GABA等,它们对情绪,焦虑,专注等至关重要。肠道微生物群会导致大脑反应方式发生变化。
我们知道,抑郁症不仅是精神疾病。抑郁症患者可能同时患有多种疾病,包括脑功能障碍,免疫系统失调和应激激素紊乱等。
研究表明,肠道菌群在免疫,激素平衡和神经系统功能中起着至关重要的作用。
新的理论和研究表明,肠道菌群失衡可能导致许多与抑郁症有关的疾病。前面我们知道,肠道菌群会影响HPA轴的发育,该轴调节压力反应并参与皮质醇的释放。在抑郁和长期处于压力下的人中,HPA轴可能失调,导致过量的皮质醇(一种压力激素)被循环。
肠道菌群也在免疫系统功能中发挥作用,并调节称为细胞因子的化学信使的产生。促炎性细胞因子的失衡可导致慢性炎症和自身免疫性疾病,通常与抑郁症同时发生。
肠道菌群也参与神经系统的功能。肠道菌群失衡可能会影响神经递质(例如血清素)的水平,已知这些物质与抑郁症有关。
其他研究将肠道菌群失衡与降低γ-氨基丁酸(GABA)的水平联系在一起,GABA是一种可以缓解焦虑的大脑化学物质。大脑和肠道可能通过迷走神经进行交流,迷走神经是一条遍及全身的大神经。
肠道微生物群和神经系统疾病详见之前的文章:最新研究速递 | 柳叶刀:肠道微生物群在神经系统疾病中的作用
2019年发表在《自然》杂志的一项新研究:肠道微生物对于消除小鼠的恐惧反应至关重要。该研究是对肠脑轴力学的不断探索中的最新成果。
他们发现了一组代谢产物,这些代谢产物影响大脑处理恐惧的方式。特别是,科学家们正在研究小鼠用了多长时间才能克服习得的恐惧反应。
在发出信号后,立即对这些小鼠进行小小的足部电击。学会了这种联想之后,每当听到信号时,他们都会陷入恐惧中。它们的大脑已经将听到信号的神经元与那些预期电击的神经元联系起来。这种硬连线的神经联系就是巩固大脑记忆的方式。
正常和无菌小鼠都能学会恐惧关联,但Artis和同事发现,无菌小鼠不能忘记恐惧。只要它们保持无菌,在听到信号时总是会冻僵。
如果研究人员再给无菌老鼠一些健康的微生物,他们就可以忘记这种关联,恐惧反应很快消失了。并且直到小鼠达到一定年龄才起作用。此后,即使肠道菌群好,他们也总是对这种音调做出反应。
研究人员排除了迷走神经和免疫系统的作用。相反,他们发现这种作用与四种特定的代谢产物密切相关。在正常生殖的小鼠中,这些代谢物从肠道到达大脑,并促进记忆形成和随后的遗忘。如果没有那些细菌来提供代谢产物,无菌小鼠似乎注定会永远对的音调做出反应。
研究人员已经知道,精神分裂症和肠道生物群系之间已有数年的联系。可惜他们一直无法完全理解两者之间为什么或如何相关。
一项新研究由多名中国研究人员与位于锡拉丘兹的纽约州立大学医学院研究团队共同发表的论文可能提供了一些答案。
该小组招募了63名严重程度不同的精神分裂症患者和69名健康对照者。两组的性别组成,平均年龄和平均体重指数相似。
然后,研究人员从这些组中收集了样本,并通过测序评估了每个受试者肠道微生物群。他们测试了两组之间的差异,以及可能将患者精神分裂症症状的严重程度与肠道生物群系内特定失衡联系起来的差异。
研究小组在2019年2月版的《科学进展》上撰文称,“发现两组动物肠道微生物组成存在明显差异”,两组细菌群落在门或类水平上存在差异。
此外,他们发现“全球微生物表型并没有受到性别或药物状况的很大影响。” 换句话说,与健康对照的肠道生物群系相比,精神分裂症受试者的肠道生物群具有一些特定菌群丰度增加,而其他特定菌群缺乏。
研究人员还发现,精神分裂症患者的肠道菌群失调与重度抑郁症患者不同,并且存在明显的微生物标志物,与症状严重程度密切相关。他们甚至可以非常准确地确定生物群样本是否来自精神分裂症受试者或对照受试者。
为了进一步证明肠道生物群对受试者的精神状态的影响,研究人员随后将精神分裂症受试者的肠道微生物样本转移到一组健康对照小鼠的肠道生物群中。然后,他们将这些先前健康的对照小鼠的行为与一组一直健康的对照小鼠的行为进行了比较。
令人惊讶的是,研究人员发现,将精神分裂症患者的肠道细菌移植到健康对照小鼠中会诱发某些鼠类精神分裂症的症状。这意味着研究人员仅使用精神分裂症受试者的肠道细菌就能将精神分裂症的症状转移至小鼠。
正如论文作者所写,这项研究提供了开创性的证据,表明精神分裂症与肠道菌群组成的变化有关,这种变化既与精神分裂症有关,又与其症状严重程度相关。
该研究不仅在精神分裂症和肠道生物群之间建立了明确的联系,还进一步证明了许多精神障碍是系统性问题,如果没有更全面的视野,就无法正确治疗。
压 力
一些研究表明慢性和暂时性压力均可影响肠道菌群的分布。同样,啮齿动物的研究表明子宫内的压力和母体分离会破坏肠道菌群的平衡,对心理健康产生连锁反应。
自然分娩和母乳喂养
其他研究表明暴露于母体微生物对后代健康多样的肠道发育很重要。这可以通过阴道分娩,皮肤接触和母乳喂养来实现。
漏 肠
有人提出,不健康饮食,酒精和其他因素引起的微生物群变化可使肠道内膜或上皮更易渗透。肠道上皮是一种壁或屏障,旨在防止有害菌产生的有害物质循环进入体内。当肠道变得“渗漏”时,由于炎症细胞因子的产生增加,可能会发生慢性炎症。在许多抑郁症患者中也发现了慢性炎症,这可能部分解释了抑郁症和心脏病之间的联系。
纤 维
为了使食物的口感更符合现代人的需求,很多食物在加工过程中被剥去了纤维,然而对人体有用的许多微生物都依赖纤维。
所谓垃圾食品的真正问题在于,它会使你体内的有益菌饥饿。纤维存在于洋葱,朝鲜蓟,芦笋和许多绿叶蔬菜中。地中海饮食强调这些食物,是值得效仿的典范。

抗 生 素
避免使用抗生素。这些虽然是挽救生命的药物,但是它们带来的副作用是杀死肠道微生物。请谨慎使用。
关于肠脑轴的新兴研究已经产生了关于精神疾病可能病因的新见解。而且,这一研究领域在诊断和治疗方面都具有很大的希望和潜力。如果炎症是与压力相关的抑郁症的核心,那么疗法如何起作用?
神奇的是,诸如认知行为疗法之类的治疗实际上可以降低促炎化学物质的水平。
抗抑郁药的发展引发了精神病学的巨大革命,它们是如何发挥作用呢?
最早的理论之一是它们“填补”了大脑中的神经递质。据认为,5-羟色胺在患有抑郁症的人中很少,因此任何可以增加其在大脑中水平的药物都是有益的。
选择性5-羟色胺再摄取抑制剂(SSRIs)可以使5-羟色胺在大脑中循环,因此可以做到这一点。但是,鉴于SSRI还可以减轻大脑炎症,因此可能需要对该机制进行一些重新解释。
免疫系统异常复杂,我们对此了解甚少,但最近的研究表明,它与抑郁症紧密相关,减轻炎症可以减轻抑郁症。
例如,益生菌可以提供健康的肠道细菌,而抗生素可以破坏有害的肠道细菌。同样,粪便微生物移植可用于将健康和多样的肠道细菌转移给缺乏的细菌。人们开始推测这些可以用于治疗精神疾病。
01
益 生 菌
益生菌已在动物研究中显示出减少炎症细胞因子的作用。发表在《英国营养杂志》上的一项研究描述了一项针对30名患有抑郁症志愿者的试验。
每天给这些沮丧的志愿者补充益生菌补充剂,其中包括乳酸杆菌和双歧杆菌细菌。在试验结束时,他们“显着”减轻了心理困扰的症状,包括抑郁和焦虑。
除补充剂外,酸奶和咸菜等发酵食品还含有益生菌。
02
Omega-3 补充剂
一些研究表明,omega-3补充剂可以有益地影响肠道菌群的组成并增加抗炎化合物(包括脂肪酸)的产生。
肠道微生物,脂肪酸和免疫力可以共同发挥作用,以维持肠道壁的完整性并减少肠道渗漏。
omega-3补充剂可以帮助某些类型的抑郁症。
Omega-3的天然来源包括鱼类,尤其是高脂肪鱼类,例如鲑鱼,沙丁鱼和鲭鱼。
03
益 生 元
如果饮食中不能摄取足够的纤维,那么益生元也会起到类似的作用。这些是人体无法消化的复杂糖,但是肠道微生物可以消化。
04
运 动
要知道久坐对肠和肠道微生物并不友好。如果运动是一种药物,那将价值数百万。目前尚不清楚具体作用机制,但是运动可以提高微生物群的质量。

05
规 律 饮 食
古人云:日出而作,日落而息。
这句话同样适用于你的微生物群,它也具有昼夜节律。如果这两个节奏相互同步,那么生活就是美好的事物。跟着大自然的昼夜节律,对于微生物而言也是非常舒适的节奏。
总而言之,沮丧可能源于悲伤,失落或糟糕的生活。但是它也可能是您的微生物群引起或加剧的,这意味着我们每个人都有机会摆脱困境。善待肠道微生物,你会发现自己的心情可以有所改善。
应当指出,关于精神疾病和肠脑轴的大多数研究都是在啮齿动物上进行的,加利福尼亚大学专家克莱尔·马丁及其同事正确地指出“因果关系的证据仍然很少”。
的确,缺乏双盲随机纵向研究来研究益生菌,抗生素或粪便微生物移植对精神疾病康复的影响。同样,反向因果关系的问题也没有得到充分解决。
例如,与精神疾病相关的生活方式因素和药物的副作用可能导致肠道菌群紊乱和失衡。换句话说,微生物群分布的变化可能是精神疾病的结果,不一定是原因。
但是不管是结果还是原因,精准的了解特定的肠道微生物及其特征与特定的精神疾病之间的关系可能有助于精神病患者的诊断和靶向治疗和个性化治疗有帮助。
相关阅读:
肠道微生物组如何影响运动能力,所谓的“精英肠道微生物组”真的存在吗?
参考文献
Brachman, Rebecca A., Michael L. Lehmann, Dragan Maric, and Miles Herkenham. “Lymphocytes from Chronically Stressed Mice Confer Antidepressant-Like Effects to Naive Mice.” The Journal of Neuroscience 35, no. 4 (January 28, 2015): 1530–38.
Pereira, Joana da Cruz, Kieran Rea, Yvonne M. Nolan, Olivia F. O’Leary, Timothy G. Dinan, and John F. Cryan. “Depression’s Unholy Trinity: Dysregulated Stress, Immunity, and the Microbiome.” Annual Review of Psychology 71, no. 1 (2020)
The Gut-Brain Axis: The Missing Link in Depression. Evrensel A, Ceylan ME.Clin Psychopharmacol Neurosci. 2015 Dec 31;13(3):239-44. doi: 10.9758/cpn.2015.13.3.239.
Fulling, C., Dinan, T.G., & Cryan, J.F. (2019 Mar 20). Gut microbe to brain signaling: what happens in vagus… Neuron. 101(6): 998-1002.
Scott C. Anderson, Stress, Inflammation, and Microbes: A Moody Trinity,2019, Nov, 30
Del Grande da Silva, Giovanna, Carolina David Wiener, Luana Porto Barbosa, Jaciana Marlova Gonçalves Araujo, Mariane Lopez Molina, Pedro San Martin, Jean Pierre Oses, Karen Jansen, Luciano Dias de Mattos Souza, and Ricardo Azevedo da Silva. “Pro-Inflammatory Cytokines and Psychotherapy in Depression: Results from a Randomized Clinical Trial.” Journal of Psychiatric Research 75 (April 2016): 57–64.
Martina Sgritta, Sean W. Dooling, Shelly A. Buffington, Eric N. Momin, Michael B. Francis, Robert A. Britton, Mauro Costa-Mattioli. “Mechanisms Underlying Microbial-Mediated Changes in Social Behavior in Mouse Models of Autism Spectrum Disorder.” Neuron (First published online: December 3, 2018) DOI: 10.1016/j.neuron.2018.11.018
Pan, Ying, Xu-Yang Chen, Qing-Yu Zhang, and Ling-Dong Kong. “Microglial NLRP3 Inflammasome Activation Mediates IL-1β-Related Inflammation in Prefrontal Cortex of Depressive Rats.” Brain, Behavior, and Immunity 41 (October 2014): 90–100.
Jacka, Felice N., Adrienne O’Neil, Rachelle Opie, Catherine Itsiopoulos, Sue Cotton, Mohammedreza Mohebbi, David Castle, et al. “A Randomised Controlled Trial of Dietary Improvement for Adults with Major Depression (the ‘SMILES’ Trial).” BMC Medicine 15, no. 1 (January 30, 2017): 23.
Melanie Greenberg. Feeling Depressed? Gut-Brain Dysfunction May Be to Blame, 2018, Dec 30,
Karen-Anne McVey Neufeld, John Bienenstock, Aadil Bharwani, Kevin Champagne-Jorgensen, YuKang Mao, Christine West, Yunpeng Liu, Michael G. Surette, Wolfgang Kunze & Paul Forsythe. “Oral Selective Serotonin Reuptake Inhibitors Activate Vagus Nerve Dependent Gut-Brain Signalling.” Scientific Reports (First published: October 3, 2019) DOI: 10.1038/s41598-019-50807-8
Christopher Bergland. The Vagus Nerve May Carry Serotonin Along the Gut-Brain Axis, 2019, Oct 06
Christopher Bergland. Vagus Nerve Stimulation Dramatically Reduces Inflammation. 2016, Jul 06,
Sudo, Nobuyuki, Yoichi Chida, Yuji Aiba, Junko Sonoda, Naomi Oyama, Xiao-Nian Yu, Chiharu Kubo, and Yasuhiro Koga. “Postnatal Microbial Colonization Programs the Hypothalamic-Pituitary-Adrenal System for Stress Response in Mice.” The Journal of Physiology 558, no. Pt 1 (July 1, 2004): 263–75.
Dinan, Timothy G., Catherine Stanton, and John F. Cryan. “Psychobiotics: A Novel Class of Psychotropic.” Biological Psychiatry 74, no. 10 (November 15, 2013): 720–26.
Bravo, Javier A., Paul Forsythe, Marianne V. Chew, Emily Escaravage, Hélène M. Savignac, Timothy G. Dinan, John Bienenstock, and John F. Cryan. “Ingestion of Lactobacillus Strain Regulates Emotional Behavior and Central GABA Receptor Expression in a Mouse via the Vagus Nerve.” Proceedings of the National Academy of Sciences 108, no. 38 (September 20, 2011): 16050–55.
Chu, Coco, Mitchell H. Murdock, Deqiang Jing, Tae Hyung Won, Hattie Chung, Adam M. Kressel, Tea Tsaava, et al. “The Microbiota Regulate Neuronal Function and Fear Extinction Learning.” Nature 574, no. 7779 (October 2019): 543–48.
Samoon Ahmad. Schizophrenia and the Gut. 2019, Mar 05,
Xu C, Lee SK, Zhang D, Frenette PS. The Gut Microbiome Regulates Psychological-Stress-Induced Inflammation [published online ahead of print, 2020 Jul 24]. Immunity. 2020;S1074-7613(20)30280-6. doi:10.1016/j.immuni.2020.06.025
VanElzakker, Michael B., M. Kathryn Dahlgren, F. Caroline Davis, Stacey Dubois, and Lisa M. Shin. “From Pavlov to PTSD: The Extinction of Conditioned Fear in Rodents, Humans, and Anxiety Disorders.” Neurobiology of Learning and Memory 113 (September 2014): 3–18.
Scott C. Anderson,Unending Fear and the Gut-Brain Axis,2019,Oct 24
欢迎关注: 谷禾健康
——让你和你的家人更健康


本文由谷禾健康整理编译自:VIOME TEAM, 2020 Gut Microbiome Health and Cellular Health are the Keys to Optimizing Physical Performance AND How Athletic Performance is affected by your Gut Microbiome, Cellular, and Mitochondrial Health.
想象一下如果有一天,您可以模仿最优秀的运动员的微生物组和细胞健康状况,从而可以跑得更快,跳得更高,也就是说可以提高自己的运动能力。是不是感觉很神奇?

每个人身上都充满了微生物,而运动员身上的微生物可以为他们的比赛提供优势。
这要从肠道微生物组开始说起。我们都知道,每个人都有数万亿种细菌,病毒和真菌,它们生活在人体的多个微生物生态系统中,称为微生物组。
对于运动员来说,肠道微生物在他们的表现和恢复速度方面起着更加重要的作用。

肠道微生物负责帮助我们的身体分解碳水化合物,纤维,蛋白质,调节能量[1]。这些微生物影响身体的炎症反应,压力适应力,神经功能,甚至影响精神力量,所有这些对于运动都很重要[2]。
设想如果你的细胞健康状况不佳或不佳,这可能意味着您的细胞功能无法有效发挥,能量产生低下或细胞由于氧化应激,炎症或环境毒素而处于应激状态。

基因组和转录组测序技术的发展以及人工智能处理围绕能量调节和运动恢复的大量数据的能力使研究人员探索了以下问题:
“微生物组分析可以帮助我们预测下一位伟大的运动员吗?”
“将来,我们能否从精英运动员身上收获微生物,并将高性能的微生物能力传递给其他人?”
“食物和营养会通过在细胞乃至线粒体水平上积极影响他们的身体,从而影响运动员的恢复能力,增强免疫力以帮助他们训练和比赛吗?”
“基于性能的益生菌和益生菌会被广泛使用吗?”
尽管没有人能预测未来,但探索这些问题的过程会带来许多可能性。
哈佛大学的研究人员发现“精英肠道菌群”
运动表现,恢复能力,甚至运动员从事的运动类型都与特定的微生物有关。现在,这些发现促使研究人员寻找增加肠道中有益细菌的多样性和丰富度的方法,以获得更好的运动能力和更快的恢复能力。
在一项研究中,哈佛大学的研究人员从波士顿马拉松比赛训练的运动员的肠道微生物样本中取样。研究人员在马拉松比赛后再次对参与者进行了测试,发现人体分解乳酸所需的一种细菌数量激增。这些科学家认为,这种特定细菌的增加是对体内乳酸水平升高的一种反应,因为它是它们的主要食物来源[3]。
他们的发现引出了一个问题:这种细菌将来是否可以用于降低体内乳酸水平并可能加快恢复时间?

在另一项研究中,哈佛大学的科学家将赛艇运动员的肠道微生物组与超级马拉松运动员进行了比较,发现其组成存在明显差异,这表明特定的运动可能会促进特定的微生物生态系统[2]。
这些研究的发现,不仅使公司寻求创造基于性能的益生菌和益生菌,更是促使一些科学家们相信,将来他们将能够挖掘精英运动员的微生物组来帮助他人。
看到这里,或许你会有这样的疑问,微生物组是如何准确地影响运动能力?以下是微生物组影响运动表现的九种方式:
01 减 少 炎 症
肠道微生物组在炎症中起着重要作用:升高或降低炎症水平。炎症会干扰运动表现,减慢恢复速度,并且是许多慢性疾病的根本原因。
肠道微生物组失衡或营养不良与炎性疾病相关(我们前面的文章有阐述过:炎症性肠病中宿主与微生物群的相互作用),因此保持健康的微生物组有助于减少全身炎症至关重要[6]。
此外,现在可以更容易地看到人体细胞的炎症水平,这可能是由肠道微生物群和环境因素(病原体、压力、运动等)共同触发的。
02 提 高 能 量 水 平
当肠道微生物组保持平衡和健康时,它可以帮助提高能量水平,并且线粒体可以调节和产生细胞能量,因此,对人体和微生物健康都至关重要。通过以下方式,可以转化为更好的性能:
通过更好的乳酸分解来减轻疲劳[7]
控制氧化还原功能,可以延迟疲劳症状[8]
增加ATP水平,你的分子能量[9]
调节新陈代谢[4]
向线粒体提供必需的代谢产物–细胞的动力源[9]
调节能量的收集,存储和消耗[4]
03 增 强 精 神 力 量
这个听起来有点不可思议,其实我们的肠道微生物会沿着迷走神经与大脑对话。肠道微生物在心理健康状况中起着重要作用。
肠道微生物失衡,则可能导致精神疾病。肠道失衡或营养不良甚至与焦虑和抑郁有关(我们前面的文章有阐述过:深度解读 | 肠道菌群和中枢神经系统的关系)。肠脑轴是塑造精神力量中看不见的手,对于那些承受不起压力的职业运动员来说,这是必不可少的。
04 塑 造 理 想 的 身 体 成 分
肠道微生物组有助于身体更有效地运转。平衡的肠道会影响以下方面,因此通常更容易健康:
身体成分
白色与棕色脂肪
膳食对血糖的反应
05 强 健 骨 骼
微生物组通过激素和免疫系统调节帮助建立骨骼质量和强度。均衡的肠道菌群也可以增加钙和镁的矿物质吸收。在运动相关的创伤中,功能正常的微生物组可以加速骨骼愈合[11]。

06 帮 助 营 养 吸 收 和 使 用
平衡的微生物组对于更好地吸收和使用营养至关重要。如果您的肠道微生物组有炎症且不平衡,则微生物是为了努力存活下来,而不是提取必需的维生素,蛋白质和酶。
此外,肠道菌群还通过将消化道无法加工的食物转化为生存所需的营养来提供营养[12]。为了使运动员在巅峰时期表现出色,他们需要拥有蓬勃发展的肠道微生物群。
07 提 升 水 合 状 态
肠道微生物组与运动过程中适当的水分调节有关,这意味着人体可以更有效地利用水。另外,肠内膜的完整性是适当水合作用的关键因素,健康的肠道微生物组也有助于维持水分[13]。
08 改 善 睡 眠
肠道微生物组失衡(功能失调)与睡眠质量差和认知灵活性降低有关,因为肠道微生物组控制着各种激素(例如皮质醇,5-羟色胺和GABA)的水平,所有这些激素都会影响睡眠质量[14]。微生物群还影响褪黑素的产生,这对于适当的睡眠-觉醒周期是必不可少的[15]。

高质量的睡眠、良好的肠道健康、能量水平和表现都存在于一个强化循环中,这些循环既可以相互促进,也可以拖累你。运动员知道他们需要适当的睡眠才能表现良好。但是,许多人可能没有意识到,有一种促进睡眠神经递质的药是在自己的肠道内产生的。
09 抗 氧 化 防 御 系 统
人体内有一个强大的系统,称为抗氧化防御系统,即氧化还原信号,它使用抗氧化酶使您保持健康。运动员需要这套系统处于良好的工作状态,才能始终保持出色的表现并保持比赛的最高水准。

健康的氧化还原状态与均衡的肠道微生物组有关。这个肠道微生物组调节的抗氧化酶系统[8,16]:
防止运动引起的组织损伤
防止剧烈运动引起的氧化损伤
与运动员的身体状况有关
减轻身体疲劳
改善运动表现
通常,密集和持续的运动训练以及高水平的竞争会产生大量的自由基,这些自由基可能超过典型人体的能力。这使运动员容易受到氧化应激的影响,并更有可能累积有害的炎症。
肠道微生物组学和人类细胞科学的未来是性能科学的未来
健康饮食(多样性饮食)是一个很好的开始。你可以通过饮食把你的运动表现提升到一个新的水平,特别是支持属于你独特的微生物群和身体的饮食。
谷禾健康肠道菌群健康检测服务结合了高通量测序技术,大数据和人工智能,可以帮助您了解健康的独特需求以及独特的食物并补充建议。帮助您微调肠道微生物组的功能,以最大程度地减少有害代谢物的产生并最大化有益代谢物的产生。
参考文献
1. Blaser MJ. The microbiome revolution. J Clin Invest. 2014;124(10):4162-4165. doi:10.1172/JCI78366
2. Christopher Bergland Does Gut Microbiome Influence Mindset and Mental Toughness?Harvard researchers link specific gut microbiota with peak athletic performance. 2017
3. Torrice M. A Conversation with Jonathan Scheiman. ACS Cent Sci. 2017;3(10):1057-1058. doi:10.1021/acscentsci.7b00470
4. Monda V, Villano I, Messina A, et al. Exercise Modifies the Gut Microbiota with Positive Health Effects. Oxid Med Cell Longev. 2017;2017:3831972. doi:10.1155/2017/3831972
5. Endurance exercise and gut microbiota: A review.Author links open overlay panelNúriaMachabDolorsFuster-Botellaa June 2017, Pages 179-197
6. Clemente JC, Manasson J, Scher JU. The role of the gut microbiome in systemic inflammatory disease. 2018 BMJ ;360:j5145
7. Pessione E. Lactic acid bacteria contribution to gut microbiota complexity: lights and shadows. Front Cell Infect Microbiol. 2012;2:86. Published 2012 Jun 22. doi:10.3389/fcimb.2012.00086
8. Neish AS. Redox signaling mediated by the gut microbiota. Free Radic Res. 2013;47(11):950-957. doi:10.3109/10715762.2013.833331
9. Clark A, Mach N. The Crosstalk between the Gut Microbiota and Mitochondria during Exercise. Front Physiol. 2017;8:319. Published 2017 May 19. doi:10.3389/fphys.2017.00319
10. Davis CD. The Gut Microbiome and Its Role in Obesity. Nutr Today. 2016;51(4):167-174. doi:10.1097/NT.0000000000000167
11. Xu X, Jia X, Mo L, et al. Intestinal microbiota: a potential target for the treatment of postmenopausal osteoporosis. Bone Res. 2017;5:17046. Published 2017 Oct 4. doi:10.1038/boneres.2017.46
12. Krajmalnik-Brown R, Ilhan ZE, Kang DW, DiBaise JK. Effects of gut microbes on nutrient absorption and energy regulation. Nutr Clin Pract. 2012;27(2):201-214. doi:10.1177/0884533611436116
13. Colgan SP. Swimming through the gut: implications of fluid transport on the microbiome. Dig Dis Sci. 2013;58(3):602-603. doi:10.1007/s10620-013-2575-3
14. Galland L. The gut microbiome and the brain. J Med Food. 2014;17(12):1261-1272. doi:10.1089/jmf.2014.7000
15. Anderson JR, Carroll I, Azcarate-Peril MA, et al. A preliminary examination of gut microbiota, sleep, and cognitive flexibility in healthy older adults. Sleep Med. 2017;38:104-107. doi:10.1016/j.sleep.2017.07.018
16. Neish AS. Redox signaling mediated by the gut microbiota. Free Radic Res. 2013;47(11):950-957. doi:10.3109/10715762.2013.833331

谷禾健康
高脂饮食会诱发肥胖、胰岛素抵抗、葡萄糖耐受不良和脂肪变性,而脂肪性肝炎向纤维化和肝细胞癌(HCC)的进展主要与高膳食胆固醇有关。非酒精性脂肪性肝病(NAFLD)是一类代谢疾病,可从脂肪肝进展至脂肪性肝炎,进一步导致肝硬化甚至肝细胞癌(HCC),胆固醇被认为是疾病发展过程中主要的脂毒性分子。
近日,香港中文大学于君团队及其团队研究人员在《Gut》上发表了题为“Dietary cholesterol drives fatty liver- associated liver cancer by modulating gut microbiota and metabolites(膳食胆固醇通过调节肠道微生物群和代谢产物驱动脂肪肝相关的肝癌,DOI:10.1136/gutjnl-2019-319664)”的研究论文,阐述了膳食胆固醇在NAFLD–HCC发生中的作用及其相关的分子机制。这项研究证明了长时间的高胆固醇饮食可以引起小鼠自发性NAFLD-HCC(非酒精性脂肪肝肝细胞癌)。抑制胆固醇可以恢复肠道微生物群,完全阻止NAFLD-HCC的发展。

背景
非酒精性脂肪性肝病(NAFLD)是代谢综合征的肝脏表现,包括从单纯性脂肪变性到非酒精性脂肪性肝炎(NASH)的一系列肝脏病理。NASH可进展为肝硬化、终末期肝功能衰竭和肝细胞癌(HCC)。目前,非酒精性脂肪肝是世界范围内发病率和医疗负担的主要原因。脂肪毒性会导致NASH、纤维化/肝硬化,甚至HCC。在肝脏脂质种类中,胆固醇被认为是NASH发生过程中的主要脂毒分子。肝脏是调节全身胆固醇稳态的中心。膳食胆固醇对血浆和肝脏胆固醇稳态有重要影响。虽然膳食胆固醇在NASH进展中的作用已被论述,但长期胆固醇治疗在自发性和进行性NAFLD-HCC发展的作用和致病基础尚不清楚。一些研究表明,肠道菌群是一个有助于NAFLD发生和其发展为NAFLD-HCC的环境因素。微生物衍生的代谢物,如胆汁酸、短链脂肪酸和三甲胺和它们影响的信号通路可能有助于NAFLD的发展。特别是肠道菌群代谢产物3 -(4 -羟苯基)乳酸,对肝脏脂肪变性和肝纤维化具有共同的基因效应。
实验设计
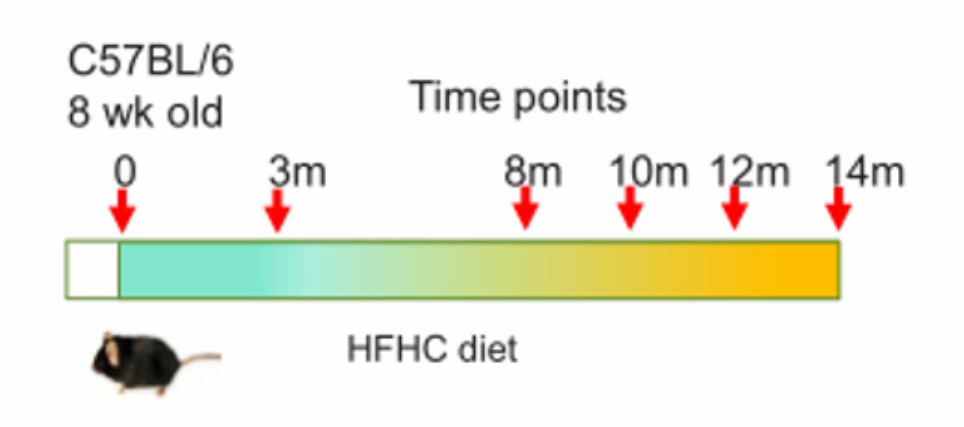
普通饲料(NC,18%脂肪,58%碳水化合物,24%蛋白质,0%胆固醇);高脂/低胆固醇饲料(HFLC,43.7%脂肪,36.6%碳水化合物,19.7%蛋白质,0.013%胆固醇);高脂/高胆固醇饲料(HFHC,43.7%脂肪,36.6%碳水化合物,19.7%蛋白质,0.203%胆固醇)
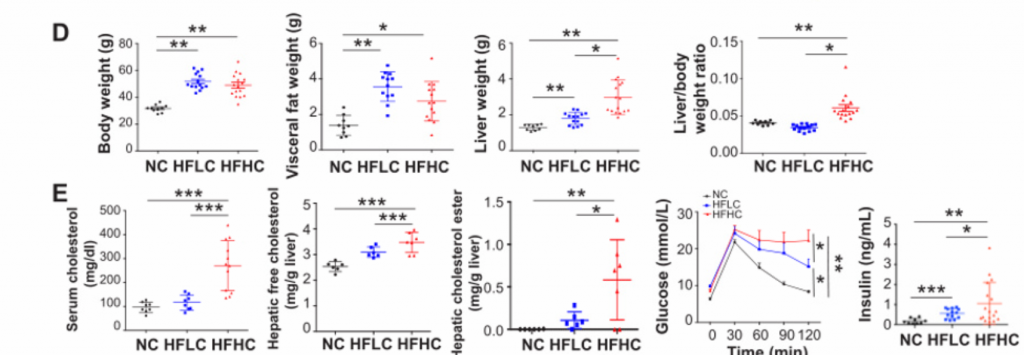

如图所示,以上饲料用于随机喂养8周龄雄性C57BL/6野生型同窝出生幼鼠14个月。阿托伐他汀(降胆固醇药)药物被用于喂食HFHC膳食7个月后的小鼠,持续7个月。然后分别在3、8、10和12个月龄处死。
粪移植实验:
取被喂养不同饲料的小鼠的1 g粪便样品用5ml磷酸盐缓冲生理盐水(PBS)混匀,然后将该混悬液2 0 0μL灌胃移植给无菌小鼠。于移植后8、10、14个月各组中随机抽取小鼠处死。
实验结束时,小鼠被禁食,取血清/组织。记录体重和内脏脂肪重量。肝脏被迅速切除并称重,评估表面结节的存在和大小。分离肝肿瘤,在液氮中快速冷冻,并将其保存在−80°C进行进一步实验。肠道菌群分析采用16SrRNA测序,血清代谢物分析采用液相色谱-质谱(LC-MS)代谢组学分析。
主要结果
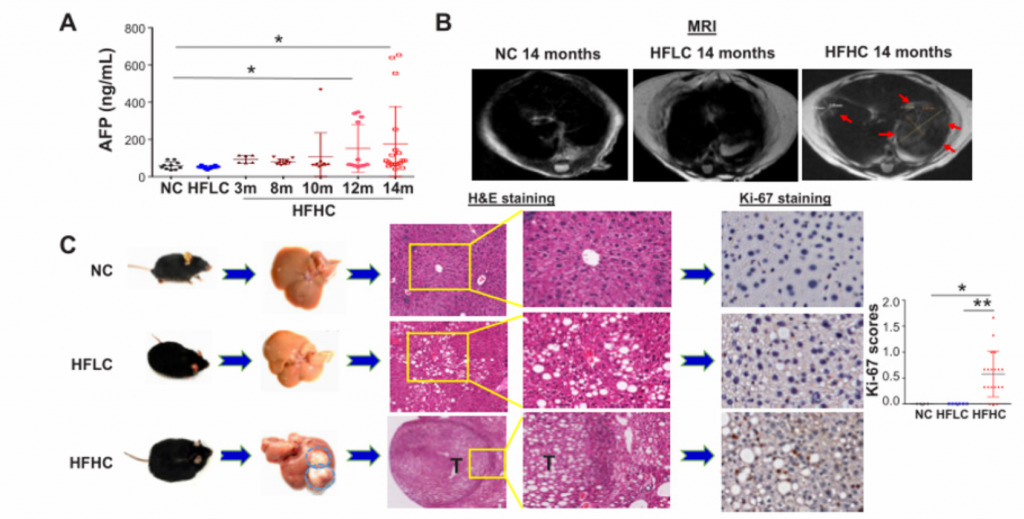
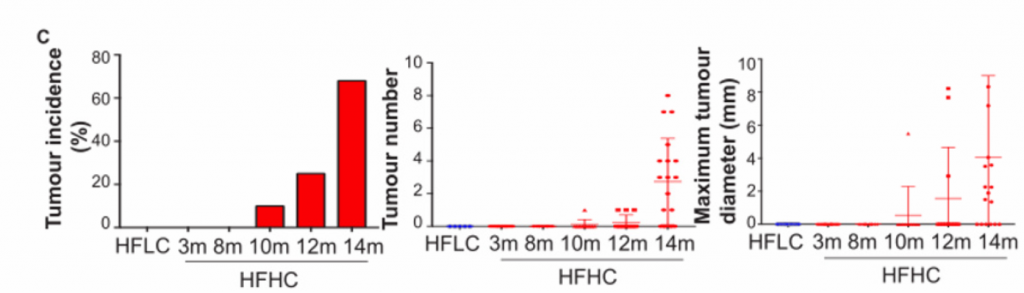
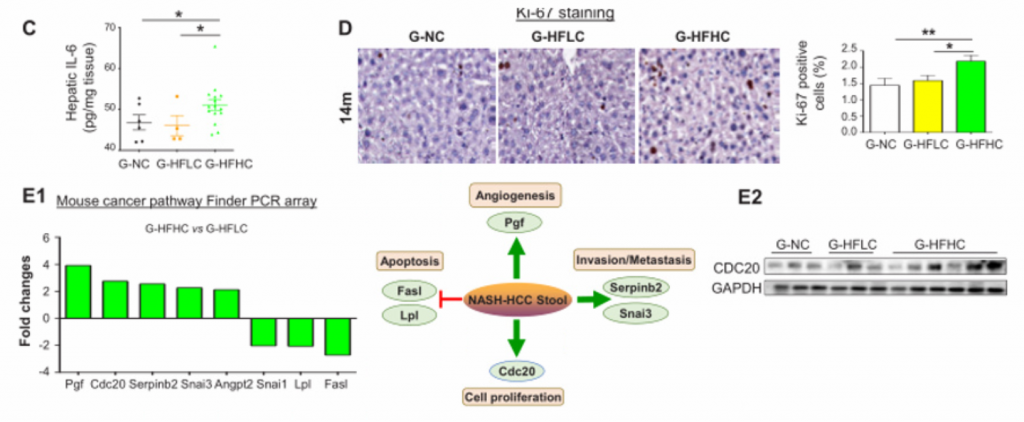
1. 膳食胆固醇可自发驱动NAFLD-HCC的发生。


A)[endif]喂养HFHC饲料的小鼠在第3、8、10、12、14个月检测的血清AFP水平以及喂养NC和HFLC饲料的小鼠在第14个月检测的血清AFP水平。在第10个月观察到AFP水平升高,在第12个月和14个月,与HFLC或NC饲料喂养的小鼠相比,HFHC饲料喂养的小鼠的AFP水平逐渐升高。
B) [endif]肝脏MRI影像。在第14个月时,喂养HFHC饲料的小鼠出现肝肿瘤,而其余两个没有
C) [endif]依次为小鼠肝脏的大体形态、镜下结构以及Ki67染色的免疫组化图像。HFHC小鼠肝脏切片组织学检查均为肝细胞癌,平均每只小鼠肝细胞癌细胞数为2.7±2.6,最大直径为4.1±5.0 mm。与HFLC喂养的小鼠相比,HFHC喂养的小鼠肝脏切片中Ki-67阳性细胞明显增多,表明HFHC喂养的小鼠细胞增殖增加。
D) [endif]依次为体重,内脏脂肪,肝脏重量,肝脏与体重的比率。随着肝癌的形成,喂养HFHC饲料的小鼠的这些指标的水平显著高于喂养NC饲料的小鼠。同时,喂养HFLC饲料的小鼠也表现出这些指标水平的上升。
E) [endif]依次为血清胆固醇、肝游离胆固醇、肝胆固醇酯含量、糖耐量试验及空腹胰岛素水平。与HFLC和NC相比,HFHC小鼠的这些指标的水平都有所上升。与NC相比,HFLC小鼠的葡萄糖耐量和空腹胰岛素也有所增加。
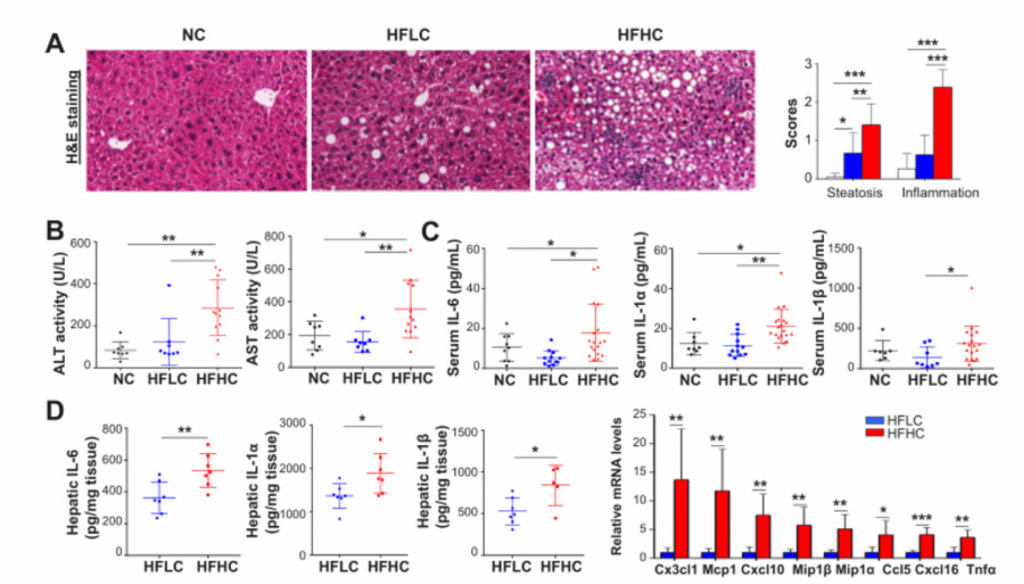
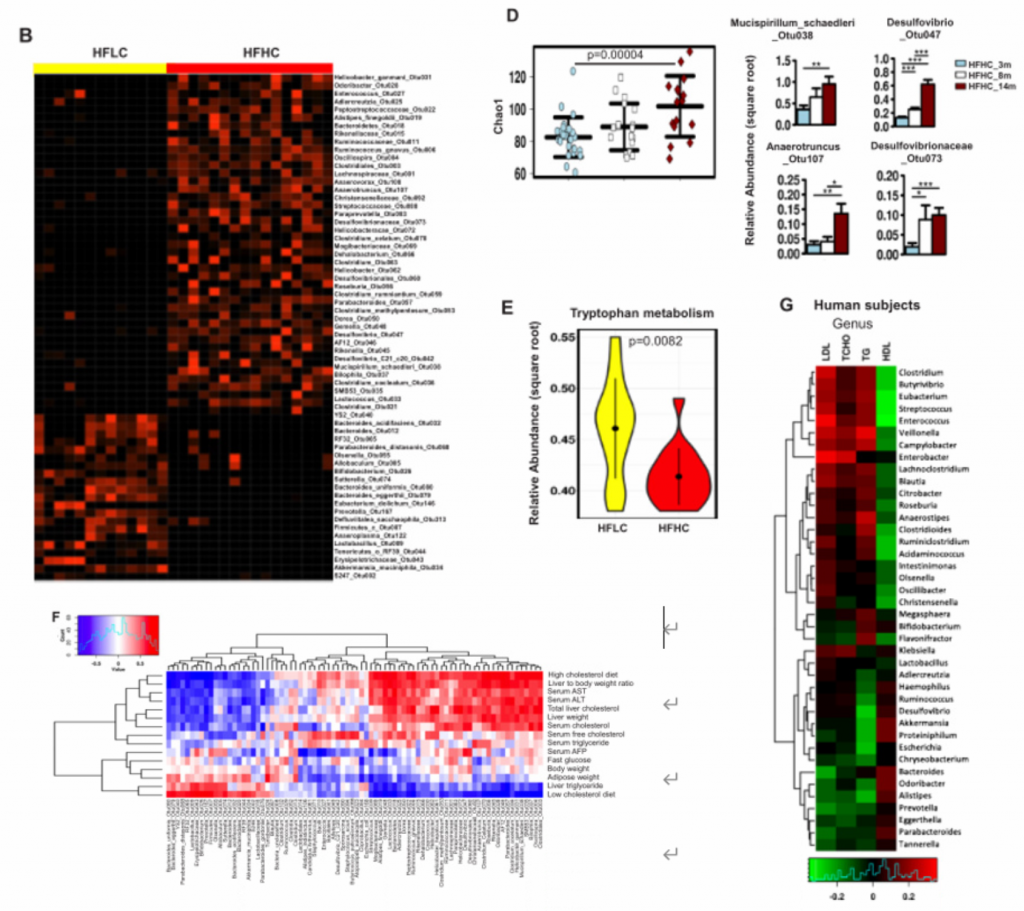
2. 高胆固醇膳食喂养的小鼠的非肝癌肝组织形成了NASH和纤维化。


A)HE染色后的肝脏切片组织检查。HFHC小鼠出现以脂肪变性和小叶炎症为特征的脂肪性肝炎,而在HFLC小鼠中只观察到脂肪变性。
B)血清ALT和AST水平。HFHC小鼠ALT水平和AST水平显著高于HFLC和NC小鼠。
C) 细胞因子谱法测定喂养NC、HFLC和HFHC饲料的小鼠第14个月的血清IL-6、IL-1抑制因子和IL-1蛋白水平。这些指标的水平均有所上升。
D)在喂养NC、HFLC和HFHC饲料的小鼠第14个月时用ELISA法测定小鼠肝脏促炎细胞因子IL-6、IL-1α和IL-1β蛋白水平,RNA测序测定Cx3cl1、Mcp1、Cxcl10、Mip1β、Mip1α、CCL5、Cxcl16、TNFαmRNA水平。与NASH相关的促炎细胞因子Cx3cl1等在HFLC小鼠肝组织中显著上调。
E) 从上至下依次为天狼星红染色和免疫组化染色。分别观察胶原沉积和α-SmA蛋白。RT-PCR法测定mRNA表达水平。HFHC小鼠的肝组织表现出严重的纤维化损伤,胶原分布面积明显增加。肝星状细胞活化表现为α-平滑肌肌动蛋白(α-SMA)的基因和蛋白水平升高。
F) 肝羟脯氨酸含量。其在HFHC小鼠中含量明显升高。
G) 肝脏的氧化应激检测。喂养HFHC饲料的小鼠的肝脏NAD+/NADH比值和SOD活性显著降低,提示膳食胆固醇可诱导肝脏的氧化应激。
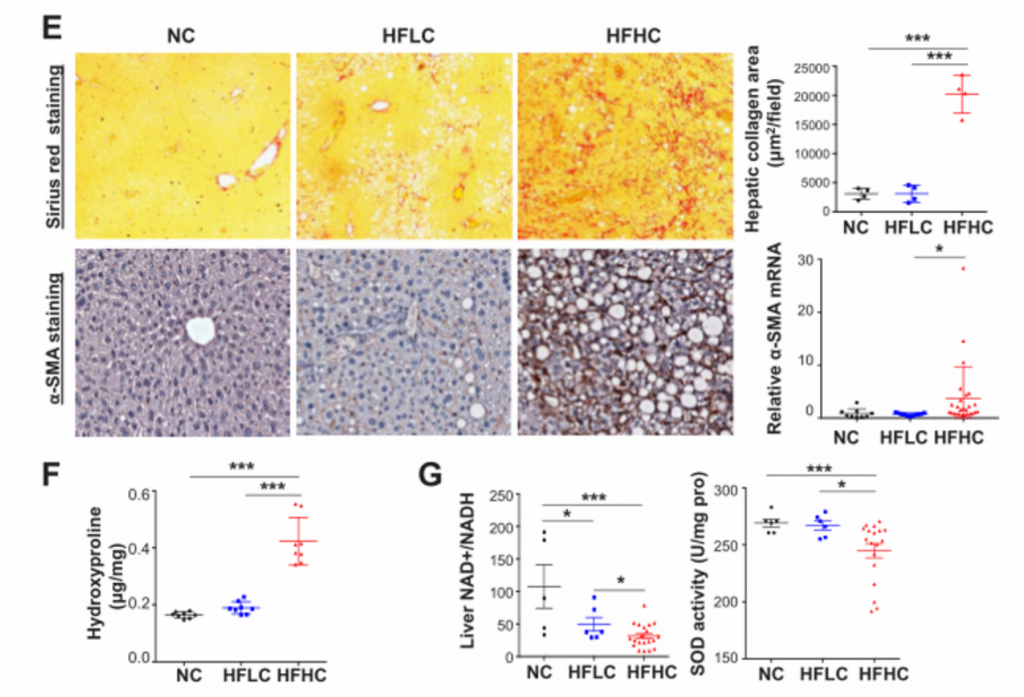
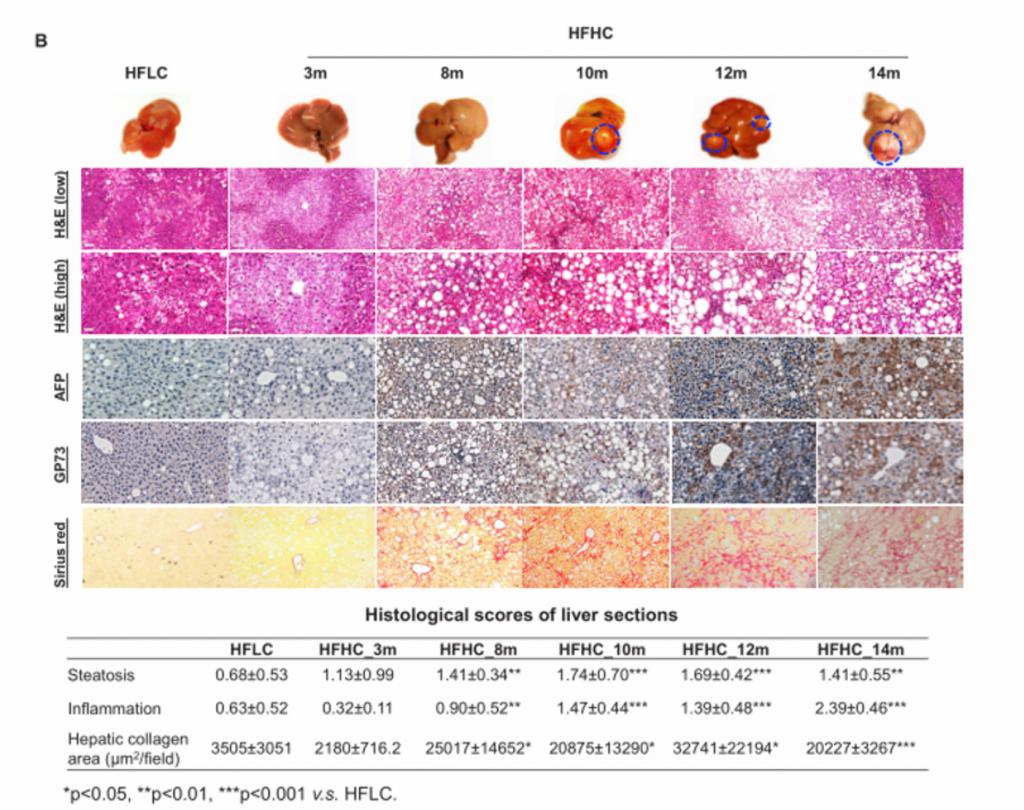
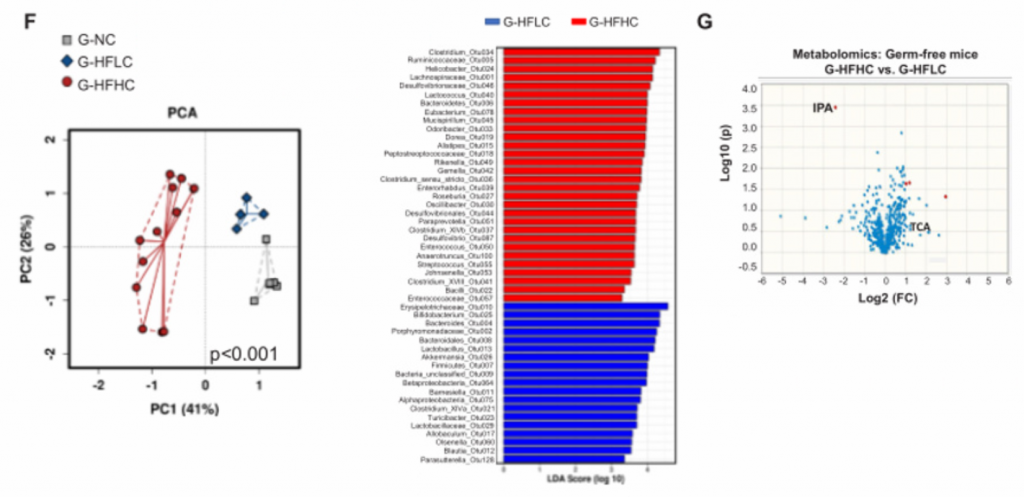
3. 高胆固醇膳食引起小鼠肝脏依次发展为脂肪肝、脂肪性肝炎、纤维化和NAFLD-HCC。


B)从上至下依次为HFLC喂养14个月和HFHC喂养3、8、10、12和14个月小鼠肝脏大体、肝脏肿瘤标志物AFP和GP73的H&E染色和免疫组化染色、天狼星红染色,脂肪变性和炎症的组织学评分以及天狼星红染色定量的肝胶原面积。HFHC小鼠肝脏组织学在3个月时表现为轻度炎症性脂肪变性,8个月时脂肪性肝炎伴纤维化,10、12、14个月时肝癌形成,而HFLC小鼠在3、8、10、14个月时仅表现为脂肪变性而没有进一步发展成肝癌。HFHC小鼠的肿瘤标志物免疫组化染色观察到阳性染色,HFLC小鼠没有。
C)在HFHC膳食控制下的不同时间点的小鼠的最大肝肿瘤的肿瘤发病率、肿瘤数量和最大肿瘤直径。
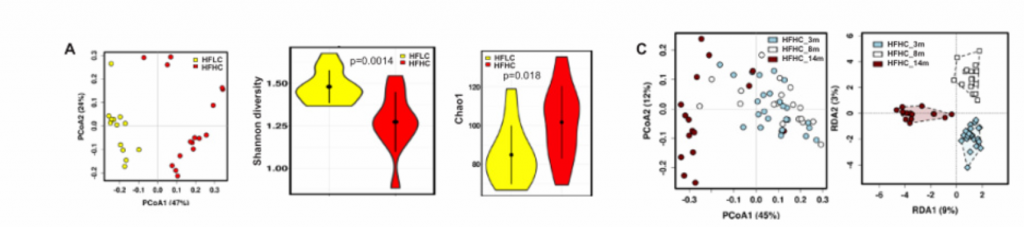
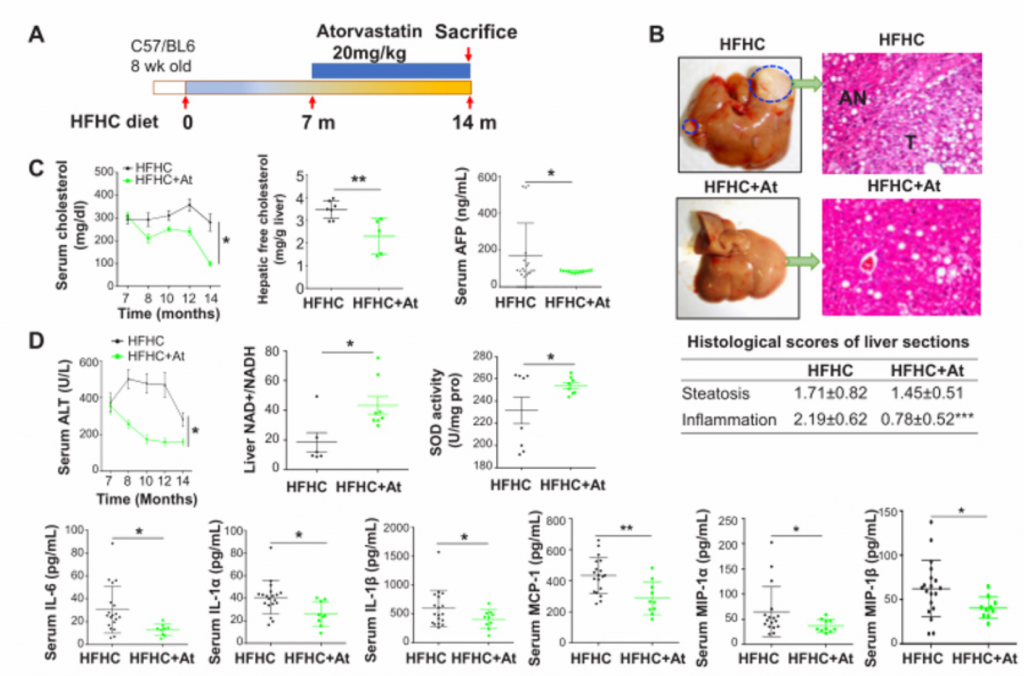
4. NAFLD-HCC的肠道菌群失调与胆固醇水平相关。






A)14月龄的HFLC和HFHC小鼠之间的肠道微生物的PCoA分析、香浓多样性和丰富度分析,PCoA分析中P<0.001. HFHC小鼠的微生物多样性更低,丰富度更高。
B)14月龄的HFLC和HFHC小鼠的粪便细菌热图。
C)HFHC膳食控制3、8和14个月后的肠道微生物的PCoA和冗余分析。发现HFHC_3m、HFHC_8m和HFHC_14m的小鼠的肠道微生物群落明显聚类,这表明肠道微生物组成随NAFLD-HCC进展阶段的变化而变化。
D)在HFHC饲养3、8 ~ 14个月,用Chao1指数测定微生物群落丰富度,发现其逐渐升高。随着NAFLD-HCC进展,细菌丰富度依次增加。
E)喂养HFLC和HFHC的小鼠肠道菌群的色氨酸代谢能力。HFHC小鼠肠道菌群的色氨酸代谢能力降低。综上,高胆固醇膳食导致肠道菌群失调和微生物色氨酸代谢受损。
F)细菌丰度与小鼠表型的相关性。
G)在59例高胆固醇血症患者和39例健康人的研究中,人血清总胆固醇、甘油三酯、低密度脂蛋白胆固醇和高密度脂蛋白胆固醇(横轴)与细菌(纵轴)基因组测序的相关性。双歧杆菌和类杆菌与血清总胆固醇、低密度脂蛋白胆固醇呈负相关,与高密度脂蛋白胆固醇呈正相关。
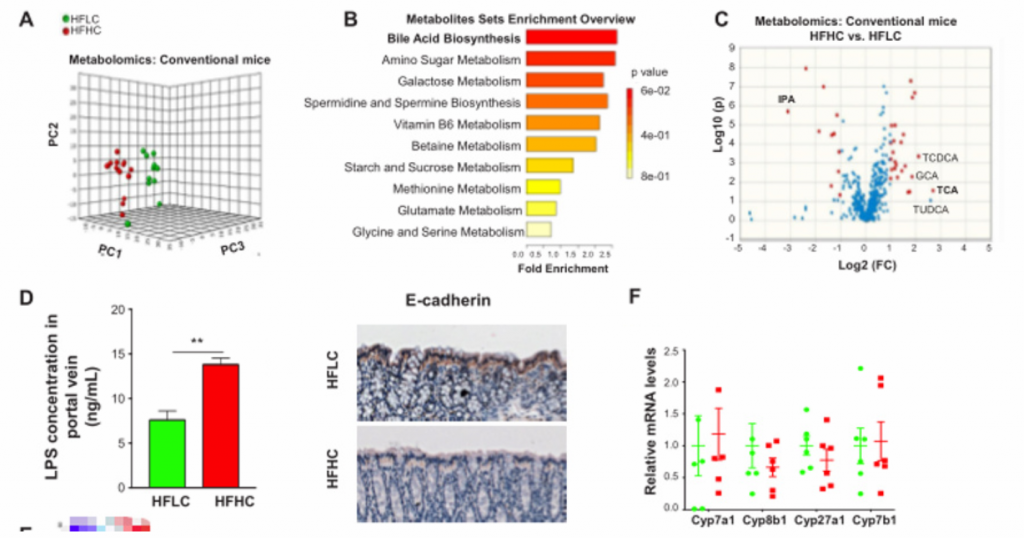
5. 膳食中的胆固醇通过诱导代谢产物的改变而促进NASH-HCC的进展。胆固醇通过调节宿主血清代谢物,以及至少部分通过增加TCA和降低IPA,促进了NASH-HCC的进展。


A) 主成分排序分析表明,膳食胆固醇含量对血清代谢产物有显著影响
B)HFHC小鼠的具有差异的富集代谢物的通路分析。胆汁酸合成是关键。
C) [endif]血清代谢组学火山图,指示了HFLC和HFHC小鼠的代谢产物的异常值。初级胆汁酸包括牛磺胆酸(TCA)、TUDCA、甘胆酸(GCA)和TCDCA,这些也是HFHC小鼠中异常上调的代谢物。IPA是微生物色氨酸代谢的产物,也是HFHC小鼠中异常下调的代谢物。
D) [endif]HFLC和HFHC饲料喂养3个月的小鼠门静脉LPS浓度和14个月的小鼠结肠组织E- cadherin表达。两者相比,HFHC小鼠门静脉血清脂多糖(LPS)浓度升高,结肠E-钙粘蛋白丢失。提示膳食胆固醇可损害肠道屏障功能
E) [endif]微生物与代谢物的相关性分析。红色呈正相关,蓝色呈负相关。
F) [endif]HFLC和HFHC饲料喂养小鼠肝脏组织中Cyp7a1、Cyp8b1、Cyp27a1和Cyp7b1 mRNA水平。高胆固醇膳食不能改变肝脏中细胞色素P450 (Cyp)7a1、Cyp8b1、Cyp27a1、Cyp7b1等胆汁酸合成酶的mRNA表达。
G) [endif]油红O染色显示,TCA加重胆固醇诱导的人LO2细胞甘油三酯的积累,IPA抑制胆固醇诱导的肝癌细胞、HKCI-2和HKCI-10细胞甘油三酯的积累。
H) [endif]IPA抑制NASH-HCC细胞系的细胞增殖。
6. 膳食胆固醇调节的肠道菌群通过诱导代谢产物的改变,促进NAFLD和肝细胞的增殖,从而促进胆固醇诱导的NAFLD-HCC的形成。
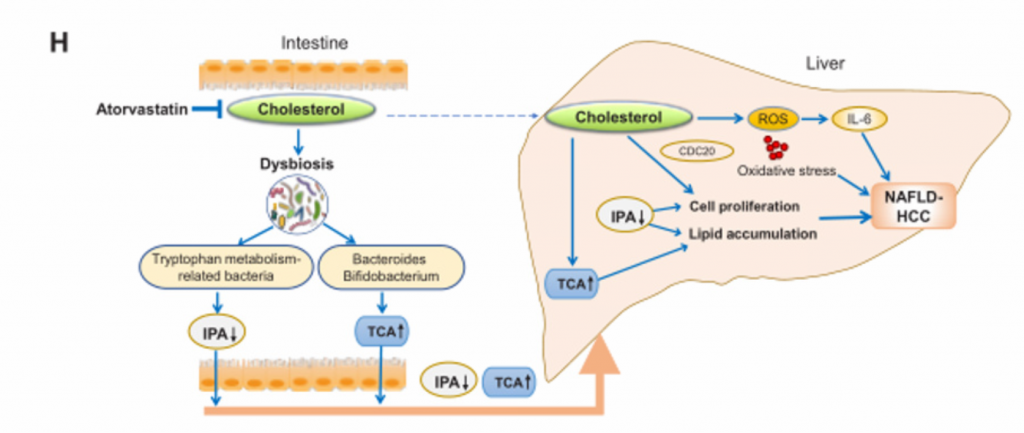



A)将喂养NC、HFLC和HFHC饲料的小鼠(14个月)的粪便移植到NC条件下的无菌小鼠(G- NC、G-
HFLC和G- HFHC)中。图中为G- NC组、G- HFLC组和GHFHC组灌胃无菌小鼠肝脏的大体形态和组织学检查。G- HFHC小鼠在14月龄时出现一个肝结节,而在8月龄和10月龄时未出现肝结节。组织学检查证实该结节发育不全,有小球状增生,细胞增殖增强。
B) [endif]8、10、14月龄时,G-NC、G-
HFLC和G-HFHC组无菌受体小鼠的肝脏甘油三酯含量、脂质过氧化反应和肝组织学改变。FMT后8、10、14个月,G-HFHC小鼠肝脏脂质积累明显增加,并伴有肝组织损害
C) [endif]G-NC、G- HFLC、G-HFHC组小鼠14月龄时的肝脏IL-6蛋白水平。
D) [endif]肝切片Ki-67染色。可见在14月龄时,G-HFHC小鼠中肝细胞增殖增加。但在FMT后8个月和10个月时肝细胞增殖并未见增加。
E) [endif]肿瘤通路检测。图中直方图向上表示基因表达上调,向下为下调。免疫印迹法验证CDC20(细胞增殖)表达上调。
F) [endif]G-NC、G- HFLC、G-HFHC小鼠肠道微生物群的PCA分析和LDA直方图。在不同的膳食控制下,肠道菌群的差异很大。
G) [endif]G- HFLC和G- HFHC小鼠血清代谢组学分析。G- HFHC小鼠的IPA较G-HFLC小鼠降低,与喂养HFHC饲料的常规小鼠一致。
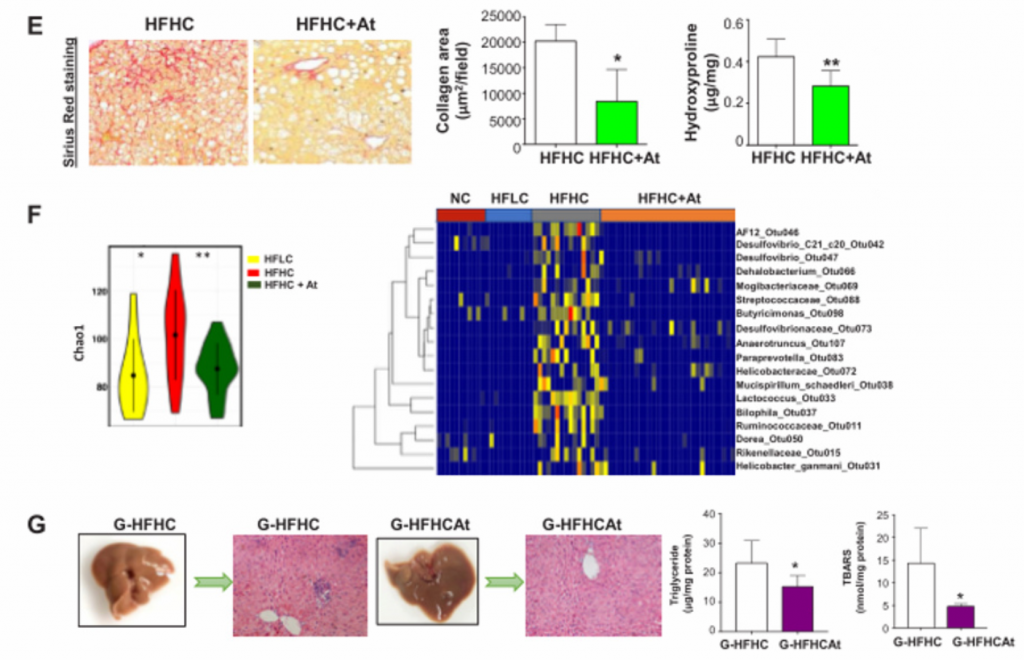
7. 降胆固醇治疗完全抑制了高脂血症小鼠NAFLD-HCC的形成。


A)阿托伐他汀治疗喂养HFHC饲料的C57BL/6小鼠的示意图。给小鼠喂食HFHC 饲料7个月,并继续喂食HFHC饲料和阿托伐他汀7个月。(HFHCAt)
B)HFHC小鼠的肝组织HE染色及肝脏大体形态,分别计算了其脂肪变性和炎症的组织学评分。在实验结束时(14个月),阿托伐他汀完全抑制了HFHC饮食诱导的NAFLD-HCC的形成,并改善了NASH的严重程度。
C)从左至右分别为血清胆固醇水平、肝脏游离胆固醇水平、血清甲胎蛋白水平,显著降低。
D) 丙氨酸氨基转移酶(ALT)、血清促炎细胞因子(IL-6、IL-1α、IL-1β、单核细胞趋化蛋白-1、巨噬细胞炎性蛋白-1α和巨噬细胞趋化蛋白-1β)显著降低。
E)阿托伐他汀通过显著减少肝脏胶原沉积和羟脯氨酸含量来改善肝纤维化
F)用NC、HFLC和HFHC饲料喂养的小鼠,在阿托伐他汀治疗或不治疗的情况下,粪便中细菌的Chao1指数和热图。经阿托伐他汀治疗后,HFHC喂养的小鼠肠道菌群丰富度显著恢复。
G)被灌服HFHCAt(阿托伐他汀治疗的HFHC喂养小鼠)粪便的无菌小鼠的肝脏大体形态、组织学检查、甘油三酯和脂质过氧化含量。与G-HFHC组相比,G-HFHCAt组小鼠肝脏组织学有所改善,肝脏甘油三酯和脂质过氧化水平降低。
H)胆固醇诱导的NAFLD-HCC发生机制示意图。胆固醇破坏胆汁酸代谢和微生物色氨酸代谢,导致血清TCA增强,IPA降低,从而促进NAFLD-HCC的发展。
结论
长时间的高胆固醇饮食通过调节肠道微生物群,诱导雄性小鼠NAFLD-HCC自发和进行性发展。胆固醇破坏胆汁酸代谢和微生物色氨酸代谢,导致血清TCA增强,IPA降低,从而促进NAFLD-HCC的发展。降胆固醇治疗完全抑制了膳食胆固醇诱导的NAFLD-HCC的形成。这项研究强调,抑制胆固醇和调控肠道菌群及其相关代谢产物可能是预防NAFLD-HCC的有效策略。

谷禾健康
发育迟缓的原因有很多,牵涉到比如母体子宫内生长迟缓、神经内分泌和激素因素、儿童早期腹泻和其他感染频繁、环境肠道功能障碍、环境毒素和遗传因素等。 环境肠道功能障碍(EED)是一种以绒毛萎缩和隐窝增生为特征的获得性小肠亚临床疾病,病因不明,可能占所有发育迟缓病例的40%以上。

近日,美国华盛顿大学医学院Jeffrey I. Gordon团队的发表在《新英格兰医学杂志》研究分析了营养不良伴肠病儿童的十二指肠微生物群(DOI:10.1056 / NEJMoa1916004 ),研究结果研支持生长发育迟缓与小肠微生物群组成和肠病之间存在因果关系,并为开发针对这些微生物对EED的作用的疗法提供了理论依据。
环境性肠功能障碍(EED)是一种神秘的小肠疾病,被认为与儿童营养不良(一个紧迫的全球健康问题)有关。由于对小肠黏膜和微生物群落直接取样较为困难,因此,难以确定这种疾病的发病率、病理生理特征及其对生长发育迟缓的影响。
该研究研究组在孟加拉国达卡市贫民窟中招募了110名没有营养干预的生长发育迟缓的儿童,平均年龄为18个月,对80例行活检的儿童进行了内镜检查, 确认的EED以及可用的血浆和十二指肠样品。
研究组对80例活检证实EED,且血浆和十二指肠样本可用的儿童进行了内窥镜检查。
研究组量化了从这些儿童获得的十二指肠活检样本中的4077个血浆蛋白和2619个蛋白的水平,确定了从每个儿童的十二指肠抽吸物中回收的微生物群中细菌菌株的水平,还从居住在同一地区的年龄匹配的健康儿童中获得了21个血浆样本和27个粪便样本。用从十二指肠抽吸物中培养出的细菌菌株定殖给孟加拉饮食的年轻无菌小鼠。

*加减值是±SD。 要将α-1抗胰蛋白酶的值转换为每升克,请乘以0.01。
由于四舍五入的关系,百分比总数可能不等于100。 IQR表示四分位距,NA不适用。
†环境肠道功能障碍(EED)的生物标志物得分是从0开始的连续测量,得分越高表明一组粪便炎症生物标志物(α-1抗胰蛋白酶,髓过氧化物酶和新蝶呤)的水平越高。
在从孩子那里获得的细菌菌株中,一组共有14个分类单元(通常不归类为肠病原体)的绝对水平与线性增长呈负相关(年龄z评分,r = -0.49; P = 0.003),并且与参与免疫炎症反应的十二指肠蛋白呈正相关。 粪便微生物群中这14个十二指肠分类群的代表与健康儿童的样本显着不同(通过方差的排列多变量分析,P <0.001)。 小肠的肠病在已被从EED儿童获得的培养的十二指肠菌株定殖的gnotobiotic小鼠中发展。
在从儿童获得的细菌菌株中,共有14个类群(通常不被归类为肠道病原体)的绝对水平与线性生长发育呈负相关,与参与免疫炎症反应的十二指肠蛋白呈正相关。
十二指肠蛋白质组成分、十二指肠细菌分类绝对水平与发育迟缓的相关性,粪便微生物群中这14个十二指肠分类群的代表与健康儿童的样本亦存在显著差异。

在从儿童获得的细菌菌株中,共有14个类群(通常不被归类为肠道病原体)的绝对水平与线性生长发育呈负相关,与参与免疫炎症反应的十二指肠蛋白呈正相关。
(EED)的研究儿童中获得的十二指肠细菌将肠病传播到致敏小鼠上。 接受EDD患儿十二指肠培养菌定殖的小鼠进展为小肠病。



随着 COVID-19 疫情的发展,相应的抗菌药物耐药性的加速确实令人担忧。疫情下,加剧了抗生素的大规模滥用,不仅用于治疗人类和动物疾病,而且也用于动物食品生产系统。
近日,BMJ发表一篇有关COVID-19的文章,消毒剂正在改变微生物群落。

幸运的是,现有的工具可用于研究抗生素耐药性的出现和蔓延,并作出“耐药”的知情预测。此外,全球和国家的具体行动计划已到位,以减轻抗生素耐药性威胁。
一个令人担忧的问题是,在Covid-19 大流行中,大量使用消毒剂,对人类、动物和环境中各种生态位的微生物群落产生了不可估量的影响。这种做法带来的结果可能是宿主-共生体相互作用中的失调,从而影响宿主的免疫功能、新陈代谢、生理参数以及对感染性和非传染性疾病的易感性。
益生菌、免疫生物制剂、合生元等都是有望治疗失调的药物。但是,由于过度使用消毒剂而引起的问题,在全球范围内已经超出了生物失调的范畴,例如,医院内的致病菌粪肠球菌出现了抗酒精性,最近报道了万古霉素耐药菌株(如超级细菌)。
困境在于,通过使用消毒剂、杀菌剂和抗生素来遏制Covid-19,我们正在给微生物群落造成不可估量的附带损害,“抹去”了不同生态位的几个共生体,并可能为新的威胁制造 “地雷”。
我们需要利用现有的工具和技术,开发新的工具和技术,以便评估对微生物生态系统的损害,密切检查人与动物之间的微生物关系,预测新的威胁,并揭示控制这些威胁的检查点。
希望从使用抗生素、消毒剂和杀菌剂来遏制Covid-19的普遍智慧中学到的所有经验教训,都将引导我们进入一个装备更完善、威胁可控的未来。
目前,人们对定义微生物群如何在癌症的背景下塑造免疫反应非常感兴趣。 在小鼠和人类中进行的各种研究都将特定的共生物种与不同癌症类型以及接受癌症免疫疗法治疗后的更好(或更坏)结果相关联。
但是,所涉及的机制仍然不确定,甚至存在争议。 在此观点下,《自然评论免疫学》邀请了该领域的六位著名科学家分享他们对该领域关键问题和挑战的看法。


来自B. Brett Finlay 的看法:
毫无疑问,微生物可以塑造免疫系统。它们最深远的影响是在生命的早期,那时它们可以将免疫系统推向过敏性或非过敏性方向时,从而影响对许多免疫介导疾病的敏感性,例如哮喘,食物过敏和湿疹。
在小鼠中也有重要的证据表明,免疫系统的许多方面可以在以后的生命中通过依赖于微生物(如分段丝状细菌)的过程进行改变。
但是,目前严重缺乏对所有这些过程机制的理解。尽管有关于潜在机制的线索,但目前我们还没有从机制上理解微生物如何影响抗肿瘤免疫力。

来自Romina Goldszmid的看法:
我同意 Brett 的看法,不同的非相互排斥的情况下,细菌,其产物和/或其代谢物通过调节免疫系统的组成部分来影响肿瘤免疫。
细菌及其产物可针对髓样细胞(例如,它们可以激活抗原呈递细胞,从而帮助引发抗肿瘤T细胞反应,调节促炎性细胞因子和活性氧的产生,或调节髓样细胞的免疫抑制活性)。
细菌也可以作为抗原源,通过分子模拟影响肿瘤免疫反应(也就是说,识别细菌表位的T细胞可以识别在肿瘤细胞上表达的相似表位),或者可以驱动旁观者T细胞活化。
其他人则提出了一种间接机制,细菌产物可通过这种间接机制诱导肿瘤中的免疫原性细胞死亡并随后进行免疫活化。对于不同的肿瘤类型和不同的治疗方式,该机制可能会有所不同。
此外,到目前为止,大多数努力都集中在细菌微生物组的作用上,我们对病毒组(病毒),真菌组(真菌)和寄生组(寄生虫)对抗肿瘤免疫的作用了解甚少。

来自Kenya Honda的看法:
多项研究表明,微生物群至少部分通过系统激活CD8+T细胞(尤其是干扰素-γ(IFNγ)+亚群)增强免疫检查点阻断。
从理论上讲,微生物介导的肿瘤浸润IFNγ+ CD8 + T细胞的诱导可能是由多种机制引起的,包括
(1)肠道诱导的CD8 + T细胞的系统循环
(2)细菌抗原或载有细菌或细菌的树突状细胞
(3)细菌代谢物(由微生物群诱导或产生)和/或细胞因子。
根据我们实验室的观察,肠道来源的CD8 + T细胞不太可能迁移到肠道外,在远端组织中全身循环并调节抗肿瘤免疫力,因为IFNγ+ CD8 + T细胞在肠道和肠道外肿瘤中蓄积。在表型上是不同的。
细菌抗原或负载细菌的树突状细胞的系统性循环也不大可能,因为在不同解剖部位的T细胞受体用途或T细胞的抗原特异性没有重叠。
最简单,最可能的解释是,肿瘤中CD8 + T细胞的积累是由于微生物群中免疫调节分子和/或代谢产物的循环所致。由于免疫调节似乎独立于主要的先天免疫信号传导途径而发生,因此代谢产物(也许是几种)而不是与微生物相关的分子模式可能负责形成CD8 + T细胞的抗肿瘤免疫应答。

来自Giorgio trinchieri的看法:
微生物群还调节单核细胞衍生的细胞和嗜中性粒细胞的肿瘤浸润。
革兰氏阴性细菌可以响应CpG寡聚脱氧核苷酸(通过脂多糖介导的Toll样受体4(TLR4)信号转导)引发髓样细胞产生免疫调节细胞因子,例如肿瘤坏死因子(TNF)和IL-12。
微生物细胞介导的骨髓细胞引发活性氧的产生会影响铂类化学疗法药物的抗肿瘤功效。
微生物代谢产物(例如短链脂肪酸)和其他微生物产物调节调节性T细胞和细胞毒性T细胞。
在小鼠肝癌模型中,肠道微生物群中存在的梭状芽孢杆菌将原发性胆汁酸转化为继发性胆汁酸,降低了肝脏中原发性胆汁酸诱导的CXC-趋化因子配体16(CXCL16)的表达;因为这种趋化因子吸引具有抗肿瘤活性的自然杀伤性T细胞,这些细菌与抗肿瘤活性降低有关。
常规和非常规T细胞的细菌识别也可能有助于抗肿瘤免疫反应,并可能通过抗原模拟来激活抗肿瘤T细胞。

来自Jennifer wargo的看法:
正如Giorgio所提到的,特定的微生物和微生物群落可能会对癌症的免疫反应产生正面或负面的影响。 对于在肿瘤本身内发现的微生物以及对于肠道菌群中存在的微生物而言,都是如此。
大量的证据与肠道微生物有关,其中肠道微生物群的多样性增加和组成差异与更有利的抗肿瘤免疫反应有关。
肠道微生物可通过肠道和肠系膜淋巴结水平上的相互作用以及代谢产物和其他循环因子介导的更远端效应来调节抗肿瘤免疫(以及一般的免疫系统)。
在过去的几年中,我们对所涉及的机制有了深刻的了解,但是仍然有很多值得学习的地方,并且存在难以置信的机会来获得进一步的机制见解。

来自Laurence Zitvogel的看法:
有几种独立的机制-有些加成机制,有些不是-当然也不能互斥,这可以解释不同的共生体对肿瘤免疫监视的免疫刺激(或抑制)作用。 我建议这里有四种主要的行动方式,其中一些已经被提及。
一,共生体可以直接为免疫系统提供抗原性和佐剂性。
二,它们确保上皮屏障的适应性。
三,共生体调节宿主的新陈代谢。
四,共生体调节微生物群生态系统(共生共存网络对于体内平衡至关重要)。
已有研究表明,肠道共生体(例如多形拟杆菌或海氏肠球菌)和自身抗原或癌细胞相关抗原出现在自身免疫性疾病和癌症中,这解释了通常维持在肠道中的T细胞池调动,通常维持在肠道内,向远处和肠外炎症性病变发展。
双歧杆菌属或脆弱拟杆菌(Bacteroides fragilis)和Akkermansia muciniphila可以介导对肿瘤引流淋巴结中存在的树突状细胞产生辅助作用,从而触发1型干扰素或IL-12指纹,这与保护性1型T辅助细胞(TH1细胞)和CD8 + T细胞抗肿瘤免疫反应相关。
通过直接影响上皮的完整性,抗癌疗法(例如化学疗法,酪氨酸激酶抑制剂或抗CTLA4免疫检查点阻断)调节了微生物群的组成,最终导致肠道上皮细胞(IEC)的凋亡,主要发生在肠道隐窝中。
根据回肠微生物群的组成,IEC的死亡可以是免疫原性或耐受性的,因此可以调节抗肿瘤免疫监测。
与微生物群落从专性厌氧菌转变为兼性厌氧菌有关的失调会导致结肠变形杆菌种类的增加。 这与抵抗免疫检查点的抵抗力相关。
IECs上微生物依赖的MHC II类表达上调也可引发移植物抗宿主病(GVHD),从而控制某些血液系统恶性肿瘤。
注:移植物抗宿主病(GVHD)是由于移植后异体供者移植物中的T淋巴细胞,经受者发动的一系列“细胞因子风暴”刺激,大大增强了其对受者抗原的免疫反应,以受者靶细胞为目标发动细胞毒攻击,其中皮肤、肝及肠道是主要的靶目标。

这仍然是微生物群领域中的主要问题。我们非常擅长将某些疾病个体中存在的不良微生物群与健康个体中的微生物群区分开来,但要确定“正常”微生物群是什么仍然难以捉摸。
问题的一部分是微生物学家强烈希望定义单个微生物物种或操作分类单位。但是,重点应该真正放在“好”菌群的作用上。物种之间的路径有很大的冗余,这意味着两个物种可以执行相同的功能,因此微生物群落在个体之间存在着主要的异质性。这方面的例子可以在抗癌研究中看到,每项研究都发现与不同的微生物有关。

关于良好的菌群是什么样还没有共识。实际上,在健康个体之间,微生物群组成存在巨大差异。
如果一个人健康,是否应该假设他/她拥有良好的微生物群?考虑到许多因素,例如宿主遗传学,饮食,地理位置和生活方式,这些因素会不断影响微生物菌群的组成和功能状态。
因此对于具有特定病理状况的个体可能有益的微生物菌群可能对另一个个体或个体都不那么有利。 即使是同一个人在不同情况下。所以我会说是的。这肯定是个体环境相关的。

良好的微生物群组成的共性仍然难以捉摸。的确,即使是单一优良菌群的概念也是模棱两可的,因为不同的成分可能有效地保护了不同宿主中的不同生物。
这是因为细菌可以适应给定的生态系统,该生态系统由宿主衍生因子和微生物群落的其他成员组成,并因此以个体环境相关的方式发挥作用。
微生物诱导的T细胞的免疫学作用类似地取决于环境,因此在个体之间可以不同。
但是,我相信有效促进抗癌免疫力的微生物群并不一定会诱发自身免疫,因为可以在不影响CTLA4和PD1表达或不影响调节性T细胞和自身反应性T细胞频率的情况下实现微生物群介导的效应T细胞活化。
因此,在我看来,良好或有效的微生物群可以增强针对非自我的免疫反应,包括肿瘤新抗原,而不会破坏免疫耐受性。

微生物群对炎症和免疫力的调节在预防癌症和治疗癌症方面有不同的作用。与抗PD1治疗有效反应相关的细菌种类已被鉴定,但在各种研究中发现不一致。这可能是由于人体肠道微生物群组成的个体差异和环境因素(例如,饮食和地理分布)的影响。
在我们的早期分析中,我们发现在某些情况下,相同的细菌种类可能与抗肿瘤免疫水平和免疫治疗后发生的免疫相关不良事件有关,尽管Laurence的小组已经报道,将抗CTLA4治疗的增强作用与自身免疫诱导分离开来的细菌组成。

现在,已在人类队列中发表了大量研究,证明了癌症类型中反应者与非反应者肠道微生物组的差异性特征。 但是,在所有这些队列中识别出的分类单元之间只有适度的重叠。
一项研究表明,具有 “1型” 肠道微生物组特征,含丰富的“有利”微生物(如瘤胃球菌科)的患者对免疫检查点阻断治疗更有反应。
但重要的是要记住,可能不是特定的分类群驱动,而是微生物群落的整体功能。研究表明,给定微生物群落的功能能力可能比给定微生物群落中微生物的分类学更为重要,从而支持“功能超过系统发育”的概念。
尽管人们可能认为一种促进抗癌免疫力的特定微生物群也可能增加自身免疫的可能性;但有证据却恰恰相反,也就是说,与更好的抗肿瘤免疫反应有关的肠道菌群实际上也与较低的自身免疫可能性有关。同时,较低的微生物多样性与治疗毒性增加有关。

是的,在我看来,我们接近于描述有益和有害的微生物群组成。
不同的肠道细菌群落与良好的健康状况有关;A. muciniphila、瘤胃球菌科和双歧杆菌属与预防胰岛素抵抗、延长寿命或对免疫检查点阻断的更好反应有关。
相比之下,其他的共生厌氧细菌(如Proteobacteriaceae科和肠杆菌科)则利用IEC压力,并具有对免疫检查点阻滞或化学疗法的抵抗力。
在抗PD1或抗PDL1治疗前1个月给药时,抗生素会降低免疫检查点阻断在肾癌和肺癌中的功效,它们会消除有益细菌,包括瘤胃球菌和梭菌科。
然而,如已经提到的,特定的共生菌株是否具有有益或有害作用将取决于个体微环境。
例如,多形拟杆菌表达的肽模拟肌球蛋白重链,它是心肌的组成部分,在自体免疫性心肌炎的背景下,这种共生体可以引发产生致病性IL-17的T辅助细胞(TH17细胞)反应。
然而,在抗CTLA4的癌症免疫治疗中,多形拟杆菌也可以促进保护性的IL-12依赖性TH1型免疫应答。
另一个例子是鸡肠球菌。当以单药疗法给予小鼠时,某些鸡肠球菌显示出抗肿瘤活性,而其他菌株与小鼠和人体内系统性红斑狼疮的发生有关。


正在进行大量的改变微生物群的工作,诸如Vedanta之类的公司正在积极地使用确定的活微生物混合物进行临床试验。
本质上,需要替换或增强生态系统,因此用抗生素(以破坏驻留的微生物)或抗炎剂(以允许传入的微生物混合物建立自身)进行预处理可以有所帮助。
毫无疑问,在接下来的几年中,将有多种微生物混合物被用作治疗疾病的生物。 几个小组正在尝试将其用于癌症治疗,并且根据小鼠的数据,如果可以建立正确的微生物混合物,则很有可能起作用。


可以使用不同的策略:
粪便移植,转移确定的细菌联合体或单个细菌分离物,使用益生元或通过饮食干预来改变现有微生态或个别菌群,以及从整体性改变菌群(使用广谱抗生素)到局部性改变(例如,使用窄谱抗微生物剂或噬菌体)。尚不清楚应单独或组合使用这些策略中的哪一个(如果有)。
然而,鉴于越来越多的证据表明微生物群会影响治疗结果,因此可以预见,考虑微生物群有一天将成为癌症治疗不可或缺的一部分。


来自健康捐献者或治疗反应者的粪便微生物菌群移植(FMT)的初步研究显示出的结果表明,可以合理地控制微生物菌群的组成。
但是,考虑到FMT的机理和成分的不确定性,我认为由定义好的一组特征良好的细菌组成的微生物疗法既可行,也更理想。
然而,实现外源细菌菌株在复杂的常驻菌群内的稳定植入可能是具有挑战性的。
研究表明,外在因素(例如营养素和抗生素)可以促进稳定的移入(尽管我们将尽可能避免使用抗生素,如下文更详细地讨论)。 补充确定的营养可以为特定的菌群提供食物,因此可以诱导肠道菌群的系统发育和功能重构,从而产生更有利于外源细菌植入的环境。


成人微生物群具有弹性,如果不消除现有微生物群,很难诱导患者微生物群发生重大变化。
但是,在与匹兹堡大学和MD安德森癌症中心合作正在进行的临床研究中,以及我们自己的小鼠研究中,我们有早期证据表明FMT以及影响微生物菌群的饮食变化可能会改善正在接受抗PD1治疗的黑色素瘤患者的抗肿瘤免疫反应。
我认为,对于改变微生物群的这类方法的广泛临床应用,我们仍然需要更好地了解有利于抗肿瘤反应的物种和微生物群组成以及潜在的机制。
但正如Romina所说,这些很可能最终成为癌症治疗的标准程序。


改变患者现有的微生物群以提高免疫疗法的效力当然是可行的。确实,在美国癌症研究协会(AACR)2019年年会上提出的两项研究表明可能是这种情况。
肠道菌群的调节可通过多种不同的策略来完成,在考虑使用最合适的策略时,必须考虑许多因素(以及相关因素,如治疗期间的饮食)


正如我的一些同事已经提到的那样,在癌症患者中使用FMT并短暂影响宿主微生物群的组成是可行的。
鉴于在PD1阻断开始后的头6-8周内进行免疫刺激至关重要,我们推测即使同种异体菌群未定植于受体,患者仍将从移植中受益。
然而,采用最佳选择粪便样本的标准(基于尚待确定的最佳成分,或基于健康状况-完全应答者与健康志愿者的比较)仍然是一个难题。
是的,我设想抗癌益生菌将有一天取代FMT,前提是单克隆益生菌或极简生态系统不会导致具有调节生物活性的α多样性降低和菌群补偿性增加(而非免疫刺激性)。
间歇性使用活体生物疗法可能比连续治疗更受青睐。

这都是抗生素更大问题的一部分-我们现在意识到,它们对癌症患者不再无害。
我们很久以来就已经意识到抗生素耐药性问题,但现在我们意识到它们还以显著的长期影响干扰了微生物群。因为癌症患者接受许多抗生素治疗,这对癌症来说是一个特别的问题。
需要记住的是,不同的抗生素会影响不同的微生物,因此不能将它们混在一起。关于影响免疫治疗的某些抗生素可能会好坏,我们只是不知道。
当我们定义免疫治疗涉及哪些微生物时,它将有助于指导抗生素治疗及其谨慎使用。

它无疑提高了人们的认识,并促使调查人员仔细观察。
最近对癌症患者的研究发现,在治疗开始前不久或短时间内使用抗生素会降低化疗和免疫疗法的反应率,而其他人则未发现临床影响。
但是,所有这些研究都有一些警告,需要进行大规模的前瞻性临床试验。抗生素在癌症治疗中的使用还带来了其他毒性作用,例如与GVHD相关的死亡率增加,以及自相矛盾的是,接受造血干细胞移植的血液系统恶性肿瘤患者感染的风险更高。及时治疗癌症患者的感染至关重要。
因此,如何在这些患者中使用抗生素以最大程度地减少对微生物群的附带损害是医生面临的挑战,需要进一步研究。

这些研究无疑使我们重新考虑了在癌症中使用抗生素的情况。这些开创性研究发表后,随后的几项研究报道,微生物群多样性是与有效的癌症免疫力相关的最重要的特征之一。
一直以来,我从临床医生那里听说,抗生素的使用会削弱免疫检查点阻断的功效。临床医生倾向于在特别害怕机会性感染的情况下预防性地使用抗生素,免疫功能低下的患者就是这样。
高度多样化的微生物群具有通过多种机制抑制病原体感染,包括通过营养竞争,抗菌产物的分泌和宿主免疫系统的激活。
因此,除了可以增强抗癌免疫力的效应菌群外,重要的是要确定抑制病原体入侵的支持性微生物群。 以活菌疗法的形式综合利用这些微生物活性,是一种在不使用抗生素的情况下同时根除肿瘤和对抗机会性感染的有希望的方法。

这些研究大量使用了几乎完全消灭肠道菌群的抗生素混合物,并不代表抗生素的临床使用。
一些研究表明,单一抗生素的施用可能会增强抗肿瘤反应,例如,通过影响胆汁酸的代谢。 用单一抗生素治疗还可以逆转肿瘤相关细菌对药物分解代谢和对免疫抑制性肿瘤微环境的作用。
但是,许多研究小组的最新发现清楚地表明,在抗PD1治疗前60天使用抗生素治疗会大大降低治疗效果。 这表明在癌症患者中应避免非必要地使用抗生素,并且在使用抗生素后我们应考虑将免疫疗法的启动延迟一定时间。

正如已经提到的,很明显,肠道微生物的有意(或无意)改变可能会影响对免疫检查点阻断的反应。
这包括抗生素的使用,现在有几项研究表明,在癌症患者中开始用免疫检查点阻断治疗之前服用抗生素,会导致生存率下降和对治疗的临床反应较差。
但是,我们对此的理解还不完整,需要做更多的研究以更好地了解影响肠道和其他部位的菌群的抗生素(以及其他药物和因素,例如饮食)如何影响癌症的发展和对肿瘤治疗的反应。
根据这些发现,在癌症患者甚至潜在的健康个体中,我们可能需要发展并承担起抗生素管理的新角色。

在12项回顾性研究中,有11项研究表明抗生素对接受PD1或PDL1免疫检查点阻滞治疗的肺癌,肾癌或黑色素瘤患者的临床结果具有负面影响。
在两个三级学术转诊中心进行的一项前瞻性,多中心队列研究招募了196名癌症患者,这些患者在2015年至2018年之间按常规临床实践(而非临床试验)接受了免疫检查点阻断疗法。
这项研究前瞻性地证实了在常规临床实践中,未经选择的接受免疫检查点阻断治疗的患者,先前(而非同时)抗生素治疗与较差的客观反应和总体生存率有关。
然而,许多问题仍未得到解答,例如用抗生素治疗后微生物群组成的表征,已使用抗生素的持续时间和光谱,以及潜在的混杂因素(例如,较差的体质)。
许多癌症中心正在考虑如何延迟首次抗PD1或抗PDL1疗法的给药,并缩短抗生素的递送时间。

尽管某些益生菌在某种程度上可对某些疾病起作用,但在临床试验中很少进行严格测试。
我非常乐观的是,第二代益生菌将会从肠道中分离出,这样它们就能够真正在肠道中定植,这与目前大多数益生菌不同。 这些将是微生物的混合物,而不是单个菌株,以帮助建立新的微生物生态系统。
我相信在不久的将来,人们会发现可以增强癌症免疫疗法的混合物。也就是说,希望我们能够超越微生物而进入影响免疫疗法的分子中,因为这样我们就可以将这些微生物产品用作常规药物,而我们的卫生系统就是为这些药物而设计的。

我们对此必须非常非常谨慎。 人们普遍认为益生菌对人体有好处,但这是相对的,并取决于多种因素,包括本地宿主微生物群,任何潜在的医疗状况和饮食。 “益生菌”一词指的是活微生物,如果给予足够的量,可以为宿主带来健康益处。
但是,没有足够的科学证据证明疗效,也没有任何监管机构(例如美国食品和药物管理局)批准将其用于任何治疗用途; 相反,这些被许可用作膳食补充剂。 例如,非处方益生菌被广泛用于帮助抗生素治疗后恢复肠道菌群。需要注意的是,最近的研究表明,它们可能会产生相反的效果,这突出了仔细研究的必要性。
通常,使用非处方益生菌与减轻胃肠道疾病有关,在许多情况下,这与它们的抗炎特性有关。重要的是,尽管研究仍在进行中,但尚未适当评估其在癌症免疫治疗中的作用。
这并不意味着新鉴定的益生菌可能不会对癌症免疫治疗有所帮助,而这些益生菌应该包含具有特定健康效应的安全性和有效性的明确证据的特定菌株(单个或联合体)。

传统的益生菌已被用于通过发酵保存食品。 因此,它们还没有出于特定目的从人的微生物群中合理地分离出来,它们也不一定是正常微生物群的一部分。
此外,单一菌株的接种不能恢复正常微生物群中通常可见的多样性,并且缺乏强有力,长期益生菌植入的证据。
因此,我认为常规的益生菌在临床上至多具有中等程度的作用。 从健康的供体或治疗反应者中分离出来的合理设计的微生物联合体可能会更有效。

传统的商业益生菌不太可能使接受免疫治疗的患者受益,因为它们会改变微生物群的组成和多样性,并可能导致营养不良,因此可能产生负面影响。
但是,某些益生菌含有细菌(例如,双歧杆菌种),它们已在实验动物模型和癌症患者中均显示出,以增强对免疫疗法的反应。
目前正在使用单一细菌菌株或已被初步确定为促进免疫疗法的细菌组合进行临床试验。一旦我们清楚地确定了有利的细菌种类并定义了它们的作用机制,它们在患者体内的定殖能力以及它们如何与现有微生物群落一起起作用,这种细菌疗法可能具有提高免疫疗法应答率的巨大潜力。

益生菌一词范围很广,是指被宣传为食用时具有健康益处的微生物。目前的非处方益生菌还没有在癌症患者中进行广泛的测试,甚至有证据表明报告服用非处方益生菌的患者在癌症免疫治疗后可能会有较少的多样性微生物群和更糟糕的结果。
临床前模型中的证据表明,大肠癌模型中非处方益生菌的施用与肿瘤发生的增加有关。因此,应警告癌症患者,非处方益生菌可能无益,在癌症治疗中应避免使用。
但是,我们需要把握机会,开发基于新见识而合理设计的下一代益生菌,但要进行广泛的测试以确保功效并证实其主张。

尽管益生菌通常被认为是一种营养素,可以补偿在使用抗生素、化疗和放疗治疗后出现的腹泻副作用,但许多益生菌菌株和制剂的结果是相互矛盾的。
临床前和回顾性研究似乎表明,使用益生菌可能会降低肠道菌群的α多样性,从而损害免疫检查点阻断的全面功效。
因此,我们仍然需要评估单个微生物菌株如何在肠道中定居,并且需要研究它们对宿主代谢和免疫系统的影响。
在任何广泛和无针对性的使用之前,这些研究需要在来自不同类型癌症患者的本土微生物群的背景下进行。

我认为动物模型和人类都有足够的初步证据表明微生物可以影响癌症的免疫治疗。 但是,还有许多其他问题。
这些包括以下内容。
涉及的机制是什么?
涉及哪些微生物?
微生物产生什么分子来影响免疫治疗?
我们能否开发出一种可靠的微生物群筛查方法,以识别哪些人会从生物素中受益,哪些人会在没有生物素的情况下做出反应?
最后,所有这些信息是否可以用来开发(1)活的细菌混合物和(2)微生物分子,它们可以可靠地用于接受免疫治疗的患者的临床。

对我来说,关键问题是这一切如何运作?
一旦我们对系统学有了更深入的了解,就可以解决一些争议(例如,不同的研究人员发现与治疗结果相关的不同细菌类群,以及有关抗生素对反应率的正面和负面影响的报道)。
另一个重要的问题是它会随着时间变化吗? 微生物群落,宿主免疫系统和癌细胞之间发生的三向相互作用不仅复杂,而且高度动态。这可能意味着在疾病/治疗的整个过程中将需要采用不同的针对微生物菌群的方法。
近年来,我们获得了大量知识; 然而,就我们对这些过程的理解而言,我们可能只是在摸索表面。

大量研究各自确定了不同的细菌,这些细菌可促进癌症患者免疫检查站阻断的功效。
关键问题是这些细菌是否通过保守的功能途径增强了对免疫检查点的阻断,还是通过简单的机制简单地促进了相同的表型。
如果保守的代谢途径起作用,则可以将功能上多余的一组细菌一起使用,以克服将单个细菌群落治疗性移植到不同患者微生物群上的可变成功率。

展望未来,我认为我们的主要目标如下:
首先,我们需要通过消除混杂因素(如早期接触,生活方式和地理差异)来预测有利于抗肿瘤免疫的微生物群组成。
其次,我们需要确定特定的分类单元和/或调节抗肿瘤免疫力的微生物群的代谢途径和产物。
第三,我们必须表征宿主免疫系统和微生物群之间发生串扰的潜在机制。
第四,我们应该将这些分析扩展到理解除肠道以外的其他屏障组织的微生物群,确定与肿瘤相关的微生物群的作用,并阐明该微生物群的其他非细菌成分的作用,如真菌、病毒、噬菌体和原生动物。
最后,一旦确定了单个菌株或联合体,我们需要评估它们在患者体内的定殖能力,以及在现有微生物群落生态中以功能状态共存的能力。

我认为,需要考虑三个关键问题:
首先,什么是“完美”的肠道菌群? 同时,这种促进癌症免疫治疗反应的完美微生物群是否也有助于预防癌症的发生呢?它甚至能增强对疫苗的反应并促进整体免疫健康吗?
第二,肿瘤微生物组的影响是什么? 我们能把这一点作为癌症预防的目标吗?
最后,我们如何协调微生物组学研究?

我认为在“免疫肿瘤学”领域中存在四个主要问题:
首先,我们如何在临床上定义肠道失调并开发适当的工具以进行准确诊断?
第二,肠道失调是晚期癌症的原因和/或后果吗? 如果答案是肯定的,那么这是对任何一种癌症还是只对特定类型的癌症有效?
第三,肠道失调是否会导致对癌症免疫疗法的“原发性耐药”?
最后,我们能通过干预癌症相关的肠道失调来治愈这个问题吗?如果答案是肯定的,那么这种治愈是短暂的还是会产生长期影响?
微生物群由共生细菌和生活在宿主上皮屏障上的其它微生物组成,影响着大量的生理功能,包括维持屏障的内稳态,调节新陈代谢、造血作用、神经系统、炎症、免疫力等。微生物群也参与了癌症的发生、进展和转移。
不断积累的研究表明,微生物群,尤其是肠道微生物群能够调节机体对癌症疗法的响应以及对毒副作用的敏感性。一旦确定了每一种临床状况所对应的最有利的微生物群组成,如何修改患者的微生物群将是下一步挑战。
科学家们普遍认为在癌症和其它疾病中靶向微生物群可能会成为精准医疗和个性化医疗的下一个前沿方向之一,肠道微生物群的弹性、稳定性以及它对生理、病理以及环境变化的响应性使得我们能够利用微生物组的组成作为一种生物标志物、一种诊断工具或者一种治疗靶标。尽管通过靶向微生物组实现治疗干预这一研究领域还处在初始阶段,但一些途径已经证明了其可能性。
相关阅读:
参考文献:
BMJ 2020 07 14;370:m2795. Epub 2020 Jul 14.
Covid-19: disinfectants and sanitisers are changing microbiomes. Department of Veterinary Microbiology, Lajpat Rai University of Veterinary and Animal Sciences, Hisar-125004, India. doi.org/10.1136/bmj.m2795
Finlay, B. B., Goldszmid, R., Honda, K., Trinchieri, G., Wargo, J., & Zitvogel, L. (2020). Can we harness the microbiota to enhance the efficacy of cancer immunotherapy? Nature Reviews Immunology. doi:10.1038/s41577-020-0374-6
01
随着 COVID-19 疫情的发展,相应的抗菌药物耐药性的加速确实令人担忧。疫情下,加剧了抗生素的大规模滥用,不仅用于治疗人类和动物疾病,而且也用于动物食品生产系统。
近日,BMJ发表一篇有关COVID-19的文章,消毒剂正在改变微生物群落。

幸运的是,现有的工具可用于研究抗生素耐药性的出现和蔓延,并作出“耐药”的知情预测。此外,全球和国家的具体行动计划已到位,以减轻抗生素耐药性威胁。
一个令人担忧的问题是,在Covid-19 大流行中,大量使用消毒剂,对人类、动物和环境中各种生态位的微生物群落产生了不可估量的影响。这种做法带来的结果可能是宿主-共生体相互作用中的失调,从而影响宿主的免疫功能、新陈代谢、生理参数以及对感染性和非传染性疾病的易感性。
益生菌、免疫生物制剂、合生元等都是有望治疗失调的药物。但是,由于过度使用消毒剂而引起的问题,在全球范围内已经超出了生物失调的范畴,例如,医院内的致病菌粪肠球菌出现了抗酒精性,最近报道了万古霉素耐药菌株(如超级细菌)。
困境在于,通过使用消毒剂、杀菌剂和抗生素来遏制Covid-19,我们正在给微生物群落造成不可估量的附带损害,“抹去”了不同生态位的几个共生体,并可能为新的威胁制造 “地雷”。
我们需要利用现有的工具和技术,开发新的工具和技术,以便评估对微生物生态系统的损害,密切检查人与动物之间的微生物关系,预测新的威胁,并揭示控制这些威胁的检查点。
希望从使用抗生素、消毒剂和杀菌剂来遏制Covid-19的普遍智慧中学到的所有经验教训,都将引导我们进入一个装备更完善、威胁可控的未来。
目前,人们对定义微生物群如何在癌症的背景下塑造免疫反应非常感兴趣。 在小鼠和人类中进行的各种研究都将特定的共生物种与不同癌症类型以及接受癌症免疫疗法治疗后的更好(或更坏)结果相关联。
但是,所涉及的机制仍然不确定,甚至存在争议。 在此观点下,《自然评论免疫学》邀请了该领域的六位著名科学家分享他们对该领域关键问题和挑战的看法。


来自B. Brett Finlay 的看法:
毫无疑问,微生物可以塑造免疫系统。它们最深远的影响是在生命的早期,那时它们可以将免疫系统推向过敏性或非过敏性方向时,从而影响对许多免疫介导疾病的敏感性,例如哮喘,食物过敏和湿疹。
在小鼠中也有重要的证据表明,免疫系统的许多方面可以在以后的生命中通过依赖于微生物(如分段丝状细菌)的过程进行改变。
但是,目前严重缺乏对所有这些过程机制的理解。尽管有关于潜在机制的线索,但目前我们还没有从机制上理解微生物如何影响抗肿瘤免疫力。

来自Romina Goldszmid的看法:
我同意 Brett 的看法,不同的非相互排斥的情况下,细菌,其产物和/或其代谢物通过调节免疫系统的组成部分来影响肿瘤免疫。
细菌及其产物可针对髓样细胞(例如,它们可以激活抗原呈递细胞,从而帮助引发抗肿瘤T细胞反应,调节促炎性细胞因子和活性氧的产生,或调节髓样细胞的免疫抑制活性)。
细菌也可以作为抗原源,通过分子模拟影响肿瘤免疫反应(也就是说,识别细菌表位的T细胞可以识别在肿瘤细胞上表达的相似表位),或者可以驱动旁观者T细胞活化。
其他人则提出了一种间接机制,细菌产物可通过这种间接机制诱导肿瘤中的免疫原性细胞死亡并随后进行免疫活化。对于不同的肿瘤类型和不同的治疗方式,该机制可能会有所不同。
此外,到目前为止,大多数努力都集中在细菌微生物组的作用上,我们对病毒组(病毒),真菌组(真菌)和寄生组(寄生虫)对抗肿瘤免疫的作用了解甚少。

来自Kenya Honda的看法:
多项研究表明,微生物群至少部分通过系统激活CD8+T细胞(尤其是干扰素-γ(IFNγ)+亚群)增强免疫检查点阻断。
从理论上讲,微生物介导的肿瘤浸润IFNγ+ CD8 + T细胞的诱导可能是由多种机制引起的,包括
(1)肠道诱导的CD8 + T细胞的系统循环
(2)细菌抗原或载有细菌或细菌的树突状细胞
(3)细菌代谢物(由微生物群诱导或产生)和/或细胞因子。
根据我们实验室的观察,肠道来源的CD8 + T细胞不太可能迁移到肠道外,在远端组织中全身循环并调节抗肿瘤免疫力,因为IFNγ+ CD8 + T细胞在肠道和肠道外肿瘤中蓄积。在表型上是不同的。
细菌抗原或负载细菌的树突状细胞的系统性循环也不大可能,因为在不同解剖部位的T细胞受体用途或T细胞的抗原特异性没有重叠。
最简单,最可能的解释是,肿瘤中CD8 + T细胞的积累是由于微生物群中免疫调节分子和/或代谢产物的循环所致。由于免疫调节似乎独立于主要的先天免疫信号传导途径而发生,因此代谢产物(也许是几种)而不是与微生物相关的分子模式可能负责形成CD8 + T细胞的抗肿瘤免疫应答。

来自Giorgio trinchieri的看法:
微生物群还调节单核细胞衍生的细胞和嗜中性粒细胞的肿瘤浸润。
革兰氏阴性细菌可以响应CpG寡聚脱氧核苷酸(通过脂多糖介导的Toll样受体4(TLR4)信号转导)引发髓样细胞产生免疫调节细胞因子,例如肿瘤坏死因子(TNF)和IL-12。
微生物细胞介导的骨髓细胞引发活性氧的产生会影响铂类化学疗法药物的抗肿瘤功效。
微生物代谢产物(例如短链脂肪酸)和其他微生物产物调节调节性T细胞和细胞毒性T细胞。
在小鼠肝癌模型中,肠道微生物群中存在的梭状芽孢杆菌将原发性胆汁酸转化为继发性胆汁酸,降低了肝脏中原发性胆汁酸诱导的CXC-趋化因子配体16(CXCL16)的表达;因为这种趋化因子吸引具有抗肿瘤活性的自然杀伤性T细胞,这些细菌与抗肿瘤活性降低有关。
常规和非常规T细胞的细菌识别也可能有助于抗肿瘤免疫反应,并可能通过抗原模拟来激活抗肿瘤T细胞。

来自Jennifer wargo的看法:
正如Giorgio所提到的,特定的微生物和微生物群落可能会对癌症的免疫反应产生正面或负面的影响。 对于在肿瘤本身内发现的微生物以及对于肠道菌群中存在的微生物而言,都是如此。
大量的证据与肠道微生物有关,其中肠道微生物群的多样性增加和组成差异与更有利的抗肿瘤免疫反应有关。
肠道微生物可通过肠道和肠系膜淋巴结水平上的相互作用以及代谢产物和其他循环因子介导的更远端效应来调节抗肿瘤免疫(以及一般的免疫系统)。
在过去的几年中,我们对所涉及的机制有了深刻的了解,但是仍然有很多值得学习的地方,并且存在难以置信的机会来获得进一步的机制见解。

来自Laurence Zitvogel的看法:
有几种独立的机制-有些加成机制,有些不是-当然也不能互斥,这可以解释不同的共生体对肿瘤免疫监视的免疫刺激(或抑制)作用。 我建议这里有四种主要的行动方式,其中一些已经被提及。
一,共生体可以直接为免疫系统提供抗原性和佐剂性。
二,它们确保上皮屏障的适应性。
三,共生体调节宿主的新陈代谢。
四,共生体调节微生物群生态系统(共生共存网络对于体内平衡至关重要)。
已有研究表明,肠道共生体(例如多形拟杆菌或海氏肠球菌)和自身抗原或癌细胞相关抗原出现在自身免疫性疾病和癌症中,这解释了通常维持在肠道中的T细胞池调动,通常维持在肠道内,向远处和肠外炎症性病变发展。
双歧杆菌属或脆弱拟杆菌(Bacteroides fragilis)和Akkermansia muciniphila可以介导对肿瘤引流淋巴结中存在的树突状细胞产生辅助作用,从而触发1型干扰素或IL-12指纹,这与保护性1型T辅助细胞(TH1细胞)和CD8 + T细胞抗肿瘤免疫反应相关。
通过直接影响上皮的完整性,抗癌疗法(例如化学疗法,酪氨酸激酶抑制剂或抗CTLA4免疫检查点阻断)调节了微生物群的组成,最终导致肠道上皮细胞(IEC)的凋亡,主要发生在肠道隐窝中。
根据回肠微生物群的组成,IEC的死亡可以是免疫原性或耐受性的,因此可以调节抗肿瘤免疫监测。
与微生物群落从专性厌氧菌转变为兼性厌氧菌有关的失调会导致结肠变形杆菌种类的增加。 这与抵抗免疫检查点的抵抗力相关。
IECs上微生物依赖的MHC II类表达上调也可引发移植物抗宿主病(GVHD),从而控制某些血液系统恶性肿瘤。
注:移植物抗宿主病(GVHD)是由于移植后异体供者移植物中的T淋巴细胞,经受者发动的一系列“细胞因子风暴”刺激,大大增强了其对受者抗原的免疫反应,以受者靶细胞为目标发动细胞毒攻击,其中皮肤、肝及肠道是主要的靶目标。

这仍然是微生物群领域中的主要问题。我们非常擅长将某些疾病个体中存在的不良微生物群与健康个体中的微生物群区分开来,但要确定“正常”微生物群是什么仍然难以捉摸。
问题的一部分是微生物学家强烈希望定义单个微生物物种或操作分类单位。但是,重点应该真正放在“好”菌群的作用上。物种之间的路径有很大的冗余,这意味着两个物种可以执行相同的功能,因此微生物群落在个体之间存在着主要的异质性。这方面的例子可以在抗癌研究中看到,每项研究都发现与不同的微生物有关。

关于良好的菌群是什么样还没有共识。实际上,在健康个体之间,微生物群组成存在巨大差异。
如果一个人健康,是否应该假设他/她拥有良好的微生物群?考虑到许多因素,例如宿主遗传学,饮食,地理位置和生活方式,这些因素会不断影响微生物菌群的组成和功能状态。
因此对于具有特定病理状况的个体可能有益的微生物菌群可能对另一个个体或个体都不那么有利。 即使是同一个人在不同情况下。所以我会说是的。这肯定是个体环境相关的。

良好的微生物群组成的共性仍然难以捉摸。的确,即使是单一优良菌群的概念也是模棱两可的,因为不同的成分可能有效地保护了不同宿主中的不同生物。
这是因为细菌可以适应给定的生态系统,该生态系统由宿主衍生因子和微生物群落的其他成员组成,并因此以个体环境相关的方式发挥作用。
微生物诱导的T细胞的免疫学作用类似地取决于环境,因此在个体之间可以不同。
但是,我相信有效促进抗癌免疫力的微生物群并不一定会诱发自身免疫,因为可以在不影响CTLA4和PD1表达或不影响调节性T细胞和自身反应性T细胞频率的情况下实现微生物群介导的效应T细胞活化。
因此,在我看来,良好或有效的微生物群可以增强针对非自我的免疫反应,包括肿瘤新抗原,而不会破坏免疫耐受性。

微生物群对炎症和免疫力的调节在预防癌症和治疗癌症方面有不同的作用。与抗PD1治疗有效反应相关的细菌种类已被鉴定,但在各种研究中发现不一致。这可能是由于人体肠道微生物群组成的个体差异和环境因素(例如,饮食和地理分布)的影响。
在我们的早期分析中,我们发现在某些情况下,相同的细菌种类可能与抗肿瘤免疫水平和免疫治疗后发生的免疫相关不良事件有关,尽管Laurence的小组已经报道,将抗CTLA4治疗的增强作用与自身免疫诱导分离开来的细菌组成。

现在,已在人类队列中发表了大量研究,证明了癌症类型中反应者与非反应者肠道微生物组的差异性特征。 但是,在所有这些队列中识别出的分类单元之间只有适度的重叠。
一项研究表明,具有 “1型” 肠道微生物组特征,含丰富的“有利”微生物(如瘤胃球菌科)的患者对免疫检查点阻断治疗更有反应。
但重要的是要记住,可能不是特定的分类群驱动,而是微生物群落的整体功能。研究表明,给定微生物群落的功能能力可能比给定微生物群落中微生物的分类学更为重要,从而支持“功能超过系统发育”的概念。
尽管人们可能认为一种促进抗癌免疫力的特定微生物群也可能增加自身免疫的可能性;但有证据却恰恰相反,也就是说,与更好的抗肿瘤免疫反应有关的肠道菌群实际上也与较低的自身免疫可能性有关。同时,较低的微生物多样性与治疗毒性增加有关。

是的,在我看来,我们接近于描述有益和有害的微生物群组成。
不同的肠道细菌群落与良好的健康状况有关;A. muciniphila、瘤胃球菌科和双歧杆菌属与预防胰岛素抵抗、延长寿命或对免疫检查点阻断的更好反应有关。
相比之下,其他的共生厌氧细菌(如Proteobacteriaceae科和肠杆菌科)则利用IEC压力,并具有对免疫检查点阻滞或化学疗法的抵抗力。
在抗PD1或抗PDL1治疗前1个月给药时,抗生素会降低免疫检查点阻断在肾癌和肺癌中的功效,它们会消除有益细菌,包括瘤胃球菌和梭菌科。
然而,如已经提到的,特定的共生菌株是否具有有益或有害作用将取决于个体微环境。
例如,多形拟杆菌表达的肽模拟肌球蛋白重链,它是心肌的组成部分,在自体免疫性心肌炎的背景下,这种共生体可以引发产生致病性IL-17的T辅助细胞(TH17细胞)反应。
然而,在抗CTLA4的癌症免疫治疗中,多形拟杆菌也可以促进保护性的IL-12依赖性TH1型免疫应答。
另一个例子是鸡肠球菌。当以单药疗法给予小鼠时,某些鸡肠球菌显示出抗肿瘤活性,而其他菌株与小鼠和人体内系统性红斑狼疮的发生有关。

正在进行大量的改变微生物群的工作,诸如Vedanta之类的公司正在积极地使用确定的活微生物混合物进行临床试验。
本质上,需要替换或增强生态系统,因此用抗生素(以破坏驻留的微生物)或抗炎剂(以允许传入的微生物混合物建立自身)进行预处理可以有所帮助。
毫无疑问,在接下来的几年中,将有多种微生物混合物被用作治疗疾病的生物。 几个小组正在尝试将其用于癌症治疗,并且根据小鼠的数据,如果可以建立正确的微生物混合物,则很有可能起作用。

可以使用不同的策略:
粪便移植,转移确定的细菌联合体或单个细菌分离物,使用益生元或通过饮食干预来改变现有微生态或个别菌群,以及从整体性改变菌群(使用广谱抗生素)到局部性改变(例如,使用窄谱抗微生物剂或噬菌体)。尚不清楚应单独或组合使用这些策略中的哪一个(如果有)。
然而,鉴于越来越多的证据表明微生物群会影响治疗结果,因此可以预见,考虑微生物群有一天将成为癌症治疗不可或缺的一部分。

来自健康捐献者或治疗反应者的粪便微生物菌群移植(FMT)的初步研究显示出的结果表明,可以合理地控制微生物菌群的组成。
但是,考虑到FMT的机理和成分的不确定性,我认为由定义好的一组特征良好的细菌组成的微生物疗法既可行,也更理想。
然而,实现外源细菌菌株在复杂的常驻菌群内的稳定植入可能是具有挑战性的。
研究表明,外在因素(例如营养素和抗生素)可以促进稳定的移入(尽管我们将尽可能避免使用抗生素,如下文更详细地讨论)。 补充确定的营养可以为特定的菌群提供食物,因此可以诱导肠道菌群的系统发育和功能重构,从而产生更有利于外源细菌植入的环境。

成人微生物群具有弹性,如果不消除现有微生物群,很难诱导患者微生物群发生重大变化。
但是,在与匹兹堡大学和MD安德森癌症中心合作正在进行的临床研究中,以及我们自己的小鼠研究中,我们有早期证据表明FMT以及影响微生物菌群的饮食变化可能会改善正在接受抗PD1治疗的黑色素瘤患者的抗肿瘤免疫反应。
我认为,对于改变微生物群的这类方法的广泛临床应用,我们仍然需要更好地了解有利于抗肿瘤反应的物种和微生物群组成以及潜在的机制。
但正如Romina所说,这些很可能最终成为癌症治疗的标准程序。

改变患者现有的微生物群以提高免疫疗法的效力当然是可行的。确实,在美国癌症研究协会(AACR)2019年年会上提出的两项研究表明可能是这种情况。
肠道菌群的调节可通过多种不同的策略来完成,在考虑使用最合适的策略时,必须考虑许多因素(以及相关因素,如治疗期间的饮食)

正如我的一些同事已经提到的那样,在癌症患者中使用FMT并短暂影响宿主微生物群的组成是可行的。
鉴于在PD1阻断开始后的头6-8周内进行免疫刺激至关重要,我们推测即使同种异体菌群未定植于受体,患者仍将从移植中受益。
然而,采用最佳选择粪便样本的标准(基于尚待确定的最佳成分,或基于健康状况-完全应答者与健康志愿者的比较)仍然是一个难题。
是的,我设想抗癌益生菌将有一天取代FMT,前提是单克隆益生菌或极简生态系统不会导致具有调节生物活性的α多样性降低和菌群补偿性增加(而非免疫刺激性)。
间歇性使用活体生物疗法可能比连续治疗更受青睐。

这都是抗生素更大问题的一部分-我们现在意识到,它们对癌症患者不再无害。
我们很久以来就已经意识到抗生素耐药性问题,但现在我们意识到它们还以显著的长期影响干扰了微生物群。因为癌症患者接受许多抗生素治疗,这对癌症来说是一个特别的问题。
需要记住的是,不同的抗生素会影响不同的微生物,因此不能将它们混在一起。关于影响免疫治疗的某些抗生素可能会好坏,我们只是不知道。
当我们定义免疫治疗涉及哪些微生物时,它将有助于指导抗生素治疗及其谨慎使用。

它无疑提高了人们的认识,并促使调查人员仔细观察。
最近对癌症患者的研究发现,在治疗开始前不久或短时间内使用抗生素会降低化疗和免疫疗法的反应率,而其他人则未发现临床影响。
但是,所有这些研究都有一些警告,需要进行大规模的前瞻性临床试验。抗生素在癌症治疗中的使用还带来了其他毒性作用,例如与GVHD相关的死亡率增加,以及自相矛盾的是,接受造血干细胞移植的血液系统恶性肿瘤患者感染的风险更高。及时治疗癌症患者的感染至关重要。
因此,如何在这些患者中使用抗生素以最大程度地减少对微生物群的附带损害是医生面临的挑战,需要进一步研究。

这些研究无疑使我们重新考虑了在癌症中使用抗生素的情况。这些开创性研究发表后,随后的几项研究报道,微生物群多样性是与有效的癌症免疫力相关的最重要的特征之一。
一直以来,我从临床医生那里听说,抗生素的使用会削弱免疫检查点阻断的功效。临床医生倾向于在特别害怕机会性感染的情况下预防性地使用抗生素,免疫功能低下的患者就是这样。
高度多样化的微生物群具有通过多种机制抑制病原体感染,包括通过营养竞争,抗菌产物的分泌和宿主免疫系统的激活。
因此,除了可以增强抗癌免疫力的效应菌群外,重要的是要确定抑制病原体入侵的支持性微生物群。 以活菌疗法的形式综合利用这些微生物活性,是一种在不使用抗生素的情况下同时根除肿瘤和对抗机会性感染的有希望的方法。

这些研究大量使用了几乎完全消灭肠道菌群的抗生素混合物,并不代表抗生素的临床使用。
一些研究表明,单一抗生素的施用可能会增强抗肿瘤反应,例如,通过影响胆汁酸的代谢。 用单一抗生素治疗还可以逆转肿瘤相关细菌对药物分解代谢和对免疫抑制性肿瘤微环境的作用。
但是,许多研究小组的最新发现清楚地表明,在抗PD1治疗前60天使用抗生素治疗会大大降低治疗效果。 这表明在癌症患者中应避免非必要地使用抗生素,并且在使用抗生素后我们应考虑将免疫疗法的启动延迟一定时间。

正如已经提到的,很明显,肠道微生物的有意(或无意)改变可能会影响对免疫检查点阻断的反应。
这包括抗生素的使用,现在有几项研究表明,在癌症患者中开始用免疫检查点阻断治疗之前服用抗生素,会导致生存率下降和对治疗的临床反应较差。
但是,我们对此的理解还不完整,需要做更多的研究以更好地了解影响肠道和其他部位的菌群的抗生素(以及其他药物和因素,例如饮食)如何影响癌症的发展和对肿瘤治疗的反应。
根据这些发现,在癌症患者甚至潜在的健康个体中,我们可能需要发展并承担起抗生素管理的新角色。

在12项回顾性研究中,有11项研究表明抗生素对接受PD1或PDL1免疫检查点阻滞治疗的肺癌,肾癌或黑色素瘤患者的临床结果具有负面影响。
在两个三级学术转诊中心进行的一项前瞻性,多中心队列研究招募了196名癌症患者,这些患者在2015年至2018年之间按常规临床实践(而非临床试验)接受了免疫检查点阻断疗法。
这项研究前瞻性地证实了在常规临床实践中,未经选择的接受免疫检查点阻断治疗的患者,先前(而非同时)抗生素治疗与较差的客观反应和总体生存率有关。
然而,许多问题仍未得到解答,例如用抗生素治疗后微生物群组成的表征,已使用抗生素的持续时间和光谱,以及潜在的混杂因素(例如,较差的体质)。
许多癌症中心正在考虑如何延迟首次抗PD1或抗PDL1疗法的给药,并缩短抗生素的递送时间。

尽管某些益生菌在某种程度上可对某些疾病起作用,但在临床试验中很少进行严格测试。
我非常乐观的是,第二代益生菌将会从肠道中分离出,这样它们就能够真正在肠道中定植,这与目前大多数益生菌不同。 这些将是微生物的混合物,而不是单个菌株,以帮助建立新的微生物生态系统。
我相信在不久的将来,人们会发现可以增强癌症免疫疗法的混合物。也就是说,希望我们能够超越微生物而进入影响免疫疗法的分子中,因为这样我们就可以将这些微生物产品用作常规药物,而我们的卫生系统就是为这些药物而设计的。

我们对此必须非常非常谨慎。 人们普遍认为益生菌对人体有好处,但这是相对的,并取决于多种因素,包括本地宿主微生物群,任何潜在的医疗状况和饮食。 “益生菌”一词指的是活微生物,如果给予足够的量,可以为宿主带来健康益处。
但是,没有足够的科学证据证明疗效,也没有任何监管机构(例如美国食品和药物管理局)批准将其用于任何治疗用途; 相反,这些被许可用作膳食补充剂。 例如,非处方益生菌被广泛用于帮助抗生素治疗后恢复肠道菌群。需要注意的是,最近的研究表明,它们可能会产生相反的效果,这突出了仔细研究的必要性。
通常,使用非处方益生菌与减轻胃肠道疾病有关,在许多情况下,这与它们的抗炎特性有关。重要的是,尽管研究仍在进行中,但尚未适当评估其在癌症免疫治疗中的作用。
这并不意味着新鉴定的益生菌可能不会对癌症免疫治疗有所帮助,而这些益生菌应该包含具有特定健康效应的安全性和有效性的明确证据的特定菌株(单个或联合体)。

传统的益生菌已被用于通过发酵保存食品。 因此,它们还没有出于特定目的从人的微生物群中合理地分离出来,它们也不一定是正常微生物群的一部分。
此外,单一菌株的接种不能恢复正常微生物群中通常可见的多样性,并且缺乏强有力,长期益生菌植入的证据。
因此,我认为常规的益生菌在临床上至多具有中等程度的作用。 从健康的供体或治疗反应者中分离出来的合理设计的微生物联合体可能会更有效。

传统的商业益生菌不太可能使接受免疫治疗的患者受益,因为它们会改变微生物群的组成和多样性,并可能导致营养不良,因此可能产生负面影响。
但是,某些益生菌含有细菌(例如,双歧杆菌种),它们已在实验动物模型和癌症患者中均显示出,以增强对免疫疗法的反应。
目前正在使用单一细菌菌株或已被初步确定为促进免疫疗法的细菌组合进行临床试验。一旦我们清楚地确定了有利的细菌种类并定义了它们的作用机制,它们在患者体内的定殖能力以及它们如何与现有微生物群落一起起作用,这种细菌疗法可能具有提高免疫疗法应答率的巨大潜力。

益生菌一词范围很广,是指被宣传为食用时具有健康益处的微生物。目前的非处方益生菌还没有在癌症患者中进行广泛的测试,甚至有证据表明报告服用非处方益生菌的患者在癌症免疫治疗后可能会有较少的多样性微生物群和更糟糕的结果。
临床前模型中的证据表明,大肠癌模型中非处方益生菌的施用与肿瘤发生的增加有关。因此,应警告癌症患者,非处方益生菌可能无益,在癌症治疗中应避免使用。
但是,我们需要把握机会,开发基于新见识而合理设计的下一代益生菌,但要进行广泛的测试以确保功效并证实其主张。

尽管益生菌通常被认为是一种营养素,可以补偿在使用抗生素、化疗和放疗治疗后出现的腹泻副作用,但许多益生菌菌株和制剂的结果是相互矛盾的。
临床前和回顾性研究似乎表明,使用益生菌可能会降低肠道菌群的α多样性,从而损害免疫检查点阻断的全面功效。
因此,我们仍然需要评估单个微生物菌株如何在肠道中定居,并且需要研究它们对宿主代谢和免疫系统的影响。
在任何广泛和无针对性的使用之前,这些研究需要在来自不同类型癌症患者的本土微生物群的背景下进行。

我认为动物模型和人类都有足够的初步证据表明微生物可以影响癌症的免疫治疗。 但是,还有许多其他问题。
这些包括以下内容。
涉及的机制是什么?
涉及哪些微生物?
微生物产生什么分子来影响免疫治疗?
我们能否开发出一种可靠的微生物群筛查方法,以识别哪些人会从生物素中受益,哪些人会在没有生物素的情况下做出反应?
最后,所有这些信息是否可以用来开发(1)活的细菌混合物和(2)微生物分子,它们可以可靠地用于接受免疫治疗的患者的临床。

对我来说,关键问题是这一切如何运作?
一旦我们对系统学有了更深入的了解,就可以解决一些争议(例如,不同的研究人员发现与治疗结果相关的不同细菌类群,以及有关抗生素对反应率的正面和负面影响的报道)。
另一个重要的问题是它会随着时间变化吗? 微生物群落,宿主免疫系统和癌细胞之间发生的三向相互作用不仅复杂,而且高度动态。这可能意味着在疾病/治疗的整个过程中将需要采用不同的针对微生物菌群的方法。
近年来,我们获得了大量知识; 然而,就我们对这些过程的理解而言,我们可能只是在摸索表面。

大量研究各自确定了不同的细菌,这些细菌可促进癌症患者免疫检查站阻断的功效。
关键问题是这些细菌是否通过保守的功能途径增强了对免疫检查点的阻断,还是通过简单的机制简单地促进了相同的表型。
如果保守的代谢途径起作用,则可以将功能上多余的一组细菌一起使用,以克服将单个细菌群落治疗性移植到不同患者微生物群上的可变成功率。

展望未来,我认为我们的主要目标如下:
首先,我们需要通过消除混杂因素(如早期接触,生活方式和地理差异)来预测有利于抗肿瘤免疫的微生物群组成。
其次,我们需要确定特定的分类单元和/或调节抗肿瘤免疫力的微生物群的代谢途径和产物。
第三,我们必须表征宿主免疫系统和微生物群之间发生串扰的潜在机制。
第四,我们应该将这些分析扩展到理解除肠道以外的其他屏障组织的微生物群,确定与肿瘤相关的微生物群的作用,并阐明该微生物群的其他非细菌成分的作用,如真菌、病毒、噬菌体和原生动物。
最后,一旦确定了单个菌株或联合体,我们需要评估它们在患者体内的定殖能力,以及在现有微生物群落生态中以功能状态共存的能力。

我认为,需要考虑三个关键问题:
首先,什么是“完美”的肠道菌群? 同时,这种促进癌症免疫治疗反应的完美微生物群是否也有助于预防癌症的发生呢?它甚至能增强对疫苗的反应并促进整体免疫健康吗?
第二,肿瘤微生物组的影响是什么? 我们能把这一点作为癌症预防的目标吗?
最后,我们如何协调微生物组学研究?

我认为在“免疫肿瘤学”领域中存在四个主要问题:
首先,我们如何在临床上定义肠道失调并开发适当的工具以进行准确诊断?
第二,肠道失调是晚期癌症的原因和/或后果吗? 如果答案是肯定的,那么这是对任何一种癌症还是只对特定类型的癌症有效?
第三,肠道失调是否会导致对癌症免疫疗法的“原发性耐药”?
最后,我们能通过干预癌症相关的肠道失调来治愈这个问题吗?如果答案是肯定的,那么这种治愈是短暂的还是会产生长期影响?
微生物群由共生细菌和生活在宿主上皮屏障上的其它微生物组成,影响着大量的生理功能,包括维持屏障的内稳态,调节新陈代谢、造血作用、神经系统、炎症、免疫力等。微生物群也参与了癌症的发生、进展和转移。
不断积累的研究表明,微生物群,尤其是肠道微生物群能够调节机体对癌症疗法的响应以及对毒副作用的敏感性。一旦确定了每一种临床状况所对应的最有利的微生物群组成,如何修改患者的微生物群将是下一步挑战。
科学家们普遍认为在癌症和其它疾病中靶向微生物群可能会成为精准医疗和个性化医疗的下一个前沿方向之一,肠道微生物群的弹性、稳定性以及它对生理、病理以及环境变化的响应性使得我们能够利用微生物组的组成作为一种生物标志物、一种诊断工具或者一种治疗靶标。尽管通过靶向微生物组实现治疗干预这一研究领域还处在初始阶段,但一些途径已经证明了其可能性。
相关阅读:
参考文献:
BMJ 2020 07 14;370:m2795. Epub 2020 Jul 14.
Covid-19: disinfectants and sanitisers are changing microbiomes. Department of Veterinary Microbiology, Lajpat Rai University of Veterinary and Animal Sciences, Hisar-125004, India. doi.org/10.1136/bmj.m2795
Finlay, B. B., Goldszmid, R., Honda, K., Trinchieri, G., Wargo, J., & Zitvogel, L. (2020). Can we harness the microbiota to enhance the efficacy of cancer immunotherapy? Nature Reviews Immunology. doi:10.1038/s41577-020-0374-6

现代生活方式通过部分改变微生物组而增加了人类患慢性病的风险。但是,对于少数族群而言,生活方式的改变将对健康产生怎样的影响,这方面的研究并不多。近日,爱尔兰国立科克大学Fergus Shanahan及其课题组的最新研究:Microbiome and health implications for ethnic minorities after enforced lifestyle changes 揭示了由生活方式改变引起的微生物组变化对少数族群健康的影响。这一研究成果发表《Nature Medicine》上。

生活方式会影响早期的微生物群,这时微生物群已经形成但免疫系统还未成熟。此外,通过对遗传相似的人群和居住在同一地理位置的不同种族的群体的研究,已经将生活方式的影响与遗传和地理因素分开。
爱尔兰旅行者是一个不同种族的亚群体,他们因为2002年的一项立法而结束了游牧生活,这改变了他们的生活方式(从游牧到定居)。
肠道微生物群的比较宏基因组学研究显示,爱尔兰旅行者群体保留了一个与非工业化社会人群相似的微生物群。他们的微生物群与非饮食因素有关,并且和与微生物相关的代谢性疾病的风险成比例的相关。研究结果表明,当少数族群的生活方式被迫改变时,会影响与微生物相关的公共卫生问题。
爱尔兰旅行者占爱尔兰人口的近1%,它们在200到1200年前从定居的社区中分离出来,在2017年获得了单独的种族身份,以表彰他们独特的文化、语言、历史和生活方式。
他们与欧洲的其他游牧民族(分别被称为罗马尼人和罗马人)不同,但在基因上与非游牧定居的爱尔兰人非常相似。
社会经济因素被认为是少数民族健康不平等的主要决定因素。
爱尔兰旅行者的预期寿命自1987年以来一直停滞不前,而非旅行者爱尔兰人的预期寿命却大幅上升。
爱尔兰旅行者的死亡率高于普通人群,这一高死亡率是由多种因素造成的,包括事故、自杀、创伤和心血管疾病、呼吸和癌症死亡。
研究对象:
118名成年爱尔兰旅行者,平均年龄39岁((±13岁,sd)。其中男性53人(44.9%),女性65人(55.1%)。
所有参与者都居住在科克市中心30公里半径内的5个地点之一。都进行了饮食评估(通过半定量的155项食物频率调查问卷(FFQ)评估)、世界卫生组织幸福指数(WHO-5)调差问卷填写、个人病史和家族史调查。
在研究开始后的1个月内,没有人服用抗生素,也没有人服用泻药、皮质类固醇、消炎药或抗凝剂。
研究人群的特征:
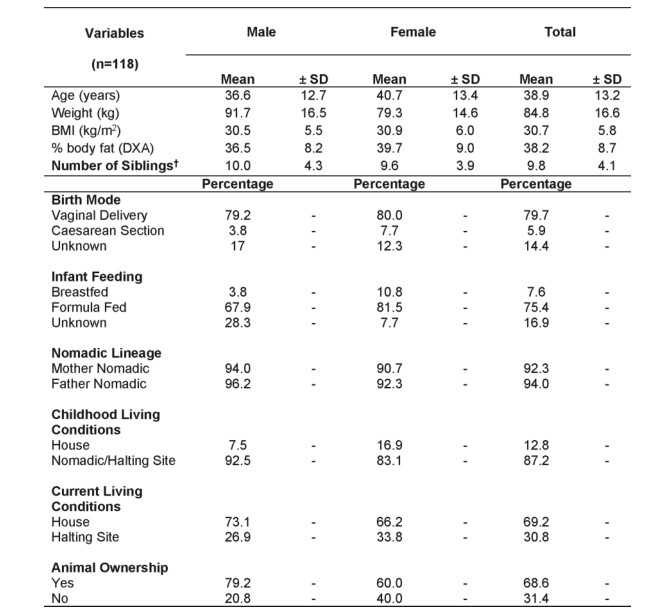
分组:
irish Travellers:爱尔兰旅行者
Elderly irish community:居住在社区住宅的爱尔兰老年人,非旅行者,137人
Elderly irish long-stay:长期接受家庭护理的爱尔兰老年人,非旅行者,53人
Young irish:青年的爱尔兰人,非旅行者,141人
international controls/ Global:从鸟枪法宏基因组公共数据集中选取了3481名成人(≥20岁)的宏基因组图谱,作为无疾病对照。这些数据覆盖五大洲的15个不同民族。包括了non-industrialized societies(非工业化群体)和“industrialized”(工业化群体)
non-industrialized-like:爱尔兰旅行者亚群体,下文称类非工业化群体
industrialized-like:爱尔兰旅行者亚群体,下文称类工业化群体
宏基因组数据分析:
采集每个参与者的粪便,提取DNA。用于Illumina NextSeq平台的宏基因组鸟枪法测序。数据预处理后,使用MetaPhlan2进行物种分析,HUMANN2进行基因功能分析。
R3.5.3进行数据的统计分析。分组间的比较使用Mann-Whitney U检验,对于组内比较,采用Mann-Whitney检验,并将丰度差异(Benjamini–Hochberg, P< 0.1) 确定为一组相对于另一组的增加或减少。I-graph包用于网络的分析,Cytoscape可视化。
1. 利用PCoA分析不同群体之间的微生物组成差异。发现爱尔兰旅行者有不同于其他队列(非工业化群体和工业化群体)的独特的微生物组成。
a. 爱尔兰旅行者群体与老年和年轻的非旅行者爱尔兰人群体间的PCoA分析。可以看见两者之间有明显的分离。(PERMANOV A F model = 4.86,R2 = 0.01, P = 0.001)
b. 爱尔兰旅行者与其他来自全球的某些地区的宏基因组元数据(international controls)之间的PCoA分析。在PCoA的第二条轴上能明显看到非工业化和工业化群体之间的分离。(ANOVA,PCoA1:P<2.2*10^-16, PCoA2:P<0.003)
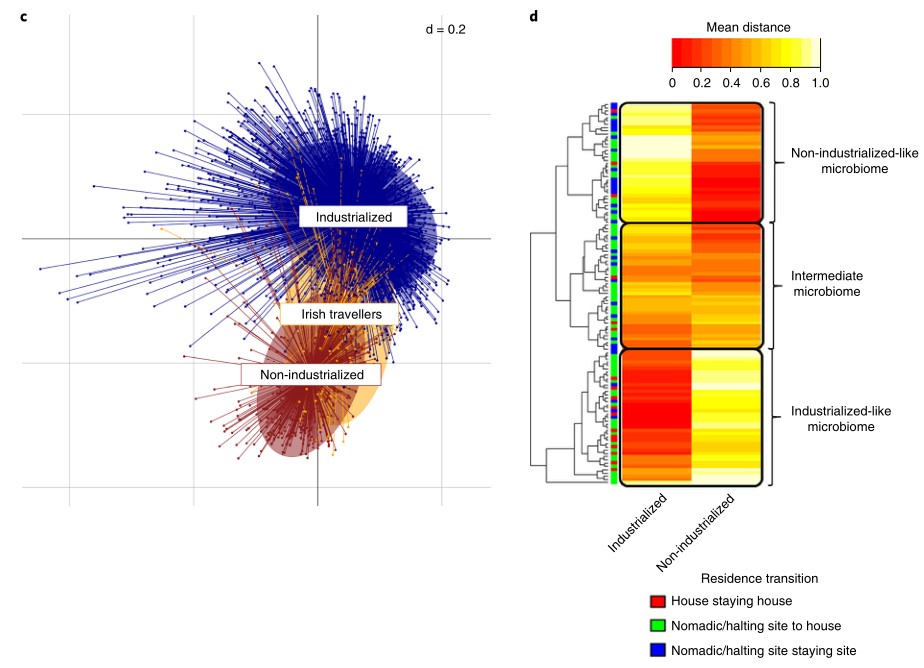
c. PCoA图。微生物群α多样性的两个指数(物种多样性和丰富度)表明,爱尔兰旅行者在工业化和非工业化群体之间处于中间位置。
d. 为了探索c图中处于中间位置的微生物群,研究人员在PCoA坐标上采用了一种基于距离的方法,通过计算每个参与者各自的肠道微生物群与工业化和非工业化人群的中位距离,确定了“中间”(Intermediate)的微生物群。
2. 在旅行者亚群体中确定了类非工业化、类工业化和“中间”的微生物群落
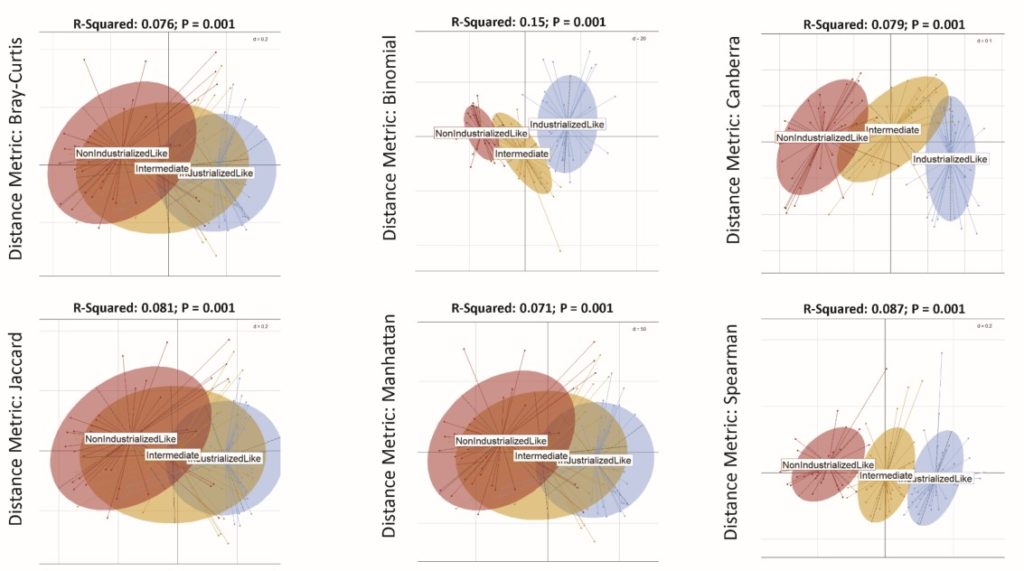
通过计算6种不同的相异距离度量,Bray-Curtis,
Jaccard, Binomial, Manhattan, Canberra 和Spearman,进行PCoA分析,在每个分析情况下都明显出现了分离,红色为类非工业化,暗黄色为“中间”,蓝色为类工业化。
3. 通过对爱尔兰旅行者的饮食模式与其它爱尔兰人队列或爱尔兰旅行者亚群之间的比较研究。结论表示爱尔兰旅行者组之间的饮食差异很小,主要的饮食差异在旅行者和非旅行者爱尔兰人之间,不在旅行者的亚群中。
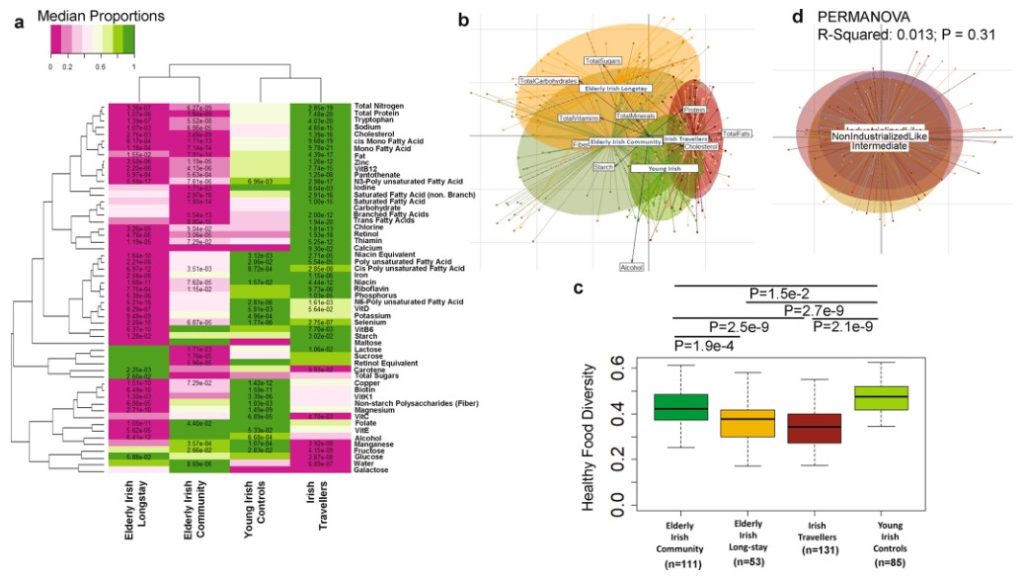
a.各种常量营养素和微量营养素的平均消耗频率的中位数的热图,横轴为不同队列,纵轴为各营养素,单元格内的值为校正后P值,仅显示P<0.1的情况。
爱尔兰旅行者饮食的主要标志是高脂肪(各种形式)和蛋白质,以及相对较高的视黄醇、氯化物、泛酸、色氨酸、碘、维生素B12和锌。
爱尔兰旅行者的纤维摄入量(由非淀粉多糖反映)低于所有其他组,除了Elderly irish long-stay组。
b.PCoA图展现了不同队列之间基于常量营养素的膳食概况和指定营养素与各队列的关联。爱尔兰旅行者用红色表示。
c. 箱线图显示爱尔兰各队列的健康食品多样性(HFD)的指数值。爱尔兰旅行者的饮食质量明显低于其他组。
d.对爱尔兰旅行者中三个亚群(非工业化组,中间组【Intermediate】,工业化组)的饮食概况进行了PERMANOVA分析,结果显示三组之间的饮食相似。
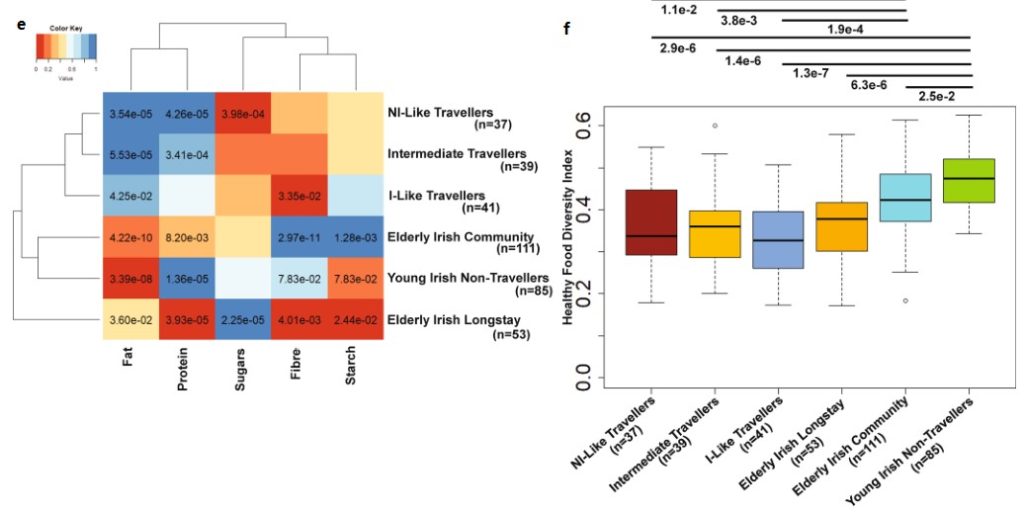
e、f. 热图显示了爱尔兰旅行者的三个亚群(*Travellers)与另外三个非爱尔兰旅行者队列中五大主要食品的平均消费水平。从图中侧边的聚类看,爱尔兰旅行者亚群之间的差异较小。箱线图指示健康食品多样性指数。
4.利用网络分析,探索非工业化群体和工业化群体的微生物群之间的关联性。
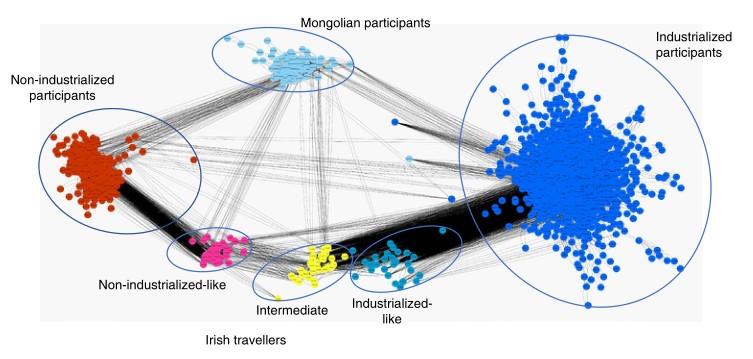
使用了international controls数据集(3481个样本),揭示了两个截然不同的群体(工业化和非工业化),其中蒙古人(Mongolian)和旅行者这两个群体都与工业化和非工业化群体有联系。
在旅行者亚群中观察到了不同的关联性,其中类非工业化群体主要与非工业化群体、“中间”和类工业化群体有关联,而“中间”和类工业化群体又与工业化群体有关联。
4. 爱尔兰旅行者儿童期到成年期生活条件变化与微生物群落的关系,论证了生活条件的变化与微生物群的差异有关。
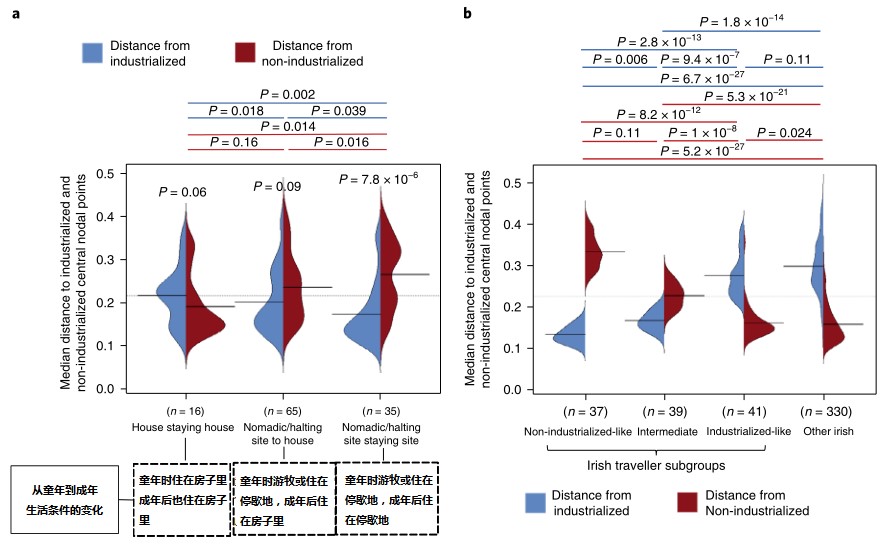
计算了工业化与非工业化微生物群的PCoA坐标与爱尔兰旅行者队列中特定居住地点组(a图横轴坐标名称所示)的样本之间的欧氏距离。然后计算平均距离。
Bean图显示了给定变量在y轴上每个值上的平滑密度分布。对于每个Bean图,在给定值y处的图的宽度指示具有该y值的变量的频率。
Bean图中的P值表示在特定居住地点组内的非工业化和工业化群体之间的中位数距离的成对比较的重要性。Bean图上的P值(颜色分别对应非工业化和工业化群体)表示特定居住地点组间的两两比较的重要性。
从a图中得出结论,在工业化群体中,对比一直住在房子里的旅行者,童年时就住在停歇地的旅行者的微生物群变化更大。
b. 爱尔兰旅行者亚群与非旅行者爱尔兰群体微生物群的比较。Bean图显示了3个爱尔兰旅行者亚群和非旅行者爱尔兰人的微生物群与非工业化和工业化队列的微生物群的平均距离的分布。
5. 与旅行者保留祖先或非工业化微生物群有关的主要的生活方式因素是住房条件、兄弟姐妹数量和动物拥有率。饮食并不是将旅行者的微生物群分成类非工业化的和类工业化的两个组的决定因素。
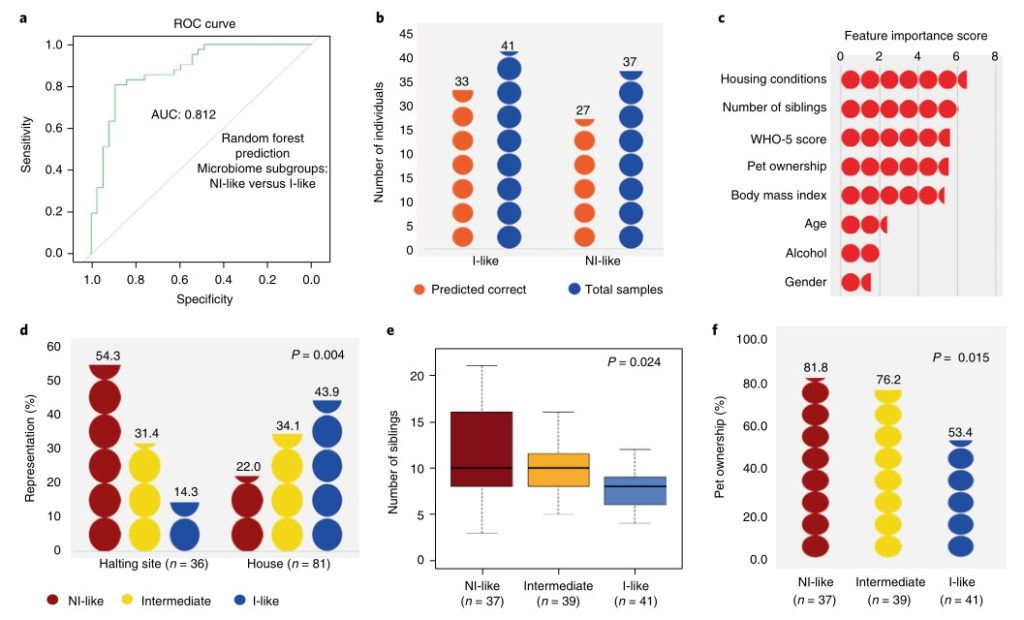
a.b. 曲线下面积(AUC)为0.82,总体准确率>77%的旅行者中,有8个因素可以解释非工业化的微生物群与工业化的微生物群
c. 有3个因素使爱尔兰旅行者有别于其他爱尔兰人:住房条件(Housing conditions)、兄弟姐妹数量(Number of siblings)和动物所有权(animal ownership)。
d.e.f 在住房条件为停歇地的群体中,保留非工业化类微生物群的旅行者的个体比例明显更高(Fisher’s
exact test, P = 0.004),而这些保留非工业化类微生物群的群体都存在显著的兄弟姐妹数量增加和拥有动物所有权的个体比例增高。
6. 爱尔兰旅行者微生物群的组成变化与基因功能代谢变化有关,而这些功能变化与工业化(西方)社会的慢性病风险的增加有关,如SCFA途径减少、TMA增多。
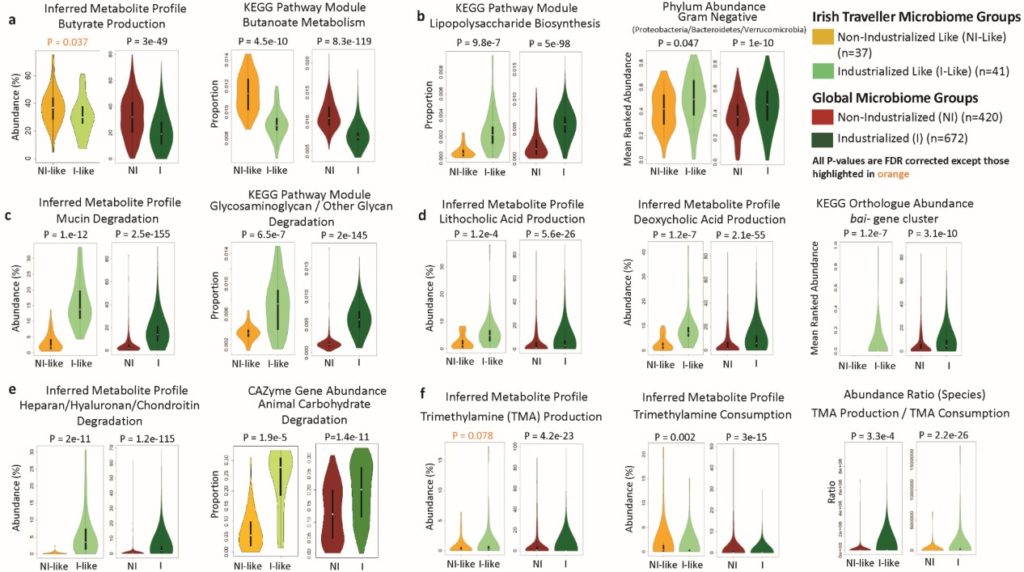
类非工业化(NI-Like)和类工业化(I-Like)旅行者之间的微生物群功能差异与全球队列(Global)中的微生物群功能差异情况,结果证明这两者是相似的。通过观察功能预测和已知的种水平的功能分析之间的一致性,用来识别具有差异的代谢途径或功能。
图中abcdef分别为丁酸盐、脂多糖生物合成、粘蛋白降解、次级疏水胆汁酸生成、动物碳水化合物、三甲胺转化率。
左边为产物的生产丰度,右边为与该产物相关的KEGG代谢基因的丰度。
生产丰度的变化趋势也反映在与其相关的功能代谢基因的丰富度上。所有P值都经过FDR校正,出了橙色高亮的P值。
TMA被认为是动脉粥样硬化、脂肪性肝病和2型糖尿病的危险因素。血清TMA水平受包括肠道微生物在内的因素调控。
爱尔兰旅行者的微生物群的特征表现为缺乏在非工业化群体中典型丰富的物种,同时又表现出对工业化社会典型丰富的物种的较低的、可变的获取。
旅行者微生物群差异的主要驱动因素是非饮食的生活方式和环境因素。虽然旅行者的饮食是西式的,但他们的生活条件与非工业化社会相似。
与工业化相关的微生物群的功能变化可能使旅行者更容易患上慢性病,包括次级胆汁酸的生成增加,LPS的生物合成增加,TMA转化率增加。产甲烷古菌的丧失,这也是一个特征,预计会导致氢水平和氧化还原通量的改变。
研究人员认为要预防西方的慢性病,需要了解肠道微生物群如何随着现代化的发展而变化。由于环境是宿主与微生物相互作用的一个深刻决定因素,因此在现代工业化社会中拥有一个非工业化的类似微生物群落的这一意义需要引起大家的关注。
相关阅读:
随着高通量技术的发展,我们对微生物的认识逐步深入,微生物在临床以及科研上的应用越来越多,而微生物采样的正确与否直接关系到微生物数据的准确性。
因此,微生物的采样方法就显得格外重要。

首先,我们需要在取样的过程中,注意以下几个方面:
(1) 所有实验用到的取样用具必须是经过灭菌的(如样品袋、取样器、采血管、试管、铲子、匙、刀类工具等)。
(2) 收集样品信息,取样前或取样后应在盛装样品的容器或样品袋上立即贴上标签,每件样品必须标记清楚(例如品名、来源、数量、取样地点、取样人及取样日期等)。
(3) 请尽量将样本分装 3-5 份备份;建议每组取 6 个及以上重复较为合适,至少 3 个重复。
(4) 用到谷禾取样管的实验样品,可以常温运输;如果不用取样管,则必须低温-80℃保存,保存期间切忌反复冻融;低温寄送时请将样品置于泡沫箱中,用干冰寄送。
然而样品种类繁多,不同的样品取样有不同的方案,下面详细介绍每类样品对应的取样方案。
(1)去除表面浮土、凋落物等,根据土壤情况,选择是否过2mm筛网,每个样品从3个及以上采样点采集并混合而成,建议将样本分装 3-5 份备份;
注:采样时需注意每个采样点的取土深度及采样量应均匀一致,土样上层与下层的比例要相同。
(2)保存于无菌离心管中,置于泡沫箱(冰袋或干冰)中,0°C以下运回实验室;
(3)如果条件不允许立即提DNA,可将样品置于-80°C冰箱中保存,并通过干冰寄送至公司后提取样品总 DNA。

根际,是指受植物根系活动影响,在物理、化学和生物学性质上不同于土体的那部分微域环境。部分研究是针对土壤微生物与植物之间的相互关系,因此需要对根际土进行取样。
(1) 采集植株,去除根周较松散的土,仍附着在根系表面的视为根际土。将植物置于装有冰袋或干冰的保温箱中,避光条件,0℃以下运回实验室;
(2)准备无菌容器,将植物根系部分置于缓冲液(PBS或生理盐水)中震荡,震荡时间和强度依据样本实际情况调整,洗下根际土;
(3)将洗涤液进行高速离心,收集土壤沉淀,若沉淀较少,也可过滤膜,同一样方取样的样本进行多点等量混合;
(4) 将样本分装至离心管中,密封,标记样本信息后液氮速冻,置于-80℃冰箱保存,干冰寄送。
采集水底沉积物5~10 g(具体深度依据实验目的确定),保存于冻存管中,标记好样本信息后液氮速冻或置于装有干冰的泡沫盒中,黑暗环境下,尽快运回实验室,-80℃冰箱保存,干冰寄送。
多点取样并等量均匀混合至约10 ml的悬浮污泥样本(5~10 g沉降物),保存至冻存管中,标记好样本信息后液氮速冻或置于装有干冰的泡沫盒中,黑暗环境下,尽快运回实验室,-80℃冰箱保存,干冰寄送。
(1) 采集水样应注意无菌操作,以防止杂菌混入。
(2) 根据实验设置,选取适当孔径的滤膜,使用水体抽滤机抽滤 1~100 L 水体(不同水质间差异极大),或抽滤至滤膜上可见明显覆盖物。
对于浑浊水体,建议过滤前静置分离悬浮颗粒物,先用大孔径滤膜过滤一遍,再用小孔径滤膜过滤。
(3) 标记好样本信息后液氮速冻或置于装有干冰的泡沫盒中,黑暗环境下,尽快运回实验室,-80℃冰箱保存,干冰寄送。
注:根据水体中微生物含量不同,取样量有所不同:污水等菌群丰富的水体用量一般为200-1000 ml,湖泊、河流等自然水体一般为 1-2 L,自来水等菌群含量较低的水体则一般为 1-5 L,海洋水体随着地理位置、季节和取样深度浮动较大。
尽可能取原包装,检验前不要开封,以防污染。
大块冷冻食品,应从几个不同部位用灭菌工具取样,使样品具有充分的代表性;
在将样品送达实验室前,要始终保持样品处于冷冻状态。样品一旦融化,不能再冻,保持冷却即可。
通常情况下,液态食品较容易获得代表性样品。
盛放在大罐中的液态食品(如牛奶、奶昔、饮料等),取样时可连续或间歇搅拌;
盛放在小容器的液态食品,可在取样前将液体上下颠倒,使其完全混匀。较大的样品要放在已灭菌的容器中送往实验室。实验室在取样检测之前应将液体再彻底混匀一次。
对于牛奶、葡萄酒、植物油等,常采用虹吸法(或用长形吸管)按不同深度分层取样,并混匀。如样品粘稠或含有固体悬浮物或不均匀液体应充分搅匀后,方可取样。
用无菌操作开启包装,此类样品无法用吸管吸取,可用无菌勺子从不同部位挖取样品,放入无菌盛样容器。若样品是粉末,应边取边混合。
面粉或奶粉等易于混匀的食品,其成品质量均匀、稳定,可以抽取小样品(如100g)检测。但散装样品必须从多个点取大样,且每个样品都要单独处理,在检测前要彻底混匀,并从中取一份样品进行检测。
肉类、鱼类或类似的食品既要在表皮取样又要在深层取样。深层取样时要小心不要被表面污染,同时避免使之暴露在空气当中。
有些食品,如鲜肉或熟肉可用灭菌的解剖刀和钳子取样;冷冻食品可在未解冻的状态下可用锯子、木钻或电钻(一般斜角钻入)等获取深层样品;全蛋粉等粉末状样品取样时,可用灭菌的取样器斜角插入箱底,样品填满取样器后提出箱外,再用灭菌小勺从上、中、下部位取样。

注:
· 注意不要使固体样品过度潮湿,以防食品中固有的细菌滋生。
· 食品如为非冷藏易腐食品,应迅速将所取样品冷却至0℃~4℃;若样品是冷冻的,应保持冷冻状态,低温寄送。
植物内生细菌是植物微生态系统中的重要组成部分,直接对植物组织样本进行测序分析,可以研究植物内生细菌或真菌的多样性。
(1) 根据研究对象选取新鲜植物组织(如果是植物根部,需去除根表粘附土壤);0℃以下运回实验室;
(2) 根据组织大小剪成小块或小段(至少两边不超过0.5cm),在无菌工作台内,用无菌水对植物组织进行洗涤30s;
(3) 表面无菌化处理:
第一次消毒:75%无水乙醇浸泡1min;第二次消毒:2%次氯酸钠处理3min,转入75%无水乙醇浸泡30s,接着用无菌水漂洗3次;
(4) 进行核酸提取实验或液氮速冻,放置-80℃冰箱保存。干冰寄送。
(1) 用无菌手术刀挖取(或无菌镊子挤出,也可用无菌生理盐水冲洗出)肠道内容物;
(2) 直接用谷禾取样盒里的棉签沾取约黄豆大小的肠道内容物,将沾取内容物的棉签浸入取样管液体搅拌洗脱内容物进入取样管保存液内;搅拌约30-40s,然后丢掉棉签,将管子盖子拧好,快速摇晃取样管至少30秒;
(3) 观察到管内液体带有明显肠道内容物颜色即取样成功,常温寄送。
(1) 使用前请洗净双手,防止杂菌干扰。将取样管取出,拧开盖子。
(2) 用取样棉签沾取少量新鲜粪便(需要量为黄豆一绿豆大小);如粪便较干,可先将棉签在取样管中浸湿后沾取;
(3) 将沾取粪便的棉签浸入取样管液体搅拌洗脱粪便进入取样管保存液内,搅拌约30-40s,然后丢掉棉拭子,将管子盖子拧好,快速摇晃取样管至少30秒;
(4) 观察到管内液体带有明显粪便颜色即取样成功,常温寄送。

注:
· 如3天内使用过抗生素类、质子泵类胃药、阿片类精神药物请停药3天后再检测。
· 感冒、腹泻或其他症状期间不影响取样,拉稀或稀便可以用棉签反复沾取粪便至取样管。
· 如长期便秘无排便可使用开塞露等辅助手段获取粪便样本。
·取样无时间和饮食限制,但是取样前最好正常饮食。
· 完成取样后样本可常温有效存储一周,为保证检测时效请完成取样后尽快送回。
(1) 采样前30分钟禁食、禁烟酒等;
(2) 准备一杯清水(约150ml),将清水含入口中,充分洗漱口腔约10秒,重复2-3次。
(3) 将拭子伸入口腔,使拭子头充分接触左侧口腔内壁,上下牙床处粘膜,用适当力度上下擦动并旋转拭子15-20圈。用同样方法在右侧口腔内壁及上下牙床粘膜处进行另一根拭子的采样。
(4) 将拭子置入取样管保存液内,搅拌约30-40s,然后丢掉拭子,将管子盖子拧好,快速摇晃取样管至少30秒;常温寄送。
(1) 采样前30分钟禁食、禁烟酒等;
(2) 准备一杯清水(约150ml),将清水含入口中,充分洗漱口腔约10秒,重复2-3次。
(3) 将采集对象舌头外拉,使悬雍垂尽可能向外牵引。同时,将拭子越过舌根到咽后壁或者悬雍垂后侧,反复擦拭15次以上,避免接触舌、口腔粘膜和唾液。
(4) 将拭子置入取样管保存液内,搅拌约30-40s,然后丢掉拭子,将管子盖子拧好,快速摇晃取样管至少30秒;常温寄送。
(1) 采样前30分钟禁食、禁烟酒等;
(2) 准备一杯清水(约150ml),将清水含入口中,充分洗漱口腔约10秒,重复2-3次。
(3) 用舌尖抵住上颚或下颚齿根以富集唾液,向取样管中轻轻吐入唾液,直至液体唾液(非气泡)达到2ml 以上。搅拌约30-40s,快速摇晃取样管至少30秒;常温寄送。
(1) 采样前30分钟禁食、禁烟酒等;
(2) 准备一杯清水(约150ml),将清水含入口中,充分洗漱口腔约10秒,重复2-3次。
(3) 用棉卷隔湿所采集的牙齿,再以无菌生理盐水冲洗牙面,用无菌挖匙或探针采集窝沟菌斑或唇颊面菌斑;以牙线置于两邻牙之间,紧贴牙面采集邻面菌斑。采集后放入含有保存液的取样管内,搅拌约30-40s,快速摇晃取样管至少30秒;常温寄送。
一、样本筛选
如果受试者存在以下情况,则需剔除:
(1) 采样前7天,脸部,头皮,脖子,胳膊,前臂或者手使用过抗生素或者萜类物质者;
(2) 脸部,胸部,背部或者肩膀有痤疮者;
(3) 头皮,脸,脖子,手臂,前臂或者手上有水泡,脓疱,疮,脓肿,腐烂或者溃疡者;
(4) 在取样部位处有一处水泡,脓疱,疮,脓肿,腐烂,溃疡,藓,伤口,裂纹或者色素及粉红色的皮肤修复区者;
(5) 身上有多处粉色或红色皮肤区域者(暗指牛皮癣或者湿疹患者);
(6) 手掌或者脚掌皮肤增厚或者有裂纹者;
(7) 两周之内,每天使用去屑洗发水都不能清理干净头皮屑者;
(8) 身体一处或者多处有散发型皮疹者。
二、收集方法
(1) 将拭子在无菌生理盐水或清水中浸湿,在实验目的区域擦拭10s;
(2) 样本采集后立即将拭子置入取样管保存液内,搅拌约30-40s,然后丢掉拭子,将管子盖子拧好,快速摇晃取样管至少30秒;常温寄送。
(1) 取相应部位组织样品,于无菌操作台上解剖,用灭菌剪刀、解剖刀切取指甲盖大小左右组织块;
(2) 将组织块放入冻存管中并盖紧盖子,标记好样本信息。样品液氮速冻后放入-80℃冰箱保存。干冰寄送。
(1) 口咽部进行雾化利多卡因麻醉后,滴注120-300mL无菌生理盐水进行BAL液体标本的收集。取样后离心,去掉上清,取沉淀物。
(2) 用液氮速冻-80℃保存,干冰寄送;或取沉淀物洗脱置取样管,搅拌约30-40s,快速摇晃取样管至少30秒,常温寄送。
(1) 收集肠道灌洗液,过膜;或者将肠道灌洗液离心,弃上清,取沉淀物。
(2) 用液氮速冻-80℃保存,干冰寄送;或取沉淀物洗脱置取样管,搅拌约30-40s,快速摇晃取样管至少30秒,常温寄送。
(1) 采用无菌棉拭子,沿着阴道口、阴道中部和后穹窿内腔壁旋转拭子做圆周运动数次。
(2) 擦拭后立即将拭子浸入取样管中的保存液中,搅拌约30-40s,快速摇晃取样管至少30秒,常温寄送。
(1) 成人10-20ml(儿童1-3ml)全血收集到真空采集管(已加入抗凝剂),上下颠倒两次。
(2) 标记信息,低温保存,干冰寄送。
(1) 洗净双手,母乳期用无菌水擦拭乳头,取乳汁5-10ml 3管(除掉第一管)。
(2) 低温(-20℃)保存,干冰寄送;或者用拭子沾取乳汁后,立即将拭子浸入取样管中的保存液中,搅拌约30-40s,快速摇晃取样管至少30秒,常温寄送。
(1) 收集腹水10-20ml,离心,弃上清,取沉淀物。
(2) 用液氮速冻-80℃保存,干冰寄送;或取沉淀物洗脱置我们的取样管,搅拌约30-40s,快速摇晃取样管至少30秒,常温寄送。
(1) 胆汁样本由十二指肠引流法或胆囊穿刺法或手术中留取胆汁,采集胆汁量为5ml,置无菌管内,离心,弃上清,取沉淀物。
(2) 用液氮速冻-80℃保存,干冰寄送;或取沉淀物洗脱置我们的取样管,搅拌约30-40s,快速摇晃取样管至少30秒,常温寄送。
(1) 采集中段尿液40 ml左右。(中段尿:在排尿过程中,弃去前、后时段排出的尿液,以无菌容器收集);
(2) 一般留取清晨第一次尿液,最好憋尿6小时。住院患者可以直接取晨尿,患者将晨尿的中段尿收集至无菌杯中40 ml为宜;门诊患者或健康人等无尿意者,可以适量饮水,在有尿意时尽可能的多憋一段时间,将尿液的中段尿收集至无菌尿杯中40 ml为宜;
(3) 离心,去上清,加入我们的液体保存,常温寄送。
注:
· 应在抗生素使用前或停用抗生素药5天后留尿液标本。
· 对于小便轻度失禁患者或者憋尿时间过长患者,对中段尿比较难控制的情况下,我们可以告知患者在排尿过程中3 s后采集尿液。
·如果是病危或昏迷病人,可由相关医务人员用导尿方法采集尿液,此操作过程中要严格执行无菌操作,避免因导尿不慎而导致的泌尿系统感染。也可以采用经下腹前壁膀胱穿刺取尿培养的方法。
以上是各类样品的取样方法,可供参考。
也许有人问,是否一定要用我们的取样管保存液,能不能用生理盐水或者别的来替代。下面是我们进行的关于保存液的测试:
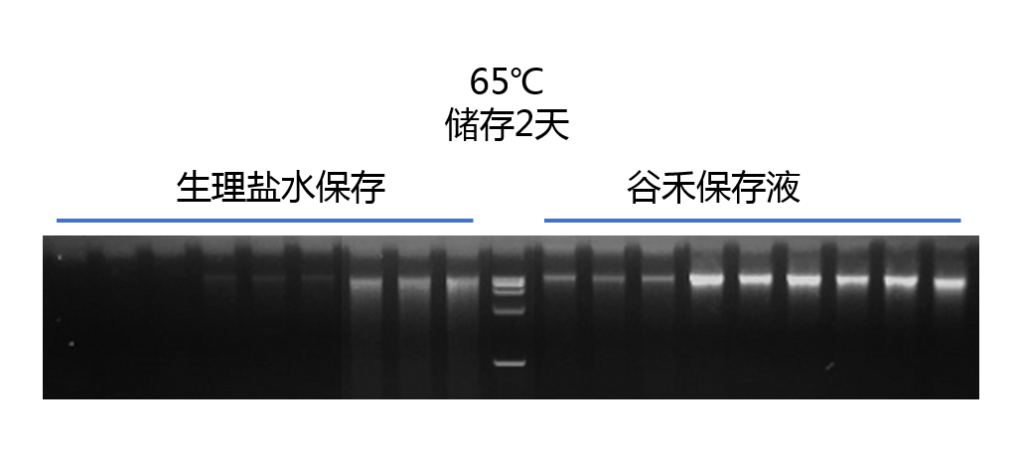
经测试,使用谷禾取样管保存液,能在-80℃ ~ 65℃ 的条件下保持DNA完整性,室温有效储存长达30天,尽可能保证与新鲜样本具有一致的菌群构成特征。
要想得到较为理想的测序结果,首先样品的质量需要得到保障,当然样本提取、建库、分析对最终结果都会产生一定的影响,而取样同样重要。如果没有取好,后续分析再多,可能也得不到想要的结果。
另外,严谨的研究设计对于获得准确且有意义的结果也很重要,研究设计中应注意考虑混杂因素,如果在不确定的情况下,也可以适当咨询我们的技术顾问,结合我们的经验,给您合适的建议。
当然实验过程中,也可能会受到多种因素的影响,我们会采用阴性和阳性对照,尽可能减少这些不必要的影响。
总之,做好前期的准备工作,包括研究设计,取样等,后续的提取、测序、分析交给我们~
相关阅读:
参 考 文 献
[1] Edwards J , Johnson C , Christian Santos-Medellín, et al. Structure, variation, and assembly of the root-associated microbiomes of rice[J]. Proceedings of the National Academy of Sciences, 2015, 112(8).
[2] Bulgarelli D, Rott M, Schlaeppi K, et al. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota[J]. Nature, 2013, 501(7468): S25.
[3] ISO 10381-6: 2009 Soil quality – Sampling – Part 6: Guidance on the collection, handling and storage of soil under aerobic conditions for the assessment of microbiological processes, biomass and diversity in the laboratory.
[4] Ruiz-González C, Niño-García J P, Kembel S W, et al. Identifying the core seed bank of a complex boreal bacterial metacommunity[J]. The ISME journal, 2017, 11(9): 2012.
[5] Wang Y, Ma L, Mao Y, et al. Comammox in drinking water systems[J]. Water research, 2017, 116: 332-341.
[6] Chow C E T, Winget D M, White III R A, et al. Combining genomic sequencing methods to explore viral diversity and reveal potential virus-host interactions[J]. Frontiers in microbiology, 2015, 6: 265.
[7] Yang Y, Li B, Zou S, et al. Fate of antibiotic resistance genes in sewage treatment plant revealed by metagenomic approach[J]. Water research, 2014, 62: 97-106.
[8] Pesant S, Not F, Picheral M, et al. Open science resources for the discovery and analysis of Tara Oceans data[J]. Scientific data, 2015, 2.
[9] Ma J, Wang Z, Li H, et al. Metagenomes reveal microbial structures, functional potentials, and biofouling-related genes in a membrane bioreactor[J]. Applied microbiology and biotechnology, 2016, 100(11): 5109-5121.
[10] Liu J, Yang H, Zhao M, et al. Spatial distribution patterns of benthic microbial communities along the Pearl Estuary, China[J]. Systematic and applied microbiology, 2014, 37(8): 578-589.
[11] Mason O U, Scott N M, Gonzalez A, et al. Metagenomics reveals sediment microbial community response to Deepwater Horizon oil spill[J]. The ISME journal, 2014, 8(7): 1464.
[12] Bråte J, Logares R, Berney C, et al. Freshwater Perkinsea and marine-freshwater colonizations revealed by pyrosequencing and phylogeny of environmental rDNA[J]. The ISME journal, 2010, 4(9): 1144.
[13] Li J, Sung C Y J, Lee N, et al. Probiotics modulated gut microbiota suppresses hepatocellular carcinoma growth in mice[J]. Proceedings of the National Academy of Sciences, 2016, 113(9): E1306-E1315.
[14] Gebhart D, Lok S, Clare S, et al. A modified R-type bacteriocin specifically targeting Clostridium difficile prevents colonization of mice without affecting gut microbiota diversity[J]. MBio, 2015, 6(2): e02368-14.
[15] Hänninen A, Toivonen R, Pöysti S, et al. Akkermansia muciniphila induces gut microbiota remodelling and controls islet autoimmunity in NOD mice[J]. Gut, 2018, 67(8): 1445-1453.
[16] Fujisaka S, Avila-Pacheco J, Soto M, et al. Diet, genetics, and the gut microbiome drive dynamic changes in plasma metabolites[J]. Cell reports, 2018, 22(11): 3072-3086.
[17] Agus A, Denizot J, Thévenot J, et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation[J]. Scientific reports, 2016, 6: 19032.
[18] Stanley D, Moore R J, Wong C H Y. An insight into intestinal mucosal microbiota disruption after stroke[J]. Scientific reports, 2018, 8(1): 568.
[19] Rabbi M F, Munyaka P M, Eissa N, et al. Human catestatin alters gut microbiota composition in mice[J]. Frontiers in microbiology, 2017, 7: 2151.
[20] Rabbi M F, Eissa N, Munyaka P M, et al. Reactivation of intestinal inflammation is suppressed by catestatin in a murine model of colitis via M1 macrophages and not the gut microbiota[J]. Frontiers in immunology, 2017, 8: 985.
[21] McInnes P, Cutting M. Manual of procedures for human microbiome project: Core microbiome sampling, protocol A, HMP protocol no. 07-001, version 11. 2010[J]. Current version: https://www.hmpdacc.org/hmp/resources/HMP_MOP_Version12_0_072910.pdf
[22] Jie Z, Xia H, Zhong S L, et al. The gut microbiome in atherosclerotic cardiovascular disease [J]. Nature communications, 2017, 8(1): 845.
[23] Mar J S, LaMere B J, Lin D L, et al. Disease Severity and Immune Activity Relate to Distinct Interkingdom Gut Microbiome States in Ethnically Distinct Ulcerative Colitis Patients [J]. mBio, 2016, 7(4).
[24] Sze M A, Baxter N T, Ruffin M T t, et al. Normalization of the microbiota in patients after treatment for colonic lesions [J]. Microbiome, 2017, 5(1): 150.
[25] Flemer B, Lynch D B, Brown J M, et al. Tumour-associated and non-tumour-associated microbiota in colorectal cancer [J]. Gut, 2017, 66(4): 633-43.

微生物研究领域在过去的几十年里发展迅速,已经成为一个重大的科学和公众利益的话题。然而我们对“微生物组”一词缺乏一个公认的明确定义。
近日,奥地利格莱兹科技大学Gabriele Berg教授和亥姆霍兹慕尼黑中心Schloter教授究国际专家组成的小组会议讨论关于微生物和微生物组到最新的技术发展和研究结果,明确区分了“微生物群”和“微生物组”两个术语,并就微生物群的组成、微生物群在时间和空间上的异质性和动态性、微生物网络的稳定性和恢复力、核心微生物群的定义进行了全面的讨论,以及功能相关的关键物种以及微生物宿主和微生物群落内物种间相互作用的共同进化原理。

这些宽泛的定义以及所建议的统一概念将有助于今后改进微生物组研究的标准化,并可能成为综合评估数据的起点,从而使知识从基础科学更快地转移到实践中。此外,微生物组标准对于解决与地球健康领域人为驱动的变化相关的新挑战非常重要,对微生物组的理解可能会起到关键作用。
本文是基于2019年3月6日在奥地利图尔恩举行的研讨会的讨论而形成的,该研讨会是Microbiome Support项目的一部分,该项目旨在建立国际研究标准。
研讨会汇集了来自学术,政府和行业团体的领先微生物组研究人员,他们代表了不同领域的专业知识。 在研讨会之前,通过发送在线调查问卷,收集了来自世界100多位专家的建议。调查的结果和随后的研讨会讨论构成了术语“微生物组”的拟议定义的基础,此处介绍了包含微生物组研究规则和基准修订。
许多技术发明推动了微生物的研究,导致我们对健康和疾病的理解发生了范式转变。
图1 历史回顾

显微镜的发明,让我们得以发现一个全新的未知的微生物世界。
罗伯特·科赫(Robert Koch)对由于微生物感染而导致的人类和动物疾病起源的解释以及病原性概念的发展是微生物学的重要里程碑。
DNA的发现,测序技术,PCR和克隆技术的发展使人们能够使用与培养无关,而是基于DNA和RNA的方法研究微生物群落。
这些新的可能性彻底改变了微生物生态学,因为以高通量的方式进行基因组和宏基因组分析提供了有效的方法来解决单个微生物以及整个自然栖息地中整个群落的功能潜能。
多项综述已强调了结合多种“组学”技术分析宿主微生物相互作用的巨大潜力和巨大潜力。
通常将微生物群落定义为生活在一起的微生物的集合。更具体地说,微生物群落被定义为多物种集合,其中(微生物)有机体在连续的环境中彼此相互作用。
微生物组的首次定义
1988年,Whipps及其同事研究了根际微生物的生态学,首次定义了微生物组。他们将“微生物组”描述为“微生物”和“生物组”的组合,并在“具有明确的理化特性的合理定义的栖息地”中将“特征微生物群落”命名为“活动剧场”。
该定义代表了微生物群落定义的实质性进步,因为它定义了具有不同特性和功能的微生物群落及其与环境的相互作用,从而形成了特定的生态位。
微生物组的常用定义
但是,在过去的几十年中,还发布了许多其他的微生物组定义。当前最常引用的定义是Lederberg将生态环境中的微生物群落描述为生物体空间或其他环境中的共栖,共生和致病微生物群落。
Marchesi和Ravel集中在对特定环境下的基因组、微生物(和病毒)基因表达模式和蛋白质组以及生物和非生物条件的定义。所有这些定义暗示着宏观生态学的一般概念可以很容易地应用于微生物-微生物以及微生物与宿主的相互作用。
然而,这些为大型真核生物开发的概念在多大程度上可应用于原核生物,这些原核生物具有不同的生活方式,如休眠,表型变异和水平基因转移以及目前尚不清楚的微型真核生物。这就提出了一个挑战,即要考虑一种全新的微生物组生态学概念生态模型和理论体系,特别是在微生物相互之间以及与宿主生物和非生物环境相互作用的不同层次上。
许多当前的定义未能捕捉到这种复杂性,并且将术语“微生物组”描述为仅涵盖微生物的基因组(下表)。 例如,Merriam Webster发布平台提出了两种微生物组定义:一种描述宏基因组,另一种是微生物群落,但仍然无法将宿主和环境作为微生物组的整体生态成分而不是一个独立的实体来捕获。
表1 微生物组的定义
修订后的概念框架将使我们能够从对微生物分类的方法转变为更全面的微生物功能及其与环境相互作用的观点。
然而,这将需要跨不同领域工作的科学家之间更多的跨学科互动。关于微生物组的各种观点是Microbiome Support研讨会中讨论的中心部分。
根据在线调查和研讨会讨论中获得的答复,参与者得出的结论是Whipps等人的原始定义仍然是最全面的,它描述了微生物组的复杂性及其生态学和进化生物学的各个方面。 研讨会的参与者讨论了许多关键点,并提出了一些建议,以澄清和修正原始的Whipps和同事的定义。
这些修正案涉及(1)微生物组的成员,(2)微生物组的成员之间以及现有微生物网络内部的相互作用,(3)环境中微生物群落的时空特征,(4)核心微生物群,(5)从功能预测到物种表型,以及(6)微生物组-宿主或环境相互作用和协同进化。 下面将详细讨论这些方面。
微生物群包括形成微生物组的所有活的成员。 下表解释了这两个词源的词源学和差异。细菌,古细菌,真菌,藻类和小的原生生物应被视为微生物组的成员。大多数微生物组研究人员都同意这一定义。
表2 微生物组/微生物群词源

噬菌体,病毒,质粒和移动遗传元件的整合是微生物组定义中最具争议的问题之一。 参与者在“微生物组定义”在线调查中的评论也证实了这一点。 受访者对病毒和噬菌体是否应属于微生物组这一问题的回答没有给出明确的答案,并且从“无论如何”延伸到“绝对没有”。
关于来源于死细胞的细胞外DNA(所谓的“遗留DNA”)是否属于微生物组还没有明确的共识。在更广泛的生境分析中,残留DNA最多可占土壤中测序DNA的40%,平均占细菌总DNA的33%,在某些样品中最高比例为80%。
有意思的是,尽管它非常丰富且无处不在,但遗留DNA对分类学和系统发育多样性的估计影响很小。 当使用特定术语时,微生物组和微生物群之间的清晰区分有助于避免有关微生物组成员的争议(下图)。微生物群通常被定义为存在于特定环境中的活微生物的集合。由于噬菌体,病毒,质粒,病毒,类病毒和游离DNA通常不被视为活微生物,因此它们不属于微生物群。
图2 强调微生物组(微生物群落)及其“活动区”组成的示意图
最初由Whipps及其同事提出的微生物组一词不仅包括微生物群落,还包括微生物的“活动区”。后者涉及微生物产生的所有分子,包括其结构元素(核酸、蛋白质、脂类、多糖)、代谢物(信号分子、毒素、有机和无机分子),以及共存宿主产生的并由周围环境条件构成的分子。因此,“微生物组”术语应包括所有可移动的遗传元件,例如噬菌体,病毒,“遗物”和细胞外DNA,但不属于微生物群(上图)。
此外,在这方面,重要的是要考虑方法方面的差异,以区分DNA与活生物体及其环境(请参阅技术标准章节)。术语微生物组有时也与宏基因组混淆。 然而,宏基因组被明确定义为来自微生物群成员的基因组和基因的集合。
微生物组研究有时关注特定微生物群的行为,通常与明确的假设有关或由其明确证明。虽然越来越多的术语如“细菌组”,“古细菌组”,“霉菌组”或“病毒组”在科学文献中开始出现,但这些术语并非指生物群落(一个具有独特的(微生物)集合的区域生态系统,而物理环境往往反映一定的气候和土壤)作为微生物群本身。因此,最好使用原始术语(细菌,古细菌或真菌群落)。
与可以单独研究的微生物群落相反,微生物组始终由所有成员组成,这些成员彼此相互作用,生活在相同的栖息地,并共同形成其生态位。
公认的术语“病毒组”衍生自“病毒”和“基因组”,用于描述由一系列与特定生态系统或全生命周期相关的核酸组成的病毒Shotgun基因组。但是,这里也可以将“病毒基因组”作为语义和科学上更好的术语来建议。
每个微生物组成员应以何种分辨率进行研究?
对于真核生物,在大多数情况下,“生殖单位”是一个适当的水平,可以测量生物的动态,尽管物种定义仍在争论中。
但是,对于原核生物,尚不存在基于繁殖的定义:当前的物种定义基于生物之间的DNA同源性,例如DNA-DNA杂交揭示的“ 70%以上的DNA相似性”或根据Goris及其同事的建议。 与真核生物相似,微生物菌株或生态型是分类和功能的基础。 此处定义的菌株的稳定性是最关键讨论的问题,主要是由于水平基因转移(HGT)的频繁发生。
后者是由诸如质粒,噬菌体和转座子之类的可移动遗传元件从一种菌株转移到另一种菌株而引起的,导致不可避免的基因组变化并严重影响菌株的稳定性。然而,由于微生物菌株之间的本质功能差异,忽略菌株水平可能会导致对数据的误解。
在微生物菌株中定义具有生态意义的种群对于确定其在环境微生物和与宿主相关的微生物群落中的作用很重要。
最近,作为一种解决方案,引入了一种新的衡量最近基因流量的指标,该方法可确定与近亲相邻的,被强基因流不连续性隔开的一致的遗传和生态单位。
微生物彼此相互作用,这些共生相互作用对微生物组内的微生物适应性,种群动态和功能能力具有不同的影响。这些相互作用可以在相同物种的微生物之间,也可以在不同物种,属,科和生命域之间。
这些网络中的交互模式可以是积极的(互惠的、协同的或共栖的)、消极的(偏害共栖[包括捕食、寄生、对抗或竞争])或中性的,即对相互作用物种的功能能力或适应性没有(或没有观察到)影响。
微生物生命策略的概念可以影响相互作用的结果。 例如,争夺相同来源的微生物在不同营养水平争夺相同化合物时也可以从彼此受益。复杂微生物生态系统的稳定性取决于同一底物在不同浓度水平下的营养相互作用。
重要的是要强调指出,到目前为止,人们对自然界中微生物社会适应的研究还不够。在这里,分子标记可以通过支持自然微生物群系中利他主义者和作弊者等理论来提供社会适应的见解。
次生代谢产物在介导复杂的种间相互作用并确保竞争环境中的生存中起着至关重要的作用。群体感应(QC), 由小分子(如N-酰基高丝氨酸内酯或肽)诱导的细菌诱导细菌控制合作活动,并使它们的表型适应生物环境,从而导致例如细胞间粘附或生物膜形成。
直接种间电子转移(DIET)是大多数厌氧生态系统中交流的重要机制。此外,挥发性化合物还可以作为长距离信使,实现长距离跨界交流。
此外,所谓的“真菌高速公路”是细菌以及水和养分的运输系统,因此可以在构建微生物网络中发挥重要作用。
尽管有这些例子,但微生物组内的交流和相互作用仍未得到充分研究,将从对所有微生物组成员的代谢相互作用的更多了解中受益。在这里,还原论实验模型和微生物组模型可以帮助鉴定参与复杂相互作用的微生物和分子机制。
细菌的群体行为调控机制
群体效应(Quorum sensing) 是近来日益受到广泛关注的一种细菌群体行为调控机制, 很多细菌有这种能力, 即分泌一种或多种自诱导剂(Autoinducer) ,细菌通过感应这些自诱导剂来判断菌群密度和周围环境变化, 当菌群数达到一定的阀值(quorum , 菌落或集落数) 后, 启动相应一系列基因的调节表达, 以调节菌体的群体行为。
不同类型的细菌具有不同的群体效应调节系统, 很多细菌分泌同一种诱导剂, 以此调控不同种类细菌间的作用行为。群体效应系统在自诱导剂与受体之间存在专一性, 同时又在调节基因和信号传递系统中体现出多样性和复杂性。由于不少人或植物的病原菌的致病机制等受群体效应的调控, 该机制已成为医学等领域的研究热点。
图3 通过微生物共生网络可视化微生物相互作用
生物信息网络和共现分析给出了关于微生物相互作用模式的复杂性的想法,但它们不适合揭示这些相互作用的性质(上图b)。 尽管存在这一限制,但对微生物网络的分析仍使研究人员能够识别中心物种,并探索微生物组内各种类型的物种相互作用的潜力。在微生物共生网络中,中枢物种以与其他物种的连接程度最高的节点(上图b)为代表。
共现分析也可以在不同的规模上应用,例如,群落规模的生态系统之间的共现模式,群落内共生微生物的模块以及嵌套在微生物群落内的模块内共生对。
它们可以与定植抗性联系起来,这决定了异源微生物入侵本地群落的潜力,并且可以被认为是产生假说的重要工具。 但是,特定类型的微生物相互作用的存在及其对种群动态或功能的后果,需要在相关的模型系统中进行测试(上图c)。
此外,技术方法,如使用稳定同位素的交叉喂养实验或荧光原位杂交和共焦激光扫描显微镜(FISH-CLSM)与双重培养分析相结合,对于检验在硅胶中产生的假设非常有用。
微生物相互作用可能是微生物群落内部进化和共同进化动力学的重要基础
微生物群落成员之间的交流产生了一个复杂的景观,其中细胞的适应性或功能不仅取决于单个细胞的遗传潜能和化学环境,还取决于感测到的生物环境。 网络中的中心物种通常被假设为关键物种,这一概念已从宏观生态学转移到微生物组学研究中。
与其他物种相比,基石物种在各种物种相互作用中起着至关重要的作用,并且对生态系统的性能和动态影响更大。 但是,共同关联网络中的枢纽物种不一定充当基石物种。 后者的特征还必须通过适当的方法加以确认和补充。
枢纽和基石分类单元绝对需要对它们的功能有更好的了解; 此外,它们可以集成到计算方法中以联系微观和宏观生态学问题。 如果可以将关键物种视为“指示分类群”,那么另一个术语(已被定义为高度指示特定实验处理或环境条件的那些类群)仍不清楚。
指示分类群的概念从实践的角度引起了人们的极大兴趣,并从宏观生态学转移到了微生物组研究中,现在被广泛使用,例如,用于评估农业实践对微生物群落的影响或疾病对微生物的影响的研究。 人类微生物群; 如此处所示,可以使用简单且高度标准化的基于qPCR的方法进行分析。
微生物群落的时间和空间结构问题对于总体上了解微生物组的功能很重要。对于理解特定过程,例如在生物技术和食品加工应用中病原体的暴发,以及预测和控制微生物群落,它也具有重要意义。总的来说,最初针对大型生物描述的大小多样性关系也被证明存在于各种生态系统中的微生物群落中。
微生物组内的时间动态可以从秒或分钟的尺度进行评估,这反映了信使RNA的时间跨度到几个世纪和一千年的尺度,在此期间微生物与其宿主或在特定环境中共同进化。细菌mRNA的半衰期取决于转录的基因,但通常在数分钟的范围内,而古细菌基因的转录本较长,并且已报道了数小时的时间。
重要的是,尽管过去许多作者将微生物活性与rRNA含量联系在一起,但最近的研究表明该概念存在严重局限性,只有mRNA可被视为代谢状态的可靠指标。 在这一时间尺度范围内理解研究的适当维度对于任何微生物组的操作都是至关重要的,例如在人类微生物组研究中的治疗策略或在环境研究中使用生防制剂。仔细考虑感兴趣宿主的具体特征,例如昼夜节律,季节性变化或与宿主生物的生理学相关的生长阶段,可能有助于确定评估时间动态的最佳尺度。
图4 微生物群在时间和尺度上的动态变化
Stegen等建议考虑三类:(i)生物和非生物历史,(ii)内部动力学,和(iii)外部强迫因素作为影响微生物群落时间动态的因素。
大多数自然生态系统的特征是高度的空间结构,这被认为对许多生态系统服务都很重要。 考虑到空间规模,可能意味着要比较远处区域之间的微生物模式,以及相同生境的生存空间之间的区别不明显(图4b)。
植物多样性与土壤微生物组多样性的相互影响
土壤主要由微团聚体(<0.25毫米)组成,这些团聚体结合土壤有机碳并保护其免受侵蚀,而大团聚体(0.25到2毫米)则限制了氧气的扩散并调节了水的流量。 每个聚集体都具有独特的生态位,并具有其独特的微生物组结构。 实际上,由于在小空间尺度上存在如此众多的生态位,土壤被认为是地球上微生物群落组成最多样化的生态系统。 例如,农业耕作引起的生态位的减少会导致微生物多样性的丧失。 由于植物和耕作在很大程度上影响土壤结构的发展,植物多样性的丧失也对土壤微生物组的生物多样性产生了强烈影响。
但是,对于“先生鸡还是蛋”的答案尚不清楚(土壤微生物组的变化会引起植物多样性的改变,反之亦然)。微生物群落对宿主的定殖也不均匀。
众所周知,例如,叶子的根部与根部相比具有不同的菌群,并且根部本身被微生物异质地定殖,沿着根际的长度以及根部表面与根部内部的微生物群不同。
最近,种子微生物群作为核心微生物群从一代到另一代的垂直传播的一种可能模式引起了人们的关注。
与植物相似,人体也不是被微生物均匀地定殖的:每个人体隔室都包含自己的微生物群,甚至来自一个身体部位的微生物群也可能因采样面积而异(例如皮肤微生物群)。
微生物热点和热点时刻通常紧密相连。例如,土壤的特征是存在所谓的微生物热点(包括根际,小球层或碎屑层在内的空间分离)和热时刻(时间动态)。在热点中,活跃代谢微生物的比例是非热点中微生物的2-20倍,与微生物活性较低的部位相比,热点中微生物组结构和功能的时间变化更具动态性。
基于共现分析和捕获微生物群落时空动态的实验数据,研究人员寻求定义核心微生物群落。这确实是有帮助的,因为本地微生物群通常非常复杂,包括来自不同王国的数千种物种。
定义核心微生物群可以帮助区分微生物群的稳定成员和永久成员,这些种群可能是断断续续的,仅与特定微生物组状态相关或仅限于特定环境条件的种群。
Shade和Handelsman提出了第一个建议,他们将核心微生物组定义为来自相似栖息地的微生物群落之间共享的一组成员,以鉴定复杂微生物群中稳定,一致的成分。
当前,核心微生物群主要是根据具有分类学信息的DNA序列来定义的。考虑到基于DNA的分析(主要是标记基因的扩增子测序)的分辨率极限,但是很明显,核心微生物群主要是通过群体的属级鉴别来定义的,并且菌株特异性和功能变异是不考虑的。
功能性核心微生物群包含携带复制子的媒介
相比之下,Lemanceau及其同事提出了一个功能性核心微生物群,该微生物群包含携带复制子(基因)的微生物媒介,而复制子具有对整体生命的基本功能。
最近,Toju等人提出了核心微生物群的概念,专门用于将农业生态系统管理为物种丰富的社区; 他们将核心定义为“形成相互作用核心的微生物集,可用于在各个植物和生态系统级别优化微生物功能。”
“核心”微生物群似乎保持相当恒定
Astudillo-García及其同事在审视高度多样化的海洋海绵微生物系统时,评估了不同核心微生物群定义的影响。虽然在定义核心社区时必须谨慎,但是,总体结果似乎对核心定义的变化相对不敏感。瞬时微生物群随时间变化,这取决于环境条件,营养的可获得性和/或宿主的生长和健康阶段,甚至昼夜节律。相反,“核心”微生物群似乎保持相当恒定。就时间动态而言,核心微生物群描述了与给定宿主基因型或特定环境持续相关的微生物群落(图4a)。
这一概念的例外情况也被描述过,例如,在最佳适应再次发生水合/脱水循环的微生物组中,不同的细菌群落在循环中发挥了不同的功能:两者均属于核心。类似地,在空间尺度上,例如,考虑在同一地理区域或同一地理区域的一系列土壤中生长的植物,核心微生物群不变(图4b)。
研究微生物组的当前可用方法,即所谓的多组学方法,范围从高通量分离(培养组学)和可视化(显微镜)到靶向分类学组成(宏条形码)或解决代谢潜能(功能基因的宏条形码,宏基因组学) 分析微生物活性(代谢组学,代谢组学,代谢组学)(下图)。
图5 评估微生物功能的方法
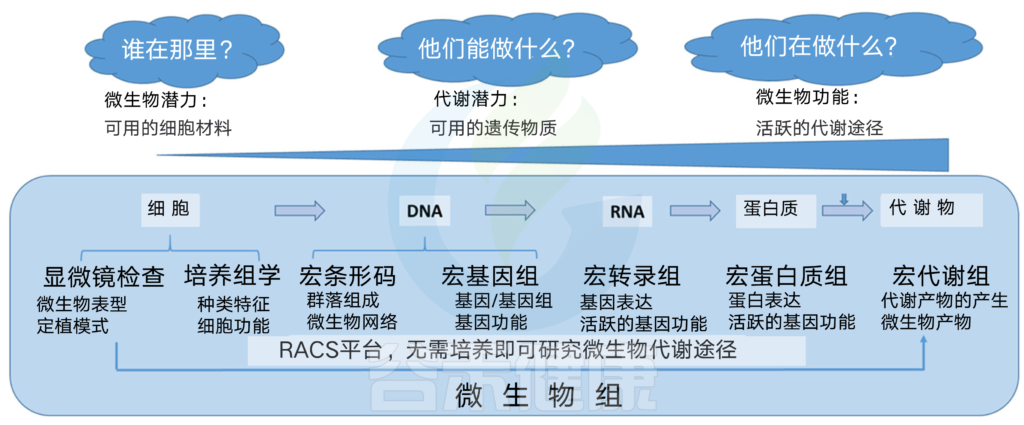
基于宏基因组数据,可以重建微生物基因组。 虽然从环境样品中重建了第一个由基因组组装的基因组,但近年来,数千个细菌基因组在没有培养其背后的生物体的情况下被组合在一起。 例如,最近从9,428个宏基因组中重建了全球154,723个微生物基因组。
然而,由于一方面微生物基因组DNA序列数据的大量可利用性与另一方面确认基因功能的宏基因组学预测所需的微生物分离物的可利用性之间缺少联系,理解仍然受到很大限制。
宏基因组数据为新的预测提供了场所,但是需要更多的数据来加强序列与严格的功能预测之间的联系。 考虑到一个氨基酸残基被另一个残基取代可能导致自由基功能改变,从而导致给定基因序列的功能分配不正确,这一点变得显而易见。
此外,需要培养新菌株以帮助鉴定从宏基因组学分析中获得的大部分未知序列,对于研究不足的生态系统,这些序列可能超过70%。根据应用方法的不同,即使在经过充分研究的微生物组中,完全测序的微生物基因组中有40-70%的注释基因没有已知或可预测的功能。
另外,目前的估计预测,具有未知功能的域将很快超过已知功能的族。显然需要更经典的微生物学,包括结合使用靶向突变体和微生物生物化学来应对这一挑战。
从已经发现的具有未知功能的蛋白家族的全面功能表征中获得的好处,远不止于进一步扩展这些家族的列表。尽管长期以来,结合(广泛的)培养和独立于培养的分析相结合的多阶段方法一直是环境和植物微生物学领域的最新技术,但医学微生物学通常并非如此。 近年来,高通量培养组学建立了人类肠道菌群的参考基因组和培养集。
了解原核生物的功能多样性非常具有挑战性
因为目前建立的118种门中有85种迄今尚未描述过单一种。最后,原核门的数量可能达到数百个,而古细菌门的研究最少。这个问题需要通过收集尚未培养的原核生物有意义的分类学和功能信息来解决。纯培养中细菌和古细菌的多样性与通过分子方法检测到的细菌和古细菌的多样性之间的差距日益扩大,这导致人们提议主要根据序列信息为尚未培养的分类单元建立正式的命名法。
原核生物国际命名法的建议修改
根据这项建议,念珠菌属的概念将扩展到密切相关的基因组序列的组,并按照已建立的细菌命名规则公布其名称。对原核生物国际命名法的建议修改引起了以下方面的关注:
(1)命名法的可靠性和稳定性;
(2)技术和概念上的局限性以及参考基因组的可用性;
(3)计算机功能的信息内容预测;
(4)识别微生物多样性的进化单位。
这些挑战需要克服,以达到一个有意义的分类学尚未培养的原核生物,目前尚不清楚的表型。 在这种背景下,已经做出了巨大的努力来培养来自不同环境的细菌。
Staley和Konopka在1985年确定了“大平板计数异常”,它描述了一个事实,即90至99.9%的细菌物种无法在标准实验室条件下生长。对于某些微生物栖息地,尤其是那些具有较高养分含量和微生物活性的微生物,如肠道微生物群所述,相对于通过测序检测到的分子种类,培养物中可利用的代表性菌株的比例从35%增长至65%。
来自其他自然栖息地的微生物种群以及微生物组的真核成员也需要类似的进展。微型真核生物,如原生动物、真菌和藻类的成员,通常可以更好地培养和显微镜下研究;然而,它们的系统发育和分类学更复杂,研究较少。有趣的是,来自不同环境的16S和18srRNA基因的无引物测序表明,在微真核生物中,有大量以前未检测到的类群。
同位素探测技术
除了当前使用的计算机模拟比较和培养方法外,还可以使用一组同位素探测技术直接测试复杂微生物群落中的功能假设。这些方法包括DNA,RNA,蛋白质和脂质稳定的同位素探测(SIP)以及FISH显微放射自显影,FISH-NanoSIMS和FISH-Raman显微光谱学,后三种方法仅提供一种细胞分辨率。
RACS平台无需培养即可研究微生物代谢途径
最近,开发了一种微流体拉曼活化细胞分选(RACS)平台。在最近的一项研究中,Lee和他的同事在存在氘水的情况下,允许小鼠结肠菌群中的细胞代谢未标记的目标化合物(粘蛋白)。随后,使用RACS平台从复杂的微生物组中选出了活跃地代谢粘蛋白的氘标记细胞,并通过单细胞基因组学和培养方法进行了进一步分析。
这种方法允许以一种新型的无栽培方式将微生物的代谢表型与其基因型联系起来,从而使从微生物的潜能到微生物的功能成为可能(图5)。
尽管取得了一些进展,但这种功能分类平台的产量仍然有限,而FISH和生物正交非标准氨基酸标记(BONCAT)等互补的新技术解决方案将有助于微生物组研究中更迫切需要的以表型为中心的研究。
宿主及其相关微生物之间的密切关系引起了宿主及其相关微生物群共同进化的理论。协同进化是沿袭相互适应的谱系。
一个例子是建立早期的陆地植物,这是由共生真菌联合体促进的,这表明植物自从首次出现在土地上以来就已经与微生物共同进化了。
另一个例子是真核生物。 线粒体和质体是真核细胞中的细胞器,来源于内共生细菌,在整个进化过程中,它们完全取决于宿主,反之亦然。 为了促进对微生物群的全面了解,需要考虑宿主微生物的协同进化(下图)。
图6 对微生物-宿主协同进化的理解从“分离”理论转向整体方法
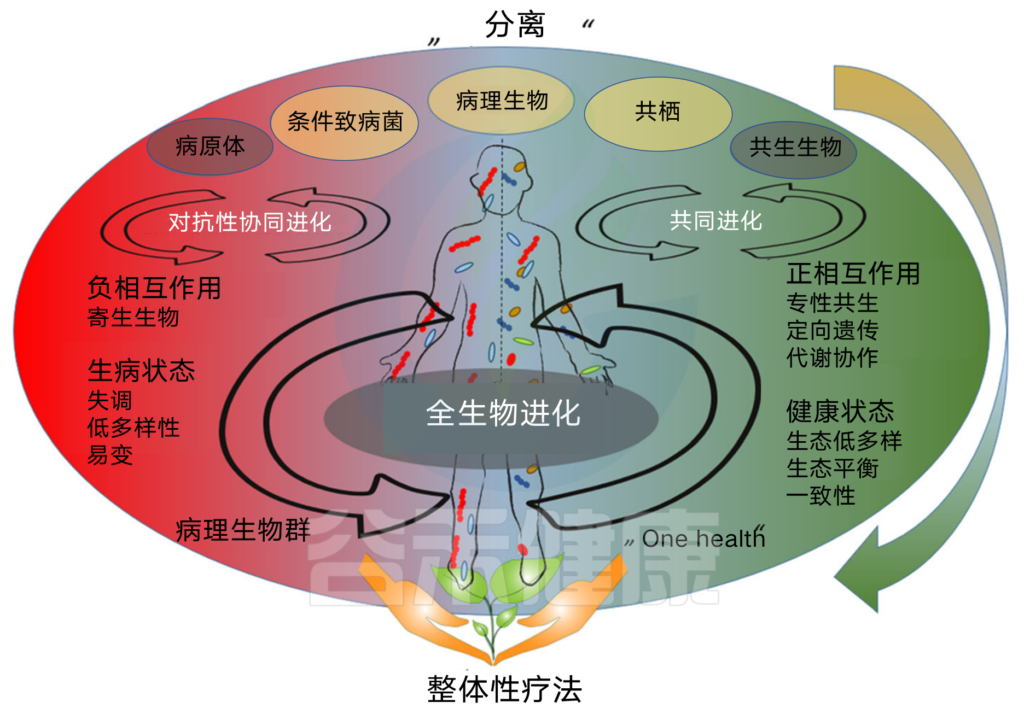
基于微生物与其宿主之间的相互作用,将微生物分为有益的,致病的和中性的,是广泛引用的Lederberg和McCray关于微生物组定义的一部分。
根据他们的解释,拮抗协同进化包括宿主-寄生虫的相互作用,而当积极相互作用占优势时,则存在互助协同进化。这种积极的相互作用可能演变成专性共生,垂直遗传和代谢协作(上图)。
根据致病微生物,有益微生物和中性微生物与宿主之间的相互作用,对致病微生物,有益微生物和中性微生物进行分类可能对微生物宿主相互作用在调节宿主适应性中起核心作用的研究(例如医学研究)有用。 然而,对致病性数据的解释应谨慎。
对条件致病菌的最新研究表明,宿主与微生物的相互作用不仅取决于宿主,还取决于整个微生物组。自然抑制疾病的土壤表明植物和许多土壤传播条件致病菌的情况类似。
此外,在许多环境研究中,可能没有长时间可利用的特定宿主,这使得划分致病菌和有益菌变得无关紧要。
与其研究一种特定微生物与其宿主之间的相互作用,不如考虑基于全环素理论的整体方法(图6)。 按照这种方法,宿主及其微生物组之间的有益相互作用负责维持整体生物的健康,而疾病通常与微生物失调有关。
在失调的背景下,建立了“病原体组”概念(图6),该概念代表了整合在其生物环境中的病原体,并应用于多种病理系统。这种方法表明,微生物多样性是预防植物和人类肠道疾病的关键因素。
尽管进行了众多研究,定义了“健康的微生物群”,但是在未来,平衡和失调之间的界限仍然是一个重大挑战。
微生物与宿主之间相互作用的另一种有趣解释是所谓的“安娜·卡列尼娜原理” 。声明指出,与列夫·托尔斯泰的格言类似,“所有幸福的家庭看起来都一样; 每个不幸的家庭都会以自己的方式感到不幸。”
与健康的个体相比,菌群失调个体的微生物群落组成差异更大
例如,健康珊瑚的微生物群比患病珊瑚的微生物群更均匀。 同样,最近的一项研究发现,炎症性肠病患者的群落组成和免疫反应的稳定性明显低于健康人。
进化过程和选择压力极大地推动了宿主-微生物的相互作用
因此,区分为人造的,以人为中心的类别可以随时间变化。一个例子是幽门螺杆菌,它是二十世纪初期几乎所有人胃中的优势微生物,在短短100年内几乎消失了。 幽门螺杆菌一方面是消化性溃疡和胃癌的危险因素;另一方面,这种细菌的丢失与儿童时期的哮喘,花粉症或皮肤过敏有关。
微生物与其预期宿主之间复杂的进化相互作用的另一个例子是病原体的出现,如嗜麦芽单胞菌,这种植物相关起源的细菌与全球人类条件致病菌株没有区别。 总而言之,自然选择被认为有利于塑造群落组成的宿主,从而促进有益的宿主微生物共生,但是许多因素可以使这种动态失衡,从而诱导宿主微生物组内对病原性或拮抗性微生物的选择。
基于计算模型,Lewin-Epstein等人提出,操纵宿主以利他行为行动的微生物可能会受到选择的青睐,并可能在利他主义的广泛发生中发挥作用。
宿主微生物共同进化导致与植物和动物相关的特定微生物群。这种特异性的程度受许多因素影响,并且在系统发生分支之间也有所不同。 对于代表地球上最古老的陆地植物的苔藓而言,其植物特异性异常高。 这种特异性与宿主的地理来源无关,并且从子孢子垂直传递至配子体,反之亦然。
驯化和育种活动也可以显着影响宿主微生物组
在某些情况下,其程度也超出预期。消失的微生物群系的理论表明,流行的慢性病是由人为微生物群向多样性降低的转变所引起的。考虑到这一点,重要的是重新考虑我们的行为(例如,对家庭环境进行“过度清洁”)并评估作物管理方法(例如育种策略),以避免丧失共同进化的有益宿主微生物相互作用。
在过去的十年里,面对微生物学领域显著而持续的技术进步,研究界未能为微生物组研究制定一致的标准。这导致了许多缺点,包括缺少实验室间数据交叉比较的可能性,或者在最近的研究中使用“旧”技术生成的数据的实现。
有大量出版物表明DNA提取和加工程序对微生物组的后续分析存在偏差。使用定义的模拟群落一方面可以帮助确定最佳的提取方法,另一方面可以作为内部控制来估计整个工作流程和分析中的可能偏差。
但是,即使将模拟群落实施到分析中也不能解决所有问题,这会给从环境样本中进行微生物群落的分子分析带来偏差。 例如,不能通过将模拟群落应用于土壤来反映土壤微生物对土壤颗粒的自然吸附过程。
此外,在复杂的栖息地(例如土壤)中,多达80%的提取DNA可能包含遗物DNA(死细胞的细胞外DNA以及活细胞的分泌DNA)。这种残留的DNA可能会增加观察到的原核和真菌的多样性,并导致对分类单元相对丰度的估计不准确。
PMA法有效地抑制了革兰氏阴性菌和革兰氏阳性菌死亡细胞中DNA的PCR扩增
Nocker等人提出了一种可能的解决方案使用叠氮溴化丙锭(PMA)的方法,该单叠氮化物仅穿透膜受损的细胞,其中光诱导的叠氮化物基团与DNA共价结合; 这种交联有效地抑制了革兰氏阴性菌和革兰氏阳性菌死亡细胞中DNA的PCR扩增。该方法已成功应用于微生物组研究,尤其是微生物丰度低和活性低的研究,但这还不是标准。
DNA处理中的一个众所周知的例子是通过分析引物组和引物选择和PCR诱导的选择性扩增或大量未知读物。Karst等人开发的无引物rRNA基因测序方法促进了未知微生物多样性的发现。
作者建议将SSU rRNA分子的poly(A)尾和逆转录与合成的长读测序结合起来。使用这种方法,他们能够以高通量产生高质量,全长的SSU rRNA序列,而没有引物偏倚,并且在他们的研究中观察到了迄今未描述的多样性中的很大一部分。
对获得的序列数据进行生物信息学分析既不是标准化的,也不是明确表达的偏见。为了进行16S rRNA基因序列的数据评估,可以使用免费软件解决方案和完善的论坛,例如Qiime2和Mothur。
操作分类单元(OTU)长时间用于分析高通量条形码,例如使用标记基因(如16S rRNA和/或ITS区域)获得高通量测序数据。
另外,可以使用基于精确解析的扩增子序列变体(ASV),低至测序基因区域上的单核苷酸差异水平。立即获得更好的分辨率的好处显而易见,ASV作为具有固有生物学意义的一致标签的状态独立于参考数据库而被确定。可重用性,可再现性和全面性方面的改进足够大,以至于ASV可以取代OTU作为标记物基因分析和报告的标准单元。
为了比较不同的工具和管道,已经进行了一些研究和联合项目,结果表明,根据工具或管道以及所使用的设置,结果存在显着差异。显然,除了足够的测序深度之外,用于注释读段的数据库的选择对结果有很大影响。
此外,通常会自动完成的参数选择(例如截止值,嵌合体过滤等)对结果有很大影响。例如,由于数据评估方法仅允许调查代表性超过1%的分类单元,因此经常忽略稀有分类单元。
然而,这种罕见的分类单元最多占所有微生物的28%,可以代表某些生境中的关键角色,并且可能对构建社区具有重要意义。它们在生物地球化学循环中可能起着过比例的作用,并且可能是微生物组功能的隐性驱动因素。而且,携带抗生素抗性基因的微生物通常属于天然微生物群中的稀有分类群。 在压力条件下,它们为健康和生存提供了保障,并确保了生态系统的可塑性。
但是,在动物和人类肠道微生物组中,这些方面对于疾病暴发和治疗至关重要,因此在WHO对抗生素耐药性扩散的预测中得到了应用。
同样,在这里,计算资源仍然是使生物信息学成为微生物组分析瓶颈的问题。Ten Hoopen和他的同事描述说,例如,塔拉海洋微生物群项目的一个子集,已经对原核生物进行了尺寸划分,包括135个样本。对于这些样本的分析,需要248次运行,其中包含288亿次读取,分析输出代表大约10 TB的数据。这项广泛的研究产生了232亿个预测的蛋白质编码序列。为了处理如此大的数据,在过去的10年里出现了几个全球微生物组学项目,如人类微生物组群项目和地球微生物组群项目。
此外,宏数据方面的巨大差距,通过阻碍研究的可比性和综合分析,限制了微生物组分析。确实有一些数据存储库,例如序列读取档案SRA(NCBI)、欧洲核苷酸档案(ENA)或CNGB核苷酸序列档案(CNSA)。然而,在许多情况下,数据只“按要求提供”,这与可查找、可访问、可互操作和可重用(公平)原则相矛盾。即使数据是可用的,宏数据往往缺乏重要的信息,并且没有得到包括实验和统计研究设计信息在内的知识库的充分审查。
Schloss和他的同事发表了一篇综合性的综述,关于识别和克服微生物组学研究中再现性、可复制性、稳健性和普遍性的威胁。因此,迫切需要标准化和开发正确和全面的宏数据存储库平台,如Proctor所示,他开发了人类病原体/载体基因组序列的项目和样本应用标准。Ten Hoopen和他的同事描述了一个设计良好的策略,允许制定和应用标准,并根据公平原则从微生物组研究中获得可比较和可重用的数据。
在经济学技术进步的推动下,微生物组数据的不断增加,使得我们对微生物组群在提高不同系统的生产力和可持续性方面的潜力的理解有了极大的提高。应用微生物研究的宏伟愿景是改善人类、动物、植物和整个生态系统的健康。
一般来说,微生物组可以通过以下方式直接管理:(i)微生物组移植
(ii)具有有益特性的微生物
(iii)微生物群活性代谢物,或通过改变环境条件,使微生物群也将其结构和功能从失调状态转变为健康状态。
在比较人类、动物和种植系统中基于微生物的应用时,可以看到显著的协同作用(下图)。
虽然各个领域还没有很好地联系起来,但所有领域的一致趋势已变得明显。这一趋势涉及到对定制治疗的关注,例如,“下一代”精确农业或个性化药物(下图)。这一概念认识到,并不是所有的个体宿主及其相关的本土微生物群都会以相同的方式对特定的引入微生物、微生物群落移植或代谢物作出反应。
图7 跨领域的微生物组应用趋势
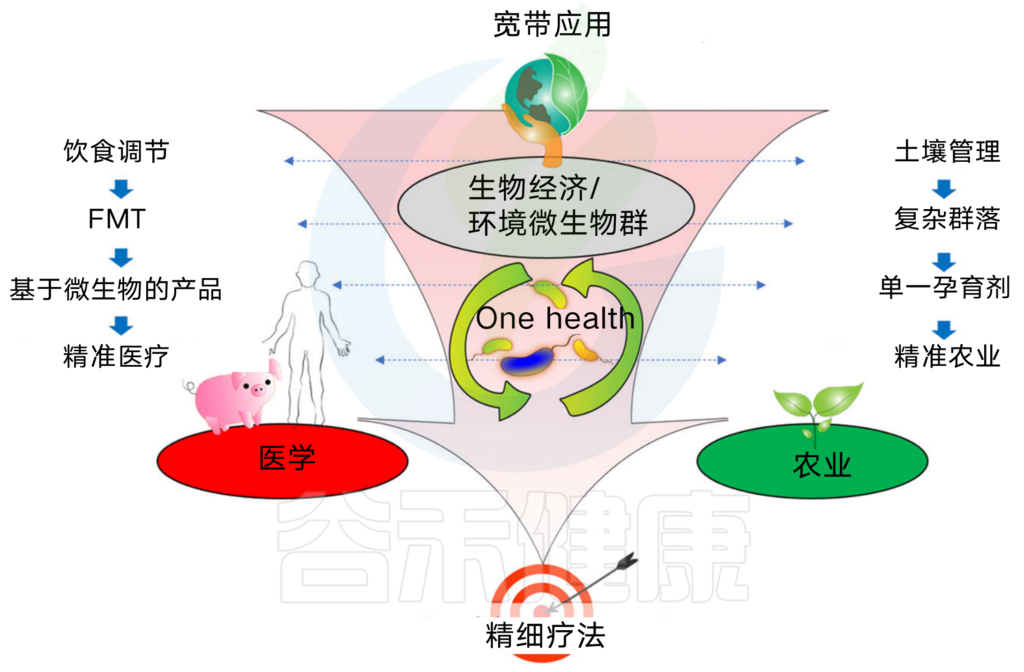
相反,它依赖于对那些特定宿主微生物、环境微生物和微生物相互作用的基本理解,这些微生物在不同的环境中调节微生物组合和功能能力。
Stegen等人在其微生物组管理的概念框架中,建议在不同领域进行更大的串扰,例如,利用环境微生物组学中的特定生态概念来指导优化策略,以操纵人类微生物群,从而改善健康状况。
通过为各种环境和代谢疾病提供有趣的解决方案,人类微生物群正逐渐成为个性化医学的一个关键目标。不仅有助于疾病各个方面的个体差异,也代表了潜在的治疗目标,可通过治疗,饮食变化,使用前,益生元或合生元,与之相对应的生命生物治疗产品来调节物种或物种混合(合成微生物群)和微生物组移植。
植物生物群落路线图
以此类推,植物微生物组被认为是下一次绿色革命(到2030年的科学突破)的关键。结合植物育种,精准农业,农业管理和微生物组研究的系统方法,为在不断变化的世界中改善可持续作物生产提供了强有力的战略。
跨学科团队将这种方法包含在特定环境中可能影响植物生产的许多生物和地球物理成分,称为“植物生物群落路线图” 。
特定于物种和栖息地的植物微生物区系对植物整体生物的功能有多个方面,例如(i)种子发芽和生长,(ii)营养供应,(iii)对生物和非生物胁迫因素的抵抗力,以及(iv)产生生物活性代谢物。 由于其对作物健康的重要性,对植物微生物组进行了长期的研究。
此外,在农业领域开发了广泛的微生物组管理策略和产品清单,包括(i)微生物组移植(粪便和生物动力添加剂),(ii)微生物接种剂,(iii)微生物和植物提取物以及(iv)改变环境条件的方法。
在过去的几十年中,管理密集型农业主要依靠合成化学物质,导致严重的环境和健康问题以及生物多样性丧失。另一方面,对植物微生物组学的研究将支持针对田间特定条件的针对性和预测性管理方法,从而可以提高可持续性。
种子/根际微生物组对植物至关重要,与我们的食物微生物群紧密相连
与人类肠道微生物组相似,种子/根际微生物组对植物至关重要,种子是下一代微生物的理想靶标。 Pérez-Jaramillo及其同事提出了种子的“回归根源”方法,这为揭示野生亲缘种和古代传家宝品种的种子微生物群提供了一个有趣的机会,以节省有益的种子微生物群用于农业。
利用农作物野生近缘种的种子微生物群或利用有前途的生物资源,有可能实现植物与其特定种子微生物群之间的匹配共生。 收获后微生物群与我们的食物微生物群紧密相连,也可以针对食品的所需功能特性进行管理,例如安全性和保存性,感官或健康特性。
这代表了一个相对尚未开发的基于微生物组的应用,它正在受益于食品生态系统中新出现的大量数据。
生活方式-微生物-人类-健康的联系
微生物群的重要性超出了个体宿主的健康。来自不同宿主和生态系统的微生物可以相互作用并相互影响。这些观察结果导致了“健康环境促进健康人类”的口号,支持“One Health”概念。
【注:“One Health”意指多学科共同合作,为人类健康,动物健康,环境健康三者共同成为一个健康整体而进行的工作和努力.“One Health”是一个新的名词,但是这一概念是很早之前已经存在的.2003年4月7日,华盛顿邮报的Rick Weiss引证了兽医博士William Karesh的话“人类和家禽或者野生动物的健康再也不能分开谈论了,世界上只有‘One Health’,所有的问题解决办法需要各个学科的工作人员共同努力,在不同的层面为问题的解决作出贡献.”关于“One Health”,目前国内翻译的名称非常不一致,比如“唯一健康”,“共同健康”,“一体化健康”, “大健康”等,编者在此不做翻译。
引自:Xiao Zheng, 陆家海, Sarah K.White,等. 应用”One Health”策略解决复杂健康问题[J]. 中华预防医学杂志, 2014, 000(012):1025-1029.】
根据世界卫生组织的说法,“One Health”是一种设计和实施方案、政策、立法的方法,以及多个部门进行交流和合作以取得更好的公共卫生成果的研究。“One Health”概念的扩展,包括环境健康及其与人类文化和习惯的关系表明,在社会决策和政策制定过程中,应考虑生活方式-微生物-人类-健康的联系(图7)。例如,城市化与过敏、哮喘和其他慢性病的增加有关。此外,在城市地区观察到总体污染模式,即微生物多样性的显著丧失,这与疾病的发展有关。
Dominguez Bello等人表明,伴随工业化而发生的人类微生物群的变化可能是代谢、免疫和认知疾病急剧增加的潜在因素,这些疾病包括肥胖、糖尿病、哮喘、过敏、炎症性肠病和自闭症。多样性的丧失反过来又与细菌对抗生素的耐药性增加相关,因此,需要实施策略来恢复建筑环境中的细菌多样性。了解不同宿主和栖息地的微生物群之间的复杂联系及其与人类、动物和植物健康的关系,为在One Health概念的背景下进行诊断、治疗和干预提供了创新和整体方法的潜力。
类似One Health,存在着将人类与环境健康联系起来的不同概念;行星健康概念是最流行的概念。许多国家和国际生物经济战略也承认这一主题,在这些战略中,可持续的生物产品生产满足了经济的需求。毫无疑问,要使这些概念成为成功的故事,不仅需要自然科学不同学科之间的相互关联战略,而且需要社会科学和利益相关者的整合。
然而,我们现在所面临的往往是相反的:生物多样性丧失、污染、臭氧消耗、气候变化、生物地球化学循环边界的跨越,都是人类世这个时代的特征。上新世也反映在行星边界概念中。
人类活动已经跨越了九个行星边界中的四个:气候变化、生物圈完整性的丧失、陆地系统的变化和生物地球化学循环的改变。
首先,研究表明,它们改变了整个微生物群落的功能和遗传多样性;然而,关于这些人为因素对不同微生物群落的影响及其对我们地球的影响的更多知识是绝对必要的。在这里,研究人员认识到迫切需要更多的研究,特别是关于环境微生物组和人为驱动变化的机制性见解。
了解海洋和陆生微生物群落及其相互作用可能是找到解决与上流新世相关的巨大挑战的关键。微生物群管理和基于微生物群的高潜力创新的开发在各个应用领域都是有希望的,但同时也应仔细评估这些新的和有前途的技术对环境的影响。
基于该领域的最新进展,研究人员建议恢复Whipps等人提出的微生物组术语的原始定义。该定义包含了1988年发表后30年内仍然有效的所有要点,并通过两个解释性句子对微生物群和微生物组进行了区分,并宣布了其动态特征。
微生物组
微生物组被定义为一个具有独特理化性质的、占据合理、明确的栖息地的微生物群落。
微生物组不仅指所涉及的微生物,还包括其活动区,从而形成特定的生态位。
微生物组是一个动态的、互动的微生态系统,在时间和规模上都会发生变化,它整合在包括真核生物宿主在内的宏观生态系统中,对它们的功能和健康至关重要。
微生物群
微生物群由属于不同领域的微生物组成(原核生物[细菌、古生菌]、真核生物[例如原生动物、真菌和藻类]),而“它们的活动区”包括微生物结构、代谢物、可移动遗传元素(例如转座子、噬菌体和病毒)以及嵌入到栖息地的环境条件。
此外,作者认为以下几点对于微生物组研究至关重要:
1.核心微生物群是一组来自相似栖息地的微生物群落之间共享的成员,这对于了解稳定性,可塑性以及在复杂微生物组合中的功能非常重要。
2.从宏观生态学改编的理论可能有助于理解不同环境中微生物组动力学的模式,但需要验证其一般应用。
3.每个微生物组研究的基础都是适当的实验,方法和统计设计。 在设计中应通过以下方式实现空间,时间和发育的整合:(i)根据系统特性选择适当的采样频率,以捕获完整的核心和瞬时微生物区系;(ii)通过考虑系统的适当空间规模 认识到规模的子集也与微生物群评估有关,并且(iii)对于强动力系统,应考虑研究微生物分布的时空连续体,而不是捕获一个特定的矩/空间单位。
4.方法学的进步极大地推动了微生物组的研究。 尽管在这方面取得了所有进展,但是还没有完美的通用方法。技术工具箱将减少每种技术带来的偏差,并从整体上更完整地了解生物系统。
5.微生物功能在生态系统中起着重要作用。 因此,我们建议在微生物组研究中包括几种当前可用方法的组合,以便对微生物功能有更深入的了解。
6. 尽管在过去十年里,我们获得了大量的组学数据,但我们仍然缺乏关于其背后的生物信息。因此,在微生物组学研究中实施更多基于培养的方法需要大力的努力,这样可以描述特定微生物群的生态类型和适应环境的模式。
7.微生物相互作用是微生物群落功能和进化动力学的基础。 因此,我们提倡考虑研究设计中的相互作用。
8.宿主与微生物的相互作用决定了相互适应性,表型和新陈代谢,从而提出了微生物群及其宿主的协同进化理论。 我们建议基于进化的全基因组理论的整体方法。 整体人的疾病状态以生物失调(病原体组)为特征,而尤物病是指平衡的宿主-微生物相互作用(“健康”微生物组)。
9.根据微生物与宿主之间的相互作用,将微生物分为有益的,致病的和中性的是基于人类中心观。确实,宿主和整个微生物组的生理学在很大程度上影响相互作用的结果。
这些澄清和建议的应用将有助于研究人员以整体的方式设计其微生物组学研究,这将有助于开发微生物模型和预测,进而加快我们在生活各个领域设计应用程序的能力。
【参考文献】
Berg et al. Microbiome (2020) 8:103
人类微生物组正逐渐成为个性化医学的一个关键目标
植物微生物组是下一次绿色革命的关键
我们一直在微生物领域助力科研,也希望为每个人的健康出一份力
欢迎关注谷禾健康
让你和你的家人更健康
近日,美国布兰代斯大学Piali Sengupta和Michael P. O’Donnell研究小组发现,肠道细菌产生的神经递质调节宿主的感觉行为(DOI:10.1038/s41586-020-2395-5)。相关论文于2020年6月17日在线发表在《自然》杂志上。该研究结果表明,肠道细菌产生的神经递质模拟了同源宿主分子的功能从而越过宿主调控感觉决定,这促进了宿主和微生物的适应性。


动物与包括微生物在内的多种生物体存在互惠共生关系。过去认为一些细菌产生的具有生物活性的神经递质可以调节其宿主的神经系统的活动和行为。然而这种“微生物-脑”的信号传输机制和它的生理相关性在很大程度上是未知的。在这篇文章中,研究人员通过使用秀丽隐杆线虫为模式生物,结果显示在肠道内定居共生菌普罗维登斯细菌产生的神经调节剂酪胺,绕过了宿主酪胺生物合成的要求并操纵了宿主的感觉行为。细菌产生的酪胺大概率被宿主酪胺β羟化酶转化为章鱼胺。反过来,章鱼胺靶向ASH伤害感受神经元上的OCTR-1章鱼胺受体以调节厌恶性嗅觉反应。研究人员确定了普罗维登斯细菌酪胺生物合成所需的基因,并表明这些基因对于调节宿主行为是必需的。而在普罗维登斯菌的定殖实验中发现,秀丽隐杆线虫在进行食物选择时,优先选择这类细菌,这种选择偏好性需要细菌产生的酪胺和宿主章鱼胺信号。结果表明,肠道细菌产生的神经递质模仿宿主同源分子的功能,以超越宿主对感觉决定的控制,从而促进宿主和微生物的适应性。
秀丽隐杆线虫(Caenorhabditis elegans,C. elegans)是一种无毒无害、可以独立生存的线虫。其个体小,成体仅1.5mm长,为雌雄同体(hermaphrodites),在20℃下平均生活史为3.5天,平均繁殖力为300-350个。自1965年起,科学家Sydney Brenner利用线虫作为分子生物学和发育生物学研究领域的模式生物。进入21世纪以来,已经有六位科学家利用秀丽隐杆线虫为实验材料解开了生命科学领域的重大秘密而获得了诺贝尔奖。2013年12月17日,据国外媒体报道,科学家认为,如果延长蠕虫生命的科学应用到人类身上,人类活到500岁将不只是梦想。秀丽隐杆线虫的两个基因通路,使通过遗传相互作用进行抗衰老治疗的成为可能。
背景
肠道定植细菌和宿主神经系统之间的化学沟通的调节途径大部分还是未知的。秀丽隐杆线虫是近年来出现的一种研究宿主-微生物化学沟通的强大实验系统。野外线虫的主要食物来源于其肠道内定殖的不同的致病菌和非致病菌菌群。接触致病菌会改变秀丽隐杆线虫的嗅觉行为,但肠道内的共生菌是否也能调节宿主行为还不清楚。
实验设计
线虫的制备:
所有的实验都使用的雌雄同体的秀丽隐杆线虫,所有应用于线虫的菌株都保持在在NGM平板上20℃条件下。分别以10
ng/μl注射SRV-11p::octR-1(PMOD110)和SRG-47p::ocTR-1(PMOD111)质粒。以20 ng/μl注射tbh-1p::tbh-1a::sL2::mCherry(PMOD115)质粒。以30 ng/μl注射UNC-122p::mCherry共注射标记,获得转基因菌株。接着将线虫培养在NGM平板上,以获得不同细菌型或基因型的线虫。
主要的实验:
长时程趋化实验和短时程趋化实验分别用于分析线虫对食物和环境选择的偏好性。响应1-辛醇和2-壬酮的SOS分析用于测定其对100%1-辛醇和100%2-壬酮的反应。肠道细菌细胞的量化、定量测定肠道菌落形成单位、荧光强度测量反应了细菌的变化趋势。通过细菌宏基因组测序得到的组装序列,使用普罗维登斯菌株JUb39 的TyrDC和adcA基因序列blastp比对至Nr数据库初始筛选,然后以粪肠球菌TyrDC和线虫tdc-1基因组序列作为tblastn搜索查询序列,最后使用从这些tblastn查询得到的氨基酸序列构建最终的系统发育树。Metaboseek MS分析软件分析高效液相色谱质谱法数据,并集成代谢网络分析。所有的统计分析都是在原始的、非归一化的数据上进行的。
主要结果:
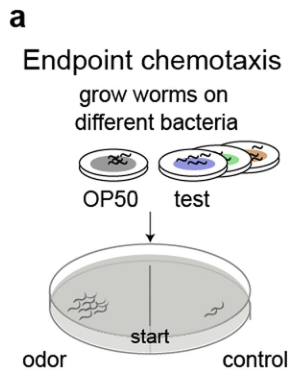
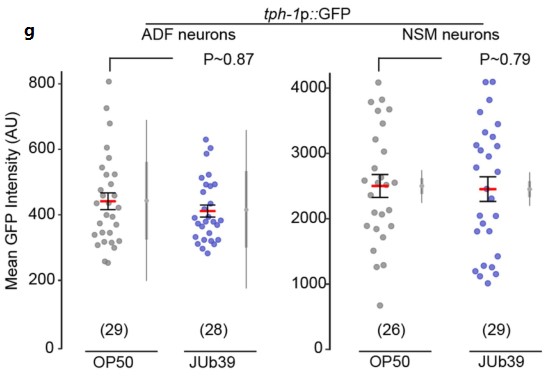
1肠道细菌改变宿主嗅觉行为。普罗维登斯菌定殖于秀丽隐杆线虫肠道内,定植程度与对辛醇的回避行为正相关。
注:以下的辛醇调节表示为对辛醇的回避行为减少的现象
a. 在长时程趋化实验分析中,秀丽隐杆线虫与这些指定的细菌菌株共培养,对一组有吸引力的挥发性气味表现出强烈的吸引力。纵坐标的chemotaxis index表示趋化指数,在辛醇(1-octanol)和壬酮(2-nonanone)的实验中,与在大肠杆菌OP50菌株共培养的线虫相比,与普罗维登斯JUb39菌株共培养导致线虫对辛醇的回避行为减少,但对壬酮的回避行为不减少。
b.c. Modulation index表示线虫对100%辛醇的调节指数。Dead指用庆大霉素预处理过的细菌。PYb007指从堆肥中的线虫中分离出来的与普罗维登斯JUb39菌株有较远亲缘关系的 Providencia Rettgeri菌株。灰色虚线指在辛醇环境下生长的线虫与在不同环境下独立生长的线虫的对照。正值表示辛醇回避减少。使用庆大霉素预处理过JUb39菌株喂食线虫可以消除辛醇调节。而线虫必须摄取活的JUb39菌株才能诱导辛醇调节。
d.在成年线虫的后肠中使用红色荧光蛋白mCherry标记细菌。箭头指示完好的细胞。星号指示漫反射肠道荧光。虚线指示肠边界。喂食OP50的成虫的肠道只显示弥漫的肠道荧光而喂食JUb39菌株的成虫肠道内通常含有大量完整的杆状细胞,表达mCherry。JUb39细菌倾向于富集在后肠,此外被JUb39菌株定植的线虫没有表现出病原性感染的表型特征(如肛门肿胀),进一步证实JUb39菌株对线虫基本无致病性
g.在OP50或JUb39菌株环境下生长的年轻成虫的头部神经元tph-1p::GFP的荧光报告基因检测。发现tph-1p::GFP的表达没有明显变化。且a图中线虫在细菌含量较低的食物来源B. megaterium菌中的生长并没有改变辛醇调节。由此推断普罗维登斯菌对辛醇的调节不太可能是由于摄食状态的改变。
e.被喂食指定菌株生长的线虫的肠道细菌载量。点表示10条线虫的菌落形成单位中的细菌载量。在喂食JUb39菌株的线虫可以分离出大量的细菌菌落,但在喂食OP50菌株的线虫不能分离出大量的细菌菌落。这表明JUb39菌株很可能活在线虫肠道中。
f.在趋化试验中, 朝1-Octanol(100%辛醇)方向的线虫始终含有更多的肠道细菌。括号中的数字表示线虫的数量。
2. 发现了一种可能性即线虫产生的章鱼胺,而不是酪胺,其对于JUb39菌株介导的辛醇调节是必要的。细菌产生的酪胺在功能上补充了宿主衍生的酪胺在驱动感觉行为决定方面的损失。
a.线虫体内酪胺(TA)和章鱼胺(OA)的生物合成。酪胺通过酪胺β-羟化酶(由TBH-1编码)转化为章鱼胺。
b.线虫对100%辛醇的调节指数。括号内的数字表示至少3个独立日内进行独立化验的次数。发现在JUb39菌株环境下生长的tdc-1和tbh-1突变体线虫都表现出辛醇调节,但tbh-1突变体线虫表现出的辛醇调节减弱。cat-2和tph-1的突变并不影响辛醇调节。WT表示野生型。
c.短程急性回避行为试验SOS分析。评估辛醇反应的单胺能调节。
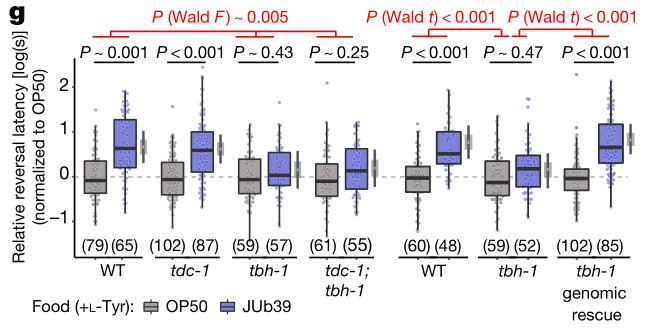
d.g.使用SOS分析进行了所有的辛醇行为实验。
d图中OP50菌株环境下生长的tdc-1突变线虫比野生型线虫对30%辛醇的反应更快,但在JUb39菌株环境下受到抑制。
g图中tbh-1的突变完全抑制了辛醇的调节,而tdc-1突变的线虫对辛醇的调节一如既往。此外tdc-1和tbh-1双突变线虫的辛醇回避行为与只有tbh-1突变的线虫相似。这些结果表明,缺乏宿主来源的章鱼胺是tbh-1突变线虫辛醇调节减少的原因。红色的P值指示指定基因型对JUb39菌效应大小的影响。
e.f. HPLC-MS进行代谢组成分析和N-琥珀酰酪氨酸的定量分析。喂食OP50菌株的tdc-1突变线虫缺乏N-琥珀酰酪氨酸。而5-羟色胺(N-acetyl serotonin)代谢不变。当喂食JUb39菌株时,这些酪胺衍生的代谢物被恢复。结果表示普罗维登斯细菌与线虫联合产生的酪胺,可以弥补tdc-1突变线虫内源性酪胺产生的不足。
3. 在真核生物和细菌中,生物胺通常由AADC产生。普罗维登斯细菌的多个AADCs产生的酪胺对于调节野生型线虫回避辛醇的行为是必要的,也是理由充分的。
a.从上至下分别描述了乳杆菌的tryDC位点,Morganella菌的adcA位点,对JUb39菌株的tyrDC和adcA位点(分别为ΔtyrDC::cmr和ΔadcA)中设计了缺失。革兰阳性菌肠球菌和乳杆菌的酪胺生成主要是通过位于操纵子的两个基因tyrDC和tyrP介导的,它们分别编码AADC
tyrDC和酪氨酸通透酶。该操纵子可被l-Tyr诱导。全基因组测序证实JUb39菌株和PYb007菌株中均含有tyrDC和tyrP的同源操纵子。在Morganellaceae科的其它成员中也发现了tyrDC同源物。
b.系统发育分析。Morganellaceae科的菌株以前曾被报道在某些条件下产生酪胺,尽管没有可辨别的tyrDC同源物。但在摩根氏菌中鉴定了一个编码AADC的基因(这里命名为adcA),分别位于编码Tyt1家族酪氨酸渗透酶基因上游的操纵子中,与tyrDC肠球菌和人类GAD67有大约29%和27%的序列同源性。在普罗维登斯菌基因组中也存在adcA同源物(包括JUb39),但不与酪氨酸转运蛋白相邻。结论是,普罗维登斯菌编码至少两个可能产生酪胺的AADC,系统发育不一致提示,摩根科植物中的tyrDC和adcA基因可能都是通过水平基因转移而丢失或获得的。
c. 以乳杆菌tyrDC的晶体结构为指导进行蛋白质模拟,结果表明JUb39 tyrDC与乳杆菌tyrDC具有大多数已知的催化位点。JUb39 tyrDC在A600处含有一个替换(乳杆菌tyrDC中的S586),能增强酪氨酸乳酸杆菌对酪氨酸的特异性催化活性。由此推测JUb39 tyrDC可能由酪氨酸产生酪胺。
e.在不同基因型细菌环境下生长的野生型线虫在NGM+0.5%l-Tyr培养基中生长期间对100%辛醇回避行为的SOS测定。h图为在此基础上补充了指定浓度的酪胺。
在单个细菌突变体的培养中,辛醇调节减弱,而野生型线虫在双敲除ΔtyrDC::cmrΔadca基因JUb39菌株环境下生长则取消了辛醇调节。
g.f. N-琥珀酰酪胺的含量测定(f);(g)检测到的其它酪胺衍生物的平均丰度,1指示OP50,2指示JUb59,3指示ΔtyrDC::CmrΔADCA JUb39。与野生型JUb39相比,比较代谢组学分析表明,在双敲除ΔtyrDC::cmrΔadca基因JUb39菌株环境下生长的tdc-1突变线虫不产生N-琥珀酰酪胺或其他酪胺衍生代谢物。
h.在双敲除ΔtyrDC::cmrΔadca基因JUb39菌株环境下生长的野生型线虫在补充酪胺后辛醇调节得以恢复。此外,虽然JUb39菌株环境下生长的野生型线虫不会因为补充酪胺而增加对辛醇的回避行为,但补充酪胺足以诱导在OP50菌株环境下生长的野生型线虫的辛醇调节。
4. 普罗维登斯细菌的酪胺靶向感觉神经元。线虫头部两栖器官中的双侧ASH伤害性神经元表达多种酪胺和章鱼胺受体,这些单胺对辛醇的调节需要一部分受体。普罗维登斯细菌酪胺的产生和宿主章鱼胺的信号都是发生食物偏好所必需的。
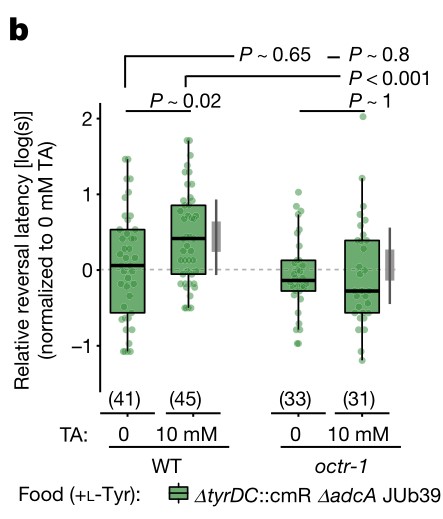
a. SOS测定。在ASH神经元表达的章鱼胺受体编码基因中,octr-1而不是ser-3的突变在不改变肠道定植的程度下取消了JUb39菌株介导的辛醇调节。但在ASH神经元表达的octr-1互补DNA完全恢复了辛醇调节。
b. SOS测定。当线虫在添加了酪胺的ΔtyrDC::cmR ΔadcA JUb39菌株环境中生长时,octr-1突变线虫也会缺乏辛醇调节。
c. JUb39菌株和其他普罗维登斯菌株会产生酒精异戊醇,这种酒精在浓缩时对线虫是有害的。线虫在食物选择实验中可能会优先选择对这类酒精产物的回避行为的减少的这些细菌。
d.e. 在指定菌株环境下生长的野生型和突变型线虫对测试菌株JUb39的相对偏好指数,事实上,在JUb39菌株环境下生长的线虫更喜欢JUb39菌株,而在OP50菌株环境下生长的线虫在短期食物选择试验中只显示出对JUb39菌株的轻微偏好,在octr-1和tbh-1突变的线虫中也是如此。
f. JUb39菌株在线虫肠道内产生的酪胺(TA)通过宿主章鱼胺(OA)和octr-1章鱼胺受体发挥作用,以减少ASH神经元介导的对JUb39菌株产生的回避行为,并改变摄食选择。
结论:
研究人员认为,综上所述,肠道普罗维登斯细菌产生的神经递质酪胺可以推翻宿主依赖的酪胺合成,从而调节线虫的多条单胺能途径,改变线虫的行为和生理。研究人员推测,普罗维登斯菌一旦定植于秀丽隐杆线虫肠道内的,就会促进更多的消耗,从而导致细菌稳定的联系和传播。由于普罗维登斯菌是线虫的主要食物来源,这种联系可能是互惠互利的。
这些结果描述了一种途径,即由天然共生细菌产生的神经递质通过补充或补偿关键的宿主生物合成酶的活性来指导宿主的感觉行为决定,从而改变宿主和微生物的适应性。