 国家高新企业 | ISO9001认证
国家高新企业 | ISO9001认证 二级病原微生物安全实验室
二级病原微生物安全实验室- 联系电话:+13336028502
- +400-161-1580
- service@guheinfo.com
国家高新企业 | ISO9001认证 二级病原微生物安全实验室

谷禾健康
新冠肺炎是一种由新型冠状病毒(SARS-CoV-2)引起的呼吸道疾病,截至2020年5月1日,全球已有330多万人感染,致病和死亡率高,人类生命受到了极大威胁。近日,来自香港中文大学医学院Siew C. NG教授及其团队在国际顶级期刊《Gastroenterology》发表的一篇题为:Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization (COVID-19患者住院期间的肠道菌群变化),揭示了COVID-19患者住院期间的肠道菌群特征,及其与疾病程度和粪便病毒载量的关联。虽然此项研究样本量有限,而且只包括中/重度疾病的住院患者,但是研究人员认为这些发现表明个体的肠道微生物组结构可能会影响宿主对SARS-CoV-2感染的敏感性和反应。这些数据突出了一个新的概念,即新颖和有针对性的肠道菌群调节方法可能代表着新冠肺炎及其共病的一条治疗途径。
摘要
虽然知道SARS-CoV-2可以感染胃肠道组织,但关于肠道微生物对感染SARS-CoV-2的易感性和感染严重程度的作用却知之甚少。在本文中,研究人员对15名新冠肺炎患者的粪便样本进行了宏基因组测序分析,并比较了6名社区获得性肺炎患者和15名健康对照的微生物数据,用以调查新冠肺炎患者住院期间的粪便微生物群的变化以及感染严重程度与病毒的粪便排出之间的关联。研究人员发现15名新冠肺炎患者与对照组相比,新冠肺炎患者在入院时和住院期间各时间点的粪便微生物群均有明显变化,主要表现为条件致病菌增多,有益共生菌减少。即使清除了SARS-CoV-2(通过咽拭子确定)和呼吸道症状缓解后,耗竭的共生菌和肠道菌群失调仍然存在。就结果而言,粪便微生物群的改变与粪便中SARS-CoV-2的含量和新冠肺炎的严重程度相关。疾病程度与基线时的粪芽孢菌属、多枝梭菌、哈氏梭菌丰度正相关,与普氏粪杆菌负相关;改变肠道菌群的策略可能会降低疾病的严重程度。

背景
来自武汉的早期报告显示,2-10%的新冠肺炎患者有胃肠道(GI)症状,包括腹泻,但最近的一项Meta分析报告称,高达20%的患者有胃肠道症状。研究发现,在近50%的新冠肺炎患者的肛拭子和粪便样本中都检测到了SARS-CoV-2病毒,这表明消化道可能是病毒复制和活动的肺外部位。此外,有研究证明SARS-CoV-2利用血管紧张素转换酶2(ACE2)受体进入宿主,该受体在呼吸道和胃肠道都高度表达。ACE2在控制肠道炎症和肠道微生物生态中起重要作用。肠道中的共生微生物生态系统是动态的,可以受入侵的病毒调节以促进刺激或抑制反应。研究表明,呼吸道病毒感染可能与肠道微生物群改变有关,这使患者容易受到继发性细菌感染。最近对支气管肺泡灌洗液的宏转录组测序显示,SARS-CoV-2感染患者的微生物群以病原菌或口腔和上呼吸道共生菌为主。了解SARS-CoV-2感染背后的宿主微生物扰动,这可能会影响对感染的应对和未来各种免疫干预措施(如疫苗)的有效性。
实验设计
1.受试者筛选
研究涉及15名经实验室确诊感染SARS-CoV-2并因此住院的新冠肺炎患者和6名社区获得性肺炎住院患者(肺炎对照)以及15名健康人(健康对照)。
其中:
新冠肺炎患者是指对RdRp基因的不同区域进行连续两次RT-PCR检测确认。肺炎对照是指在两份呼吸道样本中SARS-CoV-2的PCR检测结果均为阴性。健康对照是指在过去3个月中没有病史或抗生素摄入史的个体,且SARS-CoV-2的PCR检测结果为阴性。
2.粪便样本采集
新冠肺炎患者每周连续采集粪便标本2~3次,直至出院。
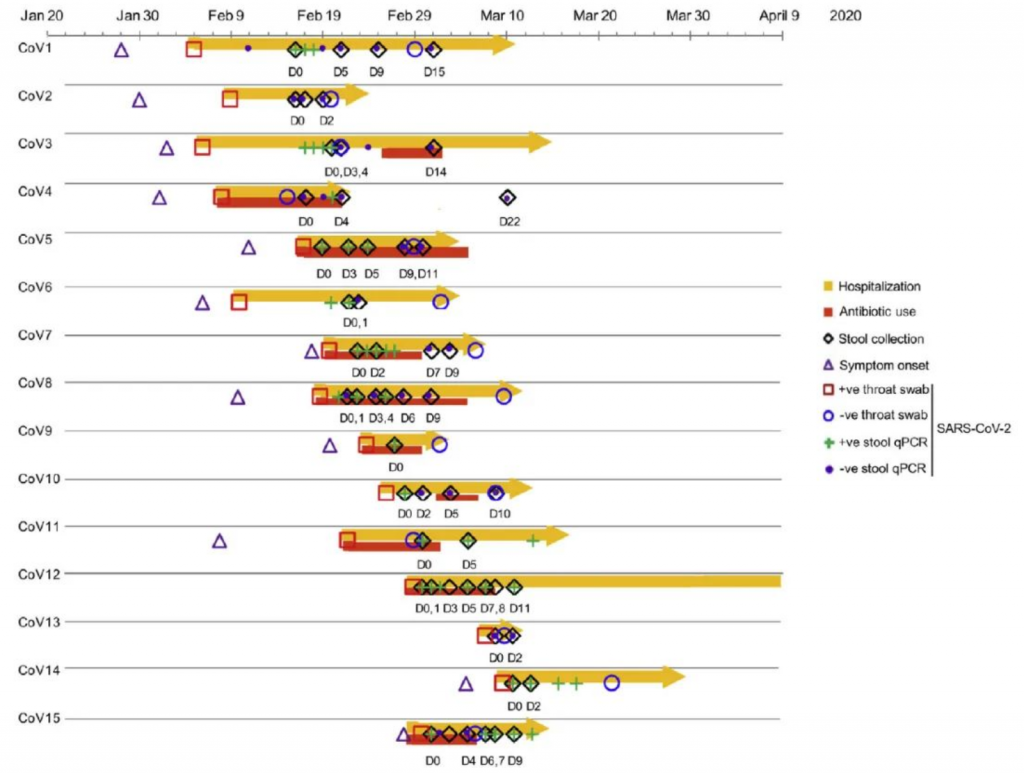

“Cov”指新冠肺炎患者;“D0”表示住院后收集第一次大便的基线日期,以下以“D”开始的时间点表示自基线收集粪便后的天数;“+ve throat swab”表示鼻咽/咽喉/混合拭子检测中首次出现SARS-CoV-2病毒阳性结果;“-ve throat swab”表示在连续两次鼻咽/咽喉/混合拭子检测中首次出现SARS-CoV-2病毒阴性结果,患者随后出院。“Symptom onset”表示首次出现症状;
3.样本分析手段
利用RT-PCR检测技术对粪便样品中的SARS-CoV-2定量。再利用宏基因组测序技术对粪便样品进行微生物分析。
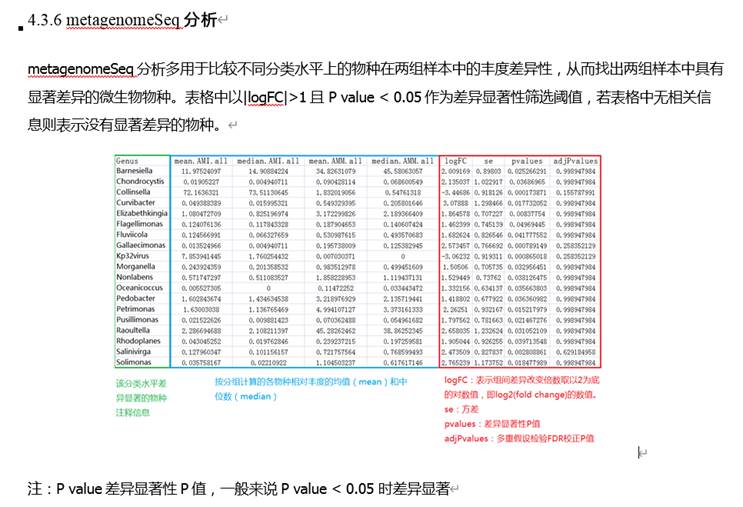
主要结果
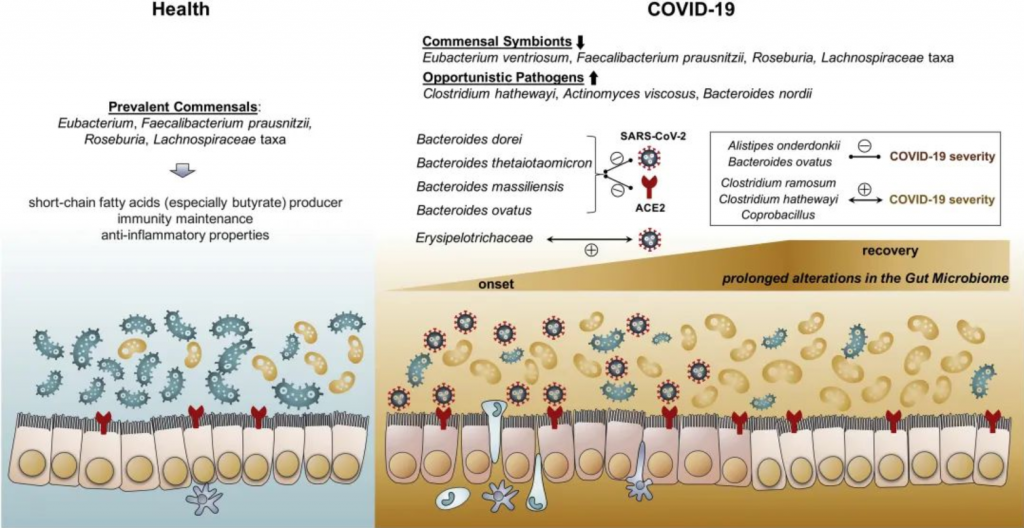
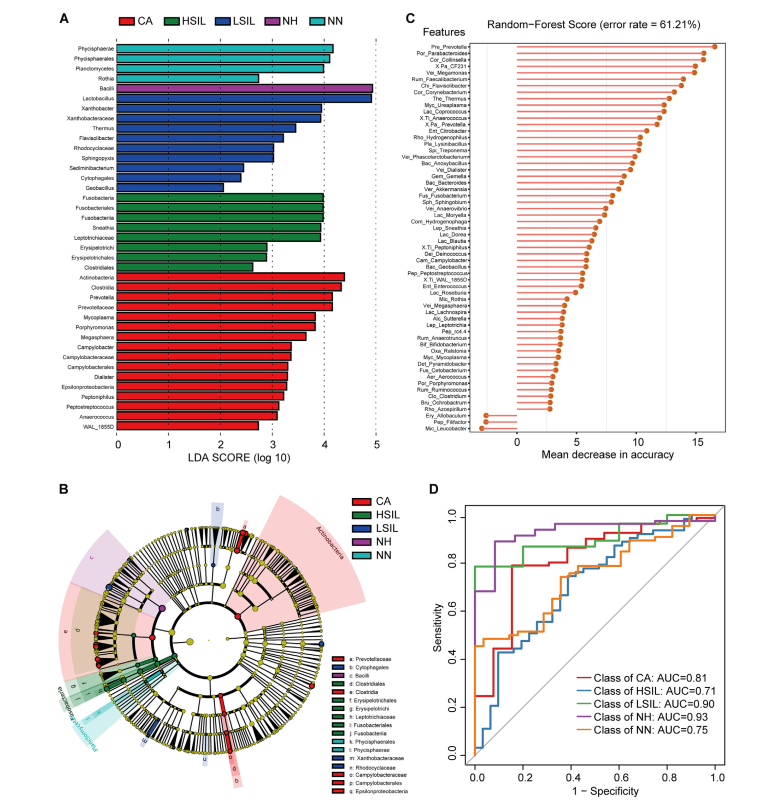
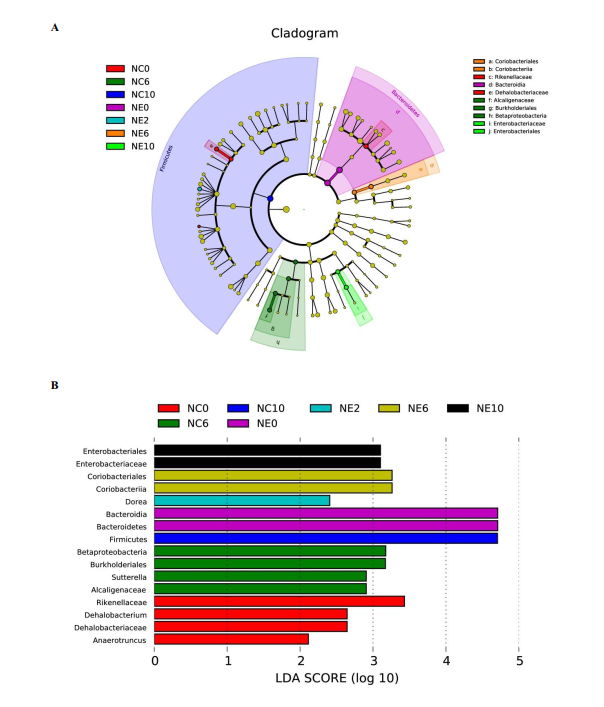
1. 新冠肺炎患者肠道菌群变化及病程纵向变化。新冠肺炎患者肠道菌群的特点是条件致病菌富集,而有益共生菌减少。并且这种菌群失调在新冠肺炎病程中持续存在,即便是在SARS-CoV-2清除/恢复之后。
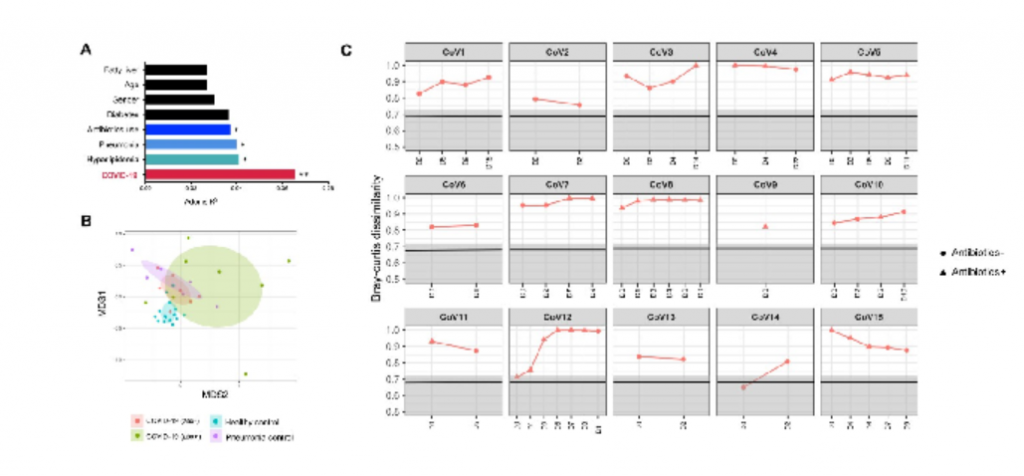

15名新冠肺炎患者中,7名未使用抗生素药物[COVID-19(abx-)],8名在基线(定义为入院后第一次粪便采集之日)接受经验性抗生素使用[COVID-19(abx+)]
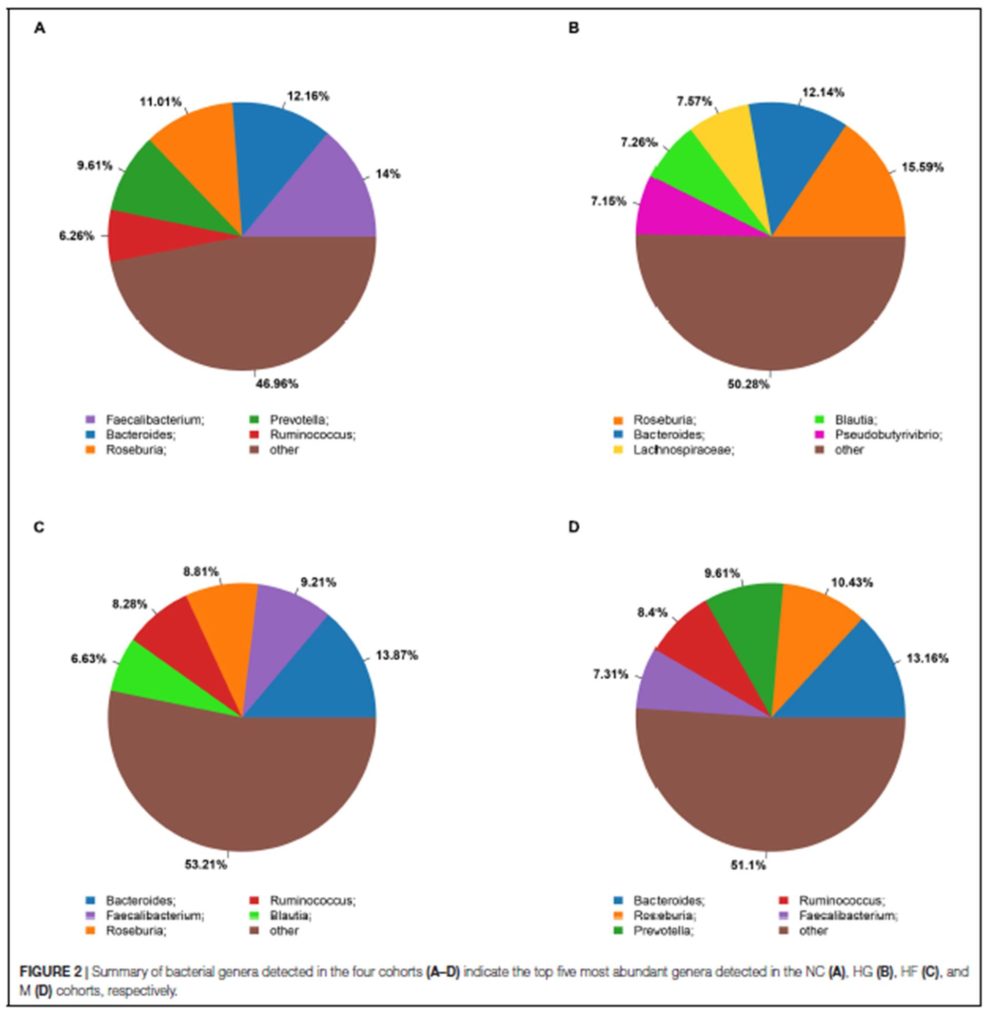
A).通过PERMANOVA检验确定脂肪肝、年龄、性别、糖尿病、抗生素使用、高脂血症在肠道菌群组成中的影响大小。结果表示新冠肺炎感染对肠道菌群组成的影响最大(P=0.002,** p<0.01,* p<0.05)
B). 基于Bray-Curtis相异度的NMDS(非度量多维尺度)图观察COVID-19微生物群落的变化。从整个微生物群落水平看,健康对照组和COVID-19(abx-)的粪便微生物群各自聚集,且COVID-19(abx-)的微生物群落更具异质性。新冠肺炎患者伴随着抗生素的使用,其微生物群落组成与健康对照组差异越来越大。
C).新冠肺炎患者在病程中的肠道菌群与健康对照组的不同。Bray-Curtis计算。灰色区域表示健康对照者肠道菌群的Bray-Curtis差异范围,黑色实线表示健康个体之间的中位数差异。“圆点”表示使用了抗生素,“三角形”表示未使用抗生素。总体而言,所有新冠肺炎患者的肠道菌群都保持着稳定,但无论是在疾病进展中还是在SARS-CoV-2清除后,其肠道菌群都与健康对照组明显不同。值得注意的是,患者CoV4在第5天出院,但他在第22天的肠道微生物群与健康人还是持续不同。
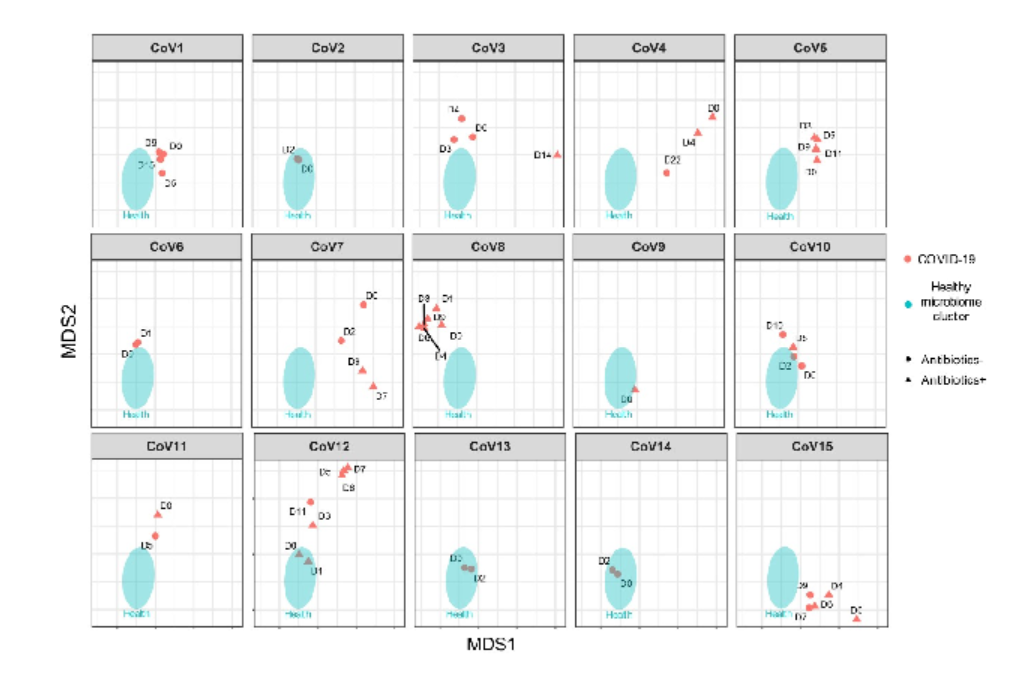

上图所示5名新冠肺炎患者的微生物群(CoV1,4,7,11,15)随着时间的推移显示出与健康微生物群更接近,而患者CoV3,5,8,10和12的微生物群随着时间的推移与健康微生物群变得更加不同
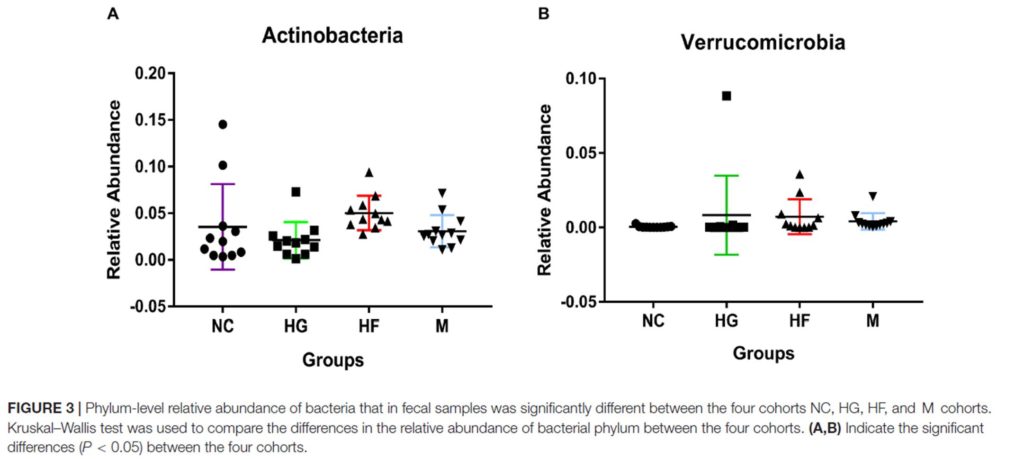
2.肠道菌群与新冠肺炎病情严重程度的相关性。肠道厚壁菌门细菌与COVID-19严重程度的相关性突出表明,细菌成员在调节人类对SARS-CoV2感染的反应和感染严重程度方面具有潜在作用。
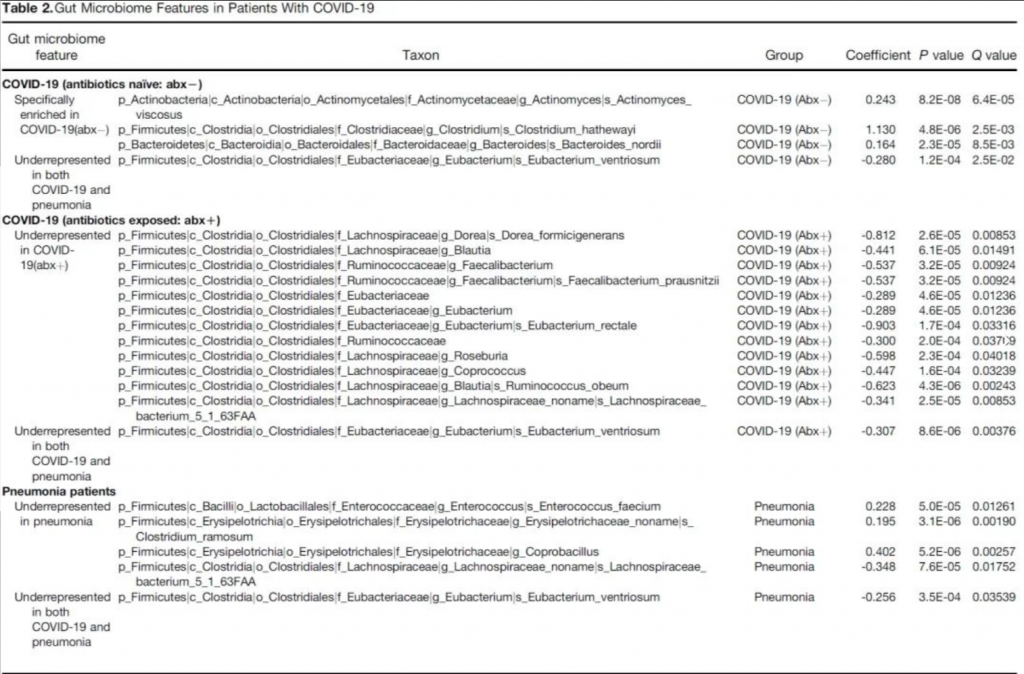

总共发现23个细菌分类群与新冠肺炎病的严重程度密切相关,其中大部分(15个)来自厚壁菌门。与新冠肺炎疾病严重程度最具正相关的细菌有三种,分别为来自厚壁菌门的Coprobacillus菌属,Clostridium
ramosum和Clostridium hathewayi菌种,且Coprobacillus菌已被证明能显著上调ACE2在小鼠结肠中的表达。与此相反,两种最常见的益菌Alistipes onderdonkii和Faecalibacterium
prausnitzii,它们与新冠肺炎的疾病严重程度呈负相关。“correlation coefficient
Rho”表示Spearman相关系数。
3.拟杆菌物种可能通过阻碍SARS-CoV-2通过ACE2进入宿主,而在抗击SARS-CoV-2感染方面具有潜在的保护作用。肠道Erysipelotrichaceae科可能在增强宿主肠道感染SARS-CoV-2方面发挥作用。
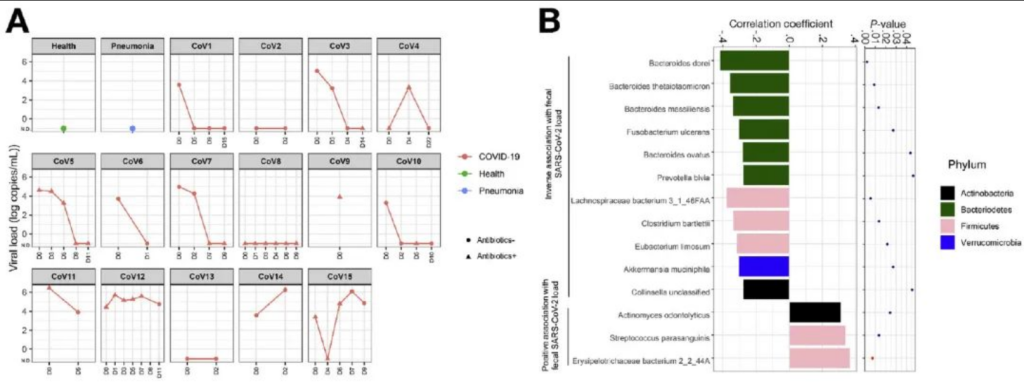

A). 新冠肺炎患者粪便中SARS-CoV-2病毒含量的纵向变化。15名患者中有11名患者住院期间在粪便中检测到SARS-CoV-2核酸,其中5名患者体内的SARS-CoV-2病毒随着时间的推移被清除了。
B).病程期间细菌与粪便病毒含量的Spearman相关分析。在所有粪便样本中,共有14种细菌与粪便中的SARS-CoV-2病毒含量显著相关。4种拟杆菌种包括Bacteroides dorei、Bacteroides thetaiotaomicron , Bacteroides massiliensis和Bacteroides ovatus,与粪便中SARS-CoV-2含量呈显著呈负相关。值得一提的是,这4种物种都与小鼠结肠中的ACE2表达下调有关。Erysipelotrichaceae bacterium
2_2_44A菌种与粪便中SARS-CoV-2含量呈显著正相关。一些研究表明Erysipelotrichaceae科与胃肠道炎症疾病有关。
结论
综上所述,研究发现尽管清除了SARS-CoV-2病毒,但大多数患者仍然在COVID-19中损失了有益的物种,这表明接触SARS-CoV-2感染和/或住院治疗可能对肠道微生物群有更持久的有害影响。在新冠肺炎患者肠道中发现的致病菌中,Clostridium hathewayi的基线丰度越高,新冠肺炎病情越重。而以带有能够抑制结肠ACE2表达的几种拟杆菌与SARS-CoV-2粪便病毒含量呈负相关。这些发现表明个体的肠道微生物组结构可能会影响宿主对SARS-CoV-2感染的敏感性和反应。但此项研究仍具有局限性,研究人员认为最主要的问题是样本量有限,且由于实验设计中只包括中/重度疾病的住院患者,这些发现可能并不适用于所有的新冠肺炎病例,包括轻度或无症状的新冠肺炎。而利用住院后收集的粪便样本进行的微生物群分析并不能代表新冠肺炎发病时真实的基线微生物群,也不能代表发病前的基线微生物群。但研究人员表示他们的研究提供了新冠肺炎会导致长期肠道菌群失调的证据及其与粪便中SARS-CoV-2病毒含量和疾病严重程度的关系。这些数据突出了一个新的概念,即新颖和有针对性的肠道菌群调节方法可能代表着新冠肺炎及其共病的一条治疗途径。
TIPs:
轻度,如果没有肺炎的影像学证据
中度,如果肺炎伴随发烧和呼吸道症状出现
严重,如果呼吸频率≥30次/分钟,呼吸环境空气时血氧饱和度≤93%,或动脉血氧分压/氧体积分数≤300mmHg(1mmHg=0.133kPa)
危重,如果休克或呼吸衰竭需要机械通气或器官衰竭需要重症监护。

近日,美国加州理工的一项研究表明,当防止啮齿动物食用粪便时,它们的小肠微生物群与人类肠道中的微生物群落更为相似。
越来越多的人研究表明肠道微生物群参与人身心健康的许多方面,因为大多数营养和药物都是在这个位置被身体吸收的。为了研究肠道微生物群,研究人员通常使用小鼠和大鼠,因为这些动物很容易照顾、繁殖迅速,并且与人类有许多生物学上的相似之处。 但人类和这些动物之间存在着显著的差异。 其中一个差异-实验室啮齿动物吃自己粪便的倾向-可能对与小肠微生物群有关的研究有重大影响。
近日,发表在“Microbiome”杂志上的一项研究中,RustemI smagilov实验室的研究人员发现标准实验室小鼠(消耗粪便)在其小肠微生物群落中与人类相比可能有重要的差异。
研究界几十年来一直意识到实验室啮齿动物消耗粪便,这是一种被称为共养的做法,但尚未理解的是,这种活动实际上是如何影响小肠内的条件的。大多数研究人员通过将老鼠安置在允许粪便通过电线的地板上来解决共生问题。 然而,老鼠和老鼠非常擅长吃便便,因为它出来了,因此研究人员怀疑电线地板实际上可能没有那么有效。
在这项研究中,进一步实验室小鼠小肠中的微生物群,以了解当这些小鼠被阻止食用自己的粪便时,微生物群及其功能是否不同,如果这样做会使这些实验室小鼠更类似于人类。为了找出答案,研究小组给老鼠安装了“尾巴杯”-基本上是小老鼠尿布,捕捉动物的繁殖力,防止它们吃。研究人员分析这些尿布症小鼠的肠道含量和微生物群落时,他们发现与标准小鼠相比,小肠的肠道存在显着差异。
Bogatyrev称通过共同预防将粪便细菌带入小肠的过程是“自我重新接种”。通过消耗它们自己的粪便,小鼠将细菌从大肠重新引入小肠,并改变上消化道的状况和微生物群落。
遗憾的是,该研究中Bogatyrev博士和他的同事并未试图确定自我再接种一般如何影响涉及小鼠的研究,但他们怀疑,如果小鼠模型的消化系统的行为不像人类的消化系统,那么可能会对许多研究领域产生广泛的影响。
但是研究人员指出:“一个领域可能是饮食研究。” “如果小肠中有更多微生物,反过来会影响那里的胆汁酸成分,饮食中的营养成分可能会被不同地吸收。例如,脂肪。另一个区域可能涉及益生菌和肠道微生物生态自我再接种可能会在控制给药方案中导致不一致的结果,因为您不知道动物本身如何将益生菌重新引入肠道。
此外,对共同预防至关重要的另一个大领域可能是药物研究。研究人员在临床前模型中使用啮齿动物,所施用的药物通常被小肠吸收,在那里它们可能会受到小肠微生物群的影响。这项工作表明,自我再接种的效果需要经过严格控制排除,这将为未来肠道微生物研究带来很多新发现和机会。
参考文献
1. Said R. Bogatyrev, Justin C. Rolando, Rustem F. Ismagilov. Self-reinoculation with fecal flora changes microbiota density and composition leading to an altered bile-acid profile in the mouse small intestine. Microbiome, 2020; 8 (1) DOI: 10.1186/s40168-020-0785-4
2. California Institute of Technology. “Making the mouse gut microbiome more human-like.” ScienceDaily. ScienceDaily, 13 February 2020.

谷禾健康 原创


孩子出现偏瘦小,不太会说话等状况有可能是发育迟缓的信号。
调查显示,有97%的家长不知道宝宝生长发育的基本规律;30%的家长即使发现宝宝出现生长发育迟缓时,也会盲目乐观;还有部分家长甚至认为发育迟缓太少见,根本不会发生在自己孩子身上。
但其实孩子出现发育迟缓的概率比我们想象的更高,关于目前发育迟缓的人数,我们来看一组最新的统计数据:


也就是说,世界上有大约四分之一的五岁以下儿童受发育迟缓的困扰。
此外,儿童营养状况与死亡率的变化也有着密切的关系。据世界卫生组织报告,全球5岁以下儿童死亡归因于营养不良的比例达35%,急性重度营养不良儿童的死亡风险是非营养不良儿童的9倍。
那么发育迟缓究竟是什么?会带来什么后果?所谓的最关键时期是哪个阶段?与哪些因素有关呢?
发育迟缓/矮小是指在生长发育过程中出现速度放慢或是顺序异常等现象。生长发育迟缓表现往往是多方面的,多有体格发育、运动发育及智力发育落后但也可以某一方面为突出表现。
如果家长不引起重视,有可能导致错过了最关键的时期。
《中国0-6岁儿童营养发展报告》指出:儿童早期特别是从胎儿期到出生后2岁(生命早期1000天),是决定其一生营养与健康状况最关键时期。
在这个关键时期的营养不良,可能导致儿童不可逆转的生长和认知发育迟缓,影响智力潜能的发挥,降低学习能力和成年后的劳动生产能力,导致成年后患肥胖、高血压、冠心病、糖尿病等诸多慢性疾病的风险增大。
判断营养状况指标:
MAZ——微生物群 / 年龄 Z评分
HAZ(LAZ)——身高 / 年龄 Z评分
WHZ——体重 / 身高 Z评分
WAZ——体重 / 年龄 Z评分
世界卫生组织定义的生长发育标准:
发育迟缓:年龄在0-59个月的儿童中低于身高/年龄中位数两个标准差的百分比,HAZ ≤ -2.
消瘦:年龄在0-59个月的儿童中低于体重/身高中位数两个标准差的百分比,WHZ ≤ -2.
严重消瘦:年龄在0-59个月的儿童中低于体重/身高中位数三个标准差的百分比,WHZ ≤ -3.
临床评估
母亲怀孕妊娠史、生产过程、新生儿健康指标、家族史、发育史、体格检查、神经系统检查等。
量表评估
从运动、语言、社会适应能力、社交能力等多维度评估。
另外还可以结合影像学评估、基因评估、代谢评估、肠道菌群评估等。
注:早期如需自行观察评估,可对照参考文末附录——发育健康表。
发育迟缓的原因有很多,牵涉到比如母体子宫内生长迟缓、神经内分泌和激素因素、儿童早期腹泻和其他感染频繁、环境肠道功能障碍、环境毒素和遗传因素等。
在20%到25%被认为“发育不良”的婴儿和儿童中,生长发育不良始于母体子宫内:早产和宫内生长受限,尤其是两者的结合,会使产后孩子发育迟缓的风险增加2倍到7倍。
母亲的健康程度、成熟度、经济和社会地位在LBW发病中起着核心作用,并且伴随着孩子出生后的成长。易导致LBW和儿童发育迟缓的因素包括:
有中度或重度产妇营养不良、发育迟缓病史或早孕的病史;
妊娠体重增加不理想;
产妇吸烟;
不合适的婴儿喂养等。
肠道感染带来的影响包括明显的腹泻,以及一系列潜在的长期影响,如生长衰竭、认知障碍和后来的生活代谢综合征,这些可能伴随着‘无症状’(指没有明显的腹泻)或症状性肠道感染。
除了腹泻与认知受损的潜在关联之外,平克顿等人在巴西东北部的研究也报告了腹泻对生长的显著影响。Kvestad等人同时也描述了北印度儿童腹泻与独立于生长发育的认知障碍的关系。
导致这种感染的几种特定的肠道原生动物和细菌病原体包括肠凝性大肠杆菌、隐孢子虫、弯曲杆菌和贾第虫。
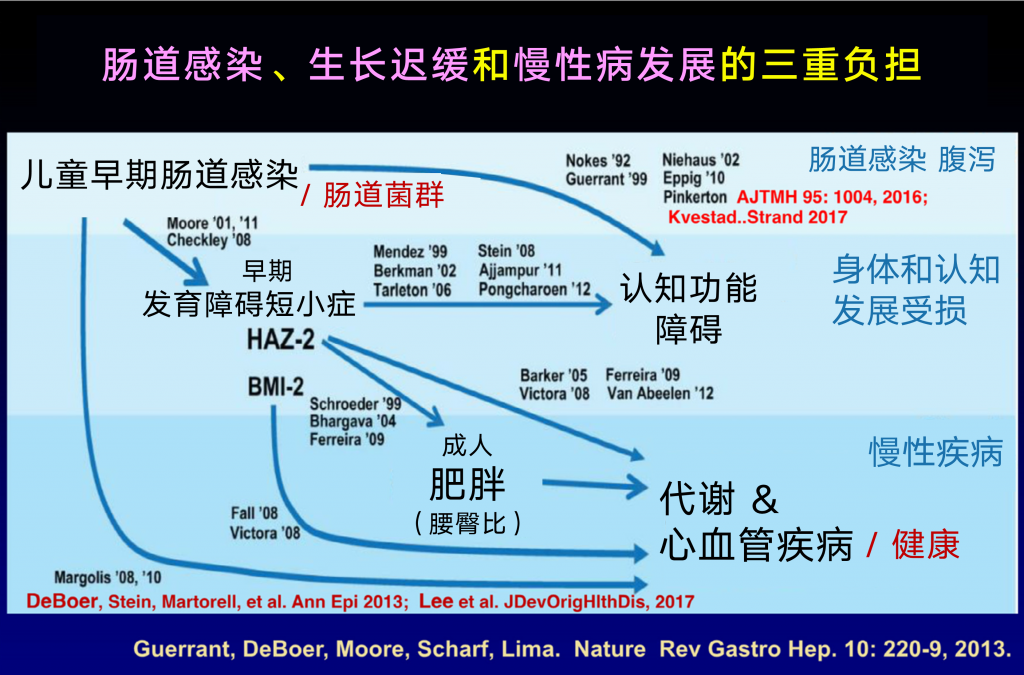

肠道感染,腹泻等状况不能忽视,有可能发展为发育迟缓,从而损害认知功能,甚至伴随着肥胖和慢性病的风险。
即使在没有腹泻的情况下,小肠对碳水化合物和α1-抗胰蛋白酶的通透性也会增加,营养素吸收不良。 许多患有环境肠道功能障碍的儿童也缺乏被小肠吸收的微量营养素,如铁和锌,如果耗尽,会降低食欲、减少绒毛表面积和胃肠吸收能力。
小肠炎症与C反应蛋白水平高有关,伴随细胞因子的释放,从而降低食欲和食物摄入量,并阻碍软骨细胞生长因子的产生和作用。发育迟缓儿童白细胞介素6(IL-6)水平升高,锌的摄入不足和吸收不良也可能减弱线性生长。因此适当进行锌的补充也是有必要的,具体关于食物的干预将在后面的章节详细阐述。
小A —— 1岁,发育迟缓
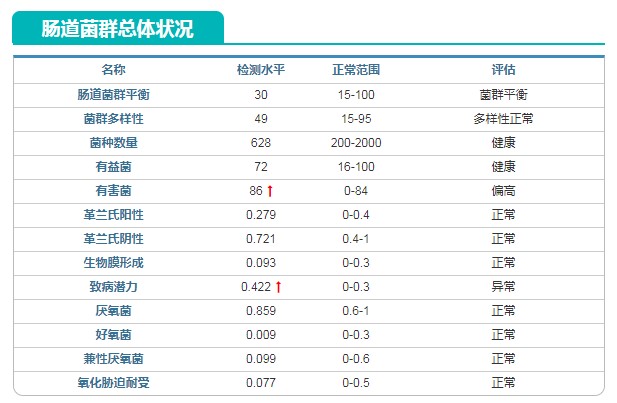
——本案例数据取自我们的肠道菌群健康检测数据【谷禾健康数据库】
对应的和发育迟缓相关的最主要因素从疾病风险中可以看到,肠炎和过敏性腹泻提示较高风险。其中过敏性腹泻是导致营养不良的很重要因素,提示存在较为严重的过敏情况,需要进行过敏原检查。
肠炎部分通过对照炎症指标发现降钙原素PCT和高敏C反应蛋白都超标,提示存在炎症情况。

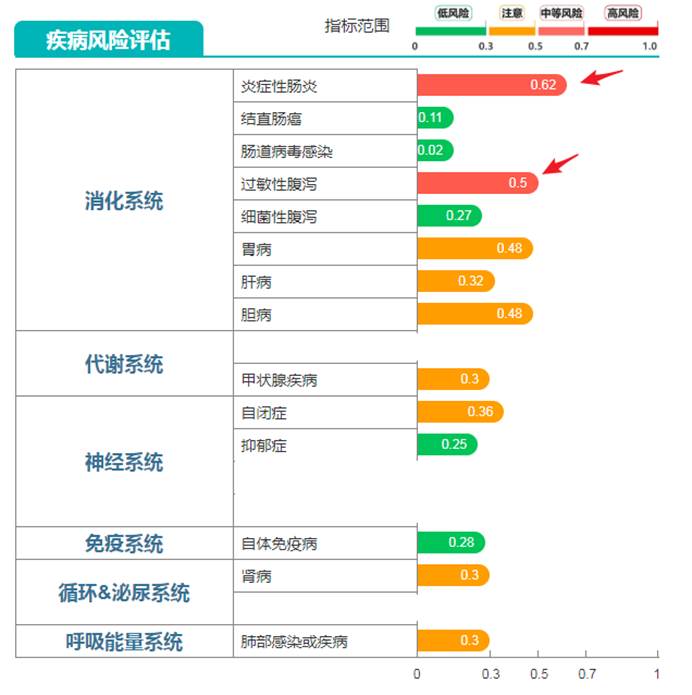
针对过敏的情况,检查肠道屏障和LPS等指标并未发现异常,表明免疫状况并未有异常,主要的可能是之前经历过病原物感染,且肠道炎症一直存在。
炎症本身也会改变肠道菌群,这在后面章节会详细阐述。
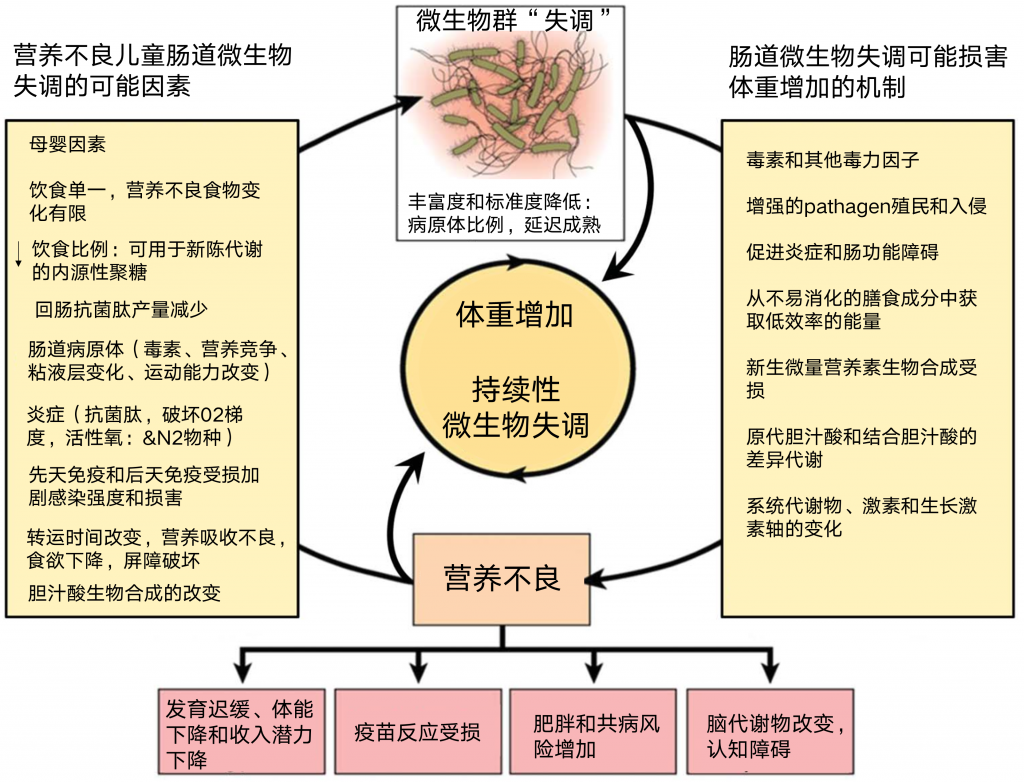
在婴儿期或幼儿期出现环境肠道功能障碍(EED)可能会放大宫内和围产期持续的生长缺陷。
环境肠道功能障碍(EED)是一种以绒毛萎缩和隐窝增生为特征的获得性小肠亚临床疾病,病因不明,可能占所有发育迟缓病例的40%以上。


在生活在不卫生条件下的发育迟缓儿童中普遍存在环境肠道功能障碍,与食品和水中的微生物和寄生虫污染有关。对患有环境肠道功能障碍的儿童进行的生物标记研究表明,存在亚临床炎症,这可能是由于暴露于病原体、饮食变化或微生物群的干扰所致。除了炎症,微生物引起的厌食症也可能是一个因素。
决定环境肠道功能障碍的因素
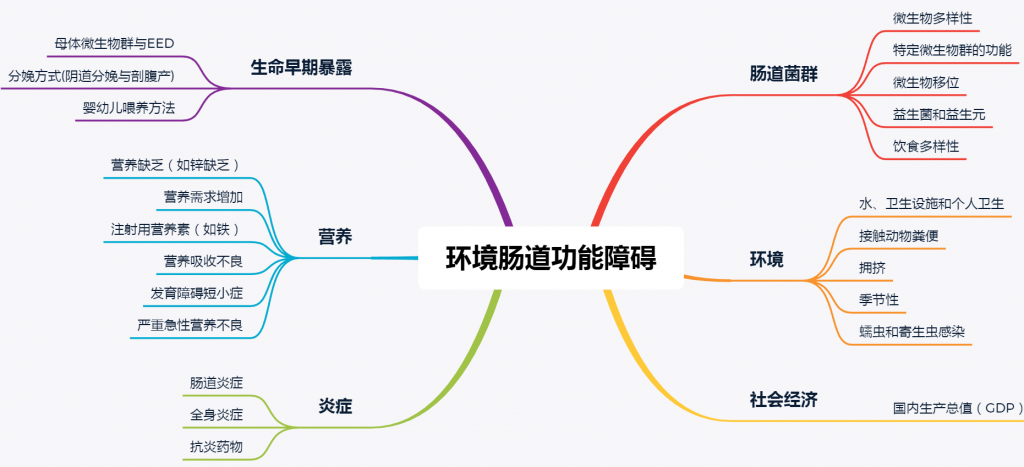

发育迟缓的儿童LAZ值从出生到18至24个月之间通常会下降到最低点,这可能是因为快速成长的婴儿和蹒跚学步的孩子特别容易受到营养、感染和有毒的环境毒害。
结合胎儿、围产期和产后早期的营养缺陷,环境肠道功能障碍可能会限制营养的传递和利用,从而损害小肠上皮细胞、肾单位、胰腺β细胞、骨骼肌细胞和生长板软骨细胞的成熟和增殖。
导致发育迟缓的因素可能包括肠道微生物群的不成熟和某些肠道微生物和/或母乳成分的缺乏,如唾液化低聚糖,这些低聚糖促进肠道屏障完整性、营养利用和组织合成代谢。
肠道微生物群代表了几十万亿个微生物,这些微生物在生命早期是从母体和其他来源获得的,被长期保留,并在生命的第一年经历一种生态演替。这些微生物对宿主生物学有广泛的影响,包括对膳食成分代谢、胆汁酸转化和保护肠道病原体的影响。 在没有腹泻的情况下接触微生物可能会对吸收上皮造成直接损害,从而损害热量的吸收。
最近的一项研究表明,贾第虫感染可能导致肠道异常通过直接破坏肠道微生物群。多项研究表明,一个不成熟的微生物群可能导致生长迟缓。尤其是,肠杆菌科的异常高患病率持续超过6个月的似乎与生长迟缓有关。已发表的研究表明,其中一些细菌可能直接损伤上皮。肠杆菌科以外的细菌,如链球菌,也可能直接造成伤害。越来越多的证据表明,不同的常驻微生物群对宿主既有有益的影响,也有有害的影响。
此外,微生物群培养免疫系统并诱导粘膜IgA的产生。
一项来自美国弗吉尼亚大学的研究,在271名0-24个月的出生队列中,每月对生长、腹泻发病率、疾病、病原体感染和抗生素接触情况进行评估。通过对6、12、18和24个月粪便样本进行16s rRNA测序,来量化肠道菌群多样性和特定菌群的丰度。
腹泻频率、持续时间和严重程度与细菌多样性和丰富性呈负相关(P<0.05)。
出生时发育不良的儿童(LAZ ≤ -2)在取样时也严重发育不良(LAZ ≤ -3),其腹泻相关的细菌多样性减少程度最大,腹泻后菌群多样性恢复最慢。菌群多样性的增加预示着6-18个月后腹泻的减少。
营养不良的儿童与非营养不良儿童的肠道菌群落有显著差异
我们来看一项来自印度南部对发育迟缓幼儿肠道微生物群的纵向分析:
该研究针对印度南部 每3个月至2岁出生队列中10名低出生体重和发育迟缓儿童(病例)和10名出生体重正常、无发育迟缓儿童(对照组)检测其肠道微生物群落组成和粪便微生物多样性。
发现所有儿童的多样性指数均随年龄增长而增加(P<0.0001),但发育不良的儿童的拟杆菌门相对丰度百分比高于对照组(P=0.043)。对照组儿童的微生物群中富含益生菌,包括双歧杆菌和粘膜乳杆菌,而发育不良儿童的微生物群中富含致病菌群,包括脱硫弧菌属和弯曲杆菌目。
在营养不良的情况下,特定细菌群的比例表达发生了改变——致病菌增加,有益菌减少
再来看一组87名6-59个月的儿童的粪便检测数据。
营养不良的两个队列的粪便微生物群落,包括肠杆菌、大肠杆菌、克雷伯氏菌和志贺氏菌在内的变形菌中致病菌群比例增加,在其他地方也已得到证实。不过需要注意与炎症性肠病的区分。
另一方面,在营养不良的肠道中潜在有益菌属被耗尽。在营养不良儿童肠道中缺乏罗斯氏菌、粪杆菌和丁酸梭菌(结肠细胞丁酸的重要来源)以及乳酸杆菌和双歧杆菌(在某些条件下可以减少炎症、增强肠道屏障功能、抑制病原体和介导其他有益作用)。
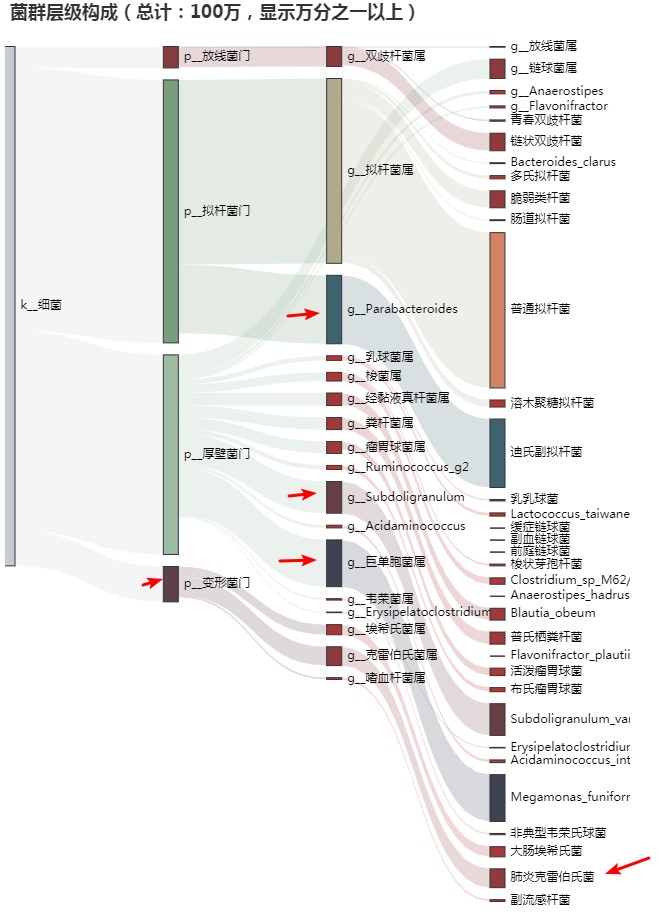
小A —— 1岁,发育迟缓
——本案例数据取自我们的肠道菌群健康检测数据【谷禾健康】
还是小A的报告,可以看到其有害菌比例偏高。

主要菌群中,正常菌以普通拟杆菌为主,此外他的双歧杆菌水平偏低一些。其他的菌均不是健康人的常见菌,且肺炎克雷伯氏菌也存在一定比例,需要对肠道菌群进行一定的干预。

中度或重度急性营养不良与肠道微生物群有因果关系,并与肠道微生物群的持续不成熟有关
美国华盛顿大学基因科学与系统生物学中心的Michelle Smith及其同事对非洲东南部内陆国家马拉维的317对双胞胎婴儿进行了研究,观察到3岁后发现,恶性营养不良(kwashiorkor)的病因除了营养摄入不足,还与肠道菌群有关。
为了验证肠道微生物群正常发育中的干扰与营养不良有因果关系的假设,研究人员首先将随机森林(RF)这一机器学习方法应用于16SrRNA数据集,该数据集是从健康马拉维婴儿和儿童在他们出生后的头三年内的粪便中连续采集的细菌。
基于该模型的指标MAZ用于定义营养不良程度不同的婴儿和儿童粪便微生物群的发育(成熟)状态。
将从6个月和18个月大的健康生长模式或不同程度营养不良的儿童身上获得的粪便样本移植到喂食代表性马拉维饮食的无细菌幼鼠体内。
结果发现:马拉维出生队列中营养不良的儿童肠道微生物群不成熟。与健康儿童的微生物群不同,未发育成熟的微生物群能将生长受损、骨形态改变和肌肉、肝脏和大脑中的代谢异常传递给无菌小鼠。
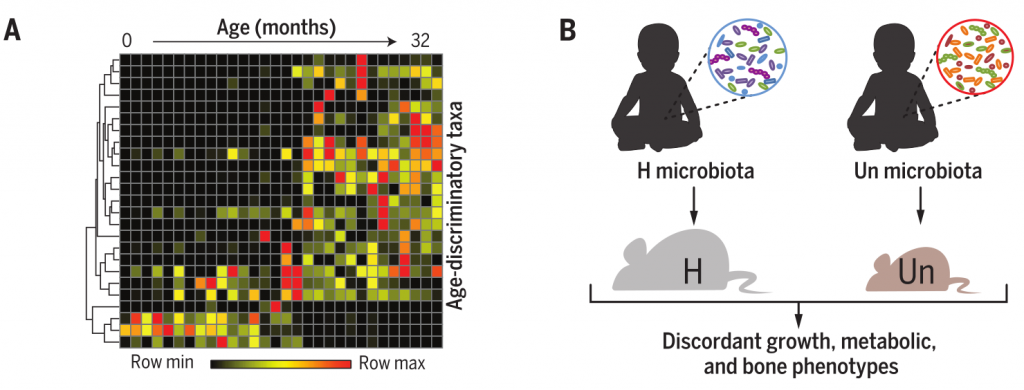

(A) 马拉维婴幼儿正常肠道微生物群落发育模型,基于25个细菌类群的相对丰度,它们提供了定义个体(粪便)微生物群“年龄”或成熟状态的微生物特征
(B)健康(H)或发育不良和体重不足(Un)的婴儿和儿童的粪便样本移植到喂养马拉维饮食的无菌幼鼠的单独组中。营养不良捐献者的未成熟微生物群向小鼠传递了受损的生长表型。
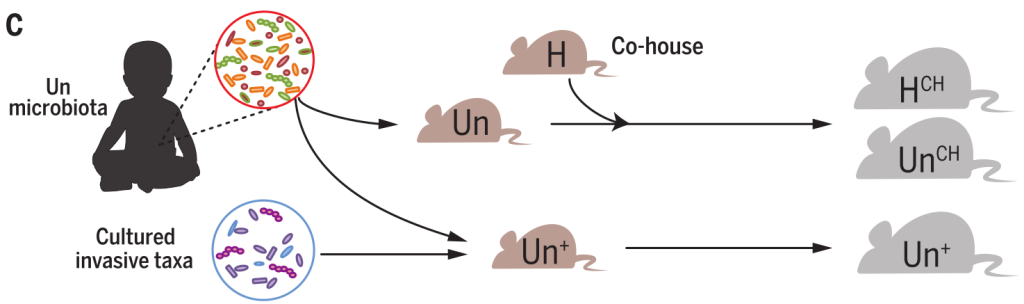

(C)不成熟的微生物群的其中一部分也有生长发育迟缓的证据。接受了6个月宝宝的健康或营养不良的菌群的小鼠们不久后混在一起同居,导致健康供体微生物群(HCH)中的菌群群侵入营养不良供体在受体动物中的菌群,并防止生长障碍。
肠道菌群的能量收获对宿主代谢有很大的贡献。其影响食物中能量的获取、新的微量营养素合成和胆汁酸的稳态。
菌群通过将不可消化的饮食成分转化为上皮细胞可能吸收的能量形式,微生物群对成人热量需求的估计为10%。然而,体重不足的印度儿童粪便缺乏发酵复杂植物低聚糖和肽聚糖的微生物基因。 同样,在新生小鼠蛋白质-能量营养不良模型中观察到盲肠和结肠细菌的丰度下降,以及它们能够代谢肽聚糖的基因丢失。因此,菌群失调会导致饮食中能量提取效率较低。
一些微生物可以通过从各种饮食或内源性来源(如尿素、氨)中回收氮来合成氨基酸,尽管这些微生物对个人总蛋白质需求的贡献程度尚不清楚。然而,氨基酸代谢是严重营养不良儿童和营养不良小鼠、大鼠和猪代谢组学分析中最严重干扰的生物途径之一。
同样,维生素k和水溶性b族维生素的一部分是由肠道菌群合成的。这些菌株的耗竭在理论上可能影响宿主的维生素状态。营养不良儿童中发现了大量的维生素缺乏症,和蛋白质-能量营养不良的多项研究中的代谢组学分析表明,维生素代谢途径中中间体的浓度发生了变化。
当甘氨酸或牛磺酸结合胆汁酸进入小肠时,其乳化特性促进了膳食脂类和脂溶性维生素的摄取,其抗菌性能调节肠道微生物群落。 一些细菌基因组编码增强其在胆汁中生存能力的酶。这些酶包括胆盐水解酶(BSH),将初级胆汁酸转化为次级胆汁酸。这些微生物活动可能对宿主生理产生深远的影响,包括胃肠道转运和脂质代谢。
姜黄通过增加共轭胆汁酸、牛磺去氧胆酸和牛磺-胆酸硫酸盐的浓度来减缓无菌小鼠的胃肠道转运。此外,克隆BSHs的肠道表达改变了血浆、肝脏和粪便中胆汁酸的浓度,影响了参与脂质代谢的宿主基因的转录,降低了血清胆固醇,肝脏甘油三酯和体重增加。
肠道微生物可以通过降低牛磺酸结合的β-鼠胆酸的浓度来影响回肠胆汁酸转运和代谢相关基因在肝脏中的表达,这是一种核芳烃类x受体拮抗剂。虽然目前尚不清楚改变的微生物种群在多大程度上导致儿童和儿童胆汁酸谱异常,但随着蛋白质-能量不足或环境肠道功能障碍的出现,改变的胆汁酸可能影响能量代谢、膳食脂肪和脂溶性维生素的吸收,并最终影响体重增加。
研究人员熟悉使用标准化生长曲线来评估儿童生长与正常轨迹相比的进展。 Planer博士指出,类似的方法可以应用于肠道微生物的发展,产生一个Z评分(MAZ)。 通过对三组健康孟加拉婴儿和儿童的粪便取样,验证了这一假设。
为了阐明哪些细菌是健康婴儿和儿童IgA反应的靶点,居住在圣路易斯地区的40对双胞胎和他们的母亲被招募为健康对照组,从这些母亲和儿童中获得多个粪便样本。结果表明,儿童IgA靶向性的成熟,24个月大时,儿童的模式与母亲的模式相似。
然后将该方法应用于恶性营养不良综合征(蛋白质缺乏)的马拉维双胞胎,数据显示,与健康的双胞胎相比,患恶性营养不良综合征的双胞胎肠杆菌具有很强的IgA靶向性。给药后,肠杆菌科的IgA靶向性降低。
此外,阿克曼菌(Akkermansia)是健康儿童微生物群中IgA的靶点。进一步的研究表明,从营养不良儿童的肠道微生物群中纯化的IgA阳性细菌群的转基因小鼠受体表现出肠道组织病理学,其特征是肠上皮屏障破坏和体重减轻;可通过从一个健康的微生物群中给药两种IgA靶向细菌来防止这些影响。

如上所述,随着儿童年龄的增长,肠道微生物群落存在着相当可重复的演替。普雷沃菌属在发展中国家正常儿童出生后的头五年中数量普遍增加。相比之下,肠杆菌科减少,是生命前六个月最丰富的。在最初的两年后,韦荣球菌属、链球菌和乳酸杆菌同样减少。
一项研究中对1300名中重度腹泻儿童的粪便中的微生物进行了分析,与来自1735名无腹泻对照儿童的样本相比。这些儿童年龄不到60个月,来自肯尼亚、冈比亚等。 研究表明,某些特定的菌群与生长不良有关。最引人注目的是链球菌和大肠杆菌与生长发育不良的关系。 此外,普雷沃菌属和粪杆菌属在所有年龄类别中都与良好的生长有关。在定植志贺氏菌严重的患者中,发现某些乳酸菌对腹泻有保护作用。唾液乳杆菌菌株在体外培养时抑制了所有志贺氏菌和腹泻大肠杆菌的生长。
此外,唾液乳杆菌的丰度升高与感染志贺氏菌时出现症状的风险之间有很强的相关性。具体来说,粪便中没有唾液乳杆菌的儿童有很高的感染志贺氏菌症状的风险;只要唾液乳杆菌存在,甚至丰度较低时,儿童也会受到保护,免受志贺氏菌引起的疾病。有趣的是,对照组粪便中唾液乳杆菌的水平并不影响微生物群中其他属的丰度,这表明可能产生直接抗菌作用。此外,乳酸菌对其他病原体有预防保护作用。
国际著名临床诊断实验室创始人Stephen Barrie博士对肠道失调定义为“具有有害影响的肠道菌群的生存状态”。
肠道失调有四种类型:腐败、发酵、缺陷和致敏
参考自:Stephen Barrie ND. Intestinal Dysbiosis and the Causes of Disease. healthy.net
一、 腐败
腐败失调是由于饮食中高脂肪和动物肉,低不溶性纤维。这种饮食结构会使粪便中拟杆菌的浓度升高,双歧杆菌的浓度降低。增加胆汁流量,诱导细菌脲酶活性。这种饮食引起的菌群动态变化主要发生在厌氧菌之间,但其影响可通过胆汁或尿胆素原中粪便pH值的升高(部分原因是氨生成量升高)和短链脂肪酸(特别是丁酸)的减少来衡量。
腐败失调有关的菌与疾病风险:
1、拟杆菌、变形杆菌和克雷伯氏菌中发现尿素酶,由高肉含量的饮食诱导,将尿素水解成氨,提高粪便pH值。相对较高的粪便pH值与结肠癌的发病率较高有关。
2、氨基酸的细菌脱羧作用产生血管活性和神经毒性胺,包括组胺、八胺、酪胺和色胺;这些胺通过门静脉循环吸收并在肝脏中脱氨。在严重的肝硬化中,它们进入系统循环,导致脑病和肝功能衰竭低血压。
3、细菌色氨酸酶将色氨酸降解为致癌酚类物质,与脲酶一样,是由高肉类饮食引起的。
4、细菌酶如β-葡萄糖醛酸酶能水解结合的雌激素和胆汁酸。肝脏结合和胆汁排泄是调节体内雌激素水平的重要机制。细菌去连接增加雌激素的肠肝循环。西方饮食会增加粪便中去结合酶的水平,降低粪便中的雌激素水平,提高血液和尿液中的雌激素水平,可能会导致乳腺癌的发生。
5、β-葡萄糖醛酸酶和其他水解细菌酶也能解结合胆汁酸。去结合胆汁酸对结肠上皮有毒并引起腹泻。它们或其代谢物似乎致癌,被认为有助于结肠癌和溃疡性结肠炎的发展。肠道菌群也会将原代胆汁酸(如胆酸和鹅脱氧胆酸盐)还原为次级胆汁酸(如脱氧胆酸(DCA)和石胆酸)。继发性胆汁酸的吸收效率低于原发性胆汁酸,更有可能导致结肠癌的发生。结肠癌的患病率与粪便中DCA的浓度成正比。
二、 发酵
这是由于内源性细菌在胃,小肠和盲肠中过度生长而引起的碳水化合物不耐症。小肠细菌过度生长的原因和影响已得到很好的表征。
胃酸过少,运动异常,手术盲环引起的淤滞,免疫缺陷或营养不良都会促进细菌的过度生长。小肠寄生虫病也可能导致细菌过度生长。小肠细菌过度生长引起的某些损害是由细菌蛋白酶的作用引起的,这些蛋白酶降解胰腺和肠道刷状缘,导致胰腺功能不全,粘膜损害和吸收不良。
在更严重的情况下,肠绒毛变钝变宽,单核细胞浸润固有层。细菌过度生长引起的内毒素血症会导致实验动物的肝损害。
胃细菌过度生长会增加全身感染的风险。胃细菌可以将饮食中的硝酸盐转化为亚硝酸盐和亚硝胺。因此,胃酸过少的人患胃癌的风险增加。小肠的一些细菌感染会增加肠的通透性。
碳水化合物不耐受可能是细菌过度生长的唯一症状,无法与肠道念珠菌病区分开。无论哪种情况,都可以将膳食糖发酵产生内源性乙醇。小肠长期暴露于乙醇本身可能会损害肠道通透性。糖的细菌发酵的另一产物是D-乳酸。小肠发酵可能是这些患者D-乳酸性酸中毒的原因。
根据治疗结果,研究肠道发酵综合症的英国医生初步得出结论,多数病例是由于酵母菌过度生长引起的,大约20%的细菌起源于细菌。症状包括腹胀,碳水化合物耐受不良,疲劳和认知功能受损。
三、 缺陷
缺乏接触抗生素或饮食中的可溶性纤维不足可能会导致正常粪便菌群(包括双歧杆菌,乳杆菌和大肠杆菌)的绝对缺乏。缺乏症和营养不良是相辅相成的疾病,通常一起发生并接受相同的治疗。
四、 致敏
对正常本地肠道菌群成分的异常免疫反应的加剧可能会导致炎症性肠病,脊椎关节病,其他结缔组织病和皮肤病(如牛皮癣或痤疮)的发病。负责任的细菌成分包括内毒素,它们可以激活替代补体途径和抗原,其中一些可能与哺乳动物抗原发生交叉反应。对强直性脊柱炎和炎症性肠病的治疗研究表明,致敏作用可以补充发酵过量,类似的治疗方法对这两种情况都可能有益。
临床研究表明细菌性营养不良与肠内或涉及皮肤或连接组织的多种炎症疾病有关。
炎症可能通过三种关键机制破坏微生物群。
首先,炎症触发免疫反应,将抗菌肽释放到肠腔;这种反应内在地防御病原体,但也可以针对共生微生物群。
第二,炎症增加腔内氧气水平。 氧气通常从粘膜毛细血管网络向管腔扩散,形成一个氧梯度,严格调节氧、微氧和缺氧区内的微生物。这一梯度有助于塑造微生物生态,粘膜表面附近有兼性厌氧菌,缺氧腔中有严格的厌氧菌,并影响细菌转录。炎症过程中腔氧的增加选择性地促进耐气微生物的生长,特别是肠杆菌科及其病原体的生长。
第三,炎症产生活性氧和氮物种,通过促进某些细菌之间的呼吸来塑造微生物群。活性氧与腔内硫化合物结合形成四硫酸氧化产物。 如斑疹伤寒杆菌等能利用四硫磷酸盐作为呼吸电子受体的细菌,比不能利用四硫磷酸盐的细菌具有选择性生长优势。鉴于炎症是环境肠道功能障碍的标志,这些机制可能有助于塑造营养不良儿童观察到的微生物失调。
在这个恶性循环中,肠道微生物群(通常含有来自不卫生环境的肠道病原体)的改变,会引发包括炎症、屏障功能障碍、易受病原体侵袭、转运改变等在内的肠道亚临床病理群,以及营养吸收不良。
这些病理学促进生长衰竭和持续性失调。当这种恶性循环出现在一个关键的早期发育期时,儿童的终身共病风险增加,包括身材矮小、健康和收入潜力下降、认知障碍、肥胖、2型糖尿病和心血管疾病。

饮食与营养
首先,从饮食习惯来说,宝宝在断奶后,食物供应情况和饮食传统在世界范围内各不相同。发展中地区的个人饮食习惯通常会选择食用富含复杂植物多糖的谷类和植物饮食,而不是西方饮食中的那些动物衍生食品和加工碳水化合物。
来自撒哈拉以南的非洲和欧洲的健康儿童之间的粪便微生物群落差异可能是饮食中碳水化合物的类型和数量的部分原因。与健康的意大利儿童相比,布基纳法索(非洲国家)健康儿童的粪便中含有更大比例的拟杆菌门,具有特定富集的属(普雷沃菌和木聚糖),它们含有代谢不可消化的膳食纤维素和木聚糖的酶,这是布基纳法索饮食中的关键成分。因此,一个培养物的饮食碳水化合物成分可以驱动含有基因组库的细菌的选择,将这些营养物质作为能量来源进行代谢。
小B —— 2岁,发育迟缓
——本案例数据取自我们的肠道菌群健康检测数据【谷禾健康数据库】
小B的报告,营养饮食部分提示的问题更大一些,首先主要营养中蛋白质摄入不足。
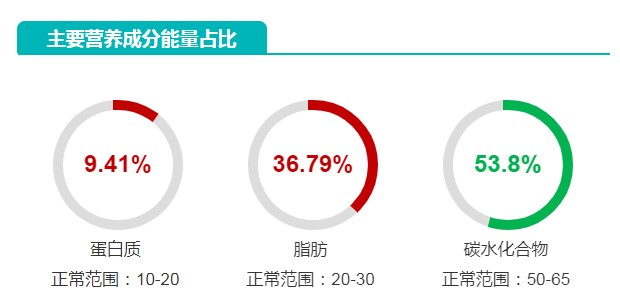

此外,膳食纤维摄入严重不足,这也是双歧杆菌和其他正常菌群占比不高而变形菌门占比很高的主要原因之一。顺带的也导致短链脂肪酸部分出现了两种缺乏。

发育迟缓的治疗充满困难。首先,在缺乏生物标志物的情况下,在单个儿童中识别环境肠道功能障碍是有难度的。 此外,临床试验没有强有力的证据表明,特定的干预措施可以治愈或改善。不过好在我们发现有很多相关因素,可以着手干预加以改善。比如说上面的例子我们看到营养饮食部分的问题(主要是三大类营养素),肠道菌群检测报告中的内容则可以提示我们再从更细微的层面去了解缺乏的状况,结合微量元素甚至代谢产物,更容易找到问题的核心所在。
为什么可以通过饮食干预?
从机理上讲,沿着肠道纵轴建立微生物群落是一个多因素甚至部分随机的过程,并在一定程度上受膳食碳水化合物的影响。
简单的碳水化合物被小肠吸收,留下不可消化的复合多糖,多糖是作为结肠微生物群的关键决定因素。膳食多糖、宿主和肠道微生物之间形成复杂的相互作用,它们代谢的多糖类型差异很大。例如,拟杆菌属含有大量的多糖降解酶,将抗性淀粉、植物细胞壁多糖、菊粉和纤维素代谢成短链脂肪酸和其他肠道可吸收的产物。
拟杆菌是结肠中动物源性糖蛋白最有效的降解菌之一,这就可以解释为什么该属在食用西方动物性饮食的个体中富集,而在食用植物性饮食的个体中富集较少,同时增加了这样一种可能性:如果缺乏拟杆菌,那么微生物群无法将不可消化的膳食成分转化为宿主可利用的能量形式,对生长产生不利影响。
母乳低聚糖(HMO)可以塑造婴儿肠道微生物群
HMO是有益菌的代谢基质。研究人员分析了88名马拉维母亲的母乳,与严重发育不良的6个月的婴儿相比,健康婴儿的母亲母乳中的总HMO、岩藻糖基HMO和唾液酸HMO浓度更高,在215名母亲的第二个队列中,健康婴儿和中度发育不良婴儿的母亲的母乳中总的HMO和唾液化的HMO浓度增加。作者又用小鼠模型将唾液酸化乳低聚糖与瘦体重增加联系起来。
不同膳食淀粉类型的影响
膳食淀粉酶抗性淀粉(RS)的健康益处来自肠道微生物发酵和短链脂肪酸(SCFA)的产生,研究人员比较了印度南部发育迟缓和非发育迟缓(“健康”)儿童的肠道发酵能力,使用两种类型的RS:高直链淀粉玉米淀粉(HAMS)和乙酰化HAMS(HAMSA)。
这两种类型的RS导致健康和发育迟缓的儿童粪便pH值显著降低,粪便中乙酸和丙酸增加,但健康儿童在服用HAMSA后的乙酸和丁酸明显高于发育不良儿童。这表明这些儿童的结肠生理发生了改变。HAMS能增加发育不良儿童的粪便丁酸,丁酸对结肠健康,特别是对结肠上皮细胞,非常重要。(这在我们之前的文章里也有详细讲过 你吃的膳食纤维对你有帮助吗) 。
可以看到发育不良儿童发酵某些类型RS的能力受损,这种发现为改善儿童发育迟缓的生理影响而补充RS类型的选择具有重要意义。RS干预被认为是可以改善肠道健康的一种方法。
腐败失调通常是通过高可溶性和不溶性纤维,低饱和脂肪和低动物蛋白的饮食来解决的。
乳制品


乳制品的影响是可变的。发酵乳制品,如新鲜酸奶有时会有所帮助。这些饮食变化有助于降低结肠中拟杆菌的浓度,增加乳酸产生菌(双歧杆菌、乳酸杆菌和乳酸链球菌)的浓度。
定期摄入酸奶可降低粪便中脲酶阳性菌和拟杆菌的浓度可能有助于致癌的RIA酶。发酵乳制品和嗜伊乳杆菌制剂被证明在治疗和预防沙门氏菌病、志贺氏菌病、抗生素引起的腹泻和抑制肿瘤生长方面是有用的。
纤维
在饮食中补充一定来源的纤维可以对结肠失调产生不同的影响。不溶性纤维降低细菌浓度和微生物酶活性。另一方面,可溶性纤维在提高有益的短链脂肪酸水平的同时,往往会提高细菌浓度和酶活性。这种差异可能解释了不溶性纤维在预防结肠癌方面的优越性。
低聚糖
在洋葱和芦笋等蔬菜中发现的含有果糖的低聚糖,已经发展成为一种食品补充剂,用于提高双歧杆菌的粪便水平和降低粪便pH值。在一些蔬菜中发现的低聚糖,特别是胡萝卜,能抑制肠道细菌与肠粘膜的结合。胡萝卜汁和浓缩胡萝卜低聚糖在欧洲用于细菌性腹泻已有近一个世纪。
其他
相比之下,在发酵失调中,淀粉和可溶性纤维可能会加剧肠道生态的异常。当上小肠受累时,单糖也是禁忌。不吃谷类食品和加糖的饮食通常是最有益的。水果、脂肪和淀粉类蔬菜在不同情况下都有不同程度的耐受性。
微生物群重塑可以很快帮助治疗儿童期的环境肠道功能障碍和营养不良。以代表性马拉维饮食为基础并由严重营养不良的马拉维儿童粪便微生物定植的侏儒小鼠中,通过将这些小鼠与接受健康微生物群的动物共住、通过共食促进微生物转移或通过灌胃五种菌群来改善生长障碍。
这就提示了我们可以从益生菌益生元的角度去思考干预措施。

在一项研究中,含有益生菌德氏乳杆菌保加利亚亚种、嗜热链球菌和干酪乳杆菌DN-114-001的发酵乳
改善营养不良小鼠的生长。益生菌混合剂也改善了肠道组织病理学和在营养不良状态下改变的各种免疫反应。
正如施瓦泽等人的一项研究所证明的那样,个别益生菌菌株也可以改善体重增加。无菌小鼠轻度体重不足与常规饲养的小鼠相比发育迟缓,胰岛素样生长因子-1(IGF-1)和IGF-1结合蛋白3的浓度降低。表明肠道微生物群通过生长激素轴促进生长。
长期以来,乳酸菌给药一直被用于改善肠道微生物生态。
双歧杆菌是结肠的主要乳酸菌,其浓度是乳杆菌的1000倍。人和动物服用短双歧杆菌可降低粪便中梭状芽孢杆菌和肠杆菌种类、氨和产毒细菌酶(包括β-葡萄糖醛酸酶和色氨酸酶)的浓度;尿液指标也会降低。在欧洲,为了改变肠道菌群而使用特定的大肠杆菌和肠球菌菌株已经很流行了。
侧孢芽孢杆菌(Bacillus laterosporus)是一种新的非致病性微生物,具有独特的代谢产物,具有抗菌、抗肿瘤和免疫调节活性。这种菌在美国作为食品补充剂已经有5年了。我们发现它是一种有效的辅助治疗方法,可以控制一些患者的小肠功能障碍相关症状。
几十年来,欧洲一直在使用酵母布拉氏酵母菌(Saccharomyces boulardii)来控制非特异性腹泻,这一点同样值得关注,而且研究得更为深入。布拉氏酵母菌最初是从印度荔枝果中分离出来的,在法国作为药物进行种植和包装。对照研究已证明其在预防抗生素相关性腹泻和艰难梭菌性结肠炎方面的有效性。
酿酒酵母(S. boulardii)还被证明可以刺激大鼠分泌性IgA的产生。患有反应性关节炎和其他疾病的患者应禁用此类免疫增强疗法,以防过度的肠道免疫反应疾病。
以代表性马拉维饮食喂养的无菌小鼠接受了发育不良的马拉维婴儿中分离的25个菌株群落(其中19个菌株成功定植)。与未添加益生元或添加菊粉的小鼠相比,添加唾液酸化牛乳低聚糖(S-BMO)的小鼠体重增加、消瘦改善和骨骼形态改善。然而研究人员同样给无菌小鼠添加S-BMO之后并没有增重效果,这就说明了其促进生长的作用机制是依赖菌群的。
抗生素药物可能导致或有助于控制失调,这取决于药物和失调的性质。如果厌氧菌污染小肠是问题所在,甲硝唑或四环素类药物可能是有益的。当肠道细菌过度生长占优势时,环丙沙星通常是首选药物,因为它倾向于保留厌氧菌。
草药抗生素可能是首选,在细菌过度生长综合征中需要长期的抗菌治疗,它们相对更安全。柑橘种子具有广谱的抗菌、抗真菌和抗原生动物作用,可能是理想的一线治疗手段。
草药药典列出了许多具有天然抗生物活性的物质,草药治疗肠道失调的潜力实际上是无限的。
其他潜在干预措施包括:
(1) 通过改善水、环境卫生和个人卫生等方案减少粪便接触和与动物接触;
(2) 母乳喂养和增强饮食多样性;
(3) 营养补充剂,包括锌、多不饱和脂肪酸和氨基酸;
(4) 抗炎剂,如5-氨基环酸;
(5) 急性营养不良和感染情况下的抗生素;
(6) 注意疫苗的使用,减毒活细菌疫苗可能以与非致病性亚临床感染相同的方式诱发生长迟缓;此类疫苗的临床试验应将生长作为一项结果指标。
考虑到可能导致生长障碍的饮食、环境和宿主因素的范围,在所有儿童营养不良的情况下,单一的治疗剂不太可能完全恢复微生物功能、全身代谢物以及微量和微量营养素的健康平衡。
从饮食、环境卫生条件等各方面去完善,结合肠道微生态的最新研究,特别是其发病机制和潜在原因,了解为什么某些菌群在某些个体中的代表性过高或过低,以及它们如何影响肠道健康。从这些研究中获得的信息来指导未来新疗法的发展。
相关阅读:
主要参考文献
Nataro J P, Guerrant R L. Chronic consequences on human health induced by microbial pathogens: Growth faltering among children in developing countries[J]. Vaccine, 2017, 35(49): 6807-6812.
Velly H, Britton R A, Preidis G A, et al. Mechanisms of cross-talk between the diet, the intestinal microbiome, and the undernourished host.[J]. Gut microbes, 2017, 8(2): 98-112.
Dinh D M, Ramadass B, Kattula D, et al. Longitudinal Analysis of the Intestinal Microbiota in Persistently Stunted Young Children in South India[J]. PLOS ONE, 2016, 11(5).
Hoffman D J, Camposponce M, Taddei C R, et al. Microbiome, growth retardation and metabolism: are they related?[J]. Annals of Human Biology, 2017, 44(3): 201-207.
Balamurugan R, Pugazhendhi S, Balachander G M, et al. Effect of native and acetylated dietary resistant starches on intestinal fermentative capacity of normal and stunted children in southern India[J]. International Journal of Environmental Research and Public Health, 2019, 16(20).
Mahfuz M, Das S, Mazumder R N, et al. Bangladesh Environmental Enteric Dysfunction (BEED) study: protocol for a community-based intervention study to validate non-invasive biomarkers of environmental enteric dysfunction[J]. BMJ Open, 2017, 7(8).
Blanton L V, Charbonneau M R, Salih T, et al. Gut bacteria that prevent growth impairments transmitted by microbiota from malnourished children[J]. Science, 2016, 351(6275).
Rouhani S, Griffin N W, Yori P P, et al. Diarrhea as a Potential Cause and Consequence of Reduced Gut Microbial Diversity Among Undernourished Children in Peru[J]. Clinical Infectious Diseases, 2019.
Harper K M, Mutasa M, Prendergast A J, et al. Environmental enteric dysfunction pathways and child stunting: A systematic review.[J]. PLOS Neglected Tropical Diseases, 2018, 12(1).
Owino V O, Ahmed T, Freemark M, et al. Environmental Enteric Dysfunction and Growth Failure/Stunting in Global Child Health[J]. Pediatrics, 2016, 138(6).

谷禾健康 原创
转移性肾细胞癌(RCC)长期以来被认为是一种“免疫性恶性肿瘤”,对不同的免疫疗法敏感。免疫检查点阻断疗法(ICB)的发展,使肾细胞癌(RCC)的临床预后发生了革命性的变化。虽然如此,但治疗响应的持续时间以及预测的改善仍旧没有满足医疗需求。虽然人们已经认识到抗生素(ATBs)会降低ICB在各种恶性肿瘤中的临床活性,但对于不同的肠道非致病菌(共生菌)对ICB在肾细胞癌中的疗效的直接影响却知之甚少。
近日,来自法国科学家的一项发表在《Eur Urol (欧洲泌尿学杂志)》期刊的一篇题为“Gut Bacteria Composition Drives Primary Resistance to CancerImmunotherapy in Renal Cell Carcinoma Patients” 研究发现:微生物群的组成受TKI和ATB的影响,并影响免疫治疗的成功, 未来研究人员表示在开始治疗前对粪便的分析可能可以指导临床医生用药,以防止肾细胞癌患者对免疫治疗的原发性耐药。
简称/名词介绍:
ORR:客观缓解率,指肿瘤缩小达到一定量并且保持一定时间的病人的比例,也就是在任何时候完全缓解和/或部分缓解的患者数量。
PFS:无进展生存期,指从随机分组开始到第一次肿瘤进展或死亡时间。
BOR:本文中,研究者评估的从nivolumab开始治疗之日到治疗结束的最佳肿瘤反应(完全缓解、部分缓解、疾病稳定或疾病进展)。
文章中使用RECIST疗效评价标准1.1评估肿瘤反应
完全缓解【complete response,CR】:除结节性疾病外,所有目标病灶完全消失或所有目标结节须缩小至正常大小(短轴<10mm)
部分缓解【partial response,PR】:所有可测量目标病灶的直径总和低于基线≥30%。目标结节总和使用短径,而所有其它目标病灶的总和使用最长直径。
疾病进展【progressive disease,PD】:可测量目标病灶的直径总和增大≥20%超过观察到的最小总和,如治疗期间未观察到总和降低,则定义为超过基线。
疾病稳定【stable disease,SD】:介于PR和PD之间。
摘要
文章的主要目的是评估在晚期肾细胞癌患者中,其肠道菌群组成对ICB疗效的预测价值。研究人员收集了69例晚期肾细胞癌患者的粪便样本和2994例健康志愿者的粪便样本。同时,研究人员还进行了临床前研究,将对ICB具有抗性的肾细胞癌患者粪便微生物移植到荷瘤小鼠上,以此证明了肠道细菌组成与ICB临床结果之间的因果关系。研究人员还评估了在开始nivolumab治疗前使用TKl对微生物群组成的影响。通过WGS-MG测序分析识别其微生物群组成。研究发现,近期抗生素使用可改变肠道菌群组成,并降低患者的客观缓解率,促进了不同优势物种类群的形成,如Clostridium hathewayi菌在肾细胞癌患者的粪便中较为富集。还有一个重要的点,就是在接受nivolumab药物前服用酪氨酸激酶抑制剂(TKI)能够改变肠道菌群的组成。最后,研究人员表示微生物群的组成受TKI和ATB的影响,并影响免疫治疗的成功。未来的研究将有助于提高这些特定细菌的作用,以及它们作为新的生物标志物的潜力。
实验设计
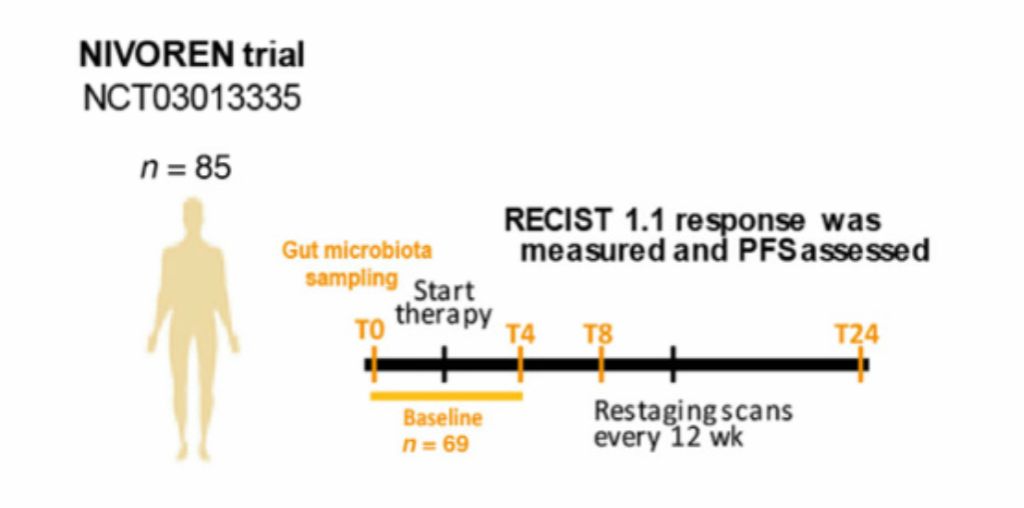
受试者纳入标准:
对IV期肾细胞癌患者和疾病进展的患者每2周注射3mg/kg的nivolumab(纳武单抗),直到疾病进展或在NIVOREN
GETUG-AFU 26第二阶段试验中出现无法忍受的毒性为止。
细节:
第一年在基线和每8-12周进行一次CT扫描,然后每12-15周进行一次CT扫描直到疾病进展。根据国际人体微生物组标准指南(SOP_03_V1) 进行粪便样本收集,在第一次注射nivolumab前(T0,<1mo);第二次(T4,4wk);第四次(T8,8wk);第12次(T24,24wk)
宏基因组学和统计学分析:
从粪便标本中提取总DNA,并按照MetaGene Polis(INRA)工作流程使用IonProton技术进行测序。从丰度矩阵开始,只考虑在所有样本中至少有20%存在的分类群,然后对原始数据进行归一化和标准化。监督偏最小二程判别分析(PLS-DA)和随后的变量对模型的重要性图(VIP)找出与BOR相关的组中的差异物种。双尾Mann-Whitney U和Kruskal-Wallis检验分别评估成对比较和多重比较的意义,p<0.05。计算用于网络相关分析的Pearson矩阵。所有的p值都经过FDR矫正。
主要结果
1. ATBs影响了ICBs对肾细胞癌患者的临床疗效,并改变了肠道菌群的β多样性和组成
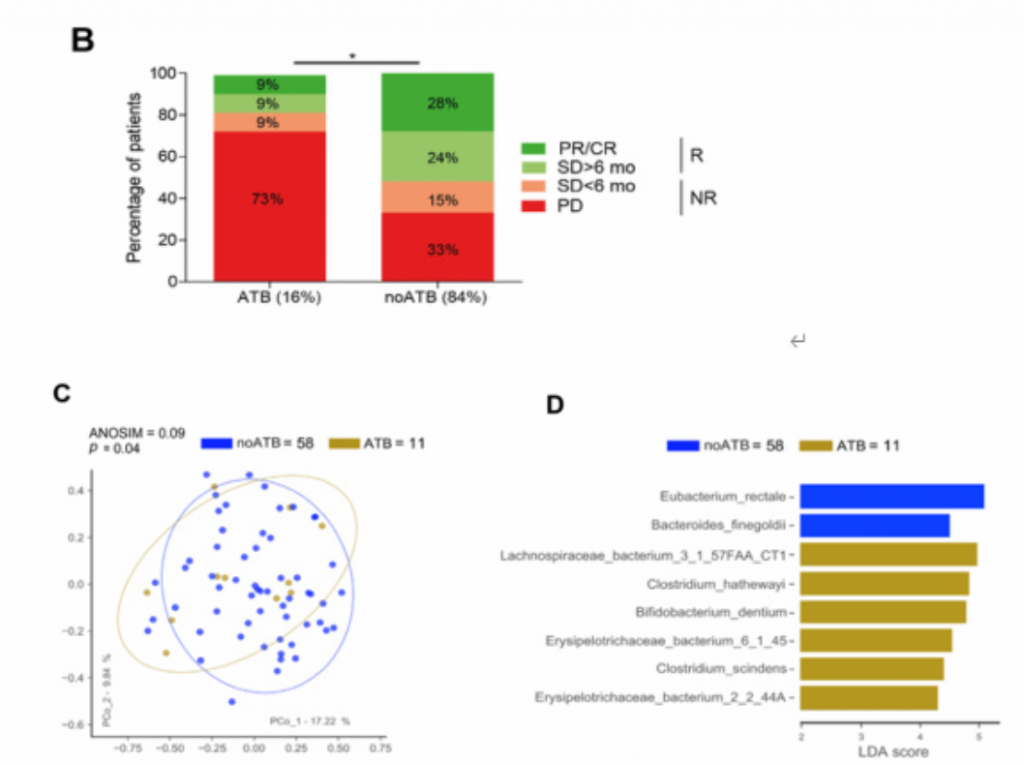

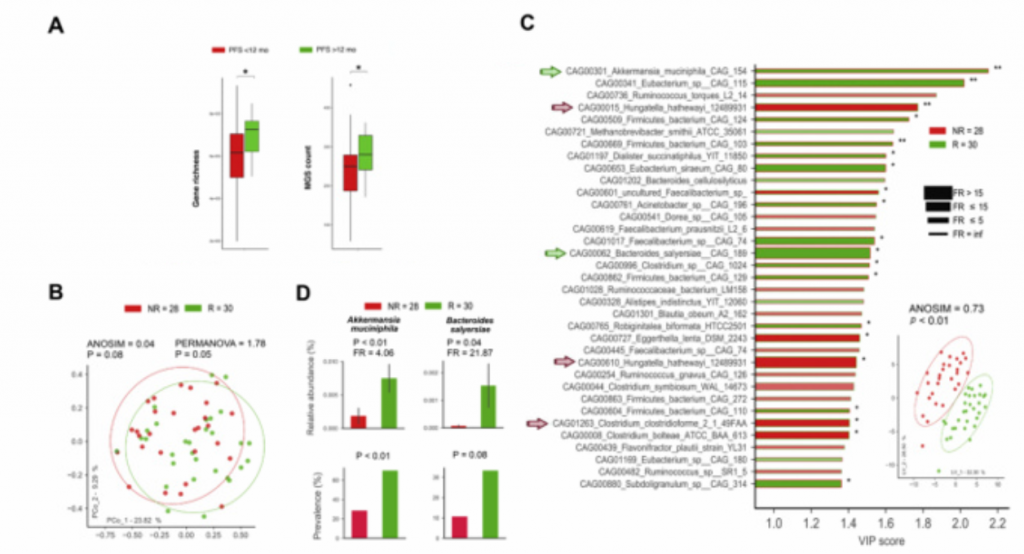

ATB:抗生素;noATB:未使用抗生素;NR:无肿瘤反应;PR:部分缓解;PD:疾病进展;
PFS:无进展生存期;R:有肿瘤反应;SD:疾病稳定;ORR:客观缓解率
(B).使用和不使用抗生素肾细胞癌患者人数占总患者人数的比例以及不同RECIST疗效结果占使用或不使用抗生素肾细胞癌患者人数的比例。ATB组11例,在抗PD-1阻断后2个月内服用抗生素;noATB组58例,未服用抗生素。接受ATBs治疗的患者的ORR低于未接受ATBs治疗的患者(9%
vs 28%, p < 0.03,p值采用双尾卡方检验)
(C). 基于PCoA分析的预处理样品粪便微生物群组成标准化和归一化数据的β多样性排序图。95%置信度。ANOSIM指标通过999次置换来评估ATB组和noATB组之间的差异。ATB个体与noATB个体的从粪便中分离出的细菌种类有显著差异。
(D).
Lefse图,基于LDA得分为每个组识别出差异物种。在这些差异物种中,如Eubacterium rectale菌在noATB粪便中富集,而其他细菌类群,如Erysipelotrichaceae bacterium_2_2_44A和Clostridium hathewayi在ATB粪便中富集。
2. 在noATB队列中,肠道菌群组成可以预测ICBs的临床结果。
利用粪便中的微生物组成的Alpha和Beta多样性结果可以对肾细胞癌患者人群中的R和NR个体进行分层,并可用于预测PFS>
12个月的患者。

mAb:单克隆抗体;MGS:metagenomic species;SEM:标准误
(A).对noATB基线样本(n=58)中的粪便微生物使用鸟枪法测序,图中展示了PFS<12个月和PFS>12个月的所有癌症患者的基因丰富度和MGS计数。ICBS开始后12个月,丰富度计数(GC)或MGS水平与以无疾病进展为标准的临床反应相关。
(B). noATB预处理样本的PCoA分析(使用细菌流行率>=20%的阈值)的β多样性排序图。95%置信度。ANOSIM和PERMANOVA指标使用999次置换,评估R(完全缓解或部分缓解或病变稳定>6个月)和NR(死亡或疾病进展或病变稳定<6个月)之间的差异。两组间有明显的分离。
(C).使用偏最小判别分析的变量对模型的重要性图,按重要程度由大到小描述了35个组间差异物种。箭头突出重要物种。条形填充颜色表示物种平均相对丰度最高的队列,条形框颜色表示物种平均相对丰度最低的队列。条形的宽度表示FR的高低。*表示Mann-Whitney
U检验的显著性。Akkermansia muciniphila, Bacteroides
salyersiae,和 Eubacterium siraeum菌种比例过高。
(D).被选中的Akkermansiamuciniphila和Bacteroides salyersiae菌种的相对丰度和流行率条形图。在肾细胞癌患者粪便中这两种菌的流行率和相对丰度在R和NR队列中都较高。相对丰度的p值由双尾Mann-Whitney U检验得到,流行率的p值由卡方检验得出。
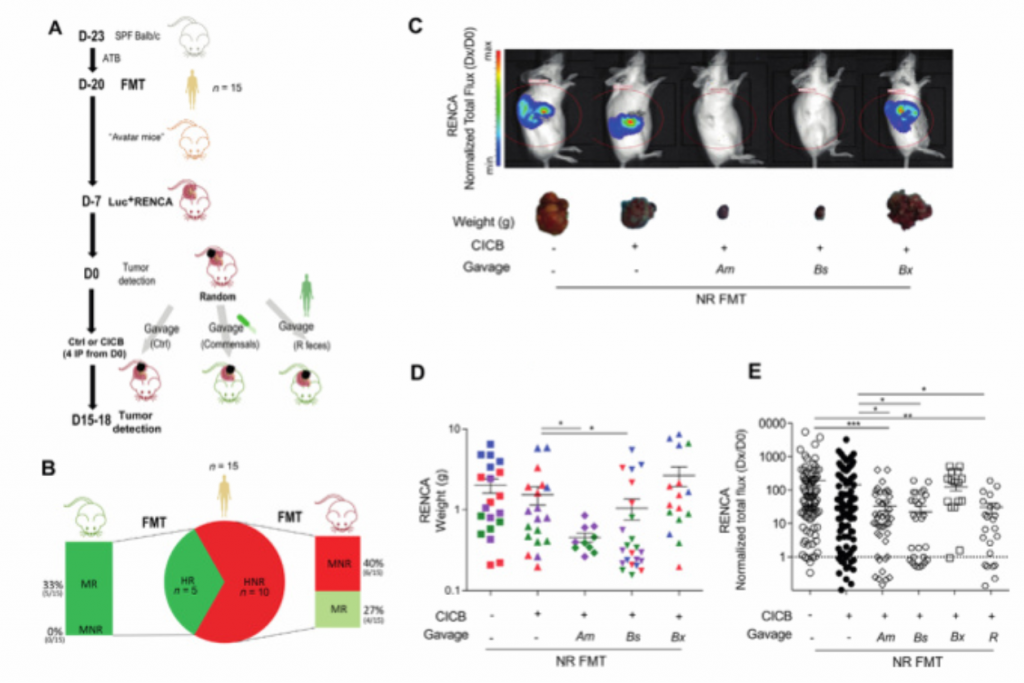
3.在肾细胞癌患者中,在使用nivolumab前先使用TKI和ATB药物与肠道菌群组成改变密切相关。在69个RCC队列中,R和NR的细菌比较分析,弥补了用NR-FMT在小鼠中观察到的反应性的不足,建立了粪便中有利的细菌组成与临床结果之间的因果关系。

CICB:联合ICB治疗;D0:随机日期;Dx:最后一次IVIS测量;
(A). 在无特定病原体(SPF)的BALB/c小鼠体内注射ATB3天后进行粪便微生物移植(FMT)。2周后,原位接种荧光素酶工程肾癌(Luc+RencA),从第7天开始每4天腹腔接种抗PD-1+抗CTLA-4单抗(CICB)或同型对照单抗(Ctrl)。此外,在第4天,每隔3天给接受CICB的受体小鼠口服Akkermansia muciniphila菌(Am)、Bacteroides salyersiae 菌(Bs)细菌B.xylanosolvens(Bx)或应答者患者(R)的粪便。
(B). 15份FMT供体粪便的比例. (人类有肿瘤反应者[HR]和人类无肿瘤反应者[HNR])在BALB/c小鼠(小鼠有肿瘤反应者[MR]和小鼠无肿瘤反应者[MNR])。观察到患者的反应和小鼠受体对ICBS的反应之间有27%的一致性:只使用了4份高于15FMT的粪便样本。
(C)(E).使用荧光素酶活性的生物发光成像监测ATB治疗小鼠在FMT后的Renca进展。在每个ICB周期前口服具有免疫刺激作用的Am或Bs或R-FMT来补偿NR-FMT(不含Am或Bs菌),恢复了对治疗的敏感度(牺牲时的肾脏重量)且荧光亮度降低。
(C)(D).使用肿瘤重量的生物发光成像监测ATB治疗小鼠在FMT后的Renca进展。在每个ICB周期前口服具有免疫刺激作用的Am或Bs或R-FMT来补偿NR-FMT(不含Am或Bs菌),恢复了对治疗的敏感度(牺牲时的肾脏重量)。
4. TKIs诱导肠道菌群改变。TKIs诱导了显著的和典型的微生物群落改变,包括免疫刺激共生物种,可以利用这些共生物种来提高肾细胞癌患者ICBS的疗效。[if !vml]
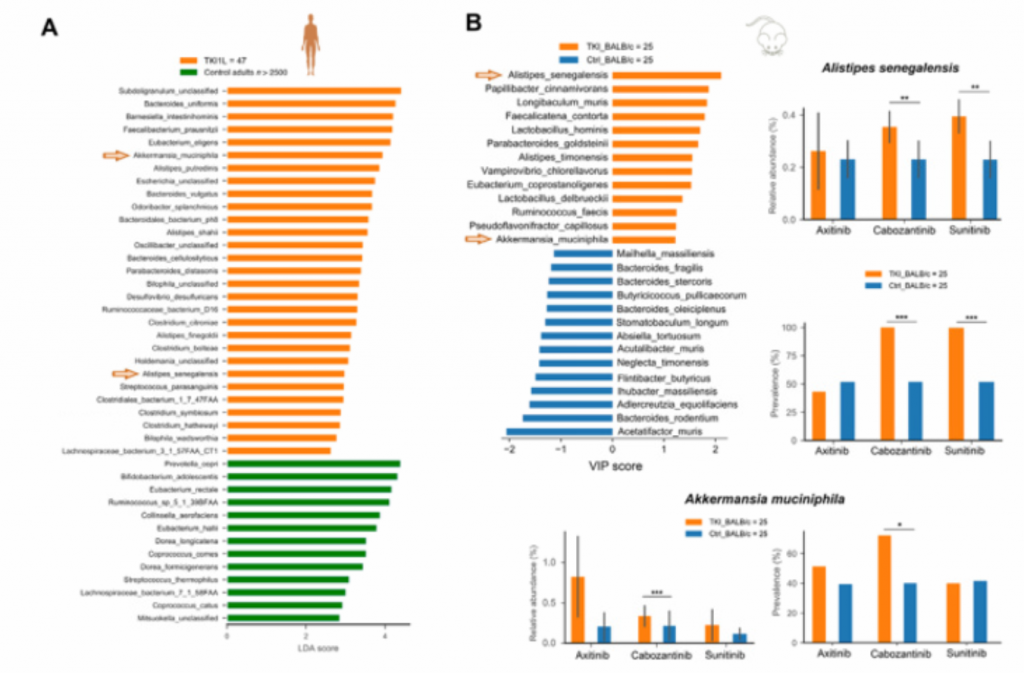

(A).接受TKI一线治疗的患者与对照成人粪便菌群的差异。按LDA分值排序的组间差异物种。观察到在TKI一线治疗组中Akkermansia muciniphila和Alistipes senegalensis富集。
(B).对接受TKI治疗(阿西替尼、舒尼替尼和卡博替尼)的BALB/c小鼠进行分析,利用偏最小二乘判别分析,差异物种按重要性从大到小排序。在BALB/c小鼠中,随着时间的推移,所有三种TKI都显著地诱导了菌群的α和β多样性的变化。
箭头表示相关的细菌种类。
柱状条形图分别描述了TKI治疗组中最具判别意义的两个菌种的相对丰度和流行率,即Akkermansia muciniphila和Alistipes senegalensis. 其中,观察到在人类和小鼠身上,Sunitinib和Cabozantinib有效的刺激了Alistipes senegalensis的丰度增长。对于三种不同的TKI(axitinib(阿西替尼)、sunitinib(舒尼替尼)和Cabozantinib(卡博替尼)),使用Mann-Whitney U检验来评估统计学差异. (*p<0.05,**p<0.01,*p<0.001)
结论
通过应用不同的生物信息学和组间差异分析手段(LEfSe, PLS-DA, VIP, and networks),研究人员确定了一组与原发耐药相关的物种(厚壁菌门,梭菌科,Clostridium clostridioforme种和Clostridium hathewayi种),并且由于使用ATB和癌症转移的状况。这些物种变得丰富。据以往的一些研究报告,Clostridium hathewayi菌种是与结肠癌诊断相关的致病因子的一部分,并且可以减轻小鼠的抗原特异性T细胞反应。
研究人员还发现了一些与预后良好和肠道稳态状态相关的共生物种,它们分别属于优杆菌科(Eubacterium
rectale和Eubacterium siraeum种),毛螺菌科(Dorea longicatena种),Verrucomicrobioaceae科(Akkermansia muciniphila种),而且都属于拟杆菌目。
结果表示微生物群的组成受TKIs和ATBs的影响,并影响免疫治疗的成功。ATB显著影响微生物群的β多样性,导致如前所述的优杆菌科家族成员(如Eubacterium rectale)的优势不突出,而有利于致病菌种(Erysipelotrichaceae bacterium_2_2_44A和Clostridium hathewayi)。这种微生物群的改变与ICB治疗期间ORR的降低有关(ATB组73%的原发耐药,noATB 33%,p<0.03)。在使用nivolumab之前使用TKI也可导致肠道菌群组成的变化,可能抑制ICB的疗效。该研究的局限性在于,比如研究的结论依赖于69名肾细胞癌患者的单一队列,其中只有11名患者服用ATBs和二线治疗,并受到许多混杂因素(既往治疗、发病和其他因素,如血红蛋白)的干扰。
展望
研究人员表示在开始治疗前对粪便的分析可能可以指导临床医生用药,以防止肾细胞癌患者对免疫治疗的原发性耐药。而更好地了解肠道菌群组成与局部、全身和肿瘤免疫系统之间的机制联系,将有助于为肾细胞癌患者的肠道菌群失调设计最佳的补充治疗方案。
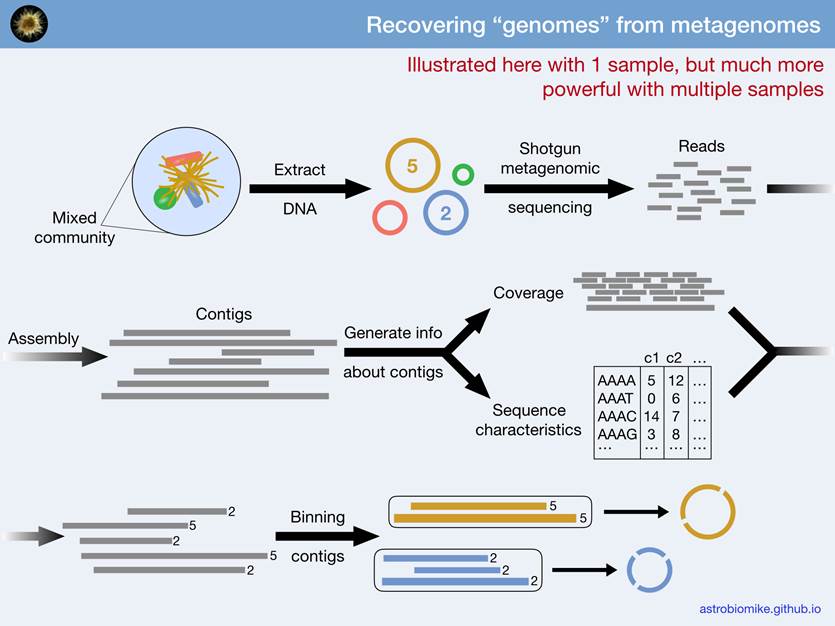
近年的研究热点集中于环境和生物体相互作用的微生物群体,而大量复杂的微生物群体存在培养困难,构成复杂(包括细菌、古菌、真菌、原生生物、病毒甚至小型真核生物)。 为了克服传统纯培养技术的不足,充分挖掘此类未培养微生物所蕴涵的巨大潜能,发展了 宏基因组学的研究方法,该方法绕过纯培养技术来研究微生物的多样性及功能。
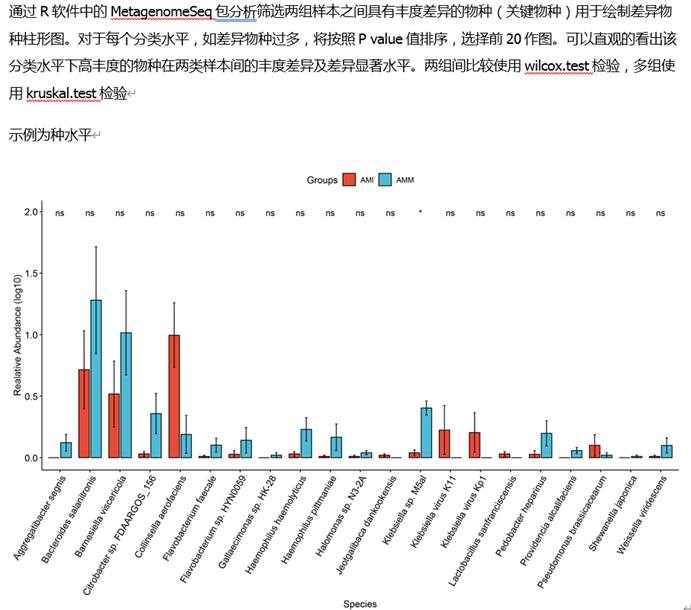
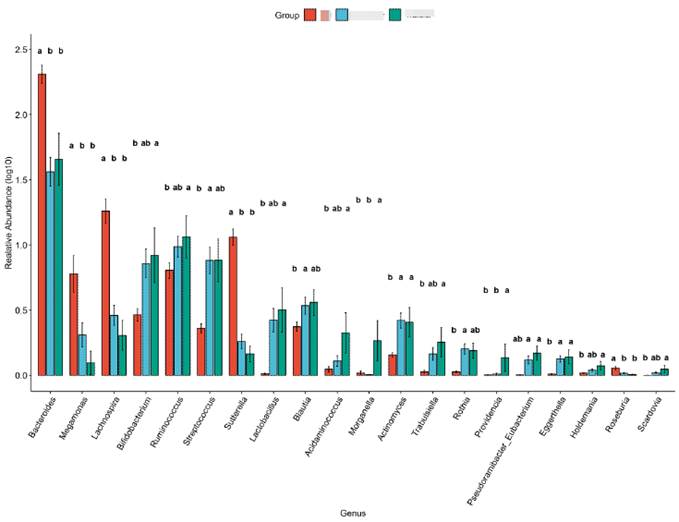
宏基因组测序也就是shotgun测序, 以环境中所有微生物基因组为研究对象,通过对环境样品中的全基因组DNA进行高通量测序,获得单个样品的饱和数据量,基于denovo组装进行微生物群落结构多样性, 深度全面的了解微生物群体的构成,甚至获得单个菌株的完整基因组,在这些高质量基因组序列的基础上可以更加精细化开展其基因构成、分布,次生代谢合成,抗生素耐药基因及其演化, 微生物群体基因组成及功能,特定环境相关的代谢通路等分析 甚至菌株的生态演变和水平转移事件等研究。 进一步发掘和研究具有应用价值的基因及环境中微生物群落内部、微生物与环境间的相互关系。
此外对于缺乏深度研究和高质量参考基因组的样本,如土壤和特殊环境下的样本,宏基因组获得的较为完整的基因组不仅可以丰富参考基因组数据库,同时可以提供更加准确的物种分类。
建库流程
1) 将检测合格的基因组DNA样品用酶切随机打断成400bp-500bp的片段并末端修复加A;
2) 将测序引物连接到DNA片段上;
3) 进行PCR扩增引入测序index
4) 对构建的文库进行质量检测;
5) 将质量检测合格的文库上机测序。
粪便、动物肠道内容物、皮肤、组织、痰液、血液、唾液、牙菌斑、尿液,阴道分泌物、发酵物,瘤胃,废水,火山灰,冻土层、病害组织、淤泥、土壤、堆肥、污染河流,养殖水体、空气等有微生物存在的样本都可以用于宏基因组测序。
测序平台:Illumina Novaseq,PE150,默认:5-6G/样,每增加一个G加100元。
周期:2-3周
粪便、动物肠道内容物、皮肤、组织、痰液、血液、唾液、牙菌斑、尿液,阴道分泌物、发酵物,瘤胃,废水,火山灰,冻土层、病害组织、淤泥、土壤、堆肥、污染河流,养殖水体、空气等有微生物存在的样本都可以用于宏基因组测序。
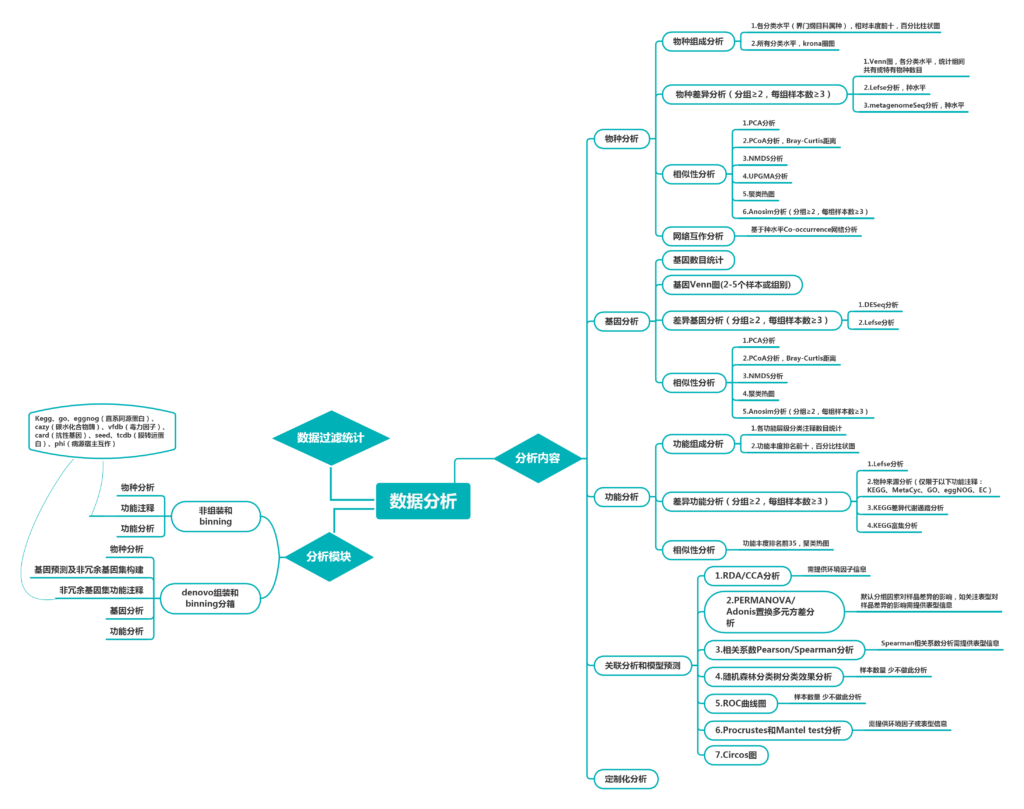




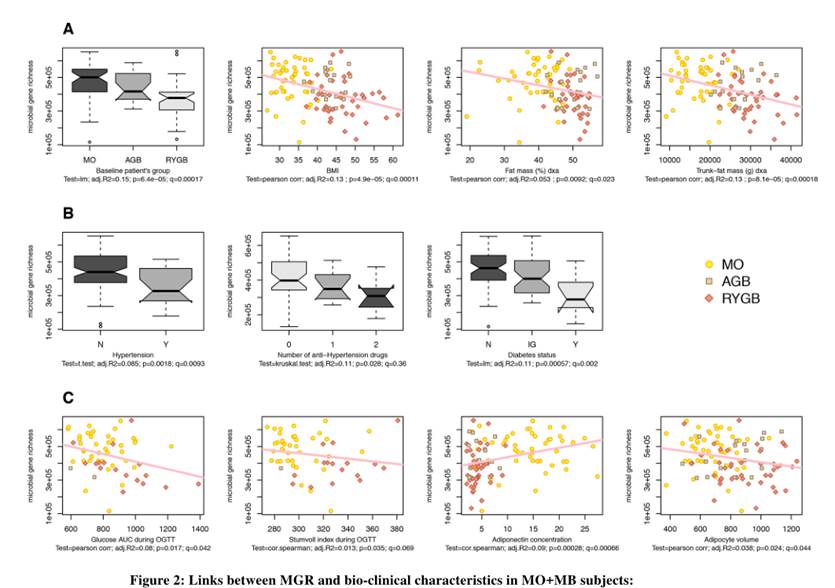
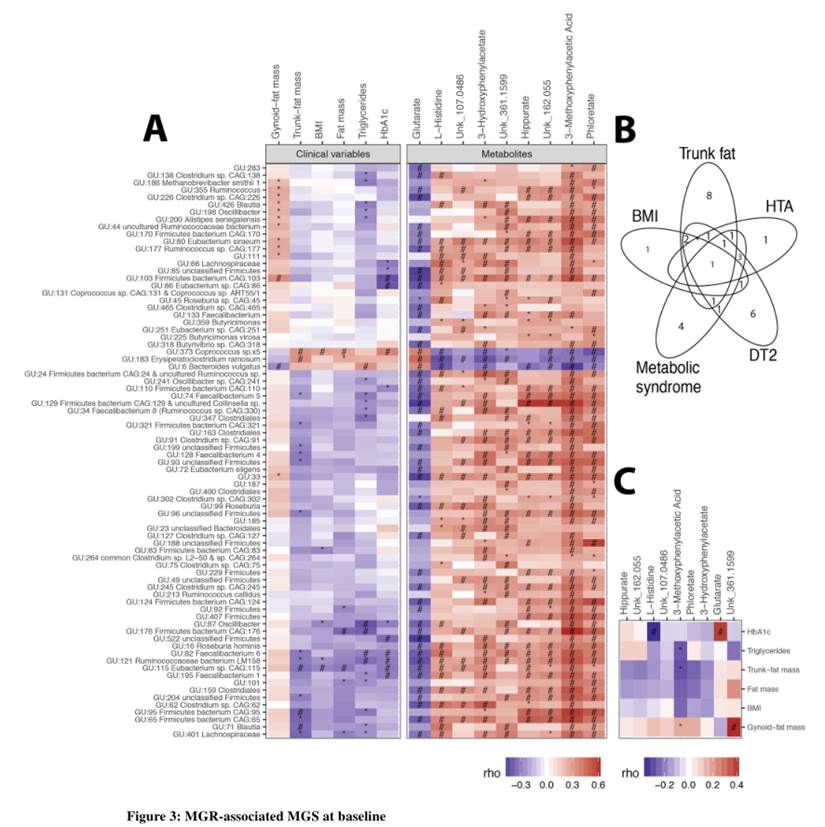
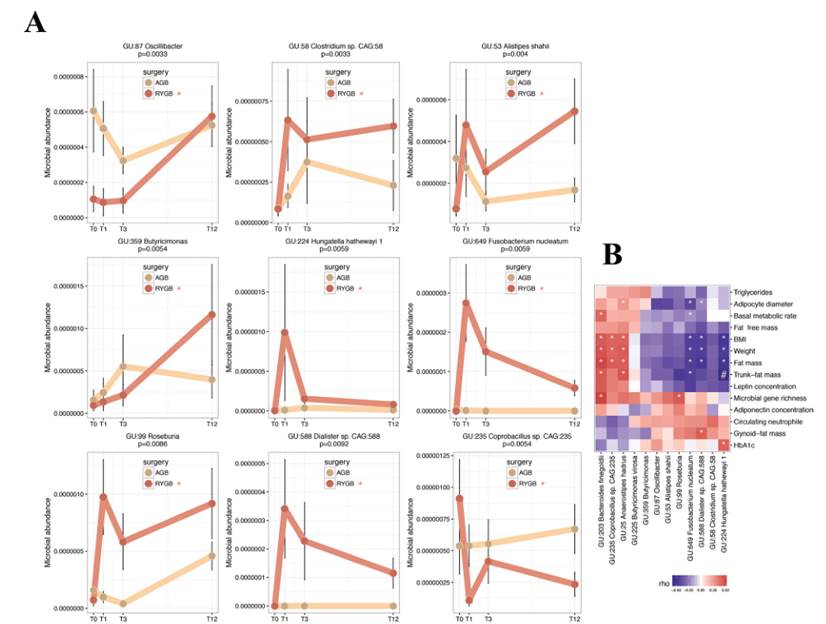
第一项研究是关于肥胖患者减肥手术后的宏基因组和代谢数据的分析研究。
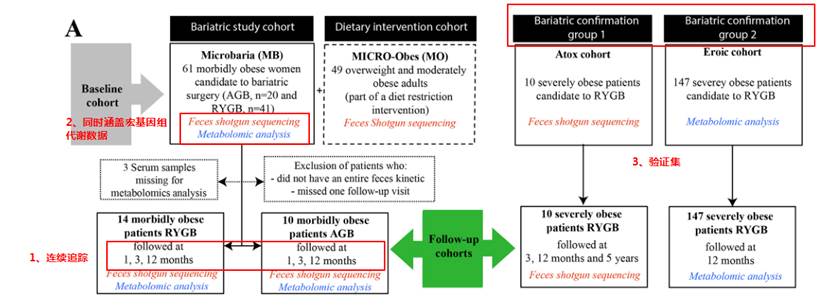
研究纳入了61名严重肥胖的受试者,他们是可调节胃束带术(AGB,n = 20)或Roux-en-Y胃旁路术(RYGB,n = 41)的候选人。减肥手术后1、3和12个月随访24名受试者。使用宏基因组学测序和LC-MS分析肠道菌群和血清代谢组。另外纳入了10人和147人分别作为宏基因组和代谢检测的验证集。
研究思路
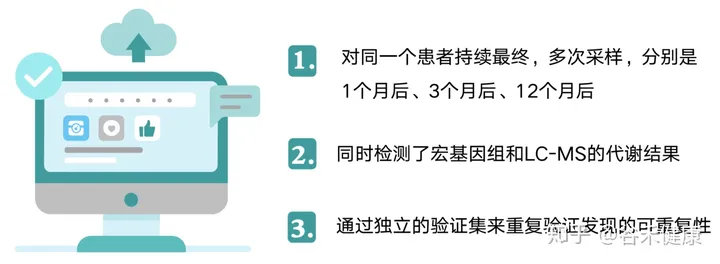

这样的设计分别有什么作用?
第一点持续的动态采样可以获得持续变化情况,尤其是在一个特定变化后(减肥手术),持续的最终采样有助于确认菌群的变化出现和特定事件或生理病理变化的前后,尤其是在确定因果中有重要帮助。
第二点获得多维的数据有助于帮助我们全方位的了解菌群变化背后的带来的生理和代谢变化以及之间的关联。
第三点独立验证集的存在将大大增强研究的可信度,尤其是该研究纳入的样本量并不多,无法全面有效的控制无关因素,使得很多统计检验的效力无法显现。这也导致该研究仅在基因总量和多样性上获得较好的重复效果,而更多的菌群精细特征以及具体基因和代谢通路没有得到深入分析。但是独立验证集保证了核心结论的可靠性和重复性,这点在宏基因组研究中非常重要。
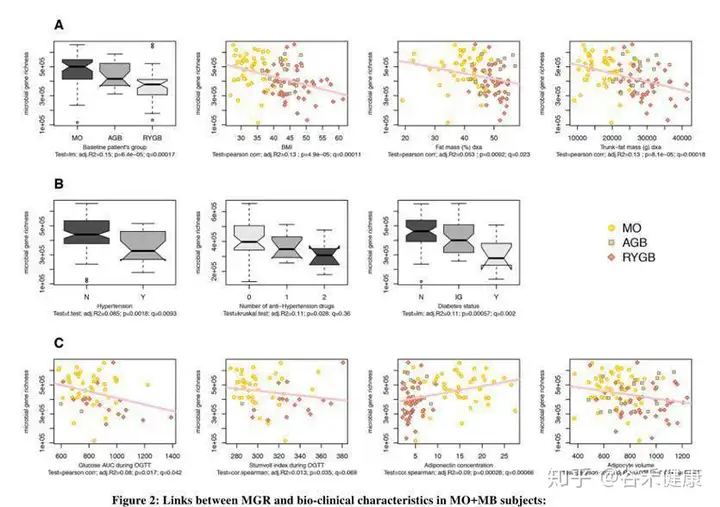
从下图可以看到研究针对样本的总基因多样性水平与生理指标和疾病状态进行相关性分析和组间差异分析,图中给出了显著相关和差异的指标。
使用的统计检验方法是pearson和sperman相关和t-test以及Kruskal-Wallis检验。
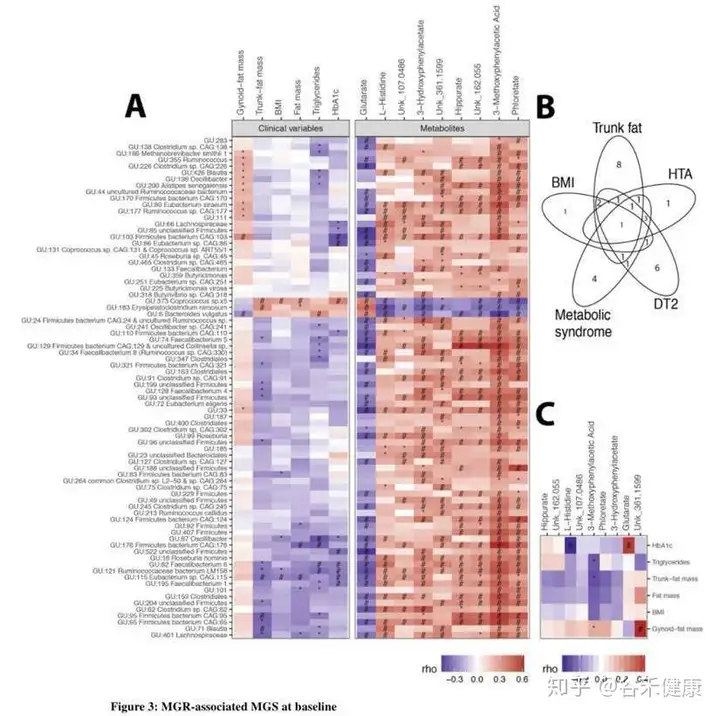
下图是研究将MAGs与各项生理和代谢值进行相关性分析后的热力图。该研究由于测序较早,并未独立拼接,而是直接使用了之前一项人类肠道菌群研究获得组装基因组参考序列。
进一步研究分析了术后特定变化模式的MAGs以及它们与代谢生理指标的相关性,见下图:
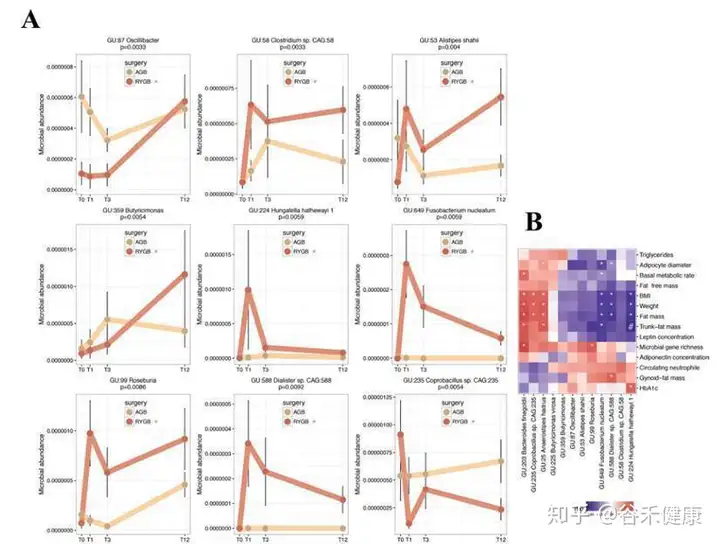
上图的研究可以通过pattern search的方法寻找特定变化模式的菌种。
研究的主要结论发现是低基因丰富度(LGC)存在于75%的患者中,并且与躯干脂肪质量和合并症(2型糖尿病,高血压和严重程度)增加相关。LGC改变了78种宏基因组种(MGS),其中50%与不良的身体成分和代谢表型有关。九种血清代谢产物(包括谷氨酸盐,3-甲氧基苯基乙酸和L-组氨酸)和含有参与其代谢的蛋白质家族的功能模块与低MGR密切相关。术后一年,BS会增加MGR,但尽管RYGB患者的代谢改善比AGB患者大,但术后一年的MGR仍然很低。
总体而言该项研究可以使用浅宏基因组来完成所有测序和分析,进一步扩大样本数量,如果能同时获得人的转录组数据甚至能更加明确的找到菌群变化与特定代谢通路的关联关系。
< 案例二 > 食物与人类肠道微生物组
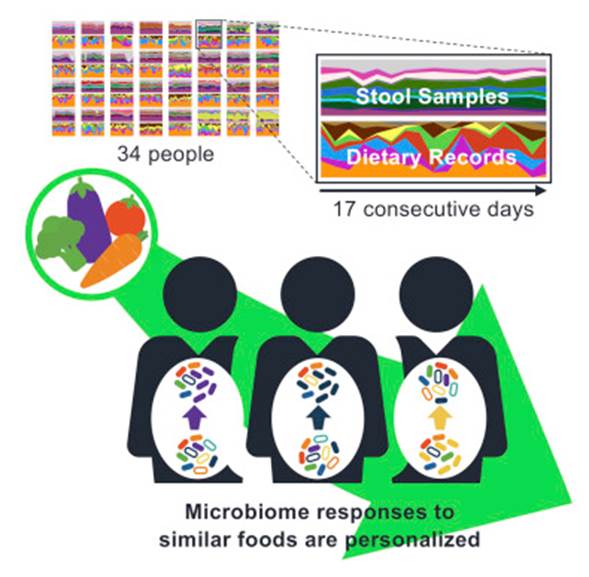
第二项研究是Dan Knights实验室发表在Cell Host & Microbe,2019的一篇针对34个人17天每日饮食和菌群变化的相关研究,试图揭示日常食物选择与人类肠道微生物组组成之间的精细关系。
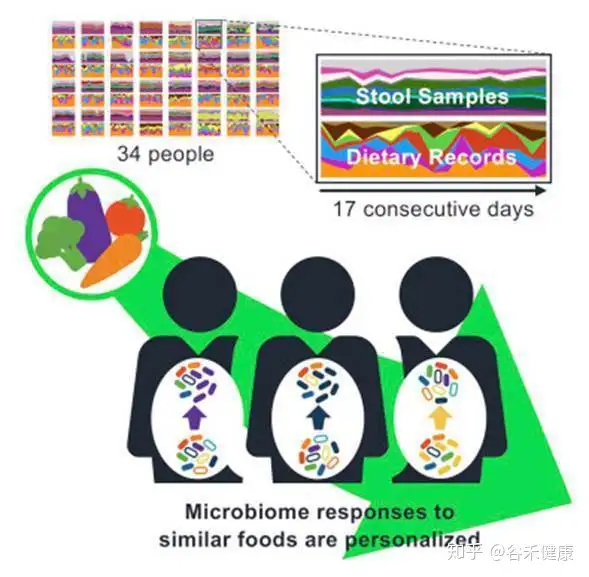
可以看到,研究同时记录了粪便样本的菌群宏基因组和每日的饮食记录。研究的核心在于将每日饮食的食物通过营养构成进行量化,并构建类似物种进化树的食物物候树。
此外由于有每日的数据,可以通过前一日的食物与第二日的菌群数据进行时间序列分析,构建食物与菌之间的关联以及时间相关性。
最后基于菌群数据和前一日饮食来构建模型预测判断后一日的菌群状态,帮助我们了解食物对于个体菌群的影响因素并实现定量和预测。
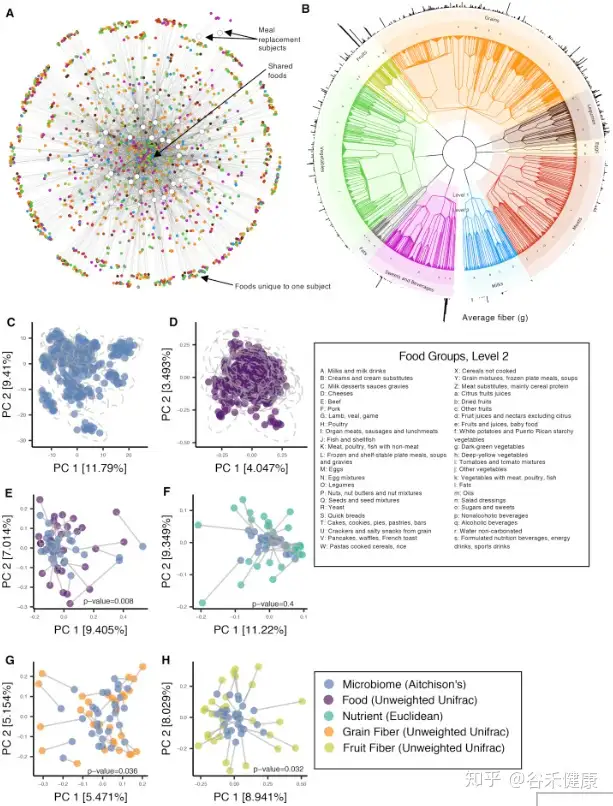
研究中对数据的处理过滤标准如下:删除所有具有低读取计数(每个样品<23,500个读取)的样品。物种级别的分类表仅限于研究对象中至少存在25%的研究日,且在10%的研究样本对象中发现的那些物种。
最后,相对丰度<0.01%的稀有物种被丢弃,将物种数量限制为290个注释。将得到的分类表汇总到较高的分类级别(即属,科,门等),以进行下游分析。
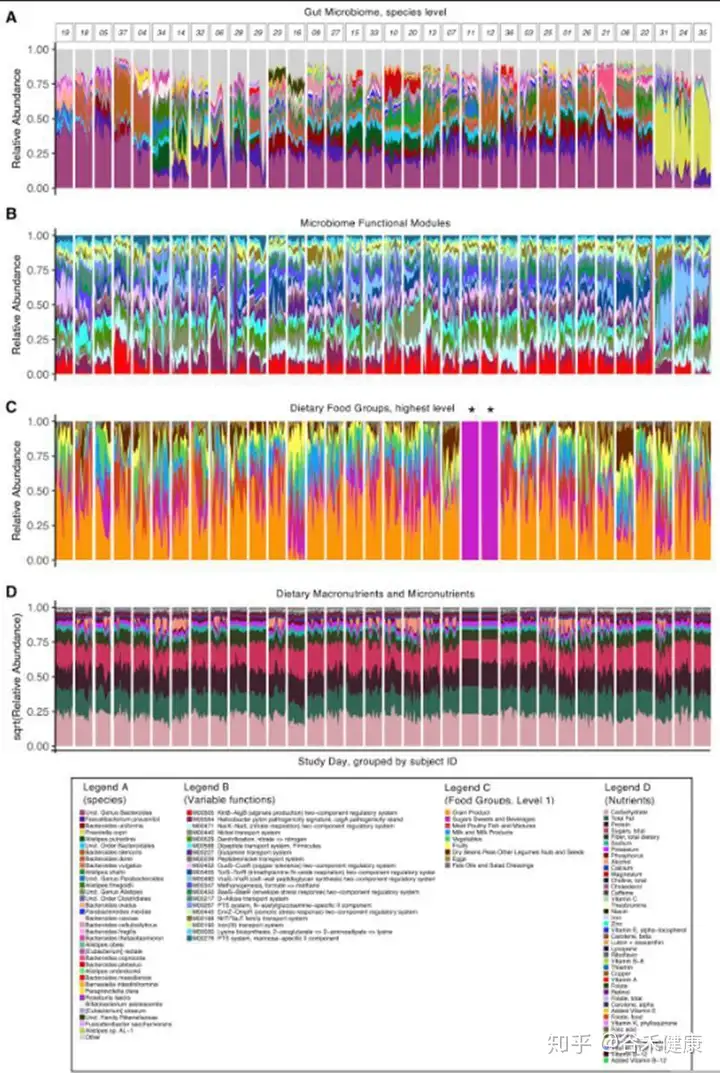
菌群和饮食以及营养构成的堆叠图很好展现了变化和对应。
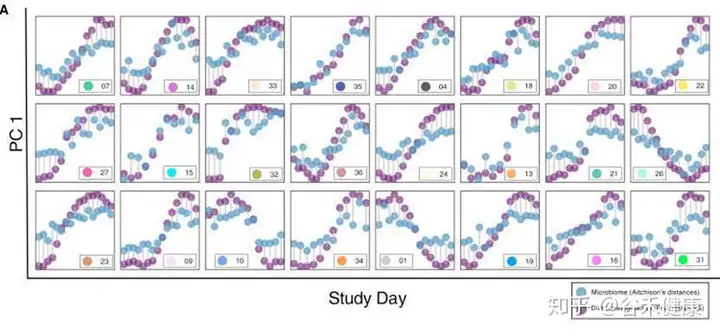
下面这张图很好的显示了饮食食物的变化与菌群变化之间的时间变化关系:
下面这张图通过对每个人单独进行菌属与食物的Spearman相关,展现了菌与食物之间的关联的个体化差异,在特定菌属对应相同食物不同人会出现完全不同方向的变化,这也正是这项研究所揭示的,这种关联关系的复杂性。
本研究虽然有大量样本,但并未进行组装,而是直接使用了Refseq的细菌完成基因组序列作为参考。研究由于样本数量众多,测序深度也很有限,类似研究也可以使用浅宏基因组方式完成。
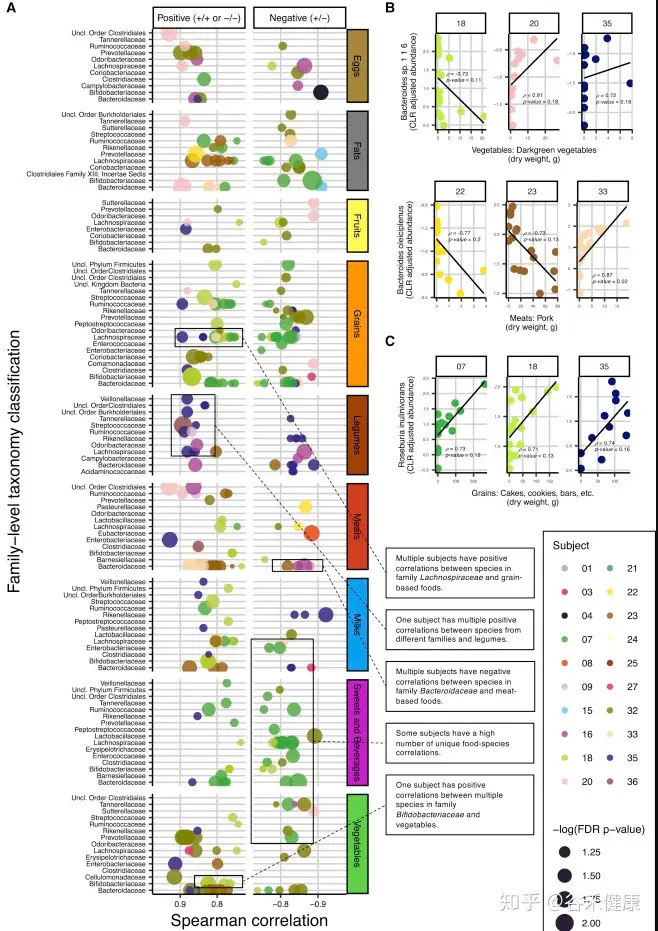
接下来的一个研究是比较典型的宏基因组组装并与疾病进行关联分析的案例,研究的是类风湿关节炎人群的肠道微生物组的全基因组关联研究。
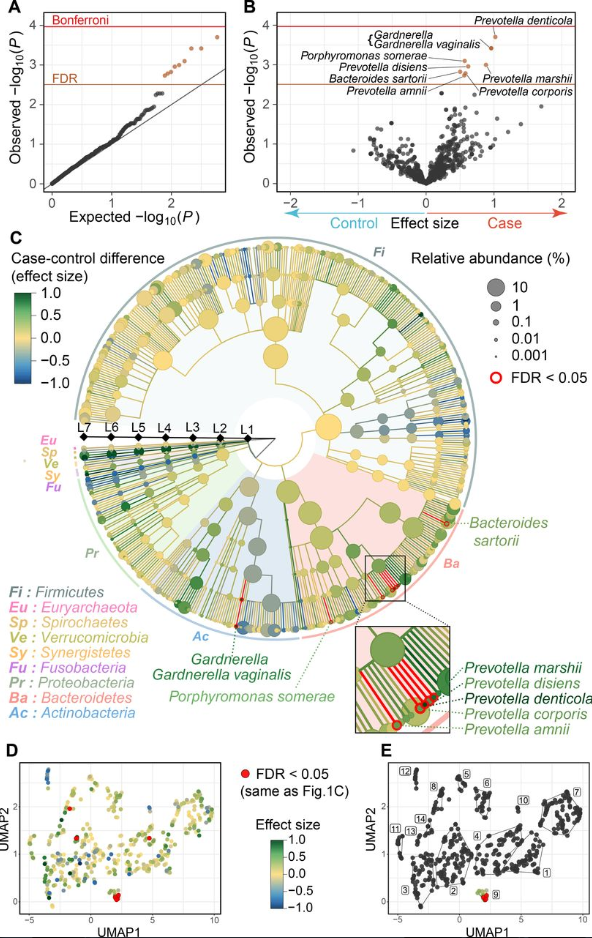
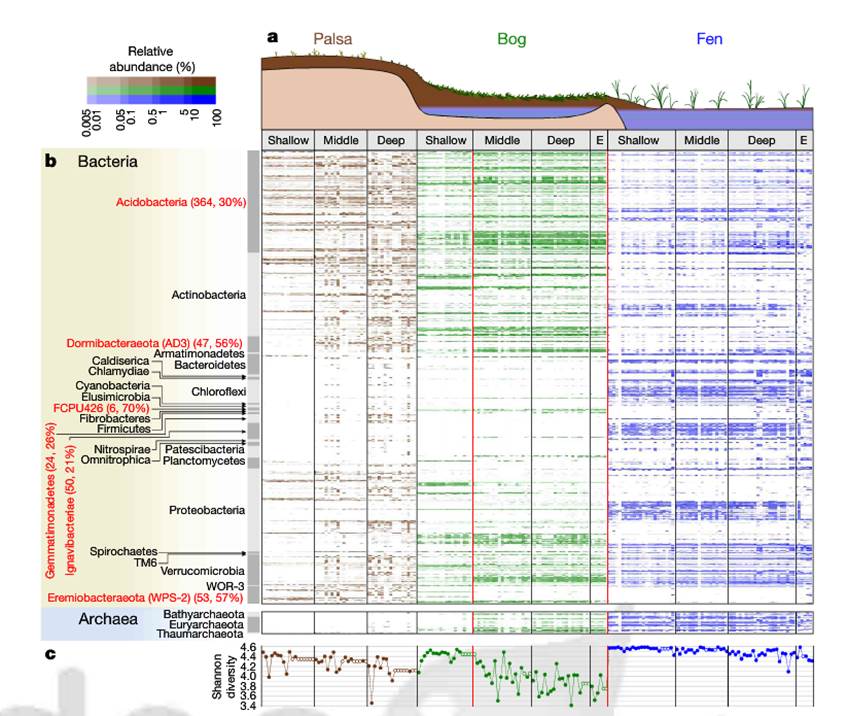
研究使用较高深度的宏基因组shotgun测序(每个样本平均13 Gb)对日本人群(病例 = 82,对照 = 42)进行了RA肠道微生物组的MWAS分析。MWAS由三个主要的生物信息学分析渠道(系统发育分析、功能基因分析、途径分析)组成。
使用了之前研究中6139个完成拼接日本人肠道宏基因组作为参考序列以及其他几项研究的参考基因组,在过滤部分种过多的基因组之后,最后一共使用了7881个参考基因组。
将QC后的序列直接比对到参考基因组,并根据基因组长度计算对应物种的相对丰度。
基因方面选择denovo组装,使用MegaHIT,然后再contig上完成ORF预测和CD-HIT聚类去冗余,最后与UniRef和KEGG数据库比对。
最后使用bowtie2将测序序列比对到注释后的unigene序列上获得基因丰度,经过KEGG注释得到代谢途径的丰度。研究的数据处理流程图如下:
在数据分析流程和方案选择上人体肠道菌群由于研究众多,以及有多个深度测序拼接完成的Binning参考基因组数据集,确实可以直接使用参考基因组直接比对。
对于其他一些环境或来源的样本这个深度的数据量可以考虑独立拼接和分箱。该研究中使用已有参考基因组,大概88%的序列能比对到参考基因组,如果直接组装这个比例应该可以更高一些。另外在获得基因丰度是可以考虑使用Salmon,比对获得基因丰度更为方便。
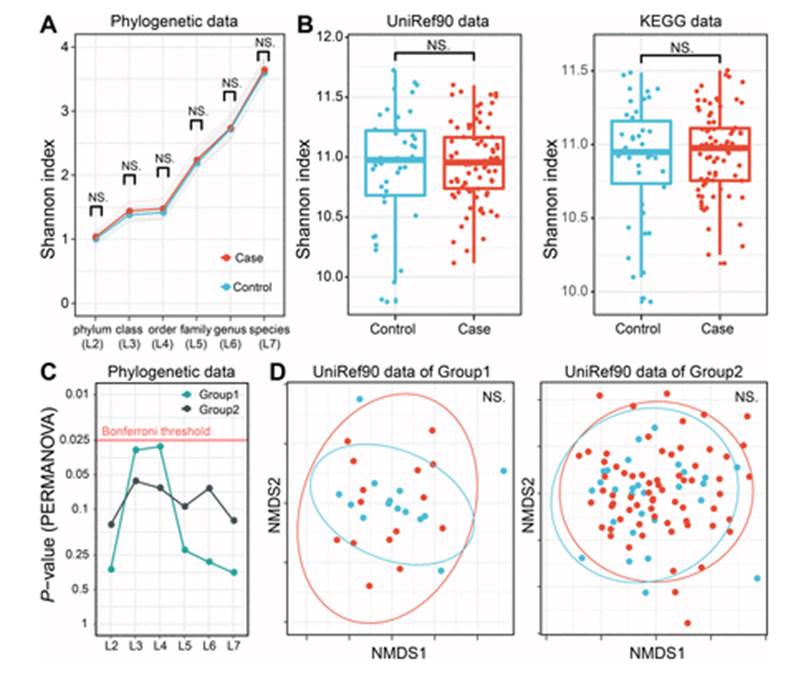
获得相应数据后对相对丰度,该研究使用Box-Cox transformation对数据进行标准化,并过滤了一些低丰度的菌属。Case-control的相关性分析使用的R的glm2模块,将年龄、性别和测序上机分组作为协变量。
对于菌属的关联分析,最终将显著性结果以火山图和GraPhlAn图的形式展现如下:
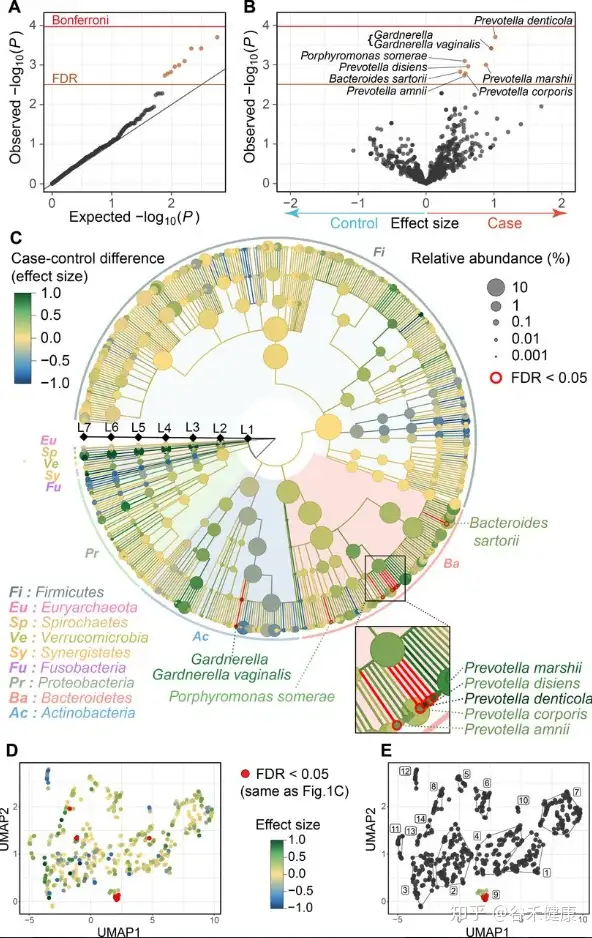
上面其中D图使用每个菌的丰度进行UMAP分析,并映射关联效应的展示比较有意思。
不过在基因层面上并未找到相应的关联,可以看到下图UniRef90的NMDS分布图两组之间无法有效区分,多样性也没有显著差异。
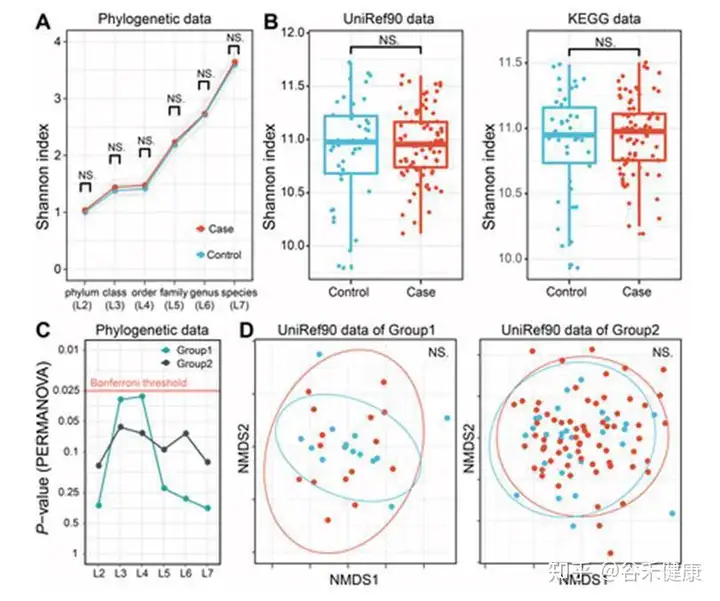
这项研究在菌层面发现了多个普雷沃氏菌属的菌在日本人群中与类风湿性关节炎存在关联,不过除此之外其他方面的发现并不多,仅找到一个基因存在显著关联,涉及的临床调查也相对有限,且人群队列数量不算多,并无独立验证集,因此亮点并不多。如果能纳入免疫相应指标可能能研究的更细致一些。
最后这项研究是对来自永冻土融化梯度的214个样品的宏基因组测序组装了1,529个基因组,揭示了参与有机物降解的关键种群,包括其基因组编码先前未描述的木糖降解真菌途径的细菌。
通过宏基因组denovo组装和分箱Binning,最终获得了1529个永冻土菌群基因组。基于这些数据描绘了永冻土融化梯度下的菌群构成特征,如下图。
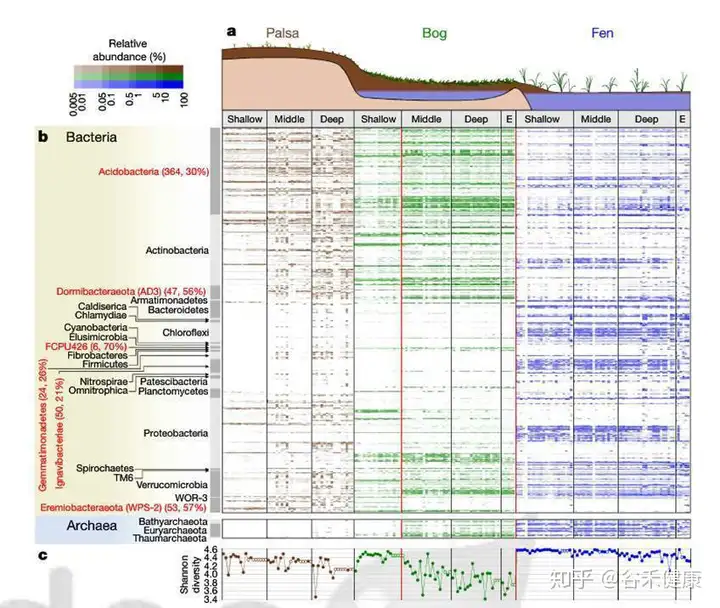
论文是2018年发表的,测序是在2011和12年测的,使用的是CLC Genomics Workbench 较早的4.4版分单样本组装,然后使用MetaBAT进行分箱,最后的标准是70%完成度和低于10%的污染。
其中糖苷水解酶基因使用dbCAN数据库的HMM进行预测,碳代谢使用KEGG数据。
研究还同时选择了部分样本进行了宏转录组和宏蛋白组的测序,对碳代谢同时结合转录组和蛋白组的数据,展现了特定通路下不同永冻土的菌群构成和表达丰度差异。
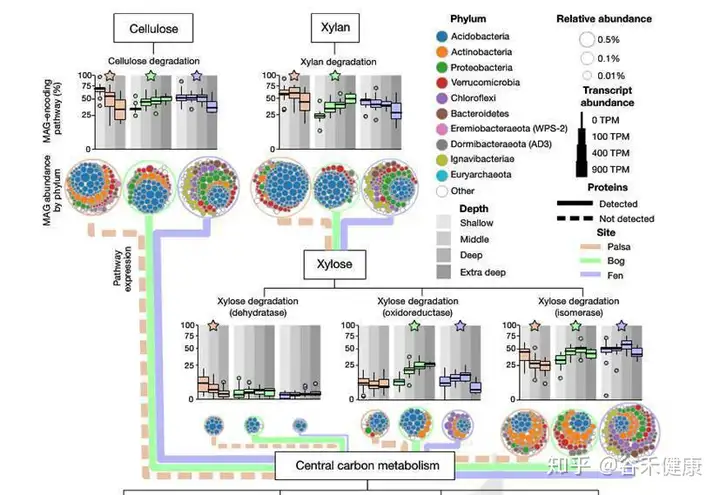
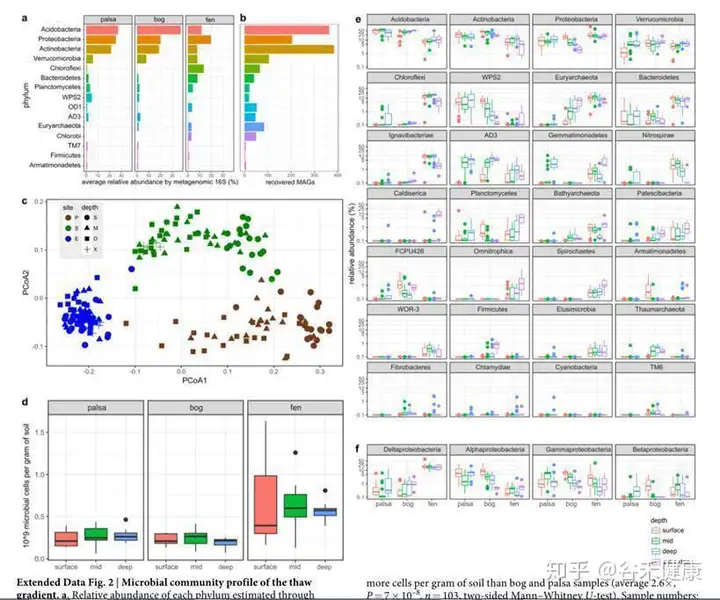
基因组拼接的分布情况,以及不同地域样本分布和菌属丰度情况如下:
木糖降解途径在每个样本中的分布和维恩图,另外详细的展现了主要门对每个代谢途径的贡献和基因表达丰度,如下图:
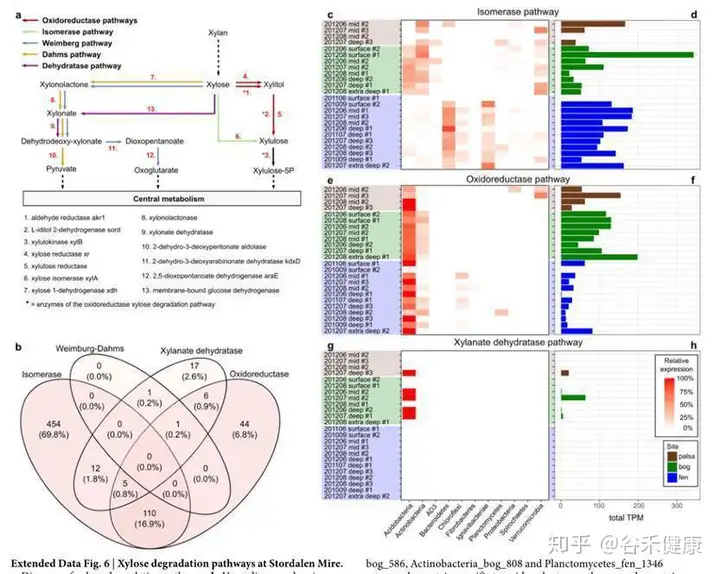
这张图分析了特定菌与地理位置和CO2以及甲烷的浓度的关联性,如下图:
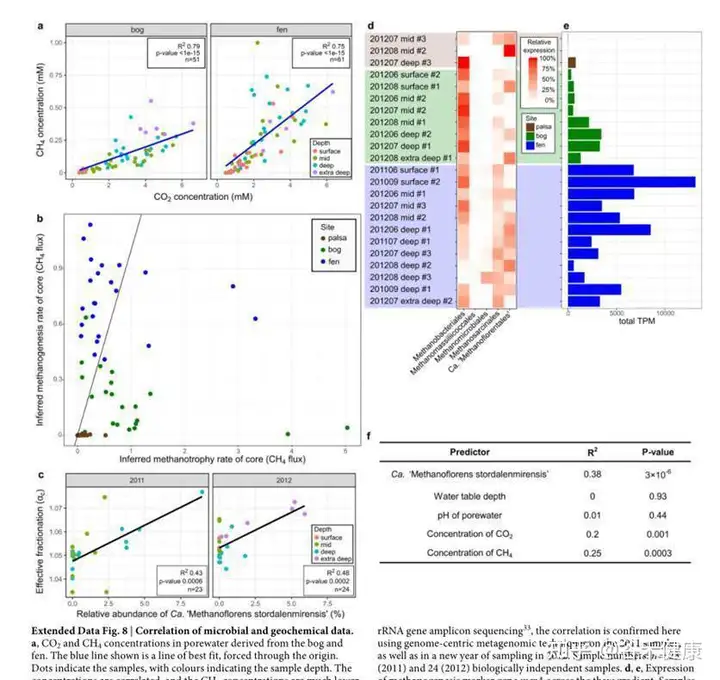
对关键物种的CH 4 :CO2浓度比相关代谢途径的重建,以及相应基因的基因家族分析。
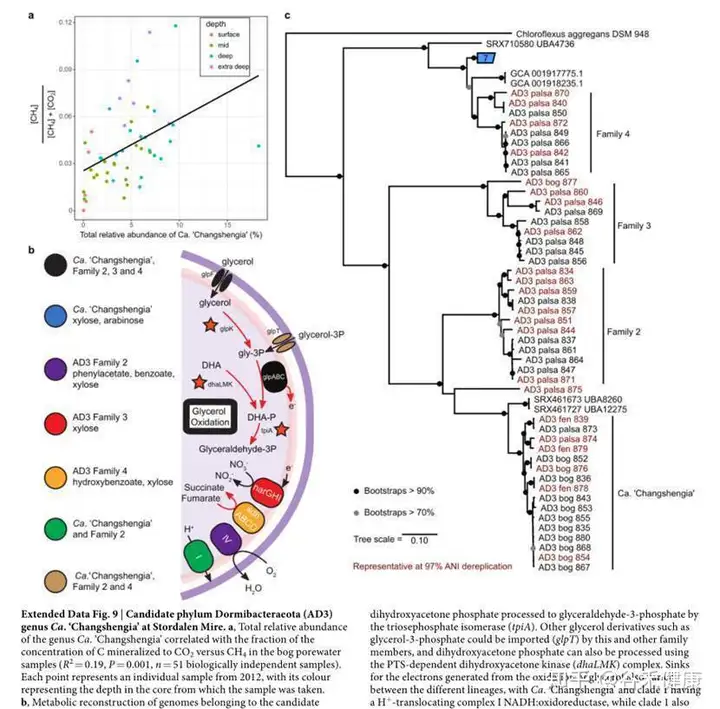
总结一下这项研究,永冻土的菌群参考基因组数据缺乏,该研究从大量地点采集样本重建了1500多个参考基因组。
首先从物种构成特征上与永冻土融化阶段特征进行分析,并与重要环境因子进行分析,锁定重要的特征菌。
然后针对重要的代谢途径和关键基因结合宏转录组和宏蛋白组全面解析代谢途径的分布和差异变化。对关键的物种重建了相关代谢途径并对其相关基因家族进行分析。
研究基本上从头构建了一个生态环境下的菌群结构数据,并利用获得的基因组深度解析特定代谢途径和基因的构成和表达变化,应该说既宽又深。很多样本采集和测序是2011年和 2012年就开展的,虽然测序技术远不如现在成本低和成熟,但是其独特的研究对象和全面深入的分析仍然使整项研究和目前的一些研究相比完成的更加出色。



样本需求量低:常规宏基因组建库建议样本量在500ng以上,公司研发实现了低当量微生物样本提取和建库,保证提取丰度以及片段完整性同时,样本量需求低于同行其他公司要求;对于样本获取困难的样本,也可以选择微量建库,样本量可低至10ng。
免费取样盒和针对性取样建议:粪便及环境样本提供取样盒助力临床/科研取样,人体口腔、痰液、腹水、脑脊液、尿液、皮肤等高寄主细胞含量样本可根据我们的处理方案简单处理后大幅降低宿主DNA比例。
严格标准的实验流程:自动化样品处理平台辅助,每轮设置阳性对照,上轮检测样本对照,阴性对照。评估污染,轮次比对,最大化减少误差,保证样本重复性和稳定性
Illumina测序平台:宏基因组测序(PE150)采用先进的Illumina Novaseq测序平台,快速、高效地读取高质量的测序数据、结合样品特点和数据的产出,充分挖掘环境样品中的微生物菌群和功能基因
大数据分析流程质量流程控制严格:优化的数据质量控制,包括过滤比对质量低、非特异性扩增、覆盖度低、低复杂度的序列,从而快速准确获得样本中微生物信息及其丰度信息,最大化提高质量数据
分析内容丰富全面:物种分析,基因预测与分析,多样性和相似性分析,功能分析,网路互作分析,代谢网络,关联分析等
完整详细的报告:提供质检实验报告,分析统计报告,分析报告解读,原始数据
高效个性化服务:在线项目系统方便您及时查看项目动态和下载报告以及与分析人员高效交流,免费支持个性化图表修改以及重新分组出报告。
价格低,周期快:包括提取,测序到分析,最快2周出报告。
大数据分析团队和多中心大项目分析经验(团队主要源自浙江大学,包括生物信息学,计算机,微生物以及统计分析等专业,积累了多年的大健康项目多中心项目分析经验,有助于宏基因组大数据,多样本,多表型,多组学联合分析
兼容性强的合作模式:有专门团队负责,提供切实可行的项目方案,兼顾临床和科研双需求模式。

16S rRNA 基因是编码原核生物核糖体小亚基的基因,长度约为1542bp,其分子大小适中,突变率小,是细菌系统分类学研究中最常用和最有用的标志。
16S rRNA基因序列包括9个可变区和10个保守区,保守区序列反映了物种间的亲缘关系, 而可变区序列则能体现物种间的差异。
16S rDNA是细菌染色体上编码16SrRNA相对应的DNA序列, 16S rRNA是由16s rDNA转录来的 , 一般扩增检测和分析的对象都是16s rDNA 。
16S rRNA基因测序以细菌16S rRNA基因测序为主,核心是研究样品中的物种分类、物种丰度系统进化、功能预测以及微生物与环境互作关系等。
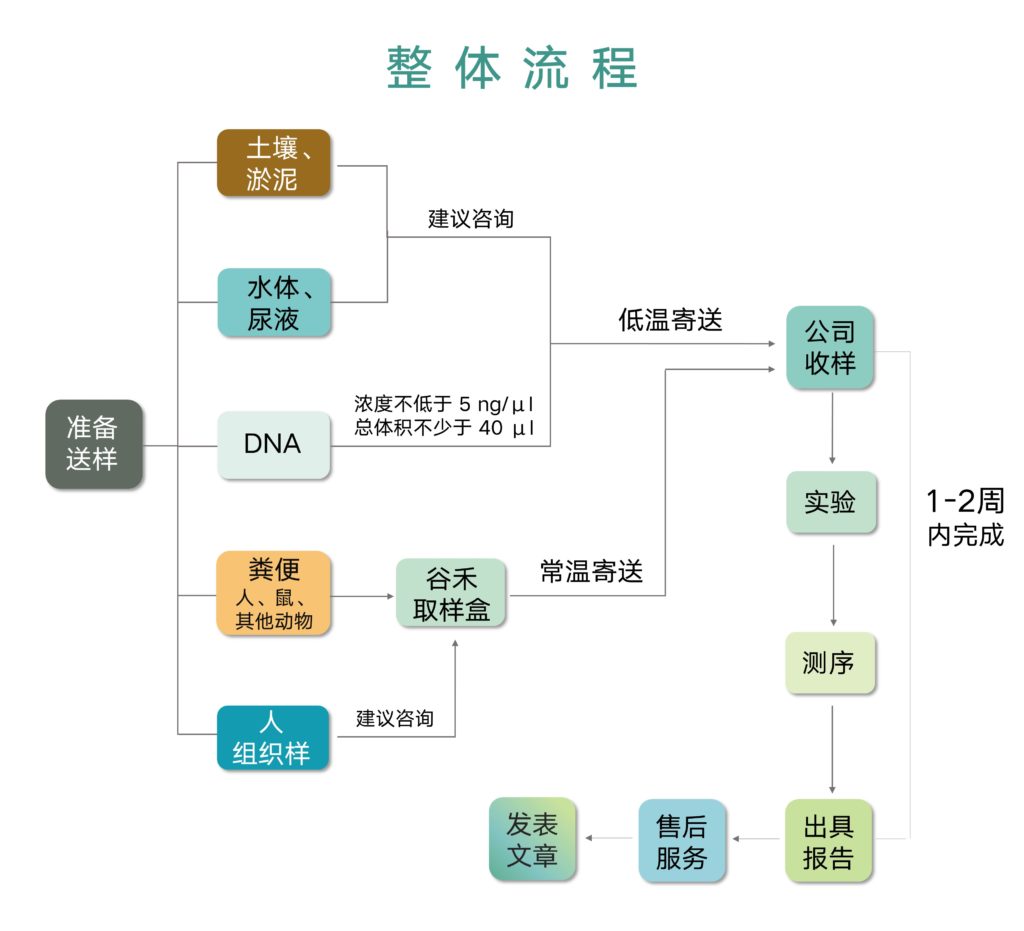
粪便、动物肠道内容物、皮肤、组织、痰液、血液、唾液、牙菌斑、尿液,阴道分泌物、发酵物,瘤胃,废水,火山灰,冻土层、病害组织、淤泥、土壤、堆肥、污染河流,养殖水体、空气等有微生物存在的样本都可以用于16s测序分析
取样:人/动物粪便,口腔,唾液,阴道,尿液,皮肤等可提供免费常用保存运输取样盒,其他样本可直接送样
测序平台:Illumina Novaseq
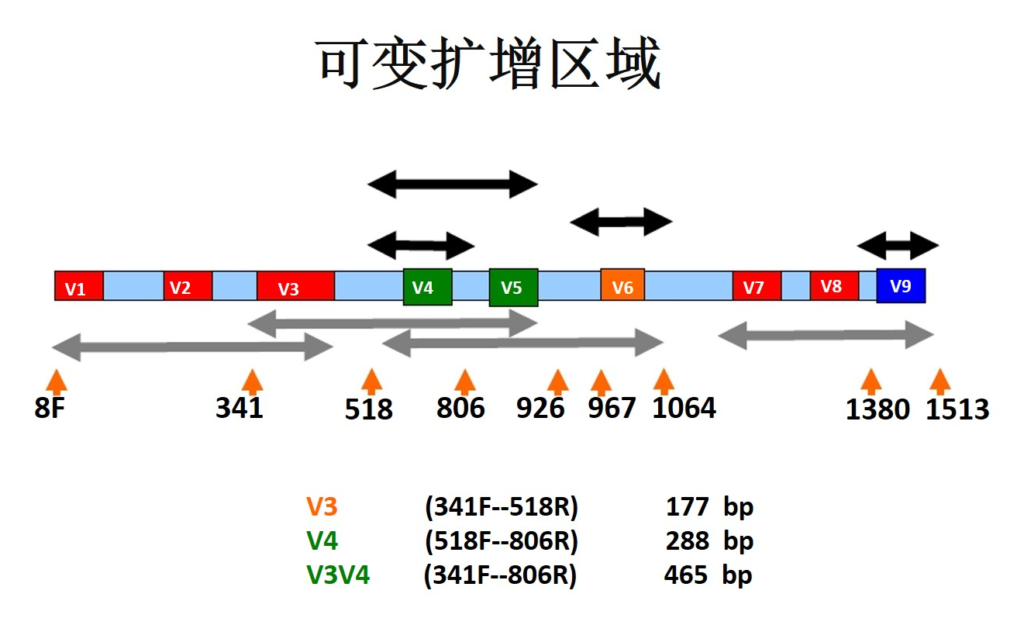
测序区域:V4,V3V4
测序数据量:10万 reads(V4); 5万 reads(V3V4)
周期:1-2周(V4);2-4周(V3V4)
①粪便样本包括肠道内容物:我们提供专门取样盒(免费)。人、大鼠、猪等,直接用取样盒里的棉签沾取约绿豆至黄豆大小的粪便至粪便保存液即可。颗粒状粪便,如小鼠,可根据粪便大小取几颗至粪便保存液即可。
备注:取样盒里有详细的粪便取样操作说明。
②人或者动物其他部位:例如口腔,鼻腔,阴道等:我们提供专门取样盒(免费)。取样方式也是用棉签沾取相应部位菌至保存液。但是根据研究项目,取样部位以及方式略有不同,这个不能一概而论,特殊项目最好单独咨询便于提供最佳方案。
③土壤,底泥水,污泥:需要5-10g的鲜样,土壤,底泥样若有沙石等需要先过筛后再送样。
④水体样,包括河流,湖畔,自来水等:需要先过滤膜,根据水体中含菌量选择一定体积的水体过滤膜,如自来水,一般需要5-15升水过滤膜,然后将滤膜送过来即可。
⑤DNA:浓度不低于5ng/ul, 总体积不少于40ul。


解决了从前期准备到怎么看报告、如何利用数据等问题,包括个性化图表的制作,离发表文章也就不远了,就像长跑已经能看到终点。
但仍然会有零星小问题,如何 “跑赢最后一公里”?
我们能做的就是为大家创建一个良好的交流环境,提供的交流平台致力于用最少的时间,最高效地解决问题。

售后服务:


售后服务:



样本需求量低:常规宏基因组建库建议样本量在500ng以上,公司研发实现了低当量微生物样本提取和建库,保证提取丰度以及片段完整性同时,样本量需求低于同行其他公司要求;对于样本获取困难的样本,也可以选择微量建库,样本量可低至10ng。
免费取样盒和针对性取样建议:粪便及环境样本提供取样盒助力临床/科研取样,人体口腔、痰液、腹水、脑脊液、尿液、皮肤等高寄主细胞含量样本可根据我们的处理方案简单处理后大幅降低宿主DNA比例。
严格标准的实验流程:自动化样品处理平台辅助,每轮设置阳性对照,上轮检测样本对照,阴性对照。评估污染,轮次比对,最大化减少误差,保证样本重复性和稳定性
Illumina测序平台:宏基因组测序(PE150)采用先进的Illumina Novaseq测序平台,快速、高效地读取高质量的测序数据、结合样品特点和数据的产出,充分挖掘环境样品中的微生物菌群和功能基因
大数据分析流程质量流程控制严格:优化的数据质量控制,包括过滤比对质量低、非特异性扩增、覆盖度低、低复杂度的序列,从而快速准确获得样本中微生物信息及其丰度信息,最大化提高质量数据
分析内容丰富全面:物种分析,基因预测与分析,多样性和相似性分析,功能分析,网路互作分析,代谢网络,关联分析等
完整详细的报告:提供质检实验报告,分析统计报告,分析报告解读,原始数据
高效个性化服务:在线项目系统方便您及时查看项目动态和下载报告以及与分析人员高效交流,支持个性化图表修改以及重新分组出报告。
价格低,周期快:包括提取,测序到分析,最快一周出报告。
大数据分析团队和多中心大项目分析经验(团队主要源自浙江大学,包括生物信息学,计算机,微生物以及统计分析等专业,积累了多年的大健康项目多中心项目分析经验,有助于宏基因组大数据,多样本,多表型,多组学联合分析
兼容性强的合作模式:有专门团队负责,提供切实可行的项目方案,兼顾临床和科研双需求模式。

研究背景:全球塑料产量飞速增长,而且呈持续上升的趋势,因此导致大量塑料废物排放到环境中,从沿海河口到大洋环流,从东大西洋到南太平洋海域。塑料废弃物具有化学稳定性和生物利用率低的特点,可长期存在于海洋中,从而影响海洋环境包括海洋生物的生存。
作为一个独特的底物,塑料碎片可以吸附海洋中的微生物并形成个“塑性球”。以生物膜形式存在于塑料碎片上的微生物群落。许多研究表明,无论是在海洋还是淡水生态系统中,附着在塑料碎片上微生物群落的组成明显不同于周围环境(水和沉积物),而且易受位置、时间和塑料类型的影响。
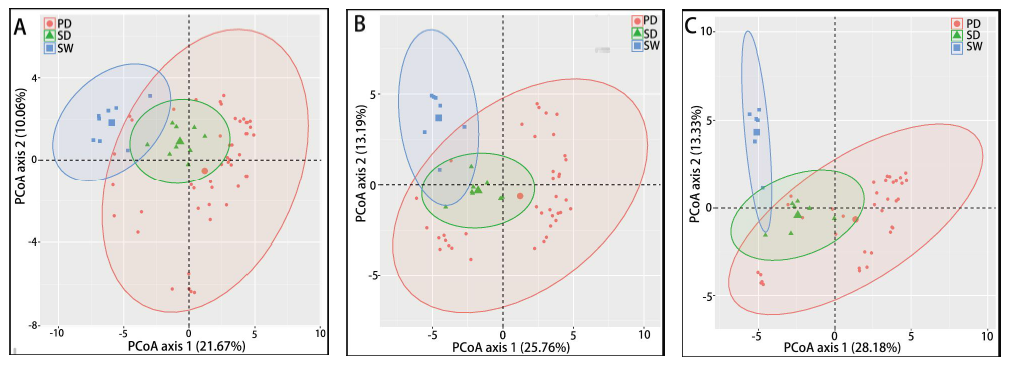
主要图表
两两群落差异指数的PCoA图
PCoA 图可以清楚地看到,SW区细菌群落的置信椭圆与pd和sd的置信椭圆有显著的偏差(p<0.05),而sd上细菌群落的置信椭圆几乎覆盖了pd的置信椭圆(p>0.05),这表明pd和sd上的细菌群落有相似之处。
不同样本和处理下的细菌群落( 前 10 位)丰度分布
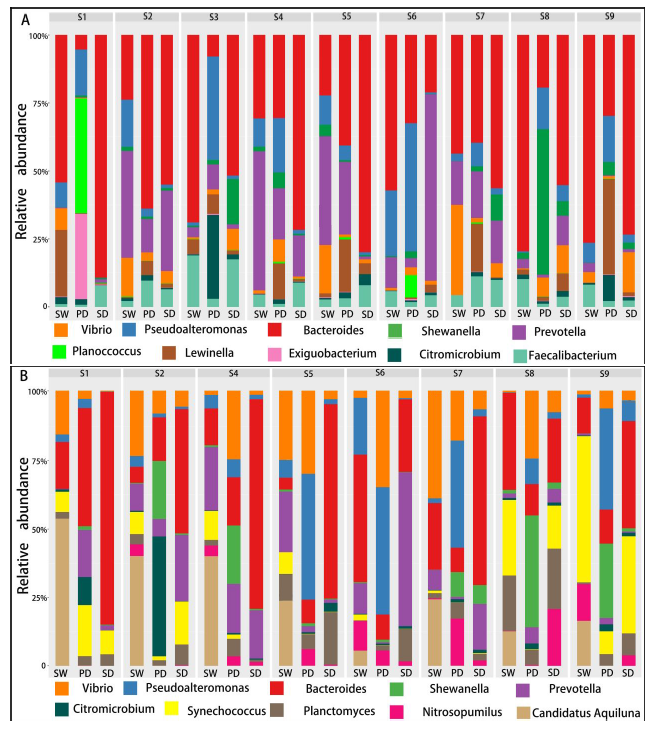
底物(SW、SD和Pd)上的主要属为细菌和假互斥单胞菌,暴露两周后,这些菌可能是分布广泛和适应性强的三种底物(SW、SD和PD)。暴露4周后,弧菌相对丰度增加.此外,暴露6周后,自养细菌(如扁平菌和硝酸菌)的数量增加。这三种底物上个细菌群落的生长模式也与3.2的结果一致。图5还显示,在6个星期内,在429个原位点中,假单胞菌在pd上的相对丰度高于sw和sd(anova,p<0.05)。
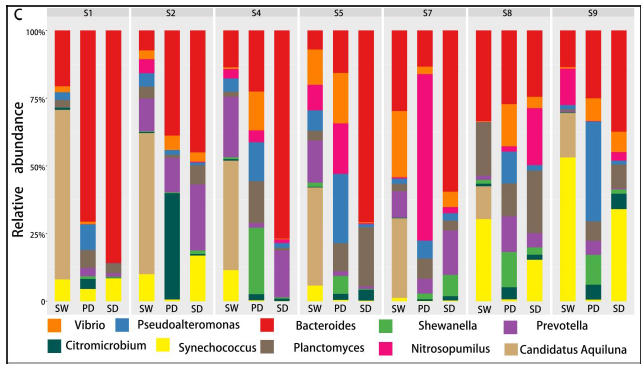
研究结论:首先,营养物质 (TN 和 TP) 与生物膜的平均生长速率呈正相关,而盐度与生物膜的平均生长速率呈负相关。盐度是影响PD的个细菌多样性的主要因素,而温度、溶解氧和养分(TN和TP)在类似的盐度条件下可能具有二次效应。尽管种聚合物类型对PD上的细菌群落的多样性具有较少的影响,但是在细菌群落中的一些属显示对PD的聚合物类型的选择性,并且倾向于将其优选的基质定殖。大的相对丰度SW、PD、SD间属显著差异。盐度是改变河口地区Pd条件致病菌富集的主要因素。另外,在种病原物种丰富的基础上,PD具有较高的致病性。

研究背景:研究表明遗传和环境影响都在I型糖尿病的发展中起作用,增加的遗传风险不足以引起疾病,环境因素也是需要的,而且起着至关重要的作用。肠道菌群也许就是这个重要的环境因素,肠道菌群在免疫系统的成熟中起重要作用,此外还影响自身免疫疾病发展。

不同遗传风险儿童的LDA差异菌群
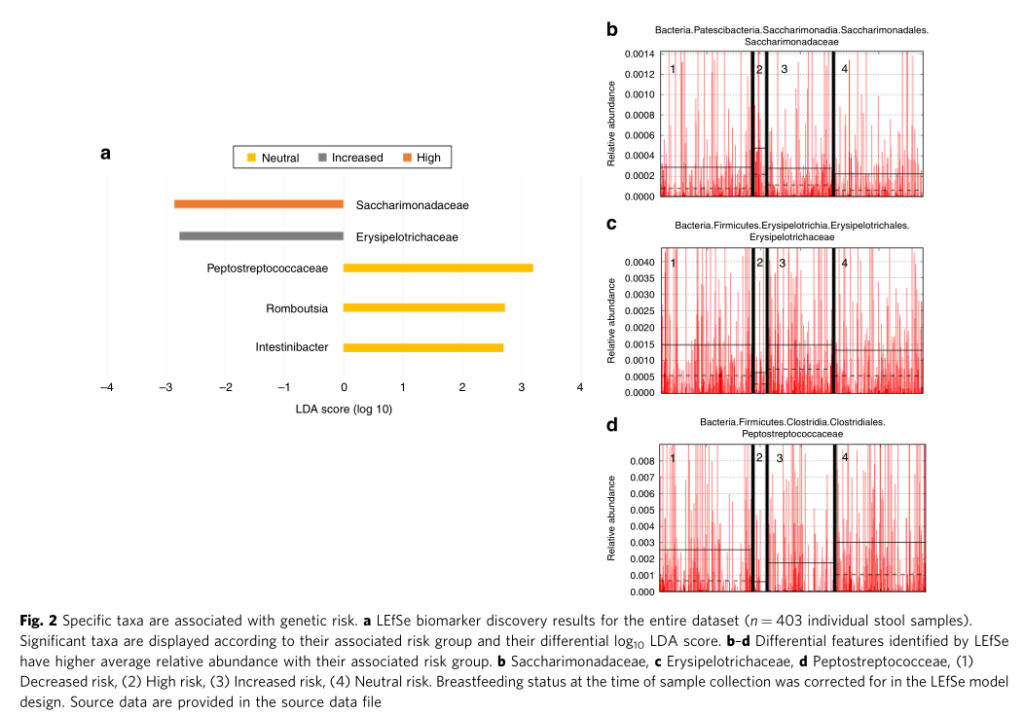
不同遗传风险分组中包含的常见菌属,部分存在特定分组中
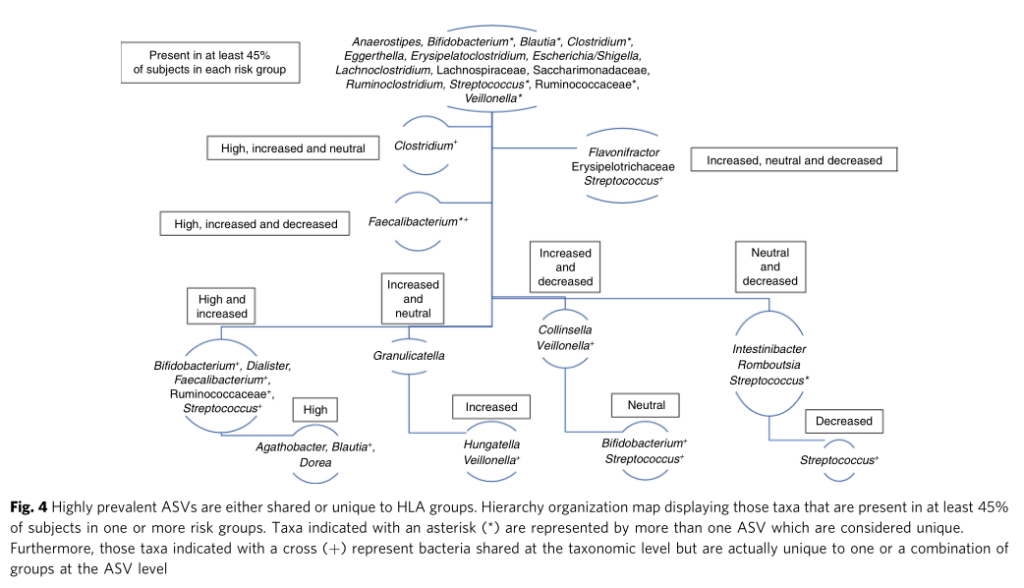
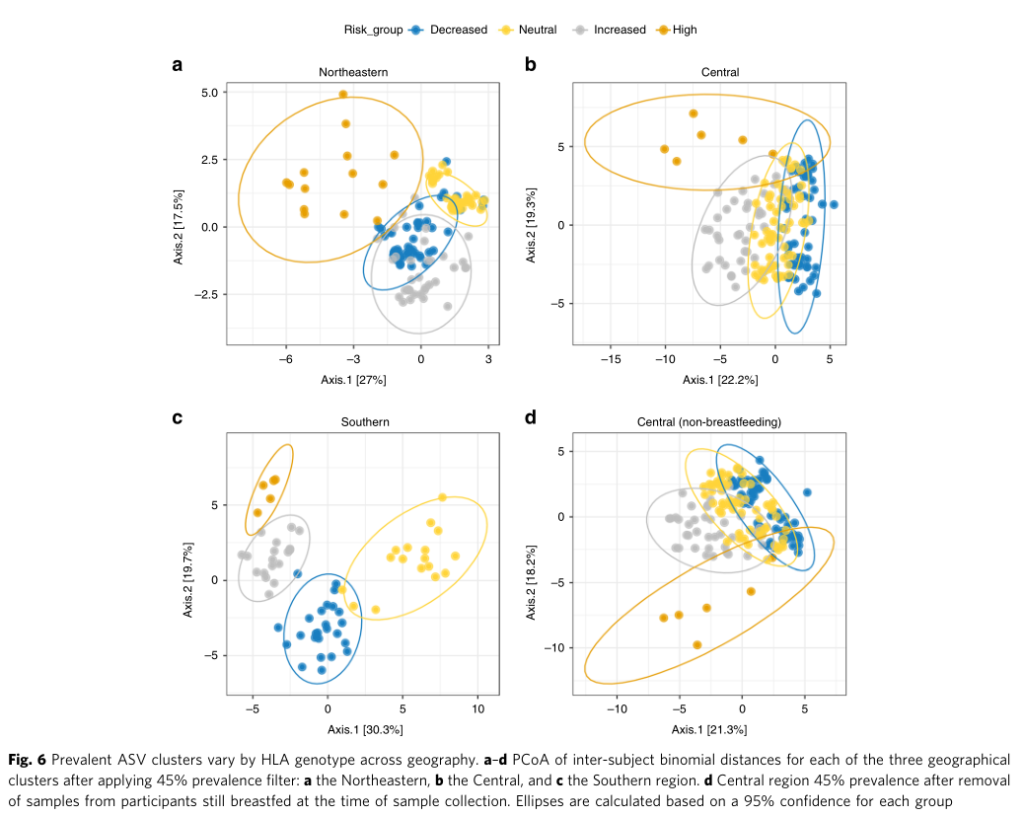
PCoA分析揭示不同遗传风险儿童肠道菌群的在不同地域样本中均存在显著差异

点评:针对I型糖尿病疾病发生过程中遗传HLA分型风险和对应肠道菌群菌的关联分析,揭示了特定肠道菌群与宿主特定遗传风险共同作用推进疾病发生。某些特定菌属可能无法在遗传高风险儿童肠道内定植,可能对疾病发生存在特定作用。此外对于其他遗传风险的自身免疫疾病也具有重要提示意义,例如乳糜泻和类风湿性关节炎。
Khan A, Rehman A, Ayub Q, et al. The composition of the blood microbiota and its relationship to osteoporosis-related clinical parameters [J]. Medicine in Microecology, 2023, 100097.
Xu S, Liu W, Gong L, et al. Association of ADRB2 gene polymorphisms and intestinal microbiota in Chinese Han adolescents [J]. Open Life Sciences, 2023,18(1), 20220646.
Wang X, Weng Y, Geng S, et al. Maternal procymidone exposure has lasting effects on murine gut-liver axis and glucolipid metabolism in offspring
[J]. Food and Chemical Toxicology, 2023,174, 113657.
Liu H, Xing Y, Wang Y, et al. Dendrobium officinale Polysaccharide Prevents Diabetes via the Regulation of Gut Microbiota in Prediabetic Mice [J]. Foods. 2023 , 8;12(12):2310.
Yan X, Yan J, Xiang Q, et al. Early-life gut microbiota in food allergic children and its impact on the development of allergic disease [J]. Ital J Pediatr. 2023, 9;49(1):148.
Xiang Q, Yan X, Lin X, et al. Intestinal Microflora Altered by Vancomycin Exposure in Early Life Up-regulates Type 2 Innate Lymphocyte and Aggravates Airway Inflammation in Asthmatic Mice [J]. Inflammation. 2023, 46(2):509-521.
Qiu J, Zhao L, Cheng Y, Chen Q, et al. Exploring the gut mycobiome: differential composition and clinical associations in hypertension, chronic kidney disease, and their comorbidity [J]. Front Immunol. 2023, 14;14:1317809.
Jiang S, Zhang B, Fan X, et al. Gut microbiome predicts selenium supplementation efficiency across different Chinese adult cohorts using hybrid modeling and feature refining [J]. Front Microbiol. 2023, Oct 17;14:1291010.
Liu T, Liu J, Wang P, et al. Effect of slurry ice on quality characteristics and microbiota composition of Pacific white shrimp during refrigerated storage
[J]. Journal of Agriculture and Food Research, 2023,14, 100792.
Chen B, Xu J, Lu H, et al. Remediation of benzo[a]pyrene contaminated soils by moderate chemical oxidation coupled with microbial degradation [J]. Sci Total Environ. 2023 , 1;871:161801.
Shouhua, Z., & Meilan, L. Microbial Diversity and Abundance in Pulmonary Tissue of Patients with Early-Stage Lung Cancer
[J]. Jundishapur Journal of Microbiology, 2023, 16(5).
Wei T, Liao Y, Wang Y, et al. Comparably Characterizing the Gut Microbial Communities of Amphipods from Littoral to Hadal Zones [J]. Journal of Marine Science and Engineering, 2023, 11(11), 2197.
Ji C, Luo Y, Yang J, et al. Polyhalogenated carbazoles induce hepatic metabolic disorders in mice via alteration in gut microbiota [J]. J Environ Sci (China). 2023, 127:603-614.
Han B, Yang F, Shen S, et al. Effects of soil habitat changes on antibiotic resistance genes and related microbiomes in paddy fields [J]. Sci Total Environ. 2023, 15;895:165109.
Li S, Niu Z, Wang M, et al. The occurrence and variations of extracellular antibiotic resistance genes in drinking water supply system: A potential risk to our health [J]. Journal of Cleaner Production, 2023, 402, 136714.
Gao X, Zhao J, Chen W, et al. Food and drug design for gut microbiota-directed regulation: Current experimental landscape and future innovation
[J]. Pharmacol Res. 2023,194:106867.
Han B, Shen S, Yang F, et al. Exploring antibiotic resistance load in paddy-upland rotation fields amended with commercial organic and chemical/slow release fertilizer [J]. Frontiers in Microbiology, 2023,14, 1184238.
Alsholi DM, Yacoub GS, Rehman AU, et al. Lactobacillus rhamnosus Attenuates Cisplatin-Induced Intestinal Mucositis in Mice via Modulating the Gut Microbiota and Improving Intestinal Inflammation [J]. Pathogens, 2023, 11;12(11):1340.
Xie X, Xu H, Shu R, et al. Clock gene Per3 deficiency disrupts circadian alterations of gut microbiota in mice [J]. Acta Biochim Biophys Sin (Shanghai). 2023,15; (12):2004-2007.
Xiao W, Zhang Q, Zhao S, et al. Citric acid secretion from rice roots contributes to reduction and immobilization of Cr(VI) by driving microbial sulfur and iron cycle in paddy soil [J]. Sci Total Environ. 2023, 1;854:158832.
Li R, Liu R, Chen L, et al. Microbiota from Exercise Mice Counteracts High-Fat High-Cholesterol Diet-Induced Cognitive Impairment in C57BL/6 Mice [J]. Oxid Med Cell Longev. 2023, 20;2023:2766250.
Liao J, Dou Y, Yang X, et al. Soil microbial community and their functional genes during grassland restoration [J]. Journal of Environmental Management, 2023, 325, 116488.
Liu F, Du J, Lin H, et al. The bladder microbiome of chronic kidney disease with associations to demographics, renal function, and serum cytokines
[J]. medRxiv, 2023-05.
Ullah H, Deng T, Ali M, et al. Sea Conch Peptides Hydrolysate Alleviates DSS-Induced Colitis in Mice through Immune Modulation and Gut Microbiota Restoration [J]. Molecules. 2023, 28;28(19):6849.
Zou S, Liu R, Luo Y, et al. Effects of fungal agents and biochar on odor emissions and microbial community dynamics during in-situ treatment of food waste [J]. Bioresour Technol. 2023; 380:129095.
Wu J, Zhang D, Zhao M, et al. Gut Microbiota Dysbiosis and Increased NLRP3 Levels in Patients with Pregnancy-Induced Hypertension
[J] . Curr Microbiol. 2023, 6;80(5):168.
Li R, Liu R, Chen L, et al. Microbiota from Exercise Mice Counteracts High-Fat High-Cholesterol Diet-Induced Cognitive Impairment in C57BL/6 Mice
[J]. Oxid Med Cell Longev. 2023, 20:2766250.
Xiao W, Zhang Q, Zhao S, et al. Citric acid secretion from rice roots contributes to reduction and immobilization of Cr(VI) by driving microbial sulfur and iron cycle in paddy soil [J]. Sci Total Environ. 2023, 16:158832.
Wang X, Weng Y, Geng S, et al. Maternal procymidone exposure has lasting effects on murine gut-liver axis and glucolipid metabolism in offspring [J]. Food Chem Toxicol. 2023, 174:113657.
Liao J, Dou Y, Yang X, et al. Soil microbial community and their functional genes during grassland restoration [J]. J Environ Manage. 2023, Jan 1;325(Pt A):116488.
Li S, Niu Z, Wang M, et al. The occurrence and variations of extracellular antibiotic resistance genes in drinking water supply system: A potential risk to our health [J]. Journal of Cleaner Production. 2023, 20, 136714.
Hu Y, Li J, Ni F, et al. CAR-T cell therapy-related cytokine release syndrome and therapeutic response is modulated by the gut microbiome in hematologic malignancies[J]. Nature communications, 2022, 13(1): 1-14.
Lou M, Cao A, Jin C, et al. Deviated and early unsustainable stunted development of gut microbiota in children with autism spectrum disorder [J]. Gut, 2021 Dec 20:gutjnl-2021-325115.
Chen C, Du Y, Liu Y, et al. Characteristics of gastric cancer gut microbiome according to tumor stage and age segmentation[J]. Applied Microbiology and Biotechnology, 2022, 106(19): 6671-6687.
Chen C, Shen J, Du Y, et al. Characteristics of gut microbiota in patients with gastric cancer by surgery, chemotherapy and lymph node metastasis[J]. Clinical and Translational Oncology, 2022, 24(11): 2181-2190.
Shen J, Jin CL, Zhang YY, et al. A multiple-dimension model for microbiota of patients with colorectal cancer from normal participants and other intestinal disorders[J]. Applied Microbial and Cell Physiology, 2022, February 26
Wang YB, Shang LC, Zhong CH, et al. Transplantation of feces from mice with Alzheimer’s disease promoted lung cancer growth [J]. Biochemical and Biophysical Research Communications. 2022, 600: 67-74
Zhang G, Pang Y, Zhou Y, et al. Effect of dissolved oxygen on N2O release in the sewer system during controlling hydrogen sulfide by nitrate dosing [J]. Science of The Total Environment. 2022, Apr 10;816:151581.
Zhao Y, Huang J, Li T, et al. Berberine ameliorates aGVHD by gut microbiota remodelling, TLR4 signalling suppression and colonic barrier repairment for NLRP3 inflammasome inhibition[J]. Journal of Cellular and Molecular Medicine, 2022, 26(4): 1060-1070.
Hang, ZC, Cai SL, Lei T, et al. Transfer of Tumor-Bearing Mice Intestinal Flora Can Ameliorate Cognition in Alzheimers Disease Mice[J]. J Alzheimers Dis. 2022, 86(3):1287-1300.
Zhu W, Yan M, Cao H, et al. Effects of Clostridium butyricum Capsules Combined with Rosuvastatin on Intestinal Flora, Lipid Metabolism, Liver Function and Inflammation in NAFLD Patients[J]. Cellular and Molecular Biology. 2022, 68(2): 64-69.
Yang F, Wang Z, Zhao D, et al. Food-derived Crassostrea gigas peptides self-assembled supramolecules for scarless healing[J]. Composites Part B: Engineering, 2022, 246: 110265.
Yu T, Ji L, Lou L, et al. Fusobacterium nucleatum Affects Cell Apoptosis by Regulating Intestinal Flora and Metabolites to Promote the Development of Colorectal Cancer[J]. Frontiers in microbiology. 2022, 13.
Qin Z, Yuan X, Liu J, et al. Albuca Bracteata Polysaccharides Attenuate AOM/DSS Induced Colon Tumorigenesis via Regulating Oxidative Stress, Inflammation and Gut Microbiota in Mice [J]. Frontiers in pharmacology. 2022, 13: 833077.
Wang R, Deng Y, Zhang Y, et al. Modulation of Intestinal Barrier, Inflammatory Response, and Gut Microbiota by Pediococcus pentosaceus zy-B Alleviates Vibrio parahaemolyticus Infection in C57BL/6J Mice[J]. Journal of Agricultural and Food Chemistry. 2022, 70(6): 1865-1877.
Hu L, Sun L, Zhou J, et al. Impact of a hexafluoropropylene oxide trimer acid (HFPO-TA) exposure on impairing the gut microbiota in mice. Chemosphere, 2022: 134951.
Yang T, Tang G, Li L, et al. Interactions between bacteria and eukaryotic microorganisms and their response to soil properties and heavy metal exchangeability nearby a coal-fired power plant. Chemosphere. 2022, 302: 134829.
Chen G, Zeng R, Wang X, et al. Antithrombotic Activity of Heparinoid G2 and Its Derivatives from the Clam Coelomactra antiquata. Marine Drugs, 2022, 20(1): 50.
Khan A I, Rehman A U, Farooqui N A, et al. Effects of shrimp peptide hydrolysate on intestinal microbiota restoration and immune modulation in cyclophosphamide-treated mice. Molecules. 2022, 27(5): 1720.
He L, Xing Y, Ren X, et al. Mulberry Leaf Extract Improves Metabolic Syndrome by Alleviating Lipid Accumulation In Vitro and In Vivo[J]. Molecules, 2022, 27(16): 5111.
Rehman A U, Siddiqui N Z, Farooqui N A, et al. Morchella esculenta mushroom polysaccharide attenuates diabetes and modulates intestinal permeability and gut microbiota in a type 2 diabetic mice model[J]. Frontiers in Nutrition, 2022, 9.
Wang X, Hu L, Wang C, et al. Cross-generational effects of maternal exposure to imazalil on anaerobic components and carnitine absorption associated with OCTN2 expression in mice[J]. Chemosphere, 2022, 308: 136542.
Zhang G, Wang G, Zhou Y, et al. Simultaneous use of nitrate and calcium peroxide to control sulfide and greenhouse gas emission in sewers[J]. Science of The Total Environment, 2022, 855: 158913.
Ma C J, He Y, Jin X, et al. Light-regulated nitric oxide release from hydrogel-forming microneedles integrated with graphene oxide for biofilm-infected-wound healing[J]. Biomaterials Advances, 2022, 134: 112555.
Dong Y, Sui L, Yang F, et al. Reducing the intestinal side effects of acarbose by baicalein through the regulation of gut microbiota: An in vitro study[J]. Food Chemistry, 2022, 394: 133561.
Ji C, Luo Y, Yang J, et al. Polyhalogenated carbazoles induce hepatic metabolic disorders in mice via alteration in gut microbiota[J]. Journal of Environmental Sciences, 2022, 127: 603-614.
Hu J, Wei S, Gu Y, et al. Gut Mycobiome in Patients With Chronic Kidney Disease Was Altered and Associated With Immunological Profiles[J]. Frontiers in immunology, 2022, 13.
Xiao W, Zhang Q, Zhao S, et al. Citric acid secretion from rice roots contributes to reduction and immobilization of Cr (VI) by driving microbial sulfur and iron cycle in paddy soil[J]. Science of The Total Environment, 2022, 854: 158832.
Gu Y, Chen H, Li X, et al. Lactobacillus paracasei IMC 502 ameliorate type 2 diabetes by mediating gut microbiota‐SCFAs‐hormone/inflammation pathway in mice[J]. Journal of the Science of Food and Agriculture, 2022.
Sun D, Wang C, Sun L, et al. Preliminary Report on Intestinal Flora Disorder, Faecal Short-Chain Fatty Acid Level Decline and Intestinal Mucosal Tissue Weakening Caused by Litchi Extract to Induce Systemic Inflammation in HFA Mice. Nutrients. 2022, 14(4): 776.
Zhou S P, Zhou H Y, Sun J C, et al. Bacterial dynamics and functions driven by bulking agents to enhance organic degradation in food waste in-situ rapid biological reduction (IRBR)[J]. Bioprocess and Biosystems Engineering, 2022, 45(4): 689-700.
Wen Y, Feng S, Dai H, et al. Intestinal dysbacteriosis-propelled T helper 17 cells activation mediate the perioperative neurocognitive disorder induced by anesthesia/surgery in aged rats[J]. Neuroscience Letters, 2022, 783: 136741.
Gu Y, Li X, Chen H,et al. Antidiabetic effects of multi-species probiotic and its fermented milk in mice via restoring gut microbiota and intestinal barrier. Food Bioscience. 2022, Volume 47
Wang R, Deng Y, Zhang Y, et al. Modulation of Intestinal Barrier, Inflammatory Response, and Gut Microbiota by Pediococcus pentosaceus zy-B Alleviates Vibrio parahaemolyticus Infection in C57BL/6J Mice. J Agric Food Chem. 2022, 16;70(6):1865-1877.
Zhang M, Sun Q, Chen P, et al. How microorganisms tell the truth of potentially toxic elements pollution in environment[J]. Journal of Hazardous Materials, 2022, 431: 128456.
Wang Q, Guan C, Han J, et al. Microplastics in China Sea: analysis, status, source, and fate[J]. Science of The Total Environment, 2022, 803: 149887.
Sun D, Wang C, Sun L, et al. Gooneratne R. Preliminary Report on Intestinal Flora Disorder, Faecal Short-Chain Fatty Acid Level Decline and Intestinal Mucosal Tissue Weakening Caused by Litchi Extract to Induce Systemic Inflammation in HFA Mice. Nutrients. 2022, Feb 12;14(4):776.
Sun Y, Ling C, Liu L, et al. Effects of Whey Protein or Its Hydrolysate Supplements Combined with an Energy-Restricted Diet on Weight Loss: A Randomized Controlled Trial in Older Women. Nutrients. 2022, 28;14(21):4540.
Ye S, Wang L, Li S, et al. The correlation between dysfunctional intestinal flora and pathology feature of patients with pulmonary tuberculosis. Front. Cell. Infect. Microbiol. 2022,
Ma CJ, He Y, Jin X, et al. Light-regulated nitric oxide release from hydrogel-forming microneedles integrated with graphene oxide for biofilm-infected-wound healing. Biomater Adv. 2022, 134:112555.
Wang X, Hu L, Wang C, et al. Cross-generational effects of maternal exposure to imazalil on anaerobic components and carnitine absorption associated with OCTN2 expression in mice. Chemosphere. 2022, 308(Pt 3):136542.
Xiao W, Zhang Q, Zhao S, et al. Citric acid secretion from rice roots contributes to reduction and immobilization of Cr(VI) by driving microbial sulfur and iron cycle in paddy soil. Science of The Total Environment, 2022, 16:158832.
Gu Y, Chen H, Li X, et al Lactobacillus paracasei IMC 502 ameliorates type 2 diabetes by mediating gut microbiota-SCFA-hormone/inflammation pathway in mice. J Sci Food Agric. 2022, 11.
Liao J, Dou Y, Yang X, et al. Soil microbial community and their functional genes during grassland restoration. J Environ Manage. 2023, 1;325(Pt A):116488.
Ou-Yang YN, Yuan MD, Yang ZM, et al. Revealing the Pathogenesis of Salt-Sensitive Hypertension in Dahl Salt-Sensitive Rats through Integrated Multi-Omics Analysis. Metabolites. 2022, 7;12(11):1076.
Dong Y, Sui L, Yang F, et al. Reducing the intestinal side effects of acarbose by baicalein through the regulation of gut microbiota: An in vitro study. Food Chem. 2022, 15;394:133561.
Zhang G, Wang G, Zhou Y, et al. Simultaneous use of nitrate and calcium peroxide to control sulfide and greenhouse gas emission in sewers. Sci Total Environ. 2023, 10;855:158913.
Fang Z, Chen Y, Li Y, et al. Oleic Acid Facilitates Cd Excretion by Increasing the Abundance of Burkholderia in Cd-Exposed Mice. Int J Mol Sci. 2022, 25;23(23):14718.
Siddiqui NZ, Rehman AU, Yousuf W, et al. Effect of crude polysaccharide from seaweed, Dictyopteris divaricata (CDDP) on gut microbiota restoration and anti-diabetic activity in streptozotocin (STZ)-induced T1DM mice. Gut Pathog. 2022, 17;14(1):39.
Hu J, Wei S, Gu Y, et al. Gut Mycobiome in Patients With Chronic Kidney Disease Was Altered and Associated With Immunological Profiles. Front Immunol. 2022, 16;13:843695.

肥胖和营养不良是普遍面临的健康问题,而肠道菌群在其中扮演的什么样的角色?最近一项研究通过交叉过量、过低饮食干预以及随机双盲万古霉素抗生素实验,揭示了不同热量摄入和抗生素会破坏肠道菌群特定代谢产物(如:丁酸和其他短链脂肪酸)影响肠道屏障,并改变特定微生物群落(如阿克曼菌),这种变化是粪便热量流失的原因,表明肠道菌群在饮食能量吸收中起着因果的作用。

单位:美国国家卫生所
期刊: Nature Medicine 《自然医学》
摘要
文章揭示了营养不良和口服万古霉素对人体肠道菌群和营养吸收的影响。以往的研究发现,营养不良显著增加粪便热量损失,且降低了拟杆菌门和厚壁菌门的相对丰度,这是人类肠道中的两个主要门。还有各种证据表明,早期使用抗生素与儿童体重增加有关,在营养不良中,抗生素促进生长的证据好坏参半。在本文中,研究人员进行了一项长期的住院研究,使用了两种干预措施。在每个实验中,研究人员都测量了过量饮食和饮食不足期间的粪便卡路里流失,这是营养吸收的直接表征,第一阶段是随机交叉饮食干预,所有受试者按随机顺序接受3天的过量和不足饮食喂养。第二阶段是随机、双盲、安慰剂对照的药物干预,使用万古霉素或安慰剂(NCT02037295)。研究人员观察到,肠道菌群结构在营养不良和饮食过量的情况下有轻微的变化,但口服万古霉素后群落结构伴随着多样性降低发生了更显著的变化。两种干预措施中嗜粘蛋白艾克曼菌的丰度都增加了,这导致了更多的粪便热量流失。这些结果表明,营养吸收对环境扰动敏感,并与临床前模型相关,表明肠道菌群在饮食能量吸收中可能起因果作用。
背景
全球肥胖症的流行促使人们努力确定影响能量平衡的环境和宿主因素,能量平衡的定义是能量摄入和消耗之间的平衡。虽然卡路里消耗是能量摄入量的关键决定因素,但个体消化和吸收所消耗的饮食底物的能力的不同程度也可能影响能量平衡。在过去的十年中,利用啮齿动物模型得到的研究成果,支持了数以万亿计的微生物在人类胃肠道(肠道菌群)的因果作用,由于它们对能量摄入和消耗的广泛影响,形成了能量平衡的个体间变化。但可惜的是,尚无直接证据表明微生物群对营养吸收有影响.
实验设计
实验对象:
选取了除葡萄糖耐受和肥胖症以外的27名健康志愿者(男17名,女10名,年龄35.1±7.3,BMI 32.3±8.0),其中25名完成了整个实验。在整个研究过程中,参与者都是住院患者,每天都接受监测,没有被报告过副作用,特别是腹泻或腹部症状。
设计方案:
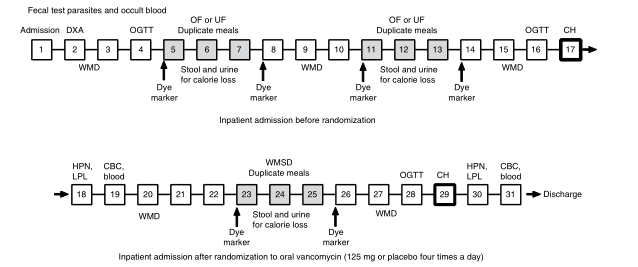
第一阶段:随机的交叉饮食干预
所有受试者均以过量(OF)和节食(UF)三天的随机顺序进行饮食,期间会有3天的洗脱期。OF为WMD的150%的饮食,UF为WMD的50%的饮食。
第二阶段:药物干预
同样的受试者被随机分为安慰剂组和口服万古霉素组
注:
(WMD)体重维持饮食【20%大卡蛋白质;30%大卡脂肪;50%大卡碳水化合物】;(DXA)双能X线吸收法,用于骨密度测定;(OGTT) 口服葡萄糖耐量试验;(Dye marker)表示粪便收集所使用的染料标记物;(CH) 间接量热法,用于评估24小时能量消耗;(HPN,LPL) 肝素诱导的LPL测定;(CBC) 全血细胞计数
主要结果
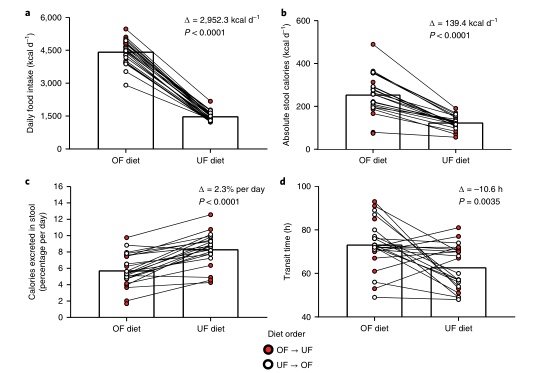
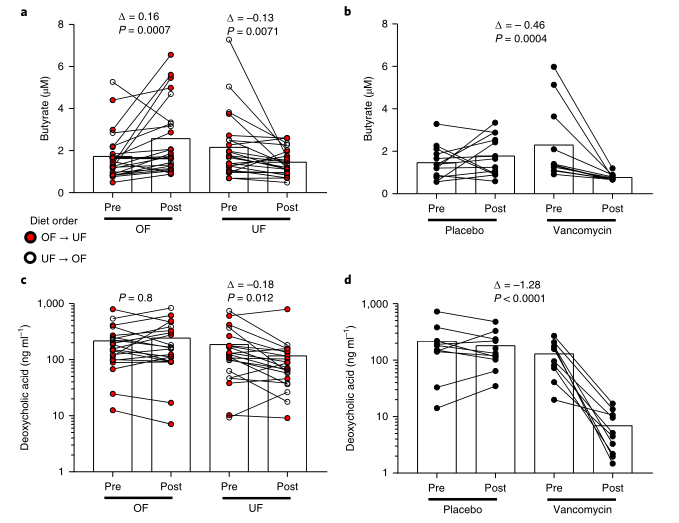
“ Δ ”表示组间的平均差异;“ P ”表示双边配对学生t检验; 红点表示开始饮食阶段为OF的受试者,白点表示开始饮食阶段为UF的受试者
a):过量饮食组(4,446.5 ± 547.8 kcal d−1)的每日摄入热量显著高于营养不良组(1,494.2 ± 211.0 kcal d−1),
b):与营养不良的人相比,过量饮食的人每天消耗的卡路里绝对数量明显更高。
c):当以每天摄入的卡路里的百分比来表示时,UF期间的粪便热量流失相对于OF明显更大,且有很强且显著的体内相关性(Pearson’s r = 0.70, P = 0.004)
d):UF比OF转运时间快,转运时间被定义为每次干预期间染料标记第一次出现和最后一次出现之间的时间(以h为单位)。
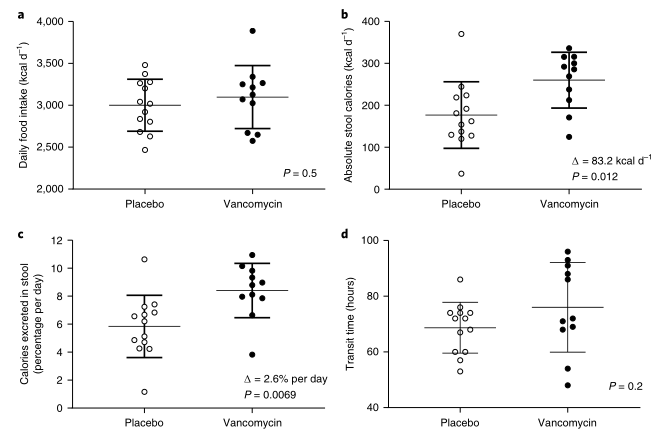
Δ ”表示组间的平均差异;“ P ”表示双边非配对学生t检验;误差条表示95%置信区间的均值
a):3天的平均摄入热量。安慰剂组(Placebo)和万古霉素组(Vancomycin)每天摄入的热量是相当的。
b):万古霉素导致粪便热量流失显著增加。
c):粪便平均热量流失百分比。
d):万古霉素组和安慰剂组的转运时间没有差异(P=0.2)
2.使用扩增子和宏基因组测序相结合的方法,评估了饮食干预和药物干预对肠道微生物群落结构的影响。在饮食干预组间分析中发现了几种对饮食敏感的细菌物种。在药物干预组间分析中也发现了几种显著富集的物种。且扩增子和宏基因组测序结果具有一致性。尽管存在这些差异,但在这两种干扰下,有两种细菌的种类都发生了一致的变化。嗜粘蛋白艾克曼菌和一种隶属于Lachnospiraceae NK4A136群的一个未确认的种。
饮食干预:
a):整体肠道菌群在OF和UF期间的定植。在UF期间,肠道菌群由于相对地缺乏营养,这可能导致细菌定殖的减少。但是在UF期间与OF相比,总的定殖水平显著增加(P=0.01,双边配对的Wilcoxon检验)。
b):两种饮食措施的Bray-Curtis距离的PCoA第一主坐标的差异。受试者按饮食顺序分开;c):在两种饮食措施期间每天从10个受试者中取样,其PcoA分析结果。每个点代表一个样本,其中颜色表示受试者身份,形状表示饮食方式(WM,保持体重);d):随着时间的推移,微生物群落到基线样本(第一天)的距离。颜色代表饮食,每个点代表一个样本字母表示受试者身份。发现在整个3天的UF和OF干预过程中,总体微生物群落结构保持不变,在基线上保持显著的个体间差异。
e):在被评估的10个个体中,有2个个体被发现在UF组中的微生物群落总体结构上有显著差异,这可能表明肠道菌群的基线关系到其对饮食干预的敏感性。(对每个受试者进行Adonis分析,R2值量化不同饮食上的方差,“*”表示P<0.05,纵坐标的字母表示受试者身份)
f):在整个第一阶段的16天内,微生物的总体Shannon多样性相对稳定。
g):通过16SrRNA鉴定出的4个与饮食显著相关的物种:嗜粘蛋白艾克曼菌、Bacteroides coprocola、毛螺旋菌、瘤胃球菌。纵坐标为相对丰度,横坐标为时间(d)。
(经FDR调整后的P值,Q < 0.1,双侧Wald检验的DESeq2)。线代表mean ± s.e.m.
h):基于宏基因组测序的物种分类,发现了9种细菌在不同饮食组间的显著差异(FDR < 0.05, 配对的Wilcoxon检验),每个点代表一个样本,纵坐标表示相对丰度。这与16S rRNA基因测序的分析一致。
药物干预:
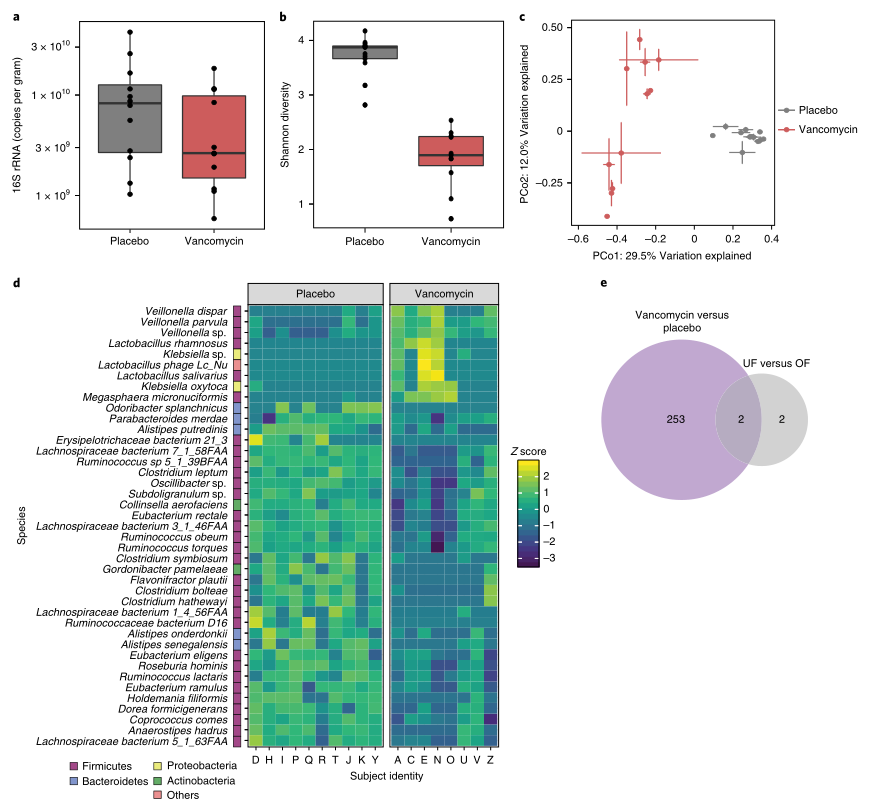
a):不同治疗组的肠道菌群的定植水平无明显差异(P = 0.26,双侧非配对Wilcoxon检验)。每个点代表每个受试者样本的平均值。
b):不同治疗组的微生物群落Shannon多样性。(P <0.0001,双侧非配对Wilcoxon检验)。每个点代表每个受试者样本的平均值。
c):不同治疗组间的基于Bray-Curtis距离的PCoA分析。发现万古霉素对肠道微生物群落结构也有显著影响,超过了先前存在的个体间差异。(每个数据点都显示了每个受试者样本的平均值和标准差)
d):通过宏基因组测序得到的不同治疗组间差异丰富物种的热图。横坐标代表受试者样本,纵坐标代表物种(FDR<0.05,双侧非配对Wilcoxon检验)。其中万古霉素组的物种相对丰度有下降的趋势,31种下降,10种增加。与16SrRNA基因测序数据一致,在万古霉素处理的个体中,有3个Veillonella菌属显著富集,还检测到多种乳杆菌和克雷伯氏菌以及感染乳杆菌的强毒噬菌体(LcNu)的富集。
e):万古霉素和安慰剂与UF和OF之间的16S rRNA序列变异的Venn图。
3. 对UF和OF组、万古霉素组和安慰剂组与代谢疾病相关的宿主-微生物相互作用的机制研究。丁酸盐是肠道细菌代谢的主要终产物。脱氧胆酸,一种次级胆酸,较低的浓度可能意味着肠道屏障的保护和较低的营养吸收。
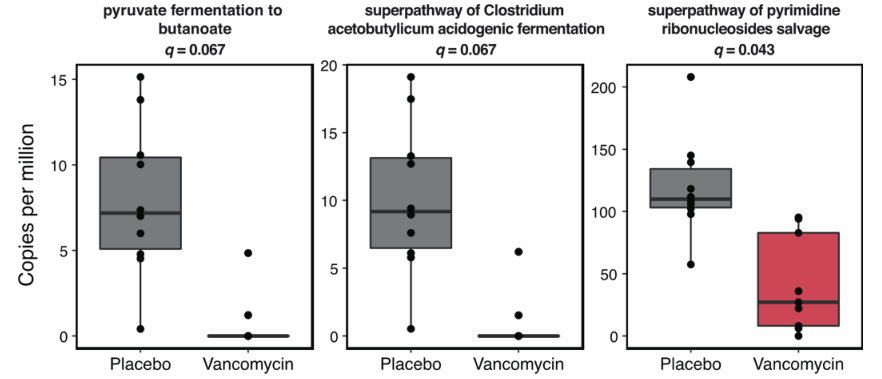
通过分析KEGG代谢途径,发现了三条在万古霉素和安慰剂之间差异丰富的代谢途径。其中的两条(丙酮酸发酵为丁酸和乙酰丁酸梭菌产酸发酵超途径)与糖发酵为主要短链脂肪酸丁酸盐(又名丁酸)有关。说明万古霉素治疗期间肠道菌群可能会减少丁酸的产生或细菌代谢。
“ Δ ”表示组内干预措施前后的差异
a):OF和UF期间丁酸盐的平均浓度;b):万古霉素组和安慰剂组的丁酸盐的平均浓度。UF和万古霉素处理组的丁酸盐平均浓度均显著下降,这支持肠道菌群在这些干预过程中获取营养的能力下降的说法。
c):OF和UF期间脱氧胆酸的平均浓度;d):万古霉素组和安慰剂组的脱氧胆酸的平均浓度。脱氧胆酸的平均浓度在UF和万古霉素处理组也显著降低了。
结论
在这项分两个阶段的研究中,研究人员直接测量了摄入的和粪便的卡路里,证明了限制热量摄入和口服万古霉素都会导致粪便热量流失增加,血浆丁酸水平降低。这种影响的幅度约为摄入卡路里的2.5%,这将转化为100公斤受试者在1年内体重减轻约1.2公斤。另一方面,研究人员观察到UF和OF对肠道微生物群落结构的轻微干扰,而口服万古霉素引起了广泛的变化,降低了肠道细菌的多样性,并使肠道细菌的相对丰度发生了显著变化。这两种扰动都导致了嗜粘蛋白艾克曼菌的相对丰度增加。而在人一项基于人体的随机、双盲、安慰剂对照的先导研究显示,补充嗜粘蛋白艾克曼菌有降低体重和脂肪量的趋势。此外,体外研究表明,丁酸和其他短链脂肪酸可以刺激肠道屏障的形成,从而保护肠道免受LPSs的破坏。根据研究人员的观察表明,热量摄入和抗生素可能会破坏这些代谢产物和其他调节屏障功能的微生物代谢产物之间的平衡,导致营养吸收的改变。研究中也有一些局限性,比如尽管肠道微生物群落结构和代谢物浓度发生广泛变化,但未发现口服万古霉素对粪便热量的影响、不知道其他抗生素是否会对粪便卡路里产生影响、任何关于营养吸收的机制都是推测出来的,因为没能直接评估营养在肠道中的传输等。

原创 谷禾健康


近年的研究热点集中于环境和生物体相互作用的微生物群体,而大量复杂的微生物群体存在培养困难,构成复杂(包括细菌、古菌、真菌、原生生物、病毒甚至小型真核生物)。因此如何用高通量精准的了解这些群体的构成,基因功能分布以及具体的表达活性和代谢状况成为首要问题。
高通量测序技术的发展,让我们可以不经过培养,一次性了解微生物群落构成甚至基因代谢组成。
随着技术的进步,检测方法也逐渐丰富,对应的分析手段和软件算法也逐步完善,使我们可以根据研究需要选择不同的检测和分析策略来获得海量的数据并进行相应的研究分析。


免于培养的微生物学研究方法主要基于测序,高通量测序使我们一次可以获得整个微生物群体的数据信息,简单来说包括两种策略:
1、基于特定标记基因的扩增测序方案(常见的16s,ITs,18s或特定功能基因)
2、对整个群落DNA进行测序,获取全部微生物基因组进而进行分类和功能分析的策略(鸟枪法宏基因组测序shotgun metagenomics)。
基于16s基因的分析方法
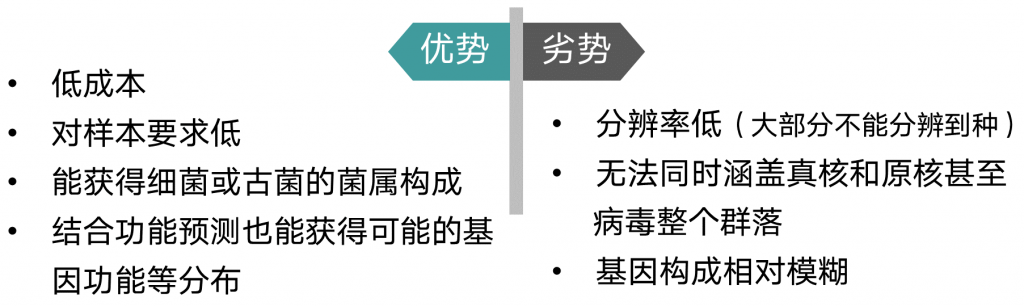

由于其极低的成本,对于样本DNA的低要求非常适合于大规模群体样本的调查和分析,随着DADA2等分析方法的改进,物种分类精度和准确度也有所提升,加上PICRUST等功能预测方法一定程度上弥补了基因信息的缺失,因此16s这类基于基因的微生物研究方法仍然是不可或缺的方案。
下表列了16s常见的分析软件,目前QIIME2作为整合包使用最为方便,VSEARCH也作为UPARSE的开源版本使用也非常广泛。
16s测序的分析流程如下图,获得序列经过聚类后获得OTU或ASV,并得到相对丰度。
经过PICRUSt可以得到预测的基因分类丰度,进而进行alpha多样性和Beta多样性以及组间差异和相关性分析。
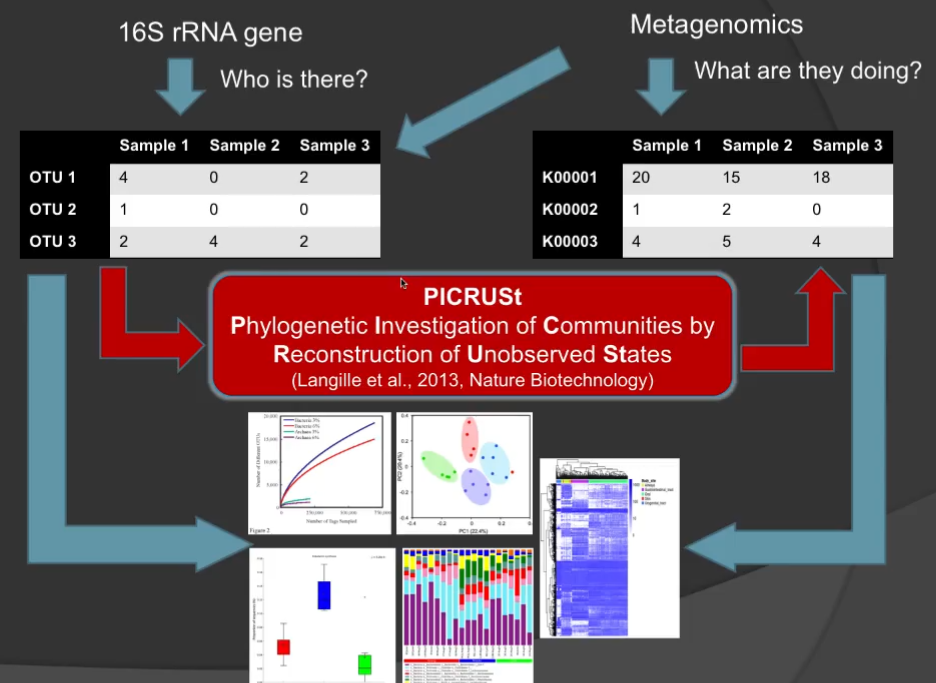
PICRSt的工作原理如下图,将OTU表内16s序列进行对应物种16s拷贝数标准化后,将物种丰度乘以已经整理好的物种的基因注释数表就获得基因的预测丰度。
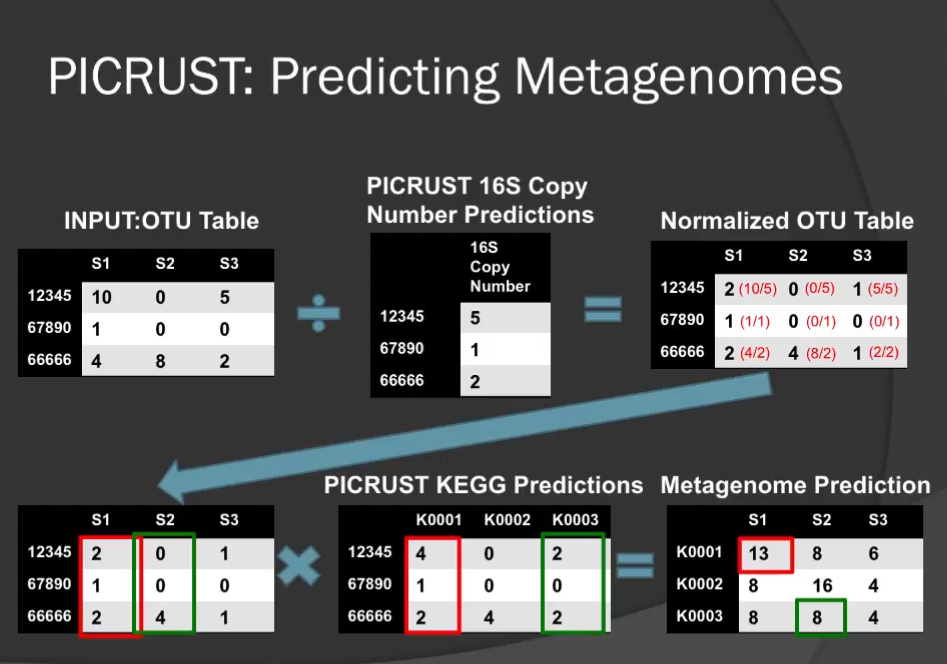
浅宏基因组测序方案是去年knights-lab在msystems上发表的针对16s分辨率和宏基因组高成本之间的一个折中方案,通过降低测序深度,每个样本50万reads,但是物种的分辨率并没有低于一般宏基因组(普遍5~10G数据量)。
不通过拼接组装,直接基于kraken2等kmer,或MetaPhlAn2等标记基因的参考基因组方法进行种属丰度分类。结合其到菌株的物种分类和丰度数据可较16s方案下的PICRUST更加准确的预测基因构成。

Hillmann B, Al-Ghalith GA, Shields-Cutler RR, Zhu Q, Gohl DM, Beckman KB, Knight R, Knights D. 2018. Evaluating the information content of shallow shotgun metagenomics. mSystems 3:e00069-18. https://doi.org/10.1128/mSystems.00069-18.
我们发现有些小伙伴的需求是:
想要获得更全和更精细分类精度同时不需要获得完整基因组序列和重建菌群基因的。
那么这时候,我们提供的浅宏基因组测序就可以成为很好的选择,其成本低(快要接近16s测序分析的价格了,文末有福利),分析简便快速,同样能获得宏基因组的基本丰度数据。不过浅宏基因组也有其适用范围,根据样品类型的不同,一些样品可能包含 >99%的人类宿主DNA,这不仅增加了序列成本,而且给测量带来了不确定性。
在许多研究中也会采取在进行宏基因组测序文库的准备之前去除宿主DNA的方法。但是,在去除宿主DNA后,可能没有足够的微生物基因组DNA用于宏基因组测序,这通常需要最少50ng的输入。因此浅宏基因组较适合于宿主DNA含量较低的样本,如人类粪便、水体、土壤等;而如口腔唾液、肺泡灌洗液、血液等人体体液类样本就不太适合。
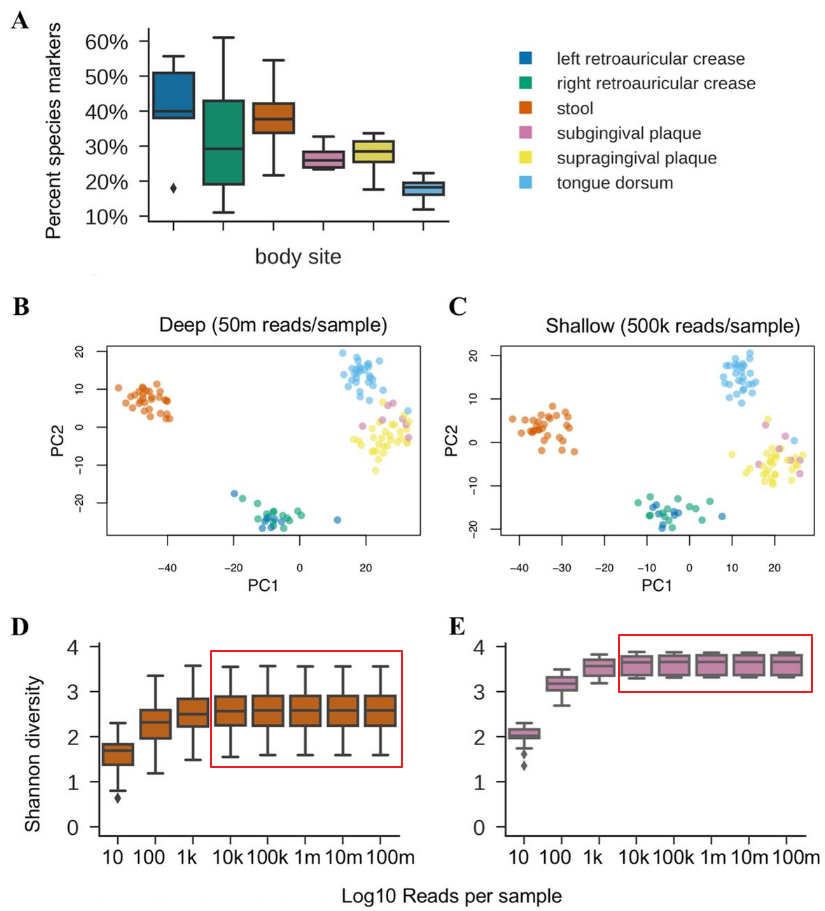
下图是宏基因组测序数据中比对到人类基因组的序列比例,根据样本类型不同而不同。

我们可以免费提供针对粪便及环境样本助力临床/科研取样。
人体口腔、痰液、腹水、脑脊液、尿液、皮肤、阴道分泌物等高寄主细胞含量样本可根据我们的处理方案简单处理后大幅降低宿主DNA比例。
高宿主含量DNA样本(包括唾液、血液、肺泡灌洗液、腹水、阴道分泌物和黏膜类样品)的取样前处
将200微升唾液等体液样本以10,000g离心8分钟
弃去上清液,通过移液将细胞沉淀重悬于200μl无菌水中,短暂涡旋,然后在室温下静置5分钟,以渗透压裂解哺乳动物细胞
添加终浓度为10μm的PMA(叠氮溴化丙锭)(向200μl样品中添加10μl的0.2 mM PMA溶液),并将样品短暂涡旋,然后在黑暗中于室温温育5分钟
然后将样品从标准台式荧光灯放置在<20cm的冰上水平放置25分钟,短暂离心并每5分钟旋转一次
完成后,可将样品冷冻在−20°C或转移到取样管的储存液中
Marotz CA, Sanders JG, Zuniga C, Zaramela LS, Knight R, Zengler K. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome. 2018;6(1):42. Published 2018 Feb 27. doi:10.1186/s40168-018-0426-3
本处理方案以后宿主DNA可以降低8%以下。
说起宏基因组,对于熟悉宏基因组或者打算做宏基因组的同学可能已经迫不及待想知道这个怎么分析啊,怎么看结果啊之类的问题… 但在这之前,首先你应该了解的是宏基因组是什么,做宏基因组你能得到什么。


此外,对于缺乏深度研究和高质量参考基因组的样本,如土壤和特殊环境下的样本,宏基因组获得的较为完整的基因组不仅可以丰富参考基因组数据库,同时可以提供更加准确的物种分类。
因此,深度宏基因组测序是解析新环境样本的核心方法,不过从单一样本中重建出完整的菌株基因组有相当困难,一般需要较多样本或设置梯度样本从而利用更高深度和共同变化来获取分箱信息,当然对应测序和分析成本会更高。
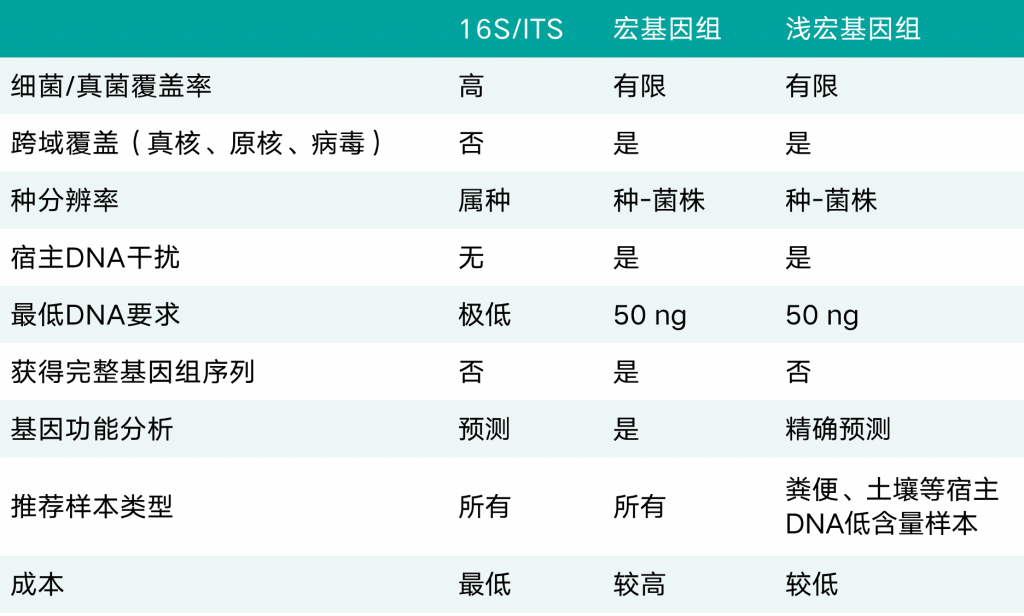
至此,我们了解了16s、浅宏基因组、宏基因组三种方式,我们将它们各自的特点总结如下表,便于你更直观地去了解(文末有福利~)。

宏基因组报告中有哪些分析内容?
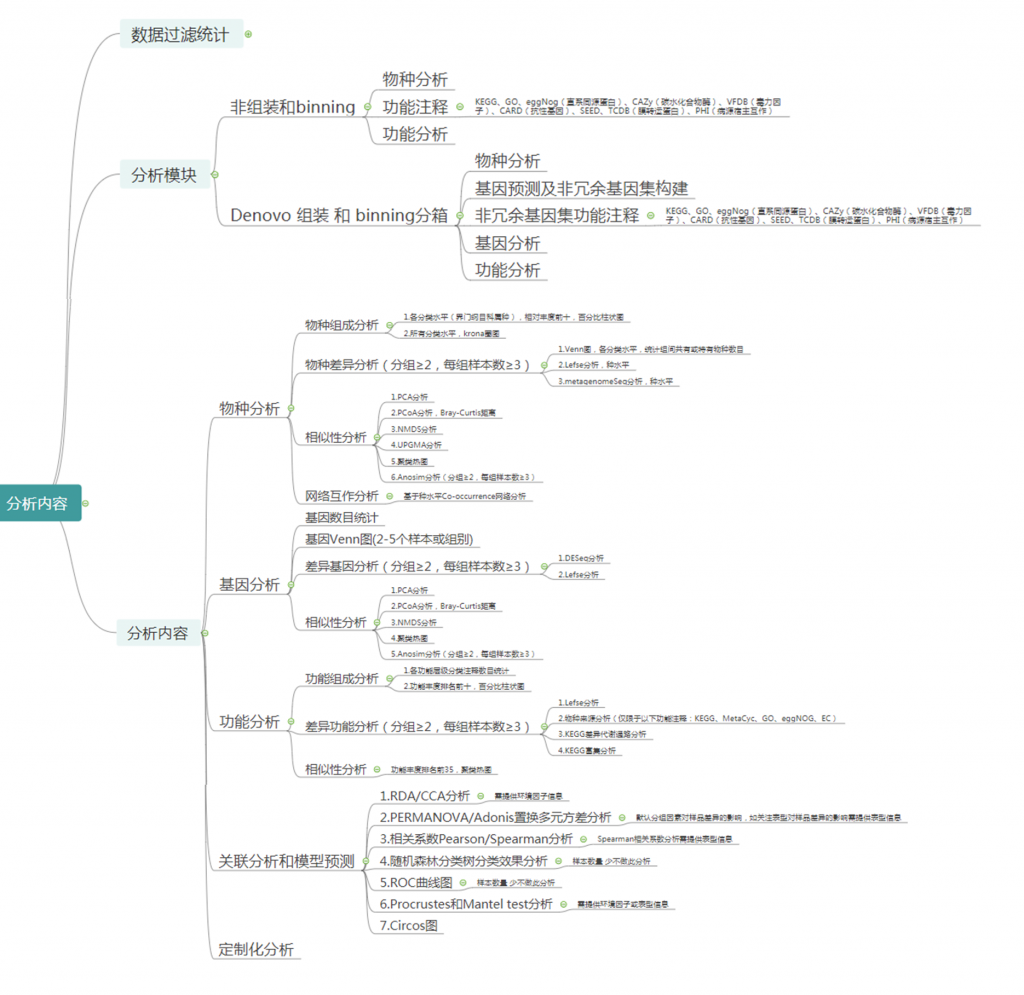

上图可以快速预览一下我们报告中的分析内容。
接下来,我们会详细介绍这些内容是如何从原始数据开始一步步实现的,同时也会选取一些文章案例来给大家做详细解读,希望给大家带来一些思路。
测序数据需要经过质检,去除接头和低质量序列,一般还会进行一步过滤人的基因组序列,然后分为两个路径,使用参考数据的比对方法和从头组装的方法,下图是一个完整的宏基因组分析流程:

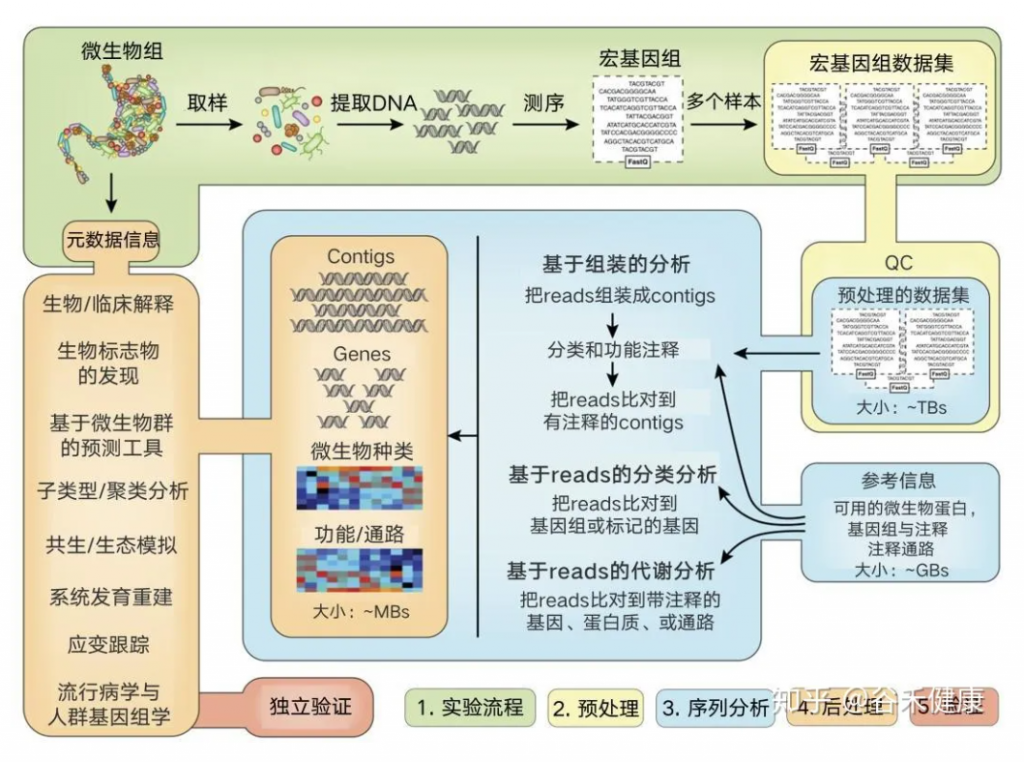
看完上图,可以对宏基因组测序的基本流程有个大致了解。
对于宏基因组测序而言,最重要的就是获得微生物群准确的物种构成及其丰度。
首先你需要了解的是无论16S测序还是宏基因组测序获得的均是相对丰度,即每种菌占所有菌属的比例。
要获得绝对的丰度需要在取样时做好取样量的计量,并在提取和建库中加入已知绝对量的参照DNA。
宏基因组测序获得物种构成及其丰度有以下两条路可以走:


我们先讲其中之一: 直接比对 。
直接比对是基于参考数据的,那么基于参考数据的物种构成分析主要有两类方法:
一类是基于Kmer和LCA比对特征来分析对应物种丰度,如kraken2等。
另一类是基于特征标记基因进行分析的,如MetaPhlAn2等。
基于参考基因组的分析工具如下表:
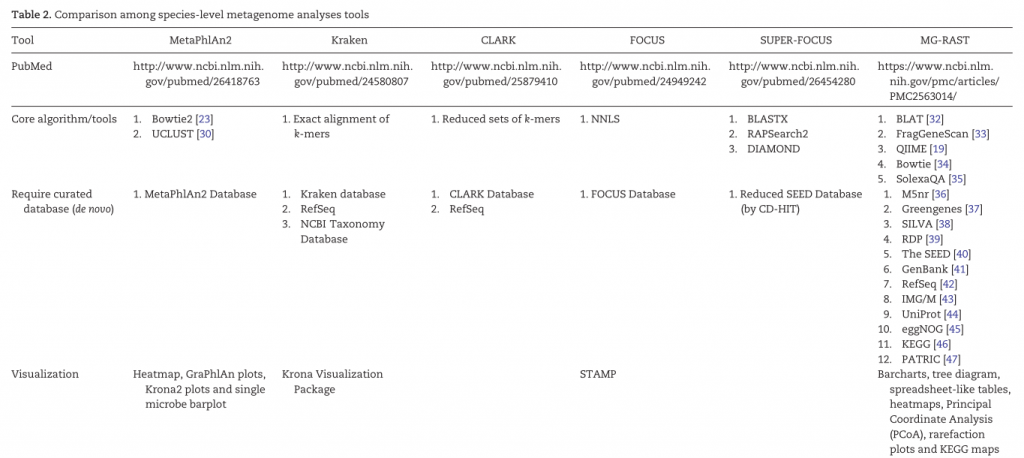

除了上面表中列出来的,另外还有
Centrifuge:比kraken2慢2x,内存使用少很多
Sourmash:类似CLARK,可以使用整个refseq作为数据库。
主流的kraken2——快速、准确度高、内存要求高
目前主要使用kraken2为主,因为快速,准确度也相当不错。不过,对于内存的要求较高,另外受数据库本身质量影响较大,默认kraken2的参考数据库只包括了细菌、古菌、病毒和人,还需要添加其他域的参考基因组。但涵盖的测序参考种仍然有限,对于菌株水平的鉴定受一定影响。后续使用Bracken可以针对kraken2的比对结果进行计算相对丰度。
MetaPhlAn2——物种跨度大、实用
MetaPhlAn2首先从全基因组数据库中找出clade-specific marker genes,然后利用这个marker genes的数据库对高通量测序得到的shotgun序列进行注释,目前主要用于后面直接使用reads获得基因和代谢通路丰度的HUMANn2的流程中,其物种跨度较大,速度也可以接受。
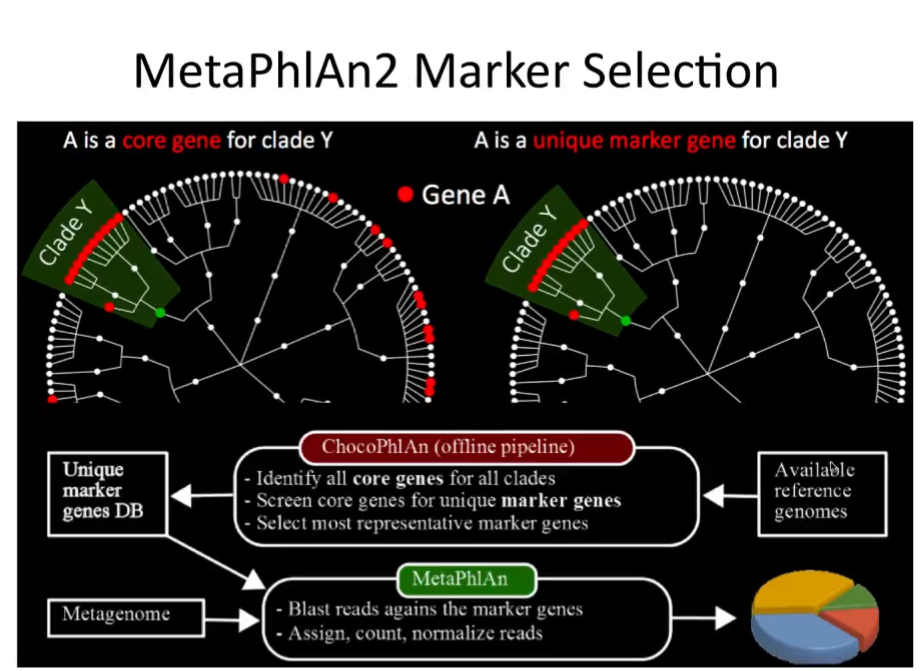

以上我们了解了直接使用reads获得丰度。
如果有足够测序深度和样本数量还可以通过组装出参考基因组来鉴定获得。该部分我们在下面的组装和分箱流程部分详细讲。
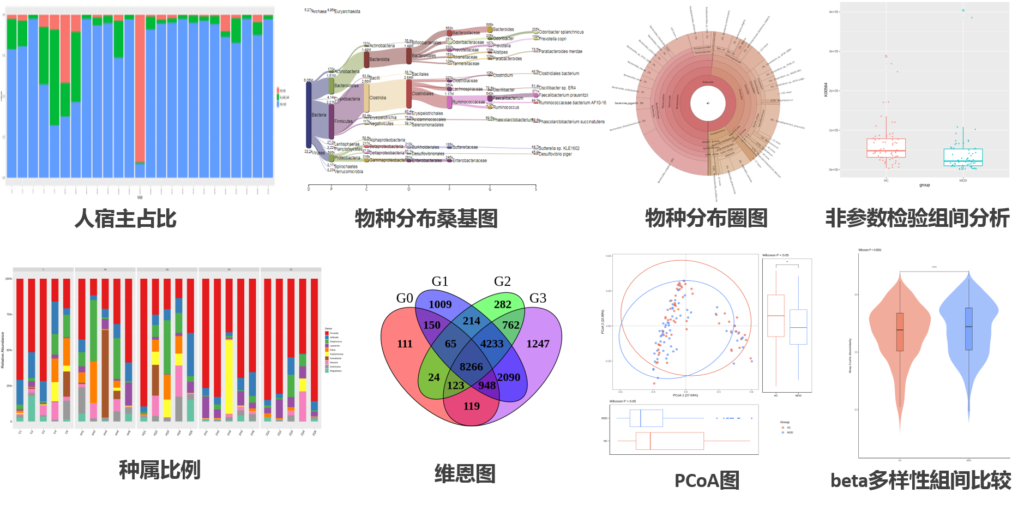
接下来,看一下我们报告中获得的结果和图:
使用Kraken2对其中的微生物进行物种注释。我们的Kraken2使用的数据库是由Refseq(2020.04.20)细菌,古细菌、真菌、原生动物和病毒库以及GRCh38人类基因组构建的。
通过查询数据库序列中的每个k-mer,然后使用所得的LCA分类单元集确定序列的适当标签,对序列进行分类。数据库中没有k-mers的序列不会被Kraken2分类。这里我们是在使用k-mer=35的条件下进行物种注释。
使用Bracken对物种注释结果计算相对丰度。Bracken是一种高度精确的统计方法,可从宏基因组学样本计算DNA序列中物种的丰度。Braken使用Kraken2分配的分类标签来估计源自样本中每种物种的读数数量。
对物种注释结果使用 KRONA 进行可视化展示。
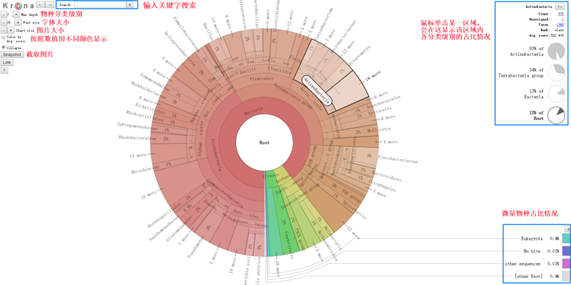

注:圆圈从内到外依次代表不同的分类级别(界门纲目科属种),扇形的大小代表不同注释结果的相对比例。
上面的是使用KRONA对单个样本的构成图形化,所有样本合并使用柱状图就可以了解具体的样本构成丰度,从门-纲-目-科-属-种-甚至菌株每个层次都可以进行显示(下面是截取我们报告中的相关图)。

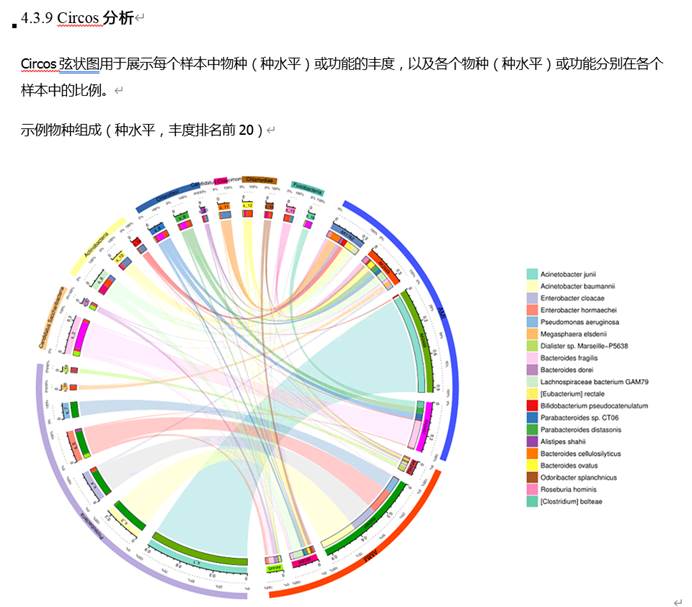
如果嫌柱状图的展示方式单一,当然也可以有别的选择。比如说以Circos的环图形式展现:

也可以进行聚类分析:
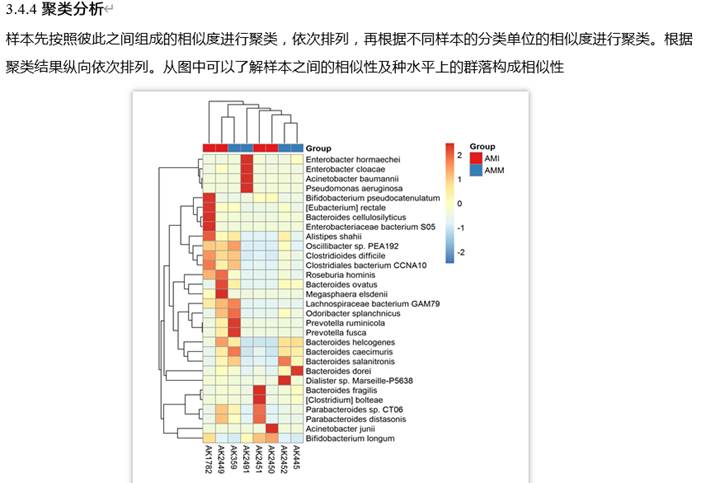

有了这些数据我们就可以进行alpha多样性(指每个样本内部菌群多样性)的分析了。

各样本和多组之间也可以进行Beta多样性的比较分析:

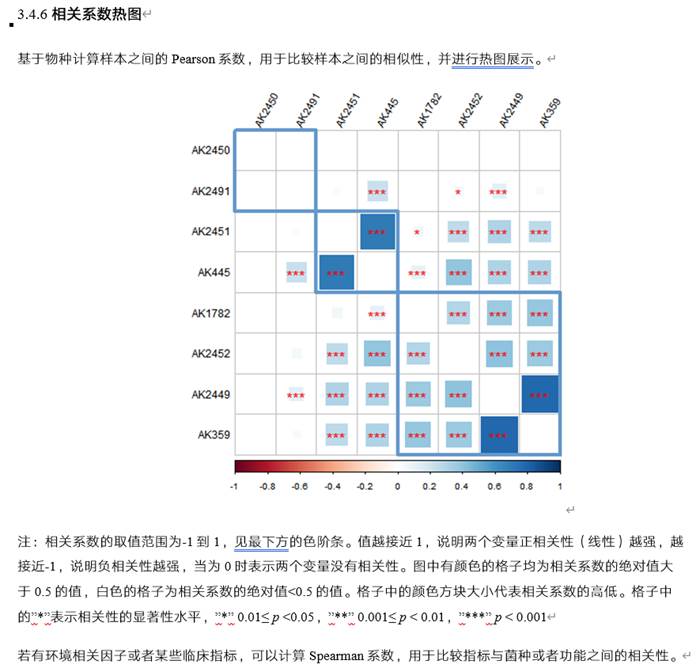
计算样本之间的菌属构成相似度:

组间的差异分析:寻找差异或代表性菌属,如下:


Trukey多组间检验

LefSe分析
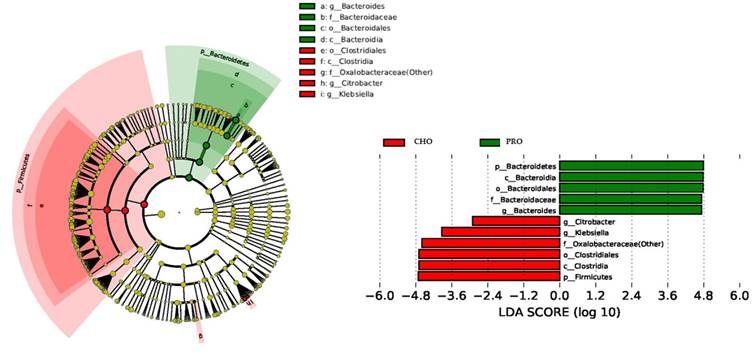

其中LEfSe基于线性判别分析(Linear discriminant analysis,LDA)的分析方法,筛选组与组之间生物标记物Biomarker(基因、通路和分类单元等),即组间差异显著物种或基因。当分组较多时较难获得每个分组独特的Biomarker。
以上是关于物种组成部分,但是有些小伙伴会有这样一些疑惑:物种构成变化很大怎么办?个体差异也很大?之类的诸多疑问。
是的,微生物群落一般对应特定的环境,其物种构成有时候变化迅速,而且个体或不同地点的构成差异极大。如人体的肠道菌群,个体之间的菌群构成差异很大,仅少量核心菌在绝大部分人的肠道内出现,个体特异性菌株也非常常见。那么如此多样性和复杂的构成如何应对相似的环境呢?
研究显示不同的菌属可能有着相似的基因或代谢能力,差异极大的种属在基因功能层面可能有着相似的构成。因此,获得微生物群的基因和功能代谢构成及分布对于解释和了解微生物如何响应和适应环境就尤为重要。
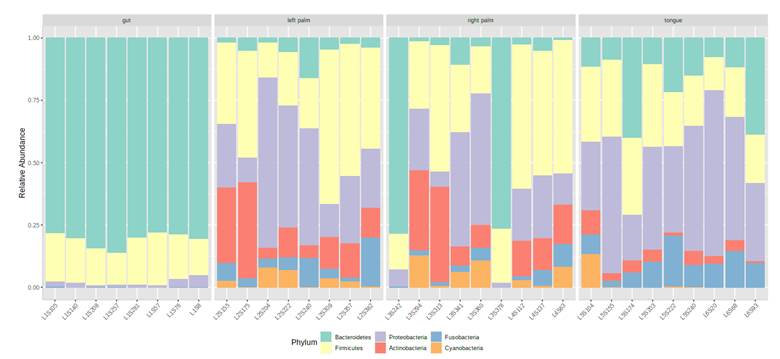
下图可以帮你更好地理解上面这段话。从图中我们可以看到,舌背样本和粪便样本虽然在种属上有很大差异,但它们在基因功能层面却有着相似的构成。
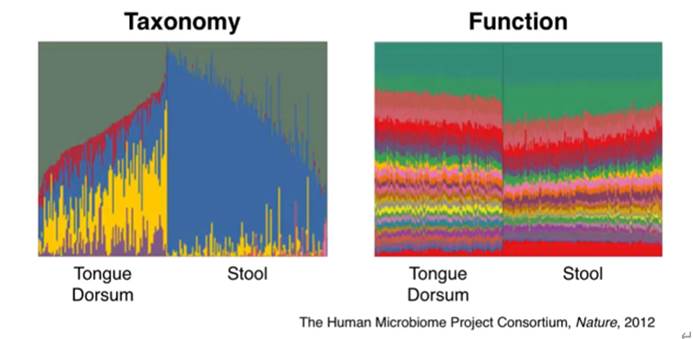

与物种构成丰度的分析类似,基因功能构成分析也同样可以包括两种方法:
方法一、通过直接基于reads的参考数据库方法获得
方法二、通过组装后预测注释基因并得到丰度
在具体展开方法之前,我们需要先了解关于基因功能的基本概念。
基因功能
每个菌的基因组中都包含大量的编码基因(ORF)以及非编码的RNA。这些基因之间又存在同源或序列相似性,达到一定相似程度的称为同源基因(一般通过CD-hit聚类为unigene,gltA这类基因名称,而数据库中一般聚类为如uniref90,eggNOG_ortholog等不同相似度的非冗余基因),这些同源基因除了序列相似同样也有着相似的功能,基于其功能或具备的蛋白功能域可以进一步分类为基因家族(Pfam),酶(EC 1.4.1.13),代谢通路(ko:K00266),更进一步层层分类为GO或顶层代谢通路Metacyc或COG等。
我们先来看方法一,具体是如何操作的?
主流的HUMAnN2——获得基因和代谢通路丰度的同时可直接进行下游分析
基于测序原始序列直接获得基因构成丰度的软件目前最主要的是HUMAnN2,其首先使用MetaPhlAn2进行物种分类(关于这个软件我们在前面物种组成部分已经讲过),并提取相应物种参考基因组用于比对,未比对上的用于进一步和uniref数据库进行蛋白质序列比对。原理见下图:
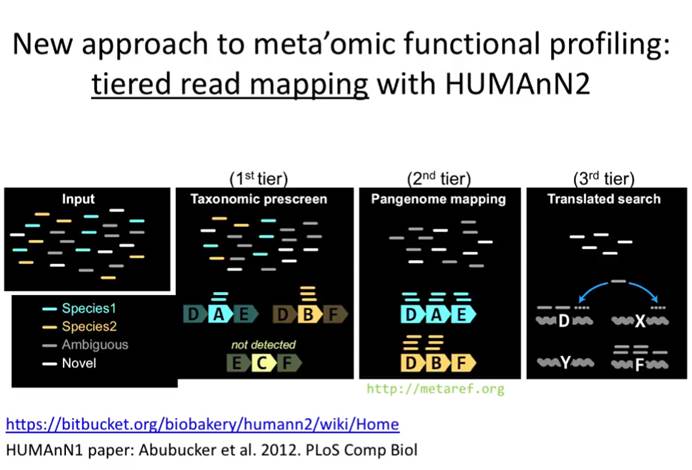

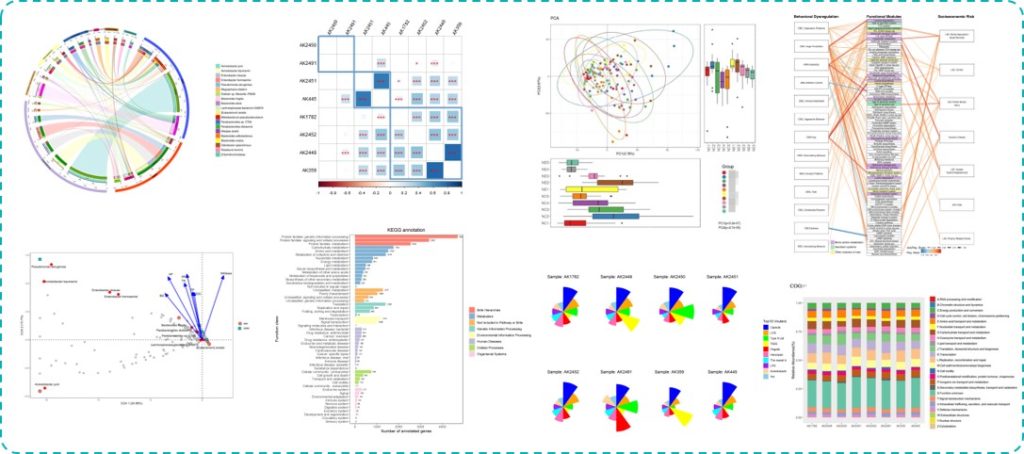
HUMAnN2的便利之处在于获得基因和代谢通路丰度的同时可以直接进行下游分析,将导出的表用于如LEFSE等差异分析,此外还可以反向给出不同样本中每个基因或代谢通路里的物种贡献。
下图是基于HUMAnN2的不同代谢通路的菌贡献比例图:
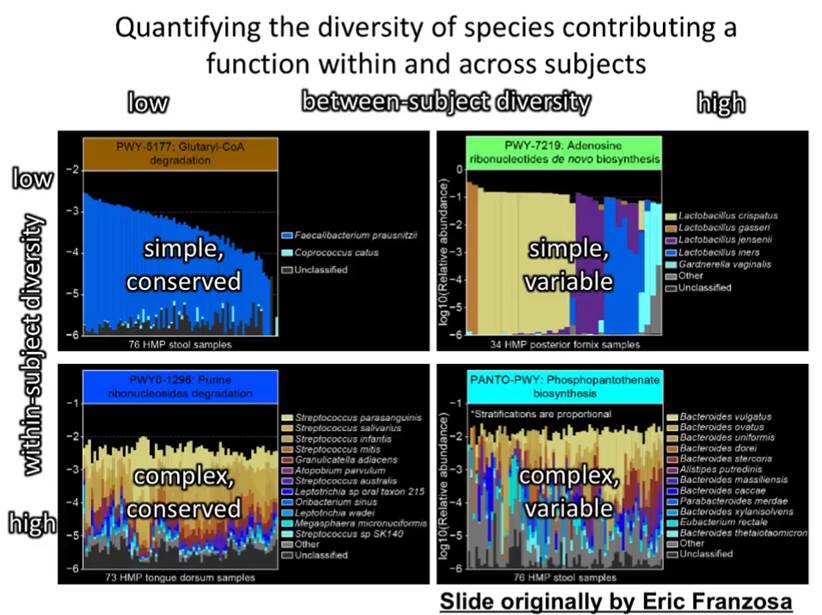

在我们的宏基因组报告中获得的是这样的:

而另外一种方法是通过组装获得,我们在前面物种构成小节也已经提到过组装分析,那么这里我们就组装拼接分析这部分展开讲解一下。
什么样的条件下可以进行组装分析?
当测序深度足够的情况下,目前illumina二代和Pacbio以及Nanopore等长片段测序技术已经足以组装出高质量的细菌基因组草图,结合Binning方法可以一次性获得大量物种的高质量接近完成基因组。此外还有Hi-C等手段可以进一步完成基因组以及对应质粒的完整拼接。
组装的流程是什么样的?
来看一下整个基于组装的流程:
① 提取、测序
首先从样本中提取基因组DNA,进行测序,可以使用Illumina的段片段深度测序也可以辅助三代长片段测序。
② 获得contig序列
接着对序列经过质检过滤处理后直接使用序列进行拼接,获得contig序列,这时通常每个菌的基因组会有几十到数千个contig片段,由于构成复杂,很多近缘菌之间的基因组存在大量相似序列,以及每种菌丰度都不一致,所以contig阶段的片段仍然较多。

③ Binning分析
基于序列构成特征如GC含量、核苷酸多态性、覆盖度以及基因的物种相似度等多种数据,如果有多个样本或梯度可以同时结合样本丰度变化来进行分箱也就是Binning分析,将具有相同特征和变化的contig聚类归为同一个来源的箱,每个bins通常来自单一菌也就是一个菌株的基因组(我们的数据分析中包含这部分分析内容)。
④ 进一步质检评估
之后会进行进一步的质检,如checkM等评估每个Bin的完整度(核心基因以及rRNA等的完整性)和污染比例(如错误拼接,不同物种来源等)。一般要求50%以上的完整度以及10%以下的污染,当然样本数量越多,测序深度越高,测序读长越长理论上binning的质量也会更好,能获得更多高质量的单一菌完整基因组。
借用一张分箱的说明PPT:
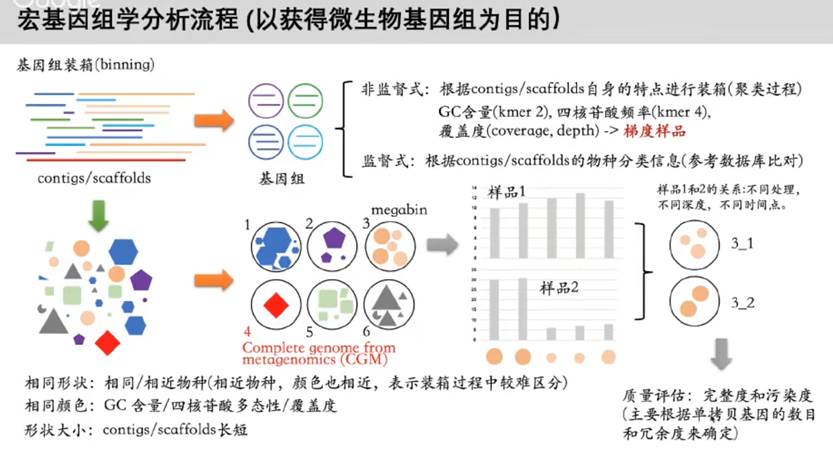

目前组装contig方面比较好的软件主要是SPAdes和MegaHIT。分箱方面MetaWRAP流程可以将整个组装和分箱优化全部完成,包括前期质检到组装以及使用三种分箱方法concoct, metabat2和maxbin2,并最终进行合并提纯优化,输出最终的分箱。
同时还可以对每个分箱bins进行物种鉴定和定量,这样我们就可以获得基于拼接组装后的物种丰度构成表,开展上述的物种多样性和样本差异统计分析。
⑤ 注释
最后使用PROKKA进行基因预测,获得的编码序列我们经过进一步CD-Hit聚类去冗余,然后使用eggNOG-mapper对其进行进一步的功能比对注释。使用salmon完成基因的定量,这样我们就得到基于组装注释的基因丰度数据了。之后就可以进行基因和功能层面的多样性、构成以及样本和组间差异分析。
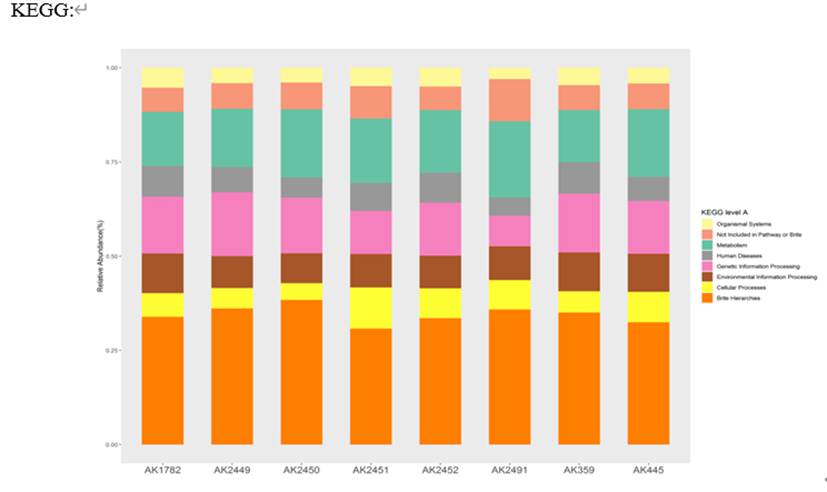
我们获得的最基础的uniref,eggNOG,KEGG和GO等注释如下:
KEGG

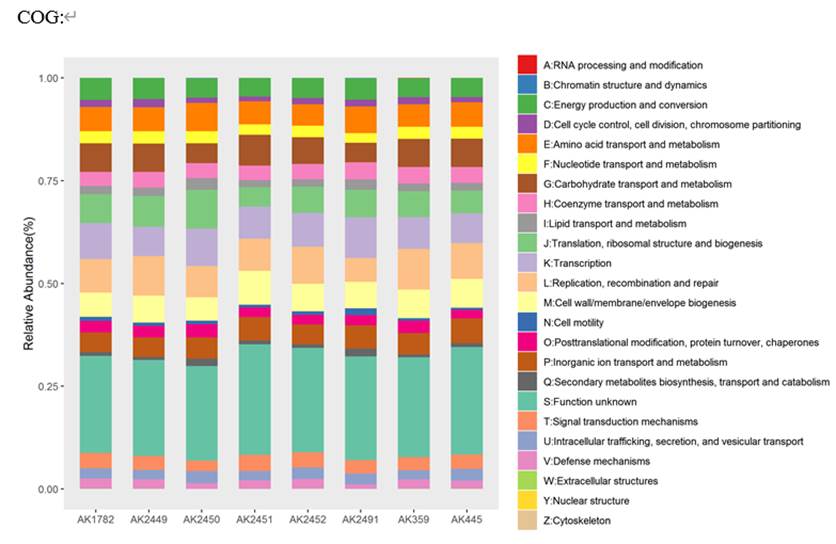
COG

eggNOG

组间差异分析,如KEGG途径:

除此之外,还可以使用其他的功能基因数据库来进行进一步的基因注释和分析。比如:
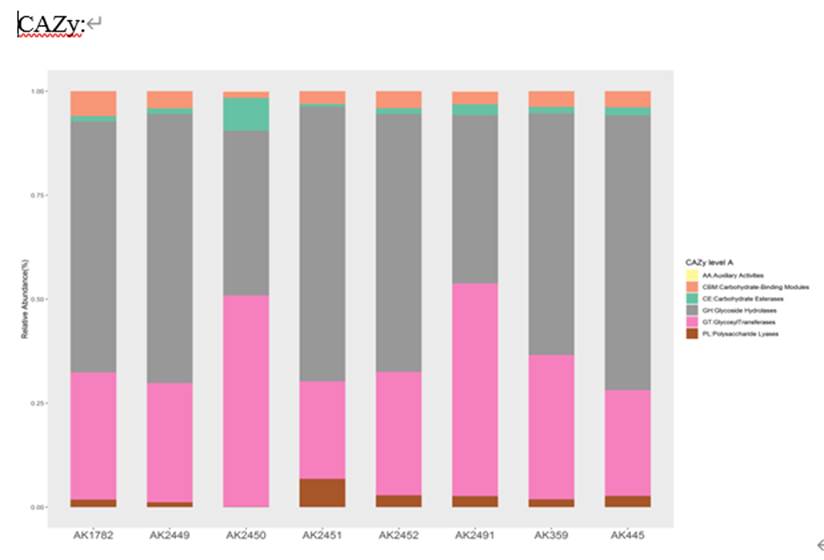
CAZy:

VFDB毒力因子注释:


抗性基因注释:




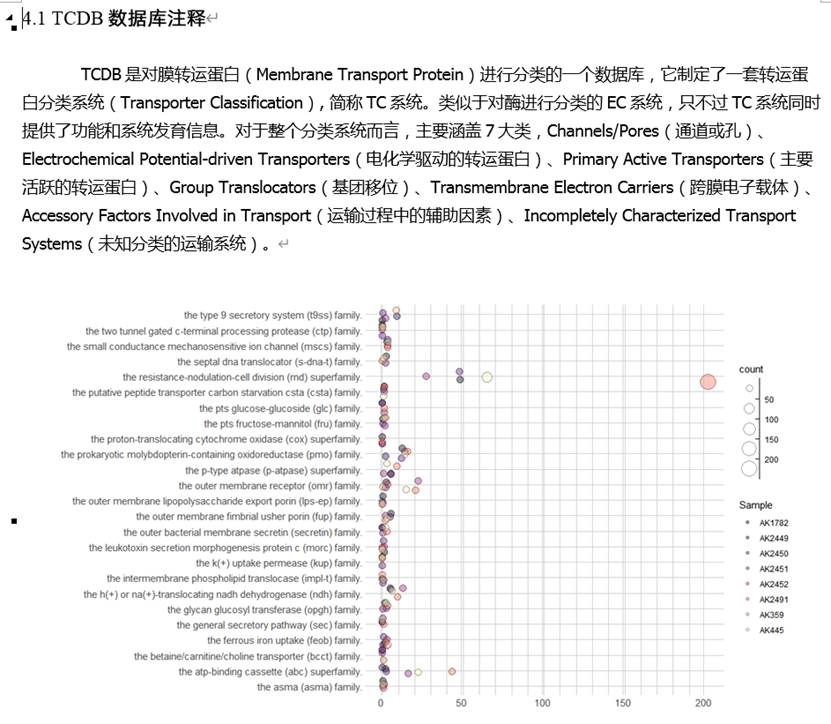
TCDB数据库注释:

PHI数据库注释:
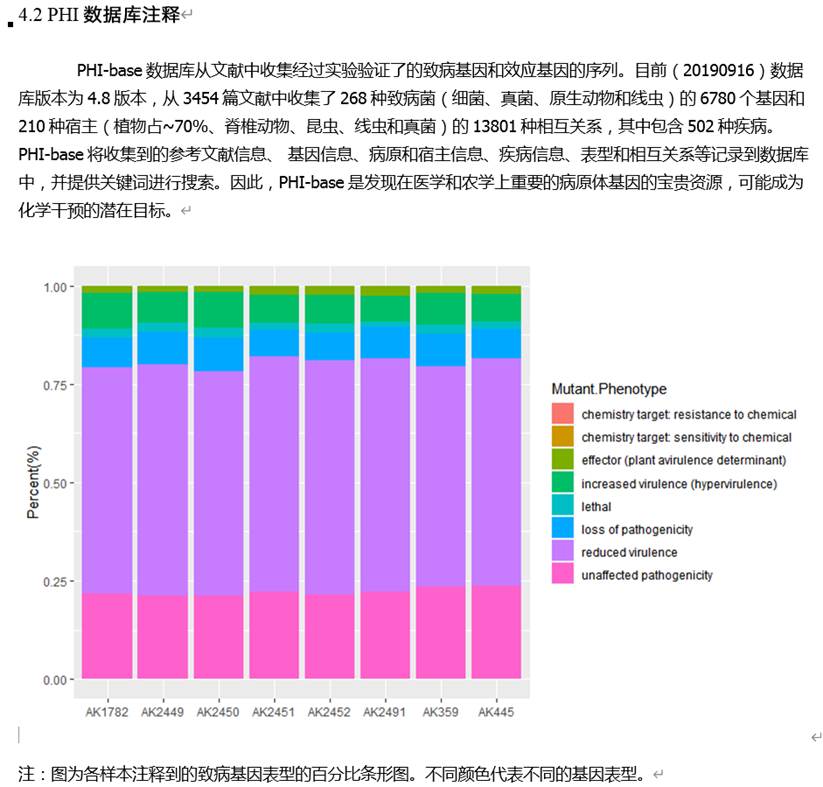

BCGs分析:
以及基于antiSMASH和BiG-SCAPE来对代谢物的合成生物基因簇BCGs进行分析。

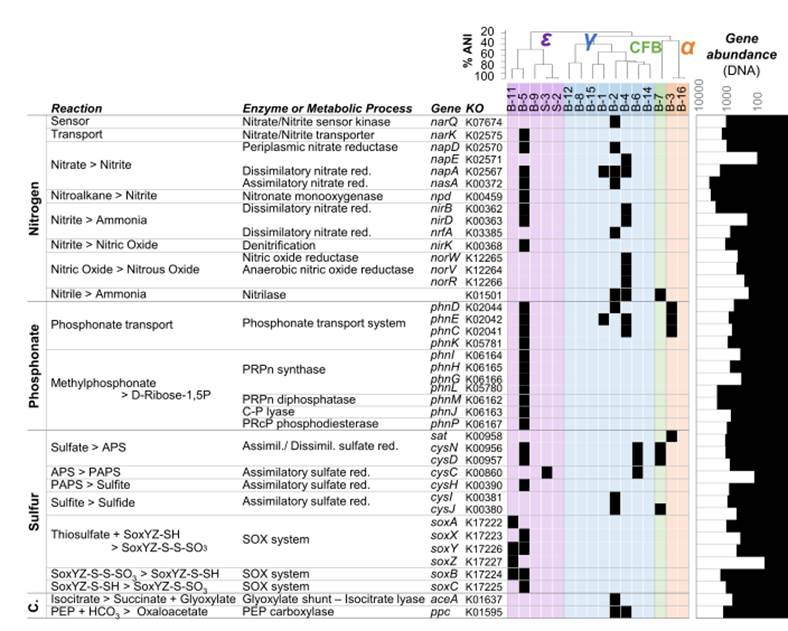
固定代谢能力评估:
或更聚焦于特定代谢的如下图中的氮、磷、硫和碳固定代谢能力和水平的评估:

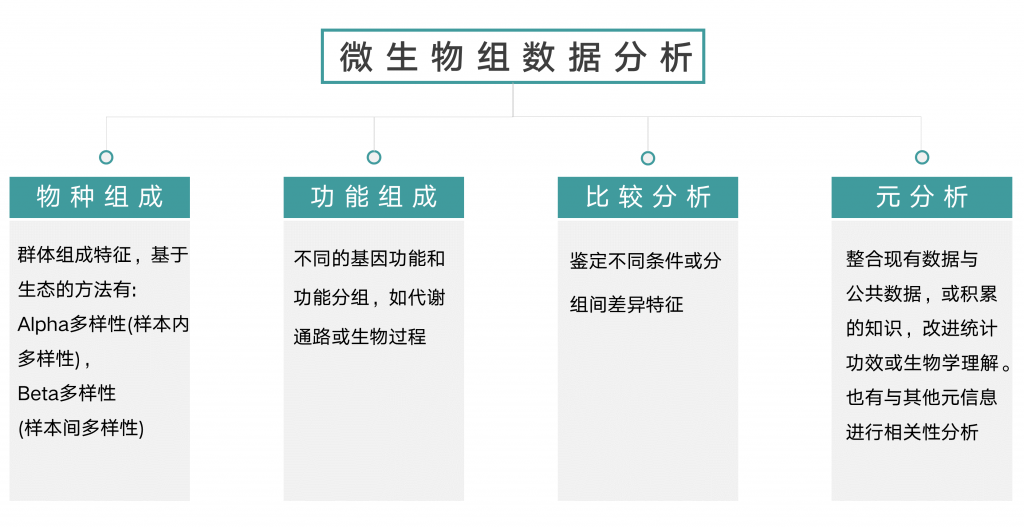
当有了大量样本的菌群构成丰度信息,以及各种基因和代谢丰度数据后,我们需要根据样本的meta信息,基于不同分组,时间或环境因子等数据进行统计分析和检验,进而发现和探索可能的关联以及背后的生物学意义。
那么在面对宏基因组这类数据时在进行统计检验分析时需要注意什么呢,应该采用哪些分析,并如何解读这些结果呢?
首先,微生物组数据分析分为四大类:

在对所有数据进行统计检验前一般建议对数据进行基本的质量过滤。一类是去除绝大部分样本都不存在的物种和基因,如Prevalence in samples (20%),还有一类是去除变异度过小的Percentage to remove (10%)基于Inter-quantile range。
为什么可以过滤这两类?
上述的两类由于其携带的信息量和变化过小在进行组间比较统计检验的时候都建议过滤,因为要么是污染,要么与差异无关。
宏基因组数据具有一些独特的特征,例如测序深度的巨大差异,稀疏性(包含许多零)和分布的巨大差异(过度分散)。在进行后续的统计检验之前建议针对不同的分析方法进行相应匹配的标准化处理。标准化包括:
Rarefaction和缩放方法:这些方法通过将样本放到相同的比例进行比较来处理不均匀的测序深度;
转换方法:包括处理稀疏性,组成性和数据中较大变化的方法。
那么各种标准化方法是什么,应该选择哪种方法?
参考MicrobiomeAnalyst网站提供的信息,以下是一个简短的介绍:


请注意,数据标准化主要用于可视数据探索,例如beta多样性和聚类分析。有时候不使用标准化也能获得最佳结果,比如:单变量统计和LEfSe。
同时,其他比较分析将使用其自己的特定标准化方法。例如,对metagenomeSeq使用累积总和定标(CSS)标准化,对edgeR应用M值的修剪均值(TMM)。
经常有小伙伴问,这个数据是用的什么标准化?没有做标准化怎么办?这类问题。
目前,尚无关于应使用标准化的共识性指南。建议大家可以探索不同的方法,然后目视检查分离模式(即PCoA图)以评估不同标准化程序对实验条件或其他感兴趣的宏基因组数据的影响。
有关这些方法的详细讨论,请参考使用者最近发表的两篇论文
① Paul J. McMurdie等
(https://doi.org/10.1371/journal.pcbi.1003531)
② Jonathan Thorsen等
(http://doi.org/10.1186/s40168-016-0208-8)
以上是关于标准化的这部分内容需要了解的知识,接下来我们来看具体如何操作,怎么得到那些图表?它们分别代表什么?
一般我们需要先进行探索性分析,也就是不设预订的假设,首先从主成分分析结果中了解样本的菌属和基因的大概分布。
主成分分析是根据不同距离算法计算样本之间的距离矩阵,然后进行降维,最终形成一个三维的空间分布。样本之间在空间上分隔越远表明样本之间的差异越大。
比如我们报告中的下图,疾病和正常样本可以较好的区分,一般此处我们还会进行一个统计检验,来判别PC1和PC2这几个维度上两组之间是否真的存在统计差异。
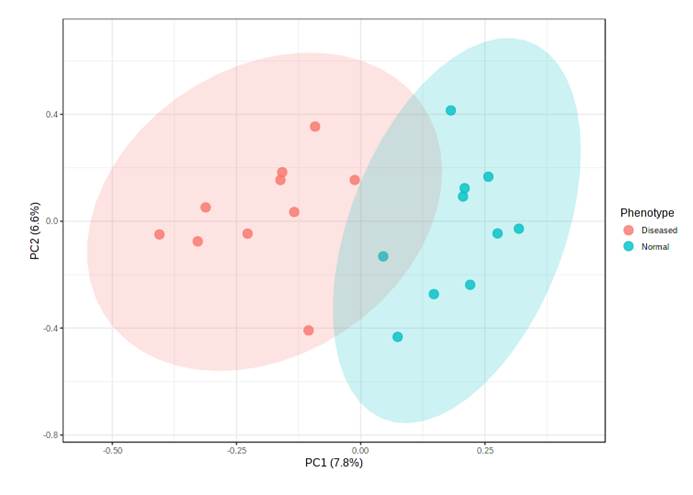

基于丰度图来评估各样本和分组的基本构成,如:

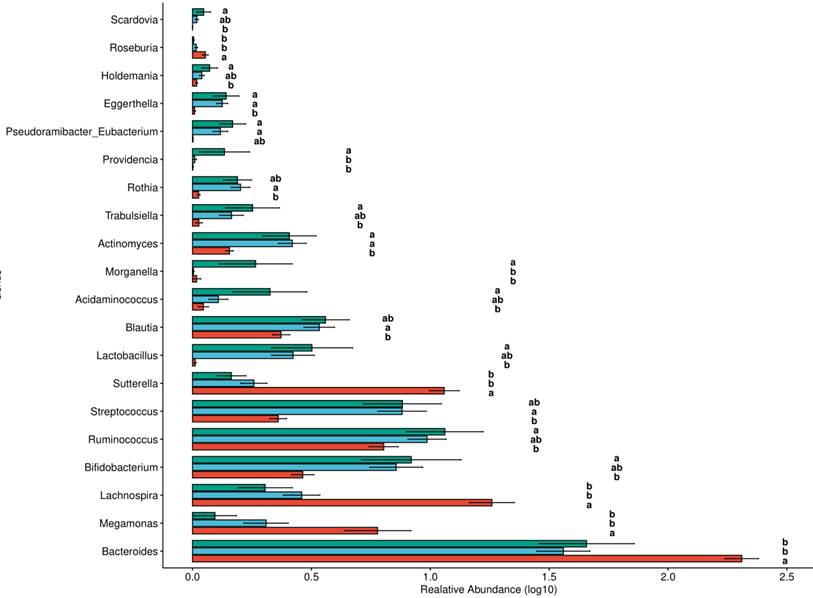
之后我们可以针对不同分组或处理之间的样本进行统计检验,可以使用的检验方法包括两组间的非参数统计检验T-test/ANOVA,3组以上组间统计检验可以使用Tukey test,其直接生成各组将的统计差异,并提供字母标注,直观简便,如:

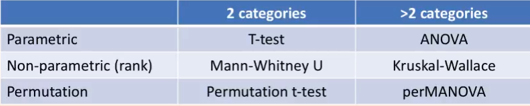
具体的统计方法选择可以参考下表:

除了常规的非参数检验外,包括metagenomeSeq和DEseq以及edgeR等统计方法包可以很好的分析组间差异特征。LEfSe则一般用于寻找特征标志物。
那么有了大量的差异特征菌属或基因之后,我们是否能基于这些差异菌属有效的区分不同的分组呢,或构件一个模型来预测或分类呢?
这时候可以使用随机森林(Random Forest)一类的决策树机器学习模型,来利用这些差异特征构建分类模型,并使用AUC等指标来评估基于这些模型的预测有效性和准确度(我们报告中如下图)。
当然也可以使用其他更复杂的如深度学习等方法来构建分类模型。

除了性别、疾病、地点等分类差异之外,我们通常还有很多元数据,包括临床指标或环境因子等信息,这些数据通常是连续型数值,对于这类数据我们可以进行相关性分析。
当然反过来,将菌群特征作为表型也可以和如基因组的基因型或SNP构成来进行相关性分析。
对于菌群数据的相关性分析比较推荐:
SparCC方法,可以构建菌种或菌属之间的相关性网络,相对稳定。
对于与疾病或环境变量进行相关性分析可以使用:
Sperman秩相关分析。
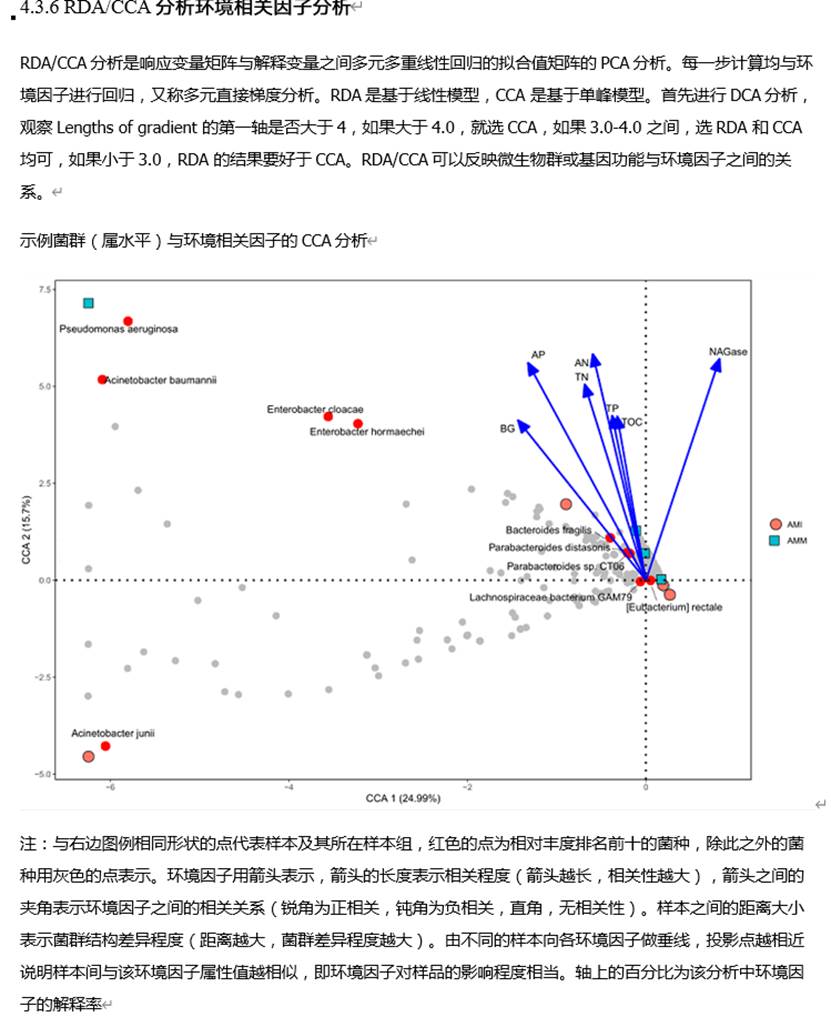
另外RDA/CCA分析也可以有效的反映菌属与环境因子等指标直接的关系(我们报告中如下图)。

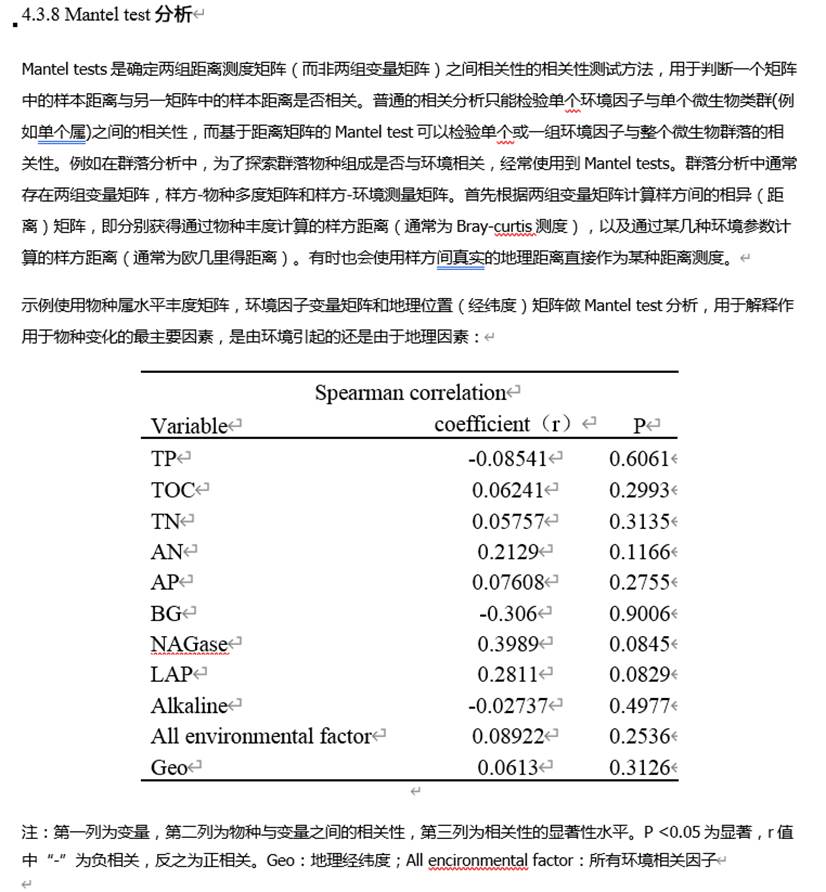
Mentel检验也可以用于判断菌群构成特征与单个或一组环境因子之间是否存在显著相关。

要 点
宏基因组从大量菌群和基因构成中寻找关联是需要足够的样本量才能达到有效的统计效力,因为一次性获得了大量的特征数据,样本量过少会带来统计结论的无效,越是组内差异大,组间差异小的研究足够大的样本量才能得到可靠的结论。
一般动物样本具有较好的背景可控,组内样本数量建议至少6个,而人群研究由于背景复杂,个体多样性高,一般建议组内50例以上较好。
以上看完后,你应该对宏基因组的数据分析流程有了整体的认识,也学会了相应的一些操作,但是不一定能直接从自己的这些数据、图表中真正探索到和实际生物学相关的有价值的研究成果。
所以,我们又选取了一些已发表的研究作为案例,结合实际问题来具体分析,从实验设计到具体分析流程方法和图表的展示,再到相应的结论,掌握这类文章的总体思路。
之后无论是刚开始的实验设计,还是后面的分析,都会更加得心应手。
建议想好整个实验思路再开始(或者也可以咨询我们,我们专业的数据分析团队会为你提供切实可行的项目方案)。
第一项研究是关于肥胖患者减肥手术后的宏基因组和代谢数据的分析研究。
文献来源:Aron-Wisnewsky J, Prifti E, Belda E, et al. Major microbiota dysbiosis in severe obesity: fate after bariatric surgery.Gut. 2019;68(1):70–82. doi:10.1136/gutjnl-2018-316103
研究纳入了61名严重肥胖的受试者,他们是可调节胃束带术(AGB,n = 20)或Roux-en-Y胃旁路术(RYGB,n = 41)的候选人。减肥手术后1、3和12个月随访24名受试者。使用宏基因组学测序和LC-MS分析肠道菌群和血清代谢组。另外纳入了10人和147人分别作为宏基因组和代谢检测的验证集。
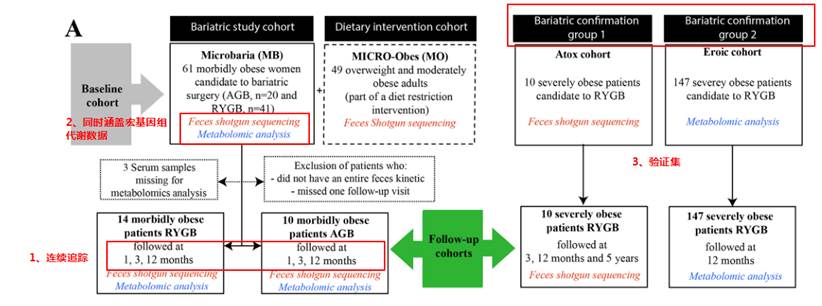

研究思路

这样的设计分别有什么作用?
第一点持续的动态采样可以获得持续变化情况,尤其是在一个特定变化后(减肥手术),持续的最终采样有助于确认菌群的变化出现和特定事件或生理病理变化的前后,尤其是在确定因果中有重要帮助。
第二点获得多维的数据有助于帮助我们全方位的了解菌群变化背后的带来的生理和代谢变化以及之间的关联。
第三点独立验证集的存在将大大增强研究的可信度,尤其是该研究纳入的样本量并不多,无法全面有效的控制无关因素,使得很多统计检验的效力无法显现。这也导致该研究仅在基因总量和多样性上获得较好的重复效果,而更多的菌群精细特征以及具体基因和代谢通路没有得到深入分析。但是独立验证集保证了核心结论的可靠性和重复性,这点在宏基因组研究中非常重要。
从下图可以看到研究针对样本的总基因多样性水平与生理指标和疾病状态进行相关性分析和组间差异分析,图中给出了显著相关和差异的指标。
使用的统计检验方法是pearson和sperman相关和t-test以及Kruskal-Wallis检验。

下图是研究将MAGs与各项生理和代谢值进行相关性分析后的热力图。该研究由于测序较早,并未独立拼接,而是直接使用了之前一项人类肠道菌群研究获得组装基因组参考序列。

进一步研究分析了术后特定变化模式的MAGs以及它们与代谢生理指标的相关性,见下图:

上图的研究可以通过pattern search的方法寻找特定变化模式的菌种。
研究的主要结论发现是低基因丰富度(LGC)存在于75%的患者中,并且与躯干脂肪质量和合并症(2型糖尿病,高血压和严重程度)增加相关。LGC改变了78种宏基因组种(MGS),其中50%与不良的身体成分和代谢表型有关。九种血清代谢产物(包括谷氨酸盐,3-甲氧基苯基乙酸和L-组氨酸)和含有参与其代谢的蛋白质家族的功能模块与低MGR密切相关。术后一年,BS会增加MGR,但尽管RYGB患者的代谢改善比AGB患者大,但术后一年的MGR仍然很低。
总体而言该项研究可以使用浅宏基因组(在文章开头第二部分详细介绍过)来完成所有测序和分析,进一步扩大样本数量,如果能同时获得人的转录组数据甚至能更加明确的找到菌群变化与特定代谢通路的关联关系。
第二项研究是Dan Knights实验室发表在Cell Host & Microbe,2019的一篇针对34个人17天每日饮食和菌群变化的相关研究,试图揭示日常食物选择与人类肠道微生物组组成之间的精细关系。
文献来源:Johnson Abigail J,Vangay Pajau,Al-Ghalith Gabriel Aet al. Daily Sampling Reveals Personalized Diet-Microbiome Associations in Humans.[J].Cell Host Microbe, 2019, 25: 789-802.e5.
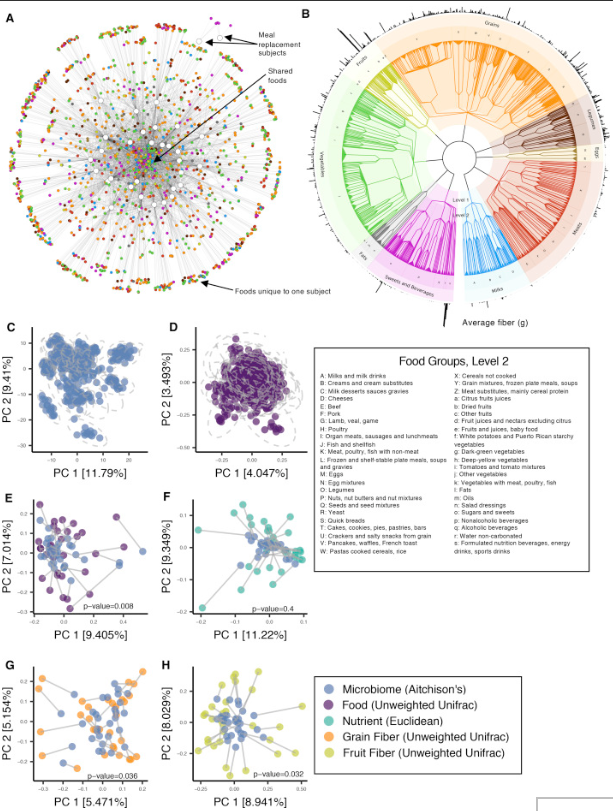

可以看到,研究同时记录了粪便样本的菌群宏基因组和每日的饮食记录。研究的核心在于将每日饮食的食物通过营养构成进行量化,并构建类似物种进化树的食物物候树。
此外由于有每日的数据,可以通过前一日的食物与第二日的菌群数据进行时间序列分析,构建食物与菌之间的关联以及时间相关性。
最后基于菌群数据和前一日饮食来构建模型预测判断后一日的菌群状态,帮助我们了解食物对于个体菌群的影响因素并实现定量和预测。
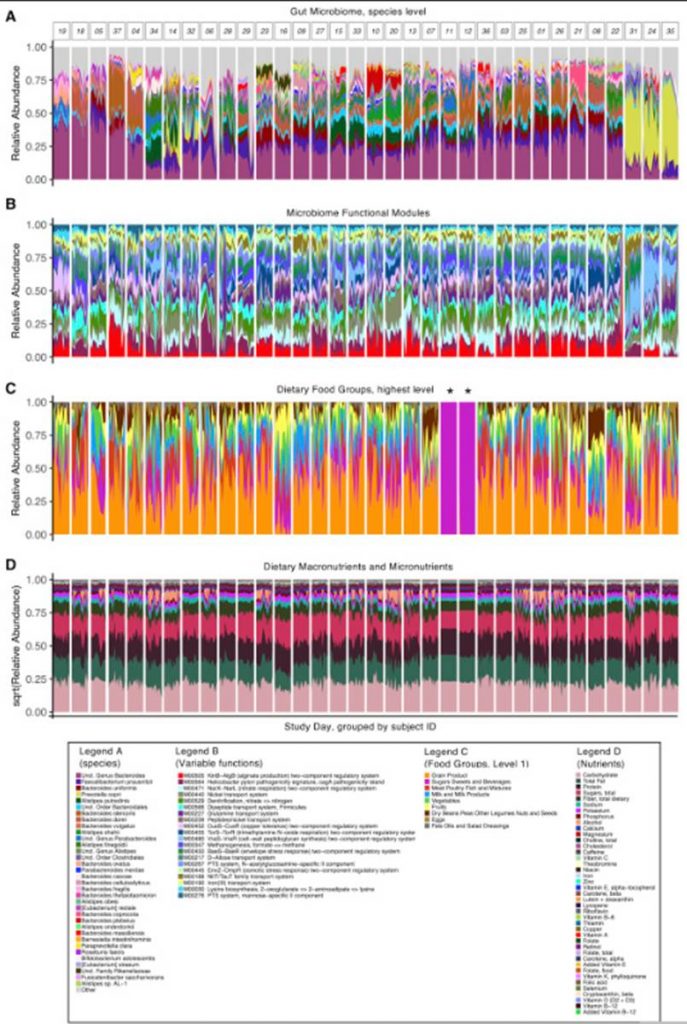

研究中对数据的处理过滤标准如下:删除所有具有低读取计数(每个样品<23,500个读取)的样品。物种级别的分类表仅限于研究对象中至少存在25%的研究日,且在10%的研究样本对象中发现的那些物种。
最后,相对丰度<0.01%的稀有物种被丢弃,将物种数量限制为290个注释。将得到的分类表汇总到较高的分类级别(即属,科,门等),以进行下游分析。
菌群和饮食以及营养构成的堆叠图很好展现了变化和对应。
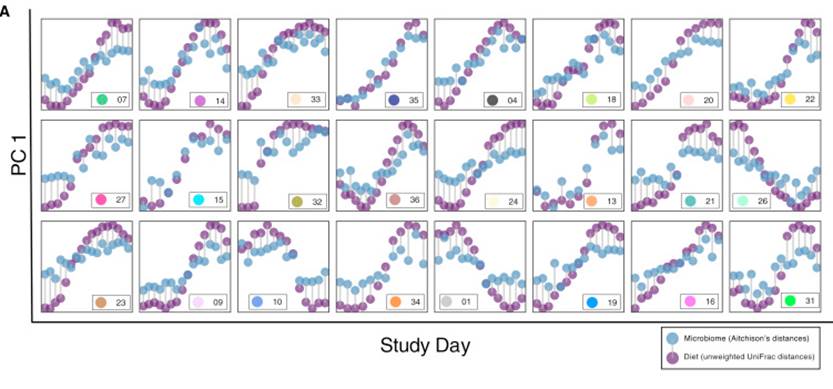

下面这张图很好的显示了饮食食物的变化与菌群变化之间的时间变化关系:
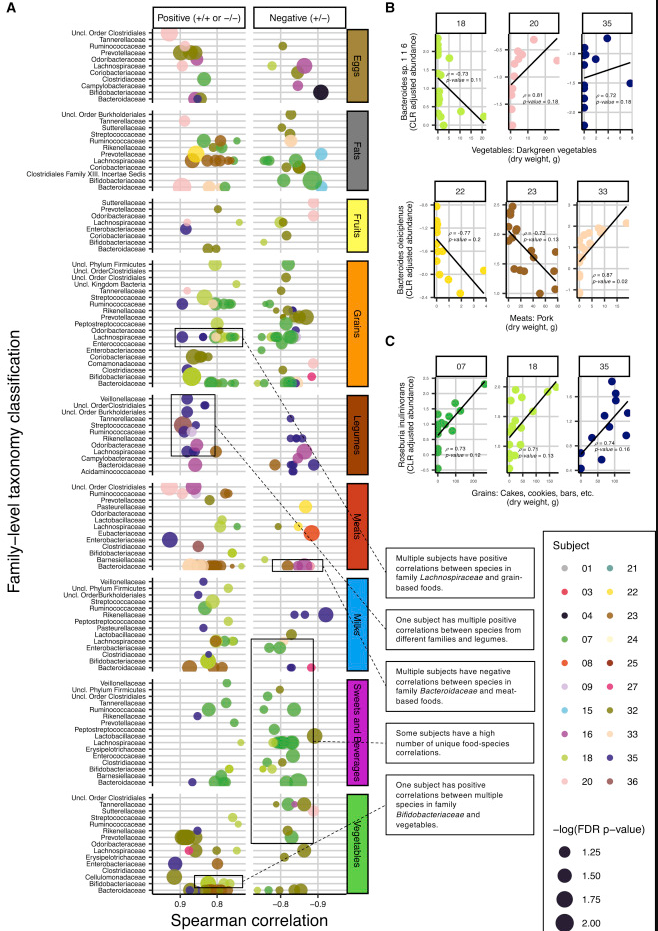

下面这张图通过对每个人单独进行菌属与食物的Spearman相关,展现了菌与食物之间的关联的个体化差异,在特定菌属对应相同食物不同人会出现完全不同方向的变化,这也正是这项研究所揭示的,这种关联关系的复杂性。

本研究虽然有大量样本,但并未进行组装,而是直接使用了Refseq的细菌完成基因组序列作为参考。研究由于样本数量众多,测序深度也很有限,类似研究也可以使用浅宏基因组方式完成。
接下来的一个研究是比较典型的宏基因组组装并与疾病进行关联分析的案例,研究的是日本人群类风湿关节炎的肠道微生物组的全基因组关联研究。
文献来源:Kishikawa Toshihiro, Maeda Yuichi,Nii Takuroet al. Metagenome-wide association study of gut microbiome revealed novel aetiology of rheumatoid arthritis in the Japanese population.[J] .Ann. Rheum. Dis., 2020, 79: 103-111.
研究使用较高深度的宏基因组shotgun测序(每个样本平均13 Gb)对日本人群(病例 = 82,对照 = 42)进行了RA肠道微生物组的MWAS分析。MWAS由三个主要的生物信息学分析渠道(系统发育分析、功能基因分析、途径分析)组成。
使用了之前研究中6139个完成拼接日本人肠道宏基因组作为参考序列以及其他几项研究的参考基因组,在过滤部分种过多的基因组之后,最后一共使用了7881个参考基因组。
将QC后的序列直接比对到参考基因组,并根据基因组长度计算对应物种的相对丰度。
基因方面选择denovo组装,使用MegaHIT,然后再contig上完成ORF预测和CD-HIT聚类去冗余,最后与UniRef和KEGG数据库比对。
最后使用bowtie2将测序序列比对到注释后的unigene序列上获得基因丰度,经过KEGG注释得到代谢途径的丰度。研究的数据处理流程图如下:

在数据分析流程和方案选择上人体肠道菌群由于研究众多,以及有多个深度测序拼接完成的Binning参考基因组数据集,确实可以直接使用参考基因组直接比对。
对于其他一些环境或来源的样本这个深度的数据量可以考虑独立拼接和分箱。该研究中使用已有参考基因组,大概88%的序列能比对到参考基因组,如果直接组装这个比例应该可以更高一些。另外在获得基因丰度是可以考虑使用Salmon,比对获得基因丰度更为方便。
获得相应数据后对相对丰度,该研究使用Box-Cox transformation对数据进行标准化,并过滤了一些低丰度的菌属。Case-control的相关性分析使用的R的glm2模块,将年龄、性别和测序上机分组作为协变量。
对于菌属的关联分析,最终将显著性结果以火山图和GraPhlAn图的形式展现如下:

上面其中D图使用每个菌的丰度进行UMAP分析,并映射关联效应的展示比较有意思。
不过在基因层面上并未找到相应的关联,可以看到下图UniRef90的NMDS分布图两组之间无法有效区分,多样性也没有显著差异。

这项研究在菌层面发现了多个普雷沃氏菌属的菌在日本人群中与类风湿性关节炎存在关联,不过除此之外其他方面的发现并不多,仅找到一个基因存在显著关联,涉及的临床调查也相对有限,且人群队列数量不算多,并无独立验证集,因此亮点并不多。如果能纳入免疫相应指标可能能研究的更细致一些。
最后这项研究是对来自永冻土融化梯度的214个样品的宏基因组测序组装了1,529个基因组,揭示了参与有机物降解的关键种群,包括其基因组编码先前未描述的木糖降解真菌途径的细菌。
文献来源:Woodcroft Ben J,Singleton Caitlin M,Boyd Joel Aet al.Genome-centric view of carbon processing in thawing permafrost.[J] .Nature, 2018, 560: 49-54.
通过宏基因组denovo组装和分箱Binning,最终获得了1529个永冻土菌群基因组。基于这些数据描绘了永冻土融化梯度下的菌群构成特征,如下图。
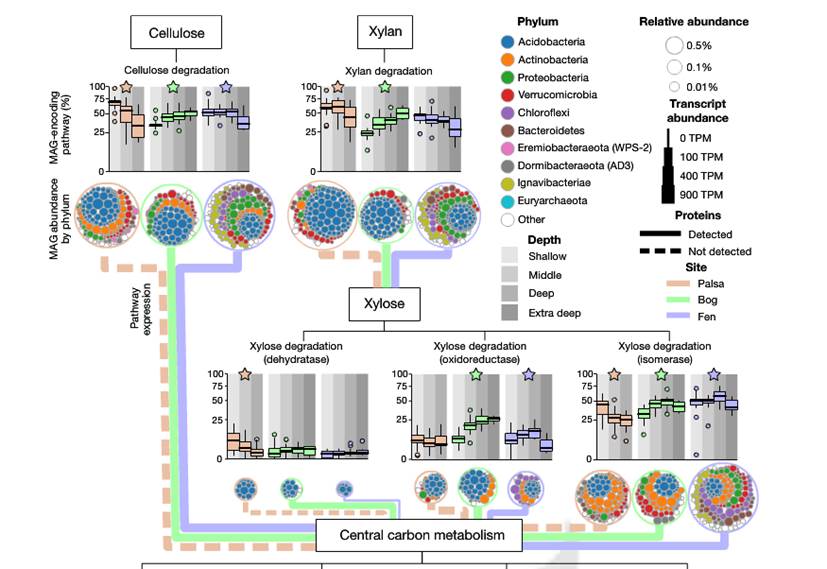

论文是2018年发表的,测序是在2011和12年测的,使用的是CLC Genomics Workbench 较早的4.4版分单样本组装,然后使用MetaBAT进行分箱,最后的标准是70%完成度和低于10%的污染。
其中糖苷水解酶基因使用dbCAN数据库的HMM进行预测,碳代谢使用KEGG数据。
研究还同时选择了部分样本进行了宏转录组和宏蛋白组的测序,对碳代谢同时结合转录组和蛋白组的数据,展现了特定通路下不同永冻土的菌群构成和表达丰度差异。

基因组拼接的分布情况,以及不同地域样本分布和菌属丰度情况如下:

木糖降解途径在每个样本中的分布和维恩图,另外详细的展现了主要门对每个代谢途径的贡献和基因表达丰度,如下图:

这张图分析了特定菌与地理位置和CO2以及甲烷的浓度的关联性,如下图:
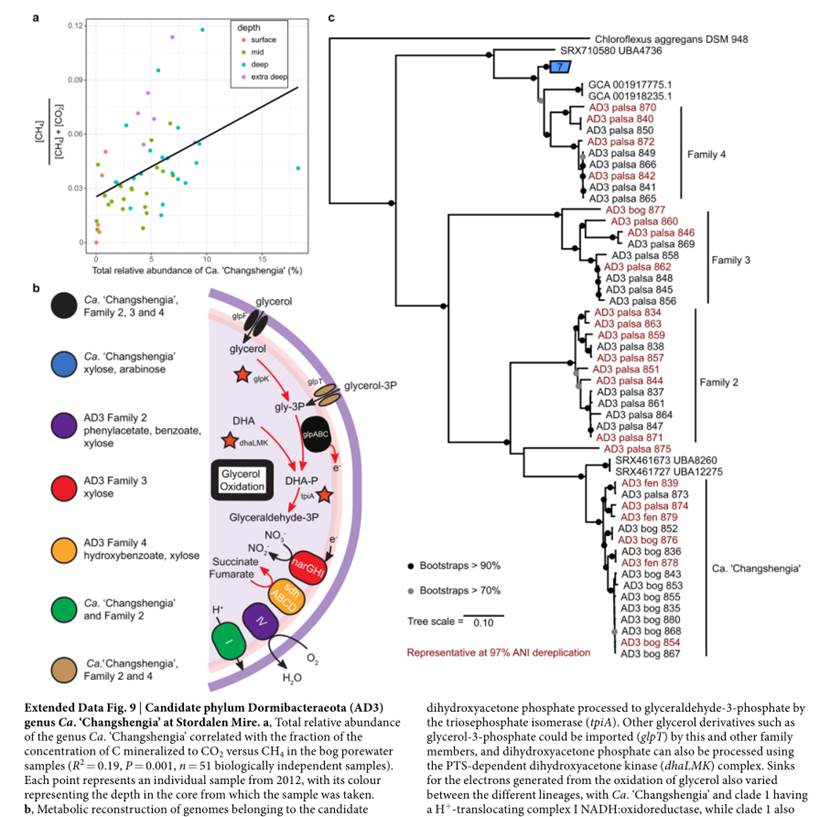

对关键物种的CH 4 :CO2浓度比相关代谢途径的重建,以及相应基因的基因家族分析。

总结一下这项研究,永冻土的菌群参考基因组数据缺乏,该研究从大量地点采集样本重建了1500多个参考基因组。
首先从物种构成特征上与永冻土融化阶段特征进行分析,并与重要环境因子进行分析,锁定重要的特征菌。
然后针对重要的代谢途径和关键基因结合宏转录组和宏蛋白组全面解析代谢途径的分布和差异变化。对关键的物种重建了相关代谢途径并对其相关基因家族进行分析。
研究基本上从头构建了一个生态环境下的菌群结构数据,并利用获得的基因组深度解析特定代谢途径和基因的构成和表达变化,应该说既宽又深。很多样本采集和测序是2011年和 2012年就开展的,虽然测序技术远不如现在成本低和成熟,但是其独特的研究对象和全面深入的分析仍然使整项研究和目前的一些研究相比完成的更加出色。
p.s. 以上展示的图表,我们都可以帮你实现~
网址:https://www.microbiomeanalyst.ca/,只需要biom文件或丰度表就可以进行绝大部分统计检验分析,而且生成图表完整,可以直接使用。偶尔会有服务器不稳定,上传提示错误的情况。
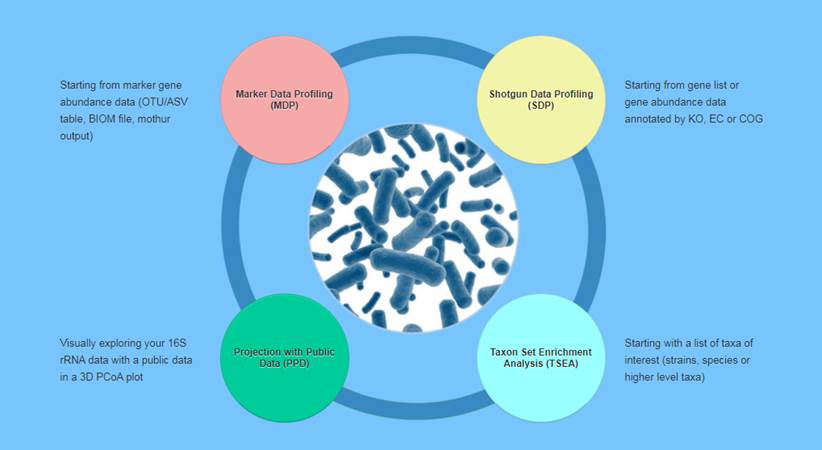

特别推荐其中的Taxon Set Enrichment Analysis模块,直接提交物种列表(一般是找到的差异物种列表),可以直接在各种已有的相关性(人体基因-菌属相关,生活方式-菌属相关,疾病-菌属相关)中进行富集分析,能很好的帮助判断和提供差异菌群的具体关联和证据支持。
完整的支持分析包括:


可以直接生成下面的图:


基本上常见的分析和图都能在线实现。
另一个是https://gcmeta.wdcm.org/,是中科院微生物研究所搞的平台,里面包括了宏基因组的样本数据和在线分析平台,可以直接上传原始数据,直接使用工具进行在线分析,大部分常见工具都有,也有一些流程。



对于缺乏计算资源或想自己动手分析的朋友挺友好的,非常推荐试试看。
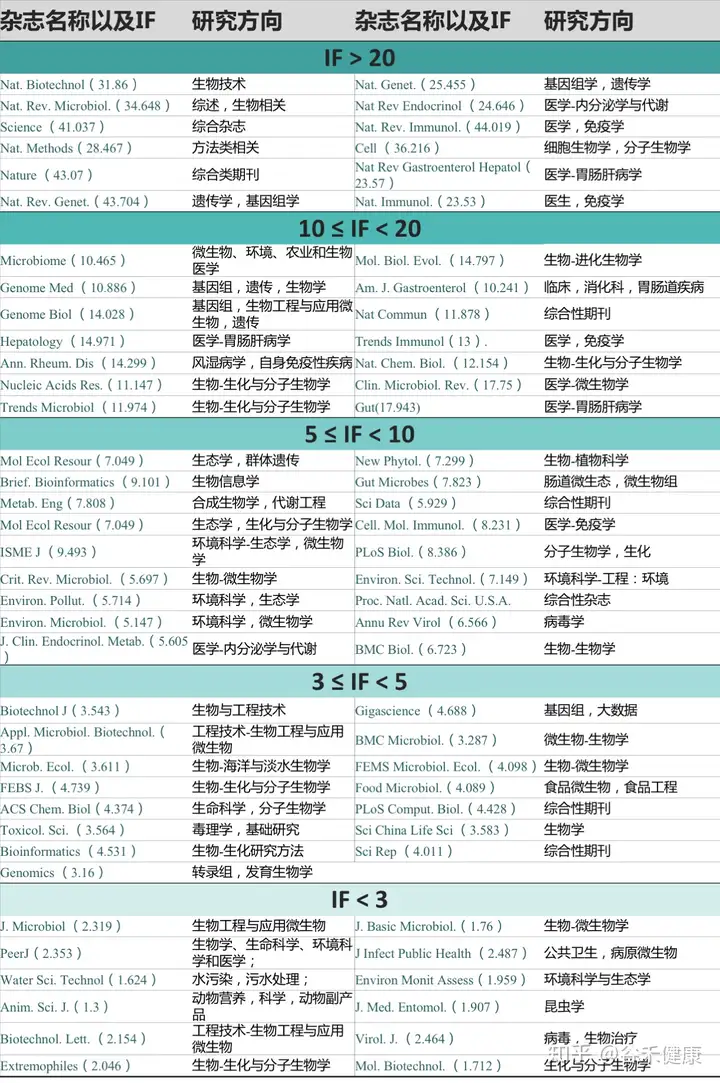
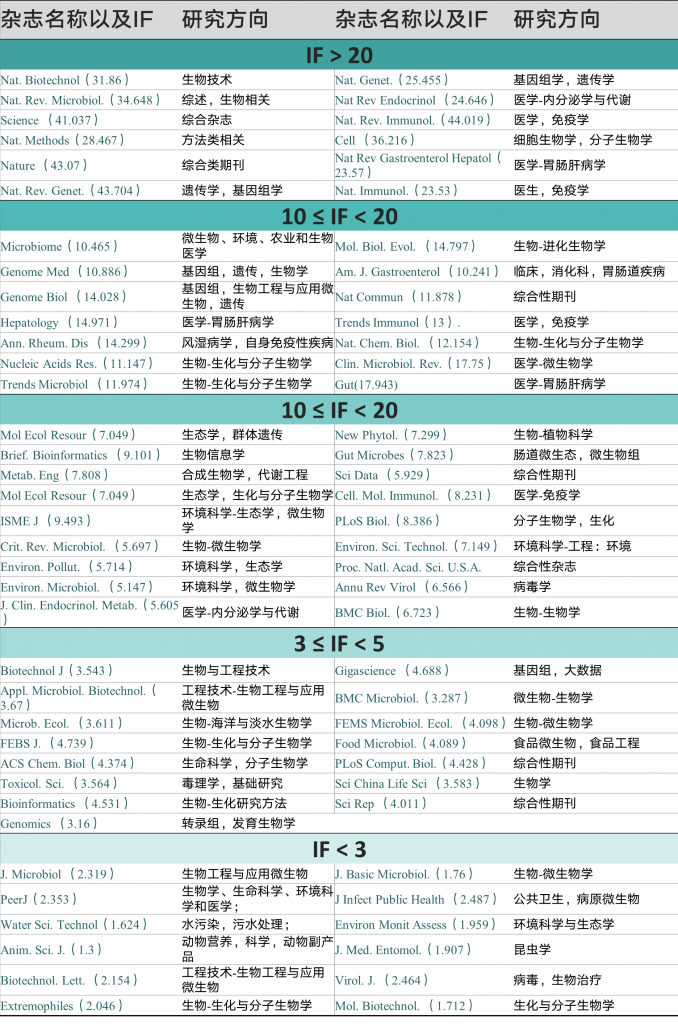
最后,帮大家整理了宏基因组可投稿的期刊,具体研究方向和影响因子见下表:



来源: 谷禾健康

肥胖到底美不美? 我不知道。
我只知道肥胖有可能会影响到健康。
肥胖威胁到健康的范围很广,包括2型糖尿病、冠状动脉疾病、中风、睡眠呼吸暂停和诱发某些癌症等严重疾病,甚至加速衰老,影响寿命。
肥胖不仅会产生这些生理上的不良影响,也会对患者心理造成一定影响,比如说由于行动不便导致无法工作,无法正常社交,甚至抑郁…


肥胖给很多人的生活带来困扰。但是,我们会发现这样一些现象,有些人是所谓的“吃货”,却不怎么胖,而有些人说自己“连喝水都会胖”。
看来肥胖背后的原因恐怕不简单。
今天,我们来详细讨论一下关于肥胖。
首先来看一下,肥胖的定义。
肥胖给很多人的生活带来困扰。但是,我们会发现这样一些现象,有些人是所谓的“吃货”,却不怎么胖,而有些人说自己“连喝水都会胖”。
看来肥胖背后的原因恐怕不简单。
今天,我们来详细讨论一下关于肥胖。
首先来看一下,肥胖的定义。


世界衡量标准
BMI全称身体质量指数,是世界卫生组织(WHO)确认的,衡量成人肥胖/超重的金标准。我们平常知道的都是以 BMI>25 算超重。
但由于人种差异带来的体脂比、肥胖形态的不同,亚洲许多国家和地区有自己的标准。
亚太地区标准
亚太地区肥胖和超重的诊断标准:

*亚太地区肥胖和超重的诊断标准专题研讨会依据亚洲人的情况制定。在BMI相对较低时,就易出现腹型或内脏肥胖,并显示患者高血压、糖尿病、高脂血及蛋白尿的危险性明显增加。
《中国成人肥胖患病率的地理变化:2013-2014年国家慢性病和危险因素监测调查》(以下简称《肥胖调查》)中有写到我国肥胖现状。
*《肥胖调查》由中国疾病预防控制中心张晓博士及其同事们共同完成。

该团队利用了中国慢性病和危险因素监测数据库,评估了2013-2014年全国和各省肥胖率。
将“中国肥胖标准”定义为:
普通型肥胖:BMI ≥28;
腹型肥胖:女性腰围大于85cm,男性腰围大于90cm
按照这个标准,该调查报告显示:2004年-2014年间,中国肥胖症患病率增加3倍多。腹型肥胖增长超50%。
从地区来看,“京津冀”的肥胖发生率很高。以天津为例,女性腹型肥胖率达49.4%,男性为54.4%。
儿童肥胖率也在日趋上涨
据经济合作与发展组织的调查数据显示:
至少有五分之一的儿童患有肥胖和超重。
《中国儿童肥胖报告》显示:
1985-2014年,我国7岁以上学龄儿童超重肥胖率:

肥胖已经是一种全球流行病,而且肥胖人数正越来越多。
3.1 能量摄入/消耗
我们知道,要维持健康的体重,需要保持食物摄入和能量消耗的平衡。

而这种平衡是由控制进食行为和能量代谢的中枢神经系统来负责。
饮食
饮食是影响体重非常重要的因素。饮食通常与饥饿感和饱腹感有关。我们饿不饿并不是自己能决定的,而是听大脑发出的信号,告诉我们饿了(产生饥饿感),该吃东西了,也就产生了食欲。那么这个食欲是怎么调节的呢?
这里有一个理论叫作“葡萄糖稳态理论”,这也是短期调节食欲的基础。


这就好比是特定脑区是一个指挥中心,它整合来自大脑、外周循环和胃肠道的信号,以调节能量的摄入和消耗。
神经元
在下丘脑弓形核(ARC)中,含有两类调节能量代谢的神经元:促进食欲神经元和抑制食欲神经元。

此外,pvn神经元进一步处理信息投射到其他回路,从而控制能量摄入和消耗。
小分子信号
当我们吃东西之后会诱发一系列的信号,包括肠道激素、肽、代谢物和营养素,进而影响能量摄入。大脑又会激活身体的不同机制,来控制食欲,或利用吃的食物和营养来开启发酵过程,合成代谢产物。
图1 体重调节的关键代谢机制
此外,在大脑中,传出信号被产生并发送到不同的组织,以促进产热和/或能量储存,并有助于能量消耗。(对应图中红线部分)
胃肠激素调节——饱腹感的生理介质
胃肠激素通过对胃肠运动和分泌的局部影响来优化营养物质的消化和吸收过程。在这方面研究最多的是胆囊收缩素(CCK)、胰腺多肽、肽YY、胰高血糖素样肽-1(GLP-1)、氧调节蛋白和胃饥饿素ghrelin。
它们通过各种不同的机制来增加你的饱腹感,让你别再吃了(具体方式在后面碳水化合物的小节展开)。
神经递质
许多肠肽既是激素又是神经递质。CCK和GLP-1等肽在投射到中枢神经系统内外对能量平衡至关重要的区域的神经元中表达。
大脑皮层高级神经活动,通过神经递质影响下丘脑食欲中枢,在调节饥饿感和饱腹感方面发挥一定作用。神经递质种类很多,包括五羟色胺、多巴胺等。
基于谷禾人群队列,我们选择了146例超重和肥胖人群,以及179例对照正常体重人群。其中超重标准为BMI>28,队列年龄分布为35~75岁。
根据菌群测序数据和相关营养及代谢调查进行KS统计检验,发现:
神经递质的检测数据显示与神经兴奋有关的多种神经递质和代谢物质肥胖人群普遍偏高,包括:
谷氨酸、五羟色胺、色胺和多巴胺。
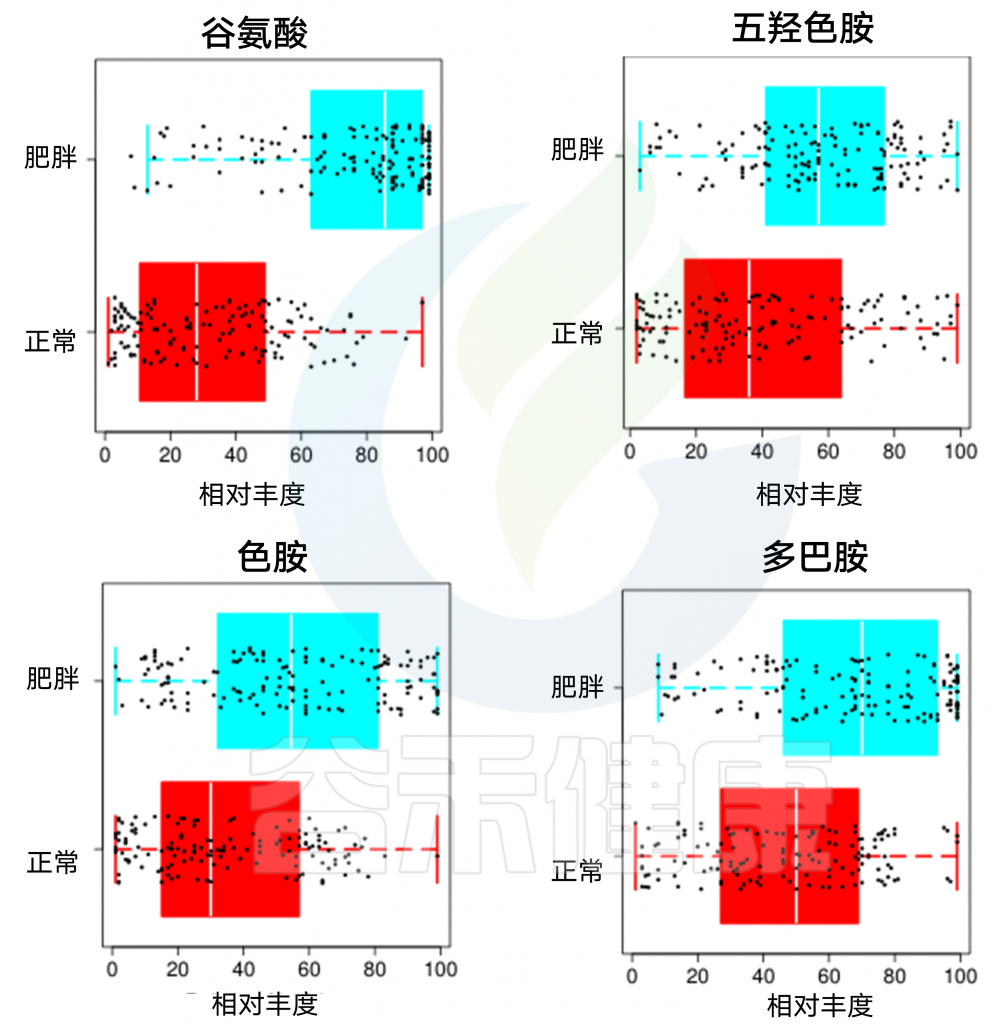

而神经抑制类的γ-氨基丁酸(GABA)则偏低:
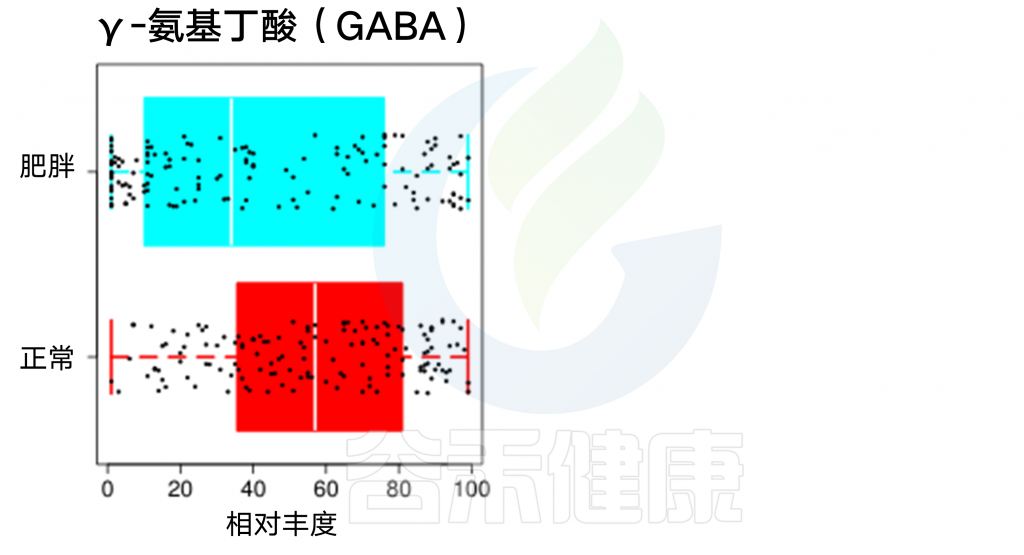

这些结果与食欲和饮食产生的神经兴奋是否有直接联系还需要进行更深入的生理分子机制的研究。
短链脂肪酸
短链脂肪酸影响与之相关的短期途径,调节食欲和食物摄入。能量收集上的差异似乎是由细菌产生的短链脂肪酸引起的,它提高了消化残余物的能量利用率。
短链脂肪酸有助于血糖稳定以及饥饿和饱腹激素的平衡。结肠输注短链脂肪酸混合物增加血浆PYY水平,这是由上皮G蛋白偶联受体(GPR41和GPR43)的激活触发的。
[具体关于短链脂肪酸的文章:你吃的膳食纤维对你有帮助吗?点此复习]

睡眠改变会影响饱腹感
这就涉及到“生物钟系统”。饮食脂肪和碳水化合物与生物钟系统相互作用,在生物钟系统中,特定的脂肪酸和葡萄糖可以影响行为和分子的昼夜节律,通过改变能量状态传感器影响中枢神经系统、非中枢神经系统和外周器官。这一假设符合葡萄糖稳态理论。
代谢的昼夜节律振荡受日常喂养模式和能量利用模式的影响,影响内分泌系统(瘦素、胰岛素或ghrelin分泌)与睡眠模式之间的昼夜节律排列。睡眠改变(如睡眠不足)可以改变内分泌昼夜节律,影响饱腹感和/或饥饿感,以及与主观幸福感有关的其他方面。

运动改善肥胖者的代谢灵活性
代谢灵活性的程度,即在骨骼肌和脂肪组织中将葡萄糖和脂肪酸之间的燃料选择转移的能力。
体育锻炼已被证明在改善肥胖患者的代谢灵活性方面具有关键作用。研究报告说,随着饮食脂肪的增加,肥胖者开始运动后增强了肌肉脂肪酸氧化。


3.2 三大类营养物质
人体摄入的营养物质中,有包括碳水化合物、脂肪、蛋白质在内的宏量营养素,也有微量营养素诸如维生素和矿物质等。但人新陈代谢的最核心内容是三大营养物质的代谢及其相互转化。
我们先看看这三大营养物质是如何直接或间接产生热量。注意热量不等同于能量。能量加上氧气参与,转化为热量。
在三大营养物质的代谢中,起枢纽作用的是糖类代谢,蛋白质(身体携带氧气的关键)要转变成为热量,需要先转变成为糖类,才能够燃烧产生热量,脂肪的燃烧,也需要糖类的参与才能够顺利完成。也就是说,蛋白质、脂肪是能量的两个储藏仓库,而糖类,会直接释放热量。
接着我们具体来看每种营养素与肥胖之间的关系。
3.2.1 碳水化合物
碳水化合物包括不同类型的分子,从复杂的多糖和淀粉到单糖,如单糖或双糖,以及不同的纤维,能量值和消化率不同。


这些结构差异导致它的物理性质、消化率和功能的不同。碳水化合物对食物摄取和饱腹感的影响是通过与胃肠-脑神经内分泌信号、肠道发酵(由短链脂肪酸或其他代谢物诱导)和碳水化合物的物理或化学性质(如膨胀和粘度)相关的几种途径介导的。
碳水化合物通过调节胃肠道肽释放来影响饱腹感
与维持血糖水平有关的食欲和饱腹感调节由胰岛素和胰高血糖素调节,胰岛素和胰高血糖素由胰腺分泌,分别导致导致糖原生成和糖原分解。 事实上已证实,包括纤维在内的碳水化合物,对饱腹感相关胃肠道肽(CCK,ghrelin,GLP1,PYY和GIP)餐后释放有影响。单糖和双糖,如蔗糖、果糖和乳糖,是人类消耗的最常见的糖,它们在食欲和食物奖励中的作用与甜味受体有关。
纤维和淀粉是饮食中最复杂的碳水化合物结构。
它如何产生饱腹效应?
纤维通过调节胃肠道转运、葡萄糖吸收和粪便膨胀起作用。
低能量密度(2kcal/g)和水化能力产生饱腹效应,它们提供粘度和膨胀,影响肠道中的机械感受器,影响血糖反应和饱足信号,增加肠道蠕动能力,并作为肠道微生物的主要底物。
3.2.2 脂肪
脂类具有较高的能量密度,其消耗可能通过间接机制直接影响食物摄入和能量消耗以及代谢,从而影响肥胖和相关疾病。


脂类在能量积累中起着核心作用,在内分泌系统中起着重要的作用。 它在食欲和饱腹感调节中的作用包括通过感觉受体和化学感受器。
「瘦素」——脂肪与下丘脑饱腹调节中心沟通的桥梁
瘦素是由脂肪组织分泌的一种激素,分泌过程依赖胰岛素。
当体内瘦素水平较高时,会告诉大脑身体有足够的能量储备,不要再吃了。瘦素和胰岛素共同作用于下丘脑和其他脑区,从而抑制食物摄入,增加能量消耗。
可想而知,当瘦素长期处于低水平时,往往伴随着肥胖。低水平的瘦素在不断告诉大脑,身体正处于饥饿状态,那么很有可能出现暴饮暴食,且身体在不停地储存能量。
脂肪的质量比数量更重要
越来越多研究表明,除了饮食中的脂肪或碳水化合物的数量外,膳食脂肪的质量(主要指天然不饱和脂肪而不是反式或饱和脂肪)比数量更能决定饮食对维持体重的影响。
具体来看,目前研究的重点是脂肪质量与脂肪酸饱和度之间的关系。饮食中omega-6脂肪酸与omega-3脂肪酸比例的增加与通过脂肪生成、脂肪组织稳态、脂肪褐变和炎症机制增加体重,和肥胖的风险增加有关;这种增加是由脑-肠-脂肪轴通过二十烷类代谢物和大麻素系统的多动介导的。
在这种情况下,高脂肪消耗在与脂肪沉积有关的胃肠道调节喂养途径上的调节作用影响脂质的吸收和利用,减弱饱腹信号并促进超重,其中CCK、ghrelin、GLP1和其他神经肽与食欲和/或饱腹有关。此外,中链甘油三酯、共轭亚油酸和多不饱和脂肪酸引起不同的饱腹和胃排空效应。
多不饱和脂肪酸导致能量摄入和消耗变化
此外,多不饱和脂肪酸在调节身体成分中起着重要作用;它们导致能量摄入和支出的定向变化,以及脂质周转和脂肪生成的改变,这是由神经内分泌系统介导的。 此外,一些饮食脂质可能通过促进棕色和米色脂肪的生成来刺激产热功能。脂类也在食物质地中起作用,这可能会使食物更可利用。
当然,脂肪也可能表现出不受欢迎的口感,这可能导致食物的排斥,从而干扰能量的摄入,而不是餐后机制。
肥胖也与诱导的食物偏好和减少底物氧化、酮体保留糖代谢机制以及脂溶性和/或脂肪生成活动或炎症途径的变化有关。
要想躲避肥胖,建议多选不饱和脂肪酸
单不饱和脂肪酸摄入量与BMI呈负相关,突出了脂肪分布的重要性,而不仅仅是脂肪总量,而且影响的差异取决于遗传背景。
食用不饱和油不易导致肥胖,这表明脂肪质量比数量更重要(例如,在那些遵循地中海饮食模式的人中观察到的或食用树坚果)。
平时食用油的选择,动物油脂,橄榄油,菜籽油等都要好于色拉油、调和油。
3.3.3 蛋白质
蛋白质分子结构的多样性,决定了蛋白质分子有多种重要功能,它们也负责酶的功能,并具有内分泌调节作用。
膳食蛋白质和蛋白质衍生分子表现出食欲和厌食效应,这些效应是由不同的神经肽和肠道激素介导的,如胃饥饿素、CCK、GLP1、PYY、瘦素、胰岛素和特定氨基酸。

蛋白质有助于保持无脂质量和骨骼肌,从而促进能量消耗的维持。
高蛋白饮食对食欲抑制作用
蛋白质在饮食中的比例影响食欲调节和饱腹感的诱导。例如,干预性研究显示了高蛋白饮食对食欲抑制的饮食作用。富含蛋白质的饮食导致饥饿感减少,能量消耗和β-羟丁酸水平增加,这可能有助于食欲下降。
蛋白质具有明确的产热性质,由氧化磷酸化过程、尿素合成、糖异生和能量产生的中间代谢途径介导。然而,对于特定的蛋白质,如明胶gelatin,在食欲抑制中也有独特的作用。
此外,与蛋白质诱导饱腹感有关的机制已被归因于氨基酸(如酪氨酸、色氨酸或组氨酸)、影响胃迷走神经传入的鲜味物质(包括味精)或来自甜味剂的阿斯巴甜(asp-phe)氨基酸有关的钙传感受体,它们调节食欲和餐后发生热。
长期动物蛋白的摄入对体重的维持有不利的影响
然而,一些长期的纵向分析表明,动物蛋白的摄入对随访5年后体重的维持有不利的影响。 事实上,每天食用250克肉类,与肉类含量较低的等热量饮食相比,每年增加422克体重。因此,以动物蛋白和糖为代价增加植物蛋白的摄入量,可能是在人群水平上对抗超重和肥胖的一种方法。
这些结果突出了营养素比例的重要性。但进一步的研究还应侧重于蛋白质来源和血糖指数的体重维持。
4.1 遗传机制
些人哪怕吃正常量的食物就是比一般人容易发胖,这可能与肥胖基因有关。
接下来我们来看看遗传因素在决定个体对肥胖的易感性方面起了什么样的作用。
不同的研究报道了总脂肪比例与食物摄入调节相关基因(如FTO)、脂肪代谢相关基因(如APOA5)和脂肪细胞分化相关基因(如PPARG)在不同人群中的相关性。
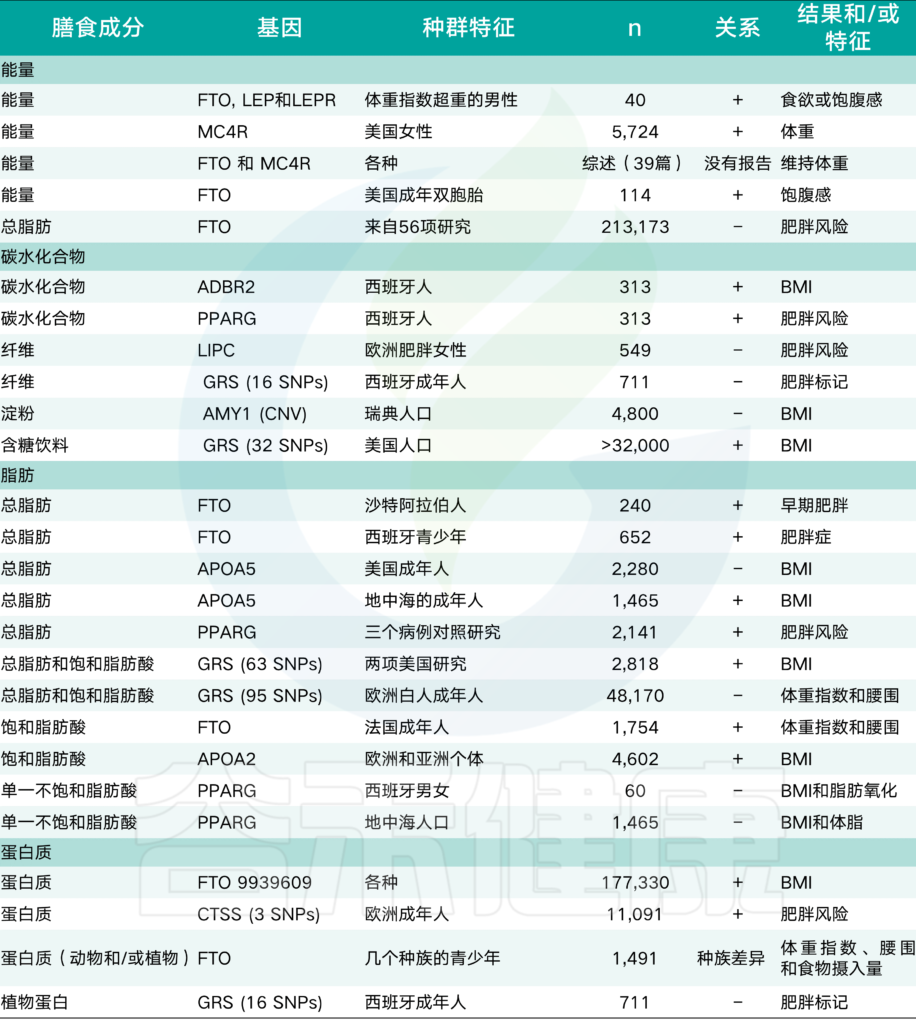
肥胖基因——FTO基因(减弱饱腹感)
与没有风险等位基因的参与者相比,那些具有FTO变异型rs9939609的风险等位基因的人报告显示,主观饱腹感较低,能量密集的食物消耗较高,这表明具有遗传风险的个体已经损害了中枢神经系统的饱腹感。
此外,有FTO危险等位基因的个体也会消耗较高比例的能量作为脂肪,显示出肥胖风险的加剧。
APOA2等位基因的存在通过调节参与支链氨基酸和色氨酸代谢的途径,在饱和脂肪酸摄入时增加的肥胖风险。
与之相反,APOA5基因变异rs662799的C携带者,在饮食中消耗更多的脂肪,则可避免肥胖。

然而需要注意的是,即便是检测出了携带肥胖基因,也不意味着必然会永久肥胖下去;反之,不携带肥胖基因,也不意味着可以胡吃海喝不运动。要知道决定肥胖的不止基因这一个因素,例如还有肠道微生物群的影响。如果说基因难以改变,那么肠道菌群是你可以改变的。
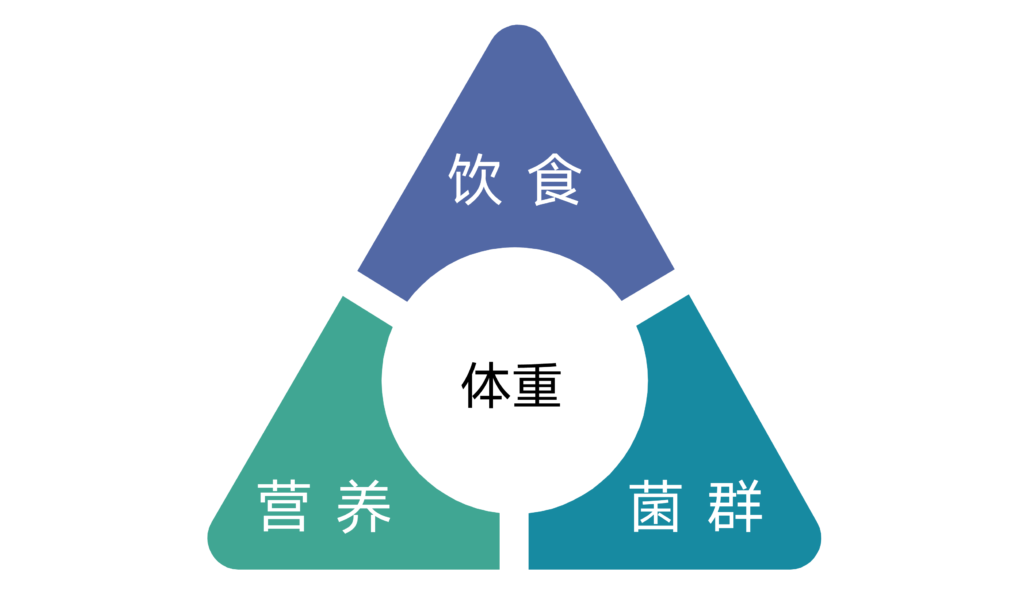
4.2 菌群介入,影响肥胖
易患肥胖的个体的肠道微生物群可以通过饮食、益生菌等来改变。
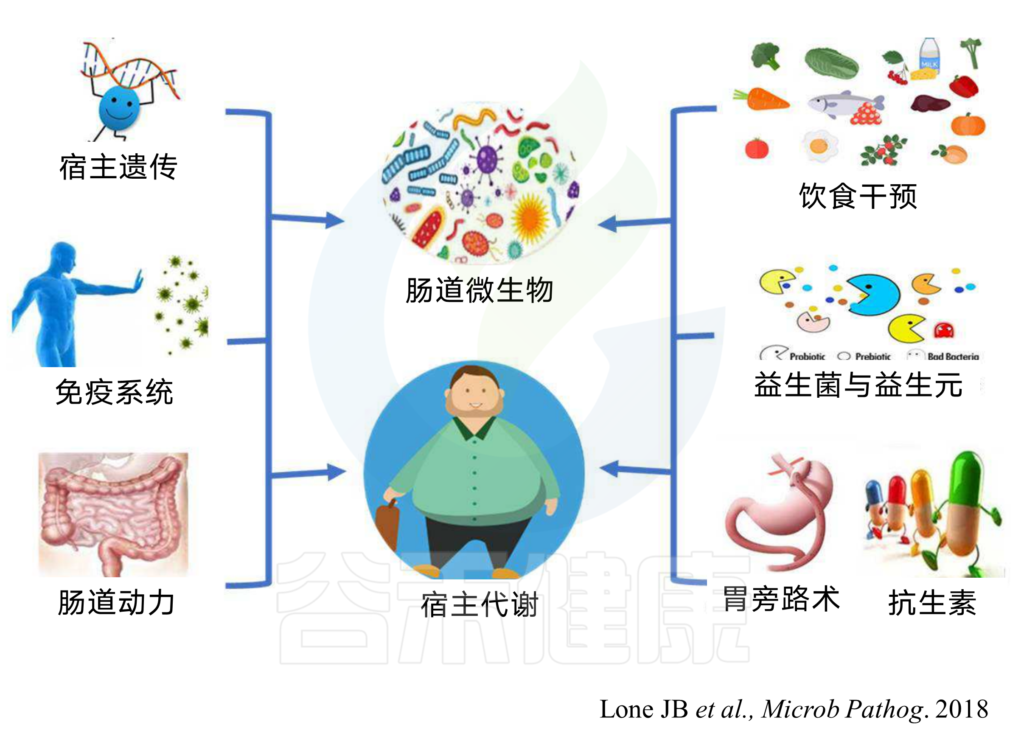
碳水化合物(即纤维)、蛋白质和脂肪的摄入会影响肠道微生物群,从而使宏量营养素与微生物群相互作用。具体在前面 深度解读|饮食、菌群和健康 一文有详细阐述。
个体微生物群的组成是独特的,是个体代谢和体重状况的代表性特征。
人体摄入宏量营养素、肠道菌群与肥胖的关系
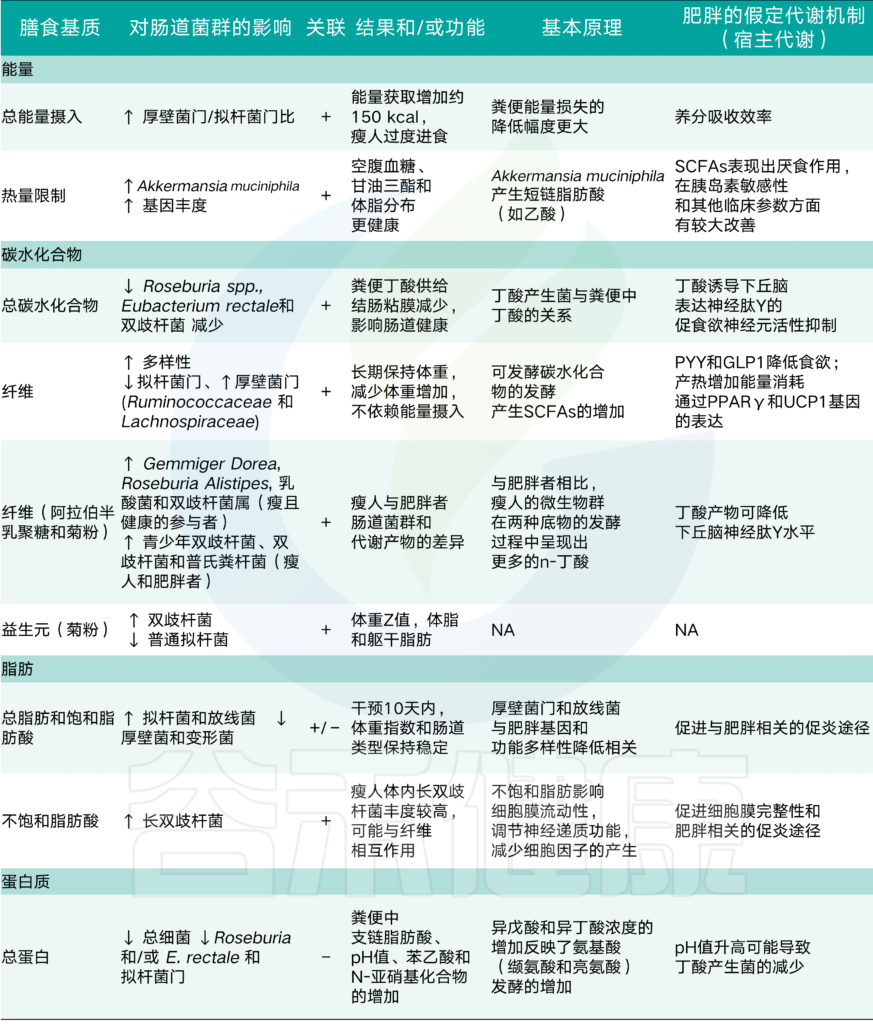
↑, 增加;↓,减少;NA,不可用;第三列:营养物质和/或饮食基质与肠道微生物群之间的关系
4.2.1 菌群和肥胖之间的关系
菌群丰度和种类
菌群丰度和多样性与肥胖呈负相关
开创性的研究已经描述了微生物群与肥胖之间的关系。数据显示肥胖小鼠的厚壁菌数量增加,拟杆菌数量减少,这表明这些微生物对宿主代谢可能有影响。这些结果随后在人类研究中得到证实,研究人员在研究中比较了来自瘦弱或肥胖人群的样本。
一些研究描述了肥胖者从食物中获得的能量的增加,并确认了这些人粪便样本中的普雷沃氏菌科和古细菌的增加。此外,菌群丰度和多样性与肥胖呈负相关,即菌群丰度低的人会提高肥胖、胰岛素抵抗和血脂异常,以及低度全身炎症风险。
基于谷禾人群队列数据发现:
肠道菌群的种类数量肥胖人群要显著低于对照人群(下左图),均值分别为850比1150,这一现象与之前的研究是符合的。
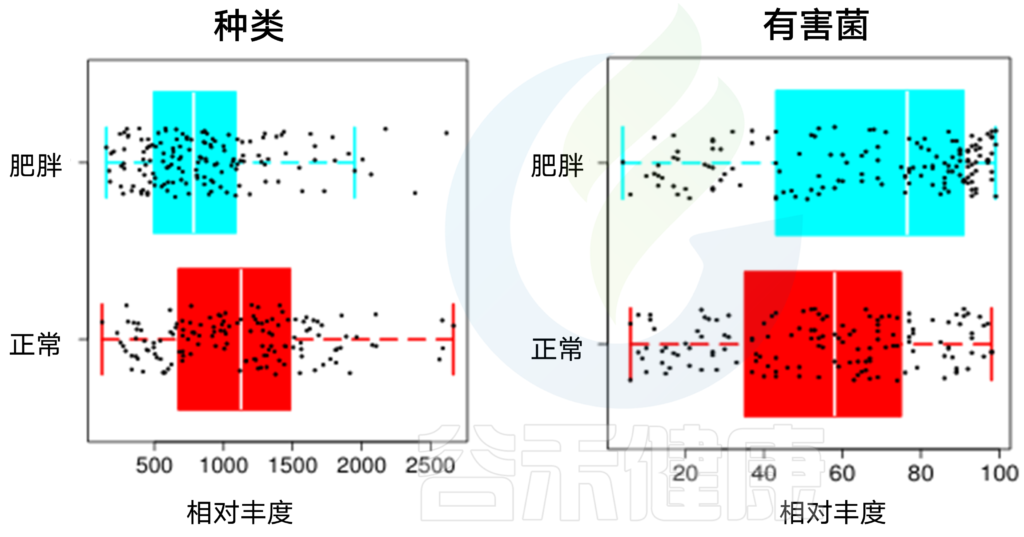
有害菌
在有害菌属的丰度水平分布上,肥胖人群也要高于对照人群(上右图)。进一步对具体菌属进行分析,发现肺炎克雷伯氏菌的丰度水平肥胖人群更高(下左图)。
有益菌
而双歧杆菌属的水平肥胖人群普遍较低。
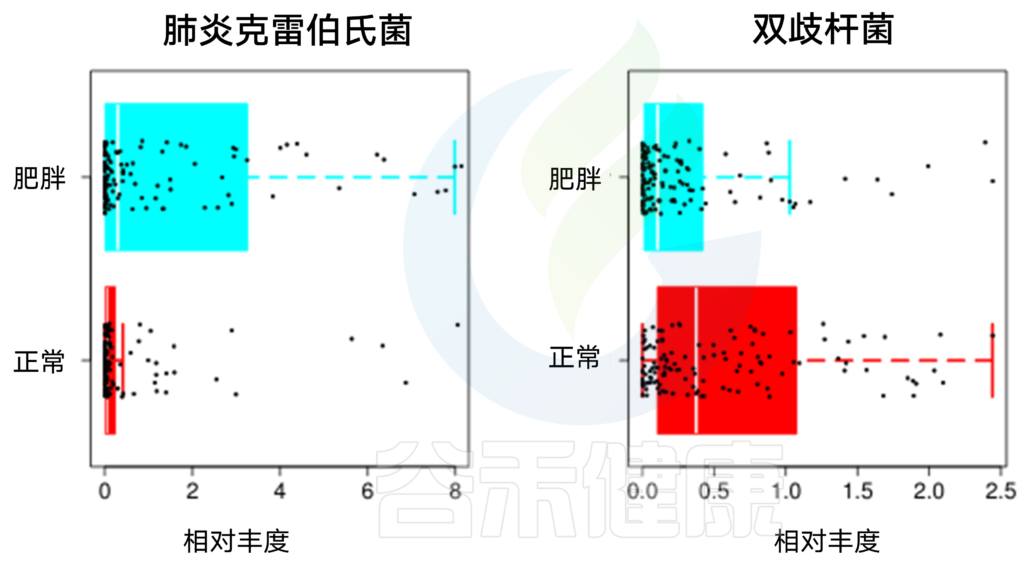
一项调查同卵双胞胎脂肪摄入质量的研究发现,单不饱和脂肪酸与双歧杆菌和拟杆菌的丰度正相关,以及n-3多不饱和脂肪酸和乳酸杆菌之间存在正相关关系,而n-6多不饱和脂肪酸与双歧杆菌是负相关。
一些学者认为在饮食中添加益生菌可以减轻饮食中脂肪对体重维持和脂肪积累的有害影响。
4.2.2 菌群如何影响肥胖
肠道微生物群影响超重和肥胖个体体重和代谢的两种可能机制。
(1)肠道微生物群将不可消化的膳食纤维发酵为短链脂肪酸,这些脂肪酸可能由肠道上皮代谢或是葡萄糖和脂质从头合成的一部分。
(2)由于紧密连接处的薄弱导致肠道通透性增加,来自肠道细菌表面的脂多糖(LPS)能够进入循环,并可能在包括肌肉、肝脏和脂肪组织在内的多个组织中引起免疫反应、炎症、巨噬细胞浸润和胰岛素抵抗。胰岛素抵抗和厌食激素(包括胰高血糖素样肽1[GLP-1]和肽酪氨酸[PYY])在下丘脑的表达降低刺激了食物摄入的增加。
此外,微生物蛋白水解可产生支链氨基酸和芳香烃氨基酸,可通过微生物交叉喂养(微生物物种的营养相互依赖性)进一步代谢。这些交叉喂养途径的紊乱导致这些氨基酸的吸收增加,并与肠道完整性和胰岛素抵抗的损害有关。
糖化和蛋白水解代谢物生产中涉及的微生物群落和微生物网络
肠道微生物的功能群、主要代谢途径和生产碳水化合物和蛋白质发酵产生的代谢物的中间体,包括糖解产物短链脂肪酸,琥珀酸盐和乙醇以及蛋白质水解代谢产物,包括氨、吲哚和酚类化合物、硫化氢、胺和支链脂肪酸(BCFAs)。
4.2.3 饮食干预菌群
越来越多的研究者开始对饮食模式感兴趣,以研究食物营养素、肠道微生物群组成和遗传背景之间复杂相互作用。
对人类的一项观察研究表明,肥胖人群中存在小肠细菌生长,他们的饮食中富含碳水化合物和精制糖,缺乏纤维。饮食蛋白质和碳水化合物对肠道微生物群的调节作用改变了蛋白质消化的衍生物。这些衍生物通过改变芳香族和支链氨基酸的发酵以及作为短链脂肪酸的前体而影响能量代谢。
基于谷禾肠道菌群的营养量化算法可以有效反映和量化饮食及营养元素的摄入状况。数据显示,主要饮食结构中精制糖摄入肥胖人群显著偏高,而乳制品摄入普遍偏低。
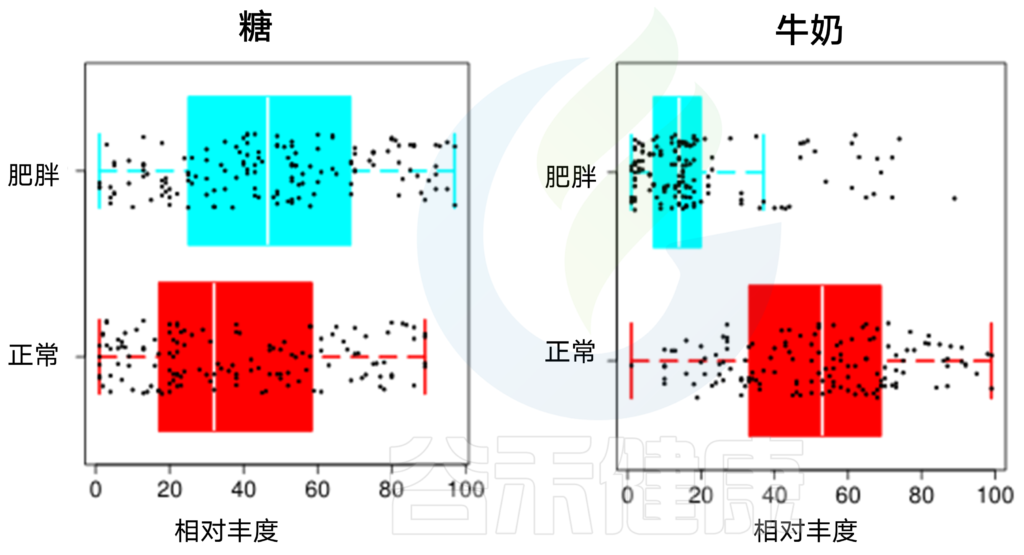
低聚果糖调节菌群,与短链脂肪酸相关
膳食宏量营养素发酵产生的代谢产物对肥胖及合并症的影响已被广泛研究。此外,碳水化合物的摄入被认为是调节肠道微生物群的主要因素。纤维通过调节胃肠道转运、葡萄糖吸收和粪便膨胀起作用,影响血糖反应和饱足信号。
可发酵膳食纤维在调节微生物方面具有关键作用肠道菌群。益生元,如低聚果糖,对肠道微生物群的影响已经被一些研究证明,因为低聚果糖调节了粪便细菌和其他细菌的丰度,并与丁酸盐的产生相关,通过降低下丘脑神经肽Y的表达而减少食物摄入。
还研究了富含糖和人工甜味剂的“西方”饮食对肠道微生物组成的影响。不幸的是,甜味剂和人体肥胖的肠道微生物群之间的联系仍然不清楚。

富含脂肪的饮食可以通过抗菌作用影响肠道微生物群
其他研究者认为,饮食中脂肪摄入对体重增加的影响,除了燃料正平衡外,还有其他作用。富含脂肪的饮食可以通过抗菌作用影响肠道微生物群,从而促进与肥胖相关的全身促炎症状态相关的失调的发展。对健康人进行的横断面研究发现,在习惯性饮食中,拟杆菌和放线菌与脂肪摄入呈正相关。此外,与体重指数相关的现有分类群也与饱和脂肪酸的热量摄入相关。
一些作者认为在饮食中添加益生菌可以减轻饮食中脂肪对体重维持和脂肪积累的有害影响。
饮食干预后的菌群改变与脂肪改变有关
一项包括一组健康人的对照喂养试验发现,在饮食干预24小时后,肠道微生物群的组成发生了变化,这些变化在10天内保持稳定。
研究人员还报告了长期饮食模式和菌群之间的关系。更具体地说,他们报告了拟杆菌与蛋白质、动物脂肪之间的关系,普雷沃菌与碳水化合物之间的关系。同样,其他营养干预措施也发现,大鼠高脂高糖和低脂高糖饮食后,菌群多样性下降。这些饮食与肠道炎症的增加和迷走神经-肠道-大脑连接有关,与身体脂肪沉积的增加有关。
综合分析,健康饮食模式下菌群丰富,炎症减少
以往有多项研究显示肥胖人群在一些炎症指标上较高,存在长期的炎症反应,我们针对主要炎症指标的统计检验同样发现以下炎症指标确实存在偏高:
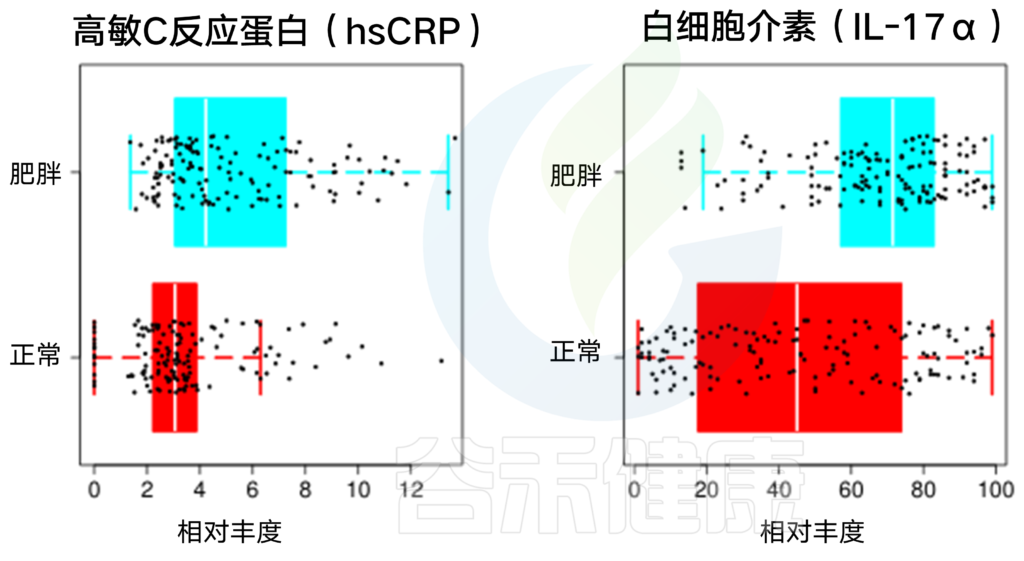
一些作者强调了使用肠道微生物群的综合分析和代谢组学分析,客观评估对坚持健康的饮食模式,帮助阐明这些模式的明确饮食组成,如地中海饮食模式,以及它们在心脏代谢紊乱的复杂相互作用中的假定作用。例如,一项试验报告了肥胖人群中与健康饮食模式相关的肠道微生物群组成的差异。更具体地说,那些饮食摄入更健康的人微生物丰富,炎症标志物浓度降低。
在这些研究的基础上,数据表明饮食因素之间的相互作用,如宏量营养素分布、肠道微生物群组成和遗传背景,与肥胖中发现的代谢障碍有关。对个体特征和对肠道菌群代谢物相关敏感性的整体研究有助于制定新的策略,提高肥胖防治效果。
现在我们知道各种因素会改变肠道微生物,肠道微生物的改变会导致代谢功能的改变,从而导致肥胖。
要精确的了解每个人具体的问题所在,就要了解自己的肠道微生物,请进行肠道菌群检测,不仅了解肠道菌群,更重要的是能找出肥胖背后的真正问题,确定适合你身体的方式,科学减肥。
相关阅读:
主要参考文献:
San-Cristobal R, Navas-Carretero S, Martínez-González M Á, et al. Contribution of macronutrients to obesity: implications for precision nutrition[J]. Nature Reviews Endocrinology, 2020: 1-16.
de Clercq, Nicolien C., et al. “Gut microbiota in obesity and undernutrition.” Advances in Nutrition 7.6 (2016): 1080-1089.
Canfora, Emanuel E., et al. “Gut microbial metabolites in obesity, NAFLD and T2DM.” Nature Reviews Endocrinology15.5 (2019): 261-273.
Diabetes Care 2015 Jan; 38(1): 150-158.https://doi.org/10.2337/dc14-2391
Lancet 2004; 363: 157–63
Linfeng Zhang, Zengwu Wang, et al. Prevalence of Abdominal Obesity in China: Results from a Cross-Sectional Study of Nearly Half a Million Participants.Obesity.2019 Sep. DOI:10.1002/oby.22620.
Hou X, Lu J, et al. Impact of waist circumference and body mass index on risk of cardiometabolic disorder and cardiovascular disease in Chinese adults: a national diabetes and metabolic disorders survey. PLoS One. 2013,8(3): e57319.DOI: 10.1371/journal.pone.0057319.
Geographic variation in prevalence of adult obesity in China: results from the 2013-2014 National Chronic Disease and Risk Factor Surveillance. Ann Intern Med. 29 October 2019.2.
Lone Jameel Barkat,Koh Wee Yin,Parray Hilal A et al. Gut microbiome: Microflora association with obesity and obesity-related comorbidities.[J] .Microb. Pathog., 2018, 124: 266-271.

本文由谷禾健康整理编译自:VICTORIA FRANKEL, Give Your Gut a Chance: Microorganisms and Your Immune System和VIOME,19 Science-Backed Ways to Improve Your Gut Health
每当一种传染病威胁到许多人的健康时,免疫系统如何运作的话题就经常出现。
从整体上看,人类的免疫反应有助于对抗病原体。在生病的时候适当地使用免疫系统是保持我们健康的一个重要部分。由于我们的免疫系统很大一部分位于肠道内,肠道微生物群在决定我们的免疫反应何时以及如何运作方面起着关键作用。
大家都希望保持自身免疫系统的最佳运作,特别是随着呼吸道疾病的流行和病毒感染影响我们的生活。随着人们对季节性传染病如流感和新近出现的新型冠状病毒(又名COVID-19)的日益关注,许多人正在寻找是否有办法提高自身持续免疫能力。
但最新的科学表明,你的肠道微生物群、你吃的食物和整体免疫力之间可能存在持续的有益关系。
商店里或者仓库中,衣服上,书桌上…到处都是微生物。只要正常生活,身体就不可能处于一个无菌的环境。
事实上,我们的身体内充满了细菌,这些微生物帮助我们维持体内平衡和免疫酶活性——通常是通过与我们的免疫细胞相互作用。这已经被证明会影响自身免疫、炎症、癌症和我们对疾病的易感性。
读过我们前面文章的小伙伴都知道,体内的微生物群对人体而言十分重要,例如肠道微生物群对我们的消化系统、神经系统、免疫系统等等都有重要影响。
肠道微生物群
把你的肠道微生物群想象成一种工具。当功能失调时,它会在执行正常任务时造成无意伤害,就像一把刀。然而,当保持在合适的条件下,它可以精确地执行。
一个健康的肠道微生物群功能良好,不太可能损害你的肠道。这个薄薄的屏障可以保护微生物在它们喜欢的环境中安全健康,并且防止它们出来探索我们不愿看到的其他领域,比如你的血液。
当肠壁受损时,我们的肠道微生物群就可以逃逸到不属于它们的地方,就像其他器官一样。当它们在这些区域累积时,它们分泌的毒素和代谢物会导致炎症反应,损害这些组织并导致慢性疾病的发展。
因此,我们需要重视肠道微生物群健康平衡。它也在感染时控制我们的免疫反应中发挥作用。
排除病原菌
许多共生或有益的微生物有助于保持肠粘膜的健康。
当我们的肠道微生物群能正常产丁酸(关于短链脂肪酸详见之前的文章 你吃的膳食纤维对你有帮助吗?),它有助于我们的肠道保持强大和完整。相反地,我们的肠壁在感应到病原体时,通过保护这些共生细菌群落而得到回报。事实上,我们的免疫细胞可以非常“聪明”地识别特殊的肽。
可见,肠道微生物组在人体对感染的免疫反应和维持整体健康中起着至关重要的作用。
从排便看肠道健康
“定期”是描述良好排便习惯或正常排便功能的一种方式。
我们经常谈论排便要规律,但这常常被误解为需要每天上厕所排便。有些人两天排便一次就很焦虑,觉得好像不健康,其实没必要过于担心。
但只要保持规律,轻松的排便,每天排便1-3次至每周3次都是正常的。饭后约30分钟内排便(通常是早餐),但是这可能因人而异。
怎么知道我是否有肠道健康问题?
导致日常生活中许多因素导致肠道微生物群失衡。这些包括睡眠质量差,压力大,不健康的食物以及过度使用抗生素。肠道健康问题有很多物理指标。这些包括:
胃部不适
腹胀,腹泻,便秘等都是肠道健康问题的明显症状。当肠道微生物群达到平衡时,人体可以更有效地消化食物。
自身免疫问题
一些科学家认为肠道健康不良会加剧体内炎症。这会阻止免疫系统正常运行。结果可能是自身免疫性疾病。
对甜食的渴望
如果你总是想吃甜食类食物,那么肠道细菌可能会失衡。高糖饮食和加工食品会减少肠道的有益菌,然后你会渴望更多的糖分,从而导致炎症,而炎症会导致多种疾病,甚至被证明会导致癌症。


体重变化
在没有改变运动习惯或饮食习惯的情况下,体重是否正在增加或减轻?
肠内失衡会阻止人体适当调节血糖水平,储存脂肪和吸收营养,从而影响体重变化。
睡眠质量差
如果你有患有失眠症,那么长期疲劳是不可避免的结果。要知道90%的血清素是肠道产生的,而不是大脑。由于血清素是调节睡眠所必需的,因此肠道不健康就会影响你的睡眠质量。
皮肤问题
如果患有湿疹或其他皮肤疾病,则可能源于肠道不健康。肠道内部发炎可能导致蛋白质泄漏到您的体内。这会刺激您的皮肤并引起皮肤问题。
食物不耐受
如果您的身体无法消化某些食物,则可能是由于肠道中有害菌所致,可能导致腹痛,腹胀,恶心和腹泻。肠道菌群问题也可能引起食物过敏。
呼吸困难 / 哮喘
不同的肺部疾病会受到肠道微环境变化的影响,反之亦然。由此研究人员提出了“肠-肺轴”连接的概念。
在哮喘中,微生物群是导致这两者之间相互作用的重要因素。在人类和小鼠的研究中,肺和肠道之间的联系已经被反复证明。这种相互作用可以是双向的;肠道微生物可以影响肺部的免疫反应,而肺部刺激可以导致肠道反应。来自肠道菌群的短链脂肪酸对肺部的促炎反应有抑制作用。
详细可见之前的文章「哮喘」最新研究已逐步渗透到更精细层面
口臭
如果患有口臭(长期口臭),则可能会导致肠道菌群不平衡。口臭是微生物在牙龈,舌头和牙齿之间生活的结果。这些微生物产生难闻的气味。有时,这可能是由于牙龈疾病引起的。

心情变化
肠道和大脑通过迷走神经相连。肠道微生物组会产生或消耗大量的神经递质。微量营养素缺乏会损害您的肠道功能,并导致肠道渗漏。
除了许多其他慢性疾病外,肠漏也被认为是抑郁症和焦虑症的潜在病源。认为肠漏是因为肠道没有健康的边界。发生这种情况时,可能会出现疾病。当肠道屏障破裂时,它会开始连锁反应,引起低度的慢性炎症。
肠道功能受损也会影响您的身体利用多巴胺和血清素的能力。这些激素对你的快乐感知是必不可少的,也是积极精神态度中的另一个关键要素。肠道可以产生大约一半的人体多巴胺和90%的血清素。因此维持肠道健康平衡可以直接支持你的心理健康。
详细可见之前的文章 深度解读 | 肠道菌群和中枢神经系统的关系
另外,还可以通过粪便的颜色,形状,频率等去判断,详细了解可移步文末附录部分。
如果你在搜索引擎或者网站上搜索改善肠道健康时,会发现大量建议。遍历所有内容时,你会发现大多数内容都是模棱两可的,甚至是冲突的。所以我们需要基于更科学的方法来改善。
完美的饮食神话
我们有时候往往会有这样一种想法,一种饮食方式比另一种饮食方式更好,其实对于个人的肠道微生物群来说是不可取的。同样,某些食物可以改善每个人的肠道健康的想法也不是完全正确的。
肠道微生物组科学揭示出一个事实,那就是没有普适的健康饮食。每个人(及其肠道微生物群)都需要不同的食物。 所以不要想着光吃某几种所谓的健康食物来拯救你的肠道健康。
在改善肠道微生物组方面,适用于所有人的唯一饮食建议是,每个人都应尝试食用多种食物。这是因为饮食多样性更有利于菌群维持健康平衡。
试想一下,你的微生物就好比一群挑剔的孩子,每个孩子都会去吃自己喜欢的食物。当您吃各种食物时,就相当于喂食了各种微生物。
有时候,谷禾的消费者会对拿到的报告中的饮食建议感到不可思议。
比如说他们发现自己的列表里不推荐的有类似酸菜和泡菜这样的食物,这些食物对他们来说是非常传统,家常,甚至不可或缺的食物。为什么是不推荐的呢?这可能是因为这两种食物中的组胺含量都很高,而他们的肠道无法很好地处理。
什么肠道健康建议适用于大多数人?
你可以采取多种措施来改善肠道健康。下面列出的20种是以科学为基础的方法。这些结果不仅基于科学文献,而且还基于我们在大数据检测内容。

我们将建议分为两部分。首先,您可以通过避免破坏性因素来改善肠道健康。
避免这8种破坏肠道的食物和行为
1. 糖
很多女生爱吃甜食,实际上甜食对肠道微生物组不是很友好,因为像白色念珠菌这样的有害细菌和真菌会增多,已显示高糖饮食可改变肠道微生物组的组成和功能。

2. 人造甜味剂
人造甜味剂也不是一个很好的选择,因为它们会导致代谢异常,生物失调或生态系统失衡。
人造甜味剂会改变肠道微生物组的组成和功能,从而促进葡萄糖不耐症,从而导致疾病。尽管它们似乎可以代替糖,但最好是避免使用人造甜味剂。
3. 转基因生物(GMO)食品
许多人都知道转基因生物不好,但并不知道为什么。这很大程度上与肠道微生物组有关。
对转基因食品进行了改良,使其能够在草甘膦等苛刻的杀虫剂中存活下来。本质上,当食用转基因食品时,是在增加这些化学药品的摄入量,这些化学药品会损害肠道。杀虫剂是专门为清除细菌而开发的,因此它们在肠道中起到一定作用也就不足为奇了。
4. 防腐剂
我们知道加工食品对我们的健康不利,它也会影响肠道微生物群。具体来说,防腐剂,比如聚山梨酯80和羧甲基纤维素(CMPF),它们是许多加工食品中常见的乳化剂,直接改变了肠道微生物群的组成。所以尽可能避免吃加工食品,以改善肠道健康。
5. 抗生素
抗生素会导致肠道微生物群发生很大变化,因为它们无法区分有益菌和致病菌。有些情况其实没必要使用抗生素。总的来说,在服用抗生素时,我们需要格外小心。
详细可见之前的文章 细菌的天敌抗生素,如何用好这把救命的双刃剑?
6. 非甾体抗炎药
非甾体抗炎药(NSAID),例如某些药物,会改变肠道微生物群的组成,并导致肠道渗漏。当胃肠道内壁在其连接处有缝隙时,会发生漏肠,这会使颗粒泄漏到血液中。这可能导致炎症,最终导致自身免疫性疾病。
当我们受到轻伤或不适时,我们会习惯性地拿出这些药物。在知道它们会损坏肠道的情况下, 是否值得用药需要慎重考虑。
7. 压力
肠道和大脑通过迷走神经不断交流-通过肠道激素信号传导,神经递质产生以及其他微生物代谢产物。压力会影响肠道微生物群,而肠道微生物组会影响压力,因此这是双向的。 也许你知道减轻压力对健康有益,但更具体地说,它对肠道健康有益。

8. 吸烟
有研究表明吸烟会改变肠道菌群的组成。研究人员发现,吸烟者的肠道菌群与肥胖者相似,并具有炎症性肠病等症状。
因此建议不吸烟。
1. 肠道微生物群检测
肠道健康在整体健康中起着关键作用。它会影响免疫系统,神经系统,皮肤和大脑。随着科学家对微生物群的了解越来越多,他们越认识到找到平衡是必不可少的。
每个人都有自己独特的肠道菌群,处于自己独特的平衡中。不同的食物将以不同的方式影响我们所有人。
也许令你感到不适的食物会使别人充满活力和健康,对你有效的药物对别人无效。因此,通过肠道微生物检测,可以更加准确了解自己的肠道健康状况并制定营养计划以满足自身需求。人们会越来越倾向于采用更加个性化的健康方法。
2. 出去接触大自然
在过去的几十年中,人们普遍从农村搬到城市。这意味着我们不再像以前那样接触大自然。平时生活里,我们不会经常在户外玩耍或在农场工作。因此,生活中遇到大量菌的机会减少了。
你可以在户外玩耍时有意地将自己暴露于多种微生物中,从而使菌群加以平衡。安排远足,海滩时间和自然之旅,以扩大对不同生态系统的接触,尤其是在旅行时!(当然疫情期间,还是建议大家尽量减少出行或者戴口罩去人少的地方)

3. 尝试间歇性禁食
间歇性禁食对肠道微生物组非常重要。虽然9到12个小时不吃东西可能听起来是很长一段时间,但如果在晚上9点左右停止进食并且直到早上9点才吃早餐,就可能已经很接近了。某些细菌在卡路里密集的环境中繁殖,而另一些细菌在卡路里缺乏的环境中繁殖。
因此,偶尔可以在晚餐和早餐之间适当延长些时间不吃东西——这样你的微生物组可能会感谢你。
4. 充足的睡眠
没有充足的睡眠是一件很糟糕的事,不良的睡眠也会影响肠道微生物组的组成。
每晚7~8个小时睡眠很重要,但保证睡眠质量也同样重要。高质量意味着你正在进入深度的恢复性睡眠阶段。如果你每天醒来时感到不安,或者每天下午很困,那么就值得考虑一下您的睡眠质量,因为它会影响健康的许多方面。
5. 运动适量
锻炼对肠道微生物组也非常有用。运动被证明可以丰富多样性,并增加有益菌。
可以尝试通过快步走,慢跑甚至是瑜伽来提高整体肠道健康。不过凡事需要有个度,别把自己当成是极限运动员那样疯狂运动,过量的运动可能会带来不利影响。

6. 尊重自然规律
许多肠道菌群都以24小时昼夜节律运行。遵守自然规律可以帮助一切顺利进行。我们知道轮班工作和时差会导致疾病,这可能就是原因之一。
7. 养宠物
研究发现,养宠物的孩子过敏和肥胖的几率反而较低。普遍认为这是因为动物身上的微生物会增加孩子的整体微生物多样性。这意味着你那些心爱的宠物也可能有助于保持肠道健康。听信谣言盲目抛弃宠物不可取。

8. 保持家庭微生物组健康
有些家庭非常爱干净,用洗碗机洗碗,经常对屋子全面消毒,包括孩子的玩具,衣服等等。在这样的环境下长大的孩子出去后反而更容易过敏。
在家里接触的微生物太少,菌群多样性下降。实际上孩子的生长发育过程中,在不断构建起自身免疫系统,需要有外界的微生物参与刺激体内的免疫细胞。过度清洁相当于干涉了免疫系统的正常构建,脆弱的免疫系统抵御不了外界的细菌,一出家门便容易生病。
9. 选择当季的有机蔬菜
当季的有机蔬菜不仅可以避免农药,而且还可以吸收土壤中的污物,产生有益的微生物。而且,其实用各种清洁剂洗那些蔬菜没有必要。
10. 有针对性的吃对自己有益的食物
每个人都是独特的,所以我们都有不同的营养需求,无法一劳永逸地解决所有饮食问题。有时候可以相信直觉,它知道哪些食物会帮助您,哪些食物现在对您有害。
没有普遍的健康饮食,可能普遍认为“健康”的食物实际上对您不健康。比如说建议您多吃蔬菜,蔬菜和坚果具有抗炎作用。但不一定适合你。
菠菜,麸皮,大黄,甜菜,坚果和坚果黄油都含有草酸盐。你体内如果没有那些能够将草酸盐代谢成无害物质的微生物,那么这些含草酸盐的食物对你来说就可能是有害的。
换句话说,像菠菜这样的“健康食品”实际上对这类人来说并不健康。同理,食品中的多酚通常被认为非常健康,但对某些人来说也不一定合适。
11. 如果可能,选择自然分娩
婴儿出生时会获得第一批微生物。当婴儿通过阴道出生时,其最初的微生物与母亲的阴道相似。通过剖腹产出生的婴儿,其微生物组中有来自母亲的皮肤微生物,这与以后生活中某些疾病的高风险相关。
当然有些危急时刻,剖腹产是必须的。这里要表达的是:阴道分娩对建立强大的健康微生物群很重要。当然,研究人员寻找能够抵消剖腹产负面影响的解决方案也很重要。

12. 母乳喂养超过配方
母乳喂养有助于婴儿的微生物群的构建,并奠定坚实的基础。有趣的是,母乳中含有人乳低聚糖,科学家最初认为这是没用的,因为婴儿无法消化它们。现在我们知道它们是专门用来滋养微生物的。
毫无疑问,肠道微生物群几乎在您健康的各个方面都可以发挥作用。这意味着我们必须开始照顾微生物群。
疫情面前首先要遵循国家预防COVID-19的指导方针:出门戴上口罩,避免聚众,勤洗手,避免触摸脸部,以减少感染病毒的机会。
同时,通过增强免疫系统从内部增强防御能力。而一个健康的肠道微生物组不仅能对冠状病毒等传染性病原体做出反应,而且还有助于防止潜在的具有危险性的过度免疫反应。
适当选择合适的措施,科学改善肠道健康。也许在防新冠的同时,也悄悄预防了某些慢性疾病的发生。
附录一: 粪便颜色和性状
你吃的食物会影响大便的颜色。例如,如果你正在吃很多深绿色的多叶蔬菜,则您的大便可能看起来有点绿色。根据你的便便,需要注意以下几点:
浅色便便
如果您发现浅色的,几乎是粘土或黄棕色的便便,则可能是粪便中脂肪浓度较高的迹象。这可能与胆囊,肝脏或胰腺的炎症有关。它也可能与胆汁释放不足有关,胆汁用于帮助饮食中的脂肪处理。
黑色便便/红色便便
假如你最近没有吃过几份黑甘草或蓝莓、黑莓或甜菜等深色食物,则可能表明肠道出血。这可能是由多种不同的原因引起的:直肠或肛门出血,胃壁肿胀,食物或消化系统中附着的异物或癌症迹象。尽管原因可能有所不同,但如果您发现粪便中有血迹,最好咨询医生。
通常,健康的排便应该呈现出自然的棕色阴影。但是,大便的形状也可能表示饮食或健康状况下降。
附录二: 粪便形状和大小
比较便便的最简单方法之一就是查阅“布里斯托尔粪便图表”,这是一种易于理解的图表,可帮助您直观地观察便便的变化。该图表已在全国各地的医生办公室使用,可以通过描绘您可能遇到的7种不同类型的粪便,可靠地帮助您评估消化过程中健康指标的普遍差距。
便秘(1-2型)
类型1:单独的硬块
如果您发现肠蠕动似乎很小,粪便呈硬块状,很可能是您患有严重的便秘。这可能是由于脱水或低纤维饮食引起的。许多不同的药物也会引起便秘,与医生讨论如何减轻便秘症状可能会有所帮助。
类型2:香肠状但块状
比严重的症状好一级,但很可能您仍在遭受轻度的便秘。大便比小肿块大是一个明显的指标,表明您已接近足够的水分,但仍有改善的空间。增加运动量,水分摄入和吃富含纤维的食物可能会改善粪便的硬度,并有助于调节肠蠕动。
普通(3-4型)
类型3:香肠形但有裂纹
定期排便的一个很好的例子,可以保持其形状并易于通过。这表明您的饮食相对稳定,消化过程相当正常。
类型4:像香肠或蛇
理想的例子:易于通过并保持其形态。最佳排便。
可能表明腹泻(5-7型)
类型5:柔和的斑点
随着大便开始软化,这可能是营养吸收不良,消化道中气体过多或大便中脂肪浓度升高的迹象。如果持续存在,也可能是胃肠道感染或胰腺炎的征兆。它本身可能并不重要,但是如果同时有任何其他症状,寻求专业医护人员可能会有所帮助。
类型6:蓬松的碎片
在这种情况下,这可能是食物不耐受或过敏的迹象。此外,如果您正受到胃部感染或在使用通便药的情况下,可能会遇到这种糊状的大便。
近期服用抗生素也可能导致大便稀疏。如果症状持续,请寻求专业帮助。
类型7:水状,无固体碎片
腹泻的经典症状会对您的健康造成严重影响,并增加您脱水的风险。这不是正常的大便,长时间的剧烈腹泻不只是令人不舒服,还可能是有害的迹象。
腹泻表明您的消化系统正在排斥某些东西,并且可能与您可能遇到的细菌或寄生虫或病毒或近期旅行相关。此外,长期症状可能提示其他疾病,例如肠易激综合症,克罗恩氏病或其他消化系统疾病。如果您长时间腹泻,请立即就医。
相关阅读:
【部分参考文献】
Kruis W, Forstmaier G, Scheurlen C, Stellaard F. Effect of diets low and high in refined sugars on gut transit, bile acid metabolism, and bacterial fermentation. Gut. 1991;32(4):367–371. doi:10.1136/gut.32.4.367
Suez J, Korem T, Zilberman-Schapira G, Segal E, Elinav E. Non-caloric artificial sweeteners and the microbiome: findings and challenges. Gut Microbes. 2015;6(2):149–155. doi:10.1080/19490976.2015.1017700
Aitbali Y, Ba-M’hamed S, Elhidar N, Nafis A, Soraa N, Bennis M. Glyphosate based- herbicide exposure affects gut microbiota, anxiety and depression-like behaviors in mice. Neurotoxicol Teratol. 2018;67:44–49. doi:10.1016/j.ntt.2018.04.002
Lange K, Buerger M, Stallmach A, Bruns T. Effects of Antibiotics on Gut Microbiota. Dig Dis. 2016;34(3):260–268. doi:10.1159/000443360
Lange K, Buerger M, Stallmach A, Bruns T. Effects of Antibiotics on Gut Microbiota. Dig Dis. 2016;34(3):260–268. doi:10.1159/000443360
Rogers MAM, Aronoff DM. The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin Microbiol Infect. 2016;22(2):178.e1–178.e9. doi:10.1016/j.cmi.2015.10.003
Foster JA, Rinaman L, Cryan JF. Stress & the gut-brain axis: Regulation by the microbiome. Neurobiol Stress. 2017;7:124–136. Published 2017 Mar 19. doi:10.1016/j.ynstr.2017.03.001
Savin Z, Kivity S, Yonath H, Yehuda S. Smoking and the intestinal microbiome. Arch Microbiol. 2018;200(5):677–684. doi:10.1007/s00203-018-1506-2
Tasnim N, Abulizi N, Pither J, Hart MM, Gibson DL. Linking the Gut Microbial Ecosystem with the Environment: Does Gut Health Depend on Where We Live?. Front Microbiol. 2017;8:1935. Published 2017 Oct 6. doi:10.3389/fmicb.2017.01935
Jantscher-Krenn E, Bode L. Human milk oligosaccharides and their potential benefits for the breast-fed neonate. Minerva Pediatr. 2012;64(1):83–99.
Pannaraj PS, Li F, Cerini C, et al. Association Between Breast Milk Bacterial Communities and Establishment and Development of the Infant Gut Microbiome. JAMA Pediatr. 2017;171(7):647–654. doi:10.1001/jamapediatrics.2017.0378
Voigt RM, Forsyth CB, Green SJ, Engen PA, Keshavarzian A. Circadian Rhythm and the Gut Microbiome. Int Rev Neurobiol. 2016;131:193–205. doi:10.1016/bs.irn.2016.07.002
Monda V, Villano I, Messina A, et al. Exercise Modifies the Gut Microbiota with Positive Health Effects. Oxid Med Cell Longev. 2017;2017:3831972. doi:10.1155/2017/3831972