 国家高新企业 | ISO9001认证
国家高新企业 | ISO9001认证 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 二级病原微生物安全实验室
冠状动脉疾病是世界范围内最常见的健康问题,也是导致发病率和死亡率的主要原因。肠道菌群在人类新陈代谢、免疫力等方面扮演重要角色,与冠状动脉疾病之间也存在相关性。
1、肠道微生物群与CAD发展之间的因果关系尚未得到证实。
2、必须通过微生物代谢产物和与免疫系统的相互作用,了解肠道微生物群与CAD发展之间潜在的直接和间接因果关系。
3、动态因素包括我们的饮食和人口因素,如年龄、性别和种族,也会影响我们的肠道微生物群和CAD的发展,并使这一问题复杂化。
4、需要跨学科的方法来揭示肠道微生物群的调节所涉及的因素及其与CAD发展的关系。
5、阐明系统层面的多方面因素参与微生物介导的机制和人类健康与疾病,可以进一步指导新的预防和治疗CAD的干预措施。
高血清胆固醇(高胆固醇血症)是最常见的心血管疾病(简称CVD,下同)的危险因素,称为冠状动脉疾病(简称CAD,下同)。
动脉壁内胆固醇沉积(斑块)的累积可导致动脉粥样硬化。

高胆固醇血症可能有遗传来源,并影响主要负责体内胆固醇稳态的身体功能,包括从头合成、肝脏分解代谢和胆汁分泌,以及肠道吸收。
胆固醇的来源
人体内的胆固醇来源于两种来源,在肝脏中从头合成,或通过饮食和富含胆固醇的食物进入人体。人体内大约四分之一的胆固醇来自膳食摄入(外源性),其余的是通过甲羟戊酸途径从头合成(内源性)。
冠状动脉疾病的形成
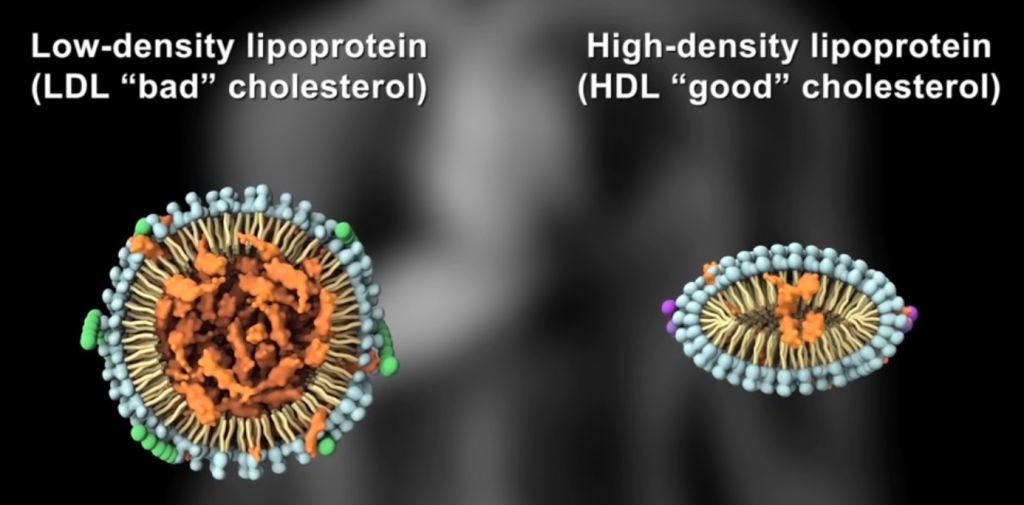

体内合成的胆固醇分为高密度脂蛋白胆固醇(简称HDL,下同)和低密度脂蛋白胆固醇(LDL),后者可进入循环系统,成为冠状动脉疾病的重要标志。
我们看看LDL如果多了会发生什么:
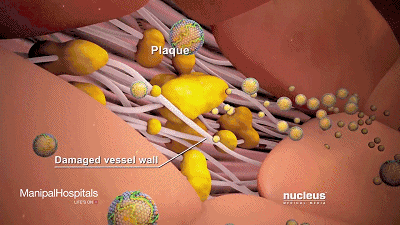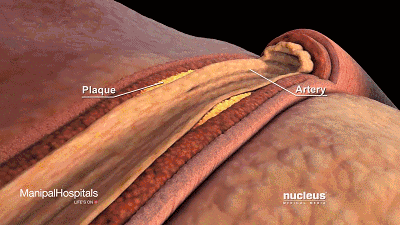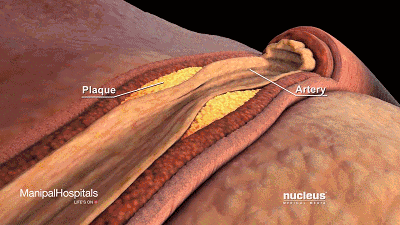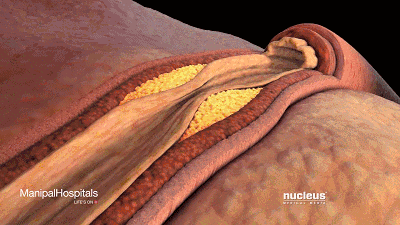

可以看到它聚积在动脉壁动脉,随着时间的推移脂肪沉积形成斑块,使动脉狭窄。
高密度脂蛋白胆固醇与CAD呈负相关,也就是说它在阻止CAD的形成。通过发挥抗炎和抗氧化作用以及促进反向胆固醇转运(RCT)而具有抗动脉粥样硬化的功能,RCT可以消除低密度脂蛋白胆固醇。
然而,在炎症、糖尿病和氧化应激等条件下,高密度脂蛋白可能会失去其抗动脉粥样硬化特性,并成为促动脉粥样硬化(功能失调)物质。此外,低密度脂蛋白胆固醇升高是冠状动脉疾病的危险因素,这可能是由于巨噬细胞摄取低密度脂蛋白胆固醇颗粒导致泡沫细胞和动脉粥样硬化。
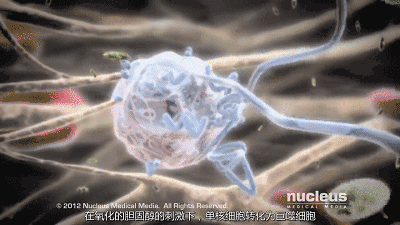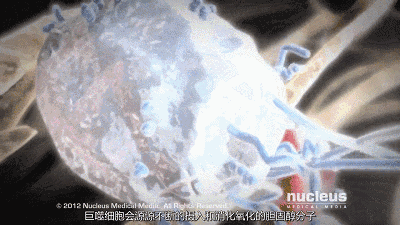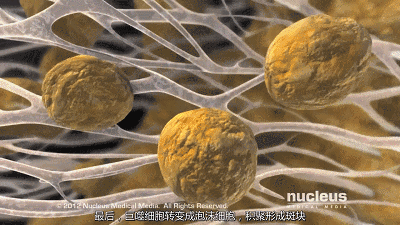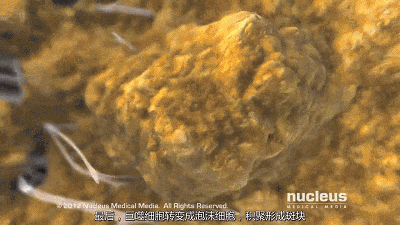

肠腔在控制体内胆固醇平衡方面起着重要作用,并通过胆固醇吸收负责外源性摄入。管腔内胆固醇可以来自不同的来源,主要来源于(i)我们的饮食,(ii)通过肝胆途径的胆汁,(iii)通过经肠胆固醇流出途径的新胆固醇。
图1 腔内胆固醇的外源性和内源性来源
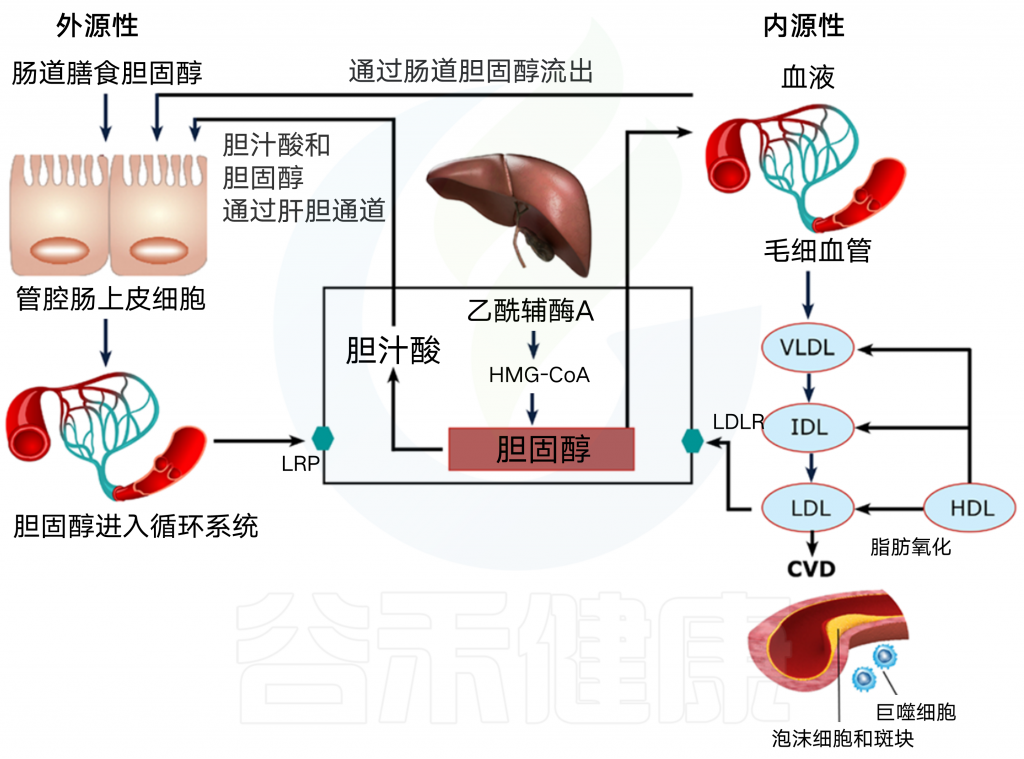

在肝脏中,胆固醇被代谢成胆汁酸,并通过肝胆途径分泌到胆汁中,其中ATP结合盒转运体G5/ATP结合盒转运体G8(ABCG5/G8)在胆固醇从肝细胞流出到胆汁中起关键作用。
TICE是肝胆通路的另一种途径,血液中的胆固醇可以通过低密度脂蛋白受体(LDL-R)直接进入肠细胞,并通过ABCG5/G8和ATP结合盒转运体B1(ABCB1a/b)进入肠腔。
然后,肠腔中的胆固醇通过Niemann-Pick C1-like 1(NPC1L1)被吸收到肠细胞中,并与乳糜微粒结合进入循环系统,或者被肠道微生物群还原为低吸收的粪烷醇(5B胆固醇-3B-ol),大部分排出体外。
其他因素影响胆固醇平衡和CAD发展
除了体内多种胆固醇来源的复杂相互作用外,还有许多其他因素可以影响胆固醇平衡和CAD的发展,包括肠道微生物群。
到目前为止,已经有人认为肠道微生物群的改变与代谢紊乱如肥胖、糖尿病和心血管疾病(与年龄、性别和宿主遗传学无关),包括动脉粥样硬化、血脂异常、高血压和心力衰竭之间的关系。
这种联系可以通过直接(通过代谢物)和间接途径(通过免疫系统)实现。
肠道微生物群在维持宿主健康方面发挥着多重关键作用,包括帮助宿主获得营养和能量、肠上皮内稳态、药物代谢和毒性、免疫系统反应以及抵御病原体。
微生物代谢产物影响
这些微生物还可以产生微生物产物,如尿毒症毒素、胆汁酸、氧化三甲胺(TMAO)、短链脂肪酸(SCFA)、脂多糖(LPS)、一氧化氮、维生素K、维生素B复合物、肠道激素和神经递质,它们可以改变宿主的新陈代谢和在健康和疾病状态下影响身体功能。例如,对动脉粥样硬化的易感性已经被证明可以通过小鼠模型中的微生物群移植进行转移。
迄今为止,许多传染源与动脉粥样硬化有关,包括幽门螺杆菌、巨细胞病毒、丙型肝炎病毒、肺炎衣原体和牙龈卟啉单胞菌。
Mitra等人的一项研究结果表明,在有症状和无症状的动脉粥样硬化斑块之间,微生物群表现出差异,无症状斑块有更多的宿主微生物群,包括卟啉单胞菌科、拟杆菌科、微球菌科和链球菌科。与此相反,有动脉粥样硬化斑块中含有越来越多的致病菌,包括螺旋杆菌科、奈瑟菌科和Thiotrichacaea.
此外,由于肠道微生物群整体状态的破坏而导致的肠道微生物群失调与炎症的增加有关,炎症与动脉粥样硬化的发展有关。
肠道菌群与高血压、心力衰竭、中风等有关
最近,肠道微生物群及其代谢产物的改变也与高血压和血管功能障碍有关。心力衰竭还与特定的肠道微生物种类有关,如增加的大肠杆菌、肺炎克雷伯菌、病毒性链球菌等。
一项研究表明,有症状的中风和短暂性脑缺血发作患者的肠道微生物群发生了改变,增加了机会致病菌,包括肠杆菌、巨球菌、Oscillibacter和脱硫弧菌。
此外,肠道微生物群有能力导致血脂成分的实质性变化,从而影响冠状动脉疾病的发展。例如,厚壁菌如罗伊氏乳杆菌与高密度脂蛋白相关,而Eggerthella属与低密度脂蛋白相关。
目前,肠道微生物群与CAD发展之间的因果关系尚不清楚,因为许多其他人口因素,如年龄、性别和种族,不仅会影响肠道微生物群和胆固醇水平,而且还会影响我们的饮食,这是影响肠道微生物群和全身胆固醇水平的另一个组成部分。因此,体内胆固醇的调节是一个复杂的机制,其中的各种因素在一个多方面的系统中交织在一起。
图2 CAD发展中涉及的多方面机制
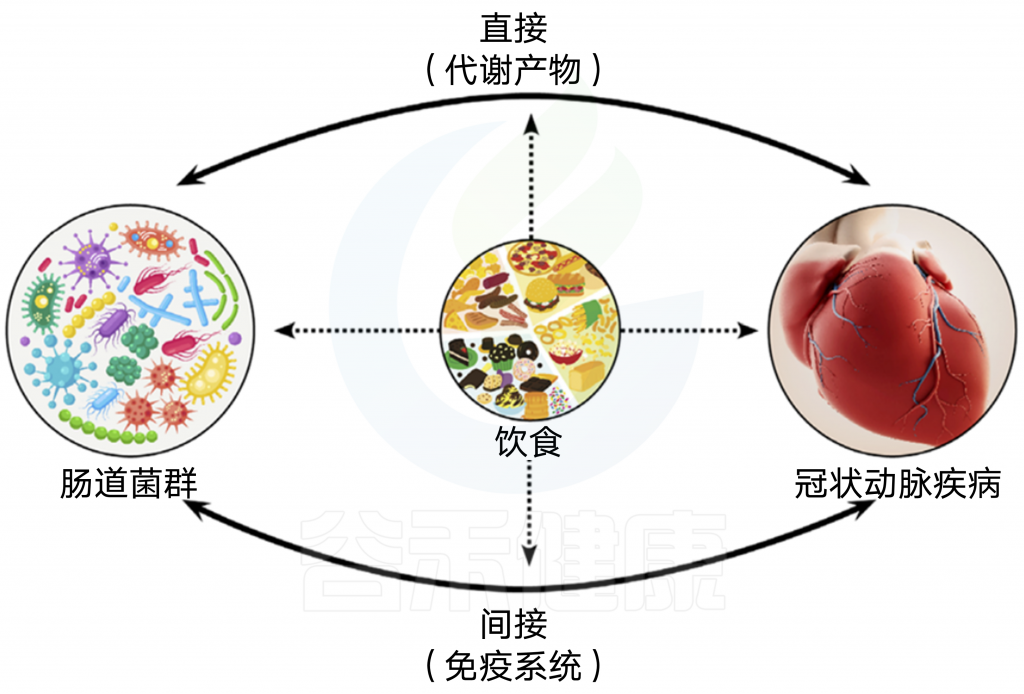

肠道微生物群可以直接(通过代谢物)和间接(通过免疫系统)导致CAD。
因此,需要进一步的研究,以了解潜在的机制,并确定哪些微生物菌株或其代谢物负责CAD的发展。
接着,将通过直接和间接途径探讨肠道微生物群的动态因素及其对高胆固醇血症和冠状动脉疾病发展的影响。
肠道微生物可以通过代谢产物如胆汁酸、粪甾醇、短链脂肪酸和三甲胺-N氧化物的产生直接影响高胆固醇血症和CAD的发育。
胆汁酸调节
肠道微生物可以影响肝脏胆固醇代谢的调节,并在改变胆汁酸中发挥作用,从而影响全身胆固醇水平(图3)。
图3 影响CAD的多方面机制
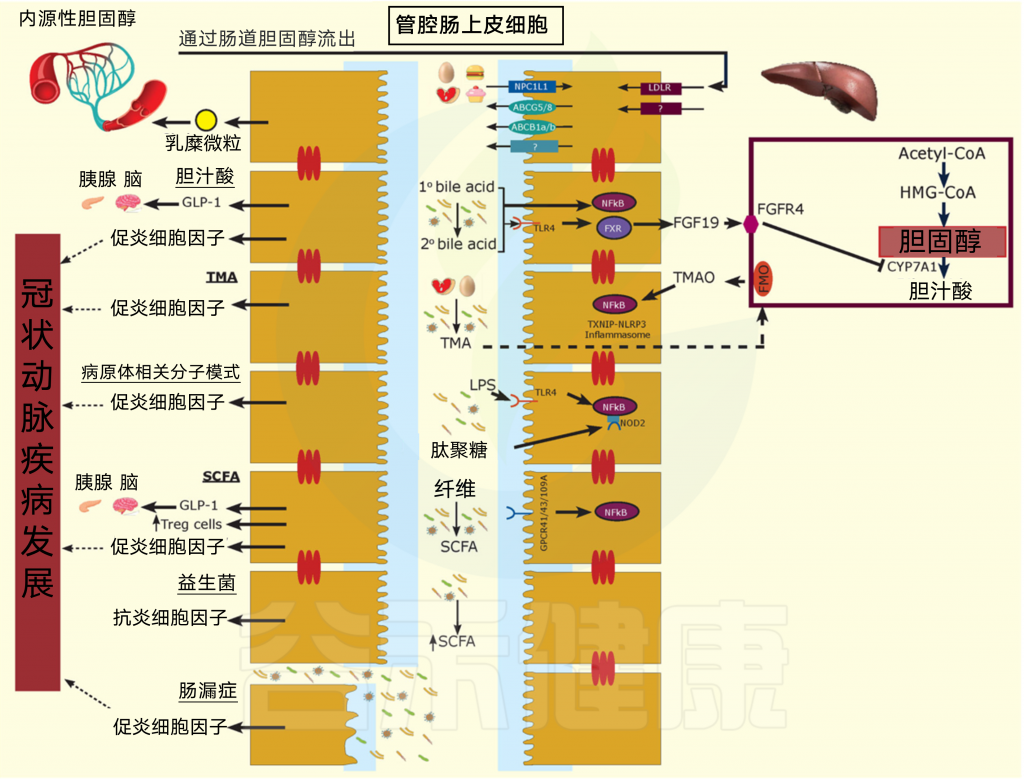

肠腔胆固醇和饮食的外源性和内源性来源,以及影响免疫系统和CAD发育的肠道微生物机制。
胆汁酸由限速酶胆固醇7-α-羟化酶(CYP7A1)形成,是肝脏中胆固醇的主要代谢产物,有助于脂肪、营养物质和亲脂维生素的吸收,也有助于调节脂质、葡萄糖和能量代谢。
初级胆汁酸
初级胆汁酸与氨基酸、牛磺酸或甘氨酸结合形成胆汁盐,分泌到胆汁中并储存在胆囊中,直到它们释放到小肠,在小肠中乳化脂肪并形成胶束,这些胶束被吸收到肠细胞中。
在肠道中,初级胆汁酸,如胆酸(Ca)和鹅去氧胆酸(CDCA)被肠道微生物和胆盐水解酶(BS H)去结合,形成次级胆汁酸,包括脱氧胆酸(DCA)、石胆酸(LCA)和熊去氧胆酸(UDCA)。
胆固醇水平如何升高
腔内所有共轭和未结合的胆汁酸可被重新吸收(95%)并转运回肝脏,但UDCA和LCA除外,它们大多在粪便中排出。肠道内胆汁酸等信号分子也能激活核受体法尼样X受体(FXR)和膜G蛋白偶联的胆汁酸受体Gpbar-1(aka TGR5)。通过这一机制,胆汁酸可以下调胆汁酸的合成,从而导致胆固醇水平升高。
胆汁酸激活FXR的顺序为CDCA>DCA>LCA>CA,FXR可诱导成纤维细胞生长因子19(FGF19)激活成纤维细胞生长因子受体4(FGFR4),抑制CYP7A1下调胆汁酸合成。
FXR还可以通过增加ATP结合盒B亚家族成员11(ABCB11)的表达,减少肝细胞对胆汁酸的摄取,增加胆汁酸的分泌。
原发性和继发性胆汁酸比值可能与高胆固醇血症和CAD的发生有关。例如,在Myerhofer等人的一项研究中,心力衰竭患者的血浆原发胆汁酸减少,继发胆汁酸与原发胆汁酸的比例更高,胆汁酸也可以通过以下途径在心血管功能中发挥作用。
肠道菌群调节胆汁酸比率
通过调节心房和心室心肌细胞的通道传导和钙动力学及调节血管张力来降低心率。此外,研究人员还提出,肠道微生物群调节胆汁酸比率,如果不平衡和处于不健康状态,可能导致继发胆汁酸减少,从而增加初级胆汁酸,如CDCA,激活FXR,降低胆汁酸生成,从而增加胆固醇和CAD的发展。因此,肠道微生物群及其相关的潜在机制需要进一步研究。
粪甾醇生产
某些肠道微生物群长期以来一直被认为具有将可吸收胆固醇转化为粪甾醇的能力,这是一种还原的不可吸收粪甾醇,可在粪便中排出。
人体内的粪甾醇的产生开始于生命第一年的后半段,并依赖于性别,年轻女性比年轻男性具有较高的转化能力。
此外,目前认为,人类群体中微生物胆固醇到粪甾醇的转化率是双峰的,高转化率显示几乎完全的胆固醇转化,而低转化率显示粪甾醇含量不到粪便中性固醇含量的三分之一。
迄今为止,分离出的降胆固醇菌株仅限于真杆菌属和拟杆菌属,但仍有许多有待发现。
使用动物模型,口服E. coprostanoligenes导致饮食诱导的高胆固醇血症兔的血浆胆固醇浓度显著降低,并在最后一次细菌喂养后持续至少34 天。
对于人体模型,已经有许多关于肠道胆固醇代谢的研究,并且已经提出了人类血清胆固醇与人类粪便中粪烷醇/胆固醇比率之与粪便中粪甾醇/胆固醇的比值呈反比。
然而,这些研究采用了非常小的样本量,样本群体的变化有限,缺乏不同的人口背景,包括未能成功地分离出导致粪甾醇/胆固醇转化的特定微生物菌株。此外,参与胆固醇在肠道内转化为粪甾醇的基因或酶仍然未知。
短链脂肪酸生产
SCFAs是一种微生物衍生的代谢物,由复杂的碳水化合物发酵形成,影响宿主的一系列过程,如宿主微生物信号、能量利用和结肠pH的控制,从而影响微生物群的组成和肠道运动。我们在前面的文章中也多次提到短链脂肪酸主要有乙酸、丙酸和丁酸。
拟杆菌门可以产生乙酸、丙酸,厚壁菌门可以产生丁酸。SCFAs与Alistipes putredinis、拟杆菌、罗斯氏菌(产丁酸盐的菌)、直肠真杆菌和普氏粪杆菌 呈正相关。
此外,它们通过调节紧密连接蛋白的表达,在维持肠屏障完整性方面起着不可分割的作用。
SCFAs还可以通过阻止胆固醇合成和/或将其转移到肝脏来降低血脂水平;因此,它们被认为是CAD发展中的一个保护因素。
在某些CAD患者和高血压患者的肠道失调中,通过激活G蛋白偶联受体41(GPR41),产生SCFA的细菌也减少了。因此,他们在体内的作用和目标需要进一步的研究。
三甲胺-N-氧化物(TMAO)生产
膳食胆碱、甜菜碱、磷脂酰胆碱、卵磷脂和左旋肉碱参与了TMAO的产生,TMAO是冠状动脉疾病发展的危险因素。
平时我们吃的包括红肉、鸡蛋、鱼、芸苔类蔬菜、花生和大豆等都有这种TMAO。


TMAO升高带来风险
具体来说,TMAO水平的升高与死亡和非致命性心肌梗死或中风的风险增加有关。肠道微生物通过(A)胆碱的产生和(B)中间分子三甲胺(TMA)的产生也在TMAO的产生中起作用。
最近才发现肠道微生物通过磷脂酶D(PLD)酶产生胆碱的能力。微生物介导的TMA分子可以进入宿主循环并进入肝细胞,在那里它被含黄素单加氧酶(FMO)代谢为TMAO,该酶由肝脏、肾脏和其他组织中的FMO基因编码。
高TMAO的产生会影响脂质,并导致43%的CAD风险,因为胆固醇转运的减少和胆汁酸运输、组成和细胞池大小的改变。TMAO还与C-反应蛋白(CRP)和内皮功能障碍有关,在肠道通透性增加的情况下,与LPS内毒素水平升高有关。此外,它还会导致钙的释放和血小板的高反应性,从而影响CAD。
与TMAO产生的有关菌群
肠道微生物群对TMAO的产生有重要影响。 健康人体内有大量产生TMAO的菌群,厚壁菌门与拟杆菌门的比例为2:1。在覆盖36个物种的102个基因组中发现了TMA的产生,TMA的产生者包括厚壁菌门,变形菌门,放线菌门,在拟杆菌门中不存在。


一项研究发现,来自于厚壁菌门和变形菌门的8种细菌,为生产TMA消耗了60%的胆碱,分别是:
产氢厌氧菌、天冬酰胺梭菌、C. hathawayi,
C. sporogenes, Escherichia fergusonii,
Proteus penneri, Providencia rettgeri, Edwardsiella tarda.
其他与高TMAO产生相关的肠道微生物包括:
阿克曼菌、孢子杆菌、普雷沃菌和瘤胃球菌,它们与动脉粥样硬化性冠心病相关。
因此,代谢物包括胆碱、TMA和甜菜碱可以帮助预测CAD的发展。 例如,益生菌或药理学干预可以用来抑制或阻断特定的微生物代谢途径,以减少产生TMAO的微生物。
肠道微生物也可以通过间接途径导致CAD的发展,如操纵我们的免疫系统。
动脉粥样硬化是由动脉粥样硬化血栓形成引起的一种慢性炎症性疾病,在这种疾病中:
(A)表面侵蚀可导致血栓形成
(B)细胞因子损伤的斑块破裂,从而导致暴露的凝血系统,导致血流抑制并诱导CAD。
因此,炎症引起的巨噬细胞和免疫系统与CAD有关。例如,最近高白细胞(WBC)计数被认为是CAD发展的危险因素。
此外,一项研究确定IL-22途径可作为代谢性疾病治疗干预的新靶点,因为IL-22能提高胰岛素敏感性,保护肠粘膜屏障和内分泌功能,减少内毒素血症和慢性炎症,调节肝脏和脂肪组织的脂质代谢。
在我们体内,氧化低密度脂蛋白(oxLDL)也可以通过激活内皮细胞、巨噬细胞和T细胞来发挥促动脉粥样硬化和促炎症作用。
巨噬细胞可吞噬oxLDL,导致炎性细胞因子如肿瘤坏死因子α(TNF-α)、白细胞介素1β(IL-1β)、IL-6、IL-18、IL-37和泡沫细胞,从而加重CAD。
TNF-α还通过激活蛋白激酶c(PKC)参与包括糖尿病在内的CAD危险因素,PKC增加胰岛素受体底物的磷酸化,导致其失活。
T细胞还可导致促炎细胞因子IL-2、IL-12和干扰素γ(I FN-γ),这与动脉僵硬有关。 泡沫细胞、T细胞和巨噬细胞一起会导致出现脂肪条纹,从而促进CAD的发展。
肠道菌群群落结构影响免疫系统
肠道微生物群的群落结构可以极大地影响我们的免疫系统。 例如,肠道微生物群的低基因计数(LGC)与高WBC计数有关,正如前面所说的,这是CAD的危险因素。 在我们的肠道微生物群中,Lactobacillus reuteri 的存在与高WBC计数有明确的联系。
低基因计数的个体患有代谢紊乱,导致脂蛋白异常和促炎状态,这可能导致CAD。 LGC也与高的CRP水平相关,而与高CRP水平相关的则是低Oscillibacter、粪便杆菌 和瘤胃球菌。
类似于TLRs的模式识别受体(PRRS)在肠道中的表达也受到肠道细菌的调节,这些细菌帮助宿主通过病原体相关的分子模式(PAMP)和共生细菌在病原体之间导航,以及激活免疫感觉细胞。
此外,我们的菌群会影响调节性T细胞(Treg),它们的减少会加剧感染结果,增加自身免疫性疾病、过敏和癌症的风险。例如,Prevotella可以通过Toll样受体2(TLR2)激活介导炎症反应,从而导致炎症和细胞17(Th17)免疫反应。
心肌炎
心肌炎(一种炎症性心脏病)进展为致死性心肌病,可能依赖于肌球蛋白特异性Th17细胞在肠道中由半乳糖苷酶模拟肽共同作用于多形拟杆菌和粪杆菌,可促进炎症性心肌病的发生。
梭菌簇IV增强Treg细胞丰度,并导致抗炎分子的产生。因此,TLR2与CAD发病机制有关。
NOD/CAD是另一类PRRS,通过激活炎症细胞因子和/或激活免疫系统转录因子NF-κb来识别应激反应并激活炎症半胱氨酸蛋白酶,从而产生炎症分子。
肠漏也会导致肠道微生物衍生成分如PAMP的易位,包括LPS,从而导致促炎细胞因子的产生。
SCFA既能抗炎也能导致炎症
肠道微生物代谢物,如SCFA,也可以影响免疫系统,通过激活G蛋白偶联受体41(GPR41)、GPR43和GPR109A,通过诱导由叉头盒P3(Foxp3)启动子控制的Treg细胞,发挥抗炎作用。
此外,它们还能产生抗炎的肠道激素,如胰高血糖素样肽1(GLP-1)。 虽然SCFAs有许多积极的作用,但它们的产生也可以通过激活Toll样受体4(TLR4)来改变细菌平衡并导致炎症。因此,它们在免疫系统中的作用有待进一步研究。
TMAO激活炎症小体影响免疫系统
肠道微生物衍生的TMAO也可以通过激活TXNIP-NLRP3炎症小体影响我们的免疫系统,导致炎症标记物如TNF-α、IL-6、IL-18和IL-1B的表达,这些炎症标记物可以通过产生胆固醇填充的泡沫巨噬细胞促进动脉斑块的形成,最终导致CAD。
TMAO还能促进PKC/NF-κb激活,促进血管细胞黏附分子1(VCAM-1)的表达和单核细胞的黏附。 除了影响高密度脂蛋白胆固醇和抗炎特性外,肠道微生物及其相关代谢物还可以通过非炎症诱导途径影响免疫系统。
胆汁酸抑制NF-κb转录
初级(肠道菌群解偶联)和次级胆汁酸,例如,可以通过FXR和TGR5受体抑制促炎细胞因子的NF-κb转录。激活TGR5也能保护LPS诱导的炎症和动脉粥样硬化。
细胞因子如IL-10的积极作用
此外,某些细胞因子,如IL-10,可以产生积极的作用,如通过诱导RCT增加动脉粥样硬化病变中乙酰化和oxLDL的摄取和流出,从而降低小鼠血清胆固醇和动脉粥样硬化斑块。
这种细胞因子还可以通过增强肝脏驻留的Kupffer细胞的吞噬作用来降低总胆固醇。 这些细胞代表体内80%-90%的巨噬细胞,可能是治疗的新靶点。
剖析免疫系统和代谢系统之间的复杂相互作用,将有助于深入了解CAD的生物学基础,以及当前和未来的治疗方法如何影响代谢。
饮食影响整个系统
如前所述,我们身体胆固醇的四分之一来自饮食摄入。 这导致了关于饮食胆固醇是否会影响CAD发展的越来越多的争论。
我们的饮食可以通过直接食用富含胆固醇的食物和间接地通过改变肠道微生物及其群落结构、胆汁酸的产生、粪甾醇的产生、SCFA的产生和TMAO的产生来影响胆固醇的调节和CAD的发展,从而使问题复杂化。例如,地中海饮食引起的肠道微生物群的有益修饰已被证明能改善肥胖、炎症、CAD和其他相关代谢改变。
图4 微生物群、饮食和CAD
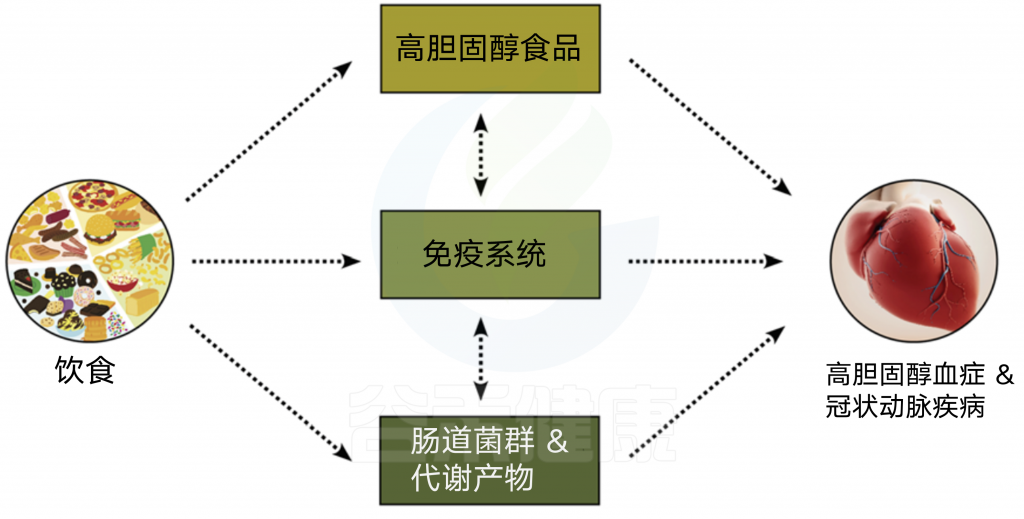

饮食通过食用富含胆固醇的食物直接或间接地影响胆固醇水平和CAD的发展,影响免疫系统,并导致肠道微生物群及其代谢产物如胆汁酸、黄连醇、短链脂肪酸和TMAO的调节。
这种饮食更加强调水果、蔬菜和豆类,并且与增加SCFA水平有关。此外,饮食可以通过改变与胆固醇有关的炎症反应来影响免疫系统调控和CAD发展。
一项结果表明,高盐摄入会影响肠道微生物群,特别是通过消耗鼠李糖乳酸菌和增加Th17细胞和高血压。补充L.Murinus 可以抑制Th17的激活,改善高血压。
此外,与高饱和脂肪组成的饮食相比,由高不饱和脂肪组成的西方饮食可导致拟杆菌增多、厚壁菌和Bilophila wadsworthia(亚硫酸盐还原微生物)减少,而由高饱和脂肪组成的饮食可导致LDL胆固醇和B.Wadsworthia菌增加,这种菌与血脂异常和炎症增加有关。
高蛋白和高脂肪饮食也与瘤胃球菌的增加和拟杆菌、梭状芽孢杆菌、双歧杆菌、直肠杆菌、赤霉菌的减少有关,并增加粪便中的胆汁酸浓度,包括DCA浓度的升高,这可能导致肝癌。
此外,这些饮食可以激活与炎症反应相关的TLR4,如促炎细胞因子、Th1、CD4和T细胞,从而导致Treg细胞的下调。在高脂饮食诱导的糖尿病中,来自肠道的细菌被转移到组织和血液中,这取决于CD14和NOD1。
通过益生菌逆转
然而,这种菌血症可以通过益生菌(双歧杆菌)逆转,这可以减少糖尿病期间细菌、脂肪组织和炎症的粘附和移位。
在另一项研究中,益生菌给药干酪乳杆菌可减少细菌易位,并通过增加Clostridium coccoides, C. leptum 和总乳酸杆菌来改变肠道微生物群。与纯素相比,杂食动物产生更多的TMAO,高纤维饮食导致更高的SCFA和增加肠道菌群多样性。
饮食影响人体胆固醇的概念是一个持续的争论,需要进一步研究。尽管许多研究表明高饮食胆固醇与CAD之间存在直接关系,但其他研究表明饮食中胆固醇的临床影响在疾病发展中可能很小或可忽略不计。
这场争论可能是由于对胆固醇水平管理中涉及的身体系统机制缺乏了解,以及由于人群和环境因素而异的正常肠道菌群所致。
饮食的抗炎作用
我们的饮食也可以通过与转录因子NF-κB和PPAR-Y相互作用的omega-2(n-3)多不饱和脂肪酸产生抗炎作用,下调促炎基因,抑制TLR4的激活,从而产生抗炎反应。例如,我们饮食中的花青素(如蓝莓)是一种抗氧化剂,可以通过增加其多样性影响肠道微生物群,从而减少炎症反应。
益生菌和益生元也被深入研究,并证明通过增强肠道屏障、调节免疫功能和预防致病性感染来改善肠道环境。它们与炎症减少和SCFA、拟杆菌、双歧杆菌和厚壁菌减少有关。口服益生菌甚至可以降低22-33%的胆固醇,这是由于BSH的活性。
例如,益生菌乳酸杆菌和双歧杆菌可以通过(a)增加胆固醇对从头合成胆汁酸的需求,或(b)降低胆固醇溶解度并减少其吸收,从而解除胆汁酸的结合并增加排泄。
尽管益生菌和益生元越来越受欢迎,但关于它们可能对健康和疾病产生的特定免疫和生理影响的问题仍然存在,因此需要进一步研究。
微生物群、人口因素和CAD
在精确医学时代,一个关键的挑战是弥合人口统计学因素、肠道微生物组成和心血管系统病理生理学之间相互作用的知识差距。
除了男性和女性之间的环境和社会差异(如职业危害、生活方式、社会压力和获得医疗保健)外,在疾病发展、性染色体和性激素方面的差异也会导致CAD患者的性别差异。更具体地说,最近已经显示了血脂和脂蛋白代谢的性别差异,以及针对血脂异常的性别特异性考虑因素。
尽管CAD被认为是一种“男性疾病”,但越来越多的证据也显示了CADs在女性中的重要性,并提高了在CADs的发生、诊断、治疗和预后方面性别相关差异的认识。
例如,女性在生命后期更容易患上这种疾病。 这可能是由于激素和更年期的变化,这可能会影响胆固醇的比率,随着雌激素的停止产生,脂蛋白向低密度脂蛋白转移,并远离高密度脂蛋白胆固醇的女性。
性别差异也与肠道微生物群的整体结构有关
正如前面所讨论的,这与CAD的发展有关。 例如,在一项研究中,分别在男性和女性中发现普氏菌属、巨单胞菌属、梭杆菌属、巨球型菌属、双歧杆菌属、瘤胃球菌属和阿克曼菌属的显著增加。
然而,男性和女性在微生物多样性方面没有显著差异。基于性别和性别相关的肠道微生物组成和CAD发展差异的研究仍然很少,需要在数量和深度上进行扩展。
种族差异影响高胆固醇血症和CAD发展
种族差异,虽然在研究中经常被忽视,但已知影响高胆固醇血症和CAD的发展。 种族差异可以捕捉来自社会、经济和文化差异、人类遗传变异、生物地理祖先差异以及生活方式和饮食差异的生物变异。
包括吸烟、血压、肥胖和胆固醇在内的CAD发展的危险因素也可能在不同的种族群体中有所不同,导致某些群体的CAD发病较早,预后较差。例如:
南亚人是一个高风险族裔群体,他们的体育活动率较低。
居住在美国的非裔美国人也有更高的CAD发展风险,这可能是由于生活方式、环境因素和社会经济因素。非裔美国人有相对较高的糖、较高的钠和较低的钾含量的饮食,这会导致更高的血压。
此外,种族和饮食差异与微生物组成和丰度的变化有关,甚至与肠道微生物群的变化相关性比遗传、年龄、性别和BMI等其他因素更强烈。例如,对健康个体中农村和城市地区微生物群的比较研究报告显示,与美国和欧洲的人口相比,居住在非西部和/或农村地区的人口具有更高的菌群多样性。
在Deschasaux等人的另一项研究中,在荷兰人口中观察到较高的肠道微生物群多样性,在南亚的肠道微生物多样性最小,加纳人、土耳其人和非洲人在中间。 在荷兰人群中也观察到了厚壁菌增多,拟杆菌减少。而在南亚人群中观察到了放线菌增多。
人口因素之间的相互作用,如性别、年龄和种族,以及它们与我们的饮食、肠道微生物组成和CAD发展的联系,说明了我们身体因素在健康和疾病状态中的复杂性。 因此,需要更多的研究努力来理解这些因素涉及肠道微生物群的变化和CAD的发展。
图5 微生物群、老化和CAD
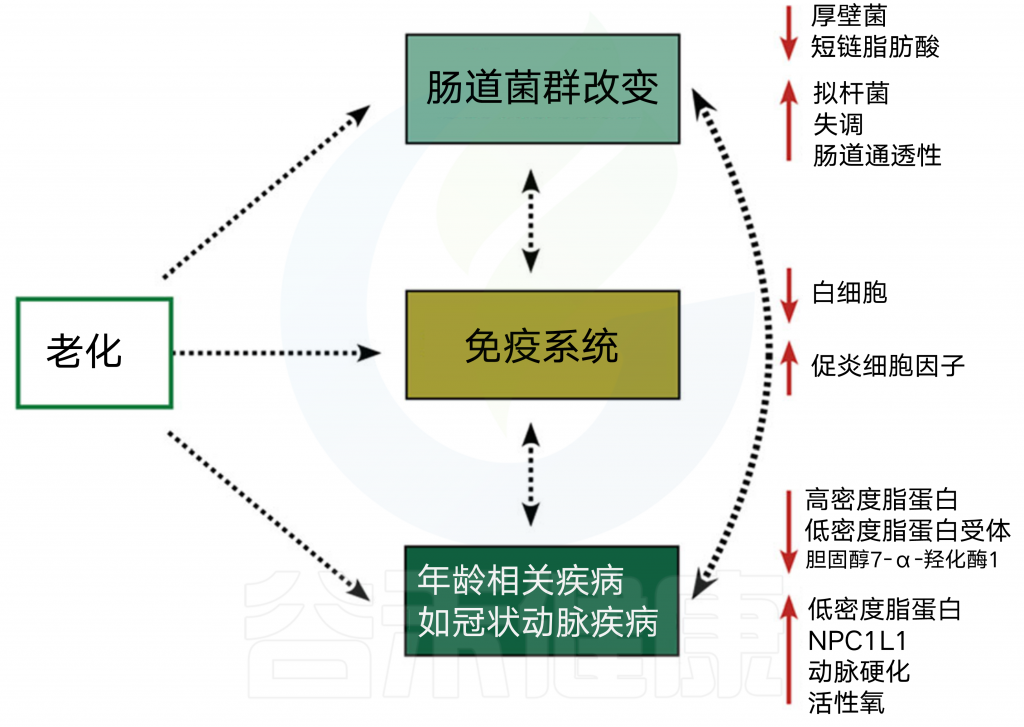

与全身炎症和不良健康结果相关的衰老机制。
身体中的胆固醇也会受到自然衰老过程的影响,这是一个无法控制的危险因素,会导致全身胆固醇代谢失调(图5)。到2030年,预计将有10亿人超过65岁。一般来说,衰老过程与心脏结构和功能的逐渐恶化有关,以及促进CAD发展。此外,通过衰老过程,LDL胆固醇水平可以增加,HDL胆固醇水平可以降低,这可能导致CAD的发展速度增加。
衰老过程引起的其他因素包括降低CYP7A1酶活性(减少胆汁酸合成的调节)、降低肝脏LDL胆固醇受体(降低LDL胆固醇清除率)和增加NPC1L1(胆固醇吸收介质)。衰老还影响肠道微生物群落,因为疾病的积累,饮食的变化,运动和行动能力的减少,以及某些药物的使用。然而,也有相反的发现表明不同年龄组参与者的肠道微生物结构没有显著差异。
总的来说,衰老与肠道失调增加有关,与肠道微生物多样性呈负相关。还有,参与SCFA生产的基因数量也随着年龄的增长而降低。 衰老影响免疫系统,全身炎症是衰老的标志之一,也是许多年龄相关疾病(包括CAD、糖尿病和癌症)风险增加的原因之一。
此外,衰老是由一个正反馈回路调节的,其中老年人的慢性全身炎症与发展和年龄相关的疾病有关,然后通过这些条件也导致炎症反应的增加。 由于这些原因,在精确的时代,包括年龄、性别和种族等人口因素的研究是有必要的。
精准医疗中的微生物群
目前,许多技术可以用来分析肠道微生物群与人类免疫学、神经学和内分泌学的关系。 由于这种关联及其在精确医学中的潜力,人类微生物群正在被广泛研究,作为粪便微生物移植、益生菌和益生菌的治疗靶点。.
尽管如此,对于大多数疾病,机械洞察力和翻译应用仍然很少。 人体微生物群在组成和空间上是动态变化的,微生物群个体内和个体间的变化可以通过药物的直接生物转化或微生物与宿主免疫系统的相互作用等间接机制影响药物的疗效和副作用分布。在此讨论了多种新出现的策略,以精确操作复杂的微生物群落,以改善心血管病的治疗结果。
预计在未来将朝着包含人类和微生物基因组以及它们的综合代谢活动的精确医学的包容性观点积极转变。
微生物群与药物治疗
目前治疗高胆固醇血症和CAD的方法包括能有效降低胆固醇水平的药物,并用于治疗高胆固醇血症和CAD预防。
他汀类药物可通过影响胆固醇合成中的限速酶达到疗效
羟甲基戊酰辅酶a(HMG-CoA)还原酶抑制剂,又称他汀类药物,可影响胆固醇合成中的限速酶,并已彻底改变了高胆固醇血症的治疗方法。在各种研究中,这类药物已被证明了显著降低总胆固醇、LDL胆固醇和甘油三酯的能力,并使HDL胆固醇增加18%、25%、11%和5%。
尽管他汀具有疗效,但它们对非LDL胆固醇的影响是有限的;因此,其他针对非LDL胆固醇的药物可能补充他汀类药物以降低心血管风险。例如,依泽替米贝是另一种降低LDL胆固醇的药物,它通过阻断NPC1L1来减少饮食和胆汁胆固醇的肠道吸收,从而降低LDL胆固醇。在一项随机对照的人类试验中,Ezetimibe(10毫克/天)与安慰剂相比,胆固醇吸收减少54%,总胆固醇和低密度脂蛋白胆固醇分别减少15%和20%。
药物副作用
虽然许多药物可以降低胆固醇,但它们往往是次优的,昂贵的,并带来许多不必要的副作用。例如,他汀类药物与骨骼肌、代谢和神经效应以及其他可能的副作用有关。他汀类药物治疗的停止也与心血管预后不良有关。此外,Ezetimibe还表现为肝脏内源性胆固醇合成的代偿性反馈上调,并可增加TICE,从而导致血清胆固醇升高。抑制肝NPC1L1还可提高胆汁中胆固醇饱和指数,并有可能导致胆结石。因此,虽然这些常规治疗提高了许多患者的生活质量和预后,但CAD和高胆固醇血症仍然是一种进行性疾病(注:进行性疾病是指症状不断加重、患者状况不断恶化的疾病。)。
另一个挑战是肠道微生物群可以通过干扰药物的药代动力学或药效学直接和间接地影响药物。例如,辛伐他汀、罗舒伐他汀和阿托伐他汀(3种常用的他汀类药物)显示了肠道微生物群调节的证据。 代谢物如胆汁酸,也可以影响药物的药代动力学,通过竞争药物运输机制跨越肠腔,或通过影响肝脏的摄取。 进一步研究肠道微生物群参与CVD和药物反应的分子机制将改善CVD患者的预后,并朝着微生物精确医学的方向发展。
接下来将提出纳米技术在揭示参与CAD开发的潜在机制以及针对微生物组的治疗工具方面的潜在作用。
基于微生物群和纳米医学的方法
纳米医学被美国国家卫生研究所(NIH)定义为纳米技术在控制生物系统、治疗、诊断和疾病监测方面的应用。这一新的医学分支是一个多学科的科学领域,其重点是开发至少在一个维度上位于0.1-100nm范围内的诊断和治疗纳米物体。
图6 纳米医学、微生物群和计算机辅助设计
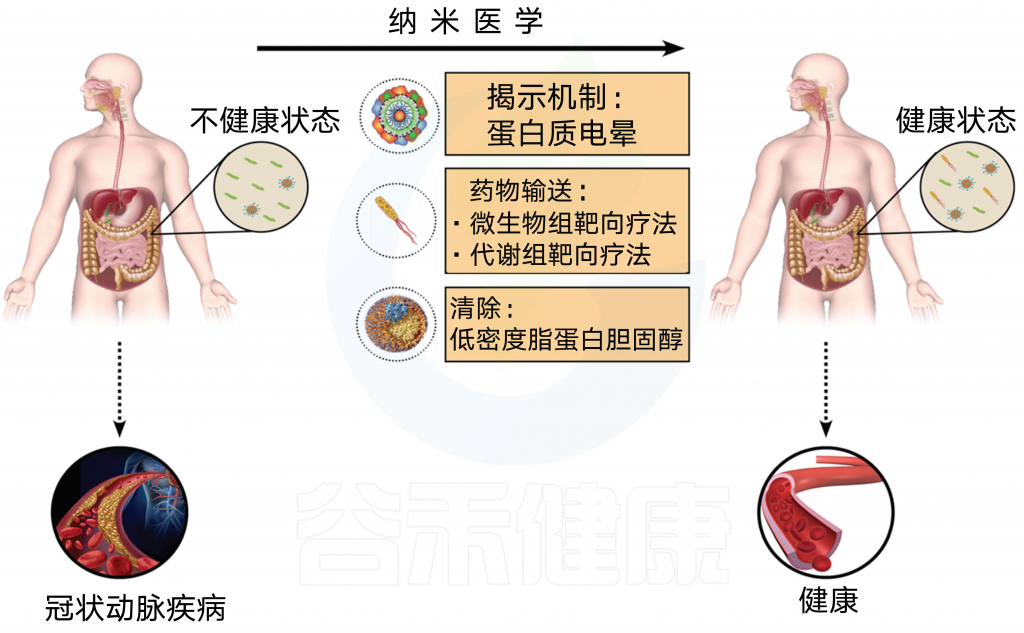

纳米粒子在纳米医学中有许多应用,可以帮助预防、诊断和治疗CAD。利用纳米颗粒了解潜在的身体机制(即蛋白质电晕分析)、药物传递(即微生物组和代谢组靶向治疗)和清除颗粒(即对于低密度脂蛋白胆固醇调节免疫系统)可以导致一个更健康的肠道微生物群和免疫系统,从而导致更好的整体健康状态,排除CAD的发展。
纳米医学的进一步发展也可能为现代医学中许多悬而未决的问题提供解决方案,包括高胆固醇血症和CAD(图 6)。 肠道微生物群与疾病发病机制之间关系的研究已被证明是一项困难的任务,特别是在找出原因方面。纳米医学中的纳米粒子可以帮助我们理解CAD发展的潜在机制。纳米粒子在体内应用的一个有用方面是生物分子/蛋白质电晕的形成(即一层生物分子,在纳米粒子与生物流体相互作用时覆盖其表面)。

在2014年,研究人员发现不同疾病患者的蛋白质电晕分布有很大的不同,尽管传统的血浆分析显示变化可以忽略不计。这种效应被称为“疾病特异性蛋白电晕”,已在其他地方复制,并用于早期发现疾病,包括神经退行性疾病。
最近发现,使用具有不同物理化学性质的纳米粒子(即称为蛋白质电晕传感器阵列技术)可以提高蛋白质电晕疾病检测的灵敏度、特异性和预测精度。
另一种更好地分析血浆蛋白并获得有关CAD发展的有用信息的潜在方法可能是磁悬浮。 研究人员最近使用超顺磁性氧化铁纳米粒子悬浮血浆蛋白,发现悬浮血浆蛋白产生椭球图案。
机器学习和液相色谱质谱法证明有用信息
利用机器学习和液相色谱质谱方法,研究人员证明悬浮血浆蛋白的模式包含了有关血浆供体健康光谱的有用信息。这一策略对于监测肠道微生物模式与CAD之间的相互作用是非常有帮助和可行的。
先进的数据分析技术提供机会
利用先进的数据分析,可以定义蛋白质/生物分子模式,与肠道微生物剖面的变化和CAD的发生和/或进展有很强的关联。关于重要生物分子变异的作用的知识可能提供一个宝贵的机会,不仅是根据特定的肠道微生物模式早期检测CAD(这反过来又会影响血浆生物分子的组成),但也用于开发新的治疗方法,基于肠道微生物的操作使用口腔纳米技术。
前瞻性诊断和治疗应用显示巨大潜力
目前的前瞻性诊断和治疗应用包括成像、组织工程、常规药物、蛋白质和遗传物质的传递以及清除低密度脂蛋白胆固醇。例如,肝素和壳聚糖共轭磁性纳米粒子在从血浆中去除LDL胆固醇方面显示出巨大的潜力。
纳米粒子还能调节免疫系统,并已被用于诱导抗炎作用。例如,广谱ROS清除纳米粒子已被用于小鼠研究,以有效地减少氧化应激和局部和全身炎症。
此外,壳聚糖纳米粒通过降低肠上皮单层的通透性和促炎细胞因子的分泌而诱导抗炎作用。而且,基于纳米粒子的TLR信号抑制剂已被用于减少炎症和治疗炎症性疾病。

纳米技术用于菌群调节来影响CAD发展
虽然纳米医学在CAD的诊断和治疗方面显示出相当大的和日益增长的能力,但其在调节肠道微生物群中的应用仍在研究中。 最近,研究人员提出了几种基于纳米技术的策略来控制肠道微生物的组成。通过调节肠道微生物有利于健康状态,可以直接(通过代谢物)和间接(通过免疫系统)以积极的方式影响CAD的发展。
为此,纳米粒子可用于传递与HDL增加、SCFA增加、LPs减少和促炎细胞因子减少有关的特定肠道微生物群。清除纳米粒子也可以优化(I)LDL胆固醇、(II)LPS、(III)促炎细胞因子和(IV)TMAO的摄取和去除。这些机制在预防、诊断和治疗CAD方面有很大的潜力,可用于替代目前具有各种负面副作用的药物。
然而,在设计安全有效的纳米粒子以预测和治疗CAD方面仍然存在挑战。此外,蛋白质电晕还会影响纳米载体的药物释放谱。因此,为了诊断和治疗CAD,需要进一步研究这些新的治疗平台的生物学特性。
临床微生物群研究的其他挑战
将人类肠道微生物群整合到临床设计和设置中并不是一项容易的任务,而且可能面临许多挑战。 通常,人类微生物群多年来保持稳定。尽管肠道环境中具有长期的稳定性和可塑性,但个体间变异和个体内变异是很重要的。
内部变异可能是由于婴儿过渡期(即出生胎龄、分娩类型和喂养方法)、年龄和抗生素使用等环境因素造成的。此外,肠道微生物群的变异可能是由于性别、肠型、体重指数(BMI)和生活方式、运动频率、种族、饮食和文化习惯等外部因素造成的。
这种个体间和个体内变异的研究可能会使旨在识别生物标志物和研究肠道微生物群组成和作为群体比较的功能的研究复杂化。因此,将微生物群科学纳入临床实践,可以通过考虑CVD患者的变异来确定生物标志物和治疗方法。
用于研究肠道微生物群的样本收集(即粪便样本)也会导致许多挑战,没有标准的协议和共识可用于质量保证和下游分析。例如,肠道微生物群在唾液、上消化道、下消化道和粪便样本中含有不同的微生物群落。上消化道显示孪生球菌、韦荣球菌属、奈瑟菌、梭菌、链球菌、普氏菌、假单胞菌和放线菌增加,而下消化道显示粪杆菌、瘤胃球菌和拟杆菌增加,这可能会产生方法上的挑战。
此外,粪便细菌群落的组成可能受到实验设计和收集、储存和DNA提取等程序等因素的影响。已经证明,粪便微生物群不是粘膜微生物群的代表,因此超越单一“以粪便为中心”的观点是至关重要的。 除了样本的类型外,纵向抽样可以增加对稳态的理解,但肯定会给病人带来负担。
测序技术和“组学”发展
最后,在过去的十年里,肠道微生物群研究的激增可归因于成本效益高的下一代测序(NGS)技术和人类基因组、代谢组学和蛋白质组学数据等“组学”数据的发展。NGS技术加上生物信息学的进步,彻底改变了微生物群的领域,取代了以培养为基础的方法,从而可以分析日益复杂的微生物群特征。
然而,局限性仍然存在。 例如,16SrRNA测序可以可以导致对细菌的观点是单一领域的,必须考虑生命的各个方面,包括真菌、原生动物和病毒。


宏基因组学研究可以将科学视角扩展到一个多领域的视角,但也存在局限性。 例如,由于参考数据库缺乏密切匹配,很大一部分数据无法分配功能特别是病毒数据。 因此,这些复杂的组学数据需要专门的统计模型来考虑成分、稀疏性、批效应、技术噪声、采样噪声和时空变化等因素。
解释“组学”数据也会产生挑战,因为特定肠道微生物丰度的变化可能不会被推断为对宿主有保护或有害的影响。例如,在Vandeputte等人的一项研究中,微生物的绝对数量(用定量微生物组谱测量)比经典的相对丰度谱更好,因为后者不能提供关于分类群丰度或代谢潜力变化的方向性程度的信息。
建立一个知识库,以巩固微生物群领域中不相连的知识片段,以及微生物组研究中的其他创新(包括自然语言处理,文本挖掘,分类学表示和全领域词汇标准化),可以加快理解并帮助推进因果关系。因此,为了建立肠道生态系统动力学的全球模型,需要在质量控制、方法学和使用流程方面进行进一步的研究和改进。
为了充分了解肠道微生物在人类健康中的作用,并指导高胆固醇血症和CAD发展的治疗干预措施,必须阐明相互关联的身体因素共同作用,影响肠道微生物群和疾病发展。
对这些复杂机制的进一步研究是阐明肠道菌群介导的机制的组成部分(例如,通过先进的纳米医学技术、数据科学以及种族和性别等因素),这反过来又可以导致更有效和高精度的基于微生物群的CAD预防和治疗方法,最终可以降低CAD的社会和经济成本。
相关阅读:
参考文献
Kazemian N, Mahmoudi M, Halperin F, et al. Gut microbiota and cardiovascular disease: opportunities and challenges[J]. Microbiome, 2020, 8(1): 1-17.
OHSAS 18001标准是一个国际性职业安全卫生管理体系的全球认证审核体系。为有效保障企业员工的安全与健康,为企业提供科学、有效的手段从源头预防事故发生,健康生产,谷禾主动申请了OHSAS18001的认证,审核组本着公正、公平的原则以及认真负责的态度,对照国际有关规定及标准的要求,主要针对公司职业健康安全有关的控制规程、检查规范、从业人员的职业健康证明、危险源的识别和处置、人员的能力、意识和培训等方面逐项逐条进行审核核定,通过抽查大量资料以及项目现场巡视,审核组经过内部论证评估,确认我公司职业健康安全管理体系系符合GB/T28001-2011/OHSAS18001:2007标准的要求,并于2020年3月20日,向我公司正式颁发了OHSAS18001:2007职业健康安全管理体系认证。
谷禾ISO9001:2015质量管理体系及OHSAS18001:2007职业健康安全管理体系认证证书的取得,是公司各项管理工作的规范化、标准化的全面检测,是对公司持续稳定健康发展的重要保证。我们将进一步优化和规范各项管理制度,不断提高体系运行的有效性,不断提高公司的管理水平,严格按照体系文件要求,不断提升客户服务,做到持续改进,给客户提供更有力的保障。


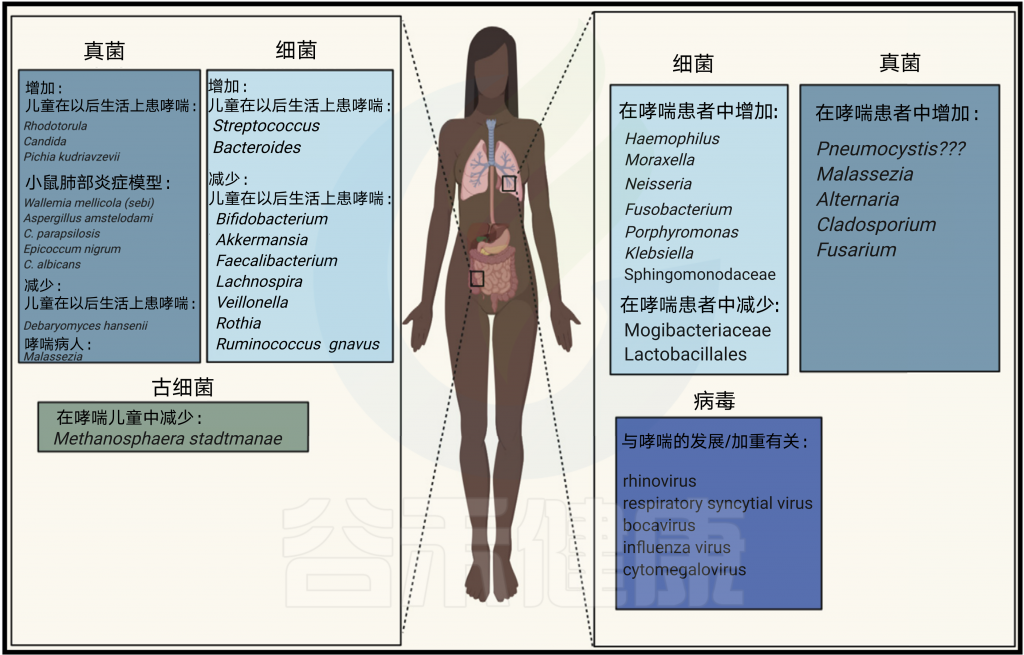
哮喘是一种常见的慢性呼吸道疾病,影响着全世界3亿多人。预计到 2025 年将增加到 4 亿人。

咳嗽、胸闷、喘息、气促等
发作起来的窒息感让人崩溃
喘息是如何发生的?
当一个人没有哮喘症状时,呼吸是顺畅的,空气可以很容易进出肺部,一旦哮喘发作,肺部就发生了变化:

气道外平滑肌收紧,气道或支气管肿胀,管内粘膜开始产生粘液,粘液堆积进一步堵塞导致喘息,哮喘的征兆开始了。
哮喘带来的危险
除了咳嗽,患者会觉得呼吸困难,哮喘发作时,炎症会导致呼气比吸气更困难,肺部过度充气,人体超负荷进行肺部空气循环,各个器官和组织氧气输送量减少,甚至无法维持人体正常运作而发生危险。
于是,人们开始积极寻找各种方式来预防或者治疗哮喘。吸入器便出现了,它可以喷雾的形式将药物传送到受感染的呼吸道,以快速缓解症状。然而,人们对哮喘的病因依然缺乏足够的了解。
目前普遍认为,出现哮喘与多种因素有关,例如遗传、环境、感染和营养因素等。
随着研究的深入,逐渐发现在生命早期就已经有了潜在危险。对哮喘的研究也逐渐渗透到更精细的层面。


哮喘不是一种简单的疾病,具有许多表型和内型,目前,对于这些复杂致病过程的最佳理解,在于它们的二元分类:分为2型哮喘或非2型哮喘。
· 2型哮喘
临床上常见的2型哮喘有早期过敏性哮喘、晚期嗜酸性哮喘或运动性哮喘。
· 非2型哮喘
肥胖相关哮喘、中性粒细胞性哮喘和寡粒细胞性哮喘
哮喘表型和内型的示意图
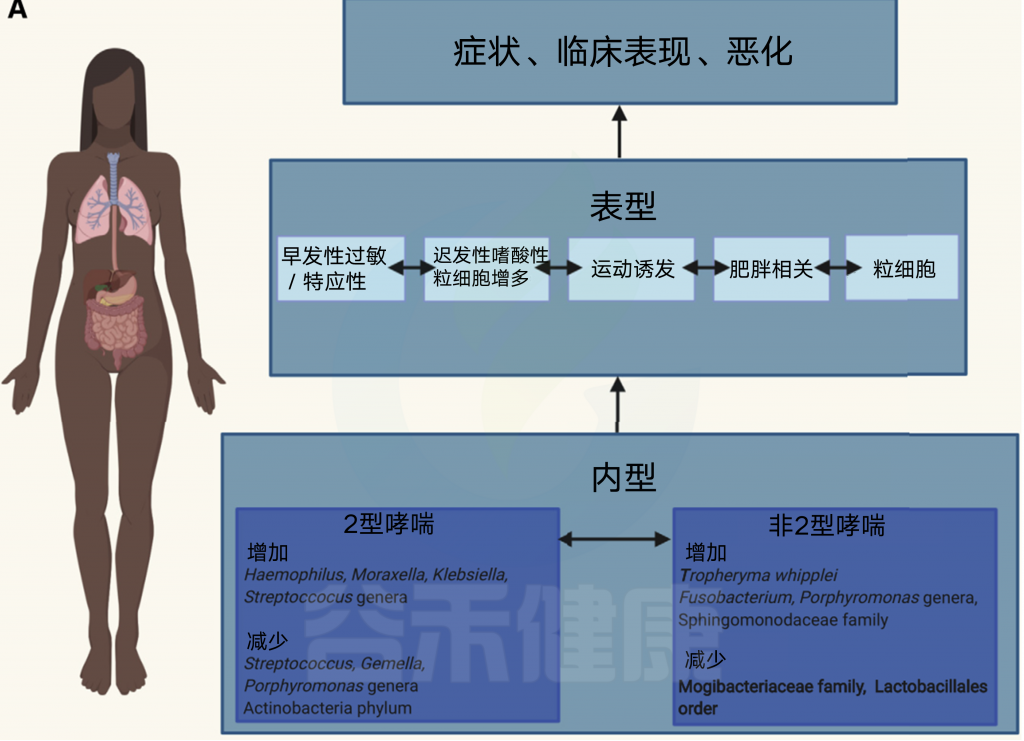

内型是根据病理生理机制对哮喘进行分类,而表型是指疾病的临床和形态描述。
涉及哮喘的简化细胞机制及其与微生物的相关性
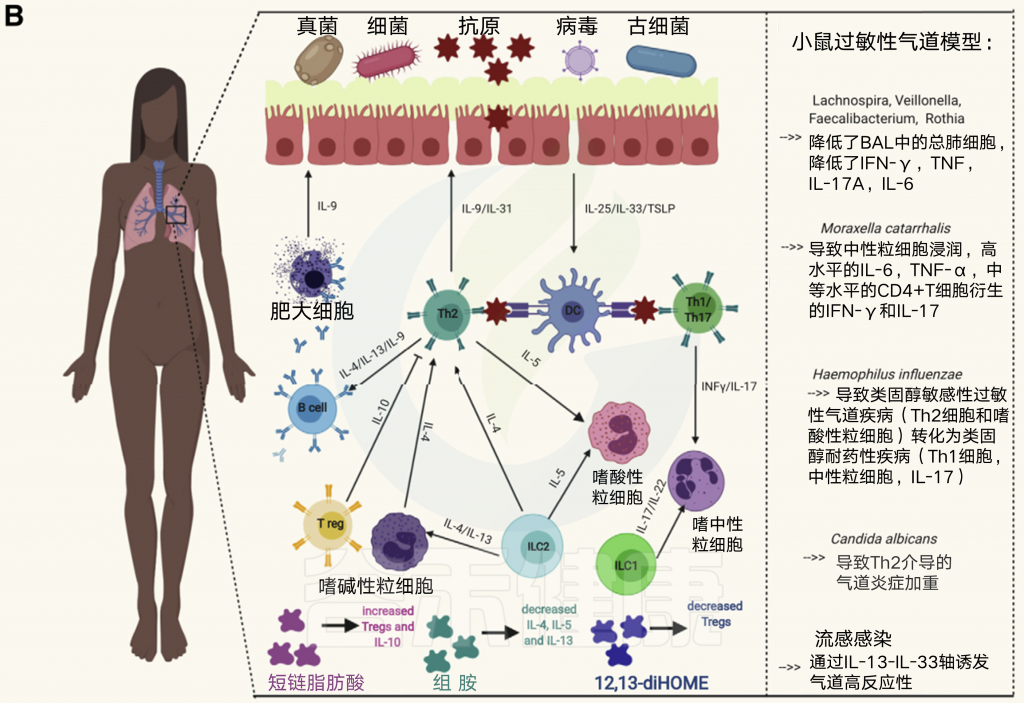

Th2细胞驱动的炎症与Th2细胞、2型固有淋巴细胞、T滤泡辅助细胞、2型B细胞、嗜酸性粒细胞和肥大细胞结合,导致痰、支气管肺泡液体、血清和支气管活检中IL-4、IL-5、IL-9、IL-13、前列腺素D2和CCR8含量增加。
非2型炎症的特点是Th1和Th17细胞和中性粒细胞浸润,I型干扰素的存在,NLRP3炎症小体的激活,以及IL-1b和IL-17的特征。
人体所有表面都充满了微生物群,当然也包括上呼吸道(URT)和下呼吸道(LRT)在内。
呼吸系统的组成
人类的呼吸系统由上呼吸道(从鼻腔开始,然后是鼻咽、口咽和声带上方的喉) 和下呼吸道(一直到声带下的喉部,包括气管、支气管、细支气管和无数肺泡)组成。
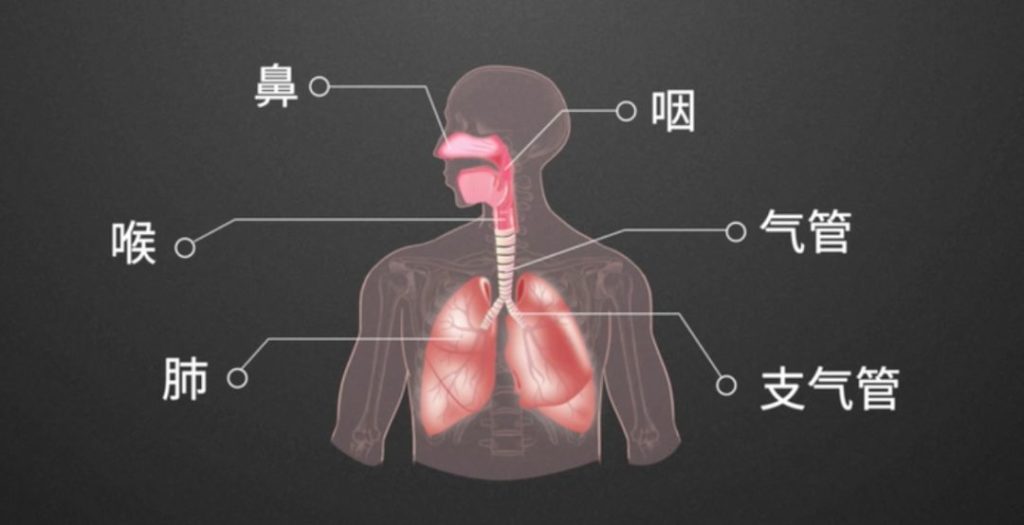
而这个上呼吸道,相当于一个看门人,守护着呼吸健康。微生物群主要通过鼻咽部的微呼吸进入肺部,口咽部则更密集、分布更广。微生物也可以通过粘膜表面的扩散进入肺部。
上下呼吸道菌群
上呼吸道和下呼吸道的健康微生物群在呼吸道和整个机体内稳态的发展和维持中起着重要作用。
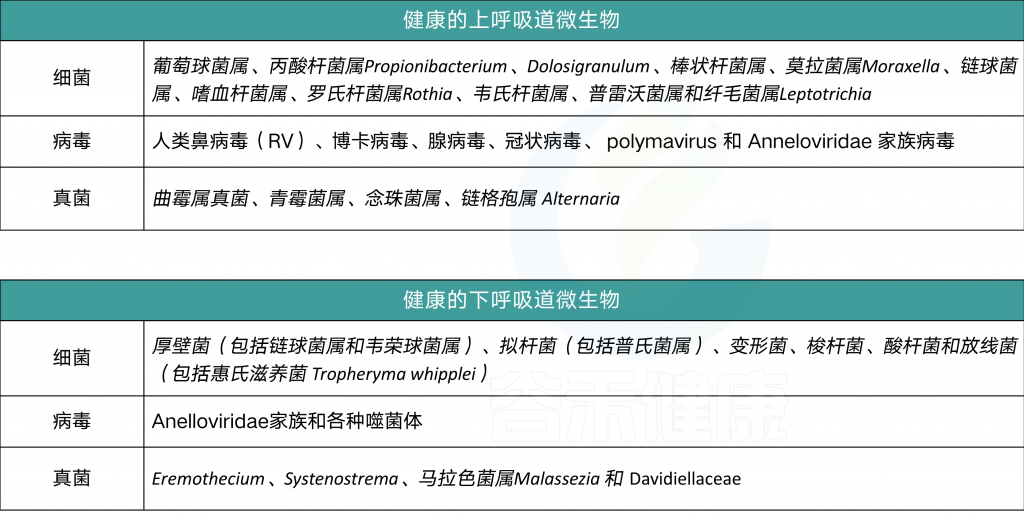

上呼吸道和下呼吸道的健康微生物群在呼吸道和整个机体内稳态的发展和维持中起着重要作用。
生命早期的“机会之窗”
从生命的第一天起,所有粘膜部位的微生物群都会发生动态变化,从而形成微生物群与人体之间适当的相互作用,并构成决定未来健康或疾病的关键“机会之窗”。
在出生和出生的第一天,母体阴道和皮肤的微生物群只能短暂地转移到婴儿身上,而母亲的肠道微生物群则成为婴儿在四个月大时获得的微生物群的主要来源。
由于这两种解剖结构代表了不同的环境,因此早期生活的背景下,有必要将上下呼吸道分开讨论。
从生命的第一刻起,上呼吸道就不断地暴露在外界环境及其伴随的环境中,从而形成了上呼吸道微生物群。
稳定状态下,下呼吸道菌群的瞬时特性取决于上呼吸道进入的菌群与被宿主免疫消除的菌群之间的平衡。 因此,上呼吸道样本无法完全反映下呼吸道中发生的相互作用。
健康呼吸道微生物群对宿主免疫的总体影响反映了活微生物及其细胞或代谢物成分对宿主局部和全身固有和适应性免疫过程的累积影响。如果健康适时的定植过程被破坏,早期生命肠道和肺微生物群的失调就成为许多呼吸系统疾病发生的重要危险因素。
不同的肺部疾病会受到肠道微环境变化的影响,反之亦然。由此提出了“肠-肺轴”连接的概念。
在哮喘中,微生物群是导致这两者之间相互作用的重要因素。在人类和小鼠的研究中,肺和肠道之间的联系已经被反复证明。
肺部炎症时的肠-肺轴
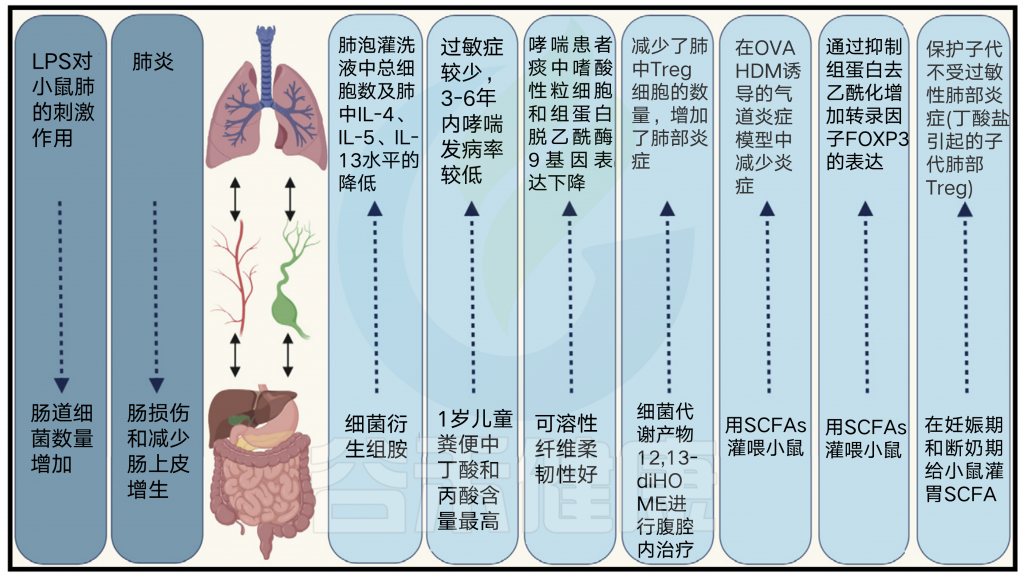

这种相互作用可以是双向的;肠道微生物可以影响肺部的免疫反应,而肺部刺激可以导致肠道反应。
此外,在慢性阻塞性肺病(COPD)中还描述了「肠-肺-肝轴」。
来自肠道菌群的短链脂肪酸对肺部的促炎反应有抑制作用。
肝脏反应增强巨噬细胞活化和肺部炎症
肠道菌群产生的短链脂肪酸(SCFAs)对肺内炎症反应有抑制作用,肝脏通过HMGCoA还原酶抑制甲羟戊酸途径或与G-蛋白受体结合抑制炎症反应。
我们知道,肠道是人体菌群最密集的定植部位,其菌群及其与哮喘的关系,无论是在早期还是在已确诊的疾病中,都需要特别关注。
在一项研究中,来自加拿大的三个月大婴儿哮喘高风险的粪便样中观察到Lachnospira, Veillonella, Faecalibacterium, Rothia显著下降。这种菌群特征在1岁时不再明显,同时伴随着粪便乙酸的减少和肝肠代谢物失调。此外,预测的细菌群落功能分析表明,哮喘高危儿童微生物群中脂多糖生物合成途径的减少。
在卵清蛋白(OVA)诱导的小鼠呼吸道炎症模型中,将哮喘婴儿的粪便浆液补充具有代表性的Lachnospira, Veillonella, Faecalibacterium, Rothia菌属后,移植到无菌小鼠体内,可以减轻呼吸道炎症。

在另一项人类出生队列研究中,厄瓜多尔儿童在3个月大时,粪便样本中链球菌和拟杆菌的相对丰度增加,双歧杆菌和Ruminococcus gnavus减少的,在5岁的时候有更高的患过敏和哮喘的风险。
最后,根据肠道微生物群的组成,在美国新生儿中,双歧杆菌属、阿克曼菌属和粪杆菌属相对丰度低,念珠菌和红酵母真菌相对丰度较高的新生儿患过敏和哮喘的风险最高。
短链脂肪酸
肠道微生物对哮喘的影响至少部分是由细菌代谢物介导的,这可能会影响身体远端的免疫反应。在人类呼吸道炎症中,最著名的具有保护性质的代谢物是短链脂肪酸。
1岁时粪便中含有大量丁酸和丙酸的儿童,其特应性敏感性明显降低,3至6岁之间哮喘的可能性较小。此外,可溶性纤维补充可降低哮喘患者痰嗜酸性粒细胞增多和痰组蛋白去乙酰化酶9基因的表达。
怀孕和断奶期间口服SCFAs保护小鼠后代免受过敏性肺炎影响
在小鼠中,短链脂肪酸通过抑制组蛋白去乙酰化而增加转录因子Foxp3的表达,从而支持T调节细胞(T调节细胞)的扩增,增加IL-10的产生。
在OVA和家庭尘螨(HDM)诱导的气道炎症模型中,短链脂肪酸还被证明可以减少炎症。
此外,在怀孕和断奶期间口服SCFAs对小鼠有保护作用,使其后代免受过敏性肺部炎症的影响,尤其是丁酸能有效诱导幼鼠肺部Tregs的产生。
哮喘粪便样中组胺分泌细菌数量增多
最近有研究表明,人体肠道细菌能够产生其他具有促炎和抗炎作用的代谢物,如生物胺(包括组胺)和氧化脂质,如12,13-di-HOME。哮喘患者粪便样本中组胺分泌细菌数量明显增多。此外,组胺分泌细菌的数量与疾病的严重程度有关。
然而,在OVA引起的过敏性气道炎症模型中,细菌源性组胺降低了支气管肺泡液中的总细胞数和肺匀浆中IL-4、IL-5和IL-13的含量,突出了细菌源性免疫调节的复杂性。相反,12,13-dihome小鼠腹腔治疗可减少肺部Treg细胞的数量,并增加蟑螂抗原小鼠气道炎症模型的肺部炎症,由肺和肠道细菌分泌的代谢物可能被证明有助于额外的治疗方法。
占主导的变形菌——潜在的致病菌
在几项人类研究中,肺部变形菌似乎是哮喘患者中最占主导地位的门。变形杆菌门以潜在的致病菌为代表,包括Haemophilus, Moraxella, and Neisseria菌属。
在哮喘炎症表型中,嗜中性粒细胞性哮喘患者,通常接受高剂量吸入性皮质类固醇(ICSs),与嗜酸性哮喘患者比起来,表现出菌群多样性较少,Haemophilus 和 Moraxella、变形菌门相对富集,链球菌、Gemella、卟啉单胞菌的相对丰度降低。
克雷伯氏菌在严重哮喘患者中富集
在严重哮喘患者中,代表非2型哮喘的Th17细胞上皮基因特征与变形菌门的表达增加有关,这进一步与哮喘控制的恶化和哮喘中性粒细胞恶化有关。克雷伯氏菌是变形菌门中的一个细菌属,在严重哮喘患者中特别富集,而放线菌门与哮喘控制有关。
哮喘治疗也可能导致肺部菌群变化
哮喘治疗也可能导致肺部微生物群落的变化,这表明在确定因果关系方面的复杂性。例如,放线菌与类固醇反应的分子证据(FKBP5表达)有关。因此,某些气道微生物可能有潜力被用作哮喘对类固醇反应的指标,使临床医生能够改进治疗方法。
重要的是,肥胖和哮喘对机体免疫反应、气道和肠道微生物群的实质性相加效应最近已被描述。
患有严重疾病的肥胖和非肥胖哮喘患者的粪便中Akkermansia muciniphila菌水平均有所降低,这可能与急性和慢性气道炎症的小鼠模型中所显示的因果关系有关。严重哮喘患者肺部的另一个属是链球菌。
特应性哮喘患者,通常表现为嗜酸性炎症表型,在梭杆菌属、卟啉单胞菌属和鞘氨醇单胞菌科的细菌中有丰富的菌群,同时单胞菌科和乳杆菌科的相对丰度降低。
此外,哮喘患者痰液中的嗜酸性粒细胞与成人体内的Tropheryma whipplei有关。与中性粒细胞性哮喘惊人的可重复性研究结果相反,嗜酸性粒细胞性哮喘和2型哮喘中的微生物群较不清晰且异质,可能再次反映了ICSs治疗引起的不同的微小型或混合型反应。
目前尚不清楚成人哮喘患者气道中特定细菌的多样性和存在在多大程度上反映了炎症的类型或微生物对皮质类固醇治疗的反应。研究表明,ICSs和口服糖皮质激素联合治疗与变形菌和假单胞菌数量增加,拟杆菌、梭菌和普雷沃菌数量减少呈正相关。
另一方面,一项未经吸入皮质类固醇治疗的轻度哮喘研究表明,嗜血杆菌、奈瑟菌、梭菌和卟啉单胞菌具有独特的富集作用,同时在其气道中的单胞菌科和乳杆菌科存在缺失。这些研究表明,经常使用皮质类固醇治疗的哮喘患者的肺微生物群组成,可能是炎症环境和药物作用之间复杂相互作用的结果。
小鼠在感染Moraxella catarrhalis菌(M. catarrhalis,Proteobacterium phyla)1天后,可导致嗜中性粒细胞浸润、IL-6、IL-1b和肿瘤坏死因子a (TNF-α)的高浓度、CD4+T细胞来源的IFN-g和IL-17的中度浓度。此外,IL-17或TNF-α而不是IL-6的中和作用,可加速卡他性支气管炎的清除,有效地预防感染引起的过敏性气道炎症的恶化。
来自变形菌门的流感嗜血杆菌Haemophilus influenzae 能够将与Th2细胞和嗜酸性粒细胞相关的类固醇敏感过敏性气道疾病(AAD)转化为与Th1细胞、中性粒细胞和显性IL-17反应相关的类固醇抗性疾病。
越来越多的证据表明细菌在哮喘中起着一定的作用,但还需要进一步的研究,以更清楚所涉及的最重要的菌群,并了解哮喘背景下的细菌失调是否是疾病的原因或影响。更详细的机理研究是必要的,以充分了解肺和肠道微生物组成和复杂的关联代谢在生活中的不同点与特定类型的哮喘炎症。未来的工作应集中在继续详细描述介导细菌与哮喘宿主之间沟通的细胞和分子机制。
目前尚不清楚真菌是在健康的成人肠道中定植,还是只是作为短暂的口腔或食物来源的真菌被检测出来,但即使是短暂的肠道定植者也有可能影响微生物群落生态和宿主免疫反应。
真菌失调是与儿童高危哮喘表型的发展相关
虽然所涉及的机制没有被探索,但最近对来自美国和厄瓜多尔的人类出生队列的两项研究已经确定真菌失调是与儿童高危哮喘表型的发展相关的婴儿肠道微生物特征的一个关键特征。
这些结果表明,真菌失调可能是一个容易检测到的哮喘风险的生物标志物。 此外,这两项研究都发现真菌失调与细菌失调有关。
肠道内的微生物影响宿主对其他微生物的免疫反应,在生理和代谢上相互作用,并争夺空间和营养。 动物模型的早期研究旨在建立真菌失调与哮喘严重程度之间的因果关系,结果表明用抗生素预处理的小鼠经常见的人类肠道共生白色念珠菌(C.albicans)灌胃后受真菌过度生长的影响。
这些小鼠进一步证明,在气道刺激后,Th2细胞介导的气道炎症加剧,这一效应被认为至少部分是由白色念珠菌产生的免疫调节前列腺素介导的。
与此相一致的是,抗生素治疗后小鼠肠道中另一种念珠菌的过度生长与前列腺素PGE2介导的肺巨噬细胞向M2表型的极化有关,并在抗原致敏和刺激后加重了过敏性炎症。
在该模型中,真菌过度生长与肺部的炎症免疫细胞浸润有关,但也需要特定的抗生素引起的细菌微生物的变化。只有引起乳酸菌属相对数量减少的抗生素才与真菌过度生长和抗原激发后气道炎症加重有关。
值得注意的是,乳酸菌和其他产乳酸的细菌因其作为益生菌而受到关注,以预防哮喘和过敏,并具有抗真菌作用。
进一步的证据表明,真菌失调发生的历史和当前微生物环境影响了哮喘期间相关的免疫后果,这一点最近才出现。
不存在支原体的小鼠在接种了失调真菌后显示出Th2和Th17细胞的混合性肺浸润,而同样的方法在有支原体的常规小鼠中只导致哮喘期间Th2细胞炎症的增加。类似地,感染白色念珠菌的真菌初生小鼠表现出与真菌特异性Th17细胞和IL-17高反应中性粒细胞的系统性扩张相关的AAD恶化。这些研究共同强调了肠道微生物群的复杂性、动态性以及跨界微生物相互作用在确定哮喘预后中的重要性。
除抗生素治疗外,抗真菌治疗还可引起特异性无病原体(SPF)小鼠肠道细菌和真菌群落的变化,并在AAD的HDM模型中加剧2型变态反应性气道炎症和嗜酸性粒细胞增多。
连续补充在该模型中鉴定为在真菌治疗后肠道中过量表达的三种真菌(安斯曲霉Aspergillus amstelodami,Wallemia mellicola [sebi]和Epicoccum nigrum)或仅在抗生素治疗后单独补充Wallemia mellicola会增加哮喘的严重程度。
在这些研究中,抗真菌治疗和向特定无病原体小鼠引入抗生素真菌群落导致肠道细菌群落的变化,包括乳杆菌科减少。然而,在未经抗生素预处理的改变的Schaedler菌群小鼠中,在细菌微生物群没有实质性变化的情况下,再补充失调真菌可使AAD的恶化作用重现。
此外,最近证明,肠道常驻CX3CR1+单核吞噬细胞直接感应抗真菌诱导的真菌失调增加固有层中Th2细胞的数量,并导致AAD HDM模型中真菌失调相关的气道炎症增加,与细菌群落的抗真菌相关变化无关。
因此,除了通过重组免疫调节肠道细菌种群或产生系统代谢物对气道炎症有潜在的间接影响外,在肠道中的哮喘相关真菌失调可对宿主天然和适应性免疫细胞群体产生直接影响。
肺中的局部真菌失调也与哮喘有关
在一个小样本的受试者中,有和没有严重哮喘的儿童在支气管肺泡液样本中表现出特定真菌的相对丰度的差异,但在支气管肺泡液体样本中,真菌的总体多样性并不存在差异。这些差异包括严重哮喘患者 Pneumocystis相对丰度的增加,这是肺中存在的一种菌,与人体和动物模型中宿主2型免疫反应的诱导有关。


Pneumocystis 在2-5个月龄时的亚临床定植被认为是婴儿期最常见的感染之一,并与粘蛋白基因表达增加有关。Pneumocystis的早期肺定植可能是诱导肺中Th2细胞偏向免疫反应的危险因素,并使婴儿容易哮喘。
一项针对成人患者的18S焦磷酸测序研究同样发现,与非特应性对照相比,哮喘患者痰中特异性真菌相对丰度存在差异。在这些成年人中,哮喘患者表现出与特应性皮炎过敏条件有关的马拉色菌Malassezia 的相对丰度增加。
总的来说,这种情况逐步浮现:哮喘患者表现出与诱导2型炎症免疫途径相关的特定真菌的相对丰度增加,一些真菌要么不存在,要么只存在于健康肺中的低丰度中。
哮喘患者肺部真菌改变的结果并不令人惊讶,因为真菌成分,如甘露聚糖、蛋白酶、几丁质和β-葡聚糖通常存在于常见的人类过敏原和室内灰尘中。虽然致敏的潜在机制尚不清楚,但许多严重和/或不受控制的哮喘患者对真菌敏感和/或在抗真菌治疗后哮喘症状可能有所改善。
通常与致敏和哮喘相关的肺部真菌包括曲霉属Aspergillus、链格孢属Alternaria和枝孢属Cladosporium。

有意思的是,一些来自动物模型的证据表明,致敏作用是继发于自然Th2细胞和嗜酸性粒细胞免疫应答之后,才能有效清除引起感染的真菌。此外,支气管肺泡液样本中真菌载量增加与严重哮喘患者的皮质类固醇治疗有关,无论是否有真菌致敏。
因此,需要进一步的研究来确定病理性真菌定植和肺部真菌失调是否会导致更严重的哮喘发作,并在严重哮喘相关的皮质类固醇治疗后发生,和/或对哮喘相关的肺部微环境变化作出反应。
气道真菌群
最近,研究已经开始精确地描述与某些哮喘内质型和临床特征相关的气道真菌群。在一项检查气管内灌洗和支气管肺泡液体的微生物群的研究中,与健康对照组和2型低哮喘患者相比,Fusarium, Cladosporium, Alternaria在哮喘患者和2型高哮喘患者的支气管肺泡液体中富集。
表明其作为疾病生物标志物的潜力,支气管肺泡液液中交互链球菌和棒孢菌的相对丰度也与强制呼气容积1呈负相关(一种肺功能的临床测量方法)。
在过去的几十年里,对肠道微生物(包括真菌)的对宿主的组成和免疫作用的研究已经爆发。 虽然还有许多有待阐明的地方,但一组强有力的证据现在支持真菌在失调-哮喘中的重要作用。
真菌-细菌和真菌-宿主相互作用在确定真菌失调对宿主健康的影响方面都起着重要作用。 许多这项工作已经在儿童出生队列和小鼠模型中完成,需要更多的研究肠道菌群与成人哮喘的相关性。
此外,早期真菌失调是否以及如何影响以后生活中当失调不再时的哮喘发病和/或严重程度,仍然需要进行机械试验。
尽管病毒感染被认为是哮喘恶化的主要原因,但病毒与哮喘之间的关系还不完全清楚,是由于不断发展的有关呼吸道健康的知识。
已观察到上呼吸道和下呼吸道病毒的组成和丰度在健康受试者和无症状和有症状的慢性呼吸道疾病患者中有所不同,但需要进行详细的纵向研究,以更好地理解因果关系。
病毒与额外的环境和内在因素之间复杂的相互作用可能是哮喘患者呼吸道中某些病毒存在时间较长但短暂的原因,或是导致严重恶化的完全感染的发生。
在人类生命的第一年,几乎所有的喘息发作都与病毒感染有关。
已知引起哮喘喘息或恶化的最常见的病毒是RV和RSV,但博卡病毒、流感病毒和巨细胞病毒也有牵连。 据报道,早期人类鼻病毒引起的喘息对以后的哮喘风险有附加作用。
RV病毒
RV毛细支气管炎患儿气道中最丰富的细胞类型是中性粒细胞,尽管嗜酸性粒细胞的浸润也起着重要作用。此外,最近已经证明肥大细胞IL-6、IL-8、TNF-α和IFN-α的浓度在RV病毒刺激后显著升高。
T细胞参与通过识别病毒抗原和随后启动TC和抗体介导的免疫应答来控制RV病毒感染。最近的一项研究表明,RV病毒在体外进入并形成B淋巴细胞病毒复制中心,并诱导B细胞增殖。
RSV病毒
另一种与哮喘风险增加有关的病毒是RSV病毒;然而,它与该疾病的关系不太清楚。 有研究表明RSV病毒引起的毛细支气管炎与哮喘之间存在联系,但其他研究尚未确定婴儿RSV病毒感染与学龄哮喘发展之间的相关性。
RSV病毒感染对哮喘的长期影响与以下事实有关:该病毒通常在肺发育过程中影响新生儿。新生儿调节性B细胞对RSV病毒感染有很高的耐受性,其频率可以预测急性细支气管炎的严重程度。 此外,RSV病毒迅速附着在人类嗜酸性粒细胞上,并可能被这些细胞灭活。 随着哮喘严重程度的增加,嗜酸性粒细胞捕获病毒的能力降低了75%。
流感病毒
流感病毒是与哮喘恶化有关的另一种病毒感染。 流感感染背景下的哮喘与更严重、更高的住院风险、需要重症监护和死亡率有关。据推测,IL-33是驱动流感病毒引起的哮喘恶化所必需的。 在哮喘小鼠模型中,IL-33通过抑制先天和适应性抗病毒反应来增强气道高反应性和气道炎症。 另一项研究表明,流感感染通过一种需要IL-13-IL-33轴的途径诱导气道高反应性。
影响儿童哮喘的不常见病毒——HBoV和巨细胞病毒
除了与哮喘相关研究最为深入的病毒和上面描述的病毒外,还有一些不太常见的病毒影响哮喘中的气道炎症。 人博卡病毒1型 (HBoV)和巨细胞病毒也与儿童哮喘发作有关。
另一项研究报告说,在一个小样本中,急性喘息儿童HBoV病毒年龄为 ≥4 岁的因急性细支气管炎住院的儿童,他们都发展为反复哮喘,其中一半在5-7岁时患有哮喘。
还有证据表明,在OVA致敏小鼠中皮下注射急性喘哮喘儿童HBoV-VP1u 或 B19V-VP1u可导致气道炎症升高。 最后,51.4%的复发性喘息患者检测到巨细胞病毒DNA,在12-36个月的哮喘预测指数阳性的患者中比在阴性的喘预测指数同一年龄组中更普遍。
呼吸道病毒感染与哮喘发展之间的联系最近已经用很多方法进行了研究。 然而,导致这种相关性的确切机制尚不清楚,需要在动物模型和人类研究中使用多组学方法进行进一步的详细研究。考虑患者全球病毒的分析也应在未来考虑。最后,科学家还应该问一个问题:肠道病毒是否可能影响肺部炎症?
虽然研究不足,但古细菌在人类微生物研究中一直被发现,它们的群落组成因身体部位而异。
古细菌主要有Methanobrevibacter smithii,Methanosphaera stadtmanae,具有免疫原性。
最近,一项针对来自荷兰考拉出生队列的472名儿童的研究表明,肠道古菌在失调-哮喘范式中的作用。6至10岁儿童粪便样本中存在M. stadtmanae(一种常见的肠道古菌,与儿童哮喘风险显著相关,提示肠道古菌可能在过敏的发生发展中起到调节作用),这种情况与被诊断为哮喘的风险降低有关。这种效应与父母哮喘状态无关。
然而,在本研究的样本中,只有8.3%的样本检测为阳性,而某些古生物蛋白实际上被认为具有促炎性或致敏特性。 因此,需要更多的队列研究和机制研究肠道古菌在哮喘背景下的免疫调节作用。
与其他微生物群的情况一样,古生物群破坏的微生物和免疫环境可能决定了宿主的最终免疫后果。随着儿童和成人中的人类肺微生物群的不断特征化,肺古菌在生物失调模式中的作用有待确定。
现在有大量的文献支持人类微生物群影响宿主免疫系统的成熟和功能。肺和肠道微生物对哮喘的发展、表型和严重程度都有影响,但这些相关性的详细细胞和分子机制尚未得到充分的表征。
一个有助于理解人类微生物群和免疫系统之间联系的机制的重要工具是用于过敏性气道炎症的人源化微生物群小鼠模型。
人类队列研究应包括明确界定的临床结果,并从关于症状、治疗和临床信息的详细和具体问题中收集纵向数据。 关于病人饮食和生活方式的完整信息也应仔细准备,以确保最好地概述病人的情况和对微生物群的潜在影响。
需要对更多患者进行纵向和前瞻性分析,以了解疾病的病程、其表型和内型、对疾病进展的易感性以及对治疗的反应之间的关系。
总之显而易见,哮喘患者的未来诊断和治疗应通过分析个体微生物群的组成和代谢活动来辅助。
参考文献
Barcik W, Boutin R C T, Sokolowska M, et al. The Role of Lung and Gut Microbiota in the Pathology of Asthma[J]. Immunity, 2020, 52(2): 241-255.

近日,日本学者发表在Science上的一项研究
(DOI: 10.1126/science.aaw8429 )为妊娠期母体肠道微生物对后代健康影响新增一项证据,该研究表明表明,孕期母体肠道环境是影响后代代谢发育以预防代谢综合征的关键因素,因此,孕鼠肠道微生物群提供了一个环境线索,可以微调后代的能量稳态以预防代谢综合征的发育起源。
早期生命期是一个关键的时期:影响胎儿发育的事件可能带来一生的影响。人类胎儿发育过程中的一点点紊乱不仅影响主要的发育结果,甚至有可能影响几十年来无法表现的表型,如心脏代谢疾病的风险。
近几十年来,抗生素使用和摄入高热量、低纤维饮食的迅速扩大导致肠道微生物群的紊乱,使人类容易患上各种疾病,如代谢综合征。虽然微生物群对出生后的影响已经有很好的文献记载,但是关于胚胎阶段肠道微生物群的影响所知甚少。 尽管越来越多的证据支持健康和疾病的发展起源(DOHaD)这一概念,但其潜在机制仍不清楚。
在这项研究中,主要探讨了母体肠道微生物群对胚胎发育和后期疾病易感性的影响。
以短链脂肪酸为代表的肠道微生物群衍生代谢产物(如乙酸、丙酸和丁酸),不仅是宿主细胞的燃料,而且还是肠道微生物群和肠外器官之间的信号分子。
GPR41和GPR43属于游离脂肪酸受体(FFAR)家族,是SCFAs的受体。
之前已经证实了FFARs在能量代谢中的生物重要性,通过与膳食成分以及肠道微生物群衍生代谢产物的相互作用。
肠道微生物SCFAs通过交感神经系统、脂肪组织、胰腺和肠道中的GPR41和GPR43调节宿主能量稳态。
最近的一项研究进一步表明,怀孕小鼠的肠道微生物群影响后代的免疫和大脑功能。在DOHaD理论的背景下,这些发现增加了母体SCFAs在调节出生后疾病易感性中起关键作用的可能性。
妊娠期间,母体肠道微生物群通过胚胎SCFA受体影响后代肥胖倾向

研究人员发现孕期母体微生物群对后代的肥胖具有抵抗力。
怀孕小鼠在特定的无病原体(SPF)和无菌(GF)条件下繁殖,之后由寄养母亲在常规条件下抚养新生儿,以调整出生后的生长环境。来自GF母亲的后代极易患代谢综合征,其特征是肥胖和葡萄糖不耐症的加重,与成年期高脂饮食消耗的能量消耗减少有关。在妊娠期喂食低纤维饲料的小鼠后代中也观察到类似的表型。
用SCFA治疗妊娠GF或低纤维饮食喂养的小鼠使成年后代对肥胖具有抵抗力。妊娠小鼠结肠腔内的SCFA通过母体肝脏和血流到达胚胎。值得注意的是,胚胎的交感神经、肠上皮和胰腺高度表达GPR41和/或GPR43,以感知来自母体肠道微生物群的SCFAs。
然而,他们观察到GPR41在无菌小鼠中的表达较低。小鼠GPR41表达的丧失导致交感神经投射到心脏的缺陷,这在无菌后代中也是明显的。
体外证实了SCFAs激活交感神经元分化的能力,丙酸具有最大的作用。在缺乏GPR43的小鼠中,作者发现SCFA和微生物群在胚胎肠内分泌和胰腺B细胞发育中的依赖性缺陷。
微生物对代谢疾病的保护

SCFA-GPR41 和 SCFA-GPR43轴 赋予后代对肥胖的抵抗力
胚胎GPR41和GPR43信号转导的缺失在产前由于交感神经功能障碍和高血糖而影响能量代谢。SCFA-GPR41和SCFA-GPR43轴促进神经细胞、表达GLP-1的肠内分泌细胞和胰脏细胞的发育,从而形成胚胎能量代谢。这个发育过程有助于维持出生后的能量平衡。
在怀孕期间,母体肠道微生物群通过 SCFA-GPR41 和 SCFA-GPR43轴 赋予后代对肥胖的抵抗力。妊娠期间,GPR41和GPR43在胚胎的交感神经、肠道和胰腺中感知来自母体肠道微生物群的SCFAs,影响代谢和神经系统的产前发育。
这些发现表明,孕期的母体肠道环境是防止代谢综合征的后代代谢程序的关键因素。
因此,怀孕小鼠的肠道菌群提供了一种环境线索,可微调子代的能量平衡,从而防止代谢综合征的发展。
补充丙酸来拯救?
事实上,另一项研究结果也表明,在怀孕期间喂养低纤维饮食的小鼠的后代增加了肥胖和胰岛素抵抗的风险。低纤维饮食的有害影响可以用丙酸补充来拯救。
当怀孕的小鼠服用抗生素时,高纤维和低纤维饮食喂养的母亲的后代的代谢参数没有差异,证实了母体微生物在介导保护方面的重要性。
高纤维喂养动物的后代出生时更重,但在以后的生活中免受肥胖的影响。这与人类是一致的,因为低出生体重与未来的肥胖有关。补充丙酸孕鼠保护后代免受未来疾病的影响。 然而,丙酸补充是否对母亲有任何影响尚未评估。
考虑到减少人类代谢性疾病的目标,确定类似的机制是否控制人类的发展是至关重要的。 迫切需要更好地理解如何、为什么以及在什么情况下应该尝试调节妊娠中的肠道微生物群或SCFA。
高纤维饮食符合现有的营养建议,包括在怀孕期间,但这些是否能有效地增加SCFA和保护后代免受未来代谢性疾病的影响尚不清楚。人类微生物组成存在很大的个体间变异,不同的微生物具有不同的SCFA生产能力。 因此,微生物群产生SCFA能力的差异可能会影响后代肥胖的风险。
虽然补充丙酸可能是一个方便的选择,但怀孕期间的安全性和有效性仍有待确定。对人类的进一步研究是有必要的,以了解调节这一途径是否可以成为改善下一代代谢健康的途径。
参考文献
I. H. L. Blackmore, S. E. Ozanne, J. Mol. Cell. Cardiol. 83, 122 (2015).
II. I. Kimura et al., Science 367, eaaw8429 (2020).
III. J Ferguson. Science 367 eaaw6481 (2020)

本文原创:谷禾健康
头痛,喉咙痛,咳嗽,流涕…不管什么症状,先吃上阿莫西林再说。感冒还没正式开始,抗生素却已经吃上了。
很多这样想当然的行为正透露着人们对抗生素的认知不足,那么该如何正确使用抗生素呢?
1、首先使用抗生素应该在医生指导下使用,在未明确是细菌感染的情况下,尽量不要自行随意服用抗生素。不应作预防用药。
2、抗菌药物疗程因感染不同而异,对于急性感染抗生素的疗程一般为5~7 天,宜用至体温正常、症状消退后72~96 小时之后方可停药,特殊情况,妥善处理。
3、如果医生嘱咐的一天吃三次,并不等于早、中、晚饭后各一次,而是每隔4-8 小时服用一次(不同种类药物间隔要求不同)。
4、长期使用抗生素容易引起体内菌群失调,并容易引起耐药性。
5、老人、肝肾功能减退者等特殊人群用药需遵医嘱。
6、头孢类药物服用后禁止饮酒。严重者半小时可危及生命。
7、如需做菌群相关的健康检测,应在服用抗生素停药至少3天 后进行。
家长给儿童使用时也应当注意:
8、不要自行随意给孩子用抗生素
9、不要要求医生给自己孩子用抗生素
10、不要拒绝医生给孩子开的抗生素
抗生素是把双刃剑,如果合理使用,可以很快治愈细菌感染的疾病;若是不合理使用,也会带来巨大的危险。
什么是抗生素?
抗生素是怎么发现的?
抗生素有哪些类型?
抗生素是如何作用于细菌?
为什么会出现耐药性?
耐药性的危害有哪些?
抗生素、菌群与疾病
三者之间会发生什么?
… …
本文将为大家一一解答。
抗生素,是指由微生物(包括细菌、真菌、放线菌属)或高等动植物在生活过程中所产生的具有抗病原体或其他活性的一类次级代谢产物,能干扰其他生活细胞发育功能的化学物质。
以上这段话为了便于理解,我们还是要从微生物说起。相信微生物这个概念对于熟悉我们公众号的你,并不陌生。

我们经常强调,它们的无处不在,存在于环境,也存在于人们体内。这些细菌并不是闲着,每天会干很多事,诸如帮助消化,合成维生素,促进营养吸收等。
这么多细菌之间也会存在相互竞争的关系,于是某些细菌发展出了一些特殊技能——“排挤”,该技能是通过分泌一种化学物质让其他细菌无法生存,从而利于自己生存。
这种分泌出来的化学物质就是抗生素。
很多年前,细菌感染是让人害怕的,一个微不足道的伤口,甚至只是虫咬都可能要人的命。
在19世纪,肺炎、结核病、腹泻和白喉被认为是儿童和成人死亡的主要原因。无数专家学者日以继夜地奋斗,希望找到感染者的治疗方式。
在20世纪初发现抗生素之后这一切都变得不一样了。特别是在亚历山大·弗莱明发现青霉素,抗生素的治疗得以在世界各地逐渐普及,可以说抗生素拯救了数百万人的生命,并使困扰人类历史数百年的大多数传染病得到控制。
毫无疑问,抗生素是过去100年中人类和动物医学中最重要的药物之一。那抗生素是何时出现?如何被发现并一步步走向今天这样大规模生产的呢?
早期抗生素的发现
早在现代医学出现之前,抗生素已经使用了很长一段时间。自古埃及以来,人们就知道面包对治疗伤口和烧伤有效果,而面包上生长着丝状真菌。


往后几十年是抗生素发现的黄金时代,发现了多种抗生素,具体每种抗生素的发现时间及来源详见下表。
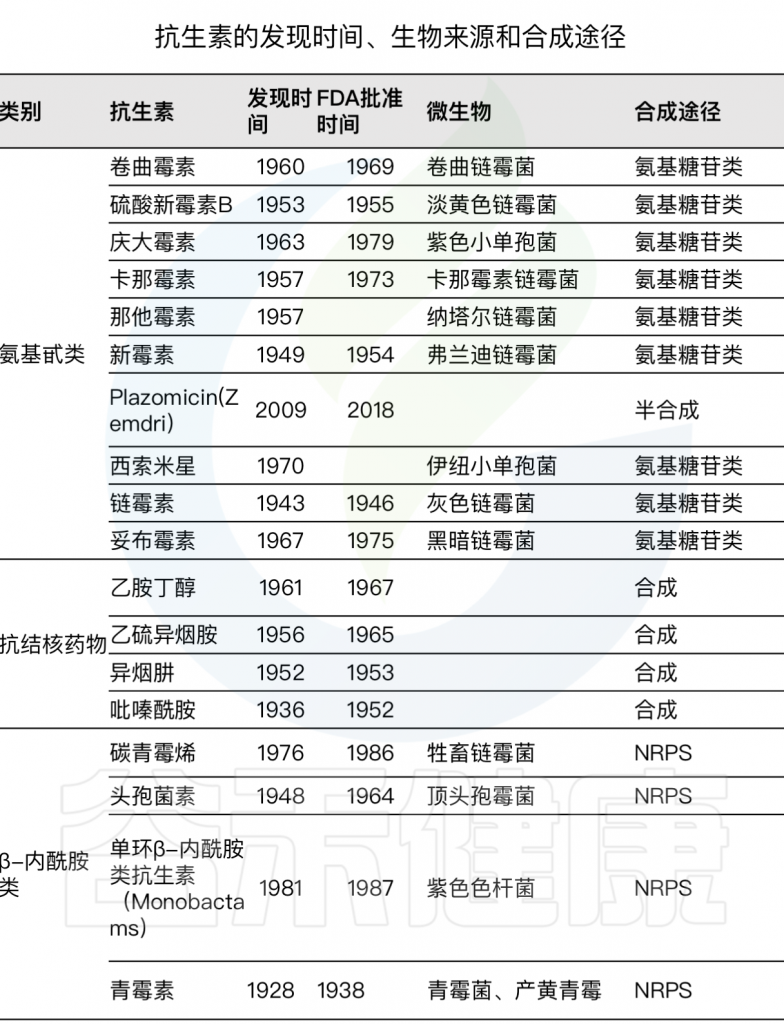
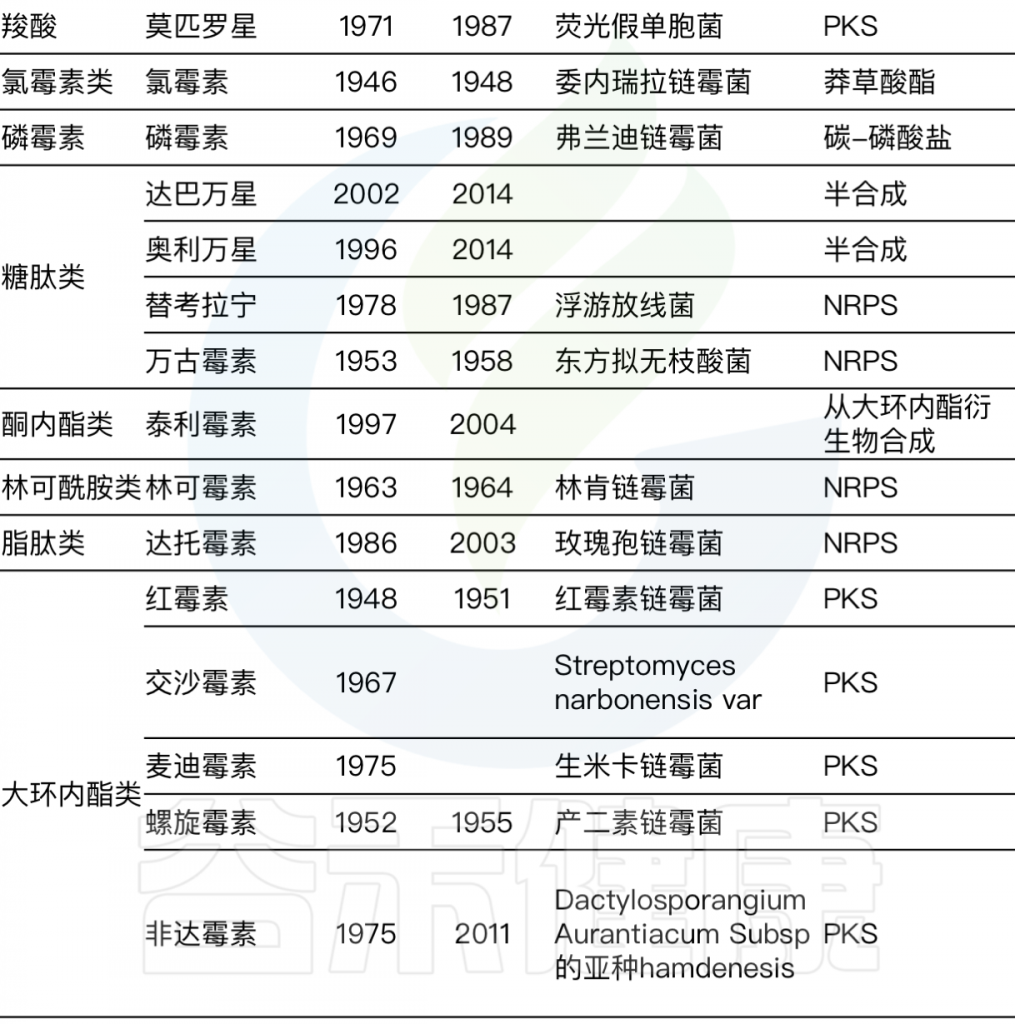
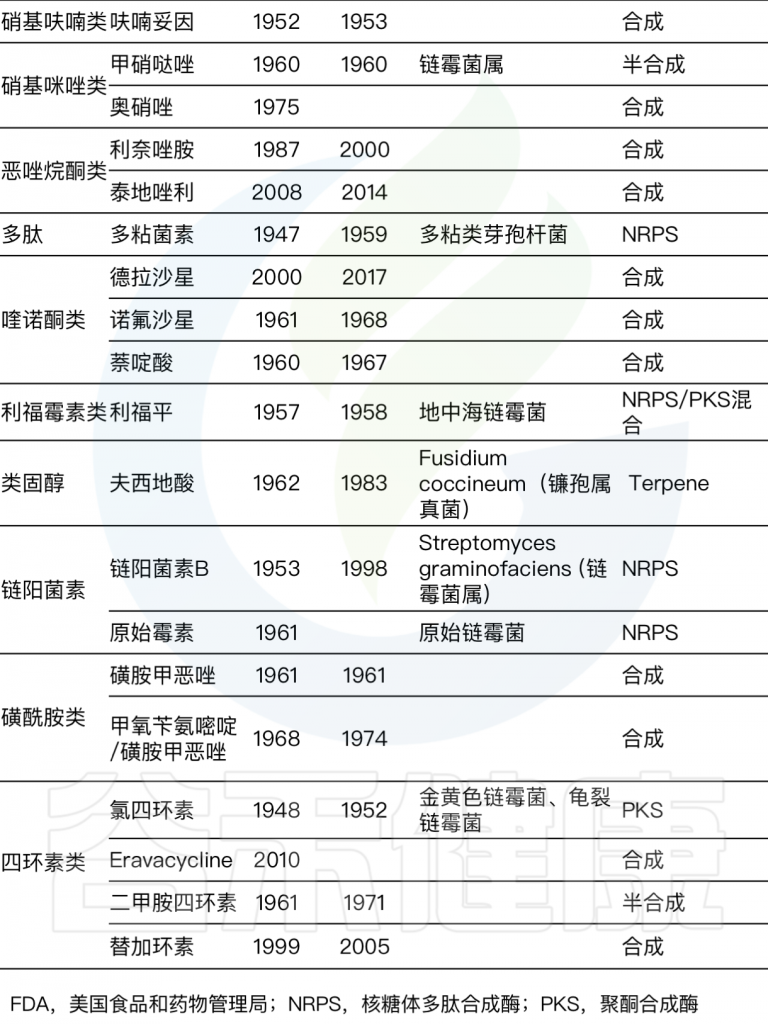



Durand G A,et al, 2019
细菌和真菌是最大的生产者。链霉菌属是目前人类医学中大约一半抗菌剂的来源。有了抗生素,致病菌能够及时清除,许多疾病得以治愈,人们生存几率大大提升。
以上是抗生素的发展史,我们可以看到抗生素的种类也非常多,那要怎么去认识它们?也许可以从抗生素的分类开始。
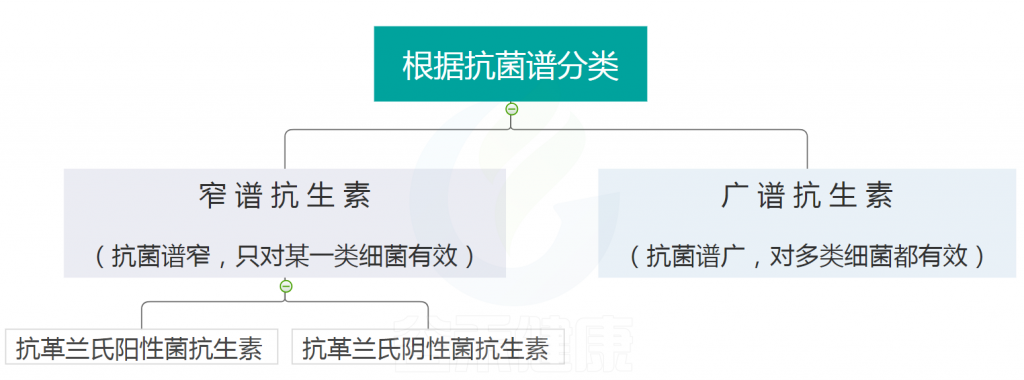
抗生素的分类,根据标准不同,分法不同。
根据抗菌谱可分为窄谱 和广谱 两大类,如下:

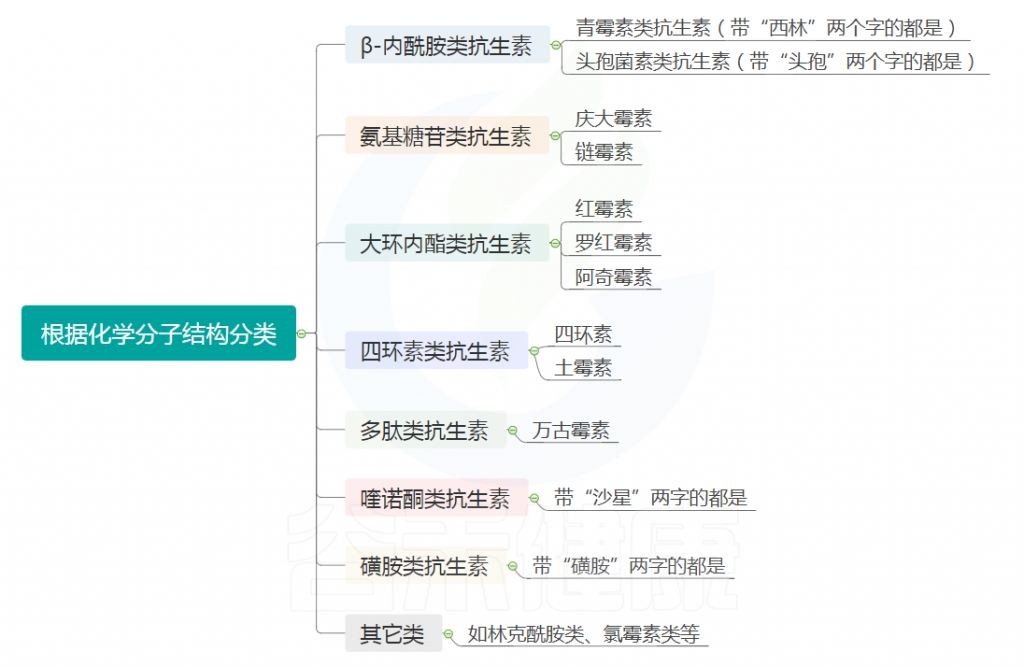
根据化学分子结构,可大致分为以下类别,列出的是该类别具有代表性的抗生素。

在了解了抗生素的历史和分类之后,我们再来了解下抗生素是如何凭一己之力力挽狂澜战胜病原菌,从而挽救那么多生命的?
总的来说,抗生素的作用原理有以下三种。

一:它们进攻的是细菌制造细胞壁所需要的部件。一旦细胞壁受损,细菌细胞就会死亡。比如青霉素就巧妙地利用了这个特点。
二:对于细胞而言,蛋白质至关重要。细菌细胞需要蛋白质来消化食物、构筑细胞壁、运动、繁殖、抵御入侵者等。抑制细菌合成蛋白质的抗生素直接作用于蛋白质合成的部件,使细菌严重受损,但它们对人体细胞的蛋白质合成没有多大影响。比如链霉素等。
三:破坏它们的增殖过程。一旦细菌的生长受到了限制,它们的威胁就会大大降低,宿主便来得及积累足够的免疫反应清除它们。
常见抗生素的具体作用详见本文末附录部分。
· 平时你熟悉的 “消炎药” 就是抗生素
相当一部分人听闻抗生素胆战心惊,认为需要远离,但是这其中很多人认为普通的感冒药都只是消炎药,与抗生素没什么关系。感冒了吃点消炎药,但其实很多你熟悉的所谓的消炎药就是抗生素,比如阿莫西林。
严格意义上来讲,消炎药并不能算医学概念。
判断是否是抗生素最简单的方法就是看名字。
这一招看着不是很高大上,但足以对付常用药。
一般药名里含有「霉素」「菌素」「沙星」「头孢」等字样的,大多是抗生素(少数是化疗药)。
名字里有「西林」的也是抗生素,像我们非常熟悉的阿莫西林,另外还有氨苄西林、苄卡西林、羧苄西林等,都属于经典的青霉素类抗生素。
名字里有「磺胺」的,一般就是磺胺类抗生素。
而消炎药,一般有两类:
一类是我们常说的激素(看名字里面的“松”),如可的松、氢化可的松、地塞米松等;
另一类是消炎止痛药,如布洛芬、阿司匹林等。
当然还有一部分人并不在乎到底是抗生素还是消炎药,认为这些药就是管用见效快,甚至有一些患者主动向医生要求使用抗生素,希望好起来快点…
· 不恰当服用抗生素可能危及生命
头孢类抗菌药物服用之后是不能喝酒的。因为服药期间饮酒很有可能引起双硫仑样反应,出现心慌,脸红,血压下降等改变,严重者危及生命。
为什么会出现这么严重的后果?简单来说就是,头孢类药物抑制了肝脏里的乙醛脱氢酶,如果这个酶的水平高意味着解酒功能强。但是这种酶功能被抑制了,使得酒精在人体内氧化为乙醛后不能再继续氧化分解,从而导致乙醛在体内蓄积,引起乙醛中毒反应。
·抗生素不是你想用就能用,想停就能停
这里仍需强调的是,抗生素不能随意使用,但是用了之后也不能随便停。因为当症状消退时可能还剩下一小部分菌没有完全消灭,这时候如果停药,那么这部分细菌生存下来之后会产生耐药性。当然如果过量,也会产生耐药性。
再比如说抗菌药物需每隔4-8小时服用(每种药物半衰期不同,所需间隔时间不同),因为如果两次服药时间间隔太近,会造成药物在血液中的浓度太高,从而导致神经或肝肾功能损伤;而间隔太远,血液中药物浓度不够,对细菌的杀灭作用就会减弱,同样会产生耐药性。
可以看到,耐药性被频繁提及,那么到底抗生素耐药性为什么可怕?滥用抗生素的后果是什么?
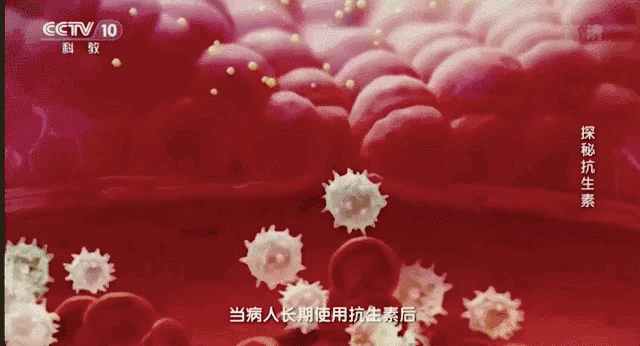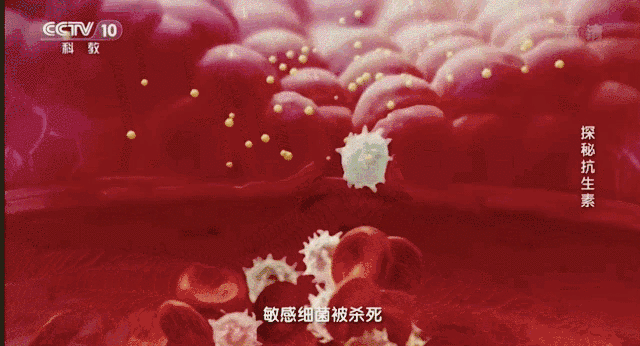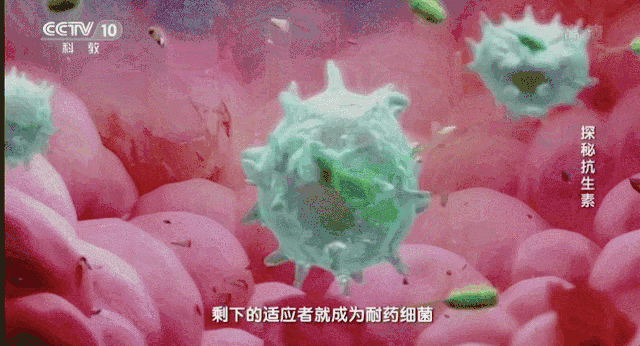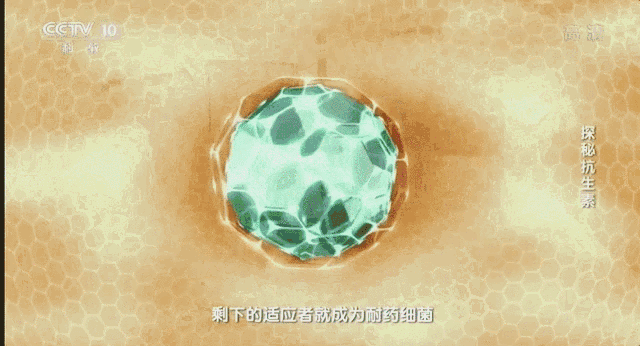

在2011年世卫组织提出:抵御抗生素耐药性。之后该组织在2015年再次向人们发出警告不要滥用抗生素。这无疑给世界敲响了警钟。
其实在过去20年中抗生素的负面性已经逐渐显现,由于抗生素滥用,问题变得逐渐复杂起来,耐药菌的出现越来越多。
· 抗生素药物的研发困难
在实际治疗中,人们对于是由何种细菌引起的感染认知较少,生产窄谱抗生素困难投入大,收效低,然而,在并不需要精确判定是什么菌引起感染的时候,只要用上广谱抗生素,就能起到作用,广谱抗生素的普适性优势使得其应用频率越来越大,随之而来的问题是,耐药菌的产生变得更为容易。
· 超级细菌的诞生
当抗生素在能力范围内将致病菌清除之后,那一小部分耐药菌留下来,不断生长繁殖,当耐药菌与药物不断接触之后,对药物的敏感性越来越低直至消失,逐渐进化成了无敌的超级细菌。


超级细菌的抗性机制可以通过获得基因来介导,使他们能够在抗生素或基因突变的作用下存活下来,作为自然选择过程的一部分进化而来。
超级细菌目前在全球范围内广泛流行:耐甲氧西林金黄色葡萄球菌、肺炎克雷伯菌、肺炎链球菌、艰难梭菌、沙门氏菌、大肠杆菌、铜绿假单胞菌和耐万古霉素肠球菌属等都是超级细菌的典型例子。
细菌可以天生对某些抗生素产生抗药性,但也可以通过染色体基因突变和水平基因转移获得对抗生素的耐药性。
耐药性的固有机制
一种细菌对一种特定抗生素的固有抗性是由于其固有的结构或功能特性而产生的对该抗生素作用的抵抗能力。
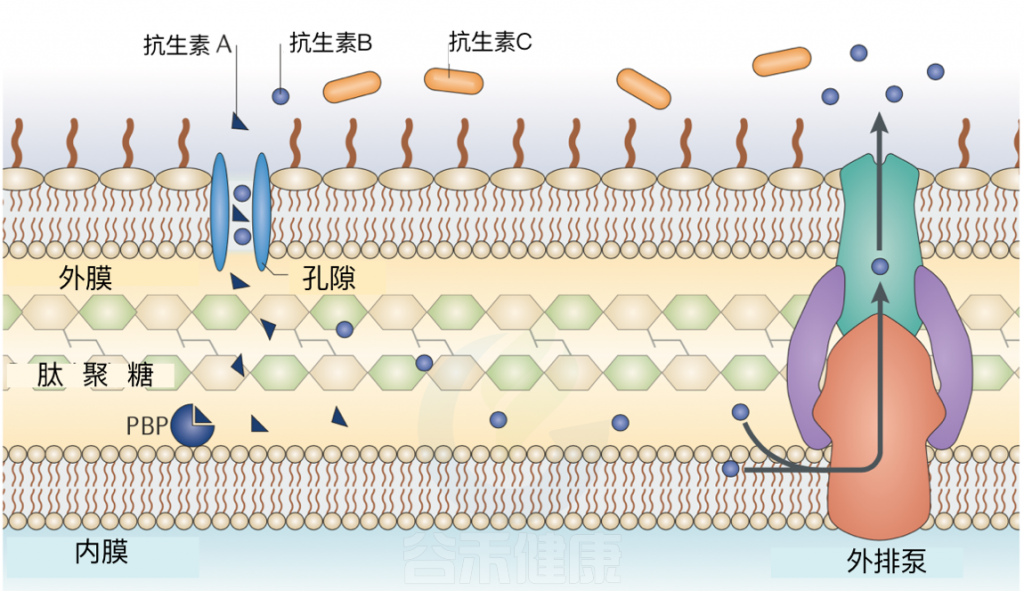

Blair J M A,et al, 2015
该图显示了固有机制的概述。所示的例子是针对青霉素结合蛋白(PBP)的β-内酰胺抗生素。
抗生素A可以通过一种跨膜孔蛋白进入细胞,达到目的,抑制肽聚糖的合成。抗生素B也可以通过孔蛋白进入细胞,但与抗生素A不同,它是通过外排有效去除的。 抗生素C不能穿过外膜,因此不能进入靶PBP。也就是说这个抗生素C想进去没门。
除了固有的耐药性外,细菌还能获得或产生对抗生素的耐药性。可以通过以下三类主要机制来调节:
由于细菌渗透不良或抗生素外排而使抗生素在细胞内的浓度降至最低的机制;
通过基因突变或靶标的翻译后修饰来修饰抗生素靶标的;
通过水解或修饰使抗生素失活的。
· 耐药性危害
毫无疑问,抗生素耐药性对生命构成了威胁,由耐药病原体引起感染的患者死亡率更高,人类遭受的痛苦也更大。由于长期住院和生产力下降,与抗生素耐药性相关的经济负担是巨大的,并在卫生保健系统中造成惊人的损失。
仅根据流行病学监测数据,对各种抗生素的耐药性比预期的要早几年到几十年。可以看到发展非常迅速。
目前主要的威胁来自于碳青霉烯类抗生素革兰氏阴性病原体,由一系列基因编码,包括ndm-1,oxa-48,kpc,vim.
而比碳青霉烯类抗生素耐药问题更为复杂的是,对粘菌素耐药的质粒介导基因(mcr-1、mcr-2、mcr-3、mcr-4、mcr-5和icr-mo)出现了。
粘菌素是一种比较古老的药物,一般不到万不得已的时候不会用到,它是用于复杂感染患者身上来对付超级细菌的,可以说是抗生素的最后一道防线。
现在对粘菌素耐药的质粒介导的基因出现可以说是最后的防线破了,那么患者的生存率就更小了。
· 动物中发现耐药菌
2016年,中国首次报道了mcr-1,它是从猪培养的大肠杆菌中分离出来的,并在从生肉、牲畜和人类培养的大肠杆菌中证实了它的存在。自那时以来,mcr-1已在全球范围内报道,并在许多细菌物种中检测到,包括肺炎克雷伯菌和肠杆菌属。
编码超广谱β-内酰胺酶(ESBL)的基因的多个变种,使大多数青霉素类、头孢菌素类和单杆菌类药物产生耐药性,目前已在全球分布。欧洲抗生素耐药性主要监测网络的报告表明,这些基因在大肠杆菌和肺炎克雷伯菌中的流行率仍然很高。
以上越来越多的证据指向一个事实,在动物身上使用抗菌药物与人类的耐药性有关。
· 动物与人之间的互相传播
上世纪40年代,人们发现给动物喂食抗生素后它们的体重飙升,为了尽快达到饲养目的,越来越多人将抗生素运用于动物饲养,那么这些抗生素首先会在动物之间进行传播,然后在不知不觉中就传播到人类中去。
Muloi及其同事的一项Meta分析表明,动物和人类之间的抗药性转移是双向的,但强度也不尽相同,这促使人们呼吁在全球范围内采用稳健的基因分型方法进行进一步的研究,世卫组织对当前证据的一项审查已经促使重新提出建议,大幅度减少动物体内的抗生素的使用,并停止使用它们促进生长。
· 抗生素耐药性也许就在身边
不要觉得自己离抗生素耐药性还相当遥远,其实稍不注意,抗生素耐药性就会发生。
案例一:5岁儿童,服用抗生素半年,他的肠道菌群检测报告显示:
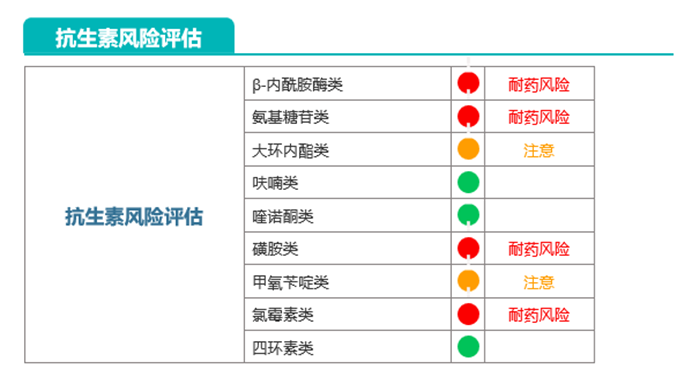
可以看到,这位5岁的小朋友已经出现抗生素耐药性高风险(红色为高风险状态)。
也就是说半年已经可以让一个小朋友产生多种抗生素耐药性。
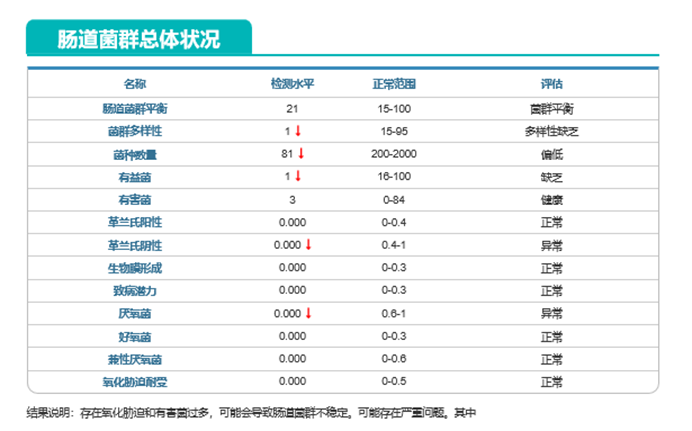
肠道菌群总体状况也提示菌群多样性减少,菌种数量减少。
尤其是有益菌数量的下降。

关于抗生素,疾病与菌群关联的这部分,我们将在下一章节详细介绍。
抗生素的另一个有可能被忽视的影响是:
抗生素的使用,无论是口服还是静脉注射,都会影响肠道微生物群。
人类的肠道微生物群代表着一个复杂的相互网络,抗生素的作用并不局限于其有限的抗菌谱:例如,由于相互依赖,利用途径,某些菌株不能在没有其他菌株的情况下有效地建立菌落。
健康肠道微生物群的稳定组成通过在空间和营养物质上的竞争,提供了对入侵病原体的自然抵抗力,这可能在抗生素暴露后受到严重影响。
抗生素引起的肠道菌群失调及其在疾病中的作用
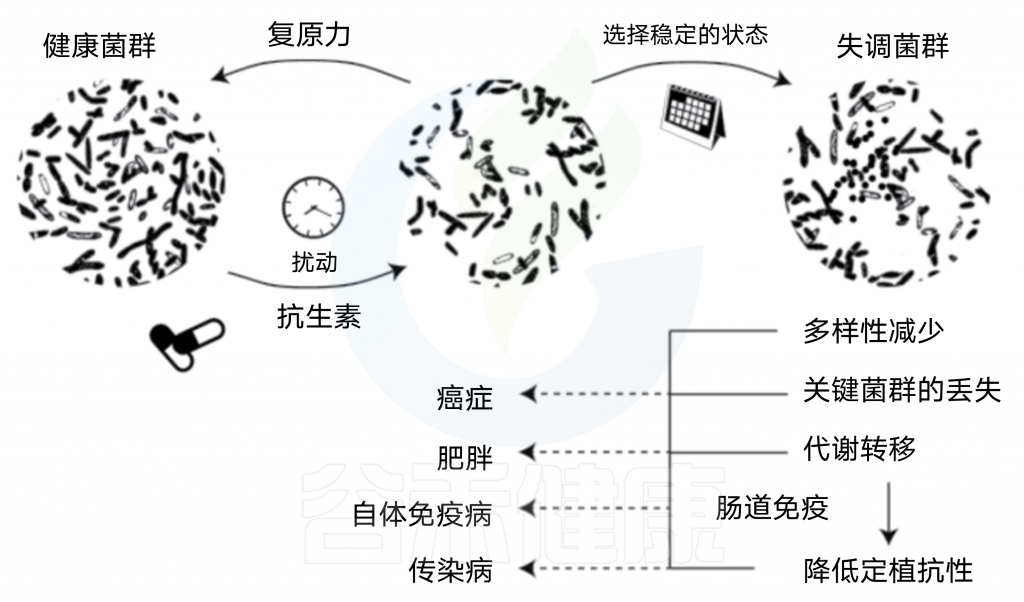

抗生素对肠道失调的影响和在疾病中的作用(即使短期使用抗生素)可能会将微生物群转变为长期失调状态,其特征是菌群多样性的变化,重要菌群的丧失,以及由此引起的代谢改变导致对肠道病原菌的定植抗性受损。
我们来看 案例二:住院患者,年龄76岁,长期服用抗生素治疗改善,其肠道菌群检测报告如下:
这是该患者的健康总分:29分。属于不健康状态。(注:总分100分制,一般健康人或亚健康普遍能在60分以上)


我们来看他的抗生素风险评估部分:
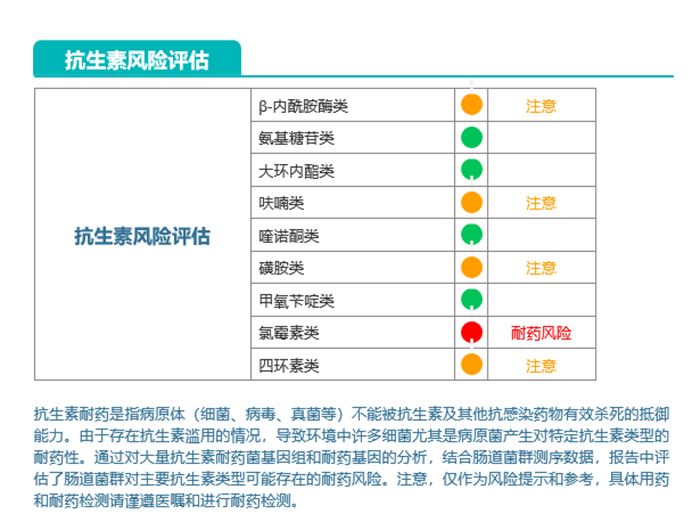
报告显示他的氯霉素类耐药风险高。再看肠道菌群的总体状况如下:

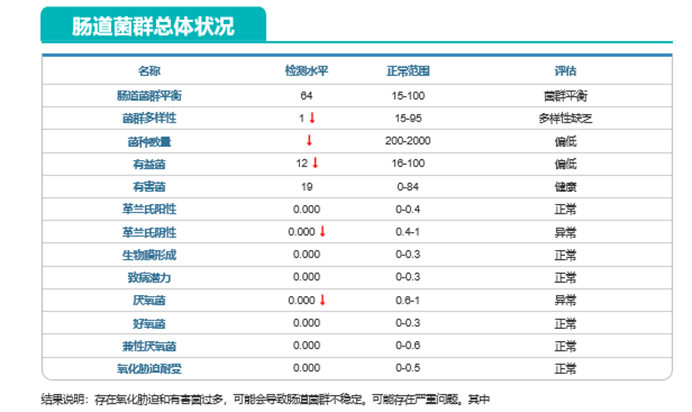
提示菌群多样性缺乏,有益菌数量明显下降。说明其菌群紊乱。而疾病风险评估这部分则显示了多种疾病高风险状态。尤其是代谢系统方面的疾病。
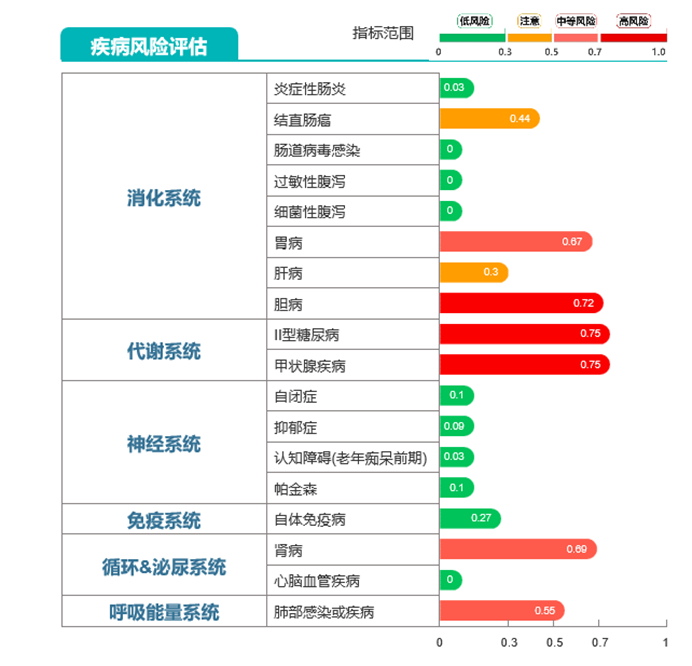

如果说你认为上述例子是长期使用抗生素的状态,短期使用没事。
那可不一定。短期使用抗生素的影响要看在什么年龄段,怎么使用法。
· 即使短期使用,也可能有长期影响
肠道免疫和微生物群的相互作用会促进代谢、自身免疫、传染病和癌症的发病机制和加重其严重程度。
即使是抗生素短期使用,特别是在生命的头两年,也可能导致肠道微生物的长期变化,从而改变与宿主的相互作用。由抗生素引起的干扰比成年人体内更剧烈,两岁内儿童的微生物群还处于一个低适应性阶段。
肠道微生物群在生命的最初几年发展,影响免疫系统的成熟。儿童期和成年期的抗生素治疗可能导致某些个体长期破坏生态系统,减少细菌多样性,导致群落不稳定,并对宿主生理产生负面影响。

Thiemann S,et al, 2016
而相对健康稳定的肠道微生物群是具有复原力的,也就是说在正常使用抗生素之后,微生物群依然可以恢复形成原来的样子。
抗生素治疗对肠道微生物的整体分类组成表现出快速和令人惊讶的持久性。
与幼儿相似,老年人(>65岁)代表了另一个群体,他们可能对抗生素引起的微生物群紊乱更敏感。
· 服用不同类别抗生素菌群变化
每一类抗生素都有不同的性质和排泄系统,导致菌群改变的模式不同。
大环内酯类抗生素现在是世界上最常见的抗生素之一,在儿童和成人中使用。许多患者由于慢性传染病而长期服用大环内酯类药物。
一项涉及142名芬兰儿童的研究显示,儿童食用大环内酯类药物会导致肠道微生物的改变,从而减少放线菌,增加拟杆菌和变形菌。有趣的是,在两岁以内开始服用大环内酯类药物的儿童与肥胖和哮喘患病率的增加之间存在正相关关系。
目前,克拉霉素已成为第一种用于根除幽门螺杆菌的抗生素。瑞典的一个研究小组报告说,根除幽门螺杆菌后放线菌和厚壁菌减少,拟杆菌和变形菌增加。即使根除没有成功,肠道微生物的改变也持续了很长一段时间。
另一组报告了口服万古霉素与阿莫西林治疗的比较。万古霉素表现出明显的减少粪便微生物多样性,因为厚壁菌减少和变形菌的增加。阿莫西林也能减少厚壁菌,但与万古霉素相比,这种减少是最小的。
此外,一个法国小组报告了一大批来自营养不良项目的尼日尔儿童。他们比较了安慰剂组和阿莫西林组的体重增加情况。虽然阿莫西林降低了住院疾病转移的风险,但也影响了患者的体重增加。在后面肥胖小节还会详细介绍。
环丙沙星减少厚壁菌和放线菌(特别是双歧杆菌),增加拟杆菌。另一组比较环丙沙星和克林霉素,环丙沙星减少双歧杆菌和克林霉素减少双歧杆菌和乳酸杆菌。然而,克林霉素组的乳酸杆菌没有恢复。
利用小鼠模型,一项研究证实了暂时服用低剂量青霉素和环丙沙星引起的微生物群变化的差异。在喂食正常食物的小鼠中,青霉素对肠道菌群的影响大于环丙沙星。此外,青霉素对微生物群的干扰在给药5天后用了5周时间来改善。相比之下,环丙沙星中正常微生物群的恢复速度更快。
肥胖已经是全球20多年来最大的健康问题之一,而且它仍在增加。
根据2010年的数据报告显示,在美国,婴儿大量接触处方抗生素。这份报告表明,抗生素在儿童中的使用,特别是在早期生活中,已经变得更加广泛。
· 生命早期服用抗生素影响体重
20世纪50年代,兽医研究人员已经报告说,在食物或水中使用抗生素可以促进哺乳动物家畜的生长。这种生长与抗生素之间没有先前的相关性。四环素,糖肽、大环内酯类和青霉素已在鸡、猪、牛等家畜中使用了70多年,以增加动物的体重。
非常重要的是要提到,抗生素在早期生活中的使用对动物的生长有更大的影响,而不是在以后的生活中使用。令人惊讶的是,研究人员已经报告说,在20世纪50年代,抗生素的使用与人类体重的增长之间存在着类似的相关性。
在20世纪60年代,抗生素的双重作用在小鼠模型中报告了体重增长,小剂量青霉素或土霉素可诱导体重增加,但大剂量相同抗生素可减轻体重。在无菌动物模型中使用抗生素并不能促进动物生长,因此假设微生物群的存在对于抗生素治疗下体重的增加或减轻是必不可少的。
·7岁儿童超重或与其婴儿时期服用抗生素有关
特拉桑德等人报道,儿童的体重增加取决于他们早期服用抗生素的时期。在生命的前6 个月内使用抗生素与肥胖有很强的相关性,但如果抗生素在生命的前6个月后使用,这种相关性就会消失。这项研究表明,在生命的6个月前接触抗生素药物的儿童在7岁时超重的风险要高得多。
在美国和加拿大进行的一项开放标签研究显示,6-18岁的儿童与对照组相比,阿奇霉素显著增加了体重增加的风险。虽然人们认为抗生素在早期生活中的暴露与肥胖有很强的相关性,但我们不知道明确的宿主-微生物相互作用,或导致这种表型的必要微生物变化。
· 并不是所有使用抗生素的研究都使体重增加,或与剂量有关
一项关于在英国成人中根除幽门螺杆菌的研究包括以下根除方案:柠檬酸雷尼替丁铋400毫克和克拉霉素500毫克,每日两次,为期两周。这项研究显示,治疗后体重显著增加。阿莫西林对体重增加的影响有两项研究。


如上所述,法国的这项研究没有发现用阿莫西林治疗的尼日尔儿童体重增加的显著差异。在另一项成人心内膜炎的研究中,患者静脉注射万古霉素庆大霉素与阿莫西林庆大霉素治疗6周。观察到万古霉素组体重明显增加,而阿莫西林组体重没有增加。
从所有这些研究中,我们可以假设,根据剂量的不同,广谱抗生素的使用与体重改变之间存在正相关关系。
当以治疗剂量使用抗生素时,对体重的影响有相互矛盾的结果。Muphy等人表明万古霉素可减轻高脂饮食的小鼠体重。其他研究表明抗生素治疗与体重减轻或代谢变化改善之间存在相关性。相反,Nobel等人指出,在小鼠生命早期给药治疗剂量的抗生素与体重显著增加有关。
以上,在人类和动物的研究中,有许多报告表明抗生素治疗与体重增加呈正相关。一些研究报告的相互矛盾的结果可能与使用抗生素的类型以及使用抗生素的时间有关。因此,医生应该更仔细地考虑什么时期的生命和他们应该开什么类型的抗生素。
有几个临床观察支持改变的肠道微生物群与炎症性肠病(IBD)的发展和病程密切相关:粪便移植改善了克罗恩病(CD)患者的临床表现。肠内细菌感染可导致IBD的恶化;一组CD患者对抗生素治疗有反应,特别是在术后情况下。
· IBD患者的菌群特征
一开始,“太多”的微生物群似乎是引发IBD的原因;但事实上,IBD的特征是胃肠道微生物群的组成发生了变化。与健康受试者相比,CD、溃疡性结肠炎(UC)和微囊炎患者的微生物菌群多样性较低。
IBD患者肠道宏基因组包含的基因比健康的肠道少25%,蛋白组学研究显示,蛋白质和功能途径也同时减少。
可以想象,抗生素引起的失调促进了IBD的发展和延续。实际上,厚壁菌门中的具有保护作用细菌数量在减少,特别是Faecalibacterium prausnitzii 和Akkermansia muciniphila的减少(这两种菌还能产生短链脂肪酸,丁酸和丙酸,从而支持粘膜屏障的完整性),并伴有粘附性侵袭性大肠杆菌的增加。
根据抗生素暴露可能导致丰度降低和某些菌群丢失,最近发表的一项Meta分析表明,抗生素暴露似乎增加了诊断克罗恩病而不是溃疡性结肠炎的几率。
· IBD中抗生素治疗有待进一步研究证实
此外,有一些证据表明,IBD中抗生素治疗的与更严重的疾病过程有关。虽然正常人类微生物组成的短期和长期变化可能影响IBD的易感性和病程,但抗生素引起的失调与IBD的发展之间的因果关系尚未得到证实。
在出现IBD和合并症的情况下,情况会变得更加复杂。肠易激综合征和伴发的原发性硬化性胆管炎(PSC)患者的肠道菌群与单纯的肠易激综合征患者不同。它们显示出Prevotella和Roseburia属减少,几乎完全不存在拟杆菌门,而大肠杆菌显著增加。与PSC中肠漏作为肝脏炎症的驱动因素的作用一致。某些研究中抗生素治疗改善了的生化或组织学活性。然而不知道的是肠道菌群组成的改变还是胆道感染的治疗引起的。
如前所述,完整的共生菌群通过多种机制(包括竞争营养物质、空间和结合位点)以及间接免疫介导作用和直接抗菌作用入侵病原体,从而保护胃肠道免受感染。
抗生素相关的继发性肠道感染易感性是众所周知的,肠杆菌科包括沙门氏菌、志贺氏菌和大肠杆菌,对于革兰氏阳性菌,包括艰难梭菌和肠球,特别是厌氧菌,似乎是定植抗性的重要介质。
· 某些菌的保护作用
某些主要来自厚壁菌门和放线菌门的菌,已被鉴定为通过各种直接和间接机制拮抗特定感染。
已有研究表明,Ruminococcus obeum菌对霍乱弧菌感染具有保护作用,多形拟杆菌和梭状芽孢杆菌相关的分段丝状细菌对革兰氏阳性菌具有保护作用。值得注意的是,在某些情况下发生肠源性感染时,这些保护性菌群自然扩大,改善了恢复,并提供了防止反复感染的优势。
· 抗生素引起的菌群变化促进结肠炎发生
抗生素治疗减少了肠道菌群的多样性和保护性分类群的数量,增加了对新感染的易感性,也产生了抗生素耐药细菌。抗生素引起的肠道微生物群分类和功能多样性的减少为孢子形成、孢子萌发和艰难梭菌毒素的产生提供了最佳条件,从而促进了肠结肠炎的复发。
因此,克林霉素和广谱β-内酰胺会对微生物组造成长期干扰,它们是最容易感染梭状芽胞杆菌相关腹泻的药物,尤其是当梭状芽胞杆菌菌株对致病药物有耐药性时。
许多研究已经调查了菌群失调与结直肠癌(CRC)的关系。抗生素治疗后某些类群的丰富或丢失可能通过与粘膜免疫细胞和上皮细胞的失调菌群相互作用而导致癌变。
再加上其他危险因素,如遗传易感性、单菌种的慢性定殖,如脆弱拟杆菌可以促进上皮细胞增殖、呼吸内皮应激、DNA损伤,并促进小鼠体内的致癌性微生物群。
然而,考虑到饮食对肠道菌群和CRC的巨大影响,这种假说很难在人类身上得到验证。 当根据饮食模式对个体进行CRC风险分层时,高风险个体显示出产生短链脂肪酸的细菌数量减少,例如抗炎性F. prausnitzii 和 Eubacterium / Roseburia spp.,产次胆汁酸的菌群增加。
几个宏基因组研究表明,结直肠癌患者肠道菌群的细菌丰度中拟杆菌、厚壁菌、变形菌减少,而梭状杆菌门富集。
· 频繁使用抗生素与CRC风险之间存在关联
这些物种的丰度减少可能是用抗生素后失调的结果。支持这一假设的是,最近的一项病例对照研究表明,频繁使用抗生素与CRC风险之间存在关联。一项包括300多万人在内的队列研究也可以证明,与使用一种或少于一种抗生素处方的人相比,过去3年内使用过6 种或更多抗生素处方的人患癌症的相对风险为1.37(95%CI 1.34-1.40),患CRC的风险为1.15(95%CI 1.04-1.26)。
即使在纠正了诸如生活方式、共患病和联合用药等潜在的混淆因素之后,在另一项研究中也报道了青霉素和CRC重复给药的关联。由于这类队列研究的性质,只能知道关联而不能解决因果关系。可以想象,免疫系统较弱的患者(因为这是恶性疾病的风险之一)更容易发生需要使用抗生素的感染。
这可以解释为什么在其他研究中也描述了恶性肿瘤的风险更高,例如非霍奇金淋巴瘤或乳腺癌,却没有说明抗生素引起的微生物群变化是这些恶性肿瘤的潜在原因。
肠道与肝脏之间的在解剖上和功能上密切联系表明,微生物群可以在多个层面上影响肝脏功能,并促进各种慢性肝病和代谢性疾病的发病。
· 菌群失调与肝脏炎症
很长一段时间以来,人们都知道,菌群失调会导致肥胖相关的非酒精性脂肪肝(NAFLD)的发展。在20世纪80年代,人们发现非酒精性脂肪性肝炎(NASH)在肠道转流后细菌过度生长后发展起来的,但在用甲硝唑进行抗菌治疗后有所改善。有证据表明,与其他因素相结合,失调可能导致门静脉循环中细菌产物的异常积聚,并导致肝脏炎症。
· 菌群可能通过不同机制驱动NAFLD
新的研究表明,肥胖的NAFLD患者粪便中的微生物群显示,厚壁菌门尤其在乳杆菌科和毛螺菌科中富集,同时也存在着瘤胃球菌科的缺失。
与没有NASH的肥胖NAFLD患者相比,有肝脏炎症表现的患者仅在变形菌门内表现出显著差异。这些结果表明,肠道微生物群可能通过不同的机制独立地驱动NAFLD的不同方面,如肝脏炎症或肝脏脂肪变性。NASH尤其涉及紊乱的肠道菌群代谢后果。
在NASH患者中观察到的血液中乙醇水平升高是否是肠道微生物群内源性生产的结果仍是一个有争议的问题。一项针对NAFLD与失调菌群组成的系统得出了有关联的结论,但没有因果关系的证据。
· 菌群与疾病的关系中发现使用抗生素的价值
动物模型显示,肠上皮的固有免疫(toll样和NOD样受体)是如何严格调控肠道微生物群,从而控制NAFLD和NASH的进展的。在这些模型中,Revotellaceae和卟啉单胞菌科(均来自拟杆菌门)是与肝脏疾病进展相关的丰富物种中的两个,可使用广谱抗生素来消除。
靶向肠道微生物群以减轻肝损伤可能是预防炎症性、代谢性和自身免疫性肝病并发症的最有希望的方法之一。
抗生素的发现是现代医学最重要的进展之一。我们看到抗生素在发挥作用的同时,其耐药性的发展将比治疗这些感染的新药物的出现更为迅速,因此需要更好地理解控制抗生素耐药性传播的分子、进化和生态机制。
当然我们也不必恐慌,DNA测序技术和基因组学研究的最新进展使我们能够跟踪抗生素耐药性的演变。有效的疫苗和更快、更敏感的诊断在内的其他策略也有助于减少耐药性的出现。其他也有很多科研成果给我们带来新的希望。
综合微生物群、抗生素和相关疾病之间的双边联系的间接证据,重要的是要认识到,抗生素的影响并不总是对宿主有害,也可能在疾病中提供治疗机会。抗菌疗法可通过长期改变肠道微生物群来调节各种疾病,包括代谢、自身免疫和传染病。
附录:
较为常见的抗生素种类作用
磺胺类药物是一种抑菌、抗代谢物药物,可抑制叶酸的合成。它们是对氨基苯甲酸(PABA)的类似物,是二氢翼状体合成酶的竞争性抑制剂。堵住叶酸的合成,诱导DNA、RNA和蛋白质合成的抑制作用。
磺胺类药物对革兰氏阳性菌和革兰氏阴性菌均具有广谱活性。它们主要用于治疗简单的尿路感染和诺卡氏菌相关感染。磺胺乙酰胺用于局部和眼科细菌感染。
磺胺类药物应用最广泛的是磺胺甲恶唑,由于其在抑制叶酸合成的后续步骤中的协同作用,通常与吡甲恶唑合用。它们是针对易感形式的链球菌、金黄色葡萄球菌的抑菌抗生素,包括来自社区获得性甲氧西林耐药葡萄球菌的皮肤感染。
此外,磺胺类药物对口腔厌氧菌和一些革兰氏阴性棒(如大肠杆菌和流感嗜血杆菌)具有活性。
青霉素类的所有成员都是6-氨基戊二烯酸的衍生物,含有β-内酰胺环结构,对抗菌活性至关重要。
青霉素以及其他β-内酰胺类抗生素具有间接杀菌活性。它们抑制肽聚糖在细菌细胞壁中的交联形成,附着在称为青霉素结合蛋白(pbps)的细菌酶上,从而激活细胞壁自溶。
青霉素的分类取决于侧链上附加的化学替代物的基础,与青霉素G相比,侧链上附加的化学替代物主要通过向革兰氏阴性菌延伸而导致生物利用度和活性谱的差异。
根据活性谱识别出4个青霉素亚类:
窄谱或天然青霉素,非常窄谱(或耐青霉素酶的青霉素),广谱或氨基青霉素,广谱或抗假性青霉素。窄谱青霉素类包括青霉素V、青霉素G、普鲁卡因青霉素和苄星青霉素。
抗药性的出现一直是开发新型青霉素的主要动力之一。
青霉素的两个最显著的作用可能是治疗风湿热和梅毒。在1945年至1975年期间,仍然是早期梅毒和潜伏梅毒的推荐治疗方法的苄星青霉素G使梅毒的发病率和死亡率下降了近90%。
耐青霉素葡萄球菌的迅速出现促使人们研究药物替代品。在20世纪50年代末,围绕6-氨基青霉烷酸的半合成提出了一类新的抗青霉素酶青霉素:苯唑西林、甲氧西林、氯唑西林和双氯唑西林。
这些抗生素也被称为抗葡萄球菌青霉素,因为它们与青霉素G相比具有窄谱的活性。它们仅用于治疗对甲氧西林敏感的金黄色葡萄球菌(MSSA)。重要的是要强调非常窄的活性谱和MRSA(耐甲氧西林金黄色葡萄球菌)菌株的迅速出现。
青霉素的大规模扩张是在1940年至1960年期间,Rolison博士在比查姆研究实验室的工作中完成的。目的是鉴定具有抗菌活性的化学结构,扩大活性谱,提高生物利用度和口服吸收。
氨苄西林和阿莫西林是第二代青霉素或广谱青霉素的第一批抗药药。氨苄西林被引入以获得对革兰氏阴性菌更好的覆盖,但很快人们认识到这些微生物的菌株具有可变的敏感性。它们呈现相同的化学结构,但口服阿莫西林后显示出较高的血清浓度。
第二代青霉素对许多细菌都有活性,包括链球菌和葡萄球菌、肠球菌、单核细胞增多性链球菌、梭状芽胞杆菌和炭疽杆菌等非青霉素产生菌。在革兰氏阴性菌中,这些青霉素对流感嗜血杆菌、大肠杆菌、沙门氏菌和志贺氏菌有活性。
阿莫西林
阿莫西林(1972)被认为是治疗由化脓性链球菌引起的急性中耳炎和上呼吸道感染的首选药物。
阿莫西林在治疗简单的尿路感染时也表现出抗粪肠球菌的活性,但考虑到大肠杆菌和肠杆菌科菌株的高耐药率,阿莫西林并未广泛用于这些病原体。

考虑到与该感染相关的致癌风险,阿莫西林在治疗和根除幽门螺杆菌方面的作用极其重要。最后,阿莫西林被广泛用于治疗孕妇尿道炎和宫颈炎。
氨苄西林仍然是治疗婴幼儿肺炎球菌性肺炎的首选药物,同时也是李斯特菌病的首选药物。
下一代青霉素的代表是羧苄青霉素、替卡西林和哌拉西林。这些广谱抗生素对革兰氏阳性球菌,厌氧菌和革兰氏阴性菌具有活性。 而且,这些药物可抵抗某些生物体的染色体β-内酰胺酶。
哌拉西林-他唑巴坦的组合于1995年推出,由于对革兰氏阳性细菌(包括链球菌和肠球菌)的高活性,因此可能是使用最广泛的组合,但对耐甲氧西林的葡萄球菌无效。
然而,这种组合的主要优点是对革兰氏阴性杆菌特别是铜绿假单胞菌具有活性。
在整个19世纪,结核病及其所有临床表现代表了年轻人最常见的死亡原因。链霉素是第一种分离的氨基糖苷类药物,也是第一种治疗结核病的抗生素。此外,链霉素为临床研究的发展开辟了道路。
链霉素是一种蛋白质合成抑制剂和杀菌抗生素。它与细菌核糖体30s亚基的小16Sr RNA结合,导致密码子误读和抑制蛋白质合成。

主要用途是治疗分枝杆菌感染,并作为第四种药物与异烟肼,利福平和吡嗪酰胺治疗结核病。
链霉素已被证明在体外和体内对革兰氏阴性微生物具有活性,如布鲁氏菌、肉芽肿性克雷伯菌和肺炎克雷伯菌、大肠杆菌、蛋白质杆菌属、产气杆菌、特氏杆菌、杜氏嗜血菌、流感嗜血杆菌和鼠疫杆菌等,以及链球菌和粪肠球菌等阳性细菌。
按时间顺序,头孢菌素是继青霉素之后发现和开发的β-内酰胺类抗生素中的第二类。头孢菌素的化学结构含有β-内酰胺环,和一个含有硫和氮原子的六元环融合。
在美国最常见的处方药和临床依赖抗生素的药物中,这些药物制剂占医院开支的很大一部分。
头孢菌素作为内酰胺类抗生素,主要具有杀菌作用,其主要作用方式是干扰细菌细胞壁的合成。
个别头孢菌素的抗菌活性是可变的,受多种因素的影响,包括抗酶降解的能力和穿透细菌细胞壁的能力。
一般来说,头孢菌素在治疗耐青霉素的肺炎链球菌、表皮葡萄球菌、粪肠球菌、肺炎支原体、艰难梭菌或肺炎衣原体方面历来不是首选抗生素。
与前一代相比,头孢菌素的渐进世代在革兰氏阴性抗菌特性方面表现出渐进性的改善,通常对革兰氏阳性生物体的活性降低。
第一代头孢菌素
在所有头孢菌素中,第一代头孢菌素,如肠外药物头孢霉素和头孢唑林以及口服制剂头孢氨苄对革兰阳性菌、甲氧西林敏感葡萄球菌和非肠道链球菌最为有效。
第二代头孢菌素
第二代半合成衍生物头孢克洛于1979年在美国上市。作为一个整体,第二代头孢菌素对革兰氏阳性菌的疗效较低,而对革兰氏阴性菌(包括流感嗜血杆菌、产气大肠杆菌和某些奈瑟菌)的临床应用效果更高。
值得注意的是,第二代头孢菌素中唯一能跨越血脑屏障的是用于治疗脑膜炎的头孢呋辛。
第三代和第四代药剂的活性通常被认为是广谱的,尽管某些成员对革兰氏阳性生物的活性降低,但与第一代和第二代相比,对革兰氏阴性生物的活性进一步提高。第三代头孢菌素可能在治疗医院获得性感染,尤其是社区感染方面具有临床应用价值,尽管越来越多的超广谱β-内酰胺酶降低了这类抗生素的临床应用。
第三代头孢菌素
也能穿透中枢神经系统,使其对肺炎球菌、脑膜炎球菌、流感嗜血杆菌、易感大肠杆菌、克雷伯菌和耐青霉素的淋病奈瑟菌引起的脑膜炎有效。更具体地说,第三代头孢菌素经常是淋病的临床治疗选择。
第四代头孢菌素
包括头孢哌罗、头孢吡肟和头孢克立定,是对肠杆菌属、柠檬酸杆菌属和粘质沙雷氏菌具有增强活性的广谱头孢菌素。同时它与氨基糖苷类药物或氟喹诺酮类药物联合治疗,为严重的铜绿假单胞菌感染也提供了更多的治疗选择。
新一代
进一步开发用于临床的新一代头孢菌素。头孢吡普(2002)和头孢洛林(2005)被认为是第五代头孢菌素,但这一术语仍有争议。头孢吡普和头孢洛林可能不易产生耐药性,从而为耐甲氧西林金黄色葡萄球菌提供治疗选择。
主要参考文献
Durand G A, Raoult D, Dubourg G. Antibiotic discovery: History, methods and perspectives[J]. International journal of antimicrobial agents, 2019, 53(4): 371-382.
Zaffiri L, Gardner J, Toledo-Pereyra L H. History of antibiotics. From salvarsan to cephalosporins[J]. Journal of Investigative Surgery, 2012, 25(2): 67-77.
Iizumi T, Battaglia T, Ruiz V, et al. Gut microbiome and antibiotics[J]. Archives of medical research, 2017, 48(8): 727-734.
Aminov R I. The role of antibiotics and antibiotic resistance in nature[J]. Environmental microbiology, 2009, 11(12): 2970-2988.
Lange K, Buerger M, Stallmach A, et al. Effects of antibiotics on gut microbiota[J]. Digestive Diseases, 2016, 34(3): 260-268.
Laxminarayan R, Van Boeckel T, Frost I, et al. The Lancet Infectious Diseases Commission on antimicrobial resistance: 6 years later[J]. The Lancet Infectious Diseases, 2020.
Blair J M A, Webber M A, Baylay A J, et al. Molecular mechanisms of antibiotic resistance[J]. Nature reviews microbiology, 2015, 13(1): 42-51.
Thiemann S, Smit N, Strowig T. Antibiotics and the intestinal microbiome: individual responses, resilience of the ecosystem, and the susceptibility to infections[M]//How to Overcome the Antibiotic Crisis. Springer, Cham, 2016: 123-146.

炎症性肠病(IBD)是一种慢性且易复发的肠道炎症性疾病。全球500多万人受其困扰。
这种疾病是由多种遗传和环境因素共同作用引起的,这些因素改变了肠道的稳态,从而引发了基因易感个体的免疫介导炎症反应。
炎症性肠病包括克罗恩病和溃疡性结肠炎。
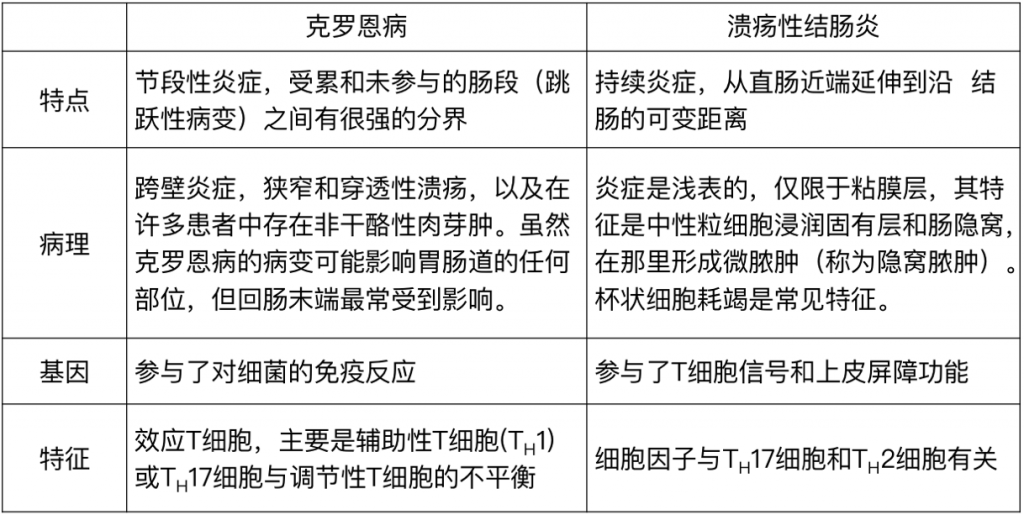
两者异同:
虽然克罗恩病和溃疡性结肠炎的发病机制都涉及肠道炎症,但这两种疾病在几个方面有所不同,包括与特定易感位点的关系、与疾病相关的免疫反应和病理类型。

这两种形式的IBD发病机制的一个关键方面是它们与生活在肠道中的共生微生物的存在有关。
寄主共生体参与了许多宿主的生理过程,包括消化和代谢功能、上皮屏障的调节、宿主免疫系统的发育和调节以及对病原体定植的保护。
IBD的一个关键特征是肠道微生物群失调,然而,失调在疾病中的确切作用仍然鲜为人知。


大量共生微生物靠近上皮表面是对粘膜免疫系统的一个独特挑战,因为它必须保持对入侵病原体的免疫反应的能力,同时避免对肠道微生物群产生有害的炎症反应。
这篇文章主要围绕以下几个问题展开讨论:
被称为“粘膜防火墙”的保护机制包含哪几种策略来维持稳态?
IBD相关的基因突变如何破坏粘膜防火墙?
IBD相关的遗传缺陷如何导致病变积累和渗透到肠道组织,从而进一步促进失调和炎症?
如何将微生物群作为IBD的潜在治疗方法?
肠道中大量的共生微生物被单层上皮细胞从宿主组织中分离出来。为了维持其与肠道菌群的稳态关系,宿主已经进化出几种策略来减少微生物与上皮表面的接触,并限制可能引发不必要的炎症反应的渗透共生体的存在。
宿主通过几种机制限制潜在有害的微生物接近粘膜表面,包括粘液分泌、抗菌蛋白和免疫球蛋白释放到肠腔。
我们先结合图1来看看大小肠的特点。
在大肠中,有两个明显不同的粘液层:
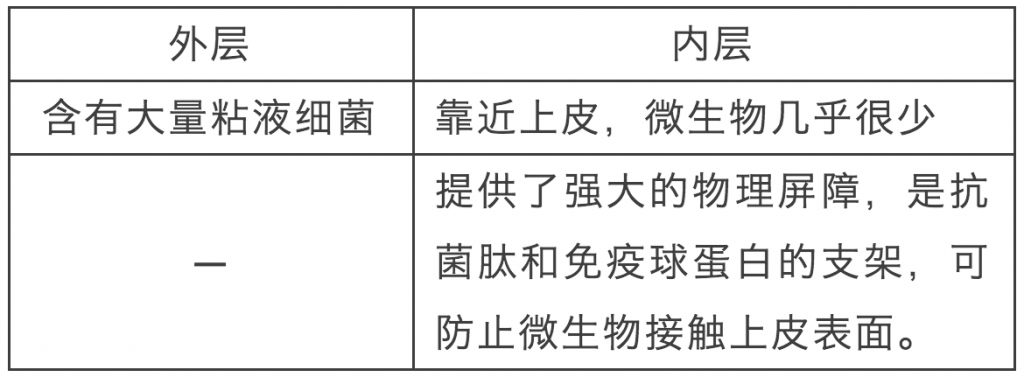

而小肠呢,没有像大肠那样清晰的内部粘液层,但它含有大量的Paneth细胞,这是一种特殊的肠道细胞,富含抗菌分子。为响应细菌刺激,Paneth细胞将富含的抗菌分子(包括α-防御素)释放到肠腔中,以限制共生菌的积累。
此外,小肠中的肠上皮细胞(IEC)分泌抗微生物凝集素,例如再生的胰岛衍生蛋白3γ(REG3γ),其在粘液层中积累并进一步促进微生物从宿主中的分离。
【名词小讲堂】
肠上皮细胞(IEC):
· 肠道上皮细胞是体内与外界之间的一个重要的保护屏障。
· 它由不同功能性的肠道干细胞群体共同维持,是具有极性的柱状上皮细胞,参与肠道的消化,吸收,分泌,免疫屏障和应激反应等。
· 粘膜上皮内含有大量的免疫细胞和免疫分子,是机体内最大的免疫组织。
再来看固有层(也就是图1 蓝色部分)
虽然固有层基本不存在急性炎症细胞,但某些嗜中粒细胞可以稳定迁移至肠腔侧,通过多种机制杀死上皮表面附近的细菌,包括诱导氧化猝发。
【名词小讲堂】
氧化猝发(Oxidative burst ):可直接杀死细菌的吞噬细胞迅速产生活性氧。
图1 粘膜防火墙
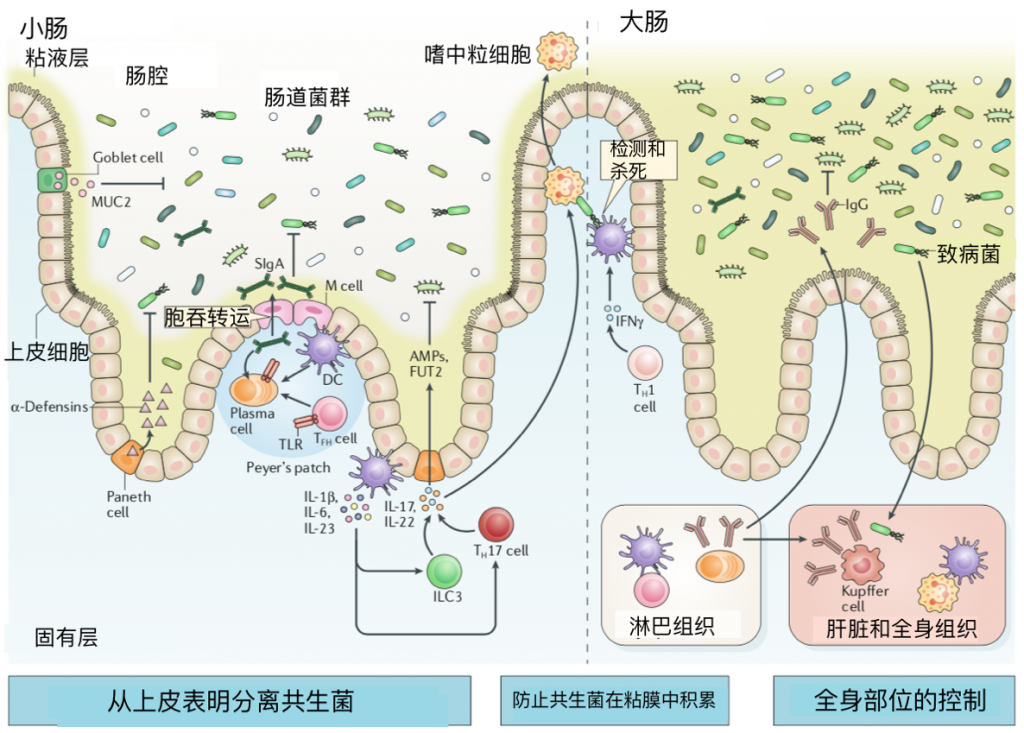

图注:杯状细胞分泌糖蛋白,包括粘蛋白2(MUC2),形成粘液屏障,防止肠腔中的微生物与上皮接触。在小肠中,松散形成的粘液层包裹上皮细胞,而在大肠中,内部粘液层基本上没有细菌,粘液共生体仅限于外部粘液层。
位于小肠隐窝的Paneth细胞构成性地表达杀微生物α-防御素。微折叠(M)细胞是一种特殊的上皮细胞,覆盖淋巴组织,如Peyer‘s斑片,便于抗原取样。树突状细胞(DCS)样本抗原在Peyer‘s斑片中由m细胞传递,或可能直接在腔内通过投射穿透上皮层。
分泌性IgA(SIgA)是由位于 Peyer‘s 斑片中的浆细胞产生的,通过转胞分泌穿过上皮进入肠腔。大多数 SIgA 是在 Toll 样受体(TLR)通过微生物抗原参与T细胞和 b 细胞后产生的,并与广泛的肠道微生物低亲和力结合,防止它们在上皮中易位。滤泡辅助(TFH)细胞依赖性反应通过支持高亲和力IgA的产生进一步促进稳态,后者与选择细菌结合。
髓系细胞衍生细胞因子,包括IL-1β、IL-6和IL-23,促进t辅助17(Th17)细胞分化和第3组固有淋巴样细胞(ILC3)的激活。由ILC3S和Th17细胞产生的IL-22通过诱导抗菌肽(AMPS)的表达,包括再生胰岛衍生蛋白3γ(Reg3γ),进一步加强共生体的分离。
il-22还能促进岩藻糖基转移酶2(fut2)介导的上皮性糖聚糖的岩藻糖基化,以支持微生物共生,防止潜在有害细菌的入侵。如果细菌能够克服这些障碍,IL-17刺激的中性粒细胞可以消除根尖上皮表面附近的病理组织或那些已经到达固有层的病理组织。
TH1细胞源性干扰素γ(IFNγ)进一步增强了巨噬细胞和DCs对微生物的杀灭作用。细菌特异性IgG的产生通过结合病理生物促进调理作用来控制全身感染。此外,渗透微生物被肝和脾巨噬细胞中的Kupffer细胞吞噬,以控制系统传播。
接着看下细胞因子IL-22的作用。
细胞因子IL-22还通过作用于上皮细胞介导屏障功能和抗微生物宿主防御,在建立宿主-微生物相互作用中发挥作用。例如,第3组固有淋巴细胞(ILC3s)分泌IL-22是抑制共生细菌所必需的,因为它诱导了抗菌肽的表达,从而阻止了Alcaligenes spp 的全身性传播。
IL-22的天然来源对于控制肠道内分段丝状菌(SFB)的增殖和限制T辅助17(TH17)细胞介导的结肠炎也很重要。
【名词小讲堂】
分段丝状菌(SFB),一种与梭菌有关的细菌,主要居住在小鼠的末端回肠,并促进辅助17细胞的发育。
IL-22还促进上皮聚糖岩藻糖基化,以支持适应岩藻糖作为营养源的共生细菌的生长。IL-22信号传导和岩藻糖基化的中断导致更易感染肠道和结肠炎,部分原因是条件致病菌的过度生长。
IgA也能促进肠道细菌从上皮表面的分离,IgA是由肠道相关淋巴组织中的浆细胞产生。
大量的聚合物IgA在上皮细胞中跨通道进入内腔,其中IgA通过包被细菌和结合微生物抗原及其毒素来维持屏障功能,并塑造微生物的组成。
IgA反应在体内平衡过程中通过T细胞独立和T细胞依赖过程发生。IgA抗体具有典型的多反应性,与微生物脂多糖、DNA和鞭毛抗原结合亲和力低。通过Toll样受体(TLR)参与的微生物感测可以直接刺激IgA的产生,从而提供针对肠道炎症的保护。
【名词小讲堂】
Toll样受体(TLR):
· TLR是参与非特异性免疫(天然免疫)的一类重要蛋白质分子,也是连接非特异性免疫和特异性免疫的桥梁。
· TLR是单个的跨膜非催化性蛋白质,可以识别来源于微生物的具有保守结构的分子。
· 当微生物突破机体的物理屏障,如皮肤、粘膜等时,TLR可以识别它们并激活机体产生免疫细胞应答。
此外,T细胞对于MYD88对微生物信号的内在感应对体内IgA稳态反应对预防营养不良和肠病非常重要。
【名词小讲堂】
髓样分化因子(MYD88):是Toll样受体(TLR)信号通路中的一个关键接头分子,在传递上游信息和疾病发生发展中具有重要的作用。
肠道微生物群中的某些菌在结肠炎期间被IgA包被,当转移到无菌动物时,可增强对结肠炎的易感性,这表明在失调期间优先结合IgA可识别与IBD相关的菌群。
而大多数共生体强烈诱导T细胞独立的IgA结合,“非典型”细菌,如SFB,M. schaedleri,Prevotella spp.和Helicobacter sp. flexispira,可以逃避T细胞独立抗体应答,使其非常接近上皮表面,在上皮表面引发抗原依赖性,高亲和性,T细胞依赖性IgA反应。
这些研究表明,IgA可以通过促进有益共生菌的定殖来强化“健康”的微生物群落,但是,一旦在失调的状态下,可能会诱发IgA对潜在致病菌的免疫反应。
前面小节提到的:促进共生体与上皮分离的保护机制并不是万无一失的。
考虑到存在于上皮表面附近的大量微生物,并不是所有的微生物都能守规矩,其中一小部分就可以在稳态条件下突破上皮屏障。为了应对这种情况,宿主免疫系统就发展出具有限制粘膜破坏性炎症和将渗透性微生物传播至全身组织的策略。
它可以让大量存在于上皮下面的特殊固有层巨噬细胞去杀死细菌共生体,通过多种机制吞噬和杀死渗透微生物,包括产生抗菌分子和活性氧。
在稳态条件下,这些驻留巨噬细胞在微生物感应方面有缺陷,因此不会引起炎症反应,这是一种通过IL-10刺激介导的调节活动。
肠道微生物也可以被固有层中的树突状细胞(DCs)吞噬并运输到肠系膜淋巴结,在那里,含有树突状细胞的细菌诱导保护性IgA和调节性T(Treg)细胞;这些树突状细胞不能到达全身的二级淋巴结结构,从而限制了它们的全身传播。
【名词小讲堂】
树状突细胞(DCs):
· 也称DC细胞,最早是由加拿大学者Steinman于1973年发现的
· 是功能最强的抗原提呈细胞,因其成熟时伸出许多树突样或伪足样突起而得名。
尽管前面强调存在坚固的粘膜防火墙,但依然会有非常小部分的肠道微生物可通过静脉门系统或血管系统扩散(图1)。 不过这些微生物到了那之后,肝脏会派出kupffer细胞或脾脏派出的巨噬细胞来把这些偷跑出来的菌吞噬和杀死。
共生菌还可以通过T细胞依赖和T细胞独立机制诱导IgG稳态反应。IgG抗体可以通过识别高度保守的蛋白质(例如murein脂蛋白)来结合多种细菌,包括变形杆菌和相关病原体,以限制细菌从肠道的全身传播。
此外,IgG2b和IgG3等型与IgA结合的共生物种具有相似的反应性,可以在母乳中转移给新生儿;这些母体IgG等型与IgA结合新生儿微生物群,以限制异常的共生特异性T细胞介导的炎症。
在缺乏IgA或MYD88和TIR结构域的动物中,系统性IgG对微生物群的反应增强,这些结构域包含用于TLR介导的细菌感应的干扰素-β(TRIF)信号适配器。
这种增强的IgG反应很可能代表由于适应性降低和渗透性共生细菌被杀死而导致的全身组织中共生菌增加的补偿性适应。
肠道病原体已经进化出多种策略来破坏和逃避粘膜防火墙,限制细菌共生体进入粘膜组织。几种病原体,包括肠沙门氏菌、Shigellaflexneri、Yersinia enterocolitica和霍乱弧菌,都会产生粘蛋白降解酶。
此外,肠道病原体已经进化出抵抗机制对抗IECs产生的抗菌蛋白。例如,肠杆菌可表达参与调节抗菌肽抗性的脂多糖修饰的基因,以及参与抗菌肽固存、外排和降解的基因。
同样,Listeria monocytogenes 使肽聚糖中的N-乙酰氨基葡糖残基脱乙酰化,从而逃脱了溶菌酶的溶菌活性,溶菌酶是Paneth细胞产生的一种酶。
与细菌共生体不同,肠道产生毒力蛋白,以逃避吞噬细胞中的溶酶体降解。进入肠细胞后,S. enterica 会破坏液泡膜并逃逸到宿主胞浆中,从而逃避自噬介导的降解,复制并在IECs之间传播。
在中性粒细胞中,S.flexneri 可以通过超氧化物歧化酶和过氧化氢酶的表达来抑制活性氧介导的杀伤。因此,肠道病原体已经进化出多种机制来逃避粘膜防火墙。也许与IBD更相关的是,在缺乏粘膜防火墙的一个或多个组件的易感宿主中,致病菌就有可能引发疾病。
在上一小节,主要解释了坚固的粘膜防火墙,它是如何来保护宿主,在这一小节我们来看,在IBD患者中这个粘膜防火墙是如何被摧毁的。
减少微生物与上皮细胞表面接触的稳态过程的破坏可能会增加IBD发病的易感性,因为观察到多个IBD易感基因编码蛋白质,它们可以限制细菌共生体进入粘膜或促进细菌杀灭。
图2 炎症性肠病粘膜防火墙的破裂
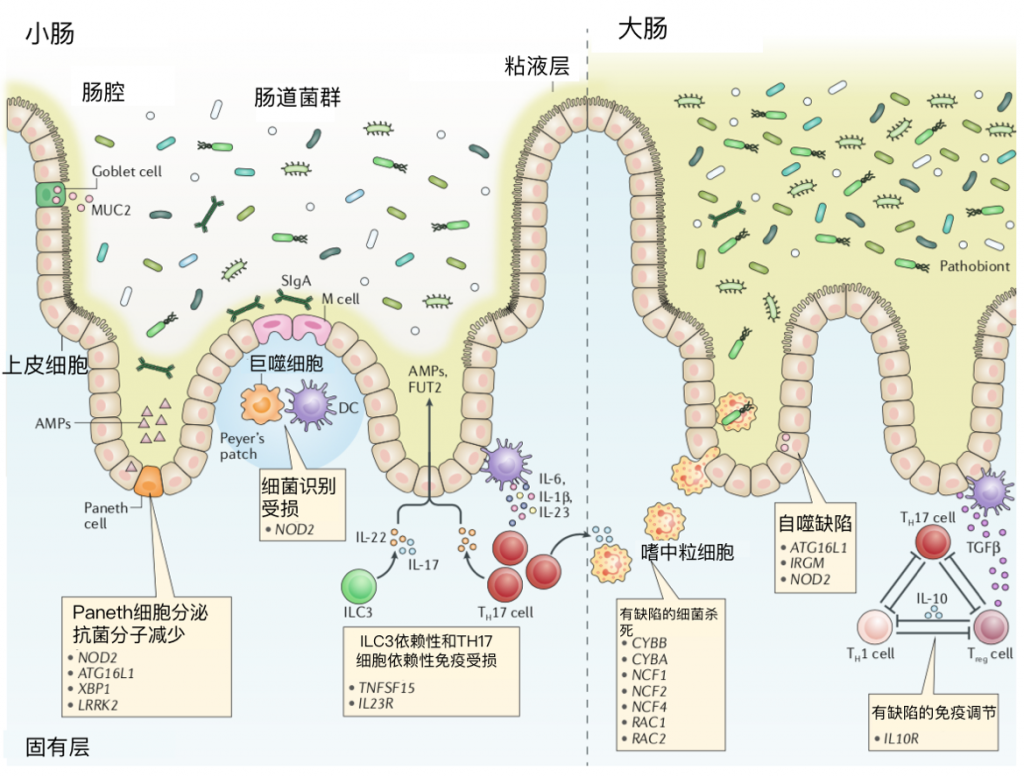

炎症性肠病易感基因的突变会损害宿主用来防止有害微生物进入固有肠板的粘膜策略。
在小肠中,核苷酸结合寡聚结构域2(NOD2)和自噬相关基因(ATG16L1, IRGM, XBP1 and LRRK2)的变异导致Paneth细胞衍生抗菌肽(AMPS)的分泌减少。
肿瘤坏死因子15和IL23R基因多态性可影响第3组固有淋巴样细胞(ILC3s)和T辅助17(Th17)细胞,通过产生IL-22和IL-17从粘膜分离细菌共生体。
克罗恩病相关的吞噬细胞(如巨噬细胞和树突状细胞(DCs)中的NOD2功能丧失突变损害细菌识别。
肠上皮细胞自噬相关基因(ATG16L1, IRGM and NOD2)的突变可能导致细菌清除缺陷。
在大肠中,吞噬体烟酰胺腺嘌呤磷酸二核苷酸氧化酶复合物(CYBB, CYBA, NCF1, NCF2, NCF4, RAC1, RAC2)组分或调节因子的遗传变异损害了活性氧的氧化爆发和产生,导致吞噬细胞中细菌的杀伤缺陷。
功能丧失突变在IL10R改变肠道免疫稳态,导致失调的T细胞反应。
FUT2,岩藻糖基转移酶2;M,微凝胶;MUC2,粘蛋白2;SIgA,分泌性IgA;TGFβ,转化生长因子β;Treg细胞,调节性T细胞。
在小鼠中,MUC2(图2左上角)的缺乏会导致粘液层异常,从而促进微生物群与肠上皮表面的紧密接近并导致自发性结肠炎。
在溃疡性结肠炎患者中观察到的共同特征是隐窝深处细菌与上皮紧密接触。
岩藻糖基转移酶2(FUT2)中的功能缺失变异,编码一种促进粘膜屏障功能的蛋白质,与克罗恩病易感性增加有关。
核苷酸结合寡聚结构域2(NOD2)的遗传变异是第一个与克罗恩病相关的基因变异,也是已知的疾病发展的最强遗传危险因素。但光靠NOD2是不会感染的,它需要辅助条件。
NOD2是一种细胞内受体,能感受肽聚糖衍生的壁酰二肽并诱导对细菌的免疫反应。NOD2在Paneth细胞中表达,可能调节其抗菌功能。然而,Paneth细胞消融或基质金属蛋白酶7(MMP7)缺乏,可将不活跃的前α-防御素转化为杀菌形式,不会导致小鼠自发性炎症。
这表明IBD的发病机制需要额外的遗传缺陷或存在特定的致病菌,而这些致病菌在绝大多数在特定无病原体(SPF)条件下饲养的小鼠微生物群中均未发现。
克罗恩病相关蛋白自噬相关蛋白ATG16L1中的突变也可能通过损害Paneth细胞内分泌颗粒的胞吐作用而导致回肠疾病,这种活动限制了细菌共生菌的渗透。
未折叠蛋白反应(UPR)转录因子X-box结合蛋白1(XBP1)的基因变异与克罗恩病风险增加有关。XBP1特异性上皮缺失导致Paneth细胞内质网应激和结构缺陷。
值得注意的是,IECs中UPR和自噬通路的损伤与自发性克罗恩病(如跨壁回肠炎)有关。
两个克罗恩病易感基因,TNFSF15和IL23R,调节ILC3s和TH17细胞,它们在通过产生IL-17和IL-22抑制共生微生物方面起着至关重要的作用。
然而,还需要进一步的研究来了解这些基因的变异是如何与克罗恩病联系在一起的。
利用固有层中的特殊大噬菌体杀灭细菌共生体,在限制黏膜损伤性炎症和防止渗透性微生物传播方面具有关键作用。
在肠道中,NOD2由吞噬细胞、上皮细胞、基质细胞和Paneth细胞表达。
值得注意的是,NOD2缺乏与CYBB(也被称为NOX2)的缺乏——吞噬体烟酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶复合物的一个组成部分,其通过氧化猝发杀死细菌-在小鼠中触发M. schaedleri的增殖和自发性克罗恩病样TH1细胞驱动性结肠炎。
由于克罗恩病相关的NOD2变异体是功能丧失突变,因此吞噬细胞或肠和/或基质细胞内产生的细菌感应减弱可能促进病理生物的管腔累积和粘膜渗透,导致T细胞介导的肠道炎症。
然而,NOD2相关的克罗恩病是否是由人类体内特定病理生物的积累引起的,还需要进一步的研究。
NOD2在自噬中也有作用,自噬是一种介导溶酶体降解和细胞内细菌清除的途径。
NOD2将克罗恩病相关蛋白ATG16L1募集到细菌进入部位的质膜上,但克罗恩病相关的NOD2变异体有缺陷的ATG16L1募集和受损的细菌诱导的上皮内自噬。
除ATG16L1外,免疫相关GTPase家族M(IRGM)和富含亮氨酸重复激酶2(LRRK2)也调节自噬途径,这些基因的变化与克罗恩病风险有关。
由于自噬的缺陷损害了细胞内细菌的清除能力,可以想象,自噬相关基因的突变可能导致病理生物在肠道中的渗透和炎症。
在极早发性IBD患者中,细菌感染与IBD易感性之间的联系是一个明显的例子,它与吞噬细胞NADPH氧化酶复合物的组分和调节基因的遗传变异有关。
同样,高达40%的慢性肉芽肿性疾病,由NADPH氧化酶组分的功能突变导致的原发性免疫缺陷病,发展为克罗恩病样结肠炎。
总的来说,这些观察表明,杀死细菌共生体的缺陷促进了克罗恩病的发展。
共生细菌通过增强粘膜防火墙来促进保护性免疫,这些防火墙限制了共生体的渗透,同时控制了对微生物的异常T细胞反应。破坏这些相互作用的宿主-共生相互作用可能会促进对IBD患者的失调菌群的不适当免疫反应。
肠道微生物群在调节宿主免疫以建立和维持肠道稳态方面起着至关重要的作用。
图3 共生体的有益作用。
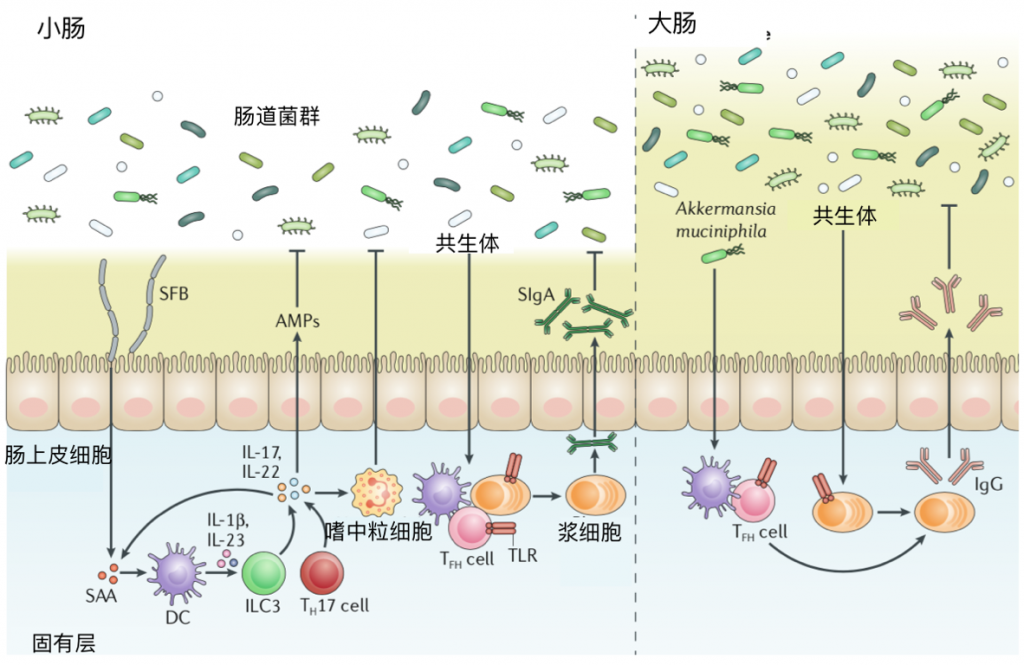

分段丝状细菌(SFB)与肠上皮的粘附会触发血清淀粉样蛋白A(SAA)的释放,后者作用于固有层的树突状细胞(DCs)上,以刺激包括IL-1β和IL-23在内的细胞因子的分泌,并诱导视黄酸受体相关的孤儿受体-γt (RORγt)+ T辅助物17(TH17)细胞分化和第3组先天性淋巴样细胞(ILC3)激活。
由ILC3S分泌的IL-22增强了上皮的SAA产生,以增强Th17细胞介导的粘膜防御,包括抗菌肽(AMP)分泌和中性粒细胞募集。
共生体还通过Toll样受体(TLRs)诱导的B细胞内和T细胞内微生物传感诱导的IgA反应促进肠道稳态。分泌的IgA(SIGA)通过与肠道微生物结合和防止上皮易位而促进屏障功能。
同样,IgG的产生可以通过TLR对B细胞的参与直接发生,而粘液阿克曼菌Akkermansia muciniphila的肠道定植可以通过抗原特异性T滤泡辅助(TFH)细胞反应诱导稳态IgG的产生。此外,IgG有助于穿透上皮屏障的病原体的全身控制(未显示)。
Treg细胞起源于两种不同的个体遗传谱系:
胸腺来源的Treg(tTreg)细胞
外周来源的Treg(pTreg)细胞
大多数pTreg细胞在共生菌的存在下在胸腺外的结肠中发育,因为在无菌小鼠中,结肠中pTreg细胞的频率明显降低。
Treg细胞通过防止诱导不适当地的T细胞对微生物抗原的反应而在维持组织稳态方面起着至关重要的作用(图4a)
动物:
例如小鼠,缺乏对其调节或功能有重要作用的Treg细胞或因子(包括IL-10、转化生长因子β(TGFβ)和αVβ8整合素)的小鼠会发生自发性结肠炎。
人类:
与动物实验数据一致,IL-10受体突变的幼童会发展成结肠克罗恩病。
此外,FOXP3是Treg细胞发育所需的转录因子,其突变的小鼠和人类都有自身免疫性疾病,包括结肠炎。
图4 调节性T细胞支持肠道稳态
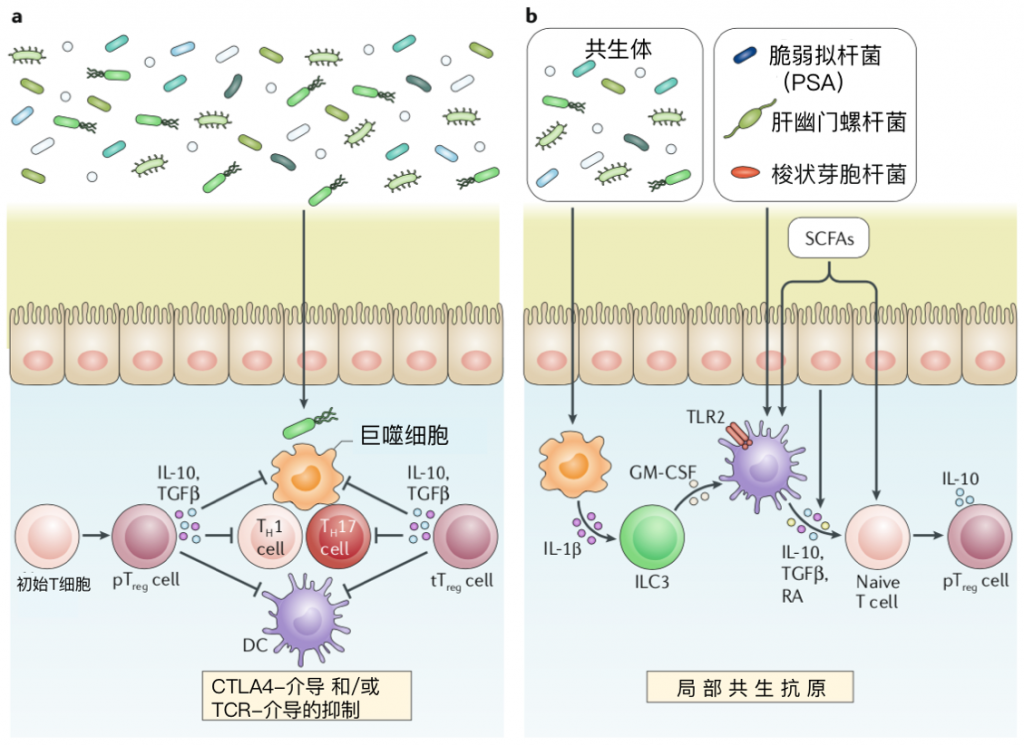

a | 外周来源的调节性T(pTreg)细胞和胸腺来源的Treg(tTreg)细胞抑制肠道异常炎症。Treg细胞产生IL-10和转化生长因子β(TGFβ),以抑制效应T辅助1(TH1)细胞和TH17细胞。另外,髓样细胞如巨噬细胞是IL-10驱动的肠内稳态的重要靶标。
肠道Treg细胞还通过抑制通过细胞毒性T淋巴细胞抗原4(CTLA4)介导的或T细胞受体(TCR)介导的抗原呈递细胞(例如树突状细胞(DCs))来抑制效应T细胞反应。自反应性tTreg细胞也可通过与微生物抗原的TCR交叉反应来促进抗原特异性免疫抑制。
b |共生体通过抗原依赖性和非抗原依赖性过程在pTreg细胞的外周培养中起着至关重要的作用。
细菌的巨噬细胞感应导致IL1β刺激的第3组先天淋巴样细胞(ILC3s)分泌粒细胞-巨噬细胞集落刺激因子(GMCSF),进而增强视黄酸(RA)和IL10的DCs表达,以支持pTreg细胞分化和在结肠中扩散。
此外,除了RA和IL10外,共生体还可以通过上皮细胞和DC促进TGFβ的表达。脆弱拟杆菌可通过与Treg细胞和DC表达的Toll样受体2(TLR2)结合的多糖A(PSA)增强Treg细胞的活性。
选择细菌种类可以直接诱导pTreg细胞分化;这些pTreg细胞对梭状芽胞杆菌和螺杆菌表达的抗原具有TCR特异性。这表明共生抗原在支持Treg细胞分化中起作用。
短链脂肪酸(SCFAs)是包括梭状芽孢杆菌在内的共生菌产生的发酵副产物,可通过表观遗传修饰或通过增强RA产生的DCs直接促进pTreg细胞群的扩张。
Treg细胞在抑制共生菌免疫反应中的作用也得到了证实,通过Treg细胞的共同转移,CD45RBhiCD4+T细胞转移到淋巴细胞减少小鼠中而引起的微生物依赖性结肠炎被清除。
pTreg细胞缺乏动物粘膜屏障的免疫病理学表明,tTreg细胞足以系统地维持对自身抗原的耐受性,而pTreg细胞在抑制肠道炎症方面具有非冗余的作用。
结肠pTreg细胞深受局部抗原的影响,TCR repertoire 不同于外周淋巴结和脾脏的类似Treg细胞,能够识别梭状芽孢杆菌和拟杆菌属表达的抗原,这进一步支持细菌对肠Treg细胞诱导的重要性(图4b)
大量的证据表明,微生物群在引发IBD方面起着至关重要的作用。此外,与IBD相关的遗传缺陷与环境因素一起,可以诱导病理离子的积累和渗透到肠道,从而进一步促进肠道菌群失调和炎症反应。
尽管没有一种病原体或致病菌被一致认为是IBD的病因,但多方面的证据支持微生物群在驱动肠道炎症中的重要作用。例如,粪便流改道可以减少或消除了回肠克罗恩氏病的炎症。
此外,在回肠切除术后,克罗恩病的复发取决于暴露于肠腔内容物。抗生素治疗可使活动期溃疡性结肠炎和克罗恩病患者病情缓解,并可预防某些克罗恩病患者复发。然而,考虑到这些药物对多种菌群的影响,抗生素研究的解释是困难的。
动物模型进一步支持肠道微生物群在IBD发病机制中的作用。例如,将结肠炎小鼠的粪便微生物群口服到健康动物体内就足以引发疾病。最重要的是,基因易感的小鼠在常规的微生物群的情况患上了结肠炎,但不是在无菌条件下。例如,SPF小鼠,而不是无菌小鼠,TCR基因缺失突变会导致结肠炎。
此外,在无菌的TCR缺陷小鼠中没有观察到结肠炎,这些小鼠被固定在一个确定的共栖群落中,这表明引发疾病需要特定的微生物。
对IBD患者体内微生物群的组成进行了广泛的研究。16SrRNA测序分析粪便和粘膜样本显示存在菌群失调,其特征是菌群多样性下降,厚壁菌门的某些属的改变和肠杆菌科物种的丰度增加。
这些改变在克罗恩病患者中比溃疡性结肠炎患者更为明显。一些研究还显示了拟杆菌属的变化,特别是在克罗恩病患者中。
研究发现,IBD患者与近亲(包括双胞胎)之间的微生物群也存在差异,这表明菌群失调与疾病有关,而不是与遗传因素有关。
然而,需要纵向研究来确定是否失调先于炎症的发生。与未治疗的克罗恩病患者在诊断时收集的粪便样本相比,粘膜样本中微生物群的失调更为明显。与溃疡性结肠炎相比,对粘膜样本的研究还显示克罗恩病中潜在有益菌的净损失更大,某些菌群包括克罗恩病中的肠杆菌科和溃疡性结肠炎中的瘤胃球菌属与疾病活动性的相关,以及克罗恩病中某些菌群与肿瘤坏死因子治疗反应的相关。
粪便样本的宏基因组测序显示,克罗恩病患者和非IBD患者之间存在明显的区分,而溃疡性结肠炎患者的差异更显著,总体上有菌群多样性丧失的趋势。
这些研究还发现IBD患者粪便中代谢物多样性的丧失与菌群多样性的丧失相当。尽管到目前为止,元基因组学的研究仅限于粪便样本的分析,但由于宿主来源的DNA在粘膜活检样本中含量很高,便于更深入理解IBD中微生物群的功能破坏。
虽然IBD中的大多数微生物组学研究都集中在细菌对疾病发病的贡献上,但也有一些研究强调了其他微生物在IBD中的重要性。例如,与健康人相比,克罗恩病患者结肠活检样本中真菌菌群多样性增加。同样,克罗恩病患者回肠粘膜标本和粪便标本中的真菌多样性增加,白念珠菌、棒状曲霉菌和新生隐球菌增加。
其他证据证实了IBD中真菌失调的存在,包括与健康受试者相比,酿酒酵母菌比例降低,白色念珠菌增加。值得注意的是,一些研究将真菌菌群多样性的增加与疾病的严重程度联系起来,并认为克罗恩病的环境有利于真菌而损害了细菌,或者抗生素治疗为真菌扩张创造了特定的生态位。
肠道真菌与宿主免疫受体dectin 1相互作用,该信号通过CARD9诱导炎症分子的产生和TH17细胞的反应。CARD9变异与发生IBD的风险增加有关。同样,Card9基因敲除小鼠改变了肠道真菌群落,增加了化学诱导结肠炎的易感性。此外,dectin 1基因的多态性与医学上难治的溃疡性结肠炎有关。
肠道中也含有大量的病毒,这些病毒可能在IBD的发病机制中起作用。例如,感染小鼠诺如病毒会导致潘氏细胞异常,出现克罗恩病易感基因ATG16L1的突变。
此外,FUT2的遗传变异与诺如病毒感染和克罗恩氏病的易感性有关。 可能与IBD相关的其他病毒是噬菌体。
IBD患者肠道病毒组的宏基因组测序显示,与对照组相比,Caudovirales 噬菌体有表达,这似乎不是继发于细菌菌群的变化;然而,这种扩增是队列特异性的,未在验证队列中得到证实。因此,需要更多的研究来了解真菌和病毒在人类IBD中的作用。
虽然在IBD早期微生物群的改变可以独立于治疗而发生,但没有直接证据表明生态失调在IBD发病机制中的因果作用。
在小鼠中,化学诱导的结肠炎和肠道感染会引发微生物群组成的强烈变化,其中一些变化与在IBD患者中观察到的变化相当,包括变形菌的增殖。
炎症导致肠腔氧合增加,硝酸盐和宿主电子受体的有效性增加,可驱动肠杆菌科的厌氧呼吸和增殖。
因此,在IBD患者的肠粘膜和肠腔中观察到的许多微生物群变化可能是继发于炎症。
在一个自发性克罗恩病的遗传模型中,纵向分析揭示了在结肠炎发病前单一或有限数量的致病菌的种群扩张,这种菌群足以在具有复杂微生物群的小鼠中引发疾病。
根据这些观察,失调可能分为早期和晚期两个阶段进行。
在早期,IBD相关的遗传和环境因素可能导致致病菌的积累,这可能先于临床疾病的发展。
尽管参与早期失调的致病菌的身份和数量尚不清楚,但在IBD动物模型中有限的证据表明,该细菌的遗传和代谢特征可能是重要的。
例如,schaedleri和Helicobacter hepaticus,这两种致病菌可以在遗传易感性小鼠中引发自发性结肠炎,产生毒力因子并生活在上皮附近,尽管这些活性在诱发结肠炎中的作用尚不清楚。当黏膜防火墙受到IBD易感基因突变的影响时,这种细菌特征可能降低局部穿透的阈值(图5)
图5 炎症性肠病中的生态失调
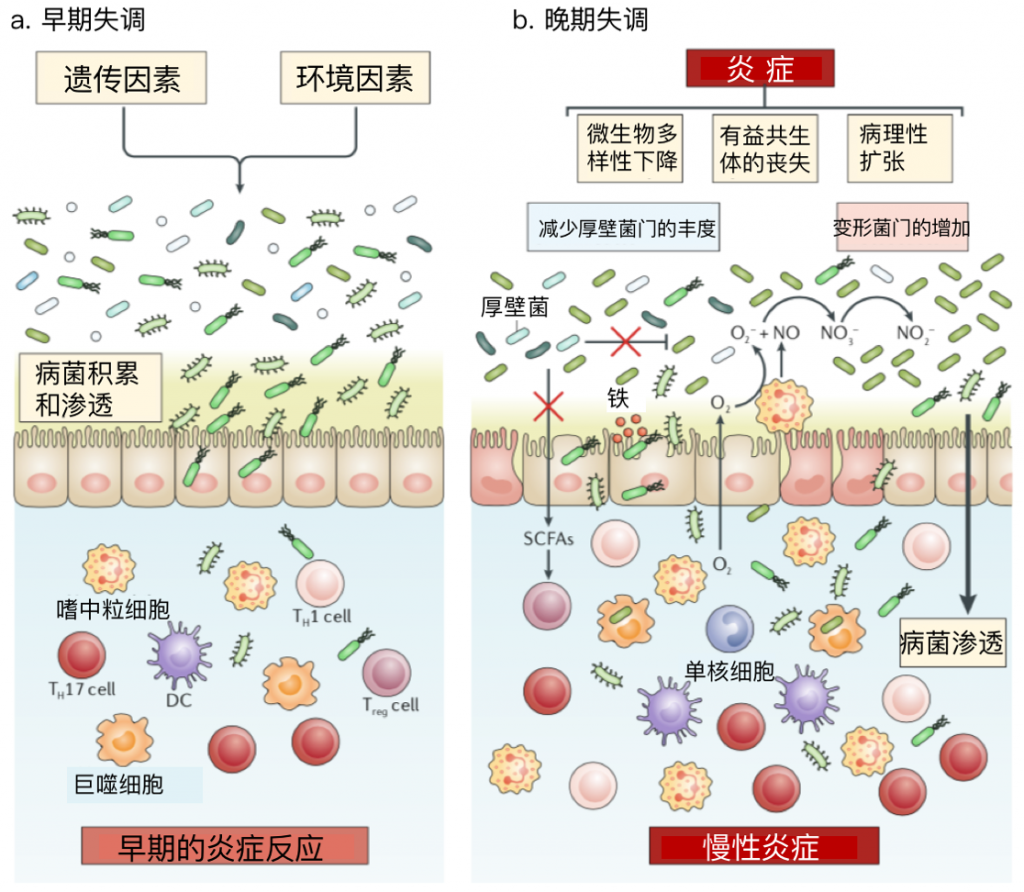

与炎症性肠病相关的遗传缺陷,再加上饮食和抗生素使用等环境因素,可能导致引起疾病的病原体积累和渗透到肠道固有层(早期失调)中,这可能先于临床显性疾病的发展。
炎症可导致细菌类群的更大变化,包括变形菌(晚期失调)的扩张,其途径是增强肠腔氧合,增加炎症肠道环境中的硝酸盐(NO3-)、宿主来源的氧受体和铁的可用性。
这种后期的失调还表现为微生物多样性全面下降,有益的共生体丧失,这可能导致粘膜粘附增加,共生微生物移位,从而引发慢性炎症。
DC,树突状细胞;TH细胞,T辅助细胞;Treg细胞,调节性T细胞;SCFAs,短链脂肪酸。
在晚期失调期间,肠道炎症推动了菌群的进一步变化,包括变形菌的增殖(图5)。鉴于肠道微生物群的不同菌群对宿主免疫系统和肠道屏障有有益的影响,某些菌群的缺失可能导致肠道炎症的加剧或消退。特定细菌的大量繁殖,如粘附性和侵袭性大肠杆菌,积聚在IBD患者的炎症粘膜中,可进一步促进炎症反应(图5)。
粘附性和侵入性大肠杆菌使用常见的1型菌毛粘附素FimH粘附到肠上皮,该粘附素可识别在克罗恩病患者回肠中异常表达的癌胚抗原相关细胞粘附分子6(CEACAM6)。 需要对来自IBD易感性增高的人群和/或IBD患者的黏膜样本进行精心设计的纵向研究,以了解其在疾病复发中的作用。
IBD患者常检测到T细胞和抗体对微量双抗原的反应增强,进一步证明了微生物群在疾病发病机制中的作用。与健康人相比,IBD患者产生大量抗共生细菌的IgG抗体,并且血清中抗真菌和/或细菌肽和聚糖的活性较高。尽管抗微生物抗体可以作为疾病和诊断的标志物,但这种抗体在IBD中的作用尚不清楚。鉴于健康的小鼠和人类产生了抗细菌共生体的抗体,IBD相关抗体高滴度的存在可能反映了IBD患者的适应性免疫反应增强或肠道内微生物抗原暴露增加。
除了体液反应外,在IBD患者中还观察到T细胞对微生物群的反应失调。早期研究表明,IBD患者炎症粘膜中的T细胞或单核细胞在肠道微生物抗原刺激后,其反应性较正常粘膜中的细胞有所增强。
然而,在IBD患者和健康人的血液和粘膜中都可以检测到CD4+T细胞对细菌共生体的反应。对微生物群有反应的人T细胞主要具有记忆表型,具有不同的TCR Vβ基因库,这与对多种微生物抗原的反应一致。
与人类数据一致的是,在结肠炎动物模型中也可以检测到对共生细菌和细菌抗原有反应的T细胞。CD4+T细胞对共有抗原的反应转移到淋巴细胞减少的动物体内足以引发结肠炎。
一些实验观察结果表明,致病性CD4+TH细胞可以在稳态条件下识别致病生物,而Treg细胞介导的免疫抑制对结肠炎的发展至关重要。然而,Treg细胞作用于效应T细胞抑制细菌引起的肠道炎症的机制尚不清楚。Treg细胞可以以抗原无关的方式抑制效应T细胞和/或通过其TCR作用于抗原提呈细胞以抑制抗原特异性效应T细胞。
目前大多数IBD的治疗方法,包括类固醇和生物药物,如抗肿瘤坏死因子或抗整合素治疗,都能抑制宿主免疫系统,但不直接针对引起或导致炎症的微生物。
鉴于炎症性肠病的治疗在不到50%的患者中可获得完全缓解,针对肠道微生物的治疗方法的发展可能为治疗炎症性肠病提供一种独特的方法。
图6 微生物治疗在炎症性肠病中的作用
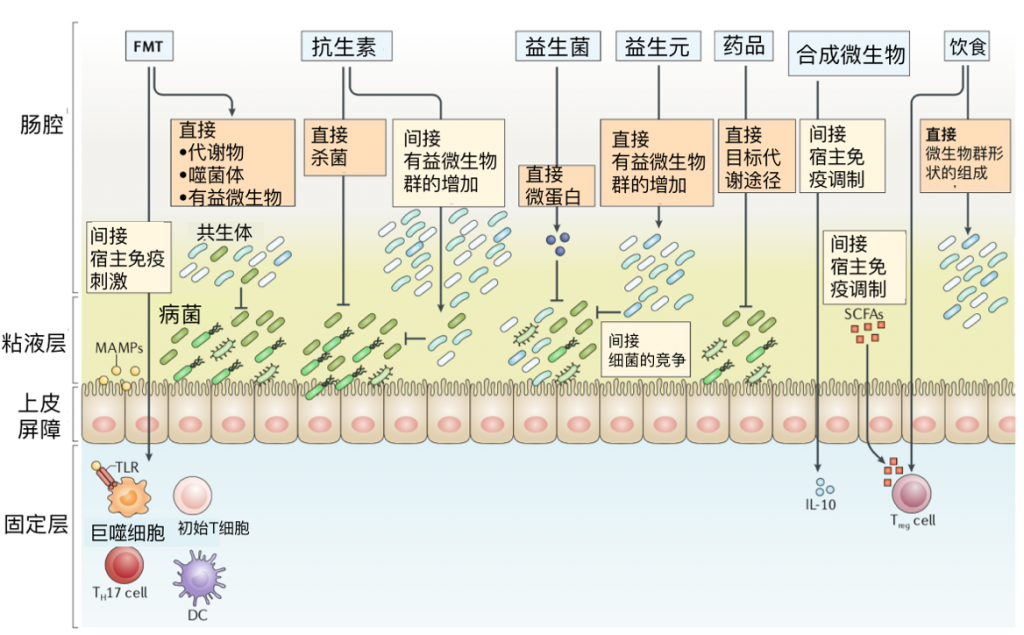

鉴于肠道微生物群在炎症性肠病发病机制中的关键作用,针对肠道微生物的治疗方法的发展可能为治疗该疾病提供一种独特的方法。
粪便菌群移植(FMT)
直接有益效果:通过转移细菌衍生的代谢产物和噬菌体或通过恢复有益微生物而产生直接的有益效果。
间接有益效果:通过细菌成分间接产生有益的效果。例如,Toll样受体(TLR)由微生物相关分子模式(MAMPs)触发宿主固有免疫刺激。
抗生素
抗生素可能通过直接杀死致病菌或间接促进有益微生物的种群扩张而产生广泛的作用。
益生菌
使用益生菌制剂,如大肠杆菌Nissle 1917,可导致微球蛋白的分泌,微球蛋白是具有抗菌活性的小多肽,可直接抑制与肠杆菌科密切相关的细菌。
益生元
补充益生元可以促进有益共生菌的种群扩张,从而战胜有害细菌。
其他
合成工程微生物的给药可以通过调节免疫系统(例如通过分泌IL-10)对宿主产生有益作用。
饮食通过促进有益微生物的生长或减少致病微生物的消耗来塑造肠道微生物群的组成。
来自膳食多糖的短链脂肪酸(SCFA)也可以支持肠道调节性T(Treg)细胞群的分化和扩展。
随着对某些微生物的失调和免疫调节功能的进一步了解,研究人员为改善肠道微生物群以预防或改善IBD提出了一些创新的策略。
IBD和反复难辨梭状芽孢杆菌感染的生物学特性:
门级多样性的丧失和肠杆菌科兼性厌氧菌(包括变形菌)的过度生长。因此研究人员想到了纠正微生物群落失衡可能会成为治疗IBD的有效方法。
通过恢复肠道微生物群落多样性,粪便微生物群移植(FMT)在解决复发性艰难梭菌感染方面的高成功率增强了微生物群调节疗法的潜力。尽管已经接受FMT治疗的IBD患者数量有限并且对治疗的反应有所不同,但大约30%的溃疡性结肠炎患者已经实现了临床缓解。抗生素预处理和多次粪便输注似乎可以改善FMT在溃疡性结肠炎患者中的有效性。
也有研究人员对此持不同看法。
Rossen等人的一项双盲,随机,安慰剂对照研究未观察到FMT对溃疡性结肠炎患者有任何有益作用,其结果发表在Gastroenterology期刊。
此外,FMT在克罗恩病患者中的有效性仍不清楚,可能是因为目前的研究报告潜在疗效在队列规模上是有限的,并且缺乏安慰剂治疗组。需要进一步的研究,了解更多机制,来制定更有效的治疗方案。
随机对照试验的Meta分析显示了抗生素在治疗克罗恩病和溃疡性结肠炎中的益处。
然而,使用抗生素来改变微生物群受到限制,因为在消灭致病菌的同时对有益菌也有影响,这可能带来不良后果。
由于相互矛盾的发现,在IBD治疗中抗生素的使用仍然有限,但使用广谱抗生素混合疗法的研究表明,它们可能重症急性结肠炎和慢性溃疡性结肠炎有效。
在未来,一旦确认了引起IBD的微生物代谢途径,就有可能用新一代抗生素专门针对它们。


活微生物可能在几个方面是有利的。
一旦达到稳定的定殖,有益的微生物因子可以持续地传递给宿主,活菌可能会触发有益的免疫反应。通过服用益生菌,包括乳酸菌或双歧杆菌,有针对性地调节微生物群,已经成功地治疗了一些肠道疾病。
此外,在维持溃疡性结肠炎患者病情缓解方面,益生菌大肠杆菌Nissle 1917口服治疗与美沙拉嗪标准治疗具有相似的疗效。益生菌大肠杆菌Nissle 1917能分泌具有抗菌活性的微胞素,抑制可能加剧肠道炎症的竞争性肠杆菌科细菌。

但是,益生菌通常对微生物组的整体组成影响有限,因为这些细菌无法持久地定居健康的成年人。 在某些适应症中,同时补充益生元可以提高外源微生物的移入效率,包括增强对艰难梭菌的定植性。
合成生物学的进展为IBD的治疗和管理提供了创新的方法(如下图6)
给小鼠口服经工程改造以表达和分泌IL-10的乳酸乳球菌,足以保护它免受DSS和IL-10缺乏引起的结肠炎的侵害。
在一小群克罗恩氏病患者中,使用限制胸腺嘧啶的遏制策略安全地生产了重组的产生IL-10的乳酸乳球菌菌株,但仅引起了很小的疾病活动性改善。
由于乳酸杆菌的惰性,这种益生菌也被修饰以表达胰岛素生长因子1、血红素氧合酶1或丝氨酸蛋白酶抑制剂;当给小鼠口服时,这些合成的益生菌可以缓解实验性结肠炎的发展。然而,这些策略在临床试验中并不成功,可能是由于工程乳酸菌菌株不能持续地将IBD患者定殖。
精确定位有害微生物在失调期间增殖的代谢途径,对于改善小鼠结肠炎是有效的。例如,变形杆菌利用增加的氮源,包括在炎症肠中产生的一氧化氮,大量繁殖。
值得注意的是,靶向钼辅因子依赖的酶,需要使用一氧化氮进行厌氧呼吸,通过减弱肠杆菌科的增殖,包括变形菌,来保护小鼠免受DSS诱导的结肠炎。因此,开发针对有害菌代谢途径的药物可能为治疗IBD提供一种新的策略。
饮食在塑造微生物群的组成中起着重要作用,并可通过调节饮食来控制IBD症状。
全肠内营养(EEN)是IBD中为数不多的已被广泛研究的膳食干预措施之一,它是一种营养完整的无固体食物的元素和聚合物配方食品。在克罗恩病的儿科患者中,甚至与皮质类固醇一样有效,但没有与皮质类固醇治疗相关的副作用。
EEN能独立于其他环境因素迅速改变微生物群的组成,有效降低克罗恩病患儿的肠道炎症。EEN诱导克罗恩病缓解的机制尚不清楚,但它可能促进有益微生物的生长或减少致病生物。
在实验性DSS诱导的结肠炎中,一项对各种精制饮食的调查确定了洋车前子纤维的有益作用。而膳食蛋白(包括酪蛋白)的增加,粪便微生物密度的增加以及肠通透性的改变加剧了结肠炎的严重性。
寄主对结肠炎的易感性很大程度上依赖于形成微生物群的特定纤维或蛋白质成分的组合,可见饮食成分在IBD中非常重要。
在过去的十年里,人们关于微生物群-宿主相互作用以及遗传和免疫系统在IBD中的作用的了解有了大幅提高。然而,对于IBD发病机制的几个方面的认识仍很局限。
一个主要的问题是缺乏对引起易感个体炎症的IBD致病菌的识别。多种致病菌可能引起疾病,这些致病菌可能因患者特定的IBD易感位点而异。因此,利用具有相同或相似遗传缺陷的个体进行研究可能对鉴定这种微生物很重要。
识别引起IBD的微生物对于理解疾病的发病机制,监测患者这些病原体的改变以及合理开发新疗法具有重要意义。
动物模型为研究微生物群和免疫系统在疾病发展中的作用提供了重要的线索。然而,除了少数之外,这些模型并不是基于与人类疾病相关的遗传缺陷,也没有完全概括IBD的病理学。小鼠模型通常使用简化的微生物群,这种简化的微生物群不能反映完整肠道中发生的复杂的微生物-宿主相互作用。
未来,如果能开发出更适合人类IBD的新模型,掌握更多机制,或许能从微生物群的角度(比如说饮食干预)带来更多的治疗甚至是预防措施。
参 考 文 献
Caruso R, Lo B C, Núñez G. Host–microbiota interactions in inflammatory bowel disease[J]. Nature Reviews Immunology, 2020: 1-16.

谷禾健康 原创
拟南芥(Arabidopsis thaliana),又名阿拉伯芥、鼠耳芥、阿拉伯草,是一种原生于欧亚大陆的小型开花植物,为一种模式生物,被广泛地应用于植物遗传学、细胞生物学、分子生物学,以及群体进化学等方面的研究中。据不完全统计,截止到2019年,已经有超过6万篇标题中含有拟南芥的学术论文发表,覆盖了几百个生物学领域,拟南芥中各种生物学过程的研究,都是最细致最深入的。当我们要研究某一课题时,最先想到的问题是“拟南芥中是怎样的”。现在,拟南芥已经成为了植物科学研究的金标准。然而目前,我们对于驱动植物在大陆规模层面的根部微生物变异和适应的因子所知甚少。
近日,一篇发表于《Nature Ecology & Evolution》来自德国科隆马克斯普朗克植物育种研究所关于拟南芥最新研究成果:Root microbiota assembly and adaptive differentiation among European Arabidopsis populations(欧洲拟南芥群落根系微生物群的组装与适应性分化),探索了这一问题。该研究表明土壤和气候条件对根系微生物群落中的细菌和真菌的分化有不同的控制作用,气候是土壤中真菌位点变异和地理分布的关键驱动因素,而土壤细菌的地理分布主要受土壤因素的控制。气候的差异比土壤和本地微生物群的差异更重要。


研究人员在3年中监测了欧洲17个地点的拟南芥和禾本科植物中与根系相关的微生物群落。观察到土壤微生物群落具有强大的地理结构,但没有观察到根部微生物群落的地理结构。一些具有系统多样性并地理分布广泛的细菌始终定植在植物根部。细菌的根系微生物群在不同地点之间和不同年份间的群落组成相似性要强于真菌。在瑞典和意大利的两个拟南芥种群之间的交互移植中,排除地理位置效应对土壤微生物群落的影响,测试了它们对根系微生物群变化和植物适应性的贡献。植物根部的群落分化主要是通过真菌的位置和细菌的土壤来源来解释,而宿主基因型的影响较小。观察到两个拟南芥种群之间存在很强的地方适应能力,土壤特性和微生物的差异对于观察到的适应性分化的影响不大。结果表明,在较大的空间尺度上,气候比土壤条件对真菌根系相关微生物群落的变异和植物适应性更重要,而土壤特性是根系细菌群落分化的主要驱动因素。
人们对引起根系微生物群和植物适应性方向在大陆尺度中变化的因素了解甚少。植物的免疫系统、根际沉积物和微生物相互作用是根系相关微生物群有别于周围土壤微生物群的已知决定因素。土壤生物群组成的大规模空间变化与土壤和气候条件的差异有关。特别是当地土壤特性,如pH值主要预测土壤细菌的地理分布,而气候条件更好的预测土壤中真菌的分布。然而,在土壤和气候条件的变化对植物根系微生物群落组成和适应性分化的影响程度方面缺乏系统的实地研究。交互移植已在许多植物物种中表现出局部适应,包括模式植物拟南芥。然而,不同的非生物和生物因素对当地适应的进化和维持的相对重要性知之甚少。在瑞典和意大利,研究了土壤条件和气候对两种海百合种群根系微生物群聚集和适应性分化的影响。
1.样本采集
在欧洲选择了17个具有不同土壤特性的地点,在2月-5月间统一发育阶段连续3年采集自然生长的拟南芥,在不影响植物根系的情况下,用手铲将植物与其周围的块状土壤一起收获,转移到7厘米×7厘米的温室花盆中 。并在距离拟南芥生长50cm的范围内采集了三种邻近的禾本科植物,共采集285株植物。而后人工将单株植物与土体分离,分离出4个微生物生态位【土壤(Soil);根际(RS);根际平面(RP);根(Root)】。whole-root是RP和Root的组合。
2.交互移植实验
2016年春季,在两个试验点收集了土壤,并将其储存在6摄氏度下。种子接种于琼脂培养皿中,在6摄氏度黑暗中冷沉积一周后移入生长室(22°C 16h,150μEm−2s−1 PAR,16°C黑暗8h),萌芽9天后,将20株本地苗和22株外地苗移植到每个土壤类型的6个区块的随机位置上,每个点×土壤组合共移植120株本地苗和132株外地苗。移栽过程中,插盘在温室内分别在~18℃和~12°C下白天放置16h,夜间放置8h。在6天内,插盘被运到意大利(IT4)和瑞典(SW4)。移栽苗与源群体中自然萌发的植株处于同一发育阶段。
3.将上述实验分离出的样本提取DNA后对细菌16SrRNA(799F-1192R)V5-V7、细菌16SrRNA(341F-806R)V3-V4、真菌ITS1(ITS1F-ITS2)、真菌ITS2(fITS7-ITS4)和卵菌ITS1(ITS1-O-5.8 s-Rev-O)区域扩增后使用MiSeq测序仪和定制测序引物在内部进行成对末端Illumina测序。得到序列后进行微生物群落组成及差异分析。
1. 17个欧洲拟南芥种群中的微生物群落结构。表明根环境促使欧洲各地的细菌群落组成趋同。
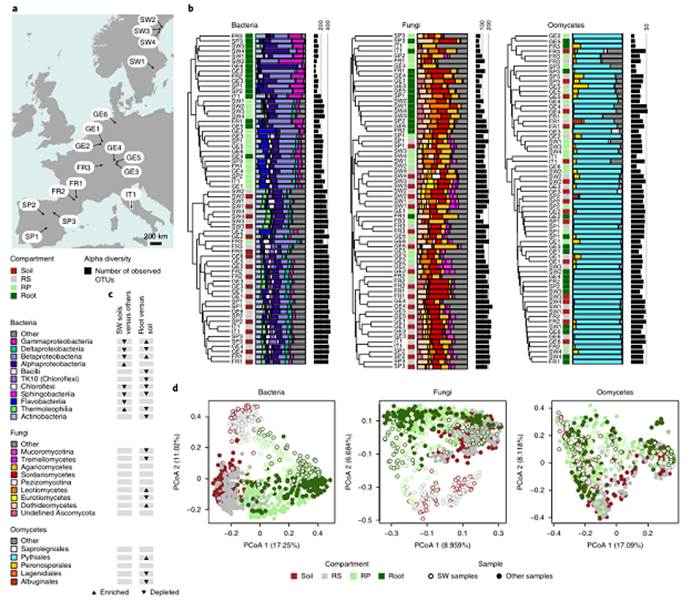

a). 欧洲地图,显示了17种拟南芥种群的名称和位置
b). 基于Bray-Curtis相似度的树状图显示了3年中在每个站点中每个采样部位的微生物群落组成,从左至右分别为细菌、真菌、卵菌。采样部位由侧边彩色方块表示(深红色:土壤;灰色:根际(RS);浅绿色:根际平面(RP);深绿色:根)。彩色条形图表示群落组成(纲或目分类水平)。黑色条形图表示微生物的α多样性。从土壤到根内圈,细菌、真菌和卵菌的α多样性逐渐下降,其中细菌的下降幅度大于丝状真核生物。Bray-Curtis距离聚类显示RS与Soil间的细菌群落组成有较高相似性,RP与Root间的细菌群落组成有较高相似性。
c). 4个瑞典土壤样品(SW soils)与其他周围13个土壤样品(versus others),以及根(Root versus)和土壤样品(soil)之间的差异丰度分析(纲或目分类水平)。三角表示差异有统计学意义(Wilcoxon秩和检验,FDR < 0.05)。与周围土壤样品相比,植物根部微生物群落的相对丰度显著增加。
d). 基于从17个地点、4个采样部位和连续三年(2015年、2016年和2017年)采集的样本之间的Bray-Curtis距离的PCoA图。列出了整个拟南芥数据集的微生物群落:细菌(n = 881)、真菌(n = 893)和卵菌(n = 875),并根据不同采样部位进行了颜色编码。空心圆圈表示瑞典样本,实心圆圈表示所有其他样本。揭示了瑞典土壤和其他欧洲土壤之间土壤细菌群落的显著差异。这些差异在相应的根系样本中大大减小,表明植物内圈细菌群落组成趋同。
2.大陆尺度中,少数地理上广泛分布的细菌丰富地定殖在拟南芥的根部,推动了细菌群落组成的趋同。
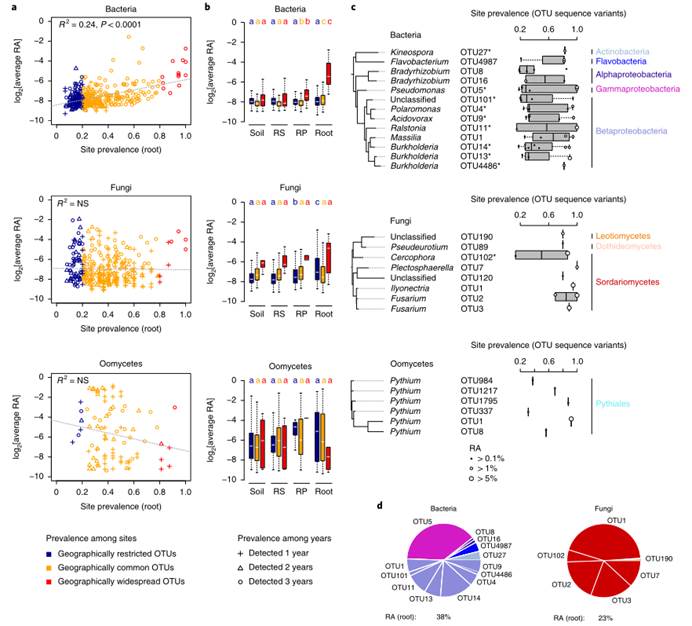

a).细菌、真菌和卵菌在不同地点(3年的平均值)普遍流行的OTU与植物根部的OTU相对丰度(3年的平均值log2后的值)之间的相关性。蓝色代表地理上受限的OTUs,流行率<20%;橙色代表地理上常见的OTUs,流行率20-80%;红色代表地理上分布广泛的OTUs,>80%。不同的形状提示在第1、2或3年间检测到的与Root相关的OTU。NS表示无统计学意义。观察到在细菌群落中两者间呈正相关。相反,在真菌和卵菌群落中两者没有相关性。
b).a图数据的盒形图展示。描述了不同采样部位之间的显著差异(Kruskal–Wallis with Dunn’s的事后检验, P < 0.01)。地理上分布广泛的细菌OTUs在RP和Root样本中比相应的土壤样本中丰富得多。
c). 根据16S rRNA V5-V7基因片段(细菌)和ITS1区域(真菌和卵菌)构建的地理上分布广泛的Root相关的OTUs的细菌、真菌和卵菌的系统发育树。星号表示,与土壤样本相比,显著富集的与Root相关的OTU。对列出的OTUs中的进行序列变异的检查,发现细菌OTUs中有大量的序列异质性。其中OTU14和OTU5的序列异质性最高。相比之下,真菌和卵菌的OTUs几乎没有序列变异。这意味着一个保守的分类单元的小子集已经进化出在大陆尺度上支配根系细菌菌群的机制,而不考虑周围土壤细菌菌群的主要差异。
d).在拟南芥根部检测到的分布广泛的细菌和真菌的OTUs的比例。展示了相对丰度大于0.1%的OTUs。这些OTUs在root样本中的总体相对丰度显示在每个饼图下面。
3. 少量地理上分布广泛的细菌可以有效地在远亲植物的根系微生物群落中定殖,并随着进化时间的推移与植物根系微生物群落建立稳定的联系。拟南芥和禾本植物之间的群落组成差异较小。
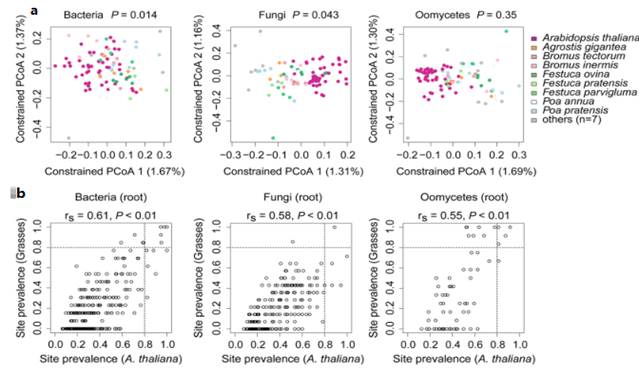


a).基于Bray-Curtis距离的禾本植物和拟南芥的细菌、真菌、卵菌群落的种分类水平的PCoA图。观察到拟南芥和共生牧草在微生物群落组成上的差异较小。
b).细菌、真菌和卵菌的拟南芥根系普遍流行的OTUs和禾本植物根系普遍流行的OTUs之间的斯皮尔曼相关性。在大陆尺度上总体是一致的。
c).共生牧草根系中广泛存在的拟南芥根系OTUs在细菌群落中占总相对丰度45%,但在真菌群落中只占总相对丰度16%。而生长在不同土壤类型中的莲花根系微生物群落中也检测到这些OTUs,所占比例RA表示。
4.大陆尺度中影响拟南芥根系微生物群落的因素。不同地点间环境条件的变化比时间变化对根系相关微生物群落的影响更大
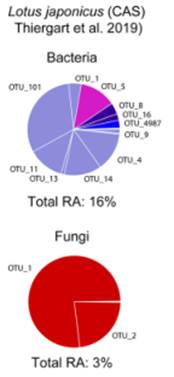


a). 在17个地点、四个采样部位和连续三年(2015年、2016年和2017年)采集的样本之间基于Bray-Curtis距离的PCoA图。从数据集中排除相对丰度 < 0.1%的OTUs。不同地点是影响拟南芥根系微生物群落组成变化的主要因素(Adonis: d.f. = 16; 细菌: R2 = 0.17; P < 0.001; 真菌: R2 = 0.19; P < 0.001; 卵菌: R2 = 0.17; P < 0.001),,但是与土壤样本中观察到的结果相比,这一效应明显降低。(Adonis: d.f. = 16; 细菌: R2 = 0.53; P < 0.001; 真菌: R2 = 0.51; P < 0.001; 卵菌: R2 = 0.27; P < 0.001)
b).采样地点、采样年份和采样地点x采样年份对细菌、真菌、卵菌群落组成的影响。其中,真菌和卵菌群落的年际变化尤为明显。
c). 在每个位置测量的环境变量或共线变量组对细菌、真菌和卵菌群落组成的影响。pH组包括pH、铁、纬度、经度、平均气温和平均相对湿度。硼组包括硼、有效钙和储备钙三个因子。P组包括有效P因子和储备P因子,NO3组包括有效NO3因子、有效Mg因子和储备Mg因子。K组包括有效K因子和储备K因子,显著统计意义用深色突出表现。两种土壤在有效钙、储备钾、有效镁、pH、铁和速效钾方面的差异是最大的。(c1图为描述了18个环境变量之间共线性的热图。定义了两个唯一变量(锰(Manganese)、铜(Copper))和五组共线变量(K组、P组、Boron组、NO3组、pH组)。土壤、RS和RP细菌群落组成的主要驱动因素是pH和/或与pH密切相关的环境变量,如铁、平均气温和平均相对湿度。细菌群落与环境变量的相关性比真菌更强。
5. 土壤、气候和基因型的差异驱动了拟南芥种群间根系微生物群落的分化;与细菌相比,气候条件等特定位置因素对根相关真菌群落分化的影响更大。土壤条件的差异对拟南芥种群间的适应性分化贡献较小
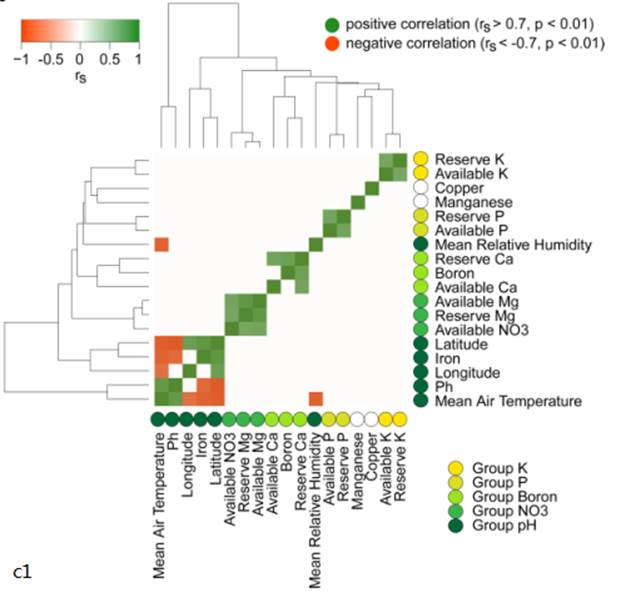

a).交互移植示意图。来自IT1和SW4地点的土壤和植株基因型在这两个地点(8个不同处理组合)相互移植。
b). 根据Bray-Curtis距离用PCoA法测定了细菌、真菌和卵菌的土壤和全部根系微生物群落结构。与细菌相比,土壤来源的真菌和卵菌的解释变异百分比(PCoA1)较弱。土壤中微生物真核组合的位点间变异主要是由地理位置决定的。在whole-root组中,位置的影响与土壤对真菌的影响一样强,比土壤对卵菌的影响更强
c). 在IT1和SW4的土壤中相互种植,并在IT1和SW4的地点生长的两者基因型的整体适合度。红色表示IT1,蓝色表示SW4。圆形表示IT1土壤,三角形表示SW4土壤。填充符号表示在IT1生长,未填充符号表示在SW4生长。在IT1位点,本地基因型的相对适合度高于外地土壤。两种土壤在有效钙、储备钾、有效镁、pH、铁和速效钾方面的差异是最大的。在SW4地点,没有一株意大利植物幸存下来繁殖。较强的本地优势是较高的存活率和繁殖力共同作用的结果。
土壤和气候条件对根系微生物群落中的细菌和真菌的分化有不同的控制作用,气候是土壤中真菌位点变异和地理分布的关键驱动因素,而土壤细菌的地理分布主要受土壤因素的控制。气候的差异比土壤和本地微生物群的差异更重要,因为这两个研究种群之间存在适应性差异。研究人员认为未来的研究应该确定在大空间尺度上影响根系微生物群组装和适应性分化的环境因子是否与在较小地理尺度上作用的环境因子不同。

谷禾健康 原创

微生物群是免疫防御的原动力,但是防止感染的特定共生微生物的特性尚不清楚。相对的,病原体如何与其他微生物群落竞争以建立其宿主生态位也鲜为人知。
鉴定对病原体有保护作用的微生物群成员可以提供一种治疗对当前抗菌治疗有耐药性的感染的替代方法。
在这些抗药性微生物中,肠杆菌科是最大的临床问题。而肠杆菌科中的肺炎克雷伯菌对人类健康构成最紧迫的威胁,因为许多菌株对多种抗生素耐药,毒性很强,在成人和婴儿中都会引起疾病,并且很容易在宿主之间传播。
因此,预防肺炎克雷伯菌的定殖和传播显得至关重要。
近日, 国际顶级微生物学期刊《Nature microbiology》发表了由英国伦敦帝国理工学院传染病系分子细菌学与感染中心和外科与癌症系综合系统医学与消化疾病科合作研究最新成果:“Commensal Bacteroidetes protect against Klebsiella pneumoniae colonization and transmission through IL-36 signalling(共生拟杆菌通过IL-36信号预防肺炎克雷伯菌定植和传播)”。
该研究从机制上提供了一种视角,何时,何地,拟杆菌如何对抗肺炎克雷伯氏菌的定植和传染,深入了解如何利用这些保护性微生物来提供人群级别的预防肺炎克雷伯氏菌感染的保护。
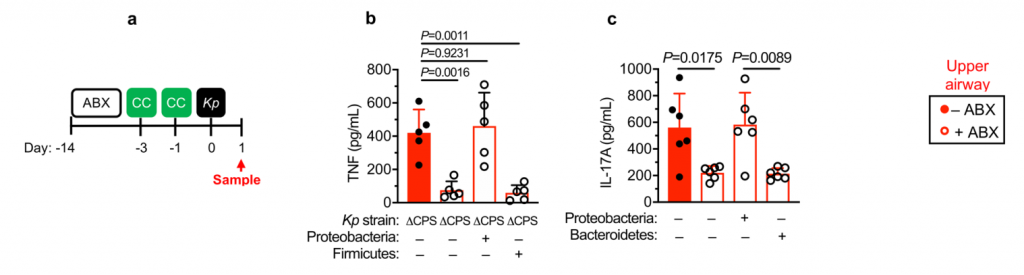
肺炎克雷伯氏菌在人群宿主中有两个主要的定植渠道:上呼吸道和肠道。肺炎克雷伯菌建立定植必须与这两个位置的微生物群和免疫系统建立的防御系统抗衡。
该研究发现,成熟的微生物群推动了不同免疫防御程序的发展,从而在上呼吸道和肠道限制肺炎克雷伯氏菌在这些生态位内的定植。
肠道免疫保护取决于拟杆菌、白细胞介素IL-36信号和巨噬细胞的发育。拟杆菌的这种作用需要其保守的共生定植因子的多糖利用位点。相反,在上呼吸道,变形菌门通过IL-17A增强免疫力,但是肺炎克雷伯氏菌通过包囊IL-17A来克服这些防御进而有效定植。
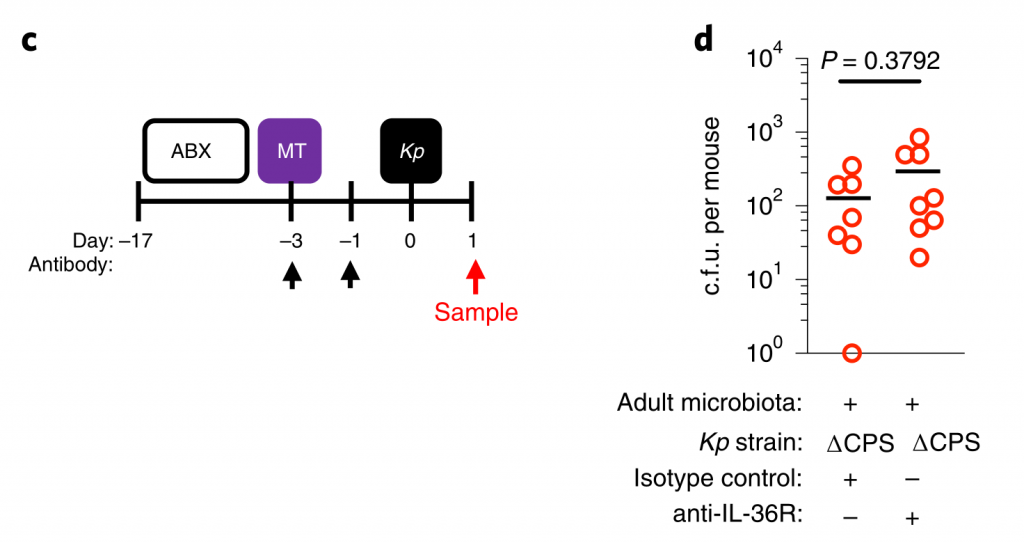
最终发现肺炎克雷伯氏菌的宿主间传播主要发生在其肠道贮存器中,而产生共生定植因子的拟杆菌足以通过IL-36阻止宿主之间的传播。
首先我们先来看看,肺炎克雷伯菌这种菌在人群中的分布情况究竟是怎样的?
我们从谷禾健康2019年近期检测人群肠道菌群样本抽取1.3万例样本,人群共计13358人,涵盖0~103岁人群。
其中肺炎克雷伯氏菌丰度占比超过1%的人群有3765例,占比28.2%。
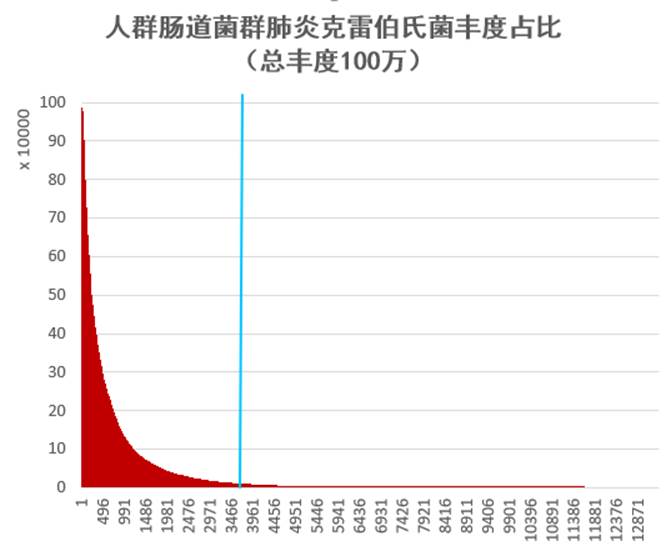
预 告
本文后面【交流探讨】章节会结合该论文的观点,对我们谷禾健康目前已测的人群数据进行整理分析。
由于肺炎克雷伯菌在婴儿和成人中引起疾病,研究人员希望研究在整个生命过程中微生物群介导的对肺炎克雷伯菌定植的防御能力的发展。为此,耗尽了新生和成年小鼠中的微生物群。
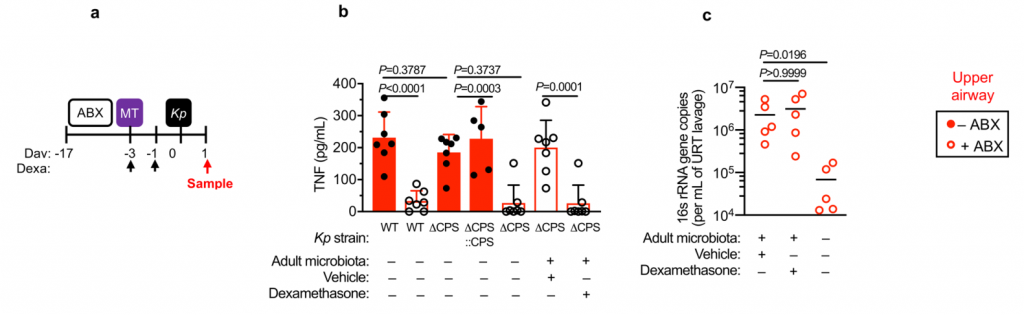
为了研究肺炎克雷伯菌在上呼吸道的定殖作用,研究人员使用了一个小的接种量,并且没有麻醉小鼠进行鼻内接种。这种方法意味着细菌保留在鼻腔和鼻咽,并不会到达肺。
在新生小鼠中,肺炎克雷伯菌可能在上呼吸道和肠道都定居,这不受微生物群耗竭的影响(图1a–c)。同样,在成年上呼吸道中,对照动物和消耗微生物的动物中肺炎克雷伯菌的定殖水平相似(图1d–f)。相比之下,在成年肠道中,肺炎克雷伯菌只有在微生物群枯竭后才能建立可检测的定植(图1g–j)。
持续的广谱抗生素治疗(图1g-j)或临床上相关的短期抗生素治疗方案,消除了微生物群介导的防御。将成年菌群而非新生儿菌群转移至新生小鼠,以保护其免受肺炎克雷伯菌的肠道定殖(图1k–m)。
图1:成年微生物菌群可抵抗抗生素抗性肺炎克雷伯菌在肠中的定植,但不能防止上呼吸道定植。
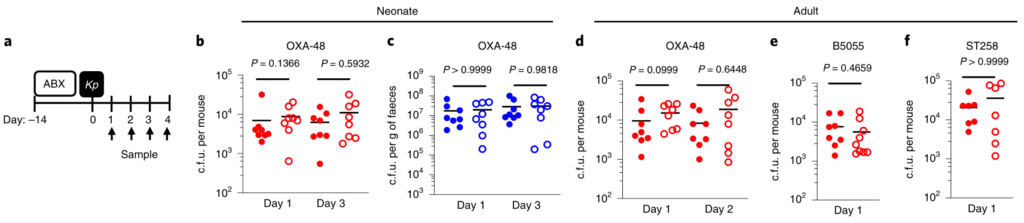
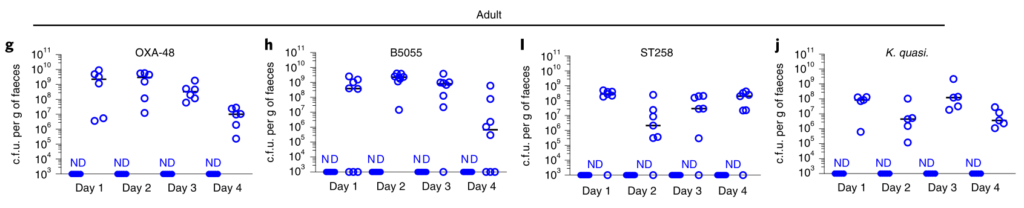
上呼吸道定植数据显示为红色,肠道定植数据显示为蓝色。所有统计比较均使用Mann-Whitney(双尾)进行;水平线表示中值;ND,未检出(肺炎克雷伯菌的检出限在粪便中= 10 3 cfu g -1)。
成人肠道菌群具有广泛的保护性,可通过临床分离株产生肺炎克雷伯菌,产生OXA-48碳青霉烯酶(图1g),流行性肺炎克雷伯菌 ST258(图1l)和近缘拟肺炎克雷伯菌亚种(图1j)。
在任一生态位定居期间均未观察到体重减轻,支持我们正在对无症状定殖进行建模的概念。这些数据表明,成年菌群的发育产生了足以阻断肺炎克雷伯菌在肠道内而不是上呼吸道定居的屏障。
这就提出了三个问题:
(1)成人肠道菌群中的哪些菌阻止肺炎克雷伯菌的定殖,其机制是什么?
(2)哪些因素可使肺炎克雷伯菌成功与上呼吸道菌群竞争,从而定居于这一生态位?
(3)在不同的粘膜贮库中,微生物群和肺炎克雷伯菌之间的竞争如何影响这种传染性病原体在宿主之间的传播?
下文将围绕这3个问题展开实验与探讨。
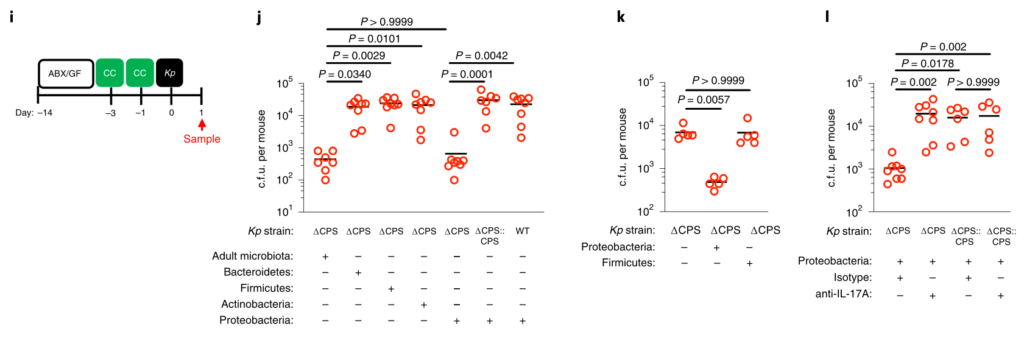
为了回答第一个问题,成人肠道菌群中的哪些菌阻止肺炎克雷伯菌的定殖,其机制是什么?
研究人员对新生小鼠和成年小鼠肠道菌群进行了测序,以确定这些允许和抑制微生物群落的各自组成。新生小鼠肠道菌群主要是来自厚壁菌门,尤其是乳杆菌科的成员(图2a,b)。相比之下,成年小鼠肠道菌群主要由拟杆菌门(拟杆菌门)和厚壁菌(梭状芽胞杆菌和乳杆菌)的共生菌组成,而放线菌门和变形菌门只占共生菌的一小部分(图2a,b)。
研究人员发现只有拟杆菌能促进肺炎克雷伯菌从肠道的清除,无论是在肺炎克雷伯菌定植之前还是之后(Fig. 2c–k)。拟杆菌在抗生素治疗(图2d-f,i,j)和无菌小鼠(图2g)中都具有保护作用,这与使用不同成分的共生体一致。
证实施用代表性的共生菌群的小鼠的肠道菌群由该菌群占主导地位,并且每个菌群的定植水平相似。
图 非抗生素处理、抗生素处理和共生联合体接种小鼠粪便中的细菌负荷和组成

此外,还发现拟杆菌属特别保护新生小鼠免受肺炎克雷伯菌的肠道定植(图2k),这支持了这种共生体的发展可以保护成年小鼠免受肺炎克雷伯菌的侵害。
图2:肠内的拟杆菌属可以预防肺炎克雷伯菌的定殖。


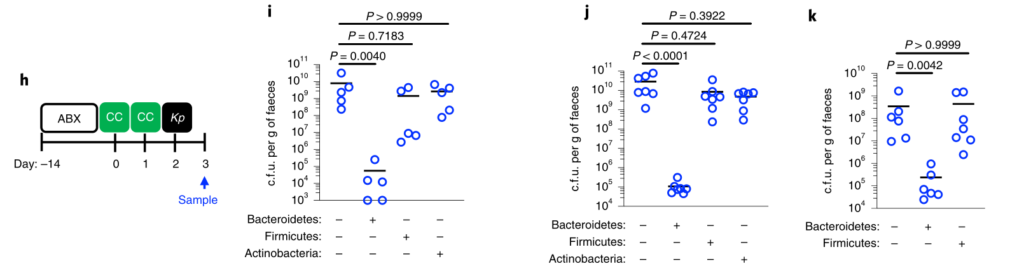
所有的统计比较都是用Kruskal-Wallis检验和Dunn对多重比较的修正进行的。水平线表示中值。
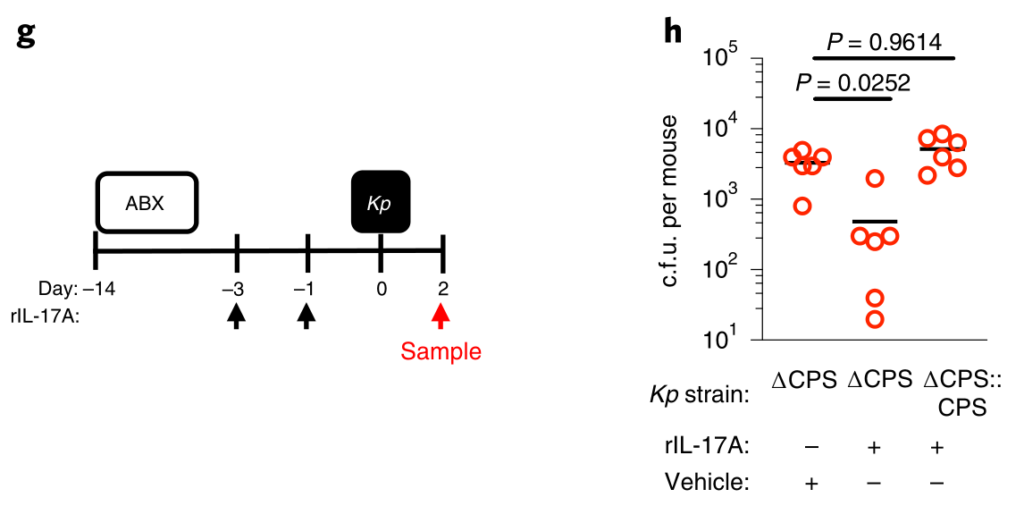
产生共生定殖因子(CCF)的拟杆菌属通过IL-36和巨噬细胞加强肠道免疫屏障,以防止肺炎克雷伯菌肠道定殖。
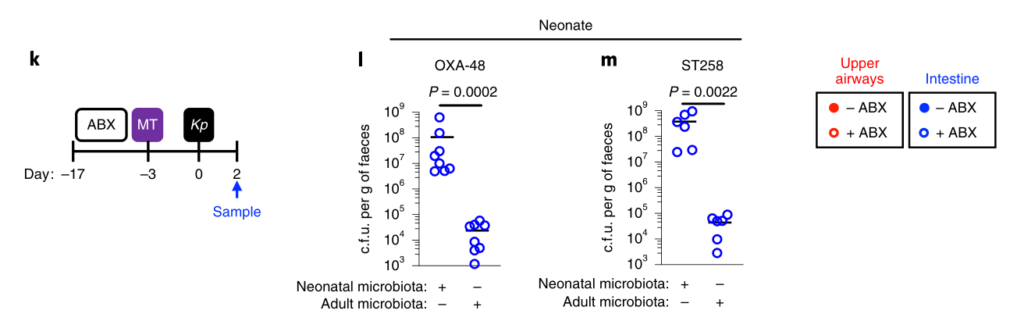
接下来,研究人员想确定拟杆菌的保护作用是否需要宿主免疫。因此,在肺炎克雷伯氏菌定植之前,将微生物群转移到施用了免疫抑制性地塞米松或媒介物对照的微生物群缺失小鼠中。地塞米松消除了微生物群对肺炎克雷伯菌定殖的抑制作用(参见图3a,b),表明该保护作用需要免疫信号。
此外,研究人员发现地塞米松对未经抗生素治疗的小鼠的治疗,使这些正常耐药的小鼠容易被肺炎克雷伯氏菌定殖。同样,拟杆菌属赋予的保护作用需要免疫信号传导。
为了确保地塞米松对免疫系统起作用而不是破坏微生物群,研究人员将微生物群从地塞米松或媒介物对照治疗的小鼠转移至微生物群枯竭的小鼠,然后研究了对肺炎克雷伯菌定植的抑制作用。这两个微生物群均提供了针对肺炎克雷伯菌定殖的同等保护作用。由此证明地塞米松不能消除微生物菌群的保护性优势,而是抑制微生物菌群对免疫系统的刺激作用。
然后,研究人员试图确定将拟杆菌属的作用转化为对肺炎克雷伯菌定植的抗性的免疫因子。在肠道中,从新生小鼠到成年的转变以许多细胞因子的稳态表达增加为标志,这些因子对维持宿主与微生物的动态平衡很重要。
为了确定拟杆菌的保护作用是否受这些细胞因子的驱动,他们取消了细胞因子的信号传导,该信号的稳定表达从新生小鼠到成年小鼠均以微生物群依赖性方式增加。
图3 拟杆菌通过IL-36信号和巨噬细胞保护肺炎克雷伯菌在肠道的定植。
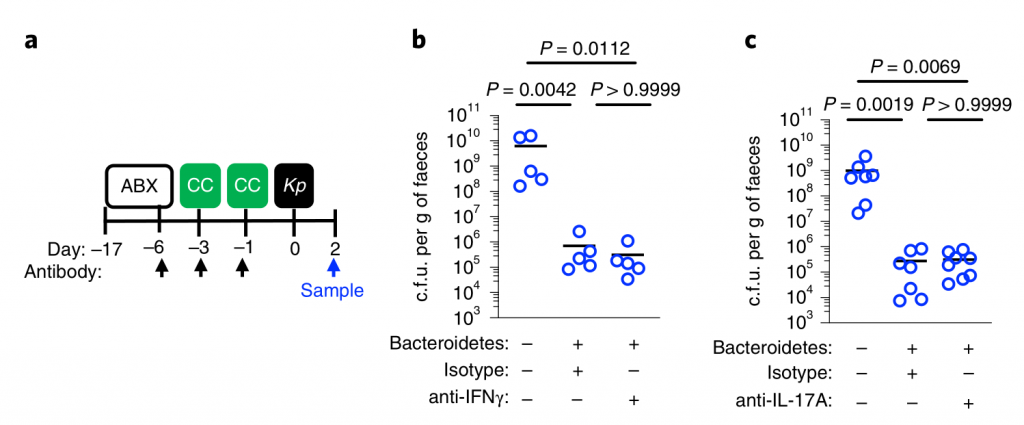
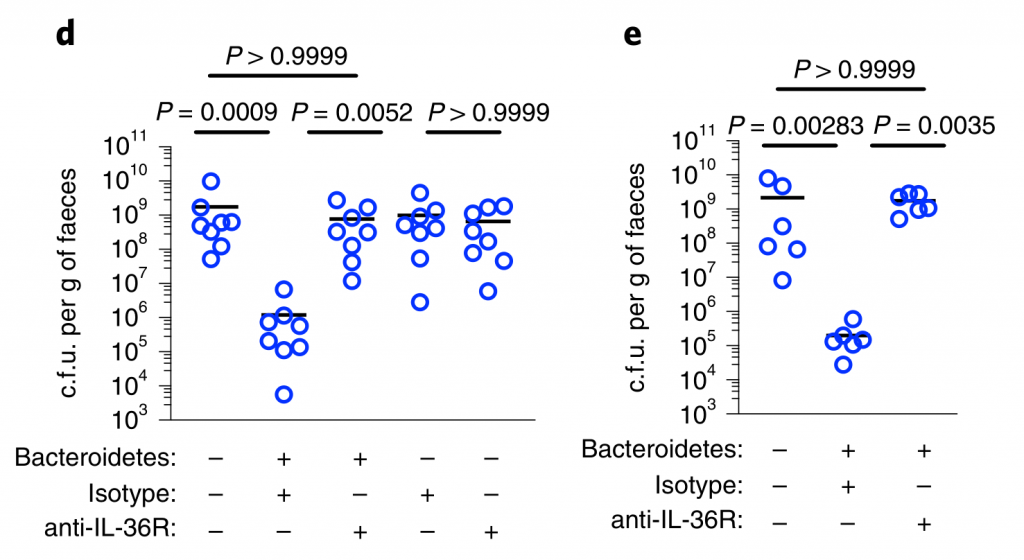



水平线表示中值,误差线为s.d.
用针对IL-36受体(IL-36R)信号的抗体治疗,但不针对IL-17A或干扰素(IFN)-γ的抗体治疗后,拟杆菌属对肺炎克雷伯菌的肠道菌落保护被取消(图3a-e)。支持IL-36的作用,重组IL-36γ处理可促进肠道肺炎克雷伯菌的清除(图3f–h),而微生物群落衰竭的动物中肠道IL-36γ的产生可通过拟杆菌特异恢复(图3i)
利用脂质体氯膦酸盐处理耗尽这些细胞来研究它们对肺炎克雷伯菌的保护作用。巨噬细胞耗竭后,拟杆菌不能预防肺炎克雷伯菌或调节IL-36γ(图3j–m)。相反,嗜中性粒细胞耗尽后,拟杆菌仍具有保护作用。
图 变形菌在定植过程中恢复了上气道的稳态IL-17A生成和肺炎克雷伯菌诱导的上气道TNF生成

此外,在用氯膦酸盐处理的小鼠中,IL-36R信号的破坏不会增加肺炎克雷伯菌的定殖,这表明IL-36和巨噬细胞沿一条共同途径起作用(图3n)。
为了了解IL-36如何通过巨噬细胞控制肠道中的肺炎克雷伯菌水平,研究人员调查了IL-36是否促进了这些细胞的杀菌活性。然后发现IL-36γ刺激促进巨噬细胞杀死肺炎克雷伯菌的多种菌株。
接下来,研究人员想了解抗肺炎克雷伯菌定殖所需的拟杆菌属中的因素。调节肠道免疫力的常见方法是与宿主粘膜紧密接触以发挥其作用。拟杆菌内高度保守的是荚膜多糖产生位点CCFs,这促进了拟杆菌与肠黏膜的关联。
图4 拟杆菌需要它们的ccf来防止肺炎克雷伯菌在肠内的定植。
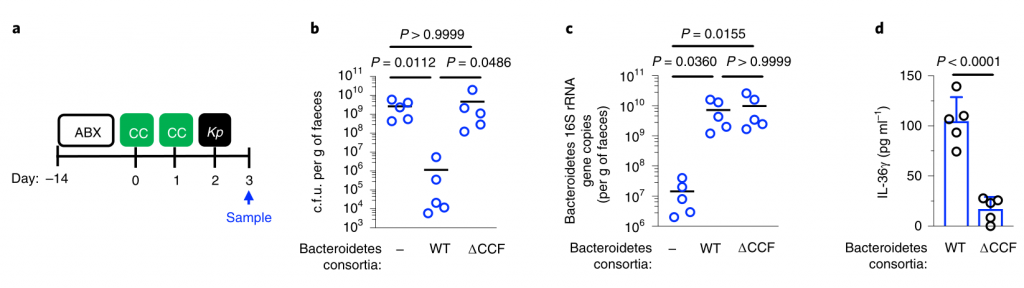
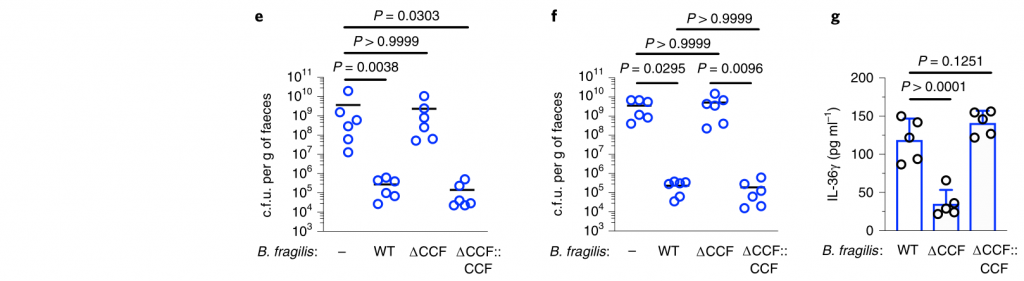



研究人员测试了假单胞菌抑制肺炎克雷伯菌定植所需的CCF的假设。与此一致,具有完整的CCF系统的拟杆菌属或单个拟杆菌属物种可以预防肺炎克雷伯菌的细菌定植和主要肠道IL-36γ的产生,但是在没有CCF的情况下会消失,并且可以通过CCF的互补来重新建立(图4a–g)。
与设想的在粘膜定植中的作用相一致,发现CCF促进了拟杆菌与粘膜的缔合(图4h),但是在存在或不存在CCF的情况下,粪便中的拟杆菌水平是相等的(图4i)。
以上,数据支持这样一个模型,其中产生细菌CCF的拟杆菌属成员与粘膜屏障结合,在肠内形成免疫屏障,从而防止肺炎克雷伯菌通过IL-36和巨噬细胞定居。
接着回答第二个问题:哪些因素可使肺炎克雷伯菌成功与上呼吸道菌群竞争,从而定居于这一生态位?
为此,研究人员检查了肺炎克雷伯菌包封的作用。传统上,包囊被视为一种毒性决定因素。然而,荚膜多糖的产生是常见的病原体和非病原体。特别是在上呼吸道中。
在具有微生物群的成年小鼠中,我们发现包封的肺炎克雷伯菌定植上呼吸道水平高于等基因的未包封的肺炎克雷伯氏菌突变体,并且,通过未封装的突变体的互补,恢复了抵抗微生物群介导的防御的能力(图5a,b)。
包囊化的这种优势在微生物群落缺乏的动物中消失了,在这些动物中,包囊化和未包囊化的肺炎克雷伯菌定殖到相似的水平(图5a,b)。
图5 变形菌通过IL-17A启动上呼吸道防御,但包封可以使肺炎克雷伯菌克服这些防御。
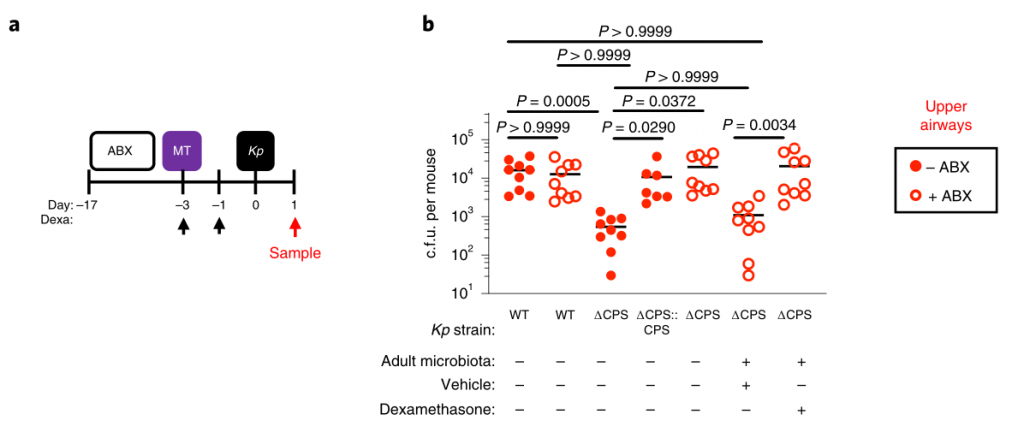


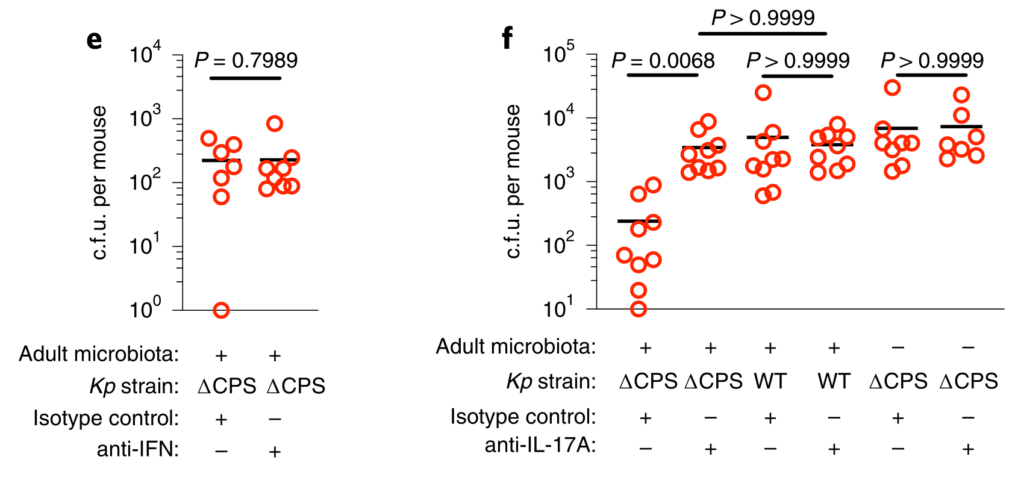
下一步,研究人员确定包封是否允许肺炎克雷伯菌抵抗上呼吸道内的直接微生物竞争,或通过免疫系统间接影响微生物群。为了解决这一问题,他们将微生物群转移回经地塞米松或载体控制治疗的微生物群衰竭的动物,然后再经鼻腔给药肺炎克雷伯菌。
微生物群转移减少未包封肺炎克雷伯菌在载体对照中的上呼吸道定植,但地塞米松治疗的动物却没有,这支持了微生物群通过免疫信号增强上呼吸道防御的观点。与此相符的是,在微生物群落衰竭的动物中,肺炎克雷伯氏菌在上呼吸道定植期间诱导的先天细胞因子产生(肿瘤坏死因子-α)减少,并通过免疫信号通过微生物群转移而恢复。
图 微生物群启动上呼吸道免疫,但封装使肺炎克雷伯菌克服这些防御

研究人员确认,微生物群转移后上呼吸道共生细菌的水平在媒介物和地塞米松治疗的小鼠之间是相同的,且微生物群的转移没有导致共生接种到肺中。
与肠道类似,上呼吸道的微生物防御启动是成年动物所特有的,因为封装和未封装的肺炎克雷伯菌都将新生的上呼吸道定居到相似的水平,并在缺乏微生物群和对照的动物中引起了同等的先天细胞因子反应。
从新生到成年的转变伴随着依赖于微生物群的上呼吸道多种细胞因子稳态表达的增加,因此,研究人员取消了这些细胞因子的信号传导,以了解成年微生物群是否需要它们才能在上呼吸道发挥其免疫调节作用。
IL-17A的破坏,而不是IFN-γ或IL-36R信号的破坏,抑制了微生物群介导的未包囊但未包囊的肺炎克雷伯菌从上呼吸道的清除(图5c–f)。
此外,用重组IL-17A处理足以促进未包封的肺炎克雷伯菌的清除(图5g,h)。小鼠上呼吸道菌群包括的菌来自放线菌门,拟杆菌,厚壁菌门和变形菌。
在这些共生体中,发现变形菌通过IL-17A信号(图5i-l)特异性地增强了抗生素治疗和无菌小鼠上呼吸道对未包被肺炎克雷伯菌的清除。变形菌还恢复了上呼吸道的稳态IL-17A生成和肺炎克雷伯菌定植诱导的细胞因子生成。
研究人员证实,给代表性共生体注射的小鼠上呼吸道微生物群主要由该共生体控制,并且每个共生体的定殖情况相似。
这些数据支持一种模型,通过该模型,变形菌可以在成年上呼吸道引发IL-17A依赖性免疫防御程序。但是,包囊可以使肺炎克雷伯菌经受住这些防御,从而成功地建立定植。
然后回答第三个问题:在不同的粘膜贮库中,微生物群和肺炎克雷伯菌之间的竞争如何影响这种传染性病原体在宿主之间的传播?
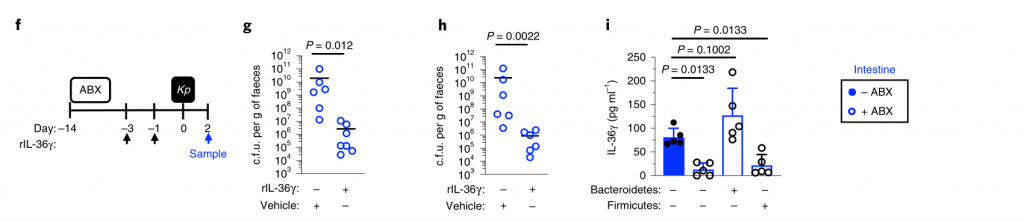
为了了解防止肺炎克雷伯菌在肠道定植的成人微生物群的发育如何影响传播,研究人员采取以下措施:
首先,将已建立肠道肺炎克雷伯菌定殖的成年小鼠与抗生素治疗或非抗生素治疗的小鼠接触,共同饲养。抗生素治疗的成年接触小鼠都获得了肺炎克雷伯菌,但对未经抗生素治疗的成年接触小鼠的传播有限。
研究人员再次确认从成年接触小鼠分离出的肺炎克雷伯菌的抗生素耐药谱与接种到索引小鼠中的肺炎克雷伯菌的抗生素耐药谱相匹配,确认从接触小鼠分离出的肺炎克雷伯菌来自索引动物。
图 接触小鼠肺炎克雷伯菌的耐药谱与对照小鼠肺炎克雷伯菌的耐药谱一致
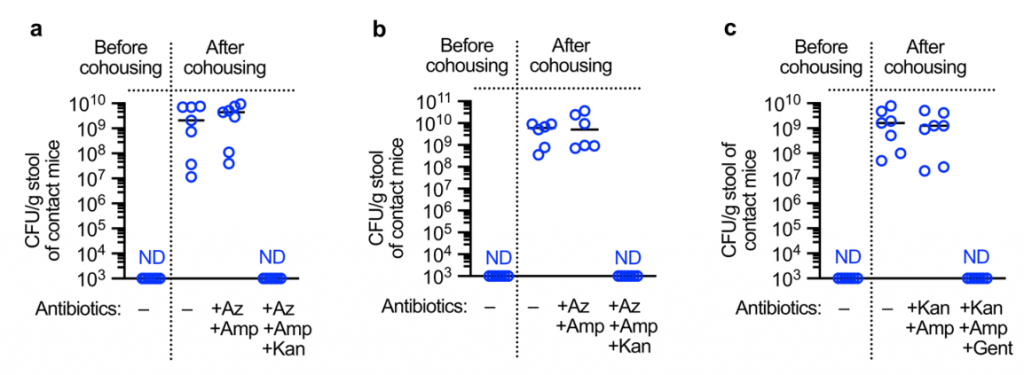


其次,在共寄居之前,将保护性拟杆菌属共接种到肺炎克雷伯菌定殖的新生指数小鼠和幼稚的新生接触小鼠中。这足以阻止宿主之间的肺炎克雷伯菌传播。为了检查传播是否需要与动物直接接触,将成年接触者关在先前装有肠道定殖的索引小鼠的笼子中。尽管在饲养前已从笼子中取出所有的索引小鼠和粪便材料,但所有接触的小鼠都被肺炎克雷伯菌定殖,这表明传播不需要动物之间或同食性的直接接触。
第三,在共寄居之前,将保护性的拟杆菌共生体口服到肺炎克雷伯菌定植的成年指数小鼠和未经抗生素处理的成年接触小鼠体内。发现拟杆菌特异性足以阻断肺炎克雷伯菌在宿主间的传播。在IL-36信号的破坏或没有CCF的情况下,拟杆菌的抑制作用丧失了。
综上所述,这些数据表明拟杆菌对肺炎克雷伯菌定植的保护作用限制了其在人群中的传播。研究人员质疑拟杆菌对肺炎克雷伯菌传播的抑制作用是否是由于拟杆菌减少了从指数动物身上脱落的肺炎克雷伯菌,或是在接触动物身上建立了针对肺炎克雷伯菌的保护屏障。
为了回答这个问题,拟杆菌只给肺炎克雷伯菌定植的索引小鼠或单纯接触者。发现,这两种方法都减少了肺炎克雷伯菌的传播,但将拟杆菌引入接触者比将拟杆菌引入索引小鼠的效果更为有效,证明拟杆菌控制传播的关键点是加强免疫屏障以防止肺炎克雷伯菌在新宿主中的建立。总之,数据显示,产生CCF的拟杆菌抑制了肺炎克雷伯菌在人群中的传播。
该文章最后提到,个体内的微生物群失调不仅会促进肺炎克雷伯菌在宿主体内的定植,而且会促进其向其他宿主的传播。因此,拟杆菌群可以通过防止肺炎克雷伯菌在宿主之间的传播提供人群级别的保护,这是控制传染病的最终手段。
在本文开头部分,我们已经看到从谷禾健康2019年样本中抽取样本的实际分布情况:
其中肺炎克雷伯氏菌丰度占比超过1%的人群有3765例,占比28.2%。
为了解感染携带肺炎克雷伯氏菌的人群和不携带人群的菌群构成,尤其是论文中拟杆菌对于肺炎克雷伯氏菌感染的防控作用在真实人类群体中的情况,我们将根据肺炎克雷伯氏菌的占比丰度分为两类人群。
其中肺炎克雷伯氏菌占比丰度5~50%的人群作为肠道感染携带者(1629例),占比<1%的人群(9593例)作为对照。
提取拟杆菌门和厚壁菌门的比例进行比较,结果如下:
肺炎克雷伯氏菌肠道感染人群菌群构成
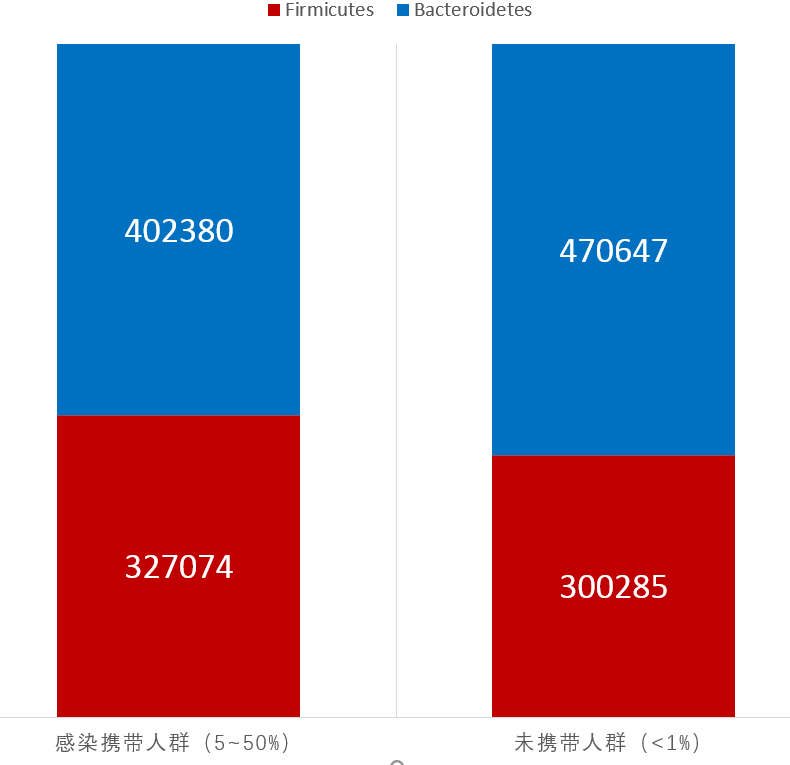

可以看到两组人群的拟杆菌门的丰度水平确实存在差异,未感染人群的拟杆菌门比例相对更高,统计检验是极显著。
论文的研究发现在人类群体中应该存在一定的作用,但是感染人群其拟杆菌并不是极低。
进一步使用MaAslin2进行检验肺炎克雷伯氏菌与其他菌属,结果如下:
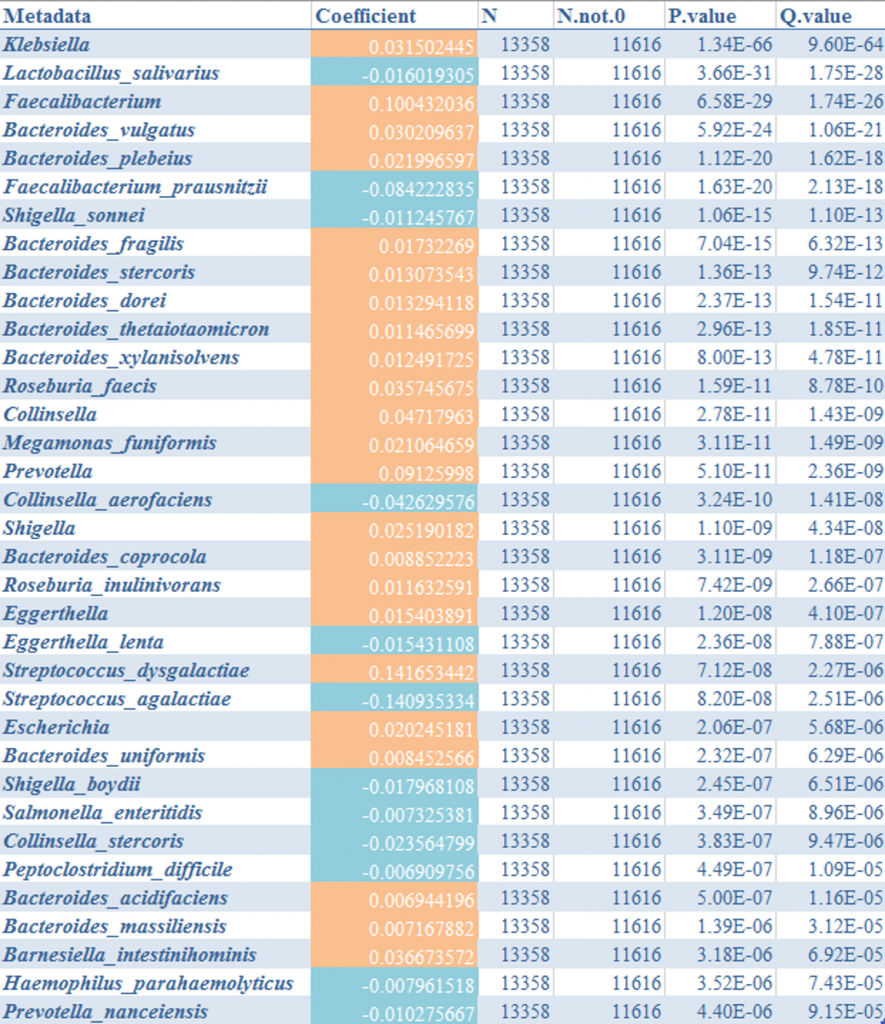

发现和预期的不太一样,拟杆菌与肺炎克雷伯菌的丰度水平之间显示是正相关,唾液乳杆菌和栖粪杆菌与其是显著负相关,另外其他病原菌与其负相关。
总之,论文的研究发现在人类群体中应该存在一定的作用,但是大样本人群肠道菌群检测结果分析表明单纯的拟杆菌存在并不能完全抵御肺炎克雷伯氏菌的定植和传播。
参 考 文 献
Sequeira, R.P., McDonald, J.A.K., Marchesi, J.R. et al. Commensal Bacteroidetes protect against Klebsiella pneumoniae colonization and transmission through IL-36 signalling. Nat Microbiol (2020)
因新型冠状病毒疫情影响,响应杭州市政府疫情控制规定,谷禾健康实验室节后恢复时间延后至2月10日以后,具体时间等待政府通知。期间数据分析与已完成测序样品报告可正常出具,新样本接收延后,实验室暂停样品处理,一旦恢复正常会及时发布通告。值此非常时刻,敬请谅解。
收样安排
春节前实验室的收样时间截止1月19日,客户的样本必须在1月18日前送到实验室会安排节前上机。春节期间1月20日至1月31日实验室无法收样。
报告交付
1月14到19日收到样本交付期限为节后2月1日;2月1号起恢复正常上机和报告出具。

杭州谷禾信息技术有限公司
2019年12月30日

我们都知道饮食对人体健康的重要性,不太合理的饮食与很多疾病相关,也包括癌症。
在过去的十年里,人们都在寻找预防癌症的饮食指南,使得饮食模式和癌症领域的迅速扩展。
多个系统回顾和Meta 分析了特定癌症类型与数据驱动饮食模式之间的流行病学关联,这些饮食模式是由实证分析决定的,研究者根据预先确定的一组饮食成分确定的饮食指标。
不同饮食模式的建议在降低风险方面的有效性,可能取决于癌症的类型或其他风险因素,如家族史、性别、年龄和其他生活方式因素或共病,以及新陈代谢特征或肠道菌群特征。
饮食是多种癌症的一个公认的危险因素。
对个别营养素或植物化学物质的研究可以揭示某些饮食因素与癌症风险之间的联系。
然而,个别的饮食成分相互关联和相互作用进而影响身体健康和疾病发生。
对饮食作为一个整体的检查,就像在饮食模式研究中所做的那样,可以产生更强的效果估计和结果,可以更容易地转化为饮食指南。
在早期的饮食模式研究中,在流行病学研究中很少观察到与癌症有实质性的联系。特别是,在1995年美国人的饮食指南的早期研究中,旨在反映美国人的饮食指南的健康饮食指数与癌症风险无关。
然而,随着美国人的饮食指南的进化和随后在健康饮食指数中包含更具体的指导,逐渐观察到例如癌症死亡率的更强的流行病学关联。制定了多种其他饮食模式和指标,以反映其他国家或组织的饮食指南,或专门关注慢性疾病预防指南。
以下内容主要总结了在过去5年中发表的有关饮食模式和癌症风险的流行病学文献。
在看后面的内容之前,我们需要在本章节先了解一下关于膳食模式的分析法,以及各类名词的定义,以便于更好地理解后面的内容。
膳食模式或指标的表征取决于对食物、饮料或营养摄入的了解,或这些因素的某些组合(例如,健康饮食指数和膳食炎症指数(DⅡ),从自我报告或谈话者使用的调查表或膳食召回或记录获得。
一般来说,饮食模式分析分为两大类,一类是根据经验确定的,另一类是根据研究者定义的标准。
后验饮食模式由研究人群中的数据驱动,并使用统计技术确定,而先验饮食模式是根据一组预定义的标准来构建的,以在特定人群的数据收集之前或独立于数据收集之前测量对特定饮食方式的依从性。
先验饮食模式或指数包括各种成分,可使用简单的二元方法对其进行评分,以满足(1)或不满足(0)指南(如地中海饮食评分(MDS))或更复杂的算法,其中考虑了基于文献的加权效应大小(如DII)。
表1 关于各种先验饮食模式和指标及其相应组成部分
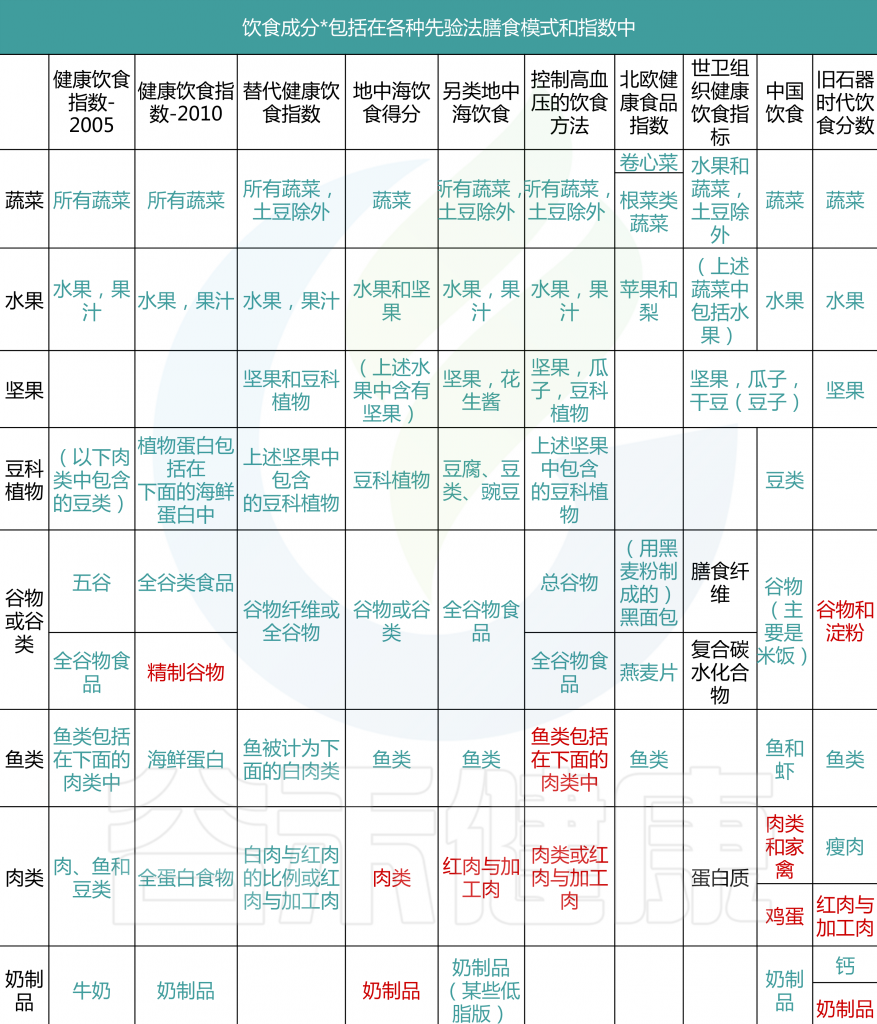


一般认为,先验饮食模式比后验饮食模式更容易在不同研究和人群之间进行比较,因为成分是预先确定的,而不是从研究人群分布中得出的。然而,先验方法在膳食成分的数量、FFQ中包含的与成分相关的食品和饮料的数量以及用于描述依从性的阈值(临界点)的研究之间可能有很大的不同。例如,MDS中使用的临界点基于特定人群的中位数摄入量,因此,尽管MDS被认为是一个先验指标,因为组成部分是预先确定的,但得分取决于所研究的人群;然而,其他具有固定切点的得分可能允许在人群之间进行更稳健的比较。
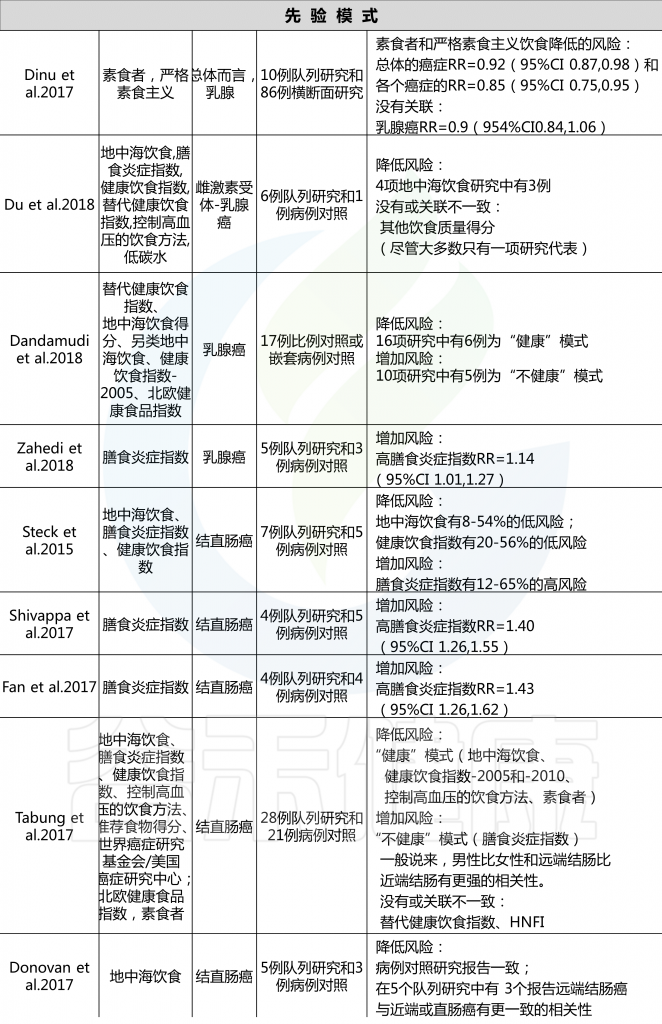
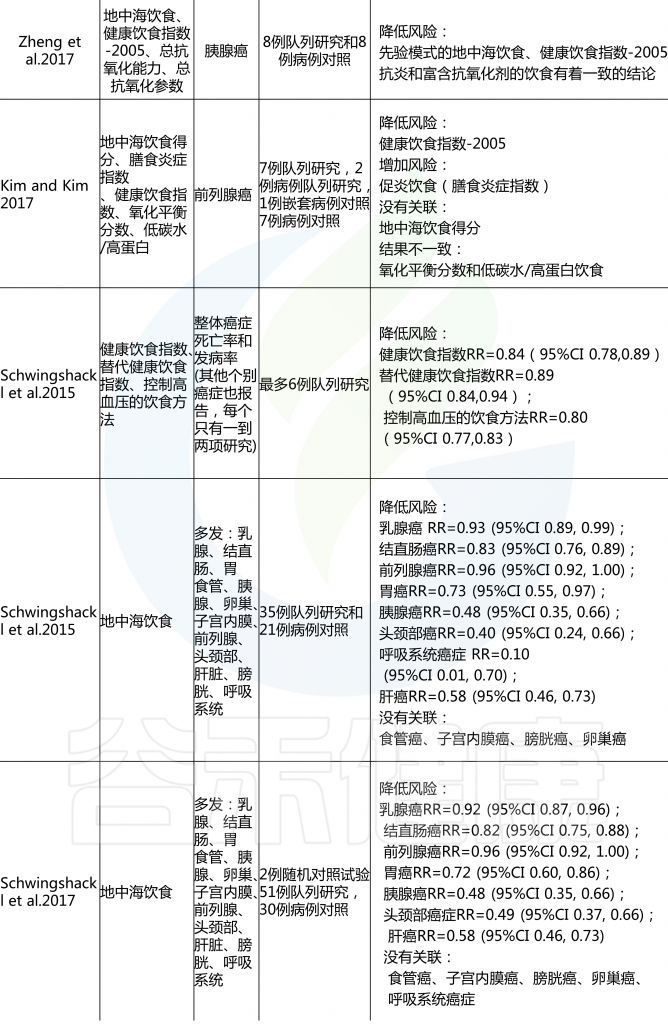
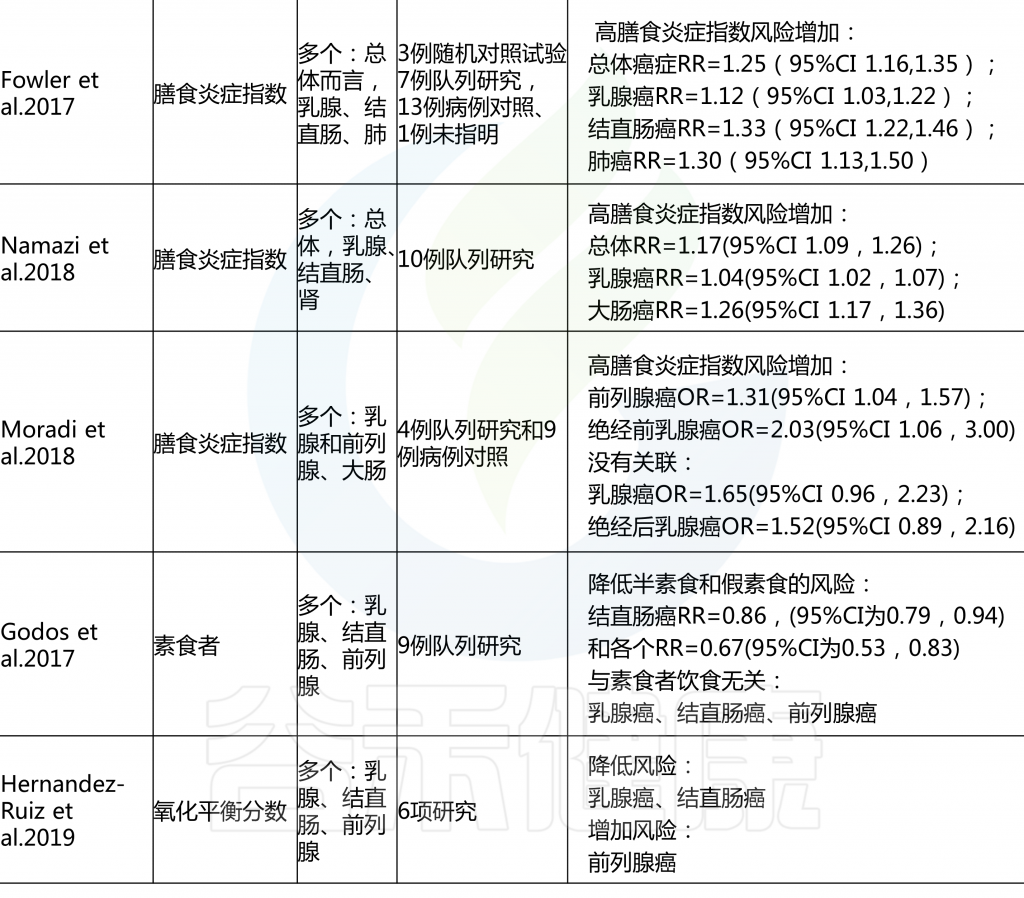




名 词 预 习


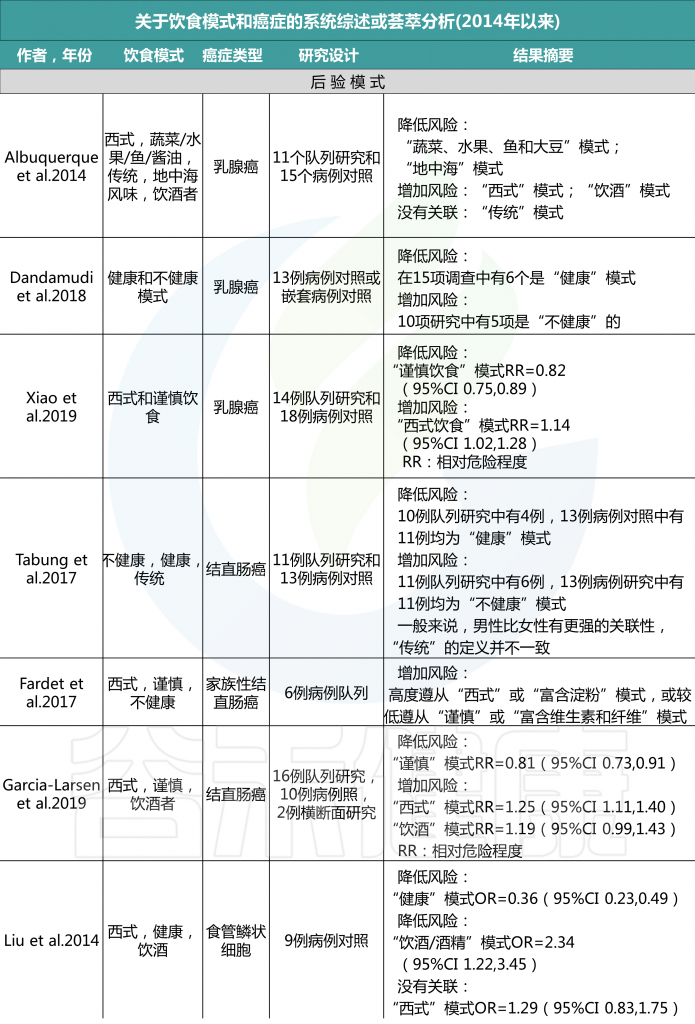
各种病例对照和队列研究报告称,后验饮食模式与各种癌症风险的增加或降低有关,一些研究报告称,没有一致的风险关联。具体看看各种饮食模式对应什么疾病的风险。
后验饮食模式与癌症风险
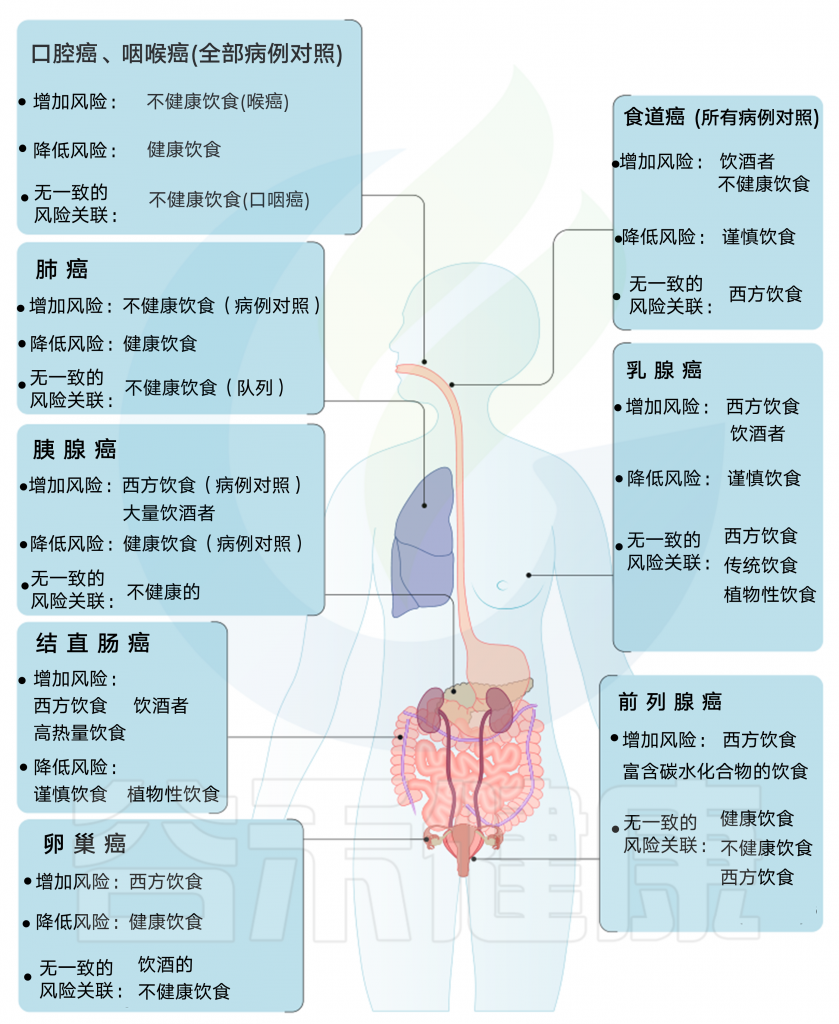

由上图可以发现,西方饮食模式与结直肠癌风险增加相关。而乳腺癌、前列腺癌和胰腺癌的证据往往因研究设计的不同而有所不同。病例对照研究报告显示正相关,但队列研究报告没有一致的相关性。
另外,谨慎或健康的饮食模式与患乳腺癌、结直肠癌与肺癌风险降低相关,而胰腺和前列腺疾病的相关性要么在研究设计上不一致,要么仅限于病例对照研究。
饮酒模式与乳腺癌、结直肠癌、胰脏癌和食道癌的风险增加有关。
雷达图可以直观地显示西方和审慎饮食模式研究中所包含的每个食物组的主要成分和相应的因子负荷。
主成分分析雷达图
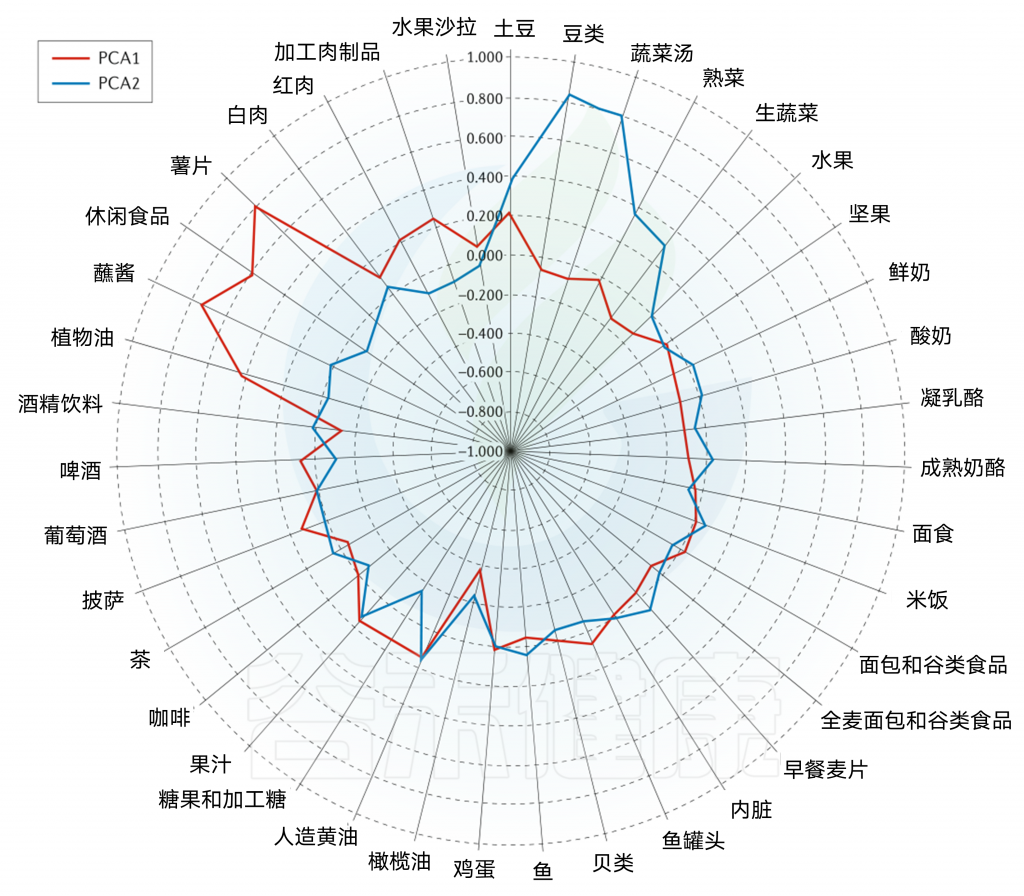

西方饮食模式(PCA1;红色);
谨慎饮食模式(PCA2;蓝色);
接近1.000的线表示正因子负荷(较高的摄入量);
接近-1.000的线表示负因子负荷(较低的摄入量)。
西方饮食模式特点:大量摄入薯条、零食、蘸酱、植物油、红肉、加工肉和土豆,而橄榄油的摄入量较低。
谨慎饮食模式特点:大量摄入豆类、蔬菜汤、土豆、熟蔬菜和生蔬菜。
许多病例-对照研究和队列研究以及这些研究的系统审查和 Meta 分析研究了各种后验饮食模式与癌症风险之间的相关性。一项关于后验饮食模式(分组为“健康”或“不健康”)和癌症风险的93项研究的全面系统审查和 Meta 分析报告如下:
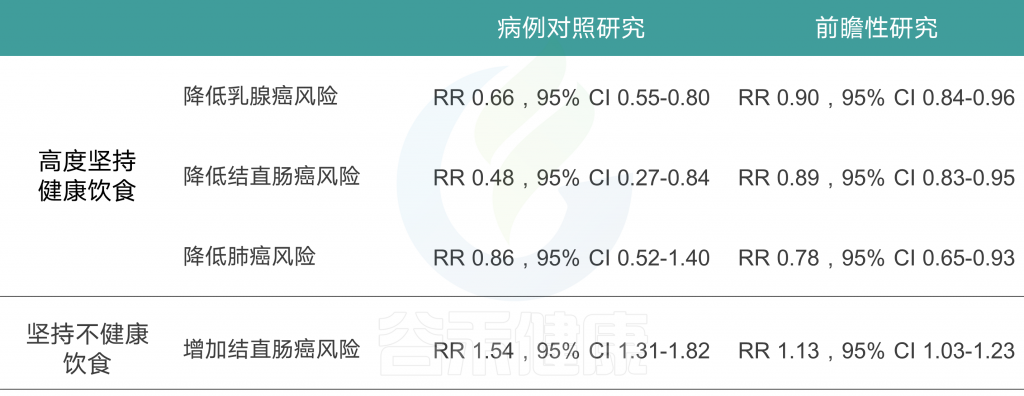

对乳腺癌的Meta分析回顾了14组和18项病例对照研究,得出结论认为西式饮食使乳腺癌风险增加(RR 1.14,95% CI 1.02-1.28,尽管这种关联仅限于病例对照研究,而不是队列研究),而“谨慎”模式的风险使其风险降低(RR 0.82,95% CI 0.75-0.89).
另一项基于经验的22项饮食模式的Meta分析显示,饮食“健康”的人患卵巢癌的风险降低(OR 0.86,95%CI 0.74-0.99;p=0.04),“西方”饮食模式者患卵巢癌的风险增加(OR 1.19,95%Ci 1.01-1.41;p=0.04)。
总体而言,系统评估和Meta分析报告显示,与“健康”或“谨慎”的饮食模式相比,结直肠癌的风险持续降低,而“西方”或“不健康”模式的风险增加,而其他类型癌症的关联则具有暗示性,但在病例对照和队列研究中都不那么一致。
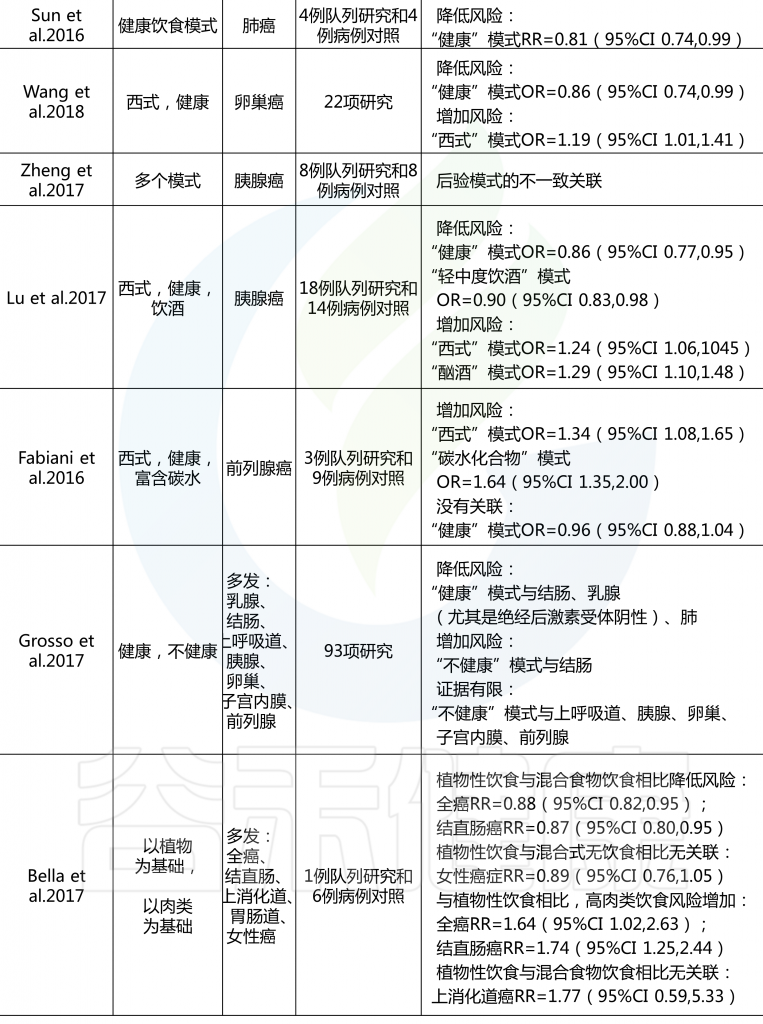
许多研究人员根据饮食指南(国家指南和慢性病预防指南)和文化饮食方式制定了饮食模式和指数。这些先验饮食与各种癌症风险之间的联系在各种研究中都有报道,系统回顾和 Meta 分析提供了广泛的见解。
重要的是,一些饮食成分,特别是乳制品和酒精,它们对癌症风险有潜在的相反影响,不是包含在先验饮食模式中,就是被认为是不同的。
先验饮食模式与癌症风险
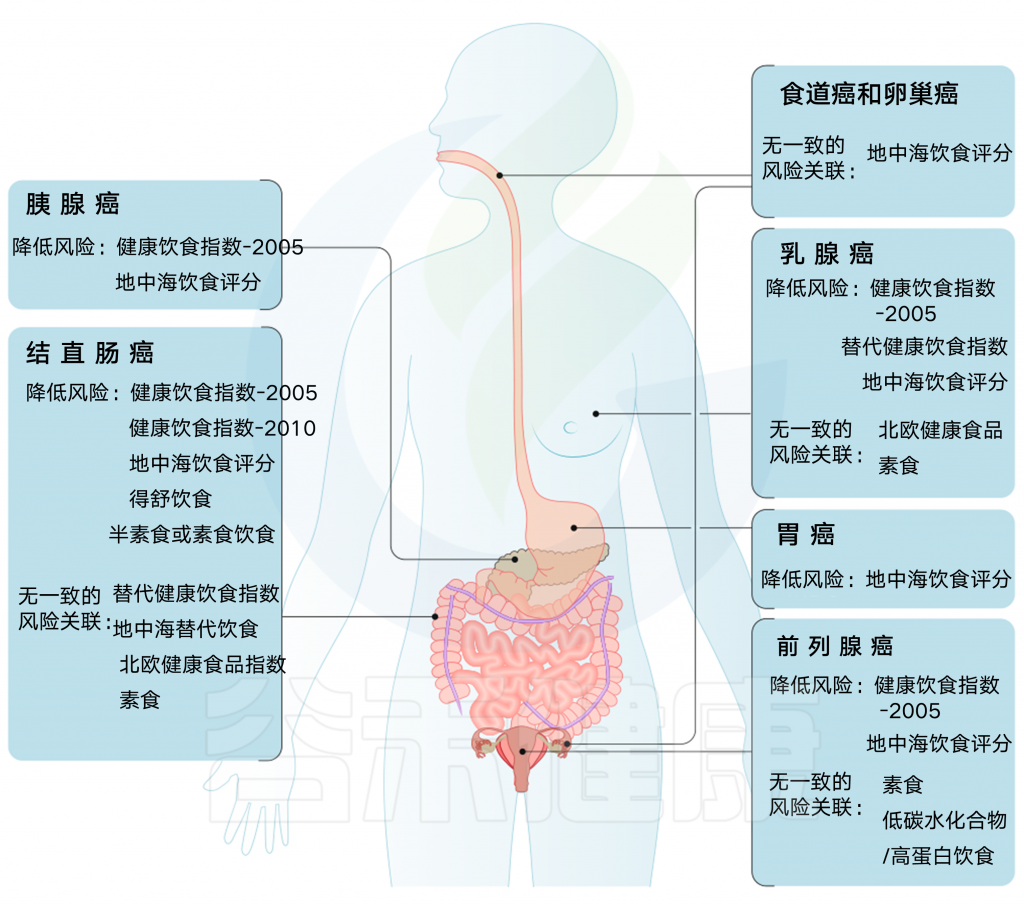

结直肠癌的证据最充分,健康饮食指数(HEI-2005或HEI-2010)、地中海饮食评分(MDS)、预防高血压的饮食方法(DASH)和半素食饮食与降低风险相关。HEI-2005、地中海替代饮食和MDS也与乳腺癌风险降低相关,HEI-2005和MDS在系统回顾中与前列腺癌和胰腺癌风险降低相关。
健康的北欧食物指数(HNFI)与结直肠癌或乳腺癌之间没有一致的证据。对于前列腺癌,很少有证据表明其与低碳水化合物和高蛋白以及素食有关。
在癌症流行病学文献中,基于特定国家指南的先验饮食模式包括健康饮食指数(HEI)、健康北欧食品指数(HNFI)、世界卫生组织健康饮食指标和中国食品宝塔,而基于慢性病预防指南的主要模式包括地中海替代饮食(aHEI)和预防高血压饮食(DASH).
· 健康饮食指数和替代健康饮食指数
该指数是为衡量对美国人的饮食指南的遵守情况而制定的,多年来不断发展,以与每5年更新一次的美国人的饮食指南相对应。
多个系统的回顾和 Meta 分析报告了不同版本的HEI与癌症风险之间的关联,风险和注意之间相对一致的反向关联。aHEI是根据慢性病预防的证据来定义饮食质量的,目的是作为健康饮食指数的替代品;显著的区别包括酒精和坚果和/或豆类的不同类别,白肉和红肉和/或加工肉的区别,以及长期使用多种维生素。
· 其他全球或国家特有的饮食指数
基于其他国家特定饮食指南的饮食指数还没有像健康饮食指数那样被广泛的研究,但是有一些证据表明它与癌症风险有关联。HANFI (也称为波罗的海饮食)反映了北欧人推荐的饮食指南。
一个版本包括六个组成部分(鱼、卷心菜、黑麦面包、燕麦片、苹果或梨和根类蔬菜),另一个版本结合饮食和体力活动(新的北欧饮食)包括9个同等重量的成分(碳水化合物、蛋白质、脂肪、酒精、纤维、盐、维生素、矿物质和体力活动)。见表1.
在丹麦队列中,HNFI与女性结直肠癌风险降低相关(RR 0.65,95%CI 0.46–0.94),而与男性无关(RR 0.87,95%CI 0.61–1.25)。但在瑞典的一项研究中与乳腺癌风险无关,在病例对照研究中,新的北欧饮食与前列腺癌风险无关。
世卫组织健康饮食指标是一个基于国际慢性病预防指南的七分制评分。在欧洲对癌症和营养的前瞻性调查(EPIC-nl)研究中,它与英国人乳腺癌风险无关,它与总体癌症风险或与吸烟或酒精相关的癌症风险无关。


中餐宝塔由10个组成部分组成(见表1),在两个上海队列中与结直肠癌风险降低相关(与第一个四分位数相比,第四个四分位数的RR为0.84,95%CI为0.73–0.96),其中未发现与改良aHEI-2010(RR为0.91,95%CI为0.79–1.05)和修正后的DASH评分(RR 0.90,95%可信区间0.78-1.03),表明针对具体国家的指南对一个国家人口内的健康结果有更强的影响。
· 控制高血压的饮食方法
得舒饮食(DASH)是一种推荐的饮食模式,可以降血压。它与 HEI 和 aHEI 略微不同,因为它不包括用于脂肪摄入或酒精消耗的组分(见表1)。
多项研究已经显示了其与癌症风险的关联。
在2018年,据报道,在健康专业人员随访研究中,得舒饮食与男性患结直肠癌的风险呈负相关(RR 0.81,95%CI 0.66-0.98,最高五分位数与最低五分位数比较),而在护士健康研究中的女性中则不是这样(RR 0.98,95%CI 0.82-1.17)。
一项前列腺癌侵袭性种族差异的个案研究中,DASH分数与侵袭性前列腺癌的几率之间存在适度的反向关联(OR 0.76,95% CI 0.55–1.06,比较高依从性和低依从性)。
根据文化饮食行为和传统对癌症风险进行调查的主要先验饮食模式是各种地中海饮食分数(如地中海饮食评分和地中海替代饮食)、素食或纯素饮食分数以及旧石器时代饮食模式评分。
· 地中海饮食评分
地中海饮食强调摄入蔬菜、水果、谷物和谷类食品、坚果、种子和豆类,适度摄入鱼类、橄榄油和酒精,少量摄入红色或加工肉类和乳制品。
这是在一项随机对照试验(PREDIMED试验)中试验过的少数几种饮食模式之一。在该试验中,与对照组(被随机分为低脂饮食)相比,干预组(被随机分为地中海饮食组和特级初榨橄榄油组)乳腺癌风险的降低是非常明显的 (RR 0.31, 95% CI 0.13–0.77)。
由于其在2003年首次描述了地中海饮食评分,因此已经创建了多个评分系统的迭代。
地中海替代饮食评分系统则将水果和坚果分开,去除乳制品,只包括全谷物 (而不是所有谷类) 以及红色和加工肉类 (而不是所有肉类) (见表1)。


对地中海饮食和健康结果的27项Meta分析的概括性回顾发现,在70篇关于不同健康结果的原始研究文章中使用了34种不同的评分系统。使用的主要分数是地中海饮食评分和地中海替代饮食,但其他分数的变化主要与酒精和脂肪酸摄入量的差异评分有关。
2019年的一项研究比较了同一西班牙研究人群中五种不同地中海饮食模式得分的值,发现不同得分之间只有中等程度的一致性。这一发现很可能是由于得分制定方法的变异性,包括组成得分的摄入单位(例如,一些使用克/天,而其他使用能量密度)、所含食物或营养素的类型(例如,乳制品包括在一些但不是全部得分中)以及得分取决于研究人群的摄入量水平(例如,使用中位数的摄入量值作为切入点,而不是预定的摄入量水平)。
Meta 分析得出结论,尽管饮食习惯与地中海饮食相近的参与者与乳腺癌风险的关联性较低,但其患结肠直肠癌的风险较低。
在五项研究中,没有关于地中海饮食模式与前列腺癌的风险关联的报道,尽管我们之前报道过在食用与地中海饮食密切相关的饮食的男性中,高侵袭性前列腺癌的几率降低。
在MDS、改良MDS或意大利地中海饮食指数定义的高依从性研究中,大肠癌风险降低的证据似乎最为一致,而美国人群中应用的aMED评分系统的相关性通常为空。如Donovan等人所述,MDS与结直肠癌风险之间的关联性会随年龄和性别而变化,并且观察到远侧肿瘤而非近端肿瘤的风险关联性更高。
· 素食或纯素食饮食
WCRF/AICR推荐以植物为基础的饮食,重点是全谷类、蔬菜、水果和豆类,并限制红肉的摄入,以预防癌症,主要是基于蔬菜、水果、全谷类和豆类的食用量增加,癌症风险增加,从而降低癌症风险的证据,特别是结直肠癌,增加了红肉和加工肉的摄入量。
在一项系统审查中,与非素食者相比,素食主义者(RR0.92,95%CI 0.87-0.98)和素食者(RR 0.85,95% CI 0.75-0.95) 患癌症的风险降低,而与乳腺癌发病率无关(RR 0.94,95%CI 0.84-1.06)。


在另一项仅对前瞻性队列研究进行的Meta分析中,与非素食饮食相比,素食饮食与降低乳腺癌、前列腺癌或结直肠癌的风险之间没有显著的关联。
然而据报道,半素食和 PESCO-素食饮食与结直肠癌的风险呈负相关(RR 0.67,85%CI0.53-0.83)。
使用传统的食物频率问卷定义素食者或纯素饮食会带来一些独特的挑战(例如,根据有限的食物清单中对动物产品没有正面反应的情况,将被访者归类为素食者或素食者,可能导致分类错误,而食物频率问卷通常不评估遵循素食或纯素饮食的持续时间或原因),观察性研究受到残留或未测量的混杂变量的困扰,因为选择素食或纯素饮食的人与杂食者相比,其他生活方式因素往往不同。
· 旧石器时代饮食模式评分
旧石器时代饮食富含蔬菜、水果、坚果、瘦肉和鸡蛋,盐含量极低,不含乳制品、谷物和精制脂肪和糖。
在加州教师的研究中,旧石器时代的饮食模式评分与乳腺癌的风险无关。
在爱荷华州妇女健康研究中,与结直肠癌的风险无关。尽管包括体重指数、吸烟和体力活动以及饮食的“进化一致”生活方式评分与结直肠癌风险降低相关。
乳制品与结直肠癌风险降低相关;因此,将乳制品排除在饮食评分中作为有益因素可能解释旧石器时代饮食与结直肠癌风险之间缺乏关联。
根据各种饮食因素对特定生物过程或途径的影响,已经开发了一些先验饮食指标来评估饮食质量。在流行病学文献中,与癌症生物学机制有关的几个指标-特别是炎症、胰岛素抵抗、氧化应激和雌激素代谢-与癌症风险有关。
基于生物过程的饮食模式与癌症风险的关系
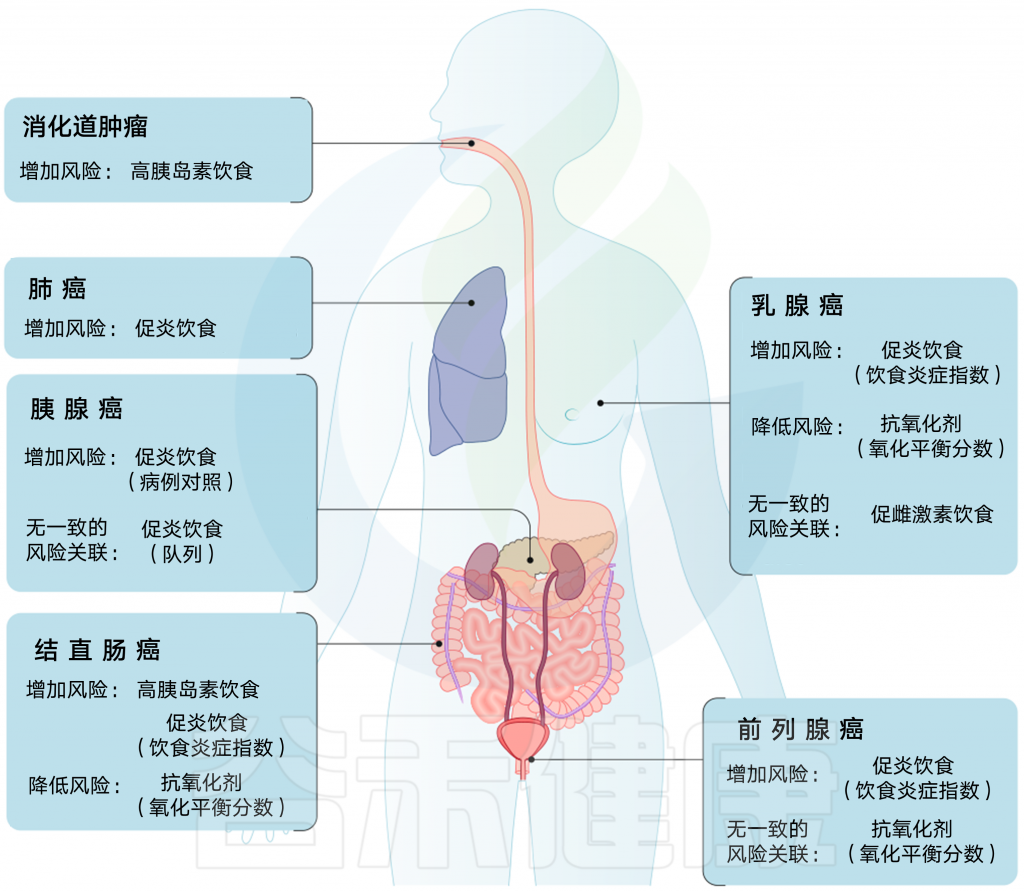

基于生物过程的膳食模式包括通过炎症指数(DII)测量的具有促炎性潜能的饮食,如抗氧化平衡能力(OBS)、高胰岛素血症潜能和亲雌激素潜能评估的抗氧化能力。根据几项 Meta 分析,较高的 DII 分数始终与结直肠、乳腺、前列腺和肺癌的风险增加相关,而胰腺癌的报告结果不一致取决于研究设计。
OBS与乳腺癌和结直肠癌的风险降低相关,而与前列腺癌的相关性不一致。在两项研究中,高胰岛素饮食与结直肠癌和消化道癌风险增加相关。在三分之一的研究中,促雌激素饮食模式与乳腺癌风险增加有关。
炎症是一种公认的癌症特征,影响恶性肿瘤的发展和发展。DII是根据摄入多达45种不同的膳食成分(其中大部分是大营养素和微量营养素)来评估整个饮食的总体炎症潜能的。见表2
在前瞻性研究(RR 1.25,95%可信区间1.16–1.35)和病例对照研究(或1.75,95%可信区间1.43–2.16)中,高DII评分(代表促炎症饮食)与癌症发病率增加有关。最常见的报道是,与结直肠癌的风险相关性最高,为男性高于女性,近端结肠高于远端结肠癌。在四分之三的研究中,膳食炎症指数与前列腺癌风险呈正相关。
氧化应激可导致DNA损伤,如果不修复,可能有助于增加癌症的风险。氧化平衡评分(OBS)已被定义为多种方式,包括3至28个组成部分,并且通常包括具有抗氧化或抗氧化作用的饮食和非饮食生活方式因素。
目前OBS与前列腺癌风险增加相关的证据有限。然而,在2019发表的一篇综合性的研究报告显示,OBS患者的“抗氧化”得分高的个体显著降低了结直肠癌(两项研究)和乳腺癌(一项研究)的风险。因此,关于OBS和个体癌症的数据是有限的,并且相关的数据通常反映了其他与生活方式相关的危险因素,如吸烟、肥胖和使用非甾体抗炎药。
胰岛素和胰岛素样生长因子调节碳水化合物和能量代谢,与癌症风险有关。新出现的证据将生酮饮食(碳水化合物含量低,脂肪含量高)与癌症预防联系起来。生酮饮食将每日碳水化合物的摄取量限制在20-50克/天,这导致胰岛素分泌减少,并通过脂肪酸转化为酮体来转化为脂肪氧化燃料。


生酮饮食已在癌症患者中进行了研究,但关于其与人类癌症预防的关系的数据有限。到目前为止,有一个随机试验,报道了生酮饮食对子宫内膜癌或卵巢癌妇女的身体机能和食欲的有利影响。
基于动物模型的研究显示其相关性:2019年对致酮饮食作为癌症治疗方法的综述显示,在大多数临床前小鼠研究中,它减缓了肿瘤生长、延长生存期、延迟肿瘤的发生和逆转癌症诱导的缓存过程。
饮食模式和癌症研究领域的新发展包括使用创新的统计技术来降低多个预测变量以及实验室工具(如代谢组学)的维数,这可以揭示生物机制,提供比自我报告数据更客观的饮食测量。
两种统计技术在癌症预防中尚未得到很好的研究,但有望有希望识别高相关性的新饮食模式,包括降秩回归 (RRR) 和分类及回归树 (CART) 分析。
RRR通常使用一个中间标记,首先根据其解释标记变化的能力来定义饮食模式,然后检查与癌症风险的关系。
a. 下图说明了在绝经后妇女的前瞻性队列研究中,用于建立与雌激素代谢物相关的饮食模式的降秩回归过程。
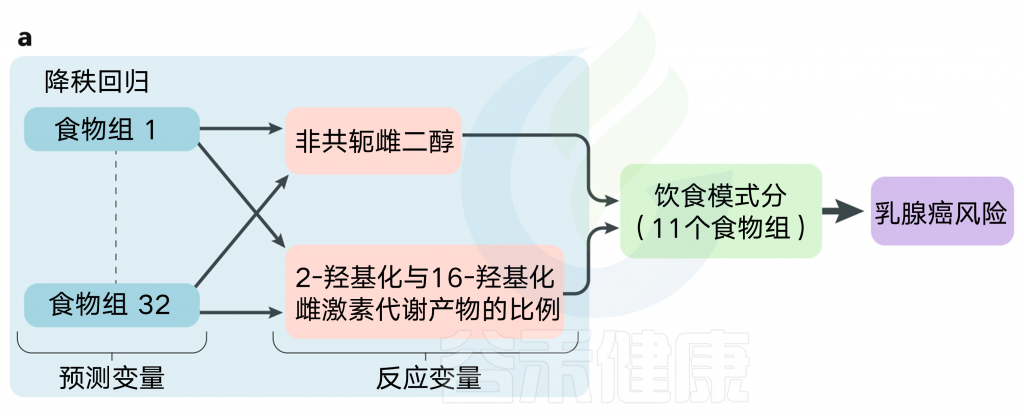
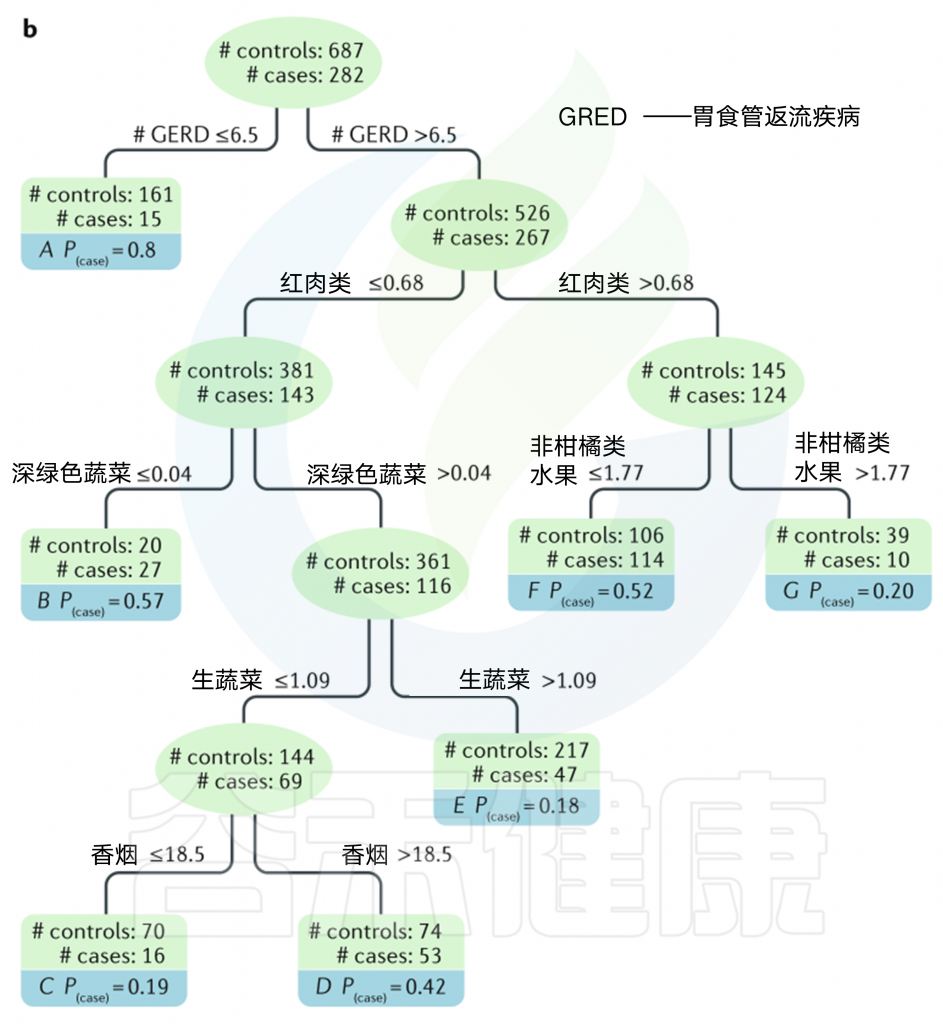


简单地说,32个食物组被用来确定哪些食物组(预测变量)解释了两个反应变量(雌激素代谢物)中的最大变化。11种食物组(非全麦/精制谷物、西红柿、十字花科蔬菜、奶酪、富含omega-3脂肪酸的鱼/贝类和法兰克/午餐肉的重量为正;坚果/种子、其他蔬菜、低omega-3脂肪酸的鱼/贝类,酸奶和咖啡的重量为负)保留在“促雌激素”的饮食模式中,然后检查与乳腺癌风险的关系。在前列腺癌、肺癌、结直肠癌和卵巢癌的研究中,更高的得分反映了更倾向于雌激素的饮食与乳腺癌风险的增加有关,但在姐妹研究中没有。
b. 下图示为食道癌之分类与回归树分析。
分类和回归树分析使用逐步回归来确定解释癌症终点变化的成分,方法是创建多个分类树(当终点是分类的)或回归树(当终点是连续变量)来将人群分成具有相似特征的子组逐步的特点。
方形框表示终端子集。每个方框或圆圈的顶部数字表示控件的数量,下面的数字表示案例的数量。每个方框右下角的数字是该组案例的概率。以食管腺癌为例,胃食管反流病(GERD)是最大的预测因子,其次是红肉摄入量。对于红肉摄入量低的个体来说,深绿色蔬菜、生蔬菜和香烟是重要的危险因素。对于高红肉摄入量的个体来说,低非柑橘类水果摄入量是一个重要的危险因素。
代谢组学领域的扩展使得使用客观的生物标记物同时检查多个与饮食相关的实验,这可以减轻人们对自报FFQ数据测量误差的一些担忧
与健康饮食指数-2010相关的前20名代谢物中有许多与维生素E、B和C等维生素的代谢物有关。在一项随访研究中,PCA用于识别一组与“健康饮食指数-2010/复合维生素评分”相关的23种代谢产物;但是,与乳腺癌风险无关(OR90例vs10百分位0.96,95%CI0.67-1.37)。
未来在代谢组学和饮食模式领域的研究有可能有助于确定某些饮食对健康益处的生物学机制,并提供比自我报告的饮食摄入数据更客观的饮食暴露测量方法。
饮食模式旨在评估整体的饮食质量,因此,它们的使用被视为营养研究的整体方法。然而,关于饮食模式中应包括或排除哪些成分的决定通常是由各种成分的个体效应的证据驱动的。因此,营养科学的还原主义方法通常是构建饮食模式的必要前提。不同饮食模式与癌症风险之间关联的生物学机制可能是由于不同饮食成分的生物效应(协同作用或添加剂)。一些相关的生物学机制已经被提出,包括肠道微生物群及其代谢物,表观遗传学,炎症和免疫功能,以及代谢或激素的破坏。
饮食与癌症关系的潜在机制
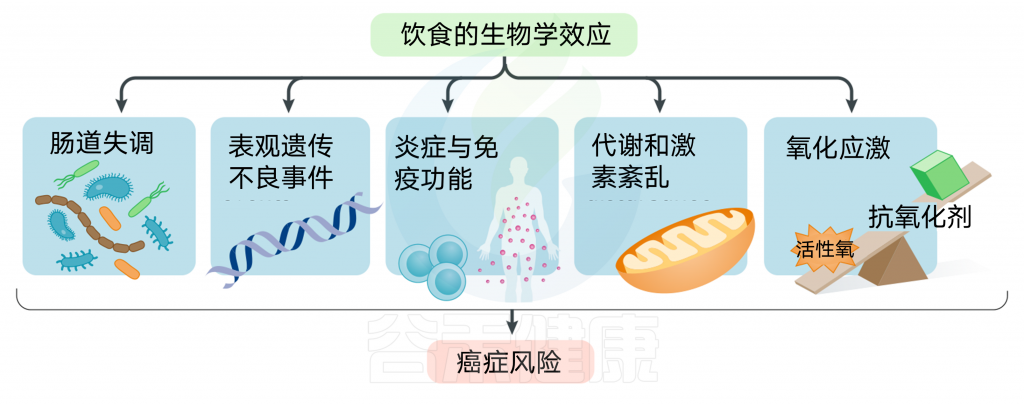

据报道,一些相关的生物学机制被特定的饮食模式所调控,包括能够显示肿瘤抑制或致癌特性的微生物群和代谢物谱;表观遗传事件,包括CpG位点的DNA甲基化模式或编码肿瘤促进基因和非编码RNAs113位点的染色质状态失调;炎症或免疫功能失调;代谢和激素反应,包括高胰岛素血症和胰岛素抵抗,性激素代谢异常,脂肪酸信号和氧化应激和氧化损伤,导致基因组DNA的修饰和导致的突变。
CpG位点DNA甲基化模式的表观遗传改变或编码肿瘤促进基因和非编码RNA位点染色质状态的失调是控制肿瘤进展的主要因素。
由于表观遗传变化可以通过营养(例如,丁酸盐介导的组蛋白脱乙酰酶抑制)来重塑,因此饮食策略在逆转癌细胞中的不良表观遗传标记方面具有巨大的潜力。
目前的研究主要局限于富含多酚的饮食,这种饮食在影响DNA甲基化,组蛋白修饰和非编码RNA在癌细胞中的表达以减轻肿瘤进展和预防转移方面取得了一些成功。
流行病学研究表明,所有癌症中至少有20%是由炎症或免疫功能失调直接引起的,因此抗炎饮食模式可能是很有吸引力的癌症预防策略。
各种炎症饮食模式评分的发明,如膳食炎症指数(DII),促进了将炎症作为饮食与癌症风险之间的生物学联系的研究。以高膳食炎症指数评分为代表的炎症饮食与多种癌症的风险增加有关,而旧石器时代和地中海饮食模式的得分与炎症的生物标志物成呈负相关。
代谢综合征-一种以腹部肥胖、胰岛素抵抗、高血糖、血脂异常和高血压为特征的疾病,常伴有促炎症状,与癌症密切相关,因为已报告增加癌症风险和癌症相关发病率。这种关系的潜在机制包括高胰岛素血症和胰岛素抵抗,性激素代谢和脂肪因子信号异常,慢性炎症和慢性多血糖症。地中海饮食和素食饮食与代谢综合征的预防和延迟发病有关,而西方饮食模式与代谢综合征的发病率增加有关。
越来越多的证据表明肠道微生物可以影响癌症的易感性和病因。饮食可以影响这一过程,因为它有能力促进具有抑瘤或致癌特性的细菌的生长,这取决于营养物质的含量。此外,一些饮食成分被共生或共生的肠道细菌代谢为具有生物活性的食物成分,可以预防癌症。
支持饮食、肠道微生物群及其代谢物与癌症风险之间联系的最有说服力的证据来自于高纤维摄入量饮食模式的研究。
膳食纤维在结肠中经过细菌发酵产生丁酸、短链脂肪酸(SCFA)和组蛋白脱乙酰酶抑制剂,抑制结直肠癌细胞的体外生长。
另外,谨慎饮食评分与结直肠癌组织中具核梭杆菌Fusobacterium nucleatum 的存在呈负相关,支持肠道微生物群介导富含纤维饮食与结直肠癌之间联系的假设。
在一项研究中显示,对地中海饮食的依从性提高与明显增加粪便SCFAs的水平有关。
相比之下,晚期结直肠癌患者的膳食纤维摄入量低于健康对照组,SCFA生成量也低于健康对照组,这些变化与条件致病菌增加和粪便样本中SCFA生成菌的水平降低有关。
在预防和治疗癌症的道路上,肠道菌群的角色已渐渐从微不足道转向不可或缺的重要力量。
流行病学文献中关于饮食模式和癌症的研究越来越多,新的饮食模式正在迅速发展。
目前一些饮食模式,例如生酮饮食或操纵宏营养成分,在动物模型中比在人类预防癌症方面得到更广泛的研究,现在需要更多的人类观察研究来为饮食指南提供依据。
大规模流行病学研究提及关于癌症的几个危险因素,其中包括日常能做到改变的饮食模式、生活方式等。如果能根据这些易感因素(例如家族史、性别、年龄、其他生活方式因素或同居关系、代谢组学特征或基于微生物的特征)为人群定制个性化的饮食建议,对于推动这一领域向前发展将是非常重要的。
幸运的是,我们也在做同样的事~
最后,希望阅读此文的你,注意饮食习惯,有个健康的身体~
相关阅读
参 考 文 献
Steck, S.E., Murphy, E.A. Dietary patterns and cancer risk. Nat Rev Cancer (2019) doi:10.1038/s41568-019-0227-4